/



























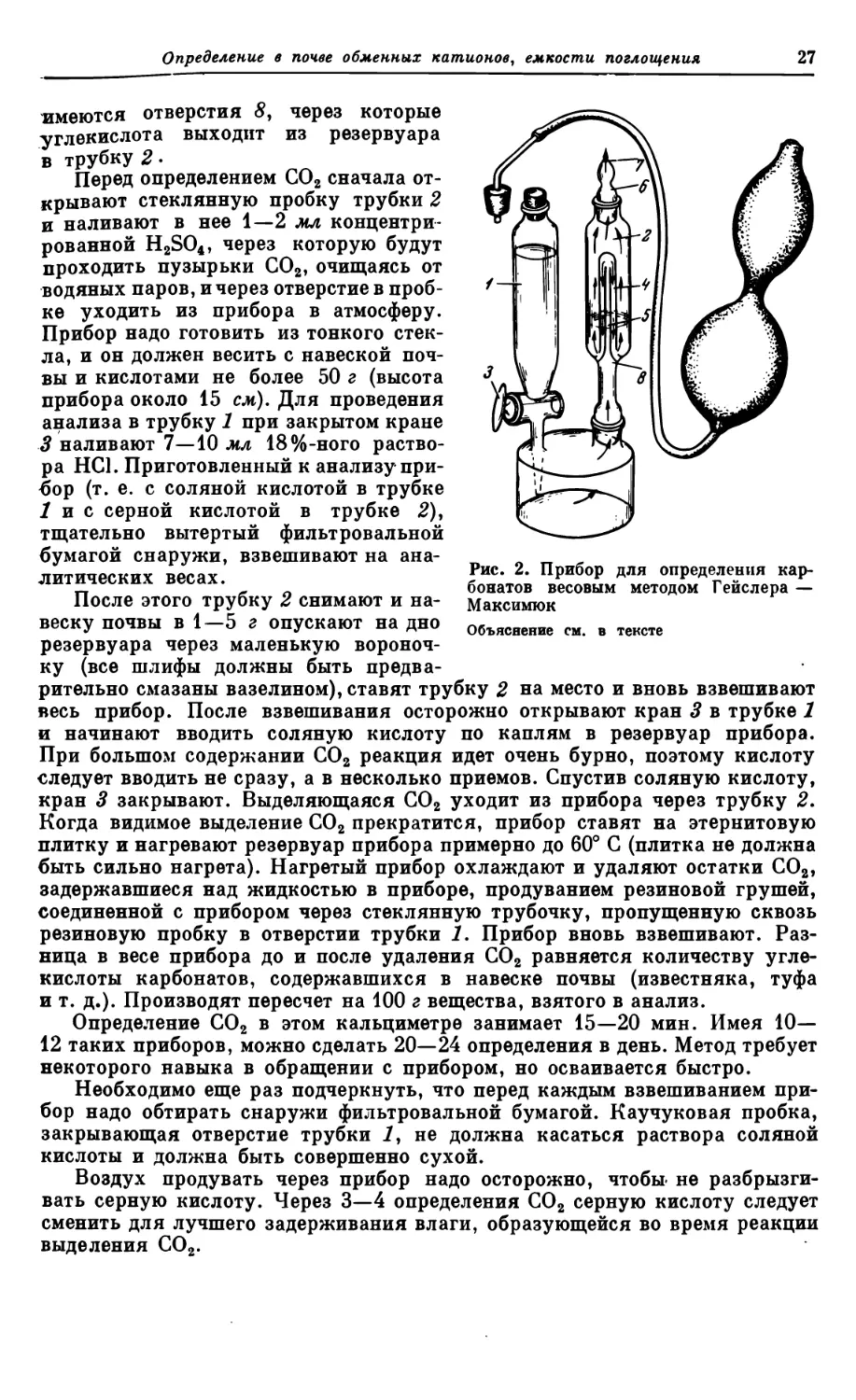

































































































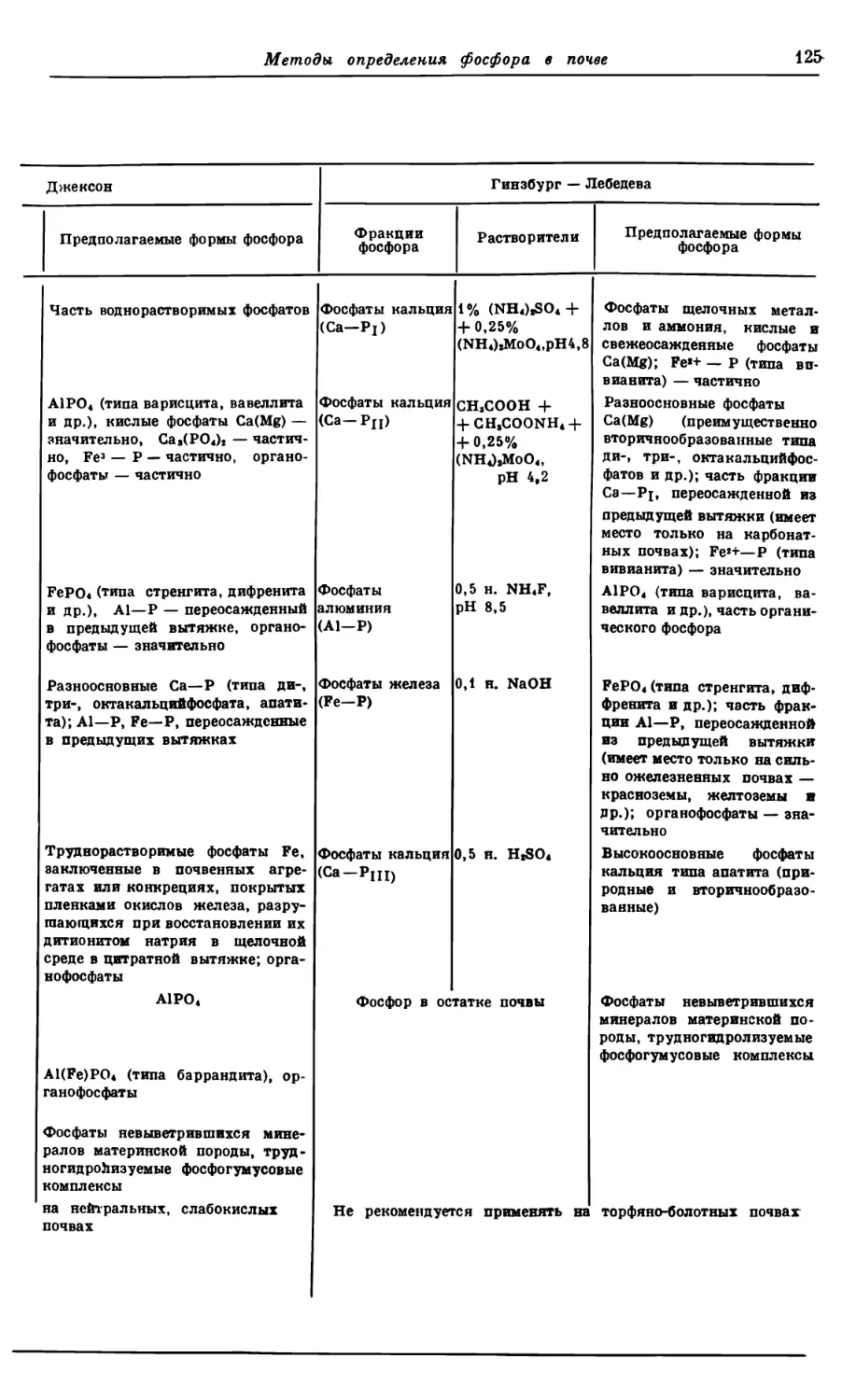






































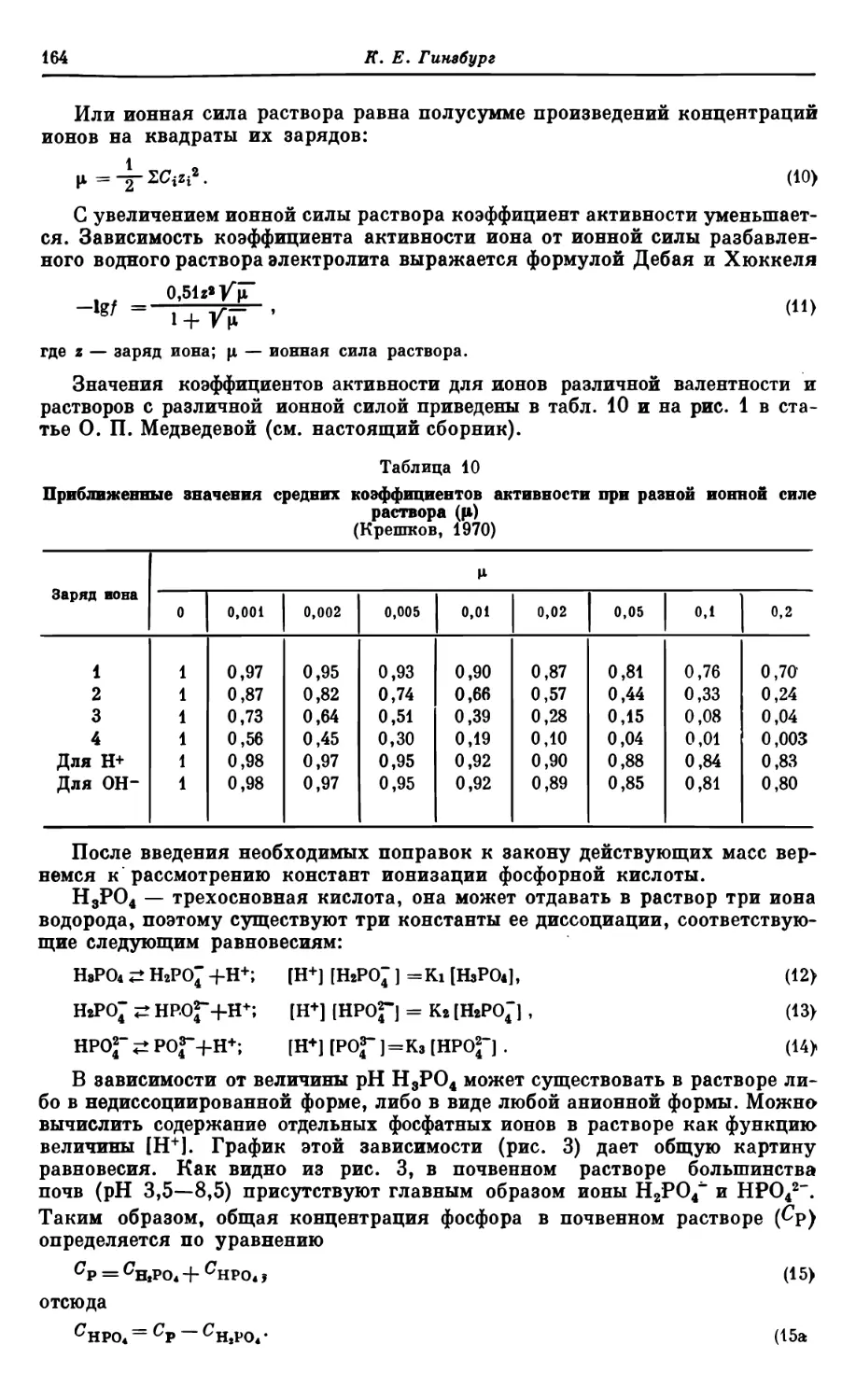






















































































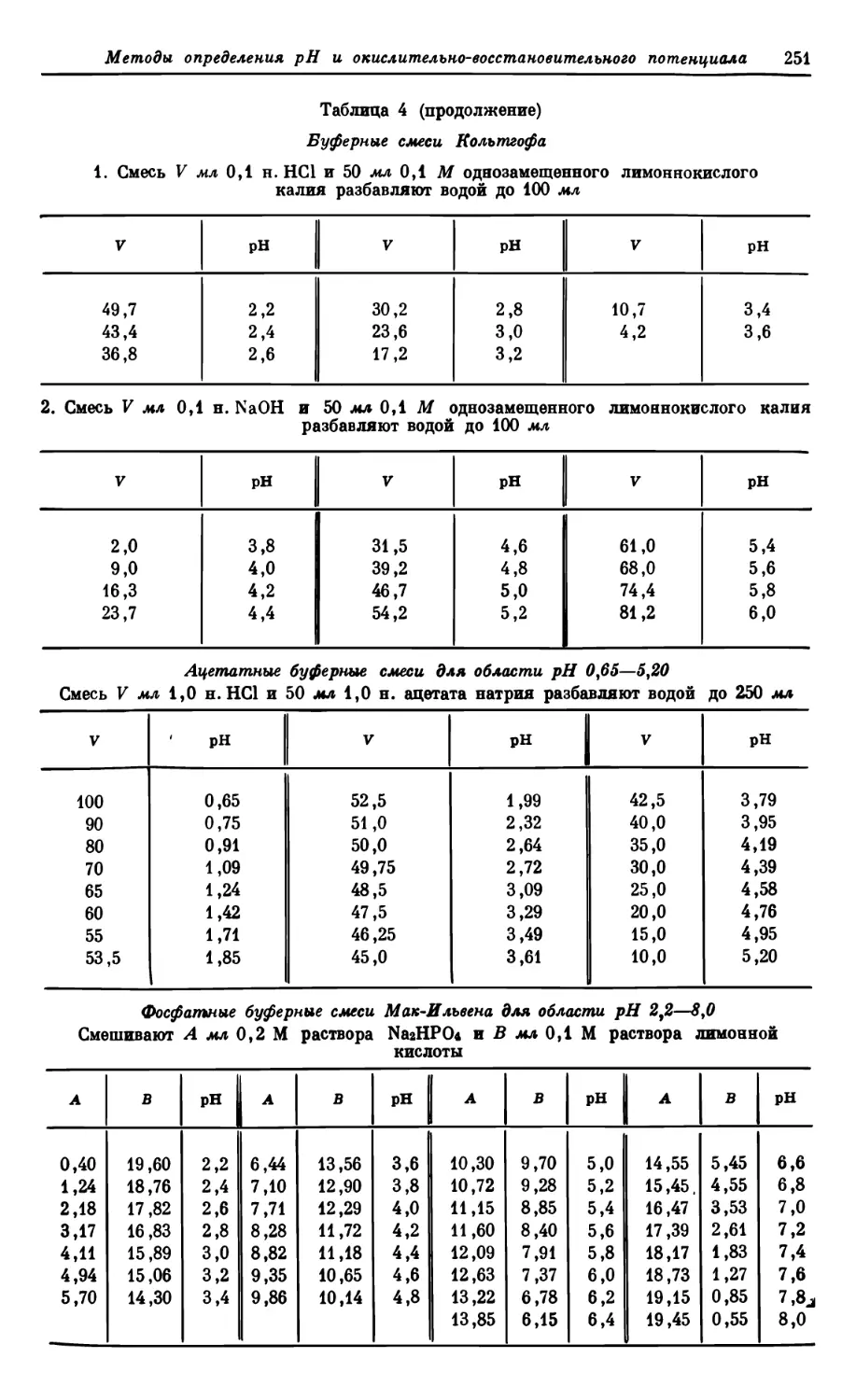




















































































































































































































































































































































































































Текст
АКАДЕМИЯ НАУК СССР
ОБЪЕДИНЕННЫЙ НАУЧНЫЙ СОВЕТ
«НАУЧНЫЕ ОСНОВЫ ХИМИЗАЦИИ СЕЛЬСКОГО ХОЗЯЙСТВА»
ВСЕСОЮЗНАЯ АКАДЕМИЯ
СЕЛЬСКОХОЗЯЙСТВЕННЫХ НАУК им. В. И. ЛЕНИНА
ПОЧВЕННЫЙ ИНСТИТУТ им. В. В. ДОКУЧАЕВА
АГРОХИМИЧЕСКИЕ
МЕТОДЫ
ИССЛЕДОВАНИЯ
ПОЧВ
Издание пятое,
дополненное и переработанное
в
ИЗДАТЕЛЬСТВО «НАУКА»
Москва 1975
УДК 631.8:631.4
Агрохимические методы исследования почв. М., «Наука»,
1975.
В работе описаны методы анализа почв, широко применяю¬
щиеся в системе агрохимического обслуживания сельского хо¬
зяйства^ а также оригинальные методы исследований, исполь¬
зуемые в отечественных и зарубежных научно-исследователь~
ских институтах.
Ответственный редактор
члеп-корреспондент АН СССР А. В. СОКОЛОВ
40306—338
A Q55 (02)—75 6*5—75 © Издательство «Наука», 1975 г.
ПРЕДИСЛОВИЕ
Пятое издание руководства «Агрохимические ме¬
тоды исследования почв» выходит в значительно
переработанном и дополненном виде.
Первое издание «Агрохимических методов» по¬
явилось еще в 1944 г. под редакцией академика
Д. Н. Прянишникова, Д. JI. Аскинази и А. В. Со¬
колова. Руководство было издано в двух выпусках.
В первом выпуске в основном приводилось описание
химических, лабораторных методов определения со¬
держания в почве питательных элементов. Кроме
того, в нем излагались методы определения свойств
почвы: pH, поглощенных оснований, карбонатов,
гумуса, засоленности почв и др. Уже тогда для
характеристики плодородия почв и степени обеспе¬
ченности их питательными для растений элементами
широко использовались биологические методы: по¬
левой, вегетационный, микробиологический, диагно¬
стика питания растений по их внешнему виду и ана¬
лизу. Описание всех этих методов и было дано во
втором выпуске руководства, опубликованном в
1947 г. Следующие издания были однотомные, но
иногда к ним дополнительно иэдавались отдельные
выпуски. Так, к третьему изданию (1960 г.) при¬
лагался составленный К. П. Магницким альбом
цветных рисунков по диагностике питания растений,
а в дополнение к четвертому изданию (1965 г.) была
выпущена «Методика полевых и вегетационных опы¬
тов с удобрениями и гербицидами» (1967 г.), в ко¬
торой были опубликованы биологические методы
определения плодородия почвы.
Содержание руководства перерабатывалось с
каждым изданием в связи с развитием агрономиче¬
ской химии и методики аналитической и экспери¬
ментальной работы по изучению плодородия почв
и обеспеченности их питательными для растений
веществами. Так, начиная с третьего издания в ру¬
ководство был включен раздел «Методика меченых
атомов».
4
Предисловие
В данном пятом издании введены новые разделы:
определение фосфатного и калийного потенциалов,
состава почвенного воздуха, фракционный анализ
гумуса, фосфора и азота и др. Существенно изме¬
нился поэтому и состав авторов пятого издания по
сравнению с предыдущим.
Для определения отзывчивости растений на
удобрения в настоящее время рекомендуются разно¬
образные методы, причем число их быстро увели¬
чивается. Из общего количества существующих ме¬
тодов в данное руководство включены, во-первых,
методы, применяющиеся в агрохимическом обслу¬
живании сельского хозяйства, и, во-вторых, методы
с оригинальным подходом к решению поставленной
задачи, в том числе еще не вошедшие широко в прак¬
тику работы агрохимических лабораторий.
Составительская и редакционная работа выпол¬
нена Д. М. Алексеевой и А. В. Соколовым. В под¬
готовке и редактировании издания принимали уча¬
стие К. В. Веригина и Д. С. Орлов.
3. Г. ИЛЬКОЛСКАЯ, А. С. КОНОВАЛОВА
ОПРЕДЕЛЕНИЕ В ПОЧВЕ ОБМЕННЫХ КАТИОНОВ,
ЕМКОСТИ ПОГЛОЩЕНИЯ, ГИПСА, КАРБОНАТОВ, СЕРЫ,
ВОДНОРАСТВОРИМЫХ ВЕЩЕСТВ И ПОДВИЖНЫХ СОЕДИНЕНИЙ
Подготовка почв для анализа. Образцы почв должны
доставляться с мест обследования в лабораторию для анализа в воздушно¬
сухом состоянии. Если эти образцы прибыли с поля недостаточно сухими*
их расстилают на столах и просушивают до воздушно-сухого состояния.
Для получения средней пробы образец почвы раскладывают равномер¬
ным слоем в виде прямоугольника и делят на четыре части по диагоналям.
Две противоположные части берут для анализа, а две другие откладывают
на хранение. Отобранную среднюю пробу почвы взвешивают и затем про¬
пускают через сито с отверстиями диаметром 1 мм1. Почву, которая не прой-
flet через сито, переносят в фарфоровую ступку и растирают деревянным
пестиком с резиновым наконечником. Растираются только почвенные комоч¬
ки. Остающиеся камни, крупный и мелкий хрящ собирают вместе, взвеши¬
вают и вычисляют процентное содержание хряща в данном образце.
Просеянную через сито почву тщательно перемешивают и складывают
в коробку, на которую наклеивается этикетка с указанием номера разреза,
глубины взятия образца, фамилии почвоведа, года и места взятия образца.
Такая же этикетка кладется в коробку с почвой.
Определение гигроскопической воды. В тариро¬
ванном бюксе отвешивают с точностью до 0,001 г около 5 г воздушно-сухой
почвы и сушат в термостате при температуре 105° С в продолжение 5 час.
Бюкс с высушенной почвой взвешивают. По потере в весе вычисляют содер¬
жание воды во взятой навеске почвы, что позволяет определить процент
воды в исследуемом образце. Для этого при навеске ровно в 5 г достаточно
полученную потерю в весе бюкса с почвой помножить на 20.
При массовых анализах удобно пользоваться коэффициентом перевода
данных анализа воздушно-сухой почвы на почву, высушенную при 105° С:
Коэффициент этот равен отношению 100 к 100 минус процент влаги в данной
почве. Например, если воздушно-сухая почва содержит 5% влаги, то по¬
лучают следующий коэффициент:
100 100
100-5 “ 95 - 1*052-
ОПРЕДЕЛЕНИЕ ОБМЕННЫХ КАТИОНОВ
Вытеснение обменных катионов
раствором уксуснокислого аммония
Из различных испытанных методов вытеснения обменных катионов ме¬
тод вытеснения их раствором уксуснокислого аммония (предложен Шоллен-
бергером) является одним из лучших. Преимущество его заключается
в том, что уксуснокислый аммоний легко разрушается в процессе выпари¬
вания фильтрата после промывания им почвы, а при прокаливании сухого
1 Вместо ручной подготовки почвы применяют также размельченпе почв на специальных
растирочных машинах.
3. Г. Илъковская, А, С. Коновалова
остатка полностью удаляется. Таким образом, для осаждения кальция и
магния получается раствор, не содержащий избытка лишних солей, как
это имеет место при вытеснении обменных катионов хлористым аммонием
(или хлористым натрием). Метод позволяет в одном фильтрате определить
кальций, магний, калий и натрий.
5—10 г почвы промывают декантацией 1,0 н. раствором уксуснокислого
аммония при pH около 6,5 до прекращения реакции на кальций. Фильтрат
выпаривают в стакане на электрической плитке до 50—60 мл. Органическое
вещество в процессе выпаривания разрушают 3%-ной перекисью водорода
и 10%-ным раствором азотной кислоты. В полученном растворе осаждают
полуторные окислы аммиаком в присутствии метилрота. Для этого при¬
бавляют 10%-ный раствор аммиака по каплям до перехода красной окраски
раствора в желтую. Затем раствор в стакане доводят до кипения и ставят
стакан на теплую плитку. Если в растворе окажутся хлопья полуторных
окислов, их отфильтровывают через рыхлый фильтр, осадок на фильтре
промывают несколько раз 1%-ным раствором азотнокислого аммония (с 1—
2 каплями аммиака до щелочной реакции по метил роту). Полученный фильт¬
рат идет на определение в нем кальция и магния.
Определение кальция. Фильтрат подкисляют 5%-ным ра¬
створом уксусной кислоты до явно кислой реакции и кипящим раствором
щавелевокислого аммония осаждают кальций: 10 дм насыщенного (4%-ного)
раствора щавелевокислого аммония по каплям прибавляют в кипящий
раствор (фильтрат) и оставляют в теплом месте на 4 часа для укрупнения
осадка. Держать осажденный кальций более 4 час. не рекомендуется во из¬
бежание оседания вместе с ним и магния. Осадок щавелевокислого кальция
отфильтровывают через фильтр (синяя лента) и промывают теплой водой
до потери реакции на ион щавелевой кислоты (проба раствором азотнокисло¬
го серебра). Промытый осадок растворяют на фильтре в 5%-ной горячей
серной кислоте и титруют нагретый до 80° С раствор 0,05 н. раствором мар¬
ганцовокислого калия до появления не исчезающей в течение нескольких
минут розовой окраски. Бросают в стакан фильтр, через который отфильтро-
вался щавелевокислый кальций, и если раствор обесцветится, снова тит¬
руют марганцовокислым калием.
1 мл точно 0,05 н. раствора КМп04 отвечает 0,05 мг-экв кальция. Чтобы
вычислить содержание обменного кальция в миллиграмм-эквивалентах
на 100 г почвы, количество миллилитров раствора КМп04, пошедшее на тит¬
рование кальция, умножают на 0,05 и на 100 и делят на навеску почвы.
Определение магния. Для определения содержания магния
фильтрат и промывные воды, полученные для выделения из раствора каль¬
ция, выпаривают до объема 200—250 мл, нейтрализуют 10%-ным раствором
аммиака. Затем подкисляют по метилроту 10%-ным раствором НС1, при¬
бавляют 20 мл 10%-ного раствора Na2HP04, доводя до кипения, нейтрали¬
зуют 10%-ным раствором аммиака и прибавляют при энергичном помешива¬
нии палочкой 20—30 мл крепкого аммиака. Раствор оставляют стоять на
18—36 час. Фильтрование производят через синюю ленту (фильтр для уп¬
лотнения следует предварительно промыть кипящей дистиллированной
водой). Осадок промывают 2,5%-ным раствором аммиака до потери реакции
на хлор (проба с азотнокислым серебром, подкисленным азотной кислотой).
Фильтр с осадком помещают в предварительно взвешенный фарфоровый
тигель, смачивают несколькими каплями азотной кислоты (чтобы получить
х>садок белого цвета) и ставят в холодную муфельную печь, установленную
в вытяжном шкафу. Прокаливание ведут при температуре 850—900° С до
постоянного веса. Чтобы вычислить содержание обменного магния в мил-
лиграмм-эквивалентах на 100 г почвы, полученный вес Mg2P207 умножа¬
ют на 0,218, получают вес магния во взятой для анализа навеске почвы и
пересчитывают на 100 г почвы, высушенной при 105° С; умножением про¬
Определение в почве обменных катионов, емкости поглощения
7
цента магния на 1000 и делением на 12,16 (эквивалентный вес магния) полу¬
чают обменный магний в миллиграмм-эквивалентах на 100 г почвы.
Калий и натрий определяют спектральным пламенно-фотометрическим
методом, при котором не требуется специальной подготовки раствора.
Трилонометрический метод
Вытеснение обменных кальция и магния ук¬
суснокислым аммонием. Метод основан на свойстве трилона Б
(двунатриевой соли этилендиаминтетрауксусной кислоты) давать устойчи¬
вые комплексные соединения с ионами двухвалентных металлов, в том числе
с кальцием и магнием.
Трилонометрическое титрование следует проводить в условиях невысо¬
ких концентраций солей. Поэтому после вытеснения обменных катионов 1 н-
раствором уксуснокислого аммония разрушают уксуснокислый аммонии
выпариванием раствора, затем прокаливают полученный остаток на кол-
бонагревателе или в муфеле при 400—600° С, причем получают кальций
и магний в форме карбонатов или окислов. Органическое вещество при этом
сгорает. Полученный осадок растворяют 10%-ной соляной кислотой и*
убедившись в его полном растворении (не видно кристаллов на дне чашки),
солянокислый раствор разбавляют горячей водой, фильтруют в мерную
колбу емкостью на 200 мл и доводят до метки водой.
Уксуснокислый аммоний вытесняет из почвы небольшое количество по-
лутораокисей, поэтому во многих случаях их не приходится выделять иа
раствора. Высокие концентрации железа мешают титрованию трилоном —
переход окраски теряет четкость; кроме того, можно получить несколько
завышенные данные. В таких случаях рекомендуют добавочное разбавле¬
ние раствора водой для уменьшения концентрации железа либо выделение
железа, если его очень много по отношению к кальцию и магнию. Это лучше
сделать перед подготовкой раствора к выпариванию — выделить полуто-
раокиси обычным способом с аммиаком, а затем довести выпаривание до
конца и прокалить осадок.
Вредное влияние марганца уничтожается прибавлением солянокислого
гидроксиламина (1—2 мл 5%-ного раствора), который препятствует обра¬
зованию перекиси марганца, мешающей титрованию. Необходимо также
устранить вредное действие меди. Все реактивы для этой цели готовятся
на дистиллированной воде, не содержащей меди. Для связывания меди и
других тяжелых металлов прибавляют несколько кристалликов диэтил-
дитиокарбамата натрия.
При титровании трилоном Б определяют сумму кальция и магния (тит¬
рование в присутствии индикатора хромогена черного) и кальций (титрова¬
ние в присутствии индикатора мурексида), магний определяют по разности.
Присутствующий в вытяжке марганец титруется вместе с кальцием и маг¬
нием и несколько завышает результаты для магния. Поскольку для боль¬
шинства почв содержание марганца по сравнению с магнием мало, им пре¬
небрегают.
Для определения суммы кальция и магния часть анализируемого раство¬
ра, содержащего обменные катионы (удобно брать 50 ли), помещают в ко¬
ническую колбу емкостью 250 ли, разбавляют водой примерно до 100 мл*
Раствор подогревают до 60—70° С, прибавляют для создания щелочной
реакции 5 мл аммиачного буферного раствора, несколько кристалликов
диэтилдитиокарбамата натрия, 1—2 мл 50%-ного раствора солянокислого
гидроксиламина, 10—15 мг индикатора хромогена черного, растертого
с хлористым натрием, и титруют 0,05 н. или 0,01 н. раствором трилона Б
при энергичном помешивании до перехода окраски раствора от вишнево¬
красной через фиолетово-синюю в чисто-голубую в точке эквивалентности.
8
5. Г. Ильковская, А. С. Коновалова
При прибавлении избытка трилона окраска не меняется. Поэтому реко¬
мендуется проводить титрование, сравнивая окраску раствора со «свиде¬
телем»—заведомо перетитрованной пробой. Сумма кальция и магния (в мг-экв
на 100 г почвы) находится по уравнению
r. + Mg- л.о,о5-лмоо
с *
где А — количество трилона, пошедшее на титрование кальция и магния, мл; 0,05 —
нормальность раствора трилона; К — поправка к титру трилона; С — навеска почвы,
соответствующая количеству раствора, взятого на титрование кальция и магния, г; 100 —
коэффициент для пересчета результатов анализа на 100 г почвы.
Определение кальция трилонометрическим методом можно про¬
изводить в присутствии индикатора мурексида
(аммонийная соль одноосновной пурпуровой кислоты). С ионами кальция
анион пурпуровой кислоты в щелочной среде образует комплекс, окрашен¬
ный в красный цвет. Этот комплекс менее стоек, чем соединение кальция с
трилоном, и при титровании происходит резкое изменение окраски от
красной к лиловой в точке эквивалентности. Вредное влияние меди и мар¬
ганца уничтожают так же, как и при титровании суммы кальция и магния.
Ход анализа. Определенный объем раствора помещают в коническую
колбу емкостью 250 мл, разбавляют раствор водой примерно до 100 мл.
Для предупреждения соосаждения кальция с магнием при прямом опреде¬
лении кальция с мурексидом в пробу предварительно (до добавления NaOH)
вводят 2 мл 0,5 н. раствора Na2C03. При этом кальций выпадает в осадок
в виде СаС03, образуя отдельную фазу, которая в ходе последующего тит¬
рования растворяется. Благодаря этому исключается возможность соосаж¬
дения кальция с Mg(OH)2 и обеспечивается полнота определения кальция.
Прибавляют 2 мл 2 н. раствора NaOH, несколько кристалликов диэтилди-
тиокарбамата натрия, 1—2 мл 5 %-ного раствора солянокислого гидроксил-
амина, затем 10—15 мг индикатора мурексида и титруют 0,05 н. или 0,01 н.
раствором трилона Б при интенсивном помешивании до перехода ярко¬
пурпурной окраски раствора в лиловую. В дальнейшем от прибавления три¬
лона окраска не меняется, поэтому титрование лучше вести в присутствии
«свидетеля»— заведомо перетитрованной пробы.
Содержание кальция (в мг-экв на 100 г почвы) рассчитывается по урав¬
нению
А- 0,05-А". 100
Са = 71 ,
где А — количество трилона Б, пошедшее на титрование кальция, мл; 0,05 — нормаль¬
ность раствора трилона; К — поправка к титру трилона; С — навеска почвы, соответ¬
ствующая количеству раствора, взятого на титрование кальция, г; 100 — коэффициент
для пересчета результатов анализа на 100 г почвы.
Из суммы кальция и магния на 100 г почвы вычитают количество каль¬
ция и получают количество магния (в мг-экв на 100 г почвы).
Полученные данные по содержанию обменного кальция и магния пересчи¬
тывают на 100 г почвы, высушенной при 105° С.
Реактивы. 1. Раствор трилона Б. Для приготовления 0,05 н. раствора 9,3 г
трилона растворяют в 1 л дистиллированной воды. 0,01 н. раствор готовится разведением
0,05 н. раствора. Титр раствора трилона устанавливают по раствору сернокислого магния,
приготовленного из фиксанала.
Для проверки титра трилона 20 мл приготовленного раствора сернокислого магния
переносят пипеткой в коническую колбу емкостью 250 мл, прибавляют 100 мл дистилли¬
рованной воды, 5 мл аммиачного буфера, 10—15 мг хромогена черного и титруют раство¬
ром трилона Б до перехода окраски раствора из вишнево-красной в голубую.
Определение в почве обменных катионов, емкости поглощения
94
2. Буферный раствор. 70 г NH4C1 растворяют в дистиллированной воде, добавляют
570 мл 25%-ного раствора NH4OH и доводят объем до 1 л.
3. Индикаторы хромоген черный и мурексид. Растирают в ступке 5 г индикатора
с 95 г NaCl или КС1 до^ равномерно окрашенного состояния. Индикаторы хранят в темной
банке с притертой пробкой.
4. 5%-ный водный раствор солянокислого гидроксил амина.
Вытеснение обменных кальция и магния хло¬
ристым натрием. Вытеснение обменных катионов 1,0 н. раствором
хлористого натрия дает возможность определить кальций и магний три-
лонометрическим методом без специальной подготовки раствора.
5 г почвы промывают декантацией 1,0 н. раствором хлористого нат¬
рия (pH 6,5) в колбу емкостью 500 мл до потери реакции на кальций. Колбу
доливают до метки дистиллированной водой и перемешивают. Две пробы
по 50 мл помещают в конические колбы на 250 мл, разбавляют водой до
100 мл и титруют трилоном Б сумму кальция и магния с индикатором хро¬
могеном черным и кальций с индикатором мурексидом, как описано выше.
По разности между суммой кальция и магния и кальцием находят магний.
Результаты выражают в миллиграмм-эквивалентах на 100 г почвы.
Определение обменного водорода методом Гедройца
Перед определением обменного водорода в почвах, не насыщенных ос¬
нованиями, необходимо определить pH этих почв (см. ниже). Анализировать
следует только почвы, у которых pH ниже 5,5. Для проведения анализов
на содержание обменного водорода необходимо иметь комнату с чистым
от паров кислот и аммиака воздухом. Вся посуда должна быть свежевымыта
и высушена. Стаканчики для навесок, фильтры с воронками и конические
колбы для фильтрата ополаскивают раствором, приготовленным для вытесне¬
ния иона водорода.
Обменный водород вытесняют из почвы 1,0 н. раствором ВаС12 при pH
около 6,5. Если продажный реактив имеет более кислую реакцию, то 1—2
каплями баритовой воды устанавливают нужное значение pH. Если реактив
щелочнее, то раствор подкисляют до требуемого pH 1—2 каплями 10%-ного
раствора соляной кислоты. Раствор хлористого бария рекомендуется при¬
готовить сразу в количестве 10—20 л. На 1 л воды берут 122 г ВаС12*2Н20
(чистый реактив для анализа).
1—10 г почвы (в зависимости от ее кислотности) обрабатывают в стакан¬
чике на 100 мл раствором хлористого бария путем декантации и фильтруют.
Фильтрат собирают в коническую колбу на 500 мл. Когда наберется 300—
400 мл фильтрата, приступают к титрованию фильтрата. Не следует остав¬
лять фильтрат на ночь неоттитрованным. Весь фильтрат титруют 0,02 н.
раствором едкого натра в присутствии 10—15 капель бромтимолблау до
появления синей окраски. Этот оттенок быстро исчезает. Для контроля при¬
бавляют 1—2 капли раствора едкого натра, и, когда вновь появляется си¬
ний оттенок,- титрование считают оконченным. Если на титрование фильтра¬
та пошло более 1 мл 0,02 н. раствора едкого натра, почву на фильтре начи¬
нают промывать дальше тем же раствором и в ту же колбу, вылив из нее
оттитрованный раствор и сполоснув ее 2 раза раствором хлористого бария.
Когда фильтрата наберется 300—400мл, его вновь титруют таким же об¬
разом. Так поступают до тех пор, пока на титрование пойдет не больше
1 мл 0,02 н. раствора едкого натра. Количество 0,02 н. раствора едкого нат¬
ра, пошедшее на титрование отдельных порций фильтрата, суммируют*
Пример. Предположим, что на 5 г почвы при первом титровании фильтрата пошло^
10 мл раствора едкого натра, при втором — 3 и при третьем — 0,8 мл. Всего на титрова¬
ние взятой в анализ навески почвы пошло 13,8 мл раствора едкого натра.
40
3. Г. Илъковская, Л. С. Коновалова
Пусть поправка к титру 0,02 н. раствора едкого натра равна 1,008; количество об¬
менного водорода (в %) будет составлять 13,8-1,008-0,00002*20 АТ, где К равно перевод¬
ному коэффициенту на почву, высушенную при 105° С.
Полученное число умножают на 1000 и получают количество миллиграмм-экви¬
валентов обменного водорода на 100 г почвы (эквивалентный вес водорода равен еди¬
нице).
Приготовление бромтимолблау. 0,1 г индикатора в порошке рас¬
тирают в агатовой ступке с 3,2 мл 0,05 н. раствора едкого натра до полного растворения
порошка. Пипеткой добавляют 10—15 мл воды, перемешивают, переносят в мерную кол¬
бу на 250 мл и доводят водой до метки. На титрование берут 10—15 капель индикатора.
Определение обменного натрия методом Гедройца
Метод основан на вытеснении обменного натрия из почвы углекислым
•кальцием при добавлении в почву СаС03 и воды, насыщенной углекис¬
лотой.
5—20 г почвы помещают в литровую колбу, прибавляют химически чисто¬
го мела в количестве 10% от веса взятой навески почвы, заливают 1 л воды
и пропускают ток углекислоты в продолжение 3 час., встряхивая колбу
с жидкостью каждые 15 мин. Для пропускания тока углекислоты через
10—12 колб удобно пользоваться установкой из 10—12 литровых колб,
соединенных между собой в две непрерывные цепи по 6 колб в каждой.
Ток углекислоты проходит по развилке в обе цепи. По длинной трубке ток
углекислоты идет до дна колбы, проходит через жидкость и по короткой
трубке поступает в соседнюю колбу и т. д. Через 3 часа прекращают про¬
пускать углекислоту. Выключать ток углекислоты следует после того,
когда колбы будут разделены все по одной, начиная с конца. Это необходимо
для того, чтобы жидкость не перебросилась из одной колбы в другую. Жид¬
кость быстро фильтруют через складчатый фильтр, фильтрат получается
совершенно прозрачный. Натрий определяют пламенно-фотометрическим
методом. В случае отсутствия бомбы с углекислотой рекомендуется вытес¬
нять обменный натрий 1%-ным раствором углекислого аммония. Навеска
почвы взбалтывается 5 мин. с 1 л 1%-ного раствора углекислого аммония
я настаивается в течение ночи, при этом обменный натрий переходит в ра¬
створ. В фильтрате натрий также определяют пламенно-фотометрическим
методом.
Обменные катионы в карбонатных почвах
Метод Шмука
Определение обменных кальция и магния в карбонатных почвах затруд¬
няется тем, что при взаимодействии их с солевыми растворами вместе с
вытеснением обменных оснований растворяется значительное количество
углекислых солей кальция и магния. Это требует применения особых мето¬
дов. Среди них наиболее широко распространен метод Шмука. Однако и он
является приближенным.
Принцип метода А. А. Шмука заключается в том, что если навеску
карбонатной почвы обработать солевым раствором в количестве, достаточном
для вытеснения всего обменного кальция, то при удвоении количества со¬
левого раствора на ту же навеску почвы последний растворит вдвое больше
углекислого кальция. Отсюда обменный кальций может быть вычислен
яю формуле
Са = 2А — В,
где А —содержание кальция в первом]растворе; В — то же, во втором растворе.
Определение в почве обменных катионов, емкости поглощения
11‘
Одну навеску почвы в 1 г помещают в колбочку на 100 мл, другую наве¬
ску, также в 1 г,— в колбочку на 200 мл. В обе колбочки прибавляют йо‘
0,2 г химически чистого мела (избыток сверх того количества, которое мо¬
жет раствориться в данном объеме и при данной температуре). Колбочки'
на три четверти заливают 1,0 н. раствором хлористого натрия (pH около
6,5 по бромтимолблау), взбалтывают, доводят до метки тем же раствором
и оставляют на ночь. На другой день суспензию отфильтровывают и в части
фильтрата (например, из 100-миллилитровой колбочки — по 50 мл, а из
200-миллилитровой — по 100 мл) определяют кальций и магний.
В 100 мл солевого фильтрата содержатся поглощенные основания 1 г поч¬
вы и растворенный карбонат кальция. В 200 мл солевого фильтрата содер¬
жатся те же количества поглощенных оснований и двойное количество ра¬
створенных карбонатов. Если результаты анализа кальция в 100 мл фильтра1
та удвоить, согласно формуле, и вычесть из полученных данных результаты
анализа кальция в 200 л фильтрата, то разность покажет количество об¬
менного кальция в данной навеске почвы. Затем вычисляют содержание’
обменного кальция в процентах и миллиграмм-эквивалентах.
По этой же формуле можно высчитать и содержание обменного магния*
В Средней Азии при определении обменных кальция и магния принята
навеска почвы в 5 г. Опускается также внесение мела, так как почвы Сред¬
ней Азии имеют высокое содержание карбонатов.
При определении обменных кальция и магния по методу Шмука удобна
пользоваться трилонометрическим методом, так как хлористый натрий
не мешает титрованию суммы кальция и магния с индикатором хромогеном
черным, а также титрованию кальция с индикатором мурексидом. Полуто-
раокисей при этом выделять не надо. Высокая концентрация солей хлори¬
стого натрия уничтожается разбавлением водой. В анализ берут 25 и 50 дел
раствора, прибавляют по 100 мл дистиллированной воды. В дальнейшем!
производят титрование 0,01 н. раствором трилона Б, как указано выше,
без всяких изменений. Сначала получают сумму кальция и магния в первой
и второй колбах. Затем титруют кальций в первой и второй колбах. Вы¬
читанием кальция из суммы кальция и магния получают магний в первой
и второй колбах. Применив формулу Шмука, получают кальций и магний
и рассчитывают их содержание на 100 г почвы, высушенной при 105° С.,
Метод Тюрина
Метод Тюрина основан на вытеснении из карбонатных почв обменных
кальция и магния раствором хлористого натрия и учете растворившегося
СаС03 титрованием общей щелочности. Метод не применим для почв, содер¬
жащих карбонат магния.
5 г почвы взбалтывают 5 мин. с 500 мл 1,0 н. раствора хлористого натрия
и оставляют на ночь. На следующий день раствор отфильтровывают и в
100 мл фильтрата определяют НСОз титрованием 0,02 н. раствором соля-
ной кислоты в присутствии метилоранжа.
Общую щелочность выражают в миллиграмм-эквивалентах на 100 г поч¬
вы. В аликвотной части фильтрата трилонометрическим методом определяют
содержание кальция (как описано выше). Общее содержание кальция также
выражают в миллиграмм-эквивалентах на 100 г почвы. Из обкцего содержа¬
ния кальция вычитают общую щелочность, выраженную в тех же величинах»
Разность выражает количество обменного кальция в мг-экв/iO0 г почвьи
Для определения обменного магния в аликвотной части вытяжки три¬
лонометрическим методом определяют сумму кальция и магния (титрование»
трилоном Б в присутствии хромогена черного). Выражают полученную ве¬
личину в миллиграмм-эквивалентах на 100 г почвы и вычитают из нее общее.*
содержание кальция (кальций обменный кальций карбонатный).
12
3. Г. Ильковская, А. С. Коновалова
Реакцию на присутствие в почве углекислого магния проводят так:
около 5 г почвы взбалтывают с 10—15 мл 1,0 н. раствора хлористого натрия,
к фильтрату прибавляют 2 капли фенолфталеина; слабое порозовение или
отсутствие окрашивания укажут на отсутствие в почве MgC03, заметное
порозовение укажет на присутствие в почве MgC03.
Метод Мелиха
Навеску почвы, отвечающую 0,5—1 мг-экв обменных катионов, поме¬
щают в стеклянный тигель с пористым дном, покрытым маленьким бумаж¬
ным фильтром. Тигель вставляют в колбу для отсасывания. Почву промы¬
вают при отсасывании 50 мл 0,2 н. раствора ВаС12 триэтаноламин с pH
8,1 и затем 50 мл дистиллированной воды. Фильтрат доводят до объема
100 мл.
Определение обменного кальция. Для определения
обменного кальция берут 25 мл фильтрата, прибавляют 25 мл 0,1 н. раство¬
ра H2S04 и 1 каплю метилоранжа. Затем прибавляют 20%-ный раствор
уксуснокислого натрия до тех пор, пока не исчезнет розово-оранжевая
окраска. Смесь нагревают до 70° С и осторожно прибавляют при помешива¬
нии 2 раза по 5 дед 4%-ного раствора щавелевокислого аммония. После 1-ча¬
сового стояния осадок, представляющий собой смесь BaS04 и СаС204, от¬
фильтровывают и промывают горячей водой. Затем из осадка растворяют
СаС204 в 50 дел 1’%-ного раствора H2S04, раствор нагревают до 80—90° С
и титруют 0,025 н. КМп04.
Определение обменного магния. Магний определяют
в форме пирофосфата магния после осаждения кальция щавелевокислым
аммонием, как это описано выше.
Реактивы. 1. 0,2 н. раствор ВаС12 + триэтаноламин с pH 8,1. 25 мл триэтанол-
амина уд. веса 1,126 (около 8 н.) доливают водой до[250 мл и нейтрализуют соляной кисло,
той до pH 8,1. Для этого требуется приблизительно 90 мл 1 н. НС1. Раствор доливают
водой до 500 мл и смешивают с 500 мл 0,4 н. раствора ВаС12. Раствор предохраняют от СО*
воздуха.
2. 0,4 н. раствор ВаС1а« 50 г ВаС1а*2Н20 растворяют в дистиллированной воде»
объем доводят до 1 л•
Обменные кальций и магний
в произвесткованных почвах по Айциняну
(Айдинян, Иванова, Соловьева, 1973)
Для вытеснения обменных оснований почву обрабатывают 0,02 н. раство¬
ром (NH4)2S04 в смеси с ацетоном в объемном соотношении 55 : 45. Вытес¬
ненный из обменного состояния кальций, реагируя с сульфатом, выпадает
в форме нерастворимого в водно-ацетоновой среде осадка CaS04. Обмен¬
ный кальций определяется по разности концентрации сульфатов в раство¬
ре вытеснителя до и после взаимодействия его с почвой путем титрования
0,02 н. раствором ВаС12 в присутствии металлоиндикатора нитхромазо.
Образовавшиеся в результате обменных реакций сульфаты магния,
алюминия и одновалентных катионов в присутствии 45'%-ного раствора
ацетона полностью растворимы. Обменный магний определяется прямым
титрованием в присутствии индикатора хромогена черного.
Подготовка почвы к анализу и приготовление
вытяжки. После тщательного перемешивания воздушно-сухой почвы,
пропущенной через сито в 1 мм, отбирают среднюю пробу. 3 г почвы или
1 г торфа и торфяно-луговой почвы помещают в колбу объемом 200 мл. Ту¬
да же приливают отмеренные пипеткой с грушей 100 мл титрованного 0,02 н.
Определение в почве обменных катионов, емкости поглощения
13
водно-ацетонового раствора (NH4)2S04, приготовленного со строгим соблю¬
дением соотношения объемов воды и ацетона 55 : 45. Колбы плотно закры¬
вают притертыми стеклянными пробками (при отсутствии стеклянных про¬
бок можно применить резиновые). Содержимое колб взбалтывают в тече¬
ние одного часа на ротаторе. Это можно заменить настаиванием в течение
18 час. с предварительным энергичным взбалтыванием вручную в течение
5—7 сек. По истечении указанного срока суспензию фильтруют через плот¬
ный складчатый фильтр диаметром 11 см (воронка диаметром 7 см) в колбу
на 100 мл. Каждый раз перед наливанием очередной порции суспецзии на
фильтр производят ее взбалтывание. Первую порцию фильтрата объемом
10—15 мл отбрасывают. В период фильтрования колбы должны быть закры¬
ты пробками, а воронки — покровными стеклами для предотвращения
улетучивания ацетона. Фильтрование следует продолжать до тех пор,
пока вся суспензия не будет перенесена на фильтр. Если предполагают
определять только обменный кальций, то достаточно набрать около
25—30 мл фильтрата. По окончании фильтрования колбы с фильтратом
плотно закрывают пробками и несколько раз встряхивают для переме¬
шивания фильтрата.
Объемно-титриметрический метод определения
обменного кальция
Из приготовленной вышеуказанным способом водно-ацетоновой вы¬
тяжки берут пипеткой с грушей 5 мл прозрачного фильтрата и помещают
в коническую колбу объемом 50 мл. Туда же добавляют 1—2 капли 0,1 %-но-
го водного раствора металлоиндикатора нитхромазо, содержимое переме¬
шивают. После появления четко выраженной фиолетовой окраски пробу
титруют, прибавляя по каплям из микробюретки 0,02 н. раствор ВаС1*
до появления голубой окраски, не исчезающей в течение 1 мин. Как правило,
при титровании параллельных проб из одного фильтрата получают одина¬
ковые результаты, а в случае расхождения, которое обычно не превышает
0,02—0,03 мл, вычисляют среднюю величину иэ двух-трех определений.
Содержание обменного кальция (в мг-экв на 100 г почвы) рассчитывают
по формуле
,v 100*0,02.100
Са — (а — b) ,
где а — объем 0,02 н. раствора ВаС1а’(приведенного к точной нормальности), пошедшего
на титрование 5 мл исходного 0,02 н. раствора (NH4)2S04, мл; Ь — объем 0,02 н. раствора
ВаС1а (приведенного к точной нормальности), пошедшего на титрование 5 мл фильтрата,
мл; 100 — коэффициент для пересчета результатов анализа на 100 мл фильтрата; 0,02 —
нормальность титрованного раствора ВаС12; 100 — коэффициент для пересчета результа¬
тов анализа на 100 а почвы; 5 — объем фильтрата, взятого на титрование, мл; В — навеска
почвы (торфа), г.
Заменяя в вышеприведенной формуле постоянные множители на соответствующий
коэффициент, получаем упрощенные формулы: а) для навески почвы в 3 а содержание
обменного Са равно (а — Ь)* 13,333; б) для навески торфа в 1 г содержание обменного Са
равно (а—Ь) *40.
Реактивы. 1. Титрованный 0,02 н. раствор ВаС12. На аналитических весах
взвешивают 2,4430 г химически чистого ВаС12*2НгО и растворяют в дистиллированной
воде в мерной колбе на 1 л. Раствор доводят до метки. Титр устанавливают по 0,02 н.
H2S04, приготовленной из фиксанала.
2. 0,02 н. раствор (NH4)2S04 в смеси с ацетоном в объемном соотношении 55 : 45.
Сначала готовят 0,1 н. раствор (NH4)2S04. Для приготовления этого раствора взвешивают
на аналитических весах 6,6070 г сернокислого аммония и растворяют в мерной колбе на
1 л в^неболыпом количестве дистиллированной воды, после чего раствор доводят до метки.
14
3. Г. Ильковская, Л. С. Коновалова
Для приготовления 0,02 н. раствора (NH4)2S04 в смеси с ацетоном (в объемном соотноше-
нии 55 : 45) 200 мл приготовленного 0,1 н. раствора (NH4)2S04 помещают в мерную колбу
на 1 л, добавляют 350 мл дистиллированной воды. Оставшийся объем (450 мл) доливают
99%-ным ацетоном небольшими порциями, осторожно перемешивая. Смесь оставляют
на некоторое время для охлаждения до комнатной температуры, после чего раствор до~
водят ацетоном до метки и снова перемешивают. Перед тем как использовать приготов~
ленный раствор, проверяют его нормальность. Титр раствора (NH4)2S04 в смеси с ацетоном
должен быть точно 0,02 н.
3. 0,1%-ный раствор индикатора нитхромазо. 50 мг индикатора нитхромаао раство¬
ряют в 50 мл дистиллированной воды.
Трилонометрический метод определения обменного магния
в водно-ацетоновой вытяжке
В коническую колбу на 250 мл отбирают пипеткой с грушей 25 мл водно¬
ацетонового фильтрата и разбавляют дистиллированной водой до 100 мл
с помощью цилиндра. Затем приливают 10 мл хлоридно-аммиачного буфе¬
ра с pH 10, перемешивают и вносят 30—50 мг индикатора хромогена черного.
Содержимое колбы еще раз перемешивают и медленно титруют 0,01 н. рас¬
твором трилона Б при непрерывном и энергичном вэбалтывании. Конец
титрования устанавливают по переходу окраски от винно-красной через
лиловую и фиолетово-синюю к чисто-голубой в точке эквивалентности.
По количеству ватраченного на титрование раствора трилона Б вычис¬
ляют содержание обменного магния (в мг-экв на 100 г почвы) по формуле
a»0,0i»K *100*100
М£ 25 В ’
где а — количество раствора трилона Б, израсходованного на титрование, мл; 0,01 —
нормальность раствора трилона Б; /«Г — поправка к титру раствора трилона Б; 100 —
коэффициент для пересчета результатов анализа на 100 мл фильтрата; 100 — коэффициент
для пересчета результатов анализа на 100 г почвы; 25 — объем фильтрата, взятого на
титрование, мл\ В — навеска почвы (торфа), г.
Расчет можно упростить, объединив постоянные множители в коэффициенты, тогда
для навески почвы в 3 г содержание обменного магния будет равно а*Т*133,3 и для на¬
вески торфа в 1 г содержание обменного магния будет равно а* Г*400.
Реактивы. 1. 0,01 н. раствор трилона Б. Растворяют 1,86 г трилона в 1 л дистил¬
лированной воды или готовят из 0,05 н. раствора путем разбавления.
Титр раствора устанавливают по сернокислому магнию с индикатором хромогенол-
черным.
2. Хлоридно-аммиачный буферный раствор с pH 10. 20 а хлористого аммония раство¬
ряют в 500 мл дистиллированной воды, добавляют 100 мл 25%-ного раствора аммиака и
доводят объем до 1 л. Раствор хранят в склянке с притертой пробкой. При длительном
хранении необходимо проверить pH буфера.
3. Индикатор хромоген черный. 0,25 г индикатора и 25 г NaCl (или KG1) растирают
в ступке до равномерной окраски. Хранят в темной банке с притертой пробкой.
Примечание. Применяемая дистиллированная вода не должна содержать
следов меди. При значительных ее количествах окраска индикатора не изменяется во
время титрования раствором трилона Б.
ОПРЕДЕЛЕНИЕ ЕМКОСТИ ПОГЛОЩЕНИЯ ПОЧВ
Когда нет надобности устанавливать количественный состав обменных
катионов или когда почва содержит гипс и поэтому затруднительно опреде¬
ление обменных катионов обычными методами, прибегают к определению
емкости поглощения почвы.
Определение в почве обменных катионов, емкости поглощения
15
Метод Гедройца
Универсальный метод определения емкости поглощения почвы, предло¬
женный К. К. Гедройцем, основан на насыщении почвы натрием, который
в дальнейшем вытесняется из почвы кальцием двууглекислого кальция при
добавлении сухого порошка мела, в присутствии достаточного количества
воды, в токе углекислоты.
5—10 г почвы (в зависимости от предполагаемой величины емкости по¬
глощения) насыщают натрием, обрабатывая навеску почвы декантацией
1,0 н. раствором хлористого натрия при pH около 6,5. Почву постепенно
переносят на фильтр и промывают до исчезновения реакции на кальций.
Чем дольше промывают почву декантацией, меньше раствора приливают
к ней и полнее сливают его на фильтр, тем меньше будет истрачено вы¬
тесняющего раствора NaCl и скорее произойдет насыщение почвы нат¬
рием.
После насыщения почвы натрием избыток хлористого натрия удаляют,
промывая фильтр небольшими количествами воды. Как только в трубке
воронки покажется окрашенный органическим веществом фильтрат, про¬
мывание водой прекращают; далее продолжают промывание почвы 88°-ным
спиртом до исчезновения реакции на хлор.
Почву с фильтром переносят в банку или бутыль для водной вытяжки,
прибавляют 1 г мела, не содержащего натрия, приливают 1 л воды и в те¬
чение 3 час. пропускают через жидкость сильный ток углекислоты. Каждые
15 мин. содержимое колбы встряхивают. После пропускания тока углекис¬
лоты раствор фильтруют и определяют натрий пламенно-фотометрическим
методом.
Емкость поглощения вычисляется в миллиграмм-эквивалентах на 100 г
почвы, высушенной при 105° С.
Легкорастворимые соли натрия (Na2S04 и NaCl) не мешают определению
емкости поглощения этим методом. Если почва содержит гипс, то он должен
быть предварительно удален, так как в его присутствии нельзя насытить
почву натрием. Вымыть из анализируемой навески почвы гипс в данном слу¬
чае нетрудно, так как растворимость гипса в растворе хлористого натрия
довольно высока (7 г в 1 л).
Этим методом можно определить емкость поглощения и в карбонатных
лочвах, если вести промывание почвы хлористым натрием достаточно долго,
так как в 1 л 1,0 н. раствора NaCl растворяется лишь 0,05 г СаС08. Раство¬
римость углекислого магния значительно выше (0,41 г в 1 л 1,0 н. раствора
хлористого натрия).
Метод Бобко и Аскинази для некарбонатных почв
10 г воздушно-сухой почвы, просеянной через сито с отверстиями в 1 мм,
обрабатывают в фарфоровой чашке небольшим количеством 1 н. раство¬
ра ВаС12 *. Полученную почвенную суспензию переносят с помощью того
же раствора ВаС12 на воронку с плотным фильтром. Почву прикрывают
куском бумаги. При промывании почвы избытком раствора ВаС12 барий
вытесняет из почвы кальций, магний и другие имеющиеся в исходной почве
обменные катионы, становясь на их место 2. Закончив эту операцию, почву
отмывают водой до исчезновения реакции на ион Ва2+, а лучше — на ион
1 Необходимо проверить нейтральность этой соли — от 1—2 капель концентрированной
HG1 раствор в присутствии метилоранжа должен порозоветь.
■* Промывание кислых почв раствором ВаС12 удобно вести до тех пор, пока pH вытекаю¬
щего из почвы раствора не перестанет заметно отличаться от pH исходного раствора
ВаСЬ.
16
3, Г. Ильковская, А. С. Коновалова
СГ, и обрабатывают 0,05 н. НС11 для вытеснения из нее поглощенного ба¬
рия, который затем определяют в фильтрате количественно в виде BaS04.
Полученные результаты анализа следует пересчитать в миллиграмм-
эквиваленты на 100 г почвы, что дает величину емкости поглощения почвы.
Метод Бобко и Аскинази для карбонатных почв
Метод заключается в следующем: 5—10 г карбонатной почвы обрабаты¬
вают декантацией 0,05 н. раствором НС1 до исчезновения реакции на каль¬
ций. Если почва содержит гипс или большое количество карбонатов, то раз¬
рушить эти соединения 0,05 н. НС1 трудно. В таких случаях навеску почвы,
взятую для определения емкости поглощения, обрабатывают 2—3 раза
50 мл 0,2 н. раствора НС1 декантацией до прекращения разрушения кар¬
бонатов (шипение, вспучивание). После этого почву отмывают 0,05 н. ра¬
створом НС1 до отсутствия реакции на кальций. Необходимы частые пробы
фильтрата на содержание в нем кальция, так как промывать почву после
удаления обменного кальция избытком соляной кислоты не рекомендуется.
После того как карбонаты будут удалены, почву полностью переносят
на фильтр и промывают 1 н. раствором ВаС12 (pH около 6,5) до полного на¬
сыщения поглощающего комплекса барием, т. е. до тех пор, пока pH про¬
шедшего через почву раствора хлористого бария не достигнет величины
около 6,0—6,5 по бромтимолблау. Механически задержанный почвой хло¬
ристый барий отмывают водой, воронку также тщательно отмывают от при¬
сохших к ней солей бария. Промывание ведут до исчезновения реакции на
хлор. Если начинает появляться мутный раствор от присутствия в нем поч¬
венных коллоидов, промывание заканчивают 88°-ным спиртом.
Затем поглощенный барий вымывают иэ почвы 1,0 н. раствором НС1
до исчезновения в фильтрате реакции на барий. Для осаждения бария при¬
бавляют к кипящему раствору, содержащему барий, 5—10 мл горячего
10?£-ного раствора H2S04 и после двухминутного кипячения оставляют
стоять в теплом месте на 18—20 час. Если при промывании почвы соляной
кислотой фильтрата окажется 300—400 мл, то предварительно следует объем
его довести выпариванием до 200 мл; избыток соляной кислоты нейтрализуют
10%-ным раствором NH4OH, избегая полной нейтрализации, и в кислой
среде осаждают барий серной кислотой. Полученный осадок BaS04 филь¬
труют через плотный фильтр и отмывают от избытка серной кислоты горячей
водой, подкисленной 10%-ным раствором НС1. Затем осадок подсушивают,
озоляют, прокаливают в муфеле при температуре не выше 600° С (во избе¬
жание перехода осадка в Ва) и взвешивают. Полученный вес BaS04 умно¬
жают на 0,588 и получают содержание бария во взятой для анализа навеске
почвы. Затем производят пересчет на содержание бария в 100 г почвы, вы¬
сушенной при 105° С. Емкость поглощения почвы выражают в миллиграмм-
эквивалентах на 100 г почвы. Для этого полученный процент бария умножают
на 1000 и делят на эквивалентный вес бария, равный 69.
Применению данного метода определения емкости поглощения не мешает
присутствие в почве гипса и растворимых солей, так как в процессе промы¬
вания соляной кислотой эти соли из почвы будут удалены.
Для более быстрого вытеснения барием ионов водорода рекомендуется
промывать почву после разрушения карбонатов и декальцирования ее бу¬
ферным раствором хлористого бария, предложенным П. Г. Грабаровым
и 3. JI. Уваровой (1940).
1 Можно пользоваться и более крепкой НС1, но перед осаждением бария солянокислый
фильтрат следует довести до слабокислой реакции ввиду того, что в дальнейшем при
осаждении бария серной кислотой в виде BaS04 полученный осадок частично растворя¬
ется в присутствии избытка соляной кислоты.
Определение в почве обменных катионов, емкости поглощения
17
Буферный раствор хлористого бария готовят следующим образом. При¬
готовляют раствор хлористого и уксуснокислого бария; 1 л такого раствора
должен содержать 6,1 г ВаС12-2Н20 и 6,8 г уксуснокислого бария. pH раст¬
вора следует установить ~6,5.
По методу П. Г. Грабарова и 3. JI. Уваровой данный раствор готовят
следующим образом. На 10 л воды берут 61,1 г ВаС12*2Н20. Затем 78,87 г
Ва(0Н)2*8Н20 нейтрализуют в фарфоровой чашке 70 мл 55%-ной уксусной
кислоты. Содержимое чашки вливают в раствор ВаС12. Приготовленный ра¬
створ по содержанию бария — 0,1 н. По универсальному индикатору ра¬
створ доводят до pH 6,5 добавлением едкого бария или уксусной кислоты.
Промывать буферным раствором рекомендуют почвы, насыщенные ка¬
тионами. Почвы, не насыщенные катионами, могут показывать завышенные
данные.
Объемно-титриметрический метод Айдиняна
для карбонатных и кислых почв
(Айдинян, Иванова, Соловьева, 1970)
Сущность метода заключается в следующем. Образцы почв, глин, торфов
осадков и их высокодисперсных фракций независимо от химического состава
насыщаются барием при обработке их забуференным раствором ВаС12 с
pH 6,5 без предварительного разрушения карбонатов соляной кислотой
и удаления солей. В процессе обработки происходит фиксация двух форм
бария: обменного и химически связанного в форме труднорастворимых
соединений в карбонатных и засоленных почвах. В последнем случае для
вытеснения только обменного бария с целью определения его содержания
применяется 0,02 н. титрованный раствор сернокислого натрия в водно¬
ацетоновой смеси при равных объемных соотношениях. В такой смеси прак¬
тически исключается возможность растворения химически Ьвяэанного ба¬
рия.
В кислых, некарбонатных, незасоленных почвах вытеснителем обменно¬
го бария служит 0,02 н. водный раствор серной кислоты, так как здесь от¬
сутствуют другие формы его соединений.
Вытесняемый натрием или водородом обменный Ва2+ образует с SO*”
нерастворимый осадок сернокислого бария. Поэтому реакция замещения
протекает очень быстро до полного вытеснения обменного бария. Количе¬
ство сульфатов, израсходованных на связывание бария, эквивалентно ко¬
личеству обменного бария. Непрореагировавшая часть сульфатов вытесни¬
телей (Na2S04 или H2S04) оттитровывается раствором ВаС12 в присутствии
металлоиндикатора нитхромазо. Емкость поглощения рассчитывается по
количеству SOсвязавшемуся с обменным Ва2+. Определение сульфатов
объемно-титриметрическим методом производится очень быстро в течение
5—7 мин.
Ход анализа. Образец средней пробы, подлежащий анализу, должен
быть предварительно измельчен и просеян через сито с отверстиями диа¬
метром 1 мм. Навеску воздушно-сухой почвы или глины в количестве 2,5 г,
а для торфов и сильно оторфованных почв 1 г, помещают в стаканчик ем¬
костью 50 млу заливают 35—40 мл вспомогательного небуферного 0,1 н.
раствора ВаС12 (в целях экономии буферного раствора) и тщательно раз¬
мешивают стеклянной палочкой. Образцы почв с высоким содержанием
соды (со значением pH свыше 10) необходимо предварительно 1—2 раза
обработать декантацией 20—25 мл 0,1 н. раствора НС1 в том же стаканчике.
После осветления, которое наступает через несколько минут, весь прозрач¬
ный раствор осторожно переносят на воронку с плотным фильтром, стараясь
не переносить частицы почвы. Воронку с фильтром предварительно взве¬
шивают с точностью до 0,01 г.
18
3. Г. Ильковская, А. С. Коновалова
Фильтрование лучше вести в мерные цилиндры объемом 250 мл, так
как они занимают на столе меньшую площадь и, кроме того, ими удобно
производить измерение израсходованного раствора.
Насыщение почвы барием проводят декантацией, каждый раз наливая
в стаканчик с почвой около 20 мл 0,1 н. раствора ВаС12. Когда наберется
250 мл фильтрата, его отбрасывают и затем профильтровывают еще 100 мл
0,1 н. раствора ВаС12. После того, как общий объем фильтрата составит
350 мл, почву переносят на фильтр и продолжают промывание на воронке
буферным 0,1 н. раствором ВаС12 с pH 6,5. Промывание удобнее вести из
полиэтиленовой промывалки, направляя струю промывной жидкости от
края фильтра к центру. Новую порцию раствора не следует наливать до
тех пор, пока не профильтруется весь раствор на фильтре до последней
капли. Фильтрование ведут до объема 150 мл. Общий объем 0,1 н. раствора
ВаС12—500 мл. Во время промывания навески буферным раствороммворонка
должна быть все время покрыта часовым стеклом для уменьшения испаре¬
ния раствора. Для уменьшения ошибки анализа в конце насыщения почвы
барием применяется менее концентрированный 0,01 н. буферный раствор
ВаС12, который снижает концентрацию механически задержанного раство¬
ра ВаС12. Промывание 0,01 н. буферным раствором ведется до объема 120—
150 мл. При таких условиях обработки почвы раствором ВаС12 поглощающий
комплекс полностью насыщается барием, происходит связывание и выпаде¬
ние осадков карбонатов и сульфатов бария, одновременно удаляются лег¬
корастворимые соли. Поэтому отпадает надобность в проверке реакции на
полноту насыщения и отмывку солей.
После отфильтрования последней порции раствора избыток его осторож¬
но отсасывают и быстро взвешивают воронку с почвой и механически задер¬
жанным раствором. Этой операцией заканчивается насыщение почвы обмен¬
ным барием. Все фильтраты отбрасывают.
Затем приступают к вытеснению обменного бария раствором сернокис¬
лого натрия или серной кислоты в зависимости от того, какую почву анали¬
зируют: засоленную, карбонатную или незасоленную и некарбонатную.
Если почва карбонатная, засолена и содержит гипс, вытеснителем слу¬
жит сернокислый натрий в водно-ацетоновой смеси. С этой целью в кониче¬
скую колбу с притертой пробкой объемом 300 мл, в которую предварительно
помещают мерной колбой 250 мл 0,02 н. титрованного водно-ацетонового
раствора Na2S04 при соотношении ацетона к воде 1 : 1 (приготовление смеси
см. ниже), быстро опускают взвешенный фильтр с почвой, тщательно раз¬
мешивают палочкой, колбу закрывают пробкой и содержимое взбалтывают
в течение 10—15 сек. Спустя 30 мин. суспензию фильтруют в колбы объемом
100 мл через плотный складчатый фильтр диаметром 11 см (воронка диамет¬
ром 8—9 см). Не допускается откладывания фильтрования после погруже¬
ния фильтра с почвой в водно-ацетоновую смесь по истечении указанного
срока или временное прекращение начатого фильтрования.
Фильтрование следует проводить непрерывно, не ожидая полного про¬
сачивания жидкости через фильтр. При фильтровании колбы с суспензией
должны быть закрыты пробками, а воронки — часовыми стеклами. Первую
порцию фильтрата отбрасывают. Когда наберется 30—40 мл фильтрата,
берут пипеткой с грушей 5 мл фильтрата, предварительно 2 раза ополоснув
пипетку фильтратом, и переносят в узкогорлую колбу на 50 мл. Туда же
добавляют 1 каплю 0,1%-ного водного раствора металлоиндикатора нит-
хромазо. Содержимое колбочки взбалтывают и титруют из автоматической
микробюретки на 10 мл 0,02 н. титрованным раствором ВаС12 до перехода
фиолетовой окраски в голубую Ч
1 Титрование следует проводить при одинаковом освещении, учитывая, что свет от лю¬
минесцентных ламп оказывает влияние на изменение окраски индикатора.
Определение в почве обменных катионов, емкости поглощения
19
Для некарбонатных и незасоленных почв вытеснителем служит водный
раствор серной кислоты.
В коническую колбу на 300 мл помещают 250 мл 0,02 н. титрованного
раствора H2S04, опускают взвешенный фильтр с почвой и энергично взбал¬
тывают, а спустя 30 мин. фильтруют, как указано выше. Образцы различных
торфов и сильно оторфованных почв после взаимодействия с серной кисло¬
той оставляют стоять до следующего дня (около 18 час.) и по истечении этого
срока еще раз взбалтывают, а затем фильтруют.
Дальнейший ход анализа отличается от предыдущего (при водно-ацето¬
новой смеси) только тем, что перед титрованием к 5 мл фильтрата добавляет¬
ся 5 мл ацетона для достижения необходимой при титровании пропорции
ацетона к воде (1 : 1).
Титрование необходимо провести сразу же после сбора указанного объе¬
ма фильтрата. В случае откладывания титрования колбы с фильтратом долж¬
ны быть плотно прикрыты пробками.
Результаты анализа выражают в миллиграмм-эквивалентах на 100 г поч¬
вы. Расчет можно вести по формуле
где G — емкость поглощения, мг-экв на 100 г воздушно-сухой почвы; V — объем фильтра»
та, взятого на титрование, мл; Vx — объем титрованного 0,02 н. раствора ВаС12, пошед¬
шего на титрование,] мл;] Ki — поправка к титру 0,02 н. раствора ВаС12; V2 — объем
титрованного 0,02 н. раствора Na2S04 (или H2S04), равный 250 мл; 0,02 — нормальность
раствора Na2S04 (или H2S04); К2 — поправка к титру 0,02 н. раствора Na2S04 (или H2S04);
V9 — объем механически задержанного 0,01 н. раствора ВаС12, приравненный к его весу^
мл; 0,01 — нормальность буферного раствора ВаС12, применяемого при окончательной
промывке почвы; К3 — поправка к титру 0,01 н. раствора ВаС12; Р — навеска воздушно¬
сухой почвы, г; 100 — коэффициент для пересчета результатов анализа на 100 г почвы.
Реактивы. 1. 0,1 н. раствор ВаС12. Для приготовления 10 л раствора берут на¬
веску 122,15 г ВаС12*2Н20 и растворяют в дистиллированной воде.
2. Приготовление забуференных растворов 0,1 н. ВаС12 (pH 6,5):
а) по Никольскому с малеиновым буфером. 10 л раствора готовят следующим образом:
122,15 г ВаС12*2Н20 и 5,8 г малеиновой кислоты (или 4,9 г малеинового ангидрида) рас¬
творяют в свежепрокипяченной и охлажденной дистиллированной воде и разбавляют до
объема 10 л. К этому раствору для установления pH 6,5 надо прибавить около 900 мл
0,1 н. Ва(ОН)2/Если раствор барита мутный, то его необходимо предварительно отфиль¬
тровать. pH раствора проверяют одним из следующих способов: предварительно с инди¬
каторной бумагой, затем более точно индикатором бромтимолсиний, универсальным инди¬
катором или электрометрически. В случае, если реакция более кислая или щелочная,
соответственно добавляют несколько миллилитров барита или малеиновой кислоты;
б) По Грабарову и Уваровой с ацетатным буфером. Для приготовления 10 л раствора
берут 61,1 г ВаС1г*2Н20 и 68 г уксуснокислого бария Ва(СН3СОО)2 и растворяют в ди¬
стиллированной воде. Бели нет готовой соли Ва(СН3СОО)2, то ее готовят следующим
образом: 78,87 г Ва(0Н)2*8Н20 помещают в фарфоровую чашку, нейтрализуют 70 мл
55%-ного раствора СН3СООН. Полученный раствор уксуснокислого бария приливают
к 10 л раствора, содержащего 61,1 г ВаС12*2Н20. Приготовленные буферные растворы
должны быть прозрачным! |
3. Титрованный буферный 0,01 н. раствор хлористого бария. Приготовляют п*утем
десятикратного разбавления 0,1 н. буферного раствора хлористого бария. Титр проверяют
по 0,02 н. H2S04, беря среднее из трех определений. К 5 мл буферного раствора добавляют
5 мл ацетона и 1 каплю 0,1%-ного раствора нитхромазо, а затем титруют 0,02 н. раствором
H2S04 до перехода окраски из голубой в фиолетовую.^
4. 0,02 н. титрованный раствор ВаС12. Для приготовления 1 л 0,02 н. ВаС12 берут
навеску 2,4430 г ВаС12*2Н20 и растворяют в 1 л дистиллированной воды. Устанавливают
титр по серной кислоте так же, как указано в пункте 3.
[VK% — ViK\)-V%-K%-Q№
УКг
100
20
3. Г. Ильковская, А. С. Коновалова
5. 0,02 н. раствор сернокислого натрия в водно-ацетоновой смеси при соотношении
ацетона к воде 1:1. Для приготовления 2 л этой смеси берут в 2-литровую мерную колбу
200 мл 0,2 н. раствора Na2S04 и 800 мл дистиллированной воды, размешивают и доливают
до метки ацетоном. При смешивании ацетона с раствором происходит некоторое умень¬
шение общего объема, поэтому раствор следует вновь довести до метки' ацетоном. Титр
раствора устанавливают по 0,02 н. раствору хлористого бария с индикатором нитхромазо*
Для этого берут пипеткой с грушей 5 мл раствора, прибавляют 1 каплю нитхромазо и
титруют 0,2 н. раствором хлористого бария до перехода фиолетовой окраски в голубую*
Для приготовления 1 л 0,2 н. раствора Na^C^ берут 14,2048 г Na^C^ или 32,2208 г Na2S04*
•ЮН20, тщательно растворяют в дистиллированной воде в мерной колбе на 1 л и доводят
до метки.
6. 0,1%-ный водный раствор нитхромазо. 0,1 г нитхромазо растворяют в 100 мл
бидистиллированной воды и переносят в капельницу.
7. 0,02 н. титрованный раствор H2S04. Раствор готовят из фиксанала.
8. 0,05%-ный раствор индикатора бромтимоловый синий. 0,1 г индикатора перети¬
рают в агатовой ступке с 3,2 мл 0,05 н. раствора едкого натра до полного растворения.
Пипеткой добавляют 10—15 мл воды, перемешивают, переносят в колбу на 200 мл и до¬
водят до метки.
Определение емкости поглощения
и обменных катионов в солонцах и солонцеватых почвах
вытеснением их реактивом Пфеффера, в модификации Беляевой (1969)х
Метод Пфеффера был предложен для определения емкости поглощения
карбонатных засоленных почв. Он был модифицирован Н. И. Беляевой
в целях совместного определения емкости поглощения и состава обменных
катионов в солонцовых почвах. Метод имеет ряд недостатков, в частности
рассматриваемая методика применима для почв с относительно невысоким
содержанием гумуса (не выше 5—6%).
Ход анализа. Навеску почвы 5 г, пропущенную через сито с отверстиями
диаметром 1 мм, помещают в широкогорлую колбу на 100 мл, заливают
25 мл спирта ректификата для отмывания почвы от легкорастворимых со¬
лей, содержимое колбы сильно взбалтывают и сразу же, возможно полнее,
сливают жидкость на фильтр (диаметр И см, белая лента). К оставшейся
в колбе почве добавляют 15 мл спирта и, снова взболтав, фильтруют. При
отмывке почвы от легкорастворимых солей время взаимодействия спирта
с почвой во избежание обменных реакций должно быть минимальным. Поэ¬
тому одновременно можно работать с одной-двумя навесками; необходимо
также, при возможно полном сливе промывной жидкости на фильтр,
стараться переносить на него как можно меньше почвы. Обработку навески
почвы спиртом повторяют 4—5 рае. Когда наберется примерно 100 мл
фильтрата, проверяют реакцию на хлор. Как правило, 100 мл спирта вполне
достаточно для отмывки почвы от легкорастворимых солей, а в ряде случаев
его требуется и меньше, но для лучшей сопоставимости данных рекомендует¬
ся придерживаться во всех случаях вышеуказанной нормы — 100 мл.
Промывание производится 70%-ным спиртом ректификатом; в случав
содовых солонцов начинать промывку следует 96%-ным спиртом (две—
три промывки), а затем 70%-ным спиртом. После окончания промывки
и в том, и в другом случае для быстрого высушивания почву в колбе и на
фильтре промывают небольшим количеством 96%-ного спирта. Отмытую,
высушенную на воздухе почву переносят вместе с фильтром в ту же колбу
и обрабатывают реактивом Пфеффера. Почву заливают 25 мл реактива Пфеф¬
фера, взбалтывают в течение часа (или интенсивно взболтав в течение 3 мин.,
1 Метод описан JI. А. Моряковой и А. Ф. Новиковой в сборнике «Разработка способов
мелиорации солонцов и солонцеватых почв в условиях орошения и на богаре» (М., 1969).
Определение в почве обменных катионов, емкости поглощения
21
оставляют стоять на ночь), после чего жидкость фильтруют и почву декан¬
тацией промывают реактивом Пфеффера 5 раз по 15 лед. Для более полного
вытеснения ионом аммония обменных катионов почвы всю партию следует
интенсивно взболтать и жидкость сливать на фильтр только после полного
оседания почвы в колбе и полной фильтрации предыдущей порции. Во вре¬
мя промывания почвы реактивом Пфеффера колбы должны быть закры¬
ты пробками, а воронки — часовыми стеклами (во избежание испарения
спирта).
Определение емкости поглощения. Для определения
омкости поглощения из полученного фильтрата отбирают 20 или 25 мл
в узкогорлую колбу на 100 мл, подогревают до появления первых пузырь¬
ков, добавляют 5 мл 35%-ного (не менее 30%-ного) раствора формальдегида,
титруют ОД н. раствором NaOH с фенолфталеином до розовой окраски и,
снова подогрев (до температуры 60—70° С) и прибавив 2—3 капли фенол¬
фталеина, дотитровывают. Точно так же титруют реактив Пфеффера. Раз¬
ность между количеством NaOH, пошедшим на титрование исходного ра¬
створа и фильтрата, соответствует величине емкости поглощения данного
образца почвы.
Бмк.ость поглощения (мг-экв) рассчитывают по формуле
0,1 (а — ai)»K *100
Е - с ,
'где 0,1 — нормальность раствора NaOH; а — количество NaOH, пошедшее на титрование
исходного реактива Пфеффера, мл; ах — количество NaOH, пошедшее на титрование филь»
трата, мл; К — поправочный коэффициент] к] титру раствора NaOH; С — навеска почвы9
соответствующая^количеству раствора, взятого на титрование, г; 100 — коэффициент для
пересчета результатов анализа на 100 г почвы.
Определение обменных катионов. Из оставшейся час¬
ти фильтрата берут для определения обменных катионов 50 лед и выпарива¬
ют в фарфоровой чашке на водяной бане досуха. Сухой остаток осто¬
рожно прокаливают при температуре 400° С (не выше) для удаления солей
аммония.
После прекращения выделения аммонийных солей в виде дыма остаток
прокаливают еще 3—4 мин. (не более!). Дают чашкам остыть, затем добав¬
ляют 2—3 капли концентрированной соляной кислоты, заливают горячей
водой и фильтруют в мерную колбу на 250 мл. Фильтрование ведут до прекра¬
щения реакции на хлор, после чего содержимое колбы доводят до метки
водой. Из полученного раствора берут 100 мл для трилонометрического
определения кальция с мурексидом, 50 мл для определения суммы кальция
и магния с хромогеном черным. Остаток используют для определения нат¬
рия и калия на пламенном фотометре.
Определение суммы кальция и магния. 50 дед раство¬
ра помещают в коническую колбу на 250 мл, разбавляют раствор водой
примерно до 100 лед и подогревают до 60—70° С. Прибавляют для создания
щелочной реакции 5 мл аммиачного буферного раствора, несколько кристал¬
ликов диэтилдитиокарбамата натрия (для связывания следов меди) и 1—2 мл
5%-ного раствора гидроксил амина, 10—15 мг индикатора хромогена чер¬
ного, растертого с хлористым натрием, и титруют 0,02 н. раствором трило¬
на Б до перехода окраски индикатора от вишнево-красной через фиолето¬
во-синюю в чисто-голубую.
Определение кальция. 100 мл раствора помещают в коничес¬
кую колбу на 250 лед, прибавляют несколько кристалликов диэтилдитио¬
карбамата натрия, 1—2 мл 5%-ного раствора гидроксиламина, 2 мл 10%-но-
го раствора NaOH и титруют 0,02 н. раствором трилона Б в присутствии
индикатора мурексида до перехода ярко-пурпурной окраски в фиолетовую.
22
3. Г. Ильковская, А. С. Коновалова
Сумма кальция и магния (в мг-экв на 100 г почвы) вычисляется по урав¬
нению
а *0,02 »К *100
Са + Mg = £ ,
где а — количество трилона Б, пошедшее на титрование кальция и магния, мл; 0,02 —
нормальность раствора трилона Б; К — поправочный коэффициент к титру трилона Б;
С — навеска почвы, соответствующая количеству раствора, взятого на титрование каль¬
ция и магния, г; 100 — коэффициент для пересчета результатов анализа на 100 г почвы.
Содержание кальция (в мг-экв на 100 г почвы) рассчитывается по той
же формуле. Содержание магния рассчитывается по разности между суммой
кальция и магния и количеством кальция.
Реактивы. 1. Реактив Пфеффера — 0,1 н. раствор NH4C1 в 70%-ном этиловом
спирте. 5,35 г NH4C1 растворяют в 270 мл воды, добавляют 730 мл 96%-ного этилового
спирта. Раствор доводят прибавлением аммиака до pH 7 по универсальному индикатору
или по бромтимолблау.
2. 10%-ный раствор NaOH.
3. 35%-ный раствор^формальдегида. Формальдегид, приобретенный в торговой сети,,
может иметь кислую реакцию; поэтому его нужно предварительно нейтрализовать NaOH
по фенолфталеину. При стоянии формальдегид, полимеризуясь, становится мутным, ме»
нее реактивоспособным, пользоваться им не следует.
4. 0,02 н. раствор трилона Б. 3,72 г реактива растворяют в 1 л воды.
5. Хлоридно-аммиачный буфер. 20 г х. ч. NH4C1 растворяют в 100 мл дистиллиро¬
ванной воды, приливают 100 мл 25%-ного раствора NH4OH, разбавляют дистиллирован¬
ной водой до 1 л и тщательно перемешивают.
6. Хромоген черный и мурексид применяются в смеси, приготовленной из 1 части
индикатора] и 100 или 200 частей] х. ч. NaCl. Смесь тщательно растирается и перемешива¬
ется в ступке.
7. 5%-ный водный раствор гидроксиламина.
ОПРЕДЕЛЕНИЕ ГИПСА
Навеску почвы в 1—5 г (в зависимости от содержания гипса), просеян¬
ной через сито с отверстиями в 0,25 мм, заливают в стакане на 100 мл 0,25 н.
раствором НС1, тщательно перемешивают и оставлгт'>* на ночь. На другой
день почву многократно отмывают декантацией этой же кислотой до прекра¬
щения реакции на S04” (проба с Bad*) в фильтрате. Фильтрат собирают
в стакан, в котором предполагается осаждение S04~» концентрируют до
150—200 мл. Избыток соляной кислоты нейтрализуют 10%-ным раствором
NH4OH. Фильтрат подкисляют 10%-ным раствором НС1 до явно кислой
реакции, подогревают до кипения и осаждают S04” 10 мл горячего 10%-но-
го раствора ВаС12, кипятят несколько минут. Осадок оставляют на 4 часа
в теплом месте. На другой день после пробы на полноту осаждения осадок
отфильтровывают черев фильтр диаметром 7—9 см с синей лентой, про¬
мывают горячей водой, подкисленной несколькими каплями 10%-ного*
раствора НС1 до прекращения реакции на Ва2+ (проба с 10%-ным раство¬
ром H2S04). При промывании осадка лучше ставить под воронку неболь¬
шой стаканчик на 100 мл и прозрачный фильтрат выливать по мере его на¬
копления.
Если же при промывании осадок BaS04 начинает проходить через фильтр,
то небольшое количество фильтрата следует еще раз перефильтровать.
Фильтр с осадком переносят во взвешенный тигель, прокаливают при 500° С
до постоянного веса и взвешивают. Полученный вес BaS04 умножают на
коэффициент 0,4114 и таким образом определяют количество S04~ в грам¬
Определение в почве обменных катионов, емкости поглощения
23
мах в данной навеске почвы. Наконец делают пересчет содержания SOil”
на 100 г почвы, высушенной при 105° С.
При необходимости выразить содержание гипса в CaS04-2H20 получен¬
ное количество BaS04 умножают на 0,737, а затем делают пересчет на 100 г
почвы, высушенной при 105° С.
Чтобы представление о количестве гипса в почве было правильным,
необходимо от полученного количества SOI* отнять количество SO^, со¬
держащееся в водной вытяжке.
Экспресс-метод Айдиняна
(Айдинян, Иванова, Соловьева, 1968)
Наряду с гипсом в почвах всегда встречаются легкорастворимые суль¬
фаты. Примесь этих солей в довольно значительном количестве обнаружена
также в друзах гипса, образовавшихся в почвенных условиях. Очень часто
легкорастворимые сульфаты, особенно Na2S04, в негипсоносных горизонтах
засоленных почв превышают содержание сульфатов, связанных с кальцием
(например, сульфатно-хлоридные солончаки-солонцы). Различная степень
растворимости сернокислых солей в растворах разного состава дает возмож¬
ность производить раздельное определение их. Экспериментально установ¬
лено, что в смеси а^зтон* с водой (1 :3) растворяются легкорастворимые
сульфаты одновалентных катионов, гипс же практически не растворяется.
Это дает основание предложить методику определения гипса в почвах при
одновременном наличии в них легкорастворимых сульфатов. При определе¬
нии ионов SO*- путем титрования раствором ВаС12 в присутствии металлоин-
дикатора нитхромазо на анализ затрачивается очень мало времени, поэтому
его мэжно отнести к экспресс-методам.
Ход анализа. Одновременно из одного образца почв приготовляются
две вытяжки — солянокислая и водно-ацетоновая.
1. 0,2 н. солянокислая вытяжка для определения общего количества
сульфатов в почвах, в том числе и гипса. Из средней пробы образца, пред¬
варительно тщательно растертого в ступке и пропущенного через сито с
отверстиями диаметром 0,25 мм, отвешивается на аналитических весах на¬
веска 3—5 г для почв, содержащих 1% и более гипса, или для почв, засолен¬
ных легкорастворимыми сульфатами. Навеска для анализа гипсовых конкре¬
ций (друзы) не должна превышать 0,5 г. При малом содержании в почве гипса
или легкорастворимых сульфатов (особенно в верхних горизонтах почв)
величину навески можно довести до 10 г.
Навеску помещают в коническую колбу объемом 300—500 мл, при¬
ливают мерной колбой 250 мл 0,2 н. НС1, содержимое взбалтывают пе¬
риодически в течение 5 мин., колбу закрывают пробкой со стеклянной
трубкой, служащей обратным холодильником. Затем суспензию нагревают
на электрической плитке до кипения, кипячение продолжают 3 мин. Колбу
снимают с плитки, вынимают пробку и колбу погружают в воду для быстро¬
го охлаждения горячей суспензии до комнатной температуры. После этого
колбу оставляют стоять в течение 30 мин., периодически взбалтывая. За¬
тем раствор фильтруют через плотный беззольный фильтр (синяя лента)
диаметром 9 см. Берут градуированной пипеткой 10—25 мл прозрачного
солянокислого фильтрата, помещают в мерную колбу и в зависимости от
содержания сульфатов в почве фильтрат разбавляют в 2—10 раз дистилли¬
рованной водой, объем доводят до метки. Разбавленный раствор пропуска¬
ют через Н-катионит (отбрасывая первые 10—15 мл), пока не наберется
30—40 мл фильтрата. Полученный раствор (10—15 мл) помещают в коничес¬
кую колбу на 50—100 мл, приливают ацетон в количестве, равном объему
испытуемого раствора, прибавляют 1 каплю 0,1%-ного водного раствора
24
3. Г. Илъковская, А. С. Коновалова
нитхромазо и титруют из автоматической микробюретки (объем на 10 мл)
0,02 н. раствором ВаС12 до перехода окраски индикатора из фиолетовой
в голубую. Титрование следует проводить вначале медленно, прибавляя
раствор ВаС12 по каплям и тщательно перемешивая. Появляющаяся в от¬
дельных случаях голубая окраска от первых капель ВаС12 через 30—40 сек.
должна перейти в фиолетовую (или сине-фиолетовую). Дальнейшее измене¬
ние окраски идет быстро. Конец титрования отмечается четким переходом
фиолетовой окраски в голубую, не изменяющуюся в течение 1—2 мин.
В некоторых случаях при титровании солянокислой вытяжки, получен¬
ной из сильнокарбонатных почв и пропущенной через Н-катионит, из-за
наличия в титруемом растворе ионов Са2+, проскочивших через Н-катионит,
переход окраски может быть затруднен. В этих случаях, если разбавление
раствора не помогает или оно невозможно, для частичного связывания ио¬
нов Са2+ перед титрованием вводят дополнительно 1—2 капли 5%-ного вод¬
ного раствора NH4F. Переход окраски становится при этом отчетливым.
Чаще всего продолжительность одного титрования составляет 5—7 мин.
2. Водно-ацетоновая вытяжка для определения легкорастворимых суль¬
фатов в присутствии гипса. Навеску почвы 3—5 г (в зависимости от предпо¬
лагаемого содержания легкорастворимых сульфатов) помещают в коническую
колбу объемом 150 мл, туда же приливают 100 мл водного раствора ацетона
(3 объема воды: 1 объем ацетона) и взбалтывают на ротаторе в течение 1 часа.
Содержимое колбы фильтруют через плотный фильтр (воронки должны быть
прикрыты часовыми стеклами) и, не дожидаясь конца фильтрования всего
объема, часть фильтрата пропускают через Н-катионит (отбрасывая первые
10—15 мл), пока не наберется 30—40 мл фильтрата. Из фильтрата пипеткой
с грушей берут необходимый объем для титрования сульфатов. Если титруе¬
мый объем составляет 5 мл, то его надо разбавить дистиллированной водой
до 10—15 мл и в таком объеме вести определение S04“. Иногда приходится
прибегать к еще большему разбавлению, особенно при анализе нижних
горизонтов засоленных почв, богатых растворимыми сульфатами. Испытуе¬
мый раствор помещают в коническую колбу объемом 50—100 мл. Туда же
приливают ацетон в количестве, равном объему титруемого раствора. При
этом необходимо учитывать количество ацетона, содержащееся во взятом
для титрования объеме вытяжки, и соответственно уменьшить количество
ацетона, прибавляемое при титровании S04“. Так, если для титрования
взято 5 мл вытяжки и объем доведен водой до 15 мл, то перед титрованием
следует прибавить 13,3 мл (15—1,66) ацетона, 1 каплю 0,1%-ного водного
раствора нитхромазо и титровать 0,2 н. раствором ВаС12, как и в случае
солянокислой вытяжки.
По количеству 0,02 н. раствора ВаС12, израсходованного на титрование,
рассчитывают содержание сульфатов (в, %) в водно-ацетоновой и соляно¬
кислой вытяжках по формуле
o,v2_ а-К- 0,00096-100
S 4 С ’
где а — количество 0,02 н. раствора ВаС12, пошедшее на титрование, мл; К — поправка
к титру 0,02 н. раствора ВаС12; 0,00096 — количество SOj", эквивалентное 1 мл 0,02 н.
раствора ВаС12, г; С — навеска почвы, соответствующая объему фильтрата, взятого н а
титрование, г; 100 — коэффициент для пересчета результатов анализа в проценты.
Разность между процентным содержанием S04” в солянокислой вытяжке
и SO4” в водно-ацетоновой вытяжке составляет процентное содержание
SO4” гипса почвы. Если количество S04” нужно перевести в форму гипса
(CaS04*2H20), то полученный процент S04“ умножают на 1,7921.
Определение в почве обменных катионов, емкости поглощения
25
При отсутствии гипса в образцах почвы количество сульфатов, извле¬
каемое солянокислым раствором и смесью ацетона с водой, должно быть
близким.
Реактивы. 1. 0,02 н. титрованный раствор ВаС12 (приготовление см. в разделе
«Определение емкости поглощения почв по Айдиняну»).
2. 0,1%-ный водный раствор нитхромазо (приготовление см. там же).
3. 1%-ный водный раствор AgN03.
4. 5%-ный водный раствор NH4F.
5. Н-катионит (КУ-2 или КУ-2-8). Ионообменную смолу помещают в химический
стакан и промывают дистиллированной водой до полного удаления пыли. После этого
смолу заливают 5%-ным раствором НС1, хорошо перемешивают и оставляют стоять до
следующего дня. На следующий день раствор соляной кислоты заменяют свежим, смолу
перемешивают, оставляют стоять, после чего кислоту сливают. Обработку катионита
соляной кислотой продолжают до исчезновения в растворе реакции на SO2' (проба с BaCJ2).
Затем катионит промывают дистиллированной водой до исчезновения реакции на С1“
•(проба с AgNOs).
Для устройства ионообменной колонки используют или бюретку, или воронку Нуча,
которую укрепляют на штативе. Если в качестве колонки используют воронку Нуча,
то на дно ее кладут бумажный фильтр (красная лента), затем переносят водную суспен¬
зию катионита (8—10 г смолы).
Испытуемый раствор пропускают через колонку без отсасывания. Первые порции
фильтрата в количестве 10—15 мл отбрасывают для того, чтобы удалить из ионообменной
колонки присутствующую там воду.
После каждого определения SO^“ катионит в колонке необходимо регенерировать.
Для этого через колонку с катионитом пропускают 5%-ный раствор НС1 в объеме при¬
мерно 150—200 мл и затем отмывают катионит дистиллированной водой до исчезновения
реакции на С1~~ (проба с AgN03). Заряженные колонки с катионитом хранят во влажном
•состоянии под водой.
ОПРЕДЕЛЕНИЕ КАРБОНАТОВ
Метод Голубева
Наиболее распространенными приборами для определения карбонатов
являются кальциметры различной системы, основанные на объемном опре¬
делении углекислоты.
Предложенный И. Ф. Голубевым вариант кальциметра для определения
карбонатов отличается большой простотой и позволяет определять карбона¬
ты с достаточной точностью (рис. 1). Он может быть рекомендован там,
где требуется массовое определение С02 при исследовании карбонатных
почв, мергелей и известковых туфов. Этим методом определение С02 произ¬
водится в течение 10—15 мин.
В основу работы кальциметра И. Ф. Голубева положено измерение до¬
бавочного давления, получаемого от С02, выделившейся при воздействии
18%-ного раствора НС1 на содержащиеся в почве карбонаты. Давление из¬
меряют по высоте поднявшегося ртутного столба в стеклянной трубке.
Отсчет производят по специальной шкале. Шкала наносится эмпирически,
путем определения показаний давления при различных навесках х. ч. и
абсолютно сухого СаС03. Для составления шкалы удобно брать следующие
навески: 0,005 г\ 0,007; 0,01; 0,0125; 0,015 и т. д. до 0,2 г.
Прибор состоит из следующих частей: шкалы 1 высотой 30 см с нанесен¬
ными на ней величинами навесок СаС03; предохранителя 2 от выбрасывания
ртути при очень больших дозах карбонатов; резервуарчика 3 для ртути;
стеклянной банки 4 емкостью 125—150 мл с каучуковой пробкой а и метал¬
лическим кольцом б, дающим возможность погружать пробку до определен¬
26
3. Г. Илъковская, А. С. Коновалова
ной глубины; тигля 5 и крана 6, изоли¬
рующего прибор от внешнего воздуха.
Определение С02 сводится к следую¬
щему. Навеску почвы от 1 до 5 г (в за¬
висимости от содержания карбонатов)
помещают в банку прибора. Так как
объем банки рассчитан на 5 г почвы,
навеску меньше 5 г (в случае большо¬
го содержания карбонатов) досыпают
до 5 г тонкорастертым песком, не содер¬
жащим карбонатов. Затем в специаль¬
ный тигель 5 наливают 5 мл 18%-ного
раствора НС1, тигель при помощи пин¬
цета с резиновыми наконечниками осто¬
рожно помещают в банку. После того,
как навеска высыпана и тигелек с НС1
помещен в банку, ее при открытом кра¬
не 6 плотно прикрывают пробкой до от¬
каза (по кольцу б) и, закрыв кран, изо¬
лирующий прибор от воздуха, а следо¬
вательно, и от атмосферного давления,,
начинают слегка встряхивать банку;
тигелек при этом опрокидывается, НС1
выливается и приходит в соприкоснове¬
ние с почвой. Вследствие выделения С02
в приборе образуется добавочное давление, которое поднимает столб ртути
на ту или иную высоту в зависимости от содержания карбонатов в почве.
Когда ртуть в трубочке перестает подниматься, производят отсчет по
шкале.
Для того чтобы банка не нагревалась от рук во время работы, верх ее
обматывают марлей и при встряхивании держат через марлю.
Шкалу можно составить таким образом, чтобы она соответствовала ко¬
личеству С02 в граммах, тогда сразу будет видно, сколько С02 находится
во взятой для анализа навеске. Затем производят пересчет содержания
С02 на 100 г высушенного вещества (почвы, известняка и т. д.). Шкала при¬
бора пригодна только для определенной температуры воздуха. Поэтому
следует иметь несколько шкал, приготовленных при разной температуре
(15, 20, 25°), которыми и необходимо пользоваться в зависимости от темпе¬
ратуры воздуха в лаборатории. Изменения барометрического давления не
оказывают заметного влияния на результаты анализа. Точность работы
прибора зависит исключительно от точности нанесения шкалы. Если шкала
составлена в пересчете на СаС03, то полученное количество СаС03 надо
помножить на коэффициент 0,44 для перевода величины СаС03 в С02, а
затем произвести пересчет С02 на 100 г почвы, высушенной при 105° С.
Метод Гейслера — Максимюк
Кальциметр для определения углекислоты представлен на рис. 2.
Прибор состоит из небольшого резервуара емкостью 50 мл, в который
вставлены две трубки. Трубка 1 для соляной кислоты, закрытая сверху
каучуковой пробкой, заканчивается краном 5, при помощи которого соляная
кислота спускается в резервуар. Вторая трубка (2) соединяется на шлифе
с резервуаром. Эта трубка имеет впаянный внутри колпачок 4, в середине
которого проходит капиллярная трубка 5, соединенная с резервуаром и
открытая сверху. Стеклянная пробка 6, закрывающая трубку 2, имеет сквоз¬
ное отверстие 7 для сообщения с внешним воздухом. На колпачке внизу
Рис. 1. Прибор для определения кар¬
бонатов по методу Голубева
Объяснение см. в тексте
Определение в почве обменных катионов, емкости поглощения
27
'имеются отверстия 8у через которые
углекислота выходит из резервуара
в трубку 2 •
Перед определением С02 сначала от¬
крывают стеклянную пробку трубки 2
и наливают в нее 1—2 мл концентри¬
рованной H2S04, через которую будут
проходить пузырьки С02, очищаясь от
водяных паров, и через отверстие в проб¬
ке уходить из прибора в атмосферу.
Прибор надо готовить из тонкого стек¬
ла, и он должен весить с навеской поч¬
вы и кислотами не более 50 г (высота
прибора около 15 см). Для проведения
анализа в трубку 1 при закрытом кране
3 наливают 7—10 мл 18%-ного раство¬
ра НС1. Приготовленный к анализу при¬
бор (т. е. с соляной кислотой в трубке
1 и с серной кислотой в трубке 2),
тщательно вытертый фильтровальной
бумагой снаружи, взвешивают на ана¬
литических весах.
После этого трубку 2 снимают и на¬
веску почвы в 1—5 г опускают на дно
резервуара через маленькую вороноч¬
ку (все шлифы должны быть предва¬
рительно смазаны вазелином), ставят трубку 2 на место и вновь взвешивают
весь прибор. После взвешивания осторожно открывают кран 3 в трубке 1
и начинают вводить соляную кислоту по каплям в резервуар прибора.
При большом содержании С02 реакция идет очень бурно, поэтому кислоту
следует вводить не сразу, а в несколько приемов. Спустив соляную кислоту,
кран 3 закрывают. Выделяющаяся С02 уходит из прибора через трубку 2.
Когда видимое выделение С02 прекратится, прибор ставят на этернитовую
плитку и нагревают резервуар прибора примерно до 60° С (плитка не должна
быть сильно нагрета). Нагретый прибор охлаждают и удаляют остатки С02,
задержавшиеся над жидкостью в приборе, продуванием резиновой грушей,
соединенной с прибором через стеклянную трубочку, пропущенную сквозь
резиновую пробку в отверстии трубки 1. Прибор вновь взвешивают. Раз¬
ница в весе прибора до и после удаления С02 равняется количеству угле¬
кислоты карбонатов, содержавшихся в навеске почвы (известняка, туфа
и т. д.). Производят пересчет на 100 г вещества, взятого в анализ.
Определение С02 в этом кальциметре занимает 15—20 мин. Имея 10—
12 таких приборов, можно сделать 20—24 определения в день. Метод требует
некоторого навыка в обращении с прибором, но осваивается быстро.
Необходимо еще раз подчеркнуть, что перед каждым взвешиванием при¬
бор надо обтирать снаружи фильтровальной бумагой. Каучуковая пробка,
закрывающая отверстие трубки 7, не должна касаться раствора соляной
кислоты и должна быть совершенно сухой.
Воздух продувать через прибор надо осторожно, чтобы* не разбрызги¬
вать серную кислоту. Через 3—4 определения С02 серную кислоту следует
сменить для лучшего задерживания влаги, образующейся во время реакции
выделения С02.
Рис. 2. Прибор для определения кар¬
бонатов весовым методом Гейслера —
Максимюк
Объяснение см. в тексте
28
3. Г. Илъковская, А. С. Коновалова
ОПРЕДЕЛЕНИЕ СЕРЫ1 ПО АЙДИНЯНУ
Валовое содержание серы
Метод основан на окислении S-содержащего вещества при нагревании
с бертолетовой солью. Выделяющийся при разложении КС103 кислород
(2КСЮ3 = 2КС1 + 302) окисляет серу и переводит ее в сульфатную форму.
Прибор для определения (рис. 3) состоит из двух пробирок длиной в 18 см
и диаметром в 2,5 см каждая; в пробирку 1 (из кварцевого стекла) помещают
навеску для сжигания, пробирка 2 (из обычного стекла) выполняет роль
«приемника».
Средние пробы исследуемых образцов должны быть предварительно очи¬
щены от корешков, пропущены через сито с отверстиями в 1 мм и растерты
Рис. 3. Схема установки для
определения серы
] Объяснение см. в тексте
в агатовой ступке. Если образец растерт недостаточно тонко, реакция будет
протекать неравномерно и может не произойти полного окисления.
Навески почв, содержащих 4—5% гумуса, должны быть не более 1—
1,5 г; навески почв из нижних слабогумусных горизонтов — от 2 до 3 г.
Для спокойного течения реакции сжигания к почве, содержащей более
4—5% гумуса, добавляют 8 г промытого (1%-ной НС1 и дистиллированной
водой) и прокаленного кварцевого песка. (Навески растений и органических
соединений берут не более 0,3 г с добавлением около 8 г песка, а навески суль¬
фидных минералов — в пределах 0,025—0,050 г без добавления песка).
В пробирку для сжигания помещают навеску почвы, 2 г КС103, в случае
необходимости добавляют песок и все тщательно перемешивают. Пробирка-
приемник на три четверти заполняется дистиллированной водой с добавле¬
нием 1—2 капель Н202. Затем на пламени газовой горелки сжигают смесь.
Нагревание начинают с конца пробирки, постепенно передвигая горелку
к ее середине. Если реакция пойдет очень бурно, то горелку на короткое
время следует отодвинуть. Может случиться, что внутренние стенки про¬
бирки почернеют (что происходит довольно часто). Тогда перед концом
сжигания пробирку поворачивают так, чтобы над пламенем пришлась та ее
часть, где имеется чернота, т. е. несгоревшее вещество.
После того, как в пробирке для сжигания прекратятся вспышки, а в
пробирке-приемнике выделение пузырьков, сжигание считают законченным.
Процесс окисления серы происходит очень быстро, примерно в течение
10 мин.
В остывшую пробирку прибавляют 2—3 капли концентрированной НС1.
Содержимое обеих пробирок соединяют вместе и фильтруют через плотный
1 Описание метода составлено 3* С* Повелягиной (Почвенный институт им. В. В. Доку¬
чаева).
Определение в почве обменных катионов, емкости поглощения
29
фильтр, перенося постепенно сухой остаток и песок на фильтр горячей
водой. Остаток на фильтре промывают 0,1 н. раствором КС1 для того, чтобы
мелкодисперсные частицы почвы не прошли в фильтрат. Установлено, что
после намывания 200—250 мл фильтрата ионы S04” полностью отмываются.
Если сжигание растительного образца не удается провести полностью,
что видно по отдельным несгоревшим частицам, то содержимое обеих про¬
бирок переносят в фарфоровую чашку (диаметром 9—10 см), прибавляют
0,2 г КСЮ3, несколько капель НС1 и выпаривают на водяной бане. Такую
обработку следует произвести 2—3 раза. Вместо КСЮ3 и НС1 можно исполь¬
зовать перекись водорода. Обработку также производят 2—3 раза 2—3 мл
30%-ной Н202. После этого проводят фильтрование вышеуказанным спо¬
собом.
Фильтрат выпаривают примерно до 100 мл, подкисляют несколькими
каплями НС1 и доводят до кипения на плитке. В кипящую жидкость при¬
ливают по частям 4—10 мл 10%-ного раствора ВаС12. Раствор с осадком
оставляют в теплом месте не менее чем на 4 часа, после этого фильтруют
через плотный фильтр с синей лентой. В конце фильтрования необходимо
проверить, полностью ли отмыт осадок от иона хлора. Сжигание осадка
производят в муфеле при температуре 450—500° С. Вес осадка BaS04 умно¬
жают на коэффициенты 0,1373, 0,3430 и 0,4115 и получают соответственно
вес S, SOs и S04 в данной навеске.
Органическая и минеральная сера
При раздельном учете органической и минеральной серы в почвах посту¬
пают следующим образом.
Навеску воздушно-сухой почвы в количестве от 3 до 5 г помещают в
стаканчик, доливают 0,2 н. НС1 и фильтруют декантацией через воронку
со взвешенным фильтром (синяя лента). Почву обрабатывают НС1 до исчез¬
новения реакции на S04~. Таким способом из почвы удаляется минеральная
форма серы. Остаток почвы вместе с фильтром высушивают при 45—50° С
в течение 1,5—2 час. По охлаждении в эксикаторе остаток почвы взвешивают,
после чего тщательно растирают и перемешивают. Из остатка почвы берут
среднюю пробу 1—1,5 г и в ней определяют серу, связанную с органическим
веществом, как описано выше при определении валового содержания серы
в почве. Минеральную серу вычисляют по разности между валовым коли¬
чеством серы и количеством серы, связанным с органическим веществом.
Указанный способ пригоден также для почв, содержащих серу в форме
гипса.
[ОПРЕДЕЛЕНИЕ ВОДНОРАСТВОРИМЫХ ВЕЩЕСТВ
Водные вытяжки из почв, не содержащих соду
Анализ водной вытяжки является одним из основных приемов при ис¬
следовании химического состава засоленных почв, а также при изучении
динамики некоторых питательных элементов почвы. Существует много раз¬
личных вариантов приготовления водных вытяжек. Наиболее распростра¬
нен следующий метод получения водной вытяжки: отношение почвы к воде
1 : 5, взбалтывание 3 мин. и фильтрование. При полном анализе водной
вытяжки обычно определяют: плотный остаток, щелочность нормальных,
карбонатов и бикарбонатов, СГ, S04‘, Са2+,* Mg2Y К+, Na+, R203, Si02,
Р206, нитраты, нитриты, органическое вещество.
Чаще в водной вытяжке определяют лишь плотный остаток, щелочность
нормальных карбонатов и бикарбонатов, СГ, S04", Са2+ и Mg2*, К+, Na+.
30
3. Г. Ильковская, А. С. Коновалова
При невозможности определить Na количество натрия в водной вытяжке
рассчитывается по разности между суммой миллиграмм-эквивалентов анио¬
нов и катионов. Недостающее количество катионов по отношению к сумме
анионов относят к натрию.
100 г почвы помещают в банку с притертой пробкой и заливают 500 мл
воды, лишенной углекислоты. Банку закрывают пробкой и, переворачивая,
встряхивают в продолжение 3 мин., после чего тотчас же производят филь¬
трование. При сильнозасоленных почвах достаточно брать 50 г почвы и
250 мл воды.
Фильтрование водных вытяжек часто представляет большие затруднения.
Если почва богата растворимыми солями и не имеет щелочной реакции,
фильтрование идет быстро и гладко, вытяжка получается без опалесценции.
Когда почва бедна растворимыми солями или имеет щелочную реакцию,
минеральные коллоидные частицы почвы легко могут проходить через
фильтр, засорять его и замедлять фильтрование. Чтобы по возможности
избежать этих явлений, водную вытяжку лучше фильтровать через склад¬
чатый фильтр из плотной бумаги. На фильтр наливают вытяжку вместе с
почвой, чтобы задержать прохождение через фильтр коллоидных частиц.
Первые порции вытяжки (25—50 мл) рекомендуется вообще отбрасывать.
Следующие порции вытяжки приходится перефильтровывать несколько
раз, пока фильтрат водной вытяжки не будет прозрачным.
Практически удобно поступать следующим образом. Воронку для вы¬
тяжки закрепляют на штативе. Под воронку подставляют маленький ста¬
канчик. Отбросив первую порцию вытяжки, последующие порции пере-
фильтровывают до тех поп, пока не будет получаться прозрачный фильтрат
водной вытяжки. После этого стаканчик отставляют и осторожно переносят
воронку на колбу, в которую производится дальнейшее фильтрование.
Если колба загрязнена мутной вытяжкой, то ее очень трудно очистить пе-
рефильтровыванием и лучше в таком случае заменить ее чистой колбой.
Если при перефильтровывании не удается получить прозрачную вытяжку,
рекомендуется употреблять двойной фильтр. Воронку желательно брать
такую, чтобы на ней поместилась вся водная суспензия. Обычно для этого
требуется воронка диаметром 15 см. Когда вытяжка полностью отфильтрует-
ся, можно приступить к анализу. Определение щелочности или кислотно¬
сти водной вытяжки необходимо производить непосредственно после при¬
готовления самой вытяжки: анализ вытяжки, постоявшей длительное время,
может дать неверные результаты.
Перед проведением анализа водной вытяжки следует сделать качествен¬
ную пробу на содержание в ней СГ, SO^ и Са2+. Это даст возможность пра¬
вильно выбрать объем вытяжки для анализа отдельных компонентов. Если
реакции на СГ, SOf и Са2+ дают слабую муть, надо брать объем вытяжки
для анализа возможно больший, например для определения щелочности и хло¬
ра — по 50 дел, SOj"—50—100 мл, а кальция и магния — не менее 100 мл.
Труднее выбрать объем вытяжки для анализа, когда в ней солей настоль¬
ко много, что качественная проба дает обильные осадки. В таких случаях
для определения хлора приходится брать 1—5 мл, для SO4” — 10 мл, а для
определения кальция, магния и плотного остатка — только 10—25 мл.
При очень концентрированных растворах водной вытяжки или грунтовой
воды удобно приготовить разведенный раствор в мерной колбе, разбавив
исходный раствор в 10 или 20 раз, и производить анализ этой разведенной
вытяжки.
Определение общей суммы воднорастворимых
веществ. 25—50 мл водной вытяжки выпаривают в тарированном су¬
шильном стакане или в платиновой чашке на водяной бане. Тщательно об¬
терев снаружи стаканчик фильтровальной бумагой, 3 часа сушат его в тер¬
Определение в почве обменных катионов, емкости поглощения
31
мостате при температуре 105° С, после чего охлаждают в эксикаторе, взве¬
шивают стаканчик с осадком и рассчитывают количество сухого плотного
остатка на 100 г высушенной почвы.
Определение щелочности водной вытяжки от
нормальных карбонатов. К 25—50 мл водной вытяжки в ко¬
нической колбочке на 100 мл прибавляют 1—2 капли раствора фенолфта¬
леина. Окрашивание жидкости в розовый цвет означает, что в ней присут¬
ствуют нормальные карбонаты; в этих случаях жидкость титруют 0,02 н.
раствором H2S04 до исчезновения окраски. Титрование щелочности надо
производить по возможности скорее после взятия пробы, чтобы избежать
добавочного насыщения вытяжки углекислотой воздуха.
Пример. Предположим, что на титрование 50 мл водной вытяжки затрачено 2 мл
0,02 н. раствора H2S04. При титровании по фенолфталеину карбонатная форма щелоч¬
ности переводится в бикарбонатную. Таким образом, при данном способе анализа оттит-
ровывается половина количества карбонатов щелочей: следовательно, на все количество
нормальных карбонатов, содержащихся в 50 мл водной вытяжки, пойдет не 2 мл, а 4 мл
0,02 н. раствора H2S04. 1 мл 0,02 н. раствора H2S04 соответствует 0,00060 г С03.
Содержание нормальных карбонатов (в %) будет равно: 0,00060-4-100-а: 10, так как
50 мл вытяжки соответствует 10 г почвы (а — коэффициент пересчета на почву, высушен¬
ную при 105° С). Процентное содержание нормальных карбонатов, умноженное на 1000
и деленное на 30 (эквивалентный вес СО|~), даст их количество в миллиграмм-эквива¬
лентах на 100 г почвы.
Определение общей щелочности водной вытяж-
к и. После прибавления фенолфталеина, если в почве на оказалось нормаль¬
ных карбонатов или они были оттитрованы до бикарбонатов, в ту же кол-,
бочку с водной вытяжкой прибавляют 2 капли раствора метилоранжа и
продолжают титровать той же кислотой до появления едва заметной розо¬
вой окраски. Титрование необходимо вести при наличии другой колбочки
такого же размера с таким же количеством вытяжки и метилоранжа. Обе
колбочки должны стоять рядом на листе белой бумаги. При титровании надо
сравнивать цвет жидкости в обеих колбочках и, как только оттенок в кол¬
бочке, к которой приливалась кислота, изменится по сравнению с оттенком
контрольной колбочки (слегка порозовеет), заканчивать титрование.
Все количество кислоты, пошедшей на титрование в присутствии фенол¬
фталеина и метилоранжа, будет соответствовать общей щелочности взятого
в анализ количества водной вытяжки; эти данные выражают в величи¬
нах НСО3.
Пример пересчета. Всего на титрование 50 мл водной вытяжки пошло 5 мл
0,02 н. раствора H2S04.1 мл этой кислоты соответствует 0,00122 г НС08. Общая щелочность
(в %) в таком случае составит: 0,00122*5• 100• а :Д0.
Для пересчета в миллиграмм-эквиваленты полученное значение общей щелочности
надо умножить на 1000 и разделить на 61 (эквивалентный вес НСО"). Следует обязатель¬
но делать контрольное определение щелочности дистиллированной воды и вносить соот¬
ветствующую поправку.
Определение щелочности, вызываемой углекис¬
лыми щелочами. Эту щелочность определяют лишь по мере надоб¬
ности. Для этого необходимо выделить из водной вытяжки углекислые
кальций и магний. 50 мл вытяжки выпаривают в небольшой платиновой
чашечке досуха, прокаливают при температуре 300° С и, не давая чашке
остыть, обрабатывают ее содержимое небольшими порциями свежепроки-
пяченной воды. Тщательно перемешав содержимое чашки палочкой с рези¬
новым наконечником, фильтруют раствор в небольшой стаканчик через
фильтр диаметром 7—9 см с белой лентой. Достаточно промыть осадок в
чашке 10—12 раз и дважды ополоснуть фильтр. Чем меньше порции употреб¬
32
3. Г. Илъковская, Л. С. Коновалова
ляемой воды, тем быстрее будет окончено растворение щелочей и тем полнее
будут отделены щелочные карбонаты от щелочноземельных. Эту операцию
выпаривания фильтрата, прокаливания и отмывания горячей водой надо
повторять до тех пор, пока при растворении осадка в чашке раствор
будет оставаться прозрачным, что указывает на отсутствие углекислых
кальция и магния.
Полученный фильтрат, содержащий щелочные углесоли как нормальных,
так и двууглекислых карбонатов, титруют по охлаждении в присутствии
метилоранжа 0,02 н. раствором H2S04 до розового оттенка. На основании
результатов этого титрования рассчитывают содержание углекислых ще¬
лочей в водной вытяжке.
Пример пересчета. На 50 .мл водной вытяжки израсходовано при титрова¬
нии углекислых щелочей 6 мл 0,02 н. раствора H2S04. Количество углекислых щелочей
(в %) будет [равно: 0,00122»6*100*а: 10, так как 50 лед водной вытяжки соответствуют
10 г почвы, а 1 мл 0,02 н. раствора серной кислоты — 0,00122 г НСО" (а — коэффициент
пересчета на почву, высушенную при 105° С).
Щелочность от щелочноземельных карбонатов равна разности между
общей щелочностью и щелочностью углекислых щелочей (в мг-экв).
Определение воднорастворимого хлор-иона. При
определении хлора титрованием раствором AgNOs требуется нейтральная
реакция. Поэтому хлор определяется в той вытяжке, в которой было произ¬
ведено титрование общей щелочности. Если из-за большого содержания хло¬
ра в водной вытяжке объем, взятый для определения щелочности, слишком
велик, то берут меньший объем и нейтрализуют взятую вытяжку 0,02 н.
раствором H2S04 в присутствии метилоранжа до розоватого окрашивания.
Затем прибавляют к титруемому раствору 1 мл 10%-ного раствора хромо¬
вокислого калия (К2Сг04) и титруют 0,02 н. раствором AgNOs до появления
красноватой окраски. Для контроля при титровании берут колбочку с та¬
ким же количеством водной вытяжки и 1 мл 10%-ного раствора хромово¬
кислого калия для сравнения жидкостей в обеих колбочках.
Умножая количество миллилитров израсходованного раствора AgN03
на 0,00071, получают содержание хлор-иона во взятом для анализа коли¬
честве водной вытяжки. Затем пересчитывают содержание хлор-иона на
100 г почвы, высушенной при 105° С.
Полученное процентное содержание хлор-иона умножают на 1000, делят
на 35,5 (эквивалентный вес хлора) и получают количество миллиграмм-
эквивалентов СГ на 100 г высушенной почвы. В применяющейся в анализе
воде должно быть определено содержание хлор-иона и внесена соответствую¬
щая поправка.
Определение воднорастворимых сульфат-ио¬
нов. Взятый для определения сульфат-ионов объем водной вытяжки, если
он слишком велик, упаривают примерно до 50 мл\ вытяжку подкисляют
10%-ным раствором НС1 до явно кислой реакции, доводят до кипения и в
кипящем состоянии осаждают сульфат-ионы кипящим 10%-ным раствором
ВаС12 (берут 2—10 мл раствора ВаС12 в зависимости от ожидаемого коли¬
чества сульфатов), кипятят несколько минут и оставляют в теплом месте
на 4 часа. На следующий день, определив полноту осаждения, раствор филь¬
труют через плотный беззольный фильтр с синей лентой, осадок промывают
горячей водой, подкисленной тем же раствором соляной кислоты, до ис¬
чезновения в промывных водах реакции на Ва2+ (проба с 10%-ным раство¬
ром H2S04). Далее осадок с фильтром переносят во взвешенный тигель,
просушивают и озоляют фильтр на электрической плитке. После озоления
прокаливают осадок в муфеле при невысокой температуре (500° С) до постоян¬
ного веса. Вес полученного осадка BaS04, помноженный на 0,4114, даст
количество SO*- в граммах во взятом для анализа количестве водной вытяж¬
Определение в почве обменных катионов, емкости поглощения
33
ки. Полученный результат пересчитывают на 100 г почвы, высушенной
при 105° С.
Для перевода полученных данных в миллиграмм-эквиваленты процент
ЭОГ умножают на 1000 и делят на 48 (эквивалентный вес S04~).
Содержание S04” в водной вытяжке можно определить также объемным
методом, как указано в экспресс-методе определения гипса по Айдиняну.
Для этого водную вытяжку пропускают через Н-катионит. К определенному
объему вытяжки приливают такой же объем ацетона или спирта и титруют
0,02 н. раствором ВаС12 в присутствии индикатора нитхромазо. Если pH
титруемой жидкости выше 2,0, то необходимая кислотность достигается
прибавлением 0,02 н. НС1.
Определение воднорастворимого кальция. Пе¬
ред определением кальция необходимо сделать пробу на присутствие в вод¬
ной вытяжке полуторных окислов. Если водную вытяжку готовят из кислых
почв, присутствие в вытяжке полуторных окислов вполне вероятно. В та¬
ких случаях к 50—100 мл водной вытяжки прибавляют 1—2 капли раствора
метилрота, и если раствор имеет красный цвет, то выделяют полуторные
окислы 10%-ным раствором NH4OH, осадок отфильтровывают через рыхлый
фильтр и промывают несколько раз 1 %-ным горячим раствором азотнокисло¬
го аммония, в который прибавлены 1—2 капли 10%-ного раствора NH4OH
(до желтой окраски с метилротом).
Если водную вытяжку готовят из карбонатных почв, то при существую¬
щей в этих почвах щелочной реакции трудно ожидать наличия полуторных
окислов. Но пробу на содержание полуторных окислов необходимо сделать
ввиду растворимости алюминия в щелочной среде, для чего вытяжку под¬
кисляют 10 %-ным раствором НС1 и в присутствии метилрота прибавляют
10%-ный раствор NH4OH до щелочной реакции.
После выделения полуторных окислов щелочную водную вытяжку под
кисляют 10 %-ным раствором уксусной кислоты до розового окрашивания
по метилроту, прибавляют 2—3 г сухого хлористого аммония и осаждают
кальций кипящим насыщенным раствором щавелевокислого аммония, при¬
бавляя его каплями в кипящую анализируемую водную вытяжку. После
4-часового стояния в теплом месте раствор отфильтровывают через фильтр
диаметром 7—9 см с синей лентой. Осадок щавелевокислого кальция промы¬
вают теплой водой с несколькими каплями 10%-ного раствора NH4OH до
прекращения реакции на щавелевую кислоту (проба с AgN03 без подкисле-
ния азотной кислотой). Затем щавелевокислый кальций растворяют в 5%-ном
растворе горячей H2S04 и титруют 0,05 н. раствором КМп04. 1 мл 0,05 н.
раствора КМп04 соответствует 0,001 г кальция. Полученный результат
пересчитывают на 100 г почвы, высушенной при 105° С. Процент кальция,
умноженный на 1000 и разделенный на 20 (эквивалентный вес кальция),
дает количество миллиграмм-эквивалентов воднорастворимого кальция на
100 г высушенной при 105° почвы.
Определение воднорастворимого магния. После
выделения кальция и промывания осадка от хлор-иона собирается не менее
150—200 мл фильтрата. Его упаривают до 100 мл. Затем уксуснокислый
раствор нейтрализуют 10%-ным раствором NH4OH, подкисляют 10%-ным
раствором НС1, прибавляют 5—10 мл 10%-ного раствора фосфорнокислого
натрия (Na2HP04) и доводят до кипения. После этого нейтрализуют раствор
до щелочной реакции 10 %-ным раствором NH4OH, а затем прибавляют
15—20 мл крепкого раствора NH4OH и оставляют его на 18—36 час. Если
ожидается много магния, раствор перед осаждением магния не следует силь¬
но концентрировать. Аммиак также рекомендуется прибавлять осторожно,
небольшими порциями, чтобы избежать выпадения гидрата окиси магния.
Если случится, что магний выпал в этой форме, осадок следует растворить
34
3. Г. Илъковская, А. С. Коновалова
в крепкой соляной кислоте при нагревании, затем переосадить магний вновь,
т. е. избыток соляной кислоты нейтрализовать аммиаком, подкислить со¬
ляной кислотой, прибавить 5—10 мл фосфорнокислого натрия, осторожно
довести до щелочной реакции 10%-ным раствором NH40H, а потом также
осторожно прибавить 15—20 мл крепкого раствора NH4OH.
Если раствор при осаждении магния становится мутным от прибавления
аммиака или в растворе появляются хлопья, характерные для полуторных
окислов, то это показывает, что полуторные окислы не были полностью вы¬
делены. В таких случаях магний осаждать нельзя и для определения магния
надо взять новую пробу вытяжки, удалить полуторные окислы, осадить,
как обычно, кальций, выделить его и затем осадить магний. Бывают случаи,
когда магний выпадает в виде кристаллического осадка фосфорнокислого
аммоний-магния и гидрата окиси магния одновременно. В таких случаях
лучше отфильтровать полученный осадок магния, затем растворить его в
горячем 10%-ном растворе НС1 и вновь осадить магний фосфорнокислым
натрием.
Осадок Mg2P207, полученный после прокаливания соли магния в муфеле
при 850—900° С, доводят до постоянного веса, умножают на 0,218, полу¬
чают вес магния во взятом для анализа количестве водной вытяжки.
Полученные данные пересчитывают на 100 г высушенной почвы.
Для перевода в миллиграмм-эквиваленты процент магния умножают
на 1000 и делят на 12,16 (эквивалентный вес магния).
Трилонометрический метод определения каль¬
ция и магния в водной вытяжке. Анализ ведется так же,
как описано выше при определении обменных катионов.
К определенному объему водной вытяжки, подогретой до 60—70° С,
в случае надобности разведенной водой до 50—100 мл, прибавляют 5 мл
аммиачного буферного раствора, 10—15 мг индикатора хромогена черного
и титруют 0,01 н. раствором трилона Б до перехода вишнево-красной ок¬
раски через фиолетово-синюю в голубую.
Вычисление суммы кальция и магния (в мг-экв) производится по урав¬
нению
А «0,01 *К *100
Са -(- Mg — ^ ,
где А—количество трилона, пошедшее на титрование, мл; 0,01 — нормальность раствора
трилона; К — поправка к титру трилона; С — навеска почвы, соответствующая количе¬
ству вытяжки, взятой на титрование, г; 100 — коэффициент для пересчета результатов
анализа на 100 г почвы.
Определение кальция в водной вытяжке. Опре¬
деленный объем водной вытяжки помещают в коническую колбу емкостью
250 мл, при необходимости разбавляют водой до 50—100 мл, прибавляю!
2 мл 2 н. раствора NaOH, вносят 10—15 мг мурексида и титруют 0,01 н. рас¬
твором трилона Б до перехода ярко-пурпуровой окраски анализируемого
раствора в лиловую. Титрование рекомендуется вести со свидетелем. Вы¬
числение кальция (в мг-экв) ведут по уравнению А • 0,01 -К- 100/С с теми же
значениями членов уравнения, как и при определении суммы кальция и
магния.
Содержание магния в водной вытяжке вычисляют по разности между
суммой кальция и магния и содержанием кальция (в мг-экв на 100 г почвы).
В случае необходимости устранить влияние меди прибавляют несколько
кристалликов диэтилдитиокарбамата натрия; вредное влияние марганца
устраняют солянокислым гидроксиламином.
Определение в почве обменных катионов, емкости поглощения
35
Водная вытяжка из почв, содержащих соду
Если водная вытяжка готовится из почвы, содержащей соду, то фильтрат
ее бывает более или менее окрашен органическим веществом. Иногда окра¬
шивание бывает настолько сильным, что определение щелочности или хлора
в фильтрате становится невозможным. В таком случае титрование водной
вытяжки нужно проводить после значительного разбавления водой. При
очень сильном окрашивании лучше воспользоваться электрометрическим
титрованием. Для этого поступают следующим образом. Из какой-нибудь
прозрачной водной вытяжки берут две пробы по 25—50 мл в два стакана
и оттитровывают в одном стакане нормальную щелочность, а в другом —
Рис. 4. Схема установки
для потенциометрическо¬
го титрования
Объяснение см. в тексте
общую. Затем берут 25—50 мл анализируемой темноокрашенной водной
вытяжки и составляют цепь, приведенную на рис. 4. Один из двух хорошо
промытых водой платиновых электродов вставляют в стакан 1 с темноокра¬
шенной вытяжкой, а другой — в стакан 2 с оттитрованной щелочностью,
по которой будет производиться электротитрование в темной вытяжке. Эти
стаканы соединяют агаровым сифоном 6, которым обычно пользуются при
потенциометрическом титровании. Один электрод присоединяют к клемме
ключа переключателя 4, а другой — через гальванометр 3 к другой клемме
ключа. В оба стакана прибавляют хингидрон. При нажиме ключа стрелка
гальванометра покажет отклонение, связанное с разностью потенциалов
в двух стаканах. Из бюретки 5, установленной над стаканом с темноокрашен¬
ной водной вытяжкой, по каплям прибавляют 0,02 н. раствор серной кисло¬
ты, перемешивают и при помощи ключа проверяют, есть ли еще отклонение
стрелки гальванометра. Серную кислоту прибавляют до тех пор, пока при
нажиме ключа стрелка гальванометра перестанет отклоняться.
После этого записывают количество миллилитров 0,02 н. раствора сер¬
ной кислоты, пошедшее на титрование нормальных карбонатов. После оп¬
ределения нормальных карбонатов проводят электротитрование бикарбона¬
тов водной вытяжки. Для этого берут другой стакан с оттитрованной общей
щелочностью, прибавляют в него хингидрон, перемешивают, соединяют
сифоном со стаканом, в котором находится титруемая вытяжка й продолжают
титровать общую щелочность в том же стакане и таким же образом, т. е.
прибавляют 0,02 н. раствор серной кислоты, перемешивают, нажимают,
ключ и смотрят, отклоняется ли стрелка гальванометра. Кислоту добавляют
осторожно, по каплям, до тех пор, пока стрелка гальванометра перестанет
отклоняться, и вновь записывают количество миллилитров 0,02 н. раствора
кислоты, пошедшее на титрование. Первый отсчет по бюретке относится к
36
3. Г. Илъковская, А. С. Коновалова
карбонатной щелочности, второй показывает общую щелочность. Расчет
производится обычным путем, как это указано выше.
Для определения хлора в темноокрашенной вытяжке надо разрушить
ее органическое вещество перекисью водорода. Для этого в фарфоровой
чашечке выпаривают 25 мл окрашенной водной вытяжки, прибавляя к ней
1—2 мл крепкой перекиси водорода. Операцию разрушения органического
вещества, т. е. выпаривание вытяжки с перекисью водорода, приходится
иногда производить 2—3 раза. После этого жидкость становится прозрач¬
ной, и титрование хлора ведется обычным способом в этой же чашке.
Определение хлор-иона в водных вытяжках
меркуриметрическим методом1
Определение хлор-иона в водных вытяжках проводится в пробе после
определения общей щелочности или в аликвоте водной вытяжки.
В случае высокого содержания хлор-иона в вытяжке и необходимости
ее разбавления определение хлор-иона производится без предварительного
определения щелочности.
В первом случае к пробе (10—25 мл) после определения общей щелочно¬
сти добавляют 10 капель двойного индикатора (дифенилкарбазон -f бром-
феноловый синий) и титруют 0,02 н. раствором Hg(N03)2 до перехода окраски
из оранжевой в сиреневую. Присутствие метилоранжа, применяемого при
определении щелочности, несколько мешает улавливать переход окраски
двойного индикатора в точке эквивалентности. В водных вытяжках, содер¬
жащих высокое количество хлор-иона, после разбавления проб необходимо
довести pH до величины 3,0—3,5, прибавляя по каплям к испытуемому
раствору, содержащему 10 капель двойного индикатора, 0,05 н. HNOs
до перехода окраски из сиреневой в желтую. При этом хлор-ион оттитровы-
вают 0,02 н. раствором Hg(N03)2. В этом случае окончание титрования улав¬
ливается яснее, так как отсутствует метилоранж. Следует отметить, что ко¬
нец титрования в обоих случаях улавливается значительно отчетливее,
чем при определении хлор-иона по методу Мора.
Содержание хлор-иона (в %) рассчитывается по формуле
А- 0,00071* 100* а
где А — количество раствора Hg(NOs)2, пошедшее на титрование, мл; 0,00071 — содержа¬
ние хлора в 1 мл 0,02 н. раствора Hg(NOs)2, г; 100 — коэффициент пересчета результатов
анализа на 100 г почвы, т. е. в проценты; С — навеска почвы, соответствующая количеству
раствора, взятого на титрование, г; а — коэффициент пересчета на высушенную почву
при 105° С.
Процент хлора, умноженный на 1000 и разделенный на 35,5 (эквивалентный вес
хлора), дает количество миллиграмм-эквивалентов хлора на 100 г почвы. $
Реактивы. 1. 0,02 н. раствор азотнокислой ртути. 16,23 г Hg(NOs)2 растворяют
в 100 мл дистиллированной воды, содержащей 1,0—1,5 мл концентрированной HNOs,
и разбавляют водой до 5 л. Можно также раствор для титрования приготовить из окиси
ртути. Для этого 10,83 г окиси ртути помещают в стеклянный стакан, смачивают водой,
растирают палочкой, приливают 15—20 мл HN03 (уд. вес 1,4) и растворяют при нагре¬
вании. Разбавляют водой, фильтруют и доводят до 5 л. Титр устанавливают по стандарт¬
ному раствору NaCl, приготавливаемому из фиксанала.
2. Двойной индикатор. Берут на аналитических весах 0,5 г дифенилкарбазона, 0,05 г
бромфенолового синего и растворяют в 100 мл спирта — ректификата. Реактив хранят
в темной склянке. Готовить более 100 мл раствора не рекомендуется, так как при хранении
он портится.
1 Описание метода дано 3. С. Власовой и В. А. Молодцовым (1966).
Определение в почве обменных катионов, емкости поглощения
37
Сводка данных анализа водной вытяжки
Все данные по водной вытяжке принято выражать в процентах и мил¬
лиграмм-эквивалентах на почву, высушенную при 105° С. После окончания
анализа водной вытяжки необходимо дать сводную таблицу величин, полу¬
ченных для всех ее ионов. В качестве примера приводится табл. 1.
Натрий в данном случае найден по разности в результате пересчета.
Для этого суммируют величины миллиграмм-эквивалентов всех анионов
(НСОз, СГ, SO4'), что составляет для нашего примера 2,57; сумма катио¬
нов (Са2+ nMg2+) составляет 0,32 мг-экв. Разница между этими числами равна
2,57 — 0,32 = 2,25 мг-экв; ее и относят к содержанию вводной вытяжке нат¬
рия. Умножая количество миллиграмм-эквивалентов натрия на 23 (эквива¬
лентный вес натрия) и разделив полученное число на 1000, получаем коли¬
чество натрия в процентах (0,052).
Следует также просуммировать проценты, найденные по анализу ионов
данной вытяжки, и сопоставить полученную сумму с выраженной в про¬
центах величиной сухого остатка вытяжки. Во взятом примере эта сумма
составляет 0,188% и отличается от полученного сухого остатка на 0,006,
что составляет 3%.
При анализе водной вытяжки иногда бывает необходимо сделать пересчет
содержания каждого ее элемента в проценты от суммы миллиграмм-экви¬
валентов, что и дается в третьей строке табл. 1.
Таблица 1
Данные анализа водной вытяжки
Разрез 201, глубина 365—375 см
Единица измерения
Сухой
остаток
СО^-
нсо;
С1-
S02-
Са^
Mg*+
Na+
по разно¬
сти
Сумма-
анионов и
катионов
% на высушенную
0,188
Нет
0,064
0,032
0,029
0,004
0,001
0,052
0,182
почву
Мг-экв на 100 г
1,05
0,91
0,61
0,21
0,11
2,25
5,14
почвы
% от суммы мг-экв
—
—
20,4
17,7
11,9
4,1
2,1
32,8
о
о
о
Определение суммы катионов водной вытяж-
к и (для проверки правильности анализа водной вытяжки). 20 мл водной
вытяжки пропускают через ионообменную колонку, заполненную Н-ка-
тионитом.
Для устройства ионообменной колонки в бюретку емкостью 25 мл поме¬
щают небольшое количество стеклянной ваты, заполняют до 15 мл дистил¬
лированной водой и осторожно всыпают 3 г смоченного водой катионита
КУ-2, предварительно насыщенного водородом. Водную вытяжку пропуска¬
ют через такую колонку со скоростью 0,5 мл/мин. Когда пройдет вся вытяж¬
ка, пропускают 30—40 мл дистиллированной воды (со скоростью 2 мл/мин).
Вытяжку и промывные воды собирают в мерную колбу емкостью 100 мл
и доводят до метки водой. Полученный раствор, в котором все катионы за¬
мещены водородом, служит для определения суммы катионов.
25—50 мл этого раствора помещают в коническую колбу, кипятят для
удаления углекислоты, охлаждают и титруют при продувании воздуха,
лишенного углекислоты, 0,05 н. раствором буры в присутствии индикатора
Гроака до перехода красновато-лиловой окраски в бледно-зеленую.
Содержание катионов (в мг-экв на 100 г почвы) равно:
0.0,05-ЯМ 00.500
33
3. Г. Ильковская, Л. С. Коновалова
где а — количество раствора буры, пошедшее на титрование, мл; 0,05 — нормальность
буры; К — поправка к титру буры; 100 — объем, до которого доведена вытяжка после
ее пропускания через ионообменную колонку, мл; 500 — коэффициент для пересчета
на 100 г почвы; b — количество вытяжки, взятой на титрование, мл; с — количество вод¬
ной вытяжки, взятой для пропускания через катионит, мл.
Содержание в водной вытяжке натрия и калия вычисляют по разности
между суммой полученных катионов и суммой кальция и магния, определен¬
ной титрованием трилоном.
Примечание. При значительной щелочности водной вытяжки перед пропуска¬
нием через обменную колонку ее нейтрализуют 0,02 н. раствором НС1 в присутствии метил”
оранжа.
Реактивы. 1. Раствор буры. Для приготовления 0,01 н. раствора буры навеску
1,907 г перекристаллизованной десятиводной буры (Na2B407* ЮН20), растворяют в мерной
колбе на 1 л водой, свободной от углекислоты. Для перекристаллизации буры 150 г про¬
дажной соли буры растворяют при нагревании в 300 мл воды и раствор фильтруют через
складчатый фильтр в чашку или стакан, которые охлаждают холодной водой или снегом*
Выпавшие кристаллы буры отфильтровывают от раствора и сушат между листами филь¬
тровальной бумаги. Перскристаллизованную соль сохраняют в склянке с притертой
пробкой.
2. Индикатор Гроака (смешанный индикатор метиленблаумегилрот). 100 мл насыщен¬
ного спиртового раствора метилрота смешивают с А мл 1%-ного водного раствора метилен-
блау. Переход окраски от красновато-лиловой в бледно-зеленую.
3. Подготовка катионита: для насыщения водородом катионит промывают на бюхне-
ровской воронке 10%-ным раствором HG1. В первый раз катионит промывают до потери
реакции на железо. После промывания катионита соляной кислотой избыток ее отмывают
дистиллированной водой.1
После использования в работе емкости катионита она восстанавливается промыва¬
нием 60 мл 10%-ного| раствора НС1 (из расчета на 3 г катионита) со скоростью 2 мл/мин%
Избыток кислоты отмывается 300 мл дистиллированной воды с той же скоростью.
ОПРЕДЕЛЕНИЕ ПОДВИЖНЫХ СОЕДИНЕНИЙ В ПОЧВЕ
Трилонометрический метод определения подвижного (доступного)
магния в бедных поглощенными основаниями почвах
по Мазаевой (1962)
Определение подвижного магния в почвах проводится в 1 н. КС1-вытяж-
ке. 100 г почвы, просеянной через сито с отверстиями в 1 мм, помещают в
бутылку, заливают 250 мл 1 н. раствора КС1, взбалтывают на ротаторе в
течение 1 часа и фильтруют через складчатый фильтр.
Для определения суммы Mg, Са и Мп в стакан емкостью 150 мл помещают
50 мл вытяжки, добавляют 5 мл аммиачной буферной смеси (20 г химически
чистого NH4C1 и 100 мл 25 %-ного раствора NH4OH в 1 л воды), 2мл1 %-ного
раствора солянокислого гидроксиламина, несколько кристаллов диэтил-
дитиокарбамата натрия, 50 мл дистиллированной воды и на кончике ножа
сухой индикатор— кислотный хром темно-синий, смешанный с NaCl в отно¬
шении 1 : 99. Затем вытяжку титруют 0,02 н. раствором трилона Б до пе¬
рехода окраски раствора из вишнево-красной в чисто-голубую. При пользо¬
вании фотоэлектрическим титриметром типа ФЭТ-УНИИЗ титрование про¬
изводят до остановки стрелки амперметра.
Для определения суммы Са и Мл берут 50 мл вытяжки, добавляют 2 мл
1 %-ного раствора солянокислого гидроксиламина, 10 мл боратной буфер¬
ной смеси (6 мл 0,05 н. раствора буры и 4 мл 0,02 н. раствора борной кисло¬
ты), 10 мл аммиачной буферной смеси и на кончике ножа сухой мурексид.
Определение в почве обменных катионов емкости поглощения
39
К вытяжке добавляют из бюретки 0,02 н. раствор трилона Б до перехода
окраски из оранжевой в малиновую. Затем приливают 2 мл 20 %-ного рас¬
твора NaOH и продолжают титрование до остановки стрелки титриметра
или перехода ярко-пурпурной окраски в лиловую при визуальном титровании.
Магний (в %) определяют по формуле:
{а — Ь)-К *0,0004032 *100
MgO g ,
где а — количество 0,02 и. раствора трилона Б, пошедшее на титрование суммы Mg,
Са и Мп, мл\ b—количество 0,02 н. раствора трилона Б, пошедшее на титрование суммы
Са и Мп, мл; К — поправочный коэффициент к титру трилона Б; С — навеска почвы,
соответствующая объему вытяжки, взятой на титрование, г; 0,0004032 — количество MgO,
соответствующее 1 мл 0,02 н. раствора^трилона Б, г\ 100 — коэффициент пересчета
результатов анализа на 100 г почвы, т. е. в проценты.
Метод Шахтъиабеля для определения доступного растениям магния
(Schachtschabel, 1954)
5 г воздушно-сухой почвы, пропущенной через сито диаметром 1 мм,
помещают в колбу на 100 мл, приливают 50 мл 0,025 н. раствора СаС12 и
встряхивают на механическом встряхивателе 2 часа. Фильтруют через плот¬
ный фильтр, свободный от следов магния.
10 мл прозрачного фильтрата помещают пипеткой в мерную колбу на
50 мл, прибавляют 5 мл 2 %-ного раствора поливиола, затем 5 мл 0,05 %-ного
раствора титанового желтого. После прибавления титанового желтого до¬
ливают приблизительно до 40 мл дистиллированной водой, затем приливают
5 мл 2 н. раствора NaOH и доливают водой до метки. Следует строго соблю¬
дать порядок приливания реактивов. После внесения каждого реактива
раствор в колбе необходимо перемешивать, но не встряхивать. Окраска
раствора развивается 30 мин. и остается стабильной в течение 1,5—2 час.
Оптическую плотность измеряют в кювете с просматриваемым слоем в 10 мм,
при 545 нм, применяя в качестве нулевого раствора холостую пробу. При
приготовлении холостой пробы берут вместо почвенной вытяжки 10 мл
0,025 н. раствора СаС12 и далее обрабатывают, как и испытуемые растворы.
Шкалу образцовых растворов готовят ежедневно для каждой серии ана¬
лизов. Помещают в мерные колбы емкостью 50 мл отдельно 2; 5 и 15 мл об¬
разцового рабочего раствора магния, содержащего в 1 мл 0,01 мг Mg, при¬
ливают 10 мл 0,025 н. раствора СаС12 и далее-обрабатывают, как описано
выше. Если содержание Mg в почве будет выше 15 мг на 100 г почвы, то
вместо 10 мл вытяжки следует брать 5 или 2 мл, доливая до 10 мл 0,025 н.
раствором СаС12.
В табл. 2 приводятся данные, характеризующие обеспеченность почв
магнием в зависимости от их механического состава.
Таблица 2
Обеспеченность почв доступным магнием по методу Шахтшабеля
(см. Bergmann, 1964)
Содержание
Mg, мг на 100 г лочвы
Легкие почвы
Суглинистые почвы
Глинистые почвы
Высокое
5,0
7,0
12,0
Среднее
го
1
сл
о
3,5—7,0
о
!
го
о
Низкое
2,5
3,5
7,0
40
3. Г. Илъковская, А. С. Коновалова
Реактивы. 1. Раствор хлористого кальция. Запасной 1 н. раствор. 110 г СаС12*
•6Н20 растворяют в воде, объем доводят до 1 л.
Рабочий 0,025 н. раствор готовят из запасного разбавлением дистиллированной во¬
дой в 40 раз. Рабочий раствор хлористого кальция должен отвечать 0,025 н., допускается
отклонение ±5 %.
2. 2%-ный раствор поливиола. Смешивают в стакане на 500 мл 20 г поливиола со
100 мл глицерина (дважды перегнанного), приливают около 400 мл горячей дистиллиро¬
ванной воды, нагревают при помешивании примерно до 90° С, пока раствор не станет од¬
нородным. После охлаждения раствор доводят водой до 1 л.
Если раствор не прозрачен, его отфильтровывают и сохраняют в темноте. При долгом
стоянии в растворе появляется муть; в этом случае раствор необходимо отфильтровать.
3. 0,05%-ный раствор титанового желтого. 125 мг титанового желтого растворяют
в воде, объем доводят до 250 мл. Раствор сохраняют в темноте; если при стоянии в растворе
появятся хлопья, его следует отфильтровать.
4. 2 н. раствор едкого натрия. 80 г NaOH (не содержащего карбонатов!) растворяют
в воде, объем доводят до 1 л.
5. Образцовый раствор магния. Запасной образцовый раствор магния. Помещают
в мерную колбу на 1 л 500 мг магниевой ленты и растворяют в 25 мл 10%-ной НС1. Когда
лента растворится, доводят до 1 д дистиллированной водой (pH приблизительно 1,5).
Запасной образцовый раствор содержит 0,5 мг Mg в 1 мл.
Для приготовления рабочего образцового раствора магния в мерную колбу на 1 л
помещают пипеткой 20 мл запасного раствора и доводят водой до 1 л (pH 3). Рабочий об¬
разцовый раствор содержит 0,01 мг Mg в 1 лед, что отвечает 1 мг Mg на 100 г почвы, если
для определения магния берут 10 мл вытяжки.
Растворы титанового желтого и поливиола при хранении в темноте могут
применяться в течение трех месяцев. Для анализа испытуемых образцов
и холостой пробы необходимо применять одни и те же реактивы.
Для получения хорошо воспроизводимых результатов необходимо при¬
менять только фильтры, свободные от следов Mg, фильтраты должны быть
прозрачными. Все части операций при определении магния должны выпол¬
няться во всей серии анализов одновременно, особенно необходимо одно¬
временное прибавление раствора титанового желтого.
Вытяжъа Тамиа для определения подвижных железа,
алюминия и кремнекисло ты
Для определения подвижных железа и алюминия, находящихся в виде
неорганических гелей в почвах, применяют оксалатные вытяжки. Раство¬
рителем для этого служит смесь щавелевой кислоты и щавелевокислого
аммония (раствор Тамма).
31,5 г щавелевой кислоты и 62,1 г щавелевокислого аммония растворяют
в 2,5 л дистиллированной воды. pH такого раствора 3,2—3,3.
К 2 г почвы приливают 100 мл раствора Тамма, взбалтывают 1 час на
ротаторе и фильтруют через фильтр с белой лентой. Затем фильтр с почвой
помещают обратно в ту же колбу, приливают еще раз 100 мл раствора Тамма,
вновь взбалтывают 1 час, после чего фильтруют и трижды промывают почву
холодной водой.
Фильтровать во второй раз надо в отдельную посуду, так как раствор
при этом часто бывает мутным и его приходится несколько раз перефильтро-
вывать до прозрачного состояния. После этого два прозрачных фильтрата
соединяют вместе и разрушают оксалаты и перешедшее в раствор органиче¬
ское вещество в фарфоровой чашке на бане 10%-ным раствором перекиси
водорода и 10%-ным раствором азотной кислоты, прибавляя их одновремен¬
но несколько раз. Затем раствор выпаривают 2—3 раза с 10 %-ным раствором
соляной кислоты и выделяют кремнекислоту обычным способом. Фильтрат
Определение в почве обменных катионов, емкости поглощения
41
от кремнекислоты собирают в мерную колбу на 200 мл. В фильтрате опреде¬
ляют железо и алюминий.
Примечание. Для карбонатных почв этот метод применим только после раз¬
рушения карбонатов и декальцирования 2%-ным раствором уксусной кислоты.
Трилонометрическое определение железа и
алюминия проводится последовательно в одной пробе.
Определение железа. Отбирают аликвотную часть фильтрата
(20—25 мл) в коническую колбу и осторожно нейтрализуют 10 %-ным раство¬
ром аммиака до начала выпадения гидроокисей. Прибавляют 10 мл 1 н. раст¬
вора соляной кислоты. Этим обеспечивают значение pH 1,0—1,4, необходимое
при определении железа. Перемешивают раствор до полного растворения
мути. Раствор доводят дистиллированной водой примерно до объема 100 мл.
Нагревают раствор до 60—70° С, прибавляют 1 мл 20%-ного раствора суль-
фосалициловой кислоты и титруют 0,05 н. раствором трилона Б до перехода
фиолетовой окраски в светло-желтую. Титрование, особенно под конец,
следует вести медленно.
По количеству израсходованного на титрование трилона Б вычисляют
содержание железа. 1 мл точно 0,05 н. раствора трилона Б соответствует
1,9965 мг Fe203 или 1,3975 мг Fe. Титр трилона Б устанавливают по 0,05 н.
раствору FeCl3, точное содержание железа в котором определяют весовым
способом после осаждения железа аммиаком.
Определение алюминия. При объемном трилонометрическом
определении алюминия используют пробу, в которой было оттитровано же¬
лезо.
К раствору после объемного трилонометрического титрования железа
с сульфосалициловой кислотой прибавляют для разрушения последней
1 мл НС1 (1 : 1) и кипятят 3—5 мин. Затем для связывания в комплекс алю¬
миния прибавляют 15 мл 0,1 н. раствора трилона Б, нагревают до 50—60° С,
вводят 2 капли спиртового раствора фенолфталеина и нейтрализуют 10%-ным
раствором аммиака до слабо-розовой окраски и затем осторожно по каплям
прибавляют НС1 (1 : 1) до исчезновения этой окраски. Приливают 20 мл
ацетатного буферного раствора с pH 5,5—5,6, кипятят 3 мин., охлаждают,
прибавляют 5 капель 0,1 %-ного водного раствора ксиленового оранжевого
и оттитровывают избыток трилона Б, не связавшийся с алюминием, 0,1 н.
раствором сульфата цинка до изменения окраски из желтой через оранже¬
вую в сиренево-фиолетовую. Это количество ZnS04 не учитывается. Затем
прибавляют 5 мл 4%-ного раствора NaF для связывания алюминия во фто-
ридный комплекс и кипятят 3 мин. Окраска раствора при этом становится
желто-оранжевой. Раствор охлаждают, прибавляют еще 2 капли ксиленово¬
го оранжевого и вновь точно оттитровывают трилон Б, выделившийся в
результате разрушения его комплекса с алюминием фторидом натрия —
0,1 н. раствором ZnS04. По количеству сернокислого цинка, израсходован¬
ному на второе титрование, вычисляют содержание А1203, считая, что 1 мл
точно 0,1 н. раствора ZnS04 соответствует 2,549 мг А1203 или 1,348 мг А1.
Реактивы. 1. 20%-ный раствор сульфосалициловой кислоты. 20 г сульфосалици¬
ловой кислоты растворяют в воде, объем доводят до 100 мл.
2. 0,05 н. раствор трилона Б. 9,3 г трилона Б растворяют в дистиллированной воде,
объем доводят до 1 л. Титр трилона устанавливают по раствору железа с известным его
содержанием.
3. ОД н. раствор трилона Б. 18,6 г трилона Б растворяют в дистиллированной воде,
объем доводят до 1 л.
4. Ацетатный буферный раствор с pH 5,6. 500 г уксуснокислого натрия (CH3COONa*
•ЗН20) растворяют в дистиллированной воде, добавляют 24 мл ледяной уксусной кислоты,
объем доводят до 1 л.
5. 0,1%-ный водный раствор ксиленового оранжевого.
42
3. Г. Илъковская, А. С. Коновалова
6. 0,1 н. раствор сернокислого цинка. 3,269 г металлического цинка помещают в мер¬
ную колбу на 1 л, приливают 50 мл бидистиллированной воды, 3 мл H2S04 (уд. вес 1,84).
Когда цинк полностью растворится, объем доливают до метки бидистиллированной во¬
дой.
0,1 н. раствор цинка можно готовить из сернокислого цинка, для чего растворяют
14,375 г ZnS04*7H20 в бидистиллированной воде, переносят раствор в мерную колбу
на 1 л и доливают до метки водой. Содержание цинка в растворе, приготовленном из суль¬
фата цинка, необходимо проверить, что можно сделать объемным трилонометрическим
методом.
Купферроновый метод определения железа и
алюминия. Этот метод дает возможность определить весовым способом
железо и алюминий. Железо и титан, если последний есть в растворе,
осаждаются купферроном в сильнокислой среде, причем алюминий оста¬
ется в растворе. Если после выделения железа довести кислотность до pH
4,5, купферрон количественно выделяет в осадок алюминий в виде сое¬
динения, которое после прокаливания дает окись алюминия.
Купферрон плохо сохраняется в растворе. Даже при хранении в твердом
виде его необходимо помещать в сосуд из желтого стекла с пакетиком из
фильтровальной бумаги, содержащим несколько кристаллов карбоната
.аммония. Водный раствор готовят на холоду предельно концентриро¬
ванным.
Купферроновый метод осаждения железа и ти¬
тана. При содержании в растворе только следов титана его можно не при¬
нимать во внимание. Раствор, содержащий железо, титан и алюминий,
должен иметь объем 100 мл; к нему добавляют 20 мл соляной кислоты уд. ве¬
са 1,19, затем 5 мл насыщенного раствора купферрона, приготовленного
перед определением; купферрон прибавляют по каплям, помешивая жид¬
кость. Комплекс железа с купферроном выделяется в виде охристого осадка.
Конец этой операции можно определить по свертыванию осадка, наступаю¬
щему после полного завершения осаждения. Если в этот момент добавить
еще немного купферрона, появляется беловатая муть, что также служит
указанием на полное осаждение железа и титана. Осадок фильтруют через
обычную воронку и фильтр с белой лентой и промывают холодной 10%-ной
соляной кислотой. Фильтрат должен быть совершенно прозрачным. По
окончании промывания дают хорошо стечь жидкости и подсохнуть фильтру,
затем его озоляют и прокаливают в фарфоровом тигле. Полученный после
прокаливания осадок Fe203 взвешивают и рассчитывают содержание же¬
леза в процентах на почву, высушенную при 105° С.
Осаждение алюминия купферроном после выде¬
ления железа. К фильтрату, объем которого должен быть около
200 мл, добавляют 5 г ацетата аммония, немного индикатора бромфенолового
синего, а затем 10%-ного раствора аммиака до перехода окраски индикато¬
ра в фиолетовый цвет. После этого прибавляют по каплям 5 мл свежеприго¬
товленного насыщенного раствора купферрона, помешивая содержимое
колбы.^Комплекс алюминия с купферроном появляется в виде белого тво¬
рожистого очень объемистого осадка. Полученный осадок рекомендуют по¬
догреть на водяной бане до 50—60° С, причем масса сжимается. Надо избе¬
гать перегрева на голом огне или газовой горелке, так как можно вызвать
прилипание осадка к стеклу и затруднить его определение. Осадок отфиль¬
тровывают, промывают холодной водой, затем сушат, прокаливают в фар¬
форовом тигле, взвешивают полученный осадок А1203 и рассчитывают со¬
держание алюминия в процентах на почву, высушенную при 105° С. Опре¬
деление железа и алюминия желательно сделать в один день.
Реактивы. 1. Насыщенный раствор купферрона. Приготавливается на холоду.
2. 0,1%-ный раствор бромфенолового синего. 0,1 г индикатора растворяют в 20 мл
спирта, объем доводят до 100 мл.
Определение в почве обменных катионов, емкости поглощения
43
фотоколориметрическое определение железа
сульфосалициловым методом. 20 мл вытяжки Тамма по¬
мещают в стакан на 100 мл, прибавляют 5—10 мл 10 %-ного раствора серной
кислоты, нагревают до 80° С и разрушают оксалат 1 н. раствором КМп04
до появления бурых хлопьев. Затем приливают небольшое количество го¬
рячей 0,05 н. щавелевой кислоты, подогревают жидкость до 80° С. При¬
бавляют 0,05 н. раствор КМп04 до слабо-розовой окраски, которая через
несколько минут исчезает. Весь раствор из стаканчика переносят в мерную
колбу емкостью 100 мл, прибавляют 10 мл 1,0 н. раствора NH4C1 и 3 мл
раствора а-динитрофенола. Если появится желтая окраска, то ее уничтожа¬
ют прибавлением нескольких капель 1,0 н. раствора НС1, затем прибав¬
ляют 2,5%-ный раствор аммиака до устойчивой желтой окраски и 10 мл
25%-ного раствора сульфосалициловой кислоты, раствор доводят до мет¬
ки дистиллированной водой и взбалтывают. Раствор окрашивается в
малиново-розовый цвет, после чего ждут 20 мин. и фотоколориметрируют
с зеленым светофильтром (536 нм).
Шкалу стандартных растворов готовят из такого расчета, чтобы содер¬
жание Fe203 в 100 мл составляло от 0,1 до 2,0 мг с интервалами в 0,1 мг.
К растворам в колбах на 100 мл добавляют 10 мл 1,0 н. раствора хлористого
аммония, 3 мл раствора а-динитрофенола, прибавляют несколько капель
1,0 н. раствора НС1 до исчезновения желтой окраски индикатора и затем
нейтрализуют 2,5%-ным раствором аммиака до устойчивой желтой окраски.
Прибавляют 10 мл 25%-ного раствора сульфосалициловой кислоты, объем
доводят водой до метки, раствор взбалтывают и оставляют стоять 20 мин.
Одновременно готовят «нулевой» раствор, содержащий все те же реактивы,
кроме железа. Этот раствор используют в качестве раствора сравнения.
Реактивы. 1. 1 н. раствор КМп04. 31,6 г КМп04 растворяют в дистиллирован¬
ной воде, объем доводят до 1 л.
2. 0,05 н. раствор КМп04. 1,58 г КМп04 растворяют в дистиллированной воде, объем
доводят до 1 л.
3. 0,05 н. раствор щавелевой кислоты. 3,15 г С2Н204-2Н20 растворяют в дистилли¬
рованной воде, объем доводят до 1 л.
4. 1 н. раствор NH4CI. 53,5 г NH4CJ растворяют в дистиллированной воде, объем
доводят до 1 л.
5. рс-динитрофенол. Приготовляют водный насыщепнын раствор.
6. 25%-ный раствор сульфосалициловой кислоты для колориметрического определе¬
ния железа. 25 г сульфосалициловой кислоты растворяют в 100 мл дистиллированной воды
и доводят 10%-ным раствором аммиака до pH 2,0 по индикаторной бумаге или тропео-
линовой бумажке (тропеолиновую бумажку приготовляют, растворяя 0,1 г тропеолина
в 100 мл воды, и смачивая этим раствором полоски фильтровальной бумаги и высу¬
шивая их). Тропеслиновая бумажка меняет при pH 2,0 фиолетовый цвет на розовый.
Опускать тропеолиновую бумажку в кислоту нельзя: она красит раствор кислоты. Поэто¬
му пробу производят, перенося стеклянной палочкой капли кислоты на бумажку.
7. Образцовый раствор железа для стандартной шкалы. Приготовляют раствор, со¬
держащий в 1 мл 0,1 мг Fe203, для чего растворяют в небольшом количестве воды 0,6039 г
железоаммиачных квасцов FeNH4(S04)2»12H20, прибавляют несколько миллилитров
НС1 (уд. веса 1,19) и переносят раствор в мерную колбу на 1 л, доводя объем до метки
дистиллированной водой.
Колориметрическое определение кремнекисл о-
ты в окса латных вытяжках Тамма (Беляева, 1962).
Так как ион С204” образует комплексное соединение с молибденом, что пре¬
пятствует образованию кремнемолибдата, выполнение анализа возможно
только после разрушения в анализируемом растворе оксалатов. Для этого
применяется раствор КМп04. От 5 до 15 мл фильтрата вытяжки Тамма по¬
мещают в мерную колбу на 50 мл, добавляют необходимое количество сер¬
44
3. Г. Ильковская, А. С. Коновалова
ной кислоты i : 1 (на 5 мл вытяжки необходимо 0,8 мл серной кислоты).
Раствор окисляют насыщенным раствором КМп04. Рекомендуется предва¬
рительно оттитровать используемый в работе реактив Тамма и рассчитать,
какое количество перманганата необходимо для окисления анализируемого
объема вытяжки. Это количество перманганата следует прибавлять не пол¬
ностью, а на 0,1—0,2 мл меньше, и конец окисления проводить более слабым
раствором (~ 0,1 н.) при нагревании. Нагревать в начале окисления не
следует во избежание слишком бурной реакции и выбрасывания раствора
из колбы. После окисления в колбах остается небольшой избыток окислителя
в виде осадка Мп02, который не мешает дальнейшему определению.
Колбы охлаждают, прибавляют 2,5 мл 20%-ного раствора молибденово-
кислого аммония и оставляют на 20 мин. Затем добавляют 4 мл насыщенного
раствора щавелевой кислоты, при этом осадок Мп02 растворяется в резуль¬
тате восстановления четырехвалентного марганца в двухвалентный. До¬
бавляют 10 мл 5%-ного раствора соли Мора. Растворы доливают до метки
водой, перемешивают и колориметрируют с красным светофильтром (660 нм)
в кювете с просматриваемым слоем в 20 мм. Для образцовой шкалы берут
от 0 до 10 мл (с интервалом в 2 мл) образцового раствора Na2Si03, содержа¬
щего 0,0175 мг Si02 в 1 мл. К образцовой шкале добавляют реактив Тамма
в объеме взятой для анализа вытяжки Тамма и окисляют перманганатом
так же, как испытуемый раствор. Дальнейший ход анализа аналогичен
описанному выше.
Реактивы. 1. Насыщенный раствор КМп04. 60—70 г КМп04 растворяют в ди-
стй л лированной воде, объем доводят до 1 л.
2. 0,1 н. раствор КМп04. 3,16 г КМп04 растворяют в дистиллированной воде, объем
доводят до 1 л.
3. 20%-ный раствор молибденовокислого аммония. 200 г (NH4)eMo7024*4H20 раство¬
ряют в 200 мл дистиллированной воды, нагретой до 60° С. Охлаждают полученный раствор»
фильтруют и объем доводят дистиллированной водой до 1 л.
4. Насыщенный раствор щавелевой кислоты. 90—100 г Н2С204*2Н20 растворяют
в дистиллированной воде, объем доводят до 1 л.
5. 5%-ный раствор соли Мора. 50 г соли Мора [FeS04(NH4)2S04*6H20] растворяют
в H2S04 (1 : 4) и доводят этой кислотой до объема 1 л.
6. Стандартный раствор Si02. Для приготовления стандартного раствора SiOa сплав»
ляют в платиновом тигле 0,175 г чистого кварца с 1,75 г безводного углекислого натрия*
Сплав растворяют в горячей воде и количественно переносят полученный раствор в мер¬
ную колбу на 1 л. После охлаждения раствор доводят до метки дистиллированной водой*
1 мл раствора содержит 0,175 мг Si02. Стандартный раствор хранят в парафинирован¬
ной склянке. Рабочий раствор приготавливают разбавлением стандартного раствора
в 10 раз.
При отсутствии чистого кварца стандартный раствор можно приготовить из Na2Si03:
растворяют в воде 0,3554 г Na2Si03, переносят раствор в мерную колбу на 1 л, слабо под¬
щелачивают NaOH и доливают до метки дистиллированной водой. 1 мл раствора отвечает
0,175 мг Si02. Стандартный раствор хранят в парафинированной склянке. Рабочий раствор
приготавливают разбавлением стандартного раствора в 10 раз. Содержание Si02 в раство¬
ре, приготовленном из Na2Si03, проверяют весовым методом.
Метод Мера и Джексона для определения свободного железа
(Mehra, Jackson, 1960)
Кроме вытяжки Тамма, для извлечения свободных гидратов окислов
железа применяют вытяжку Мира и Джексона (натрий дитионит — цит-
рат-бикарбонатный метод).
1—4 г почвы (в зависимости от ожидаемого количества железа) поме¬
щают в колбочки объемом в 100 мл, добавляют 40 мл 0,3 М раствора лимон¬
нокислого натрия, 5 мл 1 М раствора бикарбоната натрия. Колбочки поме¬
Определение в почве обменных катионов, емкости поглощения
45
щают на предварительно нагретую электроплитку с закрытой спиралью.
В одну из колбочек помещают термометр. Содержимое нагревают при еже¬
минутном помешивании до 80° С. После этого все колбочки снимают с элек¬
троплитки и помещают на асбестовый лист. В каждую колбочку вносят 1 г
дитионита натрия (Na2S204). Содержимое быстро перемешивают и все кол¬
бочки помещают на верхнюю полку термостата при температуре 125° С
на 15 мин. Каждые 5 мин. содержимое колбочек должно перемешиваться.
Ровно через 15 мин. колбочки вынимают из термостата и к суспензии добав¬
ляют 15—20 мл насыщенного раствора NaCl (для свертывания и оседания
коллоидов). Для почв, богатых аллофанами, вместе с NaCl добавляют 10 мл
ацетона. Суспензию перемешивают и нагревают на водяной бане до выпадения
осадка. Через 5 мин. содержимое фильтруют через плотный фильтр (синяя
лента) в мерную колбу на 100 мл г. Осадок на фильтре дважды промывают
горячей смесью 0,3 М раствора лимоннокислого натрия и 1,0 М раствора
бикарбоната натрия, приготовленной в соотношении 8 : 1 (5 частей) плюс
насыщенный раствор NaCl (1 часть). После промывания осадка объем рас¬
твора доводят до метки водой, перемешивают и берут аликвотную часть для
определения Fe. Для почв, содержащих больше 5% экстрагируемого же¬
леза, обработку повторяют еще 1—2 раза, причем все порции фильтрата
сливают в одну и ту же колбу и Fe определяют из одного соединенного рас¬
твора.
Конечное определение железа проводят объемным трилонометрическим
методом или фотоколориметрическим методом, как описано выше.
Реактивы. 1. 0,3 М раствор лимоннокислого натрия. 88 г Na8CeH607*2H20
растворяют в 1 л воды.
2. 1,0 М раствор бикарбоната натрия. 84 г NaHC03 растворяют в 1 л воды.
3. Дитионит натрия Na2S204 — сухая соль.
4. NaCl — насыщенный раствор.
Литература
Айдинян P. X., Иванова М. С., Соловье¬
ва Т. Г, Методы извлечения и опреде¬
ления различных форм серы в почвах и
растениях. М., 1968.
Айдинян P. X., Иванова М. С., Соловье¬
ва Т. Г. Метод определения емкости
поглощения карбонатных и кислых почв.
Инструкция к объемно-титрометрическо¬
му методу с результатами эксперимен¬
тальной проверки. М., 1970
Айдинян P. X., Иванова М. С., Соловье¬
ва Т. Г. Объемно-титрометрический ме¬
тод определения обменного кальция в из¬
весткованных кислых почвах.— Агро¬
химия, 1973, № 4.
Аринушкина Е. 5. Руководство по хими¬
ческому анализу почв. М., Изд-во МГУ,
1970.
Беляева Н. И. Колориметрическое опре¬
деление кремнекислоты в вытяжках по
Тамму.— Почвоведение, 1962, № 2.
Бобко Е. Z?., Аскинази Д. JI. Определение
емкости поглощения и насыщенности
почв основаниями.— Труды НИУ, 1925,
вып. 25.
Власова 3. С., Молодцов В. А. Определе¬
ние хлорид-иона в почво-грунтах и
грунтовых водах меркурометрическим
методом.— Почвоведение, 1966, № 7.
Гедройц Я. Я. Химический анализ почв,
т. 2. М., Сельхозгиз, 1955.
Грабаров Я. /\, Уварова 3. А. Новый ме¬
тод определения емкости обмена карбо¬
натных почв с помощью буферного рас¬
твора хлористого бария.— Почвоведе¬
ние, 1940, № 9.
Мазаева М. М., Неугодова О. В. Трилоно-
метрическое определение подвижного
магния в бедных поглощенными основа¬
ниями почвах.— Почвоведение, 1962,
№ 7.
Максимюк Г. Я. Применение кальциметра
Гейслера для определения углекислоты
карбонатов в почвах.— Почвоведение,
1948, № 1.
Методы агрохимических, агрофизических
и микробиологических исследований
в поливных хлопковых районах. Таш¬
кент, 1963.
Пршибил Р. Комплексоны в химическом
анализе, изд. 2. М., ИЛ, 1960.
Тюрин И. В. К вопросу о методике опре¬
деления поглощенных кальция и маг¬
1 Анализ можно проводить в центрифужных пробирках, заменив фильтрование центри¬
фугированием.
46
3. Г. Илъковская, А. С. Коновалова
ния в карбонатных почвах.— Почвове¬
дение, 1927, № 1.
Шварценбах Г. И., Пршибил Р. Комплек-
сометрия. М., Гостехиздат, 1958.
Шварценбах Г. И., Флашка Г. Комплоксо-
номстрическое титрование. М., «Химия»,
197°.
Bergmann W., Witter В. Eiri (jberblick
liber die Versorgung der Thiiringer-Bodeii
mit Magnesium auf Grund der Ergeb-
nisse serienmassiger Magnesiumuntcr-
suchungen.— Albcrt-Thaer-Arch., 1964,
Bd. 8, H. 8/9.
Jackson M. L. Soil chcmical analysis. Pren-
tice-Hall, 1958.
Mehra 0. P., Jackson M. L. Iron oxide
removal from soils and clays by a dit-
hionite — citrate system buffered with
sodium bicarbonate.— Clays and Clay
Minerals, 1960, v. 5, 317—327.
Schachtschabel P. Das pflanzenverfiigbare
Magnesium des Bodens und seine Bestim-
mung.— Z. Pflan7enemahr. Diing. und
Bodenkunde, 1954, Bd. 67, H. 1—2.
В. В ПОНОМАРЕВА, Г А ПЛОТНИКОВА
ОПРЕДЕЛЕНИЕ ГРУППОВОГО И ФРАКЦИОННОГО СОСТАВА ГУМУСА
ПО СХЕМЕ И. В. ТЮРИНА, В МОДИФИКАЦИИ
В. В. ПОНОМАРЕВОЙ и Т. А. ПЛОТНИКОВОЙ
*
Гумусовые вещества почв не являются индивидуальными химическими
соединениями, а представляют гетерогенную систему органических веществ
сложного состава и строения. Поэтому всякая методика разделения гумуса
на группы и фракции до известной степени условна. Сравнивать почвы по
составу гумуса можно лишь в том случае, если эти данные получены с по¬
мощью унифицированной схемы анализа, с тщательным соблюдением всех
рабочих деталей методической прописи.
В нашей стране основа развития методов изучения качественного соста¬
ва гумуса и приложения полученных результатов к познанию вопросов
происхождения, состава, свойств и плодородия почв заложена трудами ака¬
демика И. В. Тюрина, которые были начаты в начале 30-х годов. К настоя¬
щему времени схема И. В. Тюрина для фракционирования почвенного гу¬
муса известна в нескольких авторских вариантах (Тюрин, 1940, 1951) и
позднейших модификациях (Пономарева, 1957; Пономарева, Николаева,
1961; Пономарева, Плотникова, 1968, 1972).
Модификация схемы И. В. Тюрина, предложенная В. В. Пономаревой
и Т. А. Плотниковой в 1968 г., была рекомендована Совещанием по методам
изучения органического вещества почвы в 1971 г. как наиболее рациональ¬
ная. Главное ее отличие от оригинальной схемы И. В. Тюрина состоит в
том, что исключена длительная попеременная обработка почвы кислотой
и щелочью, которая приводит к частичному гидролизу гуминовых кислот
и требует неоправданно длительного времени проведения анализа. В осно¬
ве новейшей модификации схемы И. В. Тюрина лежит принцип однократно¬
го воздействия на почву применяемых растворителей гумуса. Принятая
в схеме И. В. Тюрина попеременная обработка почвы кислотой и щелочью
для выделения прочно связанных гумусовых веществ заменена однократной
обработкой слабым 0,02 н. раствором NaOH при нагревании на водяной
бане.
Модифицированная схема включает следующие операции: с первой на¬
веской почвы: непосредственная 0,1 н. NaOH вытяжка (остаток почвы вы¬
брасывается); со второй навеской той же почвы: декальцирование 0,1 н.
H2S04, 0,1 н. NaOH вытяжка, 0,02 н. NaOH вытяжка при 6-часовом на¬
гревании на водяной бане, определение нерастворимого остатка гумуса.
Ниже дается методическая пропись для выполнения анализа по данной
схеме.
ПОДГОТОВКА ОБРАЗЦОВ ПОЧВ
Воздушно-сухие образцы почв растирают и просеивают через сито с
отверстиями диаметром 1 мм. В процессе этой работы отделяют неразло-
жившиеся корешки растений, пожнивные остатки и т. п. Крупные корни
и остатки растений отбирают вручную, а мелкие — пинцетом, а также пу¬
тем осторожного сдувания их с поверхности почвы на сите, что следует пов¬
торить несколько раз. Отделять растительные остатки наэлектризованной
48
В. В. Пономарева, Т. Л. Плотникова
стеклянной палочкой мы не рекомендуем, так как при этом из почвы уда¬
ляются и некоторые гумифицированные частицы.
Для определения в почвах общего содержания гумуса из образцов,
пропущенных через сито с отверстиями в 1 мм, берут небольшие средние
пробы (10—20 г), которые дополнительно растирают и полностью пропуска¬
ют через сито с отверстиями в 0,25 мм. Определение общего гумуса или ор¬
ганического углерода должно предшествовать определению его качествен¬
ного состава, так как величина навески почвы для определения состава
гумуса устанавливается исходя из его процентного содержания в почве.
ОПРЕДЕЛЕНИЕ ФРАКЦИИ ВОСКОСМОЛ И БИТУМОВ
На методическом совещании в 1960 г. определение этой фракции было
признано необязательным для большинства минеральных почв. Оно ре¬
комендуется при анализе органического вещества торфяных почв (Поно¬
марева, Николаева, 1961), а также, если возникает надобность, при анализе
лесных подстилок. С этой целью навески почв 10—20 г экстрагируют в ап¬
парате Сокслета смесью спирта и бензола 1 : 1 до полного выделения фрак¬
ции воскосмол и битумов. Фракция в целом определяется по весу после
отгонки органического растворителя на водной бане и высушивания остатка
до постоянного веса при температуре 70—80° С. Содержание углерода в
этой фракции принимается равным 72%.
Дальнейший ход анализа органического вещества почвы разделяется
на две параллельные ветви: первая — непосредственная щелочная вытяжка
из отдельно взятой навески почвы, вторая — последовательное выделение
различных фракций органического вещества из другой навески той же почвы.
НЕПОСРЕДСТВЕННАЯ 0,1 Н. NaOH ВЫТЯЖКА № 1
В эту вытяжку переходит фракция бурых гуминовых (по прежней тер¬
минологии — ульминовых) кислот, свободных и связанных, как пред¬
полагают, с подвижными полуторными окислами. Из образца почвы, пропу¬
щенной через сито с отверстиями диаметром 1 мм, берут в конические кол¬
бочки на 300—400 мл навески от 2,5 до 20 г в зависимости от общего содер¬
жания в почве гумуса. Рекомендуем придерживаться следующих навесок:
2,5 г при содержании в почве гумуса более 10%,
5—10 г » » » » » 10—3%,
10—15 г » » » » » 3—0,5%
20 г » » » » меньше 0,5%
К навескам приливают из мерной колбы по 200 мл 0,1 н. NaOH, почву
хорошо перемешивают с растворителем и оставляют до следующего дня
(20—24 час.). В течение рабочего дня содержимое колб время от времени
перемешивают. Чтобы изолировать раствор NaOH от С02 воздуха (во избе¬
жание образования соды и растворения гуматов кальция), необходимо
колбы плотно закрыть пробками.
На следующий день прибавляют в колбы насыщенный раствор Na2S04
в количестве V4 от объема жидкости (точно пипеткой 50 мл) для коагуляции
илистых частиц и ускорения фильтрации, хорошо перемешивают растворы,
оставляют колбы в покое на 10—15 мин., снова перемешивают непосред¬
ственно перед фильтрацией и фильтруют через бумажный, простой, не склад¬
чатый фильтр диаметром 15—17 см. При фильтрации, особенно в самом
начале, необходимо перенести на фильтр раствор вместе с почвой, чтобы
Определение группового и фракционного состава гумуса
49
заполнить поры фильтра почвенными частицами. Если первые порции филь¬
трата получаются мутными, их снова выливают на фильтр, добиваясь аб¬
солютной прозрачности фильтрата. Остаток почвы на фильтре и в колбах
не промывают, а выбрасывают, так как в дальнейший анализ он не идет.
В отдельных порциях полученной щелочной вытяжки № 1 делают следую¬
щие определения:
а) содержание органического углерода по И. В. Тюрину. Для этого берут
в конические колбочки на 100 мл от 10 до 50 мл вытяжки в зависимости от густо¬
ты ее окраски, прибавляют очень немного, на кончике тонкого шпателя, про¬
каленной пемзы или прокаленной лёссовой почвы и выпаривают содержимое
колбочек на кипящей водяной бане или этернитовой плитке. В последнем
случае нужно избегать сильного кипения вытяжек и лучше выпаривать их
не совсем досуха, во избежание опасности разложения при высокой темпе¬
ратуре некоторых органических веществ и растрескивания колб от пере¬
грева в сухом состоянии. Окончательное досушивание остатка на дне кол¬
бочек производят на водяной бане или в сушильном шкафу при 80—90° С.
Определение органического С в сухих остатках производят по И. В. Тю¬
рину. Для получения точных результатов нужно проводить это определение
в двух повторностях. В случае расхождения повторностей следует повторить
определение;
б) содержание углерода бурых гуминовых кислот. Для этого опреде¬
ления в коническую колбочку на 100—200 мл берут пипеткой 50—100 мл
щелочной вытяжки и прибавляют к ней двойное эквивалентное количество
1,0 н. раствора H2S04, т. е. соответственно 10 или 20 мл, с таким расчетом,
чтобы концентрация свободной серной кислоты в подкисленной вытяжке
была приблизительно 0,05 н. и pH раствора приблизительно 1,3—1,5 (осаж¬
дение гуминовых кислот концентрированной серной кислотой недопустимо).
Содержимое колбочек нагревают приблизительно до 70—80° С (не доводить
до кипения!) и фильтруют в нагретом или охлажденном состоянии через
небольшой беззольный фильтр — белая или синяя лента. Осадок гуминовых
кислот в колбочке и на фильтре промывают 2—3 раза слабым раствором
серной кислоты (0,05—0,1 н.), после чего промывать осадок водой не нужно
во избежание частичного растворения гуминовых кислот. Воронки с осад¬
ками гуминовых кислот после того, как они отмыты слабым раствором серной
кислоты, вставляют в те же колбочки, в которых производилось осаждение
(на стенках их всегда остаются частицы гуминовых кислот) и растворяют
гуминовые кислоты из промывалки небольшими порциями горячего раство¬
ра 0,1 н. NaOH. Щелочной раствор доводят до объема 100 мл в мерной кол¬
бочке и отсюда берут пробы на определение С гуминовых кислот. Это де¬
лается совершенно так же, как определение общего С щелочной вытяжки;
в) углерод фульвокислот вычисляют по разности между общим С щелоч¬
ной вытяжки и С гуминовых кислот.
ПОСЛЕДОВАТЕЛЬНОЕ ВЫДЕЛЕНИЕ
РАЗЛИЧНЫХ ФРАКЦИЙ ГУМУСА
Декальцирование почвы
Для декальцирования берут точно такие же навески почв, какие были
взяты для непосредственной щелочной вытяжки, и помещают их в кониче¬
ские колбы на 250 мл. Приливают к навескам из мерной колбы по 200 мл
0,1 н. раствора H2S04*, хорошо перемешивают и оставляют до следующего
• При изучении состава гумуса почв гумидных областей (подзолистых, бурых лесных и
пр.) целесообразно брать для декальцирования 0,5 н. раствор H2S04 с целью опре¬
деления в этой вытяжке подвижных форм полу^рных окислов.
50
В. В. Пономарева, 7. Л. Плотникова
дня (на 20—24 часа). Колбы должны быть закрыты пробками или часовыми
стеклами во избежание попадания пыли, и в течение рабочего времени
следует несколько раз перемешать их содержимое. На другой день серно¬
кислую вытяжку фильтруют в колбы на 500—1000 мл (в зависимости от со¬
держания в почве обменного Са и длительности его отмывания) через глад¬
кий бумажный фильтр средней плотности, перенося на него вместе с раство¬
ром и почву. Кальций вытесняют из почвы сначала декантацией, а затем
переносят всю почву на фильтр и продолжают промывание почвы на фильтре.
Промывание ведут 0,1 н. раствором H2S04 до полного вытеснения Са. После
этого промывают раза 2—3 водой, слабо подкисленной серной кислотой,
для удаления из навесок почв излишней серной кислоты.
Пробу на Са в промывных водах делают так: в стаканчик на 20—25 мл
набирают свежую порцию промывных вод, нейтрализуют кислый раствор
аммиаком (если выпадет осадок полуторных окислов, его отфильтровывают),
прибавляют 1—2 мл насыщенного раствора щавелевокислого аммония,
затем оставляют стаканчик в теплом месте на 10—15 мин. Полное отсут¬
ствие белого осадка щавелевокислого кальция указывает на полноту огам •
вания Са. Пробу на Са можно делать, конечно, и трилонометрическим
методом.
После полного отмывания Са сернокислую вытяжку вместе с промыв¬
ными водами переносят в мерную колбу, доводят до определенного объема
(500—1000 мл) и хорошо перемешивают. Органические вещества кислых
вытяжек из почв неустойчивы к микробиологическому разложению, поэ¬
тому анализ их нельзя откладывать больше чем на 2—3 дня.
В вытяжках определяют:
а) содержание органического С по И. В. Тюрину в сухом остатке после
выпаривания 10—100 мл вытяжки в зависимости от ее окраски. Так как
при выпаривании кислых растворов некоторые органические вещества
могут разлагаться с выделением С02, то необходимо взятую порцию вытяжки
нейтрализовать сухой содой до начала выпадения полуторных окислов,
а затем уже выпаривать;
б) в зависимости от задач исследования в сернокислой вытяжке можно
определить содержание Са (оно будет приблизительно равно содержанию
обменного Са) и подвижных полуторных окислов — Fe, А1 (см. методику их
определения в статье В. В. Пономаревой, 1957).
При содержании в почве карбонатов для декальцирования навесок почв
приходится брать не серную, а соляную кислоту. В этом случае следует
поступать так. Сначала нужно прилить к навеске почвы приблизительно
эквивалентное содержанию карбонатов количество 1,0 н. раствора НС1
или, если содержание карбонатов неизвестно, приливать раствор соляной
кислоты небольшими порциями при постоянном помешивании до прекращения
вскипания. По окончании реакции разложения карбонатов (при помешива¬
нии, но при комнатной температуре) прибавить 200—250 мл 0,1 н. раство¬
ра НС1 и проверить, действительно ли все карбонаты разложились и кон¬
центрация свободной НС1 в растворе близка к 0,1 н. В противном случае
надо откорректировать концентрацию свободной НС1 в растворе. На сле¬
дующий день отфильтровать раствор от почвы через гладкий (не склад¬
чатый) фильтр и промывать почву в колбе и на воронке сначала 0,1 н.
раствором НС1 до удаления Са, а после этого — 0,1 н. раствором H2S04 до
удаления С1.
Наличие в солянокислой вытяжке огромного количества хлора, который
окисляется хромовой смесью, препятствует определению в ней С. Поэтому
для приблизительного учета кислотнорастворимой фракции органического
вещества в образцах почв, содержащих карбонаты, приходится отдельную
навеску почвы заливать 0,1 н. раствором H2S04 (200 мл), как это описано
выше для некарбонатных почву и определять в ней органический С. В этом
Определение группового и фракционного состава гумуса
51
случае навеска почвы на воронке не промывается и доводить фильтрат до
определенного объема не требуется. Расчет содержания в вытяжке орга¬
нического С ведется на объем H2S04, прилитый к почве. Определение это
условно, так как в солянокислую вытяжку при длительном отмывании почвы
от Са, вероятно, переходит больше органического С, чем в однократную
0,1 н. Н2304-вытяжку.
0,1 н. NaOH вытяжка № 2 после декальцированпя почвы
В эту вытяжку переходят бурые гуминовые кислоты и фульвокислоты,
извлекаемые непосредственной щелочной еытяжкой, описанной выше, плюс
гуминовые кислоты (черные) и фульвокислоты, связанные с кальцием и
растворимые в щелочи только после декальцированпя почвы.
Сразу же после операции декальцирования влажные навески почв смы¬
вают с бумажного фильтра в те же колбы, где производилось декальцирова-
ние. Замедление с этой операцией приведет к подсыханию навесок почв на
фильтрах, после чего их трудно будет количественно смыть с фильтров.
Эта операция требует большой тщательности и осторожности. Лучше всего
ее проводить так. В колбу, куда должна быть смыта навеска почвы, вставляют
воронку диаметром 10—15 см с коротким и широким горлом. В небольшую
чистую промывалку наливают точно 200 мл 0,1 н. раствора NaOH, и этим
количеством щелочи стараются смыть с фильтра, не вынимая его из воронки,
всю почву обратно в колбу. Следует тщательно избегать разбрызгивания
раствора щелочи и частиц почвы по сторонам. Незначительный налет мел¬
ких частиц почвы на фильтре иногда остается после данной операции. Одна¬
ко остатки щелочи в промывалке выливают на фильтр, и щелочь растворяет
то незначительное количество гумусовых веществ, которое могло задержать¬
ся с частицами почвы на фильтре. Остатком отмеренного количества щелочи
обмывают и широкогорлую воронку, вставленную в колбу. Если взятого
количества щелочи не хватило на смывание почвы с фильтра, следует доба¬
вить в промывалку еще 50 или 100 мл 0,1 н. раствора NaOH и продолжать
смывание. Но в принципе это нежелательно, так как нарушает принятое
соотношение почвы с растворителем.
После того, как навески почв полностью смыты с фильтра в колбы и
остатки раствора щелочи в промывалке вылиты соответственно в те же кол¬
бы, их закрывают пробками или стеклами и оставляют до следующего дня
(на 20—24 часа). В течение рабочего дня содержимое колб несколько раз
перемешивают. На следующий день в колбы прибавляют по 50 мл насыщен¬
ного раствора Na2S04 для коагуляции тонких илистых суспензий и ускорения
фильтрации. Содержимое колб хорошо перемешивают, оставляют в покое
на 10—15 мин., выжидая свертывания илистых частиц, после чего снова
перемешивают непосредственно перед фильтрованием и тотчас же фильтруют
через плотно пригнанные к воронкам бумажные фильтры, перенося на них
раствор вместе с почвой.
Остатки почвы в колбах и на воронках промывают слабым (около 1—2°0)
раствором Na2S04 до полного или почти полного просветления (обесцвечи¬
вания) промывных вод. Щелочную вытяжку вместе с промывными водами
доводят до определенного объема (400—500 мл) и хорошо перемешивают.
В отдельных порциях ее определяют:
а) содержание органического С по И. В. Тюрину, точно так же, как
описано выше для щелочной вытяжки № 1;
б) содержание С гуминовых кислот по И. В. Тюрину, точно так же,
как описано выше для щелочной вытяжки № 1;
в) содержание С фульвокислот по разности между содержанием общего С
в вытяжке и С гуминовых кислот;
52
В. В. Пономарева, 7. Л. Плотникова
г) индекс оптической плотности гуминовых кислот, определяемый с
одним синим светофильтром (Плотникова, Пономарева, 1967). Индекс ко¬
леблется в разных почвах от нескольких единиц до 30 и является дополни¬
тельным очень ярким показателем качества гумусовых веществ. Его опре¬
деление несложно, так как не требует предварительного выравнивания кон¬
центрации С в анализируемых растворах и делается очень быстро в тех
самых растворах, которые получают в ходе фракционирования гумуса.
Для этой цели используют раствор гуминовых кислот в 0,1 н. NaOH, имею¬
щий pH 12—13. В принципе можно определить индекс оптической плотности
в любом растворе гумусовых веществ после доведения его pH до 12—13,
что является обязательным условием этого определения. Оно производится
на фотоэлектроколориметре ФЭК-М или спектрофотометре с синим свето¬
фильтром при длине волны 430 нм и толщине просматриваемого слоя в 1 см.
Результаты выражают величиной Eq мг^мл, которую получают путем деления
показателя экстинкции Е на содержание в растворе органического С (в
мг/мл).
0,02 и. NaOH вытяжка № 3 при нагревании
на водяной бане
В эту вытяжку переходят гуминовые кислоты и фульвокислоты, прочно
связанные с глинистой фракцией и устойчивыми формами полуторных окис¬
лов. Остаток почвы после предыдущей щелочной вытяжки № 2 в сыром со¬
стоянии смывают в ту же колбу отмеренным в чистую промывалку количе¬
ством 0,02 н. раствора NaOH (200 мл). Смывание идет быстрее, если раствор
щелочи предварительно подогреть до 70—80° С. Затем колбы накрывают
часовыми стеклами во избежание испарения жидкости и ставят на кипящую
водяную баню на 6 час., считая от момента, когда содержимое колб доста¬
точно разогреется от водяной бани. Фильтрацию вытяжек и промывание
остатков почвы на фильтрах производят на следующий день после прибав¬
ления 50 мл насыщенного раствора Na2S04 и при комнатной температуре —
совершенно так же, как описано выше для предыдущих щелочных вытяжек.
Вытяжку вместе с промывными водами доводят до определенного объема
и в ней производят те же определения, что и в предыдущих щелочных вытяж¬
ках. Определение индекса оптической плотности гуминовых кислот может
быть исключено, хотя и представляет интерес убедиться в том, что гуминовые
кислоты фракции 3 менее оптически плотны, чем гуминовые кислоты фрак¬
ции 2.
ОПРЕДЕЛЕНИЕ ОСТАТКА ГУМУСА,
НЕРАСТВОРИМОГО ПРИ ДАННОЙ СХЕМЕ АНАЛИЗА
Прямое определение органического С в остатке почвы после всех преды¬
дущих операций, для чего требуется количественно собрать его с фильтра,
довести до воздушно-сухого состояния, взвесить, растереть и просеять через
сито, определить в нем С и ввести поправку на убыль в весе по сравнению
с исходной навеской почвы, как правило, не дает надежных результатов,
но требует затраты большого труда и времени. Поэтому в практике обычных
аналитических работ рекомендуется определять С остатка по разности меж¬
ду общим содержанием органического С в почве и суммой С всех выделенных
фракций гумусовых веществ, исключая, понятно, С непосредственной ще¬
лочной вытяжки № 1.
Определение группового и фракционного состава гумуса
53
ДОПОЛНИТЕЛЬНЫЕ МЕТОДИЧЕСКИЕ РЕКОМЕНДАЦИИ
1. По описанной схеме и методике можно анализировать одновременно
Ю—16 образцов почв, на что затрачивается в зависимости от опытно¬
сти аналитика 3—4 недели. При фракционировании гумуса можно опре¬
делять в получаемых вытяжках не только С, но, если необходимо,
и N. Однако в этом случае следует в 2 раза увеличить навески почв и
объемы растворителей против тех, которые рекомендованы в начале описа¬
ния метода.
2. Очень важно соблюдение по возможности строго одинаковых условий
фракционирования гумуса в разных почвах и разными аналитиками. Глав¬
ными условиями являются: соотношение навесок почв с растворителями,
время воздействия растворителей, концентрация растворителей, темпера¬
турные условия растворения.
3. Для определения состава гумуса по С не следует брать навески почв
более 20 г, особенно глинистых почв, так как в щелочной среде происходит
сильное набухание почв и замедляется процесс фильтрации вытяжек. Кро¬
ме того, при большой навеске малогумусной почвы выходу органического
вещества в раствор мешает узкое отношение растворителя к почве: в этом
случае ничтожное количество органического вещества «закрыто» от действия
растворителя огромной минеральной массой почвы. При очень низком со¬
держании органического С в почве, порядка 0,1—0,2%, получаемые данные
состава гумуса следует считать приблизительными.
4. Все сернокислые растворы гумусовых веществ необходимо перед
тем, как выпаривать досуха, нейтрализовать химически чистой сухой со¬
дой, а все щелочные растворы гумусовых веществ следует выпаривать без
нейтрализации их кислотой. Необходимо по возможности быстро анализи¬
ровать как кислотные, так и щелочные вытяжки из почв, так как при стоянии
кислых вытяжек их органическое вещество подвергается микробиологиче¬
скому разложению, а при стоянии щелочных вытяжек содержащиеся в них
гуминовые кислоты под влиянием щелочного гидролиза частично превра¬
щаются в фульвокислоты. Каждая вытяжка должна анализироваться не
позднее 3—4 дней со времени ее получения.
5. В зависимости от задач исследования декальцирование почв иногда
производят не 0,1 н., а 0,5 н. раствором H2S04. Это нужно делать в тех
случаях, когда хотят получить более полный выход в сернокислую вытяжку
подвижных полуторных окислов и производят их определение (Пономарева,
1957), что представляет интерес для подзолистых, бурых лесных, влажных
субтропических и тропических почв. Отметим, что 0,5 н. сернокислая вы¬
тяжка употреблялась И. В. Тюриным в его схеме 1940 г. В схе¬
ме же И. В. Тюрина 1951 г. для декальцирования почв употреблялся 1,0 н.
раствор Na2S04. В настоящее время имеются основания не рекомендовать
раствор Na2S04 для декальцирования почв.
6. Точность определения органического С в почвах и вытяжках из почв
зависит в общем от содержания С во взятой пробе. Нужно стремиться к
тому, чтобы на окисление испытуемой пробы расходовалось около половины,
но не больше, взятого количества хромовой смеси, т. е. чтобы на титрование
испытуемой пробы расходовалось не менее половины того количества соли
Мора, которое идет на холостое титрование. Если на окисление затрачивает¬
ся больше половины взятого количества окислителя, что сопровождается
позеленением раствора, результаты определения С будут заниженными;
так как при концентрации Сг03 ниже 0,2 н. уменьшается полнота окисле¬
ния гумуса. Определение С по И. В. Тюрину можно считать надежным,
если разница между холостым и испытуемым титрованием составляет не
менее 2 мл соли Мора и не более 10—12 мл при величине холостого титрова¬
54
В. В. Пономарева, Т. А. Плотникова
ния 20—22 мл. В противном случае нужно повторить определение С, взяв
для этого соответственно большую или меньшую навеску почвы или пробу
раствора.
Результаты определения группового и фракционного состава гумуса при¬
нято выражать в процентах к общему содержанию органического углерода
в почве. Сначала данные состава гумуса вычисляют в процентах к почве
с точностью до двух знаков после запятой, при очень малых цифрах — с
точностью до трех знаков. Окончательные данные состава гумуса в процен¬
тах к общему С следует вычислять с точностью до одного знака после запя¬
той — такова максимальная разрешающая способности этого метода. В не¬
которых случаях, например при выведении средних чисел из массовых
данных, можно выражать состав гумуса в процентах к общему С в целых
числах.
Описанная схема и методика дают возможность фракционировать почвен¬
ный гумус на три-фракции гуминовых кислот и четыре фракции фульвокислот,
а именно:
гуминовые кислоты:
фракция 1 — растворимая непосредственно в 0,1 н. NaOH — свободная
и связанная с подвижными полуторными окислами;
фракция 2 — растворимая в 0,1 н. NaOH только после декальцирования
почвы и связанная в основном с кальцием;
фракция 3 — растворимая в 0,02 н. NaOH при 6-часовом нагревании на
водяной бане и связанная с глинистой фракцией и устойчи¬
выми полуторными окислами
фульвокислот ы:
фракция 1а — растворимая в 0,1 н. H2S04 при декальцировании почвы,
свободная и связанная с подвижными полуторными окисла¬
ми (так называемая агрессивная фракция);
фракция 1 — растворимая в 0,1 н. NaOH из недекальцированной почвы
и связанная в почве с фракцией 1 гуминовых кислот;
фракция 2 — растворимая в 0,1 н. NaOH только после декальцирования
и связанная с фракцией 2 гуминовых кислот;
фракция 3 — растворимая в 0,02 н. NrOH при 6-часовсм нагревании на во¬
дяной баге и связанная с фракцией 3 гуминовых кислст.
Перечисленные фракции находят следующим образом:
фракции гуминовых кислот:
1 — определяется непосредственно из щелочной вытяжки № 1;
2 — вычисляется по разности между содержанием гуминовых кислот в ще¬
лочной вытяжке № 2 и № 1;
3 — определяется из щелочной вытяжки № 3.
фракции фульвокислот:
1а — определяется из 0,1 н. H2S04 вытяжки;
1 — вычисляется по разности между содержанием фульвокислот в щелоч¬
ной вытяжке № 1 и в 0,1 н. H2S04 вытяжке;
2 — вычисляется по разности между содержанием фульвокислот в щелоч¬
ной вытяжке № 2 и в щелочной вытяжке № 1 за вычетом фульвокис¬
лот 1а;
3 — определяется из щелочной вытяжки № 3.
Кроме того, получается величина нерастворимого остатка гумуса, ко¬
торая характеризует прочность закрепления гумусовых веществ с глини¬
Определение группового и фракционного состава гумуса
55
стой фракцией или слабую степень гумификации органического вещества,
например в торфе, лесной подстилке и т. п.
Ниже приводим формы вычисления данных состава гумуса и их таблич¬
ного выражения. Форма 1 является рабочей, форма 2 — окончательной.
Форма 1 (рабочая)
Содержание органического С в вытяжках из почв
^общ
почвы,
Непосредственная
0,1 н. NaOH
вытяжка, Кя 1
0,1 н.
H*S04
вытяж¬
ка
0,1 н. NaOH вытяжка
после декальцирова¬
ния, Me 2
0,02 н.
. NaOH вытяж¬
ка, М» 3
%
собщ
сгк
СфК
собщ
^общ
сгк
Сфк
^общ
СГК
СфК
% к почве
% к Собщ почвы
3,44
100
0,35
10,2
0,07
2,0
0,28
8,2
см
О СО
1,45
42,1
^ СЧ
«*н СМ
СО
0,34
9,9
0,60
17.5
0,29
8,5
0,31
9,0
Форма 2 (составляется на основании данных формы 1)
С фракций гумуса, % к Собщ почвы
Собщ
Сгк
СфК
СГК +
сГк
„мг/мл
ЕСгк
почвы,
%
1
2
3
сумма
1а
1
2
3
сумма
+ Сфк
Сфк
3,44
2,0
30,2
8,5
40,7
3,4
4,8
5,1
9,0
22,3
63,0
1,8
20,2
Принято также графическое выражение данных состава гумуса (по
Пономаревой, 1956), если он определяется в полном профиле почвы.
Литература
Плотникова Т. А., Пономарева В. В.
Упрощенный вариант определения опти¬
ческой плотности гумусовых веществ
с одним светофильтром.— Почвоведение,
1967, № 7.
Пономарева В. В. Географические зако¬
номерности подзолообразования.— Поч¬
воведение, 1956, № 3.
Пономарева В. В. К методике изучения
состава гумуса по схеме И. В. Тюрина.—
Почвоведение, 1957, № 8.
Пономарева В. В., Николаева Т. А. Ме¬
тоды изучения органического вещества
в торфяно-болотных почвах.— Почвове¬
дение, 1961, № 5.
Пономарева В. Z?., Плотникова Т. А. Ме¬
тодика и некоторые результаты изучения
состава гумуса в черноземах.— Почво¬
ведение, 1968, № 7.
Пономарева В. В.у Плотникова Т. А. Срав¬
нительное изучение принятых в СССР
схем определения группового и фракци¬
онного состава гумуса.— Почвоведение,
1972, № 7.
Решение первой Межвузовской конферен¬
ции по изучению органического вещества
почвы. МГУ, 1967.
Решение рабочего совещания по методам
изучения органического вещества почвы.
М., 1971.
Тюрин И. В. Из результатов работ по изу¬
чению состава гумуса в почвах СССР.—
Пробл. сов. почвоведения, 1940, сб. И.
Тюрин //. В. К методике анализа для
сравнительного изучения состава поч¬
венного гумуса.— Труды Почв, ин-та
им. В. В. Докучаева, 1951, т. 38.
Я. Я. БЕЛЬЧИКОВА
ОПРЕДЕЛЕНИЕ ГУМУСА 1ЮЧВЫ
ПО МЕТОДУ И. В. ТЮРИНА
*
Метод И. В. Тюрина основан на окислении органического вещества
почвы хромовой кислотой до образования углекислоты. Количество кисло¬
рода, израсходованное на окисление органического углерода, определяют
по разности между количеством хромовой кислоты, взятой для окисления,
и количеством ее, оставшимся неизрасходованным после окисления. В ка¬
честве окислителя применяют 0,4 н. раствор К2Сг207 в серной кислоте,
предварительно разбавленной водой в соотношении 1:1.
Реакция окисления протекает по следующим уравнениям:
2К2СГ2О7 + 8H2SO4 = 2K2SO4 + 2Cr2(S04)3 + 8Н2О+ЗО2 ,
ЗС + ЗОг = ЗС02.
Остаток хромовой кислоты, не израсходованной на окисление, оттитро-
вывают 0,1 н. раствором соли Мора с индикатором дифениламином или фе-
нилантраниловой кислотой. Титрование солью Мора, представляющей со¬
бой двойную соль сернокислого аммония и сернокислой закиси железа —
(NH4)2S04FeS04-6H20, идет по уравнению
К2СГ2О7 + 7H2SO4 + 6FeS04 = 7Н2О -(- K2SO4 -j- Cr2(S04)3 + 3Fe2(SC>4)3.
Полнота окисления органического вещества при соблюдении всех условий
метода, указанных ниже, составляет 85—90% величины окисления гумуса
методом сухого сжигания по Густавсону. Применение сернокислого серебра
в качестве катализатора увеличивает полноту окисления до 95% (Комаро¬
ва, 1933).
Для получения надежных результатов необходимо обратить внимание:
1) на тщательную подготовку почвы к анализу и 2) на точное соблюдение
продолжительности кипячения органического вещества; само кипение окис¬
лительной смеси должно протекать спокойно.
Метод дает хорошую сходимость параллельных анализов, быстр, не тре¬
бует специальной аппаратуры (в связи с чем может быть использован и в
экспедиционных условиях) и в настоящее время является общепринятым,
особенно при массовых анализах.
При подготовке почвы к анализу на содержание гу¬
муса особое внимание должно быть обращено на удаление из почвы кореш¬
ков и различных органических остатков растительного и животного проис¬
хождения.
Из взятого в поле и доведенного до воздушно-сухого состояния образца
почвы берут среднюю пробу в количестве 50 г, тщательно отбирают пинце¬
том корни и видимые глазом органические остатки (панцири насекомых,
семена, угольки и т. п.), раздавливают почвенные комки деревянным пести¬
ком с резиновым наконечником и вновь тщательно отбирают корни, поль¬
зуясь при этом лупой.
Затем растирают почву в фарфоровой ступке и пропускают через сито
с отверстиями диаметром 1 мм, после чего из нее снова берут среднюю пробу
весом 5 г и повторяют отбор корешков, используя для этого следующий при¬
ем: сухую стеклянную палочку энергично натирают сухой суконкой или
Определение гумуса почвы по методу И. В. Тюрина
57
шерстяной тканью и быстро проводят на высоте около 10 см над почвой,
распределенной тонким слоем по поверхности восковки или пергаментной
бумаги. Тонкие мелкие корешки и полу разложившиеся растительные ос¬
татки, которые до этого не удалось отобрать в связи с их малыми размера¬
ми, прилипают к поверхности наэлектризованной палочки и таким образом
выносятся из почвы. Их снимают с палочки при повторном ее натирании.
Не следует слишком низко проводить палочкой над поверхностью почвы
во избежание выноса из почвы не только органических остатков, но и мел¬
козема.
В процессе отбора корешков надо неоднократно перемешивать почву
и вновь распределять ее тонким слоем. Операцию следует вести до тех пор,
пока на палочке будут обнаруживаться лишь единичные корешки. Чистоту
отбора корешков контролируют, помимо того, просмотром почвы в лупу.
По окончании отбора корешков почву снова растирают в фарфоровой,
яшмовой или агатовой ступке и пропускают через сито с отверстиями диа¬
метром в 0,25 мм. Описанным выше способом должен быть подготовлен
весь образец в 5 г. Отбрасывать трудно поддающуюся растиранию часть
образца ни в коем случае нельзя.
Почву, подготовленную для анализа, следует хранить в пакетиках из
пергаментной бумаги или восковки либо в пробирках с пробками.
Ход анализа. Навеску воздушно-сухой почвы для анализа на гумус
берут на аналитических весах. Размер навески зависит от предполагаемого
содержания гумуса в почве, причем учитывается тип почвы (чернозем,
подзолистая и т. д.) и глубина взятия образца.
При содержании гумуса от 7 до 10% И. В. Тюрин рекомендует навеску
в 0,1 г, при 4—7%—0,2 г, при 2—4%—0,3 г, меньше 2%—0,5 г. В случае _
песчаных почв с малым содержанием гумуса навеску можно увеличить до 1 г.
Навески лучше брать точные — 0,1; 0,2 г, что облегчает в дальнейшем
вычисления. Для взятия точных навесок можно пользоваться тарированным
часовым стеклом диаметром 2,5—3 см, с которого навеску целиком переносят
в колбу для сжигания при помощи маленького шпателя и кисточки для
акварельных красок. Определение гумуса по Тюрину одновременно можно
вести в 20—30 навесках.
Навески помещают в сухие конические колбы на 100 мл из обыкновен¬
ного стекла, туда же добавляют на кончике ножа порошкообразное сернокис¬
лое серебро. При массовых анализах сернокислое
серебро не применяется. Для возможности сравнения полу¬
чаемых в этом случае результатов с методом сухого сжигания И. В. Тюрин
приводит коэффициент 1,17 (1936). Затем в каждую колбу приливают по
10 м г 0,4 п. раствора К2Сг207, приготовленного на смеси из одной части
H2S04 (удельного веса 1,84) и одной части дистиллированной воды.
Раствор бихромата калия следует приливать из бюретки медленно, от¬
меривая необходимый объем каждый раз от нуля и давая жидкости стекать
всегда с одинаковой скоростью. Можно пользоваться также пипеткой,
но обязательно снабженной в верхней части предохранительными шариками.
Очень удобна в данном случае делительная воронка из тугоплавкого стек¬
ла, приспособленная для работы с крепкими кислотами. Пользование такой
воронкой намного ускоряет работу и делает ее безопасной.
После приливания раствора К2Сг207 в горлышко колб вставляют воронки
диаметром около 4 см% содержимое колб осторожно перемешивают (следя,
чтобы почва не прилипала к их стенкам), после чего колбы ставят на уже
горячую этернитовую плитку или песчаную баню или на плитку с обнажен¬
ной спиралью, но прикрытую слоем асбеста. Можно пользоваться также
газовыми горелками, а в экспедиционных условиях — примусом или керо¬
синкой, помещая нагревательный прибор под песчаную баню (сковорода
с прокаленным кварцевым песком).
58
Я. П. Бельчикова
Содержимое колб доводят до кипения и кипятят ровно 5 мин. Необходимо
отмечать начало кипения жидкости, не смешивая его с появлением в начале
нагревания мелких пузырьков воздуха. Кипение должно быть равномерным
и умеренным; выделение пара из воронки и подпрыгивание последней не¬
допустимы. Сильного кипения следует избегать, чтобы не изменить концент¬
рацию серной кислоты, увеличение которой может вызвать разложение
хромовой кислоты. Во избежание слишком бурного кипения кипяче¬
ние на плитках с обнаженной спиралью недо¬
пустимо.
Титрование с дифениламином. После 5-минутного ки¬
пячения колбы снимают с нагревательного прибора, дают им остыть, обмы¬
вают воронки над колбами с внутренней и наружной стороны дистиллиро¬
ванной водой из промывалки и содержимое колб количественно переносят
в конические колбы на 250 мл, несколько раз тщательно ополаскивают колбу,
в которой производилось окисление. Объем жидкости после переноса в колбу
на 250 мл должен составлять 100—150 мл. Цвет жидкости — оранжево-жел¬
тый или зеленовато-желтый; позеленение ее свидетельствует о недостатке
окислителя; анализ в этом случае необходимо повторить, уменьшив навеску
почвы.
К жидкости прибавляют 8 капель раствора дифениламина, являющегося
индикатором, и титруют оставшуюся неизрасходованной после окисления
органического вещества хромовую кислоту 0,1 н. раствором соли Мора.
Индикатор следует вносить непосредственно перед титрованием. Титрова¬
ние ведут при комнатной температуре. Красно-бурая окраска жидкости,
появляющаяся после прибавления дифениламина, при титровании раство¬
ром соли Мора постепенно переходит в интенсивно синюю, а затем в грязно¬
фиолетовую. С этого момента титрование ведут осторожно, прибавляя соль
Мора по 1 капле и тщательно перемешивая содержимое колбы. Конец титро¬
вания — изменение грязно-фиолетовой окраски раствора в бутылочно-зеле¬
ную; посл*е некоторого стояния (10—15 мин.) окраска жидкости становится
зеленой. Появление при титровании ярко-зеленой окраски указывает на
избыток соли Мора, т. е. на то, что раствор перетитрован; анализ в этом
случае необходимо повторить.
Для устранения влияния ионов трехвалентного железа, которое окисляет
индикатор и вызывает преждевременное изменение окраски раствора, и для
более четко выраженного конца титрования вносят в колбу перед титрова¬
нием 85%-ную ортофосфорную кислоту в количестве 2,5 мл; изменение ок¬
раски в конце титрования в присутствии фосфорной кислоты очень резкое
и вызывается 1—2 каплями раствора соли Мора.
Титрование с фенилантраниловой кислотой.
В 1957 г. проф. В. Н. Симаков предложил применять в качестве индикатора
при титровании хромовой смеси раствором соли Мора фенилантраниловую
кислоту взамен дифениламина.
Многочисленные определения показали, что результаты титрования
с фенилантраниловой кислотой вполне совпадают с результатами титрования
в присутствии дифениламина. Однако применение фенилантраниловой ки¬
слоты имеет большие преимущества по сравнению с дифениламином.
Так, изменение окраски в конце титрования бихромата калия раствором
соли Мора в присутствии фенилантраниловой кислоты более резко выра¬
жено, нежели с дифениламином, и наступает от одной избыточной капли
даже такого разведенного раствора соли Мора, как 0,02 н., что свидетель¬
ствует о высокой чувствительности индикатора и позволяет с большой точ¬
ностью вести определение малых количеств гумуса (например, в водных вы¬
тяжках и при анализе природных вод).
Другим преимуществом является следующее. При использовании фенил¬
антраниловой кислоты в качестве индикатора при титровании бихромата
Определение гумуса почвы по методу И. В. Тюрина
59
калия раствором соли Мора необходима высокая концентрация серной ки¬
слоты в титруемом растворе — около 15—20 н. Это позволяет лишь незна¬
чительно разбавлять жидкость перед титрованием (после сжигания) и таким
образом проводить титрование в тех же конических колбах на 100 мл, в кото¬
рых проводилось сжигание (без переноса их содержимого для разбавления
в большие колбы), как в случае дифениламина.
Таким образом, сокращается время анализа, расход посуды и дистилли¬
рованной воды.
Ход определения содержания гумуса с фенилантраниловой кислотой та¬
ков: сжигание органического вещества в навесках почвы производят точно
по методу Тюрина в конических колбах на 100 мл с теми же реактивами. Пос¬
ле 5-минутного кипячения и остывания колб тщательно обмывают воронки
над колбами минимальным количеством воды, прибавляют 3—5 капель
0,2 %-ного раствора фенилантраниловой кислоты и титруют в этих же кол¬
бах 0,1 н. раствором соли Мора до перехода окраски из вишнево-фиолетовой
в зеленую. Так как переход окраски очень резкий — мгновенный, то раствор
соли Мора следует приливать в конце титрования по каплям.
Одновременно с основными анализами в той же последовательности про¬
водят холостой (в трехкратной повторности) для установления соотноше¬
ния между 10 мл раствора хромовой смеси и раствором соли Мора. Для рав¬
номерного кипения жидкости при холостом анализе в колбу перед прилива-
нием раствора хромовой смеси обязательно вносят около
0,1—0,2 г растертых в порошок прокаленных пемзы
или почвы. В противном случае происходит неизбежное при кипячении
чистого раствора перегревание, которое может вызвать разложение хромо¬
вой кислоты. В остальном поступают согласно описанному ходу аналиэа.
При проведении больших партий анализов на содержание гумуса по мето¬
ду Тюрина (30—60 анализов одновременно) можно делать перерывы на сле¬
дующих этапах работы: взятие навесок — один день; окисление, перенос
в колбы для титрования и титрование — на другой день. Или, что менее
желательно, взятие навесок и окисление проводить в один день, титрование —
на следующий. В последнем случае содержимое колб после сжигания долж¬
но быть разбавлено и перенесено в колбы для титрования. Титрование холо¬
стых анализов в этом случае также должно быть оставлено до следующего
дня. Титрование каждой партии необходимо всегда вести при одинаковых
условиях освещения (при дневном или электрическом свете).
Вычисление результатов анализа. Количество мил¬
лилитров раствора соли Мора, пошедшее на титрование после окисления
гумуса, отвечает тому количеству хромовой кислоты, которое осталось неиз¬
расходованным в процессе окисления.
И. В. Тюрин указывает, что устойчивые результаты получаются тогда,
когда на титрование остатка хромовой кислоты после окисления органиче¬
ского вещества идет не менее 20 мл 0,1 н. раствора соли Мора, иначе говоря,
концентрация Сг03 не должна быть к концу окисления ниже 0,2 н.
Количество соли Мора, отвечающее тому количеству хромовой кислоты,
которое пошло на окисление гумуса пробы, определяют вычитанием из ре¬
зультатов холостого титрования результатов титрования после окисления
гумуса.
При вычислении содержания органического углерода и гумуса приняты
следующие величины: 1 мл 0,1 н. раствора соли Мора соответствует 0,0003 г
органического углерода или 0,000517 г гумуса (1 г углерода соответствует
1,724 г гумуса).
Формула вычисления содержания гумуса (в % к воздушно-сухой почве)
следующая:
(а — 6)-AM),000517-100
Гумус = ■ р ,
60
Н. /7. Бельчикова
где а — количество 0,1 н. раствора соли Мора, пошедшее на титрование 10 мл 0,4 я. рас¬
твора К2Сг207 при холостом анализе, мл; b — количество 0,1 н. раствора соли Мора, по¬
шедшее на титрование после окисления гумуса, мл; (а — Ъ) — количество 0,1 н. раствора
соли Мора, отвечающее количеству хромовой кислоты, израсходованному на окисление
гумуса, мл; К — поправочный коэффициент к титру раствора соли Мора; 0,000517 —
количество гумуса, соответствующее 1 мл 0,1 н. раствора соли Мора, г; Р — навеска воз¬
душно-сухой почвы, г.
Если результаты анализа желательно выразить в углероде, то вместо
0,000517 (коэффициента перевода на гумус) следует поставить величину
0,0003, так как 1 ди 0,1 н. раствора соли Мора соответствует 0,0003 г орга¬
нического углерода.
Для пересчета содержания гумуса в процентах на почву, высушенную при
105° С, определяют влажность в отдельной навеске почвы и в вычисление
вводят соответствующий коэффициент.
Для работы удобна следующая форма записи:
1. Холостой анализ. На титрование 10 мл 0,4 н. раствора К2Сг207 идет
45,0 мл раствора соли Мора 0,102 н. (среднее иэ трех определений).
2. Результаты анализа почв:
N* анализа
М« разреза
Глубина пробы, см
0»
р
в
«
еб
К
>*
9
к
м Ь
Количество соли Мора,
пошедшее на титрова¬
ние после окисления
гумуса, мл
Количество соли Мора,
отвечающее количеству
хромовой кислоты, из¬
расходованному на
окисление гумуса, мл
Гумус, % к воздушно-сухой почве
15,0.1,02*0,000517.100 „
1
8
0—10
0,1
30,0
45,0-30,0= 15,0
о/l -7’91
Реактивы. 1. Раствор двухромовокислого калия 0,4 н. (окислитель): 40 г из¬
мельченного в ступке К2Сг207 растворяют в дистиллированной воде, фильтруют через
бумажный фильтр в мерную колбу на 1 л, доводят до черты, после чего переносят в колбу
из тугоплавкого стекла на 3—5 л или в большую фарфоровую чашку, где смешивают с1л
H2S04 (уд. вес 1,84). Во избежание сильного разогревания жидкости при смешивании
серную кислоту следует приливать к водному раствору К2Сг207 понемногу, с интервалами
в 15—20 мин. и при осторожном перемешивании. Смеси дают остыть, вновь тщательно
перемешивают и переливают для хранения в склянку с притертой стеклянной пробкой.
2. Раствор соли Мора 0,1 н. готовят из расчета: 40 г соли Мора в 1 л воды, содержащей
20 мл серной кислоты (уд. вес 1,84). Навеску продажной соли Мора растворяют на холоду
в некотором количестве дистиллированной воды, раствор фильтруют через складчатый
фильтр. К фильтрату добавляют необходимое по расчету для данной навески количество
серной кислоты, доводят раствор дистиллированной водой до заданного объема, после чего
тщательно перемешивают. Раствор соли Мора хранят в бутыли, снабженной сифоном со
стеклянным краном для подачи раствора в бюретку и склянкой Тищенко с щелочным рас¬
твором пирогаллола для предохранения раствора соли Мора от окисления кислородом
воздуха.
Титр соли Мора устанавливают по 0,1 н. раствору КМп04 следующим образом:
в коническую колбу на 100 мл вносят с помощью мерного цилиндра 1 мл H2S04 (уд. вес
1,84), затем приливают из бюретки 10 мл раствора соли Мора; содержимое колбы разбав¬
ляют дистиллированной водой до 30—40 мл и тут же титруют 0,1 н. раствором КМп04
до слабо-розовой окраски, не исчезающей в течение 1 мин. Титр раствора соли Мора вы¬
ражают в виде десятичной дроби или в виде так называемого гумусового числа, при¬
Определение гумуса почвы по методу И. В. Тюрина
61
нимая, как указывалось выше, что 1 мл 0,1 н. раствора соли Мора соответствует
0,000517 г гумуса.
В связи с тем, что в состав соли Мора входит закисное железо, титр раствора ее, не-
смотря на пирогаллоловый предохранитель, сравнительно неустойчив, и его необходимо
проверять каждый раз перед началом работы.
3. Раствор пирогаллола (предохранитель от окисления раствора соли Мора кислоро¬
дом воздуха). 12 г пирогаллола растворяют в 50 мл воды; 180 г КОН растворяют в 300 мл
воды. Оба раствора смешивают, помещают в склянку Тищенко и присоединяют при помощи
резиновой и стеклянной трубок к бутыли с раствором соли Мора.
4. Раствор дифениламина (индикатор при титровании раствором соли Мора): 0,5 г
дифениламина растворяют в 100 мл H2S04 (уд. веса 1,84); к раствору постепенно, с боль¬
шой осторожностью, добавляют 20 мл дистиллированной воды.
5. Раствор фенилантраниловой кислоты (индикатор при титровании раствором соли
Мора): 0,2 г ее растворяют в 100 мл 0,2%-ного водного раствора Na2COs; для лучшего
смачивания порошка феаилантраниловой кислоты навеску ее предварительно растирают
стеклянной палочкой в фарфоровой чашке с небольшим количеством 0,2%-ного раствора
соды до сметанообразного состояния и только после этого добавляют остальное количество
раствора соды при тщательном перемешивании.
6. Сернокислое серебро (катализатор). Применяют в порошкообразном состоянии.
Однако при массовых анализах определение углерода по методу Тюрина проводится, как
указывалось выше, без сернокислого серебра.
7. 0,1 н. раствор КМп04 (для установки титра раствора соли Мора). Раствор готовят
обычным приемом; титр его устанавливают по 0,1 н. раствору перекристаллизованного
щавелевокислого натрия (Na2C204).
8. Прокаленные пемза или почва (для равномерного кипения окислительной смеси
при холостых анализах). Малогумусную почву или продажную пемзу растирают в фар¬
форовой ступке, просеивают через сито с отверстиями в 1 мм и прокаливают в фарфоровой
чашке в муфельной печи при красном калении в течение 1 — 1,5 час. при периодическом
перемешивании во избежание спекания.
9. Ортофосфорная кислота 85%-ная, х.ч. Применяют для устранения влияния ионов
окисного железа при индикаторе дифениламине.
Заканчивая описание метода, следует указать, что присутствие в почве
карбонатов, даже в таких больших количествах, как в сероземах, не метает
определению гумуса по Тюрину и не требует никаких изменений в ходе
анализа.
Метод не применим при содержании гумуса в почве свыше 15—20%, так
как при этом не достигается полноты окисления.
Для почв, содержащих хлориды свыше 0,6%, закисные соединения желе¬
за и марганца, метод не пригоден, так как часть хромовой кислоты при этом
расходуется на окисление указанных соединений, что искажает результаты
анализа. Для таких почв следует применять метод Кнопа или Густавсона.
Тюрин указывает на возможность частичного устранения влияния хло¬
ридов путем предварительного добавления к взятой навеске почвы 0,2 г
Ag2S04 с 5 мл H2S04, разведенной водой в соотношении 1:1. После добав¬
ления указанных реагентов смесь оставляют на час при периодическом пере¬
мешивании для перевода содержащихся в почве хлоридов в AgCl. Однако
этот прием не устраняет полностью влияние хлора, так как во время даль¬
нейшего кипячения окислительной смеси происходит некоторое разложение
хлористого серебра с выделением хлора. Поэтому для почв, засоленных
хлоридами свыше 0,6%, можно рекомендовать предварительную отмывку
последних при помощи дистиллированной воды. Для этого поступают сле¬
дующим образом: 100—200 г воздушно-сухой почвы, пропущенной через,
сито с отверстиями в 1 мм, помещают в химический стакан емкостью 500—
800 мл, заливают доверху дистиллированной водой, слабо подкисленной
несколькими каплями t,0 fc. H2S04, и неоднократно перемешивают в течение
02
Н. II. Бельчикова
дня. Утром отстоявшийся прозрачный раствор сливают декантацией, не
взмучивая осадка почвы, которую затем вновь заливают подкисленной водой.
Если раствор над осадком почвы к утру продолжает оставаться мутным, воду
следует подкислять серной кислотой несколько сильнее. Операцию с отмыв¬
кой повторяют до исчезновения в промывных водах иона хлора (качествен¬
ная проба на присутствие хлора с 1%-ным раствором AgN03).
По окончании отмывки всю почву из стакана переносят при помощи ди¬
стиллированной воды в тарированную фарфоровую чашку, ставят на кипя¬
щую водяную баню и выпаривают досуха. Почву в чашке оставляют около
весов на сутки для установления воздушно-сухого состояния, после чего
взвешивают (взвешивание производят на технохимических весах). Таким
образом, определяют соотношение между весом исходной почвы, взятой
для отмывки, и весом почвы после отмывки. Из отмытой, высушенной и
взвешенной почвы берут среднюю пробу весом 5 г, растирают в ступке и
пропускают через сито с отверстиями диаметром 0,25 мм, после чего берут
обычные навески для определения гумуса. Зная соотношение между весом
почвы до и после отмывки, расчет содержания гумуса ведут на исходную
почву.
МОДИФИКАЦИЯ МЕТОДА ТЮРИНА
СО СПЕКТРОФОТОМЕТРИЧЕСКИМ ОКОНЧАНИЕМ
ПО ОРЛОВУ и ГРИНЦЕЛЬ
В модификации Д. С. Орлова и Н. М. Гриндель окисление органиче¬
ского вещества почвы проводится по методу Тюрина с раствором бихромата
калия в серной кислоте. Но количество хромовой кислоты, пошедшее на
окисление, учитывается не объемным способом, а спектрофотометрическим.
Подробное изложение метода приведено в статьях Д. С. Орлова и Н. М. Грин¬
дель (1967а, б) и Д. С. Орлова с соавт. в «Практикуме по биохимии гумуса»
1969). Метод особенно удобен при определении малых количеств органиче¬
ского углерода (например, в водных вытяжках, почвенных растворах).
Л» тсратура
Арипушкина Е.В. Руководство по хими¬
ческому анализу почв. Изд-ео МГУ,
1970. .
Бельчикова II. II. Методы определения об¬
щею содержания н состава гумуса мине¬
ральных почв.— В сб. «Органическое
вещество целинных и освоенных почв».
М., «Наука», 1972.
ГеЯройц К. К. Избранные сочинения, т. 2.
Химический анализ почвы. М., Сель-
хозгиз, 1955 (раздел «Новые методы
анализа почв»).
Комарова Н. А. Применение объемного ме¬
тода определения гумуса к исследованию
различных его форм в почвах.— Труды
Ленингр. отд. ВИУА, 1933, вып. 17.
Кононова М. М. Органическое вещество
почвы, его природа, свойства и методы
изучения. М., Изд-во АН СССР, 1963.
Кононова М. М., Бельчикова II. II. Уско¬
ренный метод определения состава гу¬
муса минеральных почв.— Почвоведе-
ниэ, 1961, № 10.
Орлов Д. С., Гриндель Н. М. Спектрофо-
томстрическое определение содержания
гумуса в почве.— Почвоведение, 1967а,
№ 1.
Орлов Д. С., Гриндель II. М. Еще о спек-
трофотометрич''ском методе опр'деления
содержания гумуса в почве.— Почво¬
ведение, 19676, № 8.
Орлов Д. С., Гришина Л. А., Ерошичева
//. Л. Практикум по биохимии гумуса.
Изд-во МГУ, 1969.
Пономарева В. В. К методике изучения
состава гумуса по схеме И. В. Тюрина.—
Почвоведение. 1957, № 8.
Пономарева В. В., Плотникова Т. А. Ме¬
тодика и некоторые результаты фрак¬
ционирования гумуса черноземов.—
Почвоведение, 1908, № 11.
Симаков В. Н. Применение флшлантра-
ниловой кислоты при определении гуму¬
са по методу И. В. Тюрина.— Почвове¬
дение, 1957, № 8.
Тюрин И. В. Новое видоизменение объем¬
ного метода определения гумуса с по¬
мощью хромовой кислоты.— Почвове¬
дение, 1931, № 5—6.
Тюрин И. В. Материалы по сравнитель¬
ному изучению методов определения
органического углерода в почвах.—
Пробл. сов. почвоведения, 1936, сб. 2.
Тюрин И. В. Органическое вещество почв.
Сельхозгиз, 1937.
В. В. ЗАМЯТИНА
МЕТОДЫ ОПРЕДЕЛЕНИЯ АЗОТА В ПОЧВЕ
*
В верхних горизонтах почв преобладающая часть азота (95—99%) связа¬
на в органических соединениях. Наблюдается зависимость между накопле¬
нием в почвах органического вещества и содержанием азота. В богатых гуму¬
сом черноземах величины общего азота составляют 0,4—0,6%, в бедных
песчаных почвах — 0,03—0,04%, в торфе может содержаться 2,5—3,0%.
Химическая природа органических соединений азота почв еще недоста¬
точно полно изучена. В последние годы метод хроматографии позволил
установить, что в пахотных горизонтах почв 40—50% азота может присут¬
ствовать в виде аминокислот. В почвах найдены пуриновые и пиримидиновые
основания, глюкозиды и некоторые другие соединения, содержащие азот,
но величины их незначительны.
Минеральные соединения азота в пахотных горизонтах составляют лишь
небольшую часть общего азота почв (1—5%). Они в основном представлены
нитратами и соединениями аммония. Содержание нитритов в почвах обычно
очень мало, заметные количества нитритов отмечены только в специфических '
условиях, например в избыточно увлажненных почвах, в щелочных почвах
после применения высоких доз аммонийного азота.
Основным источником почвенного азота, обеспечивающего питание ра¬
стений, являются нитраты и обменный аммоний.
Аммоний присутствует в почвах в форме: а) воднорастворимых солей,
б) обменного NH4 и в) фиксированного (необменного) NH4. В пахотных
горизонтах преобладает обменный аммоний.
Нитраты находятся в почвах в виде воднорастворимых солей. Они отли¬
чаются высокой подвижностью, в связи с чем содержание их в почвах под¬
вержено большим колебаниям. Из пахотных горизонтов, особенно песчаных
почв, нитраты могут вымываться атмосферными осадками (и поливными вода¬
ми) в более глубокие слои. В почвенных образцах одной и той же почвы, взя¬
тых в разные сроки, содержание N03 может значительно варьировать.
Органическое вещество почвы служит главным природным резервом,
поставляющим растениям минеральный азот. В результате жизнедеятельно¬
сти микроорганизмов, использующих органическое вещество почвы как
источник энергии, происходит аммонификация азотсодержащих органиче¬
ских соединений. Значительная часть освободившегося при этом аммония
подвергается нитрификации.
Определение минеральных соединений азота, доступных растению, в поч¬
венных образцах устанавливает величины их только для срока взятия образца,
но не дает представления об обеспеченности растения почвенным азотом в
течение вегетационного периода.
Для определения способности органических соединений азота перехо¬
дить в более подвижные формы, в основном в минеральные соединения, пред¬
ложен ряд химических и биохимических методов анализа почв. Из них пред¬
ставляет интерес определение нитрификационной способности почв, а также
кислотный и щелочной гидролиз органических форм азота в сочетании с его
минеральными соединениями, присутствующими в почве.
64
В. Б. Замятина
В почвенных и агрохимических исследованиях в настоящее время широко
применяют тяжелый изотоп азота (I5N) как индикатор. Метод основан на
определении изотопного состава азота исследуемого соединения. Это требует
выбора соответствующих методов анализа почв и соблюдения ряда условий
при их выполнении.
ПОДГОТОВКА ПОЧВ К АНАЛИЗУ
Условия хранения почвенных образцов после взятия их и до проведения
анализа, особенно при определении минеральных соединений азота, могут
существенно отразиться на результатах анализа.
В свежевзятых почвенных образцах величины нитратов, нитритов, водно¬
растворимого и обменного аммония быстро изменяются в результате биоло¬
гических процессов.
Применение консервирующих соединений (толуол, хлороформ и др.)
обычно не дает желаемых результатов по ингибированию биологической ак¬
тивности почв, но может привести к изменению содержания N03 и NH4
в почвенных образцах.
Приемы консервирования почвы (выдерживание ее при низких темпера¬
турах — порядка —10, —15°), рекомендуемые в последнее время некото¬
рыми авторами, не дали согласованных результатов в отношении возмож¬
ности их применения.
Высушивание образцов, а также хранение их при полевой влажности
даже относительно короткое время (1—2 дня) приводят к значительному из¬
менению содержания в почве воднорастворимого и обменного аммония.
В связи с этим все определения обменного и воднорастворимого NH4 прово¬
дят в свежевзятых образцах. Перед взятием навесок комочки почвы разру¬
шают стеклянной палочкой, из почвы отбирают органические остатки. Луч¬
ше, если влажность позволяет просеять почву через сито с отверстиями диа¬
метром 2—3 см.
Нитраты обычно определяют в воздушно-сухих образцах. Как показал
А. Н. Лебедянцев (1960) и другие исследователи, содержание N03 практи¬
чески не меняется при доведении почвы до воздушно-сухого состояния и хра¬
нении ее при этой влажности. Для высушивания почву распределяют тон¬
ким слоем на бумаге, помещая ее в тень, в хорошо проветриваемом помеще¬
нии. Высушивание почвы на солнце приводит к изменению содержания N03.
Состав почвы характеризуется большой неоднородностью. При опреде¬
лении в почве общего содержания азота на анализ берут небольшие навески,
особенно при микроопределении. Чтобы навески отображали среднюю пробу,
подготовка почвы для анализа осуществляется следующим образом.
Из нерастертой воздушно-сухой почвы сначала берут среднюю пробу
весом 25—30 г. Из нее удаляют видимые невооруженным глазом органические
остатки. Комочки почвы разрушают легким надавливанием фарфоровым пес¬
тиком и снова выбирают органические остатки. Затем почву растирают в фар¬
форовой ступке и пропускают через сито с отверстиями 1 мм. Из растертой
почвы берут среднюю пробу весом около 10 г и затем из нее выбирают остав¬
шиеся еще органические остатки. Для этого образец распределяют тонким
слоем на пергаментной бумаге и над ним на высоте около 3 см проводят на¬
электризованной стеклянной палочкой, к которой пристают органические
остатки. Палочку очищают и снова проводят ею над почвой. Операцию
заканчивают, когда к палочке будут приставать лишь единичные кусочки
органических веществ. В процессе их удаления почву несколько раз переме¬
шивают. Чтобы наэлектризовать стеклянную налочку, ее протирают шер¬
стяной материей. Это можно сделать одновременно с удалением приставших
к палочке органических остатков, вытирая ее куском шерстяной ткани.
Методы определения ааота в почве
65
Палочку нельзя держать очень близко от почвы, так как к ее поверхности
могут пристать илистые частицы. После удаления из почвы органических
остатков ее растирают и пропускают через сито с отверстиями 0,25 мм.
Образцы хранят в банках, закрытых пробками.
Остальные анализы (если нет специальных указаний в ходе их проведе¬
ния) выполняют с почвами, высушенными до воздушно-сухого состояния на
воздухе, растертыми и пропущенными через сито с отверстиями 1 мм.
Способы хранения воздушно-сухих образцов также могут оказать влия¬
ние на результаты анализа. Хранение их (особенно в лаборатории) в бумаж¬
ных пакетах или в картонных коробках, как это широко практикуется, мо¬
жет привести к заметному повышению содержания аммонийного азота, нит-
рификационной способности, в меньшей степени — нитратов в результате сор¬
бирования почвой аммиака из воздуха. Это было замечено давно, однако
хранению образцов в условиях, исключающих поглощение почвой NH3, до
сих пор не уделяется должного внимания. Недавно Бремнер сообщил, что при
медленном токе воздуха, содержащего меньше 0,01 мг I6NH8 в 1 м3, над по¬
верхностью воздушно-сухих почв в* почвах очень быстро были обнаружены
заметные количества меченого аммиака. Чтобы исключить поглощение поч¬
вами аммиака из воздуха, воздушно-сухие образцы следует хранить в банках
с хорошо пригнанными пробками, можно также использовать полиэтиленовые
пакеты.
ОПРЕДЕЛЕНИЕ ОБЩЕГО АЗОТА
Методы определения общего содержания азота в почвах основаны на окис¬
лении органического вещества и учете минерализовавшегося при этом азота*.
В основном применяются два метода — Кьельдаля и Дюма. Первый со¬
стоит в окислении органического вещества концентрированной серной кисло¬
той при кипячении с добавлением в конце сжигания (для полноты окисле¬
ния) перманганата калия. Образовавшийся при этом аммиак определяют объ¬
емным методом после отгона его из сильнощелочпой среды. Метод Дюма
включает сухое озоление органического вещества при повышенной темпера¬
туре в присутствии металлической меди и окиси ее. Содержание образующе¬
гося при этом элементарного азота определяют по объему в нитрометре.
Разработано много вариантов этих методов.
В настоящее время классические методы Кьельдаля и Дюма в первона¬
чальном виде в практике анализа уже не применяются, но модификации их
обычно сохраняют первоначальное наименование авторов.
При анализе почв более широко используется метод Кьельдаля. Он зна¬
чительно проще по технике выполнения, требует меньше времени и по точно¬
сти не уступает методу Дюма. Многие варианты метода Кьельдаля позволяют
значительно ускорить процесс окисления органического вещества почв по
сравнению с классическим методом, улучшить технику отгона и определения
аммиака. Ряд из них дает устойчивые и надежные результаты. Разработаны
полумикрометоды, которые по точности определения близки к макромето¬
дам. Они требуют меньше времени и реактивов для выполнения анализа, что
весьма существенно при массовых определениях. Для почв с небольшим со¬
держанием азота (меньше 0,05%) более надежны макрометоды.
Метод Кьельдаля и многие его модификации не полностью учитывают
азот нитратов и нитритов. Однако содержание нитратов (и особенно нит¬
ритов) в почвах обычно настолько небольшое, что оно не может повлиять
на результаты определения общего азота (величины N —NOs и N — NOe
лежат в пределах точности метода определения валового азота). Укаэанные
методы могут быть использованы при массовых определениях аэота боль¬
шинства почв. Если почвы содержат значительные количества нитратов (на¬
пример, образцы с полей, недавно удобренных аэотом), а также при исследо¬
66
В. Б. Замятина
ваниях с применением I6N для установления общего количества азота необ¬
ходимо использовать варианты метода, включающие определение нитратов.
Для учета азота нитратов и нитритов при определении общего N по
Кьельдалю предложен ряд модификаций: применение для сжигания органи¬
ческого вещества почв фенолсерной кислоты (метод Кьельдаля — Иодльбау-
ера), предварительная обработка почв салициловой кислотой вместе с серной
(метод Коне) или перманганатом калия и восстановленным железом (метод
Ольсена), сжигание почвы серной кислотой в присутствии цинка и восста¬
новленного железа и др. В практике анализа почв наиболее часто используют
первые два варианта, которые дают хорошо совпадающие результаты.
Ниже дается описание варианта макро- и полумикроопределения общего
азота методом Кьельдаля (не включающего азот N03 и N02), широко приме¬
няемого в практике почвенного анализа, и две модификации этого метода
с применением фенолсерной и салициловой кислот, учитывающих нитраты
и нитриты. Кроме того, дается описание полумикрохромового метода опре¬
деления общего азота, предложенного Тюриным. Метод дает хорошее совпа¬
дение с методом Кьельдаля.
Метод Кьельдаля
Метод состоит из двух операций:
1. Окисление органического вещества почвы серной кислотой при кипя¬
чении. Для более быстрого и полного сжигания органического вещества при¬
меняют сернокислый калий, повышающий температуру кипения кислоты, и
катализаторы: металлический селен и сернокислую медь. Выделяющийся
при разложении органического вещества аммиак связывается серной кисло¬
той, образуя сернокислый аммоний.
2. Разложение сернокислого аммония доведением реакции раствора до
сильнощелочной, прибавлением едкого натра. Образующийся при этом
аммиак отгоняют в раствор кислоты и определяют его количество объемным
методом.
Макрометод Кьельдаля
Ход анализа. 1. Окисление органического вещества
почвы. Навеску почвы 3—10 г (в зависимости от предполагаемого содер¬
жания азота) помещают в колбу Кьельдаля емкостью 350 мл. Чтобы избе¬
жать при этом загрязнения горла колбы, навеску берут в пробирку. Ниж¬
ний конец пробирки с помощью резиновой трубки присоединяют к стек¬
лянной толстостенной трубке. Пробирку держат наклонно за трубку и на нее
надевают колбу Кьельдаля так, чтобы открытый конец пробирки не достигал
на 4—5 см до дна колбы. Колбу переворачивают и высыпают в нее почву.
Туда же вносят 0,1 а порошка металлического селена и 8 а смеси сернокис¬
лого калия и медного купороса. Это делают с помощью небольшой воронки
с длинным концом, опущенным на 2—3 см ниже начала расширяющейся
части колбы. Мерным цилиндром туда же приливают 25 мл серной кислоты
(уд. вес 1,83—1,84). Содержимое колбы тщательно перемешивают. Чтобы
уменьшить или совсем исключить вспенивание жидкости при последующем
нагревании, колбу оставляют при комнатной температуре на 3—4 часа (лучше
на ночь). После этого содержимое ее нагревают при частом взбалтывании,
регулируя нагрев, чтобы жидкость постепенно довести до кипения. Кипение
продолжают несколько часов (3—5, а иногда и больше) в зависимости от со¬
держания и качества органического вещества. Сжигание ведут под тягой.
Его заканчивают, когда охлажденный раствор над осадком будет бесцветным
или окрашенным в голубоватый цвет от присутствия меди.
Чтобы избежать выбрасывания раствора из колбы Кьельдаля во время
кипения, особенно при толчках, которые могут возникнуть в конце сжигания,
Методы определения азота в почве
67
Рис. 1. Прибор для отгопа аммиака
Объяснение см. в тексте
Рис. 2. Полумикродистилляционный при¬
бор для отгона аммиака. Прибор Хоскинса,
модифицированный Бремнером
Объяснение см. в тексте
особенно при анализе песчаных почву колбы устанавливают в наклонном
положении на специальной металлической подставке. В процессе сжигания
нагревают только ту часть колбы, где находится жидкость. Нагревание кол¬
бы выше уровня жидкости может привести к потерям азота.
Нагрев жидкости регулируют так, чтобы выделяющиеся при кипении
пары серной кислоты концентрировались в горле колбы, примерно на г/3 пути
от его основания. При этом в колбе будет сохраняться необходимое количе¬
ство серной кислоты. Улетучивание паров серной кислоты может привести
к потерям азота из-за повышения температуры кипения жидкости в связи
с увеличением концентрации сернокислого калия. Для предотвращения ис¬
парения серной кислоты можно использовать специальные пробки, кото¬
рыми прикрывают колбы Кьельдаля. Пробки служат воздушными холодиль¬
никами и препятствуют испарению жидкости.
При большом содержании в почве карбонатов, чтобы избежать сильного
вспенивания, серную кислоту приливают в колбу в несколько приемов, каж¬
дый раз взбалтывая ее содержимое. Следующую порцию кислоты приливают
после прекращения бурного выделения С02.
2. Отгон аммиака. Для отгона аммиака используют дистилля-
ционные аппараты различных модификаций. В основе их лежит один прин¬
цип, различия лишь конструктивные. Наиболее удобны приборы с приме¬
нением водяного пара.
Один из наиболее простых приборов для макроанализа показан на рис. 1.
Он состоит из отгонной (дистилляционной) колбы 1 емкостью в 700—1000 мл
(удобно использовать колбу Кьельдаля), соединенной при помощи шлифа
с насадкой 2, в которую впаяна стеклянная трубка, одним концом доходя¬
щая почти до дна дистилляционной колбы, другим, снабженным воронкой 6
(закрывающейся стеклянной пробкой на шлифе), соединена шлифом или
тефлоновой трубкой с ловушкой S, присоединенной к парообразователю 7.
Последним служит 3—5-литровая плоскодонная колба, на 2/3 заполняемая
дистиллированной водой, доводимой до кипения. Ловушка внизу имеет кран,
68
В. Б. Замятина
при помощи которого регулируется подача пара в дистилляционную колбу.
Верхняя часть насадки 2 снабжена каплеуловителем 3, соединенным тефло¬
новой трубкой или шлифом с холодильником 4. Нижний конец холодильника
оканчивается трубкой, которую опускают в приемник 5, представляющий
собой плоскодонную или коническую колбу емкостью 400—500 мл. Во время
работы холодильник обертывают полосками фильтровальной бумаги. Это
предупреждает попадание в приемник капель, образующихся на наружных
стенках холодильника в результате конденсации водяных паров из воздуха.
Отгон аммиака проводят, пропуская водяной пар из парообразователя через
чсследуемый раствор, помещенный в дистилляционную колбу. Выделяю¬
щиеся при этом пары NH3 и воды, проходя через холодильник, конденсиру¬
ются, и далее дистиллят поступает в приемную колбу. Чтобы избежать
накопления в дистилляционной колбе большого количества жидкости в резуль¬
тате конденсации проходящего через раствор водяного пара, колбу подогре¬
вают, поддерживая раствор при слабом кипении. Перед работой воду в паро¬
образователе слабо подкисляют серной кислотой для связывания могущего
присутствовать в ней аммиака.
Полумикродистилляционный аппарат, недавно описанный Бремнером
(1965), для анализа почв показан на рис. 2. Этот прибор отличается высокой
производительностью. Он удобен при отгоне аммиака, обогащенного t5N.
Аппарат просто разбирается и благодаря этому стекло легко очищать от
эагрязнения тяжелым изотопом азота.
Прибор состоит из дистилляционного сосуда 1% снабженного кожухом,
который внизу имеет трубку, соединенную с двухходовым краном 8. К одному
отводу крана присоединяют водоструйный насос, с помощью которого осво¬
бождают дистилляционный сосуд после окончания анализа и при его промы¬
вании. Второй отвод крана служит для сообщения прибора с воздухом и спу¬
ска накапливающегося в кожухе конденсата водяных паров. К кожуху при¬
соединен парообразователь 7. Последним служит 5-литровая плоскодонная
колба. Верхняя часть дистилляционного сосуда при помощи широкого шли¬
фа соединена с насадкой 2% снабженной каплеуловителем 3 и воронкой 6,
закрывающейся стеклянной пробкой. Каплеуловитель соединен с холодиль¬
ником 4. Нижний конец холодильника оканчивается трубкой, которую
опускают в приемник 5, представляющий собой плоскодонную или кониче¬
скую колбу емкостью 250 мл.
Чтобы избежать при проведении анализа попадания капель в приемник
с поверхности холодильника (образующихся при конденсации водяных паров
из воздуха), нижний конец холодильника снабжают воронкой, служащей
для сбора и отвода конденсата. Парообразователь соединен с дистилляцион-
ным сосудом тефлоновой трубкой, остальные части прибора соединяются
с помощью стеклянных шлифов.
Для обеспечения постоянного уровня воды в парообразователе в его
пробку впаяна стеклянная трубка, подающая дистиллированную воду из
резервуара, установленного выше парообразователя. Подачу воды регулируют
зажимом. Воду в парообразователе слегка подкисляют серной кислотой для
связывания могущего быть в ней аммиака. Для нагрева парообразователя
используют газовые или электрические приборы. Пар, поступающий из
парообразователя, прежде чем попасть в дистилляционный сосуд, нагревает
его с наружной стороны. В связи с этим в дистилляционном сосуде не про¬
исходит конденсации большого количества водяного пара и уменьшается
время отгона аммиака. Работая на двух приборах, один аналитик за рабо¬
чий день может отогнать 50—60 проб.
Если во взятой навеске почвы предполагают содержание азота больше
5 мг, то для отгона аммиака можно использовать только часть раствора, по*
лученного после сжигания, применив полумикродистилляционный аппарат.
Это занимает меньше времени и в случае необходимости позволяет повторить
Методы определения азота в почве
анализ. При меньшем содержании аэота в навеске аммиак отгоняют из всега
раствора, используя макроприбор.
Отгон NH3 из всего раствора. К содержимому колбы Кьельда-
ля, охлажденному до комнатной температуры после сжигания, осторожно
(по стенкам) небольшими порциями при взбалтывании приливают 10—15 мл
дистиллированной воды. Раствор с осадком переносят с помощью воронки
в дистилляционную колбу макроприбора для отгона аммиака. Колбу Кьель-
даля 4—5 раз ополаскивают водой по 40—50 мл, присоединяя ее к основ¬
ному раствору. Если в колбе останется небольшое количество песка, его
можно не переносить, достаточно только промывки водой. Присутствие
песка в отгонной колбе может повлечь к возникновению толчков при дис¬
тилляции аммиака.
Раствору дают остыть, объем его доводят водой до 300—400 мл, после
чего колбу присоединяют к прибору. В приемник наливают 10 мл 2%-ного
раствора борной кислоты, прибавляют 2—3 капли индикатора Гроака.
Нижний конец отводной трубки холодильника погружают (примерно на
0,5—1 см) в раствор борной кислоты. Включают холодильник. Через ворон¬
ку в дистилляционную колбу прибавляют 100 мл 40%-ного раствора едкого
натра. Затем, закрыв кран ловушки 8, сейчас же пускают пар из парообра¬
зователя, вода в котором предварительно должна быть доведена до кипения.
Количество 40%-ного раствора щелочи берут в 4 раза больше, чем концент¬
рированной серной кислоты, взятой для сжигания.
Выделяющийся из раствора NH3 увлекается паром, который конденси¬
руется, проходя через холодильник. Конденсат далее поступает в приемник,
где NH3 поглощается кислотой. Ток водопроводной воды, охлаждающий
холодильник во время отгона, регулируют, чтобы температура поступающего
в приемник конденсата не превышала 25°. В начале отгона отводную трубку
холодильника держат погруженной в раствор борной кислоты. Когда будут
отогнаны основные количества аммиака (в приемнике накопится 100—150 мл
раствора), дистилляцию продолжают, держа конец отводной трубки холо¬
дильника на 1—2 см выше уровня жидкости в приемнике. Отгон NH3 закан¬
чивают, когда объем дистиллята достигнет 300—400 мл. Об этом судят по
отсутствию реакции с реактивом Несслера в последних 2—3 мл дистиллята.
По окончании отгона конец трубки холодильника ополаскивают водой, со¬
бирая ее в приемник, дистиллят титруют 0,02 н. раствором H2S04. Окончание
титрования устанавливают по переходу зеленой окраски в фиолетово-красную.
Аммиак образует с борной кислотой борат аммония по реакции
H3BO3+3NH4OH = (NH4)3B03+3H20.
При последующем титровании бората аммония раствором серной кислоты
снова образуется борная кислота:
2(NН4)зВОз+ 3H2SO4 = 3 (NH4)2S04+2H8B03.
По количеству серной кислоты, пошедшей на титрование, рассчитывают
содержание N — NH4 в растворе (1 мл 0,02н. H2S04 соответствует 0,28 мг N).
Для связывания аммиака вместо борной кислоты можно использовать
0,02 н. титрованный раствор H2S04. По окончании отгона избыток серной
кислоты оттитровывают 0,02 н. раствором NaOH; по разности устанавливают
количество связанного кислотой аммиака. Оба поглотителя по точности
дают идентичные результаты. Однако применение борной кислоты более
удобно, содержание N — NH4 устанавливают прямым титрованием, поэтому
нет необходимости применять борную кислоту точной концентрации и точно
отмеренного объема, не требуется титрованный раствор щелочи.
Чистоту реактивов проверяют холостым определением (проводят не
менее двух определений). В колбу Кьельдаля помещают 25 мл концентриро¬
ванной серной кислоты, туда же вносят K2S04 и катализаторы в количествах,
70
В. Б. Замятина
используемых при сжигании почвы. Смесь нагревают и кипятят 2 часа, далее
проводят те же операции, что и при анализе почвы. Данные N — NH4 кон¬
трольного определения вычитают из результатов анализа почвы и рассчиты¬
вают содержание азота, выражая его в процентах от веса почвы.
Отгон NHS из части раствора. Содержимое колбы Кьельда¬
ля после сжигания навески охлаждают до комнатной температуры. В колбу
небольшими порциями приливают 40—50 мл дистиллированной воды, каж¬
дый раз перемешивая раствор после добавления очередной порции. При
этом часть осадка, находящегося на дне колбы, растворится, часть перейдет
во взвешенное состояние. Если осадок трудно переходит в раствор, то колбу
дополнительно подогревают. Раствор со взвесью при помощи небольшой
воронки переносят в мерную колбу емкостью 250 мл. Колбу Кьельдаля не¬
сколько раз ополаскивают водой (по 20—30 мл), присоединяя промывные
воды к основному раствору.
Для отгона NH3 применяют полумикродистилляционные приборы. Удоб¬
но использовать аппарат, показанный на рис. 2. При работе с ним в прием¬
ник наливают 5—10 мл 2 %-ного раствора борной кислоты, прибавляют 2—
3 капли индикатора Гроака, нижний конец холодильника опускают в раствор
борной кислоты. Кран 8 (см. рис. 2) ставят в положение, обеспечивающее
доступ воздуха в прибор. Пипеткой из мерной колбы отбирают 30—50 мл
раствора, полученного после сжигания почвы, его переносят в воронку при¬
бора, вынимают пробку воронки. При этом раствор переходит в дистилля-
ционный сосуд. Воронку ополаскивают небольшим количеством воды, за¬
крывают пробкой. Затем в воронку помещают 40%-ный раствор NaOH,
исходя из следующего расчета. Вычисляют, какое количество концентриро¬
ванной серной кислоты, используемой для сжигания почвы, приходится на
объем раствора, взятого для отгона; объем щелочи берут в 4 раза больше.
Вынув пробку воронки, спускают раствор едкого натра в дистилляционный
сосуд. Оставшуюся на воронке щелочь быстро смывают 10—15 мл воды,
спуская ее в сосуд. Воронку сейчас же закрывают пробкой. Общее количе¬
ство раствора в дистилляционном сосуде не должно превышать 80 мл (для
определения его на сосуде при изготовлении ставят метку). В прибор подают
пар из парообразователя, закрыв кран 8. Во время отгона регулируют интен¬
сивность кипения воды в парообразователе, чтобы за 1 мин. в приемнике
собиралось 5—6 мл дистиллята. Ток водопроводной воды, охлаждающий
холодильник, должен обеспечивать температуру дистиллята не выше 25°.
Когда в приемной колбе накопится 15—20 мл дистиллята, отгон продолжают,
держа конец холодильника на 1—2 см выше уровня жидкости в приемнике.
Отгон NH3 обычно заканчивается, когда в приемнике наберется 35 мл рас¬
твора (для удобства на колбе делают метку несмывающейся краской, указы¬
вающую нужный объем). Окончание дистилляции проверяют реактивом
Несслера. Для этого конец холодильника обмывают водой (собирая ее в при¬
емник), под него подставляют пробирку и, набрав 1—2 мл дистиллята,
прибавляют 2—3 капли реактива Несслера. Отсутствие желтой окраски
укажет на полноту отгона. Раствор в приемнике титруют 0,02 н. H2S04,
его окончание устанавливают по переходу зеленой окраски в фиолетово¬
красную (1 мл 0,02 н. H2S04 соответствует 0,28 мг N).
Холостое определение на чистоту реактивов проводят, как указано при
описании отгона NH3 из всего раствора. Результаты холостого определения
рассчитывают, исходя из общего количества раствора (до которого доведен
раствор после сжигания) и объема его, взятого для отгона аммиака. Получен¬
ные величины вычитают из результатов анализа почв и рассчитывают со¬
держание азота в процентах веса почвы.
Реактивы. 1. Концентрированная серная кислота (уд. вес. 1,83—1,84). Приме¬
няют х. ч. реактив. Серная кислота не должна содержать аммонийных солей и нитратов.
Проверку ее на присутствие NH4 проводят следующим образом. К 15 мл дистиллирован.
Методы определения ааота в почве
71
вой воды приливают 1 мл серной кислоты. Жидкость охлаждают, нейтрализуют 10%-ным
раствором едкого натра (в присутствии лакмусовой бумажки). Затем прибавляют еще 1 мл
того же раствора щелочи и 0,5 мл реактива Несслера (приготовление см. ниже). Кислота
пригодна при наличии слабо-желтой окраски раствора. Красно-бурая окраска раствора
или [выпадение осадка указывают на большое содержание в кислоте аммонийных солей.
Применяемые при испытании кислоты дистиллированная вода и щелочь не должны содер¬
жать аммония.
Испытание серной кислоты на присутствие нитратов проводят с хромотроповой кис¬
лотой. Растворяют 0,1 г динатриевой соли хромотроповой кислоты в 50 мл H2S04. Для
ускорения растворения раствор подогревают на водяной бане не более 3—4 мин. при
температуре не выше 70°. Приобретение раствором желтой окраски указывает на присут¬
ствие нитратов в серной кислоте. Такая кислота не пригодна для работы.
2. 40%-ный раствор NaOH. В большой фарфоровой чашке растворяют 2,1 кг х. ч.
едкого натра в 2 л воды. Содержимое чашки размешивают толстой стеклянной палочкой
до полного растворения щелочи (при растворении едкого натра происходит разогревание
раствора). Охлажденный до комнатной температуры раствор переносят в бутыль, закры¬
вают резиновой пробкой. Через несколько дней прозрачный раствор над осадком сливают
сифоном и объем его разводят в 2,5 раза дистиллированной водой, не содержащей угле¬
кислоты. Процентное содержание NaOH проверяют ареометром и, если требуется, раствор
подгоняют до 40%-ного прибавлением воды (не содержащей С02) или концентрированного
раствора едкого натра. Если приготовленный реактив содержит аммиак (желтое окраши¬
вание при прибавлении реактива Несслера), его удаляют кипячением раствора, используя
для этого медную посуду (жидкость при этом приобретает голубой цвет от присутствия
меди). После удаления аммиака раствор охлаждают и доводят водой, не содержащей угле¬
кислоты, до первоначального объема. Реактив хранят в склянке, закрытой резиновой
пробкой.
3. Металлический селен (в порошке). Препарат может быть загрязнен солями аммо¬
ния. Для очистки порошок промывают горячим раствором 0,5 н. КС1 и затем водой для
удаления избытка хлора (сначала горячей, затем холодной), высушивают на воздухе
между листами фильтровальной бумаги.
4. Смесь сернокислого калия и сернокислой меди. На 10 частей х. ч. K2S04 берут
1 часть CuS04-5H20. Смесь растирают в фарфоровой ступке в тонкий порошок.
5. 0,02 н. титрованный раствор H2S04.
6. Индикатор Гроака. Смешивают равные объемы 0,4%-ного спиртового раствора
метилового красного и 0,2%-ного спиртового раствора метиленового синего. Хранят
в темной склянке с притертой пробкой.
7. Реактив Несслера. Растворяют 25 г иодистого калия (KJ) в 30 мл воды, прибав*
ляют 35 г иодистой ртути (красной) HgJ2 и в фарфоровой ступке пестиком растирают до
полного растворения. Прибавляют 870 мл 15%-ного раствора КОН, перемешивают, дают
отстояться и декантируют прозрачную жидкость. Раствор хранят в темной склянке.
Чтобы приготовить 15%-ный раствор КОН, 150 г едкого кали растворяют в 900—
950 мл воды; после охлаждения доводят до 1 л водой.
В продаже имеется готовый реактив Несслера, им можно пользоваться при выполне¬
нии анализов.'
8. Дистиллированная вода, применяемая при анализе и для приготовления реакти¬
вов, не должна давать реакцию на NH3 с реактивом Несслера.
Полумикрометод| Кьельдаля
Ход анализа. Навеску почвы от 0,2 до 1 г (взвешенную с точностью 0,001 г)
помещают в колбу Кьельдаля емкостью 50—100 мл. Туда же прибавляют
0,05 г Se, 1,5 г смеси K2S04 и CuS04, а также 5 мл серной кислоты (уд. вес
1,83—1,84). Содержимое колбы медленно доводят до кипения и продолжают
его до обесцвечивания жидкости над осадком. После сжигания к раствору
прибавляют 10 мл воды и количественно переносят в дистилляционный сосуд
полумикродистилляционного аппарата. Далее в сосуд добавляют 20 мл
72
В. Б. Замятина
40%-ного раствора NaOH и отгоняют NHS, предварительно налив в прием¬
ник 5 мл 2 %-ного раствора борной кислоты. Сжигание почвы и отгон NH3
проводят, соблюдая те же условия, что и при определении азота макромето¬
дом. Приготовление реактивов описано там же.
Модификации метода Кьельдаля,
включающие азот нитратов и нитритов
Вариант с применением фенолсерной кислоты
Этот вариант обычно называют методом Кьельдаля — Иодльбауера. Ос¬
новные операции при анализе почв те же, что и в методе Кьельдаля. Разли¬
чия заключаются в следующем.
1. Для окисления органического вещества почвы вместо серной кислоты
применяют (в тех же количествах) фенолсерную кислоту. Смесь оставляют
на 2 часа при комнатной температуре. Затем в колбу вносят 1 г цинковой
пыли и оставляют еще на час. Дальнейший ход анализа тот же. Нитраты и
нитриты при сжигании почвы фенолсерной кислотой в присутствии цинка
восстанавливаются до аммиака.
Фенолсерную кислоту готовят растворением при нагревании 40 г чистого
фенола в 1 л серной кислоты (уд. вес 1,83—1,84).
Вариант с применением салициловой кислоты
Приводится модификация анализа, предложенная Бремнером и Шоу
(Вгешпег, 1965).
Навеску почвы, содержащую около 10 мг азота, помещают в колбу Кьель¬
даля емкостью 350 мл, прибавляют 40 мл"смеси салициловой кислоты с сер¬
ной (ее готовят растворением 50 г салициловой кислоты в 2 л серной кисло¬
ты, уд. вес 1,83—1,84). Содержимое колбы осторожно взбалтывают до пол¬
ного перемешивания почвы с кислотой. Оставляют на несколько часов (или
на ночь) в покое при комнатной температуре. Затем прибавляют 5 г кристал¬
лического тиосульфата натрия (Na2S203*5H20), предварительно растертого
и пропущенного через сито с отверстиями 0,5 мм. Соль вносят при помощи
небольшой воронки с длинным носиком, доходящим до расширенной части
колбы. Смесь постепенно нагревают, доводя до кипения. Затем охлаждают.
В колбу добавляют 20 мл дистиллированной воды. С помощью той же во¬
ронки вносят 10 г K2S04, 1 г CuS04-5H20 и 0,1 г Se. Далее почву сжигают и
отгоняют NH3, соблюдая все условия хода анализа и приготовтения реак¬
тивов, как при выполнении анализа по методу Кьельдаля.
Нитраты и нитриты при взаимодействии с салициловой кислотой обра¬
зуют нитросоединения, которые в кислой среде при кипячении смеси с тио¬
сульфатом натрия восстанавливаются до аминов. Реакции нитрования ме¬
шает присутствие воды. Это недостаток метода. Однако Бремнер показал,
что метод вполне пригоден для анализа воздушно-сухих образцов почвы.
Метод Тюрина (1933)
При определении общего азота в почве по методу Тюрина органическое
вещество почвы окисляют смесью хромовой кислоты с серной. Азот органи¬
ческих соединений при этом переходит в аммонийную форму. Его определяют
объемным методом после отгона NH3 из щелочной среды.
Ход анализа. В зависимости от предполагаемого содержания азота навес¬
ку 0,2—1,0 г воздушно-сухой почвы, взятой с точностью 0,001 г, помещают
Методы определения азота в почве
73
Рис. 3. Упрощенный
прибор для отгона
аммиака
Объяснение см. в тексте
в плоскодонную или коническую колбу емкостью 250 мл. Туда же пипеткой
или из бюретки приливают 2,5 мл 25%-ного раствора хромового ангидрида
(или 10%-ного раствора К2Сг207). Содержимое колбы перемешивают легким
взбалтыванием. Цилиндром добавляют 5 мл концентрированной серной
кислоты и снова осторожно взбалтывают. В горло колбы вставляют воронку
диаметром около 4 см, которая служит воздушным холодильником. Кол¬
бу ставят на предварительно нагретую электрическую плитку с закрытой
спиралью (можно также использовать песчаную баню с толстым слоем песка).
Жидкость доводят до кипения, которое продолжают не менее 10 мин. до
приобретения раствором интенсивно зеленой окраски. Кипение ведут уме¬
ренно, чтобы предотвратить преждевременное разложение Сг03 и сильное
испарение жидкости. Во избежание толчков при падении капель конденси¬
рующейся жидкости с воронки в раствор воронку помещают наклонно, чтобы
конец ее касался стенки горла колбы. Это обеспечит плавное стекание жид¬
кости по стенкам. Позеленение раствора в начале анализа указывает на не¬
достаток хромового ангидрида. Анализ повторяют снова с меньшим количе¬
ством почвы или с большим объемом хромового раствора и соответствую¬
щим ему двойным количеством серной кислоты.
По окончании сжигания содержимому колбы дают охладиться, воронку
обмывают с внутренней и наружной сторон водой, смывая ее в колбу, в кото¬
рой проводилось сжигание, и приступают к отгону аммиака.
В оригинальном методе Тюрина отгон NH3 производят в упрощенном
дистилляционном аппарате, без водяного холодильника (рис. 3). Прибор со¬
стоит из отгонной колбы 1 емкостью 250 мл\ небольшого каплеуловителя 2
(в виде шарика диаметром 5—6 см)\ приемника 5, которым служит кониче¬
ская колба емкостью в 50 мл; стеклянной трубки 4 с оттянутым концом и
шариковым предохранителем, препятствующим перебрасыванию жидкости
из приемника в отгонную колбу.
Перед началом отгона аммиака оттянутый конец трубки 4 опускают до
дна приемника, содержащего 10—15 мл 0,02 н. раствора серной кислоты и
2—3 капли индикатора метилрота. В таком положении трубка остается до
конца отгона. Для охлаждения дистиллята приемник помещают в сосуд
с холодной водой. Колба, в которой проводили окисление почвы, обычно ис¬
пользуется в качестве отгонной. Содержимое ее доводят водой примерно
до 100 мл. Чтобы обеспечить равномерное кипение, в нее добавляют неболь¬
шое количество прокаленной пемзы (на кончике шпателя) и 2—3 зерна цин¬
ка. Осторожно, по стенкам, не перемешивая жидкость, в отгонную колбу
74
В. Б. Замятина
приливают 20 мл 50 %-ного раствора NaOH (из расчета четырехкратного ко¬
личества взятой для сжигания серной кислоты). Колбу присоединяют к при¬
бору, жидкость осторожно перемешивают покачиванием колбы и нагревают,
постепенно доводя до кипения. Для подогрева используют электрическую
плитку или газовую горелку. Если в начале кипения жидкость в дистилляци-
онной колбе сильно вспенивается (что может повлечь перебрасывание ее
в приемник), нагрев уменьшают, но не прекращают во избежание засасыва¬
ния раствора из приемника в отгонную колбу. Дистилляция аммиака обычно
заканчивается через 15—20 мин., когда отгонится около половины объема.
По окончании отгона конец отводной трубки вынимают из раствора, ополас¬
кивают водой. Дистиллят выпаривают (не доводя до кипения) на одну треть
и горячий титруют 0,02 н. раствором NaOH. Конец титрования устанавли¬
вают по переходу окраски от оранжево-розовой в бледно-желтую. По разности
между количеством серной кислоты, взятой и оттитрованной щелочью,
устанавливают количества ее, связавшиеся аммиаком, и на основании этого
рассчитывают содержание N — NH3 в анализируемой пробе (1 мл 0,02 н.
H2S04 соответствует 0,28 мг N). Одновременно проводят холостое определе¬
ние (для каждой партии не менее двух). Ход анализа и количество реактивов
сохраняют те же, что и при анализе почв, только вместо почвы используют
прокаленную пемзу (или прокаленную почву) в количестве 0,2 г. Результаты
холостого определения вычитают из данных, полученных при анализе почвы.
Содержание общего азота рассчитывают в процентах от веса почвы.
Отгон аммиака после сжигания почвы хромовой смесью можно проводить,
аспользуя обычные полумикродистилляционные аппараты. Для этого смесь
после сжигания количественно с помощью воды переносят в отгонную колбу
прибора, добавляют 20 мл 50 %-ного раствора NaOH и отгоняют NHS с соблю¬
дением условий, принятых при определении общего азота методом Кьельда-
ля. Для поглощения аммиака применяют раствор борной кислоты.
Реактивы. 1. 25%-ный раствор хромового ангидрида (Сг03). Навеску 25 г
хромового ангидрида растворяют в дистиллированной воде в мерной колбе« доводят объем
водой до] 100 мл. Вместо хромового ангидрида можно использовать 10%-ный раствор
К2Сг207, но это менее удобно. При окислении органического вещества двухромовокислым
калием во время кипения жидкости могут происходить толчки, это затрудняет проведение
анализа.
2. Серная кислота (уд. вес 1,83—1,84), х. ч.
3. 50%-ный раствор NaOH. Способ приготовления описан выше (см. метод Кьель-
даля), только слитый сифоном с осадка концентрированный раствор едкого натра разводят
водой не в 2,5] раза, а в 2.
4. Порошок] пемзы. Куски пемзы растирают в фарфоровой ступке, пропускают через
сито с отверстиями диаметром 1 мм и прокаливают в муфеле при красном калении в боль¬
ших фарфоровых тиглях в течение 1—1,5 часа. Хранят в хорошо закрытой банке.
5. Метилрот. Растворяют 0,2 г метилрота в 60 мл этилового спирта и добавляют 40 мл
дистиллированной воды.
6. 0,02 н. титрованный раствор NaOH.
7. 0,02 н. титрованный раствор HaS04.
ОПРЕДЕЛЕНИЕ СОЕДИНЕНИЙ АЗОТА ПОЧВЫ
Нитраты
Нитраты присутствуют в почве в виде воднорастворимых солей. Для из¬
влечения их применяют водные или солевые вытяжки. Как показали ис¬
следования А. Н. Лебедянцева (1960), отношение почва : вода, равное
1 : 0,75, достаточно для полного извлечения нитратов. В практике почвенных
Методы определения азота в почве
1Ъ
анализов используют обычно отношение 1 : 5 или 1 : 10, что технически
более удобно при получении вытяжек.
Нитраты в почвенных вытяжках определяют колориметрическими мето¬
дами и объемными. Более широко распространены первые, поскольку они
отличаются более высокой чувствительностью и требуют меньше времени для
выполнения анализа.
Из колориметрических методов всеобщее признание получил метод
с применением дисульфофеноловой кислоты, предложенный Грандваль-Ляжу.
Имеется несколько модификаций этого метода, которые различаются в ос¬
новном по способу приготовления реактива дисульфофеноловой кислоты.
Ниже описана одна из модификаций, предложенная Лебедянцевым. Метод
дает хорошо воспроизводимые результаты. Недостаток его заключается
в необходимости выпаривать вытяжки, что осложняет массовые определе¬
ния. Варианты без выпаривания менее чувствительны.
В последнее время предложен новый колориметрический метод определе-
ления нитратов с хромотроповой кислотой. Он прост в выполнении, практи¬
чески не чувствителен к тем концентрациям примесей, которые обычно при¬
сутствуют в почвенных вытяжках. По точности и чувствительности он не
уступает методу с дисульфофеноловой кислотой. Метод еще недостаточно
широко апробирован в практике почвенных анализов, однако его преимуще¬
ство перед другими методами позволило привести его описание.
Колориметрические методы определения нитратов можно применять
только при наличии прозрачных и бесцветных вытяжек. Эти условия особен¬
но трудно выполнить при работе с водными вытяжками. Окраска вытяжек
в большинстве случаев связана с присутствием растворимых органических
веществ. Ряд авторов для извлечения нитратов рекомендуют применять
слабые растворы солей в концентрациях, не мешающих колориметрическо¬
му определению. Это ускоряет процесс фильтрования, уменьшает мутность и
окраску вытяжек, но далеко не всегда позволяет получать бесцветные и
прозрачные растворы, особенно при анализе лесных, заболоченных, солон¬
цеватых и некоторых других почв.
Существуют способы осветления вытяжек путем осаждения органическо¬
го вещества углем, сажей, квасцами, едкой известью, полиакриламидами и
другими реагентами. Такие приемы бывают эффективными, однако приме¬
нять их надо очень осторожно, поскольку в ряде случаев это приводит к за¬
ниженным результатам.
Когда нельзя использовать колориметрические методы, применяют методы,
основанные на восстановлении нитратов до аммиака с последующей дис¬
тилляцией его водяным паром или методом диффузии. Аммиак определяют
объемным методом. Этим методом пользуются так же при работе с 15N, когда
требуется определение изотопного состава азота нитратов.
При выполнении этих анализов нитраты извлекают солевыми раствора¬
ми, применяя более высокие концентрации, чем используются при колори¬
метрических методах. Этим достигают получение быстро фильтрующихся,
прозрачных вытяжек. Солевые вытяжки одновременно с нитратами извлека¬
ют и обменный аммоний. В полученных вытяжках нитраты восстанавливают
сплавом Деварда, трехвалентным титаном, восстановленным железом и
другими реагентами. Аммиак отгоняют из щелочной среды, для создания
которой используют окись магния, едкую щелочь, буферный раствор бората
и пр. Применение NaOH или КОН менее желательно, так как они могут
вызвать гидролиз присутствующих в вытяжке органических соединений, что
приведет к завышению результатов анализа, особенно при отгоне аммиака
водяным паром.
Способ отделения аммиака после восстановления нитратов методом диф¬
фузии более прост по технике выполнения, чем дистилляция NH3 водяным
паром, но этот метод менее чувствителен. Метод диффузии дает хорошие
76
В. Б. Замятина
результаты при содержании не менее 5 лег N — N03 на 100 г почвы. Ниже
описан метод с применением дистилляции аммиака водяным паром с ис¬
пользованием для восстановления нитратов сплава Деварда в присутст¬
вии окиси магния. Метод дает хорошо воспроизводимые результаты.
В последнее время разрабатываются новые методы определения целого
ряда ионов, в том числе и нитратов, с использованием ионоселективных
мембранных электродов. На основе жидких анионообменников предложены
электроды, специфичные к N03, позволяющие определять концентрации
порядка 10"3—10"4 М. Такие электроды выпускаются зарубежными фирма¬
ми. Использование их при определении в почве нитратов описано рядом ав¬
торов (Myers, Paul, 1968;Mahenarappa, 1969). В СССР селективные электроды
для определения N03 еще не получили применения.
Колориметрический метод с дисулъфофеноловой кислотой
Метод основан на взаимодействии нитратов с дисульфофеноловой кисло¬
той.
При подщелачивании смеси раствор приобретает желтую окраску, по
интенсивности которой судят о содержании нитратов в почве. Метод позволяет
определять не менее 0,02 мг N—N03 в 100 мл раствора.
Ход анализа. Навеску 20 г воздушно-сухой почвы помещают в колбу ем¬
костью 250 мл, туда же наливают 100 мл дистиллированной воды. Содер¬
жимое [колбы взбалтывают 3 мин. и сейчас же фильтруют через складчатый
плотный фильтр. Чтобы получить прозрачные вытяжки, нужно как можно
полнее перенести почву на фильтр. Почва задержит прохождение через
фильтр мелкодисперсных частиц, создающих муть. Первые порции филь¬
трата отбрасывают. Если вытяжка мутная, ее перефильтровывают через тот
же фильтр. Раствор отфильтровывают полностью.
При определении нитратов в свежих образцах берут большую навеску
с учетом влажности почвы (влажность рассчитывают на воздушно-сухой
вес), объем же воды для приготовления вытяжки уменьшают, учитывая
влажность почвы.
Пипеткой берут 20—50 мл фильтрата, который помещают в небольшую
фарфоровую чашку и выпаривают досуха на водяной электрической бане.
Выпаривать на газовой бане не следует, это может привести к потерям нит¬
ратов. В охлажденную чашку по каплям из пипетки добавляют 1 мл ди¬
сульфофеноловой кислоты, стараясь смочить находящийся на дне и на боках
чашки остаток. Остаток тщательно растирают с кислотой оплавленным кон¬
цом стеклянной палочки. Чашку оставляют в покое на 10 мин. Затем в нее
добавляют пипеткой 25 мл дистиллированной воды, смесь перемешивают
стеклянной палочкой и доводят до щелочной реакции, прибавляя раствор
едкой щелочи или аммиака. При этом в присутствии нитратов жидкость ок¬
рашивается в желтый цвет* Щелочь прекращают добавлять, когда раствор
приобретет устойчивую желтую окраску или лакмусовая бумажка, опущен¬
ная в раствор, посинеет. Небольшой избыток щелочи не вредит окраске. Окра¬
шенный раствор с помощью воронки переносят в мерную колбу емкостью
100 мл. Чашку и стеклянную палочку несколько раз ополаскивают водой,
которую присоединяют к раствору в мерной колбе. Объем жидкости водой
доводят до 100 мл. Колбу закрывают пробкой, хорошо перемешивают. Опре¬
деляют оптическую плотность полученного (испытуемого) раствора, исполь¬
зуя фотоколориметр или спектрофотометр. По калибровочной кривой образ¬
цовых растворов находят содержание N — N03 в испытуемом растворе.
Для построения калибровочной кривой в фарфоровых чашках выпари¬
вают 5, 10, 15, 20 и 25 мл рабочего образцового раствора нитрата и далее
проводят те же операции, что и с испытуемыми растворами.
Методы определения ааота в почве
77
Оптическую плотность растворов определяют тотчас же после развития
окраски. Интенсивность окраски при стоянии испытуемых растворов сни¬
жается.
При концентрациях ниже 12 лег N — N03 в 1 л окраска растворов под¬
чиняется закону Бера. Максимум спектра поглощения лежит в области 410 нм.
При использовании фотоколориметра ФЭК-56 измерения проводят со свето¬
фильтром № 4 (эффективная длина волны 440 нм). В качестве раствора
сравнения используют дистиллированную воду, выпаренную и обработан¬
ную реактивами (дисульфофеноловой кислотой и щелочью), как при работе
с вытяжками.
Если испытуемый раствор окрашен более интенсивно, чем образцовые
растворы, его разбавляют в несколько раз водой и далее определяют опти¬
ческую плотность. Разведение учитывают при расчете содержания нитратов
в почве.
Выпаривание вытяжек, содержащих значительные количества аммония,
снижает величины N03. Чтобы предотвратить отрицательное влияние аммо¬
нийных солей, перед выпариванием.вытяжки в фарфоровую чашку добавля¬
ют 2—3 капли 10%-ного раствора сернокислого калия. Присутствие в вытяж¬
ках СГ также мешает определению. Если содержание иона хлора более
30 лег в 1 л, его осаждают серебром. Ниже описан способ, рекомендуемый
Бремнером (Bremner, 1965). К 100 мл вытяжки прибавляют 0,1 г Ag2S04
(в порошке), смесь взбалтывают 25 мин. Для осаждения избытка Ag+ в раст¬
вор добавляют, в виде сухих реактивов, 0,2 г Са(ОН)2 и 0,5 г MgC03, взбал¬
тывают 5 мин. Суспензию фильтруют через сухой складчатый фильтр. Бе¬
рут определенный объем фильтрата и выпаривают, далее проводят анализ,
как указано выше. При удалении хлора серебром подщелачивание раствора,
после обработки осадка дисульфофеноловой кислотой, проводят аммиаком.
Использование NaOH или КОН может вызвать появление бурого оттенка при
развитии желтой окраски растворов.
Большим недостатком работы с водными вытяжками являются труд¬
ность получения прозрачных и бесцветных фильтратов и длительность филь¬
трования. В связи с этим предложено для приготовления вытяжек приме¬
нять 0,03 н. или 0,05%-ный раствор K2S04 при сохранении того же отноше¬
ния почвы к раствору. При определении нитратов с дисульфофеноловой
кислотой Бремнер рекомендует к навеске почвы 50 г добавлять 0,5 г Са(ОН)2
(в порошке) и далее обрабатывать почву пятикратным количеством воды при
10-минутном взбалтывании.
Реактивы. 1. Раствор дисульфофеноловой кислоты. Навеску 30 г чистого све-
жеперегнанного фенола помещают в плоскодонную колбу емкостью 500 мл. Туда же прп.
ливают 200^мл HaS04 (уд. вес 1,83—1,84). Хорошо перемешивают, закрывают корковой
пробкой со вставленной в нее длинной стеклянной трубкой (обратный холодильник).
Колбу нагревают в течение 6 час., опустив ее в кипящую воду. После этого реактив ох"
лаждают и используют при анализе.
Реактив, хранящийся в холодном месте, может выкристаллизоваться. Кристаллы
растворяют, поставив колбу в горячую воду. Разбавлять реактив водой не следует.
В настоящее время в продаже имеется реактив — фенол-2,4-дисульфокислота. На¬
веска 50 г его, растворенная в 100 мл H2S04 (уд. вес. 1,83—1,84), соответствует реактиву
дисульфофеноловой кислоты, приготовление которой описано выше.
2. 20%-ный раствор NaOH или КОН. Можно также использовать 10%-ный раствор
аммиака.
3. Образцовый раствор нитрата. Запасной раствор. Для приготовления используют
х.ч. KNOs, его перекристаллизовывают из воды и высушивают при 100—105° до постоян¬
ного веса. На аналитических весах отвешивают 0,722 г KN03, переносят в мерную колбу
емкостью 1 л, растворяют в дистиллированной воде, объем водой доводят до 1 л, хорошо
перемешивают (1 мл раствора содержит 0,01 мг N—N03).
78
В. Б. Замятина
Рабочий раствор. Готовят разведением запасного раствора в 50 раз водой. Рабочий
раствор содержит 0,002 мг N—NOs в 1 мл. Его используют для приготовления образ¬
цовых растворов.
4. Реактив Ag2S04, х. ч. в порошке.
5. Реактив Са(ОН)2, х. ч. в порошке.
6. Реактив MgCOs, х. ч. в порошке.
Колориметрический метод с хромотроповой кислотой
При взаимодействии нитрат иона с хромотроповой кислотой [1,8-диокси-
3,6-нафталиндисульфокислота — (HO)2C10H4(SO3H)2] образуется комплекс,
окрашивающий раствор в желтый цвет. Реакция протекает в 70%-ной сер¬
ной кислоте и весьма избирательна. Метод позволяет определять от 0,2 до
20 мг N — N03 на 100 г почвы. Присутствующие в почвенных вытяжках ка¬
тионы практически не влияют на развитие окраски. Это дает возможность
для извлечения нитратов применять растворы электролитов.
Использование хромотроповой кислоты для определения нитратов в почве
предложено сравнительно недавно, и этот метод еще недостаточно широко
апробирован в практике почвенных анализов. Однако он может представлять
значительный интерес, особенно при выполнении массовых определений
благодаря своей простоте, быстроте и высокой чувствительности. Метод
дает результаты, близкие с методом дисульфофеноловой кислоты.
Ход анализа. Приводится методика, описанная Н. Н. Басаргиным и
Е. А. Черновой (1968).
Навеску 10 г воздушно-сухой почвы, растертой и просеяной через сито
с отверстиями 1 мм, помещают в колбу емкостью 100 мл. Туда же прибавляют
50 мл 0,05%-ного раствора K2S04, взбалтывают 3 мин. и фильтруют через
двойной складчатый фильтр из плотной бумаги. Если фильтрат мутный, его
перефильтровывают. В стакан емкостью 50 мл помещают пипеткой 3 мл
вытяжки, осторожно по стенкам стакана прибавляют 7 мл рабочего раствора
хромотроповой кислоты. Жидкость круговым движением стакана хорошо
перемешивают и оставляют стоять при комнатной температуре 30 мин. для
охлаждения. За это время в растворе развивается желтая окраска. По ис¬
течении указанного срока определяют оптическую плотность раствора, ис¬
пользуя фотоколориметр или спектрофотометр. Раствор сравнения, ис¬
пользуемый при колориметрировании, готовят прибавлением к 3 мл дистил¬
лированной воды 7 мл рабочего раствора хромотроповой кислоты. Содержа¬
ние N — N03 находят по калибровочной кривой. Для этого из образцовых
рабочих растворов NaN03 отбирают пипеткой по 3 мл, переносят в стаканы
емкостью в 50 мл. Туда же осторожно (по стенкам стакана) прибавляют 7 мл
рабочего раствора хромотроповой кислоты. Содержимое стакана перемеши¬
вают и через 30 мин. измеряют оптическую плотность.
Желтая окраска растворов нитрат-хромотропового комплекса имеет два
максимума спектра поглощения, в области 357 и 415 нм. При использова¬
нии фотоколориметра ФЭК-56 оптическую плотность растворов измеряют
с применением светофильтра № 4 (эффективная длина волны 440 нм). Окрас¬
ка устойчива в течение 24 час. Температурные колебания от 20 до 65° не вли¬
яют на ее интенсивность.
Если используемые растворы окрашены более интенсивно, чем образцо¬
вые, для колориметрирования берут меньший объем вытяжки. Его доводят
водой точно до 3 дел и затем прибавляют хромотроповую кислоту.
Присутствующие в почвенных вытяжках соединения практически не вли¬
яют на интенсивность окраски колориметрируемых растворов в концентра¬
циях, значительно превышающих содержание их в почвенных вытяжках,
за исключением N02 и Fe2+. Анион N02 образует с хромотроповой кислотой
комплекс, окрашивающий раствор в желтый цвет, аналогичный тому, кото¬
Методы определения ааота в почве
79
рый дают нитраты. Закисное железо в сернокислой среде (после прибавле¬
ния раствора хромотроповой кислоты) восстанавливает N03, это вызывает
снижение интенсивности окраски. По данным Симса и Джексона (Sims,
Jackson, 1971), предельно допустимые концентрации N02 и Fe2+, которые
не проявляют еще отрицательного влияния, выражаются соответственно
8,4 и 167 мг/л. Это не может служить ограничивающим условием исполь¬
зования метода, так как в почвенных вытяжках такие концентрации N02 и
Fe2+ встречаются крайне редко, только при наличии в почвах восстанови¬
тельных процессов. Ион хлора не дает окраски при взаимодействии с хромо¬
троповой кислотой, но в присутствии нитратов повышает ее интенсивность.
Отрицательное влияние хлора элиминируют добавлением НС1 при приготов¬
лении реактива хромотроповой кислоты.
Для извлечения из почвы нитратов вместо 0,05%-ного раствора K2S04
можно, как рекомендуют Симс и Джексон, применять 0,2%-ный раствор
Са(ОН)2 (отношение почва : раствор составляет 1 : 5) при 15-минутном
взбалтывании. Вытяжки быстро фильтруются и очень редко дают мутные
фильтраты.
Реактивы. 1. Раствор хромотроповой кислоты. Используют двунатриевую
соль — (HO)2C10H4(SO8Na)2. В продаже часто имеется технический препарат. Перед упо¬
треблением его очищают. Насыщенный раствор соли дважды обрабатывают углем, не
содержащим трехвалентного железа, и перекристаллизовывают из воды. По внешнему
виду очищенный препарат представляет собой белый порошок.
Сначала готовят основной 0,1%-ный раствор. Растворяют 100 мг двунатриевой соли
хромотроповой кислоты в 50—70 мл концентрированной серной кислоты. Раствор должен
оставаться бесцветным. Если он желтеет, это указывает на загрязнение серной кислоты
нитратами. Такая кислота не пригодна для анализа. Чтобы ускорить растворение соли
в кислоте, смесь можно подогреть на водяной бане при температуре не выше 70° в течение
3—4 мин. Более длительное подогревание и более высокая температура вызовут побуре-
ние раствора. После растворения соли раствор переносят в мерную колбу емкостью 100 мл
и объем его доводят концентрированной серной кислотой до метки на колбе. Из этого
основного раствора готовят рабочий раствор.
Пипеткой 10 мл основного раствора помещают в мерную колбу емкостью 100 мл,
туда же приливают 7 мл концентрированной соляной кислоты и объем раствора доводят
концентрированной H2S04 до 100 мл.
Полученный реактив используют при проведении анализов. Им можно пользовать¬
ся в течение 4 дней после приготовления. Основной и рабочий растворы хранят в
склянках с притертыми пробками в темном месте.
Перед использованием угля для осветления двунатриевой соли хромотроповой кис¬
лоты его проверяют на отсутствие трехвалентного железа. Навеску угля 2—4 в обрабаты¬
вают 2—3%-ным раствором НС1 в течение примерно 15 мин. Если полученный фильтрат
дает положительную реакцию на железо в присутствии роданистокислого аммония, уголь
необходимо подвергнуть очистке. При загрязнении небольшими количествами железа
уголь очищают промыванием 2—3%-ным горячим раствором НС1 до полного удаления
железа с последующим отмыванием хлора горячей водой. При значительном загрязнении
уголь очищают от железа следующим образом. Навеску угля дважды (а иногда и более
раз) кипятят по 3 часа на умеренном огне с концентрированной соляной кислотой, беря
10 объемов кислоты на 1 г угля. Смесь желательно кипятить в колбе с пришлифованной
стеклянной пробкой, снабженной обратным холодильником. Можно также использовать
обычную колбу, вставив в ее горло стеклянную пробку, применяемую Для прикрывания
колб Кьельдаля, или воронку с коротким загнутым кончиком. Во время кипения жидкости
необходимо, по возможности, поддерживать исходный объем раствора прибавлением кон¬
центрированной соляной кислоты. После 3-часового кипения смесь охлаждают, отфиль¬
тровывают на воронке Бюхнера, применяя разрежение воздуха в приемной колбе. Изли¬
шек соляной кислоты отмывают водой и повторяют обработку угля концентрированной
соляной кислотой, как указано выше, до полного удаления железа. Затем уголь отмывают
80
В. Б. Замятина
от хлора водой (сначала горячей, потом холодной) и высушивают в термостате при тем¬
пературе около 100°.
2. Образцовые растворы нитратов. Растворяют 0,6070 г х. ч. NaN03 в 1 л дистилли¬
рованной воды. Раствор содержит 0,1 мг N—NOs в 1 мл. Из этого запасного раствора
готовят образцовые рабочие растворы, содержащие 2, 4, 7, 10 и 15 мг азота в1л. Для этого
в пять мерных колб емкостью по 100 мл соответственно вносят следующие объемы запас¬
ного раствора: 2, 4, 7, 10 и 15 мл. Колбы доводят водой до метки, растворы используют
при анализах для построения калибровочной кривой. Запасные и рабочие образцовые
растворы хранят в темном месте.
3. Концентрированная серная кислота (уд. вес 1,83—1,84), х. ч. , не должна содержать
нитратов.
4. Соляная кислота (уд. вес 1,16), х. ч.
Аммоний
Для извлечения из почвы аммония применяют различные вытяжки. При
определении воднорастворимых соединений аммония используют водные
вытяжки. Для вытеснения из почвы обменного NH4 применяют солевые
растворы; при этом из почвы одновременно извлекают и воднорастворимые
соединения аммония, их учитывают вместе. Фиксированный аммоний опре¬
деляют после разрушения кристаллической решетки глинистых минералов,
закрепляющих его.
Для определения аммония в вытяжках из почвы используют колоримет¬
рические методы, методы дистилляции NH3 с водяным паром или путем диф¬
фузии.
Хороших методов колориметрического определения аммония для почв
нет. Ниже приводится описание двух методов: с применением реактива Нес¬
слера (наиболее часто используется при анализе почв) и с применением фено¬
ла, которые дают удовлетворительные результаты.
В том случае, когда нельзя использовать колориметрические методы оп¬
ределения аммония (мутные, окрашенные вытяжки, содержащие соедине¬
ния, создающие помехи при анализе), используют методы дистилляции ам¬
миака с последующим определением его объемным или колориметрическим
способом. Метод дистилляции также применяют при работе с 15N, когда
требуется определить изотопный состав азота присутствующих в почве соеди¬
нений аммония. Один из этих методов с применением водяного пара для
отгона NH3 описан ниже (см. определение нитратов и аммония методом
дистилляции).
Колориметрический метод с реактивом Несслера
Реактив Несслера широко используется при определении аммония
в вытяжках из почвы. Метод основан на получении оранжевого комплекса
HgO*Hg (NH2)J при взаимодействии реактива Несслера (щелочной раствор
K2HgJ4) с солями аммония. Реакция очень чувствительна. Молярный коэф¬
фициент погашения е = 6200 при А, = 400 нм. Предельная концентрация, до¬
пускающая определения аммония, не должна превышать 0,15 мг N — NH*
в 100 мл. При более высокой концентрации растворы приобретают оранже¬
вый или красноватый цвет, и через несколько минут образуется муть или
выпадает осадок. До указанной концентрации окраска растворов подчиняет¬
ся закону Вера. Определению мешают присутствующие в почвенных вытяж¬
ках Са2+ и Mg2+. Они осаждаются реактивом Несслера и вызывают опалесцен¬
цию растворов. Для устранения этого в вытяжку перед добавлением реак¬
тива Несслера вносят сегнетову соль, образующую с кальцием и магнием
растворимые комплексные соединения.
Методы определения азота в почве
81
Определение воднорастворимых соединений
аммония. Ход анализа. Из почвы аммоний извлекают водной вытяжкой,
используя почву с естественной влажностью сейчас же после взятия образ¬
цов. Параллельно определяют влажность почвы, учитывая ее при расчете
содержания N — NH4 в почве. Способ приготовления водной вытяжки опи¬
сан выше (см. определение нитратов с дисульфофеноловой кислотой).
Для установления содержания NH4 определенный объем вытяжки поме¬
щают в мерную колбу емкостью 50 мл, добавляют 2 ле^'раствора сегнетовой
соли, приливают воды примерно до 45 лед. Смесь перемешивают взбалтывани¬
ем, добавляют 2 мл реактива Несслера, снова перемешивают и объем дово¬
дят водой до 50 мл. В растворе сейчас же начинает развиваться окраска,
цвет ее должен быть чисто-желтым, светлого оттенка. Если раствор при¬
обретает оранжевый или красноватый цвет, анализ повторяют, беря мень¬
ший объем вытяжки. Оптическую плотность раствора измеряют с помощью
фотоколориметра. Содержание в растворе NH4 устанавливают по калибровоч¬
ной кривой образцовых растворов. Для построения ее одновременно с испы¬
туемыми растворами готовят образцовые растворы. В мерные колбы емко¬
стью 50 мл помещают 2, 5, 7 и 10 мл рабочего образцового раствора хлори¬
стого аммония, в колбы добавляют раствор сегнетовой соли и реактив Нес¬
слера, как это указано при приготовлении испытуемого раствора. Просмотр
окрасок образцовых и испытуемых растворов производят через 15 мин.
после прибавления реактива Несслера. Окрашенные растворы могут стоять
не более 30 мин., при удлинении срока в растворах появляется опалесценция.
При отсутствии фотоколориметра для сравнения окрасок испытуемых
и образцовых растворов можно использовать визуальный колориметр.
Реактивы. 1. Вода, не содержащая аммиака. Получают перегонкой дистилли¬
рованной воды после слабого подкисления ее серной кислотой или прибавления алюмо-
калиевых квасцов (на 5 л воды добавляют около 0,5 г квасцов). Свободную от NH3 воду
используют для приготовления всех реактивов и при получении вытяжек.
2. Реактив Несслера (см. выше метод Кьельдаля).
3. Сегнетовая соль. Готовят 50%-ный водный раствор. Его проверяют на отсутствие
солей аммония (с реактивом Несслера). При загрязнении сегнетовой соли аммонийными
солями NH4 осаждают. К 100 мл раствора сегнетовой соли прибавляют 5 мл реактива
Несслера, при этом сначала образуется муть, затем выпадает красно-бурый осадок. Осад¬
ку дают несколько дней отстояться. Прозрачный раствор декантируют и проверяют на
полноту осаждения. Для этого к 10 мл слитого раствора добавляют 0,5 мл реактнва[Нес-
слера. При отсутствии мути раствор используют для анализов, в противном случае про¬
водят дополнительное осаждение добавлением еще 2—3 мл реактива Несслера. Раствор
хранят в темном месте.
4. Образцовый раствор хлористого аммония. Растворяют в воде 0,7640 г х. ч. NH4CI,
объем в мерной колбе доводят водой до 1 л. Получают запасной образцовый раствор, в 1 мл
его содержится 0,2 мг N—NH4. Рабочий образцовый раствор получают разбавлением за¬
пасного образцового раствора водой в 20 раз. Раствор содержит 0,01 мг N—NH4 в 1 мл.
Его используют для приготовления образцовых растворов.
Определение обменного аммония методом Ко¬
нева (1929). Для извлечения обменного аммония применяется 0,05 н.
раствор NaCl при широком отношении почвы к раствору. Метод одновремен¬
но учитывает присутствующий в почве воднорастворимый и- обменный ам¬
моний. Анализу подвергают почву с естественной влажностью, сразу же
после взятия образцов. Влажность почвы учитывают при расчетах содержа¬
ния N — NH4 в почве.
Ход анализа. Навеску 20 г свежевзятой почвы помещают в литровую бу¬
тыль, туда же приливают 500 мл 0,05 н. раствора NaCl и 0,5 мл толуола
для предотвращения микробиологических процессов. Содержимое бутыли
взбалтывают на ротаторе в течение 1 часа. Суспензию фильтруют через
82
В. Б. Замятина
складчатый фильтр. Фильтр предварительно промывают горячим, а затем
холодным раствором 0,05 н. NaCl для удаления солей аммония или погло¬
щенного из воздуха аммиака, которые могут быть в фильтровальной бумаге.
Если первые порции фильтрата мутные, их отбрасывают. Когда вытяжка
полностью отфильтрована, приступают к определению аммония. Определен¬
ный объем вытяжки помещают в мерную колбу емкостью 50 мл. Раствор
водой доводят до 40 мл, в колбу добавляют 2—4 мл раствора сегнетовой соли
для связывания] присутствующих в вытяжке Са2+ и Mg2+. Затем вносят 2 мл
реактива Несслера, взбалтывают, объем водой доводят до 50 мл и далее
после 15-минутного стояния определяют оптическую плотность раствора
с помощью фотоколориметра. Растворы окрашивают небольшими партия¬
ми, чтобы определение оптической плотности закончить в течение 15 мин.
(при удлинении срока в окрашенных растворах может развиться опалесцен¬
ция).
Одновременно готовят образцовые растворы для построения калибровоч¬
ной кривой. Для этого в мерные колбы емкостью 50 мл помещают 2, 5, 7 и
10 мл рабочего образцового раствора хлористого аммония, объем доводят
водой до 40 мл,'добавляют раствор сегнетовой соли в том же количестве, ко¬
торое использовалось для приготовления испытуемых растворов, и 2 мл
реактива Несслера, соблюдая те же условия, что и при приготовлении испы¬
туемых растворов для колориметрирования. Когда в вытяжках содержится
большое количество Са2+ и Mg2+ и добавление сегнетовой соли не обеспечи¬
вает получения прозрачных растворов для колориметрирования, Са и Mg
осаждают. Для этого к 100 мл вытяжки, помещенной в стеклянный ци¬
линдр, закрывающийся пробкой, добавляют 1—2 мл осаждающей смеси.
Жидкость взбалтывают, дают отстояться в течение 12 час., затем прозрач¬
ный раствор сливают сифоном и фильтруют через фильтр, не содержащий
аммиака. К полученному раствору прибавляют реактив Несслера и опре¬
деляют NH4.
Для извлечения иэ почвы обменного аммония хлористый натрий может
быть заменен 0,1 н. раствором КС1, что позволяет более полно вытеснить
NH4 из почвы. Б. П. Мачигин (1963) рекомендует применять для извлечения
обменного аммония 1%-ный раствор КС1. По этой методике навеску 10 г
свежевзятой почвы обрабатывают 100 мл 1%-ного раствора КС1 при часовом
взбалтывании на ротаторе (или 5-минутном взбалтывании и последующем
18—20-часовом настаивании). В вытяжке аммоний определяют колоримет¬
рически с реактивом Несслера.
Реактивы. 1. Раствор хлористого натрия. Для приготовления 0,1 н. раствора
58,46 г х. ч. NaCl растворяют в,10 л воды. Реактив не должен давать желтого оттенка при
црибавлении к 5 мл 2 капель реактива Несслера.
2. Сегнетовая соль (см. выше определение аммония с реактивом Несслера).
3. Образцовый раствор хлористого аммония (см. там же).
4. Реактив Несслера (см. выше метод Кьельдаля).
5. Осаждающая смесь. Растворяют 50 г NaOH и 50 a Na2C03 в 600 мл воды. Раствор
кипятят, не допуская разбрызгивания, охлаждают, доводят водой до 500 мл. Хранят
в закрытых резиновыми пробками склянках.
Все реактивы готовят на воде, не содержащей аммиака (см. выше определение аммо¬
ния с реактивом Несслера).
Феноловый метод,, модификация Важенина (1949)
Метод основан на образовании соединения, окрашивающего раствор в
синий цвет при взаимодействии аммония с фенолом в присутствии гипохло¬
рита натрия. Реакция протекает в щелочной среде. Метод менее точен, чем
с реактивом Несслера. Присутствие в вытяжках значительного количества
Методы определения азота в почве
83
солей щелочноземельных металлов и полуторных окислов не влияет на
развитие окраски. Это позволяет получить достаточно точные данные при
определении аммония не только в водных, но и в солевых вытяжках.
Ход анализа. Определенный объем вытяжки, но не превышающий 10 мл,
помещают в мерную колбу емкостью 50 мл. Раствором, используемым для
приготовления вытяжки, объем в колбе доводят до 10 мл.
Одновременно готовят шкалу образцовых растворов, помещая после¬
довательно в мерные колбы емкостью 50 мл 1; 2; 3; 5; 7,5 и 10 мл рабочего
образцового раствора хлористого аммония. Объем в колбах доводят до 10 мл
раствором, который применяют для приготовления вытяжки.
В каждую колбу с испытуемым и образцовым растворами добавляют по
2.5 мл раствора фенола, хорошо перемешивают взбалтыванием. В том же
порядке, как прибавляли раствор фенола, в колбы из бюретки наливают по
2.5 мл гипохлорита натрия, хорошо перемешивают. Через 10—15 мин. пер¬
вые пять колб ставят на электрическую плитку и нагревают. При этом в раст¬
воре развивается синяя окраска. Как только в растворе со дна колб начнут
появляться первые струйки газа и появится слабый звенящий звук, колбы
снимают и помещают для охлаждения в холодную воду. Нагревание на элект¬
роплитке можво заменить выдерживанием колб в водяной ванне при темпе¬
ратуре 55—60° в течение 2 час. Охлажденный раствор доводят водой до ЪО мл,
перемешивают и измеряют оптическую плотность окрашенных растворов.
На основании данных оптической плотности образцовых растворов строят
калибровочную кривую; по ней определяют содержание NH4 в испытуемых
растворах.
При использовании этого метода необходимо соблюдать следующие ус¬
ловия:
1. Объем раствора перед прибавлением рективов должен быть одинако¬
вым и возможно наименьшим.
2. Строго соблюдать последовательность прибавления реактивов с обя¬
зательным перемешиванием раствора после их добавления.
3. Необходимо сохранять определенное соотношение между объемом
фенола и гипохлорита.
4. При нагревании раствора на электроплитке нельзя доводить его да
полного кипения. Это может привести к исчезновению синей Ъкраски.
5. Если при работе испольэуют давно приготовленные реактивы, то перед
началом работы устанавливают соотношение фенола и гипохлорита, дающее
наилучшее развитие синей окраски. Для этого в три колбы приливают по
10 мл рабочего образцового раствора хлористого аммония, затем прибавля¬
ют по 2,5 мл фенола, растворы взбалтывают и к ним добавляют раэличные
количества гипохлорита — 2,0; 2,5; 3,0 мл. Растворы нагревают, как эта
указано выше, охлаждают и доводят водой до 50 мл. Устанавливают объем
гипохлорита, при котором синяя окраска наиболее интенсивна и устойчива.
Реактивы. 1. 5%-ный водный раствор фенола (СбНвО). Для приготовления ре¬
актива используют препарат ч.д.а. Если он окрашен, то его предварительно очищают
перегонкой. Для этого фенол помещают в колбу, которую закрывают корковой пробкой
со вставленной в нее изогнутой под углом в 45° стеклянной трубкой (диаметром около
2 сж, длиной 20—30 см), которая служит воздушным холодильником. Колбу нагревают
на электроплитке до кипения фенола (182°). Пары фенола сгущаются в трубке и стекают
в подставленную под нее колбу. Первые порции погона отбрасывают. Если при отгоне
в трубке образуется пробка из кристаллов фенола, это место подогревают. Перегнанный
фенол бесцветен. Операцию ведут под тягой.
2. Раствор гипохлорита натрия (NaOCl). В склянку, содержащую 800 мл 1,5 н. рас¬
твора NaOH (или КОН), взвешенную с точностью до 0,1 г, через стеклянную трубку вво¬
дят из баллона газообразный хлор до увеличения первоначального веса склянки на 35,5 а.
Хлор для очистки предварительно пропускают через склянки, содержащие: а) воду,
84
В. Б. Замятина
б) концентрированную H2S04 и в) 40—50%-ный раствор NaOH. Хлор пропускают со
скоростью не более 2—3 пузырьков в секунду. После насыщения раствор охлаждают,
объем его доводят 1,5 н. раствором щелочи до 1 л.
3. Образцовый раствор сернокислого аммония. В 500 мл воды растворяют 1,836 г
х. ч. (NH4)2S04t 1 мл его содержит 1 мг NH4. Этот раствор разводят в 100 раз раствором,
используемым для приготовления почвенных вытяжек. Получают рабочий образцовый
раствор, в 1 мл которого содержится 0,01 мг NH4.
Вода, используемая для приготовления реактивов, не должна содержать аммония.
Определение нитратов и аммония
в почвенных вытяжках методом дистилляции
При использовании этого метода почву обрабатывают растворами ней¬
тральных солей, применяя 1—2 н. растворы. Это позволяет получить про¬
зрачные, быстро фильтрующиеся вытяжки и требует относительно неболь¬
шого объема раствора для извлечения обменного NH4, что очень существенно.
В вытяжку одновременно переходят нитраты, воднорастворимые соли ам¬
мония и обменный аммоний.
Присутствующий в вытяжке аммоний непосредственно отгоняют в виде
NH3 из щелочной среды, создаваемой окисью магния. Нитраты предвари¬
тельно восстанавливают сплавом Деварда до NH3 в присутствии MgO и далее
отгоняют его.
Для отгона аммиака применяют дистилляционные приборы. Удобно ис¬
пользовать прибор, изображенный на рис. 2.
Ход анализа. Приготовление вытяжки. Навеску почвы в 20 г помещают
в стакан емкостью 100 мл. Почву обрабатывают 4—5 раз небольшими порция¬
ми 1 н. раствора КС1, применяя декантацию. Затем почву переносят на
фильтр и еще несколько раз промывают раствором хлористого калия до
полного извлечения из нее обменного аммония. Это устанавливают по от¬
сутствию желтого окрашивания в последних порциях фильтрата при добав¬
лении реактива Несслера.
Для фильтрования удобно использовать небольшие воронки Бюхнера.
Небольшая муть в растворе не мешает определению. По окончании извле¬
чения фцльтрат доводят 1 н. раствором КС1 до определенного объема (лучше
это проводить в мерных колбах), стараясь как можно меньше разводить
вытяжку, и далее используют его для проведения анализа.
1.Определение аммонийного аэота. В приемную колбу
пародистилляционного аппарата помещают 5 мл борной кислоты и добавляют
1—2 капли индикатора Гроака. Колбу ставят под холодильник прибора,
опуская нижний конец его в раствор кислоты. Пипеткой отбирают опреде¬
ленный объем вытяжки (обычно 30—50 мл) и помещают его в дистилляцион-
ную колбу прибора. При помощи небольшой воронки с длинным носиком,
опущенным на 1—2 см ниже расширяющейся части колбы, прибавляют
0,3 г сухого порошка окиси магния. Колбу присоединяют к прибору и сразу
же пускают пар из парообразователя с уже кипящей водой. В процессе отго¬
на, когда в приемной колбе накопится около 20 мл дистиллята, ее опускают,
чтобы конец холодильника находился на 1—2 см выше уровня жидкости, и
продолжают дистилляцию до конца. Обычно отгон заканчивается, когда
накопится 30—40 мл дистиллята. О конце его судят по отсутствию желтой
окраски в последних 1—2 мл дистиллята при прибавлении к ним 1—2
капель реактива Несслера. По окончании отгона конец трубки холодильника
ополаскивают водой. В дистилляте определяют содержание NH4 титрованием
0,005 н. раствором H2S04 с помощью микробюретки. Конец титрования уста¬
навливают по переходу окраски раствора иэ зеленой в фиолетово-красную
(1 мл 0,005 н. H2S04 соответствует 0,07 мг N).
Методы определения азота в почве
85
2. Определение азота нитратов. Из вытяжки предвари¬
тельно удаляют присутствующий в ней аммоний (см. выше). После этого
в приемную колбу дистилляционного прибора наливают 5 мл борной кислоты
и 1—2 капли индикатора Гроака. Колбу помещают под холодильник прибора,
опуская нижний конец его в раствор кислоты. Дистилляционную колбу отсо¬
единяют от прибора и в находящийся в ней раствор при помощи воронки
с длинным носиком вносят 0,3 г тонкорастертого, просеянного через сито в
0,05 мм сплава Деварда. Колбу немедленно присоединяют к прибору, пуска¬
ют пар и отгоняют аммиак, соблюдая условия, указанные в пункте 1. Содер¬
жание NH4 определяют титрованием 0,005 н. раствором H2S04 с помощью
микробюретки. Конец титрования устанавливают по переходу окраски ра¬
створа из зеленой в фиолетово-красную (1 мл 0,005 н. H2S04 соответствует
0,07 мг N).
3. Определение суммы азота нитратов и аммония.
В приемную колбу дистилляционного прибора наливают 5 мл борной кислоты
и добавляют 1—2 капли индикатора Гроака. В отгонную колбу прибора по¬
мещают 20—30 мл вытяжки. Туда же ъ помощью воронки с длинным носиком
(опущенным на 1—2 см ниже расширяющейся части колбы) прибавляют
0,3 г тонкорастертого, просеянного через сито в 0,05 мм сплава Деварда
и 0,3 г сухого порошка MgO. Колбу присоединяют к прибору, пускают из
парообразователя пар и отгоняют аммиак, как указано в пункте 1. По окон¬
чании отгона в дистилляте определяют NH4 титрованием 0,005 н. раствором
H2S04, используя микробюретку (1 мл 0,005 н. H2S04 соответствует 0,07 мг N).
Применяемые для анализа реактивы (MgO, сплав Деварда, КС1) могут
быть загрязнены соединениями азота, влияющими на результаты анализа.
Это устанавливают холостым определением, используя вместо вытяжки
1 н. раствор хлористого калия. Данные холостого определения вычитают
из результатов анализа.
Реактивы. 1. Окись магния (MgO). Реактив х. ч. окиси магния в течение 2 час.
нагревают в электрическом муфеле при 600—700°, затем переносят в эксикатор, содержащий
КОН в кусках (палочках), для охлаждения. Реактив хранят в плотно закрытых банках.
2. Раствор борной кислоты. Растворяют 20 г х. ч. Н8В03 приблизительно в 700 мл
горячей воды. Остывший раствор переносят в мерную колбу на 1 л и доводят объем водой
до метки на колбе.
3. Сплав Деварда. Применяют в виде тонкорастертого порошка. Для этого препарат
измельчают в шаровой мельнице и просеивают через сито с отверстиями диаметром 0,05 мм»
Реактив может быть загрязнен солями аммония. Его очищают промыванием порошка 0,2 н.
раствором KG1 до отсутствия в последних порциях^фильтрата реакции с реактивом Нес-
чглера. Избыток хлора удаляют промыванием препарата водой (сначала горячей, затем
холодной). Хранят в хорошо закрытых банках.
4. 0,005 н. титрованный раствор H2S04.
5. Индикатор Гроака (см. выше метод Кьельдаля).
Фиксированный аммоний в почве1
Фиксированным обычно называют такой аммоний, который не извлекает¬
ся из почвы при обработке ее 1 н. раствором КС1.
Фиксированный аммоний присутствует практически во всех почвах. Его
содержание в пахотном слое почв СССР колеблется от 2—3 до 10—12 мг
N на 100 г почвы, что составляет от 2 до 10 % общего содержания азота в поч¬
ве. В почвах с утяжеленным механическим составом в нижних горизонтах
профиля содержание фиксированного аммония возрастает с глубиной и в
отдельных случаях достигает 15—18 мг N на 100 г почвы, или 30—50% обще¬
го содержания азота.
1 Описание составлено И. А. Могилевкиной.
86
В. Б. Замятина
Фиксация ионов NH4 обусловливается вхождением их в межпакетные
промежутки кристаллической решетки почвенных глинистых минералов
с последующим проникновением в гексагональные пустоты в сетке кислород¬
ных атомов тетраэдрических слоев. Взаимодействуя с обоими отрицательно
заряженными кислородными слоями, катионы NH4 притягивают их к себе,
в результате чего кристаллическая решетка минерала сокращается.
Способностью к фиксации обладают трехслойные глинистые минералы
с разбухающей решеткой. При этом емкость фиксации зависит от суммарного
межслоевого заряда. Наибольшей фиксирующей способностью среди поч¬
венных минералов обладает вермикулит, межслоевой заряд которого равен
216—219 мг-экв на 100 г почвы, наименьшей — монтмориллонит (заряд ра¬
вен 110—115 мг-экв на 100 г почвы).
Фиксированный аммоний может быть частично использован растениями:
доступна та его фракция, которая не является структурным элементом ре¬
шетки.
Определение фиксированного аммония в почве затруднено присутствием
в ней органических соединений азота. Поэтому во всех методах определения
фиксированного аммония в почве наибольшую сложность представляет устра¬
нение погрешностей за счет азота органических соединений. По принципу
подхода к решению этого вопроса все методы можно разделить на две груп¬
пы.
К первой группе относятся методы, по которым в первую очередь наибо¬
лее полно освобождают почву от органического вещества, а затем, разрушая
минеральную фракцию почвы, извлекают фиксированный аммоний. Таковы
методы Даривала и Стивенсона (Dhariwal, Stevenson, 1958), Шахтшабеля
(Shachschabel, 1961), И. А. Могилевкиной (1964), Сильвы и Бремнера (Silva,
Bremner, 1966).
Вторая группа методов не предусматривает освобождение почвы от орга¬
нического вещества. При извлечении фиксированного аммония этими метода¬
ми необходимо стремиться свести к минимуму переход в вытяжку органи¬
ческих соединений азота. Следует отметить, что при этом одновременно эк¬
страгируется и обменный аммоний. Ко второй группе относятся методы
Родригеса (Rodrigues, 1954), Бремнера (Bremner, 1959), В. Н. Кудеярова
(1966).
Метод Могилевкиной (1964)
Предварительное удаление из почвы обменного аммония и органических
соединений азота достигается прокаливанием почвенного образца в муфеле.
Для разрушения минеральной фракции почвы и определения аммония при¬
меняют метод Кьельдаля.
Навеску почвы 8 а, взятую на технических весах, помещают в фарфоро¬
вую чашку диаметром 5—6 см и ставят в муфель, отрегулированный на тем¬
пературу 400° (в пределах + 20°). При этом рекомендуется сделать внутри
муфеля металлический стеллаж и помещать образцы на дно муфеля и на
стеллаж, отступя от дверцы муфеля примерно на 7з его объема, поскольку
в передней части муфеля температура обычно несколько ниже заданной.
При содержании в почве гумуса до 1,5% образцы выдерживают в муфеле
1 сутки (24 часа), при 1,5 — 5% — 2 суток (48 час.), при 5% и более —
3 суток (72 часа)1.
Озоленный образец ссыпают в колбу Кьельдаля емкостью 250 мл через
широкогорлую воронку и тщательно смывают из промывалки приставшие
1 Перерывы в нагревании (например, на ночь) не имеют значения. Необходимо лишь
выдержать образцы при требуемой температуре в течение всего рекомендуемого срока*
В противном случае получаются завышенные результаты анализа из-за недостаточно
полного удаления органического вещества.
Методы определения азота в почве
87
к чашке частицы почвы. В колбу добавляют 5 г K2S04, 0,5 г CuS04, 0,05 г
Se и 15 мл концентрированной H2S04 (уд. вес 1,83—1,84) и сжигают в тече¬
ние 5 час. Аммиак отгоняют 1 в аппарате Кьельдаля (отгонные колбы на 1 л)
в 4 %-ный раствор борной кислоты, который наливают в приемные колбы при
помощи цилиндра (25 мл). При отгоне стараются набирать в приемники
примерно одинаковое количество дистиллята. Титруют 0,005 н. раствором
H2S04 с комбинированным индикатором Гроака. Если необходимо опреде¬
лить изотоп I6N, отгон аммиака производят в 0,01 н. раствор H2S04 и титруют
0,01 н. раствором NaOH.
Реактивы. 1. H2S04 (уд. вес. 1,83—1,84) не должна содержать аммонийных
солей).
2. Металлический 'селен, измельченный до мелкозернистого порошка.
3. 4%-ный раствор борной кислоты. 40 г Н3В03 растворяют в 700 мл воды при нагре
вании и доводят общий объем раствора до 1 л.
4. Индикатор Гроака (см. выше метод Кьельдаля).
Метод Сильвы и Бремнера (Silva, Bremner, 1966)
Почву освобождают от органического вещества, обрабатывая ее гипобро-
•митом калия. Фиксированный аммоний извлекают, разрушая минеральную
фракцию почвы смесью плавиковой и соляной кислот.
Навески почвы 1 а, взятые на технических весах, помещают в стаканы ем¬
костью 200 мл и приливают к ним по 20 мл гипобромита калия — КОВг.
После тщательного перемешивания содержимого стаканов их покрывают
часовым стеклом и оставляют на 2 часа. Затем в стаканы вливают по 60 мл
воды, нагревают (в закрытом состоянии) до кипения и энергично кипятят
в течение 5 мин. Для охлаждения и отстаивания суспензию оставляют некото¬
рое время (лучше на ночь) стоять, после чего раствор над осадком сливают.
Осадки переносят с помощью 0,5 М раствора КС1 в 100-миллилитровые по¬
лиэтиленовые центрифужные пробирки, на которых отмечен объем, равный
80 мл. Обмывая стаканы раствором КС1 для того, чтобы полностью перенести
весь осадок в пробирки, доводят объем суспензии в них до 80 мл. Пробирки
закрывают резиновыми пробками, взбалтывают вручную несколько секунд и
центрифугируют 10 мин. Раствор над осадком сливают. Затем вновь налива¬
ют 0,5 М раствор КС1 до метки, взбалтывают и центрифугируют.
После удаления раствора над осадком декантацией в пробирки приливают
полиэтиленовым цилиндром 20 мл смеси HF + НС1 с концентрацией этих
кислот в смеси, равной 5 н. для HF и 1 н. для НС1. Пробирки закрывают рези¬
новыми пробками и взбалтывают на ротаторе в течение 24 час.
Фиксированный аммоний, перешедший в раствор в процессе обработки,
отгоняют в аппарате Кьельдаля. Для этого в отгонные колбы на 0,5 л нали¬
вают 15 мл Юн. раствора КОН и вливают в него содержимое центрифужных
пробирок через полиэтиленовые воронки с оттянутым концом, погруженным
в раствор щелочи. Пробирки и воронки тщательно обмывают водой и доводят
содержимое колб до объема 250 мл. Колбы помещают на штатив прибора
для отгона аммиака, взбалтывают и оставляют на несколько минут. Отгон
аммиака и титрование производят так же, как при использовании предыду¬
щего метода.
Реактивы. 1 «ЗГипобромит калия (КОВг) готовят непосредственно перед употреб¬
лением. 6 мл жидкого брома вливают в 2 н. раствор КОН. Приливают бром медленно
>(0,5 мл/мин) при непрерывном взбалтывании. Колбу с реактивом охлаждают в сосуде со
льдом.
1 Бели во время отгона происходят сильные толчки (карбонатные почвы, сероземы и др.),
то необходимо фильтровать суспензию в процессе перенесения содержимого колбы
Кьельдаля в отгонную. Осадок нужно тщательно промыть водой и отбросить.
88
В. Б. Замятина
2. Смесь HF + НС1 (5 н. для HF и 1 н. для НС1). В полиэтиленовый сосуд емкостыо
2,5 л с отметкой объема 2 л наливают 1,5 л воды, 167 мл концентрированной НС1 (уд. вес
1,19) и 325 мл 52%-ной HF. Объем смеси доводят водой до 2 л и тщательно перемешивают.
3. 0,5 М раствор хлористого калия. 186 г КС1 растворяют в 5 л воды.
4. 10 н. раствор едкого кали. 3,3 кг КОН растворяют в 4,4 л воды и доводят общий
объем раствора после остывания до 5 л. Переливают раствор в бутыль и закрывают ре¬
зиновой пробкой.
Колориметрический метод определения нитритов по Гриссу
В основе метода лежит получение в кислой среде красной окраски при
обработке растворов, содержащих нитриты, смесью а-нафтиламина и суль-
фаниловой кислоты. Азотистая кислота превращает сульфаниловую кис¬
лоту в диазосоединение, которое, взаимодействуя с а-нафтиламином, дает
красную азо-краску. Нарастание окраски идет медленно, максимум ее на¬
ступает через несколько часов. Однако удовлетворительное окрашивание
растворо,в, позволяющее проводить определение, достигается через 15 мин.
Максимум спектра поглощения лежит при 520 нм.
Метод очень чувствителен. Наиболее удобны для определения количества
0,1—0,05 мг N—N02 в 1 л. При содержании 0,2 мг N — N02 в 1 л быстро
выпадает осадок. Анализу подвергают образцы почв с естественной влаж¬
ностью (свежевзятые).
Ход анализа. Для извлечения нитритов применяют водную вытяжку при
отношении почва : вода, равном 1 : 5, и 3-минутном взбалтывании. К 40 мл
прозрачной бесцветной водной вытяжки, помещенной в мерную колбу ем¬
костью 50 мл, прибавляют 8 мл нитритного реактива, объем в колбе доводят
водой до 50 мл. Сейчас же начинает появляться красная окраска, она разви¬
вается постепенно. Через 15 мин. после добавления к вытяжке нитритного
реактива (необходимо строго соблюдать этот промежуток времени) опреде¬
ляют оптическую плотность окрашенных растворов с помощью фотоколори¬
метров, используя светофильтры с областью пропускания 520—550 нм. Со¬
держание N — N02 в испытуемых растворах вычисляют по калибровочной
кривой образцовых растворов. Для построения ее в мерные колбы емкостью
50 мл помещают 3, 5, 10 мл рабочего образцового раствора, объем доводят
водой до 40 мл, прибавляют по 8 мл нитритного реактива, перемешивают и
доводят водой до 50 мл. Через 15 мин. измеряют оптическую плотность раст¬
воров.
При использовании для просмотра растворов визуального колориметра
испытуемые и образцовые растворы готовят одновременно и сравнивают
спустя 15 мин. после прибавления нитритного реактива.
Реактивы. 1. Нитритный реактив. Готовят смешиванием равных объемов рас¬
твора сульфаниловой кислоты и а-нафтил амина. Реактив неустойчив, поэтому его гото¬
вят непосредственно перед употреблением.
Раствор сульфаниловой кислоты. Навеску 0,5 г х. ч. сульфаниловой кислоты раство¬
ряют в 100 мл уксусной кислоты (уд. вес. 1,04).
Раствор а-нафтиламина. Навеску 0,1 г а-нафтиламина кипятят в течение 5 мин.
с 20 мл воды, процеживают через хорошо промытую хлопчатобумажную ткань в 180 мл
уксусной кислоты (уд. вес 1,04). Нитритный реактив должен быть бесцветным. Если он
имеет розовую окраску (от присутствия нитритов), раствор взбалтывают с цинковой
пылью и отфильтровывают.
2. Образцовый раствор нитрита. Растворяют в воде 0,2463 г х. ч. сухого NaN02,
раствор переносят в мерную колбу и объем доводят водой до 1 л. Получают запасный
образцовый раствор, в 1 мл которого содержится 0,05 мг N. Из него готовят рабочий образ¬
цовый раствор. Берут 10 мл запасного раствора и разводят его в 100 раз водой. Получают
раствор, содержащий 0,5 мг N в 1 л. Раствор хранят в холодильнике.
Методы определения азота в почве
89
Продажный реактив NaN02 не всегда бывает достаточно чист для приготовления
образцовых растворов. Для получения чистого препарата 0,1099 г х. ч. AgN02 раство¬
ряют в 100—150 мл воды; к раствору прибавляют хлористый натр до прекращения выпа¬
дения осадка AgCl; смесь в мерной колбе доводят водой до 200 мл, сильно встряхивают
и оставляют в темноте, пока не осядет осадок. Прозрачный раствор сливают, 1 мл его
содержит 0,05 мг N (запасный образцовый раствор). Для получения рабочего образцового
раствора 10 мл запасного раствора разводят в 100 раз водой; получают раствор, со дер
жзщий 0,5 мг N в 1 л. Чистое азотистокислое серебро приготовляют прибавлени м к
16 частям концентрированного горячего раствора азотнокислого серебра 10 частей концен»
трпрованного горячего раствора азотистокислого калия. Осадок AgN02 отделяют от
маточного раствора отсасыванием на воронке Бюхнера. Азотистокислое серебро снова
растворяют в возможно меньшем объеме горячей воды, раствору дают охладиться и кри¬
сталлический осадок отделяют на воронке Бюхнера (стараясь как можно больше отжать
раствора); осадок высушивают на водяной бане и хранят в плотно закрытой склянке
а темноте.
ОПРЕДЕЛЕНИЕ АЗОТА ОРГАНИЧЕСКИХ СОЕДИНЕНИИ ПОЧВЫ
Азот в верхних горизонтах почвы в основной своей массе представлен
органическими соединениями, входящими в состав гумуса.
Для извлечения из почвы азотсодержащих органических соединений ши¬
роко применяют кислотный или щелочной гидролиз (на холоду или при
нагревании).
Значительная часть органического азота почв при кислотном гидролизе
расщепляется до аминокислот. Полученные в последние годы данные амино¬
кислотного состава гидролизатов показали, что соотношение отдельных ами¬
нокислот в различных почвах довольно близко и приближается к таковому
для белка бактериальных клеток. Суммарный выход аминокислот при гид¬
ролизе почв 6 н. НС1 или H2S04 (слабое кипение в течение 24 час.) составляет
40—50 % общего азота (для верхних горизонтов).
Кроме аминокислот, при кислотном гидролизе отщепляется аммиачный
азот, выход которого при этом составляет 15—25% общего азота в верхних
слоях почвы. Относительное содержание аммонийного азота в гидролизатах
значительно возрастает в нижних горизонтах. По данным Стивенсона (Ste¬
venson, 1957), для горизонта В2 и ниже в результате кислотного гидролиза
от 40 до 70% общего азота приходится на аммонийную форму. Содержание
аммонийного азота в кислотных гидролизатах верхнего, относительно бога¬
того органическим веществом горизонта почвы может быть частично обязано
амидам, при гидролизе которых отщепляется аммиак. Но для нижних гори¬
зонтов с крайне низким содержанием органического вещества и в то же время
весьма высоким (по отношению к общему азоту) содержанием аммонийного
азота в гидролизатах доля амидных групп в общем количестве азота не мо¬
жет составлять сколько-нибудь значительных величин. Наиболее вероятно,
что в данном случае аммонийный азот присутствует в почве в форме фикси¬
рованного аммония.
Из аминосахаров в почве были найдены глюкозамипы и галактозамины.
Азот этих соединений может составлять в верхних горизонтах почвы 5—10%
общего азота. Сумма глюкозаминов и галактозаминов в почвах почти
соответствует общему содержанию аминосахаров (Bremner, 1958; Sowden,
1959).
При обработке почв растворами NaOH или КОН в щелочном гидролизате
обнаружены нуклеиновые кислоты. Почвенный азот (верхних горизонтов),
представленный этими соединениями, не превышает 10 %.
Методы определения в почве аэотсодержащих органических соединений
{аминокислот, гексозаминов и др.) представляет собой модификации методов,
принятых при анализе биологических объектов.
90
В. Б. Замятина
Ниже приведено описание методов определения основных азотсодержа¬
щих органических соединений почвы с применением для гидролиза их 6 н.
раствора НС1 (метод Бремнера) и двухступенчатого гидролиза путем обра¬
ботки почвы 0,5 н. и 5 н. раствором H2S04 (вариант Шконде и Королевой).
Для определения состава аминокислот в почвах описывается методика,,
предложенная Турчиным с применением ионообменной и бумажной хрома¬
тографии.
Метод Бремнера (Вгетпег, 1965)
Метод состоит в получении гидролизата в результате обработки почв 6н.
раствором НС1 при 12-часовом кипячении с последующим определением
в нейтрализованном гидролизате: а) общего азота, б) N — NH4, в) суммы
азота NH4 и гексозаминов, г) N серина и треонина, д) N аминокислот, е) сум¬
мы азота аммония, гексозаминов и аминокислот.
Ход анализа. Получение гидролизата. Для анализа ис¬
пользуют воздушно-сухую почву, растертую и пропущенную через сито
с отверстиями диаметром 0,15 мм. Навеску почвы, содержащую около
10 мг N, помещают в коническую колбу емкостью 125 мл. Колба должна
закрываться пришлифованной стеклянной пробкой, снабженной обратным
холодильником типа Либиха. К почве добавляют 2 капли октилового спирта
(для предотвращения образования пены при получении гидролизата) и 20 мл
6 н. раствора НС1. Смесь хорошо перемешивают круговыми движениями кол¬
бы. Затем колбу ставят на электроплитку (с регулятором нагрева), включают
водопроводную воду для охлаждения холодильника и постепенно доводят
смесь до умеренного кипения, которое продолжают в течение 12 час. По окон¬
чании гидролиза содержимое колбы охлаждают, раствор отфильтровывают
через воронку Бюхнера. При фильтровании используют широкогорлую отса¬
сывающую колбу на 1 л, в которую для сбора фильтрата вставляют плоско¬
донную широкую пробирку с отметкой на 60 мл. Для сбора фильтрата также
может быть использована высокая отсасывающая колба емкостью 250 мл.
Остаток почвы промывают небольшими порциями воды (5—10 мл) до тех пор,
пока объем фильтрата не достигнет 60 мл. Раствор охлаждают и нейтрали¬
зуют щелочью (pH 6,5 +0,1). Сначала к гидролизату осторожно, при по¬
стоянном перемешивании добавляют 5 н. раствор NaOH, доводя гидролизат
примерно до pH 5. Для окончательной нейтрализации используют 0,5 н. рас¬
твор едкого натра. Ход нейтрализации контролируют измерением pH со
стеклянным электродом.
Необходимо очень внимательно следить за прибавлением щелочи. Если
в процессе нейтрализации раствор приобретет щелочную реакцию, это
может повлечь к потерям аммиака и разложению гексозаминов. Нейтрали¬
зованный гидролизат с помощью небольшой воронки переносят в мерную
колбу емкостью 100 мл. Пробирку и электрод несколько раз ополаскивают
небольшими порциями воды, доводя объем гидролизата до 100 мл. Колбу
закрывают пробкой, раствор перемешивают, переворачивая колбу вверх
дном.
Гидролизат (до прибавления щелочи) может храниться в течение долгого
времени без изменений, вызываемых микробиологическими процессами.
В случае необходимости на этой стадии анализ можно приостановить.
Нейтрализовать можно только часть гидролизата. При этом фильтрат
удобно довести до 50 мл в мерной колбе, откуда взять часть его (20—25 мл)
и довести щелочью до pH 6,5, как указано выше. Нейтрализованный раствор
разбавляют водой в мерной колбе до 50 мл. Этот прием позволяет, если необ¬
ходимо, повторить нейтрализацию гидролизата.
Чтобы получить хорошо воспроизводимые результаты, перед отбором
определенного объема нейтрализованного гидролизата на анализ нужно хоро¬
шо перемешивать раствор, переворачивая колбу вверх дном.
Методы определения азота в почве
91
Анализ гидролизата. Определение в гидролизате азотсодер¬
жащих соединений сводится к переводу их азота в аммиачную форму, отгону
NH3 и учету его объемным методом.
В процессе анализа соблюдают следующие условия.
1. Для отгона аммиака используют полумикродистилляционный прибор
с применением пара, у которого отгонной колбой служит колба Кьельдаля
емкостью 50 мл (или 100 мл).
2. Прибор предварительно нужно отрегулировать, так, чтобы sa 1 мин.
отгонялось 7—8 мл дистиллята и температура его не превышала 22°.
3. Перед отгоном NH3 приемник с борной кислотой помещают под ниж¬
ний конец холодильника, чтобы он был примерно на 4 см выше раствора
в приемной колбе. В ходе отгона NHS конец холодильника не опускают в
раствор дистиллята.
4. Все сухие реактивы прибавляют в дистилляционную колбу с по¬
мощью сухой небольшой воронки, нижний конец которой должен достигать
расширенной части колбы.
Общий гидролизуемый азот. В отгонную колбу дистил-
ляционного прибора емкостью 50 мл помещают 5 мл нейтрализованного
гидролизата и в ней проводят сжигание органических соединений (присутству¬
ющих в гидролизате) по Кьельдалю. К раствору прибавляют 0,5 г смеси
катализаторов и сернокислого калия и 2 мл концентрированной серной ки¬
слоты. Смесь постепенно нагревают. После удаления из раствора воды и пре¬
кращения пенообразования раствор доводят до умеренного кипения, кото¬
рое продолжают 1 час. Затем колбе дают охладиться, в нее добавляют при
постоянном перемешивании раствора небольшими порциями 10—15 мл воды.
Колбу охлаждают под струей водопроводной воды и помещают в сосуд
с мелко наколотым льдом. Охлаждение раствора льдом способствует умень¬
шению образования пены при последующем добавлении к нему щелочи. Если
эта операция вызывает трудности, ее можно не проводить.
В приемник прибора, которым служит коническая колба емкостью в
50 мл с отметкой на 35 мл, помещают 5 мл 2%-ного раствора Н3В031 содер¬
жащего индикатор, и ставят под холодильник аппарата. К прибору присое¬
диняют отгонную колбу с охлажденным раствором. Через воронку прибора
постепенно добавляют в дистилляционную колбу 10дел Юн. раствора NaOH.
Когда в воронке останется около 0,5 дм щелочи, ее быстро ополаскивают при¬
мерно 5 мл воды, спускают около 3 мл раствора и закрывают воронку. Подво¬
дят пар в дистилляционную колбу из парообразователя и отгоняют аммиак.
Отгон заканчивают, когда в приемнике наберется 35 мл дистиллята (до метки
на колбе). Конец холодильника ополаскивают водой. Раствор титруют
0,005 н. H2S04, используя микробюретку (1 мл 0,005 н. H2S04 соответству¬
ет 0,07 мг N). Конец титрования устанавливают по переходу зеленой окраски
раствора в малиново-красную. На дистилляцию затрачивают 4 мин.
Азот аммония. В приемный сосуд, которым служит коническая
колба емкостью 50 мл с отметкой на 20 мл, наливают 5 мл раствора борной
кислоты, содержащей индикатор. Колбу ставят под холодильник прибора,
как указано выше. В дистилляционную колбу помещают 10 мл нейтрализо¬
ванного гидролизата, прибавляют 0,07+0,01 г MgO, присоединяют колбу
к дистилляционному прибору и отгоняют NH3. Дистилляцию заканчивают,
когда в приемной колбе соберется 20 мл раствора (на отгон ’затрачивают
около 2 мин.). Обмывают конец холодильника водой и титруют дистиллят
0,005 н. H2S04, используя микробюретку (1 мл 0,005 н. H2S04 соответствует
0,07 мг N - NH4).
При соблюдении описанных условий (небольшое количество MgO и ко¬
роткий период дистилляции NH3) присутствующие в гидролизате глюкозами-
ны и другие неустойчивые азотсодержащие органические соединения не под¬
вергаются минерализации.
92
В. Б. Замятина
Азот аммония и гексозаминов. Метод основан на разло¬
жении глюкозаминов и галактозаминов в щелочной среде с выделением NH3.
Дистилляцию аммиака проводят при pH 11,2, создаваемым фосфатно-борат-
ным буферным раствором.
В дистилляционную колбу емкостью 100 мл помещают 10 мл гидролизата,
прибавляют 10 мл фосфатно-боратного буферного раствора. Определяют ко¬
личество отогнанного NH3 тем же способом, что и общий азот в гидролизате.
Аммиак отгоняют около 4 мин.
В процессе получения гидролизата часть гексозаминов минерализуется
с отщеплением NH3. Поэтому при расчете азота гексозаминов, присутствую¬
щих в почве, величины азота, полученные при анализе гидролизата, нужна
помножить на коэффициент 1,143.
Азот серина и треонина. После удаления из гидролизата
азота аммония и гексозаминов (дистилляцией NH3 в присутствии фосфатно-
боратного буферного раствора) от прибора отсоединяют дистилляционную
колбу, споласкивают конец трубки прибора (вынутой из дистилляционной
колбы) 3—5 мл воды, которую сливают в дистилляционную колбу. Колбу
охлаждают под струей водопроводной воды. Прибавляют 2 мл раствора
йодной кислоты, перемешивают вращением колбы в течение 30 сек., добав¬
ляют 2 мл раствора мышьяковистокислого натра и присоединяют колбу к при¬
бору. Определяют содержание аммиака, соблюдая условия анализа, описан¬
ные при определении общего азота в гидролизате.
Выделение аммиака при взаимодействии серина и треонина с ионом йод¬
ной кислоты обычно идет по реакции
R1.CH(0H).CH(NH2)R2+NaJ04 = R^CHO+R^CHO+NHs+NaJOs.
Избыток иона йодной кислоты восстанавливают мышьяковистокислым
натром.
Азот аминокислот. Определение основано на переводе азота
а-аминокислот при взаимодействии с нингидрином в аммиачную форму
(после предварительного удаления из раствора аммонийного азота и азота
гексозаминов).
В дистилляционную колбу емкостью 50 мл помещают 5 мл гидролизата,
туда же приливают 1 мл 0,5 н. раствора NaOH. Колбу опускают в кипящую
водяную баню и выдерживают там до уменьшения объема раствора до 2—3 мл
(примерно 20 мин.). Колбу охлаждают, в нее прибавляют 500 мг лимонной
кислоты, 100 мг нингидрина и снова помещают в сильно кипящую водяную
баню так, чтобы расширенная часть колбы была полностью погружена в воду.
Примерно через минуту смесь, не вынимая колбы из воды, взбалтывают и
дополнительно оставляют на 9 мин. Затем колбу вынимают из бани, охлаж¬
дают, прибавляют 10 мл фосфатно-боратного буфера и 1 мл 5 н. NaOH, при¬
соединяют к дистилляционному прибору, отгоняют NH3 и определяют содер¬
жание его (см. анализ общего азота в гидролизате).
Азот аммония, гексозаминов и аминокислот.
Помещают 5 мл гидролизата в дистилляционную колбу емкостью 50 млу
прибавляют 100 мг лимоннокислого буферного реактива и 100 мг нингидрина.
Колбу опускают в сильно кипящую водяную баню, чтобы расширенная ее
часть полностью находилась в воде. После минутного выдерживания (не
вынимая колбы из бани) содержимое ее перемешивают взбалтыванием в те¬
чение нескольких секунд и оставляют в бане еще на 9 мин. Затем колбу вы¬
нимают, охлаждают, прибавляют 10 мл фосфатно-боратного буферного рас¬
твора и присоединяют к прибору. Определяют количество отогнанного NH3,
соблюдая условия анализа определения общего азота в гидролизате.
Метод не позволяет полностью учесть азот гексозаминов; последние реа¬
гируют с нингидрином, образуя соединения, из которых азот в виде NH^
количественно не выделяется. Однако содержание в гидролизате азота гек-
Методы определения азота в почве
93
созаминов обычно намного меньше, чем азота аммония и аминокислот.
В связи с этим результаты, полученные описанным методом, немного зани¬
жены. Метод отличается простотой, быстротой выполнения и вполне пригоден
для исследовательских работ, где не требуется особо высокая точность ана¬
лизов.
Параллельно с анализом гидролизата необходимо проводить холостые
определения возможного загрязнения реактивов соединениями азота. При
расчетах анализа вносят соответствующие поправки.
Реактивы. 1. Концентрированная серная кислота (уд. вес. 1,83—1,84).
2. 6 н. раствор соляной кислоты (приблизительно). К 490 мл соляной кислоты (уд*
вес 1,19) прибавляют 300 мл воды. Раствор доводят водой до 1 л в мерной колбе, хорошо
перемешивают.
3. н-октиловый спирт.
4. Смесь катализаторов и сернокислого калия. Смешивают 100 г K2S04, 10 г CuS04«
• 5Н20 и 1 г Se после предварительного растирания в порошок отдельно каждого реактива*
Смесь тщательно перемешивают и дополнительно растирают в ступке.
5. 10 н. раствор NaOH (приблизительно). Растворяют в большой фарфоровой чашке
при размешивании 2,1 кг NaOH в 2 л воды. Охлажденный раствор переносят в склянку,
закрывают резиновой пробкой. Через несколько дней светлый раствор над осадком сли¬
вают сифоном. Разводят водой, не содержащей С02, до 5 л. Хранят в бутыли с предохра¬
нителем от С02 атмосферы.
6. 5 н. раствор NaOH (приблизительно). Разводят в 2 раза реактив 5. Хранят
в хорошо закрытой бутыли.
7. 0,5 н. раствор NaOH (приблизительно). Разводят водой 50лл реактива 5 до 1 л.
Хранят в хорошо закрытой бутыли.
8. 2%-ный раствор борной кислоты, содержащей индикатор. Растворяют 2 г х. ч.
Н3В08 в 700 мл горячей воды. Остывший раствор переносят в литровую мерную колбу,
содержащую 200 мл этилового спирта и 20 мл смеси индикаторного раствора. Последний
готовят растворением 0,33 г бромкрезолового зеленого и 0,165 г метилового красного
в 500 мл этилового спирта. После смешивания к содержимому колбы прибавляют небо ль*
шими порциями 0,05 н. раствор NaOH до изменения цвета от малиново-красного до блед¬
но-зеленого. Количество добавляемой щелочи устанавливают предварительно в отдель¬
ной пробе. Для этого берут 10 мл раствора борной кислоты с индикатором, прибавляют
10 мл воды и из бюретки по каплям добавляют 0,05 н. раствор NaOH до приобретения
раствором бледно-зеленого цвета. Рассчитывают количество щелочи, потребное для всего
раствора.
Смесь индикаторов бромкрезолового зеленого и метилового красного можно заменить
смешанным индикатором Гроака (см. выше метод Кьельдаля).
9. 0,005 н. титрованный раствор H2S04.
10. Окись магния (см. выше определение нитратов и аммония методом дистилляции)*
11. Нингидрин (СэН408*Н20), сухой реактив, растертый в порошок.
12. Фосфатно-боратный буферный раствор (pH 11,2). В 900 мл воды растворяют
100 г Na3P04*12H20 и 25 г Na2B407*10H20. Раствор в мерной колбе доводят водой до
1 л. Хранят в хорошо закрытой бутыли.
13. Лимонная кислота, х. ч. Реактив растирают до тонкого порошка. Хранят в не¬
большой широкогорлой банке.
14. Цитратный буферный реактив (pH 2,5). Смешивают 2,06 г лимоннокислого натра
(Na8CeH607*2H20) и 19,15 г лимонной кислоты (СвН807*Н20). Растирают ц тонкий поро¬
шок в фарфоровой ступке. Хранят в небольшой широкогорлой банке.
15. Раствор йодной кислоты. Готовят приблизительно 0,2 М раствор. Растворяют
4,6 г йодной кислоты (HJ04*2H20) в 100 мл воды. Раствор хранят в склянке, плотно
закрытой стеклянной пробкой.
16. Мышьяковистокислый натр (мета). Готовят приблизительно 1 М раствор. Раство¬
ряют 13 г NaAs02 (ч. д. а.) в 100 мл воды. Хранят в хорошо закрытой склянке.
94
В. Б. Замятина
Вариант Шконде и Королевой1
По этому методу азот почвы делят на четыре группы: 1) минеральный
азот (азот нитратов, нитритов и обменного аммония); 2) легкогидролизуе¬
мый (амиды, часть аминов, часть необменного аммония), извлекаемый при
обработке почвы 0,5 н. серной кислотой и представляющий отгоняемую
фракцию азота этого гидролизата; 3) трудногидролизуемый (часть аминов,
амиды, необменный аммоний, часть гуминов) — отгоняемая часть почвен¬
ного азота, извлеченного 5 н. серной кислотой; 4) негидролизуемый (боль¬
шая часть аминов, гумины, меланины, битумы, остаток необменного аммо¬
ния) — представляет сумму неотгоняемого азота гидролизата 5 н. серной
кислоты и собственно негидролизуемого азота.
Ход анализа. Определение минерального азота. 10 г
воздушно-сухой почвы, растертой и пропущенной через сито с отверстиями
диаметром 1 мм, обрабатывают 300 мл 0,1 н. КС1 (солевая вытяжка по Коневу;
Турчин, 1965). После часового взбалтывания на ротаторе вытяжку фильтру¬
ют сразу в отгонную колбу прибора для отгона через предварительно про¬
мытый от аммиака складчатый бумажный фильтр. Почву на фильтре не¬
сколько раз промывают малыми порциями горячего раствора 0,1 н. КС1.
Полнота отмыва от аммиака проверяется реактивом Несслера. В фильтрат
добавляют 0,5—1,0 г (в зависимости от содержания в почве нитратов) тон-
корастертого сплава Деварда, растертую прокаленную пемзу для равномер¬
ности кипения, 20 мл 40%-ного раствора NaOH. Затем быстро отгонную
колбу присоединяют к дистилляционному прибору' с заранее приготовлен¬
ной колбой — приемником с 20 мл 4%-ной борной кислоты, содержащей
2—3 капли индикатора Гроака. В таком состоянии прибор оставляют на
ночь. Затем отгоняют NH3 при последующем титровании дистиллята 0,02 н.
раствором серной кислоты. По количеству израсходованной кислоты
рассчитывают содержание минерального азота в вытяжке (1 мл 0,02 н. H2S04
соответствует 0,28 мг N).
Определение легкогидролизуемого дистилли¬
руемого азота. В коническую колбу емкостью 100 мл помещают
10 г воздушно-сухой почвы, растертой и пропущеной через сито в 1 мм, до¬
бавляют 50 мл 0,5 н. раствора H2S04 и закрывают воронкой, которая слу¬
жит обратным холодильником. Гидролиз проводят в течение 3 час. на
электрической плитке с закрытой спиралью. Затем суспензию отфильтровы¬
вают в отгонную колбу емкостью 1 л. Почву на фильтре промывают малы¬
ми порциями горячей (подкисленной) воды до исчезновения реакции на ам¬
миак. Общий объем жидкости в отгонной колбе должен составлять около
700 мл. Для восстановления нитратов в фильтрат добавляют 0,5—1,0 г тонко-
растертого сплава Деварда и присоединяют отгонную колбу к дистилляционно¬
му аппарату с заранее приготовленной колбой — приемником (см. опре¬
деление минерального азота). Для создания щелочной среды в фильтрат до¬
бавляют 40 мл 40%-ного раствора NaOH и оставляют на ночь. На следующий
день отгоняют аммиак, как описано выше.
В состав гидролизата входит как минеральный азот (первая группа),
так и легкогидролизуемые органические соединения (вторая группа). По¬
скольку для определения разных форм азота каждый раз берут новую на¬
веску, то при расчетах легкогидролизуемого азота содержание минерального
азота вычитают из азота гидролизата (0,5 н. H2S04).
Определение трудногидролизуемого дистилли¬
руемого азота аналогично легкогидролизуемому,но применяют 5н.
раствор H2S04 и увеличивают при отгоне аммиака объем щелочи до 80 мл.
Рассчитывая содержание трудногидролизуемого азота, из дистиллируе¬
1 Описание составлено И. Б. Королевой.
Методы определения азота в почве
95
мого азота гидролизата (5 н. H2S04) нужно вычесть содержание минерально¬
го и легкогидролизуемого азота.
Определение общего азота (см. метод Кьельдаля).
Негидролизуемый азот определяется по разности между об¬
щим азотом и суммарным содержанием азота первой, второй и третьей групп
и включает остаточный азот после 3-часового гидролиза почв с 5 н. раство¬
ром H2S04 и азот недистиллируемого остатка этого гидролизата.
Реактивы. 1. 0,1 н. КС1. 7,5 г х. ч. хлористого калия растворяют Еводе и объем»
доводят водой до 1 л.
2. Сплав Деварда (см. выше определение нитратов и аммония методом дистилляции).
3. 40%-ный раствор NaOH (см. выше метод Кьельдаля).
4. 4%-ная Н3В08. 40 г Н3ВО3 растворяют^в воде и объем доводят водой до 1 л.
5. Прокаленная пемза. Пемзу растирают в ступке, просеивают через сито с диаметром
ячеек 1 мм и прокаливают 1—1,5 часа в фарфоровой чашке при красном калении.
6. Смешанный индикатор Гроака (см. выше метод Кьельдаля).
7. 0,5 н. раствор H2S04.
8. 5 н. раствор H2S04.
9. 0,02 н. раствор H2S04 (титрованный).
Определение аминокислотного состава по Турчину (1965)
Для извлечения аминокислот применяют гидролиз почвы 6 н. раствором
серной кислоты. Из гидролизата аминокислоты выделяют Н-катионитом и
далее разделяют их с помощью бумажной хроматографии. Метод позволяет
выделить следующие аминокислоты: цистин, треонин, аланин, пролин, ти-
разин, триптофан, валин, лейцин + изолейцин, фенилаланин, аспарагин,
серии, гликоколь, глютаминовую и аспарагиновую кислоты.
Ход анализа. Навеску почв 20 г помещают в колбу, туда же прибавляют
100 мл 6 н. раствора H2S04. Содержимое колбы доводят до слабого кипения,
которое продолжают 24 час. (при кипячении применяют обратный холодиль¬
ник), затем раствор охлаждают, отфильтровывают. Для осаждения серной
кислоты к фильтрату прибавляют раствор Ва(ОН)2; осадок BaS04 отделяют
фильтрованием. Кальций и полуторные окислы, перешедшие в гидролизат
из почвы, а также избыток бария удаляют прибавлением раствора углекис¬
лого аммония. Осадок отфильтровывают и полученный фильтрат пропускают
через колонку, наполненную Н-катионитом (могут быть использованы эспа-
тит, вофатит и т. п.). В этих условиях аминокислоты полностью сорбируются
катионитом, присутствующие в гидролизате другие органические соединения
и неорганические кислотные остатки уходят в фильтрат при последующем*
промывании катионита водой.
Для извлечения сорбированных катионитом аминокислот применяют
многократное встряхивание катионита с СаО (1 г СаО на 10 г катионита) и
с 2%-ным водным раствором аммиака, добиваясь полного извлечения ами¬
нокислот. Отсутствие розовой окраски в последних порциях элюата при
прибавлении нингидрина указывает на полноту извлечения аминокислот.
Элюат выпаривают досуха в фарфоровых чашках на водяной бане. Остаток
растворяют в точно отмеренном небольшом объеме воды (3—5 мл). Для раз¬
деления аминокислот отбирают микропипеткой 0,01—0,04 мл раствора, на¬
носят его на хроматографическую бумагу № 3. Бумагу предварительно об¬
рабатывают фосфатно-цитратной буферной смесью, имеющей pH 7,8. Ее гото¬
вят, смешивая 191,5 мл 0,2 М раствора Na2HP04-2H20 и 8,5 мл 0,1 М раство¬
ра лимонной кислоты.
Для разделения аминокислот применяют два растворителя: 1) насыщен¬
ный водный раствор фенола и 2) смесь амилового спирта, изопропилового
спирта, уксусной кислоты и воды при соотношении 4:4:1 : 1. Локализо¬
96
В. Б. Замятина
ванные на бумаге аминокислоты проявляют нингидрином (0,2%-ный раствор
нингидрина в насыщенном водой н-бутиловом спирте). Рекомендуемый авто¬
ром способ весьма точно локализует отдельные аминокислоты, что позволяет
полностью отделить их друг от друга.
Окрашенные участки бумаги, на которых локализованы отдельные амино¬
кислоты, после проявления их нингидрином, вырезают из хроматограммы и
обрабатывают буферным раствором веронала (pH 7,0). Раствор готовят, сме¬
шивая 53,6 мл 0,1 М натрий-веронала (20,62 г/л) и 46,4 мл 0,1 М раствора
НС1. Окраска пятен полностью переходит в раствор. Оптическую плотность
раствора измеряют с помощью спектрофотометра или фотоколориметра.
Сравнивая полученные показания с градуировочной кривой образцовых
растворов отдельных аминокислот (предварительно подвергшихся такой же
обработке на хроматографической бумаге), определяют содержание амино¬
кислот в почве. Точность анализа ±5%.
ОПРЕДЕЛЕНИЕ ПОДВИЖНОГО АЗОТА
При современном уровне земледелия растениям не всегда хватает азота,
присутствующего в почве в минеральной форме и мобилизуемого из органи¬
ческого вещества. Недостаток его восполняют азотными удобрениями.
В связи с этим возникла необходимость в разработке лабораторных методов
характеристики почв в отношении способности их обеспечивать растения
азотом и прогноза действия азотных удобрений. Однако это встречает боль¬
шие трудности, так как в природных условиях величины азота, мобилизуе¬
мого из органического вещества, для одной и той же почвы весьма измен¬
чивы. Степень мобилизации зависит от многих факторов: температуры, влаж¬
ности почв, обработки, возделываемой культуры и т. д. В связи с этим дан¬
ные лабораторных анализов весьма условны. Из многочисленных методов
анализа наиболее широко распространены биохимические и химические.
Из химических методов наиболее простым и довольно часто используемым
является метод определения нитратов или суммы нитратов и обменного ам¬
мония перед посевом. Данные анализа дают представление об обеспеченно¬
сти растений почвенным азотом в ранние фазы развития, но не позволяют
судить о количестве доступного растениям азота почвы в течение вегетацион¬
ного периода. , , ,
Кроме того, применяются методы определения легкогидролизуемого азо¬
та почвы, которые включают определение азота минеральных и органических
соединений, легко подвергающихся гидролизу при обработке почв слабыми
растворами кислот, щелочей, окислителей (щелочной раствор марганцово¬
кислого калия и др.) и растворами солей. Эти методы основаны на допуще¬
нии, что указанные растворители извлекают (минерализуют) из почвы азот¬
содержащие органические соединения, легко подвергающиеся минерализа¬
ции в природных условиях и благодаря этому являющиеся ближайшими
источниками пополнения минеральных соединений азота, используемых
растениями. По существу, эти методы эмпирические. Пригодность их многие
авторы обосновывают удовлетворительной корреляцией результатов анализа
и данных полевых опытов.
В связи с тем, что минерализация азота органических соединений почвы
связана с биологическими процессами, предложен ряд биохимических мето¬
дов. Они включают компостирование почвы в условиях, способствующих
микробиологической деятельности, с последующим химическим определени¬
ем накопленных в почве нитратов (определение нитрификационной способ¬
ности почв) или минеральных соединений азота (N03 и NH4 — обменного).
Существует много вариантов этих методов. Однако различия модификаций
вводятся к условиям компостирования почв (сроки, температура, влаж¬
ность почв, аэробные или анаэробные условия, использование для компо¬
Методы определения азота в почве
97
стирования сырых или предварительно высушенных образцов, добавление
минеральных или органических соединений, содержащих или не содержа¬
щих азот и ряд других). Эти варианты различаются и по методам химического
анализа, который применяют для определения накопившихся в почве мине¬
ральных соединений азота (определение только N03 или N03 + NH4, ис¬
пользование колориметрических или объемных методов и т. д.).
Содержание минерального азота, накопленного при компостировании
почв в лабораторных условиях, позволяет судить о возможном количестве
его, способном мобилизоваться из почвы при благоприятных условиях.
Определение легкогидролизуемого азота
по Тюрину и Кононовой (1934)
По этому методу почву на холоду обрабатывают 0,5 н. раствором H2S04.
Отношение почвы к раствору 1 : 5. В вытяжке определяют общий азот объем¬
ным методом после перевода его в аммонийную форму.
Метод учитывает присутствующий в вытяжке азот нитратов, поглощен¬
ного аммония и легкогидролизуемых органических соединений.
Ход анализа. 20 г воздушно-сухой почвы, предварительно растертой и
пропущенной через сито с отверстиями диаметром 1 мм, в течение 3 мин.
взбалтывают с 100 мл 0,5 н. раствора H2S04 и оставляют в покое на 16—
18 час. Затем вытяжку полностью отфильтровывают через сухой складчатый
фильтр. Берут пипеткой 25—50 мл фильтрата, помещают его в коническую
колбу емкостью 100 мл, туда же прибавляют 0,5 г смеси цинковой пыли и
восстановленного железа. Колбу закрывают небольшой воронкой и раствор
постепенно доводят до кипения, которое продолжают до растворения внесен¬
ной смеси. При этом происходит восстановление присутствующих в вытяжке
нитратов до аммиака. Содержимому колбы дают охладиться, обмывают во¬
ронку в колбу водой, прибавляют 5 мл H2S04 (уд. вес 1,83—1,84) и раствор
упаривают на электроплитке до побурения и начала появления белых паров.
В упаренном растворе определяют общее содержание азота хромовым мето¬
дом по Тюрину. Для поправки на загрязнение реактивов проводят холостое
определение (не менее двух определений). При этом вместо вытяжки берут
0,5 н. раствор H2S04, в остальном соблюдают те же условия, что и при анали¬
зе почв. Результаты вычисляют в миллиграммах азота на 1 кг воздушно¬
сухой почвы.
Сопоставляя содержание гидролизуемого азота с данными полевых опы¬
тов, авторы предложили градации отзывчивости растений на азотные удобре¬
ния. Однако дальнейшее применение этого метода показало, что он далеко
не всегда может быть использован для установления потребности растений в
азотных удобрениях. Метод хорошо улавливает генетические особенности
почв в отношении азота и широко используется для этих целей.
При определении легкогидролизуемого азота по данному методу в кар¬
бонатных почвах необходимо дополнительно добавлять H2S04 для разруше¬
ния карбонатов. Для установления этих дополнительных количеств
П. В. Протасов (1963) рекомендует предварительно определять в почвах кар¬
бонаты с учетом углекислоты по методу Кнопа. Исходя из полученных дан¬
ных, при приготовлении вытяжки следует дополнительно прибавлять соответ¬
ствующее количество концентрированной серной кислоты.
Определение в почвах гидролизуемого азота методом Тюрина и Кононо¬
вой вошло в число методов, утвержденных Министерством сельского хозяй¬
ства СССР для зональных агрохимических лабораторий. Однако в анализ
внесены следующие изменения *. После окисления органического вещества
1 Межреспубликанские технические условия методов проведения агрохимических ана¬
лизов почв для зональных агрохимических лабораторий. М., «Колос», 1968.
98
В. Б. Замятина
вытяжки хромовой кислотой охлажденный раствор (для отгона аммиака)
переносят в плоскодонную колбу емкостью 500 мл. Добавляют пемзу (пемзу
предварительно растирают в ступке, просеивают через сито с отверстиями
диаметром 1,0 мм и прокаливают 1—1,5 часа в фарфоровой чашке при крас¬
ном калении). В колбу приливают 20 мл 40%-ного раствора NaOH и ее
сейчас же присоединяют к прибору для отгона аммиака. В приемник предва¬
рительно наливают 15 мл 4%-ного раствора борной кислоты. Отгон NH3
продолжают 30 мин. от начала кипения, пока в приемник не перегонится
% содержащейся в колбе жидкости. Аммиак, связавшийся борной .кислотой,
оттитровывают 0,02 н. раствором H2S04 в присутствии индикатора Гроака
(см. выше метод Кьельдаля).
При анализе допускаются расхождения между параллельными опреде¬
лениями из разных навесок почвы в одной лаборатории 10%, в разных лабо¬
раториях — 15%.
Реактивы. 1. 0,5 н. раствор H2S04.
2. Серная кислота, х. ч. (уд. вес. 1,83—1,84).
3. Смесь цинковой пыли с восстановленным железом. На 9 г цинковой пыли берут
1 г восстановленного железа, хорошо перемешивают.
Определение щелочногидролизуемого азота по Корнфилду
В основу метода положен гидролиз органических соединений почвы 1 н.
раствором NaOH. Выделяющийся при этом аммиак учитывают микродиффуз-
ным методом. Помимо аммиака, образующегося при гидролизе органиче¬
ского вещества почвы, метод учитывает обменный аммоний. Ниже приводит¬
ся ход анализа несколько измененного метода Корнфилда, описанный
А. П. Голубевой (1969).
Ход анализа. Анализ проводится в чашках Конвея, сделанных из обыч¬
ного или органического стекла (рис. 4).
Навеску 2 г воздушно-сухой почвы, растертой и пропущенной через сито
с отверстиями 1 мм, помещают в периферийную часть чашки Конвея. В цент¬
ральную часть чашки наливают 2 мл 2 %-ного раствора борной кислоты и при¬
бавляют 2 капли индикатора Гроака. Во внешнюю часть чашки вносят 5 мл
а
Рис. 4. План чашки
Конвея
- а — вид сверху; б — раз-
12 рез по линии АБ. Раз¬
меры даны в мм
1 н. раствора NaOH, не допуская смачивания почвы (этому способствует
имеющаяся в чашке перегородка С). Добавляя щелочь, чашку немного на¬
клоняют в сторону перегородки. Не изменяя положения, чашку накрывают
крышкой, края которой для лучшего прилегания смазывают вазелином.
Осторожно круговым вращением чашки в течение минуты почву смешивают
с раствором щелочи. Затем чашку ставят в термостат и выдерживают 48 час.
при 28°.
Выделяющийся из щелочного раствора аммиак, образующийся в
результате гидролиза органических соединений и за счет извлеченного
Методы определения азота в почве
99
из почвы обменного аммония, благодаря диффузии поглощается раствором
борной кислоты, которая находится во внутренней части чашки Конвея.
По истечении указанного срока чашку вынимают из термостата, осто¬
рожно снимают крышку и определяют количество аммиака, связавшегося
борной кислотой, титрованием 0,02 н. раствором серной кислоты, используя
микробюретку. Конец титрования устанавливают по изменению зеленой
окраски в фиолетово-красную.
Для поправки на возможное загрязнение реактивов проводят холостые
определения (не менее двух) бея навески почвы. Величины азота, найденные
при холостом определении, вычитают из данных анализа.
Результаты анализа выражагт в миллиграммах азота на 1 кг почвы
(1 мл 0,02 н. H2S04 соответствует 0,28 мг N).
Исследованиями ВИУА показана лыеокая положительная корреляция
между результатами, поллченакгми при щелочном гидролизе почн по Корн*
филду, и данными нитрификационной способности, особенно для почв с не¬
большим исходным содержанием нитратов (метод Корнфилда не учитывает
присутствующий в почве азот нитратов). При сравнении этих методов на поч¬
вах, содержащих большие количества нитратов, коэффициент корреляции
получен несколько ниже.
Реактивы. 1. 1 н. раствор NaOH,
2. 0,02 н. титрованный раствор HaS04.
3. 2%-ный раствор борной кислоты. Растворяют 20 г борной кислоты в 1 л воды.
4. Индикатор Гроака (см. выше метод Кьельдаля).
Определение нитрификационной способности почв
методом Кравкова, в модификации Болотиной и Абрамовой (1964)
•Метод определения нитрификационной (нитрифицирующей) способности
почв основан на определении нитратов, накапливающихся в почве в резуль¬
тате разложения азотсодержащих органических соединений, при компости¬
ровании почвы с соблюдением аэрации и оптимальных условий температуры
и влажности.
Ход анализа. 20 г воздушно-сухой почвы, пропущенной через сито
с отверстиями 2 мм, помещают во взвешенную (до 0,1 г) плоскодонную колбу
емкостью 100 мл. Почву увлажняют дистиллированной водой из расчета
60% капиллярной влагоемкости. Колбу прикрывают листом фильтровальной
бумаги и выдерживают 7 дней в термостате при 26—28°. В течение этого пе¬
риода дважды проверяют общий вес колбы (взвешиванием на технических
весах).
При уменьшении веса в результате испарения воды в почву добавляют
по каплям воду до восстановления первоначального веса. По истечении ука¬
занного срока в почве определяют содержание нитратов. Для этого в колбу
добавляют Дополнительно к воде, имеющейся в навеске, 0,05%-ный раствор
K2S04, чтобы общий объем жидкости составлял 100 мл. Суспензию взбалты¬
вают 3 мин. и фильтруют. Параллельно готовят вытяжку из исходной воз¬
душно-сухой почвы. В вытяжках определяют нитраты колориметрическим
методом с дисульфофеноловой кислотой. Разница в содержании N — N03,
полученном для почвы, которую неделю выдерживали в термостате, и исход¬
ной характеризует интенсивность нитрификационного процесса.
Метод дает хорошо воспроизводимые результаты. Благодаря своей просто¬
те может быть использован при массовых анализах нитрификационной спо¬
собности почв. Короткий срок компостирования почвы, предусмотренный
методом, не позволяет с его помощью установить общую способность почв
к накоплению нитратов. Если в этом возникает необходимость, следует удли¬
нить срок компостирования почвы в тех же условиях.
100
В. Б. Замятина
При определении нитрификационной способности почв более надежные
результаты получают, когда используют для анализа почвы, не подвергав¬
шиеся высушиванию (свежевзятые) образцы. Нитрификационная способ¬
ность почв, доведенных до воздушно-сухого состояния, почти во всех слу¬
чаях выше, чем при определении в сырых образцах.
Определение азота, минерализуемого
из органического вещества почвы методом Б ремне ра
(Вгетпег, 1965)
При определении нитрификационной способности учитывают только
азот, минерализованный из органического вещества почв и окисленный до
нитратов. В почвах, где нитрификация идет медленно, процесс мобилизации
азота органического вещества может задерживаться на стадии образования
аммиака, который не учитывают при определении нитрификационной способ¬
ности почв.
Метод, предложенный Бремнером, позволяет определять общее количе¬
ство азота (нитратов и аммиака), мобилизуемое из органического вещества
почвы. Метод основан на компостировании почвы в анаэробных условиях
при оптимальной влажности и температуре. В этих условиях выделяющий¬
ся при разложении органического вещества аммиачный азот будет накапли¬
ваться в почвах в основном в виде обменного аммония. По окончании компо¬
стирования содержание минерализованного азота определяют в 2 н. КС1-вы-
тяжке, после перевода минеральных соединений азота в NH3 и его отгона.
Общие количества азота, мобилизованного из органического вещества
почвы, как указывает Бремнер, при компостировании почв в анаэробных и
аэробных условиях (при тождестве других условий) близки.
Ход анализа. Навеску 10 г воздушно-сухой почвы, пропущенной через
сито с отверстиями 2 мм, смешивают в стакане емкостью 100 мл с 30 г квар¬
цевого песка. Смесь переносят в широкогорлые квадратные бутыли емкостью
250 мл, содержащие 6 мл воды. При переносе смесь равномерно распреде¬
ляют по дну бутыли, слегка постукивая по ее бокам на уровне смеси почвы
с песком. Бутыли завязывают полиэтиленовой пленкой и помещают в термо¬
стат, где выдерживают при 30° в течение 14 дней. Затем к смеси прибавляют
100 мл 2 н. раствора КС1, бутыли закрывают резиновыми пробками, содержи¬
мое их взбалтывают в течение часа на ротаторе. Суспензию оставляют стоять
до просветления (обычно 30 мин.). Из осветленного раствора пипеткой берут
определенный объем (20—30 мл), помещают в отгонную колбу дистилляцион-
ного аппарата (используя полумикродистилляционные приборы). Туда же
добавляют 0,2 г MgO и 0,2 г тонкоразмолотого сплава Деварда и сразу же от¬
гоняют NH3 в раствор борной кислоты, содержащей индикатор. Количество
отогнанного аммиака определяют титрованием 0,005 н. раствором H2S04
(используя микробюретку). Конец титрования устанавливают по переходу
зеленой окраски в малиновую. Подробное описание отгона аммиака дано
выше (см. определение нитратов и аммония методом дистилляции). Парал¬
лельно определяют содержание минерального азота, переходящего в 2 н.
КС1 вытяжку из смеси почвы с песком, не подвергавшейся компостирова¬
нию. По разности между результатами этих анализов устанавливают ко¬
личества мобилизованного азота.
Если раствор над почвой после отстаивания суспензии недостаточно ос¬
ветлен, его следует отбирать пипеткой с широким концом, чтобы не забить
пипетку мелкодисперсными частицами почвы. Муть в растворе не влияет на
результаты анализа.
При использовании для компостирования влажных образцов навеску
почвы увеличивают соответственно весу 10 г воздушно-сухой почвы, а коли¬
чество прибавляемой для компостирования воды уменьшают на величину, со¬
ответствующую влажности почвы.
Методы определения азота в почве
101
Автор метода указывает на хорошую сходимость результатов, получаемых
при анализе воздушно-сухих и влажных почв.
Чтобы удалить из кварцевого песка минеральные соединения азота, его
предварительно промывают соляной кислотой и водой (до полного удаления
хлора). Используют песок с размерами зерен от 0,25 до 0,50 мм. Промытый
песок нужно хранить в упаковке, исключающей доступ воздуха. Несоблю¬
дение этих условий приводит к поглощению песком присутствующего в воз¬
духе аммиака.
Реактивы. 1. Окись магния — MgO. Для анализа используют MgO, х. ч., пред¬
варительно прокаленную в электрическом муфеле при 600—700° в течение 2 час. Реактив
охлаждают в эксикаторе над кусочками КОН. Хранят в плотно закрытой склянке.
2. Сплав Деварда. Препарат размалывают на шаровой мельнице, просеивают через
сито с отверстиями в 0,15 мм (он должен содержать не менее 75% частиц меньше 0,05 мм).
Реактив хранят в тонкоразмолотом состоянии в плотно закрытой склянке.
3. 2%-ный раствор борной кислоты, содержащий индикаторы (см. выше метод Брем-
нера, стр. 90).
4. 0,005 н. титрованный раствор H2S04.
МЕТОДИКА ПОДГОТОВКИ ПОЧВЕННЫХ ОБРАЗЦОВ
ДЛЯ ИЗМЕРЕНИЯ ИЗОТОПНОГО СОСТАВА АЗОТА
При проведении почвенных и агрохимических исследований азота с при¬
менением метода меченых атомов в качестве индикатора используют ста¬
бильный изотоп азота — 16N.
Из известных шести изотопов азота два — с массой 14 и массой 15 — яв¬
ляются стабильными. Остальные четыре обладают радиоактивностью, одна-,
ко широкого практического применения они не получили в связи с коротким
периодом распада. Наиболее долгоживущий радиоактивный изотоп азота
(с массой 13) имеет период полураспада около 10 мин.
Природный азот состоит из двух стабильных изотопов — азота 14 и азота 15.
В азоте воздуха содержится 99,635 ат.% 14N и 0,365 ат.% 15N. Для почв
могут наблюдаться некоторые отклонения от этого соотношения изотопов.
По даннымБремнерас соавт. (Bremner et al., 1963), в изученных ими почвах
величины 16N составляли0,3651—0,3729 ат.%.
Методы определения концентрации I6N (ат. % I6N) основаны на измере¬
нии изотопного состава азота исследуемого образца и совершенно отличны
от тех, которые используются при применении радиоактивных изотопов,
основанных на определении интенсивности излучения.
Использование стабильных изотопов в качестве индикаторов (в том числе
и I6N) не позволяет применять таких больших разбавлений, как при работе
с радиоактивными изотопами. Это является недостатком метода. Однако при
работе со стабильными изотопами не возникает опасности в отношении влия¬
ния радиации на биологические процессы и не требуется соблюдения специ¬
альных мер предосторожности, которые необходимы при работе с радиоактив¬
ными изотопами.
Для анализа изотопного состава азота используют масс-спектрометры или
эмиссионные спектрометры. В обоих случаях наиболее удобным веществом
для анализа изотопного состава этого элемента является газообразный
азот (N2).
Чтобы получить хорошо воспроизводимые результаты с помощью масс-
спектрометра, требуется 1—3 мг азота. Эмиссионный же спектрометр (типа
NOJ-4) позволяет измерять изотопный состав при наличии около 0,01 мг
азота. Поэтому эмиссионные спектрометры более широко применяются при
проведении биохимических исследований, где получение азота порядка не¬
скольких миллиграмм часто встречает определенные трудности в связи
с ограниченностью анализируемого материала.
102
В. Б. Замятина
В агрохимических исследованиях количество подвергающегося анализу
материала (почвенные и растительные образцы) обычно не является лими¬
тирующим фактором. В то же время благодаря неоднородности раститель^
ных и особенно почвенных образцов возникают значительные трудности при
взятии небольших навесок, представляющих среднюю пробу анализируемо¬
го образца. Поэтому при анализе почвенных и растительных образцов на
изотопный состав азота, независимо от прибора, каким будет производить¬
ся измерение, целесообразно брать навески размером, рекомендуемым при
проведении химических анализов и далее уже использовать необходимые
количества Ns для измерения изотопного состава.
Измерение изотопного состава азота позволяет установить лишь кон¬
центрацию I6N в анализируемом образце. Чтобы определить содержание
I5N, присутствующего в почве в виде какого-либо соединения, помимо изо¬
топного состава, определяют величины азота этого соединения химическим
методом одновременно с подготовкой образцов газообразного азота для изо¬
топного анализа.
При анализе почвенных образцов, обогащенных I5N, газообразный азот
удобно получать в две стадии: 1) перевод анализируемых соединений азота
в аммиачную форму и отгон NH3; 2) окисление аммиака до элементарного
азота гипобромитом натрия (или лития).
Анализ изотопного состава азота описан в специальной литературе и в на¬
стоящей работе не рассматривается.
Перевод азота образцов в аммонийную форму
с последующим отгоном NH3 представляет собой ход выпол¬
нения соответствующих химических анализов. Однако не все методы могут
быть для этого применимы. Пригодны методы, которые после отгона аммиа¬
ка и окисления его до элементарного азота исключают присутствие в послед¬
нем соединений, влияющих на измерения изотопного состава азота (метил¬
амин, диметиламин, аминокислоты, СО, 02 и др.).
Значительное количество тяжелого изотопа азота может присутствовать
в почвах в форме нитратов. При определении общего содержания I6N в почве
для окисления органического вещества следует применять варианты метода
Кьельдаля, включающие нитраты, с применением для сжигания фенолсер¬
ной или салициловой кислоты. Чтобы гарантировать полноту окисления
органического вещества, сжигание ведут 15—18 час. Присутствие ряда орга¬
нических соединений в образцах N2, идущих на измерение изотопного соста¬
ва, благодаря неполноте окисления органических веществ почвы при сжига¬
нии может внести значительные ошибки в результаты измерения.
Когда предполагают измерять изотопный состав отдельных соединений
азота, например обменного аммония или нитратов, их извлекают из почвы,
применяя солевые вытяжки, и отгоняют NH3 в присутствии MgO (при оп¬
ределении нитратов их предварительно восстанавливают до аммиака). Ход
анализа описан выше (см. определение нитратов и аммония методом дистил¬
ляции).
Дистилляцию NH3 удобно проводить в аппарате, показанном на рис. 2.
Прибор позволяет отгонять до 5 мг N — NH3, обладает высокой производи¬
тельностью, легко разбирается, что представляет большое удобство в связи
с необходимостью очищать прибор от загрязнения I6NH3.
После отгона NH3 в дистилляте определяют содержание аммиачного азота
титрованием. Затем раствор доводят до слабокислой реакции, прибавляя
несколько капель 0,02 н. H2S04, и выпаривают в фарфоровых чашках на
водяной бане до 2—5 мл. При этом из жидкости удаляется растворенный
в ней N2 и этиловый спирт, внесенный вместе с индикатором. Отгон аммиака
лучше проводить в титрованный раствор серной кислоты, а не борной.
При использовании борной кислоты в конце выпаривания дистиллята из
раствора может выкристаллизоваться борная кислота.
Методы, определения азота в почве
103
Для получения надежных результатов изотопного анализа в упаренном
дистилляте содержание аммиачного азота не должно быть ниже 0,5 мг в 1 мл.
Упаренные растворы переносят в пробирки, которые закрывают пробками.
Перенесение оставшегося на стенках чашек раствора споласкиванием водой
не требуется (изотопный анализ устанавливает соотношение I6N к 14N, а не оп¬
ределяет содержание этих изотопов в образце). Далее приступают ко второй
стадии — окислению аммиака в элементарный азот.
Чтобы избежать ошибок в результатах анализа изотопного состава, при
выполнении первой стадии работы необходимо соблюдать следующие усло¬
вия:
1. Выбор химических методов для подготовки образцов газообразного азо¬
та должен обеспечить отсутствие в N2 примесей, мешающих определению
изотопного состава азота.
2. Внесенные в почву соединения азота, меченные 16N, распределяются
в ней микроочагами. Кроме того, отдельные азотсодержащие компоненты поч¬
вы могут различно обогащаться добавляемым в почву тяжелым изотопом
азота. Например, азот нитратов или обменного аммония может приобрести
более высокое обогащение, чем азот органических соединений почвы. Это
создает большую пестроту в распределении 16N, поэтому необходимо исклю¬
чительно тщательно брать навески почвы (особенно небольшие), чтобы они
представляли среднюю пробу анализируемого образца.
3. В процессе отгона аммиака в стеклянных дистилляционных аппара¬
тах некоторое количество его задерживается на стеклянной поверхности кап-
леуловителя и холодильника. Из-за этого при отгоне I5NH3 прибор загряз¬
няется тяжелым изотопом азота. При дистилляции через прибор следующего
образца часть удерживаемого стеклом 15NH3 будет освобождаться и перехо¬
дить в дистиллят. Это поведет к ошибке в изотопном составе азота послед¬
него образца. Ошибка может достигнуть значительных размеров, если отгоня¬
ется аммиак с низким обогащением после высокообогащенного.
При подготовке образцов для определения изотопного состава следует
изготовлять каплеуловители и внутреннюю часть холодильника дистилляцион¬
ных аппаратов из кварцевого стекла; оно практически не сорбирует на своей
поверхности аммиак. Стекло 46 (молибденовое) не может быть использовано.
Холодильники и каплеуловители из этого стекла сильно загрязняются I6NH3.
Для очистки стекла Бремнер (Bremner, 1965) рекомендует между отгонами
аммиака, обогащенного 15N, применять дистилляцию этилового спирта (20 мл).
Этот прием дает положительные результаты при работе со стеклом, удержи¬
вающим на своей поверхности небольшое количество аммиака. После отгона
I6NH3 с высоким обогащением (выше 50%) даже при использовании приборов
из кварцевого стекла необходимо очищать холодильники и каплеуловители
отгоном этилового спирта или длительной дистилляцией всдяного пара (не
менее часа).
Следует систематически контролировать загрязнение дистилляционных
приборов тяжелым изотопом азота путем отгона через прибор необогащенно-
го аммиака из (NH4)2S04 или NH4C1 с последующим анализом его изотопного
состава. Если при этом атомный процент будет выше, чем у природного азота,
значит прибор загрязнен и его необходимо подвергнуть очистке (дистилля¬
цией спирта или водяного пара).
4. Ошибку в данные изотопного состава азота может внести загрязнение
применяемых при анализе реактивов. Присутствие в них соединений азота,
которые в процессе анализа могут быть переведены в элементарный азот,
вызовет разбавление меченого азота исследуемого образца. Этот источник
ошибок обычно недоучитывается.
Значимость такой ошибки можно видеть из следующего примера. Допустим, что
анализируемый образец содержит 2,0 лг N с обогащением 3,00 избытка ат. % 16N. Холо¬
104
В. Б. Замятина
стое определенпе показало содержание в реактивах 0,2 мг азота. В результате разбавле»
ния образца азотом реактивов изотопный анализ покажет вместо 3,00 только 2,73 избытка
ат.% 16N, т. е. ошибка определения составит 9% (относительных). Пример очень упрощен.
Процесс разбавления образца азотом реактивов более сложен. Это не позволяет вносить
поправку на загрязнение реактивов, аналогично тому, как это делается при химическом
анализе на основании холостого определения.
Все реактивы, применяемые для анализа, должны быть проверены на
загрязнение их соединениями азота. Если применяемый NaOH содержит ам¬
миак, после приготовления щелочи ее кипятят в течение нескольких часов для
удаления NH3 (в медной посуде), затем концентрацию NaOH доводят до ис¬
ходной прибавлением воды. Чтобы уменьшить загрязнение серной кислоты,
ее перегоняют. При анализе необходимо использовать дистиллированную
воду, не содержащую аммиак. Для этого в воду добавляют алюмокалиевые
квасцы (или подкисляют ее серной кислотой) и вторично перегоняют, соблю¬
дая условия, исключающие поглощение водой аммиака из воздуха.
5. Выпаривать дистилляты на водяной бане следует в фарфоровых чаш¬
ках, а не в стеклянной посуде, которая значительно сильнее загрязняется
I5NH3 и труднее подвергается очистке.
Окисление аммиака до элементарного азота
может быть проведено непосредственно после упаривания дистиллята
или отложено. В последнем случае сконцентрированные растворы хранят
в закрытых пробирках в холодильнике.
Окисление аммиака до элементарного азота проводят гипобромитом нат¬
рия (или лития). Реакция протекает в щелочной среде:
2NH3 + ЗХаВгО — N2 + ЗН20 + ЗКаВг.
Приготовление гипобромита: в 300мл воды растворяют 200 г NaOH. Раст¬
вор охлаждают льдом. Половину охлажденного раствора помещают в широ-
когорлую коническую колбу, которую погружают в сосуд, содержащий мелко
наколотые кусочки льда, и небольшими порциями добавляют 60 мл брома,
хорошо перемешивая раствор после каждого прибавления. Скорость прибав¬
ления Вг2 регулируют, чтобы температура смеси не поднималась выше 5°
(в раствор погружают термометр). Операция занимает не менее 30 мин. Ее
проводят под тягой в резиновых перчатках. Когда весь бром прибавлен,
к смеси добавляют оставшуюся половину NaOH, перемешивают несколько
минут, затем колбу закрывают пробкой и оставляют в холодильнике на 4—6
дней. За это время на дне колбы образуется осадок NaBr. Раствор отделяют
декантацией или фильтрованием через стеклянные фильтры с применением
отсасывания. Полученный прозрачный раствор разбавляют равным объемом
раствора, содержащего 2 г KJ в 1 л воды. На окисление 5—6 мг N—NH3
расходуется 1 мл этого реактива. Раствор хранят в плотно закрытой склянке
в холодильнике. В этих условиях он не теряет своей окисляющей способности
в течение 6 мес.
Чтобы избежать разбавления получаемого N2 при окислении аммиака
азотом воздуха, реакцию проводят в специальной вакуумной установке, пред¬
ложенной Д. Риттенбергом (1948), при разряжении не хуже 10“3 мм. Газооб¬
разный азот собирают в ампулы, которые далее поступают на изотопный
анализ.
В последнее время эту операцию стали проводить в специальных при¬
ставках, непосредственно соединенных с напускной системой приборов,
измеряющих изотопный состав азота. Ее выполняют лица, проводящие изо¬
топный анализ. Это позволило значительно повысить производительность
выполнения анализа и исключить дополнительные возможности загрязне¬
ния образцов при окислении аммиака в вакуумных установках.
Методы определения азота в почве
105
Литература
Басаргин Я. Я., Чернова Е. А. Экспрес-
еионный фотометрический метод опре¬
деления нитратов в почве с применением
хромотроповой кислоты.— Ж. анал. хи¬
мии, 1968, т. XXIII, вып. 1.
Болотина Я. И., Абрамова Е. А. О ме¬
тодике определения нитрификационной
способности почв.— Агрохимия, 1964,
№ 3.
Важенин И. Г. Колориметрический метод
определения поглощенного аммония.—
Почвоведение, 1949, № 6.
Гедройц К. К. Избранные сочинения, т. II.
М., Сельхозгиз, 1955.
Голубева А. Я. Методы определения под¬
вижного азота в почве.— В кн. «Пособие
по проведению анализа почв и состав¬
лению агрохимических картограмм». М.,
Россельхозиздат, 1969.
Конев Д. А. О методах определения ам¬
миака в почве. — Научно-агрон. ж., 1929,
№ 2.
Кудеяров В. Я. Колориметрическое опре¬
деление фиксированного аммония в поч¬
вах.— Агрохимия, 1966, № 4.
Лебедянцев А. Я. Методика исследования
нитрификационного процесса в почве
колориметрическим способом Грандва-
ля — Ляжу.— Избранные труды. М.,
Сельхозгиз, 1960.
Мачигин Б• Я. Методы определения азота
в почве.— В кн. «Методы агрохимиче¬
ских, агрофизических и микробиологи¬
ческих исследований в поливных хлоп¬
ковых районах». Ташкент, 1963.
Могилевкина И. А. Фиксированный аммо¬
ний в почве и методы его определения.—
Почвоведение, 1964, № 2.
Протасов Я. В. Определение подвижных
форм азота.— В кн. «Методы агрохи¬
мических, агрофизических и микробио¬
логических исследований в поливных
хлопковых районах». Ташкент, 1963.
Риттенберг Д. Приготовление образцов
газа для масс-спектрографического изо¬
топного анализа.— В кн. «Получение
и определение меченых атомов». М.,
ИЛ, 1948.
Турчин Ф. В. Методы определения соеди¬
нений азота в почве.— В кн. «Агрохи¬
мические методы исследования почв».
М., «Наука», 1965.
Тюрин И. В. Микрохромовый метод опре¬
деления общего азота в почве.— Почво¬
ведение, 1933, № 2.
Тюрин Я. /?., Кононова М. М. О новом
методе определения потребности почв
в азоте.— Труды Почв, ин-та им.
В. В. Докучаева, 1934, т. X, вып. 4.
Bremner J. М. Amino sugars in soil.—
J. Sci. Food Agric., 1958, v. 9, 528.
Bremner /. M. Determination of fixed am¬
monium in soil.— J. Agric. Sci., 1959
v. 52, 147.
Bremner /. M. In «Methods of Soil Ana¬
lysis. 2. Chemical and Microbiologieal
Properties». C. A. Black et al. (Eds.)
Madison, Amer. Soc. Agron., Inc., 1965.
Bremner J. М., Cheng Я. Я., Edwards A. P.
Assumptions and errors in nitrogen-15
tracer research.— Report FAO/IAEA
Techn. Meeting, 1963, Braunschweig.
Dhariwal A. P. S., Stevenson F. J. Deter¬
mination of fixed ammonnium in soils.—
Soil Sci., 1958, v. 86, 343.
Mahenarappa М. К. Determination of ni¬
trate nitrogen in soil extracts using a
specific ion activity electrode.— Soil Sci.,
1969, v. 108, 132.
Myers R, J. К., Paul E. A. Nitrate ion
electrode method for soil nitrate nitro¬
gen determination.— Canad. J. Soil Sci.^
1968, v. 48, 369.
Rodrigues G. Fixed ammonia in tropical
soils.— J. Soil Sci. 1954, v. 5, 264.
Schachtschabel P, Bestimmung des fixier-
ten Ammoniums im Boden.— Z. Pflan-
zenernahr., Diing., Bodenkunde, 1961,
Bd. 93, H. 2, 125.
Silva J. A., Bremner J. M. Determination
and isotope-ratio analysis of different
forms of nitrogen in soils. 5. Fixed am¬
monium.— Soil Sci. Soc. America Proc.,
1966, v. 30, 5.
Sims J. Я., Jackson G. D. Rapid analysis
of soil nitrate with chromotropic acid.—
Soil Sci. Soc. America Proc., 1971, v. 35,
603.
Sowden F. J. Investigations of the amounts
of hexosamines found in various soils
and methods for their determination.—
Soil Sci., 1959, v. 88, 138.
Stevenson F. J. Distribution of the forms
nitrogen in some soil profiles.— Soil
Sci. Soc. America Proc., 1957, v. 21, 3.
К. Е. ГИНЗБУРГ
МЕТОДЫ ОПРЕДЕЛЕНИЯ ФОСФОРА В ПОЧВЕ
*
Общее количество фосфора на нашей планете составляет примерно 1019 т,
из них 10Гб т сосредоточено в земной коре, которая в среднем содержит
0,08—0,12 вес.% фосфора; атомы фосфора составляют 0,04—0,07 % от общего
числа атомов земной коры (Соколов, 1960; Larsen, 1967).
Основными фосфатными минералами в коре выветривания являются апа¬
титы и фосфориты.
Благодаря высокой реакционной способности фосфор в свободном состоя¬
нии в природе не встречается. В почвах он всегда содержится в пятивалент¬
ном виде, т. е. соответствует ангидриду Р205, который с водой и другими
элементами образует обширную группу кислородных соединений фосфора —
фосфорных кислот и фосфатов. В зависимости от числа молекул воды и числа
замещенных водородов, а также от количества тетраэдров РО^” и способов
их соединения между собой фосфаты образуют химические соединения, раз¬
личающиеся по растворимости в воде и различных растворителях, содержа¬
нию фосфора, физическим, физико-химическим и другим свойствам. На этом
основании их можно подразделить на две основные группы:
1. Ортофосфаты (мономеры) — построены из изолированных тетраэдров
РОГ, в вершинах которых расположены атомы кислорода.
2. Конденсированные фосфаты (ортофосфаты — полимеры) — получают¬
ся путем частичного отщепления воды («конденсации») от ортофосфатов; со¬
держат меньше конституционной воды, но больше фосфора, чем мономерные
ортофосфаты; состоят также из тетраэдров РО^”, которые через общие ки¬
слородные вершины образуют фосфатные комплексы. В зависимости от спо¬
соба соединения отдельных тетраэдров между собой конденсированные фос¬
фаты образуют различные полимерные соединения (Реми, 1963).
Почти все соединения) почвенных фосфатов (минеральных и органических
форм) являются ортофосфатами (мономерами или полимерами). Но могут
встречаться и полимерные конденсированные фосфаты. В последнее время
конденсированные фосфаты вносятся в почвы в виде удобрений, а также
в составе дезинфицирующих веществ.
Валовой фосфор почвы состоит из органических и минеральных соедине¬
ний. Соотношение между фракциями органических и минеральных форм фос¬
фора, их качественный и количественный состав в различных типах почв
различен и характерен для данных почвенных условий. Определение запаса
валового фосфора, общего "содержания минеральных и органических форм
фосфатов и отдельных фракций фосфора имеет теоретическое и практическое
значение для характеристики генетических типов почв, для обоснованной
оценки агрохимических свойств почв, балансовых расчетов и др.
Очень важным показателем, имеющим значение для прогноза эффективно¬
сти фосфорных удобрений, является способность почв поглощать фосфаты
в малорастворимой форме. Для изучения этого важного процесса применяют
методы определения емкости поглощения фосфатов почвами, фосфатной бу¬
ферной способности почв и др.
Методы определения фосфора в почве
107
ОПРЕДЕЛЕНИЕ ФОСФОРА В РАСТВОРЕ
Определение различных форм фосфатов в почвах состоит из нескольких
этапов:
1) переведение фосфора из твердых фаз почвы в раствор (сплавление на¬
вески почвы с содой, разложение фтористоводородной кислотой, обработка
при нагревании и на холоду концентрированными и разбавленными минераль¬
ными и органическими кислотами, щелочами, буферными растворами, водой,
слабосолевыми растворами и др.);
2) подготовка полученных почвенных вытяжек к анализу: обесцвечива¬
ние, окисление, выпаривание, восстановление, осаждение, хроматографиро¬
вание и др.;
3) определение фосфора в растворах различными способами: весовым9
объемным, фотометрическим и др.
Чтобы правильно определить содержание фосфатов в исследуемых образ¬
цах почв, минералах и других объектах, следует фосфор перевести в диссо¬
циируемое соединение, поэтому .необходимо знать, в какой форме (орто-,
пиро-, метафосфаты и др.) фосфор переходит в данный раствор. Как пра¬
вило, в различных почвенных вытяжках ион ортофосфата из минеральных
соединений свободно диссоциирует в раствор и может быть определен там
непосредственно общепринятыми методами. В фосфорорганических соеди¬
нениях ортофосфат связан в комплексах со сложными высококонденсирован-
ными, сильнополимеризованными органическими радикалами (в нуклеино¬
вых кислотах, инозитфосфатах, гуминовых, фульвокислотах и др.), поэто¬
му диссоциирует в раствор после разрушения этих комплексов, их минера- .
лизации. Если же в растворе присутствуют и конденсированные фосфаты
(мета-, пиро-, полифосфаты), то последние сперва выделяют из смеси экст¬
ракцией, хроматографией и другими методами, затем подвергают кислотному
гидролизу и переводят в ортофосфаты, которые и определяют в растворе
обычным способом (Аронов, 1959).
Таким образом, конечное определение различных форм фосфатов в раство¬
рах практически сводится к определению иона ортофосфорной кислоты Р043".
Почти все методы определения в растворах ортофосфата основаны на спо¬
собности фосфора в кислой среде взаимодействовать с окислами некоторых
элементов пятой и шестой побочных групп периодической системы Менде¬
леева, чаще всего Mo, Wo, V, с образованием комплексных гетерополикислот
(ГПК) или соответствующих гетерополисоединений (ГПС). В ГПК цент¬
ральным атомом-комплексообразователем является ион ортофосфата [могут
быть: As, Si, Ti, В, F, Al, Fe, S, H и др.|, а в качестве ли¬
гандов представлены чаще всего окислы Мо08, Wo08, V2O5 или смеси этих
окислов, обладающих способностью к полимеризации (Никитина, 1962).
Для определения образовавшихся в растворах ГПС применяют химиче¬
ские, физико-химические, физические способы анализа.
Важными критериями при выборе метода определения в растворах
любого химического элемента, в том числе и фосфора, является чувст¬
вительность, точность, воспроизводимость, избирательность анализа, а
также подготовка проб и стандартов, стоимость оборудования и продол¬
жительность анализа (Марченко, 1971).
Химические методы
Сюда относятся: гравиметрические (весовые) и титриметрические (объем¬
ные) методы, анализа.
108
К. Е. Гинзбург
Весовой метод Лоренца
(см. Хейфец, 1965)
Метод основан на осаждении фосфора молибдатом аммония из сильнокис¬
лого раствора в виде ГПС — фосфомолибдата аммония — кристаллического
осадка золотисто-желтого цвета (NH4)3P04.12Mo03-6H20. Чувствительность
анализа от 1 до 20 мг Р205 в анализируемом объеме. Избирательность анали¬
за—Fe8+, Si02 и небольшие количества НС1 не мешают осаждению фосфора.
Избыток НС1 удаляют выпариванием на водяной бане с последующим раст¬
ворением сухого остатка в HN03.
Ход анализа. 20 мл анализируемого раствора (если фосфора содержится
в растворе мало, то берут 50—100 мл и упаривают их до 20 мл) помещают в
химические стаканы емкостью 100 мл, приливают 30 мл смеси азотной и сер¬
ной кислот, нагревают на электрической плитке, покрытой асбестом, до по¬
явления первых пузырьков, снимают с огня, взбалтывают, чтобы стенки
стакана не были перегреты (стеклянной палочки не применяют); затем добав¬
ляют 25 мл сульфатмолибденовой жидкости, вливая ее в середину раствора,
закрывают часовым стеклом и оставляют стоять; через несколько минут (но не
более чем через 5 мин.), когда выпадает главная масса осадка, взбалтывают
жидкость вращением стакана в течение 30 сек. и оставляют стоять 15—
18 час. Фильтруют раствор с отсасыванием через тигель Нуча № 4 или тигель
с асбестом. Осадок полностью переносят во взвешенный тигель, смывая его
2%-ным раствором NH4N03 и затем им же 3—4 раза. После этого промы¬
вают осадок 2 раза спиртом, затем эфиром. Промывание спиртом и эфи¬
ром можно заменить двухкратным промыванием ацетоном. Промытые осадки
в тиглях ставят на час в термостат при 45—50°, охлаждают в пустом экси¬
каторе (без СаС12 или H2S04) и взвешивают.
Для подготовки тиглей к новому определению осадки в них растворяют
5%-ным раствором аммиака, затем, после промывания горячей водой, тиг¬
ли высушивают при 110° до постоянного веса.
Реактив ы9 1. Сульфатмолибденовая жидкость. 100 г сернокислого аммония в
мерной колбе на 1 л растворяют в 900 мл концентрированной HNOs (уд. вес 1,4) и доводят
той же кислотой до метки. В химическом стакане емкостью на 1 л подогревают 700 мл
дистиллированной воды до 60—80° С и растворяют в ней 300 г молибденовокислого аммо¬
ния при тщательном помешивании стеклянной палочкой и дальнейшем нагревании; по
охлаждении раствор переносят в мерную колбу на 1 л и доводят водой до метки. Раствор
молибдата аммония вливают при перемешивании тонкой струей в раствор сульфата ам¬
мония. Через 48 час. смесь отфильтровывают через плотный фильтр. Сохраняют раствор
в прохладном месте в склянке из темного стекла.
2. Азотная кислота, содержащая серную. 30 мл H2S04 (уд. вес 1,84) приливают к
1 л HN03 (уд. вес 1,2), получаемой разбавлением 424 мл крепкой HN03 (уд. вес. 1,4)
водой до 1 л.
3. 2%-ный раствор азотнокислого аммония подкисляют несколькими каплями кон¬
центрированной HN03.
4. Этиловый спирт (ГОСТ 5962-67), 85—90%-ный (об.%).
5. Эфир этиловый.
6. Ацетон (ГОСТ 2603-63).
Расчет Р2Об на воздушно-сухую почву (у, %):
(Ь—а) *0,03295.о100
у = л
У Z
где а — вес тигля без осадка, г\ Ь — вес тигля с осадком, г; (Ь — а) — вес осадка желтой
соли, г; с — общее разбавление исследуемого раствора; 0,03295 — переводный коэф¬
фициент веса осадка на Р2Об; z — исходная навеска почвы, г\ 100 — коэффициент пере¬
счета на 100 г почвы, т. е. в проценты.
Методы определения фосфора в почве
109
Комплексом метрический (объемный) метод
(Краснощеков, Максимова, Мыгиляева, Столбов, 1969)
В кислом растворе фосфор с молибдатом образует фосфорномолибденовую
кислоту (ФМК), которую экстрагируют бутиловым спиртом, затем разру¬
шают, реэкстрагируют ФМК щелочью, а выделившийся при этом молибден
оттитровывают комплексонометрически. Постоянство отношения Р : Мо =
= 1 : 12 в ФМК, образовавшейся в данных условиях анализа, позволяет по
молибдену косвенно определять фосфор.
Ход анализа. 10—20 мл исследуемого раствора, содержащего 7—15 мг
Р2Об, помещают в делительную воронку емкостью на 250 мл, приливают
3 мл концентрированной H2S04, 100 мл дистиллированной воды и 25 мл 5%-
ного раствора молибдата аммония. Растворы перемешивают и спустя 10—
15 мин. экстрагируют ФМК 50 мл н-бутилового спирта. Смесь встряхивают
20—ЗОсек. и отстаивают 3—5 мин., а затем осторожно спускают нижнюю вод¬
ную фазу. В этих условиях ФМК извлекается бутиловым спиртом нацело при
однократной экстракции (концентрация H2S04 в растворе ~1 н.).
Аликвотную часть ФМК в н-бутаноле (10—15 мл), содержащую 60—70 мг
молибдена, переносят в коническую колбу емкостью 250 мл, добавляют 10—
15 капель 25%-ного NH4OH и дают постоять 1—2 мин. (для более полного
разрушения ФМК); при этом раствор становится мутным. Добавляют сюда же
20 мл дистиллированной воды, 5 мл солянокислого гидразина (N2H4*2HC1)
с титром около 0,05 г/мл, 3 мл концентрированной НС1 и избыток 0,02 М
раствора комплексона III и кипятят на плитке 3—5 мин. (раствор при этом
окрашивается в интенсивно синий цвет). Горячий раствор нейтрализуют
25%-ным NH4OH по индикатору метиловому оранжевому, прибавляют 30 мл
аммиачной буферной смеси с pH 10 и снова кипятят 3—5 мин. При этом появ¬
ляется золотисто-оранжевая окраска комплексоната пятивалентного молиб¬
дена. Не охлаждая, разбавляют содержимое стакана дистиллированной во¬
дой в 3—4 раза, прибавляют дндикатор хромоген черный (ЕТ-00) в смеси с
хлоридом натрия (1 : 200) в таком количестве, чтобы окраска раствора стала
чисто-зеленой (30—50 мг смеси) и титруют избыток комплексона III 0,02 М
раствором сульфата цинка при температуре раствора 40—60° С до перехода
зеленой окраски в коричнево-красную.
По содержанию молибдена, входящего в состав ФМК, рассчитывают со¬
держание фосфора.
Титр раствора комплексона III по молибдену устанавливают в аналогич¬
ных условиях по стандартному раствору молибдата аммония. Раствор молиб¬
дата стандартизируют весовым методом, определяя молибден в форме РЬМо04.
Реактивы. 1. Концентрированная H2S04 (уд. вес 1,83—1,84).
2. Концентрированная НС1 (уд. вес 1,17—1,19).
3. 5%-ный водный раствор молибденовокислого аммония.
4. н-бутиловый спирт.
5. 25%-ный раствор NH4OH.
6. Буферная смесь с pH 10 (NH4OH + NH4C1). 25 г NH4C1 растворяют в 100 мл воды,
приливают 200 мл 25%-ного раствора NH4OH, доливают водой до 1 л, перемешивают,
проверяют pH. Хранят в склянке с притертой пробкой.
7. Солянокислый гидразин с титром 0,05 г/мл.
8. 0,02 М раствор комплексона III (трилона Б).
9. 0,02 М раствор сульфата цинка.
10. Индикатор метил оранжевый.
11. Индикатор хромоген черный (ЕТ-00). 0,25 г индикатора растирают в фарфоровой
ступке с 50 г NaCl до тонкого однородно окрашенного порошка. Храня1 в темной банке
в притертой пробкой.
110
К. Е. Гинзбург
Физико-химические (инструментальные) методы
Сюда относятся спектральный, активационный, атомно-абсорбционный*
радиохимический, электрохимический, кинетический, фотометрический и
другие методы анализа (Дыбина, Куртасова, 1972; Бабко, Пилипенко, 1974;
Плескач, 1969; Крешков, 1970; Марченко, 1971; Яцимирский, 1971; Акопян,
1974; «Аналитическая химия элементов», 1974).
Из перечисленных способов определения фосфора в растворах в настоя¬
щее время в агрохимических и почвенных исследованиях используются глав¬
ным образом фотометрические методы.
В основе этих методов лежит способность фосфора (Р043_) в кислой среде
при определенной концентрации ионов водорода 1 образовывать с молибда-
том желтый комплекс — фосфорномолибденовую гетерополикислоту (ФМК)
состава Н7[Р (Mo207)e] *nH20. В случае добавления восстановителя к раствору
ФМК входящий в ее состав шестивалентный молибден частично восстанавли¬
вается до пятивалентного; при этом образуется так называемая фосфорномо¬
либденовая синь и раствор окрашивается в голубой цвет. По интенсивности
образовавшейся голубой окраски определяют содержание фосфора в исследу¬
емом растворе.
Для того чтобы получить большую точность определения фосфора, следует
подбирать такие условия анализа, при которых восстанавливался бы молиб¬
ден, связанный с фосфором, и не восстанавливалась остальная часть молибде¬
на, которая находится в растворе в избытке в форме молибденовокислого
аммония. Это достигается определенным соотношением реактивов: минераль¬
ной кислоты (H2S04, НС1, НС104), молибденовокислого аммония и восста¬
новителя (двухлористого олова, аскорбиновой кислоты, сернокислого гид¬
разина, гидрохинона и др.)
Условия образования ГПС при колориметрическом определении фосфора
(значение величины pH, влияние ингибирующих веществ, значение восста¬
новителя) описаны в литературе (Гинзбург, 1958; Никитина, 1962; Никули¬
на, 1965).
Различные колориметрические методы определения фосфора отличаются
по концентрации Н-ионов (от 0,1 до 1 н., чаще всего 0,4—0,5 н.), по соотно¬
шению кислоты и молибдата (от 9 до 30, чаще всего 15—20), по окислительно¬
восстановительным свойствам используемых восстановителей (слабые, нор¬
мальные, сильные). Эти методы характеризуются различной чувствительно¬
стью, точностью, избирательностью анализа (Гинзбург, 1958; Никулина, 1965).
В зависимости от целей и задач исследования используется тот или иной
способ определения фосфора. Так, при определении общего содержания
фосфора в почвах, растениях, удобрениях желательно применять менее чув¬
ствительные колориметрические методы, чтобы снизить ошибку анализа за
счет большого разбавления растворов (ванадатно-молибдатный метод, методы
с использованием слабых восстановителей — гидрохинона, метола, эйконо-
гена и др.). Наоборот, в водных, слабосолевых, слабокислых и др. почвенных
вытяжках требуется применение более чувствительных способов коло¬
риметрического определения фосфора с использованием сильных и нормаль¬
ных восстановителей (SnCl2 аскорбиновой кислоты, сульфат-гидразина,
сульфита Na и др.) или их смесей.
Чувствительность и избирательность анализа можно повысить, экстра¬
гируя желтый или синий комплекс ФМК с помощью органических жидкостей:
спиртов, эфиров и др. (Алексеев, 1945).
Ниже приводим описание некоторых наиболее часто применяемых мето¬
дов колориметрического определения фосфора в растворах.
1 Следует учесть, что ГПК не образуются в нейтральной, щелочной среде, а некоторые
ГПК (например, ФМК) разрушаются в сильнокислых растворах (Никитина, 1962).
Методы определения фосфора в почве
111
Метод Труога — Мейера
(Truog, Meyer, 1929)
Метод основан на определении ортофосфата по восстановленной в голубой
цвет фосфорномолибденовой кислоте.
Условия анализа х: кислотность 0,4 н. H2S04; отношение кислоты к Мо03
равно 21; восстановитель сильный — SnCl2*2H20. Время фотометрирования
окрашенных растворов не ранее 5—7 мин. от развития окраски и в течение
15—20 мин. Чувствительность 0,002—0,003 и не более 0,04 мг Р2Об на 50 мл.
Точность ±5%.
Избирательность анализа: ] Fe3+ мешает определению при содержании
> 2 мг/50 мл; Si02 — >4 мг/50 мл; мешают органические кислоты, гуму¬
совые вещества, F*, As5* и др. (Гинзбург, 1958).
Ход анализа. Аликвоты испытуемых растворов, содержащих 0,003—
0,04 мг Р2Об, переносят в мерные колбы на 50 мл, разбавляют водой пример¬
но до 30—40 мл и, если необходимо, доводят до определенного pH. Для
этого к раствору прибавляют индикатор бесцветный в кислой и окрашенный
в щелочной среде (фенолфталеин, а-, р-, у- или /?-нитрофенолы и др.) Если
раствор при этом окрашивается в розовый или желтый цвет, то прибавляют
10%-ную НС1 или H2S04 до его обесцвечивания; если раствор бесцветен, то
прибавляют 10%-ный раствор NaOH или NH4OH до появления соответствую¬
щей окраски, которую снимают обратным титрованием кислотой (1—3 кап¬
ли).
После этого в колбы приливают 2 мл кислого раствора молибдата аммо¬
ния (комплексообразователя), доводят до метки, перемешивают и прили¬
вают 3 капли восстановителя — 2,5%-ного раствора SnCl2, немедленно вновь
перемешивают и спустя 5—7 мин. и в течение 15—20 мин. просматривают ок- *
рашенные растворы на фотоколориметре с красным светофильтром при
X ~650 нм или спектрофотометре при X ~ 780—825 нм.
При использовании мерных колб емкостью 100 мл расход указанных ре¬
активов увеличивают в 2 раза; для мерных колб емкостью 25 мл количество
реактивов, наоборот, уменьшают в 2 раза.
Реактивы. 1. Приготовление комплексообразователя. 25 г молибденовокислого
аммония (желательно по ГОСТ 3765-64, х. ч. или ч. д. а.) растворяют в 200 мл дистилли¬
рованной воды при нагревании. Одновременно в мерную колбу емкостью 1 л приливают
500 мл воды и очень осторожно, по стенкам колбы, без перемешивания, вливают неболь¬
шими порциями, с перерывами] 280 мл H2S04 (уд. вес 1,83—1,84) по ГОСТ 3765-64, х. ч.
или ч. д. а.
После остывания обоих растворов в серную кислоту небольшими порциями, при по*
стоянном помешивании, приливают раствор молибденовокислого аммония; после вторич¬
ного остывания общий объем доводят точно до 1 л, перемешивают и переливают в склянку
из темного стекла, где он может сохраняться длительное время (приблизительно в течение
года).
2. Восстановитель. 0,25 с (или 0,13 г) SnCl2*2H20 (ГОСТ 36-68), ч. д. а., всыпают в
стеклянную пробирку емкостью 10—15 мл, приливают сюда же 10 мл (или 5 мл соответ¬
ственно) 10%-ной НС1 (ГОСТ 3118-67, х. ч. или ч.[д. а.). Погружают пробирку в химиче¬
ский стакан емкостью 100 или 200 мл, наполненный водой, и кипятят на электрической
плитке до полного растворения восстановителя.
1 А. М. Мещеряков (1965) проверял ряд колориметрических методов определения фосфора
и установил, что только в методах Левицкого и Труога — Мейера количества кис¬
лоты и молибдата, необходимые для образования синей ФМК, близки расчетным,
теоретическим показателям, поэтому в этих условиях в меньшей степени влияют
возможные колебания в величине pH исследуемых растворов, обусловленные предва¬
рительной их подготовкой к колориметрированию (окисление вытяжек, осаждение
железа и др.).
112
К. Е. Гитбург
После этого раствор охлаждают, перемешивают и используют для окрашивания ис¬
пытуемых и стандартных растворов. Раствор готовят ежедневно.
3. Индикаторы: а-, (3-, у-, р-нитрофенолы. Одноцветные кислотно-основные индикато¬
ры. Интервал перехода от бесцветной окраски к желтой и наоборот находится в пределах
pH 2,5—5,0. Применяют их ~0,1%-ные водные растворы. Фенолфталеин применяют
в виде 0,1%-ного спиртового раствора.
4. Образцовые растворы фосфата. А. Крепкий раствор фосфата. 0,1917 г КН2Р04
(ГОСТ 4198-65, х. ч.) растворяют в дистиллированной воде в мерной колбе емкостью 1 л,
добавляют какой-нибудь антисептик (тимол, толуол, несколько капель уксусной, серной,
0,8 М борной кислот), затем доливают водой до метки, перемешивают, переливают в склян¬
ку из темного стекла, где он сохраняется длительное время. В 1 мл этого раствора содер¬
жится 0,1 мг Р2Об.
Б. Рабочий раствор фосфата. 20 мл крепкого раствора фосфата в мерной колбе раз¬
водят водой до 200 мл. Полученный раствор содержит 0,01 мг Р205 в 1 мл. Он использует¬
ся для построения шкалы стандартных растворов.
Построение шкалы стандартных растворов.
Аликвоты рабочего образцового раствора Б, соответствующие 0,005; 0,01;
0,02; 0,03 и 0,04 мг Р2Об в 50 мл, окрашиваются точно так же, как испытуе¬
мые растворы, и просматриваются затем на фотоколориметре.
Согласно полученным отсчетам фотоколориметра (оптическая плотность
D), соответствующим известным концентрациям рабочего образцового
раствора фосфата, на миллиметровой бумаге составляют калибровочный
график (рис. 1 ,А). При фотометрировании испытуемых растворов, сделав на
приборе отсчет, на графике находят, какому содержанию фосфора в данном
объеме отсчет соответствует.
Н а п р и м е р, при просмотре в фотоколориметре испытуемого раствора, содержа¬
щего в 50 мл колбе 10 мл вытяжки, полученной при соотношении почва: раствор = 5 : 125,
получили отсчет по шкале фотоколориметра — 0,12; на калибровочном графике этому от¬
счету соответствует 0,02 мг Р205. При этом расчет содержания Р205 в 100 г почвы выразит¬
Ц005Ц01 0,02 ЦОЗ 0,04 0,05 0,0В 0,05 0,1 0,2 0,3 0,9 0,5 0,6 0,7 0,8 0,9 У,О
мг Р^в 50 мл
Рис. 1. Зависимость оптической плотности D растворов фосфорномолнбденовой кислоты
(ФМК) от концентрации фосфора
А — калибровочный график восстановленных синих комплексов ФМК. Светофильтр красный, X
~ 655 нм, толщина кюветы d = 10 мм. I — метод Мерфи — Райли (Murphy, Riley, 1962), II — методы
Труога — Мейера (Тгио?, Меуег, 1929) и Понса — Гутри (Pons, Guthrie, 1946). Б — калибровочный
график для желтых комплексов фосфованадомолибденовой кислоты. Светофильтр синий, А, ^ 415 пм.
Толщина кюветы* 1 — d = 10 мм; 2 — d = 20 мм; 3 — d = 30 мм; 4 — d = 50 мм
Методы определения фосфора в почве
113
ся следующим образом:
v 0,02*с-100
— »
Ьа
где X — количество Р2Об, мг на 100 г почвы; а — навеска почвы при приготовлении вы¬
тяжки, г; с — количество раствора при приготовлении вытяжки, мл; b — количество вы¬
тяжки, взятое в колбу для колориметрирования, мл, 100 — коэффициент пересчета
яа 100 г почвы.
В данном конкретном примере содержание Р2Об в мг на 100 г почвы выразится:
v _ 0,02 -125-100 *
10*5
Калибровочный график можно составлять с пересчетом в мг Р2Об на 100 г почвы (За¬
мятина, 1969).
Метод Мерфи — Райли
(Murphy, Riley, 1962)
Образующийся в кислой среде желтый комплекс ФМК восстанавливают
аскорбиновой кислотой в присутствии катализатора сурьмы. Голубая ок¬
раска достигает максимума через 10 мин. и устойчива в течение 24 час.
Условия анализа такие же, как в методе Труога — Мейера (см. выше).
Ход анализа. Аликвотную часть исследуемого раствора, содержа¬
щего от 0,003 до 0,04 мг Р2Об, помещают в мерные колбы на 50 мл, разбав¬
ляют водой примерно до 20—30 мл, нейтрализуют по р-нитрофенолу или дру¬
гому индикатору (как описано в методе Труога — Мейера), добавляют 8 мл
реактива Б для образования синей ФМК, доводят до метки, перемешивают и
спустя 10 мин. в течение 24 час. фотометрируют окрашенные растворы с*
красным светофильтром х.
Реактивы: 1. Реактив А (смесь комплексообразователя и катализатора). 12 г
молибдата аммония растворяют в 250 мл дистиллированной воды при нагревании.
В 100 мл дистиллированной воды растворяют 0,2908 г сурьмяно-виннокислого ка¬
лия 2.
Два этих растворенных реактива вливают в 1 л 5 н. H2S04 (140 мл концентрированной
H2S04 в 1*л), тщательно перемешивают и доводят дистиллированной водой до 2 л, сохра¬
няют в склянкё из темного стекла.
2. Реактив Б (смесь комплексообразователя и восстановителя). Растворяют 1,056 г
аскорбиновой кислоты в 200 мл реактива А п перемешивают. Реактив годен в течение 24
час.
Метод экстракции Понса и Гутри
(Pons, Guthrie, 1946)
Метод основан на экстракции желтого комплекса ФМК смесью изо¬
бутил ового спирта и эфира, а затем его восстановлении двухлористым
оловом.
Предполагается, что экстракция предотвращает возможный гидролиз
фосфорных эфиров, увеличивает чувствительность определения и снижает
влияние сопутствующих ионов (Si02, органических веществ и др.).
Чувствительность определения 0,002 мг Р2Об на 50 мл и не более 0,04 мг
Р205 в 50 мл. Точность ± 5%.
Ход анализа. 50 мл прозрачного фильтрата почвенной вытяжки помещают
в стеклянную пробирку с притертой пробкой емкостью на 100 мл или в дели¬
тельную воронку такой же емкости, приливают 20 мл смеси изобутилового
спирта и бензола и 5 мл молибденовокислого аммония. Закрывают пробирки
1 Для получения большей чувствительности может быть использован спектрофотометр ‘с
кюветой d = 7—10 см и светофильтром с X = 882 нм.
2 K(Sb0)C4H406-l/2 Н20 — антимонил-калпй-виннокислый (рвотный камень).
414
К. Е. Гинзбург
пробками, перемешивают 2 мин. и отстаивают 5 мин. После разделения фаз
пипеткой отбирают Юлмизобутилового слоя и переносят в мерную колбу на
25 мл, многократно промывая пипетку раствором серной кислоты и этилового
спирта, сливают все в ту же мерную колбу, доливают реактивом 4 до ~ 22 мл,
добавляют 0,5 мл SnCl2, доводят до метки реактивом 4, тщательно переме¬
шивают и спустя 5—7 мин. фотометрируют окрашенные растворы с красным
светофильтром с К = 650 нм (чувствительность метода выше при X = 730—
:840 нм).
Приготовление шкалы образцовых растворов.
Аликвоты рабочего образцового раствора КН2Р04 — реактив Б (приготов¬
ление — см. метод Труога — Мейера), соответствующие 0,005; 0,01; 0,02;
0,03; 0,04 мг Р206, помещают в мерные колбы на 50 мл, доливают дистилли¬
рованной водой до метки, перемешивают, затем экстрагируют и окрашивают
точно так же, как испытуемые растворы.
Реактивы. 1. Экстрагирующая смесь. Изобутиловый спирт и бензол (без тио-
фена) смешиваются в равных объемах (1 : 1).
2. Комплексообразователь. В мерную колбу емкостью 1 л вливают 400 мл 10 н. H2S04.
В 200—300 мл дистиллированной воды при нагревании растворяют 50 г молибдата аммо¬
ния. После охлаждения раствор молибдата аммония небольшими порциями, при поме¬
шивании приливают к раствору серной кислоты. Смесь вновь охлаждают, доливают до
1 л, перемешивают и хранят в склянке из темного стекла.
3. Восстановитель. За. 10 г SnCl2*2H20 растворяют в 25 мл концентрированной НС1.
36. 1 мл реактива За разбавляют до 200 мл 1 н. раствором H2S04, перемешивают и исполь¬
зуют для анализа. Реактив 36 готовят ежедневно.
4. Растворитель. Раствор этилового спирта в серной кислоте. 20 мл H2S04 (уд. вес
1,83—1,84) небольшими порциями осторожно по стенкам колбы приливают к 980 мл эти¬
лового спирта (99,5%).
5. Концентрированная H2S04 (уд. вес 1,83—1,84), ГОСТ 4204-66, х. ч.
6. Концентрированная НС1 (уд. вес 1,17—1,19), ГОСТ 3118-67, х. ч. или ч. д. а.
7. 10 н. H2S04. 280 мл концентрированной H2S04 осторожно вливают в 600 мл дистил¬
лированной воды, раствор охлаждают, доливают водой до 1 л, перемешивают.
8. 1н. H2S04— 28 мл концентрированной H2S04 в 1 л.
Ванадомолибдатный метод
(«Инструкция для зональных агрохимических лабораторий...», 1968)
Метод основан на определении ортофосфата в кислой среде в виде окра¬
шенной в желто-оранжевый цвет фосфорнованадомолибденовой гетерополи¬
кислоты (HgPO^HVOg-llMoOg-mHjjO, в которой Р : V : Мо = 1:1: 11).
Окраска устойчива во времени. Реакцию следует проводить в 0,5—0,9 н. (до
1,5 н.) раствора HN03 (можно H2S04, НС104).
Концентрация ванадия в конечном растворе должна быть 0,002 М, мо¬
либдата ~ 0,01 М. В оптимальных условиях реагенты сами дают желтое
окрашивание, поэтому необходимо измерить величину светопоглощения
растворов относительно раствора холостого опыта.
Избирательность анализа: кремнекислота, арсенаты, пирофосфаты прак¬
тически не мешают определению. Fe3+ в количествах ^>2 мг на 50 мл
мешает определению. Чувствительность определения от 0,1 мг до 1,0 мг Р205
в 50 мл. Точность ± 2—5%.
Метод рекомендуется для определения валового фосфора в почвах, фос¬
фора в удобрениях, растениях. При определении подвижных фосфатов в
почвенных вытяжках этот метод не пригоден для практического использо¬
вания, так как является малочувствительным.
Ход анализа. Аликвоты исследуемого раствора, содержащие 0,1—1,0 мг
Р206, помещают в мерные колбы на 50 мл, разбавляют водой до 30 мл, нейт¬
Методы определения фосфора в почве
115
рализуют 10%-ным NH4OH по фенолфталеину или другому индикатору (см.
метод Труога — Мейера), затем добавляют 1—2 капли разбавленной HNOa
(1 : 2) до исчезновения розовой или желтой окраски, приливают еще 5 мл
HN03 (1 : 2) и 15 мл реактива 4. Раствор доводят до метки, перемешивают.
Через 30 мин. колориметрируют, используя синий светофильтр с X = 460—
470 нм; метод более чувствительный в ультрафиолетовой области при X —
315 нм.
Приготовление шкалы образцовых растворов
фосфата (см. рис. 1 Б). Аликвоты крепкого образцового раствора
КН2Р04 — реактив А, соответствующие 0,05; 0,1; 0,2; 0,3... 1,0 мг Р205 в
50 мл окрашивают точно так же, как испытуемые растворы.
Пример вычисления содержания фосфора в исследуемых растворах при¬
веден выше в описании метода Труога — Мейера.
Реактивы. 1. Разбавленная HN03 (уд. вес 1,14). Один объем концентрированной
HN03 (уд. вес 1,40) разводят двумя объемами дистиллированной оды.
2. 0,25%-ный раствор ванадокислого аммония. 2,5 г ванадата аммония растворяют
в кипящей воде, охлаждают, добавляют 20 мл концентрированной HN03 и доливают водой
до 1 л в мерной колбе.
3. 5%-ный раствор молибденовокислого аммония. 50 г молибденовокислого аммония
растворяют в 500 мл воды при нагревании, охлаждают и доливают водой до 1 л
4. Комплексообразователь. Растворы 1—3 смешивают в отношении 1:1:1. В тем¬
ном прохладном месте смесь сохраняется длительное время (более полугода).
ОПРЕДЕЛЕНИЕ ВАЛОВОГО ФОСФОРА В ПОЧВЕ
Валовой фосфор почвы состоит из органических и минеральных соедине¬
ний. В соответствии с этим методы его определения должны включать такую
обработку почвы, чтобы как можно полнее разрушить минеральную и орга¬
ническую часть почвы и таким образом перевести в раствор входящий в их
состав фосфор.
Классические методы определения валового фосфора в почве: сплавле¬
ние со щелочами и разложение почвы фтористоводородной кислотой, трудо¬
емки, длительны и требуют наличия платиновой посуды. Другие методы
определения валового фосфора почвы (не требующие наличия платиновой по¬
суды): обработка почвы царской водкой, азотной кислотой и перманганатом
по Лебедянцеву, прокаливание почвы с азотнокислым магнием, также дли¬
тельны и не способны обеспечить полное разложение органической и мине¬
ральной части почвы. В последнее время широкое распространение получили
ускоренные методы разложения навески почвы хлорной кислотой, смесью
хлорной и азотной, смесью хлорной и серной кислот, смесью серной кислоты
и хлората калия и др. (Гинзбург и др., 1963).
Указанные методы дают результаты, очень близкие к данным, полу¬
ченным при помощи классических методов анализа, но значительно менее
трудоемки и разлагают навеску] почвы за сравнительно короткий срок
(от 15—30 мин. до 2—4 час.).
Разложение прокаленной навески почвы
смесью фтористоводородной и азотной кислот
(Добрицкая, 1973)
Навеску почвы для окисления органических веществ предварительно про¬
каливают в муфеле при температуре не выше 500—550° С, чтобы избежать по¬
терь калия и фосфора вследствие их улетучивания. В дальнейшем до окисле¬
ние органических веществ3 проводят, обрабатывая прокаленную навеску
почвы концентрированной HN03.
116
К. Е. Гинзбург
Для наиболее полного разложения минеральной части почвы используют
38%-ный водный раствор фтористоводородной кислоты — плавиковую кис¬
лоту. Плавиковая кислота очень активно взаимодействует с большинством
металлов, растворяя их; исключение составляют «благородные» металлы —
золото, платина; свинец разъедается только с поверхности. Помимо этого,
HF агрессивно действует на кремневую кислоту и силикаты, образуя при этом
тетрафторид кремния — SiF4 — газообразное летучее соединение. Поэтому
при взаимодействии почвы с HF происходит почти полное разрушение ее
силикатной части, сопровождающееся выделением кремневой кислоты
в виде газообразного продукта. В связи с этим в полученном растворе после
обработки почвы плавиковой кислотой, помимо фосфора, можно определять
и другие элементы (S, К, Na, Мп и др.), кроме кремневой кислоты.
Ход анализа. 0,5—2,0 г почвы (сито d 0,25 мм) помещают в платиновые чаш¬
ки 1 и прокаливают 2—3 часа в муфельной печи при 500—550° С.
Чашку с прокаленной почвой охлаждают, прибавляют 1 мл воды, 5 мл
концентрированной HN03 и примерно 15 мл HF, не загрязненной Р205, ста¬
вят на электрическую плитку с закрытой спиралью или этернитовую плитку
и выпаривают досуха, избегая пересушивания.
Остаток в чашке повторно обрабатывают 5 мл концентрированной HN03
и 15 мл HF, выпаривают досуха. Для полного удаления HF из остатка почвы
приливают в чашку 5 мл воды, 2 мл концентрированной НС1 и смесь выпа¬
ривают. Приливают еще 5 мл воды, вновь выпаривают. Приливают 5 мл кон¬
центрированной НС1, закрывают чашки часовым стеклом и нагревают
5—10 мин. до растворения остатка. Затем добавляют около 15 мл горячей
воды и смесь количественно переносят в мерные колбы на 100—200 мл, раст¬
вор охлаждают, доливают до метки водой, перемешивают, фильтруют
(раствор А).
В аликвоте раствора А определяют фосфор весовым, объемным, фотомет¬
рическим и другими методами. При использовании колориметрических ме¬
тодов из раствора А необходимо удалить Fe3+, мешающее определению фос¬
фора. Для этого существуют несколько способов (Гинзбург, 1958), но наибо¬
лее надежный из них метод Уоррена и Пью.
Осаждение Fe3+ по Уоррену и Пью
(Warren, Pugh, 1930)
Из нейтральной или кислой среды Fe3x осаждается раствором
К4 [Fe(CN)e] в виде берлинской лазури — осадка интенсивно голубого или си¬
него цвета
3 [Fe2'*' (CN)6]4~-f-4Fe3+ -> Fef [Fe2+ (GN)e]3 .
Оставшийся в растворе избыток ферроцианида калия связывают рас¬
твором соли двухвалентного марганца, которая образует белый осадок же¬
лезистосинеродистого марганца — Mn2 [Fe (CN)e]. В щелочной среде обе соли
нерастворимы, поэтому подщелачивание смеси аммиаком способствует более
полному выделению осадка из раствора.
Ход анализа. В мерную колбу на 100 мл помещают 10—20 мл раствора А,
разбавляют водой примерно до 30 дед и прибавляют по каплям при помеши¬
вании 3 мл 10%-ного раствора К4 [Fe (CN)e] и спустя 5 мин. 2,5 дед 10%-ного
1 В случае отсутствия платиновой посуды можно пользоваться чашками из тефлона или
фторопласта-4 (термоустойчивы при 200°), но предварительное прокаливание в муфеле
исходной навески почвы в данном случае проводят в фарфоровых тиглях, затем коли¬
чественно переносят прокаленную навеску дистиллированной водой в чашки и дальше
проводят обработку почвы точно так же, как описано выше в случае использования
платиновых чашек.
Методы определения фосфора в почве
117
раствора MnS04. После стояния в течение нескольких минут смесь титруют
10%-ным раствором аммиака до резкого перехода голубой окраски в сире-
невато-лиловатую; значение pH устанавливается 6,8—6,9, при котором ком¬
плексные соединения железа и марганца удерживаются в осадке. В связи
с тем, что при этой реакции осаждается и фосфор, для его растворения до¬
бавляют 3,5 мл 2,0 н. раствора H2S04, доливают затем водой до метки, пере¬
мешивают и фильтруют через плотный фильтр (раствор Б).
5—20 мл прозрачного раствора Б, содержащего 0,005—0,04 мг Р205, по¬
мещают в мерные колбы на 50 мл и определяют фосфор колориметрически.
Примечание I. В оригинальной прописи анализа Уоррен и Пью (Хейфец,
1960) для осаждения Fe3+ рекомендовали добавлять 6 мл 10%-ного раствора K4[Fe(Ci\)e]
и 5 мл 10%-ного раствора MnS04. Использование такого количества реактивов при неболь¬
шой исходной навеске почвы часто бывает излишним и мешает нормальному ходу осаж¬
дения Fe3**-. Так, в растворе то остается в большом избытке соль ферроцианида калия,
то раствор соли марганца. В первом случае фильтрат бывает окрашен в желтый цвет,
а во втором — выпадает бурая гидроокись марганца. В связи с этим было предложено
использовать половинную дозу реактивов при осаждении Fe3+ (Гинзбург, 1969).
П римечание II. Посуду после осаждения окисного железа лучше всего промы¬
вать раствором щавелевой кислоты, которая хорошо растворяет и берлинскую лазурь
(образуются растворимые чернила), и бурые хлопья гидроокиси марганца.
Реактивы. 1. Фтористоводородная (плавиковая) кислота, ГОСТ 10484-63, х. ч.
2. Концентрированная HNOs (уд. вес 1,3—1,4), ГОСТ 4461—48.
3. Концентрированная НС1 (уд. вес. 1,17—1,19), ГОСТ 3118—46.
4. 2,0 н. H2S04. 56 мл концентрированной H2S04 (уд. вес 1,83—1,84), ГОСТ 4204—66,
приливают осторожно к ~ 700 мл дистиллированной воды, охлаждают, доливают водой"
до 1 д, перемешивают.
5. 10%-ный раствор NH4OH. Крепкий аммиак (ГОСТ 3760-64) разбавляют водой в
соотношении 1 : 1 (по объему).
6. 10%-ный раствор MnS04*7H20 (ГОСТ 435-41). Сохраняется не более 6 месяцев.
7. 10%-ный раствор K4[Fe(CN)e] *ЗН20 — желтая кровяная соль. Сохраняется не бо¬
лее 6 месяцев.
Метод хлорной кислоты Шермана
(Sherman, 1942)
Метод был описан и проверен на некоторых типах почв Э. И. Шконде
{1954).
Навеску почвы сжигают в 30—72 %-ной НС104 при t ~ 205° С. Вр емя сжи¬
гания одного образца 1—4 часа. В полученном растворе осаждают Fe3+ и
определяют фосфор колориметрически.
Ход анализа. 0,5—1,0 г почвы (сито d 0,25 мм) всыпают в колбу Кьель¬
даля емкостью 100—150 мл (из огнеупорного стекла) и заливают 20 мл 30 —
72 %-ной хлорной кислоты. Спустя один час (можно оставлять на ночь) смесь
ставят на электрическую плитку и кипятят до исчезновения темной окрас¬
ки органического вещества, затем кипятят еще 10—15 мин. для более полного
разложения минеральной части почвы и удаления из раствора накапливаю¬
щихся газообразных продуктов реакции, например хлора. Рекомендуемое
в оригинальной прописи (см. Хейфец, 1965) последующее кипячение смеси
с дистиллированной водой для удаления из нее хлорной кислоты излишне:
во-первых, таким путем хлорную кислоту из раствора удалить невозможно;
во-вторых, эта кислота не мешает определению фосфора, так как исследуе¬
мые растворы перед их окрашиванием нейтрализуют, доводя до необходи¬
мого pH.
Таким образом, смесь после озоления охлаждают и количественно пере¬
носят в мерные колбы на 100—250 мл, вновь охлаждают, доливают до метки,
перемешивают и либо фильтруют, либо отстаивают.
118
К. Е. Гинзбург
В аликвотах прозрачного [раствора осаждают Fe3+ и определяют фосфор
колориметрически.
Реактив. 30—72%-ная НС104.
Мокрое озоление почвы в смеси серной
и хлорной кислот по Гинзбург и др. (1963, 1971)
Навеску почвы сжигают в смеси концентрированной H2S04 с добавлени¬
ем НС104 в качестве катализатора. В данном случае разложение серной
кислоты ускоряется и происходит уже при температуре не 338° С, а
~ 215° С. Кроме того, хлорная кислота активно участвует и в окислении
органических веществ почвы. Разложение обеих кислот происходит по сле¬
дующим уравнениям реакции:
H2SO4 ^ SO3+H2O,
2SO3 ^ 2SO2+O2,
4НС104 ^ 2С12+702+2Н20,
2НС104 ^ С12+ЗО2+Н2О2.
Поэтому при использовании указанной смеси кислот значительно сокра¬
щается время озоления навески почвы. Обычно для сжигания одного образца
требуется 15—30 мин.
Ход анализа. 0,5 г воздушно-сухой почвы (сито d 0,25 мм) помещают в
плоскодонные колбы емкостью 100 мл из жаростойкого стекла или в колбы
Кьельдаля, смачивают водой, приливают 8 мл концентрированной H2S04 и
0,5—0,8 мл ~ 50 %-ной НС104, оставляют на 30—60 мин. (или на ночь). После
этого колбы закрывают маленькими воронками, ставят на электрические плит¬
ки, покрытые асбестом, и кипятят смесь при t > 200° до полного обесцвечи¬
вания раствора, после этого кипячение продолжают еще 5—10 мин.
В зависимости от химического состава анализируемой навески почвы цвет
смеси после сжигания может быть белесый, сероватый, желтоватый, иногда
с вкраплениями черных мелких частичек, возможно окислов тяжелых ме¬
таллов, угля.
После сжигания смесь охлаждают, затем доливают 20—30 мл дистил¬
лированной воды и количественно переносят в мерные колбы на 200—250 мл,
обмывая при этом воронки, горлышко и стенки колб. Раствор вновь охлаж¬
дают, доводят дистиллированной водой до метки, перемешивают, фильтру¬
ют. В аликвоте прозрачного раствора осаждают Fe3r по Уоррену и Пью и
определяют фосфор колориметрически.
Реактивы. 1. H2S04 концентрированная (уд. вес 1,83—1,84).
2. 30—72%-ная НС104.
ОПРЕДЕЛЕНИЕ ОБЩЕГО СОДЕРЖАНИЯ МИНЕРАЛЬНЫХ
И ОРГАНИЧЕСКИХ ФОСФАТОВ ПОЧВЫ
Органические и минеральные фосфаты почвы представляют различную
ценность для питания растений, поэтому очень важно знать количественное
соотношение двух этих групп соединений в общем запасе фосфатов почвы.
Для этих целей чаще всего используют методы экстракции или прокалива¬
ния, включающие косвенные и прямые способы определения фракций орга¬
нических и минеральных фосфатов почвы. В последнее время для ускорения
извлечения минеральных, и органических фосфатов рекомендуют обрабаты¬
вать почву ультразвуком.
Сравнение различных методов определения органофосфатов в почвах и их
критическая оценка приводятся в работах Дж. Андерсона (Anderson, 1967)г
К. Е. Гинзбург (1969), О. Г. Ониани (Oniani et al., 1973) и др.
Методы определения фосфора в почве
119
Кислотно-аммиачная экстракция.
Метод Хейфец (1948, 1965)
Метод основан на] последовательной обработке навески почвы 4 н. НС1
и горячим 4%-ным NH4OH с последующим определением органического и
минерального фосфора отдельно в кислотном и аммиачном экстрактах. Дан¬
ным способом извлекается до 80% валового фосфора почвы.
Ход анализа. 4 г воздушно-сухой почвы (сито d 0,25 мм) помещают в кони¬
ческие колбы емкостью 250 мл, обрабатывают 100 мл 4,0 н. НС1 в течение
1 часа с периодическим перемешиванием суспензии, оставляют на ночь, за¬
тем фильтруют в мерные колбы на 250 мл. После этого почву промывают на
фильтре 0,01 н. НС1, а затем горячей водой от кислоты (проба на СГ с
AgN03), доводя общий объем раствора до 250 мл, перемешивают, получают
раствор А. В аликвотах раствора А определяют содержание общего и
минерального фосфора.
Определение общего фосфора в 4 н. НС1 вытяжке. 25 мл
раствора А помещают в 100-ми л ли литровые плоскодонные колбочки, прибав¬
ляют 2 мл 5%-ного раствора КМп04, перемешивают и нагревают на элек¬
трической плитке, покрытой асбестом, до полного растворения бурого осад¬
ка перекиси марганца (—30 мин.), охлаждают и осаждают Fe3+ по Уор¬
рену и Пью. 10—20 мл фильтрата после осаждения Fe3+ переносят в мерные
колбы емкостью 50 мл и после нейтрализации раствора определяют фосфор коло¬
риметрически. 4
Определение минерального фосфора в4н. НС1 в ы-
тяжке. 25 мл раствора А сливают в мерные колбы на 100 мл, осаждают
Fe3+. 10—20 мл фильтрата после осаждения Fe8+ переносят в мерные колбы
на 50 мл и определяют фосфор колориметрически. Органический фосфор
вычисляют по разности между содержанием общего и минерального фосфора
в солянокислой вытяжке.
Приготовление и анализ 4 %-н о й NH4OH вытяжки
(вариант Гинзбург, 1969). Остаток почвы после ее обработки 4,0 н. НС1 пе¬
реносят в те же исходные конические колбы, заливают 40 мл 4 %-ного NH4OH,
закрывают перевернутыми маленькими коническими колбами (или пробками
с двумя бунзеновскими клапанами *) для предотвращения улетучивания ам¬
миака и упаривания раствора и нагревают на водяной бане в течение 4—
5 час. при периодическом перемешивании, затем оставляют суспензию на
16—18 час. После этого смесь количественно переносят водой в мерную кол¬
бу на 200—250 мл, доливают водой до метки, перемешивают, отстаивают и
фильтруют при разрежении через плотный фильтр с синей лентой на воронке
Бюхнера или через фильтр Нутча № 4, погружая последний в аммиачную
вытяжку. Прозрачный фильтрат аммиачной вытяжки (раствор Б) исполь¬
зуют для определения общего и минерального фосфора.
Определение общего фосфора в NH4OH в ы т я ж-
к е. 10—20 мл раствора Б переносят в плоскодонные колбы на 50 мл, добав¬
ляют по каплям 2 н. H2S04 до выпадения хлопьев гуминовой кислоты, по¬
мещают на электрические плитки с закрытой спиралью, выпаривают смесь до¬
суха, избегая пересушивания. К сухому остатку приливают 3—5 мл НС104
(30—72%) и нагревают на плитке до полного обесцвечивания раствора,
охлаждают, переносят водой количественно в мерные колбы на 50—100 мл,
еще раз охлаждают, доливают водой до метки, перемешивают (раствор В).
5—20 мл раствора В сливают в другие мерные колбы на 50—100 мл, раз¬
бавляют водой, нейтрализуют и определяют фосфор колориметрически.
Определение минерального фосфора в NH4OH вы¬
тяжке. 40—50 мл раствора Б сливают в конические колбы на 100 мл, поме¬
1 Один для впускаф другой для выхода газов, образующихся в условиях анализа.
120
К. Е. Гинзбург
щают их на этернитовые плитки, нагревают до исчезновения запаха аммиака,
затем охлаждают, подкисляют 2 н. H2S04 до выпадения осадка гуминовых
кислот, затем меркой всыпают активированный уголь (~0,15—0,30 г) для
поглощения окраски фульвокислот и настаивают 10—20 мин. Если раствор
не обесцветился, то добавляют еще порцию угля (~ 0,1 г), повторно наста¬
ивают 10—20 мин. и фильтруют через небольшой беззольный фильтр (си¬
няя, белая лента) в мерные колбы на 50—100 мл, промывая стенки колб
и фильтр 0,01 н. НС1, доводят до метки водой, перемешивают (раствор Г).
20—40 мл раствора Г сливают в другие мерные колбы на 50 мл, нейтра¬
лизуют и определяют фосфор колориметрически.
Органический фосфор вычисляют по разности между содержанием общего
и минерального фосфора в аммиачной вытяжке.
Реактивы. 1. 4 н. HCJ.
2. 4%-ный раствор NH4OH.
3. 30—50%-ная НС104.
4. Очищенный активированный уголь. Приготовление см. ннже в методе Мачигина*
Кислотно-щелочная экстракция.
Метод Мета (Metha et al., 1954),
вариант Гинзбург (1969)
Метод основан на последовательной обработке навески почвы концентри¬
рованной НС1 и 0,5 н. NaOH при комнатной температуре и при нагревании.
Обе вытяжки объединяют и в полученной смеси, после осаждения Fe3+,
определяют содержание общего и минерального фосфора, а по разности —
фосфор органических соединений. Данным способом извлекается до 100%
валового фосфора почвы.
Ход анализа. 0,5 г почвы (сито d 0,25 мм) помещают в центрифужные
пробирки на 50 мл из жаростойкого стекла, добавляют 20 мл концентри¬
рованной НС1, перемешивают, настаивают 20 мин., вновь перемешивают и
погружают на 30 мин. в водяную баню, нагретую до 70° С. После этого смесь
охлаждают, добавляют 25 мл воды, перемешивают, центрифугируют (5—
10 мин. со скоростью 2—3 тыс. об/мин) и раствор сливают в мерные колбы
на 200—250 мл. К остатку почвы в пробирке добавляют 30 мл 0,5 н. раство¬
ра NaOH, перемешивают, настаивают 1 час., центрифугируют, раствор до¬
бавляют к солянокислой вытяжке. К остатку почвы добавляют еще 30 мл
0,5 н. NaOH, суспензию перемешивают, оставляют на 16—18 час., затем
центрифугируют, раствор добавляют к предыдущим вытяжкам, а остаток
почвы вновь заливают 30 мл 0,5 NaOH и пробирки погружают в водяную ба¬
ню, нагретую до 90° С, закрывают их перевернутыми коническими колбоч¬
ками, чтобы избежать упаривания раствора, и нагревают 4—5 час., затем
смесь охлаждают, центрифугируют, центрифугат добавляют к общему рас¬
твору. После этого раствор заливают до метки водой, перемешивают (полу¬
чают раствор А).
Определение общего фосфора. Раствор А, содержащий
осадок гуминовых кислот, тщательно «перемешивают и пипеткой немедленно
отбирают 10 мл суспензии, которую сливают в плоскодонные колбы на 50 мл,
ставят их на электрические плитки с закрытой спиралью и раствор выпари¬
вают досуха, избегая пересушивания остатка. После этого приливают 3—
4 мл НСЮ4 (30—72 %-ной) и смесь нагревают на электрических плитках до
полного обесцвечивания раствора. Окисленную вытяжку количественно
переносят водой в мерные колбы на 100 мл так, чтобы общий объем раствора не
превышал 40 мл, и осаждают Fe3+ (по Уоррену и Пью). В растворе после
осаждения Fe3+ определяют фосфор колориметрически. Получают Р0бщ-
Определение минерального фосфора. Раствор А вновь тща¬
тельно перемешивают и немедленно сливают примерно 40—60 мл раствора
Методы определения фосфора в почве
121
в конические *:ли плоскодонные колбы на 100 мл. Сюда же для полного
обесцвечивания раствора, находящегося над осадком гуминовых кислот,
добавляют меркой 0,15—0,5 г очищенного активированного угля. Смесь
перемешивают, оставляют стоять 15—20 мин. и фильтруют через плотный
фильтр (белая, синяя лента). 20—25 мл обесцвеченного раствора переносят
в мерные колбы на 100 мл, доливают дистиллированной водой до 30 мл и осаж¬
дают Fe3+. 10—40 мл раствора после осаждения Fe3+ сливают в мерные
колбы на 50 мл и определяют фосфор колориметрически. Получают Р мине¬
ральный. По разности между Р бщ и РМин. находят содержание Рорг*
Реактивы. 1. Концентрированная НС1 (уд. вес 1,17—1,19).
2. 30—72%-ная хлорная кислота.
3. 0,5 н. раствор NaOH.
4. Очищенный активированный уголь (см. метод Мачигина).
Ускоренный метод Стеварда
и Оадиса с применением ультразвука
(Steward, Oades, 1973)
Навеску почвы последовательно обрабатывают кислотой и щелочью при
комнатной температуре. Для ускорения диспергирования почвы используют
ультразвук.
В кислотно-щелочном экстракте определяют общий, минеральный и
органический фосфор. Метод позволяет извлекать до 80% от валового фосфора
почвы.
Ход анализа. 0,5—2,0 г почвы (сито d 1—2 мм) помещают в полиэтиле¬
новые пробирки емкостью 25—50 мл, приливают 15 мл 1 н. НС1, взбалтывают
1 час, центрифугируют (2—3 тыс. об./мин.), раствор сливают в мерные
колбы на 200—250 мл. К остатку почвы приливают 15 мл дистиллированной
воды, перемешивают, центрифугируют. Центрифугат добавляют к соляно¬
кислому экстракту. Почву повторно промывают 15 мл воды, центрифуги¬
руют, центрифугат добавляют к общему раствору. К остатку почвы приливают
6 мл 0,5 н. раствора NaOH, тщательно перемешивают и подвергают смесь
действию ультразвука в течение 3 мин., затем приливают 10 мл воды, пере¬
мешивают, центрифугируют. Прозрачный центрифугат добавляют к соляно¬
кислому экстракту. Оставшуюся в центрифужных пробирках почву дважды
промывают по 15 мл 0,5 н. NaOH с последующим центрифугированием сме¬
си. Все центрифугаты сливают в одну и ту же мерную колбу, доливают до
метки водой, перемешивают. Получают кислотно-щелочный экстракт (рас¬
твор А). Почву из пробирок выбрасывают. В растворе А определяют содержа¬
ние общего и минерального фосфора точно так же, как описано выше в методе
Мета, исключая процедуру осаждения железа. По разности между Р0бщ и
Рмин вычисляют содержание Рорг.
Реактивы. 1. 1 н. раствор НС1.
2. 0,5 н. раствор NaOH.
3. 30—50%-ная НС104.
4. Очишенный активированный уголь (см. метод Мачигина).
Метод прокаливания Сэндерса и Вильямса
(Saunders, Williams, 1955)
Прокаленную и исходную навески почвы обрабатывают 0,2 н. раствором
H2S04. По разности между содержанием фосфора в прокаленной навеске
(Робщ) и содержанием фосфора в исходной навеске (РМИн) определяют Р0рг. Ме¬
тод позволяет выделять 75—95% валового фосфора почвы.
122
К. Е. Гинзбург
Ход анализа. 1 г почвы (сито d 0,25 мм) помещают в фарфоровый тигель и
прокаливают 2—3 часа при температуре 500—550° С. После этого прокален¬
ную навеску количественно с 50 мл 0,2 н. раствора H2S04 переносят из тигля
в плоскодонную колбу емкостью 100 мл, закрывают пробкой, взбалтывают
2 часа, настаивают 16—18 час., фильтруют, получают раствор А.
Отдельную навеску почвы в 1 г помещают в плоскодонную колбу на
100 мл, приливают 50 мл 0,2 н. раствора H2S04 и обрабатывают точно так же,
как прокаленную навеску почвы; получают при этом раствор Б. В аликвотах
растворов А и Б после их предварительной нейтрализации определяют фос¬
фор колориметрически.
Органический фосфор вычисляют по разности между содержанием фосфора
в растворах А и Б.
Реактив. 0,2 н. раствор H2S04.
ОПРЕДЕЛЕНИЕ МИНЕРАЛЬНЫХ ФОРМ
ФОСФАТОВ ПОЧВЫ
В зависимости от генетического типа почв состав минеральных форм фос¬
фатов в них различен. В карбонатных почвах предполагают преимущественна
содержание различных форм фосфатов кальция (ди-, октакальцийфосфа-
тов и др.)» в слабокислых почвах — фосфатов кальция и полутораокисей
(гидроксил-, фторапатитов, варисцита и др.) в кислых почвах — фосфатов,
полутораокисей (варисцита, стренгита, баррандита и др.) Исследователями
было выделено и идентифицировано около 200 различных минеральных сое¬
динений фосфора, устойчивость которых зависит от различных почвенных ус¬
ловий, в частности от pH, активности различных катионов (особенно Са,
Mg, Al, Fe), примененных удобрений, известкования, гипсования, оро¬
шения и др. Поэтому одна и та же форма фосфорного соединения в различных
почвенных условиях может иметь различную ценность для питания расте¬
ний (Везер, 1962; Larsen, 1967).
Наглядное представление об основных формах минеральных соединений
фосфора в почвах дает рис. 2.
Для определения минеральных форм фосфатов у нас в стране широко ис¬
пользуют методы Чирикова (1939,1947), Чанга — Джексона (1957) и в послед¬
нее время метод Гинзбург — Лебедевой (1971). В табл. 1 приводится схема
выделения минеральных форм фосфора с помощью указанных трех методов.
Метод Чирикоеа (1947),
вариант Шконде (1952)
Отдельные навески почвы многократно обрабатываются различными
кислотами — слабой 0,006 н. Н2С03, более крепкой 0,5 н. СН3СООН и еще
более сильной 0,5 н. НС1 (или H2S04) до почти полного вытеснения фосфора
из почвы данным растворителем. В результате в раствор переходят отдельные
группы соединений почвенных фосфатов, различающихся соответственно по
растворимости и доступности растениям (см. табл. 1). Одновременно после
обработки почвы 0,5 н. раствором НС1 выделяют с помощью 3 н. раствора ам¬
миака фракцию органических фосфатов.
По разности между содержанием валового фосфора в почве и суммой из¬
влеченных органических и минеральных форм фосфора определяют содержа¬
ние группы труднорастворимых фосфатов почвы.
При применении метода Чирикова следует учесть, что он в значительной
степени недоизвлекает фракцию минеральных фосфатов на кислых ожелез-
ненных почвах (красноземы, желтоземы, пойменные ожелезненные почвы
и др.). На карбонатных почвах в III группу (0,5 н. НС1 вытяжка) попадают
Методы определения фосфора в почве
123
Отдельнь/е частицы
(отчасти усвояемые)
Отдельные фосфаты,
осажденные на повер¬
хности, включая хими¬
чески поглощенную
Р0Ч /А/ (большей час¬
тью усвояемые)
Окклюдированные фос¬
фаты (малоусвояемые)
Фосфаты кальция
(апатит f двухкаль-
циевь/и фоарот и др.)
СГ?
Фосфаты кальция, оса¬
жденные на карбонате
кальция
О
Фосфаты кальция, ок¬
клюдированные карбо¬
натом кальция, сила-
катами и т.п.
Фостаты алюминия
(варисцитообразныеf
вавеллитообразные и
т.п.)
Фосфаты алюминия ,
осажденнь/е на алюмо¬
силикатах или гиб -
бейте
Фосфаты алюминия,
окклюдированные окис¬
лами железа
Фосфаты железа
(стренгито образные,
дифренитообразнь/е
и т.п.)
восстановленно-рас -
творимые фосфаты
железа, окклюдирован¬
ные окислами железа
Фосфаты железа, оса¬
жденные на окислах
железа
Фосфаты алюминия-
железа (баррандито-
образные и т. о.)
Фосфаты алюминия,
железа и алюминия-
железа, окклюдиро -
ванные окислами же¬
леза
\gJJ
Рис. 2. Формы минеральных соединении фосфора и почве (Chang, Jackson, 1957)
труднорастворимые фосфаты кальция (типа октакальцийфосфатов, апатитов
и др.), что часто не учитывается исследователями и делаются ошибочные
выводы о накоплении в этих почвах фракции фосфатов полутораокисей.
Примечание. В оригинальном методе Чирикова (Хейфец, 1960, 1965) исходная
навеска почвы соответствовала 10 г, ее обрабатывали соответствующими кислотами по
250 мл, а далее промывали на фильтре до отсутствия в промывном растворе ионов Mg2+,
Са2+, Р043+ (обычно до объема ~1 л).
В варианте Шконде (Хейфец, 1965) навеску 2,5 г заливали также 25-кратным количе¬
ством соответствующих кислот (т. е. 62,5 мл), затем промывали ее точно до определенного
объема (250 мл) без контроля на полноту выделения ионов Mg, Са, Р04.
В обоих методах перед колориметрическим определением фосфора уксуснокислую и
солянокислую вытяжки предварительно выпаривали и окисляли, что значительно услож-
няло ход их анализа.
124
К. Е, Гинзбург
Таблица 1
Схема выделения минеральных форм фосфора из почвы с помощью различных методов
Ч и р и к о
Группа
фосфора
Растворители
Предполагаемые формы фосфора *
Чанг —
Фракции
фосфора
Растворители
II
III
IV
0,04—0,06 н.
н,со,
0,5 н. СН,СООН,
pH 2,5
0,5 н. HG1 или
H,S04, pH < 1
3,0 и. NH4OH
Фосфор в остатке
почвы
Фосфаты щелочных металлов и
NH4, кислые фосфаты Ca(Mg), часть
Mg,(P04)„ Са,(Р04), (преимущест¬
венно свежеосажденные)
Разноосновные фосфаты Са (типа
ди-, три-, октакальцийфосфатов и
др.) и А1Р04 — частично
Высокоосновные фосфаты Са типа
апатита (природного и вторичнооб¬
разованного), разноосновные
А1Р04, FeP04 Часть фосфорных
эфиров
Фосфоинозиты, нуклеиновые кис¬
лоты, нуклеопротеиды, фосфогуму-
совые комплексы, немного мине¬
ральных фосфатов — А1Р04, FeP04,
продукты гидролиза W фосфорных
эфиров
Фосфаты невыветрившихся минера¬
лов материнской породы, трудно-
гидролизуемые фосфогумусовые
комплексы
Рыхлосвязанные
фосфаты
Фосфаты
алюминия (А1—Р)
1 н. NH4C1
0,5 н NH.F,
pH 8,5
фосфаты
железа (Fe—Р)
Фосфаты
кальция (Са—Р)
0,1 н. NaOH
0,5 н. HtS04
В осста нов ленно-
растворимые
Fe-фосфаты
(окклюдирован¬
ные окислами
железа)
Окклюдирован¬
ные Al-фосфаты^
0,3 н
NaaCeH»07 -}-
+1М NaHCOs +
+ 1 в Na*S,04
0,5 н. NH4F,
pH 8,5
0,1 н. NaOH
Не рекомендуется применять на кислых субтропиче¬
ских почвах (красноземах, желтоземах, ожелезненных
пойменных почвах, на карбонатных черноземах).
Окклюдирован*
ные
Al-Fe-фосфаты
Фосфор в остатке почвы
Не рекомендуется применять
• Приводятся не по оригинальной схеме Чирикова, а с учетом современного состояния данного воп¬
роса.
Методы определения фосфора в почве
12 S
Джексон
Гинзбург — Лебедева
Предполагаемые формы фосфора
Фракции
фосфора
Растворители
Предполагаемые формы
фосфора
Часть воднорастворимых фосфатов
А1Р04 (типа варисцита, ва вел лита
и др.), кислые фосфаты Ca(Mg) —
значительно, Саа(Р04)2 — частич¬
но, Fe3 — Р — частично, органо-
фосфаты — частично
FePO* (типа стренгита, дифренита
и др.), А1—Р — переосажденный
в предыдущей вытяжке, органо¬
фосфаты — значительно
Разноосновные Са—Р (типа ди-,
три-, октакальцийфосфата, апати¬
та); А1—Р, Fe—Р, переосажденные
в предыдущих вытяжках
Труднорастворимые фосфаты Fe,
заключенные в почвенных агре¬
гатах или конкрециях, покрытых
пленками окислов железа, разру¬
шающихся при восстановлении их
дитионитом натрия в щелочной
среде в цятратной вытяжке; орга¬
нофосфаты
А1Р04
Al(Fe)PC>4 (типа баррандита), ор¬
ганофосфаты
Фосфаты невыветрившихся мине¬
ралов материнской породы, труд-
ногидро)шзуемые фосфогумусовые
комплексы
' на нейтральных, слабокислых
почвах
Фосфаты кальция
(Са—Р!)
Фосфаты кальция
(Са— Р1Х)
Фосфаты
алюминия
(А1-Р)
Фосфаты железа
(Fe-P)
Фосфаты кальция
(Са— Рщ)
1% (NH4)*S04 +
+ 0,25%
(NH4)jMo04,pH4,8
сн„соон +
+ CHsCOONH4 +
+ 0,25о/*
(NHJaMoO,,
pH 4,2
0,5 н. NH4F,
pH 8,5
0,1 н. NaOH
0,5 h. HtSO*
Фосфор в остатке почвы
Фосфаты щелочных метал¬
лов и аммония, кислые и
свежеосажденные фосфаты
Ca(Mg); Fe*+ — Р (типа ви¬
вианита) — частично
Разноосновные фосфаты
Ca(Mg) (преимущественно
вторичнообразованные типа
ди-, три-, октана л ьцийфос-
фатов и др.); часть фракции
Са—Pj, переосажденной из
предыдущей вытяжки (имеет
место только на карбонат¬
ных почвах); Fe*+—Р (типа
вивианита) — значительно
А1Р04 (типа варисцита, ва-
веллита и др.), часть органи¬
ческого фосфора
FeP04 (типа стренгита, диф-
френита и др.); часть фрак¬
ции А1—Р, переосажденной
из предыдущей вытяжки
(имеет место только на силь¬
но ожелезненных почвах —
красноземы, желтоземы н
др.); органофосфаты — зна¬
чительно
Высокоосновные фосфаты
кальция типа апатита (при¬
родные и вторичнообразо¬
ванные)
Фосфаты невыветрившихся
минералов материнской по¬
роды, трудногидролизуемые
фосфогумусовые комплексы
Не рекомендуется применять на торфяно-болотных почвах:
126
К. Е. Гинзбург
Ф. В. Чириковым (см. Хейфец, 1965) была предложена также довольно громоздкая
процедура обработки аммиачной вытяжки для определения IV группы фосфора (много¬
кратное выпаривание растворов с HN03, прокаливание в муфеле с KN03 и т. п.).
Э. И. Шконде (1952) для определения IV группы фосфора применял сжигание навески
почвы с Н202. В данном случае менее полно определяется фракция органофосфатов почвы,
а кроме того, требуется сложная процедура предварительной очистки перекиси водорода
от постоянно присутствующей в ее составе в качестве стабилизатора Н3Р04.
Нами проводилась методическая проработка метода Чирикова, в результате которой
предлагается менее громоздкий ход определения фосфора в различных вытяжках. В част¬
ности, для более точного и быстрого приливания реактивов (Н2С03, СН3СООН, НС1) мы
рекомендуем исходную навеску почвы брать 2 г (вместо 2,5 г) и обрабатывать ее, 50 мл
(вместо 62,5 мл) соответствующих растворов кислот с последующим доведением! общего
объема раствора точно до 200 мл вместо 250 мл, принятых в варианте Шконде.
Нами было также установлено, что уксуснокислую и солянокислую вытяжки перед
колориметрическим определением в них фосфора не требуется предварительно ни выпари¬
вать, ни окислять (Гинзбург, 1958). Предлагается также более простой ход обработки (окис¬
ление, обесцвечивание) аммиачной вытяжки перед колориметрическим определением в ней
•фосфора (Гинзбург, 1969; см. также метод Хейфец).
Ход анализа. 1. Углекислая вытяжка (I группа Р). 2 г почвы (сито d
0,25 мм) заливают 50 мл воды, насыщенной 0,05—0,06 н. С02. Взбалтывают
2 часа (или один час и настаивают 24 часа) и фильтруют в мерные колбы на
200 мл. Почву на фильтре промывают малыми порциями (по 10 мл) того же
растворителя точно до 200 мл, перемешивают (раствор А).
2. Уксуснокислая вытяжка (II группа Р). 2 г почвы (сито d 0,25 мм) об¬
рабатывают 50 мл 0,5 н. раствора СН3СООН точно так же, как в случае при¬
готовления углекислой вытяжки. Получают раствор Б.
3. Солянокислая вытяжка (III группа Р). 2 г почвы (сито d 0,25 мм)
заливают 50 мл 0,5 н. раствора НС1 или H2S04 и далее поступают точно так
же, как при приготовлении углекислой и уксуснокислой вытяжек. Полу¬
чают раствор В.
В аликвотах растворов А и Б определяют фосфор колориметрически. Так
же поступают с раствором В, но предварительно его нейтрализуют (по 13-
динитрофенолу или другому индикатору).
Если же в солянокислой вытяжке (раствор В), помимо фракции минераль¬
ного фосфора, требуется определить и органические фосфаты, то поступают
так, как описано в методах Хейфец при анализе 4 н. НС1 вытяжки.
4. Аммиачная вытяжка (IV группа Р). Остаток почвы, после промывания
его 0,5 н. НС1, переносят вместе с фильтром в плоскодонные колбочки на
100 мл, приливают к ним 50 мл 3,0 н. раствора NH4OH, энергично встряхи¬
вают, чтобы фильтры разорвались на части, затем закрывают перевернутыми
маленькими коническими колбами и ставят на 5 час. на водяную баню, на¬
гретую до 70—80° С. После этого почву с раствором количественно перено¬
сят на фильтр в мерные колбы на 200 мл, промывают тем же раствором амми¬
ака порциями по 10—15 мл точно до 200 мл и перемешивают. В полученном
растворе определяют общее f содержание фосфора и фосфор минеральных
соединений, по разности вычисляют содержание фракции органофосфатов в
почвах. Ход обработки аммиачной вытяжки (окисление, обесцвечивание ра¬
створов) рекомендуем проводить по Гинзбург (1969), см. также метод Хейфец.
Вычисление содержания групп фосфора по Чи¬
рик о в у. Фосфаты I группы определяют по содержанию фосфора в Н2С03
вытяжке.
Фосфаты II группы определяют по разности между содержанием фосфора
в уксуснокислой и углекислой вытяжках.
Фосфаты I + II групп соответствуют содержанию фосфора в уксусно¬
кислой вытяжке.
Методы определения фосфора в почве
127
Фосфаты III группы определяют по разности между содержанием фос¬
фора в 0,5 н. НС1 и 0,5 н. СН3СООН вытяжке.
Фосфаты IV группы. Сюда относится фракция органических фосфатов,
найденная в аммиачной вытяжке. Если имеются данные по содержанию ор¬
ганических фосфатов в солянокислой вытяжке, то их приплюсовывают.
Фосфаты V группы вычисляют по разности между содержанием валового
фосфора почвы и суммой фосфатов, перешедших в солянокислую и аммиачную
вытяжки.
Реактивы 1. 0,004—0,006 н. Н2С03. Приготавливают, пропуская ток С02 в гер¬
метической системе через воду, охлаждаемую льдом.
2. 0,5 н. раствор СН3СООН. 30 мл ледяной уксусной кислоты доводят дистиллирован¬
ной водой до 1 л.
3. 0,5 н. раствор НС1. 41 мл концентрированной НС1 (уд. вес 1,17—1,19) доводят дш>
тиллированной водой до 1 л.
Концентрации приготовленных кислот проверяют титрованием по фенолфталеину
соответствующим раствором щелочи, приготовленным из фикс нала. В случае необходимо¬
сти искомые концентрации растворов получают добавлением соответствующих количеств
воды, кислоты, щелочи.
4. 30-72%-ная НС104.
5. 3 н. (~5%-ный раствор) NH4OH (уд. вес 0,977). 216 мл крепкого аммиака разбав¬
ляют водой до 1 л, концентрацию проверяют по удельному весу.
Метод Чанга — Джексона
(Chang, Jackson, 1957),
вариант Аскинази, Гинзбург, Лебедевой (1963)
Метод основан на последовательной обработке одной навески почвы раз¬
личными растворителями, каждый из которых извлекает определенные
фракции минеральных фосфатов почвы (Са—Р, А1—Р, Fe—Р и др.; см. так¬
же табл. 1). Соотношение почва : раствор = 1 : 50.
П римечание. Выделение из почвы с помощью дитионит-цитратной вытяжки в
щелочной среде фракций восстановленно>растворимых и окклюдированных форм фосфатов
железа и алюминия является громоздкой аналитической процедурой, которая практиче¬
ски себя не оправдывает, так как в этот растворитель переходит и некоторая часть органи¬
ческих фосфатов, отделить которые от минеральных соединений фосфора не представляем
ся возможным. В результате данная фракция фактически представляет смесь труднорасм
воримых форм фосфора неопределенного химического состава и малопригодна^ для ха¬
рактеристики фосфатного режима щ>чв.
Следует также учесть, что во фторидную вытяжку наряду с А1—Р переходят и неко¬
торые соединения Са—Р типа моно-, дикальцийфосфатов и др. (см. табл. 1 и приготовле¬
ние фторидных вытяжек). Многие исследователи этот факт не учитывают и делают оши¬
бочные выводы о том, что основным источником питания растений фосфором] на различ¬
ных типах почв являются фосфаты алюминия.
В этом методе отмечается также значительное вторичное осаждение фосфора в по¬
следовательных почвенных вытяжках, поэтому каждая последующая вытяжка может быть
васорена фосфатами, извлеченными в предыдущей вытяжке. Все это следует учитывать
при интерпретации данных, получаемых по методу Чанга — Джексона.
Ход анализа. 1. Приготовление 1 н. NH4C1 вытяжки (рыхлосвязанные
фосфаты). 0,5 г почвы (сито d 0,25 мм) помещают в центрифужные пробирки
емкостью 40—50 мл, приливают 25 мл 1 н. раствора NH4C1, закрывают проб¬
ками, взбалтывают 30 мин. на ротаторе, центрифугируют (~10 мин. со ско¬
ростью 2—3 тыс. об/мин), прозрачный раствор сливают в плоскодонные кол¬
бы на 50—100 мл, затем 10—20 мл центрифугата берут в мерные колбы на
25—50 мл для колориметрического определения фосфора.
128
К. Е. Гинзбург
2. Приготовление 0,5 н. NH4F вытяжки (фракция А1—Р).
К остатку почвы в центрифужной пробирке приливают 25 мл 0,5 н. ра¬
створа NH4F с pH 8,5. Пробирки закрывают пробками, взбалтывают один час
и центрифугируют. Прозрачный центрифугат сливают в плоскодонные кол¬
бочки на 50 мл, сюда же добавляют для обесцвечивания вытяжки активиро¬
ванный уголь (0,15—0,25 г), перемешивают и оставляют стоять 15—20 мин.
Если раствор над осадком полностью не обесцветился, то добавляют еще пор¬
цию угля (~0,1 г), настаивают дополнительно 10 мин. и фильтруют через
плотный фильтр (белая, синяя лента) в полиэтиленовую или пропарафини-
рованную посуду.
Хранить в стеклянной посуде фтораммонийную вытяжку не рекомендует¬
ся, так как NH4F способен выщелачивать из стекла кремневую кислоту,
которая может повысить результаты колориметрического определения
фосфора в вытяжке.
5—20 мл обесцвеченной вытяжки сливают в мерные колбы на 50 мл, раз¬
бавляют водой примерно до 30 мл и добавляют 10 мл 0,8 М раствора Н3В03
для связывания иона фтора в боратный комплекс NH4[BF4]*. После[этого при¬
ливают реактивы для окрашивания фосфора по Труогу — Мейеру.
Шкалу образцовых растворов фосфата готовят с добавлением к ней 0,5 н.
NH4F в таком же количестве, какое берется для определения испытуемого
раствора; затем также приливают 10 мл Н3В03 и реактивы для окрашивания
фосфора (см. метод Труога — Мейера).
Для удаления механически задержанного раствора фторидной вытяж¬
ки к остатку почвы в центрифужной пробирке приливают 25 мл насыщенного
раствора NaCl, взбалтывают 15 мин., центрифугируют и раствор выбрасы¬
вают. Почву используют для приготовления щелочной вытяжки.
3. Приготовление 0,1 н. NaOH вытяжки (фракция Fe—Р).
К промытому остатку почвы приливают 25 мл 0,1 н. раствора NaOH,
взбалтывают 2 часа, настаивают 18—20 час., центрифугируют. Прозрач¬
ный центрифугат сливают в плоскодонные колбочки, приливают сюда же
10 капель (~0,5 мл) концентрированной H2S04 для коагуляции гуминовой
кислоты и активированный уголь (~ 0,15—0,25 г) для поглощения окраски
фульвокислот, смесь перемешивают, настаивают 10—20 мин., фильтруют
через плотный фильтр.
5—20 мл фильтрата сливают в мерные колбы на 50—100 мл, разбавляют
водой, нейтрализуют и определяют фосфор колориметрически. Остаток почвы
в пробирке промывают 25 мл NaCl, как описано выше во фторидной вытяжке.
4. Приготовление 0,5 н. H2S04 вытяжки (фракция Са—Р).
К промытому остатку почвы приливают 25 мл 0,5 н. H2S04, взбалтывают
1 час, центрифугируют и раствор сливают в плоскодонные колбочки.
5—20 мл прозрачного центрифугата сливают в мерные колбы на 50—100 мл,
нейтрализуют и определяют фосфор колориметрически.
В отдельных навесках определяют валовой и органический фосфор поч¬
вы (см. выше).
Реактивы. 1.1 н. раствор NH4C1. 53,5 г соли растворяют в дистиллированной
воде, доливают до 1 л, перемешивают.
2. 0,5 н. раствор NH4F. 18,5 г соли растворяют в дистиллированной воде, до¬
ливают до 1 л, перемешивают.
3. 0,5 н. раствор H2S04. 14 мл H2S04 (уд. вес 1,84) осторожно приливают к 700 мл
воды, охлаждают, доливают до 1 л, перемешивают.
4. Насыщенный раствор NaCl. 400 г в 1 л воды.
•* Это необходимо, так как F может образовывать с молибденом соединения различного
состава (оксифториды — MoOF, Mo02F2; триоксотрифтормолибдаты — (NH4)3Mo03F3 и
др.) и мешать образованию фосфорномолибденовой кислоты, а следовательно, и колори¬
метрическому определению фосфора (Реми, 1963).
Методы определения фосфора в почве
129
5. 0,8 М раствор Н3В03 — 50 г в 1 л горячей воды.
6. 0,1 н. раствор NaOH.
7. Активированный уголь (см. метод Мачигина).
Метод Гинзбург — Лебедевой (1971)
Навеску почвы последовательно обрабатывают различными растворите¬
лями (соотношение почва: раствор = 1 : 50), в результате сначала выделяют
фракцию наиболее растворимых форм фосфатов щелочных и щелочноземель¬
ных элементов (фракцию Са—Pi), затем фракцию менее растворимых форм
фосфатов Ca(Mg), преимущественно вторичнообразованных и фосфатов за¬
писных форм железа (фракцию Са—Ри). После этого выделяют фракции Al—Р,
Fe — Р и наконец фракцию высокоосновных труднорастворимых фосфатов
Са типа апатита (природного и вторичнообразованного) — фракцию Са — Рщ.
Таким образом, наиболее растворимые соединения фосфатов Ca(Mg)
выделяются до обработки почвы раствором NH4F, т. е. не попадают
во фракцию фосфатов алюминия, как это имеет место в методе Чанга —
Джексона. В данном методе выделяются три фракции фосфатов кальция, ко¬
торые различаются по своей основности и степени окристаллизованности,
а следовательно, по растворимости и доступности растениям, что позволяет
давать более обоснованную агрохимическую оценку фосфатного фонда иссле¬
дуемых почв.
Добавление молибдата аммония в первые две вытяжки значительно сни¬
жает переосаждение фосфора в последовательных почвенных вытяжках
вследствие связывания фосфора молибдатом в недиссоциируемую фосфорно¬
молибденовую гетерополикислоту, что позволяет более чисто и полно вы¬
делять отдельные формы фосфора из почвы.
Ход анализа. 1. Приготовление аммонийно-молибдатной вытяжки (фрак¬
ция Са—Pi).
0,5 г почвы (сито d 0,25 мм) помещают в центрифужные пробирки емко¬
стью 40—50 мл и приливают к ним 25 мл смеси 1 %-ного раствора (NH^SC^ +
+ 0,25 %-ного раствора (NH4)2 Мо04 с pH 4,8. Пробирки закрывают проб¬
ками, взбалтывают 15 мин. и центрифугируют (примерно 10 мин. со ско¬
ростью 2—3 тыс. об!мин.). Прозрачный раствор сливают через воронку с
фильтром** в плоскодонные колбы на 50—100 мл (раствор А). Остаток поч¬
вы в пробирке обрабатывают 2Ъ мл насыщенного раствора NaCl (для удале¬
ния из почвы механически задержанного раствора аммонийно-молибдатной
вытяжки), смесь взбалтывают в течение 15 мин. и центрифугируют. Центри-
фугат выбрасывают, а остаток почвы используют для получения следующей
ацетатно-молибдатной вытяжки.
Аликвотную часть раствора А берут для колориметрического определения
фосфора, но с учетом содержащегося в ней количества молибдата аммония
(подробнее см. ниже в методе Гинзбург и Артамоновой).
2. Приготовление ацетатно-молибдатной вытяжки (фракция Са—Ри) ***.
К промытому остатку почвы в центрифужных пробирках приливают
25 мл смеси CH3COONH4 + СН3СООН + 0,25 %(NH4)2Mo04, pH ~ 4,3,
взбалтывают 15 мин., центрифугируют. Прозрачный центрифугат сливают в
* Приводим простейшую формулу молибдата аммония (см. ниже метод Гинзбург и
Артамоновой).
** Это необходимо, чтобы очистить раствор от всплывших на поверхность жидкости
мертвых корней.
*** На сильнокарбонатных почвах аммонийно-молибдатную и ацетатно-молибдатную вы¬
тяжки (для избежания выбрасывания растворов от выделяющейся газообразной
СОа) готовят, не закрывая пробирки пробками, а осторожно, периодически переме¬
шивая их от руки в течение 30 мин.
130
К, Е. Гинзбург
плоскодонные колбочки (раствор Б). В аликвоте раствора Б определяют фос¬
фор колориметрически с учетом содержащегося в нем молибдата аммония
(см. метод Гинзбург и Артамоновой).
Остаток почвы промывают 25 мл раствора NaCl (см. выше) и используют
для приготовления следующей фторидной вытяжки.
3—5. Вытяжки 0,5 н. NH4F;0,1 н. NaOH и 0,5 н. H2S04 готовят точно так
же, как описано выше в методе Чанга — Джексона.
Реактивы 1. Смесь 1% (NH4)2S04+ 0,25% (NH4)2Mo04 (pH ~ 4,8). Приготов¬
ление см. ниже (метод Гинзбург и Артамоновой).
2. Смесь CH3COONH4 + СН3СООН + 0,25% (NH4)2Mo04, pH 4,2-4,3.
2а. Приготовление смеси CH3COONH4 + СН3СООН с pH 4,2—4,3. 30 мл ледяной ук¬
сусной кислоты вливают в мерную колбу на 1 л, содержащую около 800 мл дистиллирован¬
ной воды. Смесь перемешивают, добавляют 10 мл крепкого раствора аммиака,
вновь перемешивают и в небольшом стеклянном стаканчике измеряют pH стеклянным
электродом. После измерения pH раствор из стаканчика сливают в ту же мерную колбу.
Процедуру повторяют до доведения pH до нужной величины (~4,2). После этого раствор
доливают до метки водой, тщательно перемешивают. Этот исходный раствор смеси сохра¬
няется длительное время.
26. Смесь CH3COONH4 + СН3СООН + 0,25% (NH4)2Mo04 (pH 4,2—4,3). Для при¬
готовления указанной смеси в другую мерную колбу на 1 л сливают примерно 700 мл при¬
готовленного раствора CH3COONH4 + СН3СООН, всыпают 2,5 г соли (NH4)2Mo04 и
взбалтывают до полного растворения навески молибдата аммония. Можно смесь оставить
для растворения на ночь, после чего доливают колбу до метки этим же раствором
CH3COONH4 + СН3СООН (pH ~4,2), перемешивают. Реактив сохраняется в течение
двух недель.
3—5. Приготовление растворов 0,5 н. NH4F; 0,1 н. NaOH; 0,5 н. HaS04 описано выше
в методе Чанга — Джексона.
6. Активированный уголь (см. ниже метод Мачигина).
ОПРЕДЕЛЕНИЕ ПОДВИЖНЫХ СОЕДИНЕНИЙ ФОСФОРА В ПОЧВЕ
Действительное, эффективное плодородие почв в отношении фосфатов
определяется запасом растворимых, подвижных форм фосфора. К этой
группе относятся различные формы почвенных фосфатов, участвующих в ди¬
намическом равновесии между твердыми фазами почвы и ее раствором, т. е.
в процессах перехода фосфора из твердых фаз в раствор и обратно. Степень
доступности растениям запаса подвижных фосфатов зависит от химических,
физико-химических, физических свойств данного типа почвы, сезонной ди¬
намики ее водного, воздушного и теплового режимов, биологической актив¬
ности почв, биологических особенностей произрастающих растений и дру¬
гих факторов.
Согласно современным представлениям (Кук, 1970; Блэк, 1973), мине¬
ральные фосфаты почвы по степени их участия в фосфорном питании растений
можно подразделить на три большие группы, которые находятся между
собой в постоянном обмене и динамическом равновесии:
1. Фосфаты, осажденные или адсорбированные на поверхности твердых
фаз почвы, способные в определенных условиях, главным образом при на¬
рушении фосфатного равновесия между твердыми и жидкими фазами почвы
(при выносе фосфора растениями, внесении фосфорных удобрений и др.), пере¬
ходить путем самодиффузии в почвенный раствор. Эта фракция фосфора ха¬
рактеризует общее «количество», «запас» подвижных фосфатов почвы, ее фос¬
фатную «емкость». Это резерв почвенных фосфатов, рассчитанный на дли¬
тельное снабжение растений фосфором. Количественно данную группу
фосфора почвы определяют экстракцией разведенными кислотами и щело¬
чами, радиоизотопным, хроматографическим методами и др.
Методы определения фосфора в почве
131
2. Фосфаты почвенного раствора, полностью доступные растениям. Пред¬
полагают, что эта фракция фосфора почвы^ интенсивно используется расте¬
ниями в начальный период их роста и развития.
Очень важный показатель этой фракции фосфатов почвы — фактор «ин¬
тенсивности», который характеризует степень подвижности запаса раство¬
римых фосфатов почвы. Количественно этот показатель выражается или ве¬
личиной активной концентрации фосфат-ионов в слабосолевых или водных
вытяжках из почв, или энергией, необходимой для перехода фосфатов из
твердых фаз почвы в раствор (фосфатный потенциал почв), или коэффициен¬
том использования радиоизотопа фосфора растениями по методу Соколова
(1960а).
3. Труднорастворимые фосфаты, заключенные в минеральном скелете
почвы в первичных и вторичных минералах (окклюдированные гидратами
полутораокисей, карбонатами, гипсом и др.). Эта фракция почвенного фос¬
фора фактически не имеет практического значения для питания растений.
Определение количества фосфатов, доступных растениям, является одной
из важных проблем агрохимической науки. Для ее решения применяют раз¬
личные методы. Наиболее надежные из них — биологические, основанные
на результатах полевых и вегетационных опытов с растениями. Но этот спо¬
соб является дорогостоящим, трудоемким, и с его помощью не представляется
возможным оценивать фосфатный режим различных почв на большой терри¬
тории. Более дешевы и доступны химические методы, которыми широко
пользуются для оценки степени обеспеченности почв фосфором и прогнози¬
рования эффективности фосфорных удобрений. Данные содержания раство¬
римых, подвижных фосфатов, полученные химическими анализами, по аб¬
солютным значениям не совпадают с содержанием доступных, усвояемых
растениями фосфатов. Поэтому для характеристики химических методов
необходимо результаты анализов сопоставить с данными по эффективности
фосфорных удобрений, установленных в полевых опытах с растениями. На
основании таких сопоставлений определяются так называемые предельные
числа («лимиты», «индексы») для каждого химического метода, которые по¬
казывают степень нуждаемости почв в фосфорных удобрениях.
Определение запаса подвижных фосфатов
(фактор «емкости»)
Для определения запаса подвижных фосфатов почвы используют кис¬
лотные, щелочные, буферные растворители, анионно-обменные смолы, радио-
изотопный метод и др.
Применение различных экстрагентов для извлечения подвижных соеди¬
нений почвенных фосфатов получило обоснование при изучении их взаимо¬
действия с фосфатными препаратами и природными минералами в широком
диапазоне pH. Установлено, что переход в раствор фосфора из разнооснов¬
ных фосфатов кальция и полутораокисей имеет два максимума: при pH <
< 5,5—5,0 и pH —9,0—11,0 (Аскинази, 1949). Этот факт предполагает воз¬
можность использования на различных типах почв в равной степени как кис¬
лотных, так и щелочных вытяжек. Но вследствие ряда специфических поч¬
венных условий (содержание карбонатов, гумусовых веществ и др.) практи¬
чески это не получается. Так, при применении кислотных вытя&ек на карбо¬
натных почвах с невысоким содержанием в них карбонатов кислота может
растворять и значительную часть труднорастворимых соединений фосфатов*
кальция, магния, фактически недоступных растениям. При высоком содер¬
жании карбонатов часть кислоты расходуется на их нейтрализацию, что мо¬
жет привести к недоизвлечению растворимых почвенных фосфатов.
На кислых и слабокислых почвах можно было бы применять щелочные
вытяжки, но в данном случае наряду с подвижными фосфатами в раствор пе¬
132
К. Е. Гинзбург
реходят гумусовые вещества. Поэтому щелочные вытяжки часто бывают ок¬
рашены в темно-бурый цвет, и перед колориметрическим определением в них
фосфора их необходимо предварительно обесцвечивать (окислять, обраба¬
тывать активными углями и т. п.), что усложняет ход анализа и снижает его
точность.
Водные и слабосолевые вытяжки являются универсальными раствори¬
телями, пригодными для определения подвижных фосфатов на разных ти¬
пах почв, однако использование их для массовых определений представ¬
ляет методическую трудность из-за того, что почти невозможно получить про¬
зрачный фильтрат водной вытяжки. Кроме того, концентрация фосфора в
этих вытяжках бывает очень низкая и часто требуется сгущение исследуе¬
мых растворов перед колориметрическим определением в них фосфора,
что также увеличивает продолжительность хода анализа и снижает его
точность.
Существенным недостатком почти всех предложенных методов опреде¬
ления подвижных фосфатов в почве является вторичное осаждение (пере-
осаждение) растворившихся в данном экстрагенте фосфатов компонентами
твердых фаз почвы и раствора, в результате в вытяжке определяется только
часть экстрагируемого фосфора.
Метод изотопного разведения по Соколову (1960а) позволяет учесть рас¬
пределение метки Р32 между раствором и почвой. Это дает возможность уста¬
новить общее количество фосфатов, способное переходить в данный раство¬
ритель из почвы, т. .е. определить «запас» подвижных фосфатов почвы.
Другой способ учета вторичного поглощения фосфора при приготовле¬
нии почвенных вытяжек — это удаление фосфора из равновесной системы:
твердая фаза — раствор. Сюда относится применение анионно-обменных
смол для поглощения фосфора, переходящего в раствор, или связывание
фосфора в комплексные, недиссоциированные соединения с помощью молибда-
та аммония, ванадата-молибдата аммония и др. (Гинзбург, Артамонова, 1966,
1968).
Таким образом, для определения подвижных фосфатов на кислых, слабо¬
кислых почвах, как правило, применяют кислотные вытяжки и различные
буферные смеси с исходным pH, колеблющимся в пределах — 1—5, а на кар¬
бонатных почвах — буферные смеси с pH 3,2—5,0 и щелочные вытяжки с
pH 8,5-11.
Водные, слабосолевые вытяжки, анионообменные смолы, радиоизотоп-
ный методы применяются на различных типах почв (кислых, слабокислых,
нейтральных, карбонатных).
Кислотные вытяжки
Метод Кирсанова (1935)
Метод основан на извлечении подвижных фосфатов и калия из почвы 0,2 н.
раствором НС1 (pH < 1) при соотношении почва : кислота = 1:5, взбалты¬
вании 1 мин., настаивании 15 мин., температуре 18—20° С.
Метод принят стандартным для почв нечерноземной зоны («Инструкция
по проведению массовых анализов почв.. .», 1973).
Индексы обеспеченности по фосфору даны в табл. 2.
Ход анализа. 10 г воздушно-сухой почвы (сито d 1—2 мм) помещают в
плоскодонные колбы на 100 мл и заливают 50 мл 0,2 н. НС1, перемешивают
1 мин. (желательно с помощью электромеханической мешалки), настаивают
15 мин., фильтруют. 2 мл прозрачного фильтрата помещают в мерные колбы на
50 мл (или 5 мл в мерные колбы на 100 мл) и определяют фосфор колориметри¬
чески. В оставшейся части фильтрата определяют калий на пламенном фо¬
тометре.
Методы определения фосфора в почве
133
Таблица 2
Обеспеченность почв подвижными фосфатами по содержанию их в вытяжке Кирсанова
Обеспеченность
Р*Ов, мг на 100 г почвы
зерновые, зернобобовые
корнеплоды, картофель
овощные, технические
культуры
Очень низкая
<3
<8
<15
Низкая
<8
<15
<20
Средняя
ю
1
ОС
15—20
20—30
Высокая
>15
>20
>30
Для наиболее полного извлечения из почвы остаточных фосфатов удоб¬
рений рекомендуют применять 0,2 н. НС1 вытяжку с широким соотношением
почва: кислота (1 : 100) и более продолжительным временем взаимодействия:
5 мин. взбалтывание, 18—20 час. настаивание в термостате при температуре
25 + 1° С (Похлебкина, 1973).
Автором метода установлено, что данные, полученные по предлагаемой
ею прописи, и запас подвижных фосфатов, вычисленный по методу Соколова
(1960а) с помощью метки Р32, в этой же вытяжке дают близкие абсолютные
значения.
Реактив. 0,2 ± 0,01 н. НС1. 16,4 мл НС1 (уд. вес 1,19) доливают водой до 1 лч
перемешивают. Концентрацию кислоты проверяют титрованием.
Мепод Чирикова (1947)
Метод основан на извлечении подвижного фосфора и калия из почвы 0,5 н.
раствором уксусной кислоты (pH 2,5) при температуре 18—20° С и соотно¬
шении почва : раствор = 1 : 25.
Метод принят стандартным для почв черноземной зоны — некарбонатных
черноземов («Инструкция по проведению массовых анализов почв...», 1973).
Индексы обеспеченности по фосфору представлены в табл. 3.
Таблица 3
Обеспеченность почв подвижными фосфатами по содержанию их в вытяжке Чирикова
Обеспеченность
Р*Ов, мг на 100 г почвы
зерновые, зернобобовые
корнеплоды, картофель
овощные, технические
культуры
Очень низкая
<2
<5
<10
Низкая
<5
<10
<15
Средняя
5-10
10—15
15—20
Высокая
>10
>15
>20
Ход анализа. 4 г воздушно-сухой почвы (сито d 1—2 мм) помещают в пло¬
скодонные колбы емкостью 200—250 мл, приливают 100 мл 0,5 н. СН3СООН,
взбалтывают 1 час, настаивают 18—20 час. (или 2 часа взбалтывают без на¬
стаивания) и фильтруют. 5—40 мл прозрачного фильтрата переносят в мер¬
ные колбы на 50—100 мл, разбавляют водой и приливают реактивы для ко¬
лориметрического определения фосфора. В оставшейся части фильтрата оп¬
134
К. Е. Гинзбург
ределяют калий на пламенном фотометре. Уксуснокислую вытяжку перед
колориметрическим определением в ней фосфора не требуется ни нейтрали¬
зовать, ни выпаривать, ни окислять (Гинзбург, 1958).
Реактив. 0,5 н. раствор СН3СООН (см. выше метод Чирикова, вариант Шконде).
Метод Труога (1930)
Метод основан на извлечении подвижного фосфора и калия из почвы
0,002 н. раствором H2S04, забуференным сернокислым аммонием до pH 3,0,
при температуре 18—20° С. Соотношение почва: раствор = 1 : 200, взбал¬
тывание в течение 30 мин.
Метод в настоящее время используется при анализах некоторых черно¬
земных почв Сибири и Молдавии.
Индексы обеспеченности по фосфору представлены в табл. 4.
Таблица 4
Обеспеченность почв подвижными фосфатами по содержанию их в вытяжке Труога
Обеспеченность
PtO„ мг на 100 г почвы
зерновые, зернобобовые
корнеплоды, картофель
овощные, технические
культуры
Очень низкая
<3
<7
<12
Низкая
<7
<12
<18
Средняя
7-12
12—18
18—25
Высокая
>12
>18
>25
X од анализа. 1 г воздушно-сухой почвы (сито d 1—2 мм) помещают в пло
скодонные колбы емкостью 500 мл, приливают 200 мл 0,002 н. H2S04 +
+ (NH4)2S04, pH 3,0, взбалтывают 30 мин., фильтруют, отбрасывая первые
порции фильтрата. 20—40 мл прозрачного фильтрата помещают в мерные
колбы на 50 мл (или 25—50 мл в мерные колбы на 100 мл), раствор разбав¬
ляют водой, перемешивают и приливают реактивы для окрашивания фос¬
фора. В оставшейся части фильтрата определяют калий на пламенном фо¬
тометре.
Реактив. 0,002 н. раствор H2S04 с pH 3,0. 20 мл точно установленного 0,1 н. раст¬
вора H2S04 (лучше всего приготовленного из фиксанала) разбавляют дистиллированной
водой примерно до 700—800 мл, прибавляют 3,0 а сернокислого аммония, перемешивают
до полного растворения соли, затем доливают до 1 л дистиллированной водой и вновь
перемешивают.
Метод Аррениуса (Arrhenius, 1933),
вариант Гинзбург (1952)
Метод основан на извлечении подвижного фосфора из почвы 1 %-ным ра¬
створом лимонной кислоты, pH 2,3, отношение почвы к раствору составляет
1 2 10, взбалтывание 4 часа, настаивание в течение 18—20 час. при темпе¬
ратуре 25 + 2° С.
Метод используется для определения подвижных фосфатов в субтропи¬
ческих почвах (красноземах, желтоземах) и некарбонатных горных поч¬
вах Армении.
Индексы обеспеченности по фосфору приведены в табл. 5.
Ход анализа. 5 г воздушно-сухой почвы (сито d 1—2 мм) помещают в
плоскодонные колбы на 100 мл, заливают 50 мл 1 %-ного раствора лимонной
Методы определения фосфора в почве
435
Таблица 5
Обеспеченность почв подвижными фосфатами ио содержанию их в 1%-ной лимоннокислой
вытяжке по Аррениусу — Гинзбург и 0,1 н. H2SO4 вытяжке по Ониани
Обеспеченность
Р2Ов, мг на 100 г почвы
зерновые, чай
корнеплоды
овощные культуры
Очень низкая
<8
<15
<30
Низкая
<15
<30
<45
Средняя
15-30
30-45
45—60
Высокая
>30
>45
>60
кислоты, взбалтывают 4 часа и настаивают в течение 18—20 час. при тем¬
пературе 25 +2° С (желательно настаивание проводить в термостате), затем
суспензию фильтруют, отбрасывая первые порции фильтрата.
10 мл прозрачного фильтрата переносят в плоскодонные колбы на 50—
100 мл и окисляют лимонную кислоту, которая мешает колориметрическому
определению фосфора. Для этого добавляют 3 мл концентрированной НС1
и 6 мл 5 %-ного раствора КМп04, перемешивают, настаивают 30 мин., затем
ставят на горячую этернитовую плитку, перемешивая время от времени.
Нагревание следует вести до полного исчезновения бурого осадка перекиси
марганца. После охлаждения окисленную вытяжку переносят в мерную
колбу на 50—100 мл, доливают до метки водой, перемешивают (раствор А).
5—25 мл раствора А переносят в другие мерные колбы на 50 мл, добавляют
2 капли индикатора p-динитрофенола и осторожно титруют до желтой окрас¬
ки 15—20%-ным раствором NH4OH; желтую окраску уничтожают добавле¬
нием 2—3 капель 10 %-ного раствора H2S04(HC1). После этого определяют
фосфор колориметрически.
Лимоннокислую вытяжку • можно окислять раствором КМп04 в серно¬
кислой среде точно так же, как это описано ниже в методе Мачигина. В дан¬
ном случае не требуется последующей нейтрализации окисленной вытяжки.
Метод Аррениуса (Arrhenius, 1933),
вариант ВИУА (см. Хейфец, 1960)
Принцип определения и ход анализа точно такие же, как описано выше
в варианте Гинзбург. Различие состоит в том, что лимоннокислую вытяжку
перед колориметрическим определением в ней фосфора не окисляют. Для
восстановления фосфорномолибденовой кислоты используют смесь восста¬
новителей сульфита натрия и гидрохинона. Этот способ анализа лимонно¬
кислой вытяжки в настоящее время принят стандартным при определении
подвижных соединений фосфора в некарбонатных горных почвах Армении
(«Инструкция по проведению массовых анализов почв...», 1973).
Ход анализа. 6 мл прозрачного фильтрата лимоннокислой вытяжки (при¬
готовление см. выше) переносят в узкогорлые конические колбы емкостью
50 мл и приливают к ним 24 мл окрашивающего раствора. Колбы закрывают
пробками и ставят в термостат на 6 час. при температуре 55 ±’2° С для раз¬
вития окраски. На следующий день окрашенные растворы колориметрируют
на электрофотоколориметре с красным светофильтром.
Реактивы. 1. 1%-ный раствор лимонной кислоты — Н3(СвНБ07)-Н20.
2. НС1 концентрированная (уд. вес 1,19—1,17).
3. 5%-ный раствор КМп04.
136
К. Е. Гинзбург
4. Сульфатмолибденовый реактив. 25 г молибденовокислого аммония (ГОСТ 3765-64,
X, ч. или ч. д. а) растворяют в 825 мл горячей дистиллированной воды, после охлаждения
в раствор небольшими порциями прибавляют 175 мл H2S04 (уд. вес 1,83—1,84). Реактив
сохраняется длительное время в склянке из темного стекла.
5. Раствор гидрохийона. 0,25 г гидрохинона (по ГОСТ 2549-60) растворяют в 80 мл
дистиллированной воды в мерной колбе емкостью 100 мл, В раствор прибавляют 1—2 кап»
ли концентрированной H2S04, доливают до метки дистиллированной водой, перемешивают*
Раствор готовят ежедневно.
6. Раствор сульфита натрия. 20 г сульфита натрия (кристаллического, ГОСТ 429-66,
ч. д. а.) растворяют в 80 мл дистиллированной воды и фильтруют через фильтр, не со¬
держащий фосфора.
7. Приготовление окрашивающего раствора (смесь комплексообразователя и восста¬
новителя). Смешивают 300 мл сульфатмолибдена, 18 мл гидрохинона и 18 мл раствора
сульфита натрия с 2064 мл дистиллированной воды. Окрашивающий раствор готовят в
день проведения анализа.
Метод Ониани (1964)
Метод основан на извлечении подвижных соединений фосфора и калия из
почвы 0,1 н. серной кислотой, pH < 1, при отношении почвы к раствору
1 I 25 и кратком времени взаимодействия.
С 1968 г. метод принят стандартным для почв субтропической зоны
(красноземов, желтоземов).
Индексы обеспеченности по фосфору см. в табл. 5.
Ход анализа. 4 г воздушно-сухой почвы (сито d 1—2 мм) помещают в пло¬
скодонные колбы емкостью 200—250 мл, приливают 100 мл 0,1 н. раствора
HaS04, перемешивают 1 мин. (желательно электромеханической пропеллер¬
ной мешалкой) и фильтруют.
2—5 мл прозрачного фильтрата помещают в мерные колбы на 50 мл (или
5—10 мл берут в мерные колбы на 100 мл), разбавляют 20—30 мл дистилли¬
рованной воды и приливают реактивы для окрашивания фосфора. В остав¬
шейся части фильтрата определяют калий на пламенном фотометре.
Определение подвижных фосфатов
в торфяно-болотных почвах
(Вельский, Кулаковская, Розина, 1961)
Метод основан на извлечении подвижных фосфатов из торфяно-болотных
почв с помощью 0,2 н. раствора НС1 при соотношении почва : раствор = 1:50.
Авторы провели сравнение методов извлечения фосфора 1 %-ной лимон¬
ной кислотой, 0,2 н. НС1 и 0,05. н. НС1 и радиоизотопным методом по
А. В. Соколову, который был принят как эталон. В результате установлено,
что 0,2 н. НС1 давала наилучшую корреляцию с радиоизотопным методом
для слабо- и среднеокультуренных торфяно-болотных почв.
Ход анализа. 2 г воздушно-сухой почвы (сито d 1—2 мм) помещают в пло¬
скодонные колбы емкостью 250 мл, приливают 100 мл 0,2 н. раствора НС1,
взбалтывают один час, настаивают 16—18 час., фильтруют, сливая первые
порции фильтрата.
Фильтрат часто бывает окрашен гумусовыми веществами и содержит ио¬
ны Fe3+ в количествах, мешающих колориметрическому определению фос¬
фора. Поэтому перед колориметрированием испытуемых растворов их необ¬
ходимо предварительно окислить и удалить Fe3+.
В целях ускорения обесцвечивания данной вытяжки мы предлагаем много¬
кратное выпаривание ее на водяной бане с царской водкой и НС1 (как это было
предложено авторами прописи) заменить на окисление хлорной кислотой. Для
этого берут 50 мл фильтрата вытяжки, сливают в 100-миллилитровые плоско¬
Методы определения фосфора в почве
137
донные колбы жаростойкого стекла, выпаривают на электрической плитке
с закрытой спиралью досуха, избегая пересушивания. К сухому остатку
приливают 3—5 мл 50%-ной НС104 и нагревают смесь на электрической плит¬
ке до полного ее обесцвечивания. Раствор охлаждают, переносят количе¬
ственно водой в мерные колбы на 100 мл так, чтобы общий объем раствора не
превышал 40 мл, и приливают реактивы для осаждения Fe3+.
5—40 мл прозрачного фильтрата после осаждения Fe3+ сливают в мер¬
ные колбы на 50—100 мл и определяют фосфор колориметрически.
Реактивы. 1. 0,2 н. НС1 (см. выше метод Кирсанова).
2. Хлорная кислота, 30—72%-ная.
Авторы предлагают следующие индексы обеспеченности по фосфору для
торфяно-болотных почв (в мг Р2Об на 100 г абсолютно сухого торфа):
< 20 мг — низкая обеспеченность, 20—40 — средняя, 40—60 — выше сред¬
ней, ]> 60 мг — высокая обеспеченность.
Буферные растворители
Метод Эгнера — Рима
(Egner et al., 1938, I960), или D—L метод1
Метод основан на извлечении из почвы подвижных соединений фосфора и
калия 0,04 н. раствором молочнокислого кальция, забуференного соляной
кислотой до pH 3,5—3,7, при соотношении почвы к раствору 1 : 50.
Метод принят стандартным для почв республик Прибалтики.
Ход анализа. 4 г воздушно-сухой почвы (сито d 1—2 мм) всыпают в плоско¬
донные колбы емкостью 250—300 мл, приливают 200 мл рабочего лактат-
ного раствора, взбалтывают на ротаторе в течение 1,5 час. и суспензию либо
фильтруют, либо отстаивают в течение 18—20 час.
Определение фосфора. 25 мл прозрачного фильтрата или от¬
стоя лактатной вытяжки сливают в плоскодонные колбы емкостью 50 мл, при¬
ливают 2 мл окрашивающей смеси, перемешивают и добавляют 1 мл раство¬
ра SnCl2, снова перемешивают и колориметрируют окрашенные растворы не
ранее чем через 30 мин. и не позднее чем через 3 часа после прибавления
раствора SnCl2.
Если в почве много фосфора и в колбу для определения берут меньше
25 мл вытяжки, то дополнительно вносят недостающее до 25 мл количество
рабочего лактатного раствора. Общий объем колориметрируемой жидкости
должен быть 28 мл. Колориметрируемую жидкость водой разбавлять нельзя.
При приготовлении шкалы образцовых растворов следует к точно изме¬
ренному количеству образцового рабочего раствора добавить такое количе¬
ство отфильтрованного рабочего лактатного раствора, чтобы общий объем
равнялся объему взятого для сравнения испытуемого раствора, и произвести
окрашивание точно так же, как это описано выше для определения фосфора
в фильтрате почвенной вытяжки.
Колориметрическое определение фосфора в лактатных вытяжках можно
проводить по методу Труога — Мейера с одним восстановителем — SnCl2,
но при этом лактатную вытяжку следует разбавить водой в 2—4 раза для
устранения влияния лактатного раствора на образование фосфорномолиб¬
деновой кислоты. Необходимо снизить также концентрацию фосфора в ко-
1 По оригинальному методу Эгнера, концентрация исходного раствора лактата кальция
была принята 0,02 н. Римом концентрация лактата кальция была увеличена в 2 раза,
соответственно было увеличено количество добавляемой соляной кислоты/для заоуфе-
рения этого раствора до pH 3,5—3,7, как это было принято в методе Эгнера. В настоя¬
щее время этот метод известен как двойной лактатный метод Эгнера — Рима — D —
(Double Lactate) метод.
138
К. Е. Гинзбург
лориметрируемом растворе, чтобы она не превышала 2 мг Р2Об на 1 л (Замя¬
тина, 1969). Получаемая в данном случае синяя окраска устойчива в тече¬
ние 15—30 мин.
Определение калия. 15 мл фильтрата или отстоя лактатной
вытяжки помещают в стаканчики на 25—50 мл или в пенициллиновые бу¬
тылочки, приливают 3 мл раствора щавелевокислого аммония и оставляют
на 16—18 час. На следующий день осторожно декантируют раствор в другие
стаканы и определяют калий на пламенном фотометре.
Шкалу образцовых растворов калия готовят с добавлением лактата
кальция в такой же концентрации, что и в испытуемых растворах. Осажде¬
ние кальция проводят так же, как и в испытуемых растворах.
Реактивы. 1. Экстрагирующая смесь — смесь лактата кальция и НС1. 1а. За¬
пасный раствор. 120 а лактата кальция (СН3СН0НС00)2Са*5Н20 (по ГОСТ 6-09-3380-67,
х. ч.) растворяют в 700—800 мл кипящей воды; к теплому раствору прибавляют 80 мл
5 н. раствора НС1 и после охлаждения доливают дистиллированной водой до 1 л, переме¬
шивают. Этот запасный раствор имеет pH 3,2. К нему прибавляют несколько капель кон¬
сервирующей жидкости (хлороформа ксилола, формалина). В темной склянке в холодиль¬
нике раствор сохраняется 7—14 дней.
16. Рабочий раствор служит для получения вытяжек. Его готовят разбавлением раст¬
вора 1а в 20 раз. Для этого 50 мл раствора 1а разбавляют водой до 1 л. Значение pH раст¬
вора 3,6 ±0,1. Рабочий раствор готовят в день приготовления вытяжки, он не должен
содержать фосфора и калия.
2. Реактив для восстановления. 2а. Запасный раствор. 2 а метола, 10 г безводного
сульфита натрия (Na2S08) и 300 г сухого метабисульфита натрия (Na2S206) растворяют при
слабом нагревании примерно в 800 мл дистиллированной воды. После охлаждения раст¬
вор доводят водой до 1 л. Мутный раствор употребляют без фильтрования, так как муть
быстро отстаивается. В склянке из темного стекла раствор сохраняется длительное время.
26. Рабочий раствор для восстановления. Смешивают один объем раствора 2а и три
объема дистиллированной воды. Смесь готовят в день определения фосфора.
3. Молибденовый реактив. За. Запасный раствор. 50 г молибденовокислого аммония
(ГОСТ 3765-64, х. ч. или ч. д. а.) растворяют при нагревании в 400 мл дистиллированной
воды. Раствор охлаждают и приливают его небольшими порциями при помешивании
в 500 мл 10 н. раствора H8S04, смесь охлаждают, доливают до метки водой, перемешивают
и хранят в склянке из темного стекла. Раствор сохраняется длительное время.
36. Рабочий раствор. Один объем запасного раствора За смешивают с одним объемом
дистиллированной воды. Раствор устойчив в течение 5—6 месяцев.
4. Молибденово-восстанавливающая смесь. Один объем раствора 26 смешивают с од¬
ним объемом раствора 36. Смесь готовят перед употреблением.
5. Двухлористое олово. 0,1 г SnCl2 (кристаллического) всыпают в мерную колбу на
50 мл, растворяют в 1,Ъ мл 10 н. НС1 (уд. вес 1,16), доливают водой до метки, перемеши¬
вают. Если SnCl8 плохо растворяется, то подогревают раствор на водяной бане, охлаж¬
дают его и доливают до метки водой.
6* Образцовый раствор можно приготовить общий для определения фосфора и калия*
ба. Запасный раствор. 0,1917 а КН2Р04 (ГОСТ 4198-65, х. ч.) и 0,0534 а КС1 (ГОСТ
4234-69, х. ч.) растворяют в 800 мл воды и доливают затем водой до 1 л, для сохранения
к раствору добавляют несколько капель формалина или другого антисептика. Раствор
содержит в 1 мл 0,1 мг Р206 и 0,1 мг К20 и может сохраняться в течение 1—3 месяцев.
бб. Рабочий раствор. 100 мл запасного образцового раствора (6а) и 50 мл отфильтро.
ванного запасного раствора лактата кальция (1а) доводят водой до 1 л. Полученный ра¬
бочий раствор содержит 0,01 мг Р206 и 0,01 мг К20 в 1 мл; он неустойчив, его надо гото¬
вить перед употреблением.
Образцовые растворы для фосфора и калия можно готовить и раздельно. В этом слу¬
чае исходная навеска для приготовления образцового раствора фосфата остается такой
же, т. е. 0,1917 г/л КН8Р04, а для образцовых растворов калия навеску берут 1,583 г/л
Методы определения фосфора в почве
139
КС1 (х. я.)* Полученный раствор содержит 1 мг К20 в 1 мл и служит исходным для после¬
дующего разбавления.
7. 10 н. раствор H2S04 (уд. вес 1,29). 280 мл концентрированной H2S04 (ГОСТ 4204-66,
уд. вес 1,83—1,84, х. ч.) осторожно добавляют к 600 мл дистиллированной воды, раствор
охлаждают и доливают до 1 л водой, перемешивают.
8. Раствор щавелевокислого аммония. 120 a (NH4)2C204 (ГОСТ 5712-67, х. ч. или
ч. д. а.) всыпают в колбу емкостью Зли растворяют примерно в 1200 мл дистиллированной
воды при нагревании, затем добавляют 135 мл крепкого аммиака (ГОСТ 3760-64, ч. д. а.),
смесь перемешивают, охлаждают и доливают водой до 3 л, перемешивают. Раствор сохра¬
няется длительное время.
Эгнер и Рим считают лактатный метод универсальным и рекомендуют его
для анализа кислых (pH до 5,5) и нейтральных почв легкого и тяжелого ме¬
ханического состава. Индексы обеспеченности по фосфору для D — L метода
приведены в табл. 6.
Таблица 6
Обеспеченность почв фосфатами по данным лактатного метода Эгнера — Рима,
мг РгОб на 100 г почвы
Обеспечен¬
ность
Легкие почвы при pH
Суглинистые почвы при pH
Тяжелые почвы при pH
до 5,5
5,6—6,5
6.6
в более
до 5,5
5,6-6,5
6,6
и более
до 5,5
5,6-6,5
и более
Плохая
<7,0
<8,5
<10,0
<5,0
<6,0
<7,5
<4,0
<5,0
<6,0
Средняя
<14,0
<17,0
<20,0
<10,0
<12,0
<15,0
<8,0
<10,0
<12,0
Хорошая
>14,0
>17,0
>20,0
>10,0
>12,0
>15,0
>8,0
>10,0
>12,0
Метод Эгнера — Рима — Доминго
(Egner, Riehm, Domingo, 1958), или A — L метод
Метод основан на извлечении из почвы подвижных соединений фосфора и
калия смесью молочной и уксусной кислот, забуференных уксуснокислым ам¬
монием до pH 3,7. Полученная лактатно-ацетатно-аммонийная смесь — А — L
— является 0,1 н. по лактату аммония и 0,4 н. по уксусной кислоте.
Соотношение почвы : раствор = 1 : 20, взбалтывание 4 часа при температуре
20 ± 1° С. Метод используется для определения подвижных соединений
фосфора и калия в почвах республик Прибалтики.
А — L метод позволяет одновременно с фосфором определять в вытяжке
не только калий, но и другие катионы (кальций, магний и бор).
Введение лактата аммония вместо лактата кальция (при этом в вытяжку
не вводится нежелательный ион Са) дает возможность применять этот извле¬
кающий раствор и Для почв, содержащих более 4% СаС03, без резкого сниже¬
ния pH равновесного раствора после его взаимодействия с карбонатной
почвой. В случае почв, содержащих свыше 20% СаС03, рекомендуется пов¬
торное извлечение.
Ход анализа. 5 г воздушно-сухой почвы (сито d 1—2 мм) помещают в пло¬
скодонные колбы емкостью 200—250 мл, приливают 100 мл А — L смеси с
pH 3,7, взбалтывают 4 часа при температуре 20 ± 1° С и фильтруют суспен¬
зию через двойной фильтр, не содержащий фосфора и калия.
Определение фосфора. 10 лед прозрачного фильтрата поме¬
щают в плоскодонные колбы емкостью 50 мл, приливают 15 мл дистиллиро¬
ванной воды, 2 мл молибденово-восстанавливающей смеси и i мл двухлори¬
стого олова (см. выше D — L метод), раствор перемешивают и не ранее чем
через 1 час просматривают в фотоколориметре с красным светофильтром.
140
К. Е. Гинзбург
Определение калия для карбонатных почв проводят точно так
же, как описано выше в D — L методе, для других почв предварительное
осаждение ионов кальция не требуется.
Реактивы. 1. Экстрагирующая смесь — раствор аммоний-лактат-уксуснокислый
(А—L смесь). 1а. Крепкий раствор молочной кислоты. 1 кг молочной кислоты (ГОСТ
6-09-3198-66, х. ч.) заливают 2 л дистиллированной воды. С целью гидратации этот раст¬
вор в закрытой колбе настаивают 48 час. при температуре 95° С в термостате. Раствор ох¬
лаждают, отбирают 5 мл в мерную колбу на 100 мл, доливают дистиллированной водой до
метки, перемешивают. 10 мл (разбавленного в 20 раз) раствора молочной кислоты пере¬
носят в конические колбы на 100 мл, приливают несколько капель индикатора^фенолфта-
леина и титруют 0,1 н. титрованным раствором NaOH до появления розовой окраски. Та¬
ким образом устанавливают концентрацию гидратированного раствора молочной кислоты,
она должна быть примерно 3,0 н.
Для того чтобы определить необходимое количество молочной кислоты для приготовле¬
ния А—L смеси, делают соответствующие расчеты.
Пример. Для приготовления 10 л запасного A—L раствора требуется 10 же мо¬
лочной кислоты. Если раствор крепкой молочной кислоты оказался 3,2 н., то 10 же ее
будут содержаться в 10000:3,2 = 3125 мл молочной кислоты.
16. Запасный раствор A—L смеси. К 3125 мл молочной кислоты добавляют 1795 мл
96%-ной уксусной кислоты (ГОСТ 61-69, х. ч. или ч. д. а.), перемешивают, растворяют
в этих двух кислотах 770 г уксуснокислого аммония (ГОСТ 3117-68, х. ч. или ч. д. а.)
и общий объем полученной смеси доливают дистиллированной водой точно до 10 л, пе¬
ремешивают.
1в. Рабочий раствор A—L смеси. 1 л запасного раствора 16 разбавляют водой до 1 лч
перемешивают. Получают аммоний-лактат-уксуснокислый раствор для приготовления
вытяжек.
Авторы предлагают следующие ориентировочные индексы обеспеченно¬
сти по фосфору (в мг Р2Об на 100 г почвы): <10- низкая; 10—20 — средняя;
20 — хорошая.
Метод Пича — Инглиша
(Pe?ch, English, 1944), вариант Хейфец (1962)
Метод основан на извлечении подвижного фосфора из почвы буферной
смесью уксусной кислоты и уксуснокислого натрия (раствор Моргана) с pH
4,8, соотношение почва : раствор = 1 : 20, время взаимодействия 30 мин.
Метод рекомендуется для анализов черноземных почв.
Ход анализа данного метода подробно описан в предыдущем издании кни¬
ги «Агрохимические методы исследования почв» (Хейфец, 1965).
Метод Бурриеля — Гернандо
(Burriel, Hernando, 1951)
Метод основан на извлечении подвижных фосфатов почвы с помощью
буферного раствора с pH 3,25, содержащего ионы Са, Mg, S04 и С03 в соот¬
ношениях, соответствующих, по мнению авторов метода, соотношению этих
ионов в системе почва — растение. Вытяжку готовят при соотношении почва:
раствор = 1 : 100 и врэмени взаимодействия 5 мин.
Метод рекомендуется для анализа кислых, слабокислых и карбонатных
почв.
При работе этим мэтодом на серых лесных почвах и черноземах можно
пользоваться лимитами Эгнера — Рима (см. выше табл. 6).
Ход анализа. 2 г воздушно-сухой почвы (сито d 1—2 мм) помещают в пло¬
скодонные колбы на 300—500 мл, приливают 200мл буферной смеси с pH 3,25,
взбалтывают 5 мин. и фильтруют, отбрасывая первые порции фильтрата.
Методы определения фосфора в почве
141
5—20 мл прозрачного фильтрата переносят в мерные колбы на 50 мл, разбав¬
ляют водой до 30—40 мл и приливают реактивы для колориметрического
определения фосфора.
Реактивы. 1. Экстрагирующий раствор. 0,1 г СаС03 и 0,088 г MgC03 помещают
в мерную колбу на 1 л, приливают около 700 мл воды, 0,5 мл 20%-ной H2S04 (уд. вес 1,14)
и 2,45 мл 98%-ной уксусной кислоты, доливают водой до метки, перемешивают. Полу¬
чают раствор с pH 3,25.
2. 20%-ный раствор H2S04. 129,9 мл H2S04 (уд. вес 1,83—1,84) осторожно вливают
в 800 мл дистиллированной воды. Раствор охлаждают, доливают водой до 1 л, перемеши¬
вают.
Метод Гинзбург и Артамоносой
(Гинзбург и др., 1968, 197Ь)
Метод основан на извлечении из почвы подвижных соединений фосфора
и калия с помощью смеси солей сернокислого аммония и молибдата аммония
с pH 4,8. Соотношение почва : раствор = 1 : 25 для кислых и слабокислых
почв и 1 : 50 для карбонатных почв. Время взаимодействия 30 мин., темпе¬
ратура 18—25° С.
Основанием для применения указанного экстрагента является способ¬
ность его переводить в раствор преимущественно наиболее доступные расте¬
ниям формы фосфора (кислые и свежеосажденные фосфаты Са, Mg, Fe2+ и др.).
Добавление молибдата аммония к солевой вытяжке на кислых и слабо¬
кислых почвах предотвращает вторичное осаждение фосфора в процессе эк¬
стракции вследствие образования недиссоциируемого соединения — фосфор¬
номолибденовой кислоты, что удерживает фосфор в растворе и обеспечивает
полноту его экстракции. На карбонатных же почвах молибдатная вытяжка
взаимодействует с почвой так же, как солевая вытяжка со слабокислым pH.
Обменный калий вытесняется из почвы указанной смесью с помощью
иона аммония, который по валентности и размерам радиуса иона близок к ио¬
ну калия.
Ход определения подвижных соединений фосфора и калия разработан для
лабораторных исследований и для массовых определений с использованием
полуавтоматического оборудования, имеющегося в зональных агрохимиче¬
ских лабораториях.
Ход анализа для лабораторных исследований. 2 г воздушно-сухой поч¬
вы (сито d 1—2 мм) помещают в колбы емкостью 100—200 мл, приливают
50 или 100 мл смеси растворов 1 %-ного (NH4)2S04 и 0,25%-ного (NH4)2Mo04*,
взбалтывают 30 мин. и фильтруют через два плотных фильтра, сливая первые
порции фильтрата.
5—20 мл прозрачного фильтрата сливают в мерные колбы на 50 мл (или
5—40 мл в мерные колбы на 100 мл) и приливают реактивы для окрашивания
фосфора, но с учетом количества молибдата аммония, содержащегося в алик¬
воте испытуемого раствора, взятого для колориметрирования (табл. 7
и 8)**. В оставшейся части фильтрата определяют калий на пламенном фото¬
метре.
Ход анализа для массовых определений фосфора. 2 г воздушно-сухой почвы
(сито d 1—2 мм) помещают в банки емкостью 200 мл (ГОСТ 5717—51), нахо¬
дящиеся в стандартных кассетах. С помощью десятипозицирнного дозатора
* Приводим простейшую формулу молибдата аммония. Торговый препарат этой соли
обычно представляет собой полимолибдат аммония, чаще всего это тетрагидрат гекса-
аммоний — гептамолибдат (NH4)eMo7024*4H20.
♦♦В молибдатных вытяжках образуется желтый комплекс фосфорномолибденовой гете¬
рополикислоты. Поэтому чем выше содержание фосфора в фильтратах, тем в более ин¬
тенсивно желтый цвет они окрашены и тем меньшая аликвота исследуемого раствора
отбирается для колориметрического определения фосфора.
Таблица 7
Реактивы, необходимые для окрашивания вытяжки, содержащей 0,25%-ный раствор (NH4)2Mo04 но Труогу — Мейеру
Реактивы для окрашивания вытяжки *
Необходимо добавить
реактивов для колори-
Первый
вариант
Второй вариант
Взято
вытяжки
на определе¬
В исследуе¬
мом объеме
содержится
(NH<),MoO<,
мг
метрирования
Реактив¬
Добавить на окрашива¬
ние
Добавить на окраши¬
вание
ние фосфора,
мл
(NH4),MoO«,
мг
H,so4,
0
ная
смесь
(NH4).Mo04,
г/л
H*S04
(уд. вес
1,84), мл/л
реактивы
А, . . . ,
Д, мл
SnCl t,
капель
(NH4)2Mo04,
г/л
H,S04
(уд. вес
1,84), мл/л
реактивы
Д, мл
SnCl«,
капель
Мерные колбы на 50 мл
5
12,5
37 ,5
0,945
А
18,8
280
2
3
0,833
12,5
45
3
10
25,0
25,0
0,945
Б
12,5
280
2
3
0,625
14,0
40
3
20
50,0
0
0,945
В
0
280
2
3
0
18,7
30
3
Мерные колбы на 100 мл
5
12,5
87,5
1,89
Г
21,9
280
4
6
0,921
11,8
95
6
10
25,0
75,0
1,89
А
18,8
280
4
6
0,833
12,5
90
6
20
50,0
50,0
1,89
Б
12,5
280
4
6
0,625
14,0
80
6
25
62,5
37,5
1,89
д
9,4
280
4
6
0,500
14,9
75
6
40
100,0
0
1,89
в
0
280
4
6
0,000
18,7
60
6
*1. Комплексообраэователь. 1а. Указанные в соответствующих графах количества миллилитров H{SO« (уд. вес 1,83—1,84) осторожно сливают в 600 мл дистиллиро-
ванной воды в мерную колбу емкостью 1 л. Раствор охлаждают.
16. Навески соли молибдата аммония в граммах растворяют ~ в 300 мл дистиллированной воды при нагревании. Раствор охлаждают.
Реактивные смеси А,Д. Раствор*1б осторожно при помешивании небольшими порциями сливают в раствор 1а, охлаждают, доливают до метки водой, перемсши
в а ют, сохраняют в склянке из темного стекла (в течение года).
2. Восстановитель. Готовят 2,5%-ный раствор SnCl* в 10%-ной НС1 при нагревании на водяной бане. Реактив готовят в день определения.
Дозирование реактивов. Вариант первый. На одно определение фосфора приливают соответствующий реактив А, Б, ..., Дпо 2 мл ъ мерные колбы на 50 мл и
по 4 .мл в 100 мл.
Вариант второй. Для аликвоты исследуемого раствора вЪмл приливают 45 мл реактива А, для 10 мл — 40 мл реактива Б, для 20 мл — 30 мл реактива В и т.п.
Для окрашивания вытяжки для обоих вариантов в мерные колбы емкостью 50 мл приливают 3 капли раствора SnCl* и соответственно 6 капель — в 100 мл.
3. Шкала образцовых растворов фосфата (приготовление см. выше, метод Труога — Мейера или массовые определения фосфора в молибпатных вытяжках).
Таблица 8
Реактивы для окрашивания вытяжки, содержащей 0,25%-ный раствор (NH^MoO* по Мерфи — Райли
go
а К
II
Н
Wtt
Реактивы для колориметрирования
о
о
Я
И
ъ
о
со
if
II
I
я
х
к
св
*
О
§*>
§
«
ев
н
в
ё
$
рц
Реактивы для окрашивания вытяжки *
Первый вариант
IV (смесь реактивов I—III
в 1 л)
Аскорбиновая кис¬
лота, г в 200 мл
реактива IV (реак¬
тивы А, ..., Д)
Добавить А—Д на
окрашивание, мл
IV (смесь реактивов I—III
в 1 л)
О
о
Я
т
И£
5Г
H.S04,
(уд. вес 1,84),
мл (II)
сурьмяно¬
виннокислый
калий, г (III)
HsS04
(уд. вес 1,84),
мл (I)
о
о
я
is
5Т
сурьмяно¬
виннокислый
калий, г (III)
Второй вариант
о i Я
Bag
К S
§:iL
В" «
о <V»> „
Р. J-* Г
2 «в «в :
|8В-г
св
н*
I »
«В
н в
Р
о£
5*
Рчо
Общий объем 50 мл
5
12,5
35,5
0,998
1,1632
42,24
А
17,8
280
0,5816
4,224
2
13,4
0,710
0,026
0,942
10
25,0
23,0
0,998
1,1632
42,24
Б
11,5
280
0,5816
4,224
2
15,0
0,460
0,029
1,056
20
50,0
0
0,998
1,1632
42,24
В
0
280
0,5816
4,224
2
20,1
0,00
0,039
1,411
Общий объем 100 мл
5
12,5
83,5
1,996
2,3264
84,48
Г
20,9
280
0,5816
4,224
4
12,7
0,035
0,0244
0,887
95
10
25,0
71,0
1,996
2,3264
84,48
А
17,8
280
0,5816
4,224
4
13,4
0,710
0,0260
0,942
90
20
50,0
46,0
1,996
2,3264
84,48
Б
11,5
280
0,5816
4,224
4
15,0
0,460
0,0290
1,056
80
25
62,5
33,5
1,996
2,3264
84,48
д
8,4
280
0,5816
4,224
4
16,0
0,335
0,0310
1,123
75
40
100,0
0
1,996
2,3264
84,48
в
0
280
0,5816
4,224
4
20,1
0,000
0,0390
1,411
60
*1. Комплексообразователь. I. Указанные выше в соответствующих графах количества миллилитров H,S04 (уд. вес 1,83—1,84) осторожно сливают в 600 мл ди¬
стиллированной воды в мерную колбу емкостью 1 л. Раствор охлаждают.
II. Навески соли молибдата аммония в граммах растворяют в 300 мл дистиллированной воды при нагревании. Раствор охлаждают.
III. Соответствующие навески соли сурьмяно-виннокислого калия в граммах растворяют в 100 мл воды.
IV. Растворы II и III вливают последовательно (небольшими порциями при помешивании) в раствор 1. Полученную смесь доводят водой до метки (до 1 л),
перемешивают.
2. Реактивные смеси А,Д (комплексообразователь + восстановитель). В 200 мл или в 1 л реактива IV растворяют соответствующую навеску аскорбиновой кис¬
лоты (готовят в день определения).
Дозирование реактивов. Вариант первый. На одно определение фосфора приливают соответствующих реактивов А, ..., D по 2 мл в мерные колбы на 50 мл и
по 4 мл — в 100 мл.
Вариант второй. Для аликвоты исследуемого раствора в 5 мл приливают 45 мл реактива А, для 10 лсд — 40 мл реактива Б, для 20 мл—30 мл реактива В и т. п.
3. Шкала образцовых растворов фосфата (приготовление см. выше, метод Труога.— Мейера или массовые определения фосфора в молибдатных вытяжках).
144
К. Е. Гинзбург
приливают 50 лед* аммонийно-молибдатной смеси, pH 4,8. Банки закрывают
крышками, взбалтывают на ротаторе 30 мин. и фильтруют через два плотных
фильтра, не содержащих фосфора и калия.
Шприц-дозатором отбирают 5—20 мл прозрачного фильтрата и сливают
в банки емкостью 200 мл, сюда же дозатором приливают соответствующие
реактивы для окрашивания вытяжки (см. приготовление реактивов А — Д
в табл. 7 и 8).
Дозирование реактивов для окрашивания в объеме 50 мл: кЬ мл аммоний¬
но-молибдатной вытяжки приливают 45 мл реактива А, к 10 мл аммонийно-
молибдатной вытяжки приливают 40 мл реактива Б, к 20 мл — 30 мл реак¬
тива В.
Шкала образцовых растворов для определения
Рг^б- 0,1917 г КН2Р04 (ГОСТ 4198-65, х. ч.) растворяют, доливают водой
до 1 л, перемешивают. Раствор содержит 0,1 мг Р2Об в I мл и является ис¬
ходным крепким раствором для приготовления шкалы образцовых раство¬
ров.
В мерные колбы на 500 мл (1—6) отбирают следующие количества крепкого
исходного раствора фосфата и доводят их до метки экстрагирующим раство¬
ром 1% (NH4)2S04 + 0,25% (NH4)2Mo04:
1 2 3 4 5 6
Исходный раствор фосфата, мл |0,5 1 1,5 2 3 4
Р205, мг на 1 кг почвы 2,5 5,0 7,5 10,0 15,0 20,0
Из полученных образцовых растворов в кассету с банками отбирают та¬
кое же количество миллилитров, как из почвенных вытяжек. Окрашивают
образцовые растворы аналогично почвенным вытяжкам.
Шкала образцовых растворов для определе¬
ния К20. 0,7915 г КС1 (ГОСТ 4234-69, х. ч.) растворяют и доливают точно
до 1 л дистиллированной водой, перемешивают. Раствор содержит 0,5 мг
К20 в 1 мл (исходный крепкий).
Для приготовления шкалы в мерные колбы на 500 мл (1—8) отбирают сле¬
дующие количества исходного крепкого раствора КС1 и доводят его до метки
раствором 1% (NH4)2S04 + 0,25% (NH4)2Mo04:
1 2 3 4 5 6 7 8
Исходвый раствор, мл 0,5 1 2 4 5 10 20 40
К20, мг на 1 кг почвы 12,5 25 50 100 125 250 500 1000
Реактивы. Экстрагирующий раствор — смесь растворов 1%-ного (NH4)2S04 и
0,25%-ного (NH4)2Mo04, pH 4,8. 2,5 г соли (NH4)2Mo04 растворяют в 150—200 мл дистил¬
лированной воды при нагревании. Одновременно в мерной колбе на 1 л растворяют в не¬
большом объеме воды 10 г соли (NH4)2S04. Приготовленный охлажденный раствор молиб-
дата аммония приливают к раствору сернокислого аммония, смесь доводят до метки во¬
дой, перемешивают. Если раствор не имеет pH 4,8, то его доводят до нужной величины
крепким раствором аммиака. Экстрагирующий раствор должен иметь температуру в пре¬
делах 18—25° С.
2. Реактивы А — Д для окрашивания молибдатных вытяжек (см. табл.. 7, 8).
При колориметрическом определении фосфора необходимо учитывать то количество
молибдата аммония, которое содержится в аликвоте раствора, взятой для окрашивания мо-
либдатной вытяжки. Это требуется для того, чтобы сохранить установленное в той или
иной прописи анализа соотношение реактивов: молибдата аммония, кислоты, восстанови¬
теля, необходимых для получения окраски фосфорномолибденовой сини, пропорциональ¬
ной концентрации фосфора в растворе.
* В случае карбонатных почв приливают 100 мл экстрагента.
Методы определения фосфора в почве
145
Для колориметрического определения фосфора в молибдатных вытяжках из почв
рекомендуется использовать метод Труога — Мейера или Мерфи — Райли (см. выше).
В методе Мерфи — Райли на одно определение фосфора в 50 мл раствора требуется
48 мг молибдата аммония, 1,1632 мг сурьмяно-виннокислого калия, 0,998 г H2S04 и
42,24 мг аскорбиновой кислоты. При колориметрировании в 100 мл раствора расход ука¬
занных реактивов увеличивается в 2 раза. В методе Труога — Мейера на одно определе¬
ние требуется 50 мг молибдата аммония, 0,945 г H2S04 и 3 капли 2,5%-ного раствора SnCla
в объеме 50 мл.
Независимо от аликвоты исследуемого раствора, взятой на окрашивание фосфора,
количество кислоты и восстановителя сохраняется такое же, как предусмотрено в ука¬
занных прописях. Количество же молибдата аммония изменяется. Например, если на
колориметрическое определение фосфора взяли 5 мл фильтрата в объеме 50 мл, то в нем
будет содержаться 12,5 мг молибдата аммония. Следовательно, если окрашивать этот раст¬
вор по прописи Труога — Мейера, то необходимо приготовить реактив, в котором молиб¬
дата аммония содержалось бы уже не 50 мг, а 50—12,5 = 37,5 мг на 50 мл, что потребует
18,8 г соли (NH4)2Mo04 в 1 л вместо 25 г!л, как это предусмотрено в прописи автора. По
методу Мерфи — Райли, потребуется уже не 48 мг молибдата аммония, а 48—12,5 =
= 35,5 мг на 50 мл. Аналогичные пересчеты делают для аликвоты исследуемого раство¬
ра в 10, 20 мл и т. п. (табл. 7, 8).
Приготовление окрашивающих смесей для колориметрического определения фосфора
в молибдатных вытяжках приведено в двух вариантах (см. табл. 7 и 8).
Первый вариант. Необходимое количество кислоты и молибдата аммония для раз¬
личных аликвот исследуемых растворов содержится в объеме 2 или 4 мл, поэтому точно
до объема 50 или 100 мл растворы доливают водой.
Второй вариант. Необходимое количество кислоты и молибдата аммония содержится
в 45 мл для аликвоты испытуемого раствора, разной 5 мл, в 40 мл — для 10 мл и т. п. Таким _
образом, аликвоты испытуемых растворов точно до объема 50—100 мл доливают не водой,
а соответствующими реактивами А—Д.
Авторы метода приводят следующие ориентировочные индексы обеспе¬
ченности по фосфору для кислых, слабокислых, нейтральных почв (в мг
Р205 на 100 г почвы): < 4 — очень низкая, <10 — низкая, 10—20 — сред¬
няя, >20 — высокая.
Индексы обеспеченности почв калием по данным аммонийно-молибдат-
ного метода на почвах различных почвенно-климатических зон хорошо сов¬
падают с показателями, установленными для соответствующих стандартных
методов.
Щелочные вытяжки
Щелочные растворители — K2C03, NaHC03, (NH4)2C03, NH4HC03, смесь
(NH4)2C204 с NH4HC03 и др.— используются преимущественно для опре¬
деления подвижных фосфатов на карбонатных почвах. Указанные экстра¬
генты содержат карбонатный, бикарбонатный (гидрокарбонатный) ионы,
которые подавляют активность ионов кальция в растворе, что способствует
переходу в раствор фосфора, связанного с ионом кальция. Эти растворители
способны также извлекать фосфор, связанный с алюминием и железом, вслед¬
ствие образования алюминатов и гидратов окисей железа и освобождения в
раствор фосфора, связанного с этими ионами. Трудность использования ще¬
лочных растворителей заключается в том, что они диспергируют органиче¬
ские вещества, в результате получаются окрашенные вытяжки. Поэтому
вытяжка 1%-ного раствора К2С03 с pH 11 (по Дасу) в настоящее время прак¬
тически не используется. Менее щелочные и менее окрашенные вытяжки
1 %-ного раствора (NH4)2C03, pH 9,0 (по Мачигину) и 0,5 н. раствора NaHC03,
pH 8,5 (по Олсену) в последнее время нашли широкое применение при ана¬
лизах почв.
146
К. Е. Гинзбург
Метод Мачигина (1952)
Метод основан на извлечении подвижных соединений фосфора и калия из
почвы 1%-ным раствором углекислого аммония, pH 9,0, при 25 ± 2° G. Со¬
отношение почва : раствор равно 1 : 20, взбалтывание 5 мин., настаивание
20 час. Метод принят стандартным для карбонатных черноземов, каштановых,
«бурых, коричневых почв и сероземов. Индексы обеспеченности по фосфору
даны в табл. 9.
Таблица 9
Обеспеченность почв подвижными соединениями фосфора по содержанию их в вытяжке
Мачигина
Обеспеченность
Р*Ов, мг на 100 г почвы
зерновые, хлопчатник
корнеплоды
овощные
Очень низкая
<1,0
<1,5
<3,0
Низкая
<1,5
<3,0
<4,5
Средняя
1,5-3,0
СО
0
1
сл
4,5-6,0
Высокая
>3,0
>4,5
>6,0
Ход анализа. 5 г воздушно-сухой почвы (сито d 1—2 мм) помещают в пло¬
скодонные колбы емкостью 200 мл, приливают 100 мл 1%-ного раствора
(NH4)2C03, взбалтывают 5 мин., настаивают 18—20 час. при 25 ± 2° С (же¬
лательно в термостате). На следующий день суспензию перемешивают и филь¬
труют. Если углеаммонийная вытяжка бесцветна или слабо окрашена, то
фосфор в ней определяют колориметрически без предварительной обработки.
Если вытяжка окрашена, то ее предварительно обесцвечивают. Для этого
окисляют органические вещества перманганатом или поглощают их активи¬
рованным углем.
Следует отметить, что на почвах, богатых органическими соединениями
фосфора (южные, карбонатные черноземы, темно-каштановые почвы и др.)»
более правильно обесцвечивать вытяжки углем, так как в этом случае учиты¬
вается юлько минеральный фосфор. Окисление же вытяжек может давать по¬
вышенное представление о содержании в почвах подвижного фосфора, так как
таким способом определяется сумма его минеральных и органических со¬
единений.
В оставшейся части фильтрата (без предварительного его обесцвечивания)
определяют калий на пламенном фотометре.
Обесцвечивание щелочных вытяжек
Окисление перманганатом. 5—20 мл окрашенного фильтрата поме¬
щают в мерные колбы емкостью 50—100 мл, приливают 2 мл 27%-ного раствора H2S04
(реактив 2) и 4 мл 0,5 н. раствора КМп04 (реактив 3). Колбы помещают на электрические
плитки и кипятят 2 мин. с момента закипания смеси. Избыток КМп04 обесцвечивают,
прибавляя к горячему раствору 1 мл 10%-ного раствора глюкозы После охлаждения в
растворе определяют фосфор колориметрически, но с учетом добавленной для окисления
H2S04 (см. ниже приготовление реактивов).
Обесцвечивание углем. Способ I. Примерно 30—40 мл фильтрата сли¬
вают в другие колбочки емкостью 50—100 млу всыпают 0,1—0,3 г угля (берут меркой),
перемешивают и настаивают 10—20 мин. (если раствор не обесцветился, добавляют еще
* Глюкоза медленно обесцвечивает избыток КМп04, более быстро восстановление идет в
присутствии аскорбиновой кислоты (Самохвалов, Соколова, 1974).
Методы, определения фосфора в почве
147
0,1 г угля), затем фильтруют. В аликвоте бесцветного фильтрата определяют фосфор ко¬
лориметрически .
Способ II. 2,5 г воздушно-сухой почвы (сито d 1—2 мм) помещают в плоскодонные кол¬
бы емкостью 100 мл, приливают 50 мл 1%-ного раствора (NH4)2C03, взбалтывают 5 мин.,
настаивают 18—20 час. при 25 ±2° С. Перед фильтрованием в вытяжку всыпают 0,3—1,0 г
активированного угля (берут меркой), перемешивают от руки и настаивают суспензию
15—20 мин. (если вытяжка не обесцветилась, добавляют еще небольшую порцию угля).
Обесцвеченную вытяжку фильтруют. В аликвоте прозрачного фильтрата определяют фос¬
фор колориметрически. В оставшейся части фильтрата определяют калий на пламенном
фотометре. Используемый для обесцвечивания вытяжки уголь не должен выделять в
вытяжку и поглощать из нее фосфор и калий (см. приготовление реактива 8).
Колориметрическое определение фосфора в
обесцвеченных вытяжках. При обесцвечивании углеаммоний¬
ной вытяжки углем в фильтрате определяют фосфор колориметрически обыч¬
ным способом.
При обесцвечивании вытяжки окислением перед колориметрическим опре¬
делением фосфора необходимо избыток добавленной H2S04 или ней¬
трализовать (громоздкая, длительная процедура), или учесть ее при приго¬
товлении реактивов для окрашивания фосфора. Нами были сделаны перес¬
четы и внесены соответствующие поправки в состав окрашивающих смесей,
приготовляемых по прописи Труога — Мейера и Мерфи — Райли.
Окрашивание вытяжки по Труогу — Мейеру. Окисленный раствор ох¬
лаждают, доливают водой до 40 мл, приливают 2 мл молибденового реактива
(реактив 4в), доливают водой точно до 50 мл% перемешивают, приливают
3 капли восстановителя — SnCl2 (реактив 4г), вновь перемешивают, спустя
7 мин. в течение 15—20 мин. просматривают окрашенные растворы на фото¬
колориметре с красным светофильтром.
Окрашивание вытяжки по Мерфи — Райли. Окисленный раствор разбав¬
ляют водой до 40 мл, приливают 2 ^докрашивающей смеси (реактив 5а),
доливают водой до 50 мл, перемешивают и спустя 15 мин. и в течение 24 час.
просматривают окрашенные растворы на фотоколориметре.
Реактивы. 1. 1%-ный раствор (ГШ4)2С0з *. 10 г измельченной, хорошо перемешан¬
ной соли (NH4)2C03 (ГОСТ 3770-64, х. ч.) растворяют в дистиллированной воде, доводя
затем объем раствора точно до 1 л, В связи с непостоянством химического состава соли
углекислого аммония •• концентрацию и pH приготовленного экстрагирующего раство¬
ра предварительно проверяют и, если необходимо, вносят соответствующие поправки.
Выполняют это следующим образом.
Измеряют pH исходного раствора экстрагента. Затем устанавливают концентрацию
приготовленного раствора (NH4)2C03. Для этого 10 мл приготовленного раствора
(NH4)2C03 помещают в конические колбочки емкостью 50—100 мл, добавляют 2—3 капли
индикатора метилоранжа и титруют 0,1—0,2 н. НС1 с установленным титром до появления
оранжево-розовой окраски.
По объему кислоты, пошедшей на титрование (а, мл), нормальности кислоты (Ь, н.)
и объему раствора, взятого на титрование (с, мл), вычисляют концентрацию (NH4)2C03 в
растворе : х = аЪ/с. Допустим, установили, что исходный раствор (NH4)2C03 имеет
pH 8,0, а концентрацию 0,121 н. (0,58%). Тогда поступают следующим образом.
Вначале доводят раствор крепким аммиаком до pH 9,0 ± 0,2. Затем повторно оп¬
ределяют концентрацию полученного раствора (NH4)2C03 с pH 9,0 титрованием.
Допустим, концентрация экстрагента оказалась равной 0,178 н. (0,85%). Необходимо
ее довести до 0,208 н. (1%), т. е. увеличить на 0,208—0,178 = 0,03 н. Для этого проводят
* Приготовление 1%-ного раствора (NH4)2C03 приводим по прописи, указанной в «Инст¬
рукции по проведению анализов почв...» (1973).
•* Обычно углекислый аммоний выделяет на воздухе аммиак и поэтому по химическому
составу не соответствует формуле (NH4)2C03, а является смесью (NH4)2COg и NH4HC08.
148
К. Е. Гинзбург
следующий расчет:
10 г соли (NH4)2C03— 0,121 н.
у — 0,03 н.
Отсюда у = 10*0,03:0,121 = 2,48 г.
Таким образом, на каждый литр приготовленного раствора необходимо добавить до¬
полнительно 2,48 г соли (NH4)2C03. Полученную при этом концентрацию проверяют по¬
вторным титрованием. Допустимо применять 0,9—1,05%-ный раствор (NH4)2C08.
2. 27%-ный раствор H2S04. 150 мл концентрированной H2S04 (уд. вес 1,84) осто¬
рожно вливают в 600 мл дистиллированной воды., охлаждают, доливают водой до 1 л%
перемешивают.
3. 0,5 н. раствор КМп04. 15,8 г соли КМп04 (ГОСТ 4527-48, х. ч. или ч. д. а.)
растворяют, доливают водой до 1 л, перемешивают.
4. Окрашивающие реактивы по Труогу — Мейеру. 4а. 25 г молибдата аммония (ГОСТ
3765-64, х. ч. или ч. д. а.) растворяют в 200 лед дистиллированной воды при нагревании.
46. 130 мл концентрированной H2S04 осторожно вливают в 600 мл дистиллированной
воды.
4в. После охлаждения реактив 4а небольшими порциями при помешивании вливают
в реактив 46, смесь вновь охлаждают, доливают до 1 л водой, перемешивают. В склянке
из темного стекла реактив сохраняется длительное время.
4г. Восстановитель. 0,25 г кристалл, соли SnCl2*2HsO (ГОСТ 36-40, ч. д.а.) раство¬
ряют в 10 мл 10%-ной НС1 в пробирке, которую погружают в стакан с кипящей водой.
5. Окрашивающие реактивы по Мерфи — Райли. 5а. 24 а молибдата аимония раство¬
ряют при нагревании в 200 мл дистиллированной воды. Раствор охлаждают.
56. 130 мл концентрированной H2S04 вливают осторожно в 500 мл дистиллированной
воды. Раствор охлаждают.
5в. 0,5816 г сурьмяно-виннокислого калия (ВТУ МХП 2928-51) растворяют в 100 мл
дистиллированной воды.
5г. Растворы 5а и 5в вливают последовательно небольшими порциями при помешива¬
нии в раствор 56. Смесь охлаждают, доливают до 1 л водой, перемешивают. В склянке
из темного стекла реактив сохраняется длительное время.
5д. 4,224 г аскорбиновой кислоты растворяют в 200 мл реактива 5г. Реактив годен в
течение 24 час.
6. Образцовый раствор фосфата (см. выше метод Труога — Мейера).
7. 10%-ный раствор глюкозы (ГОСТ 6038-51, ч. д. а.). Раствор сохраняется в темном
месте 2—3 недели.
8. Очищенный активированный уголь. 8а. Бели уголь незначительно загрязнен фос¬
фором, то его промывают несколько раз (2—4) 1%-ным раствором (NH4)aC08 (или другого
экстрагента), контролируя фильтраты на содержание в них фосфора. Отмытый от фосфора
уголь просушивают на фильтре, затем растирают до порошка и используют для обес¬
цвечивания вытяжки.
86. Если уголь значительно загрязнен фосфатами, то его очищают обработкой кон¬
центрированной НС1 следующим образом. Продажный препарат древесного угля, жела¬
тельно крупночешуйчатый, марки КАД-иодированный *, насыпают в фарфоровые стака¬
ны или стеклянные сосуды емкостью 1—3 л, заливают концентрированной НС1, накры¬
вают часовым стеклом и оставляют стоять в течение 1—2 суток, периодически перемеши¬
вая смесь стеклянной палочкой. Затем кислоту сливают декантацией, предварительно
разбавив ее водой. Уголь в стаканах многократно заливают горячей водой, декантируя
раствор на большие воронки с фильтрами или на воронки Бюхнера. Промывают уголь до
отсутствия в промывных водах иона хлора (проба с AgNOs). Промытый уголь просу¬
шивают затем между листами фильтровальной бумаги либо при комнатной температуре,
либо в термостате при 30—40° С, затем его растирают в фарфоровой ступке или на мель¬
нице типа «Пируэт» до тонкого порошка.
1 В лаборатории агрохимии Почвенного института им. В. В. Докучаева с помощью Рза
было установлено, что уголь этой марки не поглощает фосфор из раствора.
Методы определения фосфора в почве
149
Производится проверка активированного угля на способность выделять и поглощать
фосфаты из раствора.
Проверка на отсутствие фосфатов, переходящих в различные вытяжки — 1%
<NH4)2C03; 0,5 н. NaHC03£0,5 н. NH4F; 0,1 н. NaOH и др. 0,1—0,2 или 1 г угля зали¬
вают 50 мл соответствующего раствора, взбалтывают 1 час и настаивают 18—20 час.,
фильтруют. 15—40 мл фильтрата помещают в мерные колбы на 50 мл и определяют фос¬
фор колориметрически. Если раствор не окрашивается, то уголь очищен и пригоден для
анализов. Если уголь загрязнен, его очищают, как описано выше.
Некоторые марки древесного угля обладают способностью поглощать фосфаты из
раствора. В связи с этим уголь проверяют также на его способность сорбировать фосфаты
из почвенных вытяжек. Для этого в 50 мл раствора различных экстрагентов приливают
1, 2, 3, 4 мл рабочего образцового раствора фосфата (см. метод Труога — Мейера), содер¬
жащего 0,01, .., 0,04 мг Р206 соответственно, всыпают в одну партию колб 0,1—0,3—1 а
угля, взбалтывают 1 час, настаивают 18—20 час., фильтруют. Другую партию колб под¬
вергают точно такой же обработке, исключая добавление угля. В фильтрате определяют
фосфор колориметрически. Если содержание фосфора в колбах с добавлением и без добав¬
ления к ним угля совпадает, то уголь может быть использован в анализах для обесцве¬
чивания вытяжек.
Если обесцвечивание вытяжки происходит в присутствии почвенной пробы (способ II),
то уголь необходимо проверять на содержание и поглощение не только фосфора, но и ка*
лия. Выполняют это точно так же, как было описано выше для фосфора.
Метод Олсена (Olsen et al., 1954)
Метод основан на извлечении подвижных фосфатов почвы с помощью 0,5 н.
раствора NaHCOs (pH 8,5), соотношение почва : раствор = 1 : 100, время
взаимодействия 30 мин., температура 25 ± 1° С. Автор рекомендует метод
для анализа кислых, нейтральных и карбонатных почв.
Ход анализа. 5 г воздушно-сухой почвы (сито d 1—2 мм) помещают в пло¬
скодонные колбы емкостью 200 мл, приливают 100 мл 0,5 н. раствора NaHC03
(реактив 1), добавляют 1—3 г (берут меркой) активированного угля (см. ме¬
тод Мачигина, реактив 8), взбалтывают 30 мин., затем фильтруют. Если филь¬
трат не обесцветился, то добавляют к нему еще порцию угля, перемешивают,
«настаивают 10—15 мин. и вновь фильтруют. Чтобы уменьшить расход угля,
эдожно обесцвечивать не всю вытяжку, а только часть ее, как это описано
выше в методе Мачигина в способе I.
5—40 мл бесцветного прозрачного фильтрата сливают в мерные колбы на
50—100 мл, осторожно нейтрализуют по p-динитрофенолу 10%-ной НС1,
дают раствору постоять, помешивая его время от времени от руки, чтобы как
можно полнее удалить выделяющиеся пузырьки С02, и приливают реактивы
для колориметрического определения фосфора, при этом следует вновь
проследить за удалением пузырьков С02, чтобы при перемешивании растворы
не разбрызгивались и С02 не накапливалась на стенках кюветы фотоколори¬
метра, что может исказить величину светопоглощения окрашенных растворов,
а отсюда и результаты определения фосфора.
Обесцвечивание вытяжки NaHC03 можно проводить и путем ее окисления
0,5 н. раствором КМп04 с последующим колориметрическим определением
фосфора, как описано выше в методе Мачигина.
Реактивы. 1. 0,5 н. раствор NaHC03 с pH 8,5. 42 г соли NaHC03 (ГОСТ 4201-66)
растворяют в 500—600 мл теплой воды, охлаждают и доливают до 1 л водой, перемеши¬
вают, проверяют значение pH и, если необходимо, доводят до нужной величины с помощью
0,1—0,5 н. раствора NaOH.
Концентрацию 0,5 н. раствора NaHCOs проверяют точно так же, как описано в методе
Мачигина. Реактивы для окисления, обесцвечивания и колориметрического определения
.фосфора описаны там же.
150
К. Е. Гинзбург
Автор приводит следующие ориентировочные индексы обеспеченности
почв фосфором (мг Р206 на 100 г почвы) для различных сельскохозяйственных
культур: < 2,5 — низкая; 2,5—5,0 — средняя; 5—9 — хорошая; > 9 —
высокая.
Метод Мещерякова (1971)
Метод основан на извлечении подвижных соединений фосфора и калия из
сероземных почв с помощью смеси оксалата и гидрокарбоната аммония при
pH 8,7. Соотношение почва : раствор = 1 : 20. На количество извлекаемого
фосфора мало влияют колебания температуры в пределах 15—25° С и содер¬
жание СаС03 до 20% от веса почвы. Отмечается также незначительное пере-
осаждение фосфора в процессе экстракции.
Данные, получаемые этим методом, лучше (чем методом Мачигина) отра¬
жают эффективность фосфорных удобрений на сероземных почвах с доста¬
точным обеспечением хлопчатника азотными и калийными удобрениями и с
повышенным содержанием сульфатов и хлоридов кальция и магния, карбона¬
тов, гидрокарбонатов и обменного магния.
Ход анализа. 5 г воздушно-сухой почвы (сито d 1—2 мм) помещают в пло¬
скодонные колбочки емкостью 200 мл, приливают 100 мл извлекающей смеси
0,126 н. NH4HC03 + 0,025 н. (NH4)2C204, взбалтывают 10 мин., настаивают
24 часа, фильтруют через фильтр, не содержащий фосфора и калия. Первые
порции фильтрата отбрасывают. В аликвоте прозрачного фильтрата, после
его обязательного предварительного окисления (окисляют гумусовые веще¬
ства и оксалаты), определяют фосфор колориметрически. В оставшейся части
неокисленного фильтрата определяют калий на пламенном фотометре.
Окисление вытяжки перманганатом. Вариант окисления оксалатно-гид-
рокарбонатной вытяжки, предложенный автором метода, несколько громозд¬
кий, поэтому рекомендуем ход окисления и окрашивания вытяжки такой же,
как описан выше в методе Мачигина.
Реактивы. 1. Извлекающий раствор оксалата — гидрокарбоната аммония*
1а. Запасный раствор гидрокарбоната аммония (бикарбоната или двууглекислого аммония).
20 г NH4HC03 (ГОСТ 3762-64) растворяют в 600 мл дистиллированной воды и доливают
водой точно до 1 л, перемешивают. Установление концентрации полученного раствора
производят точно так же, как описано выше в методе Мачигина. На основании полученных
данных вычисляют, сколько требуется взять раствора 1а, чтобы получить 1 л 0,126 н.
(1%-ного) раствора NH4HC03.
16. Рабочий раствор оксалата — гидрокарбоната аммония. Определенное число мил¬
лилитров раствора 1а, соответствующее 0,126 н. (1%-ному) раствору NH4HCOs, вливают
в мерную колбу емкостью 1 л\ сюда же всыпают 1,78 г соли (NH4)2C204 оксалата (щавеле¬
вокислого) аммония (ГОСТ 3774-51). После растворения навески соли объем раствора
доводят до 1 л водой, перемешивают. Полученный раствор примерно 1%-ный по гидро¬
карбонату аммония и 0,18%-ный по оксалату аммония.
Автор приводит следующие групповые градации по содержанию подвиж¬
ных фосфатов в сероземах (мг Р2Об на 1 кг почвы): первая группа (очень бед¬
ные почвы) — до 30 мг/кгу вторая — от 31 до 60, третья — от 61 до 90, чет¬
вертая — от 91 до 120, пятая — от 121 до 150 и шестая группа (высокая
обеспеченность) — ]> 150 мг. Градации по содержанию калия (мг К20
в 1 кг почвы): первая группа — до 104, вторая — от 105 до 204, третья — от
205 до 304, четвертая — от 305 до 404, пятая — от 405 до 504.
Фторидные вытяжки
Теоретическим обоснованием использования фторидных вытяжек для
определения растворимых фосфатов в почвах является способность ионов
F* в кислой среде образовывать с ионами Fe8+ и А13+ недиссоциируе-
Методы определения фосфора в почве
151
мые комплексные соединения и таким образом вытеснять в раствор фосфор,
связанный с этими ионами.
В нейтральной и щелочной среде ионы F+ образуют комплексы преиму¬
щественно с ионами А13+. На этом основано выделение из почвы фракции
А1 — Р по методу Чанга — Джексона (Аскинази и др., 1963; см. также
табл. 1). Схематически образование фторидных комплексов с А13+ и Fe8+
можно представить следующим образом:
3NH4F+3HF+AlP04HJbP04+(NEU)3AlFet
3NH4F+3HF+FePOd — HaPOi+CNHOaFeFe.
Здесь А1Р04 и FeP04 представлены фосфатами различной основности и
степени окристаллизованности, включая фосфаты адсорбированные и осаж¬
денные на поверхности алюмосиликатов и полуторных окислов. Ионы фтора
могут осаждать из раствора и ионы Са2+, образуя труднорастворимый
CaF2. Это обусловливает переход в раствор фторидных вытяжек некоторых
соединений фосфатов кальция типа дикальцийфосфатов, частично трикаль-
цийфосфатов (по-видимому, аморфных форм) и др.
Таким образом, фторидные вытяжки могут извлекать наиболее раствори¬
мые соединения фосфора из различных фракций минеральных фосфатов поч¬
вы (Са — Р; А1 — Р; Fe — Р).
Метод Соколова
(см. Хейфец, 1960)
Метод основан на извлечении подвижных и обменных фосфатов почвы
ОД н. раствором NH4F с pH 6,3 ± 0,1 при соотношении почва : раствор =►
= 1 : 100, времени взаимодействия 30 мин.
Метод можно использовать для анализов различных типов почв как для
пахотных горизонтов, так и по их профилю.
Ход анализа. 2 г воздушно-сухой почвы (сито d 1—2 мм) помещают в
плоскодонные колбы емкостью 300 мл, приливают 200 мл 0,1 н. раствора
NH4F, взбалтывают на ротаторе 30 мин. и фильтруют, отбрасывая первые
порции фильтрата.
Обработку вытяжки и определение в ней фосфора проводят так же, как
описано выше в методе Чанга — Джексона.
Реактив. 0,1 н. раствор NH4F с pH 6,3. 3,7 г NH4F (ГОСТ 4518-60) растворяют и
доливают до 1 л водой, перемешивают. Если необходимо, pH раствора доводят до нужной
взличины раствором аммиака или водой. Раствор сохраняют в полиэтиленовой или про-
парафинированной изнутри склянке.
При применении метода Соколова на дерново-подзолистых почвах можно
пользоваться лимитами, установленными для метода Кирсанова.
Метод Брейя и Куртца
(Bray, Kurtz, 1945)
Метод основан на извлечении подвижных и обменных фосфатов почвы
смесью 0,03 н. раствора NH4F и 0,025 н. НС1, pH 2,9, при соотношении поч¬
ва : раствор = 1:7, времени взбалтывания 1 мин.
Чтобы снизить вторичное осаждение и получить более высокие абсолют¬
ные значения содержания фосфора в вытяжках, У Мью Тхант (1969) предло¬
жил использовать более широкое соотношение почва : раствор — 1 : 30.
Метод рекомендуется для анализов кислых, нейтральных, карбонатных
почв.
Ход анализа. 5 г воздушно-сухой почвы (срто d 1—2 мм) помещают в
плоскодонные колбочки емкостью 50 мл, приливают 35 мл экстрагирующей
152
К. Е. Гинабург
смеси 0,03 н. NH4F и 0,025 н. НС1, взбалтывают 1 мин., желательно электро¬
механической пропеллерной мешалкой, фильтруют. 5 мл прозрачного филь¬
трата сливают в 50 мл мерные колбы, разбавляют водой примерно до 40 мл,
приливают реактивы для колориметрирования фосфора. В данном случае
ионы F+ содержатся в количествах, не мешающих колориметрическому оп¬
ределению фосфора.
Реактив. Извлекающий раствор — смесь 0,03 н. NH4F и 0,025 н. НС1. pH 2.9.
1.11 г соли NH4F (ГОСТ 4518-60, х. ч. или ч. д. а.) растворяют в 500 мл дистиллированной
воды, приливают сюда же 2,5 мл концентрированной НС1 (уд. вес 1,18—1,19), доливают
водой до 1 л, перемешивают. Раствор сохраняется более года.
Авторы установили хорошее соответствие между показаниями содержания
растворимых фосфатов в данной вытяжке и эффективностью фосфорных удоб¬
рений. Для предлагаемого ими метода они рекомендуют следующие индексы
обеспеченности по фосфору (мг Р2Ов на 100 г почвы) : <3 — очень низкая,
3—7 — низкая, 7—20 — средняя, ^>20 — высокая.
Вариант Миллера и Акслей
(Miller ' Axley, 1956)
Метод основан на извлечении подвижных и обменных фосфатов почвы
смесью 0,03 н. раствора NH4F и 0,03 н. H2S04, pH 2,5, соотношение почва :
: раствор = 1:7, время взбалтывания 1 мин. Для снижения вторичного осаж¬
дения при приготовлении вытяжки рекомендуется более широкое соотноше¬
ние почва : раствор — 1 : 30 (У Мью Тхант, 1969).
Авторы на основании проведенных исследований пришли к заключению,
что смесь H2S04 и NH4F более эффективна для извлечения фосфора, чем смесь
NH4F и НС1.
Метод рекомендуется для анализов кислых, слабокислых, нейтральных
почв.
Ход анализа. 5 г воздушно-сухой почвы (сито d 1—2 мм) помещают в пло¬
скодонные колбочки емкостью 50 мл, приливают 35 мл экстрагирующей смеси
0,03 н. NH4F и 0,03 н. H2S04, а далее поступают точно так же, как в методе
Брея — Куртца.
Реактив. Извлекающий раствор — смесь 0,03 н. NH4F и 0,03 н. H2S04, pH 2,5.
1.11 г соли NH4F растворяют в 500 мл дистиллированной воды, приливают 0,85 мл кон¬
центрированной H2S04 (уд. вес 1,84), доливают водой до метки, перемешивают. Раствор
сохраняется более года.
Ионообменная хроматография
Ионообменная хроматография основана па обратимом эквивалентном
обмене содержащихся в растворе ионов на ионы, входящие в составе ионо-
обменника. При этом смолы, обладающие свойствами кислоты, способны к
обмену катионов (катиониты), а свойствами оснований — к обмену анионов
(аниониты). Поглощение фосфат-ионов из раствора осуществляется ани¬
онитами. Схематически этот процесс можно представить следующим образом:
R - ОИ+С1- ^ R[— С1+ОН- ,
3R;- Cl+PO^ ^ R — РО4+ЗСГ ,
где R — макромолекула, к которой присоединены активные группы ионо-
обменника (ОН”, СГ, НС03“; НСОСГ и др.).
Использование водных вытяжек с добавлением к ним анионитов яв¬
ляется наиболее обоснованным способом определения запаса подвижных
фосфатов в почвах. Это обусловлено тем, что аниониты практически не оказы¬
Методы определения фосфора в почве
153
вают никакого химического воздействия на почву. Они не изменяют ее pH, не
влияют на ионный состав равновесного раствора. В присутствии анионитов
не происходит вторичного осаждения (переосаждения) фосфора в процессе
экстракции.
Механизм взаимодействия фосфатов почвы с анионитами несколько схо¬
ден с теми процессами, которые происходят в почвах под растениями. Так,
при добавлении анионитов в водную вытяжку ортофосфаты, перешедшие в
раствор, поглощаются ими, в результате равновесие между твердыми и жидки¬
ми фазами почвы нарушается, что обусловливает переход в раствор новых
порций фосфора и т. п. Количество фосфора, извлекаемого из почвы аниони¬
том, зависит от времени взаимодействия, соотношения между почвой и ани¬
онитом, температуры. В зависимости от целей и задач исследования можно
подобрать такие условия анализов, при которых будет получен необходи¬
мый запас растворимых фосфатов, в наибольшей степени отражающий коли¬
чество усвояемых растениями фосфатов почвы.
В настоящее время аниониты используются как для лабораторных иссле¬
дований, так и для изучения динамики подвижных фосфатов и обменного
калия в почвах под растениями в полевых условиях.
Лабораторный метод с применением анионитов
(Amer et al., 1955; Карпинский, Замятина, 1958; Сидорина, 1961)
Метод основан на извлечении подвижных и обменных фосфатов почвы
анионитами в водной вытяжке. Соотношение почва: анионит может быть в
пределах 1 : 0,5; 1 : 1; 2 : 1; 3 : 1; 4 : 1 и т. п. Соотношение почва: вода =
= 1:5; 1 : 10; 1 : 25; 1 : 100 и т. п. Время взаимодействия от нескольких _
минут до нескольких суток и более. Температура взаимодействия чаще всего
комнатная, но может быть и повышенная до 30—40° С, но не выше пределов
термической устойчивости ионообменника (не выше 40—60° С).
Ниже приводим описание методики определения подвижных фосфатов
анионитами, используемой в Почвенном институте им. В. В. Докучаева (Чжан
Фу-чжу, 1962) и ВИУА (Карпинский, Замятина, 1958) на основных типах
почв Советского Союза (дерново-подзолистых, черноземе, красноземе, серо¬
земе). Количество фосфора, извлеченное в данных условиях анионитом, было
очень близко к запасу усвояемых фосфатов, определенных в вегетационных
опытах с растениями с Р32 (Соколов, 1960а).
Ход анализа. Вариант Почвенного института. Ь г воз¬
душно-сухой почвы (сито d 0,25 мм) помещают в плоскодонные колбы емкостью
100 лед, приливают 50 мл дистиллированной воды и погружают в суспен¬
зию капроновый мешочек с 1 г анионита (сито 0,5 мм). Был использован ани¬
онит ЭДЭ-10П (можно АВ-17 и др.)1 в форме С03. Смесь взбалтывают 2 часа
при комнатной температуре и настаивают 16—18 час. Эту процедуру повто¬
ряют еще 2 раза. После этого мешочек с анионитом вынимают, отмывают сна¬
ружи водой от частичек почвы, затем анионит из мешочка количественно пе¬
реносят в химический стакан емкостью 50—100 мл, заливают 15—20 мл 0,5 н.
раствора НС1, перемешивают, настаивают 30—60 мин. и сливают раствор
декантацией через небольшие воронки с фильтрами в мерные колбы
на 100 мл. Это повторяют несколько раз до полного извлечения фосфора
из анионита, после чего доливают объем раствора до метки водрй, перемеши¬
вают.
5—40 мл прозрачного фильтрата переносят в другие мерные колбы на 50—
100 мл, раствор нейтрализуют по p-динитрофенолу или другому индикатору
и определяют фосфор колориметрически.
1 Характеристика ионообменных смол отечественного производства приводится в книге
Е. В. Аринушкиной (1970).
154
К. Е. Гинзбург
Навеску анионита (фракции 0,5 мм) можно добавить непосредственно в сус¬
пензию (без мешочка), а по истечении установленного срока взаимодей¬
ствия перенести смесь на сито с отверстиями диаметром 0,25 мм и отделить
на нем анионит от навески почвы промыванием водой. Фосфор из анионита
выщелачивают затем НС1 и определяют его колориметрически точно так же,
как описано выше при использовании запакованных в мешочек анионитов.
Добавление в суспензию анионитов в мешочках значительно облегчает его
последующее отделение от почвы, что сокращает ход анализа, повышает его
точность, воспроизводимость.
Вариант ВИУА. 2 г воздушно-сухой почвы (сито d 0,25 мм)
помещают в плоскодонные колбы емкостью 200 мл, приливают 100 мл дистил¬
лированной воды и погружают капроновый мешочек (материал 26) с 0,5 г
анионита (сито d 0,5 мм) ЭДЭ — 10П или АВ — 17 в Cl-форме. Взбалтывают
суспензию в течение 5 час., затем вынимают мешочек и поступают далее точ¬
но так же, как описано выше в варианте Почвенного института.
Полевой метод с применением анионитов
(Карпачевский, 1971; Карпачевский, Киселева, 1969)
Метод основан на изучении динамики подвижных фосфатов почвы с по¬
мощью ионообменников в полевых условиях.
Ход анализа. В целлофановые пакеты, выполняющие роль полупрони¬
цаемой мембраны, насыпают 1—2 г анионита ЭДЭ — 10П в ОН-форме.
В исследуемой почве ножом или другим приспособлением делают щели
глубиной 4—6 см, куда вставляют в вертикальном положении пакет с анио¬
нитом так, чтобы один конец его оставался снаружи щели. Через 10 дней
пакеты вынимают из почвы, укладывают в коробки или другие пакеты (бу¬
мажные, полиэтиленовые), сопроводив их этикеткой, на которой надписы¬
вают дату и место взятия образца. Одновременно с выемкой инообменников»
вставляют новые пакеты с анионитами также на 10 дней, но в другие щели,
сделанные на расстоянии 2—3 см от предыдущих.
Анионит после экспозиции высушивают при комнатной температуре да
воздушно-сухого состояния, что соответствует абсолютной влажности анио¬
нита ~ 30%. После этого из каждого пакета берут навеску 100—200 мг ани¬
онита, помещают ее в плоскодонные колбы или стаканы емкостью 50—100 мл,
выщелачивают из анионита поглощенный из почвы фосфор с помощью 0,5 н.
раствора НС1 точно так же, как описано выше для лабораторных опытов.
В определенной аликвоте прозрачного фильтрата, после его предварительной
нейтрализации, определяют фосфор колориметрически.
Результаты анализов авторы выражают в мг Р206 на 100 г анионита, делая
допущение, что анионит и растения поглощают из почвы примерно одина¬
ковые количества фосфора. Для пересчета фосфора в мг Р205 на 100 г почвы
применяют уравнение Доннана: Св почве = 0,5 Сваниошшл где С — со¬
держание фосфора, мг Р2Об на 100 г почвы. Но в этом случае не учитывается
диффузия фосфора в почве под воздействием градиента концентрации, соз¬
даваемого анионитом. Поэтому данные, пересчитанные на 100 г почвы, могут
быть несколько завышены.
С помощью этого метода авторы получили интересные данные по дина¬
мике фосфора в дерново-подзолистых и светло-каштановых почвах под дре¬
весной растительностью.
Используя катионит КУ-2 в Н-форме, авторы точно так же, как описано
выше для фосфора, изучали динамику обменного калия в почвах (Карпачев¬
ский, Киселева, 1969).
Материалы. 1. Очистка и насыщение ионообменных смол (Самуэльсон, 1966).
Товарные иониты содержат органические и неорганические примеси, коллоидные вещест.
ва, мелкую пыль и другие примеси, которые не были полностью удалены в процессе И8-
Методы определения фосфора в почве
155
готовления ионитов. Поэтому перед употреблением иониты следует очистить. Для этого
продажный препарат смолы помещают в стеклянные стаканы, заливают дистиллирован¬
ной водой, перемешивают стеклянной палочкой, настаивают 30 мин. и сливают воду де¬
кантацией. Эту процедуру повторяют несколько раз до осветления промывных вод, что
свидетельствует о том, что ионит отмыт от мелких фракций.
Далее катиониты промывают несколько раз при настаивании 4—5 н. НС1, которую
сливают декантацией, а затем отмывают смолу от избытка НС1, многократно обрабатывая
ее горячей дистиллированной водой до отсутствия в промывных водах иона хлора (проба
с AgN03). Так получают очищенный катионит в Н-форме. Если катионит требуется полу¬
чить в другой форме, то после очистки НС1 его обрабатывают соответствующими реакти¬
вами (СаС12; NH4C1; КС1 и др.).
Для очистки анионитов рекомендуется попеременная их обработка щелочью, водой,
затем кислотой и опять водой и т. д. Выполняют это следующим образом. Промытый водой
анионит заливают 0,5 н. раствором NaOH, перемешивают, настаивают 30—60 мин., затем
раствор щелочи сливают на большие воронки с фильтрами или воронки Бюхнера декан¬
тацией. Эту процедуру повторяют несколько раз, после чего анионит отмывают от избыт¬
ка щелочи дистиллированной водой до pH воды (проба с индикаторами фенолфталеином, ме¬
тилоранжем или универсальной индикаторной бумагой). Таким образом получают анио~
нит в ОН-форме. Анионит можно перевести в С 1-форму, насыщая его 1 н. раствором НС1.
Процедура эта выполняется точно так же, как описано выше в случае обработки анионита
щелочью. Избыток НС1 затем тщательно отмывают теплой водой (не выше 40—50° С)
до отсутствия в промывных водах иона хлора (проба с AgN03). Если требуется получить
анионит в С03-форме, то анионит в ОН-или Cl-форме обрабатывают раствором Na2COs
до установления в промывных водах pH этой соли, затем смолу тщательно отмывают от
избытка соли Na2C03 до j)H воды (проба с соответствующими индикаторами) и т. п.
После очистки и насыщения ионообменных смол соответствующими ионами их дово¬
дят до воздушно-сухого состояния между листами фильтровальной бумаги при комнатной
температуре. В ряде случаев смолы сохраняют в дистиллированной воде.
В дальнейшем, если требуется, смолы просевают через сита с отверстиями определен¬
ного диаметра или растирают в фарфоровой ступке, или размалывают на кофейной или
шаровой мельницах и т. п.
2а» Изготовление] целлофановых пакетов. Целлофановый лист размером 5 X 7 см
складывают] пополам, дважды отгибают полоску сбоку, а затем с двух противоположных
боков складывают лист в виде треугольника, как это делают в случае изготовления бу¬
мажных пакетов для хранения образцов почв и растений.
26* Изготовление капроновых мешочков (Похлебкина, 1973). Используют прозрачную
капроновую ленту шириной 5—6 см. Разрезают ее на отрезки длиной 6—8 см, складывают
вдвое и края с двух сторон осторожно заплавляют иди на газе, или горячим ножом, или
обжигая их горящей спичкой и т. п. Можно ленту с двух сторон прошить нитками.
Определение степени подвижности фосфатов почвы
(фактор «интенсивности»)
О степени подвижности фосфатов в почвах («интенсивности») можно судить
по способности твердых фаз почвы отдавать в раствор ионы фосфора. Мерилом
этой способности является установление концентрации фосфора в почвен¬
ном растворе. Но выделение почвенного раствора практически сложно, по¬
этому исследователями (Aslyng, 1954; Schofield, 1955; Карпинский, Замятина,
1958, и др.) предложено использовать водные и слабосолевые вытяжки (Н20;
0,01 М СаС12; 0,03 н. K2S04) при узком отношении почвы к раствору, что поз¬
воляет получить данные, близкие к концентрации фосфора в почвенном ра¬
створе.
«Интенсивность» может быть выражена различными способами: 1) как
общая концентрация фосфора в водных и солевых вытяжках (Р2Об, мг/л);
2) концентрация отдельных фосфат-ионов (ЩРС^1-, НР02“4, моль/л); 3) ак¬
тивность отдельных фосфат-ионов (ан,Р04> Днро4> М0ЛЬ!л\ 4) суммарная ак¬
156
К. Е. Гинзбург
тивность ионов кальция и отдельных фосфат-ионов в логарифмической
форме — фосфатный потенциал почв — 0,5рСа + (рН2Р04 + 0,5рНР04), где
р — знак отрицательного логарифма.
Применение того или иного показателя для характеристики фактора
«интенсивности» зависит от целей и задач исследования. Установлено, что
наиболее удовлетворительную корреляцию со степенью обеспеченности
почв фосфором, доступным растениям, показывает общая концентрация его
в слабосолевых и водных вытяжках (Блэк, 1973).
Блэк (1973), Н. П. Карпинский, В. Б. Замятина и Н. М. Глазунова (1960)
показали, что в группе почв, сходных по свойствам и генезису, содержание
фосфора в кислотных, щелочных, буферных вытяжках (фактор «емкости»)
хорошо коррелирует с концентрацией фосфора в водных и слабосолевых ра¬
створах (фактор «интенсивности»). Поэтому в данном случае каждый из этих
методов может быть использован для характеристики обеспеченности почв
фосфатами.
В группе несходных почв корреляция между показателями «емкости» и
«интенсивности» обычно отсутствует и приходится в каждом конкретном слу¬
чае устанавливать, какой тип определения более подходит для установления
степени обеспеченности фосфором той или другой почвы. Некоторые почвы
в силу своих природных особенностей, обусловленных характером почво¬
образовательных процессов в них, содержат высокие количества кислотно¬
растворимого фосфора, которые не соответствуют фактической степени обес¬
печенности этих почв фосфатами. В данном случае для характеристики пло¬
дородия в отношении фосфора более приемлемы водные и слабосолевые вы¬
тяжки (Карпинский и др., 1960).
Водные вытяжки
Метод основан на извлечении ортофосфата из почв водными вытяжками.
Использование водных вытяжек с узким отношением почва : вода
(1 : 1; 1 : 5; 1 : 10) дает представление о концентрации фосфора в почвенном
растворе. Водные вытяжки с широким отношением почва : вода (1 : 50;
1 : 100; 1 : 200) дают представление об общем содержании воднорастворимых
фосфатов в почве.
Метод пригоден для анализов различных типов почв.
Ход анализа. 20 г воздушно-сухой почвы (сито d 1—2 мм) помещают в
плоскодонные колбы емкостью 250 мл, приливают 100 мл дистиллированной
воды, взбалтывают в течение 30 мин. и фильтруют. Получить прозрачную
водную вытяжку из почвы во многих случаях весьма трудно. Первые порции
фильтрата приходится отбрасывать, прибегать к заиливанию фильтра, пе¬
ренося на него возможно больше почвенной суспензии, к центрифугированию
при большом числе оборотов или к ультрафильтрации полученной вытяжки.
Получить прозрачный раствор водной вытяжки можно также, добавляя в
фильтрат 1—5 г (если требуется, то больше) соли NaCl или Na2S04, или соли
другого электролита для коагуляции коллоидных частиц. После этого раствор
вновь фильтруют, желательно через плотный беззольный фильтр (синяя, бе¬
лая лента).
Если вытяжка окрашена воднорастворимыми гумусовыми веществами,
то ее перед колориметрическим определением фосфора обесцвечивают точно так
же, как описано выше в методе Мачигина.
20—40 мл прозрачного бесцветного фильтрата помещают в мерные колбы
на 50—100 мл и определяют фосфор колориметрически.
Для повышения концентрации фосфора в колориметрируемых растворах
водные вытяжки, если требуется, упаривают до небольшого объема. Для по¬
вышения чувствительности колориметрическое определение фосфора прово¬
дят в кюветах диаметром 30—50 мм.
Методы определения фосфора в почве
157
Полученные данные выражают в миллиграммах Р2Об на 1 л при узком от¬
ношении почва : вода, а при широком отношении — в миллиграммах P2Os
на 100 г почвы.
Метод Скофилда
(Schofield, 1955)
Метод основан на определении концентрации ортофосфата в вытяжке
0,01 М (0,02 н.) СаС12. Соотношение почва : раствор = 1:5 или 1 : 10 (по¬
лучаемые при этом цифры близки между собой). Метод предложен для опре¬
деления «интенсивности» на кислых, нейтральных и карбонатных почвах.
Ход анализа. 20 г воздушно-сухой почвы (сито d 1—2 мм) помещают в пло¬
скодонные колбы емкостью 250 мл, приливают 100 мл 0,02 н. раствора СаС12,
взбалтывают 5 мин., фильтруют через плотные беззольные фильтры (синяя,
белая лента), перенося на фильтр как можно больше почвы, чтобы ваилить
его. Первые порции фильтрата сливают.
20—40 мл прозрачного фильтрата берут для колориметрического опре¬
деления фосфора. Полученные данные выражают в миллиграммах Р206 на
1 л раствора. Солевые вытяжки при фильтровании дают прозрачные раст¬
воры. В этом их преимущество перед водными вытяжками. Но в солевых
вытяжках концентрация фосфора, как правило, ниже, чем в водных, что
в свою очередь затрудняет их анализ.
Если требуется, солевые вытяжки упаривают до небольшого объема.
Колориметрируют их в кюветах диаметром 30 или 50 мм.
Р^е активы. 1. Экстрагирующий раствор — 0,01 М (что соответствует 0,02 н.)
СаС12. 1а. Запасный раствор 0,2 н. СаС12. 22 г солиСаС12*6Н20 растворяют в 300 мл дистил¬
лированной воды, доливают затем точно до 1 л, перемешивают. В полученном растворе оп¬
ределяют концентрацию кальция объемным трилонометрическим методом1.
Пример расчета. Расход трилона Б составил 4 мл; его нормальность —
0,05 н.; поправка к титру —0,985. Концентрация запасного раствора СаС]2 равна: CGaGlt =
= 4*0,05*0,985 = 0,197.
16. Для приготовления рабочего раствора 0,02 н. СаС12 надо взять 20 : 0,197 =
= 101,5 мл запасного раствора СаС12, долить водой до 1 л, перемешать. Раствор исполь¬
зуют для приготовления почвенных вытяжек.
Метод Карпинского и Замятиной (1958)
Метод основан на определении концентрации ортофосфата в вытяжке
0,03 н. раствора K2S04. Соотношение почва : раствор = 1:5. Данная вы¬
тяжка извлекает в 1,5—2 раза больше фосфора из почвы, чем раствор 0,01 М
СаС12.
Метод предложен для определения степени подвижности фосфатов («фос¬
фатного уровня») на кислых и нейтральных почвах.
Ход анализа. 20 г воздушно-сухой почвы (ситой1—2 деле) помещают в пло¬
скодонные колбы емкостью 250 мл, приливают 100 мл раствора K2S04, взбал¬
тывают 5 мин., фильтруют и определяют фосфор колориметрически.
Если необходимо, фильтрат концентрируют, упаривая его до небольшого
объема, но не более, чем в 2 раза, чтобы избежать выпадения солей из ра¬
створа. Колориметрирование проводят в кюветах диаметром 30 или 50 мм.
Результаты анализов выражают в миллиграммах Р2Об на 1 л раствора.
Реактив. Извлекающий раствор — 0,03 н. K2S04. 2,6 г соли pacreoj яют
в дистиллированной воде, доливают водой до 1 л, перемешивают.
1 См. статью 3. Г. Ильковской и А. С. Коноваловой в настоящем сборнике.
158
К. Е. Гинзбург
Для почв нечерноземной полосы авторы установили следующие ориен¬
тировочные индексы обеспеченности по фосфору для 0,03 н. K2S04 вытяжки
(мг Р2Об на 1 л): 1) очень низкая — 0,01—0,03; 2) средняя — 0,06—0,08;
3) высокая — > 0,2.
Метод Францесона
(см. Замятина, 1969)
Метод основан на извлечении ортофосфатов почвы 0,006 н. раствором НС1
с pH 3,1. Соотношение почвы к раствору 1 : 10.
Метод предложен для изучения динамики растворимых фосфатов в чер¬
ноземных почвах. Метод нуждается в испытании на других типах почв.
Ход анализа. 10 г воздушно-сухой почвы (сито d 1—2 мм) помещают в пло¬
скодонные колбы емкостью 250 мл, приливают 100 мл 0,006 н. раствора НС1
и взбалтывают от руки 2—3 раза через каждые 10 мин. в течение часа, филь¬
труют.
20—40 мл прозрачного фильтрата помещают в мерные колбы на 50 мл и
определяют фосфор колориметрически.
Содержание фосфора в вытяжке выражают в миллиграммах Р206 на 100 г
почвы.
Реактив. Экстрагирующий раствор — 0,006 н. НС1.
МЕТОД КРИВЫХ РАСТВОРИМОСТИ ФОСФАТОВ ПОЧВЫ
[(БОБКО, МАСЛОВА, 1926)
Метод основан на извлечении фосфатов почвы кислотой (0,1 н. НС1) и
щелочью [0,03 н. Са(ОН)2] в интервале pH от 2 до 8. Одновременно определяется
способность почв изменять величину pH под влиянием добавленных к ней
кислоты и щелочи, т. е. устанавливается буферная способность почв.
Известно (Аскинази, 1949), что различные соединения фосфатов кальция
наиболее устойчивы при слабощелочном и близком к нейтральному значениях
pH. Фосфаты же полуторных окислов, наоборот, более устойчивы в интер¬
вале pH 2,5—4,5. При подщелачивании переход их в раствор увеличи¬
вается.
Определение кривых растворимости может помочь установить формы ра¬
створимых фосфатов, участвующих в фосфатном равновесии в различных
почвенных условиях.
Ход анализа. К восьми отдельным навескам почвы, по 10 г каждая,
прибавляют по 100 мл воды (освобожденной от С02), содержащей возра¬
стающие количества 0,1 н. раствора НС1 —2, 4, 6, 9 и 12 мл и 0,1 н. раствора
Са(ОН)2— 4, 7, 10 мл. Кислые вытяжки оставляют на сутки, периодически
встряхивая; через щелочные вытяжки, помещенные в склянки Дрекселя,
пропускают воздух или слабый ток углекислого газа, пока в растворе Са(ОН)2
контрольного сосуда, поставленного за склянками с почвой, не обнаружится
полного осаждения кальция в виде СаС03 (проба на фенолфталеин). По истече¬
нии суток сосуды открывают, раствор фильтруют через беззольный фильтр
и фильтрат анализируют на содержание фосфора, а при более углубленных
исследованиях также и на содержание ионов Са2+, Mg2+, Fe3+, А13+, Н+.
Реактивы. 1. 0,1 н. раствор НС1.
2. 0,1 н. раствор Са(ОН)2. 4,6 г соли Са(0Н)2*Н20 растворяют в 900 мл прокипяченной
(для удаления СОг) дистиллированной воды и доливают до 1 л водой, перемешивают.
Методы определения фосфора в почве
159
ОПРЕДЕЛЕНИЕ ЕМКОСТИ ПОГЛОЩЕНИЯ ФОСФОРА
В ПОЧВАХ ПО АСКИНАЗИ И ГИНЗБУРГ (1957)
Метод основан на взаимодействии навески почвы с растворами монокаль-
цийфосфата или 0,01 н. фосфорной кислоты. Соотношение почвы : раствор =
= 1 : 10; время взаимодействия — 1 час взбалтывания, 24 часа настаива¬
ния; температура — 20 ± 5° С.
Величина емкости поглощения фосфат-ионов вычисляется по разности
между количеством добавленного к почве и найденного в фильтрате фос¬
фора. Выражают ее в миллиграмм-эквивалентах Р2Об на 100 г почвы, причем
1 мг-экв фосфора (по Р04~)= 23,7 мг Р2Об.
Полученные по данному способу величины емкости поглощения фосфат-ио¬
нов были наибольшими на красноземных почвах (в среднем около 26 мг-экв
Р2Об на 100 г почвы), примерно в 2 раза они были ниже на желтоземных
почвах (около 15 мг-экв).
Для более подробной характеристики поглотительной способности почв
в отношении фосфора, кроме величины емкости поглощения фосфат-ионов,
Д. JI. Аскинази (1957) предложил использовать соотношение емкости погло¬
щения фосфат-ионов и емкости поглощения катионов. Было установлено, что
чем выше это соотношение, тем большей способностью поглощать фосфат-
ионы обладают почвы.
Ход анализа. Способ I. 2,5 г воздушно-сухой почвы (сито d 0,25 мм) по¬
мещают в плоскодонные колбочки на 50—100 мл, приливают 25 мл раствора
монокальцийфосфата (реактив 1а), содержащего около 21 мг Р2Об. Смесь
взбалтывают 1 час, настаивают 24 часа и фильтруют. В фильтрате опреде¬
ляют фосфор колориметрически. Колориметрическое определение фосфора
одновременно проводят и в исходном растворе фосфата (реактив 16).
Способ II. 2,5 г воздушно-сухой почвы (сито d 0,25 мм) помещают в пло¬
скодонные колбочки на 50—100 мл, приливают 25 мл 0,01 н. раствора Н3Р04
(реактив 26), содержащего около 21 мг Р206. В дальнейшем поступают точно
так же, как описано выше в способе I.
Абсолютные величины емкости поглощения фосфатов, определяемые спо¬
собом II, обычно несколько выше (иногда в 1,5 раза), чем полученные спо¬
собом I.
Чтобы избежать ошибок определения, обусловленных многократным
разбавлением исходных и испытуемых растворов, содержащих высокие
концентрации фосфора, рекомендуем для колориметрического определе¬
ния фосфора использовать менее чувствительный ванадатно-молибдатный
метод.
Реактивы. 1а. Крепкий раствор монокальцийфосфата. 4,46 г соли
Са(Н2Р04)2*2Н20, х. ч. или ч. д. а., всыпают в 800 мл дистиллированной воды, раст¬
воряют и доливают водой до 1 л, перемешивают; если необходимо, раствор отфильтровы¬
вают.
15. Рабочий раствор монокальцийфосфата. К 700 мл раствора 1а приливают 1800 мл
дистиллированной воды, перемешивают. 25 мл этого раствора добавляют к почве, он со¬
держит примерно 21 мг Р205, что соответствует 35,3 мг-экв Р205 на 100 г почвы. Содержа¬
ние фосфора проверяют колориметрически.
2а. 1 н. раствор Н3Р04. 7 мл концентрированной Н3Р04 (уд. вес 1,7) вливают осторож¬
но в 500 мл воды, охлаждают, доливают водой до 1 л, перемешивают.
26. Рабочий раствор 0,01 н. Н3Р04. 20 мл раствора 2а доливают до 2 л водой, переме¬
шивают. 25 мл этого раствора добавляют к почве, он содержит примерно 21 мг Р2Об,
или 35,3 мг-экв Р206, на 100 г почвы. Содержание фосфора проверяют колориметрически.
160
К. Е. Гинзбург
ТЕРМОДИНАМИКА И КИНЕТИКА ПОЧВЕННОГО ФОСФОРА
Определение фосфатного потенциала в почвах
В последнее время в агрохимии для характеристики степени подвижности
(«интенсивности» растворения) почвенных фосфатов используют законы
химической термодинамики, которая изучает энергетические соотношения в
различных системах.
Одним из наиболее важных применений химической термодинамики яв¬
ляется нахождение закошу управляющих равновесием химических реакций,
установление констант равновесия' этих реакций. Любая химическая реакция
(обмена, растворения, осаждения и др.) сопровождается выделением или пог¬
лощением тепла. Эти эффекты обусловлены содержанием в каждом веществе
определенного количества энергии, накапливаемой веществом при его обра¬
зовании, так называемой «внутренней», «свободной», «потенциальной» энергии.
Поэтому при постоянных условиях опыта (температура, давление и др.) один
моль каждого индивидуального вещества, находящегося в любой фазе (твер¬
дой, жидкой, газообразной), обладает определенным содержанием этой энер¬
гии (равно, как и определенной массой).
Определить абсолютную величину свободной, потенциальной энергии
для отдельных веществ практически невозможно, но подсчитать ее изме¬
нение (AZ) для любой равновесной обратимой реакции, в которой участвует
это вещество, можно. Выраженная в килокалориях или других условных
значениях эта величина и характеризует термодинамический потенциал дан¬
ной химической реакции или данной системы.
В общем виде химический потенциал (свободная энергия 1 моля веще¬
ства) выражается уравнением.
AZ = AZ°-)-/?7’ In ах , (1)
где AZ — свободная энергия 1 моля вещества, или изобарно-изотермический потенциал,
сокращенно — изобарный потенциал; AZ° — то же, в стандартном состоянии (при актив¬
ности данного вещества в 1 моль, давлении — 1 атом, температуре — 25° С или 298° К);
R = 1,987 кал!град — универсальная газовая константа; Т = 298° К— абсолютная тем¬
пература в градусах Кельвина; In ах — логарифм активности компонента х.
Например, для реакции
изменение свободной энергии в процессе превращения реагентов с актив¬
ностями аА и аВ в продукты реакции с активностями aCnaD равно разности
между изобарными потенциалами конечных продуктов и потенциалами ис¬
ходных веществ:
где а, 6, с, d — стехиометрические коэффициенты веществ А, В, С, D.
Используя уравнение (1) для данной реакции, получаем:
где отношение активностей ионов А, В, С, D есть константа равновесия —
Кх- реакции (2). Величина A Z° представляет собой константу, характери¬
зующую рассматриваемую реакцию при данной £, и называется изменением
стандартной свободной энергии рассматриваемой реакции.
(2)
AZ — 2(яг Zt) конечное — ^ начальное
(3)
или
AZ = cZoc+dZ°D—aZ0A—bZ°B ,
(4)
(5)
Методы определения фосфора в почве
161
Имеется определенная связь между константой равновесия и свободной
энергией. Так, для любой равновесной обратимой реакции при AZ < О
реакция (2) протекает самопроизвольно в прямом направлении, при AZ ^> О
реакция самопроизвольно протекает в обратном направлении, при AZ = О
система находится в равновесии. При равновесии уравнение (1) примет вид:
AZ° = — RT\nKx. (6)
Для комнатной температуры при переходе к десятичным логарифмам для
AZ° получается следующее выражение (в ккал/молъ):
AZ0 = —1,987-298-2,3 lg = —1364 lg . (7)
Таким образом, стандартная свободная энергия всех веществ, а также
ионов определяется как стандартная свободная энергия реакций образования
их из простых веществ или гидратированных ионов водорода (для водных ра¬
створов).
Величина свободной энергии позволяет предсказать возможность проте¬
кания той или иной реакции в данной системе, характеризовать состояние
равновесия этой реакции и приближенно рассчитать значение константы рав¬
новесия.
Почва представляет собой сложную многофазную систему, поэтому ми¬
неральный фосфор в почвенном растворе принимает участие во многих рав¬
новесных реакциях:
1. Реакции, происходящие только между жидкими фазами или только
между твердыми фазами,— гомогенные равновесия:
р —► р р —> р
х раствор 1 раствор , гтв. фаза ^тв. фаза
реакции, идущие с большой реакции, идущие с малой
скоростью скоростью
Формы, в которых фосфор находится в растворе, определяются реакция¬
ми протонирования (реакции, в которых хотя бы один из ионов воды, Н+ или
ОН", принимает участие в равновесии) и комплексообразования. В резуль¬
тате этих реакций Н8Р04 и соответствующие ей ионы Н2Р07, НРО4”, РО4""
образуют простые и сложные соли, растворимые комплексные соединения,
которые и находятся между собой в равновесии.
2. Реакции, происходящие между твердыми и жидкими фазами — гете¬
рогенные равновесия:
Р р
1 почва«— г раствор*
Реакции фосфора в данной равновесной системе включают растворение и
осаждение труднорастворимых фосфорных солей, которые контролируются
произведением растворимости этих соединений и адсорбцией фосфора на по¬
верхности почвенных частиц.
Поскольку концентрация фосфора в растворе обусловлена реакциями его
с твердыми фазами, то состояние равновесия между ними может быть исполь¬
зовано для характеристики энергетического состояния фосфора во всей си¬
стеме. Это положено в основу работ по определению химического потенциала
•фосфатов в почве. Известен принцип физической химии, что в любой много¬
фазной системе при равновесии химические потенциалы или молярные «сво¬
бодные» энергии для всех диффузноспособных химических компонентов рав¬
ны (при отсутствии скачка потенциала). Таким образом, в системах, состоящих
из жидких фаз, находящихся в равновесии с твердыми фазами, химические
потенциалы всех компонентов одни и те же. Поэтому потенциалы твердых
фаз почвы могут быть вычислены по активностям ионов, находящихся
в растворе.
Скофилд (Schofield, 1955), Аслинг (Aslyng, 1954), Ульрих (Ulrich, 1961)
и другие исследователи предложили величину термодинамического химиче¬
ского потенциала моно- и дикальцийфосфата в равновесной гетерогенной
162
К. Е. Гинзбург
системе почва — почвенный раствор использовать для оценки обеспеченно¬
сти почвы фосфором, т. е. для определения «интенсивности» самопроизволь¬
ного перехода фосфора из твердых фаз почвы в раствор или вычисления
энергии химической реакции растворения фосфатов почвы. Полученная та¬
ким образом величина не является показателем абсолютного содержания
растворимых фосфатов в почвах, но дает им некоторую качественную оценку
и служит показателем степени подвижности почвенных фосфатов. Вывод
формулы фосфатного потенциала основан на использовании закона дей¬
ствующих масс с внесением соответствующих поправок на истинную, эффек¬
тивную концентрацию фосфора и других ионов в растворе, т. е. с учетом ак¬
тивной концентрации веществ, принимающих участие в равновесных реак*
циях.
Установлено, что на кислых, слабокислых и нейтральных почвах в поч¬
венном растворе содержатся преимущественно монофосфатные ионы, и опре¬
деление фосфатного потенциала в данных условиях проводится с учетом ак¬
тивности в растворе этих ионов. На карбонатных почвах в равновесных ра¬
створах содержатся моно- и дифосфатные ионы, поэтому при вычислении
фосфатного потенциала необходимо учитывать сумму двух этих ионов (Ulrich,
1961).
В первом случае при значении Рпотенциала > 7 отмечается плохой фосфат¬
ный режим ПОЧВ. На карбонатных же почвах значение Рпотенциала ]> 7
не свидетельствует об их плохом фосфатном режиме.
Метод Аслинга (Aslyng, 1954)
и Скофилда (Schofield, 1955)
Метод основан на определении активной концентрации ионов Н ,Р04, Са,
Mg, а на кислых почвах — ионов А1 и Fe в вытяжке 0,01 М СаС12; соотношение
почва : раствор = 1:2; время взаимодействия 15—30 мин.; температура —
25 ± 1° С.
Ход анализа. 50 г воздушно-сухой почвы помещают в плоскодонные кол¬
бочки емкостью 250 мл или центрифужные пробирки, заливают 100 мл 0,01 М
раствора СаС12 и взбалтывают на ротаторе в течение 30 мин. Для карбонат¬
ных почв время взбалтывания следует сократить до 15 мин., чтобы избежать
влияния растворяющихся карбонатов.
Взбалтывание следует проводить в открытой посуде, чтобы предотвратить
накопление С02. Лучше всего процедуру взбалтывания заменить пропуска¬
нием воздуха через суспензию с помощью компрессора типа КВМ-3 или др.
После этого суспензию фильтруют или центрифугируют. В фильтрате опре¬
деляют pH потенциометрически, кальций и магний комплексонометрически,-
фосфор колориметрически, алюминий и железо спектрофотометрически
(Беляева, 1966).
Реактив. 0,01 М раствор СаС12. Приготовление см. выше в разделе «Определение
степени подвижности фосфатов почвы».
Метод Ульриха (Ulrich, 1961)1
Метод основан на определении активной концентрации ионов Н, Р04, Са,.
Mg, а на кислых почвах — ионов А1 и Fe в водной вытяжке при узком соот¬
ношении почва : раствор — 1,5 : 1; t = 25 ± 1° С.
1 Ход анализа описан в статье О. П. Медведевой в настоящем сборнике.
Методы, определения фосфора в почве
163
Определение фосфатного потенциала
в почвенном растворе (Кобзева, 1969)
Метод основан на определении активной концентрации ионов Н, Р, Са,
Mg, а на кислых почвах — ионов А1 и Fe в почвенном растворе.
Ход анализа. Почвенный раствор получают давлением сжатого азота в
8—10 атм на почву, увлажненную до 70% от полной влагоемкости. Выделение
лочвенного раствора проводят на приборе Рейтмейера и Ричардса, который
описан Д. В. Федоровским (1964). Соотношение почва : вода составляет при¬
мерно 2,5 : 1.
Полученный раствор следует считать более близким к истинному почвен¬
ному раствору и ближе к состоянию равновесия, чем при соотношении
почвы к раствору 1 : 2 и 1,5 : 1, принятых Скофилдом, Аслингом
и Ульрихом.
Вычисление фосфатного потенциала
Способ Ульриха (Ulrich, 1961)
В почвенном растворе фосфатное равновесие обусловлено содержанием
фосфорной кислоты с соответствующими ионами — Н2Р07, НРО^-, РО^-.
Распределение фосфора между фосфорной кислотой и фосфатными ионами
определяется преимущественно значениями pH в следующем равновесии
(реакции протонирования):
Фосфорная кислота ^ фосфорное основание + Н+ (протон).
Согласно закону действующих масс, константа диссоциации Н3Р04 в дан-
„ [РоснННЧ
ной равновесной системе выражается уравнением К = —гр -—, где квад-
[*кисл]
ратные скобки обозначают концентрацию соответствующих ионов.
Для более точных расчетов вместо равновесных концентраций пользуются
активностями. Эта величина введена для учета взаимного притяжения ионов,
взаимодействия растворенного вещества с растворителем и других явлений,
изменяющих подвижность ионов и не учитываемых теорий электролитиче¬
ской диссоциации. Активность является эффективной, действующей концен¬
трацией, проявляющей себя в химических процессах в качестве реально дей¬
ствующей массы в отличие от общей концентрации вещества в растворе.
Численно активность (а) равна:
a = Cf , (8)
где С — концентрация вещества; / — коэффициент его активности.
Коэффициент активности отражает все изменения подвижности ионов,
.происходящие в данной системе. При бесконечном разбавлении а = С, а
/ = 1; для реальных систем а Ф С, а / < 1 или / > 1 (в очень концентриро¬
ванных растворах).
Коэффициент активности ионов (/) зависит не только от концентрации
данного электролита в растворе, но и от концентрации посторонних ионов,
присутствующих в этом растворе. Мерой электрического взаимодействия меж¬
ду всеми ионами в растворе является так называемая ионная сила, величина
которой зависит от концентрации и зарядов всех ионов, присутствующих
в растворе. Ионная сила (\i) раствора, содержащего, например, ионы Kt,
Ап ит. п., может быть определена по формуле
у=4' (zht[Kt] + zin[An] + •••)» (9)
где [Kt], [Ап] — концентрация ионов Kt, Ап, г-ион/л', ZKt. «ап — заряды этих
ионов.
164
Я*. Е. Гинзбург
Или ионная сила раствора равна полусумме произведений концентраций
ионов на квадраты их зарядов:
,i=4-2С<г*2- <10>
С увеличением ионной силы раствора коэффициент активности уменьшает¬
ся. Зависимость коэффициента активности иона от ионной силы разбавлен¬
ного водного раствора электролита выражается формулой Дебая и Хюккеля
0,51
-18' ' i + Vir ■ (И>
где z — заряд иона; ц — ионная сила раствора.
Значения коэффициентов активности для ионов различной валентности и
растворов с различной ионной силой приведены в табл. 10 и на рис. 1 в ста¬
тье О. П. Медведевой (см. настоящий сборник).
Таблица 10
Приближенные значения средних коэффициентов активности при разной ионной силе
раствора (р)
(Крешков, 1970)
Заряд иона
0
0,001
0,002
0,005
0,01
0,02
0,05
0,1
0,2
1
1
0,97
0,95
0,93
0,90
0,87
0,81
0,76
0,70
2
1
0,87
0,82
0,74
0,66
0,57
0,44
0,33
0,24
3
1
0,73
0,64
0,51
0,39
0,28
0,15
0,08
0,04
4
1
0,56
0,45
0,30
0,19
0,10
0,04
0,01
0,003
Для Н+
1
0,98
0,97
0,95
0,92
0,90
0,88
0,84
0,83
Для ОН-
1
0,98
0,97
0,95
0,92
0,89
0,85
0,81
0,80
HaPO* ^ HBOf+Н+;
HPOf^PO|“+H+;
После введения необходимых поправок к закону действующих масс вер¬
немся к рассмотрению констант ионизации фосфорной кислоты.
Н3Р04 — трехосновная кислота, она может отдавать в раствор три иона
водорода, поэтому существуют три константы ее диссоциации, соответствую¬
щие следующим равновесиям:
Н8Р04 ^ Н2Р(>7 +Н+; [Н+] [HaPOJ ] =Ki [Н3РО4], (12)
[Н+] [HPOf] = К* [H2POJ , (13>
[H+][POf ]=K3[HPOf]. (14>
В зависимости от величины pH Н8Р04 может существовать в растворе ли¬
бо в недиссоциированной форме, либо в виде любой анионной формы. Можно
вычислить содержание отдельных фосфатных ионов в растворе как функцию
величины [Н+]. График этой зависимости (рис. 3) дает общую картину
равновесия. Как видно из рис. 3, в почвенном растворе большинства
почв (pH 3,5—8,5) присутствуют главным образом ионы Н2Р04~ и НР042'.
Таким образом, общая концентрация фосфора в почвенном растворе (Ср)
определяется по уравнению
Ср _ CHtр04 + CKVOa 3 (15)
отсюда
^нро* =cv~ • (15а
Методы определения фосфора в почве
165
Согласно (8), выражаем концентрации через активности соответствующих ио¬
нов, и тогда уравнения (12—14) примут вид:
0н*с?н,ро4*/1 = ^-^НзРОг/з* (16)
ан • ^нро4 * /2 = * ^н,ро4 • h, U7)
ан • ^ро4 • /3 = к* • ^нро4 • /2 , (18)
где /j, /2, /3— коэффициенты активностей соответственно одно-, двух-, трехзарядных ионов
Н3Р04.
Выражаем Снро4 в (17) через (15а), получаем
аН * f2 (^р СН,Р04) = K*-CHtV04-h • (19)
После соответствующих алгебраических преобразований в (19) (откры¬
вание скобок, группировка членов, умножения обеих частей уравнения на
%
Рис. 3. Распределение содер¬
жания ионов фосфорной кисло¬
ты при различных значениях
pH (Larsen, 1967)
Сплошная линия — при бесконеч¬
ном разбавлении, пунктирная ли¬
ния — при ионной силе |i= 0,03
рк
одинаковый множитель и т. п.) получаем значение активности монофосфатного
иона:
ан,ро4
Ст)
ан-/г
Кг + ан д
(20)
Аналогично из (15) и (17) выводят формулу для определения активности
НР042-:
Кг-h
(21)
"НРО, — СР
h
В (20) и (21) Ср — общая концентрация фосфора в растворе {моль/л),
определяется колориметрически.
aH'f* / (*« + *Н -£-) и Kt.f! I (ан + Кг Jl) — корректирующие фак¬
торы, устанавливающие долю содержания ионов Н2РО“4 и НР02~4 в общей
концентрации фосфора в растворе. Вычисляются из (20) и (21).
ав — активность ионов водорода, определяется из значений pH; Д и /2 —
коэффициенты активности соответственно одно- и двухзарядных ионов. Вы¬
числяются по (И) или берутся из табл. 10; К2 = 6,53* 10“8 — вторая констан¬
та диссоциации Н3Р04.
Для получения линейной зависимости концентрацию фосфора и других
ионов, содержащихся в растворе, обычно выражают в логарифмической
форме. Но так как абсолютные значения концентраций ионов в почвенном
растворе крайне низки, то для удобства проведения различных расчетов ис¬
пользуют отрицательные логарифмы активностей этих ионов (Батлер, 1973).
166
К. Е. Гинзбург
Таким образом, для вычисления фосфатного потенциала определяют в
водных и слабосолевых вытяжках из почв активности ионов acai ан,
«н2ро4> Днро4 > яаь «Fe, затем находят их отрицательные логарифмы
(р): рСа, pH, pH2P04, pHP04, pAl, pFe. Далее вычисляют фосфатный и изве¬
стковый потенциалы по следующим уравнениям:
0.5.Ca+pH2P(V—для кислых, слабокислых, нейтральных почв; (22)
0f5pCa-f-0e5pHPO4+pH2PO4 — для карбонатных почв; (23)
1
— рА1+рНгР04 — для кислых, слабокислых почв; (24)
1
-g- pFe-f-pH2P04 — то же; (25)
pH -(-0,5 (Ca-f-Mg) — для кислых, слабокислых, нейтральных, карбонатных почв. (26)
^Пример расчета фосфатного потенциала по уравнению (22). Почва дерново»
подзолистая, среднесуглинистая, вариант — без удобрений.
1. Определение значений fi и /.
Как указывалось выше, значения коэффициентов активностей (/) различных ионов
зависят от ионной силы данного раствора (ц).
Для определения ц в почвенной вытяжке 0,01 М СаС12 учитываем концентрации пре-
обладающего катиона Са2+ и эквивалентного ему количества одновалентного аниона С1~»
В исследуемом растворе найдено содержание ионов Са = 520 мг/л = 0,52 г/л. Чтобы ус»
тановить концентрацию ССа, необходимо содержание его в граммах на 1 л перевести в мо¬
ли на 1 л. Для этого значения в граммах на 1 л делим на молекулярный вес кальция и по*
лучаем: ССа = 0,52 : 40 = 0,0130 = 1,3* 10“2 моль/л.
По (10) определяем \i: fi =1/2 2 С\ -z\ = V2 (CCSi*22 + CG1* l2). Так как на один ион кал!*»
ция приходятся два иона хлора, то практически получаем: = Va (1,3*10“2*22 +
+ 2.1,3-10-М2) = 1/2 (5,2.10-4- 2,6* 10“2) = 3,9* 10“2^ 0,04.
Из табл. 10 для ц = 0,04 находим соответствующие]коэффициенты активностей /н =
= 0,89; 0,82; /2= 0,46/Для более точных расчетов / вычисляют по (11). Пример такого
расчета дан в табл. 11.
2. Определение Сн и ан.
В данном растворе установлено значение pH = 4,85. Отсюда pH = —lg [Н+] =
= 4,85; [Н+] = —4,85 = (—5 + 0,15). По таблице антилогарифмов находим: 0,15 (ман¬
тисса) = 1413 [Н+] = 0,00001413 г-ион/л или = 1,41 «10“? моль/л. = £н*/н =
= 1,41-Ю"5*0,89 = 1,25.10-® моль/л.
3. Определение аСа и рСа.
В исследуемом растворе Сс& = 1,3*10“2 моль/л (см. определение |д и / раствора).
Отсюда аСа = £(*а*/2 = 1,3• 10~2-0,46 = 0,598*10“2 молъ/л, рСа = —lg 0,598«10~2 =
= “lg 5,98*10“8 = —(lg 5,98 + lg 10“8). По таблице логарифмов находимгр Са =*(0,7767—
- 3) = (3-0,7767) = 2,22; 0,5рСа = 2,22 : 2 = 1,11.
4. Определение ян,ро4 и рНаР04. Содержание Р206 в вытяжке установлено колори¬
метрически: Р206 = 1,29 мг/л. Переводим Р2Об в Р. Для этого используем пересчетный
коэффициент 2,3 (Р206 -> 2Р или 142 -> 62; 142: 62 = 2,3). Р = 1,29 Р2Об мг/л : 2,3 =
= 0,56 Р мг/л, или 0,00056 Р г/л Для перевода Р в моли на 1 л содержание его в граммах
на 1 л делим на молекулярный вес фосфора, получаем: Ср = 0,00056 : 31 = 0,00001807 =
= 1,81 *10“6 молъ/л.
Активность монофосфатного иона определяем по (20):
AV/i 6,53-10-*-0.82
вН2Р04 = Ср~ /Г = 1>81,10'5 046“ =1-4610~* моль/л.
г + а*Т 6,53-ю-* + 1,25-10-»
pHjP04= -lg 1,46-10-*= —(lg 1,46 + lg 10-») = -(0,1644-5) = (5-0,1644) = 4,84.
5. Определение фосфатного потенциала по (22):
0,5рСа+рН2Р04 = 1,11+4,84 = 5,96.
В табл. 11 приводятся несколько примеров расчета фосфатного потенциала почв.
Таблица 11
Расчет фосфатного потенциала (0,5 рСа + рНгР04) дерново-подзолистой почвы в вытяжке 0,01 М СаСЬ (1:2) по Ульриху
(см. Кудеярова, 1970)
Варнант
по фону
NK
почвенной
вытяжки
lg [Н+]
сн
ан
Са2+*
мг/л
Саг+'
г/л
сСа»
•10-ямоль/л
сСа‘2’
10-2
ССГ2*
10-*
SC*.г.*
1 г
ю-*
V-
10-*
П
ю-*
0,51 У у.
ю-*
•10-*дс оль/л
Без Р
4,85
5,15
1,41
1,25
520
0,52
1,30
5,20
2,60
7,80
3,90
1,975
1,01
Рс0,1 *
4,85
5,15
1,41
1,25
548
0,55
1,37
5,48
2,74
8,22
4,11
2,027
1,03
Рс°.4
4,95
5,05
1,12
1,00
580
0,58
1,45
5,80
2,90
8,70
4,35
2,086
1,07
Вариант
по фону NK
1 + Vt
-1 &/» =
(при
0,51 W-z*
i+ Vjl
г = 1)
/.
0,51 W-г*
—lg ft = -fZT-
i + W
(при г = 2)
/,
аСа,
• 10-Кчоль/л
рСа
/*//*
ан •/„
ю-‘
Без Р
1,198
-0,0845
1,9155
0,82
-0,338 1,662
0,46
0,598
2,22
0,55
0,65
Рс0,1
1,203
—0,0855
1,9145
0,82
—0,342 1,658
0,46
0,653
2,18
0,55
0,64
рс0,4
1,209
—0,0885
1,9115
0,82
-0,354 1,646
0,44
0,643
2,19
0,54
0,50
Вариант
по фону NK
<*Н *ft/ft
ю-»
к + аН */г//|
ю-§
/н,ро4
Р*Об, мг/л,
Р, мг/л
сР,
•10 -ъмоль/л
аН2Р04,
•10 ~ь1мольл
рН2ро4
0,5рСа + pH,POi
Без Р
0,79
0,80
0,82
1,29
0,56
1,82
1,49
4,83
5,94
РСМ
0,77
0,78
0,83
2,12
0,92
2,98
2,47
4,61
5,71
Рс0,4
0,61
0,61
0,81
9,60
4,17
13,40
10,82
3,97
5,07
* Рс — суперфосфат.
168
К. Е. Гинзбург
Способ Аслинга (Aslyng, 1954)
Для ускорения расчетов фосфатного потенциала по формуле (22) Аслинг
приводит таблицу, в которой при различных значениях pH дана вычисленная
уже поправка (А) на долю содержания Н2Р04 в общей концентрации фосфора
(Ср) в вытяжке 0,01 М СаС12 (t ~ 20° С).
Вычисляется поправка А из уравнения рНаР04 = рР + p-R^H (вы¬
водится из уравнения диссоциации Н3Р04, где р — энак отрицательного
логарифма, Р — общая концентрация фосфора в вытяжке (определяется ко¬
лориметрически); рК2 = 6,99 при 20° С (вторая константа диссоциации
Н8Р04); р [Н/(К2 + Н)] — поправка на содержание иона Н2Р04” в общей
концентрации фосфора в вытяжке (приводится в табл. 12 для разных значе¬
ний pH).
Таким образом, пользуясь данными табл. 12, значительно проще прово¬
дить расчеты фосфатного потенциала. В частности, достаточно определить
колориметрически общую концентрацию фосфора в вытяжке (Р), перевести
ее затем в логарифмическую форму (рР) и, исходя из значений pH этой же
вытяжки, по табл. 12 найти поправку А (содержание рН2Р04 в общем рР
при данном pH), которую приплюсовывают к установленному значению рР,
а далее проводят вычисления фосфатного потенциала по (22).
Из данных табл. 12 видно, что в случае кислых, слабокислых почв (pH ^ 5)
рН2Р04 = рР, т. е. А = 0; для почвенных вытяжек с pH > 5
рН2Р04 = рР + А.
Таблица 12
Поправка А для вычисления доли рНгРО* в общем количестве фосфатов (Ср), содержащихся
в 0,01 М СаСЬ вытяжке из почвы при различных значениях pH
(Aslyng, 1954)
pH
Н*Р0С
в Ср, %
А
pH
н,ро4-
в Ср, %
А
pH
Н.РО4-
В Ср, %
А
8,00
9,1
1,04
6,95
52,9
0,28
5,90
92,6
0,03
7,95
10,1
1,00
6,90
55 Л
0,25
5,85
93,4
0,03
7,90
11,2
0,95
6,85
58,5
0,23
5,80
94,1
0,03
7,85
12,4
0,91
6,80
61,3
0,21
5,75
94,7
0,02
7,80
13,7
0,86
6,75
64,0
0,19
5,70
95,2
0,02
7,75
15,1
0,82
6,70
66,6
0,18
5,65
95,7
0,02
7,70
16,6
0,78
6,65
69,1
0,16
5,60
96,2
0,02
7,65
18,3
0,74
6,60
71,5
0,15
5,55
96,6
0,02
7,60
20,1
0,70
6,55
73,8
0,13
5,50
96,9
0,01
7,55
22,0
0,66
6,50
76,0
0,12
5,45
97,3
0,01
7,50
24,0
0,62
6,45
78,0
0,11
5,40
97,6
0,01
7,45
26,2
0,58
6,40
79,9
0,10
5,35
97,8
0,01
7,40
28,5
0,55
6,35
81,7
0,09
5,30
98,0
0,01
7,35
30,9
0,51
6,30
83,4
0,08
5,25
98,2
0,01
7,30
33,4
0,48
6,25
84,9
0,07
5,20
98,4
0,01
7,25
36,0
0,44
6,20
86,3
0,06
5,15
98,6
0,01
7,20
38,7
0,41
6,15
87,6
0,06
5,10
98,8
0,01
7,15
41,5
0,38
6,10
88,8
0,05
5,05
98,9
0,01
7,10
44,3
0,35
6,05
89,9
0,05
5,00
99,0
0,00
7,05
47,1
0,33
6,00
90,9
0,04
7,00
50,0
0,30
5,95
91,8
0,04
Методы определения фосфора в почве
169
Значения фосфатного потенциала, рассчитанные по Ульриху и по Аслин-
гу, близки между собой, но по второму способу затрачивается меньше вре¬
мени на соответствующие вычисления.
Поскольку фосфатный потенциал выражается отрицательным логариф¬
мом активностей ионов Са2+ и Н2РО~4, то при улучшении фосфатного режима
почвы снижается значение фосфатного потенциала и наоборот, что видно из
табл. 11.
Определение минеральных форм фосфатов почвы
по константам произведения растворимости
Для определения минеральных форм фосфатов в почвах в последнее время
используют константы произведений растворимости продуктов реакции. Из¬
вестно, что в насыщенном растворе труднорастворимого электролита проте¬
кают два взаимно противоположных процесса: растворение, т. е. переход ио¬
нов из осадка в раствор, и кристаллизация — переход ионов из раствора в
осадок.
Например, в насыщенном растворе труднорастворимой соли Са3(Р04)ь
между осадком Са3(Р04)2 и находящимися в растворе ионами Са2+ и Р043~
устанавливается подвижное равновесие:
Саз(Р04)г _»ЗСа*++2РОГ'
осадок раствор
Применяя к этому гетерогенному равновесию закон действующих масс
и зная, что концентрация твердой фазы не входит в выражение константы
равновесия, можно написать:
[Са2+]8* [Р03‘]2 = const.
Из этого уравнения следует, что в насыщенном растворе труднораствори¬
мого электролита произведение концентраций его ионов есть величина по¬
стоянная при данной температуре. Эта величина и называется произведением
растворимости и обозначается ПР. Для Са3(Р04)2 ПР = 1,5* 10“32 при 25° С.
Следует также учесть, что при [Са2+]3- [Р043*]2 < ПРсаа(Р04ь получается не¬
насыщенный раствор и для установления равновесия часть осадка перейдет
в раствор, при [Са2+]3*[Р043"]2 > ПРсаа(Р04)а получается перенасыщенный
раствор и электролит из раствора выпадает в осадок и при [Са2+]3 • [Р043“]2=
= ПРса8(Ро*)* получается насыщенный раствор, т. е. система находится в ди¬
намическом равновесии.
Для более точных расчетов, как указывалось выше, необходимо вместо
произведения концентраций ионов данного электролита применять актив¬
ности ионов.
По аналогии с чистыми препаратами фосфатов в гетерогенной системе
почва ^ почвенный раствор по составу ионов в равновесном почвенном рао
творе можно характеризовать соединения фосфатов, содержащиеся в твердых
фазах почвы, вычислив константы произведения растворимости продуктов
реакции. Сопоставляя затем величины ПР, полученные на чистых препа¬
ратах фосфатов и фосфатных минералах с соответствующими ПР, получен¬
ными при анализе почвенного раствора или почвенных вытяжек, устанавли¬
вают, какие примерно фосфорные соединения содержатся в твердых фазах
почвы.
В табл. 13 приводятся ПР наиболее распространенных фосфатных мидора--
лов и искусственно приготовленных чистых фосфатных препаратов, которые
предположительно содержатся в почве. На этой таблице все ПР даны в виде
отрицательных логарифмов активностей ионов, т. е. в виде рКпр.
Таким образом, если исходить из того что состав почвенного раствора ре¬
гулируется реакциями растворения — осаждения только минеральных фос¬
/70
К. Е. Гинзбург
фатов твердых фаз, то описанный подход может быть использован для опре¬
деления минеральных форм фосфатов в почвах.
Метод основан на определении активной концентрации ионов Н, Р04, Са,
Mg, Al, Fe и др. в почвенном растворе или водной, слабосолевой вытяжках при
узком отношении почва : раствор, при постоянной температуре 25° С.
Ход анализа точно такой же, как описано выше при определении фосфат¬
ного потенциала в почвах.
Вычисление констант произведения растворимости
фосфорных соединений (рКПр)
Для расчета рКПр необходимо определить активности всех ионов, на кото¬
рые диссоциирует в раствор то или иное фосфорное соединение, взять их отри¬
цательные логарифмы и подставить в соответствующие уравнения, известные
для чистых фосфатных препаратов (табл. 13).
Величины рКПр, найденные для почвенных фосфатов, сравниваются с тео¬
ретически вычисленными для соответствующих фосфатных препаратов.
Сходимость значений рКпр указывает на то, что данное соединение фосфора
участвует в равновесии в системе почва — почвенный раствор.
Пример расчета произведения растворимости фосфатов почвы.
Произведение растворимости гидроксил апатита (рКНА) выражается уравнением
(см. табл. 13):
РКНА = РСаю (Р04)в (ОН)2 = ЮрСа + 6рР04 + 2рОН. (27)
1. Находим значение рОН:
рОН + pH = pKHi0, где рКн#0 = 14,
рОН = РКН20 -pH-14- pH, (28)
2рОН = 28 — 2рН. (29)
2. Находим значение рР04.
Из уравнения диссоциации фосфорной кислоты (14):
рРО* = рНРО* — pH + рК3, (30)
где рК8 = 12,02 — третья константа диссоциации Н3Р04.
Из уравнения (13):
рНР04 = рНгРО* - pH + рК2, (31)
где рК2= 7,2 — вторая константа диссоциации Н3Р04.
Подставляя значения рНР04 из (31) в (30), получаем:
РР04 = рШРОл — pH + рКг - pH + рКз = pHaPOi — 2рН+7,2 +12,02. (32)
3. Находим значение рКод*
Подставляем (29) и (32) в (27):
РКНА = 10РСа + 6 (РН2РО4 - 2рН + 19,22) +28 - 2рН = ЮрСа+брШРО* — 14рН +
+143,32.
В связи с тем что растворимость фосфатов кальция зависит от концен¬
трации в растворе ионов водорода и кальция, Аслинг (1954) предложил про¬
изведения растворимости этих соединений выражать через их фосфатный и
известковый потенциалы, что упрощает также расчеты их рКПр. Тогда уравне¬
ние (27) примет вид:
РКНА = (3РСа + 7РСа) + брНаРО* - 14рН+143,3 = 6 (0,5рСа+рНаР04) -14 (pH -
— 0,5рСа) + 143,3.
Таблица 13
Произведения растворимости и константы диссоциации некоторых химических соединений
(Кудеярова, 1970; Lindsay, Moreno, 1960)
Препарат
Химическая формула
Произведения растворимости при ajjto = 1
Величина рКпп
при 25° С р
Дикальцийфосфат безводный (DCP)
СаНР04
рК = рСа + рНР04
6,66
Дикальцийфосфат водный (DGP)
СаНР04-Н20
рК = рСа + рНР04
6,56
Октакальцийфосфат (ОСР)
Са4(Р04)3Н-ЗН20
рК = 4рСа + pH + ЗрР04
46,91
Гидроксилапатит (НА)
Са10(Р(ШОН)г
рК = ЮрСа + 6рР04 + 2рОН
113,7
Фторапатит (FA)
Caio(P04)eF2
рК = ЮрСа + 6рР04 + 2pF
118,4
Варисцит (А1Р)
А1Р04-2Н20
рК = рА1 + 2рОН + рН2Р04
Ь0,5
Стренгит (FeP)
FeP04-2H20
рК = pFe + 2рОН + РН2Р04
35,0
Таранакит аммония (NH4P)
He(NH4)3Al5(P04)e.18H20
рК = 6рН + 3pNH4 + 5рА1 + 8рР04
175,5
Таранакит калия (КР)
НвК3А1б(Р04)в.18Н20
рК = 6рН + ЗрК + 5рА1 + 8рР04
178,7
Гиббсит
А1(ОН)3
рК = рА1 + ЗрОН
33,8
Гетит
Fe(OH)3
рК = pFe + ЗрОН
38,1
Флюорит
CaF2
рК = рСа + 2pF
9,84
Фосфорная кислота
H3P04
рК = pH + рН2Р04 - рН3Р04
2,12
Одновалентный фосфат-ион
н2ро4-
рК2 = pH + рНР04 — рН2Р04
7,19
Двухвалентный фосфат-ион
НРОа*-
рК3 = pH + рР04 - рНР04
12,32
Вода
Н20
рК = pH + рОН
14,00
172
К. Е. Гинзбург
Аналогично составляют уравнения для других соединений фосфатов
кальция:
рКНА = 6 (0,5рСа + рНгРОл) —14 (pH — 0,5рСа) -f- 143,32 — гидроксилапатит, (33)
рКрА = 6 (0,5рСа + рНгРОл) —12 (pH — 0,5рСа) + 126,96 — фторапатит, (34)
рК0ср = 3(0,5рСа + рНгРОл) — 5 (pH — 0,5рСа) -f- 57,66 — октана л ьцийфосфат, (35)
pKDCp = (О^рСа+рНгРО*) — (РН-0,5РСа) + 7,20 — дикальцийфосфат, (36)
pKAip = pAl-f-рНгРОА—2рН+28 — варисцит. (37)
Чтобы узнать, какие соединения фосфора определяют равновесные ра¬
створы в почвах, рассчитывают произведения растворимости каждого образ¬
ца, подставляя найденные в исследуемых почвенных вытяжках значения pH,
рСа, рН2Р04, рА1 в уравнения с (33) по (37).
Приводим пример расчета для дерново-подзолистой суглинистой почвы,
удобренной фосфором (Синягина, 1969).
Анализ водной вытяжки из этой почвы показал значения: pH — 5,36; рН2Р04— 4,81;
рА1 — 4,74; pH — 05р (Ca+Mg) = 3,72; 0,5р (Ca+Mg) + рН2Р04= 6,45. Подставляем
эти значения в уравнения с (33) по (37), получаем: рКНА = 6*6,45—14*3,72 + 143,32 =
= 38,7 + 143,32 — 52,08 = 129,9, рКРА = 6-6,45 — 12*3,72 + 126,96 = 121,0, рК0СР =
= 3*6,45 — 5*3,72 + 57,66 = 58,4, pKDGP = 6,45 — 3,72 + 7,2 = 9,93, рКА1Р =
*= 4,74 + 4,81 — 2*5,36 + 28 = 26,83.
Полученные значения рКпр сравним с рКпр чистых соединений фосфатов (см. табл. 13).
Результаты этого сравнения показывают, что для исследуемой почвы найденные значения
рКпр наиболее близки^варисциту (рКА1р = 30,5) и фторапатиту (pKFA = 118,4). Согласно
правилам произведения растворимости, исследуемый раствор перенасыщен по отношению к
фторапатиту, который, следовательно, будет находиться в осадке в твердых фазах почвы
и ненасыщен по отношению к варисциту, т. е. в данных почвенных условиях образуются
более растворимые соединения, чем варисцит.
Определение форм фосфатов почвы
по изотермам растворимости
Формы почвенных фосфатов могут быть определены также с помощью
изотерм растворимости чистых фосфорных соединений. Известно, что раст¬
воримость фосфатов зависит от pH. Используя данные табл. 13, Линдсей и
Морено (Lindsay, Moreno, 1960) выразили растворимость чистых фосфорных
соединений графически в виде функции рН2Р04 от pH. Для выражения ра¬
створимости варисцита, стренгита и фторапатита делается допущение, что
активности ионов Al3+, Fe3+ и F" в почвенном растворе определяются
условиями равновесия с содержащимися в твердых фазах гиббситом, гетитом
и флюоритом как наиболее стабильными соединениями в почвах. При постро¬
ении изотерм растворимости фосфатов кальция следует учитывать концентра¬
цию кальция в растворе. Если активности ионов определяются в вытяжке
0,01 М СаС12, то концентрация кальция будет равна 0,01 М/л (рСа = 2,0).
С учетом этих допущений уравнения, используемые для построения изотерм
растворимости фосфатов, имеют такой вид:
для варисцита рНгРО* = 10,7—pH, (38)
для стренгита рНгРО* = 10.,9— pH, (39)
для дикальцийфосфата рНзРОд = pH—2,53, (40)
5
для октакальцийфосфата рНгРОл = “3 pH—6,24, (41)
7
для гидроксилапатита рНгРО* = -g- pH—8,26, (42)
для фторапатита рНгР04 = 2рН—4,12. (43)
Методы определения фосфора в почве
173
Путем подстановки различных значений pH в уравнения (38—43) нахо¬
дится величина рН2Р04, которая откладывается по оси ординат, по оси
абсцисс откладывается величина pH. По нескольким точкам строятся изо¬
термы растворимости чистых фосфорных соединений (рис. 4). Используя
данные рН2Р04 и pH, определенные экспериментально в 0,01 МСаС12 вытяж¬
ках из почв, и нанеся их на диаграмму растворимости, можно сравнить раство¬
римость почвенных фосфатов с растворимостью известных фосфорных сое¬
динений. На рис. 4 показано существенное различие в формах фосфатов,
содержащихся в различных типах почв. Так, в дерново-подзолистой почве и
Рис. 4. Изотермы растворимо¬
сти фосфатных минералов и фос¬
фатов почвы в зависимости от
величины pH (Кудеярова, По¬
лякова, 1973) (*=25° С; актив¬
ность Са2+ = 0,001 М/л;
рСа = 2)
1 — дерново-подзолистая почва;
2 — краснозем; з — чернозем; 4 —
серозем
красноземе фосфатное равновесие устанавливается с фосфатами алюминия
типа варисцита, в черноземе — с фосфатами кальция, растворимость которых
больше, чем у гидроксилапатита, но меньше, чем у октакальцийфосфата.
В сероземе основным фосфорным соединением был октакальцийфосфат.
Изотермы растворимости различных фосфорных соединений можно вира-
зить также в виде функции рР от pH; для соединений фосфатов кальция —
с помощью отношения фосфатного и известкового потенциалов, для соедине¬
ния фосфатов алюминия и железа — в виде функций рН2Р04 от 2рН — рА1
или 2рН — pFe соответственно и т. п. (Ulrich, 1961; Larsen, 1967).
В литературе (Ларсен, 1967) имеются данные, указывающие на то, что нет
хорошего соответствия между концентрацией фосфора в почвенном растворе
и концентрацией, предсказываемой из значений произведений раствори¬
мости. Исследователи объясняют это тем, что существуют, например, две
константы рКпр для гидроксилапатита и октакальцийфосфата, которые соот¬
ветствуют случаю достижения равновесия при их осаждении и при раство¬
рении.
Дальнейшее осложнение при установлении рКпр обусловлено сильным
влиянием примесей на растворимость различных фосфорных соединений.
Так, было показано, что чистый гидроксилапатит подчиняется правилу про¬
изведения растворимости, но в присутствии небольших количеств карбоната
кальция правило нарушается. Большое влияние оказывает также возмож¬
ное комплексообразование в растворе. Поэтому несоответствие между кон¬
центрацией фосфора в почвенном растворе и рКпр чистых препаратов фосфатов
еще не означает, что эти соединения не определяют концентрацию фосфора
в растворе.
Использование констант и изотерм произведения растворимости не дает
абсолютных величин содержания различных форм фосфорных соединений в
почвах, но дает им качественную оценку, показывающую степень их рас¬
творимости и возможные превращения в различных почвенных условиях.
PW
174
К. Е. Гинзбург
Определение
потенциальной буферной способности почв
в отношении фосфора
Адсорбция фосфора на поверхности твердых фаз почвы является другим
вероятным механизмом, который может определять концентрацию фосфора
в растворе и отсюда фосфорное питание растений.
Практически при внесении фосфатов в почвы адсорбционная система на¬
сыщается фосфором, что может сопровождаться повышением концентрации
фосфора в растворе до осаждения умеренно растворимого фосфорного соеди¬
нения. Наоборот, если концентрация фосфора в растворе понижается, на¬
пример в результате выноса фосфора урожаем растений, то умеренна
растворимый фосфор твердых фаз почвы будет растворяться до тех пор,,
пока адсорбционный комплекс не станет насыщенным в такой мере, которая
соответствует растворимости наименее устойчивого фосфорного соединения,,
находящегося в системе.
В результате этих реакций почвы способны поддерживать свой фосфат¬
ный потенциал («интенсивность») на относительно постоянном уровне при уве¬
личении или уменьшении в них общего запаса подвижных фосфатов.
Свойство почв поддерживать концентрацию фосфора на постоянном уров¬
не С. М. Драчев (1928) назвал фосфатной буферной способностью почв*
Для определения этой величины он предложил использовать соотношение
показателей а/е, где а — количество Р2Об (мг на 1 кг почвы), извлекаемое
из почвы водной вытяжкой при широком соотношении почва : вода (запас
подвижных фосфатов); в — количество Р2Об (мг на 1 кг почвы), извлекаемое
водной вытяжкой при узком отношении почва: вода («интенсивность»). В ре¬
зультате проведенных исследований им было установлено, что чем больше
величина а/в, тем сильнее выражена способность почвы поддерживать кон¬
центрацию фосфора в растворе на постоянном уровне.
Аналогичным образом способность почвы противостоять изменению
фосфатного потенциала Бекетт и Уайт (Beckett, White, 1964) назвали «по¬
тенциальной буферной» способностью почв в отношении фосфатов — РВСР.
РВСр = <?//, (44)
где Q — общий запас подвижных фосфатов почвы (фактор «емкости»); / —
равновесная активность Н2Р04~, или равновесный фосфатный потенциал почв
(фактор «интенсивности»). Отношение Q/I показывает, какое количество
подвижных фосфатов должно перейти из их общего запаса в почвенный ра¬
створ или должно быть внесено в почву для изменения активности Н2Р04“ на
единицу.
Аналитическими методами определить величину РВСР нельзя, но можно
определить ее изменение в данных условиях опыта (± ДР). Величина ± АР
представляет собой количество подвижного фосфора, которое почва отдает
(— АР) или поглощает (+ ДР) к моменту установления равновесия между
фосфатами твердых фаз почвы и раствора.
РВСР учитывает не только общее содержание подвижных фосфатов в поч¬
вах, но и степень их подвижности, зависящей от поглотительных свойств поч¬
вы (емкости поглощения фосфора и прочности его закрепления в твердых
фазах), поэтому этот показатель более полно характеризует фосфатный режим
почв, чем фосфатный потенциал (Кудеярова, 1971).
Метод основан на определении сорбции и десорбции фосфатов, добавлен¬
ных в слабосолевые почвенные вытяжки. Количество сорбированных или де¬
сорбированных фосфатов (± ДР) определяется по разности между концен¬
трацией фосфора в исходном растворе и концентрацией фосфора в равновес¬
ном растворе после взаимодействия с навеской почвы.
Методы определения фосфора в почве
175
Ход анализа. Серию] навесок почвы по 5 г заливают 50 мл раствора 0,01 М
СаС12, содержащего от 0 до 5 • 10“4 Р моль/л в виде монофосфата кальция. Смесь
взбалтывают в течение часа при 25 + 1° С, фильтруют, в фильтрате определя¬
ют pH потенциометрически и фосфор колориметрически, а затем вычисляют
активность иона Н2Р04~ по (20).
Вычисление РВСР. По изменению концентрации фосфора в раство¬
рах, после взаимодействия их с почвами, высчитывают количество сорбиро¬
ванных или десорбированных почвами фосфатов (+ АР, молъ/г почвы). От¬
кладывая на графике (рис. 5) по оси ординат найденные значения АР, а по
Рис. 5. График определения
потенциальной буферной спо¬
собности почв (Кудеярова,1971)
I — равновесная активность иона
Н*Р04- в почвенном растворе дерно¬
во-подзолистой супесчаной почвы;
Q — количество фосфатов в твер¬
дых фазах, находящихся в равно¬
весии с раствором
оси абсцисс соответствующие им величины активности иона Н2Р04 в молях/л,
определенные в вытяжках, строят кривые, характеризующие фосфатную по¬
глотительную способность почв. При невысоких концентрациях Р2Об в исход¬
ных растворах адсорбция фосфора почвами возрастает прямо пропорциональ¬
но концентрациям. Пересечение прямых отрезков изотерм сорбции фосфатов
с осью абсцисс дает значение равновесной активности фосфат-иона (/).
Продолжив прямые до пересечения с осью ординат, определяют количество
Таблица 14
Расчет равновесной активности фосфат-иона и потенциальной буферной способности
дерново-подзолистой супесчаной почвы
(Кудеярова, 1970)
Вариант
оныта
Добавление фосфата
в почву с исходными
растворами 0,01 М
СаС1,
Ср в рав¬
новесном
растворе
вытяжки,
моль- 10~*/г
почвы
Поглощено
почвой
фосфатов
из раство¬
ров, ДР.
молъ»10-*/г
почвы
ан,Р04
в вытяж¬
ке, моль •
• 10-»/*
Равновес¬
ная
°Н,РО.
(I)*, моль•
• 10~*/л
О* моль•
•10-»/г
почвы
Потен¬
циальная
буферная
способ¬
ность
(Q/0
Р, моль •
Ю-о/л
Р, моль•
•10-в/г
почвы
NK
0
0
2,0
-2,0
0,16
0,24
10,0
0,041
160
160
28,2
131,8
2,37
310
310
98,0
212,0
8,24
NPK
0
0
6,3
-6,3
0,53
0,66
23,0
0,035
160
160
50,0
110,0
4,20
310
310
131,0
179,0
11,00
* Данные найдены с помощь» графика определения равновесной активности иона ЩРОд (рис. 5).
176
К. E. Гинзбург
фосфатов (Q) в твердых фазах, находящееся в равновесии с раствором, по
(39) определяют величину потенциальной буферной способности почв.
Пример вычисления РВСР приведен в табл. 14.
Определение скорости перехода фосфатов из почвы в раствор
(кинетический фактор 22)
Метод Кука (Cooke, 1966)
Как указывалось выше, при снижении концентрации фосфора в почвен¬
ном растворе (в результате выноса фосфора растениями, поглощения, вымыва¬
ния и т. п.) динамическое равновесие между твердыми и жидкими фазами поч¬
вы нарушается. Для восстановления нарушенного равновесия новые порции
фосфатов почвы переходят в раствор в результате самодиффузии. Скорость
этого процесса имеет большое значение для фосфорного питания растений.
if мин
в 10
/Г; мин
Рис. 6. Зависимость между количеством фосфора и временем извлечения его анионитом
по Куку (Cooke, 1966)
а — количество фосфатов, извлеченных из почвы анионитом за время t мин.; б — то же, за времяУТмин.
Так, если концентрация фосфатов в растворе восстанавливается медленно, то
поглощение фосфора растениями будет ограниченно. Только в том случае, если
скорость перехода фосфатов в раствор является достаточной, растения могут
поглощать необходимое для их нормального роста и развития количество фос¬
фатов.
При измерении суммарной скорости перехода фосфатов из твердых фаз
почвы в раствор Кук (Cooke, 1966) использовал принцип Амера и др. (Amer
et al., 1955), которые показали, что если в почвенную суспензию добавляют
анионообменную смолу, то фосфор поглощается смолой со скоростью, которая
не зависит от свойств смолы, а зависит только от скорости растворения фос¬
фатов твердых фаз почвы.
При этом Кук установил, что для промежутков времени примерно до 2-х
час. между количеством фосфора (Р), извлеченного из почвы, и временем (*)
существует взаимосвязь, показанная на рис. 6 а.
Для получения этой зависимости в линейной форме в качестве независимой
переменной взята величина У t (рис. 6 б)1 и тогда
Р =RVr+B, (45}
где В — константа, представляющая количество фосфатов в исходном растворе до при¬
бавления к суспензии анионита (при t = 0); R — константа скорости извлечения фосфатов
m почвы (в указанном интервале времени). В и R — параметры, постоянные для данной
почвы, но различные для разных почв.
Методы определения фосфора в почве
177
JI. П. Похлебкина (1973) по методу Кука определяла скорость перехода
фосфатов в раствор из дерново-подзолистой почвы. Она установила, что вели¬
чина В возрастает с повышением количества внесенных в почву фосфатов, а
величина Р повышается с увеличением времени (t) взаимодействия почвы с
анионитом.
Метод основан на извлечении фосфатов из почвы анионитом в С1-форм&
(анионит марки ЭДЭ-10П или АВ-17) в течение 2-х часов с интервалами вре¬
мени по 10 мин. при t = 30 + 1° С. Соотношение почва : вода = 1 : 25; поч¬
ва : анионит = 1:1.
Ход анализа. 12 навесок по 1 г воздушно-сухой почвы (сито d 0,25 мм) по¬
мещают соответственно в 12 плоскодонных колбочек емкостью 50—100 мл,
приливают по 25 мл дистиллированной воды и взбалтывают 2 часа. После это¬
го в каждую колбочку, за исключением одной контрольной колбы, погружают
капроновый мешочек с 1 г анионита (сито d 0,5 мм) и продолжают взбалтыва¬
ние: одной колбочки — 10 мин., второй — 20 мин., третьей — 30 мин.,...,
одиннадцатой — 120 мин. После этого анионит отделяют от почвы и выщела¬
чивают поглощенный фосфор с помощью 0,5 н. НС1, как описано выше (см.
ионообменную хроматографию). Вытесненный из смолы фосфор определяют
колориметрически, получают значение Р (в мг Р205 на 100 г или на 1 кг поч¬
вы) при времени t = 10, 20, ..., 120 мин.
Контрольную колбу (без добавления анионита) сразу же фильтруют, а в
фильтрате определяют фосфор колориметрически. Данные содержания фосфо¬
ра (мг Р206 на 100 г или 1 кг почвы) при времени взаимодействия навески поч¬
вы с анионитом t = 0 дают значение константы — В. Зависимость извлечения
из почвы фосфора от времени t (кинетический фактор В) вычисляют из урав¬
нения (45).
Реактивы и материалы. Приготовление анионита в Cl-форме и капроно¬
вых мешочков описано выше (см. ионообменную хроматографию).
Таким образом, для характеристики фосфатного состояния почв в послед¬
нее время используют следующие факторы:
1) «интенсивность» (/) — концентрация фосфат-ионов почвенного раство¬
ра, фосфатный потенциал почв;
2) фосфатную «емкость» (@) — потенциальный запас растворимых фосфа¬
тов почвы, фосфаты поверхности твердых фаз почвы;
3) фосфатную буферную способность почв (РВСР), характеризующую спо¬
собность почв противостоять изменению фосфатного потенциала при внесении
фосфатов или их извлечении из почвы;
4) кинетический фактор (В) — скорость, с которой фосфат-ионы посту¬
пают с поверхности твердых фаз почвы в почвенный раствор путем самодиф-
фузии.
5) взаимосвязь одного или нескольких этих факторов между собой, урожа¬
ем и выносом фосфора растениями.
БИОХИМИЯ ПОЧВЕННОГО ФОСФОРА
Органические фосфаты почвы происходят из остатков растений, животных
и микроорганизмов. Они являются одним из потенциальных источников пи¬
тания растений фосфором. Представлены органические фосфаты двумя раз¬
личными по природе группами соединений: продуктами биологического син¬
теза (соединения индивидуальной природы, неспецифические органофосфа¬
ты) и продуктами гумусообразования (специфические соединения).
Фосфорные эфиры индивидуальной природы, обнаруженные в почвах,
относятся к трем основным классам соединений: фосфолипиды (до 1 % от об¬
щего органического фосфора), нуклеиновые кислоты (5—10%) и различные
178
К. Е. Гинабург
изомерные формы инозитфосфатов (30—60%). Имеются указания о наличии
фосфопротеинов и сахарофосфатов, но практически они не были выделены из
почв. Более половины фосфорорганических соединений почвы составляют
новообразованные специфические фосфогумусовые соединения невыясненной
природы.
Для выделения, идентификации и количественного определения отдельных
групп и соединений органических фосфатов используют ряд последователь¬
ных приемов: диспергирование почв (декальцирование, обработка ультра¬
звуком), селективную растворимость (спирты, эфиры, кислоты, щелочи и др.)»
гидролиз, осаждение, хроматографию, ионофорез, спектрофотометрию и др.
Ниже приводим описание наиболее распространенных методов определения в
почвах индивидуальных (неспецифических) органических соединений фосфа¬
тов (фитиновых, нуклеиновых кислот и фосфолипидов).
Инозитфосфаты (фитиновые кислоты, фосфоинозиты)
Инозит (циклогексангексол) — циклический насыщенный шестиатомный
,спирт. Он может находиться в девяти стереоизомерных формах (рис. 7). Пять
из этих изомеров инозита найдены в природе (нео-, мио-, сцилло-, dl- и лево-).
Общим изомером является миоинозит.
При присоединении к инозиту с помощью эфирных связей шести молекул
фосфорной кислоты образуется 12-основная инозитгексафосфорная кислота —
С6Нв (0Р03Н2)в. Двойная кальциево-магниевая соль этой кислоты, так
называемый фитин, широко представлена в растениях, особенно в семенах.
При неполном фосфорилировании инозита или дефосфорилировании инозит-
гексафосфата могут образоваться инозитпентафосфаты — 10-основные кис¬
лоты — СвНв(ОН) (0Р08Н2)б и более низкие фосфорные эфиры инозита (тет-
ра-, три-, ди- и моноинозитфосфаты), которые соответствуют 8-, 6-, 4-,
2-основным фосфорным кислотам инозита. Эти кислоты с разной скоростью и
прочностью фиксируются на анионообменных смолах, что в настоящее вре¬
мя широко используется для их идентификации и хроматографического раз¬
деления.
Свободные кислоты инозитфосфатов и их соли с щелочными металлами
^егкорастзоримы в воде, но большинство других их солей имеет ограничен¬
ную растворимость; слабо растворимы в абсолютном спирте, нерастворимы
в эфире, бензоле и хлороформе.
Рис. 7. Изомерные формы цик¬
лического спирта инозита
(Anderson, 1967)
1 — цис;
II — эпи;
III — дило (dl);
IV — нео;
V — мио;
VI — муко;
VII — сцилло;
VIII — декстро;
IX — лев*
ОН ОН ОН
IV V VI
он он но
VII VIII (X
Методы определения фосфора в почве
179»
Основным компонентом полифосфатной смеси инозитов являются пента- и
гексафосфаты нео-, сцилло-, dl-, миоинозитов (до 90% от общего содержания
фракции фосфоинозитов), и незначительное их количество представлено низ¬
комолекулярными фосфорными эфирами (моно-, ди-, три- и тетраинозитфос-
фатами).
Ранее существовало представление о том, что фитиновые кислоты извле¬
каются из почвы только кислотными, а нуклеиновые кислоты и нуклеопротеи-
ды — щелочными вытяжками. В настоящее время установлено, что инозитфос-
фаты в почве находятся не в свободном состоянии, а связаны в комплексах с
белками, углеводами и некоторыми минеральными компонентами, поэтому
наиболее полно они растворяются в щелочах, тогда как даже жесткая кис¬
лотная экстракция извлекает только от 25 до 60% от их общего количества
(Halstead, Anderson, 1970).
Метод Андерсона
(Anderson, 1964,1967)
Метод основан на извлечении органических фосфатов 3 н. NaOH при на¬
гревании, после предварительного декальцирования почв 0,1 н. НС1. Иа
щелочного экстракта удаляют гуминовые кислоты и полуторные окислы, а за¬
тем для очистки фитатов и разрушения комплексов с полисахаридами и про¬
теинами их осаждают ацетатом бария в присутствии алкоголя. Осадок фита¬
тов Ва промывают и растворяют катионообменной смолой в Н-форме. Полу¬
ченный фильтрат фитиновых кислот пропускают через анионообменную смолу,
насыщенную муравьиной кислотой. Из смолы инозитфосфаты элюируют
нейтральным раствором муравьинокислого аммония (HCOONH4, pH 7,0) с
повышающимся градиентом концентраций: 0,24; 0,41; 0,55; 0,62; 1,2 М (с
объемами 100; 300; 300; 100 и 100 мл соответственно). При этом последователь¬
но вытесняются: 1) ортофосфаты и моноинозитфосфаты, 2) диинозитфосфаты;
3) триинозитфосфаты; 4) тетраинозитфосфаты; 5) смесь пента- и гексаинозит-
фосфатов. В целом, чем больше молей фосфата входит в молекулу инозита,
тем большая концентрация ГШ4-формиата требуется для вытеснения (элю-
ции) инозитфосфатов из смолы.
Так как фосфаты в 1—4 фракциях содержатся в незначительных количест¬
вах, то, пропуская через колонку растворы HCOONH4 с концентрацией
0,6 М и 1,2 М, можно разделить инозитфосфаты на две большие фракции:
ортофосфаты и низкомолекулярные эфиры инозитфосфатов (моно-, ди-, три-*
тетраинозитфосфаты) и сложные эфиры инозитфосфатов (пента- и гексаино-
зитфосфаты, которые практически неразделимы).
Ход анализа. 5 г воздушно-сухой почвы (сито d 0,25 мм) помещают в стек¬
лянные (жаростойкого стекла) центрифужные пробирки емкостью 200 мл,
приливают 100 мл 0,1 н. НС1 и взбалтывают в течение часа при 20° С. Суспен¬
зию центрифугируют, раствор выбрасывают, а остаток почвы в центрифужной
пробирке обрабатывают еще раз 100 мл 0,1 н. НС1. Если при этом карбонаты
не полностью разрушены (продолжается выделение пузырьков С02), то обра¬
ботку 0,1 н. НС1 повторяют третий раз. Все солянокислые растворы выбрасы¬
вают. После этого остаток почвы промывают от кислоты 100 мл дистиллиро¬
ванной воды, взбалтывают от руки, центрифугируют. Раствор выбрасывают,
остаток почвы заливают 40 мл 3 н. NaOH и нагревают на кипящей водяной
бане в течение 12-ти час. с периодическим перемешиванием суспензии. Чтобы
избежать выпаривания растворов при нагревании, центрифужные пробирки
закрывают сверху перевернутыми маленькими коническими колбочками.
После нагревания растворы охлаждают, центрифугируют, прозрачный рас¬
твор сливают в другие центрифужные пробирки. Остаток почвы промывают
дважды водой, порциями по 20 мл, центрифугируют. Промывные воды добав¬
ляют к щелочному экстракту, получают раствор А. Остаток почвы выбрасы¬
вают.
480
К. Е. Гинабург
Раствор А подкисляют 90 %-ной НСООН до pH 4, при этом выпадают гу-
зшновые кислоты, затем подщелачивают до pH 8—9 крепким NH4OH, осажда¬
ются полуторные окислы. Спустя 15 мин. суспензию центрифугируют. Раст¬
вор сливают в другие центрифужные пробирки, получают раствор Б. Осадок
промывают 20 мл воды, центрифугируют, промывные воды добавляют к раст¬
вору Б, осадок выбрасывают. К раствору Б приливают равный объем этанола
и 10 мл 30%-ного раствора (СН3СОО)2Ва, выпадает осадок фитатов бария,
♦его коагулируют 10 мин. на кипящей водяной бане и настаивают в течение
ночи в закрытых пробирках, затем смесь центрифугируют, центрифугат вы¬
брасывают, а осадок дважды промывают 10—20 мл 50 %-ного этанола, центри¬
фугируют и центрифугаты выбрасывают. Осадок фитатов высушивают и ре-
растворяют, взбалтывая с 50 мл воды и 25 мл катионообменной смолы (амбер-
лит IR-120) в Н-форме. После растворения осадка смолу отделяют от раствора
фильтрованием. Фильтрат затем используют для хроматографического ана¬
лиза.
Ионообменная хроматография. Раствор, содержащий
инозитфосфаты, пропускают со скоростью 0,5 мл/мин через анионообменную
колонку (160 х 12 мм), наполненную смолой Дауэкс-1 (сито d 0,25—0,5 мм)
в формиатной форме. Осажденные фосфаты затем элюируют, также со ско¬
ростью 0,5 мл/мин, сперва с помощью 300 мл 0,6 М HCOONH4, pH 7,0 (элюат
I), а затем в другую колбу собирают элюаты с 85 мл 1,2 М HCOONH4. Спустя
один час пропускают еще 5 мл 1,2 М HCOONH4, затем через 30 мин. еще 5 мл
и через 30 мин. последние 5 мл 1,2 М HC00NH4. Таким образом, элюируют в
100 мл все инозит пента- и гексафосфаты (элюат II). В 300 мл элюата I пере¬
ходят ортофосфаты и все низкомолекулярные фосфорные эфиры инозита.
Аликвоты элюатов I и II выпаривают на водяной бане и окисляют с добав¬
лением раствора Mg(N03)2 при сжигании в муфеле, точно так же, как в слу¬
чае определения фосфолипидов (см. ниже). В окисленных растворах опреде¬
ляют фосфор колориметрически.
Мы рекомендуем окисление фракций органических фосфатов в элюатах I
м II проводить после их предварительного выпаривания, с помощью 5—10 мл
Рис. 8. Хроматограмма распределения содержания различных изомерных форм ино-
зитфосфатов в щелочном экстракте краснозема (Oniani et al., 1973)
I — мио-ияозитпентафосфат; II — мио-инозитпентафосфат -f сцилло-инозитпентафосфат -f- нео-инозит-
тексафосфат; III — мио-инозитгексафосфат + дило-инозитгексафосфат; IV — сцилло-инозитгексафос-
фат
Методы определения фосфора в почве
181
50% НСЮ4 (см. выше определение органических фосфатов по Хейфец). На
рис. 8 приведены результаты хроматографического разделения на колонке ани¬
онита (формиатная форма) различных изомерных форм инозит пента- и гекса¬
фосфатов. Как следует из рис. 8, наибольшее количество в смеси сложных
фосфорных эфиров падает на долю мио- и dZ-инозитгексафосфатов.
Для более подробного изучения состава полифосфатной смеси инозитов
проводят их хроматографическое разделение на бумаге без предварительного
гидролиза и после их гидролиза до инозитов (McKefcher, Anderson, 1968а, б).
Реактивы. 1. 0,1 н. НС1 и 6 н. НС1.
2. 3 н. NaOH.
3. 0,6 и 1,2 М HCOONH4. Соответственно 37,8 и 75,6 г соли формиата аммония ра¬
створяют в 800 мл воды, доливают водой до 1 л, перемешивают.
4. 90%-ная НСООН.
5. 30%-ный (СН3СОО)2Ва (ацетат бария).
6. Этанол 96° и 50%-ный водный раствор.
7. Катионит Н-форма и анионит — СООН (формиатная) форма. Приготовление смФ
«выше в разделе ионообменной хроматографии.
Нуклеиновые кислоты (полинуклеотиды)
Нуклеиновые кислоты (НК) — сложные высокомолекулярные соедине¬
ния, состоящие из большого количества нуклеотидов. Их можно рассматри¬
вать как полимеры мононуклеотидов или полинуклеотиды с очень высоким
молекулярным весом (^> 1 млн.). Наипростейший мономер НК — мононук¬
леотид — содержит три основных структурных элемента: азотистое основа¬
ние, углевод и фосфорную кислоту. Связь между основанием и углеводом —
глюкозидная (С—N) и углеводом и кислотой — эфирная (С—О—Р). НК по
входящему в их состав сахару пентозы и составу пиримидиновых оснований
разделяются на две группы: дезоксирибонуклеиновую кислоту — ДНК и
рибонуклеиновую кислоту — РНК.
ДНК (тимонуклеиновая кислота) содержит углевод — d-2-дезоксирибозу
и пиримидиновое основание — тимин (2,6-диокси-5-мети л пиримидин). РНК
(дрожжевая кислота) содержит сахар — рибозу и пиримидин — урацил (2,6-
диоксипиримидин). Пуриновые основания — аденин (6-амино-пурин), гуанин
(2-амино-6-оксипурин) и пиримидиновое — цитозин (2-окси-6-аминопирими-
дин) входят в состав и ДНК и РНК (рис. 9).
Оба типа НК содержатся в растениях, животных, микроорганизмах, при¬
чем чаще всего они находятся не в свободном состоянии, а в соединениях с
•белками (протеинами), образуя сложные белки — нуклеопротеиды. В обра¬
зовании нуклеопротеидов непосредственное участие принимают аминокисло¬
ты. Присутствие в почвах соединений НК доказано рядом тонких современных
методов исследований (хроматографией, электрофорезом, по спектрам погло¬
щения в ультрафиолете и др.), но найденные их абсолютные количества (5—
10% от Рорг общего) значительно меньше, чем это считалось раньше (40—
60% Р0рг). НК в почвах представлены преимущественно ДНК, содержание
РНК очень ограничено (Anderson, 1967).
Идентификация ДНК и РНК основана на их различной устойчивости в
щелочных растворах. Так, в щелочном растворе РНК медленцо разлагается
до составляющих ее нуклеотидов, а ДНК остается в полимерной форме. При
подкислении щелочного почвенного экстракта с фракцией гуминовых кислот
осаждается ДНК. Осаждение РНК вариабельно и зависит от степени ее по¬
лимеризации и времени нахождения в растворе. Так, высокомолекулярная
РНК может осаждаться с гуминовой кислотой. Андерсон (Anderson, 1967)
в некоторых гидролизатах гуминовых кислот идентифицировал пиримидин
урацил, что было доказательством наличия РНК в гуминовых кислотах почв.
182
К. Е. Гинзбург
Через 16 час. после настаивания в 0,1 н.
NaOH при 37° С РНК полностью раз¬
лагается и количественно переходит
в фульвокислотную фракцию (Ander¬
son, 1967).
Мягкий кислотный гидролиз, спо¬
собствующий высвобождению пурино¬
вых и пиримидиновых оснований из
РНК, в случае ДНК высвобождает лишь
пуриновые основания, образующие апу-
риновые кислоты. В последних исход¬
ные полинуклеотидные структуры не
полностью распадаются. При более
жестком кислотном гидролизе ДНК бы¬
стро распадается до пуриновых и пи¬
римидиновых оснований. Подобные ус¬
ловия и были использованы для иден¬
тификации азотистых оснований в ДНКГ
выделенных из разных источников (An¬
derson, 1961).
Многие пуриновые и пиримидиновые
основания, ионизируясь, приобретают
свойства катионов или анионов в зави¬
симости от условий опыта. Так, в кис¬
лой среде гуанин, аденин, цитозин —
строгие катионы (благодаря присутствию аминогрупп), которые быстро сор¬
бируются катионообменниками. Тимин, урацил в этих условиях, наоборот*
сорбируются на катионите очень слабо. Этим способом их можно выде¬
лить из сложной почвенной смеси и, сконцентрировав, исследовать при
отсутствии примесей.
В почвах НК определяют по азотистым основаниям. Количественные из¬
мерения пуринов и пиримидинов в гидролизатах НК проводят спектрофото¬
метрически и хроматографически.
Рис. 9. Химический состав и структура
нуклеиновых кислот (Anderson, 1967)
1 — звено цепи РНК; 2 — звено цепи ДНК*
R,R',R" — пурины или пиримидины
Метод Андерсона
(Anderson, 1967; Halsted, Anderson, 1970)
Метод основан на декальцировании почв 0,1 н. НС1, экстракции орга¬
нофосфатов 0,3 н. NaOH при 20° С 16 час., осаждении ДНК и части РНК с
гуминовыми кислотами при pH 1, их гидролизе в 50% НСЮ4, реосаждении и
рерастворении продуктов гидролиза ДНК и РНК в целях их очистки, иден¬
тификации и определения азотистых оснований (пуринов, пиримидинов) с по¬
мощью ионообменной и бумажной хроматографии, спектрофотометрии (при
250—290 нм) и др.
Ход анализа. 20 г воздушно-сухой почвы (сито d 1—2 мм) помещают в
центрифужные пробирки емкостью 250 мл, приливают 100 мл 0,1 н. НС1.
взбалтывают 1 час, центрифугируют и растворы выбрасывают. Остаток почвы
в пробирке заливают 100 мл 0,3 н. NaOH и настаивают при 20° С 16 час. с
периодическим перемешиванием суспензии. После этого суспензия центрифу¬
гируется, а к почве добавляют еще 50 мл 0,3 н. NaOH, взбалтывают несколько
минут от руки и центрифугируют. Прозрачные щелочные экстракты объединя¬
ют и сливают в другие пробирки, затем их подкисляют концентрированной
НС1 до pH 1 и оставляют на 10 мин. для коагуляции осадка гуминовых кис¬
лот. Суспензию центрифугируют, промывают водой, вновь центрифугируют.
Растворы выбрасывают, а отмытые осадки гуминовых и нуклеиновых кислот
подвергают промораживанию при —5, —15° С в течение ночи, затем их от¬
Методы определения фосфора в почве
183
0,9
ЦВ
0,7
Ц6
V
0,3
—/
10 20
/V* франции (по У О ил)
30
таивают и высушивают в вакууме при D
50° С. Сухой осадок обрабатывают
2 мл 50 % НСЮ4 (или несколько боль¬
ше кислоты, если больший размер
осадка) и нагревают на кипящей во¬
дяной бане в течение 1 часа. Образую¬
щийся при этом осадок разбивают
время от времени стеклянной палоч¬
кой. Суспензия разбавляется затем
водой до 15 мл у настаивается 10 мин.,
центрифугируется. Осадок промыва¬
ют один раз водой (5—10 мл), цен- Ц5
трифугируют. Центрифугат и про¬
мывная жидкость объединяются и
сливаются в другие пробирки, к ним
осторожно, по каплям приливают
2 н. КОН до pH 4,0, при этом осаж¬
дается перхлорат калия, полуторные
окислы, органические вещества.
Суспензию далее нагревают на во¬
дяной бане в течение 5 мин., охлаж¬
дают., центрифугируют и промывают
водой (5—10 мл). Прозрачный цен-
трифугат и промывную жидкость объ¬
единяют и сливают в пробирки, упа¬
ривают на слабокипящей водяной
•бане примерно до 10 мл, добавляют
1 каплю NH4OH до pH 10. Если при
этом выпадает осадок, то его удаляют
центрифугированием, промыванием,
м объем исследуемого раствора вновь
упаривают до — 10 мл. После этого
раствор, содержащий пуриновые и пиримидиновые основания, пропускают
.со скоростью 0,5 мл/мин через колонку (13 X 1 см), заполненную ани¬
онитом ДАУЭКС-1 (100—200 меш.) в формиатной форме (СООН-форма).
Элюцию проводят со скоростью также 0,5 мл/мин сначала с 20 мл буфер¬
ного раствора HCOONH4 с pH 9,4, а затем 400 мл того же раствора, но с pH 6,2.
С помощью автоматического устройства в кюветах собирают фракции по
10 мл элдоата. Все собранные фракции пропускают затем через спектрофото¬
метр, сначала при длине волны 265 нм, а затем при 250, 260, 270, 280, 290 нм
на фоне буферного раствора HCOONH4. С помощью известных кривых иденти¬
фицируют выделенные соединения азотистых оснований. На рис. 10 пред¬
ставлены результаты ионообменной хроматографии азотистых оснований,
выделенных из гидролизатов нуклеиновых кислот.
Из полученных на хроматограммах пиков, соответствующих определенно¬
му номеру выделенной фракции, отбирается аликвотная часть элюата и либо
с дополнительной очисткой, либо без нее переносится на хроматографическую
бумагу для установления присутствия отдельных форм НК. Цитозин элюиро¬
вали буфером при pH 9,4; тимин, аденин, гуанин — буфером,при pH 6,2.
Перед хроматографированием на бумаге фракции цитозина очищали на ка¬
тионите. Растворы, содержащие аденин, гуанин, тимин, выпаривали, сухие
остатки растворяли в 0,1 н. НС1 и наносили на бумагу. Цитозин и тимин раз¬
гоняли нисходящим способом в течение 16 час. в растворителе н-бутанол —
NH40H. Гуанин и аденин разгоняли на восходящей хроматограмме 16 час. в
растворителе пропанол : НС1 (Аронов, 1959).
Рис. 10. Хроматограмма распределении
и содержания пуриновых и пиримидино¬
вых оснований в гуминовой кислоте крас¬
нозема при К = 265 нм (Oniani et al.,
1973)
I — цитозин; II — тимин; III — гуанин;
IV — аденин
184
К. Е. Гинзбург
Реактивы. 1. 0,1 н. НС1 и НС1 (уд. вес 1,19).
2. 0,3 н. NaOH.
3. 50%-ная НС104 (или 30—72%-ная НС104).
4. 2 н. КОН.
5. 25%-ный NH4OH.
6. 1 М раствор НСООН (муравьиной кислоты).
7. Буферный раствор HCOONH4 с pH 9,4. 10 мл 1 М раствора НСООН разбавляют
водой до 950 мл и добавляют по каплям крепкий NH4OH до pH 9,4.
8. Буферный раствор HCOONH4 с pH 6,2. 5 мл 1 М раствора НСООН доводят во¬
дой до 1 л, перемешивают. Смешивают растворы 7 и 8 в таких соотношениях, чтобы получить
раствор HCOONH4 с pH 6,2.
Фосфолипиды (фосфатиды)
Фосфатиды-фосфоглицериды — вещества липоидного характера, жиропо¬
добные, которые по химическим и физическим свойствам сходны с жирами, но
отличаются от последних наличием азота и фосфора. В состав фосфолипидов-
входят глицерин, фосфорные кислоты, азотистые основания и остатки жир¬
ных кислот. Различное положение остатков жирных кислот, их состав, а
также состав и местоположение фосфорной кислоты и азотистого основания,
дают целую серию разнообразных фосфатидов.
В составе фосфолипидной фракции почвы были идентифицированы преи¬
мущественно два фосфатида:
CH2OOCR1 CH2OOCR1
I I
CHOOCR2 CHOOCR2
I ОН I ОН
/ I /
СНгО—Р=0 СН20-Р=0
\cH2CH2N(CHs)» \c№CH*NH2
лецитин (фосфатидил-холин) кефалин (фосфатидил-этаноламин)
Ri и R2— остатки жирных кислот.
В почвах могут содержаться низкомолекулярные эфиры инозитола —
фосфоинозитиды, которые отличаются от фосфоглицеридов тем, что содержат
вместо глицерина моно- или диинозиты.
Фосфатиды нерастворимы в воде, но хорошо растворяются в органичес¬
ких растворителях (спиртах, эфирах, хлороформе, бензоле или их смесях)»
Но в почвах фосфатиды находятся не в свободном состоянии, а могут быть
связаны в комплексах с некоторыми органическими и минеральными компо¬
нентами, что снижает их растворимость. Чтобы разрушить эти комплексы и.
более полно извлечь фракцию фосфатидов, Андерсон (Anderson, 1967) пред¬
ложил предварительную обработку навески почвы смесью минеральных кис¬
лот (НС1 + HF), а затем экстракцию фракции фосфолипидов органическими,
растворителями.
Метод Хенса и Андерсона
(Напсе, Anderson, 1963а)
Метод основан на двукратной обработке навески почвы смесью минераль¬
ных кислот (НС1 + HF) в течение ночи с последующим отмыванием избытка
кислот дистиллированной водой. Полученные центрифугаты выбрасывают.
Проводят затем последовательную экстракцию остатка почвы ацетоном, пет-
ролейным эфиром, смесью этанола и бензола, смесью метанола и хлороформа.
Полученный раствор перемешивают, выпаривают и из сухого остатка реэкст-
рагируют фракцию фосфолипидов рядом последовательных обработок смесью*
этилового эфира и петролейного эфира, хлороформа и др. После выпаривания,
Методы определения фосфора в почве
185
реэкстракта липоидный остаток окисляют и в растворе определяют фосфор
колориметрически. Полученные Хенсом и Андерсоном значения фракции фос¬
фолипидов колебались от 0,31 до 0,70 мг Р на 100 г почвы, что составляло 0,6—
<0,9% от общего содержания органических фосфатов.
Ход анализа. 1 г воздушно-сухой почвы (сито d 0,25 мм) помещают в цент¬
рифужные пробирки на 100 мл и взбалтывают в течение 6 час. с 10 мл смеси
равных объемов 2,5%-ной НС1 и 2,5%-ной HF. Суспензию центрифугируют,
центрифугат выбрасывают. К остатку почвы в центрифужной пробирке при¬
ливают еще 10 мл смеси кислот и обработку повторяют. После этого почву
промывают порциями по 10 мл воды до отсутствия кислот (проба с индикатор¬
ной бумагой). Промывные воды выбрасывают.
К промытому остатку почвы приливают 50 мл ацетона, закрывают пробир¬
ки полиэтиленовыми или стеклянными крышками и оставляют на 4 часа,
перемешивая тщательно содержимое через каждые 20—30 мин., затем смесь
центрифугируют и прозрачный раствор сливают в 500-миллилитровую бу¬
тыль, закрывающуюся стеклянными пробками (желательно притертыми).
Точно так же остаток почвы последовательно обрабатывают 50 мл петро-
лейного эфира, затем 50 мл смеси этанола и бензола (1 : 5 по объему) и 50 мл
метанола и хлороформа (1 : 1 по объему). Все три центрифугата добавляют к
ацетоновому экстракту. Получают раствор А. Раствор А выпаривают затем
досуха при низкой температуре и давлении. В полученном остатке реэкстра-
гируют фракцию фосфолипидов, последовательно обрабатывая его следую¬
щими растворами: а) 20 мл холодной смеси эфира и петролейного эфира (1 : 1
по объему) в течение 2 мин., декантируют; б) 20 мл холодного хлороформа в
течение 2 мин., декантируют; в) 20 мл холодной смеси этилового и петролей¬
ного эфиров (1 : 1 по объему), доводят до кипения на водяной бане, деканти¬
руют; г) 20 мл холодного хлороформа, нагревают до кипения на водяной ба¬
не, декантируют.
Реэкстракты а, б, в, г сливают в одну и ту же фарфоровую чашку (или
большой тигель) и выпаривают на водяной бане досуха. В полученном остатке
определяют общее содержание липоидного фосфора в почве.
Определение фосфора. Липоидный остаток в фарфоровой чаш¬
ке (или тигле) смачивают 1 мл этанола и 1 мл 12%-ного водного раствора
Mg(N08)2, смесь выпаривают на водяной бане досуха, прокаливают при 600° С
в течение 12 мин., затем растворяют в 15 мл 1 н. НС1, выпаривают досуха на
водяной бане. К сухому остатку добавляют 1 мл 1 н. НС1 и смесь количествен¬
но переносят горячей водой в мерные колбы на 50 мл, охлаждают, нейтрали¬
зуют раствор и определяют в нем фосфор колориметрически.
Мы рекомендуем выпаренные остатки фракции фосфолипидов окислять
3—7 мл 50%-ной НСЮ4 (см. определение органического фосфора по методу
Хейфец).
Хроматография на бумаге. Хенс и Андерсон (19636) пред¬
ложили идентификацию продуктов щелочного и кислотного гидролиза фрак¬
ции фосфолипидов (глицерофосфата, холина, этаноламина, серина, инозитола
и др.) проводить с помощью бумажной хроматографии с последующей элюцией
с бумаги отдельных компонентов гидролизата и их количественного опреде¬
ления.
Реактивы. 1. 2,5%-нал НС1. 58 мл концентрированной НС1 (уд. вес 1,19) доли¬
вают до 1 л водой.
2. 2,5%-ная HF. ~ 80 мл 35,5 %-нон HF (уд. вес 1,15) доливают до 1 л водой.
Раствор сохраняют в полиэтиленовой посуде.
3. Смесь 2,5%-ной НС1 и 2,5%-ной HF. Смешивают равные объемы двух кислот.
4. 1 н. НС1. 82 мл НС1 (уд. вес 1,19) доливают водой до 1 лу перемешивают.
5. 12%-ный раствор Mg(NOs)2.
186
if. Е. Гинзбург
6. Ацетон (диметилкетон) — СН3СОСНя.
7. Этанол (этиловый спирт) — СаНБОН.
8. Метанол (метиловый спирт, карбинол, древесный спирт) — СН3ОН.
9. Этиловый эфир (диэтиловый) — (С2Н6)20.
10. Бензол — СвНв.
11. Хлороформ — СНС13.
12. Петролейный эфир (t кипения 40—60° С) — светлые легкие фракции керосинам
Модификация метода Хенса и Андерсона
(Kowalenko, Me Kercher, 1970)
Весь ход анализа такой же, как в методе Хенса — Андерсона, но исполь-
вованы более эффективные растворители — гексан-ацетон (вместо ацетона),
гексан-бензол и вновь гексан-ацетон, и время взаимодействия сокраще¬
но с 4-х час. до 2-х.
Определение фосфора. Фосфор определяли колориметрически
в остатке после его прокаливания с Mg(N03)2 (см. выше) и в остатке непрока-
ленном. По разности между общим и минеральным фосфором определяли
Рорг — липоидный фосфор.
Авторы приводят данные, в которых показано, что предварительная обра¬
ботка почвы смесью кислот (НС1 + HF) увеличивает в несколько раз коли¬
чество извлеченного липоидного фосфора.
Определение органических фосфатов почвы
с помощью поливалентных катионов
Метод Тинслея (Tinsley, Ozcavaski, 1974)
Метод основан на растворении большей части минеральных и осаждении
органических фосфатов в смеси НС1 — HF — TiCl4. Осадок реэкстрагируют
затем в HCl-купферроне, при этом растворяется весь органический и часть
минерального фосфора (который может происходить из остатков почвенных
минералов или от РО|“, окклюдированной осадком Ti-инозитфосфатов, или
освобождаться в результате гидролиза органофосфатов в процессе экстрак¬
ции). Из реэкстракта реосаждают органические фосфаты солями бария из ра¬
створа этанол-этано л амина. Осадок Ва-фитатов промывают, растворяют в
Н-смоле и хроматографируют с помощью ионообменной и бумажной хромато¬
графии (см. выше определение инозитфосфатов).
Ход анализа. 5 г воздушно-сухой почвы (сито d 1 мм) помещают в 250-мил-
лилитровые центрифужные пробирки стекла «Пирекс», приливают 40 мл смеси
6МНС1 в0,5 MHF и 10 лел 15 % TiCl4 и кипятят с обратным холодильником в
течение 10 мин. После этого смесь охлаждают, центрифугируют и промывают
остаток почвы и осадок органических фосфатов с поливалентными катионами
дважды с 25 мл 6М НС1 +1 % TiCl4.
Все центрифугаты сливают в мерную колбу на 100 мл, доливают до метки
водой, перемешивают (раствор А). Отбирают аликвоту раствора А, нейтрали¬
зуют и определяют в ней фосфор колориметрически. Получают содержание
минерального фосфора. Другую аликвоту раствора А помещают в плоскодон¬
ные колбочки, выпаривают и окисляют 3—5 мл 50%-ной НС104 (общий фос¬
фор). По разности между содержанием общего и минерального фосфора уста¬
навливают содержание фосфора органических соединений.
Для растворения осадка органофосфатов в центрифужные пробирки при¬
ливают 10 мл свежеприготовленного 5%-ного водного раствора купферрона,.
перемешивают, добавляют 40 мл 3 М НС1 и взбалтывают в течение 2-х час.г
центрифугируют, промывают остаток в пробирке дважды 10 мл 1 М НС1, со¬
держащей 0,1% купферрона. Все центрифугаты сливают в мерную колбу и*?
Методы, определения фесфора в почве
187
100 мл, доливают до метки водой, перемешивают (раствор Б). В аликвотах
раствора Б определяют содержание общего, минерального и органического
фосфора точно так же, как в растворе А.
50 мл раствора Б переносят в другие центрифужные пробирки емкостью
250 мл, добавляют равный объем этанола, перемешивают и оставляют раствор
.для осветления. Добавляют 40 мл триэтаноламина для повышения pH до
8—9 (А13+ в данных условиях не образует комплексы), раствор подогревают,
нриливают 10 мл 20%-ного ВаС12«2Н20, перемешивают, оставляют на ночь,
затем центрифугируют и промывают выпавший осадок Ва-фитатов 50%-ным
водным раствором этанола (порциями по 25—50 мл) до удаления избытка куп¬
феррона. Промытый таким образом осадок нагревают с 10 мл ЗМ NaOH на
водяной бане при 90° С в течение 8 час., затем добавляют 10 мл воды и подкис¬
ляют до pH 4 НСООН, затем подщелачивают 25%-ным аммиаком до pH 8—9,
приливают равный объем этанола (96°) и 2 мл 20%-ного раствора ВаС12, на¬
гревают на водяной бане, настаивают 2 часа, центрифугируют. Реосажденные
Ва-фитаты промывают несколько раз 50%-ным этанолом (порциями по 10 мл),
рерастворяют в Н-смоле, фильтруют. Прозрачный фильтрат используют для
ионообменной и бумажной хроматографии (Me Kercher, Anderson, 1968а, б).
Реактивы. 1. 1%-ный и 15%-ный водные растворы TiCl4.
2 Смесь 6 М НС1 в 0,5 М HF. 492 мл НС1 (уд. вес 1,19) разбавляют дистиллиро¬
ванной водой до 700 мл, приливают 8 мл 35,5%-ной HF (уд. вес М5), доливают водой
до 1 л, перемешивают. Сохраняют в полиэтиленовой посуде.
3. 1 М и 3 М НС1.
4. ЗМ NaOH.
5. 0,1%-ный и 5%-ный водные растворы купферрона. Навески соли купферрона пред¬
варительно промывают эфиром от продуктов разложения.
(6. Этанол (96°) и 50%-ный водный растворы.
7. Триэтаноламин.
.8. 20%-ный водный раствор ВаС12*2Н20.
.9. 90%-ная муравьиная кислота (НСООН).
НО. 25%-ный NH4OH.
11. Катионит — Н-форма, анионит — СООН-форма. Приготовление см. выше, в под¬
разделе об ионообменной хроматографии.
Литература
Акопян Э. М. Применение кинетического Аскинази Д. Л., Гинзбург К. Е., Лебеде-
метода анализа для автоматического ва Л. С. Минеральные формы фосфора
экспрессного измерения концентрации почвы и методы их определения.— Поч-
фосфора.— Биол. ж. Армении, 1974, воведение, 1963, 5.
т. 27, № 1. Бобко Е. В., Маслова А. Л. Доступность
Алексеев Р. И. Количественное разделение фосфорита растениям в зависимости от
и определение анионов фосфорной, свойств почв.— Труды НИУ, 1926, вып.
мышьяковой и кремневой кислот по- 39.
средством избирательного извлечения.— Бобко А. К., Пилипенко А. Т. Фотомет-
Зав. лаб., 1945, т. 11, № 2—3. рический анализ. Определение неметал-
Аналитическая химия элементов. Фосфор. лов. М., «Химия», 1974.
М., «Наука», 1974. Батлер Дж. Н. Ионные равновесия. JI.,
Аринушкина Е. В. Руководство по хими- «Химия», 1973.
ческому анализу почв. Изд-во МГУ, Вельский Б. Б., Кулаковская Т. Н., Ро-
1970. зина М. С. Определение усвояемых фос-
Аронов С. Изотопные методы в биохимии. фатов в торфяно-болотных почвах.—
М., ИЛ, 1959. Почвоведение, 1961, № 11.
Аскинази Д. Л. Фосфатный режим и из- Беляева Н. И. Определенйе алюминия
весткование почв с кислой реакцией. М., в присутствии железа спектрофотометри-
1949. ческим методом с реактивом феррон.—
Аскинази Д. Л. Лабораторные исследова- Почвоведение, 1966, № 2.
ния фосфатного режима некоторых почв Блэк К. А. Растение и почва. М., «Колос»,
Ленкоранской зоны Азербайджанской 1973.
ССР.— Труды Почв, ин-та им. В. В. До- Везер Ван Дж. Фосфор и его соединения,
кучаева, 1957, т. 50. М., ИЛ, 1962.
188
Литература
Гинзбург Я. Е. К методике колоримет¬
рического определения фосфорной кис¬
лоты в лимоннокислых вытяжках из
почвы.— Почвоведение, 1952, № 12.
Гинзбург К. Е. К методике колориметри¬
ческого определения фосфора в кислот¬
ных вытяжках из почвы.— Почвоведе¬
ние, 1958, № 2.
Гинзбург К. Е. Определение общего содер¬
жания минеральных и органических
форм фосфатов почвы.— Агрохимия,
1969, № 5.
Гинзбург К. Е., Артамонова Л. Ф. Опре¬
деление подвижных фосфатов почвы
в молибдатной вытяжке.— Агрохимия,
1966, № 8.
Гинзбург К. Е.у Артамонова Л. Ф. Опре¬
деление подвижных фосфатов и обмен¬
ного калия почвы в аммонийно-молиб-
датной вытяжке.— Третий делегатский
съезд почвоведов. М., «Наука», 1968.
Гинзбург К. Е., Артамонова Л. Ф., При-
жукова В. П., Соколова Р. А. Опреде¬
ление подвижного фосфора и обменного
калия в аммонийно-молиодатной вытяж¬
ке при массовых анализах почв.— Хи¬
мия в сельском хоз-ве, 1973, № 5.
Гинзбург К. Е., Лебедева Л. С. Методика
определения минеральных форм фосфа¬
тов почвы.— Агрохимия, 1971, № 1.
Гинзбург К. Е., Щеглова Г. М. Опреде¬
ление азота, фосфора и калия в расти¬
тельном материале из одной навески.—
Почвоведение, 1960, № 5.
Гинзбург К. Е., Щеглова Г. МВульфиус
Е. А. Ускоренный метод сжигания почв
и растений.— Почвоведение, 1963, № 5.
Добрицкая Ю. И. Экспресс-Метод полного
валового анализа почв. М., 1973.
Драчев С. М. К изучению мобильности
фосфатов почвы.— Научно-агроном. ж.,
1928, № 9.
Дыбина U, В., Куртасова Л. 3. Способ
пламенно-фотометрического определе¬
ния фосфора. Описание изобретения
к авторскому свидетельству 326494.—
Бюлл. Госкомитета по изобретениям и
открытиям, 1972, № 4.
Замятина В. Б. Методы определения под¬
вижного фосфора в почвах.— В кн.
«Пособие по проведению анализов почв
и составлению агрохимических карто¬
грамм». М., Россельхозиздат, 1969.
Инструкция по проведению массовых ана¬
лизов почв в зональных агрохимических
лабораториях. М., «Колос», 1973.
Инструкция для зональных агрохимиче¬
ских лабораторий по анализу кормов
и растений. М., «Колос», 1968.
Карпачевский Л. О. Применение анионита
ЭДЭ-10П для изучения динамики Р2Об
в дерново-подзолистых почвах.— В сб.
«Московский университет сельскому хо¬
зяйству». Изд-во МГУ, 1971.
Карпачевский Л. О., Киселева Н. К. При¬
менение КУ-2 для изучения динамики
калия в почве.— Агрохимия, 1969, № 1.
Карпинский Я. Я., Замятина В. Б. Фос¬
фатный уровень почвы.— Почвоведение,
1958, № 11.
Карпинский Я. Я., Замятина В. Б., Гла¬
зунова Я. М. Подвижный фосфор почвы
и использование его растениями.—
Докл. сов. почвоведов к VII Междун.
конгр. в США. М., Изд-во АН СССР,
1960.
Кирсанов А. Т. Методы определения по¬
требности почв в фосфорнокислых удо¬
брениях.— Труды Почв, ин-та, 1935, т. 12^
Кобзева И. И. Определение фосфатного по¬
тенциала в почвенном растворе.— Вест¬
ник с.-х. науки, 1969, № 3.
Краснощеков В. В., Максимова Т. ГМи-
шляева Л. В., Столбов В. Ф. Комплек-
сонометрический метод определения фос¬
фора.— Агрохимия, 1969, № 7.
Крешков А. Я. Основы аналитической хи¬
мии, т. I—III. М., «Химия», 1970.
Кудеярова А. Ю. Определение фосфатного
потенциала почв и активности фосфат-
ионов в почвенных растворах для оценки
обеспеченности растений доступными фос¬
фатами и изучения превращения фосфор¬
ных удобрений в почвах. (Автореф. канд.
дисс.), М., 1970.
Кудеярова А. Ю. Использование показа¬
теля фосфатной буферной способности
почв для изучения их фосфатного режи¬
ма и обоснования эффективного приме¬
нения фосфорных удобрений.— Агрохи¬
мия, 1971, № 11.
Кудеярова А. Ю., Полякова Г. В. Срав¬
нение некоторых методов определения
форм минеральных почвенных фосфа¬
тов.— Агрохимия, 1973, № 10.
Кук Дж, Регулирование плодородия почвы.
М., «Колос», 1970.
Марченко 3. Фотометрическое определе¬
ние элементов. «Мир», 1971.
Мачигин Б. Я. Методы агрохимических,
агрофизических и микробиологических,
исследований в поливных хлопковых
районах. Ташкент, Изд-во АН УзбССР,
1952.
Мещеряков А. М. Значение соотношения
между кислотой и молибдатом при ко¬
лориметрическом определении фосфора.—
Почвоведение, 1965, № 2.
Мещеряков А, М., Артамонова Н. В. К ме¬
тодике извлечения метастабильных фос¬
фатов из сероземов.— Агрохимия, 1971,
№ 9.
Никитина Е. А. Гетерополисоединения.
М., Гостеххимиздат, 1962.
Никулина Г, Н. Обзор методов колори¬
метрического определения фосфора по
образованию «молибденовой сини». М.—
JI., «Наука», 1965.
Ониани О. Г. Определение подвижных со¬
единений фосфора и калия из одной вы¬
тяжки из красноземных и подзолистых
почв Грузии.— Агрохимия, 1964, № 6.
Плескач Л. И. Методы пламеннофотомет¬
рического определения фосфора. (Авто¬
реф. канд. дисс.). Алма-Ата, 1969.
Похлебкина Л. Я. Взаимодействие фосфа¬
тов с дерново-подзолистой почвой и до¬
ступность для растений остаточных фос¬
фатов. (Автореф. канд. дисс.). М.г
1973.
Литература
189'
Рем и Г. Курс неорганической химии, т. 1.
М., «Мир», 1963.
Самохвалов С. Г., Соколова Ю. В, Уско¬
ренное определение фосфора в вытяжке
по Мачигину. Химия в сельском хоз-ве,
1974, № 9.
Самуэльсон О. Ионообменные разделения
в аналитической химии. М.— JI., «Хи¬
мия», 1966.
Сидорина Л. В. Применение анионитов для
определения содержания в почве под¬
вижных фосфатов.— Почвоведение, 1961,
№ 2.
Синягина М. Г. Определение фосфатного
потенциала в почвах.— Агрохимия,
1966, № 10.
Синягина М. Г. Определение минераль¬
ных форм фосфорных соединений по
ионному составу почвенного раствора.—
Агрохимия, 1969, № 2.
Соколов А. В. Методика определения за-'
паса растворимых и легкоусвояемых
фосфатов в почве при помощи Р82.—
Докл. сов. почвоведов к VII Междуна¬
родному конгрессу в США. М., Изд-во
АН СССР, 1960а.
Соколов А. В. Фосфор. В кн. «Справочник
по минеральным удобрениям». М., Изд-
во хим. лит-ры, 19606.
У Мью Тхант, Формы фосфатов и методы
их' определения в рисовых почвах
Бирмы. (Автореф. канд. дисс.). М., 1969.
Федоровский Д. В. Выделение почвенного
раствора давлением сжатого ’ газа.—
Агрохимия, 1964, № 3.
Хейфец Д. М. Методика определения и
содержание минеральных и органиче¬
ских соединений фосфора в некоторых
почвах Советского Союза.— Почвоведе¬
ние, 1948, № 2.
Хейфец Д. М. Методы определения фос¬
фора в почве.— В кн. «Агрохимические
методы исследования почв». М., Изд-во
АН СССР, 1960.
Хейфец Д. М. Определение содержания
подвижных фосфатов в ацетатно-буфер¬
ных вытяжках из почв.— Почвоведение,
1962, № 5.
Хейфец Д. М. Методы определения фос¬
фора в почве.— В кн. «Агрохимические
методы исследования почв». М., «Нау¬
ка», 1965.
Чжан Фу-чжу. Использование анионитов для
определения подвижных фосфатов в почвах.
— Почвоведение, 1962, № 10.
Чириков Ф.В. К методике учета форм фосфа¬
тов в почве. — Химиз. соц. земледелия,
1939, № Ю—11.
Чириков Ф. В. Инструкция по учету форм
фосфатов в почве. Программно-методи¬
ческие указания по географической сети
опытов с удобрениями. М., Сельхозгиз,
1947.
Шконде 9. И. Система удобрения и фос¬
фатный режим черноземных почв УССР.—
Почвоведение, 1952, № 8.
Шконде Э. И. К методике определения
валового фосфора в почве.— Почвове¬
дение, 1954, № 3.
Яцимирский К. Б. Физико-химические ме¬
тоды анализа. JI., «Химия», 1971.
Атет F., Bouldin D. i?., Black С. А.,
Duke F. R. Characterisation of soil pho¬
sphorus by anion exchange resin adsorp¬
tion and P32 equilibration.— Plant and
Soil, 1955, v. 6, N 4.
Anderson G. /. Estimation of purines and
pyrimidines in soil humic acid.— Soil
Sci., 1961, v. 91, N 3.
Anderson G. /. Investigations on the ana¬
lysis of inositol hexaphosphate in soils.—
Trans. 8-th Internat. Congr. Soil Sci.
Bucarest, 1964, v. 4, 563—72.
Anderson G. /. Nucleic Acids, Derivatives
and Organic Phosphates. Soil Bioche¬
mistry. Marcel Dekker, N. Y., 1967, 67.
Arrhenius O. Die Phosphatfrage. VI. Be-
stimmung des Phosphatbedarfs auf che-
mischen Wege. — Z. Pflanzenemahr., Diing.
und Bodenkunde, 1933, 32.
Aslyng H. C. The lime and phosphate po¬
tentials of soils.— Yearbook Roy. Ve-
terin Agric. Coll., Copenhagen, 1954,
v. 1, 1—50.
Beckett P., R. White. Studies on the phos-
hate potentials of soils.— Plant and
oil, 1964, v. 21, N 3.
Bray R. H., Kurtz L. T. Determination of
total organic and available forms of
phosphorus in soils.— Soil Sci., 1945,
59, N 1.
Burriel F., Hernando V. New method for
determining available phosphorus in so¬
ils.— Soils and Fertilizers, 1951, N 3.
Chang S. С., Jackson M. L. Fractionation
of soil phosphorus.— Soil Sci., 1957,
v. 84, N 2.
Cooke J. J. A kinetic approach to the
description of soil phosphate status.—
J. Soil Sci., 1966, v. 17, N 1.
Desjobert A., Petek F. Chromatographie sur
papier des esters phosphoriques ae Г ino¬
sitol. Application а Г etude de la degra¬
dation nydrolytique de Tinositolh^xa-
phospate. —Bull. Soc. chim., biol., 1956,
v. 38.
Dickman S. R., Bray R. H. Replacement
of adsorbed phosphate from kaolinite
by fluoride.— Soil Sci., 1941, 52, N 4.
Egner Kohler G., Nydahl F. Die Lak-
tat methode zur Bestimmung, leichtlosli-
cher Phosphorsaure in Ackerboden.—
Kgl. Landbrukshogsk. ann., 1938, v. 6.
Egner #., Riehrn, Domingo W. R. Unter-
suchungen liber die chemische Bodenana-
lyse als Grundlage fur die Beurteilung
des Diinger-Bedurfnisses des Ackerboden.
Bestimmung von leichtlosslichen Phos¬
phor, 1958.
Egner H., Riehm H., Domingo W. R. Un-
tersuchungen iiber die chemische Boden-
analyse als Grundlage fur die Beurtei¬
lung des Nahrstoff zustandes der Boden.
II. Chemische Extraktionsmethoden zur
Phosphor- und Kaliumbestimmung. Uppsa-
tionsmethoden zur Phosphor- und Kalium¬
bestimmung. Uppsala, 1960.
Jackson M. L. Soil chemical analysis. Ma¬
dison, Wisconsin, 1958*
190
Литература
Halstead R. L., Anderson G. Chromatograp¬
hic fractionation of organic phosphates
from alkali, acid and aqueous acetyla-
cetone extracts of soil.— Canad. J. Soil
Sci., 1970, v. 50, N 2.
Лапсе R. Anderson G. Extractions and
estimation of soil phospholipids.—Soil Sci.,
1963a, v. 96, N 2.
Лапсе R. Anderson G. Identification
of hydrolysis products of soil phospho¬
lipids.— Soil Sci., 1963b, v. 96, N 3.
Kowalenko C. G., Me Kercher R. В. An
examination of methods for extraction
of soil phospholipids.— Soil. Biol, and
Biochem., 1970, v. 2, 263—273.
Larsen S. Soil phosphorus.— Advances in
agronomy, 1967, v. 19, 151—209.
Lindsay W. L., E, С. Moreno. Phosphate pha¬
se equilibria in soils. Soil Sci. Soc. Ame¬
rica Proc., 1960, v. 24, N 3.
Me Kercher R. 2?., Anderson G. Content of
inositol penta- and hexaphosphates in
some Canadian soils.— J. Soil Sci., 1968a,
v. 19, N 1.
Me Kercher R. /?., Anderson G, Characteri¬
zation of the inositol penta- and hexa-
phosphate fractions of a number of Cana¬
dian and Scotish soils.— Soil Sci., 1968b,
v. 19, N 2.
Me Kercher R. B., Anderson G. Observations
on the accuracy of an ignition and an
extraction method for measuring orga¬
nic phosphorus in some Canadian soils.—
Soil Sci., 1968b, v. 105.
Metha N. С., Legg /. ОCaring C. A.,
Black C. A. Determination of organic
phosphorus in soils, p. I.— Soil Sci.
Soc. America Proc., 1954, v. 18, N 4.
Miller J. Л., Axley J. H. Correlation of
chemical soil tests for available phos¬
phorus with crop response, including a
proposed method.— Soil Sci., 1956, v. 82,
N 2.
Murphy Riley J. P. A modified single
solution method for the determination
of phosphate in natural waters.— Ana-
lyt. chim. acta, 1962, v. 27, N 1, 31 —
36.
Ю1$еп S. R. et al. Estimation of available
phosphorus in soils by extraction with
sodium bicarbonate.— U. S. Dept Agric.
.Stat., 1954, N 939.
Oniani O. G., Margaret Chater A., Mat¬
tingly G. A. M. Some effects of ferti¬
lizers and Farmyard manure of the orga¬
nic phosphorus in soils.— J. Soil Sci.,
1973, v. 24, N 1.
Pons W. Л., Guthrie /. D. Extraction me¬
thod for the colorimetric determination
of phosphorus.— Industr. and Engien
Chem. Analyt. Ed., 1946, v. 18, 184—
186.
Saunders W. M. H., Williams E. G. Obser¬
vations on the determination of total
organic phosphorus in soils.— Soil Sci.,
1955, v. 6, 254-267.
Schofield R. К. Can a precise meaning be
given to «available» soil phosphate.—
Soil and Fertilizers, 1955* v. 18, N 5.
Sherman M. S. Colorimetric determination
of phosphorus in soils. Industr. and Eng.
Chem. Analyt. Ed., 1942, v. 14, 182—
185.
Steward J. H., Oades J. M. The determi¬
nation of organic phosphorus.— Soil Sci.,
1973, v. 23, N 1.
Tinsley Qzcavasci C. Studies of soil
organic phosphorus, using titanic chlo¬
ride.— Trans. 10-th Internat. Congr. Soil
Sci., 1974, v. II, 332—341.
Truog E. The determination of the readily
available phosphorus of soils.— J. Amer.
Soc. Agron., 1930, 22; N 10.
Troug E., Meyer A. The determination of
available phosphorus in soils.— J. Amer.
Soc. Agron., 1929.
Ulrich В. Die Wechselbezichungen von Bo-
den und Pflanzen. Stuttgart, 1961.
Wade H. E., Morgan D. M. Fractionation
of phosphates by paper ionophoresis
and chromatography.—Biochem. J., 1955,
v. 60, 167—70.
Warren R.G., Pugh A. L. The colorimetric
determination of phosphoric acid.— J.
Agric. Sci., 1930, v. 20, pt. 4, 532.
Watanabe F. S., S. R. Olsen. Test of an
ascorbic acid method for determining
phosphorus in water and NaHCOs extracts
from soil.— Soil Sci. America Proc.,
1965, v. 29, N 6.
И. Г. ВАЖЕНИН
МЕТОДЫ ОПРЕДЕЛЕНИЯ КАЛИЯ В ПОЧВЕ
*
Содержание калия в почве определяется прежде всего и в основном мине¬
ралогическим составом, а именно — наличием калийсодержащих минералов:
слюд, гидрослюд, полевых шпатов и др. Влияние гранулометрического соста¬
ва почв на содержание калия проявляется постольку, поскольку это отража¬
ет перераспределение по гранулометрическим фракциям калийсодержащих
минералов. В общем случае этих минералов (особенно гидрослюд) мало в сос¬
таве песка и крупной пыли и больше в составе мелкой пыли и ила. Поэтому,
как правило, в песчаных почвах содержание валового калия в несколько раз
меньше, чем в почвах глинистых. Правда, в некоторых случаях, например в
почвах на молодом элювии гранитов, отмечается высокое содержание калия
и в крупнопесчаных фракциях, за счет остаточных зерен ортоклаза и слюд.
Колебания в содержании валового калия в почвах, кроме того, обусловлены
и другими причинами: химическими изменениями первичных минералов в
процессе выветривания и почвообразования, в том числе — образованием,
накоплением или потерей вторичных минералов.
Среднее содержание валового калия в почвах СССР, по нашим подсчетам
(Важенин, 1965), выражается величинами, представленными в табл. 1.
Таблица 1
Валовое содержание калия в пахотном слое различных почв
Почвы
Число
Содержание
Число
Содержание
разрезов
к,о, %
(М +т)
Почвы
разрезов
К,0, %
(М -f т)
Дерново-подзолистые:
Черноземы:
песчаные и су¬
47
1,20 ±0,14
типичные
53
2,15 ±0,09
песчаные
оподзоленные и
41
2,23 ±0,10
легкосуглинястые
61
1,77 ±0,07
выщелоченные
среднесуглинистые
53
2,17 ±0,09
обыкновенные и
53
2,01 ±0,08
тяжелосуглини¬
55
2,23 ±0,10
южные
стые и глинистые
Каштановые
22
2,27 ±0,17
Серые лесные:
Сероземы Средней
33
2,29 ±0,09
светло-серые и
32
1,92 ±0,10
Азии
82
2,09 ±0,09
серые
Бурые
темно-серые
20
2,03 ±0,15
Красноземы Запад¬
12
0,70±0,11
ной Грузии
Для дерново-подзолистых почв, формирующихся на рыхлых отложениях
ледникового, водно-ледникового и водного происхождения, содержание ва¬
лового калия тем выше, чем тяжелее почва по механическому составу. При
одинаковом гранулометрическом составе почв различного типа (дерново-
подзолистые, серые лесные, черноземы) валовое содержание калия примерно
одинаковое. Резко отличаются по содержанию калия красноземы, формиру¬
192
И. Г. Важенин
ющиеся на сильновыветрелых породах, содержащие незначительные количест¬
ва первичных минералов, в том числе калийсодержащих полевых шпатов и
слюд.
В агрономических целях, при характеристике плодородия почв в отноше¬
нии калия, имеет значение определение не валового калия, а той части его,
которая является наиболее растворимой и доступной растениям. Эта часть
составляет лишь небольшую долю общего (валового) калия почвы.
Чем выше подвижность калия (растворимость и гидролиз соединений, со¬
держащих калий), тем меньше его в почве. Наименьшее количество калия со¬
держится в почвенном растворе, больше его — в обменной форме: в среднем
0,5% (от валового) в дерново-подзолистых, 1% в серых лесных, 1,5—2% в
черноземах, 2—3% в каштановых и сероземных почвах; еще больше калия
в необменной форме (2—5% от валового количества); основное количество
калия (90—95%) сосредоточено в составе почвенного скелета.
Все формы калия взаимно связаны между собой, все они в различной сте¬
пени участвуют в калийном питании растений. Даже калий кристаллической
решетки минералов (например, ортоклаза, если созданы условия для разру¬
шения — измельчения минерала до состояния пыли и ила, гидролиза и раст¬
ворения частиц) становится усвояемым для растений.
Растения в процессе питания усваивают прежде всего наиболее подвиж¬
ные формы: калий почвенного раствора, затем обменный (через почвенный
раствор), далее, по мере развития растения и возрастания усвояющей способ¬
ности и потребностей в калии, в процесс питания растений вовлекаются необ¬
менные (экстенсивно обменные) формы. Поэтому при характеристике плодо¬
родия почв в отношении йалия следует учитывать не только калий почвен¬
ного раствора и обменный, но и необменные формы его, являющиеся резервом
плодородия почвы в отношении калия.
Ниже излагаются различные методы определения калия в растворах и ме¬
тоды извлечения и определения различных форм калия в почвах.
ОПРЕДЕЛЕНИЕ КАЛИЯ В РАСТВОРАХ
Прежде чем определить содержание калия в почве, независимо от формы
'Соединения, его необходимо перевести в раствор (водный, солевой, кислот¬
ный). Переведенный в раствор калий определяется химическими методами или
физическими: потенциометрически, пламенной фотометрии, атомной абсорб¬
ции и др.
Хлороплатинатный метод
Метод основан на том, что хлороплатинат калия — (C2(PtCle) нераст¬
ворим в 80%-ном спирте, а хлороплатинаты других щелочей растворимы.
Ион аммония аналогично калию образует нерастворимый в спирте хлоро-
платинат аммония, поэтому он должен быть полностью удален из анализа
руемого раствора. Из раствора должны быть также удалены сульфаты (осаж¬
дением в форме BaS04), так как сульфаты щелочных земель трудно растворя¬
ются в 80%-ном спирте. Испытуемый раствор выпаривают и остаток прока¬
ливают.
Прокаленный от аммонийных солей остаток растворяют в чашке в неболь¬
шом объеме горячей, подкисленной НС1 воды, фильтруют, промывают фильтр
водой и упаривают раствор до объема 10—12 мл. Подкислив раствор 1 мл
10%-ной НС1, осаждают калий небольшим избытком 10%-ного раствора пла-
тино-хлористоводородной кислоты (PtCl4 + Н20). Цвет раствора должен со¬
ответствовать цвету хлороплатината: слабая окраска указывает на недоста¬
точное количество реактива (на 100 мг К20 вполне достаточно 3 мл реактива).
Методы, определения калия в почве
193
Фарфоровую чашку с раствором помещают на почти кипящую водяную
баню и содержимое медленно выпаривают до сиропообразного состояния.
Выпаривать досуха не следует, так как это способствует образованию нераст¬
воримой в спирте формы хлороплатината натрия.
После охлаждения жидкости в чашку приливают 8—10 мл 80%-ного спир¬
та для растворения в нем хлороплатината натрия, перемешивают и растирают
осадок стеклянной палочкой. Если цвет раствора при этом окажется слабо¬
желтым (недостаток реактива), то добавляют еще хлороплатината и, подкис¬
лив соляной кислотой, повторяют выпаривание до сиропообразного состоя¬
ния.
По охлаждении в чашку приливают 10 мл 80%-ного спирта, осадок расти¬
рают палочкой, жидкость декантированием переносят во взвешенный стек¬
лянный тигель-фильтр № 3, фильтруют под вакуумом; приливание спирта,
растирание и декантацию повторяют 2—3 раза. Затем осадок из чашки пере¬
носят на фильтр и промывают его спиртом до полного обесцвечивания
фильтрата. Промытый осадок приобретает чистый золотисто-желтый цвет
без оранжево-красных кристаллов хлороплатината натрия. Тигель с осадком
просушивают при 130° С в продолжение 1 часа; тигель охлаждают и взве¬
шивают. 1 часть K2(PtCle) соответствует 0,608 весовым частям калия.
Анализ считается более точным, если осадок хлороплатината калия восста¬
новить и взвесить металлическую платину. В таком случае промытый в
тигле осадок растворяют горячей водой; фильтрат собирают в колбу для от¬
сасывания, а затем переносят в стакан на 250—300 мл, в котором производят
восстановление хлороплатината калия до металлической платины. Восста¬
новление удобнее всего вести металлическим магнием в виде ленты. Ленту
магния, намотанную на загнутый конец стеклянной палочки, опускают на
дно стакана. При отсутствии магниевой ленты для восстановления хлоропла¬
тината используют магний в виде стружки или порошка, металлический алю¬
миний (в палочках) или муравьинокислый натрий, прибавляемый порциями.
Жидкость доводят до кипения, приливают 10 мл 10%-ной НС1, стакан на¬
крывают часовым стеклом и кипятят 2—3 мин., затем дают постоять до окон¬
чания бурной реакции; приливают 5 мл 10%-ной НС1 и раствор оставляют в
покое на 3—4 часа до полного восстановления хлороплатината калия. Прове¬
ряют полноту восстановления, опустив в раствор кусочек ленты магния. Если
появится муть, то восстановление повторяют. Убедившись в полноте восста¬
новления хлороплатината, прибавляют в раствор 5—6 мл 10%-ной НС1 (для
разрушения основных солей магния, что ускоряет фильтрование) и фильтру¬
ют раствор через беззольный фильтр (диаметр 7 мм) с белой обмоткой. Оса¬
док металлической платины промывают горячей водой до полного удаления
хлора (проба с AgN03).
Промытый осадок вместе с фильтром переносят во взвешенный фарфоро¬
вый тигель, сжигают, а затем прокаливают на горелке или в муфеле до по¬
стоянного веса; 1 мг платины соответствует 0,481 мг К20.
Хлороплатинатный метод наиболее точный, но дорогой, в связи с дефицит¬
ностью реактива, и может использоваться как арбитражный метод.
Кобалътнитритный метод
Различные варианты кобальтнитритного метода основаны на осаждении
калия кобальтнитритом натрия в виде труднорастворимой комплексной соли
[K2NaCo(N02)e]. Определение калия этим методом возможно в присутствии
значительных количеств кальция и магния, анионов азотной, соляной и сер¬
ной кислот и небольших количеств полутораокисей; фосфорная кислота ме¬
шает определению калия. Раствор, в котором определяют калий, должен быть
свободным от аммонийных солей, с которыми кобальтнитрит дает также нера¬
створимый осадок, и от органических веществ, на окисление которых расхо¬
194
И. Г. Важенин
дуется КМп04. Поэтому в случае наличия аммонийных солей и органических
веществ раствор предварительно выпаривают с 5 мл концентрированной
HNOs и остаток прокаливают при температуре не выше 400—450° С до полного
удаления аммонийных солей и органического вещества. К обработанному су¬
хому остатку прибавляют 1 мл концентрированной НС1, содержимое чашки
выпаривают досуха и сухой остаток вновь слабо прокаливают. Состав осадка
[K2NaCo(N02)e] в значительной мере определяется условиями осаждения ка¬
лия: меняется соотношение между К и Na, изменяется содержание воды в
осадке. Поэтому обязательно строгое соблюдение определенных условий
осаждения калия. Содержание калия в составе комплексной соли устанав¬
ливается весовым, объемным, колориметрическим или седиментометрическим
и нефелометрическим методами.
Осаждение калия (по Друшелю). К прокаленному остатку хло¬
ристых солей в чашке, т. е. после разрушения органического вещества и уда¬
ления аммония, приливают 10 мл воды и раствор нагревают на водяной бане
до 80—90° С. К нагретому раствору понемногу приливают 35%-ный раствор
кобальтнитрита натрия. На 20—30 мг калия прибавляют 5 мл реактива. За¬
тем содержимое чашки упаривают до пастообразного состояния (не допуская
образования корочки и пересушивания осадка на стенках). К упаренному ос¬
татку дважды приливают по 1—2 мл 15%-ной уксусной кислоты, каждый раз
концентрируя содержимое до пастообразного состояния. К охлажденной сме¬
си приливают 10—15 мл воды для растворения реактива и раствор отфильт¬
ровывают.
Весовой метод. Раствор от осадка отфильтровывают через просу¬
шенный взвешенный стеклянный тигель-фильтр № 4. Осадок в чашке и на
фильтре промывают 2,5%-ным раствором Na2S04 или 1%-ным раствором ук¬
сусной кислоты, насыщенной осадком кобальтнитрита калия-натрия. Про¬
мывание ведут сначала декантацией, а затем на фильтре до полного обесцве¬
чивания промывной жидкости. Во время промывания следует избегать про-
сасывания воздуха через осадок, так как это способствует его растворению.
После окончания промывания жидкость тщательно отсасывают, и тигель с
осадком сушат 1^2 часа при 110° С. Высушенный осадок должен иметь
желтый цвет; бурая окраска указывает на неполноту отмывания кобальт¬
нитрита натрия. Эмпирический коэффициент пересчета осадка [K2NaCo-
•(NO^el на К20 различен в зависимости от веса осадка, а именно:
Вес осадка, мг Коэффициент Вес осадка, мг Коэффициент
5 0,210 40 0,206
10 0,209 50 0,204
15 0,208 100 0,192
20 0,207 200 0,187
30 0,206 500 0,178
Объемный метод. Осадок кобальтнитрита калия-натрия, отмытый
от примесей и избытка реактива, разрушают в кислой среде титрованным
раствором перманганата калия:
5KaNaC0(N0a)e+llK.Mn04+14H2S04 = 15KN084-5NaN03+3K2S04+llMnS04+
+5CO(N Оз)а+ 14ШО.
Из уравнения реакции следует, что И г-молей КМп04 соответствуют 10 г-
атомам калия в комплексной соли; следовательно, каждый миллилитр 0,1 н.
раствора КМп04, затраченный на разрушение осадка, отвечает 0,856 мг
К20.
В стакан емкостью 500—700 мл приливают 200—250 мл воды и точно от¬
меренный объем (25—50 мл) 0,1 н. раствора КМп04; раствор нагревают на
кипящей водяной бане до 80—90° С. В горячий раствор опускают тигель-
Методы определения калия в почве
195
фильтр с осадком, осторожно вращают тигель в жидкости, а затем добавляют
10 мл разведенной (1 : 7) серной кислоты и раствор хорошо перемешивают.
Нагревание жидкости на бане продолжают до полного растворения желтого
осадка. Если цвет станет бледно-фиолетовым, то в стакан немедленно добавля¬
ют из бюретки 2—4 мл 0,1 н. раствора КМп04. Во время окисления кобальт-
нитрита калия-натрия в стакане образуется двуокись марганца (Мп02) чер¬
ного цвета.
Когда желтый осадок в тигле полностью растворится, в стакан приливают
(из бюретки) точно отмеренный объем 0,1 н. раствора щавелевой кислоты до
полного растворения двуокиси марганца и обесцвечивания раствора. Избыток
щавелевой кислоты обратно оттитровывают 0,1 н. раствором КМп04 до слабо¬
розового окрашивания. По количеству миллилитров ОД н. раствора КМп04,
израсходованного на окисление кобальтнитрита калия-натрия, вычисляют
содержание калия в анализируемом растворе.
Реактивы. 1. 35%-ный раствор Na3Co(N02)e. а) 25 г азотнокислого кобальта
Co(N03)2‘6H20 растворяют в 50 мл воды и прибавляют 12,5 мл ледяной уксусной кислоты;
б) 120 г азотистокислого натрия (NaN02) растворяют при осторожном нагревании в 180 мл
воды. За день до употребления смешивают одну часть раствора «а» с тремя частями ра¬
створа «б» и спустя 1—2 часа через смесь продувают воздух для удаления окислов азота»
По истечении 5—6 час. раствор отфильтровывают от выпавшего осадка. Реактив хранят в
склянке, защищенной от света; он годен к употреблению в продолжение 3—4 дней.
2. 2,5%-ный раствор Na2S04«10H20 или 1%-ный раствор уксусной кислоты, насы»
щенной осадком [K2NaCo(N02)e].
3. 0,1 н. раствор КМп04.
4. 0,1 н. раствор Н2С204.
5. Серная кислота (уд. вес 1,84), разбавленная 1 : 7.
Тетрафенилборатный метод
Тетрафенилборат натрия NaB(CeH6)4 с ионами калия дает белый объемис¬
тый мелкокристаллический осадок состава КВ(СвН5)4, нерастворимый в раз¬
бавленных (0,1 н.) минеральных кислотах. Реакция эта более специфична,
чем при осаждении калия другими реактивами. Аналогично калию этим ре¬
активом осаждаются NH4, Rb, Cs, Hg, Ag, ТЬ. Ионы Al, Ca, Mg, Li, Na, N03,
Cl, Br, S04 мешают только при содержании их свыше 2 мг/мл, а ионы Ni, Со,
Fe — при концентрации свыше 1 мг/мл (Pflaum, Howick, 1956).
Весовой метод. Осаждение калия ведут в кислой среде. Раствор
подкисляют обычно уксусной кислотой до pH 3—4. Некоторые авторы про¬
изводят более сильное подкисление, используя для этого минеральные кисло¬
ты. Однако установлено, что сильное подкисление приводит к завышенным
результатам вследствие выпадения продуктов разложения самой кислоты
Н[В(С6Н6)4]. В большинстве модификаций метода осаждение калия ведут при
комнатной температуре. Но считается, что состав осадка наиболее устойчив
при осаждении в ледяной ванне (0° С). Если осаждение ведут при комнатной
температуре или выше, то к фильтрованию осадка приступают немедленно
(не позднее, чем через 10 мин.).
Ход анализа — общий для всех модификаций. К определенному объему
анализируемого раствора — обычно около 50 мл и содержащему около 5 мг
К20, подкисленному соответствующей кислотой до pH 3—4, при медленном
взбалтывании каплями прибавляют осаждающий реактив тетрафенилбората
натрия (на каждые 5 мг К20 вносится 10 мл реактива). Через 5—10 мин. оса¬
док переносят во взвешенный тигель-фильтр № 4, каждый раз полностью от¬
сасывая. Промывающую жидкость приливают 4—6 раз в объеме по 2—3 мл.
Промытый осадок высушивают в продолжение 30 мин. при температуре
110—120° С и после охлаждения взвешивают. Коэффициент пересчета осадка
на К20 = 0,1314.
196
И. Г. Важенин
Модификации весового метода. А. Ф. Иевинып и Э. Ю»
Гудриниеце (1954) добавляют к анализируемому раствору уксусную кислоту
в таком количестве, чтобы к концу осаждения концентрация ее была окола
1%. Жидкость подогревают до 40° С и каплями прибавляют 3—4%-ный раст¬
вор тетрафенилбората натрия.
Тендилл (Tendille, 1955) к раствору калия в объеме 40 мл прибавляет 1 —
2 мл НС1 (1 : 1). Затем при взбалтывании по каплям приливает осаждающий
реактив (на каждые 5 мг К20 — 10 мл 0,6%-ного раствора тетрафенилбората
натрия).
К. Ф. Спорек (Sporek, 1956) применяет осаждение калия на холоду. К ана¬
лизируемому раствору в объеме 50 мл прибавляют 3 мл концентрирован¬
ной НС1, закрывают колбу пробкой и ставят в ледяную баню (0° С). К охлаж¬
денному раствору медленно приливают 30 мл охлажденного (0° С) 1 %-нога
раствора тетрафенилбората натрия и ставят колбы на несколько минут в ле¬
дяную ванну.
А. А. Резников и А. А. Нечаева (1956) разработали вариант этого метода,
позволяющий вести определение калия в щелочной среде. К анализируемому
раствору (в объеме 100—200 мл) добавляют 1—2 капли 1%-ного раствора фе¬
нолфталеина, 250 мг сухого трилона Б для связывания в комплекс кальция и
магния, каплями приливают 0,1 н. раствор NaOH до покраснения раствора
(pH 8,0—8,5) и затем 10 мл 3%-ного водного раствора тетрафенилбората нат¬
рия. Осадок отфильтровывают, промывают водой, насыщенной осадком тетра¬
фенилбората калия, и высушивают.
Энгельбрехт и Мак-Кой (Engelbrecht, Мс.Соу, 1956) разработали метод
определения калия в присутствии аммония. Влияние аммония устраняется
добавлением формальдегида. К определенному объему раствора, содержащего*
до 45 мг калия и до 150 мг аммония, прибавляют 130 мл 37%-ного раствора
формальдегида, энергично перемешивают и оставляют на 5 мин. в покое. За¬
тем добавляют 6 г NaOH. Концентрация щелочи становится — 1,0 н. После
растворения щелочи раствор нагревают до кипения. К еще горячему раствору
медленно, при взбалтывании приливают 50 мл 1%-ного раствора тетрафенил¬
бората натрия в 0,01 н. NaOH. Смесь хорошо перемешивают, охлаждают и
фильтруют через взвешенный тигель № 4. Осадок промывают насыщенным
водным раствором соли тетрафенилбората калия, высушивают в течение 1 ча¬
са прц температуре 110° С.
Если одновременно определяют калий и аммоний, то вначале поступают
так же, как описано выше, но реактива приливают больше, с учетом содержа¬
ния аммония. Взвешенный осадок смеси калия и аммония растворяют в чис¬
том ацетоне. К раствору добавляют 20 мл 2%-ного раствора соды и медленно
выпаривают ацетон на водяной бане. Тетрафенилборат калия оседает, а нат¬
рий остается в растворе. Охлажденный осадок отфильтровывают через взве¬
шенный тигель, промывают, высушивают и взвешивают. Разница во взвеши¬
ваниях дает вес тетрафенилбората аммония. Коэффициент пересчета на NH4
равен 0,0528.
Объемный метод. Модификация А. Ф. Иевинып и Э. Ю. Гудри¬
ниеце (1954). Калий осаждают титрованным раствором тетрафенилбората нат¬
рия, избыток которого связывают хлористым аммонием, определяемым фор-
мальдегидным методом.
Ход анализа. К 5—10 мл раствора, содержащего около 20 мг калия, при¬
бавляют 2 мл 0,05 н. НС1 и 15 мл 0,05 н. раствора NaB(CeH5)4. Выпавший оса¬
док КВ(С6Н5)4 через 10 мин. отсасывают и трижды промывают водой по 5 мл.
К фильтрату вместе с промывными водами добавляют 20 мл примерно 0,03 н.
раствора NH4C1 и взбалтывают. Через 10 мин. выпавший осадок тетрафенил¬
бората аммония отсасывают и 3 раза промывают водой по 5 мл. Фильтрат,
содержащий неосажденную часть аммония, нейтрализуют по метилроту,
прибавляют 5 мл 40%-ного раствора формалина и титруют 0,05 н. раствооом
Методы определения калия в почве
197
NaOII до красной окраски по фенолфталеину. Титр раствора NH4C1 устанав¬
ливают по образцовому раствору хлористого калия, осаждаемого тем же ра¬
створом тетрафенилбората натрия.
Пример расчета анализа. 1. Определение соотношения между концентрациями
растворов NH4C1 и NaB(CeH6)4.K 20 жл^-0,03 н. раствора NH4C1,подкисленного 2 мл0,0Ь н*
НС1, добавляют 15 мл ~0,05 н. раствора NaB(CeH5)4. Осадок тетрафенилбората аммония
отфильтровывают, фильтр промывают 3 раза водой по 5 мл (насыщенной тетрафенилбо-
ратом калия). Фильтрат нейтрализуют в присутствии индикатора метилрота 0,05 н. ра¬
створом NaOH, добавляют 5 мл нейтрального раствора 40%-ного раствора формалина и
титруют освободившуюся кислоту 0,05 н. раствором NaOH, определяя таким образом из¬
быток NH4C1. Например, на титрование 20 мл раствора NH4C1 израсходовано 13,6 мл
раствора NaOH. На оттитровывание избытка NH4C1 израсходовано 2,85 мл раствора
NaOH. Следовательно, 15 мл раствора NaB(CeH5)4 эквивалентны 13,60—2,85 = 10,75 мл
раствора NaOH или 15,81 мл раствора NH4C1.
2. Расчет анализа. К образцовому раствору, содержащему, например, 18,67 мг К2Ог
прибавили 2 мл 0,05 н. НС1 и 15 мл ~ 0,05 н. раствора NaB(CeH6)4. После отделения осад¬
ка КВ(СвН5)4 к фильтрату прибавили 20 мл ~ 0,03 н. раствора NH4C1, а на оттитровыва¬
ние избытка NH4C1 израсходовали 12,7 мл 0,05 н. раствора NaOH, что эквивалентно
18,68 мл раствора NH4C1. Отсюда следует, что на осаждение избытка NaB(CeH6)4 израсхо¬
довано 20,0 — 18,68 = 1,32 мл раствора NH4C1. На осаждение калия израсходовано
NaB(CeH6)4: 15,81 — 1,32 = 14,49 мл раствора NH4C1, что соответствует 18,67 мг К20.
Следовательно, 1 мл ~ 0,03 н. раствора NH4C1 соответствует 18,67 : 14,49 = 1,29 мг
калия. Зная этот титр, по израсходованному количеству раствора NH4C1 рассчитывают
содержание калия в растворе. Титр раствора следует проверять каждые 2—3 дня.
Проверкой тетрафенилборатного метода определения калия в растворах
чистых солей в Почвенном институте (Русакова, 1963) установлено, что наи¬
более благоприятной средой для осаждения калия в растворах оказалась сла¬
бокислая (pH 4,2—4,4); при комнатной температуре обеспечивается достаточ¬
ная полнота осаждения; время взаимодействия осадителя и раствора 10 мин.
вполне достаточно; в качестве промывной жидкости лучше использовать на¬
сыщенный водный раствор тетрафенилбората калия; совместное присутствие
ионов Са, Mg, А1 и Р04 в количествах, обычно встречающихся в почвенных
вытяжках, существенно не мешает определению калия.
Реактивы. 1. 0,05 н. НС1 (готовится из фиксанала).
2. 0,05 н. NaOH (готовится из фиксанала).
3. ~0,05 н. раствор тетрафенилбората натрия. 17,4 г NaB(CeH6)4 растворяют в 1 4
дистиллированной воды. Для коагуляции нерастворимого осадка и повышения устойчиво¬
сти раствора при хранении перед фильтрованием к раствору добавляют 5 мл 0,1 н. раство¬
ра А1(ОН)3.
4. ~0,03 н. раствор NH4C1. Растворяют 1,9 г NH4CI в 1 л воды. Содержание NH4CJ в
растворе устанавливают следующим образом. К 20 мл раствора NH4C1 прибавляют каплю
индикатора метилрота и нейтрализуют 0,05 н. раствором NaOH; затем прибавляют 5 мл
40%-ного раствора формалина и титруют освободившуюся кислоту 0,05 н. раствором
NaOH с фенолфталеином.
5. 35—40%-ный раствор формалина, нейтрализованный по фенолфталеину.
6. Промывная жидкость (вода), насыщенная тетрафенилборатом калия. 20—30 мг
осажденного, промытого и высушенного в эксикаторе КВ(СвНь)4 встряхивают в продол¬
жение 30—40 мин. с 250 мл дистиллированной воды. Пользуются прозрачным раствором
над осадком.
Фотометрический метод
В практике работы агрохимических лабораторий для определения калия
в растворах широко используются пламенные фотометры следующих моде¬
лей: Цейс (ГДР), ГП-21 или ГП-21Б, ФПФ-58, ППФ-УНИИЗ.
Рис. 1. Схема пламенного фотометра типа Цейс
1 — компрессорХвоздушный;
2 — буферный баллон;
$ — фильтр для очистки воздуха;
4 — сброс излишка воздуха;
5 — манометр для воздуха;
6 — распылитель;
7 — баллон с ацетиленом;
8 — водяной затвор;
® — газосборник;
ю — осушитель ацетилена;
и — манометр для ацетилена;
S3 /9 /S
смеситель;
горелка;
светофильтр;
фотоэлемент;
гальванометр
Ме'поды определения калия в почве
191»
Главные узлы всех систем пламенных фотометров одни и те же (рис. 1)г
система возбуждения излучений — пламя; оптическое устройство — свето¬
фильтры, линзы; приемник излучения — фотоэлемент и отсчетный прибор —
гальванометр. Техника и основные правила работы на всех приборах одина¬
ковые *.
Преимущество пламенно-фотометрического метода определения калия пе¬
ред химическими состоит в простоте выполнения анализа и высокой произво¬
дительности. Чувствительность метода при массовых анализах — 1 мг/л раст¬
вора; точность определения + 3—5%. Но эти преимущества достигаются
лишь при строгом и обязательном выполнении ряда условий проведения ана¬
лиза: а) постоянство заданного режима работы прибора; б) постоянство тем¬
пературы и освещенности в помещении; в) максимальное соответствие кон¬
центраций калия в анализируемом растворе градуировочному графику; г)
возможно полное устранение мешающего влияния ионов, сопутствующих ка¬
лию в растворе.
При работе прибора на газо-воздушном (пропан-бутан или бытовой газ)
или ацетилено-воздушном пламени, имеющем температуру соответственно
1600—1800 и 21С0—2300° С, наиболее оптимальный режим работы фотометра
достигается при давлении воздуха 0,4 атм, давлении газа на уровне 40—
50 мм водяного столба по манометру. Главное в режиме работы: постоянство
соотношения между воздухом и горючим газом.
Ход работы 2. Работу на приборе начинают с установления давления воз¬
духа в системе. Сжатый воздух подают или из баллона и регулируют его при
помощи редуктора, или из помещения через компрессор; давление воздуха
при этом регулируют при помощи манометра. Созданное давление (0,4 атм)
поддерживается на одном уровне в продолжение всего времени работы при¬
бора.
Вводя через распылитель дистиллированную воду, проверяют всасываю¬
щее и пульверизирующее устройство. Струя воздуха засасывает из стакан¬
чика воду (или анализируемый раствор), превращая ее в аэрозоль, который
подается в смеситель 3. После установления давления воздуха включают в
систему газ. Газ подается из баллона через редуктор. Проходя через фильтры
и осушитель (с концентрированной серной кислотой), осушенный газ направ¬
ляется в смеситель, где смешивается с аэрозолем анализируемого раствора в
далее через каплеуловитель поступает в горелку. Газо-воздушная смесь за¬
жигается; давление газа поддерживается в пределах 40—50 мм водяного стол¬
ба. Пламя должно быть ровным, спокойным. Зеленовато-голубые язычки пла¬
мени должны быть резко очерчены и выступать над поверхностью горелки на
2—3 мм.
Построение калибровочной шкалы. Поскольку пла¬
менно-фотометрический метод является относительным методом анализа, то
точность определения калия в первую очередь зависит от тщательности при¬
готовления эталонных (образцовых) растворов и построения калибровочной
шкалы (графика). 1,583 г перекристаллизованного КС1 растворяют в 1 л
дистиллированной воды. В 1 мл этого раствора содержится 1 мг К20. В мерные
колбы емкостью 250 мл берут возрастающие количества основного образцо¬
вого раствора: 0,5; 1,0; 2,5; 5,0; 7,5; 10; 15; 20; 25 мл и доводят до метки соот¬
ветствующим данному методу экстрагирующим раствором. В пересчете на 1 л
концентрация калия (К20) в колбах будет соответствовать 2, 4, 10, 20, 30, 40,
60, 80, 100 мг К20.
1 См. статью Д. Н. Иванова и JI. А. Лернер в настоящем сборнике.
2 Техника работы описана применительно к пламенному фотометру типа Цейс, при ис¬
пользовании ацетилено-воздушного пламени.
8 Если раствор не всасывается, то это указывает на засоренность капилляра. В таком
случае, открыв пульверизатор, капилляр промывают водой; если этого недостаточно»
то капилляр прочищают очень тонкой проволочкой.
200
И. Г. Важенин
После того как будет установлен нормальный режим горения пламени,
по приготовленным образцовым растворам соли калия строят калибровочный
график. Сначала производят «утомление» фотоэлемента. Для этого под распы¬
литель на 10 мин. подставляют стаканчик с раствором хлористого калия
(100 мг К20 на 1 л). После «утомления» фотоэлемента вводят в пламя дистил¬
лированную воду, устанавливают шкалу гальванометра (при помощи макро-
и микровинтов) в нулевое положение. Затем последовательно вводят в пламя
прибора образцовые растворы, начиная с наименьшей концентрации. Глубина
погружения всасывающей трубочки в раствор 15—20 мм. Для каждого раст¬
вора записывают показания гальванометра и на основе их строят график: на
оси абсцисс откладывают значения концентрации калия (мг/л К20), на оси
ординат — соответствующие отсчеты гальванометра. По точкам, полученным
при пересечении показаний гальванометра и шкалы концентраций образцо¬
вых растворов, строят калибровочную кривую, которая в основной своей
части представляет прямую линию с небольшими изгибами при наименьших
и наибольших концентрациях. Соблюдая те же условия работы на приборе,
что и при построении калибровочной шкалы, снимают показания гальваномет¬
ра для каждого испытуемого раствора. Через каждые 20 определений произ¬
водят проверку работы прибора по образцовым растворам (обычно 10—20—40
КоО мг/л). По показаниям гальванометра по калибровочной шкале вычисляют
содержание калия в испытуемых растворах. Для этого из точки на прямой,
соответствующей показаниям гальванометра, опускают на ось абсцисс пер¬
пендикуляр и отсчитывают содержание калия в мг К20 на 1 л. или мг К20 на
100 г почвы при соотношении почва : раствор, равном 1 : 10.
Вследствие недостаточной монохроматичности интерференционных свето¬
фильтров определению калия на пламенном фотометре, при использовании
ацетилено-воздушного пламени, мешает присутствующий кальций. Это связа¬
но с тем, что кальций при возбуждении в пламени дает широкую полосу в
спектре, близко расположенную к полосе калия. Вследствие этого измеряемый
фототок является суммарным и получается завышение величины калия. Име¬
ется несколько путей устранения этого явления: удаление из раствора каль¬
ция — осаждение его в форме оксалата; снижение температуры пламени —
практически замена ацетилено-воздушного пламени пропано-бутано-воздуш-
ным; введение в раствор связывающего кальций (в пламени) элемента, а имен¬
но алюминия (например, сернокислого алюминия в количестве 5 : 1 по отно¬
шению к кальцию); весьма распространен метод компенсации.
Компенсацию фототока, вызываемого световой полосой кальция, произ¬
водят следующим образом. Перед началом работы в пламя вводят свежепри¬
готовленный раствор соли кальция, содержащий 500 мг СаО на 1 л. При по¬
мощи ирисовой диафрагмы кальциевой головки шкалу гальванометра возвра¬
щают на нуль. Чередованием введения в распылитель раствора соли кальция
и воды добиваются нулевого положения шкалы. Это положение принимают за
нуль отсчета при дальнейшем измерении интенсивности излучения калия как
в образцовых, так и в испытуемых растворах. Компенсация при концентра¬
ции кальция 300—500 мг/л позволяет определять калий в растворах, содер¬
жащих кальций от 0 до 10Э0 мг/л.
Интенсивность излучения щелочных элементов взаимно усиливается при
их совместном присутствии в пламени. При значительном содержании в раст¬
воре, например, натрия, в 10 и более раз превышающем содержание калия,
происходит значительный ионизационный эффект. В таком случае необходи¬
мо в образцовые растворы (при построении калибровочного графика) ввести
соответствующее количество натрия.
Присутствие сильных кислот в растворе (HCI, HN03, H2S04) снижает ин¬
тенсивность излучения находящихся в растворе элементов, вследствие измене¬
ния физического состояния растворов (вязкость) и условий их испарения. По¬
этому общим правилом является следующее: рабочие образцовые растворы
Методы, определения калия в почве
20Г
соли калия, используемые для построения калибровочного графика, следует
готовить на тех же растворах (солевых, кислотных, щелочных), которые ис¬
пользованы для извлечения из почв калия.
ОПРЕДЕЛЕНИЕ РАЗЛИЧНЫХ ФОРМ КАЛИЯ В ПОЧВЕ
В агрохимических исследованиях стремятся охарактеризовать почву в
отношении различных форм содержащегося в ней калия по степени подвиж¬
ности и доступности его растениям.
Легкоподвижные легкоусвояемые формы калия
Водная вытяжка
Воднорастворимый калий в почве обычно определяется для количествен¬
ной характеристики содержания калия в почвенном растворе. Но известно,
что калий почвенного раствора и воднорастворимый калий это совсем не одно
и то же. Калий почвенного раствора — это в основном калий, входящий в
состав простых солей (хлоридов, сульфатов, нитратов и др.), находящихся
в растворе в условиях естественной влажности, т. е. очень узкого соотноше¬
ния раствор : почва; калий воднорастворимый — это калий тех же простых
солей и, кроме того, калий сложных солей, силикатов и алюмосиликатов,
перешедших в водную вытяжку (при широком соотношении вода : почва) в.
результате гидролиза и растворения этих соединений в воде.
Для почв незасоленных, вследствие кратковременности взаимодействия
воды с почвой, водная вытяжка в определенной мере отражает содержание
калия в почвенном растворе; в почвах засоленных водная вытяжка не отража¬
ет концентрацию и состав солей в почвенном растворе и используется для
решения специальной задачи: установления степени и характера засоления
почв.
Содержание воднорастворимого калия в незасоленных почвах очень не¬
значительно — обычно менее 1 мг на 100 г почвы, и оно не характеризует пло¬
дородия почвы в отношении калия. Однако анализ этот широко используется
при исследовании форм соединений калия в почве и при изучении динамики
калия в почве, степени окультуренности и удобренности почв.
Ход анализа. 50 г воздушно-сухой почвы, просеянной через сито с отверс¬
тиями диаметром 1 мм, помещают в коническую колбу или склянку емкостью
0,5 л; приливают 250 мл дистиллированной воды, закрывают пробкой и взбал¬
тывают 3 мин. Вода должна быть освобождена от углекислоты путем предва¬
рительного кипячения (30—40 мин.) или пропускания через воду тока возду¬
ха, лишенного С02. Это достигается путем протягивания воздуха через две-
склянки Дрекселя, наполненные 50%-ным раствором H2S04 и 50%-ным раст¬
вором КОН. Вода хранится в бутыли, закрытой резиновой пробкой с
вставленной в нее хлоркальциевой трубкой, наполненной натронной из¬
вестью.
Суспензию почвы отфильтровывают через складчатые беззольные фильтры.
Перед фильтрованием содержимое склянки энергично взбалтывают и суспен¬
зию быстро переносят па фильтр. Это ускоряет фильтрование и помогает по¬
лучению прозрачных фильтратов. Если первые порции фильтрата все же мут¬
ные, то их переносят обргпно на фильтр с почвой. Лучше даже в первые ми¬
нуты фильтрования поронки с почвой вставлять в те же склянки, в которых
производилось взбалтывание, и переносить их на чистые приемники (колбы)
только убедившись, что фильтруется прозрачная жидкость. В случае очень
медленного фильтрования во избежание испарения воды воронки накрывают
часовыми стеклами.
202
И. Г. Важенин
Концентрация калия в водной вытяжке обычно столь низка, что для
определения его любыми из приведенных выше методов требуется предвари¬
тельное концентрирование в 5—10 раз. 100—200 мл прозрачного фильтрата пе¬
реносят в большие фарфоровые чашки; чашки ставят на водяную баню и
жидкость выпаривают полностью. К сухому остатку прибавляют 10—20 мл
0,1 н. HG1 при определении калия на пламенном фотометре или 10—20 мл
2%-ного раствора формалина при определении калия химическими методами
(кобальтнитритным, тетрафенилборатным), при которых требуется устране¬
ние влияния аммония. Стеклянной палочкой-пестиком растирают осадок,
тщательно обмывают стенки чашки и раствор отфильтровывают через малень¬
кий беззольный фильтр. Раствор поступает в анализ.
В случае очень высоких требований к точности анализа (химическими ме¬
тодами) рекомендуется водную вытяжку до выпаривания пропустить через
колонку с Cl-анионитом, чтобы удалить из раствора анионы SO^“ и РО^~.
Углекислотная вытяжка
(метод Сердобольского, 1939)
Метод Сердобольского принципиально отличается от других подобных
'(углекислотных) методов, так как им одновременно определяются две формы
калия: легко- и труднорастворимая (в углекислотном растворителе), т. е.
определяется легкоусвояемая форма и ее ближайший резерв. Извлечение ка¬
лия из почвы производят водой, насыщенной углекислотой, при разном соот¬
ношении почва : растворитель — от 1 : 2,5 до 1 : 40.
Ход анализа. В шесть банок или колб емкостью 500 мл берут шесть наве¬
сок воздушно-сухой почвы: 80, 60, 40, 20, 10 и 5 г. Приливают в колбы по
200 мл дистиллированной воды. Колбы закрывают пробками с вставленными
Рис. 2. График уравнения Сер¬
добольского для вытеснения
калия из почвы водой, насы¬
щенной углекислотой
Объяснение см. в тексте
в них Г-образными стеклянными трубками. Нижние концы трубок доходят
почти до дна колб. На наружные концы надевают резиновые трубки, подсое¬
диненные (через коллектор) к баллону с углекислотой. Взболтав содержимое
колб, пропускают через суспензии в течение 3 час. ток углекислоты со ско¬
ростью 2—3 пузырька газа в 1 сек. За это время колбы с содержимым перио¬
дически взбалтывают (4—5 раз за 3 часа). Через 3 часа ток газа прекращают, и
<сэдержимое колб фильтруют через складчатый беззольный фильтр. Для ана¬
лиза берут 100 мл фильтрата. Фильтрат выпаривают в фарфоровых чашках
•яа водяной бане и далее поступают так, как описано выше.
Калий определяют на пламенном фотометре или одним из описанных хи¬
мических методов. Результаты анализа выражают в миллиграммах К20 на
100 мл вытяжки.
Чтобы определить труднорастворимые и легкорастворимые соединения
калия (в углекислотной вытяжке), результаты анализа изображают графи¬
чески, как показано на рис. 2. Для этого по оси абсцисс откладывают величи¬
Методы определения калия в почве
20
ну навесок почвы, соответствующую 100 мл вытяжки, а по оси ординат —
количество миллиграммов калия (К20), определенное в 100 мл вытяжки. На¬
несенные на рисунок точки соединяют двумя прямыми, из которых одна (ОА}
выходит из начала координат (точка О), а другая проходит через точки, соот¬
ветствующие большим навескам почвы (АВ). Полученная ломаная линия име¬
ет точку излома А. Участок ломаной О А служит для определения всего коли¬
чества калия, переходящего в углекислотную вытяжку, а участок ломаной
АВ — для определения труднорастворимых и легкорастворимых соединений
раздельно. Такое разделение проводят по уравнению
У = тр + Ь,
где у — количество калия в 100 мл вытяжки, полученное экспериментально, мг; т — ко»
личество калия легкорастворимых соединений (КС1, KN03 и др.), перешедшее в вытяжку
из 1 а почвы, мг; р — навеска почвы, соответствующая 100 мл вытяжки, г; Ь — количест¬
во калия труднорастворимых соединений в 100 мл вытяжки, мг.
Пример. Пусть, согласно анализам, получены данные на 100 мл вытяжки, при¬
веденные в табл. 2.
Таблица 2
Результат анализа углекислотных вытяжек из почвы
Разведение
Навеска почвы,
соответствующая 100 мл
вытяжки, г
К*0, мг на 100 мл
вытяжки
Обозначение
точек на рис. 2
1
1,25
80
12,7
е
1
2,5
40
7,2
д
1
5
20
4,6
г
1
10
10
3,4
е
1
20
5
1,6
б
1
40
2,5
0,9
а
Данные таблицы изображены на рис. 2. Точка перегиба А указывает, что общее коли¬
чество калия, переходящего в углекислотную вытяжку, может быть рассчитано по дан¬
ным в — а. Для этих точек, согласно таблице, имеем: 3,4; 1,6 и 0,9 мг К20 при навесках
почвы 10; 5 и 2,5 г. После пересчета на 100 г почвы получим 34,0; 32,0; 36,0 мг К20 или в
среднем 34,0 мг К20 на 100 г почвы. Количество калия соединений, легко- и труднора¬
створимых в углекислотной вытяжке, определяют по точкам а, д, е.
Составим для каждой точки упомянутое выше уравнение у = тр + Ь:
4,6 — 20т-|-&, (г)
7,2 -= 40т+Ь, (д)
12,7 -= 80т-|- Ь. (е)
Решая попарно эти уравнения, получим: Ь = 0,128; 0,133; 0,137; в среднем 0,133 мш
легкорастворимого калия (К20), или 13,3 мг на 100 г почвы. Количество труднораствори¬
мых соединений калия получим в результате вычитания: 34,0—13,3 = 20,7 мг К20 на
100 г почвы.
Метод Сердобольского, к сожалению, используется мало. ,Он заслуживает
большего внимания, особенно при анализе сероземных, каштановых и черно¬
земных почв.
204
И. Г. Важенин
Хлоркалъциевая вытяжка
(метод лаборатории агропочвоведения ВИУА — см. Голубева, 1969)
Поступление обменного калия в почвенный раствор зависит не только от
количеств обменного калия, но и от степени его подвижности. Количество об¬
менного калия в почве (запас его) принимается как фактор емкости, степень
подвижности —как фактор интенсивности.
В данном методе показателем «степени подвижности» калия служит кон¬
центрация калия (мг/л) в 0,005 н. растворе хлористого кальция, после его
взаимодействия с почвой при соотношении раствор : почва = 2:1.
Предложенная методика обеспечизает получение сравнимых результатов
по определению степени подвижности обменного калия для дерново-подзо-
листых, серых лесных почв и выщелоченных черноземов.
Ход анализа. В склянку на 200 мл берут 40 г воздушно-сухой почвы,
приливают 80 мл 0,005 н. раствора хлористого кальция и взбалтывают
на ротаторе в продолжение 1 часа. Раствор отфильтровывают через без-
зольныа фильтр. В фильтрате определяют калий на пламенном фотометре
илз одним из химических методов. Шкялу образцовых растворов хло-
риз того калия — при определении калия на пламенном фотометре — го¬
товят на 0,005 н. растворе хлористого кальция. Содержание калия в мил¬
лиграммах К20 на 100 г почвы (я) рассчитывают по формуле
а•200 а
ж== 1000“ =5*’
где а — концентрация калия (К20, мг/л) в испытуемом растворе, найденная по калибро¬
вочной кривой; 200 — объем вытяжки, соответствующий 100 г почвы, мл; 1000 — коэффи¬
циент для пересчета содержания калия (К20) в 1 мл.
Величина а, рассчитанная в миллиграммах на 1 л, выражает «фактор интенсивности».
Расчет содержания калия в миллиграммах на 100 г почвы сделан в целях сопоставления
с величинами ’обменного калия («фактора емкости») в почвах г.
Реактивы. 1. 2,0 н. раствор хлористого кальция (запасный раствор). 219,2 г
хлористого кальция (СаС12*6Н20) растворяют в 500—600 мл дистиллированной воды и за¬
тем доводят раствор до объема 1 д. В полученном растворе определяют концентрацию
кальция объемным методом (титрованием с трилоном Б).
2. 0,005 н. раствор хлористого кальция (рабочий раствор). Из запасного раствора
<>ерут 2,5 мл, переносят в мерную колбу на 1 л и доливают водой до ме’ки.
Подвижные формы калия
В этот раздел методов определения подвижных форм калия включены три
труппы методов: 1) определение обменного калия, извлекаемого нейтральной
солью; 2) определение калия, извлекаемого слабыми растворами кислот; 3) оп¬
ределение калия, извлекаемого кислотно-солевыми буферными растворами.
Всеми этими методами может извлекаться из почв разное, а иногда и равное
количество калия, но характер связи извлекаемого калия с соединениями поч¬
ли в определенной мере различный.
Извлечение калия солевыми растворами
Определение общего количества обменного калия
Метод основан на полном вытеснении обменного калия из почв путем по¬
следовательного промывания их 1,0 н. раствором уксуснокислого аммония
(77,1 г соли СН3СООГШ4 растворяют в 1 л воды).
1 Более подробно см* в статье О. П. Медведевой в настоящем сборнике.
Методы определения калия в почве
205
Ход анализа. 5—10 г воздушно-сухой почвы (в зависимости от содержания
в почве обменного калия) помещают в склянки емкостью 100—200 мл, при¬
ливают 10-кратное количество 1,0 н. раствора уксуснокислого аммония и
взбалтывают на ротаторе в продолжение 1 часа. Раствор отфильтровывают.
•Затем новыми порциями того же реактива почва из склянок полностью пере-
лосится на фильтр; почва на фильтре промывается до полного вытеснения всех
обменных оснований; контроль на полноту вытеснения производят с помощью
качественной реакции на кальций (со щавелевокислым аммонием при кипяче-
лии или с мурексидом).
В целях ускорения вытеснения обменных оснований каждую новую пор¬
цию реактива (примерно 10 мл) приливают после отфильтровывания преды¬
дущей.
Вытеснение обменного калия (и других оснований) можно проводить и
«способом декантации: навески почвы берут не в склянки, а в фарфоровые
чашки; почву обрабатывают небольшими порциями раствора вытеснителя; со¬
держимое чашки тщательно перемешивают и после оседания почвы прозрач-
лую жидкость сливают на фильтр и т. д. Убедившись в полноте вытеснения
лз почвы обменных оснований (по отсутствию реакции на кальций), объем
«фильтрата доводят водой до метки, тщательно перемешивают и анализируют.
Полное вытеснение калия из почвы достигается, как правило, 50-кратным
•объемом раствора уксуснокислого аммония.
При определении калия на пламенном фотометре фильтрат анализируется
лепосредственно; при определении калия одним из химических методов про¬
изводят разрушение реактива. Определенный объем фильтрата (50—100 мл)
помещают в фарфоровые чашки и выпаривают на водяной бане. В процессе
выпаривания реактив разрушается, уксусная кислота и аммиак улетучива¬
ются. К сухому остатку для разрушения органического вещества приливают
2 мл 30%-ной перекиси водорода л 2 мл 10%-ного раствора HN03; раствор
осторожно выпаривают; чашки с сухим остатком прокаливают в муфельной
печи при температуре 400—450° С для удаления следов органического веще¬
ства и солей аммония. В охлажденные чашки приливают по 5 лед 10%-ного
раствора НС1, с помощью стеклянных палочек кислотой обмывают стенки
чашек; растворы переносят на фильтры воронок, вставленных в мерные кол¬
бы на 25 или 50 мл. Чашки и фильтры промывают горячей водой, подкислен¬
ной соляной кислотой. В прозрачном фильтрате определяют калий одним из
описанных выше методов.
Метод Масловой
(Маслова, Чернышева, 1934)
Метод основан на извлечении из почв обменного калия 1,0 н. раствором
уксуснокислого аммония при соотношении почва: раствор = 1 : 10 и 1-часовом
взбалтывании на ротаторе. Как показали специальные исследования (Важе-
нин, Карасева, 1959), этим методом извлекается из почв в среднем 75% от
общего количества обменного калия; метод рекомендуется для определения
обменного калия в некарбонатных почвах (дерново-подзолистых, серых лес¬
ных, черноземах, красноземах и желтоземах).
Ход анализа. 5 г воздушно-сухой почвы, просеянной через сито с отвер¬
стиями диаметром 1 мм, помещают в склянки емкостью 100 мл, заливают
50 лед 1,0 н. раствора уксуснокислого аммония (pH 7,0) и ставят на ротатор.
После 1-часового взбалтывания вытяжку отфильтровывают через складча¬
тый фильтр и в прозрачном фильтрате определяют калий на пламенном фото¬
метре. Благодаря летучести слагающих соль компонентов (аммиак и уксус¬
ная кислота) разложение ее в пламени происходит легко и быстро, что обеспе¬
чивает высокую производительность метода и большую точность определе¬
ния калия.
206
И. Г. Важенин
Реактив. 1,0 н. раствор уксуснокислого аммония. 77,1 г соли CH3COONH4 раст¬
воряют в 900—950 мл воды, перемешивают и определяют величину pH. Обычно раствор
бывает слабокислый. Добавляя по каплям 10%-ный раствор аммиака, устанавливают
реакцию среды (pH 7,0). Объем раствора водой доводят до 1 л и взбалтывают.
При отсутствии пламенного фотометра анализ ведут по методу, предло¬
женному А. А. Масловой. К навеске почвы в 20 г, помещенной в сухую
склянку на 400—500 мл, приливают 200 ди 1,0 н. раствора CH3COONH4f
имеющего pH 7,0 ; склянку закрывают резиновой пробкой и взбалтывают на
ротаторе в продолжение 1 часа. Полученную вытяжку фильтруют через
складчатый фильтр; 50—100 мл прозрачного фильтрата выпаривают в фар¬
форовых чашках на водяной или воздушной бане. К остатку в чашке прили¬
вают 2 мл 30 %-ной перекиси водорода и 2 дел 10 %-ной HN03 для разрушения
органического вещества и осторожно просушивают на этернитовой плитке;
после этого чашки прокаливают в муфельной печи при температуре 400—
450° С (до полного разрушения остатков органического вещества и удаления
аммонийных солей); при этом осадок приобретает пепельно-серый цвет.
Прибавляют в чашку 2—3 капли 25 %-ного раствора НС1 для перевода оки¬
сей в хлориды; содержимое чашки обрабатывают горячей водой, переносят
на маленькие фильтры и отфильтровывают в мерную колбу на 50 мл.
Калий определяют объемным кобальтнитритным или любым другим хими¬
ческим методом.
Метод Мачигина (1952)
Метод основан на извлечении калия (и фосфора) из почвы 1 %-ным раство¬
ром углекислого аммония при соотношении почва : раствор = 1 : 20; реко¬
мендован при анализе карбонатных почв (черноземов, каштановых, бурых и
сероземов). Если в почвенной вытяжке одновременно определяют фосфор
и калий, то извлечение их производят при температуре 25 + 2° С. Если опре¬
деляют только калий, то допустима комнатная температура (18—20° С).
Приготовление реактивов и вытяжки изложено выше, при описании опре¬
деления подвижных фосфатов в статье К. Е. Гинзбург в настоящем сборнике.
В вытяжке калий определяют с помощью пламенного фотометра. Шкалу
образцовых растворов готовят на 1%-ном растворе углекислого аммония.
При отсутствии пламенного фотометра 50 дел фильтрата досуха выпаривают
в фарфоровой чашке на кипящей водяной бане, а затем разрушают аммоний¬
ные соли на открытой электроплитке. Сухой остаток смачивают 5-ю каплями
концентрированной азотной кислоты (растирая ее по стенкам оплавленной
стеклянной палочкой). Чашку помещают на кипящую водяную баню, выпа¬
ривают кислоту досуха и остаток опять прокаливают на открытой электро¬
плитке или в муфеле. Остаток в чашке должен стать белым. При необходи¬
мости прибавление азотной кислоты и выпаривание повторяют. К прокален¬
ному остатку прибавляют 4—5 капель концентрированной соляной кислоты
(растирая ее палочкой по стенкам); затем прибавляют 20—25 мл горячей дис¬
тиллированной воды и раствор отфильтровывают через беззольный фильтр в
мерную колбу на 50 мл. Определение калия производят любым из химичес¬
ких методов.
Извлечение калия слабыми кислотами
Метод Кирсанова (1933)
Метод основан на извлечении из почвы калия (и фосфора) 0,2 н. раствором
соляной кислоты. Соотношение почва : раствор =1:5. Рекомендован для
определения калия (и фосфора) в некарбонатных (подзолистых, дерново-
подзолистых и серых лесных) почвах.
Метподи определения калия в почве
207
Приготовление реактивов и вытяжек описано выше, в разделе определе¬
ния подвижных форм фосфатов в статье К. Б. Гинзбург в настоящем
сборнике.
В вытяжке определяют калий с помощью пламенного фотометра. Шкалу
образцовых растворов готовят на 0,2 н. растворе соляной кислоты.
При отсутствии в лаборатории пламенного фотометра калий в вытяжке
определяют одним из химических методов. 50 мл фильтрата выпаривают на
водяной бане, добавив к вытяжке 5 мл концентрированной HN03; смачивают
сухой остаток (по 10 капель) концентрированными кислотами HN03 и НС1,
размазывая их стеклянной палочкой по стенкам чашки; выпаривают раствор
и прокаливают остаток в муфеле при 400—450° С, не допуская красного на¬
кала дна чашки. После остывания осадок растворяют в подкисленной (HG1)
воде, раствор отфильтровывают в мерную колбу (25—50 мл); в фильтрате оп¬
ределяют калий.
Метод Чирикова
Метод основан на извлечении подвижного калия (и фосфора) из почвы
0,5 н. раствором уксусной кислоты при соотношении почва : раствор = 1 :
: 25. Метод рекомендован для определения подвижных форм калия (и фосфора)
в некарбонатных черноземах.
Приготовление реактивов и вытяжки описано выше, в разделе определе¬
ния подвижных форм фосфатов в статье К. Б. Гинзбург в настоящем сбор¬
нике.
В вытяжке калий определяют с помощью пламенного фотометра. Шкалу
образцовых растворов готовят на 0,5 н. растворе уксусной кислоты.
При отсутствии пламенного фотометра 50 мл фильтрата помещают в фар¬
форовую чашку, выпаривают жидкость на кипящей водяной бане досуха;
остаток смачивают 2 мл 30%-ной перекиси водорода и 2 мл 10%-ной
HN03 для разрушения органического вещества и затем осторожно вы¬
паривают на закрытой (этернитовой) плитке; затем чашку прокаливают в
муфельной печи при температуре 400—450° С; остаток приобретает светло¬
серый цвет. В чашку прибавляют 2—3 капли концентрированной HG1, раз¬
мазывая кислоту по стенкам чашки. Приливают 20—25 мл горячей воды, все
перемешивают и жидкость отфильтровывают в мерную колбу на 50 мл. Чаш¬
ку, палочку и фильтр смывают водой, после охлаждения объем раствора до¬
водят до метки и определяют калий любым химическим методом. *
Метод Ониани (1964)
Метод основан на извлечении подвижных форм калия (и фосфора) из поч¬
вы 0,1 н. раствором серной кислоты при соотношении почва : раствор = 1 :
: 25. Рекомендован для определения подвижных форм калия (и фосфора) в
красноземах, желтоземах и субтропических подзолистых почвах Грузии.
Приготовление реактивов и вытяжки описано выше, в разделе определе¬
ния подвижных форм фосфатов в статье К. Е. Гинзбург в настоящем
сборнике.
В вытяжке калий определяют на пламенном фотометре. Шкалу образцо¬
вых растворов готовят на 0,1 н. растворе серной кислоты.
При отсутствии в лаборатории пламенного фотометра калий определяют
химическим методом. Для этого 50 мл фильтрата выпаривают в фарфоровой
чашке на кипящей водяной бане до появления белых паров, а затем медленно
досуха. Сухой остаток смачивают 5-ю каплями концентрированной азотной
кислоты, растирая ее по стенкам оплавленной стеклянной палочкой. Чашку
помещают на кипящую водяную баню; выпаривают кислоту досуха, а затем
чашку прокаливают в муфеле при температуре 400—450° С. К прокаленному
остатку (он должен быть светло-серым или желтоватым) прибавляют 1 мл
208
И. Г. Важенин
концентрированной НС1. Кислоту растирают по стенкам и чашку ставят на
кипящую водяную баню; раствор выпаривают досуха. К сухому остатку при¬
ливают 20—25 мл горячей подкисленной (НС1) дистиллированной воды, пере¬
мешивают и раствор отфильтровывают в мерную колбу на 50 мл. Чашку, па¬
лочку и фильтр обмывают водой в ту же мерную колбу. После охлаждения
объем доводят до метки и раствор анализируют.
Извлеч]е|н;и[е [калия буферными растворами
Ниже приводится описание двух методов: 1) Эгнера — Рима —калий
извлекается раствором лактата кальция, имеющего pH 3,6—3,7; 2) Шахт-
шабеля — калий извлекается смесью растворов уксуснокислого аммония и
щавелевокислого аммония с исходным pH 6,0.
Метод Эгнера — Рима является основным официальным методом при агро¬
химической характеристике почв в ГДР, ФРГ; он распространен в Польше,
Венгрии. Метод Шахтшабеля принят в Чехословакии. Все эти методы хоро¬
шо изучены в указанных странах, получили производственную проверку и
сравнительную характеристику с другими агрохимическими методами, в том
числе и с полевым опытом. На основании изучения предложены «предельные
числа» обеспеченности почв калием в зависимости от результатов анализа.
В Советском Союзе метод Эгнера — Рима при определении в почвах под¬
вижных форм калия и фосфора используют агрохимические лаборатории в
республиках Прибалтики.
Метод Эгнера —Рима (Egner et al1960)
Калий (и фосфор) извлекают из почвы буферным раствором лактата каль¬
ция Са(С8Нб03)2-5 Н20 при соотношении почва : раствор = 1 : 50, при взбал¬
тывании на ротаторе в продолжение 1,5 час.
В вытяжке определяют калий на пламенном фотометре, предварительно
удаляя из нее кальций в форме оксалата путем добавления к 25 мл фильт¬
рата 2 мл 10%-ной щавелевой кислоты.
Приготовление реактивов и вытяжки описано выше, в разделе определе¬
ния подвижных форм фосфатов в статье К. Б. Гинзбург в настоящем сбор¬
нике.
В ГДР и ФРГ принята группировка почв по обеспеченности их калием на
основании анализа по Эгнеру — Риму, представленная в табл. 3.
Таблица 3
Обеспеченность почв калием по результатам анализов по Эгнеру — Риму,
мг КгО на 100 г почвы
Класс
' Обеспеченность
Кислотность почв (pH в 0,1 н. растворе КС1)
<5,0
5,1-6,0
6,1—7,0
>7,0
III
Плохая
<10,0
<9,0
<8,0
<7,0
II
Средняя
10,1-20,0
9,1—18,0
8,1-16,0
7,1—14,0
I
Достаточная
20,1-40,0
18,1-36,0
16,1—32,0
14,1-28,0
В ГДР, ФРГ, Швеции, Нидерландах распространен разработанный Эгне-
ром, Римом и Доминго метод уксуснокислого лактата аммония, сокращенно
называемый «А — L метод». Преимущество его перед лактатным методом в том,
что при определении калия на пламенном фотометре не требуется предвари¬
тельное осаждение кальция из раствора. Шкалу образцовых растворов для
Методы определения калия в почве
209
определения калия готовят на AL-смеси. Приготовление реактивов и вытяж¬
ки описано выше в разделе определения подвижных форм фосфатов в статье
К. Е. Гинзбург в настоящем сборнике.
На основании полевых опытов и лабораторных исследований в ГДР уста¬
новлены классы обеспеченности почв калием, определяемым по методу Эгне-
ра — Рима — Доминго, которые даны в табл. 4.
Таблица 4
Обеспеченность почв калием по результатам анализов по Эгнеру — Риму — Доминго,
мг КгО на 100 г почвы
Класс обеспеченности
Содержание
Почвы
по механическому составу
легкие
суглинки
тяжелые
III
Низкое
<6
<12
<18
II
Среднее
6-12
12—18
18—24
I
Высокое
>12
>18
>24
Метод Шахтшабеля(8скасЫ$скаЬе19 1941)
Калий извлекают из почв буферным ацетатно-оксалатным раствором, ко¬
торый в отношении уксуснокислого аммония является 0,8 н., в отношении
оксалата аммония 0,2 н., а в отношении ацетатно-оксалатного аммония 1,0 н.
раствором. Щавелевокислый аммоний вводят в раствор для того, чтобы пере¬
вести в осадок обменный кальций. Определение калия производят на пламен¬
ном фотометре.
10 г воздушно-сухой почвы обрабатывают в колбе на 100 мл 25 мл буфер¬
ного ацетатно-оксалатного раствора, имеющего pH 6,0+0,2. Продолжитель¬
ность взаимодействия раствора с почвой 15—20 час. За это время суспензию
периодически энергично взбалтывают (вращательным движением) по 2—3
мин.: 2 раза после приливания раствора, через 1 и через 2 часа после прили-
вания реактива, и 2 раза на следующий день за 1—2 часа до фильтрования»
Раствор фильтруют и в фильтрате определяют калий на пламенном фото¬
метре.
Автор считает, что при соотношении почва : раствор = 1 : 2,5 обменива¬
ется на аммоний 70—90% обменного калия. По нашим исследованиям (Ва-
женин, Карасева, 1959), при таком узком соотношении переходит в раствор
60—70% обменного калия.
Метод Шахтшабеля отличается простотой выполнения, быстротой ана¬
лиза и общедоступностью реактивов.
Реактив. Ацетатно-оксалатный раствор. 61,6 г CH3COONH4 растворяют в 700 мл
воды; 15,2 г (NH4)2C204*H20 растворяют в 200 мл горячей воды и вливают его в раствор
уксуснокислого аммония. После охлаждения объем раствора доводят водой до 1 л. Про¬
веряют pH; прибавлением аммиака или уксусной кислоты устанавливают pH 6,0± 0,2*
Необменные (экстенсивнообменные и кислотнорастворщнле)
формы калия
Для определения необменного калия используются две группы методов,
которые различаются по механизму воздействия реактивов на почву: 1) из¬
влечение экстенсивнообменного калия на осн,ове образования специфичной
комплексной соли калия с тетрафенил бор атом натрия (метод Магницкого и
Малкова); 2) извлечение необменного калия в результате растворения и гид¬
210
И. Г. Важенин
ролиза кислотами калийсодержащих минералов почвы (метод Пчелкина, ме¬
тод Гедройца). Первая группа методов характеризует почву в отношении бо¬
лее подвижных и более легкоусвояемых форм необменного калия, а вторая —
связана с разрушением почвенного поглощающего комплекса и ряда первич¬
ных минералов (слюд, полевых шпатов и др.).
Метод Магницкого и Малкова (1968)
Почва обрабатывается раствором ацетата натрия, содержащим формалин,
для связывания аммония. Затем добавляется щелочной раствор тетрафенил-
бората натрия (ТФБ — Na). При этом обменный и часть необменного калия,
извлеченного реагентом, связываются в форме нерастворимого тетрафенил-
бората калия. После фильтрования почвенной суспензии в фильтрате опреде¬
ляют остаточное количество ТФБ — Na титрованным раствором цетилтриме-
тиламмоний бромида (ЦТАБ) в присутсгвии индикатора бромфенолового си¬
него. Реакция почвенной вытяжки должна быть слабощелочной или нейт¬
ральной.
Ход анализа. Навеску воздушно-сухой почвы в 20 г переносят в склянку
емкостью 250 мл, приливают 20 мл вытесняющего раствора (реактив 9) и тща¬
тельно взбалтывают. Через 10 мин. добавляют 20 мл 0,02 н. раствора ТФБ —
Na 1 и суспензию взбалтывают на ротаторе в продолжение 1 часа; затем вы¬
тяжку отфильтровывают через плотный фильтр. 5 мл фильтрата переносят в
коническую колбу емкостью 250 мл, добавляют 50 мл воды, 10 капель инди¬
катора бромфенолового синего и при перемешивании производят титрование
(из микробюретки) 0,01 н. раствором цетилтриметиламмоний бромида, до мо¬
мента перехода окраски от красно-фиолетовой к голубой.
Количество калия (мг К на 100 г почвы) вычисляется по формуле
х — (40/£т—8л*/1Гц)*0,391 *5,
где 40 — объем взаимодействующего с почвой раствора (20 мл раствора ТФБ-Na + 20 мл
вытесняющего раствора); КТ — поправка к титру 0,02 н. раствора ТФБ-Na; 8 — коэффи¬
циент пересчета с 5 мл фильтрата на общий объем 40 мл; п — количество 0,01 н. раствора
ЦТАБ, израсходованного на титрование 5 мл фильтрата, мл; Кц — поправка к титру
0,01 н. раствора ЦТАБ; 5 — коэффициент пересчета с 20 г на 100 а почвы; 0,391 — коэффи¬
циент пересчета с мг-экв калия на мг в 1 мл 0,01 н. раствора.
Разность между калием, определенным в вытяжке уксуснокислого нат¬
рия 4- ТФБ-Na, и обменным калием (по Масловой) характеризует вели¬
чину экстенсивнообменного калия в почве.
Следует сказать об условности определения этой формы калия описанным
методом. Исследованиями авторов (1968) показано, что при прочих равных
условиях количество извлеченного из почвы калия увеличивается с повыше¬
нием концентрации ТФБ-Na в растворе; то же самое происходит с увели¬
чением времени взаимодействия реактива с почвой, т. е. равновесие в системе
почва — раствор устанавливается медленно (через много суток).
Реактивы. 1. 0,02 н. раствор тетрафенилбората натрия. 6,84 г ТФБ-Na растворя¬
ют в 800 мл дистиллированной воды, добавляют 4 мл 1 %-ного раствора А1С13 (для осажде¬
ния примесей), раствор перемешивают 10 мин. и отфильтровывают в мерную колбу ем¬
костью 1 л. К фильтрату добавляют 10 мл 1,0 н. NaOH (для стабилизации раствора) и
доводят водой до метки.
2. 0,01 н. раствор цетилтриметиламмоний бромида (ЦТАБ). 3,641 г соли растворяют
в воде и объем доводят до 1 д.
1 Если содержание необменного калия в почве свыше 70—80 мг! 100 г, то следует или
уменьшить навеску почвы до 10 г, или приготовить раствор ТФБ-Na более высокой
концентрации, например 0,04 н.
Методы определения калия в почве
211
3. Стандартный 1,0 н. раствор КС1. 37,278 г х. ч. просушенного КС1 растворяют в ди¬
стиллированной воде в мерной колбе на 500 мл, объем доводится до метки.
4. Рабочий стандартный 0,02 н. раствор КС1. 20 мл раствора КС1 (реактив 3) в мерной
колбе водой разбавляют до 1 д.
5. Индикатор бромфеноловый синий: 40 г тетрабромфенолсульфофталеина растворя¬
ют в 3 мл 0,1 н. раствора NaOH и объем доводят водой до 100 мл.
6. 35—40%-ный раствор формалина.
7. 1,0 н. раствор NaOH. 40 г NaOH (ч. д. а.) растворяют в дистиллированной воде,
раствор доводят водой до объема 1 л.
8. 3,5 н. раствор CH3COONa. 476 г кристаллического уксуснокисного натрия
(CH8C00Na-3H20) растворяют в дистиллированной воде; объем доводят водой до 1 л.
9. Вытесняющий калий реактив. К 600 мл 3,5 н. раствора CH3COONa (реактив 8)
добавляют 20 мл 1,0 н. раствора NaOH (реактив 7), 100 мл 40%-ного раствора формалина
(реактив 6), 280 мл воды (общий объем 1 л). Реактив готовят перед приготовлением вы¬
тяжек и проверяют на содержание калия: при приливании ТФБ-Na к отдельной пробе
раствор должен оставаться прозрачным.
Установление титров 0,02 н. раствора ТФБ-Na и 0,01 н. раствора ЦТАБ. В мерную
колбу на 100 мл приливают 20 мл стандартного 0,02 н. раствора КС1, 1 лед 1,0 н. раствора
NaOH, добавляют 25 мл 0,02 н. раствора ТФБ-Na, тщательно перемешивают и доводят
до метки. Через 10 мин. раствор фильтруют. В коническую колбу на 250 мл берут 25 мл
фильтрата, добавляют 25 мл воды, 10 капель индикатора бромфенолового синего и титру¬
ют при непрерывном взбалтывании из микробюретки 0,01 н. [раствором ЦТАБ. Далее
устанавливают соотношение между 0,02 н. раствором ТФБ-Na’n 0,01 н. растворомЦТАБ^
Для этого в коническую колбу на 250 мл к Ъ мл 0,02 н. раствора ТФБ-Na приливают 50 мл
воды, 10 капель бромфенолового синего, перемешивают и титруют 0,01 н. раствором
ЦТАБ. Поправку к титру 0,02 н. раствора ТФБ-Na вычисляют по формуле
где 20 — количество стандартного 0,02 н. раствора КС1, мл; 25 — количество фильтрата,
взятого для титрования, раствором ЦТАБ, мл; 5 — количество 0,02 н. раствора ТФБ-Na,
мл; 4 — коэффициент для пересчета результатов анализа на весь объем фильтрата
(100 мл); п — количество 0,01 н. раствора ЦТАБ, израсходованное на титрование 25 мл
фильтрата, мл; т — количество 0,01 н. раствора ЦТАБ, израсходованное на титрование
5 мл 0,02 н. раствора ТФБ-Na, мл.
Поправку к титру 0,01 н. раствора ЦТАБ вычисляют по формуле
В основу метода положено известное положение о различной раствори¬
мости в кислотах первичных калийсодержащих минералов; легче всего рас¬
творяются слюды (биотит лучше, чем мусковит), труднее — гидрослюды, не¬
фелин и труднорастворимые полевые шпаты (ортоклаз, микроклин). В качест¬
ве показателя степени подвижности почвенного калия автор предложил ис¬
пользовать две параллельные вытяжки из почв: 2,0 н. НС1 и 0,2 н. HG1. Чем
больше разница между величинами калия, получаемого при обработке почвы
2,0 н. и 0,2 н. НС1, тем больше имеется возможность мобилизации его; в этих
случаях можно будет рассчитывать на более полное использование растения¬
ми калия почвы (Пчелкин, 1966).
Ход анализа (Пчелкин и др., 1969). В склянки емкостью 200 мл помещают
2 г воздушно-сухой почвы, приливают 50 мл 2,0 н. раствора соляной кислоты.
Содержимое склянок взбалтывают на ротаторе в продолжение 1 часа. Затем
склянки с суспензией ставят в термостат на 2 суток при температуре 24° С.
Метод Пчелкина (1966)
212
И. Г. Важенин
После термостатирования раствор отфильтровывают. Флльтрат разбавляют
водой в 4 раза (25 мл переносят в колбу на 100 мл, водой объем доводяг до
метки) и калий определяют на пламенном фотометре. Шкалу образцовых ра¬
створов калия (КС1) для построения калибровочной кривой готовят на 0,5 н.
растворе соляной кислоты.
Одновременно производят определение содержания обменного калия в
той же самой почве. Обменный калий определяют или в выгяжке 0,2 н. HG1
{Пчелкин, 1966) или по Масловой в вытяжке 1,0 н. раствора уксуснокислого
аммония (Пчелкин и др., 1969). По разнице межцу содержанием калия, из¬
влекаемого 2,0 н. НС1, и содержанием обменного калия рассчитывают вели¬
чину необменного калия.
Величина необменного калия, определяемая по В. У. Пчелкину, обычно
в несколько раз превосходит содержание обменного калия. По исследованиям
авторов метода, наименьшие резервы необменного калия содержатся в почвах
легкого механического состава и значительно бэлыпие — в почвах тяжелого
механического состава; мало необменного калия в торфяных почвах и очень
много в черноземах. Кроме того, этим методом выявляется удобренность почв
калийными удобрениями.
Извлечение калия по Гедройцу (1955)
Растворимость и кислотный гидролиз почвенных минералов, как извест¬
но, резко возрастают при нагревании суспензий. Поэтому при характеристике
почвенных запасов (резервов) в отношении калия и других минеральных ве¬
ществ в почвоведении и агрохимии давно уже пользуются горячими кислот¬
ными вытяжками. В этом отношении очень обстоятельные исследования были
выполнены еще К. К. Гедройцем. Наибольшее распространение в свое время
получила 10%-ная солянокислая вытяжка.
X од анализа. Навеску воздушно-сухой почвы (для общего анализа вытяж¬
ки рекомендуется навеска 50 е) помещают в коническую колбу соответству¬
ющей емкости; в колбу приливают 10-кратное количество 10%-ной соляной
кислоты (уд. вес 1,050). Бели почва содержит карбонаты, то прибавление
соляной кислоты должно производиться осторожно, небольшими порциями
во избежание выползания пенящейся жидкости; в этом случае в колбу сле¬
дует прибавить, кроме рассчитанного объема 10%-ной HG1, столько концент¬
рированной НС1 (уд. вес 1,19), сколько соответствует количеству углекисло¬
та, выделяющейся из карбонатов во взятой навеске.
Навески почвы хорошо перемешивают с 10%-ной кислотой, накрывают
небольшими воронками с короткими трубками и ставят на кипящую водяную
баню на 10 час., в течение которых содержимое колб время от времени (при¬
мерно каждый час) перемешивают. Затем вытяжку отфильтровывают и фильт¬
рат используют для анализа.
Поскольку от режима, способа приготовления 10%-ной НС1 вытяжки в
значительной мере зависит «выход» растворимых элементов из почвы, способ
приготовления горячих вытяжек должен быть унифицирован, чтобы полу¬
ченные результаты были сопоставимы.
В настоящее время большое распространение получили кислотные вы¬
тяжки, концентрация которых выражается не в процентах HG1, а в нормаль¬
ностях, в частности 2,0 н. солянокислая горячая вытяжка (Важенин,1965).
2,0 н. соляная кислота по концентрации HG1 является более слабой, чем
10%-ная НС1 (в ней содержится 7,3% НС1). Однако она оказывается болеэ
удобной, так как дает возможность сравнивать результаты определения ка¬
лия в этой вытяжке и в распространенных кислотных вытяжках (без нагрева¬
ния), также выраженных в нормальностях: 2,0 н., 1,0 н., 0,2 н.
Ход анализа. 5 г воздушно-сухой почвы помещают в коническую колбу на
200 мл; приливают 50 мл 2,0 н. НС1 и, закрыв колбу пробкой с обратным хо¬
Методы определения калия в почве
213
лодильником, ставят ее на электрическую закрытую (этернитовую) плитку.
Умеренное кипячение поддерживают 30 мин. Убыль жидкости обязательно
восполняют приливанием горячей дистиллированной воды до метки (нанесен¬
ной термостойким лаком). При массовых анализах, когда трудно соблюсти
«одинаковый режим кипения жидкости в колбах, предпочтительнее выдержи¬
вание колб в термостате в продолжение 1 часа при температуре 98 + 1° С.
Холодильники в таком случае заменяются маленькими воронками или
коническими стеклянными пробками. Через каждые 15 мин. колбы встряхи¬
вают.
Жидкость в горячем состоянии отфильтровывают в сухую мерную колбу
«емкостью 50 мл. После охлаждения объем жидкости доводят до метки 2 н.
раствором НС1. Определенную часть фильтрата (5 мл при анализе черноземов
и каштановых почв, 10 мл при анализе серых лесных, дерново-подзолистых»
красноземных почв и 20 мл при анализе песчаных и супесчаных почв разного
типа) разбавляют дистиллированной водой в 5 раз (перенося указанные коли¬
чества в колбы на 25, 50, 100 мл) и калий определяют на пламенном фото¬
метре. Шкалу образцовых растворов калия готовят на 0,4 н. HG1.
В основу метода положено вытеснение калия из почвы кислотой возраста*
«ощей концентрации (от 0,05 до 0,4 н.). На основании данных анализа вытя¬
жек строят изотермы десорбции калия и вычисляют константу десорбции (вы¬
теснения) калия для любой равновесной концентрации кислоты (в пределах
от 0,001 до 0,4 н.) й устанавливают соответствующий показатель подвижности
калия для данной почвы.
Ход анализа. Пять навесок воздушно-сухой почвы по 10 г для черноземов
(почв с высокой емкостью поглощения) и по 25 г для подзолистых почв по¬
мещают в склянки емкостью 250 или 500 мл и заливают десятикратным объе¬
мом (100 и 250 мл) раствора соляной кислоты возрастающей концентрации:
0,05; 0,10; 0,15; 0,20; 0,40 н. Склянки закрывают пробками, содержимое
взбалтывают на ротаторе в продолжение 2 час. и оставляют в покое на сутки.
Затем раствор отфильтровывают через плотный фильтр. Ъ 2Ъ мл фильтрата
определяют остаточную концентрацию кислоты титрованием 0,1 н. щелочью
(по фенолфталеину); часть фильтрата берут для определения калия на пла¬
менном фотометре или после выпаривания 50 мл фильтрата и прокаливания
остатка калий определяют по одному из описанных выше химических мето¬
дов. Результат определения остаточной (равновесной) концентрации кислоты
— Сн в мг-экв на 1 л — и количество вытесненного калия — (я-100): т в
мг-экв на 100 г — должны отвечать (по автору) уравнению Фрейндлиха
•где т — навеска почвы, г; К и п — константы.
Нанеся lg Сн на ось абсцисс, a lg (я-100/т) на ось ординат, получим пря¬
мую (рис. 3). Точка пересечения прямой с осью ординат (т. е. при lg С7Н = 0
или при концентрации Сш = 1 мг-экв) будет соответствовать:
Найденный графически lg К перечисляют в миллиграмм-эквиваленты (или
миллиграммы) на 100 а почвы.
Дифференцированное вытеснение калия
Солянокислотный метод Антипова-Каратаева
(Антипов-Каратаев и др., 1935)
£•100 1
или ^ т =
214
И. Г. Важенин
Показатель i/n равен тангенсу угла между прямой и осью абсцисс (или
равного ему угла а, см. рис. 3):
1 ВС
-=tg«=w.
Для примера вычисления констант 'у равнения десорб¬
ции приводим результаты дифференцированного вытесненияj подвижного] калия из
подзолистой почвы (Антипов-Каратаев и др., 1935). Для определения было взято восемь
Рис. 3. График уравнения
Фрейндлиха для вычисления
константы десорбции вытесне¬
ния калия кислотой различ¬
ной концентрации
Объяснение см. в тексте
навесок, которые заливались 10-кратным количеством раствора НС1 в концентрации
от 0,05 до 0,40 н. В табл. 5 приведены результаты анализов, логарифмы равновесных кон¬
центраций (Сн) и вытесненных количеств калия х» 100/т.
Для подзолистой почвы из Калитино найдено: Кл = 0,192, К = 0,377, а 1/л = 0,165*
Таблица 5
Десорбция калия из подзолистой почвы растворами НС1
при отношении почва : раствор = 1 :10
Концентрация НС1, мг-эпв/л
Вытеснено К
на 100 г почвы,
х-100
мг-экв
т
(найдено)
ЪС*
lg= -100
т
Вытеснено К
на 100 г почвы,
мг-экв (вычислено)
начальная
равновесная
51
44
0,369
1,664
-0,433
0,376
102
93
0,391
1,970
-0,408
0,415
151
140
0,445
2,148
-0,352
0,439
199
189
0,456
2,276
-0,341
0,456
252
237
0,476
2,375
-0,322
0,468
303
289
0,494
2,462
-0,306
0,481
357
338
0,509
2,529
-0,293
0,491
402
390
0,509
2,604
-0,293
0,500
Метод позволяет сравнивать любые почвы по содержанию калия для оди¬
наковых равновесных концентраций вытеснителя (например, для 0,001 в.г
0,2 н. или 0,4 н.) и дает возможность характеризовать почвы по степени под¬
вижности калия в зависимости от поставленной задачи..Например, динамика
калия в почве лучше характеризуется калием наиболее подвижных фракций,
соответствующих наименьшим концентрациям равновесного раствора, вплоть
до 0,001 н. НС1; общие запасы подвижного калия будут характеризоваться
количеством калия, определяемого при наибольшей равновесной концентра¬
ции кислоты (0,4 н. и выше), а степень подвижности или степень прочности
связи калия с поглощающим комплексом — величиной показателя i/n.
Методы, определения калия в почве
215
ОПРЕДЕЛЕНИЕ ВАЛОВОГО СОДЕРЖАНИЯ КАЛИЯ В ПОЧВЕ
Для перевода калия алюмосиликатов почвы в растворимое состояние на¬
веску почвы или спекают со смесью углекислого кальция и хлористогс^аммо-
ния или разлагают почвенные минералы плавиковой и серной кислотами.
Спекание почвы по Смиту
Основная масса калия в почве представлена в форме труднорастворимых
первичных минералов — алюмосиликатов: ортоклаза и микроклина, муско¬
вита и биотита, лейцита, нефелина и др. При сильном прокаливании почвы в
•смеси с хлористым аммонием и углекислым кальцием силикаты разлагаются
л о следующей схеме:
2K.AISi308-f-6CaC0*+2NH4Cl - 6CaSi03 -f-AU03+2KCl f 2NH3+6C02-(-H20.
В процессе прокаливания вначале разлагается хлористый аммоний с об¬
разованием аммиака, который улетучивается, и хлористого водорода, кото¬
рый взаимодействует с углекислым кальцием, образуя хлористый кальций;
хлористый кальций и соляная кислота, воздействуя на алюмосиликаты, раз¬
лагают их с образованием силиката кальция, хлоридов щелочных металлов и
воды. В результате спекания калий (и натрий) переходят в форму легкорас¬
творимых хлоридов, а кремнекислота, алюминий, железо, марганец, магний,
фосфорная кислота — в соединения (спек), нерастворимые в воде.
В оригинальной методике по Смиту почва в смеси с хлористым аммонием
и углекислым кальцием в соотношении 1:1:4 спекается в удлиненных паль¬
цеобразных платиновых тиглях и в специальных печах или на газовой горел¬
ке при температуре 900° и выше. Во избежание потерь калия тигель до высо¬
кой температуры нагревают только в самой нижней части, а верхняя часть
защищается от перегрева асбестовыми кольцами. П. Г. Грабаровым и Б. Я.
Квитко (1962) разработан вариант метода Смита, позволяющий спекание вес¬
ти в обычных муфельных печах при температуре 750° С.
Ход анализа. На аналитических весах берут навеску 0,5 г почвы, тонко
растертой («до пудры») в агатовой или яшмовой ступке. Готовят (смешивают и
растирают) смесь хлористого аммония и углекислого кальция в соотношении
1 : 4. Отвешивают (на технических весах) 2,5 г смеси. Часть смеси помещают
на дно платинового тигля, большую часть тщательно перемешивают с почвой
и смесь переносят в тигель; сверху всыпают оставшуюся часть смеси NH4C1 +
4- СаС03. Тигель закрывают крышкой и ставят в холодную муфельную печь.
Включают муфель в сеть. После достижения температуры 750° С, на что ухо¬
дит около 1 часа, спекание продолжают еще 30 мин. По исследованиям П. Г.
Грабарова и Б. Я. Квитко, при температуре ниже 725° С не достигалось пол¬
ного разложения, а при температуре выше 825° С отмечены потери калия.
Н. Б. Шарошкина (1962) считает, что недостатком изложенного выше ме¬
тода спекания (при соотношении NH4C1 : СаС08 = 1:4) является сильное и
прочное загрязнение стенок и дна тиглей, очистку которых производят про¬
каливанием со смесью карбонатов калия и натрия. Автор рекомендует произ¬
водить спекание 0,5 г почвы со смесью 1,5 г NH4C1 и 1 г СаС03. При этом до¬
стигается полное разложение алюмосиликатов, а очистка тиглей производит¬
ся кипячением их в разбавленной соляной кислоте.
Если спекание ведут на газовой горелке, то ход анализа рекомендуется
следующий. Тигель, желательно продолговатой формы, со смесью почвы и
реактивов при соотношении 1:1:4 помещают в круглое отверстие асбесто¬
вой пластинки так, чтобы в верхней части тигля, находящейся над пластин¬
кой, не было смеси. Асбестовая пластинка, не давая накаливаться верхней
части тигля, предохраняет хлористые соли от улетучивания. Тигель закры¬
вают плотно крышкой и нагревают на голом огне горелки, сначала очень
слабо, примерно 15 мин., до прекращения выделения аммиака. После пре¬
216
И. Г. Важенин
кращения выделения аммиака тигель нагревают сильно (t > 900° С) в продол¬
жение часа, после чего операция перевода щелочей в растворимое состояние*
може*.считаться законченной. Получившаяся в тигле спекшаяся масса долж¬
на легко отставать от стенок тигля при охлаждении. Всю массу шпателем пере¬
носят в фарфоровую чашку; крышка и тигель обмываются горячей водой в ту
же чашку.
Если спекшаяся масса при охлаждении сама не отстала от стенок тигля,
то в тигель наливают горячей воды, в которой масса через несколько минут
расплывается. Чашку покрывают стеклом и нагревают 2 часа на кипящей во¬
дяной бане; спекшиеся комочки растирают агатовым пестиком. Затем горячую
жидкость отфильтровывают в мерную колбу на 250 мл через быстро фильтру¬
ющий фильтр (красная лента, диаметром 9 см); чашку и фильтр промывают
10—15 раз небольшими объемами горячей воды в ту же колбу. В фильтрате
находятся: хлористые калий, натрий и кальций в виде хлорида и гидрата
окиси, а также сульфаты. Для осаждения кальция в колбу добавляют 25 мл
10%-ного раствора углекислого аммония. Колбы нагревают на водяной бане в
продолжение 20 мин. После охлаждения объем жидкости водой доводят да
250 мл, перемешивают и фильтруют. Полученный фильтрат используется для
определения калия на пламенном фотометре или одним из указанных выше
химических методов.
Разложение плавиковой и серной кислотами
Разложение почвенных силикатов плавиковой кислотой в присутствия
серной кислоты происходит по следующей схеме:
2KAlSi308-b32HF = 6SiF4+2KF+2AlF8-M6H20.
В присутствии серной кислоты фториды металлов превращаются в суль¬
фаты:
2KF+H2SO4 — K2SO4+2HF,
2AIF3+3H2SO4 -> Ab(S04)3+6HF.
Образующийся летучий фторид кремния (SiF4) при выпаривании улетучи¬
вается из раствора в газообразном виде. Некоторые минералы, например то¬
паз, силлиманит, турмалин, андалузит и другие, не полностью разлагаются
плавиковой кислотой, особенно в случае недостаточно тонкого растирания
почвы.
Ход анализа. Берут на аналитических весах навеску 0,5 г почвы, растертой
в агатовой или халцедоновой ступке до состояния пудры, помещают в плати¬
новую чашку емкостью 50—75 мл, смачивают 1 мл дистиллированной воды.
Осторожно выпарив воду на плитке, чашку помещают на 1,5—2 часа в муфель
для сжигания органического вещества при температуре 400—450° С. Предва¬
рительное удаление органического вещества ускоряет последующее разложе¬
ние почвы. При анализе карбонатных почв производят сначала разрушение
карбонатов разбавленной (1 : 1) соляной кислотой. В противном случае про¬
исходит выделение труднорастворимого фторида кальция, который захваты¬
вает значительное количество щелочей. Отмытая от хлоридов почва подсуши¬
вается, помещается в муфель для прокаливания. Кислота и фильтрат, дове¬
денные до определенного объема, поступают в анализ.
В охлажденную чашку приливают 1 мл Н20, после впитывания воды до¬
бавляют 1 мл концентрированной серной кислоты (уд. вес 1,84) и содержимое
тщательно перемешивают платиновым или фторопластовым (тефлон) шпате¬
лем; затем к смеси осторожно (!) приливают (обязательно в вытяжном шка¬
фу!) 15 мл фтористоводородной (плавиковой) кислоты; при этом происходит
сильное разогревание смеси. Смесь вновь перемешивают и чашку ставят на
закрытую этернитовую плитку (под тягой!) для разрушения минеральной час¬
Методы определения калия в почве
217
ти и удаления SiF4. Нагревание и выпаривание продолжают до полного разру¬
шения почвенных силикатов, что узнается по исчезновению «хрустения» при
помешивании и появлению густых белых паров серного ангидрида (S03).
Затем вновь приливают 10 мл HF и выпаривают досуха. Приливание плави¬
ковой кислоты производят в охлажденные чашки. После последнего прилива-
ния HF и появления белых паров S03 чашку нагревают более сильно (на от¬
крытой электроплитке или на пламени газовой горелки) для полного удаления
HF и паров S03.
К остатку в чашке приливают 5 мл соляной кислоты (1 : 1), закрывают ча¬
совым стеклом и нагревают на этернитовой плитке 10—15 мин. до полного ра¬
створения остатка; затем добавляют 10—15 мл горячей дистиллированной во-
ды и раствор отфильтровывают в мерную колбу на 100 мл; чашку и фильтр
промывают горячей водой, подкисленной HG1. После охлаждения объем жид¬
кости доводят до метки (100 мл) и содержимое взбалтывают. Калий определя¬
ют на пламенном фотометре или одним из химических методов.
Если поставлена задача определения калия в растворе, освобожденном
от всех катионов, кроме щелочных, что бывает необходимо при точных ана*
лизах химическими методами, то анализ заканчивают следующим образом.
В охлажденную чашку (после разложения силикатов) приливают 30—40 мл
горячей воды, 2—3 капли фенолфталеина и добавляют небольшими порциями,
при помешивании, свежепрокаленную окись кальция до устойчивой рововой
•окраски; после этого прибавляют еще 0,4—0,5 г СаО, чашку закрывают часо¬
вым стеклом и на 2 часа ставят на кипящую водяную баню. Добавляют в чаш¬
ку бумажную массу (кашицу из фильтровальной бумаги) и раствор отфильт¬
ровывают через фильтр с белой обмоткой; остаток на фильтре промывают 7—
'8 раз горячим разбавленным раствором гидроокиси кальция. К подогретому
фильтрату прибавляют насыщенный раствор углекислого аммония для осаж¬
дения кальция. Жидкость в чашке выпаривают досуха и слегка прокаливают
15—20 мин. для удаления следов аммония и перевода СаС03 в кристалличе¬
скую форму. Затем небольшими порциями горячей воды хлориды щелочей
вымывают из осадка, фильтруя раствор через маленький фильтр; чашку и
•осадок на фильтре промывают 5—6 раз холодной водой до объема 50 или
100 мл. В фильтрате содержатся только хлориды калия и натрия.
Литература
Антипов-Каратаев И. Я., Клейн Ю. Я. и
Красиков К. Я. К методам исследова¬
ния подвижных форм калия в подзоли¬
стых, каштановых и черноземных поч¬
вах.— Труды Почв, ин-та им. В. В. До¬
кучаева, 1935, т. XII.
Важенин И. Г. Методы определения калия
в почве.— В сб. «Агрохимические методы
исследования почв». М., «Наука», 1965.
Важенин И, Г., Карасева Г. И. Об агро¬
химических методах определения под:
вижных форм калия в почвах.— Почво¬
ведение, 1959, № 8.
Тедройц К. К. Химический анализ почв.
М., Сельхозгиз, 1955.
Голубева А. Я. Определение степени под¬
вижности обменного калия.— В сб.
«Пособие по проведению анализов почв
и составлению агрохимических карто¬
грамм». М., Россельхозиздат, 1969.
Грабаров Я. /\, Квитко Б. Я. К вопросу
о режиме спекания почв с хлористым
аммонием и углекислым кальцием при
•определении валового калия.— Изв.
АН КазССР, серия ботаника и почвове¬
дение, 1962, вып. 2 (14).
Иевиньш А. Ф., Гудриниеце Э. Ю. Опре¬
деление калия тетрафенилборатом нат¬
рия.— Ж. аналит. химии, 1954, т. IX,
вып. 5.
Кирсанов А. Т. Химическое определение
потребности почв в калийных удобре¬
ниях.— Химиз. соц. земледелия, 1933,
№ 6.
Коренман И. М. Аналитическая химия
калия. М., «Наука», 1964.
Левина Я. Д., Пантелеева Л. И. Приме¬
нение тетрафенилбората натрия для опре¬
деления калия в стекле.— Заводская
лаборатория, 1957, № 3.
Магницкий К. Я., Малков В. К. Опреде¬
ление запасов усвояемого растениями
калия в почве.—Агрохимия, 1968, №3.
Маслова А. Л., Чернышева 3. В. К мето¬
дике определения подвижного калия
в почве.— Труды ВИУА, 1934, вып. 5.
Мачигин Б. Я. Методы определения фос¬
фора в почве.— В сб. «Методы агрохи¬
218
И. Г. Важенин
мических, агрофизических и микробио¬
логических исследований в поливных
хлопковых районах». Ташкент, Союз-
НИХИ, 1952.
Ониани О. Г. Определение подвижных со¬
единений фосфора и калия из одной
вытяжки из красноземных и подзоли¬
стых почв Грузии.— Агрохимия, 1964,
№ 6.
Пчелкин В. У. Почвенный калий и калий¬
ные удобрения. М., «Колос», 1966.
Пчелкин В. У.у Забавская К. Л/., Козлова
К. М. Определение необменного гидро¬
лизуемого калия почв.— В кн. «Посо¬
бие по проведению анализов почв и
составлению агрохимических карто¬
грамм». М., Россельхозиздат, 1969.
Резников А. А.у Муликовская Е. П. Ана¬
лиз природных вод и рассолов.— В кн.
«Анализ минерального сырья». М., Гос.
научно-технич. изд-во хим. лит-ры,
1956.
Резников А. А., Нечаева А. А. Полевые
методы определения калия в природных
водах.— Сб. научно-технической ин¬
формации ВСЕ ГЕИ, № 5. М., Гос.
научно-технич. изд-во хим. лит-ры,
1956.
Руководство по составлению почвенных и
агрохимических карт. М., «Колос»,
1964.
Русакова Г. Г. Изучение условий осажде¬
ния калия при помощи тетрафенилбора-
та натрия.— Ж. аналит. химии, 1963,
т. XVIII, вып. 2.
Сердоболъскии Я. П. Сравнительная оцен¬
ка некоторых методов определения за¬
паса усвояемого калия в карбонатных
почвах Средней Азии (совхоз Пахта-
Арал).— Труды Почв, ин-та им. В. В.
Докучаева, 1939, т. XX.
Шарошкина Н. Б. Новые соотношения
между хлористым аммонием и углекис¬
лым кальцием при спекании почв для
определения валового калия.— Изв.
АН Каз. ССР, серия ботаника и почво¬
ведение, 1962, вып. 2 (14).
Янсон д. Ю., Иевиньш А. Ф. Тетрафенил-
бораты и их применение в аналитиче¬
ской химии.— Успехи химии, 1959,
вып. 8.
Bondorff К. А. Rapid methods for the
determination of potassium. Kalium
Symposium, Zurich, 1954.
Egner H., Riehm H., Domingo W. Я. Un-
tersuchungen iiber die chemische Noden-
analyse als Grundlage fiir die Beurteilung
des Nahrstfoffzustandes der Boden. II.
Chemische Extraktionsmethoden zur Phos¬
phor und Kaliumbestimmung.— KgL
Lantbrukshogsk. ann., 1960, v. 26, 199—
215.
Engelbrecht R. M., McCoy F. A. Determi¬
nation of potassium by a tetraphenil-
borat method.— Analyt. Chem., 1956r
v. 28, N 11.
Penceff N. P. Desprc unelc metode noi de-
microdozare a potassiu.— Rev. chinu
(RPR). 1959, v. 10, N 4.
Pflaum /?., Howick L. Spectrophotometricr
determination of potassium with sodium
tetraphenylborate.— Analyt. Chem., 1956r
N 28.
Pratt P. F. Potassium removal from Iowa
soils by greenhouse and laboratory pro¬
cedure.— Soil Sci., 1951, v. 72, N 2.
Reiteweier /?. F., Holmes R. S., Brown
J. C., Klipp L. W., Parks R. Q. Release of
non-exchangeable potassium by greenho¬
use. Neubauer and Laboratory Methods.—
Soil Sci. Soc. America Proc., 1947, v. 12.
158—163.
Riehm H. Flammenphotometrische Kalium-
bestimmung im Laktatextrakt ohne Aus-
fallung des Kalziums.— Bodenkunde und
Pflanzenernahr., 1945, Bd. 36, H. 1/2.
Schachtschabel P. Die Bestimmung des Kali-
bodarfs in Boden.— Bodenkunde und
Pflanzenernahr., 1941, Bd. 24, H. 1*
Schachtschabel P. Fixierung und Nachlie-
ferung von Kalium- und Ammonium —
lonen Beurteilung und Bestimmung des
Kalium — Versorgunsgrades von Boden.—
Landwirtsch. Forschung, 1961, Sonder-
heft, 15, N 8.
Sporek K. F. The gravimetric determination
of potassium in sea water as the potassium
tetraphenylboron salk.— Analyst, 1956r
v. 81, N 966.
Tendille C. Dosage du potassium par le
tetraphenyl borate de sodium.— Ann.
agron., 1955, N 7.
Thun R. Handbuch der landwirtschaft-
lichen Versuchs- und Untersuchungsme-
thodik (Bestimmung von Phosphorsaure
und Kalium in den nach Riehm erhal-
tenen Laktatbodenauszugen), Bd. 1. Ra-
debeul — Berlin, 1955.
Wood L. К., Turk E. E. The absorption
of potassium in soils in non-replaceable
forms.— Soil Sci. Soc. America Proc,,.
1940, v. 5. 152-161.
О. Я. МЕДВЕДЕВА
ОПРЕДЕЛЕНИЕ КАЛИЙНОГО ПОТЕНЦИАЛА
И ПОТЕНЦИАЛЬНОЙ БУФЕРНОЙ СПОСОБНОСТИ ПОЧВ
В ОТНОШЕНИИ КАЛИЯ
*
ОПРЕДЕЛЕНИЕ КАЛИЙНОГО ПОТЕНЦИАЛА
Б практике сельского хозяйства основным показателем обеспеченности
растений калием в настоящее время принято считать содержание в почве об¬
менного калия. Однако растворы нейтральных солей и слабых кислот, ис¬
пользуемые для определения обменного калия, по составу далеки от того поч¬
венного раствора, в который переходит калий почвенного поглощающего
комплекса в результате обменных реакций в почве.
Правильная характеристика калийного режима почвы должна включать
не только количественное определение содержания подвижных соединений
калия в почве, но и отражать степень его доступности растениям.
В последние 30 лет за рубежом начали развиваться представления о новом
способе оценки обеспеченности почвы доступным калием и разработаны ме¬
тоды, позволяющие производить такую оценку. Эта оценка основана на физи¬
ко-химической взаимосвязи между ионами калия и кальция в системе «поч¬
ва почвенный раствор» и выражается через термодинамический потенциал
калия, или так называемый калийный потенциал почвы.
Калийный потенциал не является показателем абсолютного содержания
калия в почве в той или иной форме. Он характеризует способность катионов
калия почвенного поглощающего комплекса переходить из твердой фазы поч¬
вы в почвенный раствор, т. е. дает им некоторую качественную оценку.
В отличие от воднорастворимого, обменного, необменного и других форм
калия почвы, которые Вудруфф, Ульрих и Бекетт (Woodruff, 1955а; Ulrich,
1961; Beckett, 1964 а, б; Beckett et al., 1966) называют «факторами емкости»,
калийный потенциал рассматривается ими как «фактор интенсивности» поч¬
венного калия.
Переход калия из почвенного поглощающего комплекса в почвенный рас¬
твор, откуда он непосредственно поступает в растения, зависит не только от
содержания катионов калия в твердой фазе почвы, но и от содержания в ней
других катионов, которые могут влиять на этот переход и которые поэтому
необходимо учитывать при определении калийного потенциала. К числу та¬
ких катионов относится доминирующий катион поглощающего комплекса
большинства почв Са2+ и сопутствующий ему Mg2+, на долю которых прихо¬
дится до 90% обменной емкости поглощающего комплекса почвы. Однако при
высокой насыщенности твердой фазы почвы катионами алюминия и натрия
содержание этих элементов также необходимо принимать во внимание при
определении калийного потенциала.
Согласно гипотезе о том, что в почве устанавливается динамическое рав¬
новесие между любой парой катионов поглощающего комплекса и почвенного
раствора, реакцию обмена между кальцием поглощающего комплекса почвы и
калием почвенного раствора можно представить следующим образом:
Са2+^ 2К++Са2р\ (1)
где KJ и Са2+ — катионы калия и кальция почвенного раствора; К+ и Са2+ — катионы
калия и кальция поглощающего комплекса почвы.
220
О. П. Медведева
Интенсивность поступления элементов питания из твердой фазы почвы в
почвенный раствор в результате обменной реакции зависит от энергии реак¬
ции обмена. Расчет энергии любой реакции обмена при постоянной температу¬
ре, равной 25° С, и постоянном давлении, равном атмосферному, дает возмож¬
ность охарактеризовать эту реакцию с помощью изобарно-изотермического
потенциала Z*, который сокращенно называется изобарным потенциалом или
свободной энергией.
Определить абсолютную величину свободной энергии вещества нельзя, но
подсчитать ее изменение AZ* для любой равновесной обратимой реакции,
в которой участвует это вещество, можно.
Изменение свободной энергии в обменной реакции (1) и будет калийным
потенциалом почвы, который выражается уравнением
AZ° = —2,3RT (рК—0,5рСа), (2>
где AZ°— изменение свободной энергии в реакции обмена между калием и кальцием в си¬
стеме «почва ^ почвенный раствор» в стандартных условиях *; R — газовая постоянная,,
равная 1,987 кал/град; Т — температура в градусах Кельвина (273 + 25°); рК — отри¬
цательный логарифм активности ионов калия в почвенном растворе; рСа — отрицатель¬
ный логарифм активности ионов кальция в почвенном растворе.
Подставив значения В и Т в уравнение (2), получим:
AZ0 = —1364 (рК—0,5рСа) кал.
Исходя из того, что катионы Са2+ и Mg2+ поглощающего комплекса почвы
обладают сходными обменными свойствами, предлагается выражать калийный
потенциал не через отношение ак+/у^Са«*. , а через отношение aK+/]/flca*++Mg*+ ,
т. е. рассматривать сумму активностей ионов Са2+ и Mg2+ как активность ио-
hjb одного вида.
Поэтому формулу калийного потенциала более правильно записать сле¬
дующим образом:
AZ° = -1364 [рК—0,5р (Ca+Mg)] кал **.
Часто, для облегчения расчетов, калийный потенциал выражают через от¬
влеченную величину, т. е. выражение (рК — 0,5 рСа) не умножают на
—1364 кал.
Формула калийного потенциала выводится из закона действующих масс,
уравнения изотермы полярной адсорбции Е. Н. Гапона и из химических по¬
тенциалов калия и кальция. Вывод формулы подробно изложен в работах
Вудруффа и Ульриха (Woodruff, 1955а; Ulrich, 1961).
По величине изменения свободной энергии химической реакции можно
оценить реакцию с точки зрения ее интенсивности. Чем больше отрицательное
эначение величины AZ°, тем с большей энергией кальций почвы спонтанно за¬
мещает калий почвенного раствора, переводя его в состояние, менее доступное
растениям (в калий твердой фазы), тем интенсивнее протекает^ реакция (1)
слева направо.
Калийный потенциал как индекс калийного режима почвы является ве¬
личиной универсальной и применим ко всем видам почв. Этот показатель по¬
стоянен для данного вида почвы и до определенных пределов не зависит от
ее влажности и концентрации почвенного раствора (Beckett, 1964а; Moss,
1963). Каждая почва имеет свое оптимальное значение калийного потенциала,
соответствующее наиболее благоприятным условиям калийного питания рас¬
тений*
* Стандартным состоянием для растворов принято считать: активность, равную 1 мсль/л,
давление — 1 атм, температуру — 25° С или 298° К.
*'•* Для краткости при дальнейшем изложении материала под величиной рСа всюду будет
подразумеваться величина р(Са + Mg).
Определение калийного потенциала и потенциальной буферной способности почв 221
Согласно градациям Вудруффа (Woodruff, 19556), изменение величины ка¬
лийного потенциала от 2,5 до 2,9 (от —3500 до —4000 кал) свидетельствует о
недостатке калия для нормального развития растений, изменение рК — 0,5 рСа
от 1,8 до 2,2 (от —2500 до—3000 кал) соответствует оптимальным услови¬
ям калийного питания растений, потенциал около 1,5 (—2000 кал) и ниже
свидетельствует об избытке калия («люкс-питание»).
В исследованиях отечественных авторов величины калийного потенциала,
соответствующие недостатку, избытку и оптимальному содержанию калия в
почве, отличаются от приведенных выше значений (Медведева, 1968).
Ряд исследователей определяет калийный потенциал в однократной вод¬
ной или слабосолевой (0,002—0,01 М раствор СаС12) вытяжке. Предполагает¬
ся, что активность ионов Са2+ в таких растворах соответствует активности
ионов Са2+ в почвенном растворе, которая определяется растворимостью
кальциевых солей почвы и переходом кальция из почвенного поглощающего
комплекса в почвенный раствор. Концентрация используемых для определения
калийного потенциала растворов хлористого кальция так мала, что она не вли¬
яет на физико-химические свойства почвы. Кроме того, слабые солевые вытяж¬
ки по сравнению с водными значительно облегчают отделение жидкой фазы
почвы от твердой и позволяют заменить центрифугирование фильтрованием.
Однако водные вытяжки при узком отношении почвы к воде в боль¬
шей степени имитируют почвенный раствор, чем слабые солевые вытяжки*.
Водная вытяжка (Ulrichy 1961)
Ход анализа. 30 г воздушно-сухой почвы помещают в полиэтиленовую или
стеклянную центрифужг ую пробирку на 50—100 мл и добавляют 20 мл
0,01%-ного водного раствора азида натрия NaN3 *.
Азид натрия по Ульриху необходим для подавления микробиологических
процессов во время определения потенциала. Взаимодействие почвы с раство¬
ром осуществляют при 25 + 1° С путем пропускания через пробирку
в течение 15 мин. тока воздуха, для чего используют компрессор типа КВМ-3.
В целях экономии времени можно пропускать воздух сразу через несколько
пробирок. Замена взбалтывания пропусканием атмосферного воздуха для
достижения равновесия между почвой и раствором способствует поддержанию
постоянного парциального давления СО.г. Последнее исключает образование
иона НСОз, который может перевести в раствор дополнительное количество
ионов Са2+ за счет образования Са(НС03)2. После 15-минутного пропускания
воздуха содержимое пробирок центрифугируют в течение 20 мин. при 15 тыс.
оборотов в минуту и пропускают через плотный фильтр для удаления корней
и других органических остатков, не осаждающихся при центрифугировании.
Фильтраты двух или трех параллельных пробирок смешивают для получения
достаточного количества «почвенного раствора». Последнее может быть до¬
стигнуто увеличением навески почвы до 60 г, а количества воды до 40 мл, для
чего, однако, потребуются центрифужные пробирки большего размера. Филь¬
трат анализируют на содержание калия, кальция и магния.
Содержание калия определяют на пламенном фотометре и пересчитывают
в моль на литр:
В
СК+-39д.юоо’
где Ск+— содержание калия в почвенном растворе, моль/л; В—содержание калия в поч¬
венном растворе, мг/л; 39,1 — атомный вес калия; 1000 — коэффициент для пересчета
миллиграммов в граммы.
* В лаборатории агрохимии Почвенного института им. В. В. Докучаева азид натрия»
заменяли тимолом, который добавляли в виде порошка в небольшом количестве на кон¬
чике ланцета в пробирку с 30 а почвы и 20 мл дистиллированной воды.
222
О. U. Медведева
В случае повышенного содержания кальция в почве (карбонатные или из¬
весткованные почвы) определение калия в фильтрате почвенного раствора
производят после осаждения кальция щавелевокислым аммонием по обще¬
принятой методике или вводят компенсацию на кальций.
Суммарное содержание кальция и магния в фильтрате определяют трило-
нометрическим методом, путем титрования определенного объема фильтрата
0,02 н. раствором трилона Б г. Полученные результаты пересчитывают в мо¬
ли на литр:
В-0,02 К
Са2+ Ж2 ’
где Са2+— содержание кальция в почвенном растворе, моль/л; В — количество трилона
Б, пошедшее на титрование, мл\ 0,02 — нормальность раствора трилона Б; К — поправ¬
ка к титру трилона Б; А — количество фильтрата, взятое на титрование, мл; 2 —
валентность Са.
Расчет величины калийного потенциала производят следующим образом:
1. Вычисляют ионную силу раствора по формуле 2
М* = З^Саа+ + CK+t
2. Рассчитывают коэффициенты активности ионов калия и кальция по
уравнению Дебая-Гюккеля
0,51.1* УК
gf~~ 1+tfjT ’
где / — коэффициент активности соответствующего катиона (Са2+ или К+); z — валент¬
ность катиона (для Са2+ z = 2, для К+ z = 1); (г — ионная сила раствора.
3. Вычисляют активности ионов калия и кальция:
аК+ = ^К+*/к+, аСа*+ — ^Са*+#/са*+>
где ак+ — активность ионов калия; аСа«+ — активность ионов кальция; Ск+ — содержа¬
ние калия в почвенном растворе, моль/л; CGa,f — содержание кальция в почвенном рас¬
творе, моль/л; /к+—коэффициент активности ионов калия; /са*+ — коэффициент активности
ионов кальция.
4. Вычисляют величину рК — 0,5 рСа,
где рК — — lg ак+, а рСа = — lgaCa*+.
Для облегчения расчетов Ульрих (Ulrich, 1961) дает график, по которому,
эная ионную силу раствора, можно найти коэффициенты активностей одно-,
двух- и трехвалентных катионов (рис. 1).
Трилонометрический метод определения кальция и магния в водной вытяжке описан в
настоящем руководстве в статье 3. Г. Ильковской и А. С. Коноваловой.
Ионная сила раствора (|л) равна полусумме произведений концентраций всех присутст¬
вующих в растворе ионов (С\) на квадрат их валентности (ф
2 Сг2;
я-2-’
При определении ионной силы почвенного раствора Ульрих считает достаточным
учитывать только концентрацию преобладающего катиона Са2+ и катиона К + и экви¬
валентное количество одновалентных анионов:
„ _(CCa»+-2i + 2CCl-,1S) + (CK+-12 + CCl--li) nr _i_ п
г— 2 Л1/Са*+"Г
Определение калийного потенциала и потенциальной буферной способности почв 223
Вытяжка раствором хлористого кальция
(Acquaye, Mac Lean, 1966)
Навеску почвы, равную 5 г, заливают 50 мл 0,002 М раствора СаС12 и
взбалтывают в течение 12 час. при 25 + 1° С. После взбалтывания суспензию
фильтруют и в фильтрате определяют калий на пламенном фотометре, а каль¬
ций и магний трилонометрически, как и в случае определения калийного по-'
тенциала в водной вытяжке. Шкалу образцовых растворов для определения
калия готовят на 0,002 М растворе СаС12.
Вычисление калийного потенциала производят так же, как при определе¬
нии его в водной вытяжке по методу Ульриха.
Реактивы. 1. 0,002 М раствор СаС12. Растворяют 0,438 г СаС12*6Н20 в 1 д дистил¬
лированной воды.
Остальные реактивы те же, что при определении кальция и магния в водной вытяжке
(см. выше).
ОПРЕДЕЛЕНИЕ ПСТЕН1ШАЛЬНСЙ БУФЕГНГЙ СПОСОБНОСТИ ПСЧВ
В ОТНОШЕНИИ КАЛИЯ
Снабжение растений калием зависит не только от «фактора интенсивности^
доступного калия, т. е. калийного потенциала, но и от способности почвы под¬
держивать калийный потенциал на относительно постоянном для данной поч¬
вы уровне при уменьшении или увеличении содержания доступного калия в
почве, что может иметь место при выносе калия растениями и при внесении
калийных удобрений. Способность почвы противостоять изменению калийно¬
го потенциала Бекетт назвал потенциальной буферной способностью почвы &
отношении калия — РВСК. Эта величина, по Бекетту, выражает зависимость
между «фактором интенсивности» (калийным потенциалом почвы) и общим
количеством подвижного калия в почве, которое Бекетт рассматривает как
«фактор емкости».
Зависимость между указанными величинами выражается уравнением
РВС*=2-,
где Q — «фактор емкости», количество подвижного калия в почве, мг-экв на 100 а почвы;
I — «фактор интенсивности» (AR), отношение активности ионов калия к корню квадрат¬
ному из активности ионов кальция (як+/У ®са*+") *
224
О. П. Медведева
РВСК более полно характеризует калийный режим почв, чем одид калий¬
ный потенциал. В зависимости от потенциальной буферной способности почв
в отношении калия одна и та же величина калийного потенциала может сви¬
детельствовать как о хорошей обеспеченности растений калием, так и о не¬
достатке этого элемента в почве.
Для нахождения потенциальной буферной способности почвы в отноше¬
нии калия определяют величину ДQ, которую чаще обозначают ДК. Вели¬
чина + ДК представляет собой количество подвижного калия, которое почва
отдает (—ДК) или поглощает (+ДК) к моменту установления равновесия
между калием почвы и калием раствора.
Если дать провзаимодействовать навеске почвы с серией растворов СаС12
одной и той же концентрации, но содержащих разные количества калия, и в
полученных равновесных растворах определить + ДК и ак+/|Л*Са*+ ,
которое Бекетт обозначает через AR, а затем отложить на оси абсцисс значе¬
ния ARy а на оси ординат соответствующие им значения + ДК и соединить
точки пересечения этих показателей, то получится характерная линия АВ —
прямая в верхней и изогнутая в нижней части (рис. 2). Пересечение линии с
осью абсцисс даст некоторую величину AR0, соответствующую отношению ак¬
тивностей ак+/у аСа*+ в растворе СаС12 — КС1, при котором почва не погло¬
щает и не отдает калия, т. е. отношению активностей, свойственному самой
почве. Форма полученной линии не является случайной. Ионы подвижного ка¬
лия занимают неравноценные обменные позиции в поглощающем комплексе
почвы и поэтому удерживаются почвой с различной силой (Beckett, Nafady,
1967).
При экстраполяции участка АВХ кривой АВ на ось ординат получают не¬
которую величину — ДК0, которая, по Бекетту, характеризует наиболее до¬
ступную часть подвижного калия — непосредственно доступный калий. Ионы
этой части подвижного калия занимают так называемые неспецифические
обменные позиции в поглощающем комплексе почвы и удерживаются в нем
силами электростатического поля, похожими по характеру, но отличными по
степени воздействия от сил, удерживающих другие катионы. С минералоги¬
ческой точки зрения, ионы непосредственно доступного калия — это катионы
калия, расположенные на внешних поверхностях кристаллитов.
Интерполяция участка ВгВ кривой АВ на ось ординат даст величину Klv
которая представляет собой общие запасы подвижного калия в данной почве,
близкие, но не эквивалентные содержанию в ней обменного калия. Разность
между Kl и — ДК0 дает количество труднодоступного подвижного калия
(Кх), ионы которого занимают «специфические» обменные позиции, распо¬
ложенные по краям кристаллической решетки или в ее гексагональных ячей¬
ках. Эти катионы удерживаются в твердой фазе почвы особыми силами, не¬
способными удержать другие обменные катионы почвенного поглощающего
комплекса.
Катионы подвижного калия, занимающие специфические обменные пози-
ции, переходят в почвенный раствор гораздо медленнее, чем катионы непо¬
средственнодоступного подвижного калия. Наличие в поглощающем комплек¬
се почвы этих катионов калия объясняет, почему не весь подвижный почвен¬
ный калий является обменным, т. е. экстрагируется 1 н. раствором
CH3COONH4, и почему нельзя точно определить величину Q, а можно лишь
установить ДQ (+ ДК).
Отношение — ДК0!ARQ, т. е. tg а, характеризует наклон участка
АВХ кривой АВ к оси абсцисс и выражает, по Бекетту, потенциальную ка¬
лийную буферную способность почвы (РВСК).
Потенциальная калийная буферная способность почвы является величи¬
ной постоянной для данной почвы в течение длительного периода времени и
мало меняется при внесении в почву калийных удобрений и при интенсивном
Определение калийного потенциала и потенциальной буферной способности почв 225
Рис. 2. Диаграмма определения потенциа^ной буферной способности почв в отношении
калия (РВСК)
Объяснение см. в тексте
использовании калия сельскохозяйственными культурами в течение вегета¬
ционного периода.
Установлено, что РВСК не зависит от соотношения почва: раствор при ис¬
пользовании для ее определения растворов СаС12 различной концентрации (не
выше 0,06 молъ/л), но зависит от времени уравновешивания калия почвы с
калием раствора СаС12 + КС1 и от температуры, при которой достигается
состояние равновесия.
Слишком длительное время уравновешивания может привести как к ис¬
кусственному увеличению количества непосредственно доступного калия за
счет диффузии ионов межслоевого необменного калия в раствор и истирания
граней и углов кристаллов глинистых минералов при длительном встряхива¬
нии почвы с раствором, так и к фиксации доступного калия. Недостаточное
время уравновешивания может не обеспечить полной дезагрегации всех поч¬
венных частиц. Увеличение температуры на 10° С, по данным Бекетта, при¬
водило к увеличению AR0 на 15%, что в свою очередь изменяло РВСК.
С помощью РВСК можно косвенно судить об участии необменного калия
почвы в питании растений. Когда в почве с высокой РВСК запасы доступного
калия истощаются за счет интенсивного выноса и достигают определенного
«минимального уровня», растения начинают получать калий из запасов резерв¬
ного необменного калия, который пополняет и поддерживает фонд доступ¬
ного калия. Такой способностью не обладают почвы с низкой РВСК, на кото¬
рых при достижении свойственного им «минимального уровня» подвижного
калия во избежание калийного голодания необходимо вносить калийные удоб¬
рения. Поэтому, исходя из РВСК, можно судить о способности почвы снаб¬
жать растения калием в большей степени, чем исходя из данных экстракцион¬
ных методов определения доступного калия.
Определение РВСК по Беккету
(Beckett, 1964а, б;Beckett et al., 1966)
Ход анализа. К серии навесок влажной почвы, соответствующих 5 г сухой
почвы, приливают по 50 мл 0,002 М раствора СаС12, содержащего 0; 0,2; 0,4;
0,6; 0,8 и 1 мг-эке/л КС1. Почву с раствором непрерывно взбалтывают в тече¬
ние 30 мин. при температуре 25 + 1° С и фильтруют. В фильтрате определя-
8 Агрохимические методы исследования почв
226
О. П. Медведева
ют калий на пламенном фотометре по шкале стандартов, приготовленной на
0,002 М растворе СаС12, и сумму кальция и магния — трилонометрическим
методом.
Для каждого раствора СаС12 + КС1 находят значения + ДК и А Л, по
которым строят линию А В (см. рис. 2), откладывая на оси абсцисс значения
AR, а на оси ординат соответствующие им значения + ДК. На графике нахо¬
дят значения AR0 (точка пересечения линии АВ с осью абсцисс) и — ДК0
(путем экстраполяции участка АВг кривой АВ на ось ординат). По получен¬
ным значениям ARQ и — ДК0 рассчитывают потенциальную калийную бу¬
ферную способность почвы
рвгк — “1^2
рвс = ЛЯо
Расчет величины РВСК производят следующим образом:
1. Вычисляют + ДК для каждого равновесного раствора:
± ДК* = Кисх .Дравн’
где ДК — изменение содержания калия в растворе СаС12п- КС1 после его взаимодей¬
ствия с почвой в мг-экв!л, что при соотношении между почвой и раствором СаС12т КС1,
равном 1 : 10, соответствует мг-экв на 100 г почвы; Кисх — содержание калия в исходном
растворе СаС12+ КС1, мг-экв/л\ Кравн — содержание калия в равновесном растворе СаС12+
+КС1, мг-экв/л.
2. Вычисляют AR для каждого равновесного раствора СаС12 4- КС1:
AR =
У аса^
где a j^+ — активность ионов калия в равновесном растворе, моль/л] дСа»+ — активность ио¬
нов кальция в равновесном растворе, моль/л.
Значения ак+ и аса*+ вычисляют через ионную силу равновесного раст¬
вора, коэффициенты активности калия и кальция и их концентрации — в рав¬
новесном растворе (см. вычисление калийного потенциала).
3. Вычисляют РВСК по значениям — ДК0 и AR0, определенным графичес¬
ки:
РВС* =
к _ —АКо
ARo *
Реактивы. 1. 0,002 М раствор СаС12. Растворяют 0,438 г СаС12-6Н20 в 1 л дистил¬
лированной воды.
Поскольку растворы СаС124- КС1 готовятся на 0,002 М растворе СаС12, удобно при¬
готовить сразу 10 л 0,002 М раствора СаС12, для чего 4,38 г СаС12 6Н20 растворяют в 10 л
дистиллированной воды.
2. Растворы, содержащие 0,2; 0,4; 0,6; 0,8 и 1 мг-экв КС1 в 11 0,002 М раствора
СаС12‘6Н20. Растворяют 14,9; 29,8; 44,7; 59,6 и 74,5 мг перекристаллпзованной соли КС1
в 1 л 0,002 М раствора СаС12.
Навески хлористого калия необходимо брать на аналитических весах. Содержание
калия в приготовленных растворах должно быть проверено на пламенном фотометре
перед использованием этих растворов для определения РВСК и должно быть равным
следующим величинам:
Содержание КС1 в 0,002 М
растворе СаС12, мг-экв! л 0,2 0,4 0,6 0,8 1,0
Содержание К+ в 0,002 М
растворе СаС12, мг!л 7,8 15,6 23,5 31,3 39,1
Определение калийного потенциала и потенциальной буферной способности почв 227
Литература
Медведева О. П. Об использовании калий¬
ного потенциала для оценки обеспечен¬
ности почвы доступным калием. (Канд.
дисс.). М., 1968.
Acquaye D. К., Мак Lean А. J. Potassium
potential of some selected soils.— Canad.
J. Soil. Sci., 1966, v. 46, N 2.
Beckett P. H. T. Studies on soil potassium.
Part. 1. Confirmation of the ratio law:
measurement of potassium potential.—
J. Soil Sci., 1964a, v. 15, N 1.
Beckett P. H. T. Studies on soil potassium.
II. The «immediate» Q/l relations of
labile potassium in the soil.—J. Soil Sci.,
1964b, v. 15, N 1.
Beckett P. H. T., Craig J. 2?., Nafady
M. H. М., Watson J. P. Studies on soil
potassium. V. The Stability of Q/I rela¬
tions.— Plant and Soil, 1966, v. 25, N 3.
Beckett P. H. 7*., Nafady М. H. M. Potas¬
sium — calcium exchange equilibria in
soils: the location on non-specific (Gapon)
and specific exchange sites.— J. Soil. Sci.,
1967, v. 18, N 2.
Moss P. Some aspects of the cation status
of soil moisture. Part. I. The ratio law
and soil moisture content. Part. II. The
effect of dilution and calcium ions on
the releas of potassium. Part. III. The
effect of potassium on the soil system.—
Plant ana Soil, 1963, v. 18, N 1.
Ulrich В. Boden und Pflanze. Stuttgart,
1961.
Woodruff С. M. Ionic equilibria l etweea
clay and dilute salt solutions. Soil Sci.
Soc. America Proc., 1955a, v. 19,
N 1.
Woodruff С. M. The energies of replace¬
ment of calcium by potassium in soils.—
Soil Sci. Soc. America, Proc., 19556,
v. 19, N 2.
Д. Л. ЛСКИНАЗИ
МЕТОДЫ ОПРЕДЕЛЕНИЯ НУЖДАЕМОСТИ ПОЧВ
В ИЗВЕСТКОВАНИИ
*
Действие извести на почву и растение весьма многообразно; она влияет на
физические, химические и биологические свойства почв. При известковании
почв, кроме ее кислотности, являющейся основным показателем для уста¬
новления необходимости известкования, необходимо учитывать механический
состав, содержание гумуса, глубину залегания карбонатов и ряд других
свойств почв, состав культур в севообороте и их особенности (например, чув¬
ствительность их к избытку в почвенном растворе ионов Н, А1, Мп, Са и др.)»
степень насыщенности севооборота минеральными и органическими удобре¬
ниями и их состав (кислый или нейтральный набор удобрений, содержание в
последних бора, магния) и пр.
Формы почвенной кислотности. Кислотность почв в ес¬
тественных условиях обычно возникает в ходе почвообразовательного про¬
цесса, но подкисление почвы может произойти и в результате внесения в нее
некоторых физиологически кислых минеральных удобрений — (NH4)0S04,
NH4C1, КС1 и др.
Основным признаком кислотности почв является наличие в почвенном
растворе свободных водородных ионов, обусловливающих его кислую реак¬
цию. Содержание водородных ионов в растворе обычно характеризуют ве¬
личиной pH, которая численно равна отрицательному логарифму активности
водородных ионов.
В почвенном растворе мы встречаемся обычно со следующими значениями
pH:
pH Реакция почв
8—9 щелочная (почвы засоленного ряда)^
7 нейтральная
6 слабокислая
5 кислая
3—4 сильнокислая
В природных условиях почвы приобретают слабокислую реакцию даже за
счет углекислоты почвенного воздуха (или атмосферной С02), которая раство¬
ряется в жидкой фазе почвы, подкисляя последнюю. Поэтому, строго говоря,
нейтральная реакция свойственна только тем почвам, в которых идет процесс
накопления оснований. Кислая или слабокислая реакция характерна для
подзолистых, дерново-подзолистых, серых лесных, бурых лесных, некоторых
луговых, торфянистых и других почв.
На основании одной величины pH почвенного раствора (или водной вы¬
тяжки из почвы) нельзя делать выводы о количестве извести, необходимой для
нейтрализации кислотности почв. Водородные ионы, присутствующие в поч¬
венном растворе, составляют только незначительную часть всего количества
водородных ионов, способных переходить в почвенный раствор или реагиро¬
вать с основаниями. Различные значения pH соответствуют следующим коли¬
чествам Н+ в почвенном растворе (в расчете на пахотный слой мощностью
Методы определения нуждаемости почв ш известковании
229
20 см и считая объемный вес практически равным единице):
pH 7 6 5 4 3
Н+, г/га 0,5 5 50 500 5000
Так как 1 г Н+ эквивалентен 50 г СаС08, то можно было бы полагать, что
для нейтрализации даже наиболее кислой почвы с pH 3 потребуется только
250 кг/га СаС03. Однако такая доза извести не даст ожидаемого эффекта, и
почва после ее внесения останется сильнокислой. Основная часть Н-ионов поч¬
вы находится в твердой фазе почвы, в ее илистой фракции, в поглощенном
состоянии. Поэтому дозы извести, рассчитанные по кислотности (pH) почвен¬
ного раствора, недостаточны для нейтрализации всей кислотности поглощаю¬
щего комплекса почвы: после нейтрализации известью почвенного раствора
Н-ионы (или Al-ионы) из поглощающего комплекса вновь перейдут в поч¬
венный раствор и подкислят его. Для правильной оценки степени кислотности
почв следует учесть общее количество ионов водорода (и алюминия), находя¬
щихся в почве в поглощенном состоянии. Различают формы почвенной кис¬
лотности: активную и потенциальную — обменную и гидролитическую (Ас-
кинази, 1926).
Под активной кислотностью понимают активную концентрацию (актив¬
ность) водородных ионов в почвенном растворе или в водной вытяжке из поч-
Потенциальная кислотность измеряется количеством ионов водорода (и
алюминия), находящихся в почвенном поглощающем комплексе в скрытом,
поглощенном состоянии. При известных условиях эти ионы могут быть пере¬
ведены в раствор: более подвижная часть ионов водорода (или алюминия)
почвы может быть переведена в раствор при обработке почвы избытком нейт¬
ральной соли, например КС1, NaCl, ВаС12; остальная менее подвижная часть
Н-ионов может быть переведена в раствор лишь при дальнейшей обработке
почвы солями, дающими в водном растворе (благодаря гидролизу) щелочную
реакцию. Обычно для этого применяют ацетат натрия (CH3COONa).
Взаимодействие кислой почвы с разными солями можно изобразить двумя
следующими схемами:
Как видно из схем, происходит обменная реакция катионов солей (К, Na)
с водородными ионами почвы; в результате этого обмена в солевой вытяжке
образуются свободные кислоты (в первой схеме — НС1, а* во второй —
СН3СООН), которые учитываются обычными лабораторными методами. По
количеству найденной соляной кислоты при обработке почвы раствором
нейтральной соли, например КС1, судят об обменной кислотности почвы;
вы (pH) К
Схема I
— Н
— К
-н
-н
Схема 2
Почвенный
поглощающий
комплекс
— Н
— Н
- Na
— Н
— Н
-Na
1 Методы определения pH, технические приемы и приборы описаны в статье Д. С. Ор¬
лова в настоящем сборнике.
230
Д. Л. Аскинази
по количеству же освободившейся уксусной кислоты при обработке почв,
например раствором CH3COONa, судят о гидролитической кислотности
почвы.
Как видно из приведенных схем, реакция почвенного поглощающего комп¬
лекса с нейтральной солью KG1 не идет до конца и в поглощенном состоянии
остается некоторое количество поглощенных ионов Н+. Это объясняется тем,
что в результате реакции образуется сильная нацело диссоциирующая кисло¬
та НС1, и избыток свободных ионов Н+ в растворе препятствует их полному
вытеснению из поглощающего комплекса. Иначе обстоит дело в случае гидро¬
литически щелочной соли. Образующаяся уксусная кислота прочно связывает
водородные ионы, и реакция смещается вправо вплоть до полного вытеснения
Н+-ионов.
Величина гидролитической кислотности равна разности между количест¬
вом кислоты, обнаруженной, например, цри обработке почвы CH3COONa и
КС1, но на практике обычно за величину гидролитической кислотности при¬
нимают все количество уксусной кислоты, которое находят в фильтрате при
обработке почвы избытком ацетата натрия.
Следует иметь в виду, что кислотность КС1 вытяжки обусловливается не
только поглощенными ионами водорода, перешедшими в вытяжку, но в зна¬
чительной мере и ионами алюминия. Последние появляются в почвенном раст¬
воре или в солевой вытяжке в результате двух реакций. Во-первых, возможен
обычный обмен ионов алюминия на катионы вытесняющих солей. Во-вторых,
образующаяся при определении обменной кислотности соляная кислота мо¬
жет растворять некоторые соединения алюминия.
При наличии в почве поглощенного алюминия взаимодействие его с раст¬
вором нейтральной соли может быть представлено следующей схемой;
В случае обработки раствором КС1 почвы, содержащей поглощенный алю¬
миний, в результате обмена в фильтрате на первом этапе появляется не сво¬
бодная кислота — НС1, а гидролитически кислая соль — А1С13. При тех зна¬
чениях pH, которые обычны для солевых вытяжек из почв, А1С13 гидролизу¬
ется по уравнению:
А1С1з+ЗНаО — А1(ОН)8+ЗНС1.
Гидроокись алюминия выпадает в осадок, и система практически ничем не
отличается от той, в которой содержатся только поглощенные ионы водорода.
Но если даже А1С13 и остается в растворе, то практически это не влияет на
результаты анализа, что видно из следующего. Кислотность солевого фильтра¬
та учитывается путем его титрования щелочью в присутствии фенолфталеина
(при предварительном кипячении фильтрата для удаления углекислоты) или
индикатора бромтимолблау (без кипячения, на холоду) по следующему урав¬
нению:
AlCl8+3NaOH — Al(OH)8+3NaCI,
что равноценно реакции:
3HCl+3NaOH — 3NaCl+3H20
Схема 3
-К
-К
— К
Методы определения нуждаемости почв в известковании
231
Поглощенные ионы алюминия вытесняются и в результате обработки поч¬
вы гидролитически щелочной солью при определении гидролитической кис¬
лотности. В этом случае весь вытесненный алюминий переходит в осадок в
виде гидроокиси.
ОПРЕДЕЛЕНИЕ КИСЛОТНОСТИ, ПОДВИЖНОГО АЛЮМИНИЯ
И ПОГЛОЩЕННЫХ ОСНОВАНИЙ ПОЧВЫ
Наибольший вред культурным растениям наносится в тех случаях, когда
почва обладает высокой обменной кислотностью. При внесении в почву мине¬
ральных удобрений в форме нейтральных солей поглощенные почвой ионы во¬
дорода (или алюминия) способны к обмену, в результате чего возможно силь¬
ное подкисление почвенного раствора. Вытесняемые ионы алюминия могут
оказывать и прямое токсичное влияние на растения и микроорганизмы. По¬
этому учет величины обменной кислотности необходим для определения по¬
требности почв в извести в целях нейтрализации или осаждения вредных для
растений ионов.
Гидролитическая кислотность не вызывает столь вредных для растений
последствий. Ионы водорода, обусловливающие гидролитическую форму кис¬
лотности почв, неспособны к вытеснению нейтральными солями и не образуют
в растворе свободных кислот или гидролитически кислых солей (НС1>
А1С13).
Для более полной оценки вероятного отрицательного действия кислотнос¬
ти почв важно установить не только разные ее формы, но и соотношение между
суммой обменных оснований S и количеством ионов водорода (и алюминия) в
почвенном поглощающем комплексе. Если через S обозначить сумму всех"
способных к обмену катионов в почве (обычно Са + Mg), а через Н — вели¬
чину гидролитической кислотности (все величины выражены в миллиграмм-
эквивалентах на 100 г почвы), то отношение [S:(S+H)]-100 будет выражать
степень насыщенности почв основаниями. Эта величина обозначается буквой
V. Сумма S + Н определяет величину обменной способности почвы. Эту ве¬
личину обычно называют емкостью поглощения или емкостью обмена почвы.
Отношение S:(S + Н) показывает, в какой степени поглощающий комплекс
почвы насыщен основаниями, и является весьма важной характеристикой поч¬
вы в отношении ее кислотности.
К. К. Гедройц преимущественно исследовал сильнокислые почвы (сильно¬
подзолистые, торфянисто-подзолистые). Поэтому при вычислении степени на¬
сыщенности почв основаниями по приведенной выше формуле он применял
величины Н, соответствующие обменной кислотности.
В дальнейших исследованиях было показано, что более целесообразно
при расчете степени насыщенности почвы основаниями принять за Н величину
гидролитической кислотности в ее практическом понимании и дозу извести
определять, исходя из величины гидролитической кислотности почв.
Почвы, содержащие в поглощающем комплексе Н-ионы, тем кислее или
тем менее насыщены основаниями, чем большая доля от общей емкости погло¬
щения представлена Н-ионами.
Определение pH солевой вытяжки из почвы -
Из каждого хорошо перемешанного образца берут две навески по 20 г
воздушно-сухой почвы и помещают в конические колбы емкостью 100 мл. В
каждую колбу приливают по 50 мл 1,0 н. раствора хлористого калия. Содер¬
жимое колб хорошо взбалтывают и оставляют на ночь или на более короткий
срок до полного оседания почвы и просветления находящейся в колбе жид¬
кости. По окончании указанного срока осторожно, чтобы не взмутить жид¬
232
Д. JI. Аскинааи
кость, из каждой колбы берут пипеткой порцию [отстоявшегося прозрачного
раствора и определяют в нем pH.
Для приготовления 1,0 н. раствора хлористого калия отвешивают на тех¬
нических весах 75 г соли, растворяют в 400—500 мл дистиллированной воды и
доливают дистиллированной водой до 1 л. Реакция раствора должна быть
около pH 5,5—6,0 и устанавливается, в случае необходимости, прибавлением
нескольких капель разбавленной /Соляной кислоты или разбавленной щелочи
(если pH раствора меньше 5,5).
Определение обменной кислотности почвы
40 г почвы взбалтывают в течение 1 часа со 100 мл 1,0 н. раствора КС1 и
фильтруют; 50 мл фильтрата кипятят для удаления С02; горячий раствор тит¬
руют 0,01 н. раствором NaOH с фенолфталеином в качестве индикатора до не
исчезающей в течение 1—2 мин. розовой окраски при кипячении (во избежа¬
ние выщелачивания стекла при кипячении рекомендуется применять стаканы,
длительно обработанные паром).
Для более полного вытеснения Н-ионов, способных к обмену с растворами
нейтральной соли, необходимо многократно обработать ими почву. Для рас¬
чета полной обменной кислотности на 100 г почвы израсходованное на титро¬
вание 50 мл фильтрата количество 0,01 н. раствора NaOH, соответствующее
20 г почвы, следует умножить на 5 (для перевода результатов титрования на
100 г почвы), затем на 0,01 (1 мл 0,01 н. раствора соответствует 0,01 мг-экв)
и на условный коэффициент 1,75 (поправка на неполноту вытеснения Н-ионов
хлористым калием при однократной обработке почвы этим реактивом), а всего
на коэффициент 0,0875 = 5*0,01*1,75. Применение коэффициента оговари¬
вается при опубликовании данных. 1 мл 0,01 н. раствора NaOH соответствует
0,01 мг-экв СаС03. Обычно данные обменной кислотности недостаточны и но
используются для расчета доз извести.
Определение гидролитической кислотности почвы
по Каппену
Из тщательно перемешанной средней пробы воздушно-сухой почвы берут
навеску в 40 г (на технических весах с точностью до 0,1 г). Навеску переносят
в бутыль или банку емкостью 200—300 мл и приливают 100 мл 1,0 н. раствора
уксуснокислого натрия (исходный раствор этой соли должен давать слабо¬
розовую окраску с фенолфталеином). Затем банки с содержимым взбалтывают
в течение 1 часа или оставляют на сутки, взбалтывая за это время 4—5 раз.
Полученную таким образом суспензию фильтруют через сухой складчатый
фильтр. Первые 10—12 мл фильтрата отбрасывают. Когда наберется доста¬
точное количество фильтрата, берут пипеткой 50 мл фильтрата, переносят в
коническую колбочку или в стаканчик и прибавляют туда 1—2 капли фенол¬
фталеина; далее эту порцию фильтрата титруют 0,1 н. раствором NaOH или
Ва(ОН)2 без предварительного кипячения фильтрата (во избежание улетучи¬
вания уксусной кислоты) до не исчезающей в течение 1 мин. розовой окраски
и записывают количество миллилитров NaOH, пошедшее на титрование. Так
как в большинстве случаев фильтраты получаются слегка окрашенными в
желтый цвет, то рекомендуется вести титрование в присутствии колбы с та¬
ким же раствором, сравнивая его окраску с цветом титруемой жидкости.
Количество щелочи (а), пошедшее на титрование 50 мл фильтрата, вы¬
ражают в миллилитрах NaOH точно 0,1 н. концентрации. Для перевода по¬
казаний в миллиграмм-эквиваленты на 100 г почвы найденное при титрова¬
нии 50 мл фильтрата количество 0,1 н. щелочи с поправкой на титр (/£)
надо умножить на 0,875 = 5-0,1-1,75. Получим 0,875 аК.
Методы определения нуждаемости почв в известковании
233
Для вычисления дозы извести в центнерах СаС03 на 1 га следует величи¬
ну кислотности почвы в миллилитрах 0,1 н. раствора щелочи, пошедшей на
50 мл фильтрата, после умножения на поправку к титру, умножить на коэф¬
фициент 13.
Коэффициент 13 получен на основании следующего расчета: 1 мг-экв щелочи соответ¬
ствует 0,001 г Н или 0,100/2 г СаС03, или 0,050 г СаС03. Принимая вес пахотного слоя
1 га (подзолистой почвы) в 3 млн. кг, или 3 • 10е г, и величину найденной гидролитической
кислотности в миллиграмм-эквивалентах на 100 г почвы, равной 0,875 а/if, получаем
количество СаС03 в центнерах на 1 га: (0,875^*3-109-0,05):(100-100 000) = 13,1 аК.
В приведенных расчетах принято во внимание, что при однократной об¬
работке раствором ацетата из почвы удалены еще не все Н-ионы, способные
к обмену с катионом соли, так как для этого требуется многократная об¬
работка почвы раствором уксуснокислого натрия.
Если желательно внесение в почву не углекислой извести, а гашеной,
то вместо множителя 13 применяют множитель 9,6; полученная величина
будет соответствовать] числу центнеров чистого Са(ОН)2 на 1 га.
Для полноты определения величины гидролитической кислотности с
целью доведения почвы до pH 7 было предложено применять коэффициент
1,75 (как поправку на полноту взаимодействия кислой почвы с ацетатом).
Для доведения почв до pH, равного 7,5; 8,0 и 8,5, Каппен предложил коэф¬
фициент 1,75 увеличить соответственно до 2; 2,5; 3,25.
Количество извести, расходуемое для доведения реакции почвы до опре¬
деленной величины pH в лабораторных условиях, всегда оказывается не¬
достаточным для получения той же реакции почвы в поле, так как в полевых
условиях часть извести теряется из пахотного слоя вследствие вымывания
ее в нижележащие слои почвы, часть вынссится с урожаем растений и, на¬
конец, часть ее остается в почве в первоначальном виде и т. д. Вследствие
этого возникает необходимость в применении особого множителя для пере¬
хода от доз извести, рассчитанных на основании лабораторного анализа почв,
к дозам довести, которые необходимо внести в поле для получения такого
же эффекта в отношении изменения реакции почвы. Величина этого множи¬
теля, обычно называемого полевым коэффициентом, значительно колеблет¬
ся по данным разных авторов, например по Н. И. Алямовскому (1936) —
от 1,8 до 4,1, по Иенсену — от 2,0 до 3,0. По данным М. Ф. Корнилова, этот
коэффициент более устойчив.
Определение суммы поглощенных оснований
по ускоренному методу Каппена — Гильковица 1
Из^лорошо перемешанного почвенного образца на технических весах
отвешивают навеску почвы в 20 г с точностью до 0,1 а. Навеску помещают
в колбу емкостью 200—300 мл. Приливают из бюретки 100 мл 0,1 н. раство¬
ра НС1 точно установленного титра.
Для приготовления 0,1 н. НС1 берут 80 мл концентрированной соляной
кислоты (уд. веса 1,19) и разводят дистиллированной водой до 10 л\ точный
титр полученной таким образом соляной кислоты устанавливают по титро¬
ванной 0,1 н. щелочи.
Содержимое колбы взбалтывают в течение 1 часа и оставляет на 24 часа.
По истечении этого срока фильтруют через сухой складчатый фильтр; пер¬
вые порции фильтрата отбрасывают. Из фильтрата берут пипеткой 50 мл в
коническую колбочку емкостью 200 мл, кипятят 1—2 мин., после чего при¬
1 Подробное]описание определения поглощенных оснований трилонометрическим методом
дано в статье 3. Г. Ильковской и А. С. Коноваловой в настоящем сборнике.
234
Д. JI. Аскинааи
бавляют 2 капли фенолфталеина; титруют этот горячий раствор ОД н. рас¬
твором едкого натра до неисчезающей слабо-розовой окраски.
При наличии большого количества полуторных окислов в вытяжке,
выпадающих при ее титровании в виде осадка, целесообразно дать осадку
осесть и протитровать затем прозрачную жидкость над осадком до слабо¬
розового окрашивания с фенолфталеином.
Полученную величину расхода ОД н. NaOH при титровании вычитают
из количества миллилитров ОД н. раствора NaOH, идущего на титрование
50 мл исходной соляной кислоты; разность, умноженная на поправку к
титру ОД н. щелочи, выражает сумму поглощенных оснований в милли¬
грамм-эквивалентах на 100 г почвы.
Метод этот неточен и при незначительном содержании в почве поглощен¬
ных оснований может их не уловить (в таком случае разность чисел при тит¬
ровании иногда близка к нулю или даже имеет отрицательный знак). Для
почв, богатых поглощенными основаниями (S ]> 15 мг-экв на 100 г), берут
на 20 г почвы не 100, а 200 мл ОД н. раствора HG1; в этом случае при под¬
счете конечный результат умножают на 2.
Вычисление степени насыщенности почв осно¬
ваниями. Как уже указывалось выше, степень насыщенности почв ос¬
нованиями вычисляется по формуле
где V — степень насыщенности почв основаниями; S — сумма поглощенных оснований,
мг-экв на 100 а почвы; Я —величина гидролитической кислотности, мг-экв на 100 г почвы.
Определение буферности почвы по Ремезову (1930)
Буферностью почвы называется способность устойчиво удерживать свою
реакцию (pH) при добавлении к почве разбавленных минеральных кислот
или щелочей. Буферность нейтральных почв (в особенности карбонатных) зна¬
чительна при добавлении к ним кислот, а буферность кислых почв —при
добавлении щелочей (NaOH, КОН), щелочноземельных оснований — Са(ОН)2,
Ва(ОН)а и др., а также так называемых гидролитически щелочных солей —
CH3COONa, Са(НС03)2 и др. На буферности почв основаны методы Иенсена,
Ремезова и других исследователей.
Буферность почв по отношению к кислотам или щелочам обусловлена в
основном количеством содержащихся в почвах поглощенных оснований (Са,
Mg и др.), карбонатов, а также ионов водорода (или алюминия) или свобод¬
ных органических кислот.
Приведем следующие реакции, ’поясняющие схематически сущность
буферности почв:
TJ
I. Почва нейтральная: (почва) Са + 2НС1 (почва) g + СаС12.
II. Почва кислая: (почва) ^ + 2СаС03 ^ (почва) Са + Са(НС03)2.
В примере I добавленная соляная кислота нейтрализовалась почвой, во
II]— кислую почву нейтрализовал добавленный к ней углекислый кальций.
В том и другом случае полученные результаты обусловлены буферностью
этих почв в отношении кислот и оснований.
Ход анализа. Берут несколько конических колб емкостью по 100 мл с
каучуковыми пробками или того же объема несколько реактивных банок с
притертыми пробками. В каждую колбу помещают по 10 г почвы, отвешенной
на технохимических весах, прибавляют по 25 мл 1,0 н. раствора хлористого
кальция и затем последовательно возрастающие количества 0,04 н. раство¬
Методы, определения нуждаемости почв в известковании
235
ра Са(ОН)2*. Обычно в первую колбу раствор гидроокиси кальция не при¬
бавляют, и она служит для определения pH в солевой суспензии; во вторую
колбу прибавляют 5 мл щелочного раствора Са(ОН)2, в третью — 10 мл,
в четвертую — 15 мл и т. д. до 25—30 мл. Следует иметь в виду, что
каждые 2,5 мл 0,04 н. раствора Са(ОН)2 соответствуют 1 мг-экв на 100 г поч¬
вы.
После прибавления щелочи колбы закрывают пробками, взбалтывают и
оставляют на 24 часа для установления равновесия. По прошествии этого
времени в суспензиях определяют pH электрометрическим методом. Полу¬
ченные данные наносят на миллиметровую бумагу таким образом: по вер¬
тикальной оси откладывают величины pH, по горизонтальной — число мил¬
лилитров прибавленого раствора Са(ОН)2. От точки пересечения кривой,
соединяющей найденные величины pH, с линией, соответствующей желаемой
величине pH (например, pH 7,0), опускают прямую, перпендикулярную го¬
ризонтальной оси. Точка пересечения этой линии с горизонтальной осью
будет соответствовать тому количеству щелочи, которое следует прибавить к
суспензии почвы, чтобы сделать ее насыщенной при избранном pH, или, дру¬
гими словами, будет соответствовать величине кислотности почвы при дан¬
ной реакции.
Найденное количество миллилитров щелочи умножают на поправку к
титру для пересчета на точный 0,04 н. раствор Са(ОН)2 и затем делят на 2,5;
частное от деления будет соответствовать величине кислотности в милли-
грамм-эквивалентах на 100 г почвы.
При установлении дозы извести найденную величину кислотности поч¬
вы в миллиграмм-эквивалентах умножают на 15, что дает необходимое ко»
личество чистого СаСОэ в центнерах на 1 га.
Коэффициент 15 вычислен на основании следующего расчета: 1 мг-экв соответствует
0,001 гН или 0,100:2 г СаСОа, или 0,050 г СаС08 на 100 г почвы, что в пересчете на 1 кг
дает 0,5 г СаСОа. Принимая, как обычно, вес пахотного слоя 1 га в 3 млн кг, мы получа¬
ем, что 1 мг-экв кислотности на 100 г почвы соответствует 15 ц СаС03 на 1 га. Следует
указать, что вес пахотного слоя колеблется обычно от 2 до 3 млн кг в зависимости от
механического состава почвы, содержания органического вещества, глубины пахоты
и т. д. Поэтому рекомендуется производить его определение для более или менее резко
разнящихся между собой почвенных районов.
Величина pH, до которой производится нейтрализация почвы, устанав¬
ливается в зависимости от особенностей возделываемых в данном районе
растений и их отношения к извести.
Автор не затрагивает вопроса о величине полевого коэффициента в ус¬
ловиях предложенного им метода.
Реактивы: 1. Нормальный раствор хлористого кальция. Кристаллический
хлористый кальций (СаС12*6Н20) имеет молекулярный вес 219. Для приготовления 1,0 н.
раствора берут 110 г этой соли на 1 л. Отвешенное на технических весах количество
соли сначала растворяют в химическом стакане в небольшом количестве воды, фильтруют
через складчатый фильтр в мерный цилиндр и доливают до метки дистиллированной во¬
дой. Хранить раствор надлежит в склянке, снабженной сифоном; удобно приспособить
к склянке автоматически наполняющуюся пипетку на 25 мл.
2. 0,04 н. раствор Са(ОН)2. Раствор приготовляется из чистого СаО. На технических
весах отвешивают 40—50 г СаО и обливают в фарфоровой чашке равным по весу количе¬
ством дистиллированной воды, после чего, закрыв кристаллизатором, оставляют стоять.
Когда дно чашки остынет, содержимое смывают дистиллированной водой в склянку ем¬
костью 10 л; заполнив водой и снабдив сифоном (конец которого на 4—5 см не доходит до
дна), склянку оставляют стоять. Отстоявшийся прозрачный раствор сливают при помощи
* При комнатной температуре раствор Са(ОН)2 обычно получается около 0,033 ы.
236
Д. JI. Аскинааи
описанного выше сифона в склянку с бюреткой, из которой предварительно удалена уг¬
лекислота воздуха. Удаление углекислоты достигается продуванием воздуха, прошед¬
шего предварительно через две промывные склянки с 50%-ным раствором КОН. Титр
приготовленного раствора определяют согласно требованиям аналитической химии.
Примечание. Следует указать, что метод Н. П. Ремезова существенно отлича¬
ется, например, от метода Каппена тем, что определение кислотности производится тит¬
рованием щелочью солевых вытяжек из почв, доведенных до разных pH. В этом методе
в большей мере учитывается буферность, а значит, и кислотность почвы. В методе от¬
падает необходимость пользования переводным коэффициентом Каппена 1,75 и др.
Определение подвижного алюминия в почве
по Соколову (1939)
На многих кислых почвах растения страдают не только от повышенной
концентрации Н-ионов, но и от повышенного содержания в них подвижного
алюминия, легко переходящего в раствор при применении нейтральносоле¬
вых вытяжек из почв. Некоторые растения относительно даже более чувст¬
вительны к повышенной концентрации подвижного алюминия в почве, чем
к концентрации Н-ионов (ячмень, в меньшей степени лен).
Поэтому при изучении вредного действия кислотности почв большой
интерес представляет выяснение роли алюминия как фактора кислотности
(удельный вес алюминия в титруемой кислотности солевой вытяжки, по-ви-
д им ому, различен для разных почв).
Простой метод учета количества легкоподвижного, активного алюминия
в почве предложен А. В. Соколовым.
Обычный метод определения в почве обменной кислотности с применением
1,0 н. раствора КС1 дает возможность установить лишь суммарное количе¬
ство ионов водорода и алюминия, перешедших в солевую вытяжку в форме
НС1 и А1С13.
Принцип метода А. В. Соколова заключается в следующем: если к по¬
лученному раствору добавить достаточное количество фтористого натрия
дли калия, то содержащиеся в фильтрате ионы алюминия выпадают в оса¬
док в виде нейтральной комплексной соли — криолита Na3AlFe по реакции
AlCl8+6NaF _> Na3AlFe+3NaCl.
Следовательно, получается возможность определить в растворе в отдель¬
ности содержание ионов свободной кислоты (Н+) и алюминия (А18+), так как,
титруя сначала вытяжку в присутствии фенолфталеина (или бромтимолблау)
без прибавления фторида натрия, определяют общую обменную кислотность,
а потом, протитровав вторую порцию той же вытяжки, после осаждения
алюминия фторидом, устанавливают кислотность, вызываемую только пог¬
лощенным Н-ионом. По разности легко определить содержание ионов алю¬
миния в нейтральносолевой вытяжке, а следовательно, и в самой почве— по¬
глощенного алюминия.
Определение общей обменной кислотности. На¬
веску воздушно-сухой почвы в 100 г помещают в бутыль. Туда же прили¬
вают 250 дед 1,0 н. раствора КС1. Содержимое бутыли взбалтывают на ро¬
таторе в течение 1 часа. Для торфяных почв отношение навески к раствору
берут не 1 : 2,5, а 1 : 5.
Из отфильтрованной через беззольный сухой фильтр вытяжки отмери¬
вают пипеткой 50 дед, которые переносят в коническую колбочку или стакан¬
чик и после 5-минутного кипячения для удаления С02 титруют в горячем
состоянии 0,01 н. раствором NaOH в присутствии 3—о капель фенолфталеи¬
на до слабо-розового окрашивания. Количество 0,01 н. раствора NaOH,
израсходованное при титровании, соответствует суммарному содержанию
ионов водорода (свободной кислоты) и ионов алюминия во взятых 50 дед вы¬
Методы определения нуждаемости почв в известковании
237
тяжки, что отвечает общей обменной кислотности в навеске почвы, прихо¬
дящейся на данный объем фильтрата.
Определение свободной кислоты в солевой вы¬
тяжке. Берут второй объем (50 мл) той же отфильтрованной вытяжки,
помещают в коническую колбочку и кипятят в течение 5 мин. для удаления
С02. Прибавляют 3 мл 3,5%-ного раствора фтористого натрия 1 для связы¬
вания в комплекс иона алюминия. Затем вытяжку титруют на холоду 0,01 н.
раствором NaOH в присутствии 3—5 капель фенолфталеина до слабо-ро¬
зового окрашивания или синего окрашивания с бромтимолблау. (В этом
случае 0,01 н. раствора NaOH должно пойти меньше, чем при определе¬
нии общей обменной кислотности.) Количество щелочи, израсходованное
на титрование 50 мл вытяжки, соответствует содержанию в ней свободных
водородных ионов без алюминия.
Для выражения обменной кислотности в миллиграмм-эквивалентах на
100 г почвы количество миллилитров 0,01 н. раствора NaOH, израсходован¬
ное на титрование, умножается на коэффициент 0,05.
Приме чание. Коэффициент 0,05 получается следующим образом. Для титрования
было взято 50 мл вытяжки, отвечающей 20 г почвы. Чтобы перейти к 100 г почвы, надо
результат умножить на 5, а так как для титрования применяли 0,01 н. раствор NaOH, то
для перехода к нормальному (1 мл нормального раствора содержит 1 мг-экв) полученный
результат надо разделить на 100. Получаем коэффициент 0,05.
Вычисление содержания алюминия. Содержание алю¬
миния в миллиграмм-эквивалентах на 100 г почвы устанавливается по раз¬
ности результатов первого и второго титрований, выраженных в миллиграммг
эквивалентах. Для выражения содержания алюминия не в миллиграмм-
эквивалентах, а в миллиграммах на 100 г почвы количество миллиграмм-экви¬
валентов умножают на 9 (эквивалентный вес алюминия).
Описанный метод определения свободной кислоты относится к случаю
титрования солевых вытяжек, не содержащих больших количеств железа.
В некоторых почвах, однако, возможно наличие железа, переходящего в со¬
левую вытяжку. В присутствии железа результаты определения алюминия
не совсем точны, поэтому железо рекомендуется осаждать 6,0 н. раствором
NaOH. По указанию автора, в этом случае титрование свободной кислоты
можно производить без осаждения железа следующим образом: к определен¬
ному объему вытяжки добавлять 3 мл 3,5%-ного раствора NaF; при этом
происходит выпадение осадка. По прошествии 15—20 мин., т. е. после окон¬
чания выпадения осадка, добавляют 2—3 капли метилрота и титруют 0,02 н.
раствором NaOH до начала ослабления красной окраски и перехода ее в
оранжевую; если при этом вновь наблюдается образование осадка, то снова
добавляют 3 дел NaF и ожидают несколько минут, после чего окончательно
эттитровывают раствор в присутствии бромтимолблау.
Титрование свободной кислоты с фенолфталеином дает преувеличенные
результаты; кроме того, окраска от фенолфталеина при стоянии обес¬
цвечивается, так как комплексные соединения железа малоустойчивы при
щелочной реакции. По данным автора, пользуясь его методикой, можно все
же оттитровать в 1,0 н. КС1 вытяжке из почвы до 5мл 0,1 н. НС1 в присутст¬
вии железа с точностью до 2%; поэтому при титровании содевых вытяжек,
в которых железо присутствует в небольших количествах, можно вполне
пренебрегать ничтожной ошибкой, вызываемой наличием в растворе железа.
Количество свободной кислоты можно определить в растворе, в котором
определялась обменная кислотность; для этого в раствор добавляют 0,01 н.
1 Исходный раствор фтористого натрия должен давать слабо-розовую окраску с фенол¬
фталеином: в противном случае он доводится до этой реакции добавлением слабого
раствора NaOH (или НС1).
238
Д. JI. Аскинааи
HG1 в количестве, точно равном количеству щелочи, израсходованной при
первом титровании. После растворения гидратов полуторных окислов, вы¬
павших при первом титровании, добавляют 3 мл 3,5%-ного раствора NaF
и далее титруют, как описано выше.
Особенности различных почв хорошо характеризуются этим методом.
Торфянистые почвы, по данным автора, отличаются содержанием большого
количества свободной кислоты в солевой вытяжке, между тем как подзоли¬
стые почвы и особенно красноземы содержат много алюминия; в соле¬
вых вытяжках из торфянистых почв, кроме Н+ и А13+, присутствует и Fe3+.
Согласно устному сообщению И. Ф. Саришвили, при использовании ме¬
тода А. В. Соколова на почвах, богатых подвижным (обменным) алюминием
(красноземы и др.)> недостаточно прибавить к хлор-калиевой вытяжке из
этих почв 3 мл 3,5%-ного раствора фтористого натрия для связывания в ком¬
плекс ионов алюминия; по-видимому, необходимо увеличить количество
миллилитров добавляемого раствора.
ИСПОЛЬЗОВАНИЕ ПОКАЗАТЕЛЕЙ КИСЛОТНОСТИ
ДЛЯ ОПРЕДЕЛЕНИЯ НУЖДАЕМОСТИ ПОЧВ В ИЗВЕСТКОВАНИИ
Характеристика почв по их кислотности. Руковод¬
ствуясь показаниями pH, почвы по кислотности, как уже указывалось,
можно разделить на сильнокислые, среднекислые, слабокислые и близкие к
нейтральным. Обычно чем кислее почвы по данным pH солевых вытяжек
тем выше эффективность известкования.
Кислотность почв может быть охарактеризована также степенью их на¬
сыщенности основаниями V. В соответствии с этим М. Ф. Корнилов (1937)
предложил схему потребности в извести различных по механическому сос¬
таву почв в зависимости от их насыщенности основаниями (табл. 1).
Таблица 1
Оценка потребности в извести различных почв в зависимости от pH и степени
насыщенности основаниями V
(по Корнилову, 1937)
Потребность в известковании
Почвы
сильная
средняя
слабая
отсутствует
pH
в КС1
V, %
в^1
V, %
pH
в КС1
У ,%
в*КС1
V, %
Подзолистые тяжело- и
5,0
45
5,0—5,5
45-60
5,5-6,0
60-70
6,0
70
среднесуглинистые
4,5
50
4,5-5,0
50—65
5,0-5,5
65-75
5,5
75
4,0
55
4,0-4,5
55-70
4,5-5,0
70-80
5,0
80
легкосуглинистые
5,0
35
5,0-5,5
35-55
5,0-6,0
55-65
6,0
65
4,5
40
4,5-5,0
40-60
5,0-5,5
60—70
5,5
70
4,0
45
4,0-4,5
45-55
4,5-5,0
65-75
5,0
75
супесчаные и песчаные
5,0
30
5,0-5,5
30—45
5,5-6,0
45-55
6,0
55
4,5
35
4,5-5,0
35-50
5,0—5,5
50-60
5,5
60
4,0
40
4,0-4,5
40-55
4,5-5,0
55-65
5,0
65
Заболоченные торфянистые
и торфянисто-болотные
3,5
35
3,5-4,2
35—55
4,2-4,8
55-65
4,8
65
1 Здесь и ниже «pH солевых вытяжек» выражает значение pH в вытяжках из почвы,
приготовленных на 1,0 н. растворе КС1 при отношении почвы к раствору, равном
1 : 2,5. Для торфяных почв берут более высокое отношение навески почвы к раствору
(1 : 10 и выше).
Методы, определения нуждаемости почв в известковании
239
Приведенная в таблице 1 характеристика почв была сопоставлена Кор¬
ниловым с результатами полевых опытов, проведенных на почвах северо-
западной зоны. Эти опыты показали сходимость данных о степени насыщен¬
ности почв основаниями с полевыми опытами в 52—88% случаев; по дан¬
ным же величины pH солевых вытяжек эта сходимость составляла от 39 до
86%, причем более или менее отчетливо обособляются лишь почвы, не тре¬
бующие известкования (с pH > 5,5). Наибольшее расхождение между ре¬
зультатами полевых опытов с известкованием и данными pH солевых вытя¬
жек отмечено Корниловым для слабокислых почв с pH солевой вытяжки от
5,0 до 5,5. Поэтому для таких почв полезно определять, кроме pH солевых
вытяжек, также степень их насыщенности основаниями. В табл. 2 дана
характеристика кислотности почв с учетом обоих факторов, т. е. величины
pH солевой вытяжки и степени насыщенности основаниями (Корнилов,
1947).
Таблица 2
Характеристика кислотности почв по данным pH солевых вытяжек и степени
насыщенности почв основаниями
(по Корнилову, 1947)
Степень на<
почв основ;
легкие почвы
сыщенности
1НИЯМИ, %
тяжелые
почвы
pH в КС1
суспензии
Обменная
кислотность,
мг-экв на 100 г
почвы
Степень кислотности почв
<4,5
>1 ,о
Очень сильнокислые
—
—
<4,5
<1,0
Сильнокислые
—
—
4,6-5,0
—
Среднекислые
<60
<75
5,1-5,5
—
Слабокислые
>60
>75
5,6-6,0
—
Близкие к нейтральным
>6,0
Нейтральные
Известкование следует проводить с учетом не только отдельных культур
севооборота, но и интересов всего хозяйства в целом, т. е. с учетом комп¬
лекса природных и хозяйственных факторов, свойств почв, состава культур
в севообороте, биологических особенностей сельскохозяйственных растений,
их чувствительности к почвенной кислотности, применяемой системы удоб¬
рений, экономических факторов и т. д.
Подвижные соединения алюминия и марганца в
почве. Вредное влияние кислотности на развитие растений проявляется
сильнее в почвах, содержащих много подвижного (обменного) алюминия.
Токсическое действие алюминия, по данным вегетационных опытов, особен¬
но сильно проявляется на льне, ячмене, яровой пшенице; очень чувстви¬
тельны и к А1-, и к Н-ионам сахарная свекла, горчица; наименее токсичны
ионы алюминия для овса.
При содержании подвижного алюминия до 2 мг на 100 г почвы клевер
развивается нормально; при 3 и 4 мг алюминия начинается угнетение кле¬
вера. Это угнетение весьма значительно при 6—8 мг подвижного алюминия.
Дальнейшее увеличение количества активного алюминия в почве вызывает
резкое снижение процента клевера в травосмеси и в большинстве случаев
его полное выпадение.
Наряду с алюминием токсическое действие на растения оказывает пог¬
лощенный и воднорастворимый марганец, появляющийся в почвах в усло¬
виях недостатка аэрации при pH солевой вытяжки менее 5,0. При содержа¬
240
Д. JI. Аскинааи
нии в воднорастворимой форме более 1—2 мг и в поглощенном состоянии бо¬
лее 6 мг на 100 г почвы марганец оказывает токсическое действие на клевер
и особенно на люцерну.
При характеристике почв по их кислотности имеет значение глубина за¬
легания в них карбонатов. При одинаковых показателях кислотности в пахот¬
ном слое почвы с близким залеганием карбонатов (до 100—125 см) должны
в общем рассматриваться как менее кислые, чем почвы с более глубоким за¬
леганием карбонатов.
Известкование по гидролитической кислотно¬
сти. При правильной системе удобрения известкование должно нейтрали¬
зовать главным образом обменную кислотность почв; но обычно вследствие
плохого перемешивания извести с почвой и в особенности при наличии в се¬
вообороте растений с повышенной чувствительностью к кислотности почв
(см. ниже) приходится вносить известковые удобрения в больших количе¬
ствах, чем рассчитано теоретически. Поэтому дозы извести исчисляются
исходя из величины гидролитической кислотности почв (0,75 гидролитиче¬
ской кислотности, 0,5 и т. д.) или по кривым титрования (см. выше метод
Ремезова).
Нередко высказывается предположение, что определение доз извести по
кривым титрования является наиболее точным, позволяющим дозировать
известь в соответствии с требованиями отдельных сельскохозяйственных
культур к реакции почвы. Приходится, однако, отметить, что точность ме¬
тода определения доз извести по кривым титрования несколько снижается
вследствие необходимости введения и в данном случае условного полевого
коэффициента, величина которого колеблется. Требование высокой точности
дозировки извести вообще трудно выполнимо хотя бы из-за наличия в сево¬
обороте различных по отношению к извести растений.
Применение в севообороте в отдельные годы разных удобрений, различ¬
ная степень перемешивания извести с почвой в разные сроки после извест¬
кования, различия по годам метеорологических условий и другие причины,
несомненно, по-разному влияют на эффективность извести и, следовательно,
также не дают возможности предъявлять слишком большие требования к
точности установления доз извести в практических условиях. По-видимому,
при установлении доз извести необходимо стремиться к точности около
5—10 ц/га.
Известкование почв в севообороте. Сильнокислые поч¬
вы надо известковать в обязательном порядке, во всех севооборотах, неза¬
висимо от их специализации, так как без этого на них нельзя обеспечить
получение высокого и устойчивого урожая сельскохозяйственных культур.
Широко следует применять известкование также на среднекислых почвах;
это дает значительные прибавки урожая трав и последующих культур сево¬
оборотов и облегчает освоение на этих почвах севооборотов с правильным
травосеянием. При небольших затратах на известкование следует известковать
и слабокислые почвы. Лишь на легких слабокислых почвах в специализиро¬
ванных картофельных севооборотах (особенно с посевом сидератов на зеле¬
ное удобрение) известкование излишне, так как, не оказывая сильного
положительного влияния на урожай трав, может ухудшить рост сидератов
(люпина, сераделлы) и оказать отрицательное влияние на урожай и качество
картофеля. Урожаи злаковых повышаются от извести и на среднекислых
почвах, а при полных и более высоких дозах извести — и на слабокислых,
главным образом вследствие мобилизации питательных веществ в подзо¬
листых почвах под влиянием извести.
Отношение отдельных групп сельскохозяйственных растений к кислот-
ности почвы и известкованию показано в табл. 3.
При установлении доз извести и способов ее внесения должна при¬
ниматься во рнимание специализация севооборота. В настоящее время дозы
Методы определения нуждаемости почв в известковании
241
Таблица 3
Отношение растений к кислотности почв и известкованию
Группа сельскохозяйственных культур
Отношение
к кислотности почв
и известкованию
зерновые
кормовые
техниче¬
ские
овощные
и картофель
плодово-
ягодные
сидераты
Переносят слабую, сред¬
нюю и сильную кислот¬
ность почв/ отзываются
главным образом на из¬
весткование очень силь¬
нокислых поч в
—
—
—
Картофель,
редька
Малина
Люпин
однолет¬
ний, сера¬
делла
Переносят слабую и
Овес,
Турнепс,
Подсол¬
Морковь,
Земляниъа,
-
среднюю кислотность
озимая
брюква,
нечник,
огурцы,
крыжовник,
почв, отзываются на из¬
весткование сильнокис-
лых почв
рожь,
кукуруза,
просо,
гречиха
тимофеевка,
овсяница
лен
томаты, репа
груша,
яблоня
Переносят слабую кис¬
Яровая и
Кормовая
Конопля,
Столовая
Вишня,
—
лотность почв, отзыва¬
ются на известкование
главным образом силь¬
но- и среднекислых почв
особенно
озимая
пшеница,
ячмень
свекла,
кормовая
капуста,
лядвенец
коксагыз
свекла,
капуста, лук
слива,
смородина
Понижают урожаи и от¬
зывают я на известкова¬
ние даже при слабой
кислотности почв
Клевер и
особенно
люцерна,
донник, рапс
Сахарная
свекла,
горчица,
мак
извести устанавливаются, в основном исходя из размера гидролитической
кислотности почв или по разработанной в ВИУАА таблице доз извести,
доводящих почвы до оптимальной для большинства сельскохозяйственных
растений слабокислой реакции (pH солевой вытяжки 5,6—5,8 и водной
6,0—6,5). Эти дозы извести указаны в табл. 4 для разных по степени кислот¬
ности и по механическому составу почв при содержании в них органичес¬
кого вещества не выше 2—3%.
Указанные в таблице дозы составлены с учетом зависимости между ве¬
личиной pH* солевых вытяжек и обменной и гидролитической кислотностью
для почв с близким механическим и минералогическим составом при близ¬
ком содержании в них органического вещества и соответствуют примерно
гидролитической кислотности подзолистых почв.
Пользование приведенной таблицей упрощает определение доз извести
в производственных условиях колхозов и совхозов. В работах научно-ис¬
следовательских учреждений при установлении доз извести можно исхо¬
дить также из данных обменной и гидролитической кислотности или из
кривых титрования почв.
Указанные в табл. 4 дозы извести (полные дозы) вносят на поля и участ¬
ки, на которых в ближайшие одну-две ротации не будет возделываться лен,
а на легких почвах и картофель. Дозы извести в льняных севроборотах на
участках, где будет возделываться лен, должны быть уменьшены до V2 — У3
от полных доз, так как более высокие дозы извести могут понизить урожаи
льносемени, ухудшить качество льноволокна и его выход. Примерно в та¬
ких же дозах известь должна применяться на легких почвах в севообороте
с большим процентным содержанием картофеля. Наконец, в специализи¬
рованных картофельных севооборотах известь следует вносить на сильно¬
кислых легких почвах в малых дозах (5—10 ц/га СаС03) под многолетние
242
Д. JI. Аскинааи
Таблица 4
Дозы углекислой извести, mha
Механический состав почв
Реакция
почв и pH
солевой вытяжки
сильнокислая
среднекислая
слабокислая
4,5 и меньше
4,6
4,8
5,0
5,2
5,4 —5,5
Супесчаные и легкосугли-
5,0
4,5
4,0
3,5
3,0
2,5
нпстые
Среднесуглинистые
6,0
5,5
5,0
4,5
4,0
3,5
Тяжелосуглинистые
8,0
7,5
6,5
5,5
5,0
4,5.
травы, а на среднекислых почвах с семенами клевера в микродозах (до 3/4
ц/га СаС03); в смеси же с минеральными удобрениями — в количествах,
необходимых для их нейтрализации1. Слабокислые легкие почвы в специа¬
лизированных картофельных севооборотах, как правило, не известкуют.
Влияние механического состава почвы, емко¬
сти обмена и органического ве*щества на действие
извести. Данные вегетационных опытов показывают, что на почвах с
малой величиной емкости катионного обмена, в частности на легких мало¬
буферных почвах, вредное влияние кислотности проявляется сильнее, чем
на почвах с большей емкостью обмена, в частности на более тяжелых поч¬
вах. В соответствии с этим действие извести на урожай культур, наиболее
чувствительных к почвенной кислотности, при низких величинах pH соле¬
вых вытяжек проявляется сильнее на почвах с меньшей емкостью обмена
(например, на более легких почвах), чем на почвах с противоположными
свойствами. При более высоких величинах pH такая зависимость не наб¬
людается. При культуре малочувствительных к кислотности растений известь
дает более высокие прибавки урожаев на тяжелых кислых почвах по
сравнению с легкими, так как в этом случае большое положительное значе¬
ние имеет усиление мобилизации питательных веществ и некоторое улуч¬
шение физических свойств почв.
При одинаковых величинах pH солевых вытяжек присутствие в почвах
подвижного алюминия увеличивает токсическое действие кислотности на
растения; однако интенсивность этого токсического действия алюминия
зависит от содержания органического вещества в почвах: последнее, связы¬
вая алюминий, уменьшает его подвижность и ослабляет токсичность для
растений.
Отмечается, что органическое вещество ослабляет вредное влияние кис¬
лотности и на почвенные микроорганизмы.
Известкование и система удобрений. Помимо механи¬
ческого состава почв и содержания в них органического вещества, токсич¬
ность почвенной кислотности для растений зависит также от применения
удобрений и от количества содержащихся в почвенном растворе оснований —
кальция и магния. Известно, что присутствие кальция ослабляет токсич¬
ность для растений ионов водорода и алюминия в физиологических опытах,
а присутствие фосфора, связывающего А13+, Мп2+ и другие ионы, снижает
вредное действие Н+ и А13+.
1 При более высоких дозах извести на легких почвах может понизиться урожай картофеля
и ухудшиться его качество (может увеличиться пораженность паршой и понизиться
процент крахмала).
Методы определения нуждаемости почв в известковании
243
На сильнокислых почвах легкого механического состава, особенно при
систематическом внесении кислых форм минеральных удобрений, может
проявиться недостаток магния для растений. Поэтому на указанных почвах
особенно высокий эффект дают магнийсодержащие известковые удобрения.
Значительные прибавки урожаев культур, малочувствительных к почвен¬
ной кислотности (овса, картофеля и др.), могут давать на этих почвах и ней¬
тральные соли магния, внесение которых на легких кислых почвах ни в ка¬
кой мере не может заменить известкование.
Значительное влияние на эффективность известкования оказывает вне¬
сение в почву органических и минеральных удобрений. По данным полевых
опытов, эффективность внесения извести под озимую рожь, овес и некоторые
другие малочувствительные к кислотности растения на фоне NPK может сни¬
зиться по сравнению с действием извести на навозном фоне или без удобрений.
Повышение эффективности известкования под эти растения при внесе¬
нии NPK возможно главным образом на сильнокислых почвах и в случаях
систематического внесения кислых форм минеральных удобрений.
Под наиболее чувствительными к кислотности растениями (клевер, лю¬
церна, кормовые корнеплоды, озимая пшеница, капуста и свекла) на кислых
и особенно сильнокислых почвах при внесении NPK действие извести на
урожай усиливается. В известной степени это зависит от нейтрализации
почвенной кислотности, возникшей под влиянием применения кислых форм
минеральных удобрений. Кроме того, улучшение условий питания растений
при внесении удобрений увеличивает их отзывчивость на нейтрализацию
избыточной кислотности. Поэтому и при внесении навоза (а не только NPK)
эффективность известкования под наиболее чувствительные к кислотности
культуры нередко повышается. Этому содействует и усиление в почве полез¬
ных для растений микробиологических процессов при внесении в нее навоза
и извести. Повышение эффективности известкования может быть обуслов¬
лено и содержанием в навозе основных питательных элементов (Р, К и др.).
Заметное влиянйе на оптимальную для сельскохозяйственных растений
реакцию почв и, следовательно, на дозы вносимой извести оказывает при¬
менение борных удобрений. При малом содержании подвижных соединений
бора на неокультуренных кислых почвах дозы извести должны быть ниже,
чем на окультуренных почвах, так как на первых увеличение доз изве¬
сти без внесения борных удобрений приводит иногда к снижению уро¬
жая льна, картофеля, кормовых корнеплодов и некоторых других сель¬
скохозяйственных культур и к ухудшению их качества, вероятно, вследст¬
вие недостатка усвояемого бора. При пониженной растворимости извести
(например, при недостаточной тонкости ее частиц) оптимальные дозы пос¬
ледней, естественно, должны быть увеличены.
Литература
Алямовский Н. И. Упрощенный способ
определения извести по методу титро¬
вания почвенных суспензий.— Химиз.
соц. земледелия, 1936, № 4.
Алямовский Н. И. Ориентировочное опре¬
деление доз извести по величине pH
KCl-вытяжек из почв.— Труды ВИУАА,
1949, вып. 29.
Алямовский Н. И. Известковые удобре¬
ния. Сельхозгиз, 1963.
Аринушкина Е. В. Руководство по хими¬
ческому анализу почв. Изд-во МГУ,
1970.
Аскинази Д. JI. Кислотность и емкость
поглощения почв в связи с их известко¬
ванием и фосфоритованием.— Труды
НИУ, 1926, вып. 38.
Аскинази Д. Л., Дружинин Д. В., Реме-
aoe Н. П. Определение потребности почв
в извести. Сельхозгиз, 1931.
Афанасьев И. А. Известкование почв по
данным полевых опытов в СССР. Из¬
весткование под картофель. Сельхоз-
гиз, 1941.
Болотина Н. И. О почвенных условиях
для развития красного клевера.— Труды
Почв, ин-та им. В. В. Докучаева, 1950,
31.
Виноградов В. Я. Рыхлые пресноводные
известняки и доломитовая мука как при*
244
Д. JI. Аскинази
родные источники известковых удобре¬
ний.— Вестн. с.-х. науки. Удобрение,
агротехника и агропочвоведение, 1941,
№ 2.
Гедроиц К. К. Химический анализ почв.
— Избр. соч., т. 2. Сельхозгиз, 1955.
Голубев Б. А., Петербургский А. В. Свой¬
ства почв и отношение растений к реак¬
ции среды. Из результатов вегетацион¬
ных опытов и лабораторных работ. М.,
1935.
Дьякова Е. В. Влияние кислотности под¬
золистых почв и подвижного алюминия
на развитие клевера и люцерны.— Поч¬
воведение, 1948, № 3.
Дьякова Е. В. Отношение клевера, люцер¬
ны и других кормовых трав к избытку
подвижного марганца в подзолистых
почвах.—Докл. ВАСХНИЛ, 1950, вып. 9.
Журбицкий 3. И. Эффективность извест¬
кования в овощеводстве.— В сб. «Из¬
весткование почв по данным полевых
опытов в СССР». Сельхозгиз, 1941.
Карпинский Н, П. Характеристика почв
и почвенное районирование дерново-
подзолистой зоны в связи с химизацией.
— Вестн. с.-х. науки, 1940, № 5.
Кедров-Зихман О. К. Известкование дер¬
ново-подзолистых почв. М., «Моск. ра¬
бочий», 1950.
Кедров-Зихман О. К.у Землемерская А. Н,
Лсаров X. К. Отношение картофеля к из¬
весткованию почвы.— Вестн. с.-х. науки.
Удобрение, агротехника и агропочвове¬
дение, 1941, № 2.
Кедров-Зихман О. /Г., Ярусов С. С. и др.
Отзывчивость сельскохозяйственных рас¬
тений на известкование в связи с поч¬
венной кислотностью и степенью насы¬
щенности почв основаниями.— Труды
ВИУАА, 1934, вып. 6.
Корнилов М. Ф. Известкование почв Ле¬
нинградской области.—Труды ЛОВИУА,
1937, вып. 52.
.Корнилов М. Ф. Сравнительная оценка
некоторых методов определения нужда¬
емости почв в известковании.— Докл.
ВАСХНИЛ, 1947, вып. 7.
Магницкий if. Я. Магнезиальные удобре¬
ния. Сельхозгиз, 1952.
Мещеряков А. М. Опыты по изучению
влияния алюминия на рост растений.—
Химиз. соц. земледелия, 1937, № 9.
Лейве Я. В. Известкование под лен по
данным полевых опытов. Сельхозгиз,
1941.
Петербургский А. В. Практикум по агро¬
химии. Сельхозгиз, 1963.
Ремезов Н. П. Почвенные коллоиды и по¬
глотительная способность почв. Сель¬
хозгиз, 1957.
Ремезов Н. П. Определение поглощенного
водородного иона.— Труды НИУ, 1930,
вып. 77.
Ремезов Н. /7., Щерба С. В* Теория и
практика известкования почв. Сельхоз¬
гиз, 1938.
Соколов А. В. Определение в почве по¬
движного алюминия.— Химиз. соц.
земледелия, 1939, № 7.
Турчин Ф. В., Соколова В. И. Об активном
марганце в почве и его токсичности
в связи с применением кислых форм
азотных удобрений.—Почвоведение, 1950,
№ 9.
Церлинг В. В. Известкование под кормо¬
вые травы.— В сб. «Известкование почв
по данным полевых опытов». Сельхозгиз,
1941.
Ярусов С. С. Известь как фон для мине¬
ральных удобрений.— В сб. «Известко¬
вание почв по данным, полевых опытов».
Сельхозгиэ, 1941а.
Ярусов С. С. Действие извести на моби¬
лизацию питательных веществ в под¬
золистых почвах.— Вестн. с.-х. науки.
Удобрение, агротехника и агропочво¬
ведение, 19416, № 2.
Ярусов С, С. О применении извести в по¬
ниженных дозах.— Труды ВИУАА,
1948, вып. 28.
Ярусов С. С. Известкование в травополь¬
ном севообороте.— Сов. агрономия,
1950, № 2.
Д. С. ОРЛОВ
МЕТОДЫ ОПРЕДЕЛЕНИЯ pH
И ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОГО
ПОТЕНЦИАЛА ПОЧВ
*
Определение реакции почв относится к числу наиболее распространенных
анализов как при теоретических, так и при прикладных исследованиях почв.
Наиболее полная картина кислотных или основных свойств почв склады¬
вается при одновременном измерении нескольких показателей, в том числе
титруемой кислотности или щелочности (фактор емкости) и величины pH,
равной отрицательному логарифму активности водородных ионов (фактор
интенсивности). Фактор емкости в данном случае характеризует общее со¬
держание кислот или оснований в почвах; от него зависят буферность почв,
устойчивость реакции во времени и по отношению к внешним воздействиям.
Фактор интенсивности характеризует силу мгновенного действия кислот или
оснований на почву и растения; от него зависит поступление минеральных
компонентов в растения в данный отрезок времени. При определенных усло¬
виях по реакции почв можно судить и о некоторых формах кислотности.
Окислительно-восстановительный потенциал (ОВП) почв измеряют пока
реже, чем pH, однако его уровень чрезвычайно важен как для оценки ус¬
ловий питания растений, так и для изучения многих происходящих в поч¬
вах процессов. Величина ОВП, обозначаемая бь, как и величина pH, явля¬
ется фактором интенсивности, а фактор емкости определяется содержанием
веществ, участвующих в окислительно-восстановительных реакциях.
Механизмы кислотно-основных и окислительно-восстановительных реак¬
ций во многом сходны; еще более близки принципы и техника измерения pH
и 6h. Поэтому их удобно рассматривать совместно. Обе величины могут быть
найдены различными независимыми методами, среди которых наибольшее
значение приобрела потенциометрия. Методы колориметрии, еще недавно
широко распространенные, отошли на второй план. В особых (хотя и редких)
случаях применяют специальные приемы и методы, например полярогра¬
фию, спектрофотометрию и т. п.
АКТИВНОСТЬ ВОДОРОДНЫХ ИОНОВ И ШКАЛА pH
Согласно представлениям Брёнстеда — Лаури, кислоты рассматривают¬
ся как доноры протонов, а основания — как вещества, способные присоеди¬
нять протоны. В водных растворах кислоты НА осуществляется реакция:
НА+НгО ^ НзО++А-
которая обычно рассматривается как реакция диссоциации, хотя она вклю¬
чает и гидратацию протона. Реально в таких растворах присутствует не про¬
тон, а ион гидроксоний (или гидроний); согласно расчетам, в 1' М растворах
одноосновных сильных кислот концентрация Сн3о+ = 1 г-ион!л, а Сн+ =
= 10-1зе г-ион/л. Но для удобства и простоты изложения материала вполне
допустимо считать, что реакция среды обусловлена простыми водородными
ионами, а водородный показатель — pH — является функцией концентра¬
ции водородных ионов. Однако в тех случаях, когда важную роль приоб¬
ретают пространственные факторы (в минералогии почв, кристаллохимии
лонного обмена), пренебречь гидратацией протона уже не удается.
246
Д. С. Орлов
Концентрация водородных ионов очень часто бывает малой. Поэтому
ради удобства Зёренсен предложил в практических целях использовать от¬
рицательный логарифм этой концентрации, обозначив его рш. В дальнейшем
этот символ был заменен символом pH, более простым для типографского
набора.
В настоящее время используют разные модификации символа pH в зави¬
симости от способа выражения концентрации раствора и выбранного стан¬
дарта. Если, например, речь идет о нормальной концентрации, пишут рсн
= — lg Сн; в случае моляльной концентрации — ргпц = — 1 g ти. В аг¬
рохимии, почвоведении, биологии обычно пользуются нормальными кон¬
центрациями и под величиной pH подразумевают, как правило, отрицатель¬
ный логарифм активности водородных ионов — ан (их активной, или эффек¬
тивной, концентрации), т. е.
pH = ран]=: —1 gaH = —
Эту единицу предложили также Зёренсен и Линдестрём — Ланг, и хотя она
несколько условна из-ea того, что, строго говоря, коэффициент активности
у отдельно взятого иона определить невозможно, она получила широкое рас¬
пространение.
Величина коэффициентов активности ионов i-то рода по теории Дебая
и Хюккеля определяется уравнением
Аг*У~
-IgTi = 1
i+pVj
где А и р — параметры, зависящие от температуры и диэлектрической по-
стоянной растворителя; z — заряд иона, a J— ионная сила раствора, равная
0,52z\.
Коэффициент активности, таким образом, учитывает концентрацию
раствора, заряды ионов, диэлектрическую постоянную среды, степень гид¬
ратации ионов и влияние других действующих в растворе сил. В резуль¬
тате активность является мерой эффективной, проявляющейся в действии
концентрации, а замена концентрации ионов на их активность позволяет
применять к реальным системам законы, выведенные для идеальных раство¬
ров. Коэффициент активности, как и другие показатели, зависит от способа
выражения концентрации (молярная, моляльная и др.), но различия между
активностями в молярной и моляльной шкалах зависят только от плотности
растворителя. В почвенно-агрохимической практике растворителем, как пра¬
вило, служит вода с плотностью, мало отличающейся от единицы, что в
подавляющем большинстве случаев позволяет пренебрегать различиями
между упомянутыми шкалами.
Однако следует иметь в виду, что почти все почвенно-агрохимические
анализы основаны на молярных концентрациях (число молей в 1 л раствора),
поскольку стандарты и анализируемые растворы (или их аликвоты) доводят¬
ся до определенного объема с помощью мерной посуды. Таким образом, даже
эти обстоятельства могут внести некоторую неопределенность в теоретическое
истолкование величины pH, если не сделаны соответствующие оговорки.
Второй источник неопределенности связан с выбором стандартных со¬
стояний и условий измерения pH. Первая шкала pH была составлена Зёрен-
сеном на основе измерения электродвижущей силы (э.д.с.) элемента:
Pt; Нг, раствор х | солевой мост 10,1 н. каломельный электрод.
В результаты измерений были внесены экспериментальные поправки на
диффузионный потенциал. В данном случае для расчетов нужны стандартные
растворы с известными значениями pH. В качестве таковых были взяты раст¬
Методы определения pH и окислительно-восстановительного потенциала 247
воры HG1 и НС1 + NaCl, концентрация водородных ионов в которых была
рассчитана по степени диссоциации. Концентрацию [Н] других растворов
можно в этом случае определить по известному уравнению
„ „ RT I lHJ
Ест-Е* - F 1п [Н]х ’
где R — газовая постоянная; Т — температура, °К; F — число Фарадея.
Этим способом Зёренсеном была составлена шкала, получившая обозна¬
чение ps Н, которая не является строгой мерой ни концентрации, ни актив¬
ности ионов водорода (Бейтс, 1968). Условность шкалы вытекает из подста¬
новки в уравнение Нернста не активностей, а концентраций, использования
«степени диссоциации» сильного электролита для расчета pH в стандартных
растворах и недостаточного учета диффузионного потенциала.
Теоретические расчеты приводят к выводу о простой связи между шкалой
ps Н и рац:
Ран = Р*н+°№.
Поскольку по указанным выше соображениям ни одна из шкал не может
считаться строго термодинамической, были разработаны условные стандарт¬
ные шкалы активности. Последнее отнюдь не является существенным недо¬
статком, ибо, как говорил Мак-Иннес: «Вероятно во всех случаях, за исклю¬
чением одного из тысячи, вовсе нет необходимости рассматривать значения
pH в понятиях теории растворов, а нужно только принимать числа pH как
характеристику кислотности или щелочности в практической шкале» (цит.
по Бейтсу, 1968, стр. 38). Такой подход полностью отвечает требованиям
агрохимиков и почвоведов и делает вполне достаточным и возможным опре¬
деление pH из следующей формулы:
(Ех—Ee) F
рНя = рН,+ дПп{о ,
где рНв — это pH, приписанный (условно) стандартному раствору; Е* и
Е8 — значения э. д. с. элемента с электродом, погруженным в исследуемую
х и стандартную s жидкости. Очевидно, что электрод должен быть селектив¬
но обратим к Н+-ионам.
Условные стандартные шкалы pH могут быть построены или на основе
условно выбранных потенциалов стандартных полу элементов, или на стан¬
дартных буферных растворах. Последний способ предпочтительнее,посколь¬
ку он легче обеспечивает требуемую воспроизводимость результатов.
Стандартные шкалы, принятые в разных странах, в целом удовлетво¬
рительно согласуются, хотя несколько и различаются. Расхождения состав¬
ляют, как правило, тысячные доли единиц pH и лишь иногда достигают
0,01—0,015. В табл. 1 приведены значения pHs первичных стандартов На¬
ционального бюро стандартов (НБС), принятые в США. Эта шкала отлича¬
ется от шкалы британского стандарта и японского метода не более чем на
0,005—0,013 единиц pH.
Первичные стандарты охватывают довольно узкий диапазон pH, тогда
как в сильнокислой и сильнощелочной средах возникают дополнительные
трудности. Поэтому для надежной работы со стеклянными электродами
используют вторичные стандарты (табл. 2). Вторичные стандарты могут
быть использованы для приближенных измерений, но в точных исследо¬
ваниях их значения pH должны контролироваться по первичным стандар¬
там.
В СССР принята и используется шкала pH, основанная на пяти образцо¬
вых буферных растворах (ГОСТ 10170-62 и 10171-62), в значительной части
она совпадает с американской и британской шкалами. Состав и условия при-
ст]
248
Д. С. Орлов
Таблица 1
Значения pH* первичных стандартов НБС (США)
Температура,
Битартрат
калия, насы¬
щенный
при 25° С
Бифталат
калия, 0,05 т
0,025 т КН2РО.
и 0,0025 т
Na2HPO;
0,008695 т КН,РО<
и 0,03043 т
Na,HP04
0,1 т
бура
0
4,003
6,984
7,534
9,464
5
—
3,999
6,951
7,500
9,395
10
—
3,998
6,923
7,472
9,332
15
—
3,999
6,900
7,448
9,276
20
—
4,002
6,881
7,429
9,225
25
3,557
4,008
6,865
7,413
9,180
30
3,552
4,015
6,853
7,400
9,Ш
35
3,549
4,024
6,844
7,389
9,102
38
3,548
4,030
6,840
7,384
9,081
40
3,547
4,035
6,838
7,380
9,068
45
3,547
4,047
6,834
7,373
9,038
50
3,549
4,060
6,833
7,376
9,011
55
3,554
4,075
6,834
—
8,985
60
3,560
4,091
6,836
—
8,962
70
3,580
4,126
6,845
—
8,921
80
3,609
4,164
6,859
—
8,885
90
3,650
4,205
6,877
—
8,850
95
3,674
4,227
6,886
—
8,833
Таблица 2
Значения pH вторичных стандартов для калибровки стеклянных электродов по методу
британского стандарта
Стандартный раствор
12° С
25° С
38° С
КН3(С204)2* 2НаО, 0,1 М
1,48
1,50
НС1(0,01 М) + КС1 (0,09 М)
—
2,07
2,08
СН3СООН (0,1 М) + CH3COONa (0,1 М)
4,65
4,64
4,65
СН3СООН (0,01 М) + CH3COONa (0,01 М)
4,71
4,70
4,72
КН2Р04 (0,025 М) + Na2HP04 (0,025 М)
—
6,85
6,84
Na2B407-10H20 (0,05 М)
—
9,18
9,07
NaHC03 (0,025 М) + Na2C03 (0,025 М)
—
10,00
—
готовления этих растворов даны по «Справочнику химика», т. III. (М.— JL,
«Химия», 1964).
I. Раствор, содержащий в 1 л при 20° С 12,70 ± 0,02 г тетраоксалата ка¬
лия КНС204*Н2С204*2Н20 (0,05 молъ/л).
II. Насыщенный при 25 °С раствор калия виннокислого кислого
КС4Н5Ов.
III. Раствор, содержащий в 1 л при 20° С 10,21 ± 0,02 г калия фтале-
вокислого кислого КС8Н504 (0,05 моль/л).
IV. Раствор, содержащий в 1 л при 20° С 3,40 ± 0,01 г калия фосфор¬
нокислого однозамещенного КН2Р04 (0,025 моль/л) и 3,55 ± 0,01 г натрия
фосфорнокислого двузамещенного Na2HP04 (0,025 молъ/л).
Методы определения pH и окислительно-восстановительного потенциала 249
V. Раствор, содержащий в 1 л при 20° С 3,81 ± 0,01 г натрия тетрабор-
нокислого Na2B407*10H20 (0,01 моль/л).
Для приготовления образцовых растворов должна применяться дистил¬
лированная вода с удельной электропроводностью не более 2*10“6 ом^смГ1.
Стандартные вещества должны быть квалификации для рН-метрии; сушат
их до постоянного веса при следующих условиях:
Тетраоксалат калия 57 ± 2°
Калий фталевокислый 110 + 5°
Калий фосфорнокислый однозамещенный. ... 110+5°
Натрий фосфорнокислый двузамещенный. . . . 120 ± 5°
Натрий тетраборнокпслый выдерживают до постоянного веса при комнатной тем*
пературе в эксикаторе над смесью влажного хлористого натрия и сахара. Калий вин*
нокислый кислый предварительно не сушат.
Приготовление и хранение образцовых буферных растворов необходимо
проводить так, чтобы в них не попадала углекислота из воздуха (также
аммиак и т. п.). Щелочные растворы надо хранить в предварительно пара¬
финированной стеклянной посуде.
Стандартные буферные растворы можно хранить не более трех месяцев,
а фосфатные — не более двух. В насыщенный раствор виннокислого калия
часто добавляют тимол для предупреждения развития микроорганизмов.
Значения pH образцовых буферных растворов приведены в табл. 3.
Таблица 3
Значения pH образцовых буферных растворов
Темпера¬
тура, ° С
Номер образцового раствора
Темпера¬
тура, °С
Номер образцового раствора
I
II
III
IV
V
I
II
III
iv!
! -
0
1,67
4,01
6,98
9,46
45
1,70
3,55
4,04
6,83
9,04
5
1,67
—
4,01
6,95
9,39
50
1,71
3,55
4,06
6,83
9,01
10
1,67
—
4,00
6,92
9,33
55
1,72
3,56
4,08
6,84
8,99
15
1,67
—
4,00
6,90
9,27
60
1,73
3,57
4,10
6,84
8,96
20
1,68
—
4,00
6,88
9,22
70
1,75
3,59
4,12
6,85
8,92
25
1,69
3,56
4,01
6,86
9,18
80
1,77
3,61
4,16
6,86
8,88
30
1,69
3,55
4,01
6,84
9,14
90
1,80
3,64
4,20
6,88
8,85
35
1,69
3,55
4,02
6,84
9,10
95
1,81
3,65
4,22
6,89
8,83
40
1,70
3,54
4,03
6,84
9,07
В практической работе можно пользоваться образцовыми растворами
для градуировки электродов, хотя с этой целью чаще применяют самые раз¬
нообразные буферные смеси. В табл. 4 приведен состав различных буферных
смесей, позволяющих работать в широком диапазоне pH. Следует только
иметь в виду, что в этой таблице значения даны по шкале Зёренсена, т. е.
в единицах ps Н. Переход к шкале pH может быть осуществлен по формуле
pH s ps Н + 0,04. Отсюда, в частности, вытекает, что если при анализе
экспериментальных данных не указана шкала pH, то уже одно это огра¬
ничивает возможность сопоставления результатов в пределах ± 0,04 еди¬
ниц pH.
250
Д. С. Орлов
Таблица 4
Приготовление буферных растворов
Буферные смеси Кларка и Лвбса (20° С)
1. Смесь V мл 0,2 н. НС1 и 25 мл 0,2 н. RC1 разбавляют водой до 100 мл
V
pH
V
pH
V
pH
47,5
1.0
13,15
1,6
5,3
2.0
32,25
1,2
8,3
1,8
3,35
2,2
20,75
1,4
2. Смесь 50 мл 0,1 М бифталата калия, и V мл 0,1 н. НС1 разбавляют водой до 100 мл
V
pH
У
pH
V
pH
46,70
2,2
26,42
2,8
9,90
3,4
39,60
2,4
20,32
3,0
5,97
3,6
32,95
2,6
14,70
3,2
2,63
3,8
3. Смесь V мл 0,1 н. NaOH и 50 мл 0,1 М бифталата калия разбавляют водой
до 100 мл
У
pH
У
pH 1
У
pH
0,40
4,0
17,70
4,8
39,85
5,6
3,70
4,2
23,85
5,0
43,00
5,8
7,50
4,4
29,95
5’2
45,45
6,0
12,15
4,6
35,45
5,4
4. Смесь V мл 0,1 н. NaOH и 50 мл 0,1 М КН2РО4 разбавляют водой до 100 мл
У
pH
У
pH 1
У
PH
5,70
6,0
23,45
6,8
42,80
7,6
8,60
6,2
29,63
7,0
45,20
7.8
12,60
6,4
35,00
7,2
46,80
8,0
17,80
6,6
39,50
7,4
/
5. Смесь V мл 0,1 н. NaOH и 50 мл 0,1 М НзВОз в 0,1 М КС1 разбавляют водой
до 100 мл
У
pH
У
PH
У
pH
2,61
7,8
12,00
8,6
32,00
9,4
3,97
8,0
16,30
8,8
36,85
9,6
5,90
8,2
21,30
9,0
40,80
9,8
8,50
8,4
26,70
9,2
43,90
10,0
Методы определения pH и окислительно-восстановительного потенциала 251
Таблица 4 (продолжение)
Буферные смеси Колътгофа
1. Смесь V мл 0,1 н. НС1 и 50 мл 0,1 М однозамещенного лимоннокислого
калия разбавляют водой до 100 мл
V
pH
У
pH
V
pH
49,7
2,2
30,2
2,8
10,7
3,4
43,4
2,4
23,6
3,0
4,2
3,6
36,8
2,6
17,2
3,2
2. Смесь V мл 0,1 н. NaOH и 50 мл 0,1 М однозамещенного лимоннокислого калия
разбавляют водой до 100 мл
Ацетатные буферные смеси для области pH 0,65—5,20
Смесь V мл 1,0 н.НС1 и 50 мл 1,0 н. ацетата натрия разбавляют водой до 250 мл
V
pH
V
pH
V
pH
100
0,65
52,5
1,99
42,5
3,79
90
0,75
51,0
2,32
40,0
3,95
80
0,91
50,0
2,64
35,0
4,19
70
1,09
49,75
2,72
30,0
4,39
65
1,24
48,5
3,09
25,0
4,58
60
1,42
47,5
3,29
20,0
4,76
55
1,71
46,25
3,49
15,0
4,95
53,5
1,85
45,0
3,61
10,0
5,20
Фосфатные буферные смеси Мак-Илъвена для области pH 2,2—8,0
Смешивают А мл 0,2 М раствора Na2HPO* и В мл 0,1 М раствора лимонной
кислоты
А
в
pH
А
в
pH
А
в
pH |
1 -
в
pH
0,40
19,60
2,2
6,44
13,56
3,6
10,30
9,70
5,0
14,55
5,45
6,6
1,24
18,76
2,4
7,10
12,90
3,8
10,72
9,28
5,2
15,45,
4,55
6,8
2,18
17,82
2,6
7,71
12,29
4,0
11,15
8,85
5,4
16,47
3,53
7,0
3,17
16,83
2,8
8,28
11,72
4,2
11,60
8,40
5,6
17,39
2,61
7,2
4,11
15,89
3,0
8,82
11,18
4,4
12,09
7,91
5,8
18,17
1,83
7,4
4,94
15,06
3,2
9,35
10,65
4,6
12,63
7,37
6,0
18,73
1,27
7,6
5,70
14,30
3,4
9,86
10,14
4,8
13,22
6,78
6,2
19,15
0,85
7,8j
13,85
6,15
6,4
19,45
0,55
8,0
гъ2
Д. С. Орлов
Таблица 4 (окончание)
Буферные смеси Зёренсена (18° С)
Смешивают А мл 0,05 М Na2B*07 и В мл 0,1 н. НС1
А
в
pH
А
в
pH
А
в
pH
А
в
pH
5,25
4,75
7 ,62
6,0
4,0
8,29
7,5
2,5
8.80
9,0
1,0
9,09
5,5
4,5
7,94
6,5
3,5
8,51
8,0
2,0
8,91
9,5
0,5
9,17
5,75
4,25
8,14
7,0
3,0
8,68
8,5
| 1.5
9,01
10,0
0
9,24
Смешивают А мл 0,0b М ЫагШО? и В мл 0,1 н. NaOH
А
в
pH
А
в
pH
А
в
pH
А
в
pH
10,0
0
9,24
8,0
2,0
9,50
6,0
4,0
9,97
4,0
6,0
12,37
9,0
1,0
9,36
7,0
3,0
9,68
5,0
5,0
11,07
Универсальные буферные смеси для области pH 1,81—11,98 (18° С)
К 100 мл смеси равных объемов 0,04 М Н3РО4, 0,04 М СНзСООН и 0,04 М НзВОз
добавляют V мл 0,2 н. NaOH
У
PH
У
pH
У
pH
У
pH
0
1,81
25,0
4,10
50,0
6,80
75,0
9,62
2,5
1,89
27,5
4,35
52,5
7,00
77,5
9,91
5,0
1,98
30,0
4,56
55,0
7,24
80,0
10,38
7,5
2,09
32,5
4,78
57,5
7,54
82,5
10,88
10,0
2,21
35,0
5,02
60,0
7.96
85,0
11,20
12,5
2,36
37,5
5,33
62,5
8,36
87,5
11,40
15,0
2,56
40,0
5,72
65,0
8,69
90,0
11,58
17,5
2,87
42,5
6,09
67,5
8,95
92,5
11,70
20,0
3,29
45,0
6,37
70,0
9,15
95,0
11,82
22,5
3,78
47,5
6,59
72,5
9,37
97,5
100,0
11,92
11,98
ЭЛЕКТРОДЫ ДЛЯ ОПРЕДЕЛЕНИЯ pH
Для потенциометрического определения pH в почвенных вытяжках, сус¬
пензиях, растворах, а также в биологических средах и других объектах при¬
годны любые индикаторные электроды, потенциал которых прямо или кос¬
венно зависит от активности водородных ионов или является функцией ве¬
личины pH: е = / (pH). Вместе с тем электрод должен быть достаточно се¬
лективен, т. е. на его потенциал не должны заметно влиять другие компонен¬
ты изучаемой системы. Наибольшее практическое значение имеют в этом от¬
ношении водородный, хингидронный и стеклянный электроды. Благодаря
развитию потенциометрической техники определение pH со стеклянным
электродом стало основным методом в агрохимических и других исследова¬
Методы определения pH и окислительно-восстановительного потенциала 25&
ниях. Хингидронный электрод употребляют лишь при отсутствии ламповых
потенциометров. Водородный электрод редко применяется для непосредст¬
венных аналитических целей, но он остается основным стандартным элект¬
родом.
Водородный электрод (рис. 1, А). Потенциал водородного электрода
обусловлен реакцией:
Н2 ^ 2Н+ + 2 в;
величина его может быть выражена уравнением
НТ
е — —2,303 pH или, при 20°С, е = —0,0581 pH.
Потенциал нормального водородного электрода принимается равным нулю
при любых температурах, а его величина лежит в основе шкалы электрод¬
ных потенциалов.
Если имеется электрод сравнения с точно известным потенциалом, то
измеренная величина потенциала водородного электрода позволяет непос¬
редственно вычислить величину pH. При 20е С значение pH находят:
Рн = 2-Д’с-~Ч
F 0,0581 ’
где э.д.с. — электродвижущая сила элемента, состоящего из электрода срав¬
нения с потенциалом еср, и водородного электрода.
Коэффициент 2,303 RT/nF, входящий в вышеприведенное уравнение,
является коэффициентом уравнения Нернста и используется во всех анало¬
гичных формулах. Его величина при различных температурах приведена
в табл. 5.
Таблица 5
Значения коэффициента Ф = 2,303 RT/F в
*,° с
ь
t, °С
&
<, °С
а
t, °с
&
5
0,0551
14
0,0569
23
0,0587
32
0,0605
6
0,0553
15
0,0571
24
0,0589
33
0,0607
7
0,0555
16
0,0573
25
0,0591
34
0,0609
8
0,0557
17
0,0575
26
0,0593
35
0,0611
9
0,0559
18
0,0577
27
0,0595
36
0,0613
10
0,0561
19
0,0579
28
0,0597
37
0,0615
И
0,0563
20
0,0581
29
0,0599
38
0,0617
12
0,0565
21
0,0583
30
0,0601
39
0,0619
13
0,0567
22
0,0585
31
0,0603
40
0,0621
Для приготовления водородного электрода берут гладкую платиновую
проволоку или пластинку, впаянные в стеклянные трубки. Поверхность
платины тщательно очищают и промывают горячим раствором хромовой
смеси, а затем дистиллированной водой. Промытый водой электрод остав¬
ляют в чистой дистиллированной воде на ночь. Затем приступают к плати¬
нированию электрода. С этой целью в 100 мл воды растворяют ,2—3 г PtCl4
и 0,02 г (СН3СОО)2РЬ. В этот раствор пускают подлежащий платинированию
электрод и соединяют его с отрицательным полюсом аккумулятора на 2—4 в.
Анодом служит также платиновая пластинка или проволока. Через раствор
пропускают ток, регулируя его силу таким образом, чтобы на аноде не¬
прерывно, но не слишком быстро, выделялись пузырьки газа. Через 4—6
мин. поверхность электрода покрывается ровным бархатисто-черным слоем
254
Д. С. Орлов
Рис. 1. Индикаторные электроды для измерения pH
А — грушевидный водородный электрод: 1 — платинированная платиновая проволока; 2 — раствор;
3 — винтовой контакт. В — хингидронныйХэлектрод в сосуде Михаэлиса: 1 — платиновая пластинка;
г — раствор; з — винтовой контакт. В — датчик со стеклянным электродом к потенциометру ППМ«
03М1: 1 — шарик из электродного стекла; 2 — внутренний вспомогательный электрод; 3 — электроли¬
тический ключ; 4 — отверстие для доливки раствора, закрытое резиновой пробкой; 5 — асбестовая нить;
б — электрод сравне шя; 7 — колпачок; 8 — соединительный кабель. Г — стеклянный электрод типа
ЭСЛ-417-04: 1 — корпус; 2 — шарик из электродного стекла; 3 — хлорсеребряный электрод; 4 — раст¬
вор; 5 — выводной провод; в — наконечник. Д — стеклянный электрод типа ЭСЛ-417-05: 1 — корпус;
2 — шарик из электродного стекла; з — хлорсеребряный электрод; 4 — раствор; 5 — выводной кабель;
в — штеккер. Е — стеклянный электрод типа ЭСЛ-40-07: 1 — корпус; 2 — шарик из электродного стек¬
ла; з — вспомогательный электрод; 4 — раствор; 5 — выводной кабель; 6 — штеккер; 7 — экран
губчатой платины. Платинированный электрод промывают дистиллированной
водой и снова подвергают катодной поляризации, но уже в 0,05 н. H2S04,
чтобы удалить адсорбированные ионы С1. Готовый электрод тщательно про¬
мывают дистиллированной водой и хранят погруженным в воду (рис. 1).
Для проведения измерений в электродный сосуд наливают буферный
раствор с известным значением pH или исследуемый раствор с неизвестным
pH так, чтобы полностью покрыть раствором платинированную поверхность.
Методы определения pH и окислительно-восстановительного потенциала 255
Этот раствор насыщают водородом, пропуская последний с такой скоростью,
чтобы можно было считать пузырьки газа. После насыщения водородом из¬
меряют потенциал электрода; затем повторно насыщают водородом и пов¬
торяют всю процедуру до тех пор, пока не получат двух постоянных зна¬
чений э. д. с.
Необходимый для насыщения электрода водород получают с помощью
аппарата Киппа. С этой целью в аппарат Киппа загружают гранулирован¬
ный металлический цинк и наливают 25 %-ную H2S04. Для очистки выделяю¬
щийся водород пропускают через склянки Тищенко или Дрекселя, после¬
довательно заполненные следующими растворами: 1) 5%-ный КМп04 в
5 %-ной H2S04; 2) 10%-ный РЬ(СН3СОО)2; 3) 10%-ный раствор пирогаллола
в 10 %-ном КОН; 4) 0,1 н. H2S04.
В нормально работающем аппарате Киппа серная кислота покрывает
только нижний слой гранулированного цинка. Выделяющийся в результате
бурной реакции водород поддерживает требуемое давление. Если кислота
полностью покрывает слой цинка, а реакция идет вяло, то аппарат Киппа
необходимо перезарядить. Свежезаряженному аппарату Киппа необходима
дать поработать вхолостую 15—20 мин., чтобы полностью заместить водоро¬
дом находившийся в нем воздух.
Теоретически водородный электрод пригоден для измерения pH в диа¬
пазоне от 0 до 14. Однако этот электрод очень чувствителен к присутствию
многих соединений, часто встречающихся в почвах и биологических средах.
Присутствие сильных окислителей и восстановителей искажает показания
электрода; отрицательно влияют соли азотной, хромовой, хлорноватой кис¬
лот, соединения закисного и окисного железа, сера, сульфиды, сулема, ам¬
миак, амины. Большие ошибки возникают в присутствии поверхностно-ак¬
тивных и коллоидных веществ. Особенно сильное влияние можно ожидать в
почвенных суспензиях, всегда содержащих гумусовые вещества и минераль¬
ные коллоиды. Все это заставляет пользоваться водородным электродом
преимущественно для целей стандартизации. В частности, при проверке
потенциалов электродов сравнения (полуэлементов).
Хингидронный электрод (см. рис. 1, Б). Хингидронный электрод пред¬
ставляет собой платиновую пластинку или проволоку в насыщенном раство¬
ре хингидрона. Строго говоря, его потенциал является потенциалом окис-
лительно-восстановительной системы:
СвШОГ ^ СвН402+2е .
анион хинои
гидрохинона
Но поскольку концентрация аниона гидрохинона зависит от активности
водородных ионов, то в конечном итоге потенциал электрода является функ¬
цией pH. В полной форме потенциал хингидронного электрода можно вы¬
разить уравнением:
ИТ ац+ “I" ХхК2
е = е° + ~^Г1п кЛ1 ’
где е0 — потенциал нормального хингидронного электрода, а Кг и К2 —
первая и вторая константы диссоциации гидрохинона. Относя величину
КХК2 из знаменателя в постоянную уравнения и пренебрегая малыми вели¬
чинами Кхап+ и КХК2 в числителе, получим:
е = ео — ФрН.
Хингидронный электрод, как и водородный, имеет ряд ограничений.
Прежде всего предел измеряемых с его помощью величин pH ограничен
величинами pH около 8. Это обусловлено как упомянутыми выше упроще¬
ниями при выводе формулы, так н окислением гидрохинона в щелочной среде
кислородом воздуха.
256
Д. С. Орлов
Большие ошибки могут быть вызваны присутствием окислителей и вос¬
становителей, что и понятно, так как потенциал электрода по своей природе
является окислительно-восстановительным. По указанным причинам хин¬
гидронный электрод нельзя применять для анализа щелочных почв с pH бо¬
лее 8 (солонцы, содовые солончаки) идляглеевыхилисильнооглеенныхпочв.
Несмотря на указанные недостатки, хингидронный электрод еще совсем
недавно был основным электродом при определении реакции почв, а в не¬
которых случаях он используется еще и сейчас. Как правило, он находит
применение в полевых условиях, когда нет возможности воспользоваться
электронными потенциометрами. В полевых условиях он может быть иногда
весьма удобен и как электрод сравнения, например при измерении окисли¬
тельно-восстановительных потенциалов почв.
Стеклянный электрод (см. рис. 1,2?—Е). Стеклянный электрод меньше все¬
го зависит от состава раствора, не отравляется как водородный электрод, не
боится присутствия окислителей и восстановителей и пригоден для работы
в значительно более широком интервале pH, чем хингидронный электрод.
Он прост в обращении и удобен в работе. По этим причинам стеклянный
электрод стал сейчас основным индикаторным электродом при измерениях
pH, хотя он и требует обязательной градуировки.
Распространению стеклянных электродов немало способствовало созда¬
ние современных электронных потенциометров. Использовавшиеся в началь¬
ный период конденсаторные и баллистические схемы с зеркальными гальва¬
нометрами были крайне неудобны в работе, что долгое время сдерживало
внедрение стеклянных электродов в практику почвенно-агрохимических ра¬
бот. Сейчас эти трудности полностью преодолены.
Теория возникновения скачка потенциала на границе стекло — раствор
довольно сложна. Сейчас установлено, что стеклянные электроды можно
рассматривать как катионообменные мембраны, высокоселективные к раз¬
личным катионам. В зависимости от состава стекло может обладать различ¬
ными функциями: или простой Н+-функцией, или функциями щелочных,
щелочноземельных и даже органических катионов («Ионоселективные
электроды», 1972).
Электроды, чувствительные к водородным ионам, были открыты в начале
века Кремером и Габером и Клеменсиевичем. Общая теория их была разра
ботана Б. П. Никольским, затем развита М. М. Шульцем и Дж. Эйзенманом.
Согласно взглядам Б. П. Никольского, в простом случае потенциал стеклян¬
ного электрода может быть выражен уравнением
е = во + fllg (ан+ + ATaNa+),
где К — константа обмена ионов Н+ и Na+ между стеклом и раствором.
Если Капа+ ^ Дн+, то
е ^ео + 01gaH+ = во ~ *>рН.
Аналогично рассматриваются уравнения и для электродов с другими функ¬
циями. Функция электрода определяется составом стекла и в известных
пределах может быть предсказана теоретически.
К недостаткам стеклянного электрода относится потенциал асимметрии,
который может быть сведен к минимуму длительным вымачиванием. Второй
недостаток связан с тем, что всякий стеклянный электрод до некоторой сте¬
пени полифункционален, как это, например, видно из приведенного выше
уравнения. Поэтому интервал измеряемых величин pH и прямолинейность
функции е — pH зависят от состава стекла и состава исследуемого раствора.
Наконец, к числу недостатков электрода следует причислить и высокое оми¬
ческое сопротивление.
Промышленность выпускает большой набор простых и комбинирован¬
ных стеклянных электродов, а состав стекол меняется в довольно широких
Метода определения pH и окисли телно-восстановительного потенциалй 257
Таблица 6
Состав некоторых электродных стекол, мол.%
Стекло
SiO,
LifO
С8,0
LfltOi
BaO
Na,0
97В
69
24
3
4
УСТ
65
26
—
—
5
4
20
69
27
2
2
—
—
пределах. Состав некоторых стекол приведен в табл. 6 (по Сердобольскому,
1965).
Стеклянные электроды различаются не только по составу стекол, но и
многими другими характеристиками: формой, размерами, типом вспомога¬
тельного электрода, гибкого вывода и т. п. Конструктивно стеклянный элек¬
трод чаще всего изготовляют в виде шарика, толщина стенок которого сос¬
тавляет 0,05—0,1 мм; шарик выдувается на конце стеклянной трубки. Стек¬
лянный электрод заполняют 0,1 н. НС1 или подходящим буферным раствором.
В этот раствор погружают вспомогательный стандартный электрод (чаще
всего хлорсеребряный), вывод от которого подсоединяется к потенциометру.
В комбинированных электродах внутри стеклянного электрода размещают
и каломельный полуэлемент сравнения. Контакт полуэлемента с раствором
осуществляется через микротрещину. В этом случае выводной кабель закан¬
чивается штекером для подключения к потенциометру.
Характеристики некоторых типов электродов отечественного и зарубеж¬
ного производства приведены в табл. 7 и 8. Наименование типа электрода,
приведенное в табл. 7, включает следующие обозначения. Первые три буквы
обозначают тип и назначение электрода (ЭСЛ — электрод стеклянный лабо¬
раторный). Четвертый знак указывает марку электродного стекла (1 — стек-
Таблица 7
Характеристики некоторых стеклянных электродов отечественного производства
Тип электрода
Координаты изопотенциаль-
ной точки относительно
насыщенного хлорсереб-
ряного электрода сравнения
Темпера¬
турный
ЯНТбрВЯЛ)
Пределы
измерения
pH при 20° С
Электрическое
сопротивление
при 20° С,
Кртгнэна
характерис¬
тики, мв/рН
pH
Е, мв
° с
Мом
ЭСЛ-41Г-04
ЭСЛ-41Г-05
ЭСЛ-11Г-04
ЭСЛ-И Г-05
-33 ±24
-5; 40
0-12,7
50 ±40
59,1 ±0,5
3,28 ±0,2
15-100
—0,5; 12,7
500±250
59,1 ±0,5
Стеклянный Б
и стеклянный
М
—
—
0-40
0-12,7
50 ±40
—
ЭСЛ-43-07
ЭСЛ-63-07
ЭСЛ-44Г-04
7.0 ±0,2
7.0 ±0,2
4,15
—10 ±12
—10 ±12
—184 ±24
0-50
20—100
10-30
0-12
0-14
0-12,7
50 ±40
700 ±300
50 ±40
59.1 ±0,5
59.1 ±0,5
59.1 ±0,5
9 Агрохимические методы исследования почв
258
Д. С. Орлов
Таблица 8
Характеристики стеклянных электродов зарубежного производства
(по Бейтс, 1968)
Показатели
Бекман
общего назначения
Бекман
Е-2 Амбер
Лидс и Норструп
лабора¬
торные
промыш¬
ленные
лабора¬
торные
промыш¬
ленные
№ 44, 49
36, 33
№ 30,
50, 37, 32
Название стекла
Прозрачное
Синее или янтар¬
ное
Синяя
точка
Червая
точка
Температурный диапазон, °С
от —5 I
до 50
от —5
до 100
15-50 I
от —5
до 100
0-30
20-60
Диапазон pH в 1 н. растворе
Na+ при 25° С
0-
10
0-12,5
0,2-9
0,2-14
Внутренний электрод
Hg, HgCl04 или
Hg, HgCl04 или
Hg,
Hg,
Ag, AgCl
Ag, AgCl
Hg2Cl2
Hg2ci*
pH при Е — 0
7,7 или 7,0
7,7 или 7,0
-7
~7
Состав стекла
Li20, BaO, Si02
Li20, BaO, Si02
Na20,
CaO, Si02
Li20T
La203,
SiOs
Сопротивление при 25° С,
Мом
50-200 |
10-100
100—300|
50-150
30-100
200-1000
ло УСТ, 4 — стекло № 20 и т. д.). Пятый знак — тип вспомогательного
электрода (1 — хлорсеребряный; 3 — то же, в ацетатном буферном растворе
и т. д.). Шестой знак указывает на особенности заполняющего раствора.
Буквой Г обозначены морозостойкие до —25° С растворы. Седьмой и вось¬
мой знаки обозначают типы конструктивного исполнения.
Устройство современных промышленных электродов ясно из рис. 1. Та¬
кие электроды поступают в продажу с подробным описанием устройства ж
паспортом, в котором приведены все важнейшие характеристики.
Стеклянные электроды для измерения pH нетрудно и самостоятельна
изготовить в любой лаборатории, но мы не приводим соответствующих ре¬
комендаций, так как они были изложены ранее (например, Зырин, Орлов,
1964), а промышленность обеспечивает практически полностью потребности
в электродах всех производственных и научно-исследовательских организаций.
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЙ ПОТЕНЦИАЛ ПОЧВ
Окислительно-восстановительный потенциал (ОВП) служит мерой нап¬
ряженности окислительно-восстановительных процессов, а его уровень от¬
ражает преобладание процессов окисления или восстановления, протекаю¬
щих в почвах. Любая почва (как и произвольная химическая система) содер¬
жит одновременно окисленные и восстановленные формы соединений, соот¬
ношение которых и создает экспериментально обнаруживаемый уровень
ОВП. Величину ОВП обозначают гп и ее можно формально выразить или
через концентрацию в системе свободных электронов, или через отношение
активных концентраций окисленной (Ох) и восстановленной форм (Red);
например, для системы Ох + пе Red:
RT «о\
8h-«0+ nF ln а
Red
где е0 — нормальный потенциал данной окислительно-восстановительной
систэмы, определяемый условием aQX : aRed = 1; п — число электронов,
Методы определения pH и окислительно-восстановительного потенциала 259
участвующих в реакции; R — газовая постоянная; Т — абсолютная тем¬
пература; F — число Фарадея.
При 20° С уравнение можно записать в виде:
0,0581 t « х
Это то же уравнение, что и в случае потенциалов электродов, обратимых
к Н+-ионам, но только учитывающее две — окисленную и восстановленную —
формы соединения. Отсюда, в частности, следует, что нельзя говорить об
ОВП какого-либо вещества, а можно рассматривать только потенциал сис¬
темы (реакции). Например, в кислой среде перманганат-ион создает потен¬
циал ~|-1,52в, если восстанавливается до Мп2+:
Мп(Г+5е+8Н+ ^ Мп,++4Н20.
Тот же ион может дать и более высокий потенциал, например в реакции
МпО~+Зе -|-4Н+ Мц0*(тв)+2Н,0, е0 = 1,67
Если же в слабокислой среде ион перманганата восстанавливается до
манганата:
МпСГ+е ^ МпО*~,
4 1 4
то потенциал такой системы составляет всего +0,54 в.
Таким образом, мы должны рассматривать конкретные окислительно¬
восстановительные системы. При одновременном присутствии нескольких
окислительно-восстановительных пар (систем) потенциал приобретает не¬
которое среднее значение. В почвах одновременно сосуществуют многие'
пары окисленных и восстановленных компонентов. В их число входят:
Fe$+—Fe,+, Mn4+—Mn*+—Mn‘+, Cu*+—Cu+,
SOr~H,S, NOr-NOr—NH„ C0,-CH4.
Почти каждая такая система в чистом виде обратима, однако в почвах,
когда главными факторами, изменяющими окислительное состояние, явля¬
ются микробиологическая деятельность и кислород воздуха, об обратимости
окислительно-восстановительных процессов не приходится и говорить.
Многие авторы неоднократно отмечали, что измеренные в почвах окисли¬
тельно-восстановительные потенциалы количественно не могут быть оха¬
рактеризованы равновесным состоянием в соответствии с уравнением Нерн-
ста (например, Bohn, 1971). В то же время наблюдались некоторые удовлет¬
ворительные полуэмпирические зависимости. Например, устойчивые связи
между величинами ОВП, pH и [Fe2+] удалось выразить уравнением:
zh = 1,058 — 0,059 lg [Fe,+] - 0,177 pH,
\
причем было найдено, что равновесие определялось метастабильными фор¬
мами соединений железа (Ponnamperuma, Tianko, Loy, 1967). Вообще мно¬
гие авторы считают, что ОВП в почвах определяется соотношением ферри —
ферро (Breemen, 1969). Некоторые полуэмпирические зависимости были
установлены также И. П. Сердобольским (см. Зырин, Орлов, 1964).
Величина потенциала в почвах, как нормального, так и равновесного,
зависит не только от соотношения активностей окисленных и восстановлен¬
ных форм соединений, но также от величины pH и способности рассматри¬
ваемых компонентов давать комплексные или труднорастворимые соедине¬
ния.
Длительное время в почвоведении и агрохимии существовало представле¬
ние, что изменение pH почвы на единицу вызывает сдвиг ОВП на 58 мв (при
9*
260
Д. С. Орлов
20° С). В действительности существуют более сложные связи, что было вы¬
ведено теоретически и подтверждено экспериментально. Реальная степень
влияния pH на величину зависит от числа водородных ионов, участвую¬
щих в окислительно-восстановительной реакции. Так, для вышеприведенной
реакции Mn04“ zf Мп2+ потенциал выразится уравнением
. .0,0581 омпо;вн+ 0,0581, «МпО^ 0,0581-8
•»-S+-5— U— е„ + —— te „мо„ + J—Ц.н.=
0,0581 , аМпо7 0,0581-8
= е»+ “5“:5— Р«-
Обозначая число участвующих в реакции электронов через пу а число
ионов водорода через /п, можем записать:
еь = ео+^42^-^РН-
и «Red п
Если водородные ионы не участвуют в реакции, то т = 0 и весь послед¬
ний член уравнения обращается в нуль. Таким образом, вопрос о влиянии
реакции среды на ОВП должен решаться конкретно в каждом отдельном
случае.
Столь же конкретный анализ требуется и тогда, когда необходимо уста¬
новить преобладание в почве элемента той или иной степени окисления.
Например, если нормальный потенциал системы ферри — феро равен +0,77 в,
то в почвенных условиях при потенциалах порядка 500—600 мв можно
было бы ожидать преобладания ионов закисного железа над окисным. На
самом деле накопление закисного железа в заметных количествах происхо¬
дит только при потенциалах ниже 400—450 мв. Последнее объяснимо обра¬
зованием комплексных соединенйй. Комплексы с Fe3+, возникающие в поч¬
вах, вероятно более прочны, чем комплексы с Fe2+, и поэтому нормальный
ОВП комплексных соединений железа ниже, чем для простых ионов. Такое
явление имеет место, например, в случае хорошо известной системы
Fe(CN)e8“/Fe(CN)e4“, значение е0 для которой равно +0,36 в. При рассмот¬
ренных условиях обычно наблюдаемые в автоморфных почвах потенциалы
должны приводить к количественному преобладанию соединений окисного
железа, что обычно и наблюдается.
Значения ОВП в автоморфных почвах колеблются в довольно уз¬
ких пределах. Резкие сдвиги в сторону развития восстановительных процес¬
сов может вызвать временное переувлажнение почв, особенно при избытке
органических остатков.
В дерново-подзолистых целинных и пахотных почвах величины обычно
находятся в пределах 450—550 мв, причем максимальные значения нередко
приурочены к горизонтам ВА и В2, тогда как в пахотных (или Ах) горизон¬
тах с повышенным содержанием органического вещества величины не¬
сколько понижены. Как абсолютные значения потенциалов, так и их профиль¬
ное распределение зависит от погодных условий. Длительное переувлажне¬
ние снижает ОВП, причем особенно сильно в оглеенных разностях почв. По
данным В. Н. Кураева (1969), в оглеенных разностях дерново-подзоли¬
стых почв величины eh снижаются в периоды избыточной влажности до 200—
250 мв. Кратковременное увлажнение за счет проходящих дождей, по нашим
данным, существенно не влияет на потенциал, но все же в периоды гроз
возможны резкие падения ОВП в верхнем слое почвы с последующим быст¬
рым возвращением к первоначальному уровню.
Значения eh тесно связаны с микро- и мезорельефом. В понижениях
условия менее окислительные; особенно сильно снижаются потенциалы в
понижениях на периферии торфяников и торфяно-болотных почв, что под¬
Методы определения pH и окислителъно-восстановителъносс потенциала 261
тверждают также наблюдения И. С. Кауричеваи B.C. Шишовой (1967). При¬
мерно тот же уровень окислительно-восстановительного потенциала, что и
в дерново-подзолистых почвах, свойствен серым и бурым лесным почвам, но
по мере обогащения их гумусом величина eh может несколько снижаться,
хотя абсолютные сдвиги потенциалов невелики. Даже в южных черноземах
в течение всего вегетационного периода диапазон окислительно-восстанови¬
тельных потенциалов не превышал 420—650 мв (Вишневская, Емельянов,
1970). Те же пределы потенциалов найдены для непереувлажненных темно¬
каштановых почв, солодей, коричневых и лугово-коричневых лесных почв,
красноземов и желтоземов, хотя иногда публикуются и резко пониженные
величины; причина последнего пока остается неясной, но возможно, что это
связано со способом расчета величин ел (Панов, Шардаков, 1968 а,б; Латария,
1967; Тотев, Трънков, 1964; Шишов, 1969).
Диапазон окислительно-восстановительных потенциалов почв в течение
года может быть значительно шире, чем за время вегетационного периода.
На псевдоглеевых почвах величины ел, например, колебались от 250—280
до 590—690 мв (Resulovic, 1970).
Общий окислительно-восстановительный режим большинства обычных
автоморфных пахотных и целинных почв характеризуется сравнительно
равномерными во времени и оптимальными по величине потенциалами.
Резкие отклонения от таких величин при ярко выраженной динамике свой¬
ственны болотным почвам, многим почвам пойм и орошаемым (или затопля¬
емым) почвам. Именно в таких почвах измерение ОВП представляет наи¬
больший интерес. Установлено, что на различных почвах под культурой
риса ОВП довольно быстро снижается до 350—400 мв, а при длительном стоя¬
нии воды величины eh приобретают даже отрицательные значения. Минималь¬
ные значения, судя по модельным опытам с добавлением глюкозы, могут
быть порядка —200, —250 мв (Баженов, Рубцова, 1969; Ларешин, Каури-
чев, 1969; Степанец, Самолина, 1968; Ларешин, Илюхин, 1970; Горшкова,
Дементьева, 1971). По единичным наблюдениям, конечный eh при затопле¬
нии рисовых почв с pH 7 достигает —320 лее. Снижение потенциала в затопляе¬
мых почвах стимулируется высоким содержанием органического вещества,
внесением растительных остатков, например, при запашке рисовой соломы
(Востров, Алешин, Долгих, 1969). Внесение растительных остатков, снижаю¬
щее ОВП, может ухудшить развитие корневых систем и снизить урожай
(Кураев, 1967).
Близкую картину дают многие почвы поймы. Если эти почвы связаны с
близко расположенными грунтовыми водами, то ОВП в них понижается
до 400—450 лее, а избыток органических веществ, встречающийся на перифе¬
рии торфяников, способствует снижению потенциалов до 200—300 мв и ниже.
Перепад потенциалов от 350 до 450 мв служит обычно показателем зоны,
в которой совершаются переходы Fe2+ ^ Fe3+ (Орлов, Розанов, Саакян, 1970;
Patrick, Turner, 1968). Хотя в других исследованиях область перехода окиси
железа в закись связывается с потенциалом 200 мв (Patrick, 1964), такого рода
разночтения могут быть вызваны влиянием органического вещества и реак¬
циями комплексообразования, о чем говорилось выше. Одна из вероятных
причин быстрого падения потенциала почв, богатых органическим вещест¬
вом,— образование Н2 за счет разложения легкорастворимых углеводов
РУатапе, Sato, 1968). Те же причины могут играть главную роль и на пери¬
ферии торфяников.
В наибольшей степени от ОВП почв зависит поведение соединений азота,
железа, марганца и серы, формы которых прямо связаны с направленностью
окислительно-восстановительных процессов (см. Зырин, Орлов, 1964). Од¬
нако не меньшую роль может сыграть и косвенное действие окислительно¬
восстановительного режима на свойства почв. Например, снижение ОВП
повышает растворимость почвенных фосфатов, видимо, за счет большей ра¬
262
Д. С. Орлов
створимости фосфатов записного железа по сравнению с окисным (Нгуен Ви,
Алешин, 1966). Положительное влияние затопления на подвижность фосфа¬
тов отмечал Ю. И. Ежов (1968).
Общие вопросы измерения ОВП изложены в ряде учебных пособий, ста¬
тей и монографий (Соломин, 1964; Захарьевский, 1967; Зырин, Орлов, 1964).
Исследование окислительно-восстановительных процессов не ограничивает¬
ся, конечно, простыми измерениями ОВП, но эти измерения — первый шаг
такого исследования.
ЭЛЕКТРОДЫ ДЛЯ ИЗМЕРЕНИЯ ОВП
Для измерения окислительно-восстановительных потенциалов исполь¬
зуют различные электронно-обратимые химически инертные электроды.
Это могут быть графитовые, платиновые или специальные стеклянные элек¬
троды. Кроме того, используются методы фотобумажной автографии, кото¬
рые дают четкое представление о расположении восстановленных зон и
скорости накопления восстановленных продуктов (Востров, 1970; Востров,
Алешин, Долгих, 1969). Многие полезные сведения о распределении в поч¬
венном профиле восстановленных продуктов можно получить с помощью
простых цветных реакций. Для оценки степени восстановления рисовых
почв предложен метод осаждения сульфида германия на синтетической бу¬
маге (Утома, 1972). Однако основным приемом остается измерение ОВП с
помощью различных электродов. Эти измерения дают количественную меру
окислительно-восстановительных процессов и позволяют следить за дина¬
микой процессов.
Графитовые электроды. Графитовые электроды не получили пока широ¬
кого распространения. Имеются сведения, что ОВП, измеренные графито¬
выми электродами, удовлетворительно характеризуют соотношение между
FeO и Fe203 (Кауричев, Тарарин, Тарарина, 1969), хотя Бон (Bohn, 1968)
нашел, что графитовый (как и золотой) электрод нестабилен и не отражает
изменения окислительно-восстановительных условий в почве. Противопо¬
ложное мнение высказали Г. М. Шпейзер и JI. А. Чарчиди (1970), согласно
которым графитовые электроды, сохраняемые в воде или в 0,1 н. КС1, дают
стабильные результаты, а время установления потенциала не превышает
5—10 мин. Методические исследования применительно к графитовым элект¬
родам не завершены и окончательные рекомендации были бы преждевремен¬
ны; многое может зависеть от качества самих электродов: их размеров, пори¬
стости, чистоты и т. п.
Платиновые электроды. Обычные платиновые электроды являются клас¬
сическими датчиками при измерении ОВП и рекомендованы многими авто¬
рами. В последнее время появляется все больше работ, в которых рекомен¬
дуется проводить измерения с помощью платиновых электродов, постоянно
погруженных в почву. Высказывались, правда, мнения, что на таких элект¬
родах происходят химические реакции, снижающие электропроводность и
чувствительность (Bailey, Beauchamp, 1971). Однако при исследовании па¬
хотных дерново-подзолистых почв нам не удавалось наблюдать каких-либо
изменений (отложений) на поверхности электродов, более месяца находив¬
шихся в почвенном слое. Показания таких электродов в стандартных раство¬
рах не отличались от показаний свежеизготовленных электродов. Снижение
чувствительности, вообще говоря, не представляет существенной опасности
при потенциометрических способах измерений; существенные ошибки могли
бы возникать за счет различных пленок на поверхностях электродов, но
такие пленки нами не наблюдались. Следует также отметить, что высказы¬
вались предположения о том, что в разбавленных растворах и почвенных
суспензиях потенциал инертных электродов из платины, золота или графи¬
та является смешанной функцией pH и eh (Bohn, 1969).
Методы определения pH и окислительно-восстановительного потенциала 263
ж
в
слительно-восстановительного потенци¬
ала
А — игольчатый;
В — копьевидный;
В — платинированный.
1 — платиновая проволока;
2 — платиновая пластина;
3 — медная проволока;
4 — пробка из менделеевской
замазки;
5— ртуть;
I— платиновое покрытие;
S— ватная пробка.
I — датчик для измерения
ОВП к потенциометру
ППМ-03М1:
1 — платиновая пленка;
2 — корпус;
8 — асбестовая нить;
4 — внутренний полуэлемент;
5,6 — резиновые пробки;
7 — насыщенный раствор КС1;.
8 — асбестовая нигь;
(— отверстие для долива рае-
твора, закрытое пробкой;
ю — колпачок;
и — соединительный кабель
Подробные рекомендации по использованию платиновых электродов дали
Ямане и Сато (Yamane, Sato, 1968). По их данным, платиновые электроды
должны быть одинаково обработаны перед началом измерения, включая вре¬
мя прокаливания и вид пламени. Рекомендуются электроды с площадью не
менее 100 деле2, причем измерения должны проводиться через 24 часа после
погружения электрода в почву. Измеренные значения потенциалов не долж¬
ны быть ниже — 0,3 в.
Платиновые электроды для измерения ОВП изготавливаются в различ¬
ных вариантах. Это могут быть проволочные электроды, пластинчатые
электроды с гладкой поверхностью или так называемые платинированные
электроды, изготовленные в виде стеклянных трубок, поверхность которых
покрыта тонкой пленкой металлической платины.
Классическим вариантом являются проволочные или пластинчатые пла¬
тиновые электроды. Техника их изготовления и применения практически
одинакова, но пластинчатые электроды предпочтительнее, поскольку они
имеют большую площадь и более высокую механическую прочность, что
особенно важно при работе в полевых условиях.
Проволочные, или игольчатые, платиновые электроды (рис. 2, А) исполь¬
зуют в тех случаях, когда предстоит измерение ОВП в небольшой по объе¬
му пробе (отдельном почвенном агрегате, тканях и т. п.), а также для изме¬
рений в равновесных буферных смесях. В частности, они получили рас¬
пространение в хингидронных электродах, в том числе и в хингидронных
электродах сравнения. Для изготовления игольчатого электрода берут глад¬
кую, желательно отполированную платиновую проволоку; при работе в
264
Д. С. Орлов
почвенных суспензиях и естественных почвах диаметр проволоки должен
быть больше. Отрезок проволоки длиной 10—15 мм сваривают с медной
проволокой и впаивают в стеклянную трубку так, чтобы снаружи остался
отрезок платиновой проволоки длиной 5—7 мм, а место спая двух проволок
находилось внутри стеклянной трубки. К концу медной проволоки припаи¬
вают медный колпачок с винтовым зажимом; этот колпачок затем с помощью
специальной замазки или битума надевают на верхний конец стеклянной
трубки. Перед использованием электрод тщательно промывают, высушивают
и платиновую проволоку тщательно прокаливают в пламени спиртовки.
Пластинчатые электроды изготавливают аналогичным образом (см. рис. 2).
Разница заключается лишь в том, что к концу медной проволоки
приваривается полированная платиновая пластинка размером 25—100 мм2
прямоугольной или треугольной формы.
Для работы в густых пастах и в полевых условиях удобнее треугольные
копьевидные электроды (рис. 2Б), которые легко погружать даже в срав¬
нительно твердые и сухие почвы. Изготавливая копьевидные электроды, сле¬
дует подбирать стекло с таким же коэффициентом расширения, как и у пла¬
тины. В противном случае электрод не будет достаточно прочен, а стеклян¬
ные трубки будут часто раскалываться в месте впаивания платины.
Платиновые электроды описанных видов наиболее удобны и надежны в
работе. Их существенный недостаток — большой расход дорогостоящего
металла. В целях экономии часто можно пользоваться платинированными
электродами; на изготовление 100 таких электродов расходуется не более
0,5 г хлорной платины. Платинированные электроды хороши в тех случаях,
когда надо производить массовые измерения ОВП на стационарных площад¬
ках. При этом электроды заранее устанавливаются на требуемую глубину и
остаются в почве на весь срок измерений.
Платинированные электроды были выпущены промышленностью, но их
легко изготовить и самостоятельное любой лаборатории. Ниже мы даем
описание способа изготовления платинированных электродов по методу
В. А. Рабинович и О. В. Куровской. Метод неоднократно проверен в лабора¬
тории факультета почвоведения МГУ и при сравнительно небольшом навы¬
ке дает прекрасные результаты. Ход изготовления следующий. Сначала
подбираются стеклянные трубки, длина которых, диаметр и толщина стенок
меняются в зависимости от поставленной задачи. Длина трубок зависит от
того, на какую глубину предполагается погружать электрод и ограничива¬
ется только механической прочностью стекла и удобствами транспортировки.
Толщина стенок должна обеспечить требуемую прочность электрода. Диа¬
метр может быть выбран любой, но при слишком большом диаметро электро¬
ды получаются громоздкими и слишком тяжелыми, так как на их заполне¬
ние идет большое количество ртути. Удобны практически толстостенные
стеклянные трубки с внешним диаметром от 4 до 10 мм и толщиной стенок
1—2 мм.
Для платинирования трубок готовят специальную смесь. С этой целью
канифоль растворяют в скипидаре при отношении 1:1, нагревая эту смесь
на песчаной бане. Канифоль вносят небольшими порциями при постоянном
помешивании. После охлаждения полученную маслообразную коричневую
жидкость смешивают с равным количеством этилового спирта. Затем 1 г
хлорной платины растворяют в 12 мл спирта, добавляют равный объем на¬
сыщенного спиртового раствора борной кислоты и полученный раствор сме¬
шивают с 25 мл ранее приготовленного раствора канифоли.
Конец стеклянной трубки погружают на 2—3 см в жидкость для плати¬
нирования и сразу вводят в коптящее пламя газовой горелки. Трубку дер¬
жат в наклонном положении платинируемым концом вниз, а нагревание
начинают несколько выше границы жидкости, постепенно вводя в пламя
всю платинируемую поверхность. При обжиге трубку вращают, не давая
Методы, определения pH и окислительно-восстановительного потенциала 265
жидкости скапливаться на нижнем срезе, иначе там образуется слишком
толстый, непрочный и неровный слой платины. После того как растворитель
испарится и на внешней поверхности трубки появится блестящий слой вос¬
становленной платины, продолжают обжиг, но уже в окислительном пла¬
мени горелки, до тех пор, пока внутренняя поверхность трубки не покроется
ровным серым налетом. Трубке дают остыть, снова погружают в раствор
для платинирования и повторяют обработку, пока вся наружная поверхность
не будет покрыта ровным слоем металлической платины. Платинированная
поверхность должна быть ровной и блестящей. При слишком быстром,
сильном и неравномерном обжиге поверхность может получиться матовой,
пузырчатой; такие электроды бракуют.
Конец платинированного отверстия заливают менделеевской замазкой
или битумом. Для этого подогретый платинированный конец погружают
в расплавленную замазку и быстро вынимают. По охлаждении образуется
прочная пробка. Внутрь электрода наливают немного ртути и погружают
медную проволоку для соединения с потенциометром. Чтобы предотвратить
выливапис ртути, верхний конец закрывают ватной пробкой и заливают мен¬
делеевской замазкой.
Промышленные платинированные электроды устроены несколько иначе.
Один из вариантов показан на рис. 2, В. Это комбинированный электрод,
внутри которого помещен и электрод сравнения; для осуществления контак¬
та последнего с почвой в стеклянный корпус впаяна асбестовая нить. Про¬
мышленные платинированные электроды в отличие от вышеописанного
имеют не блестящую, а матовую поверхность.
Независимо от вида электрода и способа его изготовления все электроды
перед началом работы должны быть проверены. Проверку удобнее всего
проводить по буферным растворам с точно известным значением ОВП. Та¬
кой смесью может служить смесь 1,0 М или 0,1 М растворов красной и жел¬
той кровяных солей (К3 [Fe(CN)e], мол. вес 329,26; K4[Fe(CNe)]-3H20, мол.
вес 422,41). Во многих случаях удобен обычный хингидронный электрод,
заполненный буферной смесью с известным значением pH; иными словами,
хингидронный электрод, используемый как электрод сравнения. Потенциал
1 : 1 смеси кровяных солей равен -(-0,36 в, потенциалы хингидронного
электрода сравнения приведены ниже (см. табл. 10, 11).
ЭЛЕКТРОДЫ СРАВНЕНИЯ
Потенциал отдельно взятого электрода измерить невозможно. Поэтому в
качестве нуля шкалы потенциалов принят потенциал нормального водород¬
ного электрода, а в качестве электродов сравнения (полуэлементов) исиоль-
зуют как водородный электрод, так и другие электроды со стабильными или
легковоспроизводимыми потенциалами. Такими электродами чаще всего
служат каломельный электрод, хлорсеребряный электрод и хингидронный
электрод.
Водородный электрод, взятый в качестве полуэлемента, ничем не отлича¬
ется от обычного водородного электрода, но заполнен он буферным раство¬
ром с известным значением pH. Водородный электрод мало удобен при мас¬
совых измерениях и поэтому его используют преимущественно для одно¬
кратного установления потенциала вторичного полуэлемента, например, ка¬
ломельного электрода. Потенциалы водородного электрода при различных
значениях pH приведены в табл. 9.
Чаще используется хингидронный полуэлемент, который служит и как
индикаторный электрод, и как электрод сравнения. В последнем качестве он
обычен в схемах, когда величины pH измеряют хингидронным электродом
на потенциометрах типа П-4, П-5 или на сборных схемах. В этом случае сос-
266
Д. С. Орлов
Таблица 9
Потенциалы водородного электрода при различных pH и температуре 20° С, <?•(—!)
Единицы
Десятые доли pH
pH
0
0,2
0,4
0,6
0,8
0
0
0,012
0,023
0,035
0,047
1
0,058
0,070
0,081
0,093
0,105
2
0,116
0,128
0,139
0,151
0,163
3
0,174
0,186
0,198
0,209
0,221
4
0,232
0,244
0,257
0,267
0,279
5
0,291
0,302
0,314
0,325
0,337
6
0,349
0,360
0,372
0,384
0,395
7
0,407
0,418
0,430
0,442
0,453
8
0,465
0,476
0,488
0,500
0,511
9
0,523
0,535
0,546
0,558
0,569
10
0,581
0,593
0,604
0,616
0,628
тавляется хингидрон-хингидронный элемент. Хингидронный полуэлемент
часто бывает удобен и при измерении ОВП, особенно в полевых условиях,
когда, имея запас буферного раствора и сухой хингидрон, можно в любой
момент приготовить электрод сравнения с достаточно хорошо воспроизво¬
димым потенциалом.
Хингидронный электрод, используемый в качестве полуэлемента, ничем
не отличается от обычного хингидронного электрода. Разница лишь в том,
что исследуемый раствор заменяется стандартным буферным раствором с
известным значением pH. Потенциал такого электрода может быть легко
вычислен или найден по таблицам. Буферный раствор для заполнения хин¬
гидронного электрода сравнения выбирается с таким расчетом, чтобы в ходе
измерений как можно реже приходилось переключать полюса элемента.
Поскольку в почвенно-агрохимических исследованиях значения pH редко
бывают ниже 2,5—3, то для таких целей наиболее удобен буферный раствор
Вайбеля с pH 2,04 или Кларка и Лэбса с pH 2,0 или 2,2 (см. табл. 4). Тот же
буферный раствор удобен и тогда, когда хингидронный полуэлемент исполь¬
зуется как электрод сравнения для измерения ОВП. Его потенциал при pH
2,0 равен 0,587 в при 20° С, что, как правило, выше реально измеряемых
ОВП почв в природе или модельных опытах.
Потенциал хингидронного электрода можно вычислить, если известен
потенциал нормального хингидронного электрода, pH буферного раствора и
температура. Потенциал нормального хингидронного электрода зависит
только от температуры и определяется формулой
ео, хинг = °’6990 + О’00074 (25° -20-
Значения нормального потенциала для некоторых температур приведены
в табл. 10. Потенциал любого хингидронного электрода вычисляется по
приведенной выше формуле и приведен в табл. 11 для 20° С.
Наиболее распространен в электрохимических работах каломельный
электрод сравнения. Это ртутный электрод, находящийся в равновесии с
катионами Hg22 в насыщенном растворе каломели Hg2Cl2; в раствор каломели
добавляют хлористый калий, влияющий на растворимость каломели, так как
имеет с ней одноименный ион. В зависимости от концентрации KG1 разли¬
чают насыщенный, нормальный и децинормальный каломельные электроды,
Методы определения pH и окислительно-восстановительного потенциала 267
Таблица 10
Потенциал нормального хингидронного электрода при различных температурах
°с
Потенциал, в
°с
Потенциал, в
°с
Потенциал, в
5
0,7138
17
0,7049
29
0,6960
6
0,7131
18
0,7042
30
0,6953
7
0,7123
19
0,7034
31
0,6946
8
0,7116
20
0,7027
32
0,6938
9
0,7108
21
0,7020
33
0,6931
10
0,7101
22
0,7012
34
0,6923
11
0,7093
23
0,7005
35
0,6916
12
0,7086
24
0,6997
36
0,6909
13
0,7079
25
0,6990
37
0,6901
14
0,7071
26
0,6983
38
0,6894
15
0,7064
27
0,6975
39
0,6887
16
0,7057
28
0,6968
40
0,6879
Таблица 11
Потенциал хингидронного электрода при различных значениях pH и 20° С, в
Единицы
Десятые доли pH
pH
0
0,2
0,4
0,6
0,8
1,0
0,645
0,633
0,621
0,610
0,598
2,0
0,587
0,575
0,563
0,552
0,540
3,0
0,528
0,517
0,505
0,494
0,482
4,0
0,470
0,459
0,447
0,436
0,424
5,0
0,412
0,401
0,389
0,377
0,366
6,0
0,354
0,343
0,331
0,319
0,308
7,0
0,296
0,284
0,273
0,261
0,250
различающиеся величинами потенциалов, а также устойчивостью потенциа¬
лов во времени и по отношению к изменению температуры. Наиболее устой¬
чив во времени насыщенный каломельный электрод, концентрация КС1 в
котором остается постоянной даже при частичной потере воды за счет испа¬
рения. Но поскольку растворимость КС1 сильно зависит от температуры, то
у насыщенного каломельного электрода и наибольший температурный
коэффициент (табл. 12). Потенциал каломельного электрода определяется
концентрацией ионов ртути в растворе, но так как последняя связана с
концентрацией ионов хлора, то потенциал каломельного электрода можно
выразить и как функцию активности хлор-ионов:
е = е0 — 0,0581 lgaG1_,
т. е. фактически он является электродом второго рода.
Приготовление каломельных электродов несложно и легко осуществимо
в любой лаборатории. Для различных по характеру исследований было
предложено несколько конструкций каломельных электродов. Ниже приве¬
дено описание только одного вида электрода (рис. 3), который можно исполь¬
зовать в самых различных условиях как при потенциометрических, так и
при полярографических и других работах. Описываемый электродный сосуд
268
Д. С. Орлов
Таблица 12
Потенциалы каломельного электрода, в
•с
Концентрация КС1
°С
Концентрация КС1
насыгц.
1,0 н.
0,1 н.
насьпц.
1,0 и.
0,1 н.
10
0,2536
0,2864
0,3374
21
0,2464
0,2838
0,3367
И
0,2529
0,2862
0,3373
22
0,2458
0,2835
0,3367
12
0,2523
0,2859
0,3373
23
0,2451
0,2833
0,3366
13
0,2516
0,2857
0,3372
24
0,2445
0,2830
0,3366
14
0,2510
0,2854
0,3372
25
0,2438
0,2828
0,3365
15
0,2503
0,2852
0,3371
26
0,2431
0,2826
0,3364
16
0,2497
0,2850
0,3370
27
0,2425
0,2823
0,3364
17
0,2490
0,2847
0,3370
28
0,2418
0,2821
0,3363
18
0,2483
0,2845
0,3369
29
0,2412
0,2818
0,3363
19
20
0,2477
0,2471
0,2842
0,2840
0,3369
0,3368
30
0,2405
0,2816
0,3362
имеет в нижней части шарообразную форму. В дно шарообразной части со¬
суда впаяна платиновая проволока, через которую осуществляется контакт
со ртутью. Другой конец платиновой проволоки сваривается с медным про¬
водником; последний подводится к потенциометру. Перед заполнением сосуд
и платиновый контакт тщательно промывают концентрированной серной кис¬
лотой и дистиллированной водой. Затем часть платиновой проволоки, на¬
ходящуюся внутри сосуда, амальгамируют. Для этого в сосуд наливают
раствор Hg2(N03)2, подкисленный азотной кислотой, а наружный конец пла¬
тиновой проволоки подсоединяют к отрицательному полюсу аккумулятора
на 2 в. В качестве анода используют также платиновую проволоку, которую
присоединяют к положительному полюсу того же аккумулятора, а конец ее
опускают в сосуд. Через раствор пропускают ток примерно в течение 1 мин.;
за это время платиновая проволока, впаянная в стенку сосуда, покрывается
серым налетом ртути. По окончании амальгамирования раствор из сосуда
выливают, а сосуд тщательно промывают дистиллированной водой, ополас¬
кивают бидистиллированной водой и высушивают.
В сухой сосуд наливают ртуть с таким расчетом, чтобы тонкий слой рту¬
ти находился и в цилиндрической части сосуда. Поверх ртути наливают
немного суспензии Hg2Cl2 в растворе хлорида калия; концентрация КС1
должна соответствовать виду приготовляемого электрода. Толщина слоя
каломели на поверхности ртути должна быть 1—2 мм. Затем отверстие соеди¬
нительного отростка закрывают и наливают в сосуд (если готовят насыщен¬
ный каломельный электрод) насыщенный раствор KG1, предварительно на¬
гретый до 60°. Сосуд закрывают притертой пробкой. Осторожно приоткрывая
пробку, заполняют жидкостью соединительный отросток и пробку снова за¬
крывают. Отверстие соединительного отростка осторожно закрывают пробоч¬
кой из туго свернутой фильтровальной бумаги; часть пробочки, оставшуюся
вне отростка, срезают лезвием бритвы.
По охлаждении раствора из него выделяются кристаллы КС1, которые
оседают поверх слоя каломели. Таким образом, в готовом электроде на по¬
верхности ртути расположен слой каломели, затем слой кристаллов КС1
и наконец насыщенный раствор хлорида калия. Спустя сутки электрод при¬
годен для работы. В сосудах иной формы электрод готовится аналогично,
несколько меняется только техника заполнения сосуда.
Суспензию каломели для заполнения электрода готовят из препарата
квалификации х. ч. С этой целью растирают Hg2Cl2 с насыщенным раствором
Методы определения pH и окислительно-восстановительного потенциала 269
хлорида калия и несколькими капельками ртути (для вос¬
становления следов ртути-11) в агатовой ступке до полу¬
чения гомогенной массы, переносят с помощью насыщен¬
ного раствора КС1 в склянку и 6 час. взбалтывают на ро¬
таторе. Раствор хлорида калия над осадком Hg2Cl2 сли¬
вают, и осадок несколько раз промывают свежими пор¬
циями насыщенного раствора КС1.
Существенное значение для приготовления каломель¬
ных электродов с потенциалами, максимально близкими
к табличным, имеет чистота ртути и КС1. Хлорид калия
перед началом работы дважды перекристаллизовывают
и затем уже готовят насыщенный раствор. Растворимость
КС1 сильно зависит от температуры и составляет при
20° С примерно 34,4 г на 100 мл, а при 60° С — около 46 г
на 100 мл. Поэтому для приготовления горячего насы¬
щенного раствора КС1 берут 45—50 г КС1 и при нагрева¬
нии растворяют в 100 мл воды. Этим раствором заполня¬
ют электрод. При остывании раствора за счет уменьшения
растворимости выпадают кристаллы КС1. Не следует гото¬
вить насыщенный раствор слишком высокой концентра¬
ции (при 80—90°), иначе при остывании выпадает слиш¬
ком много кристаллов КС1 и весь электрод, включая со¬
единительные отростки, оказывается забитым ими.
Очистка ртути описана во многих руководствах (Зы-
рин, Орлов, 1964; Сердобольский, 1965, и др.). Обычно
бывает^достаточным «фильтрование» ртути через тонкое
отверстие в бумажном фильтре. Через такое отверстие
ртуть вытекает мелкими каплями в 5%-ный раствор
HNOs. После двух-трехкратного промывания ртуть пропускают через ди¬
стиллированную воду. Часть примесей задерживается фильтровальной бума¬
гой, часть растворяется азотной кислотой. Если ртуть очень грязная, ее
предварительно несколько раз промывают азотной кислотой, встряхивая в
делительной воронке. Промытую ртуть собирают в сухую склянку с притер¬
той пробкой и высушивают, проводя по ее поверхности полоской фильтро¬
вальной бумаги.
ПОТЕНЦИОМЕТРИЧЕСКИЕ ИЗМЕРЕНИЯ
Потенциометрическое определение pH или eh сводится к измерению
электродвижущей силы (э. д. с.) элемента, состоящего из двух полуэлемен-
тов: индикаторного электрода и электрода сравнения. Техника измерения не
зависит от вида электродов.
Следует напомнить, что электродвижущая сила равна разности потен¬
циалов на концах разомкнутого источника тока и поэтому не зависит от
внутреннего сопротивления самрго источника. Согласно этому определению,
для измерения э. д. с. пригодны или схемы с очень высоким входным сопро¬
тивлением, или схемы, в которых измерение осуществляется в момент, когда
сила тока в цепи элемента равна нулю (это равноценно разомкнутой цепи).
В потенциометрах используется второй принцип, тогда как многие вольт¬
метры (в том числе ламповые) основаны на первом.
Простейшей схемой потенциометра является схема Поггендорфа. Эту
схему легко самостоятельно собрать в лаборатории, она очень проста, безот¬
казна в обращении и позволяет получать прекрасные результаты даже в
полевой экспедиционной обстановке. Эта схема лежит в основе потенциомет¬
ров П-4, П-5 и др.
Рис. 3. Устройство
простого ка л оме л ь-
ного электрода
1 — раствор КС1;
2 — кристаллы КС1,
8 — каломель;
4 — ртуть;
5 — платиновый кон¬
такт;
в — отросток;
7 — бумажная про-"
бочка
270
Д. С. Орлов
В настоящее время эта схема, как и простейшие потенциометры,
все больше вытесняется электронными полуавтоматическими и запи¬
сывающими потенциометрами, однако последние требуют специальных ус¬
ловий и квалифицированного обслуживания.
Простейшая схема Поггендорфа (рис. 4) включает аккумулятор на 1,5—
2 ву потенциометрическую проволоку (реохорд) с сопротивлением порядка
Анн
Рис. 4. Компенсационная схе¬
ма Поггендорфа
Акк — аккумулятор; Вк — выклю¬
чатель источника тока; R — рео¬
стат регулировки напряжения;
АВ — реохорд; С — скользящий
контакт; Пк — переключатель; К—
ключ; Г— гальванометр; Ев — эле¬
мент Вестона; Ех — исследуемый
элемент
*"8
20 оде, гальванометр, нормальный элемент Вестона, ключи и переключа¬
тели.
Потенциометрическая проволока со скользящим контактом — основной
рабочий узел схемы. Вся проволока должна иметь одинаковое сечение, од¬
нородный состав, малый температурный коэффициент сопротивления и вы¬
сокую механическую прочность. Проволоку или натягивают на линейку с
делениями, или наматывают на барабан.
Аккумулятор можно использовать любых марок, но желательно с высо¬
кой емкостью, что значительно удлиняет срок его непрерывной работы.
Если напряжение аккумулятора (табл. 13) превышает 1,5—2 в, то последо-
Таблица 13
Характеристики некоторых аккумуляторов
Обозначение
Номинальное
напряжение, в
Емкость
Электролит
Кислотные (свинцовые) аккумуляторы
ЗМТ-14
6
10
H2S04, d = 1,12 при 15° С
ЗСТ-60
6
60
H2S04, d = 1,25 при 15° С
6СТ-98
6
98
То же
ЗСТ-68
12
68
»
6СТ-110
12
110
»
10АС-20
17,5
16
H2S04, d = 1,24 при 25° С
ЗНС-110
5,4
110
H2S04, d = 1,28 при 30° С
Щелочные аккумуляторы
НКН-10
1,25
10
Калиево-литиевый (КЛО)
2НКН-10
2,5
10
раствор: КОН (d =1,19—1,21 при 20° С) +
НКН-60
1,25
60
+ 20 гЬЮН*Н20 на 1 л раствора
10НКН-60
12,25
60
10НКН-100
12,5
100
ЖН-45
1,25
45
ЖН-100
1,25
100
Методы определения pH и окислительно-восстановительного потенциала 271
вательно с ним включают реостат, на котором гасится избыточное напряже¬
ние. Используют и сухие батареи.
В схемах со сравнительно невысоким сопротивлением используют стре¬
лочные гальванометры типа ГМП, М-122 и другие, имеющие чувствитель¬
ность порядка 10“б—10“7 а на деление шкалы. Эти гальванометры имеют
арретир, предохраняющий прибор от повреждений при транспортировке, и
корректор нуля (рис. 5).
Нормальный элемент Вестона применяется во всех потенциометрических
схемах для периодического контроля за показаниями прибора. Фактически
с его помощью устанавливается цена деления шкалы. Элемент Вестона при¬
нят в качестве международного стандарта сравнения, э. д. с. которого очень
мало меняется с температурой (табл. 14). Конструктивно элемент Вестона
изготавливается в разных формах, одна из которых показана на рис. 6.
Таблица 14
Э. д. с. элемента Вестона при различных температурах
°с
Э.д.с.
°С
Э.д.с.
°С
Э.д.с.
15
1,01850
18
1,01838
22
1,01822
16
1,01846
19
1,01834
23
1,01818
17
1,01842
20
1,01830
24
1,01814
21
1,01826
25
1,01810
Принцип действия схемы Поггендорфа, как и потенциометров, основан
на противопоставлении э. д. с. изучаемого элемента Ех величине падения
напряжения на участке реохорда. Если (см. рис. 4) падение напряжения на
участке АС равно э.д.с. элемента, то сила тока, протекающего через галь¬
ванометр, равна нулю. Точку С, или точку компенсации, нетрудно найти
передвигая скользящий контакт вдоль реохорда и периодически замыкая
ключ. По мере приближения к точке компенсации отклонения стрелки галь¬
ванометра уменьшаются, а в точке компенсации она остается неподвижной.
Положение точки компенсации проверяют, несколько раз смещая подвижный
контакт. Если в измеряемую цепь был включен элемент Вестона, то в точке
272
Д. С. Орлов
компенсации справедливо следующее равенство:
Ев АС
^акк *
где АВ — число делений на всей потенциометрической проволоке; АС —
число делений от начала до положения подвижного контакта; Ев — э. д. с.
элемента Вестона; 2?акк — напряжение аккумулятора. Это позволяет по
э. д. с. элемента Вестона найти падение напряжения на реохорде, обуслов¬
ленное аккумулятором или сухой батареей. Аналогично можно найти э.д.с.
элемента с индикаторным электродом Ех:
п г» АС\ R ACi
х акк ~~АВ Ь*-АС'
где АСг — отсчет по реохорду в момент, когда э. д. с. изучаемого элемента
полностью компенсирована падением напряжения на этом участке реохорда.
Схему работы можно упростить, если взять реохорд с 1018 делениями*
Поскольку э.д .с. элемента Вестона равна 1,018 то подключив его к обоим
концам проволоки и погасив реостатом избыточное напряжение аккумуля¬
тора, получим, что каждому делению реохорда отвечает 1 мв. Этот принцип
использован во многих потенциометрах. Тогда шкалу реохорда можно гра¬
дуировать не в условных делениях, а непосредственно в вольтах или милли¬
вольтах, а э. д. с. элемента с индикаторным электродом считывать прямо
по этой шкале.
Потенциометр П-4. Потенциометр П-4 можно применять для измерения
э.д.с. цепей со сравнительно невысоким сопротивлением. В основе прибора
лежит обычная схема Поггендорфа; некоторая особенность схемы прибора
заключается в том, что реохорд представлен двумя узлами: десять последо¬
вательно соединенных катушек сопротивления для грубой компенсации и
один виток проволоки — для точной компенсации. Два сопротивления имеют¬
ся для грубой и точной настройки прибора по нормальному элементу. Чтобы
провести измерения э. д. с. на П-4, сначала включают нормальный элемент
Вестона и, меняя сопротивления настройки, добиваются, чтобы гальвано¬
метр показывал нулевую силу тока. Затем включают изучаемый элемент и,
действуя рукоятками реохорда (сначала грубо, через 100 мв, затем плавно),
снова добиваются нулевого положения гальванометра. Отсчет равен сумме
показаний двух реохордов.
Этот прибор сейчас сравнительно редко применяется, но он настолько
прост и удобен в работе, не требует сложного ремонта и юстировки, что им
выгодно пользоваться в тех случаях, когда работа ведется с низкоомными
цепями (водородный, хингидронный электроды, ОВП) и не стоит задача мас¬
совых серийных анализов. Потенциометр П-4 представляет интерес и для
полевых работ, особенно когда нет возможности использовать электронные
потенциометры, питаемые переменным током.
В полевых и стационарных условиях этот потенциометр удобен также для
измерения ОВП почв, особенно когда измерения проводятся непосредствен¬
но в разрезе или на делянке. Однако для измерений еЛ потенциометр П-4
надо несколько видоизменить. В схему потенциометра вместо гальваномет¬
ра включается конденсатор, а гальванометр подключается параллельно
конденсатору (рис. 7). При замыкании ключа теперь протекающий в цепи
ток идет на заряжение конденсатора; когда разность потенциалов на обклад¬
ках конденсатора сравняется с э. д. с. источника, ток прекращается. Это
позволяет избежать поляризации электродов. При размыкании ключа кон¬
денсатор практически мгновенно разряжается через гальванометр, откло¬
нение стрелки которого указывает на прохождение тока при замыкании
ключа. Таким образом, наблюдение за током (гальванометром) в этом случае
ведут не при замыкании, а при размыкании ключа. Схема с конденсатором
Методы определения pH и окислительно-восстановительного потенциала 273
позволяет вместе с тем несколько повысить чувствительность прибора. Ес¬
ли почва, в которой измеряется ОВП, слишком суха, а ее электропроводность
низка, то сила тока может быть настолько мала, что вместо «точки компенса¬
ции» на реохорде обнаруживается целый отрезок, в пределах которого стрел¬
ка гальванометра практически не отклоняется. Чтобы повысить надежность
измерений в этом случае, можно увеличить время заряжения конденсатора.
г
Рис. 7. Схема потенциометра
П-4 с дополнительным конден¬
сатором для измерения окис¬
лительно-восстановительных
потенциалов почвы
Rt и Я2 — сопротивления для на¬
стройки потенциометра грубо и
плавно; Я* и R4 — реохорд; Б —
батарея; Eg — элемент Вестона;
Ех — измеряемый элемент; Г —
гальванометр; С — конденсатор;
К — ключ
Потенциометр П-5 (П-6). Потенциометр П-6 позволяет измерять э. д. с*
в пределах от 0 до 1300 мв с погрешностью ±5 мв. Можно также брать
отсчеты непосредственно в величинах pH, работая с одним из приложенных
к прибору электродов: хингидронным или сурьмяным. Для измерения э. д. с.
в цепях со стеклянным электродом потенциометр соединяют с ламповым
усилителем. Схема прибора принципиально не отличается от вышеизложенной,
но дополнена такими приспособлениями, как температурный компенсатор,
корректор сурьмяного электрода.
Потенциометр ЛП-4 (ЛП-5). Ламповый потенциометр ЛП-4 (ЛП-5)
состоит из трех важнейших узлов: собственно потенциометра, усилителя и
источника питания. Схема потенциометрической части принципиально не
отличается от обычной компенсационной схемы, но вместо гальванометра
использована мостовая схема усилителя. В усилителе два плеча моста пред¬
ставлены электронными лампами, в диагональ моста включен обычный
гальванометр типа ГМП или др. Напряжение, подаваемое от потенциометра
на одну из ламп, смещает равновесие моста, и в диагонали появляется ток,
фиксируемый гальванометром. После компенсации с помощью обычного рео¬
хорда сила тока, как и в простых потенциометрах, падает до нуля и тогда
по шкале потенциометра можно взять отсчет в милливольтах или в pH. Эти
потенциометры сейчас почти полностью вытеснены показывающими лабора¬
торными рН-метрами или высокоомными вольтметрами.
Лабораторный рН-метр ЛПУ-01. Этот прибор может применяться как
многопредельный рН-метр для нахождения активности водородных ионов, а
также как высокоомный милливольтметр или нуль-индикатор. Показывающий
прибор рН-метра отградуирован в единицах pH и милливольтах и позволяет
производить непосредственный отсчет измеряемой величины. Пределы изме¬
рений pH от —2 до +14, причем вся шкала может быть разбита на следую¬
щие диапазоны: от —2 до +2, от 2 до 6, от 6 до 10 и от 10 до 14. Соответствен¬
но пределы измерения э. д. с. составляют от —200 до +1400 мв или от +200
до —1400 мв. Пределы измерений потенциалов могут быть изменены, и вся
шкала может охватывать диапазон в 200 мв. К прибору прилагается стек¬
лянный электрод для измерения pH и вспомогательный хлорсеребряный
электрод. Заводские электроды могут быть заменены любыми другими, что
274
Д. С. Орлов
р НАСТРиЙКА ПО
!&УФ.РЛСТ©С*»У
КРУТИЗНА
■жыт**ш
ШЯ&СРА
^ Р
*то
Рис. 8. Потенциометр ЛПУ-01
А — передняя панель прибора. Б — датчик со стеклянным электродом и простым каломельным электро*
дом сравнения: 1 — штатив; 2 — экран; з — столик для стаканчика с раствором; 4 — каломельный
электрод; 5 — стеклянный электрод
требует только или специальной градуировки прибора, или выполнения из¬
мерений не в единицах pH, а в милливольтах.
Все элементы! измерительной схемы и усилителя рН-метра смонтированы
в металлическом корпусе, а органы управления выведены на переднюю па¬
нель (рис. 8). На передней панели размещены тумблер включения прибо¬
ра, индикаторная лампочка, шкала, переключатели вида работ и пределов
измерений, рукоятки настройки прибора по буферному раствору, крутизны
и температурного компенсатора, а также клеммы для подключения датчика
и клемма заземления. С левой стороны прибора под крышкой помещены
потенциометры, изменяющие координаты изопотенциальной точки, на кото¬
рую настроен прибор. На задней панели прибора выведены клеммы для
автоматического термокомпенсатора, клеммы для подключения регистри¬
рующего прибора (типа ЭПП), заземления, предохранитель, а также вход
от сети переменного тока 220 в.
Датчик ДЛ-01, прилагаемый к прибору, или другой элемент подключают
к прибору с помощью коаксиального кабеля со штеккером. Последний вклю¬
чается в гнездо на передней панели с надписью «датчик»; затем клемму за¬
земления на задней стенке прибора соединяют с линией заземления датчика.
На подготовленном таким образом приборе устанавливают механический
нуль показывающего прибора. Для этого поворачивают отверткой корректор
нуля, расположенный в нижней части показывающего прибора. После этого
включают прибор в сеть. В дальнейшем проводится настройка прибора.
Если предполагается измерение pH, то настройка ведется по буферным
растворам; если прибор используется как милливольтметр, то его настраи¬
вают с помощью потенциометров ППТВ-1, Р-307 и других, которые включают
на вход ЛПУ-01.
Методы определения pH и окислительно-восстановительного потенциала 275
Для настройки прибора по буферным растворам включают прибор в
сеть и дают ему прогреться в течение 30 мин. В это время берут стандартный
буферный раствор с pH 4,00, тщательно промывают им электроды, а затем
опускают электроды в свежую порцию буферного раствора. Включают при¬
бор на диапазон измерений pH от 2 до 6, рукоятку температурного коррек¬
тора устанавливают на температуру раствора и через 1—2 мин. снимают
отсчет. Если показания прибора отличаются от 4,00, то рукояткой «настрой¬
ка по буферному раствору» устанавливают стрелку прибора точно на деле¬
ние, отвечающее pH 4,00. Аналогично проверяют показания прибора по
другим буферным растворам для каждого диапазона в отдельности. Рас¬
хождения по всем диапазонам не должны превышать 0,04 pH. Если расхож¬
дения оказываются более высокими, то проводят дополнительную настрой¬
ку ручкой «крутизна». С этой целью электроды помещают в буферный раст¬
вор с pH 1,68, переключатель пределов измерений устанавливают на диа¬
пазон от —2 до +2 pH и ручкой «настройка по буферному раствору» уста¬
навливают стрелку прибора на деление 1,68. Затем проверяют показания
прибора по буферному раствору с pH 9,22 в диапазоне от 6 до 10 pH. Если
отклонения в последнем случае превышают 0,04 pH, то с помощью специаль¬
ного ключа ослабляют цанговый зажим переменного сопротивления «кру¬
тизна» и поворачивают ось переменного сопротивления на 5—10° по часовой
стрелке, если показания прибора занижены, или против часовой стрелки,
если показания прибора завышены. Снова затягивают цанговый зажим.
После этого снова возвращаются к буферному раствору с pH 1,68 и повторя¬
ют настройку до тех пор, пока не будут найдены положения ручек «крути¬
зна» и «настройка по буферному раствору», при которых расхождения
показаний не будут превышать 0,02—0,04 pH.
После настройки прибора можно приступать к измерениям pH или гн.
Следует иметь в виду, что рН-метр ЛПУ-01 очень чувствителен ко всякого
рода внешним наводкам. Даже движение руки вблизи электродов может
существенно повлиять на показания. Поэтому хорошее заземление и экра¬
низация электродов — главное условие достоверности получаемых резуль¬
татов.
При измерениях pH на приборе ЛПУ-01 следует руководствоваться
следующими общими правилами. Перед каждым погружением электродов в
исследуемый раствор их надо тщательно промывать дистиллированной во¬
дой; избыток воды удаляется фильтровальной бумагой. Если есть возмож¬
ность, то после промывания водой желательно ополоснуть электроды еще и
исследуемым раствором и только после этого погружать их в свежую порцию
раствора для производства измерений. Показания pH снимают не сразу со
шкалы прибора, а лишь после того, как показания станут стабильными.
Как правило, время установления показаний не превышает 0,5—1 мин.
Однако в сильнокислых и сильнощелочных растворах, а также в малобу¬
ферных растворах (дистиллированная вода, водные вытяжки из незасоленных
почв) показания не всегда устанавливаются и за несколько минут.
При работе обязательно пользуются температурным компенсатором;
иногда для этого применяют автоматический компенсатор, но значительно
проще пользоваться ручной установкой компенсатора на соответствующую
'температуру. Температура исследуемых растворов может несколько отли¬
чаться от температуры воздуха, поэтому для контроля на лабораторный стол
помещают цилиндр с водой, в которую опускают термометр; его показания
принимают равными температуре испытуемых растворов.
Более детально возможности, настройка и использование прибора опи¬
саны в прилагаемой к нему заводской инструкции, которую следует внима¬
тельно изучить перед началом работы на рН-метре.
Лабораторный pH-метр-милливольтметр pH-340. Этот прибор, как и
ЛПУ-01, предназначен для определения pH и OBI1. При наличии электро-
276
Д. С. Орлов
/ 2 9
<Л> юо о о о~
£и гМб° О+20 мв и
рНи
Рис. 9. Потенциометр pH-340
Л — передняя панель. 1 — показывающий прибор; 2 — переключатель рода работы; 3 — переключа¬
тель пределов измерений; 4 — ручной термокомпенсатор; 5 — индикатор включения; 6 — потенциометр
настройки Еи\ 7 — потенциометр настройки S\ 8 — потенциометр установки нуля нуль-индикатора и
включатель сети; 9 — переключатель размаха шкалы. Б — задняя панель* 1 — клеммы для автомати¬
ческого терм оком пенсатоюа; 2 — перемычка; 3 — переменный резистор для грубой регулировки Еи;
4,5 — выходные гнезда; 6,7 — переменные резисторы для грубой и плавной регулировки рНи; 8 — пере^
меняые резисторы для заводской градуировки прибора; 9 — гнездо для подключения индикаторного
электрода; 10 — гнездо для вспомогательного электрода; 11 — зажим заземления; 12 — сетевой разъем;
13 — держатель предохранителя
грубо pHи
дов, обратимых к ионам натрия (и другим), он может быть использован и
для измерения их активности. Так же как и ЛПУ-01, прибор рН-340 в со¬
четании с блоком Б АТ-12 ЛМ позволяет проводить автоматическое титрова¬
ние, а при подключении автоматических записывающих потенциометров
(типа ЭПП) кривые титрования могут быть записаны на специальной диаг¬
раммной ленте.
Пределы измерения э. д. с. на рН-340 от—100 до +1400 мв или от
+ 100 до —1400 мв. Этот интервал может быть разбит на диапазоны по
Методы определения pH и окислительно-восстановительного потенциала 277
300 мв. Погрешность измерений достигает ±0,5 единиц pH и 29 мв; при
работе в узких диапазонах погрешность снижается в 10 раз.
Прибор смонтирован в металлическом корпусе размером 337 X 292 х
X 164 мм, на переднюю наклонную панель которого выведены рукоятки
управления и показывающий прибор. Шкала последнего дана в единицах pH
и в милливольтах. Кроме показывающего прибора, на передней панели рас¬
положены переключатель рода работ, переключатель пределов измерений,
ручной термокомпенсатор, потенциометры настройки, потенциометр уста¬
новки нуля нуль-индикатора и включатель сети, переключатель размаха
шкалы, индикатор включения. Гнезда для подключения датчиков, выходные
гнезда, зажим заземления и другие расположены на задней стенке прибора
(рис. 9).
Чтобы начать работу на приборе рН-340, к прибору подключают датчик
к соответствующим гнездам на задней стенке. Провод заземления датчика
соединяют с соответствующей клеммой прибора и линией заземления. Затем
устанавливают механический нуль показывающего прибора; для этого от¬
верткой поворачивают корректор нуля, подводя стрелку на начальную отмет¬
ку шкалы. Ручки переключателей «род работы», «размах», «температурный
компенсатор» устанавливают в соответствии с выбранной схемой измерения.
Прибор подключают к сети 220 *, 50 гц с помощью сетевого шнура и включают
прибор, для чего поворачивают соответствующую ручку по часовой стрелке.
Когда напряжение питания на прибор подано, загорается контрольная лам¬
почка на передней панели.
При использовании прибора в качестве милливольтметра для измерения
э. д. с. могут быть применены любые системы датчиков и вспомогательных
электродов, причем следует иметь в виду, что при отсчете показаний на
участке шкалы от 0 до 1400 мв знак, соответствующий положению ручки
переключателя «род работы», соответствует знаку потенциала индикатор¬
ного (измерительного) электрода. Если переключатель «размах» установлен
в положение «1500 мв», то отсчет показаний производится по нижней шкале
прибора, оцифрованной от —1 до 14. Если переключатель установлен на один
из узких диапазонов, то отсчет берут по верхней шкале, принимая во
внимание положение переключателя. Например, если переключатель диа¬
пазонов установлен в положение «11 -т- 14», а стрелка показывающего при¬
бора остановилась на делении 0,25, то измеряемая э. д. с. равна:
(И + 0,25).100 = 11,25.100 = 1125 мв,
множитель 100 вводится для того, чтобы перевести показания шкалы в мил¬
ливольты.
Измерения pH на приборе рН-340 мало чем отличаются от измерений на
других потенциометрах. Прибор соединяют с соответствующим датчиком
(индикаторный электрод — вспомогательный электрод), устанавливают
переключатели в положение, соответствующее виду работы, и настраивают
по буферным растворам. С этой целью прибор включают в сеть, переключа¬
тель «размах» устанавливают в положение «15 pH» и дают прибору прогреть¬
ся в течение 60 мин. Стеклянные электроды предварительно вымачивают в
0,1 н. НС1 в течение 8 час., а перед погружением в буферный раствор стек¬
лянный электрод и электрод сравнения промывают дистиллированной во¬
дой, удаляя остатки воды фильтровальной бумагой. Затем буферным раст¬
вором с pH 1,68 ополаскивают электроды и погружают их в свежую порцию
того же буферного раствора. Переключатель «пределы измерения» устанав¬
ливают в положение «1 -ч- 2», температурный корректор переводят в поло¬
жение, отвечающее температуре раствора, а переключатель «размах» — в
положение «3 pH». Установив ручку потенциометра Еи примерно в среднее
положение, подводят стрелку показывающего прибора к отметке 1,68 pH,
используя последовательно потенциометры «Еи грубо» и «Еи». Переключатель
278
Д. С. Орлов
«размах» переводят в положение «15 pH». Затем проводят проверку показаний
прибора по другим буферным растворам, пользуясь соответствующими под¬
диапазонами шкалы. Отклонения показаний прибора от pH буферных раст¬
воров не должны превышать 0,05 pH на всех узких диапазонах шкалы и
0,5 pH на диапазоне —1 -г- 14 pH.
Если отклонения показаний превышают допустимые ошибки, то прово¬
дят настройку по двум буферным растворам, используя потенциометр S.
С этой целью сначала проводят настройку по раствору 1,68 pH, как указано
выше. Затем погружают электроды в раствор с pH 9,22; если показания
отличаются более чем на 0,05 pH в диапазоне 8 -н 11 pH, то ослабляют
цанговый зажим потенциометра S и устанавливают его положение так,
чтобы прибор показывал 9,22 pH. Затем повторно проверяют показания с
буферным раствором 1,68 pH и настройку продолжают, пока показания
прибора окажутся в допустимом интервале отклонений. После этого можно
приступить к измерениям pH исследуемых растворов. Более подробно схема
настройки и другие ее варианты изложены в инструкции, прилагаемой к
каждому прибору.
Следует иметь в виду, что показания на приборе устанавливаются не
сразу. Время установления показания обычно не превышает 3 мин. и за¬
висит от температуры и состава раствора. Как и в случае прибора ЛПУ-01,
стабильность показаний сильно зависит от качества экранизации датчиков
и заземления.
Переносный pH-метр-милливольтметр ППМ-03М1. Этот небольшой и
простой в обращении прибор до сих пор иногда используется в полевых
работах, хотя он не отличается устойчивостью показаний и надежностью в
эксплуатации. Прибор позволяет измерять pH от 2 до 12 и э. д. с. от 0 до
1000 мв. По паспортным данным, погрешность измерений не превышает
0,1 pH и 10 мв. Удобство прибора определяется батарейным питанием; он
не зависит от электросети, работая от элемента «Сатурн». Кроме того, при¬
влекает небольшой вес — всего 1,3 «г и небольшие размеры: 200 х 130 X
X 70 мм. Предварительно отлаженные в лаборатории рН-метры при ак¬
куратном обращении могут обеспечить работу в течение всего полевого
сезона.
Другие рН-метры и потенциометры. В агрохимических и почвенных
лабораториях сейчас можно встретить большой набор отечественных и за¬
рубежных приборов и других марок, кроме перечисленных выше. Дать их
полное описание невозможно, да в этом и нет смысла. Конструкции прибо¬
ров довольно быстро меняются, часто появляются новые модели, а полные
и подробные описания представлены в инструкциях. Изложенные выше тре¬
бования и принципы имеют отношения ко всем потенциометрам.
Кроме перечисленных приборов, в лабораториях еще можно встретить
старые модели типа ЛП-3 и ЛП-4, иногда работают с усилительными при¬
ставками типа ЛУ-2. Для целей массовых анализов был выпущен рН-метр
«Агрохимик». Центральным опытно-конструкторским бюро приборострое¬
ния МСХ СССР разработан автоматический рН-метр с ценой деления 0,2 pH
и диапазоном измерения от 3 до 10 pH. Показания регистрируются на диаг¬
раммной ленте.
Во многих лабораториях используются и потенциометры зарубежного
производства. Например чехословацкий рН-метр модель РНК-1 с чувстви¬
тельностью до 0,01 pH или 0,5 мв. Выполнение приборов может быть весьма
различным. Часть приборов снабжена стрелочными указателями, некото¬
рые имеют записывающие на диаграммной ленте устройства. Наконец, ста¬
ли появляться потенциометры и рН-метры, у которых результаты измере¬
ний появляются в виде цифровой записи на светящейся шкале. Разнообраз¬
ны и пределы работы, а также чувствительность и точность измерений, что
позволяет подобрать наиболее подходящие для каждого вида работ приборы.
Методы определения pH и окислительно-восстановительного потенциала 279
Все чаще выпускаются потенциометры, погрешность которых не превышает
0,1 мв или 0,001 pH. Такие приборы пригодны не только для измерения
pH и ОВП, но также для измерения активности металлических и других
ионов с помощью специальных ионселективных электродов.
СПОСОБЫ ОПРЕДЕЛЕНИЯ pH ПОЧВ И ПОЧВЕННЫХ СУСПЕНЗИЙ
Прежде чем приступать к определениям, необходимо выбрать условия
измерений; сюда входят выбор стандартного состояния (или подготовка поч¬
вы), выбор электрода и измерительного прибора.
Подготовка почвы и приготовление суспензии.
В практике принято определять pH образцов почв, предварительно собран¬
ных в поле и траспортированных в лабораторию. При этом неизбежно на¬
рушается естественное состояние почвы; один из сильно влияющих факто¬
ров — высушивание почвы. Для приготовления водных вытяжек и суспензий
многие авторы рекомендуют пользоваться воздушно-сухими образцами. В ча¬
стности, White (1969) нашел, что высушивание щелочных почв в термоста¬
те снижает pH на 0,6 единицы. По другим данным, сушка образцов разными
способами и хранение их в течение 3 месяцев не повлияли на результаты
(Piskula, 1971). Есть также наблюдения, что и измельчение почв, во всяком
случае щелочных, снижает pH (White, 1969). В связи с этим предлагалось
вместо сушки и растирания прибегать к просеиванию еще сырых почв через
сито с отверстиями 2 мм. Методические исследования в этом направлении
еще не закончены.
Для получения сопоставимых результатов в настоящее время наиболее
правильно пользоваться воздушно-сухими образцами, просеянными после
разминания через сито с отверстиями диаметром 1 мм. Эти стандартные
условия могут быть изменены при специальных исследованиях, когда важно
уловить тонкие изменения pH под влиянием быстро сменяющихся факторов.
В таких случаях (но не при массовых обследованиях) следует отдать предпоч¬
тение сырым образцам почвы. Влажность сырых почв существенно не изме¬
няет соотношения почва: раствор в ходе приготовления суспензии; поэтому,
как правило, ее можно не принимать во внимание.
Различные рекомендации даются также по таким вопросам, как соот¬
ношение почва: раствор и время взаимодействия почвы с раствором. Данные
о влиянии разбавления довольно противоречивы. По последним работам
Шахтшабеля (Schachtschabel, 1971), различия pH при изменении отношения
почва: раствор от 1 : 2,5 до 1:1 составляют всего 0,1 единиц pH. Более значи¬
тельные сдвиги отмечались для более узких соотношений. Так же как и в
предыдущем случае, для массовых стандартных измерений следует принять
обычное соотношение почва: раствор = 1 : 2,5, хотя в специальных иссле¬
дованиях это отношение может быть изменено, но отклонения от стандарта
должны быть оговорены особо.
Результаты измерения pH зависят, наконец, и от времени взаимодейст¬
вия почвы с раствором. Было, например, показано что в суспензии 0,01 М
СаС12 величина pH повышается от 5,4 до 6,1, если время встряхивания суспен¬
зии увеличить от 2 до 168 час. (Bache, 1970); последний автор рекомендует
определять pH в течение часа после приготовления суспензии, хотя были
высказаны и многие другие рекомендации: 5-минутное взбалтывание, на¬
стаивание 10—15 мин. при периодическом встряхивании (White, 1969), суточ¬
ное взаимодействие. Вполне удовлетворительные и сопоставимые резуль¬
таты получаются при 5-минутном встряхивании на ротаторе, что и можно
рекомендовать в качестве стандарта.
Требует стандартизации и характер самой вытяжки. В частности, в каких
случаях следует пользоваться вытяжкой, в каких — суспензией. Известно,
280
Д. С. Орлов
что в практике почвенно-агрохимических исследований производят следую¬
щие виды измерений: 1) pH водной вытяжки; 2) pH водной суспензии;
3) pH солевой вытяжки; 4) pH солевой суспензии.
Различие результатов, получаемых при анализе вытяжек и суспензий,
ранее связывалось с так называемым суспензионным эффектом (иногда на¬
зывают эффектом Вигнера). Предполагалось, что в почвенной суспензии
концентрация ионов Н+ выше, чем в растворе за счет Доннановского равно¬
весия. Подобного рода явления действительно могли иметь место при двух
условиях: наличие полупроницаемой мембраны (границы раздела двух фаз)
и возможность определения концентрации, а не активности Н+-ионов. При
анализе почвенных суспензий граница между фазами (принимая во внимание
макроскопические размеры электродов) не может быть обнаружена. Кроме
того, в состоянии равновесия активность Н+-ионов должна быть одинаковой
во всем объеме жидкой фазы, а при отсутствии скачка потенциала между
жидкой фазой и частицами твердых фаз — и во всех фазах системы. Поэтому
«суспензионный эффект» не должен возникать при анализе почвенных сус¬
пензий. Наблюдаемые иногда различия результатов измерений следует от¬
нести не за счет суспензионного эффекта, а за счет диффузионного потен¬
циала, поскольку заряженные частицы суспензии неодинаково влияют на
диффузию ионов К+ и С1~ из каломельного электрода (Olsen, Robbins, 1971).
Не наблюдали суспензионный эффект при работе со стеклянным элек¬
тродом и другие исследователи (Schachtschabel, 1971). Более того, в ряде
случаев pH суспензии оказывался выше, чем pH центрифугата (Bache, 1970;
White, 1969). Отсюда следует, что рационально проводить только одно из¬
мерение.
Наиболее точные результаты могут быть получены для вытяжек, тогда
как при анализе суспензий необходимо применять меры для устранения
влияния диффузионного потенциала. Это легко осуществимо для солевых
вытяжек, когда избыток соли способствует коагуляции коллоидов и позволяет
получить совершенно прозрачные вытяжки. Прозрачные водные вытяжки
получить очень трудно, и для массовых измерений можно рекомендовать
ограничиться стандартным приемом: центрифугирование суспензии при
6000 об/мин в течение 15 мин. Полученные таким способом вытяжки из мно¬
гих почв заметно опалесцируют.
Принято считать, что водная вытяжка характеризует реакцию почвен¬
ного раствора, а солевая — обменную (потенциальную) кислотность. Поэ¬
тому солевая вытяжка естественно пригодна лишь для почв, содержащих
обменные ионы Н> или А13+, тогда как в присутствии СаС03 и сильных кис¬
лот может оказаться, что водная вытяжка будет более кислая, чем солевая
(Piasecki, Kotowska, 1966). Есть также небезинтересные данные, согласно
которым, между величинами pH в водных и солевых вытяжках существует
строгая зависимость. Шахтшабель (Schatschabel, 1971), сопоставляя pH в
суспензиях почв на основе 0,01 М СаС12, 0,1 М КС1, 1,0 М КС1 и водной
суспензии, установил следующие зависимости. В лёссовых почвах pH вод¬
ных суспензий были выше pH суспензий в 0,01 М СаС12 на 0,56 единиц; в
суспензии 0,1 М КС1 — меньше на 0,02 единицы pH и в суспензии 1,0 М КС1
— меньше на 0,27 единиц pH, чем в суспензии 0,01 М СаС12. В супесчаных
почвах различия оказались несколько иными по абсолютной величине. Высо¬
кую корреляцию величин pH в водных и 0,01 М СаС12 вытяжках отмечали и
другие (Davies BriaD, 1971). Если такие связи окажутся устойчивыми и функ¬
ционально обусловленными, это, может быть, позволит в будущем ограни¬
читься анализом только одной какой-либо вытяжки.
Подводя итоги сказанному, можно прийти к следующим стандартным
условиям приготовления вытяжек. Для анализа берут воздушно-сухие об¬
разцы почвы, разминают и просеивают через сито с отверстиями диаметром
1 мм. К навеске почвы приливают такой объем воды или 1,0 н. КС1, чтобы
Методы, определения pH и окислительно-восстановительного потенциала 281
отношение почва : раствор составило 1 : 2,5. Колбочку со смесью закрывают
пробкой и встряхивают на ротаторе 5 мин. Затем, если определяется pH вод¬
ной вытяжки, суспензию переносят в центрифужные пробирки, последние
попарно уравновешивают и суспензию центрифугируют 15 мин. при
6000 об/мин. В случае солевой вытяжки суспензии просто дают отстояться и
для определения pH берут часть надосадочной жидкости. Для приготовления
вытяжек используется только бидистиллированная вода, прокипяченная
для удаления С02 и хранящаяся изолированной от атмосферного воздуха.
Выбор электрода и измерительного прибора.
Два основных условия ограничивают применение отдельных видов электро¬
дов и потенциометров: диапазон измеряемых величин pH и состав почвен¬
ных вытяжек или суспензий. Реакция почв, а точнее почвенных растворов и
вытяжек, обусловлена избыточным содержанием ионов Н+ или ОН”, проис¬
хождение которых может быть различным. Источниками водородных ионов
могут быть самые простые системы, например угольная кислота, образую¬
щаяся при растворении С02 в почвенном растворе. Исходя из того, что С02
всегда присутствует в атмосферном и почвенном воздухе, мы должны рас¬
сматривать значение pH, равное 7, не как характеристику нейтральной
почвы, а как почвы, в которой осуществляется слабое накопление основа¬
ний. Другими источниками Н+ служат неспецифические органические кис¬
лоты, фульвокислоты и гуминовые кислоты. Концентрация кислот в почвен¬
ных растворах обычно невелика, а степень их диссоциации, как правило,
низка, и поэтому обусловленная органическими кислотами реакция среды
редко бывает ниже 3—4. Подкисляющее действие могут оказать поглощен¬
ные водородные ионы, а также различные гидролитически кислые соединения;
широко известна, например, роль ионов алюминия:
А13+ + ЗШО — А1(ОН)3 + ЗН+.
В довольно редких случаях (месторождения серы, горные выработки и от¬
валы) могут накапливаться минеральные кислоты, особенно серная. В этих
случаях pH даже водных вытяжек бывает сильно понижен.
Нейтральная или слабощелочная среда в почве связана чаще всего с
карбонатами кальция. Накопление соды приводит к повышению pH до 8,5—10;
значительное подщелачивание иногда возможно и за счет органических
оснований. Теоретический и практический опыт показывают, что при иссле¬
довании естественных почв диапазон измеряемых значений pH водных и
солевых суспензий находится примерно в пределах от 2—3 до 10—11. Более
кислые или более щелочные условия в природной обстановке редки, но в
лабораторной практике встречаются довольно часто, что приходится учи¬
тывать при выборе инструмента исследования. Для широких агрохимических
и почвенных анализов вполне можно ограничиться диапазоном pH от 2 до
11 и с этим расчетом подбирать электроды. Указанному диапазону pH
удовлетворяют почти все типы стеклянных электродов, причем в диапазоне
от 2 и примерно до 7—8 pH их показания практически не зависят от присут¬
ствия металлических катионов. Хингидронный электрод для всего указан¬
ного диапазона не пригоден, им можно пользоваться лишь в нейтральных
или очень слабощелочных средах. Поэтому анализ солонцов и содовых со¬
лончаков хингидронным электродом проводить нельзя.
Второе условие выбора электродов связано с составом анализируемой
среды. Водные и солевые вытяжки и суспензии почв содержат разнообраз¬
ные органические соединения, включая высокомолекулярные (белки и т. п.),
затем неорганические ионы, включая NH4+, коллоидные частицы органиче¬
ского и минерального происхождения. Концентрация неорганических солей
в случав засоленных почв может достигать 1 п. и выше. Естественно, что да¬
леко не всякий метод и не всякий электрод пригоден для изучения по¬
добных систем. Особенно многочисленны ошибки при определении pH ко-
282
Д. С. Орлов
Рис. 11. Соединение исследуе¬
мого раствора (1) через элек¬
тролитический ключ (2) и насы¬
щенный КС1 (3) со вспомога¬
тельным хингидронным элек¬
тродом (4)
лориметрическим методом и при использовании хингидронного электрода*
На показания последнего влияют все окислители и восстановители, коллоид¬
ная фаза, избыток солей. Поэтому хингидронный электрод практически не
пригоден, кроме уже перечисленных выше почв, также при анализе сильно-
восстановленных почв (глеевых, оглеенных), почв и горизонтов, обогащен¬
ных как закисными, так и окисными соединениями железа. В наибольшей:
степени всем требованиям, предъявляемым при определении pH почв, от¬
вечает стеклянный электрод.
Выбор потенциометра связан с особенностями используемых электродов.
Для измерений с хингидронным электродом пригоден практически любой
потенциометр, равно как и простая компенсационная схема Поггендорфа.
В случае стеклянного электрода приходится прибегать к электронным по¬
тенциометрам (ЛПУ, рН-340 и т. п.). Конечно, потенциал стеклянного
электрода можно измерить и на значительно более простых приборах (см.
например, Зырин, Орлов, 1964), но практически это нецелесообразно.
Определение pH с хингидронным электродом.
Для определения pH почвы с хингидронным электродом готовят вытяжку
или суспензию, как описано выше. Часть вытяжки отбирают в пальчико¬
образную пробирку (рис. 10). Пробирки должны быть чистыми и совершенно
сухими, количество взятой вытяжки не имеет значения, поскольку описывае¬
мыми методами измеряется не общее содержание, а концентрация Н+-ионов;
существенно только, чтобы взятый объем был достаточен для погруже¬
ния в него платинового электрода и электролитического ключа. К раствору
(вытяжке) добавляют немного (на кончике ножа) хингидрона, слегка встря¬
Методы определения pH и окислительно-восстановительного потенциала 283
хивают, чтобы хингидрон равномерно распределился по всему объему. За¬
тем через электролитический ключ вытяжку соединяют с пробиркой, запол¬
ненной насыщенным раствором КС1, и далее со вспомогательным полуэле-
ментом; обычно это тот же хингидронный электрод, приготовленный на бу¬
ферном растворе с известным значением pH (рис. 11). В пробирку с вытяж¬
кой опускают платиновый электрод; для этой цели пригодны игольчатый и
копьевидный электроды, описанные в разделе об электродах для измерения
ОВП. Поскольку во вспомогательном полуэлементе используется буфер¬
ный раствор с pH около 2, то последний в собранной цепи имеет знак плюс,
а индикаторный электрод знак минус; соответственно знакам полуэлементы
подключают к потенциометру или схеме Поггендорфа и измеряют э.д.с.,
как описано выше.
Найденная э. д. с. равна разности потенциалов индикаторного и
вспомогательного электродов:
э.д.с. - евсп - еинд = е0 - 0,0581 рНвсп - е0 + 0,0581 рНх (при 20°С)
Отсюда:
э.д.с.
Унх= 0,0581 + РНВСП-
В случае, когда pH почвенной суспензии оказывается ниже, чем pH вспо¬
могательного электрода, то меняют полюсность на приборе, а расчет ведут
по формуле
Э.Д.С.
pH* = рНвсп — о,0581 •
Определение pH со стеклянным электродом. Цепь
для измерения pH со стеклянным электродом может быть составлена по-
разному в зависимости от вида применяемых электродов. В случае комби¬
нированного электрода последний просто погружают в вытяжку или сус¬
пензию, а выходной штеккер подключают к потенциометру. Если исполь¬
зуется внешний вспомогательный электрод, то в зависимости от типа послед¬
него его или непосредственно погружают в испытуемый раствор вместе со
стеклянным электродом, или соединяют, как описано выше, через электро¬
литический ключ.
Непосредственное нахождение величины pH также возможно двумя
способами. В первом из них прямо берут отсчет по предварительно отградуи¬
рованной шкале pH на потенциометре. Во втором измеряют только э.д.с. в
милливольтах; сначала проводят измерения для серии буферных растворов
и строят график в координатах э.д.с. — pH, затем находят э.д.с. для цепи
с испытуемым раствором и по графику находят величину pH.
СПОСОБЫ ИЗМЕРЕНИЯ
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОГО ПОТЕНЦИАЛА ПОЧВ
Окислительно-восстановительные потенциалы измеряют платиновым или
платинированным электродом в свежесобранных образцах почв или непосред¬
ственно в поле. Предпочтительнее проводить непосредственные полевые из¬
мерения, поскольку даже в свежесобранных образцах нарушается сложив¬
шееся окислительно-восстановительное состояние. Но если транспортировка
образцов все же неизбежна, то следует брать значительный по размеру обра¬
зец с ненарушенной структурой и плотно упаковывать его в мешочек из по¬
лиэтилена, стараясь как можно больше сократить время контакта образца
•с атмосферным воздухом. В лаборатории такой образец полностью не распа¬
ковывают, а лишь освобождают часть поверхности, необходимую для по¬
284
Д. С. Орлов
гружения электродов. В лабораторных условиях с успехом можно вести
наблюдения за динамикой ОВП в модельных опытах.
Перед измерением ОВП все электроды проверяют по буферным смесям,
затем тщательно отмывают дистиллированной водой и высушивают. Сухие
электроды погружают в почву так, чтобы почва плотно прилегала к поверх¬
ности электрода и полностью изолировала его от воздуха. Этого можно дос¬
тичь, если после-погружения электрода вокруг него слегка уплотнить (обжать)
почву. Если возможно, то желательно измерения выполнять с тремя па¬
раллельными индикаторными электродами.
В тех случаях, когда применяется комбинированный платинированный
электрод с внутренним электродом сравнения, дополнительный внешний
вспомогательный электрод не требуется. Обычные электроды требуют вспо¬
могательных электродов. Таковыми могут быть или каломельные электро¬
ды, или хингидронные электроды, как это описано выше в разделе об элект¬
родах сравнения. Если применяется насыщенный каломельный электрод,
то его отросток можно непосредственно погрузить в почву рядом с индика¬
торным электродом, затем оба электрода подсоединить к потенциометру и
измерить э.д.с. Надо только следить, чтобы отросток электрода не был за¬
бит почвой и не вытекал из него хлорид калия. Для других типов вспомога¬
тельных электродов необходимо воспользоваться переходной пробиркой
с насыщенным раствором хлорида калия и электролитическим ключом. Пос¬
ледний готовят или на агар-агаре или просто U-образную стеклянную трубку
заполняют насыщенным раствором хлорида калия, а концы трубки закрывают
пробками из фильтровальной бумаги.
В полевых условиях измерения ОВП проводят или единовременно (разо¬
вые измерения) или наблюдают за динамикой ОВП. Разовые измерения
чаще всего проводят при площадных обследованиях или изучении достаточ¬
но длинных почвенно-геохимических профилей. Наблюдения за динамикой
удобнее вести на стационарных площадках, где можно обеспечить сохран¬
ность электродов.
При разовых измерениях электрод погружают в почву на глубину 5—7 см
или с поверхности, или в середину генетического горизонта на передней
стенке разреза. Электрод сравнения или электролитический ключ погружа¬
ют в почву на 1—2 см на расстоянии 8—10 см от индикаторного электрода.
После этого измеряют величину э. д. с. Следует иметь в виду, что ОВП не
остается постоянным при повторных измерениях, происходит так называемое
сползание, или дрейф, потенциала. Природа дрейфа недостаточно ясна.
В лабораторных условиях при исследовании сравнительно гомогенных
почвенных паст и исключения возможности поляризации электродов (на¬
пример, на конденсаторной схеме) потенциал, как правило, не ползет. В по¬
левых условиях изменения происходят довольно быстро при любых условиях,
что следует объяснить естественной динамикой окислительно-восстанови¬
тельных условий. Представление о том, как и в каких пределах изменяется
ОВП пахотного горизонта дерново-подзолистой почвы, дает рис. 12. Поэтому
при разовых измерениях наиболее целесообразно брать отсчеты через рав¬
ные промежутки времени после погружения электрода в почву; может быть
выбран любой интервал примерно от 5 до 15 мин. Такие измерения по¬
зволяют получить вполне устойчивую картину профильного изменения ОВП
в различных типах почв.
Изучение динамики ОВП обязательно проводится на одних и тех же ин¬
дикаторных электродах, постоянно погруженных в почву. Электрод срав¬
нения устанавливать постоянно необязательно. Измерения э. д. с. прово¬
дятся в определенные часы по заранее составленному графику, и электрод
сравнения может быть установлен непосредственно перед измерением. Если
же используется хингидронный электрод сравнения, то он обязательно за¬
меняется ежедневно на свежеприготовленный.
Методы определения pH и окислительно-восстановительного потенциала 285
В результате измерений исследователь получает значения э. д. с., рав¬
ные разности потенциалов между индикаторным и вспомогательным элект¬
родами. Для вычисления значения ОВП надо знать полярность электро¬
дов, которую определяют по положению переключателя на потенциометре
или обозначениям у входных клемм.
Рис. 12. Динамика окисли¬
тельно-восстановительного по¬
тенциала в пахотном горизон¬
те дерново-подзолистой почвы
Если платиновый индикаторныи электродимел знак «+», т. е. его
потенциал был выше, чем потенциал вспомогательного электрода, то
э.д.с. = ех бвсп,
где е* — потенциал платинового индикаторного, а евсп — потенциал элект¬
рода сравнения.
Тогда:
ел = ех = э.д.с. +евсп.
Если индикаторный электрод имел знак «—», то
э.д.с. =евсп -е* и
e/i = е* = евсп “ Э-Д-С-
Значения потенциалов различных вспомогательных электродов приведе¬
ны выше.
Кроме величины ОВП, часто вычисляют еще величину гН2, которая
численно равна такому давлению молекулярного водорода в системе, кото¬
рое может создать данное значение ОВП. Величина гН2, вычисляется по
формуле
гН.=-§- + 2 pH,
если eh выражен в милливольтах, а температура равна 20° С. Величина гН2,
вообще говоря, лишена строгого физического смысла, и ее использование не
дает каких-либо реальных преимуществ по сравнению с обычным значением
ОВП. Исключение могут составить только некоторые чисто биологические
системы.
КОЛОРИМЕТРИЧЕСКИЕ ОПРЕДЕЛЕНИЯ pH и ОВП
При развитой технике потенциометрических измерений колориметрия
крайне редко применяется для измерения pH и ОВП. Способы соответству¬
ющих определений описаны И. П. Сердобольским (1965).
Не возвращаясь к этим способам, мы приводим только данные некоторых
важнейших кислотно-основных и окислительно-восстановительных индика¬
торов (табл. 15, 16).
I 111 И
Дсадки
600
Л -
^ / v /' /
600 h 'ч~' ч\ /•-''■Л /
v - it
V
// /6 19 23 27 2 6
Июль Август
286
Д. С. Орлов
Таблица 15
Важнейшие кислотно-основные индикаторы
Кя
Индикатор
Интервал pH перехода
окраски индикатора
Концентра¬
ция, %
Растворитель
1
Метиловый фиолетовый
1-й переход
0,13-0,5
желтая—зеленая
0,1
Вода
2
Малахитовый зеленый,
1-й переход
0,13-2,0
желтая—голубовато¬
зеленая
0,1
3
Метиловый фиолетовый,
2-й переход
1,0-1,5
зеленая—синяя
0,1
См. № 1
4
Тимоловый синий,
1-й переход
1,2-2,8
красная—же лтая
0,1
а) 20%-ный спирт;
б) вода с добавлением
4,3 мл 0,05 н. NaOH
на 100 мг индикатора
5
Крезоловый красный,
1-й переход
1,9—3,1
оранжевая—желтая
0,1
а) 20%-ный спирт;
б) вода с добавле¬
нием 5,3 мл 0,05 н.
NaOH на 100 мг
индикатора
6
Метиловый фиолетовый
3-й переход
2,0-3,0
синяя—фиолетовая
0,1
См. № 1
7
р-динитрофенол
2,8-4,0
бесцветная—желтая
0,1
Вода
8
а-динитрофенол
2,8-4,4
бесцветная—желтая
Насыщенный
9
Метиловый желтый
2,9-4,0
красная—желтая
0,1 п 0,01
90%-ный спирт
10
Метиловый оранжевый
3,0-4,4
красная—оранжево¬
желтая
0,1
Вода
11
Ализариновый красный
S, 1-й переход
3,7-5,2
желтая—фиолетовая
0,1
»
12
у-динитрофенол
4,0-5,4
бесцветная—желтая
0,1
»
13
Метиловый красный
4,4-6,2
красная—желтая
0,1 и 0,2
Спирт
14
Бромкрезоловый
пурпуровый
5,2-6,8
желтая—пурпурная
0,1
а) 20%-ный спирт;
б) вода с добавлением
3,7 мл 0,05 н. NaOH
на 100 мг индика¬
тора
15
Бромтимоловый синий
6,0-7,6
желтая—синяя
0,05
а) 20%-ный спирт;
б) вода с добавле¬
нием 3,2 мл 0,5 н.
NaOH на 100 мг
индикатора
16
Феноловый красный
6,8—8,0
желтая—красная
0,1
а) 20%-ный спирт;
б) вода с добавле¬
нием 5,7 мл 0,05 н.
NaOH на 100 мг
индикатора
17
Крезоловый красный,
2-й переход
7,2—8,8
янтарно-желтая—
пурпурно красная
0,1
См. № 5
18
Тимоловый синий,
2-й переход
8,0-9,6
желтая—синяя
од
См. № 4
19
Фенолфталеин
8,2-10,0
бесцветная—пур¬
пурная
0,1 и 1,0
60%-ный спирт
Методы, определения pH и окислительно-восстановительного потенциала 287
Таблица 15 (продолжение)
JVft
Индикатор
Интервал pH перехода
окраски индикатора
Концентра¬
ция, %
Растворитель
20
Тимолфталеин
9,4-10,6
бесцветная—синяя
0,1
90%-ный спирт
21
Ализариновый красный
S, 2-й переход
10,0-12,0
фиолетовая—бледно-
желтая
0,1
См. № 11
22
Ализариновый синий БС
11,0-13,0
оранжево-желтая—
зелено-синяя
0,05
Спирт
23
Малахитовый зеленый,
2-й переход
11,5-13,2
голубовато-зеленая—
бесцветная
0,1
См. № 2
24
1,3,5-тринитробензол
12,2—14,0
бесцветная—оранже¬
вая
0,1 и 0.5
Спирт
Таблица 16
Важнейшие окислительно-восстановительные индикаторы
Л. Индикаторы, мало зависящие от pH и ионной силы раствора
Индикатор
е0, в
Окраска
окисленной формы
восстановленной
формы
2,2'-дипиридил (комплекс с руте¬
+1,33
Бесцветная
Желтая
нием)
Нитрофенантролин (комплекс с Fe2+)
+ 1,25
Бледно-голубая
Красная
н-фенилантраниловая кислота
+1,08
Фиолетово-красная
Бесцветная
п-этоксихризоидин
+1,00
Красная
Желтая
2,2'-дипиридил (комплекс с Fe2+)
+0,97
Бледно-голубая
Красная
Дифенил бенз и дин
+0,76
Фиолетовая
Бесцветная
Дифениламин
+0,76
»
»
Б. Индикаторы, чувствительные к изменению pH и ионной силы раствора
еф, • ирж pH, равном
Окраска
Индикатор
0
7
окисленной
формы
восстановлен¬
ной формы
2,6-дибромбензолиндофенол
+0,64
+0,22
Синяя
Бесцветная
Тионин (диаминофенотиазин)
+0,56
+0,06
Фиолетовая
»
Метиленовая синяя
+0,53
+0,01
Синяя
»
Индиготетрасульфоновая кислота
+0,37
-0,05
»
Индигокармин (индигодисульфоновая
кислота)
+0,29
-0,13
ь
Нейтральный красный
+0,24
-0,33
Красная
»
288
Д. С. Орлов
Литература
Баженов Я. if., Рубцова И. Г. Динамика
окислительно-восстановительных усло¬
вий и потенциала сероземно-луговых
почв под рисом.— Почвоведение, 1969,
№ Ю.
Бейтс Р. Определение pH. Теория и
практика. JI., «Химия», 1968.
Виноградова Е. Я. Методы определения
концентрации водородных ионов. Изд-
во МГУ, 1950.
Вишневская Б. Я., Емельянов И. И, Окис¬
лительно-восстановительные свойства
целинных и освоенных южных черно¬
земов Кустанайской области.— Изв.
АН КазССР, серия биол., 1970, № 1.
Вострое И. С. Методика определения ин¬
тенсивности восстановительных процес¬
сов в затопленных почвах.— В сб.
«Микроорганизмы в сельском хозяйст¬
ве». Изд-во МГУ, 1970.
Вострое И. С\, Алешин Е. Я., Долгих
Ю. Р, Влияние внесения рисовой соломы
в затопляемую почву на напряженность
в ней окислительно-восстановительных
процессов.— Изв. АН СССР, серия биол.,
1969, № 4.
Горшкова Е. И., Дементьева Т. Г, Дина¬
мика окислительно-восстановительного
потенциала сухо-степных почв при за¬
топлении.— Научн. докл. высшей шко¬
лы, серия биол. науки, 1971, № 9.
Ежов Ю. И. Влияние затопления на по¬
движность фосфора в почве рисового по¬
ля.— Труды Кубанск. с.-х. ин-та, 1968,
вып. 17 (45).
Захарьевский М. С. Оксредметрия. Л.,
«Химия», 1967.
Зырин Я. Г., Орлов Д. С. Физико-хими¬
ческие методы исследования почв. Изд-
во МГУ, 1964.
Ионоселективные электроды. М., «Мир»,
1972.
Кауричев И. С., Тарарин С. Я., Тарари-
• на Л. Ф. Сезонная динамика окисли-
тельно-восстановительного потенциала
серых лесных почв.— В сб. «Вопросы био¬
логии», вып. 2. Тула, 1969.
Кауричев И. С., Шишова В. С. Окисли¬
тельно-восстановительные условия почв
легкого механического состава Мещер¬
ской низменности.— Почвоведение, 1967,
№ 5.
Кураев В. Н. О влиянии восстановитель¬
ных процессов на свойства почв и рост
растений.— Агрохимия, 1967, № 7.
Кураев В. Н. Особенности окислительно¬
восстановительных условий в оглеенных
дерново-подзолистых почвах.— Почво¬
ведение, 1969, № 3.
Ларешин В. Г., Илюхин М. Я. Окисли¬
тельно-восстановительные условия за¬
соленных почв при избыточном увлаж¬
нении— Докл. ТСХА, 1970, вып. 160.
Ларешин в. А, Кауричев И. С. Окисли¬
тельно-восстановительный режим почв
солонцового комплекса под культурой
затопляемого риса в Сарпинской низ¬
менности.— Изв. ТСХА, 1969, вып. 6.
Латария В. Я. Окислительно-восстанови¬
тельные свойства лугово-коричневых
почв Грузии.— Труды Груз. с.-х. ин-та,
1967, т. 71-72.
Нгуен Виу Алешин С. Я. Математический
способ выражения окислительно-восста¬
новительного состояния почв и раствори¬
мости фосфатов.— Ивв. ТСХА, 1966,
вып. 4.
Орлов Д. С., Розанов Б. Г., Саакян С. Г.
Образование железистых аккумуляций
в долинах малых рек южной тайги.—
Почвоведение, 1970, № 7.
Панов Н. Я., Шардаков А. Я. Сравни¬
тельная характеристика окислительно¬
восстановительных процессов в почвах
солонцовых комплексов степной зоны.—
В сб. «Мелиорация солонцов», ч. 1. М.,
1968а.
Панов Я. Я., Шардаков А. Я. Динамика
окислительно-восстановительных про¬
цессов в почвах легкого механического
состава солонцовых комплексов При¬
иртышья.— Докл. ТСХА, 19686, вып.
133.
Сердобольский И. Я. Методы определения
pH и окислительно-восстановительного
потенциала при агрохимических иссле¬
дованиях.— В сб. «Агрохимические ме¬
тоды исследования почв», изд. 4-е. М.,
«Наука», 1965.
Соломин Г. А. К методике определения
окислительно-восстановительного потен¬
циала и pH осадочных пород. М., «Нау¬
ка», 1964.
Степанец И. Г., Самолина М. А. Окис¬
лительно-восстановительные условия со¬
лонцов при культуре риса.— В сб.
«Мелиорация солонцов», ч. 1. М., 1968.
Тотев Г., Трънков И. Окислително-редук-
ционният потенциал в излужена кане-
лена горска почва при поливни и непо-
ливни условия. Научни тр. Висш. сел-
скостоп. ин-т Г. Димитров.— Агрон.
фак. Сер. общ. земед., 1964, 13.
Шишов Л. Л. Влияние режима увлажне¬
ния на окислительно-восстановительное
состояние красноземов и желтоземов
Аджарии.— Труды Ун-та дружбы наро¬
дов им. П. Лумумбы, т. 35, вып. 2. М.,
1969.
Шпейзер Г. М., Чарчиди Л. А, Приме¬
нение графитовых электродов для изме¬
рения величины окислительно-восста-
новительного потенциала природных
вод.— Труды Иркутск, ун-та, ч. I.
1970, т. 50, серия хим., вып. 3.
У тома Таюки. Метод измерения степени
восстановления почв рисовых полей.
Японск. патент, кл. 113 El (G01n),
№ 8279, заявл. 5.04.1968, опубл.
9.03.1972, 1972.
В ache В. W. Determination of pH, lime
potential and aluminium hydroxide po¬
tential of acid soils.— J. Soil Sci., 1970,
y. 21, N 1.
Bailey L. D.y Beauchamp E. G. Nitrate re¬
duction and redox potentials measured
r
Методы определения pH и окислительно-восстановительного потенциала 289
with permanently and temporarily placed
platinum elecrodes in saturated soils.—
Ganad. J. Soil Sci., 1971, v. 51.
Bohn H. L. Redoox potentials. — Soil Sci.,
1971, v. 112, N 1.
Bohn H. L. Electromotive force of inert
electrodes in soil suspensions.— Soil Sci.
Soc. America Proc., 1968, v. 32. N 2.
Bohn H. L. The EMF of platinum electro¬
des in dilute solutions and its relation
to soil pH.— Soil Sci. Soc. America Proc.
1969, v. 33, N 4.
Breemen AT. van. The effect of illdefined
ferric oxides on the redox characteristics
of flooded soils.— Netherl. J. Agric.
Sci., 1969, v. 17, N 4.
Cremers A. Chemical and physico-chemical
aspects of soil and clay acidity.— Agri-
cultura (Bek), 1972, v. 20, N 2.
Csakvari B. The significance and applica¬
tion of glass electrodes for pH measure¬
ments.— Hung. Sci. Instrum., 1965, v.
9—16, N 3.
Dahlgreen G., Goodfriend M. J. Quinhyd-
rone electrode drift.— Analyt. Chem.,
1970, v. 42, N 1.
Davies Brian E. A statistical comparison
of pH values of some English soils after
measurement in both water and 0,01 M
calcium chloride.— Soil Sci. Soc. Ame¬
rica Proc. 1971, v. 35, N 4.
Galster H. Neure Ent wick lung der elektro-
metrischen pH-Messung mit Glaselektro-
den.— Glas- und Instrum. Techn., 1970,
Bd. 14, H. 4.
Jamane Sato K. Some problems in
the measurement of Eh of flooded soils.—
Sci. Repts. Res. Inst. Tohoku Univ.,
D, 1970, v. 21, 65—77.
Me George W. T. Isohydric pH, pH of
soil paste, and pH excnange neutrality.—
Soil Sci., 1945, v. 59, 231-237.
Olsen R. A.у Robbins J, E. The cause of
the suspension effect in resin-water sy¬
stems.— Soil Sci. America Proc., 1971,
v. 35, N 2.
Patrick W. H. Extractable iron and phos¬
phorus in a submerged soil at controlled
redox potentials.— Trans. 8th Internat.
Congr. Soil Sci. Bucharest, 1964, v. 4,
605-609.
Patrick W. #., Turner F. T. Effect of
redox potential on manganese transforma¬
tion in water-logged soil.— Nature, 1968,
v. 220, N 5166.
Piasecki Kotowska J. Proba wyjasnie-
nia dlaczego pHkc[ bywa niekiedy
wyzsze niz pHHs0 — Zesz. nauk. Wyzsza
Szkola roln. Szczecinie. 1966, N 21, 87—
97.
Piskula К. Wplyw roznych sposbow susze-
nia i przechowywania probek glebowych
na zawartosc przyswajalnego fosforu i
potasu у glebie oraz jej. Pr Inst. upr.
nawoz. i glebozn. 1971, N 42, 145—158.
Ponnamperuma F. W., Tianco E. ЛГ., Loy
T. Redox equilibria in flooded soil. 1.
The iron hydroxide systems.— Soil Sci.,
1967, v. 103, N 6.
Resulovic H. Dinamika vode, vazduha i
oksido-redukcionug potencijala u para-
podzolu sjeverne Bosne. Rad. Poljopr.
fak. Univ. Sarajevu, 1970, N 21.
Schachtschabel P. Methodenvergleich zur
pH-Bestimmung von Boden.— Z. Pflan-
zenernahr., Diing. und Bodenkunde, 1971,
Bd. 130, N 1.
Schofield R. K.y Taylor A. W. The measu¬
rement of soil pH.— Soil Sci. Soc. Ame¬
rica Proc., 1955, v. 19, N 2.
Simon W., Levi E, L’elettrodo di vetro
e le sue applicazioni per la misurazione
dela pH. Ina. vemice, 1966, v. 20, kg. 12.
Tschapek Л/., Santamaria R., Carlson R. M.
Junction potentials and pH determina¬
tion in het^erogeneouse systems.— Agro-
chimica, 1966, № 3, 230—243.
White R. E. On the measurement of soil
pH.— J. Austral. Inst. Agric. Sci., 1969,
v. 35, № 1.
Yamane Ichiro, Sato Kazuo. Initial rapid
drop of oxidation. — reduction potential
in submerged air-dried soils.— Soil Sci.
and Plant Nutr., 1968, v. 14, № 2.
А. М. АЛЕКСАНДРОВА
ОПРЕДЕЛЕНИЕ АКТИВНОСТИ ИОНОВ КАЛИЯ
В ПОЧВАХ
*
Изменение активности вещества в растворе обусловлено как изменением
числа частиц в растворе, так и изменением энергии этих частиц, т. е. как хи¬
мическим, так и физическим взаимодействием. Следовательно, в реальных рас¬
творах, в том числе и в почвенных растворах, данное конкретное вещества
проявляет свои свойства в зависимости от окружающих условий. Таким об¬
разом, одно и то же вещество при одинаковых концентрациях может действо¬
вать различно и, наоборот, одинаково при различных концентрациях в зави¬
симости от совокупности факторов. Связано это с тем, что активность вещест¬
ва в этих различных условиях будет также различной.
Метод активности применяется во всех случаях, когда нужно количест¬
венно из данных опыта охарактеризовать состояние любого компонента в ре¬
альном растворе.
Размерность активности выражают в тех же единицах, что и концентра¬
цию. Эти две величины — активность (а) и концентрация (С) связаны между
собой равенством:
<* = £./, (1)
где / — средний коэффициент активности.
В бесконечно разбавленных растворах, когда концентрация стремится к
нулю, а активность при этом будет стремиться к величине концентрации,
коэффициент активности, естественно, будет стремиться к единице, и в пре¬
деле, когда а = С, / = 1.
Так как коэффициент активности суммарно учитывает как физические,
так и химические процессы, имеющие место в данном растворе, т. е. учитыва¬
ет все формы взаимодействия, которые происходят в данной системе в ее рав¬
новесном состоянии, то его величина может значительно изменяться и быть
как меньше, так и значительно больше единицы. Соответственно этому и ак¬
тивность может быть как меньше, так и больше аналитической концентра¬
ции.
В настоящее время метод определения активности нашел самое широкое
распространение. Достаточно назвать величину pH, получившую универ¬
сальное распространение как в сельском хозяйстве, биологии, медицине,
химии, так и в промышленности. Величина pH является отрицательным лога¬
рифмом активности ионов водорода, а не их концентрации.
Метод активности в почвоведении исключительно перспективен, он вно¬
сит новые качественные понятия при оценке физико-химических процессов,
протекающих в почвах. Кроме того, электрометрический метод определения
активности ионов позволит, пользуясь переносными портативными иономе-
рами, непосредственно в поле производить определение элементов.
Развитие теории стеклянного электрода позволило создать электроды
с металлической функцией для измерения активностей ионов натрия и ка¬
лия (Шульц. 1964). В настоящее время стеклянные электроды, обратимые
к этим ионам, выпускаются нашей промышленностью.
Ионселективный К-стеклянный электрод работает согласно закону Нерн-
ста, как и стеклянный водородный электрод. В цепи с К-стеклянным
Определение активности ионов калия в почвах
291
электродом:
У? Т
Е = Ео + 2,303 пр lgaK+, (2)
где Е — электродвижущая сила цепи со стеклянным калиевым электродом; Е0 — стан¬
дартная электродвижущая сила цепи, которая объединяет все слагаемые электродвижу¬
щей силы цепи, не зависящие от состава исследуемого раствора; R — универсальная
газовая постоянная; Т — абсолютная температура; п — валентность определяемого ио¬
на; F — число Фарадея; а^+ — активность ионов калия в растворе.
Электродвижущая сила цепи будет пропорциональна величине активности
ионов калия в исследуемом растворе, так как Е0 является величиной посто¬
янной для данной цепи, а множитель RT/nF зависит только от температуры,
которая практически за время измерения не изменяется.
Строгая калиевая функция имеет место у стеклянных электродов из спе¬
циальных электродных стекол. Такие электроды в настоящее время выпус¬
каются под шифром ЭСЛ-96-10 и малогабаритные — ЭСЛ-96-11 из электрод¬
ного стекла марки № 26. Эти электроды выпускаются в виде шарика, как и
для измерения величины pH. Они обладают, по сравнению с электродами
с другими функциями, наименьшим сопротивлением — 15 + 10 Мом при
20° С и могут быть применимы для измерений в средах до температуры +60° С.
Морозостойкость у них низкая, всего до +5° С. Это нужно учитывать при
их транспортировке.
Согласно проспекту, пределы измерений калиевого электрода 0—3 рК,
однако, согласно нашим измерениям, его калиевая функция сохраняется -
в более широких пределах концентраций при комнатной температуре — до
4—4,2 рК (рК = — lg aK+).
Для работы с почвами большое значение имеет селективность применяе¬
мых электродов. По нашим данным, преобладание кальция, вплоть до 50-
кратного превышения по отношению к калию, не сказывается на показаниях
этих электродов. Содержание ионов лития в растворе допустимо до пятикрат¬
ного преобладания, что не может лимитировать применение этих электродов
в почвенных средах. Катион аммония начинает оказывать некоторое влия¬
ние на показания К-электрода при одинаковом содержании с ионом калия.
Однако этот катион присутствует в почвах обычно в незначительных коли¬
чествах, так что такой низкий предел не будет являться помехой для приме¬
нения электродов ЭСЛ-96-10.
Иначе дело обстоит с присутствием в почве ионов натрия. В этом случае
предельным также является соотношение К : Na как 1 : 1. Это значительно
суживает область прямого применения стеклянного электрода с К-функцией
из электродного стекла N 26, которое на сегодняшний день является луч¬
шим. Таким образом, применение К-электрода ограничивается кругом почв
с содержанием натрия, не превышающим содержание калия. Однако если
проводить измерение одновременно Na- и К-электродами, то в этом случае,
пользуясь методом и таблицей, предлагаемыми Н. К. Крупским, А. М. Алек¬
сандровой и Л. А. Раппопорт (1974), можно определять активность ионов
калия в почвах, в которых содержание натрия превышает содержание калия
в 13 раз.
Вновь полученные калиевые электроды следует предварительно выдер¬
жать в 0,1 н. растворе КС1 не менее 14—15 суток. При этом шарик электрода
должен быть полностью погружен в раствор. В дальнейшем, в нерабочее вре¬
мя, электроды также должны храниться в 0,1 н. растворе КС1.
Электродная функция из К-стеклянных электродов проверяется калиб¬
ровкой по серии стандартных растворов, т. е. по растворам с точной концен¬
трацией и известной активностью ионов калия. Для этого целесообразно
10*
292
А. М. Александрова
использовать следующую серию растворов хлористого калия:
м
рК
м
рК
0,2000
0,84
0,0050
2,33
0,1000
1,11
0,0010
3,01
0,0500
1,39
0,0005
3,32
0,0250
1,67
0,0003
3,52
0,0100
2,04
0,0001
4,00
Хлористый калий обязательно должен быть предварительно перекристал-
лизован (из соли квалификации х. ч.) или использован из фиксанала. Вели¬
чины рК, которые здесь приведены, рассчитаны по данным для коэффициен¬
тов активности.
Пример такого расчета мы приводим для 0,2 М раствора КС1. В этом растворе
средний коэффициент активности хлористого калия равен 0,718 при температуре 20° С.
Соответственно равенству (1) активность ионов калия ак+ в данном растворе будет равна
0,2 X 0,718 = 0,1436 М. Логарифмируя полученное выражение, получим — 1 g
= рК = 0,8428 пли, округляя, рК = 0,84.
Для проведения калибровки стеклянного К-электрода и при последую¬
щих измерениях используется цепь:
Электродное
стекло
Ag, AgCl I К Cl
I раствор
внутренний электрод
сравнения
К-стеклянный индикаторный
электрод
Раствор
для измере¬
ния
Электроли¬
тический
ключ с
СН3С001л
КС1
насыщенный
раствор
AgCl, Ag
Хлорсеребряный
электрод сравнения
Для составления такой измерительной цепи стандартный раствор нали¬
вается в небольшой стаканчик, в него полностью погружается шарик стек¬
лянного К-электрода, который подключен клеммой к любому современному
потенциометру (ЛПУ-01, ЛПМ-60, рН-340 и т. п.). В этот же стаканчик опу¬
щен один конец электролитического ключа, второй его конец погружен в ста¬
канчик с насыщенным раствором хлористого калия, в который, в свою оче¬
редь, погружен кончик хлорсеребряного электрода сравнения (например,
типа ЭВЛ-1М).
Обычный агар-агаровый ключ с КС1 для этих измерений не пригоден из-за
возможной диффузии в исследуемую среду ионов калия. Для этой цели может
быть использован солевой мостик с уксуснокислым литием. Изготавливается
он аналогично хлоркалиевому: в 100 мл дистиллированной воды растворяется
при нагревании 3 г белого агар-агара до получения однородной массы, добав¬
ляется 10 г уксуснокислого лития (х. ч.) и после его растворения еще теп¬
лая масса затягивается резиновой грушей в сухие стеклянные сифоны. Хра¬
нятся эти мостики в растворе уксуснокислого лития (60—65 г соли на 100 мл
воды). Перед использованием мостик должен быть тщательно ополоснут и осу¬
шен фильтровальной бумагой.
Для построения калибровочного графика электрода последовательно во
всех растворах производится измерение э. д. с. цепи и данные откладываются
на миллиметровой бумаге в координатах: по оси ординат — величина э. д. с*
в милливольтах, по оси абсцисс — величины рК стандартных растворов
(рис. 1).
Калибровка электрода и построение калибровочного графика должны
быть выполнены очень тщательно, так как по этой прямой, во-первых, судят
о рабочих качествах электрода и, во-вторых, находят искомые величины ак¬
тивности ионов калия в исследуемых средах.
Определение активности ионов калия в почвах
293
Если электрод обладает нормальной электродной функцией, то коэффи¬
циент 0 = 2,303 RTInF в уравнении (2) будет соответствовать своему теоре¬
тическому значению (возможно, с небольшим отклонением). Этот множитель
зависит от температуры и для широкого диапазона температур приводится
в справочниках. Здесь мы приводим эту величину лишь в интервале комнат¬
ной температуры:
t, °с
р т
2,303 —4г , мв
nF
t, °G
2,303 r
nb
18
57,77
22
58,56
19
57,97
23
58,76
20
58,16
24
58,96
21
58,36
25
59,16
Для нахождения угла наклона калибровочной кривой, соответствующего,
множителю в уравнении (2), подсчитывается отношение величин Д£УДрК
Например, при 25° С э. д. с. цепи со стандартным раствором 0,1 М КС1 (рК =
= 1,11) равна 101,0 мв, а с 0,01 М раствором КС1 (рК = 2,04) — 46,0 мв.
55
Таким образом, АЕ = 55 мв, ДрК = 0,93. Их отношение Д2?/ДрК = ^=
0,Уо
= 59,1 мв, В данном случае мы имеем идеальное совпадение, однако весьма
часто может быть отклонение до 1—2 мв. Удобнее расчет углового коэффи¬
циента производить непосредственно по калибровочному графику, в этом
случае берется разность между двумя
величинами э. д. с. по ординате для
целых значений рК, отложенных по
абсциссе (например, между рК 2 и 3).
Каждый день перед измерениями
следует проверять стабильность пока¬
заний электрода хотя бы по трем стан¬
дартным растворам и строить калибро¬
вочную прямую, по которой и будет
определяться активность ионов калия
в соответствии с измеренной величиной
э. д. с.
Для определения активности ионов
калия в исследуемой среде ее помеща¬
ют в небольшой стаканчик, полностью
погружают в нее шарик К-стеклянного
электрода. Та же цепь, с теми же ~т'це 1 ^ 1 ^ ' з,о ' з,в ' Рк
электродами, которые были использо- ;
ваны и ДЛЯ калибровки, замыкается тем Рис. 1. Калибровочный график калие-
же электролитическим мостиком, и сни- вого стеклянного электрода ЭСЛ-96-10
маются показания на потенциометре
после достижения постоянного значения
э. д. с. Обычно на это нужно 5—7 мин. В соответствии с найденной величи¬
ной э. д. с. цепи по калибровочному графику на оси абсцисс находится вели¬
чина рК.
Если нужно иметь данные активности, а не рК, то последние рассчиты¬
ваются согласно равенству рК = — lg0K+* Например, найдено, что рК =
= 3,47. Следовательно, рК = — lgajt+ = 3,47, или lgaK+ = —3,47 = 5,53.
Откуда а = 0,00034 М (исходя из выражения концентрации стандартных
растворов) или, переходя к другим единицам,—0,34 мг-экв в 1 л.
Определение активности ионов калия надо производить при возможно
близких условиях к естественным, непосредственно в поле. Для этого на
сегодняшний день уже есть прочные малогабаритные электроды для измере¬
100
60
ZO
о
-20
-60
294
А. М. Александрова
Рис. 2. Переносной иономер
И-102
Рис. 3. Общий вид стеклянных
электродов с калиевой функ¬
цией типа ЭСЛ-96-10 и ЭСЛ-
96-11
ния рК — ЭСЛ-96-11 и переносный иономер И-102, который с 1973 г. выпу¬
скается нашей промышленностью (рис. 2).
Переносный иономер И-102 весьма портативен, его габариты с футляром
составляют 225 х 135 х 75 мм. Вместе с иономером поставляется футляр
для транспортировки электродов и необходимых для измерения предметов:
штативчика, стаканов и т. п. При этом его размер точно подогнан к габаритам
футляра (225 х 85 X 75 мм), он удобно к нему пристегивается, и весь комп¬
лект вместе с иономером можно носить через плечо. Вес такого комплекта,
включая иономер И-102, всего 2,8 кг.
Определение активности ионов калия в почвах
295
Питание иономера может быть осуществлено как от сети переменного тока
напряжением 220 в, 50 гц, так и от батареи типа «Рубин». Таким образом,
он может быть использован как в стационарных условиях, так и в поле. Од¬
нако при использовании сухих батарей, т. е. в нестационарных условиях,
дрейф нуля иономера несколько увеличивается — о^ 2 мв/час при питании
от сети переменного тока до 4 мв/час.
Пределы измерения э. д. с. у этого иономера — от 0 до + 1200 мв и от
0 до + 400 мв (со сдвигом начала шкалы в пределах —400 1-400 мв).
Прибор может быть использован при температуре окружающего воздуха
до 50° С и относительной влажности до 95%. Для предотвращения ошибок,
которые могут быть вызваны изменением температуры, в иономере предусмо¬
трена ручная температурная компенсация изменения э. д. с. электродной
системы. Точность температурной компенсации определяется выбором тем¬
пературного диапазона и настройкой иономера по калибровочным растворам.
К иономеру И-102, предназначенному для определения активностей одно¬
валентных и двухвалентных ионов и окислительно-восстановительного по¬
тенциала, прилагается набор малогабаритных ионселективных электродов
и электроды для определения окислительно-восстановительного потенциала
(рис. 3).
Литература
Крупский Н. К., Александрова А. М., Pan- ионов калия в почвах.— Агрохимия
попорт Л. А. Электрометрические ме- 1974, № 3.
тоды определения активности ионов Шульц М. М. Электродные свойства сте-
в почвах. V. Определение активности кол. (Автореф. докт. дисс.). Л., 1964.
Д. В. ФЕДОРОВСКИЙ
ОПРЕДЕЛЕНИЕ ВОДНЫХ И ФИЗИЧЕСКИХ СВОЙСТВ ПОЧВЫ
ПРИ ПРОВЕДЕНИИ ПОЛЕВЫХ И ВЕГЕТАЦИОННЫХ ОПЫТОВ
*
Наблюдения за динамикой питательных веществ в почве и поступлением
их в растение не могут дать исчерпывающей и правильной картины состояния
почвы, если параллельно не учитывать такие важные факторы плодородия,
как влажность, аэрацию, плотность, механический и минералогический со¬
став и другие свойства почвы. Ниже приведены в сжатом изложении методы,
которые наиболее часто применяются при агрохимических исследованиях
пахотных почв. Более полное освещение существующих методов и описание
специфических приемов исследования болотных и каменистых почв можно
найти в руководствах, указанных в списке литературы.
Содержание воды в почве — важнейший фактор плодородия, нередко опре¬
деляющий эффективность удобрения, многих операций по обработке почв,
уровень развития биологических процессов. В свою очередь минеральное
питание воздействует на использование влаги растениями. При проведении
полевых опытов агрохимику приходится многократно определять влажность
почвы, чтобы учесть изменение запасов доступной для растений воды во вре¬
мени.
Непосредственно на участке полевого опыта определяют объемный вес,
наименьшую (предельную полевую) влагоемкость, уровень грунтовых вод
и для орошаемых условий — водопроницаемость. Кроме того, в лаборатории
исследуют взятые в поле образцы и определяют механический состав, удель¬
ный вес, порозность, агрегатный и микроагрегатный состав и недоступную
для растений воду.
Устойчивые во времени свойства почвы: влагоемкость, удельный вес, ме¬
ханический состав, влажность завядания, максимальную гигроскопичность
достаточно определить 1—2 раза за период севооборота и иметь данные об из¬
менении их с глубиной почвы. Объемный вес, глубину грунтовых вод определя¬
ют 3—4 раза за время вегетации.
При проведении вегетационных опытов также желательно иметь полную
характеристику водно-физических свойств почвы. Совершенно необходимо
при закладке опыта определить влажность почвы, влагоемкость (полную,
капиллярную или наименьшую), недоступную влагу (влажность завядания
или максимальную гигроскопичность) и механический состав. Постоянный
контроль за изменением влажности почвы в вегетационном опыте осущест¬
вляется путем взвешивания сосудов при поливе.
НАБЛЮДЕНИЯ ЗА ВЛАЖНОСТЬЮ ПОЧВЫ
В ПОЛЕВЫХ опытах;
В полевых опытах для сопряженных наблюдений за изменением влажно¬
сти и режима питательных веществ на делянке выбирают по возможности
более однородную по микрорельефу и состоянию растений площадку разме¬
ром 2x3 м или несколько более. Границы площадки отмечают натянутым
шнуром. В пределах этой площадки производят бурение в течение опыта, пе¬
редвигаясь по строкам от одного края к другому. Сроки наблюдения приуро¬
Определение физических и водных свойств почвы
297
чивают к фазам развития культуры, а если участок орошаемый — то и к
поливам. Эта площадка в конце опыта исключается из учетной площади де¬
лянки, так как при бурении неизбежно некоторое повреждение растений.
Отдельные скважины на площадке следует располагать не ближе 50 см
одна от другой и тщательно забивать их почвой после отбора проб, чтобы из¬
бежать затекания в них воды и искажения водного режима. Забитые скважи¬
ны следует отмечать колышком, чтобы не попасть в эту точку при повторном
бурении.
Выбор числа площадок и числа скважин на каждой площадке зависит от
однородности поля и рельефа. При выровненных площадях для характери¬
стики одного варианта опыта достаточно иметь две площадки, заложенные
Рис. 1. Рабочая часть буров
А — бур Измаильского (саратовс¬
кий);
В — деркульекий;
В — малый бурик для взятия проб
почв из вегетационных опытов
на делянках разных повторностей, и с каждой делянки в каждый срок брать
образцы из трех — пяти скважин. Влажность почвы определяется по слоям
из индивидуальных скважин, содержание^питательных веществ — из сме¬
шанного образца одной повторности.
Следует учитывать, что в широкорядных и гнездовых посевах происходит
неоднородное использование всей площади корневыми системами, а также
наблюдается перераспределение осадков и неодинаковое увлажнение укры¬
тых растениями участков почвы и междурядий. В этих случаях наблюдения
приходится проводить и в междурядьях, и в рядах или вблизи гнезда.
Отбирать пробы почвы до глубины 1 м удобно почвенными бурами неболь¬
шого диаметра (25—30 мм), например саратовским системы Измаильского
или деркульским (рис. 1, А и Б). Производительность труда при отборе проб
зависит от качества бура, который должен быть удобным, легким и прочным.
У хорошего бура рабочая часть (цилиндр) изготовлена из листовой (рессор¬
ной) стали не толще 2 мм, а режущие края остро заточены.
В вегетационных опытах отбор проб из сосуда во время роста растений
нежелателен, так как повреждает корневые системы. В тех случаях, когда
взять пробу все же необходимо, применяют небольшой бурик с боковой про¬
резью, изготовленный из латунной трубки диаметром около 20—24 мм, за¬
крепленной на деревянной ручке. Длина бурика должна соответствовать вы¬
соте сосуда (рис. 1, В).
Наблюдения за режимом питания растений включают обычно определе¬
ние влажности в слоях 0—5, 5—10, 10—20 и далее через 10 см до глубины
100 см. В почвах с ясно выраженной дифференциацией глубину взятия образ¬
цов приурочивают к генетическим горизонтам. 2—3 раза за лето определяют
влажность и запасы влаги в 1—2-метровом слое, а в остальные сроки наблю¬
дения можно ограничить 40 или 60-ю см. В слоях, где свойства почвы и влаж¬
ность изменяются мало, допустимо более редкое взятие проб (например, для
298
Д. В. Федоровский
мощного чернозема 0—10, 10—20, 20—40 и 40—60 см). На поливных участ¬
ках перед поливом и после него образцы берут на большую глубину, отвечаю¬
щую глубине промачивания.
Удобно брать пробы почвы в алюминиевые стаканчики диаметром 40 мм%
с четкими номерами на крышке и самом стаканчике. Пробу почвы в 30—
40 г помещают в стаканчик и быстро закрывают крышкой, чтобы избежать
потерь влаги при переноске. Во время дождя взятие проб почвы на влажность
прекращают. Недопустимо переворачивать стаканчики при переноске, так
как при этом часть почвы попадает под крышку и теряется при высушивании,
искажая результаты определения.
Определение влажности высушиванием
Наиболее распространен и достаточно точен весовой метод определения
влажности почвы высушиванием. Для изучения динамики влажности допол¬
нительно используют влагомеры-тензиометры, приборы с нейтронным излу¬
чателем и др.
Доставленные в лабораторию стаканчики с почвой взвешивают на техно-
химических весах с точностью 0,05 г и высушивают до постоянного веса в те¬
чение 6—10 час. после разогревания шкафа. Температура сушки почвы с вы¬
соким содержанием гумуса С>8%), торфяных и солончаковых, не должна
превышать 105°, для малогумусных почв допускается повышение температу¬
ры до 125—150°, что значительно ускоряет процесс.
Наиболее удобны для сушки электрические шкафы с терморегулятором,
но в экспедиционных условиях применяют также сушильные шкафы на га¬
зовой горелке или керосинке, регулируя нагрев силой огня.
Стаканчики с почвой после сушки закрывают крышками и охлаждают на
столе, после чего взвешивают. Контрольное взвешивание после 2-часовой
повторной сушки не должно давать расхождения веса более чем на 1 % опре¬
деляемой величины. Очень удобны для взвешивания быстродействующие весы
BJITK-500. Влажность почвы вычисляют в процентах на сухую почву до пер¬
вого знака после запятой.
В журнале определения влажности записывают следующие данные:
Номер образца, место взятия пробы, глубина.
Номер стаканчика.
Вес сырой почвы + вес тары стаканчика (А).
Вес высушенной почвы + вес тары (В0).
То же, контрольное взвешивание (2?).
Вес испарившейся воды (А — В).
Вес тары стаканчика (С).
Вес сухой почвы (В — С).
Влажность (W, вес. %) определяется по формуле
, А — в
w — в — С *100 *
Если определяют влажность в образце подсушенной и просеянной через
сито почвы, то удобнее употреблять стеклянные сушильные стаканчики и
брать небольшую навеску — 4—5 а, а взвешивание производить на анали¬
тических весах с точностью до 1 мг. Охлаждать высушенную почву в этом
случае следует в эксикаторе над хлористым кальцием. Сушка малых навесок
требует обычно 5—6 час.
При всех анализах почвы следует определять ее влажность для перерасчета
содержания веществ в процентах на сухую почву. Коэффициент, на который
следует умножить содержание любого вещества в миллиграммах на 100 г
Определение физических и водных свойств почвы
299
влажной или воздушно-сухой почвы для перевода на абсолютно сухую поч¬
ву, вычисляется по формуле
100 4- w
К- 100 •
Иногда применяют другую формулу: К = однако следует
помнить, что в этом случае влажность Wx выражена в процентах от веса
исходной сырой почвы.
Нейтронный влагомер и гамма-плотномер
Выпускаемые промышленностью приборы — нейтронный индикатор влаж¬
ности (НИВ) и глубинный гамма-гамма-плотномер (ГГП) — удобно при¬
менять при многолетних стационарных наблюдениях за изменением влаж¬
ности почвы естественного сложения до глубины 3—4 м и более. На засолен¬
ных почвах прибор НИВ невозможно использовать, на щебнистых почвах не
всегда удается бурение.
Зонд-датчик НИВ имеет источник быстрых нейтронов (10б нейтрон/сек)
в ампуле и блок из четырех счетчиков СТС-5, экранированных кадмием. ГГП
имеет цезиевый источник активностью 1,5 г-экв Ra и блок из четырех счетчи¬
ков СТС-5. Аппаратура поставляется фирмой «Изотоп». Вес зонда влагомера
1 кг, гамма-плотномера 3 кг, пересчетного устройства 5,5 кг и контрольно-ка¬
либровочного устройства 13 кг. Для перевозки приборов необходимо иметь
тележку. Питание прибора осуществляется от кассетных батарей
87 ПМЦГ-0,15.
Показания приборов характеризуют влажность и плотность почвы в сфе¬
ре диаметром 15—30 см. Глубинные приборы не пригодны для проведения из¬
мерений в слое 0—20 см. Пределы измерения влаж¬
ности 2—40 об.% и плотности 1—2,5 г!смг.
В намеченном месте бурят скважину и вдвигают
в нее тонкостенную дюралюминиевую обсадную тру¬
бу с заглушкой нижнего отверстия. К прибору при¬
лагается набор труб длиной 10 м при диаметре
50 мм. Можно использовать также пластмассовые или
железные трубы, причем следят, чтобы их диаметр
точно соответствовал диаметру имеющихся буров,
так как образование зазора между трубой и стен¬
кой скважины недопустимо. Во избежание затека¬
ния осадков вдоль трубы, поверхность почвы, при¬
легающая к трубе, уплотняется и в радиусе 10—
15 см примазывается глиной, цементным раствором Рис. 2. Зависимость спо¬
или смолой. Верхнее отверстие трубы закрывают рости счета тепловых
крышкой, предупреждая от попадания осадков внутрь д^вы°Н°В °Т влажности
трубы. На каждой делянке закладывают не менее
трех скважин.
Для каждой точки работу начинают и заканчивают определением скоро¬
сти счета на контрольно-калибровочном устройстве. Затем зонд-счетчик на
гибком кабеле закрепляют последовательно на разных глубинах в скважине
и производят отсчеты скорости счета регистрирующего прибора. Вычисляют
отношение скорости счета на испытуемом образце к скорости на калибровоч¬
ном устройстве и по калибровочным кривым определяют влажность почвы.
Калибровочная кривая стандартного прибора НИВ показана линией 1\ не¬
большая переделка прибора (Бескин и др., 1970) позволяет увеличить диапа¬
зон измерения (линия 2 на рис. 2).
Калибровочные кривые приложены к каждому прибору, однако перед
началом массовых определений следует произвести корректировку кривой
Д. В. Федоровский
для конкретного типа почвы. В этих целях параллельно определяют влаж¬
ность при помощи прибора и весовым методом в момент закладки скважин.
Одновременно определяют при корректировке и плотность почвы по гори¬
зонтам гамма-гамма-плотномером или с помощью бурика Качинского. Эти
данные необходимы для перевода весовых процентов в объемные.
При соблюдении указанных условий погрешность определений влажно¬
сти не превышает 1—2% (абс.). Одно определение требует 3—5 мин.
Полевое определение наименьшей
влагоемкости почвы
Наименьшая (или предельная полевая) влагоемкость показывает коли¬
чество воды, удерживаемое почвой в практически неподвижном состоянии
после обильного полива и просачивания избыточной воды под влиянием силы
тяжести. Определение делается в природных условиях. При залегании грун¬
товых вод глубже 3 м определение показывает «истинную наименьшую вла¬
гоемкость», а при более близких грунтовых водах — более высокое содержа¬
ние, достигающее величины «капиллярной влагоемкости». Глубину грунто¬
вых вод следует указывать при определении.
Влагоемкость, определяемая описанным ниже методом, называется раз¬
личными исследователями: общая влагоемкость (Качинский, Вадюнина),
предельная полевая влагоемкость (Астапов, Розов, Долгов), наименьшая по¬
левая влагоемкость (Березинь, Рыжов, Зимина), полевая влагоемкость (Ре¬
вут, Гречин).
Порядок определения наименьшей влагоемкости. Выбирают ровный, ти¬
пичный для данного поля участок и на нем окружают земляным валиком
высотой 30—40 см площадку размером 1,5 X 1,5 м. Землю для насыпания
валиков берут вне площадки, поверхность площадки оберегают от затапты¬
вания. Для ограждения площадки вместо земляных валиков иногда приме¬
няют деревянные или железные рамы. Поблизости от площадки закладывают
и описывают почвенный разрез, в стенке которого берут образцы почвы по
генетическим горизонтам для определения влажности, объемного и удельного
веса почвы.
Для промачивания почвы до 1,5 м на каждый квадратный метр площадки
надо приготовить 200—300 л на суглинистых или 200 л воды на супесчаных
почвах. Во избежание размыва поверхности под струю воды, подаваемой на
площадку, необходимо подложить кусок фанеры или слой соломы. Вода по¬
дается постепенно, так чтобы не создавать слоя воды на поверхности выше
6 см.
Когда вся поданная на площадку вода впитается в почву, ее покрыва¬
ют для предохранения от испарения с поверхности клеенкой или пластиком
и толстым слоем соломы (до 0,5 м), которую прижимают сверху землей.
Просачивание излишней воды из первого метра почвы в основном закан¬
чивается на песчаных почвах за 1—2 суток, на суглинистых — 3—5 и
глинистых — 5—10 суток. Однако и после этого срока почвенная влага про¬
должает медленно просачиваться вниз. Поэтому рекомендуют определение
наименьшей влагоемкости в три срока — через 1,3 и 10 суток, обозначая их ин¬
дексами НВ1, НВ3 и НВ10. Для песчаных и супесчаных почв достаточно оп¬
ределить НВ1 и НВ3.
Почвенные пробы для определения влажности отбирают буром с трех-
пяти мест послойно через 10 см. Для этого на площадку кладут доску и, стоя
на ней и не снимая покрытия почвы, производят бурение в центральной части
площадки 80 х 80 см. Отверстия скважин после взятия проб плотно забива¬
ют почвой.
Наименьшую (предельную полевую) влагоемкость можно определить во
всех случаях обильного увлажнения почвы — ранней весной после полного
Определение физических и водных свойств почвы
301
оттаивания почвы и впитывания талых вод или после полива орошаемых
участков. После увлажнения выбранную площадку закрывают клеенкой,
соломой и через соответствующие интервалы бурят и определяют влажность
почвы площадки.
Наименьшая влагоемкость зависит от механического состава — от 20%
объема супесчаных до 40% от объема суглинистых и глинистых почв, и не¬
сколько уменьшается с глубиной. Наименьшая влагоемкость тяжелой почвы
зависит также от сложения, приемов обработки, структурности, внесения
извести.
Вычисляют наименьшую влагоемкость послойно для каждых 10 см в про¬
центах от объема почвы, поэтому необходимо определять объемный вес почвы.
Если наименьшая влагоемкость составляет 70—80% общей порозности, то
зто считается благоприятным для сельскохозяйственных культур, при 80—
90% — посредственным, а свыше 90% — неудовлетворительным из-за недо¬
статочного содержания воздуха. Изменение влагоемкости по профилю см.
на рис. 6.
Определение уровня грунтовых вод
Грунтовые воды оказывают сильное влияние на водный режим растений
в том случае, если они находятся ближе 4—5 м от поверхности. Поэтому по¬
левые агрохимические опыты следует сопровождать контрольными замерами
грунтовых вод при их близком залегании.
На наблюдательной площадке высверливают буром скважину глубже
обычного уровня вод на 0,5—1 м. В скважину вставляют обсадную трубу с
с фильтром, засыпают нижнюю часть зазора вокруг фильтра крупным пес-*
ком, а выше зазор вокруг трубы плотно забивают почвой. Верхний конец
Рис. 3. Нижняя часть сква¬
жины для наблюдения за уров¬
нем грунтовых вод
1 — стенка скважины;
2 — часть скважины, уплотненная
грунтом;
3 — засыпка песком;
4 — сетка фильтра;
5 — стенка обсадной |трубы с от¬
верстиями U),
6 — отстойник,
7 — пробка,
9 —^ожидаемый уровень грунтовых
вод
трубы должен возвышаться над уровнем почвы примерно на 50 см. Диаметр
трубы должен быть не менее эО мм. На нижнем конце трубы, в 20 см от ее
края, для пропуска воды просверливают шесть рядов отверстий диаметром
около 10 мм и закрывают их прочно закрепленной латунной сеткой фильтра
с ячейками диаметром 0,25—0,5 мм. Нижний конец трубы забивают проб¬
кой (рис. 3).
Простейлим прибором для замера уровня воды в трубах является сталь¬
ная мерная лента, на конце которой прикреплена «хлопушка»— переверну¬
302
Д. В. Федоровский
тый вверх дном металлический стакан диаметром 30 мм, ко дну которого
прикреплена лента. Прикосновение хлопушки и отрыв ее от воды дает гром¬
кий хлопок. Отсчет по ленте ведут от верхнего края трубы и вносят поправки
на превышение трубы над уровнем почвы и высоту стакана хлопушки.
Определение водопроницаемости почвы
При проведении опытов на орошаемых участках нередко приходится
определять водопроницаемость почвы, так как поливной режим в значитель¬
ной мере определяется водопроницаемостью и водоудерживающей способно¬
стью почвы.
Для определения водопроницаемости почвы с 1962 г. выпускается ком¬
плект оборудования под названием ПВН. В комплект входят два цилиндри¬
ческих кольца высотой 200—300 мм и диа¬
метром 226 и 450 мм, а также два металли¬
ческих резервуара с приспособлением Мари-
отта, которое обеспечивает автоматически за¬
данную высоту слоя воды над поверхностью
почвы (рис. 4).
На ровной площадке в почву врезают два
стальных кольца, располагая одно в центре
другого, и утрамбовывают землю вокруг
наружной стенки внешнего кольца. В обоих
цилиндрических кольцах поддерживается
постоянная высота слоя воды в 5 см. Внутрен¬
ний цилиндр служит для наблюдений, внеш¬
ний является защитным и предохраняет от
растекания воды из внутреннего цилиндра в
стороны.
Начиная определение, открывают водо¬
спускные и воздушные трубки и записывают
время начала опыта, уровень воды в ба¬
чках и температуру, при которой происхо¬
дит инфильтрация. Наблюдения за впиты¬
ванием проводят каждые 5 и 10 мин. в течение
первого часа, затем с интервалом в 30 мин. и
1 час и каждый раз регистрируют расход,
воды из бачка. Если вода в бачке израсхо¬
дуется, то его без промедления вновь зали¬
вают водой и, установив на место, продолжают наблюдения. Для уменьше¬
ния испарения воды рамы во время опыта прикрывают фанерными листами.
Опыт продолжают до установления постоянной скорости впитывания
(когда отклонения отдельных замеров будут менее 10% от средней величины).
На это требуется обычно от 6 до 12 час. после залива площадки.
При определении записывают:
Время замера.
Интервал между замерами, мин.
Количество вылившейся воды, мл.
Водопроницаемость характеризуется величиной слоя воды в милли¬
метрах, проникающей в почву за единицу времени; изменение водопрони¬
цаемости во времени показывают графически. Почвы слабой водопрони¬
цаемости имеют среднюю скорость впитывания за первый час наблюдения ме¬
нее 50 мм, средней водопроницаемости — от 50 до 150 мм и значительной
водопроницаемости — свыше 150 мм (Астапов, Долгов, 1959).
Рис. 4. Прибор для определения
водопроницаемости грунтов *ПВН
2 — резервуары; 2 — водомерное сте¬
кло; 3 — отвес; 4 — гайка; 5 —штатив;
6— воздушная трубка; 7 — водоспуск¬
ная трубка; 8 — внутреннее кольцо;
9 — внешнее кольцо
Определение физических и водных свойств почвы,
303
На основании данных, полученных при постоянной скорости проникно¬
вения воды в почву, вычисляют коэффициент фильтрации:
1QQ
к - ts »
где К — коэффициент фильтрации, мм водного столба за 1 мин.; Q — расход воды, мл,
t — продолжительность наблюдения, мин.; S — площадь малого кольца, см8 (при диа¬
метре кольца 226 мм площадь 400 см2); 10 — перевод в миллиметры.
Вычисление запасов воды в почве
При проведении полевых опытов встречается необходимость подсчитать
запасы легкодоступной и труднодоступной воды в почве в тот или иной мо¬
мент развития. Запасы воды могут выражаться как в миллиметрах водяного
олоя, так и в кубометрах воды на 1 га (1 мм водяного слоя на 1 га соответству¬
ет 10 м3 воды).
Имея данные о влажности почвы, влажности завядания и предельной
полевой влагоемкости (выраженные й весовых процентах на сухую почву),
можно вычислить запас различных категорий влаги для каждого слоя. Для
этого данные влажности в весовых процентах (W) умножают на объемный
вес (D) и толщину слоя в сантиметрах (Н) и делят на 10. В результате по¬
лучаем запас воды слоя почвы, выраженный в миллиметрах водяного слоя:
WDH
10
Например, запас влаги в слое почвы 0—15 см при объемном весе 1,18 и влаж
ности 22,5% будет равен: В = 22,5*1,18-15:10 = 40 мм (или 400 м*/га).
Запас воды в заданной почвенной толще (например, 0—100 см) подсчиты¬
вается по формуле:
В--=0,1 (WtD1Hi+WtD2H*+... +W4o£io # ю),
где WiDiHi . . . Wi0D10H10 — запас воды каждого 10-сантиметрового слоя.
Таблица 1
Наибольший запас доступной влаги и запас влаги при НВ и ВЗ для некоторых почв
(в мм водного столба для слоя в 50, 100 и 200 см)
Почва
Запас доступной
влаги при НВ
Запас влаги при НВ
Запас
влаги при ВЗ
0—50
0—100
0—200
0—50
0—100
0—200
0—50
0—100
0—200
1. Дерново-среднеподзоли¬
стая на тяжелом покров¬
ном суглинке
109
189
347
158
325
695
49
136
348
2. Лугово-черноземная
84
164
302
186
367
710
102
203
408
3. Мощный тучный черно¬
зем на тяжелом лёссовид¬
ном суглинке
124
223
407
195
360
680
71
137
273
4. Обыкновенный чернозем
94
157
278
189
358
675
95
201
397
5. Южный солонцеватый
чернозем на легкой по¬
кровной глине
84
159
295
177
355
695
93
196
400
6. Темно-каштановая
95
190
378
150
306
616
55
116
288
7. Типичный серозем на
среднем лёссовидном
суглинке
72
148
289
119
242
477
47
94
188
Д. В. Федоровский
11111I 11II I** I Us
Рис. 5. Хроноизоплеты влаж¬
ности почвы, % от веса почвы
(данные А. А. Роде и Агрофи¬
зического института)
А — дерново-подзолистая почва;
Б — южный чернозем
Рис. 6. Профили влажности
дерново-подзолистой почвы
1 — июнь; 2 — июль; 3 — август;
4 — сентябрь; 5 — октябрь; 6 —
ноябрь
влажность, % от объема
10 20 30
90
Определение физических и водных свойств почвы
305
Приведенными методами вычисляют:
1) запас воды в почвенной толще в момент работы (В);
2) максимальный запас почвенной влаги, соответствующий наименьшей
влагоемкости (НВ);
3) запас воды, соответствующий влажности завядания (ВЗ);
4) запас доступной для растений влаги (В—ВЗ) и
5) дефицит почвенной влаги (НВ—В).
В табл. 1 приведены величины наибольшего возможного запаса доступной
влаги и общего запаса влаги при влажности почвы, соответствующей наи¬
меньшей влагоемкости, а также при влажности завядания для наиболее рас¬
пространенных почв. Данные для почв 1, 3, 5 и 7 заимствованы нами из кни¬
ги А. А. Роде (1965). а для почв 2, 4, 6 из книги П. В. Вершинина с соавтора¬
ми «Основы агрофизики» (1959).
Графическое изображение динамики изменений
влажности почвы
Результаты многочисленных определений влажности, которыми сопровож¬
дается большинство агрохимических опытов, часто приводят в виде громозд¬
ких и трудночитаемых таблиц. Графическое изображение того же материала
в виде хроноизоплет наглядно и более компактно. Этот метод позволяет сра¬
зу оценить особенности изменения влажности в почвенно-грунтовой толще
любой мощности и за любой, даже очень продолжительный, период. На
этом же графике сопряженно может быть показано выпадение осадков, из¬
менение температуры воздуха, изменение уровня грунтовых вод (рис. 5),
а также увеличение надземной массы растений и т. д.
При небольшом числе сроков наблюдений данные об изменении влажно¬
сти можно изобразить в виде профилей влажности. По оси ординат на графике
откладывают глубину от поверхности, а по оси абсцисс — влажность. Каж¬
дый срок наблюдения изображается отдельной линией, по уклону линии
можно судить о величине градиента влажности. Полезно нанести на этом же
графике основные почвенные константы — величину полной и наименьшей
влагоемкости и влажности завядания.
Профили наглядно выявляют закономерные изменения влажности во
времени, однако не отражают дробного многократного увлажнения почвы
осадками. Рассматривая поочередно линии графика, можно получить пред¬
ставление о том, как высокая по всему профилю влажность в первый срок
наблюдения снижалась с поверхности и в августе упала ниже влажности
завядэния в пахотном слое. В более поздние, 5 и 6, сроки происходило уве¬
личение влажности во всей толще почвы, начиная с поверхности (рис. 6).
ИССЛЕДОВАНИЕ НЕКОТОРЫХ ФИЗИЧЕСКИХ СВОЙСТВ ПОЧВЫ
Объемный вес почвы
Определение объемного веса почвы дает возможность высчитать запас
питательных веществ в определенном объеме или 1-метровом слое почвы, что
при сравнении обеспеченности растений питательными веществами на раз¬
ных почвах более правильно, чем подсчет запасов на 1 кг почвы. Кроме того,
зная объемный и удельный вес почвы, можно подсчитать суммарный объем
пустот в единице объема, называемый скважностью (порозностью); зная
скважность и влажность почвы, легко высчитать воздухообеспеченность.
Определение объемного веса почвы с нарушенным строением может пона¬
добиться для вычисления общей порозности и влагоемкости при проведении
306
Д. В. Федоровский
вегетационного опыта. Его производят при набивке пробного сосуда. Зная
навеску и влажность, вычисляют вес сухой почвы в сосуде. Объем цилиндри¬
ческого сосуда V = лг2/*. Разделив вес сухой почвы на объем, получают
объемный вес.
В полевых условиях определенный объем почвы с ненарушенным строе¬
нием вырезают металлическим цилиндром-буром. Для пахотного слоя упот¬
ребляют буры большой емкости. В комплект почвенного бура БП-50 входят
два буровых стакана емкостью 500 и 250 см8 при высоте 10 см и 20 штук вкла¬
дывающихся в них приемных цилиндров, в которых образцы почвы достав¬
ляются в лабораторию. Распространены также буры Дояренко, Некрасова,
Качинского. Бур Качинского на 100 см3 имеет диаметр 5,65 см и высоту 4 см.
Острая нижняя часть цилиндра имеет несколько меньший диаметр, что поз¬
воляет без деформации извлекать пробу почвы. Разные экземпляры буров
могут иметь различные объемы, поэтому до выхода в поле должен быть изме¬
рен и записан точный объем каждого бура, объем и вес каждого вкладного
цилиндра.
Для взятия проб по профилю почвы в поле выкапывают почвенный раз¬
рез и на стенке его размечают границы генетических горизонтов или слоев
определенной величины. На намеченном уровне в стенке приготовляется го¬
ризонтальная площадка, на которой устанавливают бур с надетой на него
насадкой-оголовком. Для направления бура рекомендуется применять на¬
кладку, через отверстие которой бур врезают или вбивают деревянным мо¬
лотком.
Из каждого горизонта берут два-три образца, а с поверхности, если об¬
разцы отбирают малым буром,— не менее 10 проб. Для пахотного слоя поч¬
вы лучше использовать буры большого размера, повторность при этом можно
уменьшить до 5.
Сняв накладку, подрезают ножом почву под буром и вынимают его из поч¬
вы. Аккуратно срезают ножом излишек почвы, добиваясь плоского среза
вровень с краями бура, а затем удаляют прилипшую к наружным стенкам
бура почву и переносят содержимое бура в салфетку из тонкой клеенки или
пленки и тщательно в нее заворачивают, во избежание потерь влаги при
транспортировке. Удобно иметь салфетки одного веса.
Для торфяных почв употребляют буры с зубчатым краем, которые вре¬
заются поворотным движением.
Доставив образцы с поля в лабораторию, взвешивают образец, переме¬
шивают почву и берут одну — две пробы на определение влажности. Опре¬
делив потерю воды высушиванием пробы в стаканчике, рассчитывают коли¬
чество воды во всем объеме почвы в буре и вес сухой почвы, находившейся
в буре.
Вес находившейся в буре сухой почвы в граммах, деленный на объем бура
в кубических сантиметрах, дает объемный вес. В зависимости от удельного
веса содержащихся в почве минералов, плотности сложения почвы и содержа¬
ния в ней гумусовых веществ объемный вес пахотного слоя варьирует от 0,6
до 1,4 и может доходить в подпахотных слоях почвы до 1,6—1,8. Объемный
вес торфа — 0,3—0,9 в зависимости от зольности.
При высокой плотности и почвенная влага, и питательные вещества оста¬
ются недоступными для растений. Корни пшеницы вообще не проникают в
в слои почвы с объемным весом 1,65, а корни огурцов — 1,45. Яблони недол¬
говечны, если один из слоев двухметровой толщи суглинистой почвы имеет
объемный вес более 1,55, а на глинистой почве угнетение наступает при плот¬
ности выше 1,30. Слишком рыхлое состояние почвы может быть неблагоприят¬
ным, замедляет прорастание семян и в некоторых случаях снижает урожай
пшеницы.
Объемный вес в пахотном слое подвержен значительным изменениям в те¬
чение вегетационного периода; для почв, обладающих способностью к на¬
Определение физических и водных свойств почвы
307
буханию, объемный вес, определенный для влажной почвы, всегда будет
меньшим, чем для сухой почвы; для подпахотных слоев изменения не так
заметны.
При определении записываются:
Бысота бура (Л, см).
Внутренний диаметр режущей части бура (d, еле).
Объем цилиндра (см3) V = [я (lL)2h].
Номер образца, характеристика его: поле, глубина и т. д.
Вес салфетки с сырой почвой, вырезанной буром.
Чистый вес сырой почвы, без салфетки (Е).
Номер и вес сушильного стаканчика с пробой сырой почвы (4).
Вес стаканчика с пробой после сушки (В).
Вес тары стаканчика (С).
Вес испарившейся воды (Л — В).
Вес сухой почвы (В — С).
Влажность (вес. % на сухую почву): W = ^ ^ *100.
(В — С)
Объемный вес (г/см3) вычисляют по формуле
£.100
D= V(\oo+wbec%) •
Для каменистых и щебневатых почв определить объемный вес металли¬
ческими бурами невозможно. Методика исследования физических свойств,
в том числе объемного веса, каменистых почв разработана Ф. Р. Зайдельма-
ном (1957).
Удельный вес почвы
Под удельным весом почвы понимают средний вес всех составных эле¬
ментов в единице объема твердой фазы. Его величина дает некоторое представ¬
ление о составе почвы, так как зависит от количества скелетных частиц, ми¬
нералогического состава и содержания органических веществ. Удельный
вес почвы необходимо знать для правильного выбора времени отстаивания
частиц при механическом анализе, а также для вычисления порозностп
почвы.
Определение удельного веса производят при помощи пикнометра на 50
или 100 мл, с меньшей точностью можно выполнить определение в мерной
колбочке.
Чисто вымытый пикнометр наполняют остуженной свежепрокипяченной
дистиллированной водой и погружают его на 15—20 мин. в кристаллизатор
(или другой сосуд), наполненный водой, для выравнивания температуры.
Вынув пикнометр из воды, его обсушивают фильтровальной бумагой (беря
пикнометр только за горлышко и стараясь не нагревать его рукой), взвеши¬
вают с точностью до 0,01 г и записывают температуру, при которой сделано
определение, и вес пикнометра с водой.
Воздушно-сухую, просеянную через сито с отверстиями диаметром 1 мм>
почву рассыпают топким слоем на бумаге; из разных мест слоя берут среднюю
пробу в два сушильных стаканчика: в один 10—15 г для определения удель¬
ного веса и в другой 4—5 г для определения влажности, так как удельный
вес вычисляют на абсолютно сухую почву.
Отливают около половины воды из пикнометра и через сухую воронку
всыпают из стаканчика почву (около 10 г), предварительно взвешенную с точ¬
ностью до 0,01 г. Приставшие к стенкам стаканчика и воронки частицы почвы
смывают водой в пикнометр, который затем нагревают на плитке до кипения
308
Д. В. Федоровский
и осторожно (чтобы не выбросило воду с почвой) кипятят 30 мин. После ки¬
пячения пикнометр охлаждают и доливают свежепрокипяченой водой, а за¬
тем ставят в кристаллизатор на 15—20 мин. Температура воды в кристалли¬
заторе должна быть одинаковой с температурой ее при первом измерении.
Пикнометр доливают до метки, вынимают из кристаллизатора и, обтерев до¬
суха фильтровальной бумагой, взвешивают.
Вместо кипячения пикнометр можно поместить на 1—2 часа в вакуум —
под колпак вакуум-насоса, где удаление воздуха из почвы происходит без
нагревания. Лучшие результаты получены при разрежении 0,85 атм.
При определении записываются в журнал:
Номер образца, его характеристика: поле, глубина взятия и т. д.
Номер пикнометра.
Вес почвы, взятой в пикнометр.
Влажность почвы.
Вес абсолютно сухой почвы (В, г).
Вес пикнометра с водой (А).
Вес пикнометра с водой и почвой (С).
Температура, при которой проведено определение.
Удельный вес вычисляют по формуле
В
d = (А+В) — С ■
Определение удельного веса ведется в двух-трехкратной повторности,
расхождение параллельных определений не должно превышать 0,02.
Величина удельного веса пахотного слоя суглинистых почв составляет
2,4 для типичных и обыкновенных черноземов, 2,6 для каштановых, подзо¬
листых, серых лесных и красноземов, 2,65 для сероземов, а также легких по
механическому составу почв. Торфяные почвы имеют удельный вес 1,5—2,0.
В нижних горизонтах почвенного профиля он может достигать 2,7—2,9.
Определение удельного веса с водой дает преувеличенные результаты для
засоленных, тяжелых глинистых и таких богатых органическим веществом
почв, как торфяно-болотные.
Для засоленных почв (сухой остаток водной вытяжки выше 0,5%) обра¬
зец перед определением высушивают до постоянного веса, а само определение
проводят с неполярной жидкостью: керосином, толуолом, бензолом и др.
Навеску сухой почвы переносят в пикнометр, заливают жидкостью и остав¬
ляют на ночь. Вместо кипячения для удаления воздуха пикнометры выдержи¬
вают в течение 1 часа в вакуум-эксикаторе при разрежении. Для облегчения
удаления воздуха почва должна быть хорошо измельчена (до 0,25—0,1 мм).
Извлеченные из вакуума пикнометры доливают до метки, обтирают с внеш¬
ней стороны и взвешивают.
Удельный вес жидкости определяют, наполняя пикнометр и взвешивая
его. Разделив вес жидкости на объем пикнометра, получают удельный вес
жидкости при данной температуре.
Расчет удельного веса почвы производят по формуле:
где d0 — удельный вес неполярной жидкости; А — вес пикнометра с жидкостью; С — вес
пикнометра с почвой и жидкостью; В — вес абсолютно сухой почвы.
Определение физических и водных свойств почвы
309
Вычисление общей порозности (скважности)
и воздухообеспеченности почвы
Общая порозность (скважность, или пористость) почвы показывает сум¬
марный объем пустот, различных по величине и форме. Скважность зависит
от механического, агрегатного состава и структуры почвы, формы частиц
и т. д. Она изменяется при набухании почвы.
Величина общей порозности определяет в основном водный и воздушный
режим почвы, полную влагоемкость и воздухоемкость; от нее зависит пере¬
движение воды в почве: водопроницаемость, глубина промачивания, подъем
грунтовых вод, испарение.
Если известны объемный вес (D) и удельный вес (d) почвы, то можно вы¬
числить общую порозность (скважность) в объемных процентах:
Робщ= 100 (l
Воздухосодержание подсчитывается по разности: общая скважность ми¬
нус влажность (в объемных процентах). Влажность в объемных процентах
получают умножением величины влажности в ве¬
совых процентах на величину объемного веса. Ре¬
зультаты можно изобразить графически подобно
тому, как это сделано на рис. 7. Этот график дает
наглядное представление о величине общей скваж¬
ности и количестве воды и воздуха, заполняющих
поры на разной глубине.
Порозность разных почв весьма различна: от
30 % у плотных глин до 90 % у рыхлых торфяных
почв. Благоприятные условия создаются при об¬
щей порозности пахотного слоя 55—65% для су¬
глинистых или 45—50% для песчаных почв; не¬
удовлетворительной является порозность сугли¬
нистых почв ниже 50 %.
С агрономической точки зрения важно, чтобы
при высоком содержании воды в почве воздуха
было не менее 20—25% от общей порозности. По Конецкому, растения пше¬
ницы начинают страдать от недостатка кислорода при содержании возду¬
ха ниже 15% от объема почвы, а сахарной свеклы — ниже 20%.
Структурность почвы
Почва слагается из частиц различной крупности, различных по минерало¬
гическому и химическому составу, обладающих неодинаковой активностью
в отношении проходящих в почве физико-химических и биологических про¬
цессов. Водный, пищевой, воздушный режим почвы и условия роста расте¬
ний в сильной степени связаны с гранулометрическим составом почвы.
Элементарные частицы почвы в естественном состоянии соединены в слож¬
ную систему микро- и макроагрегатов. Это агрегатное состояние динамично,
а изменение его влечет за собой изменения и физических свойств и микробио¬
логических процессов почвы. Механический состав почвы значительно менее
динамичен, изменения его можно отметить лишь за более или менее продол¬
жительный период.
Для характеристики состава почвы, кроме структурного (макроагрегат-
ного) анализа, проводят механический и микроагрегатный анализ.
Широко применяемая для структурного анализа почв методика Н. И. Сав-
винова позволяет дать оценку почв по общему содержанию водопрочных
20 90 60 80 100
Содержание, об %
Рис. 7. Соотношение воды
(^4), воздуха (Б) и твердой
части почвы (В)
310
Д. В. Федоровский
агрегатов, сравнить водопрочность агрегатов разного размера и проследить
за изменением структурного состояния на полях разных севооборотов, под
действием удобрения и т. д. Пропись метода включает «сухой рассев» образ¬
ца почвы, приготовление средней пробы, размачивание образца и рассев на
ситах, погруженных в воду.
Предложенное Почвенным институтом (1966 г.) изменение подготовки
образцов почвы делает метод более простым и быстрым, не снижая точности
характеристики почв.
Приготовление средней пробы. Для правильного опре¬
деления необходимо брать для рассева большую навеску — до 2,5 кг, но не
менее 1,5 кг почвы. Почву подсушивают на воздухе, отдельные глыбы осто¬
рожно пальцами разламывают на отдельности менее 1 см, ни в коем случае
не растирая почву.
Всю навеску просеивают через набор сит (диаметр сит 20 см) с отверстия¬
ми диаметром в 10; 5; 3; 2; 1; 0,5 и 0,25 мм, перенося почву на верхнее сито
порциями в 100—200 г. При просеивании на колонку сит надевают крышку
и поддон во избежание потерь. Колебательные движения колонке сит сооб¬
щают вручную или закрепляют колонку на горизонтальной площадке аппа¬
рата для встряхивания. При амплитуде колебаний 2—3 см и частоте около
100 в минуту на рассев одной порции уходит около 1 мин.
Закончив рассев очередной порции почвы, пересыпают отдельные фрак¬
ции из сит в коробки или чашки и, собрав колонку сит, приступают к рассеву
следующей порции. По окончании просеивания всей навески почвенные фрак¬
ции взвешивают (с точностью до 1 г), суммируют вес всех фракций и вычи¬
сляют процентное содержание каждой фракции в почве (при данной влаж¬
ности).
Из отсеянных фракций для определения водопрочности почвенных агре¬
гатов составляют навеску в 50 г таким образом, чтобы в нее вошли все выде¬
ленные фракции, пропорционально процентному содержанию каждой иа
них. (Вес отбираемой фракции численно равен половине процентного содер¬
жания.) Полезно проводить контрольное взвешивание средней пробы, чтобы
своевременно устранить возможные ошибки. Для размывания на ситах при¬
готовляют две средние пробы. Если расчет будет вестись на абсолютно сухую
почву, то для определения влажности почвы приготовляют третью пробу.
Определение водопрочности агрегатов. Состав¬
ленную таким способом среднюю пробу почвы высыпают в стакан с водой
емкостью около 200 мл и оставляют в покое на ночь.
Через 15—18 час. воду с почвенным образцом из стакана осторожно пе¬
реносят на верхнее сито колонки, полностью погруженной в ведро с водо¬
проводной водой. Колонку составляют из пяти сит с отверстиями 0,25; 0,5;
1, 2 и 3 мм без поддона и крышки и скрепляют за наружные ушки, чтобы ко¬
лонка не распалась во время определения. Над верхним ситом при погруже¬
нии колонки должен быть слой воды около 10 см. Затем приступают к просеи¬
ванию под водой. Сита медленно поднимают, но так, чтобы не обнажать ко¬
мочки почвы на верхнем сите, и затем быстрым отрывистым движением
опускают на 3—4 см. Через 2—3 сек. движения повторяют.
Такое просеивание продолжают до тех пор, пока на каждом сите останут¬
ся только комочки, не проходящие через отверстия сита. Обычно для этого
достаточно 10 встряхиваний всего комплекта, после чего снимают верхние
два сита и нижние три сита встряхивают дополнительно еще 5 раз. Остав¬
шиеся на ситах водопрочные агрегаты смывают струей воды из промывалки
в большие фарфоровые чашки. После оседания почвы воду сливают и пере¬
носят агрегаты на фильтры в воронках.
Предварительно на фильтре надписывают номер образца, размер агрега¬
тов и вес фильтра в воздушно-сухом состоянии. После подсушивания на во¬
ронке фильтры с фракциями одного образца нанизывают на нитку и выве¬
Определение физических и водных свойств почвы
311
шивают для окончательной подсушки в хорошо проветриваемом помещении.
Пачку фильтров периодически взвешивают. После достижения постоянного
веса пачки взвешивают каждый фильтр с почвой. Вычитая вес фильтра из
веса фильтра с почвой, получают вес фракции. Умножив вес фракции на 2,
получают процентное содержание водопрочных агрегатов. Количество пыли
(частиц менее 0,25 мм) вычисляют по разности: 100 минус сумма процентного
содержания всех фракций водопрочных агрегатов.
Может применяться и ускоренная сушка. Для этого агрегаты с сита пере¬
носят в маленькие чашечки или алюминиевые стаканчики, избыток воды сли¬
вают и выпаривают и высушивают почву на водяной бане или в термостате
при 60—80°. Высушенные агрегаты оставляют стоять на ночь, чтобы почва
пришла в воздушно-сухое состояние, а затем взвешивают на технических
весах. Умножив на 2 вес каждой фракции, получают процентное содержание
в почве.
Метод Саввинова
По предложенной Н. И. Саввиновым (1931 г.) методике, не рекомендуется
длительное размачивание почвы. Согласно прописи, среднюю пробу всыпают
в заполненный водой цилиндр емкостью 1 л. Через 10 мин. доливают цилиндр,
закрывают пришлифованным стеклом и переворачивают его 10 раз в одну
сторону и 10 — в другую, причем каждый раз агрегаты проходят весь столб
жидкости в цилиндре. Затем цилиндр опрокидывают над ситами, погружают
горловиной в воду и сдвигают стекло, открыв цилиндр. Агрегаты проходят
воду в цилиндре и падают на верхнее сито. Рассев почвенных агрегатов на
колонке сит, погруженных в воду, и последующие операции производят,
как описано выше.
Следует отметить, что любое изменение ме¬
тодики, замена сит, изменение энергии и числа
встряхивания почвы на ситах и т. п., может
существенно отразиться на результатах анали¬
за. Для получения сравнимых данных порядок
работы должен строго выдерживаться. Стан¬
дартизация встряхивания сит в воде достигает¬
ся с помощью приборов Цыганова или Бакше-
ева.
Агрегатный анализ по Цыга¬
нову (1957). Навеску почвы на ситах встря¬
хивают на специальном приборе (рис. 8) при
постоянной скорости движения и глубине погру- s
жения, что исключает влияние оператора на ре¬
зультаты определения.
Прибор состоит из резервуара с водой (1),
в котором помещен набор сит (2), закреплен¬
ный на штоке (3). На крышке резервуара (4)
укреплена стойка с механизмом, сообщающим
движение вверх и вниз штоку и ситам. Вращая
рукоятку (5), поворачивают эксцентрик (6), ко¬
торый медленно поднимает ролик (7), закреп¬
ленный на штоке. В верхней точке эксцентрик
освобождает ролик и силой пружины (8) сита
вместе со штоком резко опускаются вниз, встря¬
хивая агрегаты почвы. Число встряхиваний
фиксируется счетчиком (9).
Среднюю пробу (50 г) для определения водо-
прочности агрегатов готовят, как описано вы¬
ше. Перед рассевом на ситах по Цыганову про-
Рис. 8. Прибор Цыганова для
определения водопрочности
структуры почвы (модифика¬
ция прибора Венгерского при¬
боростроительного завода «La¬
bor»)
Объяснение см. в тексте
312
Д. В. Федоровский
бу в течение 30 мин. капиллярно насыщают на влажной фильтровальной
бумаге в чашке с плоским дном. Для образцов солонцовой почвы время на¬
сыщения должно быть увеличено до 15 час.
Для определения водопрочности агрегатов купанием их в воде колонку
составляют из сит с отверстиями 3, 1, 0,5 и 0,25 мм. Образец почвы, прошед¬
ший капиллярное насыщение, переносят на верхнее сито, собирают прибор
и начинают определение. Вращением рукоятки встряхивают сито 15 раз при
постоянной высоте подъема 30 мм, интервал между встряхиваниями около
3 сек. Затем колонку сит разбирают, почву переносят в фарфоровые чашки
или сушильные стаканчики, высушивают и взвешивают фракции, как описа¬
но выше.
Определение необходимо проводить с двухкратной повторностью. Раз¬
ница между параллельными определениями обычно не превышает 3—4%.
Для характеристики почвы обычно приводят данные о содержании водо¬
прочных агрегатов. Содержание их (крупнее 1 мм) в структурном черноземе
может достигать 50—60%, в то время как на старопашке количество их может
понижаться до 4—10%.
При сравнении результатов анализов, проводившихся в разное время,
необходимо иметь в виду, что содержание различных фракций за вегетацион¬
ный период подвергается некоторым изменениям. Структурность почвы су¬
щественно изменяется под влиянием посева трав, внесения извести, орошения»
Механический состав почвы
Механический состав почвы определяет многие агрономически важные ее
свойства: водный, воздушный и тепловой режим, и влияет на запас питатель¬
ных веществ в почве, подвижность и эффективность действия вносимых удоб¬
рений. От механического состава зависят сложение, порозность, влагоем-
кость, влажность завядания и другие физические свойства.
Задача механического анализа — определить содержание элементарных
механических частиц в почве после искусственного расчленения микроагре¬
гатов, ранее сцементированных карбонатами, склеенных органическими и
и минеральными соединениями. Результаты механического анализа зависят
от метода подготовки почвы, так как разные методы разрушают цементи¬
рующие вещества различной степени устойчивости.
При агрохимических исследованиях чаще всего довольствуются грубой
характеристикой механического состава почвы (суглинистая, легкосуглини¬
стая и др.)» для которой достаточно определить содержание частиц физичес¬
кой глины < 0,01 мм, ила < 0,001 мм и скелетной части. Поэтому во многих
случаях можно применить быстрый метод определения механического соста¬
ва по удельному весу суспензии. Рекомендованный ранее метод Качинского
дает большую точность и воспроизводимость данных и сохраняет значение
для углубленных исследований.
Быстрый метод определения механического состава
Метод рекомендован для зональных агрохимических лабораторий («Меж¬
республиканские технические условия...», 1969). Исследование почвы вклю¬
чает обработку раствором пирофосфата натрия, отстаивание взмученных
частиц в литровом цилиндре и определение содержания частиц в суспензии
по удельному весу. **
Подготовка почвы растиранием с раствором
пирофосфата. Для механического анализа берут навески мелкозе¬
ма — почвы, прошедшей через сито с отверстиями диаметром 1 мм. Одновре¬
менно берут две навески: для определения влажности и механического ана¬
лиза.
Определение физических и водных свойств почвы
313
Отвешивают 20—30 г воздушно-сухой почвы (для супесчаной почвы навес¬
ку увеличивают до 40 г, песчаной — до 50 г), помещают в фарфоровую чаш¬
ку и смачивают определенным объемом 4%-ного раствора пирофосфата нат¬
рия (40 г безводной или 67 г кристаллической десятиводной соли Na4P207
в 1 л воды).
Для почв незасоленных, некарбонатных на каждые 10 г почвы необходимо
добавить 5 мл раствора пирофосфата, карбонатных — 10 мл, засоленных
и гипсоносных — 20 мл. Нужный объем отмеряют в стаканчик и из него по¬
степенно увлажняют почву. Смочив образец до тестообразного состояния, его
осторожно без нажима растирают в течение 10—15 мин. пестиком с резино¬
вой головкой. (Такой пестик можно изготовить, насадив резиновый наконеч¬
ник для палки на фарфоровый пестик.) Растирать образец тонкой палочкой
с резиновым наконечником или пальцем в резиновом напалечнике не реко¬
мендуют, так как это снижает выход физической глины.
Затем приливают остаток раствора пирофосфата, добавляют воду и, раз¬
мешивая тем же пестиком, доводят почву до состояния жидкой суспензии.
Суспензию переносят через сито с отверстиями диаметром 0,25 мм в литро¬
вый цилиндр и доводят объем до 1 л. Фракцию 0,25—1,0 мм переносят с сита
в сушильный стаканчик и взвешивают после сушки.
Взбалтывают суспензию в течение 1 мин., энергично опуская и поднимая
мешалку (стеклянная палочка с резиновым кружком на конце). Цилиндр ос¬
тавляют в покое в помещении с ровной температурой, до отбора пробы пи¬
петкой. Следует избегать нагрева суспензии, который вызывает конвекцион¬
ные токи и замедляет оседание частиц.
При механическом анализе засоленных и богатых гипсом почв иногда
наступает частичная или полная коагуляция суспензии, причем наблюдается
осветление всего столба жидкости и выпадение твердой фазы в виде рыхлого
осадка.
Для прекращения частичной коагуляции в большинстве случаев доста¬
точно добавить некоторое количество пирофосфата. При явных признаках
коагуляции суспензии в цилиндре пробы не отбирают, но оставляют суспен¬
зию стоять на 1—2 суток до полного осветления жидкости. Прозрачную жид
кость с солями, вызвавшими коагуляцию, сливают возможно полнее (не ме¬
нее 800—950 мл) и определяют содержание солей (плотный остаток). Вес плот¬
ного остатка вычитают из веса взятой для анализа почвы и расчет содержа¬
ния фракций производят в процентах на бессолевую навеску.
К осадку в цилиндре вновь приливают 20 мл 4%-ного пирофосфата нат¬
рия, тщательно перемешивают с осадком, доводят объем до 1 л и отбирают
пробы суспензии.
При высоком содержании в почве СаС12 внесение пирофосфата вызывает
образование большого нерастворимого осадка. В этом случае приходится
почву перед добавкой пирофосфата промыть дистиллированной водой. Рас¬
чет фракций ведется на бессолевую навеску.
Пипетка для отбора проб емкостью 100 мл (4), отсекаемой трехходовым
краном (2), имеет ствол длиной 15—16 см. Нижний конец ствола запаян сни¬
зу и имеет 4—6 отверстий по окружности, через которые идет заполнение
пипетки (5). Засасывание суспензии происходит при помощи резиновой гру¬
ши (6) емкостью не менее 200 мл. Шарик (2) выше крана пипетки емкостью
10 мл принимает избыток жидкости при заборе пробы (рис. 9).
Глубину и сроки взятия суспензии устанавливают соответственно скоро¬
сти падения частиц определенного диаметра и удельного веса, пользуясь дан¬
ными табл. 3. (см. ниже)
Техника отбора проб. Поставив кран в положение 1 (рис. 10),
нажимают грушу и выдувают остатки воды из верхней части пипетки над
краном. Сжимают грушу и запирают кран, переведя его в положение 2. Мед¬
ленно опускают ствол пипетки в цилиндр и закрепляют на необходимой глу¬
314
Д. В. Федоровский
бине. Учитывая, что заполнение пипетки емкостью
100 мл требует около 1 мин., забор пробы начина-
ют за 30 сек. до истечения срока отстаивания. По¬
вернув кран в положение «?, заполняют пипетку до
появления жидкости над краном, после чего кран
перекрывают, переводя в положение 2. Сливают из¬
лишки суспензии над краном обратно в цилиндр, пе¬
реводя кран в положение/, и, продувая пипетку на¬
жимом на грушу, удаляют капли суспензии из крана
и отростка. Извлекают ствол пипетки из цилиндра,
погружают его в сухой, предварительно взвешенный
стакан емкостью 150 мл. Переводят кран в положе¬
ние 4 и сливают суспензию. Для удаления осевших
на стенках пипетки частиц переводят кран в поло¬
жение 5, засасывают небольшой объем суспензии из
верхней части стакана и выдувают его обратно в ста¬
кан, нажимая на грушу. Очень важно полностью
удалять суспензию из нижнего окончания ствола
с мелкими отверстиями, так как задержка лишних
0,1 мл жидкости дает ошибку содержания фракции
на 5% от веса почвы.
Стаканчик с суспензией взвешивают на техниче¬
ских весах с точностью 0,01 г и определяют вес су¬
спензии в объеме пипетки (Q). После взвешивания стакана суспензию выли¬
вают обратно в цилиндр, перемешивают и после отстаивания производят по¬
вторное определение или определение другой фракции.
Вес воды в объеме пипетки при данной температуре определяют 3—5 раз
подряд, причем расхождение параллельных определений не должно превы¬
шать 0,03 г. Для расчета принимают среднюю величину (q).
] ] Рис. 10. Положения
трехходового крана пи¬
петки
Объяснение см. в тексте
Содержание каждой фракции в процентах на абсолютно сухую почву
определяют по формуле
Г dn V Л
ш т\
где dn — удельный вес почвенных частиц; dB — удельный вес воды (принимается = 1);
V — объем всей суспензии (1000 мл); v — объем пипетки, мл; М — навеска абсолютно
сухой почвы, взятой для анализа, г; Q — вес суспензии в объеме пипетки, г; q — вес воды
в объеме пипетки, г.
При серийных анализах одной почвы (постоянный удельный вес, навеска
и объем пипетки) вычисляют коэффициент пересчета, заменяя им часть, взя¬
тую в квадратные скобки.
Для повышения точности полученных данных необходимо производить
два — три повторных определения содержания фракций по удельному весу
суспензий для каждого срока. Точность единичного определения содержания
Рис. 9. Пипетка на 100мл
для определения меха¬
нического состава почвы
но удельному весу су¬
спензии
Объяснение см. в тексте
Определение физических и водных свойств почвы
315
фракций невысока, нередки расхождения параллельных определений на
2—3% от веса почвы. Однако это не мешает оценке механического состава
по классификационной шкале, имеющей градации через 10—15%.
Межреспубликанские технические условия проведения анализов почвы
для зональных агрохимических лабораторий (МРТУ № 46-16-67) предлагают
использовать единую классификационную шкалу механического состава для
всех типов почв (табл. 2).
Таблица 2
Классификация почв по механическому составу
(для всех типов почв)
Содержание физической
глины (частиц < 0,01 мм), %
Основная характеристика почв
Дополнительная оценка
по преобладающей фракции
0-5
Рыхлопесчаная
Песчаная или крупнопылева¬
5—10
Связнопесчаная
тая
10-20
Супесчаная
20-30
Легкосуглинистая
Песчаная, крупнопылеватая,
30-40
Среднесуглинистая
пылеватая или иловатая
40-50
Тяжелосуглинистая
50—65
JI егкоглинистая
Пылеватая или иловатая
65-80
Среднеглинистая
80-100
Тяжелоглинистая
Содержание частиц менее 0,01 мм (физическая глина) определяет основ¬
ную характеристику почвы по механическому составу. Более подробное
определение показывает преобладание одной из фракций: песка (1—0,05 мм),
крупный пыли (0,05—0,01 мм), мелкой и средней пыли (0,01—0,001 мм) или
ила (<0,001 мм).
Определение механического состава почв
по Качинскому
Образец воздушно-сухой почвы, просеянной через сито с ячейками 1 мм
рассыпают на бумаге и ложечкой из разных мест отбирают понемногу и состав¬
ляют три средних пробы весом 10—15 г для суглинистых и 20—30 г для супес¬
чаных почв.
В первой пробе определяют влажность высушиванием в термостате, во
второй — потерю растворимых веществ при обработке НС1, третью исполь¬
зуют для приготовления суспензии.
Для определения потери от промывания заранее подготовляют фильтр,
помещают в сушильный стаканчик и сушат до постоянного веса, затем кла¬
дут его на воронку и производят промывание почвы. Фильтр с почвой после
промывания возвращают в тот же сушильный стаканчик, высушивают при
105—110° С до постоянного веса. Вычитая из последнего вес сухого фильтра
вместе с сушильным стаканчиком, получают вес промытой навески и вычи¬
сляют потерю от промывания. Второй фильтр, подготовляемый для обработ¬
ки почвы и дальнейшего приготовления суспензии, взвешивать не нужно.
Промывание обеих навесок почвы производится одинаково: навеску поме¬
щают в фарфоровую чашечку и испытывают на присутствие карбонатов нес¬
колькими каплями 10%-ного раствора НС1. Если почва вскипает, ее неодно¬
кратно обрабатывают в чашечке небольшими порциями 0,2 н. раствора НС1
до прекращения всякого выделения пузырьков газа; жидкость каждый раз
сливают на фильтр. После разрушения карбонатов и прекращения выделе-
316
Д. В. Федоровский
ни я С02 почву переносят при помощи 0,05 н. раствора НС1 из чашечки на
фильтр.
Почвы, в которых не обнаружено присутствие карбонатов, обрабатывают
и переносят на фильтр 0,05 н. раствором НС1. Почву на фильтре продолжают
промывать этим раствором до исчезновения кальция в фильтрате. Для этого
5—10 мл фильтрата помещают в пробирку и нейтрализуют 10%-ным раство¬
ром NH4OH до появления запаха аммиака, подкисляют несколькими капля¬
ми 10% -ной уксусной кислоты и после добавления насыщенного раствора
щавелевокислого аммония нагревают до кипения; помутнение указывает на
присутствие кальция.
Освобожденную от кальция почву промывают водой до прекращения в
фильтрате реакции на хлор; в случае появления в фильтрате мути от про¬
хождения коллоидов промывание почвы водой прекращают, несмотря на на¬
личие хлора в промывных водах.
Один фильтр с промытой почвой высушивают, как указано выше. Второй
фильтр с промытой почвой переносят в фарфоровую чашку и с фильтра стру¬
ей дистиллированной воды из промывалки тщательно смывают всю почву,
а приставшие к фильтру частицы счищают стеклянной палочкой. Получен¬
ную суспензию почвы переносят в коническую колбу на 750 мл и доливают
водой до 250 мл.
По методу Н. А. Качинского, в зависимости от емкости поглощения и ге¬
нетических особенностей почвы к суспензии добавляют для диспергирования
1 н. раствор NaOH в следующем количестве: для тучных черноземов — 6 мл,
обыкновенных черноземов — 5, серых почв — 3 и для подзолистых — 1 мл.
Колбы оставляют стоять 2 часа, встряхивая их от руки каждые 15 мин. Кол¬
бы закрывают пробкой с обратным холодильником (отрезок стеклянной труб¬
ки, вставленной в пробку) и суспензию кипятят на электрической плитке
1 час, не доводя ее до бурного кипения и вспенивания.
После охлаждения необходимо проверить реакцию суспензии, для чего,
переболтав содержимое колбы до появления пены, вносят в нее 1—2 капли
фенолфталеина; розовое окрашивание укажет на слабощелочную реакцию.
Если порозовения нет, необходимо добавить 1 мл щелочи, переболтать и оста¬
вить на ночь, после чего вновь проверить реакцию.
По охлаждении суспензию пропускают через сито с размером ячеек 0,25 мму
помещенное в воронку, опущенную в литровый цилиндр. Задержавшуюся
на сите почву промывают водой. Оставшиеся на сите частицы почвы размером
от 0,25 до 1 мм переносят струей воды во взвешенный предварительно су¬
шильный стаканчик или фарфоровую чашку, воду выпаривают, содержимое
(песок) высушивают в термостате и взвешивают. Суспензию в цилиндре доли¬
вают до 1 л и анализируют методом пипетки.
Вполне допустимо использование мерных цилиндров емкостью 500 мл
при диаметре их не менее 4 см; при этом нижний конец пипетки должен быть
настолько удален от дна цилиндра, чтобы осадок не взмучивался при взятии
проб. В противном случае пробы забирают с меньшей глубины, например
20 см. При изменении глубины с 25 до 20 см время отстаивания уменьшают
на х/5-
Отбор проб пипеткой Качинского. Схема установки по-
казана на рис. 11. Для отбора проб можно использовать также пипетки Ва¬
сильева или Майсуряна. Все пипетки в нижней части ствола имеют четыре —
шесть боковых отверстий, через которые засасывается проба. Вместо аспи¬
ратора для забора проб используют иногда резиновую грушу.
Суспензию почвы в цилиндре перемешивают, поднимая и опуская мешал¬
ку не менее 60 раз, затем вынимают мешалку, включают секундомер и остав¬
ляют до момента отбора проб. Продолжительность отстаивания частиц опре¬
деленного диаметра для соответствующей температуры и удельного веса час¬
тиц находят в табл. 3.
Определение физических и водных свойств почвы
317
Рис. 11. Схема установки для определения механического состава почвы по Качинскому
А — пипетка, градуированная на 24—26 см9; Z— двухходовой кран для соединения пипетки с аспира¬
тором и воздухом атмосферы;* С — капилляр-стопор для задержки капель воды после обмывания пипет¬
ки; У — кран для пуска воды из колбы О и обмывания пипетки; D —штатив с держателями промывал-
ки (М), пипетки (JV); К — кольцо, нормирующее глубину погружения пипетки в суспензию; В— аспи¬
ратор
Для промежуточных значений удельного веса или температуры суспен¬
зии срок отстаивания определяют интерполяцией от приведенных величин
или рассчитывают по формуле Стокса:
0,222 r*(dT-dm)g
v= Л
где V — скорость падения шарообразных частиц, см/сек; г — радиус частиц, см; dT —
удельный вес твердых частиц почвы; dm — удельный вес жидкости; г) — вязкость жид¬
кости при данной температуре; g — ускорение силы тяжести, равное 981 см/сек.
Вязкость воды заметно изменяется в зависимости от температуры и влия¬
ет на скорость оседания частиц. Температуру суспензии нужно измерять
в каждом цилиндре. Если удельный вес образца неизвестен, берут средние
величины для данного типа почвы.
Незадолго до истечения срока отстаивания в суспензию вводят на нуж¬
ную глубину пипетку и закрепляют стопором. На пипетке для отмеривания
глубины погружения должны быть отмечены тонкими резиновыми кольцами
расстояния 7, 10 и 25 см от нижнего конца. Суспензия засасывается в пипет¬
ку в течение 20—30 сек., поэтому начинать засасывание надо за 10 сек. до
истечения срока.
В журнале записывают объем взятой пробы, так как взять точно 25 мл бы¬
вает трудно. Суспензию сливают из пипетки в тарированный сушильный ста-
318
Д. В. Федоровский
Таблица 3
Интервалы времени от взмучивания до взятия проб суспензии при определении механи¬
ческого состава почвы
Удельный
вес
почвы
Дваметр
частиц, мм
Глубина
взятия
пробы,
см
Температура суспензии, ° С
15
20
25
30
Время отстоя до взятия проб
2,4
<0,05
25
149 сек.
132 сек.
117 сек.
105 сек.
<0,01
10
24 м. 51 с.
21 м. 59 с.
19 м. 32 с.
17 м. 27 с.
<0,005
10
1 ч. 39 м.
1 ч. 28 м.
1 ч. 18 м.
1 ч. 10 м.
<0,001
7
29 ч. 00 м.
25 ч. 28 м.
22 ч. 48 м.
20 ч. 23 м.
2,5
<0,05
25
139 сек.
123 сек.
109 сек.
98 сек.
<0,01
10
23 м. 12 с.
20 м. 31 с.
18 м. 15 с.
16 м. 19 с.
<0,005
10
1 ч. 33 м.
1 ч. 23 м.
1 ч. 13 м.
1 ч. 05 м.
<0,001
7
27 ч. 04 м.
23 ч. 56 м.
21 ч. 17 м.
19 ч. 02 м.
2,6
<0,05
25
130 сек.
115 сек.
103 сек.
92 сек.
<0,01
10
21 м. 45 с.
19 м. 15 с.
17 м. 06 с.
15 м. 16 с.
<0,005
10
1 ч. 27 м.
1 ч. 17 м.
1 ч. 08 м.
1 ч. 01 м.
<0,001
7
25 ч. 22 м.
22 ч. 26 м.
19 ч. 57 м.
17 ч. 50 м.
2,7
<0,05
25
122 сек.
108 сек.
97 сек.
86 сек.
<0,01
10
20 м. 28 с.
18 м. 06 с.
16 м. 06 с.
14 м. 23 с.
<0,005
10
1 ч. 22 м.
1 ч. 12 м.
1 ч. 04 м.
0 ч. 57 м.
<0,001
7
23 ч. 53 м.
21 ч. 06 м.
18 ч. 49 м.
16 ч. 47 м.
канчик или тигель. Пипетку обмывают водой в этот же сосуд. Пробу выпари¬
вают на песчаной бане и затем высушивают при 105° до постоянного веса.
Взвешивание ведут на аналитических весах.
После взятия каждой пробы содержимое в цилиндре вновь тщательно пе¬
ремешивают и, после оседания почвы, отбирают следующую пробу.
Институт почвоведения КазССР и Гидрометслужба рекомендуют прово¬
дить подготовку почвы к механическому анализу растиранием с пирофосфа¬
том, а отбор проб 25 мл пипеткой с последующим выпариванием. При подсче¬
тах и8 веса фракции менее 0,001 мм следует вычесть вес пирофосфата: при
внесении 20 мл 4 %-ного раствора на 1 л в объеме 25 мл содержится 0,020 г
соли.
Расчет результатов механического анализа
Результаты анализа выражают в процентах от веса абсолютно сухой поч¬
вы или от веса мелкозема, т. е. части почвы, прошедшей через сито с отвер¬
стиями диаметром 1 мм. Для большинства равнинных почв, не содержащих
отдельностей более 1 мм, процентное содержание фракций в мелкоземе рав¬
но содержанию во всей почве. Для почвы хрящеватой расчет производят на
мелкоземную часть почвы, но таблицу механического состава мелкозема со¬
провождают данными о содержании скелетной части почвы.
Процентное содержание фракций, выделенных на ситах (1—0,5 и 0,5—
0,25 мм), вычисляют по формуле
а. 100
С ’
где а — вес фракции, г; С — навеска мелкозема (сухой вес), г.
Определение физических и водных свойств почвы
319
Процентное содержание суммы фракций менее определенного размера
в пробах, отбираемых пипеткой, вычисляют по формуле
glNlOO
Ь с *
где а — вес фракции в объеме пипетки, г; b — объем суспензии в пипетке, мл; с — вес
абсолютно сухой навески мелкозема, взятой для анализа, е; v — объем суспензии в ци¬
линдре, мл.
В 25 мл суспензии содержится от 1 до 6 мг щелочи, что может дать су¬
щественную ошибку при анализе легких и суглинистых почв. Необходимую
поправку или вычисляют, или определяют в «холостом» опыте: принятое для
анализа данной почвы количество щелочи вносят в литровый цилиндр, добавля¬
ют водой до метки, перебалтывают. Почва в цилиндр не вносится. Отбирают
25 мл раствора пипеткой для механического анализа, помещают в тарирован¬
ный бюкс и высушивают. Вес осадка даст необходимую поправку, которую
надо вычесть из веса фракции менее 0,001 мм.
Содержание фракции определенного размера находят, вычитая из данных
предыдущей фракции данные последующей фракции, и при вычислении по¬
правку на вес щелочи не вводят. Процентное содержание фракции
0,25—0,05 мм определяется по разности 100% минус сумма процентного со¬
держания всех фракций (частиц 1—0,25 мм, выделенных на ситах частиц
менее 0,05 мм и потери от обработки).
Механический анализ желательно проводить в двух повторностях. Дан¬
ные механического анализа приводят в процентах с точностью до десятых
долей; в особой графе отмечают потерю при промывании.
Каменистые почвы классифицируются по мелкозему, так же как некаме¬
нистые, но дополнительно дается оценка каменистости. Для этого среднюю
пробу почвы 300—400 г просеивают через сито с отверстиями 1 мм, крупно¬
зернистую часть переносят в стакан и кипятят в воде 1 час. Отделившиеся
глинистые частицы смывают. После высушивания на ситах выделяют камни
0>3 мм) и гравий (1—3 мм). Если суммарное содержание их превышает 10%
от веса почвы, то при названии механического состава добавляют слово каме-
Таблица 4
Классификация почв по механическому составу
Механический состав
Содержание физической
глины (частиц <0,01 лис)
для почв
подзолистого
типа *
для почв степ*
ного типа,
красноземов
и желтоземов *
для солонцов
и сильносолон¬
цеватых почв*
для сероземов
и карбонатных
почв Средней
Азии **
Песок рыхлый
0-5
0-5
0—5
связный j
5-10
5—10
5-10
< 14
Супесь
10-20
10-20
10-15
14-25
Суглинок легкий
20-30
20-30
15-20
25—32
средний
30-40
30-45
20—30
32-40
тяжелый
40-50
45-60
30-40
40-50
Глина легкая
50-65
60-75
40-50
> 50
средняя
65-80
75—85
50-65
—
тяжелая
> 80
> 85
> 65
—
* По Качинскому (1965). ** «Методы агрохимических, агрофизических и микробиологических иссле¬
дований...» (1963).
320
Д. В. Федоровский
нистый или гравелистый по преобладающей фракции. В таблице всегда при¬
водят данные содержания фракций в процентах от абсолютно сухого мелко¬
зема. При наличии фракций более 1 мм их содержание приводят в таблице
рядом с данными механического анализа.
Основная характеристика механического состава почвы (легкосуглинис¬
тая, среднесуглинистая) определяется содержанием частиц менее 0,01 мм,
указание на преобладающую фракцию уточняет характеристику (см. пример
расчета). Для почв подзолистых, черноземных и солонцов Н. А. Качичский
(1958, 1965) предлагает использовать три разные шкалы классификации, в
в зоне орошаемых сероземов по предложению С. Н. Рыжова пользуются своей
шкалой (табл. 4).
Для агрохимика наибольший интерес имеют данные о механиче¬
ском составе пахотного и подпахотного слоя, однако для полноценной
характеристики почвы полезно иметь данные для всех горизонтов поч¬
венного профиля (рис. 12).
Рис. 12. Диаграмма изменения
механического состава по про¬
филю почвы
1 — потеря от обработки НС1;
2 — песок средний;
3 — песок мелкий;
4 — пыль крупная;
5 — пыль средняя;
6 — пыль мелкая;
7 — ил
Следует иметь в виду, что на почвенных картах механический состав поч¬
вы нередко указывается по составу почвообразующей породы и необязатель¬
но отражает состав самых верхних слоев.
Пример вычисления данных механического анализа
почвы. Из почвы, просеянной через сито с отверстиями в 1 мм, было взято 10 а воз¬
душно-сухой (влажность 5,08%) или 9,52 г абсолютно сухой почвы. После обработки на
сите с отверстиями в 0,25 мм были отделены частицы размером 1—0,25 мм, вес которых
равнялся 0,047 г, что составляет 0,047:9,52-100 = 0,5% на сухую почву. Потеря в весе
при обработке 0,05 н. раствором НС1 была равна 0,343 г, или 0,343:9,52-100 = 3,6%.
При выделении фракций пипеткой (температура 20°, уд. вес твердой фазы 2,60) были
получены следующие результаты:
Глубина
взятия
пробы, см
Время отстаивания
Диаметр
частиц, мм
Объем суспензии
в пипетке (Ь), мл
Вес
высушенной
пробы (а), г
% от веса
почвы
25
115 сек.
<0,05
25
0,2226
93,6
10
19 мин.
<0,01
26
0,1590
64,4
10
77 мин.
<0,005
24
0,1185
52,0
7
22 ч. 26 м.
<0,001
25
0,0838
35,2
<0,001
Без NaOH
0,0792
33,2
Определение физических и водных свойств почвы
321
Содержание отдельных фракций определяют по разности:
Фракции, мм Содержание. %
Пыль крупная 0,05—0,01 93,6—64,4=29,2
средняя 0,01—0,005 64,4—52,0=12,4
мелкая 0,005—0,001 52,0—35,2=16,8
Ил < 0,001 (с поправкой на NaOH) 33,2
Песок средний 1—0,25 (на сите) 0,5
Потери при обработке НС1 3,6
Итого 95,7
Песок мелкий 0,25—0,05 (100—95,7%) = 4,3
Всего 100
Черноземная почва при таком механическом составе будет называться, по классифи¬
кации Н. А. Качинского, чернозем легкоглинистый иловатый.
Микроагрегатный состав почвы
10—30 г почвы (большую навеску берут при легком механическом составе
почвы), пропущенной через сито с ячейками в 1 мм, помещают в склянку
емкостью 500 мл. При взятии навески следует обратить особое внимание на
отбор действительно средней пробы. Астапов, например, рекомендует пробу
почвы составлять из фракций после сухого рассева почвы на ситах пропор¬
ционально содержанию каждой фракции, как это делается при агрегатном
анализе по Саввинову. Одновременно берут пробу почвы на влажность.
В склянки вливают по 250 мл дистиллированной воды и оставляют почву
размокать 24 часа. После этого закрытые резиновыми пробками склянки по¬
мещают на прибор для взбалтывания, где онич подвергаются интенсивному
(200 толчков в минуту) встряхиванию в течение 2 час. Содержимое склянок
переносят через сито с отверстиями диаметром 0,25 мм в цилиндры, доводят
до 1 л и затем пипетируют 25 мл суспензии, точно так же, как при механичес¬
ком анализе по Качинскому.
В случае засоленных почв применение дистиллированной воды вызывает
пептизацию и может исказить результат. Чтобы избежать этого, предвари¬
тельно готовят водную вытяжку из отдельной навески почвы в 40 г в 1 л воды.
Через 24 часа прозрачную вытяжку сливают и используют для микроагре-
гатного анализа, заменяя ею дистиллированную воду.
При учете результатов микроагрегатного анализа, по Н. А. Качинскому,
скорость падения микроагрегатов принимают такой же, как для элементар¬
ных частиц, и применяют определенные сроки отстаивания (см. табл. 3).
Таблица 5
Время отбора проб при микроагрегатном анализе по Астапову
(уд. вес почвы 2,60)
Диаметр
агрегатов,
мм
Глубина
взятия
образца, см
Температура, °С
15
20
25
30
Время до взятия проб
1
<0,05
1
25
3 м. 43 с.
3 м. 17 с.
2 м. 55 с.
2 м. 37 с.
<0,01
10
37 м. 15 с.
33 м. 00 с.
29 м. 20 с.
26 м. 07 с.
<0,005
10
2 ч. 29 м.
2 ч. 12 м.
1 ч. 57 м.
1 ч. 45 м.
<0,001
7
43 ч. 20 м.
38 ч. 25 м.
34 ч. 12 м.
30 ч. 30 м.
322
Д, В. Федоровский
По данным Астапова, скорость падения в воде микроагрегатов значитель¬
но меньше скорости падения элементарных частиц. Это объясняется наличи¬
ем микропор в агрегатах, заполненных водой, вследствие чего их объемный7
вес значительно меньше, чем удельный вес отдельных частиц. Поэтому Аста¬
пов рекомендует расчет скоростей падения в спокойной воде для микроагре¬
гатов (табл. 5) производить по формуле Стокса с измененным коэффициентом
в числителе (0,13 вместо 0,222).
ИССЛЕДОВАНИЕ ВОДНЫХ СВОЙСТВ ПОЧВЫ
ПРИ ПРОВЕДЕНИИ ВЕГЕТАЦИОННЫХ ОПЫТОВ
Поддержание оптимального режима влажности в вегетационных опытах
требует большого внимания и знания водных свойств почвы. Хорошо разви¬
тые растения могут исчерпать запас доступной влаги в небольшом объеме
почвы сосуда. Завядание растений недопустимо в опытах, так как глубоко
травмирует растения и нарушает поступление питательных веществ и течение
многих биохимических процессов. Не менее опасен даже временный «пере¬
лив сосудов», когда влажность превышает полную влагоемкость и недостаток
воздуха в почве подавляет развитие растений.
При подготовке вегетационного опыта исследуют водные свойства насып¬
ных образцов почвы с нарушенным строением. Для расчета поливной нормы
определяют одну иэ форм влагоемкости почвы и нижний предел доступной
влаги.
Оптимальные для роста растений условия создаются в том случае, если
влажность почвы при поливе доводится до 60—70% величины полной влаго¬
емкости, 70—80% капиллярной или 100% наименьшей влагоемкости.
Влажность почвы в сосудах должна поддерживаться в пределах содержа¬
ния легкоподвижной влаги, ни в коем случае не снижаясь до влажности за¬
нял ания.
Полная влагоемкость почвы
Полная влагоемкость, определяемая в трубках, всегда бывает несколько
меньше общей порозности, так как при погружении в воду образца почвы
в нем сохраняется около 8% защемленного воздуха.
Полную влагоемкость почвы с нарушенным строением определяют в ме¬
таллических цилиндрах с сетчатым дном или в стеклянных трубках, обвя¬
занных с одного конца марлей. Диаметр трубки 5—6 см, высота 15—18 см*
На сетчатое дно накладывают кружок фильтровальной бумаги и смачивают
водой. После стекания излишка воды взвешивают трубку на технических ве¬
сах с точностью 0,05 г (удобны весы ВЛТК—500).
Цилиндр наполняют на 8/4 высоты просеянной через грохот почвой. Почву
вносят небольшими порциями и уплотняют постукиванием трубки или осто¬
рожным уминанием, добиваясь того же уплотнения, которое принято для со¬
судов вегетационного опыта. Одновременно берут пробу для определения
влажности исходной почвы.
После наполнения почвой цилиндр взвешивают и по разности между
весом цилиндра с почвой и пустого цилиндра определяют навеску исходной
почвы. Зная влажность почвы, вычисляют вес абсолютно сухой почвы в ци¬
линдре.
Цилиндр с почвой прикрывают сверху стеклом, ставят в сосуд с водой,
доводят уровень ее до уровня почвы в цилиндре и оставляют на сутки. Через
сутки вынимают цилиндр из воды, обтирают фильтровальной бумагой и взве¬
шивают. Еще через сутки повторяют взвешивание. При получении близких
данных насыщение прекращают.
Определение физических и водных свойств почвы
323
Влагоемкость выражают в весовых или объемных процентах. Для перево¬
да в объемные весовые данные следует умножить на объемный вес. Отношение
веса поглощенной воды к весу сухой почвы определяет полную влагоемкость
в весовых процентах.
Запись результатов определения:
Вес цилиндра с увлажненной обвязкой (а).
Вес цилиндра с почвой (Ь).
Навеска исходной почвы (Ъ — а).
Навеска абсолютно сухой почвы (d).
Вес трубки с почвой после насыщения (с).
Вес поглощенной воды (с — а — d).
Полную влагоемкость (в % на абсолютно сухую почву) определяют по
формуле:
Капиллярная влагоемкость почвы
Капиллярная влагоемкость показывает наибольшее количество воды, ко¬
торое способна поглотить почва при капиллярном ее насыщении снизу. Ве¬
личина капиллярной влагоемкости зависит от объема капилляров почвы и
высоты почвенного столба при насыщении.
Определение капиллярной влагоемкости можно производить в образцах
с нарушенным и ненарушенным строением почвы. Образцы с ненарушенным
строением берут специальным буром (например, буром Васильева емкостью
500 мл для определения объемного веса) непосредственно с полевых уча¬
стков.
Определение в образцах с нарушенным строением применяется при за¬
кладке вегетационных опытов. Размер цилиндров и порядок их заполнения
почвой тот же, что при определении полной влагоемкости (см. выше).
Цилиндры ставят на кювету с водой таким образом, чтобы почва через
сетку соприкасалась с поверхностью воды. Взвешивание производят 1 раз
в сутки. Постоянство веса укажет окончание насыщения.
Содержание воды в почве после насыщения определяют по разности веса
насыщенной и абсолютно сухой почвы и рассчитывают влагоемкость, как
это описано для полной влагоемкости. Возможен и другой путь: почву из
цилиндра переносят в чашку, перемешивают и отбирают пробу в сушильный
стаканчик. Влажность насыщенной водой почвы в процентах к весу сухой
почвы выражает капиллярную влагоемкость.
Наименьшая влагоемкость почвы
В лабораторных условиях можно определить наименьшую влагоемкость
образца насыпной почвы по С. И. Долгову (1948). Определение производят
в стеклянных трубках диаметром 3 см и длиной 60—80 см. Нижний конец
трубки обвязывают полотном или марлей. При подготовке почвы ее дово¬
дят до воздушно-сухого состояния и пропускают через грохот^(2—3 мм), но
не растирают.
При заполнении трубок принимают меры против образования слоистости:
засыпают почву через воронку, на носик которой надета широкая каучуковая
трубка, доходящая до дна стеклянной трубки. При насыпании почва запол¬
няет воронку и всю каучуковую трубку. При постоянном постукивании и вра¬
щении стеклянной трубки начинают медленно поднимать воронку, не отни¬
мая нижнего конца каучуковой трубки от высыпавшейся почвы. Почва сплош¬
324
Д. В. Федоровский
ным столбом, без сортировки, заполняет стеклянную трубку. Этот прием
позволяет избежать образования слоистости.
Однократный полив производят с расчетом, чтобы почвенный столб был
промочен не до дна; нижняя сухая зона может быть небольшой. Ход прома-
чивания отмечается раз в сутки — продвижение увлажненной зоны вниз
хорошо заметно через стеклянные стенки. Для предотвращения подсыхания
поверхности почвы трубку закрывают часовым стеклом.
После прекращения передвижения воды (через 30—40 дней) трубку раз¬
резают и послойно, в каждых 2—4 см, определяют влажность. Верхние 4—
6 см обычно переувлажнены, в нижней части длиной 20—25 см, прилегаю¬
щей к сухой почве, влажность заметно изменяется. Во всех слоях выше пере¬
ходной зоны (кроме самых верхних) сохраняется постоянная влажность, ко¬
торая соответствует наименьшей влагоемкости почвы.
Определение недоступной растению влаги в почве
Не вся вода почвы одинаково доступна растениям: часть ее находится
в связанной, малодоступной для растений форме. Для вычисления количест¬
ва активной, доступной растениям воды, находящейся в данный момент в поч¬
ве, определяют общее количество воды и вычитают из него воду «влажности
завядания»— содержание воды малоподвижной и не обеспечивающей нор¬
мального развития и роста растений.
Количество недоступной для растений воды в почве зависит прежде всего
от состава самой почвы. В первом приближении эту величину можно рассчи¬
тать по максимальной гигроскопичности почвы. Массовые определения влаж¬
ности завядания удобно проводить, применяя метод проростков на неболь¬
ших объемах почвы. Но более правильным, хотя и более громоздким, яв¬
ляется определение влажности завядания хорошо развитых растений, про¬
израстающих на таком объеме почвы, которое обеспечивает нормальное их
развитие. При таких исследованиях было установлено, что растения разных
видов и разных фаз развития оставляют в почве при завядании разыне коли
чества неиспользованной воды (Федоровский, 1948).
В полевых условиях во время засушливого периода также можно опреде¬
лить в корнеобитаемом слое почвы влагу, оставшуюся недоступной расте¬
ниям.
Максимальная гигроскопичность почвы (по Мит-
черлиху). Около 10 г суглинистой или 20 г супесчаной воздушно-сухой почвы,
пропущенной через сито с отверстиями 1—2 мм, насыпают в широкий низкий
сушильный стаканчик с известным весом. После взвешивания стаканчика с
с почвой его помещают с открытой крышкой в эксикатор, на дно которого
налито 200—300 мл 10 %-ного раствора серной кислоты для поддержания
в нем относительной влажности воздуха 96 %. При такой относительной влаж¬
ности воздуха удобнее насыщать почву влагой, чем при более высокой (влаж¬
ность над насыщенным раствором K2S04 по Николаеву или 3%-ным раство¬
ром H2S04 по Францессону равна 98%), так как при большом содержании
паров в воздухе небольшое понижение температуры легко вызывает обра¬
зование росы в эксикаторе и увлажнение почвы, что затрудняет опреде¬
ление.
Стаканчики выдерживают в эксикаторе до тех пор, пока почва перестанет
прибавлять в весе. Значительно скорее насыщение идет в том случае, если
в эксикаторе создать разрежение (например, водоструйным насосом). Экси¬
катор надо ставить в такое место лаборатории, где температура воздуха ме¬
няется менее всего, и оберегать его от нагревания и сотрясений.
Первое взвешивание производят через 3—4 дня. Если насыщение проис¬
ходит при разрежении, кран эксикатора соединяют с двумя промывными
Определение физических и водных свойств почвы
325
склянками с 10%-ной H2S04 и, осторожно открывая кран, впускают в экси¬
катор воздух. После выравнивания давления с эксикатора снимают крышку j
вынимают стаканчики, быстро закрывают их крышками и взвешивают с точ¬
ностью до 0,01 г. В эксикаторе сменяют 10 %-ный раствор H2S04.
После взвешивания стаканчики снова помещают в эксикатор и через 5—
6 дней снова взвешивают. Насыщение почвы влагой в эксикаторе и взвеши¬
вание повторяют до получения постоянного веса (при разрежении обычно не
ранее, чем через 20 дней). По достижении постоянного веса стаканчики с поч¬
вой высушивают в сушильном шкафу при температуре 100—105°.
Максимальную гигроскопичность (вес. %) вычисляют по формуле:
(В-С)-ЮО
с —л »
где А — вес пустого стаканчика, г; В — вес стаканчика с почвой после насыщения, г;
С — вес стаканчика с почвой после высушивания, г.
Вода в почве при влажности ее, равной максимальной гигроскопичности,
совершенно недоступна для растения и не используется клетками микроорга¬
низмов. Для расчета недоступной влаги, или влажности завядания, разные
авторы предлагают умножать максимальную гигроскопичность на 1,34; 1,5
или 2,0. Однако такой расчет дает весьма приблизительную величину влаж¬
ности завядания, так как при этом не учитывается все многообразие свойств
растений. Влажность завядания пшеницы колебалась в наших определениях
от 1,3 до 1,6 величины максимальной гигроскопичности, а льна-долгунца —
от 1,7 до 2,3.
Определение максимальной гигроскопичности сильнозасоленных почв
невозможно, так как изменение веса образца почвы при насыщении не пре¬
кращается очень долго. Поглощение паров воды будет продолжаться за счет
различия в упругости паров над раствором, из которого происходит насыще¬
ние, и над пленкой засоленной воды на поверхности почвенных частиц. Про¬
изводить предварительную промывку солей из образца и затем определять
максимальную гигроскопичность не следует, ибо содержание недоступной
для растений влаги в засоленной почве зависит также и от количества солей
в почве.
Определение влажности завядания
методом проростков по Долгову
40—60 г воздушно-сухой почвы помещают в обычный алюминиевый
стаканчик для определения влажности почвы (диаметр 4 см, высота 6 cm)v
Почву постепенно увлажняют, для чего стаканчик ставят наклонно и смачи¬
вают почву по каплям, по мере впитывания воды, чтобы вытеснить из почвы
воздух и достичь полного смачивания почвы.
С первым поливом вносят также питательные соли, если почва малопло¬
дородна. После полива в каждый стаканчик сажают пять — шесть наклю¬
нувшихся зерен ячменя или другого растения. До появления всходов все
стаканчики должны быть прикрыты плотной бумагой для предотвращения'
испарения воды с поверхности почвы и ее подсыхания. После появления всхо¬
дов растения следует держать в хорошо освещенном помещении и поливать*
не менее 3-х раз в день по весу.
В каждом стаканчике оставляют по четыре одинаковых растения. Влаж¬
ность завядания определяют в то время, когда второй лист по длине начинает
превосходить первый настоящий лист. (Обычно это бывает через 2 недели
после посева). Поверхность почвы заливают расплавленной мастикой из па¬
рафина (2—3 части) и вазелина (1 часть). Температура мастики должна быть
близка к точке ее застывания. Затем все стаканчики помещают в светлое,
но защищенное от прямого солнечного света помещение и оставляют там до
326
Д. В. Федоровский
завядания растений. В зимнее время выращивание ведут в камерах с осве¬
щением лампами дневного света.
Начало завядания заметно по подвяданию верхушек нижних (первых)
листьев; затем наступает явное завядание — все листья подвядают и опу¬
скаются до половины своей длины; третья фаза завядания характеризуется
полным завяданием и опусканием всех листьев на всю их длину.
При ежедневном осмотре утром, днем и к вечеру все стаканчики с расте¬
ниями, достигшими второй фазы явного завядания, помещают под поставлен¬
ный на стол стеклянный колпак или плотный деревянный ящик с влажной
ватой или опилками, которые поддерживают в замкнутом пространстве вы¬
сокую влажность воздуха. Если завядание растений под колпаком сохранит¬
ся до следующего утра, это будет означать, что почва достигла влажности
устойчивого завядания. Если же к утру растения восстановили тургор, их
выставляют на несколько часов на открытое место до появления признаков
завядания, а затем убирают во влажную атмосферу. Соблюдение этой тех¬
ники установления влажности почвы устойчивого завядания очень важно
для получения устойчивых, достаточно объективных данных.
Определение влажности в почве проводят в тех же стаканчиках. Почвен¬
ный комок обычно легко извлекается из стаканчика, так как почва при иссу¬
шении уменьшает свой объем. Верхнюю часть почвенного образца в 1—2 см
вместе с парафиновой корочкой и остатками семян удаляют; почвенную пробу
разламывают, по возможности освобождают от корней и затем возвращают
в тот же стаканчик, в котором производят сушку почвы и определение влаж¬
ности. Определение влажности завядания ведут с трехкратной повторностью.
Подобное определение недоступной влаги при помощи живых растений
является более точным, чем определение, например, по максимальной гигро¬
скопичности; однако и этот метод не учитывает специфичности свойств расте¬
ний на разных фазах их развития.
Определение влажности завядания вегетационным методом
с хорошо развитыми растениями
Как было установлено опытами, проведенными в лаборатории агрохимии
Почвенного института, наиболее точное определение влажности завядания
может быть получено при хорошо развитых растениях с мощной корневой
системой. Количество недоступной растениям влаги изменяется с возрастом
растения; наибольшее иссушение почвы отмечено под растениями в фазе буто¬
низации и цветения. Как показали опыты с хорошо развитыми растениями,
влажность завядания различных видов растений для одной и той же почвы
также неодинакова.
Почву набивают в сосуды.емкостью около 1 л. Удобно употреблять для
этих опытов железные крашеные сосуды. Как в обычных вегетационных опы¬
тах, на дно сосуда кладут 100—150 г битого стекла для обеспечения дренажа
и устанавливают трубку для полива. При необходимости в почву вносят удоб¬
рения. На сосуд оставляют от 1 до 10 растений, в зависимости от вида. По¬
ливку растений вплоть до бутонизации (или выколашивания) проводят как
обычно, причем стремятся поддержать оптимальную для растений влажность.
По достижении растениями стадии бутонизации или колошения сосуды
подготавливают к определению влажности завядания. Поверхность почвы
в сосудах заливают расплавленной при температуре около 40° легкоплавкой
мастикой из смеси парафина (2 части) и вазелина (1 часть). Избыток мастики
сливают через край при наклонении сосуда. Тонкий слой застывшей мастики
должен покрывать всю поверхность почвы и плотно приставать к стенкам
сосуда и стеблям растения, предотвращая испарение влаги с поверхности
почвы. Можно использовать с этой целью и холодную невысыхающую замаз¬
Определение физических и водных свойств почвы
327
ку из смеси мела (или зубного порошка) и касторового масла. Отверстие по¬
ливной трубки закрывают ватным тампоном.
После такой подготовки растения используют до возможного предела
только доступную воду из почвы в сосуде. Растения должны находиться под
непрестанным наблюдением. При первых признаках завядания расте¬
ния немедленно переносят в темную камеру с относительной влажностью воз-
духа около 95%. Такую камеру можно соорудить на одной из вагонеток:
каркас сбивают из деревянных брусков, стены из картона обтягивают мар¬
лей. На крыше камеры устанавливают большие кристаллизаторы с водой
и опускают в них концы фитилей из ваты. Второй конец каждого фитиля сое¬
диняют с марлей камеры, которая постепенно увлажняется.
Оправившиеся от временного завядания растения вынимают из камеры
и выставляют на открытый воздух, где они обычно уже через 2—4 часа подвя-
дают, после чего их снова помещают в камеру. После двух-трех перестановок
растения не восстанавливают своего тургора в камере за сутки и остаются
завядшими.
В момент наступившего устойчивого завядания растения срезают, почву
извлекают из сосудов, быстро перемешивают (причем отбрасывают крупные
корни) и берут по две пробы из каждого сосуда для определения влажности
высушиванием. Определение влажности завядания проводят в трехкратной
повторности (три параллельных сосуда).
Влажность завядания зависит от свойств почвы, свойств почвенного рас¬
твора (в первую очередь от осмотического давления его), вида и возраста рас¬
тений. Величина влажности завядания для супесчаных почв колеблется око¬
ло 3—5% на сухой вес почвы, для суглинистых черноземных почв — 15—
18% и еще выше для торфяных почв.
Определение недоступной растениям воды
в почве бед 8авядавия растевий.
Метод Соколова и Федоровского
Описанные выше методы определения количества недоступной растению
воды в почве имеют существенный недостаток: корни растений сконцентри¬
рованы в одном небольшом объеме почвы, который ими иссушается, а исчер¬
пание доступной воды определяется по завяданию. Не только при самом за-
вядании, но даже еще до его наступления растения испытывают недостаток
воды, и жизненные процессы и свойства растений в опыте значительно из¬
меняются (падает обводненность тканей, возрастает сосущая сила и т. д.)»
В отличие от этого в поле имеется разная обеспеченность слоев почвы влагой;
растения не завядают, иссушив верхние горизонты почвы, так как продол¬
жают получать воду из более глубоких слоев.
Более правильным методом определения максимального иссушения поч¬
вы корневой системой в вегетационном опыте будет постановка опыта, при¬
ближающего нас к условиям поля, в котором иссушение производится нор¬
мально развитыми и незавядшими растениями. Такие условия соблюдаются
в методе Л. В. Соколова и Д. В. Федоровского, заключающемся в следующем.
Почву в сосудах разделяют на две половины; верхнюю половину отделяют от
нижней горизонтальной прослойкой из гравия или крупного песка толщиной
около 3 см (песок должен быть отсеян от примеси пыли). Такая прослойка
изолирует верхнюю половину почвы от капиллярного поднятия воды снизу,
если влажность нижней половины ниже полной влагоемкости (рис. 13).
Первоначально, после посева, полив производят по весу сверху и через
трубочку снизу и влажность всей почвы доводят до оптимальной. F орни рас¬
тений проникают до дна сосуда, после чего можно приступить к определе¬
нию. Поверхность почвы заливают тонким слоем изолирующей мастики^и с
328
Д. В. Федоровский
этого момента производят поливку только ниж¬
ней половины почвы через трубочку. Поливную
норму снижают и доводят до количества, поддер¬
живающего влажность нижней половины почвы
в 60% от полной влагоемкости; при этом учиты¬
вают вес воды мертвого запаса в верхней полови¬
не, который прибавляют при расчете веса воды
для полива.
Растения, продолжая развиваться, извлекают
воду своей корневой системой из обеих половин
почвы, иссушают верхнюю половину почвы до
той влажности, при которой вода является недо¬
ступной. Как показали опыты, влажность эта
близка к влажности завядания, найденной други¬
ми методами. Влажность почвы определяют дней
через десять после прекращения полива верхней
части почвы; некоторой задержки определения
в этом случае опасаться не приходится, так как
в верхнем слое почвы не должно происходить ни
потерь воды с поверхности почвы, ни притока во¬
ды снизу по капиллярам (возможен лишь приток
парообразной воды снизу). Показателем правильно¬
сти применения метода является равномерность иссушения верхней полови¬
цы почвы в сосуде; поэтому всегда следует брать две пробы на влажность —
одну из верхней и одну из нижней части иссушаемой половины почвы в сосуде
Рис. 13. Схема набивки
сосудов при определении
недоступной растению влаги
1 — почва; 2 — песок; з — гра¬
вий; 4 — замазка; 5 — трубка
для полива
Вышслелие запасов доступной растениям воды
в вегетационном опыте
Поливной режим вегетационного опыта устанавливают на основании дан¬
ных о содержании воды разной степени доступности растениям.
Вся вода, находящаяся в почве сверх влажности завядания, доступна
для растений, однако увлажнение выше наименьшей влагоемкости вызы¬
вает недостаток аэрации и ухудшает условия питания растений. Поэтому
верхний предел для полива сосудов устанавливают по наименьшей влагоем¬
кости (или соответственной величине 60—70% полной, или 70—80% капил¬
лярной).
Легко поступает в растение лишь часть всего запаса доступной воды, при¬
мерно его половина. Использование второй половины запаса вызывает за¬
медление роста растений. Нижний предел допустимой в опыте влажности поч¬
вы рассчитывают по формуле (НВ — В3):2 или принимают его равным 66%
НВ по рекомендации А. А. Роде. Пример расчета приведен в табл. 6.
В таблицу вписывают данные о влагоемкости почвы, МГ и ВЗ в весовых
процентах. Зная навеску и влажность почвы, находящейся в сосуде, вычис¬
ляют вес абсолютно сухой почвы, вес воды каждой категории и рассчиты¬
вают суммарный вес сосуда. Затем устанавливают вес к поливу и допусти¬
мую потерю влаги перед поливом.
При развитии большой надземной массы (200—300 г/сосуд) ее вес необхо¬
димо добавить к расчетному поливному весу, иначе содержание легкодоступ¬
ной воды после полива окажется пониженным, а заданный режим влажности
в опыте будет нарушен.
Определение физических и водных свойств почвы
329
Таблица 6
Примеры расчета содержания влаги разных категорий для определения поливного режима
в вегетационном опыте
Показатели
Черноземная легкоглшшстая
почва
Дерново-подзолистая супесчаная
почва
Влажность,
вес %
Вода,
мл! сосуд
Вес влаж¬
ной почвы, г
Вес сосу¬
да, г
Влажность,
вес. %
Вода,
мл/сосуд
Вес влажной
почвы, г
Вес сосу¬
да, г
Тара сосуда
1.500
1.500
Навеска почвы при
12
360
3.360
4.860
3
120
4.120
5.620
набивке
Навеска абсолютно
—
—
3.000
4.500
—
—
4.000
5.500
сухой почвы
МГ
10
300
3.300
4.800
2
80
4.080
5.580
При
пттяжтгпгти
ВЗ
15
450
3.450
4.950
3
120
4.120
5.620
НВ
40
1200
4.200
5.700
21
840
4.840
6.340
ол a/xVoUv 1 л
КВ
50
1500
4.500
6.000
24
960
4.960
6.460
. пв
60
1800
4.800
6.300
30
1200
5.200
6.700
Максимальный
вес
5.700
6.340
при поливе (НВ)
Нижний предел лег¬
5.300
6.000
кодоступной влаги
Предел доступной
4.950
5.620
влаги (ВЗ)
Допустимые колеба¬
5.700-5.300
6.340
-6.000
ния веса сосуда
Литература
Агрофизические методы исследования почв.
М., «Наука», 1966.
Астапов С. £., Долгов С. И. Методы изуче¬
ния водно-физических свойств почв и
грунтов.— В кн. «Почвенная съемка.
Руководство по полевым исследованиям
и картированию почв». М., Изд-во АН
СССР, 1959.
Бескин Л. И., Карманова Л. А., Ромаш-
кевич А. И. Использование нейтронного
влагомера НИВ-1 для определения влаж¬
ности красноземов.— Почвоведение, 1970,
№ 2.
Вадюнина А. Ф., Корчагина 3. А. Мето¬
ды исследования физических свойств
почв и грунтов. М., «Высшая школа»,
1965.
Вершинин Я. В., Мельникова М. К, и др.
Под ред. А. Ф. Иоффе и И. Б. Ревута.
Основы агрофизики, М., Физматгиз,
1959.
Гречин И. Я., Кауричев И. С. и др. Прак¬
тикум по почвоведению. М., «Колос»,
1964.
Долгов С. И. Исследования подвижйости
почвенной влаги и ее доступности для
растений. М., Изд-во АН СССР, 1948.
Зайдельман Ф. Р. Методика исследования
некоторых физических и водно-физиче¬
ских свойств каменистых почв.— Почво¬
ведение, 1957v № 1.
Зайдельман Ф. Р. Особенности изучения
физических свойств заболоченных и бо¬
лотных почв.— В кн. «Агрофизические
методы исследования почв». М., «Наука»,
1966.
Инструкция по изучению водно-физиче¬
ских свойств заболоченных и болотных
почв на объектах осушения. М., Росги-*
проводхоз, 1964.
Качинский Н. А. Механический и микро-
агрегатный состав почвы, методы его
изучения. М., Изд-во АН СССР, 1958.
Качинский Н. А. Физика почвы. М., «Выс¬
шая школа», т. I, 1965; т. II, 1970.
Косицын В. Н. Некоторые результаты про¬
изводственных испытаний радиоизотоп-
ных влагомеров и плотномеров.— Изото¬
пы в СССР, 1967, № 7.
Крупский Н. К. Методическое пособие па
330
Д. В. Федоровский
лабораторным и полевым анализам при
обследованиях почв колхозов и совхозов
УССР. Харьков, 1957.
Межреспубликанские технические условия
методов проведения агрохимических ана¬
лизов почв для зональных агрохимичес¬
ких лабораторий. М., «Колос», 1968.
Методическое руководство по изучению поч¬
венной структуры. (Описание методов
определения физических свойств почвы,
применяемых в социалистических стра¬
нах). JI., «Колос», 1969.
Методы агрохимических, агрофизических и
микробиологических исследований в по¬
ливных хлопковых районах. Изд. 3-е.
Ташкент, 1963.
Пособие по проведению аналиаов почв и
составлению агрохимических картограмм.
М., Россельхозиздат, 1965.
Ревут Я. Б. Физика почв. JL, «Колос»,
1972.
Роде Л. Л. Основы учения о почвенной вла¬
ге. Л., Гидрометеоиздат, т. I, 1965; т. II,
1969.
Толковый словарь по почвоведению. Физи¬
ка почв. М., «Наука», 1972.
Федоровский Д. В. Определение в почве во¬
ды, не доступной для корней растений.—
Почвоведение, 1939, № 4.
Федоровский Д. В. Зависимость коэффици¬
ента завядания от вида растений и осмо¬
тического давления почвенного раство¬
ра.— Почвоведение, 1943, № 10.
Физические условия почвы и рост расте¬
ний. Сб. под ред. Б. Шоу. М., ИЛ, 1955.
Цыганов М. С. Прибор для автоматическо¬
го купания почвы при мокром агрегатном
анализе.— Почвоведение, 1957, JSTs 3.
Б. Н. МАКАРОВ
МЕТОДЫ ОПРЕДЕЛЕНИЯ СОСТАВА ПОЧВЕННОГО ВОЗДУХА,
ИНТЕНСИВНОСТИ ДЫХАНИЯ почвы
И ГАЗООБРАЗНЫХ ПОТЕРЬ АЗОТА ПОЧВЫ И УДОБРЕНИЙ
*
Газообразная фаза почвы — почвенный воздух — имеет большое зоаче-
ние для почвенных процессов и роста растений: принимает участие в хими¬
ческих и биохимических процессах, протекающих в почве, оказывает влия¬
ние на окислительно-восстановительные условия в почве, на ее реакцию,
растворимость химических компонентов почвы. Почвенный воздух обеспечи¬
вает кислородом корни растений и живые организмы, населяющие почву,
а также является важным фактором в углеродном питании растений (более
половины углекислого газа, идущего на построение урожая сельскохозяй¬
ственных культур, доставляется растениям из почвы).
Отдельные газы и их смеси (воздух) могут находиться в почве в свободном
состоянии (в порах почвы и пустотах, не занятых водой), адсорбированном на
поверхности почвенных частиц и могут быть растворенными в почвенном рас¬
творе. Наибольшее значение имеет свободный почвенный воздух, содержание
которого зависит от порозности и влажности почвы.
По составу почвенный воздух сходен с атмосферным, но отличается от
него большим содержанием углекислого газа и водяного пара и меньшим
содержанием кислорода. В зависимости от типа почвы и условий водно-воз¬
душного режима в почве могут образовываться и накапливаться метан, серо¬
водород, аммиак, закись азота и другие газы.
Состав почвенного воздуха значительно изменяется как во времени (сезон¬
ная и суточная изменчивость), так и по профилю почвы в зависимости от био¬
логической деятельности, гидротермических условий, адсорбции газов твер¬
дой фазой почвы и интенсивности газообмена между почвой и атмосферой*
В результате биологических процессов в почве идет постоянное поглощение
кислорода организмами и выделение углекислого газа, вследствие чего поч¬
венный воздух обедняется кислородом и обогащается углекислым газом. Вы-
деление С02 из почвы в атмосферу в процессе диффузии зависит от продуци¬
рования С02 почвой, физических и химических свойств почвы, изменения
гидротермических условий. Решающая роль в продуцировании углекислого
газа почвой принадлежит биологическим факторам, поэтому выделение С02
из почвы может характеризовать интенсивность биологических процессов
в почве. Вследствие этого интенсивность дыхания почвы является одним из
показателей биологической активности почвы.
Для почвенных, агрохимических и микробиологических исследований
большое значение имеет изучение газового (воздушного) режима почвы. Га¬
зовый режим почвы — совокупность всех взаимосвязанных явлений: посту¬
пление газов в почву и их передвижение по профилю почвы, изменение содер¬
жания газов в почвенном воздухе в результате поглощения или выделения
отдельных газов при биологических и биохимических процессах и обмена га¬
зами между почвой и атмосферой, а также между почвенным воздухом и твер¬
дой и жидкой фазами почвы. Газовый режим почвы складывается из следую¬
щих элементов: содержание (запас) воздуха в почве, его состав, аэрация и ин¬
тенсивность выделения газов (С02, N20, N02, NH3) из почвы. При стацио¬
нарных почвенных исследованиях необходимо периодическое одновременное
определение всех перечисленных элементов. Определения проводятся каж-
332
Б. Н, Макаров
мм
Рис. 1. Трубка для из-
влеченияпочвенного воз¬
духа
Объяснение см. в тексте
Зегера
дые 10—15 дней или приурочиваются к фазам раз¬
вития растений. Одновременно проводятся наблюде¬
ния за давлением и температурой воздуха, темпера¬
турой и влажностью почвы.
ОПРЕДЕЛЕНИЕ СОСТАВА ПОЧВЕННОГО ВОЗДУХА
Для характеристики газового режима почвенно¬
грунтовой толщи на одних и тех же участках (де¬
лянках) в течение длительного времени целесооб¬
разно устанавливать в почву постоянные трубки для
периодического отбора проб почвенного воздуха
(Макаров, Мацкевич, 1966). Наиболее пригодны
латунные, медные (или из другого мало окисляюще¬
гося в почве материала) трубки с внутренним диа¬
метром 3—5 мм (рис. 1,2). К нижнему концу труб¬
ки припаивается латунное или медное кольцо (2) для
укрепления ее в почве. На нижнем конце трубки
просверливается несколько отверстий, которые пре¬
дохраняются от загрязнения отрезком стеклянной
трубки (3) и стеклянной ватой (4) или металличес¬
кой сеткой.
При выборе глубин, на которые устанавливают
трубки, необходимо учитывать все генетические го¬
ризонты почвы и неоднородность механического со¬
става. Верхнюю трубку устанавливают на глубину
10—15 см, затем с интервалом примерно в 25 еле уста¬
навливают трубки до глубины 1,5 м; от 1,5 до2,5ле
интервал увеличивается до 50 см и глубже 2,5 м ин¬
тервал устанавливается в 1 м. Расстояния между
трубками должны быть не менее 20—25 см. Трубки
устанавливают в скважину, пробуренную почвенным
буром. Затем почву, взятую из скважины, засыпают
обратно в том же порядке. Нижние 10—20 см сква¬
жины засыпают рыхлой почвой, а затем каждый слой
плотно утрамбовывают. Верхний конец трубки дол¬
жен выступать на 5—10 еле над поверхностью почвы
(соответственно длина трубки должна быть на 10 см
больше заданной глубины) и закрываться каучуко¬
вой пробкой. На каждую глубину трубки устанав¬
ливают не менее чем с двойной повторностью. Для
отбора пробы почвенного воздуха к трубке присое¬
диняют приемник с помощью каучукового шланга.
Перед взятием пробы воздух, находящийся в трубке,
необходимо заменить почвенным воздухом. Для этого
через трубку протягивают небольшой объем воздуха,
равный емкости трубки и соединительной системы.
Объем пробы почвенного воздуха не должен превы¬
шать 150—200 см3, так как при отборе пробы создает¬
ся некоторое разряжение и взамен взятого воздуха
поступает воздух из прилегающих слоев почвы. Про¬
бы почвенного воздуха отбирают в газовые пипетки
Зегера (рис. 2), предварительно заполненные насы¬
щенным раствором NaCl, подкисленным НС1.
Методы определения состава почвенного воздуха, интенсивности дыхания почвы 333
Рис. 3. Колба Макарова для отсасывания пробы почвенного воздуха и определения в нем
С02 и 02
Объяснение см. в тексте
Анализ почвенного воздуха наиболее часто проводят абсорбционными ме- .
тодами, в основе которых лежит способность отдельных компонентов почвен¬
ного воздуха быстро абсорбироваться некоторыми реагентами. По измене¬
нию концентрации реагента или объема, или давления воздуха судят о
содержании определяемого компонента в исследуемой пробе воздуха. Эти ме¬
тоды разделяются на титриметрические, волюмометрические и манометри¬
ческие. Для определения состава почвенного возуха может быть применен
метод газо-адсорбционной хроматографии. Выбор метода анализа зависит от
задач исследования, концентрации исследуемого компонента и имеющегося
объема воздуха. Если в почвенном воздухе нужно определить содержание
только С02, удобно пользоваться титриметрическим или манометрическим
методом. Волюмометрический метод применяется для одновременного опре¬
деления содержания углекислого газа и кислорода. Титриметрические мето¬
ды основаны на определении изменения концентрации поглощающего рас¬
твора после взаимодействия последнего с определяемым компонентом поч¬
венного воздуха.
Для определения концентрации С02 в почвенном воздухе определенный
объем его пропускают через раствор Ва(ОН)2. Гидрат окиси бария связывает
С02 в нерастворимую в воде соль, а избыток Ва(ОН)2 оттитровывают соляной
кислотой в присутствии индикатора фенолфталеина.
Нами был предложен простой титриметрический метод определения С02
и 02 с помощью колб, позволяющий проводить анализ непосредственно в по¬
ле. Применяют колбы емкостью на 150—200 мл с притертыми пробками
(рис. 3). В пробку впаяны стеклянная трубка (а) с краном (1), и тубус (б) с
с притертой стеклянной трубкой (в) с краном (2); трубка доходит до дна кол¬
бы (г).
Отбор пробы почвенного воздуха производят следующим образом. Колбу
полностью заполняют дистиллированной водой, вставную стеклянную труб¬
ку присоединяют каучуковой трубкой к буру для отбора почвенного воздуха
или к латунной трубке, установленной в почве, и открывают кран 7, затем 2.
Чтобы систему (бур — каучуковая трубка) заполнить почвенным воздухом,
33,4
Б. Н. Макаров
из колбы выливают 20—40 мл воды, а затем снова доливают ее водой. Откры¬
вают краны, вода из колбы вытекает, а почвенный воздух поступает в колбу.
Когда вся вода из колбы вытечет, краны 1 и 2 закрывают.
Колбу с почвенным воздухом отсоединяют от иглы-бура или латунной
трубки. Вынимают вставную трубку (я), быстро вносят 10—50 мл (в зависи¬
мости от содержания С02) 0,02 н. Ва(ОН)2 и отверстие тубуса закрывают кау¬
чуковой пробкой. Для поглощения С02 барит в колбе интенсивно взбалты¬
вают 3—5 мин., затем избыток барита оттитровывают по фенолфталеину
0,02 н. раствором НС1. По разнице между холостым титрованием 10—50 мл
барита и титрованием этого же количества барита после поглощения СО*
судят о содержании углекислого газа в объеме почвенного воздуха, соответ¬
ствующем объему колбы с учетом взятого количества барита (10—50 мл).
Концентрацию С02 (в мг/л) в почвенном воздухе рассчитывают по формуле
(а — Ъ)-0,44.1000 (273 + 0*760
С— VK~Vp ' 273-(W-tv) ’
где С — содержание С02 в почвенном воздухе, мг/л; а — количество 0,02 я. раствора НС1
пошедшее на титрование взятого объема барита, мл; b — количество 0,02 н. раствора
НС1, пошедшее на титрование барита после поглощения С02, мл; 0,44 — количество СОа»
эквивалентное 1 мл 0,02 н. НС1, мг; 1000— коэффициент для пересчета на 1 л воз¬
духа; VK — объем колбы, мл; 7р — объем взятого раствора барита, мл; t — температура
воздуха; W — давление воздуха; w — упругость водяных паров при температуре t°.
Для пересчета в объемные проценты необходимо полученную величину
С (в мг!л) умножить на 0,051.
Результаты определений концентрации С02 в почвенном воздухе с помо¬
щью метода колб и метода газоанализатора близко совпадают.
При работе с колбами необходимо следить, чтобы пробка и краны были
хорошо притерты во избежание попадания атмосферного воздуха. Объем колб
и вносимых реактивов должны быть точно измерены.
Волюмометрические методы анализа почвенного воздуха основаны на из¬
мерении изменения объема исследуемого воздуха после поглощения анали¬
зируемого компонента в результате абсорбции, сожжения или каталитическо¬
го окисления.
Для поглощения С02 применяют 15—40%-ный раствор КОН (раствор
готовится за несколько дней до анализа). Для кислорода лучшим поглотите¬
лем является щелочной раствор пирогаллола. Последний готовят, смешивая
два раствора: 1) 600 г чистого КОН растворяют в воде с последующим дове¬
дением объема до 1 л и 2) 300 г пирогаллола растворяют в 800 мл воды с по¬
следующим доведением объема до 1 л. Перед употреблением смешивают рас¬
творы в соотношении: 1 объем раствора пирогаллола и 3 объема раствора ще¬
лочи.
При волюмометрическом анализе почвенного воздуха применяют газоа¬
нализаторы, различающиеся конструкцией отдельных частей.
Газоанализатор Гальдана (рис. 4) состоит из измерительной бюретки 7,
системы поглотителей 10 и 12, термобарометра 8 и трех уравнительных скля¬
нок 6,9 и 14. В верхней части трубок 10, 11 и 12 примерно на одном уровне
наносят метки, по которым устанавливают уровни поглотительных раство¬
ров. Метки (М) наносят при открытых кранах 3, 4 и 5 (положение II). Бю-
, ретка 7 объемом 10 или 20 мл в нижней части представляет собой капилляр¬
ную трубку с градуировкой до 0,01 мл, что позволяет с помощью лупы
делать отсчеты до 0,001 мл. Термобарометрическая трубка 8 имеет объем, рав¬
ный объему бюретки, и позволяет выравнивать изменения температуры и дав¬
ления во время анализа. Бюретка и термобарометрическая трубка заключены
в стеклянную муфту, заполненную водой для уменьшения влияния коле¬
баний температуры. Поглотители 10 и 12 заполнены капиллярами для уве-
Методы, определения состава почвенного воздуха, интенсивности дыхания почвы 335
Рис. 4. Схема газоанализатора Гальдана
Объяснение см. в тексте
личения поглощающей поверхности, груша 6 служит аспиратором для за¬
полнения бюретки исследуемым воздухом и перевода его в поглотительную
систему. Она заполняется ртутью или насыщенным раствором хлористого
натрия, подкисленным НС1, с индикатором розоловой кислотой или метило¬
ранжем. Для поддержания уровня в поглотителе со щелочью 10 служит гру¬
ша 9, соединенная с трубкой с натронной известью 13, а в поглотителе с пиро¬
галлолом 12 — груша 14. Для предохранения от соприкосновения пирогал¬
лола с атмосферой служит система трубок 15, заполненная вазелиновым мас¬
лом, которое также наливается на поверхность пирогаллола в груше 14.
Ход анализа. При положении / крана 1 заполняют бюретку запирающим
раствором, поднимая грушу 6 вверх. Раствор не должен подниматься выше
крана 2. Закрывают двухходовой кран 16. Кран 1 переводят в положение II.
Присоединяют к боковому отводу приемник с исследуемым воздухом. От¬
336
Б. Я. Макаров
крыв кран 16 и опуская грушу 6, заполняют бюретку исследуемым воздухом
до нижней метки бюретки. Кран 16 закрывают. Кран 1 переводят в положе¬
ние / для выравнивания давления в бюретке с атмосферным воздухом и быст¬
ро переводят кран 2 в положение, соединяющее бюретку с поглотительной
системой. Выравнивают уровень щелочи в поглотителе 10 с помощью микро¬
винта 17. Записывают отсчет объема воздуха в бюретке (V\). Закрывают
кран 4 (положение III) и производят поглощение С02. Для этого, открыв
кран, грушу 6 поднимают и опускают 8—10 раз, переводя воздух в поглоти¬
тель со щелочью; жидкости не должны касаться кранов. Устанавливают уро¬
вень щелочи в трубках 10 и 11 на метке. Закрывают кран 16. Переводят кран
4 в положение II. Проверяют уровни. Уровень в трубке 11 выравнивают
грушей 5, в трубке 10— микровинтом 17. Записывают объем воздуха без С02
в бюретке (F2).
Содержание С02 (в об. %) вычисляют по формуле1
Vx — к2-100
Гг *
После перевода крана в положение III повторяют поглощение для про¬
верки полноты поглощения. Кран 4 закрывают. Кран 3 переводят в положе¬
ние I (кран 5 — в положение IV). Производят поглощение 02, переводя воз¬
дух из бюретки в поглотитель с пирогаллолом 20—30 раз, поднимая и опу¬
ская грушу 6. С помощью груши 6 и микровинта 17 устанавливают уровень
пирогаллола. Переводят краны 3 и 4 в положение II и выравнивают уровни
щелочи. Записывают отсчет объема воздуха без С02 и 02 в бюретке (F3). Пов¬
торяют поглощение кислорода для проверки на полноту поглощения.
Содержание кислорода (в об.%) вычисляют по формуле
C = Z-ZL.iC0.
После окончания анализа термобарометр соединяют с атмосферой (поло¬
жение I крана 4), проверяют уровни щелочи, переводят кран 2 на соединение
бюретки с атмосферой и заполняют бюретку запирающей жидкостью.
Для заполнения поглотителей щелочь или пирогаллол наливают в соот¬
ветствующую грушу (9 и 14) при положении II кранов 3 я 5. При загрязне¬
нии бюретку периодически промывают хромовой смесью, смесью спирта с
с эфиром (1 : 1) и водой. Особое внимание следует обращать на смазку кра¬
нов. Лучше применять вакуумную смазку (6 частей натурального каучука,
нарезанного мелкими кусочками, смешивают с 5-ю частями белого вазелина
и 1-й частью парафина). Смесь держат в течение 180—200 час. при температу¬
ре 150—160° и постоянном помешивании.
Аналогичное устройство и принципы работы имеют и другие газоанализа¬
торы (АФИ, ГХП-3, ВТИ).
ОПРЕДЕЛЕНИЕ ИНТЕНСИВНОСТИ ВЫДЕЛЕНИЯ С02 ИЗ ПОЧВЫ
(ДЫХАНИЯ ПОЧВЫ)
Все методы определения дыхания почвы можно делить на три группы: 1)
методы обогащения С02 в изолирующем устройстве (колоколе, домике и т. п.);
при этом определяют начальную и конечную концентрацию С02 в воздухе
изолятора, установленного на поверхности почвы (Lundegardh, 1927; Ма-
1 Приведения к нормальным условиям здесь не требуется, так как по условиям расчета
газов каждый объем отдельно должен быть приведен к нормальным условиям. В прибо¬
ре Гальдана все измерения производят при одних и тех же условиях. Поэтому коэф¬
фициент пересчета одинаков как в числителе, так и в знаменателе и при сокращении
дает единицу.
Методы, определения состава почвенного воздуха, интенсивности дыхания почвы 337
Рис. 5. Определение дыхания почвы (по методу Макарова)
1 — стеклянный шцик-изолятор; 2 — поглотитель Рихтера; з — аспиратор; 4 — мерная кружка
gers, 1929; Макаров, 1952, 1957); 2) методы проветривания. Ток воздуха
протягивается через изолятор (колокол, цилиндр и т. п.), поставленный на
поверхность почвы, и углекислый газ непрерывно поглощается (Humfeld,
1930; Koepf, 1953); 3) методы абсорбции. Под изолятор над почвой поме¬
щается сосуд со щелочью, которая непрерывно абсорбирует С02 (Borneman,
1920; Штатнов, 1952; НаЬег, 1958; Скрипкин, 1959).
Принцип метода Люндегорда (Lundegardh, 1927) состоит в том, что на по¬
верхность почвы ставят колокол из оцинкованного железа и через определен¬
ный промежуток времени из него отсасывают пробу воздуха. По изменению
концентрации С02 внутри колокола судят о количестве углекислого газа,
выделившегося с поверхности почвы.
В методе Б. Н. Макарова (1952) люндегордовский колокол заменен боль¬
шим изолятором — ящиком без дна (домик) объемом 80—82 д, что позволяет
избежать возможной ошибки из-за падения градиента концентрации С02
в изоляторе. Размер ящика 60 X 30 X 50 см. Нижняя рама стеклянного
ящика (ящик может быть сделан из оцинкованного железа или оргстекла)
обита тонкой железной полосой для врезания ящика в почву на глубину 2 —
3 см. Для отбора газовой пробы в ящик на каучуковой пробке вставляют
стеклянную трубку с разветвлениями (в жестяной ящик впаивается медная
трубка с тройниками), наружный конец которой закрывают пробкой, а при
отборе газовой пробы соединяют с поглотителем Рихтера. Все пазы и стекла
должны быть тщательно промазаны замазкой и покрашены.
Определение дыхания почвы производят следующим образом (рис. 5).
Ящик ставят на поверхность почвы, свободную от растительности. Если поч¬
ва покрыта растительностью, то надземные части растений перед самым опре¬
делением срезают. Ящик врезают в почву, которая вокруг него уплотняется,
чтобы предотвратить газообмен с наружным воздухом. Через 20—30 мин.
(в зависимости от интенсивности выделения С02) из ящика аспиратором
отсасывают пробу воздуха объемом 2 л со скоростью 0,5 л/мин. С02 погло¬
338
Б. Н. Макаров
щается в поглотителе Рихтера, для чего в колбочку наливают 25 мл 0,01 н.
Ва(ОН)2 и 2 капли изоамилового или изобутилового спирта для понижения
поверхностного натяжения жидкости. Титрование производят в той же кол¬
бочке 0,01 н. раствором НС1 по фенолфталеину. Перед определением дыхания
почвы в домике определяют начальную концентрацию С02. Практически она
всегда равна концентрации С02 в приземном (до 1 м) слое воздуха.
Поглотитель Рихтера представляет собой стеклянную трубку диаметром
30 мм и длиной 400 мм. На нижний конец трубки припаивают воронку Нутча
№ 1 с длинной стеклянной трубкой, верхняя часть поглотителя имеет рас¬
ширение диаметром 45—50 мм и горло для соединения с аспиратором.
Рис. 6. Опрзделение дыхания почвы по методу Штатнова
I — установка на почве, II — коятрэль. 1 — сэзуд-люлчтор; 2 — ч1лзчка]со щелочью, 3 — поддон¬
ник; 4 — подставка
Интенсивность выделения С02 из почвы рассчитывается по формуле
(а —Ь)*0,22* Ki*10 030*63
D~ K2-S-M000 1000
где D — количество С02, выделившееся из почвы, кг/га в час; а — количество 0,01 и»
НС1, пошедшее на титрование барита после поглощения С02 в начале определения, мл;
b — количество 0,01 н. НС1, пошедшее на титрование барита после поглощения С02 в
конце определения, мл; 0,22 — количество С02, эквивалентное 1 мл 0,01 н. HG1, мг;
Vi — объем воздуха в ящике, л; V2 — объем воздуха, взятого для определения С02, л;
10000 — коэффициент для пересчета на 1 га; S — площадь почвы под ящиком, м*, 60
— коэффициент пересчета на 1 час; / — экспозиция, мин.; 1000-1000 — коэффициент
пересчета в кг.
Для дальнейшего упрощения этого метода было предложено определять
(Макаров, 1957) дыхание почвы с помощью широкогорлых колб.
Методы абсорбции определения дыхания почвы, как наиболее простые
и не требующие громоздкого оборудования, применяются давно и в настоя¬
щее время широко распространены. Принцип метода заключается в том, что
поверхности почвы изолируют от окружающего воздуха сосудом, под кото¬
рый помещают чашечку со щелочью для поглощения С02. Через определен¬
ный промежуток времени сосуд-изолятор снимают, щелочь оттитровывают
кислотой. Одновременно производят контрольное определение, для чего изо¬
лятор и щелочь ставят не на почву, а в какой-либо плоскодонный сосуд и изо¬
лируют от внешнего воздуха (водой, слабым раствором H2S04, пластилином).
По разности между титрованием контроля и почвы определяют количест¬
во выделившегося углекислого газа из почвы. В связи с тем, что в Советском
Союзе широко применяют этот метод, известный под названием «метод Штат¬
нова», опишем его не в первоначальном виде (Штатнов, 1952), а с теми допол¬
нениями, которые были сделаны позднее (Макаров, 1970). На поверхность
почвы на подставке (треножнике) ставят чашку Петри или Коха с 10 мл 0,2 н.
раствора щелочи. Чашку накрывают сосудом-изолятором, края которого
заглубляют в почву на 1,5—2 см (рис. 6). Для предохранения от нагревания
Методы, определения состава почвенного воздуха, интенсивности дыхания почвы 339
стенки сосуда снаружи обертывают белой бумагой или закрашивают белой
краской. После экспозиции (экспозиция устанавливается в зависимости от
интенсивности выделения С02 из почвы; для дерново-подзолистых почв она
должна быть не менее 1 часа) сосуд-изолятор снимают и избыток щелочи
оттитровывают 0,05 н. кислотой по фенолфталеину. Титрование производят
непосредственно в поле в чашках Петри. Одновременно определяют исходное
содержание С02 в воздухе сосуда-изолятора, для чего чашку со щелочью ста¬
вят под сосуд-изолятор на поддоннике.
Интенсивность дыхания почвы рассчитывают по формуле
(а—Ь)* 1,1-10 ООО
D — S-l-1000-1000 *
где D — количество С02, выделившееся из почвы, кг/га в час; а — количество 0,05 н.
кислоты, пошедшее на титрование щелочи при определении содержания СО2 в воздухе
сосуда-изолятора, мл; b — количество 0,05 н. кислоты, пошедшее на титрование щелочи
в опыте, мл; 1,1 — количество С02, эквивалентное 1 мл 0,05 н. раствора кислоты, мг;
S — площадь почвы под сосудом-изолятором, м2; t — экспозиция, час.
Большое значение при определении дыхания почвы методом абсорбции
имеет соотношение между диаметрами сосуда-изолятора и поглотителя. Так,
при увеличении вдвое диаметра сосуда-поглотителя показания интенсивности
дыхания почвы повышаются на 40—60%.
Определения дыхания почвы одновременно методом абсорбции по
В. И. Штатнову (1952) и методом обогащения по Б. Н. Макарову (1957) пока¬
зали, что во всех случаях интенсивность выделения С02 из почвы по методу
Штатнова в 1,5—2 раза, а в периоды максимального выделения С02 в 3—
3,5 раза ниже, чем при определении по методу обогащения. Причины такого
большого расхождения в показаниях двух методов объясняются неполным
поглощением С02 щелочью и нарушением естественных условий.
При изучении дыхания почвы следует иметь в виду, что интенсивность вы¬
деления С02 из почвы меняется как в течение года, так и в течение суток.
Отсюда следует, что единичные измерения дыхания почвы могут быть мало¬
показательны и случайны. В связи с этим определять дыхание почвы нужно
периодически в течение всего вегетационного периода в различные часы су¬
ток. При сравнении нескольких опытных участков определения должны про¬
водиться одновременно на всех участках и в одни и те же часы (лучше с 9
до 13 час. дня) в часы максимального выделения С02. Чтобы иметь надежные
средние значения, определения необходимо проводить с большой повтор¬
ностью.
ОПРЕДЕЛЕНИЕ ГАЗООБРАЗНЫХ ПОТЕРЬ АЗОТА ПОЧВЫ
И УДОБРЕНИИ
В процессах денитрификации, нитрификации и аммонификации, а также
при внесении в почву минеральных азотных и органических удобрений воз¬
можны газообразные потери азота из почвы и внесенных удобрений. В резуль¬
тате биологических процессов в почве при соответствующих гидротермичес¬
ких условиях из почвы могут выделяться свободный азот, закись и двуокись
азота и аммиак. Поэтому при изучении круговорота азота и его баланса
в земледелии большое значение имеет определение потерь азота в газообраз¬
ной форме.
В полевых исследованиях наиболее легко определить газообразные потери
азота в форме аммиака и двуокиси азота. Определение потерь в форме моле¬
кулярного азота и закиси азота требует применения сложной аппаратуры.
340
Б. Н, Макаров
Определение аммиака и двуокиси азота,
выделяю дихся из почвы (Макаров, 1966)
Интенсивность выделения аммиака и двуокиси азота из почвы можно
определять двумя методами: методом обогащения и методом абсорбции.
Метод обогащения. Улавливание аммиака или двуокиси азота
производят с помощью колб : широкогорлую (d = 5 см) плоскодонную кол¬
бу емкостью 1—1,2 л ставят на поверхность почвы, края колбы, обернутые
бумагой для предохранения от загрязнения, присыпают почвой. После экспо¬
зиции, которую устанавливают в зависимости от интенсивности выделения
NH3 или N02 из почвы, колбу приподнимают и ее горло быстро закрывают
пробкой. Через небольшое отверстие в пробке в колбу вносят 10 мл 0,01 н.
раствора H2SO4 для поглощения NH3 или поглотителя Зальцмана (для погло¬
щения N02). Колбу с раствором интенсивно взбалтывают в течение 5—10 мин.
Затем 10 мл раствора переносят из колбы в пробирку. При определении NH3
в пробирку вносят 0,5 мл реактива Несслера. Через 15 мин. визуально срав¬
нивают окраску испытуемого и образцового раствора (0,3142 г NH4G1 рас¬
творяют в мерной колбе на 100 мл, 20 мл исходного раствора вносят в литро¬
вую колбу, в 1 мл этого раствора содержится 0,02 мг NH3).
При определении двуокиси азота поглотитель Зальцмана розовеет. Полу¬
ченную окраску сравнивают с образцовым раствором (0,1500 г NaN02 рас¬
творяют в мерной колбе на 1 л, 10 мл исходного раствора доводят до 1 л; 1 мл
этого раствора содержит 0,001 мг N02).
Одновременно таким же образом определяют содержание NH3 или N02
в воздухе, для чего колбу несколько раз взбалтывают в вытянутой руке (что¬
бы атмосферный воздух вошел в колбу) и закрывают пробкой. По разнице
между содержанием NH3 или N02 в первом (почва) и втором (воздух)
случаях подсчитывают количество выделившихся из почвы NH3 или N02
по формуле
(а — &)*0,02 (или 0,001)
D= sTt ’
где D — количество NHS или N02, выделившееся из почвы, мг с 1 м2 в час; а — содер¬
жание NH3 или NOa в воздухе колбы после ее снятия с почвы, мл; b — содержание NHs
или N02 в воздухе колбы, мл; 0,02 — количество NH3, эквивалентное 1 мл образцового
раствора NH4C1, мг; 0,001 — количество N02, эквивалентное 1 мл образцового раствора
NaN02, мг; t — экспозиция, час; S — площадь горла колбы, м2;
При расчете количества N02, выделившейся из почвы, по данной формуле
полученную величину умножают на 2, так как при поглощении двух объемов
N02 связывается один объем N02 (2N02 + Н20 = HN03 + HN02).
Метод абсорбции. На треножник или узкие палочки (чтобы не
закрывать поверхность почвы) ставится чашка Петри или Коха с 10 мл 0,01 н.
H2S04 или поглотителя Зальцмана. Почва изолируется стеклянным сосудом
или сосудом из оцинкованного железа диаметром 13—15 см. Сосуды-изоля-
торы обертывают белой бумагой или окрашивают белой краской для предо¬
хранения от нагревания. Если почва занята растительностью, наземную
часть ее перед началом определения срезают. После экспозиции сосуд сни¬
мают, растворы из чашки Петри переносят в пробирку. При определении ам¬
миака в пробирку вносят 0,5 мл реактива Несслера. Полученную окраску
сравнивают с образцовым раствором. Одновременно таким же образом опре¬
деляют начальное содержание NH3 или N02 в воздухе сосуда-изолятора, для
чего чашки Петри и сосуд ставят не на поверхность почвы, а на поддонник.
Поддонник имеет желоб, в который наливают воду для изоляции воздуха
сосуда от атмосферного.
Методы определения состава почвенного воздуха, интенсивности дыхания почвы 341
Расчет количества выделившихся NH3 или N02 из почвы производится
по вышеприведенной формуле. Метод абсорбции дает заниженные результа¬
ты по сравнению с методом обогащения, но является более простым.
При определении газообразных потерь азота из почвы в полевых услови¬
ях необходимо проводить сопутствующие наблюдения за температурой
и влажностью почвы, содержанием аммиака и нитратов в почве. В связи
с тем, что интенсивность выделения NH3 или N02 из почвы изменяется в те¬
чение суток, определения на всех сравниваемых вариантах следует вести
одновременно всегда в одни и те же часы (с 9 до 13 час. в период максималь¬
ной интенсивности). Для расчета общего количества выделившихся NH3
и N02 за вегетационный период следует 3—4 раза в течение вегетационного
периода определять суточный ход интенсивности выделения NH3 и N02 из
почвы. Повторность определений на каждом варианте 3—4-кратная.
Приготовление реактиваЗальцмана:5г сульфаниловой кислоты
растворяют при нагревании в 1 л дистиллированной воды, содержащей 140 мл ледяной
уксусной кислоты. К раствору добавляют 20 мл 0,1%-ного раствора альфа-нафтил-эти-
лендиамина солянокислого. Раствор хранят в темной бутыли.
МЕТОД ГАЗОВОЙ ХРОМАТОГРАФИИ
ПРИ ИЗУЧБНИИ ГАЗОВОГО РЕЖИМА ПОЧВ
В последнее время для определения состава и концентрации газовых сме¬
сей широко применяется метод газо-адсорбционной хроматографии. Прин¬
цип метода состоит в том, что смесь газов, проходя через колонку с опреде-„
ленным адсорбентом, в результате адсорбции разделяется на составные ком¬
поненты. Эти компоненты газом-носителем подаются в детектор, который
последовательно регистрирует содержание каждого компонента.
Было установлено (Макаров, Патрикеева, 1972), что метод газо-адсорб¬
ционной хроматографии можно применять в полевых и лабораторных ис¬
следованиях. Он дает возможность одновременно определять в воздухе не¬
сколько компонентов (С02, 02, N2, СН4, N20 и др.) и улавливать изменения
в содержании С02 и 02 как по профилю почвы и срокам определений, так
и по вариантам.
В Советском Союзе выпускается несколько типов газовых хроматографов
(Фроловский, 1969), которые с теми или иными изменениями можно приме¬
нять для почвенных исследований.
Для определения состава почвенного воздуха на выбранных участках
на заданную глубину устанавливают латунные или медные трубки по обще¬
принятой методике (см. выше). Интенсивность выделения С02 из почвы опре¬
деляют методом обогащения. Для этого на поверхность почвы ставят колпак-
изолятор и из него через определенное время отбирают пробы воздуха для
определения концентрации С02. Пробы почвенного воздуха или воздуха из
колпаков-изоляторов отбирают в газовые пипетки Зегера, заполненные на¬
сыщенным раствором NaCl или СаС12. Пипетки доставляют в лабораторию,
где установлен хроматограф.
Для определения состава почвенного и надпочвенного воздуха применя¬
ют две хроматографические колонки, в которых воздух разделяется на со¬
ставные компоненты. В колонке 1 диаметром 3 мм и длиной 30 см из воздуха
выделяется С02 и N20. Колонка 1 наполняется активированным углем (мар¬
ка СКТ или АГ-3) или силикагелем, предварительно просушенным при 150° С
в течение 5 час. Разделение N2 и 02 проводят на колонке 2 диаметром 6 мм
и длиной 120 см, наполненной молекулярными ситами СаХ, прокаленными
в течение 5 час. при 375—400°. Обе колонки после наполнения адсорбентами
продуваются газом-носителем в течение 5 час. при 150° С.
342
Б. Н. Макаров
Чтобы при одновременном разделении N2, 02, С02 и N20 не менять ко-
лонки, в хроматографе установлен специальный четырехходовой кран, ко¬
торый позволяет переключать ток газа-носителя через колонку 1 или череа
1 и 2. Для предохранения попадания паров воды в разделительные колонки,
перед дозатором устанавливают стеклянную трубку, наполненную хлорис¬
тым кальцием или пемзой, смоченной концентрированной H2S04. Пробы воз¬
духа из газовых пипеток вводят в прибор через дозатор объемом 3—5 см*
или медицинским шприцем через мембрану. При этом необходимо точно учи¬
тывать объем вводимой пробы воздуха.
Разделение газов проводится при температуре 30° С, силе тока 130 ма
и скорости газа-носителя 50 мл/мин. Для определения концентрации всех
компонентов воздуха необходимо пропускать через прибор две пробы воздуха:
одну через колонку 1, вторую через колонки 1 и 2. При разделении газов на
колонке 1 через 30 сек. появляется общий пик 02 и N2, через 4 мин.— пик
С02, через 6 мин. 30 сек.— пик N20. При прохождении пробы воздуха одно¬
временно через колонки 1 и 2 пик кислорода выходит через 1 мин. 30 сек.*
пик азота — через 2 мин. 2 сек.
Для расчета концентрации газовых компонентов в пробах воздуха при¬
бор калибруют по известным концентрациям 02, N2, С02, N20 и других га¬
зов (в зависимости от того, какие газы требуется определять) и строят кали¬
бровочные графики. Проще расчет производить по сумме площадей пиков
всех исследуемых компонентов.
Ход определения на хроматографе. При отборе пробы воздуха пипет¬
ку Зегера соединяют с аспиратором, наполненным насыщенным раство¬
ром NaCl или СаС12, и с осушительной трубкой. Воздух из пипетки Зегера?
пропускают через осушительную трубку и дозатор, а затем в месте соедине¬
ния осушительной трубки с дозатором отбирают медицинским шприцем
пробу воздуха. Для определения С02 и N20 кран-переключатель колонок
ставят так, чтобы ток газа-носителя шел через колонку 1, а когда устано¬
вится нулевая линия самописца, через мембрану хроматографа вводится
проба воздуха в 10 мл. Самописец прибора регистрирует сложный пик О*
и N2 и пики С02 и N20. Чтобы каждый раз не переключать кран и не уста¬
навливать нулевую линию самописца, сначала пропускают все пробы для
определения С02 и N20, а затем пробы для определения N2 и 02 или нао¬
борот. Анализ проводят в один и тот же день. Для определения N2 и 02 проба
воздуха составляет 1 мл. Разделение С02 и N20 проводят при первой чув¬
ствительности, 02 и N2 — при восьмой и шестнадцатой.
Подсчет результатов ведут следующим образом: подсчитывают плохцадиг
пиков (S) каждого газа. Для этого измеряют высоту пика (К) и его ширину
(d) на середине высоты. Площадь пика умножают на чувствительность ре¬
гистратора (г), при которой производили разделение газа, и поправочный
коэффициент (К) и делят на взятый объем пробы (У).
hdrK
S=—-
Коэффициенты имеют следующие значения (газ-носитель — гелий): для*
N2, С02 и N20 — 2,02, для 02 — 2,13.
Содержание компонента в объемных процентах рассчитывают по формуле
£i-100
^ 1 -j— iS* 2 —J— з - . .
где Sv S2, S9 — площади пиков каждого компонента. Интенсивность выде^
ления С02 рассчитывают по формуле
(fl_b). V-1,97-МО 000 (а-Ь)*Г.0,197
D— lOO-S-t-lOOO - S-t
Методы, определения состава почвенчого воздуха, интенсивности дыхания почвы 343
еде D — интенсивность выделения С02 из почвы, кг/га в час; а — содержание СОа в воз¬
духе изолятора после экспозиции, об. %; Ь — исходное содержание С02 в вэздухе изо
лятора, об. %; V — объем изолятора, л; S — площадь изолятора, м2; t — экспозиция,
час.; 10000 — коэффициент пересчета на 1 га; 1000 — коэффициент пересчета на 1 кг;
1,97 — вес 1 л С02.
При лабораторных исследованиях часто необходимо бывает определять
интенсивность продуцирования С02 почвой. Для этих целей также можно
лрименять метод газо-адсорбционной хроматографии. Определение проводят
•следующим образом.
В широкогорлую колбу емкостью 1,2—1,5 л подвешивают капроновый
мешочек с почвой. Колбу плотно закрывают каучуковой пробкой, в которую
вставлены две стеклянные трубки. Одна из них доходит до дна колбы,
а вторая короткая, наружные концы трубок герметически закрыты. Одно¬
временно ставят колбу без почвы и так жэ плотно ее закрывают (контроль на
исходное содержание С02 в воздухе колбы). Через определенное время
(24—48 час.) из колбы отсасывается проба воздуха шприцем или в газовую
пипетку, для чего пипетку, наполненную насыщенным раствором Nad, при¬
соединяют к короткой трубке, вставленной в колбу. Длинную трубку соеди¬
няют с аспиратором. При отборе пробы открывают краны газовой пипетки,
жидкость из пипетки вытекает и в нее поступает воздух из колбы, а в колбу
лостунает раствор из аспиратора. Отбор пробы воздуха можно производить
непосредственно шприцем. Дальнейшее определение содержания С02 на
хроматографе проводят, как описано выше.
Интенсивность продуцирования С02 рассчитывается по формуле
^ (а — 6) • К. 1,97 • 100 • 1000
D- 100. n-t
где D — интенсивность продуцирования СОа, мг на 100 г почвы в час; а и b — конечное
и исходное содержание С02 в воздухе колбы, об. %; V — объем колбы, л; п — навеска
почвы, г; t — экспозиция, час.
При микрокэнцентрациях газов в воздухе (например, закиси азота)
производится предварительное концентрирование газа в колонке с адсор¬
бентом с последующей десорбцией его в хроматографе.
Н. И. Борисова и В. Н. Родионов (1970) предложили следующую мето¬
дику определения микроконцентраций N20 в почвенном воздухе (меньше
10~3 объемного процента). 1—5 л воздуха, предварительно очищенного ас-
каритом от С02, пропускается со скоростью 30—50 мл/мин через охлажден¬
ную до —73° С смесью сухого льда с этанолом U-образную трубку диамет¬
ром 5 мм и длиной 0,5 м, наполненную силикагелем (КСМ № 5). Концент¬
рирование газа может производиться непосредственно в поле. Затем обога¬
тительную колонку присоединяют к хроматографу, нагревают в электро-
воздушной бане до температуры +60° С и через нее пропускают в течение
1,5 мин. гелий, который переносит десорбированные газы в хроматографи¬
ческую колонку.
Литература
Борисова Н. ИРодионов В. Н. Оарэде-
ление микроконцентраций закиси азота
в почвенном воздухе методом газо-адсорб-
ционной хроматографии.— Почвоведе¬
ние, 1970, № 7.
Макаров Б. Я. Динамика газообмена меж¬
ду почвой и атмосферой в течение вегета¬
ционного периода под различными куль¬
турами.— Почвоведение, 1952, № 3.
Макаров Б. Н. Упрощенный метод опреде¬
ления дыхания почвы (биологической ак¬
тивности).— Почвоведение, 1957, № 9.
Макаров Б. Н. Газообразные потери азо¬
та.— В сб. «Баланс азота в дерново-подзо¬
листых почвах». М., «Наука», 1966.
Макаров Б. Н. К методике определения ин¬
тенсивности выделения С02 из почвы (ды¬
344
2>. Н, Макаров
хания почвы).— Почвоведение, 1970,
№ 5.
Макаров Б. //., Мацкевич В. Б, Методы
определения состава почвенного воздуха
и интенсивности газообмена между поч¬
вой и атмосферой.— Б сб. «Физико-хими¬
ческие методы исследования почв. Аб¬
сорбционные и изотопные методы». М.,
«Наука», 1966.
Макаров Б. Н., Патрикеева Т. А. Изуче¬
ние газового режима почв методом га¬
зовой хроматографии.— Почвоведение,
1972, № 9.
Скрипкин А. А. Прибор для определения
диффундирующей из почвы углекисло¬
ты.— Почвоведение, 1959, № 4.
Штатное В. И. К методике определения
биологической активности почвы.—Д окл.
ВАСХНИЛ, 1952, вып. 6.
Фроловский П. А. Хроматография газов.
М., «Наука», 1969.
Вогпетап Я. Kohlensaure und Pflanzenwa-
chstum, Berlin, 1920.
Haber W. Okologische Untersuchung der
Bodenatmung.— Flora oder allgemeine
botanische Zeitung, 1958, Bd. 146, H. 2.
Humfeld H. A method for measuring carbon-
dioxide evolution from soil.— Soil Sci.,
1930, v. 30, N 1.
Koepf H. Die Verwendung des Ultrarotab-
sorptionsschreibers (URAS) fur die konti-
nuierliche Registrierung der Bodenatmung
im Freiland.— Landwirtsch. Forschung,
1953, Bd 5, N 1.
Lundegardh H. Carbondioxide evolution of
soil and crop growth.— Soil Sci., 1927,
v. 23, N 6.
Magers H. Untersuchungen iiber die Produk-
tion der Kohlensaure im Ackerboden und
ihre Diffusion in die Atmosphare.— Pflan-
zenbau, 1929, Bd. 23, 447.
Д. Н. ИВАНОВ, Л. А. ЛЕРНЕР
СПЕКТРАЛЬНЫЕ МЕТОДЫ АНАЛИЗА
*
Методы атомного спектрального анализа основаны на использовании
спектров испускания (эмиссии) и спектров поглощения (абсорбции) атомов
элементов, входящих в состав пробы.
В основе количественного определения элементов эмиссионными мето¬
дами лежит измерение интенсивности излучения линий спектра определяе¬
мого элемента, а атомно-абсорбционными методами — величины поглоще¬
ния линий спектра внешнего источника света атомами определяемого эле¬
мента, находящимися в парах пробы поглощающей ячейки.
При выполнении спектрального анализа приходится иметь дело с нала¬
гающимися в различной степени друг на друга тремя спектрами: линейча¬
тыми, полосатыми и сплошными. Линейчатые спектры излучаются свобод¬
ными атомами элементов, полосатые — молекулами и сплошные — в основ¬
ном твердыми и жидкими частицами вещества. Чаще всего при спектраль¬
ном анализе применяют методы, основанные на использовании линейчатых
спектров.
Основными источниками света для спектрального анализа служат: -
электрические дуги переменного и постоянного тока, искровой разряд,
различного рода пламена и специальные лампы.
Спектр пробы содержит линии излучения как нейтральных атомов, так
и ионов. Принадлежность линии к спектру атома или иона обозначается в
таблицах спектральных линий числовым индексом, стоящим рядом с хими¬
ческим символом элемента. Например, Fel означает, что указанную в табли¬
цах линию излучает нейтральный атом железа, а символы Fell и Felll и т. д.
показывают, что данные линии излучают однократно, двукратно и т. д.
ионизированные атомы железа.
Спектральные методы, как и другие, характеризуются в первую очередь
чувствительностью, сходимостью, воспроизводимостью, точностью и произ¬
водительностью, определения которых приводятся ниже *.
Различают абсолютную и относительную чувствительность метода. Под
абсолютной чувствительностью понимается минимальное количество данного
элемента, выраженное в весовых единицах (мг, мкг), а под относительной
чувствительностью — минимальная концентрация данного элемента в пробе,
выраженная в процентах, которая может быть обнаружена применяемым
аналитическим методом. В обоих случаях за минимальную концентрацию
часто принимают такую, которая дает полезный сигнал, в 3 раза превышаю¬
щий «шумы» установки (J = За). Последнее определение придает понятию
«чувствительность метода» статистический характер и вполне конкретное
содержание.
Сходимость измерений — качество измерений, отражающее близость
друг к другу результатов измерений, выполняемых в одинаковых условиях.
Сходимость спектральных методов, как правило, высокая, особенно пла-
менно-фотометрических и атомно-абсорбционных. Сходимость метода нель¬
зя принимать за его воспроизводимость и точность.
1 Известны и другие наименования основных характеристик метода. Не вдаваясь в оцен¬
ку имеющихся расхождений, авторы придерживаются терминологии, рекомендованной
ГОСТом 16263-70.
346
Д. Н. Иванов, Л. А. Лернер
Воспроизводимость измерений — качество измерений, отражающее бли¬
зость результатов измерений, выполняемых в различных*, условиях (в раз¬
личное время, в различных местах, различными методами и средствами).
Воспроизводимость спектральных методов ниже, чем их сходимость. Ее так
же нельзя принимать за точность метода.
Точность измерений — качество измерений, отражающее близость их
результатов к истинному значению^измеряемой величины.
Правильность измерений — качество измерений, отражающее близость
к нулю систематических погрешностей в их результатах. Достижение высо¬
кой правильности спектральных методов является наиболее трудно решае¬
мой задачей.
Точность спектральных методов удовлетворяет в большинстве случаев
требованиям почвенно-агрохимического анализа, предъявляемым к определе¬
нию отдельных элементов, при выполнении исследовательских и производ¬
ственных работ.
Под производительностью спектральных методов понимается объем по¬
лезной информации о составе вещества, полученной за определенный про¬
межуток времени и удовлетворяющей целям поставленных исследований.
Опыт показал, что, как правило, спектральные методы анализа значительно
производительнее химических.
Производительность метода в значительной мере определяется требо¬
ваниями, предъявляемыми к его точности: чем выше требование к точности
анализа, тем больше времени затрачивается на его выполнение. Поэтому
правильный выбор метода анализа и рациональная схема его проведения
возможны лишь при разумных требованиях к точности результатов.
АНАЛИТИЧЕСКИЕ ПОМЕХИ
Одним из факторов, существенно влияющих на результаты спектраль¬
ного анализа, является процесс образования новых соединений определя¬
емых элементов в применяемом источнике света (поглощающей ячейке).
Если образуются термически устойчивые соединения в излучающем об¬
лаке паров, уменьшается число свободных атомов определяемого элемента
и тем самым ухудшается чувствительность анализа. Примером может служить
уменьшение интенсивности излучения (или величины поглощения) спектра
атомов щелочноземельных элементов в присутствии атомов алюминия и не¬
которых других элементов в излучающем (поглощающем) облаке паров.
Другим фактором, имеющим важное значение в спектрально-аналитичес-
кой практике, является фракционное испарение пробы, обусловленное раз¬
личием в температуре кипения разных соединений элементов. Поэтому, на¬
пример, при дуговом источнике света элементы, входящие в соединения,
имеющие более низкую температуру кипения, поступают быстрее в дуговой
промежуток, чем элементы из соединений с более высокой температурой
кипения. На первых стадиях горения дуги в разрядный промежуток посту¬
пают наиболее летучие соединения, которые на этой стадии определяют ре¬
жим горения дуги и условия возбуждения спектра определяемых элементов.
Явление фракционного испарения используют для повышения чувствитель¬
ности определения, регистрируя спектр на разных стадиях горения дуги и
выбирая для анализа пробы те спектрограммы, на которых линии тех или
иных определяемых элементов наиболее интенсивны.
В пламенных методах анализа применение органических растворителей
вместо воды приводит к значительному увеличению чувствительности.
Следует иметь в виду, что интенсивность спектральных линий ряда эле¬
ментов зависит от характера аниона, связанного с ними в пробе или в излу¬
чающем облаке. Например, у щелочноземельных элементов интенсивность
Спектральные методы
347
спектра кальция уменьшается в зависимости от аниона в последовательности
-Jcl > /nos > /so.. Поэтому, в частности, необходимо обеспечить соответ-
ствие между анионами солей анализируемого и эталонных растворов.
Присутствие H2S04 и Н3Р04 в растворах приводит также к уменьшению
интенсивности излучения спектра щелочноземельных элементов. Наиболь¬
ший эффект наблюдается при соотношении молярных концентраций
Са (Sr, Ва) : S = 1 : 1 и Са (Sr, Ва) : Р = 3 : 2. В данном рассматриваемом
случае уменьшение интенсивности вызывается образованием новых соеди¬
нений, изменение физического состояния и условий испарения растворов
играют подчиненную роль.
Для щелочных элементов наблюдается независимость интенсивности их
излучения от природы связанных с ними анионов (Cl, N03, S04), что объяс¬
няется полной диссоциацией в воздушно-ацетиленовом пламени соответст¬
вующих солей и вторичных соединений указанных элементов, которые мо¬
гут образоваться в пламени.
Значительную погрешность в результаты определения концентрации эле¬
ментов может внести собственное излучение источника света при эмиссион¬
ном методе анализа и собственное излучение поглощающей ячейки при атом-
~яо-абсорбционном методе. Излучение источника света, которое регистри¬
руют в процессе анализа, состоит из собственного излучения источника и
лзлучения, зависящего от состава пробы. Собственное излучение источника
при атомно-абсорбционном методе действует в сторону занижения результа¬
тов анализа, а при эмиссионном пламенно-фотометрическом методе — в сто¬
рону завышения данных анализа. Чтобы устранить влияние собственного
излучения поглощающей ячейки (пламени), проходящее через нее излучение
лампы модулируют. Сигнал от лампы прерывают с определенной частотой,
лользуясь, например, вращающимся диском с отверстиями, расположенными
между лампой и поглощающей ячейкой, или изменяя силу тока, питающего
лампу. Прерывистый фототок, вызванный спектральной линией определяе¬
мого элемента, частично поглощенной атомами этого элемента, находящи¬
мися в пламени, отделяют радиотехническими приемами от постоянного
^ототока, созданного собственным излучением поглощающей ячейки. Вели¬
чина измеренного прерывистого фототока в этом случае не зависит от интен¬
сивности фона пламени и может служить для более точного определения кон¬
центрации введенного в него элемента.
При атомно-абсорбционном анализе установлено, что присутствие в ана¬
лизируемом растворе посторонних элементов в больших концентрациях
вызывает поглощение резонансных линий тех элементов, которых нет в раст¬
воре. Это явление получило название неселективного поглощения. Были
лредложены методы учета величины неселективного поглощения, которые
позволили надежно вносить соответствующие поправки при выполнении ана¬
лиза атомно-абсорбционным методом.
Выполняя анализы пламенным методом, следует учитывать, что интен¬
сивность излучения спектра атомами элемента зависит от степени его иони¬
зации, а также от ионизации всех остальных элементов, входящих в пробу.
Ионизация приводит к уменьшению числа нейтральных атомов и к уменьше¬
нию интенсивности их излучения. Степень ионизации атомов возрас¬
тает с уменьшением величины ионизационного потенциала элемента, с умень¬
шением концентрации элемента и повышением температуры источника.
В низкотемпературных пламенах эффект ионизации проявляется только
у элементов с невысоким потенциалом ионизации (калий, рубидий, цезий).
Влияние ионизации можно устранить путем введения в пламя одновременно
•с определяемым элементом другого легко ионизируемого элемента. В этом
случае равновесие реакции ионизации сдвигается в сторону увеличения чис¬
ла нейтральных атомов определяемого элемента (Иванов, 1941; Полуэк-
тов, 1967).
348
Д. II. Иванову JI. А. Лернер
Влияние ионизационного эффекта следует особенно тщательно учитывать
при определении элементов с низким потенциалом ионизации (щелочные и
щелочноземельные) и при использовании высокотемпературных пламен (за¬
кись азота — ацетилен). Концентрация ионов указанных элементов в этом
пламени может быть очень высокой и достигать десятков процентов. Прие¬
мы устранения влияния ионизационного эффекта сводятся к введению в про¬
бы и эталоны добавок легко ионизируемых элементов.
Значительное влияние на интенсивность спектральных линий оказывает
также самопоглощение. Проходя через плазму источника, излучение опре¬
деляемого элемента частично поглощается атомами того же элемента и не
достигает спектрального прибора. Самопоглощение приводит к уменьшению
интенсивности центра спектральной линии. Наибольшим самопоглощением
обладают резонансные линии, поэтому их использование ограничено опре¬
делением малых концентраций.
ЭТАЛОНИРОВАНИЕ
Количественный спектральный анализ является относительным методом*
Определение неизвестной концентрации какого-либо элемента производится
путем сравнения интенсивности его аналитических линий в спектре пробы
с интенсивностью этих же линий в спектрах эталонных образцов, в которых
известна концентрация определяемых элементов. Однако зависимость ин¬
тенсивности спектра элемента не только от его концентрации, но и от общего
химического состава и структуры пробы создает существенные трудности в
осуществлении прецизионного спектрального анализа — требует соответ¬
ствия состава и структуры анализируемых проб и эталонов.
При анализе почв, растений и других биологических объектов обычна
нелегко подобрать или создать эталоны, в достаточной мере удовлетворяю¬
щие этому требованию. В практике спектрографического анализа почв до¬
пускают значительное несоответствие состава и структуры в эталонах и про¬
бах почв: в качестве эталонов применяют искусственные смеси, состав кото¬
рых соответствует некоторому «среднему» составу почв (Зырин и др., 1971;
Иванов, 1974).
Допустимость применения усредненных эталонов определяется задача¬
ми исследования и требованиями к погрешности анализа. Например, при
выяснении общих закономерностей распространения микроэлементов в поч¬
вах применение таких эталонов вполне оправдано. В ряде других исследо¬
ваний применение усредненных эталонов не обеспечивает необходимой
правильности результатов анализа.
Проблема эталонирования не менее сложна при анализе растений, состав
которых очень изменчив и оказывает сильное влияние на результаты ана¬
лиза. Состав эталонов для анализа растений, применяемых различными ав¬
торами, будет приведен при рассмотрении конкретных методик анализа.
Исследование зависимости результата анализа от состава пробы принад¬
лежит к наиболее трудным задачам эмиссионного спектрального анализа.
Один из способов уменьшение влияния состава пробы на результаты анали¬
за состоит в применении специальных добавок к пробам и эталонам (буфер¬
ных смесей). Состав добавок зависит от характера анализируемой пробы и
определяемых элементов.
Приведенными примерами далеко не исчерпываются возможности при¬
готовления эталонов при решении конкретных аналитических задач.
Спектральные методы
349
ПОТРЕБНОСТИ АГРОХИМИИ
В ОПРЕДЕЛЕНИИ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
В почвенно-агрохимических исследованиях имеют дело с рядом качест¬
венно разнородных объектов: почвами, породами, растениями, органическим
веществом, почвенными растворами и вытяжками, естественными водами,
анализу которых предъявляются специфические требования. Почвенно¬
агрохимический анализ, имеющий целью определение элементного состава
вещества, можно представить отдельными видами анализа: валовой анализ
почв, зольный анализ растений, определение обменных оснований, анализ
водной вытяжки, определение основных элементов питания растений —
азота, фосфора и калия.
Определение микроэлементов имеет существенные особенности по сравне¬
нию с определением макроэлементов. Содержание микроэлементов в поч-
венно-агрохимических объектах в ряде случаев находится на границе чув¬
ствительности спектрального метода или даже ниже ее; определение микро¬
элементов происходит в присутствии макроэлементов, концентрации кото¬
рых в 1000—10 ООО раз превосходят концентрации определяемых элементов.
Это обстоятельство может оказать влияние на результаты анализа, особен¬
но при определении концентраций, близких к пороговым.
Первое обстоятельство требует предварительного концентрирования оп¬
ределяемых элементов или разработки методов резкого повышения чувстви¬
тельности определения, а второе — разработки приемов устранения влия¬
ния на результаты анализа элементов, присутствующих в пробе в больших
концентрациях. Потребности почвенно-агрохимических исследований в
определении макро- и микроэлементов в агрохимических объектах и воз¬
можности их определения атомным эмиссионным и абсорбционным методами
спектрального анализа представлены в табл. 1 и 2.
В настоящее время эмиссионные и атомно-абсорбционные методы не
обеспечивают возможности определения ряда элементов, представляющих
значительный интерес для агрохимии. Содержание некоторых микроэле¬
ментов может быть определено спектральными методами только после их
предварительного концентрирования.
Таблица 1
Возможности определения макроэлементов спектральными методами при некоторых видах
почвенно-агрохимического анализа
Метод, вид анализа
Si
А1
Fe
р
Ti
Са
Mg
к
Na
Мп
1
S
1
С1
N
с
н
О
Спектрографический (кванто¬
_L
+
+
+
+
+
+
+
+
метрический)
Пламенно-фотометрический
—
—
+
—
—
“Г
+
+
+
+
—
Атомно-абсорбционный
+
+
+
*
+
+
+
+
+
+
*
Валовой анализ почвы
X
X
X
X
X
X
X
X
X
X
X
Определение поглощенных ос¬
нований
X
X
X
X
Водная вытяжка
X
X
X
X
Определение элементов пита¬
X
X
ния растений
(NPK) в почве
X
X
X
Зольный анализ растений
X
X
X
X
X
X
X
X
X
X
X
X
Обозначения: Н— определение возможно; определение невозможно; х — определяют при данном
виде анализа;* — определение возможно при использовании косвенных методов.
350
Д. Н. Иванов, ^4. Лернер
Таблица 2
Возможности определения микроэлементов в почве спектральными методами
Метод, вид анализа
Мп
Zn
Си
Со
Мо
в
I
Ni
Сг
V
Se
РЬ
Спектрографический (квантомет¬
рический):
Определение общего содержа¬
+
X
+
X
—
X
—
+
+
+
ния *
Определение в почвенной вы¬
+
+
—
тяжке **
Пламенно-фотометрический:
Определение общего содержа¬
+
—
—
—
—
—
—
—
+
—
—
—
ния **
Определение в почвенной вы¬
+
тяжке ***
Атомно-абсорбционный:
Определение общего содержа¬
+
+
+
X
—
—
—
+
+
+
+
X
ния **
Определение в почвенной вы¬
+
+
+
—
—
—
—
+
+
—
—
—
тяжке ***
* Определение непосредственно в почве. ** 1 г почвы разложен, остаток растворен в 25 мл.*** Экстра¬
гент условный, соотношение 1:10; предположено, что в вытяжку перешло 5% от обшего содержания
элемента в почве. Обозначения* Н— определение возможно; определение невозможно; х — оп¬
ределение возможно в некоторых объектах.
Спектральные методы позволяют полностью выполнять определенные ви¬
ды анализа (обменные основания) или подавляющие его части (зольный ана¬
лиз). При определении микроэлементов в почвах и растениях спектральные
методы могут занимать ведущее положение. Вопросы разработки системы
анализа, включающей в себя качественно различные методы, приобретают
насущное значение.
' СПЕКТРОГРАФИЧЕСКИЙ МЕТОД
Аппаратура
Основным (наиболее массовым) спектральным прибором, применяющим¬
ся в аналитической практике, является спектрограф — прибор, позволяю¬
щий фотографировать спектры.
Спектральные приборы с кварцевой призмой позволяют регистрировать
область 2000—7000 А, со стеклянной призмой — область 4000—10000 А,
а с дифракционной решеткой можно регистрировать обе указанные области.
Основными характеристиками спектральных приборов являются дис¬
персия и светосила.
Дисперсия спектрального прибора характеризует его способность про¬
странственно разделять пучки лучей близких длин волн. Это качество спект¬
рального прибора оценивают обратной величиной отношения (К2 —
:(l2 — ZJ, где и Х2 — близкие длины волн в спектре, 1Х и 12 — границы ли¬
нейной протяженности спектра между линиями указанных длин волн. Об¬
ратная линейная дисперсия выражается числом ангстремов, приходящихся
на 1 мм протяженности спектра.
Под светосилой прибора понимают величину, пропорциональную отно¬
шению (d/F) 2, где d — диаметр камерного объектива и F — его фокус¬
Спектральные методы.
351
ное расстояние. Светосила прибора определяет яркость спектра и экспо¬
зицию.
Наиболее широкое распространение получили спектрографы средней дис¬
персии ИСП-28 и ИСП-30, их используют при работе в ультрафиолетовой
и коротковолновой видимой областях спектра.
Спектрографы ДФС-8 и ДФС-13 относятся к дифракционным спектраль¬
ным приборам, оии имеют дифракционные решетки с 600 и 1200 штрих/мм.
Приборы обладают большой дисперсией, практически постоянной по всему
спектру, и позволяют работать в интервале 2000—10000 А. Фотографирова¬
ние спектра производится по частям.
Спектрографы с большой дисперсией ооеспечивают значительное повы¬
шение чувствительности определения элементов, аналитические линии ко¬
торых расположены в области интенсивного фона. Это объясняется преиму¬
щественным ослаблением фона и лучшим отделением аналитических линий
определяемого элемента от других близких линий.
Спектральный прибор ДФС-10 является многоканальным прибором с
большой дисперсией и фотоэлектрической регистрацией спектра. Входная
щель и штрихи решетки (1200 штрих/мм) расположены горизонтально, а
выходные щели расположены в вертикальной плоскости. Прибор имеет
36 выходных щелей, после прохождения которых выделенные монохрома¬
тические пучки света направляются на фотоэлементы. Прибор позволяет со¬
ставить ряд аналитических программ. Каждый измерительный канал имеет
накопительный конденсатор, и оценка интенсивности спектральной линии
производится по величине накопленного на нем заряда. Измерение зарядов
автоматизировано. Прибор чувствителен к температурным колебаниям,
приводящим к смещению световых пучков с выходных щелей. Это обстоя¬
тельство вынуждает обеспечивать стабилизацию температуры в помещении
спектральной квантометрической лаборатории. Опыт применения кванто-
метров к определению химического состава почв еще очень мал.
Правильное освещение щели спектрографа является важной частью
спектрально-аналитической техники. Наибольшее распространение имеет
трехлинэовая конденсорная система, обеспечивающая равномерное освеще¬
ние щели светом, идущим от любого участка источника, и равномерное рас¬
пределение интенсивности вдоль спектральной линии.
В качестве источника света для спектрального анализа интересующих
нас объектов следует в первую очередь указать дуговой разряд. Благодаря
высокой температуре в дуге испаряются тугоплавкие непроводящие ток
вещества, такими являются почвы, породы и другие биологические объекты.
Широкое распространение имеют активизированные дуги переменного тока,
поджигаемые маломощными искровыми разрядами. В другом источнике
света — конденсированной искре — возбуждается больше линий, чем в ду¬
ге, главным образом за счет спектров ионизованных атомов. Поэтому искра
создает большие возможности для выбора аналитических линий. Однако
применение искрового разряда к анализу непроводящих ток материалов, в
том числе почв и растений, имеет существенные ограничения. Искру при¬
меняют при анализе почв и растений в тех случаях, когда пробы целесообраз¬
но предварительно переводить в раствор.
Раствор анализируют, вводя в разряд посредством пористого электро¬
да или вращающегося диска.
Среди приемов введения пробы в источник возбуждения спектра наиболь¬
шее распространение имеют следующие:
набивка порошка пробы в кратер электрода или в специально приготов¬
ленную камеру;
сжигание в разряде предварительно приготовленных брикетов;
нанесение порошка пробы на торец нижнего стержневого электрода или
на поверхность вращающегося дискового электрода;
352
Д. Н. Иванов, Л. А, Лернер
просыпка порошка пробы через источник света и вдувание порошка про¬
бы или аэрозоля анализируемого раствора в источник.
Для решения большинства аналитических задач применяют угольные
электроды различной степени чистоты:
спектральные угли С-1, С-2, С-3, С-4; наиболее чистые и не содержащие
бора — С-1; следующими по чистоте являются угли С-2.
Форму фасонных электродов выбирают с учетом характера аналитической
задачи.
Методы фотометрирования и анализа
Напомним весьма кратко основы количественного спектрального анали¬
за и схемы его проведения фотографическим методом.
Эмиссионный количественный спектральный анализ основан на зависи¬
мости интенсивности линий спектра элемента от его концентрации в пробе.
Эта зависимость хорошо описывается выражением
1=аСь (1)
или (2)
lg/ = MgC + \$а,
где / — интенсивность аналитической линии; С — концентрация элемента в пробе;
а — параметр, характеризующий] поступление вещества в зону разряда; Ъ — величина,
характеризующая само поглощение спектральных линий, зависящее от концентрации
элемента.
Выражение (2) есть уравнение прямой линии в координатах (lg /, 1 g С)
указывающее на прямую пропорциональную зависимость логарифма ин¬
тенсивности линии логарифму концентрации элемента. Отклонения от ли¬
нейной зависимости наблюдаются при больших и малых концентрациях
определяемого элемента.
Так как интенсивность спектра элемента зависит от условий его возбуж¬
дения, то по абсолютной интенсивности линий спектра нельзя сделать точ¬
ные количественные заключения о содержании элемента в пробе. Учитывая
это обстоятельство, концентрацию элемента связывают с относительной ин¬
тенсивностью пары линий, одна из которых принадлежит спектру элемента
сравнения (обычно основа пробы или элемент, специально введенный в про¬
бу)» а другая — спектру определяемого элемента. Эти линии получили наз¬
вание аналитической пары линий. Их подбирают, руководствуясь рядом со¬
ображений.
Главные из этих соображений можно приближенно представить
в виде (Зайдель, 1965):
ДЕ<1 ое: Xi-X2< 100 A; J- <-^<10,
10 /2
где ДЕ — разность энергии возбуждения линий; — Х2 — разность их длин волн;
/х и /2 — интенсивности линий.
Спектральный анализ выполняют по предварительно установленной за¬
висимости между относительной интенсивностью аналитической пары линий
и концентрацией определяемого элемента в эталонных образцах. Наука
об измерении световых величин называется фотометрией.
В основе методов фотографической фотометрии лежит принцип, заклю¬
чающийся в том, что равные по величине спектральному составу потоки из¬
лучения, в течение одного и того же времени падающие на данную фотоэмуль¬
сию, вызывают равные почернения.
Спектральные методы
353
Зависимость почернения S на фотопластинке от интенсивности излучения
/, вызвавшего это почернение, может быть представлена выражением
где у — фактор контрастности фотопластинки; / — ее инерция.
Величину почернения фотопластинки S измеряют микрофотометром,
пропуская свет от его лампы через непочерненный участок фотопластинки и
через почерненный участок (спектральную линию). Измеренные при этом
значения интенсивности прошедшего света i0 и i связаны с почернением фор¬
мулой S = lg у*.
Почернение на фотопластинке зависит не только от интенсивности па¬
давшего на нее света, но и от свойств пластинок данного типа, условий по¬
следующей их обработки и длины волны света, освещавшего фотоэмульсию.
Поэтому только лишь по величине почернения, без учета указанных факто¬
ров, нельзя судить об интенсивности света, вызвавшего почернение.
Рассмотрим наиболее распространенный фотографический прием опре¬
деления относительной интенсивности пары спектральных линий.
На пластинку фотографируют спектр пробы, в которой необходимо ус¬
тановить относительную интенсивность аналитической пары линий. На ту
же пластинку при помощи ступенчатого ослабителя с известной пропускае-
мостью ступенек (в процентах), расположенного непосредственно перед щелью
спектрографа, наносят марки интенсивности, фотографируя, например,
спектр железа. Затем микрофотометром измеряют почернения S отдельных
ступенек выбранной линии спектра железа. По значениям почернения сту¬
пенек и значениям логарифма интенсивности света, прошедшего через дан¬
ную ступеньку ослабителя, строят характеристическую кривую (кривую
почернений) данной фотопластинки. Пользуясь этой кривой и измеренными
значениями почернений линий аналитической пары в спектре пробы, находят
значения lg 1г и lg /2. Отсюда логарифм относительной интенсивности пары
линий:
В аналитической практике пользуются и другим приемом, позволяющим
определить величину, пропорциональную относительной интенсивности ана¬
литической пары линий. Из выражений (3) и (4) для двух спектральных линий
следует:
где £оп — почернение линий определяемого элемента; Scp — почернение линий эле¬
мента сравнения.
Таким образом, разность почернений двух линий равна логарифму их
относительной интенсивности, умноженному на фактор контрастности фотопла¬
стинки (y)> который в пределах одной и той же пластинки можно считать
постоянной величиной, хотя он и зависит от других параметров, в том слу¬
чае, когда к точности анализа предъявляются не очень высокие требования.
Метод постоянного- графика. Выражения (2) и (4) поз¬
воляют получить следующую зависимость между относительной интенсивно¬
стью аналитической пары линий и концентрацией определяемого элемента:
(3)
(4)
(5)
U г— = MgC + lg/l,
ср
где А — некоторая постоянная величина.
(6)
12 Агрохимические методы исследования почв
354
Д. Н. Иванов, Л. Лернер
На практике эти зависимости для каждого определяемого элемента строят
графически по данным фотометрирования спектров эталонных образцов с
известным содержанием определяемых элементов. Для анализа пробы фото-
метрируют аналитические пары линий ее спектра, вычисляют относительные
интенсивности и по постоянным градуировочным графикам находят искомые
концентрации. Постоянные графики периодически контролируют, пользуясь
эталонами, служившими для их построения.
Метод трех эталонов. Из выражений (2 и 3) следует*
AS = Son _ £ср = С + lg Я, (7)
где В — некоторая величина, постоянная для данной фотопластинки.
Эта зависимость справедлива только в пределах одной фотопластинки,
так как в выражение (7) входит коэффициент контрастности у фотоплас¬
тинки.
Зависимость, представленная выражением (7), лежит в основе широко
применяемого в практике количественного спектрального анализа метода
трех эталонов. Спектры трех эталонов, охватывающих прямолинейную часть
графика, фотографируют на фотопластинку совместно со спектрами анали¬
зируемых проб. По графику (AS, lgC), построенному для эталонов и раз¬
ности почернений пары линий анализируемой пробы, определяют неизвестную
концентрацию.
Метод абсолютных интенсивностей (почернений).
Широкое применение в практике спектрального анализа получили методы,
основанные на измерении абсолютных интенсивностей линий (почернений)
определяемых элементов. Зависимость между почернением S линии на фото¬
пластинке и концентрацией С определяемого элемента следует из выражений
(2 и 3)
S = 'rb\gC + \gD, (8)
где D — некоторая величина, постоянная для данной фотопластинки.
Между выражениями 7 и 8 существует принципиальное различие.
В основе первого лежит принцип измерения по относительной интенсивности,
а в основе второго — принцип измерения по абсолютной интенсивности.
Метод абсолютных интенсивностей (почернений) целесообразно применять
в тех случаях, когда по характеру задачи исследования не нужна высокая
точность результатов анализа. Он может найти большое применение при ана¬
лизе почв для выяснения общих закономерностей распространения элементов.
Выполнение этих работ связано с обследованием больших территорий, на
которых наблюдается значительная естественная пестрота содержания изу¬
чаемых элементов.
Характеристика метода
Ориентировочные данные о чувствительности спектрографического ме¬
тода приведены в табл. 3.
Применение метода к анализу почв н растений
Методы эмиссионного спектрального анализа можно свести в несколько
групп, отличающихся общностью выполнения.
Общее содержание микроэлементов в почвах и растениях определяют
следующими способами.
1. Анализируют почвы без всякой предварительной химической подготов¬
ки и без введения элементов сравнения.
Спектральные методы
355
Таблица 3
Чувствительность определения элементов при испарении из канала угольного электрода
Элемент
Чувствительность определения, %
Аналитические линии, А
Спектрограф ИСП-28
Спектрограф ДФС-13
Алюминий
3082,05
3092,71
0,001
0,0001
Барий
3071,98
о
о
J,
о
©
о
0,001
4939,08
Бериллий
2348,61
0,0003
0,0001
3130,41
3131,06
Бор
2497 ,79
0,001
—
Ванадий
3183,40
3185.49
0,001
0,0001
Висмут
3067 ,71
0,001
0,00003
Вольфрам
2946,98
0,008-0,01
0,01
2895,45
3024,92
3215,56
Галлий
2943,63
0,0005
—
Гафний
2638,71
0,01
0,003
Германий
2651,17
0,001
0,0001
3039,06
Гольмий
3456,00
0,001
0,0001
3398,98
Гадолиний
3100,50
0,03
0,01
3032,85
Диспрозий
3407,79
0,03
0,01
3319,88
Европий
3115,17
0,3
0,03
Железо
3000,64
2979,35
0,0005
—
Золото
2427,95
0,001
0,001
2675,95
3122,78
Индий
3256,69
0,001
0,0001
3039,35
Иридий
3220,78
0,01-0,005
0,0003
Осмий
2909,06
3058,66
0,01
0,001
Палладий
3404,60
0,0005
0,0001
3242,70
Платина
2659,45
3064,71
0,001
0,0003
Рений
3460,47
0,005
• —
Родий
3434,89
0,0005
0,0001
3306,85
Ртуть
2536,37
3125,66
3131,54
©
о
о
о
0,03
Рубидий
4201,85
0,01
—
Рутений
3436,73
0,01—0,003
0,05
12*
356
Д. Я. Иванову Л. А. Лернер
Таблица 3 (продолжение)
Элемент
о
Чувствительность определения, % *
Аналитические линии, А
Спектрограф ИСП-28
Спектрограф ДФС-13
Самарий
3152,09
3218,61
0,1
0,03
Свинец
2833,06
0,001
—
Серебро
3280,68
3382,89
0,0003—0,0001
0,00001
Скандий
3353,73
3368,94
0,001
0,0001
Эрбий
3230,59
3312,42
0,01
0,003
Иттербий
3289,37
0,001-0,0001
0,00003
Иттрии
3242,28
3195,61
0,01-0,003
0,0001
Кадмий
3261,05
0,001
0,0003
foroft
шм
4047,20
396Я ,46
0,5-0,5
0,3
Кальций
4226,72
0,001
—
Кобальт
3953,51
0,001
0,0001
Кремний
2881,57
0,001
—
Лантан
3337,48
3245,13
0,01—0,003
0,001
Литий
6707,84
0,0003
—
Лютеций
3281,75
3312,12
0,001
0,001
Магний
2852,12
0,001
—
Марганец
2801,06
3044,56
0,001
0,001
Медь
3247,54
3274,96
0,0001
0,00001
Молибден
3132,59
3170,33
0,001
0,0001
Мышьяк
2349,84
0,1
3032,85
—
Натрий
5889,95
5895,92
0,0003
Неодим
3259,23
0,3
0,03
Никель
3090,81
0,001
0,0001
Ниобий
3163,40
0,01
—
Олово
2839,98
3175,04
0,001
0,0001
Стронций
3464,45
4607,33
0,001
0,003
Сурьма
2311,47
2598,06
3267.51
3232.52
0,01
0,003
Таллий
3349,03
3162,57
3229,75
0,001-0,01
0,005
Спектральные методы
357
Таблица 3 (окончание)
Элемент
Аналитические линии, А
Чувствительность определения, %
Спектрограф ИСП-28
Спектрограф ДФС-13
Тантал
2685,11
3311,16
0,03-0,01
0,01
Теллур
2385,76
0,03
—
Тербий
3324,40
0,03
0,003
Титан
3349,03
3162,57
3088,02
3372,80
0,001
0,0001
Торий
2837.29
3108.29
3111,43
0,01
0,003
Уран
2865,67
3270,12
0,1
0,03
Фосфор
2535,67
0,1
—
Хром
4254,34
3014,91
0,001
0,0003
Цезий
4555,39
0,3
__
Церий
3201,71
0,1
0,01
Цинк
3345,02
0,003-0,01
0,003
Цирконий
3391,97
3279,26
3273,04
0,003-0,001
0,0003
Аналитические графики строятся в координатах (S, 1 g С) или (AS, 1 g С),
где AS := Sл+ф —
2. Анализируют почвы без предварительной химической подготовки, без
введения элемента сравнения, но с введением спектрального буфера. Ана¬
литические графики строят в тех же координатах, что и в предыдущем
случае.
3. Анализируют почвы без предварительной химической подготовки,
но с введением элемента сравнения (одного или нескольких). При этом может
быть введен и спектральный буфер.
Анализ выполняют по постоянному, контролируемому графику, по¬
строенному в координатах (lg-j^, lg6’) или по методу трех эталонов и графику
в координатах (AS, 1 g С), где AS = Son — Sep-
4. Анализируют почвы после предварительной химической подготовки
(разложение почвы, удаление некоторых групп макроэлементов и др.).
Одновременно может быть введен элемент сравнения и спектральный бу¬
фер. Графики строят в координатах (lg , ig с) или (AS, lgC). ,
5. Определение общего содержания микроэлементов в почвах, почвен¬
ных вытяжках и естественных водах производят после их предварительно¬
го концентрирования. Концентрат смешивают со спектральной основой,
содержащей элемент сравнения и служащей спектральным буфером. Анали¬
тические графики строят в координатах (lg-j^-, lg С) или (AS, lg С).
358
Д. Я. Иванов, Л, А• Лернер
6. Проводят озоление растений, вводят в золу спектральный буфер и
элемент сравнения. Графики строят в координатах (lg , lg С) или (AS, \g С).
Выше было указано, что изготовление стандартов при анализе почв
и растений представляет собой очень трудную задачу. При анализе растений
на содержание в них микроэлементов применяют стандарты, состав которых
отражает видовой состав растений или их групп.
Ниже приводятся описания некоторых частных методик, относящихся
к той или иной из перечисленных схем анализа почв и растений. Мы рас¬
смотрим лишь небольшую часть опубликованных по этому вопросу работ.
В работах часто отсутствует достаточно строгая метрологическая оцен¬
ка применяемого метода. В ряде случаев имеется лишь ссылка на то, что дан¬
ные спектрального метода удовлетворительно совпадают с результатами ко¬
лориметрического метода или что данные анализа удовлетворяли задачам
почвенно-агрохимических исследований. Подобное положение затрудняет
оценку применяемых методов и их сопоставление между собой в целях вы¬
бора наиболее оптимального для данной задачи метода.
А. А. Кветкина и 3. И. Шлавицкая (1963) предложили количественный
спектральный метод определения валового содержания ряда микроэлемен¬
тов в почвах и растениях. Метод рассчитан на анализ засоленных и карбо¬
натных почв, учтен также зольный состав анализируемых растений.
Эталоны для анализа почв готовились на следующей основе: Si02 —
63%, СаС03 - 5,5%, Na2C03 - 3%, K2S04 - 5,5%, КС1 - 1,0%, MgO -
2%, Fe203 - 5%, A1203 - 15%.
Эталоны для анализа золы растений имели следующий состав: Si02 —
20%, K2S04 - 20%, КС1 - 15%, Na2C03—15%, СаНР04-Ю%, K2HP04-
15%, MgO — 5%. Определяемые микроэлементы вводились в основу в виде
растворов чистых солей. Смесь выпаривалась и прокаливалась при 450—
500° С. По-видимому, после прокаливания эталоны в основном состояли из
окислов элементов.
К анализируемым почвам, золам растений и эталонам добавляли буфер —
сульфат натрия в количестве 20% от веса пробы. Вместе с сульфатом натрия
вводили Pd, Ge и Т1 соответственно в количестве 0,1; 0,5 и 1% от веса суль¬
фата натрия. Эти элементы служат элементами сравнения при анализе почв
и растений. Анализируемые пробы и эталоны сжигали в кратере угольного
электрода диаметром 4,5 мм и глубиной 6 мм в дуге переменного тока при
Таблица 4
Аналитические пары линий, применяемые при анализе почв и растений
Определяемый
элемент
Длина волны, А
Внутренний стандарт
Длина волны, A
Ч
3280,7
Ge
3269,5
В
2497,7
Ge
2498,0
Со
3453,5
Pd
3421,2
Сг
3014,8
Pd
3027,9
Си
3273,9
Ge
3269,5
Мп
2798,3
Pd
3421,2
Мо
3170,3
Pd
3114,0
Ni
3414,8
Pd
3421,2
РЬ
2833,1
T1
2767,9
Sn
2840,9
Ge
2754,6
V
3185,4
Pd
3114,0
Zn
3282,3
Zn
2767,9
Спектральные методы
359
его силе 15— 17 а до полного испарения пробы. Аналитические пары линий,
используемые в данной работе, приведены в табл. 4.
Определение Zn, Мо и Со рассмотренным методом можно выполнять при
повышенных содержаниях их в почвах.
М. А. Риш и Я. М. Приев (1960) предложили метод определения ряда
микроэлементов в растениях без предварительного их озоления. Навеску
в 400 мг воздушно-сухого измельченного растения набивают в папиросную
гильзу и пропитывают раствором сернокислого аммония. После просушива¬
ния гильза сверху вводится в горизонтальную дугу между угольными элект¬
родами при токе 18 а, продолжительность введения пробы — 2 мин. 15 сек.,
дуговой промежуток — 6 мм. Интенсивность линии измеряется по отноше¬
нию к фону.
Анализ выполняют по методу трех эталонов, градуировочный график
строят в координатах (AS, lg С).
Точность метода зависит от правильного приготовления эталонов, мате¬
риалом которых могут служить соответствующие растения с известной кон¬
центрацией определяемых элементов или искусственные эталоны, пригото¬
вляемые из беззольных фильтров с втиранием в них минеральной основы,
отражающей зольный состав некоторых групп растений. Авторы предлагают
четыре типа минеральных основ эталонов (табл. 5).
Таблица 5
Состав минеральных основ эталонов для анализа растительного материала
(в весовых единицах)
Реактив
Основа
мясистые солянки
сухие солянки
злаки
бобовые и полыни
NaCl
38,0
10,0
10,0
15,0
Na2S04
22,0
9,0
—
—
Na2C03
23,0
50,0
17,0
2,0
СаС03
9,0
10,0
18,0
27,0
К2С03
5,0
12,0
16,6
16,0
MgO
2,5
3,0
5,0
8,0
ai2o3
0,3
0,6
0,3
0,5
Fe203
0,2
0,4
0,2
1,0
Si02
0,5
3,0
40,0
10,0
кн2ро4
0,7
2,5
19,2
9,0
k2so4
—
21,0
11,0
Т. Ф. Боровик-Романова и др. (1962, 1966, 1967) разработали методы
определения ряда микроэлементов в растениях. Большое внимание в этих
работах уделено изучению влияния, состава пробы на результаты анализа.
Эталоны готовились на основе искусственных смесей, соответствующих по
своему составу разнотравью, злакам и полыням (табл. 6).
Установлено, что изменение состава эталонов вызывает сдвйг градуиро¬
вочных графиков для определения Мо, Си, Со и РЬ. Применив буфер, состоя¬
щий из смеси угольного порошка и LiF (3 : 1), авторы значительно ослабили
влияние состава пробы, и практически оказалось возможным выполнять
анализы растений на указанные элементы по одному виду эталонов.
Градуировочные графики строили в координатах (AS, lgC),TRe
A S — $л+ф —
360
Д. Н. Иванов, Л% А. Лернер
Таблица 6
Химический состав основ для изготовления эталонов
(в весовых единицах)
Реактив
Разнотравье
Злаки
Полыни
Реактив
Разнотравье
Злаки
Полыни
КН2Р04
18,0
13,3
10,0
NfljCOj
7,0
7,0
11,0
K*S04
19,0
15,0
9,0
СаО
—
—
8,5
к2со,
14,0
7,1
13,0
СаСО,
19,0
16,7
—
КС1
—
11,5
—
MgO
4,0
3,1
2,0
NaCl
"
41,5
Si02
19,0
25,0
5,0
Приведем примеры применения спектрохимических методов, представ¬
ляющих собой сочетание химической подготовки анализируемых проб и ко¬
личественного определения элементов спектральными методами. При опре¬
делении микроэлементов задача химической подготовки состоит в удалении
основных составляющих пробы, концентрировании определяемых элементов
и приведении пробы к более однородному химическому составу. Подготовка
пробы представляет собой трудоемкую химическую процедуру, но она пол¬
ностью оправдывает себя при групповом концентрировании микроэлемен¬
тов и групповом их спектральном определении.
В указанных работах изложено также определение микроэлементов в поч¬
вах. Эталонами служили искусственные смеси, отражающие «средний» со¬
став некоторых типов почв. В эталоны и пробы вводился спектральный бу¬
фер, который ослаблял или практически устранял влияние состава проб на
результаты анализа. График строился в координатах (AaS, 1 gC), где AS =
:= *^л+ф
P. JI. Митчелл (Mitchell, 1961) предложил следующий спектрохимический
метод. При анализе растений 20—40 г сухого вещества растений озоляют
в течение ночи при 450°, золу обрабатывают соляной кислотой, осадок от¬
фильтровывают, прокаливают, сплавляют с 1 a Na2C03, растворяют в соля¬
ной кислоте и отфильтровывают для удаления возможных остатков крем¬
ния.
При анализе объектов животного происхождения пробы озоляли тем же
способом —после сушки вымораживанием. Для большинства определений тре¬
буется около 1 г золы. При анализе крови перед осаждением удаляют железо эк¬
стракцией эфиром. Анализ таких объектов, как кости и сухое молоко, требует
осаждения фосфорнокислого кальция. Рекомендуется работать при сравни¬
тельно большом разбавлении как определяемых компонентов, так и исполь¬
зуемых реагентов и кислот.
При определении подвижных форм микроэлементов в почвах готовят поч¬
венные вытяжки. Экстрагентами служат 2,5%-ная уксусная кислота, 1 н.-
раствор уксуснокислого аммония и разбавленная соляная кислота. Вытяж¬
ку из 20 —50 г почвы при соотношении почвы и раствора 1 : 25 высушивают
досуха, органическое вещество удаляют обработкой HN03 и осадок раство¬
ряют в HG1. Азотнокислые соли удаляют повторным выпариванием, чтобы
они не мешали при последующем осаждении.
При анализе воды выпаривают 1—5 л ее до сухого остатка, который затем
обрабатывают так же, как и сухой остаток почвенной вытяжки.
Анализируемые растворы должны содержать приблизительно 30 мг
А1203, 1—4 мг Fe203 и 0,4 мг Cd. Алюминий служит носителем осаждаемых
микроэлементов, кадмий — внутренним стандартом при определении цинка,
Спектральные методы
361
железо является внутренним стандартом при определении всех остальных
элементов.
Концентрирование микроэлементов достигается их осаждением смесью
8-оксихинолина, танниновой кислоты и тионалида (р-аминонафталид тио-
гликолевой кислоты) при pH 5,1—5,2. При этом осаждаются Со, Mo, Sn, Zn,
Pb, V, Сг, Ag, Au, Ga, Си и некоторые другие элементы.
Осаждение производят при комнатной температуре из 150 мл раствора
приблизительно! н. НС1. После добавления 10—15 мл 5%-ного раствора
8-оксихинолина в 2 н. уксусной кислоте прибавляют по каплям Юн. растгор
аммиака до тех пор, пока цвет раствора не начнет изменяться от желтого до
изумрудно-зеленого, pH 1,8—1,9. Изменение цвета раствора, вероят¬
но, обусловлено образованием комплекса железа с 8-оксихинолином при
pH 1,8—1,9. Необходимое для осаждения значение pH 5,1—5,2 достигается
добавлением 50 мл 2 н. раствора ацетата аммония. Однако на этой стадии
подготовки пробы добавляют при перемешивании только 30 мл. Образую¬
щийся большой осадок А1 и Fe быстро коагулирует. Затем к смеси добавляют
при непрерывном перемешивании 2 мл 10%-ного раствора танниновой кисло¬
ты, 2 мл 2 и. раствора уксуснокислого аммония и 2 мл 1%-ного раствора
тионалида в ледяной уксусной кислоте. После этого немедленно добавляют
эквивалентное количество аммиака для нейтрализации уксусной кислоты и
общее количество уксуснокислого аммония доводят до 50 мл. Танниновая кис¬
лота и тионалид должны быть свежеприготовленными.
Количество аммиака, эквивалентное 2 мл ледяной уксусной кислоты,
предварительно определяют путем титрования с индикатором бромтимоло-
вым синим. Рекомендуется добавлять аммиак в объеме 20—25 мл.
После отстаивания в течение ночи осадок отфильтровывают через фильт¬
ровальную бумагу и озоляют в фарфоровом тигле при 450°. Полученный оса¬
док взвешивают. Если осадок меньше 30 мг, то к нему добавляют А1203
до веса 40 мг. Полученная смесь поступает на спектральный анализ. Содер¬
жание железа определяют колориметрически с салицилатом натрия в алик¬
вотных частях пробы.
Концентрат весом 3—4 мг, перемешанный с двойным количеством уголь¬
ного порошка, вводят в кратер угольного электрода диаметром 1 мм и глуби¬
ной 8 мм, диаметр самого электрода 2,8 мм. Электроды с пробой, включен¬
ные в качестве катода, сжигают в дуге постоянного тока при 9 а и 250 в.
Анодом служит угольный стержень диаметром 5 мм. При зажигании дуги
сила тока должна быть равна приблизительно 3 а, через 10 сек. электроды
медленно разводят до получения промежутка в 10 мм и постепенно увеличи¬
вают силу тока до 9 а. Размеры дугового промежутка поддерживают постоян¬
ными до полного сгорания пробы, которое наступает приблизительно через
2 мин.
Спектрограммы получают на большом кварцевом спектрографе (типа
КСА-1) со сферическим конденсором у щели для проектирования дуги на
объектив колиматора. Между линзой и щелью располагают 6-ступенчатый
сектор, вращающийся со скоростью 500 об/мин. Диафрагму колиматора
устанавливают так, чтобы она пропускала свет ст вершины катода и приле¬
гающей к нему одной трети столба дуги.
Существенная особенность описываемого метода состоит в использовании
железа в качестве внутреннего стандарта, хотя оно и входит в пробы в пере¬
менных концентрациях, поэтому был предложен метод переменного графика
(эталона), который заключается в следующем.
По эталонам, содержащим различные концентрации железа, строят се¬
рию градуировочных графиков, которые оказываются смещенными парал¬
лельно на величину, зависящую от содержания железа. Принимая один из
графиков за основной (автор рекомендует полученный при 5% Fe^Og), мож¬
но построить график поправок на содержание железа в пробе. Анализ выпол¬
362
Д. Н. Иванов, Л. А. Лернер
няют по основному графику, внося поправку на содержание железа в пробе.
Эталоны готовят на основе смеси окислов А1203 и Fe203.
В табл. 7 приведены аналитические пары линий, применяемые в описан¬
ном методе.
Таблица 7
Аналитические пары линии
(по Митчеллу)
Микро¬
элемент
Длинаo
волны, A
Внутрен¬
ний
стандарт
Длина
волны, A
Микро¬
элемент
Длина
волны, A
Внутрен¬
ний
стандарт
Длина
волны, A
Сг
4254,4
Fe
4250,8
Be
3130,4
Fe
3116,6
Со
3453,5
Fe
3451,9
Ge
3039,1
Fe
3116,6
Ni
3414,8
Fe
3413,1
Ga
2943,6
Fe
2929,0
Zn
3345,0
Cd
3261,1
Sn
2840,0
Fe
2838,1
Ag
3280,7
Fe
3306,4
Pb
2833,1
Fe
2838,1
V
3185,4
Fe
3196,9
Au
2675,9
Fe
2680,5
Mo
3170,4
Fe
3196,9
Д. Н. Иванов и др. (1965, 1967) предложили спектрохимический метод
определения ряда микроэлементов, применяя для их предварительного кон¬
центрирования органические соосадители.
При определении общего содержания Со, Си, Ni, Pb, Sn, Zn, Мо выпол¬
няют предварительное их концентрирование. 1 г тонкорастертой и просеян¬
ной через шелковое сито почвы помещают в платиновую чашку и прокаливают
в муфеле в течение 4—5 час. при температуре 400—450° С. Прокаленную на¬
веску разлагают смесью H2F2 и H2S04 обычным способом. После разложения
пробы к остатку приливают 20 мл НС1 (1 : 3) и нагревают до полного рас¬
творения осадка. Раствор фильтруют горячим через беззольный фильтр «бе¬
лая лента» в стаканы на 250 мл. Фильтр промывают несколько раз подкис¬
ленной горячей водой и доводят водой объем до 150 мл. Прибавляют 2,0 мл
ацетатного буфера и 2,5 мл 25%-ного раствора цитрата аммония. Добав¬
ляя NH4OH, доводят pH исследуемого раствора до значения 6,0—7,0, конт¬
ролируя потенциометром или индикаторной бумагой. Объем раствора дово¬
дят до 200 мл, добавляют 10 мл ацетонового раствора дитизона, тщательно
перемешивают и прибавляют при перемешивании 2,5 мл 7%-ного раствора
2,4-динитроанилина в ацетоне. После 30—40-минутного стояния осадок от¬
фильтровывают через фильтр «белая лента». Осадок на фильтре промывают
4—5 раз промывным раствором, подсушивают и переносят в кварцевый ти¬
гель. Тигли с осадком подсушивают под лампой, смачивают несколькими
каплями разбавленной H2S04 и озоляют в муфеле при температуре 400—
450°.
При работе по этой прописи олово частично захватывается цитратом и
соосаждается не полностью. Чтобы количественно определить всю перечис¬
ленную выше группу микроэлементов, проводят двойное последовательное
соосаждение. Для этой цели pH солянокислого раствора после разложения
пробы прибавлением аммиака доводят до значения 2,5, а затем проводят со¬
осаждение способом, описанным выше. Через 30 мин., не отфильтровывая,
в тот же стакан добавляют ацетатный буфер, 2,5 мл раствора цитрата аммо¬
ния и доводят pH до 6—7. Тщательно перемешивают и проводят повторное
соосаждение, добавляя 10 мл ацетонового раствора дитизона и 2 5 мл 7%-
ного раствора 2,4-динитроанилина в ацетоне. После 30-минутного стояния
осадок отфильтровывают и озоляют в муфеле (Кузнецов и др.. 1964).
Спектральные методы
363
После концентрирования меди, кобальта, цинка и отделения осадка филь¬
трат и промывные воды переносят в стакан и доводят анализируемый раствор
водой до 200 мл. К этому объему раствора добавляют 6 мл 6 М НС1, чтобы
значение pH раствора было равно ~1. Затем прибавляют 5 мл 2 %-ного вод¬
ного раствора таннина, тщательно перемешивают и добавляют по каплям при
перемешивании 10 мл 4%-ного ацетонового раствора «окисленного» краси¬
теля Стенгауза (Кузнецов, Большакова, 1967).
В течение 30—40 мин. содержимое стакана перемешивают время от
времени, а затем фильтруют через фильтр «белая лента» и промывают 3—
4 раза промывным раствором. Фильтр с осадком подсушивают на воронке,
переносят в фарфоровый тигель, смачивают несколькими каплями разбав¬
ленной серной кислоты и озоляют в муфельной печи при температуре 400—
500° С (Орлова, 1969).
В случае большого содержания железа и алюминия в пробе производят
двухкратное осаждение молибдена.
Прямое спектрографическое определение микроэлементов в почвенных вы¬
тяжках и естественных водах при реально существующих уровнях концент¬
раций определяемых элементов, как правило, практически невозможно.
Предварительное индивидуальное концентрирование элементов и последую¬
щее определение их спектрографическими методами при массовых анализах
нецелесообразно. Однако применение групповой вытяжки для концентри¬
рования и спектрографического определения элементов дает значительный
выигрыш в производительности анализов по сравнению с колориметрически¬
ми методами.
При определении цинка, меди, кобальта и некоторых других элементов
в кислотных почвенных вытяжках (1 н. НС1, 1 н. HN03 и др.) на каждые"
200 мл вытяжки вводят 2—3,5 мл 25%-ного раствора цитрата аммония. При
анализе буферной ацетатно-аммонийной вытяжки с pH = 4,8, как правило,
вводят 1 мл цитрата аммония на тот же объем. После этого добавляют по кап¬
лям раствор аммиака до установления величины pH анализируемого раство¬
ра, равной 6,0—7,0. Затем на каждые 200 мл вытяжки прибавляют 10 мл
дитизона, тщательно перемешивают и далее прибавляют 2,5 мл 2,4-динит¬
роанилина. Последующий ход концентрирования такой, как и при определе¬
нии общего содержания микроэлементов.
При определении молибдена в оксалатной вытяжке 100—200 мл вытяжки
помещают в термостойкий стакан и выпаривают ее досуха. Для разруше¬
ния оксалатов и органических веществ остаток обрабатывают 30%-ной пе¬
рекисью водорода и концентрированной HN03. Остаток растворяют в 6 мл
6 М НС1 и доводят дистиллированной водой до 200 мл при pH 1. К раствору
добавляют 5 мл раствора таннина, после перемешивания вводят небольшими
порциями 10 мл «окисленного» красителя Стенгауза. Последующий ход кон¬
центрирования такой, как и при концентрировании общего содержания мо¬
либдена (Кузнецов и др., 1969). В указанной работе рекомендуется вариант
метода концентрирования молибдена непосредственно в оксалатном раство¬
ре, минуя стадию разрушения оксалатов.
При определении микроэлементов в растениях 5—10 г растительного ма¬
териала озоляют сухим методом, остаток переводят в раствор. При определе¬
нии цинка, меди и кобальта к анализируемому раствору прибавляют 0,2—
0,5 мл раствора двузамещенного цитрата аммония, 1 мл ацетатного буфера и
после этого прибавляют небольшими порциями аммиак до установления ве¬
личины pH раствора 6—7. При непрерывном помешивании вводят 20 мл аце¬
тонового раствора дитизона и 5 мл ацетонового раствора 2,4-динитроанили¬
на. Далее поступают так же, как было рекомендовано при анализе почв.
При определении молибдена фильтрат и промывные растворы объединя¬
ют, переносят в стакан, добавляют дистиллированной воды до объема 200 мл
и проводят соосаждение молибдена таннином совместно с «окисленным»
364
Д. Н. Иванов, Л. Л. Лернер
красителем Стенгауза, как и при определении общего содержания молиб¬
дена в почвах (Орлова, 1967).
Полученные описанными методами озоленные остатки проб поступают на
спектральный анализ. Можно объединять озоленные остатки, полученные по¬
сле применения различных мэтодов концентрирования.
Чтобы уменьшить влияние общего состава остатков на результаты ана¬
лиза, применяют буферную основу следующего состава: С — 52%, К2С03 —
24%, Fe203 — 16%, А1203 — 8%. На этой основе составляют эталоны опре¬
деляемых элементов (путем введения растворов чистых солей) и ту же основу
добавляют к анализируемому остатку. Элементом сравнения служит железо,
анализ выполняют по методу постоянного градуировочного графика.
Для анализа берут 50 мг эталона и делят эту навеску на три части. К ана¬
лизируемому остатку добавляют 50 мг основы, тщательно перемешивают
смесь, а затем также делят ее на три части. Эти части служат для повторного
спектрально-аналитического определения микроэлементов. Небольшие от¬
клонения содержания железа в эталонах и анализируемых пробах практи¬
чески пе влияют на интенсивность аналитических линий определяемых эле¬
ментов и линий спектра внутреннего стандарта — железа.
Пробы и эталоны сжигают в кратере угольного электрода при силе тока
10—12 а. Внешний диаметр электрода — 3 мм, диаметр кратера — 2 мм,
глубина его — 5 мм. Кратер закрывают угольной крышечкой с небольшим
отверстием. Время экспозиции — 2,5—3 мин. до полного сгорания пробы
и эталонов. Аппаратура: спектрограф ДФС-8, микрофотометр МФ-2, гене¬
ратор дуги ДГ-2. Аналитические пары линий приведены в табл. 8. На ту же
фотопластинку со спектрами при помощи 9-ступенчатого ослабителя наносят
марки почернения.
Таблица 8
Аналитические пары линий
Определяемый элемент
Длина волны, А
Внутренний стандарт
Длина волны, А
Медь
3247,54
Fe
3265,05
Медь
3273,96
»
3265,05
Цинк
3345,02
»
3355,22
Кобальт
3453,50
»
3452,27
Никель
3414,76
»
3442,88
Свинец
2833,07
»
2840,42
Олово
2339,99
»
2840,42
Молибден
3170,35
»
3180,75
3132,59
»
3180,75
По спектрам эталонов строят градуировочный график в координатах
(lg-^-,lgC) и по нему определяют искомую концентрацию. Коэффициент
вариации метода составляет 10—15%. Метод применим к определению
общего содержания микроэлементов в почвах, растениях и других биоло¬
гических объектах, а также в почвенных вытяжках и водах.
Фотографирование спектров предпочтительнее производить через 3-сту¬
пенчатый ослабитель. Дело в том, что в некоторых случаях почернения
линий определяемых элементов без ослабления излучений могут быть на¬
столько велики, что вызовут недопустимо большие ошибки фотометрирова-
ния и обесценят работу, затраченную на подготовку проб. Применение сту¬
пенчатого ослабления позволяет избежать этих трудностей.
Спектральные методы
365
Применение крышечки над пробой в электроде имеет существенное зна¬
чение при выполнении анализа. Она предупреждает выброс пробы и фонта¬
нирование мелких частиц из кратера электрода, что способствует повышению
точности анализа.
Из изложенного следует, что методы эмиссионного спектрографического
анализа представляют широкие принципиальные возможности для определе¬
ния состава почвенно-агрохимических объектов. Однако применение этого
метода к решению конкретных аналитических задач в ряде случаев сопря¬
жено с вполне определенными трудностями, устранение которых требует
выполнения специальных методических исследований.
ЭМИССИОННЫЙ ПЛАМЕННО-ФОТОМЕТРИЧЕСКИЙ МЕТОД
Пламенно-фотометрические методы анализа предусматривают приме¬
нение различного рода пламени в качестве источника возбуждения спектра
определяемых элементов, фотоэлектрической системы измерения интенсив¬
ностей аналитических линий и введения в источник анализируемых проб в
виде растворов.
Пламенно-фотометрические установки
В настоящее время разработаны и выпускаются различные варианты пла¬
менно-фотометрических установок, нашедшие широкое применение в анали¬
тической практике.
Пламенно-фотометрические приборы разделяются на два класса: пламен- -
ные фотометры со светофильтрами и пламенные спектрофотометры с моно¬
хроматором в качестве спектрального прибора. Пламенные фотометры наи¬
более просты и применяются для анализа объектов с несложными спектра¬
ми, т. е. при решении более простых аналитических задач. Эти приборы имеют
широкое применение в почвенно-агрохимическом анализе для определения
калия, натрия и некоторых других элементов. Пламенные спектрофотомет¬
ры — более сложные и совершенные приборы. Они обладают большими ана¬
литическими возможностями, нежели пламенные фотометры.
В зависимости от применяемого измерительного устройства фотометры
делят на визуальные и более совершенные — регистрирующие. Выбор способа
измерения определяется особенностями конкретной аналитической задачи
и объемом предстоящей работы. Наиболее широко применяются отечествен¬
ные пламенные фотометры: ФПФ-58, ГП-21, ППФ-УНИИЗ.
Существенной особенностью указанных пламенных фотометров являет¬
ся то, что в них осуществлена компенсационная схема измерения интенсив¬
ностей аналитических линий определяемых элементов.
Широкое применение получил также пламенный фотометр Модель III,
выпускаемый в ГДР. Прибор конструктивно прост и надежен в работе, поз¬
воляет определять в растворах содержание натрия, калия, кальция и не¬
которых других элементов. Фотометр легко перестроить для работы по ком¬
пенсационной схеме. В настоящее время выпускается ФЛЯФО-4.
Во многих лабораториях успешно работают самодельные пламенные
спектрофотометры, изготовленные на базе ряда монохроматоров. Индиви¬
дуальное изготовление такого рода фотометров не представляет трудностей.
Методы фотометрирования и анализа
При эмиссионном пламенно-фотометрическом анализе применима зави¬
симость интенсивности спектральной линии от концентрации элемента (1) и (2).
Градуировочные графики строятся обычно в координатах (/, С) или
(lg Л 1? О» т- е- практически по абсолютным значениям интенсивности ана¬
366
Д. Н. Иванову Л. А. Лернер
литических линий определяемого элемента в спектрах стандартов. Такое
упрощение метода возможно благодаря высокому постоянству условий горе¬
ния пламени и непрерывному обновлению в нем вещества пробы и стапдар-
тов, что исключает влияние фракционного испарения пробы на результаты
анализа.
Выполнение анализа можно начинать только после стабилизации режима
работы установки, давления газа и воздуха, чувствительности приемника
и др. Дальнейший порядок работы следующий. Вводят в пламя «нулевой рас¬
твор», т. е. растворитель, на основе которого составлены эталоны и полу¬
чены анализируемые растворы, при этом устанавливают гальванометр на
нуль. Вводят в пламя эталонные и анализируемые растворы, записывают
соответствующие показания гальванометра, которые позволяют определить
искомую концентрацию элемента в растворе. Это можно выполнить различ¬
ными приемами.
Метод градуировочного графика. По показаниям (/)
гальванометра и соответствующим им концентрациям (С) определяемого
элемента в эталонах строят градуировочный график. Наиболее удобен для
выполнения анализа прямолинейный участок графика. Пользуясь этим гра¬
фиком, по показанию гальванометра для анализируемого раствора опре¬
деляют концентрацию содержащегося в нем элемента. График может служить
для анализа значительного числа однотипных растворов. Метод градуиро¬
вочного графика наиболее производителен и поэтому нашел наиболее широ¬
кое применение.
Градуировочные графики зависимости показаний измерительного прибора
от концентрации элемента могут быть построены в линейном и логарифми¬
ческих масштабах. Концентрации определяемого элемента могут быть выра¬
жены в миллиграммах на единицу объема (например, мг/л) с последующим
пересчетом в проценты от взятой навески. В этом случае нет надобности брать
всегда одну и ту же навеску почвы и доводить объем анализируемого рас¬
твора всегда до одной и той же величины.
Аналитические данные можно получить и непосредственно в процентах
окислов от взятой навески, но тогда следует брать одни и те же навески почвыг
доводить объем анализируемого раствора до одних и тех же величин и вы¬
ражать концентрации элементов в эталонах в процентах окислов. Например,
при валовом анализе следует брать 1 г почвы, доводить объем анализируе¬
мого раствора до 200 мл и вводить в 200 мл эталонов такое количество опре¬
деляемого элемента, которое составляло бы нужное нам число процентов его
в 1 г почвы (1, 2, 4 и т. д.). При сохранении отношения навески почвы к объе¬
му раствора 1 : 200 абсолютные значения этих величин могут быть произ¬
вольными.
Данные анализа водной вытяжки принято выражать либо в миллиграмм-
эквивалентах на 100 г почвы, что при соотношении 1 : 5 соответствует 500 мл
раствора, либо в процентах содержания определяемого элемента в почве.
Поэтому градуировочные графики необходимо строить так, чтобы получить
результаты анализа непосредственно в этих величинах без каких-либо до¬
полнительных расчетов. Этому условно отвечают эталоны, приготовленные
в миллиграмм-эквивалентах на 500 мл раствора (1, 2, 4 и т. д. мг-экв). Дан¬
ные анализа, полученные по графикам, построенным по таким эталонам, бу¬
дут соответствовать содержанию определяемого элемента в миллиграмм-эк¬
вивалентах в 500 мл вытяжки или в 100 г почвы. Те же концентрации элемен¬
та в эталонах, выраженные в процентах, дают возможность сразу узнать по
графику содержание определяемого элемента в почве в процентах.
Н апример, 1,2, 4 мг-экв натрия на 100 г почвы соответствуют содержанию 0,023;
0,046; 0,092% в почве. Поэтому по оси абсцисс градуировочного графика можно нанести
миллиграмм-эквиваленты или проценты и по одному графику получать значения иско¬
мой концентрации в двух единицах ее измерения.
Спектральные методы
367
Такая система отсчета требует постоянства отношений величин навески
почвы и объема воды, но не их абсолютных значений.
Метод ограничивающих эталонов. В этом методе поль¬
зуются градуировочным графиком, построенным по двум эталонам с концент¬
рациями определяемого элемента С\ и С2, ограничивающими интервал воз¬
можных значений концентраций определяемого элемента в пробе: Сх < Сх <
< С2. В пламя вводят анализируемый раствор и находят соответствующие
ему показания гальванометра ах. Подбирают два близких эталона, дающих
показания и а2, больше и меньше ах. Строят график по значениям эталон¬
ных концентраций и полученным для них отсчетам. Тогда концентрация оп¬
ределяемого элемента в пробе:
Сх = Ci + (Сг — Ci) . (9)
Рассмотренный прием справедлив для прямолинейных участков графика;
на криволинейных участках графика требуется, чтобы концентрации в эта¬
лонах были близки к ожидаемой концентрации элемента в пробе.
При выполнении трех замеров режим работы практически сохраняется,
в результате чего может быть достигнута более высокая точность определе¬
ния, чем по методу градуировочного графика, но этот метод более трудоемок.
Метод добавок. Метод добавок основан на фотометрировании
аналитической линии спектра раствора с неизвестной концентрацией опре¬
деляемого элемента Сх и фотометрировании той же линии после введения в
раствор некоторой добавки с заранее известной концентрацией С.
Пусть показания гальванометра для растворов с концентрацией Сх и
Сх+ С будут соответствовать ах и а. Воспользуемся системой координат а
= ф (С) и восстановим в произвольной точке на оси абсцисс параллельно оси
ординат показания гальванометра ах и на некотором произвольном расстоя¬
нии а. Проведем через вершины ординат ах и а прямую до пересечения с осью
абсцисс. Если точку пересечения А принять за начало координат, то отрезок
по оси абсцисс между ах и а будет изображать концентрацию добавки С,
а отрезок по той же оси между ординатой ах и точкой пересечения А—неиз¬
вестную концентрацию Сх. Определяемую концентрацию можно вычислить
по формуле
_Са*_ 0
X а—ах v 1
Практически добавки вводят, смешивая анализируемый раствор с кон¬
центрацией Сх в объеме Vx и эталонный раствор с концентрацией С0 в объе¬
ме V0. Конечная концентрация смешанного раствора определяется соотно¬
шением взятых объемов. После измерения интенсивности аналитической
линии определяемую концентрацию Сх можно вычислить по формуле
(п — 1) Српх
х па — ах ’ v '
где п = (Vx + Vq)\V — коэффициент увеличения объема при введении добавки.
Метод добавок менее производителен, чем метод градуировочного графи¬
ка или ограничивающих эталонов, но во многих случаях позволяет, не имея
эталонов, выполнить анализ.
Метод прямого отсчета концентраций. Проиллю¬
стрируем этот метод конкретным примером. Предположим, что при опреде¬
лении обменного калия в почвах в 90% случаев калий содержится в коли¬
чествах до 50 мг/л раствора. Тогда целесообразно установить максимальное
показание по шкале гальванометра для 50 мг/л, а показание нуль — для
«нулевого» раствора (растворителя). Затем следует отградуировать шкалу
368
Д. Н. Иванов, Л. А. Лернер
от нуля до максимального показания по эталонам непосредственно в еди¬
ницах концентраций (мг/л или мг-экв). При выполнении анализа этим мето¬
дом надлежит строго следить за сохранением постоянства граничных по¬
казаний гальванометра. Исходные растворы с концентрациями большими,
чем 50 мг/л, анализируют после соответствующего разбавления.
Характеристика метода
Рассматриваемый метод обладает довольно высокой чувствительностью
определения некоторых элементов (табл. 9) и точностью, удовлетворяющей
требованиям агрохимического анализа. Основные преимущества пламенно¬
фотометрических методов по сравнению с химическими состоят в высокой
производительности и простоте выполнения анализа.
Таблица 9
Чувствительность определения элементов методом эмиссионной пламенной фотометрии
(по Полуэктову, 1967)
Элемент
Длина волны, А
Чувствитель¬
ность, мкг/мл
Элемент
Длина волны, А
Чувствитель¬
ность, мкг/мл
Литий
6708
0,01
Иттербий
3988
0,5
Натрий
5890-96
0,001
Алюминий
3962
0/1—10,0
Калий
7665-99
0,01
Галий
4172
5,0
Рубидий
7880, 7948
0,01
Индий
4511
1,0
Цезий
8521
0,01
Таллий
3776
0,3
Медь
3248
1,0
Титан
4970—5180
0,2-0,3
Серебро
3284
2,0
Олово
3490
1,0
Магний
2852
5,0
Свинец
4058
14,0
Кальций
4227
0,06
Ванадий
5500-5760
0,2-0,3
Стронций
4607
0,05
Ниобий
4500
2,0
Барий
8700
0,6
Фосфор
5480
25,0
Цинк
5000
2,00
Хром
4254
2,0
Кадмий
2288, 3261
—
Молибден
6000
3,0
Скандий
6070
0,006
Марганец
4033
0,1
6132
5,0
Железо
3720
1,0
Иттрий
5972
0,5-1,0
Кобальт
3873
0,5
Лантан
7940
10,0
Никель
3525
2-3
Европий
4594
5,0
Рутений
3728
10,0
Гадолиний
4617
3,0
Пламенные источники света обладают высокой стабильностью горения,
в них можно равномерно вводить исследуемый раствор и обеспечить по-
стоянство состава газового облака. Эти обстоятельства позволяют выполнять
анализ в широких пределах концентраций по абсолютной интенсивности ана¬
литической линии, что значительно упрощает технику анализа.
Применение метода к анализу почв и растений
Пламенно-фотометрический метод обычно предполагает введение пробы
в источник только в жидком состоянии, поэтому анализу почв и растений
предшествует химическая или физико-химическая подготовка проб, которая
Спектральные методы
369
заключается в переведении пробы в раствор или извлечении из нее тех или
иных соединений определяемых элементов.
При выполнении анализа следует обеспечить соответствие состава этало¬
нов составу анализируемых проб, стабильность условий горения пламенц
и поступления в него раствора (Иванов, 1949, 1953, 1962, 1965; Иванов,
Русакова, 1968).
Валовой анализ почв. Определение щелочных
элементов. Щелочные элементы в почвах присутствуют в различных
содержаниях, однако их можно разделить на две группы: к первой принад¬
лежат натрий и калий; ко второй — литий, рубий и цезий. Содержание эле¬
ментов первой группы относится к содержанию второй приблизительно как
1000 : 1. Отношение концентраций натрия и калия в большинстве случаев
ограничено узким интервалом значений и находится в пределах 1 : 10, при¬
чем натрий и калий в этом интервале могут меняться местами.
Аналитическая практика показала следующее:
1) литий, рубидий и цезий не оказывают влияния на определение натрия
и калия в почвах;
2) натрий и калий не оказывают существенного взаимного влияния на
точность определения, так как абсолютные концентрации этих элементов
велики, а отношение их концентраций ограничено узким интервалом значе¬
ний;
3) щелочноземельные элементы не вызывают заметного ионизационного
эффекта при определении натрия и калия;
4) натрий и калий могут оказать большое влияние на определение лития,
рубидия, цезия, завышая результаты анализа. Определение этих элементов
требует применения специальных методических приемов.
Приведенные замечания справедливы для большей части почвенных проб
и принятых методов анализа. Однако в практике анализа встречаются также
растворы, в которых определяемый элемент, например калий, содержится
в очень малых количествах (около 1—2 мг/л), а натрий — в значительных
количествах. В этих случаях превышение концентрации одного из щелоч¬
ных элементов над концентрацией определяемого даже в 10 раз мсжет выз¬
вать значительный ионизационный эффект. Чтобы получить правильные
результаты анализа, потребуется введение в эталоны влияющего элемевта в
соответствующей концентрации или выполнение анализа по методу добавок.
Этот прием применяют и при определении лития, рубидия и цезия в присут¬
ствии натрия, калия и щелочноземельных элементов.
Для определения щелочных элементов навеску почвы разлагают H2F2
в присутствии H2S04, остаток переводят в раствор. Значительные концент¬
рации натрия и калия, а также высокая чувствительность метода допускают
большое разведение раствора. Анализ такого раствора можно выполнять
по водным эталонам чистых солей натрия и калия.
Наличие в растворе кальция может оказать существенное влияние на
точность определения натрия и калия, особенно при анализе карбонатных,
загипсованных и известкованных почв.
Малые концентрации лития, рубидия, цезия не допускают большого раз¬
ведения анализируемых растворов, а присутствие натрия и калия в больших
концентрациях вызывает резко выраженный ионизационный эффект для всех
редких щелочных элементов. Поэтому в эталоны следует вводить соответ¬
ствующие концентрации натрия и калия. Кроме того, на точность определе¬
ния лития сильно влияет присутствие в растворе стронция, который поэтому
необходимо предварительно удалить из анализируемого раствора. Рубидий
и калий имеют близкие длины волн аналитических линий, поэтому раздель¬
ное определение их на пламенном фотометре со светофильтром встречает за¬
труднения. Этот анализ следует выполнять на пламенном спектрофотометре.
370
Д. Н. Иванову Л. Л. Лернер
При определении кальция фотометрическим методом возникают специ¬
фические трудности: интенсивность излучения спектра кальция сущест¬
венно снижается, если в анализируемом растворе присутствует алюминий,
фосфор или сера. Поэтому часто при определении кальция получают зани¬
женные данные. Кальций, а также стронций и барий рекомендуется опреде¬
лять после их предварительного осаждения и последующего перевода в ра¬
створ или же после удаления из первичного раствора мешающих элементов.
Анализ почвенных вытяжек. Водную вытяжку готовят
при соотношении почвы и воды 1 : 5. Отфильтрованный раствор непосред¬
ственно направляют на анализ. При малом содержании воднорастворимых со¬
лей определение натрия и калия можно выполнять по водным эталонам.
Водные вытяжки, содержащие большое количество натрия и калия, ре¬
комендуется перед анализом разбавлять до концентрации 10—100 мг/л.
Соотношение концентрации натрия и калия в водных вытяжках может
сильно варьировать и служить причиной непостоянного от пробы к пробе
ионизационного эффекта. Это заставляет следить за соответствием состава
эталонов составу анализируемых проб и в надлежащих случаях вводить в
эталоны добавки калия и натрия.
Если водные вытяжки содержат значительные количества кальция, то
это приводит к завышению результатов определения натрия и калия. В по¬
добных случаях следует осуществлять метод компенсации или вводить в
пробу элемент, связывающий кальций, или удалять кальций из раствора.
Определение лития, рубидия и цезия в водных вытяжках из почв пла¬
менным методом практически исключается по причине их малого содержа¬
ния в вытяжках.
Удовлетворительные результаты определения кальция в водных вытяж¬
ках, имеющих небольшой сухой остаток, можно получить, пользуясь вод¬
ными эталонами кальция. Сильноминерализованные вытяжки следует раз¬
бавлять, если определяемая концентрация кальция не слишком мала — это
повышает точность определения. Кальций можно определять и по другой
прописи: после осаждения его и переведения осадка в раствор.
Следует учитывать, что точность анализа водных вытяжек, содержащих
органическое вещество, ухудшается вследствие фона в спектре пламени и
изменения вязкости анализируемого раствора. При больших концентрациях
органического вещества вытяжки следует разбавлять, если концентрации
определяемых элементов не слишком малы, или удалять мешающее органи¬
ческое вещество.
Анализ солевых вытяжек. При определении обменных
оснований, вытесненных 1 н. раствором уксуснокислого или хлористого
аммония, полученная вытяжка без какой-либо предварительной подготов¬
ки поступает на пламенно-фотометрический анализ. Концентрация фильт¬
рата по аммонию практически совпадает с его исходной концентрацией, по¬
этому эталонные растворы предпочтительнее готовить на 1 н. растворе ук¬
суснокислого или хлористого аммония. Анализ аммонийных вытяжек по вод¬
ным эталонам приводит к некоторому занижению аналитических данных.
Анализ кислотных вытяжек. При анализе кислотных вы¬
тяжек проявляется влияние кислоты (концентрация, характер аниона) и зна¬
чительных количеств полуторных окислов, особенно алюминия. Кислоты,
как правило, понижают интенсивность излучения спектра. Этот эффект за¬
висит от характера аниона и возрастает с концентрацией кислоты. Анализ
таких вытяжек выполняют по эталонам, составленным с учетом концентра¬
ции кислоты и ее природы. Определение щелочных элементов не представ¬
ляет трудностей. Вытяжки следует разбавлять, если концентрация опреде¬
ляемых элементов не слишком мала. Анализ слабокислых вытяжек (до 3%)
проводят по водным эталонам. Определение кальция можно выполнять пос¬
ле его осаждения и растворения осадка.
Спектральные методы
371
Определение емкости поглощения почв. При
определении емкости поглощения почвы по универсальному методу Гедрой-
ца почвы насыщают натрием, который затем вытесняют кальцием (пользуют¬
ся двууглекислым кальцием) в присутствии воды и в токе углекислоты. Из¬
мерение емкости поглощения сводится к определению вытесненного натрия
спектральным методом.
Определение емкости поглощения почв по Бобко и Аскинази основано на
насыщении почв хлористым барием с последующим вытеснением его 1 н.
раствором соляной кислоты. Определение бария спектральным методом вы¬
полняют по его полосе в области 8700 А. Эталоны составляют на основе 1 н.
соляной кислоты.
Мешающие влияния кальция, алюминия и других элементов на опре¬
деление бария устраняются предварительной обработкой почвы соляной
кислотой перед насыщением хлористым барием.
Анализ вод. Методы пламенно-фотометрического определения
натрия, калия, кальция и других элементов в естественных водах принци¬
пиально не отличаются от описанных выше методов анализа почвенных вы¬
тяжек. Анализ слабоминерализованных вод можно выполнять по водным
эталонам. В случае высокой минерализации вод их следует разбавлять. Опре¬
деление щелочных элементов в водах, как правило, не встречает трудностей.
Наличие в водах алюминия, фосфора, серы осложняет определение
щелочноземельных элементов и заставляет прибегать к приемам, изложен¬
ным выше.
Анализ растений. Растения озоляют сухим или мокрым ме¬
тодом. Золу обрабатывают соляной кислотой и переводят в водный раствор,
который и поступает на анализ. Определение калия можно выполнять по
водным эталонам, без добавления других элементов. Ионизационный эффект
при определении калия в присутствии натрия не проявляется, так как нат¬
рий входит в состав золы в количествах, значительно меньших, чем калий.
При определении натрия ионизационный эффект может проявляться, поэ¬
тому в эталоны для определения натрия необходимо вводить калий. Содержа¬
ние кальция в золе не оказывает заметного влияния на определение калия г
но существенно при определении натрия. В этом случае применяют методы
компенсации, удаления кальция и другие приемы.
В состав растений входят фосфор, сера и алюминий, причем содержание
фосфора сравнимо с содержанием кальция. Присутствие перечисленных эле¬
ментов в анализируемом растворе приводит к занижению результатов опре¬
деления кальция. Поэтому при определении кальция в растениях применяют
следующие дополнительные операции: предварительное осаждение кальция
и последующее переведение его в раствор, предварительное осаждение ме¬
шающих элементов, введение значительных количеств мешающих элементов
в анализируемые растворы и эталоны в целях выравнивания состава рас¬
творов.
Анализ удобрений. Выбор приемов анализа удобрений и ис¬
ходного сырья для его производства определяется основной их компонентой.
Так, в калиевых удобрениях основной их компонентой служит калий, а нат¬
рий и кальций — примеси. Высокая концентрация калия допускает очень
широкое разведение анализируемого раствора; анализ может быть выполнен
по водным эталонам калия. Натрий и кальций, ввиду их малых концентра¬
ций, не оказывают влияния на результаты анализа. Однако на определение
натрия значительное влияние может оказать калий (ионизационный эффект) г
поэтому в эталоны вводят соответствующие количества калия или же выпол¬
няют анализ по методу добавок.
Присутствие кальция в значительных концентрациях может повлиять
на результаты определения натрия. В этом случае прибегают к методу ком¬
пенсации или другим приемам, описанным выше.
372
Д. Я. Иванов, Л. А. Лернер
В этой статье изложены лишь основные принципы пламенно-фотометри¬
ческого метода и его практического применения при выполнении наиболее
распространенных видов почвенно-агрохимического анализа. Этим далеко
не исчерпывается круг вопросов пламенно-фотометрического анализа. Хо¬
тя существующие методы позволяют простыми средствами определять ряд
химических элементов в агрохимических объектах, интересующих иссле¬
дователей, однако в аналитической практике часто встречается необходимость
разработки новых методов или уточнения существующих, исходя из особен¬
ностей объектов исследования, назначения анализа, имеющейся аппаратуры
и т. п. Успешное решение подобных задач требует знания физических ос¬
нов метода.
АТОМНО-АБСОРБЦИОННЫЙ МЕТОД
Аппаратура
Атомно-абсорбционные аналитические приборы (спектрофотометры) име¬
ют следующие основные узлы: источник света, поглощающую ячейку, опти¬
ческое устройство, приемное и регистрирующее устройство.
Источники света. Основными источниками света при атомно¬
абсорбционном анализе служат лампы с полым катодом. Полый катод лам¬
пы изготовляется в виде цилиндра из чистого металла или из трудно распы¬
ляемого вещества с нанесенным на его внутреннюю поверхность материалом,
в состав которого входят те элементы, излучение спектра которых необхо¬
димо получить для проведения анализа.
Лампы с полым катодом питают постоянным, высокочастотным или им¬
пульсным током. Чр езмерное увеличение силы тока, питающего лампу, при¬
водит к нежелательному увеличению самопоглощения в лампе и доплеров-
скому уширению линий испускания.
На аналитические возможности метода неблагоприятное влияние оказы¬
вают паразитные сигналы, регистрируемые совместно с полезным сигналом,
вызванные излучением посторонних линий спектра материала катода и га¬
за, заполняющего лампу. Чувствительность метода снижается также тем,
что на приемник излучения поступает не только аналитическая линия, ин¬
тенсивность которой зависит от концентрации элемента в пробе, но и излуче¬
ние участков сплошного фона, расположенного по обе стороны от аналити¬
ческой линии.
Кроме ламп с полым катодом, в практике атомно-абсорбционного ана¬
лиза применяются высокочастотные безэлектродные лампы, разряд в кото¬
рых возникает при введении лампы в магнитное поле катушки высокочас¬
тотного генератора. Применяются и другие высокоинтенсивные стабильные
лампы.
Выпускаются одноэлементные и многоэлементные лампы. Одноэлемент¬
ные лампы позволяют определять один элемент в пробе — тот, линию
спектра которого излучает лампа, облучающая пробу.
Многоэлементные лампы позволяют последовательно определять ряд
элементов. Полый катод в такой лампе изготовлен из сплава этих элементов
или из отдельных колец тех металлов, которые надлежит определять в про¬
бах.
Поглощающая ячейка. Назначение поглощающей ячейки
состоит в том, чтобы перевести анализируемое вещество в такое состояние,
при котором определяемые элементы будут находиться в виде свободных
атомов, способных поглощать (абсорбировать) свет внешнего источника.
В практике атомно-абсорбционного метода анализа в качестве поглощаю¬
щей ячейки наиболее широкое применение получили различного рода газо¬
Спектральные методы,
373
вые пламена. Для этого используются горючие газы, светильный, пропан,
бутан, ацетилен, водород и др. Окислителями при горении служат кислород,
который поступает в чистом виде или как составная часть атмосферного
воздуха, закись азота и некоторые другие газы.
В настоящее время усиленно разрабатываются непламенные методы ато-
мизации анализируемых проб, повышающие чувствительность определения
по сравнению с пламенем и позволяющие анализировать непосредственно,
без предварительной обработки, твердые образцы.
Оптическое устройство. Оптическое устройство в общем
случае включает в себя спектральный прибор (монохроматор или свето¬
фильтры), служащий для выделения аналитической линии источника, осве¬
тительные линзы, диафрагмы и вспомогательные зеркала, удлиняющие путь,
который проходит световой пучок источника через поглощающую ячейку.
К дисперсии спектрального прибора целесообразно предъявить требо¬
вания, основанные на реальной необходимости в ряде аналитических задач
выделять участки спектра в 1 А. Тогда при ширине щели 0,01 мм величина
обратной дисперсии прибора не должна превосходить 50 А /мм. От дисперсии
спектрального прибора зависит отношение интенсивности «шумового» сиг¬
нала к интенсивности аналитической линии. При возрастании дисперсии
спектрального прибора это отношение будет уменьшаться.
Приемно-регистрирующее устройство. Приемное
устройство включает в себя приемник света — фотоэлектронный умножитель
и электрические схемы его питания и усиления фототока; регистрирующее
(отсчетное) устройство может предусматривать визуальный отсчет, содержать
самописец или цифровой указатель с соответствующими электрическими схе¬
мами их питания. Сюда можно отнести и печатающее устройство для выда¬
чи результатов анализа.
При измерении малых оптических плотностей (малых концентраций)
применяют метод расширения шкалы (в 5—100 раз), позволяющий компен¬
сировать большую часть сигнала с помощью стабильного источника тока,
а регистрацию оставшейся части сигнала растянуть на всю шкалу прибора,
что уменьшает цену деления, увеличивает отсчет, соответствующий единице
концентрации определяемого элемента. Следует иметь в виду, что расшире¬
ние шкалы приводит к повышению точности анализа лишь в той мере, в ка¬
кой уменьшаются погрешности отсчета показаний прибора.
В настоящее время выпускается значительное число типов атомно-аб-
сорбционных спектрофотометров. Некоторые из них пригодны и для эмис¬
сионной пламенной фотометрии. Большинство атомно-абсорбционных спект¬
рофотометров рассчитано на применение в видимой и ультрафиолетовой об¬
ластях спектра приблизительно до 2000 А. Это не позволяет выполнять пря¬
мое определение инертных газов, водорода, кислорода, галогенов, углерода,
азота, серы и фосфора, так как длины волн их резонансных линий короче
2000 А.
Методы фотометрирования и анализа
В основе атомно-абсорбционного метода лежит явление селективного по¬
глощения света атомами определяемого химического элемента.
Линии испускания и поглощения не чисто монохроматические, они имеют
некоторую ширину Av. В пределах этого интервала частот (длин волн)
имеется определенное распределение энергии.
Уширение линий, в первую очередь линий испускания, снижает чув¬
ствительность атомно-абсорбционного метода анализа.
Атомное поглощение света подчиняется закону Бугера — Ламберта —
Бера.
I ^1ое-к>*п, (12)
374
Д. Н. Иванов, Л. А, Лернер
где /0 — интенсивность падающего света; I — интенсивность света, прошедшего через
поглощающий слой вещества; — коэффициент поглощения, зависящий от длины вол¬
ны падающего света; С — концентрация атомов в пламени элемента, поглощающего из¬
лучение данной длины волны; I — длина поглощающего слоя; е — основание натураль¬
ных логарифмов.
Выражение (12) можно представить в виде:
0 = 0,43А\С/, (13)
где D = lg (/o'-/). Величина D называется оптической плотностью вещества.
Согласно выражению (13), оптическая плотность поглощающего слоя (пламе¬
ни) должна быть пропорциональна концентрации атомов содержащегося
в нем элемента, поглощающего данную длину волны. Опыт показывает, что
пропорциональность сохраняется в некотором интервале концентраций, за¬
висящем от характера определяемого элемента и свойств лампы, применяе¬
мой в качестве источника излучения.
Градуировочный график строят в координатах (Z), С), по этому графику
и определяют неизвестную концентрацию С.
Атомно-абсорбционный анализ можно выполнять не только по методу
градуировочного графика, но и по методам ограничивающих стандартов,
добавок, прямого отсчета концентраций. Следует иметь в виду, что метод
добавок применим в интервале концентраций определяемого элемента, для
которого справедлива формула (13), т. е. прямая пропорциональная зависи¬
мость между концентрацией и оптической плотностью.
Характеристика метода
Существуют различные способы выражения чувствительности атомнсг-
абсорбционного метода анализа. Чаще всего за чувствительность метода при¬
нимают такую концентрацию определяемого элемента, которая вызывает
1% поглощения светового пучка лампы (табл. 10).
Часто для характеристики метода пользуются величиной «предел обна¬
ружения», под которым подразумевают концентрацию определяемого эле¬
мента, вызывающую сигнал, в 2 раза превышающий шумовой сигнал атом¬
но-абсорбционной установки. Чувствительность и предел обнаружения
обычно выражают в микрограммах растворенного элемента на миллилитр
растворителя (мкг/мл).
Нельзя утверждать, что чувствительность атомно-абсорбционного метода
во всех случаях принципиально значительно выше, чем у спектрографичес¬
кого и пламенно-фотометрического метода. Однако атомно-абсорбционный
метод создает более широкие возможности решения аналитических задач,
чем пламенно-фотометрический метод, и часто позволяет выполнять анали¬
зы с большей точностью, чем спектрографический метод.
Почва и растения содержат некоторые микроэлементы в концентрациях,
близких или ниже границы чувствительности метода, поэтому их определе¬
ние требует предварительного обогащения пробы. Концентрирование малых
примесей с применением органических растворителей и соосадителей может
быть весьма эффективным. Выбор применяемых органических растворителей
зависит от особенностей решаемой аналитической задачи и от намеченного
плана проведения анализа данной пробы. Следует в первую оче¬
редь рекомендовать метод экстракции, позволяющий одновременно скон¬
центрировать несколько определяемых элементов в органическом раство¬
рителе.
Весьма перспективно, особенно при определении очень малых примесей,
концентрирование микроэлементов с применением органических соосади¬
телей (Иванов и др., 1965). В случае концентрирования величину анализи¬
руемой навески необходимо брать в зависимости от предполагаемых содер-
Спектральные методы
375
Таблица 10
Чувствительность определения элемента атомно-абсорбционным методом
Элемент
Линия, A
Чувствитель¬
ность,
мкг/мл/i %
Газовая
смесь *
Элемент
Линия, A
Чувствитель¬
ность,
мкг/MAjl %
Газовая
смесь *
Ag
3280,68
0,06
11
Na
5889,95
0,03
III
А1
3092,71
1,00
I
Nb
3349,06
—
I
As
1890,42
2,00
II, IV
Nd
4634,23
35,00
I
Au
2427,95
0,30
II
Ni
2320,03
0,10
II
В
2497,73
50,00
I
Os
2909,06
—
I, II
Ва
5535,48
1,00
I, II
Pb
2833,06
0,50
II
Be
2348,61
0,02
I
Pd
2476,42
0,30
II
Bi
3067,72
0,50
II
Pr
4951,36
72,00
I
Са
4226,73
0,03
I, II
Pt
2659,45
3,00
11
Cd
2288,02
0,02
11
Rb
7800,23
0,10
III
Со
2407,25
0,15
II
Re
3460,46
12,0
I
Сг
3578,69
0,15
II
Rh
3434,89
0,40
II
Cs
8521,10
0,15
III
Ru
3498,95
2,00
II
Си
3247,54
0,07
II
Sb
2175,89
1,00
II
»У
4211,72
1,50
I
Sc
3907,49
1,00
I
Ег
4007,97
1,50
I
Se
1960,26
0,50
II, IV
Ей
4594,03
1,40
I
Si
2516,11
5,00
I
Fe
2483,27
0,15
II
Sm
4296,74
20,00
I
Gd
3684,12
38,00
I
Sn
2863,33
2,00
II, IV
Ga
2874,24
2,00
II
Sr
4607,33
0,06
I, II
Ge
2651,58
2,50
I
Та
2647,47
11,00
I
Hf
2866,58
2,50
I
Те
4326,47
—
I
Ho
4103,84
2,20
I
Те
2142,75
0,30
II
In
3039,36
0,40
II
Ti
3642,68
3,50
I
Ir
2924,78
1,00
II
Т1
2767,87
0,50
II
К
7664,91
0,03
III
Тш
4094,19
—
I
La
3574,91
100,00
I
и
3584,88
120,00
I
Li
6707,84
0,03
III
V
3183,98
1,50
I
Lu
3359,56
12,00
I
W
2551,35
5,30
I
Alg
2852,13
0,01
II
Y
4102,38
5,00
I
Mn
2704,82
0,05
II
Zn
3987,98
0,25
I
Hg
2536,52
2,00
II
Zn
2138,56
0,02
II
Mo
3132,59
0,40
I, II
Zr
3601,19
15,00
I
“*1 — закись азота — ацетилен; II — воздух — ацетилен; III — воздух — пропан (бутан); IV —
воздух — водород.
жаний, чувствительности и числа определяемых элементов. Соответственно
величина объема концентрата должна позволять выполнить определения
одного или нескольких элементов. Указанные обстоятельства взаимосвя¬
заны.
Количество раствора, необходимое для выполнения одного определения,
равно приблизительно 1 мл. При введении в атомизатор твердых проб, а так¬
же при работе с поглощающими ячейками некоторых конструкций требует¬
ся несколько миллиграмм анализируемого вещества.
Точность анализов, выполняемых атомно-абсорбционным методом, зави¬
сит от ряда факторов. К ним относятся стабильность излучения источника
376
Д. Н. Иванов, Л. А. Лернер
света (лампы), стабильность работы поглощающей ячейки, величина шумов
приемно-усилительной аппаратуры и регистрирующего устройства, вариа¬
ции физико-химических свойств анализируемых растворов, физико-химичес-
кие процессы в пламенщ д уровень определяемых концентраций элемента.
Важной практической характеристикой метода является также его про¬
изводительность. Обычно она лимитируется не конечными операциями ана¬
лиза, а продолжительностью подготовки пробы. В рассматриваемом ва¬
рианте метода переведение пробы в раствор занимает существенную часть
времени анализа.
Применение метода к анализу почв и растений
Рассмотрим некоторые общие вопросы применения атомно-абсорбцион¬
ного метода к определению макро- и микроэлементов при некоторых видах
почвенно-агрохимического анализа. В табл. 1 приведены данные о возможно¬
стях этого метода при определении макроэлементов, когда их высокие кон¬
центрации в анализируемых пробах облегчают выполнение анализа. Однако
во всех случаях исследуемые образцы содержат, помимо определяемых,
мешающие элементы в значительных концентрациях, влияние которых не¬
обходимо устранить.
В табл. 2 приведены данные, относящиеся к определению микроэлемен¬
тов в почвах и почвенных вытяжках. Из таблицы видно, что необходимо
проводить предварительное концентрирование некоторых микроэлементов,
без чего они в ряду анализируемых объектов или пробах не могут быть
определены.
В настоящее время разработаны основные приемы атомно-абсорбционно¬
го определения многих макро- и микроэлементов в почвах. Общие положения
методик справедливы при использовании различных атомно-абсорбционных
спектрофотометров, но вполне возможно, что в практике работы возникнет
необходимость в уточнении методики в зависимости от применяемого прибора
и объекта анализа. Приведем некоторые частные методики анализа, в кото¬
рых применены различные приемы устранения влияния общего состава про¬
бы на результаты анализа.
Определение макроэлементов. Д. Н. Иванов,
JI. А. Лернер и Э. И. Тихомирова (1975) определяли общее содержание Si,
Al, Fe, Са, Mg, Мп после сплавления пробы со смесью буры и соды, которое
позволяет полностью разложить почвенную пробу и получить устойчивый
раствор всех элементов, в том числе и кремния.
6 г почвы, прокаленной в платиновом тигле при 600° С, смешивают
со смесью буры и соды (1 : 2) и сплавляют в муфеле при температуре 950—
1000° С в течение 40 мин. Горячий тигель вынимают из муфеля и вращатель¬
ными движениями распределяют плав по стенкам тигля. Охлажденный ти¬
гель помещают в стакан емкостью 500 мл, заливают 250 мл кипящей воды,
нагревают на водяной бане до полного выщелачивания плава. При энергич¬
ном помешивании приливают 100 мл НС1 (1:3). Раствор доводят до объема
500 мл.
Состав пробы влияет на результаты определения всех указанных эле¬
ментов, поэтому для устранения влияния применяют буферирование, фо-
тометрирование в строго определенном виде пламени и в тех его областях,
где помехи от посторонних элементов минимальны. Содержание буры и соды
в эталонах делают равным содержанию в пробах.
Определение Si, Al, Са, Mg выполняют в пламени закись азота — ацети¬
лен, Мп — в пламени воздух — ацетилен, определение Fe возможно и в
том, и в другом пламени.
При определении кремния производят буферирование алюминием, ко¬
нечную концентрацию которого устанавливают в пробах и эталонах равной
Спектральные методы
377
2 мг/мл. Вследствие весьма критической зависимости чувствительности оп¬
ределения от условий анализа применяют четырехкратные измерения од¬
ного образца по методу ограничивающих стандартов. Алюминий определяют
после буферирования лантаном в концентрации 4 мг/мл. При определении
железа в пламени ацетилен — воздух применяют буферирование лантаном в
концентрации 500 мкг/мл, при измерении в пламени ацетилен — закись
азота определение проводят по эталонам, содержащим только железо. Оп¬
ределение кальция и магния проводят после буферирования лантаном в
концентрации 2 мг/мл. Буферирование стронцием не полностью устраняет
влияние кремния. Марганец определяют без буферирования, но измерения
проводят в области пламени, на расстоянии 10—14 мм от верхней пластины
горелки.
Определение титана при соотношении пробы и раствора (1 : 500) невоз¬
можно из-за небольшого содержания его в почвах и низкой чувствительности
определения.
В указанных работах проводили определение общего содержания Al, Fe,
Ti, Ga, Mg также после кислотного разложения почвы, при этом выбор]кислот
обусловливается аналитической задачей. При определении алюминия, же¬
леза возможно разложение смесью плавиковой и серной кислот. При необ¬
ходимости определения в одном и том же растворе дополнительно кальция,
магния и титана почву разлагают плавиковой и соляной кислотой.
При использовании пламени закись азота — ацетилен определение ти¬
тана проводят после буферирования алюминием в концентрации 2 мг/мл,
алюминия — после буферирования калием в концентрации 1 мг/мл. Опре¬
деление кальция и магния выполняют после буферирования стронцием в
концентрации 10 мг/мл, возможно буферирование лантаном. Измерения про-"
водят в пламени воздух — ацетилен.
Определение обменных кальция и магния в почвенных вытяжках и во¬
дах выполняют в присутствии в пробах и эталонах 1,5—2 мг/мл стронция
(лантана). Атомно-абсорбционный метод вполне допускает определение нат¬
рия, калия и других щелочных элементов, но при наличии пламенного фо¬
тометра оно, как правило, нецелесообразно. Применение атомно-абсорб¬
ционного метода вполне оправдано, например, при определении малых ко¬
личеств рубидия в присутствии больших количеств калия, которые имеют
близкие длины волн спектральных линий, и их разделение фильтровым пла¬
менным фотометром просто невозможно
Определение микроэлементов. А. М. Юр и P. JI. Мит¬
челл (Ure, Mitchell, 1967) определяли кобальт в 0,5 н. уксуснокислой вы¬
тяжке из почв. Применяли воздушно-бутановое пламя и высокоинтенсивную
лампу с полым катодом, обеспечивающую большую чувствительность ана¬
лиза и широкий прямолинейный участок градуировочного графика. В при¬
боре предусмотрена модуляция светового потока.
При малых концентрациях кобальта (< 1,25 мкг/мл) авторы наблюдали
возрастание его поглощения в присутствии кальция и алюминия, причем
кажущееся поглощение излучения по линии Со 2470 А наблюдали и при вве¬
дении в пламя растворов, содержащих только кальций и алюминий. При
больших концентрациях кобальта кальций уменьшает абсорбцию линии
спектра кобальта. В общих случаях влияние возрастает с повышением кон¬
центрации кальция.
Анализы проводили по серии эталонов, содержащих 0,3% кальция,
0,5% алюминия и 0,03 н. соляной кислоты, 50% этанола и 50% воды (по
объему).
Необходимость корректировки при определении кобальта устанавливает¬
ся также по содержанию кальция, которое в свою очередь приближенно оп¬
ределяют пламенным фотометром, работающим одновременно с атомно¬
абсорбционным спектрофотометром.
378
Д. Н. Иванов, Л. А. Лернер
Л. А. Лернер и Д. Н. Иванов (1970 а, б) определяли общее содержание цин¬
ка, меди, кобальта и марганца в почвах. Работа выполнялась на однолучевом
атомно-абсорбционном спектрофотометре АА-4 фирмы «Техтрон». Растворы
атомизировались воздушно-пропановым и воздушно-ацетиленовым пламе¬
нами; горелка — щелевая (длина щели 10 см)\ источники света — лампы с
полым катодом с электрической модуляцией светового потока. Почва (2 г)
прокаливалась и разлагалась смесью серной и плавиковой кислот. Остаток
растворялся и объем пробы доводился до 50 мл.
Градуировочные графики прямолинейны в интервале концентраций ме¬
ди 0—20 мкг/мл, кобальта 0—15 мкг/мл, марганца 0—10 мкг/мл, цинка
0—2 мкг/мл.
Присутствие в растворе эталонов посторонних элементов (Са, Mg, Nar
Al, Fe) в больших концентрациях оказывает двоякое влияние на положение
градуировочных графиков. Во-первых, главным образом, вследствие не¬
селективных помех, происходит параллельное смещение градуировочного
графика относительно построенного по эталонам, не содержащим посторон¬
них элементов. Во-вторых, изменяется наклон градуировочного графикаг
что связано с возникновением физических и химических помех (изменение
физических свойств раствора, а следовательно, и процессов поступления
раствора в пламя, изменение дисперсности аэрозоля и т. д.).
При определении кобальта, если его содержание в пробе находится в ин¬
тервале концентраций 0—0,5 мкг/мл, отсутствие учета неселективного пог¬
лощения, вызванного посторонними элементами, может быть источником
ошибки, доходящей до 100%, в интервале концентраций кобальта 0,5—
1,0 мкг/мл ошибка еще велика — 30%, в интервале 1,0—2,0 мкг/мл кобальта
она еще заметна — до 8%.
При относительно больших содержаниях цинка и марганца в пробе по¬
грешность анализа, вызванная посторонними элементами, становится пре¬
небрежимо малой.
Влияние неселективного поглощения в принципе можно было бы уст¬
ранить применением эталонов, по составу близких к пробам, однако поч¬
венно-агрохимические исследования требуют проводить анализ проб неиз¬
вестного и переменного состава.
Методы химического концентрирования позволяют отделить определяе¬
мые элементы от значительной части основы пробы, однако эти приемы не-
сколька удлиняют подготовку пробы к анализу.
В обсуждаемой работе был применен оптический метод учета неселектив¬
ного поглощения. Например, в области Со 2407 А это производится следую¬
щим способом.
Раствор распыляют в пламя п измеряют его оптическую плотность Dx по линии Со
2407 А. Полученное значение есть сумма оптических плотностей, соответствующих атом¬
ному поглощению определяемого элемента D и неселективному поглощению света D2*
При этом измерении источником света служит лампа с полым катодом, излучающая спектр
определяемого элемента. Затем заменяют источник света — ставят водородную лампу
ДВС-25, свет которой прерывают механическим модулятором. Снова распыляют тот же
раствор в пламя и измеряют его оптическую плотность D2> соответствующую неселектив¬
ному поглощению в той области спектра, где расположена аналитическая линия Со
2407 А. Все остальные условия измерений: оптическая схема, напряжение на фотоумножи¬
теле, расход газа и воздуха — остаются без изменения.
Оптическая плотность пламени D, соответствующая неизвестной концентрации оп¬
ределяемого элемента — С, равна:
D=Di — D2. (14)
Искомая концентрация определялась по графику, который построен в координатах
(D, С) по эталонным растворам. Измерения показали, что при соотношении почвы и раст¬
Спектральные методы
379
вора 1 : 25 ошибки, связанные с изменением наклона графика, несущественны по срав¬
нению с ошибками, обусловленными неселективным поглощением; ими можно пренебречь
и считать, что в области концентраций 0—1 мкг/мл графики, построенные по чпстым эта¬
лонным растворам и по растворам с введенными посторонними элементами, расположены
параллельно.
Впоследствии для учета неселективных помех авторами была применена
многоэлементная лампа с полыми катодами из железа, кобальта и меди, ко¬
торая позволила учитывать неселективное поглощение по результатам изме¬
рения интенсивности непоглощаемых в пламени линий спектра определяе¬
мого элемента или линий спектра другого элемента, например железа. При¬
менение многоэлементной лампы для учета неселективных помех имеет бес¬
спорные преимущества по сравнению с дейтериевой или водородной лампой
(Лернер и др., 1972). Таким путем исключаются погрешности фотометри-
рования, связанные с возможностью просвечивания не точно одного и того
же аналитического участка пламени излучением от двух различных источни¬
ков света.
Было показано, что соляная и азотная кислота до 1 н. в интервале кон¬
центраций 0—4 мкг/мл не оказывает заметного влияния на поглощение ана¬
литических линий меди, цинка, марганца и кобальта. Серная кислота
увеличивает поглощение кобальта и марганца и уменьшает поглощение
цинка.
Полученные данные показывают, что если перед распылителем стоит
шарик для увеличения дисперсности аэрозоля и отсекания крупных капель,
влияние уксусной кислоты на поглощение указанных элементов до 3,5 н. не
проявляется, а при дальнейшем повышении концентрации кислоты поглоще¬
ние определяемых элементов уменьшается. Если перед соплом не установлен
шарик, то поглощение линий определяемых элементов в присутствии уксус¬
ной кислоты возрастает.
Ацетатно-аммонийный буферный раствор в случае распылителя без ша¬
рика повышает поглощение всех указанных элементов.
При использовании распылителя с шариком происходит уменьшение пог¬
лощения всех четырех определяемых элементов. Уксуснокислый аммоний
понижает приблизительно на 10% поглощение всех определяемых элемен¬
тов независимо от того, применяется ли шарик перед распылителем или нет.
Авторы определяли содержание марганца, цинка, меди и кобальта не
только в почвах, но и в почвенных вытяжках. Были взяты верхние горизон¬
ты пяти широко распространенных типов почв: чернозема, серой лесной,
солонца, дерново-подзолистой и светло-каштановой. Изучались следующие
восемь вытяжек: 0,1 н. НС1, HN03, H2S04; 1 н. НС1, HN03; ацетатно-аммо¬
нийный буферный раствор (pH 4,8); 0,5 н. СН3СООН и 1 н. СН3СООХН4.
Соотношение между почвой и реагентом было 1 : 10, время встряхивания —
60 мин. Подготовка вытяжки к анализу заключалась только в фильтровании.
При определении кобальта в сильных вытяжках производился учет неселек¬
тивного поглощения.
Полученные данные анализа вышеприведенного ряда вытяжек, хотя и
имеют ориентировочный характер, показывают, что марганец можно опре¬
делять во всех этих вытяжках, цинк — в семи (кроме уксуснокислого аммо¬
ния), медь и кобальт только в двух (1 н. НС1 и 1 н. HN03). Таким образом,
определение меди, кобальта и частично цинка в слабых вытяжках потребует
предварительного концентрирования.
Результаты анализа почвенных вытяжек показали, что содержания мик¬
роэлементов в различных по характеру, но равной нормальной концентрации
вытяжках сильных кислот близки между собой, поэтому для выполнения
атомно-абсорбционного анализа нет необходимости приготовлять индиви¬
дуальные вытяжки для каждого определяемого элемента.
380
Д. Н. Иванов, Л. А. Лернер
JL А. Лернер и В. В. Недлер (1970), а также А. И. Бусев, В. М. Бырько
и JI. А. Лернер (1972) изучали условия предварительного концентрирования
меди и кобальта в целях их определения атомно-абсорбционным методом.
При выборе органических растворителей предъявлялись следующие тре¬
бования: растворитель не должен давать большого собственного поглощения
или излучения света; при его распылении пламя должно оставаться стабиль¬
ным; физические свойства растворителя должны быть такими, чтобы не ухуд¬
шать процесс распыления; растворитель должен иметь низкую растворимость
в воде; внутрикомплексные соединения определяемых элементов должны
быть хорошо растворимы и устойчивы в органической фазе.
Авторами использовались амилацетат, метилизобутилкетон (МИБК), ме-
тилбутилкетон (МБК) и изоамиловый спирт.
В качестве реагентов в работе служили дитизон, диэтилдитиокарбами-
нат натрия (ДЭДТК натрия) и гексаметилендитиокарбаминат гексаметилен-
аммония (ГМДТК-ГМА). Оптимальные условия экстракции приведены в
табл. 11.
Таблица 11
Оптимальные условия экстракции внутрикомплексных соединений меди и кобальта
X
Интервалы pH экстракции определяемых элементов
Концент¬
Маскиру¬
ющие ве¬
щества
&
Си
Со
Реагент
рация
реагента,
%
Время экс
ции, мин.
Изоами¬
ловый
спирт
Амилаце¬
тат
МБК
МИБК
Изоами¬
ловый
спирт
Амил¬
ацетат
МБК
МИБК
Дитизон
од
Тартрат
аммония
5-20
5-8
4—9
to
1
СО
2-9
00
1
со
6-8
6-8
00
1
ю.
ДЭДТК
натрия
0,17
Цитрат
аммония
10
—
2-9
—
2-9
—
2-9
—
2-9
ГМДТК-
ГМА
0,1-0,2
То же
15
2-10
2-10
2-10
00
1
со
6,5-9
Присутствие в растворе больших количеств кальция, магния, алюминия
и натрия практически не влияет на экстракцию указанных внутрикомплекс¬
ных соединений рекомендуемыми растворителями. При экстракции дитизо-
натов меди и кобальта из солянокислых и им подобных вытяжек с сильным
экстрагирующим действием в делительную воронку помещают 50—100 мл
вытяжки. Приливают 10—25 мл 1 М раствора тартрата аммония (объем ко¬
торого следует изменять в зависимости от предполагаемого суммарного со¬
держания макроэлементов в вытяжке). Затем добавляют по каплям раствор
аммиака, пока значение pH раствора не станет равным 7. Далее приливают
10 мл 0,1% раствора дитизона в амилацетате или метилизобутилкетоне,
встряхивают делительную воронку 10 мин., дают в течение 2—3 мин. вод¬
ной и органической фазам расслоиться, сливают водную фазу. Оставшийся
органический слой поступает на атомно-абсорбционный анализ.
Для анализа уксусно-аммонийного буферного раствора (pH 4,8) и подоб¬
ных ему вытяжек со слабым экстрагирующим действием следует использо¬
вать 150—300 мл вытяжки. Дальнейший ход экстракции такой же, как и
для сильных вытяжек.
При экстракции диэтилдитиокарбаминатов и гексаметилендитиокарба-
минатов меди и кобальта из вытяжек, отличающихся сильными экстракцион¬
ными свойствами, 50—100 мл вытяжки переносят в делительную воронку,
приливают 15—20 мл 25 %-ного раствора цитрата аммония и доводят аммиа¬
ком величину pH водного раствора до 8.
Спектральные методы
381
Если экстракция происходит с помощью ДЭДТК натрия, то вводят
0,3 %-ный водный раствор указанного реагента. Количество реагента устанав¬
ливают в зависимости от объема анализируемой водной фазы так, чтобы ко¬
нечная концентрация реагента в водной фазе равнялась 0,001 М. Приливают
10 мл амилацетата или метилизобутилкетона. При использовании для эк¬
стракции ГМДТК-ГМА вводят 0,1 %-ный раствор указанного реагента в
амилацетате или 0,2 %-ный раствор ГМДТК-ГМА в метилизобутилкетоне.
Проводят экстракцию в течение 10—15 мин. Полученные органические эк¬
стракты поступают на атомно-абсорбционный анализ. При экстракции ме¬
ди и кобальта из слабых вытяжек анализируемый объем водной фазы уве¬
личивают. Дальнейший ход экстракции такой же, как и для сильных вытя¬
жек.
В рассматриваемых работах установлено, что чувствительность опре¬
деления меди и кобальта в органических растворителях выше, чем в водных
растворах. В метилизобутилкетоне она возрастает для меди в 2,8 раза и для
кобальта в 3,4 раза.
В результате изучения дисперсности аэрозоля с применением каскад¬
ного импактора (Лернер, Русанов, Недлер, 1971) было установлено, что решаю¬
щим фактором, вызывающим увеличение чувствительности анализа при ис¬
пользовании органических растворителей, является увеличение степени
дисперсности анализируемого аэрозоля; увеличение скорости распыления
и количества раствора, поступающего в пламя в единицу времени, не всегда
сопутствует увеличению чувствительности определения.
JI. А. Лернер, Л. П. Орлова, Д. Н. Иванов (1971) для определения ряда
микроэлементов атомно-абсорбционным методом разработали ускоренный
метод разложения почвенной пробы в герметически закрытом фторопластовом
стакане смесью НС1 : HN03 (3 : 1) и H2F2. Авторы показали, что при спе¬
циально подобранных условиях предложенным методом можно разлагать
навески почвы до 0,9 г. Время разложения 40 мин., температура — 110° С.
После разложения пробы в раствор вводят борную кислоту для связывания
оставшейся свободной плавиковой кислоты и образования хорошо раствори¬
мых в воде фтороборатов металлов.
Мы кратко рассмотрели возможность определения элементов в почвенно¬
агрохимических объектах атомно-абсорбционным методом. Опыт показывает,
что некоторые элементы можно определять сравнительно легко, определе¬
ние других связано с некоторыми трудностями, а третьи пока вообще не мо¬
гут быть определены. Отнесение элементов к одной из этих трех групп имеет
условный и переходящий характер, оно отражает современное состояние уров¬
ня развития атомно-абсорбционного анализа и его техники, которые быст¬
ро развиваются и совершенствуются.
В заключение следует отметить, что в настоящее время разработаны тео¬
ретические и экспериментальные основы спектрального анализа, имеются
многочисленные частные методики определения спектральными методами
различных элементов в сплавах, породах и минералах.
Атомно-абсорбционные методы значительно расширяют возможности
и почвенно-агрохимического анализа. Хотя в почвоведении и агрохимии
спектральный анализ получил значительное применение, однако оно далеко
не соответствует имеющимся возможностям этого метода. Здесь предстоит
решить ряд сложных вопросов: изготовление стандартных образцов, созда¬
ние методов определения форм соединений элементов, разработка метроло¬
гии, рациональное сочетание с другими методами и др. Необходимы проверка
и улучшение существующих методов с учетом особенностей исследуемых
объектов, вида анализа и предъявляемых к нему требований.
382
Д. Я. Иванов, Л. Л. Лернер
Литература
Боровик-Романова Т. Ф. Введение, гл. I—
II.— В сб. «Спектральное определение
редких и рассеянных элементов». М., Изд-
во АН СССР, 1962.
Боровик-Романова Т, Ф., Грибовская И. Ф.
О спектральном определении лития, меди
и молибдена в растениях.—Агрохимия,
1966, № 9.
Боровик-Романова Т. Ф., Грибовская И. Ф.
Влияние валового состава пробы на ин¬
тенсивность линий исследуемых элемен¬
тов при спектральном анализе.— Агро¬
химия, 1967, № 5.
Брицке М. Э. Анализ металлургических
продуктов методом эмиссионной фотомет¬
рии пламени. М., «Металлургия», 1969.
Бусев А. И., Бырько В. М., Лернер Л. ^4.
Атомно-абсорбционное определение об¬
щего содержания и подвижных форм мик¬
роэлементов в почвах после концентриро¬
вания их экстракцией гексаметилендитио-
карбаминатом гексаметиленаммония. —
Ж. анал. химии, 1972, т. 27, вып. 3.
Зайдель А. Я. Основы спектрального ана¬
лиза. М., «Наука», 1965.
Зайдель А. Я., Прокофьев В. К., Райский
С. Л/. Таблица спектральных линий.
М.— Л., Гостехиздат, 1952.
Зырин Я. Г., Обухов А. //., Белицина
Г. Д. Методические указания по спектро¬
графическому определению микроэлемен¬
тов в почвах и золе растений. Изд-во
МГУ, 1971.
Иванов Д. Я. Взаимное влияние щелочных
элементов на интенсивность их спектра
в пламени.— Заводская лаборатория,
1941, т. 10, № 4.
Иванов Д. Я. Применение электронного
умножителя для определения щелочных
элементов.— Почвоведение, 1949, № 7.
Иванов Д. Я. Опыт применения интерфе¬
ренционных светофильтров для определе¬
ния натрия и калия в почвах.— Почвове¬
дение, 1953, № 1.
Иванов Д. Я. Некоторые вопросы пламен¬
но-фотометрического анализа.— Почво¬
ведение, 1962, № 4.
Иванов Д. Я. Пламенно-фотометрические
методы анализа растворов.— В сб. «Агро¬
химические методы псследования почв».
М., «Наука», 1965.
Иванов Д. Я. Методические указания по
определению калия, натрия, цинка, ме¬
ди, кобальта, молибдена и некоторых дру¬
гих элементов в почвах эмиссионным
спектральным методом. М., 1973а.
Иванов Д. Я. Методические указания по
определению кальция, магния, марган¬
ца, цинка, меди, кобальта и других эле¬
ментов атомно-абсорбционным методом.
М., 19736.
Иванов Д. Я. Спектральный анализ почв.
М., «Колос», 1974.
Иванов Д. Я., Иванова Я. Я., Орлова
Л. Я. Применение органических сооса-
дителей при определении микроэлемен¬
тов Со, Си, Ni, Sn, Cr, Pb, Mo, V, W в поч¬
вах.— Почвоведение, 1965, № 1.
Иванов Д. Я., Лернер Л. А., Тихомирова
Э. И. Методические указания по опреде¬
лению кремния, алюминия, железа, каль¬
ция, магния, титана и марганца в почвах
атомно-абсорбционным методом. М., 1975.
Иванов Д. Я., Орлова Л. Я., Филиппов
А. Я. Спектрохимический метод опреде¬
ления некоторых микроэлементов в поч¬
вах, растениях и водах.— Агрохимия,
1967, № 1.
Иванов Д. Я., Русаковаг Г. Г. К вопросу об
определении щелочноземельных эле¬
ментов пламенно-фотометрическим мето¬
дом. — Почвоведение, 1968, № 6.
Калинин С. /Г., Фаин 3. А. Спектральный
анализ минерального сырья. Алма-Ата,
Изд-во АН КазССР, 1962.
Калинин С. /Г., Явнель А. А., Алексеева
A. //., Марзуванов Б. Л., Неймарк
Л. 5. Атлас спектральных линий для
кварцевого спектрографа. М., Госгеол-
техиздат, 1959.
Кветкина А. А., Шлавицкая 3. И. Спект¬
ральный метод определения микроэле¬
ментов в почве и золе растений.— Вест¬
ник с.-х. науки, № 3. Алма-Ата, 1963.
Кузнецов В. И., Большакова Л. Jf., Без-
зольные органические соосадители.— Аг¬
рохимия, 1967, № 9.
Кузнецов В. И., Горшков В. В., Орлова
Л. Я. Суммарное соосаждение Со, Си,
Ni, Pb, Sn, Zn при их определении в поч¬
вах.— Агрохимия, 1964, № 2.
Кузнецов В. //., Орлова Л. Я., Синани
Т. И. Концентрирование молибдена при
его определении в оксалатных вытяжках
из почв.— Агрохимия, 1969, № 10.
Лернер Л. А,, Безлепкин А, И., Иванов
Я. Я., Баранов С. Б. Сравнительные ис¬
следования различных конструкций
спектральных ламп для атомно-абсорбци¬
онного анализа и применение двухраз¬
рядной многоэлементной лампы для учета
неселективных помех.— Ж. прикл. спект¬
роскопии, 1972, т. 16, № 15.
Лернер Л. i4., Иванов Д. Я. Определение
общего содержания цинка, меди, кобаль¬
та, марганца в почвах методом атомно¬
абсорбционной спектрофотометрии.— Аг¬
рохимия, 1970а, № 3.
Лернер Л. А., Иванов Д. Я. Определение
содержания подвижных форм меди, ко¬
бальта, цинка, марганца в почвах методом
атомно-абсорбционной спектрофотомет¬
рии.— Агрохимия, 19706, № 7.
Лернер Л. А., Недлер В. В. Атомно-аб-
сорбционное определение подвижных
форм меди и кобальта в почвах.— Почво¬
ведение, 1970, № 11.
Лернер Л. А., Орлова Л. Я., Иванов Д. Я.
Применение экспрессного метода разло¬
жения пробы при атомно-абсорбционном
определении валового содержания Си,
Zn, Ма в почвах.— Агрохимия, 1971,
№ 5.
Лернер Л. Д., Русанов А. Л., Недлер
B, В, К вопросу о влиянии органического
растворителя на чувствительность атом-
Спектральные методы
383
но-абсорбционного анализа.— Ж. анал.
химии, 1971, т. 26, вып. 9.
Лернер Л. А.у Тихомирова Э. И. Атомно¬
абсорбционное определение общего со¬
держания железа и алюминия в почвах.
— Почвоведение, 1974, № 6.
Лернер Л. А,, Тихомирова Э. И., Плотни¬
кова Л. Ф. Атомно-абсорбционное опре¬
деление общего содержания кальция,
магния и марганца в почве, разложенной
сплавлением со смесью буры и соды.—
Почвоведение, 1975, № 1.
Лернер Л, А., Тихомирова Э. И., Шостак
Р. В, Определение общего содержания
кремния в почвах атомно-абсорбционным
методом.— Почвоведение, 1973, № 12.
Львов Б. В. Атомно-абсорбционный спект¬
ральный анализ. М., «Наука», 1965.
Мандельштам С. Л. Введение в спект¬
ральный анализ. М.— JJ., Гостехиздат,
1946.
Митчелл Р. Л, Эмиссионный спектрохи¬
мический аналнз. Определение следов
элементов в растениях и других биологи¬
ческих объектах.— В сб. «Анализ следов
элементов». Пер. с англ. М., ИЛ, 1961.
Орлова Л. П. Применение органических
соосадителей для концентрирования мик¬
роэлементов Си, Со, Zn и Мо при их опре¬
делении в растениях.— Агрохимия, 1967.
№ 9.
Орлова Л. Я. Концентрирование Zn, Си,
Со и Мо с органическими соосадителями
при анализе почв, растений и природных
вод. (Автореф. канд. дисс.). М., 1969.
Полуэктов Н. С. Методы анализа по фото¬
метрии пламени. М., «Химия», 1967.
Прокофьев В, К. Фотометрические методы
количественного спектрального анализа
металлов и сплавов, ч. I, т. 2. М.— Л.,
Гостехиздат, 1951.
Риш М. А., Приев Я. М, Спектрально¬
аналитическое определение растительно¬
го материала без предварительного озо-
ления.— Труды Ин-та каракулеводства,
т. 10. Ташкент, 1960.
Русанов А. К. Основы количественного
спектрального анализа руд и минералов.
М., «Недра», 1971.
Славин В. Атомно-абсорбционная спектро¬
скопия. Л., «Химия», 1971.
Ure А. М., Mitchell R. L. The determina¬
tion of cobalt in soil extracts by atomic
absorption.— In «А study of interference
effect».— Spectrochim. acta, 1967, v. 23B„
79-96.
К. В. ВЕРИГИНА
МЕТОДЫ ОПРЕДЕЛЕНИЯ В ПОЧВЕ МЕДИ,
ЦИНКА, КОБАЛЬТА И БОРА
*
ВЗЯТИЕ ПОЧВЕННЫХ ОБРАЗЦОВ
И ПОДГОТОВКА ИХ К АНАЛИЗУ
При взятии в поле образцов почв для определения микроэлементов и
подготовке их к анализу необходимо соблюдать ряд предосторожностей. Ре¬
комендуется брать их или в полиэтиленовые мешочки или в восковку. Бу¬
мага «крафт» неприменима, когда предполагается проводить определение
бора, так как клей содержит бор и возможно значительное загрязнение
почвы бором. Мешочки из белой ткани перед взятием почвенных образ¬
цов должны быть тщательно промыты в бидистиллированной воде.
Высушивание почвенных образцов следует производить в чистом, сво¬
бодном от пыли помещении. Растирают почвенные образцы в агатовых или
фторопластовых ступках. Если образцы растирают механическими измель¬
чителями, то предварительно надо установить, не вносится ли при этом
загрязнений микроэлементами. Просеивают почву через шелковое или кап¬
роновое сито. При хранении образцы помещают в пластмассовые коробочки
или стеклянные банки.
ОПРЕДЕЛЕНИЕ ВАЛОВОГО СОДЕРЖАНИЯ МЕДИ, ЦИНКА
И КОБАЛЬТА ИЗ ОДНОЙ НАВЕСКИ ПО ВЕРИГИНОЙ (1974)
ПОСЛЕ РАЗЛОЖЕНИЯ ПОЧВЫ КИСЛОТАМИ
В настоящее время для определения меди, цинка и кобальта исполь¬
зуется как метод их последовательной экстракции дитизоном после разло¬
жения навески почвы кислотами и перевода остатка в раствор, так и опре¬
деление их в аликвотных частях раствора, полученного после разложения
почвы.
Преимуществом первого способа является то, что для его выполнения тре¬
буется разложение меньшей навески почвы (1—2 г), на что расходуется мень¬
ше времени; это важно, когда имеется ограниченное количество анализируе¬
мого материала (например, при анализе коллоидов). Кроме того, не всегда
можно качественно выполнить определение микроэлементов в аликвотной
части раствора без их предварительного отделения от присутствующих в
растворе мешающих элементов (Fe, Са и др.). Особенно это относится к оп¬
ределению цинка и кобальта.
Метод основан на разложении почвы после удаления из нее органичес¬
кого вещества прокаливанием, плавиковой кислотой в присутствии серной
кислоты. Остаток от разложения переводят в солянокислый раствор и из¬
влекают из него медь, цинк и кобальт в виде их дитизонатов: медь при pH
2,0, цинк и кобальт при pH 8,2. После разрушения дитизоната меди ее опре¬
деляют колориметрически в форме комплекса с диэтилдитиокарбаматом.
Цинк отделяют от кобальта разрушением дитизоната цинка слабой соляной
кислотой и определяют колориметрически в форме дитизоната цинка по ме¬
тоду одноцветной окраски. Кобальт после разложения дитизоната кобальта
определяют в форме комплекса с нитрозо-И-солью.
Почву высушивают до воздушно-сухого состояния и растирают в агатовой
ступке. Для почв глинистых и суглинистых берут навески в 1,5 г, для су¬
Методы определения в почве меди, цинка, кобальта и бора
385
песчаных и песчаных — 2—3 г. Навеску помещают в платиновую чашку и
прокаливают в течение 2 час. в муфеле при 500—550° С. Более высокой тем¬
пературы следует избегать, так как при этом возможна потеря микроэле¬
ментов. После прокаливания почве дают остыть, затем смачивают ее биди-
стиллированной водой, приливают 1 мл H2S04 (уд. вес. 1,84) и 20 мл трижды
перегнанной HF1. Чашку ставят на закрытую этернитовую плитку и держат
на ней до появления белых паров S03. Нагревание на плитке не должно быть
очень сильным во избежание разбрызгивания содержимого чашки.
После появления белых паров чашку снимают и почву обрабатывают но¬
вой порцией HF (10—15 мл). Затем чашку снова помещают на плитку и на¬
гревают до тех пор, пока остаток в чашке не станет совершенно сухим. Пос¬
ле этого переносят чашку на более сильно нагревающуюся плитку и про¬
должают нагревание до полного прекращения выделения белых паров. За¬
тем чашку снимают с плитки, дают остыть и для растворения остатка прили¬
вают 10 мл дважды перегнанной 22 %-ной НС1 и 10 мл бидистиллирован-
ной воды. Чашку снова помещают на плитку (не очень мощную) и держат на
ней в течение 30—40 мин.
Когда остаток растворится, раствор фильтруют через фильтр 2 (белая
лента) и промывают фильтр горячей бидистиллированной водой, подкислен¬
ной НС1, до исчезновения следов железа. Объем фильтрата и промывных вод
составляет обычно 40—50 мл. Если объем фильтрата и промывных вод будет
больше 50 мл, их следует упарить до этого объема. Из приготовленного таким
образом раствора выделяют сначала медь, а затем цинк и кобальт.
Определение меди
Полученный солянокислый раствор переносят в капельную воронку на
100 мл и приливают 10 мл 25 %-ного раствора лимоннокислого натрия. Ра¬
створ доводят аммиаком до pH 2,0 (по универсальной индикаторной бумаге).
Приливают из бюретки 10 мл 0,05%-ного раствора дитизона в СС14 и поме¬
щают воронку в специальный вертикальный встряхиватель. Встряхивают в
течение 15 мин., затем снимают с встряхивателя и ставят воронки в штатив.
Дают отделиться органической фазе от водной и после разделения фаз сли¬
вают органическую фазу, в которой находятся дитязон и дитизонат меди,
в стаканчик емкостью 50 мл. Приливают в воронку новую порцию дитизона
(5 мл) и опять встряхивают на встряхивателе 15 мин. Затем воронку ставят
в штатив, дают слоям разделиться и сливают дитизоновый слой в тот же
стаканчик. Обычно для извлечения меди бывает достаточно двух обработок
раствором дитизона в СС14. Водный раствор, оставшийся в капельной ворон¬
ке, употребляют для выделения цинка и кобальта, а собранные в стаканчик
дитизонатные экстракты в СС14 — для определения меди.
К собранному в стаканчик раствору дитизоната меди в СС14 прибавляют для
разложения дитизона 1 мл HN03,1 мл H2S04 и 2млН202 (или НС104, свободную
от следов меди) и помещают стаканчик на закрытую этернитовую плитку. На¬
гревают очень осторожно и постепенно, так как реакция протекает бурно.
Нагревание продолжают до тех пор, пока из стаканчика не испарится весь
раствор. Остаток в стаканчике вторично обрабатывают 1 мл HN03 и 2 мл
Н202 (или НС104) После окончания обработки очень небольшой'остаток, на¬
ходящийся на дне стаканчика, должен быть совершенно белым.
1 Одновременно с анализом почвы необходимо производить контрольный опыт со всеми
реактивами, употребляемыми в анализе, так как даже при самой тщательной очистке
реактивов приходится вносить поправку на загрязнение цинком и реже — кобальтом и
медью.
2 Фильтр предварительно промывают водой, подкисленной НС1, для удаления следов
микроэлементов.
13 Агрохимические методы исследования почв
386
К. В. Веригина
Остаток в стаканчике смачивают несколькими каплями (5—10) дважды
перегнанной HG1, приливают 10 мл бидистиллированной воды и нагревают
на плитке до полного его растворения. Раствору дают остыть, затем количе¬
ственно переносят его в капельную воронку на 100 мл, обмывая стаканчик
бидистиллированной водой. Приливают 1 мл 5 %-ного раствора лимоннокис¬
лого аммония и нейтрализуют по фенолфталеину аммиаком (до получения
бледно-розовой окраски). Приливают из бюретки точно 15 мл раствора ди-
этилдитиокарбамата свинца в СС14 и встряхивают на встряхивателе 10 мин.
После разделения фаз слой СС14, содержащий диэтилдитиокарбамат меди и
имеющий желтую окраску, сливают в пробирку с притертой пробкой. Так
как раствор диэтилдитиокарбамата меди часто бывает мутным из-за капелек
воды, то перед фотоколориметрированием его фильтруют через сухие малень¬
кие фильтры (белая лента) непосредственно в кювету фотоколориметра и фо-
токолориметрируют с синим светофильтром (453 нм), употребляя в качестве
нулевого раствора чистый СС14. При указанной навеске, применении 15 мл-
диэтилдитиокарбамата свинца в СС14 фотоколориметрирование производят
в кювете с толщиной просматриваемого слоя 10 мм.
Шкалу стандартных растворов меди приготовляют
из раствора, содержащего 10 мгк/мл Си. Для этой цели 0; 0,3; 0,5; 1,0; 1,5;
2,0 мл стандартного раствора меди (что соответствует 0; 3,0; 5,0; 10,0; 15,0-
и 20,0 мкг Си) помещают в капельные воронки. Приливают бидистиллнро-
ванную воду до объема 10 мл, 1 мл 5 %-ного раствора лимоннокислого аммо¬
ния, нейтрализуют аммиаком по фенолфталеину, приливают из бюретки точ¬
но 15 мл диэтилдитиокарбамата свинца в СС14 и далее извлекают медь таким
же образом, как и в случае испытуемых растворов.
По полученным для стандартных растворов оптическим плотностям строят
градуировочную кривую, по которой определяют содержание меди в испы¬
туемых растворах. Для этой цели на миллиметровой бумаге в системе
координат по оси абсцисс откладывают взятые количества меди, а по оси
ординат — оптическую плотность и соединяют полученные точки. Для опре¬
деления содержания меди в испытуемых растворах находят по градуировоч¬
ной кривой, какому количеству меди соответствует полученная для данного-
раствора величина оптической плотности. Для расчета содержания меди в
миллиграммах на 1 кг почвы найденное количество меди (в мкг) делят на взя¬
тую для анализа навеску почвы (в г).
Определение цинка и кобальта
Для экстракции цинка и кобальта к водному раствору, оставшемуся в
капельной воронке после выделения из него дитизоната меди, прибавляют
3—5 мл 25 %-ного раствора лимоннокислого натрия и раствор нейтрализуют
аммиаком по фенолфталеину до бледно-розовой окраски. После нейтрализа¬
ции к раствору прибавляют 10 мл 0,05 %-ного раствора дитизона в СС14
и встряхивают на вертикальном встряхивателе в течение 3 мин. Дают фазам
разделиться, сливают слой СС14 с растворенными в нем дитизонатами цинка
и кобальта, имеющими красную окраску, в чистую капельную воронку на
100 мл и повторяют обработку раствором дитизона в СС14 сначала порциями
по 10 мл, а затем, когда окраска дитизона будет изменяться мало, по 5 мл.
Дитизонаты цинка и кобальта сливают в ту же капельную воронку. Обработ¬
ку ведут до тех пор, пока дитизон не перестанет изменять свою первоначаль¬
ную окраску. Для полного выделения дитизонатов цинка и кобальта доста¬
точно 30—40 мл раствора дитизона в СС14. В некоторых случаях окраска
дитизона при выделении дитизонатов цинка и кобальта очень долго изменяет¬
ся (при 5—6 кратной обработке дитизоном), переходя в серую, что может
быть объяснено неполным связыванием железа в лимоннокислый комп-
Методы определения в почве меди, цинка, кобальта и бора
387
леке. В этом случае следует прибавить еще 3—5 мл 25%-ного раствора ли¬
моннокислого натрия и произвести экстракцию с раствором дитизона в
СС14; если окраска дитизона при этом останется зеленой, экстракцию можно
заканчивать.
Для разделения дитизонатов цинка и кобальта к собранным в капельную
воронку дитизонатам прибавляют пипеткой 50 мл 0,02 н. HG1 и встряхивают
в течение 3 мин. При этой обработке дитизонат цинка разрушается, и цинк
в форме хлористого цинка переходит в водную фазу. Дитизонат кобальта
0,02 н. НС1 не разрушается и количественно остается в органической фазе.
Раствор дитизоната кобальта в СС14 сливают в чистую капельную воронку
и повторяют обработку 0,02 н. НС1. Водную фазу после первого и второго
извлечения цинка сливают в одну капельную воронку и для удаления сле¬
дов дитизоната кобальта дважды промывают небольшими порциями (3—5 мл)
чистого СС14, сливая органическую фазу в термостойкий стакан на 100 мл.
Сюда же количественно переносят дитизонат кобальта в СС14 из капельной
воронки.
К собранным в стаканчик растворам дитизоната кобальта в СС14 прили¬
вают 1 мл HN03 (уд. вес. 1,4), 1 мл H2S04 (уд. вес. 1,84) и 2 мл 30%-ного рас¬
твора Н202 (или 2 мл НС104) и выпаривают досуха на закрытой этернитовой
плитке. Если после выпаривания остаток в стаканчике будет темноокрашен-
ным, необходимо повторить обработку Н202 или НС104.
После того как обработка будет закончена и остаток в стаканчике будет
совершенно белым, в стаканчик приливают пипеткой 1 мл 0,01 н. НС1, 1 мл
40%-ного раствора лимоннокислого аммония и 1 мл 0,2%-ного раствора нит-
розо-К-соли и нагревают на водяной бане в течение 5 мин., накрыв стаканчик
часовым стеклом. Температура раствора должна быть 60° С. Затем снимают
стаканчик с бани и приливают 1 мл HNOs (уд. вес 1,4) и 2 мл НС1 (уд. вес.
1,19). Накрывают стаканчик часовым стеклом и нагревают на водяной бане
при 60° С в течение 40 мин. для разрушения избытка нитрозо-К-соли. Через
40 мин. снимают стаканчик с бани и после охлаждения раствор из стаканчика
переносят в мерную колбу емкостью 25 мл, обмывая стаканчик бидистилли-
рованной водой.
Фотоколориметрируют окраску раствора комплекса кобальта с нитрозо-
R-солью, употребляя в качестве нулевого раствора бидисти л лированную
воду. При фотоколориметрировании пользуются кюветами с толщиной
просматриваемого слоя 20 мм и зеленым светофильтром (536 нм). Мы реко¬
мендуем пользоваться зеленым светофильтром, несмотря на то, что максимум
светопоглощения комплекса кобальта с нитрозо-И-солью лежит при длине
волны 413 нм, так как при длине волны 536 нм (зеленый светофильтр) влия¬
ние окраски нитрозо-К-соли минимально.
Для приготовления шкалы стандартных растворов
разбавляют стандартный раствор кобальта, содержащий 100 мкг/мл
Со, в 100 раз, чтобы получить раствор, содержащий 1 мкг/мл Со. В стакан¬
чики вводят пипеткой 2,0; 5,0; 7,5; 10,0; 15,0 и 20,0 мл этого раствора и вы¬
паривают досуха на водяной бане. Приливают по 1 мл 0,01 н. НС1, 40 %-ного
раствора лимоннокислого аммония и 0,2%-ного раствора нитрозо-К-соли.
Нагревают, как и в случае испытуемых растворов, на водяной бане 5 мин.
при 60° С, затем прибавляют по 1 мл HN03 и 2 дм НС1 и опять нагревают при
той же температуре в течение 40 мин. Снимают стаканчики с бани, охлаждают,
переносят их содержимое в мерные колбочки на 25 мл, доливая до метки би-
дистиллированной водой и фотоколориметрируют в тех же кюветах и с тем
же светофильтром, как и испытуемые растворы.
По полученным для стандартных растворов оптическим плотностям строят
градуировочную кривую, находят содержание кобальта в испытуемых ра¬
створах и рассчитывают содержание кобальта в почвах, как и при определе¬
нии меди.
13*
388
К. В. Веригина
Определение цинка в полученном растворе хлористого цинка производят
при помощи дитизона, методом одноцветной окраски. Для этого берут 5 или
10 мл раствора, полученного после отделения цинка от кобальта (для почв
глинистых и суглинистых достаточно 5 мл, для почв легкого механического»
состава — 10 мл), и помещают в капельную воронку на 100 мл; приливают
2 капли метилоранжа, перемешивают и приливают по каплям из градуиро¬
ванной пипетки 5%-ный раствор уксуснокислого натрия до перехода окрас¬
ки индикатора в желтую. Добавляют еще 0,2 мл раствора уксуснокислого
натрия (5 капель), перемешивают, приливают для связывания следов микро¬
элементов 5 мл 50%-ного раствора гипосульфита. Приливают 2 мл 0,05%-
ного раствора дитизона в СС14 и встряхивают на вертикальном встряхивате-
ле в течение 5 мин. Сливают дитизоновый слой в чистую капельную ворон¬
ку. Прибавляют новую порцию дитизона в СС14 (1 мл) и снова встряхивают
5 мин., сливая дитизоновый слой в ту же капельную воронку, куда был поме¬
щен первый экстракт. Обработку раствором дитизона продолжают до тех
пор, пока первоначальная зеленая окраска дитизона не перестанет изменять¬
ся. Собранные экстракты (растворы дитизоната цинка в СС14) для удаления
из них избытка дитизона обрабатывают 0,01 н. раствором аммиака. Для это¬
го в капельную воронку с дитизонатами цинка приливают 50 мл 0,01 н. раст¬
вора NH4OH и немедленно встряхивают 3 мин. Дают органическому слою
отделиться и сливают окрашенный в красный цвет раствор дитизоната цинка
в СС14 в сухую мерную колбочку на 25 мл, доливая до метки СС14. Измеряют
оптическую плотность раствора, помещая раствор в сухую кювету с толщи¬
ной просматриваемого слоя в 10 мм и применяя зеленый светофильтр
(536 нм). В качестве нулевого раствора применяют СС14.
Для приготовления шкалы стандартных растворов
разбавляют раствор сульфата цинка, содержащий 100 мкг/мл цинка, в
100 раз, чтобы разбавленный раствор содержал 1 мкг/мл цинка. В капельные
воронки помещают пипеткой 0; 0,5; 1,0; 2,0; 3,0; 4,0 мл раствора цинка, до¬
водят бидистиллированной водой до объема 10 мл, приливают 2 капли метил¬
оранжа, перемешивают, приливают по каплям из градуированной пипетки
5%-ный раствор уксуснокислого натрия до перехода розовой окраски ин¬
дикатора в желтую, добавляют еще 0,2 мл раствора уксуснокислого натрия,
приливают 5 лел50%-ного раствора гипосульфита. Экстрагируют дитизоиом,
как и в случае испытуемых растворов. Собранные дитизонаты цинка в СС14
обрабатывают 0,01 н. NH4OH, затем дитизонат цинка переносят в мерные
колбочки на 25 мл, доливают до метки СС14 и фотоколориметрируют, как и
испытуемые растворы. Шкалу стандартных растворов необходимо пригото¬
влять при каждом определении цинка. При приготовлении шкалы следует
пользоваться тем же раствором дитизона, который применяли для определе¬
ния цинка в испытуемых растворах.
Градуировочную кривую для определения цинка строят так же, как и
при определении меди и кобальта. Для вычисления содержания цинка в мил¬
лиграммах на 1 кг почвы найденное по градуировочной кривой содержание
цинка в испытуемом растворе в микрограммах умножают на фактор разве¬
дения (в том случае, если для определения брали 10 мл раствора из 100 мл.—
на 10, если 5 — на 20) и делят на взятую навеску (в г).
Реактивы. При определении микроэлементов необходимо обращать особое вни¬
мание на чистоту реактивов и бидистиллированной воды. При наличии реактивов «особой
чистоты» надобность в их очистке отпадает, но все же следует контролировать их чистоту.
При работе с «химически чистыми» и «чистыми для анализа» реактивами требуется ях
предварительная очистка.
При определении микроэлементов желательно вести работу в кварцевой посуде,
однако можно работать с посудой из бесцинкового стекла, безборного стекла и стекла
«Пирекс». Неприменима посуда из стекла марки «Шотт», так как в этом случае происходит
Методы определения в почве меди, цинка, кобальта и бора
389
загрязнение растворов цинком. Надо с большой осторожностью относиться к полиэтиле¬
новым сосудам, так как часто они вносят загрязнения цинком и медью.
1. Бидистиллированная вода. Для приготовления всех реактивов и для всех анали¬
тических операций употребляют бидистиллированную воду, которую получают перегон¬
кой обычной дистиллированной воды в стеклянном приборе с пришлифованным холодиль¬
ником.
2. Плавиковая кислота. Плавиковая кислота обычно сильно загрязнена микро¬
элементами, особенно цинком, медью и свинцом. Для очистки ее трижды перегоняют в
специальном перегонном аппарате, изготовленном из платины или палладия.
3. Соляная кислота. Соляную кислоту очищают двойной перегонкой в стеклянном
аппарате с пришлифованным холодильником. В колбу для перегонки кислоты вливают
550 мл соляной кислоты (уд. вес 1,19) и 450 мл бидистиллированной воды. Перегоняющаяся
кислота имеет концентрацию около 22%-ной, что соответствует уд. весу 1,1 и тем¬
пературе кипения 110° С.
4. Аммиак. Для очистки аммиака от загрязнений раствор 25%-ного аммиака нали¬
вают на дно большого эксикатора. На стекло, помещенное в верхней части эксикатора,
ставят стеклянные чашки (объемом 200 дел), до половины заполненные бидистиллированной
водой, плотно закрывают эксикатор крышкой и оставляют стоять не менее чем на 48 час.
Через этот или несколько больший срок сливают содержавшийся в чашках раствор аммиа¬
ка в парафинированную склянку с притертой пробкой. Концентрация полученного раст¬
вора аммиака бывает различной, и ее определяют по удельному весу. Когда желательно
иметь аммиак более высокой концентрапии, налитый на дно эксикатора аммиак заменяют
новой порцией 25%-ного аммиака и повторяют процедуру настаивания.
5. Четыреххлористый углерод. Химически чисгый СС14 в большинстве случаев можно
применять для анализа без предварительной очистки, однако имеются случаи, когда СС14
загрязнен окислителями и требует перегонки. СС14 наливают в стеклянную реторту и^
присыпают около 1 г дитизона на 1 л СС14. Ставят реторт у на кипящую водяную баню
и собирают перегоняющийся СС14 в колбу, которую охлаждают снегом или льдом. Пе¬
регонку повторяют, не внося дитизона.
СС14, бывший в употреблении, может быть очищен обработкой соответствующими
реактивами и перегонкой. Для очистки бывшего в употреблении СС14 к 500 мл СС14 при¬
бавляют 12,5 мл разбавленной НС1 (1 : 1) и встряхивают в делительной воронке. После
разделения фаз водный слой отбрасывают. К органическому слою прибавляют 12,5 мл
разбавленного аммика (1 : 10) и содержимое встряхивают в делительной воронке. После
разделения слоев водный слой отбрасывают. Повторяют обработку кислотой и аммиаком.
К очищенному таким образом СС14 прибавляют несколько кусочков безводного хлористо
го кальция, встряхивают и фильтруют. Отфильтрованный СС14 перегоняют в реторте на
водяной бане, как описано выше.
6. Раствор дитизона в СС14. 0,2 г дитизона помещают в капельную воронку на 250 мл,
приливают 200 мл СС14 и встряхивают 15 мин. на вертикальном встряхивателе. Переносят
раствор дитизона в СС14 в делительную воронку емкостью 1—1,5 л, приливают 500 мл
N Н4ОН (2,5 мл 25%-ного раствора аммиака на 500 мл воды) и встряхивают 3—5 мин.
Дитизон переходит в аммиачный слой, окрашивая его в оранжевый цвет. Отбрасывают
слой СС14 и промывают аммиачный раствор дитизона небольшими порциями (по 5—10 мл)
СС14 пока СС14 не будет окрашиваться в зеленый цвет. Затем приливают 12,5 мл раз¬
бавленной H2S04 (1 : 5), встряхивают, приливают 400 мл СС14 и сливают раствор дитизона
в CCU в чистую делительную воронку. Полученный раствор дитизона в СС14 2—3 раза
промывают бидистиллированной водой для удаления остатков кислоты. Затем раствор
дитизона в СС14 фильтруют через промытый соляной кислотой и бидистиллированной во¬
дой и высушенный фильтр в склянку темного стекла. Раствор дитизона хранят в тем¬
ноте в холодильнике. Не следует хранить его долго. Особенно важно следить за качест¬
вом дитизона и употреблять свежеприготовленный раствор для конечного определения
цинка в форме дитизоната. При использовании долго хранившихся или плохо приготов¬
ленных растворов дитизона дитизонат цинка приобретает вместо нормальной розовой
окраски бурую, что делает невозможным определение цинка.
7. 25%-ный раствор лимоннокислого натрия. 125 г двузамещенного лимоннокислого
390
К. В. Веригина
натрия растворяют в воде, доводя объем раствора бидистиллированной водой до 500 мл.
Раствор помещают в делительную воронку, подщелачивают аммиаком до слабощелочной
реакции по фенолфталеину и очищают от следов микроэлементов, встряхивая с неболь¬
шими порциями дитизона в СС14 (5—10 мл) и отбрасывая дитизоновую фазу. Когда рас¬
твор дитизона перестанет менять окраску, промывание дитизоном прекращают и раствор
лимоннокислого натрия несколько раз промывают СС14 до тех пор, пока последний не
будет оставаться бесцветным. После промывания раствор фильтруют через фильтр (бе¬
лая лента), предварительно промытый дважды перегнанной НС1 и бидистиллированной
водой для удаления следов микроэлементов. Раствор хранят в холодильнике.
8. Раствор диэтилдитиокарбамата свинца в СС14. 664 мг диэтилдитиокарбамата нат¬
рия взбалтывают в делительной воронке в 1 л СС14, прибавляют раствор нитрата свинца—
489 мг Pb(NOs)2 в 100 мл бидистиллированной воды—и встряхивают в течение 5 мин.
Дают слоям разделиться и фильтруют слон СС14 с растворенным в нем диэтилдитиокарба-
матом свинца через сухой фильтр в сухую склянку темного стекла. Раствор диэтилдитио¬
карбамата свинца в СС14 хранят в холодильнике.
9. 0,02 н. раствор НС1. 3,2 мл дважды перегнанной НС1 (уд. вес 1,1) разбавляют до
1 л бидистиллированной водой.
10. 0,01 н. НС1. 1,6 мл дважды перегнанной НС1 (уд. вес 1,1) разбавляют до 1 л би¬
дистиллированной водой.
11. 40%-ный раствор лимоннокислого аммония. 40 г двузамещенного лимоннокис¬
лого аммония растворяют в бидистиллированной воде. Раствор подщелачивают аммиаком
по лакмусовой бумаге до ее посинения и доводят объем раствора бидистиллированной
водой до 100 мл. Лимоннокислый аммоний обычно не содержит кобальта в аналитически
определимых количествах, поэтому операцию его очистки можно опустить.
12. 0,2%-ный раствор нитрозо-И-соли. Растворяют 0,2 а нитрозо-И-соли в 100 мл
бидистиллированной воды и фильтруют. Раствор следует приготавливать непосредствен¬
но перед определением кобальта.
13. 0,01 н. раствор аммиака. Разбавляют 0,7 мл 25%-ного раствора аммиака (так
как очищенный раствор аммиака обычно имеет более низкую концентрацию, берут его со¬
ответственно больше) до 1 л бидистиллированной водой.
14. 5%-ный раствор лимоннокислого аммония. 5 г двузамещенного лимоннокислого
аммония растворяют в бидистиллированной воде, доводя объем раствора до 100 мл. В бо¬
льшинстве случаев лимоннокислый аммоний свободен от следов меди и не требует очистки.
В тех случаях, когда лимоннокислый аммоний содержит медь (дает желтую окраску с
диэтилдитцокарбаматом свинца вСС14), раствор лимоннокислого аммония подщелачивают
аммиаком по фенолфталеину и встряхивают в делительной воронке с несколькими мил¬
лилитрами раствора дитизона в СС14, отбрасывая органический слой. Экстракцию раство¬
ром дитизона в СС14 продолжают до тех пор, пока зеленая окраска дитизона не пере¬
станет изменяться. После окончания экстракции дитизоном раствор лимоннокислого
аммония промывают чистым СС14 до полного удаления следов дитизона.
15. 5%-ный раствор уксуснокислого натрия. 5 г уксуснокислого натрия растворяют
в бидистиллированной воде, доводя объем до 100 мл. Приливают 3—5 мл раствора дити¬
зона в СС14, встряхивают в делительной воронке и отбрасывают органическую фазу.
Экстракцию раствором дитизона в СС14 продолжают до тех пор, пока раствор дитизона
не перестанет изменять свою первоначальную окраску. Затем промывают раствор уксус¬
нокислого натрия небольшими порциями СС14, пока СС14 не будет оставаться бесцветным»
16. 50%-ный раствор гипосульфита. Растворяют 250 г гипосульфита в бидистилли¬
рованной воде, доводя до объема 500 мл, переносят раствор в делительную воронку на
1 л, приливают 10 мл раствора дитизона вСС14 и тщательно встряхивают несколько минут.
Отбрасывают органическую фазу. Обработку дитизоном в СС14 повторяют до тех пор,
пока раствор дитизона не перестанет менять свой цвет. Гипосульфит обычно значительно
8 а грязнен микроэлементами и для их удаления необходимо многократно повторять об¬
работку раствором дитизона в СС14. Остатки дитизона извлекаются из раствора гипосуль¬
фита промыванием его чистым СС14. Раствор гипосульфита фильтруется через свободный
от следов микроэлементов фильтр в склянку из бесцинкового стекла.
17. Метилоранж. Растворяют 0,1 г метилоранжа в 100 мл бидистиллированной воды.
18. Фенолфталеин. Растворяют 0,1 г фенолфталеина в 100 мл этилового спирта.
Методы, определения в почве меди, цинка, кобальта и бора
391
Стандартные растворы
1. Стандартный раствор цинка. 0,1 г чистого металлического цинка помещают в
мерную колбу емкостью 1 л и растворяют в небольшом количестве бидистиллированной
воды, содержащей 1 мл HaS04 (уд. вес 1,84). После того, как весь цинк растворитсяг
раствор доливают до 1 л бидистиллированной водой. Этот раствор, содержащий в 1 мл
100 мкг цинка, перед приготовлением шкалы стандартных растворов разбавляют
в 100 раз, чтобы получить раствор, содержащий 1 мкг!мл цинка.
2. Стандартный раствор меди. 3,93 г сульфата меди (CuS04*5H20), содержащего 1 г
меди, растворяют в бидистиллированной воде, прибавляют несколько капель серной кис-
лоты, переносят в мерную колбу на 1 л и доливают до метки бидистиллированной водой.
Приготовленный раствор содержит 1 мг!мл меди. Для приготовления шкалы стандарт¬
ных растворов этот раствор разбавляют в 100 раз (2,5 мл до 250 мл) так, чтобы рабочий
раствор содержал 10,0 мкг/мл меди.
Перед приготовлением стандартного раствора меди необходимо определить содержа¬
ние меди в исходной соли (CuS04*5Ha0). Для этого 0,5 г соли растворяют в 50 мл бидис¬
тиллированной воды, прибавляют 3 мл уксусной кислоты, 3 мл разбавленной серной кис¬
лоты (1 : 5), затем 3 г KJ, перемешивают, разбавляют водой и титруют выделившийся иод
0,1 н. раствором гипосульфита, прибавляя в конце титрования раствор крахмала. 1 мл
0,1 н. раствора гипосульфита эквивалентен 0,006354 г меди. Если содержание меди в
сульфате меди отвечает формуле, то в 1 г соли должно находиться 0,2545 г меди и на тит*
рование навески в 0,5 г расходуется точно 20 мл 0,1 н. раствора гипосульфита. Если со¬
держание меди не соответствует теоретическому, необходимо внести поправку при взятии
навески сульфата меди при приготовлении исходного стандартного раствора.
3. Стандартный раствор кобальта. 0,4770 г сульфата кобальта (CoS04 -7H20) раст¬
воряют в бидистиллированной воде. Раствор переносят в мерную колбу емкостью в 1 л,
подкисляют 1 мл серной кислоты (уд. вес 1,84) и доводят до метки бидистиллированной
водой. Полученный раствор содержит 0,1 мг/мл Со. Для приготовления рабочего раствора
исходный раствор разбавляют бидистиллированной водой в 100 раз. Рабочий раствор
содержит 1 мкг/мл Со.
ОПРЕДЕЛЕНИЕ ПОДВИЖНЫХ ФОРМ МИКРОЭЛЕМЕНТОВ
Определение подвижных форм микроэлементов в почвах состоит из двух
операций: извлечение микроэлементов из почвы соответствующими вытяж¬
ками и их определение в полученном растворе.
Для определения подвижных форм микроэлементов в почвах предло¬
жено большое количество вытяжек, которые различаются как по силе воз¬
действия на почву, так и тем, что в одних случаях вытяжка применяется как
групповой экстрагент, в котором определяется ряд биологически важных
микроэлементов, в других — для вытеснения каждого микроэлемента приме¬
няются отдельные вытяжки.
В большинстве зарубежных стран для определения меди принята вытяж¬
ка по Вестерхоффу (Westerhoff, 1955), в Дании — вытяжка по Хенриксену
(Henriksen, 1957). Для определения подвижного цинка в качестве стандарт¬
ной используется вытяжка 0,1 н. НС1 (Wear, Sommer, 1947). Для определе¬
ния подвижного кобальта применяют вытяжки 1,0 н. НС1 и HN03.
В СССР наиболее широкое распространение для некарбонатных почв
нашла система вытяжек, предложенная Я. В. Пейве и Г. Я. Ринькисом (Ринь-
кис, 1963), где для извлечения каждого элемента применяют индивидуальную
вытяжку. Конечное определение микроэлементов по Ринькису производят
колориметрическим пробирочным визуальным способомг. На основании
Г Метод, предложенный Я. В. Пейве и Г. Я. Ринькисом, был модифицирован в лабора¬
тории химии почв Почвенного института им. В. В. Докучаева. По этой модификации
все конечные определения подвижных форм производятся фотоколориметрически.
392
К. В. Веригина
полевых опытов с микроудобрениями, проведенных на дерново-подзолистых
и дерново-карбонатных почвах Латвии, для метода Пейве и Ринькиса были
разработаны шкалы обеспеченности почв микроэлементами. Для ряда райо¬
нов страны эти шкалы были в дальнейшем уточнены (Даутов, Минибаев, 1968;
Макеев, 1968).
При выборе экстрагента для определения подвижных форм микроэле¬
ментов необходимо учитывать состав почвы: так, вытяжки, разработанные
Я. В. Пейве и Г. Я. Ринькисом для некарбонатных почв, малопригодны для
анализа карбонатных почв, особенно без учета содержания в почвах карбо¬
натов.
В качестве вытяжек для карбонатных почв предложены буферные рас¬
творы уксуснокислого аммония (Крупский, Александрова, 1964) и уксусно¬
кислого натрия (Круглова, 1972). Буферные солевые растворы применяют
в качестве групповых экстрагентов для извлечения подвижных форм микроэле¬
ментов как из карбонатных, так и некарбонатных почв. Являясь слабыми экст¬
рагентами, эти буферные растворы извлекают очень небольшие количества
микроэлементов (особенно из некарбонатных почв), что затрудняет конечное
определение обычно применяемыми фотоколориметрическими методами.
К. Боратынским (Boratynski, Zieteoka, 1970) предложено в качестве груп¬
пового экстрагента для извлечения из почв подвижных форм микроэлементов
использовать лактат-уксуснокисло-аммонийную вытяжку по Эгнеру — Ри¬
му — Доминго (А — Ьметод), которая широко применяется также для оп¬
ределения доступных растениям форм калия и фосфора.
Уксусно-аммонийная вытяжка
по Крупскому и Александровой (1964)
50 г воздушно-сухой почвы, пропущенной через сито с отверстиями диа¬
метром 1 мм, помещают в коническую колбу с притертой пробкой, приливают
250 мл буферного раствора уксуснокислого аммония и встряхивают на ме¬
ханическом встряхивателе в течение 30 мин. Фильтруют через сухой фильтр
(белая лента), свободный от следов микроэлементов. В аликвотах получен¬
ного фильтрата определяют цинк и кобальт (см. ниже определение цинка и
кобальта по Крупскому и Александровой).
Определение меди в вытяжке уксуснокислым аммонием авторы не приво¬
дят, так как количество меди, экстрагируемое этой вытяжкой, очень мало
и определить его существующими методами трудно.
Реактивы. Буферный раствор уксуснокислого аммония, pH 4,8. Растворяют в
б и дистиллированной воде 77 г кристаллического уксуснокислого аммония, приливают
50 мл 98%-ной уксусной кислоты и доводят бидистиллированной водой до 1 л. Величина
pH раствора измеряется стеклянным электродом. В случае отклонения pH от значения
4,8 его доводят добавлением соответственно уксусной кислоты или аммиака. Следует
применять реактивы квалификации «особо чистые».
При отсутствии в лаборатории реактивов особой чистоты, раствор лучше готовить из
уксусной кислоты и аммиака, ;которые легче очистить от следов микроэлементов.
108 мл уксусной кислоты (98%-ной) разводят бидистиллированной водой примерно до
600—700 мл, приливают 75 мл 25%-ного раствора аммиака, перемешивают и после ох¬
лаждения доводят бидистиллированной водой до 1 л. Проверяют pH полученного раствора
потенциометрически. В случае необходимости доводят pH раствора до 4,8, как описано
выше.
Очистка реактивов. Уксусную кислоту дважды перегоняют в стеклянном приборе с
пришлифованным холодильником. При перегонке уксусной кислоты 98%-ную кислоту
наливают в колбу для перегонки и прибавляют небольшое количество безводного уксус¬
нокислого натрия (2—3 г). Концентрацию перегнанной уксусной кислоты проверяют по
удельному весу.
Методы определения в почве меди, цинка, кобальта и бора
393
Очистка аммиака (см. выше в разделе «Определение цинка и кобальта»).
Необходимые для приготовления буферного раствора уксуснокислого аммония коли¬
чества уксусной кислоты и аммиака рассчитывают, исходя из указанной выше пропор¬
ции — 108 мл 98%-ной уксусной кислоты и 75 мл 25%-ного аммиака.
Вытяжка буферным раствором уксуснокислого натрия
по Кругловой (1972)
В климатических условиях Средней Азии (при повышенной температуре)
применять аммонийные соли для приготовления вытяжек неудобно в связи
с их летучестью, поэтому там предпочтительнее использовать буферные ра¬
створы уксуснокислого натрия. Сравнительные данные, полученные при
применении буферных растворов уксуснокислого натрия и уксуснокислого
аммония (pH 3,5), показали, что они извлекают из почв равнозначные коли¬
чества микроэлементов.
Приготовление вытяжки. 30—50 г воздушно-сухой поч¬
вы, пропущенной через сито с отверстиями диаметром 1 мм, помещают в
коническую колбу на 500 мл, приливают 150—250 мл буферного раствора.
Суспензию периодически взбалтывают круговыми движениями до разру¬
шения основной массы карбонатов (около 10 мин). Колбы закрывают мале¬
нькими воронками и взбалтывают на встряхивателе в течение 30 мин. Затем
растворы сразу же фильтруют через складчатые фильтры (белая лента),
свободные от следов микроэлементов.
Если почва содержит меньше 25% СаС08, для приготовления вытяжки
применяют буферный раствор № 1, если выше 25% — раствор № 2.
В аликвотных частях полученного фильтрата определяют медь, цинк -
и кобальт, как описано ниже для вытяжек по Кругловой.
Реактивы. 1. Буферный раствор № 1. Для его приготовления необходимы
следующие исходные растворы: а) 1 н. раствор уксусной кислоты. 60 мл ледяной уксус¬
ной кислоты разбавляют бидисти л л прованной водой до 1 л. Если уксусная кислота за*
грязнена микроэлементами, ее очищают перегонкой, как описано выше; б) 1 н. раствор
уксуснокислого натрия. 82 г СН3СООКа или 136 г CH3C00Na-3H20 растворяют в
200—250 мл бидистиллированной воды, помещают в делительную воронку на 1 л, до¬
бавляют 2 капли фенолфталеина и нейтрализуют до появления розовой окраски 10%-ным
раствором аммиака, затем приливают 10 мл 0,05%-ного раствора дитизона в СС14, встря¬
хивают 2—3 мин., дают слоям разделиться и отбрасывают дитизоновый слой. Экстрак¬
цию следов микроэлементов дитизоном повторяют до тех пор, пока раствор дитизона не
перестанет изменять свою окраску. Когда обработка дитизоном закончена, раствор ук¬
суснокислого натрия несколько раз промывают небольшими порциями (по 10—15 мл)
чистого СС14 энергично встряхивают, дают слоям разделиться и отбрасывают слой СС14.
Обработку СС14 ведут, пока он не будет оставаться бесцветным. Полученный очищенный
раствор уксуснокислого натрия доводят бидистиллированной водой до 1 л.
Для приготовления буферного раствора уксуснокислого натрия JVb 1 сливают 925 мл
1 н. раствора уксусной кислоты и 75 мл 1 н. раствора уксуснокислого натрия. pH рас¬
твора должен отвечать 3,5.
2. Буферный раствор № 2. Необходимы следующие исходные растворы: а) 2 н.
раствор уксусной кислоты. 120 мл ледяной уксусной кислоты разбавляют бидистиллиро¬
ванной водой до 1л; б) 2 н. раствор уксуснокислого натрия. 164 г CH3COONa или
272 г. CH3C00Na-3H20 растворяют в 400—500 мл бидистиллированной воды, переносят
в делительную воронку, очищают, как описано выше, и доводят до 1 л бидистиллирован¬
ной водой.
925 мл 2 н. уксусной кислоты сливают с 75 мл 2 н. уксуснокислого натрия. Буфер¬
ный раствор № 2 имеет также pH 3,5, но буферная емкость его в 2 раза выше, чем у рас¬
твора № 1, поэтому он применим для почв с более высоким содержанием карбонатов
(>25% СаС03).
394
К. В. Веригина
Вытяжка по Эгнеру — Риму — Доминго (А — L метод)
Боратынским (Boratynski, Zietecka, 1970) предложено в качестве группо¬
вого экстрагента для извлечения из почв подвижных форм микроэлементов
использовать лактат-ацетат-аммонийную вытяжку до Эгнеру — Риму —
Доминго.
Приготовление вытяжки. 80 г воздушно-сухой почвы по¬
мещают « коническую колбу на 500 мл, приливают 400 мл рабочего лактат-
ацетат-аммоыигаого раствора и встряхивают в течение 2-х час. на механи¬
ческом встряхивателе, затем оставляют стоять 20 час. На следующий день
вытяжку фильтруют через сухой складчатый фильтр, отбрасывая первые
капли фильтрата. Одновременно ставят холостой опыт, проводя через все
аналитические операции экстракционный раствор без почвы.
Для определения меди берут 100 мл фильтрата, цинка — 5—10 мл, ко¬
бальта — 25—50 мл, бора — 10 дед фильтрата и далее проводят анализ по
Боратынскому (Boratynski, Zietecka, 1970), как описано ниже.
Реактивы. Лактат-ацетат-аммонийный раствор. В колбу на 3 л помещают 1 кг
молочной кислоты, приливают 2 л бидистиллированной воды и отмечают полученный
объем. Колбу с ее содержимым выдерживают в термостате при 95° в течение 48 час.
(процесс гидратации). После охлаждения раствор доливают бидистиллированной водой
до исходного объема. Нормальность приготовленной молочной кислоты определяют тит¬
рованием аликвота 1 н. NaOH по фенолфталеину. Молочная кислота должна быть око¬
ло 3 н.
Для приготовления 5 л запасного лактат-ацетат-аммонийного раствора требуется
Б эквивалентов молочной кислоты. Если раствор молочной кислоты будет 3 н., то 5 эк¬
вивалентов кислоты будут содержаться в 5000:3 = 1667 мл приготовленной молочной
кислоты. К 1667 мл 3 н. молочной кислоты прибавляют 892,5 мл 96%-ной уксусной кис¬
лоты и 385 г уксуснокислого аммония и разбавляют бидистиллированной водой до 5 л.
Бее реактивы должны быть квалификации ч. д. а. или особой чистоты. Запасной раствор
хранят в холодильнике при 5е С.
Рабочий лактат-ацетат-аммонийный раствор готовят в день приготовления почвен¬
ных вытяжек, разводя запасной раствор в 10 раз. pH должен равняться 3,8.
5 г почвы или 2,5 г торфа помещают в к< очку емкостью 100 мл, прили¬
вают пипеткой 50 мл 1,0 н. НС1 и взбалтывают в течение 1 часа. Вытяжку
фильтруют через сухой складчатый фильтр. Для удаления мешающих ана¬
лизу органических веществ и железа 10 мл фильтрата помещают в пробирку
или цилиндрик, градуированные на 20 мл, прибавляют 1 каплю фенолфта¬
леина и нейтрализуют 12,5%-ным аммиаком до получения розового оттенка.
Доливают бидистиллированной водой до объема 20 мл, перемешивают и
фильтруют через сухой маленький фильтр в колбочку. Затем пипеткой берут
в пробирку с притертой пробкой 5 мл полученного фильтрата и приливают
2 мл комплексного буферного раствора. Перемешивают, закрыв пробирку
пробкой. Проверяют по индикаторной бумаге pH, который должен быть близ¬
ким 2,0. Пробу производят, касаясь пробкой пробирки тонкой полоски ин¬
дикаторной бумаги. При необходимости прибавляют по каплям разбавлен¬
ную вдвое бидистиллированной водой HG1.
Когда *рН установлено, приступают к экстракции меди дитизоном. Для
этого прибавляют из бюретки 1 мл раствора дитизона в СС14 и сильно встря¬
хивают в течение полминуты, пока окраска дитизона не перестанет изменять-
Определение подвижной меди
Определение меди по Ринъкису (1963) в вытяжке lfi н. НС1
дитизоновым ме дом
Методы определения в почве меди, цинка, кобальта и бора
395
с я. Дают слоям разделиться и сравнивают окраску дитизонового слоя с про¬
бирочной шкалой. Если в растворе присутствуют окислы азота, то дитизон
окисляется и раствор дитизона желтеет. В таких случаях анализ надо пов¬
торить и перед добавлением дитизона прибавить 1—2 капли 1%-нойН202.
Если цвет дитизонового экстракта краснее последней пробирки шкалы,
то следует прибавить еще 1 мл дитизона, снова встряхнуть пробирку и, дав
слоям разделиться, сравнить со шкалой. Если при прибавлении 6—8 мл
дитизона окраска экстракта будет краснее последней пробирки шкалы, то
для анализа берут меньшее количество фильтрата. В противном случае,
когда при добавлении 1 мл дитизона его окраска оказывается более зеленой,
чем окраска дитизона во второй пробирке шкалы, объем взятого для анализа
фильтрата следует увеличить.
Для приготовления шкалы образцовых растворов 3,933 г х.ч. перекри-
сталлизованного CuS04-5H20 растворяют в бидистиллированной воде и до¬
водят водой в мерной колбе до 1 д. Такой раствор содержит в 1 мл 1 мг Си.
10 мл этого раствора разбавляют до 1 л бидистиллированной водой и полу¬
чают образцовый раствор с содержанием в 1 мл 0,01 мг Си. Последний
раствор разбавляют еще в 10 раз, получая рабочий образцовый раствор, со¬
держащий в 1 мл 1 мкг Си. Этот раствор используется для приготовления
шкалы.
В семь пробирок с притертыми пробками наливают 0; 0,5; 1,0; 1,5; 2,0;
2.5 и 3,0 мл образцового раствора, что соответствует 0; 0,5; 1,0; 1,5; 2,0;
2.5 и 3,0 мкг Си. После этого в пробирки приливают бидистиллированную
воду так, чтобы общий объем составлял 5 мл, добавляют по 2 мл комплекс¬
ного буферного раствора. Проверяют pH и доводят его до 2,0. Приливают из
бюретки 5 мл раствора дитизона в СС14. Затем пробирки сильно встряхивают
в течение 2 мин., пока окраска дитизона не перестанет изменяться.
Вычисление результатов анализа производится по формуле
где х — содержание меди на 1 кг почвы, мг; а — содержание меди в совпадающей пробир¬
ке шкалы, мкг/мл; б — объем израсходованного для экстракции раствора дитизона, мл\
е — объем вытяжки, мл; н — навеска почвы, взятая для анализа, г; 2 — коэффициент,
вводимый в связи с разбавлением исходной вытяжки.
Почва, содержащая < 0,3 мгЫг Си в 1,0 н. НС1, — очень бедная; 0,3—
1,5 — бедная; 2—3 — средняя; 4—7 — богатая; свыше 7 мгЫг — очень
богатая медью.
Реактивы. 1. Раствор дитизона в СС14. 0,01 г дитизона помещают в делительную
воронку и растворяют в 50 мл СС14, для чего энергично встряхивают 20 мин. Приливают
50 мл NH4OH (1 : 20) и взбалтывают, дают слоям разделиться и отбрасывают нижний
слои. Промывают водный раствор 2—3 раза 5—10 мл СС14, каждый раз энергично взбал¬
тывая и давая слоям разделиться. Промывание СС14 производят до тех пор, пока слой
СС14 не будет иметь зеленую окраску. Затем приливают 25 мл 1,0 н. НС1 и около 50 мл
СС14. Взбалтывают, при этом дитизон переходит в слой СС14, окрашивая его в зеленый
цвет. Раствор дитизона в СС14 фильтруют через сухой фильтр в сухую склянку темного
цвета, доливая СС14 до 1 л. Приблизительная концентрация дитизона в СС14, используе¬
мого в анализе, должна быть 0,001 %-ная.
2. 1,0 н.НС1. 82 мл концентрированной НС1 (уд. вес 1,19) разбавляют бндистиллиро-
ванной водой до 1 л. Если НС1 содержит следы меди, ее необходимо очищать перегонкой
(см. выше). Перегнанная НС1 имеет концентрацию около 22%-ной. Для приготовления
1,0 н. НС1 166,0 мл дважды перегнанной HG1 доводят бидистиллированной водой
до 1 л.
3. Комплексный буферный раствор. Растворяют в 40 мл бидистиллированной воды
15 г уксуснокислого натрия, 7,5 г двузамещенного фосфата натрия и 2,5 г роданистого
К. В. Веригина
калия, затем раствор подкисляют концентрированной НС1 до pH 2,0 по индикаторной
бумаге. Доводят бидистиллированной водой до 100 мл.
4. Фенолфталеин. 0,1 г фенолфталеина растворяют в 100 мл этилового спирта.
5. Аммиак очищают, как указано выше в разделе «Определение цинка и кобальта».
Определение меди по Ринъкису,
в модификации Почвенного института им. В. В. Докучаева
(Веригина, Добрицкая, 1974)
5 г воздушно-сухой почвы, пропущенной через сито с отверстиями диа¬
метром 1 мм, помещают в коническую колбу с притертой пробкой (на 100 мл),
приливают 50 мл 1,0 н. HG1, встряхивают 1 час на горизонтальном встряхи-
вателе и фильтруют через фильтр (белая лента).
Из приготовленной вытяжки берут пипеткой 10—25 мл (в зависимости от
ожидаемого количества меди) раствора и помещают в капельную воронку
на 50—100 мл, приливают 5 мл 5%-ного раствора лимоннокислого аммония,
1 каплю фенолфталеина и приливают по каплям, перемешивая раствор, 10%-
ный раствор аммиака до получения розовой окраски. Приливают из бюретки
точно 15 мл раствора диэтилдитиокарбамата свинца в СС14, встряхивают
10 мин. на вертикальном встряхивателе, дают фазам расслоиться и сливают
фазу четыреххлористого углерода, окрашенную в желтый цвет диэтилдитио-
карбаматом меди, через маленький сухой фильтр (красная лента) в кювету
фотоколориметра. Применяют кювету с толщиной просматриваемого слоя
20 мм. Оптическую плотность раствора измеряют с синим светофильтром
(453 нм)у применяя в качестве нулевого раствора СС14.
Шкалу стандартных растворов меди приготовляют из раствора, содержа¬
щего 10 мкг/мл Си. Для этой цели 0; 0,3; 0,5; 1,0; 1,5; 2,0 мл стандартного
раствора меди, что соответствует 0; 3,0; 5,0; 10,0; 15,0; 20,0 мкг Си, помещают
в капельные воронки. Приливают бидистиллированную воду до объема 10 мл.
1 мл 5%-ного раствора лимоннокислого аммония, нейтрализуют аммиаком
по фенолфталеину, приливают из бюретки точно 15 мл раствора диэтилди¬
тиокарбамата свинца в СС14. Встряхивают 10 мин. на вертикальном встряхи¬
вателе и далее поступают, как и в случае испытуемых растворов.
По полученным для стандартных растворов величинам оптических плот¬
ностей строят градуировочную кривую, по которой находят содержание ме¬
ди в испытуемых растворах. Для вычисления содержания меди в милли¬
граммах Си на 1 кг почвы найденное по градуировочной кривой содержание
меди в микрограммах во взятом для анализа объеме вытяжки делят на на¬
веску почвы (в г), соответствующую взятому для анализа объему раствора.
Реактивы. 1. 1,0 н. НС1. Для приготовления 1,0 н. НС1 166,0 мл дважды пе¬
регнанной 22-ной НС1 доводят бидистиллированной водой до 1 л.
2. 5%-ный раствор лимоннокислого аммония. 5 г двузамещенного лимоннокислого
аммония растворяют в бидистиллированной воде, доводя объем раствора до 100 мл.
В большинстве случаев лимоннокислый аммоний свободен от следов меди и не требует
очистки. Б тех случаях, когда лимоннокислый аммоний содержит медь (дает желтую ок¬
раску с диэтилдитиокарбаматом свинца в СС14), раствор лимоннокислого аммония под¬
щелачивают аммиаком по фенолфталеину и встряхивают в делительной воронке с не¬
сколькими миллилитрами раствора дитизона в СС14, отбрасывая органический слой.
Экстракцию раствором дитизона продолжают до тех пор, пока зеленая окраска дитизо¬
на не перестанет изменяться. После окончания экстракции дитизоном раствор лимон¬
нокислого аммония промывают чистым СС14 до полного удаления следов дитизона.
3. Раствор диэтилдитиокарбамата свинца в СС14. 664 мг диэтилдитиокарбамата нат¬
рия взбалтывают в делительной воронке с 1 л СС14, прибавляют раствор нитрата свинца—
489 мг Pb(NOs)a в 100 мл бидистиллированной воды — и встряхивают в течение 5 мин.
Дают слоям разделиться и фильтруют слой СС14 с растворенным в нем диэтилдитиокарба-
Методы определения в почве меди, цинка, кобальта и бора
397
матом свинца через сухой фильтр в сухую склянку темного стекла. Раствор хранят в
холодильнике.
4. Фенолфталеин. Растворяют 0,1 г фенолфталеина в 100 мл этилового спирта.
5. Аммиак очищают, как указано выше в разделе «Определение цинка и кобальта».
Определение меди по Шарреру и Шаумлеффелю
(Scharrer, Schaumloffel, 1958)
в вытяжке по Вестерхоффу (Westerhoff, 1955)
К 10 г почвы приливают 100 мл азотной кислоты (30 мл HN03 уд. веса
1,39 доводят до 1 л водой) и встряхивают 2 часа. Фильтруют и помещают
25 мл фильтрата в колбочку Эрленмейера. Приливают 2 мл 0,5%-ного рас¬
твора КМи04 и нагревают до кипения. К горячему раствору прибавляют
0,5 мл 5%-ной щавелевой кислоты для обесцвечивания раствора. После ох¬
лаждения раствор переносят в капельную воронку на 100 мл, смывая биди-
стиллированной водой, доводят объем бидистиллированной водой примерно
до 50 мл. Затем прибавляют 1 мл насыщенного раствора Na3P04, 4 мл 50%-
ного раствора лимоннокислого натрия и 4 мл аммиака (уд. вес 0,910); после
прибавления каждого реактива раствор тщательно перемешивают. Прибав¬
ляют точно 15 мл раствора диэтилдитиокарбамата свинца в четыреххлорис¬
том углероде и встряхивают от руки или на механическом встряхивателе
в течение 2 мин. Дают слоям разделиться и фильтруют окрашенный диэтил-
дитиокарбаматом меди слой четыреххлористого углерода через сухой фильтр
в кювету с просматриваемым слоем в 20 мм. Фотоколориметрируют с синим
светофильтром (453 нм), применяя в качестве нулевого раствора СС14.
Калибровочную кривую приготовляют из раствора, содержащего
10 мкг/мл Си. Для этого помещают в капельные воронки 0; 0,2; 0,5; 1,0;
2,0 мл раствора меди (что соответствует 0; 2; 5; 10; 20 мкг Си), доводят бидис-
тиллированной водой до 50 мл и далее ведут анализ, как и с испытуе¬
мыми растворами.
По интенсивности окраски стандартных растворов строят градуировочную
кривую, по которой определяют содержание меди в испытуемых растворах.
Для этого в системе координат по оси абсцисс откладывают количество меди
(в мкг), а по оси ординат — отсчеты по фотоколориметру и соединяют полу¬
ченные точки. Находят по калибровочной кривой, какому количеству меди
(в мкг) соответствует полученный для данного испытуемого раствора отсчет
по фотоколориметру. Для определения содержания меди в милиграммах на
1 кг почвы найденное количество меди (в мкг) делят на взятую навеску почвы
<2,5 г).
В ГДР принята следующая оценка обеспеченности почв медью (в мг/кг):
Обеспеченность Песчаные Суглинистые и
почвы глинистые почвы
Хорошая 3,5 2,5
Мало нуждаются 2,1—3,5 1,6—2,5
Сильно нуждаются 2,0 1,5
Реактивы 1. Азотная кислота. 30 мл HN03 (уд. вес 1,39) доводят до 1 л бидис¬
тиллированной водой.
2. 5%-ный раствор Н2С204‘2На0. 5 г щавелевой кислоты растворяют в бидистилли¬
рованной воде и доводят до 100 мл.
3. 0,5%-ный раствор КМп04. 0,5 г КМпО* растворяют в воде и доводят до 100 мл.
4. Насыщенный раствор Na3P04. 25 г трехзамещенного ортофосфата натрия раст¬
воряют в бидистиллированной воде и доводят до 100 мл.
5. 50%-ный раствор лимоннокислого натрия. 50 г трехзамещенного лимоннокисло¬
го натрия растворяют в бидистиллированной воде и доводят до 100 мл.
398
К. В. Веригина
6. Аммиак (уд. вес 0,910).
7. Раствор диэтилдитиокарбамата свинца (см. выше в разделе «Определение меди по
Ринькису, в модификации Почвенного института»).
8. Стандартный раствор меди (см. «Определение валового содержания...», стандарт¬
ные растворы).
Определение меди по Хенриксену
(Henriksen, 1957)
К 10 г почвы приливают 100 мл 0,02 М раствора трилона Б, содержащего
в 1 л 5 г хлористого аммония (для получения прозрачного фильтрата). Встря¬
хивают на ротаторе в течение 1 часа и фильтруют через плотный фильтр.
50 мл полученного фильтрата помещают в капельную воронку на 100 млг
приливают 10 мл 25%-ного раствора лимоннокислого аммония (pH 9,0)г
сильно встряхивают, прибавляют 1 мл 1 %-ного свежеприготовленного рас¬
твора диэтилдитиокарбамата натрия и 15 мл четыреххлористого углерода*
Встряхивают 2 мин., дают слоям разделиться и фильтруют окрашенный
диэтилдитиокарбаматом меди слой четыреххлористого углерода через сухой
фильтр в кювету фотоколориметра с просматриваемым слоем в 2 см. Фотоко-
лориметрируют с зеленым светофильтром (536 нм), сравнивая с четыреххло¬
ристым углеродом. Градуировочную кривую приготовляют из раствора, со¬
держащего 10 мкг/мл Си. Для этого помещают в капельные воронки 0; 0,2;
0,5; 1,0; 2,0; 3,0; 4,0 мл раствора меди (что соответствует 0, 2, 5, 10, 20, 30,.
40 мкг меди), доводят бидистиллированной водой до 50 мл, прибавляют 10 мл
25 %-ного раствора лимоннокислого аммония, 1 мл 1 %-ного раствора ди¬
этилдитиокарбамата натрия, 15 мл четыреххлористого углерода и далее ведут
анализ, как и с испытуемыми растворами. По оптическим плотностям стан¬
дартных растворов строят градуировочную кривую, по которой определяют
содержание меди в испытуемых растворах. Исследования показывают, что
для глинистых и песчаных почв внесение медных удобрений рентабельно при
содержании меди менее 1 мг/кг почвы, для почв, богатых органическим вещест¬
вом,— менее 3 мг!кг.
Реактивы. 1. 0,02 М раствор трилона Б. Б 1 л воды растворяют 7,4 г трилона
Б и 5 г хлористого аммония.
2. 25%-ный раствор лимоннокислого аммония. 250 г лимоннокислого аммония раст¬
воряют в 700—800 мл бидистиллированной воды, проверяют pH раствора по индикатор¬
ной бумаге и доводят его аммиаком до 9,0. Доливают бидистиллированной водой до 1 л*
3. 1%-ный раствор диэтилдитиокарбамата натрия. 1 г диэтилдитиокарбамата натрия
растворяют в 100 мл бидистиллированной воды, раствор встряхивают с 10 мл СС14, дают
слоям разделиться и сливают слой СС14. Если раствор СС14 будет окрашен в желтый цвет,,
повторяют промывание раствора диэтилдитиокарбамата натрия новыми порциями СС14г
пока последний не будет оставаться бесцветным.
4. Образцовый раствор Си. 3,933 г х. ч., перекристаллизованного CuS04*5H20 раст¬
воряют в бидистиллированной воде и доводят водой до 1л. 10 мл этого раствора, содер¬
жащего в 1 мл 1мг Си, разбавляют до 1 л бидистиллированной водой, получая образ¬
цовый раствор с содержанием в 1 мл 10 мкг Си.
Определение меди по Шарреру и Шаумлеффелю
(Scharrer, Schaumloffel, 1958) в вытяжке по Кругловой (1072)
40—50 мл вытяжки буферным раствором уксуснокислого нагрия (см.
стр. 393) помещают в капельную воронку на 1С0 мл. Приливают 5 мл 5%-ного
раствора лимоннокислого аммония, 1 каплю фенолфталеина и добавляют по
каплям аммиак до щелочной реакции (розовый цвет). Далее поступают, как
описано при определении меди в модификации Почвенною института им-
В. В. Докучаева.
Методы определения в почве меди, цинка, кобальта и бора
399
Определение меди по Боратынскому
(Boratynski, Zietecka, в вытяжке Эгнера — Рима — Доминго
В коническую колбу на 150 мл помещают 100 мл вытяжки (см. стр. 394),
прибавляют 10 мл 30%-ной Н202 и выпаривают на водяной бане приблизи¬
тельно до объема 25 мл*. После охлаждения к содержимому колбы приливают
20 мл буферной маскирующей смеси, перемешивают и приливают из бюретки
точно 15 мл раствора диэтилдитиокарбамата свинца в СС14. Колбу закрывают
пробкой из мягкой пластмассы и встряхивают в течение 5 мин. на горизон¬
тальном встряхивателе. Колбу снимают с встряхивателя, дают фазам раз¬
делиться и отсасывают водную фазу с помощью капилляра, соединенного
с водоструйным насосом (рисунок). Органическую фазу, содержащую
Отделение водной фазы отсасыванием с помощью капилляра при определении меди
<х — экстракция меди с органическом растворителем в колбе е притертой пробкой; б —отсасывание вод¬
ной фазы с помощью капилляра; в — способ держания капилляра
комплекс меди с диэтилдитиокарбаматом, фильтруют через маленький сухой
фильтр непосредственно в кювету фотоколориметра. Фотоколориметрируют
при длине волны 440 нм, применяя в качестве нулевого раствора раствор
диэтилдитиокарбамата свинца в СС14.
Шкалу стандартных растворов меди готовят из раствора меди, содержа¬
щего 1 мкг Си в 1 мл, в интервале 0—20 мкг Си, доливая до 25 мл бидистилли¬
рованной водой. Вносят маскирующую смесь, приливают из бюретки по 15 мл
раствора диэтилдитиокарбамата свинца и далее поступают, как описано выше
для испытуемых растворов.
Реактивы. 1. Буферная маскирующая смесь. 13 г Naj,P04*12H20 и 100#
Na3CeH607*2H20 растворяют в 500 мл бидистиллированной воды, приливают 200 мл ам¬
миака (уд. вес 0,910), перемешивают и доводят бидистиллированной водой до 1 л.
2. Стандартный раствор меди. 0,1965 г CuS04*5H20 растворяют в бидистиллирован¬
ной воде в мерной колбе на 500 мл. Перемешивают и доводят до метки бидистиллировав-
ной водой. 1 мл этого раствора содержит 100 мкг Си. Стандартный рабочий раствор гото¬
вят, разбавляя запасной раствор в 100 раз. 1 мл рабочего раствора должен содержать
1 мкг!мл Си.
3. Раствор диэтилдитиокарбамата свинца готовят, как описано выше в разделе «Оп
ределение меди по Ринькису, в модификации Почвенного института».
1 При наличии в лаборатории хороших капельных воронок выпаренный до объема 25 мл
раствор лучше перенести в воронки и вести дальше анализ в них.
400
К. В. Веригина
Определение подвижного цинка
Определение цинка по Ринькису (1963) в вытяжке КС1
5 г почвы или 2,5 г торфа помещают в колбочку емкостью 100 мл из квар¬
ца или бесцинкового стекла, приливают 50 мл 1 н. КС1, закрывают колбочку
пробкой и встряхивают в течение часа. Вытяжку фильтруют через свобод¬
ный от следов цинка фильтр в кварцевую колбочку.
В пробирку с притертой пробкой (из бесцинкового стекла) берут пипет¬
кой 5 мл вытяжки, приливают 2 мл комплексного буферного раствора
и встряхивают. pH раствора должен быть в пределах 5—5,5. Проверяют
pH, касаясь пробкой пробирки узкой полоски индикаторной бумаги. В боль¬
шинстве случаев необходимая реакция устанавливается при приливании
буфера; однако, если при проверке окажется, что pH отличается от нужной
величины, необходимо прибавить НС1 или NH4OH.
Цинк определяют в виде его комплекса с дитизоном. Прибавляют в про¬
бирки из бюретки 1,0 мл дитизона, закрывают пробирку пробкой и энергич¬
но встряхивают в течение 20—30 сек. Дают слоям разделиться и сравнивают
полученную окраску дитизонового экстракта со стандартной пробирочной
шкалой. Если цвет экстракта краснее последней пробирки стандартной шка¬
лы, прибавляют еще 1 мл дитизона и снова встряхивают, после чего срав¬
нивают со шкалой. Если и при прибавлении 6—8 мл дитизона окраска экс¬
тракта будет краснее последней пробирки шкалы, анализ следует повторить
с меньшим количеством вытяжки. В противоположном случае, если при
1,0 мл дитизона окраска будет зеленее, чем раствор дитизона во второй про¬
бирке шкалы, анализ следует повторить, взяв большее количество фильтрата.
Шкалу образцовых растворов готовят из раствора, содержащего в 1 мл
0,5 мкг Zn. В семь пробирок с притертыми пробками (из бесцинкового стекла)
берут пипеткой 0, 0,5; 1,0; 1,5; 2,0; 2,5; 3,0 мл образцового раствора цинка,
содержащего в 1 мл 0,5 мкг Zn. После этого в пробирки приливают бидистил-
лированную воду так, чтобы общий объем составлял 5 мл, добавляют по 2 мл
комплексного буферного раствора, 5 мл дитизона (дитизон приливают из.
бюретки) и встряхивают 30 сек. Сравнивают со стандартной пробирочной
шкалой окраску испытуемых растворов.
Вычисление результатов анализа производится по формуле
абв
где х — содержание цинка на 1 кг почвы, мг; а — содержание цинка в совпадающей про¬
бирке шкалы, мкг! мл; б — объем израсходованного для экстракции раствора дитизонаг
мл; в — объем исходного раствора или вытяжки, мл; н — навеска почвы, г; г — объем
вытяжки, взятой для экстракции цинка, мл.
Почва, содержащая <0,2 мгЫг Zn в вытяжке 1,0 н. КС1, — очень бедная;
0,2—1,0 — бедная; 2—3 — средняя; 4—5 — богатая и свыше 5 мг/кг — очень
богатая подвижным цинком.
Реактивы. 1. 1,0 н. раствор КС1. 75 г КС1 растворяют в бидистиллированнои
воде, доводя до 1 л. Доводят pH раствора до 5 и очищают от следов микроэлементов раст¬
вором дитизона, для чего раствор КС1 переносят в большую делительную воронку, при¬
ливают небольшое количество (около 10 мл) дитизона и сильно встряхивают, дают слоям
разделиться и отбрасывают нижний слой. Промывание дитизоном продолжают^до тех пор,/
пока дитизон не перестанет изменять свою окраску. Затем приливают СС14 (20—25 мл) и
промывают раствор КС1 четыреххлористым углеродом до тех пор, пока последний не будет
оставаться бесцветным.
2. Комплексный буферный раствор. 50 г уксуснокислого натрия растворяют в бидис¬
тиллированной воде, доводят pH раствора до 5,5—6,0 прибавлением 1,0 н. HCJ, раство¬
Методы, определения в почве меди, цинка, кобальта и бора
401
ряют в этом же растворе 40 г гипосульфита, доводят объем бидистиллированной водой
до 200 мл и очищают раствором дитпзона так же, как это описано при очистке КС1.
3. Стандартный раствор цинка. 0,1 г х. ч. металлического цинка растворяют в 50 мл
бидистиллированной воды, содержащих 1 мл концентрированной серной кислоты, и дово¬
дят бидистиллированной водой до 1 л. Этот раствор содержит в 1 мл 0,1 мгЪп. Из этого
раствора берут Ь мл и бидистиллированной водой доводят до1л. Раствор содержит в
1 мл 0,5 мкг Zn.
4. Раствор дитизона для определения цинка готовят так же, как при опреде¬
лении меди по Ринькису.
Определение цинка по Ринькису (1963)
в вытяжке уксуснокислым аммонием
Вытяжка хлористым калием не пригодна для определения обменного
цинка в карбонатных почвах, для которых можно применять метод из¬
влечения обменного цинка буферным раствором уксуснокислого аммония
с pH 4,8.
5 г почвы помещают в кварцевую колбочку на 100 мл, приливают 50 мл
буферного раствора уксуснокислого аммония (pH 4,8) и встряхивают в те¬
чение часа, фильтруют через сухой фильтр. Фильтр предварительно очи¬
щают от следов цинка промыванием разведенной соляной кислотой и бидистил¬
лированной водой и высушивают. В полученной вытяжке цинк определяют
в форме его дитизоната, как описано выше при определении цинка в вытяжке
КС1 по Ринькису.
Реактив. Уксуснокислый аммоний (pH 4,8). Готовят из уксусной кислоты и ам¬
миака, очищенных от следов микроэлементов (см. выше раздел «Уксусно-аммонийная
вытяжка по Крупскому и Александровой»).
Определение цинка по Ринькису,
в модификации Почвенного института им. В. В. Докучаева
(Веригина, Добрицкая, 1974) в КС1 вытяжке
5 г почвы или 2,5 г торфа помещают в колбочку емкостью 100 мл из квар¬
ца или бесцинкового стекла, приливают 50 мл 1 н. раствора КС1, закрывают
колбочку пробкой (пластмассовой или корковой). Встряхивают в течение
часа на горизонтальном встряхивателе. Вытяжку фильтруют через свободный
от следов цинка фильтр (белая лента) в кварцевую колбочку.
Берут пипеткой 5—10 мл вытяжки и переносят в капельную воронку
на 50 мл, приливают 2 капли раствора метилоранжа, прибавляют по каплям
из градуированной пипетки 5 %-ный раствор уксуснокислого натрия до пе¬
рехода окраски индикатора в желтую, добавляют еще 0,2 мл- 5 %-ного раствора
уксуснокислого натрия и 2 мл 50%-ного раствора гипосульфита. Приливают
из бюретки 2 мл 0,02%-ного раствора дитизона в СС14. Встряхивают воронку
на вертикальном встряхивателе в течение 3 мин. Дают слоям разделиться
и сливают слой дитизона в СС14 в чистую капельную воронку. Экстракцию
цинка повторяют, приливая 1 мл раствора дитизона, дитизоновый слой сли¬
вают в ту же капельную воронку. К собранным в капельную воронку дити-
зонатам цинка приливают 25 мл 0,01 н. раствора аммиака и встряхивают
1—2 мин. Окрашенную дитизонатом цинка органическую фазу переносят
в сухую мерную колбу на 25 мл и доливают до метки СС14. Фотоколоримет-
рируют в кювете с толщиной просматриваемого слоя 10—20 мм с зеленым
светофильтром (536 нм), применяя в качестве нулевого раствора СС14.
Для приготовления градуировочной шкалы в капельные воронки поме¬
щают 0; 0,5; 1,0; 2,0; 3,0; 4,0 мл раствора цинка, содержащего в 1 мл 1 мкг
Zn, доливают бидистиллированной водой до 5 мл, приливают 2 капли метил-
402
К, В. Веригина
оранжа и далее ведут анализ, как описано для испытуемых растворов. По
полученным оптическим плотностям строят градуировочный график, по ко¬
торому находят содержание цинка в испытуемых растворах.
Реактивы. 1,0 н. раствор хлористого калия. 75 г хлористого калия растворяют
в бидистиллированной воде и доводят до объема 1 л. Раствор переносят в делительную
воронку и очищают, как описано выше.
Приготовление остальных реактивов, необходимых для определения цинка, см.
выше в разделе «Определение валового содержания...».
Определение цинка по Крупскому и Александровой (1964)
в вытяжке уксуснокислым аммонием
20 мл вытяжки буферным раствором уксуснокислого аммония с pH 4,8
(см. стр. 392) помещают в капельную воронку емкостью 100 лед и приливают
1 мл комплексообразующего раствора (уксуснокислый натрий + гипо¬
сульфит, pH 5,5—5*6). При добавлении к анализируемому раствору комплек-
сообразователя воронку нужно энергично встряхивать во избежание разло¬
жения комплексообразователя. Прибавляют 2—3 мл 0,001 %-ного раствора
дитизона в СС14, после чего воронку встряхивают в течение 3 мин., дают слоям
разделиться и сливают нижний дитизоновый слой в чистую сухую капель¬
ную воронку. Экстракцию цинка дитизоном продолжают до тех пор, пока
раствор дитизона не перестанет менять свою окраску. Собранные в капельной
воронке экстракты обрабатывают 20 мл раствора аммиака (1 : 200) для уда¬
ления избытка дитизона. Обработку дитизонатов цинка аммиаком производят
2 раза.
Окрашенный в красный цвет дитизонат цинка переносят в сухую мерную
колбу на 25 или 50 мл, доливают до метки СС14, перемешивают и фотоколо¬
риметрируют в сухой кювете с толщиной просматриваемого слоя 10 мм с зе¬
леным светофильтром (536 нм), применяя в качестве нулевого раствора СС14.
Для приготовления градуировочной шкалы в капельные воронки на
100 мл берут 0; 0,5; 1,0; 1,5; 2,0; 2,5; 3,5; 5,0; 6,0; 7,5 мл рабочего раствора
цинка с содержанием цинка 1 мкг в 1 мл. Доводят бидистиллированной во¬
дой до объема 20 мл, приливают 1 мл комплексообразователя. Прибавляют
в каждую воронку по 2—3 мл 0,001 %-ного раствора дитизона в СС14 и далее
ведут анализ, как описано для испытуемых растворов.
По полученным величинам оптических плотностей строят градуировоч¬
ный график, по которому находят содержание цинка в испытуемых рас¬
творах.
С. И. Тома (1970) предложена следующая шкала обеспеченности почв
цинком для вытяжки уксуснокислым аммонием, pH 4,8 (для условий Молда¬
вии):
Культура Степень Пределы содержания
обеспеченности Zn, мг/кг почвы
Зерновые Не обеспечены <0,3
Технические » » <0,3
Зерновые Ниже среднего 0,3—0,9
Технические Слабо 0,3—0,9
Зерновые Средне 0,9—1,5
Технические Ниже среднего 0,9—1,5
Зерновые Обеспечены >1,5
Реактивы. 1. Комплексообразователь. 125 г уксуснокислого натрия и 100 е
гипосульфита растворяют в 500 мл бидистиллированной воды и переносят в делитель¬
ную воронку. Очищают от следов цинка дитизоном, для чего приливают 5 мл 0,001 %-ного
раствора дитизона в СС14 и встряхивают 2—3 мин. Дают слоям разделиться и отбрасы¬
Memodu определения в почве меди, цинка, кобальта и бора
403
вают дитизоновый слой. Экстракцию дитнзоном продолжают до тех пор, пока он не будет
сохранять свою первоначальную зеленую окраску. Промывают несколько раз чистым
СС14 для устранения следов дитизона.
2. 0,001 %-ный раствор дитизона в СС14. 20 мг дитизона растворяют в 50 мл СС14.
Раствор переносят в делительную воронку на 500 мл, прибавляют 200 мл аммиака
(1 : 200) и встряхивают 3—5 мин. Дают слоям разделиться, отбрасывают слой СС14. Аммиач¬
ный раствор дитизона снова обрабатывают СС14; обработку продолжают до тех пор, пока
раствор не будет оставаться бесцветным или слабо-зеленым. Затем приливают в делитель¬
ную воронку 100 мл СС14, подкисляют несколькими каплями 10%-ной НС1 (ос. ч.) и
встряхивают. Водная фаза становится почти бесцветной, а дитизон переходит в слой
СС14, окрашивая его в зеленый цвет. Дитизоновый слой переносят в чистую делитель¬
ную воронку и несколько раз промывают горячей бидистиллированной водой для удале¬
ния следов HG1.
Раствор дитизона сливают в темную склянку и хранят в холодильнике. 0,02%-ный
раствор является запасным, из него готовят 0,001 %-ный рабочий раствор, разводя СС14
в 20 раз.
3. Аммиак 1 : 200. 1 часть 25%-ного аммиака (ос. ч.) на 200 частей бидистиллиро¬
ванной воды.
4. Стандартный раствор цинка. 0,05 г металлического цинка отвешивают на анали¬
тических весах, переносят в мерную колбу на 500 мл, приливают 2,5 мл серной кислоты
(уд. вес 1,84) и по мере растворения цинка доливают бидистиллированную воду. После
полного растворения цинка доливают бидистиллированной водой до метки. Серная кисло¬
та и бидистиллированная вода должны быть очищены от следов цинка.
Приготовленный таким образом стандартный раствор цинка содержит в 1 мл 100 мкг
цинка. Рабочий раствор цинка готовят разведением запасного раствора в 100 раз. Со¬
держание цинка в рабочем растворе отвечает 1 мкг в 1 мл.
Определение цинка по Кругловой (1972)
в вытяжке уксуснокислым натрием
5—10 лед вытяжки уксуснокислым натрием (см. стр. 393) помещают в ка¬
пельную воронку на 50 мл, прибавляют 2 капли метилоранжа и в случае
кислой реакции нейтрализуют до желтого цвета 5%-ным раствором уксус¬
нокислого натрия. Прибавляют 2 мл 50%-ного раствора гипосульфита.
Затем приливают из бюретки 2 мл раствора дитизона в СС14 и встряхивают
3 мин., дают фазам разделиться и сливают дитизоновый слой в сухую чистую
капельную воронку. Экстракцию дитизоном повторяют, прилив из бюретки
1 мл дитизона, встряхивают и присоединяют второй экстракт к первому,
а водную фазу отбрасывают.
К собранным в капельную воронку дитизонатным экстрактам приливают
20 мл 0,01 н. раствора аммиака для разрушения избытка дитизона и встря¬
хивают. Обработку аммиаком производят 2 раза. Окрашенный дитизонатом
цинка органический слой фильтруют через маленький сухой фильтр в мер¬
ную колбу на 25 мл, доводят раствор до метки чистым СС14, перемешивают
и фотоколориметрируют в кювете с толщиной просматриваемого слоя в 10 ммt
с зеленым светофильтром (536 нм), применяя в качестве нулевого раствора
СС14.
Для нормального обеспечения хлопчатника цинком почва должна содер¬
жать в 1 кг 2,0 ± 0,5 мг цинка, переходящего в вытяжку буферным рас¬
твором уксуснокислого натрия (pH — 3,5).
Приготовление градуировочной шкалы и необходимых реактивов см*
в разделе «Определение валового содержания ...»
404
К. В. Веригина
Определение цинка по Боратынскому
(Boratynski, Zietecka, 2970^ в вытяжке Эгнера — Рима — Доминго
5—10 мл вытяжки Эгнера — Рима — Доминго (см. стр. 394) помещают
пипеткой в коническую колбу с притертой пробкой объемом 100 мл, прибав¬
ляют 15 мл буферной маскирующей смеси, перемешивают и приливают из
бюретки точно 20 мл рабочего раствора дитизона. Колбу закрывают проб¬
кой и энергично встряхивают на горизонтальном встряхивателе в течение
5 мин. Снимают колбы с встряхивателя и после разделения фаз водный слой
отсасывают стеклянным капилляром, соединенным с водоструйным насосом
(см. рисунок). Слой СС14 с растворенным в нем дитизоном и дитизонатом цинка
переносят в кювету с толщиной просматриваемого слоя 10—20 мм и колори-
метрируют с зеленым светофильтром (510 нм), используя в качестве нулево¬
го раствора рабочий раствор дитизона в СС14. К каждой серии определений
готовят шкалу стандартных растворов в интервале от 0,0 до 20 мкг Zn. Ко¬
личество цинка в испытуемых растворах определяют по градуировочной
кривой, которую строят по величинам оптических плотностей стандартных
растворов, учитывая оптическую плотность холостого опыта.
Реактивы. 1. Буферная маскирующая смесь: а) 250 г лимоннокислого натрия
ч. д. a. (Na3CeH607*2H20) растворяют в горячей бидистиллированной воде в колбе на
1 л. После охлаждения доводят до метки бидистиллированной водой; б) 272 г уксусно¬
кислого натрия ч. д. a. (CH3C00Na*3H20) растворяют в бидистиллированной воде в
колбе на 1 л и доводят бидистиллированной водой до метки. 115,6 мл уксусной кислоты
ч. д. а. доводят бидистиллированной водой до 1 л. Смешивают раствор уксуснокислого
натрия с раствором уксусной кислоты в отношении 1:1; pH приготовленного раствора дол¬
жен равняться 4,7—4,8. Если pH раствора отклоняется от этой величины, то его необ¬
ходимо довести до нужной величины прибавлением раствора уксуснокислого натрия пли
раствора уксусной кислоты; в) 250 г гипосульфита ч. д. а. растворяют в бидистиллирован¬
ной воде в колбе на 1 л. После растворения соли раствор размешивают и доводят до мет¬
ки бидистиллированной водой.
Буферную маскирующую смесь готовят, смешивая растворы а, б, в в соотношении
4:5:6; pH смеси должен составлять 5,0—5,2.
2. Раствор дитизона. 2а. Запасной раствор. 100 мг дитизона растворяют в 250 мл четы¬
реххлористого углерода в колбе на 600 мл. Подогревают до температуры 50° С и фильтруют
в делительную воронку на 1 л, прибавляют 250 мл аммиака (12 мл 25%-ного аммиака
доводят бидистиллированной водой до 250 мл) и в течение 3 мин. энергично встряхивают,
при этом дитизон переходит в водный слой. После разделения фаз органическую фазу
отбрасывают, а водную фазу с растворенным в ней дитизоном несколько раз (2—5) про¬
мывают 10—15 мл СС14, каждый раз энергично встряхивая и после разделения фаз от¬
брасывая органическую фазу. Промывание с СС14 продолжают до тех пор, пока слой СС14
не будет окрашиваться в зеленый цвет. К промытому таким способом раствору дитизона
прибавляют по каплям 20%-ный раствор НС1 до перехода окраски раствора в зеленую.
Прибавляют 250ллСС14 и энергично встряхивают, при этом дитизон переходит в органи¬
ческую фазу. Отбрасывают водную фазу и промывают несколько раз раствор дитизона
в СС14 небольшими порциями бидистиллированной воды, до тех пор, пока водный слой
не будет оставаться бесцветным. Раствор дитизона тщательно отделяют от водного слоя
и сохраняют в склянке темного стекла с притертой пробкой в холодильнике при темпе¬
ратуре 2—5° С.
26. Рабочий раствор дитизона. 100 мл запасного раствора дитизона доводят СС14 до
объема 1 л. Хранят в склянке темного стекла при температуре 2—5°.
Стандартный запасной раствор цинка. 0,2470 г ZnS04, ч. д. а.,
прокаленного до постоянного веса при температуре 400—500°, растворяют в бидистиллиро¬
ванной воде, прибавив несколько капель концентрированной H2S04 (ч. д. а.), доводят би¬
дистиллированной водой до 1 л и перемешивают. 1 мл приготовленного раствора содержит
100 мкг Zn. Стандартный рабочий раствор готовят разведением в 50 раз запасного раст¬
вора Zn. 1 мл этого раствора содержит 2 мкг Zn.
Методы определения в почве Меди, цинка, кобальта и бора
405
Проверка чистоты рабочего раствора дитизона. Несмот¬
ря на то, что реактив предварительно очищается, перед выполнением серии анализов
необходимо проводить проверку его чистоты, так как дитизон легко загрязняется. Для
этого берут 5 мл рабочего раствора дитизона, помещают в тщательно очищенную пробир¬
ку, прибавляют 5 мл аммиака (12 мл 25%-ного аммиака доводят бидистиллированной
водой до 250 мл) и встряхивают. Когда слои разделятся, слой четыреххлористого угле¬
рода должен быть бесцветным или слегка желтоватым. Если появляется красная или фио¬
летовая окраска, очистку дитизона необходимо повторить.
Проверка чистоты реактивов и бидистиллированной
воды. В предварительно тщательно очищенную пробирку (см. ниже) помещают 5 мл
реактива, прибавляют несколько миллилитров рабочего раствора дитизона и 5 мл раз¬
бавленного раствора аммиака и встряхивают. Слой СС14 должен быть бесцвет¬
ным или слегка желтоватым. Если появляется красная или фиолетовая окраска, данный
реактив необходимо очистить, встряхивая его с небольшими порциями рабочего раствора
дитизона и удаляя остатки дитизона промыванием СС14.
Очистка лабораторного стекла. Перед выполнением серии анали¬
зов употребляемую стеклянную посуду следует очистить, прополаскивая ее несколько
раз небольшими порциями рабочего раствора дитизона до тех пор, пока раствор дитизона
не будет оставаться зеленым. После промывания посуды дитизоном ее прополаскивают
•бидистиллированной водой. Посуду при определении цинка не следует мыть водопровод¬
ной водой, а применять бидистиллированную воду и дитизон. Реактивы не следует филь¬
тровать, нельзя применять также смазывания притертых пробок.
Определение подвижного кобальта
Определение кобальта по Ринькису,
в модификации Почвенного института им. В. В. Докучаева
(Веригина, Добрицкая, 1974)
10 г почвы помещают в коническую колбу с притертой пробкой (250 мл),
приливают 100 мл 1,0 н. HN03, встряхивают на горизонтальном встряхива-
теле в течение часа и фильтруют через фильтр «белая лента».
25—50 мл вытяжки помещают в термостойкий стакан (на 100 мл), прили¬
вают 1 мл концентрированнойHN03,1 мл Н202 и выпаривают.досуха на закры¬
той этернитовой плитке. Обработку азотной кислотой и перекисью водорода
повторяют еще раз и выпаривают до начала кристаллизации солей. Затем
в стакан приливают 6 мл бидистиллированной воды, подкисленной НС1
(1 : 100), и раствор доводят до кипения. Приливают 1 мл 20 %-ного раствора
лимоннокислого натрия и 1 мл 40%-ного раствора уксуснокислого натрия
и кипятят около 1 мин. pH раствора должен быть несколько выше 5,5, что
проверяется по индикаторной бумаге. В случае необходимости pH раствора
доводят до нужной величины добавлением уксуснокислого натрия. Затем
к анализируемому раствору приливают 1 мл 0,05%-ного водного раствора
нитрозо-И-соли и доводят до кипения. Если анализируемый раствор при этом
становится желто-красным, приливают еще 1 мл 0,05%-ного раствора нитро-
зо-К-соли, 5 мл бидистиллированной воды и доводят до кипения. После ох¬
лаждения анализируемого раствора (раствор должен быть еще теплым) к не¬
му приливают 3 мл смеси ортофосфорной и азотной кислот и немедленно
тщательно перемешивают.
Затем переносят раствор в градуированные пробирки; так как в процес¬
се кипячения количество раствора в стакане несколько уменьшается, то
конечный объем раствора при перенесении его в пробирку бывает несколько
меньше 10 мл при внесении 1 мл нитрозо-И-соли и меньше 15 мл при внесе¬
нии 2 мл нитрозо-Б-соли. При внесении 1 мл нитрозо-И-соли конечный объем
раствора должен быть доведен до 10 мл, при внесении 2 мл нитрозо-И-соли —
. до 20 мл.
406
К. В. Веригина
Если раствор, содержащий комплекс кобальта с нитрозо-И-солью, будет
мутным или будет опалесцировать, перед фотоколориметрированием раствор
переносят в центрифужные пробирки и центрифугируют в течение 5—7 мин.
при 3000 об!мин и только после этого измеряют его оптическую плотность.
Комплекс кобальта с нитрозо-К-солью фотоколориметрируют в кювете с тол¬
щиной просматриваемого слоя в 20 мм с зеленым светофильтром (536 нм)9
применяя в качестве нулевого раствора бидистиллированную воду.
Для приготовления градуировочной шкалы в 10 мерных колб емкостью
100 мл помещают градуированной пипеткой 0; 0,5; 1; 2; 3; 4; 5; 6; 7; 8 мл
рабочего раствора сернокислого кобальта, содержащего в 1 мл 10 мкг ко¬
бальта. Приливают бидистиллированную воду примерно до 50 мл, прибав¬
ляют 5 мл 20 %-ного раствора лимоннокислого натрия и 5 мл 40 %-ного рас¬
твора уксуснокислого натрия, приливают 10 мл 0,05-ного раствора нитрозо-R-
соли и нагревают до кипения. Охлаждают и приливают к еще теплому рас¬
твору смесь ортофосфорной и азотной кислот и доводят до метки бидистиллиро¬
ванной водой, фотоколориметрируют, какуказанодля испытуемых растворов.
Почва, содержащая <0,2 мг/кг Со, переходящего в вытяжку 1,0 н.
HN03,— очень бедная; 0,2 — 1,0 — бедная; 1,5—2,0 — средняя; 4—5 —
богатая; ]>5 мг/кг — очень богатая кобальтом.
Реактивы. 1. А отная кислота 1 ы. Для приготовления 1,0 н. раствора HN05
68,9 мл концентрированной азотной кислоты (уд. вес 1,40) доводят до 1 л бидистиллиро¬
ванной водой. Желательно применение особо чистой азотной кислоты.
2. Натрий лимоннокислый трехзамещенный, 20%-ный водный раствор. 20 г трех-
замещенного лимоннокислого натрия растворяют в бидистиллированной воде и доводят
до объема 100 мл.
3. Натрий уксуснокислый, 40%-ный водный раствор. 40 г уксуснокислого натрия
растворяют в бидистиллированной воде и доводят до объема 100 мл.
4. Нитрозо-И-соль, 0,05%-ный раствор. 0,05 г нитрозо-И-соли отвешивают на
аналитических весах и растворяют в бидистиллированной воде, доводя до объема 100 мл.
Раствор должен быть свежеприготовленным.
5. Смесь ортофосфорной и азотной кислот. К 100 дел 85%-ной ортофосфорной кис¬
лоты приливают 20 мл азотной кислоты (уд. вес 1,40).
6. Стандартный раствор кобальта. Берут на аналитических весах 0,4770 г сульфата
кобальта CoS04-7H*0, помещают в стакан и растворяют в бидистиллированной воде.
Раствор переносят в мерную колбу на 1 л, подкисляют 1 мл серной кислоты (уд. вес.
1,84) и доводят до метки бидистиллированной водой. Полученный раствор содержит в
1 мл 100 мкг Со. Для приготовления рабочего раствора берут пипеткой 50 мл этого 'рас¬
твора, помещают в мерную колбу на 500 мл и доводят до метки бидистиллированной во¬
дой.
Определение кобальта по Крупскому и Александровой
(1964) в вытяжке уксуснокислым аммонием
50 мл вытяжки (см. стр. 392) помещают в фарфоровую чашку на 100 мл
и выпаривают досуха на водяной бане. Чашку с сухим остатком помещают
в холодный муфель и прокаливают в течение 1—1,5 час. при температуре
500—600°. Температура должна регулироваться терморегулятором.
После прокаливания и охлаждения чашки осадок в чашке растворяют
в 3 мл HN03 (1 : 1) и 2 мл НС1 (1:1). Если не происходит полного раство¬
рения, т. е. раствор не полностью прозрачен, чашку ставят на кипящую во¬
дяную баню, через 2 мин. раствор становится прозрачным. Избыток кислоты
нейтрализуют аммиаком (1 : 1) до появления слабой мути, которую удаляют
добавлением нескольких капель H2S04 (1 : 1) или HN03 (1 : 1). Чтобы полу¬
чить оптимальную кислотность среды и элиминировать влияние мешающих
определению кобальта элементов, к раствору прибавляют 8 мл 50%-ного
Методы определения в почве меди, цинка, кобальта и бора
407
раствора уксуснокислого натрия и нагревают до кипения. Кипячение про¬
должают 3—5 мин., при этом в осадок выпадают железо, медь и никель.
Кипячение следует проводить не более 5 мин., так как при более длительном
кипячении могут осаждаться обезвоженные формы уксуснокислого железа,
которые не растворяются полностью при нагревании с кислотой, а оставший¬
ся осадок будет затруднять определение кобальта. Когда осадок оформится,
в чашку прибавляют 10 мл 1 %-ного раствора нитрозо-И-соли или 5 мл
2 %-ного раствора нитрозо-И-соли, т. е. добавляют избыточное количество кра¬
сителя, что определяется присутствием в почвах многих мешающих компо¬
нентов и необходимостью производить измерение экстинкции при таком из¬
бытке нитрозо-И-соли, когда увеличение ее содержания уже не сказывается
на величине оптической плотности.
После добавления нитрозо-И-соли раствор кипятят около 1 мин. до дости¬
жения оптимальных условий образования комплекса кобальта, раствор
при этом интенсивно окрашивается. Он может иметь любой цвет, вплоть
до темно-бутылочного, в зависимости от содержания кобальта, никеля,
меди и железа, которые дают окрашенные комплексы. Для разрушения об¬
разовавшихся комплексов всех металлов, кроме кобальта, и растворения
осадка в чашку добавляют 5 мл концентрированной HN08. Кипятят в тече¬
ние 1 мин. до полного растворения осадка и просветления раствора, который
теперь имеет характерный для комплекса кобальта с нитрозо-Н-солью оран-
жево-красный цвет. Содержимое чашки переносят в мерную колбу объемом
50 мл, раствор доводят до метки бидистиллированной водой и фотоколоримет-
рируют в кюветах с толщиной просматриваемого слоя 10 мм с зеленым све¬
тофильтром.
Для приготовления градуировочной шкалы в мерные колбочки емкостью
50 мл приливают стандартный раствор кобальта (табл. 1).
Таблица 1
Шкала стандартных растворов для определения кобальта
Ni колбы
Стандартный раствор Со, мл
Со в 50 мл раствора,
мкг
Со в 1 мл раствора,
мкг
1 мкг/ли
10 мкг/мл
1
2,5
2,5
0,05
2
5,0
—
5,0
0,10
3
10,0
—
10,0
0,20
А
20,0
—
20,0
0,40
5
—
3,0
30,0
0,60
6
—
4,0
40,0
0,80
7
—
5,0
50,0
1,0
8
—
10,0
100,0
2,0
К содержимому колбочек прибавляют по 5 мл 50%-ного раствора уксусно¬
кислого натрия, нагревают до кипения и кипятят 3—5 мин., затем приливают
по 7,5 мл 2%-ного раствора нитрозо-И-соли и кипятят еще 1—2 мин.; после
чего прибавляют 5 мл концентрированной HNOs и опять кипятят 1 мин.
Дают колбочкам остыть, доводят бидистиллированной водой до метки и фо-
токолориметрируют, как и испытуемые растворы. По полученным величи¬
нам оптических плотностей строят градуировочный график, по которому
находят содержание кобальта в почвах.
Реактивы. 1. Уксуснокислый натрий, 50%-ный раствор. 250 г уксуснокислого
натрия растворяют в бидистиллированной воде и доводят до объема 500 мл•
408
К. В. Веригина
2. Нитрозо-И-соль, 2%-ный раствор. 10 г нитрозо-И-соли растворяют в бидистил¬
лированной воде и доводят до объема 500 мл.
3. Стандартный раствор кобальта. Навеску 0,0808 г СоС12*6Н20 помещают в мернук>
колбу на 200 мл и растворяют в бидистиллированной воде, приливая 3—4 капли НС1
(ос. ч.). Раствор доводят до метки водой. Этот запасной раствор содержит в 1 мл
100 мкг Со.
Рабочие растворы с содержанием Со 10 и 1 мкг/мл готовят путем разбавления запас¬
ного раствора.
Определение кобальта по Кругловой (1972)
в вытяжке уксуснокислым натрием
Е. К. Круглова рекомендует определять кобальт в буферных вытяжках
уксуснокислым натрием в форме комплекса кобальта с нитрозо-И-солью
или в форме комплекса с 2-нитрозо-1-нафтолом.
При определении кобальта с нитрозо-Й-солью 80—100 мл уксуснокисло¬
натриевой вытяжки (см. стр. 393) помещают в термостойкий стакан на 100—
150 мл, приливают 1 мл концентрированной HN03 и 1 мл 30%-ного раствора
Н202 и выпаривают на закрытой плитке до начала кристаллизации солей.
Обработку HN03 и Н202 повторяют дважды. Далее анализ ведут, как опи¬
сано в разделе «Определение кобальта по Ринькису, в модификации Почвен¬
ного института».
При определении кобальта в виде комплекса с 2-нитрозо-1-нафтолом в тер¬
мостойкие стаканы помещают 50—100 мл вытяжки. Если вытяжки из почв
буферным раствором уксуснокислого натрия окрашены слабо, сжигание
органического вещества не производят, в случае сильно окрашенных вытя¬
жек необходимо производить окисление органического вещества. Для этого
к помещенной в стакан вытяжке приливают 1 мл концентрированной HN03,
3 мл 30%-ного раствора Н202 и раствор кипятят. Обработку вытяжки азот¬
ной кислотой и перекисью водорода повторяют 2—3 раза, пока вытяжка не
будет иметь слабо-желтоватую окраску.
Затем прибавляют для связывания в комплекс железа 3—5 мл 20%-ного
раствора лимоннокислого аммония, раствор перемешивают стеклянной палоч¬
кой и нагревают до 60°. Устанавливают по индикаторной бумаге pH раство¬
ра и доводят его аммиаком до 5,5—6,0. Приливают 2 мл водного раствора
2-нитроЗо-1-нафтола, перемешивают и оставляют стоять в теплом месте 30—
40 мин. По истечении этого времени переносят раствор в капельную воронку *
тщательно обмывая стакан бидистиллированной водой. Приливают из бю¬
ретки точно 10 мл толуолового раствора 2-нитрозо-1-нафтола, встряхивают
2—3 мин. и оставляют стоять 2—3 часа. Верхний органический слой при этом
окрашивается в янтарно-розовый цвет. В водной фазе возможно выпадение
белого кристаллического осадка. Для разрушения нафтолатов других ионов
и растворения осадка в водной фазе в анализируемый раствор прибавляют
10 мл 22%-ной НС1, встряхивают и дают фазам разделиться. Водную фазу
далее отбрасывают, а органическую фазу вторично обрабатывают 5 мл 22%-ной
НС1 и встряхивают. После разделения фаз водную фазу отбрасывают.
Органическую фазу промывают 20—30 мл бидистиллированной воды и для
удаления избытка реагента и остатков кислоты дважды обрабатывают 10%-
ным раствором едкого калия (по 5 мл). В заключение органическую фазу про¬
мывают 5 мл НС1 и 20—30 мл бидистиллированной воды.
Раствор нафтолата кобальта в толуоле фильтруют через сухой маленький
фильтр в кювету с толщиной просматриваемого слоя 10 мм. Раствор должен
быть прозрачным и иметь розовый цвет. Фотоколориметрирование произ¬
водят с зеленым светофильтром (536 нм). Нулевой раствор — чистый толуол.
Для приготовления градуировочной шкалы в стаканчики берут 0; 1,0;
2,0; 3,0; 4,0 мл рабочего раствора сернокислого кобальта, содержащего
Методы определения в почве меди, цинка, кобальта и бора
409
1 мкг/мл Со, доводят бидистиллированной водой до объема 25 мл и далее про¬
водят анализ, как и для испытуемых растворов, начиная с установления
в анализируемом растворе pH 5,5.
Для нормального обеспечения хлопчатника (ведущей культуры в Средней
Азии) кобальтом почва должна содержать 0,2 ± 0,05 мг/кг кобальта,
переходящего в вытяжку буферным раствором уксуснокислого натрия
(pH 3,5).
Реактивы. 1. Лимоннокислый аммоний, 20%-ный раствор. 100 г двухзамещен-
ного лимоннокислого аммония растворяют в 300 мл бидистиллированной воды, добавляют
5 капель фенолфталеина и нейтрализуют аммиаком до розовой окраски. Затем доводят
бидистиллированной водой до 500 мл.
2. 0,04%-ный водный раствор 2-нитрозо-1-нафтола. Отвешивают на часовом стекле
на аналитических весах 0,04 г 2-нитрозо-1-нафтола и растворяют его 8 каплями 5%-
ного раствора КОН и 1ли бидистиллированной воды. Количественно смывают раствор
через воронку в мерную колбу на 100 мл. После растворения осадка доводят раствор бн-
дистиллированной водой до метки и перемешивают. Раствор можно употреблять для ана¬
лиза в течение 2 недель, сохраняя его в темной склянке.
3. Толуоловый раствор 2-нитрозо-1-нафтола. Готовят 150 мл раствора толуола,
насыщенного 2-нитрозо-1-нафтолом. Помещают 20 мл насыщенного 2-нитрозо-1-нафтолом
раствора толуола в колбу на 500 мл и доводят до метки чистым толуолом.
4. НС1, 22%-ная. Дважды перегнанная НС1 (см. стр. 389) является 22%-ной.
5. Аммиак, 10%-ный раствор. Разводят 25%-ный раствор аммиака в 2,5 раза.
6. КОН, 10%-ный раствор. 10 г КОН растворяют в бидистиллированной воде, дают
раствору остыть и доводят бидистиллированной водой до объема 100 мл.
7. Запасной и рабочий растворы сернокислого кобальта для приготовления шкалы
готовят, как описано в разделе «Определение валового содержания...», стандартные рас¬
творы.
Определение кобальта по Боратынскому
(Boratynski, Zietecka, 1970)
в вытяжке Эгнера — Рима — Доминго
25 мл вытяжки Эгнера — Рима—Доминго (см. стр. 394) помещают в кони¬
ческую колбу на 100 мл, прибавляют 5 мл 50%-ного раствора лимоннокисло¬
го натрия и 15 мл 50%-ного раствора уксуснокислого натрия, нагревают на
плитке приблизительно до 90°. При появлении мелких пузырьков прибав¬
ляют 5 мл Н202 и 0,5 мл раствора 2-нитрозо-1-нафтола. Перемешивают, сни¬
мают колбу с плитки и оставляют стоять 30 мин. при комнатной темпера¬
туре. После охлаждения к содержимому колбы прибавляют из бюретки точ¬
но 7 дел СС14. Колбу тщательно закрывают пробкой из мягкой пластмассы
и встряхивают в течение 10 мин. на горизонтальном встряхивателе. После
разделения фаз водный слой удаляют, отсасывая его при помощи стеклянно¬
го капилляра, соединенного с водоструйным насосом (см. рисунок). Органи¬
ческую фазу трижды встряхивают с 10 мл 2 н. раствора NaOH (каждый раз
по минуте), отсасывая водную фазу. Горлышко колбы обмывают бидистилли¬
рованной водой, слегка встряхивают и тщательно удаляют водную фазу.
Органический слой фильтруют через сухой фильтр в кювету фотоколоримет¬
ра и колориметрируют с зеленым светофильтром (530 нм), используя в ка¬
честве нулевого раствора СС14. Для каждой серии определений готовят шка¬
лу стандартных растворов в интервале 0,0—5,0 мкг Со, добавляя к ним по
25 мл экстракционного раствора. При расчете содержания Со в вытяжке
необходимо учитывать величину холостого опыта.
Реактивы. 1. Раствор лимоннокислого натрия. 500 г трехосновного лимонно¬
кислого натрия ч. д. a. (Na3CeH607*2H20) растворяют в 1 л горячей бидистиллированной
воды.
410
К. В. Веригина
2. Раствор уксуснокислого натрия. 500 г уксуснокислого натрия ч. д. a. (CH3COONa*
•ЗН20) растворяют в 700 мл бидистиллированной воды, прибавляют 300 мл Юн. НС1
и подогревают до полного растворения.
3. Перекись водорода 5%-ная. Готовится разбавлением бидистиллированной водой
пергидроля ч. д. а.
4. Раствор 2-нитрозо-1-нафтола. 1 г 2-нитрозо-1-нафтола растворяют в 100 мл ледя¬
ной уксусной кислоты ч.д.а. Раствор каждый раз готовят свежий.
5. 2 н. раствор NaOH. 80 г NaOHJ ч.д.а. растворяют в бидистиллированной воде и
доводят до 1 л.
6. Стандартный раствор кобальта. 0,2630 г CoS04, только что прокаленного при 400—
500° до постоянного веса, растворяют в бидистиллированной воде, прибавляя 2 мл кон¬
центрированной серной кислоты в мерной колбе на 1 д. После растворения соли доводят
раствор до метки бидистиллированной водой и тщательно перемешивают. 1 мл пригото¬
вленного запасного раствора содержит 100'мкг Со. Рабочий раствор Со готовят, разбавляя
в 100 раз стандартный запасной раствор. 1 мл рабочего раствора содержит 1 мкг Со..
Определение кобальта с 2-(2-пиридилазо)-5-диэтилметааминофенолом
(ПААФ) 1
Метод основан на получении окрашенного комплекса кобальта с ПААФ,
экстракции полученного комплекса хлороформом с последующим фотоколо-
риметрическим измерением интенсивности окраски полученного комплекса*
Для устранения влияния других элементов, реагирующих с ПААФ, их
окрашенные комплексы разлагают трилоном Б при нагревании. Необходи¬
мую реакцию среды (pH около 5) создают, прибавляя к анализируемой про¬
бе лимоннокислый и уксуснокислый натрий. Лимоннокислый натрий одно¬
временно препятствует выпадению в осадок полутораокисей. Использование
ПААФ не требует предварительной обработки анализируемых вытяжек*
Для определения кобальта с ПААФ из анализируемых вытяжек берут
следующие аликвоты: из вытяжки уксуснокислым аммонием по Крупскому
и Александровой — 25 мл, вытяжки уксуснокислым натрием по Кругловой—
20 мл, азотнокислой вытяжки по Ринькису — 5 мл. Вытяжку по Кругловой
и по Ринькису забуферивают, чтобы получить pH выше 5. Для этого к взя¬
тым аликвотам приливают 5 мл 20%-ного лимоннокислого натрия и 5 мл
40%-ного уксуснокислого натрия. Окрашивание раствора в пробе из вытяж¬
ки по Крупскому и Александровой проводится без добавления этих реакти¬
вов. Последующий ход анализа для всех вытяжек одинаков: к анализируемой
пробе прилпвают 1 мл 0,02%-ного раствора ПААФ, тщательно перемеши¬
вают и через 10 мин. приливают 1 мл насыщенного раствора трилона Б.
После перемешивания колбу с раствором помещают на водяную баню, нагре¬
тую до 60—80°, и выдерживают при этой температуре 30 мин. Охлажденный
раствор переносят в капельную воронку, приливают из бюретки 5 мл хлоро¬
форма и встряхивают в течение 1 мин. Дают слоям разделиться и сливают
нижний органический слой, содержащий комплекс кобальта с ПААФ, не¬
посредственно в кювету фотоколориметра с толщиной просматриваемого слоя
в 10 мм. Фотоколориметрируют с желто-зеленым светофильтром (550 нм),
используя в качестве нулевого раствор, содержащий все реактивы, кроме
анализируемой пробы.
Для приготовления градуировочной шкалы в конические колбы берут
0; 0,5; 1,0; 1,5; 2,5 3,5 и 5,0 мл рабочего раствора кобальта, содержащего
в 1 мл 0,2 мкг кобальта, и доливают до объема, соответствующего каждой
аликвоте, экстрагирующие растворы: уксуснокисло-аммонийный раствор
с pH 4,8 до объема 25 мл, уксуснокисло-натриевый раствор до 20 мл и 1 н.
1 Метод раэработан Центральным институтом агрохимического обслуживания — ЦИНАО
(«Методические указания...», 1973).
Методы определения в почве меди, цинка, кобальта и бора
411
HN03 до 5 мл. Далее поступают так же, как при анализе испытуемых раст¬
воров. По полученным величинам оптических плотностей строят градуиро¬
вочную кривую, по которой находят содержание кобальта в испытуемых
растворах.
Реактивы. 1. Натрий трехзамещенный лимоннокислый, 20%-ный раствор. 200 г
лимоннокислого натрия растворяют в бидистиллированной воде и доводят объем до 1 л.
2. Натрий уксуснокислый, 40%-ный раствор. 400 г уксуснокислого натрия раство¬
ряют в бидистиллированной воде и доводят до объема 1 л.
3. Трилон Б, насыщенный раствор. 10 г трилона Б растворяют в 100 мл бидистил¬
лированной воды.
4. ПААФ, 0,05%-ный раствор. 50 мг ПААФ растворяют в этиловом спирте и доводят
спиртом до объема 100 мл.
5. Стандартный запасной раствор кобальта. Растворяют 0,4770 г сульфата кобальта
(CqS04*7H20) в бидистиллированной воде, подкисляют 1 мл серной кислоты (уд. вес
1,84) и доводят бидистиллированной] водой до объема 1 л. Полученный раствор содержит
100 мкг/мл кобальта.
6. Рабочие растворы кобальта: 2,5 мл запасного стандартного раствора кобальта
отбирают пипеткой в мерную колбу на 250 мл и доводят до метки соответствующим экст“
рагирующим раствором: при анализе вытяжек по Крупскому и Александровой — буфер-
рым раствором уксуснокислого аммония с pH 4,8, по Кругловой — буферным раство-
ном уксуснокислого натрия с pH 3,5, по Ринькису — 1 н. азотной кислотой. Полученный
раствор содержит 1 мкг/мл Со. Из этого раствора отбирают 10 мл в мерную колбу на 50 мл
и доводят до метки соответствующим экстрагирующим раствором. Полученный раствор
содержит 0,2 мкг/мл Со. Этот раствор непосредственно используют для -фиготовления
градуировочной шкалы.
Рабочие растворы кобальта готовят в день проведения анализа.
Определение валового содержания бора
Определение бора по Элъшлегелю
(Oelschldger, 1958) с 1,1-диантримидом
1 г тонкорастертой почвы сплавляют в течение 20—25 мин. в платиновом
тигле с 3 г смеси Na2C08 и К2С08 (1 : 1) при температуре 900° С. Горячий
тигель помещают в холодную воду и отставший от стенок тигля сплав раст¬
воряют в 30 мл бидистиллированной воды, нейтрализуют и подкисляют 10 мл
10 н. H2S04. Затем раствор выпаривают до половины на водяной бане. При
этом кремнекислота выпадает в осадок. Переносят с помощью 10 мл 10 н.
H2S04 раствор с осадком в мерную колбу на 100 мл, хорошо встряхивают
и доливают до метки водой. Оставляют стоять до следующего дня. Берут пи¬
петкой аликвот ъ2 млъ переносят в мерную колбу на 50 мл, прибавляют 5 мл
концентрированной H2S04 и немного персульфата аммония, ставят на 1 час
в термостат при 100° С. Приливают (при затемненном дневном свете) 5 мл
1,1-диантримида и выдерживают 3 часа в термостате при 90°. Охлаждают
и измеряют окраску в кювете с толщиной просматриваемого слоя в 2 см
при 630 нм, против воды.
Для приготовления стандартной шкалы 0,5715 г Н3В04 растворяют в би¬
дистиллированной воде, доводят до 1 д. 1 дед этого раствора содержит
100 мкг В.
Готовят серию стандартных растворов, для чего доводят бидистиллирован¬
ной водой:
2 мл до 200 мл = 1 мкг/мл, 10 мл до 250 мл = 4 мкг/мл,
5 мл до 250 мл = 2 мкг/мл, 10 мл до 200 мл = 5 мкг/мл,
6 мл до 200 мл = 3 мкг/мл, 12 мл до 200 мл = 6 мкг/мл.
412
К. В. Веригина
Помещают в серию мерных колбочек на 50 мл по 2 мл каждого раствора
и обрабатывают дальше, как и испытуемые растворы.
Реактив. 1,1-диантримид. Растворяют 0,2 г 1,1-диантримида в 1800 мл концент¬
рированной H2S04 и 200 мл воды.
Определение бора по Джексону (Jackson, 1962) с куркумином
Сплавляют в платиновом тигле 0,5 г тонкорастертой почвы с 3 г безводно¬
го углекислого натрия. В каждую серию анализов включают холостой опыт.
Тигель охлаждают и помещают в стакан из безборного стекла, содержащий
около 50 мл бидистиллированной воды. Покрывают стакан покровным стек¬
лом и постепенно прибавляют приблизительно 4 н. серную кислоту, пока
сплав не растворится (обычно бывает достаточно около 14 мл кислоты). Реак¬
ция раствора будет находиться в пределах 6,0—6,8 (определяют с бромти-
молблау, как с внутренним индикатором). Переносят полученный раствор
в мерную колбу на 500 мл, смывая тигель и стакан несколько раз бидистил¬
лированной водой, так чтобы общий объем не превышал 150 мл. Прибавляют
этиловый спирт, почти до объема 500 мл, и осторожно перемешивают содер¬
жимое колбы. Прибавляют небольшое количество соды, так, чтобы реакция
была слабощелочной, и снова перемешивают. Затем суспензию центрифуги¬
руют или фильтруют через плотный фильтр для получения прозрачного раст¬
вора.
Помещают 400 мл полученного прозрачного раствора в стакан из безбор¬
ного стекла и прибавляют 100 мл воды, чтобы предотвратить последующее
осаждение. Выпаривают раствор до небольшого объема и переносят в плати¬
новую чашку, где выпаривают досуха. Прокаливают чашку для разложения
органического вещества. После того как чашка остынет, прибавляют 5 мл
0,1 н. соляной кислоты и осторожно разминают остаток стеклянной палочкой.
Переносят 1 мл полученного раствора в чашку для выпаривания (лучшие
результаты получаются при содержании во взятой для анализа аликвоте
от 0,2 до 0,8 мкг В), прибавляют 4 мл куркумин-щавелевокислого реактива
и покачивают чашку, чтобы привести раствор в контакт с пробой.
Опускают чашку на 1 еле в водяную баню с температурой 55 ± 3°. Темпе¬
ратура бани должна контролироваться. Чашки после выпаривания жидко¬
сти должны простоять на бане 15 мин., затем их снимают. Смачивают остаток
спиртом, применяя промывалку с тонким носиком. Через несколько минут
переносят содержимое чашек с помощью спирта и стеклянной палочки в цент¬
рифужную пробирку на 15 мл (в этой стадии анализа безборное стекло не
является необходимым, так как окраска уже больше развиваться не будет)
и центрифугируют 10 мин. при 1,5—2 тыс.об/мин. Декантируют раствор
в мерные колбочки на 25 мл и доливают спиртом до 25 мл. Следы вытяжки г
оставшейся в центрифужной пробирке, не имеют значения, так как холостой
опыт и стандартные шкалы готовятся таким же образом.
Измеряют окраску растворов в кювете с толщиной просматриваемого-
слоя в 20 мм с зеленым светофильтром (536 нм). Окраска растворов стабильна
в течение 1—2 час.
Для приготовления стандартной шкалы помещают в чашки для выпари¬
вания 0,1,2, 3, 4 и 5 мл стандартного раствора В (в 1 мл 1 мкг В), прибавляют
5 мл суспензии Са(ОН)2 и выпаривают досуха. Охлаждают чашку и прибав¬
ляют 1 каплю фенолфталеина. Прибавляют по каплям 2,5 н. НС1, пока окрас¬
ка индикатора не исчезает, и затем добавляют 0,5 мл той же кислоты. Прибав¬
ляют 4 мл куркумин-щавелевокислого реактива и далее продолжают ана¬
лиз, как и с испытуемыми растворами.
Реактивы. 1. Натрий углекислый, безводный.
2. Серная кислота 4 н. Вливают в бидистиллированную воду 112 мл серной кислоты
(уд. вес 1,84) и доливают до 1 л водой.
Методы определения в почве меди, цинка, кобальта и бора
413
3. Соляная кислота, 0,1 н. 8,23 мл НС1 (уд. вес 1,19) или 16,6 мл дважды перегнан¬
ной НС1 (уд. вес 1,1) разбавляют до 1 д бидистиллированной водой.
4. Соляная кислота, 2,5 н. 205,75 мл соляной кислоты (уд. вес 1,19) или 413 мл
дважды перегнанной НС1 (уд. вес 1,1) разбавляют до 1 л бидистиллированной водой.
5. Куркумин-щавелевокислый реактив. Растворяют 0,04 г тонкоизмельченного кур-
кумина и 5 г щавелевой кислоты в 100 мл этилового спирта.
6. Суспензия гидроокиси кальция. 0,4 г Са(ОН)2 взбалтывают в 100 мл бидистилли¬
рованной воды.
7. Индикатор бромтимолблау. Размешивают в ступке 0,1 г реактива с 16 мл 0,01 н.
NaOH и доливают водой до 250 мл.
8. Фенолфталеин. Растворяют 0,05 г фенолфталеина в 50 мл 95%-ного спирта и 50 лм
бидистиллированной воды.
9. Стандартный раствор бора. Растворяют 0,5715 г борной кислоты в бидистилли¬
рованной воде и доливают до 1 л в мерной колбе. 1 мл этого раствора содержит 100 мкг
В. Для приготовления рабочего раствора бора 10 мл этого раствора разводят бидистилли¬
рованной водой до 1 л, получая раствор с содержанием в 1 мл 1 мкг В.
Определение бора по Биру (Bear, 1964) с хинализарином
Навеску тонкоизмельченной почвы в 3—5 г перемешивают с трех¬
кратным количеством углекислого натрия, помещают в платиновый ти¬
гель и ставят в холодный муфель, постепенно доводят температуру да
900° С. Сплав охлаждают и переносят в колбу на 250 мл, тщательно об¬
мывая тигель горячей водой. Объем жидкости в колбе должен быть око¬
ло 50—60 мл. Колбы оставляют на ночь или встряхивают до тех пор,
пока весь сплав не растворится. Затем осторожно прибавляют концент¬
рированную НС1 из расчета 2 мл на 1 г Na2C03, встряхивают в течение
5—10 мин., прибавляют небольшими порциями СаС03 до тех пор, пока
раствор не станет щелочным (качественная реакция) и не выпадет в оса¬
док гидрат железа и алюминия. Необходимо*, чтобы СаС03 был в избытке.
Колбу закрывают пробкой с обратным холодильником и осторожно кипятят
на слабом огне в течение 10—15 мин. Затем суспензию фильтруют через
Бюхнеровскую воронку, осадок 3—4 раза промывают горячей водой, каж¬
дый раз отсасывая досуха. Фильтрат переносят в стакан на 250 мл и упари¬
вают на водяной бане до объема 100 мл. Переносят раствор в мерную колбу
на 100 мл и для определения бора берут 1 мл раствора.
При определении бора с хинализарином 1 мл раствора (эквивалентный
0,05 г почвы) помещают в пробирку с притертой пробкой и добавляют из
бюретки точно 10 мл раствора хинализарина. Закрывают пробирку притертой
пробкой и быстро перемешивают. Оставляют стоять 30—60 мин., после чего
сравнивают со стандартной шкалой.
Для приготовления раствора хинализарина растворяют 5 мг хинализарп-
на в 1 л серной кислоты.
Для приготовления шкалы стандартных растворов 2,8578 г Н3В03 раст¬
воряют в 1 л воды, из этого раствора (а) приготовляют второй раствор (Ь)
с содержанием в 1 мл 10 мкг В, для чего 20 мл раствора «а» доводят бидистил¬
лированной водой до 1 л. Затем приготавливают раствор «с» с содержанием
в i мл i мкг В, для чего 100 мл раствора «Ь» доводят бидистиллированной
водой до 1 л.
Для приготовления шкалы берут в пробирки с притертыми пробками
различные количества растворов (табл. 2), доводят объем растворов
до 1 мл бидистиллированной водой и затем добавляют из бюретки по 10 мл
раствора хинализарина в серной кислоте. Окраска шкалы развивается
в течение 30 мин. и в герметически закрытых пробирках, защищенных от
дневного света, не изменяется в течение длительного срока (двух-трех не¬
дель).
414
К. В, Веригина
Таблица 2
Приготовление шкалы из растворов «Ь» и «с»
пробирки
Бидистиллиро-
ванная Н*0, мл
Раствор В,
МЛ
В, мкг
JSft
пробирки
Бидистиллиро-
ванная Н,0, мл
Раствор В,
мл
В, мкг
1
1,00
0,00
0,0
7
0,85
0,15 (Ь)
1 ,5
2
0,80
0,20 (с)
0,2
8
0,80
0,20 (Ь)
2,0
3
0,60
0,40 (с)
0,4
9
0,75
0,25 (Ь)
2,5
4
0,40
0,60 (с)
0,6
10
0,70
0,30 (Ь)
3,0
5
0,20
0,80 (с)
0,8
И
0,65
0,35 (Ь)
3,5
6
0,00
1,00 (с)
1,0
12
0,60
0,40 ib)
4.0
Определение подвижного бора
При определении подвижного бора в почвах наиболее часто применяется
его извлечение кипячением навески почвы с водой. Различные исследовате¬
ли рекомендуют при извлечении подвижного бора использовать различное
соотношение почва: вода и разное время взаимодействия.
За последнее время описано определение подвижного бора в вытяжках
буферным раствором уксуснокислого аммония (Крупский, Александрова,
1964) и лактат-ацетат-аммонием (Boratynski, Zietecka, 1970), рекомендуемых
в качестве групповых экстрагентов. Ниже дается описание некоторых из
предложенных методов.
Определение воднорастворимого бора по Ринъкису (1963)
5 г воздушно-сухой почвы помещают в колбочку на 100 мл из безборного
стекла, приливают 25 мл кипящей бидистиллированной воды и добавляют
раствор CuS04 (2 г CuS04-5H20 растворяют в 100 мл бидистиллированной
воды): для песчаных и супесчаных почв с небольшим содержанием органи¬
ческого вещества — 0,2 мл, для остальных — 0,5 мл (к холостому анализу
CuS04 не добавляют). Колбочки закрывают корковыми пробками, через кото¬
рые пропущена стеклянная трубка из безборного стекла, играющая роль
холодильника (длина трубки 25—30 см) и медленно кипятят 10 мин. Корко¬
вая пробка может быть заменена пришлифованной стеклянной пробкой
с холодильником из безборного стекла. После кипячения содержимое кол¬
бочки взбалтывают в течение еще 5 мин. и фильтруют через беззольный
фильтр. Затем берут 20—40 мл фильтрата и помещают в 50 мл колбочку
из безборного стекла, добавляют 1 мл 30%-ной Н202, медленно кипятят при¬
близительно в течение 1 мин. до обесцвечивания раствора и добавляют 1 кап¬
лю 2 н. NaOH.
Затем раствор выпаривают досуха (не должно быть влаги на горлышке
колбочки). Если остаток окрашенный, добавляют 0,5 мл Н202 и снова выпа¬
ривают досуха. В процессе выпаривания нагревание ведут осторожно, с тем,
чтобы избежать механических потерь при быстром выделении газов. Также
следует учесть, что стекло колбочек нетермостойкое и в горячем виде их нель¬
зя ставить на холодную поверхность. Сухой остаток в колбочке растворяют
в 0?5 мл раствора гипофосфита, затем добавляют 4,5 мл H2S04 (уд. вес 1,84)
и 0,5 мл рабочего раствора хинализарина, содержимое колбочки сразу пере¬
носят в сухую пробирку (из безборного стекла) с притертой пробкой.
Через 30 мин. анализируемый раствор сравнивают с образцовой шкалой.
Если раствор синее последней пробирки шкалы, проводят разбавление. Для
этого прибавляют 0,5 мл раствора гипофосфита, 4,5 мл Н2Э04(уд. вес 1,84)
Методы, определения в почве меди, цинка, кобальта и бора
415
и 0,5 дел раствора хинализарина. Если раствор синий и постепенно обесцве¬
чивается, что указывает на присутствие окислителей (остаток Н202), окис¬
ляющих хинализарин, обработку вытяжки повторяют, тщательно удаляя
остаток влаги из колбочки.
Для приготовления шкалы образцовых растворов готовят следующие
стандартные растворы.
1. Раствор «а». Растворяют 2,8578 г Н3В03 и доводят бидистиллированной водой до
0,5 |л. 1 мл этого раствора содержит i мг В.
2. Раствор «Ь» (содержит 10 мкг бора в 1 мл). Получают путем разбавления раствора
«а» в 100 раз (10 мл раствора «а» доводят бидистиллированной водой до 1 л).
3. Раствор «с» (содержит 1 мкг бора в 1 мл). Получают разбавлением раствора «Ь» в
10 раз (100 мл раствора «Ь» доводят бидистиллированной водой до i л).
Наливают в серию пробирок с притертыми пробками определенное ко¬
личество стандартных растворов (табл. 3).
Таблица 3
Шкала стандартных растворов для определения бора
пробирки
Показатели
0
1
2
3
4
3
6
7
8
9
Стандартный раствор
с
С
ь
ь
Ь
b
ь
ь
ь
Количество стандарт¬
ного раствора, мл
0
0,5
1,0
0,15
0,2
0,25
0,3
0,35
0,4
0,45»
Количество бора в про¬
бирке, мкг
0
0,5
1,0
1,5
2,0
2,5
3,0
3,5
4,0
4,5
Количество бора в 1 мл
колориметрируемого рас¬
твора, мкг
0
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
. 1»
0,45
В каждую пробирку доливают бидистиллированную воду с таким расче¬
том, чтобы общий объем жидкости в пробирке составлял 1 мл. Затем прили¬
вают 9 мл х. ч. H2S04 (уд. вес 1,84) и 1 мл раствора хинализарина. Пробирки
немедленно закрывают пробками и содержимое перемешивают. Через час
шкала готова для сравнения анализов. Хорошо закупоренная шкала сохра¬
няется в темноте без изменений в течение одного месяца.
Для вычисления результатов анализа пользуются формулой:
абв
х ~~ нг ’
где х — содержание бора в 1 кг почвы, мг; а — содержание бора в 1 мл в совпадающей
пробирке шкалы, мкг; б — объем колориметрируемого раствора, мл (добавленное коли¬
чество раствора гипофосфита не учитывается); в — общий объем вытяжки, мл; н—навес¬
ка почвы, г\ г — взятый для определения бора объем вытяжки, мл.
Реактивы. 1. Раствор гипофосфита. 10 г гипофосфита натрия (калия или каль¬
ция) растворяют в бидистиллированной воде, добавляют 5 мл концентрированной НС1
п водой доводят до 100 мл.
2. Раствор хинализарина. 2а. Исходный раствор. 50 мг хинализарина растворяют
в 100 мл х.ч. концентрированной H2S04 (уд. вес 1,84). *
26. Рабочий раствор хинализарина. 10 мл исходного раствора'хинализарина доводят
до 100 мл концентрированной H2S04.
416
К. В. Веригина
Определение воднорастворимого бора
по Никиткиной (1959) с кармином
10—20 г воздушно-сухой почвы, просеянной через сито d в 1 мм, помещают
в кварцевую колбу или колбу из безборного стекла, приливают 30—60 мл
кипящей бидистиллированной воды, 0,5 мл 3%-ного раствора CuS04, при¬
соединяют обратный холодильник и кипятят в течение 5 мин. при постоянном
помешивании. После охлаждения раствор фильтруют через плотный склад¬
чатый фильтр. Берут определенный объем фильтрата и помещают в кварце¬
вую колбочку или колбочку из безборного стекла, добавляют 1 каплю 2 н.
NaOH и 0,5—1,0 мл 30%-ной Н202, медленно кипятят 1 мин. для обесцве¬
чивания жидкости. Если вытяжка не обесцветилась, то добавляют еще
0,5 мл Н202. Содержимое колбочки выпаривают досуха на водяной бане или
этернитовой плитке. Сухой остаток в той же колбочке растворяют в 1 мл
0,5 н. H2S04, прибавляют 0,5 мл восстановителя (гипофосфита натрия или
калия) и прибавляют 19 мл 0,005%-ного раствора кармина. Содержимое
колбочки тщательно перемешивают, закрывают и по истечении 1,5—2,0 час.
фотоколориметрируют в кювете с толщиной просматриваемого слоя в 10 мм
при 610 нм, применяя в качестве нулевого раствора воду.
Для приготовления стандартной шкалы в кварцевые цилиндры или квар¬
цевые пробирки с притертыми пробками наливают из микробюреток указан¬
ное в табл. 4 количество стандартного рабочего раствора борной кислоты
и 0,5 н. H2S04, прибавляют 0,5 мл гипофосфита калия (или натрия) и 19 мл
0,005%-ного раствора кармина, закрывают пробкой и перемешивают. Через
1,5—2,0 часа фотоколориметрируют растворы, как указано для испытуе¬
мых растворов.
Таблица 4
Шкала стандартных растворов для определения бора
№ про¬
бирки
0,6 н. H.S04,
мл
Рабочий
раствор
НэВОа, мл
Количество
В, мкг
Ne про¬
бирки
0,5 н. H,S04,
мл
Рабочий
раствор
НаВОа, мл
Количество
В, мкг
1
0,95
0,05
0,5
5
0,40
0,60
6,0
2
0,90
0,10
1,0
6
0,20
0,80
8,0
3
0,80
0,20
2,0
7
0,00
1,00
10,0
4
0,60
0,40
4,0
Реактивы. 1.0,5 н. H2S04. 14 мл H2S04 (уд. вес. 1,84) вливают в бидистиллиро-
ванную воду и доводят объем до 1 д.
2. 0,005%-ный раствор кармина. 50 мг кармина растворяют в 1 л H2S04 (уд. вес
1,84).
3. 3%-ный раствор CuS04*3 г CuS04 растворяют в бидистиллированной воде и доводят
до 100 мл.
4. 10%-ный раствор гипофосфита. 10 г гипофосфита калия или натрия растворяют в
бидистиллированной воде, добавляют 5 мл НС1 (уд. вес 1,19) и доводят водой до
100 мл.
5. Стандартный раствор бора. Для приготовления запасного раствора бора 2,858 г
перекристаллизованной Н3В03 растворяют в 0,5 н. H2S04 в мерной колбе на 1 л, доводя
до метки тем же раствором кислоты. 1 мл этого раствора содержит 0,5 мг В. 20 мл этого
раствора разбавляют 0,5 н. H2S04, доводя до объема 1 л и получают рабочий раствор с
содержанием в 1 мл 10 мкг В.
Методы, определения в почве меди, цинка, кобальта и бора
417
Определение бора по Крупскому и Александровой (1964)
в вытяжке буферным раствором уксуснокислого аммония
5 г почвы помещают в колбочку на 100 мл из безборного стекла с при¬
тертой стеклянной пробкой и приливают 25 мл буферного раствора уксусно¬
кислого аммония с pH 4,8; взбалтывают в течение 30 мин. Фильтруют
в сухие колбочки из безборного стекла. 5 мл вытяжки помещают в плати¬
новые или фарфоровые чашки, прибавляют по 1 капле фенолфталеина и по
4 мл насыщенного раствора Са(ОН)2. Последний прибавляется для создания
щелочной среды. Чашку ставят на водяную баню и раствор выпаривают до¬
суха. Затем ее помещают в муфельную печь и прокаливают при температу¬
ре 500—550° до тех пор, пока осадок не станет белым.
После охлаждения к осадку в чашку прибавляют 0,5 мл смеси, состоя¬
щей из этилового спирта и H2S04. Чтобы перевести марганец в растворимую
форму, добавляют 2 капли свежеприготовленного 20%-ного раствора сер¬
нокислого железа. Прибавляют 10 мл 0,01 %-ного раствора кармина. Со¬
держимое чашки осторожно перемешивают и оставляют стоять в темном месте
на 2 часа, затем измеряют оптическую плотность растворов в кюветах с тол¬
щиной просматриваемого слоя 10 мм при длине волны 610 нм.
Нулевой раствор (раствор сравнения) готовят следующим образом: в пла¬
тиновую чашку наливают 4 мл насыщенного раствора Са(ОН)2 и каплю фе¬
нолфталеина, выпаривают и после охлаждения прибавляют 0,5 мл смеси
спирта и серной кислоты, 2 капли сернокислого железа и 10 мл 0,01 %-ного
раствора кармина.
Для приготовления калибровочной шкалы в платиновые чашки наливают
0,5; 1,0; 2,0; 3,0; 4,0; 6,0; 8,0 и 10 мл рабочего раствора бора с содержанием
в 1 мл 1 мкг В; прибавляют по 4 мл Са(ОН)2, выпаривают досуха на водяной
бане, охлаждают и добавляют по 0,5 мл смеси серной кислоты и этилового
спирта, смачивая этой жидкостью осадок. Затем прибавляют 10 мл 0,01%-
ного раствора кармина, перемешивают и оставляют стоять 2 часа до образо¬
вания устойчивой окраски, после чего измеряют оптическую плотность, как
и для испытуемых растворов.
Реактивы. 1. Раствор кармина. На аналитических весах отвешивают 100 мг
кармина, переносят в мерную колбу на 1 л, приливают 600—700 мл серной кислоты#
(уд. вес 1,84) и взбалтывают до полного растворения кармина. Доводят до метки концент*
рированной серной кислотой, взбалтывают и сохраняют в темном месте.
2. Смесь серной кислоты и этилового спирта. Сначала готовят серную кислоту 1 : 1
с этой целью к 50 мл бидистиллированной воды приливают 50 мл серной кислоты (уд.
вес. 1,84). Затем 25 мл приготовленной кислоты смешивают с 2Ь мл 96%-ного этилового
спирта.
3. Насыщенный раствор гидроокиси кальция. 1,5 г СаО помещают в мерную колбу
на 500 мл, доводят до метки бидистиллированной водой, взбалтывают, фильтруют и со¬
храняют в склянке без доступа углекислоты.
4. Сернокислое железо. 10 г сернокислого железа отвешивают на технических ве¬
сах, навеску переносят в колбу емкостью 50 мл, растворяют в бидистиллированной воде и
доводят до метки.
5. Стандартный запасной раствор бора. На аналитических весах отвешивают 0,4407 г
NajBaCVlOH^, навеску переносят в мерную колбу на 500 мл, растворяют в бидистилли¬
рованной воде и доводят до метки»
Рабочий раствор бора с содержанием в 1 мл 1 мкг бора готовят разведением запасного
раствора в 100 раз.
418
К. В. Веригина
Определение бора по Боратынскому
(Baratynski, Zietecka, 2970,)
с 1,1-диантримидом в вытяжке Эгнера — Рала — Доминго
10 мл вытяжки Эгнера—Рима — Доминго (см. стр. 394) помещают в кол¬
бу на 100 мл, прибавляют 1 мл 10%-ного раствора фосфата аммония и выпа¬
ривают досуха на водяной бане. К сухому остатку прибавляют 2 мл кон¬
центрированной серной кислоты, перемешивают вращательными движениями
и ставят на 15 мин. в термостат при температуре 100°. Затем прибавляют око¬
ло 500 мг растертого персульфата аммония и ставят в термостат еще на
20 мин. Если по истечении этого времени раствор не обесцвечивается, следует
добавитья еще 500 мг персульфата аммония. К обесцвеченному раствору
прибавляют около 100 мг раствора гидразин сульфата, перемешивают вра¬
щательными движениями и держат в термостате при 100° 20 мин. После ох¬
лаждения к содержимому колбы прибавляют (в темном помещении) 20 мл
0,02 %-ного раствора 1,1-диантримида, перемешивают несколько раз вра¬
щательными движениями и переливают в коническую колбу с притертой
пробкой на 50 мл. Колбу закрывают притертой пробкой и держат в термостате
3 часа при температуре 90° С. Спустя 3 часа колбу с раствором вынимают из
термостата и ставят в темное место, где выдерживают до полного охлаждения.
Комплекс бора с 1,1-диантримидом колориметрируют при 630 нм, применяя
в качестве нулевого раствора воду.
Для каждой серии определений необходимо приготовлять градуировоч¬
ную шкалу в интервале 0,0—5,0 мкг В. Стандартные растворы помещают
в серию колб, доливают до 10 мл бидистиллированной водой и далее посту¬
пают, как с испытуемыми растворами.
В каждую серию опытов необходимо включать холостой опыт, учитывая
его величину при расчете содержания бора в почве.
Реактивы. 1. 10%-ный раствор фосфата аммония. 10 г (NH4)2HP04, ч.д.а.,
растворяют в бидистиллированной воде и доводят до 100 мл.
2. Персульфат аммония, ч.д.а., растертый в агатовой ступке порошок.
3. Гидразин сульфат, ч.д.а., растертый в агатовой ступке порошок.
4. 0,01 %-ный раствор 1,1-диантримида. 100 лег 1,1-диантримида растворяют в серной
кислоте, разбавленной бидистиллированной водой,— 9 частей концентрированной серной
кислоты (уд. вес 1,84) и 1 часть воды. Хранить в посуде темного стекла.
5. Стандартный]раствор борной кислоты. 0,5715 г борной кислоты (ч.д.а.) растворяют
в бидистиллированной воде в мерной колбе на 1 л и после растворения доводят до метки
бидистиллированной водой. 1 мл этого раствора содержит 100 мкг В. Стандартный рабочий
раствор готовят разведением стандартного запасного раствора в 100 раз. 1 мл рабочего
раствора содержит 1 мкг В.
Вся посуда, в которой проводится определение бора, должна быть из безборного
стекла или кварца.
Литература
Веригина К. В., Добрицкая Ю. И. Опреде¬
ление в почвах и растениях микроэлемен¬
тов химическими методами.— В кн. «Ме¬
тоды определения микроэлементов в поч¬
вах, растениях и водах». М., «Колос»,
1974.
Даутов Р. КМинибаев В. Г. Картограм¬
мы содержания микроэлементов в почвах
Татарской АССР.— В сб. «Микроэлемен¬
ты в сельском хозяйстве и медицине».
Труды V Всес. совещания. Улан-Удэ,
Бурятское кн. изд-во, 1968.
Круглова Е. К. Методика определения до¬
ступных форм микроэлементов в карбо¬
натных почвах и растениях. Ташкент,
«ФАН», 1972.
Крупский Н. К., Александрова А. М. К
вопросу об определении подвижных форм
микроэлементов.— В сб. «Микроэлемен¬
ты в жизни растений, животных и чело¬
века». Киев, «Наукова думка», 1964.
Макеев О. В. Микроэлементы в Сибири.—
В сб. «Микроэлементы в сельс ком хозяй¬
стве и медицине». Труды V Всес. совеща¬
ния. Улан-Удэ, Бурятское кн. изд-во,
1968.
Методические указания по определению
подвижных форм микроэлементов в поч¬
Методы определения в почве меди, цинка, кобальта и бора
419
вах для зональных агрохимических ла¬
бораторий. М., 1973.
Никиткина Я. И. Фотометрическое опре¬
деление бора карминовым методом.—Поч¬
воведение, 1959, № 9.
Ринькис Г. Я. Методы ускоренного коло¬
риметрического определения микроэле¬
ментов в биологических объектах. Рига,
Изд-во АН ЛатвССР, 1963.
Тома С. И. Усвояемость микроэлементов
черноземной зоны для различных поле¬
вых культур.— Труды Кишиневского
СХИ, т. 64. Кишинев, 1970.
Bear Е. Chemistry of the soil. London, 1964»
Boratynski /Г., Zietecka M. On the possibi¬
lity of applying a common extraction solu¬
tion for determination in soil of trace ele¬
ments Cu, Mn, Mo, Zu available to
planjts. — Polish J. Soil Sci., 1970, v.
Jackson M. L. Soil chemical analysis. Lon¬
don, 1962.
Henriksen A. Kobberbestemmlser i iordl
sammenligning med virknigen af kobber-
gedskning.— Tidsskr. planteavl, 1957,
v. 61, N 4.
Oelschlager W. Fehlermoglichkeiten bei der
Bestimmung des pflanzenaufnehmbaren
Bors im Boden. Landwirtsch. Forschung,
1958, Bd. XI, H. 1.
Scharrer К., Schaumloffel E. Die quantita¬
tive Bestimmung kleinster Mengen Kupfer
mittels Diathyldithiocarbaminat (DDTC)
als Cu (DDTC)a. Landwirtcsh. Forcshung,
1958, Bd. XI, H. 1.
Wear /., Sommer A. Acid-extractable zinc
of soils in relation to the occurrence of
zinc deficiency symptoms of com: a met¬
hod of analysis.— Soil. Sci. Soc. America
Proc., 1947, v. 12, 143—144.
Westerhoff H. Beitrag zur Kupferbestim-
mung im Boden. Landwirtsch. Forschung,
1954/1955, Bd. 7, 190-193.
Ю. И. ДОБРИЦКАЯ
ОПРЕДЕЛЕНИЕ МОЛИБДЕНА, ВАНАДИЯ, МАРГАНЦА
И ИОДА В ПОЧВАХ
*
Молибден, ванадий и марганец рекомендуется определять из одной навес¬
ки почвы (Добрицкая, 1969 а, б) после ее разложения двумя способами:
1) при наличии платиновых чашек — фтористоводородной и серной кисло¬
тами и 2) при отсутствии платиновых чашек — разложение почвы в стеклян¬
ных колбах серной и азотной кислотами в присутствии перекиси водорода
с последующей обработкой хлорной кислотой. Оба способа дают близкие
результаты.
РАЗЛОЖЕНИЕ ПОЧВЫ ФТОРИСТОВОДОРОДНОЙ
И СЕРНОЙ КИСЛОТАМИ
Навеску почвы (2,5—3,0 г), растертой до состояния пудры, помещают
в платиновую чашку, ставят в холодную муфельную печь и после нагревания
до 500—550° С прокаливают не менее 3 час. для разрушения органических
веществ. После охлаждения в чашку с почвой прибавляют 2 мл бидистилли¬
рованной воды, 2 мл концентрированной серной и 20 мл концентрированной
фтористоводородной кислот. (Работать следует в резиновых
перчатках под хорошей тягой.) Ставят чашку на электро¬
плитку с закрытой спиралью и выпаривают до появления паров серного ан¬
гидрида, затем прибавляют еще 0,5 мл серной и 10 мл фтористоводородной
кислот и вновь выпаривают до появления белых паров SOs и третий раз обра¬
батывают 10 мл HF, выпаривая досуха. Для полного удаления фтора и кис¬
лот дважды выпаривают с 5 мл воды. Сухой остаток слегка прокаливают на
горелке или в муфеле для полноты разрушения органических веществ и уда¬
ления кислот. Чашку охлаждают и для растворения остатка прибавляют
20 мл 22%-ной НС1, прикрывают часовым стеклом, нагревают на закрытой
плитке до растворения остатка и фильтруют через фильтр средней плотности
в эрленмейеровскую колбу на 200—250 мл. Фильтр и чашку промывают
горячей водой, подкисленной НС1 (98 : 2), до отсутствия реакции на железо
с роданидом калия. Фильтрат выпаривают в той же колбе почти досуха,
прибавляют 5 мл 22%-ной НС1 и 3 мл 35—40%-ной НС104 и вновь выпа¬
ривают досуха для удаления хрома и более полного разрушения органичес-
ких веществ. Прибавляют 5 мл воды и выпаривают досуха для удаления сле¬
дов хлорной кислоты. К сухому остатку прибавляют 5 мл 22%-ной НС1 и для
переведения солей в хлориды выпаривают досуха. Сухой остаток растворяют
при нагревании в 20 мл 22%-ной НС1 и 15 мл воды, переносят, не фильтруя,
в мерную колбу на 50 дед и доводят водой до метки. В аликвотных частях
фильтрата определяют молибден, ванадий и марганец. Все работы ведутся
с бидистиллированной водой.
РАЗЛОЖЕНИЕ ПОЧВЫ СЕРНОЙ, АЗОТНОЙ
И ХЛОРНОЙ КИСЛОТАМИ
Навеску почвы (2,5—3,0 г), растертой до состояния пудры, помещают
в термостойкую широкогорлую колбу или стакан на 100 мл, ставят в холод¬
ную муфельную печь и прокаливают при температуре 500—550° С не менее
Определение молибдена, ванадия, марганца и иода в почвах
421
3 час. После прокаливания в охлажденную колбу (или стакан) с почвой при¬
бавляют по 5 мл концентрированных серной и азотной кислот и 2 мл перекиси
водорода (пергидроль) и осторожно нагревают на электроплитке с закрытой
спиралью до появления белых паров серного ангидрида, прибавляют еще
3—4 раза азотную кислоту и пергидроль, выпаривая каждый раз до паров
S08, под конец прибавляют еще 0,5 мл серной кислоты и 3 мл азотной кисло¬
ты и все выпаривают досуха. Прибавляют 5 мл 22%-ной НС1 и 3 мл 35—40%-
ной хлорной кислоты и выпаривают досуха (для удаления хрома в виде паров
хромилхлорида и для дополнительного разрушения органических веществ).
Затем дважды прибавляют по 5 мл воды и после каждого прибавления выпа¬
ривают досуха для удаления образовавшейся нитрозилсерной кислоты.
К сухому остатку прибавляют 5 мл 22%-ной НС1 и для перевода солей
в хлориды выпаривают досуха. Прибавляют 10 мл той же кислоты и 10 мл
горячей воды, нагревают и осторожно фильтруют (декантацией) через фильтр
средней плотности; к остатку прибавляют 5—6 раз по 1,5 мл НС1 и по 5 лед
горячей воды, каждый раз нагревают, фильтруют и под конец весь остаток
переносят на фильтр, промывают горячей водой, подкисленной НС1 (98 : 2),
до отсутствия реакции на железо с роданидом калия. Фильтрат упаривают
в той же колбе до объема около 50 мл, переносят в мерную колбу на 50 лед,
доводят водой до метки и в аликвотных частях фильтрата определяют мо¬
либден, ванадий и марганец.
Реактивы. 1. Фтористоводородная (плавиковая) кислота, х. ч*
2. Серная кислота, х. ч., уд. вес 1,84.
3. Соляная кислота, перегнанная, уд. вес 1,1 (22%-ная). Смесь из 550 мл концент»
рированной НС1 и 450 мл бидистиллята перегоняют в стеклянном дистилляционном ап
парате. Собирают 500 мл перегнанной НС1. Полученная кислота обычно имеет уд» вео
1,1, что соответствует 22%-ной концентрации по объему.
4. Роданистый калий, 20%-ный раствор.
5. Хлорная кислота (НСЮ4), х. ч., 40%-ная.
6. Азотная кислота, конц., уд. вес 1,4.
7. Перекись водорода (пергидроль), х. ч., 30%-ный раствор»
МОЛИБДЕН
Из химических методов определения в почвах микрограммовых количеств
молибдена в СССР наибольшее распространение получили роданидный коло¬
риметрический метод в модификации Добрицкой (1958, 1969а) или ускорен¬
ный метод Ринькиса (1963).
Методы основаны на образовании молибден-роданидного комплекса, ок¬
рашенного в слабо-оранжево-желтый цвет, образующегося в кислой среде
в присутствии роданида калия и восстановителя (хлорида олова). Определе¬
ния обычно производят на ФЭК, спектрофотометре или спектроколориметре
(«Спекол») после экстрагирования полученного комплекса органическим раст¬
ворителем (этиловый эфир, изоамиловый спирт, нормальный бутиловый
спирт и т. д.). В методе Ринькиса рекомендуется конечное визуальное опре¬
деление молибдена по пробирочным шкалам.
В последние годы, кроме того, получил распространение ме,тод определе¬
ния молибдена с помощью реакции с дитиолом (3,4-димеркаптотолуол) или
его цинковой солью (цинк-дитиол). Это соединение является высокочувст¬
вительным реагентом на молибден и ряд других элементов. Молибден в кис¬
лой среде (4 н. НС1) дает с дитиолом соединение, окрашенное в зеленый цвет,
хорошо экстрагируемое как полярными, так и неполярными органическими
раствори телями.
422
Ю. И. Добрицкая
Определение валового содержания молибдена
по Добрицкой (1969а)
Экстракция молибдена нормальным бутиловым спиртом
Аликвотную часть фильтрата после разложения почвы, соответствующую
1,5 г почвы, переносят в мерную колбу на 50 мл, прибавляют 15 мл 4%-ного
раствора фторида натрия (для связывания в комплекс титана, железа и алю¬
миния) и 5 мл 20%-ного раствора нитрата натрия (для предотвращения
восстановления Мов до более низких степеней валентности, чем Моб), доводят
водой до метки.
Содержимое колбы переносят в делительную или капельную воронку ем¬
костью 100 мл, стенки колбы смывают 2—3 мл воды, которую выливают ту¬
да же. Прибавляют 4 мл 20%-ного раствора роданида калия (для образования
молибден-роданидного комплекса), перемешивают, прибавляют 3—4 мл
10%-ного раствора SnCl2, перемешивают и после полного исчезновения
буровато-красной окраски от роданида железа (Fe3+ переходит в Fe2+) при¬
ливают 20 мл нормального бутилового спирта, предварительно промытого
водой и насыщенного хлоридом олова, и для извлечения молибден-роданид-
ного комплекса энергично встряхивают в течение 1 мин., дают жидкостям
расслоиться, сливают нижний водный слой, оставляя около 1 мл вместе с экс¬
трактом.
Экстракт при этом может опять приобрести розовый оттенок от частично¬
го окисления железа в присутствии кислорода воздуха, а поэтому его следует
промыть раствором олова. Для этой цели в воронку с экстрактом приливают
20 мл свежеприготовленного раствора SnCl2 (2 мл 10%-ного раствора SnCl2 +
+ 18 мл воды), хорошо встряхивают и после полного исчезновения розового
оттенка водную фазу сливают, а органический экстракт переносят в кювету
ФЭК-Н с просматриваемым слоем в 30 мм. Прибавляют туда же 1 мл этило¬
вого спирта для освобождения от мелких капелек воды и 1—2 капли 10%-
ного раствора олова и осторожно перемешивают тонкой стеклянной палоч¬
кой.
После того как экстракт станет совершенно прозрачным и не будет иметь
розового оттенка, его колориметрируют с синим светофильтром (длина волны
453 нм) против нормального бутилового спирта, не обработанного водой
и оловом. Измерения и отсчеты следует производить не ранее чем через 20—
30 мин. с момента прибавления нормального бутилового спирта.
Содержание молибдена в образце определяют по калибровочному графи¬
ку. Одновременно можно работать не более чем с 3—4 делительными ворон¬
ками.
Калибровочный график готовят из стандартного раствора молибдена,
содержащего в 1 мл 1 мкг молибдена. Отбирают в ряд мерных колб на 50 мл
по 0,5; 1,0; 2,0; 3,0; 4,0 и 5,0 мл, в каждую колбу прибавляют по И мл 22%-
ной НС1, по 3 мл раствора хлорного железа, содержащего в 1 мл 5 мг Fe3+f
все хорошо перемешивают, приливают по 15 мл 4%-ного раствора фторида
натрия и по 5 дел 20%-ного раствора нитрата натрия, перемешивают, доли¬
вают водой до метки. Дальнейшие операции такие же, как и при определе¬
нии молибдена в испытуемых растворах. По полученным отсчетам строят
калибровочный график, нанося на ось абсцисс количество молибдена в мик¬
рограммах, а на ось ординат — процент светопропускания или величину
оптической плотности. Все отсчеты производят 2—3 раза и записывают сред¬
ние показания. Одновременно с определением молибдена в почвенных об¬
разцах необходимо проводить контрольный (холостой) опыт со всеми^реак-
тивами, но без почвы. Результаты контрольного опыта вычитают из резуль¬
татов определения молибдена в испытуемом образце.
Реактивы. Все реактивы следует готовить на бидистиллированной воде, перего¬
няемой в стеклянном дистилляционном аппарате.
Определение молибдена, ванадия, марганца и иода в почвах
423
1. Соляная кислота, 22%-ная (6 н., уд. вес 1,1). Смесь из 550 мл концентрированной
НС1 и 450 мл воды перегоняют в стеклянном дистилляционном аппарате. Собирают 500\ил
перегнанной НС1.
2. Серная кислота, уд. вес 1,84.
3. Азотная кислота, уд. вес 1,4.
4. Хлорная кислота, 40%-ная.
5. Перекись водорода (пергидроль), 30%-ный раствор.
6. Хлорное железо. Навеску 24,2 г FeCl3*6H20 растворяют в воде, подкисляют, 10 мл
22%-ной НС1 для предотвращения гидролиза солей железа и доводят водой до 1 л. Та*
кой раствор должен содержать около 5 мг железа в 1 мл раствора. Для установления
истинного содержания железа его осаждают обычным аммиачным способом,. осадок про»
наливают и взвешивают. По весу осадка вычисляют содержание железа в растворе.
7. Фторид натрия, 4%-ный раствор. Растворяют при нагревании в воде 4 г NaF*
фильтруют и доводят водой до 100 мл.
8. Нитрат натрия, 20%-ный раствор. Растворяют в воде 20 г NaN03, фильтруют и
доводят водой до 100 мл.
9. Роданид калия, 20%-ный раствор. Растворяют 20 г KCNS в воде, фильтруют и
доводят водой до 100 мл»
10. Хлорид олова, 10%-ный раствор. Расплавляют металлическое олово в фарфо¬
ровой чашке и быстро растирают фарфоровым пестиком до получения мелкого серого
металлического порошка. Отвешивают 5,3 г порошка и растворяют при нагревании
в 20 мл 22%-ной НС1 (прикрыть колбу стеклом!). Можно раствор упарить до кристалли¬
зации (влажные соли), затем прибавить точно 20 мл 22%-ной НС1 и довести водой до 100мл*
Раствор олова готовят каждый раз свежий. Можно приготовить раствор олова из
химически чистой соли SnCl2, для чего 10 г ее растворяют при нагревании в 20 мл 22%-ной
НС1, затем на кончике ножа прибавляют металлическое олово, прикрывают стеклом
и нагревают 10—15 мин., затем доводят до 100 мл водой.
11. Фтористоводородная кислота, концентрированная. Работать с нею следует под
хорошей тягой, в резиновых перчатках, наливать в чашки запарафинированным или поли¬
этиленовым стаканом.
12. Ацетат свинца, 4%-ный раствор. Отвешивают 4 г РЬ(СН3С00)2-ЗН90, растворяют
в воде, фильтруют, доводят водой до 100 дел (раствор ядовит!).
13. Нитрат аммония, 5%-ный раствор. Растворяют 5 г NH4N03 в воде, доводят во¬
дой до 100 мл.
14. Нормальный бутиловый спирт. Промывают водой для освобождения его от окис¬
лителей: к 250 мл спирта, помещенного в большую делительную воронку, приливают
250 мл бидистиллированной воды, встряхивают 1 мин., после разделения слоев нижний
водный слой сливают, а спирт насыщают хлоридом олова (на 100 мл промытого спирта
10 мл 10%-ного раствора SnCl2), встряхивают 3—5 мин., прибавляют 5 мл этилового спи¬
рта, перемешивают и употребляют для экстракции.
15. Стандартный раствор молибдена. Для получения чистого молибденовокислого
аммония его перекристаллизовывают, растворяя в концентрированном аммиаке при про-
до лжите льном нагревании (раствор все время должен быть щелочным), и после охлаждения
осаждают молибдат аммония этиловым спиртом. Осадок ^высушивают между листами
фильтровальной бумаги. Для приготовления запасного стандартного раствора берут
5—6 г этого молибдата в фарфоровый тигель и прокаливают в муфеле при 450—480° С в
течение 40—60 мин.; отвешивают 1,5 г полученного Мо03, растворяют в 10 мл 1 н. NaOH,
прибавляют 10 мл 22%-ной НС1 и доводят бидистиллятом до 1 л. Этот раствор должен
содержать в 1 дел 1 мг молибдена.
Рабочий раствор получают соответственным разбавлением водой.
Для установления точного титра полученного запасного стандартного раствора мо¬
либдена из него отбирают две пробы по 20 дел каждая, подкисляют 5 дел 10%-ной уксус¬
ной кислоты, разбавляют водой до 100 дел, нагревают до 90°, прибавляют 3 дел 5%-ного
раствора NH4N03 и медленно приливают 4%-ный раствор уксуснокислого свинца до
полного осаждения РЬМо04, затем добавляют небольшой его избыток и фильтруют через
фильтр с синей лентой. Фильтр с осадком промывают водой, подкисленной уксусной кис¬
424
Ю, И. Добрицкая
лотой, подсушивают, прокаливают при 450—480° С в течение 30—40 мин. и осадок
взвешивают. Вес осадка умножают на 0,2614 и вычисляют точное содержание молибдена
в i мл запасного стандартного раствора молибдена.
Экстракция молибдена смесью изоамилоеого спирта
и четыреххлористого углерода — 1:1
Вместо экстракции молибден-роданидного комплекса н-бутиловым спир¬
том можно применять его извлечение смесью изоамилового спирта и четырех¬
хлористого углерода (1:1). Экстракт оседает на дно, и аналитик меньше
ощущает неприятный запах растворителей.
Для этой цели изоамиловый спирт предварительно следует обработать
растворами роданида калия и двухлористого олова. 200 мл изоамилового
спирта помещают в большую делительную воронку, прибавляют 5 мл 20%-
ного раствора KCNS и 10 мл 10%-ного раствора SnCl2, встряхивают в тече¬
ние 3 мин., прибавляют 10 мл этилового спирта для просветления раствора,
слегка перемешивают. Водную фазу сливают, а к промытому изоамиловому
спирту приливают 200 мл х. ч. четыреххлористого углерода, перемешивают
и употребляют для экстракции (смесь готовят перед началом экстракции).
Весь ход анализа остается таким же, как это описано выше для экстрак¬
ции молибден-роданидного комплекса нормальным бутиловым спиртом,
но вместо последнего прибавляют 14 мл полученной смеси (1 : 1). Воронку
с содержимым встряхивают от руки 1 мин.; экстракт молибден-роданидного
комплекса оседает на дно, поэтому его сливают в другую делительную ворон¬
ку, прибавляют для промывания экстракта 10 мл 1%-ного раствора SnCl2
(свежеприготовленного), встряхивают и после исчезновения розового оттен¬
ка переносят в кювету фотоколориметра с просматриваемым слоем в 30 мм.
Если экстракт мутный от мелких капелек воды, то прямо в кювету прибав¬
ляют 0,5 мл этилового спирта (для обезвоживания экстракта), слегка пере¬
мешивают тонкой стеклянной палочкой и после того, как экстракт станет
прозрачным и не будет иметь розового оттенка, его колориметрируют с си¬
ним светофильтром (453 нм) против смеси изоамилового спирта и четырех¬
хлористого углерода, не обработанной роданидом калия и хлоридом олова.
Содержание молибдена в пробе определяют по калибровочному графику,
приготовленному так же, как это описано выше, но с извлечением комплекса
полученной смесью.
Определение молибдена на спектроколориметре «Спекол»
с приставкой для измерения экстинкции в пробирках
«Спекол» является однолучевым спектральным фотометром с дифракцион¬
ной решеткой. Для определения экстинкции на «Спеколе» предварительно
следует подобрать пробирки с притертой пробкой диаметром 16 мм и исклю¬
чить пробирки с оптически заметными отклонениями. Для этой цели налива¬
ют в пробирки 0,001 н. раствор КМп04 и помещают в ячейку приставки;
отбирают пробирки с одинаковыми показателями экстинкции.
Ход анализа по определению молибдена остается таким же, но вместо
нормального бутилового спирта в делительную воронку с подготовленным
раствором вносят 6 мл смеси изоамилового спирта и четыреххлористого угле¬
рода, подготовленной для экстракции (см. выше). Воронку встряхивают
от руки 1 мин. После извлечения молибден-роданидного комплекса экстракт,
находящийся в нижней части делительной воронки, сливают в другую дели¬
тельную воронку, прибавляют 10 мл 1%-ного раствора SnCl2 (1 мл 10%-ного
раствора SnCl2 + 9 мл 1 н. раствора НС1), хорошо встряхивают и после ис¬
чезновения розовой окраски экстракт переливают в пробирку с притертой
пробкой. После того как он станет прозрачным, определяют экстинкцию,
Определение молибдена, ванадия, марганца и иода в почвах
425
помещая пробирку в гнездо приставки «Спекол». Измерения производят при
длине волны 475 нм. Если экстракт помутнеет от мелких капелек воды, то
пробирку на несколько секунд помещают в стакан с теплой водой и после
просветления экстракта сразу же измеряют экстинкцию.
Если экстракт не прозрачен, то можно также прибавить 0,5 мл этилового
спирта, слегка перемешать, поместить на несколько секунд в стакан с теплой
водой и прозрачный экстракт колориметрировать. Калибровочный график
готовят точно так же, как это описано выше, но молибден-роданидный комп¬
лекс извлекают смесью изоамилового спирта и четыреххлористого углерода
(1 : 1), прибавляя ее 6 мл. Дальнейший ход анализа такой же, как и в испы¬
туемых растворах.
Ускоренный метод определения валового содержания молибдена
по Ринькису (1963)
Молибден этим способом можно определить или из аликвотной части
фильтрата, полученного после разложения почвы кислотами (по Добрицкой),
или из отдельной навески почвы с кислотным разложением по Ринькису.
Навеску в 1—1,5 г почвы, растертой до состояния пудры, помещают
в широкогорлую термостойкую колбу на 100 мл и сушат при слабом нагре¬
вании плитки до тех пор, пока на стенках колбы не исчезнут капли воды.
Затем увеличивают нагрев и окисляют под тягой органическое вещество
парами азотной кислоты, поступающими из колбы с отводной трубкой. При
этом конец трубки должен находиться на расстоянии 1—1,5 см над слоем
почвы. Когда почва станет светлой, в колбу после охлаждения на каждый
грамм навески добавляют по стенкам 1,5 мл концентрированной азотной
кислоты, 0,5 мл 30%-ной Н202 и 1 мл концентрированной серной кис¬
лоты. Колбу медленно нагревают до паров S03 и затем, увеличивая нагрев,
выпаривают досуха. Если сухой охлажденный остаток совершенно бел или
слегка желтоват (в образце много железа), то приступают к растворению
полученных солей. Если остаток темный, то разложение повторяют. Для
этого прибавляют 1 мл концентрированной НС1, 1 мл Н202 и 0,5 мл концен¬
трированной Нг304. Содержимое колбы упаривают досуха. Для растворения
солей прибавляют на 1 г почвы 1 мл НС1, слегка нагревают, приливают 5 мл
воды и доводят до кипения. Раствор фильтруют декантацией, остаток в кол¬
бе нагревают с 5—10 мл воды, подкисленной НС1 (1 : 100), фильтруют и так
продолжают до обесцвечивания очередной порции воды. К остатку в колбе
снова приливают 0,5 мл концентрированной НС1, нагревают, добавляют
воды, фильтруют и под конец колбу и фильтр 2—3 раза промывают горячей
подкисленной водой.
Фильтрат упаривают до 30—40 мл, переносят в делительную воронку
(или отбирают аликвотную часть фильтрата, соответствующую 1—1,5 г
почвы — после разложения почвы по Добрицкой), прибавляют 2,5 мл кон¬
центрированной х. ч. НС1, 5 мл 10%-ного раствора KCNS и взбалтывают.
Затем прибавляют 4—5 мл 10%-ного раствора SnCl2 и хорошо перемешивают.
После обесцвечивания для экстрагирования молибден-роданидного комп¬
лекса прибавляют 6 мл изоамилового спирта, встряхивают воронку 1—2 мин. и,
если после расслоения фаз экстракт имеет розовый оттенок, добавляют еще
0,5—1,0 мл SnCl2 и хорошо встряхивают. После исчезновения розового оттен¬
ка часть водной фазы сливают и выбрасывают, а экстракт с 5—6 мл водной
фазы переносят в калиброванную пробирку с притертой пробкой и сравни¬
вают визуально с пробирочной шкалой, приготовленной в тот же день из
стандартного раствора молибдена (от 1 до 5 мкг молибдена и соответству
ющие реактивы в тех же количествах, что и в исследуемых образцах).
Шкала образцо вых растворов. В серию калиброванных
пробирок с притертой пробкой отбирают из стандартного раствора молибдена
426
Ю. И. Добрицкая
(приготовление см. в разделе «Экстракция молибдена н-бутиловым спиртом»),
содержащего в 1 мл 1 мкг Мо, 0,5; 1,0; 2,0; 3,0; 4,0 и 5,0 мл, затем в каждую
пробирку прибавляют по 1 мл концентрированной НС1 и по 2 капли 5 %-ного
раствора FeCl3, дистиллированной водой доливают до 8 мл. Затем приливают
1 мл 10%-ного раствора KCNS, взбалтывают, добавляют 1 мл 10%-ного раст¬
вора SnCl2, вновь взбалтывают и, как только полностью исчезнет красноватая
окраска, вызванная роданидом трехвалентного железа, приливают 6,0 мл
изоамилового спирта и хорошо встряхивают для экстракции молибден-
роданидного комплекса. Если после разделения фаз органический слой будет
окрашен в розовый тон, то необходимо прибавить еще раствора SnCl2, и после
исчезновения этого тона шкала пригодна для сравнения с окраской органи¬
ческого слоя в испытуемых растворах.
Содержание молибдена (мкг/г) вычисляют по формуле
А
Mo — q ,
где А — содержание Мо в совпадающей по окраске пробирке шкалы, мкг/мл; В — вес
почвы в г, соответствующий взятому для анализа объему раствора.
Реактивы. Приготовление описано выше.
Изоамиловый спирт — х. ч., хранить в темной, хорошо закрытой посуде. Работать
с ним в хорошем вытяжном шкафу.
Определение подвижных форм молибдена
Для того чтобы выяснить, насколько почва обеспечена подвижными фор¬
мами молибдена, различные исследователи применяют разные методы: поле¬
вой или вегетационный, микробиологический и химический. Последний ме¬
тод заключается в извлечении из почв подвижных форм молибдена разными
химическими реагентами.
Наиболее распространен метод вытеснения молибдена оксалатным бу¬
ферным раствором с pH 3,3 (метод Григга — Grigg, 1953), так как данные,
полученные этим методом, хорошо коррелируют с данными полевых опытов
по отзывчивости растений к применению молибденовых удобрений. Окса-
латный буферный раствор извлекает разные формы молибдена: воднораство¬
римый, обменный, частично связанный с органическим веществом, а также
со свободными окислами железа и алюминия. Поэтому следует считать, что
оксалатная вытяжка скорее характеризует потенциальный запас подвижного
молибдена в почвах, а не количество молибдена, доступного растениям
в данный момент. Существует много модификаций этого метода. Ниже
описаны варианты, которыми пользуются в Советском Союзе и некоторых
варубежных странах.
Фотоколориметрическое определение молибдена
по Григгу, в модификации Добрицкой (1973)
Навеску почвы, просеянной через сито с отверстиями диаметром 1 иш,—
15 ? для суглинистых почв и 25 г для супесчаных и песчаных почв, помещают
в колбу с притертой пробкой, приливают 150 или 250 мл (соответственно
навеске) оксалатного буферного раствора Григга, имеющего pH 3,3, и встря¬
хивают на ротаторе 1 час. Фильтруют через складчатый фильтр (белая лента),
отбирают 125 или 200 мл фильтрата, помещают в широкогорлую термостойкую
колбу или стакан и упаривают до V4 части объема, прибавляют для разру¬
шения оксалатов и органического вещества, частично перешедшего в окса-
латную вытяжку, 3 мл 30%-нойН202 (пергидроль) и 3 мл концентрированной
HN03 и продолжают упаривать досуха. Такую обработку повторяют 4—
5 раз, каждый раз выпаривая досуха.
Определение молибдена, ванадия, марганца и иода в почвах
427
Затем к остатку приливают 5 мл 22%-ной НС1 и 2 мл хлорной кислоты
(НС104, 35—40%-ной) для удаления хрома и более полного разрушения орга¬
нических веществ, выпаривают досуха. Прибавляют 5 мл бидистиллирован¬
ной воды и для удаления следов хлорной кислоты вновь выпаривают досуха.
К сухому остатку прибавляют 5 мл 22%-ной НС1 (для перевода солей в хло¬
риды) и выпаривают досуха. Остаток растворяют при нагревании в 11 мл
22%-ной НС1, переносят малым количеством воды в мерную колбу на 50 мл,
прибавляют 15 мл 4 %-ного раствора фторида натрия (для связывания в комп¬
лекс железа, алюминия и титана) и 5 мл 20 %-ного раствора нитрата натрия
(для предотвращения восстановления молибдена до более низких степеней
валентности, чем Моб), доводят водой до метки и хорошо перемешивают.
Содержимое колбы переносят в делительную или капельную воронку на
100 мл, стенки колбы смывают 2—3 мл воды, которые выливают туда же.
Дальнейший ход анализа такой же, как и при определении валового содер¬
жания молибдена.
Оксалатный буферный раствор Григга готовят на бидистиллированной
воде, растворяя 25 г щавелевокислого аммония и 12,6 г щавелевой кислоты
и доводя до 1 л водой. Раствор должен иметь величину pH 3,3.
Обязательно параллельно проводить холостой опыт со всеми реактивами,
но без почвы. Результаты определения вычитать из данных определения Мо
в испытуемых образцах.
При определении подвижного молибдена в карбонатных почвах, без учета
содержания в них карбонатов, данные оксалатнорастворимого] молибдена
могут получиться сильно заниженными, так как при взаимодействии окса-
латного раствора с кальцием почвы выпадет осадок щавелевокислого каль¬
ция, в связи с чем концентрация анионов С204 в растворе меняется настоль- •
ко, что подвижный молибден вытесняется далеко не полно. Поэтому в карбо¬
натных почвах необходимо предварительно определить в отдельной навеске
процентное содержание С02. После этого вычислить, какое количество ща¬
велевой кислоты нужно добавить к навеске почвы (например, 10 г почвы)
в сухом состоянии, чтобы связать кальций карбонатов и затем уже приба¬
вить 100 мл оксалатного буферного раствора с pH 3,3. При навеске почвы
в 10 г на каждый процент С02 следует добавить 0,29 г сухой Н2С204-2Н20.
Все дальнейшие операции по извлечению и конечному определению подвиж¬
ного молибдена остаются такими же, как и для некарбонатных почв.
Визуальное определение молибдена по Ринькису (1963)
10 г воздушно-сухой почвы, пропущенной через сито с отверстиями диа¬
метром 1 мм, помещают в колбу] на 250 мл, приливают 100 мл оксалатного
буферного раствора Григга с pH 3,3, взбалтывают на ротаторе 1 час и отфиль¬
тровывают. 75 мл вытяжки помещают в широкогорлую колбу из термостойкого
стекла (объем 100 мл), прибавляют 2 мл концентрированной HN03, 1 мл
30%-ной Н202 и выпаривают до образования пенистой желто-коричневой
сиропообразной массы. Такую обработку производят еще 2—3 раза.
За ем в колбу прибавляют 1 мл концентрированной HN03, около 0,2 г
сухого персульфата аммония, выпаривают до начала кристаллизации солей
и немедленно добавляют 5 мл воды, подкисленной HN03 (100 : 1). Раствор
нагревают до кипения и для завершения окисления добавляют по каплям
1%-ный раствор КМп04 до покраснения раствора. Затем прибавляют 3 мл
концентрированной НС1 и нагревают до кипения. Для определения молибде¬
на раствор из колбы переносят в делительную воронку (на 100 мл) с отметкой
на 50 мл, споласкивают колбу водой и доводят до объема 50 мл. К раствору
в воронке прибавляют 5 мл 10 %-ного водного раствора роданида калия,
взбалтывают, прибавляют 4—5 мл 10 %-ного раствора SnCl2, хорошо переме¬
шивают и после полного исчезновения красной окраски роданида трехвалент¬
428
Ю. И. Добрицкая
ного железа прибавляют 6 мл изоамилового спирта для экстрагирования
молибден-роданидного комплекса и хорошо встряхивают 30—40 сек.
После разделения слоев большую часть водной фазы сливают и отбрасы¬
вают, а изоамиловый экстракт с (5—6 мл водной фазы переносят в калиб¬
рованную пробирку с притертой пробкой и, если экстракт не имеет розового
оттенка, то его окраску сопоставляют со шкалой. Если же в экстракте появ¬
ляется розовый тон от окисления части железа, то в пробирку прибавляют
0,2 —0,3 мл SnCl2 и встряхивают, а затем сравнивают со шкалой.
Приготовление пробирочной шкалы и вычисление результатов описано
выше (см. метод определения валового содержания молибдена по Ринькису).
Определение молибдена по Вергманну1
Навеску почвы 30 г, просеянную через сито с отверстиями диаметром
1 мм, помещают в колбу с притертой пробкой, прибавляют 300 мл оксалатно-
го раствора Григга с pH 3,3 и встряхивают 12 час. на ротаторе.Раствор фильт¬
руют и отбирают 200 мл фильтрата, соответствующего 20 г почвы, и выпари¬
вают на водяной бане в фарфоровой чашке досуха. Для удаления органиче¬
ских веществ чашку с сухим остатком помещают в муфель и прокаливают
при 450° С в течение 4 час. Затем к прокаленному остатку прибавляют 20 мл
20%-ной НС1, нагревают на водяной бане и фильтруют через складчатый
фильтр прямо в делительную воронку. Промывают фильтр бидистиллирован¬
ной водой, причем намывают около 60 мл фильтрата. Кислотность фильтрата
должна быть в пределах 5—7%, так как только при такой концентрации НС1
молибден-роданидный комплекс стабилен.
Далее производят определение молибдена по Григгу (Crigg, 1953). В де¬
лительную воронку из бюретки прибавляют 5 мл 10 %-ного раствора рода¬
нида калия, хорошо перемешивают и затем приливают 3,5 мл 20%-ного рас¬
твора SnCl2. После перемешивания ждут полного обесцвечивания образовав¬
шегося роданида железа. Прибавляют 25 мл эфира, не содержащего перекис-
ных соединений, и сильно встряхивают 2 мин.
После разделения слоев водную фазу сливают и отбрасывают, а экстракт
промывают смесью, состоящей из 1,5 мл 20 %-ного раствора SnCl2 и 23,5 мл
7%-ной НС1, встряхивают еще 1 мин. и измеряют оптическую плотность
эфирного экстракта молибден-роданидного комплекса в кювете с просматри¬
ваемым слоем в 30 мм против чистого эфира при длине волны 465 нм.
Содержание молибдена в образце определяют по калибровочному графи¬
ку, построенному из стандартного раствора молибдена, прибавив предва¬
рительно по 1 мл 3%-ного раствора FeCl3. Параллельно ведут холостой опыт,
и данные вычитают из результатов определений молибдена в исследуемых
образцах почв.
Применение цинк-дитиола для определения подвижного молибдена
в почвах дает также хорошие результаты. Ниже дано описание двух вариан¬
тов этого метода.
Дитиоловый метод определения молибдена по Шелу (1962),
вариант Добрицкой
Для определения молибдена при помощи реагента цинк-дитиола исполь¬
зуется оксалатная вытяжка, приготовленная так, как это описано выше, в ва¬
рианте Добрицкой. Сухой остаток (соответствующий 10—15 г почвы) после
обработки вытяжки перекисью водорода и азотной кислотой, а затем хлор¬
ной кислотой растворяют в 10 мл 22%-ной НС1, переносят в колбу с притер¬
1 Методика принята в ГДР.
Определение молибдена, ванадия, марганца и иода в почвах
429
той пробкой (на 100—150 мл) и для связывания в комплекс железа прибав¬
ляют 20 мл 50 %-ного раствора цитрата натрия, хорошо перемешивают и че¬
рез 5 мин. приступают в нейтрализации раствора до pH 3,5, для чего прили¬
вают небольшими порциями при помешивании около 8 мл 6 н. раствора
NaOH, затем проверяют pH раствора, нанося тонкой стеклянной палочкой
каплю раствора на кусочек универсальной индикаторной бумаги; если вели¬
чина pH выше или ниже 3,5, то соответственно прибавляют по каплям 22%-
ную НС1 или 6 н. NaOH до установления необходимого значения pH раствора.
Затем для восстановления Fe3+ до Fe2+ прибавляют 5 мл свежеприготовлен¬
ного 20 %-ного раствора аскорбиновой кислоты и оставляют в темном месте
на 20 мин.; после этого для устранения влияния меди приливают 5 мл све¬
жеприготовленного 10 %-ного раствора тиомочевины и перемешивают. За¬
тем для создания кислотности около pH 0,6, при которой происходит полный
переход Мо6 из анионной в катионную форму и нормальное образование мо-
либден-дитиолового комплекса, прибавляют 30 мл 22%-ной НС1, переме¬
шивают и после полного обесцвечивания раствора сразу же прибавляют
2 мл спиртовой суспензии цинк-дитиола, тщательно перемешивают, закры¬
вают пробкой и помещают на 1,5—2 часа в темное место для полного образо¬
вания молибден-дитиолового комплекса. После этого в колбу прибавляют
12 мл толуола для растворения и экстракции молибден-дитиолового комплек¬
са и встряхивают на вертикальном встряхивателе в течение 10 мин. (или от
руки, энергично встряхивая колбу в вертикальном направлении: поочеред¬
но каждую колбу встряхивать 0,5 мин., дать постоять 1 мин. и так всю серию
колб — 10—15 шт.).
Затем весь раствор полностью переносят в делительную воронку и после
расслоения жидкостей нижний водный слой сливают и отбрасывают, а толуо-.
ловый слой переносят в кювету фотоэлектроколориметра с просматриваемым
слоем в 30 мм и через 5—10 мин. колориметрируют со светофильтром № 8
(красный, длина волны 660 нм). Содержание молибдена в образце вычисляют
по калибровочному графику.
Калибровочный график готовят из стандартного раствора молибдена,
содержащего в 1 мл 1 мкг молибдена. В колбы с притертой пробкой на 100—
150 мл переносят градуированной пипеткой 0,5; 1,0; 2,0; 3,0; 4,0 и 5,0 мл
стандартного раствора молибдена, прибавляют по 5 мл раствора FeCl3,
содержащего в 1 мл 5 мг Fe, 10 мл 22 %-ной НС1, перемешивают, прибавляют
20 мл 50 %-ного раствора цитрата натрия и через 5 мин. производят нейтра¬
лизацию раствором NaOH. Последующие операции такие же, как описано
выше при определении молибдена в исследуемых растворах.
Реактивы. Все реактивы готовят на бидистиллированной воде. 1. Цитрат нат¬
рия, 50%-ный раствор. Растворяют в бидистиллированной воде 500 г лимоннокислого
натрия, фильтруют и доводят до 1 л водой.
2. Натрий едкий, 6 н. раствор. 240 г NaOH растворяют в воде и доводят до 1 л.
3. Соляная кислота, 6 н. (22%-ная). Перегоняют в стеклянном аппарате смесь из
550 мл концентрированной НС1 и 450 мл воды. Собирают 500 мл дистиллята.
4. Железо хлорное. Растворяют 24,2 г FeCl8-6H20 в воде, подкисляют 10 мл 22%-ной
HG1 и доводят водой до 1 л. Раствор содержит около 5 лег Fe в 1 мл.
5. Аскорбиновая кислота, 20%-ная. Растворяют 20 г кислоты в воде и доводят до
100 мл. Раствор готовят сразу же перед употреблением.
6. Тиомочевина, 10%-ная. Растворяют 10 в тиомочевины в воде, доводят до 100 мл
водой. Раствор должен быть свежеприготовленным.
7. Толуол особой чистоты.
8. Цинк-дитиол, 0,4%-ный раствор. Помещают в колбу 0,2 г цинк-дитиола, при¬
бавляют 10 мл 2 н. раствора NaOH и 40 мл этилового спирта, все хорошо перемешивают.
Суспензию цинк-дитиола хранят в холодильнике, но лучше готовить в день определения^
430
Ю. И. Добрицкая
Дитиоловый метод определения молибдена
по Стентону (Stanton, Hardwick, 1967),
в модификации Самохвалова и Целиковой (1973)
Определения производят из оксалатной вытяжки. Реакция с цинк-дитио“
лом протекает в 4 н. растворе НС1. Влияние вольфрама устраняют лимонной
кислотой, меди — связыванием ее в йодный комплекс. Железо восстанавли¬
вают до Fe2+ аскорбиновой кислотой в присутствии лимонной.
Ход анализа. 15 г воздушно-сухой почвы, просеянной через сито в 1 мм,
помещают в колбу с притертой пробкой, прибавляют 150 мл оксалатного
раствора с pH 3,3 и взбалтывают на ротаторе 1 час. Отфильтровывают через
фильтр с синей лентой. Параллельно ведут холостой опыт со всеми реактива¬
ми, но без почвы.
Отбирают 100 мл фильтрата в стакан термостойкого стекла и выпаривают
на плитке с закрытой спиралью досуха и даже до частичной возгонки окса-
латов. Затем стакан помещают в муфель и озоляют оксалаты в течение 1 ча¬
са при 500°. К остатку приливают 5 мл 22 %-ной НС1 и 2 мл 57 %-ной НСЮ4
и выпаривают на плитке до прекращения выделения паров хлорной кислоты.
Остаток растворяют при нагревании в 20 мл 4 н. раствора НС1, охлаждают,
прибавляют в стакан 4 мл восстановительной смеси, перемешивают и через
5 мин. прибавляют 2 мл 50%-ного раствора NaJ и переносят в 100 мл дели-
тельную воронку, на которой предварительно отмечен объем в 30 мл. Ста-
кан обмывают 4 н. раствором НС1, доводят объем до 30 мл, прибавляют
2 мл раствора цинк-дитиола, перемешивают и оставляют на 2 мин. Затем
прибавляют 4 мл хлороформа, закрывают воронку пробкой и встряхивают
2 мин. После расслоения жидкостей нижний слой, окрашенный в зеленый цвет
дитиолатом молибдена, отфильтровывают через маленький сухой фильтр
с белой лентой прямо в кювету с толщиной просматриваемого слоя 20 мм.
Оптическую плотность экстракта измеряют против чистого хлороформа на
спектроколориметре «Спекол» при 675 нм, используя приставку ЕК-5.
Калибровочный график готовят из стандартного раствора молибдена,
содержащего 1 мкг в 1 мл, но для разведения запасного раствора используют
4 н. НС1. Готовят в день определений. В ряд делительных воронок отбирают
0; 0,5; 1; 2; 3; 4 и 5 мл рабочего раствора, прибавляют по 5 мл 1 %-ного рас¬
твора железо-аммонийных квасцов, доводят 4 н. раствором НС1 до 20 мл.
Далее ход анализа такой же, что и в вытяжках.
По отсчетам строят калибровочный график и рассчитывают содержание
молибдена в почве.
При отсутствии в лаборатории «Спекола» определения проводят на ФЭК,
внося в пропись следующие изменения: для определения молибдена берут
150 мл вытяжки, увеличив навеску почвы до 20 г, оксалатный раствор до
200 мл. Остаток после обработки вытяжки хлорной кислотой растворяют
при нагревании в 30 мл 4 н. раствора НС1, по охлаждении прибавляют 6 мл
восстанавливающего раствора. Через 5 мин. прибавляют 3 мл 50 %-ного рас¬
твора иодистого натрия, перемешивают и переносят в делительную воронку
на 100 мл. На воронке предварительно отмечают объем в 45 мл. Обмывают
стакан 4 н. раствором НС1 и доводят объем до метки. Прибавляют 3 мл рас¬
твора цинк-дитиола, перемешивают и через 2 мин. прибавляют 10 мл хлоро¬
форма и встряхивают 2 мин.
После расслоения жидкостей сливают нижний слой через сухой малень¬
кий фильтр с белой лентой в кювету с толщиной просматриваемого слоя
20 мм. При измерении интенсивности окраски на ФЭК-57 используют крас¬
ный светофильтр 8 (X = 660 нм), на ФЭК-М — светофильтр № 4.
Калибровочный график готовят так же, как описано выше, но объем в де¬
лительной воронке доводят 4 н. раствором НС1 до 45 мл. Далее ход анализа
такой же, что и в вытяжках.
Определение молибдена, ванадия, марганца и иода в почвах
431
Реактивы. Все реактивы готовят на бидистиллированной воде.
1. Оксалатный буферный раствор. 25аоксалата аммония и 12,6 г щавелевой кислоты
растворяют в горячей воде и по охлаждении доводят до 1 л.
2. Восстанавливающий раствор. 75 г лимонной кислоты и 150 г аскорбиновой кислоты
растворяют в воде и доводят до 1 д. Хранить в холодильнике.
3. Кислота соляная, 4 н. раствор. 330 мл х. ч. концентрированной НС1 (уд. вес 1,19)
доводят до 1 л водой.
4. Натрий иодистый, 50%-ный раствор. Отвешивают 250 г NaJ, растворяют в воде и
доводят до 500 мл. Хранить в холодильнике.
5. Цинк-дитиол, 0,3%-ный раствор. Отвешивают 0,3 г цинк-дитиола, смачивают 2 мл
этилового спирта, прибавляют 4 мл воды и 2 a NaOH, перемешивают и после полного ра¬
створения цинк-дитиола и щелочи разводят водой до 50 мл. Прибавляют 50 мл 50%-ного
раствора NaJ и перемешивают. Готовят в день определения.
6. Железо-аммонийные квасцы, 1%-ный раствор. Отвешивают 10 а реактива и рас¬
творяют в 4 н. НС1 при нагревании, после охлаждения доводят 4 н. раствором НС1
до 1 л. В 1 мл содержится 1 мг железа.
ВАНАДИИ
Из колориметрических методов определения ванадия в горных породах,
рудах, сплавах и других объектах наиболее распространен вольфраматный
метод, предложенный А. П. Виноградовым (1931) для определения ванадия
в золе растений. Метод основан на том, что при прибавлении к кислому
анализируемому раствору фосфорной кислоты и вольфрамата натрия обра¬
зуется окрашенный в зеленовато-желтый цвет фосфоро-вольфрамово-вана-
диевый комплекс, состав которого до сих пор еще изучен мало. Растворы
подчиняются закону Вера при концентрации ванадия от 10 до 200 мкг в 50 мл
раствора.
В связи с тем, что железо, медь, кобальт и хром, если они присутствуют
в значительных концентрациях (например, Сг : V более 6 : 1 или железа
больше 50 мг в 50 мл раствора), могут мешать или собственной окраской,
дли образованием своего комплекса с вольфраматом натрия (Сендел, 1964),
в ряде вариантов вольфраматного метода рекомендуют отделять ванадий от
мешающих элементов. Эта процедура очень осложняет ход анализа и требует
применения органических растворителей, работа с которыми неприятна
и часто вредна для организма аналитика.
Ю. И. Добрицкой (19696) для определения ванадия в почвах опробован
ряд реагентов, в результате чего выбрана реакция с вольфраматом натрия.
Проведено значительное количество экспериментов по применению этой
реакции для определения ванадия в почвах без его предварительного отде¬
ления от сопутствующих элементов. Выявлено влияние железа, кислотности
раствора, нагревания, маскирования фторидами и фосфорной кислотой,
окисления ванадия до пятивалентного состояния и других факторов на ре¬
зультаты определения ванадия в почвах.
Определение валового содержания ванадия
по Добрицкой (19696)
Из фильтрата после разложения почвы вышеописанными способами (по
Добрицкой) отбирают аликвотную часть, соответствующую 1 г почвы, по¬
мещают в стакан на 100—200 мл, прибавляют 3 мл раствора H2S04 (1 : 1)
и для окисления ванадия до пятивалентного состояния прибавляют 2%-ный
раствор КМп04 до устойчивой розовой или бурой окраски, не исчезающей
в течение 5 мин., затем прибавляют 2—3 капли 0,5%-ного раствора нитрита
яатрия (NaNOa) до исчезновения этой окраски.
432
Ю. И. Добрицкая
Для разрушения избытка нитрита натрия прибавляют 2—3 капли
2 %-ного раствора КМп04 до слегка розового оттенка, исчезающего в течение
1 мин. Затем для маскирования титана, железа и алюминия прибавляют
10 мл 4 %-ного раствора фторида натрия и 10 мл ортофосфорной кислоты (уд.
вес 1,7); можно добавить только одной Н3Р04, но не 10, а 15 мл. Содержимое
стакана хорошо перемешивают стеклянной палочкой, приливают 5 мл
5 %-ного раствора вольфрамата натрия, нагревают до кипения и кипятят ровно
5 мин. Охлажденный раствор переносят в мерную колбу на 50 мл и доводят
водой до метки. Если раствор помутнеет, то его следует после доведения
до объема отфильтровать через плотный фильтр и не ранее чем через 1 час
после кипячения колориметрировать в кювете с просматриваемым слоем
30 мм с фиолетовым светофильтром (№ 2, длина волны 413 нм) или колори¬
метрировать, не фильтруя, после 3—4-часового отстаивания.
Содержание ванадия в испытуемом растворе вычисляют по калибровоч¬
ному графику, приготовленному ий стандартного раствора ванадия. В ста¬
каны на 100—200 мл отбирают стандартный рабочий раствор, содержащий
в 1 лед 10 мкг ванадия (1,0; 2,5; 5,0; 7,5; 10,0 и 12,5 мл), прибавляют по 8 лсд
22 %-ного раствора перегнанной НС1, по 3 мл раствора H3S04 (1 : 1) и по
5 мл раствора хлорного железа, содержащего ъ i мл Ь мг Fe3+. После этого
для окисления ванадия прибавляют 20%-ный раствор КМп04 до устойчи¬
вой розовой или бурой окраски и далее поступают так же, как описано выше
для определения ванадия в испытуемых растворах. Для получения графика
с более высоким содержанием ванадия (от 125 до 250 мкг) измерения произ¬
водят в кювете с просматриваемым слоем 20 мм с тем же светофильтром; ис¬
пытуемые растворы колориметрируют в той же кювете. Строят график, на¬
нося на ось абсцисс содержание ванадия в микрограммах, а на ось ординат —
процент светопропускания или величину оптической плотности.
Реактивы. Приготовление всех реактивов и весь анализ производят на бидистил.
дированной воде.
1. Соляная кислота, 22%-ный раствор. Перегоняют в стеклянном аппарате смесь
концентрированной НС1 (550 мл) и воды (450 мл), собирают 500 мл перегнанной кислоты*
которая соответствует 22%-ному раствору (уд. вес. 1,1).
2. Серная кислота, 1:1.
3. КМп04, 2%-ный раствор. Растворяют 20 г марганцовокислого калия в воде и дово¬
дят до 1 л.
4. Нитрит натрия, 0,5% -ный раствор. Растворяют 0,5 г NaN02 в воде и доводят до
100 мл.
5. Вольфрамат натрия, 5%-ный раствор. Растворяют 5 г NaW04 в воде и доводят
до 100 мл.
6. Хлорное железо. Растворяют 24,2 г FeCls*6H20 в воде, подкисляют 10 мл 22%-ной
НС1 для предотвращения гидролиза солей железа и доводят водой до 1 л. Такой раствор
содержит около 5 г железа в 1
7. Стандартный раствор ванадия. Отвешивают 2,3 г ванадата аммония или 1,785 а
V2Ofi, прокаленной при 450°, растворяют при нагревании в 10 мл 5%-ного прозрачного
раствора NaOH, нейтрализуют 10%-ным раствором серной кислоты. Переносят в мерную
колбу на 1 л, доводят водой до метки. Для приготовления раствора с содержанием в 1 мл
100 мкг ванадия отбирают 10 мл этого раствора и доводят в мерной колбе до 100 мл. Рабо¬
чий стандартный раствор ванадия получают разведением 10 мл предыдущего раствора
водой до 100 мл. Этот раствор содержит в 1 мл 10 мкг ванадия.
Определение подвижного ванадия по Добрицкой (1973)
В связи с тем, что ванадий имеет довольно близкие химические и физио¬
логические свойства с молибденом, для извлечения из почв подвижных форм
ванадия автором опробован ряд вытяжек, предложенных ранее для молиб¬
Определение молибдена, ванадия, марганца и иоОа в почвах
433
дена. В результате экспериментов рекомендовано извлечение подвижного
ванадия производить оксалатным буферным раствором с pH 3,3 с конечным
определением вольфраматным колориметрическим методом, описанным выше
(см. определение валового содержания ванадия).
Экстракцию подвижных форм ванадия можно производить одновременно
с экстракцией молибдена (из одной навески почвы), но для этого необходимо
увеличить навеску почвы до 35—40 г и количество оксалатного буферного
раствора до 350—400 мл. Все дальнейшие операции по обработке вытяжки
азотной кислотой, перекисью водорода и хлорной кислотой такие же, как
это описано для подвижного молибдена. Обработанный сухой остаток под
конец растворяют при нагревании в 18 мл 22%-ной НС1 и 10 мл воды и, не
фильтруя, переносят в мерную колбу на 50 мл, доливают водой до метки
и после перемешивания отбирают 20 мл раствора для определения ванадия,
а в оставшихся 30 мл раствора определяют подвижный молибден.
Переносят 20 мл раствора в термостойкий стакан на 100 мл, прибавляют
3 мл H2S04 (1 : 1) и окисляют ванадий до V6 2%-ным раствором КМп04,
как это описано выше при определении валового содержания ванадия. Весь
дальнейший ход анализа остается без изменений.
Содержание подвижного ванадия рассчитывают по калибровочному гра¬
фику (см. определение валового содержания ванадия).
МАРГАНЕЦ
Наиболее распространенным методом определения марганца в почвах
является фотоколориметрический метод, основанный на изменении интен¬
сивности окраски марганцевой кислоты, образующейся при окислении
Мп2* до Мп04 персульфатом аммония в сернокислом растворе, содержащем
азотнокислое серебро и фосфорную кислоту. Существует несколько вариантов
определения марганца в почвах этим способом.
Определение валового содержания марганца
Колориметрическое определение марганца
по Добрицкой (1958)
Из фильтрата после разложения образца почвы тем или иным способом
(по Добрицкой) отбирают аликвотную часть, соответствующую 0,2—0,3 г
почвы, помещают в стакан на 10 мл, прибавляют 5 мл концентрированной
азотной кислоты и для удаления из раствора хлора выпаривают на этерни¬
товой плитке досуха. Такую обработку повторяют еще 3—4 раза, прибавляя
азотную кислоту по стенкам стакана для смывания с них следов хлора.
К сухому остатку прибавляют 25 мл 10%-ного раствора серной кислоты,
нагревают до растворения (10—15 мин.). Затем прибавляют 15 мл бидистил¬
лированной воды и 2 мл ортофосфорной кислоты (уд. вес 1,7) для связывания
окрашенных ионов железа, а также для предупреждения гидролиза солей,
образуемых ионами марганца высшей валентности. После перемешивания
прибавляют 2—3 мл 1%-ного раствора азотнокислого серебра (в качестве
катализатора и для осаждения возможных следов хлора). Нагревают еще
10 мин. Бели раствор помутнеет, его отфильтровывают через плотный фильтр,
промывают водой, подкисленной серной кислотой, доводят до объема около
45 мл. К нагретому раствору равномерно порциями прибавляют около 2 г
персульфата аммония, нагревают до кипения 2—3 мин., для быстрого и пол¬
ного окисления марганца до марганцевой кислоты. При этом происходит
энергичное разложение персульфата аммония и обильное выделение пузырь¬
ков озона.
434
Ю. И. Добрицкая
Стакан с раствором охлаждают, окрашенный раствор переносят в мерную
колбу на 50 мл, доводят водой до метки и колориметрируют на ФЭК-Н с зе¬
леным светофильтром (№ 5, длина волны 536 нм) в кювете с просматривае¬
мым слоем 20 мм против 5 %-ного раствора серной кислоты. Если окраска
раствора слишком интенсивна, то его можно разбавить 5 %-ной серной кис¬
лотой и тотчас же измерить или измерения интенсивности окраски сделать
в кювете с просматриваемым слоем 10 мм, соответственно измеряя стандарт¬
ные растворы в той же кювете.
Содержание марганца вычисляют по калибровочному графику, приго¬
товленному из 0,1 н. раствора КМп04 (фиксанал). Этот раствор, разбавлен¬
ный до 0,001 н., содержит в 1 мл 11 мкг Мп.
В случае анализов карбонатных почв лучшие результаты получаются,
если остаток после разложения почвы смесью серной и фтористоводородной
кислот растворить не в серной кислоте, а в 5 мл концентрированной соляной
кислоты, прибавить 25 мл горячей воды, профильтровать и промыть фильтр
горячей водой, подкисленной НС1. Весь фильтрат перенести в фарфоровую
чашку и выпарить досуха на водяной бане, прибавив к нему для удаления
хлора 10 мл концентрированной HN03. Эту обработку повторить 2—3 раза.
Сухой остаток растворить в 25 мл 10%-ной H2S04, нагреть на бане 15 мин.,
прибавить 2 мл Н3Р04 и 3 мл 1 %-ного раствора AgN03. Нагреть в течение
10 мин., отфильтровать в мерную колбу на 50 мл. Дальнейший ход анализа,
как описано выше.
Калибровочный график готовят следующим образом. Отбирают пипеткой
в мерные колбы на 50 мл по 2; 5; 10; 15; 20 и 25 мл 0,001 н. раствора КМп04,
доводят до метки свежеперегнанной бидистиллированной водой и тотчас
же колориметрируют с зеленым светофильтром (длина волны 536 нм), ис¬
пользуя те же кюветы, которые применяли для фотоколориметрирования
испытуемых растворов. Строят калибровочный график, нанося на ось абс¬
цисс количество миллилитров 0,001 н. раствора КМп04, а на ось ординат —
процент светопропускания или величину оптической плотности. Затем по
графику находят содержание марганца в испытуемых растворах.
Иногда окраска такого раствора может иметь несколько другой оттенок,
чем испытуемые растворы. В таком случае раствор КМп04 предварительно
восстанавливают, а затем окисляют персульфатом аммония. Для этого в хи¬
мический стакан на 500 мл отбирают 91 мл точно 0,05 н. раствора перманга¬
ната калия, прибавляют 250 мл дистиллированной воды, 20 мл концентри¬
рованной H2S04, раствор перемешивают и обесцвечивают несколькими кап¬
лями 10%-ного раствора сульфита натрия (Na2S03). Раствор кипятят до ис¬
чезновения запаха S02, охлаждают, доводят в мерной колбе водой до 1 л.
В 1 мл этого раствора содержится 0,05 мг Мп.
Из него готовят шкалу стандартных растворов, для чего отбирают в мер¬
ные колбы на 50 мл от 1 до 6 мл стандартного раствора, доводят водой до
25 мл, прибавляют 1 мл Н3Р04 и 1 мл AgN03, нагревают на плитке, вносят
по 3 г персульфата аммония, доводят до кипения и кипятят 2—3 мин. Охлаж¬
дают, доводят водой до метки и измеряют светопоглощение на фотоколори¬
метре при длине волны 536 нм. По отсчетам строят калибровочный график.
Реактивы. Все реактивы готовятся на бидистиллированной воде.
1. Азотная кислота, х. ч., уд. вес. 1,4.
2. Серная кислота, уд. вес. 1,84и 10%-ный раствор. 65 мл концентрированной H2S04
выливают в воду, перемешивают и по охлаждении доводят водой до 1 л.
3. Ортофосфорная кислота, уд. вес 1,7.
4. Азотнокислое серебро, 1 %-ный раствор. Растворяют 1 г AgNOa в 100 мл воды.
5. Надсернокислый аммоний (персульфат аммония), х. ч.
6. Фиксанал КМп04, 0,1 н. раствор.
7. Сульфит натрия, 10%-ный раствор. 10 г NaaSOs растворяют в 100 мл воды.
Определение молибдена, ванадия, марганца и иода в почвах
435
Визуальное определение марганца по Ринъкису (1963)
Аликвот фильтрата после кислотного разложения почвы (см. стр. 425)
в количестве 5—10 мл переносят в широкогорлую термостойкую колбу на
50 мл или термостойкий стаканчик на 100 мл и для удаления хлора прибав¬
ляют 1—2 мл концентрированной азотной кислоты и выпаривают досуха.
Такую обработку азотной кислотой повторяют еще 2—3 раза, каждый раз
выпаривая досуха.
Для растворения остатка прибавляют 0,3 мл концентрированной азотной
кислоты, 7 мл бидистиллированной воды, доводят до кипения и после раст¬
ворения и охлаждения приступают к окислению марганца. Для этого к раст¬
вору прибавляют 0,2 мл концентрированной серной кислоты, 0,3 мл 85%-
ной ортофосфорной кислоты, 0,5 мл 2 %-ного раствора азотнокислого серебра
и после перемешивания присыпают около 0,1 г сухого персульфата аммо¬
ния — (NH4)2S208 — и нагревают до кипения. Если сразу развивается
сильное окрашивание, то приливают 10—15 мл воды, содержащей окисли¬
тель. Добавление 0,1 г персульфата аммония и нагревание повторяют еще
1—2 раза до тех пор, пока окраска раствора не перестанет изменяться.
Затем, с таким расчетом, чтобы интенсивность окраски раствора находилась
в пределах шкалы, доводят объем прибавлением воды, содержащей окисли¬
тель, до 10, 25, 50 или 100 мл. Часть этого раствора (10 мл) переливают
в калиброванную пробирку и производят визуальное сравнение со
шкалой.
Приготовление шкалы образцовых растворов марганца. Перекристалли-
зовывают х.ч. сернокислый марганец, прокаливают в муфеле при 500—
600°. Отвешивают 2,75 г прокаленного сульфата марганца и растворяют
в бидистиллированной воде, содержащей 5 мл концентрированной серной
кислоты, и доводят водой до 1 л. Такой раствор является исходным и содер¬
жит 1 мг Мп в 1 мл. Для приготовления рабочего стандартного раствора с со¬
держанием 0,1 мг Мп в 1 мл исходный раствор разбавляют водой в 10 раз.
В серию (10 шт.) мерных колб на 100 мл наливают рабочий стандартный ра с -
твор по схеме (табл. 1).
Далее объем жидкости в каждой колбе доводят водой до около 50 мл,
прибавляют по 2 мл концентрированной серной кислоты, 1 мл 85%-ной ор¬
тофосфорной кислоты, 1 мл 2 %-ного раствора азотнокислого серебра, пере¬
мешивают и прибавляют 3 раза сухой персульфат аммония (всего около 0,6 г),
каждый раз доводят до кипения. После охлаждения раствор доводят до мет¬
ки (100 мл) водой, содержащей окислитель, взбалтывают и часть (10 мл)
переносят в калиброванные пробирки для пробирочной шкалы и по ней про¬
изводят визуальное сравнение содержания марганца в анализируемых рас¬
творах.
Таблица 1
Шкала стандартных растворов для определения марганца
М колб
Показатели
1
2
3
4
5
6
7
8
9
10
Стандартный раствор,
МЛ
0,5
1,0
2,0
3,0
4,0
5,0
6,0
7,0
8,0
9,0
Содержание Мп
в колбе, мкг
50
100
200
300
400
500
600
700
800
900
Содержание Мп в 1 мл
колориметрируемого
раствора, мкг
0,5
1
2
3
4
5
6
7
8
9
436
Ю. И. Добрицкая
Результаты вычисляют по формуле
r АВВ
л- IH ’
где X — содержание марганца в почве, мг/кг, А — содержание марганца в совпадаю¬
щей по окраске пробирке шкалы, мкг/мл; Б — объем колориметрируемого стандартно¬
го раствора в пробирке шкалы, мл; В — общий объем анализируемого раствора, мл\
Г — объем анализируемого раствора, взятого в пробирку для определения марганца, мл\
Н — навеска почвы, соответствующая общему объему анализируемого раствора, г.
Реактивы. 1. Марганец сернокислый, х. ч.
2. Ортофосфорная кислота, 85%-ная, уд. вес 1,7.
3. Персульфат аммония, х. ч.
4. Азотная кислота, уд. вес 1,4.
5. Серная кислота, х. ч., уд. вес 1,84 и 0,1 н. раствор — 2,8 мл H2S04 выливают в
воду и доводят водой до 1 л.
6. Серебро азотнокислое, 2%-ный водный раствор.
7. Вода с окислителем. К 100 мл бидистиллированной воды прибавляют 2 мл кон¬
центрированной серной кислоты, 1 мл 85%-ной фосфорной кислоты, 0,5 мл 2%-ного ра¬
створа азотнокислого серебра, около 0,6 г персульфата аммония и кипятят до прекра¬
щения сильного выделения пузырьков.
Определение подвижных форм марганца
Для извлечения из почв подвижных форм марганца применяют различ¬
ные вытяжки химическими реагентами, из которых наиболее распростране¬
ны вытяжки: 1) ОД н. раствором H2S04; 2) раствором сульфита натрия
(Schachtschabel, 1957); 3) раствором сульфата магния в присутствии гидро¬
хинона (Schachtschabel, 1957) и 4) ацетатно-натриевым или ацетатно-аммо-
нийным буферным раствором с pH 3,5 (по Кругловой, 1972).
Визуальное определение марганца по Ринькису (1963)
Отвешивают 5 г почвы или 2,5 г торфа в 100 мл колбу с притертой пробкой,
прибавляют 50 мл 0,1 н. раствора H2S04, взбалтывают 1 час и фильтруют.
Помещают 10—20 мл фильтрата в термостойкий стаканчик на 50—100 мл,
прибавляют 1 мл концентрированной азотной кислоты и выпаривают вы¬
тяжку досуха; для полного удаления хлора из вытяжки сухой остаток обра¬
батывают азотной кислотой еще 2 раза. Для растворения сухого остатка
прибавляют 0,5 мл концентрированной HNOa, 7 мл воды, доводят до кипе¬
ния и после охлаждения приступают к окислению марганца. Для этого к
раствору прибавляют 0,2 мл концентрированной H2S04, 0,3 мл 85%-ной
Н3Р04, 0,5 мл 2 %-ного раствора AgN03, вносят около 0,1 г сухого персуль¬
фата аммония и нагревают до кипения. Если сразу развивается сильное
окрашивание, то приливают 10—15 мл воды, содержащей окислитель. При¬
бавляют еще 2 раза персульфат аммония и нагревают. Объем доводят до 25,
50 или 100 мл бидистиллированной водой, содержащей окислитель (в зави¬
симости от интенсивности окраски исходного раствора). Часть его (10 мл)
переливают в калиброванную пробирку с притертой пробкой и затем срав¬
нивают визуально с пробирочной шкалой. Приготовление шкалы дано в
табл. 1.
Фотоколориметрическое определение марганца
по Добрицкой (1958)
Метод основан на извлечении подвижного марганца 0,1 н. раствором сер¬
ной кислоты при соотношении почва : раствор = 1 :10 и времени взаимодей¬
ствия 1 час, окислении марганца до марганцевой кислоты персульфатом ам¬
Определение молибдена, ванадия, марганца и иода в почвах
437
мония в присутствии азотнокислого серебра и фосфорной кислоты. Измере¬
ние окраски растворов производится на фотоэлектроколориметре.
Ход анализа. Помещают 5 г воздушно-сухой почвы, просеянной через
-сито в 1 лме, в колбу с притертой пробкой (на 100 мл), приливают 50 мл ОД н.
раствора H2S04, встряхивают на ротаторе 1 час и фильтруют через складча¬
тый фильтр с белой лентой.
Для определения марганца отбирают 10—15 мл вытяжки, помещают
в термостойкий стаканчик на 50 мл, прибавляют 5 мл концентрированной
HN03 и 2 мл 30%-ной Н202, выпаривают на закрытой плитке досуха.
Операцию окисления HN03 и Н202 повторяют еще 2—3 раза, затем
прибавляют по 3 мл только одной азотной кислоты и выпаривают досуха.
К сухому остатку приливают 25 мл 10%-ной серной кислоты, нагревают
до его полного растворения, приливают 15 мл воды, прибавляют 2 мл Н3Р04
•(уд. вес 1,7) и 2 мл 1%-ного раствора азотнокислого серебра и нагревают
5—10 мин. Если раствор помутнеет, то его следует довести до кипения
и отфильтровать через фильтр с синей лентой. Дальнейшие операции, при¬
готовление калибровочного графика и вычисление результатов такие же,
как это описано выше при определении валового содержания марганца в поч¬
вах.
Определение сульфитнорастворимого марганца
по Шахтшабелю (Schachtschabel, 1957)
Навеску в 5 г почвы, просеянной через сито в 1 мм, помещают в колбу
•с притертой пробкой на 100 мл, прибавляют около 0,2 г активированного
угля и 50 мл экстракционного раствора и взбалтывают на ротаторе 1 час.
Активированный уголь прибавляют для того, чтобы адсорбировать орга- -
нические вещества, переходящие в вытяжку. Затем сразу же фильтруют
через сухой складчатый фильтр с белой лентой. Для анализа употребляют
лервые порции фильтрата.
Отбирают 15—20 мл фильтрата в мерную колбу на 50 мл, прибавляют
дая маскирования восстанавливающего влияния хлора и связывания ионов
железа 2,5 мл смеси, состоящей из сульфата ртути, азотной кислоты, азот¬
нокислого серебра и фосфорной кислоты. Затем для окисления Мп2+ до
Мп7+ прибавляют 2 г персульфата аммония, колбу с раствором ставят в тер¬
мостат на 20—30 мин. при температуре 90—95° до прекращения выделения
лузырьков газа. Если окраска раствора станет коричневых тонов, то берут
новую порцию фильтрата (меньшую — 5—10 мл) и повторяют те же опера¬
ции. Раствор после охлаждения доводят водой до метки и колориметрируют,
«сак описано выше при определении валового содержания марганца по Доб-
рицкой.
Реактивы. 1. Экстракционный раствор. Отвешивают 123,3 г MgS04*7H20 и 2 г
Na2SOs, растворяют в воде и доводят до 1 л.
2. Раствор сульфата ртути. 75 г Hg2S04 растворяют в 400 мл HN03 (уд. вес 1,4) и
300 мл Н3Р04 (уд. вес 1,7), прибавляют 0,2 г AgN03 и бидистиллированной водой доводят
до 1 л.
3. Активированный уголь.
4. Персульфат аммония (NH4)2S208, х. ч.
Определение легковосстанавливаемого марганца
по Шахтшабелю (Schachtschabel, 1957)
Экстрагирование производят 1,0 н. раствором сульфата магния, в кото¬
рый прибавлено 2 г гидрохинона на 1 л раствора. Все операции по экстракции
я дальнейшему определению марганца такие же, как и при определении
сульфитного марганца.
438
Ю. И. Добрицкая
Определение подвижного марганца по Крупскому
и Александровой (1964) в ацетатно-аммонийной вытяжке с pH 4,8
Метод применим как для карбонатных, так и некарбонатных почв.
Приготовление буферного раствора. 108 мл уксусной кислоты (98%-ной)
разводят бидистиллированной водой примерно до 600—700 мл, приливают
75 мл 25%-ного раствора аммиака, перемешивают и доводят водой до 1 л.
Проверяют pH раствора по стеклянному электроду. Уксусная кислота
я аммиак должны быть предварительно очищены от следов микроэлементов.
Приготовление вытяжки. Юг воздушно-сухой почвы, про¬
пущенной через сито в 1 мм, переносят в колбу на 250 мл, приливают 100 мл
буферного раствора, встряхивают на ротаторе 1 час и фильтруют через фильтр
с белой лентой. Переносят 25 мл фильтрата в термостойкий стаканчик и вы¬
паривают 2—3 раза на закрытой плитке, прилив по 2 мл 30 %-ной перекиси
водорода и 5 мл HN03 (уд. вес 1,4). Это необходимо потому, что в вытяжку
переходит часть органического вещества, мешающего определению марганца,
и, кроме того, присутствие ацетатного иона может затруднить дальнейший
ход анализа. Остаток еще раз выпаривают досуха с 3 мл HN03. К сухому
остатку приливают 25 мл 10 %-ного раствора серной кислоты, нагревают на
плитке до его полного растворения, прибавляют 2 мл Н3Р04 (уд. вес 1,7)
и 3 мл 1 %-ного раствора AgN03 и нагревают 5—10 мин.
Дальнейшие операции, приготовление калибровочного графика и реак¬
тивов такие же, как описано при определении валового содержания
марганца по Добрицкой.
Определение подвижного марганца по Кругловой (1972)
в карбонатных почвах в ацетатно-натриевой вытяжке с pH 3,5
Приготовление буферного раствора. Исходные растворы: 1) 1 я. раствор
СН3СООН — 60 мл ледяной уксусной кислоты растворяют в воде и доводят
до 1 л, уд. вес 1,006; 2) 1 н. раствор уксуснокислого натрия — 136 г
CH3C00Na*3H20 растворяют в воде и доводят до 1 л.
Буферный раствор с pH 3,5 состоит из 925 мл 1 н. раствора уксусной кис¬
лоты и 75 мл 1 н. раствора уксуснокислого натрия. Раствор тщательно пере¬
мешивают, дают постоять 2 суток и проверяют pH. Этот раствор пригоден
для среднекарбонатных почв.
Приготовление вытяжки из почв. 10 г воздушно-сухой
почвы, пропущенной через сито в 1 мм, помещают в колбу на 250 млг
приливают 100 мл буферного раствора с pH 3,5. Суспензию периоди¬
чески взбалтывают круговыми движениями (10 мин.) для разрушения
основной массы карбонатов. Затем закрывают пробками и взбалтывают
на ротаторе 1 час. Раствор отфильтровывают через складчатый
фильтр. Отбирают 25 мл фильтрата в термостойкий стакан, при¬
ливают 2 мл концентрированной HN03 и 2 мл 30%-ного раствора Н202
и выпаривают досуха. Остаток прокаливают в муфельной печи до полного
удаления органических веществ. Охлажденный остаток растворяют при
нагревании в 25 мл 10%-ного раствора HN03. Приливают 3 мл 1%-нога
раствора AgN03, нагревают и, если выпадает осадок, то его отфильтровы¬
вают через фильтр с синей лентой. Фильтр промывают 2—3 раза водой с 3—
5 каплями 1 %-ного раствора AgN03. Прибавляют 2 мл Н3Р04 (уд. вес 1,7),
раствор нагревают до 70—80°. Прибавляют около 1 г персульфата аммония
и нагревают до кипения. Окрашенный раствор охлаждают в ванне с холодной
водой и количественно переносят в мерные колбы на 50 мл, доводят водой
до метки и колориметрируют с зеленым светофильтром (536 нм).
Рассчитывают содержание марганца по калибровочному графику, при¬
готовленному из 0,1 н. фиксанала КМп04.
Определение молибдена, ванадия, марганца и иода в почвах
439
Определение подвижного марганца формальдоксиматным методом
по Самохвалову (Самохвалов и др., 1971)
Определение подвижного марганца с помощью формальдоксима менее
сложно и значительно чувствительнее персульфатного метода. С этим реа¬
гентом образуют окрашенные комплексы не только Мп, но и Fe, Си, Со и Ni.
Окрашенный комплекс с железом разрушают трилоном Б и гидроксилами-
ном, а Си, Со и Ni в почвах мало и их комплексы окрашены более слабо.
Для образования комплекса необходима щелочная реакция, которую соз¬
дают хлоридно-аммиачным буфером или аммиаком. Использование формаль¬
доксима позволяет определять подвижный марганец непосредственно в 0,1 н.
сернокислой или ацетатно-аммонийной вытяжках с pH 3,5 (по Кругловой,
1972) без их дополнительной обработки, что делает анализ достаточно прос¬
тым и высокопроизводительным.
Ход анализа. Вытяжку 0,1 н. раствором H2S04 получают обычным спосо¬
бом при отношении почва: раствор = 1 : 1 и времени взаимодействия 1 час.
Вытяжку ацетатно-аммонийным буферным раствором с pH 3,5 получают
следующим образом: навеску 10 г воздушно-сухой почвы, пропущенной через
сито 1 мм, помещают в колбу емкостью 250 мл и заливают 100 мл ацетатного
буферного раствора с pH 3,5. Суспензию вначале периодически взбалтывают
от руки 10 мин. для разрушения основной массы карбонатов, затем закры¬
вают полиэтиленовой или стеклянной пробкой и взбалтывают на ротаторе
1 час. Сразу же отфильтровывают через фильтр с белой лентой. Обязательно
проводить холостой опыт со всеми реактивами. Результаты холостого опыта
учитывают при вычислении содержания Мп. Для определения подвижного
марганца формальдоксиматным методом из фильтрата (0,1 н. раствором
H2S04 или буферным раствором) отбирают 10 мл, помещают в сухие мерные
колбы на 50 млу прибавляют 20 мл 1%-ной аскорбиновой кислоты, 5 мл
формальдоксима и 5 мл аммиачного буферного раствора, перемешивая рас¬
твор после прибавления каждого реактива.
Одновременно работают с 10 колбами; соблюдая ту же очередность колб,
в них приливают по 10 мл маскирующего раствора и перемешивают. После
того, как в 10-ю колбу прилит маскирующий раствор, сразу же колоримет-
рируют окрашенные растворы, начиная с первой и кончая десятой колбой.
Необходимо, чтобы после прибавления аммиачного буфера и до прибавления
маскирующего раствора к данной колбе проходило не менее 2 и не более
10мин., а перед колориметрированием после прибавления маскирующего раст¬
вора — от 5 до 10 мин. Колориметрируют при 490 нм (сине-зеленый свето¬
фильтр) в кювете с толщиной просматриваемого слоя 10 мм против воды.
Содержание марганца в почве находят по калибровочному графику и вы¬
читают результаты холостого опыта.
Калибровочный график строят так. Отбирают 10 мл запас¬
ного стандартного раствора КМп04 (0,1 н.), помещают в мерную колбу на
100 мл, доливают до метки тем же раствором, которым сделана вытяжка.
Раствор содержит 110 мкг/мл Мп. В 50 миллилитровые колбы отбирают от
0,2 до 3 мл рабочего раствора Мп, доливают раствором, применявшимся для
вытяжек (0,1 н. H2S04 или буферный раствор с pH 3,5). Отсюда берут пробы
по 5 мл, помещают в 50-миллилитровые мерные колбы, окрашивают и коло¬
риметрируют так же, как и испытуемые растворы.
Реактивы. 1. Серная кислота, 0,1, н. раствор. В воду прибавляют 2,8 мл H2S04
(уд. вес 1,84) и после перемешивания доводят водой до 1 л.
2. Буферный раствор с pH 3,5. Отбирают 60 мл ледяной уксусной кислоты, прибавля¬
ют воды около 500 мл у перемешивают, прибавляют 5,7 мл 25%-ного раствора аммиака и
доводят водой до 1 л,
3. Аскорбиновая кислота, 1%-ный раствор. В день анализа 10 г реактива растворяют
в 1 л воды.
440
Ю. И. Добрицкая
4. Формальдоксим. Растворяют 12 г солянокислого гидроксиламина в 70 мл воды,,
прибавляют 6 мл 36%-ного формалина и доводят водой до 100 мл. Готовят в день анали¬
за.
5. Аммиачный буферный раствор. Растворяют в 800 мл 25%-ного аммиака 70 г хлс~
ристого аммония и доводят водой до 1 л.
6. Маскирующий раствор. Готовят 1%-ный раствор трилона Б (10 г в 1 л воды)».
В день анализа в этот раствор вносят 60 г солянокислого гидроксиламина и перемешивают»
7. Стандартный раствор марганца. Фиксанал 0,1 н. КМп04 растворяют в 500 мл
воды, в которую предварительно внесено 50 мл концентрированной H2S04. Ра»
створ затем обесцвечивают, прибавляя по каплям 10%-ный раствор сульфита натрия; за»
тем кипячением полностью удаляют сернистый газ, переносят раствор в мерную колбу на
1 л и по охлаждении доводят водой до метки. Раствор содержит 1100 мкг/мл Мп. Это за»
пасной раствор, из которого готовят рабочие растворы»
иод
Определение валового содержания иода
При определении иода в почвах и растениях применяют как объемный,,
так и колориметрический и кинетический методы. В связи с тем, что иод
в этих объектах связан в основном с органическим веществом, существует
много способов озоления и разложения проб, но наиболее распространены
сухое озоление в присутствии щелочей и минерализация кислотами. При су¬
хом озолении в муфельной печи (при определенной температуре) возможны
потери иода или неполное разрушение органических веществ, препятствую¬
щее нормальному ходу анализа. Метод кислотного озоления применяют
к материалам, бедным органическим веществом; этот способ обычно совме¬
щается с отгонкой иода в специальных аппаратах. Потери иода ничтожны.
В СССР до последнего времени был широко распространен объемный ме¬
тод определения иода в почвах и растениях по Драгомировой (1950), ос¬
нованный на открытом озолении материала в муфельной печи в присутствии
поташа (К2С03) с последующим титрованием иода (из иодидов) раствором
тиосульфата. Необходимым условием метода является чистота реактивов,
на иод.
Объемное определение иода по Драгомировой,
в модификации Глущенко и Миненковой
В литературе опубликовано несколько вариантов объемного метода опреде-
деления иода, в которых описаны некоторые изменения и дополнения метода
Драгомировой, упрощающие или ускоряющие определение.
Нами описан вариант данного метода по А. В. Глущенко и Е. П. Минен¬
ковой (1965) \ которые рекомендуют прямое открытое сжигание органичес¬
ких веществ в присутствии раствора поташа (пропитывание насыщенным:
раствором К2СОз), что предотвращает потерю иода при высокой температу¬
ре, а также исключают первую операцию в прописи Драгомировой (экстрак¬
ция разбавленной соляной кислотой и затем раствором поташа).
Ход анализа. Навеску растертой до состояния пудры почвы (2—4 г) поме¬
щают в платиновую чашку, прибавляют раствор поташа (1:1) при соотно¬
шении почва: раствор К2СОз=1:6. Чашку с содержимым ставят на водянук>
баню и упаривают до пастообразного состояния, а затем сушат в термо¬
стате вначале при 100—105°. и под конец при 170°. При выпаривании содер¬
жимое чашки периодически перемешивают круговым движением для разру¬
шения. корки, образующейся на поверхности раствора, чтобы предотвратить
1 Метод опубликован в статье Н. Г. Зырина и Т. X. Имади (1965).
Определение молибдена, ванадия, марганца и иода в почвах
441
разбрызгивание раствора при пробулькивании через пленку пузырьков газа.
Выпаривание идет медленно и иногда продолжается до 2 суток.
После высушивания производят спекание почвы с поташем в муфельной
лечи. Содержимое чашки перед спеканием покрывают сухим поташем (около
2 г), чтобы предотвратить потери иода во время спекания, и чашку ставят
в холодный муфель. Спекают 3—4 часа при постепенном повышении темпе¬
ратуры до 430°: первый час — при 300°, второй и третий — температуру по¬
вышают до 430° и при этой температуре чашку выдерживают только 5 мин.
Хорошо спекшаяся масса имеет серую или бурую окраску. При высоком
содержании гумуса в почвах время спекания удлиняют до 5—6 час. и через
каждые 1,5 часа чашку вынимают, охлаждают, окапывают 2—3 мл 10%-ного
раствора поташа, ставят в холодный муфель и опять постепенно повышают
температуру до 430°.
При таком постепенном повышении температуры при озолении происхо¬
дит более полный и равномерный отвод газов и продуктов сухой перегонки,
а также предупреждается обволакивание несгоревших органических частиц
продуктами легкоплавких компонентов золы. После полного озоления мате¬
риала приступают к экстракции иода из пека, для чего в охлажденную чашку
прибавляют 5—6 мл бидистиллированной воды, содержимое чашки тща¬
тельно перемешивают и растирают стеклянным пестиком до однородной вяз¬
кой пасты. Если воды прибавлено много, то перед растиранием для получе¬
ния пасты в чашку прибавляют сухой поташ. Затем для извлечения иода
в чашку приливают 7—8 мл 96 %-ного этилового спирта, очень хорошо рас¬
тирают и перемешивают всю массу со спиртом в течение 5—10 мин. и прозрач¬
ный спиртовый раствор над осадком сливают в коническую колбу на 100 мл
с притертой пробкой. Экстрагирование спиртом повторяют 8—10 раз. Оста¬
ток пека выбрасывают, а экстракт переносят из колбы в платиновую чашку
{можно в фарфоровую), прибавляют 2—3 капли 10 %-ного раствора поташа
и удаляют спирт осторожным выпариванием на водяной бане, не допуская
кипения спирта. Сухой остаток на дне и стенках чашки должен быть белым.
Если остаток окрашен в желтые тона, то следует дополнительно прокалить
•чашку (20—30 сек.) на открытом пламени газовой горелки или в нагретом
до 450° муфеле.
Белый остаток в чашке, содержащий весь иод, извлеченный из почвы,
количественно растворяют небольшими порциями (около 3 мл) бидистилли¬
рованной воды (5 раз), раствор каждый раз переносят в коническую колбу
•{на 50 мл) и в полученном минерализате определяют иод иодометрическим
объемным методом. Для этого раствор подкисляют серной кислотой (1:1)
до pH 2—3, кислотность контролируют по универсальному индикатору,
нанося на бумагу капли раствора капилляром. В подкисленный раствор при¬
бавляют свежеприготовленную бромную воду (3—4 капли) для окисления
иодида в иодат, перемешивают и для равномерного кипения прибавляют на
кончике ножа тальк. Колбу ставят на предварительно нагретую электро¬
плитку среднего нагрева и кипятят при частом перемешивании 10 мин.,
упаривают раствор примерно до 5 мл. Стенки колбы смывают раствором,
наклоняя и вращая колбу.
Во время кипячения бром, не вступивший в реакцию с иодидом, улету¬
чивается вместе с парами воды, а иодиды окисляются до иодатов по реакции
К J + ЗВг2 + ЗН20 = KJOs + бНВг.
После охлаждения в раствор прибавляют 2—3 кристаллика иодистого
калия (в пятикратном количестве к ожидаемому содержанию иода в навес¬
ке почвы) и 1 мл 0,5 %-ного свежеприготовленного раствора крахмала. Из¬
бытка KJ следует избегать, так как вместо синей окраски иодкрахмального
комплекса может получиться лиловая, что будет мешать определению конеч¬
ной точки титрования. В кислой среде KJ, взаимодействуя с KJOs, выде¬
442
Ю. И. Добрицкая
ляет эквивалентное количество свободного иода по уравнению: KJ03 +
+ 5KJ + 3H2S04 = 3H20 + 3K2S04 + 3J2. Выделившийся иод оттитро-
вывают из микробюретки 0,001 н. раствором тиосульфата. Реакция протекает
по схеме: J2 + 2Na2S203 = 2NaJ + Na2S4Oe. Для сравнения окраски обя¬
зательно ставят холостой опыт с дистиллированной водой, подкисленной
и обработанной бромом и всеми реактивами, с которыми работают в данный
день. Из результатов титрования вычитают данные холостого опыта.
Вычисление содержания иода по результатам титрования производят
по следующей формуле
21,15 vn
м — — ,
т ’
где М — количество J в 1 кг почвы, мг; 21,15 — по условиям реакции на 1 моль иодата
приходится 6 атомов иода, т. е. 126,9 : 6 = 21,15; отсюда 1 мл 1 н. раствора тиосульфата
эквивалентен 21,15 мг или 21 150 мкг иода; v — количество тиосульфата, пошедшее на
титрование, мл\ п — нормальность тиосульфата и т — навеска почвы, г.
Описанный вариант объемного метода определения иода в почвах позво¬
ляет определять содержание иода с погрешностью около 7—8% при малой
и 2—3% при средней и высокой концентрации его в почвах. Абсолютная
чувствительность определения 3,2 • 10~7 % в растворе; коэффициент варьиро¬
вания данных равен 1,6—1,5%.
Реактивы. Реактивы для определения иода должны быть «химически чистыми»
или «особо чистыми». Основной подготовкой к анализам является приготовление очищен¬
ных от иода реактивов.
1. Поташ. Отвешивают 3 г-экв щавелевой кислоты и 3 г-экв поташа, растворяют в поч¬
ти кипящей воде раздельно — поташ в 400 мл и щавелевую кислоту в 600 мл. Горячие
растворы фильтруют, затем смешивают в литровом стакане и выпаривают до начала выпа¬
дения кристаллов. К горячему раствору прибавляют половинный объем этилового спирта,
чтобы понизить растворимость оксалата калия. После охлаждения осадок отфильтро¬
вывают через воронку Бюхнера. Осадок дважды промывают небольшим количеством
этилового спирта и переносят в платиновую или фарфоровую чашку, подсушивают и по¬
мещают в холодный муфель, где прокаливают при 600—650° 2—3 часа. При прокаливании
выделяется СО, поэтому прокаливать необходимо в вытяжном шкафу.
2. Йодистый калий. Перекристаллизовывают из воды. Для увеличения выхода соли
в охлажденный раствор с кристаллами прибавляют этиловый спирт, выпавшую соль от¬
сасывают или фильтруют и промывают небольшим количеством этилового спирта, просу¬
шивают между листами фильтровальной бумаги и хранят в склянке из темного стекла с
притертой пробкой.
3. Этиловый спирт перегоняют, добавив поташ для связывания иода.
4. Бидистиллированная вода — перегоняют с поташем и КМп04.
5. Тальк — промывают соляной кислотой (1 : 5) и водой, затем высушивают и прока¬
ливают в муфеле.
6. Тиосульфат, 0,001 н. Готовят перед титрованием разбавлением в 100 раз 0,1 н*
раствора тиосульфата водой без С02, проверяют обычным способом его титр.
7. Бромная вода. Готовят в день анализов. Бром должен быть свободен от иода.
8. Серная кислота особой чистоты (без иода).
9. Крахмал, 0,5%-ный раствор. Свежеприготовленный из растворимого крахмала
при непродолжительном нагревании.
Колориметрическое визуальное определение иода
по Каменеву (1965)
В основу метода положена способность иода перегоняться с водяным
паром, конечное определение иода производят колориметрически визуаль¬
ным сравнением в пробирках после извлечения иода хлороформом или бен¬
золом.
Определение молибдена, ванадия, марганца и иода в почвах
443
Ход анализа. 15 г почвы, растертой до состояния пудры, помещают в пла¬
тиновую чашку, приливают 20 мл 0,1%-ного раствора поташа; хорошо пере¬
мешивают и высушивают в сушильном шкафу при 105—110°, а затем прока¬
ливают в муфеле при 450—480° в течение 4 час. Из охлажденной прокаленной
почвы производят четырехкратную экстракцию иодидов. Для этого в чашку
с почвой вводят 20 мл бидистиллированной горячей воды и после перемеши¬
вания ставят на 10 мин. на горячую электроплитку. После отстаивания фильт¬
руют через бумажный фильтр. Все четыре отфильтрованных водных экстрак¬
та собирают в одну колбу и упаривают до 20 мл. Затем приступают к извле¬
чению иода. В связи с тем, что иод легко перегоняется с водяным паром при
взаимодействии с окислителем в кислой среде, то для отгона иода раствор из
колбы переносят в перегонную колбу, нейтрализуют постепенным прибавле¬
нием по каплям 50%-ного раствора серной кислоты до прекращения вспени¬
вания и выделения углекислого газа. Затем в колбу вводят 100 мл 0,1%-но-
го раствора бихромата калия в 10%-ном растворе серной кислоты и после
тщательного перемешивания прибавляют 3 г кристаллической щевелевой
кислоты. В приемную колбу вводят 2 мл 2 %-ного раствора двууглекислого
натрия, аппарат соединяют и начинают отгонку иода. За 15—20 мин. [иод
полностью перегоняется. Собирают по 20—40 мл дистиллята, упаривают до
3—5 мл и далее в нем определяют иод.
Для визуального определения иода в ряд колориметрических пробирок
с притертыми пробками, d 12—15 мм, вводят рабочий раствор иодистого
калия(см. стр. 447), начиная со второй пробирки, по 0,1; 0,2; 0,3; 0,4жлит. д.,
что соответствует 1, 2, 3, 4 и т. д. мкг иода. В первую пробирку KJ не прибав¬
ляют. Объем в пробирках доводят до 5 мл 10 %-ной серной кислотой. В такие
же пробирки переносят испытуемые растворы, доливают до объема 5 мл
10 %-ной серной кислотой и затем во все пробирки прибавляют по 1 мл хлоро¬
форма или бензола, вводят по 4 капли (не более!) 0,5 %-ного свежеприготов¬
ленного раствора нитрита натрия и, закрыв пробирки пробками, поочередно
встряхивают 20—30 сек. После разделения жидкостей сравнивают окраски
органического слоя в испытуемых и образцовых растворах.
Вычисление результатов (в мг/кг) производят по формуле:
тде А — количество иода в совпадающей по окраске пробирке шкалы, мкг; М — навеска
почвы, взятая для определения иода, а.
Реактивы. 1. Б и дистиллированная вода, перегнанная в присутствии поташа в
КМп04.
2. Поташ (К2СОэ) должен быть освобожден от загрязнений иодидами. Очищать его
можно способом, описанным в методе Драгомировой, или более простым и вполне надеж*
ным методом автора. К 100 мл бидистиллированной воды, нагретой до 80°, прибавляют
1 мл 0,1 н. раствора азотнокислого серебра и 5 г поташа; раствор оставляют в темном месте
на 2 суток. После отстаивания прозрачную часть раствора осторожно сливают с осадка
(не взмучивать!). Для освобождения от излишка AgNOs к раствору прибавляют 1 лд 2%-
ного раствора NaCl и снова оставляют в темном месте на 2 суток, после чего раствор осто¬
рожно сливают с осадка и отфильтровывают через стеклянный фильтр № 3 или № 4. По¬
лученный раствор поташа доводят до 0,1%-ной концентрации разведением бидистиллиро¬
ванной водой до 5 л.
3. Нитрит натрия (NaN02), 0,5%-ный свежеприготовленный водный раствор.
4. Щавелевая кислота, кристаллическая, х. ч.
5. Хлороформ или бензол, х. ч.
6. Азотнокислое серебро, 0,1%-ный водный раствор.
7. Бихромат калия (К2Сг20?), 1%-ный раствор в 10%-чой серной кислоте.
8. Натрий хлористый, 2%-ный водный раствор.
444
Ю. И. Добрицкая
Фотоколориметрическое определение иода по Лапину,
Ришу и Бен-Утяевой (1959), вариант Покатилова (1968)
Как для кинетических, так и для объемных и колориметрических методов
определения иода необходимо: 1) разложение образца почвы, 2) извлечение-
иода из полученного минерализата и 3) определение выделенного иода. Пер¬
вые два этапа по Лапину выполняются точно по методу, описанному Драго-
мировой, с предварительной экстракцией навески почвы растворами поташа
и соляной кислоты и извлечением иода из сухого остатка после выпаривания
объединенных вытяжек многократной экстракцией спиртом. Заключитель¬
ная часть анализа — определение извлеченного из пробы иода — выполня¬
ется авторами фотоколориметрическим методом, основанным на высокочувст¬
вительной реакции иона J2Br” с бриллиантовым зеленым, предложенной
JI. Н. Лапиным (Лапин и др., 1959). Реакция проводится в кислой среде
в присутствии нитрита натрия с образованием полигаллоидных соединений.
К достоинствам данного метода относится большая чувствительность реак¬
ции, хорошая воспроизводимость результатов и конечное определение на
фотоколориметре.
Ход анализа. Подготовка почв к анализу по Драгомировой (1950) заклю¬
чается в том, что 1—2 г почвы, растертой до пудры, предварительно обра¬
батывают 30—40 мл разбавленной (10%-ной) соляной кислоты, а затем 30—
40 мл горячего 10 %-ного раствора поташа. Остаток почвы после обработки
промывают горячей водой, переносят в платиновый тигель и высушивают
в сушильном шкафу при 105—110°. К сухому остатку прибавляют 5-кратное-
количество поташа, постепенным нагреванием доводят содержимое тигля до
спекания и далее нагревают до 450—480°. После 3—4-минутного нагревания
операция заканчивается. Содержимое тигля смывают горячей водой в ста¬
кан и растворяют при нагревании. Фильтрат и промывные воды соединяют
с ранее полученными НС1 и К2С03 вытяжками, переносят в платиновую
чашку и выпаривают на водяной бане досуха. Затем чашку с осадком высу¬
шивают при 150—170° и осадок далее обрабатывают этиловым спиртом, как.
это описано в методе Драгомировой. Для определения содержания иодидов
в сухом остатке, полученном после выпаривания спиртового экстракта, вы¬
сушивания при 170° и последующего осторожного прокаливания, осадок
растворяют в 15 мл 0,243 н. раствора бромида натрия и количественно пере¬
носят в делительную воронку. В чашку (тигель) прибавляют 1 мл раствора
Н3Р04 (1 : 1), вносят 1 мл воды и переносят в делительную воронку, в кото¬
рую затем приливают 0,5 мл 0,25 %-ного раствора нитрита натрия и 0,75 мл-
0,5 %-ного раствора бриллиантового зеленого. Смесь тщательно перемеши¬
вают и оттитровывают Н3Р04 (1 :1 ) из микробюретки до кислой среды,,
при этом сам бриллиантовый зеленый служит индикатором, имея в кислой
среде сине-зеленую окраску. К хорошо перемешанному раствору приливают
10 мл толуола и 2 мл 85%-ной Н3Р04, тщательно встряхивают для экстрак¬
ции иода и отстаивают 5 мин. После расслоения жидкостей солевой раствор
сливают, а толуоловый экстракт переносят в кювету ФЭК объемом 10 мл
и колориметрируют с красным светофильтром. Отсчеты производят по пра¬
вому барабану. В левой кювете помещают раствор, приготовленный из 15 мл
раствора бромида натрия, 0,75 мл 0,5 %-ного раствора бриллиантового зе¬
леного, 10 мл толуола и 2 лел85%-ной Н3Р04, которым можно пользовать¬
ся только в течение 1 дня.
Калибровочный график готовят из рабочего стандартного раствора йоди¬
стого калия с содержанием в 1 мл 1 мкг иода (стр. 447). Помещают в делительные
воронки от 1 до 10 мл\ рабочего раствора, прибавляют по 15 мл 0,243 н. раст¬
вора бромида натрия, по 0,5 мл 0,25 %-ного раствора нитрита натрия и по
0,75 мл 0,5 %-ного раствора бриллиантового зеленого. Перемешивают и от¬
титровывают Н?Р04 (1 : 1) из микробюреткп до кислой среды (сине-зеленая
Определение молибдена, ванадия, марганца и иода в почвах
445
окраска). Приливают 10 мл толуола и 2 мл 85%-ной Н3Р04 и далее экстра¬
гируют иод встряхиванием, как это описано выше, и колориметрируют.
Строят калибровочный график и рассчитывают содержание иода в почве.
Р е а к т и в~ы. 1. Поташ. 1 кг К2С03 растворяют в 810 мл дистиллированной воды,
прибавляют 100 мл этилового спирта, переносят в большую делительную воронку и затем
сильно и долго встряхивают. После отстаивания отделяют спиртовый слой, а к водному
раствору вновь прибавляют 100 мл этилового спирта. Экстракцию спиртом повторяют не
менее 10 раз. Водный раствор поташа концентрируют при нагревании. По охлаждении ра¬
створа выпавшие кристаллы поташа отсасывают от маточника, затем соль высушивают
между листами фильтровальной бумаги и хранят в склянке с притертой пробкой. Для
проверки поташа на содержание иода можно использовать цветную реакцию J2Br~ с брил¬
лиантовым зеленым. Для этого в колбу на 100 мл всыпают 0,5—1 г поташа, 3 г бромида
натрия, нейтрализуют поташ концентрированной соляной кислотой и доводят общий
объем жидкости до 15 мл и затем прибавляют остальные реактивы в том порядке, как это
указано для определения иода в испытуемых образцах. Если толуоловый слой окрасит¬
ся, то экстракт помещают в кювету ФЭК и фотоколориметрируют.
2. Соляная кислота, перегнанная.
3. Бромистый натрий, 0,243 н. Растворяют 33,8 г NaBr и доводят водой до 1 л.
4. Ортофосфорная кислота, 1 : 1 и 85%-ная (уд. вес 1,7).
5. Нитрит натрия, 0,25%-ный раствор. Растворяют 0,25 г NaN02 и доводят водой до
100 мл. Раствор готовят сразу же перед употреблением.
6. Бриллиантовый зеленый, 0,5%-ный раствор. Нагревают на водяной бане 0,5 г
индикатора с 25 мл 96°-ного этилового спирта и 75 мл дистиллированной воды. После ра¬
створения кристаллов красителя горячий раствор фильтруют через складчатый фильтр,
охлаждают и переливают в склянку из темного стекла.
7 Толуол, ч.д.а.
Кинетический роданидно-нитритный метод определения иода
в почвах по Проскуряковой (1966)
Сущность метода заключается в определении скорости реакции окисле¬
ния роданида железа, зависящей от концентрации ионов иода, являющихся
катализаторами. Скорость реакции определяют по изменению светопогло-
щения раствора, окрашенного роданидом железа в оранжево-красный цвет.
При окислении роданида эта окраска исчезает, и чем больше концентрация
иода в растворе, тем быстрее происходит его обесцвечивание, т. е. быстрее
уменьшается светопоглощение или оптическая плотность раствора, измеряе¬
мая на фотоэлектроколориметре через определенные промежутки времени.
Изменение оптической плотности изображают графически, откладывая по
горизонтали время, а по вертикали — соответствующие плотности (рис. 1),
и получают кинетические прямые, угол наклона которых (tg а) зависит от
концентрации иода в растворе. По этим прямым подсчитывают изменение
светопоглощения за 1 мин., т. е. тангенс угла наклона прямой (tg а) к оси
времени. Например, для шестой прямой tg ae = АВ/ВС. Тангенсы углов
являются количественной характеристикой скорости реакции и прямо про¬
порциональны количеству иода в растворе (табл. 2).
По растворам с известной концентрацией иода строят калибровочный
график (рис. 2), а по тангенсу угла наклона кинетической прямой (tg аж),
полученной для исследуемого раствора, и калибровочному графику опреде¬
ляют содержание иода в анализируемом растворе.
Скорость химической реакции сильно зависит от температуры. Поэтому
измерение светопоглощения стандартных и анализируемых растворов сле¬
дует производить при одной и той же температуре. Для этого автор реко¬
мендует применять водяной циркуляционный термостат, металлическую ру¬
башку для кювет и сосуды-смесители. При отсутствии таких установок
446
Ю. И. Добрицкая
Таблица 2
Скорость реакции (tg а) при различном содержании иода
Содержание иода
в 5 мл, мкг
tg а
Содержание иода
в 5 мл, мкг
tg а
0,0
0,014
0,3
0,051
0,1
0,026
0,4
0,065
0,2
0,037
0,5
0,078
и сосудов-смесителей анализы можно проводить и при комнатной температу¬
ре, смешивая растворы просто в колбах на 100 мл, но обязательно соблюдая
следующую последовательность добавления реактивов: 1) испытуемый или
стандартный раствор, 2) роданид калия, 3) нитрит натрия, 4) раствор квас¬
цов в азотной кислоте. Секундомер включают после добавления последнего
раствора. В этом случае калибровочный график необходимо проверять,
если температура воздуха в комнате изменяется больше чем на 2°.
Ниже приведено описание хода анализа при наличии вышеуказанной
аппаратуры и сосудов-смесителей.
Навеску тонкорастертой почвы (1 г) помещают в фарфоровую чашку или
тигель, смачивают 1 мл 30%-ного раствора поташа, высушивают в сушиль¬
ном шкафу при 120° в течение 1 часа до абсолютно сухого состояния (посвет-
ление поверхностной пленки), переносят в муфельную печь и прокаливают
30 мин. при 420—460°. После охлаждения почву смачивают несколькими кап¬
лями воды и вновь высушивают и прокаливают. При такой обработке проис¬
ходит озоление органических веществ почвы. Прокаленный остаток взмучи¬
вают водой, переносят через воронку в мерную колбу с притертой пробкой
на 50 мл, доводят водой до черты, тщательно перемешивают и дают отстоять¬
ся в течение 12—24 час. При этом весь иод переходит в раствор в виде йоди¬
стого калия. Затем осторожно, пе взмучивая осадка, отбирают пипеткой
п
Рис. 1. Кинетические прямые при различном содержании иода в растворе (мкг в 5 мл)
1 — без иода; 2 — 0,1; 3 — 0,2; 4 — 0,3; 5 — 0,4; 6 — 0,5
Рис. 2. Калибровочный график для иода
Определение молибдена, ванадия, марганца и иода в почвах
447
5 мл осветленного раствора в один из отростков сухого сосуда-смесителя
(конструкции Бударина); во второй отросток наливают 1,5 мл роданида калия
и 1,5 мл нитрита натрия, в третий отросток — 2 мл раствора железо-аммо-
нийных квасцов в азотной кислоте. Сосуд-смеситель с растворами выдер¬
живают в течение 10 мин. при 32° и приступают к фотометрированию.
Измерения проводят с зеленым светофильтром (длина волны 500 нм)
в кювете с толщиной просматриваемого слоя 10 мм на правом барабане.
Через 15—20 мин. после включения фотоколориметра в сеть устанавливают
гальванометр на нуль нейтральными клиньями, поместив предварительно
кюветы с водой в оба пучка и поставив правый барабан на нуль оптической
плотности. Устойчивость нулевого положения проверяют в течение
5—10 мин. После подготовки прибора к измерениям переворачивают сосуд-
смеситель, одновременно включая секундомер, и перемешивают растворы
встряхиванием. Полученным раствором заполняют кювету и помещают ее
в правое плечо фотоэлектроколориметра. Первый раз измеряют светопогло-
щение через 1 мин. после начала реакции (после включения секундомера)
и затем через каждые 30 сек. Для построения кинетических прямых доста¬
точно 8—10 отсчетов. Интервалы времени зависят от скорости реакции:
чем быстрее отклоняется стрелка гальванометра, тем чаще следует произво¬
дить отсчеты с тем, чтобы за время измерений оптическая плотность умень¬
шилась не более чем на 40% своей первоначальной величины.
На основании полученных данных строят кинетическую прямую, как опи¬
сано выше, определяют изменение светопоглощения за 1 мин. (tg ах) и по
калибровочному графику находят количество микрограмм иода в 5 мл раст¬
вора.
Содержание иода в почве (в мг/кг) определяют по формуле
(а — Ь)-10
с 1
где а — количество иода в 5 мл раствора, мкг; Ъ — количество иода в контрольной пробе,
мкг; (а — Ь) 10 — количество иода в 50 мл раствора, т. е. во всей навеске, мкг; с —
навеска почвы, взятая для анализа, г.
Контрольная проба — это 1 мл раствора поташа, проведенный через все стадии ана¬
лиза параллельно образцам почвы.
Калибровочный график. В один из отростков сосудов-смесителей вносят
по 0,1 мл поташа, различные количества стандартного раствора иодистого
калия в пределах от 0,1 до 0,5 мл (раствор содержит 1 мкг J в 1 мл) и доводят
водой до общего объема 5 мл. Остальные реактивы и операции те же, что и
при анализе проб.
Реактивы. I. Поташ (КгСОз), 30%-ный водный раствор.
2. Роданид калия, 0,006 М. Растворяют 0,6 г KCNS в 1 л воды.
3. Нитрит натрия, 0,29 М. Растворяют 20 г NaN02 в 1 л воды.
4. Железо-аммиачные квасцы, 0,10 М. Растворяют 50 г NH4Fe(S04)2-12H20 в 1 л
3,0 н. азотной кислоты.
5. Стандартный раствор иодистого калия (KJ). Вначале готовят раствор, содержащий
500 мкг иода в 1 мл\ для этого 32,6 мг иодистого калия растворяют в воде и доводят в мер¬
ной колбе до 50 мл. Для получения раствора с содержанием иода 10 мкг в 1 мл разбавля¬
ют водой 1 мл первого раствора до 50 мл. Полученный раствор еще разбавляют в 10 раз и
получают стандартный раствор, содержащий 1 мкг иода в 1 мл.
6. Азотная кислота, 3,0 н. Приготовляют по удельному весу и проверяют титрованием
навески соды.
Все реактивы должны быть химически чистыми или предварительно очищены пере¬
кристаллизацией. Для приготовления растворов и во всех остальных операциях приме¬
няют бидистиллированную воду, перегнанную в присутствии поташа и марганцовокислого
калия. После приготовления новой партии реактивов необходимо проверить калибровоч¬
ный график.
448
Ю, И. Добрицкая
Ускоренный вариант кинетического
роданидно-нитритного метода определения иода
по Проскуряковой, Швейкиной и Никитиной (1973)
Метод основан на каталитическом действии иодида в реакции окисления
роданида нитрит- и нитрат-ионами. Навеску тонкорастертой почвы 0,5 г
помещают в фарфоровый тигель, смачивают 1 мл 30%-ного раствора поташа
(К2С03), высушивают в сушильном шкафу при 150° и прокаливают 30 мин.
в муфельной печи при 480°. После охлаждения смачивают 6—8 каплями
бидистиллированной воды и еще раз прокаливают. Обычно при такой обра¬
ботке сгорает вся органическая часть почвы, изменяется цвет остатка. Для
полного удаления органического вещества можно повторить смачивание,
высушивание и прокаливание до посветления остатка. После озоления оста¬
ток смывают через воронку в мерную колбу емкостью 50 мл, раствор доводят
водой до метки, перемешивают и оставляют отстаиваться до следующего
дня.
Для определения иода из мерной колбы отбирают точно 5 мл осветленного
раствора и переносят в стеклянную пробирку с притертой пробкой емкостью
10—15 мл, прибавляют 0,4 мл KCNS, 1 мл NaN02, 1,6 мл NH4Fe(S04)2.
После добавления последнего реактива включают секундомер и измеряют
оптическую плотность раствора точно через 5 мин. на фотоэлектроколоримет¬
ре (ФЭК-57) с синим светофильтром (длина волны 430 нм) в кювете с толщи¬
ной просматриваемого слоя 10 мм.
Содержание иода в испытуемом растворе определяют по калибровочному
графику.
Калибровочный график. В пять пробирок с притертыми пробками вно¬
сят по 1 мл 3%-ного раствора К2С03, различные количества (0,2; 0,6; 1,0;
1,4; 1,8 мл) стандартного раствора иодида калия, содержащего 0,2 мкг/мл J,
и доливают воду до 5 мл. Затем добавляют все реактивы, как и в испытуемых
пробах, и измеряют оптическую плотность в той же кювете. На основании
полученных данных строят калибровочный график, нанося на ось абсцисс
содержание иода в микрограммах, на ось ординат — оптическую плотность.
Содержание иода (в мг/кг) в почве рассчитывают по формуле
где А — количество иода, найденное по калибровочному графику, мкг; 50 — общий объ¬
ем раствора, мл; 5 — объем раствора, взятого для анализа, мл; 0,5 — навеска почвы, г.
При высоком содержании иода в почве вместо 5 мл отбирают 2—3 мл или
меньше из осветленного раствора и добавляют бидистиллированную воду
до 5 мл, учитывая это при расчетах.
Реактивы. 1. 30%-ный раствор поташа. Растворяют 300 г очищенного К2СОз
в 1 л воды.
2. 3%-ный раствор поташа. Растворяют 30 г очищенного К2С08 в 1 л воды.
3. 0,006 М раствор роданида калия. Растворяют в воде 0,6 г KGNS и доводят водой до
1 д.
4. 0,29 М раствор нитрита натрия. Растворяют 20 г NaN02 в 1 л воды.
5. Железо-аммонийные квасцы. Растворяют 100 г NH4Fe(S04)2*12H20 в 1 л 3,0 н
раствора HN03.
6. 5%-ный раствор нитрата калия. Растворяют в 1 л воды 50 г KN03.
7. Вода бидистиллированная. Дистиллированная вода вторично перегоняется после
добавления 5 г/л поташа в стеклянном перегонном аппарате.
8. Стандартный раствор KJ. Для приготовления запасного раствора иодида калия,
содержащего 500 мкг иода в 1 мл, растворяют 32,6 мг К J в воде, раствор доводят в мерной
колбе до 50 мл. Из этого раствора путем разведения] готовят раствор с содержанием
0,2 мкг иода в 1 мл.
Определение молибдена, ванадия, марганца и иода в почвах
449
Литература
Виноградов А. Я. Колориметрическое оп¬
ределение ванадия с фосфоровольфрамо¬
вой кислотой.— Докл. АН СССР, 1931,
серия А.
Добрицкая Ю. И. Определение валового
содержания марганца в почвах и расте¬
ниях.— В кн. «Методы определения мик¬
роэлементов в почвах и растениях». М.,
Изд-во АН СССР, 1958.
Добрицкая Ю. И. Определение ванадия,
молибдена, марганца, титана и железа иэ
одной навески почвы.— Почвоведение,
1969а, № 2.
Добрицкая Ю. И. Фотоколориметрическое
определение ванадия в почвах.—Агрохи¬
мия, 19696, № 4.
Добрицкая Ю. И. Определение подвижных
форм ванадия и молибдена из одной на¬
вески почвы.— В сб. «Микроэлементы в
биосфере и их применение в сельском хо¬
зяйстве и медицине Сибири и Дальнего
Востока». Улан-Удэ, 1973.
Драгомирова М. А. Определение малых ко¬
личеств йода в почвах, растительных и
животных организмах.— В сб. «Методы
определения микроэлементов». М., Изд-
во АН СССР, 1950.
Зырин Я. -Г., Имади Т. X. Оценка методов
определения и варьирования содержания
йода в почвах.— Агрохимия, 1965, № 4.
Каменев В. Ф. Определение йода и брома в
почве, воде и биологическом материа¬
ле.— Химия в сельском хозяйстве, 1965,
№ 1.
Круглова Е. К. Методика определения до¬
ступных форм микроэлементов в карбо¬
натных почвах и растениях. Ташкент,
«ФАН», 1972.
Крупский Я. К., Александрова А. М. К во¬
просу об определении подвижных форм
микроэлементов.— В сб. «Микроэлемен¬
ты в жизни растений, животных и чело¬
века». Киев, «Наукова думка», 1964.
Лапин Л. Я., Рейс Я. В. Определение йо¬
да в питьевой воде с бриллиантовым зеле¬
ным.— Лабораторное дело, 1960, № 8.
Лапин Л. Я., Риш;М. А., Бен-Утпяева
Г. М. Йод в почвах и растениях Зерав-
шанского бассейна и новый фотоколори-
метрический метод его определения.—
Труды Ин-та каракулеводства, № 9. Са¬
марканд, 1959.
Методические указания по определению
подвижных форм микроэлементов в поч¬
вах для вональных агрохимлабораторий.
М., 1973.
Покатилов Ю. Г. К методу определения
подвижного и валового йода в почвах и
пищевых объектах.— В сб. «Микроэле¬
менты в сельском хозяйстве и медицине».
Улан-Удэ, Бурятское кн. изд-во, 1968.
Проскурякова Г. Ф. Кинетический рода-
нидно-нитритный метод определения мик¬
роколичеств йода в почве.— Агрохимия,
1966, № 11.
Проскурякова Г. Ф., Швейкина Р. В., Ни¬
китина О. Я. Способ определения йода в
почве, воде, растениях, молоке и крови.—
Тезисы докл. второго Всес. симпозиу¬
ма по методам определения микроэлемен¬
тов в природных объектах. Самарканд.
1973.
. Ринъкис Г. Я. Методы ускоренного коло¬
риметрического определения микроэле¬
ментов в биологических объектах. Рига,
Изд-во АН ЛатвССР, 1963.
Самохвалов С. /\, Целикова Я. А. Исполь¬
зование дитиола при определении под¬
вижного молибдена в почвах и анализе
растений.— Химия в сельском хозяйстве,
1973, № 7.
Самохвалов С. /\, Чеботарева Я. Л., Жу¬
кова Л. Ф. Определение подвижного мар¬
ганца в почвах с помощью формальдок-
сима.— Химия в сельском хозяйстве,
1971, № 4.
Сендел Е. Колориметрические методы опре¬
деления следов металлов. М., «Мир»,
1964.
Grigg J. L. Determination on available mo¬
lybdenum in soils.— J. Sci. Techn., 1953,
v. 34, N 5.
Schachtschabel P. Die Bestimmung des Man-
genversorgungsgrades des Boden und seine
Beziehhung zum Auftreten der Dozzflec-
ken-Krankneit bei Hafer.— Z. Pflanzener-
nahr. Dung, und Bodenkunde, 1957,
Bd. 78, H. 2-3.
Scholl W. Chemische Bestimmung von Mo-
lybdan in Boden, Pflanzen und Gesteinen
mit Toluol-3, 4-Dithiol. Die Spurenele-
mentverborgung von Pflanze, Tier und
Mensche. Frankcfurt, 1962.
Stanton R. E.f Hardwick Mrs. A. /. The co¬
lorimetric determination of molybdenum
in soils and sediments by zinc dithid.—
Analyst, 1967, v. 92, 1095.
А. В. СОКОЛОВ, Я. Я. СЕРДОБОЛЬСКИЙ, М. Г. СИНЯГИНА
МЕТОДИКА ПРИМЕНЕНИЯ МЕЧЕНЫХ АТОМОВ
ПРИ АГРОХИМИЧЕСКИХ ИССЛЕДОВАНИЯХ1
*
Атомы одного и того же элемента могут иметь разную массу. Например,
известен в настоящее время кальций, имеющий массовые числа 39, 40, 42
и др.; известен фосфор с массовыми числами 29, 30, 31 и др. Несмотря на это,
все химические свойства, например у атомов кальция с массовыми числами
от 39 до 49, совершенно одинаковы, в связи с чем все атомы этого элемента
должны быть отнесены к одному и тому же месту в таблице элементов
Д. И. Менделеева. Чтобы отличить их друг от друга, им дали название изо¬
топов элемента. Слово изотоп греческое, оно слагается из двух слов:
isos — одинаковый, ровный, подобный и topos — место. Таким образом
кальций имеет до 10 изотопов с атомными весами 39, 40, 42, . . ., 49;
фосфор — 6 изотопов; известны элементы, например иод, имеющие 15 изо¬
топов, и т. д. В настоящее время доказано, что все элементы* имеют изо¬
топы.
Возникает вопрос, что является однозначной характеристикой элемента,
что общего у всех его иэотопов? Оказывается, что такой однозначной характе¬
ристикой элемента является эаряд ядра его атома. Ядро атомов элементов
слагается из двух видов элементарных частиц: электрически нейтральных
нейтронов, которые обозначают обычно символом и, и частиц, несу¬
щих один положительный заряд,— протонов, имеющих символ р (или
Я1). Массы нейтрона и протона равны между собой. Массовое число каждого
изотопа элемента равно общему числу нейтронов и протонов, содержащихся
в ядре его атомов. Так, ядро атомов изотопа кальция, имеющего массовое
число 39, содержит общее число нейтронов и протонов 39; для изотопа каль¬
ция с массовым числом 40 число нейтронов и протонов составит 40 и т. д.
Но для всех изотопов кальция, т. е. для элемента кальция вообще, характер¬
но, что в ядрах их содержится одинаковое количество заряженных частиц,
именно 20, которые определяют и общий заряд ядра, и строение всей элект¬
ронной оболочки атомов. Точно так же для всех изотопов фосфора, имеющих
раэные массовые числа, характерно одинаковое содержание в ядрах числа
протонов, именно 15, и т. д.
Таким образом, основными характеристиками элементов следует считать:
1) число протонов, или общий заряд ядра, т. е. число, получившее название
порядкового номера элемента, так как в порядке этих чисел элементы рас¬
полагаются в таблице Д. И. Менделеева; 2) массовое число элементов, ко¬
торое у различных изотопов элемента может быть различным.
При символическом изображении элементов принято указывать обе эти
характеристики, причем массовое число обычно указывают вправо и вверху
у символа элемента, а порядковый номер (заряд ядра, число содержащихся
в ядре протонов) — внизу и слева. Например, 15Р80; 15р31; 1бР32 — изобра¬
жают три изотопа фосфора с характерным для него числом протонов 15
и с массовыми числами 30, 31, 32. Соответственно: 20Са39; 20Са40; 20Са« -
три изотопа кальция и т. д. Символами элементарных частиц нейтрона и про¬
тона, очевидно, будут 0гЛ, хр1 (или Н1), которые указывают, что массовые
1 В подготовке работы к печати участвовали Д§ В* Федоровский, Р» А» Ширшова»
Методика применения меченых атомов
451
числа обеих частиц равны единице (значок справа), а заряд ядра нейтрона
равен нулю и протона — единице (значок слева).
Стабильные и радиоактивные изотопы. Стабильны¬
ми изотопами называют те, в ядрах атомов которых никаких превращений
ядер не происходит и структура ядра которых остается стабильной, постоян¬
ной. К стабильным изотопам относится большинство природных изотопов,
с которыми приходится иметь дело в обычных условиях. Сюда относятся,
например, хН1; 15Р31; 7N14; вС12 и др. Радиоактивные изотопы в большинстве
случаев получают искусственно, при бомбардировке стабильных изотопов
мощными потоками нейтронов, протонов, осуществляемой на сцециальных
установках: циклотронах, линейных ускорителях, бетатронах и др. Ядра
радиоактивных изотопов неустойчивы, и в них самопроизвольно происхо¬
дит ядерная реакция превращения (распада), приводящая чаще всего к из¬
менению заряда и, следовательно, к превращению самого элемента в другой,
близкий по расположению в таблице Д. И. Менделеева. Для примера приво¬
дим самопроизвольную реакцию распада изотопа натрия:
11N а24 = i2Mg44 + (К
Происходит ядерная реакция, при которой один из нейтронов ядра превра¬
щается в протон с выделением отрицательно заряженной частицы |5. При
такой реакции массовое число, равное 24, не меняется, а положительный
заряд ядра увеличивается на единицу. Это приводит к полному изменению
химических свойств изотопа натрия, т. е. к превращению его в магний.
Таким образом, внешним признаком, отличающим стабильные и радиоак¬
тивные изотопы, является наличие (у радиоактивных) или отсутствие (у ста¬
бильных изотопов) радиоактивного излучения.
Каждый элемент может иметь стабильные и радиоактивные изотопы.
Например, известны изотопы:
Стабильные: хН1, хН2, eC12, eC13, 7N14, 7N16, e0le, gO18 и т. д.
Радиоактивные: 1Н3, eC10, eC14,7N13,7N16, вО14, вО16, аО19 и т. д.
При агрохимических исследованиях в качестве метки могут быть исполь¬
зованы как стабильные, так и радиоактивные изотопы. Методы определения
их резко различны: стабильные изотопы определяют по разности их атомных
весов на масс-спектрометрах и оптических спектрографах; радиоактивные
изотопы определяют по характеру и интенсивности их излучения на особых
счетчиках, на более простых, более доступных для массовых лабораторий
установках. Ниже в статье излагаются методы работы только с радиоактив¬
ными изотопами; лиц, интересующихся методикой работы со стабильными
изотопами, отсылаем к специальным руководствам.
Радиоактивное излучение. Как известно, радий при своем
радиоактивном распаде дает лучи трех видов: 1) а-лучи — поток сравнитель¬
но крупных частиц, представляющих собой ядра атомов гелия с массовым
числом 4 — 2Не4, легко поглощаемых слоем воздуха в 1—2 см; 2) (J-лучи —
поток электронов, или, как их называют, ^-частиц с массой в 1845 раз мень¬
ше массы протонов, поглощаемых слоем воздуха в 0,5—2 ле, и 3) у-лучи —
электромагнитное излучение типа рентгеновских лучей с очень большой
проницаемостью в зависимости от так называемой жесткости их; очень «жест¬
кие» у-лучи не задерживаются заметно даже слоем свинца в 1—2 см.
При самопроизвольном распаде радиоактивных изотопов, применяемых
при агрохимических исследованиях, могут образоваться все эти три вида
излучений. Реже всего может быть обнаружено a-излучение; его можно наб¬
людать, например, при распаде лития и бериллия. Некоторые радиоактив¬
ные изотопы при самопроизвольном распаде испускают положительные
452
А. В. Соколов, if. /7. Сердобольский, М. -Г. Синягина
электроны, так называемые позитроны —р+, с очень небольшой длиной про¬
бега, например: вС10; «С11; 7N12; 7N18 и пр. Чаще встречаются случаи, когда
при самопроизвольном распаде ядер радиоактивных изотопов происходит
испускание отрицательных электронов —Р“, излучение, наиболее удобное
для учета при работе с «мечеными» атомами. Нередко при самопроизвольном
распадс изотопов наряду с р~-излучением наблюдается у-излучеьие. Кроме
указанных, можно иногда наблюдать и другие типы радиоактивного распа¬
да. Ниже мы остановимся на технике измерения только р~-излучений как
излучений, чаще всего наблюдаемых при самораспаде изотопов, применяе¬
мых при агрохимических исследованиях. Описание других типов проявле¬
ния радиоактивности можно найти в специальных руководствах.
Каждая Р~-частица, испускаемая каким-нибудь радиоактивным изотопом,
обладает своей энергией движения, .так что в потоке электронов, испускаемых
изотопом, можно наблюдать электроны различной энергии. Но оказыва¬
ется, что для каждого изотопа энергия испускаемых им электронов не может
превышать некоторой, характерной для этого изотопа величины. Эта мак¬
симальная энергия является, таким образом, определенным характерным
признаком каждого радиоактивного изотопа. В качестве примера укажем,
что наибольшая энергия ^“-частиц (электронов), испускаемых при распаде
16Р32, составляет 1,7 Мэв х, для изотопа 20 Са46 — 0,25 Мэв, для вС14 —
0,15 Мэв, для трития ХН3 — около 0,01 Мэв. Лучи с большой энергией, на¬
пример для 16Р32, называют жесткими, с малой энергией, например для 2о^а45)
вС14,—мягкими.
Электроны (^“-частицы), проходя через какое-нибудь вещество, встре¬
чаются с атомами последнего, ионизируют их и вследствие этого постепенно
теряют свою энергию. При достаточной толщине слоя вещества все р-излу-
чение может быть поглощено им полностью. Очевидно, что чем больше энер¬
гии излучения, тем толще слой вещества должен быть взят для полного пог¬
лощения излучения. Проницаемость различных веществ для р- излучений раз¬
лична и зависит от плотности вещества. Например, плотность алюминия
2,6, плотность свинца 11,4; следовательно, поглощение Р-частиц пластинами
этих металлов будет одинаковым, если толщина свинцовой пластинки будет
в 4,4 раза меньше, чем алюминиевой. Проницаемость (или пробег) для
P-излучений различных веществ измеряют количеством миллиграммов ве¬
щества на 1 см2. При таком способе количественного определения проница¬
емости эта величина будет характерной для излучения каждого изотопа
независимо от того, какое вещество задерживает излучения. Например, мак¬
симальный пробег Р“-частиц вС14 составляет 25 мг/см2, слой половинного пог¬
лощения — 2,6 мг/см2, независимо от того, будет это свинец или алюминий,
или воздух и пр. Для излучений изотопа ieS36 слой половинного поглощения
составляет 3 мг/см2, слой максимального пробега —17 мг/см2 и т. д. Для вы¬
числения толщины слоя вещества в сантиметрах следует величины проницае¬
мости разделить на плотность данного вещества, выраженную в миллиграм¬
мах на 1 см3. Например, плотность воздуха равна 1,3 мг/см*, тогда толщина
слоя воздуха, задерживающая на 50% излучение вС14, составит: 2,6 : 1,3 =
= 2 см, то же для ieS35 — 3 : 1,3 = 2,3 см, то же для 1б Р32 — около 0,6 м
и т. д.
Времяполураспада. Учет распада при измере¬
ниях. Напомним, что распад и превращение ядер радиоактивных изотопов
сопровождаются испусканием, например, р~-частицы. Так, распад радио¬
активного изотопа фосфора 16Р32 и превращения его в стабильный изотоп
1 Мэв — мегаэлектроновольт, равный миллиону электроновольт; эв — единица измерения
энергии, применяемая в ядерной физике, равная энергии, которую приобретает электрон,
проходящий в электрическом поле с разностью потенциалов в 1 в\ 1 эв = 1,6*10”19
джоулей.
Методика применения меченых атомов
453
16S32 сопровождаются излучением Р“-частиц:
15Р32 ieS82 + р".
Время распада отдельных ядер какого-нибудь изотопа весьма различно;
распад ядер одних атомов совершается через ничтожно малый промежуток
времени после их образования, продолжительность жизни других, наоборот,
чрезвычайно велика и т. д. Теоретически полный распад, т. е. распад всех
ядер вновь полученного изотопа, заканчивается только через бесконечный
промежуток времени. Поэтому говорить о времени полного распада какого-
нибудь изотопа трудно. Весьма показательным и легко определяемым яв¬
ляется время полураспада, т. е. время, за которое распадается 50% налич¬
ного количества атомов изотопа. Так как распад каждого ядра сопровожда¬
ется излучением одной Р~-частицы, то время полураспада может быть легко
определено как то время, за которое интенсивность р_-излучения уменьшится
вдвое.
Время распада или полураспада и вообще скорость самопроизвольного
распада ядер радиоактивных изотопов совершенно не зависит от обычных
условий проведения опыта. Ни нагревание, ни прокаливание при высоких
температурах, ни осаждение или возгонка, или кипячение, так же как и все
другие обычные химические и физические операции с изотопом, не могут ока¬
зать никакого действия на скорость распада и интенсивность р-излучения
изотопа. Поэтому время полураспада является характерной для каждого
радиоактивного изотопа величиной. Так, для 2(£а46 время полураспада со¬
ставляет 165 дней, для 20Са47 — 5,8 дня, для 20Са49 — 2,5 часа. Так же раз¬
лично время полураспада для изотопов других элементов; например, для
19К40 время полураспада равно 1,2- 10е лет, для 15Р29—4,6 сек.
Агрохимические опыты, особенно с растениями, занимают довольно зна- -
чительный промежуток времени, за который интенсивность р-излучения
изотопа, применяемого для опыта, может резко уменьшиться благодаря яв¬
лению самораспада. В этом случае, очевидно, возникает необходимость пе¬
ресчета всех данных по интенсивности излучения, полученных в разные
сроки, на какой-нибудь один срок. Чаще всего пересчитывают или на день
(момент) начала опыта, или на день (момент) окончания опыта. Можно вести
пересчет и на какой-либо промежуточный срок проведения опыта.
Известно, что количество р “-частиц (или других), испускаемых изотопом
в какой-то начальный период (N0), связано с числом их (7V), испускаемых
через t единиц времени, уравнением:
N=INо-еГх< ,
где X — константа распада; е]—основание натуральных логарифмов.
Константа распада, в свою очередь, связана с периодом полураспада (Т)
уравнением:
Т = 0,693 : X
Зная N, Т и tj можно по этим уравнениям рассчитать и N0, т. е. число
распадов к начальному моменту времени t0.
Проще этот пересчет делать по таблице, составляемой для каждого изо¬
топа отдельно самим экспериментатором. Для упрощения составления таких
таблиц можно предложить два уравнения:
0,301
No = KNH\gK= 'т
Например, для изотопа i5P32 период полураспада равен 14,2 дня. Тогда
0,301
\gK = 14 2~ •t = 0,0210 t.
Подставляя различные значения t, например 1, 2, 3, ..., дня, и пользуясь
обычными таблицами логарифмов, можно составить таблицу для изотопа
фосфора 1БР32 (табл. 1).
454
А. В. Соколов, Я. Я. Сердоболъский, М. Г. Синягина
Таблица 1
Таблица значений пересчетного коэффициента £ для деР32
Число дней
1 е к
к
к, %
Число дней
lg К
к
к, %
1
0,0210
1,05
105
4
0,0840
1,21
121
2
0,0420
1,11
111
5
0,1050
1,27
127
3
0,0630
1,16
116
и т. д.
Табл. 1 показывает, что для пересчета всех результатов измерений (про¬
веденных в разные сроки) на момент начала опыта следует данные, получен¬
ные, например, через 4 суток после начала опыта, умножить на 1,21; полу¬
ченные через 5 суток — на 1,27 и т. д.
Подробные таблицы изотопов можно найти в специальных руководствах.
В табл. 2 приведены данные только тех изотопов, которые наиболее часто
применяются в агрохимических исследованиях.
Каждый элемент периодической системы имеет несколько изотопов с раз¬
личным атомным весом, периодом полураспада и типом излучения. Напри¬
мер: натрий имеет пять изотопов, из которых Na21 и Na28 — стабильные,
Na22, Na24 и Na26 — радиоактивные. Калий имеет семь изотопов: К89 и К41 —
стабильные, К40 — имеет период полураспада 1,2-109 лет (в природных усло¬
виях содержится 0,01 ат.% К40 в элементе К), искусственно полученные изо¬
топы К37, К88, К42 и К48 являются радиоактивными. Из перечисленных изо¬
топов калия практически может быть использован в краткосрочных опытах
только К42 с периодом полураспада 12,36 часа. К87 и К88 имеют период полу¬
распада, исчисляемый минутами и секундами, а изотоп К48 еще не произво¬
дится х. Углерод имеет пять изотопов, из них С12 и С18 — стабильные и вхо¬
дят в состав природного углерода: первый в количестве 98,9% и второй —
1,1%; из радиоактивных изотопов С14 имеет период полураспада более 5000
лет, такой период полураспада позволяет применять С14 в различных иссле¬
дованиях в области физиологии и агрохимии. Природный азот представляет
тоже смесь изотопов N14 — 99,62% и N16 — 0,38%. Оба изотопа стабильные.
Все радиоактивные изотопы азота (N18, N1® и N17) имеют период полураспада
от нескольких секунд до 10 мин., поэтому практического значения для наших
исследований не имеют.
В качестве меченого азота применяют стабильный азот N16, но в этом
случае определение его производят не по излучению, а по атомному весу.
Для определения изотопа элемента по атомному весу применяется масс-
спектрометр.
Определение изотопного состава азота можно производить и спектраль¬
ным методом на спектрофотометре (Оболенская, Жадкова, 1964). Радиоак¬
тивные изотопы магния, алюминия и кремния имеют короткое время полу¬
распада, исчисляемое секундами и минутами, поэтому они не могут приме¬
няться при агрохимических исследованиях.
Очень удобен для агрохимических исследований радиоактивный изотоп
фосфора Р32, имеющий период полураспада 14,2 дня и высокую энергию
излучения — 1,7 Мэе.
Данные табл. 2 и 3 позволяют сделать выбор наиболее удобного для при¬
менения при агрохимических исследованиях изотопа каждого элемента.
1 В настоящее время выпускаются препараты с обогащением К40 до 2 ат. %, что позволяв
ет проводить агрохимические опыты с ним в качестве метки калия удобрений.
Методика применения меченых атомов
455
Таблица 2
Радиоактивные изотопы и меченые соединения
Элемент
Изотов
Период
Тип
Энергия излучения,
Мэв
В виде какого соединения
выпускается
полураспада
излучения
3
V
Железо
Fe60
45,1 дня
PVv
0,273
0,47
1,56
0,145
0,192
0,337
1,10
1,20
Fe203,(CeHe0e)Fe0,
Fe2(CeH507)2
Fe2(S04)s, FeCb
Иод
J131
8,06 дпя
Р~. V
0,25
0,34
0,61
0,81
0,08
0,164
0,264
0,364
0,637
0,722
NaJ, KJ, AgJ
Иттрий
58 дней
р- V
0,32
1,53
1,21
YCl, Y(N03)2
Калий
К42
К40
12,36 часа
1,2• 10е года
Р~ Y
Р~, Y
1,99
3,524
1,35
1,52
1,50
K2COs, KHC03,KC1
К Cl
Рубидий
Rb*8
18,7 дня
Р'. Y
0,68
1,77
1,08
Rb2C03, RbCl
Каль¬
ций
Са*5
165 дней
Р-
0,25
■
CaO, CaCOs, CaCl2,
CaHP04, CaS04-2H20
и др.
Кобальт
Со*а
5,25 года
Р". Y
0,309
0,630
1,478
1,17
1,332
2,158
Со металлический,
Co(NOj)j, СоС12, C0SO4
Марга¬
нец
Мп52
5,7 дня
Р+. V
0,6
1,332
1,434
1,73
МпС1г
Натрий
Мп64
313 дней
Y
0,835
MnCl2, Mn(NOa)2
Na24
15,05 часа
Р-. V
1,39
1,368
2,752
NaCl, NaHCOs, Na2C03,
Na2SiOs
Сера
Na22
2,62 года
Р+. Y
0,545
1,274
0,511
NaCl
S85
87,2 дня
р-
0,167
BaS04, CuS, FeS,
CaS04, Na2S04, H2S04l
K2S04, CuS04 и др.
Строн-
fTTTTT
Sr8»
51 день
Р-. V
1,46
0,95
SrCl2, Sr(N03)2, SrC 0 3
дни
Sr86
64 дня
у
—
0,514
SrCOs, SrCl2
Строн¬
ций +
+иттрий
Sr*>
29,3 года
р-
0,546
—
SrCl2, Sr(N03)2, SrCO,
Y0o
64,3 часа
р-
2,26
—
Тритий
H8
12,26 года
р-
0,018
—
Тритиевая вода и др.
Углерод
C“
5760 лет
р-
0,155
BaC03, CaC03, Na*C03,
мочевина, глюкоза, дру¬
гие органические соеди¬
нения
Фосфор
' pS2
14,2 дня
р-
1,71
КН2Р04, К2НР04,
Na2HP04, Са(Н2Р04)2,
Н3Р04 без носителя
456
А. В. Соколов, И. Я. Сердоболъский, М. А Синягина
Таблица 2 (окончание)
Элемент
Изотоп
Период
полураспада
Тип
Энергия излучения,
Мэв
В виде какого соединения
излучения
Э
V
выпускается
Фосфор
раз
24,4 дня
р-
0,25
_
Н3Р04 без носителя
Хлор
С1*
3,03* 10* лет
р-
0,714
—
NH4C1, К Cl, NaCl, НС1
и др.
Цезий
Cs184
2,07 года
Р“ V
0,086
0,282
0,652
0,886
0,475
0,567
0,797
1,037
1,168
1,368
1,570
CsCl, CS2CO3, CSNO3
Цезий-f
Сзш
29,6 года
Р"> V
0,51
0,661
CsCl, CsN03
+ барий
Ва187
2,6 мин.
V
1,17
0,66
Цинк
Zn®«
245 дней
Р+. Y
0,326
0,511
Zn(N03)2, Z11SO4, ZnClj
Таблица 3
Стабильные изотопы и меченые соединения
Элемент
Ивотоп
Содержание
в естественной
смеси, %
Обогащение,
ат.%
В воде какого соединения выпускается
Азот
N16
0,366
До 99
NHJbPO*, (NHihHPOi, NH4CI, KNOs,
Са (NO»)2, HNO», CO(NH*)*, NaNOa,
NHs, N2 и др.
Бор
В10
19,610
До 85—95
HsBOs, B2O3, В
Дейтерий
Н2
0,0149
До 99
NHs, (NH4)2S04, NH4CI, H20, H2SO4,
HC1, ШРО4, H2C2O4 и др.
Кислород
018
0,204
До 80
До 60
H20
CaO, HNOs, H2SO4, НзР04, NaOH
и др.
Сера
S86
0,014
До 9,8
BaSOft, S
Углерод
С18
1,107
До 70
ВаСОз, Na2C03, СО, CO2
При составлении таблиц были использованы каталоги: «Соединения и
изделия с радиоактивными изотопами« (М., Изд-во Всес. объединения «Изо¬
топ», 1972) и «Соединения и изделия со стабильными изотопами» (М., Изд-во
Всес. объединения «Изотоп», 1971).
СЧЕТЧИКИ РАДИОАКТИВНЫХ ИЗЛУЧЕНИЙ
Для того чтобы обнаружить и определить активность радиоактивного
излучения, применяются приборы, называемые детекторами. Детекторы
бывают различные: пластинки с фотоэмульсией, изменяющиеся под воздей¬
ствием радиоактивного излучения; калориметрические,*4 действие которых
Методика применения меченых атомов
457
основано на измерении тепла, выделяемого веществом при поглощении энер¬
гии излучения, и наконец электрические и сцинтилляционные, позволяющие
регистрировать отдельные элементы радиоактивного излучения в момент
распада атома.
Электрические детекторы
К электрическим детекторам относятся ионизационные камеры, полупро¬
водниковые детекторы и газонаполненные счетчики. Последние в зависимо¬
сти от режима работ подразделяются на пропорциональные и с самостоятель¬
ным разрядом (счетчики Гейгера — Мюллера). В зависимости от состава за¬
полняющих их гаэов счетчики делятся на две группы: а) «медленные», или
несамогасящие, и б) «быстрые», или самогасящие.
При агрохимических исследованиях почти исключительно пользуются
«быстрыми» счетчиками Гейгера — Мюллера, различающимися формой и
деталями конструкции. Наиболее распространены: цилиндрические счетчи¬
ки, приспособленные для подсчета более «жестких» излучений, обладающих
энергией более 0,6 Мэе, и торцовые счетчики, приспособленные для более
«мягких» излучений, с энергией менее 0,6 Мэе.
Цилиндрические счетчики изготовляют из трубок разного диаметра и
длины, материалом для них служат стекло, алюминий (вариант Троста)
или нержавеющая сталь. Схему устройства цилиндрического счетчика можно
видеть на рис. 1, а, где 1 — стек¬
лянный баллон, заполненный ге¬
лием, аргоном, азотом или други¬
ми газами с добавкой к ним в ко¬
личестве примерно 10% многоатом¬
ных газов или паров: этилена,
ацетилена, паров метилового или
этилового спирта и т. д., с целью
самогашения электрического раз¬
ряда в счетчике; 2 — клеммы для
включения в цепь; 3 — металли¬
ческая нить; 4 — металлический
цилиндрический катод. В другом
варианте цилиндрических счетчи¬
ков катодом служат металличе¬
ские стенки баллона счетчика; Рис* Цилиндрические счетчики Гейгера —
схематический рисунок такого сче- МюллеРа
тчика изображен на рис. 1, б, где Объяснение см. в тексте
1 — алюминиевый баллон, 2 —
клеммы; 3 — нить. На рис. 1 в изображен счетчик из нержавеющей стали.
В торцовых счетчиках, приспособленных для «мягкого» слабопрони¬
кающего излучения, одна из стенок изготовляется из тонкой слюдяной
мембраны, которая и служит местом проникновения в счетчик р-частиц
(рис. 2).
Принцип действия счетчика и механизм раз¬
ряда. На нить счетчика подается высокое напряжение от выпрямителя или
батареи, а корпус металлического счетчика или катод стеклянного заземля¬
ются. Напряжение на нить подается с таким расчетом, чтобы, с одной сто¬
роны, не происходило пробоя газовой прослойки между нитью и катодом и не
возникал в счетчике самостоятельный разряд и, с другой стороны, чтобы при
уменьшении сопротивления газовой прослойки, например вследствие иони¬
зации газов, возникал бы электрический разряд. Счетчики типа СТС-6,
СБТ-7, СБТ-13 работают при напряжении 300—400 в, счетчики МСТ-17,
Т-25-БФЛ. СИ-2Б и другие — 1300—2000 в. Как говорилось выше.
6 z
«0h!bsssbh®8&d
е
458
А. В. Соколов, И. Я. Сердоболъский, М. Г. Синягина
^--излучение производи! ионизацию газа, через который оно проходит, поэто¬
му при подаче указанного~напряжения на нить достаточно будет проникнуть
внутрь счетчика одной |5”-частице, как произойдет, вследствие ионизации
газа, разряд, возникнет импульс тока, который после усиления в ламповом
усилителе может быть учтен пересчетным устройством.
Механизм такого разряда весьма сложен; схематически он сводится к сле¬
дующему. При проникновении в счетчик электрона происходит ударная ио¬
низация находящихся в счетчике молекул газа и образование положитель¬
ных ионов и вторичных электронов, которые, нарастая в количестве, направ¬
ляются ускоряющимися темпами к положительно заряженной нити счет-
Рис. 2. Торцовый счет¬
чик
а, — внешний вид;
б — вертикальный разрез:
1 — стеклянный? корпус;
2 — металлическое покры¬
тие (катод);
8 — нить;
4 — контакты;
5 — слюдяное окно
чика. За короткое время, исчисляемое 10~7—10“8 долями секунды, эта лавина
электронов, количеством около 10 ООО, достигает нити счетчика, где и разря¬
жается. Одновременно с этим процессом при ионизации молекул происходит
возникновение ультрафиолетовых лучей, которые вырывают из стенок счет¬
чика фотоэлектроны; последние тоже ускоренным движением направляются
к нити, ионизируют новые молекулы газа и создают новую лавину электро¬
нов. Таким путем в счетчике возникает непрерывный разряд, который может
быть приостановлен включением в цепь большого сопротивления (несамога-
сящие счетчики). Тот же эффект получается при добавке к газам счетчика
высокомолекулярных газов или паров: этилена, метилового и этилового
спирта и пр. (самогасящие счетчики Гейгера — Мюллера). Спирт и другие
добавки поглощают ультрафиолетовое излучение и предупреждают возни¬
кновение вторичных лавин электронов. Таким образом, в самогасящих
счетчиках каждому проникшему внутрь счетчика электрону соответствует
один короткий разряд, или импульс тока.
Процесс ионизации сосредоточен вблизи нити, где электроны приобре¬
тают наибольшую скорость и образующиеся при ионизации положительные
ионы располагаются вдоль нити в виде чехла. Разряд этого положительного
заряда происходит медленнее разряда электронов, что создает так называ¬
емое мертвое время счетчика, о чем будет сказано ниже. Недостатком само-
гасящего счетчика является его ограниченный срок службы в результате
постоянного распада наполнителя — многоатомного газа. Счетчик, зареги¬
стрировавший около 109 импульсов, практически становится непригодным
к работе.
Характеристическая кривая счетчика (рабочая
характеристика счетчика, плато). Характеристическая кривая счетчика
представляет кривую зависимости числа импульсов, возникающих на счет¬
чике в единицу времени, от напряжения, приложенного к счетчику. На рис. 3
приведена в качестве примера кривая для одного из счетчиков. Начальный
потенциал счетчика, т. е. потенциал, при котором в счетчике начинают появ¬
ляться разряды, в приведенном на рис. 3 примере около 740 в. При увеличе¬
Методика применения меченых атомов
459*
нии напряжения число импульсов п сначала быстро растет, затем в интер¬
вале величины потенциала 880—1230 в практически остается постоянным,
после чего число импульсов резко возрастает вследствие возникновения
в счетчике самостоятельных разрядов. Горизонтальная часть кривой (АВ)
получила название «плато». Плато является важной рабочей характеристи¬
кой счетчика, показывающей, при каком значении поданного на нить потен¬
циала можно вести работу на счетчике, так как только в этом интервале
число импульсов практически не зависит от величины потенциала.
Каждый счетчик характеризуется своим начальным потенциалом и своим
плато, поэтому перед началом работы с новым счетчиком прежде всего следует
снять его характеристическую кривую.
Для этого измеряют число импульсов в
1 мин. от какого-нибудь источника при
разных значениях потенциала, начиная
с напряжения начала счета, указанного
в паспорте, например с 750 в. Увеличивая
потенциал каждый раз на 50 в, подсчиты¬
вают и записывают число импульсов, воз¬
никших в счетчике за 1 мин., пока не будет
достигнуто резкое возрастание числа им¬
пульсов вследствие появления в счетчике
самостоятельных разрядов. Данные изме¬
рений наносят на график, по которому
и устанавливают рабочее напряжение.
К новым счетчикам, поступающим в лаборатории, всегда прилагается
их характеристика с указанием начального потенциала и рабочего плато.
Но следует иметь в виду, что характеристическая кривая и положение на ней
плато несколько изменяются со временем, особенно при работе, почему сле¬
дует раз в 3—6 месяцев снова строить кривую и определять по ней плато.
Рабочее напряжение следует устанавливать всегда в пределах первой трети
плато. В области плато счетчики регистрируют 100% попавших в него
частиц.
Ложные, или самопроизвольные, разряды. Явле¬
ние ложных импульсов состоит в появлении в счетчиках разрядов, которые
прямо не связаны с прохождением через счетчик той или другой ионизиру¬
ющей частицы, например электрона. Появление ложных импульсов связано
с материалом катода, загрязнением его поверхности. Они всегда появляются
в виде группы импульсов, следующих один за другим через равные проме¬
жутки времени, и возникают после разряда, вызванного ионизирующей ча¬
стицей. Характер появления позволяет легко отличить их от импульсов,
вызванных ионизирующей частицей, которые возникают во времени хаоти¬
чески, неравномерно. Ложные импульсы присущи всем счетчикам, но в счет¬
чиках хорошего качества число их незначительно и не влияет на точность
подсчета. В счетчиках плохого качества наличие большого числа ложных
импульсов приводит к нарушению точности отсчета импульсов и вида харак¬
теристической кривой счетчика: более крутому подъему ее после плато и на¬
рушению горизонтальности плато. Хорошие счетчики имеют плато про¬
тяженностью 150—200 в и более и наклон плато не более 2—3%
на 100 в.
«Мертвое врем я» счетчика. Как описано выше, в счетчике
при разряде образуется вокруг нити положительный пространственный заряд»
который в связи с меньшей скоростью более крупных положительных ионов
ликвидируется значительно медленнее, чем заряд лавины отрицательных
электронов. Время, необходимое для разрушения положительного заряда,
получило название «мертвого времени», так как при наличии этого заряда
повый импульс тока в счетчике при проскоке через счетчик ионизирующей
Рис. 3. Характеристическая кривая
счетчика Гейгера — Мюллера
460
А. В. Соколов, И. J7. Сердобольский, М. Г. Синягина
частицы возникнуть не может. Продолжительность мертвого времени счет¬
чика постоянна для каждого поданного напряжения и имеет порядок 10~4 сек.
(при одном и том же числе импульсов).
Сцинтилляционные детекторы и датчики
Сцинтилляционными детекторами, или сцинтилляторами, называются
вещества, в которых при попадании ионизирующей частицы возникает све¬
товая вспышка. Это происходит за счет поглощения энергии частицы веще¬
ством, отчего атомы молекул приходят в возбужденное состояние, выделяя
фотоны. Отношение энергии световой вспышки к энергии вызвавшей ее чи-
стицы называется эффективностью сцинтиллятора. Эти короткие вспышки
(10“9—10"в сек.) лежат в области спектра от 800 до 6000 А. Каждому сцин¬
тиллятору соответствует свой максимум спектра. Все сцинтилляторы про¬
зрачны. По виду они бывают твердые, жидкие или газообразные.
В настоящее время предложено большое количество сцинтилляторов
(фосфоров). Очень удобны фосфоры в виде монокристаллов органических и
неорганических веществ: антрацена, стильбена, нафталина, флуорена, тер-
фипила, NaJ(Ti), KJ(Ti), CsJ, NaCl(AgCl), ZnS(Ag), CdW04 и др. 1
Для предупреждения от загрязнения, влаги и механических повреждений
кристаллы выпускают в герметических контейнерах с тонкой алюминиевой
крышкой и прозрачным стеклом — оптическим выходом. Из жидких сцин¬
тилляторов можно назвать: р-терфенил, РРО, РОРОР2 и др. В качестве
растворителя обычно берется толуол. Жидкие сцинтилляторы помещаются
в стеклянных илц металлических контейнерах с прозрачным оптическим
выходом.
Особого внимания заслуживают пластические сцинтилляторы, представ¬
ляющие собой включенные в полистирол органические сцинтилляторы:
стильбен, антрацен, л-терфенил с добавкой РОРОР, 2%-ный л-терфенил +
-|-0,1%-ный a-NPO 8 и др.
Выбор сцинтиллятора зависит от вида ионизирующего излучения: чем
больше поглощается энергия измеряемой частицы сцинтиллятором, тем эф¬
фективней будет его работа. Сцинтилляторы в сочетании с фотоумножителем
ФЭУ называются сцинтилляционным датчиком или сцинтилляционным счет¬
чиком (рис. 4).
Фотоумножитель — электровакуумный прибор, состоящий из фоточув-
ствительного катода и системы умножения (см. рис. 4). По внешнему виду
фотоумножитель напоминает большую радиолампу, внутри которой размещен
ряд электродов, на которые подается положительное напряжение. В качестве
катода фотоумножителя служит полупрозрачный светочувствительный слой,
который наносится с внутренней стороны плоского торца фотоумножителя.
Принцип работы ФЭУ состоит в том, что кванты света, попадая на фотокатод,
выбивают из него за счет фотоэлектрического эффекта электроны, которые,
ускоряясь, движутся на первый электрод (динод). В результате вторичной
эмиссии электроны выбивают из первого динода вторичные электроны иу
возрастая в количестве и ускоряясь, попадают на второй динод, затем на тре¬
тий, четвертый и т. д. Направление движения и их ускорение происходит
с помощью электрического поля между катодом и первым электродом, между
первым и вторым электродом и т. д. В результате происходит умножение
числа электронов, которые лавинообразно достигают анода фотоумножителя,
образуя электрический сигнал на внешнем нагрузочном сопротивлении ФЭУ.
1 В скобках (Ti, AgCl, Ag) указаны активаторы (активирующие добавки).
2 РРО — 2,5-дифенилоксазол-1,3; РОРОР — 1,4-ди—[2-(5-фенилоксазолил)]бензол.
в a-NPO - 2(4-бифеяилил)-5-фенилоксазол.
Методика применения меченых атомов
461
В сцинтилляционных датчиках используют¬
ся ФЭУ с коэффициентом усиления от 10е до
109 раз.
По конструкции фотоумножители имеют
несколько разновидностей, условно их можно
разбить на две группы: с фокусирующими
электродами и с нефокусирующими элект¬
родами. К первой группе относятся ФЭУ-19,
ФЭУ-24, ФЭУ-29, ФЭУ-35, ФЭУ-23 и др.; ко
второй — ФЭУ-11, ФЭУ-13, ФЭУ-16. Фотоум¬
ножители разных марок имеют число элект¬
родов от 8 до 14. Материалом фотокатода
служат сурмяно-цезиевые висмуто-серебря-
но-цезиевые или другие покрытия.
Сцинтилляционные датчики. Принцип
работы сцинтилляционного датчика описан
выше. Для большей эффективности работы
сцинтилляционного датчика необходимо, что¬
бы фотоумножитель имел максимальную чув¬
ствительность фотокатода в области макси¬
мума спектра сцинтиллятора. Разрешающее
время пересчетной схемы радиометра должно
быть также согласовано со сцинтилляцион-
ным датчиком. Как и счетчики Гейгера —
Мюллера, сцинтилляционный счетчик имеет
свой «фон», который следует учитывать при
подсчете результатов измерений. Правиль¬
ность работы сцинтилляционных счетчиков
контролируется эталонами. Применять сцин¬
тилляционные счетчики можно для любого
вида излучения, но о обенно широкое при¬
менение они получили при работе с а-части-
цами и в ^-спектроскопии.
Учитывая малую проникающую способ¬
ность а-частиц, для их счета используют
сцинтилляторы с тонким слоем фосфора на
стекле, плексигласе или органической плен¬
ке. Мягкое P-излучение, например С14, в ор¬
ганическом соединении можно определять
путем непосредственного введения препара¬
та в жидкие сцинтилляторы. При помощи
пластмассовых фосфоров в сцинтилляцион¬
ных датчиках подсчитываются тепловые и бы¬
стрые нейтроны. Отсутствие инертности у
сцинтилляционных датчиков и короткие сиг¬
налы позволяют регистрировать высокоактивные препараты. Тонкие слои
сцинтиллятора позволяют раздельно учитывать а-частицы на фоне |5- и у-
излучения. Поскольку интенсивность световой вспышки в сцинтилляторе
пропорциональна энергии ионизирующей частицы, можно путем введения
дополнительного прибора (амплитудного дискриминатора) регистрировать
не только частоту импульсов во времени, но определять их энергию. На этом
принципе основан метод у-спектроскопии.
Совершенным прибором для снятия спектра амплитуд импульсов, стати¬
стически распределенных во времени, являются амплитудные анализаторы
АИ-128, АИ-256. В последнее время находит все более широкое применение
метод у-спектроскопии на основе полупроводниковых детекторов (ППД)
сцинтилляционно-
Рис. 4. Схема
го датчика
1 — р-частица или v-квант; 2 — кри¬
сталл (фосфор); з — фотокатод; 4 —
экран; 5 — фокусирующий электрод;
6 — эмиттеры; 7 — фотоумножитель;
8 — анод; 9 — сопротивление нагруз¬
ки; 10 — вывод к регистрирующему
устройству
462
А. В. Соколов, if. П. Сердоболъский, М. Г. Синягина
ядерных излучений. Полупроводниковые у-спектрометры имеют повышенное
(на порядок) энергетическое разрешение. Совершенным отечественным спек¬
трометром рентгеновского и 7-излучения г германиевым полупроводниковым
детектором дзлучений является СЭГ-П2 («Лангур») в комплекте с анализа¬
тором АИ 1024-4. Амплитудные анализаторы применяются при исследовании
спектров ядерных излучений, при анализе горных пород и радиоактивных
РУД*
Счетчики ионизирующего излучения при работе должны быть защищены
от влияния посторонних излучений. Имеются разные типы свинцовых защит.
Газоразрядные счетчики (торцовые и цилиндрические) помещают в универ¬
сальный домик ДС с толщиной свинцовой защиты 40 мм. В малофоновой уста¬
новке УМФ-1500 толщина защиты 50 мм.
РАДИОМЕТРИЧЕСКИЕ ПРИБОРЫ1
Существует разнообразная аппаратура для количественного определения
интенсивности радиоактивного излучения и его спектрального анализа. Для
большинства агрохимических исследований}пригодны простые радиометри¬
ческие установки, включающие несколько блоков (рис. 5).
Рис. 5. Блок-схема радиометров для
измерения р и -у-излучения
1 — БГС; 2 — входное устройство; 3 —
пересчет ный блок; 4 — блок питания
/( сета
Импульсы*тока, возникающие в газоразрядном счетчике, усиливаются
блоком БГС и передаются на входное устройство пересчетного прибора.
В случае применения сцинтийляционного счетчика усиление* происходит
в фотоумножителе. Входное устройство преобразует импульсы в стандарт¬
ные по амплитуде и длительности и передает их на пересчетный блок. Отсчет
импульсов за определенный период времени производится по декатронам.
Высоковольтный выпрямитель блока питания может быть смонтирован
отдельно от пересчетной установки (ПП-8) или объединяется с нею (уста¬
новка ПП-16).
Применявшиеся ранее радиометрические установки типа Б, Б-2, ДА,
ПС-10 ООО конструктивно устарели и сняты с производства. В настоящее
время распространены более совершенные, надежные и удобные в работе
установки Б-3, Б-4, ПП-8 («Волна»), УМФ-1500.
Установка Б-3. Малогабаритный прибор, предназначенный для работы
с низковольтными газоразрядными счетчиками. В комплект прибора входят:
входной блок БГС-3, пересчетный прибор ПС-20 со стабилизатором напряжения
(390 ± 20 в) и комплект счетчиков. Емкость счета прибора 10е импульсов.
Система счета — десятичная, с отсчетом показаний по декатронам. Возможно
использование в установке прибора ПС-100 (рис. 6).
Установка Б-4. Малогабаритный прибор, состоящий из пересчетного
устройства ПП-16 и блока детекторов БГС-4. Предназначен для работы с
низковольтными газоразрядными счетчиками типа СТС-6, СБТ-13, СИ-22Г
и др. При наличии отдельного источника высокого напряжения (ВСВ-2,
ВСЭ-2500 или др.) возможна работа с высоковольтными счетчиками МСТ-17,
Т-25 БФЛ и др. Емкость счета прибора 10е с.прямым отсчетом показаний.
1 В составлении раздела приняли участие Н. И. Борисова, В. Н. Родионов.
Методика применения меченых атсмов
463
Рис. 6. Радиометр Б-3
Включение и остановка счета производятся вручную. Предусмотрена воз¬
можность последовательной и параллельной работы двух приборов (рис. 7).
Установка ПП-8 («Волна»). Предназначена для работы с газоразрядными
и сцинтилляционными счетчиками. В комплект установки входят высоко¬
вольтный выпрямитель ВСВ-2 (400—2000 в), пересчетный прибор ПСТ-100,
блок УГС для работы с цилиндрическими или торцовыми газоразрядными
счетчиками, блок УСС для сцинтилляционного счетчика с использованием
ФЭУ-19 и блок катодный повторитель (рис. 8).
Отсчет показаний производят по неоновым лампочкам и декатронам
ПСТ-100. Схема обеспечивает автоматическую остановку счета и электрон¬
ного секундомера по истечении 10, 100 и 1000 сек. или после того, как будет
набрано 103, 104, 106 или 10е импульсов. Средняя скорость счета показы¬
вается интенсиметром. Предусмотрена поочередная работа двух приборов
с автоматической остановкой как по набору импульсов, так и по заданному
времени. Отсчет времени производится электронным секундомером. Работать
на установке ПП-8 удобно, практика показала его надежность и стабиль¬
ность.
В настоящее время подготовлен выпуск аналогичного прибора РПС-2-ОЗТ,
включающего в комплекте два блока датчиков, пересчетную установку, элек¬
тронный секундомер и высоковольтный выпрямитель.
Установка с малым фоном УМФ-1500 М. Предназначена для измерения
малых активностей P-излучения газоразрядными счетчиками типа СБТ-13,
СИ-16 БГ, СИ-17 БГ, СТС-5 и СТС-6. Установка имеет защитный контейнер,
в котором ?находится детектор излучения, и пересчетный прибор ПП-16.
Мощная экранировка и электронная схема защиты снижают фон установки
до 3—9 имп/мин в зависимости от типа детектора (рис. 9).
Рис. 7. Радиометр Б-4
Рве. 8.^Радиометр ПП-8
Методики применения меченых атомов
465
Перспективными в агрохимических исследованиях могут стать новые
типы приборов, снабженные автоматическими и печатающими устройствами:
Декадный пересчетный прибор ПП9—2М. Предназначен для счета им¬
пульсов и снабжен электронным секундомером. Прибор состоит из отдель¬
ных блоков: усилителя-дискриминатора, пяти пересчетных декад, блока
автоматики, блока питания. Емкость пересчетного устройства 10е импуль¬
сов. Индикация счета десятичная, однострочная — в окошках прибора сме¬
няются светящиеся цифры электролюминесцентных индикаторов.
Прибор автоматически прекращает счет после набора заданного числа
импульсов или по истечении установленного срока. В первом случае фикси¬
руется прошедшее с начала счета время, во втором — число импульсов.
Предусмотрена возможность подключения к прибору цифропечатающего
устройства БЦ-1.
Установка 2154-1-1М «Протока». Предназначена для измерения а- и
P-излучений радиоактивного пятна диаметром менее 20 мм при толщине под¬
ложки не более 2 мм. Установка обладает высокой эффективностью: учиты¬
вается до 95% радиоактивности образца (рис. 10).
В комплект входят 4л-счетчик (через который пропускается метан, этан
или другой природный газ) и пересчетный прибор ПП-9-2М. Возможно под¬
ключение цифропечатающего устройства БЗ-15.
Рис. 9» Установка с малым фоном УМФ-1500М для измерения р-активности
Рис. 10. Проточный 4л-счетчик с комплектом электронной аппаратуры «Протока»
466
А. В. Соколов, И. П. Сердобольский, М. Г. Синягина
Рис. 11» Универсальный радиометр «Тисс»
Сцинтилляционная хроматографическая установка УСХ-1 может ис¬
пользоваться при биохимических исследованиях, проводимых с мечеными
соединениями Н8, С14, S86, Р32 и др. Измерение активности производится
в хроматографических пятнах на тонкослойных или бумажных хроматограм¬
мах. Для этого в состав сорбента наносимой тонкослойной пластины вводят
один из сцинтилляторов (Вдовенко, 1973) или готовую бумажную хромато¬
грамму пропитывают раствором стильбена в бензоле.
Механизм сканирования, управляемый блоком автоматики, перемещает
хроматограмму, расположенную под фотоэлектронным умножителем. Им¬
пульсы регистрируются логарифмическим интенсиметром марки БИ-3 и
электронным самопишущим потенциометром КСП-4. Предусмотрено также
подключение декадно-пересчетного прибора ПП9-2М с печатающим устрой¬
ством БЗ-15.
Бета-радиометры с жидкостными сцинтилляторами применяются для
измерения мягких p-излучений Н8, С14, S35 и др. Установка такого рода
производства Венгерской Народной Республики включает измеритель
P-излучений Nz-137 с полуавтоматической подачей образцов под сцинтилля-
ционный датчик и одноканальный амплитудный анализатор типа NC-241/B.
Заменив датчик на двухканальный анализатор NK-110, получают возмож¬
ность одновременно определять в образце радиоактивность двух изотопов
(Н3 и С14 или Р82 и С14 и т. д.).
В СССР выпускаются экспериментальные образцы спектрометров с жид¬
костной сцинтилляцией Всесоюзным научно-исследовательским институтом
медицинского приборостроения.
Контроль загрязненности а-, |}- и ^-активными веществами рабочих мест,
рук, одежды и обуви персонала производят с помощью следующих радио¬
метров.
Универсальный радиометр «Тисс» имеет три сменных датчика: сцинтил-
ляционный счетчик с фотоумножителем ФЭУ-19М для учета загрязненности
a-активными веществами малых поверхностей; пропорциональный счетчик
с воздушным наполнением для измерения загрязнения больших поверхно-
Методика применения меченых атомов
467
Рис» 12. Комплект индивидуальных фотопленочных дозиметров ИФКУ
стей (рабочая поверхность счетчика 150 см2) и блока из трех газоразрядных
счетчиков СТС-6 для измерения загрязнения поверхности р-активными
веществами (рис. 11).
Радиометр регистрирует число поступивших импульсов механическим
счетчиком, имеет прямопоказывающий прибор средней скорости поступления
импульсов — до 100 ООО имп/мин, а также световую сигнализацию преду¬
преждения о превышении установленного уровня. Питание прибора от сети
переменного тока.
Носимый универсальный $-у-радиометр «Луч-А» имеет вес около 3 кг
и питание от двух элементов 1,6 ПМЦ-У8 или от сети переменного тока.
Прибор снабжен сменными счетчиками и дифференцирующими экранами,
что позволяет раздельно регистрировать мягкое p-излучение с энергией
0,15 Мэе (S85, Са46), жесткое P-излучение с энергией 1,7 Мэе (Р32, Sr90) и
Y-излучение. Диапазоны измерения от нуля до 20, 100, 500 и 1000 имп/сек
по шкалам прямопоказывающего прибора. Звуковой резонатор позволяет
судить по частоте сигналов о радиоактивности.
Носимый универсальный радиометр РУП-1М имеет шесть сменных дат¬
чиков, при помощи которых можно определить степень загрязненности по¬
верхности а- и ^-активными веществами, мощность дозы Y-излучения и
интенсивность потоков быстрых и тепловых нейтронов.
Сцинтилляционный датчик с фотоумножителем ФЭУ-35 позволяет изме¬
рять поток а-частиц от 0,5 до 2*104 частиц!см2, в 1 мин. При помощи гало¬
генного счетчика СБТ-10 учитывают интенсивность потока Р-части^ от 10
до 5*104 и счетчиков СИ-13Г и СИ-ЗБГ — мощность дозы у-излучения. Для
регистрации нейтронов используют сцинтилляционные детекторы из по¬
рошка плексигласа и сернистого цинка, активированного серебром.
Прибор надежен в эксплуатации, удобен для экспресс-контроля лабора¬
торий.
Комплект индивидуальных фотопленочных дозиметров ИФКУ позволяет
определить суммарную дозу внешнего облучения по почернению рентгенов¬
ской пленки. Комплект используют в изотопных лабораториях для постоян¬
468
А. В. Соколов, И. Я. Сердобольский, М. Г. Синягина
ного индивидуального контроля за дозой внешнего ^-излучения, нейтронов,
рентгеновского и у-излучения, полученной каждым сотрудником, как этого
требуют санитарные правила (ОСП-72).
В комплект входят: набор кассет, оборудование для резки и проявления
пленки и фотометрический прибор ИФКУ (рис. 12).
Каждому сотруднику лаборатории выдаются пластмассовые кассеты
с рентгеновской’"пленкой для постоянного ношения в течение 2—6 недель.
Кассеты имеют четыре окна с разными экранами, что позволяет учесть вид
излучения, а степень почернения определяется дозой радиоактивного облу¬
чения. Эталоном сравнения служат контрольные пленки, получившие из¬
вестную дозу облучения — от 0,2 до^2,0 бэр. Специальный прибор, фотометр
ИФКУ, позволяет провести сравнение с высокой точностью.
Сцинтилляционний дозиметр ДРГЗ предназначен для измерения мощности
дозы рентгеновского и у-из л учений.
Приборы и оборудование, необходимые для работы с радиоактивными
изотопами, поставляет Всесоюзное объединение «Изотоп» через межобласт¬
ные конторы по заявкам научных учреждений, а также отдел снабжения
Министерства сельского хозяйства.
Приборы высылаются вместе с описанием, технической характеристикой
и подробной инструкцией по эксплуатации и устранению возможных непо¬
ладок.
АКТИВНОСТЬ И ЕДИНИЦЫ ЕЕ ИЗМЕРЕНИЯ
Под радиоактивностью или просто активностью какого-либо вещества
следует понимать способность его атомов к самораспаду; активность вещества
проявляется обычно в его излучении. Международной единицей измерения
активности принята единица, получившая название «кюри» (символ кюри),
равная активности газа радона, находящегося в равновесии с 1 г радия.
Число актов распада, соответствующее 1 кюри, равно 3,7-1010 в 1 сек. Таким
образом, эту единицу можно определить как активность, которой соответ¬
ствует 3,7• 1010 актов распада в 1 сек. В настоящее время кюри измеряют
любую активность, сопровождаемую а-, |5- и Р+-из л учением. Производными,
более мелкими единицами активности приняты милли- и микрокюри (сим¬
волы мкюри, мккюри). Отсюда число^распадов, соответствующих этим еди¬
ницам, будет равно:
1 мкюри — 3,7• 107 распадов в 1 сек., или 22* 10е распадов в 1 мин.;
1 мккюри — 3,7‘Ю4 распадов в 1 сек., или 22-106 распадов в 1 мин.
В практической работе активность какого-либо препарата часто выражают
просто в импульсах тока в 1 сек. или чаще в 1 мин. Если бы все излучение
препарата проходило через счетчик полностью, то, очевидно, число импуль¬
сов в 1 мин., отсчитанное на счетчике, отвечало бы указанным выше едини¬
цам кюри, милли- и микрокюри. Но излучаемые препаратом р~-частицы
двигаются во все стороны, и количество их, попадающее в счетчик, составляет
небольшой процент от всего излучения препарата. Поэтому для пересчета
числа импульсов в 1 .мин. в единицы кюри необходимо знать коэффициент
пересчета для каждой счетной установки. Очевидно, что коэффициент пере¬
счета будет меняться при изменении толщины слоя препарата, при смене
счетчика (у разных счетчиков проницаемость стенок различна) и при пере¬
мене положения препарата по отношению к счетчику. Обычно величина
коэффициента пересчета колеблется в пределах от 3 до 30%.
Для экспериментального определения коэффициента пересчета измеряют
в импульсах в 1 мин. активность препарата того же изотопа с известной
активностью в единицах кюри. Процентное отношение данных измерения
Методика применения меченых атомов
469
к известной активности препарата дает коэффициент пересчета. Пусть, на¬
пример, навеска препарата с общей активностью 0,001 мккюри (что состав¬
ляет 22-105-0,001 = 2200 распадов в 1 мин.) дала на счетной установке
в принятых в лаборатории условиях 110 имп/мин. Тогда коэффициент пере¬
счета будет равен: 110:2200*100 = 5%.
При определении коэффициента пересчета для изотопа Sr90 или для дол¬
гоживущего изотопа, имеющего у-излучение, можно использовать фабрич¬
ные эталоны, но в этом случае подсчет активности испытуемого образца не¬
обходимо производить в тех же условиях, что и эталоны, т. е в сухом состо¬
янии и т. д.
Коэффициент пересчета необходимо определять перед каждым новым экс¬
периментом в тех же условиях, в каких будут подсчитываться активности
препаратов. Это необходимо сделать на случай, если испортится счетчик
(что часто ^наблюдается у торцовых счетчиков) и продолжение подсчетов
препаратов опыта будет производиться другим счетчиком. Чтобы увязать
данные, полученные при подсчетах на различных счетчиках, необходимо
результаты привести к одному коэффициенту пересчета. Например: коэф¬
фициент пересчета счетчика № 1 — 7,5%, № 2 — 8,25%. Тогда после вне¬
сения поправки на фон и пересчета всех результатов на один срок измерения
следует пересчитать результаты и на единый коэффициент. Если принять
коэффициент пересчета 8,25, то все полученные результаты на счетчике №1
следует умножить на величину 8,25:7,5 = 1,1.
МЕТОД МЕЧЕНЫХ АТОМОВ
Если к раствору какого-либо нерадиоактивного элемента добавить нич¬
тожно малое количество радиоактивного изотопа того же элемента, то вне¬
сенная метка, распределившись в растворе, будет вести себя так же, как и
нерадиоактивный элемент *. Поэтому по излучению радиоактивной метки
можно проследить поведение интересующего нас элемента и определить его
не только качественно, но и количественно.
При всяких количественных определениях с помощью внесенного радио¬
активного изотопа в качестве метки необходимо определить удельную актив¬
ность исходного раствора или препарата в начале и в конце опыта.
Удельная активность. Под удельной активностью понимают
активность, отнесенную к 1 мл раствора или 1 мг (или 1 г) вещества. Удель¬
ную активность измеряют в тех же единицах, как и общую активность,—
в единицах кюри, импульсах в 1 мин. (отнесенных к единице), например:
мккюри/млу мккюри/мг, имп/мин/мл, имп/мин/мг. При расчете удельной
активности по отношению к веществу последним может быть: вес сухого
растения, вес золы растения, вес удобрения и пр.
Особый интерес представляет расчет удельной активности по отношению
к тому элементу, с изотопом которого проводится работа.
Пример. Пусть раствор КН2Р3104 с небольшой добавкой КН2Р8204, содер¬
жащий в 2 мл 3 мг фосфора, дает 360 им/мин; тогда удельная активность раствора по фос¬
фору будет 360 : 3 = 120 имп/мин/мг (фосфора). Или пусть раствор Na2C1203 с неболь¬
шой добавкой Na2Cl408, имеющий общую активность 0,1 мккюри, содержит 106 мг соли
или 12 мг углерода; удельная активность по углероду будет: 0,1 : 12 = 0,008 мккюри/
мг (углерода). ^
1 Следует~иметь в виду, что радиоактивная метка метит лишь одноименные ионы. Так,
двухвалентный марганец нельзя метить радиоактивнымТраствором КМп04, органиче¬
ские фосфаты (где фосфор находится в комплексном ионе) не'будут метиться раствором
NaHaPM04 или КН2Р3204 и т. д.
470
А. В. Соколов, И. Я. Сердобольскийу М. Г, Синягина
Радиоактивные изотопы какого-нибудь элемента поступают в лаборато¬
рию обычно в ампулах, заполненных раствором соли нерадиоактивного,
стабильного изотопа этого элемента с очень небольшой добавкой соли его
радиоактивного изотопа. Содержание в ампуле радиоактивного изотопа
составляет, таким образом, небольшую долю процента от стабильного, не¬
радиоактивного. Активность раствора ампулы бывает обычно так велика,
что счетчик не успевает регистрировать возникающие в нем импульсы, он
«захлебывается», так как не выходит из мертвого периода. Поэтому для при¬
готовления рабочего раствора берут для метки только небольшую часть рас¬
твора изотопа из ампулы и вносят в приготовленный раствор желаемой кон¬
центрации стабильного изотопа того же элемента.
Из практики можно рекомендовать другой способ приготовления мече¬
ного раствора, а именно: сначала вся ампула переносится в мерную колбу
на 100, 200 или 500 мл в зависимости от активности раствора в ампуле, а
уже из колбы берется пипеткой необходимое количество раствора для метки.
Это предварительное разведение всей ампулы дает возможность более точно
брать активный раствор не по каплям, а пипеткой и только 1 раз иметь дело
с ампулой или пенициллиновым флаконом. Активный раствор из мерной
колбы переносится в толстостенную склянку для хранения. На склянке
указываются: номер контейнера, общая и удельная активность на дату,
указанную в паспорте. Следует иметь в виду, что теперь удельная активность
будет иная, чем указанная в паспорте.
Пример. Ампула изотопа фосфора Р32 по паспорту на 10.X имела: общую ак¬
тивность — 5 мкюри, удельную активность — 2 мкюри, объем раствора в ампуле — 2,5 мл,
содержание фосфора — 7 мг Р в 1 мл.
После разведения всего раствора ампулы до 500 мл общая активность остается
5 мкюри, удельная активность на 10.X будет 5:500 = 0,01 мкюри, содержание фосфора
7*2,5:500 = 0,035 мг/мл Р.
В результате такого большого разбавления отношение радиоактивного
изотопа к стабильному (нерадиоактивному) в рабочем растворе становится
еще меньше. Таким образом, все изучаемые в опыте реакции происходят,
можно сказать, исключительно между стабильными изотопами, радиоактив¬
ный же изотоп служит лишь своеобразной «меткой» раствора соли стабиль¬
ного изотопа.
Существуют разные способы метки. Метку раствора, как было указано,
производят добавкой к раствору какой-либо соли стабильного изотопа не¬
большого количества раствора той же соли радиоактивного изотопа. Для
метки сухих препаратов, например суперфосфата или калийного удобрения
K2S04 и других, необходимо провести осаждение препарата из меченого
раствора этой соли. Для сложных веществ органического происхождения
прибегают к биологической метке. При этом соответствующее растение вы¬
ращивают на почве, удобренной меченым препаратом; после уборки из нега
выделяют требуемое для проведения опыта сложное органическое вещество.
Подсчет импульсов тока на счетной установке.
Вылет ионизирующих частиц при распаде радиоактивного изотопа происхо¬
дит беспорядочно, хаотично, не через равные промежутки времени. Поэтому
при измерении активности какого-нибудь препарата числа импульсов в еди¬
ницу времени, отсчитываемые в разные периоды, не будут равны между собой,
а будут колебаться, согласно закону больших чисел, в некотором пределе.
Если при измерении активности какого-нибудь препарата сделать под¬
счет импульсов за 1-ю, 2-ю, 3-ю, ..., 10-ю минуты, то получим ряд чисел,
неравных между собой, например: 540, 555, 535, 545, 542...; эти числа груп¬
пируются около какой-то средней, в нашем примере 543 имп/мин. Эту сред¬
нюю и принимают за экспериментальное выражение активности препарата»
Методика применения меченых атомов
471
Средняя может быть определена двумя способами: 1) как средняя между
п отсчетов числа импульсов в 1 мин; в указанном примере — среднее из 10
отсчетов: (540 + 555 + 535 + ...):10 = 543; 2) как средняя за одну еди¬
ницу времени из общего числа импульсов за п единиц времени; например,
за 10 мин. было учтено 5430 имп.; следовательно, средняя равна 5430:10 =
= 543. Оба способа определения средней одинаково законны и приводят
к одинаковой величине средней арифметической. Второй способ более прост.
Из сказанного следует, что средняя является приближенной величиной,
и величина эта при повторении определения может несколько отличаться
от величины средней, полученной при первом определении. Поэтому суще¬
ственно установить, с какой точностью было сделано наше определение,
т. е. найти ошибку средней арифметической.
Существует несколько способов определения ошибки: 1) определение
средней квадратичной ошибки, 2) определение стандартного отклонения
и др. Нахождение средней квадратичной ошибки по способу наименьших
квадратов связано с большими вычислениями, а потому неудобно при обра¬
ботке результатов измерений большого числа образцов. В этом случае целе¬
сообразно пользоваться вторым способом, основанным на законе распреде¬
ления Пуассона.
Стандартное отклонение равно корню квадратному из общего числа
набранных импульсов
A = N± ,
где А — активность образца; N — число импульсов.
Если число импульсов сосчитано за время t, то в единицу времени наи¬
более вероятная активность образца будет равна N : t импульсов, а среднее
стандартное отклонение ±YW: U
TJ . 5430 , УбШГ , 7
В нашем примере: л = ю~=Ь fo =54^±7-
Для получения большой точности результата надо, чтобы число N
было достаточно велико. Для образцов с очень малой активностью это до¬
стигается лишь за счет увеличения времени отсчета. Например, если образец
эа 2 мин. дал 100 имп., активность его будет равна:
A_jm±vm_ _ро±8>
Относительная ошибка результата составляет 10%.
При подсчете того же образца за время 50 мин. общее число импульсов
составит 2500. В этом случае:
. 2500 . /2500
А=~50“± 50 ‘ *
Относительная ошибка результата равна 2%, ^
Табл. 4 позволяет определить то количество импульсов, которое следует
набрать при подсчете, чтобы получить результаты с заданной точностью.
Фон и его измерение. При включении прибора даже в отсут¬
ствие излучающего препарата счетная трубка всегда регистрирует неболь¬
шое количество импульсов, получившее название фона. Эти импульсы воз¬
буждаются вследствие различных причин: прохождения через счетную труб¬
ку космических лучей, излучения загрязненных радиоактивными веществами
предметов, самопроизвольных разрядов в счетчике.
При определении активности препарата следует учитывать наличие фона
и всегда вычитать его из числа импульсов, зарегистрированных при подсчете
препарата. Фон подсчитывается в условиях, аналогичных условиям измере-
472
А. В. Соколов, И. Я. Сердобольский, М. Г. Синягина
Таблица 4
Определение количества импульсов при подсчете образца и фона для получения заданной
точности счета *
Относительная ошибка
1%
2%
3%
5%
10%
Необходимее число импульсов при подсчете
Й?
+
Й?
*©•
£
Й?
*
*0*
t
Й?
Й?
•&
£
Й?
£
•&
+
о
о
£
40 000
9500
14 000
2400
3500
18 000
3600
6 500
900
1600
12 000
2000
4 000
470
1000
7 600
1000
2 700
240
710
5 100
450
1 800
115
450
2 600
80
900
20
230
1 800
20
650
5
160
1 500
6
540
—
130
1 300
—
480
—
120
1 200
—
450
—
112
1,3
1,5
1,7
2,0
3.0
5.0
10,0
20,0
50,0
100,0
24 000
11 500
2 000
500
150
34
11
68 000
46 000
23 000
16 000
13 000
И 900
И 200
22 000
12 000
6 000
3 000
500
130
40
9
41 000
26 000
17 000
И 000
5 700
4 000
3 300
3 000
27 000
10 000
5 000
2 700
1 300
200
60
20
4
* К — отношение числа импульсов в 1 мин. образца -f фон к числу импульсов фона; #0б+ф— импульсы
от препарата + фон; — импульсы фона.
Примечание. Ниже черты — измерение фона не имеет значения.
ния активности препарата: та же установка, тот же счетчик импульсов и та
же подложка, помещенная на одном расстоянии от окна счетчика. Например,
если препарат представляет из себя раствор, который будет подсчитываться
в стеклянной чашечке в объеме 2 мл, то при определении фона вместо испы¬
туемого раствора в чашечку следует налить 2 мл воды.
Для каждого счетчика фон постоянен, и колебания его обычно незначи¬
тельны. Для торцовых счетчиков фон составляет около 15—30 имп/мин в зави¬
симости от толщины окна; для цилиндрических счетчиков при разной длине
фон равен 30—40 имп/мин и т. п.
Определение фона производят каждый раз перед началом работ и после
окончания всех измерений. Резкое увеличение фона счетчика показывает
наличие загрязнений внутри свинцовой камеры. В этом случае работу по
измерению следует приостановить и устранить причину повышения фона»
Среднее значение фона и его ошибку определяют также по способу стандарт¬
ного отклонения:
■±'
где <4ф — среднее значение фона обычно за время, равное 1 мин.; ЛГф— общее количество
импульсов, подсчитанное в начале и в конце работы за время tф.
Определение активности образца с поправкой
на фон. Как было отмечено выше, при измерении активности образцов
счетчиком импульсов одновременно пересчетный прибор регистрирует
и импульсы фона. Следовательно, истинное значение активности образца
в импульсах в единицу времени (обычно в 1 мин.) равно среднему значению
Методика применения меченых атомов
473
активности образца плюс фон в 1 мин. минус среднее значение фона (тоже
в импульсах в 1 мин.).
A N06 + Ф
об =
‘’об + ф *ф
где индекс «об + ф» относится к подсчетам активности образца вместе с фоном; индекс
«ф» — к определению фона.
Теперь следует подсчитать ошибку к найденному истинному значению
активности образца (без фона), т. е. к А0б. Очевидно, здесь следует учесть
ошибку, полученную при определении активности образца + фон, и ошибку
при определении фона.
Согласно теории вероятности, средняя квадратичная ошибка (или сред¬
нее стандартное отклонение) равна корню квадратному из суммы квадратов
ошибок, полученных при измерении образца + фон и измерении фона.
Если
г‘об +
У Nog + ф
об + ф
где тоб+ф и Шф — средние стандартные отклонения при подсчете активности образца
с фоном и активности фона,
тогда
т,
об
=±^
об + ф
-f-m2
В табл. 5 приводится форма записи в рабочей тетради и пример расчета
ошибки измерения.
Как указывалось выше, проскок ионизирующей частицы через счетчик
в период его мертвого времени не сопровождается возникновением импульса,
а появление ложных импульсов не вызывается проскоком ионизирующей
частицы через счетчик. Отсюда, как правило, число отсчитываемых на счет-
Таблица 5
Форма записи в рабочем журнале
М образца
Дата определения
(в начале и в
конце опыта)
JSft чашечки
Показания
пересчетного
прибора,
имп.
Время, мин.
^об+ф
*об+ф
^об+ф
Ошибка измерений
f
•&
+
0
1
0
1
-Н
*об+ф
*об+ф *ф
Фон
5.Х 9 ч. 30 мин.
1
210
10
0,9
2
297
15
и
сл
о
II
о
5
3
2027
5
"ф
4
2073
5
4100
410
390
6,4
6,5
7
5.Х 10ч. 15 мин.
5
1436
4
1838
5
3274
364
344
•
6,4
6,5
Расчет ошибки измерения тоб:
I. V*507 , 22,5 . л q т
— ± —— ± uiy» тоб+ф
тА = ±
ф 25
25
= _Vmo_ _5L=+6 4)
ю 10
*об
= ± У>2+<б+ф=± У”0,94 + 6,4* = ± V 41= ±6,5.
474
А. В. Соколов, И. П. Сердобольский, М. Г. Синягина
ной установке импульсов не совсем точно отражает число частиц, прошедших
в счетчик.
К сожалению, в настоящее время нет еще достаточно простых и удобных
способов введения необходимых поправок при учете активности. Необхо¬
димо поэтому при работе на счетчике принять меры, чтобы эти поправки
составляли минимальную, не существенную для работы величину. Возник¬
новение ложных импульсов следует предупреждать соответствующим под¬
бором счетчиков, бракуя те, в которых они возникают. Особый характер
ложных импульсов, описанный выше, позволяет проводить такую браковку.
Поправки на мертвое время счетчика тоже можно избежать, если в работе
применять препараты не очень высокой активности. С небольшой погреш¬
ностью можно принять, что при активности препарата, не превышающей
8—10 имп/сек, эти поправки составят величину, во всяком случае меньшую,
чем величина вероятной ошибки, о которой говорилось выше. Таким образом,
при проведении работы на счетчиках высокого качества, а также при исполь¬
зовании в опытах препаратов с активностью 8—10 имп/сек (до 600 имп/мин)
получаемые данные по подсчету импульсов будут с достаточной точ¬
ностью определять действительное ионизирующее излучение.
МЕТОДИКА ПРОВЕДЕНИЯ ВЕГЕТАЦИОННЫХ ОПЫТОВ С Р8*
Метод меченых атомов позволяет работать с «мечеными удобрениями»
и проследить поступление в растения питательных веществ из удобрений и
их превращения в растении и в почве. Применение радиоактивного изотопа
фосфора дало возможность получить новые данные о фосфорном питании рас¬
тений, проверить правильность многих ранее высказанных положений и
разработать новые представления о фосфорном обмене у растений.
Использование метода радиоактивных индикаторов основано на добав¬
лении к нерадиоактивному изотопу ничтожно малого количества радиоак¬
тивного изотопа этого элемента. В результате этого добавления происходит
изменение изотопного состава элемента, и так как изотопы практически оди¬
наковы по химическим свойствам, то по радиоактивной добавке можно судить
о содержании данного элемента. Добавка радиоактивного изотопа является
радиоактивным индикатором, по которому можно проследить распределение,
передвижение и накопление в растении меченого им элемента.
В радиоактивных препаратах фосфора, с которыми приходится иметь
дело при постановке вегетационных опытов, содержание Р82 ничтожно мало.
Например, когда 1 г Р206 имеет активность в 0,1 мккюри, в нем содержится
только 1,5‘10"7 мг Р32, все остальное количество фосфора этого препарата
является нерадиоактивным фосфором Р31. Таким образом, удельная актив¬
ность ничтожных количеств радиоактивных индикаторов весьма высока, что
позволяет обнаруживать при помощи их количество вещества порядка 10"14 г.
Набивка вегетационных сосудов. При проведении
вегетационных опытов с радиоактивными изотопами необходимо соблюдать
определенные правила предосторожности, чтобы не причинить вреда здо¬
ровью работающих (см. «Основные санитарные правила...», 1973).
,Опыты с радиоактивным изотопом фосфора Р32 при соблюдении элемен¬
тарных правил предосторожности не могут привести к нежелательным в от¬
ношении здоровья явлениям. Радиоактивный изотоп Р3° имеет сравнительно
короткий период полураспада, он нелетуч, применяется в относительно не¬
больших дозах, хорошо абсорбируется почвой, т. с. обладает свойствами,
которые делают его относительно безопасным элементом.
Работа с получаемыми для опытов концентрированными препаратами,
имеющими высокую удельную активность, должна проводиться в специаль¬
ной лаборатории, которая удовлетворяет санитарным правилам работы
Методика применения меченых атомов
475
с радиоактивными веществами. Все операции: вскрытие ампул, разведение
и приготовление рабочих растворов или активных удобрений, проводятся
в вытяжном шкафу на кювете во избежание возможного разлива радиоактив¬
ного раствора. Все работающие должны быть в халатах и резиновых пер¬
чатках.
Препараты с высокой активностью следует хранить в специальном хра¬
нилище, в металлическом сейфе. По получении исходных концентрированных
препаратов необходимо возможно скорее перевести их в те рабочие рас¬
творы и препараты удобрений, которые будут применяться в опытах. Радио¬
активное излучение препаратов в окружающую среду зависит от самопогло-
щения радиоактивных веществ, т. е. от поглощения излучений в толще самого
препарата. При разведении препаратов происходит увеличение самопогло-
щения; в результате этого излучение, испускаемое препаратами во внешнюю
среду, сокращается во много раз. Например, получен препарат фосфата
калия с удельной активностью 1 мкюри в 1 мл раствора. Если для постанов¬
ки опытов требуется вносить по 100 мккюри на сосуд, то легко и точно изме¬
ряемым количеством раствора будет 10 мл раствора на сосуд. Следовательно,
полученный препарат надо развести в 100 раз до удельной активности
в 10 мккюри на 1 мл раствора или до 100 мккюри на 10 мл. Самопоглощение
излучения в разбавленном растворе будет большое, так как большинству
Р-частиц надо будет проходить сквозь слой жидкости. Аналогичное увели¬
чение самопоглощения будет происходить и при внесении радиоактивных
удобрений. Внося 100 мккюри на один сосуд, в который помещается не менее
1 кг почвы, мы производим дальнейшее увеличение самопоглощения; удель¬
ная активность вещества равна теперь только 0,1 мккюри на 1 кг почвы.
Излучение поглощается теперь большим количеством почвы, стенками со¬
суда, слоем песка, насыпанным сверху, и уже не может иметь существенного
влияния на развитие растений. Таким образом, первым правилом работы
с радиоактивными изотопами является возможно более быстрое разбавление
их веществом, в котором происходит большое самопоглощение излучения.
Измерение необходимых количеств растворов радиоактивных веществ
должно производиться автоматическими пипетками или бюретками. При
набивке сосудов обычно радиоактивный изотоп фосфора добавляется к рас¬
твору немеченого фосфора, чтобы его «пометить».
При набивке сосудов почву высыпают в большую фарфоровую чашку и
в середину почвы выливают раствор, содержащий меченый фосфат. После
этого почву перемешивают с раствором руками в резиновых перчатках.
При внесении больших количеств радиоактивного изотопа, например в опы¬
тах по установлению токсических для растений доз, раствор предваритель¬
но размешивают с почвой стеклянной или фарфоровой палочкой, а затем
уже руками в перчатках.
Сначала производится набивка сосудов, в которые не вносится радиоак¬
тивный изотоп, затем набивка сосудов с возрастающими дозами Р82. При
такой последовательности необязательно тщательно отмывать чашку, пере¬
ходя к набивке следующего сосуда, а достаточно ее протереть сначала не¬
большим количеством неудобренной почвы и песка, а затем марлей. Необ¬
ходимо следить за тем, чтобы поверхность чашек была гладкая, а не шеро¬
ховатая.
Дозы 15Р32. Существенное значение при проведении вегетационных
опытов имеет установление правильной дозы радиоактивного изотопа, обес¬
печивающей и точность аналитической работы, и отсутствие токсичности
радиоактивного вещества для растений.
Чтобы получить достоверные результаты определения активности урожая
при серийных, массовых анализах, должна быть обеспечена возможность
непосредственного определения активности излучения в навеске сухого
вещества в 0,1—0,2 г. При этом количество импульсов для этой навески, за
476
А. В. Соколов, Я. П. Сердобольский, М. /\ Синягина
вычетом «фона» (т. е. числа импульсов, получающихся от такой же навески
сухого нерадиоактивного вещества), должно быть не менее как в 2—3 раза
больше, чем число импульсов для фона. Наличие в конце опыта небольших
отсчетов, близких по величине к фону, приводит к неточным определениям,
не допускающим количественных вычислений выноса фосфора урожаем.
В Научном институте удобрений инсектофунгицидов и Почвенном ин¬
ституте им. В. В. Докучаева было проведено большое количество вегета¬
ционных опытов по установлению оптимальной дозы Р82. Установлено, что
при скорости распада радиоактивного изотопа фосфора (период полураспада
Р82 — 14,2 дня) возможно проведение краткосрочных вегетационных опытов
с малыми дозами Р32, порядка 5—10 мккюри на сосуд. Но в вегетационных
опытах, в которых растения доводятся до полного созревания, к концу опыта
Р82 распадается на 90—95%.
Было показано, что в почвенных культурах внесение дозы порядка 100—
200 мккюри на сосуд обеспечивает достоверные отсчеты в конце опыта при
отсутствии токсического действия радиоактивности на растения.
Отношение растений к дозам Р32 различно. По выносливости к высоким
дозам Р82 растения располагаются в следующий ряд, начиная с менее вынос¬
ливых: гречиха, зерновые злаки, горох, лен, горчица. Токсичность Р32
в опытах с зерновыми злаками была заметна при дозе в 500 мккюри на 1 кг
почвы, в опыте с гречихой — при дозе в 360 мккюри, в опыте с горохом —
при дозе в 1000 мккюри. Для льна и горчицы не было отмечено вредного дей¬
ствия Р82 и при этой высокой дозе. Следует иметь в виду, что эти данные от¬
носятся к равномерному и тщательному перемешиванию радиоактивного
удобрения с 1-килограммовой навеской почвы. В опытах с местным внесе¬
нием следует брать меньшие дозы Р32. Вообще желательно работать с воз¬
можно более низкими дозами Р32. Нужно учитывать, что токсичность Р82
зависит от типа почвы; чем сильнее фиксирует почва фосфаты, тем более вы¬
сокие дозы легко переносят растения. Так, на черноземе токсичность высо¬
ких доз Р82 больше, чем на подзолистом суглинке. Таким образом, при уста¬
новлении дозировки Р32 для конкретного опыта следует учесть его длитель¬
ность, способ внесения фосфата, особенности растения и особенности почв.
Внесение фосфорных нерадиоактивных удобрений уменьшает токсичность Р82.
Размол урожая. Определение активности полученного в опыте
урожая производится в навеске сухого вещества. Размол вещества произво¬
дится на мельнице типа «Пируэтт».
Весьма важно, чтобы размол проводился в определенной последователь¬
ности. Сначала нужно размалывать материалы без радиоактивных веществ,
а затем — в порядке возрастания их активности. При таком порядке раз¬
мола меньше опасность засорения урожаев радиоактивными веществами.
В противном случае приходится перед размолом каждого нового образца
производить тщательную чистку мельницы и длительный размол нерадиоак¬
тивного вещества. При размоле урожаев первые порции размола отбрасы¬
ваются.
Работающие должны иметь очки-консервы и закрывать нижнюю часть
лица марлевой повязкой или надевать респиратор во избежание попадания
пыли в глаза и носоглотку. Размол и просев радиоактивной массы следует
производить под тягой. Для очистки помещения от пыли радиоактивных
веществ надо протирать стены и мебель или удалять пыль пылесосом. Все
работающие должны быть в халатах или комбинезонах.
Определение активности урожаев. Точность опре¬
деления активности зависит от строгого соблюдения во всех случаях одно¬
родности условий определения. С этой целью необходимо иметь всегда оди¬
наковые навески однородного по своим качествам вещества (солома, зерно
и т. д.) и одинаковую тару. В качестве тары при определении активности
сухого вещества и растворов цилиндрическим счетчиком в горизонтальном
домике, как показала практика, удобны фарфоровые лодочки, употребля¬
Методика применения меченых атомов
477
емые для сжигания органического вещества, или аналогичная тара с глад¬
кими блестящими, легко очищаемыми стенками. При объеме раствора 1—2 мл
и величине навески сухого вещества растений 0,1—0,2 г удобны лодочки
длиной 5—8 см и шириной около 1,0 см. Если измерения проводятся торцо¬
вым счетчйком, то для подсчета активности растительной массы и растворов
хорошо себя зарекомендовали стеклянные чашечки диаметром 25—30 мм,
высотой 10—12 мм. Все чашечки должны иметь плоское дно и одинаковый
диаметр. Такие чашечки легко приготовляет стеклодув из стеклянных
трубок одного диаметра. Навеска сухой массы в стеклянную чашечку берется
0,2 г, раствора — 2 мл.
При работе с небольшими объемами растворов лодочки необходимо мыть
эфиром, чтобы обеспечить равномерное растекание жидкости по их дну.
Очистка чашечек и лодочек от радиоактивного вещества производится про¬
мывкой раствором 10%-ной соляной кислоты, иногда их приходится проки¬
пятить с кислотой. После обработки соляной кислотой лодочки промывают
под струей воды. Этими приемами достигается очистка посуды от загрязнений
радиоактивным фосфором. Перед взятием пробы тару проверяют на счетчике
на остаточную активность. При низкой активности урожаев необходимо
иметь совершенно чистую тару. Если производятся определения активности
образцов, дающих около 1000 имп/мин, остаточная активность лодочек
порядка 3—5 имп/мин не имеет существенного значения.
При определениях небольших активностей следует обращать особое вни¬
мание на правильное установление фона. Фон должен устанавливаться
до начала определений и после их окончания. Для точного определения фона
помещают под счетную трубку лодочку с навеской нерадиоактивного ве¬
щества: зерна, соломы, раствора, в зависимости от того, активность какого
вещества далее будет определяться.
Расположение лодочки или чашечки под счетчиком должно быть точно
зафиксировано как в вертикальном, так и в горизонтальном направлении.
Расстояние лодочки от счетной трубки при массовых определениях актив¬
ности сухого вещества не должно быть очень малым, так как в этом случае
малейшее изменение расстояния от счетчика, вследствие неравномерности
поверхности вещества или его уплотнения, скажется на точности определе¬
ний. Практически удобно расстояние порядка 2—4 см. Чашечка фиксируется
на пластмассовой подставке, прилагаемой к вертикальному свинцовому до¬
мику для счетчиков.
Поправка на самопоглощение. При вегетационных опы¬
тах определяют активность урожаев в навеске сухого вещества 0,1—0,2 г.
Такое количество вещества легко разместить равномерно на дне лодочки
или чашечки, площадь которых примерно одинакова. Самопоглощение
излучений веществом будет небольшим вследствие тонкого слоя вещества.
Самопоглощение зависит от количества вещества на 1 см2 поверхности,
в данном случае оно составит около 12—25 мг/см2, что, по имеющимся данным,
почти не влияет на количество отсчетов.
Активность исходных удобрений можно определять в растворах. Мини¬
мальное количество их, которое может быть равномерно распределено по
дну тары, около 1—2 мл. При точных определениях следует измерение объе¬
мов заменить взвешиванием лодочки с раствором на аналитических весах.
При объеме раствора 1 или 2 мл его самопоглощение достигает уже замет¬
ных величин, так как на 1 см2 приходится уже 125—250 мг. Поэтому необ¬
ходимо установить коэффициент пересчета с 1 или 2 мл раствора на 0,1 или
0,2 г сухой массы для внесения поправки на самопоглощение. Этот коэффи¬
циент приходится устанавливать эмпирически, производя разведение не¬
больших количеств раствора или выпаривание 1 или 2 мл раствора. Более
точно этот коэффициент устанавливают при сопоставлении активности на¬
вески 0,2 г сухого вещества и активности раствора зольных веществ этой на-
478
Аь В. Соколов, И. Я. Сердобольский, М. Г. Синягина
Таблица 6
Изменение содержания иР32 в зависимости от времени *
t
к
t
к
X
К
t
к
t
К
1
1,05
25
3,35
49
10,69
73
34,12
97
108,9
2
1,11
26
3,52
50
11,22
74
35,81
98
114,3
3
1,16
27
3,69
51
11,78
75
37,58
99
119,9
4
1,21
28
3,87
52
12,36
76
39,45
100
125,9
5
1,27
29
4,06
53
12,97
77
41,40
101
132,1
6
1,34
30
4,27
54
13,61
78
43,45
102
138,7
7
1,40
31
4,48
55
14,29
79
45,60
103
145,5
8
1,47
32
4,70
56
15,00
80
47,86
104
152,8
9
1,54
33
4,93
57
15,74
81
50,23
105
160,3
10
1,62
34
5,18
58
16 ,52
82
52,72
106
168,3
11
1,70
35
5,43
59
17,34
83
55,34
107
176,6
12
1,79
36
5,70
60
18,20
84
58,08
108
185,4
13
1,88
37
5,98
61
19,10
85
60,95
109
194,5
14
1,97
38
6,28
62
20,04
86
63,97
110
204,2
15
2,07
39
6,59
63
21,04
87
67,14
111
214,3
16
2,17
40
6,92
64
22,08
88
70,47
112
224,9
17
2,28
41
7,26
65
23,17
89
73,96
113
236,0
18
2,39
42
7,62
66
24,38
90
77,62
114
247,7
19
2,51
43
8,00
67
25,53
91
81,47
115
260,0
20
2,63
44
8,40
68
26,79
92
85,51
116
272,9
21
2,77
45
8,81
69
28,12
93
, 89,74
117
286,4
22
2,90
46
9,25
70
29,51
94
94,19
118
300,6
23
3,04
47
9,70
71
30,97
95
98,86
119
315,5
24
3,19
48
10,19
72
32,51
96
103,8
120
331,1
* * — время от начала опыта, дня; К — коэффициент, на который надо умножить найденное количе¬
ство импульсов для установления их количества в начале опыта.
вески объемом 2 мл. Повторив определение несколько раз, получают коэф¬
фициент, позволяющий переводить количество импульсов, найденное в уро¬
жае, на количество импульсов в растворе исходного удобрения.
Выражение результатов опытов в миллиграм¬
мах меченых фосфатов. Число распадающихся атомов прямо
пропорционально числу атомов радиоактивного вещества, т. е. в определен¬
ное время всегда распадается определенная часть всех атомов. Например,
для радиоактивного фосфора Р32 всегда за 14,2 дня распадается половина всех
атомов этого изотопа, за 28,4 дня распадается 3/4 всех атомов и т. д. Скорость
распада атомов зависит только от особенностей строения атомов радиоактив¬
ного изотопа и не зависит от химических реакций и внешних условий (тем¬
пературы, давления и т. д.), в которых находится вещество. Поэтому, зная
число распадов в навеске вещества для одной календарной даты, можно точно
сказать, сколько распадов атомов будет происходить в этой навеске в любой
другой день. В табл. 6 приведены коэффициенты, которые позволяют вычис¬
лить количество импульсов радиоактивного фосфора на любой день опыта.
В табл. 7 приведены коэффициенты для радиоактивного кальция.
Чтобы избежать ослабления активности урожая, определения надо про¬
изводить вскоре после снятия урожая.
Результаты исследований в ряде случаев можно выражать числом импуль-
Методика применения меченых атомов
479
Таблица 7
Изменение содержания Са4Б в зависимости от времени *
t
К
t
к
t
к
t
К
2
1,01
44
1,20
86
1,43
126
1,70
4
1,02
46
1,21
88
1,45
128
1,71
6
1,03
48
1,22
90
1,46
130
1,73
8
1,04
50
1,23
92
1,47
132
1,74
10
1,05
52
1,24
94
1,49
134
1,75
12
1,06
54
1,25
96
1,50
136
1,77
14
1,06
56
1,26
98
1,52
138
1,79
16
1,07
58
1,27
100
1,53
140
1,80
18
1,08
60
1,29
102
1,54
142
1,82
20
1,09
62
1,30
104
1,55
144
1,84
22
1,10
64
1,31
106
1,56
146
1,85
24
1,11
66
1,32
108
1,58
148
1,86
26
1,12
‘68
1,33
110
1,59
150
1,88
28
1,13
70
1,34
112
1,60
152
1,90
30
1,14
72
1,35
114
1,61
154
1,92
32
1,15
74
1,36
116
1,63
156
1,93
34
1,15
76
1,37
118
1,64
158
1,95
36
1,16
78
1,39
120
1,65
160
1,97
38
1,17
80
1,40
122
1,67
162
1,99
40
1,18
82
1,41
124
1,69
164
2,00
42
1,19
84
1,42
* t — время от начала опыта, дни; К — коэффициент, на который над умножить найденное количество
импульсов для установления их количества в начале опыта.
сов в 1 мин. на 1 г вещества. Но в большинстве вегетационных опытов не¬
обходимо определить вынос меченого фосфора в миллиграммах Р2Об в уро¬
жае или в 1 г сухого вещества. Для такого расчета необходимо знать удель¬
ную активность внесенного меченого удобрения, т. е. какое количество им¬
пульсов при данных условиях определения соответствует 1 мг Р2Об меченого
фосфора. Это может быть сделано двумя способами.
При первом способе после соответствующего разведения исходного рас¬
твора определяют количество импульсов, приходящееся на 2 мл раствора.
Далее вычисляют, сколько импульсов приходится на 1 мг Р205 в исходном
растворе. Так как определение активности урожаев производится в сухом
веществе, то вводится поправочный коэффициент на более сильное самопо-
глощение излучений при работе с 2 мл раствора, чем при 0,2 г сухого ве¬
щества. В зависимости от условий определения этот коэффициент может
быть различным. О способах его определения сказано ранее. Неудобство
этого способа — введение коэффициента пересчета с раствора на сухое ве¬
щество урожая, которое может быть взято в разных навесках и,может зани¬
мать различный объем, т. е. иметь различное самопоглощение излучения.
Можно использовать и другой прием определения удельной активности
удобрения. Одновременно с взятием навесок опытных растений берут не¬
сколько навесок^ «чистой» массы растения, выращенного на той же почве
без внесения метки Р32, и смачивают в чашечках небольшим, точно измерен¬
ным объемом раствора «свидетеля», т. е. меченого раствора, который вно¬
сился в почву при набивке опыта. Смоченная навеска высушивается в термо¬
480
А, В. Соколов, И. П. Сердобольскийу М. Г. Синягина
стате, масса тщательно перемешивается и уплотняется, как все остальные
навески опытных растений.
Определив радиоактивность навески и зная объем раствора, отмеренный
в чашечку, и объем раствора, внесенного в сосуд, можно вычислить общее
число импульсов, характеризующее радиоактивность фосфатов, поступивших
на сосуд, и рассчитать удельную активность вносимого раствора в импульсах
на 1 мг носителя Р205 (при одинаковых условиях определения).
Второй способ представляется несколько более сложным, но он позволяет
не пользоваться пересчетными коэффициентами. Допустим, что ставится
вегетационный опыт в почвенных культурах, при котором вносится меченый
фосфат. В урожай поступает в этом случае как меченый фосфор из удобрений,
так и немеченый фосфор. Если поставить контрольный опыт в песчаных
культурах, в которых в одних сосудах внесен только меченый фосфат, а в дру¬
гих совсем не внесено фосфора, то можно определить удельную активность
меченого фосфата. По выносу фосфора растениями, выросшими на песке
без внесения фосфора, судят о поступлении фосфора из семян. На эту вели¬
чину следует уменьшить все данные о содержании фосфора в растениях, что¬
бы определить поступление из внешней среды. Разделив суммарное число
импульсов в урожае (по определению в навесках сухой массы) на вынос
фосфора удобрений, внесенных в песчаной культуре, вычисляют удельную
активность 1 мг Р20б, т. е. количество импульсов на 1 мг поступившего в рас¬
тения меченого фосфора. Разделив количество импульсов, приходящееся
на урожай, на удельную активность, устанавливают поступление меченого
фосфора в миллиграммах Р2Об. Этот способ позволяет избежать всех пересчет-
ных коэффициентов, кроме, конечно, пересчета на время опыта по табл. 5.
Для сокращения пересчетов удобнее определять удельную активность
внесенного раствора фосфата после уборки урожая, одновременно с опреде¬
лением радиоактивности растений. Все данные об активности растений и
раствора удобрений вычисляются на один день в конце опыта.
Метод избирательного поглощения фосфатов
В целях изучения доступности для растений различных форм фосфатов
и сравнительной оценки усвояемости фосфора в различных удобрениях
предложено проведение опытов по методу избирательного поглощения фос¬
фатов.
При постановке опытов по этому методу обе сравниваемые формы фосфа¬
тов — стандартная и изучаемая — вносятся в равных количествах в один
и тот же сосуд под одни и те же растения. Одна из вносимых форм (обычно
стандартная) вносится меченой, что позволяет установить поступление
в растения фосфора из каждой формы в отдельности.
При обычной постановке опытов, когда сравниваемые удобрения вносятся
в разные сосуды, результаты опыта определяются не только поступлением
в растения фосфора из различных удобрений, но и их действием на свойства
почвы. Метод избирательного поглощения фосфатов позволяет установить
сравнительную усвояемость фосфатов в идентичных условиях их исполь¬
зования.
Опыты с избирательным поглощением фосфатов в почвенных культурах
желательно проводить в двух вариантах, в которых меченой формой оказы¬
вается то одно, то другое удобрение. Например, устанавливая сравнительное
поступление Р2Об из порошковидного и гранулированного суперфосфата,
приготовляют два меченых суперфосфата — гранулированный и порошко¬
видный. В первом варианте вносят половину дозы Р205 в виде порошковид¬
ного немеченого фосфата, а другую — в виде гранулированного меченого
фосфата. Во втором варианте вносят, наоборот, гранулированный супер¬
фосфат в виде немеченого фосфата, а порошковидный в виде меченого. Ана¬
Методика применения меченых атомов
481
логично можно поставить опыт по сравнению суперфосфата и преципитата
или томасшлака и аммофоса.
При изучении нескольких новых форм фосфатных удобрений они срав¬
ниваются при внесении в половинной дозе во все сосуды меченого стандарта.
МЕТОДИКА ОПРЕДЕЛЕНИЯ ЗАПАСОВ В ПОЧВЕ
РАСТВОРИМЫХ И УСВОЯЕМЫХ ФОСФАТОВ ПРИ ПОМОЩИ Р32
Определение запаса в почве растворимых фос¬
фатов. При выделении из почвы подвижных фосфатов при помощи раз¬
личных растворителей (водные, кислотные и другие вытяжки) во время
приготовления вытяжки происходит не только растворение почвенных фос¬
фатов, но и их вторичное осаждение. Содержание фосфатов в вытяжке явля¬
ется результатом двух одновременно идущих процессов растворения и осаж¬
дения фосфатов, т. е. результатом обмена между твердой и жидкой фазами.
Добавка к растворителю ничтожного количества меченого фосфата (Р32),
которое не повлияет на реакции осаждения и растворения фосфатов почвы,
позволяет определить количество фосфатов, участвующих в обмене фосфатов
между жидкой и твердой фазами.
Соотношения количеств изотопов Р32 и Р31, содержащихся в твердой (т)
и жидкой (ж) фазах и участвующих в обменных реакциях, должны быть рав¬
ны, т. е.
р32 рЗ1 р31 р32
— - — ИЛИ т + Ж_ т + Ж
р32 р31 р31 р32
Ж Ж Ж Ж
Поэтому, зная исходную радиоактивность растворителя и определив ра¬
диоактивность вытяжки и содержание в ней Р205, можно вычислить, сколько
всего было в почве фосфатов, способных растворяться в данном реактиве.
Эта величина в отличие от количества фосфора, найденного в вытяжке,
характеризует запас в почве растворимых в данном растворителе фосфатов;
этот запас выражают в миллиграммах Р205 на 100 г почвы.
Общий запас в почве растворимых фосфатов может весьма существенно
отличаться от количества фосфатов, найденных в вытяжке. Например, опре¬
деление фосфатов по методу А. Т. Кирсанова в 0,2 н. НС1 вытяжке в дерново-
подзолистых и черноземных почвах показывает около половины общего ко¬
личества их (Рт+ж)-
Распределение запаса растворимых фосфатов между вытяжкой и почвой
зависит от способности почв фиксировать фосфаты; подзолистые и особенно
красноземные почвы отдают в вытяжку меньшую часть запаса растворимых
фосфатов, чем черноземные; зафосфаченные почвы — большую часть, чем
незафосфаченные. Фосфатный режим почвы поэтому может быть охаракте¬
ризован при помощи общего запаса растворимых фосфатов и коэффициента
фиксации фосфатов:
кф=-Ь-.т,
гт+ж
который равен процентному отношению фосфатов, задержанных в твердой
фазе во время производства вытяжки, к общему количеству их. Таким обра¬
зом, почва характеризуется не только содержанием в ней растворимых фос¬
фатов, но и способностью их фиксировать.
Определение запаса растворимых фосфатов разными вытяжками в одной
и той же почве дает различные значения. По данным Т. Д. Корицкой и
А. А. Малеиной («Фосфорные удобрения...», 1963), определение запасов
легкорастворимых фосфатов на различных почвах методами Труога, Чири-
16 Агрохимические методы исследования почв
482
Аь В. Соколов, И. П. Сердоболъский, М. А Синягина
нова, Эгнера — Рима и Кирсанова дало хорошее совпадение результатов:
содержание запасов фосфатов изменялось параллельно по всем вытяжкам,
несмотря на их различное абсолютное количество. При сравнении данных
по запасу растворимых фосфатов с запасом усвояемых фосфатов (вегетаци¬
онный опыт с Р82) также отмечается несовпадение их абсолютных количеств,
во всех случаях запас усвояемых фосфатов значительно меньше. Несмотря
на это, для выщелоченных черноземов и северных малогумусных, мощных
черноземов установлена высокая корреляция между данными по запасу
усвояемых фосфатов (вегетационные опыты) и запасом растворимых фосфатов
(методы Труога, Чирикова, Кирсанова).
Поэтому для характеристики тех или других почв на содержание раство¬
римых фосфатов с меткой Р82 следует применять метод (растворитель), ре¬
зультаты которого проверены вегетационным опытом.
Определение в почве запаса растворимых фосфатов производят следу¬
ющим образом. В раствор, взятый для приготовления вытяжки из почвы,
добавляют меченый фосфат. В полученном меченом растворе определяют
содержание в 1 мл фосфора в миллиграммах Р2Об и удельную активность
в милликюри на 1 мл. Необходимо внести такое количество Р32, чтобы в даль¬
нейшем во время работы получать достоверные отсчеты активности раство¬
ров и чтобы добавленное к вытяжке количество фосфора в виде меченого рас¬
твора не повлияло на точность определений Р2Об химическим методом. Чтобы
установить, какое количество меченого фосфора можно добавить к раствору,
взятому для приготовления вытяжки, надо знать примерно ожидаемое коли¬
чество фосфора в вытяжке из почвы. Желательно, чтобы добавляемое коли¬
чество меченого фосфата увеличило концентрацию Р2Об в вытяжке не более
как на 5%.
Например, надо установить в сероземе общий запас фосфатов, растворимых в
углеаммонийной вытяжке [(NH4)2C08] по методу Б. П. Мачигина. Предположим, что со¬
держание в сероземе фосфатов, определяемых по этому методу, составляет около 2 мг
Р205 на 100 г почвы. При приготовлении вытяжек на 5 г почвы берут 100 мл раствора уг¬
лекислого аммония. При содержании в почве 2 мг Р206 на 100 г почвы в 100 мл вытяжки
(соответствующих 5 г почвы) будет содержаться 0,1 мг Р20Б; следовательно, на 100 мл
раствора углекислого аммония можно добавить в виде меченого препарата не более
0,005 мг РгОб (5% от 0,1 мг). Предположим, что в 1 л раствора меченого препарата фосфо¬
ра содержалось 20 мг Р20б и общая активность его на день приготовления вытяжки была
равна 2,0 мкюри. Следовательно, на 100 мл раствора углекислого аммония следует до¬
бавить 0,25 мл меченого препарата, в которых содержится 0,005 мг Р206 с актив¬
ностью 0,5 мккюри. При радиоактивности раствора в 0,5 мккюри на 100 дел достаточно
точные отсчеты активности можно получать, используя 2—5 мл вытяжки. Одновремен¬
но определяют активность 2—5 мл исходного меченого раствора углекислого аммония.
Определив колориметрически содержание фосфора в вытяжке, мы полу¬
чаем количество фосфатов, оставшихся в растворе; это количество, выражен¬
ное в миллиграммах Р205 на 100 г почвы, рассматривается обычно как содер¬
жание в почве растворимых фосфатов. Эта величина, однако, составляет
только часть общего запаса в почве растворимых фосфатов, который состоит
из фосфатов, оставшихся в растворе и вторично осажденных.
Вычисление общего запаса растворимых фосфатов в миллиграммах Р206
на 100 г почвы производят по формуле
р32
р р И£Х
4 запас = * ж•
Р32
ж
где Рж — содержание фосфора в вытяжке в миллиграммах Р206 на 100 г почвы; F^x —
активность исходного меченого раствора; Р*® — активность вытяжки из почвы в одина¬
ковых единицах измерения.
Методика применения меченых атомов
483
В отсутствие препарата Р32 достаточно большой активности приходится
либо повышать количество добавляемого меченого фосфора до 10% от воз¬
можного содержания фосфора в вытяжке, либо прибегать к длительным
отсчетам радиоактивности растворов, но и то и другое снижает точность
работы; поэтому следует пользоваться возможно более активными препара¬
тами меченого фосфора.
Определение запаса в почве усвояемых фос¬
фатов путем вегетационных и полевых опытов.
Для определения общего запаса в почве усвояемых фосфатов проводят обыч¬
ный вегетационный опыт с внесением необходимого количества всех удобре¬
ний (фон), кроме фосфорных. Внесение фона желательно производить в виде
NH4N03 и KN03 как солей, практически не влияющих на реакцию почвы
и не содержащих добавочных компонентов. Дозы удобрений должны обес¬
печивать нормальное развитие растений. При постановке опыта с зерновыми
культурами (пшеница, овес, ячмень и т. д.) в сосудах, вмещающих 3—6 кг
почвы, достаточно внести по 0,5 г N и 0,5 г К20 на сосуд; при постановке же
опыта в сосудах, вмещающих 1 кг почвы,— по 0,25 г N и К20 на сосуд.
При постановке опыта с зерновыми культурами внесение радиоактивного
изотопа Р32 производят в виде меченого препарата с активностью около
100 мккюри и с содержанием фосфора не более 1 мг Р2Об на сосуд. Радиоактив¬
ность препарата можно менять в зависимости от условий проведения опыта,
но она всегда должна быть в несколько раз меньше токсической дозы для
опытного растения. Содержание меченого фосфора в препарате должно быть
настолько мало, чтобы оно не могло заметным образом отразиться ни на фос¬
форном питании растений, ни на содержании в почве усвояемых фосфатов;
меченый фосфор в этих опытах вносится только как «радиоактивная метка».
Внесение препарата меченого фосфата должно обеспечивать его равно¬
мерное распределение в почве и взаимодействие его со всей массой почвы.
С этой целью меченый фосфат вносят в навеску почвы с тем количеством воды,
которое обычно вносится при набивке сосудов. Можно вносить меченый
фосфат вместе с фоновыми удобрениями (NK), если они не содержат веществ,
могущих повлиять на растворимость фосфата. После внесения меченого рас¬
твора почву тщательно перемешивают руками в резиновых перчатках и пере¬
носят в пустой вегетационный сосуд. Сосуд закрывают для уменьшения испа¬
рения и оставляют на 3 дня, после этого почву высыпают, снова тщательно
перемешивают и помещают в тарированный песком и стеклом сосуд, в ко¬
тором проводится опыт.
По окончании опыта производят, как обычно, учет надземной массы,
а также тщательно отделенных и отмытых от почвы корней растений. Урожаи
надземной массы и корней взвешивают и анализируют на содержание в них
фосфора. Кроме того, в навеске сухого вещества растений определяют, по
общим правилам, радиоактивность.
На основе полученных результатов анализа вычисляют общее содержание
в растениях фосфора и общую активность урожая. Вычитая из общего со¬
держания в растении фосфора количество фосфора, бывшее в семенах, полу¬
чают поступление в растение фосфора из почвы. Сравнивая общую актив¬
ность всего урожая с общей активностью всего внесенного в сосуд количества
меченого фосфора, устанавливают процентное использование Р32, или коэф¬
фициент использования К.
Вычисление общего зайаса усвояемых фосфатов в почве производят по
формуле:
Р р.100
запас = —^ ,
где Р — поступление фосфора в растения из почвы в миллиграммах Р205, пересчитанное
ка 100 г почвы; К — коэффициент использования Р32 в процентах_от внесенного.
16*
484
А. В. Соколов, И. П. Сердобольский, Af. /\ Синягина
Эта формула основана на предположении, что растение одинаково исполь¬
зует из почвы Р32 и Р31.
При получении достаточно высокого урожая надземной массы в опытах
с растениями, у которых корневая система составляет около 10% веса уро¬
жая, можно ограничиться учетом и анализом только одной надземной массы
растений.
При небольших урожаях и малом содержании в растениях фосфора на точ¬
ности опытов сказывается неправильный учет количества фосфора, поступив¬
шего в растения из семян. В этом случае правильнее вычитать не все количе¬
ство фосфора, бывшего в семенах растений, а только некоторую его часть
(50—80%), в зависимости от имеющихся данных об использовании фосфора
семян в опытах, поставленных в чистых условиях песчаных культур. Этот
источник ошибки не имеет значения для растений, которые содержат мало
фосфора во взятом для посева количестве семян.
Определение запаса усвояемых фосфатов может быть произведено и в
полевом опыте с внесением меченого удобрения или более точно — при вне¬
сении радиоактивной метки. Предполагая, что усвояемые фосфаты почвы
используются так же, как фосфаты меченого удобрения, имеем
р Р31
запас растение
^удобрение Р32
* растение
или
Р31
_ # растение
*запас ^ удобрение р32 *
растение
где Рудобрение Д°за меченого фосфора; Растение ” поступление в растение неме¬
ченого фосфора; Ррастение — поступление в растение меченого фосфора.
Но отношение Рркстение/Ррастение остается одинаковым, вычислим ли
МЫ Ррастение И Рр|Стение На. общий урожай ИЛИ на 1 2 урожая ИЛИ В ОТ"
носительных величинах; отсюда возникает возможность определения запаса
усвояемых фосфатов (Рзапас) по анализу только средней пробы урожая.
Для этого достаточно проанализировать среднюю пробу урожая на содер¬
жание в ней общего фосфора и определить ее радиоактивность. Зная удель¬
ную активность внесенного меченого фосфора, т. е. количество импульсов
в 1 мин, приходящееся на 1 мг Р205 меченого удобрения, можно установить,
сколько миллиграммов Р205 поступило в анализируемую навеску растения
из меченого удобрения, а по разности установить, сколько миллиграммов
Р205 поступило в растения из почвы.
Вычисление проводят ПО последней формуле, НО ПОД Ррастение И Ррастение
понимают в данном случае не общее поступление меченого и немеченого
фосфора в растение, а содержание их в навеске в 1 г вещества или количество
их, выраженное в процентах.
Например, в полевом опыте с овсом было внесено меченое удобрение в количе¬
стве 30 кг Р2Об на 1 га. При анализе средней пробы овса в навеске в 10 г сухого вещества
содержалось 85 мг Р2Об, из которых, судя по активности урожая и активности удобрения,
17 мг приходилось на долю меченого фосфата и 68 мг на долю фосфатов почвы (85—17 =
= 68). Тогда запас усвояемых фосфатов в почве составит (в кг Р206 на 1 га): Раапас =
= 30-68:17 = 120.
Аналогичные вычисления можно произвести в обычном вегетационном
опыте, беря дозу меченого фосфора равной обычной дозе фосфорного удо¬
брения (в мг Р206 на 100 г почвы). Но такое определение величины общего
Методика применения меченых атомов
485
запаса недостаточно точно, так как внесение фосфорных удобрений влияет
на использование растениями почвенных фосфатов. Поэтому более надежные
показания получаются при внесении радиоактивной метки или минимальных
количеств меченого фосфата, но с активностью, достаточной для точного
определения коэффициента использования К.
Для характеристики фосфатного режима почвы определение только ве¬
личины запаса усвояемых фосфатов, без определения коэффициента исполь¬
зования фосфора удобрений, недостаточно. Запас в почве усвояемых фосфа¬
тов может быть велик, но степень его подвижности незначительна, и, наобо¬
рот, запас может быть мал, а подвижность большая. Например, в краснозе¬
ме имеем большие общие запасы фосфатов, но малую их подвижность, ко¬
торая характеризуется низким коэффициентом (процентом) использования
радиоактивной метки (К). Определение же величины К нельзя произвести,
не зная общего урожая и его общей активности.
Применение радиоактивной метки при химическом анализе почвы и при
постановке вегетационных опытов позволяет получить не только правильное
представление о фосфатном режиме почвы, но и решить практические во¬
просы о зафосфаченности почв, о пригодности различных методов определе¬
ния усвояемых фосфатов и т. д.
Литература
Вдовенко В. М., Боброва В. Н., Рысьев
О. А. и др. Сцинтилляционная хромато¬
графическая установка УСХ-1 для опре¬
деления радиоактивности в пятнах на
тонкослойных и бумажных хроматограм¬
мах.— Изотопы в СССР, 1973, № 30.
Верховская И. Н., Габелова Н. А., Зиновь¬
ева Е. Г. и др. Метод меченых атомов в
биологии. Изд-во МГУ, 1955.
Голубцов И. ВЗаборенко К. Б., Иоффе
Б. 3. и др. Практическое руководство по
радиохимии. Под ред. А. Н. Несмеяно¬
ва, т. 2. М., Г осх им из дат, 1961.
Клечковский В. М. Изотопы и излучения в
агрономии.— Труды Всес. научно-техн.
конф. по применению радиоактивных и
стабильных изотопов и излучений в на¬
родном хозяйстве и науке. Физиология
растений. Агрохимия. Почвоведение. М.,
Изд-во АН СССР, 1958.
Матвеев В. В., Соколов А. Д. Фотоумно¬
жители в сцинтилляционных счетчиках.
М., Атомиздат, 1962.
Методы приготовления препаратов и обра¬
ботка результатов измерений радиоактив¬
ности. М., Атомиздат, 1973.
Несмеянов А. Баранов В. И., Заборен¬
ко К. Б. и др. Практическое руководство
по радиохимии. М., Госхимиздат, 1956.
Нормы радиационной безопасности НРБ-
69. М., Атомиздат, 1972.
Оболенская Л. И., Жадкова Н. Т. Приме¬
нение спектрального метода для опреде¬
ления изотопного состава азота в биоло¬
гических и почвенных пробах.— Химия
в сельском хоз-ве, 1964, № 1.
Основные санитарные правила работы с ра¬
диоактивными веществами и другими ис¬
точниками ионизирующих излучений
ОСП-72. М., Атомиздат, 1973.
Радиоактивные изотопы и меченые соедине¬
ния. Каталог. М., Атомиздат, 1964.
Сиборг /\, Перлман И., Холлендер Дж.
Таблица изотопов. М., ИЛ, 1956.
Спицин В. И., Кодочигов П. Н., Голутви¬
на М. М.у Кузина А. Ф., Соколова 3. А.
Методы работы с применением радиоак¬
тивных индикаторов. М., Изд-во АН
СССР, 1955.
Техника измерения радиоактивных препа¬
ратов. Сборник статей под ред. В. В. Боч¬
карева. М., Атомиздат, 1962.
Фосфорные удобрения и питание растений.
Сборник экспериментальных и методи¬
ческих работ под ред. А. В. Соколова.
М., Изд-во с.-х. литературы, журналов и
плакатов, 1963.
В. В. ЦЕРЛИНГ
ДИАГНОСТИКА ПИТАНИЯ РАСТЕНИЙ
ПО ИХ ХИМИЧЕСКОМУ АНАЛИЗУ
*
Для контроля обеспеченности растений доступными им соединениями
питательных элементов почвы по фазам вегетации в настоящее время наряду
с другими агрохимическими методами все чаще используются методы расти¬
тельной диагностики. В сочетании с другими методами агрохимии резуль¬
таты растительной диагностики позволяют уточнить агрохимическую харак¬
теристику почв, полнее расшифровать результаты опытов и ряд других
явлений, основанных на роли взаимодействия почвы и удобрений со свой¬
ствами сельскохозяйственных культур.
Визуальная диагностика питания растений строится на учете изменения
ряда внешних признаков их состояния в течение вегетации в каждый кон¬
тролируемый срок вследствие нарушения питания: изменение окраски
листьев, их формы, появление характерных пятен или полос бледной окраски,
отмирание некоторых частей растений, изменение всего габитуса растений.
Химические анализы проб растений составляют методы химической диа¬
гностики. Наряду с этим в каждый срок диагностирования необходимо иметь
данные по фенологии, биометрии главнейших органов и их морфологии, так
как только сопоставление с ними результатов химической и визуальной
диагностики позволит сделать правильное заключение о состоянии питания
культуры.
Кроме названных двух видов растительной диагностики, используются
еще методы контроля питания по изменению цвета листьев, их формы и всего
состояния растений, наступающего через 7—10 дней после инъекции в ствол,
в ветвь или опрыскивания, или нанесения на часть листьев растения слабого
раствора одного из элементов, недостаток которого подозревается (Roach,
1952). Как и при любых других агрохимических методах, при исполь¬
зовании методов растительной диагностики необходимо иметь сведения
об условиях выращивания сельскохозяйственной культуры: почве, погоде,
агротехнике и др.
Растительная диагностика является комплексным методом, и ни в коем
случае нельзя дать правильного заключения по одним только химическим
анализам растений, особенно если определяется только концентрация эле¬
мента. Известна зависимость такой концентрации от массы растения. Наи¬
более наглядно эту зависимость можно выразить в виде кривой на рис. 1.
Одно и то же процентное содержание элемента может иметь совершенно
разное физиологическое и агрономическое значение (табл. 1). Так, на рис. 1
в случаях 1 и 5а растения близки по концентрации элемента, но в первом
случае причиной была малая масса растения вследствие того, что какой-то
иной фактор, а не питание, судя по процентному содержанию элемента,
задержал рост растения. В случае 5а имеются оптимальные сочетания при¬
роста массы и поступления элемента. Случай 3 свидетельствует об остром
голодании, а при случае 4 растения использовали улучшенные условия на
прирост массы, но питания не хватило на повышение ее качества. При даль¬
нейшем, но умеренном применении удобрений прирост урожая шел одно¬
временно с повышением концентрации элементов в растении. Это случай 5.
Здесь будет наибольшая корреляция химизма растений в вегетацию с уро¬
жаем. Пункт 5а соответствует оптимуму питания. Последующее неоправ¬
Диагностика питания растений по их химическому анализу
48 Г
данное увеличение удобрений или, еще хуже, одного элемента приводит
к «роскошному» питанию — случай 6. Если найдены факторы, ограничива¬
ющие дальнейший прирост урожая, и они устранены, то это характеризуется
случаем 6а. Чрезмерный избыток элемента вызывает снижение урожая
при одновременном весьма высоком токсичном содержании элемента в расте¬
нии — случай 7.
Таким образом, сопоставление данных химической диагностики с раз¬
мерами растений является первым правилом при составлении заключения/
о потребности сельскохозяйственной культуры
в питании.
Имеется немало данных о взаимодействии
элементов как при их поступлении в корни,
так и при дальнейшем их использовании в жиз¬
ненном цикле растений. Поэтому вторым пра¬
вилом растительной диагностики является необ¬
ходимость одновременного определения не менее
трех главнейших элементов — N, Р и К.
Третьим правилом растительной диагностики
будет учет условий выращивания сельскохо¬
зяйственной культуры.
Залогом успешного диагноза питания рас¬
тений является правильный выбор индикатор¬
ного органа. Этим термином обозначаются те
части растения, которые наиболее сильно из- ^ ^ В8аимосвязь процент_
меняют количества содержащихся в них пита- ного содержания элемента в
тельных элементов при изменении условий индикаторном органе с весом
возделывания культуры. Например, при недо- растения или его урожаем
статке N—Р—К—Mg питания вегетативные ор- объяснение см. в тексте
ганы сильнее обедняются, чем репродуктивные.
Поэтому их состав более показательный, инди¬
каторный. Взрослые части также сильнее обедняются в этих условиях,,
чем молодые. Поэтому индикаторными органами обычно бывают взрос¬
лые листья, нижние части стеблей, а при очень крупных листьях (кукуруза,,
капуста и др.) — верхние части таких листьев. При недостатке всех остальных
элементов питания рекомендуется анализировать, наоборот, молодые части
растений, так как эти элементы обычно не реутилизируются и потому моло¬
дые органы сильнее испытывают их недостаток.
Более сложным является выбор индикаторного органа у древесных мно¬
голетних растений. В специальных исследованиях как советских, так и за¬
рубежных ученых предпочтение отдано листьям нижней части средней трети
побегов текущего прироста. При отборе проб листьев и хвои необходимо
строго следить за тем, одинаковы ли их положение на дереве, на кусте,
возраст ветви, расположение в кроне, освещенность солнцем и ориентация
к странам света.
На выбор органа для анализа влияет и сам метод химической диагности¬
ки: для валовых анализов отбирают либо листья, либо всю наземную часть
(последнее рекомендуется для мелких растений в ранние фазы и при малых
размерах листьев — в фазу кущения у злаков, в фазу «елочки» у льна и т. д.);
для анализов неорганических соединений элементов (нитратов, фосфатов и
других, которых, как известно, больше бывает в сосудисто-проводящих
системах и в нижних ярусах растений) отбираются черешки, крупные глав¬
ные жилки листьев и стебли.
По результатам изучения распределения элементов и их соединений по
органам растений известно, например, что азот больше концентрируется
в пластинках, а калий, наоборот, в черешках. Поэтому имеются предложения
в зависимости от задач диагноза брать для анализа отдельно ту или иную
488
В. В. Церлинг
Таблица 1
Сопоставление состояния растения с содержанием в нем питательных элементов
как показатель обеспеченности ими посевов
№
Состояние растения
Концентрация
питательных
элементов в
растениях
Причина состояния
растений
Потребность
в удобрениях
1
Плохое
Равна или выше
оптимального
Другие факторы роста, кро¬
ме питания
Не требуется
2
Среднее
Ниже оптималь¬
ного
Улучшены ограничивающие
факторы роста
Начало недостатка удоб¬
рений
3
Очень плохое
Очень низкая
Острое голодание (крити¬
ческий минимум)
Усиленное удобрение
4
Слабое или близко к
среднему
Низкая
Недостаток питания
Полные дозы удобрений
5
Среднее или близко к
хорошему
Немного ниже
оптимального
Некоторый недостаток пи¬
тания
Уменьшение дозы удоб¬
рений
03
Хорошее
Оптимальная
Обеспеченное питание
Не требуются
6
«Роскошный» рост
вегетативных органов
Выше оптималь¬
ной
Избыточное питание
Необходимо улучшить дру¬
гие факторы роста или из¬
менить соотношение эле¬
ментов в составе удобрений
Изменить агротехнику
и систему применения
удобрений
6а
Хорошее
Оптимальная и
выше
Сняты ограничивающие фак¬
торы роста. Следует контро¬
лировать питание растений
В данный момент не тре¬
буются
7
Среднее или плохое с
признаками токсич¬
ности
Очень высокая
Вредный избыток элементов
питания
Не требуются
часть листа и далее сопоставлять результаты, что позволит уточнить пере¬
движение и использование питательных элементов.
Для тех культур, для которых еще не установлен индикаторный орган,
следует путем изучения распределения элементов по органам разноудобрен-
ных растений — голодающих, слабо-, средне- и высокообеспеченных — вы¬
явить тот орган, изменения которого при разных дозах будут наибольшими.
Для этого необходимо сравнить различия в составе каждого из органов раз-
ноудобренных растений с их приростом на дату анализа и с урожаями, т. е.
вычислить коэффициенты корреляции между изменениями этих величин.
Орган, состав которого будет наиболее сильно коррелировать с ходом роста
и урожаем, будет индикаторным на условия питания.
Этому же служит и «показатель различий» (Дзикович, 1970), который
вычисляется как отношение содержания элемента в каждом органе растения,
обеспеченного этим элементом, к его содержанию в таком же органе необес¬
печенного растения. Чем выше это отношение, тем достовернее выбор инди¬
каторного органа.
В. В. Панков (1970) рекомендует для той же цели учитывать отношение
разностей в процентном содержании элемента в каждом органе удобренного
и неудобренного растения к их ошибкам по формуле t = (К2 — KJ/Y’k* + /c|f
где К1 и К2 — концентрации элемента в органе удобренного и неудоб¬
ренного растений, а кх и к2 — их ошибки. Чем выше величина ty тем более
индикаторным будет данный орган растения.
Химическая диагностика основывается главным образом на валовом ана¬
лизе и анализе растворимых форм питательных элементов в растительных
пробах (анализы пасоки, вытяжки, выжатого сока из свежих растений и др.).
Важными этапами работы являются: 1) взятие и подготовка проб расте¬
ний для анализа, 2) учет хода роста и формирования урожая, 3) учет сопут¬
ствующих условий: почвенных, агротехнических, погодных и др., 4) химиче¬
ский анализ проб растений и 5) обработка всех данных и составление заклю¬
чения о нуждаемости растений в удобрениях.
Диагностика питания растений по их химическому анализу
489»
ВЗЯТИЕ ПРОБ РАСТЕНИЙ ДЛЯ АНАЛИЗА
Взятие проб растений является весьма ответственным моментом в ре¬
зультативности диагностики питания растений и оценки доступности им
почвенных ресурсов. Всю площадь исследуемого посева визуально делят на
несколько участков в зависимости от ее размера и состояния растений.
Если в посеве выделяются участки с явно худшими растениями, то на
карте поля отмечаются эти участки, выясняют, не является ли плохое со¬
стояние растений следствием энто- или фитозаболевания, местного ухудше¬
ния свойств почвы или других условий роста. Если все эти факторы не объяс¬
няют причины плохого состояния растений, то можно предположить, что
нарушено питание. Это проверяется методами растительной диагностики.
Берут пробы с участков с самыми худшими и самыми лучшими растениями
и почвы под ними и по их анализам выясняют причины ухудшения растений
и уровень их питания.
Если по состоянию растений посев однороден, то при отборе проб следует
добиваться, чтобы образцы соответствовали среднему состоянию растений
на данном участке поля. С каждого выделенного массива по двум диагоналям
берут растения, выдергивая их с корнями. Они используются: а) для учета
прироста массы и хода образования органов — будущей структуры урожая
и б) для химической диагностики.
В ранние фазы (при двух — трех листьях) в пробе должно быть не менее
100 растений с 1 га. Позже для зерновых, льна, гречихи, гороха и других —
не менее 25—30 растений с 1 га. У крупных растений (взрослых кукурузы,
капусты и др.) берут нижние здоровые листья не менее чем с 50 растений
(Церлинг, Горшкова, 1972). Чтобы учесть накопление по фазам и вынос уро
жаем, берут в анализ всю надземную часть растения.
У древесных пород — плодовых, ягодников, винограда, чая, цитрусовых,
декоративных и лесных — в связи с особенностями их возрастных измене¬
ний, двухлетности развития репродуктивных органов, периодичности пло¬
доношения и т. д. взятие проб несколько сложнее, чем у полевых культур.
Выделяют следующие возрастные группы: сеянцы, дички, привитые двухлетки,
саженцы, молодые и плодоносящие (начавшие плодоносить, в полном и в за¬
тухающем плодоношении) деревья. У сеянцев в первый месяц их роста
в пробу входит целиком все растение с последующим разделением его на
органы: листья, стволики и корни. Во второй и следующие месяцы отбирают
вполне сформировавшиеся листья, обычно — первые два после самых мо¬
лодых, считая от верхушки. У двухлетних дичков также берут первые два
сформировавшихся листа, считая от верхушки ростового побега. У приви¬
тых двухлеток и саженцев берут, так же как и у взрослых, средние листья
ростовых побегов.
Детальное изучение состава листьев разноудобренных яблонь, взятых
с ростовых и плодоносящих побегов, показало, что в качестве индикаторного
оказался лист, расположенный в нижней части средней трети продолжения
скелетных ветвей, т. е. побегов прироста текущего года.
Во всех случаях средняя проба составляется из 100 листьев с черешками,,
отобранных с 20—25 одноименных (одного сорта, возраста, в пределах одной
почвенной разности) деревьев, с 5—7 побегов одного типа с каждого дерева
по периферии всей кроны на высоте 1,5—2,0 м от уровня почвы. Поскольку
о влиянии стран света на состав листьев дерева имеются противоречивые
данные, то листья рекомендуется брать по всей окружности кроны. Дина¬
мика N, Р и К в листьях разновозрастных деревьев яблони оказалась одно¬
типной, что позволяет брать одинаковые пробы у разновозрастных деревьев
(Церлинг, Егорова, 1971).
У ягодников — крыжовника, смородины и других — отбирают с по-
бегов текущего прироста по 3—4 листа с 20 кустов с тем, чтобы в пробе было-
490
В. В. Церлинг
не менее 60—80 листьев. У земляники в том же количестве отбирают взрос¬
лые листья. У винограда до конца цветения берут листья против грозди,
позже — при созревании ягод, кроме того, берут второй и третий (считая
сверху) нормально развитые листья или листья 16—17-го узла, считая снизу
(Лагутинская, 1970). У чая в пробу отбирают флеши (Бурчуладзе, 1970).
У хвойных деревьев рекомендуется брать хвою из мутовок текущего года
из средней трети бокового побега (Баннов, 1972).
Общим требованием является унификация техники отбора, обработки
и хранения проб: взятие со всех растений строго одних и тех же частей по
их ярусности, возрасту, расположению на растении, отсутствию заболеваний
и т. д. Имеет значение также, находились ли листья на прямом солнечном
свете или в тени, причем во всех случаях должны быть отобраны листья
одинакового размещения по отношению к солнечному освещению, лучше
на свету.
Пробы в поле, саду перед отправкой их в лаборатории упаковываются
таким образом, чтобы избежать потери их влаги: это необходимо как для
учета их сырого веса, так и для анализа неорганических соединений в свежих
образцах, т. е. необходимо исключить их ферментативные изменения при
завядании. Желательно иметь портативные холодильные сумки для сохра¬
нения свежих проб до их доставки к месту анализа.
Сроки проведения диагностического контроля питания растений уста¬
навливают исходя из следующих положений.
1. Условия питания молодых растений во многом предопределяют бу¬
дущий урожай. Поэтому важно диагностировать молодые растения.
2. Компоненты структуры урожая формируются в разные фазы, начиная
‘С самой ранней. Поэтому важны ранние и последующие сроки диагноза,
причем чем раньше дана подкормка, тем больше возможности воздействовать
на большее число компонентов структуры урожая.
3. Чтобы уточнить систему применения удобрений (в т. ч. состав под¬
кормок), необходимо знать требования возделываемой культуры к питанию
по периодам формирования урожая. Проростки почти всех растений со сла¬
боразвитыми корневыми системами и малой ассимилирующей поверхностью
весьма чувствительны к высоким концентрациям питательных растворов и
"чаще всего требуют некоторого преобладания фосфора над азотом.
Основываясь на этих соображениях, следует контролировать питание
растений несколько раз за вегетацию. Однако бывают и другие подходы,
жогда берут пробу 1 раз за вегетацию, чаще в середине сезона.
Для полевых культур сроки отбора проб следующие:
I срок: у льна — начало фазы «елочки»; у злаковых, бобовых, хлопчат¬
ника, помидоров, огурцов и других однолетних — фаза трех листьев; у двух-
летников — тоже фаза трех листьев.
II срок: у однолетних — начало развития соцветия: у яровых зерновых
злаков — начало трубкования, т. е. развертывание 4-го листа; у озимых
зерновых, многолетних злаковых трав, кукурузы — при 5—6 листьях на
главном побеге; у остальных однолетников — начало образования первых
мелких бутонов; у корнеплодов и капусты — развертывание 6-го листа.
III срок: у большинства растений — фаза цветения; у гречихи, огурцов,
помидоров — цветение и начало образования первых зерен (у гречихи)
'й плодов; у корнеплодов — фаза развертывания 8—9 листьев; у капусты —
начало завязывания кочана.
IV срок: в уборку урожая — для учета выноса питательных веществ и
влияния питания на структуру урожая и его качество.
V древесных растений пробы берут в следующие фазы:
I срок: в начале периода сильного роста, до цветения.
0 II срок: в период сильного роста, цветения, физиологического осыпания
Завязей.
Диагностика питания растений по их химическому анализу
49Г
III срок: развитие плодов, окончание роста побегов.
IV срок: созревание плодов и ягод, уборка урожая.
V срок: после уборки урожая, перед осенним листопадом, в расцвечива¬
ние листьев.
У чая — перед каждым сроком уборки урожая флешей.
При отборе проб и выборе срока этих работ, а также при оценке получен¬
ных результатов следует иметь в виду, что состав растений с возрастом изме¬
няется, снижаясь к созреванию.
По мере старения всего растения лист для анализа надо брать из следу¬
ющего яруса, чем обеспечивается одновозрастность листьев. Каждый раз
надо отмечать, с какого яруса взята проба листьев. Например, у яровой
пшеницы в фазу 3-х листьев (начало кущения) берут всю надземную часть,
а при листовой диагностике — 2-й лист; в фазу 5—6-ти листьев (трубкование) —
3-4-й листья; в фазу 7-ми листьев (начало колошения) — 4—5-й листья.
У картофеля, томатов, табака всегда берут закончившие рост листья: да
бутонизации — 2—3-й, во время цветения и позже — 4—5-й листья (счет
листьев идет снизу). У хлопчатника в начале вегетации берут всю надземную*
часть, в фазу 5—6 листьев— нижние здоровые листья, в базу бутонизации —
2—3-й листья, в цветение-плодообразование — листья главного стебля
у основания 3—5 ветвей (считая снизу). При анализе клеточного сока его
отжимают из утолщенных участков листовых черешков, прилегающих к
стеблю. У свеклы, турнепса, капусты в молодом возрасте отбирают пробу
из наружных (неповрежденных) листьев розетки. Позднее, когда листья
наружного яруса подсыхают, отбирают листья из среднего яруса, которые
стоят полувертикально и уже закончили рост. У кукурузы и сорго в молодом
возрасте берут 3—4-й листья, а позже, к выходу метелки,— нижние припо-
чатковые листья.
У древесных многолетних пород отмечен период некоторого стабильного
состава листьев, который приходится на окончание интенсивного роста по¬
бегов. Поэтому когда имеют в виду однократное взятие проб листьев у этих
культур, то рекомендуется отбор в этот период: для средней полосы у пло¬
довых — в конце июля — начале августа, у смородины и крыжовника —
в фазу начала созревания ягод, у земляники — в фазу массового цветения
и формирования ягод.
Во всех случаях пробы отбирают в одни и те же часы суток, лучше утром
(в 8—9 час.), чтобы избежать изменений состава растений за счет суточного
режима их жизнедеятельности.
ПОДГОТОВКА ПРОБ К АНАЛИЗУ
Чтобы избежать завядания растений, все операции по подготовке их
к анализам проводят быстро. Если проб много, то необходимо держать их
в холодильнике и обрабатывать постепенно.
Операции состоят в проведении биометрических учетов: высоты растений,
числа побегов на одно растение, фазы развития, сырого, а затем сухого веса
с пересчетом числа растений в этом весе. Специальными трехлетними иссле¬
дованиями по установлению необходимого и достаточного числа промеров
для получения достоверных средних нами и М. А. Горшковой было найдено,
что с 1 га хозяйственного посева и с делянки в 300 м2 полевого опыта при
5%-ной точности достаточно иметь промеров высоты растений 8, учета об¬
щего кущения 19—20, продуктивного кущения 30, длины колосьев 11—12
отдельных учетов. Для выполнения экспресс-анализа, по В. В. Церлинг,
при точности в 10% надо иметь 10—20 индивидуальных определений, а при;
пестроте посева не менее 20—30 цифр (Церлинг, Горшкова, 1972).
После промеров и подсчета кустистости корни обрезаются (если они не
идут в анализ,- то отбрасываются). Части растений для анализа тщательно
492
В. В. Церлинг
протирают влажной марлей от пыли, грязи и химикатов по борьбе с вреди¬
телями. Пробы делятся на образцы для валового анализа и образцы для тка¬
невой или экспрессной диагностики. Для валового анализа либо берут всю
надземную часть, либо взрослые зеленые листья. Неорганические соединения
содержатся в наибольших количествах в стеблях, листовых черешках и глав¬
ных жилках, поэтому эти части берут для таких анализов. Анализы можно
проводить тут же в свежих растениях или в консервированных сухих образ¬
цах, из которых позже готовят вытяжки.
Консервацию можно проводить любым из известных способов. Например,
выдерживают пробы в термостате в течение 30—40 мин. при температуре
около 80—100° С и затем досушивают на воздухе, избегая прямого солнеч¬
ного света и загрязнения. Для валовых анализов пробы можно не консер¬
вировать. Высушенные и измельченные образцы растений лучше хранить
в банках с притертыми пробками и обязательно в сухом месте. Перед про¬
ведением химического анализа определяют влажность образца для последу¬
ющего пересчета результатов на абсолютно сухое вещество.
ХИМИЧЕСКИЙ АНАЛИЗ РАСТЕНИЙ
Валовой анализ
Валовой анализ проводится либо на листьях определенного положения
на растении, либо во всей надземной части, либо в иных индикаторных
органах.
Диагностика по валовому анализу листьев — зрелых, закончивших рост,
но активно функционирующих, получила название «листовая диагностика».
Она была предложена французскими учеными Лагатю и Момом (Lagatu,
Maume, 1926) и поддержана Люндегордом (Lundegardh, 1945). В настоящее
время этот вид химической диагностики широко используется как за рубе¬
жом, так и у нас в стране, особенно для растений, в корнях которых почти
полностью восстанавливаются нитраты и потому по этой форме в надзем¬
ных частях невозможно контролировать азотное питание (яблоня и другие
семячковые и косточковые, хвойные, богатые дубильными веществами,
луковичные и др.).
При валовых анализах листьев или иных частей растений используются
обычные методы озоления органического вещества для определения в нем
N, Р, К, Са, Mg, S и других элементов. Чаще определение ведут в двух на¬
весках: в одной определяют азот по Кьельдалю, в другой — остальные эле-
мены после мокрого, полусухого или сухого озоления. При мокром озолении
используют либо крепкую H2S04 с катализаторами, либо в смеси с HN03,
либо с НС104, либо с Н202. При сухом озолении необходим тщательный кон¬
троль за температурой, так как при сжигании при температуре свыше 500° С
могут быть потери Р, S и других элементов.
По инициативе Франции в 1959 г. был организован Межинститутский
комитет по изучению техники химической листовой диагностики в составе
13 французских, 5 бельгийских, 1 голландского, 2 испанских, 1 итальян¬
ского и 1 португальского институтов. В 25 лабораториях этих институтов
были проведены химические анализы одних и тех же проб листьев 13 культур
(полевых и садовых) на валовое содержание в них N, Р, К, Са, Mg, Fe, Mn,
Си и Zn. Это позволило комитету после математической обработки данных
рекомендовать способы получения стандартных проб листьев и дать стан¬
дартные методы их химического анализа для контроля точности таких ана¬
лизов при листовой диагностике.
Озоление образцов листьев рекомендуется проводить следующим обра¬
зом: для определения общего азота по Кьельдалю озолять с H2S04 (уд. вес
Диагностика питания растений по их химическому анализу
493
1,84), с катализаторами K2S04 + CuS04 и селеном. Для определения других
элементов используют сухое озоление пробы в платиновой посуде при посте¬
пенном (за 2 часа) нагреве муфеля до 450° С; по охлаждении в муфеле за
2 часа золу растворяют в 2—3 мл воды + 1 мл НС1 (уд. вес 1,19). Выпари¬
вают на плитке до появления первых паров. Добавляют воду, фильтруют
в мерную колбу емкостью 100 см3. Осадок с фильтром озоляют при 550° С
(максимум), добавляют 5 мл плавиковой кислоты. Высушивают на плитке
при температуре не выше 250° С. После охлаждения приливают 1 мл той же
НС1 и снова фильтруют в ту же колбу, смывая теплой водой. Фильтрат, до¬
веденный до 100 мл водой, используют для анализа на содержание макро-
и микроэлементов (Pinta, 1968).
Имеется довольно большое варьирование в методах озоления раститель¬
ных проб, которые различаются главным образом по видам растений —
богатые жирами или кремнием и т. д., и по задачам определения тех или иных
элементов. Достаточно подробное описание техники использования этих
методов сухого озоления дано польским ученым Новосильским (Nowosiel¬
ski, 1968). Им же даны описания различных способов мокрого озоления
с помощью тех или иных окислителей: H2S04, НС104, HN03 или Н202 в том
или ином сочетании в зависимости от определяемых элементов.
Для ускорения анализа, но не в ущерб точности, изыскиваются пути
такого способа озоления растительной пробы, который позволил бы опре¬
делить в одной навеске несколько элементов. В. В. Пиневич (1955) использо¬
вал для определения в одной навеске N и Р озоление H2S04 и в последующем
добавлял 30 %-ную Н202 (проверяя ее на отсутствие Р). Этот принцип озоления
с некоторыми уточнениями нашел широкое применение во многих лабора¬
ториях Советского Союза (Бондаренко и Харитонов, 1967).
Другой широко применяемый метод кислотного озоления навески для
определения в ней одновременно нескольких элементов был предложен
К. Е. Гинзбург, Г. М. Щегловой и Е. А. Вульфиус (1963) и основан на ис¬
пользовании смеси H2S04 (уд. вес 1,84) и НС104 (60%) в отношении 10 : 1,
причем смесь кислот предварительно готовится на всю партию анализируе¬
мого материала.
При необходимости определять серу в растениях описанные методы озо¬
ления не годятся, так как включают серную кислоту.
P. X. Айдинян с сотрудниками (1968) предложил сжигание растительной
пробы для определения в ней серы, смешивая ее с бертолетовой солью и
чистым песком. Метод В. И. Кузнецова с сотрудниками (1968) представляет
собой несколько переработанный метод Шёнигера (Schoniger, 1956). Принцип
метода заключается в быстром озолении пробы в колбе, заполненной кисло¬
родом, с последующим титрованием образовавшихся при этом сульфатов
раствором хлористого бария с нитхромазо-металлиндикатором на барий.
Чтобы обеспечить большую точность и воспроизводимость результатов ана¬
лиза, нами рекомендуется пропускание полученного раствора через колонку
с ионообменной смолой в Н+ форме с целью освобождения раствора от кати¬
онов. Полученный таким образом раствор сульфатов следует упаривать
на плитке до объема в 7—10 мл и по охлаждении титровать (Церлинг, Еро¬
феев, 1973).
Новосильский (Nowosielski, 1968), указывая на большие потери серы
при сухом озолении, приводит рецепты озоления растений для этих ана¬
лизов. Автор считает одним из наиболее простых и быстрых метод озоления
по Буттерсу и Ченери (см. Nowosielski, 1968) с азотной кислотой.
Определение содержания каждого элемента в озоленной тем или иным
способом пробе проводится разнообразными методами: колориметрическими,
комплексонометрическими, спектрофотометрическими, нейтроно-активаци-
•онным, с помощью автоанализаторов и др.
494
В. В. Церлинг
Определение растворимых форм питательных элементов
Наряду с валовыми анализами проб растений широко используется диагно¬
стирование питания сельскохозяйственных культур по растворимым сое¬
динениям элементов в тканях свежевзятых растений. Анализируются либо
вытяжки из них, либо срезы, выжатый сок из них, либо пасока растений.
Группа этих методов известна под названием «тканевая диагностика».
В Советском Союзе работы по использованию анализа растений для целей
диагностики начал Д. А. Сабинин (1928), который наряду с другими мето¬
дами еще в 1925 г. предложил использование для этой цели также пасоки
растений и разработал оригинальный метод ее сбора и анализа.
Анализ пасоки по Сабинину (1928)
Метод, разработанный Д. А. Сабининым и его сотрудниками, приме-
няется для определения снабжения растения питательными веществами. Он
может служить также для оценки развития и функциональной активности
корневых систем. Применение его вполне возможно как в условиях вегета¬
ционного опыта, так и в полевых условиях.
Пасока — это сок, выделяющийся из стеблей и стволов растений при сре¬
зании. Технически метод прост и в то же время достаточно точен, так как
основан на результатах анализа пасоки («плача растений»), т. е. естествен¬
ного и неизмененного продукта жизнедеятельности растений и их корневых
систем.
Все растения: травянистые, кустарниковые и древесные, в большей или
меньшей степени выделяют пасоку при срезании стебля, ствола. Но количе¬
ство выделяемой пасоки неодинаково. Оно зависит от возраста растения,
степени развития корневых систем и в очень значительной мере от времени
суток, а также условий произрастания. Например, злаковые образуют мак¬
симальное количество пасоки в начале кущения. В более поздние фазы раз¬
вития растений, в связи с падением влажности почвы при одновременном
усилении транспирации, подача пасоки корневыми системами замедляется,
становится неравномерной. В условиях орошаемого земледелия этот недо¬
статок легко устраняется.
При работе с растениями из вегетационного домика для сохранения влаж¬
ной атмосферы можно применять стеклянные колпаки или сосуды. При
этом надо иметь в виду то обстоятельство, что при наличии в сосуде 8—10
растений необходимо срезать все растения, если даже сбор пасоки произво¬
дится с одного-двух растений. В противном случае может произойти
остановка «плача» или засасывание уже выделенной пасоки под влиянием
несрезанных растений, продолжающих транспирировать.
Техника сбора пасоки заключается в следующем. Стебель растения сре¬
зается на некоторой высоте от поверхности почвы (1,5—2,0 см) и на срезан¬
ный конец, на пенек, немедленно натягивается и закрепляется ниткой тонко¬
стенная или винтильная чистая резиновая трубка. Последняя должна охва¬
тывать пенек настолько плотно, чтобы не пропускать протекания пасоки.
В свободный конец резиновой трубки вставляется подходящего диаметра
стеклянная трубка, нижний кончик которой немного оттянут для облегчения
соединения. Стеклянная трубка сверху затыкается пробкой с тонким отвер¬
стием для выхода воздуха (рис. 2). Нужно отметить продолжительность вы¬
деления пасоки от времени надевания до времени снятия трубки, что должно
составлять приблизительно 24 часа. При сборе пасока по возможности полно
сливается пипеткой в сухие, чистые, предварительно взвешенные весовые
стаканчики с номерами. Количество пасоки определяется по весу.
Для установления динамики выделения пасоки или для большей точности
учета можно рекомендовать в качестве приемника узкую пробирку с деле¬
Диагностика питания растений по их химическому анализу
495
ниями до 0,1 мл, соединенную через стеклянный си¬
фон с резиновой трубкой и пеньком срезанного ра¬
стения.
Собранная пасока до анализа хранится в холо¬
дильнике, в качестве антисептика следует прибавить
2—3 капли толуола. Последний желательно вво¬
дить и раньше в пробирку при сборе пасоки.
Основой использования метода пасоки для суж¬
дений о ходе снабжения растения питательными ве¬
ществами служит концепция Д. А. Сабинина о су¬
ществовании определенного равновесия между со¬
держанием минеральных соединений в пасоке и в на¬
ружной среде, окружающей корни.
При анализе пасоки устанавливается концентра¬
ция, т. е. процентное содержание и абсолютное ко¬
личество, т. е. произведение процента определяемого
вещества на вес всей собранной пасбки за известное
время. Для этого и требуется точный учет в едини¬
цу времени количества собранной пасоки по весу
или по объему, как указано выше. Произведение кон¬
центрации определяемого соединения на количество
собранной пасоки дает именно ту величину, которая
может характеризовать интенсивность снабжения
растений питательными веществами.
В пасоке анализируются как минеральные со¬
единения элементов питания, так и их общее содер¬
жание после ее кислотного озоления. В первую оче¬
редь определяют азот, фосфор, калий и кальций.
Преимущество этого метода по сравнению с дру¬
гими заключается в том, что здесь анализируется
не какая-то смесь различных продуктов жизнедея¬
тельности, вроде вытяжки или выжатого сока рас¬
тения, а определенная органическая часть растения,
физиологически понятная,— это ток питательных веществ, который корне¬
вая система подает в надземные органы.
Все другие методы анализа растворимых соединений элементов представ¬
ляют в той или иной мере анализ сока растений, полученного непосредствен¬
но из них или при определенном разведении его тем или иным экстрагиру¬
ющим раствором. Они менее громоздки, более быстры и более доступны лю¬
бой лаборатории, чем изложенные выше валовые анализы. Неорганические
соединения элементов представляют, по-существу, резервы питания расте¬
ний, еще не использованные клеткой на синтез сложных органических со¬
единений, и как резервы могут изменяться в очень широких интервалах —
от отсутствия до значительных количеств; поэтому они дают более четкую
картину при нарушении питания.
Кроме того, сопоставление данных по валовому анализу и по содержанию
неорганических соединений того же элемента указывает на способность рас¬
тения использовать поглощенные вещества.
Поскольку определяются растворимые, подвижные формы соединений,
то особое значение имеет способ сохранения проб в неизмененном состоянии
до анализа, чтобы исключить подвядание и активацию ферментативных пре¬
вращений этих соединений. Для этой цели французский ученый Рутченко
(Routchenko, 1967) предлагает иметь портативное льдохранилище с сухим
льдом, в которое помещать этикированные пробирки или иные емкости с про¬
бами растений, снабженные пробками, чтобы не потерять влагу тканей.
Дальнейшее содержание проб, отжим сока и его хранение В. П. Рутченко
рекомендует проводить в холодильной камере.
Рис. 2. Прибор для сбо¬
ра пасоки по Д. А. Са¬
бинину
а — пенек растения; б — ка¬
учуковая трубка; в — стек¬
лянная труба для сбора па¬
соки; е — накопившаяся
пасока; в — пробка
496
В4 В. Церлинг
Обычно анализ производят сейчас же после взятия пробы с растения и
очищения ее от грязи сырой марлей (мыть не следует, чтобы не выщелочить
соединения элементов).
Перед взятием навески для получения вытяжки пробу измельчают сначала
острыми 1 ножницами, а затем навеску растирают в ступке, приливая отме¬
ренное количество экстрагента. Вытяжку отделяют либо декантацией, либо
фильтрованием через плотный фильтр, ускоряя процесс отсасыванием. Мож¬
но пользоваться специальными приборами — «измельчителями тканей»,
являющимися одновременно и измельчителями, и экстракторами.
В качестве экстрагентов для получения вытяжек из растений исполь¬
зуются дистиллированная вода, 2%-ная уксусная кислота, раствор Моргана
и даже раствор первого реактива для определения элемента (например, рас¬
твор молибденовокислого аммония при определении фосфатов). Сравнение
первых трех экстрагентов показало преимущество 2%-ной уксусной кислоты
перед другими растворами (Церлинг, 1962).
Анализ вытяжек из растений
по Магницкому (1954, 1959, 1972)
Определение в растениях содержания элементов питания в неорганиче¬
ских соединениях проводят в 2 %-ной уксуснокислой вытяжке или в ацетатно¬
буферном растворе Моргана. Пробу листьев разрезают ножом на мелкие
кусочки и из нее берут навеску (10 г) в измельчитель тканей, куда добавляют
Таблица 2
Критические уровни содержания элементов питания в неорганической форме в черешках
листьев, в листьях (пшеница, чай) и стеблях вместе с листьями (лен)
Культура *
Фаза развития
Нитрат¬
ный N
Р
К при
низком
содержа¬
нии Na
К при высо¬
ком содержа¬
нии Na
Mg
мг/кг сырого вещества
Картофель
Бутонизация
1200
3 000
90
Цветение
450
—
2 800
—
80
Клубнеобразование**
150
—
2 300
—
60
Сахарная свекла
5—8 листьев
500
40
3 000
2000
120
9—14 листьев
150
25
2 800
2000
80
20—23 листа
40
25
2 100
1100
60
Озимая пшеница
3 листа
—
577
7 800
«Безостая-1»
Кущение
—
586
7 600
—
—
Трубкование
—
484
10 000
—
—
Лен
Ёлочка
—
—
3 600
2200
—
Бутонизация
—
—
3 700
1800
—
Чай (флеши)
Весна (V)
—
90
5 700
—
320
Лето (VII)
—
150
4 200
—
320
Осень (IX)
—
135
3 900
—
560
Хлопчатник
Бутонизация
300
100
4 500
—
—
Цветение
300
90
4 400
—
—
* Уровни урожаев (в ц/га): картофель — 310—
50—60, лен-соломка — 60, чай — 80.
** Через две недели после цветения.
400, caxapi
юя свей
(ла — 250—450
, озимая пшен;
ица —
* Ножницы или ножи должны быть острыми, чтобы не было выдавливания сока растений
Диагностика питания растений по их химическому анализу
497
экстрагирующий раствор при соотношении 1 : 20 и активированный уголь,
для осветления вытяжки. Можно пользоваться вытяжкой из сухого, пред¬
варительно фиксированного, измельченного растительного материала при
соотношении 1 г пробы к 100 мл 2 %-ного раствора ускусной кислоты.
В отдельных порциях вытяжки определяют азот нитратный, фосфор фос¬
фатов, серу сульфатную, хлор, магний, кальций, калий, натрий, железо*
алюминий и бор. Калий, натрий и хлор переходят в вытяжку полностью*
а кальций и магний — частично.
Фильтрат до анализа сохраняют в холодильнике либо добавляют в него
хлороформ. Для ускорения анализа рекомендуется иметь по 4 пробирочных
штатива с 50 гнездами, набор автоматических пипеток и заранее подготов¬
ленные реактивы.
Для использования результатов анализа в оценке минерального питания
надо знать нормальное (оптимальное) содержание элементов в листьях, или
критические уровни. Эти данные по содержанию неорганических соединений
элементов даны в табл. 2. Для некоторых культур критические уровни не
Таблица 3
Содержание элементов питания в неорганической форме в листьях голодающих (числитель)
и здоровых (знаменатель) растений. Нечерноземная зона
Фаза развития и анализируемый орган
Азот
нитратный
Р
Mg
К,г/кг сырого
вещества
мг/кг сырого вещества
Озимая рожь
Три листа, надземная масса
40—80
20-120
0,5—2 ,6
160-300
400-800
6,0-9,2
Кущение, листья
50-30
80-135
40-50
1,6-3,0
140-480
350-450
100-120
5,3—7,2
Трубкование, листья
60-105
50-90
1 ,2-2.3
200-700
180-400
5,2—6,2
Озимая пшеница
Кущение, листья
28-80
1 ,9—2,4
150—300
7,0-9,4
Трубкование, листья
40—70
50- 80
1,9—2,4
Ячмень
190—550
100-220
5,8—9,0
Кущение, листья
10-50
1,3-3,3
120—360
3,7-6,0
Трубкование, листья
0-50
80—120
0,7—1,2
50—130
Objc
180-530
3,6—5,5
Кущение, листья
0
40—60
30-60
1,8
240-720
Кукуруза
200—700
200-400
4,6-5,4
До выметывания метелки, листья
0—100
20
30-50
0,6-1,5
800—1500
70-100
3,0-4,0
498
В. В. Церлинг
Таблица 4
Нормальный состав листьев тепличных огурцов (при урожае 20—28 кг/м2) и томатов (при
урожае 10—15 кг/м2) в фазу плодоношения (в числителе — содержание в пластинках
листа, в знаменателе - в черешках).
Элемент
Огурцы
Томаты
Элемент
Огурцы
Томаты
мг/кг сырого веса
мг/кг сырого веса
Азот нитратный
Фосфор фосфатов
Сера сульфатов
Хлор
200-400
300—500
Калий
Магний
Кальций
Натрий
3000—4500
3600-6000
900—1200
250-360
1000—1500
300—400
3500-5000*
600-1200
3600-7600
220-560
160-240
400-600
120—250
900—1300
160—240
2000—4000
190—240
2400-3500
40-80
400—1200
60—160
200—1000
400-1200
1000-1600
сл.—26U
400—1800
1100—1600
190—250
установлены, но известен состав листьев высокопродуктивных растений и
состав листьев голодающих растений (табл. 3). Для тепличных огурцов и
томатов дан нормальный состав высокопродуктивных растений (табл. 4).
Анализ соков, экстрагированных из npoeodnufux тканей
по Рутченко (Routchenko, 1967)
При диагностике питания кукурузы в фазу 5—6 листьев от растений,
выдернутых с корнями, отделяют корни (они не учитываются). Часть стебля,
соответствующая первым двум междоузлиям без влагалищ, составляет про¬
бу для анализа: ее разрезают на кусочки по 0,5—1,0 см и сразу же поме¬
щают в пробирки, находящиеся в леднике. Остальные части используют для
учета веса растений, причем стремятся сохранить влагу.
В пробу в фазу цветения метелки поступает половина нижней части
главной жилки листа, в пазухе которого развивается початок. Всегда в про¬
бирки, где положены пробы для анализа, влит эфир.
Пробы растений взвешивают, не открывая тару 1, где они помещены.
Чтобы избежать активации ферментов, не следует допускать нагрев проб
свыше 1—2° С. По достижении 1,0° С пробу переносят в чашку; туда вли¬
вают эфир, который консервирует ткани, помогает экстракции и получению
осветленного сока. Чашку с пробой помещают под пресс с давлением не более
1 — 1,5 кг/см2 с тем, чтобы из 25 г вещества получить 8 см3 сока. Полученный
сок фильтруется через плотный фильтр с насосом. Фильтрат используется
для анализа. Хранится в холодильнике: при —15, —20° С можно хранить
2 недели, а при более низкой температуре несколько дольше. При отсутствии
холодильника следует хранить пробы в кристаллизаторе, наполненном
льдом.
Проба сока делится на две партии: 1) в количестве 2 см3 — для опреде¬
ления общей серы и 2) в количестве 5 см3 — для осаждения белков и после¬
дующего анализа на все искомые элементы.
Порцию сока для определения общей серы смешивают с каплей Н202
и 5 г очищенного кварцевого песка и высушивают при 80° С. Фиксированная
на песке сера затем переводится в сульфатную с помощью бертолетовой соли
при Мп02 в качестве катализатора.
1 Вес тары известен заранее.
Диагностика питания растений по их химическому анализу
499
Во второй порции сока белки осаждаются с помощью 75 %-ного спирта
(к 5 см3 сока приливают 15 мл спирта) и отделяются на центрифуге.
Надосадочную жидкость тут же сливают в градуированную колбу, помещен¬
ную в холодильную камеру. Осадок после центрифугирования разбавляют
5 мл смеси спирт + уксусная кислота + вода (подкисляют, чтобы высво¬
бодить ионы, поглощенные белком) и снова центрифугируют. Жидкость при¬
ливают к предыдущей. Белковый осадок оголяется по Кьельдалю серной,
кислотой и Н202. В дальнейшем в нем определяют белковый азот, нераство¬
римый в спирте фосфор и кальций, связанный с органическими кислотами..
Однако в первую очередь рекомендуется проанализировать надосадоч¬
ную жидкость, в которой пропусканием через катионную смолу отделяют
анионы от катионов и определяют ряд соединений: сумму N03 + N02 по
Гриссу, минеральный и растворимый органический фосфор, сульфатную серу
и хлор, которые составляют сумму анионов. Сумма катионов, вымытых из
смолы соляной кислотой, складывается из NH4, К, Са, Mg и Na. Сумму амин-
ного и амидного азота получают после вычитания аммиачного азота из ре¬
зультатов определения азота после озоления с H2S04 части надосадочной
жидкости. Величина общего азота получается по сумме всех форм азота, то
же и для фосфора, серы и кальция. Автор считает одним из важных критериев
контроля питания растений соотношение минеральных форм к валовому
количеству того же элемента, а также долю элемента в сумме анионов или
катионов и отношение суммы анионов к сумме катионов и другие взаимосвя¬
зи элементов и форм их соединений как показатели уровней активности
обмена веществ.
Быстрые анализы на срезах растений
или в выжатом соке из них
Самыми быстрыми методами (экспресс-методами) анализов растений
являются качественно-количественные или полуколичественные определе¬
ния содержания в растениях питательных веществ по реакциям на срезах
свежих растений или в капле сока, выжатого из черешка листа или стебля
растения. В этом случае определяют минеральные формы питательных ве¬
ществ, т. е. резервы питания, еще не использованные растением на построе¬
ние сложных соединений. Следовательно, это будут те формы веществ, ко¬
торые полнее всего могут дать ответы на вопросы о доступности растениям
питательных веществ почвы и удобрений в данный момент и о запасах в ра¬
стении этих веществ, необходимых для получения высокого урожая.
При использовании этих методов следует учитывать факторы, которые
могут усилить или ослабить способность растения быстро синтезировать
сложные органические соединения и тем самым повлиять в ту или другую
сторону на запас минеральных форм питательных веществ. Известно, на¬
пример, что недостаток фосфора затормаживает использование нитратов
на синтез азотных органических соединений, в результате чего может обра¬
зоваться избыток нитратов в тканях.
Под влиянием засухи происходит замедление поступления питательных
веществ из почвы в растение, что приводит к уменьшению их запасов в рас¬
тениях. Похолодание, недостаток солнечного света и другие условия, за¬
трудняющие фотосинтез, приводят, наоборот, к накоплению в тканях мине¬
ральных соединений питательных веществ. Таким образом, при проведении
данного типа анализов даже в большей мере, чем при других, необходимо
учитывать: 1) время — месяц, день и час взятия пробы для анализа, а также
фазу развития растения; 2) условия погоды, 3) состояние растения и внешние
признаки недостатка питания и снабжения водой, заболевания и т. д.;
4) условия агротехники и другие особенности внешних и внутренних усло¬
вий роста.
500
В. В. Церлинг
Основные преимущества разбираемых методов — их быстрота и простота
выполнения (более 100 анализов в день) — дают возможность проводить
массовые определения. В связи с этим предложены портативные полевые
приборы, содержащие необходимые для анализа реактивы и материалы.
Определение нитратов с дифениламином. Опре¬
деление основано на цветной весьма чувствительной реакции нитратов с ди¬
фениламином, в результате которой даже при наличии следов HN03 появ¬
ляется синее окрашивание. Этот синий цвет обусловливается образованием
хиноидной соли азота — N-дифенил-бензидина в результате окисления
нитратами дифениламина — слабого основания, растворимого в H2S04.
Метод Гоффера (Hoffer, 1930). Отрезают у свежевзятых с посева расте¬
ний толстые срезы (куски) стеблей или черешков листьев и кладут их на
стекло или на полоску восковой бумаги. Добавляют несколько капель
1%-ного раствора дифениламина в крепкой серной кислоте и отмечают
синюю быстро появляющуюся окраску по трехбалльной шкале: 1) отсутствие
окраски — недостаток азота, 2) светло-голубая окраска — достаточно азо¬
та, 3) синяяокраска — избыток азота.
Метод Меллера-Арнольда (1929 г.). В углублении палетки
помещают несколько капель сернокислого раствора дефиниламина. Конец
только что срезанного стебля растения опускают в раствор дифениламина и
отмечают в баллах интенсивность и быстроту появления в тканях стебля на
плоскости среза синей окраски, придерживаясь шкалы (табл. 5).
Таблица 5
Шкала для определения нитратов
Балл
Интенсивность реакции
Внесение азотных удобрений
в почву
4
Конец стебля в растворе дифениламина темно-синий
Неэффективно
3
Срез стебля темно-синий
Мало эффективно
2
Проводящие пучки темно-синие
Эффективно
1
Проводящие пучки вначале синие, а затем стано¬
вится красными и коричневыми
Сильно эффективно
0
Весь срез сразу окрашивается в красный или ко¬
ричневый цвет
Сильно эффективно
Определение проводят не менее чем в 10-кратной повторности и затем вы¬
водят средний балл.
Кранц, Нельсон и Бёрхат (Krantz, Nelson, Burhart, 1948) рекомендуют
проводить определение прямо в поле, не срезая растение, а надсекая либо
главную жилку у основания листа кукурузы, либо узел соломины. На над¬
рез наносят 1—2 капли 1%-ного сернокислого раствора дифениламина. Сте¬
пень посинения ткани указывает на степень обеспеченности нитратами.
Оценку они дают по 5-балльной восходящей шкале: 0 или 1 — нет нитратов
(отметить, имеются ли также симптомы азотного голодания); 2 — нитратов
очень мало, следы; 3 — нитратов мало, они присутствуют в небольших ко¬
личествах лишь в главных жилках вторых и третьих листьев и отсутствуют в
других частях растения; 4 — среднее количество нитратов, они присутст¬
вуют в средних жилках листьев на уровне молодого початка (например, в ку¬
курузе) и нигде выше; 5 — много нитратов, они обнаруживаются в главных
жилках листьев выше уровня початка (у кукурузы). Такой порядок опреде¬
ления нитратов авторами проверен, кроме кукурузы, также на сое и хлоп¬
чатнике.
Диагностика питания растений по их химическому анализу
501
Гоффер и Кранц предлагают этот способ и для других полевых культур.
Метод Давтяна (1939). Прибор состоит из небольшого ящика, в
котором имеются капельница с дифениламином, пипетка, фарфоровая палет¬
ка с углублениями и цветовая шкала. Предназначен для определения по¬
требности хлопчатника и других сельскохозяйственных растений в азотных
удобрениях.
Определение производят в поле, непосредственно у растущего хлопчат¬
ника. Из ящика вынимают фарфоровую палетку и в одну из ее ячеек нали¬
вают пипеткой 0,15 мл реактива дифениламина. Затем бритвой, несколько
наискось, срезают стебель, черешок листа или какую-либо другую молодую
часть растения.
Погрузив срезанный кончик отделенной части растения в реактив, нали¬
тый в ячейку палетки, легким кругообразным движением помешивают им
реактив (не размазывая) в течение 20 сек., после чего срезанную часть расте¬
ния удаляют. При этом реактив принимает определенную окраску. Ее срав¬
нивают с цветом приложенной к прибору цветовой шкалы, на которую нане¬
сены баллы, указывающие на содержание нитратов в испытуемом растении.
Нуждаемость растений в азоте определяют по табл. 6.
Таблица 6
Шкала потребности хлопчатника в азоте по Давтяну
Балл
Окраска капли
реактива
Потребность хлопчат¬
ника в азоте
Балл
Окраска капли
реактива
Потребность хлопчат¬
ника в азоте
0
Светло-серая,
Острая нуждаемость
3
Синяя
Средняя обеспечен¬
грязно-бурая
ность
1
Светло-голу¬
Сильная потребность
4
Темно-синяя
Достаточная обеспе¬
бая
ченность
2
Светло-василь¬
Средняя потребность
ковая
Окраску реактива надо определять немедленно после удаления срезан¬
ной части растения из реактива, используя 10—20 или более растений дан¬
ного участка. Средний балл выводится путем деления суммы баллов на число
определений. Ополаскивать палетки следует водой, не дающей синего окра¬
шивания с реактивом.
Для диагностических анализов следует использовать черешки листьев
главного стебля хлопчатника, причем в фазе бутонизации и в начале цвете¬
ния — листьев нижних ярусов, а в период массового цветения и плодообра-
зования — листьев верхних ярусов. Отбор проб растений желательно произ¬
водить в утренние часы, на 5 и 6-й день после полива, а при других задачах
исследования — с различной периодичностью, например через каждую неделю.
При помощи прибора Давтяна можно изучать: а) содержание нитратов в
различных частях растений на корню (хлопчатник, картофель, томаты,
свекла, табак и др.) в различные фазы их развития; б) различные вопросы
нитратного режима растущих растений (поступление, передвижение, распре¬
деление, накопление или расходование); в) условия азотного питания расте¬
ний в полевой производственной обстановке для установления сроков свое¬
временной подкормки их азотными удобрениями.
Реактив. Раствор дифениламина в серной кислоте. 0,3 г дифениламина раство¬
ряют в 25 мл крепкой серной кислоты (уд. вес 1,84) и осторожно приливают этот раствор
по стенке стакана к раствору, содержащему 25 мл такой же серной кислоты и 15 мл воды.
Получается 0,3%-ный раствор дифениламина в 80—85%-ном растворе H2S04.
502
В. В, Церлинг
И. М. Липкинд (1939) использовал прибор Давтяна для анализа хлоп¬
чатника в условиях Средней Азии (Вахшская оп. станция Тадж. ССР).
Оценку количества нитратов он проводил по окраске раствора л]среза по
следующей шкале: 5 — интенсивно-синяя окраска раствора и среза; 4 —
ясное, но слабое посинение раствора; 3 — раствор не окрашен, окрашен
срез; 2 — раствор не окрашен, окрашены только проводящие пучки; 1 — то
же, но через 5 мин. синяя окраска исчезает, побурение; 0 — нет синей окрас¬
ки, побурение.
Определение нитратов, фос'фатов, калия иТ а мм и а к а
на срезах растенийпо В. В. Церлинг (1950, 1956а, б, 1958,
1959, 1963а, б). Анализ проводят на грубых срезах (лучше поперечных) раз¬
личных органов растений: стеблей, листовых черешков, краев листовых плас¬
тинок, веток, почек, бутонов, цветков и их частей, корней, корнеплодов,
клубней, клубеньков бобовых и любых других частей растения. Метод про¬
верен на многих растениях: хлебных злаках, картофеле, корнеплодах, овощ¬
ных, бобовых, зерновых, многолетних травах, древесных многолетних садо¬
вых и лесных породах, декоративных цветах, сорняках и др. Для унифика¬
ции толщи среза можно пользоваться ручным металлическим микротомом,
который состоит из цилиндрической ручки — держателя для эажима части
растения и вращающегося кольца на верхнем конце этого держателя; движе¬
нием бритвы по этому кольцу производят срез; поворотом кольца по метке на
держателе регулируют толщу среза.
Для определения нитратов срезы кладут непосредственно на стеклянную
пластинку, а для определения фосфора и калия — на кусочки (около 2 см2)
фильтровальной бумаги («синяя» или «белая лента»), положенные на стекло.
Последнее может быть либо белое «молочное», либо обычное, но подстеленное
белой бумагой. Размер стеклянной пластинки 10—15 см X 30—40 см. Мож¬
но пользоваться несколькими предметными стеклами (для микроскопа).
Результатами служат средние величины из анализов 5—10 растений, взя¬
тых с одного и того же участка. В каждом случае срез перед анализом раз¬
давливается стеклянным пестиком. На пятно сока и остатки среза наносят
по капле реактива. Следующий по прописи реактив наносится после просы-
хания капли предыдущего реактива.
При определении характера распределения питательных веществ по ор¬
ганам растения срезы делают с разных частей растения, от кончика корня до
верхушечной почки и до частей цветка включительно.
Если требуется определить нуждаемость растения в подкормке, можно
ограничиться анализом лишь той части растения, где искомое вещество ло¬
кализуется в наибольших количествах. Если в этой части растения искомое
вещество отсутствует или находится в весьма малом количестве, то это ука¬
зывает на необходимость подкормки.
Какую же часть растения следует анализировать? Разбираемый метод оп¬
ределяет минеральные формы питательных веществ — те формы, в которых
они обычно поступают из корней в растение, в связи с чем эти формы соеди¬
нений обнаруживаются в больших количествах в нижних ярусах растений и
в тех органах, в которых преобладает сосудисто-проводящая система, т. е.
в стеблях, черешках листьев, в главных жилках листьев и т. д. Следователь¬
но, для диагностического анализа следует брать поперечные срезы черешка
зеленого взрослого здорового листа или узла стебля из нижних частей расте¬
ния, расположенных над начавшими засыхать листьями.
При оценке результатов анализа следует иметь в виду, что с возрастом
растений количество минеральных форм питательных веществ в них умень¬
шается (особенно сильно — нитратов). Например, малое количество нитра¬
тов в начале вегетационного периода указывает на недостаток азотного
питания, тогда как отсутствие их в фазу цветения является нормальным
состоянием растения. Поэтому приводимые шкалы соответствуют составу
Диагностика питания растений по их химическому анализу
503
молодых растений. При анализе более взрослых — в конце бутонизации
или трубковании (у злаков) — оценка должна быть снижена по нитратам
на 2 балла, а по фосфатам и калию — на 1 балл (Церлинг, 1956а, б, 1958).
Прибор Церлинг (ОП-2, Церлинг) содержит все необходимые реактивы
для определения нитратов, фосфатов и калия как в растворах (в капельни¬
цах), так и запасные сухие реактивы в склянках. Там имеются цветные шкалы
стандартов окрасок пятен сока при реакциях на нитраты, фосфаты и калий;
фильтры («синяя» или «белая лента»), бритва, стеклянные пестики и плас¬
тинки.
Определениенитратов. На свежий срез, положенный на стек¬
ло, наносят 1 каплю 1 %-ного раствора дифениламина (в H2S04 уд. веса
1,84). Полученную окраску пятна среза и выдавленного из него сока оцени¬
вают по шкале цветных пятен, имеющейся в приборе, а также в процентах в
сыром веществе или в баллах, пользуясь табл. 7.
Таблица 7
Шкала потребности молодых растениц в азоте по анализу срезов
N—N0,, % в
сыром
веществе
Балл
Характер окрашивания
Потребность в азоте
0,0705
±0,0094
6
Срез и раствор быстро и интенсивно
окрашиваются в иссиня-черный цвет.
Окраска устойчивая
Не нуждается. Избыток
нитратов
0 ,0221
±0,0005
5
Срез и раствор сразу окрашиваются
в темно-синий цвет. Окраска сохра¬
няется некоторое время
Не нуждается. Достаточное
количество нитратов
0,0174
±0,0007
4
Срез и раствор окрашиваются в синий
цвет. Окраска наступает не сразу
Слабо нуждается
0,0151
±0,0061
3
То же. Окраска светло-синяя, исчезает
через 2—3 мин.
Средне нуждается
0,0067
±0,0004
2
Окрашиваются главным образом про¬
водящие пучки в светло-голубой цвет.
Окраска быстро исчезает
Нуждается
0,0028
±0,0006
1
Следы голубой, быстро исчезающей
окраски
Сильно нуждается
0
0
Нет синей окраски. Наступает порозо-
вение и почернение ткани вследствие
ее обугливания от H2S04 реактива
дифениламина
Очень сильно нуждается
►
Содержание нитратов убывает от нижних частей растения к верхним. Чем
моложе ткань, тем меньше в ней нитратов. Их не бывает совсем в меристема-
тических тканях точек роста, в почках, бутонах и цветках; там азот присут¬
ствует в других соединениях. Определение азота нитратов по реакции с ди¬
фениламином вполне оправдано и должно найти широкое использование в
агрономической практике.
Определение фосфатов. На пятно выжатого сока на кусочек
фильтра и отдельно на отодвинутый в сторону срез последовательно наносят
по каплям три реактива. Интенсивность синей окраски сравнивается с цвет¬
ной шкалой образцовых растворов КН2Р04, приложенной к прибору, и с
табл. 8. Результаты записываются либо в баллах шкалы, либо в соответст¬
вующем им количестве процентов Р2Об в сыром веществе пробы.
Когда требуется выявить ткани, наиболее богатые неорганическими фос¬
фатами, то их определение проводят путем получения отпечатка среза того
или иного органа растения на кусочках фильтровальной бумаги, предвари¬
тельно пропитанных раствором молибденовокислого аммония и высушенных.
504
В. В. Церлинг
Таблица 8
Шкала потребности молодых растений в фосфоре по анализу срезов
Р20», % в сы¬
ром веществе
Балл
Окраска (пятна сока) на фильтровальной
бумаге
Потребность растения
в фосфоре
0,0692
±0,0050
5
Отпечаток всего среза темно-синий.
Отпечатки сосудистых пучков иссиня-
черные
Не нуждается
0,0415
±0,0044
4
Отпечаток всего среза синий, а сосу¬
дистых пучков — темно-синий
Не нуждается или слабо,
нуждается
0,0225
±0,0024
3
Отпечаток всего среза светло-синий,
а сосудистых пучков — синий
Средне нуждается
0,0174
±0,0014
2
Отпечаток всего среза серо-голубой, а
сосудистых пучков — немного темнее
Нуждается
0,0121
±0,0007
1
Отпечаток всего среза слабо-серо-го¬
лубой, а сосудистых пучков — серо¬
голубой
Сильно нуждается
0
0
Нет синей окраски ни тканей, ни сосу¬
дистых пучков
Очень сильно нуждается
Необходимо сделать строго прямой поперечный срез стебля или черешка
листа. Если растение несочное (например, соломина злаковых), то на срезан¬
ный конец следует нанести каплю молибденовокислого аммония или на мгно¬
вение коснуться этим срезом капли раствора молибдата аммония, налитого
на стекло или в чашечку. Срез растения прямо перпендикулярно (как печать)
прижимают к центру кусочка обеззоленного фильтра, пропитанного раство¬
ром молибдата аммония. Дав просохнуть отпечатку, на него последовательно
наносят по одной капле раствора бензидина и насыщенного раствора уксус¬
нокислого натрия. После этого появляется синяя окраска участков отпечатка
среза на фильтровальной бумаге, где была выделена из растения фосфорная
кислота. При аккуратном проведении всех операций легко удается полу¬
чить картину локализации неорганических соединений фосфора по сосудам,
тканям и клеткам растения.
Реактивы. 1. Раствор молибденовокислого аммония. 5 г молибденовокислого
аммония растворяют в 100 мл холодной воды и добавляют 35 мл азотной кислоты (уд. вес
1,2). При появлении желтого осадка реактив требуется обновить.
2. Раствор бензидина. 0,1 г бензидина растворяют в 10 мл концентрированной ук¬
сусной кислоты, приливают 10 мл насыщенного водного раствора уксуснокислого натрия
и 50 мл дистиллированной воды. Реактив со временем теряет чувствительность и через 1—2
месяца его следует обновлять.
3. Насыщенный водный раствор уксуснокислого натрия.
Определение калия проводится по реакции с дипикриламином,
которую впервые предложил в 1933 г. Н. С. Полуэктов.
Срез растения после отжима из него пестиком сока отодвигают на бумаге
в сторону от пятна сока. Если растение сочное, то можно сделать отпечаток
поперечного среза так, как это было описано при определении фосфора. На¬
носят на пятно сока и на срез по 1 капле сначала раствора дипикриламината
магния, а затем НС1. Дипикриламинат калия красно-оранжевого цвета, не
растворим в НС1, тогда как дипикриламинат магния разлагается под дейст¬
вием НС1, выделяя дипикриламин желтого цвета. Окраску сравнивают с
цветной шкалой, приготовленной по образцовым растворам КС1 и имеющейся
в приборе, и с табл. 9. Результаты записывают либо в процентах К20 в
сыром веществе, либо в баллах. Лимонно-желтый цвет указывает на отсут¬
ствие калия.
Диагностика питания растений по их химическому анализу
505
Таблица 9
Шкала потребности молодых растений в калии по анализу срезов
К,О, % в сыром
веществе
Балл
Окраска пятна сока на
фильтровальной бумаге
Потребность в калии
0,54
5
Красно-суриковая
Не нуждается
±0,023
0,37
4
Красно-оранжевая
Слабо нуждается
±0,013
0,33
3
Оранжевая
Средне нуждается
±0,018
0,24
2
Желто-оранжевая
Нуждается
±0,012
0,13
1
Соломенно-желтая
Сильно нуждается
±0,035
0
0
Лимонно-желтая
Очень сильно нуждается
Реактивы. 1. Раствор дипикриламината магния. 3 г дипикриламина и 1,3 г окиси
магния растворяют в 100 мл воды и через 15—20 час. фильтруют. Обращаться осторожно,
так как раствор может вызвать слабый ожог кожи.
2. 2 н. раствор НС1.
Определение аммиака. Нитратов в меристемах точек
роста и в цветках не бывает, а азот здесь представлен в виде аммиака,
аминокислот, белков и других азотистых соединений с той или иной степенью
восстановленности или сложности строения. Так как обеспечение азотом этих
органов очень важно для получения высокого урожая, то для контроля азот¬
ного питания был применен метод микроопределения аммиака на срезах
этих и других частей растений путем возгонки его из тканей в микрокамерах,
причем приемником служит висячая капля реактива Несслера на верхней
крышке камеры.
Микрокамеры составляют следующим образом. На предметное стекло ста¬
вят цилиндрики высотой 1 см, нарезанные из стеклянной трубки диаметром
0,8—1,0 см. Верхние и нижние края этих цилиндриков (микрокамер) при¬
терты и смазаны вязким чистым вазелином. Таких микрокамер на одно пред¬
метное стекло можно поставить 3 шт. (с промежутками около 1 см). В ка¬
честве крышки служит покровное стекло, на которое нанесена крутостоя¬
щая (нерасползающаяся) капля реактива Несслера.
Порядок анализа следующий: 1) подготавливают микрокамеры на пред¬
метном стекле; 2) размещают на специальной подставке покровные стекла и
наносят на них по 1-й капле реактива Несслера; 3) на дно каждой микро¬
камеры кладут срез растения (для унификации толщины среза рекомендует¬
ся пользоваться ручным микротомом); 4) на каждый срез наносят 1 каплю
30%-ного раствора NaOH и тут же накрывают предметным стеклом, пере¬
вернутым вниз каплей реактива Несслера.
Промазка вазелином создает герметичность камеры. Под действием щело¬
чи из ткани выделяется аммиак, который окрашивает каплю Несслера от
желтого до красно-коричневого цвета. Запись производят по шкале образ¬
цового раствора NH4C1, который готовят в концентрациях от 0,001 до 0,1 мг
NH4 в 1 мл таким же путем, как и со срезами ткани, только на дно камеры
вместо среза помещают 1 каплю образцового раствора соответствующей кон¬
центрации.
Отсчет необходимо производить в первые 5 мин. после приливания щело¬
чи на срез, так как позже этого срока образуется более интенсивная окраска
506
В. В. Церлинг
капли Несслеровского реактива за счет гидролитического расщепления под
действием щелочи амидов, р-аминокислот и некоторых других азотистых сое¬
динений с выделением из их состава аммиака, что может также характери¬
зовать до некоторой степени богатство тканей азотом. Поэтому рекомендует¬
ся делать два отсчета: через 5 и 25 мин.
Определение аммиака может быть использовано не только для анализа
точек роста, но и для анализа стеблей, черешков листьев и других частей,
когда по свойствам почв можно предполагать наличие в них восстановитель¬
ных условий (например, на почве, длительно залитой водой,— рисовые поля,
болота и др.) и когда, следовательно, азотное питание растений из почвы обес¬
печивается не нитратной, а главным образом аммиачной формой.
Анализ сока растений в полевой лаборатории
по Магницкому (1954, 1959, 1972). Прибор «полевая лаборатория»
позволяет проводить упрощенные количественные определения азота
нитратов, фосфора, калия и хлора в соке растений. Эти определения основаны
на свойстве давать с определенными реактивами цветные растворы или осадки;
интенсивность окраски их сравнивают со шкалой цветных пятен, приложен¬
ной к прибору, или со шкалой образцовых растворов. Результаты выражают¬
ся в миллиграммах элемента на 1 кг сока или условно в баллах, причем
величина балла будет соответствовать номеру образцового раствора (стандар¬
та). Если окраска исследуемого сока интенсивнее окраски последнего стан¬
дарта, сок разбавляют водой вдвое (на 1 каплю сока 1 каплю воды), тщатель-
но размешивают и используют для анализа разбавленный сок, а показатели
анализа удваивают.
Таблица 10
Расчет результатов анализа на отдельные элементы при сравнивании со шкалой стандарт¬
ных растворов или с бумажной шкалой цветных пятен
№ стандарт¬
ного
раствора
Балл
Содержание
элемента
Азот
нитратный
Фосфор
Калий
Магний
мг/кг сока
1
1
Очень низкое
100
16
600
40
2
2
Низкое
250
40
1500
100
3
3
Умеренное
500
80
3000
200
4
4
Высокое
1000
160
6000
400
Примечание. Для фосфора и магния содержание в мг указано при предварительном разбавлении сока
(на 1 каплю сока 3 капли воды).
Бумажная цветная шкала при приборе разработана для одинакового
размера капель (около 0,035 мл) сока и реактива при диаметре углубления
на палетке 10 мм, поэтому надо обращать внимание на выполнение этого тре¬
бования. Размер капель пипеток капельниц с образцовыми растворами и
сока должен быть одинаковым.
Для удобства и упрощения выполнения анализов в полевой лаборатории
имеются смешанные образцовые (стандартные) растворы, содержащие все
определяемые элементы в известной концентрации. Номера стандартных рас¬
творов от 1 до 4-го идут в порядке возрастания концентрации всех элементов.
Концентрация нитратного азота и калия в стандартных растворах будет соот¬
ветствовать концентрации этих же элементов и в соке растений. Содержание
фосфора и магния в стандартных растворах в 4 раза ниже, чем указано в
табл. 10 для содержания их в соке, так как для анализа на эти элементы берут
сок, предварительно разбавленный водой в отношении 1 : 3.
Диагностика питания растений по их химическому анализу
507
Если анализ сока проводят в помещении, то в поле пробы и этикетку
помещают в пакетик из вощеной бумаги. Можно также прямо в поле выжать
сок из проб и перелить его в пробирки, соответственно этикетированные.
Получение сока. Черешки листьев каждой пробы обтирают ва¬
той; крупные, толстые черешки (капусты, свеклы и других растений) разре¬
зают вдоль и для получения сока этих растений используют половину или
четвертую часть черешка. Затем каждый черешок обрезают с краев так,
чтобы остались кусочки длиной 3—4 см (если черешок длинный, то для про¬
бы используют нижнюю часть), и укладывают их в ручной пресс. Сдавлива¬
нием рычагов пресса выжимают сок, стекающий в специальные углубления
пресса. Если сок выжимается плохо, то необходимо перевернуть массу в прес¬
се и снова нажимать. Выжатый сок сливают в маленькие пробирки или пря¬
мо в углубления капельной пластинки.
Определение нитратов. В углубления капельной пластинки
насыпают лопаточкой сухой реактив на азот объемом, примерно равным зер¬
ну ржи, и приливают по 3 капли буферного раствора. Затем добавляют по
1 капле сока и в один из рядов углублений по 1 капле стандартных растворов;
тщательно размешивают стеклянной палочкой. Через 1 мин. сравнивают ок¬
раску исследуемых соков со шкалой стандартных растворов или со шкалой
цветных пятен и записывают результаты, согласно табл. 10 (в приборе таб¬
лица размещена под цветными пятнами).
Возможен и другой порядок добавления реактивов, но нельзя сок и стан¬
дартные растворы смешивать с сухим реактивом перед добавлением буфер¬
ного раствора.
Для получения высоких урожаев картофеля содержание нитратного азо¬
та даже во время бутонизации должно быть высоким (балл 4) и во время цве¬
тения не должно уменьшаться ниже умеренного (балл 3). Если до и во время
бутонизации содержание нитратного азота будет очень низким (балл 1) или
низким (балл 2), то необходимо провести подкормку азотными удобрениями.
Определение фосфора основано на взаимодействии фосфорной
и молибденовой кислот, которые после восстановления оловом образуют сое¬
динение, окрашивающее раствор в синий цвет. Образованию синей окраски
мешает наличие в соке растений большого количества органических кис¬
лот — щавелевой и лимонной. Разбавление сока уменьшает концентрацию
кислот и ослабляет или устраняет их мешающее влияние.
Анализ на фосфор ведут следующим образом. По 1 капле разбавленного
водой сока (на 1 каплю сока 3 капли воды) помещают в углубления капель¬
ной пластинки. Одновременно в один из рядов углублений пластинки по¬
мещают по 1 капле из четырех стандартных растворов. К соку растений и
стандартным растворам прибавляют по 2 капли реактива на фосфор и поме¬
шивают оловянной палочкой, пока окраска не станет устойчивой, на что
обычно достаточно 10—20 сек. Полученную окраску исследуемого сока срав¬
нивают со шкалой образцовых растворов или с окраской цветной бумажной
шкалы и результаты выражают в миллиграммах фосфора на 1 кг сока или
в баллах, согласно табл. 10.
Определение калия основано на образовании оранжево-крас-
ного осадка дипикриламината калия. При значительной кислотности сока,
что встречается у сока из черешков листьев ревеня и незрелых плодов, обра¬
зование осадка с калием не происходит.
В отдельные углубления капельной пластинки переносят по капле сока
и стандартных растворов. Прибавляют по капле дипикриламината магния
и по капле НС1 и перемешивают. Окраску осадков исследуемого сока срав¬
нивают с окраской пятна или с окраской шкалы стандартных растворов и
результаты сравнения записывают в миллиграммах калия на 1 кг сока или
в баллах, согласно таблице.
Калийное голодание растений проявляется при 600—1500 мг калия на
508
В. В. Церлинг
1 кг сока. Для получения высоких урожаев картофеля необходимо, чтобы
содержание калия в соке стеблей до и во время бутонизации было высоким
(балл 4) и во время цветения не уменьшалось ниже умеренного (балл 3).
Если во время бутонизации содержание калия будет очень низким (балл 1)
или низким (балл 2), следует провести подкормку калийными удобрениями,
содержащими мало хлора (калимагнезией — 2—3 ц/га или хлористым ка¬
лием — 0,5—1 ц/га, или золой — 5—10 ц/га).
Для кукурузы, сорго, проса, сои, фасоли и других культур содержание
калия в листьях в молодом возрасте и до середины вегетации, оцениваемое
баллом 2 и особенно баллом 1, указывает на калийное голодание и требует
немедленного проведения подкормки калийными удобрениями.
Определение магния основано на образовании ярко-розового
соединения при взаимодействии титана желтого с гидроокисью магния.
Помещают по 1 капле разбавленного сока (1 каплю сока смешивают с
3 каплями воды) в четыре углубления капельной пластинки и в четыре других
углубления — по 1 капле из четырех стандартных растворов. Прибавляют
по 1 капле реактива на магний, а затем, после перемешивания, по 1 капле
NaOH. Полученную окраску исследуемого раствора сравнивают с окраской
цветной бумажной шкалы или с окраской шкалы стандартных растворов на
капельной пластинке и результаты сравнения выражают в миллиграммах
магния на 1 кг сока или в баллах, согласно таблице.
Для того чтобы предотвратить свертывание окрашенного соединения маг¬
ния с реактивом, необходимо в стандартные растворы и в срок перед добав¬
лением NaOH внести по 1 капле 1 %-ного свежеприготовленного раствора
крахмала. Если в соке растений имеется значительное количество марган¬
ца (например, у картофеля, растущего на кислых почвах), то перед добавле¬
нием титана желтого вводят по 1 капле солянокислого гидроксиламина,
предотвращающего действие марганца.
Магниевое голодание проявляется при содержании 30—60 мг Mg на 1 кг
сока, причем на кислых почвах оно выражено сильнее.
Для получения высокого урожая картофеля содержание магния во вре¬
мя бутонизации и цветения должно быть умеренным или высоким (балл
3—4). Если до и во время бутонизации содержание магния будет очень низ¬
ким (балл 1), необходима подкормка магнийсодержащими удобрениями.
На 1 га вносят 1—2 ц калимагнезии, а на кислых почвах — обожженную до¬
ломитовую муку 5—10 ц. Содержание магния в соке других растений, оце¬
ниваемое баллом 1, будет указывать также на необходимость проведения
подкормки магниевыми удобрениями.
Определение хлора основано на титровании его каплями рас¬
твора азотнокислого серебра определенной концентрации. Индикатором ^по¬
казывающим конец титрования, является хромовокислый калий, который
образует красный осадок хромовокислого серебра лишь после того, как весь
хлор будет осажден азотнокислым серебром. Титрование хлора надо прово¬
дить в нейтральной среде. При наличии в соке растений значительного
количества свободных органических кислот (сок из черешков растений, незре¬
лых плодов и др.) установить конец титрования нельзя, так как осадок хро¬
мовокислого серебра не будет образовываться. В этом случае для нейтрали¬
зации сока перед титрованием надо добавить сухого тонко измолотого
порошка мела и тщательно размешать его с каплей сока.
В углубления пластинки или в маленькую фарфоровую чашку помещают
кружочки индикаторной бумаги и добавляют по 1 капле сока. Затем при¬
бавляют из капельника по каплям реактив на хлор, производя каждый раз
помешивание острием стеклянной палочки, пока от 1 капли цоявится неис¬
чезающее коричневое окрашивание. Записывают количество капель реак¬
тива, пошедшее на титрование хлора в 1 капле сока, и определяют содер¬
жание хлора по табл. И.
Диагностика питания растений по их химическому анализу
Таблица 11
Расчет результатов анализов на хлор
Число капель реактива
1
2
3
4
5
6
Содержание хлора, г на 1 кг
сока
0-1
1-2
2—3
1
со
4-5
СЛ
1
Среднее
0,5
1,5
2,5
3,5
4,5
5,5
Определение хлора можно проводить и в разбавленном соке (1 : 3) или
в вытяжке из листьев, но в этом случае надо разбавить водой в том же отно¬
шении и реактив. Расчеты остаются в этом случае без изменения.
При расходе на титрование 1 капли сока 1—3 капель раствора азотнокис¬
лого серебра содержание хлора для сока черешков картофеля является нор¬
мальным, при 4-х каплях — повышенным, близким к порогу появления
внешних признаков токсичности; при 5 и более каплях — обычно по¬
являются внешние признаки токсичности хлора, ведущие к понижению
урожая.
При содержании 3,5 г хлора в 1 кг сока в период бутонизации и одновре~
менно низком содержании нитратов необходима подкормка азотными удоб¬
рениями — аммиачной селитрой или навозной жижей. Нитраты уменьшают
дальнейшее поступление хлора (антагонизм между анионами), вызывают уси¬
ление прироста, ведущее к перераспределению хлора. Вредное действие
избытка хлора у картофеля проявляется при содержании его более 5 г
на 1 кг сока.
Реактивы. 1. Рабочий образцовый смешанный раствор. 7,22 г азотнокислого ка¬
лия, 0,18 г фосфорнокислого калия однозамещенного, 6,05 г хлористого калия и 1,01 г
сульфата магния растворяют в 1 л воды. 10, 25, 50 и 100 мл этого раствора (в первых трех
случаях разведенные водой до 100 мл) будут соответствовать четырем стандартам на каж¬
дый элемент. Хранить растворы надо в темноте (не более 1 года); позже этого срока они дол¬
жны быть либо вновь разведены из крепкого образцового раствора, либо вновь приготов¬
лены из солей.
2. Крепкий образцовый смешанный раствор готовится из тех реактивов, что и рабочий
образцовый раствор, но указанные выше навески солей растворяют не в 1 лу а в 100 мл
воды, т. е. он в 10 раз концентрированнее рабочего раствора.
3. Сухой реактив на нитратный азот по Брею: а) сульфат бария — 100 г; б) сульфат
марганца — 10 г; в) цинковая пыль — 2 г; г) лимонная кислота — 75 г; д) сульфаниловая
кислота — 4 г и е) альфа-нафтиламин — 2 г. Каждый реактив до смешивания надо тонко
растереть в ступке. Реактивы «б», «в», «д» и «е» смешивают каждый в отдельности с реакти¬
вом «а». Затем тщательно смешивают все реактивы, включая и реактив «г». Правильно
приготовленный реактив имеет светло-сероватую окраску. Появление розовой или крас¬
ной окраски после смешивания свидетельствует о том, что какой-нибудь из реактивов
(чаще всего сульфат бария) содержит незначительные примеси солей азотной кислоты,
и такой реактив употреблять нельзя. Хранят смешанный реактив в зачерненной склянке
и тщательно закрывают пробкой. Реактив при длительном хранении (более 1 года) ча¬
стично изменяется, и окраска при взаимодействии с нитратами сока и стандартных ра¬
створов появляется медленно, не через 1 мин., а через 2—4 мин., поэтому| лучше поль¬
зоваться свежеприготовленным реактивом или с небольшим сроком хранения.
4. Крепкий и рабочий буферные растворы ледяной уксусной кислоты. К 100 мл ле¬
дяной уксусной кислоты добавляют 5 г уксуснокислого натрия. При разбавлении этого
раствора водой 1 : 4 получают рабочий буферный раствор.
510
В. В. Церлинг
5. Реактив на фосфор. Растворяют 1 г молибденовокпслого аммония при нагрева¬
нии в 20 мл воды, раствор фильтруют горячим, по остывании к нему при помешивании
добавляют 20 мл концентрированной соляной кислоты и 160 мл воды.
6. Реактив на калий — дипикриламинат магния, 3 г дипикриламина и 1,3 г окиси
-магния растворяют в 100 мл воды, оставляют стоять на 15—20 час. и фильтруют.
7. Соляная кислота, разбавленная. Концентрированную кислоту разбавляют водой
ъ отношении 1 : 5/
8. Реактив на магний. Растворяют 100 мг титана желтого в 50 мл воды и 150 мл эти¬
лового спирта. При отсутствии спирта реактив растворяют в 200 мл воды, но в этом случае
-сравнение надо вести только со стандартными растворами, а не со шкалой, так как оттенок
красок будет несколько иной.
9. Едкий натр, 10%-ный водный раствор.
10. Крахмал, 1%-ный раствор. К 100—150 мг растворимого крахмала добавляют не¬
сколько капель воды и перемешивают палочкой до состояния пасты; добавляют медленно
при помешивании 10 мл кипящей воды.
11. Гидроксил амин, 5%-ный раствор.
12. Реактив на хлор. Растворяют 4,8 г азотнокислого серебра в 1 л дистиллированной
воды; 1 мл этого раствора осаждает 1 мг хлора. Раствор хранят в склянке оранжевого от¬
тенка.
13. Индикаторная бумага на хлор. Фильтровальную бумагу насыщают 10%-ным ра¬
створом хромовокислого калия, сушат и нарезают кусочками или квадратом со стороной,
равной 5 мм.
14. Уголь активированный, кислый, осветляющий. Очистка угля: насыпают уголь до
половины объема стакана и заливают разведенной соляной кислотой (1 : 3) примерно на
•3/4 высоты стакана. Смесь кипятят под тягой в течение 30 мин., оставляют на ночь, раз¬
вешивают, переносят на фильтр и тщательно промывают водой 10—15 раз до удаления
хлора, затем переносят уголь в фарфоровую чашку и прокаливают на плитке.
Анализ выжатого сока из р а с т е н и й п о Б е л о у с о в у
и Торопкиной (1960). Для контроля питания хлопчатника установ¬
лены нижние границы содержания трех элементов в соке стеблей или череш¬
ков листьев, достаточные для обеспечения нормального роста растений. Та¬
кие значения приведены в табл. 12.
Таблица 12
Оптимальное содержание нитратного азота, фосфора и калия в соке хлопчатника, лег/л
Фаза развития
N-NO,
Р*Ов
К,0 1
Фаза развития
N—N0,
Р*Ов
К,0
1—2 листа
500
90
1500
Бутонизация
300
100
2400
3—4 листа
400
90
1800
Цветение
300
90
2000
5—6 листьев
300
100
1800
ГГ лодообразование
100
70
1500
В начальной фазе анализируют стебли проростков. Начиная с фазы 5—6
•листьев анализируют черешки: в бутонизацию берут черешки со второго-
третьего листьев, в последние две фазы — с листьев, расположенных у ос¬
нования третьей — пятой плодовых ветвей (считая снизу). Собирают по
юдному листу с 15—20 растений. Для анализа используют утолщенную часть
черешка, примыкающую к стеблю. Их измельчают до 0,2—0,3 еле, помещают
в пресс, на дно которого ранее были положены три кусочка (2 см2) фильтро¬
вальной бумаги. Выжатый сок пропитывает бумажки, из которых затем де¬
лают вытяжки для анализа на нитраты, фосфор и калий. Установлено, что
таким способом от,жима каждая бумажка удерживает 0,05 мл сока.
Диагностика питания растений по их химическому анализу
51 ±
Если анализ не может быть проведен cpasy, то бумажки сушат тут же в
поле, нумеруют и убирают для последующего анализа в лаборатории.
Для получения вытяжки каждую бумажку кладут в пробирку с 2 мл
воды, тщательно встряхивают. Если растения очень богаты, то берут поло¬
вину или четверть бумажки, а результат помножают на 2 или 4.
Определение нитратов. К вытяжке прибавляют 3—4 капли
уксусной кислоты (реактив А2) и крупинку сухого реактива* на нитраты (па
Брею) величиной с рисовое зерно. Перемешивают и дают отстояться. Ма¬
линовую окраску сравнивают с цветной шкалой или со шкалой образцовых
растворов.
Определение фосфора. В пробирку с вытяжкой вносят
3 капли молибденового реактива и перемешивают оловянной палочкой до
появления постоянной синей окраски, которую сравнивают либо с цветной
шкалой, либо с окраской образцовых растворов.
Определение калия. В пробирку с вытяжкой добавляют кру¬
пинку сухого кобальтнитритного реактива. Количество желтого осадка ус¬
танавливают по интенсивности мути путем сравнения с цветной шкалой или
с осадком образцового раствора.
Возможен иной способ. Делают поперечный тонкий срез утолщенного*
места черешка листа. Помещают срезы на фарфоровую или стеклянную плас¬
тинку и окрашивают по Церлинг последовательно каплями двух реактивов:
дипикриламината магния и соляной кислоты. Сравнивают с соответствую¬
щей шкалой по Церлинг (см. выше).
Реактивы. А. Для нитратов. 1. Сухой реактив по Брею для определения нитра¬
тов (см. выше метод К. П. Магницкого).
2. 100 г уксусной кислоты и 10 г уксуснокислого натрия растворяют в 1 л воды.
Б. Для фосфора. 1. Растворяют 8,46 г молибденовой кислоты или 8,83 г окиси молиб¬
дена при нагревании в 150 мл серной кислоты (уд. вес 1,785). По охлаждении доводят во¬
дой до 1 л. Хранят в темной склянке.
2. Оловянную палочку толщиной в 2—3 мм и длиной 3 см впаивают в стеклянную
трубку. При почернении промывают в соляной кислоте.
В. Для калия. Сухой реактив. Используется готовый кобальтнитрит натрия либо*
его готовят путем растворения 150 г азотнокислого натрия в 150 мл горячей воды.Ох¬
лаждают до 40°. Прибавляют 50 г азотнокислого кобальта и при помешивании понемногу
приливают 50 мл 50%-ного раствора уксусной кислоты. После энергичного взбалтывания
оставляют стоять на несколько часов. Декантируют, после чего добавляют 208 мл 96°-
ного спирта для осаждения кобальтнитрита натрия. Фильтруют и промывают спир¬
том, сушат на воздухе.
Смешанный образцовый раствор. В 1 л воды растворяют 3,6 г калия азотнокислого,
0.19 г калия фосфорнокислого однозамещенного, 4,42 г калия хлористого.
Приготовление шкалы исходных стандартных
растворов. Из смешанного образцового раствора в мерные колбы на
Таблица 13
Расчет результатов анализа
Рабочий раствор
для приготовле¬
ния шкалы
N—N0,
Р*0.
KtO
Рабочий раствор
для приготовле¬
ния шкалы
2
1
о
т
P.O.
К20
мг/л
мг/л
Колбы № 1
100
20
900
№ 4
600
120
5400
№ 2
200
40
1800
№ 5
800
160
7000
№ 3
400
80
3600
•
512
В. В. Церлинг
100 мл (1—5) отмеривают следующее количество миллилитров раствора и
затем доливают дистиллированной водой до метки:
1 2 3 4 5
Смешанный образцовый раствор, мл 10 20 40 60 80
Для приготовления рабочих стандартных растворов отдельно на азот,
фосфор и калий в пробирки берут по 2 капли раствора из каждой колбы,
добавляют по 4 мл воды и окрашивают соответствующими реактивами.
Расчет результатов анализа клеточного сока хлопчатника производят
по табл. 13.
Автор дает дозы удобрений для хлопчатника в зависимости от обеспе¬
ченности растений и величины урожая (табл. 14).
Таблица 14
Расчет доз удобрений
Обеспеченность растений
Дозы удобрений при урожае в
25—30 ц/га
Дозы удобрений при урожае в
35—40 ц/га
N
Р20.
к,о
N
Р*0.
К20
Низкая
135
90
75
175
120
100
Недостаточная
125
75
75
150
100
75
Средняя
100
50
50
125
75
60
Высокая
50
20
100
20
Ускоренный хроматографический метод определения
содержания свободных аминокислот и амидов
в тканях растений, в модификации Церлинг
и Зинкевич (1967)
Контроль азотного питания растений может быть проведен несколькими
способами: по его валовому содержанию, по количеству нитратов, аммо¬
нийного азота, свободных аминокислот и амидов. Последние формы азота
особенно необходимо использовать при контроле питания тех групп расте¬
ний, корневые системы которых характеризуются высокой восстановитель¬
ной способностью, что исключает возможность контроля их азотного пита¬
ния по содержанию нитратов в надземных частях. Кроме того, нередки ус¬
ловия существования растений, когда исключается возможность образова¬
ния нитратов в почве. Известно, что аминокислоты могут накапливаться в
тканях как при прогрессивном этапе азотного обмена, так и регрессивном.
Поэтому при контроле питания с помощью этих соединений особенно важно
убедиться в здоровом состоянии растений — показателе нормального обме¬
на веществ.
При изменении условий питания изменяется не качественный, а коли¬
чественный состав свободных аминокислот и амидов, причем наибольшие раз¬
личия получаются у амидов, дикарбоновых аминокислот, пролина, аланина
и у-аминомасляной кислоты, т. е. соединений с низкими /?/. Это позво¬
ляет использовать малые хроматограммы. Нами была уточнена и несколько
изменена техника хроматографии на малых хроматограммах, предложенная
японским ученым Одзаки (1964) для аспарагина в листьях риса. Изучением
распределения свободных аминокислот и амидов по органам различных ра¬
стений нами было установлено^, что для этого рода диагностики следует ис¬
пользовать самые молодые, не вполне развернувшиеся листья. Было уста¬
новлено, что у ряда растений при усилении азотного питания увеличива¬
Диагностика питания растений по их химическому анализу
513
ется не только и не столько содержание одного аспарагина, сколько кон¬
центрация многих свободных аминокислот и амидов, что и было положено в
основу нашего метода.
Для хроматографирования использовались круглые беззольные плотные
фильтры, которые разрезались на шесть сегментов. С одной стороны каждого
сегмента надрезалась узкая полоска-фитиль для подачи растворителя на хро¬
матограмму (рис. 3). Камерами служат чашки Нейбауэра, в центре которых
помещают небольшой сосуд с растворителем. Края чашки смазаны вазели¬
ном; после размещения в ней хроматограмм камера плотно закрывается
Рис. 3. Сегмент фильтра для
хроматографирования свобод¬
ных аминокислот в растениях.
Надрезана узкая полоска-
фитиль для подачи раствора
на хроматограмму
стеклом. В каждой камере помещаются четыре хроматограммы: три с испы¬
туемыми пробами и одна с образцовым раствором аминокислот («метчиками»),
чем обеспечивается лучшая сравнимость хроматограмм с метчиками, находя¬
щимися в одних и тех же условиях. Каждая проба растений испытывалась в
шестикратной повторности, причем на сегмент с образцовым раствором в
каждую из этих повторностей наносились капли разной концентрации со¬
гласно принятой шкале, чтобы сравнить одну и ту же пробу со всей шкалой
метчиков. Вид камеры дан на рис. 4.
Рис. 4. Камера для хромато¬
графирования свободных ами¬
нокислот в растениях на ма¬
лых хроматограммах
Проба сока молодых листьев наносится следующим образом. В верхний
угол сегмента бумаги, положенного на стекло,' выдавливается стеклянным
пестиком (<d = 0,6 см) сок из пучка листьев (в пучке — 5 самых молодых
листочков, а для льна — 10 листочков). Размер пятна сока не должен пре¬
вышать диаметра пестика, так как только в этом случае будет обеспечено оди¬
наковое количество сока на хроматограмме, равное 9,66 мг +0,82 мг, что уста¬
новлено специальными замерами и тем подтверждены наблюдения М. А. Бе¬
лоусова и A. JI. Торопкиной (1960). Пятно образцового раствора наносится
аналогичным образом. На каждой хроматограмме простым карандашом ста¬
вится номер образца.
Сегменты с пробами сока и образцового раствора помещаются в камеры,
полоски-фитили которых опускаются в центральный сосуд с растворителем.
Камера закрывается стеклом и оставляется на 4 часа. Предложенный Од-
заки одночасовый срок для разгона хроматограммы нами был проверен и
514
В. В. Церлинг
была доказана его недостаточность. После высушивания на воздухе хрома¬
тограммы проявлялись в термостате при 70° С в течение 20—30 мин. до появ¬
ления четких окрасок.
Проверка показала, что можно разделить во времени нанесение капель
сока листьев на сегмент бумаги и хроматографический анализ аминокислот:
после нанесения сока на сегмент бумаги каплю следует быстро высушить
(например, «феном»). Таким образом подготовленные пробы могут сохра¬
няться более месяца. Это позволяет в течение лета собрать значительное чис¬
ло проб, которые осенью можно прохроматографировать.
Идентификация аминокислот и амидов проводится путем визуального
сравнения размеров и интенсивности окраски пятен на испытуемых и об*
разцовых хроматограммах. Чтобы избежать субъективности в оценке цве¬
тов и оттенков пятен, была нами использована «шкала цветов» А. С. Бон-
дарцева (1954).
Связь интенсивности окраски и размера пятна с количеством амино¬
кислот и амидов проверялась на шкале образцовых растворов «метчиков»
путем измерения планиметром размеров пятен разных концентраций «мет¬
чиков». Сопоставление всех этих данных, а также результаты количествен¬
ных анализов пятен образцовых растворов на больших хроматограммах
позволили дать наряду со шкалой в баллах (табл. 15) количественную
характеристику этих баллов (табл. 16).
Таблица 15
Шкала для визуальной оценки содержания аминокислот в пятнах на малых
хроматограммах
Интенсивность окраски пятна
Наличие амино¬
кислот и амидов
Балл
Концентрация
в пятне, v
Хроматограмма совершенно не окрашена
Нет
0
0
Бледные серо-голубые пятна
Очень мало
1
7,5
Пятна небольшие, светло-фиолетовые, слабо раз*
личаются по окраске
Мало
2
15,0
Пятна средние по размерам, заметно окрашены и
различаются по оттенкам
Есть
3
22,5
Пятна большие, интенсивно окрашены, хорошо
различаются по оттенкам
Много
4
30,0
Пятна очень большие, очень интенсивно окра¬
шены, четко различаются по оттенкам
Очень много
>4
60,0
Содержание свободных аминокислот и амидов в самых молодых листьях
зависит от фазы развития (с наступлением репродуктивного развития их ко¬
личества уменьшаются) и особенностей растения: чем интенсивнее, раньше
начинается и более длительно проходит образование новых бутонов (напри¬
мер, у гречихи), тем сильнее и быстрее снижается содержание этих соедине¬
ний в тканях молодых листьев. Эти выводы получены на основании много¬
кратных результатов в опытах с удобрениями разных сельскохозяйственных
культур. В качестве примера в табл. 17 приведены данные по наиболее био¬
логически разным трем культурам: льну, овсу и гречихе. I срок — начало
бутонизации (у овса — выход в трубку), II срок — цветение. Характеристи¬
ка оптимального азотного питания по содержанию свободных аминокислот в
самых молодых листьях для первых двух фаз развития соответствует данным,
приведенным в нижних двух строках табл. 15 при полном обеспечении азот¬
ным питанием.
Диагностика питания растений по их химическому анализу
515
Таблица 16
Количественная и цветовая характеристика баллов оценки содержания свободных
аминокислот и амидов на малых хроматограммах
Амид или
аминокислота
Концент¬
рация,
балл
Цвет пятна по А. С. Бондарцеву (1954)
Концент¬
рация, v
Размер
пятна, см*
*/
Аспарагин
1
Серо-желтоватый
7,5
0,99
2
Фиолетово-желтоватый
15,0
1,10
3
Розовато-желтоватый
22,5
1,69
0,098
4
Розовато-оранжевый
30,0
1,80
>4
Розово-оранжевый
60,0
2,04
Глютамин
1
Серовато-фиолетовый
7,5
0,54
2
Фиолетовый
15,0
0,56
3
Розовато-фиолетовый
22,5
0,82
0,10
4
Розово-фиолетовый
30,0
1,04
>4
Сливяной
60,0
1,12
Аспарагиновая
1
Слабо-серовато-фиолетовый
7,5
0,60
кислота
2
Серовато-фиолетовый
15,0
1,08
3
Серо-фиолетовый
22,5
1,44
0,135
4
С л або-фио л етовый
30,0
1,80
>4
Фиолетовый
60,0
2,16
Глютаминовая
1
Серовато-фиолетовый
7,5
1,08
кислота
2
Серо-фиолетовый
15,0
1,43
3
Серо-розовато-фиолетовый
22,5
1,50
0,275
4
Розовато-темно-фиолетовый
30,0
2,10
>4
Розово-темно-фиолетовый
60,0
2,40
Пролин
1
Слабо-желтый
7,5
0,55
2
Бледно-желтый
15,0
0,85
3
Бледно-лимонно-желтый
22,5
1,26
0,440
4
Лимонно-желтый
30,0
1,54
>4
Ярко-лимонно-желтый
60,0
2,16
Таблица 17
Содержание свободных аминокислот и амидов в тканях неразвернувшихся листьев льна9
овса и гречихи как показатель обеспеченности азотным питанием
Степень обеспечен¬
ности азотом
Отклонения урожая
от оптимума, %
Фаза
Лен
Овес
Гречиха
балл
мг-%
балл
мг-%
балл
мг-%
Слабая
10-20
I
2
72
2
72
0
0
10-20
II
1
36
2
72
0
0
Средняя
50—70
I
3
144
3
144
3
144
50-70
II
2
72
2,5
100
0
0
Полная
100
I
4
300
4
300
3,5
222
100
II
2,5
100
3
144
0
0
Все аминокислоты и амиды с увеличением концентрации образуют более
крупные пятна, главным образом сине-фиолетового или красновато-фиоле¬
тового цвета. Аспарагин при высоких концентрациях, а иминокислота про-
лин при всех концентрациях дают желтый оттенок их пятен на хроматограм¬
мах, причем пятна аспарагина при высоких концентрациях становятся оран¬
жевыми.
516
В. В. Церлинг
Реактивы. 1. Растворитель для разгона пятен сока на хроматограммах. 0,2%-
ный раствор нингидрина в н-бутиловом спирте и 50%-ной уксусной кислоте при соотноше¬
нии спирта к кислоте как 2:1.
2. Образцовый раствор аминокислот. Смешанный раствор из следующих аминокислот
и амидов: аспарагиновой, глютаминовой, пролина, аспарагина и глютамина. 7,5 мг каж¬
дой сухой аминокислоты или амида растворяют в 10 мл 10%-ного изопропилового спирта,
подкисленного из расчета 10 мл НС1 (уд. вес 1,19) на 1 л спирта. Смешивают по 1 мл каж¬
дого раствора аминокислот и амидов в пробирке — рабочий раствор. В угол сегмента
микропипеткой наносят точно 1 каплю, содержащую 0,05 мл смеси метчиков, в которой
содержится по 7,5 у каждой аминокислоты и амида — это шкала № 1. Шкалу № 2 готовят
путем нанесения второй капли на первую после высушивания феном первой. Три капли
составят № 3, четыре капли — № 4 и пять капель — № 5.
3. Фильтр «синяя лента» диаметром 15 см. Делится на шесть сегментов.
СОСТАВЛЕНИЕ ДИАГНОСТИЧЕСКОГО ЗАКЛЮЧЕНИЯ
Как указывалось в начале статьи, метод растительной диагностики являет¬
ся комплексным и выполняется на основе трех главных правил: 1) сопостав¬
ление химизма растений с данными хода роста и формирования урожая;
2) одновременное определение не менее трех главнейших элементов; 3) сбор
сведений об условиях выращивания культуры.
При составлении диагностического заключения все эти данные исполь¬
зуются путем их взаимного сопоставления и разбора взаимовлияния. В част¬
ности, сравнение химического состава с ростом растений проводится с по¬
мощью рис. 1 и табл. 1, в результате чего выясняется причина обнаруженного
содержания элемента. Если установлено, что причиной состояния растений
является питание, то сравнение с таблицами оптимумов 1 содержания эле¬
ментов (которые даны в тексте для отдельных методов анализа растений и,
кроме того, в таблицах 18 — 21) покажет глубину недостатка (или, наоборот,
вредного избытка) элемента.
Сравнение оптимального содержания элемента в одних и тех же культу¬
рах и в одинаковые фазы, но полученного разными авторами в разных стра¬
нах мира, показывает его стабильность. Это говорит о том, что данные вели¬
чины являются физиологическими характеристиками растения: столько
должно быть в его тканях элементов для нормального развития и, как тако¬
вые, они не зависят от условий произрастания. Отклонения же свидетельст¬
вуют о нарушении питания и о том, насколько не были созданы требуемые
условия питания.
Взаимодействие элементов при их поступлении в корни и в последующем
использовании в реакциях синтеза выясняется при рассмотрении их соотно¬
шений в растении. Для этого используются или отношение одного элемента
к другому (например, N : Р), или один принят за единицу и к нему отнесены
другие, или доля каждого выражается в процентах к сумме других (напри¬
мер, доля азота в сумме N + Р + К, равной 100%), или доля отдельного
аниона в сумме анионов или катиона в сумме катионов, а также отношение
суммы анионов к сумме катионов. Выбор того или иного способа расчета
зависит от задачи исследования.
Процентная доля элемента в сумме их была впервые предложена фран¬
цузскими учеными Лагатю и Момом (Lagatu, Maume, 1926) и названа «качест¬
вом питания». Все эти отношения являются дополнительными критериями и
свидетельствуют о степени равновесности питания. Они используются также
при расчете доз удобрений (Журбицкий, 1963; Болдырев, 1958; 1972; Baier,
1 Оптимумом питания является такое содержание элемента в индикаторном органе,
которое обеспечивает получение наилучшего урожая.
Диагностика питания растений по их химическому анализу
517
Таблица 18
Оптимальное содержание азота, фосфора и калия в растениях
(общее содержание, % в сухом веществе)
Вся надземная часть
Индикаторные листья
Растение
Фаза
N
Р,Ов
К,0
N
РаО*
к,о
Озимая
Кущение
0
1
СЛ
©
1,2-1,5
6,0
4,0-5,0
1,2-1,5
6,0
рожь
Трубкование
3,5
0,8
3,5
3,8-4,2
0,9-1,0
4,0
Колошение — цве¬
тение
1,3-1,4
0,6
to
со
1
to
00
2,2-3,1
0,6-0,7
2,3-3,1
Озимая
Кущение
4,0-5,9
1,0-1,5
4,0-5,0
4,0-5,9
1,0-1,5
4,0-5,0
пшеница
Трубкование
3,5-4,5
0,9-1,4
4,0-4,8
3,8—5,0
1,2
3,0-4,0
Колошение —
цветение
1,5-2,5
0,6-1,0
2,3-3,0
3,0-3,6
0,5-0,9
3,8
Яровая
Кущение
4,0-5,5
0*9—1,3
4,0-5,3
4,0-5,5
0,9-1,3
4,0-5,3
пшеница
Трубкование
3,0-4,3
0,9-1,0
3,5-4,5
3,5-4,4
0,9-1,2
3,5-4,5
Колошение-
цветение
1,6-2,0
0,7-0,9
> 2,5
2,9-3,0
0,8-1,0
3,0-4,0
Ячмень
Кущение
4,7-5,0
1,2-1,8
5,0
UN
i
СЛ
О
1,2-1,8
5,0
Трубкование
2,3-3,8
0,9-1,0
4,6-4,9
4,7
1,2
—
Колошение —
цветение
1,3-2,0
0,6—1,0
2,0-2,2
Ю
со
1
со
©
1
Ъ
to
00
1
СО
Овес
Кущение
5,0-6,0
2,0-2,4
6,0-7,0
5,0-6,0
2,0-2,4
6,0-7,0
Трубкование
3,2-4,0
1,0-1,5
4,0-5,0
3,5-4,5
1,6—2,2
3,5-4,0
Выметывание —
цветение
1,3-2,2
0,7-0,9
2,0-2,4
2,2-3,0
1,0-1,7
2,5-3,0
Кукуруза
Всходы
4,3
1,2
6,2
4,3
1,2
6,2
3—5 листьев
3,0-3,6
0,7-1,5
3,4-4,0
3,8-4,0
0,8-1,3
3,8-5,0
6—10 листьев,
выход метелки
2,9-4,0
0,7-0,8
2,4
3,5-4,0
0,7-1,2
4,0-5,0
Выход рылец —
цветение
—
—
—
3,3
0,6-1,1
со
0
1
ъ
Гречиха
3 листа
4,0-5,0
1,7
6,0
6,2
1,7
4,4
Бутонизация
3,2-3,6
1,2-1,6
6,0
4,0-5,0
1,1-1,5
4,0-5,0
Начало цветения
3,0-3,5
1,0-1,5
4,0-6,0
3,5—4,5
1,2-1,6
3,0-4,7
Начало созрева¬
ния
1,5-2,2
0,7-1,3
2,1-3,0
2,4-3,4
0,9-1,3
2,3-2,8
Горох
3—6 листьев
4,0-5,5
0,8
3,0-4,2
—
—
—
Бутонизация
3,2—3,8
0,8
3,0-3,6
—
—
—
Цветение
3,0—3,7
0,5-0,8
2,4-3,6
—
—
—
Начало созрева¬
ния
3,0-3,5
0,5-0,7
2,0-2,5
1
©
см
0,3
*1
<м
1
00
Кормовые
бобы
5—8 листьев —
бутонизация
3,0-4,5
0,5-0,9
2,0-3,0
4,0—5,0
0,5-0,6
1,0-1,6
Цветение
2,8—3,2
0,6-0,8
2,5-3,0
4,6-4,9
0,6-0,8
1,4-1,6
Плодообразование
3,2
0,6—0,7
2,9
4,5
0,7
1,7
Силосная спелость
3,2
0,6
1,8
3,0-4,6
0,5-0,6
1,1-1,6
Подсолнеч¬
4—5 листьев
4,5-5,5
0,9-1,0
5,6—6,0
5,7-6,0
—
—
ник
Бутонизация
1,4-1,7
0,4-0,5
2,7-3,0
3,0-3,6
—
—
Цветение
1,2-2,0
0,4-0,5
2,3—2,7
3,0-4,0
0,6
<5
1
©^
со
Леа
Елочка
3,6-4,8
0,9-1,6
3,7—4,0
—
—
—
Бутонизация
2,6-2,9
0,8-1,1
2,5-3,0
СЛ
0
1
Сл
"сл
1,0-1,7
<N
1
00
со
Цветение
1,9-2,5
0,6-0,7
2,5—2,8
3,5-4,0*
и 4,8 **
0,9-1,0
2,7
518
В. В. Церлинг
Таблица 18 (окончание)
Вся надземная часть
Индикаторные листья
Растение
Фаза
N
Р*0»
К,О
N
P.O.
К,О
Люцерна
Бутонизация
3,5—4,0
1,2-2,0
5,0
4,5-5,5
2,5-2,8
5,0-6,0
синяя
(посевная)
Цветение
3,0-3,5
1,0
со
00
1
О
3,6-4,0
1,1—2,6
2,0-4,0
Клевер
Бутонизация
3,5-4,0
0,6-0,9
3,5
—
—
—
красный
Цветение
2,5-3,5
0,4-0,6
2,7
3,8
0,5
3,5
Тимофеевка
Трубкование
2,6-2,8
0,8—1,0
3,6-4,0
—
—
—
луговая 4
Колошение
2,5
0,5-0,8
—
—
—
—
* Для урожая волокна льна.
** Для урожая семян льна.
Примечание к таблицам 18 и 20. Урожаи (ц/га): зерновых — свыше 40, кукурузы на зерно — 50, на
зеленую массу — 500, гречихи — 18—20, гороха — 16—20, подсолнечника — свыше 18—20, льна
волокна — 6, сена люцерны — 80, сена клевера — 50—60, сена тимофеевки луговой — 40—60.
Таблица 19
Оптимальное валовое содержание азота, фосфора и калия в овощных растениях
(% в сухом веществе)
Растение
Фаза, месяц
Часть растения
N
Р.О.
к,о
Картофель
До бутонизации
Надземная
5,2—6,0
0,9-1,4
5,0
Листья
4,5-5,0
0,6-1,3
5,0-6,6
Бутонизация
Надземная
4,0-5,0
0,6-1,0
3,5-4,5
Нижние листья
2,0-4,0
0,5-0,7
4,4-5,0
Верхние листья
5,0-5,8
1,0-1,6
3,3-4,0
Цветение
Нижние листья
3,8
0,5
3,0-5,0
Сахарная
До прорывки
Взрослые листья
5,2-5,5
1,1-2,2
5,0-7,0
свекла
Смыкание рядков
То же
4,0-4,4
0,6-0,9
2,5-4,2
Морковь
До прорывки
Надземная
3,5-3,7
0,8-1,0
4,2-4,5
Пучковый товар
»
2,6
0,7
3,5-4,0
Сентябрь
»
2,0-2,3
0,6
2,6-4,0
Огурцы
4 листа
4,6—4,9
>1 ,о
3,8—4,0
Бутонизация
»
3,5—4,0
1,0
2,9-3,2
То же
Взрослые листья
4,7-5,3
1,6-2,0
4,5
Плодоношение
То же
2,2—2,8
0,5
1,0—1,4
Томаты
Бутонизация
Листья
4,5-5,1
1,6-2,0
3,6-4,4
Плодоношение
»
3,0-3,3
1,0-1,6
>2,0
Примечание к таблицам 19 и 21. Урожаи (ц/га): картофеля — 200—300, сахарной свеклы — 300—400,
овощей — 180—300.
1972; Levy, 1968; Кенуорти, 1964; Прево и Олланье, 1964; Chapman, 1967 и
др.)-
Для лучшей наглядности полученных результатов можно пользоваться
диаграммой, предложенной Прево и Олланье (1964) и представляющей со¬
бой многоугольник (треугольник при трех анализированных элементах),
Диагностика питания растений по их химическому анализу
519
Рис. 5. Графическое изобра¬
жение результатов химической
диагностики листьев земля¬
ники из опытов с удобрениями
Радиус равен оптимуму содержания
азота, фосфора и калия: N — 2,25%,
Р,0» = 0,60%, К,0 = 1,50%. Ли¬
стовые пластинки земляники через
12 дней после подкормки.
1 — без удобрений, острый недо¬
статок азота и фосфора, сла¬
бый — кадия;
2 — Р 4* NK, слабый недостаток
калия, слабый избыток азота,
сильный избыток фосфора;
3 — NPK + NK, сильный избыток
фосфора, избыток азота сла¬
бый, близкий к оптимуму из¬
быток калия
вписанный или описавший окружность, радиус которой равен оптимуму со¬
держания каждого элемента (рис. 5).
Следующим моментом при составлении заключения является учет поч¬
венных, погодных, агротехнических и других условий выращивания сель¬
скохозяйственной культуры. Сравнение химического состава растений с со¬
держанием тех же элементов в почве позволило дать уровни содержа¬
ния элементов питания в растениях хлебных злаков, характеризую¬
щие степень нуждаемости в удобрениях (табл. 22) (Церлинг, Горшкова, Зин¬
кевич, 1967; Горшкова, 1968; Церлинг, Горшкова, 1971, 1972).
При расчете доз удобрений с использованием данных анализов растений
поступают так же, как и при использовании результатов анализов почвы, а
именно учитывают коэффициенты использования растениями элементов пи¬
тания почвы и удобрений, свойства почвы, особенности сельскохозяйствен¬
ной культуры, погоды, величины желаемого урожая и его качества и ряд
других.
3. И. Журбицкий (1963), базируясь на соотношениях элементов в расте¬
ниях N : Р2Об : К20 по фазам, уточняет потребность в удобрениях в каждую
из них.
Американский ученый А. Кенуорти (1964) рекомендует учитывать коэф¬
фициент вариации состава листьев отдельных проб при вычислении показа¬
теля обеспеченности растений в удобрениях и дает две формулы для такого
расчета — одну, когда элемент в растении ниже оптимума, и вторую — когда
выше оптимума. Сначала вычисляется процентная доля (Я) содержания эле¬
мента в образце (X) от стандарта (С), т. е. Х-100:С = Я. Влияние варьиро¬
вания учитывают по вариационному коэффициенту (У) состава отдельных
проб.
При недостатке питательных элементов влияние вариации (И) рас¬
считывается по формуле: И = (100 — Я)-У:100. При избытке питания —
по формуле: И = (Я — 100)-У: 100. Показатель обеспеченности (5) вычис¬
ляется для первого случая по уравнению Я + И = В; а для второго Я —
— И = В. Уточнение доз удобрений на основе этих величин (В) автор ре¬
комендует проводить по следующим интервалам: недостаток питания при
В = 17 — 50% от оптимума содержания в растении элемента; слабый недо¬
статок приВ= 50 — 83%; норма питания при В — 83 — 117%; выше
нормы при2? = 117 — 150% и избыток при В = 150 — 183%.
Уточнение системы применения удобрений при комбинированном ис¬
пользовании почвенных и растительных анализов подробно разбирает аме-
520
В. В. Церлинг
Таблица 20
Оптимальное содержание нитратов, фосфатов и калия в растениях
(в баллах шкалы В. В. Церлинг и в % в сыром веществе) *
Растение
Фаза развития
N—N0
э
Лист
Стебель
балл
%
балл
%
Озимая пше¬
Кущение
>6,0
>0,071
ница
Трубкование
1,0
0,0028
5-6
0,022—0,071
Начало колошения
0
0
3
0,015
Цветение
0
0
0
0
Озимая рожь
Кущение
—
—
6,0
0,071
Трубкование
1-2
0,0028-0,0067
5,0
0,022
Начало колошения
0
—
4,0
0,017
Цветение
0
—
0
0
Яровая пше¬
Кущение
—
—
6,0
0,071
ница
Трубкование
2,0
0,0067
4-6
0,017-0,071
Начало колошения
—
—
2,0
0,0067
Цветение
0
0
0
0
Овес
Кущение
—
—
6,0
0,071
Трубкование
3,0
0,015 '
4-6
0,017-0,071
Начало выметыва¬
0
0
2-3
0,0067-0,015
ния
Цветение
0
0
0
0
Кукуруза
Перед колошением
5-6»*
0,022-0,071**
4—6
0,017-0,071
Сахарная
4—5 листьев
5—6»*
0,022—0,071**
—
—
свекла
9—10 листьев
5**
0,022**
—
—
Рост корнеплода
4-5**
0,017-0,22**
—
—
* См. сноску к табл. 18 (урожаи). ** В черешках или в главных жилках.
риканский ученый X. Д. Чэпман (Chapman, 1967). Анализ листьев апельси¬
новых промышленных садов на содержание в них N, Р, К, Са, Mg, S, Cl,
Na, В, Си, Fe, Mn, Мо и Zn сравнен с оптимальным содержанием тех же еле-
ментов, что дало возможность установить, чем обеспечены и чего не хватает
деревьям конкретного сада. Почва анализировалась по слоям через 15—
20 см до 125 см на содержание в ней обменных Са, Mg, К и Na, степень насы¬
щенности основаниями, pH, Р по Бингэму (см. Chapman, 1967) (Н20 вытяж¬
ка), и эти данные также сопоставлялись с нормативами. Такое сопоставление
данных почвенных и листовых анализов позволило: во-первых, проконтро¬
лировать азотное питание по составу листьев; во-вторых, по содержанию в
листьях серы и С1 уточнить лучшую форму азотного и калийного удобрений;
в-третьих, установить причину низкого содержания фосфора в листьях не¬
которых садов при высоком количестве подвижного фосфора в почве; в-чет-
вертых, выяснить причину низкого поступления калия из-за высокого pH
и Са, Mg; в-пятых, выявить потребность в Mg, Mn и других микроэлементах
и, в-шестых, указать, в каких садах имеется опасность засоления почвы и
требуется систематический контроль этого процесса по составу листьев.
В заключение были составлены программы наиболее рационального приме¬
нения удобрений и необходимого контроля состава листьев и почвы (на опре-
Диагностика питания растений по их химическому анализу
521
Р20.
к,о
Лист
Стебель
Лист
Стебель
балл
%
балл
%
балл
%
балл
%
_
5,0
0,069
5,0
0,54
—
—
3,5
0,033
—
—
4,0
0,37
—
—
2,5
0,020
—
—
4,0
0,37
—
—
2,0
0,017
3,0
0,33
4,0
0,37
—
—
5,0
0,069
—
—
5,0
0,54
—
—
3,5
0,033
3,0
0,33
4,0
0,37
—
—
2,5
0,020
—
—
4,0
0,37
—
—
2,0
0,017
—
—
4,0
0,37
—
—
5,0
0,069
—
—
>5,0
>0,54
3,5
0,033
4,0
0,042
4,0
0,37
5,0
0,54
—
—
3,5
0,033
—
—
5,0
0,54
—
—
2,5
0,020
—
—
4-5
0,37—
-0,54
—
—
>5,0
>0,069
—
—
>5,0
>0,54
5,0
0,069
5,0
0,069
4,5
0,46
5,0
0,54
4,5
0,055
5,0
0,069
3,5
0,35
5,0
0,54
—
—
4,0
0,042
—
—
4,0
0,37
3,0*»
0,022**
>5,0
>0,069
4,5**
0,46**
>5,0
>0,54
4-5**
0,042—0,069**
—
>5,0**
>0,54**
—
—
4,5**
0,055**
—
—
>5,0**
>0,54**
—
—
4,0**
0,042**
—
5,0**
0,54**
—
деленную глубину) по наиболее важным для каждого сада элементам и дру¬
гим свойствам. Кроме того, было подчеркнуто, что ряд элементов (в т. ч.
азот) были получены только по анализу листьев, а наибольшая корреляция
состава листьев апельсиновых деревьев с составом почвы отмечается от глу¬
бины 0,5 м и глубже.
В Чехословакии широко используется метод определения доз удобрений
на основе анализа растений, разработанный в Научно-исследовательском
институте растениеводства Байером (Baier, 1972). Он, как и многие другие, в
том числе и советские ученые, исходит из следующих положений: действует
прежде всего элемент, находящийся в минимуме. Причем если одновремен¬
но не хватает нескольких элементов, то в первую очередь урожай ограничи¬
вает элемент, находящийся в самом глубоком минимуме. Поэтому важно соз¬
дать уравновешенное питание, которое контролируется по соотношению эле¬
ментов в составе растения. Важен анализ также и потому, что действуют
только элементы, поступившие в растение. Он рекомендует анализировать
всю надземную часть, чтобы иметь не только процент элемента, но и накопле¬
ние (вынос) элементов в определенную фазу. Байер подчеркивает особую важ¬
ность анализа растений (озимой пшеницы) в фазу 5—6 листьев (III этап он¬
тогенеза) для решения вопроса о потребности в удобрениях, в то время как
Таблица 21
Оптимальное содержание нитратов, фосфатов и калия в овощных растениях
(баллы шкалы В. В. Церлиег и % в сыром веществе) *
Растение
Фаза развития, месяц
Исследуемый орган растения
Нитраты (N—N0»)
Фосфаты (Р*0*)
Калий (К,О)
балл
%
балл
%
балл
Го
Картофель
6—7 листьев
Край пластинки нижних и
средних листьев
4-5
0,017—0,022
2,5
0,020
3
0,33
Бутонизация, начало
цветения
Край пластинки нижних
листьев
Черешки средних и верхних
листьев
4-4,5
6,0
0,017-0,020
0,071
4
0,042
5
0,54
Начало образования
клубней
4—5 листьев
Черешки средних листьев
5-6
0,022—0,071
2-2,5
0,017-0,020
4—5
0,37—0,54
Столовая
Черешки взрослых листьев
5-6
0,022-0,071
4-5
0,042-0,069
>5
>0,54
свекла
9—10 листьев
То же
5
0,022
4-5
0,042-0,069
>5
>0,54
Рост корнеплода
»
4-5
0,017-0,022
4,0
0,042
5
0,54
Морковь
3—4 листа
Пластинка зачаточного листа
Черешки взрослого листа
3,5-4
>6
0,016-0,017
>0,071
4-5
0,042—0,069
>5
>0,54
Пучковый товар
Пластинка зачаточного листа
Черешок взрослого листа
2-3
>6
0,007—0,015
>0,071
3-4
0,022-0,042
5
0,54
Капуста
Сентябрь
То же
5
0,022
3
0,022
4—5
0,37-0,54
До завязывания кочана
Главная жилка и черешок
взрослых листьев
6
0,071
3,5
0,033
4
0,37
Образование кочана
То же
4
0,017
3
0,022
3,5-4
0,35-0,37
Огурцы
4 листа
Край нижнего листа
Черешок нижнего листа
3-4
>6
0,015-0,017
>0,071
3-4
3,5
0,022—0,042
0,033
3
4-5
0,33
0,37-0,54
Начало цветения
Черешок среднего листа
6
0,071
4,5
0,055
4,5
0,46
Начало плодоношения
То же
5-6
0,022—0,071
4
0,042
3,5
0,35
Томаты
Июнь
5-й лист сверху
>6
>0,071
3
0,022
—
—
Июль
То же
4,5
0,020
3
0,022
—
—
Август
»
3,5
0,016
2,5
0,020
* См. сноску к табл. 19 (урожаи).
Диагностика питания растений по их химическому анализу
523
Таблица 22
Содержание питательных веществ в почве и в листьях хлебных злаков в фазу колошения
как показатель нуждаемости в удобрениях
Группа по
обеспечен¬
ности
питанием
Урожай,
% от
оптималь¬
ного *
Степень нуждае¬
мости в удобре¬
ниях
Содержание элементов
в почве, мг/кг **
Содержание в листьях злаков,
% в сухом веществе
N-N03
Р
К
N
р
К
I
30
Очень сильная
<5
8,7
25
<1 ,6
<0,18
<1,2
II
31-40
Сильная
8
22,0
83
1,6—2,2
0,18-0,23
1,2-1,4
III
41—50
Средняя
9-15
23-44
84-125
2,3-2,6
0,24-0,27
1,5-1,9
IV
51-90
Слабая
16-30
45—66
126—166
2,7-2,9
0,28-0,29
2,0-2,4
V
100
Отсутствует
31—60
67—87
167—250
in
со
1
о
со
0,30-0,36
2,5-2,9
VI
<100
Избыток
>60
>87
>250
>3,5
>0,36
>2,9
* За 100% принят урожай 40 ц/га зерна.
** N—N0* по интенсивности нитрификации, Р — по Ф. В. Чирикову, К — по A. JI. Масловой.
Люндегорд (Liindegardt, 1945) рекомендовал анализировать хлебные зла¬
ки в фазу цветения. По Байеру, следует вносить удобрения не менее чем за
20 дней до отбора проб. Он также рекомендует учитывать условия произрас¬
тания. Анализирует общее содержание в растениях N, Р, К, Са и Mg. Для
оценки результатов анализов об уравновешенности питания вычисляет сле¬
дующие отношения: N : Р, 100 К : N, 100 Са : Р и 100 Mg : Р, величины ко¬
торых наряду с процентным содержанием элементов приводит в таблицах.
Последние используются и при расчете доз удобрений. Он считает, что чем
сильнее относительный недостаток какого-либо элемента, тем хуже ис¬
пользуются растением другие элементы и, следовательно, тем ниже урожай.
Байер на основе многочисленных вегетационных и полевых опытов и
производственных испытаний составил инструкцию по практическому ис¬
пользованию анализов молодой пшеницы, пробы которых, отобранные аг¬
рономами, поступают в специализированные лаборатории для их анализа
и составления заключения о потребных дозах удобрений.
Не менее широко развертываются работы по использованию анализов
растений для рекомендаций по внесению удобрений в ряде других стран.
Так, в ГДР эти работы возглавляются Институтом питания растений (Berg-
mann, 1968; Gollmick, Neubert, Vielemeyer, 1970), где,так же как и в других
странах, отрабатываются все этапы работы: стандартизация отбора проб,
применение наиболее совершенных и быстрых методов анализа растений с
последующей обработкой результатов на ЭВМ для получения более быстрых
и обоснованных выводов о потребных удобрениях.
Н. К. Болдырев (1958, 1972) называет предлагаемый им метод листовой
диагностики комплексным, так как тоже строит заключение о нуждаемости
в удобрениях на сопоставлении результатов исследований свойств почвы,
анализов листьев и зерна, вводит ряд коэффициентов на условия погоды,
агротехники и др. Известно положение Люндегорда (Lundegardt, 1945),
что в создании урожая участвуют только те количества элементов, которые
находятся в оптимальном и равновесном соотношении; Болдырев считает,
что из внесенных удобрений прежде всего действует элемент, находящийся
в минимуме. Им обоснован ряд математических зависимостей между хими¬
ческим составом растений в какую-либо иэ фаз и величиной будущего уро¬
жая. Болдыревым рассчитана динамика химического состава и связь ее с
урожаем; это позволило автору утверждать, что, имея данные анализа в одну
524
В. В. Церлинг
из фаз, можно установить химический состав в любую другую фазу, а сле¬
довательно, и потребность растений в удобрениях.
Поиски математических зависимостей между условиями питания в тече¬
ние вегетации, свойствами почвы, приемами агротехники, погодой и величи¬
ной и качеством урожая продолжаются многими исследователями. Материалы
таких исследований, собираемые нами, позволят найти оптимальное
решение этого важнейшего вопроса агрохимии и всей агрономии в целом.
Литература
Айдинян P. X., Иванова М. С., Соловьева
Т. Г. Методы извлечения и определения
различных форм серы в почгах и расте¬
ниях. М., 1968.
Баннов М. Г. Диагностика потребности
сеянцев и саженцев лиственницы сибир¬
ской в азоте, фосфоре и калии в условиях
приобских боров. (Автореф. канд. дисс.).
Омск, 1972.
делоусов М. А., Торопкина А. Л. Инст¬
рукция для определения потребности
хлопчатника в питательных элементах по
анализам почвы и клеточного сока расте¬
ний. Ташкент, 1960.
Болдырев Я. К. Диагностирование потреб¬
ности яровой пшеницы в азоте и фосфоре
в период цветения по общему химическо¬
му анализу листьев.— Почвоведение,
1958, №11.
Болдырев Я. К. Комплексный метод листо¬
вой диагностики условий питания, вели¬
чины и качества урожая сельскохозяйст¬
венных культур. (Автореф. докт. дисс.).
М., 1972.
Бондаренко А. А., Харитонов О. К. К ме¬
тодике определения общих азота, фосфора
я калия в растительном материале в од¬
ной навеске. —В сб. «Проблемы азота и уро¬
жай в Полесье». Киев, 1967.
Бондарцев А. С. Шкала цветов. М.— JI.,
Изд-во АН СССР, 1954.
Бурчуладзе С. Т. Определение потребности
чайного куста в удобрениях.— В сб.
«Диагностика потребности растений в
удобрениях». М., «Колос», 1970.
Гинзбург К. Е.у Щеглова Г. М., Вульфиус
Е. А. Определение азота, фосфора п ка¬
лия в растительном материале из одной
навески.— Почвоведение, 1963, № 5.
Горшкова М. А. Агрохимическая характе¬
ристика почв по данным анализов почв и
растений.— Труды III делег. съезда поч¬
воведов. М., «Наука», 1968.
Давтян Г. С. Прибор для полевого качест¬
венного определения нитратов в хлопчат¬
нике.— Сов. хлопок, 1939, № 7.
Дзикович К. А. Диагностика борного пита¬
ния подсолнечника и сахарной свеклы.—
В сб. «Диагностика потребности растений
в удобрениях». М., «Колос», 1970.
Журбицкий 3. Я. Физиологические и агро¬
химические основы применения удобре¬
ний. М., «Наука», 1963.
Кенуорти А. Истолкование показателей
состава листьев плодовых деревьев.— В
сб. «Анализ растений и проблемы удобре¬
ний». Пер. с англ. М., «Колос», 1964.
Кузнецов В. И., Басаргин Я. Я., Мясище-
ва Л. Г. Сера в овощах Московской об¬
ласти.— Агрохимия, 1968, № 8.
Лагутинская Я. А. Листовая диагностика
минерального питания винограда.— В
сб. «Диагностика потребности растений в
удобрениях». М., «Колос», 1970.
Липкинд И. М. Новый метод определения
потребности хлопчатника в азотном удоб¬
рении.— Сов. хлопок, 1939, № 7.
Магницкий К. Я. Упрощенные полевые ме¬
тоды определения потребности растений в
удобрениях по химическому анализу их
сока.— В сб. «Агрохимические исследо¬
вания почв». М., Изд-во АН СССР,
1954.
Магницкий К. Я. Полевой контроль пита¬
ния растений. М., «Знание», 1959.
Магницкий К. Я. Диагностика потребнос¬
ти растений в удобрениях. М., «Москов¬
ский рабочий», 1972.
Магницкий К. Я., Щугаров Ю. Л., Мал-
ков В. К, Новые методы анализа расте¬
ний и почв. М., Сельхозгиз, 1959.
Одзаки К. Аспарагин как критерий необхо¬
димости подкормки риса в полевых усло¬
виях.— В сб. «Анализ растений и проб¬
лемы удобрений». Пер. с англ. М., «Ко¬
лос», 1964.
Панков В. В. Некоторые вопросы методи¬
ки листовой диагностики питания расте¬
ний.— В сб. «Диагностика потребности
растений в удобрениях». М., «Колос»,
1970.
Пиневич В. В. Определение N и Р в расти¬
тельном материале из одной навески.—
Докл. ВАСХНИЛ, 1955, вып. 1.
Полуэктов Я. С. Капельная реакция на ка¬
лий.— Калий, 1933, № 10.
Прево Я., Олланье М. Закон минимума и
сбалансированное минеральное пита¬
ние. — В сб. «Анализ растений и пробле¬
мы удобрений». Пер. с англ. М., «Колос»,
1964.
Сабинин Д. А. Принципы и методика изу¬
чения минерального состава пасоки.—-
Бюлл. отдела землед. Гос. ин-та опытной
агрономии, 1928, № 15.
Церлинг В, В. Влияние азотного питания
на обмен веществ, развитие репродуктив¬
ных органов и урожай яровых злаков.—
В сб. «Памяти акад. Д. Н. Прянишнико¬
ва». М., Изд-во АН СССР, 1950.
Диагностика питания растений по их химическому анализу
525
Церлинг В, В, О диагностике потребности
растений в калии.— Почвоведение, 1956а,
№ 6.
Церлинг В. В, Метод микрохимической ди¬
агностики потребности растений в фос¬
форе.— Почвоведение, 19566, № 9.
Церлинг В. В, Методика диагностирования
потребности растений в азоте.— Почвове¬
дение, 1958, № 1.
Церлинг В. В, Исследования по проверке
методов растительной диагностики.— В
сб. «Методические указания по географи¬
ческой сети полевых опытов с удобрения¬
ми», вып. 2. М., Сельхозгиз, 1959.
Церлинг В. В. Метод химической диагнос¬
тики потребности растений в удобрени¬
ях.— Труды Эстонской с.-х. академии,
1962, т. 24.
Церлинг В, В, О химической диагностике
потребности в удобрениях садовых куль¬
тур.— В сб. «Методические указания по
географической сети полевых опытов с
удобрениями», вып. 9. М., Сельхозгиз,
1963а.
Церлинг В. В. Роль азотного питания в
формировании урожая.— Химия в сель¬
ском хоз:ве, 19636, № 1.
Церлинг В. В. Принципы применения ме¬
тодов растительной диагностики для оп¬
ределения потребности многолетников в
удобрении.— Агрохимия. 1970, № 4.
Церлинг В. В., Горшкова М. А. Исполь¬
зование анализов растений для характе¬
ристики доступности азота, фосфора и
калия почвы. — Почвоведение, 1971,
№5.
Церлинг В. В., Горшкова М. А. Методи¬
ческие указания по диагностике мине¬
рального питания кормовых, овощных и
полевых культур. М., «Колос», 1972.
Церлинг В. В., Горшкова М. Л., Зинке¬
вич А. С. Современные задачи диагности¬
ки потребности растений в удобрениях.—
В сб. «Д. Н. Прянишников и вопросы
химизации земледелия». М., «Колос»,
1967.
Церлинг В. В., Егорова JI. А. Листовой
анализ как показатель урожайности и
обеспеченности взрослых яблонь пита¬
нием.— Тезисы докл. Всес. совещания
по удобрению садовых культур. Киши¬
нев, 1971.
Церлинг В. #., Ерофеев А. А. Диагности¬
ка обеспеченности серой злаковых, бо¬
бовых и крестоцветных культур.— Агро¬
химия, 1973, № 7.
Церлинг В. В., Зинкевич А. С. Содержа¬
ние свободных аминокислот в тканях как
показатель обеспеченности растений азо¬
том.— Агрохимия, 1967, № 9.
Baier J. Zavislost vynsu zrnaovsa na odberu
Najeho pomeri k P205.— Rostlinna vyro-
ba, 9 1965. J
Baier J. Vyuziti moznosti dohnojovani pse
nice podle vysledky.fanorganickeho rosbo-
ru roslin.— Arch. VMRV-MRV. Praha —
Rusyne, 1972.
Bergmann W. Bedeutung der Mikronahr-
stoffe in der Landwirtschaft. Fortschrifts-
berichte fur die Landwirtschaft, 1968. Bd.
6, H. 2/3.
Chapman H. D, Plant analysis value sugges¬
tive of nutrient status of selected crops.—
In «Soil testing and plant analysis». Part.2,
Soil Sci. Soc. America, 1967, N 2,
Gollmick F., Neubert P., Vielemeyer H. P.
Moglichkeiten und Grenzen der Pflanzen-
analyse bei der Ermittlung des Minerals-
toffbedarfs landwirtschaftlicher Kulturpf-
lanzen. Fortschrittsberichte fur die Land¬
wirtschaft und Nahrungsgiiterwirtschaft.
Bd. 8, H. 4, 1970.
Hoffer С. N. Testing corn stalks chemically
to aid in determining their plant food
needs.— Purdue Univ. Agric. Exper. Stat.,
Bull., 1930, № 298.
Krantz В. A.у Nelson W. £., Burhart L. F.
Plant-tissue tests as a tool in agronomic
research. — In «Diagnostic techniques for
soil and crops», Amer. Potash Inst. Was¬
hington, 1948.
Levy J. F. Les bases physiologiques du diag¬
nostic foliare de la vigne. «Le controle de ltf
fertilisation des plantes cultivees». 2. Co-
loq. Europeo у Mediterraneo, Sevilla, Es-
paria, 1968.
Lagatu Maume L. Diagnostic de l’ali-
mentation d un vegetale par evolution chi-
mique d une feuille convenablement choi-
see.— C. r. Acad. sci. Paris, 1926, t. 182.
Lundegardt H. Die Blattanalyse. Jena, 1945.
Nowosielski О. Metody oznaczania potrzeb
nawozenia. Warszawa, 1968.
Pinta M. Methodes de reference pour la de¬
termination des elementes mineraux dans
les vegetaux. «Le controle de la fertilisa¬
tion des plantes cultivees». 2 Coloq. Euro¬
peo у Mediterraneo. Sevilla, Espana, 1968.
Roach W. A. Preliminary diagnosis of mi¬
neral deficiencies by plant analysis and
plant injection.— Commissions II and IV.
Internat. Soc. Soil Sci., v. 1, 1952.
Routchenko W. Appreciation des conditions
de la nutrition minerale des plantes basee
sur Г analyse des sues extraits des tissus
conducteurs.— Ann. agron., 1967, t. 18, 4.
Schoniger W. Die mikroanalytische Schnell-
bestimmung von Halogenen und Schwefel
in organischen Verbindungen.— Mikro-
chim. acta, 1956, «N® 4—6.
С. В. ЩЕРБА, Ф. А. ЮДИН
МЕТОДИКА ПОЛЕВОГО ОПЫТА С УДОБРЕНИЯМИ
*
Для правильного применения удобрений нужно знать, как действуют раз¬
личные удобрения на урожай культурных растений в определенных поч¬
венно-климатических и культурно-хозяйственных условиях; какие виды и
формы удобрений, в каких дозах и сочетаниях дают наибольшие прибавки и
улучшают качество урожая; когда и какими способами лучше вносить удоб¬
рения? Для получения ответа на эти вопросы необходимо проводить поле¬
вые опыты с удобрениями.
Полевой опыт — это метод исследования, проводимого в природной по¬
левой обстановке на специально выделенном участке с целью установления
количественного воздействия условий или приемов возделывания на урожай
сельскохозяйственных растений и его качество.
Полевой опыт относится к методам биологическим: в основе лежит реак¬
ция живого организма на изменение тех или иных факторов его развития.
Он в наибольшей степени отвечает тимирязевскому правилу: «спрашивать мне¬
ние самого растения». Реагирующим организмом-индикатором является в
опыте то самое культурное растение, для которого предназначаются полу¬
чаемые данные, а реакция опытного растения изучается в нем в естественных
почвенных и метеорологических условиях.
Полевой опыт дает количественную, приложимую к производственным
условиям, характеристику эффективности действия удобрения. Поэтому он
справедливо рассматривается как конечное звено в системе агрохимических
исследований. Полевой опыт является не только завершающим этапом иссле¬
дования, но и мостом, соединяющим сельскохозяйственную науку с сельско¬
хозяйственной практикой.
Однако зависимость результатов полевого опыта от конкретных почвен¬
ных и метеорологических условий является в то же время моментом, огра¬
ничивающим его значение. Прежде всего результаты полевого опыта без
точной характеристики почвы, на которой он проведен, приложимы, строго
говоря, лишь к тому участку, на котором они получены. Другой ограничи¬
вающий момент — трудность, а иногда и невозможность такого детального
аналитического расчленения отдельных природных факторов и их искусст¬
венного регулирования, которые допускают другие методы, в частности ве¬
гетационный.
Поэтому необходимо и целесообразно комбинировать полевой опыт с дру¬
гими методами исследования: почвенными, химическими и биологическими.
Применение почвенных исследований дает возможность устанавливать ти¬
пичность участка полевого опыта для определенного района или зоны, а
следовательно, и распространять на них полученные в полевом опыте резуль¬
таты. Химические методы (анализы почвы и растения) позволяют судить не
только о конечном результате изменения питательного режима почвы, от¬
ражаемом величиной урожая, но и о самих изменениях форм и количества
отдельных элементов пищи растения в почве, а также о влиянии удобрений
на качество урожая. Наконец, вегетационный метод, благодаря детальному
расчленению факторов и возможности искусственного их регулирования.
Методика полевого опыта с удобрениями
527
позволяет быстрее подметить определенные закономерности, получающие
затем количественное выражение в естественных условиях полевого опыта.
В зависимости от цели, места постановки, длительности опыта и размера
делянок полевой опыт делится на несколько видов. Так, возможно деление
полевого опыта в зависимости от того, ставится он в условиях специально
приспособленного опытного поля или в условиях сельскохозяйственного
производства.
Первый вид опыта имеет целью углубленное изучение основных факторов
и приемов культуры сельскохозяйственных растений и комбинаций этих
факторов. Он строится с детальным расчленением отдельных факторов и
обычно тесно увязывается с другими методами исследования. Выводы его
предназначаются для значительного района или почвенной зоны. Обычной
(но не обязательной) особенностью этого опыта является проведение его
на делянках сравнительно небольшой величины.
Опыт в производственных условиях в большинстве случаев ставится по
более упрощенным схемам, включающим лишь основные, наиболее важные
варианты. Эти опыты особенно важны для экономической и организационно¬
хозяйственной оценки технически уже разработанных приемов или их ком¬
бинаций.
В особую категорию можно выделить так называемые географические и
коллективные опыты. Они необходимы для решения вопросов, имеющих
значение для ряда почвенных и климатических районов, но требующих диф¬
ференцированного решения применительно к условиям каждого отдельного
района. Они могут охватывать как очень большие территории, иногда даже
целые страны, так и отдельные, сравнительно небольшие, но разнообразные
по условиям области.
По географическому принципу могут вестись опыты и в стационарных
опытных учреждениях, и в производственных условиях. Они ставятся в ряде
пунктов, наиболее полно охватывающих все разнообразие изучаемой облас¬
ти, по одинаковой, заранее согласованной схеме.
Под коллективными массовыми опытами понимают обычно опыты в про¬
изводственных условиях, поставленные по одинаковой схеме в ряде хозяйств.
Они дают возможность, во-первых, повысить относительно малую точность
каждого отдельного опыта за счет их многочисленности и, во-вторых, уста¬
новить влияние на эффективность изучаемых приемов почвенных, климати¬
ческих и организационно-хозяйственных особенностей отдельных хозяйств
или их групп.
В зависимости от длительности периода наблюдения за действием удобре¬
ний различают однолетние и многолетние, или длительные, полевые опыты.
В однолетних опытах учитывается действие удобрения только на ту культуру,
под которую оно внесено. В многолетних опытах действие удобрений просле¬
живается в течение ряда лет на нескольких следующих одна за другой куль¬
турах. В них может учитываться либо действие раз внесенных удобрений
(чистое последействие), либо накапливающееся действие систематически
повторяемого через определенные сроки внесения удобрений. Многолетние
опыты, как правило, ведутся в определенном севообороте, имеющем в натуре
несколько клиньев. Опыт, закладываемый в течение нескольких лет каж¬
дый год на новом участке, но учитываемый на каждом участке только один
раз, нужно считать все же однолетним опытом. Опыт, учитываемый в тече¬
ние ряда лет на одном участке, лишь при чередовании на нем культур дол¬
жен считаться многолетним, хотя и неудовлетворительно организованным с
точки зрения методики.
В зависимости от размера делянки различаются крупноделяночные и
мелкоделяночные опыты. Существенной для этого подразделения является,
однако, не величина делянок, а возможность применения нормальной поле¬
вой агротехники. Мы относим к мелкоделяночным опыты с таким малым
528
С. В. Щерба, Ф. Л. Юдин
размером делянок, который не позволяет поставить изучаемый фактор в ус¬
ловия нормальной сопутствующей агротехники и заставляет прибегать к ис¬
кусственным приемам, которые могут существенно изменить высоту урожая
и эффективность изучаемого фактора. Все опыты, проводимые с соблюдением
нормальных приемов полевой агротехники, мы относим к обычным, или нор¬
мальным, полевым опытам, независимо от абсолютных размеров делянки.
ОСНОВНЫЕ ПОНЯТИЯ,
ВСТРЕЧАЮЩИЕСЯ В МЕТОДИКЕ ПОЛЕВОГО ОПЫТА
Полевой опыт всегда закладывается по определенной схеме.
Схема полевого опыта — совокупность определенного числа
вариантов. Каждый из них характеризуется видоизменением того фактора,
который изучается в данном опыте. Примером простейшей схемы опыта мо¬
жет быть схема из двух вариантов; например, первый вариант — без удобре¬
ний, второй — с удобрением.
Вариант опыта — определенная совокупность приемов возделы¬
вания растений, осуществляемая на одной делянке или на нескольких так
называемых повторных делянках. Вариант есть составная часть схемы опыта,
обозначаемая тем фактором, который изучается в опыте. Один из вариантов
схемы опыта, с которым сравнивают результаты, полученные в других ва¬
риантах, называется контрольным (стандартным) или контро¬
лем. Он позволяет определить степень чувствительности растений к изу¬
чаемому в опыте фактору. Варианты опыта размещаются на делянках опыт¬
ного участка по определенному плану.
Опытная делянка — элементарная составная часть опытного
участка определенного размера и формы, на которой осуществляются все
изучаемые приемы возделывания растений согласно какому-нибудь одному
из вариантов схемы опыта.
Каждый из вариантов схемы принято размещать повторно на нескольких
делянках, отсюда возникает понятие «повторность».
Повторностью опыта в пространстве называют число одно¬
именных делянок каждого варианта. Часть площади опытного участка, за¬
нятую полным набором делянок всех вариантов схемы опыта, расположенных
рядом друг с другом, называют повторением опыта.
Блок — часть площади участка полевого опыта, поделенного на де¬
лянки, на котором размещают варианты схемы опыта случайными методами.
Блок может быть полным, тогда он равнозначен повторению, или неполным —
когда в блок входит лишь часть вариантов, в последнем случае несколько
блоков составляют одно повторение.
ОСНОВНЫЕ МЕТОДИЧЕСКИЕ ТРЕБОВАНИЯ
К КАЧЕСТВУ ПОЛЕВОГО ОПЫТА
К любому полевому опыту предъявляется ряд основных методических
требований: 1) наличие сравнимости и соблюдение принципа единственного
различия; 2) типичность опыта; 3) точность количественных результатов
опыта; 4) достоверность.
Наличие сравнимости и соблюдение принципа
единственного различия — одно из требований методики по¬
левого опыта, которое следует учитывать при разработке программы и пост¬
роения схемы полевого опыта. Программа и схема должны быть составлены
так, чтобы на основании сравнения урожаев и наблюдений за развитием
растений на делянках разных вариантов можно было сделать определенный
Методика полевого опыта с удобрениями
529
вывод, получить ответ на поставленный вопрос, имеющий практическое
значение для сельскохозяйственного производства.
Одним из условий методически правильно поставленного опыта явля¬
ется соблюдение принципа единственного логического различия, т. е. тре¬
бования, чтобы сравниваемые варианты различались одним изучаемым
в опыте фактором. Другие факторы, оказывающие влияние на урожай,
у сравниваемых вариантов должны быть одинаковыми. Так, при изучении
действия удобрений необходимо, чтобы обработка почвы на всех делянках
опыта была одинаковой, посев проведен в один срок семенным материалом
одинакового качества, чтобы на всех делянках применялась одна и та же
система ухода за растениями и т. д. Цель этого требования — обеспечить
сравнимость данных, полученных в разных вариантах опыта, например для
суждения об эффективности удобрений. В опытах с удобрениями соблюдение
принципа единственного различия требует не только, чтобы все остальные
факторы жизни растений и приемы агротехники были одинаковыми, но что¬
бы разные формы удобрений испытывались при одинаковых дозах, эффектив¬
ность различных доз какого-либо удобрения изучалась в одной и той же фор¬
ме и т. д.
Логический принцип единственного различия на первый взгляд кажется
совершенно ясным, но соблюдение его подчас вызывает огромные трудности.
Невозможность строгого сохранения одинаковыми всех прочих условий,
кроме изучаемого, определяется тесной связью и взаимозависимостью раз¬
ных факторов жизни растений и почвы и действующих на них агротехни¬
ческих приемов. Дело в том, что если при изучении жизни растений в при¬
роде мы выделим какой-либо один фактор и, варьируя его, захотим просле¬
дить его влияние на рост, развитие и урожай растения при равенстве всех
прочих факторов, то неизбежно будем в какой-то мере затрагивать и другие*
факторы. Например, меняя влажность почвы, мы обязательно изменяем ее
воздушный и температурный режимы. Последние, в свою очередь, вызовут
те или иные изменения в микробиологической деятельности почвы, которая
тесным образом связана с пищевым режимом. Вообще, изучая влияние того
или иного фактора на жизнь растения, мы можем лишь в определенном до¬
пустимом интервале изменять степень его воздействия на растение (вода,,
воздух, элементы питания и т. д.), а не исключать полностью.
В связи с этим надо указать, что действительное сохранение без измене¬
ния всех прочих условий, кроме исследуемых по схеме, при проведении по¬
левого опыта в полной мере не всегда оказывается возможным. Иногда стрем¬
ление к формальному соблюдению этого требования может приводить к за¬
ведомо неправильным методам сравнения. Например, действие азотного и
фосфорного удобрений на урожай озимых формально, казалось бы, надо
изучать в одинаковых условиях, при одном и том же сроке и способе приме¬
нения. Но такой путь мог бы быть верным, если бы не было известно значение-
срока и способа внесения этих удобрений для озимых культур. Агрономи¬
чески более правильным и целесообразным надо признать такое построение-
схемы опыта, при котором каждое удобрение применяется в наиболее под¬
ходящий для него срок, например в опытах с озимыми фосфорное удобре¬
ние — до посева (или в рядки при посеве), а азотное — весной в подкормке.
Это не нарушение принципа единственного различия, так как здесь вид удоб¬
рения, оптимальный срок и способ его внесения составляют единый комплекс,,
который и будет вариантом опыта. Сравнение вариантов, включающих ком¬
плекс условий, будет проводиться при тождестве всех прочих условий, кото¬
рые не являются составными элементами сравниваемых комплексных приемов.
Требование необходимости соблюдения тождества всех факторов, кроме-
исследуемого в опыте, не должно вести к искусственному ограничению усло¬
вий, при которых тот или иной прием, изучаемый в полевом опыте, может
проявить свое наибольшее действие. Приведем пример, наглядно иллюстри¬
530
С. В. Щербйу Ф. Л. Юдин
рующий высказанное положение. При постановке полевых опытов по срав¬
нительной оценке разных форм минеральных удобрений чаще всего сравне¬
ние ведут при одинаковых сроках внесения удобрений. Допустим, что срав¬
ниваются две формы калийных удобрений (калий хлористый и калий серно¬
кислый), применяемых под культуру, чувствительную к избытку хлора.
Если обе формы вносить только осенью или только весной, результаты срав¬
нения могут дать неравнозначный ответ. Для получения объективной оцен¬
ки по каждой форме удобрения надо в схеме опыта иметь дополнительные
варианты с разными сроками внесения.
Игнорирование взаимной связи между испытываемыми приемами и прочи¬
ми условиями проведения опыта может привести к тому, что при кажущемся
тождестве всех прочих условий опыта оценка изучаемых приемов получится
неправильной. Во всех случаях, когда неравенство сопутствующих агротех¬
нических условий вызывается изучаемыми приемами, оно должно быть пре¬
дусмотрено программой опыта и точно зафиксировано в документации по по¬
левому опыту. Если же отклонение от принципа единственного различия не
предусмотрено программой или схемой опыта, то оно недопустимо и нали¬
чие его при оценке результатов опыта должно рассматриваться как серьез¬
ное нарушение методики полевого опыта. Таким образом, вопросы построе¬
ния схемы и программы полевого опыта не должны решаться формально:
способ решения их зависит от цели и задачи опыта, степени изученности воп¬
роса и конкретных условий, в которых проводится опыт.
Требование типичности, или, как иногда говорят, репрезентатив¬
ности, опыта включает соответствие условий проведения опыта той ок¬
ружающей обстановке, где предполагается использовать его результаты.
Различают типичность опыта в отношении природных, а также организа¬
ционно-хозяйственных, агротехнических условий. Требование природной
типичности заключается в соответствии условий проведения опыта почвен¬
ным и климатическим условиям района или хозяйства, для которого пред¬
назначаются результаты опыта.
Для полевых опытов с удобрениями почвенно-климатические условия
имеют исключительно важное значение. Отсюда вытекает необходимость
изучения многих вопросов удобрения в географическом разрезе. Кроме того,
даже в одном хозяйстве мы встречаемся с большим разнообразием почвен¬
ного покрова, поэтому полевые опыты надо закладывать на типичных поч¬
венных разновидностях с учетом механического состава, содержания гу¬
муса, степени кислотности, обеспеченности подвижными формами питатель¬
ных веществ. Требование типичности климатических условий обычно вызы¬
вает необходимость проведения опыта по одной теме в течение 3—4 лет, так
как в отдельные годы могут наблюдаться значительные отклонения в погоде
(сильная засуха, холодная дождливая погода).
Требование типичности организационно-хозяйственных условий опыта
более сложно, чем природной типичности. Не всегда удается четко выявить,
что считать типичным в производственном отношении при возделывании оп¬
ределенной культуры в хозяйстве. Для опытов с удобрениями несомненно
большое значение имеют севооборот и предшественники, уровень плодоро¬
дия, степень обеспеченности навозом, условия обработки и ухода за посе¬
вами. Легче и полнее выдержать условие производственно-агротехнической
типичности можно в полевых опытах, проводимых в условиях производства,
чем в опытах стационарных, закладываемых на опытном поле, где иногда
при первоначальном исследовании того или иного вопроса допустимо неко¬
торое отступление от типичных производственных условий.
Производственно-агротехническая типичность не всегда будет соответ¬
ствовать той агротехнике и тем организационно-хозяйственным условиям,
которые в данный момент наблюдаются в хозяйстве при возделывании той
или иной культуры. Каждый полевой опыт должен давать перспективный от¬
Методика полевого опыта с удобрениями
531
вет с учетом того, что когда результаты опыта будут внедрены в производст¬
во, будет достигнут известный прогресс в агротехнических и организацион¬
но-хозяйственных условиях зоны, района или хозяйства. Поэтому типичный:
агротехнический фон для опытов с удобрениями должен быть достаточно вы¬
соким.
В опытах с удобрениями надо особенно тщательно выбирать соответст¬
вующий фон, от которого зависит эффективность удобрений. Иногда следует
создавать два или несколько фонов (без навоза и с навозом, без известкова¬
ния и по фону извести и т. д.). В понятие типичности входит также пригод¬
ность фона для исследования того или иного вопроса. Неверным и нетипич¬
ным будет изучение эффективности фосфоритной муки на почве, незадолго
до этого произвесткованной.
Большое значение имеет закладка опыта по лучшим и типичным для дан¬
ной культуры предшественникам (пласт для льна и яровой пшеницы, пары
чистые или занятые для озимых и т. д.). Не менее важную роль в отношении
типичности для опытов с удобрениями играет выбор типичных для данной зо¬
ны (района) культур, а также районированных сортов.
Правильный выбор культуры — очень ответственная задача. При изу¬
чении разных по растворимости и доступности новых форм фосфатов не сле¬
дует брать растения, обладающие высокой растворяющей способностью корне¬
вых выделений; при изучении форм азотных удобрений с различной физио¬
логической кислотностью надо выбирать культуры, чувствительные к изме¬
нению реакции среды (клевер, ячмень, свекла, некоторые овощные и т. д.).
Иногда следует закладывать опыты с несколькими биологически различ¬
ными растениями. Безусловно, необходимо для соблюдения агротехни¬
ческой типичности при проведении опытов строго придерживаться раз¬
работанной агротехники культур (обработка почвы, время посева, норма вы¬
сева, борьба с сорняками, своевременные меры ухода за посевами, чисто¬
сортные семена, отсутствие болезней и вредителей и т. д.).
Точность количественных результатов — обяза¬
тельное требование к качеству полевого опыта. Результат его всегда выра¬
жается количественно и служит объективным показателем эффективности:
изучаемого в опыте приема или фактора. Большинство агротехнических
приемов и факторов, кроме влияния на величину урожая, оказывает также-
определенное действие на его качество (например, содержание белка в зер¬
не, крахмала в клубнях картофеля, сахара в сахарной свекле). Оно может
быть установлено химическим анализом урожаев с разных вариантов и срав¬
нением полученных результатов, как и при определении различий в вели¬
чине урожаев.
Оценка влияния того или иного приема на качество урожая также может
быть выражена количественно (в центнерах белка, крахмала, сахара на 1 га).
Количественные результаты полевого опыта, проведенного в строгом соот¬
ветствии с задачами исследования, с соблюдением требований методики и
техники, всегда оказываются лишь некоторым приближенным выражением
истинных результатов. Степень соответствия результатов, полученных в-
опыте, истинным результатам действия изучаемого приема или фактора опре¬
деляет точность опыта. Чем меньше разница между результатами, получен¬
ными в опыте (учетные данные), и истинными, тем выше точность опыта и
тем меньше его ошибка.
Причины расхождения фактически полученных в полевом опыте данных с
истинными связаны с неизбежными погрешностями, которые имеют место в
любом полевом опыте. Одной из причин погрешностей или ошибок в полевом
опыте является неточность измерений, которая допускается при определе¬
нии площади делянки, взвешивании удобрений перед их внесением и конеч¬
ного урожая при учете. Эти ошибки могут зависеть от неточности применяе¬
мых инструментов и приборов (мерная лента, угломерные приборы, весы в
532
С. В. Щерба, Ф. Л. Юдин
разновесы). Чем меньше делянка, тем точнее должны проводиться все изме¬
рения и взвешивания, так как одинаковая абсолютная ошибка будет отно¬
сительно большей при меньшей делянке.
Вторая и наиболее существенная причина ошибок в полевом опыте — не-
выравненность исходного почвенного плодородия опытного участка, которая
обусловлена пестротой в распределении почвенных разновидностей, влия¬
нием рельефа и микрорельефа участка, а также неодинаковой предшествую¬
щей историей участка (обработка, удобрение, посев разных сельскохозяй¬
ственных культур).
Выбор формы и величины делянок, их расположения, а также необходи¬
мой в опыте повторности направлен главным образом на максимальное сни¬
жение ошибки, обусловленной исходной пестротой в почвенном плодородии
опытного участка.
Наконец, ошибки опыта могут быть также вызваны чисто случайными
причинами: огрехами и просевами при закладке опыта, повреждением опыт¬
ных делянок (потрава скотом, вытаптывание и т. п.), потерями урожая.
Для устранения их влияния на результаты опыта чаще всего применяют
выбраковку некоторых делянок или выключку частей делянок, подвергшихся
случайному повреждению.
Для установления точности полевого опыта результаты его математиче¬
ски обрабатывают с использованием методов вариационной статистики.
Требование к точности полевого опыта зависит от задач и темы опыта,
вида полевого опыта, а также величины ожидаемого эффекта. От опытов ста¬
ционарных, основных, длительных требуется более высокая точность, чем
от исследований рекогносцировочных, краткосрочных, проводимых в ус¬
ловиях производства.
Если в полевом опыте при изучении эффективности различных видов удоб¬
рений ожидаются большие прибавки урожая, то требования к точности мо¬
гут быть меньшими, чем тогда, когда приходится устанавливать небольшие
различия в вариантах опыта, например при сравнении различных форм удоб¬
рений. В последнем случае требования к точности опыта должны быть бо¬
лее высокими.
Полевой опыт должен также отвечать требованиям достоверно с-
т и. Достоверность и точность опыта — понятия, тесно связанные между со¬
бой, но не идентичные. Принято различать достоверность полевого опыта по
существу, т. е. соответствие опыта поставленным задачам исследования. Кро¬
ме того, различают понятие достоверности, или существенности, результатов
полевого опыта.
Для оценки достоверности полевого опыта по существу проводят агроно¬
мический анализ его материалов, т. е. критический разбор и проверку пра¬
вильности схемы полевого опыта, данных сопутствующих наблюдений и ис¬
следований, результатов учета урожая. Проверяют соответствие методики
опыта задачам исследования, тщательно анализируют методику и технику
проведения полевого опыта.
Если полевой опыт проведен методически и технически доброкачественно и
нет оснований для выбраковки полученных в нем данных, результаты его
подвергают математической обработке для установления величины случай¬
ной ошибки и степени точности, а также достоверности, или существенности,
полученных результатов. Под существенностью результатов понимают мате¬
матическую (статистическую) доказанность получаемой в опыте разницы в
урожаях сравниваемых между собой вариантов опыта. Статистическая обра¬
ботка результатов полевого опыта позволяет определить границы возможных
случайных отклонений полученных данных и установить наличие сущест¬
венных различий между средними урожаями по вариантам опыта.
Чтобы показать различие между точностью и достоверностью результа¬
тов опыта, приведем два примера. В первом опыте сравнивалась эффектив¬
Методика полевого опыта с удобрениями
533
ность двух форм фосфорных удобрений, которые при сравнительно высокой
точности опыта не обнаружили заметных различий между величинами сред¬
них урожаев, полученных по разным вариантам. Различие в средних урожа¬
ях сравниваемых вариантов оказалось меньше, чем вычисленная средняя
ошибка опыта. Следовательно, в данном случае разница между изучаемыми
формами несущественна. Полученный в опыте результат математически не¬
достоверен, находится в пределах возможной ошибки опыта. В другом опыте,
где сравнивалось действие удобрения с неудобренным контролем, ошибка
опыта была вдвое выше, чем в первом опыте, но различие в урожаях двух
сравниваемых вариантов оказалось очень большим и во много раз превос¬
ходило вычисленную среднюю ошибку опыта. Следовательно, здесь при мень¬
шей точности опыта получена математически доказанная достоверная раз¬
ность.
Статистическая обработка результатов полевого опыта имеет чрезвычай¬
но большое значение; однако нужно ясно представлять себе, что она дает
лишь объективную оценку точности опыта, но не повышает сама по себе
эту точность, всецело зависящую of методики постановки и тщательности
проведения опыта г.
МЕТОДИКА И ТЕХНИКА ПОЛЕВОГО ОПЫТА
Говоря о методике и технике полевого опыта, мы будем иметь в виду
прежде всего методику стационарного опыта в условиях опытного поля, для
которого, в сущности, только и имеется разработанная методика, и лишь
попутно, касаясь отдельных элементов методики, будем отмечать некоторые
упрощения и видоизменения, вызываемые условиями опыта в производстве.
Выбор участка
При выборе участка для отдельного опыта нужно стремиться к его воз¬
можно большей однородности.
Рельеф. Наличие ровной поверхности — одно из основных условий
.пригодности участка для опыта. При постановке опытов на склонах делянки,
расположенные в различных частях склона, попадают в неодинаковые поч¬
венные условия и условия увлажнения. При значительном склоне возмо¬
жен, кроме того, смыв почвы и внесенных удобрений с верхних делянок на
нужние. Особенно недопустима постановка многолетних опытов с озимыми
на склонах, подвергающихся действию весенних вод. Однако выбор идеаль¬
но плоского горизонтального участка сколько-нибудь значительной площа¬
ди возможен лишь в условиях степи. В северной части СССР наличие той или
иной степени склона типично для очень значительной части пахотных пло¬
щадей. Идеально ровная поверхность встречается здесь почти исключитель¬
но на осушенных болотах. Поэтому не только трудность выбора участка, не
имеющего склона, но и соображения типичности заставляют допускать здесь
наличие на опытном участке умеренного склона (2,5 впадения на 100 пог.м).
Однако этот склон должен быть односторонним и равномерным. Недо¬
пустимо расположение в пределах участка опыта склонов, обращенных к раз¬
личным странам света, резких изменений крутизны склона и особенно нали¬
чие замкнутых понижений (западин, блюдец).
При расположении опытного участка на склоне делянки должны вытя¬
гиваться длинными сторонами вдоль склона, с тем чтобы каждая делянка по
1 Подробнее о точности и достоверности результатов опыта сказано в статье А. В. Соко¬
лова «Определение точности опыта» в настоящем сборнике.
534
С. В. Щерба, Ф. Л. Юдин
возможности полно и одинаково с другими охватывала разнообразие условий
в разных частях склона. Эти требования к рельефу не относятся, конечно,
к тем случаям, когда влияние рельефа само собой является предметом изу¬
чения в опыте (опыты по изучению влияния склонов различной крутизны и
экспозиции, опыты по изучению влияния эрозии и т. п.).
Для изучения рельефа участка в условиях опытного поля желательна
подробная нивелировка с горизонталями через 0,2 м. При постановке опы¬
тов в условиях производства приходится обычно пользоваться значительна
более грубыми планами, с горизонталями не чаще чем через 1 м или даже
определять направление и крутизну склона на глаз.
Почва. Почвенное обследование опытного участка может иметь двоя¬
кую задачу: а) дать почвенную характеристику участка в целом для того,
чтобы сделать возможным перенесение результатов опыта на сходные почвы;
б) помочь наилучшим образом расположить опыт, разместив его целиком в
пределах одной почвенной разности или, при невозможности этого, в пре¬
делах комплекса наиболее близких разностей при условии возможного
однообразия этого комплекса для всех вариантов опыта.
Первая задача обязательна не только в условиях опытного поля, но и
при постановке опытов в условиях производства. Результаты тщательнейшим
образом проведенного опыта теряют ценность, если неизвестна почва, на ко¬
торой он был поставлен.
Детализация почвенной карты, необходимая для разрешения второй за¬
дачи, зависит от пестроты почвенного покрова и размера делянок, но во вся¬
ком случае должна быть очень велика. Ошибки в нанесении почвенных раз¬
ностей на такой карте не должны превышать наименьшего измерения (т. е.
ширины) делянок, что требует почвенного обследования в масштабе 10—
50 м в 1 еле, осуществимого обычно лишь в опытных полях.
Возникает вопрос, насколько строго должно выполняться требование от¬
сутствия почвенной пестроты в пределах участка опыта. К этому вопросу от¬
носится все, что было уже сказано относительно рельефа. Идеальным будет,
конечно, расположение всех делянок опыта строго в пределах одной почвен¬
ной разности. Это возможно при наличии сравнительно крупных массивов,
занятых одной почвенной разностью, и при небольшой общей площади под
опытом в целом. Однако в северной половине Европейской части СССР, для
которой комплексность почвенного покрова столь же характерна, как и рас¬
члененность рельефа, это оказывается часто невозможным. В этом случае
приходится ограничиваться лишь требованием отсутствия в пределах раз¬
мещения опыта резко различных почвенных разностей. Так, для подзолистых
почв можно мириться с наличием в пределах опыта средне- и слабооподзо-
ленных разностей, но наличие среди этого комплекса заболоченных (вг-
леенных) пятен будет уже недопустимо. В районах частичного засоления не¬
допустимо наличие засоленных пятен среди общего массива незасоленных
почв.
При необходимости закладки опыта на комплексе хотя бы и близких почв
следует так расположить делянки (вытянутой формы), чтобы каждая из них
охватывала весь комплекс почвенных разностей, представленных в пределах
размещения опыта.
Предшествующая история. Севооборот, система обработки
и особенно степень предшествующей заправки навозом и минеральными
удобрениями определяют типичность опытного участка для обслуживаемого
района не в меньшей степени, чем природные условия. Большое значение
имеет предшествующая история также и в отношении однородности опыт¬
ного участка.
Наибольшее значение имеет неоднородность предшествующей истории
при постановке опытов в производственных условиях, где выбор участка
обычно непосредственно предшествует закладке опыта и участок не подвер¬
Методика полевого опыта с удобрениями
535
гается никакой специальной подготовке. В этих условиях непременным тре¬
бованием к участку является однородность обработки, удобрения и пред¬
шествующих культур по крайней мере за 2—3 года, а еще лучше — за пос¬
леднюю ротацию севооборота. Важную роль для однородности и типичности
участка играет строгая однообразность в проведении таких приемов агро¬
техники, которые резко изменяют плодородие почвы и обладают длительным
и значительным последействием. К ним относятся: известкование и гипсова¬
ние; заправка почвы навозом; применение повышенных доз минеральных
удобрений, особенно фосфорных; посевы многолетних бобовых трав; углуб¬
ление пахотного слоя.
Последействие всех этих приемов, конечно, рано или поздно затухает.
Однако длительность этого последействия во всяком случае превышает 2—
3 года, а в некоторых случаях (например, при известковании) может растя¬
гиваться на десятилетия. Поэтому при наличии сведений о применении од¬
ного из этих приемов на какой-то части участка нельзя использовать его
под закладку опыта без предварительного дробного учета, хотя бы со вре¬
мени применения этого приема прошло и более 2 лет.
На точность результатов опыта значительное влияние оказывает сильная
и неравномерная засоренность участка. Сильно засоренные участки, особенно
с ясно выраженными пятнами злостных сорняков (пырея, осота и др.), могут
быть использованы под опыт лишь после предварительной подготовки с
уничтожением сорняков химическими и агротехническими способами. В не¬
которых случаях на участках с естественной растительностью (залежь и
целина) составляют карту распределения растительных сообществ, отражаю¬
щую природные условия роста растений. Эта карта наряду с почвенной и^
планом нивелировки служит для установления пестроты участка при нарез¬
ке делянок.
При изучении истории участка следует также обращать внимание на
случайные факторы, которые сильно нарушают его однородность
и снижают точность результатов будущего опыта. На участке не должно
быть следов земляных работ, засыпанных ям и канав, воронок от снарядов и
бомб, раскорчевок и крупных пней, остатков от строений, бывших токов,
стоянок скота, мест вывозки и хранения навоза, бывших грунтовых дорог.
Не следует располагать опытный участок вблизи водоемов, древесных насаж¬
дений, построек, изгородей, которые создают неравномерность освещения
вследствие затенения, неодинаковые условия влажности почвы и воздуха
из-за повышенного испарения или излишнего накопления снега, задержки
ветра, а также возможности повреждения и засорения опыта.
Участок должен находиться на расстоянии не менее чем 200 м от водоемов,
40—50 м от сплошного леса и отдельных построек, 25—30 м от отдельных
деревьев и 10 м от плотных изгородей. Во избежание повреждений опыта и
влияния на него дорожной пыли участок размещают на расстоянии 10—
20 м от проезжей дороги и изолируют засеянной защитной полосой.
Изучение истории опытного участка диктуется также необходимостью
иметь характеристику типичности участка — природной (рельеф, почвенно¬
генетические особенности) и хозяйственной (степень окультуренности, обес¬
печенность усвояемыми формами питательных веществ, реакция почвы и
т. д.).
Требования к выбору опытного участка в отношении однородности, обес¬
печивающей высокую точность, и в отношении типичности не всегда совпа¬
дают. Например, для нечерноземной зоны Советского Союза, которой свой¬
ственны значительная пестрота почвенного покрова и наличие той или иной
степени склона пахотных угодий, выбор участка спокойного, выровненного
по рельефу и однородного по почвенным условиям, дающего возможность
получения точных данных, не всегда будет достаточно типичен. И наоборот,
участок, полностью удовлетворяющий требованиям типичности, может не
536
С. В. Щерба, Ф. А. Юдин
вполне обеспечивать требуемую точность. Поэтому в каждом конкретном слу¬
чае необходимо согласовывать требования типичности и точности, посту¬
паясь, в допустимых пределах, либо типичностью условий, либо точностью
результатов.
Почвенная карта, результаты химических анализов почвы, нивелировоч¬
ный план, хозяйственная история поля, а в некоторых случаях (целина или
залежь) карта распределения естественной растительности позволяют путем
их сопоставления установить степень пестроты почвенного покрова и соста¬
вить план расположения полевого опыта на определенном участке, наме¬
тить величину, форму и расположение делянок и размещение повторений.
При выборе опытного участка для производственного опыта часто этим и
ограничиваются.
Подготовка участка
Специальная подготовка участка для постановки опытов обычно прово¬
дится лишь в условиях стационарного опытного поля. Подготовка участка
включает две самостоятельные задачи: выравнивание неодинакового плодо¬
родия участка при помощи одного или нескольких сплошных по всему участ¬
ку, так называемых уравнительных посевов и изучение распределения на
площади участка исходной пестроты почвенного плодородия путем дробно¬
го учета рекогносцировочных или разведочных посевов.
Уравнительные посевы. Уравнительные посевы издавна при¬
меняются опытными станциями; тем не менее многие исследователи отри¬
цают их значение, считая, что они ничего не дают в отношении выравнива¬
ния почвенной пестроты. Если это верно или почти верно по отношению к
пестроте, обусловленной природными факторами (рельеф и почва), то по
отношению к пестроте, обусловленной предшествующей историей участка,
значение уравнительных посевов несомненно (за исключением особенно дли¬
тельно действующих приемов вроде известкования).
Кроме выравнивания пестроты участка, уравнительные посевы могут
иметь еще одну важную, но обычно забываемую задачу: доведение плодоро¬
дия и окультуренности участка до заданного уровня. При исключительном
значении, придаваемом в опытах с удобрениями фону, на котором они про¬
водятся, очень часто возникает необходимость искусственного создания или
изменения этого фона как в сторону повышения окультуренности, так иног¬
да и в сторону некоторого понижения исходного плодородия.
Для достижения этих целей подготовительные посевы, являющиеся в то
же время и уравнительными, могут продолжаться несколько лет и включать
самые разнообразные культуры. Возможно создание специальных подгото¬
вительных севооборотов или звеньев севооборота. В этих севооборотах могут
вноситься, в зависимости от исходного и создаваемого уровня плодородия,
навоз или минеральные удобрения или, наоборот, севообороты могут про¬
водиться в течение ряда лет без всякого удобрения. Так, на ДАОС для
участков, подготовляемых под однолетние опыты с удобрениями, роль урав¬
нительных посевов выполняет специальный севооборот со следующим чере¬
дованием культур: 1) чистый пар (20 тп навоза); 2) озимые с подсевом кле¬
вера; 3) клевер (один год пользования); 4) картофель; 5) яровые (половина
поля отводится под опыты). В следующей ротации опыты размещают на дру¬
гой половине поля. Севооборот имеет в натуре все пять клиньев. Следова¬
тельно, опыты накладываются друг на друга только через 10 лет. Такая под¬
готовка погашает последействие однократно внесенных удобрений даже в
высоких дозах. В промежутке между двумя опытами участок подвергается
двукратному воздействию основных приемов агротехники: чистого пара,
клевера, внесения умеренных доз навоза. Это позволяет создавать выше
чем средний уровень плодородия почвы для однолетних опытов и широ¬
Методика полевого опыта с удобрениями
537
кую типичность участка для тяжелых почв северной части нечерноземной
зоны.
Систематический и тщательный осмотр уравнительных посевов дает воз¬
можность провести глазомерную оценку пестроты в развитии растений, вы¬
делить для опыта наиболее выровненную часть опытного участка и исключить
из него те места, которые отличаются большой пестротой. В некоторых слу¬
чаях, когда этого требуют условия проведения опыта, последний уравни¬
тельный посев можно совместить с рекогносцировочным, подвергнув его
дробному учету.
Подготовку опытного участка проведением уравнительных посевов сле¬
дует считать обязательным приемом для любых опытов, закладываемых как
в стационарных условиях, так и в колхозах и совхозах, чтобы обеспечить
получение достоверных по существу и точных результатов.
Некоторые случаи специальной подготовки
участка. Помимо перечисленных общих приемов подготовки участков для
опыта, возможны некоторые специальные приемы подготовки, связанные с
задачами опыта или с особенностями самого участка. Так, в опытах с оро¬
шаемыми культурами необходимым приемом подготовки является плани¬
ровка участка, обеспечивающая равномерность орошения делянок и воз¬
можность тщательной регулировки и учета распределения воды между
делянками.
Так как планировка, т. е. снятие части пахотного слоя в одних местах
и подсыпка в других, сама по себе служит источником пестроты плодородия,
особенно в первые годы после ее осуществления, то дополнительным требо¬
ванием при выборе участка для опытов с орошаемыми культурами является
рельеф, позволяющий ограничиться минимальной планировкой.
В районах избыточного увлажнения необходимым приемом подготовки
некоторых участков под опыты будет их осушение при помощи открытых ка¬
налов или закрытого дренажа (гончарных труб и других типов дрен). В пер¬
вую очередь это относится к опытам на болотах, но может требоваться и на
участках с минеральными почвами, в частности на тяжелых глинах. Устрой¬
ство осушительной системы должно быть точно и тесно увязано с предпола¬
гаемыми величиной, формой и направлением делянок, с тем, чтобы избежать
различного влияния дрен или канав на отдельных делянках. С этой целью
делянки желательно вытягивать поперек дрен, под каждой делянкой должно
проходить по одной или по одинаковому числу дрен (или они должны про¬
ходить по границам делянок). При осушении открытыми канавами пос¬
ледние должны быть расположены так, чтобы все делянки одинаково примы¬
кали к канавам своими узкими концами.
Один из особых случаев представляет подготовка под опыты участков,
раскорчеванных из-под леса. В этом случае основной задачей подготовки
является скорейшее выравнивание пестроты, создаваемой ямами от выкор¬
чеванных деревьев. Для этого необходима тщательная засыпка ям после рас¬
корчевки и уборки пней.
В первые годы после корчевки участки должны получать возможно боль¬
шее число глубоких обработок, что обеспечивает скорейшее выравнивание
почвенной пестроты. Обработка должна производиться на достаточно мощ¬
ной тракторной тяге, с тем чтобы не оставалось непропаханных мест, и соп¬
ровождаться каждый раз тщательной выборкой корней.
Рекогносцировочные посевы и дробный учет. Сущ¬
ность дробного учета заключается в том, что участок засевается сплошь ка-
кой-либо культурой, которая учитывается по отдельным, возможно более
мелким, площадкам. Таким образом устанавливается пестрота плодородия
внутри опытного участка.
Для рекогносцировочного посева чаще всего используют зерновые хле¬
ба, иногда картофель или корнеплоды. Из зерновых удобнее для дробного
538
С. В. Щерба, Ф. А. Юдин
учета яровые культуры (овес), так как на пестроту стояния озимых накла¬
дываются, помимо плодородия почвы, условия перезимовки.
Величина элементарных делянок дробного учета зависит от культуры, ме¬
тода учета и технических возможностей опытного учреждения. Чем меньше
делянки дробного учета, тем детальнее охватывают они пестроту участка и
тем более гибко и разнообразно их можно комбинировать при проектирова¬
нии величины, формы и расположения опытных делянок. При небольших
площадях, подлежащих учету, и значительной пестроте участка можно ре¬
комендовать размер элементарной делянки в 10 м2. При более крупных и
более однородных площадях и при возможности постановки опытов на боль¬
ших делянках (несколько сот квадратных метров) можно допустить и более
крупные делянки дробного учета.
Техника дробного учета сводится к следующему.
Отведенный для дробного учета участок засевают или засаживают с
весны возможно равномернее. Необходимо обратить особое внимание на
правильную работу сеялки, отсутствие просевов и огрехов для зерновых и:
на правильное размещение растений для пропашных. Для пропашных куль-
тур учетные делянки можно выделить осенью простым отсчетом борозд и
растений в рядах при условии, конечно, правильного размещения растений.
Каждую делянку убирают отдельно и тут же на поле взвешивают урожай.
Для зерновых при ручной уборке желательно выделение элементарных де¬
лянок заранее, пока растения еще малы (до выхода в трубку), так как раз¬
бивка по шнуру спелых хлебов очень затруднительна и неточна.
Учетные делянки разбивают колышками и ограничивают бороздками ши¬
риной 10—20 еле, проведенными ручным планетом или мотыгой. Еще лучше
отделять делянки натянутой между колышками проволокой или шпагатом.
По бороздкам или натянутой проволоке делянки восстанавливаются осенью
легко и с полной точностью. Урожай каждой делянки сжинают серпом, свя¬
зывают в один или несколько снопов, которые сейчас же взвешивают.
Чрезвычайно желательно, конечно, обмолачивать урожай каждой делян¬
ки отдельно и определять урожай зерна. Однако при значительной площади
дробного учета это настолько усложняет работу, что приходится ограничи¬
ваться учетом общей массы. Нужно только, чтобы дробный учет производил¬
ся в сухую погоду, когда нет росы.
Значительно упрощает и ускоряет работу дробный учет жнейкой, техника
которого разработана опытным полем Сельскохозяйственной академии им.
К. А. Тимирязева (Дояренко, 1912) и широко применялась на Долгопрудном
опытном поле НИУИФ. Для учета жнейкой участок или часть участка реког¬
носцировочного посева (чаще всего овса) окашивают перед уборкой со всех сто¬
рон для придания ему прямоугольной формы. Обе короткие стороны разби¬
вают колышками на отрезки, соответствующие ширине захвата жнейки. Вдоль
длинной стороны, с колышка на колышек натягивают веревку, по которой
идет рабочее колесо жнейки. После каждого прохода жнейки веревку пере¬
носят на следующую пару кольев, а жнейка возвращается вхолостую. Жней¬
ку устанавливают на сбрасывание четвертой грабли и каждый раз начинают
проход при положении сбрасывающей грабли над ножом.
Каждый сноп, сбрасываемый жнейкой, немедленно связывают и взвеши¬
вают. При аккуратном выполнении работы каждый сноп представляет собой
урожай с совершенно одинаковой площадки, длина которой определяется
проходом жнейки за время полного оборота граблей (около 6 ле), а ширина —
захватом жнейки (около 1,5 м). Изменяя последнюю величину при разбивке
проходов жнейки, можно в известных пределах изменять площадь элемен¬
тарной делянки.
Дробный учет элементарных делянок большого размера можно быстро
проводить самоходным комбайном. В этом случае площадь элементарной де¬
лянки, с которой намолачивают зерно, определяют умножением ширины ре¬
Методика полевого опыта с удобрениями
539
жущего аппарата комбайна (4 м) на принятую длину гона (20—25 м). Для
этого между рядками элементарных делянок, т. е. через каждые 20—25 м,
делают прокосы в поперечном направлении. После уборки каждой элемен¬
тарной делянки комбайн при выходе на новый прокос надо останавливать,
чтобы промолотить сжатый на делянке хлеб. При урожае 20 ц и выше с 1 га
комбайном удается удовлетворительно раздельно обмолачивать делянки в
80—100 м2 (примерно такого же размера, как в большинстве полевых опы¬
тов с удобрениями).
Использование данных дробного учета может идти по нескольким путям.
Прежде всего непосредственные результаты взвешивания наносят на
план. Для этого весь цифровой материал разбивают на группы с интервала¬
ми 0,5—1,0 кг и для каждой группы подбирают определенную интенсивность
окраски (обычно более темную с повышением урожая), которой и закраши¬
вают на плане каждую ячейку, соответствующую элементарной делянке.
Такой план позволяет довольно хорошо ориентироваться в характере пест¬
роты участка и выделить в его пределах более однородные площадки и, на¬
оборот, выключить резко отличающиеся пятна.
Для правильного анализа характера и степени пестроты участка жела¬
тельно вычертить для каждого участка кривую Гаусса. Чем больше кривая,
построенная на основании данных дробного учета, отклоняется от нормаль¬
ной кривой Гаусса, тем более необходима выбраковка отдельных пятен
участка или разделение его на несколько самостоятельных частей. В част¬
ности, двухвершинная форма кривой может служить указанием на целе¬
сообразность разделения участка на две самостоятельные, более выровнен¬
ные внутри себя части.
Насколько сильно понижается ошибка при правильном разделении неод¬
нородного участка на отдельные, более однородные части, показывает сле¬
дующий пример из материалов Долгопрудного опытного поля.
На основании данных дробного учета для одного из участков опытного
поля была вычислена процентная ошибка элементарных делянок (по 10 м2)
при четырехкратной повторности (т4, %). Она оказалась равной 18%,
т. е. была очень высока. После того, как участок на основании нанесенных
на план данных дробного учета был разделен на две самостоятельные части,
ошибки, вычисленные для каждой из этих частей в отдельности, составили
8,3 и 11,3%, т. е. были уже более или менее приемлемыми.
Следующая стадия работы заключается в чисто эмпирическом комбини¬
ровании элементарных делянок по две, три и т. д. и суммировании их уро¬
жаев с тем, чтобы подобрать размер и форму опытной делянки, при которых
в наибольшей степени погашалась бы пестрота элементарных делянок.
Одновременно производят математическую обработку материала, уста¬
навливающую среднюю квадратическую ошибку для элементарной делянки
и для комбинированных делянок разной величины. Исходя из того, что сред¬
няя ошибка т прямо пропорциональна величине квадратического отклонения
6 и обратно пропорциональна корню квадратному из числа повторений п:
вычисляют число повторений9 необходимое для того, чтобы ошибка опыта
при данном размере делянок не превосходила заданной величины:
Все эти предварительные вычисления позволяют при минимальной зат¬
рате площади заложить опыт с заранее определенной точностью.
Наиболее существенным возражением против дробного учета является
утверждение, что пестрота поля не остается неизменной в различные годы и
т=—т=
У п
540
С. В. Щерба, Ф. Л. Юдин
для разных культур. Например, делянки, расположенные в понижениях,
наименее урожайные в сырые годы, могут оказаться наиболее урожайными в
засушливые годы и наоборот. Опыт показывает, что хотя некоторые измене¬
ния пестроты по годам и культурам и наблюдаются, однако общий характер
ее сохраняется, и выбор наиболее однородных площадок, по данным дроб¬
ного учета, достаточно надежен. В частности, работы С. В. Щербы показали,
что хотя абсолютная величина ошибки уже заложенного опыта далеко не
всегда совпадает с предварительно вычисленной на основании данных дроб¬
ного учета, относительная величина ошибки для различных участков опре¬
деляется этим способом безошибочно.
Таким образом, данные дробного учета дают возможность выбирать для
опытов наиболее однообразные по плодородию участки и даже приближенно
предсказывать точность будущих опытов. Изучение пестроты участка дроб¬
ным учетом элементарных делянок малого размера является наиболее пра¬
вильным и наиболее теоретически обоснованным приемом и должно приме¬
няться при постановке многолетних стационарных опытов.
Для однолетних опытов, занимающих большие площади, проведение
дробных учетов элементарными делянками невозможно из-за чрезвычайной
трудоемкости и громоздкости работы. Проще проводить предварительный
учет рекогносцировочных посевов делянками такого размера и формы, ка¬
кие проектируются для будущего опыта, исходя из задачи и цели его, и по
их урожаям рассчитывать повторность.
Часто вместо дробного учета рекогносцировочного посева выгоднее при
постановке опытов с однолетними культурами на новых землях увеличить
повторность, а затем по результатам первых экспериментов судить о пест¬
роте почвенного плодородия поля и устанавливать необходимую повторность
для последующих опытов (Доспехов, 1968).
Размещение опыта на участке
Основной задачей размещения опыта на участке является возможное
уменьшение различий в исходном плодородии сравнимаемых делянок, выз¬
ванных пестротой участка.
Обычное представление о постепенности изменения почвенного плодоро¬
дия в определенном направлении приложимо лишь к очень ограниченным
протяжениям. Более общей чертой почвенной пестроты является наличие
чередующихся пятен более высокого и более низкого плодородия, связан¬
ных переходами, характеризующимися постепенно изменяющимся плодоро¬
дием. Для таких отдельных элементов площади (пятен) со сравнительно вы¬
ровненным внутри них плодородием некоторыми опытниками (Сапегин)
предложен термин «связная площадка».
Существует пестрота сравнительно мелкого масштаба, обусловленная
микрорельефом, мелкими изъянами агротехники, неравномерностью пред¬
шествующего распределения удобрений и т. п. Эта пестрота, которую можно
назвать пестротой первого порядка, может быть перекрыта величиной от¬
дельной делянки. Кроме пестроты первого порядка, может существовать
пестрота второго порядка, обусловленная расчлененностью макрорельефа,
старым делением на клинья севооборота и тому подобными причинами, соз¬
дающими пятна более крупного масштаба. В пределах этих более крупных
сравнительно однородных пятен, но включающих пестроту первого порядка,
располагаются уже целые опыты, самостоятельные звенья или, по крайней
мере, целые повторения опытов.
Точность опыта определяется не только правильным соотношением пло¬
щади делянки (и повторности) с пестротой первого порядка, но и правильным
соотношением площади размещения всего опыта с пестротой второго поряд¬
Методика полевого опыта с удобрениями
ка. Недооценка второго положения часто служит источником неправильных
представлений о желательной величине делянки.
Величина делянки. Повышение точности опыта (уменьшение
ошибки) с увеличением площади делянки идет не пропорционально этому
увеличению, а постепенно затухая. За известным пределом увеличение пло¬
щади делянки может привести к понижению точности.
Дело в том, что увеличение площади каждой делянки означает и увеличе¬
ние площади, занимаемой опытом в целом. До тех пор, пока весь опыт ос¬
тается в пределах однородной площадки, одного пятна пестроты второго по¬
рядка, точность опыта с увеличением площади делянки повышается. Но как
только общая площадь опыта выходит за пределы однородной площадки,
точность его резко падает. Таким образом, максимальный размер делянки
ограничивается необходимостью уложить весь опыт в пределах однородной
площадки, второго порядка.
Совершенно ясно, что поскольку масштаб пестроты и первого и второго
порядка может быть совершенно различным на разных почвах, абсолютный
размер делянок, обеспечивающих достаточную точность опыта, также будет
различен для разных почв. Так, в нечерноземной полосе с ее сильно расчле¬
ненным рельефом и малыми размерами однородных площадок второго по¬
рядка увеличение площади делянки очень скоро из фактора положительного
может превратиться в отрицательный. Наоборот, в степных районах можно
-идти очень далеко в сторону увеличения площади делянки.
Кроме приведенных соображений, связанных с пестротой участка, уста¬
новление площади делянки связано еще с возможностью и удобством проведе¬
ния на ней необходимых сельскохозяйственных работ, прежде всего обра¬
ботки почвы. Обычно для полевого опыта проектируют величину делянки,
равную минимальной площади, обеспечивающей необходимую точность опы¬
та и допускающей проведение всех работ по опыту, включая учет урожая с
максимальной механизацией. Предел, меньше которого не должна быть
площадь делянки, зависит от возможности соблюдения нормальной агротех¬
ники возделываемой культуры с использованием требующихся для нее
сельскохозяйственных машин и орудий.
Размер делянки часто тесно связан со схемой опыта и количеством
вариантов в ней. В условиях опытных учреждений выровненные по плодоро¬
дию и рельефу участки, отводимые под отдельные опыты, часто имеют огра¬
ниченную площадь. Для того чтобы на постоянном опытном участке раз¬
местить схему с большим числом вариантов при принятой для опыта повтор¬
ности, неизбежно приходится уменьшать площадь делянки.
В производственных опытах, где требование типичности распространяет¬
ся на соответствие не только природным, но и хозяйственным условиям, раз¬
мер делянки может варьировать в значительных пределах. Однако сильное
увеличение его повышает затраты труда и средств и может снизить точность
опыта; площадь делянки должна быть минимальной, позволяющей проводить
все работы на уровне агротехники и механизации, типичном для данного
района. При выполнении важнейших операций на тракторной тяге наи¬
меньший размер делянки, по многочисленным данным практики, состав¬
ляет 100 м2.
На величину делянки влияет продолжительность опыта. Как правило,
площадь делянки должна быть в многолетних опытах больше, чем в однолет¬
них. В длительных опытах может возникнуть необходимость разделения де¬
лянок на части для введения дополнительных вариантов или наложения но¬
вого фона на имеющиеся варианты.
Размер делянок зависит также от биологических и агротехнических
особенностей изучаемой культуры. Для определения влияния на урожай ин¬
дивидуальной изменчивости растений на учетной площади делянки должно*
быть не менее 200 растений. Для культур сплошного посева (зерновых, льна,.
542
С. В. Щербау Ф. А. Юдин
трав), в котором на 1 м2 приходится несколько сотен растений, делянки мо¬
гут быть меньше, чем для пропашных (сахарной свеклы, картофеля, куку¬
рузы, подсолнечника), где на 1 м2 находится от 2 до 6 растений. Величина
делянки будет изменяться в зависимости от темы опыта. Если обработка,
посев и уход проводятся однородно на всем участке, т. е. поперек делянок,
то они могут быть меньшего размера, чем в случае, когда по характеру
темы каждую делянку обрабатывают, засевают и ведут на ней уход за по¬
севами раздельно (изучение способов внесения удобрений, эффективность
рядкового удобрения, подкормок во время вегетации пропашных куль¬
тур и т. д.).
Необходимость проведения всех работ по опыту, связанных с закладкой,
посевом, уходом (прополка, подкормка, полив и т. д.), одновременно и на вы¬
соком уровне агротехники, а также строгий учет урожая в короткие сроки
ограничивает возможность увеличения площади делянки сверх оптималь¬
ной, обеспечивающей требования точности и типичности опыта. Кроме того,
расширение размера делянки неоправданно, так как приводит к большому
расходу удобрений, семян, дополнительным затратам труда, что значительно
удорожает опыт.
Какие же площади делянок следует считать оптимальными для опытов с
удобрениями? На основе многолетней практики опытных учреждений ре¬
комендовать можно средние размеры делянок 50—100 м2 для растений сплош¬
ного посева и 100—200 м2 для пропашных культур. От этих величин могут
быть отклонения. В опытах с раздельной обработкой и посевом каждой де¬
лянки, с техникой внесения удобрений площадь делянки увеличивается до
300 ле2, иногда и больше. В многолетних опытах рекомендуются делянки от
200 до 300 м2. В лабораторно-полевых опытах, где соблюдение типичности в
производственном отношении необязательно, при применении конной обра¬
ботки для культур сплошного посева размер делянки может быть 20—25 ле2,
а при ручной обработке и еще меньшим.
Отсутствие специальных малогабаритных машин и орудий заставляет
увеличивать делянки, что нежелательно, так как снижается качество рабо¬
ты. Указанные выше размеры примерные, они требуют уточнения в каждом
отдельном случае.
Форма делянки. Точность опыта может быть также повышена в
результате правильного выбора формы делянки. Наилучшую для данного
участка форму делянки можно определить непосредственно, комбинируя дан¬
ные дробного (участка) учета. В случае отсутствия данных дробного учета
приходится исходить из следующих общих соображений.
Вытянутая форма делянки обеспечивает обычно большую точность опыта,
так как чем длиннее делянка, тем полнее она охватывает пестроту участка.
Особенно необходимы вытянутые делянки при наличии явно выраженного
изменения плодородия участка в каком-либо одном направлении. Делянки
должны быть вытянуты в том направлении, в котором изменяются свойства
участка. Так, при закладке опыта на склоне делянки обязательно должны
быть вытянуты вдоль склона, равномерно захватывая все его части. При
наличии на участке каких-либо полос различного плодородия делянки долж¬
ны располагаться поперек этих полос. Однако это правило приложимо лишь
к делянкам достаточно крупного размера.
Вытянутую форму делянки следует предпочесть, если обрабатывают и за¬
севают каждую делянку отдельно (техника внесения удобрений, рядковое
удобрение). В таких случаях вытянутые делянки располагаются в один
ряд, чтобы машины могли делать развороты за пределами делянки для пере¬
хода на следующие повторения.
Для пропашных культур делянки удобнее размещать параллельно рас¬
положению рядков и ширину делянки брать кратную ширине междурядий.
В опытах с рядковым удобрением ширина делянки должна быть кратной
Методика полевого опыта с удобрениями
54$
ширине рабочего захвата комбинированной сбялки. Но такая форма годится
лишь для более крупных делянок.
Недостаток делянок вытянутой формы, у которых отношение длины к ши¬
рине более 10,— их большой периметр. Чем он больше, тем сильнее сказы¬
вается влияние края и соседей на результаты; отсюда возникает необходи¬
мость обязательного введения защитных полос. Площадь их значительно
больше на узких длинных делянках, поэтому при ограниченной площади
участка и малых размерах делянок (меньше 50 м2) им следует придавать фор¬
му, близкую к квадрату, а повышения точности опыта добиваться увеличе¬
нием повторности. Считаются приемлемыми такие величины и формы деля¬
нок, при которых на защитные полосы приходится около 25% площади
опытного участка.
В условиях производства безусловно следует предпочесть вытянутую
форму делянки. Форма самого опытного участка или севооборотного клина
не имеет особенного значения и определяется формой наличной однородной
площадки для опыта и удобством обработки.
Повторность. Наиболее действенным способом повышения точ¬
ности опыта является введение нескольких повторных делянок для каждого
варианта схемы. Повторные делянки можно рассматривать как части одной
более крупной делянки, но размещенные в различных местах опытного участ¬
ка. Такое размещение дает возможность более полно охватить каждым вариан¬
том всю пестроту участка. Поэтому при одинаковом увеличении суммарной
площади, нужной для одного варианта, введение повторных делянок при уве¬
личении их числа дает большее повышение точности, чем соответствующее*
увеличение площади одной делянки. Это доказано экспериментально рядом
исследователей и, в частности, хорошо видно из табл. 1 и рис. 1, взятых из
книги Ремера.
Таблица 1
Влияние площади делянки и числа повторений на величину ошибки по Ремеру
Общая площадь на
один вариант, м*
Увеличение площади делянки без
увеличения повторности
Увеличение повторности при неизмен¬
ной площади делянки (25 л»2)
площадь делянки, м2
ошибка т, %
число повторений
ошибка in, %
25
25
10,0
1
10,0
50
50
8,3
2
7,1
75
75
7,6
3
5,8
100
100
7,1
4
5,0
125
125
6,7
5
4,5
150
150
6,4
6
4,1
175
175
6,1
7
3,8
200
200
5,9
8
3,5
225
225
5,7
9
3,3
250
250
5,6
10
3,2
Наличие нескольких параллельных делянок для каждого варианта
опыта не только повышает его точность, но и дает возможность количест¬
венно определить эту точность (вычислить величину ошибки). Повторность
одноименных делянок нужно считать обязательной для всякого полевого
опыта.
Необходимая повторность должна устанавливаться для каждого кон¬
кретного случая и участка в зависимости от его пестроты. Лучше всего руко¬
544
С. В. Щерба, Ф. А. Юдин
водствоваться при этом данными дробного учета, а при отсутствии — опы¬
том работы на сходных площадях.
Для ориентировочных подсчетов при отсутствии данных дробного учета
Б. А. Доспехов (1968) приводит, по материалам разных авторов, следующие
значения вариационного коэффициента: для делянок в 100 м2 — около 10%
(с колебаниями от 5 до 25), на более окультуренных участках при тщательном
проведении опыта — 5—8% и для делянок 300—500 м2 — 5—6%. Кроме то¬
го, для уточнения пестроты участка следует вести глазомерную оценку урав¬
нительного посева.
Требуемая точность опыта (т, %) определяется размерами тех разниц
между сравниваемыми вариантами (Z)), которые предполагается получить в
опыте. Если ожидают получить большую разницу между вариантами, на¬
пример между урожаем с полным минеральным удобрением и урожаем без
Рис. 1. Влияние размера пло¬
щади делянки и числа повто¬
рений на величину ошибки
опыта (по Ремеру)
1 — при увеличении площади де¬
лянки (при неизменной по¬
вторности);
2 — при увеличении числа повто¬
рений (при .неизменной пло¬
щади)
удобрения на бедной почве, где прибавки могут составлять 30—50% от кон¬
троля, вполне достаточна точность опыта 10—15%. Если же в опыте изу¬
чают эффективность разных форм удобрения или сравнивают разные спосо¬
бы внесения удобрений, где приходится устанавливать сравнительно неболь¬
шие различия между вариантами опыта и где прибавки в урожаях могут
достигать 6—10% по сравнению с контролем, требования к точности опыта
должны быть высокими, а ошибка не превышать 2—3%. В соответствии с
требованиями точности опыта определяется и необходимая повторность —
тем большая, чем более высокую точность опыта мы хотим иметь.
При установлении желаемой точности опыта, в котором могут быть по¬
лучены разницы неодинаковой величины, следует ориентироваться на воз¬
можные наименьшие разницы. Считают, что разницы в урожаях меньше 5%
полевым опытом не улавливаются. В стационарных условиях, как правило,
полевые опыты не закладывают с повторностью меньше чем 4-кратная.
Большинство полевых опытов при размерах делянок 50—100 ле2, а иногда и
больше ставят в 4-кратной, реже в 6-кратной повторности; это дает возмож¬
ность иметь точность опыта около 2—4%. При постановке опытов на делян¬
ках 20—10 м2 повторность повышают до 6—8-кратной. Минимальная
повторность — 2-кратная. Ее недостатком, даже когда она обеспечивает необ¬
ходимую точность опыта, является риск выпадения одной делянки по слу¬
чайным причинам, что ведет к выбраковке из опыта всего варианта. Кроме
того, при значительном расхождении результатов двух параллельных деля¬
нок невозможно судить о том, какой из результатов ближе к истинной ве¬
личине урожая: отсюда желательно иметь, как минимум, 3-кратную повтор¬
ность. 2—3-кратная повторность применяется в предварительных, реког-
юносцировочных и демонстрационных опытах, а также в производственных
опытах на площади делянки свыше 1000 м2.
I : I I : I I L
/ 13450789 *0
Чисум повторении
Методика полевого опыта с удобрениями
545
Наиболее объективно требующуюся повторность устанавливают на ос¬
новании данных дробного учета (находят v, %) и желательной точности (тпу
%) по формуле
( v,% V
Л — \ т»% / ‘
Возьмем числовой пример: на опытном участке, по данным дробного учета, для
делянок 100 м2 вариационный коэффициент 9,4%, заданная точность опыта 5%. Чтобы
определить необходимую повторность будущего опыта, подставляем в формулу найденное
значение v и заданную величину т, находим: п = (9,4:5)2 = 3,53 или, округляя, 4 по¬
вторности.
При установлении повторности, кроме пестроты участка и заданной точ¬
ности, надо также учитывать технические возможности проведения опыта.
I Ж ЛГ Ж
Z
3
Ч
/
2
3
Ч
/
Z
3
Ч
/
2
3
Ч
а
Рис. 2. Схемы систематичес¬
кого расположения вариан¬
тов и повторений в опыте
а — однорядное последователь¬
ное;
б — двухрядное;
в — многорядное ступенчатое
расположение.
1 — 8 — номера вариантов.
1 — IV — повторения
/ 2Г
/
Z
3
Ч
/
Z
3
ч
3
ч
1
Z
3
ч
1
Z
/
Z
3
Ч
5
6'
7
д
7
в
1
Z
3
Ч
5
5
5
6
7
8
1
Z
3
Ч
3
У |У
5
7
в
/
2
6
Увеличение повторности, а следовательно, и количества делянок значитель¬
но усложняет работу по закладке и учету опыта; оно требует увеличения чис¬
ла мешков для пробных снопов, дополнительной площади сноповых сараев
и сушилок, дополнительных анализов и т. п.
Более высокая повторность применяется обычно только в стационарных
опытах, закладываемых в опытных учреждениях.
Общее расположение опыта. Способы расположения пов¬
торений и вариантов в опыте преследуют цель охватить каждым вариантом
более полно пестроту почвенного плодородия опытного участка, соэдать ус¬
ловия наилучшей сравнимости между вариантами, что в конечном счете обес¬
печивает большую репрезентативность и точность опыта, повышает досто¬
верность его результатов. На выбор способов размещения полевого опыта в
пространстве оказывают влияние величина и степень пестроты плодородия
опытного участка, число вариантов в схеме, а также технические условия
постановки и проведения опыта.
В полевых опытах с удобрениями чаще всего все повторения располагают
компактно на одном участке, имея общие границы между отдельными пов¬
торениями. Такое расположение повторений носит название сплошно-
г о. При сплошном расположении повторения опыта могут быть размещены на
участке в один, два и несколько рядов (рис. 2).
Однорядное расположение повторений применяется при неболь¬
шом числе вариантов в опыте и постановке его на делянках удлиненной фор¬
мы, которые размещаются перпендикулярно длинной стороне опытного участ¬
546
С. В. Щерба, Ф. Л. Юдин
ка. Такой способ размещений очень удобен для всех работ по технике за¬
кладки и проведения опыта. Однорядное расположение обязательно в опытах
по изучению техники внесения удобрений, рядкового удобрения, где удобре¬
ния заделываются туковыми или комбинированными сеялками и требуются
раздельные обработка и посев каждой делянки. Однорядное размещение
повторений и вариантов позволяет иметь широкие полосы вдоль длинных сто¬
рон опытного участка для разворота машин и орудий, используемых при
закладке и проведении опыта. Однорядное расположение предпочтительно
при систематическом изменении плодородия участка в одном направлении.
Делянки должны быть вытянуты в том направлении, в котором изменяются
свойства участка.
При большом числе вариантов и делянок в опыте и сравнительно неболь¬
шом размере делянок укороченной формы удобнее двухрядное или мно¬
горядное расположение повторений (см. рис. 2, б, в). При двух- и много¬
рядном размещении повторений в каждом ряду их помещается целое число.
Значительно реже используют разбросанное расположение пов¬
торений, когда отдельные повторения по одному или по нескольку размеще¬
ны на отдельных опытных участках, разбросанных по разным частям одного
поля или даже находящихся на равных полях. При таком расположении
повторений варьирование плодородия разных повторений должно быть не¬
зависимым друг от друга.
Разбросанное расположение повторений чаще всего вызывается отсут¬
ствием однородного опытного участка достаточного размера для сплошного
размещения всех повторений опыта. Иногда разбросанное расположение при¬
меняют намеренно, например при постановке производственных опытов,
когда необходимо оценить эффективность удобрений при широкой амплиту¬
де почвенных и агротехнических условий.
При расположении вариантов схемы на делянках внутри повторения
различают систематическое и случайное (рендомизированное) размещение.
Кроме того, существуют стандартные методы расположения, характеризуе¬
мые увеличенным числом контрольных (стандартных) вариантов в каждом
повторении и способом вычисления прибавок.
Систематическое расположение вариантов на делянках внутри
повторений, при котором предусматривается возможно равномерное разме¬
щение одноименных вариантов по всему опытному участку, предполагает
расположение вариантов в определенном, заранее установленном экспери¬
ментатором порядке. При однорядном размещении повторений наиболее
распространено последовательное расположение вариантов, при котором
установленный порядок размещения вариантов на делянках первого повто¬
рения далее в неизмененном виде повторяется во всех остальных повторе¬
ниях (см. рис. 2,а).
При двух- и многорядном расположении повторений на делянках вариан¬
ты чаще всего размещают ступенчато; они идут в одном направлении, но в
каждом следующем ряду начало схемы сдвигается на одну, две или больше
делянок, а конец ее переносится в начало ряда (см. рис. 2 б, в). Такое распо¬
ложение опыта иногда называют шахматным.
При многорядном расположении повторений могут быть и другие методы
размещения вариантов. Однако при любых способах расположения повторе¬
ний и вариантов нельзя допускать территориального сближения одноимен¬
ных делянок, т. е. помещать их рядом как в вертикальном, так и в горизон¬
тальном направлении. Следует стремиться одноименные делянки максималь¬
но удалять друг от друга, а при многорядном расположении повторений
делянки каждого варианта в вертикальных столбцах помещать однократно.
При неизбежности повторного размещения делянок одного варианта в одной
вертикали необходимо располагать между ними по крайней мере две делян¬
ки других вариантов.
Методика полевого опыта с удобрениями
547
В большинстве полевых опытов с удобрениями, проводимых в Советском
Союзе, применяется систематическое размещение вариантов на делянках
внутри повторений. Оно удобнее и проще для проведения всех работ по за¬
кладке, уходу и уборке урожая в опыте.
При регулярном изменении плодородия почвы недостатком последова¬
тельного систематического размещения вариантов является вероятность на¬
копления систематических ошибок, так как всегда возможно, что система
изменения плодородия будет коррелировать с системой расположения ва¬
риантов. При этом одни варианты будут находиться внутри повторения на
делянках, расположенных рядом или близко, а другие — на делянках, уда¬
ленных одна от другой, что приводит к неравноточным сравнениям вариан¬
тов друг с другом и с контролем.
При многорядном расположении повторений и ступенчатом размещении
вариантов внутри повторений неравноточность сравнения вариантов сгла¬
живается, так как систематическое изменение плодородия внутри отдельных
повторений опыта не будет коррелировать с системой расположения вариан¬
тов.
Случайное, или рендомизированное(от англ. rendom —
случайный, беспорядочный), расположение вариантов на делянках предло¬
жено Р. А. Фишером. Оно заключается в случайном (рендомизированном)
размещении вариантов на делянках каждого повторения путем жребия или
же по специально составленным таблицам случайных чисел. Этот способ ос¬
нован на том, что все методы вариационной статистики приложимы в полной
мере только к случайным явлениям, и поэтому статистическая обработка
результатов опыта наиболее обоснованно применима при случайном распо¬
ложении вариантов в пространстве.
При рендомизации значительно меньше возможностей корреляции между
изучаемыми в опыте вариантами, что делает более равноточными их попар¬
ные сравнения. При систематическом изменении плодородия почвы рендоми-
зация уравновешивает его влияние внутри каждого повторения и тем самым
предотвращает накопление систематических ошибок, превращая их в слу¬
чайные.
Среди случайных методов размещения вариантов наибольшее распростра¬
нение получили метод случайных блоков (повторений) и метод латинского
квадрата.
Метод случайных блоков (повторений) — наиболее
простой способ размещения вариантов. Их объединяют в несколько блоков,
число делянок в каждом повторении равно числу вариантов схемы. Общее
количество блоков определяется принятой в опыте повторностью. В блоке
варианты по делянкам располагают в случайном порядке по жребию. В пре¬
делах каждого блока почвенные условия должны быть по возможности од¬
нородными. Форму блоков желательно иметь близкую к квадрату, чтоб!ы
улучшить сравнимость вариантов при любом размещении делянок в прост¬
ранстве.
Форма делянок может быть удлиненная, укороченная и квадратная.
Блоки на опытном участке располагают компактно в один, два или несколько
ярусов, реже их размещают разбросанно поодиночке или группами. Делян¬
ки внутри блоков также располагают в один, два или несколько рядов, иног¬
да блокам придают ступенчатую форму.
Метод случайных блоков (повторений) называют также рендомизацией с
одним ограничением. Оно состоит в том, что в каждом блоке (повторении)
должен быть полный набор вариантов схемы, и рендомизация здесь осуще¬
ствляется в пределах каждого блока (повторения), а не на всем опытном
участке.
Примеры рендомизированного расположения блоков (повторений) при*
ведены на рис. 3. При постановке опытов методом случайных блоков не ис-
548
С. В. Щерба, Ф. А. Юдин
92/3/93293/23/29239/923/
3
/
2
9
2
3
9
/
/
г
9
3
9
/
2
3
9
3
/
2
3
2
/
9
Рис. 3. Схемы размещения
вариантов методом случай¬
ных блоков (повторении)
ключена возможность того, что по жребию одноименные варианты будут
размещены рядом. В этом случае для более равномерного распределения ва¬
риантов на опытном участке допустимо введение еще одного ограничения, па
которому одноименные делянки не должны примыкать друг к другу ни в го¬
ризонтальном, ни в вертикальном направлениях.
Метод латинского квадрата состоит в том, что число повто¬
рений (п) в опыте равно числу вариантов, а общее число делянок равно м2.
Варианты на плане обозначают буквами латинского алфавита. При разме-
А
В
С
D
Б
F
В
С
D
Б
F
А
С
D
Б
F
А
В
D
Б
F
А
В
С
Е
F
А
С
С
D
F
А
В
D
D
Б
С
Б
В
А
D
F
В
F
Б
D
А
С
А
D
F
С
В
Б
F
В
D
Б
С
А
D
А
С
F
Б
В
Б
С
А
В
F
D
Рис. 4. Схемы размещения ва¬
риантов методом латинского
квадрата
а — систематическое;
б — рендомизированное
щении опыта методом латинского квадрата опытный участок квадратной или
прямоугольной формы разбивают на горизонтальные и вертикальные ряды
по числу вариантов. В горизонтальном и вертикальном ряду помещают пол¬
ный набор всех вариантов; это возможно только тогда, когда одноименные
делянки не повторяются дважды ни в горизонтальном, ни в вертикальном
ряду. Внутри этих рядов варианты на делянках расположены по жребию;
здесь мы имеем рендомизацию с двумя ограничениями. В пределах латинского
квадрата возможно и систематическое ступенчатое размещение вариантов на
делянках. Схемы расположения опыта по методу латинского квадрата при¬
ведены на рис. 4.
Расположение вариантов на делянках методом латинского квадрата очень
удачное, так как оно позволяет при соответствующей математической обра¬
ботке результатов опыта исключить влияние изменения плодородия почвы]в
двух взаимно перпендикулярных направлениях и снизить ошибку опыта.
Метод латинского квадрата используется при числе вариантов от 4 до 7.
В случае увеличения количества вариантов потребовалась бы очень боль¬
шая повторность в опыте. Чтобы сохранить возможность путем соответ¬
ствующей математической обработки вычисления влияния систематического
изменения плодородия почвы в двух взаимно перпендикулярных направле¬
ниях на точность опыта, не прибегая к очень большой повторности, варианты
на делянках размещают по методу латинского прямо¬
Методика полевого опита е удобрениями
549
угольника. При этом методе число вариантов должно быть кратным
числу повторностей, т. е. делиться без остатка на число повторностей. Част¬
ное от деления даст количество полос, на которое надо разделить каждый
вертикальный ряд соответствующего латинского квадрата. Например, в
опыте 8 вариантов при четырехкратной повторности. Разделив 8 на 4, полу¬
чим 2. Находим, что каждый вертикальный ряд надо разбить на две полосы,
и мы будем иметь полевой опыт, заложенный по методу латинского прямо¬
угольника, по схеме 4x4x2 (рис. 5). В этой схеме первая цифра озна¬
чает принятую в опыте повторность, произведение двух последних цифр —
количество изучаемых вариантов, а всех трех цифр 4 X
X 4 X 2 = 32 — число делянок в опыте. Существуют
и другие методы расположения вариантов с использо¬
ванием рендомизации.
Число и расположение контро¬
ле й. Стандартные методы. В схеме опыта
вариант, с которым сравнивают другие варианты, на¬
зывают контрольным или контролем. В опытах с удоб¬
рениями им может быть делянка варианта без удоб¬
рений; она называется чистым контролем. Но в опы¬
тах по изучению действия удобрений (виды, формы,
способы и сроки внесения) такой контроль не всегда
обязателен. Чаще всего для сравнения действия како-
го-либо одного из трех основных питательных веществ
(NPK) используется парная комбинация двух других
элементов питания (фон); в этом случае фоновая делян¬
ка служит основным контролем. В опытах с новыми
формами удобрений сравнение обычно ведут с делян¬
кой, на которой размещен вариант с хорошо изучен¬
ной формой удобрения (стандартное удобрение). При
исследовании различных способов и сроков внесения Рис. 5. Размещение
удобрений за контроль принимают вариант с наиболее полевого опыта мето-
освоенными способами и сроками внесения удобрения. Д°м латинского пря-
„ * моугольника по схе-
В каждое повторение опыта может быть включена ме 4 х 4 х 2
либо одна контрольная делянка, либо несколько. В пер¬
вом случае общее число контрольных делянок равно
числу делянок по любому другому варианту опыта, во втором — число кон-
тролей повышенное и повторность их больше, чем повторность других ва¬
риантов опыта.
Введение в опыт повышенного по сравнению с другими вариантами числа
контролей преследует цель: точнее охватить ими пестроту опытного участка
и дать более надежный масштаб для сравнения всех других вариантов.
Кроме того, повышенное число контролей позволяет оценить пестроту пло¬
дородия участка при отсутствии данных дробного учета.
Обычно один контроль должен приходиться внутри каждого повторения
на 6—8 вариантов (не больше). При 10—12 и более вариантах следует обя¬
зательно вводить дополнительные контроли; это даст возможность полу¬
чать более точные средние как по контролям, так и по всем остальным ва¬
риантам.
Для методики важно знать, что больше повышает точность опыта —
увеличение повторности для всех вариантов или же увеличение контролей
за счет некоторого сокращения общей повторности. Подсчеты С. В. ГЦербы
показали, что сначала с возрастанием числа контролей ошибки разности
снижаются, достигая минимума при отношении числа контролей к общему
числу делянок от 1 : 6 до 1 : 4, а затем при числе контролей больше V4 обще¬
го числа делянок вновь начинают расти.
Кроме обычного расположения контрольных вариантов, при системати-
в
5
/
3
6
У
2
7
S
3
7
У
5
2
в
f
f
У
6
2
в
7
S
3
2
7
в
S
/
3
'
6
550
С. В. Щерба, Ф. Л. Юдин
а
6
S
ш
'Ш
т
ш
1
i
i
ш
ш
ш
ш
щ
щ
й
!
1
V
ш
1
ш
й
УА
i
i
i
г
ж
ш
ш
щ
Ш
Ш
i
1
щ
!
i
i
Ш
I
щ
i
I
Ш
i
A
Ш,
i
i
i
Рис. 6. Схемы стандартных методов расположения контроль¬
ных (стандартных) вариантов (заштрихованные клетки) на де¬
лянках опыта
а — контроль через одну изучаемую делянку (ямб-метод); б — конт¬
роль через две изучаемые делянка (дактиль-метод);] в — в шахматном
порядке; г — по методу измерительных делянок; д — по ходу шахмат¬
ного коня
ческом зли рендомизированном размещении вариантов опыта внутри повто¬
рения существуют стандартные методы.
Стандартные методы расположения вариантов характеризуются более
частым равномерным распределением контрольных вариантов через один,
два, три и более опытных вариантов. Эти методы рассчитаны на то, что
изменение плодородия участка происходит постепенно систематически в
определенном направлении. В этом случае контроль (или стандарт) приме¬
няют для повышения точности опыта.
При стандартных методах размещения опыта исходят из предположе¬
ния, что между урожаями соседних или ближайших делянок существует
определенная корреляция и поэтому они лучше сравнимы между собой,
чем урожай делянок, удаленных друг от друга.
Различают несколько стандартных методов расположения вариантов на
делянках повторения. Если контрольные (стандартные) делянки размещены
через одну изучаемую делянку, то такой метод называют ямбическим или
ямб-методом (рис. 6,а); если контрольные делянки расположены через две
изучаемые — дактилическим или дактиль-методом (рис. 6,6). В этих мето¬
дах прибавки урожая вычисляют для каждой изучаемой делянки, срав¬
нивая со своим контролем, который определяется как среднее из двух со¬
седних контролей для ямб-метода или по интерполяции между соседними
контролями для дактиль-метода. В опытной работе широко распространен
метод размещения контрольных вариантов через два опытных.
Методика полевого опыта с удобрениями
551
При многорядном расположении опыта контрольные делянки разме¬
щают в шахматном порядке (рис. 6, в). В этом случае контроли (стандарты)
размещены в ряду через одну делянку и сдвинуты на одну в каждом сле¬
дующем ряду. Шахматный способ расположений дает очень высокую точ¬
ность определения прибавок, так как каждая изучаемая делянка сравни-
вается со средним из четырех или трех контролей. Этот прием чаще исполь¬
зуют в мелкоделяночных опытах, потому что он требует значительного
числа добавочных делянок. Метод измерительных делянок Гольстмарка и
Ларсена отличается от шахматного расположения тем, что в каждом ряду
контроль (или стандарт) помещен через две изучаемые делянки. Сравнение
ведут со средним из трех соседних контролей, два из которых соприкасаются
с изучаемой делянкой сторонами, а третий — лишь углом (рис. 6, г). Сле¬
дует отметить еще способ размещения повышенного числа контролей при
много рядном расположении опыта, по ходу шахматного коня (рис. 6, д).
Сравнение можно вести со средним из двух соседних контролей (один при¬
легает стороной, а другой соприкасается углом). При этом способе пло¬
щадь, занятая дополнительными контролями, значительно меньше. Все
описанные методы расположения повышенного числа контролей могут быть
использованы как для стандартного вычисления прибавок путем сравнения
со средним соседних контролей, так и для обычного вычисления прибавок
сравнением каждого варианта со средним контролем для всего опыта.
Стандартные методы размещения опыта и вычисления прибавок при¬
меняют сравнительно редко. Они повышают точность опыта лишь в том
случае, когда на опытном участке происходит закономерное постепенное
изменение плодородия и наблюдается прямая корреляционная зависимость
между урожаями близлежащих делянок, однако подобная закономерность
на практике встречается не часто. При выравненности плодородия на
участке или же пестроте, изменяющейся случайно, стандартные методы
никаких преимуществ перед обычными методами расположения вариантов
не имеют.
К недостаткам стандартных методов следует отнести громоздкость и не¬
которые технические сложности в связи с введением в опыт большого числа
дополнительных контрольных (стандартных) делянок. Последнее обстоя¬
тельство приводит также к нерациональному использованию земельной
площади. При размещении стандарта через одну или две опытных делянки
50% или 40% всей площади опытного участка занято контролями. Кроме
того, при однорядном расположении повторений стандартные методы за¬
трудняют сравнение опытных вариантов, далеко расположенных друг от
друга, что бывает при большом числе (свыше 10—12) изучаемых вариантов.
Значение числа вариантов в схеме опыта. Число
вариантов в схеме опыта зависит от содержания темы опыта и определяется
задачами, которые в нем разрешаются. Число вариантов оказывает влияние
на точность опыта. Исследования Н. Ф. Деревицким (1962) материалов
многих опытов выявили четкую закономерность, что с увеличением числа
вариантов в схеме относительная ошибка опыта возрастает. Это наглядно
иллюстрируют данные табл. 2.
С увеличением числа вариантов возрастает количество делянок, а сле-
довательно, и общая площадь опытного участка. Это ведет к усилию пестро¬
ты плодородия, так как опыт или отдельные повторения его выходят за
пределы однородного по плодородию участка; в результате ошибка опыта
повышается.
В тех случаях, когда площадь опытного участка ограничена, при увели¬
чении числа вариантов и общего числа делянок в опыте приходится умень¬
шать размер отдельной делянки, что также повышает ошибку опыта. Чем
больше площадь делянки, тем сильнее возрастает ошибка при увеличении
числа вариантов. Например, в одном из опытов с картофелем при размерах
552
С. В. Щерба, Ф. А. Юдин
Таблица 2
Относительная ошибка опыта в зависимости от числа вариантов при одинаковой
площади делянки
(по Н. Ф. Деревицкому)
Сахарная свекла
Картофель
Сахарная свекла
Картофель
число ва¬
риантов
относительная
ошибка опыта
(w, %)
число
вариантов
относительная
ошибка опыта
(т, %)
число
вариантов
относительная
ошибка опыта
(т, %)
число
вариантов
относительная
ошибка опыта
(т, %)
2
4,57
2
8,04
12
6,24
12
9,87
3
5,03
3
8,38
18
6,31
16
10 ,Я1
4
5,38
4
8,51
27
6,76
24
10,65
Ь
5,40
6
8,76
54
7,21
48
14,61
9
СП
-1
4^
9
8,89
108
7,93
96
16,41
делянки 1 х 1; 3 X 1 и 6 X 1 увеличение числа вариантов с 2 до 12 повы¬
сило ошибку опыта соответственно в 1,2; 1,3 и 1,89 раза.
При увеличении числа вариантов возрастают расстояние между вариан¬
тами и ошибка сравнения между ними; различия в расстоянии между отдель¬
ными делянками и попарные сравнения вариантов, размещенных на делян¬
ках, лежащих близко и далеко одна от другой, делаются неравноточными.
Этого не наблюдается, когда схема имеет немного вариантов, расстояния
между делянками, занятыми разными вариантами, мало отличаются и по¬
парные сравнения вариантов приблизительно равноточны.
Считается нормальным, когда вариантов в схеме не более 8—12. Если
их 16—20, то необходимо увеличивать повторность контрольных вариантов.
В тех случаях, когда схема содержит много вариантов, например в много¬
факторных опытах, надо стремиться к тому, чтобы ее разбить на ряд эвеньев,
включающих отдельные части схемы с меньшим числом вариантов. Каждое
звено закладывается как самостоятельный опыт в пределах более однород¬
ного участка, чем достигается большая точность опыта. Для возможности
сравнения эвеньев между собой в них вводят по несколько одинаковых,
стандартных вариантов. Для многовариантных схем имеются также спе¬
циальные методы постановки опытов, рассматриваемые в специальных
руководствах.
Повторение опыта во времени и увязка его
с севооборотом. Полевой опыт с удобрениями, кроме соответствую¬
щей повторности в пространстве, должен иметь повторность во времени.
Цель повторения опыта во времени — не только повысить точность и досто¬
верность выводов о средней эффективности удобрений за несколько лет,
но и установить зависимость действия удобрений от различных метеороло¬
гических условий (годы нормальные, сухие, влажные и т. д.). Кроме того,
многие приемы удобрения имеют длительное последействие, для учета кото¬
рого необходимо проводить опыт в течение ряда лет. В опытах с удобре¬
ниями (например, с формами, дозами и т. д.) приходится также считаться
с эффектом повторного их наложения, т. е. с суммарным эффектом действия
и последействия, учет которого возможен только при повторении опыта
во времени.
В зависимости от темы опыты с удобрениями могут быть разной продол¬
жительности: краткосрочные и многолетние. В краткосрочных при изучении
разных видов, форм, доэ, способов и сроков внесения удобрений, обладаю¬
щих коротким последействием, для получения достаточно достоверной
Методика полевого опыта с удобрениями
553
характеристики их эффективности необходимая повторность во времени
зависит от метеорологических условий. Однако при составлении плана
таких опытов для получения исчерпывающего ответа требуется не менее
3—4 лет.
Опыты, в которых изучаются приемы, отличающиеся длительным по¬
следействием (известкование, дозы навоза), а также разрабатывается систе¬
ма удобрения, должны продолжаться не менее 10 лет.
В многолетних опытах, где приходится в течение ряда лет повторно учи¬
тывать ранее и вновь внесенные удобрения (например, с формами удобре¬
ний), а также в опытах, ежегодно закладываемых на участках, для выявле¬
ния эффективности по отдельным годам необходима одинаковая последова¬
тельность в чередовании культур. Для этого подобные опыты ставят в оп¬
ределенном севообороте.
Существуют два способа проведения многолетних опытов с удобрениями
в севообороте. Первый состоит в том, что все опыты закладывают в полях
одного большого севооборота. Каждый опыт ежегодно ставят на новом поле
в соответствии с чередованием культур. Основной недостаток способа —
удаленность отдельных клиньев и культур каждого опыта друг от друга;
это снижает сравнимость данных за разные годы. Однако при этом способе
можно проводить сплошную механизированную обработку одинаковых кли¬
ньев всех опытов, что дает экономию рабочей силы и времени. Применяется
при закладке многолетних опытов в производственных условиях.
Большинство опытных учреждений при проведении стационарных много¬
летних опытов используют второй способ, при котором для каждого много¬
летнего опыта выделяют специальный севооборот. Это создает большую од¬
нородность всех клиньев и позволяет лучше приспособить севооборот к осот
бенностям опыта. Однако такой способ вызывает некоторые затруднения с
обработкой отдельных полей.
В опытах с удобрениями, в которых приемы обработки почвы не исследу¬
ются, возможна комбинация из двух описанных выше способов, которая по¬
зволяет сохранить достоинства их и избежать присущих им недостатков.
При такой комбинации для нескольких опытов отводят один опытный се¬
вооборот с полями удлиненной формы, расположенными параллельно друг
ДРУГУ- Поля отдельных опытов проходят в перпендикулярном направлении
через все поля севооборота.
Комбинированный способ дает возможность проводить одновременно оди¬
наковую механизированную обработку одноименных клиньев севооборота
всех опытов. В то же время он территориально объединяет все клинья каж¬
дого опыта, что создает лучшую сравнимость его данных за равные годы иэ-за
их большей однородности.
При закладке многолетнего опыта в севообороте в натуре необходимо
иметь столько клиньев, сколько полей (и культур) в севообороте. Это осо¬
бенно важно при постановке опытов по изучению различных вариантов си¬
стемы удобрения в севообороте. Развертывание такого опытного севооборота
должно проходить постепенно, введением ежегодно в опыт нового клина,
начиная каждый год с определенной культуры. Методически указанный
способ закладки опыта более правилен, а в опытах по изучению системы
удобрения обязателен, так как здесь наложение отдельных удобрений долж¬
но идти в строго определенной последовательности. Такая постановка опы¬
та диктуется необходимостью изучения как эффективности отдельных эле¬
ментов изучаемых вариантов системы удобрении, так и действия различ¬
ных вариантов системы удобрения за севооборот в целом.
При необходимости экономии площади следует проводить опыт в специ¬
альных коротких севооборотах или же с отдельными эвеньями севооборота,
включающими наиболее важные для данного опыта культуры в их правиль¬
ном чередовании.
554
С. В. Щерба, Ф. А. Юдин
Иногда в многолетних опытах с удобрениями в процессе работы возника¬
ет необходимость введения новых вариантов. Чтобы не снизилась точность
опыта, следует заранее предусмотреть возможность его расширения тем или
иным путем. Первый путь — расщепление первоначальных делянок попо¬
лам; для этого исходные делянки берут большей площади и такой формы,
чтобы технически было возможно их деление и оно не влияло на точность
опыта. Второй путь — наложение дополнительного приема не на все повто¬
рения, а лишь на часть их, для этого при закладке опыта должна быть пре¬
дусмотрена повышенная повторность опыта в пространстве. Третий путь,
позволяющий вводить дополнительные варианты9— это выделение части
делянок при постановке опыта в качестве запасных. Иногда они до введения
дополнительных вариантов используются как контрольные, но лучше их
занимать каким-либо «среднеоптимальным» вариантом, например при изу¬
чении форм удобрения какой-либо нейтральной формой, которую впослед¬
ствии можно заменить на новую.
Закладка опыта
Разбивка опыта. Прежде чем приступить к разбивке опыта в
натуре, необходимо нанести намеченное размещение клиньев и делянок опы¬
та на схематический план участка и уже по нему вести разбивку. Разбивку
основных линий в натуре лучше производить стальной землемерной лентой
(длиной 10 или 20 м) или цепью. Клеенчатые рулетки менее точны и в усло¬
виях полевой работы быстро портятся. Отбивка прямых углов чаще всего
производится эккером, причем восьмигранный эккер удобнее в работе и точ¬
нее, чем зеркальный. Существуют способы отбивки прямых углов и без уг¬
ломерного инструмента, но рекомендовать их не следует, во всяком случае
в условиях опытного поля. Вешки должны быть совершенно прямыми; очень
желательна окраска их в красный цвет или полосами для лучшей видимости.
Наиболее простой является отбивка участка под однолетний опыт, не имею¬
щий севооборотных клиньев.
Первая разбивка участка производится с наибольшей точностью, так как
установленные при ней точки будут служить исходными при всех последую¬
щих разбивках. Допустимая неувязка при разбивке общего контура не долж¬
на превышать 5—10 см в зависимости от общей длины периметра. После
отбивки общего контура приступают к разбивке участка опыта на делянки.
Эта работа проводится уже <без эккера — лентой или рулеткой. При одно¬
рядном расположении достаточно отложить ширину делянок по обеим сто¬
ронам участка и отметить их границы колышками. Ширина последней де¬
лянки должна оказаться одинаковой с остальными, в противном случае ра¬
боту нужно проделать заново. При многорядном расположении сначала от¬
кладывают ряды делянок. Бели между рядами намечены дорожки, одновре¬
менно выделяют и их. Последней операцией является опять разбивка на де¬
лянки. Можно производить ее для каждого ряда в отдельности или же от¬
ложить делянки только по краевым линиям, а на промежуточных границах
или дорожках расставить колышки, провешивая прямые линии с одной сто¬
роны на другую.
При закладке стационарного опыта в севообороте сущность работы оста¬
ется той же, но сначала отбивают общий контур всего севооборота, затем его
разбивают на клинья с выделением дорожек между ними и, наконец, каждый
севооборотный клин разбивают на делянки так же, как это описано для од¬
нолетнего опыта. Бели опыт не закладывают сразу на всех клиньях севообо-
рота, то на делянки разбивается только тот клин, на котором опыт заклады¬
вается в данном году.
Закрепление границ опыта. Сделанную разбивку необхо¬
димо закрепить, так как угловые (временные) колья могут быть выпаханы
Методика полевого опыта с удобрениями
555
или сдвинуты при обработке. Существует много систем закрепления границ
опытов, но в основном все они сводятся к тому, что по крайней мере две ос¬
новные линии участка опыта продолжают по прямой в обе стороны эа пре¬
делы обрабатываемой площади (на края канав, обочины дорог и т. п.) и на
этих продолжениях устанавливают уже постоянные колья (реперы, фикси-
ровочные колья). Расстояния от постоянных (фиксировочных) до угловых
временных кольев тщательно измеряют и записывают с тем, чтобы в случае
утери угловых кольев их всегда можно было восстановить промером от по¬
стоянных. Постоянные колья могут быть сделаны из самого разнообразного
материала. Чаще всего для них употребляются толстые деревянные колья
с перекладиной или крестовиной внизу, зарываемые до 50—75 см в землю.
Могут быть также использованы отрезки железных труб, рельсов и т. п.
Употребляются и каменные столбики. Непосредственно на углах севооборот¬
ного клина или участка однолетнего опыта также забиваются колья, но уже
не такие капитальные. Они всегда рано или поздно повреждаются при па¬
хоте. Поэтому необходимо тщательно проверить их после каждой обработки.
Внесение удобрений. Внесение удобрений представляет один
из ответственных моментов полевого опыта — как в опытах, в которых удоб¬
рение само является изучаемым фактором, так и тех, в которых оно служит
лишь общим фоном для других сравниваемых приемов. Сделанная при вне¬
сении удобрений ошибка впоследствии никак не может быть исправлена,
а большей частью и не бывает обнаружена.
Минеральные удобрения рассчитываются по содержанию в них основ¬
ного питательного вещества (N, Р2Об и К20). Необходимое количество каж¬
дого удобрения (в килограммах на делянку) определяется формулой
а•100с ас
6-10000 = ТооГ »
где X — количество удобрений на делянку, кг; а — доза питательного вещества, кг!га;
Ь — питательное вещество в удобрении, %; с — площадь делянки, м2.
При небольших делянках (меньше 50 м2) удобнее иметь эту величину в
граммах, для чего приведенное выражение надо умножить на 1000:
v iOac
Навеску менее 1 кг отвешивают с точностью до 1 а, от 1 до 10 кг — с точ¬
ностью до 10 г и выше 10 кг—с точностью до 100 г. В зависимости от величины
навески взвешивание производится на технохимических, на столовых ча¬
шечных или на десятичных весах.
Навески изготовляют в лаборатории или в сарае заблаговременно, но
гигроскопические удобрения (нитрат аммония и др.) не должны храниться
в развешенном виде больше 2—3 дней. Все удобрения перед развеской долж¬
ны быть тщательно измельчены и просеяны. Развешивают удобрения в бу¬
мажные пакеты, в матерчатые мешочки или мешки. Бумажные пакеты вы¬
брасываются после однократного употребления, мешки и мешочки можно
каждый раз стирать. На пакетах должны быть написаны вид удобрения и ве¬
личина навески, в мешки вложена или привязана к ним соответствующая
этикетка. Очень удобно иметь пакеты или мешочки различного цвета для
отдельных видов удобрения: азотных, фосфорных и калийных. В поле паке¬
ты или мешочки раскладывают по всем делянкам оныта (или клина севообо¬
рота), после чего еще раз проверяется правильность раскладки. Число на¬
весок должно точно соответствовать потребности; при этом условии ошибка
в раскладке сейчас же обнаруживается недостатком или, наоборот, излиш¬
ком пакета с каким-то удобрением.
Техника рассева удобрений должна обеспечивать возможно равномерное
распределение их по делянкам. Ручной рассев удобрений производится из
556
С. В. Щерба, Ф. А. Юдин
ведер или, что удобнее, из круглых или продолговатых железных тазов.
При небольших дозировках рекомендуется предварительно смешивать удоб¬
рения с землей, взятой с той делянки, для которой предназначается удобре¬
ние. При этом желательно доведение всех удобрений до одинакового объема,
что позволяет сеятелю лучше приспособиться к равномерному высеву на
делянку. Не очень опытному сеятелю рекомендуется рассевать отвешенное
количество на делянку в два приема, проходя ее вдоль и поперек; рассевать
надо так, чтобы иметь остаток удобрений, который всегда можно разбросать
по всей делянке; в случае нехватки удобрений на какую-то часть делянка
должна считаться испорченной. При рассеве удобрений вручную на большой
делянке желательно деление ее на несколько равных частей и внесение соот¬
ветствующей доли удобрения на каждую часть в отдельности.
При внесении на делянку нескольких удобрений следует строго соблюдать
все правила смешения удобрений, излагаемые в курсе агрохимии и практи¬
ческих руководствах. В тех случаях, когда смешение удобрений допустимо,
их лучше вносить в смеси, особенно при ручном внесении, так как при этом
лучше обеспечивается одинаковое соотношение питательных веществ по всей
площади делянки.
Равномерный высев удобрений вручную — работа, требующая большого
навыка и аккуратности. Поэтому чрезвычайно желательно внесение удобре¬
ний на опытные делянки сеялкой. Помимо более равномерного распределе¬
ния, сеялка устраняет необходимость в предварительной развеске удобре¬
ний для каждой делянки, так как нужно лишь точно установить ее на оп¬
ределенный высев. Однако механизированное внесение возможно лишь на
делянках достаточно крупного размера (не менее 100 м2) и требует вытянутой
формы и однорядного расположения делянок. Кроме того, отсутствие не¬
больших и достаточно точно регулируемых сеялок для удобрений часто за¬
ставляет прибегать к ручному внесению удобрений даже на делянках, пло¬
щадь которых вполне достаточна для механизированного внесения.
При механизированном внесении удобрений, так же как и при высеве
различных сортов, засевают сразу все повторные делянки одним удобрением
(или одной комбинацией удобрений), после чего сеялку тщательно очищают
от остатков этого удобрения, производят установку на новую норму и после
этого приступают к высеву следующего удобрения.
При унесении удобрений комбинированными сеялками, в которых высев
регулируется изменением быстроты поднятия подвижного дна тукового ящи¬
ка и которые высевают определенный объем удобрений, рекомендуется до¬
водить все удобрения при помощи какого-либо балласта ((земли, песка и т. п.)
до одинакового для всех вариантов опыта объема на единицу площади. В этом
случае отпадает необходимость изменения нормы высева сеялки каждый раз
при переходе от одного удобрения к другому.
При ручном внесении минеральных удобрений необходимо раздельно вно¬
сить их на каждую делянку даже в тех случаях, когда они не являются изу¬
чаемым фактором и вносятся одинаково во всех вариантах, так как ручное
внесение на больших площадях не обеспечивает равномерного распределения.
Наоборот, при применении сеялки в этих случаях вполне возможно сплош¬
ное Гвнесение через все делянки.
Навоз и другие органические удобрения (торф, компосты) обычно вносят
по общему весу на единицу площади (в тоннах на 1 га), но правильнее вносить их
по расчету на сухое вещество. Расчеты по сухому веществу делаются так же,
как и по содержанию питательных веществ для минеральных удобрений.
В некоторых случаях, при изучении эффективности отдельных питательных
веществ навоза, его можно вносить по расчету на содержание соответствую¬
щего питательного вещества. Бели навоз в навозохранилище или в куче
недостаточно однороден, то перед вывозкой его необходимо тщательно пере¬
мешать в таком количестве, которое достаточно для всего опыта.
Методика полевого опыта с удобрениями
557
Для больших делянок допускается взвешивание навоза на возовых ве¬
сах и вывозка непосредственно на делянку. В этом случае необходимо тща¬
тельно следить за тем, чтобы не было никаких потерь при вывозке и перевоз¬
ке навоза от весов до делянки. Для небольших делянок навоз вывозится в
одну или несколько куч на дорожки, окружающие участок опыта. Здесь
производится тщательное перемешивание, после чего навоз отвешивается и
разносится по делянкам. Взвешивание удобнее производить непосредственно
на носилках, приспособленных для установки на десятичные весы.
Пробы на влажность или на содержание питательных веществ берутся
по возможности незадолго до внесения навоза, так как и то, и другое может
довольно быстро меняться. Правила взятия средних проб излагаются в ру¬
ководствах по сельскохозяйственному анализу. Если анализ нужен не для
расчета навески навоза на делянку9 а для каких-либо последующих расчетов,
пробы лучше брать в момент самого взвешивания из каждого вова или носи¬
лок, соединяя затем эти отдельные пробы и отбирая из них средние. Для
навоза не в меньшей степени, чем для минеральных удобрений, имеет значе¬
ние равномерность распределения по делянке. Распределяют навоз вилами,
а иногда, при мелком навозе и малых делянках, руками. Необходимо об¬
ращать особое внимание на то, чтобы не оставалось излишнего количества
навоза на тех местах, где лежали кучки. На малых делянках лучше разбра¬
сывать навоз непосредственно с носилок, не сбрасывая его предварительно
на делянку. На больших делянках, так же как и при внесении минеральных
удобрений, рекомендуется деление делянки на части для внесения на каж¬
дую часть делянки соответствующей отдельно взвешенной части навоза.
Это совершенно необходимо при больших делянках (200 м2 и больше). Вне¬
сение навоза и других органических удобрений при помощи навозоразбра¬
сывателей из-за несовершенства последних возможно пока только в произ¬
водственных опытах на больших площадях. Навоз обязательно нужно
вносить поделяночно, даже тогда, когда он вносится одинаково во всех вари¬
антах опыта.
При закладке опытов по подкормкам, по химическим способам борьбы
с сорняками, борьбы^с сельскохозяйственными вредителями на больших уча¬
стках в производственных] условиях наиболее совершенно применение
авиации. Участки в этом случае должны быть доступны для самолетов;
вблизи участков необходимо оборудовать посадочную площадку для загрузки
самолета, а в случае применения препаратов в растворах — иметь недалеко
источник водоснабжения. Предварительно необходимо установить секунд¬
ный расход удобрений или препаратов, провести инструктаж сигнальщиков
и организовать механизированную загрузку самолета во избежание длитель¬
ных его простоев. Перед началом работы должен быть составлен план, пре¬
дусматривающий последовательность работ на разных участках, число по¬
летов над каждой опытной делянкой, количество заходов самолета, а также
направление полета и движение сигнальщиков. Предельная скорость ветра
при авиаопрыскивании устанавливается 4 м/сек, причем если скорость рав¬
на 2 м/сек, то полеты производятся в любом направлении; если же скорость
ветра больше 3—4 м/сек, то полеты допустимы только с попутно-встречным
ветром.
Направление полета определяется с помощью сигнальщиков с флажками.
Ширина перехода сигнальщиков от 10 до 15 м в зависимости от рабочего за¬
хвата самолета.
Обработка опытных делянок. Обработка опытных деля¬
нок, помимо обычных требований, предъявляемых к ее качеству в хозяйст¬
венных условиях, должна отвечать требованию полной однородности на всех
делянках опыта. При небольших размерах делянок необходимо, чтобы на
них не было развальных борозд и свальных бугров, создающих неоднород¬
ность внутри делянки. В опытах с удобрениями, в которых обработка сама
558
С. В. Щерба, Ф, А. Юдин
не является изучаемым фактором, применяется, как уже указывалось, обыч¬
но сплошная обработка всего участка опыта или севооборотного клина (в
многолетнем опыте). Для того чтобы огрехи, разница в глубине отдельных
борозд и другие подобные дефекты не нарушили сравнимости между делян¬
ками, направление обработки ориентируют обычно таким образом, чтобы
каждая борозда проходила через делянки одного повторения или серии опыта-
При вытянутой форме делянок обработка производится, как правило, по¬
перек делянок (по длине участка). Нужно лишь, чтобы пласт отваливался
при первой обработке в одну, а при следующей — в обратную сторону. При
квадратной форме делянок и многорядном их расположении обработку мож¬
но производить в обоих взаимно перпендикулярных направлениях. Само
собой разумеется, что на опытном участке фигурная пахота недопустима,
а применима только загонная. Повороты любых орудий должны производить¬
ся за пределами участка опыта. Для этого по коротким концам участков или
клиньев должны оставаться свободные дорожки не менее 6 м шириной при
конной обработке и 10—12 м при тракторной. Для продольных дорожек,
если поперечная обработка не производится, достаточна ширина от 3 до 5 м*
Для того чтобы свалы и развалы не попадали на учетные делянки, про¬
изводят одновременно вспашку двух рядов делянок (или двух половин мно¬
горядного участка опыта), пригоняя свал или развал на их границу. При
достаточной ширине защитных полос (не менее 2 м) и аккуратной работе
свал или развал не захватывают учетную часть делянок. Лучше, однако,
иметь для этой цели специальную дорожку шириной 1—2 м посередине опыт¬
ного участка. Вспашки участка всвал и вразвал должны чередоваться между
собой. При однорядном расположении делянок или многорядном, но при ма¬
лой величине делянок и отсутствии средней дорожки на участке опыта при¬
ходится прибегать к вспашке в одну сторону.
Посев и посадка в опытах. Посев на опытных участках тре¬
бует большой тщательности. Так же как и пахоту, посев производят обычно
через все делянки повторности, перпендикулярно их длинным сторонам, с тем
чтобы случайные дефекты, например забившийся сошник, влияли одинаково
на все варианты опыта. Исключение представляет посев комбинированной
сеялкой, когда на отдельные делянки вносят при посеве различные удобре¬
ния. В этом случае каждую делянку засевают отдельно, и посев производят
вдоль делянки и поперек участка. Такого рода опыты требуют вытянутой
формы делянок и их однорядного расположения. Включение и выключение
сеялок производят за пределами собственно опыта, не ближе 1 м от границы
(линии угловых кольев). Обсев краев (поперечный) не производят или про¬
изводят за границей участка опыта.
Первый ход сеялки делают либо по шнуру, натянутому с кола на кол по
длинной стороне участка, либо по предварительно сделанной по шнуру бо¬
роздке; последнее удобнее. Совершенно недопустимы всякие остановки се¬
ялки во время хода: при небольших делянках остановка сеялки на делянке
и связанные с нею просев или двойной высев могут чувствительно повлиять
на урожай этой делянки.
При посадке пропашных культур нужно, чтобы число растений на всех
делянках было строго одинаково. Для этого ширина междурядий и расстоя¬
ния между растениями в рядках должны быть рассчитаны таким образом,
чтобы на делянку приходилось целое число борозд и кустов (или, наоборот,
длину и ширину делянки надо брать кратными стандартным расстояниям
между растениями в том или другом направлении). Борозды, так же как и
рядки, при посеве лучше располагать вдоль участка и поперек делянок, но
при достаточно больших и вытянутых делянках возможно и обратное рас¬
положение, т. е. нарезка бороэд по длине делянки.
Ко всем работам, производимым на опытных участках, относится требова¬
ние одновременного выполнения их на всех делянках опыта.
Методика полевого опыта е удобрениями
559
Желательно, чтобы каждая работа — внесение удобрений, вспашка,
посев — выполнялась в опыте в течение одного дня.
Защитные полосы. При сплошной обработке участков на грани¬
цах различно удобренных делянок постоянно происходит некоторый перенос
почвы, а с ней и внесенных удобрений с одной делянки на другую. Кроме то¬
го, сами растения, расположенные по краю делянки, могут использовать
своими корнями питательные вещества не только со своей, но и с соседней,
более богатой питательными веществами делянки. Поэтому по краям неудоб¬
ренных (или более слабо удобренных) делянок, граничащих с удобренными,
Рис. 7. Опытная и учетная
площадь делянкп и защитные
полосы
АВ CD и DCEF—опытные делянки
по 120 м*; abed и d'e'e/—учетные
делянки по 75 м*
/л/-
В
«в<
2м
2-я де/гямка
Защитная полоса
d'
“'V'—
8 м
всегда наблюдается некоторая полоса растений, развитых лучше, чем во внут¬
ренней части делянки.
Краевые растения, соприкасающиеся с незасеянными дорожками по краю
поля, также развиваются сильнее остальных растений, так как они допол¬
нительно используют влагу и питательные вещества с этих дорожек и, кроме
того, находятся в лучших условиях освещения. Влияние такого усиленного
развития краевых растений на общий урожай с делянки тем сильнее, чем
меньше ее площадь. Чтобы избежать ошибки в урожае эа счет переноса удоб¬
рений и более сильного развития краевых растений, учитывают обычно не
всю засеянную площадь делянки, а лишь ее центральную часть, обрезав бо¬
лее или менее широкие полосы по краям делянки.
Эти полосы носят название защитных. Первоначальная площадь, вклю¬
чающая защитные полосы, называется опытной делянкой, а площадь, ос¬
тающаяся после обрезки защитных полос и служащая собственно для учета
урожая,— учетной делянкой (рис. 7). Так как на границе двух делянок за¬
щитные полосы отрезаются по обе стороны границы, то общая ширина неучи¬
тываемой полосы, разделяющей учетные делянки, равна удвоенной ширине
защитной полосы.
Необходимая ширина защитных полос определяется тем, насколько да¬
леко в глубь делянки может простираться влияние дополнительной площади
питания и дополнительного освещения от обнаженного края делянки и на¬
сколько далеко могут переноситься удобрения с соседней делянки. Как по¬
казывают некоторые проведенные С. В. Щербой методические исследования,
влияние дополнительного питания и освещения на обнаженном краю делян¬
ки не простирается в сколько-нибудь значительной степени дальше двух-трех
крайних рядков для зерновых и одного рядка для пропашных культур.
560
С. В. Щерба, Ф. А. Юдин
Перенос удобрений на границе двух делянок может захватывать более ши¬
рокую полосу, особенно в многолетних опытах.
В ряде старых опытов Долгопрудного поля при ширине защитных полос
0,3 сажени от каждой делянки (в сумме обеих соседних делянок 0,6 сажени,
или 1,25 м) уже после 15 лет сплошной обработки поперек делянок наблю¬
дается явное «наплывание» удобренных делянок на неудобренные. Поэтому
нужно считать минимальной ширину защитных полос в многолетних опытах
по 1 м от каждой делянки, или 2 м в сумме от обеих соседних делянок. Для
однолетних опытов допустимы более уэкие защитные полосы, но все же же¬
лательно иметь их не меньше 75 см (1,5 м в сумме). Иногда защитные полосы
по узким сторонам делянок, совпадающим с краями участка, делают шире
остальных с тем» чтобы они могли служить также защитой от потрав и дру¬
гих случайных повреждений.
Защитные полосы обрабатывают, удобряют и засевают вместе со всей
делянкой. Растения убирают на них непосредственно перед уборкой учет¬
ных делянок. Для того чтобы можно было точно выделить защитные полосы
перед уборкой, необходимо заранее зафиксировать их границы. Чаще всего
для этого пробивают по шнуру мотыгой или ручным планетом узкую полосу
(15—20 см) по границе между учетной делянкой и защитной полосой (в сто¬
рону защитной полосы). Такую отбивку производят для яровых культур
через 2—3 недели после появления всходов (обычно после полного окон¬
чания посевной и некоторого освобождения рабочих рук). Для озимых
отбивку защитных полос производят поздно осенью или весной до выхода
растений в трубку. Существует и ряд других способов выделения защит¬
ных полос. Удобно, особенно для небольших делянок, натягивать по гра¬
ницам защитных полос проволоку.
Для. пропашных культур необязательно предварительно фиксировать
границы защитных полос. Нужно только, чтобы как на всей делянке, так
и отдельно на защитной полосе и учетной делянке умещалось целое число
рядов (борозд). Чаще всего на защитную полосу (от каждой делянки) при¬
ходятся две борозды, на сдвоенную защитную полосу (от двух смежных
делянок) — четыре борозды. При нечетном числе борозд, приходящихся
на сдвоенную защитную полосу (три или пять), одну борозду помещают
как раз на границе опытных делянок, посередине сдвоенной защитной по¬
лосы.
При достаточно аккуратной посадке выделить защитные полосы перед
уборкой можно простым отсчетом борозд или рядов. Нужно стремиться
к тому, чтобы площадь как опытной, так и учетной делянки составляла
какую-то целую часть гектара (0,01; 0,02 и т. д.). Это очень упрощает рас¬
четы удобрений на делянку и пересчет урожаев на 1 га.
Кроме защитной полосы делянки, можно выделить еще защитную полосу
вокруг всего участка опыта или севооборотного клина (защитная полоса
опыта). Эта полоса уже не входит в площади опытных делянок. Ее можно
засевать как одновременно с опытом (при сплошном посеве всех делянок),
так и отдельно. Ширина ее может варьировать в широких пределах, от одно¬
го до двух десятков метров. Убирают ее перед уборкой деляночных защитных
полос или одновременно с ними.
Программа полевого опыта
В программу должно быть включено детальное описание всех условий
проведения опыта, имеющих существенное значение с точки эрения при¬
годности опыта для решения поставленной задачи, а также обеспечивающих
точность и достоверность получаемых в опыте результатов, но не являю¬
щихся предметом непосредственного изучения в данном опыте.
Методика полевого опи/па с удобрениями
561
Основной результат полевого опыта — урожай по вариантам. Кроме
того, существенное значение для оценки результатов опыта приобретают
сопутствующие наблюдения и аналитические исследования. Сюда отно¬
сятся наблюдения за ростом и развитием растений на опытных делянках*
состоянием почвы, фенологические наблюдения, учет метеорологических
факторов в течение года и т. д. Эти наблюдения предусматриваются про¬
граммой опыта.
В программу должно быть включено описание всех агротехнических
приемов, которые выполняют в опыте и которые создают его производствен¬
ную типичность. Это так называемый агротехнический фон опыта.
Агротехнический фон на опытном участке должен быть оптимальным
для проявления эффекта от изучаемого приема и, как правило, с макси¬
мальным использованием техники. При составлении программы сопутствую¬
щих наблюдений и учетов в период вегетации растений, а также аналитиче¬
ских исследований очень важно правильно запланировать методику отбора
образцов растений, почв по вариантам опыта и указать методику проведения
тех или иных исследований и анализов.
Программа опыта должна также включать методы и элементы учета ре¬
зультатов опыта. В ней необходимо предусмотреть методику статистической
обработки данных полевого опыта, статистической оценки наиболее сущест¬
венных для понимания полученных в опыте результатов сопутствующих на¬
блюдений и аналитических исследований (например, в опытах с удобрения¬
ми — показатели качества урожая, некоторые данные агрохимических ана¬
лизов почв и растений и т. д.).
Уход за растениями и сопутствующие наблюдения
в течение вегетационного периода
Уход эа растениями на опытных делянках не отличается, по существуг
от ухода за соответствующими культурами в хозяйственных условиях. Нуж¬
но лишь, чтобы все работы по уходу (полка, пропашка и т. п.) выполнялись
постоянно и совершенно одинаково по всем делянкам и не растягивались во
времени. Желательно, чтобы эти работы, по крайней мере в пределах каждо¬
го повторения, производились в течение одного дня.
В опытах могут и должны применяться все существующие способы борьбы
с вредителями и болезнями растений, не влиющие на питательный режим поч¬
вы. Конечно, нельзя применять в опытах с фосфатами суперфосфат для борь¬
бы с полевым слизнем или томасшлак для борьбы с блошкой.
К специальным работам по уходу на опытном поле относятся: поддержа¬
ние в чистоте дорожек и запольных участков, обрезка по шнуру концов по¬
сле появления всходов и т. п.
Дорожки и дороги на опытном поле, как между отдельными участками
опыта, так и между клиньями севооборотов, либо поддерживаются в черном
пару, либо засеваются после окончания весенних работ какой-нибудь куль¬
турой, чаще всего вико-овсяной смесью. Некоторыми опытными учреждения¬
ми проектируется содержание дорожек под дерниной. Это удобно для езды
и ходьбы, но задернелые дорожки могут служить рассадником всевозможных
вредителей, в частности проволочного червя.
При проведении опытов в условиях производства опытные делянки не
отделяются обычно до момента уборки от общего массива.
К специальным работам на опытном поле относится также расстановка
этикеток по делянкам. Наличие этикеток не только облегчает демонстрацию
и наблюдение над ними, но и уменьшает возможность ошибок при таких ра¬
ботах, как внесение удобрений, высев различных сортов и, наконец, учет
урожая. Поэтому желательно, чтобы этикетками снабжались все делянки с
самого начала вегетационного периода.
562
С. В. Щерба, Ф. Л. Юдин
Метеорологические наблюдения
Правильное объяснение результатов полевого опыта возможно лишь
при наличии сведений о метеорологических условиях проведения опыта.
В кратковременных опытах, однако, нет возможности организовать основ¬
ные инструментальные наблюдения, и необходимые метеорологические дан¬
ные приходится получать с ближайшей метеорологической станции. На по¬
стоянных опытных участках организуются инструментальные наблюдения
за погодой. Объем этих наблюдений определяется их задачами и близостью
метеорологической станции, откуда можно получать необходимые сведения,
например в виде декадных сводок. При этом приходится учитывать степень
изменчивости показателей тех или иных элементов погоды по территории.
В большей степени изменчивы показатели количества осадков, а также ми¬
нимальной температуры почвы в периоды наступления весенних и особенно
зимних заморозков. Поэтому на постоянных опытных участках организуются
прежде всего наблюдения за осадками (в течение всего года), за снеговым
покровом (от выпадения до схода), за минимальной температурой на поверх¬
ности почвы (в периоды, когда возможны понижения температуры, отрица¬
тельно сказывающиеся на посевах). Ведутся наблюдения за такими условия¬
ми погоды, которые могут вызвать повреждения посевов: засуха, суховеи,
градобитие, ледяная корка и т. п. При необходимости организуются наблю¬
дения за температурой и влажностью воздуха, температурой почвы, ее
промерзанием и т. д. В отдельных случаях ведутся также микроклиматиче¬
ские наблюдения на посевах.
Метеорологические наблюдения производят по местному среднему сол¬
нечному времени. Чтобы определить его, разность долгот между средним
меридианом пояса и меридианом данного пункта переводят на время (один
градус долготы равен 4 мин., и 1 мин. долготы равна 4 сек. времени).
Наблюдения за осадками (при помощи дождемера), температурой воз¬
духа, минимальной температурой на поверхности почвы, влажностью и
давлением воздуха ведут на постоянной площадке, которую выбирают в не¬
посредственной близости от опытного участка, на открытом месте, в услови¬
ях рельефа, соответствующих положению опытного участка, не ближе 50—
100 м от ближайших построек и деревьев.
Все наблюдения проводят в определенные часы дня.
Количество атмосферных осадков измеряют при помощи дождемера еже¬
дневно в 7 и 19 час., причем это количество относят ко дню наблюдения.
Измерение высоты снегового покрова производят при помощи постоянных
или переносных снегомерных реек. Одновременно регистрируют состояние
снегового покрова (уплотненность, равномерность и пр.), указывая, на
какую землю лег устойчивый снеговой покров: талую или промерзшую, и ха¬
рактеризуют сход снега весной. Измерение высоты снегового покрова при
помощи постоянной рейки производят ежедневно в 7 час. утра, а измерения
при помощи переносной рейкп — ежедекадно, а также после сильных сне¬
гопадов, метелей и т. д. Для характеристики залегания снегового покрова
на всей площади опытного участка во время максимального залегания снего¬
вого покрова можно производить разовую маршрутную снегосъемку.
Для измерения температуры и влажности воздуха используют психро¬
метр Августа («смоченный» и «сухой» термометры) и минимальный и макси¬
мальный термометры. Наблюдения производят ежедневно в 1, 7,13 и 19 час.
и все данные относят ко дню наблюдения. Абсолютная и относительная влаж¬
ность воздуха, а также «дефицит влажности» вычисляют по исправленным
показаниям психрометров Августа при помощи психрометрических таблиц.
Наблюдение минимальной температуры на поверхности почвы ведут при по¬
мощи минимального термометра, устанавливаемого на той же метеорологиче¬
ской площадке. Для введения поправок на давление при вычислении влаж¬
Методика полевого опыта с удобрениями
563
ности воздуха по психрометрическим таблицам необходимы наблюдения за
атмосферным давлением (по анероиду). В некоторых опытах могут представ¬
лять специальный интерес определения температуры пахотного горизонта в
период вегетации. Для этой цели используется комплект ртутных термомет¬
ров Савинова, при помощи которых определяется температура почвы на глу¬
бинах 5, 10, 15 и 20 см.
Определение глубины промерзания почвы производят почвенным буром
Качинского или способом вырубки.
Совокупность перечисленных выше метеорологических наблюдений дает
возможность охарактеризовать агрометеорологические условия сельскохо¬
зяйственного года по периодам.
Учет засоренности
При характеристике засоренности применяются как глазомерные, так
и количественные приемы учета. При более детализированных системах уче¬
та засоренности различают учет сорной растительности по надземным веге¬
тативным частям и учет засоренности (вегетативные зачатки, семена и пло¬
ды) в пахотной почве.
Учет вегетативных надземных частей в основном включает глазомерное
определение распределения сорняков на учетной площади и количественный
учет сорняков. При глазомерной характеристике засоренности может оп¬
ределяться видовой состав сорняков, степень покрытия ими площади (оп¬
ределение процента площади, занятой тем или иным видом или группами
их), обилие их (количество экземпляров), определение массы сорных расте¬
ний, а также характеристика встречаемости (характер распределения сор- -
няков на площади), ярусности (расположение сорных растений по отношению
к культурным) и т. д.
Степень распространения сорняков оценивается по пятибалльной шкале:
1) до 1% общей площади; 2) 1—5%; 3) 5—25%; 4) 25—50%; 5) более 50%
площади; при распространении сорняков куртинами применяются двойные
баллы для характеристики распространения сорняка и в куртинах. Ярус-
ность сорняков устанавливается по отношению к культурным растениям*
(выше, наравне, до половины высоты, не попадающие под нож уборочной
машины). Высота сорняков определяется по главной массе сорняков. Обо¬
значаются также фазы развития сорняков (всходы, стеблевание, цветение,
засыхание растений, розетки, бутонизация, колошение, созревание семян)».
Глазомерная оценка засоренности может сопровождаться учетом количества
и веса сорняков на метровых площадках, накладываемых на делянке через
определенные расстояния.
Учет засоренности пахотного слоя почвы заключается в определении'
семян и плодов сорняков в почве при помощи взятия проб буром Шевелева
с последующим выделением семян при помощи химических растворов. Для
определения содержания и расположения в почве корневищ, корней сорня¬
ков, способных укореняться и отрастать, применяются специальные методы
учета. В опытах с многолетними травами имеет значение определение за¬
соренности урожая, которое обычно проводится по пробному снопу (весом
5—6 кг), отбираемому для определения выхода сена.
Фитопатологические1 и энтомологические наблюдения
Фитопатологические и энтомологические исследования в полевых опытах
очень важны, так как болезни и вредители сильно влияют на рост, развитие
и урожай сельскохозяйственных культур. Однако проведение их требует
специальных знаний и навыков.
Распространенность болезней и вредителей можно учитывать по двум
564
С. В. Щерба, Ф. А. Юдин
показателям: а) по проценту пораженных растений, колосьев, метелок,
початков и пр.; б) по проценту площади, занятой пораженными культурами,
чаще всего глазомерно. Степень поражения растений болезнями и вредите¬
лями обычно ведут выборочным методом на небольшом числе растений. От¬
сюда важно правильно отбирать пробы. Все учеты болезней растений прово¬
дят по фазам развития растения. Самый простой способ оценки поражения
растений суммарно всеми болезнями и вредителями по пятибалльной шка¬
ле: 0 — отсутствие повреждений и поражений; 1 — повреждены единичные
растения (до 10%); 2 — повреждено 10—25% растений; 3 — повреждено
25—50% растений; 4 — повреждено 50—75% растений; 5 — повреждено
свыше 75% растений. При обнаружении заболеваний или вредителей на
растениях надо определить характер заболевания и вид вредителя и применять
рекомендуемые меры борьбы с ними.
Наблюдения над зимующими культурами
и учет зимостойкости культур и сортов
Наблюдения над зимующими культурами в осенний, зимний и весенний
лериоды имеют наибольшее значение в тех районах, где эти культуры часто
гибнут или сильно изреживаются при перезимовке. Наблюдения ведутся так¬
же за приемами агротехники, которые оказывают значительное влияние на
зимостойкость культуры, например удобрение, сроки посева и др. Наиболее
употребительными приемами наблюдения за состоянием зимующих культур
являются: периодические взятия проб на отрастание в течение зимы, подсчет
живых и погибших за зиму растений и глазомерные оценки травостоя перед
уходом в зиму и в начале весенней вегетации
Сроки взятия проб озимых культур на отрастание приурочиваются к
25 числу каждого месяца, начиная с 25 декабря, и для многолетних трав —
к 10 числу каждого месяца, начиная с 10 декабря; при неблагоприятных ус¬
ловиях — и чаще. Пробы берут на неучетных частях делянок или на за-
щитках в виде монолитов размером 25 X 25—30 (ширина) X 25 см. Учет
результатов отращивания проб выражается числом (процентом) живых и
погибших растений.
Процент погибших за зиму растений определяют весной после того, как
живые растения тронутся в рост и процесс отмирания поврежденных расте¬
ний в основном закончится. Чаще рекомендуется не сопоставление числа рас¬
тений, имевшихся осенью на закрепленных для учета площадках, с числом
растений, зарегистрированных на тех же площадках при весеннем подсчете,
я весенний подсчет живых и погибших растений на выделенных весной пло¬
щадках. Площадки для учета выделяются на концевых защитках. Все на¬
блюдения на посевах зимующих культур имеют целью не только простую
регистрацию гибели или повреждений озимых культур и многолетних трав,
но и выяснение причин гибели или повреждений. Поэтому существенное
значение имеет правильная классификация этих причин.
Наиболее часто учитывают следующие виды повреждений посевов в зим¬
ний период: а) вымерзание вследствие низких температур; б) выпревание,
имеющее место при большом снеговом покрове, сравнительно мягкой 8име
и талой почве, когда растения ослабляются в результате усиления дыхатель¬
ных процессов и расхода накопленных с осени веществ и подвергаются вес¬
ной поражению снежной плесенью; в) повреждения от ледяной корки (глав¬
ным образом притертой); г) вымокание посевов от застоя дождевых или та¬
лых вод; д) выпирание посевов в результате смены оттепелей и морозов и об¬
разования ледяной прослойки, вызывающей разрыв корней; е) высыхание
посевов, в частности при вегетации надземной части растений весной, когда
корни растений находятся еще в замерзшей почве; ж) выдувание посевов с
обнажением узлов кущения и повреждением их морозами.
Методика полевого опыта с удобрениями
565
Фенологические наблюдения
Фенологические наблюдения имеют целью установить различия в ходе
развития растений по отдельным вариантам опыта. Эти наблюдения, правиль¬
но поставленные, могут дать ценнейший материал для объяснения причин
того или иного характера действия изучаемых приемов или удобрений. От¬
сутствие увеличения окончательного урожая не всегда доказывает неэффек¬
тивность примененного приема. Часто оно обусловлено тем, что благоприят¬
ное действие того или иного приема, проявлявшееся в начальный период
развития, в дальнейшем было подавлено или ограничено какими-то неблаго¬
приятными условиями или внешними воздействиями. Фенологические на¬
блюдения позволяют обнаруживать эффекты, не сохраняющиеся до учета
урожая, и искать причины их дальнейшего затухания.
В зависимости от степени детальности наблюдений, программы и схемы
опытов и культуры можно регистрировать начало фазы и массовое наступле¬
ние ее (полная фаза). За начало фазы принимают первый день, в который она
-зарегистрирована не менее чем у 10% растений, а за массовое наступление —
день, в который фаза отмечена не менее чем у 50% (или 75%) растений.
В системе агрометслужбы у зерновых злаков отмечаются всходы, третий
лист, кущение, выход в трубку (стеблевание), колошение, цветение, молоч¬
ная спелость, восковая спелость, полная спелость; кроме того, отмечается
укоренение, а у озимых культур и начало вегетации.
У гречихи, подсолнечника, конопли отмечаются всходы, начало роста
-стебля (у гречихи не отмечается), образование соцветий, цветение, созрева¬
ние. У льна: всходы, начало роста стебля, образование соцветий, цветение,
конец цветения, зеленая и полная спелость семян. У картофеля: всходы,
образование соцветий, конец цветения, увядание ботвы. У сои: всходы,
появление первых настоящих листьев, появление третьего настоящего ли¬
ста, образование боковых побегов, цветение, созревание. У гороха, фасоли,
бобов, чечевицы, вики: всходы, начало образования боковых побегов, об¬
разование соцветий, цветение, созревание. У хлопчатника: всходы, образо¬
вание третьего листа, образование бутонов, цветение, раскрытие первых ко¬
робочек, прекращение вегетации. У табака: всходы, первый настоящий лист,
третий настоящий лист, образование соцветий, цветение. У сахарной свеклы
и других корнеплодов: всходы, первая пара настоящих листьев, третий на¬
стоящий лист, начало утолщения подсемядольного колена, увядание наруж¬
ных листьев (в сортоиспытании сахарной свеклы регистрируются фазы:
1 — вилочка, 2 — 1-я пара настоящих листьев, 3 — 3-я пара листьев, 4 —
смыкание листьев в рядках, 5 — смыкание листьев в междурядьях, 6 —
отмирание 5-й пары листьев, 7 — размыкание листьев в междурядьях).
У клещевины: всходы, первый лист, третий лист, образование соцветий,
цветение, созревание. У злаковых трав: всходы, кущение (начало кущения в
год посева), выход в трубку, колошение (выметывание), цветение, хозяйст¬
венная спелость, отмирание (конец вегетации). У бобовых трав: всходы,
образование боковых побегов, образование соцветий, цветение, хозяйствен¬
ная спелость, отмирание (конец вегетации). У трав отмечается также во¬
зобновление вегетации весной.
Наблюдения за временем наступления отдельных фаз развития и полной
или хозяйственной спелости позволяют установить как длительность отдель¬
ных периодов между фазами, так и общую длину вегетационного периода,
которая обычно определяется от полных всходов до полной или хозяйствен¬
ной спелости. Соответствующие вычисления удобно вести по специальным
таблицам, в которых все дни года занумерованы и по которым подсчеты ве¬
дутся путем вычитания номера, соответствующего исходной дате, из но¬
мера, соответствующего последующей дате.
Данные фенологических наблюдений используются при разбивке всего
566
С. В. Щерба, Ф. Л. Юдин
периода вегетации растений (начиная от посева) на отдельные отрезки, перио¬
ды, применительно к которым суммируются и показатели метеорологические.
Так, для хлебных злаков устанавливаются такие периоды между названными
выше фазами: посев — всходы — выход в трубку — колошение — спелость.
Важно, однако, не столько установление самих сроков наступления тех
или иных фаз, сколько констатация смещения этих сроков на удобренных
делянках по сравнению с контролем. Этими фазами удобно также пользовать¬
ся как определенными этапами для записи видимых различий в характере
развития растений (окраска, рост, полегание и т. п.). Очень важно немедлен¬
но отмечать появление этих различий, а также их исчезновение, не дожидаясь
определенных сроков.
Все различия в развитии делянок должны быть охарактеризованы со¬
вершенно конкретно и по возможности количественно. Из количественных
показателей развития можно отметить прежде всего число растений на 1 м2
(густоту всходов) и число стеблей на одно растение (энергию кущения). Пер¬
вое определение делают после появления полных всходов, второе для ози¬
мых — поздно осенью, для яровых — весной, по окончании кущения или
при начале выхода в трубку. Так как озимая пшеница продолжает куститься
и весной, то в этом случае интересно не только осеннее, но и весеннее опре¬
деление кущения.
Для обоих определений берут некоторое число рядков по 1 ле, составляю¬
щих, в зависимости от пестроты поля и величины делянок, один или несколько*
квадратных метров (при обычной ширине междурядий наших зерновых сея¬
лок на 1 м2 приходится 8 метровых рядков). Эти рядки располагаются в раз¬
ных местах делянки, например по диагоналям. Для определения энергии
кущения число побегов, подсчитанное на погонный метр, делят на число рас¬
тений (кустов). Из величин, полученных для каждого погонного метра, вы¬
водится среднее на 1 м2 (обычно с одним десятичным знаком).
Такие же рядки применяются для определения процента перезимовки
озимых. На выделенных и отмеченных колышками рядках подсчитывают с
осени число растений, идущих в зиму, а весной после начала вегетации —
число растений, оставшихся в живых. Вторая величина выражается в про¬
центах от первой. Вымочки, а также пятна выпревания, вымерзания и т. п.,
захватывающие более или менее значительную часть делянки, обмеривают
и наносят на схематический план опыта.
Из позднейших количественных определений наиболее общими являются
высота растений и густота стояния (колосоносных побегов). Высота опреде¬
ляется как среднее из промеров значительного числа растений (20—50).
Особое значение имеет определение высоты для прядильных культур (лен,,
конопля). Густота стояния определяется по пробным рядкам или площад¬
кам (на 1 м2).
В течение всего вегетационного периода представляют интерес наблюде¬
ния за приростом сухого вещества. Нарастание массы сухого вещества оп¬
ределяется либо по фазам развития, либо по календарным срокам (например,
по декадам). Пробы для определения прироста сухой массы берут также с
пробных рядков (метровок) или площадок (0,25—1,0 м2). Для пропашных
культур (свекла, картофель) взятие проб с определенной площадки заменяют
обычно взятием определенного числа растений (100 растений свеклы вначале
и 10—20 корней свеклы или кустов картофеля на более поздних стадиях
развития). Эти же пробы могут быть использованы в дальнейшем для хи¬
мических анализов, в частности для определения хода поступления питатель¬
ных веществ в растения. Возможность взятия растительных проб должна
быть предусмотрена при определении площади делянки.
Совершенно обязательна своевременная запись всякого рода случайных
повреждений (потрав и т. п.) с нанесением их на план участка опы¬
та. Тщательно отмечаются также повреждения растений вредителями и бо¬
Методика полевого опыта с удобрениями
567
лезнями. Желательно отмечать начало и конец заболевания, а также давать
количественную характеристику степени поражения. Имеются специальные
инструкции для определения степени повреждения насекомыми и болез¬
нями.
В последнее время большое значение придается определению так называе¬
мой структуры урожая. Наиболее важными показателями ее для хлебных
злаков являются: 1) число растений на единицу площади; 2) число побегов
колосоносных и пустых; 3) число колосьев на растении; 4) число колосков
в колосе; 5) число зерен в колосе; 6) вес 1000 зерен. Структура урожая, ус¬
танавливаемая обычно уже для зрелых растений, так же как фенологические
наблюдения за ходом развития растений в течение вегетационного периода,
имеет целью выяснить, за счет каких именно изменений в развитии растений
происходит то или иное изменение конечного урожая.
При всем значении фенологических наблюдений нельзя забывать, что
окончательным критерием для оценки всякого агрономического приема яв¬
ляется все же конечный урожай продуктивной (используемой) части расте¬
ния. Поэтому данные фенологических наблюдений в полевом опыте не само¬
цель, а лишь подсобный материал, помогающий уяснить условия получения
тех или иных прибавок окончательного урожая.
Совершенно необязательно во всех опытах проводить все возможные фе¬
нологические наблюдения. Они должны вытекать из задач опыта или из
необходимости расшифровки ранее полученных урожайных данных. Неболь¬
шое число хорошо продуманных и целеустремленных наблюдений, действи¬
тельно характеризующих особенности развития растений, обусловленные
влиянием изучаемого в опыте фактора, гораздо ценнее, чем груды шаблон¬
ных описаний фаз развития, весьма трудоемких, но в дальнейшем обычно не
используемых.
Учет результатов опыта
Подготовка к учету. Прежде чем приступить к учету урожая,
надо удалить растения с тех частей делянки, которые не поступают в учет.
Прежде всего убирают защитные полосы. Для культур сплошного посева
защитные полосы выжинают или выкашивают по заранее пробитым бороз¬
дам или по натянутой проволоке. Бели растения слишком густы, полегли
или перепутались, так что границы защитных полос плохо видны, их пред¬
варительно проходят и разбирают руками. Снопы с защитных полос выносят
на дороги, чтобы их нельзя было смешать с урожаем учетных делянок. Для
пропашных культур отсчитывают число борозд или рядков, приходившихся
на защитные полосы; выкапывают и урожай увозят с поля.
После этого производят пооледний осмотр учетных делянок, при котором
отмечают выключки или производят полную выбраковку отдельных делянок.
Выключку и выбраковку целых делянок делают с учетом всех предыдущих
записей и наблюдений и лишь в том случае, если есть совершенно объектив¬
ные данные, говорящие о вымочке, повреждении, ошибке в работе или ка¬
кой-либо другой причине, заведомо изменившей урожайность делянки или ее
части. Делать выключки и браковать делянки на основании чисто субъек¬
тивного впечатления о неоднородности повторений или частей делянки
ни в коем случае не следует.
Для простоты последующих расчетов выключки следует делать прямо¬
угольными или, еще лучше, выключать определенную часть делянки: поло¬
вину, треть, четверть и т. д. При небольших делянках (10—20 м2) и достаточ¬
ной повторности лучше вообще не делать выключек, а выбраковывать де¬
лянки целиком. Выключки обмеривают, отмечают колышками и убирают по
шнуру. Одновременно убирают делянки, забракованные целиком. Урожай
с тех и других также удаляют за пределы участка опыта. Для пропашных
568
С. В. Щерба, Ф. Л. Юдин
делают одновременно подсчет наличных кустов или корней на каждой учет¬
ной делянке для внесения в дальнейшем поправок на недостающие растения.
Прямой и косвенный методы учета урожая. Для
учета опыта может быть использована либо вся масса урожая, полученного
с учетной делянки, либо ее часть, представляющая составленную тем или
иным способом среднюю пробу из урожая всей делянки. Первый метод учета
урожая может быть назван прямым, второй — косвенным. В применении
к зерновым культурам прямой метод учета называется учетом по обмолоту
всей делянки. Наиболее распространенным косвенным методом учета зер¬
новых является учет по пробному снопу.
При учете по обмолоту всей делянки урожай с каждой учетной делянки
целиком увозят в сарай или под навес, где хранят до момента обмолота.
Взвешивание на поле в этом случае необязатально, хотя и желательно; ос¬
новная задача состоит в том, чтобы избежать потерь и перемешивания по-
деляночных урожаев до обмолота. Для достижения этой цели, а также для
обеспечения полной однородности самой уборки по делянкам опытные уч¬
реждения разработали ряд технических приемов, сводящихся в основном к
следующему.
Уборку учетных делянок можно производить как вручную — серпами
или косой с грабельками (крюком), так и уборочными машинами — жней¬
кой и др. При уборке вручную для достижения однородности работы по делян¬
кам (одинаковой высоты жнивья и одинаковой чистоты уборки) все жницы или
косцы убирают вместе каждую делянку опыта, постепенно переходя от одной
делянки к другой. При очень малых делянках, когда невозможно поставить
на одну делянку несколько человек, каждая жница убирает урожай всех
повторностей опыта. Один косец убирает при этом обычно весь урожай опыта.
Теоретически убирать жнейкой можно делянки любой величины, но прак¬
тически применять жнейку имеет смысл при величине делянки не меньше
200—300 м2. При очень большой площади делянок (0,1 га и больше) жнейкой
убирают каждую делянку в отдельности. При меньшем размере делянок (не¬
сколько сот квадратных метров) удобнее убирать подряд (одним проходом
жнейки) несколько делянок, например одно повторение. При этом на грани¬
цах делянок стоят рабочие, следящие эа тем, чтобы урожай не перетаски¬
вался с делянки на делянку. Некоторые авторы рекомендуют, во избежание
смешения урожаев, убирать делянки через одну. При исправной машине,
хорошей организации труда и неполегших хлебах применение жнейки зна¬
чительно ускоряет уборку больших делянок и имеет еще то преимущество,
что качество уборки получается строго одинаковым на всех делянках.
Урожай с каждой учетной делянки сейчас же связывают, снопы пересчи¬
тывают и складывают в бабки или копны. Продолжительность выстаивания
на поле зависит от погоды и влажности урожая, но чем оно короче, тем луч¬
ше. При перевозке урожая в сарай на повозку обязательно должны подсти¬
латься брезенты. Желательно, чтобы на каждый воз укладывался урожай
лишь с одной делянки. При перевозке на одном возу урожаев с нескольких
делянок их обязательно нужно перестилать брезентами. Некоторые опытные
станции применяют специальные повозки или фургоны, обшитые материей
для-устранения потерь зерна.
При отправке с поля к урожаю прикладывают этикетку с номером опыта,
клина и делянки; такая этикетка следует за урожаем до последнего взвеши¬
вания зерна. Рекомендуется записывать на ней также число снопов. В сарае
урожай с каждой делянки помещается в особое отделение или закром, где и
хранится до обмолота.
Необходимость иметь обширные сноповые сараи с большим числом от¬
делений или закромов представляет основное затруднение при учете по об¬
молоту всей делянки. На юге это затруднение смягчается возможностью до¬
статочной просушки снопов на поле и подвозки их прямо к молотилке. На
Методика полевого опыта с удобрениями
569
севере постройка специальных сараев, где снопы могли бы не только хранить¬
ся, но и досушиваться, неизбежна.
Общий урожай взвешивают перед самым обмолотом, причем проверяют
число снопов, записанное на этикетке. Обмолот урожая делянок произво¬
дят на любой небольшой молотилке возможно простой конструкции, так как
чем сложнее молотилка, тем больше в ней застревает зерна и тем труднее ее
очищать. Особое внимание должно быть обращено на хорошее отделение зер¬
на от соломы, но целесообразность применения соломотряса, особенно при
обмолоте урожая с малых делянок и пробных снопов, спорна; наиболее про¬
стым заменяющим его приспособлением служит длинный желоб из тща¬
тельно подогнанных досок (иногда обшитый железом) с высокими дощатыми
или фанерными бортами, устанавливаемый перед барабаном. По этому же¬
лобу прогоняют солому с тщательным многократным перетряхиванием ее
граблями. После уборки каждой делянки молотилку тщательно прочищают
(если нужно, с остановкой барабана), все насорившееся кругом зерно под¬
метают и собирают. Отвеивают зерно на веялке с возможно меньшим чис¬
лом сит.
Взвешивают все зерно как крупное, так и мелкое. Разделение зерна на
сорта и определение его качества представляют уже отдельную операцию
(не всегда обязательную), которую удобнее выполнять на пробах в лаборатор¬
ной обстановке. Многие авторы считают обязательным взятие проб на влаж¬
ность для приведения урожая зерна к стандартной влажности (14—15%).
Солому (и мякину) обычно не взвешивают, а количество ее определяют по
разности между общим весом урожая перед обмолотом и весом зерна.
Особого внимания при учете урожая заслуживает применение самоходно¬
го комбайна. Уборка урожаев опытных участков комбайном дает возможность
обойтись без специальных помещений для хранения поделяночных урожаев
до обмолота, избежать перевозки их и, главное, устранить ряд промежуточ¬
ных операций при учете. Изучение вопроса о применении самоходного ком¬
байна для уборки опытов показало (Ткачев, 1950), что комбайн С-4 обеспе¬
чивает возможность получения вполне достоверных результатов даже при
уборке на сравнительно небольших делянках (100—300 м1).
Для учета урожая комбайном необходимо, чтобы ширина учетной делян¬
ки была равна рабочему захвату жнеи комбайна (3,8 м). Механизм комбайна
должен быть предварительно отрегулирован. Необходимо установить также
оптимальный режим работы комбайна на данной культуре и определить про¬
должительность работы комбайна вхолостую между уборкой двух делянок.
Установленный режим работы комбайна следует выдерживать в течение убор¬
ки на всем участке опыта.
Чтобы создать больше удобства для разворотов комбайна, в опытах с не¬
большим числом вариантов уборку целесообразно проводить на одноименных
делянках по повторениям, а при более широких схемах — в пределах одного
повторения, через шесть — восемь делянок. Ширина защитных полос по
узким сторонам делянок должна быть при этом не менее 15 ле, что необходимо
предусмотреть при закладке опытов. Продольные защитные полосы могут
быть произвольной ширины, но не шире рабочего захвата жнеи комбайна.
При уборке урожая самоходным комбайном имеется полная возможность
организовать сортирование и взвешивание зерна в поле. Учет урожая соломы
и мякины также следует производить в поле и, таким образом, сразу же по¬
лучать результаты опыта.
Иногда самоходный комбайн используют лишь при обмолоте урожая,
убранного жнейками с участка опыта. Урожай при этом не перевозят с де¬
лянок к молотилке, а наоборот, комбайн, обмолотив на месте урожай с од¬
ной делянки, переезжает на другую и т. д. Этот способ можно применять
в тех случаях, когда расположение опыта не позволяет использовать ком¬
байн для уборки урожая.
570
С. В. Щерба, Ф. А. Юдин
Сущность учета по пробному снопу заключается в том, что в
сушку и на учетный обмолот поступает не весь урожай учетной делянки, а
лишь средняя проба из него — пробный сноп. Определив урожай зерна и
соломы в подобном снопе и зная соотношение весов урожая всей делянки и
пробного снопа, высчитывают тот урожай зерна и соломы, который получился
со всей делянки.
Уборку делянок производят так же, как и при учете по всей делянке,
но перед тем как связать снопы из каждого приготовленного жницами снопа,
из каждого сброса жнейки, а при уборке косами — через определенное чис¬
ло шагов из ряда берут пробные горсти, которые и составляют пробный сноп*
При очень больших делянках горсти берут не из каждого снопа, а из второ¬
го, третьего и т. д. Пробный сноп берет со всех делянок одно лицо, обычна
техник или опытная работница. После взятия пробного снопа весь урожай
делянки взвешивают здесь же на поле на десятичных или сотенных весах.
Пробный сноп также кладут на большие весы или же его вес в дальнейшем
присчитывают к весу урожая делянки. Затем пробный сноп взвешивают от¬
дельно на специальных весах с точностью до 10 а и укладывают в мешок. После
этого остальная масса урожая уже не нужна для дальнейшего учета и посту¬
пает в хозяйственное использование. Желательно брать с каждой делянки не
один, а два параллельных снопа, что гарантирует учет урожая в случае утери
или порчи одного из них и позволяет контролировать точность самого оп¬
ределения урожая по пробному снопу. В случае взятия двух пробных снопов
каждый из них набирают независимо от другого со всей площади делянки.
Пробные снопы должны составлять не меньше 1—2% от общей массы уро¬
жая, так как иначе они представляют недостаточно характерную среднюю
пробу. Слишком большие снопы тоже неудобны, так как плохо сохнут в
мешках. На Долгопрудном опытном поле пробные снопы берут обычно ве¬
сом 4—5 кг. Некоторые авторы рекомендуют более крупные снопы — 5—
7 кг и больше.
Мешки с пробными снопами увозят с поля и подвешивают для просушки
в специальном снопосушильном сарае. Если снопосушильного сарая нет,
их можно развешивать под навесом, на чердаке или в любом другом защи¬
щенном от дождя и хорошо вентилируемом помещении. Пробные снопы вре¬
мя от времени взвешивают. Когда убыль в весе прекратится, снопы дойдут
до постоянного веса, они готовы к обмолоту.
При поздней уборке и сырой осени применяется иногда искусственная
сушка пробных снопов в различного рода сушилках. При такой сушке сно¬
пы перед окончательным взвешиванием необходимо 1—2 дня продержать
вновь в сарае, чтобы они дошли до постоянного веса. Некоторые опытные
станции сушат пробные снопы не в мешках, а в особых матерчатых или про¬
волочных ящиках — призмах, устанавливаемых в несколько ярусов в сно¬
посушильном сарае.
Обмолот пробных снопов производится либо с возможными предосторож¬
ностями на маленькой молотилке, либо палками в мешках. Зерно отвеивают
на веялке возможно меньшего размера. Все указания относительно желатель¬
ной простоты устройства молотилки и веялки, сделанные при описании
обмолота урожая всей делянки, в еще большей степени относятся к обмолоту
пробных снопов.
Сухой пробный сноп взвешивают перед обмолотом вместе с мешком. После*
обмолота взвешивают только зерно и мешок с точностью до 1—5 г. Вес соло¬
мы определяют, как и при общем обмолоте, по разности. Ход пересчета с
пробного снопа на делянку и далее на гектар подробно изложен ниже.
Обмолот и отвеивание, выполняемые не при помощи специально приспо¬
собленных молотилок и веялок, являются наибольшим источником погреш¬
ностей при этом методе. Тем не менее учет по пробному снопу, как показал»
методические работы (Лебедянцев, 1960), не только не уступает по точности
Методика полевого опыта с удобрениями
571
учету по обмолоту всей делянки, но дает обычно несколько более высокие
абсолютные цифры, что свидетельствует о меньших суммарных потерях.
Основное преимущество учета по пробному снопу заключается в возмож¬
ности обходиться без больших помещений для хранения поделяночных уро¬
жаев до обмолота. Он незаменим при проведении большого числа опытов
в производственных условиях, дает возможность перевозить пробные снопы
на значительное расстояние и производить их обработку в приспособленной
обстановке. В условиях стационарных опытных учреждений он позволяет
при ограниченности помещений учитывать очень большое число делянок.
Само собой разумеется, что при очень малых делянках (10—20 м2), весь
урожай с которых составляет два-три снопа, учет по пробному снопу теряет
смысл и должен заменяться учетом всей делянки.
Другие косвенные методы учета урожая основаны на взятии в пределах
опытной делянки пробных площадок, полос, борозд и т. п., с большей или
меньшей точностью характеризующих урожай всей делянки. Принципиаль¬
ное отличие их от метода пробного снопа заключается в том, что в них от¬
сутствует не только обмолот, но и взвешивание общей массы урожая с делян¬
ки.
По существу эти методы сводятся к уменьшению размера делянки. Все
они менее точны, чем учет по обмолоту всей делянки или по пробному снопу,
и в условиях стационарных опытов почти не применяются.
Особенности учета незерновых культур. Учет
трав (клевера, вики и луговых трав), так же как и зерновых, можно произ¬
водить как по всей делянке, так и пробному снопу. В первом случае укос¬
ную массу оставляют на делянке, высушивают и сено взвешивают на месте.
Во втором случае из скошенных рядов набирают пробный сноп, сырую мас¬
су урожая всей делянки (вместе с пробным снопом) взвешивают на месте,
а выход сухой массы определяют по пробному снопу.
Сушат пробные снопы не в мешках, а на полу или на особых стеллажах.
Очень удобен способ сушки пробных снопов трав в сетках. Для этого служат
квадратные рамки 75 х 75 см2 с натянутой на них проволочной сеткой с
ячейками в 1—2 см. Пробный сноп раскладывают и зажимают между двумя
сетками, сетки связывают и подвешивают в сушильном сарае. Для ускорения
сушки сетки можно выносить и расставлять на солнце, а также помещать в
сушилку. В пробных снопах (до высушивания) определяют также ботаниче¬
ский состав естественного травостоя и соотношение компонентов в травосме¬
сях.
Учет картофеля и корнеплодов, как правило, производят путем взве¬
шивания всей массы урожая. Взвешивать урожай можно и на месте, и в са¬
рае, куда весь урожай привозят в мешках с этикетками. Более удобным
следует считать взвешивание урожая непосредственно на делянке. В случае
большой влажности почвы клубни или корни раскладывают до взвешивания
на несколько часов нетолстым слоем на делянке для подсушки, а затем про¬
тряхивают на ручном грохоте. Взвешивать удобно в корзинах или специаль¬
ных носилках с ящиком. Во многих случаях определяют и урожай ботвы,
которую также взвешивают на месте. При различной степени подсыхания
ботвы по делянкам из нее берут средние пробы для определения влажности.
Эти пробы чаще всего приходится сушить в огневой сушилке или определять
влажность лабораторными методами.
Очень часто приходится брать пробы из урожая корней и клубней (для
определения крахмала в картофеле, сахара — в сахарной свекле, сухого
вещества — в кормовой свекле и т. д.). Пробы собирают обычно в количест¬
ве 10—15 кг картофеля и нескольких десятков корней корнеплодов и хранят
в мешках с этикетками. Эти пробы составляют таким образом, чтобы соотно¬
шение крупных, средних и мелких экземпляров в пробе по возможности со¬
ответствовало их соотношению во всем урожае делянки.
572
С. В. Щерба, Ф. Л. Юдин
При большой загрязненности урожая пробы служат также для определе¬
ния количества земли в нем. Для этого их взвешивают в первый раз при взя¬
тии, а во второй — после отмывки и обсушки. Из этих проб отбирают также
материал для химических анализов.
Учет урожая прядильных культур (льна, конопли) в общем сходен с уче¬
том зерновых культур и тоже может производиться как по всей массе, так и
по средней пробе (пробному снопу). Однако, если в этой средней пробе пред¬
полагается определять выход волокна, она должна быть не меньше 30 кг
(в сыром состоянии) и связываться не в один, а в несколько снопов. Поэтому
учет по пробному снопу (вернее, по пробным снопам) имеет смысл для этих
культур лишь при сравнительно больших делянках — 100 м2 и больше.
Соломку, как весьма ценную часть урожая, взвешивают непосредственно
после обмолота, а не определяют по разности.
При учете урожая редкостоящих культур большое значение имеет вве¬
дение поправок на недостающие растения, число которых подсчитывают перед
уборкой. Применение поправок на переживание допустимо, если оно не свя¬
зано с изучаемым фактором и если оно не превышает 20—30%. При выпаде¬
нии растений выше указанной величины нужно исключить из учета делянку
целиком.
Простейший способ введения поправки на недостающие растения заклю¬
чается в следующем: фактический урожай с делянки делят на фактическое-
число кустов (корней), подсчитанных при уборке, находят средний вес уро¬
жая с куста (корня, кочана и т. д.). Затем его умножают на число недостаю¬
щих растений, вычисляют их предполагаемый вес и его прибавляют к факти¬
ческому урожаю. Допустимость введения поправки указанным методом тем
законнее, чем позднее произошло выпадение растений. При раннем выпаде¬
нии отдельных растений остающиеся соседние растения получают лучшее раз¬
витие, так как они как краевые растения получают дополнительную площадь
питания, поэтому введение поправки на недостающие растения по среднему
весу одного растения может внести в окончательный результат ошибку.
В связи с этим предложены другие, более сложные методы исправления уро¬
жая.
Наиболее правильным является удаление перед учетом всех растений,,
граничащих с пустыми местами в рядках; учитывают только нормальные
растения. По среднему весу одного учтенного растения, умноженному на
нормальное число растений, и восстанавливают истинный вес урожая с де¬
лянки. Этот метод поправок наиболее обоснован, но он осложняет технику
учета. Сравнительно легко он осуществим в том случае, если выпады располо¬
жены целыми пятнами или значительными отрезками рядков, а не разброса¬
ны по всей делянке.
Другие методы поправок на изреженность менее точны, так как основаны
на условных эмпирических коэффициентах, величина которых может менять¬
ся в зависимости от развития растения, плодородия почвы и целого ряда дру¬
гих факторов.
Поправки следует применять лишь при относительно небольшом числе*
выпавших растений и случайном характере этих выпадений. При выпадении
растений целыми пятнами или рядками правильней делать обычные выключ¬
ки.
Первичная обработка цифрового материала.
Полученные в результате уборки и учета тем или иным методом урожайные
данные по каждой делянке пересчитывают в центнерах на гектар. Затем для
каждого варианта опыта выписывают результаты всех повторностей и из
них выводят средние. Далее вычисляют прибавки урожая; разница между
урожаем отдельных опытных вариантов и урожаем контроля показывает
величину прибавки урожая от действия, изучаемого в данном варианте фак¬
тора. Для установления точности полученных в опыте результатов и оценки
Методика полевого опыта с удобрениями
573
достоверности полученных в опыте разниц все полученные цифровые данные
подвергают математическому анализу методами вариационной статистики.
Необходимым условием правильности проведения всех расчетов по опыту
является наличие доброкачественных и находящихся в порядке исходных
данных, полученных непосредственно при взвешивании урожаев на поле и в
сарае.
Все результаты (взвешивания общей массы урожая с делянки, веса проб¬
ных снопов, взвешивания зерна после обмолота и пр.) записываются непосред¬
ственно в полеврй дневник. Запись учета урожая следует проводить по за¬
ранее составленной форме. В этой форме предусматриваются соответствую¬
щие графы для первичной обработки полученного цифрового материала.
Образцы записей и расчетов при сплошном учете урожая по обмолоту всей
делянки и при учете урожая по пробному снопу приведены в табл. 3 и 4.
Таблица 3
Образец записи и пересчета при учете по обмолоту всей делянки
делянки
Удобрение
Учетная
площадь (за
вычетом
выключек), jh*
Коэффициент
пересчета
на 1 га
Урожай на делянку, к?
Урожай, ц/га
общей
массы
зерна
соломы
общей
массы
зерна
соломы
1
Фон
100
100
58,2
18,1
40,1
58,2
18,1
40,1
Таблица 4
Образец 8аписи и пересчета при учете по обмолоту пробного снопа
т
«ч
09^
w X
1
Сырой вес
в поле, кг
Отношение веса урожая
всей делянки к весу
пробного сноиа (коэф¬
фициент пересчета с
пробного снопа на де¬
лянку)
Сухой вес
пробного
снопа при
обмолоте, кг
Сухой вес
урожая
Урожай, ц/га
tfl 4>
|Е
1:
урожая всей
делянки (вклю¬
чая пробный
сноп)
S
дшлалв,
JSft делянки
Удобрение
О X
§1
в я
ч
^ Ё
|Я
ни
1!
о «
Ко
пробного снос
общей массы
верна
соломы
общей массы
зерна
СОЛОМЫ
общей массы
зерна
СОЛОМЫ
1
Фон
50
200
24,2
3,85
6,3
3,100
1,215
1,885
19,5
7,6
11,9
со
со
о
15,2
23,8
Дневник полевых работ и наблюдений служит по каждому полевому опы¬
ту первичным документом. Вспомогательными первичными документами
являются рабочие тетради, где ведутся все необходимые пересчеты массовых
наблюдений анализов и учетов.
Записи в дневнике полевых работ и наблюдений ведут простым каранда¬
шом, все поправки необходимо обязательно оговаривать.
Сводным документом, включающим все основные сведения о полевом опы¬
те: тема, обоснование опыта и задачи, поставленные на разрешение, схема,
программа, план, характеристика участка, методика исследований, записи
всех агрономических работ, обработанные результаты наблюдений и анали¬
зов, урожайные данные, результаты статистической обработки урожайных
данных и другие сведения, является журнал полевого опыта. Журнал хра¬
нят в помещении, все записи в нем ведут темными чернилами, своевременно
заполняя его на основе первичной документации.
574
С. В. Щерба, Ф. Л. Юдин
Некоторые особенности проведения опытов
в условиях производства
Как указывалось выше, специальная методика полевого опыта в услови¬
ях сельскохозяйственного производства еще не может считаться разработан¬
ной. Существуют и предлагаются лишь отдельные приемы и видоизменения
обычных приемов, имеющие целью наиболее экономное и в то же время точ¬
ное выполнение тех или иных операций полевого опыта в условиях практиче¬
ского хозяйства. Некоторые из таких необходимых видоизменений огова¬
ривались нами при изложении общих приемов методики. Здесь мы остановим¬
ся лишь на нескольких специальных приемах, рекомендуемых для постановки
опытов в условиях сельскохозяйственного производства.
Метод длинных полос. В 1930—1931 гг. некоторые опытники
(Вельский, 1931) предложили метод постановки простейших опытов с удоб¬
рениями в условиях практического хозяйства на длинных узких полосах.
Сущность его заключается в том, что на хозяйственном поле намечают не¬
сколько параллельных полос, проходящих через весь массив и захватываю¬
щих по возможности все разнообразие его рельефа и почвы. Ширина полос
соответствует одному-двум захватам туковой сеялки. Перед обработкой поля
на полосы вносят необходимые удобрения. Для того чтобы полосы были стро¬
го прямыми, направление ходов сеялки предварительно провешивают.
Концы полос отмечают кольями по краю поля. Контрольную полосу или ос¬
тавляют между двумя удобренными полосами, или даже совсем не выделяют
заранее, а берут при уборке с любой стороны параллельно удобренной поло¬
се. Обработку, посев и прочие работы производят сплошь по всему полю.
При уборке посередине каждой полосы делают один проход жнейкой или дру¬
гой уборочной машиной; урожай с этого хода собирается отдельно, обмола¬
чивается и взвешивается. Учетная площадь определяется произведением
длины полосы на ширину захвата уборочной машины.
Для того чтобы проход жнейки строго совпадал с направлением полос,
необходимо сделать один прокос вручную с вешки на вешку по краю первой
полосы, а дальше косить полосы подряд жнейкой, выбирая для учета нуж¬
ные проходы, приходящиеся примерно на середины полос. Схема опыта долж¬
на быть, конечно, возможно проще и может даже состоять лишь из двух де¬
лянок — удобренной и контрольной. Не исключается возможность введения
повторности, конечно небольшой.
Подобный прием может также применяться для учета хозяйственного
эффекта от удобрений. В этом случае на сплошь удобренном поле оставляют
контрольную полосу (пропускается один-два хода туковой сеялки). Так же,
как и в предыдущем случае, производят учет одного прохода жнейки на конт¬
рольной полосе и одного параллельного прохода на удобренном массиве.
Необходимым условием является только прямизна контрольной полосы.
Небольшая методическая работа, проделанная С. В. Щербой в 1932—
1933 гг. на Долгопрудном опытном поле, показала возможность некоторого
дальнейшего упрощения этого метода за счет выборочного учета пробных
полос. При взятии для взвешивания каждого третьего сброса жнейки полу¬
чались результаты, очень близкие к результатам сплошного учета всего
хода машины при уменьшении взвешиваемой массы в 3 раза.
Учет урожая, убранного комбайном и други¬
ми уборочными машинами. При больших площадях делянок,
закладываемых в условиях производства, и высокой степени механизации
нашего сельского хозяйства естественно возникает вопрос о применении для
учета опытов сложных сельскохозяйственных машин и, в частности, ком¬
байна.
Учет урожая при помощи комбайна имеет то преимущество, что дает
возможность сразу получать конечный результат учета — урожай зерна —
Методика полевого опыта с удобрениями
575
и устраняет все промежуточные операции, а также необходимость специаль¬
ных помещений для хранения поделяночных урожаев до обмолота. Для
учета комбайном учетные делянки должны иметь вытянутую форму и шири¬
ну, кратную ширине захвата комбайна. Перед началом и по окончании
уборки каждой делянки должна производиться очистка бункера комбайна.
Намолоченное с делянки зерно ссыпают в мешки, которые снабжаются эти¬
кетками и взвешиваются. Необходимо взятие проб на влажность зерна, так
как уборка может проводиться при весьма различной влажности.
Уборка урожая с больших делянок в условиях производства жнейкой
или сноповязалкой не требует особых пояснений, так как принципиально ни¬
чем не отличается от обычного учета по обмолоту всей делянки.
Некоторые особенности постановки опытов
на орошаемых землях1
Основным, решающим фактором урожайности в условиях искусственного
орошения является вода. Поэтому при постановке опытов на поливных зем¬
лях особое внимание должно быть уделено равномерности снабжения всей
площади опыта водой и возможности точного регулирования количества во¬
ды, попадающей на каждую делянку. Этими требованиями и определяются
прежде всего особенности методики полевого опыта в условиях орошения.
Выбор ипланировка участка. К рельефу поливных зе¬
мель, выбираемых для опытов, должны предъявляться значительно более
строгие требования, чем к рельефу неполивных участков. Ничтожная разни¬
ца в уровне поверхности, порядка 10—15 см, едва улавливаемая при обыч¬
ной нивелировке, может быть здесь причиной резкой разницы в урожае,
так как поливная вода стекает с повышенных и накапливается в пониженных
частях участка опыта. Разница в увлажнении способна иногда перекрыть
эффект от всех испытуемых приемов.
Так, в опытах с сахарной свеклой, проведенных в 1942 г. совместно с со¬
трудниками Сельскохозяйственной академии имени К. А. Тимирязева и Сред¬
неазиатского филиала НИИУИФ в Самарканде, при плохо спланированном
участке две одноименные делянки (90 кг N, 135 кг Р20Б и 90 кг К20) при по¬
ливных нормах в 70—80% от полной влагоемкости почвы дали такие урожаи
свеклы: делянка 9—612,7 ц/га, делянка 20—269,3 ц/га. Параллельные де¬
лянки отличались между собой по полученному урожаю приблизительно в
2,5 раза, в то время как эффект от удобрений не превышал 40—60%. Когда
стали искать причины такого расхождения, то оказалось, что делянка 20
была расположена на повышении, с которого вода скатывалась, а делянка
9 — в понижении, где вода собиралась.
Если нельзя выбрать для опыта заранее спланированный участок, необ¬
ходимо до закладки опыта произвести его планировку. Постановка опытов
с поливом на неспланированном или плохо спланированном участке может
оказаться бесполезной затратой средств и времени, особенно если в опыте
поливной режим является изучаемым фактором.
При поливе по бороздам (что рекомендуется и обычно применяется в опы¬
тах) поливная площадка должна иметь небольшой уклон, но уклон этот дол¬
жен быть равномерным и очень незначительным. Во всяком сдучае он не
должен превышать 0,01—0,02 (1—2 м падения на 100 пог. м). В противном
случае вода не будет равномерно впитываться при прохождении по полив¬
ным бороздам. При большом уклоне возможен и размыв поливных борозд.
Особенности расположения опыта в условиях
орошения. Опыт в условиях орошения должен располагаться таким>
1 6 составлении этого раздела статьи принимал участие Я. Ю. Старосельский.
576
С. В. Щерба, Ф. А. Юдин
образом, чтобы вода могла быть подведена независимо и в любом количестве
к каждой делянке. Пропуск воды через одну делянку на другую недопустим,
особенно в опытах с удобрениями, так как может повлечь за собой вымыва¬
ние и перенос питательных веществ. По этой же причине в опытах с удобре¬
ниями нежелателен вообще «полив со сбросом», при котором избыток полив¬
ных вод выпускается (сбрасывается) за пределы поливной площадки.
Желательно, чтобы вся вода, попадающая на делянки, впитывалась в ее пре¬
делах. Этим условиям в наибольшей степени удовлетворяют однорядное рас¬
положение делянок и их вытянутая форма. Поливные борозды должны на¬
резаться при этом вдоль делянки. Между рядами делянок (вдоль их корот¬
ких сторон) должны проходить непосредственно временные оросители, из
которых вода поступает на любую делянку.
Регулирование полива и измерение поливных
норм в опытах. Следующее условие постановки опытов при ороше¬
нии — возможность регулирования и достаточно точного измерения коли¬
чества воды, попадающей на каждую делянку. Измерение производят спе¬
циальными водомерными приборами, водосливами и др. Правильной счита¬
ется такая система, при которой измеряется количество всей воды, попадаю¬
щей на участок опыта (головным водосливом), и количество воды, даваемой
на каждую делянку. Иногда опытные учреждения не ведут точного учета
воды, подаваемой на каждую делянку, ограничиваясь замером общего ко¬
личества воды, поступающей на участки опыта или группу делянок с оди¬
наковым поливным режимом. Равномерное распределение воды между де¬
лянками регулируется в этих случаях временем полива и опытом квалифи¬
цированных поливальщиков. Поделяночный замер воды обязателен в специ¬
альных опытах по вопросам орошения.
В опытах при поливе должна применяться современная техника полива,
разработанная для данного орошаемого района, в частности сифонный метод
полива и др.
К комплексу наиболее необходимых наблюдений в течение вегетационно¬
го периода в поливных условиях обязательно добавляются учет и запись
поливного режима для каждой делянки и наблюдения за влажностью почвы
в межполивные периоды. Помимо важности этих наблюдений для понимания
результатов опытов и, в частности, действия удобрений, сами поливы ведут
для поддержания определенной влажности почвы и проводят их при паде¬
нии влажности до известного минимума (70—80% от полевой влагоемкости).
ХАРАКТЕРИСТИКА КАЧЕСТВА УРОЖАЯ
В полевых опытах с удобрениями очень часто, кроме учета влияния
изучаемого приема на величину урожая, важно также иметь оценку его влия¬
ния на качество урожая.
Учет качества урожая в полевом опыте представляет существенное зна¬
чение, особенно в опытах с техническими культурами. Учет качества уро¬
жая, полученного на делянках разных вариантов опыта (как выход и ка¬
чество волокна у льна, содержание сахара в сахарной свекле, эфирного мас¬
ла у эфиромасличных растений и т. п.), в значительной степени повышает
практическую ценность результатов опыта.
Для зерновых хлебов, трав и некоторых других культур основным пока¬
зателем качества продукции (зерна, сена) является влажность. Определение
влажности продукции необходимо проводить во всех случаях, когда урожаи
перед обмолотом или взвешиванием не были доведены до постоянного веса.
Для того чтобы можно было сравнивать урожаи, полученные на разных де¬
лянках и разных вариантах, необходимо их веса приводить к одинаковой,
стандартной влажности.
Методика полевого опыта е удобрениями
577
Пробы зерна для определения влажности и других показателей качества
берут после обмолота в момент взвешивания с каждой делянки в стеклянные
банки с притертыми пробками весом 1—2 кг. Влажность определяют одним
из методов, предусмотренных стандартом на зерно, и выражают в процентах
к сырой навеске. Вес урожая, приведенный к стандартной влажности (14%-
ной), вычисляют по формуле
100 —
А — в 9 100 — 14 ’
где А — вес урожая зерна, приведенного к стандартной 14%-ной влажности, кг; В — вес
урожая зерна, полученного в опыте, кг; W — влажность зерна при взвепшвснии, %; 14 —
стандартная влажность зерна, %.
Из других показателей качества зерна чаще всего определяют вес ты
сячи зерен и натуру (объемный вес). Путем химического анализа определяют
содержание в зерне белка и крахмала.
Для определения влажности в сене трав, пробы из урожая или из проб¬
ного снопа берут в момент взвешивания, режут ножницами возможно мельче
и помещают до анализа в банки с притертыми пробками.
Влажность определяют высушиванием проб в широких бюксах в термо¬
стате при температуре 65° до постоянного веса. Важным показателем качест¬
ва сена является ботанический состав. Его определяют по отдельным бота¬
ническим группам, например для сеяных трав (злаковый компонент, бобовый
компонент и разнотравье). Для ботанического анализа служит пробный
сноп в сыром виде. После общего взвешивания пробу разбирают на бумаге
по отдельным ботаническим группам, связывают в пучки или помещают в-
сетки и высушивают. Необходимо избегать потерь при разборке и сушке
проб. После доведения проб до постоянного веса определяют сухой вес от¬
дельных составных частей и общий суммарный вес. Вес каждой ботанической
группы выражают в процентах от общего веса пробы.
Путем химического анализа в сене определяют содержание «сырого»
протеина, клетчатки, а иногда и минеральный состав (зола, кальций,
фосфор).
Для картофеля важнейшим показателем его качества является содер¬
жание крахмала. Определяют его чаще по удельному весу. Для определения
крахмала среднюю пробу отбирают в поле после взвешивания урожая и хра¬
нят в мешках с этикетками. Важно, чтобы в средней пробе соотношение
клубней различной крупности соответствовало их соотношению в общем уро¬
жае, так как содержание крахмала в клубнях различной крупности может
быть различным. Показателем качества картофеля служит также товарность
урожая, т. е. процентное отношение веса товарных клубней больше 50 г
к общему весу клубней.
Для сахарной свеклы основной показатель ее качества — это содержание
в корнях сахара, которое определяют поляриметрически или же химическим
анализом. Среднюю пробу для определения сахара отбирают из кучи после
взвешивания. При взятии пробы из кучи берут каждый пятый, десятый ко¬
рень и т. д. Абсолютное число корней в пробе 20—40—60 штук.
В кормовых корнеплодах для оценки качества обычно определяют со¬
держание сухого вещества. Методика отбора проб та же, что и.для сахарной
свеклы. Содержание сухого вещества резко колеблется в зависимости от
размера корня. При отборе средней пробы из кучи надо предварительно ее
рассортировать по крупности на 3—4 фракции и брать пробу из каждой фрак¬
ции отдельно, пропорционально ее численности, а затем взятые пробы соеди¬
нить.
Для прядильных растений основной показатель качества — выход во¬
локна. Кроме* того, для оценки качества льна определяют техническую или
578
С. В. Щерба, Ф. А. Юдин
продуктивную длину, т. е. длину стебля от подсемядольного колена до пер¬
вого разветвления на стебле. Измерение производят на 50—100 растениях
и из них выводят среднюю длину.
Для подсолнечника показателем качества является содержание жира в
семенах и лузжистость семячек.
В опытах с кукурузой важное значение имеет показатель вызреваемости
початков по разным вариантам опыта, средний вес початка, вес 1000 зерен,
соотношение веса стеблей, листьев и початков.
При вэятии проб для определения качества урожая любой культуры
возникает вопрос, нельзя ли ограничиться для характеристики качества
урожая меньшим числом повторений, чем для определения его количества.
В принципе на этот вопрос должен быть дан безусловно отрицательный от¬
вет. Для того чтобы определение качества урожая соответствовало по своей
точности определению количества, оно требует не меньшей, если не большей,
повторности, тем более что оно производится не во всем урожае, а лишь в
части его (средних пробах). Необходимость анализа всех повторений совер¬
шенно очевидна по отношению к тем анализам, которые служат в дальней¬
шем для введения поправок к урожаю отдельных делянок на стандартную
влажность. Однако сложность или громоздкость некоторых анализов застав¬
ляет отступать от общего правила и ограничиваться меньшим числом ана¬
лизов, чем число повторений. Так, например, обстоит дело с анализом пря¬
дильных растений на волокно, с ботаническим анализом, часто с анализами
на крахмал и сахар. В этих случаях при невозможности проанализировать
по отдельности все повторения следует брать общие пробы из нескольких
повторений или соединить пробы, ввятые из каждого повторения в отдельно¬
сти, а не ограничиваться выборочным анализом части повторений. Так, при
4-кратной повторности можно соединять для анализа по две делянки, при
шестикратной — по две или по три и т. д. При выборочном анализе лишь
части повторений качественная характеристика урожая будет относиться
к иной, меньшей совокупности материала, чем количественная, и сопостав¬
ление их будет очень условным. При анализах объединенных проб резуль¬
таты их будут характеризовать весь опыт. При всех анализах на качество
урожая, помимо повторности по делянкам, крайне желательно иметь по¬
вторность определений, которая дает возможность судить о точности самого
анализа.
АГРОХИМИЧЕСКИЕ И АГРОФИЗИЧЕСКИЕ ИССЛЕДОВАНИЯ
В ПОЛЕВЫХ ОПЫТАХ С УДОБРЕНИЯМИ1
Почвенные исследования в полевых опытах производятся как при заклад¬
ке опыта, так и во время его проведения.
Описание почвенного разреза и анализы почвы по горизонтам позволяют
отнести почвы опыта к определенному типу и виду почв. Классификация и на¬
звания почв меняются со временем. Например, черноземы ранее делили на
деградированные, мощные, обыкновенные и предкавказские, позднее — на
оподзоленные, выщелоченные, типичные, обыкновенные, южные и т. д. Под¬
золистые почвы сравнительно недавно разделили на подзолистые и дерново-
подзолистые, причем именно те почвы, которые в агрономических книгах
прежде назывались подзолистыми, получили название дерново-подзолистых.
Все это приводит к тому, что одно название почвы без описания разреза
и почвенных анализов не позволяет установить, на какой именно почве, по
современной классификации, был поставлен опыт. Аналогичные перемены
происходят и в названии почв по их механическому составу. Поэтому в от¬
1 Раздел составлен А. В. Соколовым.
Методика полевого опыта с удобрениями
579
четах об опытах, особенно в отчетах о многолетних опытах, следует приво¬
дить данные по анализу почв и по морфологическому описанию почвенных
разрезов.
Почвенные разрезы нельзя закладывать на территории опытных делянок,
поэтому для больших опытов желательно иметь несколько почвенных разре¬
зов, расположенных вокруг опыта. Дополнительно закладывают малые
разрезы («прикопки»), которые захватывают обычно два верхних горизонта
почвы; эти прикопки можно делать и на территории опыта, на защитных по¬
лосах и дорогах. Данные «прикопок» позволяют установить, насколько удач¬
но был сделан разрез для характеристики почвы опыта.
Точное установление почвенной разновидности требует проведения не
только морфологического обследования почв, но и аналитического изучения
состава почвы и ее свойств. Для анализа почвы берут как индивидуальные
образцы, взятые из различных горизонтов почвенного профиля, так и сред¬
ние пробы пахотного слоя опытного участка.
В некоторых случаях задачи опытов требуют проведения определенных
анализов почвы, без которых результаты опытов неполноценны. Большой
ряд приемов земледелия направлен на изменения каких-либо свойств почвы.
При изучении этих приемов необходимо знать, обладает ли почва, на которой
ставится опыт, свойством, подлежащим исправлению. Типичным примером
такого рода опытов являются опыты с известкованием, которые проводятся
для устранения кислотности почвы. Постановка таких опытов требует ана¬
лиза почвы на формы кислотности, сумму поглощенных оснований и pH,
без чего нельзя ни сделать вывода о пригодности участка для постановки
опыта, ни дать правильного анализа результатов опыта.
Опыты с минеральными удобрениями требуют проведения определенных
анализов почвы, например при постановке опытов с фосфатами необходимо
определение в почве количества легкорастворимых форм Р2Об по одному из
принятых методов (Кирсанов, Чириков, Мачигин и др.). То же относится
и к опытам по мелиорации засоленных почв; в этом случае необходим учет
состава и количества солей в почве, а также степени солонцеватости — содер¬
жания в почвах поглощенного натрия.
Такие агрохимические свойства, как кислотность почвы и содержание в
нец усвояемых фосфатов, могут сильно варьировать на территории опыта.
В этом случае без данных поделяночного анализа трудно судить о пригод¬
ности участка для постановки опыта и о достоверности выводов. Средние
данные об агрохимических свойствах почвы опытного участка желательно
приводить с указанием интервала колебаний, вариационного коэффициента
или стандартного отклонения для показаний поделяночных анализов почвы.
Вопрос о том, какие именно анализы почвы необходимо производить при
постановке и проведении полевых опытов, служил предметом обсуждений
секции удобрений и агрохимии ВАСХНИЛ и методических совещаний при
Почвенном институте им. В. В. Докучаева. В «Методических указаниях...»
(1965) приведены основные виды анализов, которые следует проводить для
установления агрономической характеристики почв и наименования почвы
по установленной классификации, но не в целях изучения генезиса почв и
специальных проблем почвоведения и агрохимии.
Списки анализов следует изменять применительно к условиям проведения
и задачам отдельных опытов. При наличии оборудования для определений
радиоактивного фосфора следует определить общий запас в почве раствори¬
мых фосфатов. Желательно также проведение вегетационных опытов для
определения величины влажности завядания и запаса усвояемых питатель¬
ных веществ. Для многолетних опытов с удобрениями важно обеспечить ре¬
гулярное взятие по ротациям образцов почвы, которые в дальнейшем могли
бы быть использованы для анализов теми методами, которые будут считаться
наиболее точными в момент производства анализов.
580
С. В. Щерба, Ф. Л. Юдин
Если хранение образцов почвы не обеспечено, то следующие новые об¬
разцы надо анализировать не только новыми, но и старыми методами, чтобы
обеспечить сравнимость результатов прежних анализов, проведенных при
закладке опыта, с данными анализа новых образцов почвы с тех же делянок.
Внесение удобрений приводит к изменению свойств почвы. Общеизвестна
влияние физиологически кислых^ удобрений на подкисление почвы, навоза
на уменьшение кислотности почвы, фосфатов на увеличение содержания в
них усвояемого фосфора («эафосфачивания почв») и т. д. Если удобрения
применялись систематически в течение ряда лет или в больших дозах, то
заметить их влияние на свойства почвы сравнительно легко, так как различия
между свойствами почв опытных и контрольных делянок стали существен¬
ными. Конечно, легко заметить действие извести на кислую почву или вне¬
сения навоза в многолетнем опыте. Но если необходимо дать не только каче¬
ственную, но и количественную характеристику действия удобрений на свой¬
ства почвы, то требуется уже проведение многочисленных анализов почвы
опытных делянок. Конечно, наиболее правильная постановка исследований
требует анализа почв всех делянок опыта до его постановки. Но, к сожа¬
лению, вопрос о влиянии удобрений на те или иные свойства почвы обычна
привлекает внимание исследователей по истечении многих лет после начала
опыта. Поэтому сравнение приходится вести не с исходными данными для
почв различных вариантов опыта, а с данными для почв делянок контроль¬
ных вариантов. Это, конечно, снижает точность работы, так как почва конт¬
рольных вариантов тоже меняется за время опыта. Поэтому при постановке
многолетних опытов с удобрениями необходимо особое внимание обратить на
агрохимический анализ всех делянок до закладки опыта.
Точность полевых опытов в среднем считается;, около 10%, т. е. при уро¬
жаях в 20 ц достоверными считаются прибавки более 2 ц. Результаты много¬
численных обработок данных полевых опытов, поставленных при 4-, 6-крат-
ной повторности на Долгопрудной агрохимической станции, показали, чта
заслуживают внимания величины разниц между вариантами порядка 10,5—
18,9% для однолетних и 8,2—10,5% для многолетних опытов. Вариационные
коэффициенты агрохимических показателей в опытах этой станции были рав¬
ны: для гумуса по Тюрину — 13%, для обменного Са по Гедройцу — 13%,
для Р2Об по Кирсанову — 21%, для гидролитической кислотности по Кап-
пену — 10%, для обменной кислотности по Каппену — 24% и для актив¬
ного А1 по Соколову — 29%. Это варьирование агрохимических показателей
наблюдается для почв параллельных делянок многолетних опытов. Следова¬
тельно, в опыте с 4-кратной повторностью при содержании в почве гумуса
около 2% различия между почвами вариантов опыта порядка 0,3% недосто¬
верны; для Р2Об по Кирсанову при 5 мг на 100 г почвы заслуживают внима¬
ния разницы для вариантов более 2,5 мг Р2Об.
Основным правилом изучения влияния удобрений на свойства почвы
является анализ образцов почв для делянок всех без исключения повторе¬
ний опыта и обязательная обработка данных анализов, полученных для па¬
раллельных делянок методами вариационной статистики.
Во многих случаях в опытах делаются попытки установить (при
помощи анализов почвы делянок различных вариантов опыта) величины,
которые не могут быть достоверно определены, так как они заведомо находят¬
ся за пределами точности полевого опыта. Например, пытаются на основе
анализа почвы заметить влияние однолетнего или двухлетнего посева трав
на накопление в почве аэота или гумуса. Простые расчеты показывают,
что даже при высоком урожае трав (порядка 100 ц/га сена) и накоплении
большого количества корней (50—75 ц/га) количества гумуса и аэота могли
измениться на величины порядка не более 5% для гумуса и 2% для’азота
от их среднего содержания в пахотном слое почвы, т. е. на величины, ле¬
жащие в пределах возможной ошибки исследований.
Методика полевого опыта с удобрениями
581
При составлении программы анализов в полевых опытах следует прежде*
всего произвести теоретические подсчеты, в какой мере изучаемый прием
может отразиться на изменении различных свойств почвы. Например, в опы¬
те на бедной дерново-подзолистой почве вносится ежегодно по 45 кг азота на
1 га в форме минерального удобрения. Примерно 2/3 азота удобрения исполь¬
зуется урожаем растений и уносится с ним с поля, следовательно, ежегодна
в почве может оставаться 15 кг азота. В течение 10 лет это составит всего толь¬
ко 150 кг азота на 1 га. Предположим, что в почве опыта содержится общего
азота всего 0,1%. Если объемный вес пахотного слоя был равен 1,34, а
глубина пахотного слоя 18 еле, то вес пахотного слоя был равен 2 412 ООО кг%
следовательно, в пахотном слое содержалось 2412 кг азота.
Математическая обработка данных анализов многолетних полевых опы¬
тов Долгопрудной агрохимической станции показывает, что доказуемые раз¬
ности в содержании азота в почве различных вариантов должны быть не ме¬
нее 13% от его количества в почве. В нашем примере возможное изменение
общего азота равно 6,2% (150 кг по отношению к 2412 кг), т. е. определение
общего азота даже через 10 лет опыта не может показать доказуемого иа-
менения содержания общего азота в почве удобренных вариантов.
В приведенном выше примере нарочно была взята почва, бедная азотом*
с неглубоким пахотным слоем; на черноземных почвах при глубине пахотно¬
го слоя в 20—25 см количество азота в почве может быть в 4—5 раз больше..
В этом случае средние дозы азота могут изменить количество в почве азота
в результате многолетнего систематического применения удобрений всего»
только на 1—2% общего количества азота в почве.
Аналогичные грубые подсчеты легко можно сделать для учета возможного
влияния различных доз органических и минеральных удобрений на коли¬
чество в почве гумуса, валовых азота, фосфора и калия. Эти подсчеты, а так¬
же прямые опыты приводят к выводу, что в большинстве полевых опытов
при внесении обычных средних доз удобрений нельзя обнаружить математи¬
чески доказуемого влияния удобрений на количество в почве гумуса, вало¬
вых азота, фосфора и калия. Но можно достоверно установить влияние удоб¬
рений на содержание в почве усвояемых форм азота, фосфора и калия.
Во многих опытах важно учитывать не только урожай надземной массы,
но и количество корней. Такие темы, как влияние многолетних трав на по¬
вышение плодородия почвы, накопление в почве биологического азота,
роль растений в накоплении почвенного гумуса или в борьбе с эрозией»
техника внесения удобрений требуют учета содержания корней в почве в
определения надземных пожнивных остатков на поверхности почвы после
снятия урожая. Если надземные пожнивные остатки можно собрать с боль¬
шой площади делянки и довольно точно количественно определить их массу,
то учесть количество корней в почве очень трудно. Урожай надземной массы
определяется со всей площади делянки, а количество корней — выборочным
методом, примерно с V100“^1Лооо части делянки. Если с одной делянки взято
по 4—10 монолитов размером 25 х 25 см, то точность такого определения
весьма невелика. Основная ошибка при определении количества корней
состоит в неправильном размещении на площади делянок мест для взятия мо¬
нолитов. Чаще всего берут пробы на участках с большим количеством расте¬
ний, в результате чего приведенные данные зачастую в 2—3 раза и более
превосходят возможное содержание корней в почве. Минимальное требова¬
ние, которое должно быть соблюдено,— это соответствие урожая надземной
массы над монолитами, взятыми для определения количества корней, урожаю
надземной массы со всей делянки.
Наиболее точно количество корней устанавливается для пропашных рас¬
тений, когда для выделения корней берется все количество почвы, соответст¬
вующее одному растению. При достаточном количестве таких проб получают¬
ся достоверные данные о накоплении корней.
*582
С. В. Щерба, Ф. Л. Юдин
В полевых опытах внесение средних доз удобрений приводит к сильному
и вполне математически доказуемому изменению содержания в почве под¬
вижных усвояемых питательных веществ.
На подзолистых почвах в полевых опытах органические и минеральные
удобрения производят вполне определенное существенное изменение кислот¬
ности почвы, причем наиболее достоверные изменения отмечаются для об¬
менной кислотности и содержания в почве активного алюминия, несмотря
на большое варьирование этих показателей. В частности, небольшие дозы
навоза, не влияя в пределах точности опыта на содержание в почве гумуса,
существенно изменяют обменную кислотность подзолистой почвы. Поэтому
во всех многолетних опытах с удобрениями, проводимых на малобуферных
почвах, необходим постоянный контроль за реакцией и кислотностью почвы,
без чего результатам опыта может быть дано совершенно неверное объяс¬
нение.
Кроме наблюдений, которые проводятся через несколько лет опыта и поды¬
тоживают результаты действия на почву исследуемых приемов, в полевых опы¬
тах изучают «динамику» свойств почвы, т. е. изменения некоторых ее свойств
в течение вегетационного сезона. Последние наблюдения проводят несколько
раз в течение вегетационного сезона. Взятие проб производится либо регу¬
лярно через определенное число дней, либо приурочивается ко времени раз¬
личных обработок почвы, к посеву растений и фазам их развития.
Влажность, аэрация и скважность почвы существенно меняются в течение
вегетационного сезона; каждая обработка изменяет ее состояние: после об¬
работки происходит слеживание и уменьшается скважность почвы. Влаж¬
ность беспрерывно меняется вследствие испарения и передвижения воды
в почве.
Следовательно, все время происходит и изменение свойств почвенного
раствора, исследование которого тоже относится к изучению почвенной ди¬
намики. Трудности выделения почвенного раствора заставляют в ряде слу¬
чаев заменить его анализом водных вытяжек, что вполне допустимо при оп¬
ределении содержания в почве нитратов, нитритов, хлоридов и сульфатов.
Агрохимические анализы в течение вегетационного сезона имеют большое
значение для понимания результатов опыта. Результативность агрохими¬
ческих исследований в полевых опытах в значительной мере зависит от пра¬
вильности взятия проб почвы с опытных делянок.
При изучении динамики свойств почвы методику взятия средней пробы
устанавливают при помощи математической статистики. На делянке опыта
почвенным буром берут большое число (50—100) проб, равномерно распре¬
деленных по площади делянки. Пробы анализируются по отдельности, и ре¬
зультаты анализов обрабатывают математически для установления точности
работы при различном числе проб. В дальнейшем при изучении динамики
свойств почвы с каждой делянки берут установленное количество проб,
которые смешивают в одну среднюю пробу. Для каждой делянки правильнее
составлять две средние пробы. Количество скважин (отдельных проб), не¬
обходимых для составления средней пробы, зависит от размера опытной де¬
лянки, пестроты почвенного покрова, определяемого элемента почвенной
динамики, желательной степени точности опытной работы.
Обычно на небольшой делянке для составления средней пробы берут не
менее пяти индивидуальных проб. Наблюдения над динамикой почвы весьма
трудоемки, поэтому часто пробы берут не со всех повторений опыта, а только
с некоторых. Это, конечно, приводит к соответствующему уменьшению точ¬
ности исследования.
Результаты агрохимических исследований сравниваются с данными уро¬
жайности растений в опыте. Если агрохимические исследования проводи¬
лись только для некоторых повторений опыта, то для сравнения с ними долж¬
ны браться и данные урожайности этих повторений. Точность агрохимиче¬
Методика полевого спита с удобрениями
583
ских исследований не превышает точности учета урожаев, поэтому сокраще¬
ние числа повторений, в которых проводятся наблюдения, нежелательно.
Выполнение наблюдений над динамикой почвы во время вегетации ра¬
стений связано с некоторым повреждением посевов, для уменьшения которого
применяются различные приемы: например, люди, берущие пробы, стоят на
переносных мостиках, перекидываемых через учетную площадь делянки,
чтобы не топтать посевы на делянке. Опасение повреждения посевов при¬
водит к сокращению числа наблюдений и перенесению изучения динамики
почвы в специальные лабораторные опыты.
При изучении свойств почвы в динамике следует учитывать возможные*
изменения ее свойств во время взятия среднего образца в поле, его доставки
и хранения в лаборатории до анализа. Поэтому разработан ряд приборов для
непосредственного определения в поле влажности, аэрации, pH и других
свойств почвы. Такие свойства, как влажность, быстро меняются, и образцы
для определения влажности берут немедленно после выемки индивидуально¬
го образца почвы, до приготовления средней пробы. Но и ряд других эле¬
ментов почвы, изучаемых в динамике, меняется во время высушивания
образца, поэтому наиболее правильно анализировать почвы при сохранении
ее полевой влажности. Если это требование технически невыполнимо, то
должно быть установлено, какие элементы почвенной динамики допускают
определение в сухих образцах. Указания по этому вопросу имеются в ру¬
ководствах и специальных работах, но они меняются в зависимости от ус¬
ловий работы.
Структура почвы при взятии средней пробы разрушается, многие же свой¬
ства почвы зависят от структуры. Поэтому разработана методика взятия
проб без нарушения естественного строения почвы, ее структуры. С этой
целью разработаны различные конструкции почвенных буров, позволяющих
брать образцы с ненарушенным строением. В этих образцах изучают вла-
гоемкость почвы, ее воздухо- и водопроницаемость. Но такие свойства почвы,
как влагоемкость, воздухо- и водопроницаемость, испарение воды почвой,
газообмен почвы и атмосферы наиболее правильно изучать непосредственно
в полевой обстановке.
Почва не является однородной средой. На поверхности почвенных агре¬
гатов протекают одни процессы, внутри — другие. В почве вблизи корней
растений идут одни процессы, а в известном отдалении от них — другие.
При внесении удобрений они распределяются в почве неравномерно, обра¬
зуя очаги накопления питательных веществ. Работа со средними величи¬
нами не позволяет выявить всего этого разнообразия почвенных условий.
Средние показания для целого горизонта дают только сумму накопления и
уменьшения в почве того или иного соединения. Изучение реальной динами¬
ки почвы требует, кроме того, знания распределения в почве изучаемого
соединения, пунктов его накопления и исчезновения.
Размещение в почве веществ и интенсивность процессов в различных пунк¬
тах почвы являются предметом изучения микродинамики почвы. С этой целью
исследуются ризосфера растений, очаги удобрений и агрегаты почвы. Ме¬
тодика зависит от объекта исследования. В основном она состоит из отбора
отдельных типичных участков почвы, их разделения и дробного анализа.
При изучении микродинамики почвы могут быть использованы и статистиче¬
ские методы определения неравномерности распределения в' толще почвы
отдельных свойств или веществ. Например, для изучения влияния удобре¬
ний на свойства почвы можно на удобренной делянке взять большое количест¬
во индивидуальных проб. Эти пробы вследствие неизбежной в полевых ус¬
ловиях неравномерности распределения удобрений дадут различные пока¬
зания для накопления в почве N03, NH4, Р205, pH, активного А1 и других
элементов динамики почвы. Между последними можно установить сопряжен¬
ность путем вычисления коэффициентов корреляции. В результате будет
584
С. В. Щерба, ф. А. Юдин
установлена зависимость изменения одного элемента динамики почвы от
другого.
Работа при помощи средней пробы иногда может дать и неверное представ¬
ление о среднем содержании в почве различных веществ вследствие их не¬
равномерного распределения. Например, фосфорная кислота, извлекаемая
водной вытяжкой из одних богатых фосфором частиц почвы, поглощается
из раствора другими частицами почвы, бедными фосфором. В результате
водная вытяжка дает приуменьшенные показания о содержании воднораст¬
воримой фосфорной кислоты в почве. Все это подтверждает большое значение
изучения в полевых опытах микродинамики почвы, т. е. реального распре¬
деления в почве различных элементов почвенной динамики.
Литература
Вельский В. Я. Метод постановки полевых
опытов в механизированных совхозах и
. колхозах.— Удобрение и урожай, 1931,
№ 8.
Вольф В. Г. Статистическая обработка
опытных данных. М., «Колос», 1966.
Деревицкий Н. Ф. Опытное дело в расте¬
ниеводстве. Кишинев, «Штиинца», 1962.
Доспехов Б. А, Методика полевого опыта,
изд. 2. М., «Колос», 1968.
Доспехов Б. А. Планирование полевого
опыта и статистическая обработка его
данных. М., «Колос», 1972.
Дояренко А. Г. Дробный учет при помощи
жнейки.— Вестник сельского хоз-ва,
1912, № 27.
Константинов Я. Я. Методика полевых
опытов с элементами теории ошибок. М.,
Сельхозгиз, 1949.
Константинов П. Я. Основы сельскохо¬
зяйственного опытного дела. М., Сельхоз¬
гиз, 1952.
Кудрявцева А. А. Методика и техника по¬
становки полевого опыта на стационар¬
ных участках. М., Сельхозгиз, 1959.
Лебедянцев А. Н. Пробные снопы как спо¬
соб учета урожая на опытных делянках.—
Избранные труды. М., Сельхозгиз,
1960.
Мейерсон Р. Ф. Рационализация уборки и
учета опытов с зерновыми при помощи
северного комбайна.— Химиз. соц. зем¬
леделия, 1937, № 6.
Методические указания по географической
сети опытов с удобрениями, вып. 1—13,
ВИУА. М., 1959—1966 гг.
Методические указания по организации и
проведению полевых опытов с удобрени¬
ями. М., «Колос», 1965.
Недокучаев Н. К. Опытное дело в полевод¬
стве. Теория и практика. М., Госиздат,
1929.
Полевой опыт. Под ред. П. Г. Найдина,
изд. 2. М., «Колос», 1968.
Рожественский Б. Я. Труды сети опытных
полей Всероссийского общества сахаро¬
заводчиков. 1902, вып. 3.
Рожественский Б. Я. Основные принципы
построения программы и методики ста¬
ционарных полевых опытов с удобрения¬
ми.— Химиз. соц. земледелия, 1938,
№ 7.
Снедекор Д. Ж. Статистические методы в
применении к исследованиям в сельском
хозяйстве и биологии. Пер. с англ. М.,
Сельхозгиз, 1961.
Ткачев Я. Я. Использование самоходного
комбайна для уборки зерновых культур
на опытных посевах.— Сов. агрономия,
1950, № 8.
Уишарт Дж.% Сандерс Г. Основы методи¬
ки полевого опыта. Пер. с англ. М.,
ИЛ, 1958.
Указания по агротехнике возделывания
сельскохозяйственных культур на оро¬
шаемых землях Ростовской области на
1952 г. Ростиздат, 1952.
Фишер Р. А. Статистические' методы
для исследователей. М., Госстатиздат,
1958.
Чернавин А. С, Учет комбайном полевых
опытов с зерновыми культурами.— Хи¬
миз. соц. земледелия, 1938, № 11.
Щерба С. В, Работы по методике мелкоде-
ляночного опыта.— Труды НИУИФ,
1941, вып. 148.
Юдин Ф. А. Методика агрохимических ис¬
следований. М., «Колос», 1971.
Ярославцев Г. М., Карпова А. И. и др.
Методика и техника определения потерь
от насекомых, вредящих полевым куль¬
турам. Л., 1935.
А. В. СОКОЛОВ
ВЕГЕТАЦИОННЫЙ МЕТОД
*
Вегетационный метод, или постановка опытов с выращиванием растений
в сосудах, применяется с различными целями. В физиологии растений и
агрохимии большое значение имеют водные, песчаные и почвенные культуры,
т. е. опыты с выращиванием растений в воде, кварцевом песке или почве.
Вегетационный метод для определения количества усвояемых питатель¬
ных веществ в почве рекомендовался уже давно как в виде обычного вегета~
ционного метода, так и в виде метода проростков. Однако вегетационный
метод не может заменить собою полевые опыты, так как условия произраста¬
ния растений и использование ими питательных веществ в вегетационных
опытах существенно отличаются от условий роста растений в поле.
При постановке вегетационного опыта берут пробы почвы с различных мест
поля из пахотного слоя почвы. Взятые пробы перемешивают, просеивают через
сито и обычно смешивают с кварцевым песком, чтобы усилить усвоение рас¬
тением питательных веществ почвы.
При вегетационном опыте высевается обычно одна культура, а данные
опыта используются для определения действия удобрений на урожай различ¬
ных сельскохозяйственных культур, возделываемых в хозяйстве. Поэтому,
используя результаты вегетационного опыта, необходимо учитывать особен¬
ности различных видов, а иногда даже и сортов растений, в отношении ис¬
пользования ими питательных веществ.
В полевых условиях растения берут питательные вещества как иэ пахот¬
ного, так и из нижних горизонтов почвы, в вегетационном же опыте иссле¬
дуется только пахотный слой почвы. В сосудах в течение всего времени веге¬
тации сохраняется оптимальная влажность почвы, следовательно, мобили¬
зация питательных веществ протекает иначе, чем в полевых условиях. Почва
сосудов находится в иных температурных условиях, чем в поле, что тоже не
может не отразиться на динамике почвенных процессов. Структура почвы,
ее водо- и воздухопроницаемость, а следовательно, и проницаемость почвы
для корней растений в сосудах также иные, чем в полевых условиях.
Таким образом, расхождения между условиями использования питатель¬
ных веществ в вегетационном опыте и в поле сводятся в основном к следую¬
щим трем моментам: 1) в вегетационпом опыте обычно используются питатель¬
ные вещества только одного слоя почвы; 2) в вегетационном опыте растения
находятся в условиях (влага, тепло), когда они могут использовать питатель¬
ные вещества почвы во много раз интенсивнее, чем в поле; 3) мобилизация
питательных веществ почвы в вегетационном опыте происходит иначе, чем
в поле.
Различия в ходе мобилизации питательных веществ в вегетационном опыте
и в поле отмечаются главным образом для азотных соединений. Поэтому
общепринятый вегетационный метод применяется преимущественно для
определения использования растениями фосфора и калия. При помощи веге¬
тационного опыта можно определить только то количество питательных ве¬
ществ, которое может быть усвоено растениями из данного образца почвы
при наличии благоприятных условий вегетации, искусственно создаваемых
с целью наиболее полного извлечения из почвы питательных веществ.
586
А. В. Соколов
Почва может быть бедной усвояемыми питательными веществами, но
прибавки урожая от внесения удобрений в поле может и не быть вследствие,
например, отсутствия нужного количества осадков. Поэтому расхождения
между действием удобрений в условиях полевого и вегетационного оныта
не говорят еще о непригодности того или другого опыта; они лишь указывают
на необходимость учитывать, что может дать тот или другой метод.
Определение общего, потенциального запаса в почве усвояемых питатель¬
ных веществ при помощи обычного полевого опыта вряд ли вообще возможно.
Результаты полевого опыта часто определяются метеороло1 ическими усло¬
виями данного года и особенностями принятой в опыте агротехники. Бели
в полевом опыте не наблюдается прибавки урожая от внесения удобрения,
можно предположить, что количество усвояемых питательных веществ в
почве было достаточным для обеспечения урожая растений, возможного в
метеорологических условиях данного года. В следующем году условия вегета¬
ции могут быть иными и эффективность удобрений будет иная.
Сопоставляя показания вегетационного опыта с результатами многолет¬
них полевых опытов, можно установить степень использования в полевых
условиях имеющегося в почве запаса усвояемых питательных веществ.
Вегетационный опыт может быть также широко использован для оценки
быстрых методов определения потребности растений в удобрениях.
Химический анализ почвы дает сведения о количестве в почве веществ,
растворимых в определенном реактиве. Но так как до сих пор еще нет ме¬
тодов, позволяющих точно определить, в форме каких соединений находятся
в почве питательные вещества, то все существующие химические методы
являются в основном эмпирическими приемами, ценность которых опреде¬
ляется корреляцией их показаний с результатами вегетационного и полевого
методов. Химические методы, так же как и вегетационный опыт, характери¬
зуют только свойства анализируемого образца почвы. Первой проверкой
пригодности этих методов является сравнение их показаний с результатами
вегетационных опытов. Если имеется корреляция между показаниями хими¬
ческого и вегетационного методов, то можно предположить, что в основе
химического метода лежит растворение именно тех форм питательных соеди¬
нений почвы, которые обеспечивают питание растений. Дальнейшей провер¬
кой химического метода является установление предельных чисел (лимитов)
на основе результатов анализа почв на делянках длительных полевых опытов.
Таким образом, комплексное использование вегетационного и полевого
методов является необходимым при разработке быстрых химических методов
определения потребности в удобрениях.
В России в дореволюционное время было стремление использовать веге¬
тационный опыт для характеристики плодородия почв различных генетиче¬
ских типов. С этой целью были предложены соответствующие изменения в
общепринятой методике вегетационного опыта. В основном они сводились
к возможно большей имитации полевых условий. Рекомендовалось брать об¬
разцы почвы не средние, а типичные, с ненарушенной структурой и с глубины,
по возможности соответствующей глубине слоя почвы, наиболее используе¬
мой корнями растений. Поливать почвы в сосудах предлагалось до влажности,
близкой к полевым условиям. Чтобы приблизиться к температурному режиму
почвы в поле, принимались меры для устранения нагревания сосудов. Подоб¬
ные вегетационные опыты мало чем дополняли показания полевых опытов.
Применение их вызвало в свое время ряд критических замечаний. Ценность
вегетационных опытов заключается не в том, что они заменяют полевой опыт,
а в том, что полученные при помощи их данные позволяют понять причины
тгех явлений, которые наблюдаются в различных полевых опытах.
Вегетационный метод
587
ТЕХНИКА ВЕГЕТАЦИОННОГО ОПЫТА
Взятие и подготовка почвы. Основные операции при
постановке вегетационных опытов с почвенными культурами следующие:
взятие почвы с поля, подготовка почвы, набивка сосудов, внесение удобре¬
ния, посев, уход за растениями, поливка и учет урожая.
При выборе почвы для вегетационного опыта необходимо заранее уста¬
новить, на какой почве должен быть поставлен опыт для разрешения стоящей
перед экспериментатором задачи, установить точное наименование почвы,
указать, откуда взят образец, культурное состояние и историю участка, с
которого взят образец (унавоживался ли и в какой степени, вносились ли
на него минеральные удобрения, когда, какие и в каком количестве, из-под
каких культур взят образец).
Нередко вегетационные опыты не дают нужных результатов вследствие
неудачного выбора почвы. При неудачном взятии образца почвы может ока¬
заться, что почва не реагирует на изучаемое удобрение. При постановке
опыта по изучению фосфатных или калийных солей необходимо брать почву
с участка, для которого уже имеются данные полевых опытов об отзывчи¬
вости его почвы на фосфор или калий. В крайнем случае, зная историю поля
и его урожайность, можно ограничиться контрольными анализами на коли¬
чество усвояемого фосфора и калия. На опытных станциях обычно нетрудно
найти участок, отзывчивость которого на внесение удобрений уже известна,
но, несмотря на это, часто берут почвы с защитных полос, дорожек между
делянками, т. е. с тех участков, на которые в прошлом могли ссыпаться удоб¬
рения.
На поле почву берут лопатами в чистые мешки. Надо следить, чтобы во
взятых для почвы мешках не было остатков удобрений: один случайно
попавший комочек удобрения может испортить весь опыт. Если почву берут
в большом количестве, то ее можно погружать навалом на подстеленный на
возу или грузовике брезент. Перевозить почву лучше всего в плотных дере¬
вянных ящиках или мешках. При большом количестве почву перевозят в
вагоне навалом.
Количество необходимой для постановки опытов почвы определяют с
учетом числа сосудов и их емкости. Так как при взятии, доставке и подго¬
товке почвы для опытов происходят большие потери, то количество почвы,
взятой в поле, должно быть не менее чем на 25% выше вычисленного на осно¬
вании числа сосудов в предстоящих опытах и емкости их. Если почва в поле
была очень влажной, то ее приходится брать на 30—40% больше коли¬
чества, необходимого для набивки сосудов.
Наиболее удобной считается такая влажность почвы, при которой почва
не пылит, но и не мажется и легко распадается на комки. Доставку, хране¬
ние и разборку почвы надо организовать так, чтобы почва не успела высох¬
нуть. Высыхание почвы приводит к повышению в ней количества усвояемых
веществ, главным образом азотных, а затем и фосфорных соединений. Опыт,
поставленный с влажной почвой, может поэтому дать другие результаты,
чем опыт, поставленный с этой же почвой, но после ее высыхания.
Время взятия в поле почвы имеет существенное значение для ее свойств.
В течение летнего периода в почве происходит нитрификация почвенного
азота и иммобилизация растворимых фосфатов. Поэтому почва, взятая ранней
весной, будет сильнее отзываться на азот и слабее на фосфор, чем почва,
взятая с того же участка летом. Такое же значение имеет предварительное
парование почвы в лабораторной обстановке. При постановке опытов с фор¬
мами фосфатов можно брать паровавшую почву и заготовлять ее летом для
опытов будущего года. При постановке опытов с азотными удобрениями же¬
лательно брать почву ранней весной, но во всяком случае не летом с парующих
участков.
588
А. В. Соколов
Весьма часто вегетационные опыты закладывают с почвами, которые бе¬
рутся с опытных делянок. Если в опыте делянки малого размера, почву
приходится брать в небольшом количестве и опыт закладывать в малых сосу¬
дах. С опытных делянок почву берут по тем же правилам, как и среднюю
пробу почвы для анализа, т. е. из разных мест делянки на глубину пахотного
слоя. Совершенно недопустимо брать почву с делянок, только что получив¬
ших минеральное или навозное удобрение. В этом случае свойства почвы будут
целиком зависеть от того, попадут случайно во взятую почву комки удобре¬
ния или нет. Лишь после неоднократной обработки удобренного участка взя¬
тый с него образец почвы может характеризовать свойства почвенного по¬
крова делянки.
Подготовка почвы для опытов заключается в приведении ее в однородную
но своему составу и свойству массу и состоит из перемешивания почвы,
пропускания ее через сита и удаления камней, корней и пожнивных остатков.
В практике опытного дела принято однократное пропускание почвы через
сито с отверстиями в 3 мм. Лучше брать проволочное сито (производитель¬
ность его больше), чем металлическое сито с круглыми отверстиями. Опера¬
ция просева производится следующим образом. Почву высыпают из мешков
на сита, стоящие на стойках над брезентом. Крупные комки почвы раздав¬
ливают руками, отбирают корни, пожнивные остатки, комки и т. п. Остав¬
шиеся на ситах включения по мере их накопления сбрасывают в сторону.
Просеянную почву ссыпают в лари для хранения. В большинстве случаев
атакой однократной обработки достаточно, чтобы иметь удовлетворительное
схождение параллельных опытов. В тех случаях, когда желают иметь боль¬
шую однородность образца почвы, перед началом опыта берут необходимое
число ведер уже разработанной почвы, высыпают на чистый брезент, тщатель¬
но перелопачивают, затем равномерно распределяют по брезенту и, беря ло¬
патой почву из разных мест, насыпают в ящик, из которого потом ее берут
при набивке сосудов. Контролем однородности почвы может служить наличие
одинаковых расхождений между урожаями в параллельных сосудах при
их набивке подряд и после набивки сосудов других вариантов.
Набивка почвы в сосуды. Сосуды для вегетационных опытов
изготовляются из стекла или оцинкованного железа. Стеклянные сосуды
имеют ряд преимуществ перед железными: они легко моются, стенки их не
разъедаются почвенным раствором, сквозь стекло видно, как набит сосуд;
подготовка к опыту стеклянных сосудов занимает меньше времени, чем
подготовка железных. Металлические сосуды имеют существенное преиму¬
щество перед стеклянными по прочности. Перед набивкой железные сосуды
надо покрыть внутри сначала эмалевой краской, затем даммаровым лаком.
Покраску лаком необходимо повторять перед каждой набивкой сосудов.
Снаружи сосуды красят белой масляной краской. Если опыт ставится не в
вегетационном домике, а под сеткой, то применяют только железные сосуды
Митчерлиха, имеющие отверстия внизу сосуда, и поддонники. Сосуды Мит-
черлиха следует употреблять эмалированные.
Размер сосудов должен соответствовать опытному растению. Для боль¬
шинства растений, кроме корнеплодов, картофеля и мощно развивающихся
лубяных растений, наиболее пригодными размерами сосудов являются:
15 х 20, 20 х 20 и 15 х 30 см 1. В сосудах этих размеров можно удачно
проводить опыты со льном, овсом, ячменем, пшеницей, просом, горохом, гре¬
чихой, горчицей, люпином, большинством огородных и другими растениями.
Точность опыта зависит от числа растений в сосуде; поэтому для таких рас¬
тений, как лен и большинство злаков, сосуды размером 20 х 20 см являются
•наиболее желательными. Сосуды размером 15 х 30 см имеют некоторое пре¬
имущество: благодаря более глубокому слою почвы они лучше сохраняют вла¬
1 вдесь и в дальнейшем первое число — диаметр сосуда, второе — его высота*
Вегетационный метод
589
гу и растения в них лучше развиваются. Но при надлежащей поливке даже
такие глубоко укореняющиеся растения, как люцерна и клевер, дают боль¬
шие урожаи и в сосудах 20 х 20 см. Стеклянные сосуды шире чем 20 см в
диаметре обычно не применяют, так как большие сосуды легко бьются.
Для корнеплодов и картофеля наиболее употребительные размеры сосудов
30 х 30 и 25 х 30 см. Сосуды размером 25 х 30 см удобнее в обращении,
хотя в них растения развиваются несколько хуже, чем в сосудах 30 х 30 см,
но все же они пригодны для постановки опытов с большинством корнепло¬
дов (сахарной свеклой и др.).
Для каждого опыта необходимо подобрать партию одинаковых сосудов.
Для этого все сосуды предварительно взвешивают с точностью до 10 г и
вес отмечают на их стенках. Сосуды, отобранные для одного опыта, должны
различаться по весу не более чем на 100 г. Отобранные по весу сосуды необ¬
ходимо проверить по диаметру и высоте. Сосуды размером 15 х 30
и 20 х 20 см должны различаться по диаметру не более чем на 0,5 см. Опыт
можно закладывать только в сосудах, одинаковых по объему.
Перед набивкой сосуды надо тщательно вымыть водопроводной водой,
я затем дистиллированной, если поливка будет производиться ею. Стеклян¬
ная трубочка, применяемая для проводки воды при поливе на дно сосуда,
должна быть на 2—4 см выше краев сосуда и иметь диаметр 1,2—1,7 см,
в зависимости от размера сосудов. Узкие трубки неудобны, так как они
задерживают поливку растения. Перед употреблением трубки моют так же,
как и сосуды.
На дно сосуда помещают для дренажа битое стекло, которое подготовляют
следующим образом. Предварительно в течение нескольких дней битое стекло
выдерживают в стеклянных сосудах с крепкой технической соляной кислотой.
Затем кислоту сливают и стекло промывают струей водопроводной воды под
краном до полного удаления соляной кислоты; воду сливают и стекло высу¬
шивают. Если поливка будет производиться дистиллированной водой, то
«ю же следует обмыть стекло перед сушкой.
Количество помещаемого на дно сосуда битого стекла должно быть доста¬
точно велико, чтобы при поливке сосудов через трубку не приходилось ждать
полного впитывания воды в почву, а можно было бы сразу влить все нужное
количество воды. Недостаточный дренаж на дне сосуда или узкая трубка
приводят к большим потерям времени при поливке сосудов. Битое стекло
должно покрывать около 2/3 дна сосуда под углом примерно в 30°. Для улуч¬
шения развития растений и экономии времени лучше давать скорее излишнее,
чем недостаточное количество стекла.
При диаметре сосудов 15 см достаточно взять 200—250 г стекла, при диа¬
метре 20 см — 300—350 г. Иногда для дренажа применяют вместо битого
стекла железный эмалированный или крытый даммаровым лаком конус,
помещаемый на дно сосуда.
Для отделения битого стекла от почвы употребляются марлевые круги,
диаметр которых на 5—8 см больше диаметра сосудов. Если вместо марли
употребляют другую ткань, то ее необходимо прокипятить в воде. Марлю
помещают на дренаж из битого стекла и сверху на нее в том месте, где нет
стекла, насыпают небольшое количество песка, что обеспечивает равномер¬
ность увлажнения почвы около дна сосуда.
Кварцевый песок, употребляемый при постановке вегетационных опытов,
предварительно отмучивают от глинистых частиц и органических примесей
водопроводной водой в вегетационных или других сосудах. Затем песок вы¬
сушивают или на солнце, или в особых сушилках. Промывку песка соляной
кислотой при постановке опытов с почвенными культурами можно не делать.
В некоторых случаях можно избежать и отмучивания песка, ограничившись
-отсеиванием его от глинистых частиц через сито с размером ячеек в 1 мм
и однократной промывкой дистиллированной водой.
590
А. В. Соколов
В дальнейшем для удобства проведения поливов сосуды необходимо та¬
рировать, т. е. привести к одинаковому весу. Тарирование производят пес¬
ком и отчасти битым стеклом, применяемым для дренажа. Опыты К. К. Гед-
ройца показали, что количество дренажа (битого стекла) влияет на высоту
урожая, поэтому при тарировании следует отбирать сосуды, близкие по весу»,
и само тарирование производить песком. В случае применения конуса тари¬
ровать можно и битым стеклом, которое помещают под конус.
При тарировании и подготовке сосудов к набивке поступают так. Сначала
насыпают в сосуд дренажное стекло, покрывают его кружком марли и сбоку
через отверстие, сделанное в марле, вставляют в горку стекла трубку для
полива так, чтобы она отстояла от стенки сосуда не менее чем на 2 см. Затем
производят тарирование сосуда кварцезым песком, насыпаемым на марлю*
с точностью до 5 г. После тарирования на сосуд наклеивают этикетку и, кро¬
ме того, на стенке сосуда обозначают черным лаком его номер.
При набивке сосудов почву аккуратно насыпают на дно, в середину лежа¬
щей марли, осторожно расправив последнюю руками и плотно прижав ее
края почвой к стенкам сосуда. Почва не должна просыпаться между марлей
и стенками сосуда. Нижний слой почвы, примерно в 3—4 см толщины, укла¬
дывают более плотно, чем остальную почву в сосуде. Такое уплотнение пред¬
охраняет от попадания почвы в дренаж и создает более равномерные условия
впитывания влаги. Если сверх марли помещен кварцевый песок, как указано
выше, почву насыпают непосредственно на песок. В дальнейшем почва равно¬
мерно уплотняется и поверхность ее выравнивается; до верхнего края сосуда
должно остаться около 1,5—2 см свободного пространства для размещения
песка и полива сверху. Как правило, следует применять равномерное уплот¬
нение почвы во все время набивки, более сильно уплотнив лишь самый ниж¬
ний ее слой. Недостаточно уплотненная почва сильно оседает за вегетацион¬
ный период и при поливе сверху размывается; при поливе сниэу в недостаточ¬
но уплотненной почве происходят разрывы почвы и корней, особенно в узких
сосудах, размером 15 х 30 см.
Чтобы равномерно набить целую серию сосудов, надо иметь некоторый
навык. Недопустимо, чтобы одни сосуды уплотнялись сильнее, а другие
слабее. Если сосуды правильно подобраны для опыта (имеют одинаковые
диаметр и высоту), то при одинаковом весе почвы и равномерном уплотне¬
нии ее во всех сосудах должно остаться одно и то же расстояние от поверх¬
ности почвы до края сосуда.
Количество почвы, вносимое в сосуд, устанавливается пробной набивкой;
при неправильном установлении количества почвы на сосуд приходится наби¬
вать сосуды заново.
Наполненные почвой сосуды следует до посева или сразу после посева
засыпать сверху слоем кварцевого песка, около 200 г на сосуд среднего
размера. После появления всходов сосуды снова засыпают песком с таким
расчетом, чтобы слой песка сверху был не более 1 см толщины. Слой кварце¬
вого песка сверху сосуда предохраняет почву от излишней потери влаги,
уменьшая испарение воды с поверхности сосудов, и от размывания поверх¬
ности почвы при поливке сверху.
Перед набивкой сосудов берут из подготовленной почвы пробу с четырех¬
кратной повторностью для определения влажности.
Во время набивки заготовленная почва должна быть предохранена от вы¬
сыхания. Количество добавляемой при набивке воды должно обеспечить опти¬
мальную для набивки влажность почвы. Взятые для отдельных сосудов на¬
вески почвы помещают в большие эмалированные тазы, в которых руками
почву перемешивают с удобрениями (последние могут вноситься в виде раст¬
воров или в виде порошков). Почва не должна пылить и мазаться по стенкам
таза, а при сжимании должна образовывать комки, которые легко распада¬
ются, если их выронить из руки. Добавляемое при набивке количество воды
Вегетационный метод
591
должно быть одинаково во всех сосудах; если часть удобрений вносится в рас¬
творах, количество воды, добавляемой в эти сосуды, должно быть соответст¬
венно уменьшено.
Чтобы не перепутать удобрения при набивке, схему опыта выписывают
на листе с точным, но кратким указанием, что добавляется в каждый сосуд.
Например, сосуд №11: воды — 175 мл, (NH4)S04 — 0,5 г, Na2HP04-
• 12Н20|— 0,25 a, K2S04 — 50 мл. При набивке все вносимое отмечают на
листе карандашом. Число отвешенных пакетов с удобрениями должно
соответствовать числу сосудов с удобряемой почвой; на каждом пакете дол¬
жен быть номер сосуда. На тазу, после того как в него высыпана навеска
почвы, подписывают восковым карандашом номер сосуда.
Если набивают сосуды, получающие одинаковые удобрения, то перед
набивкой нового сосуда можно не мыть рук и тазов, достаточно протереть
их, чтобы на них не оставалось следов почвы. При переходе к сосудам,
получающим другие удобрения, необходимо мыть и тазы, и руки. Перемеши¬
вание почвы с удобрениями надо производить не менее 3—4 мин. после того,
как почва получит совершенно однородный вид. Продолжительность време¬
ни перемешивания почвы зависит от ее свойств: песчаные почвы перемешива¬
ются быстрее, чем глинистые.
Удобрения. Удобрения можно вносить или в виде растворов, или
в виде порошков и гранул. Если не имеется каких-либо специальных заданий,
удобрения вносят в растворе. Наиболее удобны растворы, в которых на каж¬
дые 10 мл приходится 0,1 г питательного вещества (N, Р2Об или К20). Раст¬
воры, если их вносят менее чем по 50 мл, отмеривают пипеткой или бюреткой;
при внесении 50 мл и более для дозировки растворов можно употреблять мер-
ные цилиндры. Величина доз удобрений зависит от темы опыта, размера со¬
судов и вида растения. Как правило, при постановке опытов в почвенных
культурах для обеспечения нормального развития растений приходится за¬
ботиться лишь о добавке азота, фосфора и калия.
В опытах с почвенными культурами в качестве основного удобрения (фо¬
на) следует выбирать соли, не вызывающие сильных изменений свойств почвы.
Удобрения, вносимые в почву в качестве фона, должны возможно меньше
изменять реакцию почвы и концентрацию почвенного раствора, а также
не содержать балластных веществ. Если требуется фон одного азота, можно
остановиться на внесении азотнокислого аммония; последний, правда, вызы¬
вает некоторое подкисление почвы, не имеющее практического значения.
При постановке опытов на кислых песчаных почвах можно рекомендовать
смесь, состоящую из двух третей азота в виде NH4N03 и одной трети азота
в виде Ca(N03)2.
Для создания фона NK можно брать смесь NH4N03 + KNOs, в которой
количество KN03 устанавливается по потребной дозе калия.
В качестве фона NP для почв черноземного типа можно применять смесь
из NH4NOs и моно- и диаммонийфосфатов, являющуюся биологически кис¬
лой. Для подзолистых почв можно остановиться на смеси Ca(N03)2 +
+ NH4H2P04, т. е. физиологически кислого моноаммонийфосфата и физио¬
логически щелочной кальциевой селитры. Для фона РК наиболее подходя¬
щей является смесь моно- и дикалийфосфатов. Количество моно- и дикалий-
фосфатов в этой смеси подбирают такое, чтобы pH смеси было близко к pH
почвы. Если последнее нельзя сочетать с желательными дозировками КгО
и Р205, то добавку фосфатов можно делать в форме кальциевого фосфата,
а калия — в виде КС1 или K2S04.
Фон одного фосфата, который обычно создается в опытах с внесением
натриевых фосфатов, во многих случаях рекомендовать нельзя, так как вне¬
сение в почу натрия оказывает сильное действие на эффективность калия.
Поэтому часто приходится останавливаться на кальциевых фосфатах: моно-
кальцийфосфате, дикальцийфосфате или на их смеси. Внесение натриевых
592
А. В. Соколов
фосфатов может быть рекомендовано, когда добавка натрия не влияет на ре¬
зультаты опытов. В этом случае интересным является применение не чистых
однонатриевых и двунатриевых солей, а их смесей, дающих pH, близкий к
pH почвы.
При выборе форм основного удобрения и их дозировок надо тщательно
продумать, какое действие на изменение свойств почвы окажут вносимые
удобрения. При неосторожном выборе форм удобрений легко получить ре¬
зультаты, определяющиеся не столько свойствами почвы, сколько свойст¬
вами фона.
При постановке опытов на известкованных почвах в состав фона для ряда
растений — свеклы, льна, горчицы, гречихи, табака, бобовых — надо
вводить бор в форме борной кислоты или буры в количестве 1 мг бора на 1 кг
почвы. Без внесения бора на известкованных подзолистых почвах нельзя
получить нормального развития этих растений. Под зерновые злаки вносить
бор излишне.
Что касается доз питательных веществ, то для получения высоких урожаев
растений в сосудах 20 х 20 см К. К. Гедройц считал достаточным 0,75 г
N, 0,5 г Р20Б и 0,5 г К20; количества эти должны изменяться в зависимости
от вида растения и темы опыта; применение удвоенных доз дает дальнейшее
повышение урожаев.
При больших дозах вредное действие высокой концентрации солей можно
уменьшить, применяя дробное внесение удобрений, которое практикуется
также в опытах по изучению времени внесения удобрений. Во время вегетации
удобрения вносят при поливе в растворенном виде. Обычно половину вноси¬
мого количества удобрений вливают на дно сосуда через трубку, а другую вно¬
сят поливом сверху.
Посев растений и уход за ними. Зерновые злаки — овесг
ячмень, рожь, пшеница, а также зерновые бобовые — горох, люпин, фасоль,
бобы и пр.— высаживают проращенными семенами. Проращивание семян
производят на блюдах, в которые насыпают ровный слой кварцевого песка,
кладут сверху его фильтровальную бумагу в два слоя и на нее укладывают
раздельно одно от другого семена. Перед раскладкой семян бумагу и песок
увлажняют дистиллированной водой до полного насыщения песка водой.
После раскладки семена закрывают сверху двумя листами фильтровальной
бумаги. Блюдо сверху закрывают стеклом, чтобы уменьшить испарение и
предохранить семена от повреждений.
Посев производят, когда семена «наклюнутся». Перед посевом поверхность
почвы выравнивают, слегка поливают из промывалки, затем делают лунки,
в которые укладывают семена. Лунки можно делать либо сажальной доской,
имеющей соответствующее число равномерно распределенных зубьев, либо
стеклянной палочкой; в этом случае на поверхность накладывают картонный
шаблон с соответствующим числом дырочек и лунки в почве выдавливаются
стеклянной палочкой с резиновым кольцом, надетым на нее на высоте, рав¬
ной глубине посадки семян. Крупные семена при посадке заделывают на глу¬
бину 1,5—2,0 см, при посеве льна лунки делают глубиной в 1 см, при посеве
мелкосеменных трав — еще мельче (0,5 см). Если почва мажется, то полезно
посыпать ее тонким слоем кварцевого песка.
При посеве проращенными семенами пинцетом отбирают одинаково
проросшие семена, которые и укладывают в лунки по одной штуке корешком
книзу. Когда семена положены во все лунки сосуда, посев проверяют и лунки
заделывают надавливанием на их стенки, после чего почву засыпают сверху
песком (200 г песка на сосуд 15 см в диаметре). Посадка по два семени в одну
луночку недопустима, это приводит к порче посевов при последующей про¬
рывке.
При посеве сухими семенами необходимо предварительно установить их
всхожесть. Для посева годятся только семена, имеющие всхожесть, близкую
Вегетационный метод
593
К 100 %, и тщательно отобранные по величине. Семенной материал должен
быть однороден, одного сорта и одного урожая.
После окончания посева сосуды закрывают сверху листами бумаги во из¬
бежание высыхания почвы. Листы снимают при первом появлении всходов.
Если опыт ставится в стеклянных сосудах, желательно до посева или сразу
после появления всходов (не позднее!) обернуть сосуды белым картоном —
«надеть чехлы». На чехол наклеивают этикетку с указанием особенностей
варианта опыта и номера сосуда; последний надписывают и непосредственно
на чехле. Чехол должен быть надет плотно, но так, чтобы можно было его
поднимать для наблюдений за увлажнением почвы и ростом корней.
Количество семян, высеваемых в сосуд, должно быть несколько больше
желательного числа растений. Если посев производится проращенными се¬
менами, то высеваемых семян должно быть примерно на 5—10 шт. больше,
чем желательное число растений. Для таких растений, как ячмень, посевы
которого иногда сильно повреждаются шведской и гессенской мушкой, ко¬
личество высеваемых семян должно быть раза в полтора больше желательно¬
го числа растений. Зерновых злаков на сосуд 15 см в диаметре надо оставлять
после прореживания 20—25 растений, гороха 10—15, гречихи 10—12, льна
35—40, клевера 6—12. При посеве растений, которые оставляют по одно¬
му на сосуд, например свеклы, в центре сосуда высевают около 10 семян.
Когда растения уже окрепнут, минует опасность гибели всходов от вре¬
дителей, производят прореживание. Все удаляемые растения помещают в
пронумерованные пакеты, сушат и взвешивают. Если растения удаляют пин¬
цетом вместе с семенами через 2—4 дня после появления всходов, то выдер¬
гиваемые растения выбрасывают. Когда растения подрастут, для предохра¬
нения их от полегания и поломки во время полива на сосуды надевают кар- -
касы или вставляют в сосуды палочки (по четыре на сосуд), между которыми
натягивают нитки; палочки должны быть подобраны одного веса.
В течение всего времени вегетации необходимо удалять вредителей, со¬
бирать в пакеты опадающие листья и семена и делать записи наблюдений
8а развитием растений (измерение хода роста растений и фенологические
наблюдения).
При уборке урожая измеряют высоту срезанных растений на линейке
с точностью до 0,1 еле, после чего у зерновых отрезают колосья, у клевера
и льна — головки и помещают их в отдельный пакет, а стебли и листья —
в другой. Обычно взвешивания для определения сырого веса не производят,
а урожай взвешивают после сушки его в сушилке или в термостате при 60°.
Сушку до абсолютно сухого веса обычно не производят. Высушенный урожай
взвешивают с точностью до 0,01 а, обмолачивают и урожай семян взвешивают
отдельно.
Поливка. Правильная поливка растений является весьма существен¬
ным условием удачного проведения опыта. Поливку всех сосудов, участвую¬
щих в опыте, производят до одинаковой влажности почвы, за исключением
тех случаев, когда изучается значение изменения влажности почвы. Уста¬
новить желательную влажность почвы можно, лишь зная водные свойства
почвы: ее максимальную гигроскопичность, наибольшую влагоемкость и
влажность во время набивки. Максимальную гигроскопичность необходимо
знать для определения коэффициента завядания растений. Под коэффициен¬
том завядания понимается количество влаги в почве, выраженное в процен¬
тах от ее сухого веса, при котором растения впервые обнаруживают признаки
устойчивого завядания. Под устойчивым, или длительным, завяданием под¬
разумевается степень увядания растений, при которой они уже не могут оп¬
равиться даже после перенесения их в атмосферу, насыщенную водяными
парами. Понятие устойчивого завядания было введено, чтобы исключить
при определении все случаи завядания, которые вызываются лишь времен¬
ным превышением транспирации над поступлением влаги в растение и могут
594
А. В. Соколов
иметь место при относительно влажной почве, но сильном сухом ветре или
сильной инсоляции.
Практически наиболее легко выполнимым способом определения коэффи¬
циента завядания (или влажности завядания) является определение его по
максимальной гигроскопичности почвы 1. В среднем коэффициент завядания
равен полуторной максимальной гигроскопичности почвы.
С. М. Богданов предложил считать полезной влагоемкостыо почвы ее
наибольшую влагоемкость за вычетом коэффициента завядания, так как
влажность почвы меньше коэффициента завядания если и не является абсо¬
лютно неусвояемой, то практически представляет мертвый запас влаги.
Поливку растений следует производить до 60 % полезной влагоемкости почвы.
Тогда влажность почвы, до которой поливают сосуды, будет равна:
Г/(наибольшая \ * с ( максимальной \"| gn
[Iвлагоемкость; ' \ гигроскопичностиJ J* / максимальной \ _
100 ’ ^гигроскопичности)
= 0 6 Г Г наиб°льшая \ I ( максимальная \ 1
’ |Д влагоемкость j ^гигроскопичность JJ '
Иногда, в тех случаях, когда почва имеет большое количество глинистых
частиц и потому сравнительно высокую максимальную гигроскопичность,
поливка до 60 % от влагоемкости является недостаточной. Поэтому для черно¬
земных и глинистых почв часто считают оптимальной влажностью не 60, а
70% от полезной влагоемкости.
Устанавливаемая по приведенной выше формуле влажность, конечно,
считается оптимальной весьма условно. Но во всяком случав влажность поч¬
вы, определяемая в размере 60% от полезной влагоемкости, для большинст¬
ва почв и растений близка к оптимальной.
Многие растения для получения максимального урожая требуют различ¬
ной влажности в разные периоды своего развития. Как правило, необходимо
уменьшать влажность почвы во время созревания растений, так как иначе
созревание задерживается.
Обычно поливку растений производят, давая половину воды сверху
и половину — снизу. Во многих опытах можно поливать почвенные куль¬
туры не дистиллированной, а водопроводной водой. Обычно поливку сосудов
по весу производят один раз в день. В жаркие дни поливать сосуды прихо¬
дится два и даже три раза в день; в этом случае один раз поливают сосуды
по весу и другой — по объему, давая на каждый сосуд определенное коли¬
чество воды.
Вес, до которого надо поливать сосуды, вычисляют следующим образом;
Предположим, что полная влагоемкость почвы — 55,0%, максимальная
гигроскопичность — 8,2%, поливка намечена до 60% от полезной влагоем¬
кости. Следовательно, влажность почвы должна быть равна (55,0 + 8,2)*
•0,6 = 37,9%. Влажность почвы при набивке сосудов была равна 15,2%,
в сосуд вошло 5 кг сырой ’почвы или 4,340 кг абсолютно сухой почвы. Вес
тары до набивки (общий вес пустого сосуда, битого стекла, трубочки, марли
и кварцевого песка, добавленного на дно сосуда) был равен 2100 г. Вес кар¬
тонного чехла (если сосуд стеклянный) 60 г. Вес палочек 40 г плюс вес квар¬
цевого песка, добавляемого сверху почвы, 200 г. Таким образом, общий вес
набитого сосуда без веса воды и почвы 2400 а, вес почвы — 4340 г, вес воды—
37,9% от 4340, или 1645 г. Следовательно, сосуд надо поливать до 2400 +
+ 4340 + 1645 = 8385 г; округляя, имеем 8400 г.
При поливке сосудов производят их перестановку на вагонетке для
выравнивания условий освещения и нагревания солнечным светом. Если со¬
1 Более подробно об определении коэффициента завядания см. в статье Д. В. Федоров¬
ского в настоящем сборнике.
Вегетационный метод
595
суды опыта стоят в два ряда, то при поливке их меняют местами. Необходимо
перемещать сосуды и по длине вагонетки; для этого каждый раз два первых
сосуда снимают и ставят вместо последней пары, а на их место ставят сосуды
второй пары. Бели в опыте много сосудов, то перестановку производят на
две пары сосудов. Правильным следует считать такое размещение, когда
сосуды стоят по повторностям, а не по вариантам опыта.
МЕТОДИКА ОПЫТОВ С НИЗКОЙ ВЛАЖНОСТЬЮ почвы
Изучение усвояемости питательных веществ из почвы, влажность которой
близка к мертвому запасу влаги, надо проводить, обеспечивая растения
влагой со стороны. Бризель предложил метод, в котором корни растений
снабжаются влагой из резервуара с водой, после того как они пройдут сухую
почву. Чтобы по корням, как по фитилю, вода не поднималась кверху, их
смазывают вазелином, а воду покрывают слоем вазелинового масла. В опытах,
поставленных по методике Бризеля, нормального развития растений не полу¬
чается; они развиваются плохо и постепенно отмирают, после чего, несмотря
на принятые меры предосторожности, почва начинает увлажняться. Простая
методика, дающая достаточную изоляцию сухого слоя почвы и вполне здо¬
ровое нормальное развитие растений, была предложена нами.
Передвижение влаги из глинистой почвы в слой песка может происходить
только тогда, когда влажность почвы близка к ее полной влагоемкости.
Глинистая почва удерживает влагу с настолько большой силой, что слабая
всасывающая способность песка не может отнять эту влагу из почвы. Если
в сосуд насыпать слой влажной почвы с содержанием влаги около 50%
от полной влагоемкости, а затем слой кварцевого песка толщиной примерно
2 см, то отсыревает только тонкий слой песка, непосредственно прилегающий
к почве; почва же, насыпанная сверху песка, остается сухой. Таким образом,
слой песка изолирует влагу одного слоя почвы от влаги другого. Внося удоб¬
рения в верхний слой почвы и производя поливку только нижнего слоя
почвы (через вставленную до дна сосуда стеклянную трубочку), можно со¬
хранить верхний слой почвы сухим или, если он был влажным, постепенна
иссушить его в результате деятельности корней растений. Поливку сосудов
производят по расчету на увлажнение одного нижнего слоя почвы. Почва
в сосудах должна быть нормально уплотнена, как принято при набивке
сосудов. Границы между слоями почвы и песка должны быть ровными го¬
ризонтальными плоскостями. Песок не должен содержать тонких частиц,
и слой его должен быть не менее 2, лучше 3 см толщиной. Сверху сосуды за¬
ливают замазкой, состоящей из мела и касторового масла. Почва берется
глинистая с большой влагоемкостью. Проведение опытов должно быть за¬
кончено до наступления сильного ночного похолодания верхних слоев почвы.
Опыты могут проводиться при посеве растений двумя способами. В пер¬
вом случае, который имеет наибольшее значение для понимания явлений,
происходящих в природных условиях, при набивке сосудов увлажняют почву
как нижнего, так и верхнего слоя. Влажность почвы берется невысокая,
исключающая возможность ее передвижения сквозь слой песка из верхнего
слоя в нижний. Производят посев, и верхний слой почвы засыпают, как обыч¬
но, слоем песка. Растения, в зависимости от условий опыта, успевают за счет
влаги верхнего слоя почвы настолько развить свою корневую систему, что
она проникает в нижний слой почвы. Иногда приходится, в ожидании пока
корни растений проникнут в нижний горизонт, делать умеренную поливку
верхнего слоя почвы. После того как наблюдениями через стеклянную стен¬
ку сосуда будет установлено, что корни растений развиваются уже в нижнем
горизонте, поливку сверху прекращают и на почву насыпают слой песка в
2 см или, как указано выше, заливают замазкой из касторового масла с мелом.
596
А. В. Соколов
Имея данные анализа растений, относящиеся к моменту конца иссушения
верхнего слоя почвы (который легко определяется по окраске почвы), и за¬
тем данные для времени окончания опыта, можно установить, сколько пи¬
тательных веществ было взято растениями из удобрений, находящихся в су¬
хом слое почвы.
Опыты с песчаной прослойкой между слоями почвы при посеве растений
во влажный слой ее требуют, таким образом, двух учетов урожая и удвоения
аналитической работы. Это является недостатком метода; его достоинство
в том, что корни растений нормально развиваются в верхнем слое почвы;
контакт почвы с растением и все развитие корней протекают нормально.
Иссушение почвы производится самими опытными растениями.
Возможна и другая постановка опытов с песчаной прослойкой. Растения
предварительно выращиваются в водных растворах, и когда их корни до¬
стигнут длины, равной глубине опытных сосудов, производят посадку рас¬
тений. В этом случае, опустив растения в сосуд и придерживая их рукой,
насыпают сначала нижний слой почвы, затем*средний слой песка, а потом
верхний удобренный слой сухой почвы и, наконец, верхний слой песка.
Чтобы вполне устранить поверхностное испарение почвенной влаги, можно
сосуды залить сверху замазкой из касторового масла и мела. При постановке
такого опыта не требуется двойного учета урожаев. Недостатками его явля¬
ются ненормальное развитие корней растений в водных культурах (в кото¬
рых корни растений даже морфологически отличаются от корней, выросших
в почвенных условиях) и отсутствие в верхнем слое почвы нормального кон¬
такта между корнями растений и почвенными частицами^
ОПЫТЫ С ПОЧВЕННОЙ ИЗОЛЯЦИЕЙ УДОБРЕНИИ
Распределение в почве удобрений имеет большое значение для их дейст¬
вия. В вегетационных опытах можно создать различное размещение в почве
удобрений. Решение ряда вопросов — о равномерности распределения удоб¬
рений, о взаимоотношении различно удобренных растений, удобрении сме¬
шанных посевов, об использовании многосторонних и односторонних удобре¬
ний — требует постановки таких опытов, когда внутри сосуда с удобренной
почвой часть ее остается неудобренной или удобренной иначе, чем остальная
часть. Для решения этих вопросов мы разработали методику почвенной изо¬
ляции удобрений.
Постановку опытов при изоляции удобрений почвой не следует смеши¬
вать с опытами в изолированных культурах. При методе изолированных куль¬
тур корни растений делят на несколько прядей между разными сосудами;
при методе почвенной изоляции механического деления корней растений при
посадке не производят, и корни сами распределяются между различно удоб¬
ренной почвой. При набивке сосуд делят вертикальными перегородками
на несколько секторов, в которые вносят различно удобренную почву.
По окончании набивки перегородки вынимают, и все посаженные растения
могут пользоваться всей почвой сосуда и распределять свою корневую сис¬
тему по всему сосуду. При посеве растений на границе различно удобренных
секторов каждое из растений находится в условиях, когда ему предоставлены
равные возможности для развития корней в обоих секторах. Бели же посев
растений сделать не на границе, а внутри площади секторов, то получим по¬
сев растений на различно удобренных микроучастках почвы.
В первом случае, когда растения посажены на границе секторов, все они
находятся в одинаковых условиях снабжения питательными веществами, и
задача исследователя сводится к изучению того, как распределяются корни
одних и тех же растений между различно удобренной почвой и как неравно¬
мерное размещение питательных веществ отразится на урожае растений.
Вегетационный метод
597
В том случае, когда растения посажены внутри различно удобренных сек¬
торов, они находятся в неодинаковых условиях снабжения питательными
веществами, и эти различно питающиеся растения дают, как показали опыты,
различные урожаи и вступают друг с другом в целый ряд закономерных
взаимоотношений.
Техника постановки таких опытов несложна. В сосуд, после того как на
дне его сделан дренаж из битого стекла и насыпан слой кварцевого песка,
насыпают слой неудобренной почвы весом 500—1000 г. При набивке сосуда
надо следить за тем, чтобы дренаж был достаточно хорошо устроен по всему
дну сосуда, с тем чтобы во время поливки сосуда, которая делается через
трубку, идущую сверху до его дна, происходило быстрое и равномерное
распределение влаги по дну сосуда. Затем в сосуд вставляют тонкие жестя¬
ные перегородки, делящие его на несколько секторов. Обычно применяют
деление на четыре сектора, из которых два противоположных получают оди¬
наковое удобрение. В этом случае, если площадь сосуда равна 314 см2,
площадь одного сектора равна всего 79 см2. После того как в секторы насыпана
различно удобренная почва, в сосуд насыпают верхний защитный слой
неудобренной почвы. Границы секторов отмечают на стенках сосудов, пере¬
городки вынимают, насыпают тонкий слой кварцевого песка, и сосуд готов
к посеву растений. Обычно при посеве овса высевают на площади каждого
лектора по 6 растений двумя рядами, на расстоянии 2—2,5 см от границы с
соседним сектором. Таким образом, при посеве внутри секторов все растения
имеют на расстоянии всего 2—2,5 см иначе удобренную почву и на расстоянии
всего 4—5 см — соседние растения, растущие в другом секторе. В данном
случае растения могут использовать почву соседних микроучастков и должны
оказать влияние на развитие соседних растений.
Опыты ставят с адсорбируемыми почвой соединениями (NH4, Р205, К20),
с почвами тяжелого механического состава, при невысоких дозах удобрений
и при умеренной поливке сосудов сверху и снизу. В этом случае боковое пере¬
движение исключается. В сосудах под растениями накопления заметных ко¬
личеств нитратов уловить не удается, что указывает на отсутствие передвиже¬
ния азота, вносимого в форме аммонийных солей.
Дальнейшее развитие метод почвенной изоляции получил в опытах
П. И. Ромашова. Изучая вопрос о смешанных посевах бобовых и злаковых
трав и приемах удобрения чистых и смешанных посевов, П. И. Ромашов
применил метод почвенной изоляции, дополненный введением половинных
•сосудов. Если внутри одного сосуда одну половину его засеваем бобовыми,
а другую злаками, то развитие тех и других зависит не только от использо¬
вания корнями питательных веществ, но и от взаимоотношения между рас¬
тениями над поверхностью почвы. Если одни половинные сосуды засеять
злаками, а другие бобовыми, то,^объединяя эти половинные сосуды, мы со¬
храняем надземную конкуренцию растений, а разъединяя их, исключаем
•ее. Соединяя два половинных сосуда, мы должны приставить их плотно друг
к другу и надеть на них общий чехол. Так как прогревание половинных со¬
судов может быть более сильное, чем круглых, необходимо тщательно обер¬
тывать сосуды несколькими слоями белого картона или помещать их внутри
деревянного чехла.
Для изучения некоторых особенностей питания растений из неоднородной
почвы использован предложенный Д. В. Федоровским (1964) тип сосудов
для опытов с изолированным питанием растений и применена радиоактивная
метка фосфатов для одного из двух объемов почвы, питающих растение. Эта
методика позволяет фиксировать поступление воды и фосфатов из каждого
объема почвы и учитывать влияние изменения почвенных условий в одном
объеме на питание растений и поступление веществ из другого объема почвы.
Применение сосудов этого типа позволяет не только размещать корни одного
растения на изолированных участках почвы, но и ежедневно (а при желании
598
А. В. Соколов
'Ш 'W
Приемы работы с сосудами но¬
вого типа в опытах с изоли¬
рованным питанием
Объяснение см. в тексте
ежечасно) учитывать расход воды из каждого от¬
сека сосуда, из каждого участка почвы. Этот со¬
суд позволяет также изменить во время опыта
режим влажности одной части почвы, поддер¬
живая неизмененный режим влажности в дру¬
гой ее части.
Сосуд представляет собой железную коробку
с плотно закрепленной пустотелой перегороди
кой посередине. Внутри перегородки встав¬
лена трехгранная ось сосуда (рисунок, а).
^ Перед набивкой при тарировке уравнове^
шиваются части сосуда. Набивка одинаковых
навесок почвы в отсеки сосуда не нарушает
балансировки (рисунок, б). Вдоль перегородки
высаживают от 3 до 16 растений, корни каждого*
из них распределяются поровну между двумя
участками почвы.
Во время опыта определяют общий расход,
воды А по убыли веса всего сосуда, так же как
в обычных вегетационных опытах (рисунок, в)~
Затем устанавливают сосуд на подставку и опре¬
деляют расход воды из почвы каждого изолиро¬
ванного отсека сосуда. В случае разного расхо¬
да воды из участков почвы нарушается балан¬
сировка сосуда. Уравновешивая сосуд гирями,,
можно количественно учесть расход воды иа
каждой части сосуда (рисунок, г и д).
Определение это достаточно просто: еслиг
например, общий расход воды А = 150 а, а для
уравновешивания на правую часть сосуда по¬
требовалось поставить гирю в 100 г (В), то рас¬
ход воды из почвы правого отсека будет равен:
А +В
2
150 +100
2
= 125 г,
а левого отсека:
А — в 150-100
г ~ 2
= 25 г.
ПРИМЕНЕНИЕ ВЕГЕТАЦИОННОГО МЕТОДА
ДЛЯ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ В ПОЧВЕ УСВОЯЕМЫХ ДЛЯ РАСТЕНИЙ
ПИТАТЕЛЬНЫХ ВЕЩЕСТВ
Для определения потребности растений в удобрениях применяются*
кроме обычного вегетационного, также специальные методы: метод пророст¬
ков Нейбауэра — Шнейдера и метод Митчерлиха. Вегетационный метод
Митчерлиха в свое время был принят Международным обществом почвове¬
дов в качестве стандарта для оценки пригодности химических и микробиоло¬
гических методов определения количества в почве усвояемых веществ. Метод
Нейбауэра получил широкое распространение в практике сельского хозяйст¬
ва, а также при почвенных обследованиях — для установления количества
усвояемых веществ в различных почвенных горизонтах. Оба эти метода раз¬
носторонне изучались в различных странах.
Вегетационный метод
599
Метод проростков Нейбауэра — Шнейдера
Метод состоит в извлечении из почв питательных веществ корнями моло¬
дых растений. Чтобы добиться быстрого извлечения из почвы корнераствори¬
мых питательных веществ, в опытах по Нейбауэру на малое количество поч¬
вы (100 г) берется большое число растений (100 экземпляров).
Опытным растением в методе Нейбауэра — Шнейдера служит озимая
рожь из семян урожая последнего года. Семена должны быть хорошо высу¬
шены, обладать высокой всхожестью (не менее 95%) и большой энергией
прорастания. Вес 1000 зерен должен быть около 40 г и не менее 37 а. Сорт ржи
не имеет большого значения для результатов опыта, но все сравниваемые
опыты должны иметь посевной материал из зерен одного сорта. Весьма важ¬
но следить за однородностью посевного материала. Отсутствие выровненно-
сти посевного материала может привести к большому расхождению между
параллельными определениями и сделать результаты опыта недостоверными.
Перед протравливанием семена должны быть тщательно отобраны. Все
легкие, мелкие и поврежденные семена отбрасываются. Протравливание
семян производят раствором хлорфеноловой ртути или протравителем,
успулуном, в котором действующим началом является тоже хлорфеноловая
ртуть. Раствор протравителя приготовляют следующим образом: 0,5 г про¬
травителя растирают с раствором 0,15 г NaOH (или с 37 мл 0,1 н. NaOH),
после чего раствор переносят в мерную колбу на 500 мл и доводят водой до
метки. При протравливании необходимое для постановки опытов количество
семян всыпают в стакан и заливают раствором протравителя, слегка взбал¬
тывают и оставляют на 1,5 часа. По окончании указанного срока жидкость
сливают, а семена раскладывают на фильтровальной бумаге для просуши¬
вания. Сушка семян продолжается от 2 до 7 дней.
Для опытов берут партии семян по 100 зерен, которые могут быть легко
отсчитаны при помощи счетной доски Мюллера. Каждую сотню взвешивают и
вес записывают на пакете, в который ее всыпают. Расхождение в весе для на¬
весок зерна желательно иметь небольшое — не более 0,1 г; хотя в методе
Нейбауэра — Шнейдера при вычислениях и вносится поправка на вес се¬
мян, все же желательно иметь навески семян, близкие по весу, так как семе¬
на различной крупности могут быть в различной мере обеспечены Р205 и
К20.
Сосудами для опытов служат стеклянные кристаллизаторы диаметром
11—11,5 еде и высотой 7 см; кристаллизаторы должны быть хорошо промыты
перед посевом 5%-ным раствором соды, а затем горячей дистиллированной
водой. При заполнении сосуда в середину его вставляют короткую стеклян¬
ную трубку, оплавленную с двух концов, длиной немного меньше 7 см.
Через эту трубку происходит поливка сосудов. Сосуды с трубками взвеши¬
вают и при массовой работе тарируют до одинакового веса битым стеклом,
промытым 5%-ным раствором соды.
Для опытов берут кварцевый песок, который используется при поста¬
новке вегетационных опытов с песчаными культурами. Песок должен быть
промыт соляной кислотой и отмыт сначала водопроводной, а затем дистил¬
лированной водой до конца реакции на С1 (проба с AgN03).
Почва должна быть пропущена через сито с диаметром отверстий в 1 мм.
Влажность почвы должна быть определена до постановки опыта, так как
в сосуд вносится навеска почвы, соответствующая 100 г абсолютно сухой
почвы. Набивку сосудов производят следующим образом: павеску почвы
смешивают с 50 г песка, полученную смесь высыпают в кристаллизатор,
в середине которого устанавливают трубочку для поливки сосудов. Поверх¬
ность смеси выравнивают и сверху насыпают 250 г песка, смоченного водой
с таким расчетом, чтобы общее количество воды было равно 80 г на сосуд
«(вода почвы + вода, добавленная к песку, всего 80 г). Вес, до которого про¬
600
А. В. Соколов
изводится поливка сосудов, должен быть равен сумме: общий вес сосуда +
+ вес трубки + вес почвы (100 г) + вес песка (300 г) + вес воды (80 г) +
вес 100 зерен.
Для гумусных почв рекомендуется давать воды не 80 мл, а несколько-
больше; для песка можно установить и более низкую норму, в 60 мл. Кон¬
трольные сосуды без почвы набивают так же, как опытные, только сначала
насыпают на дно сосуда 150 г сухого песка, а затем 250 г песка, смоченного-
80 мл воды.
При посеве семян сажальной доской делают 100 ямочек глубиной около-
1,5 см. После этого пинцетом укладывают в ямки ростком вниз 100 зерен,
которые заделывают стеклянной палочкой. В дневник опыта записывают
номера сосудов и точный, до 1,0 мг, вес 100 посаженных в них зерен. По окон¬
чании посадки сосуды закрывают стеклом; между стеклом и краем сосуда,
вставляют кусок спички или бумаги для усиления вентиляции. Сосуды оста¬
ются закрытыми стеклом до тех пор, пока проростки растений не достигнут
его и не начнут изгибаться.
Поливку производят ежедневно, в начале опыта один раз в день через труб¬
ку, а в конце опыта два раза в день опрыскиванием сверху через маленько»
сито и через трубку. Во время роста растений сосуды должны находиться на
рассеянном свету; прямого солнечного света следует избегать. Температура
помещения должна быть около 20°. Максимальные колебания температуры
допускаются в пределах ± 2°. Колебания температуры от 13 до 23° приводят
к изменению выноса более чем на 50%. Наиболее правильным будет помеще¬
ние сосудов в специальную комнату или в специальный термостат со стеклян¬
ной стенкой.
Уборку урожая производят на 14-й день после посева растений. Содер¬
жимое сосуда переносят на сито диаметром 26 см и высотой 7,5 см, с размером
отверстий в 1 мм. При легком постукивании по стенкам сосуда удается вск>
массу целиком сразу перенести на сито. После этого, придерживая рукой рост¬
ки, начинают отмывать песок струей водопроводной воды. Когда песок и часть
почвы будут смыты, переносят растения на тарелку и обрезают побеги нож¬
ницами непосредственно под зернами. Количество проростков должно быть
не менее 94; если проростков меньше, сосуд выбраковывают. Ростки обмывают
дистиллированной водой, а корни растений отмывают далее на сите сначала
водопроводной, а затем два раза дистиллированной водой.
Если к анализу приступают непосредственно после уборки, то весь ра¬
стительный материал разрезают ножницами, помещают в платиновую чашку*
добавляют 15 мл известковой воды или раствора Са(СН3СОО)2 и выпаривают
на водяной бане. Раствор Са(СН3СОО)2 готовится следующим образом^
2 г СаС03 растворяют в небольшом количестве уксусной кислоты и доводят
до 1 л. Сожжение производят осторожно, нагревая чашку на горелке с гриб¬
ком и доводя дно чашки только до слабого покраснения. После сожжения
добавляют в чашку 10 мл 10%-ного раствора НС1 и немного горячей воды,
растворяют золу и выпаривают раствор на водяной бане. По окончании выпа¬
ривания оставляют чашку еще на некоторое время на водяной бане для пол¬
ноты отделения кремнекислоты. Золу, растворенную в горячей воде, подкис¬
ленной 4 каплями 10%-ного раствора НС1, переносят в мерную колбу на
100 мл. Последнюю наполняют примерно на три четверти. После этого в кол¬
бу добавляют одну каплю фенолфталеина и приливают известковое молоко
(приготовленное из чистой окиси кальция, полученной из мрамора) до
появления устойчивой красной окраски. Доводят водой раствор в колбе до
метки, встряхивают и фильтруют через 9-сантиметровый сухой фильтр (бе¬
лая обмотка) в сухой стакан. В фильтрате определяют калий, в осадке —
р2о5.
Ход вычисления результатов опыта можно видеть из следующего'примера, привод
димого Нейбауэром. Повторность в опыте была двухкратная.
Вегетационный метод
601
Контрольные сосуды:
а) 4,109 г семян, 21,58 мг К20 и 24,05 мг РаОб;
б) 4,327 г семян, 23,41 мг КаО и 25,91 мг Р206.
Следовательно, на 1 г семян урожаи ростков содержали:
а) 5,253 мг КаО и 5,853 мг Р2Об;
б) 5,411 мг К20 и 5,989 мг Р2Об;
%в среднем 5,33 мг КаО и 5,92 мг Р2Об.
Опытные сосуды со 100 г почвы:
а) 4,240 г семян, 49,32 мг К20 и 31,24 мг РаОБ;
б) 4,415 г семян, 49,56 мг К20 и 32,79 мг Р206*
В урожае контрольных сосудов при соответственном весе семян было бы:
а) 22,60 мг К20 и 25,11 мг Р206;
б) 23,50 мг К20 и 26,15 мг Р2Об.
Следовательно, за счет внесения 100 г почвы может быть отнесено:
а) 26,72 мг К20 и 6,13 мг Р2Об;
б) 26,03 мг К20 и 6,64 мг Р205; в среднем 26,4 мг К20 и 6,4 мг РаОб.
При четырехкратной повторности, которую следует применять, и подбо¬
ре навесок семян с расхождением их по весу не более чем на 0,1 г вычисление
может быть упрощено. Берут среднее содержание К20 и Р206 в контроль¬
ных сосудах и вычитают его соответственно из количества К20 и Р206 в урожа¬
ях опытных сосудов. Ошибку опыта вычисляют по общим правилам, приня¬
тым при обработке результатов вегетационных опытов.
В основе вычисления предельных чисел (лимитов) по методу Нейбауэра
.лежит ряд условных допущений. В вегетационном опыте по методу Нейбауэра
корни растений извлекают из почвы питательные вещества при наличии
хорошего снабжения их водой и воздухом и легкой проницаемости почвы.
В поле растения находятся в худших условиях; они менее густо пронизывают
лочву своими корнями и берут поэтому из почвы меньшее количество пита¬
тельных веществ. Нейбауэр считает, что растения в течение вегетационного
периода могут взять только некоторую часть находящихся в почве корнераст¬
воримых веществ, которая различна для разных видов растений.
Размер усвоения калия растениями в течение вегетационного периода
определяется следующими величинами (в процентах от общего количества
корнерастворимых веществ почвы):
Ячмень 12 Свекла 33
Пшеница 15 Овес, рожь, клевер, люцерна и травы
Картофель 25 на сенокосах и пастбищах 20
Степень усвоения Р2Об составляет для всех растений, кроме ячменя,
33%, для ячменя —20%.
Усвоение питательных веществ из минеральных удобрений принимается:
для калийных солей — 60 %, для усвояемых фосфатов — 33%, кроме ячменя,
для которого величина усвоения принимается в 20%.
При пересчетах Нейбауэр принимает, что 1 мг вещества на 100 г почвы
соответствует 30 кг на 1 га (принимая вес пахотного слоя в 3 млн. кг). Но
этот пересчет тоже весьма условен: объемный вес почвы в нем принят равным
1,5, тогда как обычно для пахотного слоя он ближе к 1,2.
Вычисление необходимой дозы удобрения на основе определения количества корне¬
растворимых Р2Об и К20 весьма несложно. Приведем два примера. Растение — кор¬
мовая свекла, предполагаемый урожай 600 ц/га. Кроме минеральных удобрений, под свек¬
лу вносится навоз в количестве 300 ц/га. Нейбауэр считает, что 100 ц навоза содержат в
пересчете на минеральные удобрения 30 кг Р205 и 50 кг КгО. Требуется рассчитать необхо-
.димые количества калийных удобрений. Определение корнерастворимого К20 дало со¬
ответственно 20 мг К20 на 100 г почвы.
602
А. В. Соколов
Вычисление дозы К20 К20, кг
При 20 мг К20 на 1 га приходится 600
При 33% усвояемости для свеклы это соответствует 200*
600 ц свеклы выносят с урожаем 293
Не хватает 93
При 60% усвояемости К20 в удобрениях надо 155
Вносится в виде 300 ц навоза 90*
Требуется внести 6S
65 кг К20 соответствуют 163 кг 40%-ной калийной соли
Предположим, что требуется определить дозу Р206 под ячмень. Урожай ячменя ожи¬
дается 28 ц. Навоз под ячмень не вносится. Содержание в почве усвояемой Р2Об — Змг
на 100 г.
Вычисление дозы Р205 Рг06» кг
При 3 мг Р206 на 1 га приходится 90
При 20% усвояемости Р2Об для ячменя это соответствует 1&
28 ц ячменя выносят с урожаем 28-
Не хватает 10
При 20% усвояемости Р205 в удобрениях надо * . 50
50 кг Р2Об соответствуют 278 кг 18%-ного суперфосфата
Вычисление предельных чисел (лимитов), т. е. того минимального содер¬
жания в почве усвояемых веществ, при котором еще возможно получение
определенной величины урожая, производится на основании тех же коэф¬
фициентов. Например, при урожае ячменя в 35 ц зерна с урожаем выносится
85 кг К20 и 35 кг Р206. Использование корнерастворимого К20 для ячменя —
12%, для Р205 — 20%; 30 кг на 1 га соответствуют 1 мг на 100 г почвы.
Отсюда получаем лимиты:
для К20
85*100 1 п/ #ЛЛЛ
j2— ’ зо~ ~ 24 лег/100 г/
ДЛЯ Р208
35-100 1 „ ,jnn
20 ' 30 -«г/100 г.
В таблице приведены лимиты для различных урожаев.
Во многих случаях пользуются только таблицей лимитов, не производя
детального вычисления доз удобрений. Для разных почв и районов лимиты
оказываются различными, поэтому необходимо районирование лимитов.
Вначале (1923 г.) Нейбауэр установил только два предельных числа: для
К20 — 24 мг и для Р205 — 8 мг.
Недостатком метода Нейбауэра, как и всякого физиологического опыта,
является его меньшая воспроизводимость, чем химического анализа почвы.
Для повышения точности опыта необходимо строгое соблюдение деталей ме¬
тодики и увеличение повторностей. Двукратная повторность недостаточна и
поэтому заменена четырехкратной; 18-дневный срок, который был установлен
ранее, был затем заменен более коротким — 14-дневным. Химические методы
анализа золы (Лоренца для фосфора и хлорплатинатный и перхлоратный для
калия) заменяют теперь физико-химическими. Определение калия производят
спектральным анализом фотометрически, определение Р205 — фотоэлектро¬
колориметром.
Вегетационный метод
603
Урожаи сельскохозяйственных растений, вынос питательных веществ урожаем
и предельные числа — лимиты
(по Нейбауэру)
Растение
Уро¬
жай,
ц/ra
Вынос, кг/га
Лимиты,
мг/100 г
Уро¬
жай,
ц/га
Вынос, кг/га
Лимиты,
ме/iOO г
К,О
Р*0.
К,0
Р*0,
КгО
P.O.
К,О
P.O.
Ячмень
35
85
35
24
6
28
68
28
19
5
Овес
40
125
55
21
6
30
94
41
16
4
Пшеница
40
90
4
20
5
30
68
38
15
4
Рожь
35
100
50
17
5
28
80
40
13
4
Клевер
80
150
50
25
5
60
ИЗ
38
19
4
Картофель
320
280
60
37
6
240
210
45
28
5
'Сахарная свекла
400
250
60
25
6
300
188
45
19
5
Кормовая свекла
800
390
70
'39
7
600
293
53
29
6
Рапс
35
110
90
18
9
28
88
68
15
7
Люцерна
140
210
90
35
9
100
150
64
25
7
Луговое сено
80
150
50
25
5
60
ИЗ
38
19
4
Метод Митчерлиха
Отличительная особенность метода Е. А. Митчерлиха состоит в том, что
-о содержании в почве усвояемых веществ судят не по анализу урожая рас¬
тений, а по величине прибавок урожая от внесения удобрений. В основе
вычислений лежит предположение, что чем меньше в почве питательного
вещества, тем больше будет прибавка от его внесения в виде удобрений,
если, конечно, остальные условия опыта обеспечивают благоприятные усло¬
вия для роста растений. Если удобрение вносится в той дозе, при которой
обеспечивается при данных прочих условиях максимальный эффект от его
внесения, то разница между урожаем по удобрению и без удобрения может
служить показателем обеспеченности почвы питательным веществом. Зависи¬
мость между содержанием в почве питательного вещества и прибавкой уро¬
жая от внесения удобрения в первом приближении выражается формулой
1 — У) = \% А — С (х + Ъ),
где А — максимальный урожай; у — урожай при внесении изучаемого вещества в коли¬
честве х\ С — коэффициент действия изучаемого питательного вещества; Ь — его содер¬
жание в исходной почве.
Митчерлих допускает, что коэффициенты действия питательных веществ
(С) постоянны для всех растений и почв и равны для N — 0,122, для Р205 —
Х),60 и для К20 — 0,93.
Формула Митчерлиха, выражающая зависимость роста растений от
изменения условий вегетации, вызывает много возражений. В советской ли¬
тературе вопросы применения формулы Митчерлиха к результатам полевых
опытов и математический анализ его взглядов получили подробное освеще¬
ние в работах Научного института по удобрениям, проведенных Д. Н. Бо¬
родичем и В. Н. Перегудовым под руководством А. Н. Лебедянцева («Геогра¬
фические опыты с минеральными удобрениями...», 1933), а также в работах
М. К. Домонтовича и В. М. Клечковского.
Работы эти показали неправильность формулы Митчерлиха и установили
понятие об урожае как о «функции всех факторов роста, действующих в за¬
висимости друг от друга при наличии одного или более ведущих факторов»
604
А. В. Соколов
(Лебедянцев, 1960, стр. 25). Между тем Митчерлих, правильно считая урожай
функцией одновременного действия всех факторов роста, в то же время вы¬
сказал неверное положение, что коэффициенты их действия не изменяются^
Наблюдающееся часто некоторое совпадение результатов опытов с по¬
казаниями таблиц, составленных Митчерлихом на основании большого числа
опытных данных, является лишь приблизительным эмпирическим обобще¬
нием с ограниченной сферой применения и не может считаться доказатель¬
ством правильности его формулы.
Литература
Географические опыты с минеральными
удобрениями, проведенные НИ У за время
с 1926 по 1930 г. под руководством А. Н.
Лебедянцева.— Труды НИУ, 1933, вып.
93.
Журбицкий 3. И. Теория и практика веге¬
тационного метода. М., «Наука», 1968,
Лебедянцев А. Н. Избранные труды. Сель-
хозгиз, 1960.
Митчерлих Е. А. Определение потребно¬
сти в удобрениях. Сельхозгиз, 1931.
Недокучаев Н. Я. Вегетационный методг
4-е изд. Сельхозгиз, 1931.
Соколов А. В,у Ахромейко А. И.у Панфи¬
лов В. Н. Вегетационный метод. Сель¬
хозгиз, 1938.
Федоровский Д. В. Поступление воды и пи¬
тательных веществ в растения из неод¬
нородной почвы. — Агрохимия, 1964,
№ 2.
Хьюитт Э. Песчаные и водные культуры в;
изучении питания растений. ИЛ, 1960-
А. В. СОКОЛОВ
ПОСТРОЕНИЕ СХЕМ ОПЫТОВ С УДОБРЕНИЯМИ
*
Опыты с удобрениями ставятся для установления действия на данной поч¬
ве того или другого удобрения или способа его использования для повышения
урожая и наилучшего развития растений. Первым условием успешного про¬
ведения опытной работы является точная формулировка задач опыта и кон¬
кретизации условий его проведения, т. е. составление программы и схемы
опыта. Опыт должен быть поставлен таким образом, чтобы его результаты
были возможно более точны и пригодны для решения того вопроса, ради ко¬
торого опыт ставится.
Изучаемые в опыте приемы внесения удобрений и взятые для сравнения
стандартные приемы внесения удобрений или возделывание растений без
удобрений называются вариантами опыта, а их совокупность — схемой опы¬
та. Выводы из данных, полученных в опыте, делаются путем сравнения уро¬
жаев или состояния растений в различных вариантах опыта. (Только при¬
менение меченых атомов позволяет иногда изучать действие удобрений без
сравнения вариантов.)
Сравниваемые варианты должны отличаться изменением только одного
того условия, выявление действия которого и составляет задачи этого срав¬
нения; все остальные условия жизни растения (так называемый фон) должны
быть одинаковы. Если применение того или другого удобрения связано с
изменением и других условий культуры, то в схему опыта для установления
значения последних вводятся дополнительные варианты. Например, если
внесение удобрений требует лишней обработки почвы, в схему опыта вклю¬
чается вариант с одной этой обработкой без внесения удобрений. В противном
случае действие обработки на урожай растений может быть ошибочно при¬
писано действию удобрений. Для оценки того или другого удобрения прихо¬
дится иногда сравнивать их действие не в строго идентичных условиях при¬
менения, а в условиях их максимальной эффективности. Например, селитра
может быть особенно эффективна при внесении ее по всходам или поверхностно
перед посевом, тогда как цианамид кальция надо вносить заблаговременно,
до посева. В этих случаях необходимо всегда оговаривать условия применения
удобрений и иметь экспериментальные данные, обосновывающие избранные
способы внесения удобрений как действительно оптимальные.
Во многих случаях необходимо, чтобы опытные растения проявляли
чувствительность к изменениям изучаемого фактора. Например, если мы
хотим установить степень доступности растениям фосфора из различных удо¬
брений, необходимо, чтобы растения в опыте вообще реагировали на внесение
фосфора. Если растения не отзываются на внесение фосфатов, мы можем сде¬
лать только вывод об отсутствии в условиях опыта действия фосфора. Следо¬
вательно, схема опыта должна быть построена так, чтобы можно было уста¬
новить, какие могут быть сделаны выводы из данного опыта. Те варианты опы¬
та, при помощи которых устанавливается степень чувствительности опытных
растений к изучаемому фактору и эффективность изучаемого удобрения или
способа его применения, называются контрольными. Наличие в опыте пра¬
вильно выбранных контрольных вариантов является основным отличитель¬
ным признаком научного эксперимента.
А. В. Соколов
ОПРЕДЕЛЕНИЕ ДЕЙСТВИЯ РАЗЛИЧНЫХ ВИДОВ УДОБРЕНИЙ
Контрольным вариантом в схеме опытов по определению действия удобре¬
ний служит вариант без того удобрения, действие которого изучается.
Действие удобрений на том или другом типе почв зависит от многих усло¬
вий, в том числе и от внесения одновременно с изучаемым удобрением других
удобрений. Поэтому если задачей опыта является, например, установление
эффективности азотных, фосфорных и калийных удобрений, то недостаточно
использовать в опыте только четыре основных варианта: 1) без удобрений (0),
2) азотное удобрение (N), 3) фосфорное удобрение (Р), 4) калийное удобрение
(К), а необходимо иметь и варианты с совместным внесением различных удо¬
брений.
Определение действия основных видов удобрений производится по схеме,
состоящей из следующих восьми вариантов:
1 2 3 4 5 6 7 8
0 N Р К NP NK РК NPK
Эта схема полная («ортогональная»), так как в ней имеются все возможные
комбинации из трех основных видов удобрений. Если по этому же принципу
полноты возможности сравнений, или по ортогональному построению схемы
опыта, изучать действие четырех видов удобрений, то число делянок удво¬
илось бы, так как потребовалось бы иметь все эти восемь комбинаций плюс чет¬
вертое удобрение. Применение такого удвоения количества вариантов целе¬
сообразно при изучении известкования почвы. Добавка извести существен-
вым образом влияет на плодородие почвы и на ее отзывчивость в отношении
азота, фосфора и калия. Поэтому в опыте производится наложение восьми¬
вариантной схемы на известковый и неизвестковый фон. Вообще же, как
правило, действие второстепенных, или стимулирующих, удобрений изуча¬
ется на так называемом оптимальном фоне полного удобрения NPK.
Во многих случаях стремятся сократить восьмивариантную схему и делают
ее пятивариантной;
1 2 3 4 5
0 NP NK РК NPK
и четырехвариантной, представляющей собой ту же пятивариантную схему,
в которой исключен еще и вариант без удобрений. Все эти сокращения обычно
нежелательны, так как важно установить не только ту прибавку, которую
дают азот, фосфор и калий на фоне других элементов, но и выяснить,{действуют
ли они порознь или только совместно.
В районах, где калий действует слабо, часто применяют такую схему;
1 2 3 4 5
0 N Р NP NPK
ИЗУЧЕНИЕ ФОРМ ОДНОСТОРОННИХ УДОБРЕНИЙ
Контрольными вариантами в схемах опытов с формами удобрений явля¬
ются следующие: 1) без удобрений, 2) основное удобрение (фон) и 3) основное
удобрение + стандартное удобрение в одной или нескольких дозах.
Если для данного вида удобрений еще не имеется форм, действие которых
широко изучено (стандартов), то обычная постановка опытов с формами удо¬
брений сводится к следующей схеме;
1. Основное удобрение (фон).
2. » » + 1-я форма
3. » » + 2-я форма
и т. д.
Построение схем опытов с удобрениями
607
Однако далеко не всегда эта схема удовлетворительна, так как принятое
для опыта основное удобрение может быть выбрано неудачно. Поэтому для
проверки правильности избранного фона следует вводить вариант без удобре¬
ний. Вполне возможно, что выбранный фон действовал вредно, и в этом слу¬
чае результаты опыта становятся сомнительными. Обычно в таких случаях
бывает необходимо дальнейшее изучение вопроса. Оно заключается в подыска¬
нии нового подходящего фона или в сравнении форм удобрений на неудобрен¬
ном фоне.
В общем виде схема изучения новой формы имеет следующий трехчленный
вид:
1. Фон.
2. » + стандартное удобрение.
3. » + изучаемая форма удобрения.
Без варианта со стандартными удобрениями нельзя установить, было лв
действие испытуемого удобрения нормальным или меньшим, чем следовало.
Без варианта с одним фоновым удобрением нельзя вообще говорить о том,
какую прибавку дает изучаемое удобрение, да и нельзя сказать вообще,
действовало оно или нет.
Приведенная трехчленная схема изучения новой формы удобрения не
всегда оказывается достаточной. В тех случаях, когда хотят установить срав¬
нительную эффективность питательного вещества в различных удобрениях,
в схему опыта вводят контрольные варианты с различными дозами стандарт¬
ного удобрения, показывающие значение изменения в удобрениях количества
усвояемого питательного вещества.
В развернутом виде схема опыта с формами удобрений имеет следующий
вид:
1. Основное удобрение.
2. » + Vг дозы стандартного удобрения.
3. » » + 3/4 дозы » »
4. » » + полная доза » »
5. ь » + полная доза 1-го испытуемого удобрения.
6. » » + полная доза 2-го испытуемого удобрения и т. д.
При закладке длительных опытов эта схема дополняется вариантом без
удобрений. При постановке опытов с формами фосфатов закладка опытов по
данной схеме обязательна, так как основное различие между формами фосфа¬
тов заключается в степени усвояемости их Р20Б.
Для повышения чувствительности опыта необходимо прежде всего быть
весьма осторожным в выборе основной дозы. Полная доза отнюдь не долж¬
на быть такой, чтобы дальнейшее увеличение уже не давало прибавок
урожая.
При проведении опытов с различными формами удобрений необходимо
иметь в виду, что всегда все сравниваемые формы надо ставить в совершенно
одинаковые условия техники их применения. Каждому удобрению надо да¬
вать оптимальные для него технику и время внесения; если эти оптимальные
условия внесения удобрения еще не выяснены, то необходимо производить
сравнение при идентичных условиях применения удобрения. Если изменения
в технике применения удобрений приводят к введению для различных вари¬
антов с формами удобрений разных агротехнических приемов, следует ввести
в схему соответствующие контрольные варианты для учета значения этих
агротехнических приемов.
608
А. В. Соколов
СХЕМА ОПЫТОВ СО СЛОЖНЫМИ УДОБРЕНИЯМИ
В опытах со сложными удобрениями обычно применяется трехчленная
схема.
1. Без удобрения или фоновое удобрение.
2. Сложное удобрение.
3. Эквивалентная смесь простых удобрений.
Эта схема, однако, не дает полноценных результатов и должна быть за¬
менена пятивариантной схемой:
1. Без удобрения или фон.
2. Сложное удобрение.
3. Эквивалентные количества односторонних стандартных удобрений.
4. Сложное удобрение + одностороннее стандартное удобрение для получения нор»
мальных доз питательных веществ.
5. Односторонние стандартные удобрения в нормальных дозах.
Эта пятивариантная схема позволяет не только сравнить эффективность
сложного удобрения с эффективностью простых удобрений, но и установить
целесообразность имеющегося в удобрении соотношения питательных ве¬
ществ.
УСТАНОВЛЕНИЕ ОПТИМАЛЬНЫХ ДОЗ УДОБРЕНИЙ
Если требуется установить, какая доза удобрений в данных условиях
дает максимальную оплату единицы питательного вещества в удобрении и
при какой дозе получается в данных условиях максимальный урожай, а
также какая доза хозяйственно наиболее'^выгодная, необходимо получить
кривую изменения урожаев при возрастании доз удобрений. Практически
не требуется наличия в опыте многих вариантов с различными дозировками.
Интервалы между двумя дозами должны быть достаточно велики, чтобы
прибавки гот соседних доз могли различаться на величину, превосходящую
ошибку опыта. Поэтому обычно бывает вполне достаточным наличие в опыте
трех-четырех доз. Для характеристики действия отдельной дозы с целью
установления ее хозяйственной пригодности необходимо знать абсолютную
прибавку урожая, получаемую от данной дозы удобрения. Следовательно,
в этом случае^основным контрольным^вариантом^является вариант без удоб¬
рений. Но его недостаточно; возможно, что та же прибавка урожая может
быть получена и при меньшей дозе удобрения. Возможно, однако, что даль¬
нейшее увеличение дозы удобрения окажется еще более выгодным; для вы¬
яснения этого необходимо иметь в опыте дозу большую, чем изучаемая.
Таким образом, для оценки правильности выбранной дозы необходимо иметь
две дозы,] меньшую и большую. Наиболее удобно брать половинную дозу
и полуторную. Дозы удобрений, испытанные на одном фоне, часто непригод¬
ны на другом фоне удобрений или агротехники. Например, оптимальные дозы
азота при наличии полива будут одни, без полива — другие, на фоне фос¬
фора — одни, без фосфора — другие и т. д.
Опыты с дозами естественно переходят в комплексные опыты по изуче¬
нию соотношения питательных веществ при различных дозах. Для изучения
соотношения компонентов в составе смешанных удобрений О. Шрейнер
предложил такую постановку опытов по треугольной системе, при которой
в основу всех комбинаций кладется сумма N + Р205 + К20 и все комбина¬
ции, участвующие в опытах, должны иметь одну и ту же сумму этих элемен¬
тов. При такой системе чисто механическое установление возможных комби¬
наций лишает возможности проверить пригодность даже самых ходовых ком¬
бинаций. Нельзя, оставляя нормальную дозу азота, выключить действие
Построение схем опытов с удобрениями
609
фосфора, так как азот, фосфор или калий в отдельности не могут быть даны при
системе треугольника в дозе, меньшей суммы доз всех трех элементов. В схеме
такого опыта нет даже сравнимых делянок, так как все они отличаются сра¬
зу по дозам по крайней мере двух питательных веществ.
СРАВНЕНИЕ ДЕЙСТВИЯ НАВОЗА И МИНЕРАЛЬНЫХ УДОБРЕНИЙ
Для сравнения действия навоза и минеральных удобрений в большин¬
стве случаев сопоставлялось действие обычной дозы навоза с действием ми¬
неральных удобрений, внесенных также в обычных дозах, например, по
такой схеме:
1. Без удобрений.
2. Навоз 40 т/га.
3. NPK по 45—60 кг действующего начала на 1 га1.
При таком сравнении с навозом вносится значительно большее количе¬
ство питательных веществ, чем в минеральных удобрениях. Для устране¬
ния этого недостатка было предложено сравнение навоза и минеральных
удобрений вести в длительных опытах при внесении минеральных удобрений
в дозах, эквивалентных содержанию питательных веществ в навозе. Недо¬
статком этого сравнения является наличие в навозе непостоянного соотно¬
шения между количествами азота, фосфора и калия. Поэтому схему допол¬
няют иногда вариантами с внесением минеральных удобрений в нормальном
соотношении и навоза с добавкой минеральных удобрений до создания нор¬
мального соотношения питательных веществ в навозе и минеральных удоб¬
рениях. Для выявления значения органического вещества навоза С. В. Щер-
бой предложена схема, в которой возрастающие дозы полного минерального
удобрения вносят на фоне навоза и без навоза. По характеру кривых возра¬
стания урожаев устанавливают, вызывалось ли в опыте действие навоза толь¬
ко наличием в нем питательных веществ, вносимых в минеральных удобре¬
ниях, или действием его органического вещества, щелочности и другими
свойствами.
Для определения усвояемости азота в навозе нами предложено сравнение
действия навоза с эффективностью возрастающих доз азота на фоне, обеспе¬
ченном фосфором и калием. Точность сравнения обеспечивается при этом на¬
личием вариантов с внесением азота и по навозу в двух формах — нитратной
и аммиачной. Аналогично можно построить изучение эффективности фосфора
в навозе путем, сравнения действия навоза с действием минеральных фос¬
фатов при внесении их и навоза на фоне, обеспеченном азотом и калием.
Большое практическое значение имеет совместное применение навозного
и минерального удобрений; поэтому часто ставятся опыты, в которых имеются
варианты с совместным внесением половинной дозы навоза и половинной
дозы минерального удобрения, например, по такой схеме:
1. Без удобрения.
2. Навоз.
3. NPK (полная доза).
4. NPK (V2 дозы) + навоз (*/2 дозы).
Эта схема неполная, так как в ней недостает следующих контрольных вари¬
антов:
5. Навоз (V2 дозы).
6. NPK (72 дозы).
1 Та же схема опытов применяется и при использовании компостов.
2) Агрохимические методы исследования почв
612
А. В. Соколов
1-е звено — сорт А
1. Без удобрений, малая площадь питания.
2. NxPiKx » » >
3. n2p2k2 » » >
4. Без удобрений, большая » »
5. NiPxKi » » »
6. n2p2k2 » » >
2-е звено — сорт В
1. Без удобрений, малая площадь питания.
2. NxPiKi » » >
3. n2p2k2 » » »
4. Без удобрений, большая » »
5. NjPjKx » » »
6. N2P2K2 » » »
СХЕМЫ СИНТЕТИЧЕСКИХ ОПЫТОВ
Синтетический опыт является сокращенным комплексным опытом. В схе¬
ме демонстрационного синтетического опыта сохранены только наиболее ин¬
тересные варианты комплексного опыта.
Приведем для примера следующую схему:
1. Без удобрений, без полива, старые сорта растений.
2. Малые дозы удобрений, без полива, новые сорта растений.
3. Большие дозы удобрений, полив, новые сорта растений.
Совершенно ясно, что из опыта по этой схеме нельзя делать какие-либо
выводы о том, как действуют новые сорта растений или удобрения, или полив.
Эффективность всех приемов, входящих в состав сравниваемых вариантов,
должна была быть доказана предыдущими опытами. Постановка демонстра¬
ционного синтетического опыта предусматривает предварительное проведение
опытов по различным приемам или оценку этих приемов в последовательном
синтетическом опыте со следующей минимальной схемой:
1. Без удобрений, старые сорта.
2. Малые дозы удобрения, старые сорта.
3. » » » новые »
4. Большие девы удобрений, новые »
5. » » » » » и полив.
В этом случае каждый предыдущий вариант является контрольным вари¬
антом для последующего.
Однако постановка такого последовательного синтетического опыта все же
недостаточна, и только комплексный опыт дает нам все необходимые вари¬
анты для обоснования предлагаемого синтеза агротехнических приемов.
СХЕМЫ МНОГОФАКТОРНЫХ ОПЫТОВ
Опыты с дозами азота, фосфора и калия, если в их схеме предусмотрево
изучение доз азота на фоне доз фосфора и калия, доз фосфора на фоне доз
азота и калия и доз калия на фоне доз азота и фосфора, часто называются
иногофакторными.
Если в* схеме предусмотрены все возможные варианты доз азота, фосфора
и калия, схема называется ортогональной. Простейшей такой схемой являет¬
Построение схем опытов с удобрениями
613
ся восьмерная, где имеются две дозы азота, фосфора и калия (нулевая и оди¬
нарная). Но уже при введении трех доз удобрений (нулевая, одинарная и
двойная) приходится ставить опыт с огромным количеством вариантов (27),
что делает постановку его крайне затруднительным. На основе таких опытов
устанавливают эмпирические формулы действия удобрений, которые иногда
называют «производственными функциями удобрений».
Но результаты такой математической обработки данных одного опыта,
хотя и могут правильно определять действие удобрений в этом опыте, для
характеристики действия удобрений в других опытах могут быть использо¬
ваны только при наличии тех же условий в отношении почвы, погоды и агро¬
техники.
Методической ошибкой таких многофакторных опытов часто является
применение в них высоких доз удобрений, когда действие удобрений опре¬
деляется не только содержанием в них азота, фосфора и калия, но и второ¬
степенных элементов, и объяснение действия удобрений как эффекта от вне¬
сения N, Р и К является уже недопустимым.
Использование подобных опытов со многими дозами удобрений может быть
полезно для установления зависимости действия одного вида удобрений от
обеспеченности другим, например зависимость действия фосфора от
обеспеченности почв азотом. Результаты их могут быть также использованы
для проверки различных гипотез о закономерностях действия удобрений.
А. В. СОКОЛОВ
ОПРЕДЕЛЕНИЕ ТОЧНОСТИ ОПЫТА
*
ВОЗМОЖНЫЕ ОШИБКИ И ОСНОВНЫЕ ПОКАЗАТЕЛИ ТОЧНОСТИ
Даже при самой тщательной работе урожаи на параллельных делянках
и в параллельных сосудах получаются не совсем одинаковыми.
Расхождение между урожаями в параллельных сосудах или на параллель¬
ных делянках будет характеризовать степень точности нашей работы. Если
бы в нашем опыте не было грубых или систематических ошибок, то при гро¬
мадном числе параллельных делянок или сосудов среднее арифметическое
соответствовало бы истинному урожаю, который мы должны были бы полу¬
чить при точном выполнении всех операций.
Если для той или другой делянки или для того или другого сосуда была
допущена грубая ошибка, т. е. нарушено правило постановки опыта, то уро¬
жаи этих делянок и сосудов выбраковывают и не учитывают при суммиро¬
вании урожаев параллельных делянок или сосудов и выведении средних
урожаев по вариантам опыта.
Под систематическими ошибками понимаются такие недостатки в по¬
становке опыта, которые вызывают закономерные, идущие в определенном
направлении, изменения урожаев по повторностям опыта. При помощи ста¬
тистической обработки полученных величин иногда удается выявить наличие
в опыте систематической ошибки и внести соответствующие исправления
в подсчеты его результатов. В вегетационных опытах, как правило, система¬
тических ошибок не должно быть; в полевых опытах они часто вызываются
различным плодородием опытного участка. Если изменения почвенного пло¬
дородия опытного участка происходят в определенном направлении (напри¬
мер, постепенное изменение плодородия почвы при постановке опыта вдоль
склона), то эти изменения можно выявить и до некоторой степени устранить
их влияние на статистическую обработку результатов опыта.
Статистическая обработка результатов опыта позволяет установить точ¬
ность опыта, а в некоторых случаях внести исправления и в самые подсчеты
результатов опыта, например, в тех случаях, когда вводится поправка на
систематическую ошибку опыта или производится выбраковка делянок или
сосудов, для которых была допущена грубая ошибка, оставшаяся не отме¬
ченной в журнале опыта.
Исходная предпосылка при обработке результатов опытов — предполо¬
жение, что когда нет грубых и систематических ошибок, то урожаи парал¬
лельных делянок или сосудов располагаются определенным образом вокруг
истинной величины урожая для данного варианта, образуя, при громадном
числе параллельных показаний, кривую нормального распределения пока¬
заний, и что среднее арифметическое из всех параллельных урожаев соот¬
ветствует истинному значению урожая для данного варианта. Так как мы
не можем ставить большого числа параллельных опытов для каждого вари¬
анта, то наши средние арифметические полученных урожаев не являются
точными показателями истинного уро кая для данного варианта. Но зная,
что отдельные показания определенным образом располагаются вокруг истин-
IIого среднего, мы можем установить, какова вероятность отклонения эмпи¬
рической средней на ту или другую величину от истинного результата.
Изучением этого вопроса занимается вариационная статистика; ниже
приведем из нее тот минимум сведений, который необходим для обработки
Определение точности опыта
615
результатов нормально проведенного опыта, чтобы установить степень его
точности.
На рисунке представлена кривая нормального распределения показаний
при бесконечно большом числе параллельных определений (кривая Гаусса).
На горизонтальной линии (на оси абсцисс) в обе стороны от точки Му соот¬
ветствующей средней арифметической, отложены величины отклонений от
нее отдельных показаний. Как видно из рисунка, больше всего показаний
располагается вблизи среднего арифметического — над точкой М кривая
имеет выпуклую форму, которая потом переходит в вогнутую. Следовательно,
по мере увеличения отклонения от средне¬
го число показаний, сильно разнящихся
от среднего, становится все меньше. Точка
перегиба кривой, где она переходит из вы¬
пуклой в вогнутую, соответствует так назы¬
ваемому квадратическому основному откло¬
нению, обозначаемому греческой буквой а.
Число показаний, которые отклоняются
от среднего арифметического больше чем
на +а, равно трети общего числа показа¬
ний. Отклонения больше чем на ± 2 а
имеют место только для 1/22 всех определе¬
ний, а отклонение их от среднего больше чем на ±3 о встречается только
один раз из 370 случаев. Если бы нам удалось определить основное отклоне¬
ние для полученных в нашем опыте результатов, то мы узнали бы и точность
нашего опыта, т. е. могли бы сказать, какова вероятность того, что наши
показания отклонились от истинного результата на ту или другую величину.
Например, если основное отклонение в нашем опыте было равно ± 0,5 if,
то урожаи отдельных делянок могут отклониться от истинного среднего
для данного варианта на ± 1,5 ц только 1 раз из 370 возможных случаев.
В опытах мы применяем два и больше повторений и берем среднее из них.
В этом случае достоверность нашего определения возрастает пропорциональ¬
но корню квадратному из числа повторений. Характер распределения наших
эмпирических средних вокруг истинного среднего остается тем же, что и рас¬
пределение вокруг него отдельных показаний, но кривая сжимается около
точки Л/, так как величина основного отклонения о становится меньше.
Квадратическое отклонение среднего называемся квадратической ошиб*
кой и обозначается буквой т
5
тде п — число параллельных делянок или сосудов, т. е. повторений опыта.
Например, если опыт ставится в четырехкратной повторности, то точность
его возрастает в 2 раза (Y4) по сравнению с достоверностью опыта с одно¬
кратной повторностью.
Так как величина основного отклонения вычисляется путем определения
отклонений отдельных показаний от среднего арифметического, то устано¬
вить величину основного отклонения в опыте можно только при наличии в
нем минимум двукратной повторности. Поэтому для опыта без повторения
вообще нельзя иметь представление о степени его точности и ставить такие
опыты как в стационарных, так и в производственных условиях в подавляю¬
щем большинстве случаев нецелесообразно.
В опытах нам не столько приходится показывать абсолютную величину
тех или других урожаев, сколько сравнивать между собой средние арифме¬
тические для различных вариантов опыта. Следовательно, нам надо опре¬
делить достоверность разности средних арифметических. Если ошибка сред¬
Кривая Гаусса
616
А. В. Соколов
него Мг равна ± тг, а ошибка другого среднего М2 равна ± т2, то ошибка
разности (mD) средних Мг — М, равна:
mD = ±^ + 4
т. е. она больше, чем ошибка одного среднего. В большинстве случаев ошиб¬
ка средних вычисляется сразу для опыта в целом, поэтому = т2 и ошиб¬
ка разности средних становится ошибкой опыта; следовательно,
топыта = mD = ± = ±™У2Г = ±1,41 т.
В том случае, когда определяется ошибка разности двух средних величин,
характеризующих явления, имеющие между собой определенное соотноше¬
ние, ошибка разности средних для таких сопряженных рядов определяется
по формуле
mD = i 'yf mi + т2 — 2mim2T ,
где г — коэффициент корреляции, показывающий степень сопряженности сравниваемых
явлений, например изменения плодородия почвы в зависимости от рельефа.
Способы определения величин а, тиг (табл. 1). Вычисле¬
ние величины основного, или квадратического, отклонения отдельного опре-
Таблица 1
Вычисление основного отклонения (а) и ошибки среднего (ш)
JV& сосуда
Урожай по сосудам, г
Отклонение от
среднего (vt)
Отклонение от сред¬
него с точностью
до 0,1
Квадрат отклонений
(V*)
1
10,55
+0,35
+0,4
0,16
2
10,09
—0,11
-0,1
0,01
3
10,54
+0,34
+0,3
0,09
4
9,77
-0,43
-0,4
0,16
5
10,08
-0,12
-0,1
0,01
6
11,27
+1,07
+1,1
1,21
7
9,02
—1,18
-1,2
1,44
8
10,28
+0,08
+0,1
0,01
М
= 10,20
+1,84
-1,84
«00
+ 1
2*2 = 3,09
деления производится по формуле
где v — отклонение отдельного определения от среднего арифметического; 2 v2 — сумма
квадратов отклонений.
Понятно, что соответствующая формула для ошибки среднего будет:
g ,i / Ш
У» У »(л —1) ’
3 -± У 3’^9 =± 1^0,4414 =±0,66 г,
. 0,66 . 0,66 . 0,66
т - ± у- ± yg- ± -Щ- — ± 0,28 г.
Определение точности опыта
Точность вычисления ошибки зависит от числа определений, использован¬
ных для ее вычисления; считают, что ошибка вычисления квадратического-
отклонения равна о/У~2п, т. е. при восьми показаниях она равна 0/1^16 =
= 0,25сг, для нашего примера а — ±0,66 ± 0,17.
При обычных вычислениях, если определения производились с точностью
до 0,01, отклонения от среднего вычисляются с точностью до 0,1 г, а квад¬
раты — с точностью до 0,01.
Вычисление коэффициента корреляции производится по
формуле 1
ИгпУг
V 2г^.2г>:
или по формуле
2глг>2
(п — 1)<31<32
Ошибка коэффициента корреляции вычисляется по формуле
1—Г2
тг = Лг— •
У п
Величина коэффициента корреляции изменяется от +1 до —1. Когда
корреляция отсутствует, коэффициент равен нулю. Когда изменение показа¬
ний одного ряда строго соответствует изменению показаний другого ряда,
имеем коэффициент + 1, или полную положительную связь между явления¬
ми. Если показания одного ряда всегда уменьшаются, а показания другого
ряда увеличиваются, имеем обратную корреляцию между явлениями и отри¬
цательный коэффициент корреляции.
Пример вычисления коэффициента корреляции2.
В опыте на Краснокутской опытной станции наблюдалась определенная зависимость
между урожаем пшеницы и плотностью колоса для различных сортов (табл. 2).
Таблица 2
Урожай пшеницы и плотность колоса для различных сортов
Сорт
Плотность колоса (число колосков
на 1 см)
Урожай, пуд
Гордеиформе 189
2,46
57,0
44
2,66
53,9
145
2,59
55,3
Мелянопус 69
2,41
58,5
Муршензе 171
2,46
56,4
Мх = 2,52
М2 = 56,2
Вычисляем отклонения от средних, перемножаем их попарно и суммируем, получаем
2vxv2. Возводим отклонения в квадрат и находим и Zv\ (табл. 3).
—0,698 —0,698 —0,698
? =-0,96 ±0,035. *
г =
У0.0438-12,07 1^0,527 0,727
Таким образом, в этом опыте наблюдалась вполне доказуемая и весьма высокая обрат*
ная зависимость между плотностью зерна и урожаем пшеницы.
1 Значения vx и и2 см. в табл. 3.
2 Из работы П. Н. Константинова (1952).
*618
А. В. Соколов
Таблица 3
Вычисление коэффициента корреляции
Отклонение от среднего при
определении
Произведение
Квадрат отклонения п л
определении
Сорт по порядку
плотности колоса
урожая
отклонений
плотности колоса
урожая
1
-0,06
+0,8
-0,048
0,0036
0,64
2
+0,14
-2,3
-0,322
0,0196
5,29
3
+0,07
-0,9
-0,063
0,0049
0,81
4
—0,11
+2,3
-0,253
0,0121
5,29
5
-0,06
+0,2
-0,012
0,0036
0,04
Сумма
+0,21
-0,23
+3,3
-3,2
Ui;ii>2= —0,698
2*1* = 0,0438
2*»* = 12,07
Коэффициент корреляции показывает только наличие совпадения или
расхождения между изменениями, происходящими в двух рядах различных
признаков изучаемых объектов, например: плотности колоса у пшеницы и ее
урожайности или действия фосфорного удобрения и содержания в почве
растворимых фосфатов. Но коэффициент корреляции не дает представления
о количественной зависимости между изменениями в двух рядах, т. е. о том,
какое изменение плотности колоса дает какую прибавку урожая зерна или
при каком уменьшении содержания в почве растворимых фосфатов наблю¬
дается увеличение урожая на 1 ц зерна от внесения фосфорных удобрений.
Меру изменения показаний в одном ряду в соответствии с изменением по¬
казаний в другом дает коэффициент регрессии. Он является константой, ко-
торая показывает функциональную зависимость между двумя явлениями.
Если изменения одного ряда будем рассматривать как изменения незави¬
симой переменной, то изменения другого ряда можно считать функцией из¬
менений первого или зависимой от него переменной величиной. Самым про¬
стым выражением функциональной зависимости будет уравнение прямой
линии: .
у = Ьх,
в котором х — независимая переменная; у — функция от х; b — константа, которая и
является коэффициентом регрессии.
Если Mi — среднее для первого ряда показаний, а М2 — для второго,
то имеем следующее уравнение:
гг — Л/г = Ь (г> 1 — Mi),
т. е. Ъ (коэффициент регрессии) показывает, во сколько раз отклонение (v2)
•от среднего для второго ряда больше или меньше соответствующего откло¬
нения от среднего (г^) в первом ряду. Конечно, этот коэффициент соответ¬
ствует только простейшему виду функциональной зависимости или прямо¬
линейной регрессии:
у = а + Ьх.
Возможны более сложные зависимости:
у = а + bx + ex2,
У = аЬх.
Последняя распространена в биологических явлениях, при вычислениях
обычно ее применяют в логарифмической форме, когда она получает вид пря¬
молинейной регрессии:
Определение точности опита
619
lg3/ = lg« + «lg
Коэффициент прямолинейной регрессии определяется по формуле
2vil>2
Порядок вычисления этого коэффициента регрессии приводится далее. Спо¬
собы вычисления криволинейной регрессии и множественной регрессии,
когда изменение одного признака зависит от изменения двух других призна¬
ков, можно найти в руководстве Д. У. Снедекора (1961), где имеется много
примеров вычисления коэффициентов корреляции и регрессии из практики
агрономических исследований.
ОБРАБОТКА ДАННЫХ ОПЫТА
Дробный метод обработки. Обычно в опыте имеется не¬
сколько вариантов; например, в пятичленной схеме бывают следующие вари¬
анты с различными удобрениями: О, NP, NK, РК и NPK. Для каждого ва¬
рианта имеется несколько параллельных делянок или сосудов, обычно от
двух до шести, в зависимости от условий проведения опыта. Урожаи этих
параллельных делянок или сосудов неодинаковы. Для каждого варианта
в отдельности можно вычислить квадратическую ошибку средней данного ва¬
рианта.
Предположим, что в опыте имелась четырехкратная повторность, и для
варианта NP были получены следующие урожаи зерна овса в пересчете на
1 га: 20,4; 23,0; 22,9; 28,1 ц. Средний урожай овса по NP будет 23,6 ц. Урожаи
отдельных делянок отклонялись от среднего на —3,2; —0,6; —0,7; + 4,5 ц.
Возведя величины отклонений в квадрат, сложив полученные квадраты и
разделив их сумму на (п — 1), т. е. на 3, получим после извлечения квад¬
ратного корня величину основного отклонения урожая отдельной делянки
для варианта NP. Разделив ее на 1ЛГ ^ 1^4”, т. е. на 2, получим квадратиче¬
скую ошибку среднего, равную ±1,62 ц. Следовательно, урожай по NP мож¬
но характеризовать уже не только его средней величиной, но и ее квадрати¬
ческой ошибкой, показывающей дам надежность этой средней; в данном слу¬
чае имеем 23,6 ± 1,62 ц. Ошибка выражена, как и средняя, в абсолютных
величинах — в центнерах на 1 га. Ее легко пересчитать на процент от сред¬
ней: принимая урожай в 23,6 ц за 100%, имеем яг = 1,6 if, или 6,8% от М.
Аналогичным образом можно поступить и при определении квадратиче¬
ской ошибки для других вариантов опыта. Например, при урожаях по NPK,
равных 23,1; 25,3; 26,4; 29,6 ц, находим следующий средний урожай и его
квадратическую ошибку: 26,1 ± 1,35 if. Если нужно узнать действие калия
на фоне азота и фосфора, то, вычитая из урожая по NPK, урожаи по NP,
получаем прибавку, равную 2,5 ц (разность между двумя средними 26,1 и
23,6).
Ошибка разности этих средних будет равна
mD =±V*1,35* +1,622 = 2,11 ц.
Величина ошибки разности по величине близка к самой разности, т. е.
разность между средними будет малодостоверной и прибавка урожая от вне¬
сения калия сомнительной.
Такой метод обработки данных опыта часто применяется в исследова¬
тельских работах, и во многих случаях он вполне целесообразен. Его можно
назвать дробным методом обработки результатов опыта, так как посред¬
ством этого метода выводим для каждого варианта свойственную ему одному
ошибку, полученную на основе данных только этого варианта. Если в опыте
620
А. В. Соколов
было пять различных вариантов, то при дробной обработке результатов опы¬
та будем иметь пять различных ошибок, характеризующих отдельные вари¬
анты опыта. Разности между вариантами тоже будут иметь в каждом случае
свои ошибки в зависимости от того, какие варианты сравниваются. При пяти
вариантах будут возможны десять различных сравнений или столько же
различных ошибок разностей между средними.
Обобщенный метод обработки. Большое количество квад¬
ратических ошибок указывает на необходимость характеризовать точность
опыта при помощи одной средней ошибки и для всех возможных в опыте срав¬
нений.
Результаты опыта можно обработать в целом для всего опыта и получить од¬
ну ошибку, которая затем и применяется для характеристики достоверности
различных сравнений между вариантами опыта. Для этого достаточно произ¬
вести следующие расчеты: вычислить для каждого варианта его среднюю, най¬
ти отклонения отдельных определений для каждого варианта от его средней,
возвести их в квадрат; затем, вычислив квадраты отклонений для каждого
варианта, подсчитать общую сумму всех квадратов отклонений отдельных
определений от их средних 2 (2 v2), которая и будет характеризовать точность
нашего опыта. Разделив эту сумму на /г- (лг — 1) * / (где п — число пов¬
торений, а / — число вариантов в опыте) и произведя извлечение квадратного
корня, получим среднюю квадратическую ошибку для всех вариантов опыта
в целом; умножение ее на У 2 даст ошибку разности двух средних.
Если при вычислении ошибки опыта мы будем для каждого варианта
опыта самостоятельно устанавливать ошибку среднего, то получим ряд оши¬
бок средних, достоверность вычисления которых будет невелика. Например,
при работе с четырехкратной повторностью ошибка вычисления т достигает
т/У 2-4 = m/Y 8, или около 7з т.
Поэтому прибегают к вычислению ошибки в целом для всего опыта, счи¬
тая, что отклонения от среднего арифметического для всех вариантов опыта
однотипны.
При таком обобщенном способе обработки результатов опыта для получе¬
ния суммы квадратов отклонений (2гЯ) складывают квадраты всех отклоне¬
ний для всех вариантов опыта. В этом случае основное отклонение будет
равно
где N = nl — общее число всех делянок или сосудов в опыте; п — принятая в опыте
повторность; I — число вариантов.
Знаменатель в формуле основного отклонения носит название числа сте¬
пеней свободы. Он показывает, какое число независимых показаний лежит
в основе его вычисления (урожаев отдельных делянок, сосудов); число сте¬
пеней свободы равно числу показаний, уменьшенному на единицу.
Применение формулы о = ^ —основано на следующем рассуж¬
дении. При вычислении 2 v2 для каждого варианта мы имеем п показаний,
или тг — 1 степеней свободы; так как общее число вариантов — /, то для
опыта в целом, суммируя все г^, мы используем данные, полученные на ос¬
нове / (тг — 1) степеней свободы, а так как N = In,
1(л — 1) = гл — 1 = — /.
а ошибка средних арифметических
Определение точности опыта
621
При обобщенной обработке результатов опыта для всех вариантов опыта
имеем одинаковые ошибки средних. Следовательно, ошибка опыта, или раз-
ности средних, будет равна
mD=± Vm2-f-m2 =±т УТ = +1,41 т.
Обобщенная обработка может быть применена к результатам опытов с
двукратной повторностью, при которой имеем:
m=yr: m°=±/(i%)2+(yf)s =±/-f-+-r =±3>
т. е. ошибка равна основному отклонению.
В приведенном в табл. 4 примере показана система обработки резуль¬
татов опыта, когда нет систематической ошибки.
Обобщенный метод обработки при наличии си¬
стематических ошибок. Если в опыте урожаи в различных пов¬
торностях были разновелики вследствие различного исходного плодородия,
то иногда может быть применен разностный метод обработки результатов.
Предположим (табл. 5), что урожаи по делянкам, удобренным NPK, были
23,1; 25,3; 26,4; 29,6 ц, а для соседних с ними делянок, удобренных NP, со¬
ответственно (табл. 6) 20,4; 23,0; 22,9; 28,1 ц. Разность между средними
в этом случае будет 2,5 ц, и ошибка разности средних равна ± 2,11 ц. Меж¬
ду тем в опыте наблюдается систематическое возрастание урожаев от первого
повторения к четвертому. Если произведем вычитание по парам для каждого
повторения опыта отдельно, то получим следующие разности: 2,7; 2,3; 3,5;
1.5 ц. Квадратическая ошибка средней разности будет равна всего только
± 0,40 ц. Следовательно, мы вправе предположить, что, несмотря на изме¬
нение плодородия почвы по повторениям, действие калия в этом опыте было
весьма устойчивым. Получение меньшей ошибки при этом разностном спо¬
собе обработки результатов произошло оттого, что при первом способе обра¬
ботки в число факторов, определявших расхождения между параллельными,
была включена систематическая ошибка; первый же метод обработки резуль¬
татов предусматривает отсутствие систематической ошибки. Устранить си¬
стематическую ошибку, вызванную различным плодородием в разных по¬
вторениях опыта, можно также и следующим образом. Для каждого из пов¬
торений опыта вычисляют средний урожай для делянок всех вариантов этого
повторения, затем определяют средний урожай вообще для всех делянок опы¬
та. Первая величина характеризует плодородие по отдельным повторениям,
вторая — среднее плодородие всего участка, бывшего под опытом. Предпо¬
ложим, что урожай на делянке третьего повторения второго варианта был
37.5 if, средний урожай для второго варианта по всем повторениям опыта
был 39,2 ц, тогда отклонение урожая на нашей делянке от среднего для ва¬
рианта будет 37,5—39,2 = —1,7 ц. Но в этом опыте средний урожай для всех
делянок третьего повторения был 40,1 if, а средний урожай для опыта в це¬
лом был 41,0 ц. Следовательно, плодородие для третьего повторения было
ниже на 41,0—40,1 = 0,9 ц, чем среднее плодородие участка. Таким обра¬
зом, урожай на нашей делянке был ниже в результате падения плодородия
почвы на 0,9 ц и отклонение от среднего можно считать равным — 1,7 —
(40,1 —^41,0) = —0,8 ц. Получив такие исправленные отклонения от сред¬
них, можно вести дальнейшую обработку результатов опыта обычным
способом, как это описано выше.
Сравнение различных методов обработки. Рас¬
смотрим более детально различные способы обработки результатов опыта.
Возьмем в качестве примера полевой опыт с установлением действия калия
путем вычитания величины урожая, полученного по NP, из величины уро¬
жая, полученного по NPK. При дробной обработке результатов ошибка опыта
вычисляется для каждого варианта в отдельности (см. табл. 5 и 6).
622
А. В. Соколов
Таблица 4
Схема вычисления ошибок опыта по обобщенному методу обработки при отсутствие
систематических ошибок. Вегетационный опыт с дозами азота под овес
Вариант
схемы
JV& сосуда
Урожай
по сосудам, г
Средний
урожай для
варианта
схемы, г
Отклонение
от среднего
урожая с
точностью
до 0,1 ? (v)
Квадрат отклонения
V*
Xv*
1
1
7,36
7,01
+0,4
0,16
2
6,43
-0,6
0,36
3
7,35
+0,3
0,09
4
6,90
-0,1
0,01
0,62
2
5
10,04
10,47
-0,4
0,16
6
10,35
-0,1
0,01
7
10,94
+0,5
0,25
8
10,55
+0,1
0,01
0,43
3
9
13,10
13,77
-0,7
0,49
10
13,56
-0,2
0,04
11
14,60
+0,8
0,64
12
13,80
+0,0
0,00
1,17
4
13
20,07
20,58
-0,5
0,25
14
20,98
+0,4
0,16
15
21,08
+0,5
0,25
16
20,17
-0,4
0,16
0,82
5
•17
25,92
25,03
+0,9
0,81
18
23,64
-1,4
1,96
19
25,29
+0,3
0,09
20
25,24
+0,2
0,04
2,90
6
21
10,48
10,89
-0,4
0,16
22
11,48
+0,6
0,36
23
10,80
-0,1
0,01
24
10,79
-од
0,01
0,54
7
25
14,60
13,47
+1,1
1,21
26
13,40
-0,1
0,21
27
12,54
-0,9
0,81
28
13,32
-0,2
0,04
2,27
8
29
20,74
20,60
+0,1
0,01
30
20,82
+0,2
0,04
31
20,10
-0,5
0,25
32
20,72
+0,1
0,01
0,31
9
33
28,72
29,09
-0,4
0,16
34
29,04
-0,1
0,01
35
28,72
-0,4
0,16
36
29,88
+0,8
0,64
0,97
ДГ= 36
2 (2 г?2) =
= 10,03
(3= ± 12^1 = ± VO,37 = ±0,60 в.
mD' (ошибка опыта) = ± 0,30*1,41 = ± 0,42 г.
Определение точности опыта
623-
Таблица 5
Вычисление ошибки для урожаев по NPK
Повторение
Урожай, ц
Отклонение от среднего (v)
Квадрат отклонения (г?2)
1
23,1
—0,3
9,00
2
25,3
-0,8
0,64
3
26 ,4
+0,3
0,09
4
29,6
+3,5
12,25
М = 26,1
+3,8
1 -3,8
2*2 = 21,98
mi=± =± ylf83=±lf35>
Таблица 6
Вычисление ошибки для урожаев по NP
Повторение
Урожай, ц
Отклонение от среднего (v)
Квадрат отклонения (г*)
1
20,4
-3,2
10,24
2
23,0
-0,6
0,36
3
22,9
-0,7
0,49
4
28,1
+4,5
20,25
М =23,6
-4,5
I +4,5
Iv2 = 31,34
тг=± =± V2,61 = ± 1,62.
Таблица 7
Суммарное вычисление ошибки для урожаев по NPK и NP
Вариант
Повторение
Квадрат отклонения
Вариант
Повторение
Квадрат отклонения
V
2т>»
V
IV*
NPK
1
9,00
NP
1
10,24
2
0,64
2
0,36
3
0,09
3
0,49
4
12,25
21,98
4
20,25
31,34
2 (Iv9) =
53,32
«опыт. = ± = * V'2’22 = * 1)49‘
•624
А. В. Соколов
Результаты вычислений будут выражаться следующим образом.
Урожай по NPK 26,1 ± 1,35 ц и по NP 23,6 ± 1,62 ц. Ошибка разности
по формуле + У т\ + т\ будет равна
± У 1,35*+ 1,62* =± /1,83 +2,Ы=± =±2,11 ц .
Разность между средними для NPK и NP будет: 2,5 ± 2,11 ц.
При вычислении общей ошибки для опыта по обобщенному методу
обработки производят вычисления квадратов отклонений для вариантов,
но все они сразу суммируются (табл. 7).
Ошибка разности двух средних
™опыта = ± УЧ,492 + 1,492 =± V2,22 + 2,22 =+ У4Д4 = ±2,11 .
В нашем опыте, поскольку имеется всего два варианта, оба способа обра¬
ботки должны привести к одинаковым результатам.
При этих способах вычисления получается большая ошибка разности
средних; между тем имеются основания предположить, что в данном опыте
большая ошибка вызывается различием в плодородии в разных повторениях.
В этом случае вычисления производят по обобщенному разностному методу
обработки и для каждого повторения в отдельности определяют разность
между урожаями по NPK и NP (табл. 8).
Систематическую ошибку опыта, которая в данном случае обусловли¬
вается изменением плодородия почвы при переходе от одного повторения
к другому, можно устранить, применяя и другой способ вычисления.
Ошибка разности между средними для двух сопряженных рядов опреде¬
ляется по формуле
т = i У т\ + т* — 2гпцтг »
где г — коэффициент корреляции.
Подставляя значение г в формулу ошибки разности, после сокращения
имеем:
f , / Ivl + Zvl-ZZvivz
--+К ■ .(.-»>—
Таблица 8
Вычисление ошибки разности по разностному методу
Повторение
Урожай
Разность
Отклонение от
Квадрат
отклонения
по NPK
по NP
средней разности
1
23,1
20,4
2,7
+0,2
0,04
2
25,3
23,0
2,3
-0,2
0,04
3
26,4
22,9
3,5
+1 ,о
1,00
4
29,6
28,1
1,5
-1,0
1,00
Мг=26,1
Мг= 23,6 |
2,5
+1,2
-1,2
2*а= 2,08
mD = ± У ± О-40'
Определение точности опыта
625
Таблица 9
Вычисление произведения отклонений
Повторение
Отклонение от средних для урожаев
Произведение отклонений
по NPK
по NP
1
—3,0
—3,2
+9,60
2
-0,8
—0,6
+0,48
3
+0,3
-0,7
-0,21
4
+3,5
+4,5
+15,75
-3,8
-4,5
=25,62
+3,8
+4,5
_ ± у 53,32-2.25.62_ ± j/2^ ± од
Сумма 2yi + определена ранее (табл. 7) и равна 53,32. Остается найти
сумму произведений отклонений по повторениям (табл. 9).
Таким образом, применяя формулу определения ошибки разности для
сопряженных рядов, приходим к тому же результату, что и при непосред¬
ственной обработке разностей.
Тот же самый результат может быть получен, если мы будем по повторе¬
ниям вносить исправления отклонений, что и применяется для обработки
результатов полевого опыта. С этой целью определяем средние урожаи
по повторениям и отклонения их от среднего урожая для всего опыта
(табл. 10).
Если бы плодородие почвы во всех повторениях было одинаковым, то и
урожаи их были бы близки. Взяв отклонения от средних по вариантам и
вычтя из них отклонения по повторениям, получаем исправленные откло¬
нения, которые и возводим в квадрат (табл. 11).
Таблица 10
Вычисление отклонений по повторениям
(средний урожай опыта 24,85 if)
Повторение
Урожай, ц
Средний урожай
по повторениям
Отклонения повто¬
рения от среднего
урожая по опыту
boNPK
ПО NP
1
23,1
20,4
21,75
-3,1
2
25,3
23,0
24 ,15
-0,7
3
26,4
22,9
24,65
.-0,2
4
29,6
28,1
28,85
+4,0
Мх = 26,1
Л*2=23.6
24,85
+4,0
-4,0
626
А. В. Соколов
Таблица И
Вычисление ошибки при помощи исправленных отклонений
Вариант
Повторение
Отклонение от сред¬
него
Исправленное откло¬
нение по повторениям
Квадрат исправленного
отклонения
V*
Ег>*
NPK
1
-3,0
+0,1
0,01
2
-0,8
-0,1
0,01
3
+0,3
+0,5
0,25
4
+3,5
-0,5
0,25
0,52
NP
1
-3,2
-0,1
0,01
2
-0,6
+0,1
0,01
3
-0,7
—0,5
0,25
4
+4,5
+0,5
0,25
0,52
2 (2vz)
= 1,04
Отсюда ошибка среднего для варианта, вычисленная по формуле
±У п(п — 1)(1 —.1) ’
будет равна:
"-± Vrra1-*0'29 •
Ошибка же разности двух средних вычисляется по формуле тв =
= ± Yml +m| • Так как в данном случае после исправления отклонений мы
устранили их сопряженность, то имеем:
mD= ± У0,292 + 0,292 = ±0,40 .
Этот способ вычисления ошибки опыта и применяется во всех случаях,
когда урожаи по повторениям различны по величине.
ОЦЕНКА ТОЧНОСТИ ОПЫТА
Статистическую обработку результатов опыта производят для установ¬
ления точности опыта. Ошибка среднего арифметического показывает, с ка¬
кой степенью точности мы можем судить по величине найденного в опыте
среднего о величине того истинного среднего, которое было бы получено
в результате суммирования бесконечно большого числа параллельных опре¬
делений. Согласно кривой нормального распределения всех возможных
случаев, можно утверждать, что истинное среднее будет находиться в про¬
межутке между М т я М — т в 68,3% всех возможных случаев, в про¬
межутке между М + Зт и М — Зт в 99,73% всех возможных случаев, или,
говоря другими словами, мы имеем 9973 шанса из 10 000 за то, что наше сред¬
нее не отклоняется от истинного больше чем на ± 3 т. Например, если для
урожая по NPK мы получили средний урожай в 45,7 if, а ошибка среднего
была 0,6 цу то истинный урожай по NPK с достоверностью в 99,73% не раз¬
нится от 45,7 больше чем на ±3 т — ±1,8 ц. Следовательно, наш вывод,
что истинный урожай по NPK, который мы получили бы при большом числе
повторений, находится в пределах М ± Зт = 45,7 ± 1,8, или, что то же,
в промежутке между 43,9—47,5 ц, имеет большую достоверность. Наоборот,
Определение точности опыта
627
если бы мы стали считать, что истинное значение находится в пределах М ±
± т = 45,7 ± 0,6, или между 45,1 и 46,3 ц, то в 31,7% всех случаев наше
суждение было бы ошибочно. Следовательно, чем больше будет допускаемый
нами возможный размер отклонения от среднего, тем достовернее будет наше
суждение. Степень же точности нашего определения среднего будет зависеть
от величины ошибки среднего. Если бы в нашем опыте ошибка среднего
была равна ± 0,1 if, то в пределах 45,4—46,0 ц находилось бы 9973 из
10 ООО возможных случаев. Таким образом, степень точности нашего опыта
зависит от величины ошибки среднего, а достоверность наших суждений —
от того, во сколько принятый нами размер возможного отклонения истин¬
ного результата от среднего будет превышать ошибку последнего. Обычно
считают, что истинный результат достаточно точно характеризуется проме¬
жутком М ± 3 т (тройная ошибка называется иногда предельной ошибкой
среднего). Число, показывающее, во сколько раз установленный предел от¬
клонения от среднего больше его ошибки, обозначается буквой t; чем больше t,
тем достовернее наше суждение. В табл. 12 показана зависимость досто¬
верности наших суждений от принятого при анализе результатов предель¬
ного отклонения от среднего, выраженного в частях т.
В большинстве опытов приходится не столько устанавливать абсолютные
размеры урожая по тому или другому варианту опыта, сколько определять
достоверность разности между средними для различных вариантов. Досто¬
верность разности между двумя средними определяется величиной t, которая
показывает, во сколько раз разность больше своей ошибки. Обозначая раз¬
ность через D, имеем:
_ _D_
“ mD '
Когда ошибка разности равна самой разности, истинное значение раз¬
ности между двумя средними, согласно кривой нормального распределения
показаний, выйдет за пределы D ± тв в 31,7% всех возможных случаев,
так как ошибка разности так же характеризует ее, как обычная ошибка
среднего — свое среднее арифметическое. Но в одной половине возможных
случаев истинное значение будет больше нашей эмпирической разности,
а в другой половине — меньше. Если речь идет о достоверности разности
между средними, т. е. ставится вопрос только о том, действительно ли уро¬
жаи по обоим сравниваемым вариантам отличаются друг от друга, то для
Таблица 12
Вероятность'нахождения истинного результата за пределами M+trn (в % от возможного
числа случаев) при числе наблюдений больше 30
Предел отклонения
от среднего в долях
т, t
Вероятность нахожде¬
ния за указанными
пределами, %
Предел отклонения
от среднего в долях
т, t
Вероятность нахожде¬
ния за указанными
пределами, %
0,50
61,7
2,33
2,00
0,67
50,1
2,50
1,20
0,75
45,3
2,58
1,00
1,00
31,7
2,75
0,56
1,25
21,1
3,00
0,10
1,50
13,4
3,29
0,10
1,75
8,0
3,89
0,01
2,00
4,5
4,42
0,001
2,25
2,4
4,89
0,0001
628
А. В. Соколов
характеристики достоверности разности имеют значение отклонения только
в одном направлении — уменьшения разности между средними.
Предположим, что Мг — М2 = wid, т. е. разность равна ошибке разности,
t = DjniD = 1; тогда (по табл. 13) 31,7% всех случаев будет находиться за
пределами D ± wip, но только в половине из них (15,9%) разность между
средними исчезнет или станет обратной. Следовательно, достоверность раз-
ности можно характеризовать не 68,3%, а 84,1%, так как в 84,1% всех воз¬
можных случаев Мх будет больше, чем М2. В табл. 13 приведены соответ¬
ствующие данные достоверности разности средних; их легко можно вывести
из табл. 12, если учесть, что в данном случае имеет значение только одно¬
стороннее отклонение от среднего. Из табл. 12 видно, что если разность
более чем вдвое превосходит свою ошибку, то по своей достоверности она
уже заслуживает внимания, поэтому в полевых и вегетационных опытах
обычно довольствуются двойной ошибкой разности средних в качестве по-
Таблица 13
Достоверность разности двух средних (в % от общего числа возможных случаев)
при числе наблюдений больше 30
D
тр
%
D
mj)
%
D
mp
%
D
mj)
%
0,2
57,9
0,8
78,8
1,4
91,32
2,0
97,73
0,3
61,8
0,9
81,3
1,5
93,77
2,1
98,21
0,4
65,5
1,0
84,1
1,6
94,52
2,2
98,61
0,5
69,2
1,1
86,4
1,7
95,54
2,4
99,18
0,6
72,6
1,2
88,99
1,8
96,41
2,6
99,53
0,7
75,8
1,3
90,32
1,9
97,13
3,0
99,87
Таблица 14
Таблица величин t. Определение вероятности (Р) нахождения среднего значения вне
предела М ± tm при малом числе наблюдений (п)
n— 1
50
20
10
5
2
1
0,25
1
1,00
3,08
6,31
12,70
31,82
63,66
318,54
2
0,82
1,37
2,92
4,30
6,96
6,92
22,38
3
0,76
1,64
2,35
3,18
4,54
5,84
10,24
4
0,74
1,53
2,13
2,78
3,75
4,60
7,58
5
0,73
1,48
2,02
2,57
3,36
4,03
5,90
6
0,72
1,44
1,94
2,45
3,14
3,70
5,20
7
0,71
1,42
1,90
2,36
3,00
3,50
4,80
8
0,71
1,40
1,86
2,31
2,80
3,35
4,50
9
0,70
1,38
1,83
2,26
2,82
3,25
4,30
10
0,70
1,37
1,81
2,22
2,76
3,17
4,15
11
0,70
1,36
1,80
2,20
2,72
3,11
4,00
12
0,69
1,34
1,75
2,13
2,60
2,95
3,75
20
0,69
1,32
1,73
2,09
2,53
2,84
3,55
25
0,68
1,31
1,71
2,06
2,48
2,80
3,30
30
0,68
1,31
1,70
2,04
2,46
2,75
3,05
oo
0,67
1,28
1,64
1,96
2,33
2,58
3,00
Определение точности опыта
629
казателя достоверности суждений, имея 95% вероятности, которая получает¬
ся при t = 1,64 то.
Таблица Стюдента и Фишера. Для оценки достоверности
результатов опыта мы можем пользоваться табл. 12 и 13 только в тех слу¬
чаях, когда ошибка опыта, или среднего арифметического, вычислена на
основе обработки сравнительно большого числа данных, т. е. когда в опыте
было более 30 делянок или сосудов и применялся обобщенный метод
обработки результатов опыта. Если в опыте было небольшое число
делянок или производилось вычисление ошибки среднего для каждого
варианта опыта в отдельности, то следует пользоваться табл. 14, состав¬
ленной Стюдентом и несколько видоизмененной Фишером. В этой таблице
вероятность нахождения истинного результата в пределах М ± т постав¬
лена в зависимость от числа наблюдений. В первом вертикальном столбце
приведено число так называемых степеней свободы, равное для среднего
арифметического п — 1, т. е. числу показаний урожаев делянок или сосудов,
из которого выведено среднее, уменьшенному на единицу (так как, зная
п — 1 показаний, последнее показание можно установить по разности, если
известно среднее). В верхнем горизонтальном ряду указана вероятность
(в процентах) нахождения истинного результата за пределами М ± tm.
В средней части таблицы приведены величины t — коэффициенты, показы¬
вающие, во сколько раз принятое при анализе результатов опыта отклоне¬
ние от среднего больше его ошибки. Например, если мы имеем средний
урожай для четырех делянок (п = 4), равный 34,5 if, и ошибка среднего
+ 1,1 if, то вероятность нахождения истинного результата за пределами
М ± Ът (что соответствует значению t = 3), т. е. иначе в промежутке от
31,2 до 37,8 if, будет, как показывает табл. 14, около 5% (в таблице име¬
ем для п — 1 = 3 значение I, близкое к 3, именно 3,18).
Если бы ошибка среднего была установлена на основе большого числа
определений, то вероятность отклонения истинного результата за пре¬
делы М ± 3/и была бы равна только 0,27 %. Чтобы иметь подобную досто¬
верность наших суждений при п = 4, нам надо размах возможных коле¬
баний среднего принять в М ± 10,24 т, или 34,5 ± 11,3 if, так как вероят¬
ности в 0,27% при п — 1 = 3 соответствует t = 10,24. Таким образом, при
четырехкратной повторности так называемая предельная ошибка среднего
(т. е. ошибка при t= 3) должна быть принята равной не тройной ошибке,
а десятикратной.
Этой же табл. 14 можно пользоваться и для установления достоверно¬
сти разности между средними Мх — Л/2, т. е. для определения того, во сколь¬
ких случаях из всех возможных Мг будет больше М2.
В данном случае имеют значение отклонения только в одну сторону,
направленные к уменьшению разности между Мг и М2. При отклонении
только в одну сторону, положительную или отрицательную, Р в 2 раза
меньше.
Пример. Разность между средними равна 6,6 г, повторность семикратная, ошиб¬
ка разности 2,1 г, следовательно,
6,6
*= 2J = 3’14-
При п = 7 по табл. 14 имеем Р^2%,т. е. в2% всех случаев разнбсть выходит за
пределы + 6,6 г. Исчезновение положительной разности между средними, т. е. отклонение
Р 2%
за пределы разности 6,6 г, равно = ттг= или °ДН0МУ случаю из 100 возмож-
2 &
ных.
В практике оценки результатов опытов установилась привычка брать
для оценки достоверности разности двойную или тройную ошибку, не учи¬
630
А• В. Соколов
тывая числа наблюдений, на основе которых выведена ошибка опыта. Из
разобранных примеров видно, насколько последнее может иногда при¬
вести к неверным суждениям о достоверности результатов опыта.
Метод А. А. Сапегина. В практике сельскохозяйственного опыт¬
ного дела широкое применение при обработке результатов полевого опыта
имел у нас метод приведения А. А. Сапегина. Особенностью этого метода
является то, что обрабатываются процентные отклонения от средних ариф¬
метических по вариантам. В основе его лежит предположение, что величи¬
на ошибки опыта пропорциональна величине урожаев. Экспериментальные
данные не подтвердили этого предположения, но метод А. А. Сапеги¬
на дает в конце обработки очень удобную для оценки точности опыта про¬
центную ошибку среднего (т. е. т), выраженную в процентах от среднего
урожая для опыта: Р = 100 т!М. Этот показатель относительной точности
исследования весьма удобен для сравнения различных опытов по степени
их точности. С этой целью может быть применено вычисление ошибки сред¬
него, выраженной в процентах от среднего урожая для всего опыта в целом;
/71 • 1 ОО/Afо пыта •
Метод анализа вариации Р. А. Фишера. Статистическая
обработка результатов полевых опытов при наличии в опытах системати¬
ческой ошибки, вызываемой неравенством исходного плодородия почвы,
весьма сложна. Для устранения систематической ошибки полеводы приме¬
няют не только различные приемы статистической обработки результа¬
тов полевого опыта, но и различные приемы размещения в поле опытных
делянок, что, в свою очередь, накладывает свой отпечаток на приемы ста¬
тистической обработки результатов. Постановка полевых опытов с большим
числом вариантов в условиях изменчивого плодородия почвы — весь¬
ма сложная методическая задача, различно решаемая в многочисленных
работах полеводов-опытников и математиков. Мы здесь даем анализ точности
простого полевого опыта, имеющего небольшое число вариантов и поставлен¬
ного на достаточно выровненном по плодородию участке.
Кроме установления точности опыта и степени достоверности наших
суждений, обработка результатов опыта помогает определить пригодность
его для решения поставленной задачи. Пригодность опыта определяется
прежде всего правильностью составления схемы опыта и выбором надле¬
жащих условий для его проведения. Достоверность разности между кон¬
трольными вариантами и служит одним из показателей пригодности опы¬
та для решения данной задачи. Например, если опыт ставится с целью
определения эффективности различных новых форм азота, то контроль¬
ными вариантами в опыте являются варианты без азота и с внесением стан¬
дартного азотного удобрения. Если между этими вариантами имеется досто¬
верная разность, опыт пригоден для решения поставленной задачи, если нет,
то результаты опыта не могут быть использованы. Если в условиях опыта
растения вообще не нуждались в азоте, то изучать в этих условиях эффек¬
тивность новых азотных удобрений не следует. Таким образом, пригодность
опыта (его репрезентативность) определяется путем вычисления D/mo для
контрольных вариантов схемы опыта.
В практике Ротамстедской сельскохозяйственной опытной станции в Анг¬
лии и отчасти на опытных станциях США получил распространение метод,
разработанный Р. А. Фишером и названный им анализом вариации (рассея¬
ния, дисперсии). В практике сельскохозяйственного опытного дела СССР
этот метод, несмотря на проявленный к нему в свое время большой интерес,
не получил большого распространения.
При обработке результатов опыта по методу анализа вариации (рассея¬
ния) сначала находят средний урожай в опыте, затем вычисляют откло¬
нения от него всех урожаев отдельных делянок. Эти отклонения возводят
Определение точности опыта
631
в квадрат и находят их сумму, которая соответствует общей вариации (рас¬
сеянию) опыта. Далее вычисляют средние урожаи для вариантов, их от¬
клонения от среднего урожая, квадраты отклонений и их сумму. Послед¬
нюю умножают на число повторений. Полученная величина характеризует
изменение урожаев в зависимости от вариантов. Затем определяют средние
урожаи по повторениям, их отклонения от среднего урожая для опыта,
квадраты этих отклонений и их сумму, которая после умножения на число
вариантов дает величину, характеризующую варьирование урожаев, вы¬
зываемое различным плодородием повторений. Вычитая из суммы квадратов,
характеризующей общее рассеяние в опыте, величины, найденные для рас¬
сеяния вариантов и повторений, находят величину суммы квадратов, соот¬
ветствующую случайным отклонениям, или остаточное рассеяние (вариа¬
цию) в опыте.
Вычисление величин, характеризующих рассеяние общее, вариантов
повторений и случайное, может идти с использованием квадратов не от¬
клонений, а квадратов величин урожаев по делянкам, по повторениям и
по вариантам, но это целесообразно только при наличии счетных машин.
Полученные суммы квадратов следует соответственно разделить на
число степеней свободы для вариантов на (I — 1) и для повторений на (п — 1)
и для случайного рассеяния на (п — 1) • (Z — 1). После деления сумм квад¬
ратов на число степеней свободы получаем средние квадраты.
Отношение среднего квадрата вариантов к среднему квадрату слу¬
чайного рассеяния и является критерием точности опыта и его пригодно¬
сти для решения поставленного вопроса. При этом используются таблицы
Снедекора, в которых указывается, при каком отношении средних квадра¬
тов изменение урожаев, вызываемое вариантами, имеет вероятность в 95
или 99% (см. табл. 20 и 21).
Пример использования анализа вариации приведен далее при обработке
результатов полевого опыта.
При работе методом анализа вариации данные для всего опыта обра¬
батывают совместно, так как этот метод такой же обобщенный, как и опи¬
санный ранее. Далее, и в том и в другом методе при анализе вариации все
вычисления производят с абсолютными величинами отклонений. В резуль¬
тате вычислений оба метода дают одну и ту же величину суммы квадратов
случайных отклонений; только в одном случае они определяются непо¬
средственно, а при анализе вариации — по разности. Исключение влияния
изменения плодородия почвы по повторениям опыта в том и другом случае
производится на основе одного и того же принципа.
Существенное различие между обоими методами заключается в том, что
в одном случае устанавливается ошибка опыта, т. е. ошибка разности меж¬
ду двумя средними, а в другом — критерий Фишера, характеризующий,
во сколько раз отклонения между вариантами больше случайных.
В основе этого метода лежит предположение, что опыт в том случае
пригоден, когда различие между вариантами опыта больше, чем между
урожаями параллельных делянок или сосудов. Если изменения в урожаях
по делянкам, вызываемые случайными, неучитываемыми причинами, больше,
чем изменения, вызываемые изучаемыми факторами, опыт считается непри¬
годным для установления действия последних на урожай растений.
При обычной оценке достоверности опыта устанавливают, во сколько
раз разность между средними для двух вариантов опыта превышает ее
ошибку. Но в одном и том же опыте разности между различными парами
средних обладают различной степенью достоверности в зависимости от
величины разности. Критерий, предложенный Фишером, является по¬
пыткой однозначно характеризовать достоверность действия изучаемых
факторов для всего опыта в целом.
632
А. В. Соколов
Средние арифметические для вариантов опыта образуют ряд цифр, ко¬
торый характеризует действие изучаемых в опыте факторов. Установив
для этого ряда цифр его квадратическое отклонение, мы можем сравнить
его с квадратическим отклонением, характеризующим действие в опыте слу¬
чайных причин. Так как на изменении ряда средних по вариантам сказы¬
ваются и случайные причины, то, если изучаемые факторы существенно ска¬
зались на высоте урожаев, квадратическое отклонение для средних по ва¬
риантам должно быть достоверно больше, чем квадратическое отклонение,
характеризующее действие случайных, неучтенных причин. Но в некоторых
случаях применение критерия Фишера не дает правильных результатов9
так как он учитывает изменчивость между всеми вариантами опыта, тогда
как для определения пригодности опыта необходимо учитывать различие
только между определенными контрольными вариантами опыта. При поста¬
новке опыта все варианты схемы опыта можно разбить на два вида: контроль¬
ные, которые показывают чувствительность в опыте растений к изучаемому
фактору, и испытуемые. Между последними может и не быть никакого рас¬
хождения, в результате чего уменьшится основное отклонение изменчивости
по вариантам при наличии в опыте сильного действия изучаемого фактора.
Например, изучая различные формы новых растворимых фосфатов, мы, имея
опыт с громадной чувствительностью растений к количеству усвояемой
Р206, можем получить малую изменчивость между вариантами опыта, если
число одинаково усвояемых форм будет велико.
Для анализа достоверности опыта и его пригодности мы предлагаем опре¬
делять достоверность разницы между контрольными вариантами опыта.
Под последними следует при этом понимать не только делянки без удобре¬
ний, но и все делянки опыта, при помощи которых устанавливается чувстви¬
тельность опытных растений к изучаемому фактору. В зависимости от задач
опыта эти контрольные варианты будут различными.
В практике опытного дела в СССР анализ вариации (рассеяния, диспер¬
сии) используют как способ вычисления ошибки опыта, без применения
критерия Фишера. Этот способ вычисления сложнее и менее удобен, чем
вычисления ошибки опыта с исправлением отклонений по повторениям,
хотя и дает те же результаты.
Пример обработки результатов полевого опдата
В целях освоения наиболее простых практических приемов обработки результатов
полевого опыта рассмотрим детально обработку результатов одного опыта.
В 1955 г. Центральной станцией удобрений и агропочвоведения СоюзНИХИ был
проведен полевой опыт с внесением фосфорных удобрений в подкормку под хлопчатник.
Опыт был поставлен на общем для всех делянок опыта фоне удобрений, который состоял
И8 40 кг Р206, внесенных в виде суперфосфата до посева, и 110 кг азота, внесенного в под¬
кормку в виде аммиачной селитры. В схеме опыта всего было 8 вариантов (I = 8):
1. Одно фоновое удобрение («фон»).
2. Фон + суперфосфат из апатита.
3. Фон + суперфосфат из каратауского фосфорита.
4. Фон + аммонизированный суперфосфат.
5. Фон + аммофос иэ каратауского фосфорита.
6. Фон + аммофос из апатита.
7. Фон + плавленый магниевый фосфат.
8. Фон + обесфторенный фосфат.
Фосфорные удобрения вносились в подкормку в дозе 40 кг Р20Б; при внесении ам¬
мофоса количество азота в фоновом удобрении соответственно уменьшалось на содержание
аэота в аммофосо. Опыт был поставлен в четырехкратной повторности (п = 4). Задача
математической обработки результатов этого опыта установить: было ли в опыте вообще
достоверное положительное действие фосфатов и если оно было, то были ли в опыте досто¬
верные различия между эффективностью различных форм фосфорных удобрений, внесен¬
ных в подкормку.
Определение точности опыта
633
Прежде всего необходимо составить таблицу данных опыта, по которой можно удобно
подсчитать средние урожаи по вариантам и по повторениям опыта (табл. 15). Составив эту
таблицу и подсчитав сумму урожаев для каждого варианта и каждого повторения, находим
затем общую массу урожаев, полученных со всех 32 делянок опыта (1191,8). Складывая
урожаи по вариантам или складывая урожаи по повторениям, получаем ту же величину,
что свидетельствует о правильности наших подсчетов. Затем вычисляем средние урожаи
для каждого варианта и для каждого повторения. При этом средние урожаи по вариан¬
там мы записываем с точностью до 0,1 ц/га.
Цель опыта — установить прибавки урожаев хлопчатника от внесения в подкормку
фосфорных удобрений, а задача математической обработки результатов опыта —
установить степень их достоверности. Вычитая из величин средних урожаев по вариантам
опыта (с 2-го по 7-й) величину среднего урожая По первому варианту (фон), получим при¬
бавки урожаев хлопка-сырца (в ц/га) от внесения фосфатов. Эти прибавки колеблются в
зависимости от формы фосфорного удобрения от 1,8 до 5,7 ц/га хлопка сырца (см. табл. 15)*
Таблица 15
Вычисление средних показателей для вариантов и повторений полевого опыта
Вариант
Урожай делянок по повто¬
рениям, ц/га
Сумма
урожаев
по ва¬
риантам,
ц/га
Средний
урожай
по ва¬
риантам,
Ц/га
Отклоне¬
ние вари¬
антов от
среднего
урожая
опыта,
Ц/га
Прибавка
урожая
от фос¬
форной
подкорм¬
ки, ц/га
1
2
3
4
1
31,0
33,3
37,1
35,5
136,9
34,2
-3,0
2
34,8
36,5
41,7
39,8
152,8
38,2
+1,0
+4,0
3
34,5
36,2
40,7
38,4
149,8
37,4
+0,2
+3,2
4
35,6
35,7
37,4
44,6
153,3
38,3
+1Д
+4,1
5
36,4
36,3
39,5
37,2
149,4
37,4
+0,2
+3,2
6
36,7
40,9
40,6
41,3
159,5
39,9
+2,7
+5,7
7
34,6
37,4
37,4
34,6
144,0
36,0
-1,2
+1,8
8
34,2
34,7
39,9
37,3
146,1
36,5
-0,7
+2,3
Сумма урожаев
277,8
291,0
314,3
308,7
1191,8
—
—
—
по повторениям
Средний урожай
34,72
36,37
39,29
38,58
—
37,24
—
—
по повторениям
Отклонение по¬
-2,52
-0,87
+2,05
+1,34
—
—
—
—
вторений
Разделив общую сумму урожаев на число делянок в опыте, получим средний урожай
в опыте: 1191,8 : 32 = 37,24. Разделив суммы урожаев по повторениям на число вариан¬
тов, находим средние урожаи по повторениям. Вычитая из них величину среднего урожая
для опыта, получаем отклонения для урожаев по повторениям; они равны — 2,52;—0,87;
+ 2,05 и + 1,34. Как видим, первое повторение дает более низкие урожаи, а третье —
более высокие, чем остальные повторения. Видимо, в опыте имело место изменение плодо¬
родия почвы при переходе от одного повторения к другому. Следовательно, при обработ¬
ке результатов опыта надо исключить влияние варьирования величин урожаев по повто¬
рениям. Это достигается двумя способами: во-первых, путем получения исправленных от¬
клонений для каждой делянки и, во-вторых, путем вычитания суммы квадратов отклоне¬
ний по повторениям, умноженной на число вариантов из суммы квадратов неисправлен¬
ных отклонений.
Остановимся сначала на первом способе с использованием исправленных отклонений.
С этой целью устанавливаем поправки по повторениям, они будут равны отклонениям пов¬
торений, взятым с обратным знаком, + 2,5, + 0,9, — 2,1 и — 1,3. Вычисление ошибок
опыта производим на основе отклонений величин урожаев отдельных делянок от среднего
по варианту. Эти отклонения приведены в графе 2 табл. 16. Например, для 1-го варианта,
согласно табл. 15, имеем урожаи: 31,0; 33,3; 37,1; 35,5; средний урожай по варианту равен
634
Л. В. Соколов
Таблица 16
Вычисление суммы квадратов случайных отклонений
Вариант опыта
Отклонение от
среднего для
варианта
Квадрат
отклонения
Поправка
по повторе¬
ниям
Исправлен¬
ное отклоне¬
ние
Квадрат исправ¬
ленного откло¬
нения
1
-3,2
10,24
+2,5
-0,7
0,49
—0,9
0,81
+0,9
-0,0
0,00
+2,9
8,41
-2,1
+0,8
0,64
+1,3
1,69
-1,3
-0,0
0,00
2
-3,4
11,56
+2,5
-0,9
0,81
-1.7
2,89
+0,9
-0,8
0,64
+3,5
12,25
-2,1
+1,4
1,96
+1,6
2,56
-1,3
+0,3
0,09
3
-2,9
8,41
+2,5
-0,4
0,16
-1,2
1,44
+0,9
—0,3
0,09
+3,3
10,89
-2,1
+1,2
1,44
+1,0
1,00
-1,3
-0,3
0,09
4
-2,7
7,29
+2,5
-0,2
0,04
-2,6
6,76
+0,9
-1,7
2,89
—0,9
0,81
-2,1
—3,0
9,00
+6,3
39,69
-1,3
+5,0
25,00
5
-1,0
1,00
+2,5
+1,5
2,25
-1,1
1,21
+0,9
-0,2
0,04
+2,1
4,41
-2,1
+0,0
0,00
-0,2
0,04
-1,3
-1,5
2,25
6
-3,2
10,24
+2,5
-0,7
0,49
+1,0
1,00
+0,9
+1,9
3,61
+0,7
0,49
-2,1
-1,4
1,96
+1,4
1,96
-1 ,з
+0,1
0,01
7
-1,4
1,96
+2,5
+1,1
1,21
+1,4
1,96
+0,9
+2,3
5,92
+1,4
1,96
-2,1
-0,7
0,49
-1,4
1,96
-1,3
-2,7
7,29
8
-2,3
5,29
+2,5
+0,2
0,04
-1,8
3,24
-0,9
—0,9
0,81
+3,4
11,56
-2,1
+1,3
1,69
+0,8
0,64
-1,3
—0,5
0,25
Сумма квадратов
—
175,62
—
—
71,02
34,2. Тогда отклонения равны: 31,0—34,2 = —3,2; 33,3—34,2 = — 0,9 и т.д. (см.
1'абл. 16). Эти отклонения исправлены во всех вариантах на величину попрарэк по повто¬
рениям, указанным в графе 4 табл. 16, в графе 5 приведены исправленные отклоненияf
а в графе 6 — квадраты этих отклонений. Сумма всех этих квадратов равна 71,02, ее мы
должны разделить на число степеней свободы. Общее число делянок в опыте 32, следова¬
тельно, общее число степеней свободы в опыте равно 32—1 = 31, число степеней свободы
для вариантов равно 8—1 = 7 и для повторений 4—1 = 3. При вычислении исправлен¬
ных отклонений мы уже исключили влияние повторений, а вычисляя отклонения внутри
вариантов, исключили влияние вариантов. Таким образом, на долю случайного (оста¬
точного) варьирования осталась 31—7—3 = 21 степень свободы. Эту же величину сред¬
ней свободы находим, умножив число степеней свободы вариантов на число степеней сво-
Определение точности опыта
635
Таблица 17
Вычисление варьирования по повторениям
Повторение
Отклонение от среднего
Квадрат отклонения
1
-2,52
6,35
2
-0,87
0,76
3
—2,05
4,20
4
+1,34
1,80
Сумма квадратов
13,11
боды повторений (/ — 1 = 8—1 = 7 и п — 1 = 4 — 1 = 3). Вычисление квадрают~
ного отклонения производим по формуле
0 ± j/"= ± Y3,3818 = ± 1,84 фа .
Следовательно, ошибка среднего по вариантам равна:
1,84
. 1,84
:±ут
±0,92 ц.
Ошибка разности двух средних равна:
mD ='± /(0,92)* + (0,92)* =±1,41 -0,92 = ±1,30 if.
Рассмотрим второй способ вычисления ошибки опыта. Возведем в квадрат неисправ»
ленные отклонения урожаев делянок от средних по вариантам (графа 3, табл. 16) и найдем
их сумму 175,62. Возведем в квадрат] отклонения по повторениям и найдем их сумму
(табл. 17), которая будет равна 13,11. Эта сумма квадратов характеризует варьирование
в зависимости от повторений в пределах одного варианта. Общее число вариантов равно
8; умножив на него сумму квадратов отклонений (2 v2) 13,11, получим 2 (2 у2) = 13,11 *8 =*•
= 104,88, т. е. величину, характеризующую влияние различного плодородия повторений
на варьирование урожаев в опыте.
Вычитая из суммы квадратов неисправленных отклонений 2(2р2) = 175,62 варе»
рование повторений 104,88, получаем величину варьирования исправленных отклонений
70,74. Она только на 0,28 меньше, чем найденная первым способом (71,02); расхождение
вполне понятное, так как при вычислении мы прибегали к округлению цифр до 0,1 или0,01«
Вычисление по второму способу дает те же величины, что и по первому: а = ± 1,84*
т = + 0,92 и mjy = + 1,30.
Если бы не учли изменения плодородия почвы по повторениям и в основу наших
вычислений положили бы сумму квадратов неисправленных отклонений, то величин*
квадратичного отклонения была бы равна + 2,70 и т = ±1, 35, так как
« = ±|/ =±2,70.
В результате мы получили бы неправильное представление о малой точности опыта*
Достоверность прибавок урожаев в опыте определяется тем, во сколько раз они пре»
восходят свою ошибку. Вычисление ошибки разности (mD) производим при использовании
21 степени свободы; по табл. 14 находим, что для 20 степеней свободы 5% показаний будет
находиться за пределами + 2,09 mD, 1% показаний—за пределами ±2,84 mD
Следовательно, прибавки урожаев, полученные в опыте, в 95% случаев будут от*
клониться от найденных средних не более как на ± 2,09 mD = ± 2,09*1,30= ± 2,7 tf#
а в 99% случаев не более как на ± 2,84*1,30 = 3,7 ц. Но в данном случае нас интересует
не столько возможность получения прибавок определенного размера, сколько достовер~
ность наличия разности между вариантами. В этом случае важно учесть возможный про-
636
Л. В. Соколов
цент вероятных отклонений за пределы не + mD, а только за пределы — mD. Последний
будет вдвое меньше указанного в табл. 14 для ± mD, поэтому берем для характеристики
вероятной ошибки в размере 5% случаев — 1,73 mDt а для 1% — 2,53 mD, т. е. для на¬
шего опыта имеем соответственно 2,3 и 3,3 ц.
Большинство прибавок урожая от внесения фосфорных удобрений, кроме прибавок,
полученных в 7-м и 8-м вариантах, больше 2,3 if, следовательно, заслуживают внимания;
более достоверными следует считать, однако, те прибавки, которые больше 3,3 ц.
Рассматривая внимательно данные табл. 15, видим, что урожай по одной из делянок,
именно в четвертом варианте четвертого повторения, исключительно высок и равен 44,6 ц.
Этот урожай превышает урожай среднего для этого варианта на 6,3 ц/га.
Основное отклонение в опыте было равно + 1,84 if, следовательно, отклонение этой
делянки от среднего превышает основное отклонение в 3,4 раза. Среднее для трех других
повторений этого варианта равно 36,2 if,т.е. на 8,4 if меньше, чем урожай в четвертом пов¬
торении. Такое отклонение от средних бывает весьма редко, меньше чем 1 раз в 1000 слу¬
чаев, и наличие его дает неправильное представление об эффективности четвертого вари¬
анта. Целесообразно поэтому провести обработку данных опыта, исключив эту делянку.
Обработка опыта при отсутствии одной делянки может быть сделана для оставшихся
вариантов, имеющих нормальное число повторений; обработка может быть сделана и для
всех повторений, за исключением повторения с забракованной делянкой. Можно также
сохранить обработку данных опыта в целом.
Исключив делянку с урожаем в 44,6 ц, мы уменьшаем общий урожай всех делянок*
до 1147,2 if, средний урожай в опыте становится равным 37,0 if, поправки на плодородие
повторений опыта тоже меняются. Они становятся равными + 2,3, + 0,6, — 2,3 и —0,7.
При вычислении их можно исходить из среднего урожая для 8 вариантов первых трех
повторений и 7 вариантов четвертого повторения, так как урожаи последнего варианта
несущественно отличаются от урожаев других вариантов. В противном случае пришлось
бы вычислять поправку по повторениям и средний урожай в опыте, исключив все делянки
дефектного варианта.
Произведя заново вычисления исправленных отклонений и их квадратов, находим
сумму квадратов отклонений 2 и2 = 34,11 при 20 степенях свободы. Квадратическое от¬
клонение 6 уменьшилось до 1,32 if, ошибка среднего т для варианта стала 0,66 ц и ошиб¬
ка разности mD средних вариантов + 0,93 if. Но эта ошибка разности средних не примени¬
ма в тех случаях, когда в сравнении участвует четвертый вариант, в котором только три
повторения. Ошибка разности в этом случае будет равна 1,01 ц. Так как для четвертого
1,32
варианта-m = ±~гт= = ±0,76, то, следовательно,
Считая, что существенное различие (веррятное в 95% всех случаев) между разностями
равно 1,73»mD, имеем 1,73'0,93 = 1,6 if и 1,73*1,01 = 1,75, т. е. все прибавки урожаев
в нашем опыте заслуживают внимания, а действие подкормки аммофосом сильнее, чем
другими фосфорными удобрениями.
В большинстве опытов особое значение имеют фоновые, или контрольные, делянки,
с которыми сравниваются урожаи прочих вариантов опыта. Если хотя бы одна из этих де¬
лянок окажется почему-либо поврежденной, то точность наших выводов существенно
уменьшится. Поэтому весьма важно иметь в опыте удвоенное количество фоновых (кон¬
трольных) делянок, т. е. иметь для фона два тождественных варианта. При вычислении
ошибки опыта они участвуют как отдельные самостоятельные варианты.
Но при вычислении прибавки урожаев против фона (контроля) используется уже сред-
ний^урожай для всех фоновых делянок. В этом случае ошибка фонового варианта меньше,
чем ошибка прочих вариантов. Ошибка прибавок урожая против фона равна
mD=± |/~т\+т\ = ±/0,76* + 0,66* = ± 1,01.
вместо mD = ±1,41 m, когда сравниваются другие варианты. Увеличение числа кон-
Определение точности опыта
637
трольных (или фоновых) делянок не только гарантирует сохранность опыта, но и повышает
точность основных выводов.
Приведенный способ обработки при помощи вычисления отклонений имеет преиму¬
щество большой наглядности: исследователь видит, что именно определяет точность его
работы и какие отдельные урожайные данные сомнительны и какие средние величины за¬
служивают поэтому меньшего доверия. Так, средние для вариантов, в основе которых
лежат сильно расходящиеся показания, должны быть оговорены, для таких вариантов
приводятся средние как из всех повторений, так и из меньшего числа повторений, не вы¬
зывающих сомнений. Браковка делянок, дающих сильно отклоняющиеся показания
( > 3 б), специально оговаривается при опубликовании данных опыта.
Приведенный нами способ обработки результатов опыта позволяет использовать и ме¬
тод анализа вариации Фишера. Для этого необходимо вычислить средний квадрат рассея¬
ния (вариации) вариантов опыта. Вычисляем отклонения урожаев по вариантам от сред¬
него урожая в опыте,[возводим их в квадрат, определяем сумму квадратов и умножаем ее
на£число повторений (табл. 18). Получаем, что 2 (Si;2) для вариантов равна 20,51 *4 = 82,04.
Теперь легко можно составить таблицу анализа вариации в опыте по Фишеру (табл. 19),
так как остальные необходимые величины уже были нами вычислены ранее. ЧтобьГуста
новить пригодность опыта для решения поставленного вопроса, мы должны установить
отношение среднего квадрата вариантов к среднему квадрату случайного отклонения;
оно равно 11,72 : 3,37 = 3,48.
Обратимся к данным табл. 20, учтя поправочную таблицу Снедекора. При наличии
21 степени свободы (7 — для вариантов и 3 — для повторений)]для вероятности в 95%
отношение между средними квадратами по таблице Снедекора должно быть] не меньше
2,50; наше отношение равно 3,48. Для вероятности в 99% (табл. 21) находим, что соотноше¬
ние между средними квадратами при указанных степенях свободы, по Снедекору, дол¬
жно быть не меньше 3,65; наше же отношение только немного меньше его — 3,48. Следо¬
вательно, можно считать опыт вполне пригодным для решения вопроса, который в нем
изучался.
Повторения опыта тоже существенно различались по своему плодородию. Отношение
среднего квадрата для повторений к среднему квадрату случайного рассеяния при вероят»
Таблица 18
Вычисление варьирования по вариантам
Вариант
Отклонение от
среднего
Квадрат откло¬
нения
Вариант
Отклонение
от среднего
Квадрат откло¬
нения
1
—3,0
9,00
5
+0,2
0,04
2
+1,0
1,00
6
+2,7
7,29
3
+0,2
0,04
7
-1,2
1,44
4
+1Д
1,21
8
-0,7
0,49
Сумма квад¬
20,51
ратов
Таблица 19
Анализ варьирования в полевом опыте по методу Фишера
Варьирование
Число степе¬
ней свободы
Сумма
квадратов
Средний
квадрат .
Отношение сред¬
них квадратов
Общее
31
257,66
Вариантов
7
82,04
11,72
3,48
Повторений
3
104,88
34,96
11,68
Случайное (остаточное)
21
70,74
3,37
—
638
Л. В. Соколов
Таблица 20
Таблица величии для вероятности в 95% по Снедекору]
Число сте¬
пеней сво¬
боды мень¬
шего сред¬
него квад¬
рата (Па)
Число степеней свободы большего среднего квадрата (пг)
12
20J
40
6
7
8
9
10
И
12
13
14
15
16
18
20
22
24
26
30
5.99
5.99
5,32
5,12
4,97
4.84
4,75
4,67
4,60
4,54
4,49
4,41
4,35
4,30
4,26
4,22
4,17
3.84
5,14
4.74
4,46
4,26
4,10
3.99
3,89
3,81
3.74
3,68
3,63
3,55
3,49
3,44
3,40
3,37
3,32
2.99
4,76
4,35
4,07
3,86
3,71
3.59
3,49
3,41
3,34
3,29
3,24
3,16
3,10
3,05
3,01
2,97
2,92
2.60
4,53
4,12
3,84
3,63
3,48
3.36
3,26
3,18
3,11
3,05
3,01
2,93
2,87
2,82
2,79
2,74
2,69
2.37
4,39
3,97
3,69
3,48
3,33
3.21
3,11
3,03
2,96
2,90
2,85
2,77
2,71
2,66
2,62
2,58
2,53
2.21
4,28
3,87
3,58
3,27
3,22
3,10
3,00
2,92
2,85
2,79
2,74
2,66
2,60
2,55
2,51
3,47
2,42
2,09
4,21
3,79
3,50
3,29
3,14
3.01
2,92
2,84
2,77
2,70
2,66
2,58
2,52
2,47
2,43
2,39
2,34
2.01
4,15
3,73
3.44
3,22
3,07
2,95
2,85
2,77
2,70
2,64
3,59
2,51
2.45
2,40
2,36
2,32
2,27
1,94
4,00
3,57
3.28
3,06
2,91
2,79
2,69
2,61
2,54
2,48
2,42
2,34
2.28
2,23
2,19
2,15
2,08
1,75
3,87
3,44
3,15
2.93
2,77
2,65
2,54
2,46
2,39
2,33
2,28
2,19
2,12
2,07
2,02
1,99
1.93
1,57
3,77
3.34
3,05
2,82
2,67
2,53
2,42
2.34
2,27
2,21
2,16
2,07
1,99
1,93
1,89
1,85
1,79
1,40
3,67
3,23
2,93
2,71
2,54
2,40
2,30
2,21
2,13
2,07
2,01
1,92
1,84
1,78
1,73
1,69
1,62
1,00
Таблица 21
Таблица величин F для вероятности в 99% по Снедекору
Число сте¬
пеней сво
Число степеней свободы большего среднего квадрата (п0
шего сред¬
него квад¬
рата (п*)
1
2
3
4
5
в
7
8
12
20
40
ос
6
13,7
10,9
9,78
9,15
8,75
8,47
8,26
8,10
7,72
7,39
7,14
6,88
7
12,3
9,55
8,45
7,85
7,46
7,19
7,00
6,84
6,47
6,15
5,90
5,65
8
11,3
8,65
7,59
7,01
6,63
6,37
6,19
6,03
5,67
5,36
5,11
4,86
9
10,6
8,02
6,99
6,42
6,06
5,80
5,62
5,47
5,11
4,80
4,56
4,31
10
10,0
7,56
6,55
5,59
5,64
5,39
5,21
5,06
4,71
4,41
4,17
3,91
11
9,65
7,20
6,22
5,67
5,32
5,07
4,88
4,74
4,40
4,10
3,86
3,60
12
9,33
6,33
5,95
5,41
5,06
4,82
4,65
4,50
4,16
3,86
3,61
3,30
13
9,07
6,70
5,74
5,20
4,86
4,62
4,44
4,30
3,96
3,67
3,42
3,16
14
8,86
6,51
5,56
5,03
3,69
4,46
4,28
4,14
3,80
3,51
3,26
3,00
15
8,68
6,46
5,42
4,89
4,56
4,32
4,14
4,00
3,67
3,36
3,12
2,87
16
8,53
6,23
5,29
4,77
4,44
4,20
4,03
3,89
3,55
3,25
3,01
2,75
18
8,28
6,01
5,09
4,57
4,25
4,01
3,85
3,71
3,37
3,07
2,07
2,57
20
8,10
5,85
4,94
4,43
4,10
3,87
3,71
3,56
3,23
2,94
2,69
2,42
22
7,94
5,72
4,82
4,31
3,99
3,76
3,59
3,15
3,12
2,83
2,58
2,31
24
7,82
5,61
4,72
4,22
3,90
3,67
3,50
3,36
3,03
2,74
2,49
2,21
26
7,72
5,53
4 64
4,14
3,82
3,59
3,42
3,29
2,96
2,66
2,41
2,13
30
7,56
5,39
4,51
4,02
3,70
3,47
3,30
3,17
2,84
2,55
2,29
2,01
оо
6,64
4,60
3,78
3,32
3,02
2,80
2,64
2,51
2,18
1,87
1,59
1,00
Определение точности опыта
639
ности в 95% равно 39,96:3,37 = 11,68. По таблице Снедекора для большей вероятности
(в 99%) отношение должно быть в этом случае не менее 5,78; в нашем опыте оно много
больше — 11,68.
В практике опытного дела обработку результатов опыта часто ведут специальные от¬
делы статистики. Это обеспечивает качество обработки. Но исследователь должен сам хо¬
рошо понимать, в силу каких особенностей опыта получилась та или иная степень его
точности, что собственно характеризует полученные показатели и как их следует применять
с различными целями. Это возможно только тогда, когда ему ясен весь ход обработки дан¬
ных его опыта.
ПРИМЕР ВЫЧИСЛЕНИЯ
И ИСПОЛЬЗОВАНИЯ КОЭФФИЦИЕНТОВ КОРРЕЛЯЦИИ
И РЕГРЕССИИ И УСТАНОВЛЕНИЯ ИХ ОШИБОК
На Мироновской сельскохозяйственной опытной станции с различных
делянок многолетних опытов были взяты образцы почвы северного малогу-
мусного мощного чернозема. На этих образцах были поставлены вегетацион¬
ные опыты с внесением в почву радиоактивной метки для определения за¬
пасов в почвах усвояемых для растений фосфатов. Эти же почвенные образцы
были проанализированы по методу Труога для определения содержания
в них легкорастворимых, или подвижных, фосфатов.
В результате получилось два ряда показаний, характеризующих взя¬
тые почвенные образцы: запасов в них усвояемых фосфатов и содержания
в них фосфатов, растворимых по методу Труога. Спрашивается, можно ли
на основании анализов почвы по методу Труога судить о содержании в поч¬
вах усвояемых для растений фосфатов?
Для ответа на этот вопрос надо определить коэффициент корреляции,
который в данном случае показывает степень сопряженности показаний
химического анализа почв и результатов вегетационных опытов, поставлен¬
ных на тех же образцах почвы.
В табл. 22 приведены результаты анализов и вегетационных опытов,
а также порядок вычисления коэффициента корреляции.
Содержание в почве растворимых фосфатов было значительно боль¬
ше, чем усвояемых: в среднем в образцах почвы было найдено 27,9 мг Р20§
по данным химического анализа и только 16,0 мг Р205 по данным вегетаци¬
онных опытов. Но, просматривая оба ряда цифр, видим, что они меняются
соответственно один другому. Для вычисления коэффициента корреляции
определяем для каждого ряда отклонения отдельных показаний от их сред¬
них величин. В большинстве случаев положительное отклонение в одном ряду
совпадает с положительным отклонением в другом ряду. Поэтому и произ¬
ведения отклонений в большинстве случаев — положительные величины.
Произведения отклонений, положительные и отрицательные, складываем
и определяем их сумму 2 год, в данном случае она равна 1162,10. Отклоне¬
ния отдельных показаний от соответственных средних величин 27,9 для хи¬
мических анализов и 16,0 для результатов вегетационных опытов возводим
в квадрат и суммируем квадраты отклонений, в результате получаем: =
= 1481,56 и = 1029,37.
Следовательно, коэффициент корреляции между показаниями химиче¬
ских анализов и результатами вегетационных опытов будет равен:
2V1V2. 1162,10 1162,1 _
Г ~~ V Ivl-Zvl ~ /1481,56*1029,37 ~ ^ 1234>9 “±0*941 .
Пользуясь таблицами Барлоу (1965), легко производим вычисление необ¬
ходимых квадратов и квадратных корней.
640
А. В. Соколов
Таблица 22
Вычисление коэффициента корреляции между содержанием в почве растворимых фосфатов
по методу Труога и запасом усвояемых фосфатов по данным вегетационных опытов
Анализ по Труогу
Вегетационный опыт
м
образца
P.O.,
мг
отклоне¬
ние от
среднего
(Vt)
квадрат откло¬
нения
P.O.,
мг
отклоне¬
ние от
среднего
(*.)
квадрат откло¬
нения (Vs.)
Произведение
отклонений
1
20,7
-7,2
51,84
9,2
—6,8
46,24
+48,96
2
31,6
+3,7
13,69
16,0
0,0
0,00
0,00
3
26,2
-1,7
2,89
15,9
-0,1
0,01
+0,17
4
29,8
+1,9
3,61
17,2
+1,2
1,44
+2,28
5
41,8
+13,9
193,21
27,8
+11,8
139,24
+164,02
6
46,8
+18,9
357,21
30,9
+14,9
222,01
+281,61
7
16,4
—11,5
132,25
5,6
-10,4
108,16
+119,60
8
29,6
+1,7
2,89
15,0
-1,0
1,00
-1,70
9
28,2
+0,3
0,09
23,4
+7,4
54,76
+2,22
10
24,7
-3,2
10,24
13,8
-2,2
4,84
+7,04
И
28,8
+0,9
0,81
14,4
-1,6
2,56
-1,44
12
42,8
+14,9
222,01
30,2
+14,2
201,64
+211,58
13
14,7
-13,2
174,24
10,7
-5,3
28,09
+69,96
14
16,4
—11,5
132,25
5,9
-10,1
102,01
+116,15
15
15,2
-12,7
161,29
5,3
-10,7
114,49
+135,89
16
27,9
0,0
0,00
14,8
-1,2
1,44
0,00
17
32,7
+4,8
23,04
17,2
+1,2
1,44
+5,76
Сумма
Среднее
474,3
Mi =
=27,9
2i**= 1481,56
272,3
Л/а=16,0
1029,37
2i>iv2 =
=+1162,10
Вычисление ошибки коэффициента корреляции, т. е. его стандартно¬
го, или квадратического, отклонения от возможного истинного значения
коэффициента корреляции, производим в данном случае по формуле, пред¬
ложенной Фишером для небольшого количества пар сравниваемых величин
-’-УШ-
где и — количество пар сравнений, в данном случае 17.
Это количество уменьшается на 2, так как для получения коэффициента
корреляции используются две средние величины для двух рядов; следова¬
тельно, число степеней свободы уменьшается на 2. Подставляя получен¬
ные нами величины в формулу Фишера, имеем
Таким образом, в результате вычислений:
г = + 0,941±0,087.
Для оценки степени достоверности или существенности, т. е. определе¬
ния того, в каком количестве возможных случаев корреляция между двумя
рядами будет вообще иметь место, определяем величину t, т. е. отношение
величины коэффициента корреляции к его ошибке:
Определение точности опыта
641
Пользуясь далее табл. 14, видим, что в данном случае наличие корреляции
доказано более чем для 99,75% возможных случаев. При этом пользова¬
нии табл. 14 в данном случае вместо величины п — 1 берется п — 2.
Ошибка коэффициента корреляции, как это видно из его формулы, за¬
висит от двух величин: числа степеней свободы, т. е. количества пар срав¬
нений, уменьшенного на 2, и от величины самого коэффициента корреля¬
ции. Вполне понятно, что наличие соответствия между двумя величинами
тем достовернее, чем более тесно связано изменение величин одного ряда
с изменением показаний другого ряда и чем больше пар сравнений имеет¬
ся для выявления этого соответствия. Для принятого при биологических
исследованиях установления вероятности определений в 95 и 99 % (или
«существенности на уровне 5 и 1%») составлена табл. 23, по которой на осно¬
ве величины г и п — 2 определяют степень достоверности коэффициента кор¬
реляции.
Эту таблицу приводим из руководства Снедекора (1961).
В нашем примере найденный коэффициент корреляции и степень его*
достоверности определенно говорят о том, что между содержанием в поч¬
ве растворимых фосфатов по Труогу и количеством в почве усвояемых для
растений фосфатов имеется корреляция или даже определенная причинная
зависимость, конечно, для конкретных условий проведения опытов.
Поэтому вполне целесообразно и установление коэффициента регрес¬
сии (Ь) количества усвояемых фосфатов в почве по содержанию в ней раст¬
воримых фосфатов:
Таким образом, если содержание в почве растворимых фосфатов ме¬
няется на 1 мг Р2Об, содержание усвояемых фосфатов в ней меняется на
0,784 мг Р206. Следовательно, на основе отдельных показаний для раство¬
римых фосфатов (я) можем вычислить количество в почве усвояемых для
растений фосфатов (у) по уравнению
у — Mt — Ь (х — Мг),
у —16,0 = 0,784 (х -27,9),
У = — 5,9 + 0,784а:.
Предположим, что анализ установил содержание в почве в одном слу¬
чае 41,8 мг Р206 и в другом 32,7 мг Р206. Тогда по приведенной формуле на¬
ходим запасы усвояемых фосфатов в почве; они будут соответственно равны
26,8 мг Р206 и 19,7 мг Р205. В вегетационных опытах (см. табл. 22, пока¬
зания для образцов 5 и 17) были получены соответственно следующие вели¬
чины: 27,8 мг Р206 и 17,2 мг Р206. Таким образом, отклонения величин, вы¬
численных по формуле регрессии, от величин экспериментально найденных
было — 1,0 мг и + 2,5 лег. Такое расхождение между данными, полученными
на основе химического анализа и результатами опытов, нельзя считать
значительным. Конечно, такое расхождение между данными двух хими¬
ческих анализов было бы недопустимым.
Для установления возможной степени достоверности коэффициента ре¬
грессии определим его основное квадратическое отклонение по формуле
где 2d2 — сумма квадратов отклонений экспериментально найденных величин от величин,
вычисленных по формуле регрессии.
642
А. В. Соколов
Таблица 23
Коэффициенты корреляции при вероятности в 95 и 99% всех возможных случаев
Число
степеней
свободы
95%
99%
Число
степеней
свободы
95%
99%
Число
степеней
свободы
95%
99%
1
0,997
1,000
16
0,468
0,590
35
0,325
0,418
2
0,950
0,990
17
0,456
0,575
40
0,304
0,393
3
0,878
0,959
18
0,444
0,561
45
0,288
0,372
4
0,811
0,917
19
0,433
0,549
50
0,273
0,354
5
0,754
0,874
20
0,423
0,537
60
0,250
0,325
6
0,707
0,834
21
0,413
0,526
70
0,232
0,302
7
0,666
0,798
22
0,404
0,515
80
0,217
0,283
8
0,632
0,765
23
0,396
0,505
90
0,205
0,267
9
0,602
0,735
24
0,388
0,496
100
0,195
0,254
10
0,576
0,708
25
0,381
0,487
125
0,174
0,228
11
0,553
0,684
26
0,374
0,478
150
0,159
0,208
12
0,532
0,661
27
0,367
0,470
200
0,138
0,181
13
0,514
0,641
28
0,361
0,463
300
0,113
0,148
14
0,497
0,623
29
0,355
0,456
400
0,098
0,128
15
0,482
0,606
30
0,349
0,449
500
1000
0,088
0,062
0,115
0,081
Таблица 24
Вычисление ошибки — квадратического отклонения коэффициента регрессии
м
образца
Анализ почвы
по методу
Труога (х)
Содержание
усвояемых
фосфатов,
найденное в
опытах, мг
Содержание
усвояемых
фосфатов,
вычисленное по
формуле
регрессии (у)
Отклонение
найденных
величин от
вычисленных (d)
Квадрат
отклонения (d*)
1
20,7
9,2
10,3
-1,1
1,21
2
31,6
16,0
18,9
-2,9
8,41
3
26,2
16,9
14,6
+1,3
1,69
4
29,8
17,2
17,5
+0,3
0,09
5
41,8
27,8
26,9
+0,9
0,81
6
46,8
30,9
30,8
+0,1
0,01
7
16,4
5,6
6,9
-1 ,з
1,69
8
29,6
15,0
17,3
-2,3
5,29
9
28,2
23,4
16,2
+7,2
51,84
10
24,7
13,8
13,5
+0,3
0,09
И
28,8
14,4
16,7
-2,3
5,29
12
42,8
30,2
27,7
+2,5
6,25
13
14,7
10,7
5,6
+5,1
26,01
14
16,4
5,9
6,9
-1 ,о
1,00
15
15,2
5,3
6,0
-0,7
0,49
16
27,9
14,8
16,0
-1,2
1,44
17
32,7
17,2
19,7
-2,5
6,25
Сумма
Среднее
474,3
Mi = 27,9
272,3
М2 = 16,0
—
—
2d2 = 117,86
Определение точности опыта
643
Эта сумма квадратов может быть наёдена двумя путями: во-первых, пу¬
тем непосредственного определения отклонений найденных величин от вы¬
численных для всех образцов почвы, последующего возведения их в квадрат
и нахождения их суммы (см. табл. 24); во-вторых, путем использования
следующей формулы:
Подставляя в эту формулу соответствующие величины из табл. 22, имеем:
1162, Ю2
2d* = 1029,37 - 1481 56 = 1029,37 - 911,52 = 117,85.
Отсюда вычисляем основное квадратическое отклонение для расхожде¬
ний между вычисленными и найденными величинами:
Таким образом, отклонение найденных в опытах величин от вычислен¬
ных по формуле регрессии может быть весьма существенным, хотя ошибка
вычисления коэффициента регрессии и невелика, она равна
Тот же результат получаем и при вычислении ошибки коэффициента
регрессии по табл. 24. В результате округления количества усвояемых фос¬
фатов (вычисляемых по формуле) до 0,1 мг сумма квадратов отклонений
несколько (на 0,21) отличается от вычисленной ранее, но это, конеч¬
но, не отражается на результатах определения ошибки коэффициента ре¬
грессии.
Величина t, т. е. отношение коэффициента регрессии к его ошибке Ыть,
та же, что и для коэффициента корреляции. Для оценки достоверности
коэффициента регрессии или степени его существенности можем использовать
табл. 14 так же, как это ранее делали для коэффициента корреляции. Не¬
смотря на высокую степень точности вычисления коэффициента регрессии
в нашем примере, отдельные показания, полученные в опытах, могут сущест¬
венно отличаться от вычисленных на основе формулы регрессии. Из данных
табл. 24 видно, что расхождения между экспериментально найденными
и вычисленными величинами достигали 5,1 и 7,2 мг Р2Об, т. е. были равны
32 и 45% от средней величины.
В нашем примере тройное квадратическое отклонение
За = 2,80*3 = 8,40 мг Р2О5,
следовательно, получение таких больших расхождений, как 5,1 и 7,2, воз¬
можно.
При анализе опытных данных, когда мы хотим установить соответствие
между двумя рядами показаний, необходимо прежде всего установить коэф¬
фициент корреляции и степень его достоверности.
Вполне возможно, что коэффициент корреляции будет установлен с
большой достоверностью, но величина его будет малой. Тогда не имеет
смысла вычислять коэффициенты регрессии, так как, видимо, оба ряда
Id* = — (2riy2)2.
644
А. В. Соколов
сравниваемых явлений подчиняются разным закономерностям. Если меж¬
ду этими рядами имеется высокая и достоверная корреляция, то вычисле¬
ние коэффициента регрессии поможет нам более полно осветить наблюдаю-
щиеся явления. Например, предположим, что для ряда образцов почвы
были проведены определения содержания в почве подвижных форм калия
двумя методами: более сложным и дорогим стандартным методом и более
простым и дешевым новым методом. Между показаниями этих методов най¬
дена высокая и достоверная корреляция. Тогда, вычисляя коэффициент
регрессии для нового метода, можем, работая новым методом, перечислять
его показания в показания стандартного метода, для которых уже установ¬
лены соответствующие градации отзывчивости почв на внесение калийных
удобрений. Но достаточно ли наличие высокого коэффициента корреляции
и точно определенного коэффициента регрессии, чтобы обоснованно рекомен¬
довать замену стандартного метода новым методом? Предположим, что почвы
по степени обеспеченности их калием разбиты на группы, различающиеся
на 5 мг калия, а основное квадратическое отклонение между найденными
показаниями и вычисленными по формуле регрессии равно 1 мг калия.
Тогда большое количество образцов почв, отнесенное при работе стандартным
методом в одну группу по степени обеспеченности почв калием, попадет
в другую группу, если мы будем работать новым методом, несмотря на вы¬
сокую корреляцию между показаниями этих методов и точно установленный
коэффициент регрессии. Для большинства агрономических явлений поэтому
важно определение основного квадратического отклонения для расхождений
между данными, полученными в опыте и вычисленными по формуле регрессии.
Коэффициент корреляции показывает, насколько варьирование одного
ряда показаний происходит согласованно с изменением показаний для дру¬
гого ряда. Общая же изменчивость показаний для каждого ряда характе¬
ризуется суммой квадратов отклонений отдельных показаний от среднего,
т. е. 2гЯ. Квадрат коэффициента корреляции г2 показывает долю участия
в общем варьировании (2г^) согласованных изменений двух рядов, а (1 — г2)
является остаточной частью варьирования, т. е. долей несогласованных из¬
менений.
Для наглядной характеристики степени корреляции двух рядов часто
необходимо указывать не только величину г, но и г2 или 1 — г2. Это имеет
значение при характеристике корреляции между высотой урожаев и коли¬
чеством применяемых удобрений, при бонитировке почв по данным ана¬
лизов или высоте урожаев и других агрономических вопросов. Например,
при решении вопроса о замене одного метода анализа почв на содержание
подвижных питательшх элементов другим коэффициент корреляции, рав¬
ный + 0,9 (высокий при решении многих биологических вопросов), со¬
ответствует большому количеству несогласованных изменений в показаниях
методов, так как в этом случае 1 — г2 = 1 — 0,81 = 0,19. Следовательно,
количество несогласованных изменений в показаниях составляет 19%.
Литература
Константинов П. Н• Основы сельскохо¬
зяйственного опытного дела. М., Сель¬
хозгиз, 1952.
Снедекор Д. У. Статистические методы в
применении к исследованиям в сельском
хозяйстве и биологии. М., Сельхозгиз.
1961.
Таблицы Барлоу квадратов, кубов, квад¬
ратных корней, кубичных корней и об¬
ратных величин для всех целых чисел от
1 до 15 ООО. М., 1965.
Фосфорные удобрения и питание растений.
Сборник работ НИУИФ. М., Изд-во с.-х.
лит-ры, журналов и плакатов, 1963.
Фишер Р. А. Статистические методы для
исследователей. М., Госстатиздат, 1958.
ПРИЛОЖЕНИЯ
Приложение 1
Атомные веса некоторых элементов
(Данные Международной комиссии по определению атомных весов на 1973 год)
Элемент
Обозначение
Атомный вес
Элемент
Обозначение
Атомный вес
Азот
N
14,0067
Мышьяк
As
74,9216
Алюминий
А1
26,98154
Натрий
Na
22,98977
Барий
Ва
137,34
Никель
Ni
58,70
Бор
В
10,81
Олово
Sn
118,69
Бром
Вг
79,904
Платина
Pt
195,09
Ванадий
V
50,9414
Ртуть
Hg
200,59
Водород
Н
1,0079
Рубидий
Rb
85,4678
Вольфрам
W
183,85
Свинец
Pb
207,2
Железо
Fe
55,847
Селен
Se
78,96
Золото
Au
196,9665
Сера
S
32,06
Иод
J
126,9045
Серебро
Ag
107,868
Калий
К
39,098
Стронций
Sr
87,62
Кальций
Са
40,08
Титан
Ti
47,90
Кислород
О
15,9994
Углерод
С
12,011
Кобальт
Со
58,9332
Уран
U
238,029
Кремний
Si
28,086
Фосфор
P
30,97376
Литий
Li
6,941
Фтор
F
18,99840
Магний
Mg
24,305
Хлор
Cl
35,453
Марганец
Mn
54,9380
Хром
Cr
51,996
Медь
Си
63,546
Цезий
Cs
132,9054
Молибден
Mo
95,94
Цинк
Zn
65,38
Приложение 2
Пересчетные коэффициенты для перевода процентного содержания ионов в миллиграмм*
эквиваленты
|Ион
Эквива¬
лентный
вес
Милли¬
грамм-
эквива¬
лент
Коэффициент А =
1000
ион
Эквива¬
лентный
вес
Милли¬
грамм*
эквива¬
лент
Коэффициент А =
1000
экв. вес
(для пересчета %
в мг-жв)
экв «вес
(для пересчета %
в мг-акв)
H+
1,00
0,001
992,1
С1-
35,46
0,035
28,2
39,10
0,039
25,6
N03-
62,00
0,062
16,1
Na+
23,00
0,023
43,5
NOa-
46,00
0,046
21,7
NH4+
18,04
0,018
55,4
ЗОа2-
48,00
0,048
20,8
Ca2+
20,00
0,020
49,9
НСОз-
61,01
0,061
16,4
Mg«+
12,16
0,012
82,2
СОз2-
30,00
0,030
33,3
Fe2+
27,92
0,028
35,8
РО43-
31,67
0,032
31,6
Fe3+
18,61
0,019
53,7
з2-
16,03
0,016
62,4
Al3+
9,00
0,009
111,2
ЗЮз2-
38,03
0,038
26,3
Mn2+
27,46
0,027
36,4
Пример пересчета с использованием коэффициента А. Содержание поглощенного магния равно 0,04%.
Для перевода в миллиграмм-эквиваленты процентное содержание умножают на коэффициент А ж полу¬
чают 0,04 *82,2 = 3,288. Округленно эту величину принимают равной 3,29 мг-акв.
646
Приложения
Приложение 3
Аналитические множители
Найдено
Ищут
Пересчетный
коэффициент
(множитель)
Найдено
Ищут
Пересчетный
коэффициент
(множитель)
AgCl
Cl
0,247
СО2
CaO
1,274
AgCl
AgNOs
1,185
СО2
СаСОз
2,274
АЬОз
A1
0,529
СО*
Na2COs
2,409
АЬОз
AIPO4
2,393
С02
NaHCOs
1,909
АЬОз
Ab(S04)3
3,356
СаСОз
С
0,119
BaS04
S04
0,411
СаСОз
CO*
0,439
BaS04
SOs
0,343
СаСОз
СОз
0,599
BaS04
s
0,137
СаСОз
Ca
0,400
BaS04
H2S04
0,420
СаСОз
CaS04
1,360
BaS04
Ba
0,588
СаСОз
CaS04-2H*0
1,720
BaS04
CaS04
0,583
CaS04
Ca
0,294
BaS04
CaS04*2H20
0,737
CaS04
CaO
0,412
Гумус
С
0,579
CaS04
CaCOs
0,735
Гумус
CO2
2,123
Fe
Fe*Os
1,430
С
CO2
3,664
Fe
FeO
1,286
С
Гумус
1,724
Fe
FeCl*
2,269
СО*
Гумус
0,471
Fe
FeCls
2,904
СО*
С
0,272
Fe
FeS04-7H*0
4,978
СО*
COs
1,364
Fe
Fe(S04)s
3,580
СО*
Са
0,911
Fe*Os
Fe
0,699
Fe*Os
FeO
0,900
N
NOs
4,427
Fe*Os
FeP04
1 ,889
N
N*06
3,356
Fe*Os
FeCls
2,031
N
NaNOs
6,069
Fe*Os
Fe2(S04)3
2,504
N03
N
0,226
H20
H
0,111
NO3
N2O5
0,871
H2O
0
0,888
N03
NaN03
1,371
KC1
к
0,524
N2O5
N
0,259
KC1
K20
0,632
N2O5
NOs
1,148
K2SO4
к
0,449
N2O5
NaNOs
1,574
K2SO4
к20
0,541
nh4
N
0,776
K2SO4
KC1
0,856
nh4
NOs
3,437
Mg2P207
Mg
0,218
nh4
N2O5
2,994
Mg2P207
MgO
0,362
nh4
NaNOs
4,712
Mg*p207
MgCOs
0,757
NH4
NHs
0,944
MgS04
Mg
0,202
NaCl
Na
0,393
MgS04
MgO
0,335
NaCl
Na*0
0,530
MgS04
MgCOs
0,700
NaCl
Na*COs
0,907
MgCOs
Mg(HCOs)2
1,735
NaCl
NaHCOs
1,437
Mg2p207
p
0,279
Na2S04
Na
0,323
MgiP 2O7
P04
0,853
NaHCOs
Na2C03
0,630
Mg2p207
P205
0,638
Na*COs
NaHCOs
1,585
Mn
MnO
1,291
NaHCOs
Na
0,274
Mn
МпОг
1,582
Na
NaHCOs
3,653
Mn
KMn04
2,876
S1O2
Si
0,467
MnS04
Mn
0,363
SIO2
SiOs
1,266
MnS04
MnO
0,469
Si02
Si04
1,533
MnS04
МпОг
1,575
Si02
ShO?
1,400
Приложения
647
Приложение 4
Количество кпслот и аммиака для приготовления их процентных растворов
(в мл на 1 л раствора) *
Исходное
вещество
Удельный
вес исходно¬
го вещества
при 15° С
Весовой
процент
исходного
вещества
25%-ный
20%-ный
10%-ный
5%-ный
2%-ный
1%-ный
НС1
1,19
37,23
634,8
496,8
236,4
115,2
45,5
22,6
H2SO4
1,84
95,6
167,7
129,9
60,6
29,3
11,5
5,6
HNOs
1,40
65,6
313,0
243,6
115,0
56,0
22,0
10,8
СНзСООН
1,05
99,5
247,8
196,7
97,1
48,2
19,2
9,0
NH4OH
0,91
25,0
1000,0
814,0
422,0
215,4
87,2
43,7
* Е. В. Аринушкина. Руководство по химическому анализу почв, 2-е изд. Изд-во МГУ, 1970.
Приложение 5
Удельный вес и процентное содержание уксусной кислоты при 15° С *
Удельный вес
сн.соон,
%
Удельный вес
сн.соон,
%
Удельный вес
сн,соон,
%
Удельный
вес
СНаСООН,
%
1,0000
1
1,0363
26
1,0623
51
1,0747
76
1,0020
2
1,0375
27
1,0636
52
1,0748
77
1,0037
3
1,0388
28
1,0638
53
1,0748
78
1,0052
4
1,0400
29
1,0646
54
1,0748
79
1,0070
5
1,0412
30
1,0653
55
1,0748
80
1,0083
6
1,0424
31
1,0660
56
1,0747
81
1,0098
7
1,0436
32
1,0666
57
1,0746
82
1,0113
8
1,0447
33
1,0673
58
1,0744
83
1,0127
9
1,0459
34
1,0679
59
1,0742
84
1,0140
10
1,0470
35
1,0685
60
1,0739
85
1,0157
11
1,0481
36
1,0691
61
1,0736
86
1,0171
12
1,0492
37
1,0697
62
1,0731
87
1,0185
13
1,0502
38
1,0702
63
1,0726
88
1,0200
14
1,0513
39
1,0707
64
1,0720
89
1,0214
15
1,0523
40
1,0712
65
1,0713
90
А,0228
16
1,0533
41
1,0717
66
1,0705
91
1,0242
17
1,0543
42
1,0721
67
1,0696
92
1,0256
18
1,0552
43
1,0725
68
1,0686
93
1,0270
19
1,0562
44
1,0729
69
1,0674
94
1,0280
20
1,0571
45
1,0733
70
1,0660
95
1,0298
21
1,0580
46
1,0737
71
1,0644
96
1,0311
22
1,0589
47
1,0740
72
1,0625
97
1,0324
23
1,0598
48
1,0742
73
1,0604
98
1,0337
24
1,0607
49
1,0744
74
1,0580
99
1,0350
25
1,0615
50
1,0746
75
1,0553
100
• Удельный вес уксусной кислоты в пределах 1,0553—1,0748 соответствует двум растворам с различным
процентным содержанием СН,СООН. Пользоваться таблицей в этом случае можпо лишь при условии
проведения пробы на разбавление: если удельный вес уксусной кислоты при разбавлении водой уве¬
личивается — содержание СН*СООН больше 77%, если удельный вес уменьшается — содержание
СН2СООН меньше 77%.
648
Приложения
Приложение 6
Удельный вес и содержание HaS04 водных растворов серной кислоты при 15° С
Удельный
вес
Содержание H*SO«, г
Удельный
вес
Содержание H,SO«, г
Удельный
вес
Содержание H2SO<, г
в 100 г
раствора
в 1000 мл
раствора
в 100 г
раствора
в 1000 мл
раствора
в 100 г
раствора
в 1000 мл
раствора
1,310
40,35
529
1,530
62,53
957
1,750
81,56
1427
1,320
41,50
548
1,540
53,43
977
1,760
82,44
1451
1,330
42,66
567
1,550
64,26
996
1,770
83,51
1478
1,340
43,74
586
1,560
65,20
1017
1,780
84,50
1504
1,350
44,82
605
1,570
66,09
1038
1,790
85,70
1534
1,360
45,88
624
1,580
66,95
1058
1,800
86,92
1564
1,370
46,94
643
1,590
67,83
1078
1,805
87,60
1581
1,380
48,00
662
1,600
68,70
1099
1,810
88,30
1598
1,390
49,06
682
1,610
69,56
1120
1,815
89,16
161&
1,400
50,11
702
1,620
70,42
1141
1,820
90,05
163£
1,410
51,15
721
1,630
71,27
1162
1,822
90,40
1647
1,420
52,15
740
1,640
72,12
1182
1,824
90,80
1656*
1,430
53,11
759
1,650
72,96
1204
1,826
91,25
1666*
1,440
54,07
779
1,660
73,81
1225
1,828
91,70
167&
1,450
55,03
798
1,670
74,66
1246
1,830
92,10
1685-
1,460
55,97
817
1,680
75,50
1268
1,832
92,70
169а
1,470
56,90
837
1,690
76,38
1289
1,834
93,25
1710
1,480
57,83
856
1,700
77,17
1312
1,836
93,80
1722
1,490
58,74
876
1,710
78,04
1334
1,838
94,60
1739
1,500
59,70
896
1,720
78,92*
1357
1,840
95,60
1759
1,510
60,65
916
1,730
79,80
1381
1,520
61,59
936
1,740
80,68
1404
Приложение 7
Удельный вес и содержание HNOs водвых растворов азотной кислоты при 15° С
Удельный
вес
Содержание HNO», г
Удельный
Содержание HNO», г
Удельный
Содержание НЖ)3,г-
в 100 г
раствора
в 1000 мл
раствора
вес
в 100 г
раствора
в 1000 мл
раствора
вес
в 100 г
раствора
в 10000 мл
раствора
1,000
0,10
1
1,140
23,81
266
1,280
44,41
568
1,005
1,00
10
1,145
24,08
276
1,285
45,18
581
1,010
1,90
19
1,150
24,84
286
1,290
45,95
592
1,015
2,80
28
1,155
25,60
296
1,295
46,72
605
1,020
3,70
38
1,160
26,36
306
1,300
47,49
617
1,025
4,60
47
1,165
27,12
316
1,305
48,26
630
1,030
5,50
57
1,170
27,88
326
1,310
49,07
643
1,035
6,38
66
1,175
28,63
336
1,315
49,89
656
1,040
7,26
75
1,180
29,38
347
1,320
50,71
669
1,045
8,13
85
1,185
30,13
357
1,325
51,53
683
1,050
8,99
94
1,190
30,99
367
1,330
52,37
697
1,055
9,84
104
1,195
31,62
378
1,3325
52,80
704
1,060
10,68
113
1,200
32,36
388
1,335
53,22
710
1,065
11,51
123
1,205
33,09
399
1,340
54,07
725
1,070
12,33
132
1,210
33,82
409
1,345
54,93
739
Приложения
649
Приложение 7 (окончание)
Удельный
вес
Содержание HNOs, г
Удельный
вес
Содержание HNO*, г
;ельный
вес
Содержание HNOa, г
в 100 г
раствора
в 1000 мл
раствора
в 100 г
раствора
в 10U0 мл
раствора
УД
в 100 г
раствора
в 1000 мл
раствора
1,075
13,15
141
1,215
34,55
420
1
,350
55,79
753
1,080
13,95
151
1,220
35,28
430
1
,355
56,66
768
1,085
14,74
160
1,225
36 ,03
441
1
,360
57,57
783
1,090
15,53
169
1,230
36,78
452
1
,365
58,48
798
1,095
16,32
179
1,235
37,53
463
1
,370
59.39
814
1,100
17,11
188
1,240
38,29
475
1
,375
60,30
829
1,105
17,89
198
1,245
39,05
486
1
,380
61,27
846
1,110
18,67
207
1,250
39,82
498
1
,3833
61,92
857
1,115
19,45
217
1,255
40,58
509
1
,385
62,24
862
1,120
20,23
227
1,260
41,34
521
1
,390
63,23
879
1,125
21,00
236
1,265
42,10
533
1
,395
64,25
896
1,130
21,77
246
1,270
42,87
544
1
,400
65,30
914
1,135
22,54
256
1,275
43,64
556
—
Приложение 8
Удельный вес и содержание НС1 водных растворов соляной кислоты при 15° С
Удельный
вес
Содержание НС1, г
Удельный
вес
Содержание НС1,г
Удельпый
вес
Содержание НС1, г
в 100 г
раствора
в 1000 мл
раствора
в 100 г
раствора
в 1000 мл
раствора
в 100 г
раствора
в 1000 мл
pacTJopa
1,000
0,16
1,6
1,075
15,16
163
1
,145
28,61
328
1,005
1,15
12
1,080
16.15
174
1
,150
29,57
340
1,010
2,14
22
1,085
17,13
1КГ>
1
,152
29,95
345
1,015
3,12
32
1,090
18,11
197
1
,155
30,55
353
1,020
4,13
42
1,095
19,06
209
1
,160
31,52
36С
1,025
5,15
53
1,100
20,01
220
1
,163
32,10
373
1,030
6,15
63
1,105
20,97
232
1
,165
32,49
379
1,035
7,15
74
1,110
21,92
243
1
,170
33,46
391
1,040
8,16
85
1,115
22,86
255
1
,171
33,65
394
1,045
9,16
96
1,120
23,82
207
1
,175
34,42
404
1,050
10,17
107
1,125
24,78
279
1
,180
35,39
418
1,055
11,18
118
1,130
25,75
291
1
,185
36,31
430
1,060
12,19
129
1,135
26,70
302
1
,190
37,23
443
1,065
13,19
150
1,140
27,66
315
1
,195
38,16
456
1,070
14,17
152
1,1425
28,14
321
1
,200
39,11
469
Приложение 9
Удельный вес и содержание NHs годных растворов аммиака при 15° С
Удельный
вес
Содержание NH», г
Удельный
вес
Содержание NHS, г
Удельный
вес
Содержание НС1, г
в 100 г
раствора
в 1000 мл
раствора
в 100#
раствора
в 1000 мл
раствора
в 100 г
раствора
в 1000 мл
раствора
1,000
0,00
0,0
0,958
10,47
100,2
0,920
21,75
199,9
0,998
0,45
4,5
0,954
11,60
110,6
0,914
23,68
216,2
0,996
0,91
9,1
0,952
12,17
115,8
0,910
24,99
227,2
0,994
1,37
13,6
0,950
12,74
120,9
0,908
25,65
232,7
0,992
1,84
18,2
0,948
13,31
126,1
0,906
26,31
238,2
«0,990
2,31
22,8
0,946
13,88
131,2
0,904
20.98
243,7
0,986
3,30
32,5
0,944
14,46
136 ,4
0,902
27 ,65
249,2
0,982
4,30
42,2
0,942
15,04
141,4
0,900
28,33
254.7
0,980
4,80
47,0
0,940
15,63
146 ,8
0,898
29,01
200,3
0,97't
6,30
61,3
0,938
16,22
152,0
0,894
30,37
271 ,3
0,970
7,31
70,8
0,936
16,82
157 ,3
0.892
31,05
276,7
0,968
7,82
75,6
0,934
17,42
162,6
0,890
31,75
283,3
0,966
8,33
80,4
0,932
18,03
167,9
0,888
32,50
288,3
0,964
8,84
85,1
0,930
18,64
173,2
0,886
33,25
294,3
0,962
9,37
90,1
0,926
19,87
183 ,8
0,884
34,10
301,2
0,960
9,91
95,1
0,922
21,12
194,6
0.882
34,95
308,0
Приложение 10 ^
Состав в свойства солей, употребляющихся для вегетационных опытов
Соль
Химическая формула
Молеку¬
лярный
вес
Питательные
вещества, %*
Растворимость
Гигроскопичность
Реакция
N
P*0.
К,0
СаО
безводного веще¬
ства в 100 в раст¬
вора, г
при Iе
Аммоний азотнокислый
NH4NO3
80,04
35,0
63,9
20
Сильная
Нейтральная
сернокислый
(NH4)aS04
132,16J
21,2
—
—
—
43,0
20
Негигроскопична
То же
фосфорнокислый (трех-
(NH4)sP04.3H20
203,22
20,6
35,0
—
—
19,0
25
То же
Щелочная
замещенный)
фосфорнокислый (дву-
(NH4)aHP04
132,06
21,2
53,8
—
—
40,8
20
Слабая
Слабощелочная
замещенный)
фосфорнокислый (одно-
nh4h2po4
115,03
12Д
61,7
—
—
27,3
20
То же
То же
замещенный)
1
хлористый
nh4ci
53,49
26,1
—
—
—
27,1
20
Очень слабая
Нейтральная
Алюминий сернокислый
AI2(S04)s18H20
666,42
Al = 8,09
26,6
20
Негигроскопична
То же
Борная кислота
H3BO3
61,83
В = 17,49
4,7
20
То же
Кислая
Бура (борнокислый натрий)
NaaB407 10H20
381,38
В = 11,34
2,6
20
»
Щелочная
Железо фосфорнокислое
FeP04 2Ha0
186,86
—
—
—
—
Не растворяется
20
»
Нейтральная
Fe3(P04)a -8HaO
501,60
—
—
—
—
То же
20
»
То же
хлорное
FeCls
162,21
—
—
—
—
47,9
20
Гигроскопична
»
лимоннокислое
FeCeH607 H20
262,60
—
—
—
—
Слабая
20
Слабая
»
Калий азотнокислый
KNOs
101,11
13,8
—
46,6
—
24,1
20
Негигроскопична
»
сернокислый
k2so4
174,27
—
—
54,1
—
10,0
20
То же
»
фосфорнокислый (дву-
KtHP04
174,18
—
40,8
54,1
—
Сильная
20
Гигроскопична
Щелочная
замещенный)
фосфорнокислый (одно-
KHaP04
136,09
—
52,2
34,6
—
18,5
20
Очень слабая
Слабокислая
замещенный)
хлористый
KC1
74,56
—
—
63,2
—
25,5
20
То же
Нейтральная
Кальций азотнокислый
Ca(N0a)2.4H20
236,15
11,8
—
—
23,8
56,4
20
Сильная
То же
сернокислый
CaS04-2Ha0
172,18
—
—
—
32,5
0,202
18
Негиг роскопична
»
фосфорнокислый (трет-
Ca,(PO«),
310,18
—
45,8
—
54,2
0,002
20
То же
»
замещенный)
Приложение 10 (окончание)
Соль
Химическая формула
Молеку¬
лярный
вес
Питательные вещества, %
Растворимость
Гигроскопичность
Реакция
N
P.O.
KfO
GaO
безводного веще¬
ства в 100 г ра¬
створа, г
при 1е
фосфорнокислый (дву**
СаНР04
136,06
52,2
41,2
0,136
25
Негигроскоиична
Нейтральная
замещенный)
СаНР04-2На0
172,09
—
41,2
—
32,5
0,02
24
То же
То же
Кальций фосфорнокислый
Са(Н2Р04)2Н20
252,07
—
56,7
—
22,2
Сильная, гид¬
—
Гигроскопична
Слабокислая
(однозамещенный) *
ролиз
углекислый
СаСОд
100,09
—
—
—
56,1
0,001
25
Негигроскопична
Нейтральная
ОКИСЬ
СаО
56,08
—
—
—
100
0,123
20
Гигроскопична
Щелочная
Магний сернокислый
MgS04-7H20
246,48
Mg =
9,86
26,6
20
Негигроскопична
Нейтральная
Марганец сернокислый
MnS04*5H20
241,08
Mn =
22,79
38,6
20
То же
То же
хлористый
MnCl2.4HaO
197,90
Mn =
27 ,75
42,4
20
Гигроскопична
»
Медь сернокислая
CuS04-5H20
249,68
Cu =:
25,46
17,2
20
Негигроскопична
Слабокислая
хлорная
CuC12*2H20
170,48
Cu = 37,28
42,2
20
Гигроскопична
То же
Натрий азотнокислый
NaNOs
84,99
16,4
-
1 -
—
46,8
20
Слабая
Нейтральная
иодиеты й
NaJ -2HaO
185,93
J =
68,26
64,2
20
Гигроскопична
То же
фосфорнокислый (дву-
Na2HP0412H20
358,14
—
19,8
—
—
7,2
20
То же
Щелочная
замещенный)
фосфорнокислый (одно¬
NaH2P04-2Ha0
156,01
—
45,2
—
—
46,0
20
II егигроскопична
Слабокислая
замещенный)
фтористый
NaF
41,99
F =
45,13
4,2
18
Гигроскопична
Щелочная
хлористый
NaCl
58,44
Na =
39,41
26,4
20
Негигроскопична
Нейтральная
Цинк сернокислый
ZnS04*7H20
287,54
Zn =
22,73
36,6
25
То же
То же
хлористый
ZnCl2
136,28
Zn =
47 ,97
78,6
20
Гигроскопична
»
* Однозамещенный фосфорнокислый кальций растворяется в большом объеме холодной воды (без нагревания) при взбалтывании, после растворения содержи¬
мое доливается до нужного объема.
СОДЕРЖАНИЕ
Предисловие
3. Г. Ильковская, А. С. Коновалова. Определение в почве обменных
катионов, емкости поглощения, гипса, карбонатов, серы, воднорастворимых ве¬
ществ и подвижных соединений
Определение обменных катионов: вытеснение обменных катионов раствором уксуснокислого
аммония (5); трилонометрический метод (7); определение обменного водорода методом Гедрой-
ца (9); определение обменного натрия методом Гедройца (10). Обменные катионы в карбонат¬
ных почвах: метод Шмука (10); метод Тюрина (11); метод Мелиха (12). Обменные кальций и
магний в произвесткованных почвах по Айдиряну (12): объемно-титриметрический метод оп¬
ределения обменного кальция (13); трилонометрический метод определения обменного маг¬
ния в воднс-ацетоновой вытяжке (14). Определение емкости поглощения почв (14): метод Гедрой¬
ца (15); метод Бобко и Аскинази для некарбонатных почв (15) и карбонатных почв (16); объем-
но-титриметрический метод Айдиняна для карбонатных и кислых почв (17); определение ем¬
кости поглощения и обменных катионов в солонцах и солонцеватых почвах вытеснением их
реактивом Пфеффера, в модификации Беляевой (20). Определение гипса (22): экспресс-метод
Айдиняна (23). Определение карбонатов: метод Голубева (25); метод Гейслера — Максимюк (26),
Определение серы по Айдиняну: валовое содержание серы (28); органическая и минеральная
сера (29). Определение воднорастворимых веществ: водные вытяжки из почв, не содержащих
соду (29); водная вытяжка из почв, содержащих соду (35); определение хлор-иона в водных
вытяжках меркуриметрическим методом (36); сводка данных анализа водной вытяжки (37).
Определение подвижных соединений в почве: трилонометрический метод определения подвиж¬
ного (доступного) магния в бедных поглощенными основаниями почвах по Мазаевой (38); ме¬
тод Шахтшабеля для определения доступного растениям магния (39); вытяжка Тамма для оп¬
ределения подвижных железа, алюминия и кремнекислоты (40); метод Мера и Джексона для
определения свободного железа (44). Литература (45).
В. В. Пономарева, Т. А. Плотникова. Определение группового и фрак¬
ционного состава гумуса по схеме И. В. Тюрина, в модификации В. В. Пономаре¬
вой и Т. А. Плотниковой
Подготовка образцов почв (47). Определение фракции воскосмол и битумов (48). Непосред¬
ственная 0,1 н. NaOH вытяжка Jvft 1 (48). Последовательное выделение различных фракций
гумуса: декальцированяс почвы (49); 0,1 н. NaOH вытяжка Ке 2 после декальцирования поч¬
вы (51); 0,02 н. NaOH вытяжка Jvft 3 при нагревании на водяной бане (52). Определение остат¬
ка гумуса нерастворимого при данной схеме анализа (52). Дополнительные методические ре¬
комендации (53). Литература (55).
Н. П. Бельчикова. Определение гумуса почвы по методу И. В. Тюрина
Модификация метода Тюрина со спектрофотометрическим окончанием по Орлову и Гриндель
(62). Литература (62).
В. Б. 3 а м я т и н а. Методы определения азота в почве
Подготовка почв к анализу (64). Определение общего азота (65). Метод Къельдаля (66). Моди¬
фикации метода Къельдаля, включающие азот нитратов и нитритов: вариант с применением
фенолсерной кислоты (72); вариант с применением салициловой кислоты (72). Метод Тюри¬
на (72). Определение соединений азота почвы. Нитраты (74): колориметрический метод с ди¬
сульфофеноловой кислотой (76); колориметрический метод с хромотроповой кислотой (78). Аммо¬
ний (80): колориметрический метод с реактивом Несслера (80); феноловый метод, модификация
Важенина (82). Определение нитратов и аммония в почвенных вытяжках методом дистилля¬
ции (84). Фиксированный аммоний в почве (85): метод Могилевкиной (86); метод Сильвы и
Бремнера (87). Колориметрический метод определения нитритов по Гриссу (88). Определение
азота органических соединений почвы (89): метод Бремнера (90); вариант Шконде и Короле¬
вой (94); определение аминокислотного состава по Турчину (95). Определение подвижного азо¬
та (96): определение легкогидролизуемого азота по Тюрину и Кононовой (97); определение
Содержание
65а
щелочногидролизуемого азота по Корнфилду (98); определение тарификационной способ¬
ности почв методом Кравкова, в модификации Болотиной и Абрамовой (99); определение азо¬
та, минерализуемого из органического вещества почвы методом Бремнера (100). Методика
подготовки почвенных образцов для измерения изотопного состава азота (101). Литерату¬
ра (105).
К. Б. Г и н з б у р г. Методы определения фосфора в почве 106
Определение фосфора в растворе (107). Химические методы (107): весовой метод Лоренца (108);
комплексонометрический (объемный) метод (109). Физико-химические (инструментальные) ме¬
тоды (110): метод Труога — Мейера (111); метод Мерфи — Райли (113); метод экстракции Пон¬
са и Гутри (ИЗ); ванадомолибдатный метод (114). Определение валового фосфора в почве (115):
разложение прокаленной навески почвы смесью фтористоводородной и азотной кислот (115);
осаждение Fe*+ по Уоррену и Пью (116); метод хлорной кислоты Шермана (117); мокрое озо-
ление почвы в смеси серной и хлорной кислот по Гинзбург и др. (118). Определение общего
содержания минеральных и органических фосфатов почвы (118): кислотно-аммиачная экстрак¬
ция. Метод Хейфец (119); кислотно-щелочная экстракция. Метод Мета, вариант Гинэбург (120);
ускоренный метод Стеварда и Оадиса с применением ультразвука (121); метод прокаливания
Сэндерса и Вильямса (121). Определение минеральных форм фосфатов почвы (122): метод Чи-
рикова, вариант Шконде (122); метод Чанга — Джексона, вариант Аскинази, Гинзбург, Ле¬
бедевой (127); метод Гинзбург — Лебедевой (129). Определение подвижных соединений фосфора
в почве (130). Определение запаса подвижных фосфатов (фактор «емкости») (131). Кислотные
вытяжки: метод Кирсанова (132); метод Чирикова (133); метод Труога (134); метод Аррениуса,
вариант Гинзбург (134); метод Аррениуса, вариант ВИУА (135); метод Ониани (136); опреде¬
ление подвижных фосфатов в торфяно-болотных почвах (136). Буферные растворители: метод
Эгнера — Рима, или D — L метод (137); метод Эгнера — Рима — Доминго, или А — L ме¬
тод (139); метод Пича — Инглиша, вариант Хейфец (140); метод Бурриеля — Гернандо (140);
метод Гинзбург [и Артамоновой (141). Щелочные вытяжки (145): метод Мачигина (146); метод
Олсена (149); метод Мещерякова (150). Фторидные вытяжки (150): метод Соколова (151); метод
Брейя и Куртца (151); вариант Миллера и Аксдей (152). Ионообменная хроматография (152):
лабораторный метод с применением анионитов (153); полевой метод с применением аниони¬
тов (154). Определение степени подвижности фосфатов почвы (фактор «интенсивности») (155):
водные вытяжки (156); метод Скофилда (157); метод Карпинского и Замятиной (157); метод
Францесона (158). Метод кривых растворимости фосфатов почвы (158). Определение емкости
поглощения фосфора в почвах по Аскинази и Гинзбург (159). Термодинамика и кинетика поч¬
венного фосфора. Определение фосфатного потенциала в почвах (160): метод Аслинга и Скофил¬
да (162); метод Ульриха (162). Определение фосфатного потенциала в почвенном растворе (163);
Вычисление фосфатного потенциала: способ Ульриха (163); способ Аслиига (168). Определение
минеральных форм фосфатов почвы по константам произведения растворимости (169): вычисле¬
ние констант произведения растворимости фосфорных соединений (170); определение форм
фосфатов почвы по изотермам растворимости (172). Определение потенциальной буферной спо¬
собности почв в отношении фосфора (174). Определение скорости перехода фосфатов из почвы
в раствор: метод Кука (176). Биохимия почвенного фосфора (177). Инозитфосфаты (178): ме¬
тод Андерсона (179). Нуклеиновые кислоты (181): метод Андерсона (182). Фосфолипиды (184):
метод Хенса и Андерсона (184); модификация метода Хенса и Андерсона (186). Определение
органических фосфатов почвы с помощью поливалентных катионов: метод Тинслея (186). Лите¬
ратура (187).
И. Г. В а ж е н и н. Методы определения калия в почве 191
Определение калия в растворах: хлороплатинатный метод (192); кобальтнитритный метод (193);
тетрафенилборатный метод (195); фотометрический метод (197). Определение различных форм
калия в почве (201). Легкоподвижные легкоусвояемые формы калия: водная вытяжка (201);
углекислотная вытяжка (202): хлоркальциевая вытяжка (204). Подвижные формы калия (204).
Извлечение калия солевыми растворами: определение общего количества обменного калия (204):
метод Масловой (205); метод Мачигина (206). Извлечение калия слабыми кислотами: метод Кир¬
санова (206); метод Чирикова (207); метод Ониани (207). Извлечение калия буферными рас¬
творами (208): метод Эгнера — Рима (208); метод Шахтшабеля (209). Необменные (экстенсив¬
нообменные и кислотнорастворимые) формы калия (209): метод Магницкого и Малкова (210);
метод Пчелкина (211); извлечение калия по Гедройцу (212). Дифференцированное вытеснение
калия: солянокислотный метод Антипова-Каратаева (213). Определение валового содержания
калия в почве: спекание почвы по Смиту (215); разложение плавиковой и серной кислотами
(216). Литература (217).
О. П. М е д в е д е в а. Определение калийного потенциала и потенциальной бу¬
ферной способности почв в отношении калия 219
Определение калийного потенциала (219): водная вытяжка (221); вытяжка раствором хлори¬
стого кальция (223). Определение потенциальной буферной способности почв в отношении
калия (223). Определение РВСК по Беккету (225). Литература (227).
654
Содержание
Д. Л. А с к и н а з и. Методы определения нуждаемости почв в известковании . 228
Определение кислотности, подвижного алюминия и поглощенных оснований почвы (231).
Определение pH солевой вытяжки из почвы (231). Определение обменной кислотности почвы (232)0
Определение гидролитической кислотности почвы по Каппену (232). Определение суммы по¬
глощенных оснований по ускоренному методу Каппена — Гильковица (233).$ Определение
буфсрности почвы по Ремезову (234). Определение подвижного алюминия в почве по Соколо¬
ву (236). Использование] показателей кислотности для определения нуждаемости почв в изве¬
стковании (238). Литература (243).
Д. С. О р л о в. Методы определения pH и окислительно-восстановительного
потенциала почв 245
Активность водородных ионов и шкала pH (245). Электроды для определения pH (252). Окис-
лительно-восстановительный потенциал почв (258). Электроды для измерения ОВП (262).
Электроды сравнения (265). Потенциометрические измерения (269). Способы определения pH
почв и почвенных суспензий (279). Способы измерения окислительно-восстановительного по¬
тенциала почв (283). Колориметрические определения pH и ОВП (285). Литература (288).
А. М. Александрова. Определение активности ионов калия в почвах . . 290
Литература 295
Д. В. Федоровский. Определение водных и физических свойств почвы при
проведении полевых и вегетационных опытов 296
Наблюдения за влажностью почвы в полевых опытах (296). Определение влажности высушива¬
нием (298). Нейтронный влагомер и гамма-плотномер (299). Полевое определение наименьшей
влагоемкости почвы (300). Определение уровня грунтовых вод (301). Определение водопро¬
ницаемости почвы (302). Вычисление запасов воды в почве (303). Графическое изображение
динамики изменений влажности почвы (305). Исследование некоторых физических свойств
почвы. Объемный вес почвы (305). Удельный вес почвы (307). Вычисление общей порозности
(скважности) и воздухообеспеченности почвы (309). Структурность почвы (309): метод Савви-
нова (311). Механический состав почвы (312): быстрый метод определения механического сос¬
тава (312); определение механического состава почв по Качинскому (315); расчет результатов
механического анализа (318). Микроагрегатный состав почвы (321). Исследование водных
свойств почвы при проведении вегетационных опытов (322). Полная влагоемкость почвы (322).
Капиллярная влагоемкость почвы (323). Наименьшая влагоемьость почвы (323). Определение
недоступной растению влаги в почве (324). Определение влажности завядания методом про¬
ростков по Долгову (325). Определение влажности завядания вегетационным методом с хорошо
развитыми растениями (326). Определение недоступной растениям воды в почве без завядания
растений. Метод Соколова и Фздоровского (327). Вычисление запасов доступной'растениям
воды в вегетационном опыте (328). Литература (329).
Б. Н. М а к а р о в. Методы определения состава почвенного воздуха, интенсив¬
ности дыхания почвы и газообразных потерь азота почвы и удобрений 331
Определение состава почвенного воздуха (332). Определение интенсивности выделения СО* из
почвы (дахания почвы) (336). Определение газообразных потерь азота почвы и удобрений (339):
определение аммиака и двуокиси азота, выделяющихся из почвы (340). Метод газовой хромато¬
графии при изучении газового режима почв (341). Литература (343).
Д. Н. И в а н о в, Л. А. Л е р н е р. Спектральные методы анализа 344
Аналитические помехи (346). Эталонирование (348). Потребности агрохимии в определении
химических элементов (349). Спектрографический метод (350): аппаратура (350); методы фотомет-
рировапия и анализа (352); характеристика метода (354); применение метода к анализу почв
и растений (354). Эмиссионный пламенно-фотометрический метод (365): пламенно-фотометри-
ческие установки (365); методы фотометрирования и анализа (365); характеристика метода (318);
применение метода к анализу почв и растений (368). Атомно-абсорбционный метод: аппара¬
тура (372); методы фотометрирования] и анализа (373); характеристика метода (374); приме¬
нение метода к анализу почв и растений (376). Литература (382).
К. В. Веригина. Методы определения в почве меди, цинка, кобальта и бора 384
Взятие почвенных образцов и подготовка их к анализу (384). Определение валового содержа¬
ния меди, цинка и кобальта из одной навески по Веригиной после разложения почвы кисло¬
тами (384). Определение меди (385). Определение цинка и кобальта (386). Определение по¬
движных форм микроэлементов (391): уксусно-аммонийная вытяжка по Крупскому и Алексан¬
дровой (392); вытяжка буферным раствором уксуснокислого натрия по Кругловой (393); вы¬
тяжка по Эгнеру — Риму — Доминго (А — L метод) (394). Определение подвижной меди: оп¬
ределение меди по Ринькису в вытяжке 1,0 И.НС1 дптизоновым методом (394); определение
меди по Ринькису, в модификации Почвенного института им. В. В. Докучаева (396); опреде¬
ление меди по Шарреру и Шаумлеффелю в вытяжке по Вестерхоффу (397); определениеЗ меди
Содержание
655
по Хенриксену (398); определение меди по Шарреру и Шаумлеффелю в вытяжке по Кругло¬
вой (398); определение меди по Боратынскому в вытяжке Эгнера — Рима — Доминго (399).
Определение подвижного цинка: определение цинка!по'Ринькису в вытяжке КС1 (400); опреде¬
ление цинка по Ринькису в вытяжке уксуснокислым аммонием (401); определение^ цинка по
Ринькису, в модификации Почвенного института им. В. В. Докучаева в ЕС1 вытяжке (401); опре¬
деление цинка по Крупскому и Александровой в вытяжке уксуснокислым аммонием (402); опре¬
деление цинка по Кругловой в вытяжке уксуснокислым натрием (403); определение цинка по
Боратынскому в вытяжке Эгнера — Рима — Доминго (404). Определение подвижного кобаль¬
та: определение кобальта по Ринькису, в модификации хт°чвенного института им. В. В. Доку¬
чаева (405); определение кобальта по Крупскому и Александровой в вытяжке уксуснокислым
аммонием (406); определение кобальта по Кругловой в вытяжке уксуснокислым натрием (408);
определение кобальта по Боратынскому в вытяжке Эгнера — Рима — Доминго (409); опре¬
деление кобальта с 2-(2-пиридилазо)-5-диэтилметааминофенолом (ПААФ) (410). Определение
«ялового содержания бора: определение бора по Элыплегелю с 1,1-диантримидом (411); опре¬
деление бора ио Джексону с куркумином (412); определение бора по Биру с хиналиэарином
(413). Определение подвижного бора (414): определение воднорастворимого бора по Риньки¬
су (414); определение воднорастворимого бора по Никиткиной с кармином (416); определение
бора по Крупскому и Александровой в вытяжке буферным раствором уксуснокислого аммо¬
ния (417); определение бора по Боратынскому с 1,1-диантримидом в вытяжке Эгнера — Рима —
Доминго (418). Литература (418).
Ю. И. Д о б р и ц к а я. Определение молибдена, ванадия, марганца и иода в поч¬
вах 420
Разложение почвы фтористоводородной и серной кислотами (420). Разложение почвы серной,
азотной и хлорной кислотами (420). Молибден (421). Определение валового содержания мо¬
либдена по Добрицкой (422): экстракция молибдена нормальным бутиловым спиртом (422);
вкстракция молибдена смесью изоамилового спирта и четырех хлористого углерода — 1:1 (424);
определение молибдена на спектр око лориметре «Спекол» с приставкой для измерения экстинк-
ции в пробирках (424). Ускоренный метод определения валового содержания молибдена по
Ринькису (425)» Определение подвижных форм молибдена (426) фотоколориметричсское
определение молибдена по Григгу, в модификации Добрицкой (426), визуальное определение
молибдена по Ринькису (427); определение молибдена по Бергманну (428); дитиоловый
метод определения молибдена по Шелу, вариант Добрицкой (428); дитиоловый метод
определения молибдена по Стентону, в модификации Самохвалова и Целиковой (430).
Ванадий (431). Определение валового содержания ванадия по Добрицкой (431). Определение
подвижного ванадия по Добрицкой (432). Марганец (433). Определение валового содержания
марганца: колориметрическое определение марганца по Добрицкой (433); визуальное
определение марганца по Ринькису? (43 5). Определение подвижных ферм марганца (436):
визуальное определение марганца п о ринькису (436); фотоколориметрическое определение
марганца по Добрицкой (436); определение сульфитнорастворимого марганца по Шахтшабе-
лю (437); определение легковосстанавливаемого марганца по Шахтшабелю (437); определение
подвижного марганца по Крупскому и Александровой в ацетатно-аммонийной вытяжке
с pH 4,8 (438); определение подвижного марганца по Кругловой в карбонатных почвах в аце¬
татно-натриевой вытяжке с pH 3.5 (438); определение подвижного марганца формальдокси-
матным методом по Самохвалову (439). Иод. Определение валового содержания иода (440):
объемное определение иода по Драгомировой, в модификации Глущенко и Миненковой*(440);
колориметрическое визуальное определение иода по Каменеву (442); фотоколориметрическое
определение иода по Лапину, Ришу и Бен-Утяевой, вариант Покатилова (444); кинетический
роданидно-нитритный метод определения иода но Проскуряковой (445); ускоренный вариант
кинетического родатщно-нитритного метода определения иода по Проскуряковой, Швейки-
ной и Никитиной (448). Литература (449).
A. В. Соколов, И. П. Сердобольский, М. Г. С и в я г и н а. Мето¬
дика применения меченых атомов при агрохимических исследованиях 450
Счетчики радиоактивных излучений (456): электрические детекторы (457); сцинтилляционные
детекторы и датчики (460). Радиометрические приборы (462). Активность и единицы ее изме¬
рения (468). Метод меченых атомов (469). Методика проведения вегетационных] опытов с Р**
(474): метод избирательного поглощения фосф атов (480). Методика определения запасов в поч¬
ве растворимых и усвояемых фосфатов пр омощи Р32 (481). Литература (485).
B. В. Церлинг. Диагностика питания растений по их химическому анализу 487
Взятие проб растений для анализа (489). Подготовка прсб к анализу (491). Химический ана¬
лиз растений. Валовой анализ (492)» Определение растворимых форм питательных елемен-
тов (494): анализ пасоки по Сабинину (494): анализ вытяжек из растений по Магницкому (496);
анализ соков, вкстрагированных из проводящих тканей по Рутченко (498); быстрые анализы па
656
Содержание
срезах растений или в выжатом соке из них (499); ускоренный хроматографический метод опре¬
деления содержания свободных аминокислот и амидов в тканях растений, в модификации
Церлинг к Зинкевич (512). Составление диагностического заключения (516). Литература (524).
С. В. Щ е р б а, Ф. А. Ю д и н. Методика полевого опыта с удобрениями • • 526
Основные понятия, встречающиеся в методике полевого опыта (528). Основные методические
требования к качеству полевого опыта (528). Методика и техника полевого опыта (533). Выбор
участка (533). Подготовка участка (536). Размещение опыта на участке (540). Закладка опы¬
та (554). Программа полевого опыта (560). Уход за растениями и сопутствующие наблюдения
в течение вегетационного периода (561): метеорологические наблюдения (562); учет засорен¬
ности (563); фитопатологические и энтомологические наблюдения (563); наблюдения над зи¬
мующими культурами и учет зимостойкости культур и сортов (564); фенологические наблю¬
дения (565). Учет результатов опыта (567). Некоторые особенности проведения опытов в усло¬
виях производства (574). Некоторые особенности постановки опытов на орошаемых зем¬
лях (575). Характеристика качества урожая (576). Агрохимические и агрофизические иссле¬
дования в полевых опытах с удобрениями (578). Литература (584).
А. В. С о к о л о в. Вегетационный метод 585
Техника вегетационного опыта (587). Методика опытов с низкой влажностью почвы (595). Опы¬
ты с почвенной изоляцией удобрений (596). Применение вегетационного метода для определе¬
ния .содержания в почве усвояемых для растений питательных веществ (598): метод пророст¬
ков Нейбауэра — Шнейдера (599); метод Митчерлиха (603). Литература (604).
А. В. С о к о л о в. Построение схем опытов с удобрениями , • 605
Определение действия Различных видов удобрений (606). Изучение форм односторонних удоб¬
рений (606). Схема опытов со сложными удобрениями (608). Установление оптимальных доз
удобрений (608). Сравнение действия навоза и минеральных удобрений (609). Изучение тех¬
ники применения удобрений (610). Схемы комплексных опытов (611). Схемы синтетических
опытов (612). Схемы многофакторных опытов (612).
А. В. С о к о л о в. Определение точности опыта 614
Возможные ошибки и основные показатели точности (614). Обработка данных опыта (619).
Оценка точности опыта (627). Пример вычисления и использования коэффициентов корреля¬
ции и регрессия и установления их ошибок (638). Литература (644).
Приложения 645
АГРОХИМИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ ПОЧВ
Утверждено к печати Почвенным институтом им» В. В. Докучаева
Редактор издательства А. А. Фролова
Художник H. В. Илларионова. Художественный редактор С. А. Лптвак
Технические редакторы Л. И. Куприянова, Л. Н. Золотухина
Сдано в набор 12/VI 1075 г. Подписано к печати 10/XI-1975 г. Формат 70xl08Vu*
Бумага типографская *Ye 1. Уел. печ. л. 57/». Уч.-изд. л. 60,Р. Тираж 65С0
T-1C3S5 Тип. зак. 2497 Цена 4 р. 62 к.
Издательство «Наука». 103717 ГСП, Москва, К-62, Подсосенский пер., 21
2-я типография издательства «Наука». 121099, Москва, Г-9^1, Шубинский пер., 10