































































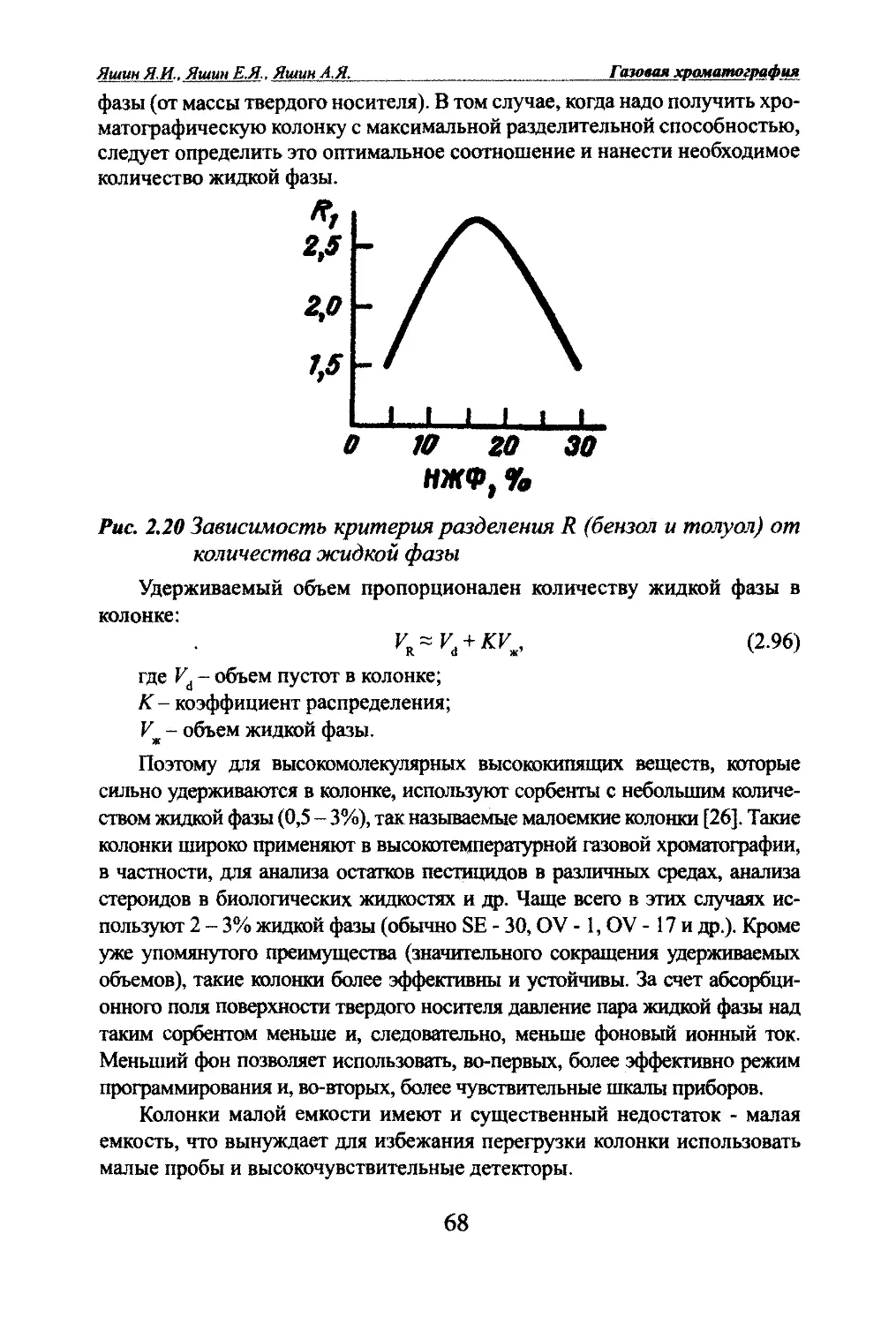
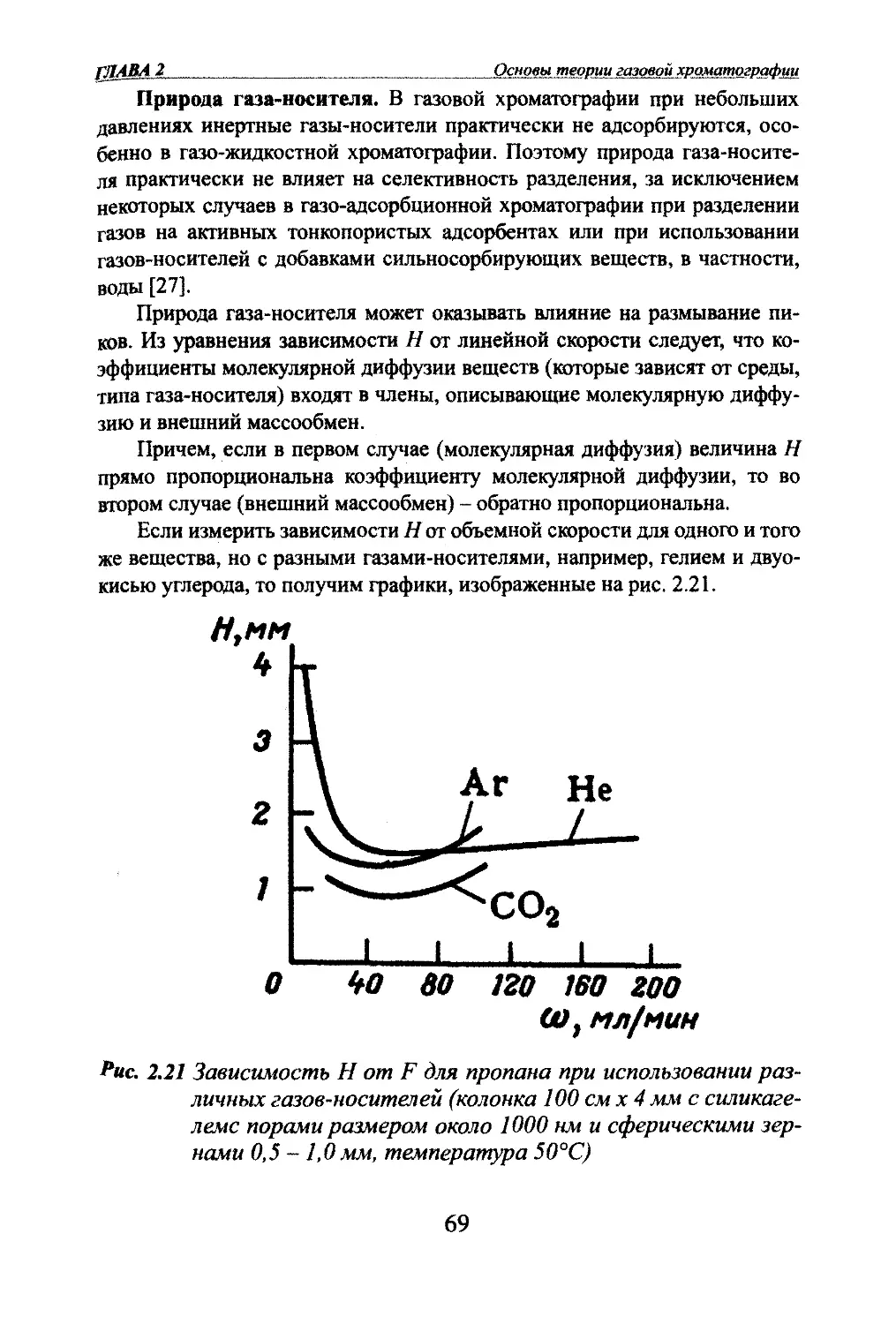










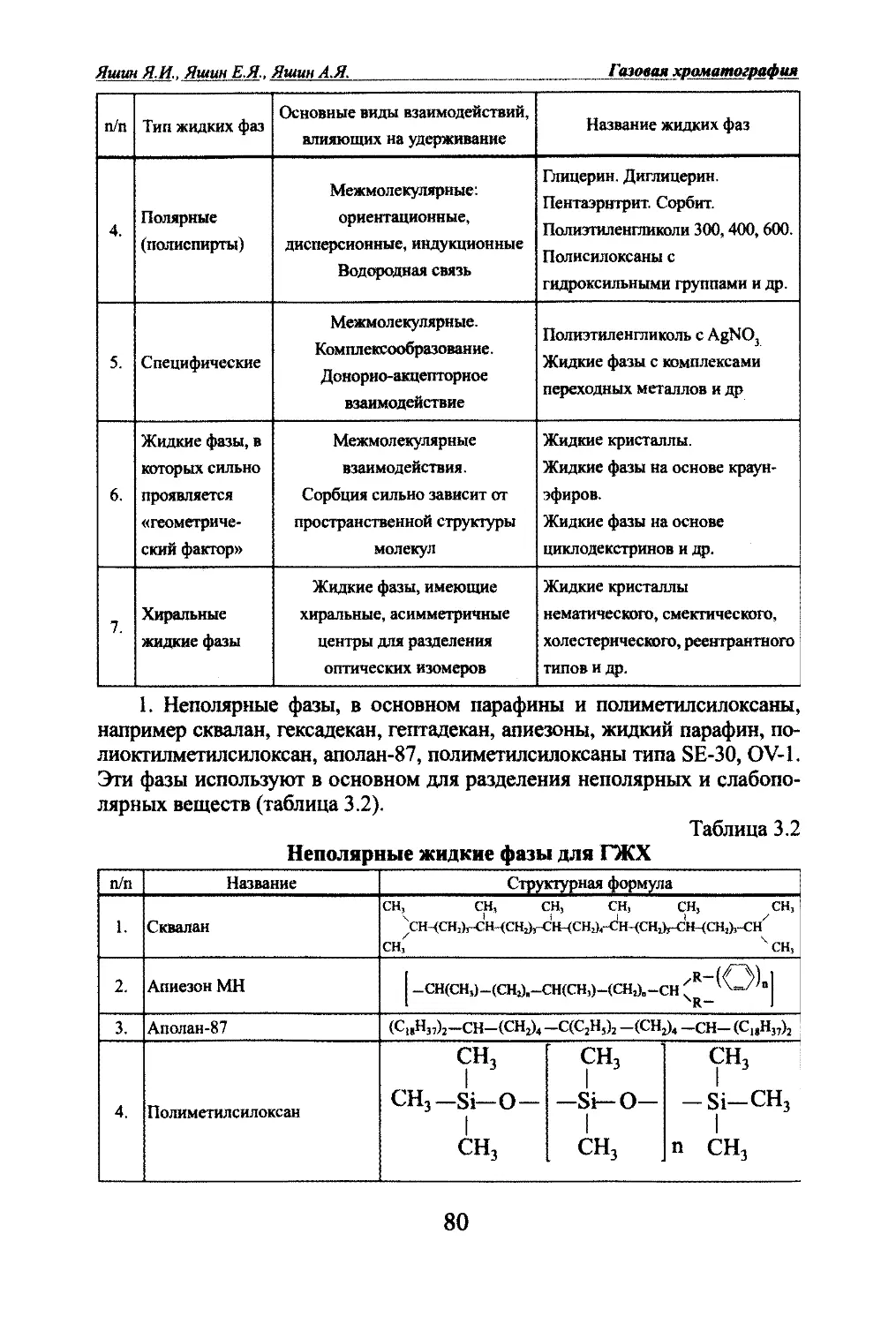


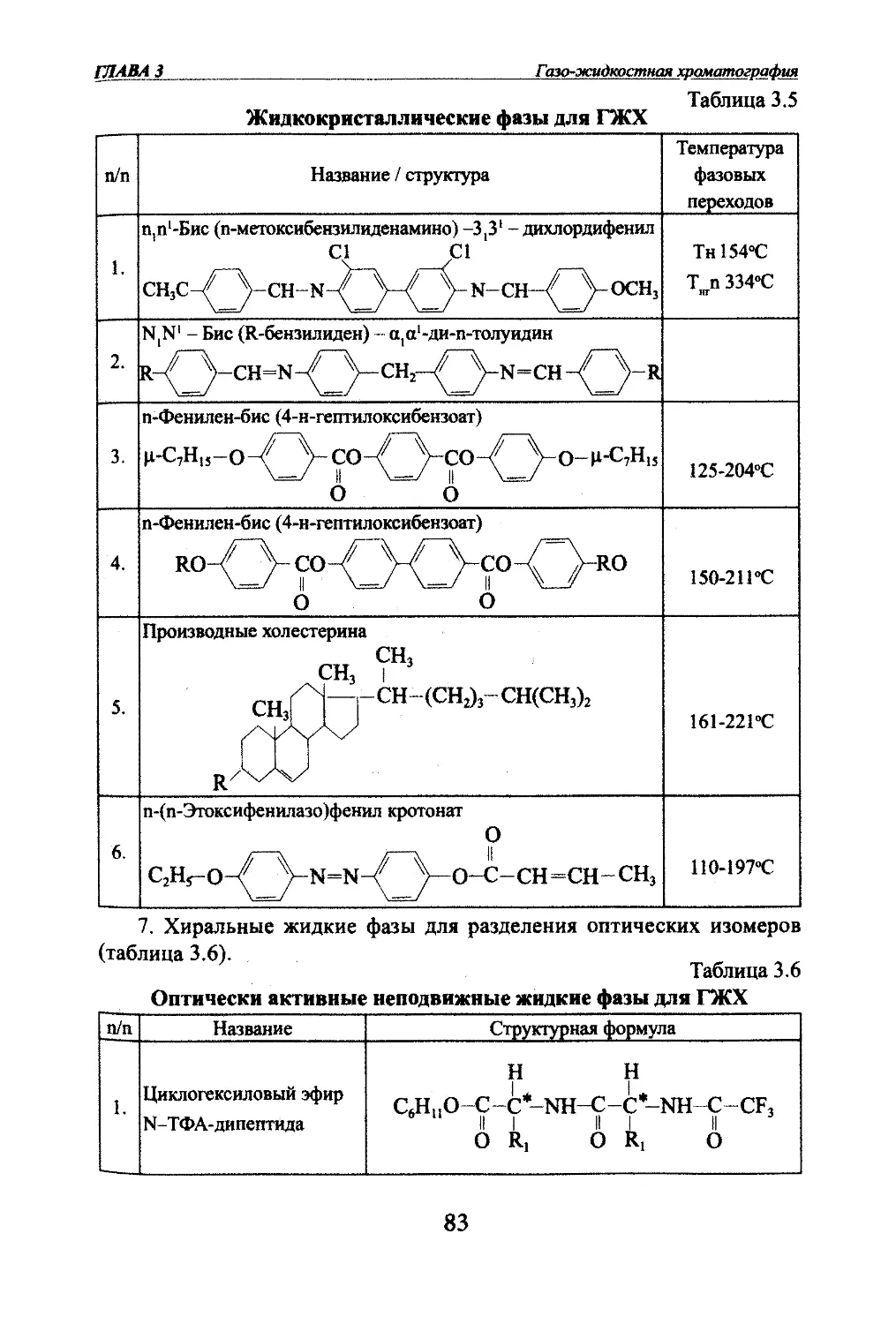



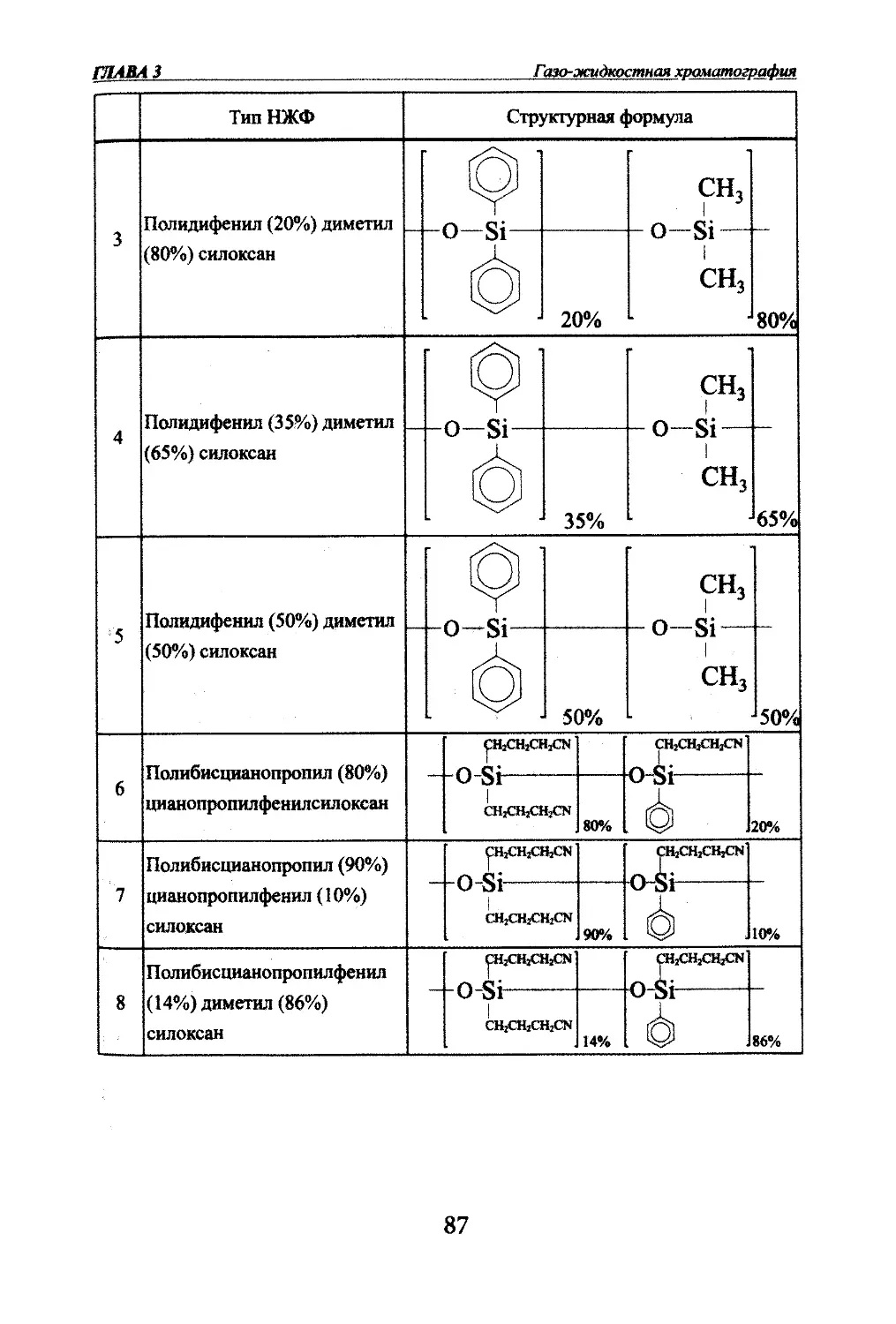





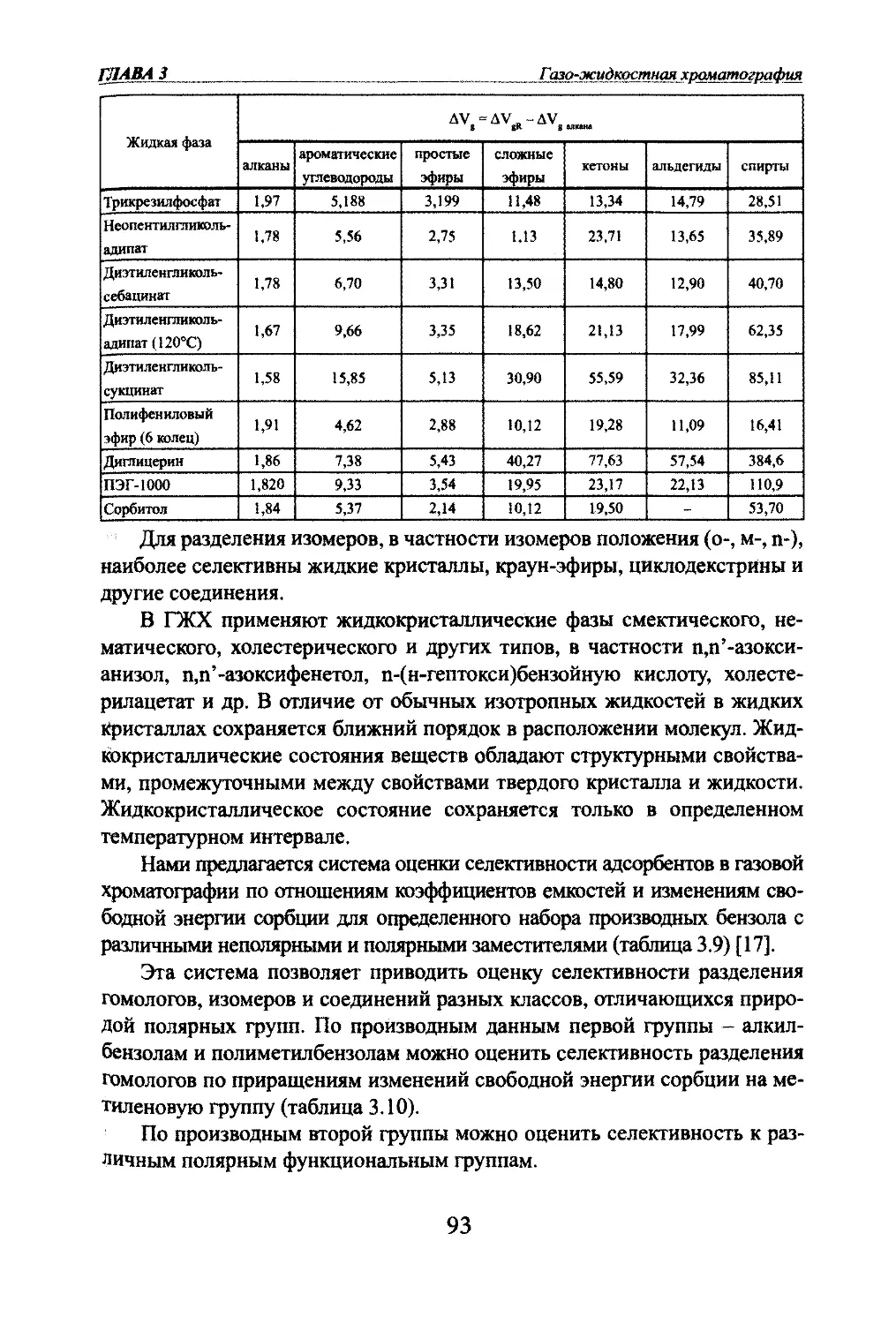








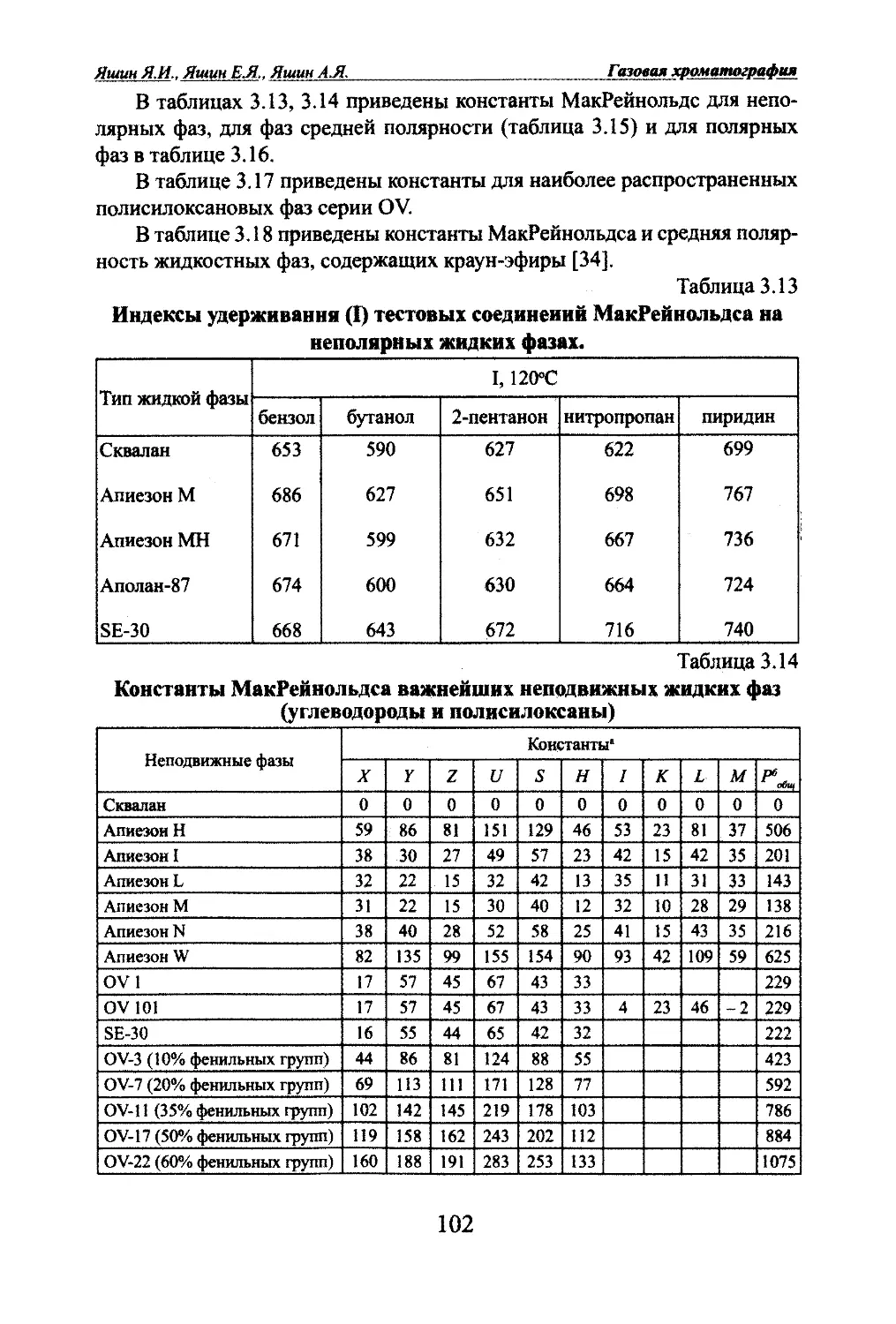

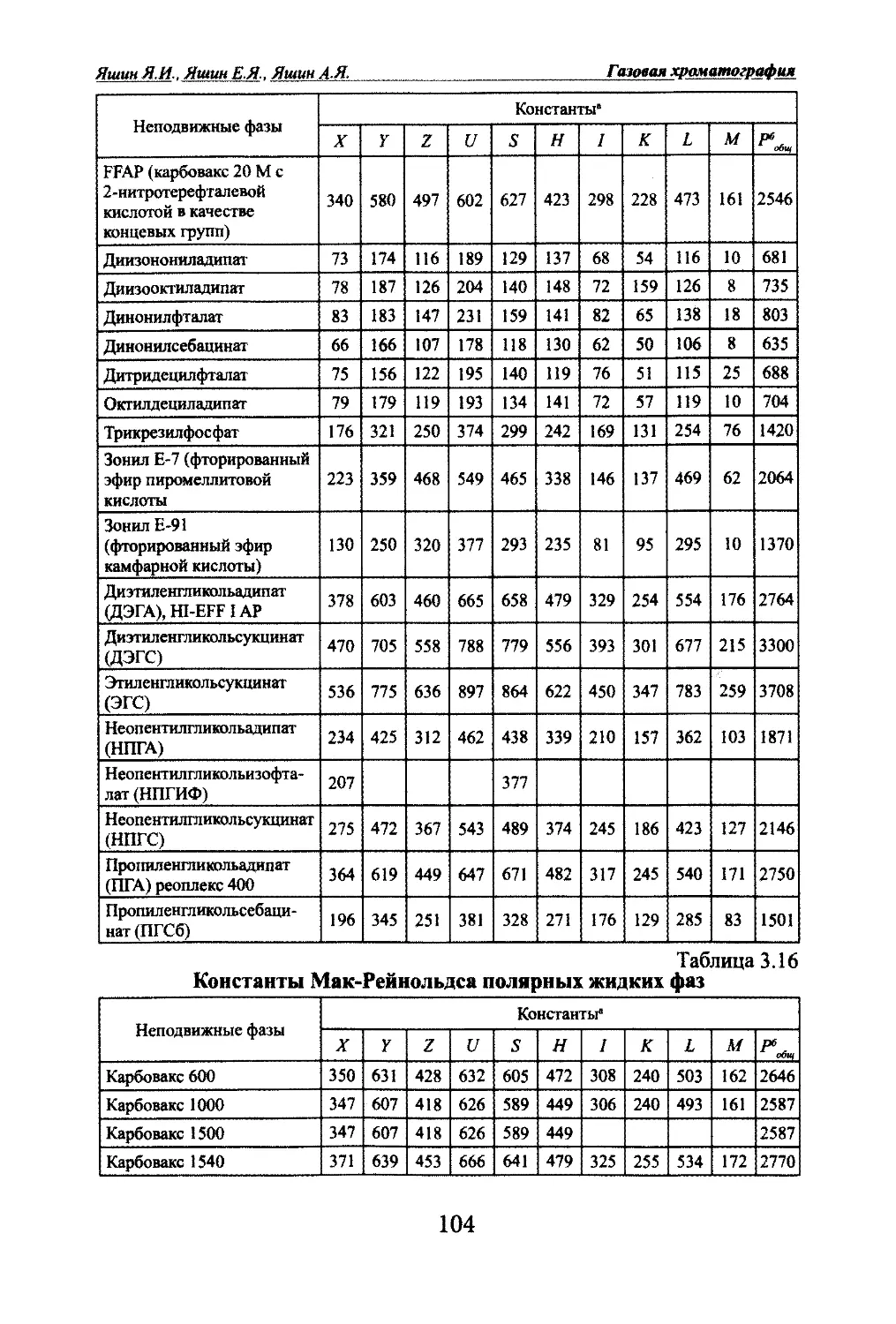

































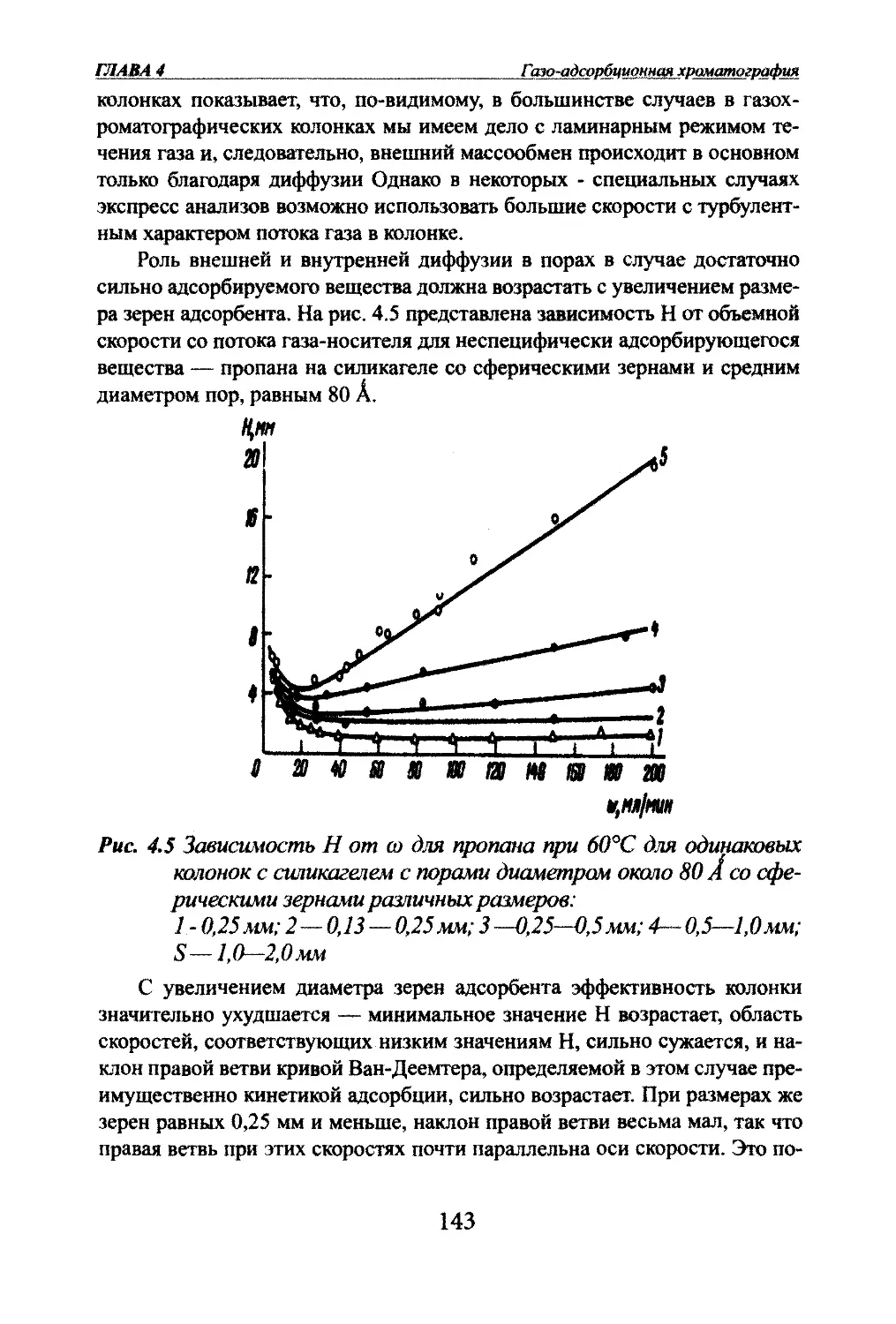

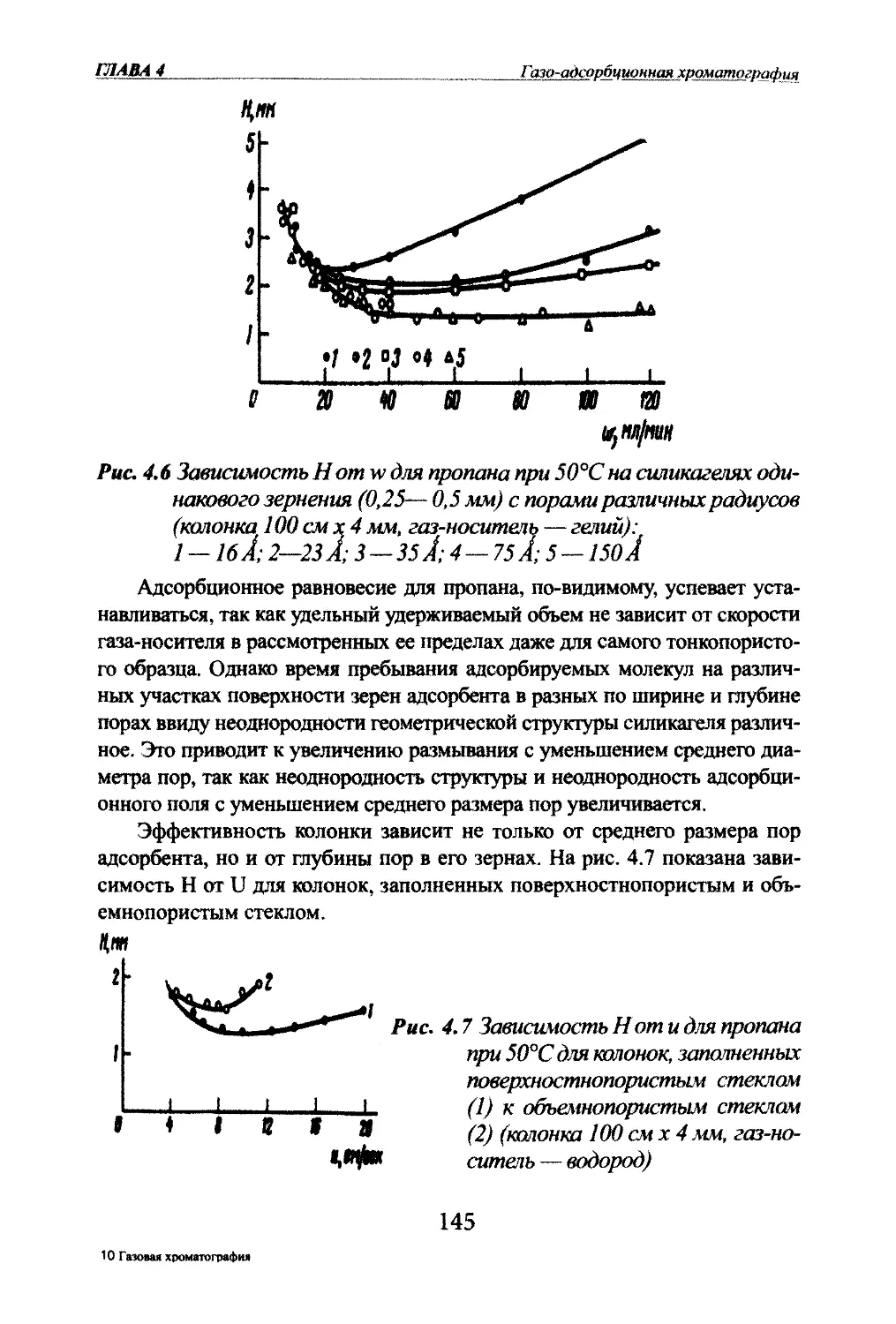





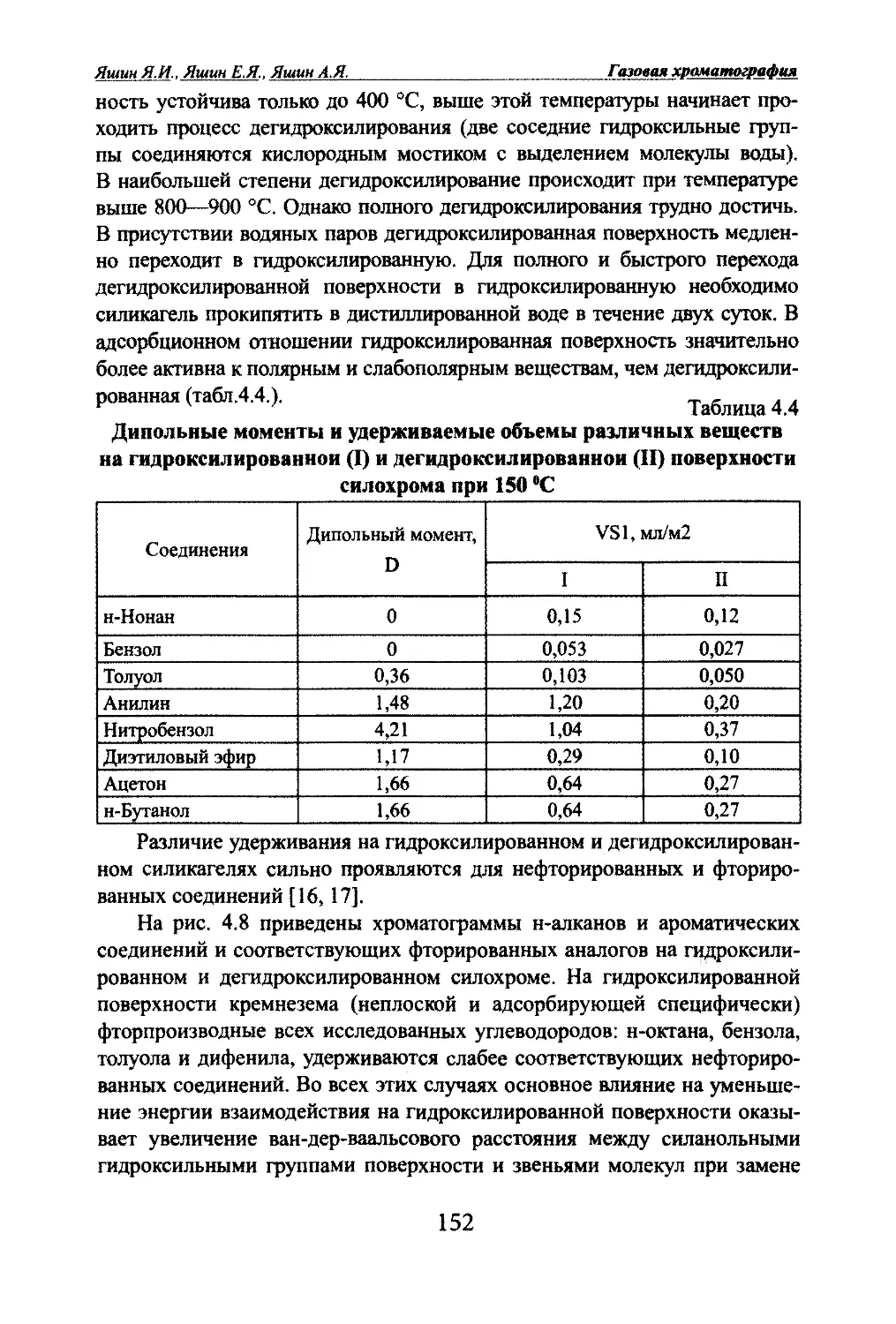



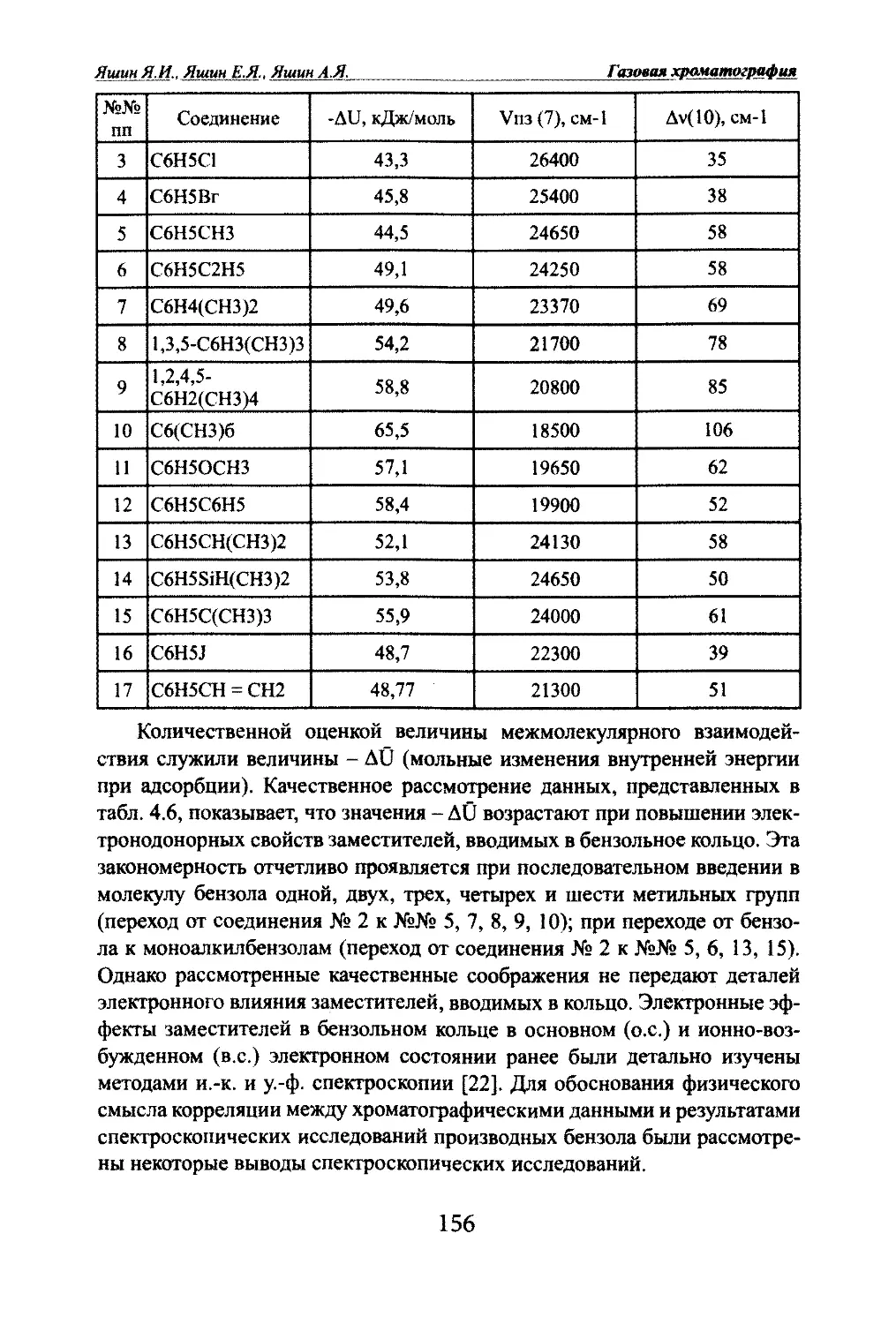

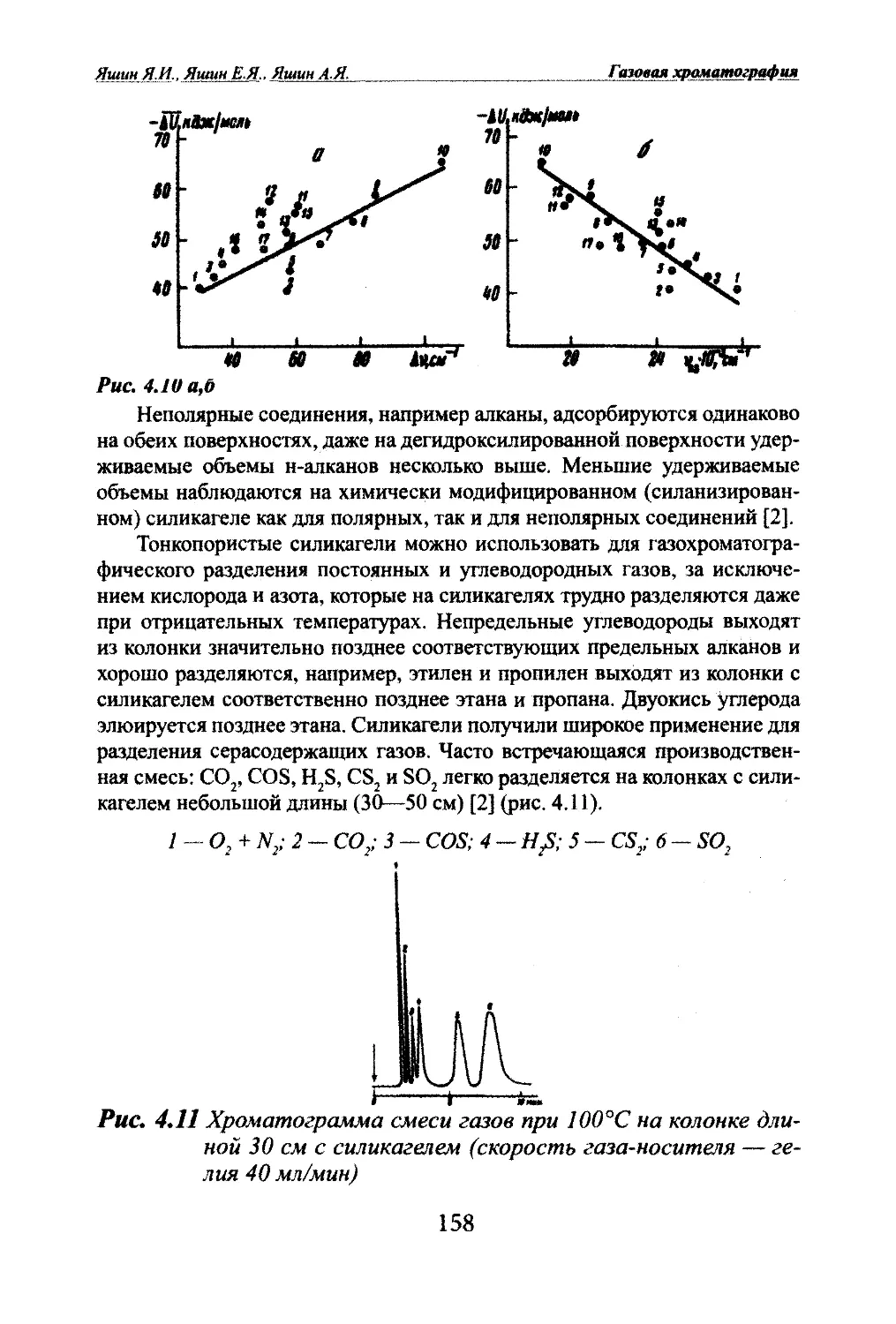
















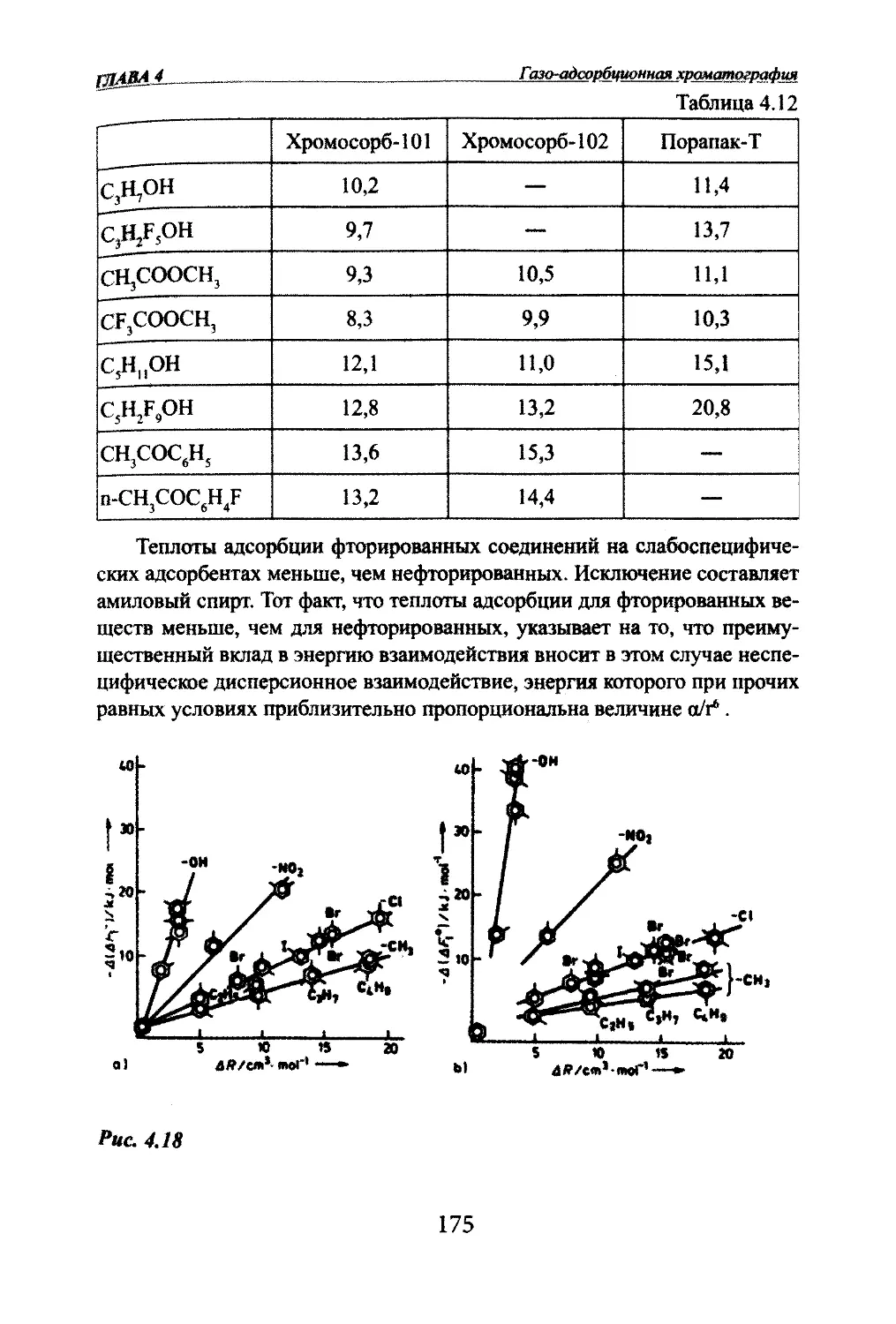
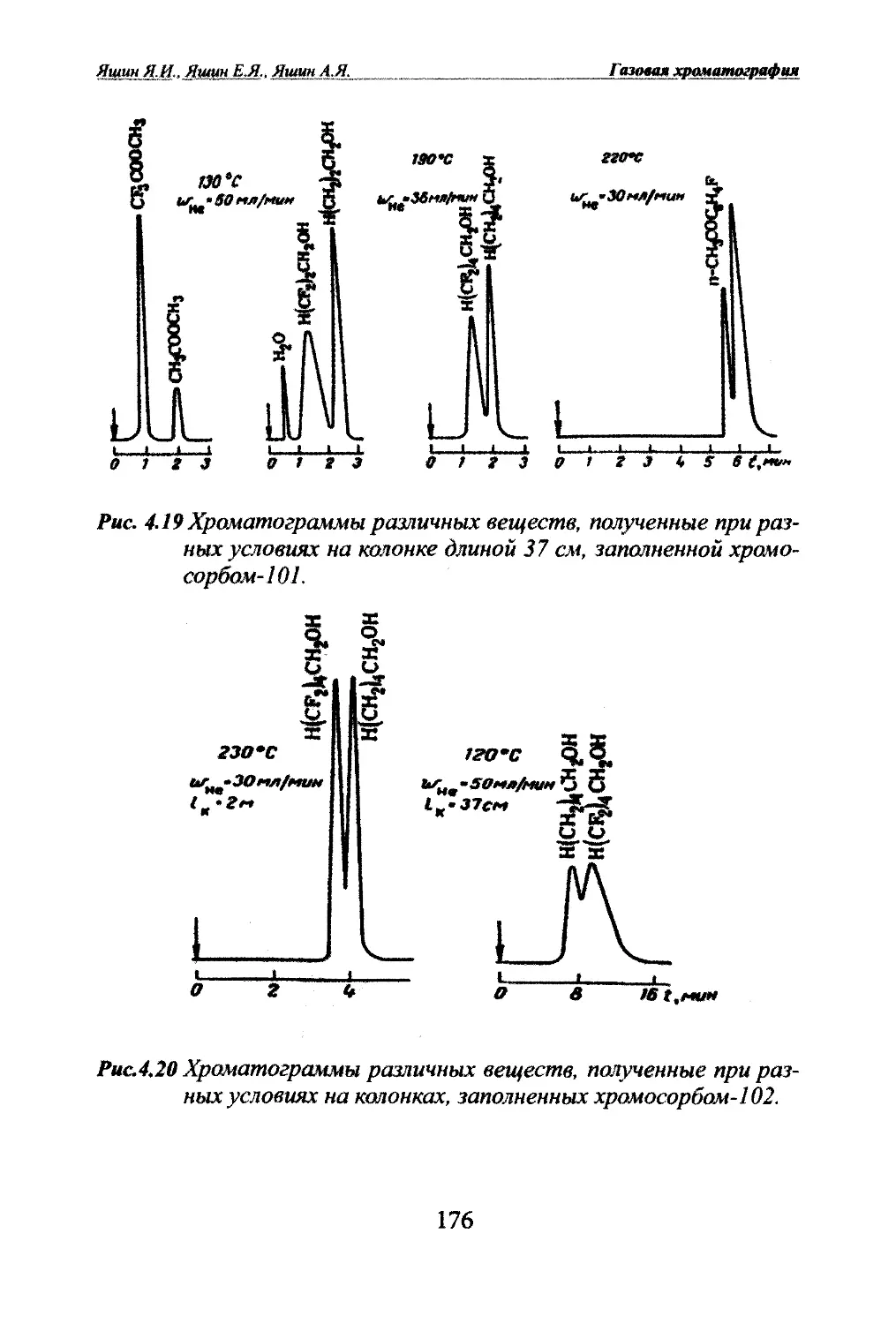

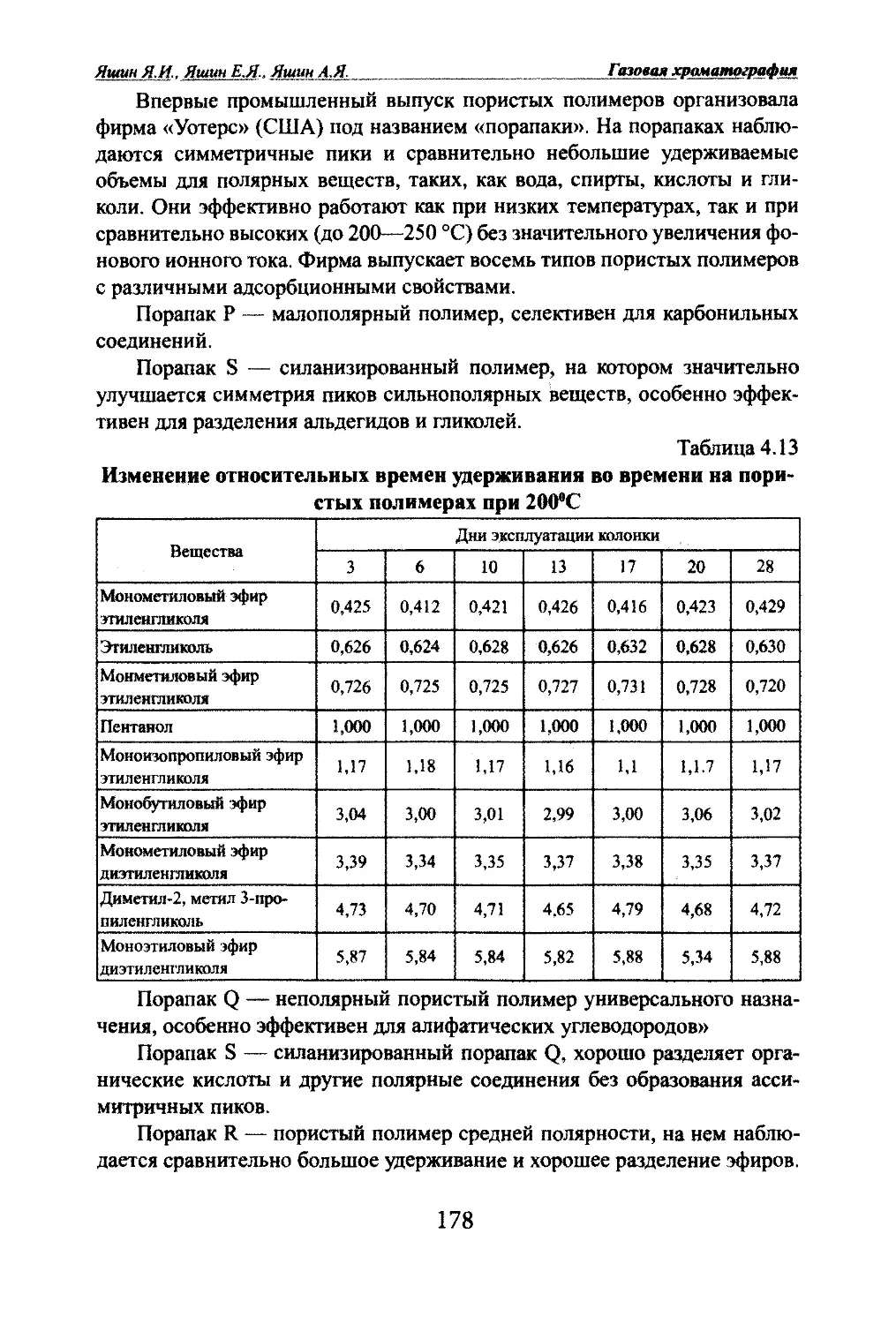


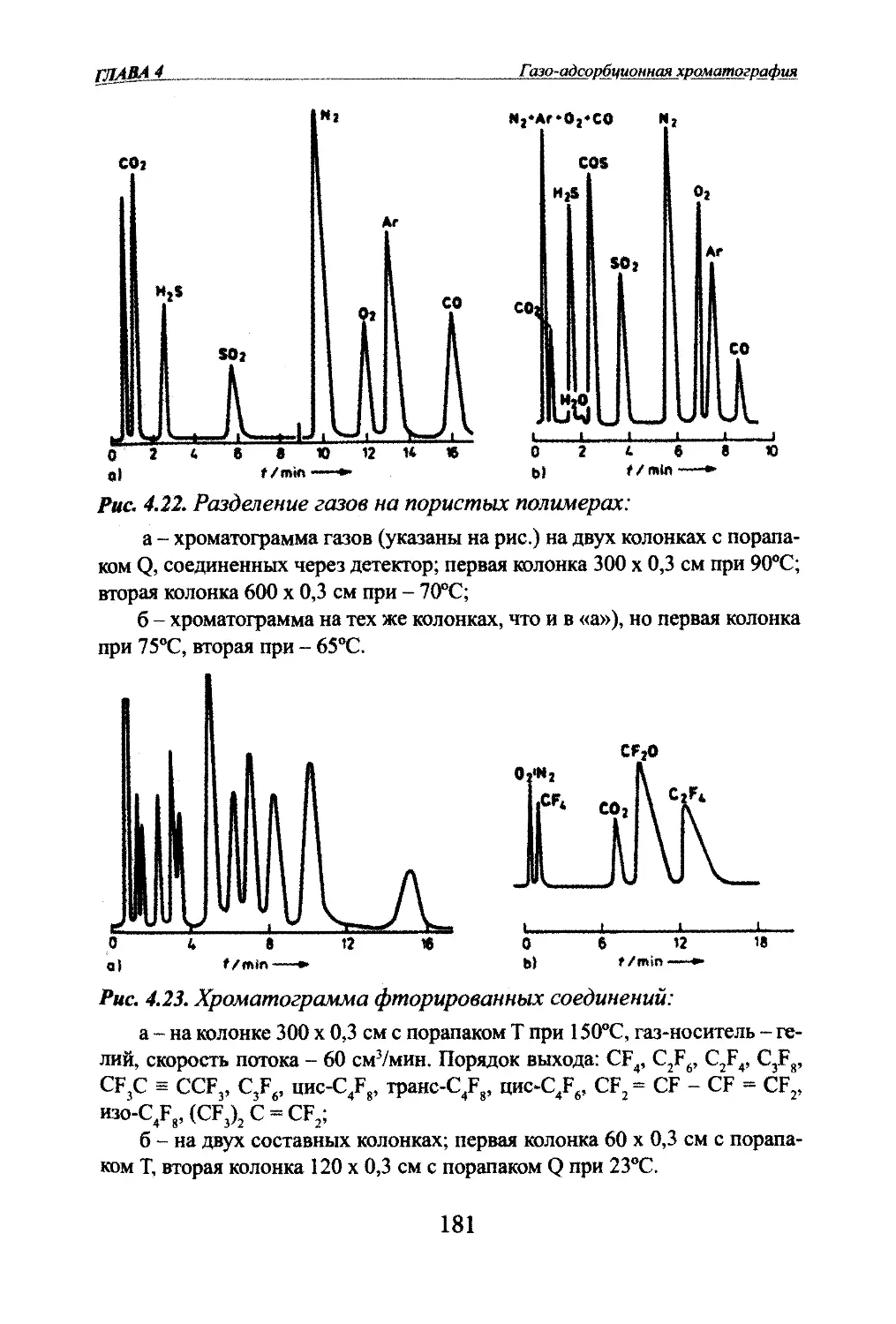
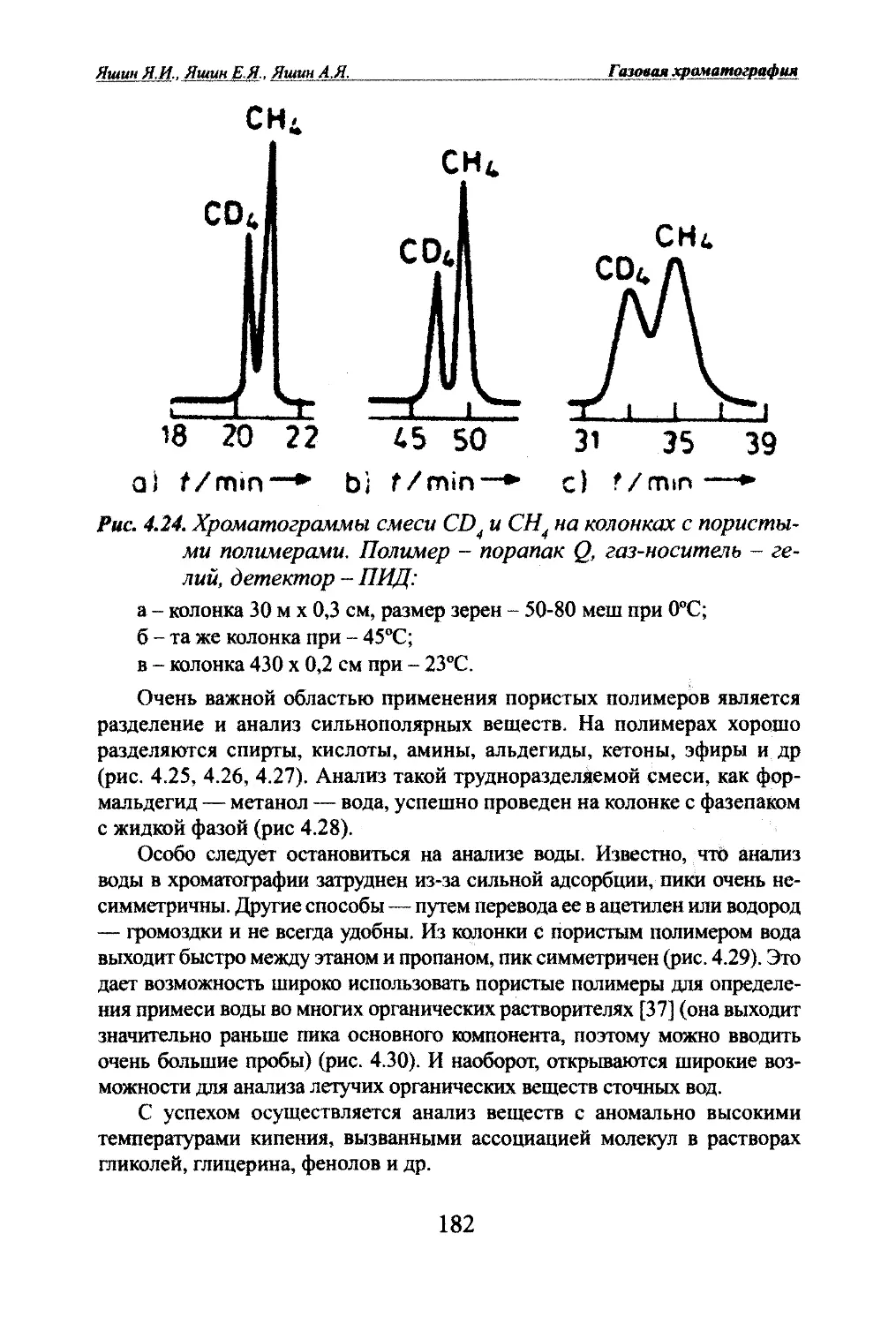

























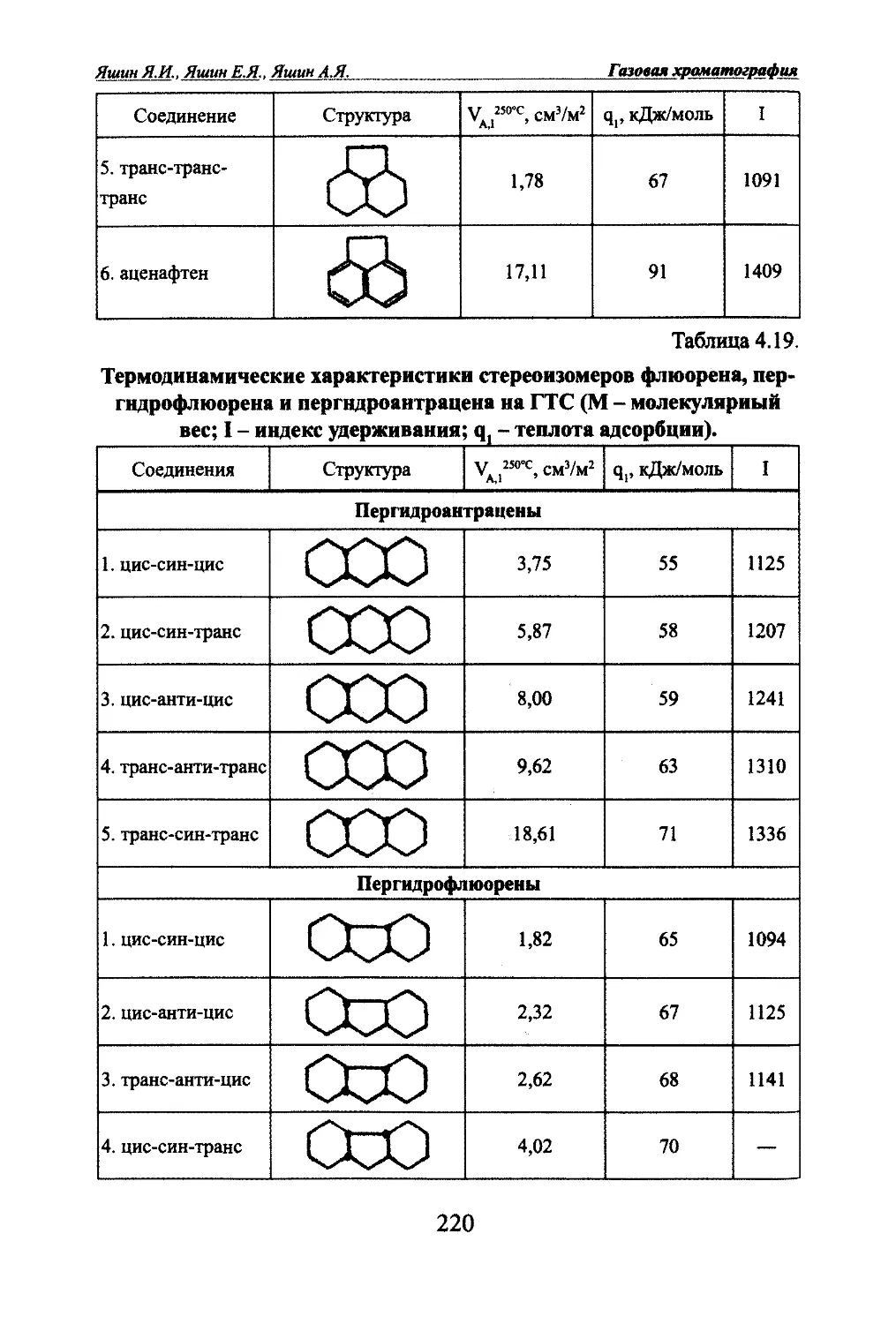

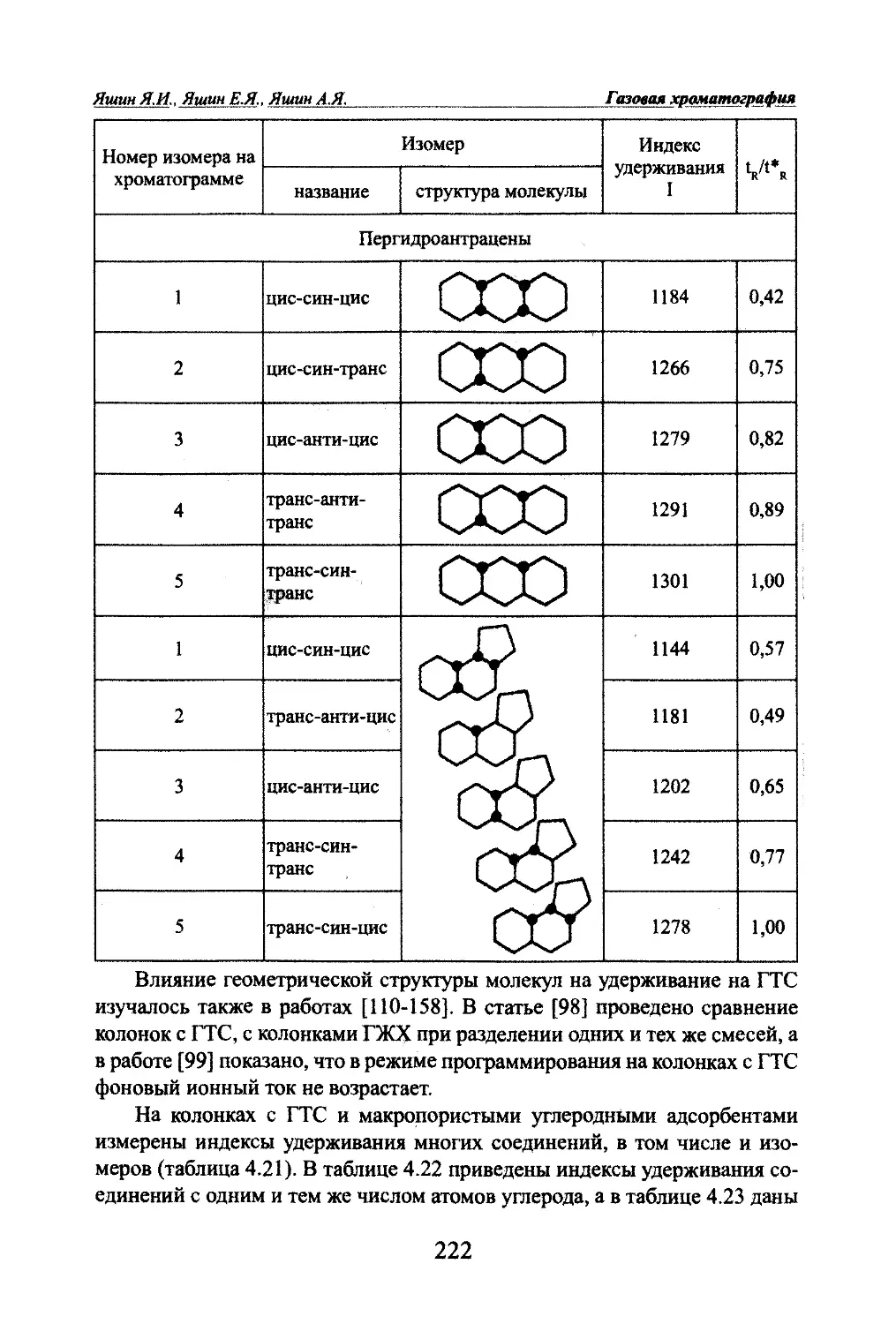

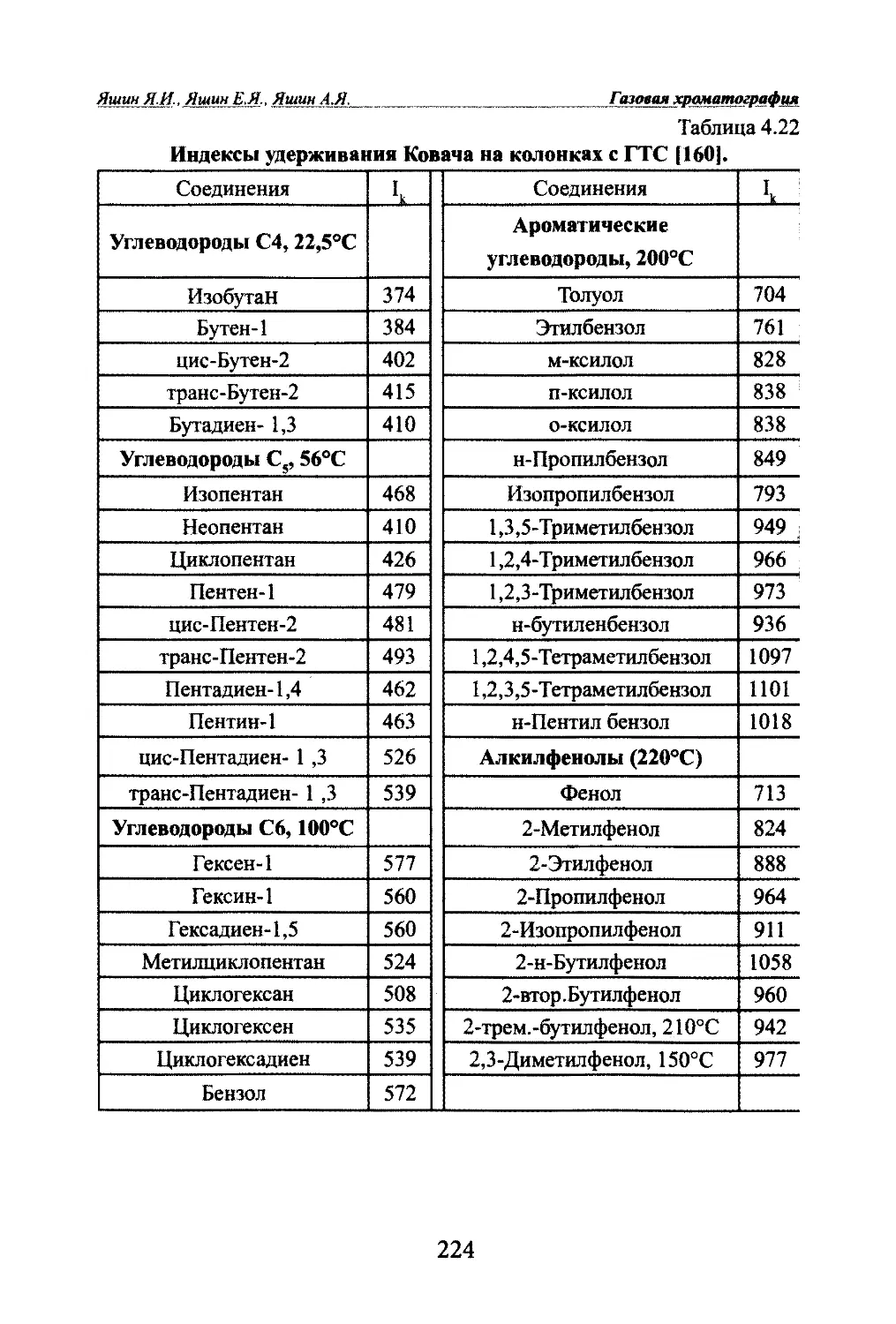
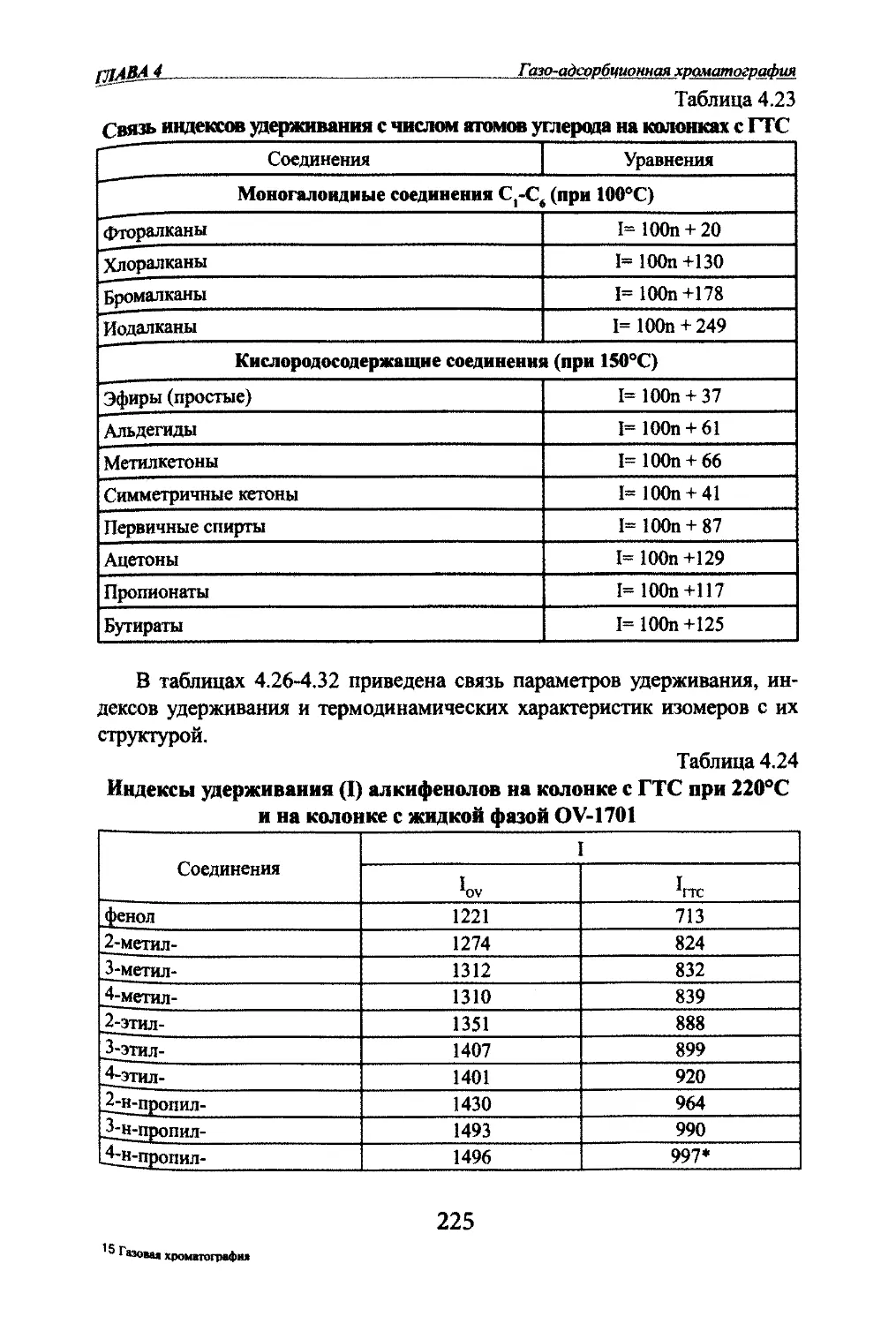

























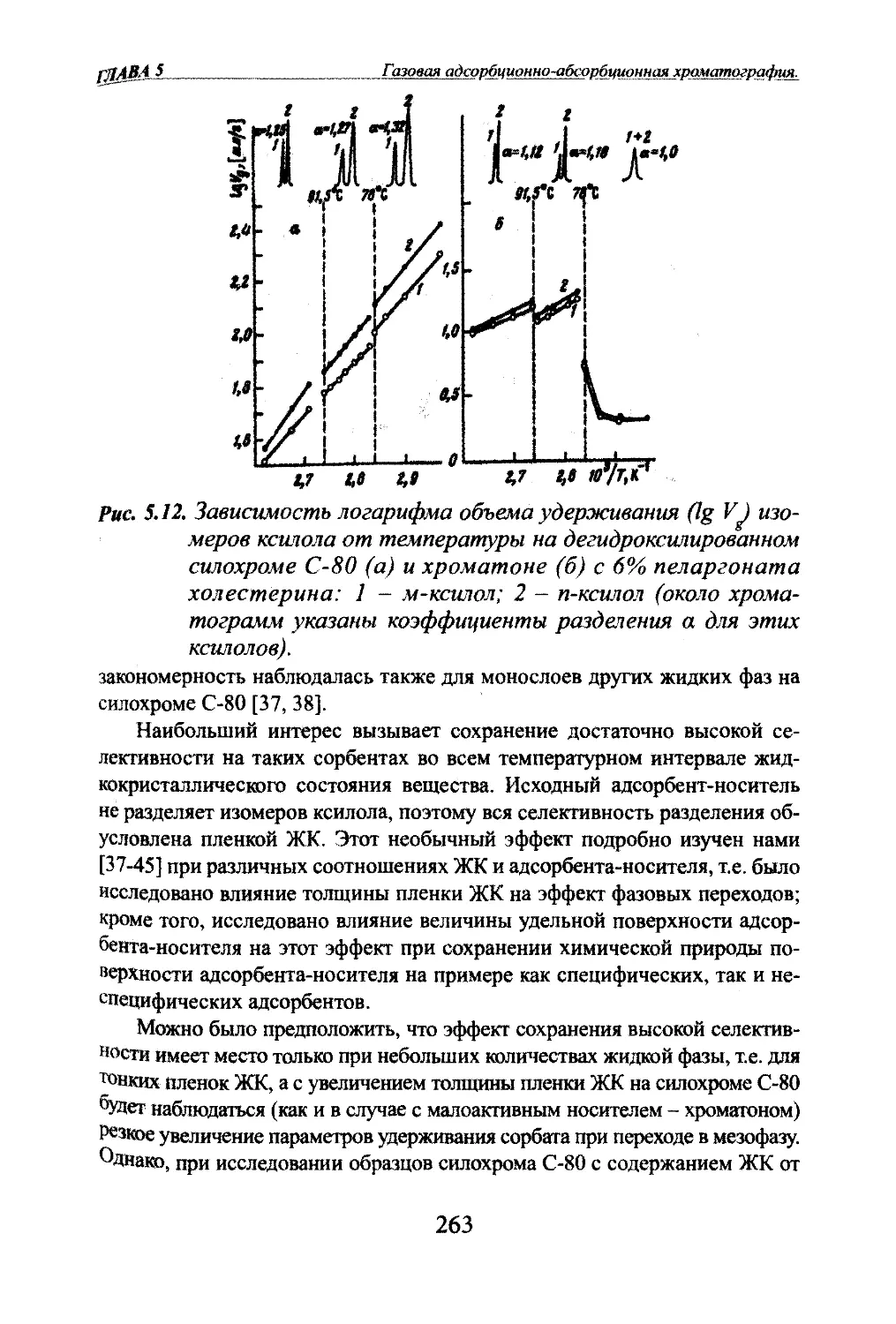
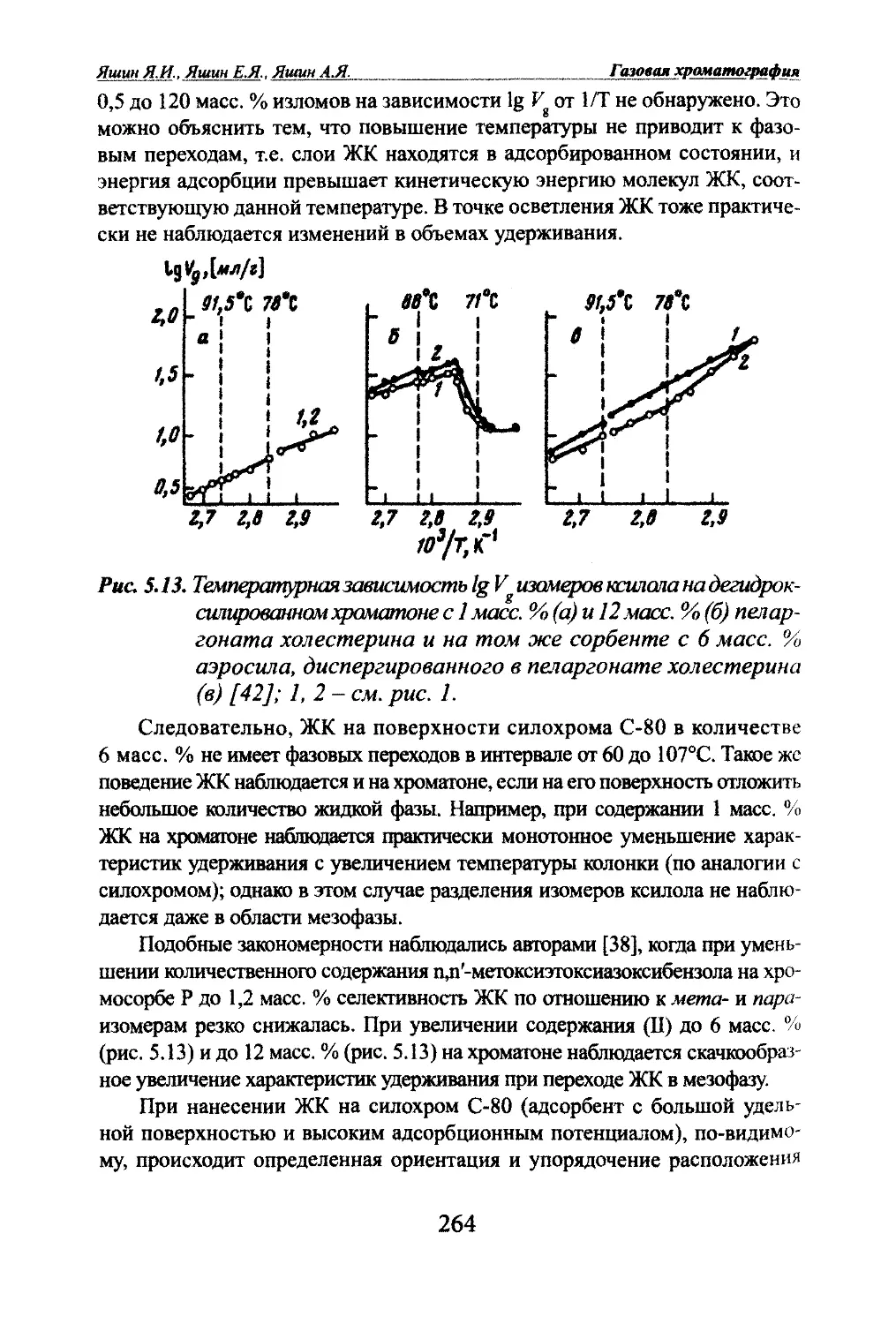

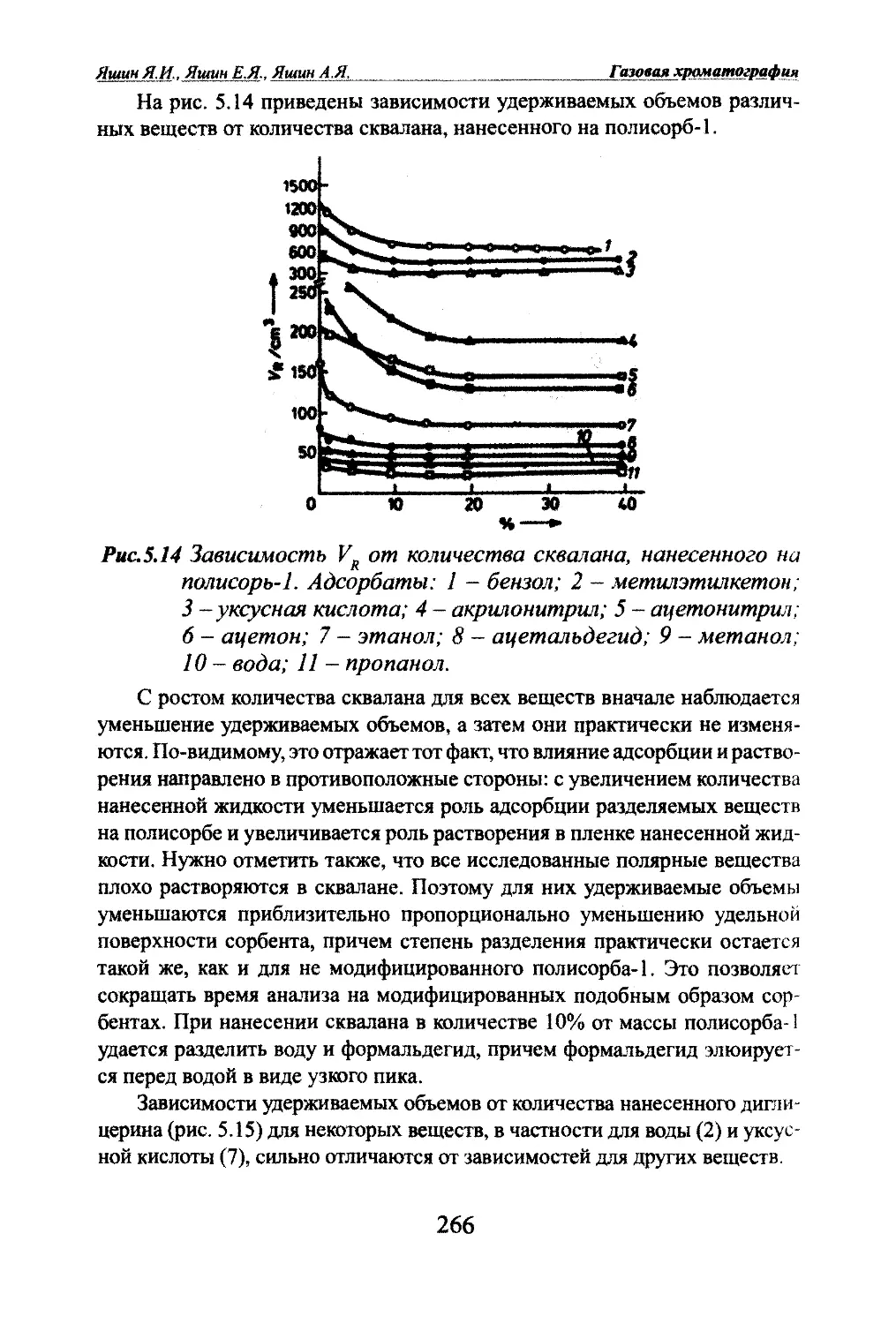
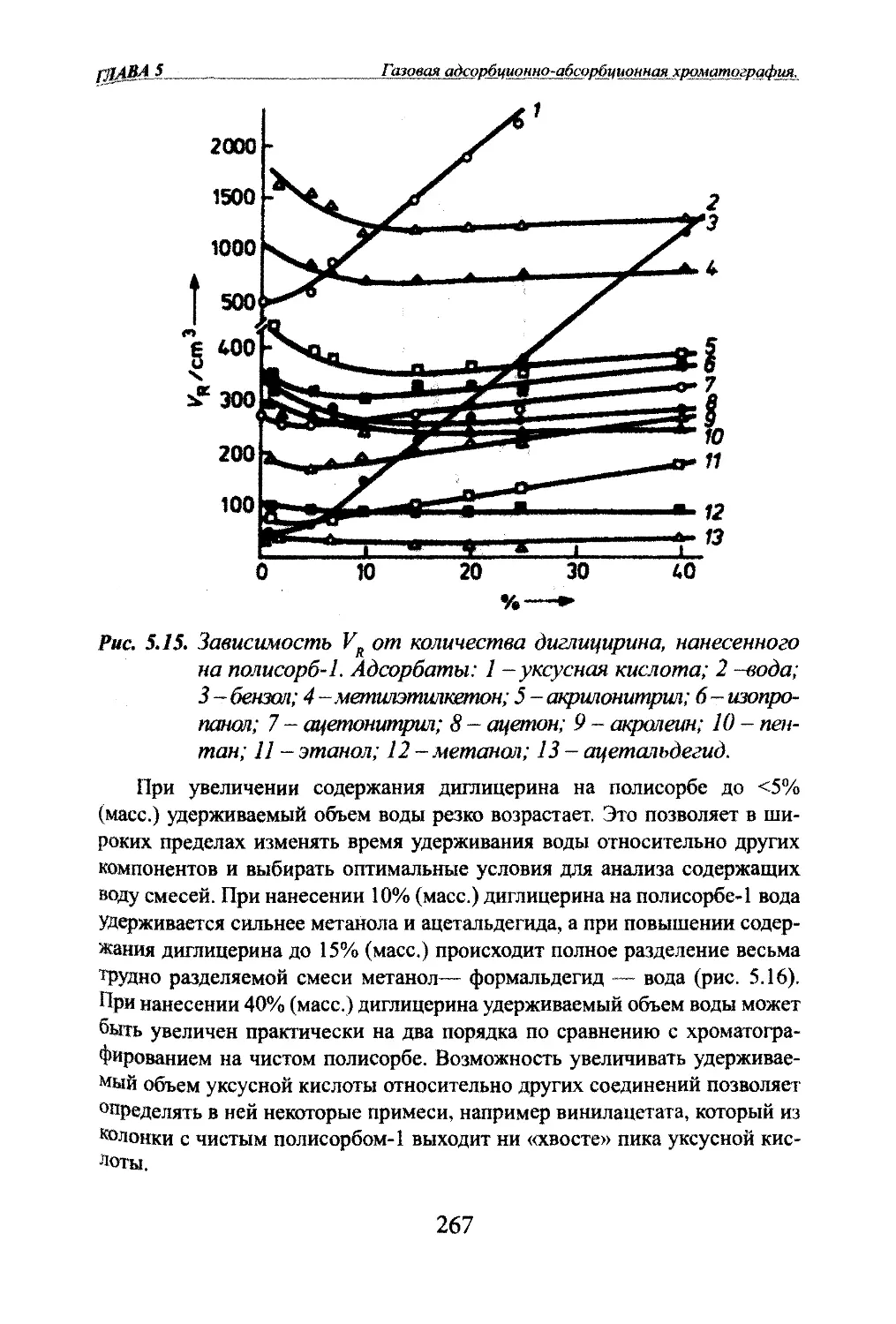

























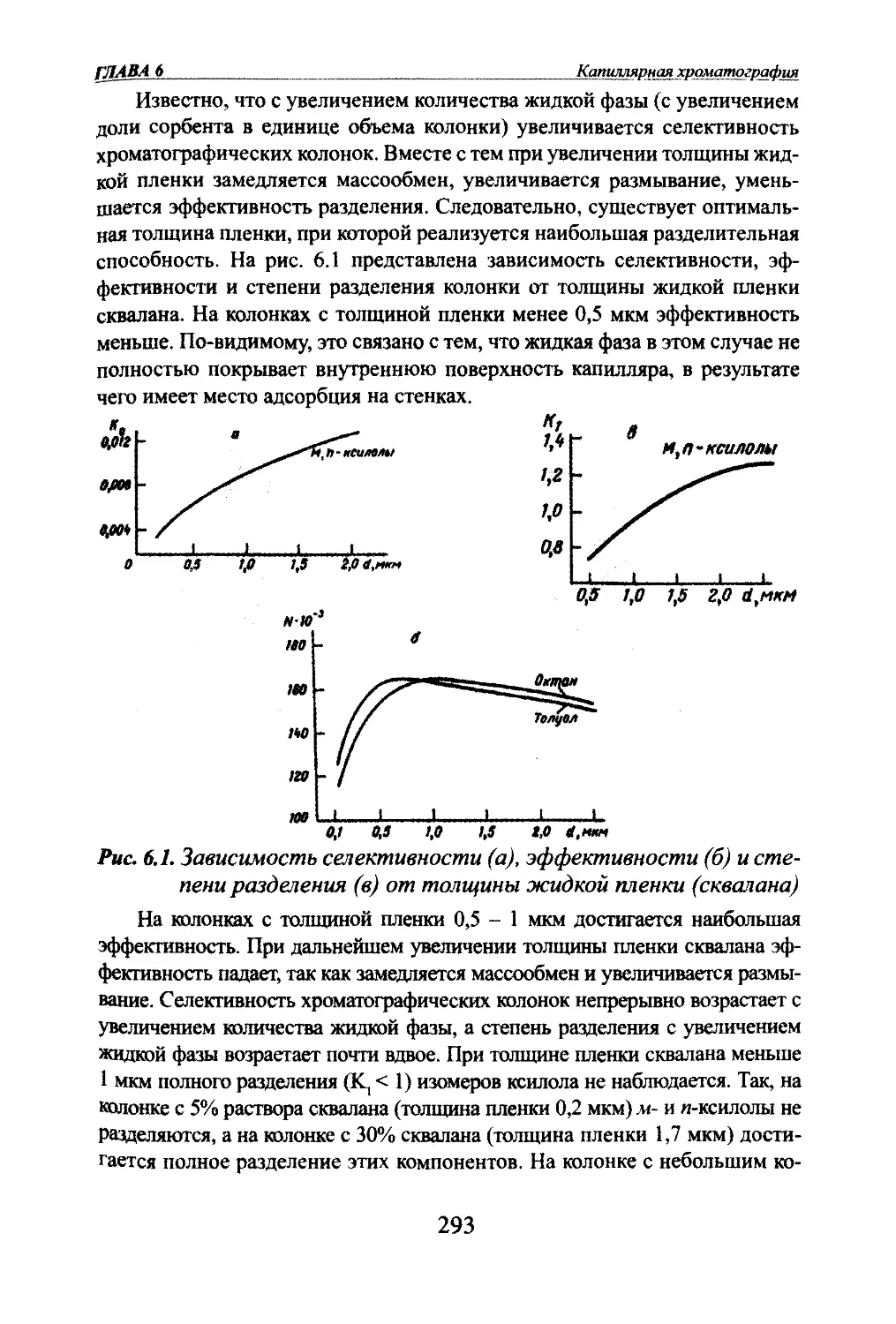




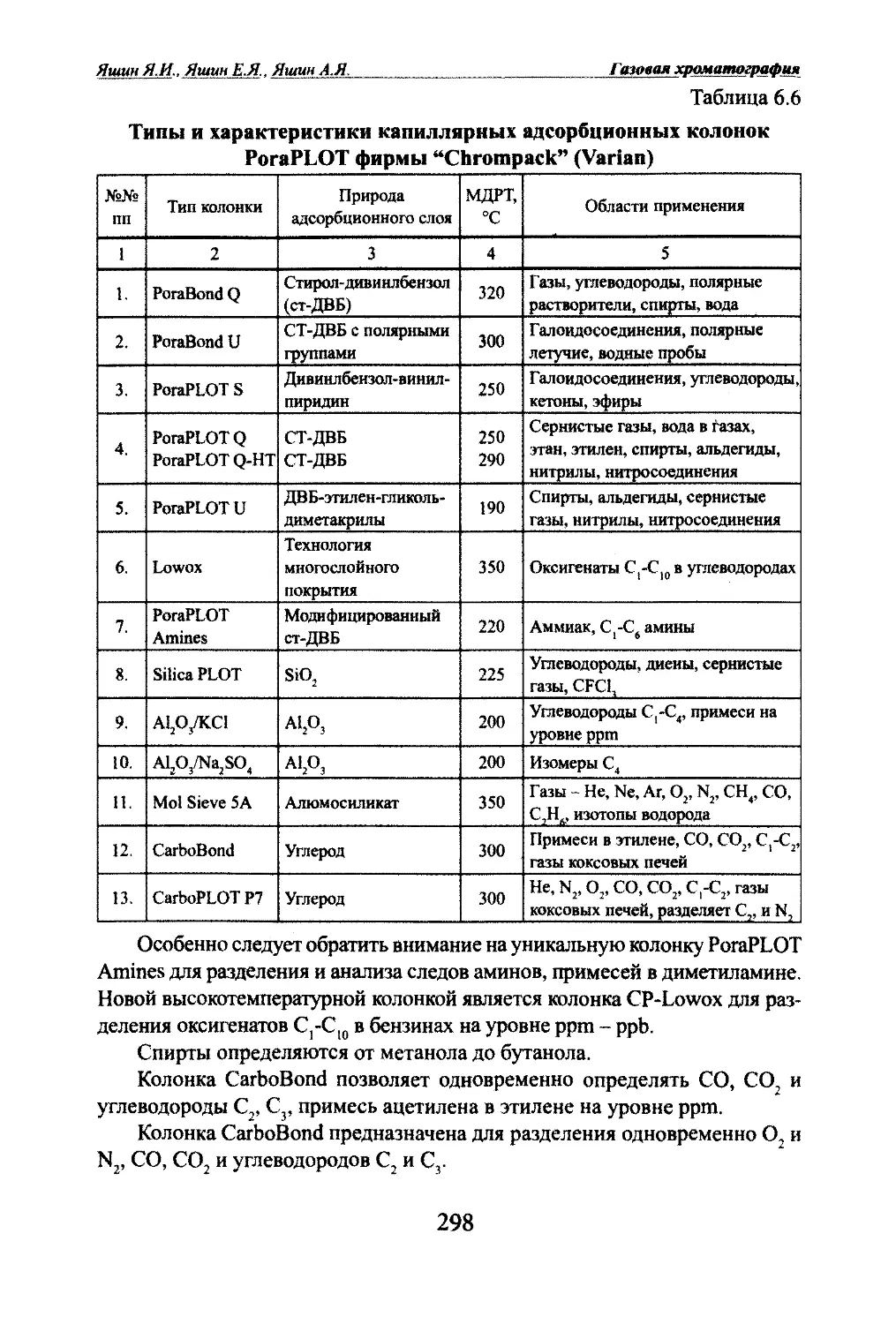
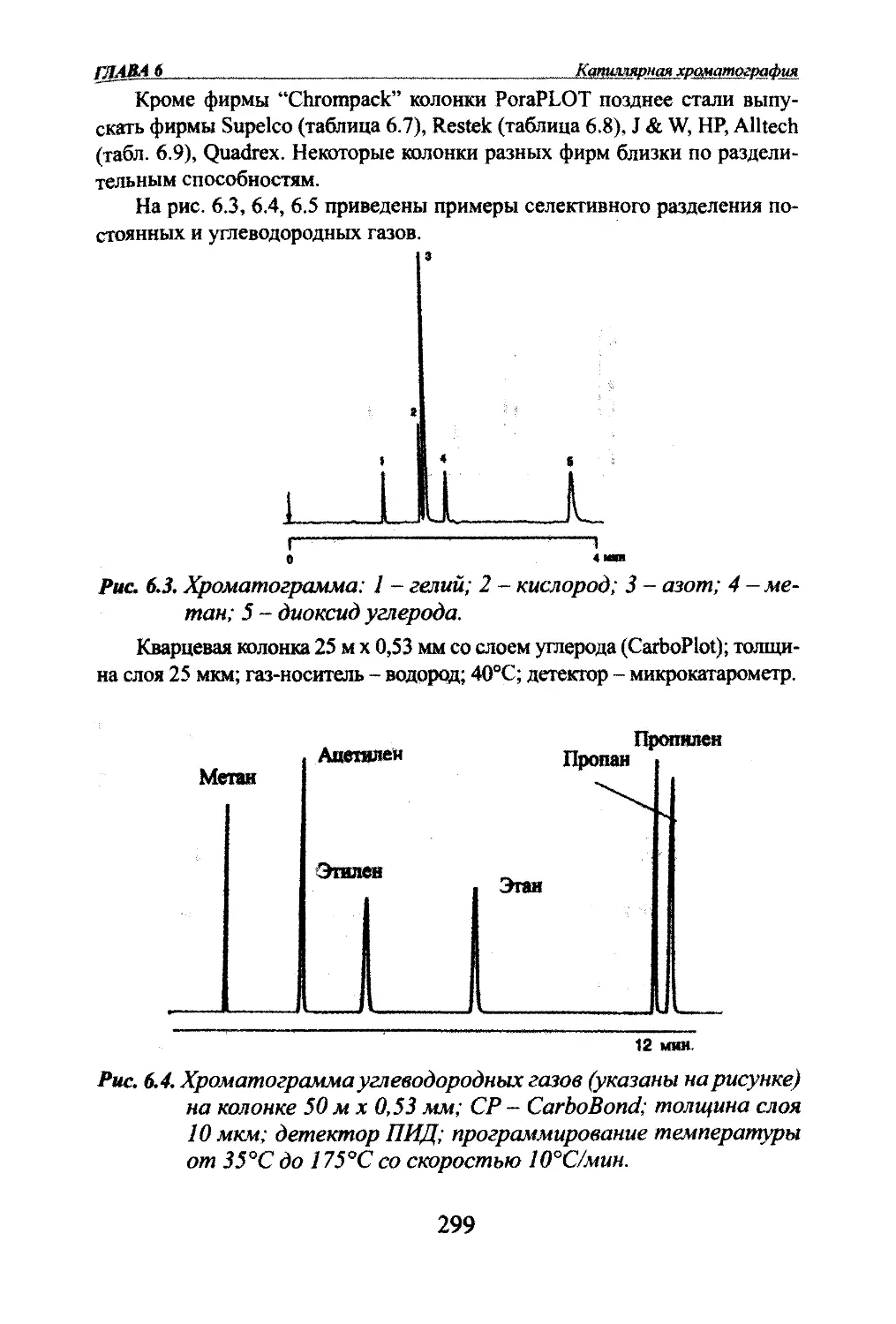

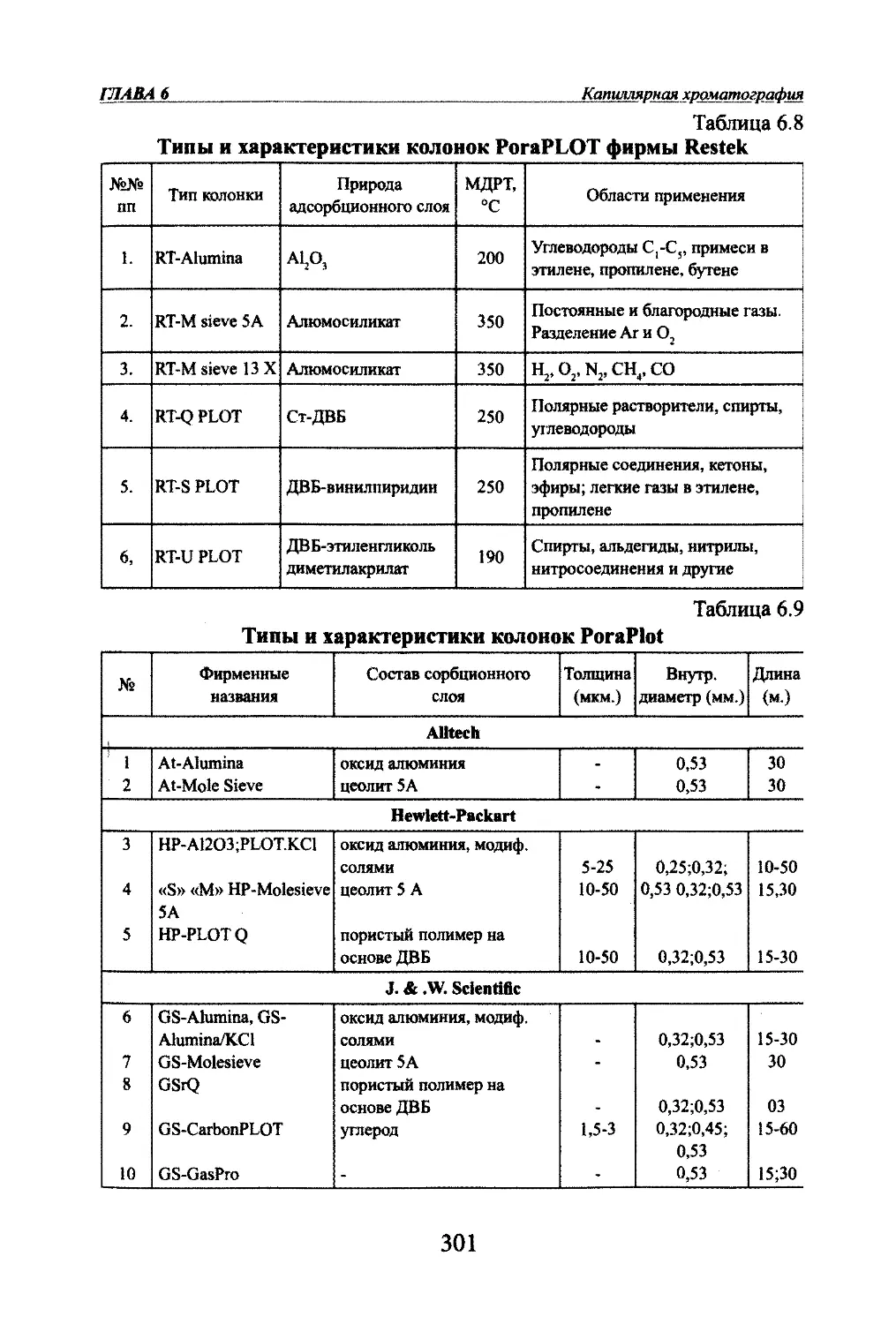
















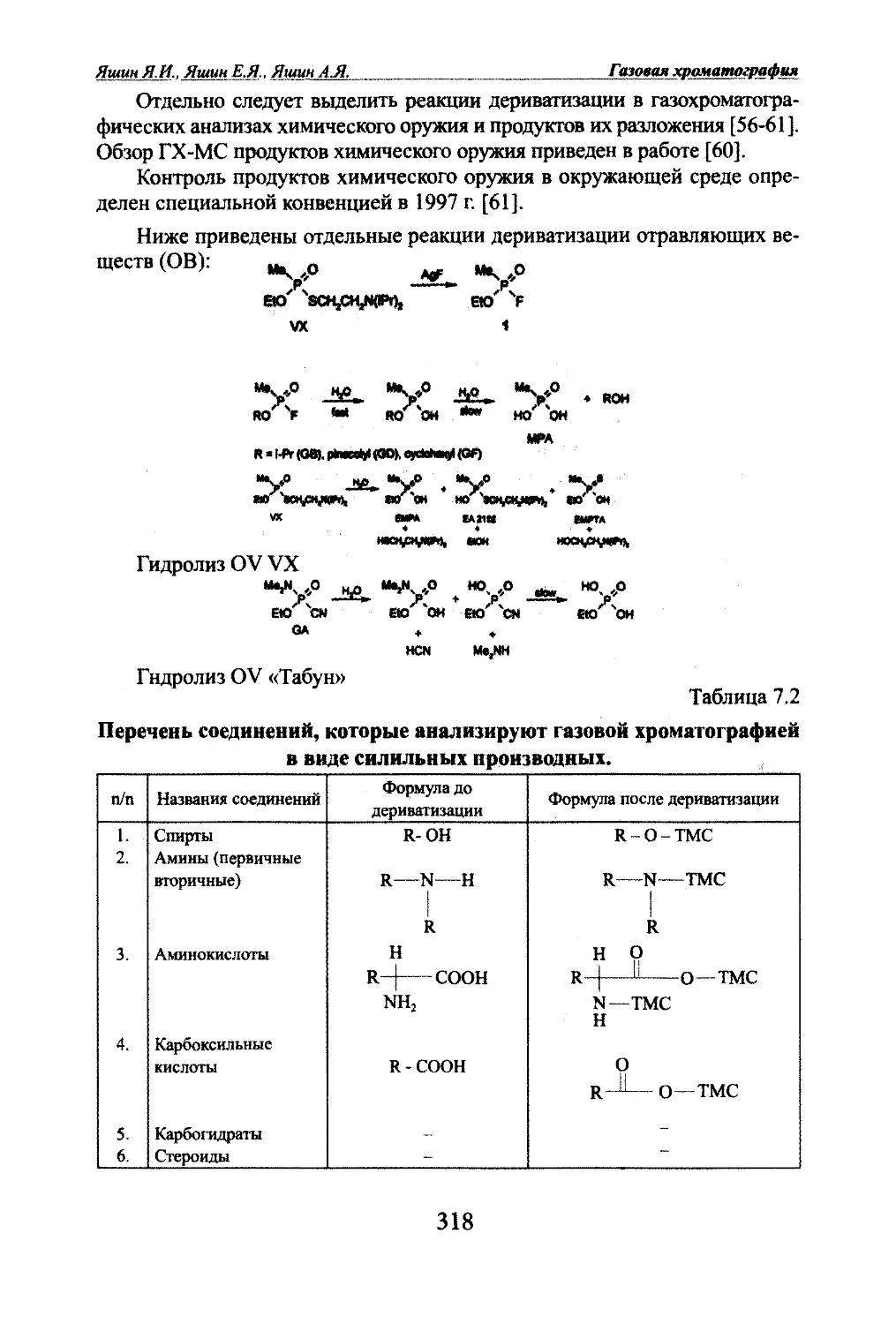








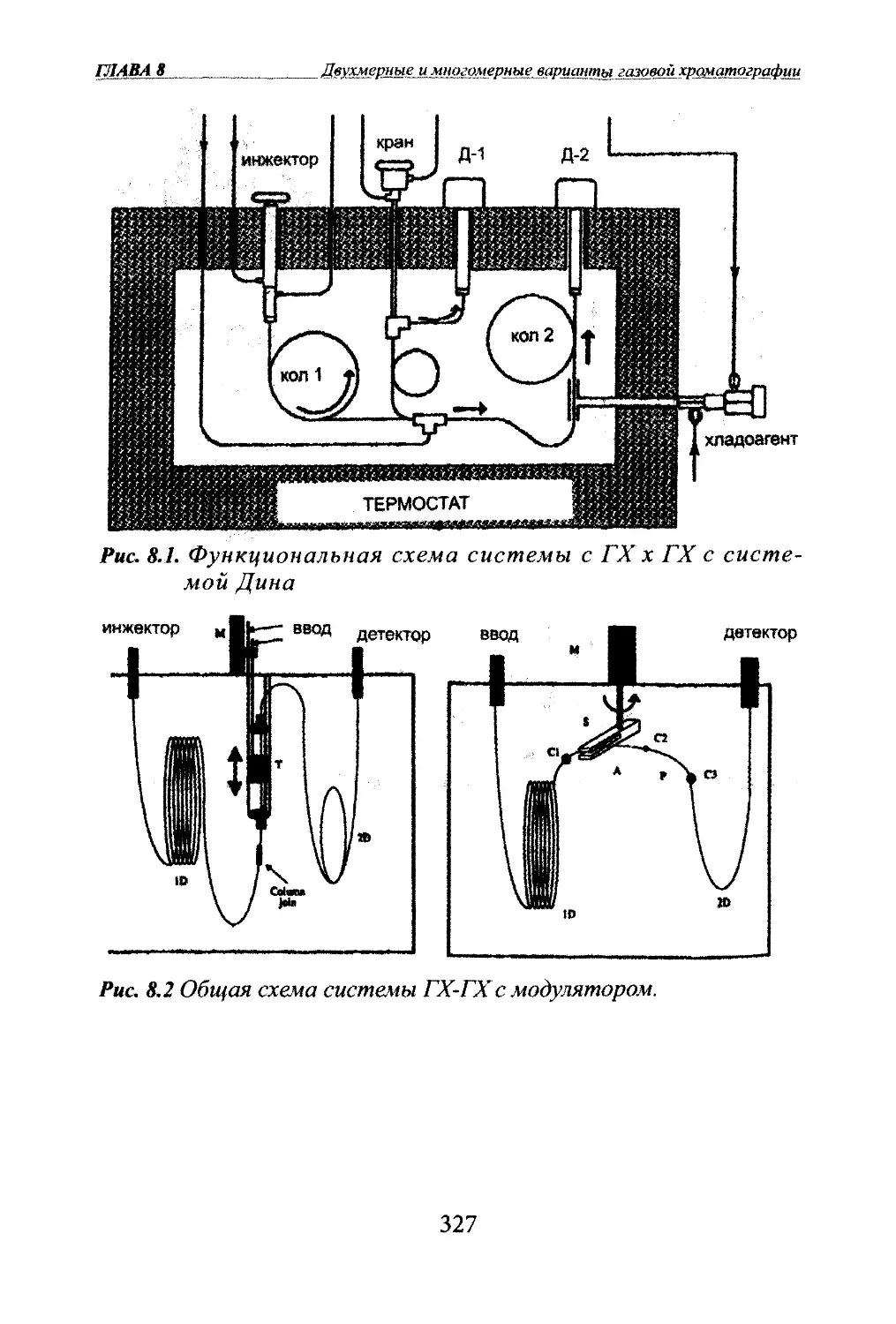


























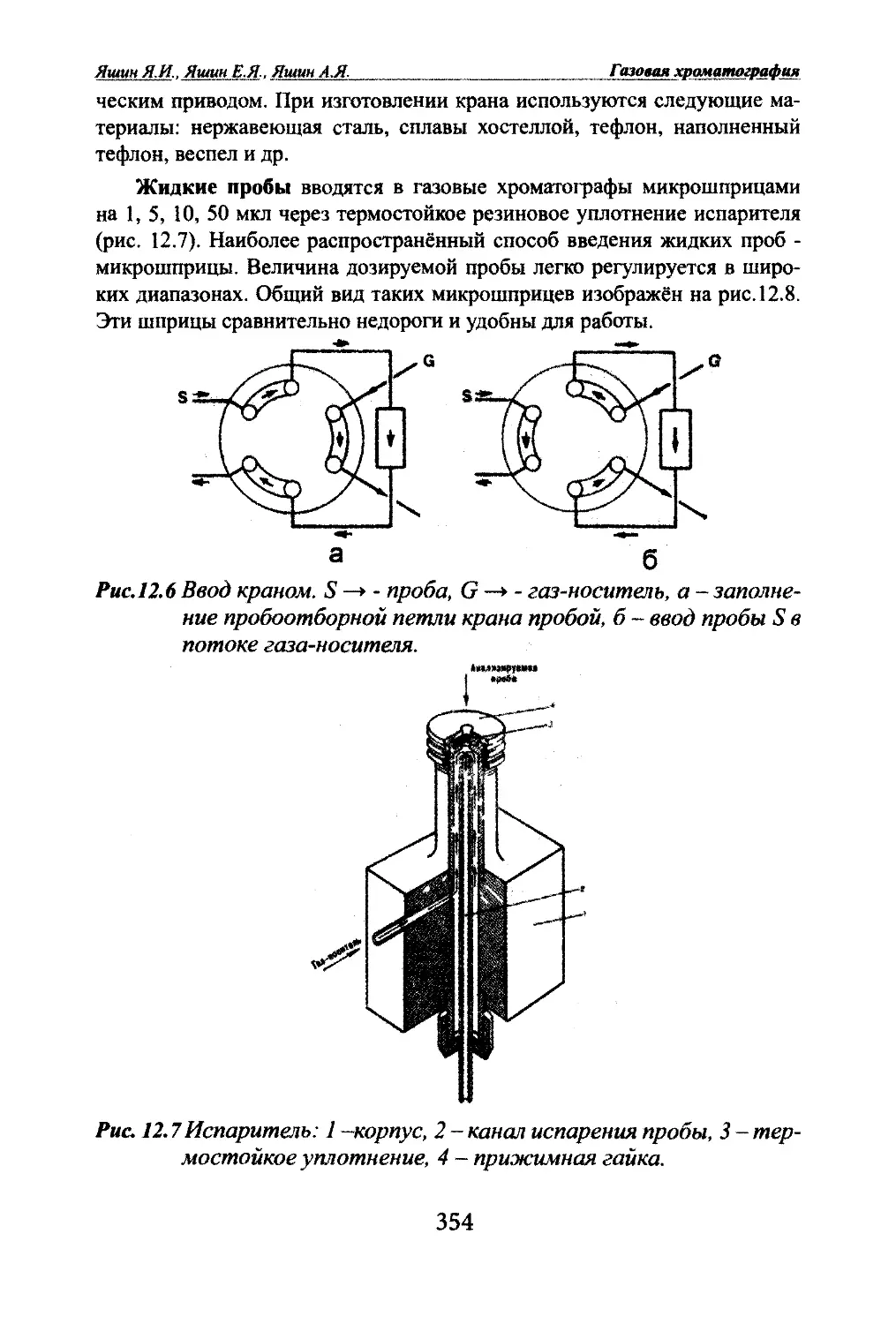
















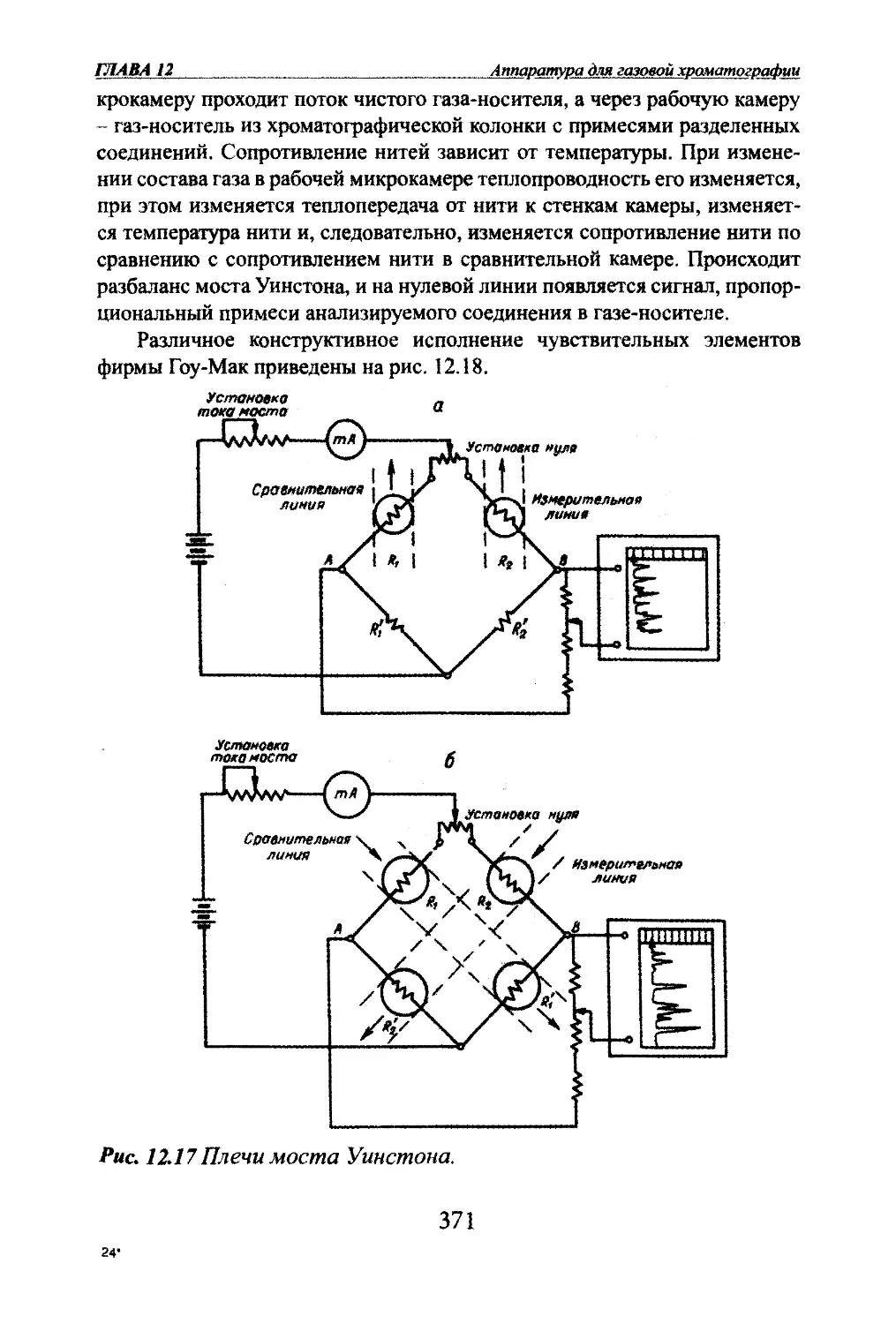



















































































































































Автор: Яшин Я.И. Яшин Е.Я. Яшин А.Я.
Теги: другие физико-химические методы анализа (кроме оптических) аналитическая химия физика химия хроматография газовая хроматография
ISBN: 978-5-94976-825-9
Год: 2009




я.и. Яшин, Е.Я. Яшин, А.Я. ЯшинГАЗОВАЯ
ХРОМАТОГРАФИЯМосква,
«ТрансЛит»,
2009 г.
УДК 543.544.2
ББК 24.4
Я 96Яшни Я.И^ Яшин ЕЛ., Яшт АЛ.Я 96 Газовая хроматография. М.: Издательство «ТрансЛит», 2009 г., 528 с.В книге изложены основы теории и практики аналитической газовой хроматографии.
Рассмотрены послшше достижения газо-жвдкостной, газоадсорбпионяой, адсорбционно-
а&!орбционной, кагаилярной, реакционной, сверхсторостной, высокотемпературной, двумер¬
ной и хиральной хроматографии. Приведены новейшие достижения в хромагографичесюм
приборостроении, в комбинированных методах, методическом и аппарпурном исполнении
методов юнцентрирования. В заключении рассмотрены применения ГХ в жизненно важных
областях (юнтроле загрязнений окружающей среды, пищевых проуктов и напитков, фарма¬
цевтике, медицине, судебной медицине, в технологическом контроле, в нефтехимии и др.).Книга предназначена для широкого круга спеш1алисгов, самостоятельно изучающих
метод газовой хроматографии. Книга может быть полезна студентам, аспирантам и слушате¬
лям разных курсов повшпения квапифншши.ISBN 978-5-94976-825-9© Яшин Я.И.,Яшин Е.Я., Яшин А.Я., 2009
© Издательство ТрансЛит, 2009
Сведения об авторахЯшин Яков Иванович - директор научно технического центра
«Хроматография» НПО «Химавтоматика», доктор химических нг^,
профессор, Лауреат Государственных премий СССР и Российской
Федерации, автор более 600 научных работ, 20 книг и сборнишв, 42
патентов и изобретений.Две книги переведены и изданы в США, Франции, Германии,
Польше и Чехии.Яшин Евгений Яковлевич - заместитель директора н^но -
технического центра «Хроматография» НПО «Химавтоматика».Его основные научные интересы в газовой хроматографии связаны
с анализом нефтепродуктов. Работая по данной теме в Раменском
филиале ВНИИЯГГ, в Московской таможне, учась в аспирантуре
Всесоюзного научно - исследовательского геологоразведочного
нефтяного института, он опублишвал более десяти научных работ,
хорошо знает современную газо-хроматографическую аппаретуру и
области ее применения.Яшин Александр Яковлевич - начальник отдела хрометографии
НТЦ «Хроматография» НПО «Химавтоматика», гандидет химических
н£^. Автор более ПО публикаций по газовой, жидкостной и ионной
хроматографии, 3 патентов, является соашором Международной
энциклопедии по аналитической химии и Нового Российского
справочника химика и технолога.
ПредисловиеКнига основана на курсе лекций авторов в Московсюм институте по¬
вышения квалифигации руководящих работников и специалистов хими¬
ческой промышленности и в Учебно-исследовательском центре повыше¬
ния квалификации РГУ нефти и газа им. И.М.Губкина и подготовлена по
многочисленным просьбам слушателей этих учреждений.С развитием промышленности, техники и научных исследований в по¬
следние годы возрастает интерес к методу газовой хроматографии (ГХ).
В связи с этим возникает необходимость подготовки квалифицированных
кадров.Пособия с систематическим изложением основ метода, анализом со¬
временного состояния методических и приборных вопросов ГХ в послед¬
ние годы не издавались.Ценность книги, на наш взгляд, в том, что в ней совмещены учебное
пособие, различные справочные материалы и обзор последних аналитиче¬
ских достижений ГХ.В книге рассмотрено современное состояние основных методов ГХ,
в том числе и новых получивших развитие в последние годы: двумерная,
высокоскоростная, высокотемпературная, хиральная и др.Достаточно подробно рассмотрено состояние хроматографического
приборостроения: лабораторные, портативные и промышленные хромато-
фафы.Проанализированы все методы экстракции: твердофазная, газовая,
жидкостная, сверхкритическая флюидная. Рассмотрены методы микро-
твердофазной экстракции, экстракции одной каплей жидкости и др.Систематизированы основные применения ГХ.Книга предназначена как для начинающих, так и для специалистов
среднего уровня.Для желающих углубить свои знания в области ГХ в книге приведены
многочисленные литературные источники.Авторы выражают благодар¬
ность Т.Ю.Рыжневой, Т.М.Дмитриевой, М.О.Осиповой, О.Н.Куминскому
за подготовку рукописи к печати.Авторы ^яут признательны читателям за замеченные недостатки и упу¬
щения в книге.
Глава 1. ВВЕДЕНИЕ1.1. Определение хроматографии.Хроматофафия - физико-химический метод разделения смесей соеди¬
нений, основанный на распределении соединений между двумя фазами:
одна фаза неподвижная, другая - подвижная, непрерывно протекающая
через слой неподвижной фазы.Хроматография как метод разделения применяется для следующих
целей: для анализа разделенных соединений (аналитическая хроматогра¬
фия); для физико-химических исследований или измерений (физико-хи¬
мические применения хроматографии, исследовательская хроматография);
для выделения разделенных веществ в чистом виде (препаративная хрома¬
тография).В связи с тем, что хроматография преимущественно применяется для ана¬
литических целей, имеется и другое ее определение: хроматофафия - физико¬
химический метод анализа сложных смесей (газов, жидкостей) путем пред¬
варительного разделения их при движении по слою сорбента за счет различий
взаимодействий и последующего определения разделяемых момпоненгов на
выходе из колонки.В отличие от других методов разделения, также основанных на рас¬
пределении соединений между фазами, хроматофафия - динамический
метод, так как разделение происходит в потоке одной из фаз. В хромато-
фафии разделение соединений происходит за счет раздичий межмолеку-
лярных взаимодействий соединений с неподвижной фазой, выраженных
в различной сорбируемости. Одно из основных требований - это обрати¬
мость взаимодействия, соединения должны сорбироваться и через некото¬
рое время полностью десорбироваться потоком подвижной фазы.Хроматофафия в большей мере основана на физичесшй адсорбции,
для которой характерны слабые межмолекулярные Ван-дер-Ваальсовы
взаимодействия. Кроме того, используются и специфические обратимые
взаимодействия, водородная связь, донорно-акцепторные взаимодействия,
слабое шмплексообразование, ионный обмен, лигандный обмен и др.Хемосорбция для хроматофафического разделения не применяется,
так как при необратимой сорбции разделяемые соединения будут оставать¬
ся в колонке и информации о них на выходе из колонки не будет.Вокруг вопроса об определении хроматофафии в последние годы раз¬
вернулась дискуссия [1-3]. Хроматофафию предложено рассматривать как
новую научную дисциплину, как новый процесс, как новое явление. Есть
попытка считать однофазные системы (фракционирование в поле сил) так¬
Яшин Я.И., Яшин Е.Я.. Яшин А.Я.Газовая хроматографияже хроматографией, но это приводит к полному отходу от классического
определения хроматографии. Необходимо в определении придерживаться
двух основных критериев хроматографии: во-первых, метод хроматогра¬
фии основан на распределении между двумя фазами; во-вторых, одна из
фаз подвижная. Этими критериями и следует руководствоваться прежде
всего при отнесении к хроматографии многих методов и вариантов, пред¬
ложенных в последнее время.1.2. Классификация методов хроматографии.Хроматография постоянно развивается. К уже предложенным несколь¬
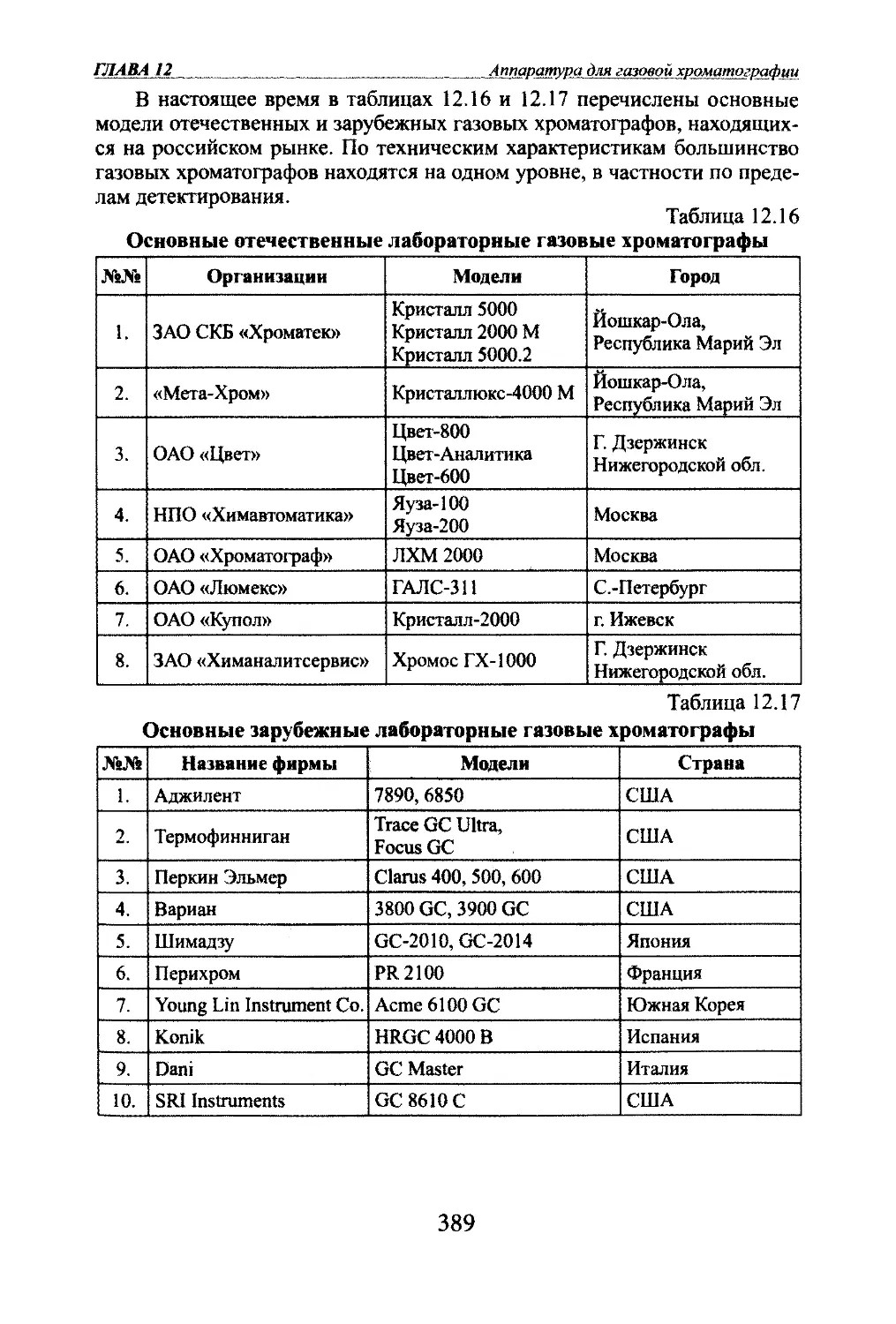
ким десяткам методов и вариантов каждый год добавляются новые.В табшцах 1.1, 1.2, 1.3 приведены некоторые методы газовой хрома¬
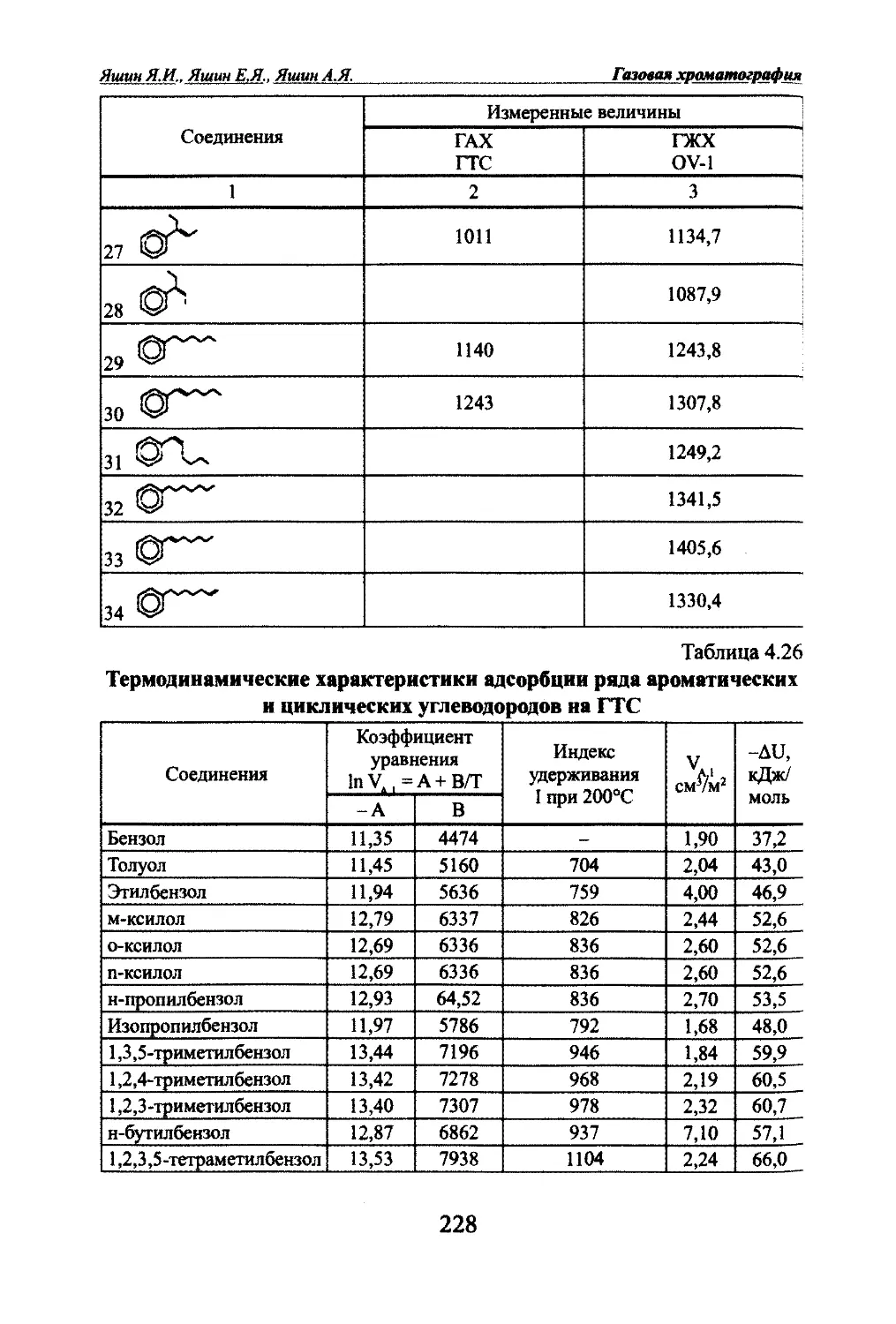
тографии.Таблица 1.1Классификация методов хроматографии по способу перемещения
разделяемых соединенийНазвание метода
хроматографииОсобенности метода1.Проявительная(элюентная)Подвижная фаза (ПФ) - газ, неподвижная фаза
(НПФ) - адсорбент или жидкость. Поток ПФ непре-
рьшно протекает через сшой НПФ. Анализируемая
смесь вводится в колонку периодически. Смесь со¬
единений может полностью разделяться на чисше
зоны2.ФронтальнаяПФ - разделяемая смесь газов или паров жид-
юстей, НПФ - адсорбент или жидкость. В этом
методе нет полного разделения, зоны соединений
примыкают друг к другу, в чистом виде только
зона первого слабосорбируемого соединения3.ВытеснительнаяПФ - газ или пары жидюсти, НПФ - жидкость или
твердое тело - адсорбент. В качестве ПФ исполь¬
зуется соединение, сорбируемое сильнее любого
из компонентов смеси, поэтому оно действует как
вытеснитель. Полного разделения нет, зоны при¬
мыкают друг к другу, но в отличие от фронтально¬
го метода каждая зона - это чистое соединение4.ИзотермическаяРазделение проюдится при постоянной температуре5.С программированием
температурыРазделение проводится с повышением температуры
одновременно всей колонки с заданной скорюстью
ГЛАВА 1Введение№№Название метода
хроматографииОсобенности метода6.ХроматермографияРазделение проводится с повышением температу¬
ры части колонки за счет движущейся по ней труб¬
чатой разогретой печки с градиентом температуры
по ее длине. В зависимости от направления гради¬
ента печки различают стационарную и нестацио¬
нарную хромагермографию7.С программированием
расхода ПФОбычно проявительная хроматография, в которой
в процессе разделения скорость расхода ПФ повы¬
шается с целью сокращения времени разделения.8.Хромадистилляция(хроматоректифика-ция)Метод газовой хроматографии, в ютором смесь
веществ перемещается потоком ПФ (газа-
носителя) вдоль {оолонки в условиях отрицатель¬
ного продольного градиента температурыТаблица 1.2Классификация методов газовой хроматографии
по агрегатному состоянию фаз№№Название мегадаОсобенности методаОбласти применения1.ГазоваяПодвижная фаза (ПФ)
-газРазделение смеси га¬
зов, летучих и устойчи¬
вых соединений2.Газо-адсорбционнаяПФ - газ, неподвижная
фаза (НПФ)
адсорбентРазделение за стег раз¬
личной адсорбируемо-
сга3.Газо-жидкостнаяПФ - газ, НПФ - жид¬
кость, нанесенная на
поверхность твердого
тела-носителяРазделение за счет раз¬
личной растворимости4.Газовая адсорбционно-
абсорбционная (газо-
жидю-твердофазная)ПФ - газ, НПФ - жид¬
кость и адсорбентРазделение за счет раз¬
личий как адс^)бируе-
моста, так и раствори¬
мости5.Газо-мезофазнаяПФ - газ, НПФ - жид¬
кость, находящаяся в
жиднокристалличесюм
(мезофазном) состоя¬
нииРазделение и анализ
летучих соединений за
счет растворимости в
жидкокристалличесдой
фазе
Яшт Я.И.. Яшин Е.Я., Яшин А.Я. Газовая хроматографияТаблица 1.3Классификация методов газовой жроматографии в зависимости
от способа оформления сорбционного слоя№№Названне методаОсобенности методаКолоночная (насадочные
или набивные юлонки)Сорбционный слой внутри колонки (трубки)
(диаметром обычно 2-4 мм). При диаметре ме¬
нее 2 мм метод называют микроколоночной хро¬
мита^ Кашшшрная1&М10н(нная хроматография, в которой в ка¬
честве колонки используются ттилляры диа¬
метром менее 0,5 мм с сорбционным слоем на
внутренней поверхности ПоликапиллярнаяКолонки диаметром 4 мм со многими парал¬
лельными капиллярами (около 900), диаметром
10-20 мк. На внутреннюю поверхность капилля¬
ров наносится пленка жидюй фазы ЦиркуляционнаяСпособ хромгггографии, в котором разделяе¬
мая смесь соединений многократно циркулирует
через одну и ту же юлонку с целью повышения
э({^ктивности разделения МногомернаяСпособ хроматографии, в котором смесь сое¬
динений разделяется сначала на одной колонке,
а затем направляется в другую юлонку или си¬
стему, гае закономерности разделения дфугие6.МногоиолоночнаяСпособ хроматографии, в котором разделяе¬
мая смесь соединений проходит через две (или
более) юлонки, соединенные как последова¬
тельно, так и параллельно с сорбентами различ¬
ной химической природыПроявительная (элюеитная) хроматография. В этом методе че¬
рез юлонку со слоем сорбента непрерывно пропускают поток инертного
газа-носителя, который сорбируется значительно меньше, чем любой из
компонентов смеси. Анализируемую смесь периодически вводят в начало
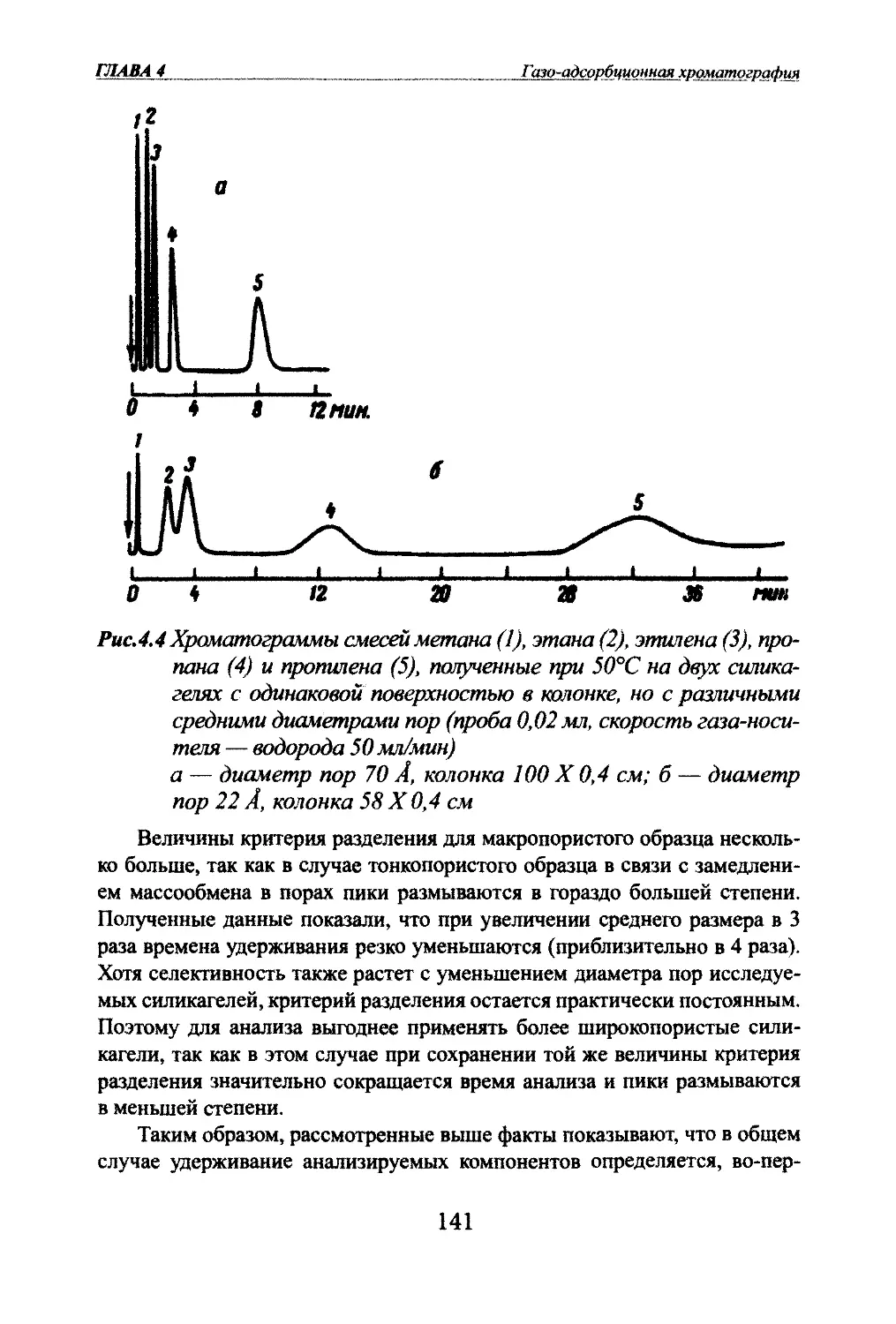
хроматографической колонки (рис. 1.1а). Компоненты разделяемой смеси
через колонку переносятся потоюм газа-носителя. Из колонки вещества
выходят в порядке возрастания их сорбируемости, т.е. первыми выходят
менее сорбируемые. На выходе из колонки детектор регистрирует полосы
разделенных веществ в виде выходных кривых.
1ШВА1Введениеjzi!Puc.1.1 Методы хроматографии
в зависимости от способа пере¬
мещения анализируемой смеси:а - проявительный; б - фронталь¬
ный; в - вытеснительныйЭтот метод получил наибольшее распространение в аналитической
практике, благодаря следующим преимуществам:1. Колонка непрерывно регенерируется потоком газа-носителя, поэто¬
му после выхода всех компонентов смеси можно повторить анализ.2. При оптимальных условиях можно полностью разделить все компо¬
ненты смеси. Это важно для точного количественного анализа и для пре¬
паративного выделения вещества в чистом виде.3. Время вытда №1ксимуш пика при постоянных условиях разщеления есть
величина поспмнная, позволяюпия проводил, вдентификащпо юсмюнешов.Количественное определение проводят по высотам или площадям пи¬
ков. Проявительную хроматографию можно проводить при постоянной
температуре (изотермический режим) и при изменении температуры в
процессе разделения (режим программирования температуры).Фронтальная хроматография. В этом методе анализируемую смесь
непрерывно подают в колонку, т.е. сама анализируемая смесь служит под¬
вижной фазой (рис. 1.1 б). При этом в колош® образуются зоны с после¬
довательно увеличивающимся числом компонентов. Наименее сорбируе¬
мое вещество будет продвигаться с большей скоростью и образует первую
зону, вторая зона будет смесью первого вещества со вторым, так как анали¬
зируемая смесь подается непрерывно. Третья зона будет уже смесью трех
веществ: первого, второго и третьего и т.д.По сравнению с проявительным методом фронтальная хроматография
имеет существенные недостатки:1. Необходима регенерация колонки после каждого анализа (нагрева¬
нием или продувкой).
Яишн ЯМ. Яшин Е.Я.. Яшин А.Я. Гюовая храттощфт2. В чистом виде выходит только полоса слабосорбирующегося ком¬
понента.Количественный анализ проводят по высоте ступени, однако на прак¬
тике они размыты, четкой границы нет.Этот метод мало используют для целей анализа. Значительно чаще его
применяют для концентрирования легких или тяжелых компонентов.Вытеснительная хроматография. В этом методе анализируемую
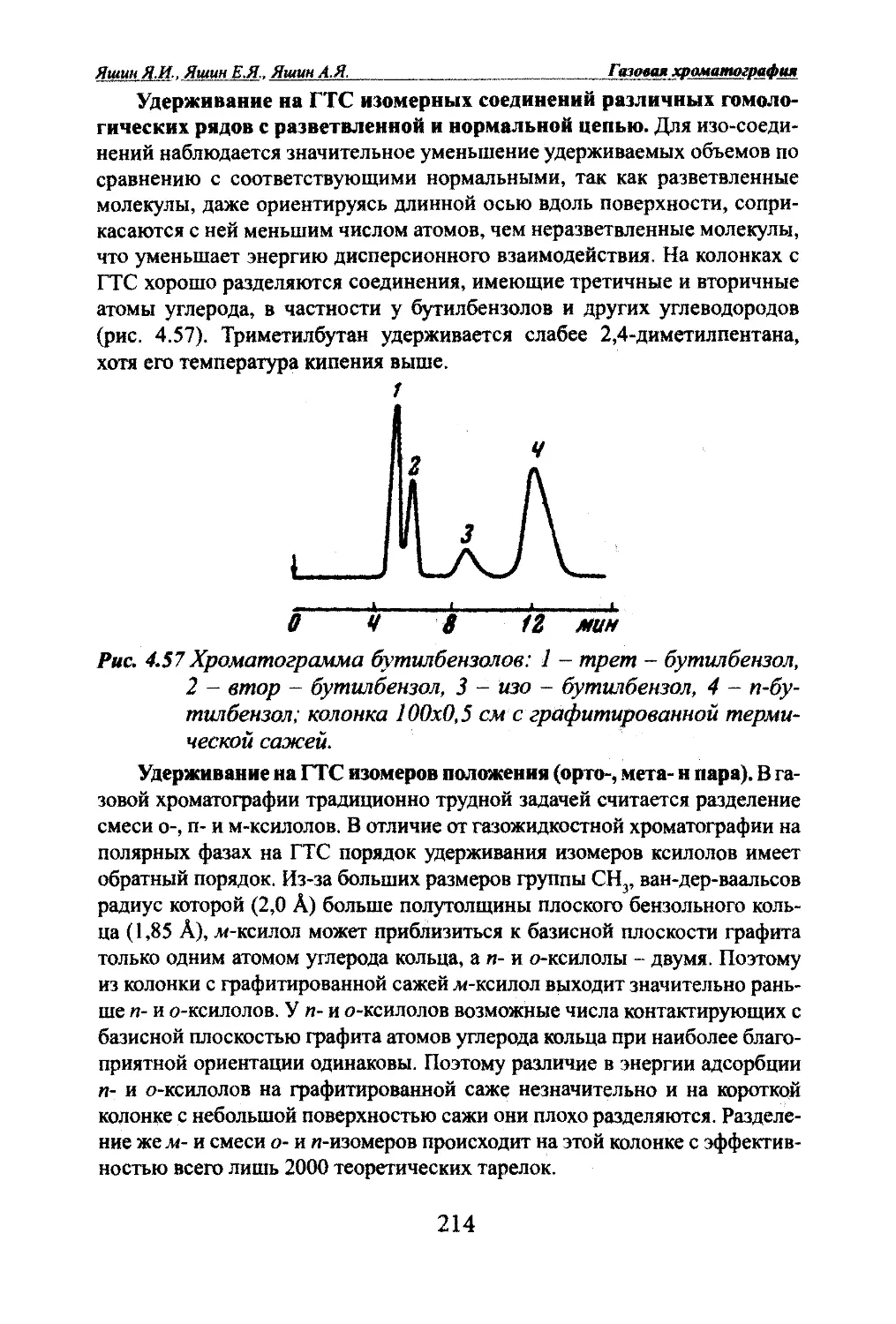
смесь вводят в начало колонки, и затем компоненты смеси по слою сорбен¬
та продвигаются потоком другого вещества, которое сорбируется сильнее
любого из компонентов смеси. Это соединение вытесняет распределяемые
в колонке вещества. По мере продвижения при создании оптимальных
условий получаются зоны, каждая из которых соответствует определенно¬
му веществу, однако они полностью не могут быть отделены друг от друга
на хроматограмме (рис. 1.1 в).По сравнению с проявительным методом в вытеснительной хромато¬
графии также имеются недостатки.1. Необходимость регенерации колонки, причем в более жестких усло¬
виях (высокая температура), чем в случае фронтальной хроматографии,
так как колонка заполнена сильно сорбирующимся веществом.2. Недостаточно полное разделение, зоны непосредственно примыка¬
ют друг к другу.Этот метод, как и фронтальный, мало использз^от в аналитической
практике. В последние годы к нему проявляется интерес в препаративной
жидкостной хроматографии.Остальные методы, приведенные в таблицах 1.2 и 1.3, будут рассмотре¬
ны подробно в соответствующих главах. Не рассмотрены методы газовой
хроматографии, не применяемые или малоприменяемые в аналитической
газовой хроматографии, в частности обращенная газовая хроматография
для исследования химической природы поверхности твердых тел по тесто¬
вым соединениям [4], жидкостно-газовая [5] и мембранно-хроматографи¬
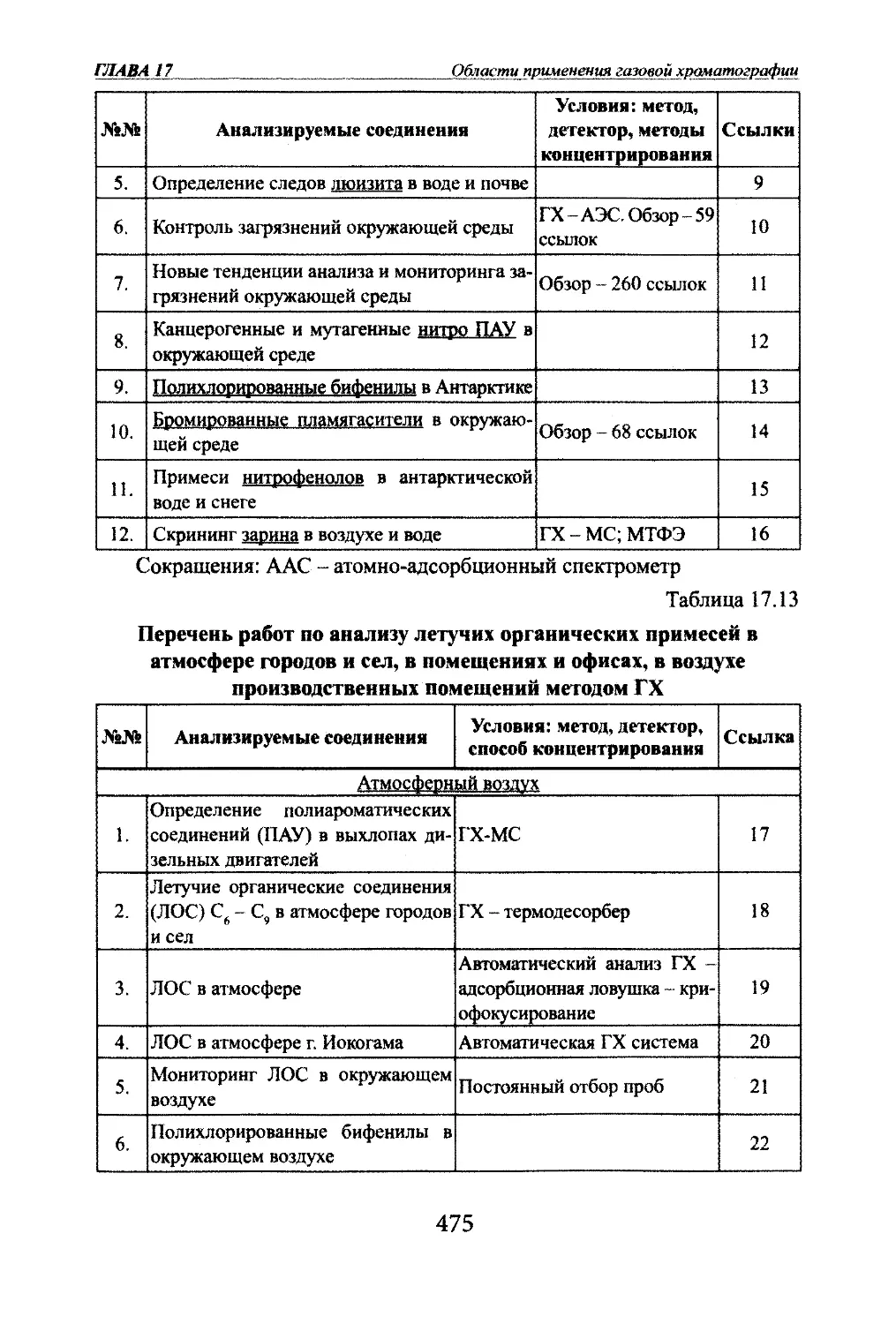
ческие методы [6].1.3 История открытия хроматографииХроматография была открыта русским ботаником М.С.Цветом в 1903 г.
[7] (фото 1).На заседании общества естествоиспытателей 14 марта 1903 г. он вы¬
ступил с сообщением об открыгии нового метода разделения сложных
смесей. Обстоятельные статьи об этом методе были опубликованы в 1906 г.
в немецких журналах [6]. В 1910 г. вышла книга М.С. Цвета «Хромофиллы10
glAMdl ВведениеВ растительном и животном мире», в
1Юторой большое внимание уделено
методу хроматографии. Всего М.С.Цветом опублиювано 62 научные
работы. М.С. Цвет был не только
первооткрывателем метода. Им были
детально разработаны методические
основы метода. Достаточно сказать,
что М.С. Цвет исследовал более 100
разных адсорбентов. Предложенная
им техника хроматографического
анализа во многих своих частях поч¬
ти не изменялась в течение пятиде¬
сяти - шестидесяти лет. Хроматогра¬
фические колонки М.С. Цвета имели ^ тт
высокую эффективность. Он также ■ ^С-Цвет
впервые продемонстрировал высокую результативность многоколоноч¬
ных вариантов хроматографии.За свои работы М.С.Цвет неоднократно награждался. Он был удо¬
стоен академической премии за н^^чную работу по хромофиллам в рас¬
тительном и животном мире, награжден орденами св. Станислава III и II
степени (1907 г., 1915 г.), св. Анны III степени (1912 г.) и юбилейной ме¬
далью в честь 300-летия дома Романовых. В 1918 г М.С.Цвет вьщвигался
на Нобелевскую премию.Первая мировая война на долгие годы затормозила и ю многих направ¬
лениях прервала научные исследования в Европе и мире. Ранняя смерть
М.С. Цвета в 1919 г. также не позволила развить метод в нашей стране.
По этим причинам хроматография мало использовалась до 1931 г. Мож¬
но выделить только единичные работы, в частности Л.Пальмера в США,Ч.Дьере в Швейцарии, Т.Липпмаа в Эстонии в университете в Тарту, в ко¬
тором М.С.Цвет некоторое время работал. В 1931 г. Р.Кун, А.Винтерштейн
к Е.Ледерер [9] практически повторили эксперименты М.С.Цвета и убе¬
дились в их огромных возможностях. Эти ученые имели в своем распо¬
ряжении немецкий перевод книги М.С.Цвета. С этого времени началось
быстрое распространение хроматографического метода в европейских
странах, чему способствовали первые опубликованные на немецком язы¬
ке книги по хроматографии Л.Цехмейстера и Л.Чолноки [10] и Г.Хессе
[11]. Последнему автору принадлежат и первые упоминания о газовой
адсорбционной хроматографии, опубликованные в 1941 г. К 40-летне¬
му юбилею хроматографии в нашей стране вышел сборник избранных11
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я. Гаювая хртйшощфштрудов М.С.Цвета в серии «Классики науки» с биографией М.С.Цвета
под редакцией А.А.Рихтера и Т.А.Красносельской [12]. Перевод книги
Л.Цехмейстера и Л.Чолноки на английский язык способствовал развитию
хроматографии в Англии и США. Не случайно с 1940 п в развитие хрома¬
тографии стали вносить огромный вклад английские исследователи, осо¬
бенно А.Мартин и его сотрудники. В 1941 г А.Мартиным и Р.Синджем
был предложен метод жидкостно-жидкостной хроматографии [И]. В 1944
г. Р.Консдан, А.Гордон и А.Мартин предложили метод бумажной хрома¬
тографии [14]. Еще раньше (1938 г) русскими учеными Н.А.Измайловым
и М.С.Шрайбер предложен метод тонкослойной хроматографии. С 1940
г. по 1944 г. шведами А.Тизелиусом и С.Классоном разработаны вариан¬
ты фронтальной, вытеснительной и проявительной хроматографии. Гра¬
диентное элюирование было предложено рядом авторов в 1952 г Идеи
аффиной (биоспецифической) хроматографии бьши предложены в 1951 г.
Д.Кемпбеллом. В последующих работах этот метод был доведен до прак¬
тического внедрения. В течение 1930 - 1970 гг. четырнадцать крупных
работ в области биологии и биохимии удостоены Нобелевских премий.
В настоящее время многими авторитетными специалистами признано,
что эти работы не были бы выполнены без применения хроматографии.
В 1953 г. в нашей стране и за рубежом было широко отмечено пятиде¬
сятилетие открытия хроматографии: 18-21 ноября 1953 г., в Москве при
участии Комиссии по хроматографии АН СССР состоялось совещание по
применению хроматографических методов. 50-летию хроматографии был
посвящен специальный выпуск «Журнала аналитической химии». К это¬
му юбилею вышли из печати публикации К.В. Чмутова, К.М.Ольшановой,
В.В.Рачинского, Т.Б.Гапон, М.М.Сенявина, Г.В.Самсонова и других.
Юбилейные совещания и публикации прошли в Чехословакии и в США.В Германии к этому юбилею выпустили на
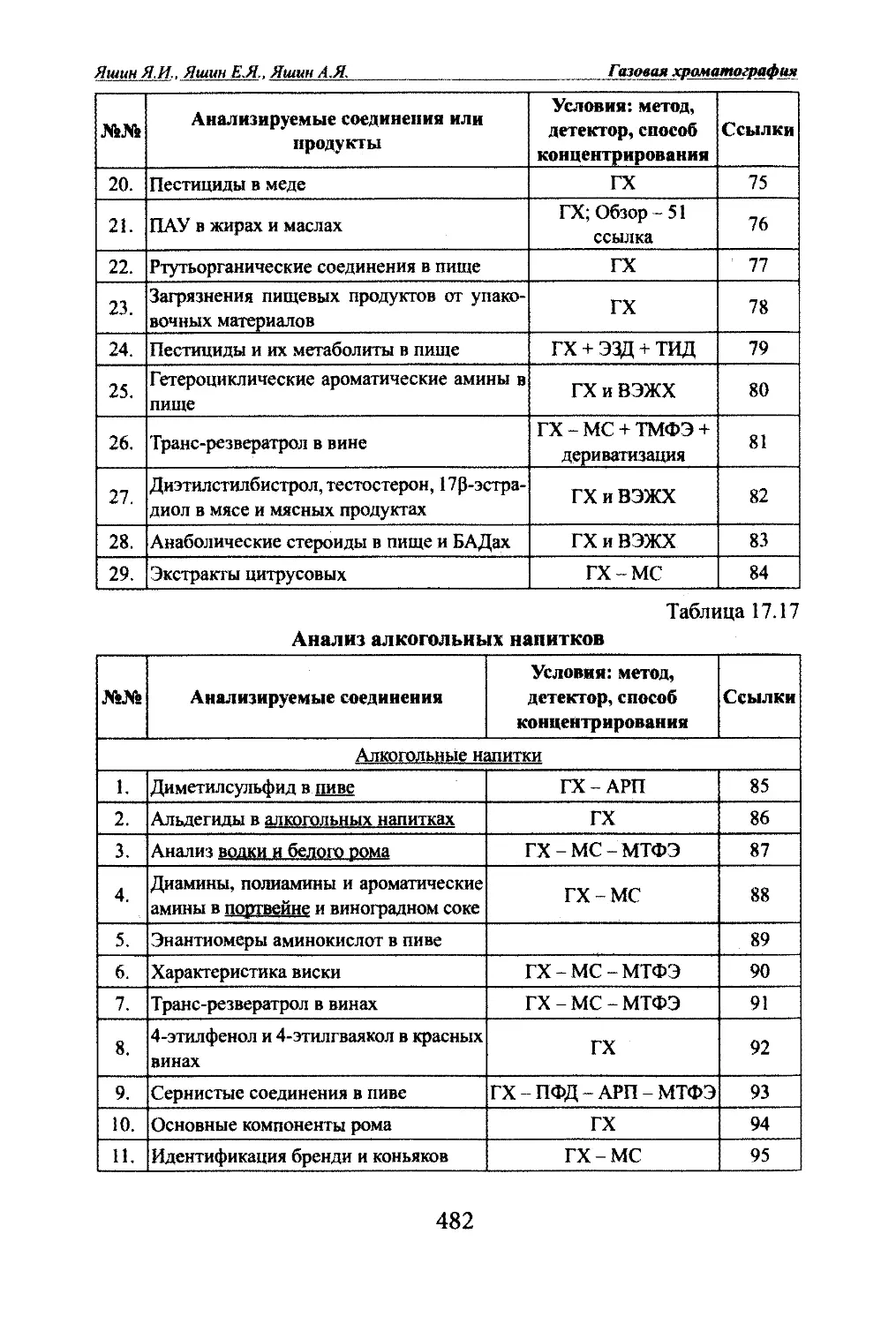
немецком и английском языках первую ра-(к боту М.С.Цвета о методе хроматографии.Особенно широко был отмечен столет-
1^^ НИИ юбилей со дня рождения М.С.Цвета
■■ в 1972 г. В этот год было проведено не-
сколько Международных симпозиумов, в
том числе и в Ленинграде. В связи с этим
юбилеем Американским химическим об¬
ществом была учреждена в СШ А Между¬
народная медаль им. М.С.Цвета «За вы-
Фото 2. Медаль «За выда- дающиеся открытия в области хромато-
ющиеся достиженш в хро- графии» (фото 2).
матографии».12
nJABA 1 ВведениеЭтой медалью награждены трое наших ученых: А.В.Киселев,
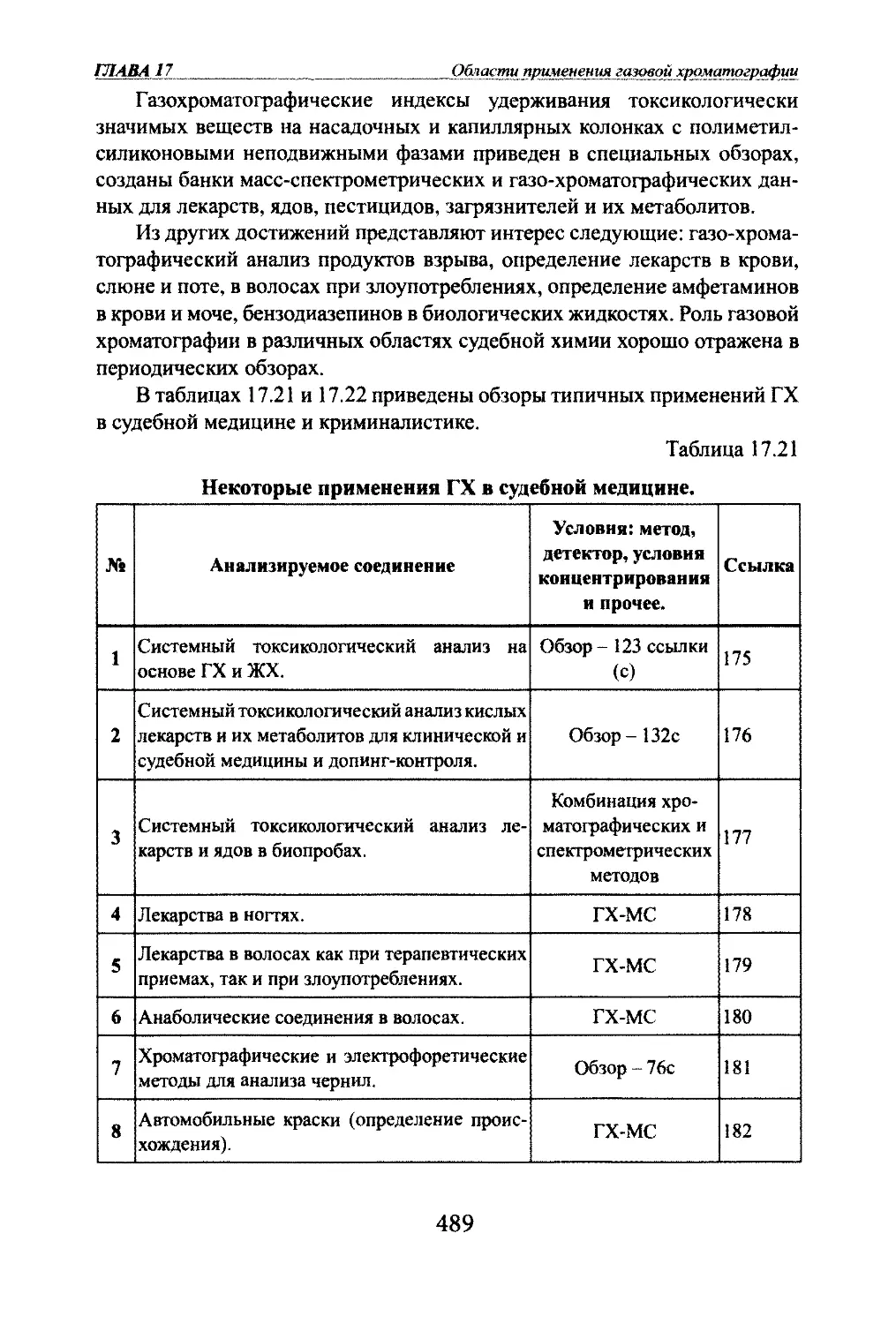
Д.Л.Жуховицкий и К.И.Сакодынский. 75-летию открытия хроматографии
был посвящен Международный симпозиум в Таллинне в 1978 г. К этому
юбилею была выпущена отечественная медаль им. М.С.Цвета, которой
была награждена большая группа советских и зарубежных специалистов
(фото 3).Фото 3. Медаль им. М. С.Цвета по случаю
75-летия открытия хроматографии.Первые биографические сведения о М.С.Цвете были опубликованы в
работах [13,14]. В поиске новых данных и в исследованиях материалов о
жизни и деятельности М.С.Цвета и истории открытия хроматографии пер¬
востепенная роль принадлежит профессору К.И.Сакодынскому [13]. Про¬
фессором К.И.Сакодынским совместно с режиссером П.А.Солуяновым
был создан н^ный документальный фильм о М.С.Цвете и о примене¬
нии хроматографии. Работы о жизни и деятельности М.С.Цвета изданы
Е.М.Сенченковой [14-16]. Истории хроматографии посвящены многие ра¬
боты Л.Эггре, М.С.Вигдергауза, В.Г.Березкина, Я.И.Яшина [18].К 75-летию открытия хроматографии в США вышла книга под редаищей
Л.Эггре и А.3лагкиса «75 лет хроматофафии - исторический диалог» [19], в
кото{юм наибояте известные спещ1алисты по хроматографии рассказывают о
том, как они пришли в хроматофафию и своих достижениях в этой области.В 2(ЮЗ г. в Москве состоялся Международный Симпозиум, посвящен¬
ный 100-летию открытия хроматофафии. Этот симпозиум открывал мэр
г.Москвы Ю.М.Лужков, который поразил всех участников своими знани¬
ями актуальных задач современной хроматофафии. К этому знаменатель¬
ному юбилею вьшши две книги, обобщающие достижении хроматофафии
в двадцатом веке [20,21].Большую роль в развитии и распространении хроматофафии в СССР
и Российской Федерации ифал и ифает Научный Совет по хроматофафии
при РАН, созданный в 1949 года.13
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я. Газовая зфоматогрдфщПервым председателем Научного Совета был К.В.Чмутов, затемО.Г.Ларионов, В.А.Даванков, Н.В.Поляков. В настоящее время председа¬
телем Совета является А.К.Буряк.В структуре Совета имеются секции по всем основным методам хро¬
матографии. Комиссии объединяют специалистов хроматографии по наи¬
более актуальным областям применения хроматографии. Самую большую
работу Научный Совет проводит по организации симпозиумов, шнферен-
ций и семинаров по хроматографии в нашей стране. Проведены десятки
общих симпозиумов по газовой хроматографии в разных городах, совеща¬
ний и конференций по применениям хроматографии в химичесюй, пище¬
вой промышленности, в медицине и биологии, по анализу примесей, по
сорбентам, по препаративной, реакционной хроматографии и другим во¬
просам.Постоянные действующие семинары по хроматофафии были в Мо¬
скве, Нижнем Новгороде, Самаре, Санкт-Петербурге.Курсы по повышению квалификации по хроматофафии действуют до
сих пор в Москве, Дзержинске.При Институте физической химии РАН создан и успешно работает На¬
учный Совет по защите докторских и кандидатских диссертаций по хрома¬
тофафии, председатель Совета - профессор О.Г.Ларионов. Высшей атте¬
стационной комиссией введена особая специальность «Хроматофафия и
хроматофафические приборы».В 1989 г. в СССР бьша создана Ассоциация хроматофафистов (первым
председателем был проф. К.И.Сакодынский, в настоящее время - проф.В.В.Помазанов), шторая сосредоточила свои основные усилия на разви¬
тии прикладных вопросов хроматофафии.В нашей стране десятки тысяч специалистов работают в области хро¬
матофафии. Более 80 специалистов защитили докторские диссертации,
свыше 1000-кандидатские.Несколько Государственных премий СССР и РФ были присуждены за
работы в области хроматофафии. Бьши выпущены два постановления ЦК
КПСС и Правительства о развитии хроматофафии в нашей стране.Краткая история развития газовой хроматофафии. Первые упомина¬
ния о юзможности реализации газоюй хроматографии относятся к 1941 г. В
1949-1950 гг. появились работы по применению газо-адсорбционной хро¬
матофафии для разделения и анализа постоянных и углеводородных га¬
зов.Быстрое развитие газовой хроматофафии началось с 1952 г., когда
Мартин и Джемс [22] предложили метод газо-жидкостной хроматофафии,
которая значительно расширила возможности газовой хроматофафии.14
ГЛАВА!ВведенжЗа разработку хроматографических методов и применение их в био-
химии Мартин был удостоен Нобелевской премии (фото 4).Фото 4. А. Мартин
В 1955-1956 гг. появились первые теории по размыванию в газохрома¬
тографических насадочных колонках.Значительным достижением явилось открытие капиллярной газовой
хроматографии М.Голэем (фото 5) [23].Фото 5 М.ГолэйПервая книга по прикладным вопросам капиллярной хроматографии
была выпущена Р.Кайзером (фото 6) [24].Фото 6. РКайзерРазвитию газовой хроматографии способствовали работы и книгаА.Кейлеманса [25] (фото 7).Фото 7. А.Кейлеманс
15
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я. Гтовая хроматографияОсновные отечественные достижения по разработке новых и развитию
уже известных методов газовой хроматографии приведены в таблице 1.4.Прежде всего необходимо отметить работы проф. А.А.Жуховицюго (фото
8) [26,27,29, 30,32-35,48] и проф. А.В.Киселева (фото 9) [36,39,40].Фото 8. А.А.Жуховицкий Фото 9. А.В.КиселевТаблица 1.4Отечественные достижения в разработке новых методов газовой
хроматографии и развитии известных.шшОткрытие новых методов и
вариантов хроматографии
или развитие известных
методовАвторыГадСсылка1.ХроматермографияЖуховицкий А. А.,
Туркельтг^ Н.М.1951262.Теплодинамический методЖуховицкий А. А.1953273.Предложен объемный метод
хроматографииВяхирев Д.А.1953284.Вакантная хроматографияЖуховицкий А.А.1962295.Ступенчатая хроматографияЖуховицкий А.А.,
Туркельтауб Н.М.1962306.Капиллярная адсорбционная
хроматографияКалмановский В.И.,
Жданов С.П.,
Киселев А.В.,Фикс М.М.,Яшин Я.И.1962317.ИтерационнаяхроматографияЖуховицкий А.А.1963328.ДифференциальнаяхроматографияЖуховицкий А.А.1966339.Хроматография без газа
носителяЖуховицкий А.А.19723410.ХромадистилляцияЖуховицкий А.А.19783511.Предложена хроматоскопияКиселев А.В.19783616
ГЛАВА 1№/№Открытие новых методов и
вариантов хроматофафии
или развитие известных
методовАвторыГодВведениеСсылка12.Жидкостно-газоваяхроматографияМосквин Л.Н.,
Горшшв А.И.,
Гумеров М.Ф.19823713.Циркуляционная
хроматография Чижков В.П.,
Стерхов Н.В.19913814.Развитие газо-адсорбционной
хроматографииКиселев А.В.,Яшин Я.И.,
Щербакова К.Д.,
Никитин Ю.С. и др.1959-19843915.Развитие газо-адсорбционно-
адсорбционной хроматогра-
фии Киселев А.В.,
Яшин Я.И.,
Березкин В.Г.1960-198040,4116.Развитие реакционной хро¬
матографииБерезкин В.Г.,
Алишоев В.Р.,
Алексеева К.В.1965-19704217,Развитие газовой хромато-
масспектрометрии для ана-
диза примесей И.А.Ревельский4318.Развитие газовой экстракции
(анализ равновесного пара)Б.В.Иоффе,А.Г.Витенберг,Б.В.Столяров4419.Развитие хроматографии на
жидких кристаллах М.С.Вшдерг^,Л.А.Онучак4520.Новые методы расчета газо-
хроматографическихиндексов
удерживания. Связь индексов
>«ержнваиия со структурой
молекул. Идеитифшощия по
индексам удерживания. Зенкевич И.Г.1990-20054621.Классификация неподвиж¬
ных жидких фаз по термоди-
намическим параметрам.Головня Р.В.1970-198047По разным экспертным оценкам хромагофафию относят к 20 выдаю¬
щимся открытиям XX века, которые в наибольшей степени преобразовали
жизнь человека.Хроматофафия несомненно способствовала профессу в XX веке мно¬
гих разных направлений н^кн, техники и промышленности.172 Гаэовы хромжгографня
Яшин ЯМ. Яшин ЕЛ, Яшин А.Я. Газовая хроматографияСледует выделить три основные области применения хроматографии:
анализ, выделение веществ в чистом виде и исследования.Хроматография в конце своего столетия представляет собой:- самый распространенный и совершенный метод разделения
сложных многокомпонентных смесей;- самый универсальный метод анализа, десятки разных методов и
вариантов, позволяющих разделять все виды смесей (разделение смесей
атомов, изотопов, молекул, всех типов изомерных молекул, вклютая и
оптические изомеры, макромолекул (полимеров и биополимеров), ио¬
нов, комплексов устойчивых свободных радикалов, мшфочастиц);- самостоятельное научное направление, физиш-химический ме¬
тод исследования и измерения;- препаративный и промышленный метод выделения веществ в
чистом виде;- мощную отрасль промышленного производства, сотни фирм во
всем мире выпускают хроматографическую аппаратуру и вспомогатель¬
ное оборудование для нее на сумму более 10 млрд. долларов, только
газовых хроматографов выпускается на сумму 1,5 млрд. долларов, жид¬
костных хроматографов высошго давления - на сумму 3 млрд. долла¬
ров, не считая аппаратуры для ионной, сверхкритичесшй, тонкослойной
хроматографии, тгиллярного электрофореза, сотни различных допол¬
нительных устройств, программного обеспечения, шпонок, сорбентов,
расширяющих аналитические возможности хромаиирафов.Во многих публикациях часто подчеркивается, что хроматография
играет большую роль в жизненно важных областях: контроле загрязнений
окружающей среды, анализе пищевых продуктов, в медицине, биологии,
производстве и др.Ни один аналитический метод не конкурентен с хроматографией по
эффективности разделения сложных многокомпонентных смесей, ни один
метод химического и физического разделения не может быть сравним с
хроматографией по универсальности и широте применения.На эффективных капиллярных колонках возможно одновременное раз¬
деление более 1000 компонентов бензиновых фракций за один ввод, опреде¬
ление супертоксикантов, например диоксинов, на уровне ppt (1х10 '®%).Хроматографические методы чаще всего применяются для целей ана¬
лиза, диапазон применения огромен: от анализа атмосферы планет, до ана¬
лиза содержимого одной клетки.При анализе сложных многокомпонентных смесей хроматографиче¬
ские методы вне конкуренции. Среди всех современных аналитических18
Введениеметодов хроматография занимает лидирующее положение. Это ярю де¬
монстрируют ежегодные Пштсбургские шнференции по аналитической
химии и прикладной спектроскопии и выставки приборов на ней.
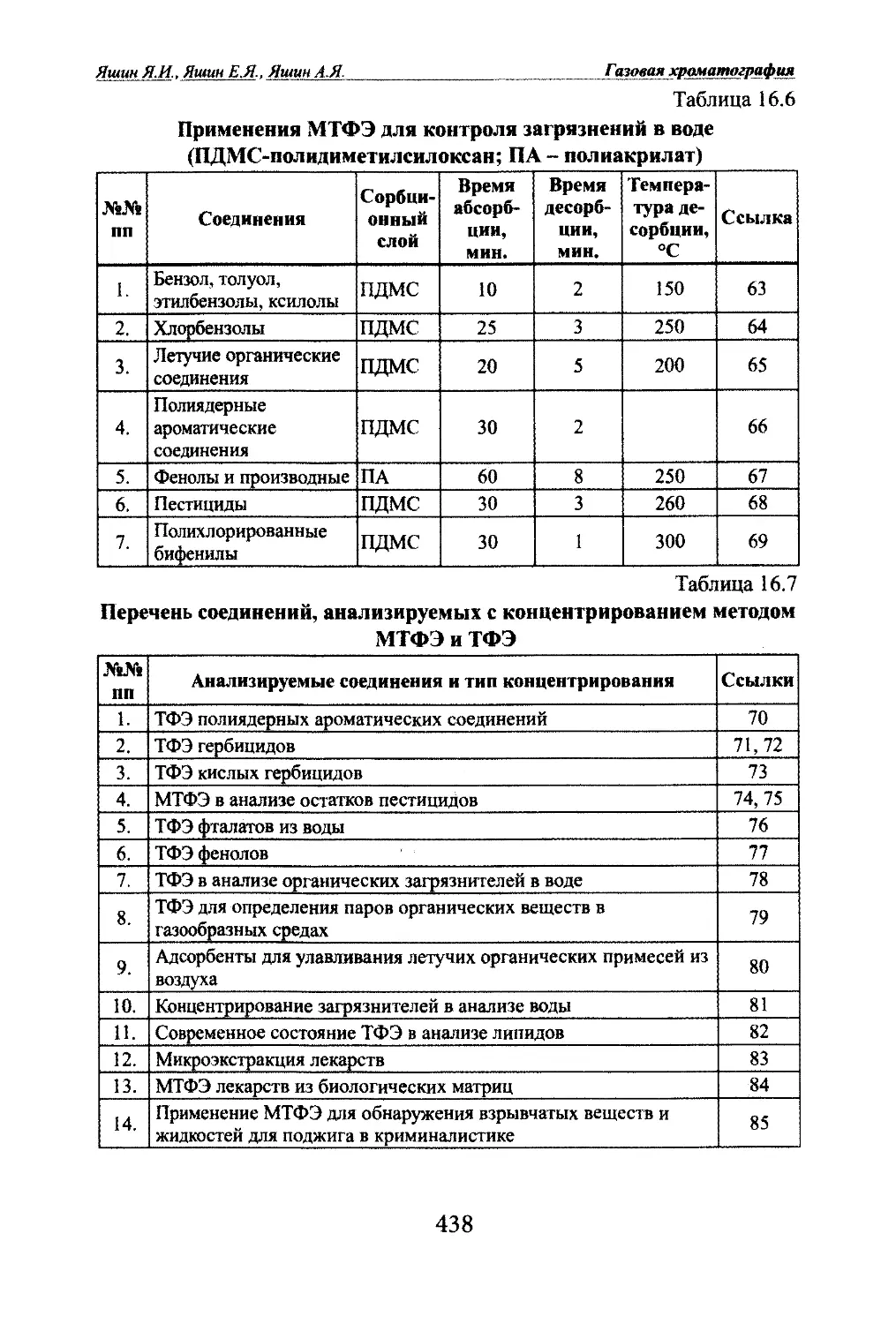
Основные применения ГХ1. Технологический контроль в химичесшй, нефтехимической, га¬
зовой промышленности.2. Контроль загрязнений окружающей среды (воздух, вода, почва).3. Сертификащ1я пищевых продуктов.4. Клинические анализы, применения в биологии и медищ1не.5. Применение в геологоразведке, в частности поиск нефти и газа.Основные аналитические достижения ГХ1. Анализ супертоксикантов (диоксинов, ПХБ, ПАУ, пестицидов,
афлатоксинов, нитрозоаминов и др.) на уровне ррЬ и ppt.2. Анализ нефтепродуктов и нефти: компонентный и групповой,
анализ ПИОНА, имитированная разгонка нефтепродуктов.3. Энантиомерный анализ.4. Анализ компонентов запахов и ароматов.5. Анализ атмосферы планет.6. Анализ феромонов (половых гормонов) насекомых.7. Анализ органических загрязнителей атмосферы городов.8. Криминалистические анализы.9. Допинговый контроль.10. Анализ микропримесей взрывчатых веществ.11. Анализ биомаркеров для ранней диагностики заболеваний.12. Анализ пищевых продуктов и напитков.13. Анализы в энергетикеи многие другие.1.5 Наукометрия газовой хроматографииПодробный анализ публикаций по газовой хроматографии проведен в
работах [49-56]. К 2007 г по газовой хроматографии опубликовано более 80
тысяч статей, тысячи обзоров и сотни книг. В настоящее время ежегодно
выходят около 2000-2200 статей. Наибольшее число публикаций относится
к капиллярной газовой хроматографии. Они составляют более 25% всех пу¬
бликаций. До 70% всех анализов с помощью газовой хроматографии прово-
датся на катгаллярных шлонках. Практически все комбинации ГХ-МС вы¬
полняются с применением капиллярных колонок. В работе [53-56] приве¬
дены данные по публикациям по следующим методам газовой хроматогра¬
фии: капиллярной, пиролизной, многомерной, реакционной, обращенной,
препаративной, промышленной и др.19
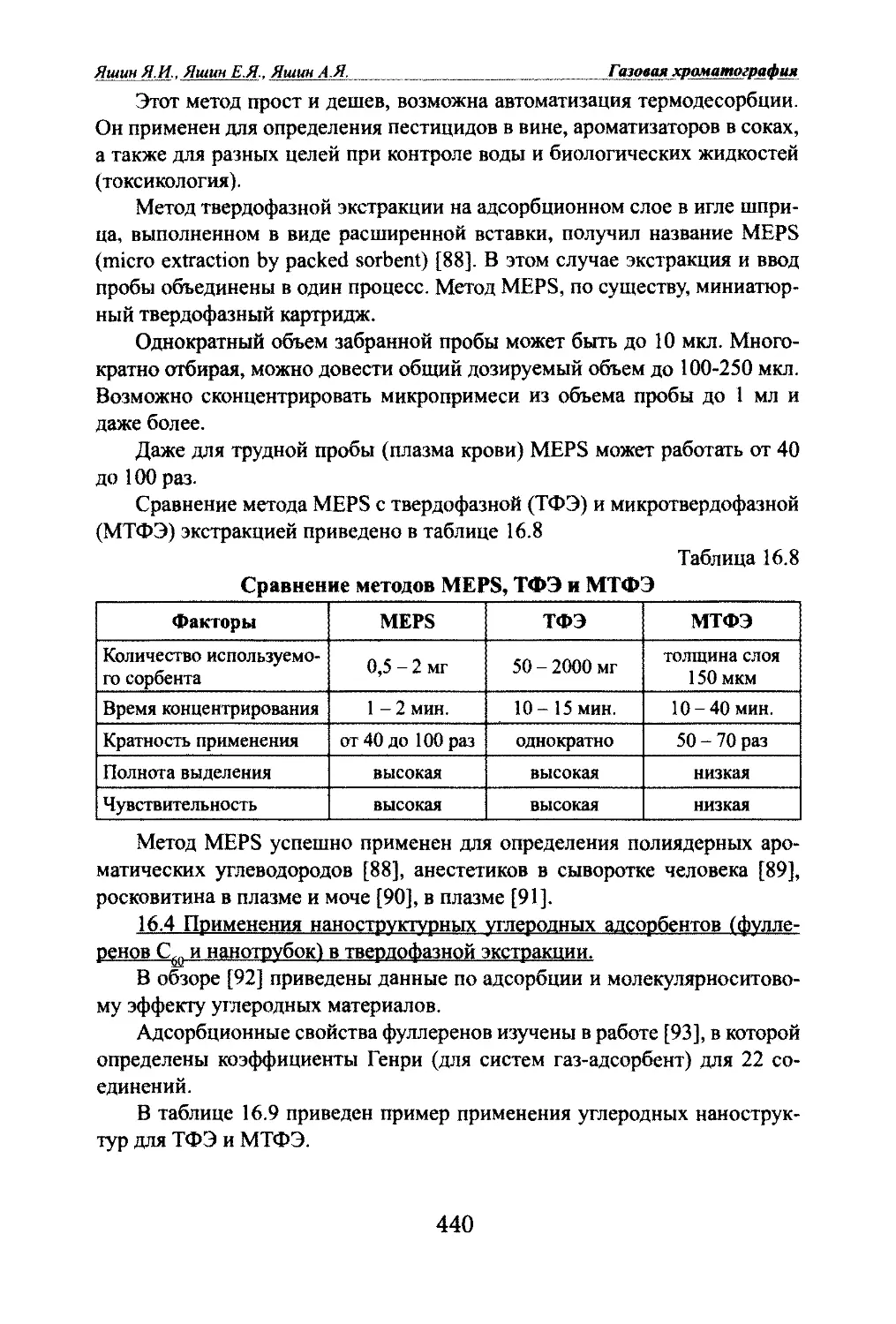
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я. Газовая хроматографияОсновные новые направления в газовой хроматографии, интенсивно
развивающиеся в последние годы, это высокоскоростные, многомерные и
высокотемпературные варианты газовой хроматографии.Темы публикаций по общим вопросам теории и аппаратуры газовой
хроматографии следующие: обзоры и книги, основы теории, общие во¬
просы, термодинамика параметров удерживания, физико-химические из¬
мерения, связь структуры молекул с параметрами удерживания, сорбенты,
комбинация ГХ с другими физико-химическими методами, детекторы, ап¬
паратура и дополнительные устройства, автоматизация и компьютериза¬
ция аппаратуры.Среди областей применения в библиографических указателях выделя¬
ются, в основном, следующие (в порядке уменьшения числа публикаций):
анализ загрязнений окружающей среды, анализ пищевых продуктов, ана¬
лиз лекарств, клинический анализ, токсикологические и судебные анали¬
зы, разделение и определение энантиомеров.Методом газовой хроматографии анализируются соединения с моле¬
кулярными массами от 2 до 500 и выше, т.е. летучие соединения и малоле¬
тучие соединения, которые при испарении не разлагаются. Публикации пО
разным классам химических соединений также приведены в обзоре [56].
Больше всего публикаций по углеводородам и их кислородосодержащим
производным. В последние десятилетия растет число публикаций по серо¬
содержащим, фосфорсодержацщм и металлоорганическим соединениям.После развития и широкого применения высокоэффективной жид¬
костной хроматографии, число публикаций по анализам соединений био¬
логического происхождения газовой хроматографией уменьшилось.1.6. Преимущества газовой хроматографии.Широкое применение газовой хроматографии в разных областях нау¬
ки, техники и промышленности, несомненно, связано с ее преимущества¬
ми по сравнению с другими методами анализа. Среди них можно вьщелить
следующие.Высокая разделительная способность газохроматографических ко¬
лонок, позволяющая за приемлемое время разделять и анализировать лю¬
бые сложные многокомпонентные смеси. Например, на капиллярных ко¬
лонках можно полностью разделять бензиновые фракции в течение часа,
состоящие из 300-500 компонентов. Подобное другими методами сделать
невозможно. Эффективность и селективность колонок постоянно возраста¬
ет, что увеличивает их разделительную способность. Внедрение двухмер-20
rjIABA 1 Введениеных и многомерных вариантов хроматографии также расширяет возмож¬
ности разделения сложных многокомпонентных смесей. За счет высокой
разделительной способности обеспечивается большая информативность
метода газовой хроматографии. Методом газовой хроматографии на селек¬
тивных сорбентах можно разделять не только разнообразные соединения,
различающиеся природой и числом функциональных групп, но и все виды
изомеров: о, - м- и п- изомеров, цис- и транс-изомеров, других простран¬
ственных изомеров и даже оптических изомеров.Методом газовой хроматографии успешно разделяют изотопозаме¬
щенные соединения (дейгеро-замещенные и др.). Описаны разделения
изотопов простых молекул (O'* и О'*, N'"* и N'^ Ne“ и Ne^^ и др.).Универсальность метода. Методами газовой хроматографии можно
разделять и анализировать благородные газы, постоянные низшкипящие
газы, летучие жидкие и твердые вещества с температурой кипения до бОСУ’С
и выше. В сырой нефти разделяют и определяют углеводороды с числом ато¬
мов углерода от С,„ до С|рд (угаеводо1юды С,(^ имеют температуру кипения
ошло 700“С). Вещества кроме летучести, должны быть термически стабиль¬
ны, т.е. при переводе в парообразное состояние они не должны разлагаться.
Для анализа нелетучих и термически неустойчивых соединений методом га¬
зовой хромагографш предложены методы реакционной хроматографии.Следует отметить еще одно преимущество - универсальность газово¬
го хроматографа, один и тот же прибор может использовать для решения
сотен - тысяч аналитических задач, меняя в основном только разделитель¬
ную колонку и детектор.Низкий предел обнаружения (высокая чувствительность). Для
газовых хроматографов разработаны высокочувствительные детектиру¬
ющие системы как универсальные, так и специфические (селективные).
Пределы детектирования нешторых детекторов около 1- 10 ’-1- 10 '^ г/см’.
Для снижения пределов обнаружения еще на 2-4 порядка, например, су¬
пертоксикантов, предложены разнообразные методы концентрирования, в
основном методы газовой, жидкостной, твердофазной и сверхкритической
экстракции. Диоксины определяют методом газовой хроматографии на
уровне ppt, т.е. 10 '®% (об.).Экспрессность анализа. Время анализа методом газовой хроматогра¬
фии (без учета времени пробоподготовки) обычно в пределах 5-30 мин., в
некоторых случаях при разделении многокомпонентных смесей - 60-90 мин.
Однако в этих случаях определяются десятки-сотни компонентов. В послед¬
нее десятилетие разработаны методы сверхсшростной газовой хроматогра¬
фии, в 30-100 раз сокращающие время разделения и анализа.21
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я. Газовая хроматографияНадежность аппщ»атурного оформления. Через 3-4 года после ся-
крытия газо-жидкостной хроматографии были созданы коммерчесвше га¬
зовые хроматографы. Приборное оформление постоянно совершенствова¬
лось, и в настоящее время десятки фирм в мире серийно выпускают 35-40
тыс. газовых хроматографов в год. В газовых хроматографах используются
последние достижения электроники, теплотехники, механики, пневматики
и компьютерного управления и обработки результатов анализа. Несмотря
на сложность газовых хроматографов, они достаточно надежны. Для их
эксплуатации не нужны специалисты высокой квалификации.Малая погрешность анализа. Воспроизводимость последовательных
измерений (случайная погрешность) может колебаться от 0,01 до 1% (отн)
в зависимости от условий разделения и типа использованной аппаратуры.
Для уменьшения систематичесшй погрешности строят градуировочные
графики.По погрешности измерения метод газовой хроматографии находится
на уровне многих других физико-химических методов.Малый размер анализируемой пробы. Для газохроматографическо¬
го анализа в большинстве случаев необходимо ввести пробу в пределах0,1 - 10 мкл (Ю'^ - 10'^ г). Это означает, что газовая хроматография-
микрометод. Это особенно важно при анализах в медицине, судебной
медицине, криминалистике и в других случаях, когда отобранные про¬
бы малы.Возможность автоматизации анализа. Газохроматографический
анализ можно полностью автоматизировать с выдачей полного протокола
как в лабораторных, так и в производственных условиях. Для промьшшен-
ного контроля разработаны автоматические хроматографы, работающие по¬
стоянно без участия человека. Для космических исследований применяются
автоматические портативные хроматографы, управляемые с «земли».Недостатки метода газовой хроматографии. Несмотря на высокие
достоинства газовой хроматографии, этот метод имеет и свои недостатки.Первый недостаток - прерывистость процесса, анализ проводится
таким образом; вводится проба, она разделяется на отдельные компонен¬
ты, на выходе из колонки компоненты детектируются и регистрируются,
рассчитывается их концентрация, затем весь процесс повторяется. Для не¬
которых химических производств необходим непрерывный анализ, кото¬
рый могут выполнить только специальные анализаторы.Второй недостаток - в газовых хроматографах измерения относи¬
тельные, т.к. на выходе измеряется концентрация, сильно отличающаяся от
начальной концентрации в пробе. При вводе в поток газа-носителя она раз-22
ГЦЛВА I Введениебавляется, затем при продвижении по слою сорбента в колонке еще допол¬
нительно разбавляется за счет диффузионных и кинетических факторов.
Поэтому необходима градуировка, т.е. установление связи между концен¬
трациями в анализируемых пробах и сигналами, полученными на выходе
из колонки. При изменении любых условий разделения необходима новая
градуировка.1.7 Высказывания о хроматографии.1. «Подобно световым лучам в спектре, различные компоненты
сложного пигмента закономерно распределяются друг за другом в
столбе адсорбента и становятся доступными качественному и количе¬
ственному определению».М.С.Цвет2. «Открытие хроматографии можно сравнить с созданием микро¬
скопа, благодаря которому открылся неведомый мир компонентов
сложных природных и синтетических смесей».А.А. Жуховицкий3. Лауреат Нобелевской премии П. Каррер, выступая на междуна¬
родном конгрессе по теоретической и прикладной химии (1997 г. Лон¬
дон), говорил: «Ни одно другое открытие не оказало столь больщого
влияния на органическую химию и не расщирило в такой мере ее об¬
ласти исследования как хроматографический адсорбционный анализ
М.С.Цвета. Исследования в области витаминов, гормонов, каротинои-
дов и многочисленных других природных соединений никогда не мог¬
ли бы так быстро прогрессировать и достичь таких успехов, если бы
не этот новый метод, который позволил обнаружить в природе наличие
невероятного разнообразия близких по природе соединений».4. «Метод Цвета осуществил заветную мечту химика - разделить и
проанализировать продукты синтеза».Цехмейстер, Чолноки, 1938 г.5. «В 1906 г. в Варшаве был предложен новый остроумный ме¬
тод химического анализа, которому предназначено оказать влияние на
жизнь человечества и всего живого мира. Он позволяет осветить слож¬
нейшие процессы природы, как-то: процессы питания, влияние гормо¬
нов на вид и характер людей и животных. Благодаря ему, в сложном
механизме живой клетки были обнаружены реакции, ранее не снив¬
шиеся и во сне».Стрэйн, 1942 г.23
Яишн ЯМ. Яшин Е.Я.. Яишн А.Я. Газовая хроматография6. «Я надеюсь, что в результате этих условий будет создано еще
более сильное стремление к внесению этой очаровательной техники,
которая имеет неоценимые заслуги в различных областях органиче-
сшй и биологической химии».проф. Гейльброн в предисловии к ангаийсшму изданию книгиЦехмейстера и Чолноки, 1993.7. «Когда я работаю с методом хромэтографии, то я чувствую, что
нахожусь в химическом раю. После проведения химической реакции я
могу сразу видеть разделенными продукты реакции».проф. Доринг, Германия
fvtARA 1 ЛитератураЛитература1. Даванков В.А. Журнал аналитической химии 2001, т. 56, с. 101,
с. 1211.2. Хроматография. Основные понятия. Терминология. Под редВ.А.Даванюва, РАН, Москва, 1997,48 с.3. Березкин В.Г. Что такое хроматография? Москва, «Наука» 2005
76с.4. Dieckmann F., Posoiech D., Uhlman P. Polymer, 1997, v.38, N23
p.5887.5. Москвин Л.Н., Горшшв А.И., Гумеров М.Ф. Докл. АН СССР, 1982
т. 265, № 2, с. 378.6. Москвин Л.Н., Родников О.В. В книге «Хроматография на благо Рос
сии» Под ред. А.А.Курганова, Москва, Граница 2007, с.215.7. Цвет М.С. Труды Варшавсшго общества естествоиспытателей. Отд
биологии, 1903, т. 14, с. 1.8. Tswett M.S. Вег. Dtsch. Botan. Ges., 1906, Bd. 24, S. 316-323, S. 384.9. Kuhn R., Winterstein A., Lederer E. Z. Physiol. Chem., 1931, Bd. 197,S. 141.10. Zechmeister L., Chohioky L. Die Chromatographische Adsorption meth-
ode, Grundlegen, Methodik Anwendungen, Wien, Springer Verlag, 1937.11. Hesse G.E. Adsorption Methodik in chemischen Laboratorium, Berlin,
1943.12. Цвет М.С. Хроматографический адсорбционный анализ. Под ред.
Рихтера А.А. и Красносельской Т.А., Москва, Изд-во АН СССР, 1946.
Сер. «Классики назш1».13. Сакодынский К.И. М.С.Цвет и хроматография. Рига: Зинайтне.
1972, 72 с.14. Sakodynskii K.I. Michael Tswett, Life and woric. Carlo Erba, 1983,
Milano, 62 p.15. Сенченкова E.M. Михаил Семенович Цвет. Москва, Наука, 1973,
306 с.16. Сенченкова Е.М. Рождение идеи и метода адсорбционной хромато¬
графии. Москва, Наука, 1991, 228 с.17. Сенченкова Е.М. М.С.Цвет - создатель хроматографии. Москва,
«Янус-К», 1997,434 с.18. Яшин Я.И. 90-летняя история хроматографии (1903-1993) Ж.аналит.
химии 1994, т.49, №10, с. 1047-1058.19. 75 Years of Chromatography - а historical dialogue. Ed. by Ettre L.S.
and Zlatkis A. Elsevier, Amsterdam, 1979.25
Яишн ЯМ, Яшин ЕЖ. Яшин А.Я. Газовая хроматография20. Chromatography а century of discovery 1900-2000 - the bridge to the
sciences/technology. Ed. by C.W.Gehrke, R.L.Wixom and E.Bayer.21. 100 лет хроматографии, Под ред. д.х.н. Б.А.Руденко Москва, Н^ка,
2003, 740 с.22. James А.Т., Martin A.J.P. Biochem, J. 1952, v.50, p.679.23. Голэй M.B. кн.: Газовая хроматография. Сб. докладов на II Между¬
народном симпозиуме в Амстердаме. Пер. с англ. Москва. Издатинлит.
1961.С.39-60.24. Kaizer R. Chromatography in Gas Phase. II Capillary Gas Chromatog¬
raphy, Mannheim. Bibliographisches Institut. 1961,345 p.25. Кейлеманс A. Хроматография газов. Пер. с аиг. М.И.Яновсшго.
М.Издатинлит 1959. 320 с.26. Жуховицкий А.А. Докл. АН СССР, 1951, т.77, с.435.27. Жуховицкий А.А., Туркельтауб Н.М., Георгиевская Т.В. Докл. АН
СССР, 1953, т.92, С.987.28. Вяхирев Д.А., Комиссаров П.Ф. Докл. АН СССР, 1953, т. 129, с. 138.29. Жуховицкий А.А., Туркельтауб Н.М. Докл. АН СССР, 1962, т. 143, с.646.30. Жуховицкий А.А., Туркельтауб Н.М. Докл. АН СССР, 1962, т. 144,C.829.31. Жданов С.П., Калмановский В.И., Киселев А.В., Фикс М.М., Яшин
Я.И. Ж. Физ. Хим., 1962, т.56, с.1118.32. Жуховицкий А.А., Туркельт^ Н.М. Докл. АН СССР, 1963, т. 150,
с.ПЗ.33. Жуховицкий А.А. Зав. лаб. 1966, т.32, с.402.34. Жуховицкий А.А. Ж. аналит. хим. 1972, т.27, с.971.35. Жуховицкий А.А., Яновский С.М., Шварцман В.П. В сб. «Хромато¬
графия» т.2, Москва ВИНИТИ, 1978, с. 49.36. Kiselev А.V. Chromatographia 1978, v.ll, №12, р.691.37. Москвин Л.Н., Горшков А.И., Гумеров М.Ф. Докл. АН СССР, 1982,
т.265, №2, С.378.38. Стерхов Н.В., Чижков В.П. Зав. лаб. 1991, т.57,с.1.39. Киселев А.В., Яшин Я.И. Газо-адсорбционная хроматография, М.
Наука, 1967,256 с; Kiselev A.V., Yashin Ya.I. Gas Adsorption Chromatog¬
raphy, Plenmn Press, New York, 1969,254 p.40. Киселев A.B., Яшин Я.И. Адсорбционная газовая и жидкостная
хроматография, М. Химия , 1979, 288 с.41. Березкин В.Г. Газо-жидко-твердофазная хроматография, М. Химия,
1986, 112 с.42. Березкин В.Г. Аналитичес1шя реакционная газовая хроматография,
М. Наука, 1966, 184 с.26
gjdMl ..JMBmemm43. Ревельский И.А. и др. в книге «100 лет хроматографии» под ред.
Б.А.Руденмо, Москва, Н^та, 2003, с.529.44. Витенберг А.Г., Иоффе Б.В. Газовая экстракция в хроматографиче¬
ском анализе. Ленинград, Химия, 1982,279 с.45. Vigdei^auz M.S., Belyaev N.F., Esin M.S. Gas-liquid chromatography,
Fres. Z. Anal. Chem. 1989, v.335, p.70.46. Зенкевич И.Г. в книге: «100 лет хроматографии» под ред. Б.А. Ру-
денш, Москва, Наука, 2003, с.311.47. Golovnya R.V., Misharina Т. А. J. High. Resolut. Chromatogr. Commun.
1980, V.33, p.51.48. Жуховицкий A.A., ТУркельт^ H.M. Газовая хрометография.
М.Гостоптехиздат. 1962.442c.49. Гришин Г.А., Коломиец Л.Н., Ларионов О.Г., Паренаго Л.А. Веста.
АН СССР 1987, №5, с.8б.50. Березкин В.Г., Ретунский В.А. Ж. аналит. хим. 1988, т.43, №1,
с. 166.51. Березкин В.Г., Викторова Е.Н. Ж. аналит. хим. 1988, т.43, №11,C.2099.52. Березкин В.Г., 1&жевник М.А. Ж. аналит. хим. 1992, т.47, №1, с.80.53. Яшин Я.И. Ж. аналит. хим. 1989, т.44, МП, с.1941.54. Яншн Я.И. Ж. аналит. хим. 1993, т.48, №3, с.415.55. Яшин Я.И., Яшин А.Я. Ж. аналит. хим. 1999, т.54, №6, с.593; 1994,
т.49, №10, С.1047; 2001, т.56, №3, с.231.56. Яшин Я.И., Яшин А.Я. Наукометрическое исследование состояния
и тенденций развития методов хроматографии и аппаратуры. В книге:
«100 лет хроматографии» под ред. Б.А.Руденко, Наука, Москва, 2003,
с.698-736.27
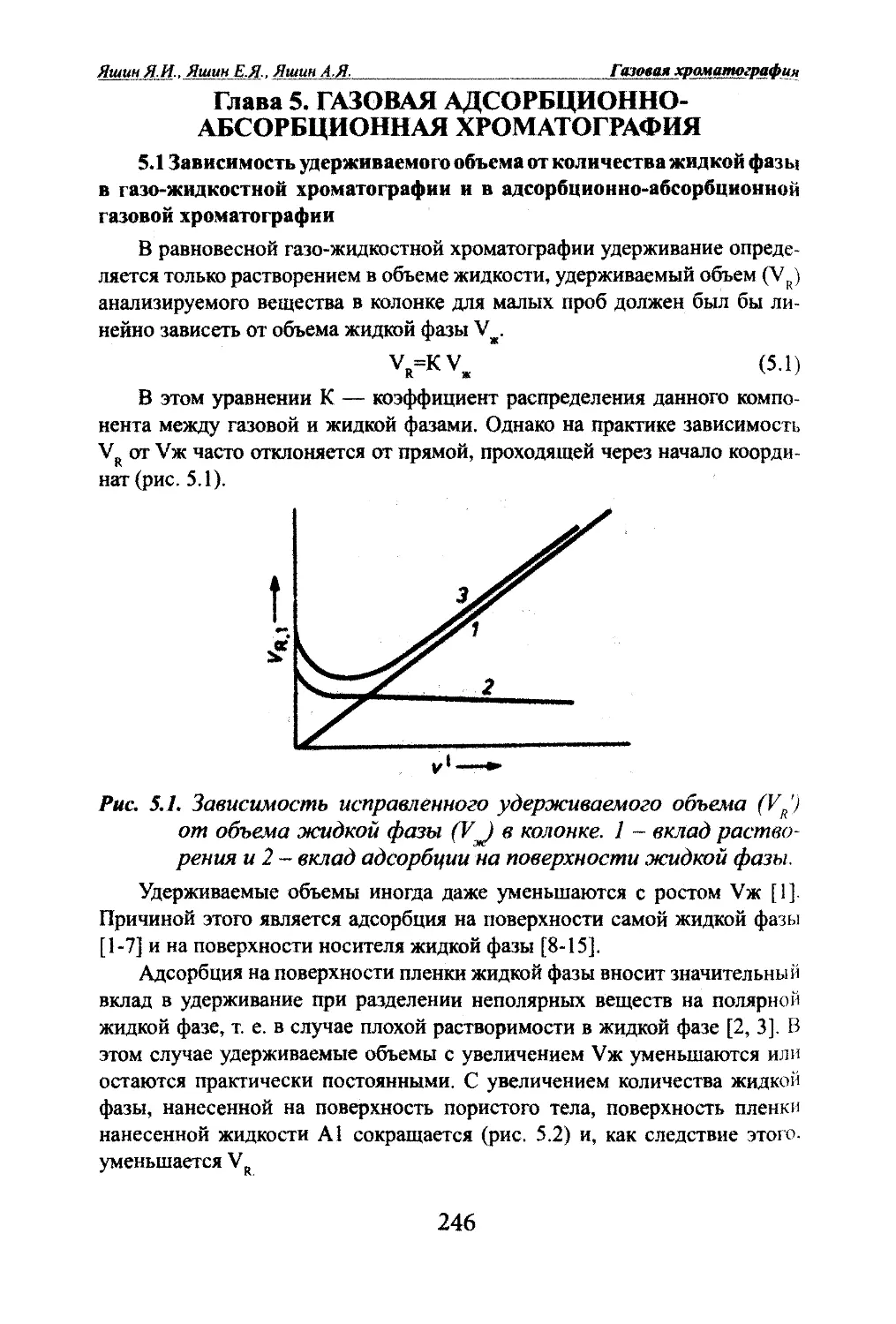
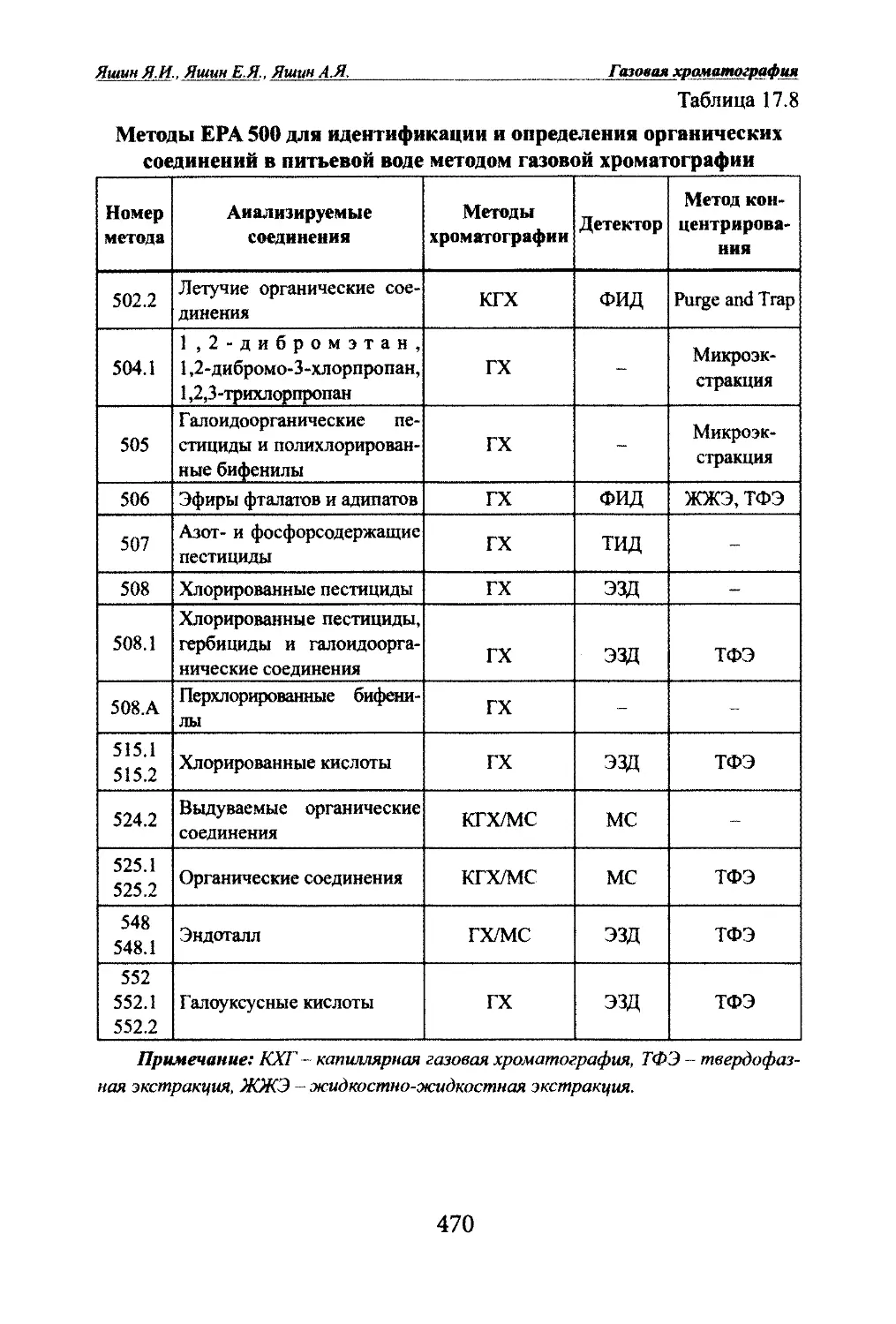
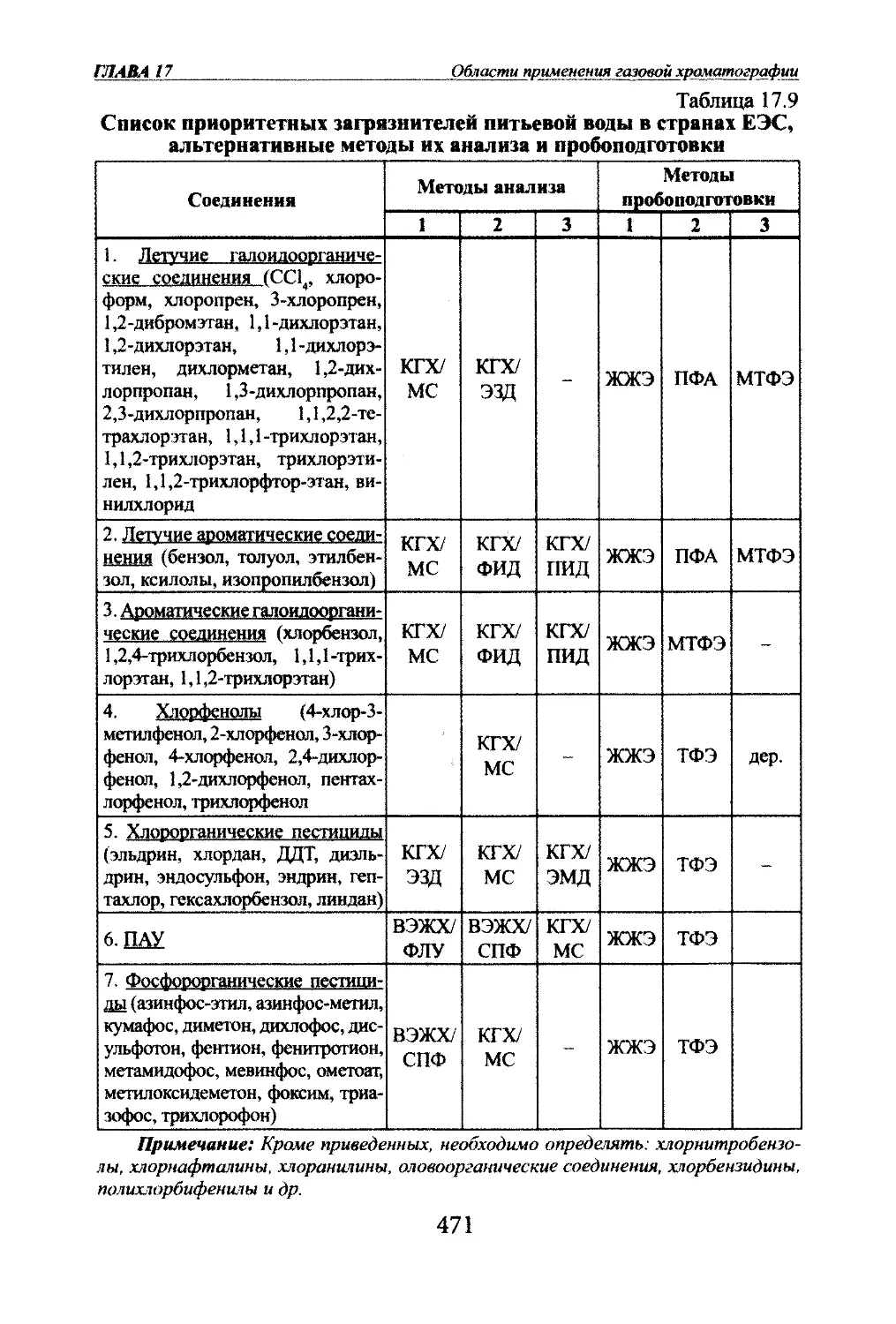
Яшт ЯМ, Яшин Е.Я.. Яшин А.Я. Газовая храчатографияГлава 2. ОСНОВЫ ТЕОРИИ ГАЗОВОЙ ХГОМАТОГРАФИИ2.1. Параметры уцерживания.2.1.1. Время удерживания.Разделение в хроматографии основано на различной скорости про¬
движения анализируемых соединений по слою сорбента в колонке. Если
соединение не сорбируется, то оно не будет удерживаться сорбентом в ко¬
лонке и будет выходить из колонки со скоростью потока - газа-носителя.
Если же соединения сорбируются, то они будут удерживаться в колонке,
причем время удерживания будет определяться их сорбционной способ¬
ностью: чем сильнее сорбируется соединение, тем меньше будет скорость
его движения по колонке и тем дольше оно будет удерживаться в колонке.Параметры удерживания, по существу, характеризуют сорбционную
способность анализируемых соединений. Различие в сорбируемости в ко¬
нечном итоге определяется различием межмолекулярных взаимодействий
вещество-сорбент.Далее обозначения параметров удерживания приводятся в соответ¬
ствии с рекомендациями ИЮПАК [1], совета по хроматографии [2] и ново¬
го справочника химика.Время от момента ввода пробы в колонку до выхода максимума пика
называется временем удерживания (рис. 2.1).Рис. 2.1 Определение времени удерживания.Это время складывается из двух величин: времени нахождения моле¬
кул соединения в газовой фазе (t^) и времени нахождения молекул соеди¬
нения в сорбируемом состоянии (t'j,):tR = to + t'a (2-1)Время нахождения молекул исследуемого соединения в газовой фазе
зависит от доли пустот в насадочной или капиллярной колонке. В разных
насадочных колонках плотность набивки различна, поэтому будет изме¬
няться и величина t„. Вследствие этого истинная удерживающая способ-28
IJIABA2. Основы теории газовой хроматографииность определяется величиной так называемым приведенным време¬
нем удерживания;t'R = tR-‘o (2-2)Величину определяют по времени от момента ввода пробы в колонку
до момента выхода несорбируемого соединения. В газовой хроматографии
эту величину определяют по времени выхода гелия или водорода в случае
применения детектора по теплопроводности и метана в случае использова¬
ния пламенно-ионизационного детектора.2.1.2. Объем удерживанияПриведенное время удерживания зависит от скорости газа-носителя:
чем больше скорость, тем меньше время удерживания. Поэтому на прак¬
тике в качестве основного параметра удерживания удобнее использовать
удерживаемый объем - произведение времени удерживания на объем¬
ную скорость газа-носителя F_,:^ V, = t,-F (2.3)Удерживаемый объем - это объем газа-носителя, который необходимо
пропустить через хроматографическую колонку, чтобы элюировать данное
анализируемое соединение.Приведенный удерживаемый объем (V'^), соответственно, равен:v; = (t, - g F = t, • F -• F = V, - V,, (2.4)где Vj - объем пустот в колонке (мертвый объем).В хроматографе реально складывается из объемов всех пустот в га¬
зовом тракте (дозатора, переходных соединений, колонок, детектора).Объемную скорость газа-носителя чаще всего измеряют на выходе из
колонки. Из-за сжимаемости газа-носителя при повышении давления объ¬
емная скорость неодинакова по длине колонки. В начале колонки она мень¬
ше, чем на выходе, поэтому для определения средней скорости в колонке
вводится специальная поправка j, учитывающая перепад давления:3 ("'/р/--где Pj - входное давление;Рд - давление на выходе колонки.Приведенный удерживаемый объем с поправкой на среднее давление
называется чистым объемом удерживания:VK = V',-j (2.6)29
Яишн ЯМ. Яшин Е.Я.. ЯшинАЯ. Газовая хроматографияЧистый yцq)живaeмый о&ьем можно считать физико-химичесюй кон¬
стантой, так как он не зависит от скорости газа-носителя при постоянной
температуре и доли пустот в колонке.Чистый удерживаемый обьем зависит от количества сорбента в колон¬
ке, поэтому для точных физико-химических измерений используют поня¬
тие удельного объема удерживания . Величина - это чистый обьем
удерживания, отнесенный к массе g сорбента в колонке или к площади
А поверхности адсорбента при усредненном давлении в хроматографиче¬
ской колонке и температуре колонки:VJ VJV, =-|- \ (2.7)Для особо точных физико-химических измерений вводят поп|шку на дав¬
ление п£фа юды, так как измерения обычно проюдят мьгаьно-пенным измери¬
телем, а также на разность темпе|шур на выходе из шлонки и в колош®.В.А.Даванков [4,5] исправил существующие неточности в определе¬
нии параметров удерживания.2ЛЗ. Относительные параметры удерживанияВсе рассмотренные выше параметры удерживания зависят от случай^
ных небольших колебаний параметров опыта, в частности, расхода газа-
носителя и температуры термостата шлонки.Относительные параметры удерживания практически не зависят от
этих влияний.При расчете относительного параметра удерживания (времени или
объема) берут отношение чистого объема удерживания исследуемого ве¬
щества к чистому объему удерживания стандартного вещества:(2.8)В качестве стандартного соединения используют н-алканы с параме¬
трами удерживания, близкими к параметрам исследуемого вещества. В
этом случае при случайных колебаниях расхода или температуры абсолют¬
ные параметры удерживания будут изменяться, а их отношения практиче¬
ски не изменяются.В канестве огаосигельнш) гщимефа ппфсш исполь^тог индекс Ковача [6]:'ig-1=100■ll.'кJHl+100П (2.9)где - приведенные времена удерживания н-алканов с числом ато¬
мов углерода в молекуле п и п + 1;t’^ - приведенное время удерживания исследуемого соединения.30
ГЦЛВА 2 Основы теории газовой хроматографииИндекс 1&вача - безразмерная величина, которая может быть подсчи¬
тана с большой точностью. Например, в капиллярных колонках - с точнос¬
тью до сотых долей процента. Индексы Ковача в первую очередь применя¬
ют для идентификации неизвестных веществ (проведение качественного
анализа). В последние годы оказалось весьма информативным для целей
идентификации сочетание масспектрометрии с индексами удерживания.Изменения индексов Ковача для соединений, ошичающихся природой
функциональной группы, используют для оценки межмолекулярных взаи¬
модействий. Индексами уцерживания определенного набора стандщттных
веществ характеризуют полярность неподвижных жидких фаз и адсорбентов.Следует еще упомянуть об одном параметре - коэффициенте емкости
равном отношению приведенного времени удерживания к времени удер¬
живания несорбируемого вещества;(2.9 а)2.1.4. Параметры хроматографического пикаВыходной сигнал анализируемого соединения имеет форму треуголь¬
ника или пика. Это обычно участок нулевой линии, на котором возникает
сигнал при выходе анализируемого соединения из хроматографической ко¬
лонки. Нулевая или базовая линия -- это участок, соответствующий нуле¬
вой шнцентрации анализируемого соединения. Запись пика исследуемого
соединения вместе с участками нулевой линии до и после пика называется
хроматограммой. Высота пика - это расстояние от максимума пика до его
основания, измеренное параллельно оси отклика детектора. Ширина пика
у основания - это отрезок основания пика, отсекаемый двумя касательны¬
ми, проведенными в точках перегибов восходящей и нисходящей ветвей
хроматографического пика. Ширина пика на полувысоте - это отсекаемый
пиюм отрезок линии, проведенной параллельно основанию пика на се¬
редине его высоты. Площадь пика - это площадь части хроматограммы,
заключенный между пиком и его основанием.Важным параметром пика является коэффициент асимметрии, кото¬
рый применяется для сравнения различных твердых носителей, адсорбен¬
тов и всей газовой системы хроматографа в целом. В идеальных условиях
пик по форме близок к кривой Гаусса, то есть симметричен. На практике
пики по разным причинам в основном несимметричны. Асимметрия пиков
ухудшает разделение и затрудняет количественную обработку.Асимметричные пики появляются при разделении на неоднородных
сорбентах, когда концентрации анализируемых соединений соответству-нелинейным участкам изотермы сорбции. Кроме того, в некоторых
случаях это может быть вызвано кинетикой сорбции (замедленный про-31
Яшин Я,И.. Яишн ЕЯ.. Яшин Л.Я.Газовая хранатографияцесс десорбции), наличием не продуваемых полостей. Асимметрию пиков
оценивают относительно полуширин пишв на половине высоты (рис. 2.2)
отношением отрезка БВ к АБ, либо отношением отрезка ДЕ к ГД на 1/10
высоты пика от основания. Точнее пользоваться отношением площадей
половин пика - отношением заштрихованной части пивш к не заштрихо¬
ванной (см. рис. 2.2).В фармацевтических измерениях используют отношение на 1/20 высо¬
ты пика от основания.Рис. 2.2 Оценка асимметрии пиков.2.2. Основные процессы в хроматографической колонкеВ хроматографической колонке происходят одновременно два про¬
цесса; разделение веществ и размывание полос разделяемых веществ. Раз¬
деление - цель хроматографичесюго процесса (т.е. полезный процесс).
Размывание всегда приводит к ухущпению разделения. Разделение оцени¬
вается различием удерживаемых объемов AV^ (рис. 2.3). Чем болыта эта
разность, тем лучше разделение.Рис. 2.3 Оценка разделительной способности хромотографической
колонки.32
ГЛАВА 2 Основы теории газовой хроматографииОднако, при одном и том же значении AV^ в некоторых случаях пол¬
ное разделение не достигается из-за большого размывания. При выборе
оптимальных условий хроматографического разделения обычно стремятся
к получению максимального значения AV^ и минимального значения ши¬
рины полосы ц (рис. 2.3). Теория хроматографии сводится к исследованию
я оптимизации этих величин.Разделение определяется природой сорбента и сорбата, температурой,
при которой происходит разделение, и в конечном итоге различием энергий
межмолекулярного взаимодействия сорбат - сорбент. На основании опре¬
деленных сведений по адсорбции, растворению разделяемых соединений
можно уже сугщть о возможности хроматографического разделения.Сорбция связана со статикой хроматографического процесса. Наибо¬
лее сушественное отличие хроматографичесшго процесса от статических
условий сорбции заключается в том, что в хроматографических колонках
обеспечивается многократность актов сорбции и десорбции, поэтому даже
при ничтожно малом различии в сорбируемости веществ можно достичь
их хроматофафического разделения.Размывание полос в хроматографической колонке - это процесс, спе¬
цифичный для хроматографии, все основные новые теории хроматографии
связаны именно с процессами размывания.На первом этапе газовая хроматография развивалась как эмпирический
метод, но, начиная с 1956 года, были многочисленные попытки разработки
теории хроматографии, описывающей скорость продвижения полос по ко¬
лонке и размывание полос. Наибольшее внимание было уделено вопросам
динамики процесса (скорости продвижения, диффузии и массообмену).
Создание теории, описывающей количественно хроматографический про¬
цесс, сопряжено со значительными, в некоторых случаях непреодолимы¬
ми трудностями. Эти трудности связаны, во-первых, с неопределенностью
многих параметров (в частности, структуры насадки) и, во-вторых, с не¬
возможностью точного решения дифференциальных уравнений, описыва¬
ющих размывание полос.Подход зависит от формы изотермы сорбции, по которой различают
линейную или нелинейную хроматографию. В зависимости от равновесно¬
сти процесса массообмена различают идеальную и неидеальную хромато¬
графию. Возможны следующие сочетания: линейная - идеальная, нели¬
нейная - идеальная, линейная - неидеальная, нелинейная - неидеальная
хроматография.Теория линейной хроматографии рассматривает процессы, которые
описываются линейной изотермой сорбции. В этом случае имеют место
симметричные пики. Теория нелинейной хроматографии рассматривает33^ Газовая хроматография
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я.Гаювая хроматографияпроцессы, которые характеризуются нелинейной изотермой (выпуклой
или вогнутой). Проведение хроматографического процесса для этого слу¬
чая приводит к несимметричным пи1ам.Теория идеальной хроматографии основана на допущении мгновенного
установления равновесия, т.е. на допущении весьма высоюй сшрости внеш¬
ней и внутренней диффузии. Теория же неидеальной хроматографии рассма¬
тривает реальный процесс и учитывает сюрость устшювяения {ктновесия.Первое сочетание наиболее простое. В случае нелинейной изотермы
матемагаческая обработка значительно усложняется. Наиболее распро¬
странена линейная - неидеальная хроматография.Для рассмотрения процессов хроматографического размывания ис¬
пользуют четыре теории: теория теоретических тарелок, молекулярно-ки-
нетическая теория, теория моментов и теория с1юростей (теория эффек¬
тивного коэффициента диффузии). Как было указано выше, из-за многих
геометрических неопределенностей в колонке теория с использованием
молекулярно-кинетических представлений не получила развития. Ниже
будут рассмотрены только теория тарелок и теория скоростей.2.2.1. Теория теоретических тарелок.Теория теоретических тарелок - общий метод описания многостадий¬
ных процессов. Представление о теоретической тарелке взято из теории
дистилляции. В дистилляции разделение происходит на отдельных сту¬
пенях, на которых осуществляется равновесие между фазами, затем фазы
разделяют. Каждая такая ступень называется теоретической ступенью
или теоретической тарелкой. В хроматографической юлонке, заполнен¬
ной сорбентом, одна из фаз находится в непрерывном движении, и полное
равновесие иногда сразу не достигается. В таких случаях длина слоя, на
котором достигается равновесие между двумя фазами, условно называется
высотой, эквивалентной теоретической тарелке (ВЭТТ).Рис. 2.4 Определение числа теоретических тарелок (N) по параме¬
трам пика34
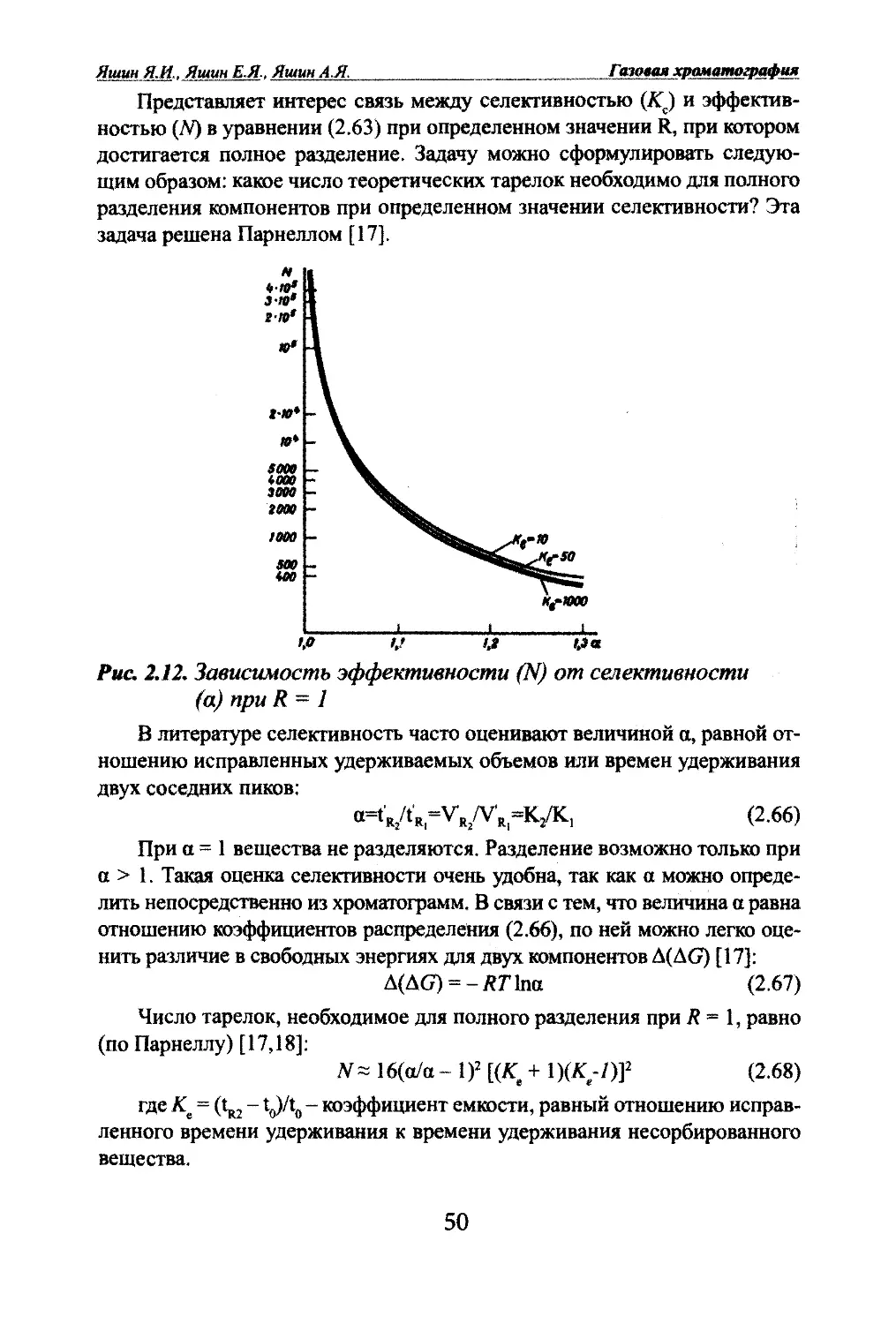
[ЖЛВАЛ Основы теории газовой хроматографииВпервые в хроматографию теория тарелок введена Мартином и Син¬
гом [6]. Хотя представление о теоретических тарелках в хроматографии
носит формальный характер, теория тарелок позволяет описывать движе¬
ние области максимальной концентрации, экспериментально оценить ши¬
рину полосы и оценить эффективность (степень размывания) хроматогра¬
фической колонки. Согласно теории тарелок вся колонка состоит из ряда
равновесных зон, т.е. теоретических тарелок. Время удерживания пропор¬
ционально числу теоретических тарелок iV [7]:(2.10)где k - коэффициент пропорциональности.Ширина полосы ц связана с Доследующим соотношением [6]:H~kHN (2.11)Из этих соотношений следует, что число теоретических тарелок легко
определить из хроматограммы, для чего достаточно измерить время удер¬
живания и ширину пика:iV= 16(Vn)^ (2.12)Ширину пика в этом случае измеряют в основании пика. Но это не¬
удобно, так как нужно проводить касательные точно к сторонам пика. Не¬
удобно использовать и расчет по следующей формуле:N=8(^1 (2.13)где |i - на высоте, равной 2,71.Поэтому на практике наибольшее применение нашла формула:5,54 (2.14)где - ширина пика на половине высоты.Величина может быть определена легко, быстро и с достаточной
точностью, (рис. 2.4).Высота, эквивалентная теоретической тарелке Я (ВЭТТ), определяет¬
ся отношением длины колонки LkN:Н = Ш (2.15)ВЭТТ имеет размерность длины и часто измеряется в миллиметрах.
Благодаря легкости определения непосредственно из хроматограмм, вели¬
чины N н Н широко используют на практике. Как будет показано ниже,
величина Н непосредственно связана с рядом других параметров колонки.2.2.2. Основные виды размыванияРазмывание полос в хроматографической колонке - сложный процесс,
он происходит по различным причинам [8]. Можно выделить три вида раз¬
мывания:35
Яшин Я.И., Яишн Е.Я., Яшин А.Я.Газовая хроматография1. Размывание, связанное с различной скоростью движения по слою
сорбента зон полосы с разными концентрациями.2. Диф4^ионные размывания (кроме молекулярной диф(^эди, в эту груп¬
пу входят вихревая диффузия, динамическое размывание, стенотный эффект).3. Кинетические размывания, связанное со скоростью внешнего и вну¬
треннего массообмена.В теории скоростей допускается, что эти виды размываний происходят
независимо друг от друга, это означает, что они аддитивны. В связи с этим
вводится понятие эффективного коэффициента диффузии, который равен
сумме всех видов размываний. Исследуется зависимость этого эффектив¬
ного коэффициента от линейной скорости. Эффективный коэффициент
диффузии непосредственно связан с величиной В ЭТТ.Размывание, связанное с нелинейной сорбцией наблюдается в том
случае, когда концентрации соответствуют нелинейному участку изотер¬
мы сорбции. При этом допускается, что равновесие устанавливается, ки¬
нетическое и диффузионное размывания отсутствуют. Нужно определить
зависимость скорости перемещения зон сорбируемых веществ разной
концентрации от параметров опыта [8]:«=(йх/ао, (2.16)в случае нелинейной изотермы:и^ = и1{Шдс\ (2.17)При линейной изотерме сорбции, когда коэффициент Генри К, равный
отношению величины оке, постоянен (не зависит от концентрации), раз¬
мывание, связанное с нелинейностью сорбвди, отсутствует.Для идеальной равновесной хроматографии начальная ширина и фор¬
ма полосы должны сохраниться. При наличии еще и диффузионного раз¬
мывания полоса будет расширяться симметрично в обе стороны, и на вы¬
ходе колонки фиксируется симметричный пик (рис. 2.5 а).tРис. 2.5 Размывание полос для различных типов изотермы сорбции36
1УЫВА_2 ^ Основы теории газовой хтшатографииВ случае нелинейной изотермы сорбции коэффициент Генри меняется
с изменением концентрации. Для выпуклой изотермы (изотермы Лэнгмюра)
с повышением концентрации коэффициент Генри уменьшается, а скорость
продвижения зон с большими концентрациями возрастает. Это приведет к ис¬
кажению формы пика. На выходе из колонки полоса регистрируется в виде
пика с резким отвесным фронтом и растянутой задней границей (рис. 2.5 б).
Для вогнутой изотермы, наоборот, с повьппением концентрации коэф¬
фициент Генри увеличивается, а скорость продвижения зон с большими
концентрациями будет уменьшаться. Форма пика исказится так, что будет
растянутый передний фронт и резкая задняя граница (рис. 2.5 в). Времена
удерживания для линейной изотермы сорбции не зависят от величины до¬
зируемой пробы (до перегрузки), в случае нелинейных изотерм сорбции
времени удерживания с увеличением пробы будут либо уменьшаться, либо
возрастать в зависимости от вида изотермы (рис.2.5).В чем физическая супцюсть этих процессов?Первый случай (изотерма выпуклая) типичен для адсорбента с энерге¬
тически неоднородной поверхностью. На такой поверхности имеются ад¬
сорбционные центры разной активности. При адсорбции молекулы в пер¬
вую очередь занимают самые активные участки поверхности. Если самых
активных центров адсорбции не хвагило для других молекул, они будут
вынуждены сорбироваться на менее активных центрах. Если же концен¬
трация сорбируемых молекул велика, то большая часть их вынуждена сор¬
бироваться на малоактивных центрах. Это означает, что на менее активных
центрах молекулы в сорбированном состоянии будут находиться меньшее
время, и, следовательно, скорость их продвижения по слою сорбента будет
выше.Второй случай (изотерма вогнутая) возможен, в частности, при адсорб¬
ции сильнополярных веществ на неполярной поверхности. При адсорбции
веществ в небольших концентрациях происходит взаимодействие сорбат -
сорбент. Когда концентрация сорбируемых веществ увеличивается и плот¬
ность адсорбированных молекул больше емкости монослоя, кроме взаимо¬
действия сорбат - сорбент будет иметь место взаимодействие сорбат - сор¬
бат. А для полярных веществ, особенно склонных к ассоциации (спирты,
вода и т.д.), такое взаимодействие значительно, поэтому зоны с большей
концентрацией двигаются по слою сорбента с меньшей скоростью.В обоих случаях получаются несимметричные пики, которые ухудша¬
ют разделительную способность хроматографической колонки и затруд¬
няют количественные анализы, так как площадь несимметричного пика
очень трудно измерить, а высота пика сложным образом меняется в зави¬
симости от концентрации.37
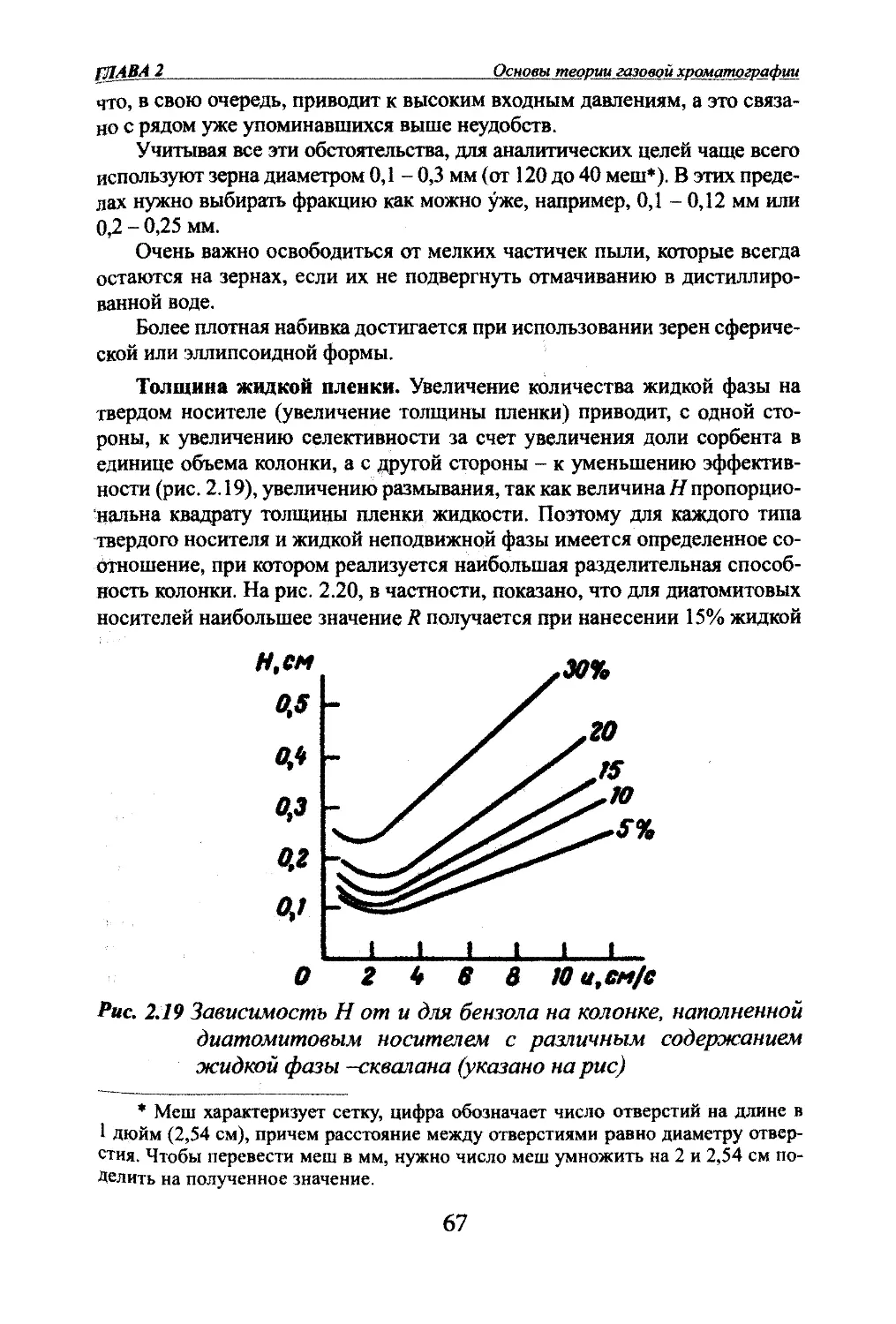
Яшин ЯМ. Яшин ЕЯ.. Яшин А.Я. Газовая хроматографияВ аналитической газовой хроматографии нужно всегда подбирать та¬
кие условия, чтобы исключить размывание, связанное с нелинейностью
сорбции. Этого можно достичь выбором адсорбентов, которые имели бы
больший линейный участок рабочих концентраций на изотерме сорбции,
^гда ташго выбора нет, то нужно работать с малыми концентрациями,
используя чувствительные детекторы. Повышением температуры разделе¬
ния можно также увеличить линейный диапазон рабочих концентраций.Диффузионные размывания. Молекулярная диффузия.
Для хроматографических колонок небольшого диаметра ди^узионное
размывание рассматривают в основном только вдоль шлонки, размывание
в других направлениях ограничено стенками шлонки, поэтому часто ис¬
пользуют термин «продольная диффузия». Тогда уравнение Фнка [9] мож¬
но записать:dc/dtD (д^с/дх^) (2.19)Одно из решений этого уравнения (при допущении, что начальная ши¬
рина полосы значительно уже шнечной) приводит к кривой Гаусса [9]:c„»=q/V^Dt (2.20)где с - шнцентрация на расстоянии х от максимума;с - шнцентрация в максимуме полосы;t - продолжительность размывания;
q - общее шличество вещества.Выражение для кривой Гаусса записывается с включением стандарт¬
ного отклонения о, равного:a^ = 2Dt(2.21)Нормальная кривая, изображающая функцию (2.21), представлена на
рис. 2.6.Рис. 2.6 Вид кривой распределения Гаусса38
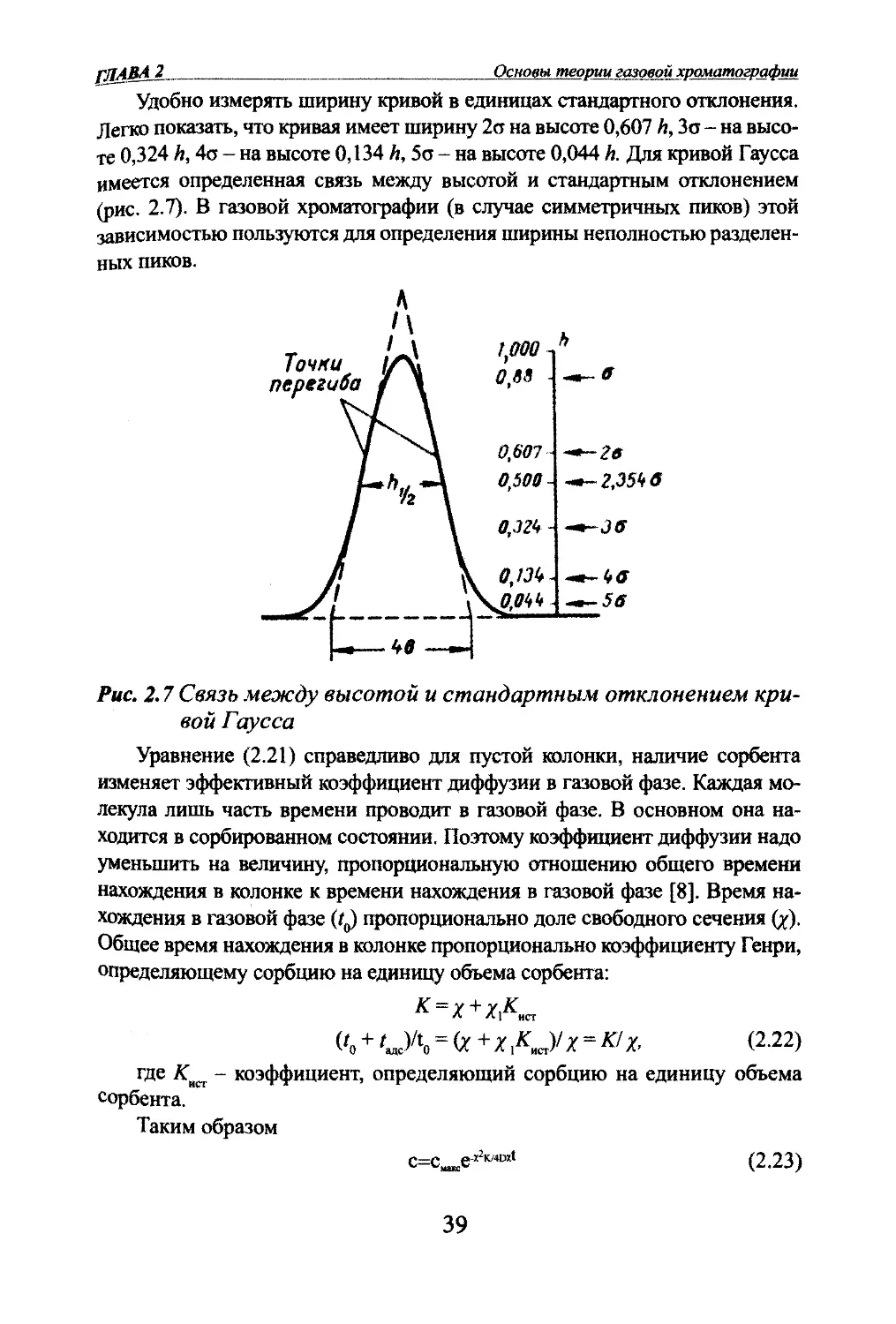
Основы теории газовой хроматографииУдобно измфять ширину кривой в единицах стандартного отклонения.
Легко показать, что кривая имеет ширину 2а на высоте 0,607 А, Зо - на высо¬
те 0,324 h, 4о - на высоте 0,134 А, 5ст - на высоте 0,044 к Для кривой Г^сса
имеется определенная связь между высотой и стандартным отклонением
(рис. 2.7). В газовой хроматографии (в случае симметричных пиков) этой
зависимостью пользуются для определения ширины неполностью разделен¬
ных пиков.Точниперегиба1,000 ^h0,88 --.-ff0,607-*~2в0,500о,т -о,т~ЖОЧк .^56Рис. 2.7 Связь между высотой и стандартным отклонением кри¬
вой ГауссаУравнение (2.21) справедливо для пустой колонки, наличие сорбента
изменяет эффективный шэффициент диффузии в газовой фазе. Каждая мо¬
лекула лишь часть времени проводит в газовой фазе. В основном она на¬
ходится в сорбированном состоянии. Поэтому коэффициент диффузии надо
уменьшить на величину, пропорциональную отношению общего времени
нахождения в колонке к времени нахождения в газовой фазе [8]. Время на¬
хождения в газовой фазе {t^ пропорционально доле свободного сечения 0^).
Общее время нахождения в шлонке пропорционально юэффициенту Генри,
определяющему сорбцию на единицу объема сорбента:(^0 + = (X +X,KJx = Klx. (2.22)где - коэффициент, определяющий сорбцию на единицу объема
сорбента.Таким образомс=с„„е-’‘':^K/4Dxt(2.23)39
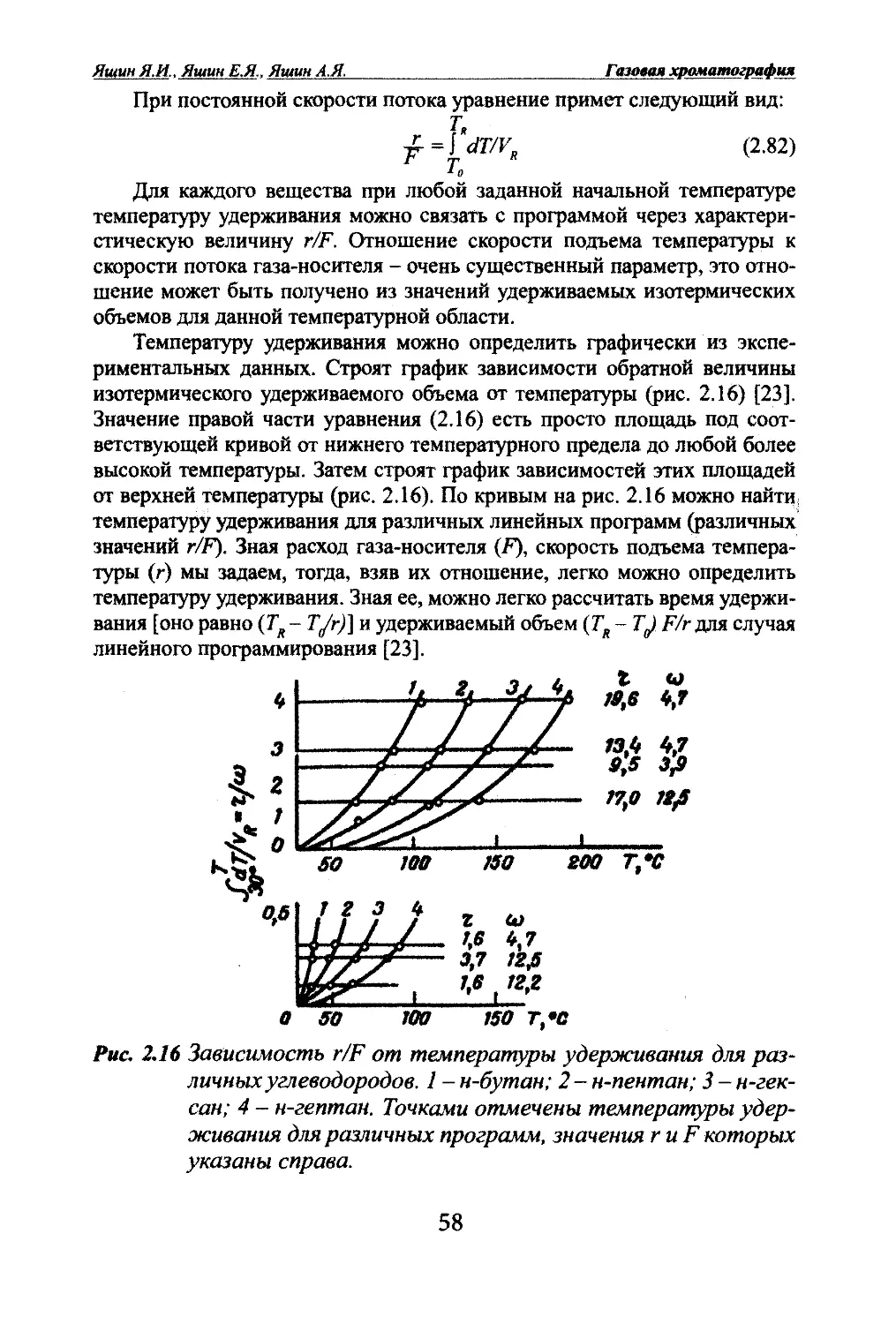
Яшин ЯМ. Яиши Е.Я.. Яшин А.Я. Газовая хроматографияЦелесообразнее исследоветь ширину полосы в зависимости не от вре¬
мени, а от длины слоя (L):t=Uu =LK/uсВ этом случае:с=с.„е-'^“ (2.24)Для определения ширины полосы на слое, необходимо сделать преоб¬
разования. Приняв \1^ = 2х, прологарифмируем обе части уравнения (2.23)
и возьмем ширину полосы на высоте е. Получим:In с = In - iiiyi6Dxt), ц>/16£^ = In(2.25)В результате получено уравнение для ширины полосы на слое. Вы¬
ражают ее в сантиметрах. Однако на практике судят о ширине полосы по
хроматограмме. Ширина полосы на хроматограмме имеет уже размерность
времени или объема. Для перехода к ширине полосы на хроматограмме
(или в газовой фазе), необходимо определить время, в течение которого по¬
лоса будет вымываться с колонки, когда один ее край приблизится к шнцу
колонки, и объем газа-носителя, необходимый для вымывания полосы со
слоя. Время вымывания равно отношению ширины полосы на слое (ц^) к
линейной скорости движения этой полосы (и^):ц/и^ = ц^К/и (так как = и/К) (2.26)Ширина полосы на хроматограмме (в газовой фазе) равна произведе¬
нию времени вымывания на объемную скорость газа-носителя:\i=\iuKS/u = \i^KS, (2.27)где S - сечение колонки.Подставляя в уравнение (2.25), получим:Ц=4К8л/ШЛ1 (2.28)С увеличением сечения колонки будет больше размываться полоса.
Чем больше коэффициент Генри, чем лучше вещество сорбируется, тем
шире полоса. С повышением температуры коэффициент Генри уменьша¬
ется, поэтому с повышением температуры размывание уменьшается.Вихревая диффузия. При движении полосы в насадочной
колонке, заполненной зернами сорбента, наблюдается еще специальное
размывание, не совсем точно названное вихревой диффузией [8]. Это раз¬
мывание в основном связано с неоднородностью насадки, приводящей к
разным сопротивлениям потоку в различных частях сечения колонки. Об¬
щий поток газа-носителя при попадании в колонку распределяются по се¬
чению на отдельные микропотоки между зернами. Если сопротивление по40
ГЛАВА 2Основы теории газовой хттатографиисечению неоднородно, то там, где сопротивление меньше, будет проходить
больший поток газа-носителя, и наоборот, там, где сопротивление больше,
поток газа-носителя будет меньшим (рис. 2.8). Это приведет к дополни¬
тельному размыванию полосы.Рис. 2.8 Размывание за счет вихревой диффузииДля определения вклада вихревой диффузии используют общее урав¬
нение диффузии А.Эйнштейна [10]. По этому уравнению всякое диффузи¬
онное размывание можно описать как результат блужданий (отклонений от
прямого пути), совершаемых молекулами:0 = (2.29)где X - величина блуждания;т - время блуждания.При вихревой диффузии величина блуждания пропорциональна диа¬
метру зерна {d)\Х~Ы^ (2.30)Время блуждания определяется отношением величины блуждания к
линейной скорости потока (и):х = Уи (2.31)Подставляя эти выражения в соотношение (2.29), получим:D ~kud (2.32)вихр в 3 ^Вихревая диффузия пропорциональна диаметру зерна. Чем больше (/,
тем больший вклад в размывание вносит вихревая диффузия.Не менее важной величиной является однородность зернения, так как
чем уже фракция используемых зерен, тем однороднее набивка. Особен¬
но вредно присутствие пыли, прилипающей к крупным частицам за счет
электростатического притяжения. В процессе работы пылинки сдуваются
потоком газа-носителя и забивают проходы.Вклад вихревой диффузии имел место даже в тех случаях, когда ко¬
лонку заполняли шариками из стекла или из свинца одинакового размера.
В этом случае упаковка была двух разных типов после тщательной вибра¬
ции при приготовлении колонки.41
Яшн ям. Ятшн ЕЯ. Яитн АЛ,_Г<аотя щттюграфшДинамическое размывание может быть в незаполненных,
в частности, в капиллярных колонках, поскольку в центре трубки скорость
газа больше, чем у стенок (рис. 2.9).Г,Шl;Ai.Рис. 2.9 Профшь скоростей потока по сечению колонки в случае ди¬
намического размывания (а) и большого вклада стеночного
эффекта (б).Время блуждания молекул в этом случае равно [11]:х = г^Ю (2.33)где г - радиус капилляра;D - коэффициент молекулярной диффузии.Величина блуждания пропорциональна времени блуждания и линей¬
ной скорости в капилляре:X~kiu (2.34)Подставляя полученные величины в выражение (2.2.2.14), имеем:D = r^tilD (2.35)ДИН ^ 'Динамическое размывание в капиллярных трубках пропорционально
квадрату радиуса трубки. Степень размывания в капиллярных колонках
пропорциональна диаметру колонки. Динамичесше размывание значи¬
тельно снижено в поликапиллярных колонках, в которых диаметр капил¬
ляров в пределах 0,02-0,04 мм.Стеночный эффект. Плотность набивки ошло стенок всеща меньше, а
доля пустот больше, осо&нно при использовании зерен крупного размера.
Стенка мешает более плотной набивке. Это приводагг к тому, что скорость
газа-носителя около стенок больше, чем в центре колонки (рис. 2.9). В связи
с этим будет дополнительное размывание [11].Воспользовавшись соотношением Эйнштейна, П)лэй установил [10,11]:~ (bdfuVW, (2.36)где Ь - постоянная, равная отношению доли пустоты единичного объ¬
ема около стенки к доле пустоты такого же объема в центре;42
fTlABA 2 Основы теории газовой хроматографии- диаметр зерен;и - линейная скорость;D - коэффициент молекулярной диффузии.По данным Голэя Ь может шлебаться от 3 до 6. Стеночный эффект
может вносить большой вклад, когда диаметр колонок значительно больше
ВЭТТ.Реальный вклад стеночного эффекта измеряли, заполняя колонку сор¬
бентом, пропитанным щелочью и пропускали газ-носитель с добавкой
кислых газов. После определенной работы брали часть насадки сорбента
около стенки и в центре и водные вытяжки их титровали. В препаративных
колонках большого диаметра стеночный эффект уменьшали конструетив-
ным способом, внутри колонки через определенное расстояние ставили
кольцевые ограждения, которые выпрямляли профиль скоростей.В аналитических колонках небольшого диаметра (внутренний диа¬
метр 2-4 мм) размывание за счет стеночного эффекта мало и им прене¬
брегают.Кинетическое размывание. Задержка массообмена с поверхностью
сорбента вследствие медленности процессов сорбции и десорбции также
приводит к размыванию полосы. Задержка при сорбции приводит к про¬
движению компонента в газовой фазе вперед, т.е. к размыванию переднего
фронта полос, а задержка при десорбции приводит к размыванию заднего
фронта. В том случае, когда скорости сорбции и десорбции неодинаковы ,
таше размывание может быть несимметричным. В частности, десорбция
может происходить медленнее, чем сорбция.Для описания кинетического размывания воспользуемся понятиями
из кинетики сорбции. В кинетике сорбции используется кинетический
юэффициент р, имеющий размерность, обратную времени [12]. Величи¬
на 1/|3 - характеристическое время, в течение которого происходит погло¬
щение единицей объема сорбента шличества вещества, находящегося в 1
см’ газовой фазы. Если вновь воспользуемся соотношением Эйнштейна,
то характеристическое время будет равно времени блуждания. Благодаря
наличию потока, молекула за это время продвигается на величину м/р, сле¬
довательно величина блуждания равна X. = и/р. ТогдаD„„ = «VP (2.37)Кинетика процесса физической адсорбции всегда определяется ско¬
ростью переноса вещества. Сам акт физической адсорбции происходит
мгновенно. Существенную роль играет процесс переноса вещества от газа
tc поверхности - внешний массообмен (внешняя диффузия), от поверхно¬
сти во внутрь зерна сорбента - внутренний массообмен (внутренняя диф¬
фузия).43
Яшин я и., Яшин Е Я., Яишн А.Я. Газовая хроматографияВ общем случае происходит сложение диффузионных последователь¬
ных сопротивлений:1/р=1/р, + 1/Рз, (2.38)где р|, Pj - коэффициенты массопередачи соответственно для внешней
и внутренней диффузии.С учетом коэффициента распределения К [8]:+ т (1/р, + 1/рз) (2.39)Для внешнего массообмена (при ламинарных потоках)(2.40)Внутренняя массопередача имеет более сложный характер. В случае мас¬
сопередачи в жидюй пленке (в случае газо-жидюстной хроматографии):(2.41)где - коэффициент диффузии в жидкости;d^- толщина жидкой пленки на твердом носителе.Необходимо помнить, что это приближенные зависимости.Подставляя полученные выражения в уравнение (2.39), имеем:~ [и^К^/2{1 + К)^] + dVDJC) (2.42)Для расчета общей ширины полосы вводят понятие эффективного ко¬
эффициента диффузии, как суммы всех видов диффузии (в случае анали¬
тической колонки):= + (2-43)(для аналитических насадочных шлонок не учитывается стеночный
эффект).Подставим в это уравнение соотношения (2.32) и (2.42) и, введя в вы¬
ражение для молекулярной диффузии (DJ коэффициент извилистости (X),
который учитывает, что молекулярная диффузия проходит не в пустой
трубке, а заполненной зернами сорбента, получим:D^~W + k^ud^ + [Л?с/“и“/2 (1 + ++ [KJ2 (1 + ^0'] (2.44)В результате мы получили сложное уравнение, хотя и учитывающее ре¬
альные процессы в колонке. Однако проводить расчеты по нему сложно, т.к.
в нем много констант, которые необходимо измерять для каждой реальной
системы. В связи с этим устанавливают связь Оэф с ВЭТТ, т.к. ВЭТТ легко
определяются из реальной хроматограммы.Связь эффективного коэффициента диффузии с Н.
Эффективный коэффициент диффузии непосредственно связан с высотой,
эквивалентной теоретической тарелке.44
fit ABA 2 Основы теории газовой хроматографииЕсли воспользоваться соотношением (2.29), то за величину блуждания
можно принять высоту тарелки:Х = Н (2.45)Тогда время блуждания:х = Н/и (2.46)Подставляя полученные выражения в соотношение (2.29), получим:= 'Л Ни, Н = ID^Iu (2.47)Уравнения зависимостей Нотскорости газа-носителя.
Подставляя выражение (2.44) в уравнение (2.47) с соответствующими пре¬
образованиями, получим:21)эф _ 2X0 , J ^ ^ 1/2 . ^ и ('> ^8)В более простом виде:Н = А+В/и + С^и^’^ + С^и (2.49)Величина А определяет вклад вихревой диффузии, В - молекулярной
диффузии, С, и Cj - соответственно внешней и внутренней диффузии в
общее размывание.Уравнение, связывающее Я со скоростью, в ташм виде получено Жу-
ховицким и Джонсом [8]. Впервые подобное уравнение (в более простом
виде) вывели Ван-Деемтер, Цнидер&рг и Клинкенберг в 1956 г. [13]:Н = А+В/и + Си, (2.50)где А = 2W , В = 2yD и С = 2К(1ЧЪ (1 + KfD^ имеют практически те
же значения, что и в уравнении (2.44). В уравнении Ван-Деемтера учи¬
тывается только внутренняя диффузия, внешняя не рассматривается, хотя
при выводе уравнения Ван-Деемтер получил выражение и для внешней
диффузии. В окончательном выражении он опустил его, приняв без экспе¬
римента, что массообмен в газовой фазе происходит достаточно быстро.Последующие экспериментальные проверки уравнения Ван-Деемтера
в течение пяти лет [14] показали, что необходимо в уравнение ввести и
четвертый член, учитывающий внешнюю диффузию.Теоретически точно подсчитать различные величины этого уравнения
не представляется возможным. Однако их можно определить графически,
для чего строят график зависимости Н от линейной скорости на основе
эксперимента. На графике (рис. 2.10) показано, как можно оценить раз¬
личные члены уравнения (2.50). Из графика же можно определить область
оптимальных скоростей газа-носителя, при которых наблюдается наиболь¬
шая эффективность колонки (минимальное значение Н). Это весьма важ¬
ная информация для специалистов-практиков, разработчиков методик.45
Яшин Я.И., Яшин Е.Я., Яшин А.Я.Газовая храчатографиянНшА * в/и* СиРис. 2.10 Определение констант уравнения Ван-Дееметра из
зависимости Н от иВ последние годы появились новые выражения для ВЭТТ. Обычно
принималось, что вихревая диффузия не зависит от скорости потока газа-
носителя. Однаш в экспериментах появился ряд аномалий, в частности, в
некоторых измерениях член А в уравнении (2.50) принимал отрицательные
значения или близкие к нулю и зависел от скорости газа-носителя. Поэто¬
му рядом исследователей предпринята попытка пересмотра классического
выражения для вихревой диффузии. Наиболее полно эта теория развита
Гиддингсом в его сопряженной теории [9]:Я = Е \m\d) + (2.51)где Н^- высота тарелки, обусловленная вкладом вихревой диффузии;X, и Ю| - величины, постоянные для данной колонки.При малых скоростях потота Я пропорциональна скорости, при вы¬
соких скоростях потока выражение переходит в классическую форму для
вихревой диффузии. Гиддингс считает, что этот переход совершается при
скоростях 10-100 см/с. Сопряженная теория экспериментально подтверж¬
дена измерениями Кизельбаха [14].Следует отметить различие в записи члена внешней диффузии в урав¬
нении А.Жуховицкого [7] и зарубежных ученых. В зарубежной литерагуре
[13] внешнюю массопередачу записьюают в следующем виде:^.иешмас = (2.52)где Ь' - константа;d - средний путь диффузии.Жуховицким показано, что более правильна следующая запись [8]:= Ь' [Ю1(\ + т(2.53)46
njjiBA_A Основы теории газовой хроматографииОбычно зависимости Я от м для одного и того же сорбента, но различ¬
ного зернения сильно отличаются. Однако зависимости так называемых
приведенных высот тарелок от приведенной скорости газа-носителя оди¬
наковы для разных зерен адсорбента [14]. Во многих случаях эта зависи¬
мость имеет следующий вид [15,16]:h = (S/V) + AV>-» + Cv, (2.54)где h = h/d^ - приведенная высота тарелки;V = udJD^- приведенная скорость подвижной фазы.Первый член представляет вклад в общее размывание продольной
диффузии, второй - определяет качество набивки колонки (для хорошо
наполненных колонок величина А около 1) [15,16], третий член отражает
скорость массообмена между подвижной и неподвижной фазами.2.3. Понятие об эффективности, селективности и степениразделения2.3.1. Основные критерии оценки степени разделенияТеория хроматографии должна быть использована в первую очередь
для решения практических задач: выбора оптимальных параметров опыта
и предсказания в нешгорых случаях возможности разделения без экспери¬
ментов. Для изучения влияния параметров на степень разделения нужны
объективные оценки, нужны надежные критерии оценки. Критерии оцен¬
ки необходимы также для сопоставления хроматографических методик и
приборов в целом.В литературе предложено и описано много различных критериев, из
которых можно выделить четыре группы [8]:1. Критфии, оценивающие общую {тделигельную способность колонок.2. Критерии оценки селективности разделения.3. Критерии, оценивающие степень размывания, эффекгивность колонки.Эти критерии характеризуют свойства колонки, определяемые кине-
тиюй и динамикой процесса, а также характеристиками дозатора и детек¬
тора.4. Критерии, оценивающие все качества хроматографической уста¬
новки, всю работу хроматографа в целом и включающие кроме степени
разделения такие важные параметры как сопротивление потоку и продол¬
жительность анализа.Степень разделения должна характеризовать чистоту фракции и опре¬
делять долю основного компонента фракции, выражаемую безразмерной
^личиной (п). Эта величина должна определяться параметрами выходной
кривой [8].47
Яшин ЯМ, Яшин Е.Я.. Яшин Л.Я. Гямвая хроматографияВ случае линейной изотермы выходная кривая определяется тремя па¬
раметрами: удерживаемым объемом (VJ, определяющим положение мак¬
симума пика, концентращ1ей в максимуме (с^) и параметром а, определя¬
ющим форму выходной кривой в соответствии с уравнением:с=с_^ (2.55)С величиной а связана ширина полосы ц:ц=2лЯ7о (2.56)Чистота фракции (т]) зависит от соотношения высот пиков или концен¬
траций в максимуме пика (с_^, и c^^j).Степень разделения двух соседних пиков на хроматограмме определя¬
ется разностью удерживаемых объемов (AF^^) и шириной пиков (ц).При полном разделении пиков эти величины можно легко определить
из хроматограммы. Критерий разделения R (для полного разделения) за¬
писывается следующим образом (рис. 2.11) [8]:Если пики имеют вид кривой Гаусса, то для целей анализа разделение
можно считать полным при R> 1. Это наиболее распространенный крите¬
рий разделения, его широко используют на практике.Жуховицкий и Туркельтауб [8] предложили критерии для неполного
разделения и оценки степени разделения пишв, сильно отличающихся по
высоте.Рис. 2.11. Оценка критериев разделения для полного разделения по
хроматограммамЧистота фракции сильно зависит от критерия разделения. Даже при
R = 1 полного разделения не наблюдается, так как левая и правая части
выходной кривой асимптотически приближаются к нулю. При расчете
площадей пиков в аналитической газовой хроматографии этим можно пре¬
небречь, но следует иметь в виду в препаративной хроматографии.Эти расчеты сделаны при допущении, что пики имеют форму кривой
Гаусса. На практике пики, как правило, отличаются от кривой Г^сса, кро-48
ГЛАВА 2 Основы теории газовой хроматографииме того, следует иногда учитывать вытесняющий эффект второго, более
сильно сорбирующего вещества.2.3.2. Связь критериев разделения с параметрами опыта.Для определения связи критерия разделения R с параметрами опыта нуж¬
но выразить величины AFj^ и ц через основные параметры разделения [8].Известно, что:V^~KSL (2.58)где К - коэффищ1ент Генри; S - сечение колонки; L - длина колонки.В этом случае:AV^ = SLAK (2.59)Для щирины полосы ц воспользуемся ранее полученным выражением
(2.28).Подставляя выражения (2.25) и (2.59) в уравнение (2.57), получим:К=АК/8Кл/Ь^ (2.60)Селективность разделения в газовой хроматографии принято оцени¬
вать соотнощением [8]:К^ = {К^-К^)Щ + К^) (2.61)где - критерий селективности.Предположив, что для двух соседних пиков абсолютные значения ко-
эффивд1ентов Генри близки (при больших удерживаемых объемах оправда¬
но, так как имеется минимальное различие коэффициентов Генри для со¬
седних пишв), м(шно записать уравнение (2.60) следующим образом:R»1/4(Kj-K0/(Kj^J>/Lu7D^==1/4KcVLu/D,* (2.62)Введя соотношение Я = 2£>^/м в выражение под корнем, получим:R,==Kc^Ш^M~^Й(KcVШ)/4=л/2(Kc^^)/4 (2.63)Это уравнение непосредственно связывает критерий разделения R,
оценивающий степень разделения или разделительную способность ко¬
лонки с селективностью (К^) и эффективностью (N) колонки (эффектив¬
ность колонки здесь выражена числом теоретических тарелок).Из уравнения (2.63) можно определить выражение для минимальной
длины слоя (минимальной длины колонки, необходимой для полного раз¬
деления):^мнн = (8Л'/Л?^)Я (2.64)При R = 1 (условие полного разделения)= (2.65)494 Газовая хроматография
Яшин ЯМ.. Яшин ЕЛ. Яшин АЛ Гтовая хртттшФтПредставляет шггерес связь между селективностью и эффектив¬
ностью (iV) в уравнении (2.63) при определенном значении R, при штором
достигается полное разделение. Задачу можно сформулировать следую¬
щим образом: каюе число теоретических тарелок необходимо для полного
разделения компонентов при определенном значении селективности? Эта
задача решена Парнеллом [17].Рис. 2.12. Зависимость эффектитости (N) от селективности
(а) при R-1В литературе селективность часто оценивают величиной а, равной от¬
ношению исправленных удерживаемых объемов или времен удерживания
двух соседних пиков:a=tR/tR,=V*^RrK-2^. (2.66)При а = 1 вещества не разделяются. Разделение возможно только при
а > 1. Такая оценка селективности очень удобна, так как а можно опреде¬
лить непосредственно из хромаготрамм. В связи с тем, что величина а равна
отношению юэффициентов распределения (2.66), по ней можно легко оце¬
нить различие в свободных энергиях для двух компонентов А(АС?) [17]:А(АС) = -ЛПпа (2.67)Число тарелок, необходимое для полного разделения при М = 1, равно
(по Парнеллу) [17,18]:N- 16(а/а 1)2 [(Л:^ + 1)(К-1)Т (2.68)где = (t^ - tg)/tj - коэффициент емкости, равный отношению исправ¬
ленного времени удерживания к времени удерживания несорбированного
вещества.50
1ШВА2Основы теории газовой храштюграФыиВ газовой хроматографии коэффициенты емкости обычно велики
(более 30 - 50), поэтому третьим множителем в уравнении (2.68) можно
пренебречь. Из рис. 2.12 и таблицы 2.1 видно, что зависимости Not а для
К^ = 50 и К^= 1000 близки.Таблица 2.1Значения N при различных аNК =1000к =500К =100К =50К-101,005647 708649 000659 409672 531782 1631,01163 542163 868166 497169 610197 4911,0241 69941 78242 45243 29750 3551,0410 83710 85911 033И 25313 0871,0650045 0145 0945 1956 0421,082 9222 9282 9763 0343 5281,121 3961 39914221 4501 6861,15942944959978I 1381,205775785875996971,254004024084164841,30301302306312363Рассмотрим графическую зависимость между Ми а (рис. 2.12). При
а = 1,02 для полного разделения веществ (/? = 1) необходимо 42 ООО теоре¬
тических тарелок. При а = 1,04 требуется уже всего 10 ООО теоретических
тарелок. Таким образом, повышение селективности на 2% (увеличение а)
снижает требования к эффективности (к необходимому числу теорети¬
ческих тарелок) в четыре раза (т.е. на 400%). При увеличении а на 10%
(а = 1,12) требуется всего 1 400 тарелок, т.е. требование к эффективности
уменьшается в тридцать раз.Число теоретических тарелок пропфционально длине хроматографиче-
сюй колонки. Таким образом, повышение селективности на 10% (повышение
а от 1,02 до 1,12) позволяет уменьшить длину колонки в тридцать раз.Необходимо это принимать во внимание при подборе сорбентов. На
селективных сорбентах можно проводить разделение на значительно бо¬
лее шротких колонках.Кроме того, из рис. 2.12 и таблицы можно сделать еще один практиче¬
ский вывод. Средняя эффективность набивных колонок колеблется от 1000
до 2000 тарелок на один метр длины, обычно используют колонки длиной
от 1 до 3 м, т.е. с максимальной эффективностью не более 6000 теоретиче¬
ских тарелок. Это означает, что на таких колонках можно разделить смеси,
компоненты которых имеют не менее а ~ 1,06. При меньших значениях а
необходимо использовать капиллярные колонки, на которых легко дости¬
гается значительно более высокая эффективность.51
Яшин Я.И.. Яшин ЕЖ. Яшин А.Я. Газовая хроматография2.4. Влияние температуры на хроматографическое разделение.Температура колонки - один из основных параметров, влияющих на
время разделения, селективность разделения, а также размывание хрома¬
тографических полос. Сорбируемость веществ сильно зависит от темпе¬
ратуры, поэтому изменение температуры позволяет в широких пределах
изменять время удерживания. Большие возможности дает изменение тем¬
пературы во времени и по длине слоя.2.4.1. Зависимость удерживаемых объемов от температуры.Удельный удерживаемый объем пропорционален коэффициенту Генри
(F^~ KSL), где S- сечение колонки, L - длина шлонки.Известно [7], что(2.69)где А - константа;Q - теплота сорбции;R - газовая постоянная;Т - температура, °К.Включив это соотношение в уравнение (2.55), можно записать:V^~SLAe^"^\ (2.70)Таким образом, удельный удерживаемый объем связан с температурой
экспоненциальной зависимостью.Возьмем натуральный логарифм (2.70), получим:In = S' + е/2,3 RT (2.71)Удобнее перейти к десятичному логарифму:lgV^ = B" + Q/2,3RT (2.72)Графические зависимости Ig от обратной температуры (1/Т) - пря¬
мые линии (рис. 2.13), таргенс угла наклона которых равен:tga = QI2,3R, e = 2,3/ftgaпри R = 8,36 Дж/(моль •K)Q~ 19,23 tg а (2.73)Следовательно, из наклона прямой температурной зависимости удер¬
живаемого объема можно определить теплоту сорбции. Для двух соседних
пиков на хроматограмме эти зависимости обычно выглядят так, как по¬
казано на рис. 2.136. Из этого рисунка можно сделать два вывода; удер¬
живаемые объемы сильно уменьшаются с температурой; селективность
разделения и разность удерживаемых объемов (А V^) падают с повышени¬
ем температуры. Однако, в нешторых случаях температурные зависимо¬
сти для двух соседних пиков могут быть такими, как на рис. 2.13а. В этом
случае наклоны для обоих веществ одинаковы (прямые параллельны), это52
ПЫВЛ2Основы теории газовой хроматографииозначает, что и теплоты сорбции для двух веществ одинаковы. Такие за¬
висимости возможны для изомеров, для которых теплоты сорбции близки,
а различие в удерживаемых объемов достигается за счет константы в урав¬
нении (2.70), в которую входит энтропия сорбции. Разделение происходит
не за счет различной энергии сорбции, а за счет различия энтропий [19].Возможны зависимости Ig от 1ЛГ с точкой перелома (рис. 2.13в), в
частности для сорбентов в точках перехода из одного агрегатного состояния
в другое или для жидких кристаллов при переходе от жидкокристалличе¬
ской фазы (нематической мезофазы) к обычной изотропной жидкости [20].№,23isaтт(}/Л¥ (уГ \1$ Vt/T, ФгтРис. 2.13 Зависимость Ig от обратной температуры колонкиИ совсем уже в редких случаях эта зависимость может быть обрат¬
ной, т.е. с повыщением температуры удерживаемые объемы не падают, как
обычно, а возрастают (рис. 2.13г). Это возможно, в частности, в том слу¬
чае, если к инертному газу-носителю добавлена примесь сильносорбируе-
мого вещества, например, водяного пара, молекулы которого на полярном
сорбенте при низких температурах блокируют наиболее активные центры,
и слабополярные молекулы анализируемых веществ не в состоянии их вы¬
теснить. С повышением температуры ориентационное взаимодействие па¬
дает, и большие молекулы анализируемых веществ за счет большего дис¬
персионного взаимодействия уже способны вытеснить молекулы воды и
сорбироваться на наиболее активных участках [20].53
Яшин ЯМ. Яши» Е.Я.. Яшин А.Я. Газовая хроматографияКомбинируя ЭТИ противоположные эффекты (рис. 2.13), теоретически
можно получить шлонку, на которой удерживаемый объемы будут слабо
зависеть от температуры.В качестве одной из мер селективности можно принять разность коэф¬
фициентов Генри или разность удерживаемых объемов:V^^KSL, AV^^AKSL (2.74)Так как К ~ Ае^’’’, то при допущении, что А близки для обоих компо¬
нентов, получим:К = А^" AQ/RT-KAQ/RT, AV^~ STKAQ/RT (2.75)Подставляя полученные выражения в уравнение (2.63), получим:R1=AQ/SRTVLu/D^ (2.76)В этом случае можно принятьK^~AQ/RT (2.77)Селективность разделения определяется разностью теплот сорбции.'
Именно теплотой (энергаей) сорбции определяется время одного акта ад¬
сорбции (т*) [21]:г' = т/й^^ (2.78)где Тц - период колебания адсорбированной молекулы в направлении,
перпендикулярном к поверхности.Величина т^, непосредственно связана с периодом колебаний молекул
или атомов поверхности адсорбента и равна 10 '^- 10“' с.Таким образом, теплота сорбции и время сорбции свгааны соотноше¬
нием (2.78).В таблице 2.2 приведены времена адсорбции при некоторых определен¬
ных значениях теплот адсорбции [21]. Величина принята равной 10 '^ с
(для графита, оксвда алюминия и оксида кремния равно 5Т0с, 7,510“' с
и 9,510 ''' с соответственно).Для газохроматографических разделений желательно использовать
только сорбенты, на которых теплоты сорбции анализируемых веществ не
более 60 кДж/моль. При больших значениях Q времена удерживания будут
весьма велики.Чаще всего используют сорбента, на которых теплоты сорбции около
40 кДж/моль, а время одного акта адсорбции - порядка 10 * с. Несмотря
на небольшое время одного акта сорбции, времена удерживания могут до¬
стигать значительных величин (порядка нескольких десятков минут) за
счет огромного числа соударений с поверхностью, огромного числа актов
сорбции.54
Таблица 2.2Время одного акта адсорбции в зависимости от теплоты адсорбцииQ, K^Ьc/MQЛЬX, с0,421,310‘’6,31,210-'214,6410-"16,8МО-'о423,2-10^631,8-10-^84Теплота адсорбции в небольших пределах температур принимается
независимой от температуры.Из уравнения (2.78) следует, что при постоянном значении Q время
адсорбции (т) будет сильно зависеть от температуры, С повышением тем¬
пературы возрастает кинетическая энергия молекулы и возрастает вероят¬
ность отрыва сорбированной молекулы от поверхности (молекула десор¬
бируется тогда, когда ее кинетическая энергия будет равна энергии межмо-
лекулярного взаимодействия или превосходить ее).2.4.2. Хроматографическое разделение с программированием тем¬
пературы колонки во времени.Повышение температуры колонки по определенной программе в про¬
цессе хроматографического разделения предложено в 1952 году [22]. Про¬
граммирование температуры - обычно повышение температуры колонки во
времени с определенной скоростью - используют для сокращения времени
разделения сложных смесей, температуры кипения компонентов которых
сильно различаются. Разделение таких сложных смесей на одной колонке
за приемлемое время в большинстве случаев невозможно, даже если се¬
лективность позволяет разделить всю смесь. Для разделения низкокипя-
щих компонентов, нужно использовать более низкую температуру, однако
при этой температуре времена удерживания высококипящих компонентов
буцут настолько велики, что элюировать их из колонки за приемлемое вре¬
мя не удается. Если использовать более высокую температуру, при которой
бы высококипящие компоненты элюировали за доступное время, то при
этой достаточно высокой температуре не буцут разделяться низкокнпящие
компоненты (рис. 2.14). Такие смеси в изотермическом режиме можно бу¬
дет разделить только в 2 - 3 приема при разных температурах или же одно¬
временно на двух-трех разных колонках на разных хроматографах.55
Яшин Я.И.. Яшт ЕЛ. Яшин ЛЯ, Гаювая хроматографияJJU_А,JIULJV_» LxaaxxРис. 2.14 Хроматограммы смеси компонентов с широким диапазо¬
ном температур кипения:
а - при низкой температуре (полное деление, но очень длительное вре¬
мя разделения и сильное размь1вание последних пиков); б - при высоких
температурах (время разделения сокращается, но первые компоненты не
разделяются); в - ^ режиме программирования температуры (полное и бы¬
строе разделение, пики узкие).В режиме программирования температуры такую задачу можно ре¬
шить на одной юлонке за один цикл анализа. Для этого достаточно во вре¬
мя разделения температуру колонки повышетъ по определенному закону
(линейному, линейно-ступенчатому, баллистическому или любому друго¬
му) (рис. 2.15).Рис. 2,15 Программирование темперащры колонки баллистическое
(1), линейное (2) и линейно-ступенчатое (3).С ростом температуры уменьшаются юэффициенты Генри и, следо¬
вательно, уменьшаются уцерживаемый объемы, ширина полосы в газовой
фазе также уменьшается, т.е. эффективность колонки возрастает.56
ГЛАВА 2 Основы теории газовой хроматографииРассмотрим этот процесс. В изотермическом режиме в процессе раз¬
деления коэффициенты (К) остаются постоянными. При вымывании поло¬
сы со слоя сорбента в газовую фазу ширина полосы возрастает примерно
в К раз (здесь К - тот же коэффициент распределения). В режиме про¬
граммирования температуры коэффициенты распределения уменьшают¬
ся в процессе разделения и к выходу полосы компонентов подходят при
значительно меньшем значении К. При вымывании со слоя в этом случае
полосы в газовой фазе расширятся значительно меньше. За счет этого эф¬
фекта ширина пиков в режиме программирования обычно меньше, чем в
изотермическом режиме для одних и тех же веществ даже при одном и том
же времени удерживания.Известно, что с повышением температуры нелинейные изотермы сорб¬
ции становятся более линейными, поэтому несимметричные пики, получа¬
емые на неоднородных адсорбентах в изотермическом режиме, в режиме
гфограммирования становятся симметричными.В изотермическом режиме каждый компонент разделяемой смеси
можно характеризовать параметрами, связанными со скоростью движения
его по колонке - временем уцерживания или удерживаемым объемом:= FL/u^ = L/u^ (2.79)где F - объемная скорость газа-носителя;L - длина колонки;- линейная скорость движения полосы.В режиме программирования температуры скорость продвижения по¬
лосы по слою непостоянна (непрерывно меняется), поэтому определение
)щерживаемого объема в режиме программирования температуры является
сложной задачей. В этом случае вводят дополнительный важный параметр- температура выхода или температура удерживания вещества Т^. Она рав¬
на температуре колонки (в режиме постоянного роста температуры колон¬
ки) в момент появления пика исследуемого вещества [23].В случае линейного программирования температуры температура ко¬
лонки растет в соответствии с простым соотношением:Т = + rt, (2.80)где Тд - начальная температура;г - скорость повышения температуры.Общее уравнение для линейного программирования, связывающее
удерживаемый объем со скоростью подъема температуры и скоростью по-
TOia, записывается следующим образом [23]:Т\=^V(F/V^dT (2.81)57
Яшин ЯМ.. Яшин Е.Я.. Яшин А.Я.Газовая хроматографияПри ПОСТОЯННОЙ скорости потока уравнение примет следующий вид:Т,f = \dT/V^ (2.82)'^0Для каждого вещества при любой заданной начальной температуре
температуру удерживания можно связать с программой через характери¬
стическую величину r/F. Отношение скорости подъема температуры к
скорости потока газа-носителя - очень существенный параметр, это отно¬
шение может быть получено из значений удерживаемых изотермических
объемов для данной температурной области.Температуру удерживания можно определить графически из экспе¬
риментальных данных. Строят график зависимости обратной величины
изотермического удерживаемого объема от температуры (рис. 2.16) [23].
Значение правой части уравнения (2.16) есть просто площадь под соот¬
ветствующей кривой от нижнего температурного предела до любой более
высокой температуры. Затем строят график зависимостей этих площадей
от верхней температуры (рис. 2.16). По кривым на рис. 2.16 можно найти
температу!^ удерживания для различных линейных программ (различных
значений r/F). Зная расход газа-носителя (F), скорость подъема темпера¬
туры (г) мы задаем, тогда, взяв их отношение, легко можно определить
температуру удерживания. Зная ее, можно легко рассчитать время удержи¬
вания [оно равно (f^j - Т/г)] и удерживаемый объем (Г^, - TJ f/r для случая
линейного программирования [23].т,*сРис. 2.16 Зависимость r/F от температуры удерживания для раз¬
личных углеводородов. 1 - н-бутан; 2 - н-пентан; 3 - н-гек-
сан; 4 - н-гептан. Точками отмечены температуры удер¬
живания для различных программ, значения ruF которых
указаны справа.58
fJIABA 2 Основы теории газовой хроматографииПри проведении хроматографического разделения в режиме програм¬
мирования температуры встречаются некоторые методические и аппара¬
турные трудности.В случае применения газо-жидкостаого варианта хроматографии и
высокочувствительных ионизационных детекторов наблюдается смеще¬
ние нулевой линии (дрейф нулевой линии) при программировании тем¬
пературы. Этот дрейф связан с ростом фонового ионного тока в связи с
ростом летучести жидкой фазы с повыщением температуры.С повышением температуры увеличивается вязкость газа-носителя.
Поэтому при постоянном давлении на входе и выходе из колонки скорость
газа-носителя уменьшается пропорционально абсолютной температуре
в степени 1,7. Чтобы сохранить постоянным расход, нужно увеличивать
входное давление, так как сопротивление колонки возрастает. Для этих це¬
лей используют специальные регуляторы расхода газа-носителя.Для компенсации ионного тока и изменения расхода применяют диф¬
ференциальный режим, т.е. используют параллельно две колонки с двумя
детекторами. Детекторы включены так, что сигналы от них взаимно ком¬
пенсируются [23].Если колонки наполнить одинаково одним и тем же сорбентом, то с
реличением температуры дрейфа нулевой линии не должно быть, так как
должна происходить взаимная компенсация фоновых ионных токов. Од¬
нако таким путем полной компенсации достичь не удается, так как абсо¬
лютно одинаковые колонки приготовить нельзя. Кроме того, в этом случае
происходит компенсация ионных токов, компенсации же флуктуаций не
происходит, наоборот, флуктуации обычно удваиваются.2.4.3. Хроматографическое разделение с программированием тем¬
пературы по длине слоя (хроматермография).Хроматермография была предложена Жуховицким, Туркельтаубом,
Соколовым и др. в 1950-51 гг [24]. В этом виде хроматографии на разделя¬
емую смесь одновременно воздействуют поток газа-носителя и движуще¬
еся во времени и в пространстве температурное поле.Вьщеляют два варианта хроматермографии: стационарный и нестаци¬
онарный.В стационарной хроматермографии направления движения темпера¬
турного поля и потока газа-носителя совпадают, а градиент температуры
имеет обратное значение.Для осуществления этого варианта хроматермографии на колонку на¬
девают цилиндрическую печь, по длине которой имеется градиент темпе¬
ратуры и которая с помощью специального мотора может с разной скорос¬
тью двигаться по колонке (рис 2.17) [24].59
Яшт ЯМ. Яшин ЕЛ. Яшт А.Я.Гтовая хроматография/UMJL!CCCSN>J»NC/lA_JUг, < г,Рис. 2.17 Схема установки для стационарной (а) и нестационарной(б) хроматермографии. 1- печь с градиентом температу¬
ры; 2 - хроматографическая колонка.В стационарной хроматермографии температура в начале печи (по
ходу движения) ниже, чем в конце. Если ввести в начало колонки анали¬
зируемую смесь и надвинуть на начало движущуюся печь, то шмпоненты
в зависимости от природы будут сорбироваться и продвигаться по слою с
разными сшростями. Вещества, которые сорбируются слабо, будут дви¬
гаться по слою с большей скоростью, уходя вперед в пределах длины печи,
они будут попадать в область более низких температур, где их сорбиру¬
емость возрастает, а скорость продвижения замедляется. Это замедление
скорости будет происходить до тех пор, пока скорость их продвижения не
станет равной скорости движения самой печи. Вещества же, которые сор¬
бируются сильно, будут двигаться по слою сначала с меньшей скоростью,
чем скорость печи; отставая, они будут попадать в область более высоких
температур (так направлен градиент температуры в печи), где их сорбиру¬
емость уменьшается, а скорость продвижения возрастает. Это возрастание
скорости будет происходить до тех пор, пока опять же она не сравняется со
скоростью движения печи.В обычном варианте хроматографии каждый разделяемый компонент
двигается со своей определенной скоростью, в хроматермографии с про¬
граммированием температуры все они двигаются со скоростью печи. Каж¬
дый компонент в соответствии со своей сорбируемостью займет опреде¬
ленное положение в пределах длины печи. Максимум концентрации по¬
лосы веществ будет группироваться около одной температуры - характери¬
стической, которая определяется по формуле [24]:Г = - (Q/R) [Шп (хК, и/и)] , (2.83)60
[ЛЛВЛ^^ Основы теории газовой хроматографиигде Q - теплота сорбции;R - газовая постоянная;X - доля пустоты в колонке;К - коэффициент Генри;- скорость движения полосы;и - линейная скорость движения газа-носителя.Стационарная хроматермография имеет ряд преимуществ перед обыч¬
ной изотермической хроматографией.1. Быстрое разделение смеси компонентов с различной со(^ируемостью.2. Возможность получения симметричных пиюв даже при нелиней¬
ной изотерме сорбции.3. Значительное повышение концентрации компонентов в центре
полосы.Стационарная хроматермография - один из немногих методов хрома¬
тографии, в котором возможно абсолютное обогащение пробы (т.е. кон¬
центрация компонента на выходе из колонки выше, чем в начале). На этом
принципе Р.Кайзером предложен «реверсивный» хроматограф фирмы «Си¬
менс», серийно выпускаемый для анализа примесей в воздухе.В нестационарной хромагермографии (рис. 2.17) процесс проходит
так же, как и в стационарной, однако градиент температуры в печи про¬
тивоположен. Это приводит к тому, что слабосорбируемые компоненты,
уходя вперед по слою сорбента в пределах печи, попадают в область более
высоких температур, где движение их ускоряется. Вещества же, шюрые
сорбируются сильно, будут отставать и попадать в область более низких
температур, где движение их будет тормозиться.Таким (Лразом, за счет градиента печи произойдет дополнительное
«растягивание» компонентов, и селективность разделения возрастет.Комбинация стационарной хромат«рмографии с фронтальным мето¬
дом (теплодинамический метод) используется для выделения первого наи¬
более слабосорбируемого компонента в чистом виде с целью концентри¬
рования и последующего анализа или же для препаративного получения
вещества в чистом виде [25].2.5. Влияние параметров на хроматографическое разделение.2.5.1. Основные группы параметров.Большой выбор параметров опыта является одним из преимуществ
хроматографического метода. Если по каким-либо причинам нельзя изме¬
нять одни параметры, то представляется возможность изменить другие и
достичь практически тех же самых конечных результатов. Однако после
выбора оптимальных параметров это достоинство становится недостат-61
Яшин Я.И., Яшин Е.Я., Яшин А.Я. Газовая хроматографияКОМ, так как для получения точных количественных результатов нужно
стабилизировать эти параметры.Параметры опыта можно разделить на четыре группы [8]:1. Параметры, связанные с разделительной колонкой - природа сор¬
бента, температура колонки, геометрия колонки (длина, сечение, форма),
зернение (размер зерна, форма зерен), толщина жидкой пленки (количе¬
ство жидкой фазы) или пористость адсорбента (удельная поверхность,
средний диаметр пор), способ набивки и др.2. Параметры, связанные со свойствами газа-носителя - природа, ско¬
рость, вязкость и давление газа-носителя.3. Параметры, относящиеся к работе дозатора - размер пробы, способ
дозирования.4. Параметры детектирующих систем - предел детектирования (чув¬
ствительность), инерционность, линейный диапазон, стабильность детек¬
тора (уровень шума, дрейф).Необходимо классифицировать все параметры по характеру их воз¬
действия на степень разделения. Большая часть параметров определяет
размывание, т.е. связана с динамикой и кинетикой процесса разделения,
и меньшая часть связана с селективностью разделения - это природа сор¬
бента, температура, в некоторых случаях природа сорбирующегося газа-
носителя и соотношения доли адсорбента и жидкой фазы в случае газовой
адсорбционно-абсорбционной хроматографии. Вопросы выбора сорбента
тесно связаны с теорией адсорбции и растворения. Вопросы же выбора
условий для получения эффективной колонки относятся непосредственно
к теории хроматографии. Знание основ теории хроматографии позволяет
сознательно подойти к выбору оптимальных условий, т.е. ускорить и об¬
легчить этот процесс выбора, в противном случае можно выбирать из бес¬
конечного числа вариаций и поэтому поиски могут длиться долго.2.5.2. Выбор оптимального режима разделения в зависимости от
характера аналитической задачи.Можно выделить две крайние аналитические задачи [8]. В первой за¬
даче - экспрессной хроматографии - необходимо получить полное разде¬
ление (определенное значение Л,) за заданное время. Эта задача типична
при использовании хроматографии для аналитического контроля в про¬
мышленности, когда необходимо разделение за короткое время.Ранее было установлено:К=:(лЙ74)(Ксл/Ш) (2.84)62
иЫВЛ 2 Основы теории газовой хштатогтФииПри переходе от длины слоя к времени разделения,t = L/u^, = u/K,t = LK/u. L = ut/K (2.85)TO получим; Rтак какtK(A/u+B/u^+C)(2.86)H/u = A/u + B/u^ + С
Уменьшение времени разделения часто связано с повышением скорости
газа-носителя. Следовательно, при больших значениях и можно записать:К^^)(К^ШС) (2.87)Экспрессная хроматография требует применения сорбентов, на кото¬
рых массообмен проходит быстро (малое значение коэффициента Q.Экспрессность всегда достигается за счет ухудшения разделения, так
как Л ~ л/1Во втором случае, наоборот, время не ограничивается, а требуется до¬
стичь наилучшего разделения. Полное разделение необходимо для получе¬
ния точных шличественных результатов, поэтому эта задача относится к
прецизионной хроматографии [8].Одним из путей улучшения разделения является увеличение длины
юлонки. Поэтому в качестве ограничивающего фактора в этом случае сле¬
дует ввести доступное значение перепада давления в колонке.Сопротивление колонки при ламинарном потоке пропорционально
длине и скорости, следовательно, необходимо рассматривать максималь¬
ное значение R^ при заданном значении произведения Lu:(2.88)При заданном uL значение Л будет максимальным при м —► О и i -♦ оо:К, = (^^/4)(KcVLu/B) (2.89)Следовательно, в прецизионной хроматографии основное размывание
будет за счет продольной молекулярной диффузии.2.5.3. Влияние основных параметров на степень разделения.Оптимальный выбор можно провести на основе критериев разделения.
В этом разделе будет рассмотрено влияние отдельных параметров на сте¬
пень разделения (критерий разделения R). В том случае, коща прямой связи
Между определенным параметром и величиной R нет, будет рассматриваться
связь с селективностью (а) и эффективностью разделения (N, Н).Природа сорбента. Сорбент в основном определяет различие параме¬
тров удерживания, в частности AV^.63
Яшт Я.И.. Яшин ЕЯ. Яшин А.Я. Газовая хроматографияСтепень разделения прямо пропорциональна критерию селективности:К~К^~ШК (2.90)Степень разделения пропорциональна разности юэффициентов Генри,
но обратно пропорциональна их абсолютному значению. Задача оптимиза¬
ции в этом случае сводится к следующему: нужно подобрать такой сорбент,
чтобы при максимальном различии коэффициентов Генри их абсолютные
значения были бы минимальными. С увеличением абсолютного значения
коэффициента Генри увеличивается ширина полосы на хроматограмме.Коэффициент Генри равен:(2.91)где А - константа;Q - теплота сорбции;R - газовая постоянная;Т - температура, °К.Поэтому, различие коэффициентов Генри сводится к различию теплот
(энергии) сорбции, т.е. к различию межмолекулярных взаимодействий.Селективность разделения в определенных пределах будет зависет
также от доли сорбента в единице объема колонки.Отношение коэффициентов Генри равно [8]:/ = ед = (X + Х,)/(Х + ,Х.) (2.92)Коэффициенты Генри (К, и К^) определяют сорбцию единицей объема
насадки. Уменьшение коэффициента за счет уменьшения количества рас¬
творителя приведет к уменьшению селективности при малых значенияхПри больших значениях в уравнении (2.92) можно пренебречь х>
тогда(2-93)и коэффициент селективности не будет зависеть от количества непод¬
вижной фазы. Однако, если х и Г^^ по абсолютному значению малы, то
уменьшение х, приведет к уменьшению коэффициента селективности, т.е.
к ухудшению разделения.Это легко проследить при сопоставлении набивных и капиллярных
газо-жидкостных колонок с одной и той же неподвижной фазой [8]. Допу¬
стим, истинные коэффициенты Генри соответственно равны 30 и 20. Для
набивных колонок х = 0,5 и х, = 0,1 (примерно одну пятую часть по объему
от твердого носителя составляет жидкая фаза). Подставляя эти величины в
уравнение (2.92), получим:/ = 0,5 + 30 • 0,1/0,5 + 20 • 0,1 = 1,4 (2.94)Для капиллярных колонок х ~ 1 и Xi ~ Ю'^, тогда:64
Основы теории газовой храматогуафии/ = 1 + 30 • 10-V1 + 20 • 10-^ = 1,09, (2.5. 3.6)а значения соответственно равны 0,17 и 0,045.Таким образом, при использовании одной и той же жидкой фазы се¬
лективность насадочных колонок выше, чем капиллярных. Поэтому при
одном значении величины Я в капиллярной хроматографии потребуется
большая длина колонки.В капиллярных колонках селективность может сильно меняться в за¬
висимости от количества жидкой фазы. На рис. 2.18 нами показано, что
при небольших количествах жидкой фазы не наблюдается полного раз¬
деления ксилолов, хотя эффективность (число теоретических тарелок)
очень высока. При увеличении количества жидкой фазы в колонке селек¬
тивность возрастает и наблюдается полное разделение изомеров.Температура сильно влияет на селективность разделения, влияние
температуры рассмотрено в разделе 2.4.Длина колонки. Степень разделения пропорциональна квадратному
корню из длины колонки (при увеличении длины колонки в 4 раза степень
разделения увеличивается только в 2 раза). Практически степень разделе¬
ния для насадочных колонок увеличивается еще в меньшей степени. Дело
в том, что при использовании колонок большой длины перепад давления в
колонке возрастает и соответственно сильно меняется скорость потока по
длине колонки. В связи с этим некоторые участки колонки буду работать
не в оптимальном режиме, и в целом эффективность колонок будет мень¬
ше, чем это следует из теоретического соотношения. Кроме того, работа
на колонке большой длины всегда связана с неудобствами (в частности,
о&спечение герметичности, потеря пробы при дозировании в испаритель,находящийся под высо-
^ КИМ давлением). Поэтомув аналитической прак-
cat тике преимущественно
используют юлонки дли¬
ной 1 - 3 м.^ис. 2.18 Хроматограммы п-, м-, о-ксшолов на капиллярной колон¬
ке длиной 50 м из нержавеющей стали с содержанием
жидкой фазы - сквалана 5 (а), 10 (б) и 30% (в).65Газовая хроматография
Яишн Я.И.. Яшин Е.Я., Яшин А.Я. Газовая хроматографияДиаметр колонки. Разделительная способность колонки обратно про¬
порциональна диаметру (сечению) колонки. Ширина пика прямо пропорци¬
ональна сечению колонки (см уравнение 2.28). С уменьшением сечения ко¬
лонки (уменьшением внутреннего диаметра колонки возрастает эффектив¬
ность и разделительная способность колонки до определенного предела.При дозировании малых проб (при использовании высокочувствитель¬
ных детекторов) наиболее оптимальными колонками для аналитических
целей являются колонки с внутренним диаметром 2- 3 мм. В некоторых
специальных случаях (в частности, анализ примесей) лучшие результаты
получаются на колонке с внутренним диаметром 4 мм.Форма колонки. С целью более компактного размещения в термо¬
стате хроматографические колонки изготавливают в виде небольших пря¬
мых, U-образных и спиралеобразных трубок. Наибольшей эффективности
можно достичь на прямых и U-образных колонках по двум причинам: во-
первых, такие колонки легче плотно наполнить (чем плотнее и однород¬
нее набивка, тем выше эффективность), во-вторых, на спиралеобразных
колонках (при большом радиусе кривизны) появляется дополнительное
размывание, связанное с неоднородностью скоростей по сечению, сопро¬
тивление потоку у ближней (к центру окружности) стенки трубки меньше,
чем у дальней, так как пути прохождения газовых потоков у ближней стен¬
ки меньше, чем у дальней.В термостате U-образные колонки желательно размещать вертикаль¬
но, так как в процессе эксплуатации в колонке происходит усадка, и сверху
образуются пустые обьемы. В вертикальных трубках легко убрать эти пу¬
стые объемы, засыпав дополнительно сорбент.Размер зерен твердого носителя или адсорбента. Размывание хрома¬
тографических полос в значительной степени определяется размером зерен
сорбента. Для всестороннего рассмотрения роли зернения нужно восполь¬
зоваться уравнением (2.48). Прежде всего из уравнения следует, что размы¬
вание за счет вихревой диффузии уменьшается с уменьшением размеров зе¬
рен (вихревая диффузия пропорциональна диаметру зерна d ). Третий член
уравнения, определяющий внешнедиффузионное размывание, также в силь¬
ной степени зависит от диаметра зерна (пропорционально d^’'^), так как пути
внешней диффузии между зернами определяются размером зерен.Внутренний массообмен в газо-жидкостной хроматографии практиче¬
ски не зависит от диаметра зерна. В газо-адсорбционной же хроматогра¬
фии диаметр зерна определяет также пути внутренней диффузии для обь-
емно-пористых адсорбентов. Таким образом абсолютный размер диаметра
зерна, а также однородность зернения сильно влияют на эффективность
колонки. Из уравнения (2.48) следует, что чем меньше диаметр зерна, тем
выше эффективность. Однако беспредельно уменьшать размер зерен нель¬
зя. Слишком мелкие зерна приводят к большому сопротивлению колонки,66
ГЛАВА 2Основы теоугт газовой хроматографииЧТО, В СВОЮ очередь, приводит к высоким входным давлениям, а это связа¬
но с рядом уже упоминавшихся выше неудобств.Учитывая все эти обстоятельства, для аналитических целей чаще всего
используют зерна диаметром 0,1 - 0,3 мм (от 120 до 40 меш*). В этих преде¬
лах нужно выбирать фракцию как можно уже, например, 0,1 - 0,12 мм или0,2 - 0,25 мм.Очень важно освободиться от мелких частичек пыли, которые всегда
остаются на зернах, если их не подвергнуть отмачиванию в дистиллиро¬
ванной воде.Более плотная набивш достигается при использовании зерен сфериче¬
ской или эллипсоидной формы.Толщина жидкой пленки. Увеличение количества жидкой фазы на
твердом носителе (увеличение толщины пленки) приводит, с одной сто¬
роны, к увеличению селективности за счет увеличения доли сорбента в
единице объема колонки, а с другой стороны - к уменьшению эффектив¬
ности (рис. 2.19), увеличению размывания, так как величина //пропорцио¬
нальна квадрату толщины пленки жидкости. Поэтому для каждого типа
твердого носителя и жидкой неподвижной фазы имеется определенное со¬
отношение, при котором реализуется наибольшая разделительная способ¬
ность колонки. На рис. 2.20, в частности, показано, что для диетомитовых
носителей наибольшее значение R получается при нанесении 15% жидкойН,ем0.S0,40,30,2o»f30%О г ^ в й Ю и,ем/еРис. 2.19 Зависимость Н от и для бензола на колонке, наполненной
диатомитовым носителем с различным содержанием
жидкой фазы -сквалана (указано на рис)* Меш характеризует сетку, цифра обозначает число отверстий на длине в
1 дюйм (2,54 см), причем расстояние между отверстиями равно диаметру отвер¬
стия. Чтобы перевести меш в мм, нужно число меш умножить на 2 и 2,54 см по¬
делить на полученное значение.67
Яшин ЯМ, Яишн Е.Я., Яшин А.Я.Газовая хроматографияфазы (от массы твердого носителя). В том случае, когда надо получить хро¬
матографическую колонку с максимальной разделительной способностью,
следует определить это оптимальное соотношение и нанести необходимое
количество жидкой фазы.Рис. 2.20 Зависимость критерия разделения R (бензол и толуол) от
количества жидкой фазыУдерживаемый о&ьем пропорционален количеству жидкой фазы в
колонке:+ (2.96)где F, - обьем пустот в колонке;К - коэффициент распределения;- объем жидкой фазы.Поэтому для высокомолекулярных высоюкипяпщх веществ, шгорые
сильно удерживаются в колонке, используют сорбенты с н^ольшим количе¬
ством жидюй фазы (0,5 - 3%), так называемые малоемкие шлонки [26]. Такие
шлонки шнрош применяют в высоштемпературной газотой хроматографии,
в частности, для анализа остагшв пестицидов в различных средах, анализа
стероидов в биологических жидшстях и др. Чаще всего в этих случаях ис¬
пользуют 2 - 3% жидшй фазы (обычно SE - 30, OV -1,0V -17 и дф.). Кроме
уже упомянутого преимущества (значительного сокращения удерживаемых
объемов), такие шлонки более эффективны и устойчивы. За счет абсорбци¬
онного поля поверхности твердого носителя давление пара жидшй фазы над
таким сорбентом меньше и, следовательно, меньше фоновый ионный ток.
Меньший фон позюляет использовать, во-первых, более эффективно режим
программирования и, ю-вторых, более чувствительные шкалы приборов.Колонки малой емшсти имеют и существенный недостаток - малая
емшсть, что вынуждает для избежания перегрузки колонки использовать
малые пробы и высокочувствительные детекторы.68
aiABA 2Основы теории газовой хтмштогтФтПрирода газа-иосителя. В газовой хроматографии при небольших
давлениях инертные газы-носители практически не адсорбируются, осо¬
бенно в газо-жидкостной хроматографии. Поэтому природа газа-носите-
ля практически не влияет на селективность разделения, за исключением
некоторых случаев в газо-адсорбционной хроматографии при разделении
газов на активных тонкопористых адсорбентах или при использовании
газов-носителей с добавками сильносорбирующих веществ, в частности,
воды [27].Природа газа-носителя может 01шзывать влияние на размывание пи¬
шв. Из уравнения зависимости Я от линейной сшрости следует, что ш-
эффициешы молекулярной диффузии веществ (которые зависят от среды,
типа газа-носителя) входят в члены, описывающие молекулярную диффу¬
зию и внешний массообмен.Причем, если в первом случае (молекулярная диффузия) величина Я
прямо пропорщ!ональна коэффициенту молекулярной диффузии, то во
втором случае (внешний массообмен) - обратно пропорциональна.Если измерить зависимости Я от объемной сшрости для одного и того
же вещества, но с разными газами-носителями, например, гелием и двуо¬
кисью углерода, то получим графики, изображенные на рис. 2.21.Wj мл/минРис. 2.21 Зависимость Н от F для пропана при использовании раз¬
личных газов-носителей (колонка 100смх4 мм с силикаге-
лемс порами размером около 1000 нм и сферическими зер¬
нами 0,5 - 1,0 мм, температура 50°С)69
Яшт Я.И.. Яшин Е.Я.. Яшин А.Я. Газовая хроматографияИз рисунка следует, что при малых скоростях, когда преобладает раз¬
мывание за счет молекулярной диффузии (второй член уравнения 2.48),
более высокой эффективности можно достичь с газом-носителем, имею¬
щим большой молекулярный вес, так как коэффициенты молекулярной
диффузии в таком газе будут меньше. При больших скоростях картина
меняется, большей эффективности можно достичь с более легким газом,
так как в этом случае размывание происходит в основном за счет медлен¬
ных процессов сорбции и десорбции, и для ускорения процесса внешней
массопередачи нужны большие коэффициенты молекулярной диффузии
(здесь уместно напомнить, что внешний массообмен при ламинарных по¬
токах происходит только за счет молекулярной диффузии).В экспрессной хроматографии, когда применяют большие линейные
скорости, выгоднее применять легкие газы-носители (например, гелий).
В прецизионной хроматографии (при использовании малых линейных
скоростей газа-носителя) выгоднее применять более тяжелые газы-носи¬
тели (аргон, азот и двуокись углерода).В капиллярной хроматографии, когда чаще всего работают при боль¬
ших скоростях газа-носителя (т.е. в кинетической области зависимости
ВЭТТ от и), выгоднее использовать легкие газы-носители (гелий).Например, замена двуокиси углерода на водород* при малых ско¬
ростях приводит к увеличению коэффициента диффузии в 4,5 раза и,
следовательно, к увеличению ширины полосы более, чем в 2 раза. При
больших скоростях, где одним из контролирующих процессов является
внешний массообмен, замена двуокиси углерода на водород уменьшит
ширину полосы примерно в 1,5 раза. В капиллярной хроматографии в
области внешнедиффузионной массопередачи и динамической диффу¬
зии Я ~ 1/D, и, следовательно, при переходе от азота к водороду ширина
полосы уменьшается почти в 4 раза.Сопротивление колонки, перепад давления колонки, кроме других
факторов, определяются также и природой газа-носителя (вязкостью га¬
зов). Это свойство газа-носителя имеет немаловажное значение при ис¬
пользовании колонок большой длины, на которых возможны большие
перепады давления. В табл. 2.3 приведены значения вязкостей некоторых
газов, которые применяют в газовой хроматографии в качестве подвиж¬
ной фазы.• Из-за взрывоопасности использовать водород в качестве газа-носителя не реко-
мендуется70
1ШВЛ2Вязкость газовОсновы теории газовой хроматографииТаблица 2.3ГазТемпература,°СВязкость,(Н-см>10^ГазТемпература,°СВязкость,(Н-см')-10-‘Азот2018,4Воздух2018,1Аммиак2010,8Гелий18,9Аргон21,0Кислород2020,9Водород209,5Метан2012,0Водяной пар159,8В последнее время в газовой хроматографии в качестве подвижной
фазы исполь^ют пары сорбирующихся веществ, в частности, водяного
пара [28-31]. Применение водяного пара в качестве подвижной фазы при¬
водит к уменьщению времени удерживания и исключает асимметрию пи¬
ков за счет блокирования акгавных центров и повышения однородности
поверхности полярных веществ. Это проявляется особенно сильно для по¬
лярных веществ, таких как жирные спирты, фенолы, амины. Кроме того,
применение всадяного пара как подвижной фазы дает возможность опреде¬
лять примеси в водных смесях (природные и сточные воды).Скорость газа-носителя. Скорость газа-носителя - один из важных
параметров хроматографического опыта. Он оказывает большое влияние
на эффективность, следовательно, и на степень разделения. Выше неодно¬
кратно уже упоминалось о влиянии скорости газа-носителя на размывание.
В частности, в теории скоростей рассматриваются вклады в Я разных ви¬
дов размываний в зависимости от линейной скорости газа-носителя (см.
уравнение (2.28). По этому уравнению вклад вихревой диффузии не зави¬
сит от скорости, вклад молекулярной диффузии обратно пропорционален
скорости, другие вклады пропорциональны в разной степени. График зави¬
симости ВЭТТ от и, построенный по экспериментальным данным, имеет
вид кривой с четким минимумом (рис. 2.22).Рис. 2.22 Зависимость Н от и.71
Яшин ЯМ. Яшин Е.Я.. Яшин А.Я. Газовая хроматографияМинимум соответствует наибольшей эффективности колонки. График
(рис. 2.22) показывает, что для каждой колонки имеются оптимальные преде¬
лы сюростей, при которых достигается максимальная эффективность. Для
вновь приготовленной хроматографической шлонки нужно получить такую
графическую зависимость и определить область оптимальных скоростей.При использовании скоростей газа-носителя меньше оптимальных
размывание будет возрастать за счет сильного вклада молекулярной диф¬
фузии в величину ВЭТТ. При использовании скоростей газа-носителя боль¬
ше оптимальных размывание будет также возрастать, но за счет задержки
массообмена (кинетическое размывание); вклад молекулярной диффузии в
этом случае будет мал. Средние оптимальные линейные скорости газа-но¬
сителя в набивных колонках могут колебаться от 2 до 5 см/с в зависимости
от типа сорбента. В капиллярных колонках оптимальные скорости состав¬
ляют 10 -15 см/с.Время удерживания, следовательно, и продолжительность анализа
прямо пропорциональны скорости газа-носителя в колонке. Поэтому для
сокращения продолжительности анализа увеличивают скорость газа-но-
сителя, так называемый режим программирования расхода газа-носителя.
Такой режим разделения желательно проводить без потери эффективно¬
сти, а это возможно только на таких сорбентах, для которых наклон правой
ветви зависимости Я от м пологий, а область оптимальных скоростей газа-
носителя широкая.Давление газа-носителя. Обычно на входе в колонку используют из¬
быточные давления около 500 - 1000 мм рг. ст. (в очень редких случаях
выше). Изменение давления в этих пределах пракгически не влияет ни на
селективность, ни на эффективность разделения. Как упоминалось выше,
более важное значение имеет перепад давления в колонке.В ряде работ применялись пониженные давления, т.е. вакуумная хро¬
матография [32] для разделения малолетучих, высококипящих соедине¬
ний. Как показано А.А.Жуховицким [8], если в вакуумной хроматографии
значения объемной скорости, а также величин и ц привести к одной
температуре и давлению на выходе, то не будет зависеть от давления.Возможен вариант газовой хроматографии при повышенных давлениях
[33]. Основная цель хроматографии при повышенных давлениях - уменьше¬
ние коэффициентов Генри. При повышении молекулярного веса разделяе¬
мых веществ коэффициент Генри (или коэффициент распределения) возрас¬
тает настолько сильно, что элюировать вещество за приемлемое время не
удается. При повышении давления возрастает сорбция подвижной фазы на
сорбенте, и значения юэффициентов Генри уменьшаются, особенно в том
случае, когда в качестве подвижной фазы используют летучие жидкости, на¬
ходящиеся в сверхкритическом состоянии. Тогда подвижная фаза - не газ и
не жидкость, по плотности имеет среднее значение между ними. По суще¬72
тЛВА2Основы теории газовой хгхшатографииству этот вариант, иногда называемый флюидной хроматографией, занимает
промежуточное положение между газовой и жидюстной хроматографией.В литературе описаны примеры разделения смесей веществ с моле-
1дглярными весами выще 1500 (обычно методом газовой хроматографии
в классичесюм варианте можно анализировать вещества с молекулярным
весом 400 - 500).В связи с аппаратурными трудностями, особенно из-за сложности соз¬
дания надежных детекторов, работающих при высоких давлениях, этот
метод не получил широкого применения.Размер пробы. Размер введенной пробы анализируемой смеси дол¬
жен быть таким, чтобы не вызыветь перегрузку шлонки. При введении
пробы больше максимально допустимой начинает увеличиваться началь¬
ная ширина полосы и начинает изменяться время удерживания. Макси¬
мально допустимое шличество дозируемого вещества можно установить
по соответствующим экспериментальным графикам зависимости времени
удерживания от размера пробы (рис. 2.23).чмтРис. 2.23 Зависимость времени удерживания от размера введен¬
ной пробыИз теории тарелок следует, что пробы не должны превышать емшсть
слоя по длине одной теоретичесшй тарелки. Практически можно превы¬
шать эту величину в зависимости от шэффициента шлонки [34]:си~ 0,5л/М(У+KV.) (2.97)где N- число теоретических тарелок;F, - объем газовой фазы в объеме одной тарелки;- объем жидкой фазы в объеме одной тарелки;К - шэффициент распределения.73
Яшин ям. Яшин Е.Я.. Яшин АЛ Газовая хроматографияОбъем нанесенной пробы не должен превышать начальной пшрины
полосы:F < ц V <4KSVDL/a (2.98)После преобразованийV <Vjn (2.99)Maix R ' ^В работе [34] установлено более определенное требование:F^=0,02FyVN (2.100)Особенно важно не пере1ружать капиллярную шлонку, так как эффек¬
тивность ее сильно падает с перегрузшй. На пршстике иногда допускается
перегрузка в насадочных шлонках, в частности, при анализе микроприме¬
сей иногда допускается перегигзка по основному веществу.Способ дозирования. Про^ можно ввести быстро в виде узшй шн-
центрированной полосы или же медленно в виде размытой полосы. Первый
способ введения - «метод поршня» - идеальный, второй способ - «способ
экспоненциального разбавления» - может приводить к дополнительному
размыванию полосы. В общем случае ширина дозируемой полосы должна
быть значительно меньше ширины полосы вещества, получаемого на вы¬
ходе из колонки.В случае дозирования жидшй смеси необходимо ее быстро испарить,
для чего используют дозаторы-испарители. Однаш при больших удержи¬
ваемых объемах иногда допускается введение жидшй пробы непосред¬
ственно в начало шлонки, так как за время испарения шмпоненты пробы
по слою сорбента продвинутся на незначительную длину.Способ детектирования. Для шличественных анализов важны сле¬
дующие характеристики детектора: предел детектирования (чувствитель¬
ность), шнейность, инерционность и стабильность детектора. Назначение
детектора - регистрация выходных кривых в виде сигналов (пишв) до¬
статочной амплитуды, необходимых для шличественного измерения. Для
точных шличественных измерений необходимо, чтобы:- детектор не искажал истинную форму полосы, образующейся на
слое сорбента, другими словами, детектор должен быть малоинерционен,
постоянная времени должна быть небольшой;- показания детектора были строго пропорциональны шнцентрации
или шличеству дозируемых веществ, т.е. детектор должен обладать доста¬
точно широкой областью линейности;- запись сигналов была устойчивой и не было флуктуаций нулевой ли¬
нии или же монотонного смещения нулевой линии в течение длительного
времени (дрейфа нулевой линии).74
ТШАЛА 2 ЛитератураЛитература1. Ettre L. Nomenclature for Chromatography. Pure and Appl. Chem. 1993,
V. № 4, p. 819-872.2. Хроматография. Основные понятия. Терминология. Под ред.
В.А.Даванкова, Москва, РАН, 1997,48 с.3. Яшин Я.И., Яшин А.Я. В «Новом справочнике химика и технолога»,
т. Аналитическая химия, С-Петербург, НПО «Мир и семья», 2002, с.
254-342.4. Davankov V.A. Pure Appl. Chem. 2001, v. 73, Лв 6, p. 969.5. Davankov V.A. Chromatographia Supplement. 2003, v. 57, S. 195.6. Wehrli A., Kovats E. Helv. chem. acta 1959, v. 42, p. 2709.7. James A.T., Martin A.J.P. Biochem. J. 1952, v. 50, p. 679.8. Жуховицкий A.A., ТУркельт^б Н.М. Газовая хроматография. М., Го-
стоптехиздат, 1962,442 с.9. Giddings J.C. Dynamics of Chromatography. P.I. Principles and Theory,
N.Y., Marcel Dekker, 1965, 380 p.10. Einstein A. Ann. Phys., 1905 v. 17, p. 549.11. Голей M. В кн.: Газовая хроматография. Сб. докладов на II
Международном симпозиуме в Амстердаме. Пер. с анга. Под ред.
А.А.Жуховицкого и Н.М.Туркельтауба. М., Издатинлит, 1961, с. 39-60.12. Тимофеев Д.Л. Кинетика адсорбции. Москва, Изд. АН СССР, 1962,
252 с.13. Van Deemter J.J., Zniderberg RJ., KJinkenberg A. Chem. Eng. Sci.,
1956, v. 15, p. 271-285.14. Kieselbach R. Analyt. Chem., 1963, v. 35, p. 1342-1345.15. Kennedy G.J., Knox J.H. J. Chromatog. Sci., 1972, v. 10, p. 549-553.16. Knox J.H., Parcher J.E Analyt. Chem., 1969, v. 41, p. 1969-1974.17. Purnell J.H. J. Chem. Soc., 1960, p. 1268.18. Современное состояние жидкостной хроматографии. Пер. с англ.
Д.Д.Новикова. Под ред. Дж. Киркленда, Москва, Мир, 1974, 325 с.19. Король А.Н. Неподвижная фаза в газо-жидкостной хроматографии.
Киев, «Паукова думка», 1969, 250 с.20. Яшин Я.И. Физико-химические основы хроматографического раз¬
деления. Москва, Химия, 1976, 216 с.21. Frenkel J.I., Zeit. f. Phisik, 1924, Bd. 26, S. 117-122.22. Griffiths J., James D.H., Phillips C.S.G. Analyst, 1952, v. 77, p. 897-
901.23. Харрис В., Хэбгуд Г. Газовая хроматография с программированием
темературы. Пер. с англ. Под ред. Б.А.Руденко. М., «Мир», 1968, 34075
Яишн ЯМ. Яшин ЕЖ. Яшин А.Я. Газовая хроматографияС.24. Жуховицкий А.А. и др. ДАН СССР, 1951, т. 77, с. 435-438.25. Жуховицкий А.А. и др. ДАН СССР, 1953, т. 92, с. 987-990.26. Ногаре С.Д., Джувет Р.С. Газо-жидкостиая хроматография. Теория
и практика. Пер. с анга. Под ред. А.Н. Александрова и М.И. Дементье¬
вой. Л., «Недра», 1966,420 с.27. Nonaka А. Analyt. Chem., 1972, v. 44, p. 271-275.28. Berezkin V.G., Zs^ainov V.F., I\mov P.B. J. Chromat 2003, v. 985, p. 57.29. Sandra R LC. GC, 1987, V.5, p. 236.30. Руденш Б.А., Байдаровцева M.A., Кучеров В.Ф. Изв. АН СССР,
Сер. хим., 1973, с. 1773-1776.31. Gvozdovich T.N., Kiselev A.V., Yashin Ya.L Chromatographia 1973,
V. 6, p. 179.32. Вяхирев Д.А. Комиссаров П.Ф. ДАН СССР, 1959, т. 129, с. 138.33. Вигдергауз М.С., Измайлов Р.И. Применение газовой хроматогра¬
фии для определения физико-химических свойств веществ. Москва,
Н^ка, 1970, 160 с.34. Кейлеманс А. Хрометография газов. Пер. с анга. Под ред. М.И. Янов-
сшго, М., Издагинлит, 1959,320 с.76
ГЛАВА 3 Газо-жидкостная хроматографияГлава 3. ГАЗО^ЖИДКОСТНАЯ ХРОМАТОГРАФИЯ3.1. Преимущества и недостатки газо-жндкостной хроматографии.В газо-жидкостной хроматографии (ГЖХ) разделение происходит за
счет различной растворимости компонентов смеси в пленке жидкой фазы,
нанесенной на поверхность макропористого твердого носителя.Метод ГЖХ впервые предложен Джемсом и Мартином в 1952 г [1]. В
настоящее время для многих аналитических задач ГЖХ - наиболее часто
используемый метод [2]. Это связано с тем, что применение жидких фаз
дает ряд преимуществ:1. возможно разделение разнообразных смесей соединений как ле¬
тучих, так и малолетучих; на жидких фазах изотерма абсорбции линейна
при обычных рабочих условиях в широком диапазоне концентраций, а это
означает, что пики в этом случае получаются симметричными;2. большой выбор достаточно селективных, разнообразных по хими¬
ческой природе жидких фаз;3. количество жидкой фазы на носителе можно легко изменять, тем
самым регулируя удерживаемые обьемы и емкость колонки;4. с одной и той же жидкой фазой можно приготовить как высокоэф¬
фективные аналитические колонки (насадочные или капиллярные), так и
препаративные шлонки;5. доступны жидкие фазы достаточно высокой степени чистоты; свой¬
ства их стабильны, т.к. они выпускаются по определенным установленным
ТУ, параметры удерживания на них воспроизводимы.Однако применение жидких фаз имеет ряд серьезных трудностей и не-
достатшв. Основные недостатки ГЖХ связаны с летучестью и нестабиль¬
ностью жидких фаз, что затрудняет применение этого метода для анализа
микропримесей и высошкипящих соединений, особенно в режиме про¬
граммирования температуры.3.2 Основные требования к жидким неподвижным фазам.Первое требование — жидкая фаза должна быть достаточно селек¬
тивной и обеспечить разделение шмпонентов анализируемой смеси за
приемлемое время.Второе требование — малая летучесть жидкой фазы при рабочих
температурах, это связано с временем жизни колонки. Жидкая фаза мо¬
жет быть очень селективной, но если она обладает большой летучестью,
то ее нельзя использовать в газовой хроматографии. Большая летучесть
жидкой фазы, с одной стороны, затрудняет работу детектирующих систем,
а с другой стороны, непрерывное уменьшение количества жидкой фазы в77
ШшыЖМ-. Яшин ЕЯ., Яшин А.Я. Газовая хроматографияколонке приводит к уменьшению удерживаемых объемов, что затрудняет
проведение количественных и качественных измерений. До настоящего
времени нет общих требований к летучести жидких фаз. Многие считают
допустимым значением давления пара жидкой фазы при рабочей темпера¬
туре порядка 10’^ мм рт. ст., хотя при средних расходах это составит около
10 ppm примесей в газе-носителе [3]. Работать с высокочувствительными
детекторами при такой летучести очень трудно, так как наиболее высоко¬
чувствительные детекторы способны реагировать на давление пара жид¬
кой фазы около 10 * мм рт. ст.В общем виде требование к летучести жидшй фазы зависит от чув¬
ствительности детектора и температуры разделения.Третье требование — жидкая фаза должна быть химически инерт¬
ной и термически стабильной. В некоторых случаях жидкая фаза может
вызывать конформационные изменения, полимеризацию, конденсацию и
другие химические превращения компонентов пробы. В этих процессах
иногда активную роль играет твердый носитель, обладающий каталити¬
ческой активностью. Термическая стабильность жидкой фазы определяет
верхний температурный предел применения жидкой фазы. Как правило,
летучесть и термическая стабильность жидкой фазы уменьшаются с по¬
вышением молекулярного веса. Поэтому по летучести некоторые фазы,
казалось бы, можно бьшо использовать при более высоких рабочих тем¬
пературах, но из-за термической нестабильности использовать их нельзя,
так как фоновый ионный ток возрастает за счет продуктов разложения.
Верхний температурный предел использования жидкой фазы зависит не
только от природы фазы, но и от природы твердого носителя и количества
нанесенной жидкой фазы. Резкое падение парциального давления жидкой
фазы в колонке наблюдается, шгда на носителе всего 1—2% жидкости.
Максимальная температура (рабочая) одной и той же жидкой фазы в ка¬
пиллярных колонках обычно на 25—50 “С ниже, чем в набивных.Четвертое требование — жидкая фаза должна иметь низкую вяз¬
кость при рабочей температуре, что увеличивает скорость внутреннего
массообмена.Пятое требование — хорошая смачиваемость поверхности носителя
или стенок капилляра жидкой фазой, так как от однородности пленки в
значительной степени зависит эффективность колонки. И, наконец,последнее требование — желательно, чтобы жидкая фаза легко рас¬
творялась в наиболее распространенных растворителях.78
ТШВАЗГазо-жидкостная хроматография3.3 Классификация жидких фазВ газо-жидкостаой хроматографии в настоящее время используют сот¬
ни различных жидких фаз. Всего описано более тысячи типов жидких фаз.
Чтобы лучше ориентироваться в их свойствах при разработке методики
разделения конкретной смеси, были предприняты многочисленные попыт¬
ки классификации жидких фаз. За основу классификации принимали или
абсолютные значения удерживаемых объемов или же относительные удер¬
живания, которые позволяют оценить селективность. В первых попытках
классификации за основу принимали не впош1е определенное понятие
«полярности» жидких фаз, которое обычно не связывалось ни с диполъ-
ным моментом, ни с поляризуемостью молекул жидких фаз.В газовой хроматофафии в качестве жидких фаз используют практи¬
чески все основные классы соединений, в частности алканы, амиды, ами¬
ны, диэфиры, простые эфиры, галогенуглеводороды, нитрилы и цианиды,
полиспирты, кислоты, полиэфиры, производные сахаров, полиметилси-
локсаны и др.Все фазы можно разделить на семь типов (табл.3.1).Таблица 3.1Классификация жидких фазп/пТип жидких фазОсновные виды взаимодействий,
влияющих на удерживаниеНазвание жидких фаз1.НеполярныеМежмолекулярные:
дисперсионные, индукционныеСкваланЛпиезоны L, М, N и ф.Аполан-87ПолиметилсилоканПолиметилоктилсшюксанПолифторметилсилоксан2,СлабополярныеМежмолекулярные:
дисперсионные,
ориентационные, индукционныеЭфирыПолиэфирыПолизтиленглиноли 20 М и 40
Ми др.3.ПолярныеМежмолекулярные:
ориентационные,
дисперсионные, индукционныеПолицианопропилсилоксаныПолицианопропилфенилсилок-саныN,N,N',N - тетракис
(2-гидроксипропил)
этнлендиаминР|Р' - оксидипропионитрил и др.79
Яшин Я.И.. Яшин Е.Я; Яишн А.Я.Гтовая хроматографияп/пТип жидких фазОсновные виды взаимодействий,
влияющих на удерживаниеНазвание жидких фазПолярные(полиспирты)Межмолекуляриые:
ориентационные,
дисперсионные, индукционные
Водородная связьГлицерин. Дигаицерин.
Пентаэритрит. Сорбит.
Полиэтиленгшшши 300,400,600.
Полисилоксаны с
гидроксильными группами и ф.СпецифическиеМежмолекулярные.Комплексообразование.Донорно-акцепторноевзаимодействиеПолиэтиленгликоль с AgNO,
Жидкие фазы с комплексами
переходных металлов и дрЖидкие фазы, в
которых сильно
проявляется
«геометриче¬
ский фактор»Межмолекулярные
взаимодействия.
Сорбция сильно зависит от
пространственной структуры
молекулЖидкие кристаллы.Жидкие фазы на основе краун-
эфиров.Жидкие фазы на основе
циклодекстринов и щ>.Хнральиые
жидкие фазыЖидкие фазы, имеющие
хиральные, асимметричные
центры для разделения
оптических изомеровЖидкие кристаллы
нематического, смектического,
холестеричесюго, реентрантного
типов и ;ц}.1. Неполярные фазы, в основном парафины и полиметилсилоксаны,
например сквалан, гексадекан, гептадекан, апиезоны, жидкий парафин, по-
лиокгилметилсилоксан, аполан-87, полиметилсилоксаны типа SE-30, 0V-1.
Эти фазы используют в основном для разделения неполярных и слабопо¬
лярных веществ (таблица 3.2).Таблица 3.2Неполярные жидкие фазы для ГЖХп/пНазваниеСтруктурная формула1.Сквалансн, он, он, сн, сн, сн,сн,"^ '' сн,2.Апиезои МН-СН(СН,)-(СНа-СН(СН,)-(СН,).-СН3.Аполан-87(C„H„)j_CH-(CHj), -C(CjH,)j -(CHj), -сн- (C„H„)j4.ПолиметилсилоксанСН,1СНз-Si-O-1СНзСН,1-Si-0-1СНзСНз1-Si-СНз1п СНз80
тллвлзГазо-жидкостная хтшатографияп/пНазваниеСтруктурная формулаПолнокгил (50%)-метил (50%)
силоксан-0-СНз
Si
пСН2СН2СН2СН2СН2СН2СН2СН32. Жидкие фазы со средней полярностью, молекулы которых содержат
слабо полярные функциональные группы (эфирные группы, двойные свя¬
зи ароматических соединений и др.). Эти фазы можно использовать для
разделения как неполярных, так и полярных веществ. К этому типу фаз
относится большое число полиэфирных жидких фаз (таблица 3.3).Таблица 3.3Эфирные и полиэфирные жидкие фазы для ГЖХп/пНазваниеСтруктура1.Дидецилфталат/^СОО(СН2)9СНз^^СОО(СН2)9СНз2.Диоктилсебацинат ^CjH, СА
1 !
СН,^(СН2)г-СН-СНг- OOG-(CH2V-COO-CH,-CH-(CHi)r-CHi3.Диокгиладипат^5 СА
CH,-(CHjV-CH-CHr-OOC-(CHJ^COO^CH^H^(CHJ,-CH,4.Полиэтил енгликоль-адипет^ СН2 - СН2 - 0 - с - (CHj)4-C -0Ь 0п5.Полидиэтиленгликоль-адипат- CHj- СИг- 0 - CH,-CHj- 0 -С - №), -С-0-1 II
0 06.Полипропиленгаиколь-себацинат- СН2-СН2-СН2- 0-C-(CH2)g -с-0-11 II0 оп7.Полибутандиол-сукцинат- CHj-CHj-CHj-CHi-O-C-CHj-CHj-C-O-
1 1
0 0п8.Полинеопентилгяиколь-адипатСНз- CHj-C-CH2-0-C-(CH2)4-C-0-1 II II
^ СНз 0 0 ^п81
9.Полициклогексан-
диметанол сукцинат-СНCHj-o-C-CHj-CHi-C-O-
Ь о10.Поли (фенилдиэтанол-
аминсукцинат)- CHj-CHj-N - CHj -CHj-O-C-CHj- CHj-C-0-ел ^ о .3. Полярные жидкие фазы, молекулы которых содержет сильно поляр¬
ные функциональные группы, например, Р-Р’-оксидипропионитрил, гекса-
метилфосфорамид, диметилсульфолан, полипропилцианметилсилоксаны,
полипропилциансилоксаны и др. Полярные фазы используют для селек¬
тивного разделения смесей слабополярных и сильнополярных компонен¬
тов (таблица 3.4).Таблица 3.4Полярные жидкие фазы для ГЖХп/пНазваниеСтруктураМДРТN|N,N‘,N' - Тетракис-(2-гидросиэтил)этилендиаминHOCHjCHj. /СН2СН2ОННОСН2СН/ XlHjCHjOH120°СN,N,N',N‘ - Тетракис-(2-гидроксипропил)этилендиаминCHjCHOHCHj..CHjCHOHCH,15(к:3.Полиамид Ро1у-Ао о-С-(СН^)„-С-Ы^Л-(СНIN-275»СCH2-0-CH2-CH2-CNCH^-O-CHa-CHj^CNCH;-0-CH2-CH;-CNJn4.1 ,2,3-Трис(2
цианоэтокси)пропан170”С4. Жидкие фазы, молекулы которых способны образовывать сильные
водородные связи с молекулами разделяемых веществ (глицерин, диглице¬
рин, полигликоли и др.).5. Специфические жидкие фазы, разделение на которых происходит за
счет слабого комплексообразования (гликоли, насыщенные нитратом сере¬
бра и др.).6. Жидкие фазы, на которых разделение происходит за счет геометри¬
ческого фактора, это фазы на основе краун-эфиров, жидких кристаллов,
циклодекстринов и др. (таблица 3.5).82
тлвАзЖидкокристаллические фазы для ГЖХГазо-жидкостная хршатогршЬияТаблица 3.5п/пНазвание / структураТемпературафазовыхпереходов1.П|П‘-Бис (п-метоксибеюилиденамино) -3,3' - дихлордифенил
С1 С1OCHjТн154“С
Т„п 334“С2.N,N' - Бис (R-бензилиден) - а,а'-ди-п-толуидин3.n-Фенилен-бис (4-н-гептилоксибензоат)M'CtHis-O-*^ У~С0-^ ^-СО-*^ '^О—H'CyHis
0 0125-204“С4.п-Фенилен-бис (4-и-гептилоксибензоат)''“Су0 0150-211“С5.Производные холестеринаСНз
СНз 1СНзП СН-(СН2)з-СН(СНз)2j^/ЧА^161-221“С6.п-(п-Этоксифекилазо)фенил кротонат0П0-197»С7. Хиральные жидкие фазы для разделения оптических изомеров(таблица 3.6). ^ ^Таблица 3.6Оптически активные неподвижные жидкие фазы для ГЖХп/пНазваниеСтруктурная формула1.Циклогексиловый эфир
N-ТФА-дипептидаН НСбН„0-С-С-NH-C-C-NH-C-CFj" II 1 II 1 II
OR, OR, 083
Яшин Я.И., Яшин Е.Я.. Яшин А.Я.Газовая хроматографияп/пНазваниеСтруктурная формула2.(+) ДодецилтартратН ОНс,2Нн-^^(р~(р”С~он:,2Н25о он н о3.Бисциклогексиловый
эфирН-ТФА-Ь-аспарагановойкислотыН НСвН„0-С- CH2-q*”NH-C-C*-NH-~C-CF3о^^-NH-C-с=о о0-QH,,СН,О4.М-Дои)заноил-Ь-валин-2--(2-метил)-н-гептадецил-амидН СНзCHr(CH2)2„-(j:-NH-(p*-C-NH-C-(CHj)H СНзоСН о
СН, СНзIСН,Жидкие фазы можно также классифицировать по природе функцио¬
нальных групп, присутствующих в составе молекулы жидких фаз и, сле¬
довательно, х^)актеру межмолекулярных взаимодействий по аналогии с
классификацией адсорбентов. Жидкие фазы можно также классифициро¬
вать по максимально допустимой рабочей температуре колонки (МДРТ).К первой группе можно отнести жидкие фазы, для которых МДРТ не
превышает 100 °С, например гексаметилфосфорамид, диметилсульфо-
лан, р-р’-оксидипропионитрил, октадекан и др. Эти фазы для некоторых
смесей обладают высокой селективностью, но применение их возможно
только для низкокипящих веществ Практически нельзя использовать эти
фазы для работы в режиме программирования температуры и для анализа
микропримесей с высокочувствительными ионизационными детектирую¬
щими системами.Ко второй группе можно отнести жидкие фазы со средними значе¬
ниями МДРТ (до 150—200 °С). Это наиболее распространенная группа
жидких фаз. В этой группе прежде всего следует выделить полигаиколи,
эфиры и полиэфиры.В ГЖХ широко используют полиэтиленгликоли с молекулярными ве¬
сами от 200 до 40 ООО [4,5]. Общая формула HO(CHjCHjO)nH, где п может
меняться от 4 до 900 Как было показано ранее, полярность полиэтиленгли-
колей, меняется с увеличением молекулярного веса; МДРТ колеблется от
120 до 220°С (для, самых высокомолекулярных), выше 220°С происходит
термическое разложение молекул полиэтиленгаиколя. В пределах рабочих
температур это одна из стабильных и устойчивых во времени жидких фаз.
Полиэтиленгликоли широко используют для разделения спиртов, эфиров84
ГЛАВА 3Газо-жидкостная хроматографияИ других полярных соединений. За рубежом полиэтиленгликоли извест¬
ны как карбоваксы. Иногда полиэтиленгликоли модифицируют (связы¬
вают концевые гидроксильные группы) обработкой соединениями типа
терефталевой кислоты. Полиэфиры такого типа (UCON) подразделяют на
водорастворимые и водонерастворимые. Водорастворимые обозначают
приставкой Н или НВ. Водонерастворимые полиэфиры ряда UCON обо¬
значают приставкой LB. Обычные продукты разложения полиэтиленглико-
лей — ацетальдегид и уксусная кислота. Полиэтиленгаиколи-2(Ю или -300
содержат много легких гликолей.Сложные эфиры и полиэфиры — наиболее распространенный класс
жидких фаз[ 5,6]. Широко используют эфиры и полиэфиры адипиновой,
изофталевой, янтарной, фталевой, себациновой, терефталевой, тетрахлор-
фталевой и других кислот.Из полиэфиров наиболее полярная фаза — диэтиленгликоль-сук-
цинат. МДРТ для всех полиэфиров обычно не превышает 225°С, выше,
как правило, идет термическое разложение. Для повышения термической
стабильности полиэфиров к ним добавляют стабилизирующие добавки (в
частности, фенилдиэтаноламин, триэтаноламин, фенилнеопентилфосфит)[7]. Удобнее их добавлять непосредственно к раствору жидкой фазы перед
нанесением. На каждый грамм жидкой фазы используют от 0,01 до 0,05 г
стабилизатора. На рис. 3.1 показаны соответствующие повышения терми¬
ческой стабильности диэтиленгаикольсукцината и этиленгликольадипата
на 50—70 °С после соответствующей стабилизации.Полиэфиры обычно стабильны к окислению и разложению, они со¬
держат мало легких примесей. Однако при высоких температурах в при¬
сутствии паров воды возможен гидролиз, особенно в присутствии сильно¬
основных или кислых веществ.т т ттт,'с то т zao т,*с
а ^Рис. 3.1 Зависимость ионного тока детектора (летучести жидкой
фазы) от температуры колонки [без стабилизатора (1) и со
стабилизатором (2): а— диэтшенгликольсукцинат; б-эти-
ленгпикодь адипат.85
Яшт Я.И.. Яшин Е.Я.. Яшин А.Я.Газовая хроматографияК третьей группе можно отнести жидкие фазы с МДРТ более 200°С.
Таких жидких фаз значительно меньше, чем фаз второй группы. Особенно
мало высокотемпературных полярных жидких фаз. В этой группе прежде
всего, следует выделить апиезоны и силиконы.Апиезоны получают путем высоштемпературной обработки специ¬
альных сортов смазочных масел [7]. Остаток после ташй термической об¬
работки подвергают ректификации и используют в качестве жидких не¬
подвижных фаз. В зависимости от температуры обработки апиезоны клас¬
сифицируют в алфавитном порядке. Наиболее широше применение нахо¬
дят апиезоны N и L. Причем апиезон L о^спечивает самое высоше число
теоретических тарелок на единиву длины колонки и имеет более высокую
МДРТ, чем апиезон N. МДРТ апиезона L при работе с ионизационно-пла¬
менным детектором — 250°С, при работе с катарометром — до 300°С.В последние годы в качестве высокотемпературных жидких фаз ши¬
роше применение находят силоксановые жидшсти [8-11]. МДРТ для них
колеблется от 200 до 350°С (даже в некоторых случаях до 380°С). Эти фазы
широш используют для разделения и анализа пестицидов, стероидов, ле¬
карств и других высошмолекулярных органических веществ. Полярность
этих фю полностью зависит от природы функциональных фупп — заме¬
стителей в бошвой цепи (см, табл.3.7), эфирный кислород между двумя
атомами кремния обычно пространственно затруднен и не проявляет себя.Таблица 3.7Полисилоксановые жидкие фазыТипНЖФСтруктурная формулаПолидиметилсилоксанСН,O^SiСН,3J 100%Полидифенил (5%) диметил
(95%) силоксанQO^Si5%СН,O^Si^СНз95%
тлвлзГаяо-жидтстная хроматографияТипНЖФСтруктурная формулаПолидифенил (20%) диметил
(80%) силоксан9О—SiЙ20%СНзО—Si—^IСНз80%Полидифенил (35%) диметил
(65%) силоксанQ-О—Si-й35%СНз
О—Si—
СНз65%Полидифенил (50%) диметил
(50%) силоксанO-Si-d50%СНзО—Si^IСНз50%Пояибисцианопропил (80%)
цианопрогошфенилсилоксан-o-st-Iсн^ж,сн^80%CHrfUCIbCNoki-20%Полибисцианопропил (90%)
цианопропилфенил (10%)
силоксан0-Si CHjCHjCHrf»!o-ii CHjCHjCHiCN10%Полибисцнанопропилфенил
(14%) диметил (86%)
силоксан^jCHjCH^N■O-Si 14%O-SiOHiCHjCHjCN86%87
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я.Газовая хроматографияТипНЖФСтруктурная формулаПолибисцианопропил (100%)
силоксанCHjCHjCHaCN^O^Si CHjCHjCHjCN100%10Полиоетил (50%) метил
(50%) силоксанСН,о—SFСН2СН2СН2СН2СН2СН2СН2СН3ИПолиалкиленгликольНОСН2-СН2-ОСНз
СН, СН-0-нтп12Полиэтиленгликольн—осасн,—он13Полиэтиленгликоль,
модифицированный
нитротерефталевой кислотойО ОНОзС-^С -^ОСНгСНгД^О-С^^СОгН
N0, ^ NO,3.4 Межмолекулярные взаимодействия, определяющие удержива¬
ние соединений в газожидкостной хроматографии.Удерживание в ГЖХ определяется природой взаимодействия сорбат-
сорбент. Основные взаимодействия, влияющие на растворимость сорбата в
жидкой фазе, а значит и на удерживание, это межмолекулярные взаимодей¬
ствия (дисперсионные, индукционные и ориентационные), донорно-акцеп-
торные взаимодействия, водородная связь и слабое шмплексообразование.Межмолекулярные силы притяжения - силы Ван-дер-Ваальса - имеют
электростатическую природу.Ориентационное взаимодействие [12] имеет место между молекула¬
ми, обладающими постоянными дипольными моментами, т.е. такими мо¬
лекулами, у которых центры положительных и отрицательных зарядов не
совпадают. Величина дипольного момента определяется произведением
значения заряда на расстояние между зарядами. Диполь ориентируется
так, чтобы положительный заряд был направлен к отрицательному заряду.Энергия ориентационного взаимодействия определяется следующим
уравнением:E„p~-%(nVliVr^)-(l/K-T) (3.1)где: и - дипольные моменты молекул; г - расстояние между цен¬
трами диполей; к - константа Больцмана; Т - температура, °К.88
ГЛАВА 3 Газо-жидкостная хроматографияИз этого соотношения видно, что ориентационное взаимодействие за¬
висит от температуры, с повышением которой оно уменьшается. Это связа¬
но с тем, что с повышением температуры возрастает кинетическая энергия
молекул, и ориентация молекул затрудняется.В газовой хроматографии при разделении полярных молекул на по¬
лярных сорбентах удерживание соединений в значительной степени будет
определяться вкладом ориентационного взаимодействия, селективность
разделения будет сильно уменьшаться с ростом температуры.Индукционные взаимодействия [11] имеют место между полярными и
неполярными молекулами. За счет зарядов молекулы, имеющей постоян¬
ный дипольный момент, в неполярной молекуле наводится индуцирован¬
ный диполь при ее приближении к полярной молекуле. При этом происхо¬
дит взаимодействие постоянного диполя с наведенным индуцированным,
такое взаимодействие всегда приводит к притяжению.Энергия индукционного взаимодействия равна:= + (3-2)где: а, и Oj- поляризуемости взаимодействующих молекул; ц, и - ди-
польные моменты (постоянный и индуцированный).Это взаимодействие не зависит от температуры, так как при любой
ориентации может возникать индуцированный заряд.В газовой хроматографии вклад индукционного взаимодействия срав¬
нительно невелик. Максимальный вклад в удерживаемые объемы индук¬
ционного взаимодействия составляет 5-10%.Дисперсионное взаимодействие [12] - взаимодействие между непо¬
лярными молекулами. Рассмотренные виды межмолекулярного взаимо¬
действия не могут объяснить взаимодействия неполярных молекул, не
имеющих постоянных диполей. На основании квантомеханических пред¬
ставлений дисперсионное взаимодействие объясняется взаимодействием
кратковременных диполей, возникающих в неполярных молекулах в очень
малые промежутки времени, например 1- 10’* с (среднее время одного акта
сорбции). Такие мгновенные дипольные моменты малы, и они постоянно
меняются по величине и направлению. Так как вероятность ориентации в
любом направлении одинакова, то средний дипольный момент равен нулю.
Для определения величины так быстро изменяющихся дипольных момен¬
тов нет технических средств.Энергия дисперсионного взаимодействия определяется следующим
уравнением:+ У (3-3)где: I, и Ij - потенциалы ионизации взаимодействующих молекул.89
Яшин ЯМ. Яшт Е.Я.. ЯштЛ.Я. Газовая хроматографияДисперсионное взаимодействие - наиболее распространенный вид
взаимодействия, оно не зависит от температуры. Вклад дисперсионного
взаимодействия во многих случаях преобладает.Кроме классических межмолекулярных взаимодействий, рассмотрен¬
ных выше, в газовой хроматографии большое значение имеет водородная
связь [12]. Водородная связь имеет место при взаимодействии протонн-
зированного ковалентносвязанного атома водорода, например водорода
гидроксильной или аминогруппы органической молекулы, с атомом, име¬
ющим электроотрицательный характер (О, N и др.) и обладающим неподе-
ленной электронной парой. Энергия водородной связи обычно находится в
пределах 20-40 кДж/моль [13].Водородная связь может происходить только при определенной ориента¬
ции взаимодейстщтощих атомов, поэтому оно так же, как и ориентационное
взаимодействие, зависит от температуры. Вклад юдородной связи в удержи¬
вание нешторых молекул так сильно уменьшается с температурой, что наблю¬
дается изменение порядка выхода соединений смеси с повьштением темпера¬
туры шпонки. Водородная связь вносит большой вклад в удерживание спир¬
тов, эфиров, кетонов, аминов и кислот на некоторых сорбентах. В нешторых
случаях весьма выгодно использовать водородную связь для селективного
разделения соединений, образующих и не офазующих водородные связи.Межмолекулярные взаимодействия определяют физическую сорбцию,
донорно-акцепторные взаимодействия имеют химическую природу.Донорно-акцепторные комплексы возникают при передаче электронов
с заполненных орбиталей на молекуле-доноре к вакантным орбиталям мо¬
лекулы акцептора.Удерживание в ГЖХ редш определяется вкладом тольш процесса рас¬
творения. Как показано Мартином [14] и Березкиным [15], в общее удер¬
живание вносит также вклад адсорбция на пленке жидшй фазы и на по¬
верхности непокрытого твердого носителя (подробно эти явления будут
рассмотрены в главе 6).3.5 Селективность жидких фаз в ГЖХВ ГЖХ селективность разделения принято оценивать величиной а,
равной отношению удерживания разделяемых соединений.Различие в удерживании соединений определяется различием в меж¬
молекулярных и других взаимодействиях.Селективность неподвижной жидшй фазы должна оцениваться по
шнкретным соединениям.Жидкая фаза может быть селективна к определенным классам со¬
единений и менее селективна к другим. Это означает, что нельзя вообще
утверждать, что жидкая фаза селективна или неселектавна, нужно обяза¬
тельно указывать, к каким классам соединений это относится.90
ГЛАВА 3Газо-жидкостная хтшатографшДля более или менее полной характеристики селективности сорбентов
в ГХ, в том числе и жидких фаз в ГЖХ, необходимо оценивать селектив¬
ность к следующим группам соединений:- селективность к неполярной метиленовой группе (-СН^-) в гомоло¬
гическом ряду н-упхеводородов, т.е. различие в удерживании двух сосед¬
них гомологов;- селективность к полярным функциональным группам при разделе¬
нии соединений, отличающихся природой функциональной группы; обыч¬
но оценивается относительно н-алкана с одним и тем же числом атомов
угаерода в молекуле;- селективность к изомерам, разделение соединений, имеющих одина¬
ковый состав, но различающихся пространственным строением молекулы;
в этом случае оценивают величину а между изомерами, например мета- и
пара-ксилолами в случае изомеров положения или цис- и транс-изомеров
одних и тех же соединений.Оценку селективности по первой и второй группам лучше проводить
по графикам зависимости логарифма удельного удерживания обьема от
числа атомов углерода в молекулах разделяемых соединений (рис. 3.2).
Эти зависимости линейны и в большинстве случаев параллельны для раз¬
ных классов соединений. Рис. 3.2 построен по данным справочника по
удерживаемым объемам [16].Рис. 3.2 Зависимость IgVg от числа атомов углерода в молекуле (п)
для SE-30:1 - спирты, 2 - эфиры, 3 - кетоны и альдегиды, 4 - арома¬
тические углеводороды, 5 - алканы.91
Яшин Я.И.. Яшин Е.Я., Яшин А.Я.Газовая хроматографияРазличие удерживания на одну метиленовую группу определяется по
зависимости 5 для н-алканов.Приращение удерживания при замене метильной или метиленовой
группы в молекуле н-алкана на полярную фушщиональную группу опре¬
деляется разностью удерживания, в частности для спиртов между зависи¬
мостями 1 и 5, например для гексилового спирта и гексана.В таблице 3.8 приведены значения приращения удельных удерживае¬
мых объемов на разные функциональные группы относительно алканов,
полученные из графиков на рис. 3.2. Приращения определяли по раз¬
ности удерживаемых объемов соединений разных классов (эфиров, спир¬
тов, кетонов, альдегидов, ароматических соединений) и н-алканов с одним
и тем же числом атомов углерода в молекуле. Приращение на мети¬
леновую группу, как указано выще, определяли по разности удерживания
двух соседних в гомологическом ряду алканов.По данным таблицы 3.8 можно оценить, какая из жидких фаз наиболее
селективна к определенным классам соединений. Например, можно заклю¬
чить, что сквалан и апиезон наиболее селективны для алканов, полиглико-
ли наиболее селективны для спиртов, так как в этих случаях наблюдаются
наибольшие приращения Д .Межмолекулярные взаимодействия определяются не только природой
функциональных групп в соединениях, но и их пространственным распо¬
ложением. Изо-, втор-, третичные производные, как правило, удерживают¬
ся слабее линейных соединений.Селективность к соединениям, различающимся строением, особенно
важна для изомеров. Для разделения изомеров в ГЖХ используется так
называемый «геометрический» фактор, т.е. применяют жидкие фазы, в ко¬
торых геометрические структуры создают разные условия для ориентации
и взаимодействия молекул разных изомеров.Таблица. 3.8Разность удельных удерживаемых объемов соединений с различными
функциональными группами и алканами.Жидкая фазаду.ду^-ду,„_алкаиыароматическиеуглеводородыпростыеэфирысложныеэфирыкетоныальдегидыспиртыСквалан2,4271,995-4,4673,8464,4674,786Апиезон L1,9971,9051,4452,9512,3992,6303,631SE-30 (120°С)1,821,821,82 W 3,163,165,56SE-311,801,661,703,302,963,475,0Диоктилфталат(120“С)2,023,022,196,767,947,4214,492
ГЛАВА 3Газо-жидкостная хроматографияЖидкая фазаароматическиеуглеводородыпростыеэфирысложныеэфирыспиртыТрикреэилфосфаг1,975,1883,19911,4813,3414,7928,51Неопентилгликоль-адипат1,785,562,751,1323,7113,6535,89Диэтиленгликоль-себацинат1,786,703,3113,5014,8012,9040,70Диэтиленгаиколь-
адипаг(120°С)1,679,663,3518,6221,1317,9962,35Диэтиленгликоль-
сукцинаг 1,5815,855,1330,9055,5932,3685,11Полифениловый
эфир (6 колец)1,914,6210,1219,2811,0916,41Диглицерии1,867,385,4340,2777,6357,54384,6ПЭГ-10001,8209,333,5419,9523,1722,13110,9Сорбитол1,845,372,1410,1219,5053,70Для разделения изомеров, в частности изомеров положения (о-, м-, п-),
наиболее селективны жидкие кристаллы, краун-эфиры, циклодекстрины и
другие соединения.В ГЖХ применяют жидкокристаллические фазы смектического, не¬
матического, холестерического и других типов, в частности п,п’-азокси-
анизол, п,п’-азоксифенетол, п-(н-гептокси)бензойную кислоту, холесте-
рилацетат и др. В отличие от обычных изотропных жидкостей в жидких
1фисталлах сохраняется ближний порядок в расположении молекул. Жид¬
кокристаллические состояния веществ обладают структурными свойства¬
ми, промежуточными между свойствами твердого кристалла и жидкости.
Жидкокристаллическое состояние сохраняется только в определенном
температурном интервале.Нами предлагается система оценки селективности адсорбентов в газовой
хроматографии по отношениям коэффициентов емкостей и изменениям сво¬
бодной энергии сорбции для определенного набора производных бензола с
различными неполярными и полярными заместителями (таблица 3.9) [17].Эта система позволяет приводить оценку селективности разделения
гомологов, изомеров и соединений разных классов, отличающихся приро¬
дой полярных групп. По производным данным первой группы - алкил-
бензолам и полиметилбензолам можно оценить селективность разделения
гомологов по приращениям изменений свободной энергии сорбции на ме¬
тиленовую группу (таблица 3.10).По производным второй группы можно оценить селективность к раз¬
личным полярным функциональным фуппам.93
Яшин Я.И.. Яшин ЕЛ, Яишн АЛ. Газовая хроматографияПо Производным третьей группы можно оценить селективность раз¬
делительных систем к изомерам, в частности к орто-, мета-, пара-изомерам
дизамещенных бензолов. Селективность к этим изомерам сильно зависит
от природы заместителей, поэтому предлагается оценивать эту селектив¬
ность по трем типам изомеров: а) оба заместителя одинаковые и их вну¬
тримолекулярные взаимодействия в орто- положении слабые; б) замести¬
тели вступают в сильную водородную связь в орто- положении; в) один за¬
меститель полярный, другой - неполярный, создающий пространственное
затруднение сорбции полярному заместителю в орто- положении.Связь удельного удерживаемого объема с давлением насьпценного
пара исследуемого вещества и коэффициентом активности определяется
выражением [18]:= const/yp°M (3.4)где: у - коэффициент активности; р° - давление насыщенного пара;
М - молекулярный вес растворителя.Как видно из этого соотношения, величина обратно пропорциональ¬
на давлению насыщенного пара. Чем больше давление насыщенного пара
(т.е. чем ниже температура кипения), тем меньше удерживаемый объем.В ГХ наибольший интерес тфедстааляет отношение уцфживгвмых объемов:Ig V ~ Ig = Ig (у,/у,) + Ig (р,/р,) (3.5)Это соотношение известно как уравнение Харрингтона [18]. Оно по¬
казывает что различие удерживаемых объемов двух веществ определяется
как различием давлений насыщенных паров, так и различием коэффици¬
ентов активности.Для оценки селективности Байер ввел параметр 5 [19]:8 = (Ра°/р,°)'(гЛ,) (3.6)В случае, когда коэффициенты активности разделяемых веществ близки,
селективность разделения будет определяться различием давлений насыщен¬
ных паров. Такие случаи имеют место при разделении углеюдородов на не¬
полярных жидких фазах. При разделении изомеров или других соединений с
близкими температурами кипения различие в удерживании и селективность
разделения буцут определяться различием шэффициентов активности.Таблица 3.9Производные бензола, по которым можно характеризовать
селективность сорбентов в газовой хроматографииНазваниеФормулы1 группа
н.алкилбензолыС,Н,(СН)„СНз, где п - 0,1,2,3,4, 5
С,Нт(СНз)„, где п - 1, 2,3,4, 5, 6 а
т-5,4, 3,2,1,094
ГЛАВА 3Газо-окидтстная хроматографияНазваниеФормулы2 группа
Производные с полярными
функциональными группамиC^HjR, где R - СООН, -ОН, -NHj, -СНрН,
-ОСН,,-СНО, -СООСН,, -CN, -NO,, -F, ~С1, -Вг, -I3 группа
Дизамещенные производные
бензола - орто-, мета- и
параизомерыC,H,R,R,, гдеа) R, и Rj-CHj, -СООСН, и другиеб) R| - ОН, Rj - ОН, - NOj и другиев) R, - СН„ R, - ОН и другиеТабшцаЗЛООценка селективности сорбентов по изменениям свободной энергии
сорбции между веществами с определенными функциональными
группами и стандартным веществомВеществоПриращение Д (ДО) на функциональные группыОценка селективности к определенным функциональным гр}шпамБензолНитробензол - NOj
Анилин - NHj
Фенол - ОН
Бензонитрил - CN
Анизол - ОСН.Л(ДО) = -ЯТ In-Оценка общей полярности сорбентовн-ПропилбензолМезителенA(AG) = -RTln-н-БутилбензолДурол A(AG) = -RTln- ibSHSL-БензолПерфторбензолД(Д0) = -КТ1п-3.6 Стандартизация жидких фазВ газовой хроматографии уже описано применение более 1000 различ¬
ных жидких фаз. Многие из этих фаз дают идентичные результаты, некото¬
рые из них нестабильны либо малодоступны. Кроме того, большое число
используемых жидких фаз в разных методиках затрудняет стандартизащ1ю
результатов разделения и анализа. В связи с этим требуется ограничение
числа применяемых жидких фаз, выбор предпочтительных жидких фаз.
Ограниченное число жидких фаз можно тщательно изучить и основные их
свойства опублиювать в справочных материалах.95
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я. Гтовая хроматографияПервая попытка стандартизации жидких фаз была предпринята в
1956 г. Было рекомендовано применять шесть жидких фаз: н-гексадекан,
сквалан, бензилдифенил, динонилфталат, диметилформамид и дипшцерин.
Эти фазы доступны в чистом виде, однаш все они имеют большую летучесть.
Бьшо много попыток по выбору предпочтительных жидких фаз [20-25].В 1964 г. на Международном симпозиуме в Брайтоне по опросам спе¬
циалистов бьшо названо всего пятьдесят восемь жидких фаз, но чаще всего
назывались десять следующих фаз (в порядке частоты упоминания): апие-
зон L, полиметилсиликон SE-30, сквалан, динонилфталат, полиэтиленгли-
коль-20000, этиленгликольадипат, полиэтиленгликоль-400, силиконовое
масло DC-550, трикрезилфосфат и полиэтиленгликоль-1500.Проблема стандартизации жидких фаз обсуждалась также на Между¬
народном симпозиуме по хроматографии в Копенгагене (1968 г.) и в Ло¬
занне (1969 г.)В 1969-1970 гг. в литературе чаще всего упоминались следующие жид¬
кие фазы: 0V-1, SE-30, DC 200, апиезон L, полиэтиленглишль-20М, диэти-
ленглишльсукцинат, QF-1, этиленгликольадипат, SE-32 и сквалан.Многие из приведенных жидких фаз несомненно можно принять в ка¬
честве стандартных. Применение только стандартных жидких фаз облег¬
чило бы внедрение хроматографических методик в аналитическую практи¬
ку и дало бы значительный эшномический эффект.В работе [26] в результате опросов потребителей в Европе было вы¬
явлено, что чаще всего применяются следующие неподвижные жидкие
фазы: полиметилсилоксаны (0V-1, OV-101 и SE-30) - 73%, полиметилфе-
нилсилоксаны (5% фенильных фупп) - 63%, полиэтиленгликоли - 53%,
полиметилфенилсилоксаны (50/50%) - 38% и полиметилцианопропилфе-
нилсилоксаны (7% циан и 7% фенил) - 25%.3.7. Бинарные жидкие фазы.При разделении многокомпонентных трудноразделяемых смесей ино¬
гда бывает сложно подобрать жидкую фазу, на которой бы разделились все
компоненты. Например, на одной жидшй фазе не разделяются более тя¬
желые компоненты (рис. 3.3), но разделяются все остальные, а на другой
наоборот - не разделяются легкие компоненты, а остальные разделяются
хорошо. Используя шмбинацию двух сорбентов (1 и 2), удается легко по¬
лучить полное разделение всей смеси.Две жидкие фазы можно комбинировать тремя путями (рис. 3.3) [27]:1. Использование двух составных последовательно соединенных шло-
нок, заполненных разными сорбентами.2. Использование одной шлонки, заполненной механической смесью
двух сорбентов (каждая жидкая фаза наносится отдельно на носитель, по¬
лученные сорбенты смешиваются).96
ГЛАВА 3Газо-жидшхтнт хроматография3. Использование колонки, наполненной твердым носителем, на кото¬
рый нанесена сразу смесь двух жидких фаз из одного раствора.Теоретически оправданы первая и третья комбинации. Во втором слу¬
чае возможно дополнительное размывание в кинетической области, так
как искусственно создаются разные сорбционные центры.Применение бинарных жидких фаз позволяет регулировать селеютв-
ность шлонки в целом в довольно широких пределах, причем из ограничен¬
ного набора жидких фаз можно получить сор&нт с любой полярностью.Оптимальное соотношение двух жидких фаз, при штором достигает¬
ся полное разделение смеси, выбирают по диаграмме, изображенной на
рис.3.4. На оси ординат откладывают времена удерживания разделяемых
веществ на первой и второй фазах. Точки на ординатах, соответствующие
временам удерживания одних и тех же шмпонентов, соединяют прямы¬
ми линиями. Затем на диаграмме выбирают таюе соотношение фаз, когда
достигается наибольшее различие во временах удерживания между всеми
шмпонентами смеси.^мбинация шлонок с разными жидкими фазами широш применяет¬
ся в двухмерных вариантах хроматографии.-[4ZнуЯАЛАЛАРис.3.3 Сопоставление разделения на колонках с одним и двумя
сорбентами./-Гт%Рис.3.4 Различные комбинации применения бинс^)ных жидких фаз на со¬
ставных колонках (I), колонка со смешанными сорбентами (II) и
колонт со смесью подвижных жидких фаз на одном носителе
(III). Диаграмма для определения оптимального соотнтиения
бинарных жидких фаз.97
Яшин ЯМ. Яшин Е.Я.. Яшин А.Я.Газовая хроматография3.8 Полярность жидких фаз по Роршнайдеру и Мак Рейнольдсу.Понятие «полярность» широко используется для классификации жид¬
ких фаз [5,10,11]. Полярность молекул обычно оценивается величиной
дипольного момента. Однако, в газовой хроматографии в понятие «по¬
лярность» вкладывается другой смысл. Оценивать значением дипольного
момента полярность высокомолекулярных жидких фаз, молекулы которых
могут иметь десятки полярных функциональных групп разной полярно¬
сти, очень трудно или невозможно.Ранее предлагалось оценивать полярность жидких фаз по отношению
полярных и неполярных функциональных групп. К неполярным группам
относятся метиленовые и метильные группы. Этим соотношением хоро¬
шо характеризовать полиэтиленгликоли (ПЭГ). В газовой хроматографии
ПЭР используются с молекулярными массами: 300, 400, 1000, 1500,
6000, 15000, 20000 и 40000. С увеличением молекулярной массы моле¬
кул ПЭГ полярность уменьшается, т.к. уменьшается число концевых по¬
лярных гидроксильных групп. С увеличением молекулярного веса ПЭГ
удерживаемые обьемы полярных веществ падают, а неполярных возрас¬
тают (рис.3.5).Рис.3.5 Зависимость Vg бензола и метанола (а) и воды (б) от моле¬
кулярного веса полиэтиленгликолей.Точнее полярность жидких фаз можно оценивать по удерживанию
стандартных соединений разной полярности. Впервые таким образом
предложил оценивать полярность жидких фаз Роршнайдер [28] и позднее
Мак Рейнольдс [29]. Роршнайдер предложил оценивать полярность жид¬
ких фаз по разности индексов удерживания Ковача на полярных и непо¬
лярных жидких фазах для пяти наиболее характерных соединений разных
классов: бензола, этанола, метилэтилкетона, нитрометана и пиридина, а
МакРейнолдс для десяти (таблица 3.11). Взаимодействия характерные для
этих тестовых соединений приведены в табл.3.12.98
шлшлзГазо-жидтктная хтматографияТаблица 3.11Эталонные соединения для оценки полярности сорбентовПо РорпгаайдеруПо МакРейнольдсуБензолБензолЭтанолБутанолМетилэтилкетонПентанон-2НитрометанНитропропанПиридинПиридин2-метилпентанол-21-иодбутанОктин-21,4-диоксанЦис-гидринданТаблица 3.12Взаимодействия, характерные для тестовых соединений Мак Рейнольдсатестовые соединения Роршнайд^>а в скобках)ОбозначенияТестовые соединенияТипы взаимодействияXбензолдисперсионное (слабое протон-
акцепторное)Yбутанол (этанол)ориентационное, водородная связьZ2-пентанон (2-%танон)ориентационное, протон-ащепторноеинитропропан(нитрометан)ориентационное, щютон-акцеоторноеSпиридинориентационное, сильное протон-
акцепторноен2-метил-2-пентанолJиодобутанк2-октинL1,4-диоксанпротон-акцепторное, но не протон-
донорноеМцис-гидриндандисперсионноеЗа фазу с нулевой полярностью принят сквалан, а за фазу со стопро¬
центной полярностью - р|Р’-оксидипропионитрил (рис. 3.6).99
Яшин Я.И., Яшин Е.Я.. Яшин А.Я.Гаювая хроматографияСкваяан оксиди-
проттионитрилРис. 3.6 Оценка полярности по Роршнайдеру.Так как тестовые вещества Роршнайдера элюируют на нешгорых ш-
лонках быстро и точность их определения индексов удерживания невысока,
Мак Рейнольдс [29] предложил оценивать полярность по десяти веществам
разных классов (бензол, бутанол, 2-пентанон, нитропропан, пиридин, 2-ме-
тил-2пе1гганол, 1-иодобуган, 2-октин, 1,4-диоксан, цис-гидриндан).Замена трех из пяти рекомендованных Роршнайдером веществ на бо¬
лее высокие гомологи получила щирокое одобрение. Однако добавленные
стандартные вещества Мак Рейнольдса редко применялись для характери¬
стики новых фаз. Многие считают, что для практических целей достаточно
пяти стандартных веществ.Замена на менее летучие (бутанол, нитропропан, 2-пентанон) позволи¬
ли более точно оценивать их индексы.Мак Рейнольдс использовал для характеристики фаз тольш разность
индексов удерживания Д1, в отличие от Роршнайдера, шторый использо¬
вал А1/1(Ю. Мак Рейнольдс охарактеризовал по своей системе более 200
жидких фаз [29, 30]. Из этих данных можно было выбирать предпочти¬
тельные жидкие фазы.Мак Рейнольдс определял индексы удерживания при температуре 120°С,
а Рорщнайдер - при 100°С.В последние годы системы оценки полярности Рорщнайдера и Мак
Рейнольдса подверглись серьезной критике. Во-первых, выбор н-алканов
в системе индексов удерживания не совсем удачен, т.к. на полярных фа¬
зах н-алканы плохо растворяются, и большой вклад в удерживание вно¬
сит их адсорбция на поверхности жидких фаз. Поэтому появилось много
альтернативных предложений, в частности вместо н-алканов предлагались100
ПЫВА 3 Гсао-жидтстная хроматографияВ качестве стандартов 2-алканоны, метиловые эфиры жирных кислот, 1-
бромалканы и другие [31].С такими стандартами удельный удерживаемый обьем будет точнее
связан с коэффициентом распределения в жидкой фазе.В последние годы широко используются полимерные жидкие фазы,
которые в производстве отличаются от одних партий к другим партиям,
что влияет на различия индексов удерживания.Много критики связано с выбором сквалана в качестве неполярной
жидкой фазы (или фазы с «нулевой» полярностью).Сквалан имеет низкое значение максимально допустимой рабочей
температуры (МДРТ), не выше 120°С, может легко окисляться и плохо де¬
активирует носитель. Вместо сквалана предлагалось использовать апиезон
МН, аполан-87, SE-30 (полиметилсилоксан) и полиметилоктилсилоксан.
Эти фазы также неполярные, индексы удерживания на них близки сквала-
ну (таблица 3.8), величина МДРТ их в пределах 250-300°С. Теоретически
Ковач рассчитал, что наиболее подходящей фазой стал бы углеводород с
молекулярным весом 2000, который бы имел МДРТ ошло 300°С.Аполан-87 имеет молекулярный вес 1222, МДРТ - 285°С, т.е. близок к
ташму теоретичесшму угаеводороду.Апиезоны имеют высокие молекулярные веса, но имеют ненасыщен¬
ные связи в своем составе, что зависит от его происхождения.Полиметилсилоксаны (SF-30, 0V-1 и др.) также не могут быть всегда
однозначными и воспроизводимыми. В этом отношении аполан-87 синте-
тичесше, хорошо характеризуемое соединение.Во многих работах понятие полярности жидких фаз связывают с ее
селективностью.В своих работах [28, 32] Роршнайдер предлагал оценивать полярность
жидких фаз для ГЖХ отношением удерживаемых параметров бутана к бу¬
тадиену. Шомбург [33] предложил определять полярность разницей индек¬
сов удерживания бензола и циклогексана на полярной фазе и на сквалане:р = А1 -Д1 (3.7)бензол циклогею: ан 'где Р - полярность.Общую полярность МакРейнольдс [30] предлагал оценивать как сум¬
му А! величин на полярной фазе и сквалана для бензола, бутанола, 2-пен-
танона, нитропропана и пиридина.Несмотря на вышеперечисленные недостатки, шнстанты МакРей-
нольда и Роршнайдера позволяют характеризовать и различать неподвиж¬
ные жидкие фазы по общей полярности и селективности к тестовым со¬
единениям.101
Яшин Я.И.. Яшт Е.Я.. Яшин АЖГазовая хроматографияВ таблицах 3.13, 3.14 приведены константы МакРейнольдс для непо¬
лярных фаз, для фаз средней полярности (таблица 3.15) и для полярных
фаз в таблице 3.16.В таблице 3.17 приведены константы для наиболее распространенных
полисилоксановых фаз серии OV.В таблице 3.18 приведены константы МакРейнольдса и средняя поляр¬
ность жидкостных фаз, содержащих краун-эфиры [34].Таблица 3.13Индексы удерживания (I) тестовых соединений МакРейнольдса на
неполярных жидких фазах.Тип жидкой фазы1,12(К:бензолбутанол2-пентаноннитропропанпиридинСквалан653590627622699Апиезон М686627651698767Апиезон МН671599632667736Аполан-87674600630664724SE-30668643672716740Таблица 3.14Константы МакРейнольдса важнейших неподвижных жидких фаз
(углеводороды и полнсилоксаны)Неподвижные фазы№кстанты*XYZиSЯ/КLМовчСквалан00000000000Апиезон Н5986811511294653238137506Апиезон I38302749572342154235201Апиезон L32221532421335113133143Апиезон М31221530401232102829138Апиезон N38402852582541154335216Апиезон W8213599155154909342109596250V 1175745674333229OV10117574567433342346^2229SE-301655446542322220V-3 (10% фенильных групп)44868112488554230V-7 (20% фенильных групп)69113111171128775920V-11 (35% фенильных групп)102142145219178103786OV-17 (50% фенильных групп)119158162243202112884OV-22 (60% фенильных фупп)1601881912832531331075102
ГЛАВА 3Газо-жидкостная хроматографияНеподвижные фазыКонстанты*XYZиSЯIКLМобщOV-25 (75% фенильных групп)17820420830528014411750V-61 (33% фенильных групп)10114314221317499773SE-52 (5% фенильных групп)327265986744334SE-54 (10% фенильных и 1%
винильных групп)337266986746336OV-210(50%
трифторпропильных
« SP 240114623835846831020613956283601520OV-10S (5% цивнэтильных
групп)36108931398674462OV-25 (25% цианпропильных
и 25% фенильных групп2283693384923862821813OV-275 (100% цианэтильных
групп)7811006885117710894938Дексил 300 ОС
(метилсиликон)374778801131031541481179655499474Дексил 400 ОС
(фенилметилсиликон)72107118168123588Дексил 410 ОС
(2-цианэтилсиликон)71286174249171951Таблица 3.15Константы МакРейнольдса эфирных и полиэфирных жидких фазНеподвижные фазыКонстанты*XГZиSЯIКLМобщБис (2-этилгексил) адипат
(флексол А-26)7618112119713414471551199709Бис (2-этилгексил) фталат92186150236167143926614026831Бис (2-этилгексил)
себацинаг72168108180125132684910711653Бис (2-этилгексил)
тетрахлорфталат112150123168181110734Дициклогексилфталат146257206316245196144104204581170Дидецилфталат136255213320235201126101202381159Дидодецнлфтаяат79158120192158120795211626707Диизодециладипат711711131851281346752114И668Диизодецилфталат84173137218155133835913024767103
Яшин Я.И.. Яшт Е.Я.. Яшин А.Я.Газовая хроматографияНеподвижные фазыКонстанты'XY7иSЯIКLМобщFFAP (карбовакс 20 М с
2-нитротерефталевой
кислотой в качестве
концевых групп)3405804976026274232982284731612546Диизонониладипет73174116189129137685411610681Диизооктиладипат78187126204140148721591268735Динонилфталат83183147231159141826513818803Динонилсебацинат6616610717811813062501068635Дитридецилфталат75156122195140119765111525688Октилдециладипат79179119193134141725711910704Трикрезилфосфат176321250374299242169131254761420Зонил Е-7 (фторированный
эфир пиромеллитовой
кислоты223359468549465338146137469622064Зонил Е-91(фторированный эфир
камфарной кислоты)1302503203772932358195295101370Диэтиленгаикопьадипат
(ДЭГА), HI-EFFIAP3786034606656584793292545541762764Диэтиленгяикольсукцинат(ДЭГС)4707055587887795563933016772153300Этиленгликольсукцинаг(ЭГС)5367756368978646224503477832593708Неопентилгликольадипет(НПГА)2344253124624383392101573621031871Неопентилгликольизофта-
лат (НПгаФ)207377Неопентилганкольсукцинат(Ш11С)2754723675434893742451864231272146Пропиленгпикольадипат
(ПГА) реоплекс 4003646194496476714823172455401712750Пропиленгликольсебаци-нат(ПГСб)196345251381328271176129285831501Таблица 3.16Константы Мак-Рейнольдса полярных жидких фазНеподвижные фазыКонстанты*XГZиSЯ/КLМобщКарбовакс 6003506314286326054723082405031622646Карбовакс 10003476074186265894493062404931612587Карбовакс 15003476074186265894492587Карбовакс 15403716394536666414793252555341722770104
ГЛАВАSГазо-жидтстная хроматографияНепо|Движные фазыКонстанты"XYZиSЯ/КLМобщКарбовакс 40003255513755825203992852244431482353Карбовакс 60003225403695775123902822224371472320Карбовакс 20 М3225363685725103872822214341482308Карбовакс 400 моностеарат2804863255124493502441913821222052Карбовакс 20 М ТРА (с
терефталевой кислотой в
качестве юнцевых груш)3215373675735203872812204351482318Дипшцерин371826560676854608245141724363287SP 1(Ю0 (полиэтиленпш-
коль с 2-нитротерефтале-
войкислотой в ючестве
конце-вых групп3325553935835464002409STAP (модифицированный
Карбовакс 20 М)3455864006106274283012354841632568Тритон Х-305 (мол. масса
1500)2624673144884303362291833661131961Твин 80 (полиоксиэтилен-
сорбитанмоностеарат)2274302834383963101774ППГ 1000 (полипропилен-
гликоль)131314185277243214110101205461150Ш1Г 2000 (полипропилен-
гаиколь)12829417326422619610698194451085Тетракис (гидроксипропил)
этилендиамин (квадрол)2145713574724894312081423791112103Таблица 3.17Характеристики неподвижных жидких фаз серии OV для газовойхроматографии.Назва¬ниеТипТемпера¬турныепределы(0)Мол.весКонстанты Роршнайдерабензолбутанол2-пен¬таноннитро¬пропанпири¬дин2-ме-тил2-пен-танол2-октинOV-1Полиметилсиликон(ПМС)100-350(75)>10*165544654232230V-3Полиметилфеиил
силикон (ПМФС)
-10% фенил20-325-3752-10*448681124885546OV-7ПМФС - 20%
фенил20-350МО*6911311117112877660V-11ПМФС - 35%
фенил20-3507-10>10214214521917810392105
Яишн Я.И: Яшин Е.Я.. Яишн АЯ.Газомя хроматографияНазва-1шеТипТемпера¬турныепределы(0)Мол.весКонстанты Роршнайдерабензолбутанол2-пен¬таноннитро¬пропанпири¬дин2-ме¬тил2-пен-танол2-окгннOV-17ПМФС - 50%
фенил20-350410>119158162243202112105OV-20ПМФС - 20%
фенил20-350OV-22ПМФС-20-350810»160188191283253133132OV-25ПМФС-20-350110*1782042083052801441470V-61ПМФС-50%фенил20-350410*1011431422131749986OV-73ПМФС - 50%
фенил20-350SIO*408676114855735OV-101пмс20-350310*17574567433323OV-105полицианопропил-неталсиликон(ПЦПМС)-25%20-250310*3610893139867429OV-202политрифторпро-пияметялсилиюн(ПТФПМС)29-250МО*14623835846831020656OV-210ПТФПМС20-250210’14624036346831020656OV-215ПТФПМС20-250310>14924036347831520856OV-225полиютнопропил
метил фенилси-
лиюн20-250810>228369338492386282150OV-275полнцианоалкилсиликон20-250510»6298727631106849686318OV-330сополимер фенил¬
метилсиликон с
карбоваксом20-250510’222391273417368284158OV-35Iполигликольнитротерефтш1ат20-250-335532382583540--OV-1701полиметилфенилшшюсилоксан20-300510*67170153228171--106
тлллзГазо-жидтстная храматогтгфшТаблица 3.18Константы Мак Рейнольдса (М) и средняя полярность неподвижных
газохроматографических фаз, содержащих краун-соединения, и фаз
сравнения при 120°С.Неподвижные фазыТестовые веществабензол 1-бутанол 1-иитропаи пиридин2-пентанонСредняяполярность18-Крчи-64.13-Диаза-18-
крщи-бБензо-18-кр|^-6
Бензо-15-1ср^-5
Дибензо-1 в-кр^^н-б*
Дибензо-24-краун-84.13-Дибензилдиаза-
18-краун-бКриптанд [2.2.2]Криптанд
[2.2.2]+4ДЗ-
дибензилдиаза-18-
краун-6Дибензо-24-кр^-
8+диаза-18-краун-бSE-30Карбомкс 20 М303201267252247271161197190285153225105024304144454542864103264995353657642549847951554825738530047964572472336416412480490259321285401415103962773383292403542022422383384436845234838937738442323531126840443462* Измерено при температ^ колонки ISff’C.3.9 Новые жидкие неподвижные фазы для газовой хроматографии
и новые технологии их производстваЗа последнее десягалетие достигнут большой прогресс в разработке и
производстве новых жидаих фаз. В настоящее время имеется в продаже боль¬
шой ассортимент стабильных жидких фаз, который постоянно расширяется.Прежде всего, необходимо отметить, что на базе полисилоксанов как
самых стабильных фаз разработана большая серия жидких фаз с разными
функциональными группами как неполярными, так и сильнополярными с
высокой максимально допустимой рабочей температурой (см. таблицу 3.7).
Достижения последних лет следующие:- разработка и выпуск химически привитых, связанных или иммо¬
билизованных жидких фаз; жидкие фазы химически прививаются к
поверхности носителя, к поверхности капиллярных колонок, одно-107
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я. Гтовая хроматографияВременно происходит и объемная сшивка жидких фаз; подобные фазы
значительно стабильнее, чем не привитые фазы;- введение в аналитическую практику жидких фаз с использованием
так называемого «геометрического фактора» для разделения изоме¬
ров и других соединений, различающихся геометрической струюу-
рой; жидкокристаллических фаз разной природы, кр^н-соединений,
щ1клодекстринов и других макроциклов; применение одновременно
смеси краун-эфиров и циклодекстринов дает синергетический эффект[35];- разработка надежных хиральных жидких фаз для разделения опти¬
ческих изомеров;- разработка «золъ-гелъ» технологии в производстве юлонок, что по¬
высило их стабильность;- применение ионных жидкостей в качестве стационарных фаз [36];- применение комплексов металлов как стационарных фаз [37];- расплавленные органические соли как стационарные фазы [38];” водные неорганические фазы как сорбенты [39];- синтез и применение фуллереновых полисилоюановых жидких фаз [40];- синтез и применение карборанполисилоксановых жидких фаз [41];- разработка жидких фаз с максимально допустимой рабочей тем¬
пературой 350-420°С для высокотемпературной газовой хроматогра¬
фии, для анализа микропримесей, для работы в составе ГХ-МС;- разработка многофункциональных жидких фаз.Смесь жидкокристаллической фазы и резоркарена изучалась для раз¬
деления дизамещенных изомеров бензола [42]. Эти смешанные фазы пока¬
зали «синергетический эффект». Синтезированы жидкокристаллические
фазы полисилоксанового типа с нафталином в цепи [43]. Эта фаза показала
высокую разрешающую способность в разделении изомеров полихлори¬
рованных дибензо-п-диоксинов, полиядерных ароматических соединений
(ПАУ) и пестицидов. Другие жидкокристаллические фазы бьши также
оценены для разделения ПАУ [44], изомеров диоксина [45].Описаны жидкие фазы на основе к{^«-эфиров [34]. Синтезированная сме¬
шанная фаза на основе щк^-эфира и циклодекстрина показала превосходящую
селективностъ при разделении энантиомеров и изомеров положения [46].В следующем исследовании было выявлено, что такая смесь также
имеет синергетический эффект в разделении изомеров [47].Исследованы фазы, содержащие полисилоксаны с каликсаренами
строением типа «корзины» [48].Были предложены новые фазы, содержащие катенантные кр^-эфиры [49]
и дибензо-24-кр^-8 [50], а также фаза на основе 4,13-диаза-18-кр^-6 [51].108
ГЛАВА 3 Газо-жидкостная хроматографияПолисилоксаны, содержащие каликсарены [52], фуллеропирролидины
[53], специально синтезированы для ГХ.По применению полисилоксанов в ГХ опублишван большой обзор [54].Новые полимерные хиральные фазы типа Перкле на основе полиси¬
локсанов с бензамидами в цепи [55].Предложена новая комплексная фаза с переходными металлами (Си,
Со) на основе производных циклодекстринов [56].В другом исследовании параметры Роршнайдера - Мак Рейнольдса
измерены для хиральных спиртов и кетонов для того, чтобы определить
термодинамическую селективность на новой фазе трис [3-(трифторметил-
гидроксиметилон)-(+)-камфорат] производные лантанидов [57].Силоксановый полимер с Си-содержащим комплексом показал хоро¬
шее разделение ряда эфиров фталата [58].Наиболее значительным достижением последних лет является разра¬
ботка золь-гель технологии производства колонок [59,60]. Этот метод был
использован для приготовления капиллярных колонок [61]. Одно из пре¬
имуществ таких колонок - их высокая термостабильность.Большинство исследований по новым жидких стационарным фазам в
КГХ связаны с производными циклодекстринов. В результате синтезиро¬
ван целый ряд стационарных фаз для разделения изомеров. Шуриг с со¬
трудниками использовали линейные декстрины также для изомерных раз¬
делений [62].Армстронг впервые докладывал об использовании ионных жидкостей в
ГХ как сорбентов [36]. Эти вещества пригодны как для хиральных, так и для
обычных разделений. Последние модификации таких жидкостей показали
термическую стабильность до 350°С и уже коммерчески доступны (фирма
Advanced Separation Technologies Inc.). Силоксановые жидкие фазы с сила-
реновыми производными показали низкую летучесть для ГХ-МС [63].Новые силоксановые фазы приведены на рис.3.7, краун-эфиры на
рис.3.8, циклодекстрины на рис.3.9.а)30-Si-i -4n_ Rem109
Яишн Я.И.. Яшт ЕЖ. Яшин АЖ.Газовт хроматографияв)Го'1кт..1Рис.3.7 Сшоксановые фазы: а) с сшареновыми функциональными
группами; б) карборановыми; в) фуллереновыми.о ооо ”и? о.IVVIVDIIXРис.3.8 Краун-эфиры и криптандыПО
ГЛАВА 3Газо-жидюжтнт хитштогтАтРис.3.9 Структурные формулы альфа, бета и гамма циклодекстри¬
нов и возможные механизмы сорбции молекул в порах цикло¬
декстринов.ЗЛО Химическая прививка (иммобилизация) жидких фаз к вну¬
тренней поверхности капиллярных колонок и к поверхности твердых
носителей.Для прививки жидких фаз, в основном полисилоксанов, исполь^ют-
ся несколыю способов, в частности термическая иммобилизация силано-
содержащих полисилоксанов [64] и поперечная сшивка, инициированная
радикалами [65].Сополимеры карбоксиалкил полисилоксанов также легко иммобили¬
зуются термически [66].Для термической конденсации деактивированная поверхность стекла
покрывается пленкой силаносодержащего полимера (возможно примене¬
ние и метоксипроизводных полимеров). Колонку нагревают до 300-370°С
в течение 5-15 часов в потоке газа-носителя. Термически индуцированные
реакции связывают полимер с поверхностью колонки и способствуют по¬
перечной сшивке полимерных цепей.При добавке нескольких процентов реактива для поперечной сшивки,
например алкилтриметоксисилана, повышается термическая стабильность
полученной полимерной пленки.Таким путем получают колонки для высокотемпературной газовой хро¬
матографии (до 325 и даже до 420°С в режиме программирования) [67].111
Яшин ЯМ. Яшин Е.Я., Яшин А.Я.Гтовая хроматографияТермическая стабильность полисилоксанов улучшается, если в цепь
полисилоксана вводятся фенильная, силареновая или карборановая группа
(рис. 3.7).Колонки с такими привитыми фазами применяются для разделения
триглицеридов и углеводородов до [68, 69].Второй способ иммобилизации жидких фаз это сшивка за счет ради¬
калов при использовании перекисей [70], азосоединений или у-облуче-
ния [64]. В этом случае сшивка происходит через связи углерод-углерод
(таблица 3.19).Всего необходимо 0,1-1,0% сшивающего агента, чаще всего использу¬
ется перекись дикумила или азо-трет-бутан.Эта обработка сравнительно проста.Основные преимущества иммобилизованных сорбентов:1. Более высокая термическая стабильность до максимально допусти¬
мой температуры (до разрыва химических связей); при этом фоновый ток
практически не растет. Это означает, что расширяется диапазон использу¬
емых рабочих температур.2. Повышенная устойчивость к растворителям: при загрязнении ко¬
лонки высококипящими соединениями колонку можно отмыть некоторы¬
ми растворителями.3. Кроме стабильности, иммобилизованные колонки интересны еще и
тем, что можно приготовить на внутренней поверхности капилляров плен¬
ки толщиной 5-8 мкм или более, тогда как при простом смачивании нельзя
получить пленку толщиной более 0,5 мкм.Таблица 3.19Реактивы для свободнорадикальной пришивки жидких фаз в ГЖХ№Х®Ш1НазваниеСтруктураСН,сн.ПерекисьдикумилаООсн.сн.оо2.ПерекисьдибензоилаоО112
ГЛАВА 3Газо-жидкостная хроматография№№ппНазваниеСтруктураСН,СН,3.nqjeKHCb ди-
трет-бутилаHjCО ОСН,СН,СН,СН,СН,4.Азо-бис-изобу-тиронитрилN=^N=N-СН,СН,СН,СН,5.Азо-трет-бутанн,с--N:СН,СН,СН,3.11 Твердые носители для жидких фазВ ГЖХ твердый носитель должен тольш удерживать жидкую непод¬
вижную фазу в виде однородной пленки, он должен служить тольш под-
ложшй жидшй фазы, поверхность самого твердого носителя не должна
принимать участие в процессе взаимодействия с молекулами анализиру¬
емых веществ. Для выполнения этой роли твердый носитель должен удо¬
влетворять следующим требованиям:1. твердый носитель должен иметь макропористую структуру и не со¬
держать микропор;2. удельная поверхность должна быть небольшой (от 0,05 до Зм^/г);3. поверхность должна быть химически инертной;4. носитель должен быть термостабилен до 400 °С и более;5. его зерна должны быть механически прочны, желательно сфериче-
сшй формы.До сих пор еще не создан ташй идеальный носитель, который удо¬
влетворял бы всем этим требованиям. В разных исследованиях указывают¬
ся различные минимальные удельные поверхности носителя, при которых
достигается оптимальная эффективность разделения, чаще всего приво¬
дятся удельные поверхности 0,5—2 mVf [71,72].1138 Газовая хроматографкя
Яшин Я.И.. Яшин ЕЯ., Яшин А.Я. Гаювая хроматографияДиатомитовые твердые носители. Обычные пористые адсорбенты
обладают большой поверхностью, а это, как известно, приводит к сильной
адсорбции. Поэтому в качестве твердого носителя в газовой хроматогра¬
фии применяют преимущественно природные диатомитовые минералы,
обладающие малой удельной поверхностью и макропористой структурой.
В природе имеются тысячи разновидностей диатомитов. Скелет всех диа¬
томитов в основном состоит из гидратированного аморфного кремнезема.
Размеры макропор диатомитов в большинстве случаев составляют около
1 мкм. Диатомиты кроме двуокиси кремния содержат примеси оксидов
других металлов. Состав диатомитов, как правило, следующий: около 90%
SiOj, 4% AljOj 1,5% FePj, 0,2% TiOj, 0,5% MgO и др. (указано минималь¬
ное содержание SiOj и максимальное— других оксидов, возможны любые
другие соотношения). В диатомитах нежелательно присутствие больших
количеств окислов алюминия и железа, так как это обычно приводит к
большой каталитической и адсорбционной активности получаемого твер¬
дого носителя.Все твфдые носигеш на основе диатомитов под[мзделяют на два типа [71].К первому типу относятся твердые носители, приготовленные из при¬
родного диатомита, предварительно размолотого, отсеянного и прокален¬
ного при 900°С. Носители этого типа имеют розовый цвет из-за присутствия
в диатомите оксидов железа. Водная вытяжка из носителей первого типа
.слабокислая за счет гидроксилированной поверхности аморфного крем¬
незема. Твердые носители первого типа обычно имеют большую удельную
поверхность и обладают каталитической активностью. К носителям этого
типа относятся хромосорб Р, диатопорт Р, газхром Р, анакром Р, фазесеп Р,
огнеупорный кирпич С-22, стерхамол, кизельгур фирмы «Мерк», шималит
А, В, С, чехасорб, ИНЗ-600, сферохром-2 и др.Ко второму типу относятся твердые носители, приготовленные путем
спекания природного диатомита со специальными флюсами (например, кар¬
бонатом натрия) и прокаливания при температуре выше 900 °С. При спека¬
нии с щелочными флюсами оксиды железа переходят в бесцветные (или бе¬
лые) соответствующие соли, поэтому все носители этого типа имеют белый
цвет, Удельная поверхность носителей второго типа, как правило, меньше,
чем у носителей первого типа, водная вытяжка их имеет щелочную реак¬
цию, и они не обладают каталитической активностью. К ним относятся хро¬
мосорб W, диатопорт W, газ-хром С, целит 545, анахром 545, шималит W,
хромосорб А, хромосорб G, хроматон N, сферохром-1, порохром и др.При прокаливании диатомитов значительно уменьшается удельная по¬
верхность вследствие спекания более тонких пор, в результате чего сильно
уменьшается адсорбционная активность. Адсорбционная активность умень¬114
ГЛАВА 3 Газо-жидкастная хроматографияшается также за счет дегидроксилирования noBq)XHocra, так как диатомит
по существу является кремнеземом (двуокиси кремния содержится до 90%).
При высоких температурах гидроксилированная (или силанольная) поверх¬
ность переходит в дегидроксилированную (силоксановую) поверхность.Наилучшими носителями являются хромосорбы W и G (фирмы «Джонс
Мэнвилл»), что объясняется в основном природой диатомита, из которо¬
го получали хромосорбы. Он обладал наибольшим объемом пор и более
однородным распределением пор по размерам. В Армении обнаружены
диатомиты, которые по своим природным, характеристикам близки амери¬
канским диатомитам. Из армянских диатомитов ранее получали твердые
носители — порохромы. Общий объем пор в них составляет 1,5—1,8 смЗ/г.
Порохром-1 по объему, размерам и однородности пор значительно превос¬
ходит выпускаемый отечественной промышленностью носитель сферох-
ром-1 и очень близок к лучшему зарубежному носителю хромосорбу W.Основным отличием порохромов является его макропористая структу¬
ра и высокая однородность пор.Следующая отличительная особенность порохромов — малое содержа¬
ние оксидов железа (в пределах 0,05—0,5%), которое приводит к юталитиче-
ской активности, и большое содержание двуокиси кремния (до 96—98%).Следует отметить твердые носители фирмы «Лахема» из Чехии; хро-
матон и инертон.По своим свойствам хроматон близок к хромосорбу W. Хроматон имеет
следующий химический состав: SiOj —^93%, Al^O, — 3,3%, Fe^Oj — 0,4%,
TiOj — 0,01%, CaO + MgO — 0,1%. Хроматон N обладает узким распре¬
делением макропор, он практически не содержит микропор, большинство
пор имеет размеры в пределах 1000 — 1500 нм.Хроматон N — универсальный твердый носитель, он может быть ис¬
пользован для разделения как полярных, так и неполярных веществ с ис¬
пользованием до 20% жидшй фазы от массы носителя. Шарообразная фор¬
ма носителя обеспечивает быстрое и равномерное заполнение хроматогра¬
фических шлонок. Узкие фракции носителя, в частности 0,2—0,25 мм, обе¬
спечивают высокую эффективность хроматографичесшго разделения.Выпускается также хроматон N, промытый кислотой. В процессе про¬
мывки с поверхности носителя удаляются все щелочные примеси, кроме
того, при этом происходит одновременно и отмучивание, т. е. удаление
с поверхности пылеобразных частичек, снижающих эффективность раз¬
деления. Хроматон N, промьггый кислотой, нужно использовать прежде
всего с жидкими фазами, чувствительными к щелочным примесям при по¬
вышенных температурах, в частности с силиконовыми полимерами, слож¬
ными полиэфирами и жидкими фазами кислого характера.115
Яшин Я.И.. Яшин Е.Я., ЯшинАЯ. Газовая хроматографияДля анализа сильнооолярных веществ (спиртов, аминов, кислот и др.)
поставляется хроматон N, химически модифицированный диметилдих-
лорсиланом или гексаметилдисилазаном.Твердый носитель — инертон, по своим техническим характеристи¬
кам близкий хромосорбу G.Кроме хромосорбов за рубежом используют газхромы, сырье для ко¬
торых производит фирма «Eagle—Picher Company» (залежи в Неваде).
На газхром Р можно наносить большие количества жидкой фазы (более
10%), тогда как газхром Z предназначен для использования с небольши¬
ми количествами жидких фаз. Газхром S — необработанный носитель,
газхром А — промыт кислотой и отмыт до нейтральной реакции, газхром
Р промыт кислотой или спиртовыми основаниями и отмыт до нейтраль¬
ной реакции, газхром Z промыт кислотой и обработан диметилдихлорси-
ланом. Газхром Р и Z можно использовать с небольшими количествами
жидкой фазы (около 3%) для анализа стероидов, эфиров жирных кислот,
терпеновых соединений. Газхром R — аналог хромосорба Р и С-22.Экспериментально установлено, что носители первого типа имеют
поры размером от 0,4 до 2 мкм (среднее значение — около 1 мкм). Носите¬
ли второго типа имеют значительно более широкие поры — от 8 до 9 мкм.
При добавлении небольшого количества жидкости к твердому носителю
жидкость располагается в основном в тонких порах, что подтверждается
резким уменьшением удельной поверхности. Более гладкие части поверх¬
ности крупных пор могут остаться при этом практически непокрытыми.
Например, при нанесении 15% жидкой фазы около 12% жидкости находит¬
ся в порах и только 2—4% на остальной поверхности.Поверхность диатомитовых носителей имеет довольно высокую спо¬
собность к специфической адсорбции и хемосорбции. Для ее подавления
используют различные методы. Обычно считается, что некоторая дезакти¬
вация происходит при нанесении жидких фаз, особенно сильнополярных,
молекулы которых вступают в водородную связь с поверхностными гидрок¬
силами. Нанесение неполярных и слабополярных жидких фаз не приводит
к дезактивации поверхности. В этом случае иногда к неполярной жидкой
фазе добавляют небольшие количества полярных жидкостей, чаще всего
поверхностно-активных, которые блокируют наиболее активные участки
поверхности. Иногда для дезактивации носителя и получения симметрич¬
ных пиков газ-носитель насыщают летучими полярными веществами, в
частности, водой для разделения спиртов, муравьиной кислотой для раз¬
деления жирных кислот, аммиаком для разделения аминов и т. д. Все эти
вещества-дезактиваторы не регистрируются ионизационно-пламенным
детектором. Для анализа основных соединений, содержащих азот (аминов.116
ГЛАВА 3 Газо-жидкостная хуоматографиядиаминов, пиридинов, хинолинов, гуанидинов, эпоксисоединений и др.),
рекомендуется подвергать носитель щелочной обработке, например, при
разделении аминов проводят обработку раствором аммиака.При промывке носителя гидратом оксида щелочного металла в водном
или метанольном растворе подавляются, каталитические центры без за¬
метного уменьшения эффективности разделения. Иногда одной промывки
недостаточно, и твердый носитель дополнительно пропитывают щелочью.
В частности, рекомендован следующий метод обработки носителя: смачи¬
вают твердый носитель метанолом и смешивают эту массу в ротационном
испарителе с 6%-ным раствором КОН в метаноле в течение 1 ч (без на¬
гревания) , затем удаляют растворитель вакуумированием и просеивают
сухую массу. Наносят, как обычно, жидкие фазы, при этом исключается
применение веществ, которые могут реагировать с КОН.Промывка кислотами значительно улучшает свойства твердого носи¬
теля при разделении низших жирных кислот. При обработке кислотами
удаляется большая часть поверхностных примесей (А1, Fe, Са, Mg) и ак¬
тивность заметно уменьшается. Однако при некоторых разделениях про¬
мывка кислотой вредна, так как приводит к химическим превращениям
разделяемых веществ.Наиболее эффективным способом дезактивации поверхности носителя
является химическое модифицирование силанами, подобное химическому
модифицированию адсорбентов. Несмотря на значительное снижение спе¬
цифичности, химическое модифицирование все же не приводит к полной
инертности поверхности. Эго связано со стерическими препятствиями при
реакции модифицирования, мешающими образованию достаточно плотно¬
го слоя привитых к поверхности инертных групп.Дезактивацию можно проводить также путем отложения на поверхно¬
сти носителя твердых тел (как органических, так и неорганических). Но¬
сители можно покрывать также тефлоном, что приводит к значительному
уменьшению активности поверхности. Удобно использовать фторопласт
Ф-42П, растворимый в ацетоне. На диатомитовом носителе с 7% этого фто¬
ропласта получено хорошее разделение углеводородов до СЗО метиловых
эфиров жирных кислот и нитрилов С;—Cj,, а также спиртов нормального
и разветвленного строения.В таблице 3.20 приведены отечественные твердые носители, а в та¬
блице 3.21 самые известные зарубежные твердые носители на основе диа¬
томита.117
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я. Газовая хроматографияТаблица 3.20Характеристики отечественных твёрдых носителей на основедиатомитовX»Марка носителя111'Ь
35 аS iillIfspHводнойвытяжки||111
в ^1Сферохром-1 (ТНД-ТС-М)II0,430,8-2,18,80,6512Сферохром-2I0,5447,50,613Сферохром-3I0,610-15---4Динохром Н10,455-87,5-80,70,425Д^нохром ПII0,5-0,61-1,58-90,60,76Порохром 1II0,2-0,40,1-1,581,5-1,83,57Порохром 2II0,3-0,40,5-281,5-1,83,58Порохром 3II0,23-0,32-4-1,5-1,8-9Цветхром 1I0,4-0,61,5-3,56-70,6-10ЦветхромЗU0,4-0,60,5-0,96,5-80,5-Таблица 3.21Основные диатомитовые носители для газовой хроматографии№№Название,фирмаSd , НМcp’V, cmVfpHНасып¬
ной весг/см’ТипносителяМаксим.
% ж.ф.mVfм^/мл1.Хромосорб A2,71,37,10,40II22.Хромосорб G0,50,29-0,36-0,457,10,47II53.Хромосорб N10,292001,88,50,18II154.Хромосорб 7500,5-1,0--0,32II75.Хромосорб Р41,88400,96,50,38I306,Хромосорб Т7-80,427.Апакгош А1,0-1,4--8,0-9,50,28-0,31--8.Апакгою AS, Q----9.Gas chrom Q10.Gas chrom P, Z,
A, RA,RZ411.Supelcoport------12.Хроматон16001,3-1,49-100,24013.Инертон0,4-0,60,4-0,67,0-8,50,55II118
ГЛАВА 3 Газо-жидкостная хроматографияПолимерные твердые носители. Наиболее распространенным но¬
сителем этого типа является тефлон. Тефлон или политетрафторэтилен
(—CF2—CF2—), является карбоцепным кристаллическим полимером
с молекулярным весом то 500000 до 2000000. Преимущество тефлона
перед многими органическими и полимерами — довольно высокая тер¬
мостойкость (до 180—220 °С). Кристаллическая структура тефлона на¬
рушается при 327 °С, при этом он становится прозрачным и переходит в
эластичное состояние. Начиная с 350 °С, тефлон разлагается; следует об¬
ратить внимание, что при этом образуются сильно токсичные соединения,
в частности перфторизобутилен, который еще более ядовит, чем фосген.
Поэтому необходимо избегать нагревания тефлона до таких температур.
Обычно в тефлоне более упорядоченным кристаллическим ядрам сопут¬
ствуют волокнистые аморфные части. Пористый тефлон может иметь
удельную поверхность до 10 м^г. Его поверхность адсорбирует многие
соединения еще более неспецифически, чем поверхность графитирован-
ной термической сажи.При наполнении тефлоном колонок трудно добиться равномерной
плотности набивки, так как тефлон обладает плохой сыпучестью и быстро
слеживается. Лучшие результаты получаются при засыпке охлажденного
тефлона (ниже 0°С), так как при низких температурах сыпучесть его воз¬
растает. На пористый тефлон (s = 10 м^г) можно наносить не более 10%
жидкой фазы, на непористый — значительно меньше.Из промышленных образцов тефлоновых носителей следует выделить в
первую очередь пористые тефлоны хромосорб Т и полихром-1. В некоторых
случаях отечественный тефлон полихром-1 по эффективности превосходит
хромосорб Т. Кроме того, в продаже имеются политетрафторэтилены под
следующими марками: тефлон-1 (s = 23 м^г), тефлон-6 (s = 1,12 м^г) фир¬
мы Дюпон, галопорт F (s = 0,64 м^г) (П)лландия), хейдефлон, хостафлон
TF (ФРГ), флуоропак 80 и др. Кроме политетрафторэтилена применяют и
политрифторхлорэтилены, которые менее инертны и менее устойчивы к на¬
греванию (экафвин, кель F, хостафлон С2, галопорт К).Тефлоновые носители используют для разделения сильнополярных
веществ агрессивных веществ спиртов, жирных кислот, аминов, сероводо¬
рода и двуокиси серы, галогенпроизводных фосфора, и др.Из отечественных полимерных твердых носителей наиболее часто
применялся полихром-1 при анализе водных растворов спиртов и особен¬
но жирных кислот. К недостаткам этого носителя следует отнести низкую
механическую прочность, высокую электризуемость, трудность нанесения
более 10%; неподвижной фазы, а также слабую смачиваемость полярными
неподвижными фазами, что вызывает неравномерность их распределения119
Яшин Я.И., Яишн Е.Я.. Яишн А.Я.Гтовая Х1шшттфшна поверхности носителя. Лучше всего поверхность полихрома-1 смачи¬
вается метилсилишновыми и фторированными жидкостями. Специальная
обработка полихрома-1 и фторопласта-4 улучшает смачиваемость поверх¬
ности носителя и повышает эффекгивность колонки. Полихром-1 легко
комкуется. Некоторые характеристики полимерных твердых носителей
приведены в таблице 3.22.Таблица 3.22Хараю-еристики полимерных носителей№№МаркиносителейНа¬сыпи.плот¬ность,г/см*Удельнаяповерх¬ность,MVrРазмерчастиц,ммОбщийобъемпор,см’/гХромосорб - Т
(J-M. США)0,427—80,25—0,42Фторопласт 4Д0,328—9100.000-140.(Ю00,25-0,50,30320до 30Попихром -10,685,60,25-0,50,25-0,3028020Попхром -20,696,6то же0,25-0,5 0,20-0,25 20020Кроме тефлона в качестве твердого носителя в газовой хроматогра¬
фии используются и другие пористые полимеры, в частности полимеры на
основе стирола и дивинилбензола.Они имеют большие удельные поверхности. Из промьшшенных образ¬
цов минимальными значениями поверхностей обладают хромосорб-101,
тенакс (19-20 м%) и хромосорб-103 (30—50 м^/г). Эти пористые полиме¬
ры по химической природе поверхности слабоспецифические. Для многих
классов соединений их считают практически неспецифическими.Стеклянные шарики. На непористые стеклянные шарики мшшо на¬
нести однородную пленку жидшй фазы, что позволяет повысить эффектив¬
ность разделения на них. В отличие от диэтомитовых носителей, стеклянные
шарики обладают малой адсорбционной и каталитичесшй активностью. Из-
за малой удельной поверхности стеклянных шаришв (ошло 0,01 м^г) на
них можно нанести тольш небольшие шличества жидшй фазы. Максималь¬
ное шличество жидшй фазы зависит от диаметра шаришв, поверхностного
натяжения и плотности жидшй фазы и изменяется в пределах 0,05—2%. На
ШЛ0Н1ШХ с таким сорбентом легш достигается высота теоретичесшй тарел¬
ки 0,5 мм. Такая высокая эффективность не уменьшается с повышением ли-120
ГЛАВА 3 Газо-жидтстная хроматографиянейной сюрости газа-носителя, так как массообмен в тонкой и однородной
пленке жидкой фазы происходит быстро.Благодаря малому количеству жидкой фазы и возможности примене¬
ния высоких линейных скоростей газа-носителя на колонке со стеклянны¬
ми шариками удерживаемые обьемы получаются значительно меньшими,
чем на колонках с диатомитовыми носителями с большими шличествами
жидкой фазы. Это обстоятельство позволяет уменьшить температуру раз¬
деления. В работах [73] показано, что в некоторых случаях можно прово¬
дить разделение при температурах на 250°С ниже температуры кипения
компонентов анализируемой смеси. Это позволяет, во-первых, разделять
термически неустойчивые соединения при пониженных температурах и,
во-вторых, .применять менее термически стабильные жидкие фазы.Но стеклянные шарики имеют и серьезные недостатки. Малая сорб¬
ционная емкость колонок со стеклянными шариками вынуждает использо¬
вать малые пробы. Таким образом, хорошие результаты на них можно по¬
лучить только при использовании высокочувствительных детектирующих
систем.Чтобы увеличить емшсть на внешней поверхности стеклянных ша-
ришв создают пористый адсорбционный слой путем травления специаль¬
ного стекла кислотами и щелочами или же путем отложения (спекания)
тоншдисперсных частичек на поверхности стеклянных шариков. На таких
сорбентах сохраняется высокая эффективность и одновременно повыша¬
ется емшсть. Для подавления адсорбционной активности стеклянные ша¬
рики можно химически модифицировать силанами.Пористые стекла. Пористое стекло — твердый носитель Coming
серии GLC-100 — химически модифицировано диметилдихлорсиланом
и предназначено для эффективного разделения смеси стероидов. Поверх¬
ность его инертна, холестерин выходит в виде симметричного пика. На нем
легко получается эффективность порядка тысячи тарелок на длине в один
фут (т.е. 3000 тарелок на 1 м). На пористое стекло рекомендуется наносить
жидкие фазы в пределах от 0,1 до 0,35%. По сравнению с диатомитовыми
носителями пористое стекло имеет значительно большую плотность (в
5 раз)—1,35 г/см’. Поэтому шличество жидкой фазы 0,1% от его массы
эквивалентно 0,55% жидкой фазы на диатомитовых носителях.В качестве твердого носителя в газо-жидкостной хроматографии мо¬
гут быть использованы также макропористые силикагели и графитирован-
ные сажи.В литературе описано применение различных материалов в качестве
твердых носителей, в частности детергентов, хлорида натрия, полиэтиле¬
на, металлических спиралей, черепицы, вермикулита и др.121
Яшт ЯМ. Яшин ЕЯ. Яшин А.Я. Газовая хрататография3.12 Способы нанесения жидких фаз на твердые носителиЭффективность газо-жидкостных колонок в сильной степени зависит
от распределения жидкой фазы на поверхности твердого носителя (от од¬
нородности жидкой пленки). Однородность распределения жидкой фазы
на поверхности носителя зависит не только от смачиваемости, количества
жидкой фазы, но и от способа нанесения жидкой фазы. В настоящее время
доступны шммерческие сорбенты с жидкой фазой и даже заполненные ко¬
лонки. Однако необходимо рассмотреть все описанные способы нанесения
жидких фаз и деть оценку каждому способу.Количество жидкой фазы обычно измеряют в процентах от массы
твердого носителя.В наиболее распространенном способе определенное количество жид¬
кой фазы растворяют в легколетучем растворителе, раствор жидкой фазы
выливают в чашку с твердым носителем. При непрерывном перемешива¬
нии легколетучий растворитель выпаривают на закрытой электроплитке
(лучше на водяной бане). Когда растворитель полностью удален, оконча¬
тельно высушивают твердый носитель с нанесенной жидшй пленшй под
инфракрасной лампой. Этот метод очень прост и удобен, однаш он имеет
недостатки. Во-первых, при перемешивании, если твердый носитель об¬
ладает небольшой механичесшй прочностью, происходит измельчение ча¬
стичек твердого носителя, во-вторых, несмотря на перемешивание, одно¬
родного заполнения трудно достичь, так как в шнце выпаривания (особен¬
но когда раствор становится вязким) шнцентрация в верхней части всегда
несшльш выше, чем в гаубине слоя.Этих недостатшв не имеет следующий способ, в котором раствор
жидшй фазы определенной шнцентрации непрерывно медленно фильтру¬
ется через неподвижный слой зерен твердого носителя. В этом случае зер¬
на твердого носителя не подвергаются механическому перемешиванию,
шнцентрация жидшй фазы одинашва во всем объеме слоя зерен твердого
носителя. При медленном прохождении раствора происходит адсорбция
молекул жидшй фазы на поверхности твердого носителя практически в
равновесных условиях. Количество адсорбированной жидшй фазы на
твердом носителе будет зависеть в основном от шнцентрации раствора
(при одинашвых прочих условиях). Меняя шнцентрацию раствора, можно
изменять шличество жидшй фазы, наносимой на твердый носитель. Сор¬
бенты, приготовленные таким способом, обычно имеют большую эффек¬
тивность, чем сорбенты, приготовленные вторым способом. Единственный
недостаток этого способа — большой расход жидкой фазы.Хорошие условия для однородного отложения жидюй фазы создаются
также в способе с использованием кипящего слоя, предложенном В. Г. Берез-122
ГЛАВА 3 Газо-жидтстная хгмшатографткиным. Определенное количество твердого ноаггеля залнвжтся раствором
жидшой фазы на фильтр» (или на спегшальном устройстве), снизу подается
поток нагретого газа-носитеп*. С помощью пузырьков газа-носшетя проис¬
ходит интенсивное перемешивание зерен носителя во взвешенном состоянии
и испарение легшлетучего растворителя. Однаш метод нанесения жидшй
фазы в кипящем слое приводит к значительньш механическим разрушениям
при использовании очень мелнспернистых или механически непрочных носи¬
телей.Во всех способах полезно раствор жидшй фазы выливать на предвари¬
тельно вакуумированный носитель или же вакуумировать непосредствен¬
но в процессе пропитки (исследования А. А. Жуховицшго). Если пропиты¬
вать без вакуумирования, то в порах могут оставшъся маленькие пузырьки
воздуха, что приводит к неоднородному отложению жидшй фазы на по¬
верхности твердого носителя.Нанесение жидшй фазы на твердый носитель можно проводить непо¬
средственно и в хроматографичесшй шлонке. Нанесение небольших ко¬
личеств (порядка емшсти монослоя) можно проводить путем дозирования
паров жидких фаз в колонку, заполненную зернами сухого твердого но¬
сителя, при условии, если жидкую фазу можно перевести в парообразное
состояние без разложения.Большие шличества жидкой фазы можно нанести при пропускании
через прямую шлонку раствора жидшй фазы до тех пор, пока шнцентра-
ция жидшй фазы на выходе из шлонки не станет равной шнцентрации на
входе в шлонку.3.13 Заполнение и тренировка колонкиПроцесс заполнения наиболее распространенных U-образных коло¬
нок очень прост. К обоим концам шлонки подсоединяют воронки (обычно
имеются в комплекте хроматографа), через которые небольшими порци¬
ями засыпают сорбент. В процессе заполнения необходима постоянная
вибрация или постукивание, если отсутствует специальный вибратор ко¬
лонки. С помощью вибрации (около двадцати колебаний в 1 с) происходит
значительно более плотная упашвка зерен сорбента.Спиральные колонки таким путем заполнить нельзя. Их нужно или вы¬
прямить, заполнить как U-образные и затем снова скрутить в спираль или
заполнять зернами сорбента под давлением. При скручивании возможно
повреждение зерен сорбента или образование зон с неплотной набивкой.
Для заполнения под давлением закрывают один конец колонки металлоке¬
рамическим фильтром, к открытому шнцу подсоединяют патрон, можно
трубку большего диаметра с зернами сорбента, и затем под давлением за¬
полняют колонку зернами сорбента.123
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я. Газовая хроматографияВ Процессе эксплуатации колонки за счет выдувания пылинок и дру¬
гих факторов набивка уплотняется и в верхней части U-образных верти¬
кальных колонок образуются пустоты, которые можно легко заполнить
дополнительно набивкой. В спиральных колонках пустоты образуются в
объеме колонки, что приводит к уменьшению эффективности.После заполнения колонки сорбентом, прежде чем проводить хрома¬
тографические измерения, необходима ее тренировка. Тренировга (заклю¬
чается в продувке колонки инертным газом-носителем при определенной
темперетуре. ^лательно, чтобы температура продувки была на 20—30°С
выше рабочей температуры. Лучше продувку проводить сразу же при тем¬
пературах близких к максимально допустимой рабочей температуре (при¬
мерно на 10—20°С ниже МДРТ), чтобы удалить все легколетучие примеси
из жидкой фазы. Тренировка колонки может продолжаться один-два дня.Нельзя проводить количественные измерения по первым же хромато¬
граммам, так как происходит процесс насыщения сорбента, в результате
чего параметры удерживания и высота пиков изменяются. Нужно сделать
несколько пробных дозировок. Обычно после пяти-десяти пробных хро¬
матограмм сорбент насыщается и можно проводить количественные из¬
мерения. При переходе к более высоким температурам процесс насыще¬
ния нужно повторить. Необходимость этого подтверждается измерениями
в режиме программирования температуры. Если перейти к режиму про¬
граммирования после длительной работы шлонки в изотермичесшм ре¬
жиме, то могут появиться «ложные пики» за счет весьма малой остаточной
сорбции анализируемых веществ.124
ГЛАВА 3 ЛитератураЛитература.1. James А.Т., Martin А. J.P.Biochem J., 1952, v.50, p.679-690.2. Horape C. Д., Джувет P.C. Газо-жидшстаая хроматография. Теория
и практика. Пер. с англ. Под ред. А.Н. Александрова, М.А. Дементье¬
вой. Л., «Недра», 1966,420 с.3. Pumell Н. Gas Chromatography, N.,Y.,L., John Wiley a. Sons, 1962,
354 p.4. Persinger H.E., Shank J.T. The chemistry of polyethylene glycols used
in gas Chromatography. J.Chromat. Sci. 1973, v.ll,p.l90.5. Король А.Н. Неподвижная фаза в газо-жидкостной хроматогра¬
фии. Киев, «Н|^аа}ва думка», 1969,250 с.6. Anderson D.G., Ansel R.E. Polyester liquid phases in gas chromatog¬
raphy. J.Chromat. Sci. 1973, v.ll3, p.l92.7. Gas Chromatography. Applied Science Laboratories Inc. News Letters
N7, April, 1964.8. Trash C.R. Methylsilicones - their chemistry and use as gas chro¬
matographic liquid phases. J.Chromat. Sci. 1973, v.ll, p. 195.9. Coleman A.E. Chemistry of liquid phases - other silicones. J.Chromat.
Sci. 1973, v.ll, p.l98.10. Rotzsche H., Stationary phases in gas chromatography, Elsevier, Am¬
sterdam, 1991.11. Baiulesu G.E., Hie V.A. Stationary phases in gas chromatography, Per-
gamon Press, Oxford, 1975.12. Курс физической химии. Под ред. Я.И. Герасимова в 3-х томах.
Т.1. Москва «Химия», 1969,614 с.13. Пиментел Дж., Мак-Клеллан О. Водородная связь. Москва, «Мир»,
1964,390 с.14. Березкин В.Г. Газо-жидкотвердофазная хроматография. Москва,
«Химия», 1986.15. Martin R.L. Anal. Chem. 1961, v.33, р.347; 1963, v.35, p.116.16. Gas Chromatography Retention Data, by W.l O. McReynolds, Preston
Technical Abstracts Company, 1966, 333 p.17. Яшин Я.И. Ж. аналит. химии, 1985, т.40, с.201-214.18. Littlewood А.В. Gas Chromatography-Principles, Techniques and Ap¬
plications, N., Academic Press, 1962,450 c.19. Байф Э. Хрш1агог1К|фия газов. Пф. с нем.. М., Издапшлит, 1 %1,279 с.20. Vigdergauz M.S., Bonkovskaya T.R. The Choice of Preferred Station¬
ary Phases for Gas Chromatography. Chromatographia, 1976, v.9, №11,
p.548.125
Яшин Я.И., Яшин Е.Я.. Яшин А.Я. Газовая хроматография21. Накеп J.K. Stationary phases and Applicability of Classifications and
Preferred Phase schemes to Fatty Esters. J.Chromat. Sci. 1975, v.l3, p.430.22. Leibrand R.J. Preferred Stationary Liquids for Gas Chromatography/
Mass Spectrometry. J.Chromat. Sci, 1975, v.l3, p.556.23. Hawkes S. et al. Preferred Stationary phase for Gas Chromatography.
J.Chromat. Sci 1975, v.l3, p.lI5.24. Marm J., Preston S.T. Selection of Preferred liquid phases. J.Chromat.
Sci. 1973,v.ll,p.216.25. Keller R.A. Stmdard liquids phases - Pandora’s Box Revisited.
J.Chromat. Sci. 1973, v.ll,p.l88.26. Muir A. LC-GC Intern. 1997, v. 10, p.250.27. Piligrim G.W., Keller R.A. Mixed Stationary Phases. J.Chromat. Sci.
1973,vll,p.206-209.28. Rohrschneider L. J.Chromat. Sci. 1963, v.32, p.2-7.29. McReynolsd W.O. J. Chromat. Sci. 1970, v.8 p.685-691.30. McReynolsd, Gas Chromatographic Retention Data. Preston Techni¬
cal Abstract Co. Evanston IL, 1966.31. Castello G. J.Chromatogr. 1999, v.842, p.51.32. Rohrschneider L. J.Chromat. Sci. 1973, v. 11, p. 160.33. Schomburg G., et al Chromatographia, 1982, v.l5, p.599.34. Карцова Л.А., Маркова О.В. Ж. аналит. хим. 1999, т.54, с.357.35. Yu X.D., Lin L., Wu C.Y. Chromatographia, 1999, v.49, p.567.36. Spanik I., Krupcik J. et al. J. High Resolut. Chromatogr. 1997, v.20, p.688.37. Shi X., Fu R. Fenxi Huaxue. 2000, v.28, p. 118.38. Kou D., Hou L„ Lu X., Sepu, 1999, v.l7, p.257.39. Березкин В.Г. Русск. хим. бюл. 1999, т.48, с. 1807.40. Fang P.-F. et ol. J. Chromatog.2000, v.867, p. 177.41. Poole, The Essence of Chromatography, Elsevier, Amsterdam.42. Zhang J. et ol. Fenxi Huaxue. 1999, v.27, p.85.43. Naikwadi K.P., Wadgaonkar P.P. J.Chromatogr. 1998, v.811, p.97.44. Lee W.S., Chang-Chien G.R Anal. Chem. 1998, v70, p.4094.45. Naikwadi K.P., Wadgaonkar P.P. Organohalogen Compd.1997, v.31,
p.268.46. Leng Z.R., Liu M. Chromatographia 1998, v.48, p.817.47. Yu X.D., Lin L., Wu C.Y. Chromato^aphia 1999, v.49, p.567,48. Zhanga L.F. et al. J.Chromatogr. 1999, v.840, p.225.49. Yan C. et al. Fenxi Huaxue 1999, v.27, p.77.50. Карцова Л.А., Маршва О.В. Ж. аналит. химии, 1999, т.54, с.227.51. Карцова Л.А., Маркова О.В., Амельченко А.И., Острянина Н.Д.Ж.
аналит. химии 2000, т.55, с.302.126
ГЛАВА 3 Литература52. Gross В., Jauch J., Shimg V.J. Microcolmnn Sep. 1999, v.ll, p.313.53. Zeng Z.R. et al. Chromatographia, 1999, v.49, p.293.54. Rotzsche H. Chromatographia, 1997, p.269.55. Juvancz Z.et al. Enantiomer, 1998, v.3, p.89.56. Wawzzyniak R., Wasiak W. Chromatographia 1999, v.49, p.273.57. Kowalski W.J. Chem. Anal. (Warsaw) 1998, v.42, p.69.58. Chuen-Ying L. et al. J. Chromatogr. 2001, v.933, p.ll7.59. Malik A., Wong D. World patent 2(Ю0011463, 2001.60. Cervini R. et al. Chem. Anal. 2001, v.68, p.l6.61. Zeng Z. et al. Anal. Sci. 2000, v. 16, p.851.62. Schurig V.J. Chromatogr. 2002, v.965, p.315.63. Wong C.C. et al. Proc. ASME Fluid Eng. Div. 2000, v.251, p. 1703.64. Seholten A.B. et al. J.High Resolut. Chromatogr. 1997, v.20, p. 17.65. Malek A. et al. Chromatographia 1997, v.46, p.79.66. Lai G. et al. Chromatographia 1991, v.32, p.241.67. Pereira A.S., de Aquino Neto F.R. J.Chromatogr. Sci. 2002, v.38,
p.369.68. Aichholz R., Lorbeer E. J.Chromatogr. 2000, v.883, p.75.69. Pereira A.S.et al. J.Chromatogr. 2002, v.947, p.255.70. Grob K., Grob G. J. High Resolut. Chromatogr. 1983, v.6, p. 153.71. Ottenstein D.M. «Advances in Chromatography» Ed. J.C. Giddings,
R.A. Keller. N.Y. Marcel Denker 1966, v.3, p. 137-185.72. Березкин В.Г., Сашдынский К.И., Пахомов В.П. Твердые носите¬
ли для газовой хроматографии. Москва, «Химия». 1972.73. Hishta С. Analyt. Chem. 1960, v.32, р.880; р.1735.127
Яшт ЯМ. Яшт Е.Я.. Яшт А.Я. Гамвая хтшатографияГЛАВА 4. ГАЗО-АДСОРБЦИОННАЯ ХРОМАТОГРАФИЯ4.1. Преимущества и нсдосгатки газо-адсорбционной хроматографии.Газовая хроматография как метод анализа начала быстро развивать¬
ся с 1952 года, когда Джемс и Мартин [1] предложили газо-жидкостный
вариант хроматографии. Преимущества газожидкостного метода перед
газо-адсорбционным в шестидесятые годы объясняются, во-первых, воз¬
можностью широкого выбора различных по химичесюму строению не¬
подвижных жидких фаз, пригодных для разных аналитических задач, и,
во-вторых, высокой чистотой и постоянством состава жидкостей, благо¬
даря чему в широкой области рабочих концентраций изотермы сорбции
практически линейны. Выбор же адсорбентов с поверхностями различного
химического состава среди выпускаемых промышленностью адсорбентов
ограничен, кроме того эти адсорбенты геометрически и химически неод¬
нородны. Однако с расширением областей применения и развитием аппа¬
ратуры для газохроматографического анализа, в частности с повышением
чувствительности ионизационных детекторов, повышением температур
работы хроматографов, а также с ростом применения газовой хроматогра¬
фии для автоматического непрерывного контроля состава смесей в про¬
мышленности и для анализа микропримесей, выявились существенные
недостатки газо-жидкостной хроматографии. Это прежде всего летучесть
и нестабильность жидких фаз, затрудняющие анализ микропримесей, а
анализы при высоких температурах и с программированием температуры;
за счет увеличения фонового сигнала, в препаративной хроматографии эти
недостатки способствуют загрязнению выделенных веществ [2-5].Газо-адсорбционный метод на неорганических и углеродных адсор¬
бентах этих недостатков не имеет. Основным его недостатком является
лишь нелинейность изотерм адсорбции, приводящая к несимметричности
пиков. Нелинейность связана с геометрической и химической неоднород¬
ностью поверхности обычных промышленных адсорбентов. Особенно
резко она проявляется в случае сильно адсорбирующихся молекул. Не¬
однородность и высокая адсорбционная, а иногда и каталитическая актив¬
ность обычных адсорбентов ранее ограничивала их применение в газовой
хроматографии. Поэтому такие адсорбенты ранее применялись и сейчас
применяются в основном лишь для анализа газообразных веществ, несо¬
держащих активных функциональных групп, изотермы адсорбции кото¬
рых при используемых в хроматофафии концентрациях и температурах
близки к линейным. Среди первых ранних работ по газо-адсорбционной
хроматофафии следует отметить работы 1947—1954 гг., в частности ра¬
бот Клессона, Филлипса, Туркельтауба, Вяхирева, Кремер, Янака и Рэя.128
ГЛАВА 4 Гаэо-адсорбционная хроматографияГазо-адсорбционный метод хроматографии до начала шестидесятых годов
рассматривали как метод, дополняюший газо-жидкостную хроматографию
для разделения газов и паров низкокипяших вешеств, так как в этом случае
разделительная способность жидких фаз вследствие малой растворимости
газов недостаточна [2]Кроме термической стабильности адсорбенты имеют и другие преиму¬
щества. Как показал Гиддингс [6], массообмен в газо-адсорбцнонных ко¬
лонках может происходить значительно быстрее, чем в газо-жидкостных,
что позволяет проводить быстрые разделения на эффективных колонках.
Вследствие большого влияния геометрической структуры молекул на энер¬
гию их адсорбции в газо-адсорбционной хроматографии имеются большие
возможности получения селективных колонок не только на основе разли¬
чий в электронной структуре молекул разных компонентов, но и на основе
различий в их геометрии при адсорбции на поверхности адсорбентов.Таким образом, в газо-адсорбционной хроматографии имеются боль¬
шие возможности получения колонок с высокой разделительной способ¬
ностью и проведения экспрессных анализов. Для реализации этих возмож¬
ностей необходимо резко снизить дополнительное размывание, связанное
с различными временами нахождения адсорбирующихся молекул на раз¬
ных участках поверхности твердых адсорбентов, т. е. с химической и ге¬
ометрической неоднородностью твердых адсорбентов. Этого можно срав¬
нительно легко добиться путем разработки непористых и макропористых
адсорбентов с однородной поверхностью.В литературе описано много способов получения симметричных пи¬
ков (благодаря выпрямлению изотерм адсорбции) в газо-адсорбционной
хроматографии путем улучшения адсорбентов [2-5] Все эти способы свя¬
заны с получением геометрически и химически однородных, чаще всего
макропористых адсорбентов, либо с геометрическим, химическим или
адсорбционным модифицированием неоднородных адсорбентов, либо с
применением сильно адсорбирующихся газов-носителей, в частности во¬
дяного пара [3].Газо-адсорбционный метод может быгь реализован и на высокоэффек¬
тивных капиллярных шлонках, имеющих на внутренней стенке пористый
слой адсорбента. Создать достаточно сильно адсорбирующий пористый
слой можно или обработкой внутренних стенок капилляра, изготовленно¬
го из соответствующего материала (например, боросиликатного стекла),
раствором щелочей [7], или же отложением на внутренней поверхности
капилляра слоя тонкодисперсного адсорбента. Такие колонки обычно име¬
ют высокую разделительную способность. Эти колонки под названием Ро-
raPlot нашли в последнее время широкое применение [8].1299 Газовая хроматография
Яишн Я.И.. Яишн Е.Я.. Яшин А.Я. Гюошы хроматографияВ последние годы налажен серийный выпуск однородных адсорбентов
для газовой хроматографии, в частности пористых полимеров разной при¬
роды, макропористых силикагелей (силохромов, порасилов, сферосилов),
пористых стекол, углеродных молекулярных сит, карбохромов и цеолитов.
Благодаря этому сфера применения газо-адсорбционной хроматографии
значительно расширилась не только в традиционных областях низких и
высоких температур, но и в области средних темперагур, например, для
анализа изомеров, агрессивных и неустойчивых вешеств, смесей сильно¬
полярных веществ, в частности водных сред [2-5].4.2. Влияние химии поверхности адсорбентов на удерживаемые
объемы и селективность разделения. Неспецифические и специфиче¬
ские межмолекулярные взаимодействия.При применении непористых или макропористых адсорбентов геоме¬
трическую неоднородность поверхности можно в значительной степени
устранить. В этих случаях основное влияние на удерживающую способ¬
ность и на селективность газо-адсорбционных колонок будет оказывать
химия поверхности адсорбента. Химия твердого тела определяет характер
и энергию межмолекулярного взаимодействия, возникающего между мо¬
лекулами разделяемых веществ и твердым телом. При адсорбции в зависи¬
мости от химической природы молекул и поверхностей могут проявляться
различные, взаимодействия.Для целей систематизации различных видов межмолекулярных взаимо¬
действий с учетом их вкладов в удерживаемые объемы в газовой хромато¬
графии целесообразно их разделить на два типа: неспецифические и спе¬
цифические. Неспецифическое, в основном дисперсионное взаимодействие
универсально, оно проявляется между любыми молекулами. Специфичесше
же взаимодействие, в основном ориентационное, вызывается особенностя¬
ми локального распределения электронной плотности во взаимодействую¬
щих молекулах. Эти особенности связаны с локальным концентрированием
отрицательного и положительного зарядов на отдельных связях или звеньях
специфически взаимодействующих молекул. Водородная свжзь представля¬
ет собой частный случай таких специфических, но еще межмолекулярных
взаимодействий. Такое подразделение взаимодействий в известной степени
условно. Однако оно помогает систематизации разрозненных фактов и по¬
зволяет дать им удобную качественную классификацию [2-4].4.3. Классификация молекул и адсорбентов по характеру межмо¬
лекулярных взаимодействийПо типу межмолекулярных взаимодействий можно выделить и объ¬
единить некоторые типичные, качественно сходные виды взаимодействия,
связав их с электронной структурой связей и звеньев молекул. Соответ¬130
ГЛАВА 4 Газо-адсоабиионшгя хтшатогтгфияствующая классификация для молекулярных (нехимических) взаимодей¬
ствий была предложена в работе [9]. В соответствии с различным характе¬
ром распределения электронной плотность на периферии связей и звеньев
молекулы удобно подразделить на следующие четыре группы; А, В, С и D
(табл. 4.1).Молекулы группы А. Прежде всего следует выделить самый простой
случай, а именно: молекулы со сферически симметричной электронной
оболочкой, как у благородных газов. Эти молекулы, сохраняя свою инди¬
видуальность (т.е. не изменяясь химически), могут взаимодействовать с
любыми другими молекулами только неспецифически, в результате про¬
явления в основном универсальных дисперсионных сил, связанных с со¬
гласованным движением электронов во взаимодействующих молекулах, а
не с особенностями локального распределения в них электронной - плот¬
ности, вызванного строением входящих в молекулу атомов и характером
химических связей между ними. К этой группе молекул следует отнести
также и молекулы насыщенных углеводородов, где между атомами углеро¬
да имеются только о-связи. У этих молекул нет вытянутых орбит, т.е. на их
периферии нет локально сосредоточенной электронной плотности.Молекулы группы В. К этой группе относятся молекулы с сосредо¬
точенной на периферии отдельных звеньев электронной плотностью (от¬
рицательным зарядом). В случае ненасыщенных и ароматических углево¬
дородов и вообще молекул, имеющих л-электронные связи, электронная
плотность локально сконцентрирована на периферии этих связей. Локаль¬
ное сосредоточение электронной плотности имеет место также и на пери¬
ферии тех звеньев функциональных групп, которые обладают свободными
электронными парами, например у атомов кислорода в воде, спиртах, эфи¬
рах, кетонах, у атомов азота в аммиаке, аминах, нитрилах и у атомов серы
в органических сульфидах.131
Яшт Я.И.. Яшт Е.Я.. Яшт А.Я. Газовая хроматографияТаблица 4.1Классификация молекул и адсорбентов по их способности к неспеци¬
фическим в специфическим межмолекуляриым взаимодействиямМолекулыАДС0|^НТЫТип I-не несущие ионов или
активных групп (гра-
фитированные сажи,
BN, поверхности,
несущие только насы¬
щенные группы)Тип П-несущие локально
сконцентрированные
положительные заря¬
ды (кислые ГИД1ЮКСИ-
лы, обменные 1штио-
ны малого радиуса)Тип III-несупше локально
сконцентрирсшанные
отрицательные з^яды
(эфирные, нитрильные,
карбонильные и ;^^тие
группы или обменные
анионы малого ра-
диуса) Группа А— со сферически
симметричными оболочками
или ст-связями (благородные
газы, насыщенные
углеводороды)Неспещ|фические взаимодействия определяемые в основном
дисперсионными силамиГруппа В— с электронной
плотностью, локально
сосредоточенной на
периферии отдельных
связей или звеньев; п-связи
(Ni, ненасыщенные и
ароматические углеводороды)
и свободные электронные
пары (эфиры, кетоны.
третичные амины, нитрилы
и т. п.)Группа С— с положительным
зарядом, локально
сконцентрированным
на периферии звеньев
(например, нешгорые
металлоорганические
соединения)НеспецифическиевзаимодействияГруппа D — сфункциональными группш^,
в которых на периферии
соседних звеньев локально
сконцентрированы как
электронная плотность, так
и положительный заряд
(молекулы с группами ОН
иНН)Неспецифические специфические
взаимодействияНаличие локально-сконцентрированной электронной плотности на
периферии отдельных связей или звеньев таких молекул создает возмож¬
ность проявления, наряду с универсальным неспецифическим взаимодей¬
ствием, также и более специфического. Это специфическое, но еще мо¬
лекулярное (нехимическое) взаимодействие может осуществиться, однако.132
ГЛАВА 4 Газо-адсорбиионная хроматографияТОЛЬКО в том случае, если другой партнер, вступающий во взаимодействие,
обладает сосредоточенным положительным зарядом, например перифери¬
ческими атомами водорода кислотного типа или выдвинутыми на перифе¬
рию катионами, особенно обменными катионами, малых радиусов.В молекулах воды, спиртов, первичных и вторичных аминов атомы
кислорода и азота со свободными электронными парами входят в функци¬
ональные группы ОН, NHj и NH вместе с частично протонизированными
атомами водорода, которые также обладают способностью к специфиче¬
скому взаимодействию.. Это осложняет молекулярные взаимодействия та¬
ких молекул и, в частности, создает возможность взаимной их ассоциации.
Поэтому целесообразно в группу В включить лишь такие молекулы, кото¬
рые обладают только связями или звеньями с локально сосредоточенной
на их периферии электронной плотностью, т. е. тс-электронными связями
(молекулы азота, ненасыщенных и ароматических углеводородов) и сво¬
бодными электронными парами (молекулы эфиров, кетонов, третичных
аминов, нитрилов, сульфидов). С молекулами группы А такие молекулы
взаимодействуют неспецифически.Молекулы группы В, в отличие от молекул группы А, способны к
специфическому взаимодействию с сосредоточенным на периферии дру¬
гих молекул или поверхностей положительным зарядом: например, они
способны к образованию водородной связи с гидроксильными группами
и аминогруппами. Однако они не способны к сильному специфическому
взаимодействию друг с другом, хотя присущие им диполи или квадруполи
могут взаимодействовать, внося некоторый вклад в общую энергию взаи¬
модействия (в дополнение к неспецифическому взаимодействию).Молекулы группы С. Сюда следует отнести молекулы, обладающие
локально сконцентрированным положительным зарядом в звеньях малых
размеров в том случае, если соответствующий избыток электронной плот¬
ности рассредоточен на соседних звеньях молекулы (т. е. если соответству¬
ющая молекула не обладает смежным звеном с сосредоточенной на пери¬
ферии электронной плотностью, как в группах ОН, NH^ и NH). К таким
молекулам, вероятно, относятся молекулы многих металлоорганических
соединений. С молекулами группы А такие молекулы взаимодействуют
неспецифически. С молекулами же группы В они должны взаимодейство¬
вать специфически.Молекулы группы D. К этой группе целесообразно отнести молеку¬
лы, обладающие соседними звеньями малого размера, в одном из которых
сконцентрирован положительный заряд, а на периферии другого — элек¬
тронная плотность, например молекулы с функциональными группами
ОН, NHj и NH. Сюда относятся молекулы воды, спиртов, первичных и вто¬133
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я. Гтовая хроматографияричных (но не третичных) аминов. Молекулы этой группы взаимодейству¬
ют с молекулами группы А неспецифически и могут взаимодействовать
специфически как с молекулами групп В и С, так и друг с другом.Многие функциональные группы обладают сложным строением. На¬
пример, карбоксильные группы или группы сложных эфиров, в которые
входят гидроксильный или алкокснльный и карбонильный кислород, каж¬
дый из которых способен к специфическому взаимодействию.Во многих молекулах и макромолекулах присутствует несколько раз¬
общенных звеньев одного или разных типов, способных к специфическо¬
му молекулярному взаимодействию, как в аминоспиргах, аминокислотах
и аминоэфирах.Неспецифическое взаимодействие проявляется во всех случаях. Спе¬
цифическое молекулярное взаимодействие проявляется как дополнитель¬
ное к неспецифическому и только в тех случаях, югда молекулы взаимо¬
действующих партнеров относятся к группам В, С и D.Особый интерес представляет взаимодействие молекул с сильно по¬
ляризующимися звеньями и связями, а также молекул, обладающих сопря¬
женными связями, как, например, дивинил, многие производные бензола,
полиядерные ароматические соединения, гетероциклы типа пиридина, фу-
рана, тиофена и т. п. молекулы.Рассмотрим теперь взаимодействия молекул выделенных четырех
групп с различными адсорбентами. Поверхности адсорбентов удобно рас¬
смотреть с этой же точки зрения как соответствующие партнеры межмоле-
кулярного взаимодействия. По этому признаку, адсорбенты так же удобно
подразделить на несколько основных типов с различным характером рас¬
пределения заряда на поверхности [2—5].I тип — неспецифические адсорбенты. Поверхность таких адсор¬
бентов не несет ни функциональных групп, ни обменных ионов. Графити-
рованная сажа и нитрид бора (BN), насыщенные углеводороды, в частно¬
сти полимерные (например, полиэтилен), являются такими неспецифиче¬
скими адсорбентами. Они взаимодействуют в основном неспецифически с
молекулами всех четырех выделенных выше групп.II тип — специфические адсорбенты. На поверхности адсорбентов
этого типа сосредоточенны положительные заряды. Эти адсорбенты вза¬
имодействуют специфически с молекулами, имеющими звенья или связи
с локально сконцентрированной на их периферии электронной плотнос¬
тью (с молекулами групп В и D). Следовательно, это, во-первых, адсор¬
бенты, несущие на своей поверхности гидроксильные группы кислотного
характера, например гидроксилированные поверхности кислых окислов,
в частности кремнезема. Вакансия d-оболочки атома кремния приводит в134
ГЛАВА 4 Газо-адсорбиионная хроматографияЭТОМ случае к соответствующему смещению электронной плотности, что
влечет за собой частичное протонизирование атомов водорода гидроксиль¬
ных групп поверхности кремнезема [3]. Подобное распределение заряда
возможно, однако, и в отсутствие кислотных гидроксильных групп. Для
этого важно, чтобы положительный заряд был выдвинут наружу и со¬
средоточен в частице малого радиуса, а отрицательный распределен по
значительно большему объему. Поэтому ко II типу следует отнести также
адсорбенты, на поверхность которых выдвинуты апротонные кислотные
центры или катионы малого радиуса, а компенсирующий отрицательный
заряд распределен по внутренним связям большого комплексного аниона.
Примером таких адсорбентов являются, в частности, цеолиты, в которых
положительный заряд сосредоточен в обменных катионах, а отрицатель¬
ный распределен по внутренним связям больших комплексных анионов
цеолитного каркаса [3,4].Таким образом, ко II типу относятся специфические адсорбенты, несу¬
щие на поверхности локально сконцентрированные положительные заря¬
ды в форме, например, частично протонизированных атомов водорода по¬
верхностных гидроксильных групп или выдвинутых обменных катионов,
компенсирующих большие, в частности, комплексные анионы.При сближении размеров кагионов и анионов, чередовании их на одной
грани кристалла и потере ионообменной спос(Лности специфичность молеку¬
лярного взаимодействия ионного адсорбента соответственно понижается.Ш тип — специфические адсорбенты. Такие адсорбенты несут на по¬
верхности связи или звенья с сосредоточенной на периферии электронной
плотностью. Адсорбирующие поверхности этого типа легко получать, от¬
кладывая на поверхности неспецифического адсорбента (т. е. адсорбента I
типа), в частности на поверхносга графитированной сажи, плотные моно¬
слои молекул или макромолекул группы В, например полиэтиленгаиколь
[3,4], или прививая соответствующие функциональные группы, например
CN, при химическом модифицировании поверхности адсорбентов [3,4].Таким образом, в зависимости от характера распределения электронной
плотности на периферии образующих поверхность связей и звеньев целесо-
(^разно вьщелить один тип неспецифических адсорбентов и два типа спе¬
цифических. В табл. 4.1 представлена [2-5] рассмотренная классификация
молекул адсорбатов и поверхностей адсорбентов по их способности только
к неспецифическому или к неспецифичесшму и дополнительному специ¬
фическому молекулярному взаимодействию. Отметим еще раз, что при этом
имеется в виду молекулярное взаимодействие, а не химическое, т. е. молеку¬
лярная (так называемая физическая) адсорбция, при которой взаимодейству¬
ющие партнеры еще не теряют свою химическую индивидуальность.135
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я. Гязовая хроматографим4.4. Влияние геометрической структуры адсорбентов на удержи¬
ваемые объемыВыбор геометрической структуры адсорбента — удельной поверхно¬
сти и среднего диаметра пор — зависит от характера разделяемой смеси.
Адсорбция молекул газов и легких углеводородов при обычных условиях
невелика, поэтому в колонке необходимо применить адсорбент с достаточно
развитой поверхностью. Вместе с тем для газов (включая и легкие углеводо¬
роды) обычные и немного повышенные температуры достаточно велики для
того, чтобы неоднородность поверхности аморфных адсорбентов с высокой
удельной поверхностью и массообмен в тонких порах не приводили к суще¬
ственному размыванию полос. Для подобных разделений применяют цеоли¬
ты, тонкопористые силикагели, тонкопористые стекла, а также капиллярные
стеклянные колонки с пористым слоем на внутренних стенках.По мере увеличения размеров молекулы необходимо уменьшение
удельной поверхности и расширение пор адсорбента, а для веществ, спо¬
собных к сильной специфической адсорбции, следует применять неспе¬
цифические адсорбенты с возможно более однородной поверхностью, а
также следует увеличивать температуру колонки.Таким образом, вопросы влияния на газо-адсорбционное разделение
геометрической структуры адсорбентов — их удельной поверхности, свя¬
занного с ней влияния неоднородности поверхности, а также формы пор
и их распределения по размерам и по глубине зерна адсорбента имеют
большое значение. Адсорбенты по их геометрической структуре целесоо¬
бразно классифицировать по системе Киселева [2]. В последовательности
уменьшения размеров пор выделены четыре основных структурных типа
(таблица 4.2).Геометрическую структуру адсорбентов принято характеризовать
удельной поверхностью S, средним диаметром пор (d^^), объемом пор
(V_j^). Удельная поверхность это поверхность одного грамма адсорбента,
выраженная в м^/т. Удельная поверхность адсорбентов для газовой хрома¬
тографии колеблется в пределах 5-1200 м^г Средний диаметр пор ранее
определялся в ангстремах, А (10 * см), в настоящее время система СИ тре¬
бует выражать в нанометрах (10 ’ см), средний диаметр пор колеблется от
0,4 до 500 нм. Средний объём пор (У_^) - это объём всех внутренних пор
одного фамма адсорбента (размерность cmVf). При допущении, что все
поры цилиндрической формы, величина S, d^, в простейшем случае
связаны между собой для адсорбентов одной природы; например силика¬
гелей, следующим образом;‘‘'р SКак видно из этого соотношения в ряду таких адсорбентов величи¬
на d^p обратно пропорциональна S, т.е. тонкопористые адсорбенты имеют136
ГЛАВА 4Газо-адсоубтонная хроматографиябольшую удельную поверхность и, наоборот, макропористые адсорбенты
имеют небольшую удельную поверхность. Адсорбенты для хроматогра¬
фии можно разделить на непористые, обьемно-пористые и поверхностно¬
пористые. В непористых адсорбентах используется зерна адсорбента, не
имеющего внутренней пор, обычно это тонко дисперсные адсорбенты (а).а)Объемно-пористые адсорбенты (б) имеют пористость во всём объёме
зерна, а в поверхностно - пористых адсорбентах имеется пористость толь¬
ко на определённую глубину зерна, а центральная часть зерна непористая.
Внутренний массообмен на таких адсорбентах происходит быстрее, чем в
объемно-пористых адсорбентах. Впервые поверхностно-пористые адсор¬
бенты предложены в работе [10, 11].Таблица 4.2Классификация адсорбентов по геометрической структуре [3].Тип адсорбентовS, м^/гdcp, нмОбъём пор,
cmVf1Непористые5-200--2Однородно макропористые5-100>50до 1,53Однородно тонкопористые500-10000,4-100,5-0,84Неоднородно пористые250-8001-200,3-1,2Непористые адсорбенты. Это непористые кристаллические адсор¬
бенты, например, графитированные сажи, а также непористые аморфные
адсорбенты, например аэросил, термические неграфитированные сажи.
Удельная поверхность таких адсорбентов составляет от десятых долей
до сотен квадратных метров на грамм. В колонку непористые адсорбен¬
ты вводят либо в макропорах носителя, либо агрегированными в шарики,
либо спрессованными в частицы нужных размеров (как, например, различ¬
ные кристаллы). При этом между первичными частицами непористого ад¬
сорбента возникают, конечно, зазоры, образующие некоторую пористость.
Размеры этих пор обычно соизмеримы с размерами самих первичных ча¬
стиц, т. е. они достаточно велики для обеспечения быстрого обмена моле¬
кул, адсорбированных на поверхности первичных непористых частиц, с
движущимся вдоль колонки газом-носителем.137
Яишн Я.И.. Яшин ЕЖ. Яишн Л.Я. Газовая хроматографияДля непористых адсорбентов с поверхностью одинакового химичесш-
го состава адсорбционные свойства единицы поверхности практически не
зависят от удельной поверхности. Это относится также и к удерживаемому
объему V^.Однородномакропористые адсорбенты. К ним относятся широко¬
пористые силикагели, крупнопористые стекла, а также спрессованные в
таблетки порошки из непористых частиц размером более 100 А и удельной
поверхностью менее 200 mVf.Однороднотоикопористые адсорбенты. Это аморфные тонкопори¬
стые стекла, многие активные угли, например утаи типа Саран и углерод¬
ных сит, а также пористые кристаллы, например цеолиты А и X. Отверстия
пор пористого кристалла одинаковы, поэтому пористые кристаллы осо¬
бенно удобны для разделений на основе молекулярно-ситового действия
Если данные молекулы не могут проникать в эти отверстия, то пористый
кристалл ведет себя по отношению к ним как непористый адсорбент [2].Неоднороднопористые адсорбенты. Это обычно неоднороднопори¬
стые силикагели, например меловидные силикагели, полученные осажде¬
нием гидрогеля из раствора силиката гидролизующимися солями сильных
кислот. Обычно они содержат много сильно адсорбирующих тонких пор,
поэтому не находят столь широкого применения в газовой хроматографии,
как адсорбенты первых трех типов.4.5. Геометрическое модифицирование адсорбентов на примере
силикагеляПри обработке тонкопористых силикагелей при высоких температурах
(800—900 °С в зависимости от размеров пор) скелет силикагеля спекается
и объем пор резко уменьшается. При гидротермальной обработке силика¬
геля в автоклаве (т. е. при воздействии на силиютель водяного пара при
температурах около 700—800 °С) объем пор меняется сначала незначи¬
тельно, а удельная поверхность резко сокращается. Это происходит за счет
исчезновения мелких глобул кремнезема и сильного роста крупных гаобул.
В результате гнщютермальной обработки в, автоклаве происходит резкое
расширение пор силикагеля. Таким путем можно легко снизить удельную
поверхность силикагеля до 50—25 м^г и ниже при расширении пор до
тысяч ангстрем [2].Промышленные силикагели содержат примеси, в частности оксиды
алюминия и железа. Присутствие этих примесей создает химическую не¬
однородность и активность поверхности, в результате чего становятся не¬
симметричными пики полярных веществ. Применение чистых кремнезе¬
мов — аэросилогелей, содержащих не более 0,1% примесей, значительно138
ГЛАВА 4 Газо-адсорбиионная хроматографияулучшает симметричность пиков. Аэросилогель представляет собой круп¬
нопористый и химически очень чистый’ кремнезем, обладающий геоме¬
трически и химически весьма однородной поверхностью [12,13].4.6. Структура пор адсорбента и удерживание различных вешествИзвестно, что адсорбция в сильной степени зависит не только от хими¬
ческой природы поверхности, но и от структуры пор адсорбентов [14, 15].
Исследования изотерм и теплот адсорбции паров углеводородов на силика-
гелж статическими методами показали, что адсорбция и теплота адсорб¬
ции угаеюдородов увеличиваются при сужении пор силикагеля, причем это
увеличение возрастает с увеличением числа атомов угаерода в молекуле. По
мере сужения пор силикагеля изотермы адсорбции сильно искривляются, а
для разных образцов с порами приблизительно одинаковых размеров они
оказываются близкими.Определение теплот адсорбции угаеюдородов на силикагелях газохрома¬
тографическим способом показало, что с уменьшением размеров пор теплоты
адсорбции угаеюдородов юзрастают. Это связано с увеличением потенциала
неспещ1фических взаимодействий (дисперсионных сил) при сужении пор ад¬
сорбента Однако для достаточно широмопористых образцов (d^ > 500 А) из¬
менение размеров пор практически уже не влияет на теплоты адсорбции и на
значения Vs, независимо от удельной поверхности адсорбента.Для достаточно широкопористых силикагелей (как и для непористых
фафитированных саж) удельные удерживаемые обьемы пропорциональ¬
ны удельной поверхности s. Величины Vs = Vg/S практически не зависят
от удельной поверхности.Теплоты адсорбции нормальных алканов на широкопористых сили¬
кагелях линейно возрастают с увеличением числа атомов углерода в
молекуле (п);Q=a+bn (4.1)причем с расширением пор величины а и Ь достигают предельных зна¬
чений, не зависящих от размеров пор, удельной поверхности и п.В случае достаточно широкопористых силикагелей значения Vs/S для
одного и того же углеводорода практически совпадают независимо от s и
зависимость величины удельных удерживаемых объемов пропорциональ¬
ны удельной поверхности S (рис. 4.1).Поэтому эти величины при данной температуре представляют собой
физико-химические константы для данной системы адсорбат - широкопо¬
ристый или непористый адсорбент. Уменьшение размеров пор силикагеля
приводит к росту Vj, Q, а и Ь, причем возрастание этих величин заметней
для углеводородов с большим числом атомов углерода в молекуле вслед¬
ствие значительного увеличения энергии их адсорбции при сужении пор
(рис 4.2).139
Яшин Я.И.. Яшин ЕЛ. Яшин А.Я.Газовая храчатографияРис 4.1 Зависимостьудепьного Рис 4.2 а, б Зависишсгт ветчш удерпшваемых
удерживаемого объема Vm объемов Vs, отн&жтых к единице тщтюсти,
н-гексана от величины для силикагелей различной пористости (с разным
удельной поверхности s ши- средним диаметром пор), от числа атомов
рокопоржтых силикагелей углерода п молекулах трмальных углеводородов
(температура KXfC). а: 1 - 2Х А; 2 - 710 А; 3 - 1000 А; 4 ~ 4100А; 5 — qxdHue данные работы [58]; 6:1 — 140 А;2 -104 А; 3 - 70 А; 4 - 46 А; 5-32 АПри использовании небольших пор молекулы адсорбируются преиму¬
щественно на наиболее выгодных участгах. В соответствии с этим при до¬
статочно сильном уменьшении среднего дааметра пор и увеличении удель¬
ной поверхности адсорбента времена удерживания возрастают (рис 4.3). Для
выяснения влияния уменьшения размеров пор на удерживание и разделение
бьши получены хроматограммы на силикагелях с одинаююй поверхностью,
но различными величинами средних размеров пор (рис 4.4) [15].2 вРис.4.3 Храматогрсммы смеси м&гшна (7), этана (2), этилена (3), про¬
пана (4), пропшена (а) и бутана (б), полученные при 80°С на
колонке 150x0,45 см с различными силикагелями (скорость газа-
носителя—водорода 50 мл/мин, объем пробы 0,02 мл, детек¬
тор ионизационно-пламенный)а — КСК-2,5 (средний диаметр пор d = 104 А);
б — КСС-3 (d = 70 А); в— КСС-4 (d = 46 А); г — КСМ-5 (d = 32 А).140
ГЛАВА 4 Газо-адсорбиионная хтматогтфияРис.4.4 Хроматограммы смесей метана (1), этана (2), этилена (3), про¬
пана (4) и пропшена (5), полученные при 50°С на двух силика¬
гелях с одинаковой поверхностью в колонке, но с различными
средними диаметрами пор (проба 0,02 мл, скорость газа-носи¬
теля — тдорода 50 мл/мин)а — диаметр пор 70 А, колонка 100 X 0,4 см; б —- диаметр
пор 22 Л, колонка 58X0,4 смВеличины критерия разделения для макропористого образца несколь¬
ко больше, так как в случае тонкопористого образца в связи с замедлени¬
ем массообмена в порах пики размываются в гораздо большей степени.
Полученные данные показали, что при увеличении среднего размера в 3
раза времена удерживания резко уменьшаются (приблизительно в 4 раза).
Хотя селективность также растет с уменьшением диаметра пор исследуе¬
мых силикагелей, критерий разделения остается практически постоянным.
Поэтому для анализа выгоднее применять более широшпористые сили¬
кагели, так как в этом случае при сохранении той же величины критерия
разделения значительно сокращается время анализа и пики размываются
в меньшей степени.Таким образом, рассмотренные выше факты показывают, что в общем
случае удерживание анализируемых компонентов определяется, во-пер¬141
Яшин Я.И., Яшин Е.Я.. Яшин А.Я. Газовая хроматографиявых, геометрической структурой пор и химической природой поверхности
адсорбента, во-вторых, молекулярным весом, геометрической и электрон¬
ной структурой молекул веществ и, в-третьих, температурой колонки. Ис¬
ходя из этого, можно сделать вывод о том, что для каждой области тем¬
ператур кипения анализируемых веществ с молекулами близкой геоме¬
трической и электронной структуры существует оптимальная пористость
данного адсорбента для получения сравнительно быстрого разделения с
минимальным размыванием полос. Так при использовании силикагелей
для разделения легких газов необходимо использовать образцы с порами
средних размеров не более 20 А, для разделения легких углеводородов (с
температурами кипения не выше 10°С) следует использовать силикагели
со средним диаметром пор от 50 до 200 А, а для достаточно быстрого ана¬
лиза более высококипящих углеводородов и некоторых их производных
использовать соответственно еще более широкопористые силикагели.4.7. Структура пор адсорбента и размывание полосРазделительная способность колонки определяется как селективнос¬
тью адсорбента, так и размыванием хроматографических полос при их
движении по слою адсорбента. Размывание полос — основной фактор,
мешающий четкому разделению. При прочих одинаковых условиях более
эффективна та колонка, в которой хроматографические полосы размыва¬
ются в меньшей степени.Для уменьшения роли вихревой и внешней диффузии в размывании
полос необходимо использовать адсорбент с наиболее однородными зерна¬
ми и распределить его равномерно по сечению колонки, чтобы создать по
возможности одинаковые промежутки между зернами. Даже при насыпа-
нии одинаковых свинцовых или стеклянных шариков в колонки число кон¬
тактов колеблется от 4 до 12, т. е. имеются различные упаковки, от рыхлой
до самой плотной. Набивка же из адсорбента, состоящего из зерен разного
размера, неправильной формы и с шероховатой поверхностью, еще более
неоднородна. Поэтому в реальных колонках всегда имеют место некото¬
рые распределения скоростей потока газа по сечению колонки.Чтобы произошла адсорбция, молекула из потока газа должна прибли¬
зиться к внешней поверхности зерен адсорбента. Это достигается путем
обычной диффузии и перемешивания газа-носителя. Поэтому в общем
случае скорость внешнего массообмена зависит как от природы и кине¬
тической энергии молекул адсорбата, так и от режима течения газа в ко¬
лонке. В ламинарном потоке внешний массообмен осуществляется только
благодаря обычной диффузии, тогда как при турбулентном течении про¬
исходит принудительное перемешивание газа-носителя. Оценка критерия
Рейнольдса для обычных скоростей в заполненных хроматографических142
ГЛАВА 4Газо-адсорбииотт храыатографшколонках показывает, что, по-видимому, в большинстве случаев в газох¬
роматографических колонках мы имеем дело с ламинарным режимом те¬
чения газа и, следовательно, внешний массообмен происходит в основном
только благодаря диффузии Однаш в некоторых - специальных случаях
экспресс аналиюв возможно использовать большие сшрости с турбулент¬
ным характером потока газа в колонке.Роль внешней и внутренней диффузии в порах в случае достаточно
сильно адсорбируемого вещества должна возрастать с увеличением разме¬
ра зерен адсорбента. На рис. 4.5 представлена зависимость Н от объемной
сшрости со потока газа-носителя для неспецифически адсорбирующегося
вещества — пропана на силикагеле со сферическими зернами и средним
диаметром пор, равным 80 А.ш,ия1тРис. 4.5 Зависимость Н от а> для пропана при 60°С для одинаковых
колонок с силикагелем с порами диаметрам около 80 А со сфе¬
рическими зернами различных размеров:1 - 0,25 мм; 2—0,13—0,25 мм; 3 —0,25—0,5 мм; 4— 0,5—1,0 мм;
S—1,0—2,0 ммС увеличением диаметра зерен адсор&нта эффеюгивность шлонки
значительно ухудшается — минимальное значение Н возрастает, область
сшростей, соответствуюнщх низким значениям Н, сильно сужается, и на¬
клон правой ветви кривой Ван-Деемтера, определяемой в этом случае пре¬
имущественно кинетишй адсорбции, сильно возрастает. При размерах же
зерен равных 0,25 мм и меньше, наклон правой ветви весьма мал, так что
правая ветвь при этих скоростях почти параллельна оси сшрости. Это по-143
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я. Гаювая хранатографияказывает, что в данных условиях вклад внешней и внутренней диффузии в
размывание полосы очень мал. Для зерен больших диаметров (0,5—1,0 мм
и особенно 1,0—2,0 мм) пики начинают сильно размываться из-за задержки
шк внешнедиффузионного обмена, так и внутридиффузионного. Это указы¬
вает на значительный вклад внешне и внутридиффузионной массопередачи
для зерен адсорбента размером более 1 мм при недостаточно высоких тем¬
пературах.Роль внешне- и внутридиффузионной массопередачи в колонке долж¬
на уменьшаться с повышением температуры. С повышением температуры
эффективность колонки повышается, расширяется область низких значе¬
ний Н и уменьшается наклон правой (кинетической) ветви кривой Ван-
Деемтера.При прочих одинаковых условиях внешнедиффузионная скорость
массопередачи зависит также от природы газа-носителя. Для более тяже¬
лого газа-носителя (с меньшим коэффициентом диффузии) можно добить¬
ся большей эффективности колонок Однако в этом случае область низких
значений Н намного уже, так как из-за небольшого значения коэффициента
диффузии газа-носителя заметная задержка массообмена происходит при
меньших его скоростях.Большое влияние на равновесие оказывает однородность набивки ад¬
сорбента в колонке. Для очень узкой фракции зерен размерами только не¬
посредственно вблизи 0,25 мм эффективность колонки выше, чем для зе¬
рен меньших размеров, но более неоднородных (фракции 0,18—0,25 мм).
Это связано в основном с улучшением однородности набивки по сечению
колонки при сужении фракции зерен и уменьшением вклада в размывание
полос, вызванного различной скоростью движения газа-носителя в разных
участках сечения колонки.Влияние пористости адсорбента на размывание хроматографических
полос видно из изменения зависимости Н от F (или от и) при изменении
средних радиусов пор силикагелей приблизительно от 150 до 1500 нм при
одинаковых размерах зерен (0,25—0,5 мм) и при других одинаковых усло¬
виях. Как, было показано на рис. 4.6, при таком зернении адсорбента роль
внешнедиффузионного массообмена мала.Из рис.4.6 видно, что эффективность колонок возрастает с увеличе¬
нием среднего радиуса пор до определенного предела. Для силикагелей с
радиусами пор 750 и 1500 нм эффективность колонок уже одинакова и при
F > 30 см’/мин практически не зависит от скорости газа-носителя. С умень¬
шением среднего радиуса пор интервал скоростей, соответствуюший низ¬
ким значениям F, сокращается, и наклон кинетической ветви кривой Ван-
Деемтера возрастает.144
ГЛАВА 4Газо-адсорбтонная хроматографиян,тш,пя/тнРис. 4.6 Зтисимость Homw для пропана при 50°Сна ститгепях оди¬
накового зернения (0,25— 0,5 мм) с порами различных радиусов
(колонка 100 смх4 мм, газ-носитель — гелий):I —16 А; 2—23 А; 3 — 35 А; 4— 75 А; 5 —150 А
Адсорбвдонное равновесие для пропана, по-видимому, успевает усга-
навливеться, так как удельный удерживаемый объем не зависит от скорости
газа-носителя в рассмотренных ее пределах даже для самого тонкопористо¬
го образца. Однаюо время пребывания адсорбируемых молекул на различ¬
ных участках поверхности зерен адсорбента в разных по ширине и глубине
порах вввду неоднородности геометрической структуры силикагеля различ¬
ное. Это приводит к увеличению размывания с уменьшением среднего диа¬
метра пор, так как неоднородность структуры и неоднородаость адсо1Лци-
онного поля с уменьшением среднего размера пор увеличивается.Эффективность колонки зависит не толью от среднего размера пор
адсорбента, но и от гаубины пор в его зернах. На рис. 4.7 показана зави¬
симость Н от и для колонок, заполненных поверхностнопористым и обь-
емнопористым стеклом.Цтпри 50°С для колонок, заполненных
поверхностнопористым стеклам(1) к обьемнопористым стеклам(2) (колонка 100 см X 4 мм, газ-но-
ситель — водород)14510 Газовая хроматография
Яшт Я.И.. Яшин Е.Я.. Яшин А.Я. Газовая хроматографияВследствие большей сшрости массообмена для поверхностнопористого
стекла получается меньшее значение Н и более широкая область низких зна¬
чений Н при изменении сшрости газа. Разрешаюшая способность, шлонки
в «линейной» газо-адсорбционной хроматографии выражается отношением
разности времен удерживания At^, отсчитываемых по максимумам пишв
(которое определяется в основном шнстантами равновесия адсорбат — ад¬
сорбент, шнстантами Генри для соответствующих шмпонентов), к сумме
полуширин выходных кривых ц1 + ц2, определяемой задержками массо¬
обмена. В общем случае размывание элюируемой полосы (ширина пика)
при прочих одинаковых условиях определяется размером и формой зерен
адсорбента, природой газа-носителя и в основном кинетишй адсорбции и
десорбции, зависящей от пр1фоды анализируемого шмпонента, от природы
и геометричесшй структуры адсорбента и от температуры. У обычных ад¬
сорбентов, например силикагелей, алюмогелей, активных углей и т. п., поры
пронизывают весь объем зерна, поэтому пути внутренней диффузии оказы¬
ваются весьма длинными. Это сильно тормозит процессы диффузии в по¬
рах и в динамических условиях может приводить к неравномерности их ис¬
пользования по глубине зерен. Применение же в качестве адсорбента стекал
тольш с поверхностной пористостью, проходящей на небольшую глубину,
позволяет значительно сократить время анализа без ухудшения разрешаю¬
щей способности. Несмотря на то, что при использовании поверхностно-по¬
ристого стекла время анализа сокращается примерно в 8 раз до сравнению с
временем анализа на обьемнопористом стекле, критерий разделения и чис¬
ла теоретических тарелок для шлонки с поверхностно-пористым стеклом
больше На таких шлонках можно работать с большой линейной сшростью
газа-носителя для еще большего сокращения времени анализа без ухудше¬
ния разрешающей способности.На основании изложенного выше можно заключить, что эффектив¬
ность колонок определяется средним диаметром и глубиной пор адсорбен¬
та, размерами зерен, однородностью зернения, природой газа-носителя,
природой используемого адсорбата и температурой.Для получения эффективных колонок в газо-адсорбционной хрома¬
тографии необходимо использовать крупнопористые адсорбенты с одно¬
родными зернами. Наиболее оптимальный размер зерен с учетом сопро¬
тивления потоку — 0,2 мм. На крупнопористых адсорбентах даже при не¬
однородном зернении 0,25—0,5 мм можно получить значение Н= 1 мм и
меньше, что соответствует более чем тысяче тарелок на 1 м колонки Для
дальнейшего увеличения эффективности таких колонок нужно использо¬
вать еще меньшее и более однородное зернение. В случае однородного и
меньшего зернения крупнопористого адсорбента значительно уменьша¬146
ГЛАВА 4 Газо-адсорбиионная хроматографтется размывание, связанное с неравномерным распределением скорости
газа-носителя по сечению, при этом можно достичь значения Н = 0,5 мм и
меньше, т. е. нескольких тысяч теоретических тарелок на 1 м колонки.4.8 Изменение химической природы поверхности адсорбентов (мо¬
дифицирование адсорбентов)Для изменения химической природы поверхности адсорбентов приме¬
няют физическое и химическое модифицирование.В одном случае, при физическом модифицировании поверхность ад¬
сорбента покрывают небольшим количеством сильносорбируемой жидкой
фазы так, чтобы для анализируемых компонентов поверхность адсорбента
была недоступна, а адсорбция происходила на слое жидкой фазы (порядка
емкости монослоя) [5]. Во втором случае поверхность адсорбента может
быть блокирована путем отложения слоя твердого тела (как органическо¬
го, так и неорганического), в частности различных неорганических солей,
фталоцианинов различных металлов [2] Кроме чисто механического от¬
ложения твердых тел на поверхности осуществляют полимеризацию на¬
несенных мономеров с образованием плотной полимерной пленки [3].Во всех случаях физического модифицирования происходит процесс
геометрического модифицирования, т. е. сокращения удельной поверх¬
ности за счет уменьшения тонких пор, которые заполняются молекулами
блокирующих соединений. Химическая однородность вновь полученной
поверхности зависит от плотности нанесенного слоя, а также его устойчи¬
вости при повышенных температурах.При химическом модифицировании адсорбента изменяется химиче¬
ская природа поверхности. В промышленных и в лабораторных условиях
получили распространение реакции силирования поверхности. Широш
используют реакции с триметилхлорсиланом, диметилдихлорсиланом и
гексаметилдисилазаном [2-5]. Триметилхлорсилан успешно используют
для уменьшения адсорбционной активности силикагелей, пористых сте¬
кол, диатомитовых твердых носителей и других кремнеземных адсорбен¬
тов и носителей.В этом случае происходит поверхностная реакция:СНзI I I- Si^-OH+ClSKCHj), ► Si—О - Si-^CH3 + НС1I I IСНзГидроксильная группа заменяется на силильный эфир. Гидрофильная
поверхность кремнезема после этого становится гидрофобной, ташй ад¬
сорбент уже не смачивается водой и плавает на поверхности воды. Это147
Яишн ЯМ. Яшин Е.Я., Яшин А.Я. Гтовт хроматографияСВОЙСТВО химически модифицированного силанами кремнезема можно ис¬
пользовать для оценки полноты и качества модифицирования. Обработку
триметилхлорсиланом можно проводить как в газовой фазе, так и в раство¬
ре бензола. Весьма существенно, чтобы и адсорбент (или носитель), и рас¬
творитель были перед реакцией обезвожены, так как триметилхлорсилан
реагирует с водой. Для полного модифицирования тонкопористых силика¬
гелей при комнатной температуре необходимо не менее восьми дней, для
модифицирования макропористы носителей — несколько часов.Модифицирование в газовой фазе проводят в динамических услови¬
ях путем пропускания потока сухого инертного газа, насыщенного парами
триметилхлорсилана, через колонку, заполненную адсорбентом (или носи¬
телем) Д ля нагрева колонки ее помещают в трубчатую печь или же процесс
модифицирования проводят непосредственно в термостате хроматографа.
В этом случае детектор должен быть отсоединен от колонки.При модифицировании в жидкой фазе адсорбент с раствором запаива¬
ют в толстостенную ампулу и выдерживают несколько часов при темпера¬
туре выше точки кипения триметилхлорсилана (57,3 °С). При этом давле¬
ние в ампуле возрастает (необходимы соответствующие предосторожно¬
сти) пропорционально содержанию гидроксильных групп на поверхности
адсорбента, так как при реакции образуется хлористый водород. После
модифицирования адсорбент (или носитель) освобождают от продуктов
реакции путем нагревания выше 150 °С, так как из-за возможного присут¬
ствия воды образуются, (СНз),810Н и (CH3)jSi-0-Si(CHj)j, которые кипят
соответственно при 98,9 и 100,4 °С.Диметилдихлорсилан (t,^=71°C) чаще всего используют для обработ¬
ки твердых носителей для газо-жидкостной хроматографии. В этом случае
атомы хлора одной молекулы реагируют с двумя соседними гидроксиль¬
ными группами:/^\н н о оII II
— о о ^ + (СНз)281СЬ ' Si ^ + 2НС1I I
Si ^SiОоДля проведения такой реакции должны быть благоприятные условия,
т.е. большая концентрация поверхностных гидроксильных групп. Если ги¬
дроксильные группы расположены не рядом друг с другом, то в реакции
участвует только один атом хлора. Второй непрореагировавший атом хло¬
ра приведет к образованию полярной поверхности другой природы Для
блокирования этого атома хлора проводят последующую реакцию с мети-148
ГЛАВА 4 Газо-адсорбиионная хроматографияловым спиртом с образованием метокси-группы. Эта группа может в бла¬
гоприятных условиях реагировать с водой В некоторых случаях проводят
модифицирование диметилдихлорсиланолом специально в присутствии
водной пленки на поверхности носителя, при этом из диметилдихлорсила-
на образуется пленка силиконового масла:^^3 СНз СНзH>C\si/CH3 Н,с^ 1,/СНз н,с^нп ? 9 он оI I I I I 'Эта пленка лучше дезактивирует носитель и блокирует все оставшие¬
ся химически несвязанные группы ОН.Для обработки диметилдихлорсиланом твердых носителей в газовой
фазе при комнатной температуре необходимо не менее двенадцати часов.
При модифицировании гексаметилдисилазаном образуется поверхность,
идентичная поверхности, получаемой при обработке триметилхлорсила-
ном. Однако процесс обработки гексаметилдисилазаном имеет некоторые
преимушества: гексаметилдисилазан менее летуч (температура кипения
126°С) и менее токсичен, с ним удобнее работать в растворах.Реакция проходит следующим образом:СНз СН,I Iн Н СН,—Si—СНз СИ,—Si—СНз
ООН О О
—Si — о—Si — +{СНз)з81Ы$1(СНз)з ► —Si О Si — +NH,Можно рекомендовать следующую методику обработки: 25 г хорошо
высушенного твердого носителя (или адсорбента) помещают в колбу, за¬
ливают его петролейным эфиром, добавляют 7 мл гексаметилдисилазана,
соединяют колбу с обратным холодильником, нагревают до 60—80 °С,
смесь перегоняется 10 ч при обработке носителей из диатомита и 6 ч для
цеолита. Кремнеземы с большой поверхностью, естественно, надо обра¬
батывать более длительное время, носитель затем отделяют декантацией
и промывают несколькими порциями н-пропанола и петролейного эфира,
после фильтрации высушивают нагреванием под вакуумом Раствор сила-
зана в петролейном эфире можно использовать и повторно, так как при
обработке носителей реагирует менее 10% реактива.149
Яшт Я.И.. Яшин Е.Я., Яшин А.Я.Газовая хроматографияЧтобы оценить количественно степень замещения гидроксильных
групп, нужно выделяющийся при реакции аммиак направлять в раствор
кислоты определенной концентрации, затем титрованием можно точно
определить число связанных гидроксильных групп.Экспериментально установлено, что химическая связь —Si—О—Si—,
получаемая при химическом модифицировании силанами, устойчива до
350°С [2]. Выше этой температуры происходит разрыв связи. Полного (на
100%) связывания групп ОН в кремнеземах (особенно тонкопористых) не
происходит из-за пространственных затруднений.4.S Ошовимб тииы адшй8иивСиликагели. Силикагели обычно получают в результате конденсации
ортокремневой кислоты, образующейся при гидролизе хлораигидрида этой
кислоты, или при реакции растворимых силикатов (жидкого стекла) с ми¬
неральными кислотами [2-4]. Однако эти способы не позволяют получать
силикагели, достаточно широкопористые для применения в газовой хро¬
матографии. Для дальнейшего направленного изменения структуры пор
силикагеля применяют гидротермальную обработку — одновременное
действие высоких температур и водяного пара. Характер изменения струк¬
туры пор силикагелей при такой обработке зависит от исходного состояния
геля (гидрогель или ксерогель), химического состава образца, его исходной
пористости, температуры и давления водяного пара. Поверхность и объем
пор тонкопористых силикагелей при термической или термопаровой обра¬
ботке сокращаются в большей степени, чем для крупнопористых образцов.
Поэтому для неоднородно-пористых образцов наблюдается более сильное
уменьшение поверхности и увеличение диаметра пор, так как в первую
очередь при спекании исчезают мелкие поры.В последние годы сначала у нас в стране, а потом за рубежом нача¬
ли серийно выпускать макропористые силикагели, которые находят более
широкое применение для разделения не только низкокипящих, но и высо-
кокипящих соединений. В табл. 4.3 приведены структурные характеристи¬
ки отечественных и зарубежных макропористых силикагелей.Таблица 4.3Основные макропористые кремнеземные адсорбенты для газовойхроматографииНазваниеУдельная пов-ть
S, mVfДиаметр пор,
нмОбьем пор,
cmVfМакропористые аэросилогели (силохромы)С-8070-9040--501,2-1,4С-120110-130--СХ-11 25-35220 - 250-150
ГЛАВА 4Газо-адсорбиионная хроматографияНазваниеУдельная пов-ть
S, мУтДиаметр пор,
нмОбъем пор,
CMVrСХ-245-60100-1301,5-1,9СХ-370-9070- 100Макропористые силикагелиМСА-115-2580- 120МСА-260-9040-700,7- 1,0мен2-6150Порасил В125-25010-20Порасил С50-10020-40Порасил D25-4540-900,6-1,0Порасил Е10-2080-150Сферосил
ХОА 200125-25010-20Сферосил
ХОВ 07550-10020-400,6-1,0Сферосил
ХОВ 03025-4540-80Сферосил
ХОС 0052-6150-200Меркосорб
Si-150150-20015МеркосорбSi-50050-80500,7- 1,0МеркосорбSi-100020-30100На поверхности кремнезема может находиться пять типов групп [16]:а) силанольная отдельно стоящая гидроксильная группа; б) силанольная
группа с адсорбированной на ней молекулой воды за счет водородной свя¬
зи; в) силоксановая группа; г) геминальные группы ОН, связанные с од¬
ним атомом кремния; д) вицинальные группы ОН, близкорасположенные
гидроксильные группы, связанные водородной связью, обычно имеются в
тонкопористых кремнеземах.он—Si —ОН
-Si —''Н—Si —О—Si-в)ОН—Si—онн/о о-Si —о—Si —б) - г;Поверхность силикагелей в основном может быть двух типов: гидрок¬
силированной и дегидроксилированной. Гидроксилированная поверх-151
Яишн Я.И.. Яшин Е.Я.. Яшин А.Я.Газовая хроматографияность устойчива только до 400 °С, выше этой температуры начинает про¬
ходить процесс дегидроксилирования (две соседние гидроксильные груп¬
пы соединяются кислородным мостиком с выделением молекулы воды).
В наибольшей степени дегидроксилирование происходит при температуре
выше 800—900 °С. Однако полного дегидроксилирования трудно достичь.
В присутствии водяных паров дегидроксилированная поверхность медлен¬
но переходит в гидроксилированную. Для полного и быстрого перехода
дегидроксилированной поверхности в гидроксилированную необходимо
силикагель прокипятить в дистиллированной воде в течение двух суток. В
адсорбционном отношении гидроксилированная поверхность значительно
более активна к полярным и слабополярным веществам, чем дегидроксили¬
рованная (табл.4.4.). Таблица 4.4
Дипольные моменты и удерживаемые объемы различных веществ
на гидрокснлированнон (I) и дегндроксилнрованнон (II) поверхностисилохрома при 150 **ССоединенияДипольный момент,
DУ81,мл/м2IIIн-Нонан00,150,12Бензол00,0530,027Толуол0,360,1030,050Анилин1,481,200,20Нитробензол4,211,040,37Диэтиловый эфир1,170,290,10Ацетон1,660,640,27н-Бутанол1,660,640,27Различие удерживания на гидроксилированном и дегидроксилирован-
ном силикагелях сильно проявляются для нефторированных и фториро¬
ванных соединений [16, 17].На рис. 4.8 приведены хроматограммы н-алканов и ароматических
соединений и соответствующих фторированных аналогов на гидроксили¬
рованном и дегидроксилированном силохроме. На гидроксилированной
поверхности кремнезема (неплоской и адсорбирующей специфически)
фторпроизводные всех исследованных углеводородов: н-октана, бензола,
толуола и дифенила, удерживаются слабее соответствующих нефториро¬
ванных соединений. Во всех этих случаях основное влияние на уменьше¬
ние энергии взаимодействия на гидроксилированной поверхности оказы¬
вает увеличение ван-дер-ваальсового расстояния между силанольными
гидроксильными группами поверхности и звеньями молекул при замене152
ШАВА4Газо-адсорбиионная хроматографияВ них етомов Н на F. В отличие от плоской поверхности графитирован-
ных термической сажи на неплоской поверхности кремнезема, покрытой
по-разному ориентированными гидроксильными группами, этот эффект
проявляется и в случае адсорбции плоских молекул фторбензолов и фтор-
толуола.На сильно дегидроксилированной поверхности кремнезема, не несу¬
щей большого количества по-разному ориентированных гидроксильных
групп, удерживание н-алканов больше, благодаря возможности более близ¬
кого контакта молекулы непосредственно с кислородным остовом крем¬
незема, что увеличивает неспецифическое взаимодействие всех молекул с
таким адсорбентом.М-НРис. 4.8. Хроматограммы различных веществ, полученные при разных
температурах на колонне 100 X 0,3 см, заполненной силохро¬
мом С-80 с гидроксшированной (а) и дегидроксилированной
(б) поверхностью (скорость газа-носителя азота 25 мл/мин,
детектор ДИП).На гифоксилированных силикагелях удерживание соединений сильно за¬
висит от распределения электронной плотности адсо1^ируюцщхся молекул.В работе [18] изучены электронные эффекты кремнезамещенных тио-
фенов методами газовой хроматографии и ядерного магнитного резонанса.
Для сравнения приведены удерживания и на сорбентах с жидкими фазами
(табл. 4.5).153
Яшин ЯМ. Яишн Е.Я.. Яшин А.Я. Газовая хроматографияТаблица 4.5Теплота адсорбции на специфическом адсорбенте и изменение
электронной плотности молекул, характеризуемое величиной хими¬
ческого сдвига 8q„., ккал/мольрастварениеадсорбцияСоединения•с’Мбапиезонрео-плекс-400силохром с
гадроксили-
рованной
поверх¬
ностьюи8484-9,17,87,5по983,49,58,98.1ги126118,53,39,99,18.0ф-8|(СН,).Н1701422,6510,49,510.51781562,7410,99.312,20-а(с.нлS2001972,779,910,813,8207217,52,4210,110,915.0(J-SC,s2451982,3612,311,813,2При сопоставлении данных величин теплот адсорбции соединений на
силикагелях с данными протонного магнитного резонанса установлено,
что величины теплот адсорбции изучаемых соединений меняются симбат-
но со значениями химических сдвигов (т) протонов в положении 3 кольца.154
ШАВЛ4Газо-адсорбиионная хроматографияа, следовательно, и с изменением электронной плотности в этом положе¬
нии (рис. 4.9). Таким образом данные таблицы 4.5 и рисунка 4.9 свидетель¬
ствуют о значительном вкладе ориентационного эффекта при межмолеку-
лярном взаимодействии адсорбат-адсорбент в общую теплоту адсорбции.
Это позволяет изучать характер распределения электронной плотности в
элементоорганических соединениях.1ш--W—Р—V—53F—?Г*Г-Рис.4.9 Зависимость теплоты адсорбции qstl от протонного хими¬
ческого сдвига д для замещенных тиофенов.В газо-адсорбционной хроматографии в ряде работ параметры удержи¬
вания на адсорбентах различной химичесшй природы сопоставляют на каче¬
ственной основе со свойствами заместителей в бензольном шльце [19,20].Как известно [21 ], в зависимости от природы заместителя в бензольном
кольце величина я-электронной плотности (т.е. величина отрицательного
заряда) на шльце заметно изменяется. Изменение заряда на кольце может
быть охарактеризовано шличественно константами типа ст-констант Гам-
мета и зарегистрировано спектросшпическими методами. С другой сто¬
роны, изменение зс-электронной плотности на бензольном кольце должно
существенным образом влиять на термодинамику адсорбции производных
бензола на адсорбентах, несущих положительные заряды на поверхности.В качестве адсорбента бьш использован гидроксилированный силох¬
ром С-80. Наличие гидроксильных групп приводит к тому, что поверх¬
ность этого адсорбента имеет сосредоточенные положительные заряды.
Изученные соединения приведены в табл. 4.6. Таблица 4 6Дифференциальные мольные изменения внутренней энергии -AU
при адсорбции на гидроксилированном силохроме, частоты переноса
заряда упз и величины Av в спектрах изученных соединений№№ппСоединение-Ли, кДж/мольУпз(7), см-1Ду(10), см-11C6H5F41,227800302С6Н640,72465058155
Яишн Я.И.. Яшин Е.Я.. Яшин А.Я.Газовая хроматографияппСоединение-Ди, кДж/мольVro(7), см-1Ду(10),см-1С6Н5С143,32640035С6Н5ВГ45,82540038С6Н5СНЗ44,52465058С6Н5С2Н549,12425058С6Н4(СНЗ)249,623370691,3,5-С6НЗ(СНЗ)354,221700781,2,4,5-
С6Н2(СНЗ)458,8208008510С6(СНЗ)б65,518500106ИС6Н50СНЗ57,1196506212С6Н5С6Н558,4199005213С6Н5СН(СНЗ)252,1241305814C6H5SiH(CH3)253,8246505015С6Н5С(СНЗ)355,9240006116C6H5J48,7223003917С6Н5СН = СН248,772130051Количественной оценкой величины межмолекулярного взаимодей¬
ствия служили величины - ДО (мольные изменения внутренней энергии
при адсорбции). Качественное рассмотрение данных, представленных в
табл. 4.6, показывает, что значения - ДО возрастают при повышении элек-
тронодонорных свойств заместителей, вводимых в бензольное кольцо. Эта
закономерность отчетливо проявляется при последовательном введении в
молекулу бензола одной, двух, трех, четырех и шести метильных групп
(переход от соединения № 2 к №№ 5, 7, 8, 9, 10); при переходе от бензо¬
ла к моноалкилбензолам (переход от соединения № 2 к №№ 5, 6, 13, 15).
Однако рассмотренные качественные соображения не передают деталей
электронного влияния заместителей, вводимых в кольцо. Электронные эф¬
фекты заместителей в бензольном кольце в основном (о.с.) и ионно-воз-
бужденном (в.с.) электронном состоянии ранее были детально изучены
методами и.-к. и у.-ф. спектроскопии [22]. Для обоснования физического
смысла корреляции между хроматографическими данными и результатами
спектроскопических исследований производных бензола были рассмотре¬
ны некоторые выводы спектроскопических исследований.156
ГЛАВА 4 Газо-адсорбиионная хроматографияПри изучении производных бензола в о.с. может быть применен методи.-к. спектроскопии [23]. Рассматриваются величины Av, которые представ¬
ляют собой сдвиг частоты валентного колебания связи О - И фенола при
образовании им межмолекулярной водородной связи типа О - Н .. .л с про¬
изводными бензола, являющимися я-основаниями. Значения Ду являются
мерой прочности водородной связи и отражают изменение л-электронной
плотности на бензольном кольце под влиянием заместителей в о.с.Изучение производных бензола в в.с. можно проводить по электрон¬
ным спектрам поглощения их комплексов с переносом заряда (КПЗ) с те-
трацианэтиленом. В КПЗ ароматическое соединение выступает в качестве
л-донора электронов (Д), а тетрацианэтилен - в качестве л-акцептора (А).
При образовании КПЗ Д и А образуют мезомерную структуру: Д,А Д*А
со значительным переносом электрона от Д к А в в.с. Для рассматриваемых
нами слабых я,л-комплексов о.с. является преимущественно незаряженной
структурой, а B.C. - структурой с переносом заряда (т.е. ионным). Переход
из о.с в B.C. приводит к появлению в электронном спектре КПЗ полосы
переноса заряда, положение которой зависит от природы заместителей
в бензольном кольце. Таким образом, если значения Av отражают влияние
заместителей в о.с., то значения v_^характеризуют это влияние в в.с.На рис. 4.10 представлена зависимость между величинами ~ АО и Av.
Данная зависимость имеет достаточно сложный характер. Это свидетельству¬
ет о том, что на величины - AU влияют не только электронные эффекты в о.с.
(характеризуемые значениями Av), а несюлью факторов. Между величина¬
ми - АО и v^ наблюдается несколью лучшая корреляция (рис. 4.106). Но и
в этом случае точки для всех соединений не описываются единым шр1кля-
ционным соотношением. Тем не менее табл. 4.6 и рис. 4.106 указьшают на
наличие следующей тенденции. При увеличении электронной плотности на
ароматичесюм юльце изменение внутренней энергии при адсорбции на ги-
дроксилированном силохроме (-АО) возрастает. Величину электронной плот¬
ности на шльце в в.с., как ясно из вышеизложенного, характеризуют значения
v^. При повышении под влиянием заместителей электронной плотности на
шльце значения v_^ уменьшаются, а при понижении - возрастают [24]. Таким
образом, наблюдаются элементы формальной аналогии между механизмом
взаимодействия произюдных бензола с поверхностью адсорбента С-80 и
механизмом донорно-акцепторного межмолекулярного взаимодействия этих
произюдных.Таким образом, сопоставление хроматофафических и спектроскопи¬
ческих данных позволяет проводить оценку относительной способности
производных бензола к специфическим и неспецифическим взаимодей¬
ствиям с положительно заряженной поверхностью адсорбента С-80.157
Яшин Я.И.. Яшин Е.Я.. Яшт А.Я.Газовая хроматографияНеполярные соединения, например алканы, адсорбируются одшаково
на обеих поверхностях, даже на дегидроксилированной поверхности удер¬
живаемые объемы н-алканов несколько выше. Меньшие удерживаемые
обьемы наблюдаются на химически модифицированном (силанизирован-
ном) силикагеле как для полярных, так и для неполярных соединений [2].Тонкопористые силикагели можно использовать для газохроматогра¬
фического разделения постоянных и углеводородных газов, за исключе¬
нием кислорода и азота, которые на силикагелях трудно разделяются даже
при отрицательных температурах. Непредельные углеводороды выходят
из колонки значительно позднее соответствующих предельных алканов и
хорошо разделяются, например, этилен и пропилен выходят из шлонки с
силикагелем соответственно позднее этана и пропана. Двуокись углерода
элюируется позднее этана. Силикагели получили широше применение для
разделения серасодержащих газов. Часто встречающаяся производствен¬
ная смесь: СО^, COS, H^S, CSj и SOj легш разделяется на шлонках с сили¬
кагелем небольшой длины (30—50 см) [2] (рис. 4.11).1-0^ + N^; 2 - СО,; 3 - COS; 4 - 5 - CS,; б — SO^lA/li i te.Puc. 4.11 Хроматограмма смеси газов при 100°С на колонке дли¬
ной 30 см с силикагелем (скорость газа-носителя — ге¬
лия 40 мл/мин)158
1ШШЛ4 Газо-адсопбтонная хрсматографшМакропористые силикагели используют для разделения слабополяр-
ных и неполярных соединений. Благодаря их термической устойчивости,
очень важно их применение для высокотемпературных разделений насы¬
щенных и ароматических угаеводородов, сложных эфиров, в частности
фталатов, используемых в качестве пластификаторов и других слабопо¬
лярных соединений [3] (рис. 4.12 а.б) [13, 16, 36].соос«^ооос,н.-f-Puc. 4.12 a Хроматограммы на сшохроме С-80, предварительно
прогретом при 100 ®С.г нтРис. 4.12 б )фоматограмма технического динсяшпфталата при 280^С накоюнт 100X 0,5 см с маиротриспшм симшепем (s = 20м^/г)159
Яшин Я.И., Яшин Е.Я.. Яшин Л.Я.Газовая хроматографияСильнополярные соединения элюируют на макропористых силикаге¬
лях в виде несимметричных пиков, особенно в тех случаях, когда силика¬
гели содержат примеси других, более активных окислов. Для уменьшения
адсорбционной активности и повышения химической однородности на
поверхность макропористых силикагелей наносят небольшие шличества
высококипящих жидкостей и твердых тел (как органических, так и неор¬
ганических) [2]. Наиболее эффективные колонки получаются, когда жид¬
кость или твердое тело наносят в количестве, соответствующем емкости
монослоя. В этом случае адсорбция происходит на новой поверхности,
создаваемой монослоем нанесенного вещества. Если количество модифи¬
цирующей жидкости больше емкости монослоя, то разделение будет про¬
исходить как за счет различий в адсорбируемости, так и за счет различий
растворимостей разделяемых шмпонентов [3].При применении юдяного пара в качестве подвижной фазы удержи¬
вания полярных соединений на силикагелях уменьшаются, а симметрия
пишв улучшается за счет деактивации активных центров.На рис. 4.13 показана возможность разделения спиртов на шлонке с
силохромом при 90°С с водяным паром в качестве подвижной фазы.Л.III-в г im й t тя
с ГРис, 4.13. Хроматограммы спиртов, полученные на колоннах раз¬
мером 100X0,3 см, заполненных силохромом, с водяным
паром в качестве подвижной фазы при 90 °С (а) и с азо¬
том при 140 °С (б): 1 — метанол; 2 — этанол; 3 — про-
панол; 4 — бутанолПрименение в обычных насадочных колонках силохрома С-80 с раз¬
мером зерен 10-20 мкм позволяет достичь эффективности до 40000 теоре¬
тических тарелок на 1 м длины шлонок. На таких шлонках в режиме про¬
граммирования можно достаточно быстро разделить смесь углеводородов
С,-С„ (рис. 4.14) [4].160
ТШВА4Газо-адсорбиионная хроматографияУс»СиСмСмс»Сис»I!Рис.4.14 Хроматограмма н-алканов С5-С17, полученная на колонне
размером 10X0,4 см с сшохромом С-80 (частицы 20 мкм),
при программировании температуры от 50 до 250°С, ско¬
рости газа-носителя азота 2,1 смЗ/минЕсли силикагели хранили в открытом виде, то на их поверхности ад¬
сорбировалась влага из воздуха (силикагели, как известно, являются хо¬
рошими осушителями). Перед заполнением хроматографической колонки
силикагели необходимо прокалить при 200 - 250°С до постоянной массы.
Адсорбированная влага быстрее удаляется, если прокаливать силикагель
под вакуумом. Эффективно проходит осушка силикагеля непосредственно
в хроматографической колонке в потоке сухого инертного газа-носителя
при высокой температуре (при этом детектор необходимо отсоединить от
колонки). Высушенные силикагели необходимо хранить в герметичных
емкостях.Пористые стекла. Из кремнеземных адсорбентов кроме силикагелей
в газовой хроматографии применяют пористые стекла [10, 11]. Было по¬
казано, что щелочно-боросиликатные стекла при определенной термооб¬
работке приобретают химическую неустойчивость к кислотам и щелочам
[10, И]. При обработке таких стекол кислотами они получаются пористы¬
ми за счет избирательной растворимости стекла. Практически растворя¬
ются толью окислы щелочных металлов и BjO,, а SiOj остается в виде
пористого остова.Структура и характеристики полученного таким путем пористого
стекла обусловлены как составом и термообработкой исходного непори¬
стого стекла, определяющими пространственные распределения B^Oj и161
Яшт Я.И., Яишн Е.Я.. Яшт Л.Я.Газовая хроматографияОКИСЛОВ щелочных металлов в кремнеземистом остове, так и условиями
выщелачивания при получении пористого стекла. Изменение химичесюго
состава исходного материала и условий предварительной термообработки
позволяет получать пористое стекло с заданными размерами пор (от 8 до
1000 А), колеблющимися в относительно небольших пределах, что пред¬
ставляет особый интерес для газовой хроматохрафии.В качестве одного из примеров получения пористых стекол можно
привести методику [2] на основе боросиликатного стекла мольного соста¬
ва; 7% Na^O, 23% BjOj и 70% SiOj. Это стекло предварительно прокалива¬
ют при 500—700 °С. После термообработки исходаое стекло дробят до зе¬
рен, имеющих размер, необходимый для заполнения хроматографической
колонки, и отсеивают фракцию 0,20—0,25 мм. Эти зерна выщелачивают в3 н. растворе НС1 при 50 °С, непрерывно перемешивая. Затем стекло от¬
мывают от ионов хлора и высушивают при 150—200 °С до постоянной
массы.При прокаливании исходного стекла при 550 °С в течение 20 ч получа¬
ются пористые стекла с порами размеров 3—6 нм.С изменением времени травления в растворе НС1 изменяется глубина пор
в зернах пористого стекла, что позволяет получать как обьемно-пористые, так
и поверхностно-пористые стекла. С{№дние размеры пор при зтсш п^иетически
не ишеняются, меняется лишь глубина пористого слоя. Поверхностно-пори¬
стые адсорбенты удобны для быстрых разделений (рис. 4.15) [10].IULг тт.А-А.IIt я вйл 1 1 1 1 1...—Jl- I III I.Ш.J.... I »nmPuc. 4.15 Хроматограммы смеси метана (1), этана (2), этилена (3), про¬
пана (4) и щюптена (5) на колонке 100 смХ4 мм с пористыми
стеклами (газ-носитель—водород) а—поверхностнопористое
стекло, скорость 68 мл/мин; б—объемно-пористое стекло, ско¬
рость 68 мл/мин; в — поверхностнопористое стекло при боль¬
шой скорости газа-носителя.162
ГЛАВА 4Газо-адсорбиионная хроматографияДля образования более крупных пор полученные травлением кислотой
пористые стекла следует подвергать дополнительному выщелачиванию в
0,5 н. растворе КОН при 20°С в течение 1—2 ч. При этом растворяется
наиболее тонкодисперсный кремнезем, в результате чего увеличивается
общий объем пор и значительно сокращается удельная поверхность.Возможность управления структурой пор боросиликатных стекол по¬
зволяет широко использовать их в газовой хроматографии. Очень мелкопо¬
ристое стекло типа молекулярного сита (с порами диаметром 10 А) может
быть использовано наряду с цеолитами для разделения низкокипящих га¬
зов. По сравнению с цеолитами пористые стекла более механически стой¬
ки и, что иногда очень важно, вполне кислотостойки.Пористые стекла с порами диаметром от 30 до 100 А можно использо¬
вать для разделения низкокипящих газов и паров, в частности для анализа
легких углеводородных газов. Благодаря однородности пористые стекла
имеют преимущества в этой области по сравнению, с силикагелем и алю¬
могелем. Высокая разделительная способность для таких смесей (напри¬
мер, СН^, CjH^ и CjH^) сохраняется даже при высоких температурах (95°С).
Более высокая по сравнению с силикагелями однородность пористого
стекла позволяет определять примеси в чистых веществах, в частности в
мономерах [25].Макропористые стекла с порами размеров более 500 А можно исполь¬
зовать для разделения жидких смесей нормальных и ароматических угле¬
водородов [8] (рис.4.16).УI''"' i i'SmРис. 4.16 )фомапюграммы определения щлшесей в техническом изопро-
пилбензоле на колонке 100 х 0,45 см с крупнопористым стеклам
(размер зерен 0,25—0,5мм, скорость газа-носителя — водорода
80 мл/мин, детектор ионизационно-пламенный) а — пропан (1),
пропилен (2), бензол (3), толуол (4), этилбензол (5), изопропилбеп-
зол (6); темперащра 120°С. 6—бензол (1), толуол (2), этилбен¬
зол (3), изопропилбензол (4); температура 150°С163
Яшин Я.И., Яшин Е.Я., Яшин АЖГазовая хроматографияХимическая природа noeqixHocra хорошо промытых пористых стекол
идентична хилгачесшй природе поверхности силикагелей [25]. Это связа¬
но с близостью химической природы гидроксилированной поверхности
этих адсорбентов.Для газохроматографических опытов пористые стекла необходимо
подготавливать так же, как и силикагели.Пористые полимеры. В последние годы в газовой хроматографии
применяют не только минеральные и природные адсорбенты, но и поли¬
мерные адсорбенты, синтезированные с такой структурой пор и химией
поверхности, что они оказались пригодными для высокоэффективного раз¬
деления многих сложных смесей [26, 27]. Эти адсорбенты различных ти¬
пов прочно вошли в практику газовой хроматографии, в особенности для
анализа газов, в том числе и агрессивных, водных смесей, смесей низкомо¬
лекулярных спиртов, кислот, аминов и других высошполярных соедине¬
ний. Пока для газохроматографического разделения применяют пористые
полимеры на основе сополимеров стирола, этилстирола и дивинилбензола.
Стирол и дивинилбензол, смешанные в определенных соотношениях, по-
лимеризуются в инертном растворителе с образованием трехмерного про¬
странственного полимера по схеме (а).СН--СН^ СН—СН,— СН--^СН,— СНСН—СН^ СН—СН,—СН—СН,- СНСхема (а)Полученные таким образом полимеры имеют на поверхности фениль-
ные группы.Полимеризацию проводят в среде инертньи, неполимеризующихся
жидкостей, которые являются хорошими растворителями мономеров и
плохими растворителями полимеров В результате образуются простран¬
ственные полимеры, пустые ячейки которых заполнены инертным раство¬
рителем После окончания полимеризации растворитель удаляется, а об¬
разовавшийся пористый каркас сохраняется.164
rmsdjLГазо-адсорбиионная хроматографияРеакции хлорметилирования с последующей обработкой подходящи¬
ми реагентами (например этилендиамином) позволяет вводить на поверх¬
ность пор этих полимерных адсорбентов самые разнообразные функцио¬
нальные группы.Для этого есть два пути. Первый состоит в том, что в саму реакцию
сополимеризации вводят мономеры, уже содержащие активные функци¬
ональные группы. Например, вместо стирола вводят винилпиридин (см.
схему б). Азот пиридинового кольца придает этому сополимеру электроно-
донорные свойства. Второй путь - химическое модифицирование поверх¬
ности уже готовых пористых полимеров.Применяются также сополимеры 2-оксиэтилметакрилата и других
мономеров с этилендиметакрилатом в качестве сшивающего агента (пори¬
стые полимеры типа сферы). На схеме (в) представлено структурное звено
полимера.Гидроксильные группы этого пористого полимера можно легко за¬
менить на другие функциональные группы, которые могут послужить, в
частности, сшивкой для иммобилизации биологически активных соеди¬
нений для аффинной (биоспецифической) хроматографии. Кроме описан¬
ных выше пористых полимеров, полученных сополимеризацией стирола
и дивинилбензола или 2-оксиэтилметакрилата с этилендиметакрилатом в
газовой хроматографии применяются более термостойкие пористые по¬
лимеры другой природы, в частности, полиакрилнитрил, полиарилат и
поли- п - 2,6 - дифенилфениленоксид (см. схему г).ЛСН2=СН + СНг=СН
СН2=СН + СН2=СНСхема (б)CHj-CH-CHj-CHйсн,—СН-СНз—СН165
Яитн Я И , Яшин, ЕМ. Яшин АЖОСН,СН,ОН ОСН.СН.ОИ
СН| io io СН,... -с-сна-с! СН,-С^ со СИ, ift соiw,iio СИ, ^ ^... _ t_CHr-t СН,—С CJ^-C- •in, do io in,Газошая хршштшФшii:iCO)CH,CHiOH hokCH/mСхема (в)ОnСхема (г)Геометрическая структура пористых полимеров. Изменяя условия
синтеза (соотношение исходных мономеров, количество и природа раство¬
рителя), можно в достаточно широких пределах регулировать геометри¬
ческую структуру пористых полимеров. Установлено, в частности, что с
увеличением содержания сшивающего агента (дивинилбензола) удельная
поверхность возрастает, а суммарный объем пор изменяется по кривой с
максимумом, одновременно наблюдается уменьшение плотности образцов
и уменьшение степени их на^ания На пористых полимерах с большим
содержанием дивинилбензола наблюдаются большие значения удельных
удерживаемых объемов и высокие значения критериев разделения Пори¬
стость возрастает также при увеличении в реакционной смеси содержа¬
ния инертного растворителя, при этом увеличивается общий объем пор и166
Газо-адсорбиионная xpoMamozpadmnснижается доля мелких пор, что, естественно, приводит к меньшему раз¬
мыванию газохроматографических полос и повышению эффективности
колонок, заполненных этим сорбентом [3].Однако в целом, как показано в ряде работ [28-30], пористые полимеры
являются неоднородно-пористыми адсорбентами. Кривые распределения
пор по обьему для порапаков Q и Т, показывают, что пористые полимеры
имеют поры разных размеров. Порапак Т обладает большей однороднос¬
тью, чем порапак Q. В отечественных полимерах распределение пор по
размерам также довольно неоднородно (от 60 до 1250 А), но с более вы¬
раженным максимумом.В табл 4.7 даны характеристики ряда зарубежных и отечественных по¬
ристых полимеров.Таблица 4.7Технические характеристики пористых полимеров, применяемых
в газоадсорбционной хроматографииТипадсорбентаХимический составS,MVrч..нмНасып-ной
вес, г/см’Предел.т-ра,°СТенаксыТенакс ORПоли (2,6-дифе-ннл-п-
фенилен-оксид35140350Тенакс ТА20--350ХромосорбыХромосорб 101СТ + ДВБ30-403500,3300Хромосорб 102350850,29250Хромосорб 103Сшитый полистирол15-253500,22275Хромосорб 104Акрилонитрил + ДВБ100 - 20060-800,32250Хромосорб 105Полиароматич. смола600 - 70040-600,34250ХрОмосорб 106СТ + ДВБ700 - 800--225Хромосорб 107Полиакриловый эфир400 -500--225Хромосорб 108100 - 200--225ПорапакяПорапак Р, PSСТ + ДВБ100-200--250Порапак Q, QS500-600--250Порапак NДВБ + винил-
пирролидон250-350--190Порапак R450 - 600-250Порапак S300-450--250Порапак ТЭтиленгликоль+диметакрилат250 - 350--190167
Яшин Я.И., Яшин Е.Я.. Яшин А.Я.Газовая хроматографияТипадсорбентаХимический составS,м^гч,-нмНасып-ной
вес, г/см’Предел.т-ра,°САмберлитыХАД-2СТ + ДВБ300200ХАД-4СТ + ДВБ750150ХАД-7Эфир подиметакрилата450150ХАД-8140150Полисорб-1СТ + ДВБ200-250130,29250Полисорб-10СТ + ДВБ300-3500,23250Объем микропор в пористых полимерах колеблется от 0,7 до 1,2 см^/г.Химическая природа поверхности пористых полимеров. Пока не
существует единого твердого представления о характере взаимодействия
молекул разделяемых веществ с пористыми полимерами Так в работах
считают [29, 30], что пористые полимеры представляют собой нечто сред¬
нее между обычным адсорбентом и пленкой жидкой фазы Другие авто¬
ры утверждают, что разделение на пористых полимерах осуществляется
за счет растворения вещества в полимере По нащим представлениям, при
низких температурах пористые полимеры ведут себя как адсорбенты, при
высоких температурах происходит их размягчение и они ведут себя как
высоковязкие жидкие фазы типа эластомеров.В ряде работ было показано, что пористые полимеры без привитых по¬
лярных функциональных групп (т.е. пористые полимеры только с фениль-
ными группами на поверхности) представляют собой слабоспецифические
адсорбенты III типа по классификации Киселева [2] Поэтому молекулы,
принадлежащие к группе D, удерживаются на них сильнее, нем молекулы
групп А и В Однако эта специфичность настолыо) мала, !что удерживае¬
мые обьемы на таких полимерах, как и на графитированной саже, прак¬
тически не зависят от дипольных моментов разделяемых веществ. Вслед¬
ствие преимущественного проявления дисперсионных взаимодействий
теплот адсорбции логарифмы удерживаемых объемов пропорциональны
электронным поляризуемостям молекул.В работе [31 ] проведено измерение времен удерживания ошло девяно¬
ста органических соединений на порапаке Q и показано, что в некотором
приближении логарифм времени удерживания является линейной функци¬
ей молекулярного веса разделяемых веществ. Это также объясняется тем,
что с ростом молекулярного веса возрастает поляризуемость молекул. На¬
блюдается также линейная зависимость логарифма исправленного удер¬
живаемого объема от числа атомов углерода в молекулах для гомологиче¬
ских рядов н-алканов, ароматических углеводородов, спиртов, кетонов и
жирных кислот и других соединений (рис. 4.17).168
ШАМА 4Газо-адсорбиионная хтиттографтВсе эти зависимости, представляющие собой параллельные прямые,
лежат выше прямой для и-алканов и «-алкенов. Кислоты имеют самые
большие времена удерживания. На основании данных рассчитаны отно¬
сительные величины изменений свободных энергий A(AG), по следующей
формуле:A(AG), = -RTln(Vt,) (4.1)где tj - время удерживания вещества определенного класса; tj - время
удерживания нормального углеводорода с тем же числом атомов угаерода
в молекуле.Индекс 1 соответствует нулеюй пробе.Значения приращений свободных энергий веществ различных классов
относительно н-алканов при 150°С слеедтощие: кислоты - (- 2280), нитрилы- (- 1705), спирты - (- 1470), сложные эфиры - (- 1240), кетоны - (- 11(Ю),
альдегиды - (- 967), амины - (- 678), алкилароматические углеводороды- (-232), алкены - 77,3.Наибольшее приращение свободной энергии адсорбции по сравне¬
нию с к-алканами для кислот составляет - 2280, а наименьшее - для
и-алкенов - 77,3 кал/моль.альвевидыз^рирыQ - митриял!
i 1 1fff пРис.4.17 Зтшшюсть логарифмов иатрстенных времен удщхжиттт
(tR) различных веществ на :qxMoa}p6e-103 от числа атомов
угпщюда (п) в молекулах этих веществ (длина колонны I м, тем¬
пература 150°С).169
Яитн ЯМ, Яшин ЕЛ. Яшин АЖ Гтш.шмтвтфшПрименение в процессе полимеризации смеси мономеров с различ¬
ными полярными функциональными группами позволяет регулировать
химическую природу поверхности и получать пористые полимеры, специ¬
фически сорбирующие полярные молекулы К таким полярным пористым
полимерам можно отнести порапаки R, S, N, Т, хромосорбы 104,105 и 107,
полисорбаг 2 и др Они различаются количеством и природой полярных
функциональных групп.На полярных пористых полимерах сильно изменяются времена удер¬
живания полярных молекул, таких, как вода, ацетилен и др. На неполяр¬
ных полимерах вода обычно элюирует между этаном и пропаном, на более
полярных порапаках N и S вода выходит между пропаном и изобутаном, а
на порапаке Т — после н-бутана.В табл. 4.8 сопоставлены удерживаемые обьемы некоторых соедине¬
ний на порапаках различной полярности и полисорбе 1 [32].Таблица 4.8Относительные удерживаемые объемы некоторых веществ в
зависимости от <^щей поляризуемости а и дипольных моментов ц.АдсорбатаА^^,DОтносительные удерживаемые обьемыполи-сорбпорапакиQQSРSNТВода1,491,841111111Метанол3,23-1,671,61,51,71.41,41.40,91Этанол5,061,703,43,04,12,72,73,31.5Ацетоншрил6,323,965,23,95,54,33,34,82,5Ацетон9,022,785,84,97,14,53,64,91,7Диэтиловый эфир9,91.177,85,510,94,84,24,70,98Пентан508,45,512,24,24.54,50,7Наиболее полярный полимер порапак Т. Из этой таблицы видно, что
молекулы, обладающие свободными электронными парами (эфир, ацетон,
ацетонитрил), удерживаются более сильно, чем. молекулы н-пентана.Исследование закономерностей удерживания различных веществ на
пористых полимерах - хромосорбах - 101,102 и 104 - проведено в работах
[33]. Значения логарифма относительных (к стандартному сорбату н-пен-
тану) удерживаемых объемов и соответствующих разностей дифференци¬
альных изменений внутренней энергии приблизительно линейно зависят
от числа атомов углерода в молекуле адсорбата (п):Ylg-^.^> = ai+biH(4.2)170
Газо-адсорбиионная хроматография(43)A(-AU,)=aj+bj
Значения Ь, и а^, bj приведены в таблице 4.9Таблица 4.9Константы al, Ы (безразмерные) и а2, Ь2 (в кДж/моль) в уравнениях
(4.2) и, соответственно, (4.3) для адсорбции на пористых полимерах
при малых (нулевых) заполненияхХрои<мврв-1<й)^вмоач>в>1М—в|&-MIjт-Алтш1.79И.Г1.8011,7~1.«11.9Прост» «фирм1.3422.21,41243-1.04Сложит афярнода2T4I1.1327Л-0J2^7КетммОМ8231.80183-0.1833.0ОшртайМ2Я.70.9831.40,014МКясяоты0.1836.80.8134.30.6249.80.Ш84,7Л»38,80,4849.0Таблица 4.10Инкременты изменения внутренней энергии А (- А01),
обусловленные специфическим межмолеклярным
взаимодействием функциональных групп.I «ротеорбцмI-О-4JS)со4JB-ад18,0<Ж»Я9м<I&0тожо-ш11,7I&8то■чю,4.Л10.0lao171
ЯшикЛЛ. Яшин Е.Я., Яшин А.Я. Газовая хроматографияТаблица 4.11Исправленные удерживаемые объемы и дифференциальные теплоты
адсорбции для очень малых (нулевых) проб ряда веществ, определен¬
ные на пористых полимерах при 150 "С с аргоном, водяным паром и
гелием в качестве газа-носителя.liI:!В•Ie.9I4.iIIilia1H.«i«8:iЩZ:?&:?iiH»0 M*—MtO^.9IIS:JHiI!49.t4в.вViiatV.BO.O24 .eIiii!sr.tS:fГрафики этих зависимостей представляют собой серии параллельных
линий, наклоны которых, т.е. коэффициенты Ы и Ь2 пропорциональны [в
случае ig^^] или равны [в случае А(-ДО,)] инкременту изменения вну¬
тренней энергии при неспецифическом взаимодействии группы СШ с по¬
верхностью адсорбента и разности между первыми членами в уравнениях
(4.2) и (4.3) для данного класса веществ и для н-алканов. Инкремент A(-AU,)
группы СН2 приблизительно одинаков для всех этих трех хромосорбов и
составляет от 6,5 до 6,9 кДж/моль [34, 35].Доступные для адсорбции нитрильные группы, находящиеся на по¬
верхности пор хромосорба-104, проявляют сильное специфическое взаи¬
модействие с функциональными группами органических кислот, спиртов,
сложных эфиров, альдегидов, кетонов, простых эфиров, т.е. с молекулами
группы В и D.Если допустить, что энергии неспецифического взаимодействия ато¬
мов углерода, кислорода и азота рассмотренных молекул с поверхностью
этих органических адсорбентов близки, то можно приблизительно рассчи¬
тать инкременты изменения внутренней энергии, обусловленные только
специфическим межмолекулярным взаимодействием функциональных
групп рассмотренных молекул с пористыми полимерами. Значения этих
инкрементов приведены в табл. 4.10.Замена атома водорода в бензоле на группу СН^ и на атом галогена вы¬
зывает приблизительно одинаковые изменения рассматриваемых термоди-172
fjjABA 4 Газо-адсорбиионная хроматографиянамических характеристик на хромосорбах-101 и 104. При замене же атома
водорода в молекуле бензола на группы NHj, NOj и особенно на группу ОН
эти изменения значительно больше при адсорбции на сильноспецифиче¬
ском адсорбенте третьего типа - хромосорбе-104 [35].В работе [35] получены также линейные зависимости инкрементов из¬
менений свободных энергий A(-AF,) = RT In (V^,,/V^, от величины
соогветствуюшего приращения молекулярной рефракции AR. Эти зависи¬
мости изображены на рис. 4.18. Величина AR, равная R- Rc<,h6> служит
удобной характеристикой изменения поляризуемости, от шторой зависит
энергия неспецифичесшго межмолекулярного взаимодействия. Получен¬
ные для слабоспецифического адсорбента хромосорба-101 прямые ха¬
рактеризуют влияние на инкремент A(-AF,) увеличения числа метильных
групп в шльце.Таким образом, зная инкременты A(-AFj) для отдельных функциональ¬
ных групп, можно приблизительно рассчитать общие величины А(-АР,) при
адсорбции на хромосорбе-101 ароматического вещества с любой шмбина-
цией функциональных групп, а следовательно, и порядок их уцерживания
на этом адсорбенте.Для сильноспецифического адсорбента хромосорба-104 аддитивность
термодинамических характеристик адсорбции по инкрементам функцио¬
нальных групп молекул наблюдается в меньшей степени. Значительно бо¬
лее крутой наклон прямых, характеризующих влияние увеличения числа
групп NOj и ОН в молекуле (рис. 4.18 б) свидетельствует о сильно спе¬
цифическом взаимодействии молекул, содержащих эти группы, с функци¬
ональными группами хромосорба-104. Кроме того, специфичность этого
пористого полимера, в отличие от хромосорба-101, проявилась также по
отношению к полиметилбензолам, для которых график исследуемой за¬
висимости представляет прямую с большим наклоном, чем прямая для
моноалкилбензолов. Эти зависимости сходны с полученными для гра-
фитированной термической сажи. Более сильное взаимодействие поли-
метилбензолов по сравнению с изомерными моноалкилбензолами может
быть объяснено более выгодной конформацией метильных групп полиме-
тилбензола на поверхности сильнее адсорбирующего хромосорба-104, а
также различием в распределении электронной плотности в бензольном
шльце при замене в нем атомов водорода на метильные группы, проявля¬
ющимся при адсорбции на ташм сильноспецифическом адсорбента, как
хромосорб-104. Изменение внутренней энергии при адсорбции о-ксилола
на 2,9 кДж/моль больше, чем для этилбензола, а для пары дурол - н-бутил-
бензол эта величина составляет уже 11,3 кДж/моль. Значения A(-AU|) для
замещающих групп при адсорбции монозамещенных бензолов на хромо¬
сорбах 101, 102 и 104 приведены в табл. 4.10.173
Яшин Я.И., Яшин Е.Я.. Яшин АЯ. Газовая зфоматографияВклад энергии неспецифического дисперсионного взаимодействия
в обо1ую энергию адсорбции молекул органических веществ различных
классов на поверхности полимеров является преобладающим. Это должно
проявиться соответствующим образом при сопоставлении удерживаемых
объемов органических соединений, молекулы которых относятся к разным
группам (А, В и D), с удерживаемыми объемами их фторированных про¬
изводных. При оценке вклада энергии дисперсионного взаимодействия в
общую энергию адсорбции важны величины электронных поляризуемо¬
стей и ван-дер-ваальсовых радиусов звеньев взаимодействующих молекул.
Как было сказано выще, поляризуемости водорода и фтора различаются
меньше, чем ван-дер-ваальсовы радиусы. Поэтому приближенно характе¬
ризующая энергию дисперсионного взаимодействия величина а/г* (г - рас¬
стояние между взаимодействующими частицами) для F меньше, чем для
Н. В связи с этим частичная или полная замена водорода в молекулах орга¬
нических соединений на фтор вызывает уменьшение времен удерживания
и теплот адсорбции (для случая преимущественного или значительного
проявления неспецифического дисперсионного взаимодействия).На рис. 4.19 - 4.20,4.21 показаны хроматограммы разделения соеди¬
нений различных классов и их фторопроизводных на пористом полимере
хромосорбе-101 (сополимер стирола и дивинилбензола). Во всех случа¬
ях (спирты, сложные эфиры и кетоны) замена атомов водорода на атомы
фтора, даже на один атом фтора у ацетофенона, приводит к уменьшению
удерживаемых объемов. Следовательно, вклад энергии неспецифическо¬
го дисперсионного взаимодействия в теплоту адсорбции на хромосор¬
бе-101 при температурах этих опытов является преобладающим. При
адсорбции на этом полимере молекул группы D должно проявляться и
ориентационное взаимодействие, при более низких температурах, уси¬
ливающееся в большей степени для фторпроизводных. В соответствии с
этим на хромосорбе-102 в интервале температур от 100 до 250°С порядок
выхода амилового и перфторамилового спиртов меняется на противопо¬
ложный. Для метилацетатов и ацетофенонов на этом полимере наблюда¬
ется явление, аналогичное наблюдающемуся на хромосорбе-102. Срав¬
нение удерживания фторированных веществ на слабоспецифических и
сильноспецифических пористых полимерах показало, что на более спе¬
цифическом полярном пористом полимере порапаке-Т нефторированный
спирт выходит гораздо раньше фторированного. Ниже приведены значе¬
ния теплот адсорбции некоторых адсорбатов, вычисленные из зависи¬
мостей удерживаемых объемов от обратной температуры для различных
пористых полимеров (табл. 4.12);174
ffdSdjГазо-е^сорбтштшя хрсшатоетиЬш
Таблица 4.12Хромосорб-101Хромосорб-102Порапак-ТСАОНсдаон10,29,711.413,7СН3СООСН,9,310,511,1CFjCOOCH,8,39,910,3С,Н„ОН12,111,015,1СДР,ОН12,813,220,8СН,СОС,Н,13,615,3п-СН,СОСДР13,214,4Теплоты адсорбции фторировашых соединений на слабоспецифиче¬
ских адсорбентах меньше, чем нефторированных. Исключение составляет
амиловый спирт. Тот факт, что теплоты адсорбции для фторированных ве¬
ществ меньше, чем для нефторированных, указывает на то, что преиму¬
щественный вклад в энергию взаимодействия вносит в этом случае неспе¬
цифическое дисперсионное взаимодействие, энергия которого при прочих
равных условиях приблизительно пропорциональна величине а/г*.-си,Рис. 4.18175
ЯышШ. .. Яитн Е.Я., Яитн Л.Я.Газовая}т^шао*сьг^‘$Опя/ттиIJLI—I I—Iв 1 г 3I LО 1 г 9Рис. 4.19 Хроматограммы различных веществ, полученные при раз¬
ных условиях на колонке длиной 37 см, заполненной хромо-
сорбом-101.tzvc ^ ^5 5ПX X1^-37смРис.4.20 Хроматограммы различных веществ, полученные при раз¬
ных условиях на тлонках, заполненных хромосорбом-102.176
тшл-Газо-адсоубиионная хроматографияРис. 4.21 Хроматограммы различных веществ, полученные при раз¬
ных условиях на колонке длиной 37 см, заполненной хромо-
сорбом-102.Аналитические возможности пористых полимеров. Пористые по¬
лимеры в процессе полимеризации получаются в виде правильных сфери¬
ческих частичек; они обладают достаточной механической прочностью, не
уступающей прочности большинства диатомитовых носителей. На хромато¬
графических шлонках с пористыми полимерами легш достигается эффек¬
тивность порядка 1300—^2600 теоретических тарелок на 1 м дайны. Пори¬
стые полимеры эффективно работают и при низких температурах (-150°С),
и при достаточно высоких (250°С), их используют для различных аналити¬
ческих задач: от анализа низшкипящих газов до анализа жидкостей с темпе¬
ратурами кипения 350°С.Критерии разделения зависят от размера частиц полимерного адсор¬
бента, скорости газа-носителя, температуры разделения и величины пробы
анализируемой смеси. Оптимальный размер фракции 0,20—0,25 мм, мень¬
ший размер зерен приводит к большим сопротивлениям колонки. Опти¬
мальные сшрости газа-носителя 30—50 мл/мин для шлонок с внутренним
диаметром 2-3 мм. При введении до 1 мкл жидшсти на шлонках с пори¬
стым полимером пики еще остаются симметричными, таким образом, не
происходит перегрузки колонки.При длительной эксплуатации пористые полимеры стабильны. Это хо¬
рошо продемонстрировано в табл. 4.13, где представлены относительные
времена удерживания некоторых глишлей и их эфиров при непрерывной
работе колонки в течение 28 дней при 200°С.Как видно из табл. 4.13, несмотря на сравнительно высокую темпера-
^у, в течение месяца, относительные удерживаемые обьемы практиче¬
ски не изменились, что позволяет решмендовать пористые полимеры для
Длительных автоматических анализов.177'2 Газоши хроматографии
Яишн Я.И-, Яшин Е.Я.. Яшин А.Я.Газовая хрйаграфияВпервые промышленный выпуск пористых полимеров организовала
фирма «Уотерс» (США) под названием «порапаки». На порапаках наблю¬
даются симметричные пики и сравнительно небольшие удерживаемые
объемы для полярных веществ, таких, как вода, спирты, кислоты и гли¬
коли. Они эффективно работают как при низких температурах, так и при
сравнительно высоких (до 200—250 °С) без значительного увеличения фо¬
нового ионного тока. Фирма выпускает восемь типов пористых полимеров
с различными адсорбционными свойствами.Порапак Р — малополярный полимер, селективен для карбонильных
соединений.Порапак S — силанизированный полимер, на котором значительно
улучшается симметрия пиков сильнополярных веществ, особенно эффек¬
тивен для разделения альдегидов и гликолей.Таблица 4.13Изменение относительных времен уцерживания во времени на пори¬
стых полимерах при 200*СВеществаДни эксплуаташш шлонки1013172028Монометиловый эфир
этиленпшшля0,4250,4120,4210,4260,4160,4230,429Этиленишколь0,6260,6240,6280,6260,6320,6280,630Монметиловый эфир
этилеигаишля0,7260,7250,7250,7270,7310,7280,720Пентанол1,0001,0001,0001,0001,0001,(ХЮ1,0(ЮМоноизопропиловый эфир
этиленгликоля1,171,181.171,161.11,1.71.17Монобутиловый эфир
этиленглишля3,043,003,012,993,003,063,02Монометиловый эфир
дютиленшижшя3,393,343,353,373,383,353,37Диметил-2, метил 3-про-
пиленглишль4,734,704,714,654,794,684,72Моноэтиловый эфир
диэтиленгликоля5,875,845,845,825,885,345,88Порапак Q — неполярный пористый полимер универсального назна¬
чения, особенно эффешнвен для алифагических углеводородов»Порапак S — силанизированный порапак Q, хорошо разделяет орга¬
нические кислоты и другие полярные соединения без образования асси-
митричных пиков.Порапак R — пористый полимер средней полярности, на нем наблю¬
дается сравнительно большое удерживание и хорошее разделение эфиров.178
fjjjiBA 4___ Газо-адсорбиионная храматограёшяПорапак N — пористый полимер средней полярности, осс^нно эф¬
фективен для разделения нормальных и разветвленных спиртов.Порапак S — полярный полимер, хорошо разделяет аммиак от воды,
ацетилен от других углеводородов сильно удерживает воду, устойчив до
190°С.Порапак Т — наиболее полярный пористый полимер, имеет наиболь¬
шее удерживание воды, применяется для определения формальдегида в
воде, устойчив до 190°С.Семь типов пористых полимеров выпускает фирма «Джон Мэнвилл»
(США).Хромосорб 101 применяется для быстрого и эффективного разделения
жирных кислот, пшколей, спиртов, эфиров (простых и сложных), альдеги¬
дов и кетонов. По своей структуре он относится к макропористым адсорбен¬
там. В изотермическом режиме может работать при температурах до 275°С
и допускает кратювременный нагрев до 325°С в [№жиме профаммирования
температуры колонки. Термогравиметрический анализ показывает, что по¬
лимер устойчив до 360°С. Хромосорб 101 можно использовать как в чистом
виде, так и с нанесенными жидкими фазами. При нанесении небольших
количеств жидкой фазы удерживаемые обьемы обычно уменьшаются. Этот
полимер имеет сравнительно слабую способность к адсорбции веществ, со¬
держащих гидроксильные группы. С ростом числа гидроксильных групп
наблюдается уменьшение удерживаемых объемов. Вода элюирует перед ме¬
танолом, этиленгликоль (Осип = 190°С) выходит раньше н-амилового спирта
(йсип = 137°С). Все спирты, имеющие температуры кипения, одинаковые с
температурами кипения алканов, будут элюировать раньше соответствую¬
щих алканов, например, н-пропиловый спирт (йсип = 97°С) элюирует рань¬
ше н-гептана (torn = 98°С). Поверхность хромосорба 101 однородна, пики,
как правило, симметричны.Хромосорб 102 решмендуется для разделения постоянных и углево¬
дородных газов, низкомолекулярных полярных веществ. Хромосорб 102
имеет сравнительно высокую удельную поверхность.Хромосорб 103 предназначен в первую очередь для быстрого и эффек¬
тивного разделения аминов, амидов, гидразинов, а также спиртов, альдеги¬
дов и кетонов. Хромосорб 103 — полиароматический пористый полимер,
синтезирован специально для разделения аминов и основных соединений.
Метиламин легко отделяется от легких газов, например от аммиака. Среди
диаминов времена удерживания уменьшаются при сближении аминогрупп,
например 1,2-диаминопропан элюирует раньше 1,3-диаминопропана. При
относительно низких температурах вода элюирует в виде несимметрично¬
го пика, при температурах выше 150°С пик становится симметричным.179
Яшин ЯМ. Яшин Е.Я., ЯшинА.Я. Гюовая хроматографияХромосорб 104 синтезирован из акрилонитрила и дивинилбензола, по¬
этому имеет полярную поверхность. Хромосорб 104 эффективно работает
при отрицательных температурах, шмнатных и высоких; очень эффективен
для разделения нитрилов, нитропарафинов, спиртов, кетонов, альдегидов и
углеводородов. Наиболее важной характеристишй хромосорба 104 является
его способность разделять серасодержащие соединения при низких шнцен-
трациях и изомеры ксиленолов. По селективности хромосорб 104 сильно от¬
личается от других полимеров этой серии. На хромосорбе 104 насыщенные
угаеводороды элюируют раньше ненасыщенных.Хромосорб 105 — полиароматический полимер с большой удельной
поверхностью (600—700 м^г), предназначен для эффективного разделения
формальдегида, воды и метанола, ацетилена и легких угаеюдородов, а так¬
же соединений других классов с температурами кипения не выше 200°С.
Полярность хромосорба 105 меньше, чем хромосорба 104.Хромосорб 106 — неполярный полимер из сшитого стирола с боль¬
шой удельной поверхностью (700—800 м^г), предназначен для разделения
газов и особенно для разделения кислот —С5 от спиртов — С,.Хромосорб 107 — пористый полимер со средней полярностью из
сшитого акрилового эфира с поверхностью 400 — 500 м^г, предназначен
для разделения различных классов соединений.Применение неполярных пористых полимеров вследствие слабой
их специфичности распространяется от анализа компонентов воздуха до
разделения гликолей. Их можно использовать при работе с программи¬
рованием температуры в интервале от —80 до 200°С. Их применяют не
только как адсорбенты, но и как носители в газожидкостной хромато¬
графии. Компоненты воздуха разделяется при —70°С на колонке дли¬
ной около 6 м с порапаком Q (рис. 4.22); порядок выхода необычный:
азот, кислород, аргон — обратный порядку выхода на цеолитах, что соот¬
ветствует увеличению электронной поляризуемости адсорбента. ?&>рошо
разделяются окислы азота, серасодержащие газы, хлористый водород и
другие агрессивные вещества (рис.4.23). Многообещающими, оказались
первые работы по использованию пористых полимеров для разделения
дейтерированных соединений (рис.4.24). Можно хорошо разделить угле¬
водороды С, — со всеми изомерами. Было показано, что на пористых
полимерах можно разделить все предполагаемые компоненты атмосферы
Марса: за 14 мин с программированием температуры от 25 до 150°С раз¬
делялись водород, азот, кислород + аргон + окись углерода, двуокись угле¬
рода, метан, вода, окись азота, аммиак, фтористый метилен, сероводород
и двуокись серы.180
ГШМА-COiH,Sso*HaГазо-адсорбиионная хроматография
N2*Ar*0]*C0 NjCOSИ,5CO,Lso,ОгAruCOДa Юf/min ——12 M0Ыa.4 et/mUi —ЮPuc. 4.22. Разделение газов на пористых полимерах:а - хроматограмма газов (указаны на рис.) на двух колонках с порапа-
юом Q, соединенных через детектор; первая колонка 300 х 0,3 см при 90°С;
вторая колонка 600 х 0,3 см при - 70°С;б - хроматограмма на тех же колонках, что и в «а»), но первая шлонка
при 75°С, вторая при - 65“С.СРгОоы6 12
f/min—<18Рис. 4.23. Хроматограмма фторированных соединений:а - на колонке 300 х 0,3 см с порапаком Т при 150°С, газ-носитель - ге¬
лий, скорость потока - 60 cmVmhh. Порядок выхода: CF^, CjF^, C^F^, CjF^,
CF3C s CCF,, C3F,, цис-С/,, транс-С/^, цис-С,Р,, CFj= CF - CF = CF^,
H30-C,F,, (СЕз)зС = СР^;6 - на двух составных колонках; первая колонка 60 х 0,3 см с порапа¬
ком Т, вторая колонка 120 х 0,3 см с порапаком Q при 23“С.181
Яшин Я.И.. Яшин Е.Я., Яшин А.Я.Гюовая хроматтаФтСН4а) f/min45 50
bi f/min-3t 35:) f/mm39Рис. 4.24. Хроматограммы смеси CD^ и СН^ на колонках с пористы¬
ми полимерами. Полимер - порапак Q, газ-носитель - ге¬
лий, детектор - ПИД:
а - колонка 30 м х 0,3 см, размер зерен - 50-80 меш при 0"С;
б - та же колонка при - 45“С;
в - колонка 430 х 0,2 см при - 23°С.Очень важной областью применения пористых полимеров является
разделение и анализ сильнополярных веществ. На полимерах хорошо
разделяются спирты, кислоты, амины, альдегиды, кетоны, эфиры и др
(рис. 4.25, 4.26, 4.27). Анализ такой трудноразделяемой смеси, как фор¬
мальдегид — метанол — вода, успешно проведен на колонке с фазепашм
с жидкой фазой (рис 4.28).Особо следует остановиться на анализе воды. Известно, что анализ
воды в хроматографии затруднен из-за сильной адсорбщ1и, пшш очень не¬
симметричны. Другие способы — путем перевода ее в ацетилен или водород— громоздки и не всегда удобны. Из шлонки с пористым полимером вода
выходит быстро между этаном и пропаном, пик симметричен (рис. 4.29). Это
дает возможность широш использовать пористые полимеры для определе¬
ния примеси воды во многих органических растворителях [37] (она выходит
значительно раньше пика основного шмпонента, поэтому можно вводить
очень большие пробы) (рис. 4.30). И наоборот, открываются широкие юз¬
можности для анализа летучих органических веществ сточных вод.С успехом осуществляется анализ веществ с аномально высокими
температурами кипения, вызванными ассоциацией молекул в растворах
гаишлей, глицерина, фенолов и др.182
тмА^Газо-адсорбиионная хроматографияНекоторые вещества, обычно используемые в газовой хроматографии
в качестве жидкой фазы, можно разделить на пористых полимерах. В част¬
ности, диметилформамид и диметилсульфолан можно разделить на полу¬
метровой колонке с хромосорбом 101 при 190 °С.В большинстве работ пористые полимеры применены для разделения
веществ, молекулярные веса которых не превышают 150. В одной из по¬
следних работ был применен порапак Q для разделения антиоксидантов,
алкалоидов, эпоксидных смол и ализариновых красителей (с молекуляр¬
ными» весами до 1500) с использованием в качестве носителя вещества в
сверхкритическом состоянии, что облегчило вытеснение тяжелых молекул
с поверхности полимера.При использовании пористых полимеров в хроматографических ко¬
лонках не требуется их предварительное прокаливание с целью осушки,
как для других адсорбентов, например силикагелей и цеолитов. Вода и
другие примеси в воздухе сорбируются на пористых полимерах слабо, по¬
этому перед анализом достаточно продуть колонку с пористым полимером
около 1—2 ч непосредственно в хроматографе.Даванковым В.А., Цюрупой М.П. разработаны сверхсшитые полисти-
рольные сорбенты «Стиросорб», их адсорбционные и хроматографические
свойства исследованы в работах [38, 39]. Эти адсорбенты перспективны
для разделения газовых смесей.t/minРис. 4.25. Хроматограмма спиртов: I - метанол; 2 - этанол; 3 -
изопропанол; 4 - н-пропанол; 5 - втор-бутанол; 6 - изобу-
танол; 7 - н-бутанол; 8 - 2-пентанол; 9 - изопентанол; 10
- н-пентанол; колонка 180 х 0,3 см с хромосорбом-101 при
200°С; газ-носитель - гелий со скоростью 50 cmVmuh.183
Яшин Я.И., Яшин Е.Я., Яшин А.Я.Газовая хршатогтФияРис. 4.26. Хроматограммы полиспиртов:а) гликоли: 1 - этиленгликоль; 2 - 4,2-пропандиол; 3 - 2,3-бу-‘
тандиол; 4 - 1,3-пропандиол; 5 - 1,3-бутандиол; 6 - 1,4-бу-
тандиол; 7-диэтшенгликть; 8-глицерин. Колонка 120х 0,3см
с хромосорбсм-101 при 21 ОХ!; гелий, 50 сл^/мин.;б) хроматограмма: 1 - глицерин; 2 - эритрит; 3 - ксилит;
4 - арабит на колонке 100 х 0,6 см с полипатм при 250Х!,
газ-носитель - азот.Рис. 4.27. Хроматограмма аминов на колонке 120 х 0,4 см с хромо-
сорбом-103:а) 1 - метиламин; 2 - этшамин; 3 - изопропиламин; 4 -
н-пропшамин; 5 - вторбутшамин; 6 - н-бутиламин; 7 -
изоамиламин; 8 - н-амиламин; 9 - н-гексиламин в режиме
программирования температуры от 200 до 250°С со ско¬184
ШМА±-Газо-адсорбиионнт хтматографияростью 15 7мин.; газ-носитель - гелий; скорость потока
35 см^/мин.;б) 1 - этилендиамин; 2 - 1,2-пропандиамин; 3 - 1,3-про¬
пандиамин; 4 - 1,4-бутандиамин; 5 - 1,5-пентандиамин
при240Т.Рис. 4.28. Хроматограмма формальдегида, воды и метанола на
колонке 100 X 0.2 см с порапаком Т при 140°С с газом-
носителем гелием, 25 см^/мин.с,н,(»С,Н«Н.0ili 4О i * t
аРис. 4.29. Хроматограммы неорганических, углеводородных газов и
воды (указаны на рис.):а) на колонке 100 х 0,4 см с пористым полимером хромо-
сорбом-102 при 57°С;б) на колонке с хромосорбом-104 при 130°С.185
Яишн ЯМ., Яшин Е.Я.. Яшин А.Я.Газовая храматографняи.ос г
аРис. 4.30. Хроматограммы на колонке 100 х 0,4 см с пористым по¬
лимером (СТ-ДВБ):а) при 60°С; б) при 120°С; в) при ISO^C. Соединения указа¬
ны на рис.Оксид алюминия. Оксид алюминия получают осаждением алюми¬
ниевых солей раствором аммиака или разложением алюмината натрия
двуокисью углерода, серной или азотной кислотой. В промышленности
активный оксид алюминия с высокоразвитой поверхностью получают из
технической гидроокиси алюминия, которую обрабатывают едким натром.
Образующийся алюминат натрия в растворе обрабатывают азотной кисло¬
той, при этом осаждается гидроокись алюминия. Осадок отфильтровыва¬
ют, промывают, сушат и прокаливают.Оксид алюминия существует в нескольких кристаллических формах,
однако устойчивыми кристаллическими формами являются только а- и
y-AljOj. a-AljOj встречается в природе в виде минерала корунда. a-AI^O,
образуется при нагревании гидроокиси алюминия до высоких температур
(900—1200°С). Вторая устойчивая кристаллическая форма—y-AljOj— ку¬
бическая, получается при нагревании других форм гщфоокиси алюминия
и алюминиевых солей до 600—900°С. Выше 900 °С y-AljOj необратимо
переходит в a-AljO, Полное превращение у-формы в а-форму заканчива¬
ется при 1200°С с сокращением о^ема на 14,3%. Известна также АЮОН в
двух модификациях; диаспор а-АЮОН, устойчивый до 350°С, в интервале
температур 350—420°С переходит в a-Al^Oj; бемит у-А100Н, устойчивый
до 600°С, при больших температурах переходит в a-Al^Oj [40].Удельная поверхность для промышленных образцов оксидов алюми¬
ния может колебаться от 170 до 300 м^г. Отечественная промышленность
выпускает активную окись алюминия двух типов: А-1 и А-2. Оксид алю¬
миния А-1 имеет насыпную плотность 0,4—0,5 г/см’ и содержит макропо-
ры, а А-2 — 0,55—0,8 г/см и не содержит макропор.186
fJIABA 4 Газо-адсорбтонная храматографгтАдсорбционная активность в сильной степени зависит от темпе¬
ратуры прокаливания. При повышении температуры оксид алюминия
постепенно теряет влагу, при этом высокоактивные полярные центры
освобождаются для адсорбции углеводородов. Однако в газовой хрома¬
тографии чаще, используют оксиды алюминия, частично дезактивиро¬
ванные адсорбированной водой, при этом сильно уменьшаются удержи¬
ваемые объемы и колонка с таким адсорбентом стабильнее во времени.
Обычно наилучшие результаты получаются при содержании влаги око¬
ло 2—3%. Для уменьшения адсорбционной активности на Al^O, наносят
различные жидкие фазы, в частности сквалан, силиконовые жидкости
и др. [2].Для высокотемпературных разделений используют оксиды алюминия,
на поверхность которых отложены различные неорганические соли и ще¬
лочи [2]. На таких адсорбеш-ах, разделяются углеводороды с числом ато¬
мов угаерода до 36 при сравнительно невысоких температурах.Волокнистый бемит имеет более однородную геометрическую струк¬
туру, чем y-AljOj. Бемит хорошо откладьтается на поверхности других ма¬
кропористых носителей и адсорбентов. Оксиды алюминия широко исполь¬
зуют для разделения легких угаеводородных газов, в частности, используя
микронабивные колонки, наполненные y-Al^Oj с небольшим количеством
сквалана (до 5—8%), можно полностью разделить все компоненты смеси
углеводородов С, - С^.Безводные y-AljOj очень селективно удерживают непредельные и аро¬
матические углеводороды. На совершенно обезвоженной сухой Al^Oj эти¬
лен элюирует даже позднее пропана. При низких температурах колонки с
оксидом алюминия позволяют разделять кислород, азот и двуокись углеро¬
да (рис. 4.31) [41].Оксид алюминия используют в газовой хроматографии для разделения
изотопов водорода (рис. 4.32) [42]. Оксиды алюминия, модифицированные
солями, как уже указывалось, применяют для разделения высококипящих
соединений как в изотермическом режиме, так и в режиме программиро¬
вания.Перед заполнением хроматографической колонки оксид алюминия
нужно прокалить при 200—300°С до постоянной массы.187
Яшт Я.И.. Яшин Е.Я.. Яшин А.Я.Газовая хроматографияРис. 4.31. Хроматограмма и СО^ на колонке 200 х 0,3 см сAiPj при 20°С.f/rain ■Рис. 4.32. Разделение изотопов водорода (указаны на рис.) на колон¬
ке 300 X 0,3 см с AiPj при 77 К, газ-носитель - неон.Оксид алюминия нанесен на внутреннюю поверхность капиллярных
колонок широкого диаметра, на которых успешно разделяется смесь угле¬
водородов С1-С10.Цеолиты (молекулярные сита). Цеолиты из-за своей уникальной
геометрической структуры — адсорбенты, обладающие ярко выраженной
избирательной адсорбцией. Селективная адсорбция на природных цеоли¬
тах была обнаружена несколько десятков лет назад, однаю из-за рассеян¬
ности и трудности выделения в чистом виде они не находили широкого
применения. В настоящее время освоено производство различных типов
синтетических цеолитов, называемых иногда молекулярными ситами, от¬
личающихся по структуре кристаллов и их химическому составу.188
fJlABA4_ Газо-адсорбиионная храматографшЦеолиты широко используют для очистки и осушки в промышленно¬
сти и лабораторной практике, а также для разделения газовых и жидких
сред, в частности и в газовой хроматографии.Синтетические цеолиты получают путем гидротермальной кристалли¬
зации в водных алюмосиликатных системах при атмосферном или повы¬
шенном давлении [2]. Таким образом, цеолиты представляют собой алю¬
мосиликаты кристалличесшй структуры Алюминий занимает в них осо¬
бое положение, он играет в цеолитах роль, близкую роли кремния, и может
быть включен в комплексный алюмокремниевый радикал. В цеолитах, как
и в других алюмосиликатах, алюминий так же, как и кремний, находится в
тетраэдрическои координации по кислороду и изоморфно замещает крем¬
ний в общем кремнеалюмокислородном каркасе. Чередующиеся (Si, О)- и
(А1, 0)-тетраэдры соединяются в трехмерных каркасных структурах цео¬
литов [2] (рис. 4.33). В таких каркасных структурах цеолитов обязательно
присутствуют катионы щелочных и щелочноземельных металлов в коли¬
чествах, строго эквивалентных содержанию алюминия. Это объясняется
необходимостью компенсации в тетраэдрических группах АЮ^,^ заряда из¬
быточного электрона, так как А1, имея только три собственных валентных
S, р-электрона, образует равноценные парноэлектронные связи с каждым
из четырех окружающих его кислородов за счет привлечения одного до¬
полнительного электрона [2].Рис.4.33 Структура цеолита типа А (а) и типа X и Y (б). S1I и
SIII - возможные места локализации катионов.Основным ядром решетки цеолитов является кубооктаэдр, состоящий
из 24 атомов алюминия и 48 атомов кислорода. В цеолитах типа А кубо-
окгаэдры связаны в простой кубической решетке, в цеолитах типа X они
упакованы более рыхло.По адсорбционным свойствам цеолиты значительно отличаются от
других адсорбентов. Благодаря однородным размерам пор они адсорбиру-189
Яшин Я.И., Яшин Е.Я., Яшин А.Я. Газовая хроматографияЮТ ТОЛЬКО те молекулы, которые могут проникнуть в эти поры. В связи
с тем что в узких порах происходит наложение силовых полей, цеолиты
обладают высокой адсорбционной способностью даже при повышенных
температурах. Так как остов пористых кристаллов цеолитов, построенный
из тетраэдров SiO^ и АЮ^ несет обменные катионы, то они обладают боль¬
шим сродством к молекулам с периферическим сосредоточением электрон¬
ной плотности в отдельных связях (т. е. молекулами, имеющими л-связи,
свободные электронные пары у атомов кислорода и азота). На этом основа¬
но применение цеолитов для очистки и осушки различных газов во многих
областях промышленности и техники, а также в газовой хроматографии.В газовой хроматографии цеолиты в основном используют для разде¬
ления и анализа низкокипящих газов Присутствие воды и двуокиси угле¬
рода сильно влияет на удерживаемые объемы на цеолитах. Поэтому для
стабильной работы колонки с цеолитами необходимо тщательно удалять
влагу и СО^ из анализируемого газа и газа-носителя. Было показано, что
при тщательной осушке газов цеолиты можно использовать и для дли¬
тельного автоматического анализа. При осушке газов (при помощи Р^О^)
колонка 400 см X 0,4 см с цеолитом NaX (13 X) проработала непрерывно
более 1000 циклов без изменения характеристик [2].Разделительная способность цеолитов зависит от выбора катионной
формы. На цеолите типа AgX может происходить необратимое поглоще¬
ние Hj, СО и ненасыщенных углеводородов, что, по-видимому, связана с
их химическим взаимодействием на восстановленном мелкодисперсном
серебре цеолита [2]. На разделительную способность цеолитов большое
влияние оказывает также количество и состав связующего материала, пол¬
нота катионного обмена, размер кристаллов и условия их синтеза и про¬
мывания. Следует отметить, что при использовании крупных кристаллов
цеолита размером 15 мкм хроматографическое разделение хуже, чем при
применении мелких кристаллов (2 — 3 мкм).Эффективность колонки с цеолитом может быть значительно увеличе¬
на (в 6 раз), если мелкие кристаллы цеолита, например типа СаА, нанести
на поверхность крупнопористого носителя хромосорба и не перемешивать
его со связующим материалом. В этом случае, по-видимому, сокращаются
пути диффузии и все кристаллы становятся доступными для адсорбиру¬
емых молекул. Высокой эффективности разделения на цеолитах можно
достичь при использовании капиллярных колонок, на внутренней поверх¬
ности которых нанесен слой мелких кристаллов цеолита [2].Если из-за влияния геометрического фактора молекулы не могут ад¬
сорбироваться в порах кристаллов цеолитов, они все же адсорбируются
на внешней поверхности цеолитов, а также в так называемых вторичных
порах, создаваемых крупнопористым связующим материалом.190
fgABA4- Гязо-адсорбтонная хттштографшМолекулярное сито — цеолит типа СаА-используют в газовой хро¬
матографии для отделения более тяжелых нормальных углеводородов
при некоторых температурах, практически необратимо адсорбирующих¬
ся, от ароматических и разветвленных углеводородов, которые не про¬
никают в поры цеолитов и поэтому свободно проходят через слой цео¬
лита. Например, цеолит СаЛ применяют для промышленного отделения
и определения нормальных углеводородов в бензиновых и керосиновых
фракциях [2].Перед вводом в колонку цеолиты необходимо обезвоживать при
450—500°С (при более высоких температурах разрушается их кристал¬
лическая структура) в течение 4—5 ч, особенно для селективного раз¬
деления смеси кислорода и азота. Для сокращения продолжительности
анализа и уменьшения температуры колонки при разделении легких
углеводородных газов и некоторых, других газов иногда целесообразнее
использовать несколько увлажненные цеолиты.Баррер выделил пять типов цеолитов по их молекулярно-ситовому
действию [2].В последнее время цеолиты типа X, в частности 13 X, используют
для количественного определения нафтенов и парафинов в нефтяных
фракциях [43]. Методом газо-жидкостной хроматографии на капилляр¬
ных колонках можно количественно установить покомпонентный со¬
став бензиновых фракций, выкипающих до 135 °С, выше этой величи¬
ны в пределах 135—200 °С число компонентов настолько сильно воз¬
растает, что точно определить состав изомеров нафтенов и н-алканов
уже нельзя. Было обнаружено, что на колонках с .цеолитом 13 X при
300—400 °С хорошо разделяются нафтены и н-алканы с одним и тем же
числом атомов углерода в молекуле, причем, парафины удерживаются
сильнее нафтенов. Предварительно нефтяные фракции нужно деаро-
матизировать. Для исключения крекинга на цеолите их обрабатывают
щелочью [43].На цеолитах СаЛ, СаХ и NaX хорошо разделяется смесь водорода,
кислорода, азота, метана и окиси углерода, но двуокись углерода десор¬
бируется только при температурах выше 200 °С (рис 4.34,4.35, 4.36, 4.37)
[44-46].Смесь аргона и кислорода на обычных цеолитах разделяется только
при отрицательных температурах (- 40°С и ниже) (рис. 4.38 а.б.).Цеолиты, подготовленные для газохроматографических опытов, необ¬
ходимо хранить в запаянных или герметичных сосудах.191
Яшин Я.И., Яишн ЕЖ. Яшин АЖ.Гаювая хроматографияи»ulРис. 4.34. Хроматограммы и на колонке 100 х 0,3 см с цео¬
литом СаА при ЗО^С, газ-носитель - гелий, 30 см^/мин.Рис.4.35 Хроматограмма разделенш смеси низкокипящих газов на
колонке 122 X 0,63 см с цеолитам СаА при программирова¬
нии температуры со скоростью 4 град/мин скорость газа-
носителя - аргона 60 мл/мин1-H^;2-0^S-N^.СО^-8-C/J^4-CH^SOjI-CO;e —СД , 7CH4 .со.о 2<.6 8f / min —Ю 12St70‘СРис. 4.36. Хроматограммы неорганических газов и метана на колон¬
ках 80 X 0,4 см с цеолитами (а - аттапульгит; 6 - сепио-
нит) в режиме программирования температуры.192
aSSAJLГазо-адсорбиионная хроматографияPuc.4.37 Разделение бутенов на капиллярной колонке, заполненной
кристаллами цеолита NaX с размерами 40—50 мкм при
21б°С (газ-носитель NJ:1 — н-бутан; 2 — транс-бутен-2; 3 — цис-бутен-2; 4— бу-
тен-1; 5 — изобутилен; б — дивинилАгАгL I124 812Рис. 4.38 а, 6. Хроматограммы разделенш аргона и кислорода на
колонке 300 х 0,3 см с цеолитом СаА при 20®С, газ-
носитель - гелий, 20 см^/мин.:а) аргон и кислород воздуха;б) искусственная смесь аргона и кислорода.193Гюоявя }р>о*иггографня
Яитн Я.И: Яшин Е.Я., Яшин А.Я.Гштмзершаттйфтill 11ЯПI r-fiЛLt t А А t t t t-Puc.4.39 a,6 Хроматограммы смеси метана (1), этана (2), пропана
(3) и этилена (4), полученные на цеолитах СаХ (а) и NaX (б)
при различных температурах и зависимости отношения ис¬
правленных времен удерживания этилена и этана к времени
удерживанш пропана от температуры колонки (колонка 50
X 0,4 см, размер зерен 0,25— 0, 5 мл, газ-носитель — гелий,
детектор — катарометр)При температурах выше 150“С из колонок с цеолитами элюируют
углеводороды С,-С,, их удерживание сильно зависит от температуры (рис.
4.39 в, г, д, е). Удерживание на колонках с цеолитами сильно зависит от
адсорбционной влаги. На рис.4.40 показаны эти зависимости. Отношение
удерживания этилена и пропана сильно падает, тогда как отношение двух
неполярных соединений (этана и пропана) остается постоянным [47].При удерживании кислорода при небольших концентрациях наблюдает¬
ся аномалия. При юнцентрациях менее 0,01% время удерживания кислорода
изменяется с изменением его концентрации [48]. На рис.4.41 представлены
хроматограммы смеси кислорода и азота на шпонке с цеолитом СаА, газ-
носитель гелий. Колонка предварительно прогревалась при 400 ®С в потоке
очищенного гелия. Хроматограмма на рис.4.41 а соответст^ет шнцентрации
кислорода 4,8x10^^% (об), при этом кислород выходит раньше азота и перед
пишм кислорода пик аргона При меньших шнцентрациях (4,6x10^ %) кис¬
лород выходит на 4 мин позже, тогда как времена выхода азота и аргона не194
mssdJLГаза-адсопбюшнная хроматографияизменяются (рис.4.41 б). При тех же условиях были исследованы цеолиты
СаХ и NaX. На цеолите СаХ получены аналогичные результата. После
предварительной продувки шлонки с СаА очищенным потошм и СО^
аномальный эффект исчезал. На цеолите NaX этот эффект проявляется в
меньшей степени.Ароматические угаеводороды не проникают внутрь цеолита СаХ, по¬
этому они разделяются за счет адсорбции на внешней поверхности [49].LЛ-в г * I t ШитJ—I ■ Iо ? 4 SHUHРис. 4.39 в, г, д, е Хроматограммы смеси углеводородов на цеолите NaX
при температурах 153°С (в). 165°С (г). 172°С (д) и 180 °С (е)
(тпонка 50 х 0,45 см, скорость газа-носителя — водорода
50 мл/мин, npcSa 0,2 мм, детектор — катарометр) [55] 1 -
СН, 2 - С3 - С Д; 4 - Cfl- 5 - С fl,м',/м1и»о,%Рис. 4.40 а,б Зависимость отношения исправленных времен удержи¬
вания от количества адсорбированной цеолитом NaX вла¬
ги (температура 150°С, величина пробы 0,2 мл) 1 — эти-
лен/пропан; 2 — этан/пропан13*195
Яшин Я.И.. Яишн Е.Я,, Яшин А.Я.Газовая хретатотгфияО 24минРис. 4.41 а Хроматограмма разделенш кислорода и азота. 111-JLШттрацш ттрШ, %Рис. 4.41 б Зависимость времени удерживания кислорода от его
концентрации для немодифицированной и модифициро¬
ванной колонок с цеолитом СаХ.196
Газо-адсорбиионная хттапографш''IlJU^diX2 J
HUHPuc. 4.41 в. Хроматограммы разделенш кислорода с концентраци¬
ями 2 X 1&^% и 8 X 10'*% на модифицированной колонке с
цеолитом СаХ.Неорганические соли как адсорбенты. Неорганические соли ис¬
пользуют в газовой хроматографии как в качестве растворителей, так и в
качестве адсорбентов. Для хроматографичесюго разделения применяют
эвтектическую смесь неорганических солей (нитратов лития, натрия и ка¬
лия) при 150—400°С. В этом случае, по существу, использовали газо-жид-
юстный вариант хроматографии. Как адсорбенты неорганические соли
были использованы для разделения о-, м- и п-терфенилов; соли в шличе-
стве 25% от массы наносились на хромосорб Р. Более детальное исследова¬
ние разделительной способности различных солей (в основном хлоридов)
по отнощению к многоядерным ароматическим соединениям было прове¬
дено в работе [50]. В этих случаях непористые соли наносили на твердые
носители, используемые в газожидюстной хроматографии.Соли также можно наносить на адсорбенты-носители: активные окси¬
дов алюминия, макропористые силикагели и пористые полимеры [51-54].
Непористые соли в тоншдисперсном состоянии без носителя, в частно¬
сти BaSO^, использовались Беляювой, Киселевым и Солояном для полно¬
го разделения изомеров ксилола [21] (рис. 4.42), изомеров в С^ (рис.4.43),
многоядерных углеводородов (рис. 4.44) [55-58].197
Яшин ЯМ: Яшин ЕЛ, Яшин АЛГазовая хртащшмфтП t,mHРис.4.42 Хроматограммы смеси шомерных ароматических углево¬
дородов при 170 ®С на BaSO^It г 3 минРис. 4.43 Хроматограмма алкинов, алкенов и алканов, полученные на
колонке 220 х 0,1 см с BaSO^ при 50 ®С; 1 - изобутан, 2 - аль¬
фа-бутилен (бутен-1), 3 - юобутилен, 4 - н-бутан, 5 - цис-
бутилен, 6 - дивинил, 7 - транс - бутилен.198
Газо-адсорбиионная жтмстогтфшIIt JJA)lMl* ^
m*—Puc. 4.44 Хроматограмма смеси конденсированных и многоядерных
ароматических углеводородов на образцах сульфата ба¬
рия, a-BaSO-2 (350 «)иб~ BaSO-2 (500 % I - нафталин,2 - бифенил, 3 - о-терфенил, 4 - фенантрен, 5 -м-терфе-
нил, 6 - п-терфенил. Детектор пламенно-ионизационный,
скорость газа-носителя азота 45 мл/мин, скорость про¬
граммирования температуры: а- 20 ”/мин иб-25 ”/мин,
длина колонки 60 см, диаметр 0,3 см.Кроме BaSO^ хорошей селективностью обладает MoSj [51]. На колон¬
ке с MoS, можно разделить цис-транс соединения (рис. 4.45), диадаман-тильные угаеводороды (рис. 4.46).MoS* , Ш*С1г«ишРис. 4.45 Хроматограмма 2,2,4-триметилпентена-4 и цис- и транс-4-
метилгептена на MoS2 колонке длиной 0,9 м, диаметром 4 мм,
газ-носитель гелий, детектор пламенно-ионизационный.199
Яшин Я.И., Яшин ЕЛ, Яшин А.Я.Гтошя хрташшфшmin 60 55 50 15 10 5 О
Рис. 4.46 Хроматограмма диадамантил полщиклических углеводо¬
родов на MoSj колонке длиной 1 м, диаметром 3 мм, газ-
носитель азот, детектор пламенно-ионизационный.В хроматографии применяли комплексные пористые соли, получае¬
мые путем частичного удаления комплексообразователей при нагревании.
В качестве комплексообразователей могут служить аммиак, вода, пиридин,
1,10-фенантролин и др.Способ нанесения солей на макропористые твердые носители. На
практике получила распространение следующая наиболее общая методика
нанесения непористь1х солей на твердые носители: 15—25% соли от мас¬
сы носителя растворяют в дистиллированной воде (для водорастворимых
солей) и в раствор помещают твердый носитель. При непрерывном поме¬
шивании выпаривают воду, затем нагревают твердый носитель с солью до
температуры на несколько десятков градусов выше температуры плавле¬
ния соли Этой операцией обеспечивают более равномерное распределение
слоя соли на поверхности твердого носителя.Способ приготовления комплексных пористых солей. Наиболее
термически устойчивыми из описанных в литерагуре оказались комплексы
различных солей меди с бипиридином и 1,1,0-фенантролином. Для приго¬
товления таких комплексных солей меди к водному 0,01 М раствору соли
меди добавляют 0,01 М раствор комплексообразователя После растворе¬
ния комплексообразователя раствор медленно охлаждают до комнатной
температуры, при этом выпадают кристаллы комплексной соли. Высу¬
шенные кристаллы вводят в хроматографическую колонку и продувают их
инертным газом при 150—200°С для частичного удаления комплексообра-
зователя. После такой обработки получаются соли следующего состава:
Cu(Phen)j(NO,)j, Cu(Bipy)(NO,)j, CH(Phen)Clj и Cu(Phen)SO,-Hp (здесь200
0ЫВА_4 Газо-адсорбиионная хроматографияgipy обозначает бипиридин, а Phen—1,10-фенантролин). Наиболее устой¬
чивый комплекс Cu(Phen)SO^-HjO не разлагается при температурах ниже
500°С. Такие комплексные пористые соли могут бьггь использованы и для
анализов с программированием температуры.ТЗК-М — адсорбент для хроматографии, изготавливают из трепела
Зикеевского карьера; его выпускала Горьковская база ВНИИ НО. Он имеет
удельную поверхность около 75—105 mVf, насыпную плотность 0,5—0,65
г/см^ ТЗК-М предназначен для газохроматографического разделения па¬
рафиновых и олефиновых углеводорода С,—С^, в том числе изо- и н-бу-
тиленов. При разделении углеводородов рекомендуется предварительно
нанести на ТЗК-М соду (2%) и прокалить при температуре 750—800°С в
течение 3 ч, после охлаждения на него наносят небольшие количества ва¬
зелиновою масла.Углеродные адсорбенты. Углеродные адсорбенты широко при¬
меняются в газовой хроматографии для разделения разных смесей от
благородных и постоянных газов до высокомолекулярных соединений.
Углеродные адсорбенты используются как в насадочных, так и капилляр¬
ных колонках. Кроме того, углеродные адсорбенты достаточно часто ис¬
пользуются для концентрирования микропримесей из газовых и жидких
сред.Существуют, по крайней мере, четыре аллотропные модификации
углерода: карбин, графит, алмаз и фуллерены [59] (рис. 4.47). Графит и
алмаз бывают как природные, так и синтетические. Фуллерены и кар¬
бин получены только синтетически, хотя есть и неподтвержденные све¬
дения о наличии фуллерена в природе. Фуллерены - полиэдрические
молекулы замкнутой структуры. По рекомендациям ИЮПАК фуллере¬
ны - замкнутые сферические многогранники, построенные из трижды
координированных атомов углерода, имеющих 12 пентагональных и
(п/2 - 10) гексагональных граней, где п>20. Например, в фуллерене С^
[60] идентичность всех углеродных атомов подтверждена с помощью
ЯМР. Молекулы фуллерена С^, - наиболее симметричная, диаметр сфе¬
ры этого фуллерена 7,1 А, доступная внутренняя полость имеет радиус
1.7 А [61].Нанотрубки впервые получены в 1991 г. [62]. Они представляют со¬
бой свернутую в цилиндр графитовую плоскость, поверхность нанотруб¬
ки выложена правильными шестиугольниками. Нанотрубки использова¬
лись в газовой хроматографии как адсорбенты для разделения газовых
смесей.201
Яшин ЯЖ. Яшт ЕЛ. Яшин АЛ Газовая хроматографияа: •Нан(УфубкиФуллерены{М)фуамра| (та]^амртРис. 4.47 Углеродные структурыАктивированные угли. Активированные угли с 1950 г применялись
для разделения постоянных газов, смеси гелия, неона и водорода, смеси по¬
стоянных и благородных газов и легких углеводородных газов С,-Сз [63,-
64]. Однаш активированные утаи сильно неоднородны по геометрической
структуре и их поверхность частично окислена, поэтому их нельзя было ис¬
пользовать для разделения полярных газов и более тяжелых углеводородов.
В работе [65] впервые предложено оценивать степень окисленности угле¬
родных адсорбентов газохроматографическим методом по тестовым смесям,
состоящим из соединений различной природы (метод обращенной газовой
хроматографии). Детально активированные угли как адсорбенты были ис¬
следованы в работе [66], в которой измерены параметры удерживания по¬
стоянных и углеводородных газов на разных активированных углях, наи¬
лучшим оказался уголь СКТ. Позднее активированные угли были заменены
углеродными молекулярными ситами на полимерной основе (карбосита).Карбосита. Однородные тонкопористые углеродные адсорбенты (угли
“саран”) впервые бьши изучены в работе [67]. В этой работе измерены
удельные удерживаемые обьемы и теплоты адсорбции постоянных и угле¬
водородных газов, включая Аг, Кг, Хе, СО, СО^, N0, N^0, N0^, NH^, H^S и
все углеводородные газы С, - Cj. Бьши измерены эти параметры и для Нр.
Бьшо показано, что этот адсорбент из-за геометрической и химической
однородности значительно эффективнее разделяет смеси указанных со¬
единений. Логарифм объёмов удерживания и теплоты адсорбции пропор¬
циональны электронной поляризуемости молекул. На этом же адсорбенте202
mssM-Гаю-адсорбииошая хроматографияхорошо разделяется смеси Не, Ne, Н^ и О^, гфи комнатной температуре
(рис.4.48). Одновременно можно разделить постоянные и угаеюдородные
в режиме программирования (рис.4.49).1* J4Рис. 4.48 Хроматограммы разделенш смеси газов; а) 1 - Не; 2 - Ne;3 - Н^; колонка размерам 300x0,4 см сугшем типа «Саран» при
25'’С, скорость газа-носителя (азота) 30 мл/мин, б) I - Не;2 - Аг; 3 - Кг; 4 - Хе: колонка размером 300x0,3 см темпе¬
ратура 15СРС, скорость газа-носителя (азота) 50 мл/мин.JiiУAJVt 12 К№««м Ни|»Рис. 4.49 )фоматограмма разделения постоянных и углеводородных
газов на колонке размером 300x0,3 см с углем «Саран» с про¬
граммированием температуры от 25 до 250 со скоростью
30 град/мин. 1-0^ 2-N- 3-СО; 4-СН^ -5-С/// 6-СОу 7-СД;
8-С^, 9-С^, 10-C/f,203
Яшин ЯМ., Яшин ЕЖ. Яшин А.Я.Газовая хроматографияВ этом же году Р. Кайзер опубликовал исследования на подобных ад¬
сорбентах [68]. Карбосита были применены для разделения газов в других
работах [69,70]. В настоящее время большой ассортимент карбосит выпу¬
скает фирма «Supelco» с удельной поверхностью от 400 до 1200 м^г (та¬
блица 4.14).Таблица 4.14Углеродные адсорбенты для газовой хроматографннАдсорбентS, м^гОбъем пор, мл/гПлотн.Vgmo-ппмикрообщийг/мл20°С!&1рбосита1.Карбоксен 5635100,200,590,537782.Карбокссн 5644000,190,500,602763.Карбоксен 5694850,070,390,582574.Карбоксен 100012000,420,850,444185.Карбоксен 10015000,170,530,612346.Карбоксен 100310000,470,800,46797.Карбостх» S III8200,380,390,613788.Карбосфера10000,500,54—779Макропористые углеродные адсорбенты1.Карботреп F5—0,020,66-2.Карботреп С10—0,020,72-3.Карботреп100—0,580,36-4.Карботреп Y250,0030,120,42-5.Карботреп X35000,630,41-6.Карбограф560— '——-7.Карбопак А и С10-15———-8.Карбопак В90-100———-9.Карбохром-88———-10.Карбохром-8080-90———-11.Графитированная
термическая сажа7———-204
Газо-адсоубтонная хроматографияАдсорбенты на основе ГТС. Как ранее было указано для разделения
jcaK легколетучих, так и высококипящих соединений как полярных, так и не¬
полярных нужны были непористые или макропористые однородные угле¬
родные адсорбенты. Такие адсорбенты созданы на основе ГТС. Внедрение
в практику газовой хроматографии адсорбентов на основе ГТС было зна¬
чительным достижением. ГТС получают прокаливанием термической сажи
при температуре около 3000“ С в инертной или восстановительной среде.
При таких темперагурах исходная сажа освобождается от всех органических
и неорганических примесей, частицы сажи образуют полиэдры, поверхность
которых образована базисной гранью графита. Электронно-микроскопиче-
свое исследование на приборах высоюго разрешения показало, что поверх¬
ность частиц ГТС имеют мало дефектов. Благодаря однородной плосшй
поверхности ГТС является идеальным адсорбентом для хроматографии. Не¬
пористые частицы ГТС размером 100-500 нм имеют удельную поверхность
в пределах 5-10 мУт.В газовой хроматографии такие тонкодисперсные частицы ГТС при¬
меняют в качестве адсорбентов следующими способами:- частицы ГТС вводят в крупные поры макропористых неполярных
носителей: силанизированного силикагеля [71], хромосорба W [72], пори¬
стого тефлона [73];- частицы ГТС вплавляют на внешнюю поверхность стеклянных [74]
или полимерных шариков [75];- ГТС откладывают на внутренней поверхности капиллярных колонок
из суспензии в тяжелом растворителе [75];- при длительном встряхивании мелкие частицы ГТС за счет гидро-
фобности агрегируются в более крупные частицы 100-300 мкм, которые
осторожно можно рассеять с помощью сит и заполнить ими колонки, проч¬
ность этих зерен мала, что снижает эффективность разделения;- на агрегированные частицы наносят пироуглерод, производя пиролиз
углеводородов в потоке газа, проходящего через слой частиц сажи. При
этом получаются механически прочные макропористые адсорбенты- кар-
бохромы А,В и С [76,77] и карбопаки [78] (таблица 4.14).Свойства поверхности карбохромов и карбопаков близки к поверхно¬
сти ГТС.Впервые ГТС как адсорбенты в газовой хроматографии бьши предло¬
жены в 1961 г. [71].В работе [79] был отмечен необычный порядок удерживания на ко¬
лонке с ГТС, связанный с неспецифичностью адсорбции. В работах [80-
^5] были исследованы адсорбционные свойства ГТС методом газовой205
Яишн Я.И.. Яшин Е.Я.. Яшин А.Я.Газовая хроматографияхроматографии: измерены удельные удерживаемые объемы, теплота ад¬
сорбции [80,81], изотерма и теплота адсорбции воды, бензола и метанола
[83], удельные удерживаемые обьемы и теплота адсорбции 68 соединений
разных классов соединений, включая и изомеры [84], проведено сопостав¬
ление констант адсорбции эфиров, измеренных газовой хроматографией и
статическим методом [85].В работах впервые [86-90] бьшо показано, что ГТС (Задает высокой
селективностью при разделении изомеров и соединений, отличающихся по
геометрической структуре. Селективность ГТС к изомерам положения, цис-
и транс-изомерам оказалась настолько высока, что насадочные шпонки не¬
большой длины (1-2 м) с ГТС разделяют многие смеси изомеров лучше, чем
капиллярные колонки с жидкими фазами [ЭД]. На колонках с ГТС сотрудника¬
ми А.В. Киселева и К.Д.Щербашвой в течение 1961 - 1984 гг. были изучены
зашномерности удерживания соединений разных классов (таблица 4.15).Таблица 4.15Перечень соединений, закономерности удерживания которых
исследованы на колонках с ГТС.п/пСоединенияСсылка1Изомеры разных классов86-902Терпеновые913Нормальные углеводороды924Дифенил и терфенилы9351,2- бис (триметилсилил) замещенные этилены и их германиевые
аналоги946Металлоорганические соединения IVB группы957Н. алкены С6-С10968Углеводороды С1-С6979Перфторированные9810Галогензамещенные алканы С1-С69911Постоянные газы10012Дейтерозамещенные углеводороды10113Ароматические углеводороды С6-С1010214Циклододекагриены- (1,5,9)10315Н. алкиныС6-С1010416Альдегиды, кетоны и спирты105206
пШШ-Газо-адсорбтонная хроматографияп/пСоединенияСсылка17Полиметилциклогексаны10618Ароматические и полициклические10719Инденуглеводороды10820Инданугаеводороды10921Трицикло (7,3,0,0 додеканы, тридеканы и тетрадеканыПО22Ароматические кислоты11123Азотсодержащие11224Терфенилы, фенилалканы и флюорен113,11425Стереоизомеры андростана и прегнана11526Стереоизомеры пергидроантрацена и пергидро фенантрена11627Пяти- и шестичленные азотсодержащие гетероциклические11728Кислородсодержащие11829Насыщенные трициклические углеводородные изомеры11930Полициклические ароматические и пергидроароматические
углеводороды12031Полихлоруглероды12132Серасодержащие аминокислоты12233Производные бицикло-2,2,1-гептанов12334Хлорбензолы12435Триангуланы125Эти работы стали классическими, многое из них признавались луч¬
шими публикациями года в зарубежных журналах. Проф. А.В. Киселев
своими выступлениями на международных симпозиумах, вызвал большой
интерес зарубежных ученых к ГТС. Этому способствовали длительные
стажировки и аспирантура зарубежных специалистов в лаборатории ад¬
сорбции и хроматографии Киселёва А.В. в МГУ, в частности специали¬
стов Германии, Англии, Франции, Австрии, Мексики, Алжира, Вьетнама,
Сирии, Болгарии, Чехословакии. В связи с этим появились серии работ
итальянских [126], французских [127], немецких [128] ученых.Основные достоинства ГТС и адсорбентов на их основе:- исключительная химическая однородность поверхности особенно
после обработки водородом при высокой температуре. ГТС не имеет207
Яшин Я.И., Яишн Е.Я., Яшин А.Я. Гаювая хроматографиявнутренних пор, адсорбенты карбохромы и карбопаки на основе гра¬
нулирования ГТС, имеют вторичную поверхность это зазоры между
частицами зерен ГТС;- поверхность ГТС неполярная, гидрофобная [2-4];- адсорбенты на основе ГТС самые универсальные на них можно раз¬
делять как неполярные, так и полярные соединения [2-4];- удерживание соединений на ГТС пропорционально электронной по¬
ляризуемости адсорбируемых молекул [149-151];- как отмечалось выше ГТС и адсорбенты на её основе обладают су¬
перселективностью к геометрическим изомерам [86-90, 152-157].
Проявление неспецифичиости адсорбции и удерживания в газо¬
вой хроматографии на ГТС. Удерживание соединений разных классов на
колонке с ГТС в преобладающей степени определяется вкладом неспеци¬
фического дисперсионного взаимодействия [2-4]. Даже в случае диполь¬
ных молекул индукционное взаимодействие остается незначительным.
Как указано выше удерживание и теплоты адсорбции хорошо коррелиру-
ются с величиной электронной поляризуемости анализируемых молекул
(рис.4.50).5Т;.гг,вг;4см5”12оса.снс1з^,уЩСгЩ)гО'ЩСЩ)^СО
С2ЩОН'^сщонJ » -вю12Of,Рис.4.50 а Зависимость IgVsl на графитированной термической
саже при 150 ®С от поляризуемости адсорбата а.208
ГЛАВА 4Газоадсорбтоннм хроматографияРис. 4.50 б Зависимость теплоты адсорбции неорганических и угле¬
водородных газов от их поляризуемости на: 1-графитиро-
ванной термической саже; 2-угле «Саран»; 3-углеродном
молекулярном сите С.Например, на колонке с ГТС циклогексанол, анилин, нитробензол,
ацетафенон выходят раньше н-декана [2,3], несмотря на наличие в указан¬
ных молекулах активных функциональных групп СО, ОН, NHj, способных
к сильному полярному специфическому взаимодействию, в частности к
образованию водородных связей. Соединения с группой NOj имеют боль¬
шой дипольный момент. В работе [83] оценена полярность ГТС по системе
Роршнайдера, ГТС имеет нулевую полярность.Величины удерживания на ГТС не связаны ни с температурой гашения,
ни с дапольным моментом. Это (^ьясняегся тем, что темпераяура кипения
зависит от юаимодействия MoneiQoi в жидюсти, где, в частности, молекулы
могут ассоциироваться за счет водородной связи, что повышает теплоту испа-
1№ния. При малых заполнениях поверхности ГТС в газовой хроматографии и
при повышенных темперэтурах колонки подобной ассоциации не происходит.
Роль молекулярного веса ослабляется различиями в геометрии молекул, влия¬
ющими на возможную их ориентацию и энфгию адсорбции на базисной шю-20914 Газовая хроматография
Яшин ЯМ. Яшин ЕЖ. Яшин А.Я. Газовая хроматографияСКОСТИ графита. Дипольный момент молекулы, вьиывающий при адсорбции
некоторую поляризацию атомов угаерода графита, не вносит значительного
вклада в энергию адсорбции и поэтому также практически не влияет на удер¬
живаемые о^мы. Молекула воды (температура кипения 100®С) элюирует на
колонке с ГТС за метаном (температура кипения метана ошло - 160“С), т.к.
электронная поляризуемость молекулы юды небольшая.1^к уже указывалось выше, основной вклад в удерживаемые объемы
на ГТС вносит дисперсионное взаимодействие. Энергия этого взаимо¬
действия определяется величинами электронных поляризуемостей и ван-
дер-ваальсовых радиусов звеньев молекул, взаимодействующих с адсор¬
бентом, а также ориентацией этих молекул на поверхности адсорбента. В
связи с этим представляет интерес сопоставление удерживаемых объемов
на ГТС для молекул ароматических и насьпценных углеводородов и их
фторированных аналогов. Электронная поляризуемость группы СН равна
1,4, а группы CF-1,7 . Ван-дер-ваальсов радиус водорода меньше, чем у
фтора в этих группах: 1,2 и 1,?А соответственно. При непосредственном
контакте с поверхностью атомов F, входящих в группы CFj и CF^ фторал-
канов, основную роль играет различие в ван-дер-ваальсовых радиусах F
и Н. В связи с этим частичная или полная замена атомов водорода в мо¬
лекулах алканов, цикланов и их производных атомами фтора должна вы¬
зывать уменьшение времени удерживания ГТС. Однаш, для плоских мо¬
лекул бензола, касающихся базисной грани графита не атомами водорода,
но атомами углерода с ван-дер-ваальсовым радиусом, равным ошло 1,8 А,
замена Н на F не изменяет расстояние от поверхности, но увеличивает по¬
ляризуемость звеньев CF по сравнению со звеньями СН. Поэтому, переход
от бензола к фторбензолу должен сопровождаться увеличением удержива¬
емого объема на графитированной термической саже.На рис. 4.51, 4.52, 4.53 показаны хроматограммы разделения соедине¬
ний различных классов и их фторпроизводных на графитированной терми-
чесшй саже и на полученном из нее механически более прочном адсор&нте
карбохроме [98]. Как и предполагалось, фторированные н-алканы (рис. 4.51)
элюируют раньше. Для фторбензолов и фтортолуола (рис. 4.52) порядок вы¬
хода обратный, т.е. фторированные молекулы этих ароматических углево¬
дородов действительно удерживаются сильнее. Атомы Н и F расположены
в плосшсти бензольного шльца, поэтому при ориентации этих молекул па¬
раллельно базисной грани графита расстояние звеньев СН и CF до поверх¬
ности одинашвое. Для более тяжелых фторированных аналогов, вследствие
большей величины поляризуемости, дисперсионное взаимодействие с фа-
фитом больше и фторированные бензолы и щ>угие ароматические углеводо¬
роды плосшго строения удерживаются адсорбентом сильнее. В молекулах
дифенила бензольные шльца повернуты друг относительно друга, поэтому
идеально плосшй ориентации этих молекул на поверхности фафита не мо-210
nidSdjLГазо-адсорбиионная хроматографиядеет быть и порядок выхода дифенила и его фторпроизводных (рис. 4.52)
таков же, как и для н-алканов (рис. 4.51), а также для их производных с
различными функциональными группами, например, для метилацетата
(рис. 4.53). Для ацетофенона же и фторированного в параположении ацето¬
фенона (рис. 4.53) порядок выхода сохраняется таким же, как у бензола или
толуола и их фторированных аналогов (рис. 4.52).При пониженных температурах на колонке с ГТС можно разделять
постоянные газы и дейтерированные соединения (рис.4.54 и 4.55). Следу¬
ет обратить внимание, что хорошо разделяется такая трудноразделяемая
смесь, как Аг, О^ и [100,101].Зависимость удерживания на ГТС от геометрической структуры
молекул. Адсорбция на плоских гранях частиц ГТС особенно чувстви¬
тельна к геометрической структуре молекул, т.к. энергия неспецифическо¬
го взаимодействия резко уменьшается с увеличением расстояния между
поверхностью и силовыми центрами (атомами, звеньями) молекулы. Поря¬
док удерживания смеси ряда циклических соединений с шестичленными
неплоскими и плоскими циклами показывает, что удерживаемые обьемы
циклогексана, циклогексена, циклогексадиена, бензола и пиридина мень¬
ше удерживаемого объема н-гексана, хотя их молекулярные массы близки,
а температура кипения н-гексана значительно ниже [2,3]. Удерживание со¬
единений с одним и тем числом атомов углерода, но с разным строением
сильно различаются для полиметилбензолов, н-алкилбензолов и фенилци-
кланов (рис.4.56).СЛ||09 *€ Г$0*С‘'7*1#1с,г„}X 1...1V ^Рис. 4.51. Хроматограммы перфторированных соединений, полу¬
ченные при разных температурах на колонке 50x0,4 см,
заполненной ГТС (скорость газа-носителя 40 мл/мин).21114-
Яшин Я.И.. Яшин ЕЛ., Яшин Л.Я.Гаювая хпаматография11»Чтч.Рис. 4.52 Хроматограммы перфторированных и нефторированных
соединений, полученные при разных температурах на ко¬
лонке 50x0,4 см, заполненной карбохромом (скорость газа-
носителя 40 мл/мин).Рсн^Го-СНзCPjСН,ггочоиСНгС-^i-Рис. 4.53 Хроматограмма на колонке 100x0,4 см с карбохромома) 1 -метиловый эфир трифторуксусной кислоты; 2-ме-
тилацетат (64°С), б) 3 - ацетофенон, 4 - фторацетофе-
нон (220PQ.212
0SMSJ—Газо-адсовбыитная хроматог/тфшо,JilРис. 4.54 Хроматограмма газов на колонке с ГТС при низких темпе¬
ратурах: а) газа воздуха при -145°С б)искусственная смесь
Аг, и СО с программированием температуры.• бРис. 4.55 Хроматограмма смесей: а) метана и дейтерометана
при-140РС и б) этилена и пердейтерометана при -90РС
на колонке с 350x0,4 см с ГТС.Рис. 4.56 Зависимость логарифма удельного удерживаемого объ¬
ема от числа атомов углерода в молекуле п для полиме¬
тилбензолов, н-алкилбензолов и фенилцикланов на ГТС.
Величина VA11 выражена в см^/м^.213
Яшт ЯМ, Яишн ЕЯ.. Яшин АЯ.1!шшхт!йттФшУдерживание на ГТС изомерных соединений различных гомоло¬
гических рядов с разветвленной и нормальной цепью. Для изо-соеди-
нений наблюдается значительное уменьшение удерживаемых объемов по
сравнению с соответствующими нормальными, так как разветвленные
молекулы, даже ориентируясь длинной осью вдоль поверхности, сопри¬
касаются с ней меньшим числом атомов, чем неразветвленные молекулы,
что уменьшает энергию дисперсионного взаимодействия. На колонках с
ГТС хорошо разделяются соединения, имеющие третичные и вторичные
атомы углерода, в частности у бутилбензолов и других углеводородов
(рис. 4.57). Триметилбутан удерживается слабее 2,4-диметилпентана,
хотя его температура кипения выше.Рис. 4.57 Хроматограмма бутилбензолов: 1 - трет - бутилбензол,2 - втор - бутилбензол, 3 - изо - бутилбензол, 4 - п-бу-
тилбензол; колонка 100x0,5 см с графитированной терми¬
ческой сажей.Удерживание на ГТС изомеров положения (орто-, мета- и пара). В га¬
зовой хроматографии традиционно трудной задачей считается разделение
смеси 0-, п- и м-ксилолов. В отличие от газожидкостной хроматографии на
полярных фазах на ГТС порядок удерживания изомеров ксилолов имеет
обратный порядок. Из-за больших размеров группы СН^, ван-дер-ваальсов
радиус которой (2,0 А) больше полутолщины плоского бензольного коль¬
ца (1,85 А), л<-ксилол может приблизиться к базисной плоскости графита
только одним атомом углерода кольца, а и- и о-ксилолы - двумя. Поэтому
из колонки с графитированной сажей л1-ксилол выходит значительно рань¬
ше п- и о-ксилолов. у и- и о-ксилолов возможные числа контактирующих с
базисной плоскостью графита атомов угаерода тольца при наиболее благо¬
приятной ориентации одинаковы. Поэтому различие в энергии адсорбции
п- и о-ксилолов на графитированной саже незначительно и на короткой
колонке с небольшой поверхностью сажи они плохо разделяются. Разделе¬
ние же м- и смеси о- и и-изомеров происходит на этой колонке с эффектив¬
ностью всего лишь 2000 теоретических тарелок.214
Газо-адсорбиионная хроматографияВ случае других заместителей порядок выхода может быть иным. В та¬
блице 4.16 приведен порядок элюирования различных о-, м- и и-изомеров.
Порядок выхода из колонки с графитированной сажей зависит от размеров,
формы и ориентации заместителей.Таблица 4.16Порядок выхода различных о-, м- и п-изомеров из колонки с графити-рованной сажейВеществоЗамещающиегруппыПорядок выхода1230, м, п- ксилолыДвеСН,МетаПара+орто0, м, п- крезолыОН, СНзОртоМетаПара0, м, п- диоксилбензолыЛвр ОНПараОртоМетанитротолуолыNO^, СНзОртоМетаПараТерфенилыДвеСДОртоМетаПараКрайние бензольные ядра в изомерах терфенила расположены не¬
сколько по-разному относительно плоскости среднего бензольного коль¬
ца. Из колонки с графитированной термической сажей первым выходит
о-терфенил [107]. Это связано с тем, что молекула о-терфенила наименее
выгодно ориентируется на плоской поверхности графитированной сажи,
так как два крайних бензольных кольца некопланарны со средним. Это
приводит к тому, что не все атомы углерода этой молекулы касаются пло¬
скости базисной грани графита одновременно. Полностью копланарные
молекулы и-терфенила наиболее выгодно ориентируются на поверхности
сажи и поэтому и-терфенил выходит последним. л<-Терфенил в этом от¬
ношении занимает, по-видимому, промежуточное положение, и поэтому
его пик выходит из колонки с графитированной сажей между пиками о- и
«-терфенилов. Разделение изомеров о-, м- и и-крезолов, диоксибенэолов
и нитротолуолов на графитированной термической саже объясняется, по-
видимому, аналогичным образом.Удерживание на графитированной термической саже цис- и транс¬
изомеров. Отмеченное влияние геометрической структуры молекул осо¬
бенно сильно проявляется при разделении цис- и отранс-изомеров. В ряду
Непредельных соединении i/мс-конфигурации имеют меньшие времена215
Яишн Я.И.. Яшин Е.Я.. Яишн А.Я. Газовая хроматографияудерживания (при более высоких температурах кипения), чем транс-пзо-
меры с тем же числом углеродных атомов [80]. В [82] также показано, что
t/мс-олефины удерживаются слабее транс-томеров с тем же числом ато¬
мов угаерода в молекуле. Так, на капиллярной колонке с графитированной са¬
жей относительный к и-пентану удерживаемый обьем цис-бутена (т. кип. 3,7®)
составлял 0,156, а у транс-бутет с т. кип. 0,86“ он равнялся 0,194. Даже
при очень небольших различиях в температурах кипения, как например,
в случае цис- и транс-пешеш-2, времена выхода заметно различались
(относительные удерживаемые объемы составляли 0,81 у ^мс-пентена-2
и 0,92 у транс-пттет-2). В случае цис- и транс-гексенов-2, имеющих
одинаковые температуры кипения (67,9“), удерживаемые обьемы также за¬
метно различались; для г/мс-изомера относительная к н-пентану величина
составляла 2,31, а для транс-изомера она равнялась 2,69. В случае цис- и
транс-гетенов-З, имеющих одинаковые температуры кипения (95,8“), ве¬
личины относительных удерживаемых объемов составляли 6,03 для цис- н
6,92 для транс-изомера.В гомологическом ряду цис- и транс-изомеров олефинов наблюдают¬
ся линейные зависимости логарифмов удерживаемых объемов и теплоты
адсорбции Q от числа углеводородных атомов п. Для г/мс-олефинов-2Q=l,0+1,32«,а для транс-олефинов0=2,1+1,32/1.Таким образом, прямая выряжающая зависимость Q от и для транс¬
изомеров олефинов, идет выше, чем для i/мс-изомеров, что объясняется
различной ориентацией молекул этих изомеров. Транс-изомеры олефинов
энергетически более выгодно располагаются на поверхности графитиро¬
ванной сажи по сравнению с ^ис-изомерами.В работе [155] хроматографически исследовали разные представители
цис- и транс-изомеров олефинов; пентадиен-1,3, гептен-3 и октен-2; все
они, благодаря различию в расположении звеньев их молекул на плоской
поверхности графитированной сажи, разделяются. Первыми выходят из
хроматографической колонки с графитированной сажей г/ис-изомеры.Нафтеновая часть бензина содержит 1,4- и 1,3-диметилциклогексаны,
разделение которых на жидких фазах затруднительно. У стереоизомеров этих
углеводородов с шестичленным кольцом и молекулярным весом 112 удержи¬
ваемые объемы на графитированной саже изменяются от 7 до 20 мл/м^ при
1(Ю°. Каждый из этих угаеюдородов, имеющих по два цис- и транс-изомера,
выходит из шлонки с графитированной сажей дщга пиками.Цис-транс изомеры разделены также в ряде других работ. Наиболее
яркий пример разделения цис-транс изомеров приведён на рис.4.58-4.61,
табл. 4.17-4.20. В работе исследованы цис-транс-изомеры ненасыщенных
сернистых соединений.216
[MSdJГазо-адсорбиионная хроматографияfODieЧЯu<?*?!«IJ ■ртштJ ; w 15 » 0 I 4 » I 10 и M и II 0 SPuc. 4.58 a) Разделение изомеров трицикло (8,4,0,(У'^) тетрадекана
на капиллярной насадочной колонке 120x0,1 см, размер ча¬
стиц 0,18-0,16 мм, скорость потока 8 см^/мин., темпера¬
тура колонки 220РСб) Разделение изомеров трицикло (7,4,0,0^-^) тридекана,
температура 200"С, остальные условш те же.в) Разделение изомеров трицикло (7,3,0,0^’^) додекана на
капиллярной насадочной колонке 200x0,1 см, размер ча¬
стиц 0,09-0,12 мм, скорость потока 4,5 см^/мин., темпе¬
ратура 24(УС.^ис. 4.59 Хроматограмма изомеров пергидрофлюоренов, полученная
при 250 ®С (условш см рис. 4.60): 1 - цис-син-цис; 2 - цис-
анти-цис; 3 - транс-анти-цис; 4 - цис-син-транс; 5 - транс-
син-транс (не зарегистрирован); б - транс-анти-транс.217
Яшин Я.И., Яшин Е.Я.. Яшин А.Я.Газовая хроматографияuXJLLXЛ. 1. L 1.О ' Ш а П № и Я tl тшРис. 4.60 Хроматограмма изомеров пергидроаценафтенов, полу¬
ченная на колонке размером 200x0,1 см, заполненной ГТС,
при 24(РС, расход газа-носителя 8 см^/мин: 1 - цис-цис-
цис; 2 - цис-цис-транс; 3 - цис-транс-цис; 4 - цис-транс-
транс (не зарегистрирован); 5 - транс-транс-транс.Таблица 4.17Термодинамические характеристики адсорбции изомеров
на ГТС (I - индекс удерживания; q, - теплота адсорбции).СоединенияV , cmWqi.1q,, кДж/мольТрицикл 0(8,4,0,0^'^тетрадекан1. цис-син-цис10,91156572. цис-анти-транс13,31179593. цис-анти-цис15,51196614. цис-син-транс27,41263645. транс-син-транс31,51290656. транс-анти-транс35,2130267218
imsddLГазо-адсорбиионная хроматографияСоединения cmVm^Iq,, кДж/мольТрицикло(7,4,0,0^^)тридекан1. ЦИС-СИН-ЦИС5,901041—2. цис-анти-цис6,171078573. транс-син-цис7.411107584. цис-син-транс9,061150605. цис-анти-транс10,71169616. транс-анти-цис13,11212637. транс-син-транс———8. тршс-анти-транс17,3127567Трицикло(7,3,0,(Р<*)додешн1, цис-син-цис2,19854472. цис-анти-цис3,43925503. цис-син-транс5,541002524. цис-анти-транс6,421027545. транс-син-транс7,211040556. транс-анти-транс8,36106857Таблица 4.18Термодинамические характеристики удерживания стереоизомеров
пергидроаценафтеновСоединениеСтруктураcmVm^q,, кДж/мольIПергидроценафтены1. цис-цис-цис661,035710152. цис-цис-трансй1,266110443- цис-транс-цисcS1,426310604- Цис-транс-транс66———219
Яшин Я.И.. Яшин Е.Я., Яшин А.Я.Газовая хроматографияСоединениеСтруктураcmVm^А,1q,, кДж/мольI5. транс-транс-
трансй1,786710916. аценафтен&17,11911409Таблица 4.19.Термодинамические характеристики стереоизомеров флюорена, пер-
гидрофлюорена и пергидроантрацена на ГТС (М - молекулярныйСоединенияСтруктураcmWА,1q,, кДж/мольIПергидроантрацены1. цис-син-цисODD3,755511252. цис-син-трансООО5,875812073. цис-анти-цисООО8,005912414. транс-анти-трансООО9,626313105. транс-син-трансООО18,61711336Пергидрофлюорены1. цис-син-цис1,826510942. цис-анти-цисОоО2,326711253. транс-анти-циссхи2,626811414. цис-син-трансиш4,0270—220
СоединенияСтруктураУд cmVm^q,, кДж/мольI5 хранс-син-трансоаи———6. транс-анти-трансOzO4,02721231флюоренооо102,5791660Таблица 4.20.Характеристики изомерных соединенийНомер изомера на
хроматограммеИзомерИндексудерживанияIV‘*Rназваниеструктура молекулыПергидрофенантрены1цис-син-цисо912020,282трмс-анти-цисо912420,383цис-анти-циса?12650,404транс-син-цисо?13250,785транс-син-транссс913620,896транс-анти-трансо913821,00221
Яшин Я.И., Яшин Е.Я., Яшин А.Я.Газовая хттатографияНомер изомера на
хроматограммеИзомерструктура молекулыИндексудерживанияIПергидроантраценыцис-син-цисссо11840,42цис-син-трансСХО12660,75цис-анти-цисССО12790,82транс-анти-трансООО12910,89транс-син-ссо13011,00цис-син-цистранс-анти-цисцис-анти-цистранс-син-транстранс-син-цис114411811202124212780,570,490,650,771,00Влияние геометрической структуры молекул на удерживание на ГТС
изучалось также в работах [110-158]. В статье [98] проведено сравнение
колонок с ГТС, с колонками ГЖХ при разделении одних и тех же смесей, а
в работе [99] показано, что в режиме программирования на колонках с ГТС
фоновый ионный ток не возрастает.На колонках с ГТС и макропористыми углеродными адсорбентами
измерены индексы удерживания многих соединений, в том числе и изо¬
меров (таблица 4.21). В таблице 4.22 приведены индексы удерживания со¬
единений с одним и тем же числом атомов углерода, а в таблице 4.23 даны222
rjjM^Газо-адсорбтонная хроматогтиЬтзависимости индексов удерживания от числа атомов углерода для разных
классов соединений. В работе [100] показано, что применение одновре¬
менно индексов удерживания одних и тех же веществ на жидких фазах
в газо-жидкостной хроматографии и индексов удерживания на ГТС дает
большую информацию о природе и структуре молекул (табл. 4.24,4.25).Таблица 4.21Соединения, для которых измерены индексы удерживания иа
графитированной термической саже (ГТС)СоединенияЧислосоединенийСсылкаИзомеры 1 ,2-циклопентинодекалина,
пергидрофлюорена и пергидрофеналена14158Изомеры трицикло(8,4,0,(Я’’)гетрадекана,
трицикло(7,4,0,0^'*)гридекана, трицикло(7,3,0,0^‘)
додекана20ПОАроматические и Вд1клические углеводороды29107Фенилэтилен (стирол), фенилацетилен, фенилцикланы,
бициклогексан, бициклогексальметан, бифенилэтилен
и бифенилацетилен11118Изомеры пергидроантраценов, пергидрофлюоренов и
флюорена12116Апициклические углеводороды С6 - С1334159Алкил фенолы25128Производные норборнаиов25160Углеводороды ряда индана и индена10108,109Стереоизомеры пергидроаценафтенов, аценафтена и
аценафтилена7120Пергидрофенантрены, пергидроантрацены и
Циклопентанодекалнны16116Апкилбензолы и алкенилбензолы34161Аякилдифенилы, терфенилы, дифенилалканы и
флюорены14113223
Яшт Я.И.. Яшт Е.Я., Яшт А.Я.Газовая хроматографияТаблица 4.22Индексы удерживания Ковача на колонках с ГТС (160).Соединения\Соединения\Углеводороды С4, 22,5°САроматические
углеводороды, 200°СИзобутан374Толуол704Бутен-1384Этилбензол761цис-Бутен-2402м-ксилол828транс-Бутен-2415п-ксилол838Бутадиен- 1,3410о-ксилол838Углеводороды С,, 56°Сн-Пропил бензол849Изопентан468Изопропилбензол793Неопентан4101,3,5-Триметилбензол949 ,Циклопентан4261,2,4-Триметилбензол966Пентен-14791,2,3-Триметилбензол973цис-Пентен-2481н-бутиленбензол936транс-Пентен-24931,2,4,5-Тетраметилбензол1097Пентадиен-1,44621,2,3,5-Тетраметилбензол1101Пентин-1463н-Пентил бензол1018цис-Пентадиен- 1 ,3526Алкилфеиолы (220°С)транс-Пентадиен- 1 ,3539Фенол713Углеводороды С6,100°С2-Метилфенол824Гексен-15772-Этилфенол888Гексин-15602-Пропил фенол964Гексадиен-1,55602-Изопропилфенол911Метилциклопентан5242-н-Бутилфенол1058Циклогексан5082-втор.Бутилфенол960Циклогексен5352-трем.-бутилфенол, 210°С942Циклогексадиен5392,3-Диметилфенол, 150°С977Бензол572224
fJt£Sd-i Газо-адсорбиионная храматограЛияТаблица 4.23Cesob индексов уддюкивания с числом атомов углерода на мшонках с ГТССоединенияУравненияМоногалоидные соединения С,-С, (при 100°С)Фгоралканы1= lOOn + 20Хлоралканы1= lOOn+130Бромалканы1= lOOn+178Иодалканы1= lOOn + 249Кислороаосодержащие соединения (при 150°С)Эфиры (простые)1= lOOn + 37Альдегиды1= lOOn + 61Метилкетоны1= lOOn + 66Симметричные кетоныI=100n + 41Первичные спирты1= lOOn + 87АцетоныI=100n+129Пропионаты1= lOOn+117БутиратыI=100n+125В таблицах 4.26-4.32 приведена связь параметров удерживания, ин¬
дексов удерживания и термодинамических характеристик изомеров с их
структурой.Таблица 4.24Индексы удерживания (I) алкифенолов на колонке с ГТС при 220°С
и на колонке с жидкой фазой OV-1701СоединенияIlovInrфенол12217132-метил-12748243-метил-13128324-метил-13108392-этил-13518883-этил-14078994-этил-1401920_2-н-пропил-14309643-н-пропил-14939904-Н-П1ЮПИЛ-1496997*22515 г,
2-изопропил-14089113-изопропил-14639424-изопропил-14629492-н-бутил-159610942-втор-бутил-—9604-втор-%тил-—10022-трет-%тил-—942*4-трет-бутил-15309902,3-Диметил-14019772,4-диметил-13599772,5-диметил-13609762,6-диметцл-14329913,5-диметил-1399978Табл. 4.25Индекс удерживания алкенил- и алкилбензолов на ГТС
и жидкой фазе OV -1СоединенияИзмерешше величишГАХГТСГЖХOV-11231 ©561663,7699767,2зОГ-762859,14СГ812885,45 0"*780870,0бСГ852949,5831938,69731018,8226
Г0М^Гсао-адая^иионнт хоаттогоафия15-227
Яитн Я.И.. Яшин Е.Я.. Яшин А.Я.Гтовйя хроматографияТермодинамические характеристики адсорбции ряда
и циклических углеводородов на ГТСТаблица 4.26
ароматическихСоединенияКоэффициентуравненияlnV.,=A+B/TИндекс
удерживания
I при 200°Ссм^м^-ди,кДж/моль-АВБензол11,354474-1,9037,2Толуол11,4551607042,0443,0Этилбензол11,9456367594,0046,9м-ксилол12,7963378262,4452,6о-ксилол12,6963368362,6052,6п-ксилол12,6963368362,6052,6н-пропилбензол12,9364,528362,7053,5Изопропилбензол11,9757867921,6848,01,3,5-триметилбензол13,4471969461,8459,91,2,4-триметилбензол13,4272789682,1960,51,2,3-триметилбензол13,4073079782,3260,7н-бутилбензол12,8768629377,1057,11,2,3,5-тетраметилбензол13,53793811042,2466,0228
тЛВА4Газо-адсовбтоннш хтшатографшСоединенияКоэффициентуравненияlnV.,=A+B/TИндекс
удерживания
I при 2(Ю°Ссм^м^-AU,кДж/моль-АВ1,2,4,5-тетраметилбензол13,47792211082,3066,01,2,3.4-тетраметилбензол13,49800011242,6066,5н-амилбеизол12,91733610353,9061,0Пентаметилбензол13,85875712516,9072,8н-гексилбензол13,07780611242,8065,0Гексаметилбензол13,989388-19,4078,0Нафталин12,82748210801,9762,2Дифенил13,298200119214,5068,1Фенантрен16,0811704-14,9097,0Антрацен15,81116(Ю-18,5097,0Тетралин12,8569969396,8058,3цис-детпин11,4958078412,1048,1транс-декалин11,6660718863,2050,3Циклогексан9,733523-0,7029,3Адамаитан10,724707-1,5039,0Диадамантан10,8358179194,4048,3Таблица 4.27Зависимость изменения внутренней энергии адсорбции -AU
от числа атомов углерода п на ГТССоединенияТип зависимостиАлканы-Ди = 5,98 + 5,44пФторалканы-Ди = 7,36 + 5,73пХлоралканы-Ди= 11,96+ 5,39пБромалканы-Ди= 14,23+ 5,3 InИодалкаиы-ди = 17,42 + 5,17пПростые эфиры-ди = 7,40 + 5,72пАльдегиды-ди = 10,31 + 5,44пКетоны-ди = 10,64 + 5,40пСпирты-Ди= 11,28 + 5,76пПропионаты-Ди=12,92 + 5,12пБутиреты-Ди=10,71+5,41п229
Яшин ЯМ. Яишн Е.Я.. Яшин А.Я. Газовая хроматографияТабл. 4.28Индексы удерживания алициклических углеводородов - С,,
на жидкой фазе OV-1 и ГТССоединения°с0V-1Iт,°сГТСIЦиклогексен80,7671,1160516,4Циклогексан83,0687,6536,6Циклогекса-1,3-диен81,5673,1539,2Циклогекса-1,4-диен88,5709,2569,5Циклогептан118,8806,9160605,0Циклогептен115,0794,2608,3Циклогепта-1,3-диен121,5820,3642,0Циклооктан151,1927,2180694,1с-Циклоокген142,0904,8680,9с,с-Циклоокта-1,3-диен141,0904,6681,2с,с-Циклоокта-1,4-диен145,2909,5681,2с,с-Циклоокта-1,5-диен148,4931,1691,2Циклононан178,41035,7220796,7с-Циклононен170,11023,6776,7t-Циклононен170,01014,5785,8с,с-Циклонона-1,5-диен1001,8750,6Циклодекан202,01133,9220912,7с-Циклодекан195,61122,5877,4t-Циклодекан193,01107,1886,4с,1-Циклодека-1,5-диен1099,8827,4Циклоундекан2211225,42601025,7с-Цнклоундецен195,61215,3993,0t-Циклоундецен193,01201,81001,61,1-Циклоундека-1,5-диен1177,6972,0с,1-Циклоундека-1,5-диен1196,0964,1Циклододекан2391323,72801154,2с-Циклододек.1н2301306,11113,1t-Циклододекан1291,51107,0с,с-Циклододека-1,5-диен1304,81176,21,1-Циклододека-1,5-диен1273,51073,9230
Газо-адсообиионная храматографшСоединения*кин’°с0V-1Iт,°СГТСI^Циклододека-1,5-диен1292,11071,8циклсяридекан1406,42801260,7с-Циклотридекан1401,31222,3t-Циклотридекан1379,11222,3Таблица 4.29Величины и -AU при адсорбции ароматических нитрилов и ами¬
нов, индолов и метилиндолов на ГТС, обработанной водородомСоединенияСтруктураУ^, при 200°СcmVm^-ди,кДж/мольНитрилыБензонитрил1,6749,6о-фенилпропионитрилн2,3853,7АминыАнилин0,9148,7tt-фенилэтиламинсн,0-C-NH,н2,1752,6N-метиланилинН3,5756,7N ,М-диметиланилин0-м-СН,
сн,7,964,1о-толуоидинсн,d""’3,3355,6231
Яшин ЯМ. Яшин ЕЖ. Яшин АЖ.Газовая хроматография232
ГЛАВА 4 Газо-адсорбиионная хроматографияТаблица 4.30Термодинамические хараю-еристики адсорбции на ГТС
алкилдифенилов, терфенилов, дифенилалкаиов и флуореиаСоединениеСтруетурамУ^,200°СcmVm^I-ликДж/мольДифенил15414,51211682-метилдифенилСНз1689,31154634-метилдифенилНзС-®-#168521374762,2 '-диметилдифенилСНз СНзА1825,31080602,6-диметилдифенилСНз&•©СНз18211,31176674,4' -диметилдифенилНзС-@-#-СНз1821921537842,6,2',6-тетраметилдифенилСН, СНз
СНз СНз2103,7103057Орто-терфенилл2305,0-85Мета-терфенил23038_95Пара-терфенил23078-95Дифенилметан©-СН2-®1684,5106658ФлуоренСНг166138149581233
Яшин Я.И.. Яишн Е.Я.. Яшин Л.Я.Газовая хроматографияСоединениеСтруктурам,200°С
cmVm^I-дикДж/мольТрифенилметан244491365741,2-дифенилэтан#-СН2-СН2-@182331318761,1-дифенилэтанщ)сн-снз1825,7109761Табл. 4.31Термодинамические характеристики адсорбции разных циклическихсоединений на ГТС
- удельный удерживаемый объем, ш - молекулярный вес,I - индекс удерживания, -AU-эне)гия адсорбции)АдсорбентыСтруктураmV200А,1СМ7М^ГТСI-ди,кДж/мольФенилэтилен(стирол)1041,783852Фенилацетилен1021,379450Фенилциклопропано-<1182,285054Фенилциклобутан1323,891056Фенилциклопентан<0^1468,098957Фенилциклогексан16016,2106760Бициклогексил16615,3106158234
ГЛАВА 4Газо-адсорбиионная хуоматографияАдсорбентыСтруктураmу200cmVm^ГТСГ-ди,кДж/мольБициклогексилметан18023,21108591,1 -бифенилэтилен18045119168Транс-1,2-бифеиилэтилен(транс-стильбен)0-с»=с,нО180[2180]91*1631*86Бифенил ацетилен
(толан)178[1260]52*1554*86Таблица 4.32Термодинамические характеристики индаи- и ииден углеводородов
на ГТС (М - молекулярный вес, I - индекс удерживания)СоединенияСтруктурамV^,200°CcmVm^I-ди,кДж/моль6ИнданSу(U>'31183,088454,11-метилиндан1324,993956,42-метилиндан1326,096257,35-метилиндан>Igо13210,1101660,41,1-диметилинданJ1464,592956,0235
Яшин Я.И., Яшин Е.Я.. Яишн А.Я.Газовая хроматографияУглеродные наночастицы как адсорбенты в ГХ.Интерес к углеродным наночастицам в аналитической химии сильно
возрос в последние года [162-164], описаны свойства и применения на¬
ноалмазов [165], фуллеренов [166], нанотрубок [167, 168], фуллерена,
заключенного в нанотрубки [169, 170], наноткани [171], юльцевых нано¬
трубок [172], нанотрубочек [173] и других [167, 169].Наночастицы могут использоваться как для химических прививок че¬
рез ковалентные связи, так и в виде исходных частиц без изменения. В не¬
которых случаях наночастицы подвергаются самосборке в определенные
ансамбли без ковалентных связей, агрегируясь в более крупные частицы.Нанотрубки и фуллерены вызывают большой интерес как новые ад¬
сорбенты в ГХ из-за их необычных адсорбционных свойств, термической
и химической стабильности.Впервые нанотрубки предложены в ГХ вместо активированных углей
и адсорбентов на основе ГТС для разделения полярных соединений: спир¬
тов, кетонов и эфиров. Из-за высокой способности к агрегации частицы
нанотрубок легш наносятся на поверхность стенок шлонок.236
—- — IjmrMm^mommxmmamoipadnm(Солонка 30x0,3 см, заполненная наночастицами, применена для раз¬
деления арометических углеводородов, алкенов, галоидоугаеводородов,
сояргов, кетонов, эфиров [174]. В работах [175, 176] волонки с нанотруб-
были применены для разделения алканов, алкенов и полиядерных
ароматических соединений. Ультра быстрое разделение угаеводородов и
gx произшдных было проведено на капиллярной шлонке длиной 50 см с
квадратными каналами 100 цм х 1(Ю цм [177] в режиме программщювания
температуры.фуллерены лж модификаторы сгшщонарных фаз в ГХ были примене¬
ны в работах [178,179].
Яшин Я.И.. Яитн Е.Я.. ЯшинА.Я. Газовая хроматографияЛитература1. James А.Т., Martin AJ.P. Biochem. J. 1952, v. 50, p. 679.2. Киселев A.B., Яшин Я.И. Газо-адсорбционная хроматография,
Москва, Наука, 1967, 256 с.3. Киселев А.В., Яшин Я.И. Адсорбционная газовая и жидкостная
хроматография, Москва, Химия, 1979, 291 с.4. Киселев А.В., Пошкус Д.П., Яшин Я.И. Молекулярные основы ад¬
сорбционной хроматографии, Москва, Химия, 1986,272 с.5. Яшин Я.И. Физико-химические основы хроматографичесюго раз¬
деления, Москва, Химия, 1976,216 с.6. Giddings J.C. Analyt. Chem. 1964, v. 36, p. 1170.7. Жданов С.П., Калмановский В.И., Киселев A.B., Фикс М.М., Яшин
Я.И. Ж. физ. химии 1962, т. 36, с. 1118.8. Березкин В.Г. Успехи химии 1996, т. 65, вып. 11, с. 991.9. Kiselev A.V. Yashin Ya.I. Inspezifische mid spezifische Adsorbenten
fur die Gas-Chromatografie, Gas-Chromatografie 1965, Akademie-Verlag,
Berlin, 1966, s. 103.10. Жданов С.П., Киселев A.B., Яшин Я.И. Ж. физ. химии 1963, т. 37,
с. 1432.11. Yaschin Ya.I. Zhdanov S.P., Kiselev A.V. Die Anwendmig por6ser
Glaser der Gas-Chromatografie, Gas-Chromatografie 1963, Lemia, 1963, s.
402.12. Бебрис H.K., Гиенко Е.Я., Зайцева Г.Е., Киселев А.В., Кустова Г.Л.,
Липкинд Б.А., Никитин Ю.С., Яшин Я.И. Нефтехимия, 1970, т. 10, с.
773.13. Bebris N.K., Kiselev A.V., Mokeev B.Ya., Nikitin Yu.S„ Yaschin Ya.I.,
Zaizeva G.E. Chromatografia, 1971, v. 4, №3, p. 93.14. КиселевА.В., Яшин Я.И. Нефтехимия, 1964, т. 4, с. 494.15. Kiselev A.V., Nikitin Yu.S., Petrova P.S., Scherbakova K.D., Yaschin
Ya.I. Anal. Chem. 1964, v. 36, №8, p. 1526.16. Лисичкин Г.В., Кудрявцев Г.В., Сердан A.A., Староверов С.М.,
Юффа А.Я. Модифицированные кремнеземы в сорбции, катализе и
хроматографии, Москва, Химия, 1986, с. 25.17. Зайцева Г.Е., Киселев А.В., Яшин Я.И. Ж. физ. химии 1973, т. 47,
Jfe6, с. 1531.18. Киселев А.В., Иогансен А.Л., Сашдынский К.И., Сахаров В.М.,
Яшин Я.И., Карнаухов А.П., Буянова Н.Е., Куркчи ГА. Физико¬
химическое применение газовой хроматографии, Москва, Химия,1973, 256 с.238
10МА — Лшвттт19. Бортников Г.Н., Егорочкин А.Н., Вязанкин Н.С., 4q)HHmeB Е.А.,
Яшин Я.И. Изв. АН СССР, сер. хим. 1970, Кй6, с. 1402.20. Gvozdovich Т.Н., Kiselev A.V., Yaschin Ya.I. Chromatogr^liia, 1973,
V. 6, p. 179.21. Kiselev A.V., Yaschin Ya.I. Gas adsorption Cromatography N.Y. Ple-
ПШП Press. 1969.22. Ишх)льд К. Теоретические основы органической химии. Москва,
Мир, 1973.23. Кузнецов В.А., Егорочкин А.Н. ДАН СССР, 1974, т. 216, с. 812.24. Егорочкин А.Н., Вязанкин Н.С. Ж. орг. химии, 1972, т. 42, с. 904.25. Кузнецов В.А., ErojKJ4KHH А.Н., Разуваев Г.А. Ж. прикладной
спектроскопии 1975, т. 22, с. 952.26. Жданов С.П., Киселев А.В., Яшин Я.И. В сборнике: «Газовая хро¬
матография», НИИТЭХИМ, Москва, 1964, с. 5.27. Гвоздович Т.Н., Ковалева М.П., Петрова Г.К., Яшин Я.И. Нефтехи¬
мия 1968, т. 8, №1, с. 123.28. Yashin Ya.L, Bebris N.K., Gwozdowitsch T.N. Gas Chromatografie
1968, Vortrage des VI Symposiimis (Iber Gas Chromatografie in Berlin,1968, s. 333-342.29. HolUs O.L. Anal. Chem. 1966, v. 38, p.309.30. СакодЕШСкий К.И., Панина Л.И. Успехи химии 1972, т. 27, с.
1024.31. Гвоздович Т.Н., Яшин Я.И. В книге: «Успехи газовой хроматогра¬
фии» вып. 2, Казань, 1970, с. 94.32. Вш^ег J.D. J. Gas Chromatog. 1968, v. 6, p. 177.33. Sakodinsky К. Chromatographia 1968, v. 1, p. 483.34. Gvozdowitsch T.N., Ki^lev A.V., Yaschin Ya.I. Chromatographia
1978, V. 11, №5, p. 137.35. Gvozdowitsch T.N., Kiselev A.V., Yaschin Ya.I. Chromatographia1969, V. 2, p. 179.36. Gvozdowitsch T.N., Kiselev A.V., Yaschin Ya.I. Chromatographia1973, V. 6, p. 179.37. Зайцева Г.Е., Яшин Я.И. Труды по химии и хим. технологии 1974,
вып. 3, с. 76.38. Гвоздович Т.Н., Гринберг Т.С., Зуева Л.В., Яшин Я.И. Нефтехи¬
мия, 1972, т 12, № 2, с. 309.39. Белякова Л.Д., Василевская О.В., Цюрупа М.П., Даваншв В.А. Ж.
физ. химии, 1995, т. 69, № 4, с. 696.40. Белякова Л.Д., Курбанбеков Э., Ларионов О.Г., Цюрупа М.П., Да-
ванков В.А. Изв. АН. Сер. хим. 1999, № 8, с. 1502.239
Яшт ям. Яшин ЕЛ. Яшин АЛ. Газотя ^тттшафия41. Краткая химическая энциклопедия. В 6 томах. Т. 1. Москва. «Со¬
ветская энциклопедия» 1961, 1261 с.42. Hoffinan R.L., List G.R., Evans C.D. Nature, 1966, v. 210, p. 965.43. Genty C., Schott R. Anal. Chem. 1969, v. 42, p. 7.44. Mc.Taggart N.G., Luke L.A., Wood D. In: “Gas Chromatography
1970”, Ed. N.Stock, London.45. Киселев A.B., Чериенькош Ю.Л., Яшин Я.И. Нефтехимия, 1965, т. 5,
Х«4,с. 589.46. Киселев А.В., Черненькова ЮЛ., Яшин Я.И. Нефтехимия, 1965, т. 5,
№11, с. 141.47. Киселев А.В., Черненькова Ю.Л., Яшин Я.И.Газовая хроматогра¬
фия, НИИТЭХИМ, вып. IV, Москва, 1966, с. 38.48. Киселев А.В., Яшин Я.И. Ж. физ. химии 1963, т. 37, с. 2614.49. Калмановский В.И., Мартынюк В.И., Чернятин А.К., Яшин Я.И.,
Шешенин В.А. Зав. лаб. 1974, № 4, с . 791.50. Киселев А.В., Яшин Я.И. Ж. физ. химии 1966, т. 40, № 4, с. 944.51. GavrilovaT.V.,KiselevA.V.,RoshchinaT.M.J.Chromat. 1980,v. 192,
р.323.52. Solomon R.W. Anal.Chem. 1964, v. 36, p.476.53. Киселев A.B., Крылов Б.К., Никитин Ю.С., Тотяшиов А.П., Яшин Я.И.
Газовая хромагоггифия, НИИТЭХИМ, Шсква, 1967, вып. IV, с. 84.54. Крылов Б.К., Яшин Я.И. Хим. промыш. 1971, № 5, с. 349.55. Belyakova L.D., Kiselev A.V., Soloyan G.A. Chromatographia, 1970,
v. 3, p. 254.56. Belyakova L.D., Kiselev A.V., Strokina L.M. Chromatographia, 1985,
v.20, p. 519.57. Belyakova L.D., Strokina L.M. J. Chromat. 1986, v. 365, p. 31.58. Белякова Л.Д., Ларионов О.Г., Строкина Л.М. Изв. АН СССР, Сер.
хим. 1988, с. 1491, с. 2454.59. Карпова А.А., Макаров А.А. Ж. анал. хим. 2001.60. Керл Ф., Смолли Р.С. В мире н^и, 1991, № 12, с. 14.61. Жариков О.В. Природа, 1992, №3, с. 68.62. lijima S. Nature (London) 1991, v. 354, p. 56.63. Жуховицкий A.A., Туркельтоуб Н.М. Газовая хроматография. Го-
стоптехиздат, 1961.64. Janak J. Chem. Listy, 1953, v. 47, p. 1348.65. Долова И. A., Яшин Я.И. Адсорбция и адсорбенты. 1972, т. 2, с. 4.66. Гвоздович Т.Н., Яшин Я.И. Газовая хроматография. Изд-во НИИ¬
ТЭХИМ. 1967, № 6, с. 71.67. Гвоздович Т.Н., Киселев А.В., Яшин Я.И. Нефтехимия. 1968, т. 8,
№ 3, с. 476.240
ГЛАВА 4 Литература68. Kaiser R. Chromatigraphia. 1969, v. 2, p. 453; 1970, v. 3, p. 38.69. Zlatkis A., Lichtenstein A., Tishbee A. Chromatographic 1973, v. 6,
№ 2, p. 67.70. Долова И.А., Киселев A.B., Яшин Я.И. Ж. физ. химии 1974, т. 48,
№ 5, с. 1285.71. Васильева B.C., Киселев А.В., Никитан Ю.С., Петрова Р.С., Щер¬
бакова К.Д. Ж. физ. химии. 1961. т. 35, № 8, с. 1889.72. FruCra J.J. Chrom. 1972, v. 65, p. 341, p. 432.73. Киселев A.B., Мигунова И.А., Яшин Я.И. Газовая хроматография
(сб.) М. НИИТЭХИМ 1967, вып. 6, с. 75.74. Halasz L, Horvath С. Anal. Chem. 1964, v. 36, p. 1178.75. Halasz I., Horvath C. Nature 1963, v. 197, p. 71.76. Бармакова T.B., Киселев A.B., Ковалева Н.В. Коллоидн. ж. 1974,
т. 36,№ 1,с. 133, №5, с. 246.77. Бортников Г.Н., Долова И.А., Вязанкин Н.С., Ковалева Н.В., Ру-
дась В.И., Яшин Я.И. Изв. АН СССР сер. физ. хим. 1973, № 3, с. 587.78. СагЬораск, Bulletin № 738, Supelco, Belltfonte Pd. 1976.79. Yashin Ya.I., In: Gas Chromatography 1964, The Institute of Petroleum,
London, 1965, p. 242.80. Киселев A.B., Пасконова E.A., Петрова PC., Щербакова К.Д. Ж.
физ. химии 1964, т. 38, № 1, с. 161.81. Belyakova L.D., Kiselev A.V., Kovaleva N.V. Analyt. Chem. 1964,
V. 36, №8, p. 1517.82. Беляюва Л.Д., Киселев A.B., Ковалева Н.В. Ж. физ. химии 1966, т. 40,
№ 7, с. 1494.83. Беляюва Л.Д., Киселев А.В., Юзвалева Н.В. Ж. физ. химии 1 %8, т. 42,
№ 9, с. 2276.84. Киселев А.В., Мигунова И.А., Яшин Я.И. Ж. физ. химии 1968, т. 42,
№ 5, с. 1235.85. Shcherbakova L.D., Kiselev A.V., Kovaleva N.V. Analyt. Chem. 1964,
v.36,Jfe8,p. 1517.86. KiselevA.V., YashinYa.I. Gas-chromatographic 1965,EdH.G.Struppe,
Verlag Deutsche Akademie der Wissen-Schaften, Berlin 1965, S. 43.87. Киселев A.B., Яшин Я.И. Ж. физ.химии, 1966, т. 40, № 2, с. 429,
№ 3, с. 603.88. Киселев А.В., Мигунова И.А., Яшин Я.И. Нефтехимия 1967, т. 7,
№ 5, с. 963.89. Киселев А.В., Мигунова И.А., Яшин Я.И. Газовая хроматография
(сб.). Москва, НИИТЭХИМ, 1969, вып. 9, с. 71.90. Киселев А.В., Щербакова К.Д., Яшин Я.И. Журнал структурной
химии, 1969, т. 10, № 5, с. 953.24116 Газовая хрометопМми
Яшин ям. Яшин Е.Я.. Яшин А.Я. Газовая хпаматография91. Киселев А.В., Петов Г.М., Щербакова К.Д. Ж. физ химии, т. 41,
№6, с. 1418.92. Кейбал В.Л., Киселев А.В., Савинов И.М., Худяков В.Л., Щербако¬
ва К.Д., Яшин Я.И. Ж. физ. химии 1967, т. 41, JSs 9, с. 2234.93. Киселев А.В., Онушка Ф., Щербакова К.Д., Мигунова И.А., Яшин
Я.И. Газовая хроматография (сб.) Москва, НИИТЭХИМ, 1967, вып. 6,
с. 75.94. Вязанкин Н.С., Бортников Г.Н., Мигунова И.А., Киселев А.В.,
Егорочкин А.Н., Яшин Я.И., Миронов В.Ф. Изв. АН СССР, сер. хим.1969, № 1,с. 186.95. Bortnikov G.N., Vyasankin N.S., Kiselev A.V., Yashin Ya.I. Chro-
matjgraphia, 1971, v. 4, № 1, p. 14.96. Eisen O.G., Kiselev A.V., Pitt A.E., Rang S.A., Shcherbakova K.D.
Chromatigraphia, 1971, v. 4, № 10, p. 448.97. Kalashnikova E.V., Kiselev A.V., Petrova R.S., Shcherbakova K.D.
Chromatographia, 1971, v. 4, № 11, p. 495.98. Долова И.А., Киселев A.B., Яшин Я.И. Ж. структурной химии,
1972, т. 13, № 1,с. 162.99. Запрометов А.Ю., Калашникова Е.В., Киселев А.В., Щербакова К.Д.
Ж. физ. химии, т. 46, № 5, с. 1230.100. Киселев А.В., Худяков В.Л., Яшин Я.И. Ж. физ. химии, 1973, т. 47
Ха 8, с. 2125.101. Киселев А.В., Худяков В.Л., Яшин Я.И. Ж. физ. химии, 1974, т. 48
№ 2, с. 448.102. Kalashnikova E.V., Kiselev А.V., Shcherbakova K.D. Chromatographia,1974, V. 7, Ка 1,р. 22.103.Engewald W., Graefe J., Shcherbakova K.D., Welsch Th. Chro¬
matographia, 1974, V. 7, № 5, p. 229.104. Rang S.A., Eisen O.G., Kiselev A.V., Meister A.E., Shcherbakova K.D.
Chromatographia, 1975, v. 8, № 7, p. 327.105. Kalashnikova E.V., Kiselev A.V., Makagon A.M., Shcherbakova K.D.
Chromatographia, 1975, v. 8, № 5, p. 399.106. Engewald W., Kalashnikova E.V., Kiselev A. V., Petrova R.S., Scherba¬
kova K.D., Shilov A.L. J. Chromat., 1978, v. 152, p. 453.107. Kalashnikova E.V., Kiselev A.V., Petrova R.S., Shcherbakova K.D.,
Poshkus D.P. Chromatographia, 1979, v. 12, № 12, p. 799.108. Kalashnikova E.V., Kiselev A.V., Shcherbakova K.D., Vasileva S.D.
Chromatographia, 1980, v. 13, № 8, p. 493.109.Dimitrov L.D., Kiselev A.V., Petrova R.S. Chromatographia, 1981,
V. 14, № 2, p. 107.242
ГЛАВА 4 Литература110. Kiselev A.V., Nazarova V.I., Shcherbakova K.D. Chromatographia,
1981, V. 14, № 3, p. 148, 1983, v. 17, № 10, p. 533.111. Гаврилова Т.Б., Киселев A.B., Криволапое С.С., Пастушеню В.Г,
Коллоидн. ж. 1981, т. 43, № 1, с. 138.112. Kiselev A.V., Polotnyak Е.В., Shcherbakova K.D. Chromatographia,1981, V. 14, №8, p. 478.113. Kalashnikova E.V., Kiselev A.V., Shcherbakova K.D., Vasileva S.D.
Chromatographia, 1981, v. 14, № 9, p. 510.114. Dimitrov L.D., Kiselev A.V., Petrova R.S. Chromatographia, 1982,
v. 15, №4, p. 245.115. Киселев A.B., Назарова В.И., Щербакова К.Д. Доют. АН СССР,1982, т. 265, №4, с. 909.116. Киселев А.В., Kjottoob Н.С. Докл. АН СССР, 1982, т. 266, № 1, с. 153.117. Киселев А.В., Полотнюк Е.Б., Щербакова К.Д. Докл. АН СССР,1982, т. 266, № 4, с. 892.118. Kiselev A.V., Markosyan D.L. Chromatographia, v. 17, № 10, p. 526.119. Kiselev A.V., Nazarova V.I., Shcherbakova K.D. Chromatographia,1983, V. 17, № 10, p. 533.120. Kiselev A.V., Nazarova V.I., Shchert>akova K.D. Chromatographia,1984, V. 18, №4, p. 183.121. Zagorevskaya E.V., Ishchenko N.V. Adsorption Sci. and Technol. 1985,
V.2, p. 219.122. Gavrilova T.B., Kiselev A.V., Kulikov N.S., Vlasenko E.V Chro¬
matographia, 1986, V. 22, № 1, p. 59.123. Kalashnikova E.V., Kiselev A.V., Petrova R.S., Shcherbakova K.D.
Advances Colloid, and Interface Sci. 1986, v. 25, № 3, p. 217.124. Киселев A.B., Ковалева H.B., Филатова Г.Н., Федотов А.Н. Ж. физ.
химии, 1986, т. 60, № 1, с. 176.125. Назарова В.И., Щербакова К.Д. Ж. физ. химии, 1999, т. 73, № 11,
с. 2044.126. Di Corcia А., Liberti А. Adv. in chromatogr. V. 14, Dekker, New York,
1976, p. 305.127. Vidal-Madjar C., Guiochon G. J. Chromatog. Sci. 1971, V. 9, p. 664.128. Engewald W., Billing U., Topalova I., Petsev N. J. Chromatogr. 1988,
V.446, p. 71.129. Kiselev A. V. Chromatographia, 1978, v. 11, № 12, p. 691.130. Kiselev A.V., Poshkus D.P. Faraday Discuss. Royal. Soc. of Chem.,
1980, V. 15, p. 13.131. Киселев A.B., Полотаюк Е.Б., Щербакова К.Д. Докл. АН СССР,
1982, т. 266, №4, с. 892.24316-
Яипт ЯЖ. Яшин ЕЛ, ЯшинА.Я. Гязоюя хтттогрвфия132.Киселев А.В., Петрова Р.С. Докл. АН СССР, 1983, т. 272, № 6,
с. 1415.133.Даллакян П.Б., Киселев А.В. Ж. физ.химии, 1985, т. 59, № 5,
с. 1277.134. Бобылева М.С., Дементьева Л.А., Киселев А.В., Куликов Н.С.
Докл. АН СССР, 1985, т. 283, № 6, с. 1390.135.Киселев А.В., Дементьева Л.А. Ж. физ.химии, 1986, т. 60, № 8,
с. 1951.136. Буряк А.К., Федотов А.Н., Киселев А.В. Вест. Моск. ун-та, сер. 2,
хим., 1985, т. 26, № 6, с. 568.137.Буряк А.К., Пошкус Д.П. Изв. АН СССР, сер. хим., 1986, № 1,
с. 223-224.138.Буряк А.К., Пошкус Д.П. Изв. АН СССР, сер. хим., 1989, № 1,
с. 12-16.139.Буряк А.К., Березин Г.И. Изв. АН СССР, сер. хим., 1989, № 8,
с. 1721-1723.140. Буряк А.К., Ульянов А.В. Изв. АН СССР, сер. хим., 1996, № 3.141. Буряк А.К. Изв. АН СССР, сер. хим., 1999, № 8.142. Кудряшев С.Ю., Онучак Л.А., Воронков А.В., Буряк А.К., Моисе¬
ев И.К. Изв. АН СССР, сер. хим., 2000, № 12, с. 2021-2025.143. Яшкин С.Н., Светлов Д.А., Курбатова С.В., Буряк А.К. Изв АН
СССР, сер. хим. 2000, № 5, с. 849.144. Яшкин С.Н., Григорьева О.Б., Буряк А.К. Изв. АН СССР, сер. хим.
2001, № 6, с. 938.145. Яшкин С.Н., Курбатова С.В., Петрова Е.И., Буряк А.К. Изв. АН
СССР, сер. хим., 2001, № 5, с. 787.146. Курбатова С.В., Яшкин С.Н., Моисеев И.К., Земцова М.Н. Ж. физ.
химии 1999, т. 73, № 9, с. 1645.147. Львова Н.Ю., Курбатова С.В., Яшкин С.Н., Колосова Е.А. Ж. физ.
химии 2002, т. 75, № 12, с. 2233.148. Курбатова С.В., Яшкин С.Н. Ж. структурной химии 2000, т. 41,
№ 4, с. 805.149. Долова И.А., Киселев А.В., Яшин Я.И. Ж. физ. химии 1972, т. 46,
№ И, с. 2935.150. Киселев А.В., Яшин Я.И. В книге «Физико-химические примене¬
ния газовой хроматографии» Москва, Химия 1973, га 1, с. 5-80.151. Киселев А.В. Межмолекулярные взаимодействия в адсорбции и
хроматографии. М., Высшая школа 1986, с. 81.152. Киселев А.В., Никитин Ю.С. Хроматография (сб.), т. 2 (Итоги нау¬
ки и техники) М., ВИНИТИ 1978, с. 5-27.244
[J0Sdl Литература153. Киселев A.B., Пошкус Д.П., Щербакова К.Д. Ж.физ. химии 1986,
т. 60, № 6, с. 1329.154. Киселев А.В., Кузнецов А.В., Филатова И.Ю., Пошкус Д.П., Щер¬
бакова К.Д. Ж. физ. химии 1970, т. 44, № 5, с. 1272.155. Бойкова А.С., Щербакова К.Д. Нефтехимия 1967, т. 7, с. 451.156. БqлникDв ГЛ., Вяшпшн Н.С., Петров Б.И., Ратушная СХ, Яшин Я.И.
1^>уцы по химии и хим. технологии 1973, вып. 2, с. 57.157. Bochkarev M.N., Bortnikov G.H., Makarenko N.P., Maiorova L.P.,
Kiselev A.V., Yashin Ya.I. J. Chromat. 1979, v. 170, № 1, p. 13.158.Kiselev A.V., Nazarova V.I., Shcherbakova K.D. J. Chromat. 1984,
V. 292, p. 97.159. Engewald W., Billing U., Welsch Т., Haufe G. Chromatographia 1987,
V. 23, № 8, p. 590.160. Kalashnikova E.V., Kiselev A.V., Poshcus D.P., Shcherbakova K.D. J.
Chrom. 1987, v. 23, № 8, p. 590.161.Engewald W., Wennrich L., Porschman J. Chromatographia 1978,
V. 11, №8, p. 434.162.Valcarcel М., Cardenas S., Simonet B.M. Anal. Chem. 2007, v. 79,
p. 4788.163. Merkoci A. Microchim. Acta 2006, v. 152, p. 157.164. Costa-Femandez J.M., Pereiro R., Sanz-Medel A. Trends Anal. Chem.
2006, V. 25, p. 207.165. Raty J.Y., Galli G. J. Electroanal. Chem. 2005, v. 584, p. 9.166. Jing G., Zheng Q. J. Appl. Polimer. 2005, v. 97, p. 2182.167. Roy D. et al. Chem. Phys. Lett. 2003, v. 373, p. 52.168. Doi H. Surf, Sci. 2002, v 500, p. 218.169.Nisha J.A. et al. Chem. Phys. Lett. 2000, v. 328, p. 381.170. Quay C.H.L. et al. Compos. Sci. Technol. 2007, v. 67, p. 2965.171. Yang Y et al. Nanotechnology 2004, v. 15, p. 1545.172. Sano M. et al. Science (Washington D.C.), 2001, v. 293, p. 1299.173. Pan H. et al. Nano Lett 2003, v 3, p. 29.174. Li Q., Yuan D. J. Chromatogr. 2003, v 1003, p. 203.175. Saridara C., Mitra S. Anal. Chem. 2005, v 77, p. 7094.176. Korva М., Mitra S. Anal. Chem. 2006, v. 78, p. 2064.177. Stadermann M. et al. Anal. Chem. 2006, v. 78, p. 5639.178. Robson M.M., Bartle K.D., Myers P. J. Microcolumn Sep. 1998, v. 10,
p. 115.179. Kartsova L.A., Makarov A.A. J. Anal. Chem. 2004, v. 59, p. 724.245
Яшин Я.И., Яшин ЕЖ. Яшин АЖ.Газовая храштошфияГлава 5. ГАЗОВАЯ АДСОРБЦИОННО¬
АБСОРБЦИОННАЯ ХРОМАТОГРАФИЯ5.1 Зависимость удерживаемого объема от количества жидкой фазы
в газо-жидкостной жроматографии и в адсорбционно-абсорбционной
газовой хроматографииВ равновесной газо-жидкостной хроматографии удерживание опреде¬
ляется только растворением в объеме жидкости, удерживаемый объем
анализируемого вещества в колонке для малых проб должен был бы ли¬
нейно зависеть от объема жидкой фазы V^.Vr=KV^ (5.1)В этом уравнении К — коэффициент распределения данного юмпо-
нента между газовой и жидкой фазами. Однако на практике зависимость
Уц от Уж часто отклоняется от прямой, проходящей через начало коорди¬
нат (рис. 5.1).Рис. 5.1. Зависимость исправленного удерживаемого объема (V^')
от объема жидкой фазы (VJ в колонке. 1 - вклад раство¬
рения и 2- вклад адсорбции на поверхности жидкой фазы.Удерживаемые обьемы иногда даже уменьшаются с ростом Уж [1]
Причиной этого является адсорбция на поверхности самой жидкой фазы
[1-7] и на поверхности носителя жидкой фазы [8-15].Адсорбция на поверхности пленки жидкой фазы вносит значительный
вклад в удерживание при разделении неполярных веществ на полярной
жидкой фазе, т. е. в случае плохой растворимости в жидкой фазе [2, 3]. В
этом случае удерживаемые обьемы с увеличением Уж уменьшаются или
остаются практически постоянными. С увеличением количества жидкой
фазы, нанесенной на поверхность пористого тела, поверхность пленки
нанесенной жидкости А1 сокращается (рис. 5.2) и, как следствие этого,
уменьшается У^246
ГЛАВА 5Газовая адсопбиионно-абсорбтотая хроматография.MQ5se-% —•Рис. 5.2. Уменьшение удельной поверхности полисорба-1 при нанесе¬
нии жидких фаз: 1 - диглицерин; 2 - теин; 3 - цианэтти-
рованный пентаэритрит; 4 - сквалан.Адсорбция на поверхности жидкой пленки, по-видимому, имеет место
также на сорбентах малой емкости, используемых для разделений при тем¬
пературах выше 200 °С. В этих случаях используют в основном, высоюмо-
лекулярные жидкие фазы, чаще всего различные полиметилфенилсилокса-
новые жидкости, в которых разделяемые вещества при высоких темпера¬
турах растворяются плохо. При малом содержании жидкости на носителе
(менее 5%) основной причиной отклонения от линейности может быть ад¬
сорбция компонента на поверхности носителя. Селективность разделения
(разность удерживаемых объемов различных компонентов) определяется в
этом случае не только различием растворимости разделяемых веществ, но
и различием их адсорбируемости на поверхности носителя.В том случае, когда эти факторы действуют в одном направлении, то
селективность такого сорбента в целом увеличивается. При этом пики во
многих случаях остаются симметричными даже на не однородных носи¬
телях, т.к. в этом случае адсорбция на твердой поверхности происходит
из жидкой фазы. Впервые эти эффекты были учтены в работах [10,11]. В
частности было показано [10], что за счет адсорбционного эффекта изо¬
мерные пишлины лучше разделяются при нанесении жидких фаз на более
активные в адсорбционном отношении твердые носители.В общем случае в удерживание вносят вклад одновременно три про¬
цесса: растворение в пленке жидкости, адсорбция на поверхности пленки
жидкой фазы и адсорбция на поверхности носителя, так что:V.= K^»+K»,A+K-A (5-2)Здесь К"д, К*д и \ представляют, соответственно, константы Ген¬
ри и площади поверхности для адсорбции на пленке жидкости из газовой
фазы и на носителе из раствора в пленке жидкости (если эта пленка покры-247
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я. Газовая хроматографиявает ВСЮ поверхность носителя). Значения К"^, К*д отнесены к единице
площади соответствующей поверхности. Вид зависимости от будет
изменяться в соответствии с относительной величиной вклада каждого из
этих факторов в общую величину V^. Следует иметь в виду, что величина,
зависит от У^и при больших обычно резко уменьшается. Уравнение,
учитывающее все эти три фактора удерживания — растворение, адсорб¬
цию на носителе и адсорбцию на поверхности пленки жидкости,— рас¬
сматривалось в работах [4,5, 16—18].Однако в реальных условиях сорбционных фаз значительно больше,
сорбент полифазный как для адсорбции, так и для растворения, т.к. свой¬
ства пленок жидких фаз разной толщины разные.Таким образом в общем случае можно записать следующее уравнение:VR=ZK,.^V,,+iK,,Aj,, (5.3)i=i j=iгде К - константы распределения в пленке жидкой фазы разной тол¬
щины; К. коэффициенты адсорбции Генри на разных поверхностях раз¬
дела, Vj^- обьемы жидких фаз; А,^- поверхность разных фаз раздела.Адсорбция на твердом .носителе в той или иной мере проявляется ю мно¬
гих системах, особенно при нанесении небольших шличеств жидких фаз. Она
мала на поверхности силанизированных диатомитовых носителей и тефлоно¬
вых носителей. В частности, как для н-алганов, так и н-спиртов, зависимость
удерживаемых объемов от количества жидкой фазы, нанесенной на тефлон
(полихром-1), линейна и практически проходагг через начало ююрдинат.Вполне оправданно применение в аналитических целях такого адсорб¬
ционно-абсорбционного варианта хроматографии, в котором на сорбенте
одновременно происходят и адсорбция, и растворение. Впервые модифи¬
цирование адсорбентов жидкой фазой для улучшения разделения было
проведено в работе [19]. Соответствующий вид хроматографии был назван
Парнелом [20] «газо-жидкостно-адсорбционной хроматографией». Нам
представляется более точным название «газовая адсорбционно-абсорбци¬
онная хроматография», которое отражает как адсорбцию — процесс, про¬
исходящий на поверхностях раздела, так и абсорбцию — поглощение объ¬
емом нанесенной пленки жидкости.5.2. Ма1фопористые iqieiuHeseMbi в качестве адсорбентов - носителей5.2.1 Макропористые кремнеземы с пленками жидких фазВ газо-жидкостной хроматографии адсорбционные эффекты обычно
исследовались и использовались на диатомитовых носителях, имеющих
малую удельную поверхность (около 1 mVf). Возможность применения ад-248
[Jl^BdA Газовая адсорбиионно-абсорбигюнная хттатография.сорбентов-носителей с большой удельной поверхностью с нанесенными
на их поверхность монослоями жидких фаз была показана впервые нами
в работе [21]. Для усиления роли адсорбции во многих случаях целесо¬
образно применять адсорбенты-носители, характеризующиеся большими
величинами К"^ и s (а, следовательно, и А^), в частности, макропористые
силикагели и силохромы [22—24]. При этом можно получить следующие
преимущества.1. Емкость и селективность сорбента в целом повышается за счет боль¬
шего вклада в удерживание адсорбции на адсорбенте-носителе.2. Пленка нанесенной жидкой фазы прочнее связывается с более ак¬
тивной поверхностью адсорбента-носителя и, следовательно, верхний
температурный предел для таких сорбентов повышается за счет снижения
летучести жидкой фазы, по сравнению с классическим газо-жидкостным
вариантом.3. Неоднородные места поверхности адсорбента-носителя экраниру¬
ются молекулами нанесенной пленки, в результате чего пики становятся
более симметричными, чем при хроматографировании на адсорбентах-но-
сителях без жидкой фазы.Новые возможности регулирования селективности появляются, когда
влияния адсорбции и растворения действуют в противоположные сторо¬
ны, т. е. порядок выхода компонентов смеси на чистом адсорбенте-носите¬
ле с большими К*д и А^ и порядок выхода на жидкой фазе, нанесенной на
твердый носитель с малыми К"^ и А^ не совпадают. В этом случае можно
сильно изменять удерживаемые объемы некоторых веществ, изменяя коли¬
чество жидкой фазы, наносимой на адсорбент-носитель с большими К*^ и
А*, причем в некоторых случаях можно изменить порядок выхода веществ
на противоположный, т. е. произвести обращение последовательности ве¬
личин удерживаемых объемов.Это можно показать на примере изменения относительных времен
удерживания спиртов С1—С4 на макропористом силикагеле с различным
количеством нанесенной жидкой фазы [25]. Малые количества спиртов
С1—С4 на неполярных жидких фазах, нанесенных на носители с малыми
значениями К*^ и s, в газо-жидкостной хроматографии и на неспецифи¬
ческих адсорбентах в газо-адсорбционной хроматографии удерживаются
в соответствии с ростом энергии дисперсионного юаимодействия. Вклад
энергии специфического взаимодействия (главным образом, водородной
связи) для спиртов С1—С4 при адсорбции на поверхности специфического
адсорбента примерно одинаков. Из табл. 5.1 видно, что увеличение удер¬
живания спиртов С1—С4 на разных адсорбентах и жидких фазах в основ-249
Яшин Я.И,. Яшин ЕЛ.. Яшин А.Я.Газовая хроматографияНОМ соответствует повышению температуры кипения спиртов этого ряда.
Исклютение составляет трет-^тганол, шгорый из колонн с неспецифическим
адсорбентом первого типа — графнггированной термической сажей,— со сла¬
бо специфическим адсорбентом т{»тьего типа — пористым полимером — и
специфическим адсорбентом второго типа — силохромом — выходит позже
и-пропанола. Это связано с особенностями геометрии молекулы трет-бу-
танола, в которой гидроксильная группа экранирована тремя метильными
группами. На полярных жидких фазах, молекулы шторых могут образовы¬
вать с молекулами спиртов водородные связи, порядок выхода спиртов из¬
меняется. В этом случае он определяется соотношением вкладов энергий
дисперсионного взаимодействия и энергии водородной связи в теплоту
растворения. Роль водородной связи при взаимодействий молекул сорбен¬
та с молекулами сорбата в ряду спиртов С1—С4 проявляется по-разному,
особенно для «изо-, втор- и трет-спиртов.Таблица 5.1.Относительные (к н-бутану) удерживаемые обьемы Уотн спиртов
С1—С4 на различных адсорбентах и жидких фазах при разных тем¬
пературах.Неподвижные фазы
и темпера1ура
разделенияVoTHClС2 1 изо-СЗтрет-С4н-СЗвтор-С4Изо - С4Н-С4(64,7)(78,3) (82,3)(82,8)(97,2)(99,5)(107,9)(117,7)АдсорбентыГТС, 100 °с0,060,120,230,380,25 1 0,690,691,0Хромосорб-101, 150°С0,110,180,330,480,43 1 0,730,821,0Гидроксилированный1силохром,llO^C0,110,250,480,710,49—0,811,0Жидкие фазыСквалан, 80 °С0,080,140,200,280,380,580,711,0Триэтаноламин,80 °С0,330,380,330,270,610,510,671,0Глицерин, 80 °С0,610,570,420,320,690,530,641,0Сорбит, 120°С2,021,180,600,311,000,560,711,0в скс^кахуказаны температуры кипения в °С. Жидкости наносились на хромосорб We количестве
15% (масс).На глицерине, триэтаноламине и сорбите, нанесенных в больших коли¬
чествах на хромосорб W (малые и s=l м^г), трет-бутанол удерживается
слабее остальных спиртов С^, а изо-пропанол и изо-бутанол значительно
слабее, соответственно, этанола и н-пропанола; метанол удерживается на
глицерине даже сильнее н-бутанола (табл. 5.1).250
0Ы§АА Газовая адсорбиионно-абсовбтонная хтшатография.При нанесении больших количеств глицерина (30—40%) на
адсорбент-носитель (макропористый силикагель с гидроксилированной
поверхностью, большие К*^, s = 60 м^/г) порядок выхода спиртов сохра¬
няется таким же, как и при нанесении на силанизированный хромосорб W
(табл. 5.1). Однако при нанесении меньших количеств глицерина (близ¬
ких к емкости плотного монослоя, 2—6%) происходит изменение поряд¬
ка выхода, так как в монослое гидроксильные группы молекул глицерина
связаны более сильной (преимущественно водородной) связью с гидрок¬
сильными группами поверхности адсорбента-носителя силикагеля.Действительно, как видно из табл. 5.2, при нанесении на поверх¬
ность макропористого силикагеля небольших количеств глицерина по¬
рядок выхода спиртов становится таким же, как при адсорбции на неспе¬
цифическом или слабо специфическом адсорбентах — графитированной
термической саже и хромосорбе-101 (см. табл. 5.1) и при растворении в
неполярной жидкости — сквалане, с которыми водородной связи не об¬
разуется.Таблица 5.2.Относительные (к н-бутанолу) удерживаемые обьемы Уотн спиртов
С1-С4 при 103°С на макропористом силикагеле МСА-2 при разных
количествах нанесенного на него глицеринаКоличествошицерина,%V отнС1С2изо-СЗн-СЗтрет-С4етор-С4ИЗО-С4Н-С410,080,200,440,470,550,730,811,0030,080,170,280,360,470,560,751,0050,140,250,360,460,400,580,801,00100,170,260,380,480,500,640,741,00250,330,380,400,560,460,620,741,00400,710,620,550,720,470,690,901,00Подобные же явления наблюдаются и при нанесении на макропори¬
стый силикагель МСА-1 (s « 30 м^г) разных количеств триэтаноламина.
Однаш в отличие от силикагеля со слоем глицерина, на силикагеле с не¬
большим шличеством (близком к плотному монослою) триэтаноламина
третбутанол удерживается слабее, чем н-пропанол.На хроматограммах метанола и трет-бутанола на силикагеле с гаице-
РИяом, нанесенным в различных количествах, обнаруживается изменение
порядка выхода этих спиртов с изменением шличества жидкой фазы на
8Дсорбенте-носителе (силохроме С-80) (рис.5.3).251
Яшин ЯМ, Яшин Е.Я.. Яшин А.Я.Газовая хроматографияРис. 5.3. Зависимость от количества глицерина, нанесенного на
микропористый силикагель МСА-2 (S ~ 60 м^/г) при 104°С
(а), и хроматограмма метанола и этанола, полученные на
колонке с силикагелем с 10% (б) и 40% (в) глицерина: 1 -ме¬
танол; 2 - этанол.Нанесение 10% гаицерина на силикагель МСА-2 соответствует в сред¬
нем приблизительно трем плотным монослоям гаицерина при плоской
ориентации молекул глицерина на поверхности силикагеля. При толщи¬
не в один плотный монослой глицерина последовательность выхода этих
спиртов такая же (рис.5.3).Изменение порядка выхода компонентов наблюдается и на силохро-
мах с разными количествами нанесенной жидкой фазы, в частности из¬
меняется порядок выхода нафталина и нитробензола на SE-30. На силох¬
роме (s= 80 mVf) с 30% (масс.) неполярного растворителя нитробензол
удерживается слабее нафталина в связи с тем, что растворимость его в
этой неподвижной фазе меньше (рис. 5.4).Эффективность колонн, заполненных сорбентами с большими К"^ и s,
наибольшая в том случае, когда количество жидкой фазы, нанесенной на
адсорбент-носитель, в среднем соответствует емкости плотного монослоя.
В адсорбционно-абсорбционной хроматографии целесообразнее исполь¬
зовать адсорбенты-носители с достаточно большими и s, в частности
силохромы и пористые полимеры. Силохромы имеют довольно однород¬
ную макропористую структуру. Однако гидроксилированная поверхность
силохрома слишком сильно адсорбирует сильнополярные вещества и хи¬
мически недостаточно однородна, так как может содержать различные
активные примесные группы, а также гидроксильные группы, способные
образовывать сильные водородные связи. Поэтому сильнополярные веще-252
ШЛВЛ5Газовая адсорбиионно-абсорбтоннш храматографш.2иг30%и%/6%1%uL ilL_ UUut t 1 i 1 I L_1 I t- 1 1U0%0 I г 0 1 го 1 г оi, тинI 21 I LО 1 гРис. 5.4. Хроматограмма нафталина (1) и нитробензола (2) на си-
лохроме С-80, модифицированном разным количеством
SE-30 (указано на рис.).
ства, особенно молекулы и макромолекулы группы D, дают несимметричные
пики. При нанесении небольших юличеств жидкой фазы на поверхность си-
лохромов удерживаемые обьемы уменьшаются и пики таких веществ стано¬
вятся более симметричными. Если количество нанесенной жидкой фазы не
превосходит емкости монослоя, то жидкая фаза за счет адсорбвд10нных сил
сильно удерживается поверхностью силохрома и фоновый ионный ток коло¬
нок, наполненных такими сорбентами (как и в случае адсорбционного моди¬
фицирования поверхности адсорбентов-носителей монослоями жидюсти),
значительно меньше, чем при использовании coj^htob с толстыми пленка¬
ми жидшй фазы. На рис. 5.5 приведена зависимость удерживаемых объемов
VR для молекул групп А, В и D от шличества нанесенной на силохром С-80
полярной жидшй фазы — ПЭГ-6000. Эффеетивность шлонок, оцениваемая
высотой теоретичесшй тарелки в минимуме кривой ван Днмтера, сначала
увеличивается, достигая максимума при -6% (масс.) нанесенной жидю¬
сти, и затем снова падает [26]. С увеличением количества нанесенной жид¬
шсти подавляется специфичность поверхности силохром и поверхность
становится более однородной. При дальнейшем увеличении шличества
нанесенной жидшсти (толщины пленки) вклад растворения возрастает и,
вместе с тем, замедляется массообмен, вследствие чего эффективность ш-
лонки уменьшается.253
Яишн Я.И.. Яишн Е.Я., Ятин А.Я.Газовая зднтатогтфияРис. 5.5. Зависимость V^^om количества ПЭГ-6000, нанесенного на
сшохром С-80.Адсорбаты: 1 - нафталин; 2 - анилин; 3 - тридекан; 4 - ди¬
фенил; 5 - тетрадекан.%Рис. 5.6. Зависимость V^om количества апиезона L, нанесенного на
силанизированный силохром С-80 при 200°С.Адсорбаты: 1 - додекан; 2 - триэтилбензол; 3 - на¬
фталин; 4 — тридекан; 5 — тетрадекан; б — дифенил.254
Газовая адсорбтонно-абсорбиионная хроматография.Характер кривой — тличество нанесенной жидкой фазы зависит
0Г адсорбционной способности исходного адсорбента-носителя. На рис. 5.6
0оказано изменение адсорбционной способности силанизированного силох¬
рома, модифицированного различными количествами неполярной жидшй
фазы — апьезоном L. Даже при нанесении небольшого шличества жидшй
фазы [1-6% (масс.)] наблюдается линейное увеличение удерживаемых объ¬
емов веществ разной полярности [27]. С увеличением содержания жидшй
фазы относительные (к алканш) удерживаемые объемы всех адсорбагов
возрастают. Это можно объяснить тем, что все исследуемые вещества хоро¬
шо растворяются в этом неполярном растворителе, влияние же поверхности
носителя в данном случае сведено к минимуму.Различие между адсорбционной способностью исходного силохрома
и силохрома, обработанного гексаметилдисилазаном, особенно заметно
проявляется при сопоставлении характера кривых зависимости удержива¬
емых объемов различных веществ от шличества ПЭГА на этих адсорбен¬
тах. Как видно из рис. 5.7, характер кривых меняется, хотя растворимость
адсорбатов в данном полярном растворителе одинакова. Для н-алканов на
исходном частично дегидроксилированном силохроме наблюдается резше
уменьшение удерживаемых объемов с увеличением шличества жидшй
фазы на исходном силохроме (примерно до 10%), а при дальнейшем уве-гоот8040в « » ао 40 п% %^ис. 5.7. Зависимость от количества полиэтиленгликольадипина-
та , нанесенного на частично дегидроксилированный (а)
и силанизированный (б) силохром С-80.Адсорбаты: 1 - додекан; 2 ~ тридекан; 3 - тетрадекан;4 - анилин; 5 - нафталин; 6 - дифенил.255
Яшин Я.И., Яшин Е.Я., Яшин А.Я. Газовая хроматографияличении количества жидкости происходит только незначительное их изме¬
нение (рис. 5.7). Относительные удерживаемые объемы для углеводородов
С13—С18 также уменьшаются с увеличением количества жидкой фазы.
Роль носителя как специфического адсорбента здесь несомненна. При по¬
давлении специфичности силохрома путем химического модифицирования
(силанизирование) основным фактором, обусловливающим удерживание,
становится растворение вещества в жидкой пленке. Так как растворимость
н-алканов в полярном растворителе, например, в полиэтиленгаикольади-
пинате, невелика, то зависимость от почти линейна и прямая имеет
небольшой наклон.В таблице 5.3 приведены изменения селективности разделения этанола
и метанола на силохромах с различным содержанием глицерина. Изменяя
содержание только глицерина, на силохроме можно получать сорбенты с
разной селективностью, аналогичной по величине а разным адсорбентам и
сорбентам с жидкими фазами, различающимися по химической природе.Таким образом, убедительно показано, что в газовой адсорбционно¬
абсорбционной хроматографии появляется дополнительный фактор,
влияющий на селективность разделения, а именно, количество жидких
фаз на активном адсорбенте-носителе, а точнее соотношение адсорб¬
ционных и абсорбционных вкладов в удерживаемые обьемы анализиру¬
емых веществ. В классической газовой хроматографии селективность раз¬
деления конкретных смесей определяется только двумя факторами - при¬
родой сорбента и температурой, при которой происходит разделение.Таблица 5.3Сорбенты на основе
силохрома и глицерина,
% содержания глицеринаАналогичные по величине
а адсорбенты и жидкие
фазыа=этанол/метанол0^2силохром2,34ГТС2,0615% сквалана на
хромосорбе W1,758хромосорб 1011,630 и выше15% глицерина на
хроматоне N0,93При химическом модифицировании поверхности силохрома три-
метилсилильными группами помимо потери специфичности адсорбции
происходит резкое ослабление неспецифического межмолекулярного
взаимодействия молекул с адсорбентом вследствие резкого уменьшения
концентрации силовых центров адсорбции на поверхности, образованной
метильными группами, по сравнению с силанольной или силоксановой256
ГЛАВА 5 Газовая адсорбиионно-абсорбиионная хроматография.поверхностью немодифицированного кремнезема. Удерживание нафта¬
лина, дифенила и анилина на силанизированном силохроме возрастает с
увеличением количества нанесенного ПЭГА, тогда как на немодифициро-
ванном силохроме удерживание этих веществ с повышением количества
ПЭГА вначале падает и только затем возрастает. На исходном силохроме
минимальное значение У|, практически совпадает с максимумом эффек¬
тивности колонны и соответствует нанесению 6% (масс.) модификатора, т.
е. величине, близкой к емкости монослоя.Эти результаты показывают, что силанизированный силохром может
служить хорошим слабо адсорбирующим носителем для газо-жидкостной
хроматографии. В отличие от общепринятых твердых носителей с неболь¬
шой удельной поверхностью (обычно около 0,5 - 4 м^/г) силанизированный
силохром имеет большую удельную поверхность (около 80 mVf). Это по¬
зволяет наносить на его поверхность тонким слоем в 10—20 раз больше
жидшй фазы, чем на обычные диатомитовые носители; эффективность
колонки при этом почти не изменяется. Однаш за счет большего шличе¬
ства отложенной жидшй фазы значительно повышается селективность и
емкость колонн, заполненных таким сорбентом.Таким образом, силанизированные силохромы с большим количеством
нанесенной в виде тонкого слоя жидшсти могут быть рекомендованы в тех
случаях, когда требуется большая сорбщюнная емшсть колонки, т. е. для
препаративной хроматографии и для анализа примесей.Нанесение жидкой фазы на силанизированный силохром в количестве
100 и даже 200% (масс.) не вызывает слипания его частиц, характерного
для обычных инертных носителей с малой удельной поверхностью при на¬
несении больших шличеств жидкой фазы. При хроматографировании на
колонках одинашвой длины с носителем хроматоном, содержащим 20%
(масс.) сквалана, и с силохромом, содержащим 100% (масс.) сквалана, во
втором случае достигается лучшая степень разделения (большая величина
критерия разделения).При меньших шличествах жидкости, сопоставимых с количествами
жидкости, наносимой обычно на диатомитовые носители с малой удель¬
ной поверхностью (15—20% от массы носителя), прочность удерживания
пленки жидшсти на большой поверхности силохрома значительно выше.
Для сопоставления летучести нанесенной жидкости бьши приготовлены
и предварительно шндициоиированы в соответствии с общепринятыми
нормами две колонки с одинашвым количеством сквалана на хроматоне и
на силохроме. На рис. 5.8 приведены зависимости фонового тока иониза¬
ционно-пламенного детектора от температуры для этих двух шлонок. Из
этих зависимостей видно, что при одной и той же температуре летучесть25717 Га'зовая хромато1"рафи)1
Яшин Я.И., Яшин Е.Я.. Яшт А.Я. Газовая хроматографиясквалана, нанесенного на силохром, в 8—10 раз меньше летучести сквалана,
нанесенного на хроматон. Предельная летучесть сквалана, нанесенного на
силохром, достигается при температуре, на 40 фадусов большей, чем при
нанесении его на обычный носитель с малой удельной поверхностью. Это
позволяет повысить предельную рабочую температуру колонки. Сопостав¬
ление динамики изменения величин фоновых сигналов этих двух шлонок
в течение 100 ч при температуре шлонны 100°С также подтверждает зна¬
чительно большую стабильность сквалана, нанесенного на силохром. Фо¬
новый сигнал от сквалана, нанесенного на хроматон, изменился в 24 раза
(с 6-10'^ до 2,5-10 ’® А). В тех же условиях фоновый сигнал колонны со
скваланом на силохроме изменился лишь в 1,2 раза (с 6,6-1 O''® до 5,7-lO"'” А),
причем сшрость его уменьшения оказалась практически постоянной, что
значительно облегчает эксплуатацию хромагофафа с ташй шлоншй.-f/*CРис. 5.8. Зависимость фонового тона от температуры: 1 - для ко¬
лонки со скваланом (15%) на хроматоне; 2 - для колонки со
скваланом (15%) на силохроме.Оценивая степени разделения в течение 100 ч, надо учесть, что одним из
следствий более прочного удерживания нанесенной жидшсти в адсорбцион¬
но-абсорбционной хроматофафии является неизменность критерия степени
разделения, который при разделении на сквалане, нанесенном на силохром,
составил 2,4—2,5 для н-алканов С^—С, j, а при разделении на ташй же шлон¬
ке со скваланом на хроматоне за 1(Ю ч работы он снизился с 2,3 до 1,2.Летучесть жидшй фазы можно уменьшить и при нанесении ее на хро¬
матон, если создать поле адсорбционных сил в объеме жидшй фазы путем
диспергирования в ней тонкодисперсных твердых тел, в частности аэроси¬
ла. На рис. 5.9 сопоставлены зависимости фонового тока от температуры
для шлонн с хроматоном, пропитанным чистым апьезоном и апьезоном,
в котором диспергировано различное количество аэросила. На сорбентах
с аэросилом сушественно уменьшается фоновый ионный ток, поскольк>
адсорбция на аэросиле резш снижает летучесть апиезона. Селективность
же разделения на шлонках со скваланом, содержащим аэросил, выше, чем
на шлонках с чистым скваланом.258
0tiSd-Газовая адсорбиионно-абсорбиионная хроматография.■тРис. 5.9. Зависимость фонового ионного тока от температуры ко¬
лонки: 1 - хроматон + 15% апиезона М; 2 - хроматон +
15% апиезона М + 20% аэросила.При использовании термически стабильных жидкостей в адсорбцион¬
но-абсорбционной хроматографии можно проводить анализы и при еще
более высоких температурах. На рис. 5.10 показаны примеры разделения
полярных и слабополярных высококипящих веществ на силохроме с нане¬
сенным на него полиметилсилоксаном (SE-30) или полиметилфенилсилок-
саном (0V-17). Следует отметить, что в этих случаях пики всех веществ
симметричны и в режиме программирования температуры нет смещения
нулевой линии [28—30].Рис. 5.10. Хроматограммы высококипящих соединений:а) Терфенилы: 1 - орто; 2 - мета; 3 - пара; колонка
100x0,3 см с силохромом + 4% 0V-17, 270°С.б) Н-алканы: С12 - С26 в реокиме программирования, усло¬
вия те же.в) Фталсты: 2 - диметилфталст; 3 - дибутшфталат; 4-ди-
октилфталат; ктонка 1(ЮхО,3 см с силохромам + 4% SE-30.259
Яшт ЯМ. Яшин Е.Я., Яшин А.Я.Газовая хроматографии5.2.2 Макропористые силикагели с полимерными пленками. Ин¬
тересный пример адсорбента-носителя с полимерной пленкой на поверх¬
ности представляет силохром, модифицированный слоем мелона. Он по¬
лучается обработкой силохрома С-80 раствором меламина в горячей воде.
После выпаривания воды пропитанный меламином силохром нагревают
в муфельной печи при 430Ч50°С. При этом происходит полимеризация
меламина в мелон, строение звена которого, по-видимому, таково:NH,пиЛ/ЦАннЭтот полимер нерастворим ни в воде и кислотах, ни в органических
растворителях. В нанесенной на силохром тонкой пленке он вполне термо¬
стоек до 450°С. Силохром с пленкой мелона на поверхности представляет
собой сильноспецифический адсорбент второго типа.В табл. 5.4 сопоставлены индексы Ковача /, определенные при 100°С,
и величины А/ для пяти стандартных веществ. Значения Л/ определялись
по формуле:А/ = (/ -I )/100мелон на силохроме сквалан''Ташй мелонированный адсорбент сильно удерживает молекулы фуппы
D и поэтому может быть использован для вьщеления веществ этой группы из
смесей. Вещества, молекулы шторых относятся к группам AviB, выходят из
шлонки с мелонированным адсорбентом узкими симметричными пиками.Структурные, термические и химические свойства мелонированного си¬
лохрома позволяют разделять и анализировать на нем как неорганические газы
и легкие угаеводороды, так и высошкипящие ароматические угаеводороды, а
также различные вещества групп AviB с близкими темпера1урами кипения.
Примеры соответстщтощих хроматограмм приведены на рис. 5. И а, б.Таблица 5.4.Сопоставление величин I и Л1 при хроматографировании на сквала¬
не и на силохромах С-80 (меланированном и исходном) при 100°С.Вещества1Д/скваланмелон на С-80С-80мелон на С-80С-80Бензол6496806630,310,14Этанол3849489185,646,34Метилэтилкетон53199610814,655,50Нитрометан4577977803,403,23Пиридин695115813744,636,79260
ГЛАВА 5Газовая адсорбтонно-абсорбтютая хроматография.Рис. 5.11. Хроматограммы на колонке с сшохромом, покрытом
пленкой мелона.а - углеводородов С1 - С4 (при 25°С): 1 - метан, 2 - этан,
3 - этилен, 4 - пропан, 5 - пропилен, б - изобутан, 7-н.бу-
тан, 8 - бутен-1 + тран-сбутен-2, 9 - цис-бутен-2.
б - примесей в галотане (при 80°С): 1 -1,2,2'- трифтор-1,-
1,2-трихлорэтан, 2 - 1,2-дибромтетрафторэтан, 3 - хло-
раллилен, 4 - 1,1,2-трифтор-2 хлор-1 бромметан, 5 -1,1,1-
трифтор-2 хлор-2 бромэтан (галотан - анестезирующее
вещество), 6 - 1,1,1-трифтор -1,2 - дибромхлорэтан.В работе [31] изучены возможности силохрома, модифицированного
сополимером бутилакрилата со стиролом. На колонке с таким сорбентом
измерены величины (при 150°С) и теплоты адсорбции соединений груп¬
пы А (углеводороды (Cj - C^^), соединения группы В (ароматические сое¬
динения, кетоны, нитро- и циансоединения) и молекулы группы D (спирты
С, - С^). Разделены цис- и транс- изомеры декалина.В следующих работах [32, 33] изучены адсорбционные и хроматогра¬
фические свойства силохромов, модифицированных разными полимерами;
сополимером винилацетата с этиленом, поливинилацетатом, сополимером
винилацетат с метилометакрилатом, поливинилхлоридами с разной моле¬
кулярной массой. Для модифицирования гидроксилированный силохром
выдерживали 24 часа в растворе полимера, избыток растворителя удаля¬
ли, а сорбент вакуумировали при комнатной температуре. Затем образцы
сорбентов нагревали в потоке гелия в колонках при 215°С до появления
стабильной нулевой линии. На всех сорбентах были измерены теплота
адсорбции элементоорганических соединений IV Б группы (производные
кремния, германия, олова в разных сочетаниях).261
Яшин Я.И.. Яишн Е.Я.. Яшин А.Я. Газовая хроматографияИнтересные результаты получены при модифицировании силохромов
фталоцианинами лития, натрия, кобальта, цинка, меди, безметального фтаци-
анина, а также тетрасульфофталоцианинов железа, никеля и кобальта [34-36].5.2.3 Макропористые силикагели с пленками жидких кристаллов.В работах [37-45] были исследованы хроматографические свойства сор¬
бентов на основе макропористых силикагелей с удельной поверхностью
10,30,50,80,120 и 150 м^г и жидких кристаллов (ЖК): олеат холестерина,
пеларгонат холестерина, п-н-нониноксибензаль-н-амиловый эфир п'-ами-
нокоричной кислоты, 4,4'-диэтоксиазоксибензол, п-н-гептилоксибензаль-
н-этиловый эфир п'аминокоричной кислоты, п-анилальаминоазобензол,
п,п'-дибензальдибензадин.На рис. 5.12 представлена температурная зависимость ig F изомеров
ксилола на пеларгонате холестерина, нанесенного на силохром (S^ = 80 mVf)
(а) и на хроматон = 1 м^г) (б) в количестве 6 масс. % (толщина плен¬
ки о = 60 нм). Как видно из рис. 5.12, характер этих кривых подчиняется
общеизвестному представлению о фазовых переходах ЖК при изменении
температуры, а именно: на графике ясно выражены три области фазового
состояния пеларгоната холестерина: твердая, жидкокристаллическая (ме-
зофаза) и жидкая (изотропная область). Переход пеларгоната холестерина
из твердого состояния в мезаморфное сопровождается резким увеличе¬
нием F сорбатов, т.е. в точке плавления при 78°С времена удерживания
возрастают в ~ 5 раз. В области мезофазы (78 - 91,5°С) при дальнейшем
увеличении температуры наблюдается уменьшение F. При переходе пе-
ларогоната холестерина в изотропную жидкость опять имеет место рост
параметров удерживания, но в меньшей степени. В точке осветления (91,5°С)
ЖК селективность разделения (величина а, равная отношению исправлен¬
ных времен удерживания пара- и л<ета-изомеров) уменьшается. Если в об¬
ласти твердого состояния пеларгоната холестерина значения F ксилолов
близки и изомеры не разделяются, то в области метафазы величины F раз¬
личаются и ксилолы разделяются.Таким образом, максимальная селективность пеларгоната холесте¬
рина по отношению к мета- и иара-изомерам проявляется в точке пере¬
хода ЖК в жидкокристаллическое состояние и затем монотонно падает
по мере приближения к точке осветления. При отложении того же коли¬
чества ЖК (6 масс. %) на активном сорбенте - силохроме С-80 (S^^ ~ 80 м%) с
толщиной пленки о = 0,7 нм характер кривых фазовых переходов сильно
изменяется. Как видно из рис. 12а в области температур перехода в ме-
зофазу не наблюдается резкого скачкообразного изменения параметров
удерживания сорбатов. Наоборот, во всем исследуемом интервале с уве¬
личением температуры наблюдается падение объемов удерживания. Эта262
ШйМ.Газовая адсорбиионно-абсорбиионная хроматографт.V и» г,9 t,7 iiРис. 5.12. Зависимость логарифма объема удерживания (Ig V^ изо¬
меров ксилола от температуры на дегидроксшированном
сшохроме С-80 (а) и хроматоне (б) с 6% пеларгоната
холестерина: 1 - м-ксилол; 2 - п-ксилол (около хрома¬
тограмм указаны коэффициенты разделения а для этих
ксилолов).закономерность наблюдалась также для монослоев других жидких фаз на
силохроме С-80 [37, 38].Наибольший интерес вызывает сохранение достаточно высокой се¬
лективности на таких сорбентах во всем температурном интервале жид¬
кокристаллического состояния вещества. Исходный адсорбент-носитель
не раделяет изомеров ксилола, поэтому вся селективность разделения об¬
условлена пленкой ЖК. Этот необычный эффект подробно изучен нами
[37-45] при различных соотношениях ЖК и адсорбента-носителя, т.е. было
исследовано влияние толщины пленки ЖК на эффект фазовых переходов;
кроме того, исследовано влияние величины удельной поверхности адсор¬
бента-носителя на этот эффект при сохранении химической природы по¬
верхности адсорбента-носителя на примере как специфических, так и не¬
специфических адсорбентов.Можно бьшо предположить, что эффект сохранения высокой селектив¬
ности имеет место только при небольших количествах жидкой фазы, т.е. для
пленок ЖК, а с увеличением толщины пленки ЖК на силохроме С-80
наблюдаться (как и в случае с малоаетивным носителем - хроматоном)
резкое увеличение параметров удерживания сорбата при переходе в мезофазу.
Однако, при исследовании образцов силохрома С-80 с содержанием ЖК от263
Яшин Я.И.. Яшин Е.Я., Яшин А.Я.Газовая храматогтФия0,5 до 120 масс. % изломов на зависимости Ig F от 1/Т не обнаружено. Это
можно объяснить тем, что повышение температуры не приводит к фазо¬
вым переходам, т.е. слои ЖК находятся в адсорбированном состоянии, и
энергия адсорбции превышает кинетическую энергию молекул ЖК, соот¬
ветствующую данной температуре. В точке осветления ЖК тоже практиче¬
ски не наблюдается изменений в объемах удерживания.Щ,[мм/г]И'л 78%мI IМ 19гл 2,в 2.9Рис. 5.13. Темгщхащ)ная зависимость Ig V ихтертшшманадегидрок-
силированнам хроматоне с 1 масс. % (а) и 12 моею. % (6) пелар-
гоната холестерина и на том же сорбенте с 6 масс. %
аэросила, диспергированного в пеларгонате холестерина
(в) [42]; 1, 2 -см. рис. 1.Следовательно, ЖК на поверхности силохрома С-80 в количестве
6 масс. % не имеет фазовых переходов в интервале от 60 до 107°С. Таше же
поведение ЖК наблюдается и на хроматоне, если на его поверхность отложить
небольшое количество жидшй фазы. Например, при содержании 1 масс. %
ЖК на хроматоне н^якщаегся практически монотонное уменьшение харак¬
теристик удерживания с увеличением температуры шлонки (по аналогии с
силохромом); однаш в этом случае разделения изомеров ксилола не наблю¬
дается даже в области мезофазы.Подобные зашномерности наблюдались авторами [38], шгда при умень¬
шении шличественного содержания п^т'-метошиэтошиюошибензола на хро-
мосорбе Р до 1,2 масс. % селекгавность ЖК по отношению к мета- и пара-
изомерам резш снижалась. При увеличении содержания (II) до 6 масс. %
(рис. 5.13) и до 12 масс. % (рис. 5.13) на хроматоне наблюдается скачкообраз¬
ное увеличение характеристик уцерживания при переходе ЖК в мезофазу.При нанесении ЖК на силохром С-80 (адсорбент с большой удель¬
ной поверхностью и высоким адсорбционным потенциалом), по-видимо¬
му, происходит определенная ориентация и упорядочение расположения264
[Щ^§ЛА гязовая адсорбиионно-абсорбиионная хроматография.молекул ЖК на поверхности адсорбента-носителя за счет его адсорбци¬
онного поля. При дальнейшем увеличении шличества ЖК на силохроме
git) молекулы ориентируются относительно тех молекул ЖК, которые уже
находятся в упорядоченном состоянии. Эта ориентация, вероятно близка
к ориентации молекул в жидкокристаллическом состоянии, поэтому се¬
лективность разделения высокая. За счет сильного адсорбционного поля
поверхности адсорбента-носителя данное упорядоченное состояние со¬
храняется в широшм интервале температур. Газовая хроматография ис¬
пользовалась как метод изучения фазовых переходов в жидких кристаллах
[45]. Нами был изучен также новый тип реентрантных жидких кристаллов
в газовой хроматографии [43].5.3. Пористые полимеры как носители жидких фазПористые сополимеры стирола с дивинилбеизолом находят применение в
газовой хроматографии как адсорбенты в чистом виде, так и в качестве адсор¬
бентов-носителей. Нанесение жидшсти позволяет изменять селективность
разделения, сократить в некоторых случаях время разделения и повысить эф¬
фективность шлонки. Пористые полимеры в большинстве случаев являются
неодаороднопористыми адсорбентами, поэтому нанесение жидшсти приво¬
дит к повышению геометричесшй однородности адсорбента тольш в тех слу-
чажс, шгда разделяемые вещества не растворяются в нанесенной жидшсти.
В раде работ [46-49] нанесение жидшсти в шличестве до 10% на пористые
полимеры проводили с целью повышения эффективности разделения. При
нанесении больших мшшчеств жидшсти можно в сильной степени изменять
селективность получаемого coj^HTa, поскольку в этом случае селективность
разделения будет определяться как адсорбцией, так и растворением.В работах [46-49] были исследованы хроматофафические свойства
сорбентов на основе полисорба-1, модифицированного различными жид¬
костями, в частности скваланом, твином-80, цианэтилированным пентаэ¬
ритритом и диглицерином. Все эти жидшсти смачивают полисорб-1 в раз¬
ной степени, краевый угол смачивания составляет в этих случаях 22,23,40
и 47® соответственно. При нанесении до 40% жидкости от массы полисор¬
ба-1 слипание его частиц не наблюдалось, однаш удельная поверхность s
сорбента при этом сильно изменялась. По изменению s можно судить о том,
каким образом жидкость распределяется в грануле пористого полимера.
Зависимость удельной поверхности полисорба-1 от шличества нанесен¬
ной жидшсти приведена на рис. 5.2. Следует отметить резше уменьшение
* при нанесении уже небольших шличеств жидкости. Наиболее сильное
уменьшение s наблюдается при нанесении до 5% (масс.) жидкости — от
240 до ~ 100 MVr Чтобы уменьшить s еще на 80—100 mVt, нужно нанести
еще 20—30% жидкости. Для всех жидкостей кривые зависимости s от на¬
несенного шличества жидкости имеют сходный вид.265
Яшин ЯМ, Яшин Е.Я., Яшин А.Я.Газовая хщшптафияНа рис. 5.14 приведены зависимости удерживаемых объемов различ¬
ных веществ от количества сквалана, нанесенного на полисорб-1.Рис.5.14 Зависимость от количества сквалана, нанесенного на
полисорь-1. Адсорбаты: I - бензол; 2 - метилэтилкетон;
3 -уксусная кислота; 4 - акрилонитрил; 5 - ацетонитрил;
6 - ацетон; 7 - этанол; 8 - ацетальдегид; 9 - метанол;
10 - вода; 11 - пропанол.С ростом количества сквалана для всех веществ вначале наблюдается
уменьшение удерживаемых объемов, а затем они практически не изменя¬
ются. По-видимому, это отражает тот факт, что влияние адсорбции и раство¬
рения направлено в противоположные стороны: с увеличением количества
нанесенной жидкости уменьшается роль адсорбции разделяемых веществ
на полисорбе и увеличивается роль растворения в пленке нанесенной жид¬
кости. Нужно отметить также, что все исследованные полярные вещества
плохо растворяются в сквалане. Поэтому для них удерживаемые объемы
уменьшаются приблизительно пропорционально уменьшению удельной
поверхности сорбента, причем степень разделения практически остается
такой же, как и для не модифицированного полисорба-1. Это позволяет
сокращать время анализа на модифицированных подобным образом сор¬
бентах. При нанесении сквалана в количестве 10% от массы полисорба-1
удается разделить воду и формальдегид, причем формальдегид элюирует¬
ся перед водой в виде узкого пика.Зависимости удерживаемых объемов от шличества нанесенного дигли-
церина (рис. 5.15) для некоторых веществ, в частности для юды (2) и уксус¬
ной кислоты (7), сильно отличаются от зависимостей для других веществ.266
Гсаовая адсорбтонно-абсорбтоннт хроматография.% »Рис. 5.15. Зависимость от количества диглицирина, нанесенного
на полисорб-1. Адсорбаты: I - уксусная кислота; 2 -вода;
3 - бензол; 4 -метшэтшкетон; 5 - акрилонитрил; 6 - изопро-
панол; 7 - ацетонитрил; 8 - ацетон; 9 - акролеин; 10 - пен¬
тан; 11 - этанол; 12 - метанол; 13 - ацетальдегид.При увеличении содержания диглицерина на полисорбе до <5%
(масс.) удерживаемый объем воды резш возрастает. Это позволяет в ши¬
роких пределах изменять время удерживания воды относительно других
шмпонентов и выбирать оптимальные условия для анализа содержащих
воду смесей. При нанесении 10% (масс.) диглицерина на полисорбе-1 вода
удерживается сильнее метанола и ацетальдегида, а при повышении содер¬
жания диглицерина до 15% (масс.) происходит полное разделение весьма
трущо разделяемой смеси метанол— формальдегид — вода (рис. 5.16).
При нанесении 40% (масс.) диглицерина удерживаемый объем воды может
быть увеличен практически на два порядка по сравнению с хроматогра¬
фированием на чистом полисорбе. Возможность увеличивать удерживае-
объем уксусной кислоты относительно других соединений позволяет
определять в ней некоторые примеси, например винилацетата, который из
колонки с чистым полисорбом-1 выходит ни «хвосте» пика уксусной кис¬
лоты.267
Яшт Я.И., Яшин Е.Я.. Яшин Л.Я.Газовом хттатогрдфияРис. 5.16. Хроматограмма: 1 - метанол; 2 - вода; 3 - формальде¬
гид. Колонка 200x0,3 см с полисорбом-1 + 15% диглицери¬
на при 5 (УС.Таблица 5.5.Суммарный коэффициент распределения К в диглицерине и вклад
растворения в удерживаемый объем на колонках с различным
количеством нанесенного на полисорб-1 диглицеринаВеществоКВклад растворения, %15% диглицерина40% дигаицеринаА1фолеин441026Ацетон521127Акрилонитрил841332Ацетальдегид211531Ацетонитрил1042146Этанол1093058Метанол864869Уксусная кислота19787787Вода42296100Формальдегид2237100100268
0^ВЛА гязовая адсорбиионно-абсорбиионная хроматография.Увеличение удерживаемых объемов воды, уксусной кислоты и неко¬
торых других веществ при повышении содержания диглицерина связано
с возрастанием вклада водородных связей в адсорбцию этих соединений.
0ри больших содержаниях нанесенной жидкости можно оценить вклады
растворения и адсорбции. Для этого надо определить удерживаемые объ¬
емы на двух сорбентах, отличающихся только количеством нанесенной
5КИДКОСТИ. Нужно выбрать такие количества жидкости, при нанесении ко¬
торых поверхность сорбента изменяется мало. Составляя уравнения типа
(5.2) для разных количеств нанесенной жидкости, можно оценить констан¬
ты равновесия для адсорбции и растворения и вклад растворения в общий
удерживаемый объем на таком сорбенте В табл. 5.5 приведены суммарные
юэффициенты распределения в диглицерине, нанесенном на полисорб-1,
и вклады растворения в суммарный удерживаемый объем.Интересно сравнить эффективность сорбента с различными количе¬
ствами нанесенной на него жидшсти. Минимум на кривых зависимости Н
от U соответствует 5—6% жидкости. Такие количества нанесенной жидко¬
сти близки к емшсти плотного монослоя на поверхности адсорбента-но-
сителя. С увеличением количества нанесенной жидшсти эффективность
колонок ухудшается, по-видимому, за счет возрастания роли внутренней
диффузии.5.4. Углеродные адсорбенты в качестве носителей жидких фазПрименение графитированных саж с нанесенными на них монослоями
жидких фаз было впервые описано в работах [50-52]. Было изучено влия¬
ние молекулярной массы полиэтиленглишлей (ПЭГ) на плотность и струк¬
туру монослоев на графитированной канальной саже (s= 80 м7г) Удержи¬
ваемые обьемы н-алканов сильно уменьшаются при нанесении небольших
количеств ПЭГ до определенного значения, равного емкости монослоя;
при дальнейшем увеличении нанесенной жидкой фазы удерживаемые обь¬
емы алканов остаются постоянными. Резкое уменьшение удерживаемых
обьемов н-алканов объясняется ослаблением неспецифического дисперси¬
онного взаимодействия с поверхностью сажи, покрытой молекулами ПЭГ.
Молекулы спиртов могут образовывать водородные связи с эфирными
группами ПЭГ. Повышение вклада этого специфичесшго взаимодействия
в общую энергию адсорбции превосходит по абсолютной величине соот¬
ветствующее уменьшение неспецифичесшго взаимодействия при тонких
покрытиях. Поэтому удерживаемые обьемы спиртов повышаются, причем
это повышение происходит до 2,3% ПЭГ-300 на единицу массы сажи, что
соответствует 75% емкости монослоя. При дальнейшем увеличении ко¬
личества отложенного ПЭГ (примерно до 3%, что соответствует полной
емкости монослоя) удерживаемые объемы спиртов уменьшаются. Очевид-269
Яшин Я.И.. Яшт ЕЯ, Яшин АЯ. Газовая хроматографииНО В ЭТОЙ области уменьшение неспецифического взаимодействия превос¬
ходит рост специфического взаимодействия. При дальнейшем увеличении
количества ПЭГ на поверхности сажи удерживаемые о&ьемы спиртов
вновь возрастают, в этом случае уже за счет дополнительного вклада рас¬
творения спирта в жидкости.Адсорбционное модифицирование поверхности ГТС было исследова¬
но во многих работах. В нешторых из них наносилось очень небольшое
количество слабо и неспецифически адсорбирующего вешества, напри¬
мер апиезона L, молекулы которого располагались на наиболее сильно ад¬
сорбирующих неоднородных местах поверхности ГТС (у мест контакта
частиц или у возможных мест разрыва графитовых сеток на поверхности
самих частиц). Такой способ модифицирования обычно сильно увеличи¬
вал прочность зерен и эффективность колонки, не влияя существенно на
селективность адсорбента [30]. В других работах стремились нанести на
поверхность ГТС более или менее плотный мономолекулярный слой моле¬
кул или макромолекул. Иногда модификатор наносили в количестве, пре¬
вышающем емкость монослоев. В этом случае, если наносилась жидкость,
получался адсорбционно-абсорбционный вариант газовой хроматографии,
а если наносилось трудно растворимое твердое вещество, то наряду с ад¬
сорбцией на модифицированной поверхности ГТС происходила также
адсорбция на поверхности кристаллов избытка модификатора. Успешное
разделение на ГТС с нанесенными на нее различными органическими ве¬
ществами было проведено Гьошоном [53-55].Изучались адсорбционные свойства самих кристаллов фталоцианина
[54], а также фталоцианина, нанесенного на графитированную термиче¬
скую сажу [56]. В игольчатых кристаллах а- и р-форм фталоцианина или
его комплексов с металлом особенно сильно развиты грани, на которые
выходят краевые группы плоских молекул фталоцианина, как бы нани¬
занных одна на другую. На грани же, образующие концы этих игольча¬
тых кристаллов, выходят плоскости молекул фталоцианина [54]. Грани
кристаллов, образованные звеньями СН, создают очень слабое поле сил
межмолекулярного притяжения. При раздроблении игольчатых кристал¬
лов возрастает роль граней, на которые молекулы фталоцианина выходя !
своими плоскостями.Дифференциальное изменение внутренней энергии при нулевом за¬
полнении -AUi при адсорбции на фталоцианине дипольных молекул груп¬
пы В (ацетон, пиридин) и молекул группы D (спирты) заметно выше, чем
для молекул группы А (н-алканы).270
Газовая адсорбтюнно-абсорбтотт хрататография.Были исследованы адсорбционные свойства фафитированных саж с
различными количествами нанесенного на них фталоцианина и фталоци-
анинов меди, никеля, кобальта и цинка и определены теплоты и энтропии
адсорбции многих классов соединений (углеводородов, фенолов, аминов,
пиридина, спиртов, эфиров и др.) [55]. Такие фталоцианины обладают се¬
лективными адсорбционными свойствами, в частности и по отношению
к изомерам ароматических угаеводородов. Например, полное разделение
всех трех изомеров ксилола возможно только на фталоцианине меди (см.
формулу). На фталоцианине никеля порядок выхода этих изомеров такой
же, как и на чистой графитированной саже: орто- и иара-ксилолы разделя¬
ются плохо. На фталоцианинах кобальта и цинка плохо разделяются мета-
и иора-изомеры. Эти два фталоцианина довольно сильно адсорбируют
ароматические многоядерные угаеводороды. На шлонке длиной 1 м с на¬
несенным на графитированную сажу фталоцианином в количестве ошло
5% можно легш разделять смесь антрацена и фенантрена, которая трудно
разделяется на газо-жидкостных шлонках, а на самой графитированной
саже - только при значительно более высоких температурах.На рис. 5.17 показаны примеры газо-адсорбционного хромагографи-
чесшго разделения некоторых смесей полишнденсированных ароматиче¬
ских углеводородов, а также стероидов на фталоцианине меди, нанесен¬
ном на графитированную термическую сажу. В первом случае изомеры
(антрацен и фенантрен) разделяются вследствие различия их электронной
конфигурации, а во втором случае (этиохоланолон и андростерон) -■ вслед¬
ствие различия их геометричесшй структуры.Порядок удерживания цис- и транс-тексетъ-2 на монослоях фтало-
Цианинов никеля и меди совпадает с порядком их удерживания на чистой
графитированной саже, тогда как на фталоцианинах кобальта и цинка по¬
рядок выхода этих веществ обратный. Таким образом, меняя форму фта-
лоцианинов, нанесенных на поверхность фафитированной сажи, можно
сильно изменять удерживаемые обьемы некоторых изомеров, причем даже
‘изменять порядок их выхода из шлонки; это открывает дополнительные
возможности при использовании модифицированного адсорбента в ана-
литичесшй хроматофафии и при установлении строения изомеров. Ад-271
Яшин Я.И., Яшин Е.Я.. Яшин А.Я.Гаювая хроматографииРис. 5.17. Хроматограммы смесей на фталоцианине меди, нанесен¬
ном на ГТС в количестве 1% (масс.) при 265 ®С; а-много¬
ядерные ароматические углеводороды (слева направо):
фенантрен, антрацен, флуорантен, пирен, хризен, на-
фтацен; б - стероиды: этиохоланолон, андростерон. Ко¬
лонка 100x0,3 см, газ-носитель - азот, скорость потока
28 cmVmuh, детектор ионизационно-пламенный.сорбционные свойства граней самих кристаллов и нанесенных монослоев
различных комплексообразующих соединений могут быть с успехом ис¬
пользованы для получения дополнительной информации уже не только о
геометрической структуре молекул, но и об их электронной конфигурации,
определяющей их донорные свойства.Состояние фталоцианина, нанесенного на поверхность адсорбента-
носителя, в значительной степени зависит от соотношения теплоты ад¬
сорбции и теплоты возгонки адсорбата [57]. По-видимому, на поверхности
графитированной сажи образуется и плотный монослой из молекул фтало¬
цианина, и мелкие его кристаллы.В работе [52] изучены графипфованные сажи с адсорбированными слоя¬
ми полимеров. Исследованы изотерлш адсорбции поливинилацетага из рас-
ТВ01ЮВ в метаноле, поливинилового спирта и полиакрилоюй кислоты из рас¬
творов в воде на поверхности графитированной сажи. Адсорбционное насы¬
щение достигается тольш в случае поливинилацетага. Полученные образцы
сажи с различным содержанием этих полимеров на поверхности исследованы
газохроматографическим методом. При адсорбции различных молекул на по¬
верхности модифицированной полимерами сажи энергия неспецифических
взаимодействий резш уменьшается. Наличие в модифицирующем слое по¬
лимеров функциональных групп COOCHj, ОН и СООН придает поверхности
таких адсорбентов способность к специфичесшму взаимодействию.272
ГЛАВА SГазовая адсорбиионно-абсорбуионная хроматография.Кроме жидкостей на графитированные сажи наносили также органи¬
ческие твердые и жидкие вещества: антрахинон [58], бензофенон [59], пи¬
криновую кислоту [60], 1,3,5-тринитробензол [61], 2,5,7-тринитрофлуорен
[62] (рис. 5.18)./♦/пLо S ш IS го
t.mnРис. 5.18 а) Хроматограмма ароматических углеводородов при 187°С
на ктонке с карбопаком С, модифицированным 2,4,5,7-те-
транитрофяуореном [63]: 1 - бензол, 2 - толуол, 3 - этил¬
бензол, 4 - изопропилбензол, 5 - м-ксилол, 6 - п-ксилол, 7 -о-
кстол, 8 - стирол, 9 - н-пропилбензол.
б) хроматограмма смеси углеводородов и С^, циклопен-
тана, изопентана и н-пентана при 50°С на колонке с кар¬
бопаком С, модифицированным 2,4,5,7-тетранитрофлуо-
реном: 1 - пропан, 2 - пропен, 3 - изобутан, 4 - бутен-1, 5- н-бутан, 6 - изобутан, 7 - цис-бутен-2, 8 - транс-бутен-2, 9 - 1,3-бутадиен, 10 - циклопентан, 11 - изопентан,
12 - н-пентан.Графитированные сажи с нанесенными монослоями жидких фаз были
использованы в аналитической адсорбционно-абсорбцинной хроматогра¬
фии [64-72], в частности для определения микропримесей полярных ве¬
ществ и повышения селективности разделения изомеров ароматических
углеводородов. Некоторые закономерности адсорбционно-абсорбционной
хроматографии на графитированной термической саже рассмотрены в ра¬
ботах [70-72]. В таблице 5.6 приведены области применения модифициро¬
ванных карбопаков фирмы “Supelco”.273
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я.Газовая хроматография
Таблица 5.6Модифицированные карбопяки и их примененияАдсорбент - жидкая фазаОбласть применения1Карбопак В+4% карбовакса 20М+0,8% КОНАмины2Карбопак B-i-5% карбовакса 20МСпирты3Карбопак B-DA-t-4% карбовакса 20МКислоты Cj-C,4Карбопак BAW+5% карбовакса 20МСпиртовые напитки5Карбопак BAW+6,6% карбовакса 20МСпиртовые напитки6Карбопак В+5% флюороколаФреоны7Карбопак В-ь2,5% оронитаСпирты, эфиры, кетоны8Карбопак В+1% SP-1000Галоидоорганика9Карбопак В+3% SP-1500Промышленные растворители10Карбопак B-t-1% SP-1510Промышленные растворители11Карбопак B-i-1.5% ХЕ-60+1%Н,РО,Сернистые газы12Карбопак С-Ю,2% карбовакса 1500Спирты, эфиры, кетоны13Карбопак С-Ю,2% карбовакса 1510Спирты, эфиры, кетоны14Карбопак С+0,3% карбовакса 20МСпирты, эфиры, кетоныПримечания:DA - деактивирован для кислых соединений;AW - промыт кислотой;Карбовакс 20М - карбовакс с молекулярной массой 20000.5.5. Силикагели с привитыми алкильными группамиВ газовой [73] и особенно в жидкостной [74] хроматографии получили
распространение адсорбенты, на поверхности которых химически приви¬
ты линейные молекулы с длинными цепями — так называемые адсорбен¬
ты типа «щеток» [75]. В основном используют кремнеземные адсорбенты:
силикагели, пористые стекла, аэросилы и др. «Прививка» идет по сила-
нольным гидроксильным группам поверхности с образованием связей:“Sr/ \ / \ /
С si-^0 -С si~-0-^SЬ-\ / \ / \-N-Наиболее устойчива связь =Si-0-C=В этом случае важно знать концентрацию гидроксильных групп на
поверхности адсорбента и площадь, занимаемую прививаемой молекулой
[73]. Адсорбенты такого типа обычно устойчивы до температуры разложс-274
fJIABA S Газовая адсорбиионно-абсорбиионная хроматография.0ИЯ привитых «щетою>, поэтому фоновый ионный ток мал. Эффективность
{(олонок с такими адсорбентами обычно выше по сравнению с исходным ад¬
сорбентом, минимальная высота тарелки составляет 0,3—0,4 мм, причем она
не меняется в широком диапазоне линейных скоростей газа-носителя [73].Механизм удерживания на «привитых» адсорбентах окончательно не
вьшснен. В некоторых работах [76] указывается, что на адсорбентах типа
«шеток» с небольшой длиной привитых цепей удерживание осуществля¬
ется по адсорбционному механизму [77], однако при увеличении длины
цепей сорбция ближе к процессу растворения [78]. Различают два типа по¬
добных сорбентов:а) «щеточные» в чистом виде, когда привитые молекулы располага¬
ются достаточно свободно и вертикально к поверхности, причем соседние
цепи химически не связываются и не полимеризуются;б) привитые цепи могут располагаться вдоль поверхности, химически
связываться друг с другом и полимеризоваться.Во втором случае удерживание обычно имеет место чисто адсорбцион¬
ный характер (когда молекулы не проникают в объем привитого вещества),
либо адсорбционно-абсорбционный характер (когда молекулы проникают
в объем набухающего полимера).5.6. Определение коэффициентов распределения методом адсорб-
ционно-абсорбционной хроматографииИз уравнения (5.2) следует, что в отсутствие заметной адсорбции на
поверхности пленки зависимость от 1Л/^ в этом случае линейна. От¬
резок, отсекаемый на оси ординат, определяет величину Ка по тангенсу
угла наклона можно оценить величину KAs при известной величине As
[18]. Соответствующие измерения проведены в работах [16]. Была также
показана возможность исследования адсорбции на носителе из жидкой
фазы (т. е., по существу, адсорбции из растворов) методом адсорбционно¬
абсорбционной хроматографии [79, 80]. Зависимости удерживаемых объе¬
мов, а также теплот адсорбции от количества жидкой фазы, откладываемой
на поверхности графитированной термической сажи, позволяют оценить
емкости первого, второго и третьего адсорбированных слоев [70 -72].Тонкие жидкие пленки, находящиеся на поверхности графитирован¬
ных саж, подвергаются воздействию со стороны адсорбционного поля
сажи, поэтому их физические свойства отличаются от физических свойств
объемных жидкостей [81, 82]:В работах [82, 83] на основе зависимостей от исследованы свой¬
ства тонких пленок жидкостей на поверхности твердого носителя, и в част¬
ности константы расклинивающего давления.275
Яшт ЯМ. Яшин ЕЛ, Яшт Л Я. Газовая хроматографииЛитература1. Fukuda Т. Japan Analyst, 1959, v.8, р 627—630.2. Martin R. L. Analyt. Chem., 1961, v. 33, p 347-351.3. Martin R. L. Analyt. Chem.1963, v.35, p 116—120.4. Pecsok R. L., de Yllana A., Abdul-Karim A. Analyt. Chem., 1964,
V. 36, p. 542—545.5. Pecsok R. L., Gimp B. A. J. Phys. Chem.,1967, v. 71, p. 2202—2204.6. Martire D. E., Pecsok R. L., Purnell J. H. Nature, 1964, v. 203,
p. 1279—1283.7. Martire D. E. Analyt. Chem, 1966, v.38, p. 244—248.8. Keller R. A., Stewart G. H. J. Chromatog., 1962, v 34, p 1834—18399. Keller R. A., Stewart G. H. J. Chromatog. Sci., 1962, v. 9, p. 1—5.10. Беленький В. Г. и др. Изв. АН СССР. Сф. хим., 1967, т. 2, с. 269—111.
И. Березкин В. Г. и др. ДАН СССР, 1968,т. 180, с 135—137.12. Березкин В, Г. и др. Нефтехимия, 1965, т. 5, с. 438—441.13. Martire D. Е., Pecsok R, L., Pumell J. H.Trans. Faraday Soc., 1965,
V. 61,p. 2496—2500.14. Березкин В. Г., Пахомов В. П., Березкина Л. Г. В кн.: Газовая хро¬
матография. Труды III Всесоюзной конференции по газовой хромато¬
графии. Дзержинск, изд. Дзержинского филиала ОКБА, 1966, с. 267.15. Martire D.E., Polara L. Z. In: Advances in Chromatography. V.I.J.C.
Giddings and С A. Keller, Ed. M. Dekker, New York, 1966, p. 335.16. . Conder J. R., Locke D. C., Pumell J. H. J. Phys. Chem., 1969, v. 73,
p. 700-706.17. Urone D., Takahashi, Kennedy G. H. J. Phys, Chem., 1970, v. 74,
p. 2326—2329.18. Martire D. E. In: Progress in Gas Chromatography. Ed. J. H. Pumell.
Interscience, New York, 1968, p 93.19. Eggertsen H. S., Knight H. S., Groennings S. Analyt. Chem., 1956,
V. 28, p. 303—306.20. Pumell H. Gas Chromatography. New York — London, John Wiley
and Sons, 1962, 375 p.21. Калмановский В. И, Шешенииа Г.В., Яшин Я.И. В кн.: Газовая
хроматография. М., НИИТЭХИМ, 1967,вып. VI, с, 45—5022. Бебрис Н. К., Зайцева Г.Е., Киселев А.В., Никитин Ю.С., Яшин Я.И.
Нефтехимия, 1968, т. 8, С. 481-485.23. Бебрис Н. К., Гиенко Е.Я., Зайцева Г.Е., Киселев А.В., Кустова ГЛ.,
Липкинд Б., Никитин Ю.С., Яшин Я.И. Нефтехимия, 1970, т 10, с 773-777.24. Bebris N.K., Kiselev A.V., Mokeev V. Ya., Nikitin Yu. S., Yashin Ya.I., Zaizeva G. Chromatographia, 1971, v. 4, p. 93—97.276
ГЛАВА 5 Литература25. Киселев А. В., Яшин Я. И. В кн.: Новые сорбенты для хроматогра¬
фии. М., НИИТЭХИМ, вып. 16, 1971, с. 4—9.26. Брук А. И. и др. В кн : Газовая хроматография М., НИИТЭХИМ,
вып. 24, 1975, с. 10—12.27. Яшин Я. И. Физико-химические основы хроматографического
разделения. М., «Химия», 1976, 216 с.28. Савинов И. М., Яшин Я. И. Труды по химии и хим. технол. Горь¬
кий, 1974, вып. 1,с. 73—75.29. Зайцева Г. Е. Яшин Я. И. Труды по Химии и хим. технол, Горький,1974, вып. 3, с 76—7830. Kiselev А.V. J. Chromatog., 1971, v. 58, p.l9—^30.31. Брук А.И., Ветрова З.П., Вяхирев Д. А., Карабанов Н.Т., Яшин Я.И.
Силохромы с полимерными пленками на поверхности в газовой хро¬
матографии. Адсорбционные свойства силохрома, модифицированно¬
го сополимером бутилакрилата со стиролом. Коллоидн. ж. 1975, т.47,
С.118.32. А.И.Брук, З.П.Ветрова, Д.А.Вяхирев, Я.И.Яшин. Адсорбционно¬
распределительная хроматография на силохроме. Процессы в хрома¬
тографических шлонках. М., НИФТИ, 1974, вып. 24, с. 22-27.33. З.П.Ветрова, Н.П.Карабанов, Я.И.Яшин. Модифицирование по¬
верхности макропористых кремнеземов адсорбированными слоями
полимеров. Сб. «Химия элементоорганических соединений» (г. Горь¬
кий), вып. 5, 92-95, 1977.34. З.П.Ветрова, Н.П.Карабанов, Я.И.Яшин. Газо-адсорбционная
хроматография на силохромах, модифицированных фталоцианинами.
Коллоидн.ж., 1975, т. 37, с. 946-949.35. А.И.Брук, З.П.Ветрова, Д.А.Вяхирев, Н.П.Карабанов, Я.И.Яшин.
Адсорбционно-распределительная хроматография металлооргани¬
ческих соединений никеля. Труды по химии и хим. технол., вып. 4,
с. 139-142, 1974.36. Vetrova Z.P., \^akhirev D.A., Karabanov N.T., Maidatzenko G.G.,
Yashin Ya.I. Gas chromatography on monolayers of liquid crystals, Chro¬
matographia, 1975, V.85, p.643.37. З.П.Ветрова, Н.П.Карабанов, Я.И.Яшин. Селективные сорбенты
на основе жидких кристаллов и макропористых силохромов, ДАН
СССР, 1980, 250, с. 1165-1167.38. З.П.Ветрова, Н.П.Карабанов, Г.Г.Майданченко, Я.И.Яшин.
Адсорбционно-абсорбционная хроматография на силохромах со слоя¬
ми жидких кристаллов. Успехи химии 1981, т. 50, № 9, с. 1678-1692.277
Яшин Я.И., Яшин Е.Я.. Яшин А.Я. Гтовая хроматография39. З.П.Ветрова, Н.П.Карабанов, Л.А.Иванова, Т.Н.Шувалова,
Я.И.ЯШИН. Изучение методами газовой хроматографии свойств пленок
жидких кристаллов, нанесенных на поверхность адсорбентов. Колло¬
идный журнал, 1984, № 2, с. 332-335.40. З.П.Ветрова, Н.П.Карабанов, Т.Н.Шувалова, Г.ПМайданченко,
Я.И.Яшин. Изучение методом газовой хроматографии свойств пленок
жидких кристаллов, нанесенных на поверхность адсорбентов. Колло¬
идный журнал, 1984, № 5, с. 1015-1018.41. Z.P.Vetrova,N.T.Karabanov,T.N.Shuvalova,L.A.Ivanova,Ya.I.Yashin.
The use of p-n-Butyl Oxybenzoic Acid as Liquid-Crystalline Sorbents in
Gas Chromatography, Chromatographia, 1985, v. 20, № 1, p. 41-45.42. З.П.Ветрова, Н.П.Карабанов, Л.А.Иванова, Г.Г.Майданченко,
Я.И.Япшн. Жидкие кристаллы в качестве сорбентов в газовой хрома¬
тографии. Межвузовский сборник, Куйбышев, 1985, с. 118-121.43. З.П.Ветрова, Н.П.Карабанов, Л.А.Иванова, О.Б.Акопова,
Г.Г.Майданченко, Я.И.Яшин. Реентрантные жидкие кристаллы. Син¬
тез и исследование мезоморфизма и газохроматографических характе¬
ристик п-акилоксибензоил-окси-бензилирен-п'-циананилинов. Ж.орг.
химии, 1987, т. 57, № 3, с. 646.44. N.T.Karabanov, Z.P.Vetrova, L.A.Ivanova, T.N.Shuvalova, Ya.I.Yashin.
Modified sorbents m binary stationary phase systems in gas chromatogra¬
phy. J. of Chromatogr. 1990, v. 508, p. 193-198.45. Ветрова З.П., Карабанов H.T., Яшин Я.И., Майданченко Г.Г., Ша-
бышев Л.Г., Газовая хроматография как метод изучения фазовых пере¬
ходов в жидких кристаллах Сб. «Жидкие кристаллы и их применение».
Иваново. 1980, с.107-111.46. Устиновская И. А., Гаврилина Л.Н., Малахов В.В., Яшин Я.И. Изв
СО АН СССР. Сер. хим., 1971, вып 2, с. 18.47. Устиновская И. А. и др. Методы анализа и контроля произв-ва в
хим. пром-ти, 1972, вып. 5, с, 3—5.48. Устиновская И. А. и др. Изв. СО АН СССР. Сер. хим 1973, вып. 2.
с. 113—116.49. Устиновская И. А. и др. Изв. СО АН СССР. Сер. хим., 1973,
вып. 2, с. 116—119.50. Белякова Л. Д. и др. ЖФХ, 1968, т. 42, с. 177—180.51. Kiselev А. V. J. Chromatog., 1970, v. 49, p. 84—104.52. Kiselev A. V, Kovaleva N. V, Nikitin Yu. S. J. Chromat., 1971, v. 58,
p. 19—24.53. Vidal-Madjar C., Guiochon G. J. Chromatog. Sci. 197Г1. V.9, p.664.54. Давыдов В.Я., Киселев A.B., Силина ТВ. Коллоидн. ж. 1972, т.34.
с.ЗО; 1974, тЗб, с.359-362; 763 - 765; 945 - 949.278
Литература55. Vidal-Madjar С., Guiochon G. Separation Sci 1967, v.2, p. 155.56. Kouznetsov A.V., Vidal-Madjar C., Guiochon G. Bull. Soc. Chim.
France 1969, p. 1440.57. Berezin G.I., Kiselev A.V., Kozlov A.A. J. Coll. Interface Sci. 1973,
v.45,p.l90.58. Vidal-Madjar C., Guiochon G. Bull. Soc. chim. France, 116, p. 1096—1099.59. Vidal-Mardjar C., Guiochon G. Separation Sci., 1967, v. 2, p. 155—162.60. Di Corcia A., Samperi R. Analyt. Chem., 1975, v. 47, p. 1853; J. Chro¬
matog., 1975, V. 107, p. 99—103.61. Di Corcia A., Samperi R. J. Chromatog., 1976, v. 117, p. 199—202.62. Di Corcia A., Liberti A., Samperi R. J.Chromatog., L976, v. 122,
p. 459—468.63. Di CorciaA., Fritz D., Bruner F. Analyt Chem., 1970, v. 42, p. 1500-1504.64. Bruner F. e. a. Analyt. Chem., 1972, v. 44, p. 2070—2074.65. Di Corcia A. J. Chromatog., 1973, v. 80, p. 69—74.66. Di Corcia A. Analyt. Chem., 1973, v. 45, p 492—496.67. Bruner F. Analyt. Chem., 1972, v. 44, p. 894—897.68. Bruner F. J. Chromatog., 1973, v. 76, p. 1-5.69. Di Corcia A., Liberti A., Samperi R. Analyt. Chem., 1973, v. 45,
p. 1228—1235.70. Bruner F. Analyt. Chem., 1973,v. 45, p. 1851—1859.71. Halasz L, Sebestian I. J. Chromatog Sci., 1974, v 12, p 161—172.72. Majors R. E. Analysis, 1975, v. 3, p. 549—558.73. Halasz L, Sebestian I. Angew. Chem, 1969,v. 81, p. 464—467.74. Rehak V, Smolkova E. Chromatographia, 1976, v. 9, p. 219—229.75. Uihlein М., Halasz I. J. Chromatog., 1973,v. 80, p. 15—18.76. Gryshka E., Kikta J. Analyt. Chem., 1974, v. 46, p. 1370—1373.77. Kirkland J. J. Analyt. Chem., 1971, v. 43, p. 36A; J. Chromatog. Sci.,
1971, V. 9, p. 206—209.78. de Stefano J. J. e. a. J. Chromatog. Sci., 1974, v. 12, p. 337—340.79. Березкин В. Г. и др. Изв АН СССР. Сер. хим., 1970, с. 19—23.80. Serpinet J. J. Chromatog., 1972, v. 68, p. 9-18; 19,73, v. 77, p. 289—299.81. Беккерова P. K. и др. ДАН СССР, 1976, т. 231, 9- 366—36882. Belenky В. G., Rusanov А. L, Turkova L. D. J. Chromatog., 1973,
V. 80, p. 147—151.83. Русанов A. И., Беленький Б. Г. ЖФХ, 1973, т. 47, с. 2046—2048.279
Яшин Я.И., Яшин Е.Я.. Яшин А.Я. Гтовая хтшшпографи»ГЛАВА 6. КАПИЛЛЯРНАЯ ХРОМАТОГРАФИЯ6.1. Введение. Основные этапы развития капиллярной хромато¬
графии.Метод капиллярной хроматографии, разработанный Голэем в
1956-1957 гг. [1], является одним из наиболее выдающихся открытий в
газовой хроматографии. Капиллярная хроматография явилась результатом
теоретических исследований работы насадочных колонок. Голэй, исследуя
размывания в наполненных колонках, рассматривал эти колонки как связку
капиллярных трубок с внутренним диаметром капилляра, близким к раз¬
меру частиц носителя. Приближенные теоретические вычисления Голэя
показали, что диаметр капилляров, который определяет сопротивление га¬
зового потока, должен приблизительно быть равен ВЭТТ. После экспери¬
ментальной проверки обнаружилось значительное расхождение: значение
ВЭТТ было больще размера зерен. Для проверки теоретических предпо¬
ложений Голэй проверил размывание в пустом длинном капилляре и обна¬
ружил полное соответствие теоретических расчетов экспериментальным
результатам. После этих экспериментов, естественно, напрашивалась сле¬
дующая мысль: почему бы в качестве хроматографической колонки не ис¬
пользовать капилляр, на внутреннюю поверхность которого нанесена жид¬
кая фаза? Таким образом была открыта капиллярная хроматография.В настоящее время капиллярная хроматография - наиболее широ¬
ко распространенный метод в газовой хроматографии, с применением
которого выполняется более 70% анализов. Самая высокая разделитель¬
ная способность юионок достигнута в капиллярном режиме в основном
за счет высокой эффективности. Потребителям доступны колонки с эф¬
фективностью несколько сот тысяч теоретических тарелок. На подобных
капиллярных юлонках разделяется до 400-500 и более компонентов при
одном вводе пробы. В частности, бензиновые фракции можно разделить
покомпонентно.Капиллярная хроматография реализуется в двух вариантах: газожид¬
костном и газоадсорбционном. В газожидшстном варианте применяют
пленку жидкой неподвижной фазы на внутренней поверхности капилляр¬
ной колонки, а в газоадсорбционном варианте - слой адсорбента. На прак¬
тике используется главным образом газожидкостной вариант капиллярной
хроматографии. Развитие газоадсорбционной капиллярной хроматографии
несколько задержалось по многим причинам. Однако сейчас капиллярные
колонки в адсорбционном варианте со слоями пористых полимеров, окси¬
да алюминия, силикагеля, углерода и цеолитов доступны потребителям
Наряду с высокой эффективностью такие колонки высокоселективны, а
поэтому обладают более высокой разделительной способностью по срав¬
нению с газожидкостными колонками.
хроматографияВначале в качестве хроматографической колонки использовались ме¬
таллические капилляры, а с 1960 г., коща Дести описал простое устройство
для вытягивания стеклянных капилляров в лабораторных условиях, стали
дрименять стеклянные капиллярные колонки [2]. Позднее были предло¬
жены кварцевые капиллярные колонки [3]. В настоящее время в аналити¬
ческой практике газовой капиллярной хроматографии в основном приме¬
няются кварцевые капиллярные колонки, особенно при анализе сложных
многокомпонентных смесей, так как поверхности стенок таких колонок
каталитически неактивны, и исключено появление посторонних соеди¬
нений (в отличие от металлических капиллярных колонок). Это особенно
важно при контроле загрязнений окружающей среды и при анализе био¬
логических веществ. Кроме того, кварцевые капиллярные колонки удобны
в работе, поскольку обладают гибкостью и механической устойчивостью,
что позволяет кварцевую капиллярную колонку подсоединять к любому
прибору.Металлические капилляры, в частности из нержавеющей стали, при¬
меняются в основном при анализе смесей стабильных соединений, на¬
пример, углеводородов в нефтехимии. В последние годы предложены
капиллярные колонки из нержавеющей стали с инертной внутренней по¬
верхностью. Опубликованы десятки книг и сотни обзоров, посвященных
капиллярной газовой хроматографии [4-16].Доля работ, выполняемых с использованием капиллярных колонок,
постоянно увеличивается. Быстрыми темпами совершенствовался метод,
аппаратура, расширялись области применения. Некоторые фирмы выпу¬
скали специализированные газовые хроматографы для капиллярной хро¬
матографии со специфическими микродетекторами и с дозирующими си¬
стемами без деления потока, которые обеспечивали получение надежных
количественных результатов. В настоящее время практически каждый га¬
зовый хроматограф имеет капиллярный режим.Можно выделить следующие основные направления в развитии ка¬
пиллярной газовой хроматографии:- расширение диапазона внутренних диаметров капиллярных хрома¬
тографических колонок (от 0,05 до 0,53 и даже до 0,75 мм);- применение коротких капиллярных колонок (от 5 до 15 м);- применение капиллярных колонок с толстыми пленками жидкой
фазы (до 5 мкм и более);- совершенствование методик приготовления кварцевых капиллярных
*®лонок;- разработка капиллярных колонок с химически привитыми жидкими
фазами на поверхности капилляров;281
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я.Газовая хроматография- разработка адсорбционных и адсорбционно-абсорбционных капил¬
лярных колонок, колонок типа РогаРШТ;- разработка капиллярных шлонок с жидкими кристаллами в качестве
неподвижной фазы;- развитие капиллярной сверхкритичесшй хроматографии;- создание капиллярных колонок для работы при высоких темперащэах;- создание капиллярных колонок для хиральной хроматографии;- применение юпиллярных шлонок в промышленных хроматографах;- разработка методов шнцентрирования и применение капиллярной
хроматографии для микроанализа.Ниже изложены основные достижения в развитии капиллярной газо¬
вой хроматографии в соответствии с перечисленными выше направления¬
ми. Обычно применяемые в практике капиллярной газовой хроматографии
шлонки имеют внутренний диаметр 0,2 - 0,3 мм. В настоящее время все
большее распространение получают капиллярные колонки большего диа¬
метра, а именно 0,32 - 0,53 мм, дозирование в которые существенно упро¬
щается. Эти шлонки с успехом могут заменить насадочные. Капиллярные
шлонки широшго диаметра изготавливают в основном небольшой длины
(от 5 до 30 м).С целью повышения эффективности шлонок применяют капилляры
меньших диаметров (0,05 - 0,1 мм).В табл. 6.1 приведены данные, характеризующие зависимость эффек¬
тивности капиллярной колонки (число теоретических тарелок, приходяще¬
еся на единицу длины шлонки) от ее диаметра.Таблица 6.1.Эффективность капиллярных колонок различного диаметраДиаметр колонки,
ммЧисло теоретических
тарелок на 1 м длины
колонкиДлина колонки,
эквивалентная 100000
теоретическим тарелкам,
м0,532100480,323400290,254500220,101100090,05210005Минимальная высота теоретической тарелки для сорбируемых ве¬
ществ составляет Н^^^ ~ 1.9 г, где г - радиус капилляра. Отсюда число те¬
оретических тарелок в расчете на 1 м длины шлонки будет определяться
соотношением N = 1000/1,9 г. Следовательно, для достижения эффектив-282
Капиллярная хроматографияности, отвечающей большему числу теоретических тарелок, целесообраз-00 переходить к колонкам небольшого диаметра (порядка 0,05 - 0,1 мм).Однако, важным достоинством обладают капиллярные колонки широ¬
кого диаметра (0,53 мм) с толстыми пленками жидкой фазы. Такие колонки
позволяют проводить анализ в 5 - 6 раз быстрее, чем при работе с насадоч-
ными колонками в сопоставимых условиях [17]. При этом эффективность
капиллярных колонок остается выше, чем насадочных. Хроматографиче¬
ские пики, полученные с помощью капиллярных колонок, значительно
уже, следовательно, чувствительность анализа повышается. Достаточно
высокая эффективность капиллярных колонок широкого диаметра позво¬
ляет решать различные аналитические задачи при меньшем числе колонок.
В настоящее время можно выделить четыре типа капиллярных колонок
(таблица 6.2).Таблица 6.2Классификация капиллярных колонокТипы колонокТолщинапленкисорбента(мкм.)Особенности колонокWCOT (Wall-Coated
open tubular)0,01-1Внутренняя поверхность капиллярных
колонок покрыта пленкой жидкой фазы.SCOT (Support-Coated
epen tubular)0,5-5Внутренняя поверхность капиллярных
колонок покрыта твердым носителем и на
него наносится жидкая фаза. PLOT(porous-layer open
tubular)5-20На внутреннюю поверхность капиллярных
колонок наносится слой адсорбента.CLOT (Carbon layer 2pen
tubular)10-20Ha внутреннюю поверхность капиллярных
юлонок наносится слой углеродного
адсорбента. Одной из актуальных задач в хроматографии, в том числе и капилляр¬
ной, является создание стабильно работающих колонок. Это исключитель¬
но важно для практики. Используя микропроцессорную технику, с помо¬
щью банка данных параметров удерживания, полученных с применением
стандартизованных колонок, по определенным программам можно прово¬
дить идентификацию неизвестных компонентов в сложных смесях.Наиболее распространенные капиллярные колонки с жидкой пленкой
на внутренней поверхности имеют ограниченный срок службы из-за лету¬
чести жидкой фазы. Кроме того, жидкую фазу в случае загрязнения часто
практически невозможно регенерировать. Этих недостатков не имеют ад¬
сорбционные капиллярные колонки со слоем твердого пористого сорбента283
Яшин Я.И. Яшин Е.Я.. Яшин А.Я.Газовая хроматографияна внутренней поверхности (адсо{Лционные незаполненные капиллярные ко¬
лонки) и капиллярные шлонки с химически привитыми жидкими фазами.Адсорбционные капиллярные колонки, как правило, обладают боль¬
шей селективностью, чем капиллярные колонки с жидшй фазой.Первой работой по адсорбционной капиллярной хроматографии была
наша работа [18], где для разделения смеси углеводородов С, - в ка¬
честве шлонки был использован стеклянный капилляр с пористым слоем
длиной 10 м (пористый слой получен за счет химичесшго травления вну¬
тренней поверхности).В работе [19] было проведено химическое модифицирование поверх¬
ности стеклянных капиллярных шлонок в процессе их вытягивания.В настоящее время в изготовлении стабильных воспроизводимых ад¬
сорбционных капиллярных колонок достигнут значительный прогресс.
Впервые голландская фирма “Chrompack” разработала четыре типа ад¬
сорбционных колонок типа PoraPLOT со слоем оксида алюминия, с по¬
лимерным пористым слоем, с цеолитами 5А и 13Х.В табл. 6.3 указаны адсорбенты, используемые в незаполненных ка¬
пиллярных хроматографических колонках в качестве неподвижной фазы,
и области применения таких шлонок (разделяемые смеси).Таблица 6.3.Неподвижная фаза адсорбционных незаполненных
капиллярных колонокАдсорбционный слой в колонкеРазделяемые смесиОксид алюминияУгаеводороды С, - С,„СиликагелиУглеводороды С, - С,„АэросилыНеполярные и слабополярные соединенияЦеолит 5АНеорганические газы и легкие
угаеводородыЦеолит 13ХГрупповое разделение углеводородовПористые полимеры (на основе
стирола и дивинилбензола)Неорганические газы, зтлеводороды С, - С|„
Кислородосодержащие органические
соединения (спирты, эфиры, альдегиды,
кетоны и др.), галоген- и серосодержащие
соединенияГрафитированные термические
сажиНеполярные и полярные соединения,
изомерыАдсорбционные капиллярные шлонки со слоем оксида алюминия
были впервые предложены в 1963 г [20]. Для нанесения адсорбционного
слоя на внутреннюю поверхность шлонки суспензию бемита динамиче-284
fllABA 6 Капиллярная хуаматографшским методом пропускали через металлический капилляр, при этом слой
частиц бемита откладывался на внутренней поверхности. На такой колонке
была разделена при комнатной температуре смесь фторированных угаеводо¬
родов. В работе [4,21] слой оксида алюминия толщиной 5 мкм был получен
окислением внутренней поверхности алюминиевого капилляра. В работе
[21] была получена кварцевая капиллярная колонка со слоем оксида алюми¬
ния, нанесенным статическим методом из стабильной суспензии. Предложе¬
ны капиллярные адсорбционные колонки со слоем силиказоля [22], графи¬
тированной сажи [23,24], цеолита [25], волокнистого бемита [26].Капиллярные колонки со слоем оксида алюминия хорошо разделяют
смеси углеводородов, однако некоторые соединения дают несимметрич¬
ные пики [21]. Высокая селективность разделения (при симметричных пи¬
ках) достигнута в колонке со слоем оксида алюминия, модифицированного
с помощью КС1.На капиллярной колонке (50 м х 0,32 мм) со слоем Al^Oj+КС1 толщиной5 мкм в режиме программирования температуры от 70 до 200°С за 30 мин
разделяется смесь предельных и непредельных угаеводородов С, - С,, при
этом полностью разделяется смесь всех изомеров С^, возможно частичное
наложение пиков изопентана и бутадиена-1,2. Порядок выхода углеводо¬
родов следующий: метан, этан, этилен, пропан, циклопропан, пропилен,
ацетилен, изобутан, пропадиен, н.бутан, транс-бутан-2, бутен-1, изобутен,
цис-бутен-2, изопентан+бутадиен-1,2, прошш, н.пентан, бутадиен-1,3,3-ме-
тил-бутен-1, винилацетилен, этилацетилен.Благодаря отсутствию фона (от колонки) дфейф нулевой линии отсутствует.
По этой же причине такие колонки выгодно применять для анализа микропри¬
месей. Так, с помощью такой капиллярной колонки (150 м х 0,32 мм) можно
определять примеси в пропилене на уровне ppm., примеси в винилхлориде
(этилен, хлорметан, винилбромид, бутаднен-1,3, хлорэтан, винилацетилен) на
Лювне 0,1 ppm при 75°С за 15 мин. Применяя такие колонки небольшой дли¬
ны, можно проводить экспресс-разделение. Так, на кгаонке 10 м х 0,53 мм с
внутренним покрытием Al^Oj+КС1 при 70°С за 5 мин была разделена сложная
смесь: изобутан, н.бутан, транс-бутен-2, fyren-l, изобутен, цис-буген-2, бутади¬
ен-1 Д, бутадиен-1,3. На колонке 50 м х 0,32 мм смесь при 130°С {шзделяется за
100 с.6.2. Размывание в капиллярных колонках.Уравнение ВЭТТ для капиллярной хроматографии имеет вид [2]H = B/u + Cu + C^u (6.1)где В = 2D определяется молекулярной продольной диффузией; и- сопротивление массопередачи в газовой и жидкой фазах.Величины и равны285
Яшт ЯМ. Яшин Е.Я.. Яшин А.Я. Гтовам зфоматографияС = [(1 + 6К + ПК/)/24(1 + К )2]C^ = [2K/3(l+K/]d^^/D^ (6.2)Коэффициент емюсти К^ вычисляют по формулеK=t'A. (6-3)где - исправленное время удерживания; - время удерживания не¬
сорбируемого компонента.Полное уравнение ВЭТТ:2D 1+6К+11К' 2К d\Ы = + S i ц + S i«_ ц (6.4)U 24(1+К/ 0 3(1+К/ D, ^ ’Вихревая диффузия не входит в это выражение.Оптимальное значение для линейной сшрости газа-носителя можно
найти, если щ)одифференцировать уравнение (6.2.1) относительно и и по¬
лученный результат приравнять нулю:(Шди = -Ви-^ + С+C^ = 0,u„„ = Vb7^CJ (6.5)Подставив это значение оптимальной линейной сшрости в уравнение
(6.1), получим выражение для минимальной ВЭТТ:= 2л/ЩТс J (6.6)Для несорбируемых веществ, шгда К^ = О и = 0:Н^«0,58г (6.7)Для сорбируемых веществ, шгда К^ достаточно велик:Н^~1,9г (6.8)В этом случае величина непосредственно определяется внутрен¬
ним диаметром тгаллярной трубки (Н^ = 1,9 г = 2г = d). Отсюда число
теоретических тарелок N в расчете на один метр длины шлонки как ранее
указывалось будет определяться соотношением:N= 1000/d (6.9)Максимально достижимое число теоретических тарелок при вну¬
треннем диаметре капилляра 0,5 мм равно 2000, при 0,33 мм - 3000 и при
0,2 - 5000 на 1 м длины колонки.Оптимальные линейные скорости, при которых реализуется макси¬
мальная эффективность 1кшиллярных шлонок, равна 4/d см/с для водорода
в качестве газа-носителя и 0,2/d см/с для других газов. Однако при таких
скоростях продолжительность разделения очень велика, поэтому на прак¬
тике применяют скорости в 10-20 раз большие.286
flJABA_6 Капиялярная хротатоггхгфия6.3. Размер пробы для капиллярных колонок.Как указывалось ранее, проба, введенная в хроматографическую ко¬
лонку, не должна сильно превосходить емкость теоретической тарелки,
иначе будет наблюдаться перегрузка колонки, и эффективность ее будет
падать.Для оценки максимальной пробы которая еще не вызывает пере¬
грузки колонки, Кейлеманс предложил следующее уравнение:q^«0,02(V+KVjA/N (6.10)где V и - объем газовой и жидюй фаз в объеме одной тарелки; К - ш-
эффициент распределения; N - число теоретических тарелок шлонки.Экспериментальными исследованиями установлено, что соотноше¬
ние (6.10) неприменимо для капиллярных юлонок. Приближенный расчет
показывает, что в капиллярные колонки нежелательно вводить пробы бо¬
лее МО ’ г, иначе шлонка %дет перегружена, и эффективности ее сильно
упадет (в частности, для н-гептана на сквалане при 25°С на капиллярной
юлонке диаметром 0,2 мм) [5]. В связи с этим фактом на раннем этапе
капиллярная хроматография не могла применятся для анализа примесей,
пока не были разработаны методы дозирования больших проб в частности
с криофокусировюй, с программированием температуры испарителя, с от-
дувюй растворителя и другие.6.4. Особенности определения числа теоретических тарелок для
капиллярных колонок.Формула, по которой определяется число теоретических тарелок для
наполненных колонок, неприменима для капиллярных колонок, так как
приводит к получению завышенных результатов. Это связано с тем, что в
формуле для насадочных колонок используют неисправленное время удер¬
живания. Для насадочных колонок время удерживания несорбируемого
компонента в большинстве случаев очень мало и гораздо меньше общего
исправленного времени удерживания. В случае же капиллярных шлонок
время удерживания несорбируемого компонента довольно значительно
(порядка 10 мин. для шлонок длиной 50 м) и в некоторых случаях сопо¬
ставимо с неисправленным временем удерживания юмпонента. В связи с
этим Парнелл предложил определять число теоретических тарелок следу¬
ющим уравнением [28]:(6.11)где t'jj - исправленное время удерживания; ^ - ширина пика на по¬
ловине высоты.287
Яшин ЯМ, Яшин Е.Я., Яшин А.Я. Газовая .хроматографииПо этому уравнению для слабосорбируемых веществ определяемое
число теоретических тарелок будет меньшим, и оно хорошо коррелируется
с числом теоретических тарелок для наполненных колонок.6.5. Материал капиллярных колонок.Колонки в капиллярной хроматографии изготавливают из стеклянных,
медных, алюминиевых, никелевых, кварцевых, стальных и полимерных
капилляров [4-10] (табл. 6.4). Стеклянные капилляры можно легко полу¬
чить на установке, предложенной Дэсти [2]. На стеклянные капиллярные
колонки хорошо наносятся неполярные фазы и некоторые силиконовые
масла. Многие жидкие фазы плохо наносятся на внутреннюю поверхность
из-за плохой смачиваемости. В общем случае бьшо замечено, что стеклян¬
ные капиллярные колонки с жидкой фазой менее долговечны, чем метал¬
лические, особенно при высоких температурах. Чтобы исключить это не¬
желательное явление, предложено на поверхность стеклянных капилляров
откладывать тонкодисперсные силанизированные частицы аэросила [8,9].
В этих условиях жидкая фаза лучше смачивает поверхность и при высоких
температурах не собирается в капли.Таблица 6.4Материал для изготовления капиллярных колонок
в газовой хроматографии.МатериалХарактеристика поверхностиСсылки1. МеталлыМедь, латунь
Алюминий
Нержавеющая сталь,
никельЗолото, сереброПоверхность активная, может окислиться
Поверхность активная,Аусгенитовая хром-никелевая сталь
Инертная поверхность
Инертная поверхность84,20529,30302. СтеклоИнертная поверхность; для улучшения
смачивания поверхность подвергают
травлению кислотами, щелочами,
химическому модифицированию3. КварцИнертная поверхность34. ПолимерыПолиамиды (нейлон,
перлон, дедерон)
ФторопластыИнертная поверхность, плохое смачивание31,3233,345. УльтиметаллКапилляры из нержавеющей стали,
внутренняя поверхность их остеклованаНа алюминиевых капиллярах происходит адсорбция на присутствую¬
щих всегда поверхностных окислах.288
[ТЫВА 6 Капиллярная хроматографияНа медных капиллярах можно получить всего лишь удовлетворитель¬
ные результаты, во-первых, из-за значительной адсорбции на стенках (как
и на алюминиевых капиллярах), во-вторых, из-за невозможности исполь¬
зования их при высоких температурах, так как в этом случае существует
вероятность разложения как жидких фаз, так и анализируемых проб.Наиболее часто используют капилляры из нержавеющей стали. Они лег¬
ко покрываются многими жидкими фазами, поверхность их относительно
инертна. Специальные добавки поверхностно-активных веществ к жидким
фазам необходимы только для разделения сильнополярных веществ.В последние годы, как указывалось ранее, в основном применяют
кварцевые капиллярные колонки. Наметилась новая тенденция - примене¬
ние стальных капиллярных колонок, на внутреннюю поверхность которых
отложен слой кварца.6.6. Приготовление капиллярных колонок.Приготовление эффективных капиллярных колонок - сравнительно
сложный и тонкий процесс. Подробные сведения по методикам приго¬
товления капиллярных колонок содержатся в монографиях [4, 5]. В этом
разделе приведены лишь самые необходимые краткие сведения практиче¬
ского характера об особенностях методов заполнения, модифицирования и
эксплуатации капиллярных колонок.Нанесение жидких фаз на стенки капиллярных колонок. Перед нане¬
сением жидкой фазы необходима предварительная промывка капиллярных
трубок органическими растворителями. Наиболее часто используемые
растворители: метиленхлорид, хлороформ, ацетон, метанол, гексан, диэ¬
тиловый эфир. Предложены два способа нанесения жидких фаз: динами¬
ческий и статический.При использовании динамического метода раствор с жидкой фазой
пропускается через капиллярную трубку под давлением сухого газа-но¬
сителя. Толщина получаемых пленок жидких фаз будет зависеть от сма¬
чиваемости, концентрации жидких фаз, скорости пропускания раствора.
Концентрации используемых растворов жидких фаз колеблются от 5 до
30%. Чтобы исключить попадание пылинок и мелких механических ча¬
стичек, раствор необходимо предварительно отфильтровать. На практике
применяют два варианта динамического метода, различающиеся объемом
пропускаемого раствора жидкой фазы, который может быть больше или
меньше объема капиллярной колонки. Последний вариант известен как
«метод поршня» [4].В обоих вариантах динамического способа нанесения жидких фаз
очень важно сохранять постоянной линейную скорость пропускания рас¬
твора через капилляр, избегать внезапных изменений скорости потока289^ Э г азовая хроьштография
Яши як. Ящн ЕЯ. Яшин А Я. Гюошхршапш1тФтпосле того, как раствор полностью выходит из шлонки. Для этого необ¬
ходимо к шнцу капиллярной шлонки подсоединить капилляр из стекла
или полимера и следить за выходом последних порций раствора. Линейная
сшрость прохождения раствора должна быть меньше 20 см/с. Динамиче¬
ский способ не требует специального оборудования и практически легко
реализуем для капиллярных шлонок, свернутых в спирали. Единственный
недостаток - трудность определения шличества жидшй фазы в юлонке.При статичесшм способе заполнения можно легш определить шли¬
чество жидшй фазы в юлонке. Этот способ заклютается в следующем:
капиллярная юлонка заполняется раствором жидкой фазы известной юн-
центращ1и, затем заполненная юлонка отключается от источника давле¬
ния, и растворитель из нее удаляется путем последовательного нагревания
отдельных участюв колонки. Недостаток этого способа связан с необходи¬
мостью выпрямления свернутых в спирали капиллярных колонок.Подготовка наполненной колонки к работе. После нанесения жидшй
фазы необходимо удалить растворитель, для чего колонку продувают пото-
шм газа-носителя в течение несюльких часов при шмнатной температуре.
Затем колонку помещают в термостат хроматографа, температуру которого
медленно (1 - 2°С/мин) поднимают до температуры на 25°С ниже так на¬
зываемой рекомендуемой максимально допустимой рабочей температуры
жидюй фазы. Колонку продувают при этих условиях несюлькЬ часов и
потом медленно охлаждают до комнатной температуры.Хранение колонок. При хранении юнцы капиллярной колонки должны
быть герметично закрыты. Как правило, разделительные свойства юлонки
сохраняются, за исключением случаев перекристаллизации и каталитиче-
сшго разложения некоторых жидких фаз.6.7. Модифицирование внутренней поверхности исходных капил¬
лярных трубок.Для исключения адсорбции на внутренней поверхности капиллярных
трубок в том случае, шгда адсорбция нежелательна и приводит к несимме¬
тричным пикам, используют химическое, адсорбционное и механическое
модифицирование.Впервые химичесюе модифицирование стеклянных капилляров три¬
метилхлорсиланом проведено А.В.Киселевым с сотр. [35].Для адсорбционного модифицирования используют небольшие добав¬
ки к жидшй фазе поверхностно-активных веществ, шторые блокируют
наиболее активные адсорбционные центры.Механическое модифицирование проводят путем отложения на стен¬
ках некоторых солей и твердых тел с меньшей адсорбционной способнос¬
тью, например, серебра или бихромата натрия, слоя углерода и др.290
ШАВА 6 Ктишюная хроматография6.8. Влияние толщины пленки жидкой фазы.Один из путей повышения разделительной способности капиллярных
колонок состоит в выборе оптимальной толщины пленки жидшй непод¬
вижной фазы, наносимой на внутреннюю поверхность катишяра. В связи
с этим нами впервые было изучено влияние толщины пленки жидшй фазы
на селективность, эффективность и степень разделения [36-38]. Было по¬
казано, что при толщине пленки сквалана, равной 0,2 мкм, разделение т- и
п-»силолов на капиллярной шлонке не наблюдается, тогда как при толщи¬
не пленки 1,7 мкм достигается полное разделение этих изомеров.Оптимальная толщина пленки жидшй фазы, обеспечивающая доста¬
точно высокую эффеютвность разделения, оп[№деляется шнкретно в за¬
висимости от аналитичесшй задачи и может меняться в широких преде¬
лах, но применение толстых пленок позволяет значительно увеличить раз¬
делительную способность капиллярных шлонок.В работе [38] подробно исследовано влияние шличества жидшй
фазы - сквалана - на эффективность колонок N, число разделения и сте¬
пень разделения R ш- и п-ксилолов на шлонке с размерами 50 м х 0,3 мм,
изготовленной из нержавеющей стали, при 100°С (сшрость газа-носите¬
ля азота - 8 cmVmhh).Известно, что степень разделения в хроматографии определяется со¬
отношением;^ ^Сгде; а - отношение исправленных времен удерживания соседних хро¬
матографических пишв;К^- шэффициент емшсти (отношение исправленного времени удер¬
живания к времени выхода неадсорбирующегося шмпонента); в формуле
относится ш второму более сильно удерживаемому шмпоненту;N - число теоретических тарелок (эффективность шлонки).В насадочных шлонках время выхода неадсорбированного шмпонен¬
та небольшое, поэтому обычно К^> 10, при этих условияхТ+Х~*‘и, следовательно, им можно пренебречь. В капиллярных шлонках
время выхода неадсорбирующегося шмпонента большое за счет большой
длины шлонки, поэтому шэффициенты емшсти для быстровыходящих
слабосорбирующихся шмпонентов невелики. При К^< Ю величина вы¬
шеприведенное соотношение заметно влияет на степень разделения R. Ко¬
эффициенты емшсти изменяются с изменением толщины пленки жидшй291
Яшт ЯК. Яшт ЕЖ. Яшт А.Я.Газовая хроматографиифазы, поэтому с уменьшением толщины пленки жидкой фазы в капилляр¬
ных колонках степень разделения для соединений с К^< 10 будет снижать¬
ся в соответствии с соотношением 6.12. Селективность жидкой фазы, т.е.
величина а, не должна изменяться с изменением количества жидкой фазы,
если поверхность стенок не обладает сильной адсорбционной способнос¬
тью. Если же адсорбционные эффекты проявляются, то возможны только
небольшие изменения величины а.В табл. 6.5 обобщены основные результаты исследования [38], пока¬
зывающие влияние толщины пленки жидкой фазы на эффективность по
октану, коэффициент емкости для октана, число разделения между окта¬
ном и гептаном и селективность разделения между трудноразделяемыми
изомерами м- и п-ксилолами. Из данных таблицы 6.5 видно, что наиболь¬
шая эффективность наблюдается при толщине жидкой пленки 0,8 мкм.Таблица 6.5.Влияние толщины пленки жидкой фазы (сквалана) на
эффективность (N и Н), коэффициент емкости (К^,
число разделения между октаном и гептаном и
селективность (а) разделения между ш- и п-ксилолами.Толщина
жидкой
пленки, мкмNпо октануН,мм
по октануКпо октануКа1+К,2,55119 0000,427,90,88401,201,52125 0000,403,60,78381,151,10135 0000,372,50,71340,800,80156 0000,321,80,64310,800,2672 0000,700,70,4114-При работе с более толстыми пленками эффективность уменьшается,
так как увеличивается размывание хроматографической зоны из-за замед¬
ления массообмена, при более тонких жидких пленках усиливается роль ад¬
сорбционных эффектов. Коэффициент емкости К^, число разделения и сте¬
пень разделения возрастают с увеличением толщины пленки. Соотношение. К1 +Кевозрастает в два раза при переходе от толщины пленки 0,26 мкм к тол¬
щине 2,55 мкм (рис. 6.1).Таким образом, для реализации наибольшей разделительной способ¬
ности капиллярных колонок необходимо использовать пленки жидкой фазы
толщиной более 2 мкм. Однако при этом возрастет продолжительность
разделения. На практике для достижения полного разделения за опреде¬
ленный промежуток времени выбирают оптимальную толщину пленки.292
ГШЛВА 6Капиллярная хроматографияИзвестно, что с увеличением количества жидкой фазы (с увеличением
доли сорбента в единице обьема шлонки) увеличивается селективность
хроматографических шлонок. Вместе с тем при увеличении толщины жид¬
кой пленки замедляется массообмен, увеличивается размывание, умень¬
шается эффективность разделения. Следовательно, существует оптималь¬
ная толщина пленки, при которой реализуется наибольшая разделительная
способность. На рис. 6.1 представлена зависимость селективности, эф¬
фективности и степени разделения колонки от толщины жидкой пленки
сквалана. На колонках с толщиной пленки менее 0,5 мкм эффективность
меньше. По-видимому, это связано с тем, что жидкая фаза в этом случае не
полностью покрывает внутреннюю поверхность капилляра, в результате
чего имеет место адсорбция на стенках.г,О й,мнмN10 'Рис. 6.1. Зависимость селективности (а), эффективности (б) и сте¬
пени разделения (в) от толщины жидкой тент (сквалана)На шлонках с толщиной пленки 0,5 - 1 мкм достигается наибольшая
эффективность. При дальнейшем увеличении толщины пленки сквалана эф¬
фекгивность падает, так как замедляется массообмен и увеличивается размы-
ваш1е. Селективность хроматографических шлонок непрерывно возрастает с
увеличением шличества жидшй фазы, а степень разделения с увеличением
жидшй фазы возрастает почти вдвое. При толщине пленки сквалана меньше
1 мкм полного разделения (К, < 1) изомеров ксилола не наблюдается. Так, на
квдонке с 5% раствора сквалана (толщина пленки 0,2 мкм) м- и и-ксилолы не
разделяются, а на шлонке с 30% сквалана (толщина пленки 1,7 мкм) дости¬
гается полное разделение этих компонентов. На колонке с небольшим ш-293
Яит! Я.И., Яшин ЕЖ, Яшин А.Я.Газовт 1тшшотФмличеством жидшй фазы (толщина пленки 0,2 мкм) времена удерживания
ксилолов значительно меньше, поэтому можно предположить, что при бо¬
лее низких темперагурах, шгда времена удерживания ксилолов увеличи¬
ваются так, что становятся сопоставимыми с временами удерживания на
шлонке с большим шличеством жидшй фазы (толщина пленки 1,7 мкм),
селективность рюделения возрастает настольш, что также можно полу¬
чить полное разделение.На рис. 6.2 пошана хроматограмма разделения изомеров ксилола при
пониженной температуре на шлонке с небольшим шличеством сквалана.
Из рисунка видно, что это разделение значительно хуже разделения на ш-
лоике с большим количеством фазы.f7/6Аf29ZJi'rРис. 6.2. Фрагменты хроматограмм бензиновой фракции.Колонка 50 мх 0,3 мм из нержавеющей стали; 100°С; жид¬
кая фаза - сквалан; расход газа-носителя (азот) 8 мл/мин.
Толщина пленки сквалана, мкм: а-0,8; б - 1,5; в-2,55294
Щ^ВЛ_6 Каттлярная хроматографияЭти результаты показывают, что разделительная способность капил¬
лярных колонок в сильной степени зависит от толщины пленки жидкой
фазы. При небольших количествах жидкой фазы полное разделение не до¬
стигается. С увеличением количества жидкой фазы в колонке достигается
полное разделение. Для достижения наилучшей разделительной способ¬
ности при минимальном времени разделения необходимо выбрать опти¬
мальную толщину жидкой фазы.6.9 Поликапиллярные колонкиПоликапиллярные шлонки - новый класс хроматографических шло¬
нок, по свойствам промежуточный между капиллярньми и насадочными
шлонками. Полшшпиллярные шлонки представляют собой стеклянные
стержни, внутри которых имеются ошло тысячи одинашвых капилляров с
внутренним диаметром 40 мкм. Размывание в капиллярных шлонках зна¬
чительно меньше, чем в насадочных, однаш формируемая проба при этом
должна быть небольшой (обычно менее МО ’ г). В поликапиллярные ш-
яонки можно дозировать пробу по крайней мере в 1000 раз большую, поч¬
ти как в насадочные колонки. Необходимо учесть, что в поликапиллярных
юлонках используются капилляры с диаметром в 6 - 8 раз меньшим, чем в
обычных, чаще всего используемых капиллярных колонках (0,25; 0,32 мм).
Известно, что эффективность капиллярных шлонок пропорциональна ди¬
аметру используемого капилляра. Коммерчески доступные поликапилляр¬
ные колонки имеют общую эффективность 10000 теоретических тарелок.Первые поликапиллярные шлонки были разработаны под рушводством
В.П.Солдатова в 80-е годы [39-41]. Идею создания поликапиллярной шлон¬
ки по крайней мере дважды высказывал М.Голэй [42,43], однаш ни ему, ни
другим специалистам реализовать эту идею не удалось. До сих пор полика¬
пиллярные колонки серийно производятся тольш в гНовосибирске. С 1996г.
фирма “Alltech”, США рекламирует и продает поликапиллярные шлонки,
не ссылаясь на то, что она закупает их в г. Новосибирске.В настоящее время спиральные поликапиллярные шлонки длиной до1 м с иммобилизованными жидкими фазами (SE-30, SE-54, Carbowax-20M,
0V-61 и др.) продает фирма ООО «Сибертех» (г. Новосибирск).Прямые поликапиллярные шлонки длиной 20 - 100 см с внутренним
диаметром 2 - 5 мм с числом капилляров 1200 шт. с диаметром 20 - 40 мкм
выпускает Институт катализа им. Г.К.Борескова СО РАН.Прямые шлонки имеют эффективность до 20000 теоретических таре¬
лок на метр.Основные преимущества поликапиллярных колонок:- сокращение времени анализа в 5 - 50 раз;- относительно высокая эффективность порядка 10000 - 20000 теоре¬
тических тарелок;295
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я. Гаювая хроматография- сохранение высокой эффективности в очень широком диапазоне рас¬
хода газа-носителя, что позволяет использовать высокие скорости, допол¬
нительно сокращая время анализа;- значительное повышение чувствительности анализа за счет умень¬
шения размывания;- возможность дозировать большие пробы по сравнению с классиче¬
скими капиллярными колонками; возможность анализировать термически
лабильные соединения за счет снижения температуры разделения.Поликапиллярные колонки рекомендуются для разделения органиче¬
ских соединений с температурами кипения до 400°С.Эти колонки можно применять в любых газовых хроматографах, в кото¬
рых есть капиллярный режим. При небольшой модернизащш дозирующих и
соединительных устройств поликапиллярные колонки можно использовать
и в газовых хроматографах, работающих с насадочными колонкши.В последующих работах [44-50] технология изготовления полика-
пиллярных колонок постоянно совершенствовалась, в частности, были
отработаны способы нанесения и иммобилизатщи пленок наиболее рас¬
пространенных жидких фаз. Разработаны методы приготовления полика-
пиллярных колонок со слоем адсорбента для газо-адсорбционной хромато¬
графии [50]. На таких колонках проведено экспрессное разделение легких
углеводородов С, - в течение 5-20 сек. с эффективностью около 10000
теоретических тарелок на метр. В кандидатской диссертации Ю.В.Патру-
шева (г. Новосибирск, 2005 г.) разработан способ синтеза пористого слоя
адсорбента непосредственно на внутренних стенках поликапиллярных ко¬
лонок с применением золь-гель химии. Для этих целей применены гибрид¬
ные органно-неорганические золи на основе алшксидов алюминия.В работе [46] поликапиллярные колонки небольшой длины успеш¬
но были применены для экспрессного определения металлорганических
вредных соединений олова, свинца и ртути с элементоселективным детек¬
тированием. Время анализа 30 - 100 сек.В следующей работе [47] подобные исследования были продолжены
на поликапиллярных колонках длиной 20 см и 5 см с детектированием МС
с индуктивносвязанной плазмой. Разделение металлорганических соеди¬
нений происходило за 30 сек. с пределами определения 110 *’ г для Hg,
510 ‘‘' г для Sn и З Ю *"' г для РЬ.6.10 Колонки PoraPLOTКолонки PoraPlot впервые предложены Голеем. В настоящее время эти
колонки получили широкое применение для разделения постоянных газов,
легких углеводородов и летучих растворителей.296
ftlABA 6 Капилляуная хроматографияКоммерческие адсорбционные капиллярные колонки впервые ввела
фирма “Chrompack” (Голландия) в 1978 г. под названием PLOT (Porous
Layer Open Tubular). По сравнению с насадочными адсорбционными ко¬
лонки PLOT обеспечивают большую разрешающую способность за более
короткое время.В последние годы фирма “Chrompack” (Varian) разработала новую
серию колонок PLOT с привитыми адсорбционными слоями, отличаю¬
щихся большой стабильностью. Прививка пористого слоя происходит
непосредственно в юлонке (“in-situ”) поэтому такие колонки отличаются
механической и температурной стабильностью. Ранее частицы пористого
слоя просто закрепляли на внутренней поверхности капиллярной колонки.
Новые колонки CP-PoraBonD Q и CP-PoraBonD U имеют максимальную
рабочую температуру 320°С и 300°С соответственно. Фоновый тон (ле¬
тучесть фазы) уменьшился при этом в 5 раз, что позволяет повысить чув¬
ствительность анализа, а также продлить время жизни колонки. В таких
колонках (в отличие от старых) нет потери микрочастиц и, следовательно,
не надо устанавливать ловушку для них. На колонках с привитым слоем
можно использовать большие линейные скорости газа-носителя и режим
программирования расхода (давления).Лучшие возможности эти колонки имеют в промышленных и пор¬
тативных газовых хроматографах, в газовых хроматографах, требующих
применения переключающих кранов и при соединении с масспектроме-
трами [51-57].В качестве материалов колонок PoraPLOT различной длины и диа¬
метра используются кварц и металл (Ultimetal). На рынке доступны ко¬
лонки длиной 10, 15, 25, 30, 50, 60 м с внутренним диаметром 0,25,0,32 и 0,53 мм. Толщина адсорбционного слоя 3, 4, 5, 6, 8, 10, 20 мкм
в зависимости от типа адсорбента. Каждая колонка тестируется, и все
производство сертифицировано по ISO 9001.Наиболее широкое применение колонки находят в химической, не¬
фтехимической, фармацевтической отраслях промышленности, а также
для контроля загрязнений окружающей среды. В таблице 6.6 приведен
перечень таких колонок, их технические характеристики и области при¬
менения.297
Яшин Я.И., Яшин Е.Я.. Яшин А.Я. Газовая хроматографияТаблица 6.6Типы и характеристики капиллярных адсорбционных колонок
PoraPLOT фирмы “Chrompack” (Varian)№№ппТип колонкиПрирода
адсорбционного слояМДРТ,°СОбласти примененияPoraBond QСтироп-дивинлбензол
(ст-ДВБ) 320Газы, угаеводороды, полярные
растворители, спирты, водаPoraBond иСТ-ДВБ с полярными
группами 300Галоидосоединения, полярные
летучие, водные пробы PoraPLOT SДивинлбензол-винил-
пиридин 250Галоидосоединения, угаеводороды
кетоны, эфиры PoraPLOTQ
PoraPLOTQ-HTСТ-ДВБСТ-ДВБ250290Сернистые газы, вода в fasax,
этан, этилен, спирты, альдегиды,
нитрилы, нитросоеяиненияРогаРШТиДВБ-этилен-гликоль-
диметакрилы 190Спирты, альдегиды, сернистые
газы, нитрилы, нитросоединенияLowoxТехнология
многослойного
покрытия 350Оксигенаты С -С.. в углеводородахРогаРШТAminesМодифицированныйст-ДВБ220Аммиак, С|-С^ аминыSilica PLOTSiO,225Угаеводороды, даеиы, сернистые
газы, CFCLА1,0,/КС1А1Д200Угаеводороды С,-С^, примеси на
уровне ppm 10.AljO,/NajSO^А1,0з200ИзомерыИ,Мо1 Sieve 5ААлюмосиликат350Газы ^ Не, Не, Аг, О,, N,, СН^, СО,
С,Н^, изотопы водорода 12.CarboBondУгаерод300Примеси в этилене, СО, СО^, С^-С^.
газы коксовых печей13.СагЬоРШТР?Углерод300Не, Nj. О3, СО, COj, C,-Cj, газы
коксовых печей, разделяет С„ и N,Особенно следует обратить внимание на уникальную юлонку ForaPLOT
Amines для разделения и анализа следов аминов, примесей в диметиламине.
Новой высокотемпературной колонкой является колонка CP-Lowox для раз¬
деления оксигенатов С,-С,д в бензинах на уровне ppm - ррЬ.Спирты определяются от метанола до бутанола.Колонка CarboBond позволяет одновременно определять СО, СО^ и
углеводороды С^, С^, примесь ацетилена в этилене на уровне ppm.Колонка CarboBond предназначена для разделения одновременно О^ и
Nj, СО, СО2 и углеводородов С^ и С,.298
пимл Каттлярная хроматогтфтКроме фирмы “Chrompack” колонки РогаРШТ позднее стали выпу¬
скать фирмы Supelco (таблица 6.7), Restek (таблица 6.8), J & W, HP, Alltech
(табл. 6.9), Quadrex. Некоторые колонки разных фирм близки по раздели¬
тельным способностям.На рис. 6.3, 6.4, 6.5 приведены примеры селективного разделения по¬
стоянных и углеводородных газов.I11 LРис. 6.3. Хроматограмма: 1 - гелий; 2 - кислород; 3 - азот; 4 - ме¬
тан; 5 - диоксид углерода.Кварцевая колонка 25 м х 0,53 мм со слоем угаерода (CarboPlot); тошщ-
на слоя 25 мкм; газ-носите® - водород; 40°С; детектор - микрокатарометр.ШтанА1ШШЮЯ•ЭгаяевГ^хиишГ^пмяеяЭган12 мин.Рис. 6.4. Хроматограмма углеводородных газов (указаны на рисунке)
на колонке 50 мх 0,53 мм; СР - CarboBond; толщина слоя10 мкм; детектор ПИД; программирование температуры
от 35°С до 175°С со скоростью 10°С/мин.299
Яшин ЯЖ. Яшин Е.Я.. Яшин АЯ.Газовом хроматография1 Метая
2Этаи3 Этен4 Пропан5 Пропен6 Ацителен7 Изобутан8 Бутан
9т-2-Бутен
10 ЬБутен
И Ц'2 Бутеи
12 Изсшентан
ВПентан14 Пронин15 1,3-БутадиенLJLLljЯJLРис. 6.5. Хроматограмма углеводородов С,- на ктонке 30 мх 0,53 мм
ValcoPlot VP-Alumina/KCl, толщина слоя 10 мкм. Газ-носи-
тель - водород; температура колонки 80°С; детектор ПИД.Таблица 6.7Типы и характеристики капиллярных адсорбционных колонок
PoraPLOT фирмы Supelco№ХаппТип колонкиПриродаадсорбционногослояМДРТ,°СОбласти применения1.Alumina Sulfate PLOTAlA180Постоянны и легкие
углеводородные газы С,-С^2.Alumina Chloride PLOTА1Д180Угаеводороды С,-С,, фреоны3.Carboxen-1006 PLOTУглерод
S ~ 750 uVt250Постоянные и углеводородные
газы C,-Cj, смесь формальдегид-
вода-метанол4.Carboxen-1010 PLOTУглерод250Hj,N,,CO,CH,,CO,hC,, с,.Разделяет Oj и Nj5,Mol Sieve 5A PLOTАлюмосиликат300Oj, Nj, СН^ и СО за 5 мин.6.Supel-Q PLOTСТ-ДВБ250COj, С,-С,, сернистые газы,
спирты, кетоны, альдегиды7.Carbopack X PLOTУглерод250Полярные и неполярные
соединения300
ГЛАВА 6 Капиллярная хтшатографтТаблица 6.8Типы и хара1стеристики колонок PoraPLOT фирмы Restek№№ппТип колонкиПрирода
адсорбционного слояМДРТ,“СОбласти применения1.RT-AluminaA1,0,200Углеводороды С,-С,, примеси в
этилене, пропилене, бутене2.RT-M sieve 5ААлюмосиликат350Постоянные и благородные газы.
Разделение Аг и 0,3.RT-M sieve 13 XАлюмосиликат350H,,0j,N,,CH,,C04.RT-Q PLOTСт-ДВБ250Полярные растворители, спирты,
углеводороды5.RT-S PLOTДВБ-винилпиридин250Полярные соединения, кетоны,
эфиры; легкие газы в этилене,
пропилене6,RT-U PLOTДВБ-этиленгаикольдиметилакрилат190Спирты, альдегиды, нитрилы,
нитросоединения и другиеТаблица 6.9Типы и характеристики колонок PoraPlot№ФирменныеназванияСостав сорбционного
слояТолщина(мкм.)Внутр.
диаметр (мм.)Длина(м.)AUtech1At-Aluminaоксид алюминия.0,53302At-Mole Sieveцеолит 5А-0,5330Hewlett-Packart3HP-A1203;PL0T,KC1оксид алюминия, модиф.солями5-250,25;0,32;10-504«8» «М» HP-Molesieveцеолит 5 А10-500,53 0,32;0,5315,305A5HP-PLOTQпористый полимер наоснове ДВБ10-500,32;0,5315-30J. & .W. ScienMc6OS-Almnina, GS-оксид алюминия, модиф.Alumina/KClсолями-0,32;0,5315-307GS-Molesieveцеолит 5А-0,53308GSrQпористый полимер наоснове ДВБ-0,32;0,53039GS-CarbonPLOTуглерод1,5-30,32;0,45;15-600,5310GS-GasPro--0,5315;30301
Яшин ЯМ. Яшин ЕЖ. Яшин АЖ. Газовая хроматография6.11 Циркуляционная капиллярная хроматографияВ работах [58-62] предложен вариант проявительной циркуляционной
газовой хроматографии с высокой разрешающей способностью на капил¬
лярных колонках. Эти авторы ранее показали, что используя циркуляци¬
онный метод, можно на насадочных колонках достигнуть эффективности
до 120000 теоретических тарелок. На капиллярных колонках достигнута
эффективность до 20 миллионов теоретических тарелок (это соответству¬
ет общей длине капиллярной колонки 4200 м). На таких системах можно
разделять не только изомеры, но и изотопозамещенные органические со¬
единения с юэффициентом разделения а = 1,005 - 1,075. Эти исследования
были реализованы в циркуляционном капиллярном газовом хроматофафе
«Биохром-27» (СКБ ИОХ АН СССР).Авторами этих работ было показано, что в ряде случаев этот вариант
газовой хроматофафии не имеет альтернативы для реализации разделения
особо сложных смесей. В работах были выбраны оптимальные условия и
режимы. Достигнуто полное разделение (с критерием разделения R > 1) изо¬
топозамещенных бензолов и циклогексанов, масса молекул которых отлича¬
ется на одну единицу.Кроме практической реализации авторы способа [60, 61] предложили
математическую модель проявительной циркуляционной газовой хромато¬
фафии, в которой впервые выявлено и учтено влияние межфазного распре¬
деления элюируемых веществ на эффект импульсно-гидродинамического
расширения хроматофафических полос при осуществлении циркуляции.Предложены три схемы установок для реализации циркуляционной
хроматофафии: с атмосферным давлением на выходе, с повышенным дав¬
лением на выходе и схема, обеспечивающая «безимпульсный режим».6.12. Монолитные капиллярные колонки.В монолитных капиллярных колонках адсорбционный слой создается
непосредственно внутри капилляра. Монолитные капиллярные колонки,
по существу - наполненные колонки.Попытки создания монолитных капиллярных колонок предпринима¬
лись в 1960-1970 гг., однако из-за неудовлетворительных результатов (низ¬
кая эффективность, невоспроизводимость) работы были прекращены.Сравнительно недавно в работах [63-69] было показано, что можно
получить удовлетворительные колонки на основе полидивинилбензола для
режима газо-адсорбционной хроматофафии.Обширные исследования способов получения монолитных колонок на
основе силикагеля и органических полимеров для газовой хроматофафии
проведены в серии работ [63-69].302
IJJAR4 6 Катшлярмая хроматографияБыли исследованы следующие параметры монолитных колонок: эф¬
фективность, емкость, проницаемость и пористость.В частности, было показано, что проницаемость монолитных шлонок
в 1000 раз ниже, чем проницаемость незаполненных капилляров. С другой
стороны, проницаемость монолитных шлонок в 10-100 раз выше, чем про¬
ницаемость наполненных капиллярных шлонок с размерами зерен сорбен¬
та, равными размерам доменов монолита [68].Важной характеристишй хроматографичесшй шлонки являются ее
нагрузочные характеристики, т.е. возможная величина максимальнодози-
руемых проб. Нагрузочные характеристики зависят от шличества сорбен¬
та (жидшй фазы или адсорбента) в колонке. Оказалось, что удельная на¬
грузка (емшсть) по анализируемым пробам для монолитных капиллярных
шлонок на порядок выше удельной нагрузки полых капиллярных колонок
при одном и том же внутреннем диаметре [63].Эта характеристика позволила применять монолитные шлонки с не¬
большой длиной (менее 0,5 м).Удельная эффективность монолитных капиллярных колонок выше
удельной эффеюгивности полых капиллярных шлонок. На монолитных
капиллярных шлонках величина ВЭТТ сильно зависит от природы газа-
носителя [67]. Наилучшие результаты достигаются для гелия в качестве
газа-носителя. Для лучших монолитных колонок величина ВЭТТ может
быть ошло 10-15 мкм.За счет низшго значения ВЭТТ и сравнительно большой нагрузочной
емшсти на монолитных капиллярных шлонках небольшой длины можно
получить высокоскоростные и эффективные разделения, возможно разде¬
ление смеси легких углеводородов за 20 сек с производительностью по¬
рядка 900 теоретических тарелок в сек для ^ана.6.13. Основные области применения капиллярной газовой хрома¬
тографии (КГХ)КГХ очень широко используется в аналитичесше практике. Более
70-80% газохроматографических методик выполняются на капиллярных
шлонках [70-72].В работе [73] проведено наукометрическое исследование по частоте
применения капиллярных колонок разной длины, разного внутреннего
диаметра и применяемых жидких фаз. В таблице 6.10 обобщены эти
данные.303
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я.Гтовая храчатографияТаблица 6.10Длинаюлонки,мДоляприменения,%Внутреннийдиаметр,ммДоляприменения,%Тип жидких
фазДоляприменения,%25-30730,22-0,2552полиметил-силоксан21,5< 100,3221полиметил-фенилсилоксан4951-100100,3316<0,2цианопропил-силоксанЧаще всего используется колонка 30 м х 0,25 мм х 0,25 мкм (47%), в
некоторой степени - стандартная колонка.На капиллярных колонках в основном используют режим программи¬
рования температуры (88%) и в меньшей степени - изотермический ре¬
жим (12%). В качестве газа-носителя в основном используют гелий (63%),
затем азот (19%) и водород (12-14%). Водород из-за взрывоопасности не
рекомендуется использовать в качестве газа-носителя.Разделения и анализы на 1шпиллярных шлонках больше всего шлпошшют
при температурах до 300°С (более 90%), по диапазонам: до 100°С - 12,5%, в
пределах 100-200°С - 30%, в пределах 201-300°С - 47%. Свыше 300°С выпол¬
няется всего 10,5% разделений.Основные области применения КГХ - разделение и анализ:- изотопов газов (N14 и N15, Ne20 и N20,016 и 018 и щ).);- дейтерировые соединения;- изомеры цис и транс; изомеры положения (орто-, мета- и пара);- оптические изомеры;- газовые шнденсаты;- бензиновые фракции;- сырая нефть;- воздух загрязненных городов;- примеси в питьевых и природных водах;- ароматы пищевых продуктов;- состав безалкогольных напитков (кофе, чай и щ).);- состав алкогольных напитков (вино, коньяк, водка и др.);- биологические жидкостии многие сложные многошмпонентные смеси.304
ГЛАВА 6 ЛитератураЛитература1. Golay M.J.E. In: Gas Chromatography, Ed. V.J.Coates et al.2. Desty D.H., Horesnope J.N., Whyman B.H.E Anal. Chem. 1960, v. 32,
p. 210.3. Dandeneau R.D. Zerenner E.H., J.High Res. Chromat. 1979, v.2, p.2.4. Kaiser R. Chromatography in Gas Phase II Capillary Gas Chromatog¬
raphy, Mannheim, Bibliographisches Institut 1961, 345 p.5. Ettre L.S. Open Tubular Columns in Gas Chromatography, Plenum
Press, N.Y. 1965,220 p.6. Jennings W. Gas Chromatography with Glass Capillary Columns, 2"“* Ed.
Academic Press, New York 1980.7. Kiselev A.V., Yashin Y.I. Gas Adsoфtion Chromatography, Plenum
Press, New York, 1969.8. Desty D.H. Advances in Chromatography Ed. J.C.Giddings, R.A.Keller,
N.Y. Dekker 1965, p. 199.9. Руценю Б.А. Капиллярная хроматография. Н^тса, Москва 1978,221 с.10. Тесаржик К., Комарек Л. Капиллярные колонки в газовой хрома¬
тофафии. Пер. с чешского, Москва, Мир 1987, 222 с.11. Высокоэффективная газовая хроматофафия. Редактор К.Хайвер.
Пер. с англ. Москва, Мир 1993, 288 с.12. Wampler T.R J. Chromat. 1999, v. 842, p. 207-220.13. Sandra P., Bicchi C. (Eds) Capillary Gas Chromatography in Essential
Oil Analysis, Huthig, Heidelberg, 1987.14. Jennings W.G.,. Nikelly J.G. (Editors) Capillary Cromatography. The
Applications, Huthig, Heidelberg, 1991, 153 p.15. Berezkin V.G. De Zeeuw J. Capillary Gas Adsoфtion Chromatogra¬
phy, Huthig Verlag, Heidelberg 1996, 320 p.16. Grob K. Split and Splitless injection in Capillary GC. Huthig, Oxford
1993,574 р.17. Ettre L.S. Anal. Chem. 1985, v. 57, p. 1419 A.18. Жданов С.П., Киселев AB., Яшин Я.И. Ж физ. химии. 1962, т.36, с. 1118.19. Калмановский В.И., Киселев А.В., Лебедев В.П. Газовая хромато¬
фафия. Москва, Наука, 1964, с. 157.20. Petitiean D.L., Leftault C.J. J. Gas Chromatog., 1963, v.I, p.18-21.21. Schwartz R.D., Brasseaux D.J., Shoemake G.R. Analyt. Chem., 1963,
(v.l,p.l8),v.35, p.496-502.22. Schwartz R.D., Brasseaux D.J., Shoemake G.R. Analyt. Chem., 1963,
(vl,p.l8), v35, p.496-502.23. Halasz I., Horvath C. Nature (London) 193, p.21624. Halasz I., Horvath C. Anal.Chem. 1963, v.36, p.349.25. Purcell J.E. Nature, 1964, v.201, p.l321.26. Kirkland J.J. Anal.Chem. 1963, v.35, p.l285.30520 Гамки хромагографИ!
Яитн Я.И., Яшин Е.Я. Яшин А.Я. Гаювая хроматография27. Кейлеманс А. Хроматография газов. Пер. англ. М. Издатинлит.
1959, 320 с.28. Pumell J.H. J.Chrom. Soc. 1960, p. 1268.29. Desty D.H., Goldup A., Whytman B.H.P. // J. Inst. Petrol. 1959. Vol. 45,
p.257.30. Zlatkis A. // Lectures on gas chromatography, 1962 / Ed. H.A. Szymanski.
N.Y.: Plenum press, 1963, p. 87.31. Scott R.PW. //Nahire. 1959. Vol. 183, p.l753.32. Scott R.P.W., Hazeldean G.S.F.// Gas chromatography, 1960 / Ed.
R.RW. Scott Wash. (D.C.):Butterworths, 1960, p. 144.33. Scott R.P.W. // Progress in industrial gas chromatography / Ed. H.A.
Szymanski. N.Y.: Plenum press, 1961, p. 229.34. Bombick D., Dinunzio J. // Chromatographia. 1981, Vol. 14, №1, p. 19-22.35. Киселев А.В. Вест. Моск. ун-та. Сер. хим., 1961, №5, с.31.36. Князева А.А., Яшин Я.И. Влияние количества жидкой фазы в ка¬
пиллярных колонках на хроматографическое разделение. Газовая хро¬
матография, НИИТЭХИМ, вып. 22, 1974.37. Князева, А.А., Яшин Е.Я., Яшин Я.И. Влияние количества жидкой
фазы в капиллярных колонках на эффективность и степень разделения
компонентов бензиновых фракций. Ж. аналит. хим. 1989, т.44, с. 166.38. Калмановский В.И., Лебедев В.Р., Полякова Л.В., Фикс М.М.,
Яшин Я.И. Применение капиллярной хроматографии в анализе угле¬
водородов. Труды по химии и хим. технологии. 1961, вып.2, с.356.39. Солдатов В.П., Кузнецов В.В., Морозов А.А. и др. Авторское сви¬
детельство № 969089 СССР, Бюллетень изобретений 1981.40. Солдатов В.П., Науменко И.И., Ефименко А.П., Вагонов B.C. Па¬
тент № 1651200, Бюллетень изобретений 1986.41. Солдатов В.П. и др. Авторские свидетельства № 1635129 (1987);
№ 1635128 (1987); № 1415060 (1986); № 1642370 (1986); № 1659838
(1986).42. Golay M.J.E. Chromatographia, 1975, v. 8, p. 421.43. Golay M.J.E. HRC CC 1988, v. 11, № 1.44. Петров B.B., Ефименко А.П., Н^енш И.И. Поликапиллярные хро¬
матографические юлонки. Новосибирск, ООО «Сибертех», 1996,30 с.45. Малахов В.В., Свдельниюв В.Н., Уткин В.А. Доклад АН. 1993, т. 39,
с. 749.46. Lobinski R., Sidehiikov V, Patrushev Y, Rodriguez I., Vasik A. Trends
in analitical chemistry, 1999, v. 18, p. 449.47. Rodriguez I., Mounicon S., Lobinski R., Sidelnikov V, Patrushev Y,
Yamaka M. Anal. Chem., 1999, v. 71, p. 4534.48. Сидельников B.H., Патрушев Ю.В., Пармон В.Н. Патент №
2205400. Приоритет 18 03 2002 г. Бюл. № 15, с. 10.306
ГЛАВА 6 Литература49. Сидельниюв В.Н., Патрушев Ю.В. Рос. хим. журнал. Журнал РХО
им. Д.И.Менделеева, 2003, т. 47, с. 23.50. Березкин В.Г., Сидельников В.Н., Патрушев Ю.В., Хотимский B.C.
Ж. физ. химии, 2004, т. 78, с. 520.51. De Zeeuw J., de Nijs R.C.M., Hemich L.T. J.Chromatog. Sci. 1987,
v.25,p.71.52. Hemich L.T, J.Chromatog. Sci, 1988, v.26, p. 198.53. Ji. Zh., Majors R.E., Guthrie E.J. J.Chromatog. 1999, v.842, p.115-142.54. De Zeeuw J., Wessels J.C.M. Int. Lab. News, 1992, Ш2, p. 18.55. Berezkin V.G. Chem. Rev. 1989, v.89, p.287.56. Березкин В.Г. Успехи химии. 1996, т.65, с.991.57. Л. Z., Mejors R LC-GC 1998, Jf»7, р.620.58. Стерхов Н.В., Равикович В.М., Литвин Е.Ф., Чижков В.П. Ж. физ.
химии 1983, т 57, с. 2897.59. Стерхов Н.В., Литвин Е.Ф., Чижков В.П. Зав. лаб. 1984, № 2, с. 17.60. Стерхов Н.В., Чижков В.П. Ж. аналит. химии 1985, т. 40, с. 865.61. Стерхов Н.В., Чижков В.П. Pto. АН СССР Сф. хим. 1986, Ns 9, с. 2019.62. Chizhkov V.P., Pavlov S.S., Sterkhov N.V., Ravicovich V.M., Litvin
E.F., Varyvonchik E.A. Talanta, 1987, v. 34, p. 227.63. Королев A.A., Попова Т.П., Ширяева В.Е., Козин А.В., Курганов А.А.
Ж.физичесюй химии, х81, ХаЗ (2007) 552-557.64. Королев А.А., Попова Т.П., Ширяева В.Е., Козин А.В., Курганов А.А.
Ж.физичесюй химии, т.81, №3 (2007) 512-520.65. Королев А.А., Попова Т.П., Ширяева В.Е., Ю)зин А.В., Дьячшв И. А.,
Курганов А.А. Высокомоялярные соединения, т.48, №8 (2006) 1373-1382.66. Королев А.А., Попова Т.П., Ширяева В.Е., Курганов А.А.
Ж.физической химии, т.80, №7 (2006) 1290-1296.67. Королев А.А., Попова Т.П., Ширяева В.Е., Курганов А.А.
Ж.физической химии, х80, №4 (2006) 709-715.68. Королев А.А., Попова Т.П., Ширяева В.Е., Курганов А.А.
Ж.физической химии, т.80, №1 (2006) 132-136.69. Королев А.А., Попова Т.П., Ширяева В.Е., Курганов А.А.
Ж.физической химии, т.79, №3 (2005) 543-547.70. Руденю Б.А., Руденш ГИ. Высокоэ4к^кгивные хроматографические
процессы. Т. I. Газовая хроматография. Москва, Наука, 2003,425 с.71. Berezkin V.G., de Zeeuw J. Capillary Gas Adsoфtion Chromatogra¬
phy, Hflthig, Heidelberg, 1996.72. Березкин В.Г. Капиллярная газотвердофазная хроматография.
Успехи химии. 1996, т. 65, с. 991.73. Berezkin V.G., Viktorova E.N. J. Chromatogr. 2003, v. 985, p. 3-10.30720‘
Яшин Я.И.. Яшин Е.Я., Яшин А.Я. Газовая хроматографияГлава 7. МЕТОДЫ РЕАКЦИОННОЙ ГАЗОВОЙ
ХРОМАТОГРАФИИ7.1. ВведениеК методам реакционной газовой хроматографии относятся все мето¬
ды, в которых химический состав компонентов исходной пробы изменяет¬
ся во время прохождения ее через газовую систему хроматографа [1]. Тер¬
мин «реакционная газовая хроматография» введен Дравертом в 1960 г. [2].
Реакционную газовую хроматографию применяют как для аналитических
целей, так и для физико-химических исследований. В последнем случае
реакционную газовую хроматографию используют в основном для изуче¬
ния химических реакций и катализаторов, в частности для исследования
влияния изменения параметров реакции на получаемые продукты, а также
для исследования активности катализатора этой реакции. Такие реакции
обычно проводят на входе в хроматографическую колонку в специальных
микрореакторах. Следует выделить три способа работы: с периодической,
импульсной и непрерывной работой микрореактора [3].7.2 Аналитическая реакционная хроматографияМетодом газовой хроматографии можно разделять и анализировать
только достаточно летучие и термически устойчивые вещества. Поэтому
многие нелетучие и термически неустойчивые соединения, например по¬
лимеры, перекиси, некоторые соли, классическим методом газовой хро¬
матографии анализировать нельзя. Больщие трудности встречаются при
хроматографическом анализе соединений, агрессивных при обычных
условиях. Однако потребность в анализе смесей таких соединений во мно¬
гих производствах очень велика. Чтобы применить метод газовой хромато¬
графии (который зарекомендовал себя как быстрый и эффективный метод
разделения) для анализа нелетучих, неустойчивых и агрессивных веществ,
их с помощью химических реакций переводят в другие, более летучие,
устойчивые и неагрессивные соединения.Аналитическая реакционная газовая хроматография - это вариант га¬
зовой хроматографии, в котором в аналитических целях проводят хими¬
ческие реакции на различных ступенях хроматографического процесса
(от ввода пробы до детектора) [1]. Основная цель проведения химических
реакций - это расширение областей применения аналитической газовой
хроматографии. В частности, реакционная газовая хроматография может
быть использована также для идентификации путем селективного погло¬
щения некоторых соединений, для улучшения разделения в случае наложе¬
ния пиков, для повышения чувствительности анализа. Первые работы по
аналитической реакционной хроматографии были опубликованы в пятиде¬
сятых годах [4-6]. Большой вклад в развитие аналитической реакционной
хроматографии внес В.Г.Березкин [1].308
ГЛАВА 7 Методы реакиионной газовой хроматографииЧаще всего химические превращения проводят на входе в хромато¬
графическую колонку, в некоторых случаях химические реакции проводят
также в самой колонке или на выходе перед детектором. Если проба под¬
вергалась химическим превращениям вне хроматографа, то, по определе¬
нию Драверта, эти варианты нельзя отнести к реакционной хроматогра¬
фии. По-видимому, это определение не совсем правильное, так как конеч¬
ный результат одинаков, независимо от того, проводят ли превращения в
самом хроматографе или вне его. Более того, во многих случаях удобнее и
надежнее проводить химические превращения вне хроматографа. Поэтому
далее будут перечислены основные типы химических превращений, ис¬
пользуемые в настоящее время, с целью расщирения областей применения
газовой хроматографии вне зависимости от места проведения химической
реакции.В первую очередь, следует выделить исключительно широкое при¬
менение химических превращений для анализа биологических смесей,
компоненты которых нелетучи либо термически нестабильны [7-9]. Это,
прежде всего, перевод в устойчивые и термически стабильные соответ¬
ствующие производные аминокислот, жирных кислот С,^ - сахаров,
стероидов. Газохроматографический анализ практически нелетучих ме¬
таллов проводят с помощью производных ацетилацетонатов (хелатов) [10].
Таким путем в настоящее время анализируют более шестидесяти видов
металлов. Неорганические соли, в частности карбонаты, анализируют по
вьщеляющейся двуокиси углерода при обработке их кислотами [1].7.3. Пиролизная газовая хроматографияКак уже отмечалось, методом газовой хроматографии нельзя анализи¬
ровать высокомолекулярные нелетучие вещества, в частности олигомеры,
полимеры, каучуки, смолы и т.д. Для таких веществ испарение можно за¬
менить пиролизом. В последние годы пиролизная хроматография как ме¬
тод изучения высокомолекулярных соединений по продуктам их разложе¬
ния получил широкое распространение [11]. Можно выделить три области
применения пиролизной хроматографии [12-19];1) определение качественного и количественного состава полимеров,
сополимеров и механических примесей гомополимеров;2) определение структурных характеристик полимеров;3) определение термостабильности, кинетики и механизма разложения;4) определение добавок в полимеры, в частности антиоксидантов.При пиролизе полимеров, как правило, образуется сложная смесь ве¬
ществ с различными молекулярными весами, поэтому применение газовой
хроматографии для разделения и исследования продуктов пиролиза осо-309
Яшин Я.И., Яшин ЕЖ. Яитн АЖ. Гтовая хроматографиябенно эффективно. Проблема сводится к установлению эмпирической кор¬
реляции между строением полимера и спеетгром образующихся при пиро¬
лизе продуктов, решить которую можно лишь при строгой стандартизации
условий пиролиза, в том числе и некоторой стандартизации образца.Пиролиз проводят с помощью обычного термического нагрева, высо¬
кочастотного нагрева (до точки Кюри), с применением коронного разряда
и лазерной техники [2, 3]. Устройство для пиролиза изготавливают в виде
приставки к стандартным газовым хроматографам. В настоящее время
многие универсальные хроматографы высокого класса снабжены пироли¬
тическими приставками, которые включают непосредственно в газовую
схему хроматографа вместо узла ввода пробы или же параллельно ему.К конструкции пиролизного устройства предъявляется несколько тре¬
бований:1) точная установка, поддержание и измерение температуры пиролиза
в широком диапазоне;2) предварительный нагрев навески при сравнительно невысокой тем¬
пературе для удаления растворителя и летучих примесей;3) полное удаление остаточных продуктов с подложки после пиролиза;4) удаление смолообразных продуктов, образующихся в результате пи¬
ролиза;5) непрерывный поток газа-носителя через пиролитическую ячейку;6) полное исключение попадания воздуха во время ввода пробы;7) хорошая воспроизводимость анализа.По конструктивному оформлению пиролитические ячейки можно раз¬
делить на три типа.Пиролизные ячейки первого типа (наиболее простые) представ¬
ляют собой нагреваемые электрическим током спирали, внутри которых
проходит пиролиз. Изучаемое вещество наносят непосредственно на спи¬
раль - нихромовую, покрытую золотом, или помещают в лодочку из инерт¬
ных материалов, вставленную внутрь спирали. После введения спирали с
анализируемым веществом в газовый поток и выхода прибора на режим
спираль быстро нагревают. Образовавшиеся продукты пиролиза вместе с
потоком газа-носителя поступают в хроматофафическую колонку, разде¬
ляются и регистрируются детектором.Ячейки этого типа имеют следующие недостатки:1) недостаточно хорошая воспроизводимость температурного режима
за счет образования пленки на спирали и изменения ее сопротивления;2) затруднен точный контроль температуры внутри пиролитической
ячейки;3) затруднено тоспроизюдимое внесение навески образца внутрь спирали.310
ШЛВА 7 Методы реакционной газовой хроматографииЯчейки второго типа - трубчатые реакторы; образец вносят в зону,
в которой постоянно поддерживается заданная температура. В этом слу¬
чае достигается лучшая стандартизация температуры. Однако в ячейках
этого типа имеется большая вероятность прохождения вторичных реакций
за счет увеличения продолжительности пребывания продуктов пиролиза в
горячей зоне по сравнению с ячейками первого типа.В последнее время широкое распространение получили пиролити¬
ческие ячейки третьего типа, представляющие собой ферромагнитный
держатель, который помещен в высокочастотное электромагнитное поле,
принцип действия их заключается в нагреве исследуемого образца до тем¬
пературы пиролиза за счет тепла, выделяемого на держателе. Конечная
температура нагрева держателя, а следовательно, и образца определяется
температурой, при которой ферромагнитный материал теряет свои магнит¬
ные свойства (точка Кюри), так что дальнейший его нагрев прекращается.
Используя различные ферромагнитные материалы, можно изменять тем¬
пературу пиролиза. Температура держателя достигается за 0,2 - 0,3 с.На рис. 7.1 представлено пиролитическое устройство, содержащее
блок пиролиза, блок питания и управления и переключающий кран, раз¬
работанное в Дзержинском филиале НПО «Химавтоматика» [20].Блок пиролиза состоит из пиролитической ячейки трубчатого типа с
кварцевой трубкой I, печи предварительного нагрева 2, устройства ввода
пробы, печи отжига пиролитической лодочки на воздухе 3 и поглотитель¬
ного устройства 4. Все элементы блока пиролиза, кроме поглотительного
патрона, установлены на кронштейне.Пиролитическая ячейка представляет собой печь, внутрь которой вве¬
дена кварцевая трубка 5 с капиллярным выходом. Кварцевая трубка по¬
мещается в металлическую 6, которая необходима для соединения деталей
печи и для выравнивания температурного поля по длине кварцевой трубки.
Для нагрева печи служит нихромовая спираль 7, навитая с переменным
шагом (в центре печи шаг шире), что позволяет создать более равномерное
температурное поле. На спираль надевается фарфоровая трубка 8, затем
цилиндр из керамики 9. Диски Ют стали закрывают печь с двух сторон.
Внутри кварцевой трубки имеется изотермическая зона (для температуры
800°С с точностью ±5°С) длиной 12 мм, в шторой и должна находить¬
ся лодочка с пиролизуемым веществом. Кварцевая трубка с капиллярным
выходом устанавливается в пиролитической печи таким образом, что изо¬
термическая зона, в которой находится лодочка, располагается у начала
капилляра. Использование пиролитической кварцевой трубки с капилляр¬
ным выходом позволяет резко уменьшить время пребывания продуктов
пиролиза в горячей зоне.311
Яшин Я.И., Яшин Е.Я.. Яшин А.Я.Газовая хранатографияРис. 7.1. Пиролизное устройство печного тгта (обозначения в тексте).Температура пиролитической ячейки может устанавливаться в преде¬
лах от 300 до 900°С.Температура предварительного нагрева может быть установлена плав¬
но в интервале от комнатной до 200°С.Нагреватель печи предварительного нагрева конструктивно выполнен
в виде полупроводникового слоя, нанесенного на участок кварцевой труб¬
ки длиной 20 мм.Печь отжига предназначена для очистки кварцевой лодочки от неле¬
тучих продуктов пиролиза путем их сжигания на воздухе. Конструктивно
печь расположена на одном кронштейне с пиролитической ячейкой и вы¬
полнена из металлической внешней 11 и фарфоровой внутренней 12 трубок
в виде щгаиндра, открытого со стороны ввода кварцевой лодочки. Внутри
фарфоровой трубки помещена нихромовая спираль электронагревателя 13.
Спираль легко заменяется.Поглотительный патрон предназначен для задержания тяжелых смоло¬
образных продуктов пиролиза во избежание загрязнения разделительной
колонки и соединительной трубки. Капиллярный выход из кварцевой труб¬
ки входит непосредственно в поглотительный патрон. Патрон выполнен из
нержавеющей стали и легко снимается для очистки. В патрон помещается
особый наполнитель, например, стеклянные шарики. Поглотительный па¬
трон может быть также использован для решения отдельных задач реак¬
ционной газовой хроматографии, например, для селективного поглощения
отдельных продуктов пиролиза.Работа с устройством производится в следующей последовательности.Навеску анализируемого вещества помещают в лодочку, отводят шток
держателя лодочки в крайнее положение так, чтобы лодочка оказалась вну¬
три патрубка устройства ввода пробы, и перемещают подвижную пласти¬312
ГЛАВА 7 Методы реатионной газовой хроматографиину с патрубком в положение ввода лодочки с образцом в кварцевую пиро¬
литическую трубку. При наличии в образце летучих примесей (мономеров,
растворителей) лодочку с образцом сначала вводят в печь предваритель¬
ного нагрева на 10 мин для удаления примесей. Для проведения пиролиза
шток перемешают в крайнее переднее положение, при котором лодочка
находится у капиллярного выхода (примерно в средней части печи). Об¬
разовавшиеся летучие продукты пиролиза в потоке газа-носителя направ¬
ляются через поглотительный патрон в хроматографическую колонку. По
ошэнчании пиролиза лодочку отводят в патрубок устройства ввода пробы
и после передвижения пластины с патрубком в другое крайнее положение
производят отжиг лодочки в печи отжига. После остывания лодочка под¬
готовлена к следующему опыту.Ячейки третьего типа имеют ряд преимуществ перед двумя первыми;
можно точнее поддерживать температуру пиролиза, температура пиролиза до¬
стигается за более юроткое время, наблюдается большая воспроизводимость
анализа и значительно меньшая вероятность шоричных реакций. К недостат-
1ШМ этого метода следует отнести ограниченный интервал температур, так как
для каждой температуры нужен свой ферромагнитный держатель.В порядке возрастания температуры разложения различают; термиче¬
ское разложение, мягкий пиролиз, нормальный (средний) пиролиз и жест¬
кий пиролиз.Степень разложения зависит от температуры и продолжительности пи¬
ролиза. При термическом разложении разрываются только некоторые наи¬
более слабые связи, образующиеся продукты по молекулярным весам не
сильно отличаются от исходного вещества. Температура термического раз¬
ложения 200 - 300°С, иногда 400°С. В этой области работают сравнительно
мало, хотя можно получить ценную информацию о структуре молекул.Мягкий пиролиз проводят при температурах не выше 500°С, чаще
всего его используют для биологических объектов, в частности для амино¬
кислот, крахмала, бактерий и т.д. При пиролизе каждой аминокислоты при
300°С образуется уникальный профиль продуктов.Нормальный (средний) пиролиз проводят при 500 - 800°С, в основ¬
ном для исследования полимеров. Если полимер нерастворим, не имеет
харатстерных функциональных групп, химически инертен, то пиролиз
становится единственным способом идентификации его структуры. При
таком пиролизе образуется богатый спектр продуктов (20 - 50 компонен¬
тов). При большой продолжительности пиролиза в этом случае возможны
вторичные процессы.В некоторых случаях для исследования полимеров применяют пиро¬
лиз в широком диапазоне температур (150 - 850°С).313
Яишн Я.И . Яшин ЕЯ.. Яшин А.Я. Газовая хрвматографияЖесткий пиролиз проводят при 800 - 1100°С. При этом разрывается
большинство связей, даже связь углерод-угаерод. Полимеры разрушаются
на небольшие фрагменты, образуется много продуктов. Поэтому иногда
предпочтительнее для исследования полимеров или сополимеров нор¬
мальный пиролиз.Кроме исследования полимеров, сополимеров и других высокомоле¬
кулярных соединений в химии полимеров пиролизная хроматография на¬
ходит широкое применение в лесохимии, криминалистике и особенно в
биохимии для исследования и идентификации биополимеров.В последние годы области применения пиролизной хроматографии
расширились, в основном, благодаря сочетанию пиролиза с капиллярными
колонками и масс-спектрометром в качестве детектора. Вышел обширный
библиографический обзор (534 ссылки) по пиролизной газовой хромато¬
графии синтетических полимеров [12].Пиролизная капиллярная хроматография широко применяется для ис¬
следования гтриродных полимеров, а также многих объектов, изготавлива¬
емых из полимеров, в частности волокон одежды, красок, лаков, покрытий
мебели, ковров, пищевых контейнеров, природных полимеров, включаю¬
щих целлюлозу (дерево, хлопок, бумагу) и белки (шерсть, шелк, пища).В последних публикациях по исследованию полибутадиена, полиэ¬
тилена, полипропилена, хлорированного полиэтилена использовалась со¬
временная комбинация методов Пир-ГХ-ИКС-МС [21-23]. В этих работах
кроме определения состава была получена информация о микроструктуре,
включая стереоспецифическую последовательность в полиэтилене. В от¬
дельных работах были изучены полиэфиры [24] и полиамид [25]. Среди
сополимеров проведен анализ стирола-метилметакрилата [26, 27], стиро-
ла-бутил-акрилата [28], бутадиен-акрилонитрила [29], акрилонитрила-бу-
тадиен-стирола (ABS), полиакрилоамида с поливиниловым спиртом [30].Резины и резиноподобные материалы, в частности полиизопрен, по¬
либутадиен, сополимер стирола-бутадиена, полидиметилсилоксан, в по¬
следние годы изучались преимущественно методами Пир-ГХ-МС [31]. На¬
полнитель в виде углеродной сажи не мешал исследованиям резин [32].В работе [29] разработана система - пиролизное устройство-газовый
хроматограф-Фурье-ИКС для исследования сополимеров бутадион-акри-
лонитрила.Для проведения этих исследований было взято пиролизное устрой¬
ство по точке Кюри. Пиролиз проводился при температуре 700°С, время
пиролиза 10 сек, величина пробы 1 мг Продукты пиролиза разделялись
на капиллярной колонке 20 м х 0,32 мм с полиметилфенилсилоксановой
фазой в режиме программирования температуры. Два образца сополимера
бутадионакрилонитрила, содержащие 33% акрилонитрила, полимеризова-
лись при разных условиях.314
ГЛАВА 7 Методы реакиионной газовой хроматографииБыло показано, что на предложенной системе можно видеть различия
в составе полимера, связанные с разными условиями полимеризации.В обзорной работе [33] описано применение пиролизной хроматогра¬
фии для идентификации предметов искусства и археологических материа¬
лов, в частности для анализа пигментов, клеев, связующих, лаков, красок.
В качестве органических связующих в темперных красках эпохи Ренессан¬
са определяются яйца и казеин [34].Несколько десятилетий методом пиролизной хроматографии изучают¬
ся биологические материалы [35]. Было выполнено много работ по иденти¬
фикации и дифференциации микроорганизмов, обнаружению химических
маркеров для отличий стрептококков ipynn А и В [36], для идентификации
бактерий в археологических объектах [37].С эшлогическими целями исследуется происхождение частиц пыли и
аэрозоли в туннелях, в помещениях, в кабинах космических кораблей [38].В пищевой промышленности с помощью пиролизной хроматографии
обычно анализируются белки, полисахариды, жиры, масла, разные расте¬
ния, специи [39,40].В сельском хозяйстве обьекгом анализа обычно являются корма для
животных и почва. В почвах методом Пир-ГХ-МС определялись различ¬
ные вещества, включая гумус [41]. В торфе изучались нерастворимые али¬
фатические вещества [42].В криминалистических лабораториях пиролизная хроматография ши¬
роко применяется для анализа фрагментов красок при автомобильных ава¬
риях [43] и фрагментов тканей [44]. В последней работе исследованы и
классифицированы десятки разных образцов тканей.С помощью пиролизной хроматографии оцениваются сложные смеси
наркотиков и наркотических средств, в частности гашиша [45].Отдельно необходимо вьщелить применение пиролизной хроматографии
в геохимии и для исследования топливного сырья, нефти и каменного угля.В топливах определялись биологические материалы как их предше¬
ственники, указываюоще на их происхождение [46].Широко исследуются керогены - нерастворимые компоненты топлив
[47]. Каменные угли классифицируются уникальной исследовательской
системой Пир-ГХ-МС-ТИД-ПИД [48].По космической программе «Викинг» был проведен пиролиз грунта
Марса с целью обнаружения органических соединений и определения
жизни на Марсе [49]. Однако органических соединений в грунте Марса не
обнаружено. Недавно органические соединения были обнаружены в мете¬
оритах [50] (таблица 7.1).В 2008 шсмический корабль США опустился на поверхность Марса,
грунт будут исследовать не только с поверхности, но и с глубины 60-100 см.315
Яшин Я.И., Яшин Е.Я., Яшин А.Я.Газовая хроматографияВ заключении этого раздела следует отметить, что создана библиотека
масспектров продуктов пиролиза (CDS analytical, Inc) и вышла вторым изда¬
нием книга Wampler Т.Р. Applied Pyrolysis Handbook, CRC Press 2006,312 pp.Таблица 7.1Основные области применения пиролизной хроматографииппОбласти примененияСсылкиИсследование и анализ синтетических полимеров1.1. Гомополимеры (полиэтилен, полипропилен, полиуретан,
силоксановые полимеры и др.).1.2. Каучуки (полибутадиен, полиизопрен и др.),1.3. Сополимеры (нитрильные каучуки - сополимеры бутадиена
с акрилонитрилом; бутилкаучуки - сополимеры изобутиленас изопреном; бутадиенметилстирольные сополимеры;
бутадиенстирольные сополимеры; этиленпропиленовые сополимеры).1.4. Смеси полимеров,1.5. Добавки к полимерам (пластификаторы, антиоксиданты и др.).21-2313,3111-19,
21-32
16,17Криминалистика (краски, адгезивы, токсические вещества, наркотики,
бактерии, волосы, волокна).43-45Биохимия, медицина, фармакология35,46Геохимия18, 19Энергетика (топлива и их источники)47,48Агрохимия (гумусовые вещества в почве)41,42Контроль окружающей среды38Определение органических веществ внеземного происхождения (грунт
Марса, Луны, метеориты) 49, 509.Археологические материалы34, 3710. Предметы искусства (картины и др.)347.4. Химическая дериватизация анализируемых проб.Цель химической дериватизации - перевод нелетучих или неустойчи¬
вых соединений в летучие и более устойчивые для разделения и анализа
методами газовой хроматографии. Основные требования к таким хими¬
ческим превращениям: экспрессность, воспроизводимость и полнота. В
некоторых случаях дериватизацию проб проводят для снижения пределов
детектирования, в частности введение атомов галоидов позволяет исполь¬
зовать селективный чувствительный электронозахватный детектор.Для дериватизации используют в основном реакции силирования, ме¬
тилирования и ацилирования.Силирование - широко используется для получения летучих и устой¬
чивых производных в газовой хроматографии, в частности для анализа
аминокислот, сахаров, стероидов и др.316
ГЛАВА 7 Методы реакиионной газовой хроматографииВ таблице 7.2 приведены основные соединения, анализируемые газо¬
вой хроматографией в виде силильных производных, а в таблице 7.3 - пе¬
речень наиболее распространенных реактивов для силирования.Подробные описания применений триметилсилильных производных в
газовой хроматографии приведены в книгах и обзорах [51-53].Для некоторых триметилсилильных производных увеличивается об¬
щий ионный ток и повышается чувствительность в МС в случае положи¬
тельных ионов [54].Алкилирование - замена активного водорода в кислотах на алкиль¬
ные группы.Метилирование - производится диазометаном и его производными.Ацилирование - замещение активного водорода на ацильную фуппу в
гидроксильных (-ОН), аминных (-NHj) и тиоловых (-SH) функциональных
группах. Получаются производные, соответственно -O-C0R, -NH-COR и
-S-COR. Ацилирование проводится ангидридами карбоновых и галогенкар-
боновых кислот в пиридине или тетрагидрофуране (табл 7.4).Этерификация - получение сложных эфиров карбоновых кислот.
Проще всего производится количественно реакцией с диазометаном CH^N^
в присутствии спиртов (метилового, этилового). Недостаток метода - ток¬
сичность реактива. Получаемые производные -COOR, где R - СН^, изо-
СзН,, С,Н,идр.В обзоре [53] дериватизация применена для определения ксенобио-
тиковв биологических пробах, в частности для определения лекарств при
злоупотреблениях и при допинг-контроле.Для определения альдегидов в окружающей среде методом газовой и
жидкостной хроматографии также используется дериватизация [55].Альдегиды относятся к опасным загрязнителям воздуха, кроме того,
они участвуют в фотохимических процессах в атмосфере.Летучие альдегиды, такие как формальдегид, ацетальдегид и акроле-
ны, воздействуют на глаза и дыхательные пути.Типичные реакции дериватизации следующие:П-СИО * -0-WH, -0-N-CH-R ♦ Н,0R-CHO + CH,-0-NH.- CH.-O-N -rH-R + Н.ОCH,-0-N»CH-R ♦ IV)Г F Г Г317
Яшин Я.И.. Яишн Е.Я.. Яишн А.Я.Газовая хтшлтогрйфияОтдельно следует выделить реакции дериватизации в газохромагогра-
фических анапизах химичесюго оружия и продуктов их разложения [56-61 ].
Обзор ГХ-МС продуктов химического оружия приведен в работе [60].Контроль продуктов химического оружия в окружающей среде опре¬
делен специальной конвенцией в 1997 г. [61].Ниже приведены отдельные реакции дериватизации отравляющих ве¬
ществ (ОВ): ^ва'VX-М.во'VR. |л (04.«»=)Гидролиз OV VX0И
еирТА>ЕЮ^Ъ
<3Aao 'oh Ш0 'cN
♦ ♦
HCNHO о
И0 СЖГидролиз OV «Табун»Таблица 7.2Перечень соединений, которые анализируют газовой хроматографией
в виде силильных производных.п/пНазвания соединенийФормула до
дериватизацииФормула после дериватизации1.СпиртыR-OHR-0-TMC2.Амины (первичные
вторичные)R—N—НR—N—ТМС3.АминокислотыRНRН О4.Карбоксильныекислоты ССЮНNHjR-COOHR-| U О—ТМСN—ТМС
н05.КарбогидратыR JJ— 0—ТМС6,Стероиды-~318
ГЛАВА 7Методы реакиионной газовой хроматографшТаблица 7.3Основные реактивы для силированияп/пНазваниеСокращенноеназваниеФормула1.ДиметилдихлорсиланDMCS(СН,), SiCl,2,ГексаметилдисилазанHMDS(СН,)зй-Ш-81 (СН,),3.ТриметилхлорсиланTMCS(СН,), SiCI4.N,0 - бис (триметилсилил)
ацетамидBSAСИ /О-s,(СИ,,.^N-Si(CH,)35.N,0 - бис (триметилсилил)BSTFA/0^8КСНз)з
' '^N^Si(CHj)36.N - метил - N - триметилсилил-
трифторацетамидMSTFA/СНзCF3CO—^8КСНз)з7.N - метил - N - триметилсилил-
гептафторбутиламидMSHFBA/СНзСзР,СО—^8КСНз)з8,N- триметилсилил-дизтиламииTMSDEA(CH,),Si-N(C,H,),Таблица 7.4Перечень реактивов для метилирования и ацилирования№№ппРеактивыСокращенноеобозначениеФормулаМетилнрованяе1.Ы,Н-диметилформамид-диметилацетальDMF-DMA0 Q_^СN=C—N-СНз-СН,2.ТриметилсульфониумгидроксидTMSHН,Сх ^S^CHjHjC^он319
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я.Газовая хроматография№N«ппРеактивыСокращенноеобозначениеФормулаАиилипование1.АнгидридтрифторуксуснойкислотыTFAA“’5°cf,Y02.ПентафторбензоилхлоридаPFBC03.АнгидридгептафторбутиловойкислотыHFBAоС.Н.ДСзН,^04.N-метил-бис (гепта-
фторбутиламид)MBHFBACF,-f07.5 Реакционная газовая экстракция в анализе объектов окружа¬
ющей среды.В обзоре Б. В. Столярова, Л. А. Филипповой [62], исследовано новое
аналитичесюе приложение парофазного анализа реакционных систем - реак¬
ционная газовая экстракция. Этим методом показана возможность опреде¬
ления общего органического углерода, азота, хлора, фосфора и серы ис¬
черпывающим окислительным разложением органических соединений из
образцов воздуха, воды и почвы. В более ранних работах [63-65] бьши от¬
работаны основы этого метода.На нижеприведенной схеме представлены обобщенные условия обеспе¬
чивающие количественные выходы целевых аналитических летучих соеди¬
нений при подчеркивающем восстановительном расщеплении (или полном
гидрогенолизе) связей углерод - углерод и угаерод - гетероатом [66].320
ГЛАВА 7c^cw,-Методы реатионной газовой хроматографииЗЮ-40ГСKi;Ni/M|0ш-mvc *H2.N/Cbnm№nN-AWS«-iS30*C *■
H2;O'Ai20j(99«%)S30-140045 nSOTСН4NHjHClH2SНа базе хроматографа «Цвет - 100» была создана установка, позволя¬
ющая проводить анализы водных обьешов на общий растворенный неор¬
ганический и органический углерод в диапазоне 5 - 500мг/л [67].32121 Газовая хроматогрМ>ш
Ямин ЯМ. Яшин Е.Я., Яшин АЖ Газовая хршштграфияЛитература1. Березкин В.Г. Аналитическая реакционная газовая хроматография.
М., «Наука», 1966, 184 с.2. Драверт Ф. В кн.: Газовая хроматография в 1961 г. М., Гостоптехиз-
дат, 1963,270 с.3. Методы-спутники в газовой хроматографии. Пер. с англ. Под ред.
В.Г. Березкина. М., «Мир», 1972, 398 с.4. Ray N.H. Analist, 1955, v. 80, p. 853-856.5. Green G.E. Nature, 1957, v. 180, p. 295-296.6. Martin A.E., Smart J. Nature, 1955, v. 175, p. 422.7. Берчфильд Г., Сторрс Э. Газовая хроматография в биохимии. М.,
«Мир», 1964, 619 с.8. Литвинов Л.Д., Руденко В.А. Газовая хроматография в биологии и
медицине. М., «Медицина», 1971, 224 с.9. Яворовская С.Ф. Газовая хроматография - метод определения микропри¬
месей в воздухе и биологических средах. М., «Медицина», 1972,207 с.10. Мошьер Р., Сивере Р. Газовая хроматография хелатов металлов.
Пер. с англ. Под ред. А.А.Жуховицкого и О.Д.Стерлигова. М., «Мир»,
1967, 175 с.11. Березкин В.Г., Алишоев В.Р. Газовая хроматография в химии по¬
лимеров. М., «Наука», 1972, 287 с.12. Накеп J.K. J. Chromat., 1998, v. 825, p. 171-187.13. Алексеева К.В. Пиролитическая газовая хроматофафия. Изд-во
«Химия», г. Москва, 1985, 256 с.14. Wang F.C.-Y. Polymer analysis by pyrolysis gas chromatography. Re¬
view. J. Chromat. 1999, v. 843, p. 413-423.15. Wampler T.P. Introduction to pyrolysis-capillary gas chromatography.
Review. J. Chromat. 1999,842, p. 207-220.16. Wang F.C.-Y., Buzanowski W.C. Polimer additive analysis by pyrolysis-
gas chromatography III Lubricants. J. Chromatog. 2000, v. 891, p. 313-324.17. Wang F.C.-Y. Polimer additive analysis by pyrolysis-gas chromatogra¬
phy IV Antioxidants. J. Chromat. 2000, v. 891, p. 325-336.18. Wampler T.P. (Ed.). Analytical Pyrolysis . Hand book. Morcel Denner,
New York, 1995.19. Tsuge S., Ohtani H. Pyrolysis Gas Chromatography of High Polymers
Fundamentals and Data Compilation, Techno-System, Tokyo, 1989, 371 p.20. В.Р.Алишоев, В.Г.Березкин, А.С.Блинов, В.И.Калмановский,
Е.П.Комарова, А.И.Малышев, Е.Е.Ростовцева, И.М.Савинов,
А.В.Чернов, И.Л.Эвентова, Я.И.Яшин и Г.А.Мирзабаев. Авторское
свидетельство №284418 Бюллетень изобретений №32, 27.01.1917.322
ПММЛ 7 Литература21. Duncan W.P. Аш. Lab. 1988, v. 20, p. 40.22. Wampler T.P. J. Anal. Appl. Pyrol. 1989, v. 15, p. 187.23. Wang F.C.-Y., Smith P.B. Anal. Chem. 1997, v. 69, p. 618.24. Oguri N. et al. JHRC, 1993, v. 16, p. 597.25. Galipo R.C. et al. J. Anal. Appl. Pyrol. 1998, v. 45, p.23.26. Ohtani H., Asai Т., Tsuge S. Macromolecules, 1985, v. 18, p. 1148.27. Wang F.C.-Y., Smith PB. Anal. Chem. 1996, v. 68, p. 3033.28. Wang F.C.-Y, Gerhart B.B., Smith RB. Anal. Chem. 1995, v. 67, p. 3536.29. Weber D. Int. Lab. 1991, v. 21, p. 51.30. Wang F.C.-Y. J. Chromat. 1996, v. 753, p. 151.31. Phair М., Wampler T.R. Rubber World, 1997, v. 215, p. 30.32. Matheson M.J. et al. J. Anal. Appl. Pyrol. 1994, v. 29, p. 129.33. Blazso M.J. Anal. Appl. Pyrol. 1997, v. 39, p. 134. Shedrinsky A.M., Wampler T.R, Baer N.S.J. Anal. Appl. Pyrol. 1989,
V. 15, p. 39335. Chiavari G., Gandind N., Russo P., Fabbri D.Chromatographia 1998,
V. 47, p. 42036. Gutteridge C.S., Norris J.R.J. Appl. Bacteriol. 1979, v. 47, p. 537. Smith C.C., Morgan S.L., Parli C.D. et al.Anal. Chem. 1987, v. 59,
p. 141038. Navale V.J. Anal. Appl. Pyrol. 1992, v. 23, p. 12139. Matney M.L., Limero T.F., James J.XAnal. Chem. 1994, v. 66, p. 282040. Huyghues-Despointes A., Yaylayon V.J. Agric. Food Chem. 1996, v. 44,
p. 67241. Hoshimoto K., Inoue T. et al.J HRC CC 1988, v. 11, p. 34742. Soiz-Jimenez C.Environ Sci. Technol. 1994, v. 28, p. 177343. Kotra R.K., Hatcher P.G.Naturwissenshaften 1988, v. 75, p. 19644. Wampler T.R., Bishea G.A., Simonsick W.J.J. Anal. Appl. Pyrol 1997,
V. 40-41, p. 7945. Aimer J.Can Soc. Forensic. Sci. 1991, v. 24, p. 5146. Hida М., Mitsui T. et al.J. Anal. Appl. Pyrol. 1995, v. 32, p. 19747. Seeley J.A., Leng Y, Uen P. et al.Anal. At. Spectrom. 1992, v. 7, p. 97948. Stankiewicz B.A., Briggs D.E., Evershed R.P.Energy Fuels 1997, v. 11,
p. 51549. Kotra R.K., Gibson E.K., Uebancic, Icarus, 1982, v.51, p.593.50. Murae T. J.Anak.Appl.Pyrol. 1995, v.32, p.65.51. Knapp D. R., Handbook of Analytical Derivatization Reactions, Wiley,
New York, 1979.52. Blan K., Halker J.M. (Eds), Handbook of Derivatives for Chromatog¬
raphy, Wiley Chichester, 2"“ Ed 1993.323
Яшин ЯМ. Яшин Е.Я.. Яшин АЛ Гюо*ая хроматографт53. Sequra J., Ventura R., Durado С. J. Chromatog. В 1998, 713, p 61.54. Wells R. J. Resent advances m non - silylatuon derivatization tech¬
niques for gas chromatography J. Chromatog. 1999, 843, p 1 - 18.55. Nishikawa H., Sakai T. Derivatization and chromatographic deter¬
mination aldehydes in gaseous and air samples, J. Chromatog. 1995, 710,
p. 159.56. Witkievich Z., Mazurek М., SzulcJ., J. Chromatog. 1990. V.503, 293.57. Kientz Ch. E. J. Chromatog. 1998. V.818, p 1.58. Mesilaakco M. Chemical Weapons Chemical analysis in: Meyers R. A.
(Ed) Encyclopedia of Analytical Chemistry, Wiley, Chichester,2000, p 899.59. Hooijschuar E. W. J., Kientz Ch. E., Brinkman U. A. Th. J. Chromatog.
2002,982, p 177.60. Wils E. R. J. in: R. A. Meyers (Ed) Encyclopedia of Analytical
Chemistry, Wiley, Chichester 2000, p 979.61. Black R. М., Miur B. Derivatization in the chromatographic analysis of
chemical warfare agents £and their degradation products, J. Chromat 2003,
v.l000,p253 -281.62. Столяров Б. В., Карцева Л. А., Филиппова О. В., Реакционная га¬
зовая экстрокция в анализе объектов окружающе11 среды. Журнал ана¬
литической химии 1987, т.61, с 1441.63. Столяров Б. В., Карпова Л. А., Успехи химии 1987, т.56, с 1024.64. Стоомров Б. В., Карпова Л. А., Журнал физической химии 1987,
т.61, с 1441.65. Карпов Ю. А., Российский химический журнал 1994, т.38, с 12.66. Столяров Б. В., Карпова Л. А., Филиппова О. В., Вестник С-Пб.
университета, серия 4,1995, выпуск 1, с 14.67. Столяров Б. В., Галев Э. Е., Химия И технология воды 1988, т. 10,
с 234.324
ГЛАВА 8 Двухмерные и многомерные варианты газовой храматографтГлава 8. ДВУХМЕРНЫЕ И МНОГОМЕРНЫЕ
ВАРИАНТЫ ГАЗОВОЙ ХРОМАТОГРАФИИО широких возможностях двухмерных вариантах хроматографии об¬
ратил внимание Дж. Гиддинг [1].Впервые двухмерная хроматография в современном ее представлении
предложена Филлипсом Дж. в 1991 г. [2]. В последующие годы вышли сот¬
ни публикаций и обзоров [3-15].Эффективные капиллярные колонки позволяют разделять одновремен¬
но сотни компонентов в смеси. Однако появляются все новые задачи, тре¬
бующие для решения колонки большей селективности и эффективности. К
таким задачам можно отнести: компонентный анализ некоторых фракций
нефтепродуктов, разделение всех диоксинов, полихлорированных бифени¬
лов (209 соединений), ароматов многих пищевых продуктов, в частности
кофе (около тысячи соединений), неюторые биологические пробы, анализ
загрязнений воздуха городов, поверхностных вод и др.Для решения таких задач эффективно использовать вторую колонку
с сорбентом другой природы, на юугорой закономерности удерживания
сильно отличаются от закономерностей удерживания на первой колонке.Ранее была предложена техника «вырезания» части фракции на хрома¬
тограмме (heart-cut) и переброски ее на вторую шлонку. Однаш в этом слу¬
чае тольш часть всей смеси подвергается более детальному разделению.В этой упрощенной системе двумерной газовой хроматографии ис¬
пользуются две шлонки (с неполярной и сильнополярной фазой), пере¬
ключатель Дина в термостате (рис. 8.1).В этом случае тольш часть анализируемой пробы передается во вторую
шлонку, а часть пробы «вырезается» и подается на первый детектор. В этих
системах обычно не используется криогенная фокусировка. Этот метод пред¬
ложен сравнительно давно, в последние годы интерес к нему возрос, т.к. по¬
явилось высокоточное управление газовыми потоками, кварцевые капилляр¬
ные шлонки. Упрощенная двухмерная газовая хроматография находит приме¬
нение для разделения и анализа сложных многошмпонентных смесей.В качестве примера можно привести методику определения кислоро¬
досодержащих (оксигенатов) и ароматических соединений в бензине в со¬
ответствии с евростандартами EN13132 и EN12177.В этих системах обычно используется и обратная продувка для удале¬
ния высошкипящих соединений.В некоторых случаях колонки размещаются в разных термостатах.Общая схема полной двухмерной хроматографии (ГХ - ГХ) показана
на рис 8.2. Между двумя колонками (обычно обе колонки капиллярные)
устанавливается дополнительное устройство - модулятор с охлаждением,
после которого только часть пробы может быть направлена на детектор325
Яшин Я.И.. Яшин Е.Я., Яшин А.Я.Газовая хроматографияИЛИ после задержки в модуляторе направлена на вторую колонку для более
детального разделения.Модулятор может фокуифовать часть или всю пробу и затем за счет «те¬
плового удара» быстро направить пробу на вторую колонку. Иногда вторая
колонка работает в режиме экспрессной хроматографии. Работа модулятора
управляется компьютером по специальной программе. За счет сжатия поло¬
сы на криомодуляторе он работает как концентратор, поэтому в некоторых
случая можно повысить чувствительность анализа. Так как используется
комбинация двух капиллярных колонок с жидкими фазами разной поляр¬
ности (обычно малополярная и сильнополярная), то ценность параметров
удерживания, в частности индексов уцерживания, сильно возрастает.На рис.8.3 показано, что неразделенный пик после криомодулирования
на второй колонке разделяется на несколько пиков. Вместо одного пика по¬
лучается серия хроматограмм с изменяющимся составом, которую можно
развернуть в двумерное и даже трехмерное изображение.Описаны сочетания и трехмерные ГХ-ГХ-ГХ. Представляют интерес
комбинации ВЭЖХ-ГХ и ГХ-ГХ-МС.Основные применения ГХ х ГХ приведены в таблице 8.1.Таблица 8.1№№ппОбласть примененияАнализируемые веществаМетоддетектированияСсылка1.Анализ атмосферылетучиеДИП162.Поверхностные водылетучие, БТЭКСДИП173.ОсадкиПАУМС184.Нефтехимиябиомаркеры,
групповое разделениеДИПДИП1920Пища:подделкаДИП195.пищевые экстрактыароматДИП.МС21,22рыбаПХБ, диоксиныДИП, ЭЗД23овощипестицидыМС24Медицина:биологические жидкостижирные кислотыДИП256.плазма кровипестицидыДИП26выдыхаемый воздухлетучиеДИП16сигаретный дымвсе соединенияМС27Примечание: БТЭКС - бензол, толуол, этилбензол, ксилолы:
ПАУ - полиараматические соединения;
ДИП - детектор ионизационно-пламенный;
МС - масспектрометр;ЭЗД - электронно-захватный детектор.326
тлвл s_ Двухмерные и многомерные варианты газовой хроматографиихладоагентРис. 8.1. Функциональная схема системы с ГХ х ГХ с систе¬
мой ДинаРис. 8.2 Общая схема системы ГХ-ГХ с модулятором.327
Яшин ЯМ. Яшин ЕЖ, Яшин А.Я.ГтмтштитощфтUUUРис. 8.3 Принцип работы полной системы ГХ-ГХ с модуляцией.328
ГЛАВА Н ЛитератураЛитература1. Gidding J.C. Two-Dimensional Separation; concept and promise. Anal,
chem. 1984, v. 56, p. 1259.2. Lm Z„ Phillips J.B. J. Chromat. Sci. 1991, v. 29, p. 227.3.BhmbejgL-M.,KleeM.S.J.Chromat(^.2001,v.933(1-2),p. 1, ll;p. 13-26.4. Blmnberg L.M. J.Chromat. 2003, v. 985, p. 29.5. Dallilge J., Beens J., Brinkman U.A.Th. J.Chromat. 2003, v. 1000,
p. 69-108.6. Ong R.C.Y., Marriott R. Trends. Anal. Chem. 2002, v. 21, p. 573.7. Phillips J.B., Gaines R.B., Blombei^ J. et al. J. High Resol. Chromatog.
1999, v. 22, p. 3-10.8. Multidimensional Chromatography: Techniques and Applications EdsH.J.Corters, Marcel Dekker, New York, 19909. Hy6tylainen Т., Riekkola M.-L. On-line Coupled liquid chromatography- gas chromatography. J. Chromatog. 2003, v. 1000, p. 357-384.10. Grob K, in: On-Une Coupled LC-GC, Hilthig, Heidelberg, Germany, 1991.11 Liu Z., Lee M.L. Comprehensive Two-Dimensional Separation using
Microcolumns. J. Micro Sep. 2000, v. 12, p. 241.12. Saskia M. von Ruth. Evaluation of two gas chromatography - olfacto¬
metry methods: the detection frequency and perceived intensity method. J.
Chromatog. 2004, v. 1054, p. 33.13. Adahchour М., Beens J., Vreuls R.J.J., Batenbui^ A.M., Brinkman
U.A.Th. Comprehensive two-dimensional gas chromatography of complex
samples by using a “reversed-type” column combination: application to
food analysis. J.Chromatog. 2004, v. 1054, p. 47.14. Phillips J.B., Xu J. Comprehensive multy-dimensional gas chromatog¬
raphy. Review. J. Chromatog. 1995, v. 703, p. 327-334.15. Schomburg G. Two-dimensional gas chromatography: principles,
instrumentation, methods. J. Chromat. 1995, v. 703, p. 309-325.16. Seeley J.V., Kramp F.J. et al. J. Sep. Sci. 2002, v. 25, p. 53.17. Beens J.,DalltlgeJ., Adahchour ML etaiJ.Microcol.Sq). 2001, v. 13, p. 134.18. Hy6tylainen T. et al. Anal. Chem. 2002, v. 74, p. 4441.19. Frysinger G.S., Gaines R.B., J. Sep. Sci. 2001, v. 24, p. 87.20. Johnson K.J., Synovec R.E. Chemoia Intell. Lab. Syst. 2002, v. 60, p. 225.21. Shellie R. et al. J. Chromat. 2002, v. 970, p. 225.22. Adahchour M. et al. Chromatographia 2002, v. 55, p. 361.23. Korytar P. et al. J. Chromat. 2002, v. 958, p. 203.24. Dalltige J. et al. J. Chromat. 2002, v. 965, p. 207.25.Westem R.J. et al. Lipids 2002, v. 37, p. 715.26. Lin Z.Y. et al. Anal. Chem. 1994, v. 66, p. 3086.27. D allUge J. et al. J. Chromat. 2002, v. 974, p. 169.329
Яшин Я.И., Яшин Е.Я.. Яшин А.Я. Газовая хттатографтГлава 9. ВЫСОКОСКОРОСТНАЯ ГАЗОВАЯ
ХРОМАТОГРАФИЯ9.1. Отлнчяе высокоскоростной хроматографии от «обычной»
хроматографии.В практике газовой хроматографии продолжительность разделения
на насадочных, микронасадочных и капиллярных колонках составляет от
5 до 60 мин. Среднее время разделения - от 15 до 30 мин. В некоторых
случаях при разделении сложных смесей время разделения может быть бо¬
лее 60 мин. В1ЖМЯ разделения в предложенных и реализованных методах
сшростной газовой хроматографии в 5 - 50 раз меньше, т.е. время разделе¬
ния может быть в пределах от десятков секунд до 5 минут. В сверхскорост¬
ных методах время разделения может быть в 400 - 4000 раз меньше.В литературе описаны разные варианты скоростной (экспрессной, бы¬
строй) хроматографии: high speed, fast, very fast и ultra fast [1-3].Для различия предложена их классификация по величине ширины пика
на половине высоты [4]. В таблице 9.1 приведены отличия этих методов.Таблица 9.1Отличие разных вариантов высокоскоростной газовой
хроматографии от «обычной» хроматографии.№№ппТипы вариантов ГХДиапазонвременианализаШирина пикаВо сколько раз
сокращается
время
разделения1.Обычнаяминуты-часы5 ^ 30 сек.12.Скоростная (fast)минуты1 ^ 3 сек.5-303.Высокосюростная (very fast)секунды30 - 200 мсек.30-4004.Сверхвысошсюростнаяменьшесекунды5-30 мсек400 - 4000Предложенная классификация признана не только правильной, но и
полезной в связи с требованиями к хроматографической аппаратуре, вы¬
двигаемыми высокоскоростными методами, в частности, требованиями к
инерционности детекторов, скорости программирования температуры, си¬
стемам дозирования, электронным схемам.Скоростная (fast) хроматография иногда может быть реализована на
обычной современной газохроматографической аппаратуре. Эффективность
колонок при этом сопоставима с эффективностью обычных колонок.В случае высокоскоростной газовой хроматографии обычная аппара¬
тура может быть использована только для простых рутинных задач при330
ГЛАВА 9 Высокоскоростная газовая хроматографияразделении простых смесей, так как снижается общая эффективность ко¬
лонок. В этих случаях используются короткие колонки 0,5 - 3 м с внутрен¬
ним диаметром 0,10 - 0,53 мм [5].Сверхбыструю (ultra fast) хроматографию на практике реализовать
очень сложно. Для этого нужна новая хроматографическая аппаратура, но¬
вая система регистрации и обработки.9.2. Преимущества высокоскоростной хроматографии и возмож¬
ные области ее применения.Быстрое разделение необходимо в контроле производственных про¬
цессов (в некоторых производственных процессах необходим непрерыв¬
ный контроль, поэтому хроматография как прерывистый метод в этих слу¬
чаях не применяется), в мониторинге некоторых процессов в природе, в
частности загрязнений окружающей среды, при необходимости быстрого
определения загрязнений на месте объекта (on-site), в анализах для про-
мыщленной гигиены. Малая продолжительность анализа необходима на
портативных (полевых) газовых хроматографах.Меньшее время анализа снижает его стоимость за счет сокращения
потребления дорогих газов-носителей, электроэнергии и пр.В некоторых случаях при контроле процессов применение быстрой хро¬
матографии позволяет лучше описывать процесс в целом, так как при этом
за отведенное время можно провести большее количество анализов. Это
было наглядно продемонстрировано при разрешении «кризиса диоксинов» в
пище в Бельгии в 1999 г. [6], когда удалось выполнить более 50(ХЮ анализов,
благодаря сокращению затрат времени на их проведение.9.3. История развития методов высокоскоростной газовой
хроматографии.Попытки сокращения времени разделения предпринимались начиная
с 1960 г. Д.Дести [7] в Англии уменьшал время разделения за счет исполь¬
зования капиллярных шлонок меньшего диаметра.Интенсивно сшростной газовой хроматографией занялись с 1990-х гг,
когда стали оценивать стоимость анализа.К этому времени бьши сформированы основные принципы и базовые
положения теории сшростной хроматографии, бьши выдвинуты необходи¬
мые требования к аналитической хроматографической аппаратуре.В ряде обзоров [8-10] были обобщены преимущества и ограничения
методов сшростной хроматографии. К.Крамерс [И] предложил уравне¬
ние, связывающее время удерживания (tj^) с параметрами газохроматогра-
фичесшй системы:331
Яшин Я.И., Яшин Е.Я., Яшин А.Я. Газовая хроматография(9,1)где: R - степень разделения; ~ коэффициент емкости; а - коэффици¬
ент селективности; Н - ВЭТТ; и^, - линейная скорость газа-носителя на вы¬
ходе колонки; f- коэффициент, учитывающий сжимаемость газа-носителя.Для быстрого разделения предлагается ошимизировагь между 1,7 - 3 [12].9.4. Основные способы сокращения времени анализа.Время удерживания в хроматографии зависит от многих параметров:
t,~f(T,,u„,K,V^,S„K,L,d,) (9.2)где: Т^ - температура колонки; - обьем жидкой фазы в колонке в
случае ГЖХ; - величина удельной поверхности адсорбента в случае
ГАХ; К - коэффициент распределения; L - длина колонки; - внутренний
диаметр колонки.Величины и К связаны с природой сорбента.В практике газовой хроматографии почти всегда основным критерием
оптимизации метода является достижение полного разделения (R - сте¬
пень разделения 1 или 1,5) за меньшее время. Время разделения без учета
времени пробоподготовки - это и есть время анализа, т.к. регистрация и
обработка хроматограмм происходят быстро с помощью компьютера.В газо-жидкостной хроматографии (ГЖХ) удерживаемый обьем пря¬
мо пропорционален количеству жидкой фазы (V^) в колонке:(9.3)где К - коэффициент распределения.Уменьшая шличество жидшй фазы на твердом носителе, можно значи¬
тельно сократить время анализа. В начале развития ГЖХ использовались сор¬
бенты с 15, 20 и даже 25% жидшй фазы на носителе. При разделении высо¬
шкипящих соединений на таких сорбентах даже при высоких температурах
время вькода бьшо очень длительным. Для сокращения времени выхода ста¬
ли уменьшать шличество жидшй фазы до 5% и даже 1 - 2%, при этом, есте¬
ственно, время выхода уменьшалось в 5 - 20 раз. В настоящее время анализ
пестицидов, в частности, проводят на шлонках с 2 - 5% малолетучей высош-
кипящей жидшй фазы. Колонки с такими сорбентами получили название ко¬
лонок мтой емкости. На шлонках малой емшсти есть еще одна юзможность
уменьшения времени анализа за счет увеличения сшрости газа-носителя.На шлонках малой емшсти зависимость высоты, эквивалентной теорети¬
чесшй тарелке (Н). от линейной сшрости газа-носителя (и) в праюй части по-332
ГЛАВА 9 Высокоскоростная газовая хроматографиялогая (рис. 2.10), в отличие от шлонок большой емшсти. Это свойство таких
шлонок позволяет работать при больших линейных сшростях газа-носителя
без снижения эффективности (числа теоретических тарелок шлонки) и позво¬
ляет реализовать {«жим программирования расхода газа-носителя.В газо-адсорбционной хроматографии время разделения можно со¬
кратить за счет применения поверхностно-пористых адсорбентов [13] по
тем же причинам, что и при снижении количества жидкой фазы. В случае
поверхностно-пористых адсорбентов уменьшается удерживание и сокра¬
щаются пути внутреннего массообмена, возрастает сшрость массообме¬
на, что позволяет также использовать большие сшрости газа-носителя без
снижения эффективности шлонки (рис. 4.15).Следующий способ снижения времени анализа - это выбор более се¬
лективного сорбента с большим значением величины а для самой трудно¬
разделяемой пары в анализируемой смеси.Для определенного разделения требуется эффективность с необходи¬
мым числом теоретических тарелок [11]:16 (9-4)При больших значениях а необходимое число N меньше, а это озна¬
чает, что для разделения нужна колонка меньшей длины, на которой про¬
изойдет более быстрое разделение.Уменьшение длины и диаметра колонок как насадочных, так и капиллярных,
также широю используется на гфактике для сокращения времени анализаВ изотермичесшм режиме время удерживания пропорционально дли¬
не шлонки. Если разделение смеси сохраняется, то можно уменьшить дли¬
ну колонки.В капиллярной хроматографии в последние годы стали использоваться
шлонки длиной 5,10,15 м. Ранее применялись шлонки длиной 25,50,100 м.Уменьшением диаметра шлонки в капиллярной хроматографии мож¬
но эффективно сократить время разделения, сохраняя степень разделения.
Например, на колонке 10 м х 0,1 мм достигается таше же разделение, как
и на колонке 25 м х 0,25 мм, но в 2,5 раза быстрее [14].Как альтернатива насадочным предлагается использовать капилляр¬
ные шлонки большого диаметра (0,32 и 0,53 мм) с большей толщиной
пленки (от 0,5 до 5 мкм). На таких колонках можно в 3 - 5 раз сократить
время разделения.Экспрессное разделение достигается на поликапиллярных шлонках
[15-17]. В трубке с диаметром обычных насадочных шлонок размещаются до
900 параллельно размещенных капилляров с внутренним диаметром 40 мкм.333
Яшин Я.И., Яшин Е.Я., Яшин А.Я. Газовая хроматографияПараметром, наиболее эффективно влияющим на время разделения,
является температура колонки, при которой происходит разделение как в
изотермическом режиме, так и в режиме программирования температуры.
Связь параметров удерживания с температурой - логарифмическая, поэто¬
му в некоторых случаях изменение температуры всего на десять градусов
изменяет время удерживания в несколько раз. Особенно эффективно при¬
менение режима программирования температуры всей колонки во времени
для смесей, компоненты которых сильно различаются по летучести.В современных газовых хроматографах скорость программирования
до 30 - 50°С/мин. В некоторых приборах рекламируются значения ско¬
ростей до 120°С/мин. и более. Однако необходимо помнить, что в наса¬
дочных колонках скорость нагрева сорбента внутри колонки следует за¬
данным значениям только до 25°С/мин., а далее наблюдается отставание с
нагревом. В капиллярных колонках максимальная скорость нагрева выше,
но она также не может быть слишком высокой.Ранее проводились работы по повышению температуры колонки из
нержавеющей стали за счет пропускания через нее электрического тока.
В 1991 г. этот способ усовершенствовали [18], нанося на поверхность ко¬
лонки проводящий слой [19], помещая нагреваемую проволоку близко к
колонке [20] или надевая на капилляр металлическую трубку-чулок [21].
Этот способ реализовала фирма Thermedics Detection в виде спещтально-
го устройства EZ Flash. На Питтсбургской конференции в 1996 г за это
устройство фирма получила Золотую медаль в номинации «Лучшие новые
технологии». Аналитические колонки длиной от 5 до Юме внутренним
диаметром от 0.1 до 0,53 мм помещаются в тонкостенную трубку из нержа¬
веющей стали. Нагревание происходит при прикладывании напряжения к
металлической трубке. Сопротивление трубки непосредственно связано
с температурой этого «термостата» и, следовательно, с температурой ко¬
лонки. При этом может быть достигнута значительная скорость подъема
температуры до 1200°С/мин. Подъем температуры от 50 до 350°С может
быгь осуществлен за 15-20 сек., а охлаждение с 300 до 50°С - за 30 сек.
Это устройство - модульное и может соединяться с любым коммерческим
газовым хроматографом [22].В работе [24] показаны возможности этой системы быстрого нагрева
колонки при контроле загрязнений окружающей среды: содержание нефте¬
продуктов в почве при разливах, скрининг полиэфирных ароматических
соединений, полихлорированных бифенилов, пестицидов. Время анализа
сокращалось в 5 - 10 раз.В заключение следует отметить, что во многих случаях время анализа
включает и время пробоподготовки. В работе [24] изучались возможности334
ГЛАВА 9 Высокостростная газовая храматогтФтзначительного со1фащения времени пробоподготовки в таких методах, как
микроволновая экстракция, твердофазная экстракция, микротвердофазная
экстракция и сверхкритическая флюидная экстракция.В некоторых случаях удалось сокрашгь время пробоподготовки до 3 мин.Фирма «Вариан» предложила шмплексную высокоскоростную га¬
зохроматографическую систему, включающую быстрое разделение и экс¬
прессное уникальное адсорбционно-десорбционное устройство [24] для
контроля окружающей среды и мониторинга промышленной гигиены.Фирма «LECO» предложила высокосшростную газохроматографиче¬
скую систему с масс-спектральным окончанием Pegasus [25]. Эта система
о&единяет газовый хроматограф и время - пролетный масс-спектрометр
и высокоскоростную технологию обработки сигнала (500 полных спектров
в секунду).Возможности этой системы были показаны на примере анализа сырой
нефти. Традиционный анализ сырой нефти занимает 133 минуты. Анализ
сырой нефти с помощью системы Pegasus II производатся всего за 6 минут
без ущерба для тх)чности количественного и качественного анализа.Вопросы высокоскоростной газовой хроматографии в узких капилляр¬
ные колонки рассмотрены в книге [26].335
Яшин ЯМ. Яшин ЕЖ. Яшин Л Я. Газовая .хроматографияЛитература1. Korytar Р., Janssen H.-G., Matisova Е., Brinkman U.A.Th. Trends Anal.
Chem. 2002, V. 21, p. 558.2. Korytar P., Matisova E. Chem. Listy. 2001, v. 95, p. 783.3. Matisova E., D6m6ter6va M.J. Chromat. 2003, v. 1000, p. 199.4. van Deursen M.M., Beens J., Janssen H.-G., Leclercq P.A., Cramers C.A.,
J.Chromatog. 2000, v. 878, p.205.5. Dagan S., Amirav A. J. Am. Soc. Mass Spectrom. 1996, v. 7, p. 737.6. Sandra P., David F. J. Chromat. Sci. 2002, v. 40, p. 234.7. Desty D.H., Goldup A., Swanton W.T. in: N Brenner et al (Eds) Gas
Chromatography, Academic Press, New York, 1962, p. 105.8. Klee M.S., Blumberg L.M. J. Chromat. Sci. 2002, v. 40, p. 234.9. Cramers C.A., Janssen H.-G., van Deursen M.M., Leclercq P.A.,
J.Chromatog. 1999, v. 856, p.315.10. Sacks R., Smith H., Novak M. Anal. Chem. 1998, v. 70, p. 29 A.11. Cramers C.A., Leclercq PA. J.Chromatog. 1999, v. 842, p.3.12. Grushka E., Guiohon G. J. Chromat. Sci. 1972, v. 10, p. 649.13. Жданов С.П., Калмановский В.И., Киселев A.B., Яшин Я.И. Ж.
физ. химии, 1962, т.36, С.1118.14. David F., Gere D.R., Scanlan F., Sandra P. J.Chromatog. 1999, v. 842, p.309.15. Солдатов В.П., Кузнецов В.В., Морозов А.А. и др. Авторское сви¬
детельство №969089, СССР, Бюллетень изобретений, 1981.16. Солдатов В.П., Науменко И.И., Ефименко А.П., Вагонов B.C. Па¬
тент №1651200 Бюллетень изобретений, 1986.17. Малахов В.В., Сидельников В.Н., Уткин НА. Докл. АН. 1993, т.39, с.749.18. Rounbehler D., MacDonald S., Lieb D., Fine D. First Int. Symp. on
Explosive Detection Tech. 1991, v. 1, p. 703.19. Jain V., Philipps J. J. Chromat. Sci. 1995, v. 33, p. 55.20. Ehrmann E., Dharmasena H., Carney K., Overton E. J. Chromat. Sci.
1996, V 34, p. 533.21. Rounbehler D. US Patent 1992, 5092217.22. Surve М., Knight D. Intern. Enviroimi. Technol. 1999, p. 22.23. MacDonald S., Jarvis G.B., Wheeler D.V. Intern. Environm. Technol.
1998, p. 31.24. Fast G.C. The Practical Solution to the Challenge of High-speed Gas
Chromatography Varian Sep-8375/371 15 M 2/95.25. Leco. Новый стандарт для скоростной газовой хроматографии и
масс-спектрального анализа. www.Leco.com26. Van Es А. High-Speed Narrow-Bore Capillary Gas-Chromatography,
Huthig Verlag, Heidelberg 1992.336
ГЛАВА 10 Высокотемпературная газовая хроматографияГлава 10. ВЫСОКОТЕМПЕРАТУРНАЯ ГАЗОВАЯ
ХРОМАТОГРАФЕ10.1. Краткий исторический обзорДо 70 г. в газовой хроматографии не было стабильных жидких фаз,
которые можно было бы использовать при температуре свыше 250°С.С введением полиметилсилоксановых жидких фаз стало возможным
работать при температурах до 300°С в изотермическом режиме и кратков¬
ременно - при температуре до 350°С в режиме программирования темпе¬
ратуры.При высоких температурах возникала проблема с применением квар¬
цевых капиллярных колонок с полиамидным покрытием. В 1983 г [1] было
предложено наносить на внешнюю поверхность кварцевых капиллярных
колонок слой алюминия, а к внутренней поверхности - химически при¬
вивать полисилоксановые жидкие фазы. На таких колонках можно было
поднимать температуру до 350-400°С. Однако позднее выявились недо¬
статки алюминированных кварцевых капиллярных колонок, связанные с
тем, что слой алюминия на полностью защищал поверхность капилляра,
и через определенное время колонка могла лопаться [2]. Для устранения
этого недостатка были разработаны более термостабильные полиамидные
лаки. Вторым значительным направлением в области высокотемператур¬
ной газовой хроматографии является разработка капиллярных колонок из
нержавеющей стали с инертной внутренней поверхностью, как у кварце¬
вых колонок.На внутренней поверхности металлических капилляров создавался
кварцевый слой и к нему химически прививались полисилоксаны, со¬
держащие гидроксильные группы, что увеличивало термостабильность и
инертность таких колонок.Термостабильность полисилоксанов оценивают по изменению фона с
повышением температуры, определяя таким образом максимально допу¬
стимую рабочую температуру (МДРТ) этих жидких фаз. Температурная
стабильность полисилоксанов зависит от степени полимеризации, от при¬
роды функциональных групп, от симметрии замещения функциональны¬
ми группами относительно силоксановой цепи [3].В работе [4] предложена в качестве высокотемпературной жидкой
фазы поли (сульфон арил эфир), которая оказалась устойчивой до 380°С.В 1988 г фирма SGE Intern, создала самую высокотемпературную
жидкую фазу карборан (5%) - полиметилсилоксан (рис. 3.7). На алюмини¬
рованных колонках с такой фазой удалось проводить разделение при тем¬
пературах 460-480°С [5].33722 Газовая я
Яшин Я.И., Яшт Е.Я.. Яшин А.Я. Гаювая хроматографияТаким образом, в настоящее время имеются колонки с неполярными и
слабополярными фазами, на которых при определенных условиях можно
работать при температурах 350-420°С.10.2. Основные области применения высокотемпературной газо¬
вой хроматофафии.В обзоре [6] обобщены все способы приготовления высокотемпера¬
турных колонок.В 1991 г. [7] вышла книга, посвященная высокотемпературной газовой
хроматографии (ВТГХ).В таблице 10.1 приведены основные области применения ВТГХ.Таблица 10.1ппОбласти примененияТипичные смесиСсылкиНефтехимияРазделение сырой нефти (С|^ - С,^)
Имитированная дистилляция
Определение маркеров нефти
Определение порфиринов9-162.Химия полимеровОпределение олигомеров173.ПромышленныепродуктыКонтроль качества неионных моющих
средств18ПищеваяпромышленностьАнализ триглицеридов в жирах и маслах
Анализ олигосахаридов19-2214-165.Химия природных
продуктовПрямой анализ флавоноидов и ротеноидов
Высокомолекулярные соединения из
растительных экстрактов 23,24
25, 266,ЭкологияАнализ высокомолекулярных ПАУ вприродных объектахАнализ почвы, загрязненной асфальтом27, 287.АрхеологияИдентификация пигментов, связующих в
предметах искусства10.3. Высокотемпературная газоадсорбционная хроматография
паров металлов и солей.В обычных газовых хроматографах, предназначенных для анализа до¬
статочно летучих соединений, используют температуры не выше 400-500°С.
Хроматографическое разделение малолетучих и практически нелетучих при
этих условиях веществ, в частности металлов, возможно только при темпе¬
ратурах выше 500°С. При таких высоких температурах (иногда до 1000°С)
условия разделения значительно усложняются, металлические стенки тру¬
бок становятся химически активными, и на них происходит необратимая ад¬
сорбция многих металлов. Наиболее трудная проблема - это детектирование
веществ при столь высоких температурах [27, 28].338
ГЛАВА 10 Высокотемпературная газовом хроматографияВпервые хроматографичесше разделение свободных металлов (Cd и Zn)
было проведено при 620°С на юлонке с морским песком, содержащим 20%
расплавленного LiCl [29]. Детектирование при этом не проводилось. В па¬
тенте [28] предложен способ хроматографической очистки многих металлов
и металлоидов: Li, Na, К, Ag, Си, Be, Mg, Cd, Sr, Ba, Zn, Hg, Al, Ga, In, Tl, Si,
Ge, Sn, Pb, As, Sb, Se, Те, Mn, Pd и Ce. Металлы очищают, пропуская их пары
при 750-2000°С через колонку с адсорбентом (силикагель, активную окись
алюминия) либо расплавленным металлом (Fe, Ni, Со, Sn), нанесенным на
твердый диатомитовый носитель. После такой предварительной очистки мо¬
жет быть проведена дополнительная очистка металлов, используемых для
полупроводниковой техники. Возможна также хроматографическая очистка
соединений металлов [30]. Описана конструкция хроматографа, работающе¬
го при температурах до 1000°С.Для разделения металлов наиболее пригодна газо-адсорбционная хро¬
матография. В качестве адсорбентов для газовой хроматографии при столь
высоких температурах использовались силикагель, окись алюминия, гра¬
фит, активный уголь, кварц, диатомит [27 - 34]. Исследовалась возмож¬
ность хроматографического разделения Cd, Zn, Hg, Na, К, S, Se, TlCl, Cdl^,
PbClj и SnClj [35]. Cd и Zn частично разделяются на многих адсорбентах
при 700-900°С [32 - 34]. Обычный активированный уголь при 850°С ад¬
сорбирует пары Zn необратимо, что связано с присутствием поверхност¬
ных загрязнений на угле. После промывания угля кислотой необратимой
адсорбции не наблюдалось. Наилучшее разделение Zn, Cd, Hg наблюда¬
ется на колонках с графитом [34]. Пары К и Na реагируют со всеми мате¬
риалами колонки. Сера и селен по-разному удерживаются на силикагеле
и графите при 450-600°С и, взятые по отдельности, элюируются в виде
симметричных пиков, однако смесь их не разделяется [35, 36].В работе [37] показана возможность определения магния в алюминие¬
вых сплавах. При температуре кипения магния (1107°С) давление пара алю¬
миния настолько мало, что он не регистрируется. При дозировании 200 мг
сплава можно определить 0,1% Mg. Методом высокотемпературной газовой
хроматофафии на колонке с углем возможно также определение состава
сплава «циркалой» [36]. Описаны возможности применения газовой хрома¬
тофафии на различных адсорбентах для анализа легких металлов [35].Редкоземельные элементы можно разделять и анализировать мето¬
дом газовой хроматофафии на адсорбционной стеклянной капиллярной
колонке небольшой длины [37]. Хлориды лантаноидов образуют летучие
комплексы с AlClj, поэтому для повышения их летучести к газу-носителю
добавляют AlClj с парциальным давлением в пределах 40-170 мм рт. ст.
[(5-10) ■ 10^ Па] [38]. Таким же образом можно проанализировать и транс¬
урановые элементы Ра, U, Np, Pu, Am, Cm [39].33922-
Яшин ЯМ. Яшин Е.Я.. Яшин А.Я. Газовая хроматографияЛитература1. Lipsky S.R., McMurray WJ. J. Chromat. 1983, v. 279, p. 59.2. Lipsky S.R., Duffy M.L. J. High Resolut. Chromatog. 1986, v. 9, p. 725.3. Pereira A.S.,de Aquino NetoF.R. Trends in anal. chem. 1999, v. 18, p. 126.4. Mathews R.G. J. Chromat. Sci. 1989, v. 27, p. 47.5. Dawes P., Cumbers M. Am. Lab. 1989, v. 21, p. 102.6. Blum W. J. High Resolut Chromat. 1985, v. 8, p. 718.7. Blum W. and Aichholz R. Hohtemperatur Gas-Chromatographie. Huthig
Heidelberg 1991,8. Aquino Neto F.R. et al. J. High Resolut Chromat. 1994, v. 17, p. 259.9. Neer L.A., Deo M.D. J. Chromat. Sci. 1995, v. 33, p. 133.10. Zeng Y., Uden PC. J. High Resolut Chromat. 1994, v. 17, p. 223.11. Heath D., Moffott B., Lowry R., Rowland S. Anal. Proc. 1995, v. 32, p. 485.12. Mradnova R., Kubinec R., Sojak L. et al. Petroleum and Coal. 2001,
v.43,p. 17.13. Park J.R., Lee D.-S. Bull. Korean. Chem. Soc. 2003, v. 24, p. 527.14. Lawrence A., Neer A., Milind D. J. Chromat. Sci. 1995, v. 33, p. 133.15. ASTM D 5307-97 Standard Test Method for Determination of Boiling
Range Distribution of Crude Petroleum by Gas Chromatography. Armual
Book of ASTM Standards. ASTM Philadelphia PA 1997, p. 582-588.16. ASTM D 5442-93 Standard Test MeAod for Analysis of Petroleim
Waxes by Gas Chromatography. Annual Book of ASTM Standards. ASTM
Philadelphia PA 1994, p. 579-584.17. Toth Т., Garay F. J. High Resolut Chromat. 1994, v. 17, p. 177.18. Castillo М., Pereira A.S. et al. Trends in anal. chem. 1999, v. 18, p. 26.19. Ulberth F., Gabemig R. J. Chromat. 1997, v. 773, p. 233.20. Aichholz R. et al. J. High Resolut Chromat. 1998, v. 21, p. 152.21. Huang A.S. et al. J. Agric. Food Chem. 1995, v. 43, p. 1834.22. Pereira R. et al. J. High Resolut Chromat. 1998, v. 21, p. 513.23. Pereira R. et al. J. High Resolut Chromat. 1998, v. 21, p. 513.24. Elias V.O. et al. J. High Resolut Chromat. 1998, v. 21, p. 87.25. Pereira A.S., Aquino Neto F.P. J. Chromat. Sci. 2000, v. 38, p. 369.26. Aichholz R., Lorbeer E. J. Chromat. Sci. 1999, v. 855, p. 601-615.27. Park J.R., Lee D.-S. Bull. Korean. Chem. Soc. 2003, v. 24, N 4, p. 527.28. Mra6nova R., Kubinec R., Sojak L. et al. Petroleum and Coal. 2001,
V. 43, p. 17.29. Sokolov D.N. J. Cromatog. 1970, v. 47, p. 320.30. Barett J.W Ger. Patent 1137222 (April 11. 1963).31. De Boer F.E. Nature, 1960, v. 185, p. 915.340
IJIABA 10 Литература32. Barett J.W., Giles C.S. US Pat. kl 55-67 № 3237380 (1.III. 1966).33. Euston C.B. Martin A.J. Chem. Eng. News 1961, v. 39, p. 46.34. Союлов Д.Н., Бойдаровцева M.A., Бакин H.A. Изв. АН СССР, сер.
хим. 1968, вып. 6, с. 1396.35. Соколов Д.Н., Сойфер Л.М. Зав. лаб. 1969, т. 35, с. 1025.36. Sokolov D.N., Vakin N.A. J. Chromatog. Sci. 1972, v. 10, p. 417.37. Becker H.J. Z. anal. chem. 1969, Bd.247, s. 301.38. Genty C. et al. Analyt. Chem. 1971, v. 45, p. 235.39. Cremer E., Deutscher F. Aluminium (BRD) 1970, v. 46, p. 174.40. Zvarova T.S., Zvara I. J. Chromatog. 1969, v. 44, p. 604.41. Zvarova T.S., Zvara 1. J. Chromatog. 1970, v. 49, p. 290.
Яшин Я.И., Яшин Е.Я., Яшин А.Я. Газовая хроматографияГлава 11. ХИРАЛЬНАЯ ГАЗОВАЯ ХРОМАТОГРАФИЯ,
РАЗДЕЛЕНИЕ ЭНАНТИОМЕРОВ11.1. Краткая история.Впервые удалось разделить рацематы (смесь оптических изомеров)
методом газовой хроматографии в 1966 г. Гиль-Ав Е. с сотрудниками [1].
Перед этим было много попыток разделить смесь оптических изомеров
хроматографическими методами.Летучие производные смеси рацемических аминокислот, в частности
оптические изомеры эфиров Н-трифторацетил-0,Ь-аминокислоты были
разделены на эффективной кштилляроной колонке длиной 100 м с нане¬
сенной на внутреннюю поверхность хиральной фазы N-трифторацетил-
L-изолейцин. Авторы считали, что причиной хиральной селективности
(дискриминации) является образование водородной связи между амидны¬
ми группами и карбонильными атомами кислорода хиральной неподвиж¬
ной фазы и разделяемыми оптическими изомерами.В 1967 г Гиль-Ав Е. и Фейбаш Б. [2] также впервые показали возмож¬
ность полупрепаративного разделения энантиомеров аминокислот мето¬
дом газовой хроматографии на насадочных колонках. Впервые оптические
изомеры методом лигандно-обменной жидкостной хроматографии были
разделены В.А. Даванковым в 1968 г. [3,4]. В настоящее время больше все¬
го разделений рацематов производится жидкостной хроматографией.Через 30 лет согласно базы данных Chirbase зарегистрировано
2200 разделений энантиомеров, включая 5500 основных хиральных
соединений, зарегистрированных в 2200 публикациях, выполненных
газовой хроматографией [5]. Статистика данных по жидкостной хро¬
матографии Chirbase в 2000 г. такова; число разных хиральных разде¬
лений 69106, число разделяемых соединений 19102, число хиральных
сорбентов 1157 [6]. В настоящее время число новых хиральных разде¬
лений 3000-5000 за каждые четыре месяца. В настоящее время пред¬
ложено более 1400 хиральных стационарных фаз, более 200 из них
получили коммерческую реализацию. Это показывает, что это направ¬
ление является одним из актуальных в настоящее время. Появилось
много книг [7-12] и обзоров [6,13-17], созданы специализированные
международные журналы; Tetrahedron;Asymmetry, Enantiomer, Chiral-
ity и Moleculor Asymmetry.11.2. Молекулярная структура и хиральность.Если атомы C,N,P,S и Si образуют тетраэдрические структуры с раз¬
ными заместителями, то у таких молекул будут оптические изомеры. Чаще
всего рассматривают углеродные структуры. Если у атома углерода три за¬
местителя разной природы, то в этом случае у такого углерода создается
асимметрический или хиральный центр. При химическом синтезе возник¬342
ГЛАВА It Хиральная газовая хроматография, разделение энантиномеровнет энантиомерная смесь 50% левого и 50% правого изомера. В живых си¬
стемах используются только оптические изомеры одной природы, например
в человеческом организме усваиваются толью левые (L) аминокислоты.Физико-химические свойства оптических изомеров практически одина¬
ковые. Однако физиологические свойства некоторых оптических изомеров
сильно различаются по запаху и вкусу. Особенно сильные фармакологиче¬
ские различия имеют некоторые лекарственные препараты - рацематы.Многие лекарственные препараты, полученные синтетически, явля¬
ются рацемическими соединениями. Тридцать лет назад считалось, что
если один изомер физиологически активен, то второй энантиомер - не
активный, не будет оказывать никакого влияния на организм. Во многих
случаях так и происходит. Однако, зафиксированы случаи, когда один из
энантиомеров может оказывать вредное воздействие.Наиболее ярким проявлением ташго действия является массовое упо¬
требление в Германии успокаивающего, снотворного и снимающего тош¬
ноту у беременных лекарства талидомида, существующего в виде двух изо¬
меров [18]. В результате употребления этого лекарства беременными жен¬
щинами родились десятки тысяч детей с серьезными уродствами. Позднее
в 1979 г. определили, что (R) - изомер талидомида обладает тератогенным
действием. Наблюдались и другие примеры нежелательных воздействий
рацемических лекарств.В настоящее время около 40% мировой продукции фармацевтиче¬
ской промышленности реализуется в виде индивидуальных оптических
изомеров и их доля продолжает расти. В 2001 г. специалистам США и
Японии присуждена Нобелевская премия по химии энантиселективного
катализа. Получила развитие препаративная хиральная хроматография.
В последние годы созданы препаративные системы, на которых можно
получить более 100 т в год оптических изомеров лекарственных препара¬
тов. Решающую роль в революционных успехах хиральных технологий
ифает хроматография, позволяющая проводить измерения энантиомер-
ной чистоты.11.3. Природа энантиомерной селе1стивности.Хиральная селективность, хиральное распознавание энантиомеров
основано на трехточечном взаимодействии хирального селектора на поверх¬
ности сорбента с одним из оптических изомеров при пространственной не¬
возможности реализации такого же числа контактов со вторым оптическим
изомером. В.А. Даванковым было показано, что при адсорбции на плоской
поверхности, например на поверхности графитированной термической сажи
достаточно двухточечного взаимодействия. Это новое для стереохимии яв¬
ление удостоено диплома открытия (диплом №372, 1990 г.).343
Ягтн Я.И.. Яшин Е.Я.. Яшин Л.Я. Гязовая хроматографияВ работе [19] обнаружена необычная кинетическая энантиоселектив-
ность. В зависимости от сшрости потока порядок элюирования может из¬
меняться на противоположный. В газовой хроматографии применяются
три вида стационарных фаз для хиральных взаимодействий [13,20]:- разделение на хиральных производных аминокислот за счет водо¬
родной связи [1,2];- разделение за счет координационных взаимодействий на хиральных
комплексах металлов [15];- разделение за счет различной адсорбции в порах производных цикло¬
декстрина [8].На первых этапах хиральные вещества использовались в растворах
малолетучих жидких фаз таких, как полисилоксаны, сквалан и др. В на¬
стоящее время хиральные селекторы химически прививают к полисилок-
санам. Коммерчески выпускаются следующие подобные сорбенты chirasil-
type с повышенной термостабильностью и эффективностью:- Chirasil-Val с диамидом ванилина в качестве селектора;- Chirasil-Metal с металл координационными соединениями;- Chirasil-Dex с модифицированными циклодекстринами;- Chirasil- Crown с хиральными краун-соединениями [21,22].11.4. Области применения газовой хиральной хроматографии.Кроме упомянутых выше хиральных разделений в фармацевтике, в на¬
стоящее время, это одно из основных применений газовой хромотографии.
Хиральная газовая хроматография находит применение в пищевой про¬
мышленности, в археологии и в разных научных исследованиях.В последние годы возникло новое направление - энантиоселектив-
ный анализ компонентов пищи. По соотношению оптических изомеров
аминокислот, оксикислот и некоторых иных соединений можно однознач¬
но установить, является ли данный продукт натуральным или содержит
синтетические имитаторы и добавки. Энантиомерный анализ показал,
что микроволновая обработка пищевых продуктов, в отличие от жесткой
термической, не приводит к рацемизации аминокислот. Однако все мо¬
лочные продукты, подвергнутые процессам брожения, содержат немало
(нетоксичных) D-аланина и D- аспарагиновой кислоты - продуктов жиз¬
недеятельности молочнокислых бактерий. Со временем L- аминокислоты
белков человека и животных приходят в D-форму По соотношению D/L
форм можно судить о сроках нахождения останков. Эта методика точнее
общеизвестного углеродного метода, используемого в археологии.В настоящее время в научных целях изучается влияние разных параме¬
тров на превращения D- и -L- форм друг в друга, в частности температуры.344
ГЛАВА 11 ЛитератураЛитература.1. Gil-Av Е., Feibush В., Charles-Sigler R. Tetrahedron lett. 1966, p. 1009.2. Gil-Av E„ Feibush B. Tetrahed. lett. 1967, p.3345.3. Рогожин C.B., Даванюв B.A. Автор, свид.308635 СССР заявл. 01.07.68.
Pat 1256007 USA.4. Даванков В.А., Рогожин С.В. Докл. АН СССР 1970, т.193, с.94.5. Schuring V. Enantiomer, 1996, v.l, p. 139.6. Piras P., Roussel C., Pierrot-Sanders J. J.Chromat. 2001, v.906, p.443.7. K6nig W.A. Time Practice of Enantiomer Separation by Capillary Gas
Chromatography, HOthig, Heidelberg, 1987.8. K6nig W.A., Enantioselective Gas Chromatography with Modified Cy-
clodextrines, Hflthig, Heidelberg, 1992.9. Jinno К (Ed) Chromatographic Separations Based on Molecular Recog¬
nition, Wiley, New York, 1996.10. Аллепмарк С. Хроматографическое разделение энантиомеров. Пер.
с англ. д.х.н. Курганова А.А. Москва Издательство «Мир» 1991, 268 с.11. Stevenson D.,Wilson D (Eds), Chiral Separations, Plenum Press, New
York, 1988.12. Zief М., Crane L.J. (Eds) Chromatographic Chiral Separation, Marcel
Dekker, New York, 1988.13. Schurig V. J.Chromat. 2001, v.906, p.275.14. Lipkowitz K.B. J.Chromat. 2001, v.906, p.417.15. Schurig V. J.Chromat. 2002, v.965, p.315.16. Maier N.M., Franco P., Lindner W. J.Chromat. 2001, v.906, p.3.17. Spanik I., Krapcik J. Chem. Listy, 2001, v.95, p.86.18. Blaschke G„ Kraft H.P, Markgraf H. Chem. Ber. 1980, v.ll3, p.2318.19. Курганов A.A., Пономарева T.M., Даванков В.А. Докл. АН СССР,
1987, т.293, С.623.20. Schreier Р., Bemreuther А., Huffer М. Analysis of Chiral Organic
Molecules, Walter de Gruyter, Berlin, 1995.21. Zhon X.C., et al. J. Hi^ Resolut. Chromatog. 1996, v.l9, p.643.22. Zhon X.C., Yan H., Chen Y.Y. et al. J.Chromat. 1996, v.753, p.269.345
Яшт ЯЖ, Яшин Е.Я.. Яшин А.Я. Газовая хроматографияГлава 12. АППАРАТУРА ДЛЯ ГАЗОВОЙ
ХРОМАТОГРАФИИ.12.1. Введение.Широкое и успешное использование хроматографических методов в
контроле загрязнений окружающей среды, пищевых продуктов и напит¬
ков, клинических анализах, в технологическом контроле в разных отрас¬
лях промышленности, в научно-технических исследованиях, было бы не¬
возможно без современной хроматографической аппаратуры.Приборное оформление основных методов хроматографии (газовой, жид-
шстной, ионной и других видов) достигло очень высокого уровня. В современ¬
ных хроматографах используются последние достижения микроэлектроники,
пневматики, теплотехники, автоматики, микроюмпьютерной техники, про¬
граммного обеспечения. Все процессы автоматизированы. В лабораторных
хроматографах оператор тольш вводит пробу и на выходе получает полный
протошл с результатами анализа. В промышленных хроматографах на потоке
анализ осуществляется автоматически без участия человека.В настоящее время хроматографичесше приборостроение - отдельная
отрасль промышленного производства (приборы, вспомогательное обору¬
дование, колонки, программное обеспечение) объемом в стоимостном вы¬
ражении более 10 млрд. долларов США.12.2.Функциональная схема газового хроматографа.Во всех аналитических хроматографах применяют проявительный
вариант хроматографии, в котором газ-носитель непрерывно пропускает¬
ся через хроматографическую шлонку. Расход газа-носителя создается за
счет перепада давления между входом и выходом колонки.Схема современного газового хроматографа приведена на рис. 12.1.Для создания перепада давления через колонку хроматограф подсо¬
единяют к источнику со сжатым газом 1 (баллоны со сжатым газом, гене¬
раторы газов). Для стабильной работы хроматографа поток газа-носителя
через колонку должен проходить с постоянной и определенной сшростью,
поэтому на входе в колонку на линии газа-носителя устанавливают регу¬
лятор и стабилизатор расхода газа-носителя 2 и измеритель расхода газа 3.
Если газ-носитель загрязнен нежелательными примесями, то в этом случае
устанавливается еще фильтр 4. Таким образом, на входе в колонку под¬
ключается ряд устройств, часто объединяемых в один блок (блок подго¬
товки газа или панель подготовки газов), назначение которого - установка,
стабилизация, измерение и очистка потока газа-носителя. Перед входом
в колонку устанавливается устройство для ввода анализируемой пробы в
колонку, так называемый дозатор-испаритель 5. Обычно анализируемую
пробу вводят микрошприцем 8 через самозатекающее термостойкое рези-346
тЛВА 12Аппаратура для газовой хроматографииf * / 1-^ /— L—1—/ I ПнэдTOп1 111}TemwfypwмтИнтвфатор(TKepmtvчистогогтмцрвтар•рйорсда■оадухаРис. 12.1. Функциональная схема газового хроматографа.новое уплотаение в дозаторе, газовые пробы вводят дозирующим шести¬
ходовым краном. Для автоматизации работы хроматографов используются
автосамплеры до 120 проб.Анализируемая проба, введенная в дозатор, захватывается потоком
газа-носителя (если анализируемая проба - жидкость, то ее предваритель¬
но за счет нагрева в дозаторе-испарителе переводят в парообразное состо¬
яние) и направляется в хроматографическую колонку 6. За счет различной
сорбируемости компоненты смеси будут с разной скоростью продвигаться
по колонке. Вещества, которые сорбируются слабо, ^ут продвигаться по
колонке медленнее. Если выбран достаточно селективный сорбент и по¬
добраны оптимальные условия, то на выходе колонки компоненты смеси
будут полностью разделены. Детектор 11 зарегистрирует присутствие раз¬
деленных компонентов в газе-носителе. Эти сигналы в случае необходи¬
мости усиливаются (усилитель 13), аналоговый сигнал преобразуется в
цифровой и подается на ЭВМ. На экране дисплея сигналы регистрируются
в виде выходных кривых (или пиков). Ранее аналоговый сигнал подавался
на самопишущий прибор. Для обеспечения стабильного режима работы
детектора используется блок питания детектора 12.Сорбируемость веществ зависит от температуры.Для исключения влияния колебания температуры на параметры удер¬
живания и результаты разделения, колонку помещают в специальную ка-347
Яшин ЯМ, Яшин Е.Я.. Яшин Л.Я. Газовая хроматографиямеру-термостат, температура которой устанавливается и поддерживается
терморегулятором 9. В режиме программирования температуры колонки в
процессе разделения температура может изменяться по определенной про¬
грамме с помощью блока программирования температуры 10.Для работы пламенно-ионизационного детектора необходимы допол¬
нительные газы: особочистые водород и воздух.Площадь или высота пика пропорциональны количеству или концен¬
трации компонента в смеси. Площадь и высота пика измеряется и реги¬
стрируется на экране ПЭВМ. При необходимости результаты могут быть
распечатаны.Перед хроматографическим анализом необходимо провести следую¬
щие операции на приборе: открыть вентиль баллона со сжатым газом и
установить по манометру или специальному измерителю определенный
расход газа-носителя; включить питание детектора; установить необходи¬
мую температуру в термостате колонок; включить ПЭВМ; после выхода
прибора на устойчивый режим (через 15-30 мин.) микрошприцем (или
краном) отобрать и ввести в дозатор-испаритель анализируемую пробу.
Все дальнейшие операции проходят без участия оператора: компоненты
пробы разделяются на колонке, регистрируются детектором, записыва¬
ются на дисплее ПЭВМ или диаграммной ленте вторичного прибора. По
градуировочной зависимости в памяти ПЭВМ рассчитывается концентра¬
ция анализируемого вещества. На принтере можно сразу получить полный
протокол - хроматограмму с распечатанной рядом таблицей концентраций
разделенных компонентов.12.3. Источники сжатых газов.Для работы на газовых хроматографах необходимы источники сжатых
газов. Это могут быть:- баллоны со сжатыми газами разного объема (25 л, 60 л и др.) и на
разные давления (до 150 атм);- лабораторные или промышленные газовые линии со сжатыми газами;- генераторы разных газов, в частности генераторы водорода, азота и
особо чистого воздуха.Тип газа определяется в основном типом детектора и аналитической
колонки. Например, при работе с катарометром лучше использовать гелий,
т.к. в этом случае обеспечивается наибольшее различие теплопроводно¬
стей; гелий чаще всего применяют с капиллярными колонками из-за более
быстрого массообмена.В таблице 12.1 приведен перечень рекомендуемых газов для работы с
разными детекторами.348
[ШВА 12Аппаратура для газовой хроматографииТаблица 12.1.ДетекторТип газа-носителяДополнительные газыПИД (или ДИП)азот, аргон, гелийводород,воздухДТПгелий, азот, аргонФИДгелий, азот, аргон, чистый
воздух ТИДгелий, азотводород,воздухПФДгелий, азотводород,воздухЭЗДазот, аргонаргон + 5% метана (только в
некоторых случаях)Примечание. Детекторы: ПИД - пламенно-ионизационный; ДТП - по
теплопроводности (катарометр); ФИД - фотоионизационный; ТИД - термо¬
ионный; ПФД - пламеннофотометрический; ЭЗД - элекгронозахватный.12.3.1. Генераторы чистого водорода.Для работы ПИД“ необходим водород высокой степени чистоты, осо¬
бенно недопустимо присутствие органических примесей, увеличивающих
значение фонового тока. Ранее для работы с ПИД°“ использовали водород,
находящийся в баллонах под давлением. Баллоны создавали опасность по¬
жара или взрыва в случае утечки больших количеств водорода. В настоящее
время чаще всего используют портативные генераторы водорода. Водород
в них получается за счет электролитического разложения дистиллирован¬
ной воды. Получаемый водород очищается, самая лучшая очистка через
палладиевую мембрану, в этом случае можно получить водород чище, чем
99,99999%, т.к. через такие мембраны проникает только водород и его изо¬
топы. Генераторы водорода совершенно безопасны, в них имеются сигна¬
лизаторы уровня воды, утечки водорода и других критических режимов
работы. Генераторы водорода удобны в эксплуатации, для их работы не¬
обходима только дистиллированная вода, потребляемая мощность обычно
50-150 Вт. Одной заправки генератора водорода дистиллированной водой
достаточно для получения более 500-700 литров водорода, что хватает на
двухнедельную работу газового хроматографа. Генераторы водорода выпу¬
скаются разной производительности от 6 до 12 л/час, что позволяет одним
прибором обеспечить работу от одного до четырех хроматографов.Генераторы водорода позволяют полностью отказаться от газов в бал¬
лонах, при использовании которых необходимо монтировать в лаборато¬
рии специальную газовую линию, а баллоны размещать вне помещения.
Генераторы водорода небольшого размера и их можно размещать непо¬
средственно рядом с прибором.349
Яшин Я.И., Яишн Е.Я., Яшин А.Я.Газовом хроматографияФункциональная схема генератора водорода приведена на рис. 12.2.Рис. 12.2 Пневмогидравлическая схема генератора водорода
ХА-Элдис/ Химавтоматика-Элдис
12.3.2. Генераторы чистого азота.Азот, наряду с гелием, часто используется в качестве газа-носителя.
Технический азот в баллонах (ГОСТ 9293-74) может содержать до 1-3%
кислорода. Кислород при высоких температурах колонки может окислять
неподвижные жидкие фазы и сорбенты и изменять их сорбционные харак¬
теристики. Кроме того, технический азот нельзя использовать с ЭЗД. При
работе с ЭЗД применяется азот особой чистоты (ТУ 6-21-39-79), который350
ГЛАВА 12 Аппаратура для газовой хроматографиинедоступен во многих регионах нашей страны. Поэтому нашли примене¬
ние разные блоки доочистки технического азота. Принцип работы таких
устройств основан на реакции окисления чистой меди кислородом при
температурах 150-160°С. Регенерация окиси меди - восстановление до чи¬
стой меди - происходит в потоке водорода при температуре 400°С. Чистота
азота может достигать 99,9999%. Продолжительность работы устройств
доочистки до регенерации около 200 часов.В последние десятилетия разработаны генераторы азота, в которых
азот особой чистоты получают из атмосферного воздуха.Принцип работы таких генераторов основан на способности специ¬
альных многослойных полимерных мембран отделять азот от кислорода.
Воздушный компрессор подает под давлением очищенный от влаги и ор¬
ганических примесей воздух в каскад специальных мембран. После двух¬
трех каскадов таких блоков получают азот чистотой 99,5%. Оставшееся
количество кислорода удаляют специальным катализатором. Генератор
азота, кроме чистого азота, выдает и чистый воздух, обогащенный кисло¬
родом. Это важно и выгодно при работе с ПИД"“.12.3.3. Генераторы особо чистого воздуха.Очищенный воздух необходим для поддержания горения водорода в
горелке ПИД*. В обычном воздухе есть органические примеси, аэрозоли,
которые увеличивают общий фоновый ионный ток и уровень флуктуаций.
Генератор особо чистого воздуха представляет собой обычный воздушный
безмасляный компрессор с набором фильтров для улавливания влаги, ор¬
ганических примесей и аэрозолей.12.4. Екпок подготовки газа.Как уже указывалось ранее, назначение блока подготовки газов
(БПГ) - это установка, регулирование, стабилизация, очистка и изме¬
рение газовых потоков; газа-носителя, водорода, воздуха и других до¬
полнительных газовых потоков, необходимых для работы некоторых
детекторов. Поддержание стабильного чистого потока газа-носителя
необходимо как для количественного, так и для качественного анализа.
Воспроизводимые значения параметров удерживания (время и объем
удерживания) и параметров пика (высота, площадь) можно получить
только при стабильных потоках газа-носителя. Колебания расходов
газа-носителя и других газов могут влиять на шумы (флуктуации) нуле¬
вой линии детектирующих систем.Основные элементы БПГ; дроссель, регулятор давления (рис. 12.3),
регулятор расхода (рис. 12.4), измеритель расхода (рис. 12.5), фильтры на
входе в колонку. Дроссель изменяет расход газа путём изменения сопро¬
тивления канала, по которому проходит газ [1, 2].351
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я.Газовая хроматографияРегулятор давления - стабилизирует давление на входе в колонку
при возможных внешних колебаниях давления газа. Специальная мембра¬
на в регуляторе давления воспринимает изменение давления газа и пере¬
даёт соответствуюшее смещению исполнительному механизму.Регулятор расхода. В режиме программирования температуры термо¬
стата сопротивление колонки повышается, а расход падает. В этом случае
для сохранения постоянного расхода в колонке используется регулятор
расхода. При падении расхода в связи с увеличением сопротивления в ко¬
лонке регулятор расхода повышает входное давление настолько, чтобы вос¬
становился первоначальный расход газа-носителя. Расход газов измеряют
мыльнопенным измерителем, реометром, ротаметром или специальным
электронным измерителем расхода на принципе теплового расходомера.
Фильтры для очистки газа-носителя заполняют адсорбентами (активиро¬
ванный уголь, силикагель, цеолит).Рис. 12.3 Схема регулятора давления газа: I-задающий элемент (винт),2 -пружина задающего элемента, 3 - мембрана, 4 - исполни¬
тельный элемент (дроссель), 5 - пружина дросселяРис. 12.4 Схема регулятора расхода газа: А - входная камера, Б -
промежуточная камера. В- выходная камера. 1 - задаю¬
щий элемент, 2 - установочный дроссель, 3 -мембрана, 4 -
регулирующий дроссель.352
ГЛАВА 12Аппаратура для газовой хроматографии
fРис. 12.5 Измерители расхода газа: а - мыльно-пенный, б - реометр,
в - ротаметр. Стрелками показано направление потока
газа-носителя, пунктиром показано исходное положение
мыльной пленки (а), уровня рабочей жидкости (б) и по¬
плавка (в).В современных хроматографах используются БПГ с электронным за¬
данием и управлением расходов газов.12.5 Дозирующие устройства.Дозаторы предназначены для ввода в хроматографическую колонку
точно выбранного количества анализируемой пробы. Общие требования к
дозаторам: воспроизводимость ввода пробы (желательно ниже 1-2%), со¬
хранение состава исходной анализируемой пробы. Кроме того ввод пробы
должен происходить быстро, без сильного размывания исходной смеси.
Различают дозаторы для ввода газообразных, жидких и твёрдых проб. Для
быстрого ввода газообразных проб используют микрошприцы, мембран¬
ные краны (чаще всего в автоматических промышленных хроматографах),
золотниковые, поршневые и вращающиеся поворотные краны. В совре¬
менных лабораторных хроматографах чаще всего применяются поворот¬
ные краны. Такой кран состоит из неподвижного корпуса со штуцерами
для подвода газа-носителя и анализируемого газа и сверху движущейся по¬
воротной втулки с каналами, соединяющими линии газа-носителя и анали¬
зируемого газа. На корпусе устанавливается трубка-доза для точного ввода
пробы. Корпус с вращающейся втулкой сильно прижаты друг к другу, их
контактирующие поверхности тщательно отполированы и при повороте
должно быть плавное скольжение этих поверхностей относительно друг
друга. Такие краны могут быть 6, 8, 10 и даже 14-ходовые (или портовые).
Чаще всего применяются для дозирования 6-ходовые краны. Схема ввода
газовой пробы таким краном показана на рис. 12.6. Поворот крана может
проводиться вручную или автоматически, электрическим или пневмати¬353
Яшин ЯМ. Яшт Е.Я, Яшин АЖГазовая хроматографияческим приводом. При изготовлении крана используются следующие ма¬
териалы: нержавеющая сталь, сплавы хостеллой, тефлон, наполненный
тефлон, веспел и др.Жидкие пробы вводятся в газовые хроматографы микрошприцами
на 1, 5, 10, 50 мкл через термостойше резиновое уплотнение испарителя
(рис. 12.7). Наиболее распространённый способ введения жидких проб -
микрошприцы. Величина дозируемой пробы легко регулируется в широ¬
ких диапазонах. Общий вид таких микрошприцев изображён на рис. 12.8.
Эти шприцы сравнительно недороги и удобны для работы.Рис. 12.6 Ввод краном. S-^- проба, G —»- газ-носитель, а - заполне¬
ние пробоотборной петли крана пробой, б - ввод пробы S в
потоке газа-носителя.Рис. 12.7 Испаритель: 1 -корпус, 2 - канал испарения пробы, 3 - тер¬
мостойкое уплотнение, 4 - прижимная гайка.354
ГЛАВА 12 Аппаратура для газовой xvmiamoepadmu Рис. 12.8 МикрошпрщМикрошприцы - самые простые и чаще всего употребляемые дозаторы
жидкостей в ГХ. Мшчюшприцы представляют собой стекляшше Щ1лш!дры с
поршнем из нержавеющей стали, который индивидуально подогаан к щшин-
дру шприца. Поэтому поршни - стержни несменные, упрочненные, износо¬
стойкие. Нашнечники шприцев заканчиваются игаами (вклеенные или смен¬
ные). Микрошприцы вьшускаются объемами 1-10 мкл, 2,5-5 мкл, 5-10 мкл,0,5-5 мкл. Мшфошприцы - средства измереюм, их обьемы калиброванные.
Микрошприцы объемами 0,5-1 мкл имеют сменные игаы и поршни. Острие
игаы чаще всего сношенное под угаом 10-12“ или игаы с боковым отверстием
для того, чтобы не вырезать резину при прокалывании.Для предотвращения изгибания и выдергивания поршня из цилиндра
используются микрошприцы со специальными направляющими. Для мно¬
гократного ввода одной и той же пробы используются адаптеры-ограничи-
тели. В этом случае улучшается воспроизводимость дозирования.Разработан широкий ассортимент специальных микрошприцев:- цифровые шприцы с электронным устройством для автоматического
дозирования;- микрошприцы для дозирования агрессивных сред;- микрошприцы с продувкой;- микрошприцы для многократного дозирования заданного объема;- многоканальные дозаторы из шести параллельно соединенных ми¬
крошприцев для ввода в луночные планшеты.Для дозирования газов выпускаются шприцы объемом 1-100 мл с
поршнями из тефлона. Для отбора больших проб воздуха используются
шприцы объемом 0,5-2 л из прозрачного акрила.Для автоматического ввода жидких проб применяют специальные
поршневые, вращающиеся и золотниковые дозирующие краны. В порш¬
невом кране движущийся поршень имеет сбоку кольцевую канавку, глу¬
бина которой определяет объём введённой пробы. Поршень двигается
между полостью, промываемой непрерывным потоком анализируемого
вещества и нагретым испарителем.Автосамплеры- это автоматические дозаторы жидких проб в газовые
хроматографы, позволяющие увеличить производительность и точность
анализов. Работа автосамплеров управляется как отдельным блоком управ¬
ления, как и с помощью персонального компьютера по определенной про¬
грамме. Пробы отбираются микро шприцами объемом от 1 до 100 мкл из35523-
Яшин Я.И., Яшин Е.Я.. Яшин А.Я.Газовая хроматографияПробирок объемом 2-2,5 мл. Пробирки устанавливаются на вращающейся
платформе (поддоне) от 8 до 120 пгг.Последовательность ввода пробы программируется и производится
автоматически до 15 шагов, включая ввод проб, промывку до и после ввода
пробы, ввод пробы с внутренним стандартом.Скорость дозирования и ввода пробы может варьироваться.Твёрдые пробы в основном вводят в пиролизных устройствах через
специальные шлюзы.12.6 Колонки для газовых хроматографовКолонки в газовой хроматографии подразделяются на препаративные
насадочные, аналитические насадочные, микронасадочные и капилляр¬
ные. В таблице 12 приведены характеристики этих колонок.Таблица 12.2Типы колонокВнутренний диаметр колонок,
ммДлина колонки,
мАналитические насадочные2-40,2-6Микронасадочные0,5-10,5-3Капиллярные широкого
диаметра 0,3-0,5310-60Капиллярные0,2-0,35-100Узкие капиллярные0,05-0,25-100Поликапиллярные0,040,2; 1В насадочных, микронасадочных колонках сорбент находится внутри
трубки и имеет форму цилиндра. Набивка должна быть плотной и одно¬
родной, без пустот. Чем плотнее и однороднее набивка, тем меньше раз¬
мывание полос и больше эффективность колонки.В капиллярных колонках слой сорбента наносится на внутреннюю по¬
верхность капилляра в виде слоя жидкой неподвижной фазы или в виде
слоя адсорбента.На рис 12.9 изображен наиболее распространенный тип колонок.По форме насадочные колонки бывают прямые, U-образные, W- об¬
разные и спиральные с разным радиусом кривизны.Прямые и U- образные насадочные колонки заполняются сорбентом
легко и наиболее плотно без специальных приспособлений. W- образные
и спиральные колонки заполняются под давлением на входе, либо с вакуу¬
мом на выходе из колонки.356
ГЛАВА 12 Аппаратура для газовой хроматографииРис.12.9 Спиральные колонки.На спиральных колонках при большом радиусе кривизны витков появ¬
ляется дополнительное размывание, связанное с неоднородностью скоро¬
стей по сечению. Сопротивление потоку у ближней (к центру окружности)
стенки трубки меньше, чем у дальней стенки, так как пути прохождения
газовых потоков у ближней стенки меньше, чем у дальней.Материал насадочных колонок. Колонки изготавливаются из ме¬
талла (нержавеющая сталь, никель, медь), стекла, тефлона и других ма¬
териалов. Чаще всего в аналитической практике применяются колонки из
нержавеющей стали (для особо агрессивных смесей применяются колон¬
ки из никеля). Для разделения неустойчивых соединений (каталитически
разлагающихся при контакте с металлической поверхностью) используют
стеклянные и тефлоновые колонки. В частности, стеклянные колонки ши¬
роко применяются при анализе пестицидов.Материал капиллярных колонок. Капиллярные колонки изготавлива¬
лись из меди и латуни, затем стали использовать стеклянные колонки (бьша
предложена специальная лабораторная установка для вытягивания стеклян¬
ных капилляров из толстостенной стеклянной трубки с внешним диаметром
6-10 мм), колонки из нержавеющей стали и позднее с 1980 кварцевые капил¬
лярные колонки, которые имеют наиболее инертную поверхность. Кварце¬
вые капилляры для предания гибкости и прочности с внешней поверхности
покрываются тонким слоем высокотемпературным полиамидным лаком (до
350®С) или слоем алюминия. Кварцевые капиллярные колонки со слоем лака
допускают изгиб до 8-10 мм. В последние годы появился вновь интерес к
металлическим капиллярным колонкам, но с инертной (пассивированной)
внутренней поверхностью.12.7 Термостаты.Сорбируемость разделяемых соединений очень сильно зависит от тем¬
пературы (см. главу 2), поэтому хроматографические колонки помещают в
специальные камеры-термостаты, в которых температура поддерживается
с высокой точностью.357
Яшин Я.И., Яшин Е.Я., Яшин А.Я. Газовая хроматографияОбычно время удерживания в ГХ уменьшается в два раза при повы¬
шении температуры на 15-20°С [1].Для газовых хроматографов конструируются термостаты обьемом от 3
до 22 л с принудительной вентиляцией воздуха. В таких термостатах на¬
ходится нагреватель (чаще всего спиральный), через который с помощью
вентилятора с большой скоростью непрерывно продувается воздух. Воздух
отбирает тепло от нагревателей и передает стенкам термостата и хромато¬
графическим колонкам, находящимся внутри термостатов. При правильном
теплообмене постоянная температура в термостате устанавливается очень
быстро. Точность поддержания температуры в термостетах современных
хроматографов колеблется в пределах ±0,5-0,01 °С в одной точке.Разделение в ГХ может происходить либо при постоянной температу¬
ре (изотермический режим), либо в режиме постоянного повышения тем¬
пературы (режим программирования температуры колонки).За счет несовершенного перемешивания нагретого воздуха в термо¬
статах возможны высокие значения градиентов температуры по высоте
термостатов, в некоторых термостатах он достигает 1°С. При разных аб¬
солютных значениях заданной температуры термостата градиенты могут
изменяться. Это различие градиентов может ухудшать воспроизводимость
параметров удерживания в режиме программирования температуры.В последние годы возрос интерес к альтернативным методам нахрева ко¬
лонок в газовых хроматографах, в частности, непосредственный нагрев только
самой колонки, нагрев колонки ИКС - источниюм, микроволновым и дф [2-8].Высокие скорости программирования достигаются при нагреве только
колонки (до 1200 ®С/мин).Важной характеристикой современных термостатов стала скорость
охлаждения после подъема температуры в режиме программирования тем¬
пературы. В лучших моделях хроматографов колонки могут охлаждаться
от 300 ®С до 50 ®С за 4,5-6 минут.12.8 Детекторы для газовой хроматографииКлассификация детекторов. Всего для газовой хроматографии пред¬
ложено более 50 типов детектирующих систем [9, 10]. По общепринятой
классификации детекторы подразделяются на дифференциальные и ин¬
тегральные по форме зарегистрированного сигнала. Дифференциальные
детекторы измеряют мгновенное различие в концентрации вещества в по¬
токе газа-носителя. Хроматофамма зарегистрированная таким детектором
представляет собой ряд пиков, площадь которых пропорциональна коли¬
честву разделенных соединений. Интегральные детекторы измеряют сум¬
марные количества соединений, выходящих из колонки. Хроматограмма
в этом случае ступенчатая, высота ступеней пропорциональна количеству
соответствующих соединений.358
ГЛАВА 12 Аппаратура для газовой хроматографииВ зависимости от однократной или многократной регистрации моле¬
кул анализируемых соединений выделяют концентрационные и потоковые
детекторы [И]. В концентрационных детекторах сигнал пропорционален
концентрации соединения в подвижной фазе (элюенте). В этом случае
имеет место многократная регистрация молекул анализируемых соеди¬
нений. В потоковых (или массовых) детекторах сигнал пропорционален
количеству пробы компонента, достигаемый ячейки детектора в единицу
времени. В этом случае происходит только однократная регистрация.По селективности детекторы классифицируются на универсальные.
селективные и специфические. В универсальных детекторах регистри¬
руются все компоненты смеси, выходящие из колонки, за исключением
подвижной фазы. Селективные детекторы регистрируют определенные
группы соединений на выходе из шлонки. Специфические детекторы ре¬
гистрируют тольш один шмпонент или ограниченное число шмпонентов
с подобными химическими характеристиками.1. Предел детектирования или чувствительность.2. Линейность.3. Инерционность (постоянная времени, быстродействие).4. Стабильность (уровень щума или флуктуация, величина дрейфа ну¬
левой линии).5. Эффеетивный объем чувствительной ячейки.Чувствительность шнцентрационных детекторов определяется следу¬
ющим выражением:SvFАк=^ (12.1)где S- площадь пика, см^; у- шкала самописца, мв/см; - сшрость
газа-носителя, мл/с; q- шличество соединения, мг; F- сшрость движения
ленты самописца, см с.Размерность чувствительности в этом случае мВ.мг/мл.Чувствительность потошвых детекторов (размерность мВ,мг/с) равна:А„-^ (12.2)Метрологи в последние годы решмендуют определять предел детекти¬
рования. Для оценки минимально определяемой шнцентрации необходимо,
кроме чувствительности, знать уровень флуктуаций (шума) нулевой линии.
Минимальным сигналом, поддающимся измерению, обычно принято считать
сигнал, высота шторого в несшяьш раз (2-5) превышает уровень шумов 8359
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я.Г . =25Газовая хранатография(12.3)Величина предел детектирования определяет предельные воз¬
можности прибора.Под линейностью детекторов понимают диапазон концентраций, в
пределах шторых наблюдается линейность зависимости «сигнал-шнцен-
трация». Для определения величины линейности строят соответствующий
график. Обычно диапазон линейности от порога детектирования до шнцен-
траций, в которой уже наблюдается отклонение от линейности на 5-10%.Под инерционностью (быстродействием, постоянной времени) под¬
разумевается сшрость реагирования детектора на быстрое изменение юн-
центрации на выходе из колонки. Детектор должен иметь такое быстро¬
действие, чтобы при регистрации не искажать формы полосы соединения,
выходящего из колонки. В современных, особенно ионизационных детек¬
торах постоянная времени менее 0,1-0,01 сек. В некоторых катарометрах,
особенно устаревших конструкциях, постоянная времени может быть око¬
ло 1 сек и даже выше.Уровень шума нулевого сигнала детектора определяется кратшвремен-
ньши флуктуациями. Дрейф - это монотонное смещение нулевой линии.
Величину смещения оценивают в течение 1 часа. Обычно требования к этим
показателям ташвы: шум 0,5% рабочей шкалы и дрейф не более 3% в час.Быстродействие сильно зависит от величины эффективного объема
ячейки. В таблице 12.3 приведены технические характеристики детекто¬
ров, применяемых в современных газовых хроматографах.Таблица 12.3Технические характеристики основных детекторов для ГХ№НазваниеПД(S/N=2)лдцТипАнализируемые соединения1ПИД2x10 '2 гС/с10’-10*селектив.Органические соединения, иони¬
зируемые в пламени водорода2Д1114х10‘“г/мл10’универс.Соединения, отличающиеся по те¬
плопроводности от газа-носителя3ЭЗД1хЮ-'Ч/с10^-10''селектив.Регис1рирует галоидоорганиче¬
ские соединения и соединения,
способные к захвату электронов4ФИД2x10 г/с10’селектив.Соединения, ионизируемые УФ
излучением с потенциалом иони-
завд1и менее 10,7 эв или 11,7 эв.360
ГЛАВА 12Аппаратура для газовой хроматографииКаНазваниеПД(S/N=2)лдцТипАнализируемые соединенияТИД4x10-'’ rN/c
2x10-'^ гР/с10^селектив.Органические гетеросоединения,
имеющие атомы N и Р в молекулеПФД2x10 " rS/c
9x10 гР/с10^10»специф.S- и Р- содержащие соединенияАЭД1x10 '’ г/с
2x10 " г/суниверс.Соединения, имеющие в своем
составе элементы: (Н, С, S, N, Р,
С1, F, Вг и др.)мед1x10 " г/с
IxlOV/c10’универс.Любые соединения, с помощью
детектора можно проводить как
количественный, так и качествен¬
ный анализ.Примечание: ПИД - пламенноионизационный дете1стор; ДТП - детек¬
тор по теплопроводности; ЭЗД - детектор электронного захвата; ФИД -
фотоионизационный детектор; ТИД - термоионный детектор; ПФД - пла¬
меннофотометрический детектор; АЭД - атомноэмиссионный детектор;
мед - масспектрометрический детектор; ПД - предел детектирования;
ЛДЦ - линейный динамический диапазон.12.8.1 Пламенно-ионизационный детекторПламенно-ионизационный детектор (ПИД) - самый распространен¬
ный детектор в ГХ. Практически каждый газовый хроматограф оснащен
им. ПИД идеален по своим техническим характеристикам для определе¬
ния органических углеродсодержащих соединений.Однако, несмотря на его массовое использование, механизм его рабо¬
ты полностью не ясен до сих пор.ПИД предложен в самом начале развития газовой хроматографии и
хроматографического приборостроения. Благодаря, в частности, ему ГХ
стала развиваться невиданными темпами, особенно в нефтехимии, в кон¬
троле загрязнений окружающей среды и пр.В 2002 г. известный специалист в ГХ Л.Эттре написал знаменатель¬
ную статью о судьбе и роли ПИД в ГХ «Изобретение, развитие и триумф
пламенно-ионизационного детектора» (LC-GC Europe 2002, № 6, р. 1).ПИД был предложен и разработан в 1957-1958 гг. практически одно¬
временно в двух местах, удаленных друг от друга на несколько тысяч кило¬
метров: МакВильямом И. и Дьюаром Р. в Центральной исследовательской
лаборатории Новой Зеландии и Австралии [12] и Харлеем Дж., Нелем В и
Преториусом В. в Отделе физической химии Университета в Претории в361
Яшин Я.И.. Яшин Е.Я. Яшин А Я. Гтовая хроматографииЮжной Африке [13]. О приоритете до сих пор идут споры. Виктор Пре-
ториус в 1973 г. организовал Институт хроматографии и возглавлял его до
1989 гВпервые в промышленный газовый хроматограф (модель 154-с) ПИД
был введен фирмой Перкин-Эльмер в 1959 г. В 1959 г [14] и 1962 г МакВи-
льямом И. бьши получены патенты на ПИД в Австралии и Новой Зеландии
[15] и в США [16].В первые 15 лет после открытия ПИД было выпущено свыше 60000
этих детекторов. К 2008 г. общее число ПИД, выпущенных в составе газо¬
вых хроматографов, составляет более миллиона штук.Впервые в нашей стране ПИД как детектор для ГХ был сконструи¬
рован уже в 1959 г. в Дзержинском филиале ОКБА. Были выпущены от¬
дельные детекторы с блоками подготовки газов ДИП-1 и ДИП-2. В 1960-61
гг. выпущен первый промышленный газовый хроматограф РХ-1 с ПИДом
во взрывозащищенном исполнении, в 1962-63 гг. - первый лабораторный
газовый хроматограф Цвет-1 с ПИДом. Начиная с 1960 г. были опублико¬
ваны исследования ПИДа, включая его применение с капиллярными ко¬
лонками [17-22].При горении чистого водорода в ПИДе фоновый ток очень мал (порядка
110 '^- 110"‘^ А). При попадании в таше пламя примесей угаеводородов из
хроматографической шлонки ионовый ток сильно возрастает. Этот сигнал
усиливается электрометрическим усилителем и регистрируется вторичным
прибором. На этом основана работа ПИДа в ГХ. При этом достигается высо¬
кая чувствительность детектора (ПДК до 1 • 10 '^ г/с) при линейном диапазоне
семь порядшв. Чувствительность ПИДа слабо зависит от температуры, ско¬
рости потока водорода, газа-носителя и других параметров.Одно из преимуществ ПИДа - предсказуемость сигнала, т.к. он про¬
порционален числу атомов углерода в молекуле. Недаром ПИД считают
«счетчиком углеродных атомов» [23].ПИД разработан эмпирическим путем без знания природы образова¬
ния ионов, т.е. без знания основного механизма его работы.Не ясен бьш механизм ионизации. Хотя температура пламени водо¬
рода очень высокая, более 1200°С, это соответствует энергии тольш около
5 eV, в то время как для ионизации большинства органических соединений
необходима энергия более 10 eV. Было предположение, что в пламени во¬
дорода имеет место хемиионизация, но непонятно, за счет каких ионов.Основной вопрос - какие ионы образуются при горении углеводоро¬
дов в пламени чистого водорода - не был ясен в продолжении 40 лет. Ио-
нообразование в пламени было научной проблемой с 1950 г., когда начали
разрабатывать ракеты с различными топливами.362
IJtABA 12 Аппаратура для газовой хроматографииМеханизм работы ПИДа начал проясняться в последние годы, когда
научились экспериментально отсасывать ионы из пламени водорода в ион¬
ный источник масспекгрометра и расшифровывать их состав.В статьях [23-25] опубликованы обзоры по механизму работы пламен¬
но-ионизационного детектора. Основные реакции в ПИДе: крекинг, раз¬
ложение, окисление и хемиионизация. Образование ионов в ПИДе проис¬
ходит в основном за счет хемиионизации радикалом СНО', возникающим
из реакций радикалов СН' и О':С„Н_^-*пСН' +(m-n)H'пСн“ + пО' пСНО' (12.4)пСНО' — пСНО*+ пе-ПИД имеет разные сигналы к соединениям, содержащим гетероатомы. Для
оценки чувствительности ПИДа к разным классам соединений Штфнберг впер¬
вые ввел понятие эффективного числа атомов угаерода в молекулах (ECNi):Am М(12.5)S 1 sгде: - число атомов углерода в стандартном соединении; М. и - мо¬
лекулярные веса исследуемого и стандартного веществ; А. и А^ - площади
хроматографических пиков исследуемого и стандартного веществ.В работах [12, 13] были определены сигналы ПИД к углеводородам,
первичным спиртам, альдегидам, органическим кислотам, эфирам, аминам
и др. Сравнительно недавно вновь тщательно измерены сигналы ПИДа к
соединениям разных гомологических рядов, а также исследовано влияние
молекулярной структуры на сигнал ПИДа [24, 25].В таблице 12.4 приведены значения ECN для н.алканов, н.спиртов,
н.аминов, кетонов и эфиров.В таблице 12.5 приведены значения ECN для соединений, отличаю¬
щихся структурой, в частности бензола и его различных производных, взя¬
тые из последних работ [24, 25].По этим данным можно прогнозировать изменение чувствительности
ПИДа как среди членов гомологических рядов, так и соединений, отлича¬
ющихся природой функциональной группы.Общая схема ПИДа приведена на рис. 12.10.В ПИД одним из электродов служит горелка, второй электрод - кол¬
лектор располагается над горелкой. Малые токи (МО’’ - 10 ‘^ А) усилива¬
ются, т.к. шумы самого детектора малы. Из-за высокой чувствительности,
большого диапазона линейности ПИД стал наиболее распространенным
детектором. В таблице 12.6 приведены атомные инкременты для показа¬
ний ПИД к соединениям разных классов.363
Яшин Я.И.. Яшин Е.Я.. Яшин Л.Я.Газовом хроматографияГоа -шштем
Рис. 12.10 Общая схема ПИДаТаблица 12.4
Значения ECN для разных классов соединенийНазваниеЧисло атомов
угаеродаСоединениесравненияECNAECNCKO %
(ECN)123456н.ал1шнын-окган8С,7,83-0,170,78н-нонан9с,8,95-0,050,89н-декан10с,10,050,050,39н-тридекан13С,о13,080,080,50м-гексадекан16С.515,28-0,721,39н-гептадекан1716,40-0,603,12н.спиртыпропанол3с,2,37-0,630,89бутанол4с,3,24-0,760,58пентанол5с,4,56-0,440,64гексанол6с,5,41-0,590,22гептанол7с.6,15-0,851,89н-аминыбутиламин4с,3,20-0,82,74пентиламин5с,4,31-0,690,81гексиламин6с,5,36-0,640,19гептиламин7с,6,11-0,890,27364
тлвл 12Аппаратура для газовой хроматогроЛииНазваниеЧисло атомов
углеродаСоединениесравненияECNAECNСКО %
(ECN)123456кетоны2-бутанон4С,3,2-0,80,872-пентанон5с»4,11-0,890,422-гею:аион6С,„4,82-1,180,172-гептанон7С,о6,01-0,990,28эфирыметилгептаноат8с*6,74-1,260,35метилпентадека-ноат1614,39-1,610,89метилпальминоат1715,17-1,830,99метил стеарат19 с.17,48-1,520,49Таблица 12.5СоединениеЧисло атомов
углеродаECNДЕСКСКО %
(ECN)Ароматические углеводородыБензол65,89-0,110,33Толуол76,88-0,120,25Этилбензол87,56-0,440,39Пропилбензол98,22-0,782,00Бзтилбензол108,50-1,502,06Пептилбензол118,92-2,081,90Хлорбензол65,81-0,190,47Бромбензол65,63-0,370,85Иодбензол65,39-0,611,28Фенол65,78-0,221,45Анилин65,74-0,260,82Ацетофенон87,21-0,790,84Метилбензоат86,84-1,492,53Хлорированные алканы1-хлорбутан43,88-0,120,041- бромбутан43,81-0,190,491-иодбутан43,87-0,131,22365
Яшин Я.И.. Яшин Е.Я.. Яшин Л.Я.Гтовая хроматографииТаблица 12.6Атомные инкременты сигналов ПИДАтомТип атомаВклад в общий сигнал (эффективное
углеродное число)Алифатический1АроматическийОлефиновый0,95Ацетиленовый1,30КарбонильныйНитрильный0,30ОПростой эфир-1оПервичный спирт-0,60ОВторичный спирт-0,75ОТретичный спирт-0,25С1У алифатического
Зтлерода-0,12С1У олефинового
углерода+0,05На рис. 12.11 приведен поперечный разрез конструкции ПИДа, впер¬
вые разработанного и изготовленного в Дзержинском филиале ОКБА [22].На рис. 12.12 приведен поперечный разрез ПИДа, работающего на
переменном токе, также впервые предложенного в Дзержинском филиале
ОКБА и запатентованном [17].При конструировании этого типа ПИДа особое внимание было обра¬
щено на уменьшение емкости между электродами 3 и 5 детектора, потому
что указанная емкость создает реактивную проводимость между электро¬
дами, шунтирующую сопротивление, на котором возникает полезный
сигнал. С целью уменьшения емкости электроды детектора выполнены в
виде двух тонких стержней 3 и 5, расположенных на одной оси и закрытых
специальными экранами 6 и 12. В указанной конструкции при расстоянии
между экранированными электродами 3 и 5 порядка 2 мм емкость между
ними составляет всего 0,01 пф, что при частоте 50 гц соответствует ем¬
костному сопротивлению 310" ом.На рис. 12.13 приведена зависимость фонового тока ПИДа от скоро¬
сти потока водорода разной степени чистоты при напряжении 105 в. Для
водорода высокой степени чистоты наблюдается очень слабая зависимость
фонового ионного тока от скорости потока водорода (зависимости 3 и 4).
При напряжении 330 в зависимость чувствительности ПИДа от скорости366
ШЛВЛ 12 Аппаратура для газовой храматогршЬшпотока водорода имеет максимум (рис. 12.14). С увеличением расстояния
между электродами чувствительность падает (кривы 1 и 2).На следующем рисунке 12.15 зависимость чувствительности от рас¬
стояния между электродами изучена в большом диапазоне (от 1 до 12 мм)
при оптимальной скорости водорода.В работе [26] изучено влияние вакуума на работу пламенно-иониза¬
ционного детектора, вакуумная газовая хроматография позволяет значи¬
тельно увеличить скорость разделения без потери степени разделения.
Было показано, что при работе при вакууме 200-400 торр чувствитель¬
ность детектора возрастала.На Pittcon - 2008 (Питтсбургская конференция по аналитической хи¬
мии и спектроскопии) был описан новый тип планарного микропламен-
ноионизационного детектора, работающего всего при расходах газовых
потоков 3-5 мл/мин [27].Рис. 12.11Рис. 12.121у///\-Lim.$ W м I» а а » J*Осягтст атш 1ШеШ,т/тиГS^ Vh: W tещктшл W ^ W\и\\/*к\вРис. 12.13Рис. 12.14Рис. 12.15367
Яшин Я.И., Яшин Е.Я., Яшин А.Я.Газовая хроматографияВарианты работы ПИДа.ПИД для анализа неорганических газов.В классическом варианте ПИД не чувствителен к неорганическим со¬
единениям. Однако при определенных условиях ПИД может чувствовать
неорганические газы:- при постоянной подаче в пламя водорода примесей метана [28, 29],- при работе с пламенями, обогащенными водородом [30].В таблице 12.7 приведены значения пределов детектирования для не¬
органических газов в условиях работы пламени с высоким содержаниемводорода [31].Таблица 12.7ГазПредел детектирования, г/секСН,210-"СО4-10-’со,4-10*H,s4.10-шNO210"NjO7-10*'NO,210’SO,410-"'О,5-10-»Ar710-‘He5'10-*В работе [31] показано, что предел детектирования к неорганическим газам
сильно зависит от конструидаи ПИДа. Различие может достигать почти двух
порядшв, в частности при определении оксида азота (МО"’ - 21№" г/сек.).Эти результаты были объяснены тем, что некоторые ячейки ПИДа
бьши более приспособлены для работы с избыточным содержанием водо¬
рода. Было замечено, что при длительной работе чувствительность ПИДа
к неорганическим газам падает из-за окисления электрода-коллектора.
Чувствительность восстанавливается при увеличении скорости водорода
или же после механичесшй очистки.На газоюм хроматографе «Термохром 23» при дозировании краном 1 мл
газовой смеси, содержащей 0,05% N0, 0,07% N0^, 0,06% SO^ и 0,06% H^S и
разделенной на шлонке с порапашм, был достигнут предел детектирования к
этим газам в пределах 1Т0 '“ г/с, для N0 - 210 " г/с.368
ГЛАВА 12Аппаратура для газовой хроматографииНаилучшие соотношения газовых потоков азота, водорода и кислоро¬
да для водород-обогащенных пламен равны 1 : 3 : 1,5.Наиболее вероятный механизм регистрации неорганических газов в
этих режимах, особенно NO, NOj, SO^ и СО - хемиионизация.ПИДы, работающие при высоких содержаниях водорода, были иссле¬
дованы и для гетероорганических соединений [32].ПИД для селективного определения кислород-содержащих со¬
единений.В последние годы в бензин стали добавлять кислородсодержащие соеди¬
нения - оксигенаты, в частности спирты (метанол, этанол) и эфиры, (третбути-
ловый эфир и дф.). В связи с этим появилась аналитическая задача - определе¬
ние оксигенатов в топливах. Эта задача ранее решалась с использованием двух
колонок: неполярной и сильнополярной со сложной системой переключения.
Для определения на одной колонке бьш специально разработан кислородспе-
цифический ОПИД [33-35]. ОПИД состоит из микрореакторов крекинга и ме-
танатора и ПИДа обычной шнструкции. Эти микрореакторы соединены по¬
следовательно перед ячейкой ПИДа. В первом реакторе угаеюдороды конвер¬
тируются до углерода, который остается в реакторе, и водорода. Если в пробе
содержатся кислородсодержащие соединения, то кислород в первом реакторе
превращается в СО. СО ю втором реакторе конвертируется в СН^, который
хорошо определяется ПИДом.Первый реакгор для крекинга углеводородов состоит из платиновородиево¬
го капилляра, второй реактор - метанатор заполнен никелевым катализатором.В качестве газа-носителя используется гелий с добавкой 1 - 3% юдорода.Периодически никелевый катализатор активируется при 375°С, время
жизни этого катализатора не менее двух месяцев.Для надежной работы всей системы необходимо исключить попадание
примесей кислорода в газовые потоки.В таблице 12.8 приведены относительные сигналы ОПИДа к кисло¬
родсодержащим соединениям.Таблица 12.8СоединениеОтносительная чувствительность (относительно
метанола)теоретическаяэкспериментальнаяметанол1,01,0этанол0,700,72изопропанол0,530,54третбутанол0,430,44метилтретбутиловыйэфир0,360,3636924 Газовая хроматография
Яшин Я.И., Яшин Е.Я.. Яшин А.Я.Газовая хроматографияПри определении оксигенатов в качестве стандартного вещества при¬
меняют изопропанол.На рис. 12.16 приведены хроматограммы бензина, содержащего окси¬
генаты, зарегистрированные обычным ПИДом и О-ПИД. На второй хрома¬
тограмме зарегистрированы только оксигенаты.УРис. 12.16 Хроматограммы бензина, содержащего оксигенаты, за¬
регистрированные обычным ПИДом и О-ПИД.12.8.2 Детектор по теплопроводности (ДТП).ДТП - один из первых детекторов в газовой хроматографии. ДТП -
универсальный детектор, в отличие от ПВДа он может определять как ор¬
ганические, так и неорганические соединения. Особенно часто ДТП при¬
меняется для анализа постоянных, благородных и углеводородных газов.ДТП сравнительно прост в исполнении, надежен, линеен в широком ди¬
апазоне, сравнительно недорог, недеструктивен к анализируемым пробам.Принцип работы чувствительных элементов ДТП связан с изменением
их сопротивления с изменением температуры. Чувствительными элемен¬
тами в ДТП являются нити (филаменты), нагретые проходящим через них
электрическим током до высоких температур. Нити диаметром 10 мкм из¬
готавливаются из специальных металлов (платина, платина-иридий, воль¬
фрам, вольфрам-рений, никель, золоченый вольфрам и др.) и помещаются
в специальные микрокамеры, продуваемые газом-носителем. Филаменты
включены в плечи моста Уинстона (рис. 12.17). Через сравнительную ми-370
ШАВА12Аппаратура dm газовой хтшатографиы1фокамеру проходит поток чистого газа-носителя, а через рабочую камеру- газ-носитель из хроматографической шлонки с примесями разделенных
соединений. Сопротивление нитей зависит от температуры. При измене¬
нии состава газа в рабочей микрокамере теплопроводность его изменяется,
при этом изменяется теплопередача от нити к стенкам камеры, изменяет¬
ся температура нити и, следовательно, изменяется сопротивление нити по
сравнению с сопротивлением нити в сравнительной камере. Происходит
разбаланс моста Уинстона, и на нулевой линии появляется сигнал, пропор¬
циональный примеси анализируемого соединения в газе-носителе.Различное конструктивное исполнение чувствительных элементов
фирмы Гоу-Мак приведены на рис. 12.18.Установка „тот мостаЯстомовкаРис. 12.17 Плечи моста Уинстона.37124*
Яшин Я.И.. Яшин Е.Я., Яшин А.Я.^ irtГазовая хроматография-ff-Рис. 12.18 Чувствительные элементы фирмы Гоу-МакЧувствшетьные элемешы, находящиеся в рабочей и qmmrroibHoft каме¬
рах, должны быть одинаювы по сопрогавяениям и по расположеншо в мшфока-
мерах, причем подбирают чувствительные элементы - плечи моста Уинстона - не
толью при комнатной TeMnqjaiype, но и при высоких температурах.Микрокамеры, микроячейки должны быть небольшого объема (поряд¬
ка 20 - 140 мкл), чтобы уменьшить размывание разделяемых полос. Ми¬
крокамеры находятся в блоках из нержавеющей стали или сплава Мопе1“
(коррозионностойких материалов). Блоки обычно достаточно массивные,
чтобы сглаживать колебания температуры. Ячейки ДТП бывают разного
типа: диффузионные, полудиффузионные, проточные (рис. 12.19). Зависи¬
мость сигнала от скорости потока в этих ячейках разная.Рис. 12.19 Типы ячеек детектора по теплопроводности: а - проточ¬
ные, б - диффузионные, в - полудиффузионные. Стрелка¬
ми показано направление потока газа-носителя.372
ГЛАВА 12Аппаратура для газовой хроматографииПри высоких скоростях газа-носителя может сильно снижаться чув¬
ствительность детектора за счет того, что газ-носитель не успевает передать
тепло стенкам, в некоторых случаях наблюдается явление инверсии пиков.
На чувствительность детектора могут влиять концевые потери тепла.С целью повышения чувствительности и стабильности работы ДТП
бьши исследованы разные схемы режима работы моста Уинстона.Наиболее успешной оказалась схема в режиме постоянной температу¬
ры чувствительных элементов [36]. Чувствительность в этом режиме мо¬
жет быть в 8 раз лучше, и, соответственно, линейный диапазон шире.В качестве чувствительных элементов используются и термисторы (рис.
12.18 б), на которых предпочитают работать при низких температурах.Чувствительность ДТП сильно зависит от разности теплопроводно¬
стей газа-носителя и анализируемого соединения.Значения теплопроводностей газов-носителей приведены в таблице
12.9, из которой видно, что в этом отношении наилучшими газами-носите¬
лями являются гелий и водород. К сожалению, водород в качестве газа-но¬
сителя в лабораторных газовых хроматографах использовать нельзя из-за
взрывоопасности. С учетом еще и вязкости водород мог бы быть самым
лучшим газом-носителем [37].Чувствительность ДТП зависит от температуры чувствительного эле¬
мента, от величины сопротивления чувствительного элемента, от темпера¬
туры корпуса детектора и расхода газа-носителя. Чувствительность ДТП
увеличивается с ростом тока в третьей степени и разности температур в
степени 3/2. ДТП чувствителен к колебаниям давления.Таблица 12.9Значения теплопроводимостей некоторых газов и паровСоединениеТеплопроводность при ЮО^С,
X10’ Вт/(м.к)Теплопроводность по
отношению к гелию, %Водород223,6128Гелий174,2100Азот31,418,0Диоксид углерода22,212,7Аргон21,812,5Этан30,617,5Бутан23,413,5Нонан18,810,8Бензол17,29,9Ацетон16,79,6Этанол22,212,7Этилацетат17,29,9Хлороформ10,56,0Метилиодид7,94,6373
Яшин Я.И.. Яшин Е.Я., Яшин А.Я.Гтовая хроматография12.8.3. Электронозахватный детектор (ЭЗД)ЭЗД предназначен для анализа веществ, обладающих электронным
сродством, в частности галоидоорганических соединений. Полезный сиг¬
нал этого детектора - это уменьщение начального фонового тока, одно¬
значно связанного с количеством анализируемого соединения.В ионизационной камере ЭЗД помещается радиоактивный источник
(например, Ni“). Под воздействием радиации молекулы газа-носителя
(азот, аргон) ионизируются с освобождением электрона;Nj-N/ + eВ камере между электродами приложено напряжение, фоновый ток
создается в основном электронами, т.к. их подвижность на три порядка
выше, чем подвижность ионов. Кроме того, большая часть ионов реком¬
бинируется, не доходя до электродов. При попадании в ячейку детектора
соединений, обладающих сродством к электрону, происходит захват ими
свободных электронов:М + е - М"Это приводит к снижению начального фонового тока.ЭЗД обладает высокой ионизационной эффективностью. В газе-носи¬
теле недопустимо присутствие кислорода, влаги и др. соединений, снижа¬
ющих количество электронов или их подвижность.Предел детектирования ЭЗД на два-три порядка ниже ПИД, он сильно
зависит от числа и положения атомов галоидов в молекулах. В таблице12.10 приведена относительная чувствительность ЭЗД к некоторым галои¬
доорганическим соединениям.Таблица 12.10СоединенияОтносительнаячувствительностьСоединенияОтносительнаячувствительностьХлорметан1Фторбензол0,3Дихлорметан11Хлорбензол10Хлороформ410’Бромбензол10Четыреххлористыйугаерод5 WЙодбензол3WЭЗД впервые разработан Ловелоком в 1957 г. [38].ЭЗД получил широкое распространение для анализа галогенсодержа¬
щих пестицидов в почве, в продуктах растительного и животного проис¬
хождения. Для этих целей разработаны и аттестованы сотни методик.374
ГЛАВА иАппаратура для газовой хртштограФюВ последние годы ЭЗД применяется для определения летучих хлорсо¬
держащих соединений в питьевой воде, хлорированных бифенилов и дру¬
гих соединений.ЭЗД обладает чувствительностью также к кислород-, азот- и металло¬
органическим соединениям, а также к ПАУ и другим соединениям с вы¬
раженным сродством к электрону. Самый серьезный недостаток ЭЗД - ма¬
лый диапазон линейности, всего два-три порядка.В последние годы [39] разработан ЭЗД с нерадиоактивным источни¬
ком ионизации.12.8.4. Термоионный детектор (ТИД).ТИД - по существу вариант ПВДа (рис. 12.20).Газы
и продуты
юреми»Sit9ч^ВодородГаз-носител»Рис. 12.20 Схема термоионного детектора: 1 - электрод-
коллектор, 2 - поляризующий электрод, 3 - изолятор
электрода-коллектора, 4 - таблетка соли щелочно¬
го металла, 5 - диффузор, 6 - изолятор питания.В пламя водорода ПИДа вводятся пары солей щелочных металлов
(К, Na, Rb и Cs), при этом появляется высокая чувствительность к азот- и
фосфорсодержащим соединениям. Селективность ТИД к азот- и фосфор¬
содержащим соединениям относительно ПИД порядаа 100-1000.Этот детектор впервые предложен в работе [40] в 1967 г. Обширный
обзор по ТИДу был опубликован В.В.Бражниковым в 1970 г. [41].ТИД обладает высокой чувствительностью также к галогенсодержа-
Щим и мыщьяксодержащим соединениям.Чувствительность ТИД увеличивается с ростом атомного номера ще¬
лочного металла.375
Яшин Я.И., Яшин ЕЖ. Яшин А.Я.Газовая хроматографииДля стабильной работы ТИДа необходимо постоянное и равномерное
поступление щелочных металлов в пламя водорода. При использовании
спрессованных цилиндров или таблеток с солями щелочных металлов
ТИД может работать несколько месяцев. Таблетка с солями нагревается
либо пламенем, либо отдельным нагревателем.Соотнощение чувствительности ТИД к соединениям, содержащим С1,
As, N и Р, примерно следующее: Cl:As:N:P- 1:20:100:1000.ТИД в зависимости от конструкции имеет линейный динамический
диапазон в пределах 10^-10*. Уровень флуктуации (щума) для ТИД на два
порядка выше, чем для ПИД и составляет 1Т0'°- МО", фоновый ток также
в 100 раз выше. ТИД весьма чувствителен к колебаниям расхода водорода.ТИД в основном применяется для анализа азот- и фосфорсодержащих
пестицидов, а также некоторых биологически активных соединений.12.8.5. Пламеннофотометрический детектор (ПФД).ПФД также разработан на основе ПИД.Механизм работы ПФД основан на селективном излучении серасодер¬
жащих и фосфорсодержащих соединений, сгорающих в пламени водоро¬
да. Эта селективность относительно углеводородов более 2000.В боковой части ячейки ПИД напротив пламени водорода размещает¬
ся окно с фильтрами на 394 нм для пропускания излучения серы и на 526
нм, пропускающее излучение фосфора. Эти сравнительно слабые излуче¬
ния попадают в фотоэлектронный умножитель, в котором сигналы усили¬
ваются (рис 12.21).376
ГЛАВА 12 Аппаратура для газовой хроматографииВ ПФД пламя больше обогащено водородом, чем в ПИДе.Предел детектирования для фосфорсодержащих соединений ниже,
чем для серасодержащих соединений.ПФД нашел большее применение для анализа серасодержащих соеди¬
нений (меркаптанов, сульфидов и дисульфидов) в природном газе и не¬
фтепродуктах.К сожалению, существенным недостатком ПФД является нелиней¬
ность его сигнала.Для улучшения стабильности работы ПФД фирма Вариан разработала
его пульсирующий вариант [42-45], в частности на 3 герца. Это позволило
увеличить его чувствительность, селективность и стабильность. Предел
детектирования ППФД: 2 10 ’^ rS/c, Ю ''* гР/с и 2 10 rN/c.В ППФД используется небольшой расход водорода 12 мл/мин и воз¬
духа 24 мл/мин. Этот детектор селективен к азотсодержащим соединениям
таким, как NOx, NH3, гидразин, амиды, нитро и нитрозо- соединения.ППФД обладает селективностью ко многим гетероатомам (S, Р, N, As,
Sn, Ge и др.), поэтому он может быть дешевой альтернативой атомно-эмис¬
сионного детектора.Основные описанные применения ППФД: серасодержащие соединения
в бензинах, фосфор и серасодержащие пестициды, ОВ (химическое оружие),
серасодержащие лекарства, мьппьяшвистые соединения в нефти, оловоор¬
ганические соединения в морсшй воде и рыбе, марганец в бензине [43].12.8.6 Фотоионизационный детектор (ФИД)В ФИДе ионизация анализируемых соединений происходит за счет
УФ-излучения в специальной камере с двумя электродами. При фотоио¬
низации молекулы анализируемых соединений диссоциируются на ион и
электрон:А + h — А^ + е-.Образуемые ионы собираются электродами. Ионизируются только те
соединения, потенциал которых ниже энергии фотонов. В зависимости от
лампы энергия фотонов может быть 9,5; 10,2 и 11,7 эВ.ФИД как и ПИД имеют высокую чувствительность ко всем органиче¬
ским соединениям. К ароматическим соединениям ФИД имеет в 10-50 раз
большую чувствительность, чем ПИД.В отличие от ПИД, ФИД может регистрировать и H^S, PHj, NH^,
AsHj и др.В таблице 2.5 приведены потенциалы ионизации некоторых веществ,
а в таблице 2.6 относительная чувствительность ФИД (относительно бен¬
зола) к соединениям разных классов [46-49].377
Яшин Я.И., Яшин Е.Я.. Яшин А.Я.Гамвая храчатографинТаблица 2.11Потенциалы ионизации некоторых соединенийСоединенияeVСоединенияеУПростые молекулыАзотосодержашиеАзот15,58соединенияКислород12,08Аммиак10,15Вода12,59Метиламин8,97Оксид углерода14,01Ацетонитрил12,22Диоксид углерода13,79Акрилонитрил10,91Оксид азота
Диокснд азота
Хлор
Иол9,259,7811,489,28Сернистые соединенияДиоксид серы12,34Сероводород10,46Нормальные и непредельныеКарбонилсульфид11,18Дисульфид углерода10,08углеводороды12,981 Л S'SМетилмеркаптан9,44МетанДиметилсульфид8,69ЭтиленАцетилен10,5211,41Диметилдисульфид8,46Бутен-1Гексан9,5810,17Альдегиды, кетоны,
спирты, кислоты, эфирыХлорированные углеводородыФормальдегидАцетальдегид10,8710,21Метилхлорид11,28Акролеин10,10Четыреххлористый углерод11,47Ацетон9,69Хлороформ11,42Метанол10,851,2-дихлорэтан11,12Этанол10,48ВинилиденхлоридВинилхлорид9,8310,00Муравьиная кислота
Уксусная кислота11,0510,37Трихлорэтилен9,45Гетероциклические иароматические углеводородыФенол8,50Пиридин9,32Бензол9,25Толуол8,82Ксилол8,45Стирол8,47Анилин7,70378
ГЛАВА 12 Аппаратутг для газовой хроматографииТаблица 2.12Относительная чувствительность ФИД к соединений разных классов
(относительно бензола)СоединенияSoTHСоединенияSoTH1234Н-алканыАльдегидыГептан0,032Бутаналь0,30Октан0,08Пентаналь0,36Нонан0,14Октаналь0,49Декан0,23СпиртыУндекан0,30Бутанол-10,023Додекан0,37Пентанол-10,053Тридекан0,46Гексанол-10,086Тетрадекан0,53Изобутанол0,029Пеитадекан0,59ЭфирыГексадекан0,71Метилпропионат0,01Гептадекан0,72Бутилацетат0,044Октадекан0,79АроматикаНонандекан0,86Бензол1,(ЮЭйкозан0,93Толуол1,09Докозан1,13Этилбензол1,16Развегвленные алканын-пропилбензол1,212,2-диметилбутан0,037н-бутилбензол1,272,3-диметилбутан0,032н-гексилбензол1,292-метилпентан0,011н-октилбензол1.52Циклогексан0,18Децилбензол1,69Метилциклогексан0,28о-ксилол1,14Декагидронафтен1,04м-ксилол1,151-алкеиып-ксилол1,201-гептен0,54Кумол1,221-октен0,55р-кумол1,271-пантадецен0,92Мезитилен1,272-гептен0,513-гептен0,581,9-декандиен1,072-октин2,75КетоныАцетон0,352-бутанон0,405-нонанон0,82379
Яшин Я.И., Яшин Е.Я., Яшин А.Я. Газовая хроматография12.8.7 Масспектрометрический детектор (МСД)В последние годы произошел большой прогресс в создании настоль¬
ных небольших МСД для газовых хроматографов. В настоящее время это
высокочувствительный детектор и самый совершенный прибор для иденти¬
фикации неизвестных веществ. Имеется библиотека масс для более 250000
соединений. МСД обычно включает вакуумный насос, ионный источник и
систему обработки. Для газовых хроматографов используются в основном
два вида ионизации: электронный удар и химическая ионизация.В качестве анализатора ионов могуг применяться: магнитные, квадру-
польные и ионные ловушки, анализаторы ионно-циклотропного резонанса, с
двойной фокусировкой (магнитные и электростатические), времяпролетные.В качестве детектора, регистрирующего пучки ионов, используются:
электронный и фотоэлектронный умножитель, коллектор Фарадея, пло¬
ская электронная матрица.МСД - это ионизационный, деструктивный, потоковый детектор. Этот
детектор универсальный и одновременно селективный, т.к. всегда можно
найти массу, типичную только для данного соединения. При исследова¬
нии МСД в режиме детектирования отдельных ионов чувствительность его
очень высока (в 1000 раз больше, чем в режиме сканирования) около lO 'V
(100 фемтограмм). Международный стандарт ионизации 70eV (1,110 ” Дж)
общепризнан, на многих современных хроматомасспектрометрах предусмо¬
трен только такой фиксированный режим ионизации. Библиотека масс соз¬
дана с этим источником.12.8.8. Другие детекторы.Как указывалось ранее всего в ГХ предложено и описано более 50 типов
детектирующих систем. В настоящее время выпускается 21 тип детекторов.Самые распространенные детекторы: ПИД, ДТП, ЭЗД, ТИД, ПФД,ФИД.
В последние годы к ним добавился масспектрометрический детектор (МСД).
Все крупные производители газохроматографического оборудования ком¬
плектуют газовые хроматографы этими детекторами. Кроме них, некоторые
фирмы выпускают еще следующие детекторы: атомно-эмиссионный, хеми-
люминисцентный, электролитической проводимости, разрядный ионизаци¬
онный, гелиевый ионизационный, пульсирующий разрядный, поверхност¬
ной ионизации, инфракрасный, флуоресцентный, плотномер, радиоактив¬
ный, ультразвуковой, ионной подвижности и другие.Представляет большой интерес детектор с пульсирующим разрядом
(фирма Valeo-Vici), который может работать в режиме электронозахватно¬
го детектора (без радиоактивного источника) и фотоионизационного.В режиме электронного захвата такой детектор селективно определяет
галоидоорганику на уровне 10 '^ - 10 '* г (пико- и фемтограммы).380
ГЛАВА 12 Аппаратура для газовой хромат>Р1тф1тВ режиме гелиевой фотоионизации разрядный детектор работает как
универсальный, неразрушающий, высокоэффективный детектор.12.8.9. Комбинации детекторов.Для многих аналитических задач представляет интерес регистрация
хроматограмм одновременно на двух и более детекторах, при этом детек¬
торы могут соединяться как последовательно, так и параллельно.На многих современных газовых хроматографах исследовательского
типа имеется возможность установки одновременно 2-4 детекторов.Впервые в СКБ аналитического приборостроения в Эстонии были
разработаны конструкции совмещенных детекторов (двух - трех) в одном
блоке. Комбинация детекторов востребована при анализе сложных смесей
пестицидов.Наиболее распространенные комбинации двух детекторов для анализа
одной смеси приведены в таблице 12.13. Возможны и другие комбинации,
в т.ч. и сразу трех детекторов.Таблица 12.13Наиболее важные комбинации детекторовКомбинация детекторовОбласти применения
(анализ одной смеси)1.ПИД-ДТПАнализ органических и неорганических соедине¬
ний, в частности природного газа2.ПИД-ЭЗДАнализ как органических, так и селективно галои¬
доорганических соединений3.ПИД-ТИДАнализ органических и селективно азот- и фос¬
форсодержащих соединений4.ПИД-ПФДАнализ органических и селективно серасодержа¬
щих соединений5.ПИД-ФИДАнализ органических и селективно ароматиче¬
ских соедшений.ФИД в 10-100 раз чувствительнее ПИДа к арома¬
тическим соединениям, поэтому при одновремен¬
ном анализе сложных соединений в нефтехимии
можно сразу отличить ароматические соединения.
Отношение сигналов ПИД/ФИД применяется для
идентификации неизвестных смесей.12.9 Дополнительные устройства для газовой хроматографииКриогенное устройство - это система термостатирования колонок от
комнатных до минус 100°С для разделения трудно разделяемых газовых
смесей. Для этих целей используется жидкий азот из сосуда Дьюара.381
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я. Газовая хроматографияСистема обратной продувки. Система, включающая шестиходовый
кран-дозатор и четырехходовый кран для включения обратной продувки
колонки для быстрого элюирования суммы тяжелых компонентов, в част¬
ности, при определении природного газа С| - С^ и ЕС^Обогатительные устройства для концентрирования тяжелых приме¬
сей из газовых потоков с последующей десорбцией и дозирования в ана¬
литическую колонку. Концентрирование примесей происходит в охлажда¬
емой небольшой обогатительной колонке. После обогащения десорбция
производится специальной разогретой печкой.Криофокусирующее устройство позволяет шицентрировагь примеси
в начале охлажденной капиллярной колонки. Сильносорбируемые высоко-
кипящие соединения сорбируются на начальном участке шлонки, а газ-но¬
ситель и легкие проходят в колонку не сорбируясь. После ошнчания процес¬
са шнцентрирования происходит быстрый нагрев (тепловой удар) для того,
чтобы при десорбции проба не размывалась, вводилась в шлонку в виде
узшй полосы с десорбированными сшнцентрированными шмпонентами.Устройство для концентрирования методом выдувания и нако¬
пления (purge and trap) - предназначено для выдувания из загрязненных
вод летучих и малолетучих примесей и нашплений их на специальной ад¬
сорбционной ловушке с последующей тепловой десорбцией и переводом в
хроматографическую колонку.Устройство парофазного концентрирования (head-space) позволяет
повысить чувствительность определения легшкипящих соединений, рас¬
творённых в воде, имеющих шэффициенты распределения менее 10. Эти
устройства позволяют также извлекать и дозировать легкие анализируемые
соединения из биологических проб, из твёрдых материалов (пород, почв, по¬
лимерных материалов и др.). Типичное устройство этого типа - парофазное
устройство «Фаза» (ОАО «Цвет).Устройство пиролизное. Пиролизная газовая хроматография приме¬
няется для анализа нелетучих высокотемпературных соединений по про¬
дуктам их разложения в инертной среде (пиролиз). Пиролизная газовая
хроматография применяется для анализа полимеров, каучуков, смол, оли¬
гомеров, биополимеров и др.Пиролиз проводят с помощью обычного термического нагрева, вы-
сошчастотного нагрева (до точки Кюри), лазерного разогрева и разряда
Устройство для пиролиза изготавливается в виде приставки к стандартном
газовым хроматографам, которые включают вместо узла ввода пробы или
параллельно ему. Пиролизные устройства бывают трёх типов: филамент-
ного, печного и высокочастотного с ферромагнитными держателями. Раз-382
дичают: мягкий пиролиз до 500®С, в основном, для биологических объ¬
ектов (бактерий, белков, крахмала и др), средний пиролиз при 500-800®С
для исследования полимеров, жёсткий пиролиз при 800-1100®С, полимеры
разрушаются на небольшие фрагменты, образуется много продуктов.Электронные измерители расходов для точного измерения расходов
газа-носителя, водорода и воздуха. Измерители расходов обычно работают
в диапазоне до 10, 100 и 1000 мл/мин. (в частности ИРГ-10, ИРГ-100 и
ИРГ-1000 ОАО «Цвет»).Приставка парофазовая «Поток-А» и фторопластовые диффузион¬
ные дозаторы типа «Микрогаз» предназначены для приготовления пове¬
рочных газовых смесей, в т.ч. и афессивных смесей HF, НС1, N0^, NHj,
меркаптанов. Динамическая установка «Микрогаз-Ф» относится к рабо¬
чим эталонам состава газовых смесей 1-2 разрядов по ГОСТ 8.578.Пробоотборники ПГО-50 и ПГО-400 предназначены для отбора сжи¬
женных углеводородных газов из стационарных емкостей, трубопроводов
и баллонов, из технологических аппаратов.Устройства каталитической доочистки газа предназначены для
очистки технического азота (ГОСТ 9293-74) от кислорода до квалифика¬
ции азота особой чистоты (ТУ 6-21-39-79), необходимого для работы с де¬
тектором электронного захвата.Автоматические дозирующие устройства (автосамплеры). Автосам-
плер включает от 8 до 120 стеклянных пробирок с пробами, которые по¬
даются к месту отбора и дозирования по специальной профамме. Кроме
пробирок с пробами имеются пробирки с промывочными растворителями
для промывки дозирующего узла после ввода анализируемой пробы. По-
следоветельность операций задаётся и контролируется микропроцессорным
блоком управления и может быть откорректирована под конкретные задачи.Метаиатор. В некоторых аналитических задачах чувствительность
детектора по теплопроводности недостаточна для определения СО и СО^.
В этих случаях проводят конверсию СО и COj до метана в специальной
трубке с Ni - катализатором в потоке водорода после разделения СО и СО^
на хроматофафической колонке. Получаемые пики метана регистрируют
ионизационно-пламенным детектором на уровне < Ippm (<10'^/о).Прочие устройства. В составе газовых хроматофафов применяют
иногда измерители потоков, разные интерфейсы (ГХ-МС, ГХ-ИКС, ЖХ-
ГХ и др.), генераторы водорода, азота, сверхчистого воздуха, фильтры очи¬
стители газов.383
Яшин Я.И., Яшин Е.Я., Яшин А.Я. Гтовая хроматография12.10 Краткая история развития хроматографического прибо¬
ростроенияПредставляет интерес история развития газохроматографичесшго пpибqю-
строения. Этому развитию способствовали организации и фирмы многих стран.Ранний период развития (1955-1965 гг.).В мае 1955 г. фирма «Перкин-Эльмер» начала серийный выпуск перво¬
го в мире газового хроматографа «Фрактометр» модель 154. Это был про¬
образ современного газового хроматографа, в нем был термостат до 150°С
(с 1956 г. термостат до 225°С), что позволяло использовать его для анализа
не только газов, но и жидкостей в варианте газо-жидкостной хроматогра¬
фии, газовый кран для дозирования газообразных проб и катарометр в ка¬
честве детектора.Нужно отметить, что макет первого лабораторного газового хромато¬
графа описали Джеймс и Мартин в 1952 г в своей классической работе, в
которой был изложен метод газожидкостной хроматографии. В этом хрома¬
тографе бьши все элементы современного хроматографического прибора:
узел ввода пробы, колонки, термостат с точным температурным контролем
и уникальный титриметрический детектор.Во Фрактометре 154 использовали U-образные стеклянные или метал¬
лические шпонки длиной 1 м. В первой модели применялись две шлонки
с разными жидкими фазами (додецилфталатом и 2-этилгексилс^ациатом).
Уже черюз год эта фирма предлагала потребителям юсемь шлонок с разными
жидкими фазами и две шлонки с адсорбентами. В приборе был предусмотрен
ввод пробы в испаритель либо ишрицем через резиновое уплотнение, либо
поворотным краном. На два месяца раньше фирмы «Перкин-Эльмер» (март
1955 п) выпустила серийный газовый хроматограф модель К-1 фирма Баррелл
Корпорейшен. Однаш этот прибор не имел термостата и применялся только
для анализа газов с шлоншй, заполненной активным углем. В 1956 г в Япо¬
нии выпустила первый газовый хроматограф фирма «Шимадзу».В Италии первый газовый хроматограф был разработан под руковод¬
ством С.Визани в 1955 г., и его серийный образец был представлен для
продажи на выставке в 1956 г во Франкфурте-на-Майне под названием
«Фрактовап».Наиболее ранние разработки английских фирм (1956-1959 гг) - это
хроматографы компаний «Гриффин и Джордж» (Лондон), «Метрополитен
Викерс» (Манчестер) и «Пай Компании» (Кэмбридж).Таким образом, к середине 1956 г в США бьшо уже 7 компаний, выпу¬
скающих газовые хроматографы. Далее к этой области приборостроения
интерес нарастал очень стремительно, и в первой половине 1961 т. уже
16 компаний производили 39 моделей хроматографов, а в конце 1962 п 23
компании выпускали 51 модель газовых хроматографов.384
ГЛАВА 12Аппаратура для газовой хроматографииБурному развитию газохроматографического приборостроения способ¬
ствовала разработка в 1958 г. ионизационных детекторов: пламенно-иониза-
ционного и аргонового, разработка режима профаммирования температуры
колонок и введение в аналитическую практику капиллярных шлонок.В СССР первыми серийными газовыми хроматофафами были хрома-
тофаф ХТ-2 и хроматермофаф ХТ-2М, выпущенные в 1958 г.. В 1960 г.
Таллиннский завод измерительных приборов изготовил газовый хромато-
фаф УХ-1, а хроматофаф УХ-2 производился на заводе газоанализаторов
в г. Выру (Эстония).В 1959-1960 г в Дзержинсшм ОКБА бьш создан первый в СССР промыш¬
ленный автоматический газовый хромагофаф во взрывозащищенном испол¬
нении с пламенно-ионизационным детектором, с 1963-1964 гг. также впервые
в СССР в Дзержинсшм ОКБА начато серийное производство лабораторного
газового хроматографа Цвет-1 с пламенно-ионизационным детектором.12.11 Основные типы газовых хроматографовЗа пятидесятилетие газохроматофафическое приборосфоение посто¬
янно развивалось. В настоящее время выпускается широкий ассортимент
газовых хроматофафов (табл. 12.14). За 53 года выпущено около миллио¬
на разных хроматофафов.Основные типы хроматографовТаблица 12.14НазваниехроматографовНазначениеПреимущества,особенностиЛабораторныехроматографыДля стационарных
лабораторийШирокий набор комплектующих
и дополнительных устройствПромышленныеавтоматическиеДля контроля производ¬
ственных процессовВ большинстве случаев
взрывозащищенное исполнениеДвухмерные,многомерныеДля полного рачлеления
сложных многокомпо¬
нентных смесейРазделение на двух колонках с
разной селективностьюХромато-масс-спектрометрыДля идентификации раз¬
деленных соединений]^М1ичественный и качественный
аиалю смесей неизвестного со¬
ставаМалогабаритныеДля передвижных и ста¬
ционарных лабораторийЭкономия денег, энергии, места,
расходных материа;юв при со¬
хранении аналитических харак¬
теристик 10-25 кгПортативные,транс-портируемые,полевыеДля анализа на месте
объектаНебольшой вес, экспрессность
анализа, автономность по элек¬
трическому и газовому питанию
5-15 кг38525 Газовая хроматография
Яшин Я.И. Яшин ЕЛ. ЯшинАЯ.ГшшштзшшФтНазваниехроматографовНазначениеПреимущества,особенностиМикрохромагографы
на чипах (крем¬
ниевая технология),
hand-held, персо¬
нальные, карманныеДля анализа на месте
объекта, переносимыеДля экспрессных анализов,
простых задач. Полная автоном¬
ность. Ограниченные аналитиче¬
ские возможности 0,2 - 3 кгСпециальныемикрохроматографыДля космических
исследованийАвтоматизация анализа
Устойчивость к ударам и тряскеТаблица 12.15Перечень научно-техиическиж достижений, способствующих разви¬
тию газохроматографического приборостроения№№ппНаименование достиженийГодАвтор, фирма1.Применение каторометра в ГХ1954Рэй2.Открытие капиллярной хроматографии
и внедрение капиллярных колонок1957- 1958Голэй3.Масспектрометрический детектор1957 (промышл.
выпуск с 1972)Холмс, Морелл4.Фотоионизационный детектор1957
(освоен с 1976)Робинсон5.Разработка плазменно-ионизаци¬
онного детеетора1958Мак-Вилльямс,
До(ф, В1фего{»|ус5а.Стеклянные капиллярные колонки1958Дэсти6.Разработка микрошприцев1958Фирма«Гамильтон»7.Внедрение программирования темпе-
ршуры1958Филипс8.Внедрение хроматермографии1958А.А.Жуховицкий,Н.Н.Туркельт^9.Электронозахватный детектор1957- 1958Ловелок, Дима10.Аргоновый ионизационный детектор1958ЛовелокИ.Термоионный детектор1964-1968
(пром. осв.
1970-1972)Кармен,Д)^нфррида12.Система переключения колонок1968Дине13.Применение микропроцессоров в ГХ1973Фирма«Хьюлетт-Паккард»386
ГЛАВА 12Аппаратура для газовой хроматографии№№ппНаименование достиженийГодАвтор, фирма14.Кварцевые капиллярные колонки1979Дондено,Зеренеем15.Испаритель с программированием тем¬
пературы1981Пой16.Адсорбционные капиллярные колонки
«ПораПлот» 1981Фирма«Хромпак»17.Поликапиллярные колонки1982Солдатов18.Многомерные варианты1985Вройт19.Инертные металлические капиллярные
колонки «Ультиметалл»1991Фирма«Хромапак»20.Разработка методов и устройств кон¬
центрирования1980-1990Многие фирмы21.Программное обеспечение1985-1995Многие фирмы12.11.1 Лабораторные газовые хроматографыЛабораторные газовые хроматографы - наиболее распространенные
хроматографы, их ежегодный выпуск составляет более 30 ООО. Эти при¬
боры предназначены для работы в аналитических лабораториях, они могут
работать в диапазонах температур 10 - 35° С при влажности более 80%.Вес лабораторных хроматографов колеблется от 30 до 50кг. Обьем
термостатов разных приборах может быть от 5 до 20л., диапазон рабочих
устанавливаемых температур в пределах от минус 99 до плюс 450°С, мак¬
симальная скорость программирования температуры колеблется от 25 до
120°/мин. В модели Flash - GS используется система нагрева до 1800°/мин.
В этом случае быстро нагревается тонкослойный металлический капил¬
ляр, через который пропускают электрический ток, в этот капилляр поме¬
щается кварцевая капиллярная колонка. Максимальный нагрев колонки до
350 - 400°С достигается очень быстро - за несколько секунд.Лабораторные газовые хроматографы комплектуют набором различ¬
ных детекторов и дозирующих устройств. Разделительные хроматографи¬
ческие колонки используются как насадочные, микронасадочные, так и
капиллярные.Для расширения аналшических юзможностей лабораторные газовые хро-
матофафы юмплектуются рядом дополнительных устройств (см. разд. 12.9).Ниже суммированы основные достижения в газохроматографическом
приборостроении за последнее десятилетие:- разработка, освоение и щирокое внедрение приборов ГХ-МС;- введение электронного контроля расхода и давления;38725-
Яшин Я.И., Яшин Е.Я.. Яшин А.Я. Газовая хроматография- разработка методов дозирования больших проб в кахшллярные шлонки;- разработка и внедрение капиллярных колонок широкого диаметра
(0,32 и 0,53 мм.), большой емкости (толщина привитой хгленки жидкой
фазы до 5 мкм.);- создание портативных промышленных хроматографов для контроля
производственных процессов;- создание портативных полевых газовых хроматографов;- разработка микрохроматографов на ЧИПах;- разработка и выпуск простых одноканальных хроматографов для ру¬
тинных аналитических задач;- разработка новых принципов нагрева и термостатирования колонок
(микроволновое, инфракрасное и пр.);- разработка методов и аппаратуры экспрессной хроматографии;- совершенствование программного обеспечения;- разработка и внедрение новых методов и устройств шнцентрирования.В газовые хроматографы в последние 5 лет разработаны, введены иулучшены следующие функции:- реализация многошлоночной хроматографии;- цифровой шнтроль газовых потоков при работе как с капиллярными,
так и насадочными колонками;- одновременная установка 3,4х инжекторов, 4х независимо термоста-
тируемых детекторов;- улучшение воспроизводимости, благодаря автоматической стабили¬
зации и контроля скоростей и давлений газовых потоков;- режим постоянной линейной скорости газа - носителя при анализе с
капиллярными колонками;- быстро охлаждаемые термостаты в режиме гфограммирования тем¬
пературы.- встроенный 16-строчный ЖК-дисплей, демонстрирующий текущую
хроматограмму и параметры работы прибора;- функции самодиагностики и валидации (отклонение от устанавли¬
ваемых параметров, изменениях в программах анализов и ошибках);- контроль и управление данными и результатами анализа с помощью
интуитивно понятного программного обеспечения GC Solution, являюще¬
гося частью системы Lab Solution для хроматографии;- система быстрой хроматографии Ultra Fast (сокращение времени ана¬
лиза в 20 -50 раз за счет скорости нагрева до 1200°С/мин.);- повышение чувствительности в 50 раз за счет ввода больших проб
(до 50 мкл.);- гибкая конфигурация прибора с автодозатором, парофазной и термо-
десорбционной приставками.388
ГЛАВА 12Аппаратура для газовой хроматографиив настоящее время в таблицах 12.16 и 12.17 перечислены основные
модели отечественных и зарубежных газовых хроматографов, находящих¬
ся на российском рынке. По техническим характеристикам большинство
газовых хроматографов находятся на одном уровне, в частности по преде¬
лам детектирования.Таблица 12.16Основные отечественные лабораторные газовые хроматографы№№ОрганизацииМоделиГород1.ЗАО СКБ «Хроматек»Кристалл 5000
Кристалл 2000 М
Кристалл 5000.2Йошкар-Ола,
Республика Марий Эл2.«Мета-Хром»Кристаллюкс-4000 МЙошкар-Ола,
Республика Марий Эл3.ОАО «Цвет»Цвет-800Цвет-АналитикаЦвег-600Г. Дзержинск
Нижегородской обл.4.НПО «Химавтоматика»Яуза-100
Яуза-200Москва5.ОАО «Хроматограф»ЛХМ2000Москва6.ОАО «Люмекс»ГАЛС-311С.-Петербург7.ОАО «Купол»Кристалл-2000г. Ижевск8.ЗАО «Химаналитсервис»Хромое ГХ-1000Г. Дзержинск
Нижегородской обл.Таблица 12.17Основные зарубежные лабораторные газовые хроматографыНазвание фирмыМоделиСтрана1.Аджилент7890,6850США2.ТермофинниганTrace GC Ultra,
Focus GCСША3.Перкин ЭльмерClaras 400, 500, 600США4.Вариан3800 GC, 3900 GCСША5.ШимадзуGC-2010, GC-2014Япония6.ПерихромPR 2100Франция7.Yomig Lin Instrument Co.Acme 6100 GCЮжная Корея8.KonikHRGC 4000 ВИспания9.DaniGC MasterИталия10.SRI InstramentsGC8610CСША389
Яшин Я.И., Яшин Е.Я., Яшин А.Я. Газовая хроматографияТенденции к упрощению лабораторных газовых хроматографов.Хроматографы по своей природе - приборы универсальные, на одном и том
же приборе можно анализировать сотни и тысячи различных смесей, меняя
только дозаторы, шлонки, детекторы и параметры разделения (температура
шлонки, расход газа-носителя и др.). До недавнего времени газовые хрома¬
тографы оснащались широким набором устройств, детекторов, дозаторов и
пр., чтобы сделать приборы универсальными, исследовательскими. Поэтому
выпускаемые газовые хроматографы как зарубежные, так и отечественные
избыточны по своим функциональным возможностям для простых (рутин¬
ных) задач, которые чаще всего встречаются в практике аналитических ла¬
бораторий разного профиля, а также в технологичесшм шнтроле производ¬
ственных процессов. В связи с этим, щирокие возможности, заложенные в
современных хроматографах, потребителями, как правило, не используют¬
ся. Потребитель несет лишние финансовые затраты за избыточность этих
приборов. Однаш, это не означает, что универсальные приборы не нужны,
просто область их применения в настоящее время меньше. На основании
опросов потребителей и других оценок для 70% потребителей нужны про¬
стые приборы, а для 30% - универсальные.В настоящее время наметились две тенденции в создании простых
хроматографов. Первое направление - разработка и выпуск одноканаль¬
ных упрощенных газовых хроматографов для рутинных задач. В послед¬
ние годы за рубежом появились такие модели: компактный одноканаль¬
ный хроматограф 6850 «Agilent», Focus GC «Thermo Finnigan», 3900 GC
«Varian». В нашей стране разработан простой одноканальный газовый
хроматограф «Яуза-100» НПО «Химавтоматика» (рис. 12.22).Рис.12.22 Газовый хроматограф «Яуза-100»390
ГЛАВА 12 Аппаратура для газовой хрпмат^г^ф^Газовый хроматограф «Яуза-100» - одноканальный газовый хромато¬
граф с электронным газовым блоком, микрокатарометром на планарной
технологии, ионизационно-пламенным детектором улучшенной конструк¬
ции и фотоионизационным детектором (прибор может работать только с
одним из указанных детекторов). Основные достоинства прибора - высо¬
кие технические характеристики, низкая стоимость, высокая надежность и
простота в эксплуатации.Вторая тенденция - это создание на основе простых приборов (с после¬
довательным наращиванием технических возможностей) хроматографиче¬
ских комплексов для решения конкретной аналитической задачи. В этот
комплекс включается специальная колонка, методическое и программное
обеспечение, градуировочные смеси (в случае необходимости) и другое. В
общем осуществляется поставка комплекса «под ключ», все необходимое
для решения определенной аналитической задачи.12.11.2 Портативные газовые хроматографы.В последние 10-летие интерес к миниатюризации газохроматографи¬
ческой аппаратуры сильно возрос. Это связано с тем, что появилось много
аналитических задач, когда необходимо оперативно провести анализ на
месте расположения объекта.Можно привести следующие основные области применения портатив¬
ных хроматографов: анализ воздуха производственных помещений, жилых
помещений, воздуха шахт, атмосферы закрытых помещений и гермокабин
(кабин и салонов автомобилей, помещений космических кораблей и под¬
водных лодок), исследования атмосферы других планет, анализ поверх¬
ностных вод, загрязненных почв, оперативный контроль за выбросами и
сбросами, анализ автомобильных выхлопов, определение отравляющих
веществ и продуктов их превращений в окружающей среде, обследова¬
ния в условиях чрезвычайных ситуаций (взрывы, аварии, пожары и др.),
экспрессный анализ для судебных экспертиз, определение взрывчатых
веществ в аэропортах, геологоразведка нефтеносных и газоносных райо¬
нов, определение утечек газопроводов, анализ алкоголя в крови водителей
транспортных средств, оперативный контроль пищевых продуктов на рын¬
ках и многие другие.Основные направления в миниатюризации хроматографов. В табл.12.10 приведена классификация портативных хроматографов. Малогабарит¬
ные хроматографы в основном предназначены для передвижных лаборато¬
рий, хотя могут использоваться и в стационарных лабораториях. Основные
технические характеристики таких приборов находятся на уровне обычных
лабораторных. К таким приборам можно отнести хроматографы Яуза-100,
Кристалл-5000, модель 6850 «Хьюлетт-Паккард» и др. Это направление до¬391
Яшт Я.И.. Яшин Е.Я., Яшин А.Я. Газовая хроматографиястаточно перспектавно, т.к. существующие технические возможности по¬
зволяют значительно уменьшить размеры газовых хроматографов без ущер¬
ба для их аналитических возможностей. Выгода очевидна из-за экономии
энергии, расходных материалов и места на лабораторных столах.Следующее наиболее распространенное направление - это созда¬
ние портативных транспортируемых газовых хроматографов (пере¬
возимых, переносных) для анализа на месте расположения изучаемого
объекта. Основные отличительные особенности таких приборов - это пол¬
ная автономность по электрическому и газовому питанию, небольшой вес,
надежность, легкость обслуживания, экспрессность анализа. По аналити¬
ческим возможностям приборы этого типа значительно уступают прибо¬
рам первого типа. Для простых задач они могут применяться и в лабора¬
ториях, поэтому во многих приборах этого типа предусмотрено питание и
от электросети.Полевые хроматографы позволяют получать результат немедленно, од¬
нако они не могут полностью заменить лабораторные хроматографы. Такие
хроматографы могут подтвердить нет ли разложения или иных изменений
пробы в промежутке времени между отбором и анализом. Однако бьюают
случаи, когда полевые хроматографы обеспечивают более точные измере¬
ния, чем лабораторные, в частности при определение летучих соединений,
которые не могут сохраниться в пробе в течение длительного времени.Третье направление - это создание сверхминиатюрных хроматогра¬
фов на ЧИП”. Пионером создания миниатюрного газового хроматографа
является фирма «Микросенсор технолоджи» (США), сотрудники которой,
работая в Стэндфордском университете, в 1975 г. создали первый макет
микрохроматографа, выполненного с помощью кремниевой технологии, с
детектором по теплопроводности для анализа состава воздуха в кабинах
космических кораблей.Четвертое направление - это портативные хроматографы для кос¬
мических исследований (см. главу 17), к которым предъявляются особые
требования. Кроме небольшого веса и низкой потребляемой мощности,
эти хроматографы должны иметь исключительно надежную конструкцию,
обеспечивающую работу при любых условиях (при атмосферном давле¬
нии, в вакууме, при повышенных давлениях и температурах), высокую ви¬
броустойчивость и ударную прочность, быстрый выход на режим, мини¬
мальное время для передачи данных, длительное время сохранения своих
характеристик (вплоть до 10 лет и более).Отечественные портативные хроматографы. Отечественные пор¬
тативные хроматографы разрабатывались с семидесятых годов. Это пор¬392
ГЛАВА 12 Аппаратура для газовой хроматографиитативные полностью автономные хроматографы ХПМ-1, ХПМ-2, ХПМ-4,
Цвет П-182, микрохроматографы серии МХ с микропроцессорным управ¬
лением и со встроенным микрогенератором водорода (Дзержинский фили¬
ал НПО «Химавтоматика»).ООО «Сибертех» (г.Новосибирск) разработал и выпускает серию пор¬
тативных полевых хроматографов «Эхо». Портативные газовые хромато¬
графы ФГХ-1, ФГХ-2 (НПП «Химприбор») с фотоионизационным детек¬
тором предназначены для анализа атмосферы производственных помеще¬
ний и санитарных зон. Малогабаритные газовые хроматографы серии МХ
(НИИХром) предназначены для анализа спиртов С, - Cj, природного газа,
для анализа ароматических соединений в окружающей среде.Для анализа атмосферы планеты был разработан портативный газо¬
вый хроматограф «Сигма», состоящий из трех детекторов и трех колонок
(программа «Венера 11,12»). В программах «Венера 13» и «Венера 14»
были использованы газовые хроматографы «Сигма 2».Зарубежные портативные хроматографы. Первые зарубежные пор¬
тативные хроматографы были разработаны в 1968 г. фирмой «Carle Instru¬
ments», затем были портативные приборы «Gow-Мак», «Analytical Instru¬
ments», «Unico Environmental Instruments».В настоящее время выпускаются следующие портативные приборы:
модель 311D фирмы HNU, Snapshot, РЕ Photovac Voyager, HP Micro GC, Varian
Micro GC, Model СР-2(Ю2Р. Портативные газовые хроматографы - масс-
спектрометры производятся фирмами «Вгикег», «Inficon» и «Viking Inst».В 2(Ю1 г широко известная Ливерморская национальная лаборатория
разработала «Hand-held» газовый хроматограф размером с книгу с двумя
детекторами: микрокатарометром (предел детектирования 1 ppm) и раз¬
рядным детектором (35 ррЬ), температура термостата 25-250® С, в режиме
программирования до 300® С, колонка 500 см х 0,1 мм с жидкими фазами
DB-1, DB-5, DB-54, дозируемые обьемы 1-2 мкл. Питание от аккумулято¬
ров 12 V DC, мощностью 24 Вт, вес около 3,5 кг, размеры 200x125x75 мм.Разработаны две модели портативных газовых хроматографов для
анализа компонентов химического оружия по программе НАТО. Приборы
устанавливаются на транспортных средствах, газ-носитель - воздух, чув¬
ствительность < 0,03 мг/м и < 0,01 мг/м, время анализа 3 мин., вес 5 кг.Наилучпше портативные газовые хроматографы, разработанные в послед¬
ние годы - это «Микро-ГХ» (Angilent) и СР-4900 GC (Varian). «Микро-ГХ»
анализирует микропримеси на уровне ppm (1*10^ %) и в 10 раз быстрее, чем
обычные хроматографы, он полностью автономен в тетение 8 часов (имеется
электричесше и газоюе питание). Прецизионный дозатор обладает исключи¬
тельной надёжностью. Все основные узлы изготовлены на основе микротехно-393
Яшин Я.И., Яшин Е.Я., Яшин А.Я.Гюовая хроматографиялогаи (монокристаллического кремния): инжектор с вентилями и с системой
обратной продувки, юлонка сравнения и рабочая, термостат и кагарометр всё
на основе планарной технологаи. Хроматограф может состоять до четырёх
независимых управляемых модулей. На коротких капиллярных колонках
можно быстро и эффекгавно разделять многае смеси, в частности природный
газ может быть разделён за 160 секунд. Также четырёх канальным прибором
является модель СР-4900 GC (Varian). В этом приборе изотермический термо¬
стат от 30 до 180“С, детектор - кагарометр с обьёмом 200 нл (4 элемента),
предел детеетирования 1*10'^% с капиллярной юлонкой, воспроизводимость- < 0,5% СКО для пропана (концентрация 1%). Размер четырёхканального
прибора: 28x15x55 см, вес 5,2 кг, потребляемая мощность 180 Вт. В приборе
два выхода: аналоговый, цифроюй (через RS-232). Прибор может комплекто¬
ваться газовым баллоном на 300 мл с давлением до 120 атм., перезаряжаемой
батареей.В таблице 12.18 приведены некоторые типичные современные порта¬
тивные газовые хроматографы. Следует выделить газовые хроматографы
для анализа выдыхаемого воздуха и портативный газовый хроматограф
для определения наркотиков и взрывчатых веществ.Таблица 12.18Портативные хроматографыМодельФирмаТехнические характеристики1ЭХО-В-ФИДКТИГЭП со РАНДетектор ФИД. Поликапиллярная
колонка 20 см (N=8000-10000) t-40“-
190“ С. Анализ кернов. Воздух газ-
носитель2ЭХО-ВКТИГЭП со РАНОпределение ОВ и ВВ3ЭХО-EWКТИГЭП со РАНСменные детекторы (ФИД, ДЭЗ,
АИД) ПКК4ФГУ-1ООО «ФизЛаб Прибор»Определение вредных веществ
в вощухе и почве. Капиллярная
колонка. ФИД5VoyagerФотовакФИД (10,6 cv) 1ррв-1000ррв
ЕРА 601,602,624, Вес 6,8 кг6СР-4900MicroGCВарианПриродный газ; параметры выхлопа;
наркотические газы, 3 детектора7GC/Saw
модель 4200
Z NoseElectronic Sensor
TechnologyДетектор поверхностной акустиче¬
ской волны. Наркотики, взрывчатые
вещества и др.8OralChromAbilit Соф/Анализ выдыхаемого воздуха394
ГЛАВА 12 Аппаратура для газовой хроматографии12.11.3 Промышленные газовые хроматографыПроизводствам, в которых необходим постоянный контроль за про¬
цессами, нужны автоматические промышленные хроматографы, в боль¬
шинстве случаев — во взрывозащишенном исполнении.К таким промышленным хроматографам на потоке выдвигаются
особые требования:- чрезвычайно высокая надежность (в некоторых случаях требует¬
ся непрерывная работа до трех месяцев и более без профилактических
осмотров);- полная автомагизация, включая забор прс^ы, разделение, детекгирова-
ние, обработку данных и передачу ре^ьтатов в режиме реального времени;- высокие требования к подготовке (освобождение от механических
примесей, влаги и тяжелых смолистых веществ);- исключительная стабильность хроматографических колонок;- взрывозащищенное исполнение.Первые отечественные промышленные хроматографы были разра¬
ботаны в СКБ АНН (Специальное конструкторское бюро по автоматике
в нефтепереработке и нефтехимии) в 1962 - 63гг.: ХПА-2, ХПА-3-150,
ХПА-Ч-133Г-84А во взрывозащищенном исполнении. Это СКБ долгое
время специализировалось в промышленных хроматографах. Из послед¬
них моделей следует выделить хроматографы серии «Микро-хром-1121»
(три модели) и «Нефтехром-1123» (восемь моделей). Дзержинские НПО
«Химавтоматика» разработало первые промышленные хроматографы с
пламенно-ионизационным детектором (1960-64 г.г.) серии РХ-1,2,3,4,5,6,7,
модели ХПУ-1, ХПУ-2, ХПИ-21, ХПУ-300. Тысячи хроматографов были
выпущены и внедрены в разные отрасли промышленности. Для автома¬
тического контроля органических примесей в жидком кислороде выпу¬
скались хроматографы «Микро-1», «Микро-2» и «Микро-3». Автомати¬
ческий хроматограф «Микрофтор» применялся для анализа органических
примесей в воздухе производственных помещений.В последние годы интерес к промышленным газовым хроматографам
сильно возрос. В течение последних лет разработаны новые отечествен¬
ные промышленные хроматографы: приборы РОС 90.50 фирмы «Бакс»
совместно с итальянской фирмой «Дани», прибор «Кристалл 7000» СКБ
«Хроматэк», прибор «Стрим» ОАО «Интерлаб», ХПУ ОАО «Цвет», Ин-
терхром-2003 (модели 2003-1, 2003-2, 2003-3).Более 20 фирм в мире выпускают промышленные хроматографы. Наи¬
более известные среди них: Иокогава, Фишер-Роземаунт, Хартман-Браун,
Сименс и др.395
Яшин Я.И., Яшин Е.Я.. Яшин А.Я. Газовая хроматографияОсновные современные тенденции развития промышленных хромато¬
графов - это миниатюризация, использование капиллярных колонок, тер¬
мостатов с программированием температуры, улучшение программного
обеспечения и удобства эксплуатации и др.Среди портативных промышленных хроматографов следует выделить
модели GCX (Роземаунт), Smart Chromatograph (Yamatake, Honeywell), Си¬
менс, модель micro SAM.Иокогава. Среди традиционных промышленных хроматографов одну
из ведущих позиций занимает фирма «Иокогава Электрик Корпорэйшн»,
принимая во внимание качество, надежность и цену приборов. Первый про¬
мышленный газовый хроматограф фирма выпустила в 1959 г. С этого време¬
ни на фирме сменилось четыре поколения промышленных хроматографов.
Последняя улучшенная модель ОС 1000 МКП разработана в 2001 г. Она
выпускается с тремя детекторами (по выбору): пламенно-ионизационным
(ПИД), пламенно-фотометрическим (ПФД) и катарометром (ДТП), в ее со¬
ставе два термостата: изотермический обьемом 40 л от 55-225®С (стабиль¬
ность ± 0,03®С), с программированием температуры от 60 до 320®С, скорость
программирования 1-30 “/мин, в приборе могут быть использованы как на¬
садочные, так и капиллярные колонки, максимальное число анализируемых
потоков более 30. По числу внедренных приборов «Иокогава Электрик Кор¬
порэйшн» занимает лидирующее положение, более 6000 штук в 40 странах.
Сотни приборов бьши внедрены в нашей стране. Наибольшее применение
эти приборы находят в нефтехимической и нефтеперерабатывающей про-
мьшшенностях: в производстве этилена, полиэтилена, полипропилена, ви¬
нил хлорида, стирола, бутадиена и многих других.Сименс. В приборе РОС 302 модели II реализован принцип «два в
одном», в одном корпусе два независимых хроматографа. Прибор RGC 202
объединяет гибкость лабораторного хроматографа с надежностью промыш¬
ленного. Детекторы: ПИД, ПФД, ДТП, ЭЗД. В 2002 г. фирма выпустила ма¬
логабаритный промышленный хроматограф Micro SAM, в котором реализо¬
вано несколько новых решений: капиллярные колонки с микро детекторами
размещены на термостатируемой пластине, в приборе нет переключающих
поворотных кранов, масса гфибора небольшая, взрывозащита щелевая.Фирма АВВ process analytics предлагает серию моделей Vista П.
Модель 2001 - это промышленный хроматограф для сверхкритической
флюидной хроматографии; модель 2002 для контроля дистилляционных
процессов; модель 2003/2004 - анализаторы PNA / PINA (парафинов, на-
фтенов, изопарафинов, ароматики); модель 2005 - с программированием
температуры; модель 2007 для анализа серасодержащих соединений в не¬
фтепродуктах; модель 2008 - анализатор олефинов.396
ГЛАВА 12 Аппаратура для газовой хоаматтрафииФирма «Daniel Industries» производит ПГХ «Danalyzer» с 1985 г. В
1997 г. модель модернизирована, сокращен цикл анализа, уменьшено по¬
требление газа. Эта модель используется в основном для определения те¬
плоты сгорания природного газа. Продано более 5000 приборов.Фирма «Фишер-Роземаунт» в 1998 г. предложила новое поколение
ПГХ. Модель GCX компактная, легкая, устанавливается ближе к процессу,
исключая транспортировку газа. Детекторы: ПИД, ДТП, термостат от 40 до
140“ С. В приборе электронный рег)отятор расхода, капиллярные колонки.Фирма «Яматаке (Хаиуэлл)» также разработала малогабаритный
ПГХ - модель SGC 302 Smart Gas Chromatograph. Этот прибор может мон¬
тироваться в полевых условиях, его вес всего 11 кг, детектор по теплопро¬
водности, термостат до 120"С, в приборе имеется самодиагностика.Широко известны следующие модели ПГХ: модель Optichrom фирмы
«Hartmann & Braun», модель 2920 On-line BTU Analyzer фирмы «EG and G
Chandler», модель 6800 фирмы «Houston Atlas», a также выпускаемые ПГХ
фирм «Baker», “Combustion Engineering”, «Bendix», «Fluid Data», «Applied
Automation», «Dani Industries», «Elsag Bailey Process Automation», «Gal¬
vanic Analytical System», «HNU Process Analyzer», «Onix Process Analysis»,
«Trace Analytical» и др.Дальнейшее развитие газохроматографического приборостроения бу¬
дет продолжено в направлении специализации моделей, совершенствова¬
ние всех узлов прибора, увеличении степени автоматизации всего анализа,
включая автоматизацию пробоотбора, систем ввода проб, совершенствова¬
ние детектирующих систем с целью снижения пределов детектирования,
создание новых многомерных вариантов хроматографии, стабильных и
селективных колонок.397
Яшин Я.И., Яшин Е.Я., Яшин А.Я. Гтовая хранатографииЛитература1. Hinshaw J.V. Термостат для газовых хроматографов - горячая тема.
LC-GC N. Amer. 2000, v. 18, p. 1142-1147.2. Hirae К. Термостаты для газовой хроматографии. Jpn. Kokai Tokuio
Koho Jp 11326301 (cl. G 01 N 30/54) 26 Nov. 1999 Appl. 1998.3. Schiewe J., Ehrfeld W., Hang Т., Lowe H., Richter Т., Yan X.L., Kur^nov A. A.
and Unger K.K. Быстрый нагревательный модуль для программирования
температуры в ГХ в микрореакторных системах. Microreact. Technol.
Ind. Prospects Proc. Int. Corf. 3"* 1999, p. 645-653.4. Gaisford S., Walters D.L. Аппаратура микроволнового нагрева коло¬
нок в ГХ. РСТ Int. Appl. WO 2000052970 Al. 8 Sep. 2000 CA Selects
Plus GC 2000, N 20, 626762.5. Van Deursen М., Janssen H., Beens J., Rutters D., Cramers C. Устрой¬
ства для нагрева. J. Microcolium Sep. 2001, v. 13, p. 337-345.6. Gordon G.B. Индукционный нагреватель-ловушка в инжекторах.
US US 5. 954860 (cl. 95-87; 861 D 115/08) 21 Sep. 1999 Appl. 956378
23 Okt. 1997 11 pp.7. Walters D.L., Gaisford S. Хроматографическая колонка с микроволно¬
вым нагревом. US US 5939614 (cl. 73-23.39) GO 1 N 30/02 17 Aug. 1999
Appl 108.297 1 Jul. 1998 14 pp.8. Ettre L.S. Некоторые замечания по программированию температуры
с использованием резистивных нагревателей. Ат. Lab. (Shelton) 1999,
v.31,N 14, p. 30.9. Бражников В.В. Дифференциальные детекторы в газовой хромато¬
графии, Москва, Атомизяат 1974,189с.10. Бражников В.В. Детекторы для хроматографии, Москва, Машино¬
строение, 1992,318с.11. Калмановский В.И. Труды по химии и химической технологии.
Горький, 1960, т.З, вып.З, с.545.12. McWilliam J.G., Dewar R.A. А Flame Ionisation Detector for Gas Chro¬
matography. Nature, 1958, v. 181, p. 760.13. Harley J., Nel W., Pretorius V. A Flame Ionisation Detector for Gas
Chromatography. Nature, 1958, v. 181, p. 177.14. Thompson A.E. A Flame Ionisation Detector for Gas Chromatography.
J. Chromatogr. 1959, v. 2, p. 148.15. McWilliam l.G. Australian patent number 224504 (issued on 21 Octo¬
ber 1959).16. McWilliam l.G. U.S. patent number 3039856 (issued 19 June 1962).17. Калмановский В.И., Фикс M.M., Яшин Я.И. Ионизационно¬
пламенный детектор. Авторское свидетельство № 134058 (приоритет
30.04.60). Бюл. изобретений 1960, № 23.398
ГЛАША 12 Литерагнщ>а18. Калмановский В.И., Фикс М.М., Яшин Я.И. Новые детектирующие
устройства для автоматических хроматографических анализаторов. Тру¬
ды по химии и хим. технологии (г. Горький) 1960, т 3, вып. 3, с. 625.19. Буров А.Н., Калмановский В.И., Яшин Я.И. Быстрое хроматогра¬
фическое определение малых примесей углеводородных газов. Труды
по химии и хим. технологии, 1961, т. 4, вып. 2, с. 345.20. Калмановский В.И., Л^дев В.Р., Полякова Л.В., Фикс М.М., Яшин Я.И.
Применение капилл^ной хроматографии в аналюе угаеводородов. Труды
по химии и хим. технологии, 1961, т. 4, вып. 2, с. 351.21. Буров А.Н., Калмановский В.И., Поляшв В. Л., Фикс М.М., Яшин Я.И.
Ионизащюнные методы определения микропримесей в газах. Труды Ко¬
миссии по аналитичесшй химии. Органический анализ, Москва, 1963,
т XIII, с. 247.22. Калмановский В.И., Фикс М.М., Яшин Я.И. Новые детекгарующие
устройства для автоматических хроматографических газоанализаторов.
В книге «Автоматические газоанализаторы» Под ред В.А.Павленш.
Москва, ЦИНТИЭПП, 1961, с. 473-486.23. Holm Т. Aspects of the mechanism of the flame ionization detector.
J. Chromatogr. 1999, v. 842, p. 221.24. Kallai М., Veres Z., Balia J. Response of Flame Ionization Detectors to
Different Homologous Series. Chromatographia, 2001, v. 54, № 7/8, p. 511.25. Kallai М., Balia J. The Effect of Molecular Structure upon the Responce of
the Flame Ionization Detretors. Chromatogr^hia, 2002, v. 56, № 5/6, p. 357.26. Bernier U.R. et al. J. Microlumn. Separ. 2000, v. 12, p. 226.27. Kuipers W.J., Mflller J. Planar micro flame ionization detector with in¬
tegrated guard. Pittcon-2008, 170-5.28. Schaefer B.A., Douglas O.M. J. Chromatogr. Sci. 1971, v. 9, p. 612;
1972, V. 10, p. 110.29. Askew W.C. Anal. Chem. 1972, v. 44, p. 633.30. Russev P., Gaugh T.A., Woolam C.J. J. Chromatogr. 1976, v. 119, p. 461.31. Russev P., Kunova М., Georev V. J. Chromatogr. 1979, v. 178, p. 364.32. Hill H.H., Aue WA. J. Chromatogr. 1976, v. 122, p. 515.33. Verga G.R. et al. HRC & CC 1988, v. 11, p. 248.34. Schneider W, Frohne J.C., Brudenwk H. J. Chromatogr. 1982, v. 245, p. 71.35. DiSanzo F.R J. Chromatogr. Sci. 1990, v. 28, p. 73.36. Witterrood P.T. Chromatographia 1972, v. 4, p. 311.37. Sandra P LC-GC 1987, v. 5, p. 236; HRC 1989, v 12, p. 82, p. 273.38. Lovelock J.E., Lipsky S.R. J. Am. Chem. Soc. 1960, v. 82, p. 431.39. Wentworth W.E. et al. Non-radioactive electron-capture detector. J.
Chromat. 1999, v. 842, p. 229-266.399
Яшин Я.И.. Яшин Е.Я., Яшин А.Я. Газовая хроматографии40. Guiffrida L. J. Ass. off Agric. Chem. 1967, v. 47, p. 293.41. Braznikov V.V., Gurev M.V., Sakodynski K.I. Chromatogr. Rev. 1970,
V 12, p. 1-41.42. Amirav A. Pulsed Flame Detectors Method and Apparatus USA Patent
No 5153673, Israel Patent No 95617, European Patent No 0475250, Japan
Patent No 2759854.43. Ator E., Cheskis S., Amirav A. Pulsed Flame - a novel concept for mo¬
lecular detection. Anal. Chem. 1991, v. 63, p. 2061.44. Frishman G., Amirav A. Field. Anal. Chem. Technol 2000, v. 4, p. 170-194.45. Bancon-Montigny C. J. Chromatog. 2000, v. 896, p. 149.46. Langhorst M.L. Photoionization detector sensitivity of organic com-
poimds. J. Chromat. Sci. 1981, v. 19, p. 98.47. Будович В.Л., Шляхов А.Ф. Успехи химии 1989, т. 58, с. 1354-1389.48. Driscall J.N. Am. Lab. 1976, v. 9, p. 71-74.49. Адамия ТВ., Будович В.Л., Шляхов А.Ф. Зав. лаб. 1994, № 7, с. 5.50. Grote С., Levsen К., Wunsh G. Автоматический анализатор органиче¬
ских соединений в воде на основе газового хроматографа и микротвер-
дофазной экстрающи (МТФЭ). Anal. Chem. 1999, v. 71, p. 4513-4578.51. Haley L.V., Thekkadath G.C. Портативная hand-held аппаратура ГХ-
МС. РСТ Int. Appl. WO 9941601 (cl. G 01 N 30/02) 19 Aug. 1999 US
Appl. PV 74431 11 Feb. 1998 32 pp.52. Haas J.S. et al. Портативный ГХ-МС для химического анализа
on-situ (на месте). РСТ Int. Appl. WO 0062054 (cl. G 01 N 30/72) 19
Oct. 2000, US Appl. 289755, 12 Apr. 1999, 18 pp.53. Crume C. ГХ на микрочипах (с 1980 г.). ГХ-МС-портативные, по¬
левые. Environ. Test. Anal. 2001, v. 10, p. 22-26.54. Eckenrode B.A. Применение полевых портативных ГХ-МС. Обзор.
J. Am. Soc. Mass Spectrum 2001, v. 12, p. 683-693.55. Frishman G. ГХ-ПИД со встроенным генератором водорода. Field
Anal. Chem. Technol. 2001, v. 5 (3), p. 107-115.56. Sacks R., Whiting J., Zellers E. Новейшая технология разработки
автономного микро газового хроматографа. Presented at the 24* Inter¬
national Symposium on Capillary Chromatography, Las Vegas NV May
20-24, 2001.57. Whiting J., Sacks R. Воздух как газ-носитель в портативных хрома¬
тографах. Anal. Chem. 2002, v. 74 (1), p. 246-252.400
ГЛАВА 13 Комбинированные методы13. КОМБИНИРОВАННЫЕ МЕТОДЫ.13.1. Основные комбинированные методы ГХ.Для расширения юзможностей ГХ, в основном для целей идентифи¬
кации, используются различные комбинированные методы. Широш приме¬
няются следующие шмбинации ГХ с другами методами анализа: ГХ-МС,
ГХ-ИКС, ГХ-АЭД (атомно-эмиссионное детектирование). Описаны при¬
менения ГХ-ЯМР, ГХ-ААС (атомно-абсорбционная спектрометрия), ГХ-
иммунный анализ, ГХ-олфактометрия, сочетания ГХ-МС-ИКС.Находят применение комбинации ГХ с другими методами хроматофа¬
фии: ГХ-ВЭЖХ, ГХ-СФХ (сверхкритическая флюидная хроматофафия),
ГХ-ТСХ (тонкослойная хроматофафия).Далее подробно будут рассмотрены самые распространенные сочета¬
ния: ГХ-МС, ГХ-ИКС и ГХ-АЭД.13.2 ГХ-МСВ последние десятилетия метод ГХ-МС получил исключительное раз¬
витие, разработаны и серийно выпускаются десятки новых моделей. Прибо¬
ры ГХ-МС стали доступными, «рутинными» для многих лабораторий. При¬
нято считать, что самая надежная идентификация неизвестных соединений
в сложных смесях может быть проведена ГХ-МС. Системы ГХ-МС могут
ушмплектовываться банками (библиотеками)масспеетров до 450000 ед.В таблице 13.1 приведена общая схема ГХ-МС систем. Указаны основ¬
ные методы ионизации, методы разделения ионов на основании различных
соотношений масса/заряд и методы детектирования ионов.Таблица 13.1Газовый 1роматографввод и подготовка пробыМетоды ионизацииэлектронный удар- химическая ионизация- ионизация лазерным излучением десорбцией в матрицеМетоды разделения ионов разной массы- магнитный- квадрупольный- ионная ловушка- времяпролетныйДетектирование- фотоэлеиронный умножитель- коллектор Фарадея- плоская электронная матрица40126 Газовая хроматография
Яшин Я.И., Яшин Е.Я., Яшин А.Я.Газовая хроматографияПервооткрывателем МС является Д.Д.Томсон, шторый установил, что
положительно заряженные частицы разной массы по-разному отклоняются в
магаитном поле. Это явление он положил в осно^ первого самодельного мас-
спектрометра в 1913 г. Д.Д.Томсон предсказал большие перспективы МС.Сочетание ГХ и МС исклкнительно плодотворно. ГХ служит гак наи¬
более совершенный метод пробоподготовки перед МС. Общая схема ГХ-МС
приведена на рис. 13.1.МшШ л.иШйСтСш1<аО»Гмтя&еOmiroUTinrafaUi» IL-0)1|имCbP9iaato|rsinРис. 13.1 Общая схема ГХ-МС.Идентификация - это установление природы соединения по опреде¬
ленным параметрам. Принцип МС заключается в ионизации соединений
с последующей регистрацией ионов. При регистрации заряженных частиц
достигаются очень низкие пределы детектирования (до Ю '* г/с).Основные методы ионизации - электронный удар и химическая иони¬
зация.В основном в масспектрометрии применяют условия, приводящие к
образованию положительно заряженных ионов катионов и катион-ради¬
калов. В некоторых специальных случаях используют детектирование от¬
рицательно заряженных анионов и анион-радикалов. В последнем случае
имеет место снижение пределов детектирования и повышение селектив¬
ности определения.МС стал наиболее распространенным детектором в ГХ. В некоторых
отношениях он идеальный детектор, т.к. позволяет проводить одновремен¬
но как качественный, так и количественный анализ.Одним из недостатков МС является необходимость вакуумирования
всей системы (интерфейс, источник ионов, масс-анализатор, детектор ио¬
нов) до давления 10 ’ Па и менее.В ГХ-МС в подавляющем большинстве случаев используется капил¬
лярная шлонка, т.к. тольш при малых расходах (0,5 - 1 мл/час) можно бы¬
стро откачать и собрать в источнике ионов давление менее 10 ^ Па.Ионизация производится потошм электронов, выделяемых из нагре¬
того катода. Электроны ускоряются за счет разности приложенных потен-402
ГЛАВА 13 Комбипиротнные методыциалов. Для ионизации большинства органических соединений достаточ¬
но электрона с энергией ионизации более 12 ev. В источнике ионизации
можно создавать электроны с энергаей ионизации до 100 ev. В настоящее
время международным стандартом энергии ионизации принят 70 ev. Все
банки данных масс - спектров получены при такой энергии ионизации.При 70 ev в основном образуется положительно заряженные катионы
и катион - радикалы. Полученная совокупность осколочных ионов после
их разделения и детектирования, представляет собой масс-спектр, характе¬
ризующий исследуемое соединение.При химичесшй ионизации происходит ион-мояекузщ)ные взаимодействия
за счет введения в источник газа - реагента. Элеюроны в основнш ионшируют
газ - реагент, штс^ый затем взаимодействует с молекупаш! ощеделяемых со¬
единений. Поэтому при химичесшй ионизации основные ионы - это протони-
рованные молекулярные ионы анализируемых соединений.В таблице 13.2 приведены используемые газы реагенты и соответству-ющие ионы реагентов.Таблица 13.2Газы реагентыИоны реагентыН,н,^щСН/нрпр*ИзоС,Н„ИзоСД*NH,Ш/Ниже приведены подробные реакции метана в качестве реагента при
химичесшй ионизации:СН^ + ё — СН/ + 2ё
СН/ + ё СН," + СН/СН/ + СН, -> СН/ + СН,"СН/ + СН^ CjH/ + Щ
СН/ + ц СН^ + цН"Аммиак протонизирует амины, но не углеводороды.Отрицетельная химическая ионизация в 1000 раз более чувствительная,
поэтому она применяется при анализе пестицидов, полихлорированных би¬
фенилов, нешторых лекарств, стероидов в биологических жидшстях.При химичесшй ионизации газ-реагент вводится в реакционную ка¬
меру объемом 1 мл и непрерывно ионизируется электронным уда1юм. Нить
лампы накаливания находится вне реакционной камеры. Выделяемые
электроны усшряются под действием разности потенциалов.403ге-
Яишн Я.И.. Яшин Е.Я.. Яшин А.Я. Газовая хрвтатографияВ квадрупольном МС масс-фильтр состоит из четырех параллельных
стержней, расположенных по углам квадрата (рис. 13.2) и создающих ги¬
перболическое поле. Только ионы с резонансной частотой покидают взаи-
моперекрывающие поля квадруполя.Ионная ловушка состоит из симметричных кожухов и кольцевого
электрода (рис. 13.3).(IVРис. 13.2Ионы остаются в ней за счет стабильного высокочастотного поля
(радиочастотный диапазона в течение короткого промежутка времени,
(миллисекунды). Для разделения ионов высокочастотное поле варьиру¬
ется так, что ионы в определенной последовательности (по значениям
отношения m/z) покидают свои траектории внутри ионной ловушки и
регистрируются электроумножителем в виде спектра. Изменения высо¬
кочастотного поля происходит очень быстро, поэтому возможно высоко¬
скоростное сканирование. Диапазон времени пребывания 30 - 250 мс.
В этом устройстве возможны системы МС-МС. Цель МС-МС изоляция
одних ионов от всей матрицы, при этом улучшается отношение сигнал/
шум и повышается чувствительность.Времяпролетный МС. Принцип работы времяпролетного МС осно¬
ван на разделении ионов за счет разной скорости прохождения области
дрейфа, в связи с этим они попадают в детектор в разное время, в зависи¬
мости от отношения массы к заряду.Время пролета занимает микросекунды, это позволяет получить ин¬
формацию о составе смеси в ультракороткое время. Наилучших дости¬
жений в разработке времяпролетных МС достигла фирма “Leco”, которая
реализовала в этом приборе и режим двухмерной газовой хроматографии
(ГХ-ГХ)(рис. 13.4).В приборе “Leco Pegasus” за счет уникального программного обеспе¬
чения и высокочастотной обработки данных возможна обработка до 500
спектров в сек. Диапазон масс прибора ГХ-МС-ВП до 1000 а.е.м.404
ГЛАВА 13Комбинированные методыРис. 13.4Этот ГХ-МС-ВП открывает новые уникальные возможности в нефте¬
химии, контроле загрязнений окружающей среды, пищевых продуктов, в
медицине, судебной медицине и др. областях.13.3. Газовая хромато1рафия - ИКС Фурье cneicrpocKoniui (ГХ-ИКС).Впервые ГХ-ИКС была реализована в 1967 г. [10], чуть позднее этими
же авторами была предложена комбинация ГХ-МС-ИКС [11]. Однако ши¬
рокого применения ГХ-ИКС не получила, пока не были разработаны чув¬
ствительные детекторы на основе теллурида ртути и кадмия и проточная
ячейка типа «light pipe» [12]. Ячейка «light pipe» представляет собой капил¬
ляр из боросиликатного стекла с позолоченной внутренней поверхностью
[12]. Ранее ячейка имела следующие размеры: внутренний диаметр 1 мм,
длина 10 см. В последние годы стали использовать капилляры с внутрен¬
ним диаметром 0,1-0,2 мм. Луч от источника ИКС-излучения направлен
в ячейку, через которую проходят полосы анализируемых соединений из
хроматографической колонки. ИКС-излучение многократно отражается от
золотой поверхности ячейки и достигает ИКС-чувствительного детектора.
Чувствительность детектора зависит от длины оптического пути. С другой
стороны, для предотвращения размывания объем ячейки по возможности
уменьшают. Объем ячейки колеблется в пределах 50-300 мкл.Предложены три типа интерфейса ГХ-ИКС: непосредственная пода¬
ча в ячейку типа «light pipe», матричная изоляция [13], прямое осаждение
(криоулавливание) [14].Сравнение всех этих методов проведено в работе [15]. Более подробно
о всех особенностях ГХ-ИКС написано в книге [16].В матричной изоляции на специальной пластине криоколлектора за¬
мораживается до 30 соединений, которые необходимо идентифицировать.
Их последовательно улавливают и регистрирует ИК-спектры с помощью405
шин я.и., Яшин Е.Я.. Яшин А.Я.Газовом хроматографияпециальных устройств. Такая матричная изоляция позволяет повысить
увствительность определения в 10-100 раз. Однако применение этого ме-
ода ограничено из-за высокой стоимости.В методе прямого осаждения соединений, которые необходимо иден-
ифицировать, их криоулавливают на пластинах из селенида цинка, про-
рачных для ИКС-излучения. Чувствительность этого интерфейса также
ысокая, но ИКС-спектры лучше расшифровываются.Предел ИКС-детектирования порядка МО * г без фокусирования. Это
начительно ниже, чем предел детектирования МС. Предел детектирова-
[ия ИКС с криофокусированием 3,5-4,0-10 '^ г.В последние годы большой интерес вызывает сочетание ГХ-МС-ИКС
Ц1Я целей идентификации неизвестных соединений, т.к. эти методы эф-
()ективно дополняют друг друга.Известно, что, с одной стороны, масспектры изомеров часто идентич-
1ы, а ИКС-спектры гомологов одинаковы. С другой стороны, МС-спектры
•омологов, так же, как и ИКС-спектры изомеров, сильно различаются. По
;пектрам ИКС можно определять структурные особенности молекул, на-
1ичие тех или иных функциональных групп непредельных связей, изоме-
юв положения, цис- и транс-изомеров.Система ГХ-МС-ИКС коммерчески доступна [17].Весьма эффективна система ГХ-ИКС с предварительным концентри-
юванием «on-line» [18].ГХ-ИКС применяется для идентификации и определения разных со-
гдинений (таблица 13.2). Особенно часто системы ГХ-ИКС находят при-
*1енение в контроле загрязнений окружающей среды, в биологических,
;удебно-криминалистических исследованиях [28].Таблица 13.2Примеры идентифи1шции соединений методами ГХ-ИКС, ГХ-ИКС-МС№№Соединения, включая изомерыМетодыСсылки1.Полихлорбифенилы (всего 209 возможных соеди¬
ненийГХ-ЖС192.Бензол, толуол, этилбензол, изомеры ксилоловГХ-ИКС203.Амфетамин, метаамфетамин (в судебной медицине)ГХ-ИКС214.Полихлордибензодиоксины, полихлордибензофу-
раны (при утилизации муниципальных отходов)ГХ-ИКС-МС225,Идентификация продуктов фиторазложения герби¬
цидов (феноксиалкановые кислоты)ГХ-ЖС-МС236.Идентификация серасодержащих соединений, от¬
ветственных за запах мясаГХ-ИКСГХ-МС247.Определение присутствия вредных, раздражающих
веществ в мылахГХ-МС-ИКС25406
ГЛАВА иКомбинированные методы№№Соединения, включая изомерыМетодыСсылкиОпределение трихотеценовых микотоксинов в
пище и кормах ГХ-ИКСГХ-МС269.Идентификация компонентов химического оружияГХ-ИКСГХ-МС2710.Бромхлордиоксииы
окружающей средеи дибензофураны вГХ-ИКС28И.Идентификация ароматов, запахов, эфирных масел
в природных продуктах ГХ-ИКС-МС2912. Идентификация компонентов эвкалиптового масла ГХ-ЖС-МС3013.4. Газовая хроматография - атомно-эмиссионное детектирование.Атомно-эмиссионный детектор (АЭД), основанный на микроволновой
индуцированной плазме, был коммерчески введен в 1989 г. [32]. В этом
детекторе элюент из колонки попадает в емкость, где создается плазма,
анализируемые соединения разлагаются до атомного состояния. Атомы
возбуждаются энергией плазмы. Спектр эмиссии диспергируется моно-
хроматорной или полихроматорной системами и измеряется фотодиодной
линейкой. На фотодиодной линейке можно детектировать разные элемен¬
ты в диапазоне 171-690 нм.АЭД может измерять разные элементы. Газовый хроматограф с АЭД
может записывать поочередно хроматограммы, на которых регистрируют¬
ся пики, содержащие атом углерода, водорода, серы, фосфора, кислорода и
многих других элементов.Основные преимущества систем ГХ-АЭД:- АЭД может регистрировать любые элементы, которые возбуждаются
в плазме гелия, а это почти все элементы периодической таблицы;- за счет измерительной системы АЭД - селективный детектор, он
может регистрировать только одни элементы, не реагируя на другие; эта
селекгавность практически абсолютная;- калибровка АЭД по какому-либо элементу не зависит от молекуляр¬
ной структуры молекулы, в которой этот элемент находится;- наряду с высокой селективностью АЭД обладает высокой чувствитель¬
ностью, в частности по угаероду АЭД в 5 раз болте чувствителен, чем НИД;- по записанным многоканальным хроматограммам, в частности по
С, Н, О, S, N, можно определять количественные соотношения этих эле¬
ментов в органических молекулах и устанавливать эмпирические формулы
исследуемых веществ.Подробнее об устройстве механизма работы АЭД и областях его при¬
менения можно прочитать в следующих работах [32-39].407
Яшин Я.И., Яшин Е.Я., Яшин А.Я. Газовая хроматографияЛитература1. Хмельницкий Р.А., Бродский Е.С. Хромато-масс-спектрометрия. М.,
Химия, 1984, 212 с.2. Карасек Ф., Клемент Р. Введение в хромато-масс-спектрометрию.
Пер. с англ. М., Мир, 1993, 237 с.3. Исидоров В.А., Зенкевич И.Г. Хромато-масс-спектрометрическое
определение следов органических веществ в атмосфере. Ленинград,
Химия, 1982, 136 с.4. Зенкевич И.Г., Иоффе Б.В. Интерпретация масс-спектров органиче¬
ских соединений. Ленинград, Химия, 1986,176 с.5. Hubschmann H.J. Handbook of GC/MS, Wiley, New York, 2000.6. Barker J. Mass Spectrometry, Wiley, Chichester, UK, 1999.7. Gerhards P., Bans V., Sawazki V., Szigan J., Westman E. GC/MS in
clinical chemistry, Wiley-VCH, Weinheim, 1999.8. Niessen W.M.A. (ed) СштеЩ Practice of Gas Chromatography - Mass
Spectrometry, Dekker, New York, 2001.9. McLafferty P.M. and Turecek F. Interpretation of Mass Spectra, 4th Edn.
University Books, Mill Valley, 1993.10. Low M.J.D., Freeman S.K. Anal. Chem. 1967, v. 39, p. 194.11. Low M.J.D., Freeman S.K. J. Agric. Food Chem. 1968, v. 16, p. S25.12. Azzaraga L.V. Appl. Spectrosc. 1980, v. 34, p. 224.13. RmiyG.T.,Boume S., Cunningham P.T. Anal. Chem. 1979,v.51,p. 1535.14. Bourne S. Am. Lab. 1998, v. 30, p. FI7.15. Norton K.L., Griffiths PR. J. Chromatog. 1995, v. 703, p. 383.16. White R. Fourier Transform Infrared Spectroscopy and its Application,
Marcel Dekker, New York, 1990.17. Demirgian J.C. Trends Anal. Chem. 1987, v. 6, p. 58.18. Hankemeier H. et al. J. High Resol. Chrom. 1998, v. 21, p. 341.19. Budzimki H., Hermange Y, Pierard C. et al. Analusis 1998, v. 20, p. 155.20. Diehl J.W., Finkbeiner J.W, Disanzo RR Anal. Chem. 1993, v. 65, p. 2493.21. Kalasinsky K.S. et al. J. Anal. Toxicol. 1992, v. 16, p. 332.22. Sommer S. et al. Anal. Chem. 1997, v. 69, p. 1113.23. Climent M.J., Miranda M.A. J. Agric. Food Chem. 1997, v. 45, p. 1916.24. Misharina T.A., Golovnya R.V. J. Anal. Chem. 1997, v. 52, p. 227.25. Tomlinson M/J., Wilkins C.L. J. High Resolut. Chromatog. 1998, v. 21,
p. 347.26. Mossoba M.M. et al. J. AO AC Int. 1996, v. 79, p. 1116.27. S6derstrom M.T. et al. J. Chromatog. 1996, v. 742, p. 191.28. Ragunathan N., Krock K.A., Klawun C. et al. J. Chromatog. 1995,
v. 703, p. 335.408
ГЛАВА 13 Литература29. Childers J.W. et al. Chemosphere 1992, v. 25, p. 1285.30. Coleman W.M., Gordon B.M. J. Chromatog. Sci. 1991, v. 29, p. 371.31. Hedges L.M., Wilkins C.L. J. Chromatog. Sci. 1991, v. 29, p. 345.32. ValenteA.L.R., Uden P.C. Analyst 1990, v. 115,p. 525, HPG2350AAtomic
emission detector for gas chromatography. Hewlett-Packard Co. 1995.6 p.33. Баффингтон P. Применение атомно-эмиссионной спектросюпии в
высокочастотном разряде для газовой хроматографии. Пер. с англ. Мо¬
сква Мир 1994. 78 с.34. Uden Р.С. Selective detectors. Environmental, industrial and biochemi¬
cal applications. Ed. by R.E.Sievers, New York, Wiley, 1995, p. 143-169.35. Anderson J.T., Schmid B. Fres. J. Analyt. Chem. 1993, v. 346, p. 403.36. Albro T.G., Dreifuss J.J., Wormsbecher R.E. J. High Resolut. Chromat.
1993, V 16, p.21.37. Quimby B.D., Sullivan J.J. Anal. Chem. 1990, v. 62, p. 1027.38. Hankemeir Th. et al. Chromatographia, 1998, v. 48, p. 273.39. Tao H. et al. Anal. Chem. 1999, v 71, p. 4208.409
Яшин Я.И.. Яшин Е.Я., Яшин А.Я. Газовая храчатографияГЛАВА 14. КАЧЕСТВЕННЫЙ АНАЛИЗ В ГАЗОВОЙ
ХРОМАТОГРАФИИ14.1. ВведениеГазовую хроматографию используют в основном как количествен¬
ный метод, причем чаще всего, особенно в химической промышленно¬
сти, перед проведением анализа уже известен качественный состав
смеси. Однако в последние годы в связи с широким применением га¬
зовой хроматографии для анализа смесей неизвестного качественного
состава в окружающей среде, в пищевой промышленности, биохимии,
медицине, санитарной и судебной химии и других областях вопросы
идентификации разделяемых компонентов в газовой хроматографии
стали весьма актуальными.В настоящее время можно, выделить следующие методы иденти¬
фикации, применяемые в газовой хроматографии:- методы ГХ-МС, ГХ-ИКС, ГХ-АЭС;- сочетание ГХ-МС с параметрами удерживания;- методы с использованием химических реакций до хроматографиче¬
ской колонки;- методы с применением специальных детектирующих систем для
идентификации;- методы с использованием качественных химических реакцийна выходе из колонки;- методы, основанные на сборе разделенных компонентов в
чистом виде и применении других физико-химических методов;- методы, основанные на селективном удалении (поглощении) неко¬
торых разделенных компонентов смеси;Методы ГХ-МС и ГХ-АЭС, описанные в главе 13.14.2 Методы идентификации, основанные на измерении параме¬
тров удерживанияВ проявительной газовой хроматографии время выхода максиму¬
ма пика компонента смеси при определенных постоянных условиях ха¬
рактеризует природу этого соединения, поэтому время удерживания
может быть использовано для целей идентификации в газовой хрома¬
тографии.Наиболее простой способ — это сравнение времени удерживания не¬
известного вещества с временем удерживания известного соединения
на той же колонке при идентичных условиях. Так как условия раз-410
ЩЛЯАМ 1Штвтши^ттш_«гтотйхрта^^^деления могут колебаться во времени даже на одной колонке, то
удобнее известное вещество добавлять к исследуемому и наблюдать
за изменением высоты и формы пика предполагаемого неизвестного
компонента. Если неизвестный пик принадлежит предполагаемому
добавляемому известному соединению, то высота пика исследуемого
вещества должна возрасти, а форма, вернее, ширина на половине
высоты, должна остаться неизменной. Если же ширина на половине
высоты изменяется после добавки, то это показывает, что добавляе¬
мое вещество другой природы. Однако даже полное совпадение времен
удерживания и неизменность ширины полосы после добавки известного
вещества еще не дает основания для идентификации с полной до¬
стоверностью, так как возможны случаи полной идентичности времен
удерживания и размывания полос двух или более соединений на одной
колонке. Для повышения достоверности следует аналогичную опера¬
цию проделать на двух или более колонках, заполненных сорбентами
разной полярности (обычно полярным или неполярным). В этом случае
вероятность полного совпадения времен удерживания двух веществ
на колонках разной полярности весьма мала. Допустим, совпадают
времена удерживания двух веществ на неполярной колонке, это могут
быть только соединения, существенно отличающиеся своей природой
или строением, так как члены гомологического ряда обычно очень хо¬
рошо разделяются на неполярной колонке. А вещества, различающие¬
ся по своей природе, должны различаться по временам удерживания на
колонках с полярными сорбентами.Для идентификации по параметрам удерживания используют линей¬
ные зависимости логарифма удерживаемого обьема от логарифма дав¬
ления пара, от числа атомов углерода в молекуле п, от температуры
кипения разделяемых веществ. Такие зависимости особенно важны, когда
предполагаемый компонент неизвестной смеси не может быть получен
в чистом виде для непосредственного сравнения удерживаемых объемов
или относительных удерживаний. Поэтому в этих случаях при наличии
двух или большего числа членов того же гомологического ряда объем
удерживания может быть найден графическим путем.Для целей идентификации широко используют индексы Ковача,
которые по существу являются относительными параметрами удержи¬
вания (применяют отношение к двум н-алканам). Индексы Ковача для
н-алканов равны 100 п, индексы Ковача для других соединений определя¬
ются соотношением (см. га. II). В справочниках и монофафиях содержат¬
ся данные по индексам Ковача многих соединений. Созданы специальные
банки данных индексов удерживания. Индексы Ковача для неизвестных411
Яшин Я.И., Яшин Е.Я., Яшин А.Я. Газовая хроматографиясоединений могут быть определены из графической зависимости логариф¬
ма удерживаемого объема от индексов Ковача н-алканов, которые равны
ШОи. Индексы Ковача могут быть определены с большой точностью, осо¬
бенно на капиллярных колонках, поэтому идентификация с их помощью
более достоверна. Для целей идентификации применяют также разность
индексов удерживания соединений на полярной фазе (например, полиэ-
тиленгликоле) и на неполярной фазе (например, апиезоне L, сквалане,
ОУ-1,8Е-ЗОидр)[1].В газо-адсорбционной хроматографии на неспецифических адсорбен¬
тах можно воспользоваться для идентификации линейной зависимостью ло¬
гарифмов удерживаемых объемов от электронной поляризуемости молекул.
Значительное влияние геометрической структуры молекул на удерживаемые
обьемы при разделении на графитированных сажах позволяет в некоторых
случаях проводить идентификацию пространственных изомеров (в частно¬
сти, ЦИС-, транс-изомеров) в методе хроматоскопии А.В.Киселева.14.3 Методы идентификации с использованием селективных
химический реакций.Реакции до колонки бывают двух типов: общие реакции, которые
применимы для широкого круга соединений, и специфические реакции,
с помощью которых можно определять только одну функциональную
группу или соединения только одного типа. Желательно, чтобы эти реакции
можно было проводить в микрограммовых количествах. При работе с ма¬
лыми количествами веществ необходимо проявлять особую тщатель¬
ность и заботу о чистоте.Наибольшее распространение получили реакции с участием водо¬
рода: гидрирование (присоединение водорода), дегидрирование (отрыв
водорода) и гидрогенолиз (распад с участием водорода). Гидрирование
широко используют для определения структуры ненасыщенных соеди¬
нений. Обычно исследуемую смесь хроматографируют перед и после ги¬
дрирования. Количественное и мгновенное гидрирование ненасыщенных
соединений можно проводить в потоке газа-носителя. Для этого перед
хроматографической колонкой нужно установить дополнительную
предколонку с катализатором гидрирования, а в качестве газа-носите¬
ля использовать водород. Идентификация непредельных соединений
в смесях углеводородов и метиловых эфиров жирных кислот с помощью
этого метода стала теперь обычной. Для этого используют палладие¬
вые, платиновые и никелевые катализаторы на твердом носителе типа
хромосорб Р. Сравнением хроматограмм до гидрирования и после ги¬
дрирования легко установить присутствие непредельных соединений.412
ГЛАВА 14 Качественный анализ в газо1Ы1гр^и^т,2,рдф,1^Дегидрирование углеводородов применяют реже. Кейлеманс впер¬
вые каталитически дегидрировал нафтеновые углеводороды С,—С, до
соответствующих ароматических соединений [2]. В этом случае исполь¬
зуют платиново-алюминиевый катализатор при 350°С. Перевод в непре¬
дельные или ароматические соединения приводит к резкому увеличению
удерживания соединений на полярных сорбентах. Путем сопоставле¬
ния хроматограмм до и после проведения реакции можно также прово¬
дить идентификацию веществ. Применение гидрогенолиза для опреде¬
ления углеродного скелета наиболее перспективно [2]. Углеродный скелет
определяют путем отщепления от молекулы всех ее функциональных
групп. Этот метод может быть использован для идентификации кислот,
спиртов, альдегидов, эфиров, кетонов, аминов, амидов, нитрилов,суль¬
фидов и др.При хроматографическом определении углеродного скелета могут
проходить все три реакции: гидрирование, дегидрирование и гидрогено-
лиз. Из них наиболее общая реакция — гидрогенолиз. Температура катали¬
затора в этом случае устанавливается 300°С, скорость потока водорода 20
мл/мин, исследуемая проба порядка 20 мкг. При этом происходит разрыв
связей в функциональных группах с образованием исходного углеводорода
или следующего низшего гомолога. Связи углерод—сера и углерод—га¬
логен (кроме фтора) разрываются и образуется исходный углеводород.
Связь углерод—кислород разрывается во вторичных и третичных спир¬
тах, вторичных и третичных эфирах и кетонах. Связь углерод—азот разры¬
вается во вторичных и третичных аминах и амидах. н-Алканы, проходя
через катализатор, не подвергаются никаким изменениям, так как все свя¬
зи в их молекулах насыщены.Альдегиды, спирты, кислоты и их производные в результате приведен¬
ных реакций дают очень небольшое количество исходного алкана, основ¬
ным продуктом реакции является следующий более низший гомолог.Углеводороды, образующиеся в указанных реакциях, хорошо разделя¬
ются и идентифицируются на колонках с неполярными жидкими фазами,
в частности со скваланом или апиезоном L. На этих фазах углеводороды
элюируют в порядке возрастания температур кипения.Кроме описанных типов реакций с водородом для целей идентифика¬
ции можно проводить и другие типы химических реакций в доколоночном
реакторе. Сводка всех юзможных типов реакций приведена в книгах (3.4).Для установления природы высокомолекулярных веществ, в частно¬
сти полимеров, используют также и реакции термического разложения, а
именно: пиролиз (без доступа кислорода, в инертной среде). Пиролиз
полимеров и других высокомолекулярных веществ при постоянных кон¬413
Яшин Я.И., Яшин Е.Я., Яшин А.Я. Газовая хроматографиятролируемых условиях дает характерный для данного вещества спектр со¬
единений (пирограмму, «отпечатки пальцев»), который во многих случа¬
ях однозначно позволяет характеризовать природу высокомолекулярного
вещества. Температуру пиролиза в зависимости от природы исследуемо¬
го вещества можно изменять в широких пределах (300—1000°С). В не¬
которых случаях пиролизная хроматография в сочетании с эффективными
капиллярными колонками и масс-спектрометром позволяет идентифи¬
цировать не только полимеры разной природы, но и однотипные поли¬
меры, различающиеся только технологией получения.14.4 Методы идентификации с применением специальных де¬
тектирующих систем.Универсальные детекторы для количественных измерений (катаро-
метр, ионизационно-пламенный) нельзя использовать для идентификации.
Однако разработаны некоторые детектирующие устройства, которые по¬
зволяют идентифицировать неизвестные компоненты непосредственно
на выходе из колонки. Показания таких детектирующих систем дают ин¬
формацию о природе разделяемых компонентов. Это прежде всего масс-
спектрометр, который регистрирует масс-спектр любого выходящего из
колонки компонента. В последние годы широкое распространение получи¬
ли приборы, хроматомасс-спектрометры, в которых на выходе хромато¬
графической колонки используют масс-спекгрометр как детектор. В системе
напуска (натекателе) от колонки в масс-спектрометр используют молеку¬
лярные сепараторы, которые селективно удаляют газ-носитель, чаще всего
гелий. Благодаря молекулярному сепаратору в масс-спектрометр попадают
практически только молекулы разделяемых веществ.Электронозахватный детектор, основанный на измерении электрон¬
ного сродства молекул, позволяет идентифицировать некоторые функ¬
циональные группы. Электронное сродство некоторых веществ сильно
различается, в частности у спиртов, эфиров и кетонов. Особенно сильное
сродство к электронам наблюдается у галогенсодержащих соединений, что
позволяет эффективно определять микропримеси галогенсодержащих пе¬
стицидов в сложных природных смесях (в пище, воде и почвах). Это
облегчается тем, что в природе органических галогенсодержащих со¬
единений не содержится.Некоторые ионизационные детекторы, в частности детекторы термоенные
(фосфорные), атомно-эмиссионые, пламенно-фотометрические и др. также мо¬
гут дать определенную информацию о природе вещества. Использование
инфракрасных и ультрафиолетовых спектрометров на выходе из колон¬
ки позволяет получить информацию о качественном составе исследуемой
смеси.414
ГЛАВА 14 Качественный анализ в газовой хроматографииДетекторы-плотномеры могут давать сведения о молекулярном весе
разделяемых компонентов. В этом случае к неизвестному компоненту до¬
бавляется внутренний стандарт с известным молекулярным весом и за¬
писываются две хроматограммы при одних и тех же условиях с раз¬
личными газами-носителями, что позволяет вычислить молекулярный вес
неизвестного компонента.Для этих расчетов знание исходного размера пробы не требуется.
В качестве газов-носителей можно применять гелий, азот и двуокись
углерода. Средняя ошибка при определении молекулярного веса состав¬
ляет около 4%.14.5 Методы идентификации с использованием качественных хи¬
мических реакций на выходе колонки.Идентифицировать неизвестные компоненты можно по специальным
селективным реакциям на функциональные химические группы в сочета¬
нии с данными по удерживанию. В работах [7] перечислены соответствую¬
щие реактивы на определенные классы химических соединений с указа¬
нием минимального обнаруживаемого анализом количества вещества и
числа испытанных гомологов.Методики приготовления реактивов можно найти в руковод¬
ствах по качественному анализу органических веществ.Для идентификации используют каталитическое разложение органиче¬
ских веществ с последующим проведением элементного анализа. В одной
из таких методик непрерывного элементного анализа органические соеди¬
нения, выходящие из колонки, разлагали путем пропускания через нагре¬
ваемую (до 725°С) спиральную кварцевую трубку длиной 60 см, заполнен¬
ную наполовину окисью меди, наполовину — восстановленным железом
на твердом носителе. При прохождении органических веществ над окисью
меди получались двуокись углерода и вода. Вода, проходя над восстановлен¬
ным железом, разлагалась с выделением водорода. Получаемая смесь двуо¬
киси углерода и водорода разделялась на вспомогательной юлонке. По от¬
ношениям площадей пиюв двуокиси углерода и водорода на хроматограмме
можно судить об отношении атомов углерода и’ водорода в исследуемом
соединении. Этот метод удобен тем, что одновременно с разделением юм-
понентов сложной смеси можно определить отношение водорода к углероду
соединений, присутствующих в сложных смесях.14.6 Методы идентификации другими физико-химическими
методами.Эти методы основаны на сборе разделенных компонентов в чистом
виде на выходе из колонки и применении других физико-химических ме-415
Яшин Я.И., Яшин Е.Я., Яшин А.Я. Газовая хромшпографияГОДОВ. Для улавливания веществ и последующего перенесения к другим
приборам используют специальные ловушки. Чаще всего выделенные в
чистом виде вещества исследуют на масс-спектрометрах и инфракрас¬
ных спектрофотометрах, поэтому ловушки изготавливают так, чтобы их
удобно было использовать в приборах. Ловушки, применяемые для
последующей идентификации с масс-спектрометром, изготавливают с
весьма герметичным краном, чтобы их можно бьшо подсоединить непо¬
средственно к вакуумной системе масс-спектрометра. Ловушки, исполь¬
зуемые для последующей идентификации с инфракрасным спектрофотоме¬
тром, изготавливают так, чтобы в приборе их можно было установить
вместо кюветы. При этом часто улавливают не вымораживанием, а
путем селективного растворения в каком-либо инертном растворителе.Для идентификации ароматических и непредельных соединений ис¬
пользуют и ультрафиолетовую спектроскопию.В последние годы применяют также и приборы ядерного магнитного
резонанса, хотя их чувствительность мала.Для идентификации соединений, содержащих галогены, азот, фос¬
фор и др., используют и эмиссионную спектроскопию, в частности пла¬
менную фотометрию.14.7 Методы идентификации, основанные на селективном удале¬
нии некоторых компонентов.В этих методах, которые иногда называют методами вычитания,
селективно удерживают один или несколько компонентов на выходе хро¬
матографической колонки. На хроматограмме частично или полностью
исчезают некоторые компоненты. Путем сопоставления хроматограмм без
вычитания и с вычитанием компонентов нетрудно идентифицировать со¬
ответствующие пики. Удаление компонентов можно проводить как за счет
необратимой хемосорбции, так и сильной физической адсорбции. Для
удаления органических кислот и оснований можно воспользоваться соот¬
ветствующими ионообменными смолами. Обычно поглотители помеща¬
ют в отдельную колонку длиной 15 см и внутренним диаметром 3-4 мм.Поглотительную колонку можно устанавливать как в начале, так и
в конце хроматографической колонки.Для удаления широш используют цеолиты. Углеводороды нормаль¬
ного строения селективно удерживаются цеолитами СаА, при этом раз¬
ветвленные и ароматические углеводороды проходят свободно; н-алканы
затем можно также элюировать при высоких температурах, ниже 100 °С
необратимо удалялись и-спирты, альдегиды и кислоты. Цеолиты можно
также использовать для удаления сильнополярных газов и паров: Н^О, H^S,
SOj, NHj, С02, HCN, (CN)j и др. Спирты селективно удерживаются также
борной кислотой, нанесенной на носитель.416
rJlAJBLA. 14 ЛишерашщюЛитература1. Ногаре С. Д., Джувет Р. С. Газо-жидкостная хроматография. Теория
и практика. Пер. с англ. Под ред. Л. П. Александрова и Л. П. Демешъе-
вой. Л.,«Недра», 1966.420 с.2. Методы-спутники в газовой хроматографии. Пер. с англ. Под ред.В. Г. Березкина. М, «Мир», 1972. 398 с.3. Гольберт К. А., Вигдергауз М. С. Курс газовой хроматографии.
М., «Химия», 1974. 376 с.4. Руководство по газовой хроматографии. Пер. с нем. Под ред.
А.А. Жуховицкого. М., «Мир», 1969. 503 с.5. Gas Chromatography Retention Data, by W. 0. Me. Reynolds, Preston
Technical Abstracts Company, 1966. 333 p.6. Яшин Я. И. Физико-химические основы хроматографического раз¬
деления, Москва, Химия, 1976г.7. Столяров Б.В., Савинов И.М., Витенберг А.Г., Карцова Л.А., Зенке¬
вич И.Г, Калмановский В.И., Каламбет Ю.А. Практическая газовая и
жидкостная хроматография С.-Петербург, Изд-во СПУ, 2002, 616с.41727 Г^вая хрошггографкя
Яшин Я.И., Яшин Е.Я., Яшин А.Я. Гтовая хранатографияГЛАВА 15. МЕТОДЫ КОЛИЧЕСТВЕННОГО АНАЛИЗА В
ГАЗОВОЙ ХРОМАТОГРАФИИ15.1 ВведениеКоличественный анализ основан на том, что площади пиков индивиду¬
альных компонентов на хроматограмме пропорщюнальны количеству или
концентрации компонента в анализируемой смеси. Кроме площади пиков
в качестве исходных величин при проведении количественного анализа
используют высоты пиков, произведения высоты пика на удерживаемые
обьемы или на время удерживания [1,2].В настоящее время времена удерживания, высоты пика и площади пи¬
ков автоматически рассчитывают программы верхнего уровня и печатают¬
ся в протоколе измерений.В аналитической практике получили наибольшее распространение три
способа количественной оценки хроматограмм: метод абсолютной градуи¬
ровки, метод внутреннего стандарта и метод нормализации с учетом по¬
правочных коэффициентов и без них [1-6].15.2 Метод абсолютной градуировкиПо методу абсолютной градуировки количество компонента в пробе
определяют по градуировочному графику зависимости площади пика или
высоты пика от дозируемого количества вещества. Для такой градуировки
необходимо использовать чистые вещества. Режим работы колонки при из¬
мерениях должен быть совершенно одинаков с режимом работы при гра¬
дуировки. Особенно жестко это условие должно выдерживаться в случае,
если градуировки проведена по высотам пиков. Метод абсолютной гра¬
дуировки чаще всего применяют в том случае, когда нет необходимости
определять все компоненты анализируемой смеси, а требуется определить
только один или два. Этот метод может быть рекомендован при введении
достаточно больших газовых паровых проб, так как в этом случае ошибка
дозирования невелика.При дозировании жидкостей ошибка больше, поэтому удобнее приме¬
нять разбавленные растворы жидкостей с точно известной концентрацией.Сходимость (правильность) результатов в основном зависит от вос¬
производимости дозирования пробы, при использовании газовых кранов
она может быть достаточно хорошей (до 0,5—1%). При введении
жидких проб микрошприцем воспроизводимость значительно хуже (в пре¬
делах 2—5%).Точность зависит от сходимости дозирования пробы и при анализе га¬
зов может быть высокой. Одно из преимуществ этого метода связано с
тем, что он может быть использован даже в тех случаях, когда некоторые
компоненты не регистрируются на хроматофамме.418
ГЛАВА 15 Методы количественного анализа в газовой хроматографии15.3 Метод внутреннего стандартаМетод внутреннего стандарта основан на добавлении известного коли¬
чества определенного вещества, называемого «внутренним стандартом».
Поправочный коэффищ1ент для него принимается равным единице. Для
градуировки строят график зависимости огаощения площадей или высот
пишв определяемого вещества и «внутреннего стандарта» от отношения
весовых шличеств или отношения процентных содержаний обоих веществ.
Ташй график строят на основании хроматограмм ряда смесей с различным
соотношением шличеств «внутреннего стандарта» и определяемого шмпо¬
нента. Если затем в процессе анализа известное шличество «внутреннего
стандарта» добавляется к неизвестной пробе, то из полученной хромато¬
граммы, определив соотношение площадей или высот исшмого шмпонента
и «внутреннего стандарта», по калибровочному графику можно определить
шнцентрацию исшмого шмпонента. Этот метод градуировки наиболее удо¬
бен и поэтому получил наибольшее распространение.При использовании этого метода градуировки нет необходимости
определять точно размер анализируемой пробы. В этом одно из основ¬
ных преимуществ метода, так как точное дозирование определенного
количества небольших жидких и газовых проб обычно вызывает затруд¬
нения (особенно при работе с ручными микрошприцами). При использо¬
вании этого метода исключается также влияние изменения скорости газа-
носителя и температуры колонки. Однаш метод применяют только тогда,
когда определяют не все компоненты.Сходимость результатов обычно хорошая, за исключением случаев,
когда возникают трудности при добавлении стандартного вещества к сме¬
си. Точность, как и в методе абсолютной калибровки, не зависит от при¬
сутствия в смеси нерегистрируемых шмпонентов.В этом методе необходимо полное разделение только анализируемых
компонентов и стандарта.Ошибка при измерении площади или высоты отдельно анализируемо¬
го пика отражается на точности определения тольш этого пика. Следу¬
ет отметить, что ошибка при введении стандарта в анализируемую смесь
будет приводить к соответствующим погрешностям определения всех ве¬
ществ. Также и ошибка в измерении пика стандарта отразится на точности
анализа всех веществ.15.4 Метод внутренней нормализацииМетод нормализации основан на том, что сумма площадей всех пиков
на хроматограмме с учетом соответствующих поправочных коэффициен¬
тов принимается за 100%. На основании этого расчет процентного содер-41927-
Яшин Я.И., Яшин Е.Я., Яшин А.Я. Газовая хроматографияжания определяемого вещества осуществляется при помощи соответству¬
ющих простых соотношении. Достоинством метода является возможность
определения процентного содержания всех компонентов смеси.Сходимость результатов в методе нормализации хорошая, погрешность
может сильно колебаться, особенно в тех случаях, когда не учитываются не¬
которые не зарегистрированные на хроматограмме вещества (систематиче¬
ская ошибка) или же неточно определены поправочные коэффициенты.Основное преимущество этого метода заключается в том, что нет не¬
обходимости в приготовлении градуировочной смеси и в воспроизводи¬
мом введении ее в хроматограф.Существенным недостатком является необходимость полного разде¬
ления и идентификации всех компонентов смеси, а также определения по¬
правочных коэффициентов для каждого пика, хотя некоторые из веществ
определять не требуется. Поэтому метод непригоден для тех случаев, ког¬
да некоторые компоненты (легкие, или, наоборот, очень тяжелые) не ре¬
гистрируют на хроматограмме. В этом случае ошибка любого измерения
одного компонента приводит к искажению результатов всех остальных.15.5 Источники ошибок в газохроматографических измеренияхОсновные источники ошибок газохроматографических измерений
следующие: ошибки при дозировании анализируемой пробы, потеря про¬
бы в колонке за счет возможной адсорбции или разложения, ошибка детек¬
тирующей системы, ошибка вторичного прибора, ошибки при измерении
площади или других параметров пика и, наконец, ошибки непосредствен¬
но в расчетах [2, 7, 8].При дозировании могут быть две причины ошибок: первая связана с
неточным определением количества анализируемой пробы, что зависит в
основном от погрешности дозирующих устройств; вторая связана с фрак¬
ционированием, разложением или адсорбцией некоторых веществ еще до
того, как проба попадет в хроматографическую колонку. При этом теряется
представительность анализируемой пробы, так как состав пробы, попада-
емой в колонку, не соответствует первоначальному истинному составу. В
литературе приведены многочисленные примеры частичной или даже пол¬
ной потери веществ, в частности за счет адсорбции и разложения в самом
дозаторе-испарителе или на переходных трубках. Особое внимание сле¬
дует обращать на материал переходных трубок и температуру испарителя
при разделении неустойчивых соединений (стероиды, пестициды, переки¬
си, металлоорганические соединения и др.).Перед проведением количественных измерений нужно убедиться, что
нет потерь, что получаемый пик соответствует всему дозируемому коли¬
честву. Если потери воспроизводимы, то погрешности, связанные с ними,
могут компенсироваться при градуировке.420
ГЛАВА IS Методы количественного анализа в газовой хроматографииПогрешности, вносимые детектирующей системой, обычно связаны с
изменением чувствительности детектора при изменении рабочих условий,
например температуры, расхода, газа-носителя, давления и др. Чтобы ис¬
ключить этот источник ошибок при точных количественных измерениях,
нужно весьма тщательно стабилизировать все эти параметры. Источником
ошибок может быть неточное определение поправочных коэффициентов.
В некоторых случаях используют поправочные коэффициенты, получен¬
ные на детекторах другой конструкции, при этом могут быть значительные
погрешности. Для прецизионных анализов нужно пользоваться поправоч¬
ными коэффициентами, полученными на том же самом детекторе.В некоторых случаях наложение нерегистрируемых компонентов, в
частности, пика воды при использовании ионизационно-пламенного де¬
тектора, приводит к изменению чувствительности детектора к анализиру¬
емому веществу.Следующим источником погрешности является измерение параме¬
тров пика (площади и высоты пика). Ранее уже обсуждались достоинства
используемых методов измерения площадей пиков. Высоты пиков могут
быть измерены во многих случаях значительно точнее, однако значения их
сильно колеблются при изменении параметров разделения. Высота пика
при отсутствии перегрузки обычно зависит от массы пробы и не зависит от
обьема пробы. Ширина же пика при отсутствии перегрузки не зависит ни
от массы, ни от обьема пробы. Поэтому площадь пика также определяется
только массой пробы и не зависит от ее обьема.15.6 Определение сходимости показанийТочность измерения определяется отклонением измеренной величины
от истинной. Для того, чтобы определить точность, нужно знать истинную
величину. Точность или ошибка измерения характеризуется относитель¬
ной ошибкой:A=[(A„-A_)/AJ100 (15.1)где А - относительная ошибка, А - истинная величина, А - изме-’ист ’ ЮМренная величина.при измерении количественного состава неизвестной смеси истинные
величины, как правило, неизвестны. В этих случаях точность выражают
через сходимость (воспроизводимость) показаний. Без хорошей сходимо¬
сти трудно достичь высокой точности. Однако и при хорошей сходимости
может быть плохая точность измерения при наличии больших системати¬
ческих ошибок, которые обычно исключаются при градуировке.Все возможные ошибки можно разделить на два типа: случайные и
систематические.421
Яшин Я.И., Яшин Е.Я., Яшин А.Я. Гтовая хроматографияСлучайные ошибки направлены как в большую, так и меньшую сто¬
рону они связаны с разбросом измеряемых показаний от средней вели¬
чины. Обычно полностью исключить эти ошибки нельзя, так как любую
величину абсолютно точно измерить в большинстве случаев невозможно,
всегда допускается определенная погрешность. Распределение случайных
ошибок соответствует кривой нормального распределения вероятностей,
из которых следует, что положительные и отрицательные отклонения рав¬
новероятны и что меньшие отклонения встречаются значительно чаще,
чем большие.Систематические ошибки обычно имеют отклонения в одну сторо¬
ну (одного знака), поэтому их можно исключить введением соответствую¬
щих поправок.В газовой хроматографии могут быть следующие источники система¬
тических ошибок [2, 6]:1) неправильный отбор проб (непредставительность пробы);2) изменение состава (потеря отдельных компонентов) анализируемой
пробы до ввода в хроматограф (фракционирование, адсорбция, разложе¬
ние и пр.), с одной стороны, и загрязнение пробы— с другой, например,
при анализе компонентов воздуха;3) различие в чувствительности разных детекторов, изменение чув¬
ствительности при односторонних изменениях условий (температуры,
скорости потока, тока детектора и т. д.);4) применение неправильного градуировочного графика, связанного с
неточным приготовлением градуировочных смесей;5) ошибки измерения площадей пиков, свойственные некоторым мето¬
дам измерения площадей.Сходимость результатов оценивают по среднеарифметической или
среднеквадратической величине.Среднеарифметическая величина — сумма результатов всех измере¬
ний, деленная на общее число измерений. Эта величина определяется до¬
вольно быстро, однако она не позволяет оценить максимально возможные
отклонения отдельных измерений.Распределение отклонений от среднеарифметической величины мож¬
но оценивать по среднеквадратичным величинам (или стандартным откло¬
нениям).Среднеквадратичная величина характеризует разброс результатов от¬
дельных измерений около среднеарифметического значения. Она может
быть использована для определения доверительного интервала в анало¬422
ГЛАВА IS Методы количественного анализа в газовой хроматографиигичных измерениях. Среднеквадратическое отклонение G измеряют по
следующей формуле:G=^I(X-X)/(N-1) (15.2)где X - измеренная величина, X - среднеарифметическая величина,
N - число измерений.Отнощение среднеквадратического и среднеарифметического значе¬
нии измеряемой величины представляет собой относительную среднеква¬
дратическую ошибку измерения, которую чаще, чем абсолютную ошибку,
используют для оценки погрешности измерения.В некоторых случаях в газовой хроматографии кроме постоянных си¬
стематических ошибок могут быть переменные систематические ошибки,
которые, в частности, связаны с наличием нерегистрируемых компонентов
при одних условиях и появлением их при других условиях.В заключение следует обратить внимание на то обстоятельство, что
погрешность газохроматографического измерения будет зависеть от ли¬
нейного диапазона детектора.Для получения точных результатов необходимо работать всегда в ли¬
нейном диапазоне. При использовании любой стандартной аппаратуры
можно проводить газохроматографические измерения с погрешностью
около 5% (отн.). На хроматографах высокого класса (с лучшей стабили¬
зацией параметров) можно проводить измерения с погрешностью 1—2%
(отн.), при дополнительной тщательной стабилизации параметров (осо¬
бенно условий работы детектора) можно проводить, по данным Кайзера,
измерения с погрешностью до 0,02% (отн.) [9].Следует отметить, что погрешность газохроматографических измере¬
ний зависит не только от аппаратуры, но и от конкретных условий разделе¬
ния (времени удерживания, степени размывания, концентрации анализи¬
руемых компонентов, выходит ли анализируемое вещество до основного
компонента или же после него и т.д.).Таким образом, для получения точных количественных газохрома¬
тографических измерений необходимо обращать внимание на качество
применяемой аппаратуры, добиваться наибольшей полноты разделения
за счет тщательного подбора сорбента и условий его работы, правильно
выбирать метод калибровки, а также способ приготовления градуировоч¬
ной смеси и пользоваться наиболее рациональными приемами измерений
параметров пика.423
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я. Газовая хранатографияЛитература1 Кейлеманс А. Хроматография газов. Пер. с анга. Под ред. М. И. Янов¬
ского. М., Издатинлит, 1969. 320 с.2 Столяров Б. В. и др. Руководство к практическим работам по газовой
хроматографии. Л., Изд-во ЛГУ, 1973. 284 с.3. Tamura Н., Hozuml К. «Japan Analyst», 1971, v. 20, p. 149—155.4. Janak 1. J. Chromatog., 1960, v. 3, p. 308—312.5. Dondi E, Betti A., Bighi J. Chromatog., 1971, v. 60, p. 1—3.6. Мак-Нейр Г., Бонелли Э. Введение в газовую хроматографию. Пер. с
англ. Под ред. А. А. Жуховицкого. М., «Мир», 1970. 277 с.7. Goedert М„ Guiochon G. J Chromatog. Sci., 1969, v. 7, p. 323—339.8. Goedert М., Guiochcn G. Analyt. Chem., 1970, v. 42, p. 962—968.9. Kaiser R. Chromatog., 1971, v. 4, p. 215-219; 362—365; 479-484Дополнительная литература1. Каламбет Ю.А. Метод внутреннего стандарта - идея и воплощение.
Партнеры и конкуренты 2004, №4, с.32.2. Зенкевич И.Г., Ещенко А.Ю., Климова И.А. Характеристика меж-
лабораторной воспроизводимости результатов количественного га¬
зохроматографического анализа методом внутренней нормализации.
Ж.анал.химии 2005, т.60, с. 137.3. Зенкевич И.Г, Макаров Е.Д., Макарова И.Ю. Количественный хро¬
матографический анализ при изменении состава образцов в ходе под¬
готовки проб. Модификация метода двойного внутреннего стандарта.
Ж.анал.химии 2007, т.62, с.829.4. Зенкевич И.Г, Климова И.О. Применение метода стандартной до¬
бавки для количественного хроматографического анализа. Ж.анал.хи¬
мии 2006, т.61, с. 1048.5. Остроухова O.K., Зенкевич И.Г. Сравнение методов внешнего стан¬
дарта и стандартной добавки для количественного хроматографиче¬
ского определения пестицидов в растительных объектах. Ж.анал.хи¬
мии 2006, т.61, С.481.6. Калмаковский В.И. Метрология для химиков. Учебное
пособие.Н.Новгород. Изд. Ю.А. Николаев,2007, 132 с.424
ГЛАВА 16 Методы кониентрирования в газовой хроматографииГЛАВА 16. МЕТОДЫ КОНЦЕНТРИРОВАНИЯ
В ГАЗОВОЙ ХРОМАТОГРАФИИ16.1. Введение.Пределы обнаружения применяемых, даже самых чувствительных, де¬
тектирующих систем не позволяют определять микропримеси некоторых
загрязнений окружающей среды, особенно супертоксикантов. В этих слу¬
чаях применяют разные методы предварительного концентрирования (обо¬
гащения) пробы. Методы концентрирования позволяют снижать пределы
обнаружения на два-четыре порядка (т.е. в 100-10000 раз) и определять
некоторые загрязнители, например диоксины, на уровне ppt (10 '®%). В по¬
следнее десятилетие интерес к методам концентрирования сильно возрос,
опубликованы книги, десятки обзоров. Старые методы концентрирования
усовершенствованы, предложены новые, разрабатывается аппаратура для
концентрирования.Концентрирование, по определению ИЮПАК, это операция или про¬
цесс, в результате которого концентрация или количество микропримесей
относительно матрицы возрастает.Развитие промышленности, транспорта, сельского хозяйства, техники
пока еще приводит к загрязнению окружающей среды. Загрязнения приоб¬
ретают всеобщий характер, для них нет границ между странами. Экологиче¬
ское равновесие в ряде регионов планеты находится под прямой угрозой.Окружающая среда - это совокупность факторов и (или) условий, вли¬
яющих на благосостояние и здоровье человечества.Окружающая среда - это не только то, что находится за пределами
человеческого жилища, но также и то, что окружает человека в процессе
его трудовой деятельности (воздух производственных помещений, офисов
и др.), и условия проживания (атмосфера внутри жилых помещений).Окружающая среда, в основном, это продукт жизнедеятельности че¬
ловека и многих организмов, находящихся в динамическом экологическом
равновесии. Если природа или человек не в состоянии нейтрализовать за¬
грязнения, то для земли это чревато катастрофическими последствиями.По определению ООН «вещества считаются загрязнителями, если они
встречаются в ненадлежащем месте и в ненадлежащем шличестве».Современные методы концентрирования позволили расширить области
применения газовой хроматографии (ГХ) на анализы примесей, микропри¬
месей и следовых концентраций. Это имело большое значение в контроле
загрязнений окружающей среды, супертоксикантов в пищевых продуетах и
напитках для оценки их качества и безопасности, биомаркеров в биологи¬
ческих жидкостях для ранней диагностики заболеваний, в судебнокримина¬
листических исследованиях. Отдельно следует выделить контроль ароматов
пищевых продуктов, феромонов насекомых, фреонов в стратосфере.425
Яшин ЯМ, Яшин Е.Я., Яшин А.Я.Газовая хроматографияИсключительное значение имели методы концентрирования для ка¬
пиллярных колонок, в частности разработка и аппаратурное воплощение
метода криофокусировки и других методов дозирования больших проб.В таблице 16.1 приведены только основные методы концентрирова¬
ния, которые находят применение в ГХ.Таблица 16.1.Основные методы предварительного концентрирования, применяе¬
мые в газовой хроматографииМетодыОсобенности методаСсылкаТвердофазная экстракция
(ТФЭ)Универсальный метод, возможно
полное извлечение из газовых и
жидких сред1-5Микротвердофазная экстракция
(МТФЭ)Сорбционное извлечение без раство¬
рителя, проста в исполнении6,7Ион-парная ТФЭПрименима для диссоциирующих
веществ8Иммуноаффинная ТФЭДля биологически активных соеди¬
нений9-12ТФЭ на полимерах с порами
молекулярных размеровСверхселективное концентрирова¬
ние соединений с определенными
размерами молекул13Жидкостная экстракцияНужны особо чистые растворители;
нет полноты выделения; необходима
регенерация или утилизация раство¬
рителя14Микро жидкостно-жидкостная
экстракцияЖидкостная экстракция небольшим
количеством (несколько мл) раство¬
рителя14Микрожидкостная экстракцияЭкстракция одной микрокаплей рас¬
творителя (2 мкл)15Жидкостная экстракция при
повышенном давленииУскорение процесса экстракции16Жидкостная микроволновая
экстракцияУскорение процесса экстракции17Сверхкритическая флюидная
экстракцияЭффективный метод экстракции из
сложных объектов; растворимость
флюидов приближается к раствори¬
мости жидкостей, а скорость массо¬
обмена - к газам18426
ГЛАВА 16Методы концентрирования в газовой хроматографииМетодыОсобенности методаСсылкаМембранная экстракцияЭффективный динамический метод,
по механизму близок к динамиче¬
ской газовой экстракции19-21Газовая экстракция (анализ
равновесного пара или head
space)Удобен для летучих примесей из
жидких и твердых сред; возможна
автоматизация анализа22,23Выдувание и улавливание
(динамическая газовая
экстракция, purge and trap)Возможен анализ как летучих,
так и малолетучих органических
загрязнителей из водных сред2416.2. Концентрирование из воздуха и газовых сред.Анализ загрязнителей в воздухе необходим для целей промышленной
гигиены, для определения загрязнения атмосферы городов, воздуха вну¬
три жилых помещений (квартир и офисов).Контроль загрязнителей проводят также с целью определения источ¬
ников выбросов, исследования влияния загрязнителей на здоровье челове¬
ка, животный и растительный мир.Необходимо определять степень загрязнения при чрезвычайных об¬
стоятельствах; авариях, взрывах, пожарах и пр. В этом случае портатив¬
ными анализаторами, в т.ч. и портативными газовыми хроматографами,
определяют состав атмосферы на месте события, а также отбирают пробы
для детального анализа в стационарных лабораториях. При этом важно,
чтобы в процессе транспортировки состав пробы не изменялся.Концентрация загрязнителей в атмосфере может изменяться как во
времени, так и в пространстве. Концентрирование из газовых сред приме¬
няется также для определения примесей в особочистых газах.Микропримесями принято считал шнцешрации ниже 1(И г/мл. В основнсм
определяют летучие фганические соединетия (ЛОС), которые присугст^ют в
юзцухе в виде пара с давлением менее 0,1 мм рт. ст. (0,0133 кРа) при 25°С.Органические соединения с меньшей летучестью обычно относят к
полулетучим. Полулетучие соединения кроме пара могут находиться в
сорбированном состоянии на аэрозолях (частицах пьши) или в микрока¬
пельках жидкости.Обогащенная концентрация определяется следующим соотношением:Г f Vc„ = -V--- (16-1)^ скгде: С„ - начальная концентрация; V„- первоначальный обьем пробы;
обьем пробы после концентрирования; - полнота извлечения ана¬
лизируемого соединения.427
Яшин Я.И., Яшин Е.Я.. Яшин А.Я. Газовая хроматографияКонцентрирование органических примесей из воздуха на адсорбцион¬
ных ловушках имеет определенные преимущества: простота в использова¬
нии, небольшие затраты на отбор, возможность использования смешанных
адсорбентов или последовательно соединенных трубок с разными адсор¬
бентами, регулирование селективности улавливания за счет изменения
природы адсорбента, обычно высокая полнота извлечения, приближающа¬
яся к 100%, многократное использование.Однако, имеются и недостатки адсорбционного концентрирования: ино¬
гда трудно сконцентрировать низкомолекулярные соединения, на некоторых
адсорбентах возможны химические превращения, нельзя использовать вы¬
сокие скорости продувки потока воздуха через адсорбционные ловушки из-
за возможного проскока, трудность десорбции тяжелых примесей.Во вредных химических производствах имеет место «персональный
мониторинг», т.е. определение количества вредных примесей, вдыхаемых
персоналом за рабочую смену. Для этих целей используют адсорбционные
ловушки с мининасосами или «пассивные» ловушки. В последних адсорб¬
ционный слой закрыт мембраной, проницаемой для воздуха и примесей,
проникновение загрязнений происходит за счет диффузии.Удаляют сконцентрированные микропримеси с адсорбционных ло¬
вушек термодесорбцией или некоторыми растворителями, в частности
дисульфидом углерода, ацетонитрилом, диэтиловым эфиром, метанолом,
гексаном, хлороформом, ацетоном и др. Чаще всего используется термо¬
десорбция.Допустимая погрешность определения примесей вредных веществ в
воздухе производственных помещений в США ±25%, в странах ЕЭС ±35%.
В США комнатную температуру принято считать 20°С, в Европе 25°С.Для улавливания микропримесей применяют адсорбенты разной хими¬
ческой природы и геометрической структуры. По классификации ИЮПАК
размер макропор >50 нм, средних мезопор - более 2 нм (но менее 50 нм) и
размер микропор <2 нм.Размер пор имеет большое значение при десорбции адсорбированных
веществ, в частности из микропор некоторые соединения десорбируются
плохо. Многие промышленные адсорбенты - активные угли, силикагели,
цеолиты и карбосита - имеют микропоры.В последние годы чаще всего применяют макропористые неполярные
полимерные и углеродные адсорбенты. На таких адсорбентах при темпе¬
ратурах выше комнатной влага практически не адсорбируется и не нака¬
пливается в ловушке, в отличие от других адсорбентов. Это исключитель¬
но важно при улавливании органических примесей из воздуха, в котором
всегда содержится относительно много водяного пара.428
ГЛАВА 16 Методы концентрирования в газовой хроматографииПри применении неполярного адсорбента с большой поверхностью из
воздуха будут улавливаться органические соединения выше С^.При десорбции будет происходить размывание, т.к. в первую очередь
будут десорбироваться легкие примеси, а потом тяжелые.Для того, чтобы это исключить, адсорбционные ловушки делают с
двумя-тремя слоями. В каждом слое адсорбенты различаются геометри¬
ческой структурой. В первом слое содержится макропористый адсорбент,
во втором - среднепористый и в третьем - тонкопористый. В первом слое
адсорбируются тяжелые примеси, во втором - среднекнпящие примеси и в
третьем - низкокипящие примеси (рис. 16.1).К.р«.ПКпС "tr(улавливание улавливания
тяжелых соединенийсоединений) Cj-Ci)Юфботреп S-I11
(для улавливания
легких
соединений), Силвнизированная.
стеклянная
ватаРис. 16.1 Трехслойные адсорбционные предколонки (ловушки).При десорбции поток инертного газа направляют в обратном направле¬
нии по сравнению с направлением потока воздуха при концентрировании.
В этом случае все примеси десорбируются практически одновременно, и
существенного размывания при десорбции не происходит.В таблице 16.2 приведены такие многослойные адсорбционные ло¬
вушки фирмы «Supelco».Таблица 16.2Применение концентрирующих колонок с многослойными сорбентамиТипыАнализируемыесоединенияОбъем пробы,лТипдетектораКарбопак - Карбосито S-IIIУглеводороды,ароматическиеуглеводороды0,35ЭЗДДИПКарботреп С/ Карботреп/
Карбосито S-IIНеполярные
и полярные
соединения1^2МС429
Яшин Я.И.. Яшин Е.Я.. Яшин Л.Я.Газовая хроматографияТипыАнализируемыесоединенияОбъем пробы,лТипдетектораКарботреп С/Карботреп;
Карботреп С/Карботреп/
Карбосито S-IIIУгаеводородыС,-С„1-2дип-мсТенакс ТА/Карботреп/
Карбосито S-IIIУглеводородыС,-С,Алкены С, - С ,I о0,5-2ДИП-МСФИД-МСТенакс ТА/амбесорбХЕ-340/активированныйугольНеполярные
и полярные
соединения1-2МСТенакс ГХ/цеолиты 5А/
Карбосито S-IIУглеводороды1-2ДИПв адсорбционных ловушках наибольшее применение нашли пористые
полимеры (тенаксы, хромосорбы 102 и 106, амберлиты XAD-2 и XAD-4,
порапаки Q и др.), углеродные адсорбенты (карбопаки, карботрепы, карбо¬
сита). Характеристики этих адсорбентов приведены ранее в главе 4.Неорганические газы и пары - важная часть промышленных загрязне¬
ний. Для их концентрирования применяют полярные адсорбенты. В част¬
ности, смесь оксидов меди и марганца (гопкалит) применяют для улавли¬
вания паров ртути, особо чистый силикагель используется для улавлива¬
ния кислых газов и паров кислот (НС1, HjSO^).В таблице 16.3 приведены основные области применения концентри¬
рования из газовых сред. В нешторых случаях концентрирование происхо¬
дило при отрицательных температурах. В последнее время вместо жидких
хладоагентов используют элементы Пельтье.Адсорбенты с нанесенными слоями кислот (силикагели, угли, диато¬
миты) применяют для улавливания газов с основными свойствами (амми¬
ак, фосфин, гидразин), угли, смоченные щелочами, применяют для улав¬
ливания диоксида серы, инертные адсорбенты со слоем триэтаноламина
используют для улавливания оксидов азота и оксидов серы.430
ГЛАВА 16 Методы кониентрироваиия в газовой хроматографииТа&лица 16.3Основные применения адсорбционного концентрирования
из газовых средОбласти примененияСсылкиОпределение органических примесей в атмосфере
промышленных городов:- Лос-Анджелеса- Хьюстона- Цюриха- С.-Петербурга
Парижа25262728
292.Загрязнения в воздухе герметичных камер:- кабины космических кораблей- помещения атомных подлодок30313.Примеси в воздухе производственных помещений324.Примеси органических соединений в выхлопных газах авто¬
мобилей335.Примеси винилхлорида в атмосфере346.Определение фреонов в стратосфере357.Загрязнения атмосферы в окрестностях г, Нагоя36Органические и неорганические примеси в чистых газах, в
частности в гелии и водороде379.Выделения продуктов разложения полимеров3810.Определение примесей органических загрязнений в выдыхае¬
мом воздухе человека для диагностических целей3911.Определение состава ароматов и запахов чая и кофе40,4112.Определение феромонов насекомых в атмосфере4213.Выделение этилена из растений4314.Анализ атмосферы биологических контейнеров4415. Примеси в сигаретном дыме45Фронтально-адсорбционное концентрирование слабо адсорбиру¬
ющихся примесей.Концентрирование примесей, адсорбирующихся хуже основного юм¬
понента, может быть проведено на основе фронтального продвижения
примеси по слою адсорбента, заполненного газом, адсорбирующимся сла¬
бее всех примесей, или предварительно вакуумированного.Метод фронтальной адсорбционной хроматографии для концентриро¬
вания был детально изучен А.А.Жуховицким [46].431
Яшин Я.И., Яшин Е.Я., Яшин А.Я. Газовая хроматографияПри фронтальном продвижении анализируемой смеси через колонку
слабо адсорбируемые компоненты продвигаются значительно быстрее. При
некоторых условиях можно из сравнительно большого (несколько литров)
исходного объема анализируемой газовой смеси сконцентрировать все лег¬
ко адсорбирующиеся примеси в начале колонки и затем с помощью крана-
дозатора направить их в аналитическую газохроматографическую колонку
на разделение. При этом легко достигается 100-1000-кратное обогащение,
что позволяет анализировать многие примеси даже с помощью такого мало
чувствительного детектора, как детектор по теплопроводности.Концентрирование легко адсорбируемых примесей в присутствии газа
описано в работах [47, 48] на примере определения примесей в этилене и
пропилене и рассмотрено в [49].Применение постороннего дополнительного газа иногда нежелательно
при анализе примесей (из-за возможного загрязнения). Кроме того, в неко¬
торых случаях трудно или даже невозможно подобрать соответствующий
газ, например при определении содержания гелия в воздухе.Основным условием осуществления фронтально-адсорбционного
обогащения является обеспечение достаточного различия констант Генри
примеси и соединений, присутствующих в больших концентрациях. Это
достигается при следующих факторах:а) подбор адсорбента, обеспечивающего достаточное различие кон¬
стант Генри для молекулярной (физической) адсорбции примесей и основ¬
ного соединения;б) более сильное удерживание основного соединения за счет специфи¬
ческих, но еще молекулярных взаимодействий, в частности водородных
связей, а также за счет слабого комплексообразования;в) полная хемосорбция основного компонента;г) подбор адсорбентов с молекулярно-ситовым эффектом, в частности
цеолитов, в случае сильной адсорбции основного соединения и малой ад¬
сорбции молекул примеси из-за пространственных затруднений.Таким образом, используя различные межмолекулярные и химические
взаимодействия, а также молекулярно-ситовые свойства адсорбентов, ме¬
тод фронтально-адсорбционного обогащения может быть достаточно ши¬
роко применен для анализа примесей во многих важных смесях.Метод фронтально-адсорбционного обогащения легких примесей на
вакуумированных колонках впервые успешно был применен для опреде¬
ления гелия, неона и водорода в воздухе [50]. Затем при снижении дав¬
ления и усовершенствовании методики проведения процесса обогащения
результаты были улучшены [51]. Фронтальный метод дает высокое обо¬
гащение уже при однократном процессе, так как из-за значительных раз¬
ностей скоростей движения по слою слабее адсорбирующийся компонент
концентрируется.432
ГЛАВА 16 Методы концентрирования в газовой хроматографииДля аналитических целей важна полнота извлечения, чего не удается
достигнуть при однократном поступлении смеси на вакуумированный ад¬
сорбент.Коэффициент извлечения у определяется уравнением [51]:у=1--Я (16.2)л,где К,' п К,"- константы Генри для адсорбции основного компонента
и примеси соответственно.Значения у можно существенно повысить, если из первой колонки с
помощью коммутирующего устройства смесь перевести во вторую так же
вакуумированную колонку с тем же адсорбентом. Извлечение может быть
продолжено на третьей колонке. Такое ступенчатое фронтальное разделение
приводит к практически полному вьщелению примеси. Таким путем, напри¬
мер, достигается полное (1(Ю%) извлечение гелия из пробы воздуха. Если
же фронтально-адсорбционное обогащение проводить сразу на тех же трех
последовательно соединенных колонках, то извлечение составит не более
60-80%. На основе этого разработан специальный хроматограф типа «Луч»
[52], который позволяет проводшъ полное извлечение примесей [53].Для повышения степени извлечения может быть использован газ или
пар, по адсорбционным свойствам близкие к основному соединению. Важ¬
ным фактором, влияющим на извлечение слабо адсорбирующихся приме¬
сей, является степень вакуумирования; оптимальна степень вакуумирова¬
ния до давления ниже 131-393 Па.Метод фронтально-адсорбционного обогащения в несколько изменен¬
ном виде (с использованием вытеснителя) может быть успешно применен
для анализа газов, растворенных в различных жидюстях. [53].В работе [54] показана возможность определения гаюв, извлеченных из
воды, которую предварительно насыщали газовой смесью, содержащей Не,
О^, Nj, СН^ и COj. Чувствительность этого метода составляет МО"’ - 110‘^%.
Этот способ анализа растворенных газов может быть широю использован в
химии, геохимии, медицине, пищевой щюмьшшенности и в других отраслях.При использовании метода фронтально-адсорбционного обогащения
для концентрирования примесей, содержащихся в этиловом спирте, было
достигнуто более чем 1000-кратное обогащение, хотя содержание приме¬
сей в этом случае было в 500 раз меньшим, чем при концентрировании
примесей в этаноле без такого обогащения.Фронтально-адсорбционное обогащение на цеолитах можно прово¬
дить, используя их молекулярно-ситовое действие [55]. Этот метод приме¬
нен для концентрирования примесей в спиртах высшей очистки и других
водочных продуктах [56, 57]. Небольшие концентрации (менее 110'*%)43328 Га'ювая хромато1-рафия
Яшин Я.И., Яшин Е.Я., Яшин А.Я.Гязовая хроматографиямикрогфимесей в этаноле высшей очистки не могут быть определены без
предварительного их обогащения. Для предварительного обогащения при¬
месей здесь успешно может быть использована жидюстная хроматография
на колонке с цеолитом СаА. Принцип обогащения на цеолите основан на
том, что основные примеси - альдегиды, кетоны, эфиры, амины, изоспир¬
ты, кислоты и другие соединения с нелинейными молекулами - не могут
проникать в поры молекулярного сита и практически им не адсорбируют¬
ся. При движении фронта жидкости вдоль хроматографической колонки,
наполненной цеолитом СаА, неадсорбирующиеся из-за геометрических
затруднений молекулы микропримесей продвигаются быстрее адсорбиру¬
ющихся молекул, в частности воды, метанола и этанола, присутствующих
в значительно ббльших концентрациях, чем микропримеси. Поэтому после
прохождения через слой адсорбента определенного объема спирта высшей
очистки из него будут извлечены практически полностью все микропри¬
меси, которые концентрируются в самом начале фронта. Отобрав первые
порции выходящей из такой колонки жидкости, примеси можно затем про¬
анализировать на эффективной колонке.16.3. Твердофазная экстракция загрязнителей из воды.В поверхностных водах содержатся сотни-тысячи органических при¬
месей на уровне микрограммов в литре и ниже.В последнее десятилетие наибольшее распространение для анализа
загрязнителей в воде получила твердофазная экстракция. Основные при¬
менения твердофазной экстракции: концентрирование, очистка пробы,
фракционирование, смена растворителя.В таблице 16.4 приведены основные типы адсорбентов, применяемые
для твердофазной экстракции неполярных, полярных и ионных соедине¬
ний из воды.Таблица 16.4Типы адсорбентов для твердофазной экстракции из растворовТипыЭкстрагируемые соединенияНеполариые адсорбенты:
силикагели с привитыми группами С^,
Г Гуглеродные адсорбенты;
пористые полимерыНеполярные и слабополярные
соединенияПолярные адсорбенты:
силикагели (= Si - ОН);
силикагели с привитыми группами
- CN, NH, = Did и др.Полярные соединенияИонообмеиникиКислоты, основания434
ГЛАВА 16_Мет2^М-!ШВ1ЩЦприрования в газовой хроматографииАдсорбенты в количестве 100-500 мг помещаются в специальные па-
троны-картриджи диаметром 5-10 мм (рис. 16.2).:¥фиямртштvytРис. 16.2 Типы патронов-картриджейДиаметр зерен адсорбента обычно 40 мк. Расход анализируемого рас¬
твора в пределах 1-10 мл/мин. Процесс концентрирования происходит
следующим образом: через картридж, например, с силикагелем С,^, про¬
пускают определенный обьем анализируемой воды. Из этого объема воды
все неполярные и слабополярные органические примеси адсорбируются.
Затем с картриджа небольшим количеством неполярного элюента (~ 1 мл),
например, гексана, смывают сконцентрированные органические примеси.
Полученный экстракт дозируют в газовый хроматограф. Таким способом
можно достичь обогащения пробы в сотни-тысячи раз. Принудительный
расход пробы создают за счет давления на входе или В81^ума на выходе.Однако применение картриджей имеет и недостатки: в некоторых слу¬
чаях большое время концентрщювания из-за малых расходов, например, по
методике ЕРА Method 525 концентрирование из 1 л питьевой воды проходит
два часа; при увеличении расхода возмшсен проскок; затруднено концентри¬
рование из мутных растворов, т.к. примеси ^спензии могут забивать патро¬
ны, при этом увеличивается сопротивление и расход уменьшается.В последние годы в связи с широким применением твердофазной экс¬
тракции для фармацевтических, биологических и клинических анализов, в
которых обьем анализируемой пробы ограничен, введены новые форматы
картриджей на основе шприцев объемом 1, 3 и 5 мл с количеством адсор¬
бентов 10, 25 и 50 мг. Для таких картриджей требуется меньший объем
растворителей для промывки и элюирования.Для облегчения процесса автоматизации пробоподготовки и анализа
картриджи стали изготавливаться в виде микропипетки.43528*
Яшин Я.И., Яшин Е.Я.. Яшин А.Я.Гаювая хранатографияДля концентрирования из больших объемов воды предложены адсорб¬
ционные мембранные диски диаметром 25 или 47 мм, толщиной 0,5 мм.Мембранные адсорбционные диски бывают разной природы:- диски на основе макропористого тефлона, содержащего в порах ми¬
крочастицы адсорбента (0 8 мк) до 90% от всего веса;- диски на основе производных целлюлозы с разными функциональ¬
ными группами.Расходы водных растворов через мембранные диски могут быть до 80
мл/мин. Мембранные диски включены в стандартные методики, например,
для выделения из воды фталатов, 2,3,7,8-тетрахлордибензо-п-диоксинов,
пестицидов, полиядерных ароматических соединений и др.Следующим этапом в развитии твердофазной экстракции стало созда¬
ние платформ с 96 и даже 384 экстракционными дисками.16.4. Микротвердофазная экстракция (МТФЭ).МТФЭ разработана Павлишиным с сотрудниками в 1990 г. [58]. Этот
метод интенсивно развивался, ему посвящены две книги [59, 60] и десятки
обзоров [61,62]. В МТФЭ концентрирование происходит на стержне с сорб¬
ционным слоем, выдвигаемым из иглы специального шприца (рис. 16.3).Рис. 16.3 Принцип работы МТФЭНа стержень, выдвигаемый из иглы шприца на 20 мм, наносятся раз¬
ные адсорбционные и сорбционные слои (таблица 16.5) разной толщины.
МТФЭ очень проста.436
ГЛАВА 16 Методы концентрирования в газовой хроматографииТаблица 16.5Перечень сорбционных слоев, наносимых на стержни
при микротвердофазной экстракцииТип сорбционного
слояТолщинаслоя,мкмМаксимальная
температура в
испарителеОбласти примененияПолиметилсилоксан(ПМС)7-100280-300°СНеполярные органические
соединения, угаеводороды,
ПАУ, летучие ароматиче¬
ские соединенияПолиакрилаты (ПА)85320°СПолярные органические
соединения (фенолы, триа-
зины) Полидиметилсилок-сандивинилбензол(ПДМС-ДВБ)60270°СРастворители, ароматиче¬
ские углеводородыКарбоксен-полидиметилсилоксан(Карб-ПДМС)75320°СЛетучие органические
соединенияПолиэтиленгликоль-
дивинилбензол (пол-
ДВБ)65265°СПолярные органические
соединения, в частности,
спиртыВыдвинутый стержень опускается в небольшой стакан или колбу с
определенным количеством анализируемого водного раствора. Шприц
удерживается на штативе. В стакане находится магнитная мешалка для не¬
прерывного перемешивания. Неполярные или слабополярные примеси ор¬
ганических соединений сорбируются на неполярном сорбционном слое на
стержне. Через 5-30 мин. (обычно достаточно 15 мин.) шприц вынимают
из раствора, втягивают стержень в иглу и затем как обычно вводят в испа¬
ритель газового хроматографа. В испарителе стержень из иглы выдвигают,
и за счет высокой температуры происходит десорбция органических при¬
месей со стержня. Поток газа-носителя направляет органические примеси
в колонку.Для построения калибровочного графика этот процесс воспроизводят
для ряда растворов с известными концентрациями анализируемых веществ.В таблицах 16.6-16.7 приведены некоторые применения МТФЭ.437
Яшин Я.И.. Яшин ЕЖ, Яшин Л.Я.Газовая хроматографияТаблица 16.6Применения МТФЭ для контроля загрязнений в воде
(ПДМС-полидиметилсилоксан; ПА - полиакрилат)ппСоединенияСорбци¬онныйслойВремяабсорб¬ции,мин.Времядесорб¬ции,мин.Темпера¬
тура де¬
сорбции,°ССсылка1.Беюол, толуол,
этилбензолы, ксилолыПДМС102150632.ХлорбензолыПДМС253250643.Ле1учие органические
соединенияПДМС205200654.ПолиядерныеароматическиесоединенияПДМС302665.Фенолы и производныеПА608250676.ПестицидыПДМС303260687.ПолихлорированныебифенилыПДМС30130069Таблица 16.7Перечень соединений, анализируемых с концентрированием методомМТФЭ и ТФЭттппАнализируемые соединения и тип концентрированияСсылки1.ТФЭ полиядерных ароматических соединений702.ТФЭ гербицидов71,723.ТФЭ кислых гербицидов734.МТФЭ в анализе остатков пестицидов74, 755.ТФЭ фталатов из воды766.ТФЭ фенолов777.ТФЭ в анализе органических загрязнителей в воде788.ТФЭ для определения паров органических веществ в
газообразных средах799.Адсорбенты для улавливания летучих органических примесей из
воздуха8010.Концентрирование загрязнителей в анализе воды8111.Современное состояние ТФЭ в анализе липидов8212.Микроэкстракция лекарств8313.МТФЭ лекарств из биологических матриц8414.Применение МТФЭ для обнаружения взрывчатых веществ и
жидкостей для поджига в криминалистике85438
ГЛАВА 16 Методы концентрирования в газовой хроматп:>рпфииДостоинством МТФЭ является также то обстоятельство, что она объ¬
единила процесс концентрирования и десорбции с дозированием в поток
газа-носителя.Однако, МФТЭ имеет и существенные недостатки: стержень со слоем
сорбента необходимо постоянно выдвигать и вдвигать, что может приве¬
сти к повреждению адсорбционного слоя; адсорбционная емкость стержня
небольшая.В 1тботах [86,87] предложен игаовой микроюнцентратор (рис. 16.4 а, б), не
имеющий вьш1еуказанных недостатков МТФЭ. Игаа заполнена адсорбентом
или абсорбентом. В качестве абсо{^нта рекомендуется использовать твердый
носитель с привитой жидкой фазой. Для концентрирования микропримесей
определенный обьем газа или жидюсти пропускают через игаоюй микрошн-
центрагор. Затем его вюдят в испаритель газового хроматографа, при этом
через него проходит дополнительный поток газа-носителя.АсбсстИгла Фуллсрснамм
сааша) б)Рис. 16.4 а, б а- схема устройства для ТФЭ, б-устройство иглового
микроконцентратораСвоеобразным вариантом МТФЭ является система «Твистер», пред¬
ставляющая палочку для магнитной мешалки с полидиметилсилоксановым
покрытием. Размер палочки: длина 15 мм, толщина 3 мм. Эта магнитная па¬
лочка (Твистер) помещается в сосуд с анализируемым образцом и переме¬
шивается несколько минут. Неполярные и слабополярные соединения, рас¬
творенные в воде, сорбируются на пленке полидиметилсилоксана и таким
образом извлекаются из раствора. Затем палочку помещают в термодесорбер
(в частности, термодесорбер Gerstel TDS 2), где соединения десорбируются,
фокусируются и переносятся в хроматографическую колонку.Пределы обнаружения примесей с помощью палочки «Твистер» по
сравнению с МТФЭ снижаются в 1000 раз за счет большего количества
сорбента.439
Яшин Я.И., Яшин Е.Я., Яшин А.Я.Газовая хроматографияЭтот метод прост и дешев, возможна автоматизация термодесорбции.
Он применен для определения пестицидов в вине, ароматизаторов в соках,
а также для разных целей при контроле воды и биологических жидкостей
(токсикология).Метод твердофазной экстракции на адсорбционном слое в игле шпри¬
ца, выполненном в виде расширенной вставки, получил название MEPS
(micro extraction by packed sorbent) [88]. В этом случае экстракция и ввод
пробы объединены в один процесс. Метод MEPS, по существу, миниатюр¬
ный твердофазный картридж.Однократный объем забранной пробы может быть до 10 мкл. Много¬
кратно отбирая, можно довести общий дозируемый объем до 100-250 мкл.
Возможно сконцентрировать микропримеси из обьема пробы до 1 мл и
даже более.Даже для трудной пробы (плазма крови) MEPS может работать от 40
до 100 раз.Сравнение метода MEPS с твердофазной (ТФЭ) и микротвердофазной
(МТФЭ) экстракцией приведено в таблице 16.8Таблица 16.8Сравнение методов MEPS, ТФЭ и МТФЭФакторыMEPSТФЭМТФЭКоличество используемо¬
го сорбента0,5 - 2 мг50 - 2000 мгтолщина слоя
150 мкмВремя концентрирования1 - 2 мин.10- 15 мин.10-40 мин.Кратность примененияот 40 до 100 разоднократно50 - 70 разПолнота выделениявысокаявысокаянизкаяЧувствительностьвысокаявысокаянизкаяМетод MEPS успешно применен для определения полиядерных аро¬
матических углеводородов [88], анестетиков в сыворотке человека [89],
росковитина в плазме и моче [90], в плазме [91].16.4 Применения наноструктурных углеродных адсорбентов (фулле-
ренов С^ и нанотрубок) в твердофазной экстракции.В обзоре [92] приведены данные по адсорбции и молекулярноситово¬
му эффекту углеродных материалов.Адсорбционные свойства фуллеренов изучены в работе [93], в которой
определены коэффициенты Генри (для систем газ-адсорбент) для 22 со¬
единений.В таблице 16.9 приведен пример применения углеродных нанострук¬
тур для ТФЭ и МТФЭ.440
ГЛАВА 16 Методы концентрирования в газовой хроматографтТаблица 16.9Применения наноструктурных углеродных адсорбентов
для ТФЭ и МТФЭ (11ТипнаноструктурАнализируемые веществаМетод и детекторСсылкиФуллерены СБензол,толуол,этилбензол,
ксилолы (БТЭКС)ГХ-МС94Фуллерены СБТЭКС, нафталины, фталатыГХ-ПИД95НанотрубкибарбитуратыГХ-МС^96НанотрубкиСоединения - пламягасителиАдсорбционные свойства фуллерена изучены по отношению к ами¬
нам, ПАУ, карбаматам, металлоорганическим соединениям (металлоценам
и свинецорганическим соединениям) [97]. В этой работе было показано,
что эффективность экстракции уменьшается с ростом полярности адсор¬
бата. Экстракция бензола, толуола, этилбензола и ксилолов проведена в
работе [30] с последующим определением ГХ-МС. В этой работе было по¬
казано, что фуллерен С^ для этих смесей показывает лучшие результаты,
чем применяемые ранее С,^ и тенакс ТА. Кроме того, выяснилось, что ад¬
сорбционные свойства фуллерена С^ не изменяются в кислых средах, что
позволяет эффективно применять его при низких pH.Фуллерен С^ обладает хорошей термостабильностью, что позволяет
использовать его в МТФЭ при десорбции при высокой температуре.Для экстракционных целей были синтезированы полимерные волокна
с включением фуллерена С^. Эти материалы были изучены для экстракции
бензола, толуола, этилбензола, ксилолов, производных нафталина, разных
диэфиров фталатов [95]. Особенно хорошие адсорбционные свойства эти
волокна показали к ПАУ [98].Угаеродные нанотрубки из-за сильной адсорбции на них многих ор¬
ганических соединений привлетпи внимание как адсорбенты для ТФЭ.
Впервые нанотрубки применили для удаления диоксинов [99], затем для
концентрирования бифенола А, 4-н-нонилфенола, 4-трет-октилфенола из
водных проб [100].Нанотрубки эффектавнее экстрагируют полярные соецинения по qmBHe-
нию с C,g, поэтому их предлсшили использовать для определения сульфонами-
дов [101], атразина [102], цианазина [103], некпторых гербищвдов [104], барби¬
туратов [96] и дахлородифенилтрихлорэтана (ДДТ) [105] из разных сред.Частицы нанотрубок наносились на адсорбционные стержни в устрой¬
ствах МТФЭ для концентрирования соединений пламягасителей [95].441
Яшин Я.И., Яшин Е.Я, Яшин А.Я. Газовая .хроматография16.5 Ион-парная твердофазная экстракцияИон-парные реагенты широко известны и применяются в ион-парной
жидкостной хроматографии. В классической обращеннофазной хромато¬
графии сильнополярные соединения слабо удерживаются и плохо разде¬
ляются. Для увеличения удерживания в элюент добавляют в небольших
количествах (порядка 0,01%) ион-парные реагенты. Ион-парные реаген¬
ты содержат неполярную часть, обычно это алкильная цепь, и полярную
функциональную группу кислотного или щелочного типа. Углеводородная
часть взаимодействует с неполярной поверхностью силикагеля С,^, а по¬
лярная часть взаимодействует с полярными группами аналита и образует
ион-парную пару.Таким образом при добавлении в анализируемые водные среды ион-пар¬
ных реагентов можно удерживать и концентрировать сильнополярные сое¬
динения на картриджах с С,^. В качестве ион-парных реагентов используют:
1-пентансульфоновую кислоту, 1-октансульфоновую кислоту и другие до 1-
додекансульфоновой кислоты. Для выделения кислых аналитов используют:
тетрагептиламмония бромид, тетраокгиламмония бромид и др.Этот метод экстракции чаще всего используется в жидкостной хрома¬
тографии [8].В ГХ ион-парная экстракция иногда применяется для выделения по¬
лярных соединений из мочи.16.6 Иммуноаффинная ТФЭ.Иммуноаффинная ТФЭ - сверхселективная экстракция, основанная на
взаимодействии антиген - антитело. Для этих целей создаются специаль¬
ные иммуносорбенты или иммуномембраны.Этот метод экстракции чаще всего используется для выделения и кон¬
центрирования малолетучих или нелетучих соединений с последующим
анализом методом ВЭЖХ или ВЭЖХ-МС [106].На иммуносорбенты наносятся биологически активные соединения
антитела, которые селективно могут удерживать только индивидуальные
соединения или отдельный класс соединений из сложной матрицы. Имму¬
ноэкстракция широко применяется для селективного выделения энзимов,
белков, вирусов, гормонов, пептидов и др.Иммуносорбентами зшюпняются картриджи, подобные картриджам с C,g.
Картриджи с иммуносорбентами должны храниться в буферном растворе.В последние годы иммуноаффинная ТФЭ стала применяться и в газо¬
вой хроматографии, в частности для определения триазинов [107]. В этом
случае иммуносорбент был на основе целлюлозы с нанесенным монокло¬
нальным антителом (анти-атризином). Высокая селективность выделения
позволяла определять триазины в речной, сточной воде, в апельсиновом442
ГЛАВА 16 Методы концентрирошнш в газовой хроматографиисоке с пределом детектирования 15-25 мг/л при пробе 10 мкл с использова¬
нием неселективного плазменно-ионизационного детектора.16.7 ТФЭ на полимерах с порами молекулярных размеров
(molecular imprinted polymers).Такие полимеры создаются при специальном синтезе с включением
молекул-шаблонов (template), которые затем вымываются, создавая поры
только размером определенных молекул. Такие полимеры обладают высо¬
кой селективностью и сорбируют только молекулы, обладающие размером
пор, либо класс соединений с молекулами таких размеров.Картриджи с такими полимерами применяются для очистки и концен¬
трирования [108, 109].Опубликованы работы по селективному выделению кофеина [110], са¬
лициловой кислоты [111], холестерина [112] и кверцетина [113].16.8 Жидкостно-жидкостная экстракция (ЖЖЭ)ЖЖЭ была и в некоторых случаях остается наиболее популярным ме¬
тодом экстракции. В работах [112] указывалось, что ЖЖЭ использовалась
среди 50% пользователей (на основании специально проведенных опро¬
сов). В этом методе экстракция основана на распределении компонентов
пробы между двумя несмешивающимися жидкостями-растворителями в
делительной воронке (рис. 16.5). В большинстве случаев одной из жид¬
костей является вода, другими жидкостями - органические растворители.
Растворители, смешивающиеся с водой (низкомолекулярные спирты, кето¬
ны, ацетонитрил, диоксан и др.), не пригодны для ЖЖЭ.Рис. 16.5 Делительная воронка для ЖЖЭВ таблице приведены типичные сочетания растюрителей для ЖЖЭ [114].Таблица 16.10ппВода и водные смесиОрганические растворители,
несмешивающиеся с водой1,Чистая водаН.углеводороды2.Вода с pH < 7Эфиры3.Вода с pH > 7Метиленхлорид443
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я.Газовая хроматография№№ппВода и водные смесиОрганические растворители,
несмешивающиеся с водой4.Водные растворы солейХлороформ5.Водные растворы с ионными парами,
хелатными, хиральными агентамиЭфиры; алифатические кетоны вышеС.6.Водные растворы с двумя или более
агентами, указанными в п. 5Н.спирты выше С^, толуол, ксилолПри ЖЖЭ органические более гидрофобные соединения из водной сре¬
ды переходят в органический растворитель. В некоторых случаях органиче¬
ский растворитель выпаривают, увеличивая степень концентрирования.Добавляя в водную среду кислоты, основания, соли или другие аген¬
ты, указанные в таблице, можно смещать равновесие в сторону увеличе¬
ния коэффициента распределения.Для лучшей экстракции необходимо тщательное перемешивание.Коэффициент распределения К - это отношение:К = ^^ (16.3)где: - концентрация экстрагируемого вещества в органической
фазе; - концентрация этого же вещества в водной фазе.Долю экстрагируемого соединения (6) можно определить, используя
соотношение:5 = С V,/(C„V + С V,) = KV/(1 + KV) (16.4)где: V^, - объемы органической и водной фазы, соответственно;
V = .О вКоэффициент распределения должен быть более 10. Для экстракции
используют 10-1000 мл каждой фазы.В некоторых случаях экстракцию делают многократно. Тогда долю
экстрагируемого соединения можно определять по соотношению:5 = 1 - [1/(1 + KV)]" (16.5)где: п - число экстракций.При небольших значениях К полезно использовать непрерывную экс¬
тракцию. при которой капли одного растворителя непрерывно проходят
через обьем другого растворителя. Весьма эффективна противоточняя экс¬
тракция.ЖЖЭ имеет много преимуществ: универсальность, возможность вы¬
бора большого числа растворителей, дешевизна, простота и доступность
аппаратуры.444
тлвл 16Методы концентрированш в газовой хроматографииЖЖЭ позволяет экстрагаровать примеси с концентрацией 10"*- 10 * %
со степенью извлечения 50-95%.Для увеличения степени извлечения применяют эффекты высаливания,
тщательного перемешивания на шейкерах, ультразвукового воздействия.В последние годы ЖЖЭ в своем классическом варианте применяется
все реже и реже в связи с тем, что нужно регенерировать или утилизиро¬
вать достаточно большие количества загрязненных растворителей. Кроме
того, особо чистые растворители дороги, а полноты ЖЖЭ иногда недо¬
статочно.Предложены новые методы ЖЖЭ:- экстракция небольшим количеством растворителя (всего несколыю мл)
в специальных колбах;- ускоренная экстракция небольшим количеством растворителя при
повышенных давлениях и температурах в динамических условиях (для
этих целей выпускается аппаратура, близкая к жидкостному хроматогра¬
фу) (рис. 16.6) [115];ПовторнаяэкстракцияРастворителиРис. 16.6 Общая схема ускоренной экстракции растворителями
- экстракция сверхгорячей водой под давлением также в динамиче¬
ских условиях; при высоких температурах возрастает растворимость не¬
полярных соединений в воде; при экстракции сверхгорячей водой нет про¬
блемы с утилизацией, т.к. вода «дружественна» к окружающей среде.Широко известна и отработана ЖЖЭ в аппарате Сокслета. особенно
из твердой матрицы, возможна многократная экстракция.В последние годы стала популярной экстракция одной каплей раство¬
рителя рис 16.7.445
Яшин ЯМ. Яшт ЕЯ.. ЯшинА,Я. Газовая хроматографияРис. 16.7В работе [115, 116, 117] предложен метод микро-ЖЖЭ одной каплей
органических растворителей, находящейся на конце иглы шприца. Микро¬
шприцем на 10 мкл отбирается растворитель (чаще всего гексан, четырех¬
хлористый углерод и др.), игла шприца опускается в исследуемый водный
раствор и штоком выдавливается обьем 2 мкл (такое количество раствори¬
теля удерживается на игае). Эта микрокапля постоянно висит на кончике
иглы и не срывается. Время экстракции 10-15 мин. После выдержки ми¬
крокапля втягивается в шприц и вводится в испаритель газового хромато¬
графа. Исследуемый раствор постоянно перемешивается микромешалкой.Для построения градуировочного графика подобный процесс прово¬
дят с растворами известной концентрации.16.9 Жидкостная микроволновая экстракция.В этом методе микроволновая энергия используется для нагрева рас¬
творителя, находящегося в контакте с анализируемой пробой, для того,
чтобы легче экстрагировать аналит из матрицы пробы за счет ускорения
массообмена. Обычно такая экстракция происходит за 15-30 мин. сравни¬
тельно небольшим объемом растворителя 10-30 мл. Это примерно в десять
раз меньше, чем при экстракции обычным методом ЖЖЭ.Для реализации этого метода используются микроволновые печи. Ин¬
терес к этому методу постоянно растет с 1990 г. Число публикаций к на¬
стоящему времени возросло на порядок [118].В этом методе применяются следуюпще растворители: вода (78,3),
ацетон (20,7), ацетонитрил (37,5), этанол (24,3), метанол (32,6), 2-пропа-
нол (19,9). В скобках указаны диэлектрические константы [119].Основные применения: экстракция ПАУ [120], полихлорированных
бифенилов [121], пестицидов [122], фенолов [123], металлов [124] и др.446
ГЛАВА 16Методы концентрировтт в газовой хроматографии16.10 Сверхкритическая флюидная экстракция (СФЭ).СФЭ введена в 1988 г. вместо Сокслет экстракции. Первые устройства
имели небольшой объем (100 мкл).Сверхкритическое состояние веществ существует при повышенных
температурах и давлении. Вещество в сверхкритическом состоянии имеет
физические свойства, промежуточные между газом и жидкостью, в част¬
ности по плотности, диффузии и др.СФЭ по растворяющей способности близка к жидкостной экстракции,
по диффузии в сложные матрицы приближается к газам. Поэтому СФЭ вред¬
ных примесей из некоторых пищевых продуктов происходит в десять или
более раз быстрее, чем классическая жидкостно-жидкостная экстракция.В таблице 16.11 приведены значения температуры и давления, при ко¬
торых существуют сверхкритические состояния для приведенных соеди¬
нений.Таблица 16.11СоединенияР^, атмПлотность в критическом
состоянии, г/млСО,31,0720,47Np36,570,60,45Этан32,347,60,20Пропан96,742,40,22Н-пентан196,632,90,23Аммиак132,5109,80,23SF,45,538,0—Вода374,2214,80,32В СФЭ в качестве растворителя в основном применяются диоксид
углерода в сверхкритическом состоянии при температурах 32 - 100 ®С и
давлении 70-400 атм.На рис. 16.8 приведена структурная схема сверхкритического флю¬
идного экстрактора, разработанного Институтом аналитического прибо¬
ростроения (гС.-Петербург). Основные области применения приведены в
работах [125-130].447
Яшин Я.И., Яшин Е.Я.. Яшин А.Я.Блок фильтровГТ1Газовая хроматографияСО,ХУМех.
фильтрх-ф-Н,0fSffiSaLFj]■#Те]М0стагРестрикторРис. 16.8 Структурная схема сверхкритического экстрактора16.11 Мембранная экстракция.В этом методе экстракция происходит за счет различия скоростей про¬
никновения разделяемых смесей через мембраны. Обычно рассматривают
три типа массообмена через мембрану [131]: пассивный, облегченный и
активный.В первом случае, который чаще всего используется для аналитических
применений, мембрана выступает в роли барьера, через который компо¬
ненты транспортируются под воздействием градиента их электрохимиче¬
ского потенциала.В облегченном варианте жидкий носитель в мембране в значительной
степени повышает проницаемость.Активное проникновение, в основном, происходит в клеточных мем¬
бранах против градиента электрохимического потенциала за счет реакции
внутри мембраны [131,132].Мембранная экстракция применяется с ВЭЖХ и ГХ [131-140]. Мем¬
бранная экстракция в сочетании с ГХ применяется для непрерывного мо¬
ниторинга летучих [136] и полулетучих [137] органических соединений в
водных пробах окружающей среды.Обычно используются плоские мембраны, однако в последних рабо¬
тах используются полые, которые обеспечивают более высокое отношение
площадь поверхности/объем [138]. Математическое моделирование экс¬
тракционных систем разработано в работе [139].Большой вклад в теорию и практику мембранной экстракции внес Мо¬
сквин Л.Н. с сотрудниками [140]. Им разработаны хроматомембранная га¬
зовая экстракция и хроматомембранная жидкостная экстракция.448
ГЛАВА 16 Методы концентрирования в газовой хроматографии16.12 Газовая экстракция (метод анализа равновесного пара,
head-space)Газовая экстракция - это способ получения качественной и количе¬
ственной информации о составе жидких и твердых проб путем анализа
выделяемых из них летучих компонентов в газовую фазу.Метод газовой экстракции в нашей стране всесторонне разработан
Б.В.Иоффе, А.Г.Витенбергом и Б.В.Столяровым [22, 23, 24]. Ими разрабо¬
тана теория метода, аппаратурное исполнение и основные аналитические
применения.Газовая экстракция может быть одноступенчатой статической равно¬
весной или непрерывной динамической. Метод, при котором выделяемые
путем динамической экстракции соединения концентрируются на адсорб¬
ционной ловушке, получил название «рш'§е and trap» - метод выдувания и
улавливания/В равновесной газовой экстракции модель извлечения основана на ве¬
личине коэффициента распределения:К = -^=-, (16.6)где: С^- концентрация анализируемого вещества в жидкой фазе;- концентрация анализируемого вещества в газовой фазе.За счет высоких значений коэффициентов диффузии паров летучих
соединений в газовых средах достаточно быстро устанавливается равно¬
весие (5-15 минут для вязких жидкостей).Процесс проведения газовой экстракции достаточно прост. В неболь¬
шую емкость помещают определенный обьем исследуемого раствора жид¬
кости V^, в котором находится измеряемое соединение в концентрации С^.
Обьем газовой фазы в этой закрытой емкости - V^. После установления
равновесия при постоянной температуре отбирают определенный обьем
газовой фазы и определяют в ней концентрацию исследуемого соединения
методом газовой хроматографии.Как обычно, предварительно проводят градуировку, т.е. устанавлива¬
ют связь величины с площадью или высотой пика на хроматограмме.Содержание соединения в исследуемом растворе определяют по урав¬
нению [22, 23]:С. = СДК + г), (16.7)где г = V^/V^.Естественно, что это уравнение справедливо, если сохраняются посто¬
янными обьемы жидкой и газовой фаз. Однако в некоторых случаях встре¬
чаются системы с неизвестными параметрами фазового распределения.44929 Газовая хроматография
Яшин Я.И.. Яшт Е.Я.. Яшт А.Я. Гюовая хроматографияДля ЭТИХ целей в статических методах газовой экстракции предложены по¬
вторные или многократные акты газовой экстракции [22].Степень снижения пределов определения летучих соединений мето¬
дом газовой экстракции по сравнению с прямым вводом анализируемого
раствора определяется соотношением;Шг 10^ ^,^04“■ = "lSr~'RTF’где; степень увеличения чувствительности парофазного анализа;
и - массы определяемого вещества, вводимые в хроматографи¬
ческую колонку при дозировании раствора с концентрацией С^ или равно¬
весного газа с концентрацией С_^Статическая газовая экстракция используется для определения соеди¬
нений с небольшими и средними значениями коэффициентов распределе¬
ния (обычно не более сотен единиц).Типичные объекты анализа этим методом - жидкости с растворенны¬
ми газами, эфирами, кетонами, меркаптанами, сульфидами, дисульфида¬
ми, летучими ароматическими и галогеносодержащими углеводородами.Методы и устройства количественной газовой экстракции основаны
на пневматичесюм отборе газовой фазы [142] и точном измерении разно¬
сти давления между емкостью с анализируемой жидкостью и испарителем
хроматографа.Нужно отметить, что эта техника отбора успешно применяется также
для градуировки хроматографов [143], приготовления парогазовых смесей
[144] и парофазного концентрирования примесей летучих веществ [145].Ранее газовую экстракцию проводили при низких температурах, позд¬
нее был разработан высокотемпературный парофазный анализ. Газовая
экстракция позволяет снизить предел детектирования, т.е. увеличить кон¬
центрацию анализируемого соединения в газовой фазе только для соедине¬
ний со значениями К < 10^ Снизить значение К можно за счет увеличения
температуры, т.к. известно, что с ростом температуры водных растворов
коэффициенты распределения уменьшаются в 1,5-2 раза на каждые 10°С
по крайней мере до 100°С. Использование высоких температур позволило
значительно расширить области применения парофазного анализа [146].Дальнейшее развитие парофазного анализа с использованием газовой
экстракции - это распространение его на гетерогенные конденсированные
фазы [147]; твердые тела, взвеси, эмульсии и пр. Основная проблема газо¬
вой экстракции трехфазных систем связана с тем, что установление равно¬
весия между двумя концентрированными фазами (между твердым телом и
жидкостью) происходит медленно. Для решения этой проблемы применя¬
ется гомогенизация конденсированных фаз.450
ГЛАВА 16Методы концентрирования в газовой хроматографииМетод анализа равновесного пара получил очень широше распростра¬
нение в практике газовой хроматографии благодаря простоте исполнения
и хорошего аппаратурного воплощения. Тысячи публикаций вышли по его
методологии и применению в контроле загрязнений окружающей среды, в
медицине, контроле пищевых продуктов, в энергетике и других областях
[22,23]. Десятки фирм выпускают устройства парофазного анализа к газо¬
вым хроматографам. Особенно часто метод АРП применяется для анализа
летучих компонентов пищевых продуктов и напитшв (см. таблицу 16.12).
Некоторые методики с использованием парофазного анализа сертифици¬
рованы на ведомственном и государственном уровнях.Таблица 16.12Некоторые примеры определения летучих компонентов в пищевых
продуктах методом анализа равновесного пара.№№Пищевые продуктыСсылки1.Аромат зеленого чая1502.Исследование свежести поджаренного кофе1513.Летучие компоненты какао1524.Летучие компоненты масла какао1535.Анализ аромата вина из винограда «Пино»1546.Летучие компоненты пива1557.Характеристика запаха свежих овощей1568.Летучие компоненты чеснока1579.Идентификация летучих компонентов черешни15810.Эфирные масла цветов яблони159И.Летучие соединения свежего апельсинового сока16012.Летучие компоненты свежих фруктов16113.Летучие компоненты картофеля161,16214.Летучие компоненты пищевых продуктов16315.Летучие компоненты растительных масел16416.Оценка прогорклости соевого масла16517.Летучие альдегиды в нагретом пищевом масле16618.Летучие компоненты нагретого свиного жира16719.Летучие продукты окисления липидов молочных продуктов16845129-
Яишн ЯМ. Яшин Е.Я., Яшин А.Я. Гтовая хроматография16.13. Динамическая газовая экстракция - метод выдувания и
улавливания.Этот метод выдувания и улавливания, в отличие от метода АРП, может
быть использован и для определения полулетучих соединений из водных
сред [148]. В этом методе анализируемая вода непрерывно продувается чи¬
стым газом, газ захватывает летучие и полулетучие органические примеси
из воды. На выходе газ с парами примесей проходит через адсорбционную
ловушку, в которой органические примеси сорбируются. В качестве сор¬
бента используются полимеры или углеродные адсорбенты, которые слабо
сорбируют пары влаги.После пропускания определенного объема газа органические примеси
с адсорбционной ловушки десорбируют под воздействием температуры.Этим методом анализируют до 110 органических примесей в природ¬
ных водах по гостированной методике в США [24].452
BMMd М ЛитератураЛитература1. Liska 1., 50 лет ТФЭ в анализе воды. J.Chromat. 2000, v. 885, N 1+2,
p. 3-162. Huck C.W., Bonn G.K. Полимерные адсорбенты для ТФЭ. J.Chromat.
2000, V. 885, p. 51-72.3. Hennion M.-I. Углеродные адсорбенты для ТФЭ. J.Chromat. 2000,
V. 885, p. 73-96.4. Rossi D.T., Zhang N. Автоматизация ТФЭ. J.Chromat. 2000, v. 885,
p. 97-114.5. Pichon V. ТФЭ для анализа органических загрязнетелей в воде.
J.Chromat. 2000, v. 885, p. 195-216.6. Snow N.H. МТФЭ лекарств из биологических жидкостей. J.Chromat.
2000, V. 885, р. 445-455.7. Lord Н., Pawliszyn J. Эволюция технологии микротвердофазной экс¬
тракции. J.Chromat. 2000, v. 885, p. 153-194.8. Carson М.С. Ион-парная твердофазная экстракция. J.Chromat. 2000,
V. 885, р. 343-350.9. Delaunay N., Pichon V., Hennion М.С. Иммуноаффинная твердофаз¬
ная экстракция (ТФЭ) в анализе следов низкомолекулярных соедине¬
ний в сложных матрицах. J.Chromat. 2000, v. 745, p. 15-37.10. Stevenson D. Иммуноаффинная ТФЭ. J.Chromat. 2000, v. 745, p. 39-48.11. Delaunay-Bertancin N., Pichon V., Hennion M.C. Иммуноэкстракция:
высокоселективный метод пробоподготовки проб. LC-GC Europe 2001,
V. 14, N2, p. 162.12. Delaunay-Bertancin N., Pichon V., Hennion M.C. Обзор no иммуноэк¬
стракции. J.Chromat. 2000, v. 745, p. 15-37.13. Ensing K., Berqyren C., Majors R.E. Селективные сорбенты для
ТФЭ, основанные на полимерах, с отпечатанными размерами молекул.
LC-GC Europe 2002, v. 15, N 1, p. 16.14. Majors R.E. Жидкостная экстракция для пробоподготовки. LC-GC
1995, V. 13, N5, р. 364.15. de Jaqer LS., Andrews A.R.J. Analyst 2000, v. 125, p. 1943.16. Ramos I., e.a. Жидкостная экстракция под давлением ПАУ из почвы
и донных отложений. J.Chromat. 2000, v. 891, p. 275-286.17. Eskilsson S.C. Аналитическая микроволновая экстракция. J.Chromat.
2000, V. 902, p. 227-250.18. McNally M.E.P. Сверхкритическая флюидная экстракция. Anal.
Chem. 1995, V. 67, p. 308.19. Jonsson J.A., Mattiasson I. Мембранная техника для обогащения
проб. J.Chromat. 2000, v. 902, p. 205-225.20. Cordeco B.M. Аналитические применения мембранной экстракции
в хроматографии и электрофорезе. J.Chromat. 2000, v. 902, p. 195-204.453
Яшин ЯМ. Яшт ЕЛ. Яшин Л.Я. Гтовая хтшатог/тфия21. Москвин Л.Н. Хроматомембранный метод и его аналитические воз¬
можности. Справочник химика и технолога т.4. «Аналитическая хи¬
мия» ММП, С. Петербург, 2002.22. Виттенберг А.Г., Иоффе Б.В. Газовая экстракция в хроматографи¬
ческом анализе. Парофазный анализ и родственные методы. Л., Хи¬
мия, 1982, с. 280.23. Kolb В., Ettre L.S. Static Headspase-Gas Chromatography. Theory and
practice. New York, Wiley-VCH, 1997, p. 298.24. Schnable J.G. Система шнцентрирования Purge-and-Trap. LC-GC
Int. 1992,v. 3,N3,p. 43.25. Lonneman W.A., Bellar T.A., Altshuller A.P. Environ. Sci. Technol,
1968, v.2, p. 17.26. Pollizzari E.D. et. al. Anal. Chem. 1976, v.48, p.803.27. Grob K„ Grob G. J.Chromat. 1972, v.62, p.l.28. lofFe B.V., Isidorov V.A., Zenkevich I,G. J.Chromatog. 1977, v.l42, p.787.29. Raymond A., Guiochon G. Environ. Sci. Technol. 1974, v.8, p. 143.30. Bertsch W. et. al. J.Chromatog. 1974, v.99, p.673.31. Eaton H.G., Umstead m.E., Smith W.D. J.Chromatog.Sci. 1973, v. 11, p.275.32. Bruner E, Bertini D., Serini C. Anal. Chem. 1978, v.50, p.53.33. Fleurat-Lessard P. et.al. J.Chem.Educ. 1999, v.76, p.962.34. Hill R. et. al. Anal. Chem 1976, v.48, p.395.35. Lovelock J.E., Watson A. J.Chromatog. 1978, v.l58, p.l23.36. Hoshika Y, lida Y. J.Chromatog. 1977, v.l34, p.423.37. Dear D.J. et.al. J.Chromatog. 1977, v.l37, p.315.38. Плешков E.H., Мальцев В.В. Полимерные строительные материа¬
лы 1976,т11,с.6.39. Gearhart H.L. et.al. J.Chromatog.Sci 1977, v.l5, p.480.40. Чаребава Л.Г. Бюллетень Всесоюзного научно-исследовательского
института чайной промышленности 1969, с.24.41. Nurok D., Anderson J.W., Zlatkis А. Chromatographia 1978, v.21, p. 188.42. Vanhaelen M. et al. J. Chromat. 1977, v. 144, p. 108.43. de Greef J., de Proff М., de Winter F. Anal. Chem. 1976, v. 48, p. 38.44. Pecka J., Feltl L. J. Chromat. 1977, v. 131, p. 179.45. Zeldes S.G., Horton A.D. Anal. Chem. 1978, v. 50, p. 779.46. Жуховицкий A.A. и др. Зав. лаб. 1963, 10, с. 1162.47. Березкин В.Г, Мирзаянов B.C. Изв. АН СССР сф. хим. 1967, с. 1202.48. Мирзаянов B.C., Березкин В.Г. Нефтехимия 1964, т. 4, с. 641.49. Березкин В.Г, Татаринский B.C. Газохроматографические методы
анализа примесей, Москва, Наука, 1970, 127 с.50. Туркельтауб Н.М. и др. Ж. аналит химии, 1964, т 19, с. 133.51. Жуховицкий А.А. и др. Зав. лаб. 1967, т. 33, с. 796.52. Крылов Б.К., Калмановский В.И. Зав. лаб. 1969, т. 35, с. 152.454
ГЛАВА 16 Литература53. Крылов Б.К., Яшин Я.И. и др. Газовый хроматограф. Авт. свид.
№ 232592, Патент Англии Ха 1265036 от 27 10 1969 г.54. Крылов Б.К., Калмановский В.И., Яшин Я.И. Зав. лаб. 1971, т. 37,
с. 133.55. Яшин Я.И. и др. Способ анализа микропримесей в этиловых спир¬
тах высшей очистки. Авт. свид. № 285319. Бюлл. изобретений № 33,
1968.56. Грязное В.П., Яшин Я.И. и др. Ферментно-спиртовая промышлен¬
ность 1968, № 8, с. 9.57. Грязное В.П., Яшин Я.И. и др. Газохроматографические методы
анализа состава примесей в пищевом этиловом спирте, Москва, ЦИН¬
ГИ, Пищепром, 1968, 93 с.58. Arthur C.L., Pawliszyn J. Anal. Chem. 1990, v. 62, p. 2145.59. Pawliszyn J. in; Solid Phase Microextraction: Theory and Practice. Wi¬
ley-VCH, New York 1997.60. Pawliszyn J. in: Application of Solid Phase Microextraction, Royal So¬
ciety of Chemistry, Cambridge, UK, 1999.61. Zhang Z., Yong M.J., Pawliszyn J. Anal. Chem. 1994, v. 66, p. 844 A.62. Pawliszyn J. Trends Anal. Chem. 1995, v. 14, p. 113.63. Martos P.A., Pawhszyn J. Anal. Chem. 1998, v. 70, p. 2311.64. Martos P.A., Soraullo A., Pawliszyn J. Anal. Chem. 1997, v. 69, p. 402.65. Valor I., Cortada C., Molto J.C. High Resolut. Chromatogr. 1996, p. 472.66. Perschmann J., Kopinke F.D., Pawliszyn J. J. Chromatogr. 1998, v. 816,
p. 159.67. Bartak P., Cap L. J. Chromatogr. 1997, v. 767, p. 171.68. Jackson G.R, Andrews A.R.J. Analyst (Cambridge) 1998, v. 123, p. 1085.69. Yang Y, Miller D.J., Hawthorne S.B. J. Chromatog. 1998, v. 800, p. 257.70. Marce R.M., Borull F. J. Chromat. 2000, v. 885, p. 273-290.71. Sherma J. J.Assoc. off Anal. Chem. Int. 1999, v.82, p.561-573 (271 c:)72. Pico Y et al. J. Chromat. 2000, v. 885, p. 251-271.73. Wells M.J.M. J. Chromat. 2000, v. 885, p. 237-250.74. Savic H. J. Chromat. 2000, v.835, p.217-236.75. Beltron J. et al. J. Chromat. 2000, v. 885, p. 389-404.76. Luns-Betley K. et al. J. Chromat. 2001, v. 938, p. 93-101.77. Rogriguez 1. et al. J. Chromat. 2000, v. 885, p. 291-304.78. Pichon V. J. Chromat. 2000, v. 885, p. 195-215.79. NamieSnik J. et al. J. Chromat. 2000, v. 885, p. 405-413.80. Нафег M. J. Chromat. 2000, v. 885, p. 129-151.81. Eisert R., Levsen K. J. Chromat. 1996, v. 733, p. 143-157.82. Perez-Camino M.C. J. Chromat. 2000, v. 885, p. 321-341.83. Lord H., Pawliszyn J. J. Chromat. 2000, v. 885, p. 17-63.84. Snow N.H. J. Chromat. 2000, v 885, p. 445-455.455
Яшт Я.И., Яшт Е.Я., Яшт А.Я. Газовая .храчатография85. Furton K.G. et al. J. Chromat. 2000, v. 885, p. 419-432.86. Березкин В.Г., Макаров Е.Д., Столяров Б.В. Нефтехимия 2002, т.42,
№3, С.232.87. Карцова А.А. и др. Ж.аналит. химии 2002, т.57.88. Abdel-Rehim М. J. Chromat. В. 2004, v. 801, р. 317.89. Abdel-Rehim М. J. Chromat. А. 2006, v. 1114, р. 234.90. Abdel-Rehim М. J. Chromat. В. 2005, v. 817, р. 303.91. Abdel-Rehim М. J. Mass. Spectrom. 2004, v. 39, p. 1488.92. Pyrynska К. Anal. Sci. 2007, v. 23, p. 63.93. Abraham M.N. et al. J. Chem. Soc. 1993, v. 24, p. 1863.94. Serrano A., Gallego M. J. Sep. Sci. 2006, v. 29, p. 33.95. Xiao C.H. et al. Chromatographia, 2000, v. 52, p. 803.96. Zhao H. et al. Anal. Chim. Acta, 2007, v. 586, p. 399.97. Ballesteras E., Gallego М., Valcarcel M. J. Chromatogr. 2000, v. 869, p. 101.98. Xiao C.H. et al. J. Chromatogr. 2001, v. 927, p. 121.99. Long R.Q., Yong R.T. J. Am. Chem. Soc. 2001, v. 123, p. 2058.100. Cai YQ. et al. Anal. Chem. 2003, v. 75, p. 2517.101. Fang G.Z., He J.X., Wang S. J. Chromatogr. 2006, v. 1127, p. 12.102. Zhou Q. et al. Talanta 2006, v. 68, p. 1309.103. Zhou Q., Ding Y, Xiao C.H. J. Chromatographia, 2007, v. 65, p. 25.104. Zhou Q. et al. Anal. Sci. 2007, v. 23, p. 189.105. Zhou Q., Xiao J., Wang @. J. Chromatogr. 2006, v. 1125, p. 152.106. Henion M.-C., Pichon V. Immtmo-based Sample preparation for trace
analysis. J. Chromatogr. 2003, v. 1000, p. 29-52.107. Dalluge J. et al. J. Chromatogr. 1999, v. 830, p. 377.108. Anderson L.l. J. Chromatogr. B. 2000, v. 739, p. 163.109. Masque N. et al. Trends Anal. Chem. 2001, v. 20, p. 477.110. Theodoridis G., Manesiotis P. J. Chromatogr. 2002, v. 948, p. 163.111. Zhang T. et al. Chromatographia 2002, v. 55, p. 447.112. Hwang C.C., Lee W.C. J. Ctaomatogr. B. 2002, v. 962, p. 69.113. Motinelli A., Weiss R., MizaikofFB. J. Agric. Food Chem. 2002, v. 50,
p. 1804.114. Majors R.E. LC-GC Intern. 1997, No. 2, p. 93.115. Richter B.E., Covino L. LC-GC, 2000, v. 18, p. 1068.116. Крылов В.A., Пылова E.B., Чернова О.Л., Лизунова Г.М., Крылов
А.В. Ж. анал. хим. 2007, т. 62, с. 1132.117. Buszewski В., Ligor Т. LC-GC Europe, 2002, No. 2, p. 92.118. Eskilsson С.S., Bj6rklund Analitical-Scule Microwave-assisted extrac¬
tion. J. Chromatogr. 2000, v. 902, p. 227-250.119. Jassie L. et al. (Eds) Microwave-Enhanced Chemistry, American
Chemical Society, Washington, DC 1997, 569 p.120. Saim N. et al. J. Chromatogr. 1997, v. 791, p. 361.456
ГЛАВА 16 Литература121. Cairo N. et al. J. Microcol. Sep. 1999, v. 11, p. 544.122. Silgoner I. et al. Fres. J. Anal. Chem. 1998, v. 362, p. 120.123. Llompart M.P. et al. J. Chromatogr. 1997, v. 757, p. 153.124. Vazguez M.J. et al. Anal. Chem. 1997, v. 69, p. 221.125. Janda V. et al. J. Cromatog. 1996, v. 733, p. 35-40.126. Turner C., King J.N., Mafliiasson L. J. Chromatog. 2001, v. 936, p. 215-237.127. Кузнецов М.П., Глазюв И.Н., Ревельский И.A., Лузянин Б.П. Зав.
лаб. 2003, т. 69, с. 7.128. Кузнецов М.П., Глазков И.Н., Ревельский И.А., Лузянин Б.П. Ж.
анал. хим. 2006, т. 61, с. 1144.129. Глазков И.Н., Ревельский И.А. и др. Сверхкрит. флюиды: теория и
практика. 2006, т. 1, с. 52.130. Smith R.M. J. Chromatog. 2003, v. 1000, p. 3-27.131. Cordaro B.M. et al. Analytical applications of membrane extraction in
chromatography and electrophoresis. J. Chromatogr. 2000, v. 902, p. 195.132. Jdnsson J.A., Mathiasson L. Membrane-based techniques for sample
enrichment. J. Chromatogr. 2000, v. 902, p. 205-225.133. Bertsch R.A., Way J.D. (Eds) Chemical Separation with Liquid Mem¬
branes, ACS Symposium Series, No. 642, American Chemical Society,
Washington DC 1996.134. Dean J.R. Extraction Methods for Environmental Analysis, Wiley,
Chichester, 1998.135. Handley A.J. (Ed) Extraction Methods in Organic Analysis, Sheffield
Academic Press, Sheffield, 1999.136. Luo Y.Z., Pawliszyn J. Anal. Chem. 2000, v. 72, p. 1058-1064.137. Matz G. et al. J. Chromatogr. 1999, v. 830, p. 365.138. Pawliszyn J. Trends Anal. Chem. 1995, v. 14, p. 113.139. Luo Y.Z., Adams М., Pawliszyn J. Anal. Chem. 1998, v. 70, p. 248.140. Москвин Л.Н. Рос. хим. журнал 1996, т. 40, № 1, с. 67-75.141. Kolb В. J. Chromatog. 1999, v. 842, p. 163-205.142. Хохенберг X., Шмидт А. Газохроматографический анализ равно¬
весной паровой фазы. Пер. с англ. Москва, Мир, 1979, 160 с.143. Mills G.A., Walker V. J. Chromatog. 2000, v. 902, p. 267-287.144. Majer H.G. J. Chromatog. 1970, v. 50, p. 329.145. Barnett M.G., Swoboda PA.T. Anal. Chem. 1962, v. 34, p. 1162.146. Lee K.Y., Nurok D., Zlatkis A. J. Chromatog. 1978, v. 158, p. 377.147. Romano S.J., Renner J.A., Leittier PM. Anal. Chem. 1973, v. 45, p. 2327.148.Berens A.R. J. Appl. Polym. Sci. 1975, v. 19, p. 203.149. Laretta J. et al. Chromatographia. 2008, v. 67, p. 93.150. Zeng X. et al. Анализ аромата зеленого чая ГХ-МС с АРП. Sepu1990, V. 8, р. 169-172.151.Holscher S. Исследование свежести поджаренного кофе АРП. Z.
Lebensm.-Unters Forsch 1992, v. 195, p. 33-38.457
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я. Гуювая хроматография152. Silwar R. Исследование летучих компонентов какао. Cofe, Cacao,
Tea 1988, v. 32, p. 243-250.153.Pino J. АРП летучих компонентов масла какао. Nohrung 1992,
V. 36, p. 175-180.154. Miranda-Lopez R. et al. Анализ аромата вина из винограда «Пино».
J. Food Sci. 1992, v. 57, p. 985-993.155. Harayama К. et al. Оценка летучих компонентов пива АРП во вре¬
мя хранения. Agric. Biol. Chem. 1991, v. 55, p. 393-398.156. Shinahara A. et al. Капиллярная ГХ с АРП для характеристики за¬
паха свежих овощей. Chromatographia 1991, v. 32, p. 357-364.157.Laakso I. et al. АРП летучих компонентов чеснока. Planta Med.1989, V. 55, p. 257-261.158. Matheis J.P., Buchanan D.A., Felhnan J.K. Идентификация летучих сое¬
динений в черешне методом АРП. Phytochemistry 1992, v. 31, р. 775-777.159. Buchbauer G. et al. АРП эфирных масел цветов яблони. J. Agric.
Food Chem. 1993, v. 41, p. 116-118.160. Lum O.L. et al. Определение органических летучих соединений АРП
ГХ в свежем апельсиновом соке. Food Chem. 1990, v. 37, p. 313-317.161.Mazza G., Petrzak E. АРП летучих компонентов картофеля. Food
Chem. 1990, v.36, p. 97-112.162. Fischer J., Mueller K. Исследование ароматических соединений
картофеля АРП. Potato Res. 1991, v. 34, p. 159-167.163. Barcarolo P., Casson P., Tutta C. Анализ летучих компонентов пищи
АРП ГХ-МС. J. High Resolut. Chromatogr. 1992, v. 15, p. 307-311.164. Synder J.M., Mounts T.L. Анализ летучих компонентов раститель¬
ных масел АРП с многократной экстракцией. J. Ат. Oil Chem. Soc.1990, V. 67, p. 800.165.Cheen H.N., Kim Z.U. ГХ с АРП - объективный метод оценки
прогорклости соевого масла. Hon’gan Nonghwa Hokhoechi 1991, v. 34,
p. 154-161.166.Yasuhara A., Shihamoto T. Определение летучих алифатических
альдегидов методом АРП в нагретом пищевом масле. J. Chromatogr.1991, V 547, р. 291-298.167. Yasumara А., Shihamoto Т. АРП летучих соединений из нагретого
свиного жира. Food Chem. 1990, v. 37, p. 13-20.168.Park P.S.W., Goins R.E. Определение летучих продуктов окисле¬
ния липидов молочных продуктов. J. Agric. Food Chem. 1992, v. 40,
p. 1581-1585.458
ГЛАВА 17 Области применения газовой хроматографииГЛАВА 17. ОБЛАСТИ ПРИМЕНЕНИЯ ГАЗОВОЙ
ХРОМАТОГРАФИИ.17.1. Введение.Диапазон применения газохроматографических методов исключитель¬
но широк: от анализа атмосферы планет до анализа содержимого одной
клетки и маркеров заболеваний в биологических жидкостях.Газовая хроматография исключительно широко применяется для ана¬
литических целей в разных отраслях промышленности, техники, а также
для разнообразных научных исследований [1-4].По распространенности в аналитических лабораториях газовые хрома¬
тографы находятся сразу же после микровесов и рН-метров, т.е. практически
каждая большая аналитическая лаборатория имеет газовые хроматографы.В течение более 50 лет области применения газовой хроматографии посто¬
янно расширяются. Можно вьщелить следующие области применения ГХ:- технологический контроль для поддержания оптимального режима
производств в химической, нефтехимической, газоперерабатывающей,
целлюлозно-бумажной, металлургической, пищевой и др. отраслях про¬
мышленности;- анализы и исследования в нефтехимии (природный газ, бензины,
керосины, дизельное топливо, групповой анализ ПИОНА, эмитированная
разгонка нефтепродуктов и др.);- контроль загрязнений окружающей среды (воздух, вода, почва, дон¬
ные отложения, осадки и др.);- контроль загрязнений пищевых продуктов и напитков, оценка каче¬
ства и безопасности пищевых продуктов;- контроль качества и подлинности лекарственных средств в фарма¬
цевтике;- определение лекарств в биологических жидкостях для исследования
задач фармакологии, исследование фармакокинетики лекарств;- ранняя диагностика заболеваний по анализу биохимических маркеров;- определение наркотиков и наркотических средств, лекарств при зло¬
употреблениях в биологических жидкостях в судебной медицине;- вещественный анализ в криминалистике для раскрытия преступлений;- разные аналитические задачи в сельском хозяйстве (контроль средств
защиты растений, контроль кормов для животных в ветеринарии, опреде¬
ление микотоксинов и др.);- энергетика (контроль в АЭС, ТЭЦ);- определение подлинности предметов искусства;- геологоразведка (поиск газо- и нефтеносных районов и пр.) и многие
другие.459
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я. Газовая хроматографтВ последнее десятилетие реальная жизнь постоянно ставит новые ана¬
литические задачи, некоторые из них являются серьезным вызовом всем
аналитическим методам, в частности это:- решение задач протеомики, гликопротеомики, метаболомики и ли-
пидомики;- терроризм (экспрессное и высокочувствительное определение взрыв¬
чатых веществ, отравляющих химических и биологаческих веществ и др.);- контроль за уничтожением химического оружия;- анализ биотоплив;- контроль супертоксикантов в пище и напитках;- метрологические вопросы нанотехнологии;- мониторинг изменения климата;- астрохимия (в частности анализ пород Марса и других планет, химия
звездообразования, идентификация молекул в космосе);- экспрессное определение «на месте» последствий чрезвычайных си¬
туаций (взрывов, пожаров, аварий, природных катаклизмов и др.)Сведения о других областях применения ГХ можно найти в 3 - 16 гла¬
вах настоящей книги, а также в специальных книгах и обзорах.Далее будут приведены ссылки о применении ГХ в наиболее жизнен¬
но важных областях (экология, пища, медицина, фармацевтика, судебная
медицина и др.).17.2. Технологический анализ ГХ для промышленного контроля.ГХ чаще всего применяется в химической и нефтехимической про¬
мышленности.Современные химические и нефтехимические производства основаны
на непрерывной технологии. На таких производствах необходим автомати¬
ческий оперативный технологический контроль для проведения процессов
в оптимальном режиме с целью обеспечения наибольшего выхода высоко¬
качественной продукции.Хроматографические методы используются для технологического
контроля за производственными процессами на всех стадиях, включая ана¬
лиз исходного сырья и готовой продукции. Кроме того, на промышленных
предприятиях хроматографы применяются для контроля воздуха произ¬
водственных помещений, выбросов в атмосферу и сбросов в водоемы.Технологические анализы осуществляются с помощью как лаборатор¬
ных, так и промышленных автоматических хроматографов во взрывозащи¬
щенном исполнении. Лабораторные хроматографы в цеховых и централь¬
нозаводских лабораториях обычно применяются для простых анализов,
определения нескольких ключевых компонентов, по которым судят о ходе460
ГЛАВА 17Области применент газовой хроматографиипроизводственных процессов. На крупных химических комбинатах (Киро¬
во-Чепецком, Нижнекамском, Салаватском, Стерлитамакском и др.) таких
хроматографов более 500-600.В таблице 17.1 приведены ГОСТы ASTM для технологического кон¬
троля разных соединений.Таблица 17.1ГОСТы ASTM для технологического контроля методом
газовой хроматографии№№Обозначение ГОСТаНазвание методики1.D3329-99Стандартный метод определения (СМО) чистоты
метилизобутилкетона2.D2593-93 (1998)СМО чистоты бутадиена3.01983-90(1995)СМО состава жирных кислот4.D5060-95 (2000)СМО примесей в высокочистом этилбензоле5.D4768-96СМО 2,6-дитретбутил п-крезола и 2,6-дитретбутил
фенола в изоляционных жидкостях6.D4059-00СМО полихлорированных бифенилов в
изоляционных жидкостях7.D3798-00СМ анализа п-ксилола8.D3760-02СМ анализа изопропилбензола (кумола)9.D3465-00СМО чистоты мономерных пластификаторов10.03452-94(1998)Стандартная практика идентификации резины
пиролизной ГХ11.D3362-93 (2000)СМО чистоты эфиров акриловой кислоты12.D2712-91 (1995)СМО следов угаеводородов в пропилене13.D4275-96СМО бутилгидроокситолуола (ВНТ)14.Е1616-94 (2002)СМО ацетангидрида15.D6159-97СМО примесей в этилене16.D6144-97СМО а-метилстирола капиллярной ГХ17.D6142-97СМО фенола капиллярной ГХ18.D5713-00СМ анализа высокочистого бензола19.D5507-99СМО следовых органических примесей в
мономере винилхлориде на капиллярных колонках
многомерной ГХ20.D5441-98СМО метилтретбутилового эфира461
Яишн Я.И.. Яшин ЕЯ., Яшин А.Я.Газовая хроматография№№Обозначение ГОСТаНазвание меюднки21.05303-92(1997)СМО следов карбонилсульфида в пропилене22.D4747-02СМО непрореагировавших мономеров в латексах23.D4735-96 (2000)СМО следов таофена в бензоле24.D4534-99СМО циклических соединений в бензоле25.D3797-00СМ анализа о-ксилола26.D3749-95СМО остатков мономера винилхлорида в
поливинилхлориде ГХ с head-space27.D3545-02СМО содержания спирта и уксусных эфиров28.D2804-02СМО чистоты метилэтилкетона29.D2505-88 (1998)СМО углеводородов и диоксида углерода в
особочистом этилене30D6526-00СМО толуола ГХ на капиллярных колонках31D4864-90 (2000)СМО следов метанола в концентрате пропилена32D4827-93 (1998)СМО непрореагировавшего мономера в латексах
на капиллярных колонках33D4424-90 (2001)СМ анализа бутилена3404322-96(2001)СМО остаточного мономера акрилонитрила в
сополимерах стирол-акрилнитрил и нитршювых
резинах ГХ с head - space3503432-89 (1996)СМО непрореагировавшего диизоциаиата толуола
в уретановых полимерах36Е1863-97СМ анализа акрилонитрила37Е475-00СМ анализа дитретбутилпероксида3805314-92(2001)СМО состава дегтярных кислот капиллярной ГХ3905135-02СМ анализа стирола капиллярной ГХ4006438-99СМО ацетона, метилацетата и
парахлорбензотрифторида в красках и покрытиях
МТФЭ-ГХ4103893-99СМ анализа чистоты метиламилового кетона и
метилизоамилового кетона4203876-96 (2001)СМО метоксил- и гидроксилпропил заместителей в
целлюлозных эфирных продуктах4302268-93 (1998)СМ анализа высокочистого н-гептана и изооктана
капиллярной ГХ462
ГЛАВА 17 Области применения газовой хроматографиитаОбозначение ГОСТаНазвание методики44D3534-85 (1995)СМО растворимости постоянных газов в
низколетучих жидкостях45Е1821-01СМО карбогидратов в биомассе ГХ46D2685-95 (1998)СМО воздуха и четырехфтористого углерода в
шестифтористой сере47Е806-99СМО тетрахлорида углерода и хлороформа в
жидком хлоре прямым вводомНиже приведен список ГОСТов по анализу чистоты газов.ОСТ 26-04-2573-80 Определение микропримесей углеводородов в
жидком кислороде н жидком возщухеГОСТ 10157-79 Аргон газообразный и жидкийОпределение объемной доли суммы углеродсодержащих соединений.
Детектор ДТПОпределение объемной доли суммы углеродсодержащих соединений.
Хроматограф с метанатором и детектором ДИП.ГОСТ 10218-77 Анализ криптон-ксеноновой смесиОпределение объемной доли ксенона и азота. Детектор ДТПОпределение объемной доли метана и двуокиси углерода. Хромато¬
граф с метанатором и детектором ДИПГОСТ 3022-80 Водород технический ТУОпределение кислорода, азота, метана, окиси углерода и двуокиси
углерода. Хроматограф с ДТПГОСТ 51673-2000 Водород газообразный чистый. ТУОпределение объемной доли азота, метана и суммарной доли кислоро¬
да и аргона. Концентрирование пробы газа на криоконцентраторе «АСК-
1». Детектор ДТПГОСТ 5583-78 Кислород газообразный технический и медицинскийОпределение водорода. Детектор ДТПГОСТ 6331-78 Кислород жидкий технический и медицинскийОпределение ацетилена и углеводородов в жидком кислороде, йжцен-
трирование пробы, детекторы ПИД, ДТПГОСТ 9293-74 Азот газообразный и жидкий. ТУОпределение суммы СО и COj в пересчете на метан. Хроматограф с
метанатором и детектором ДИП463
Яшин Я.И.. Яшин Е.Я., Яшин А.Я.т*твФшНа многих наших предприятиях методики контроля производства ат¬
тестуются на заводском или ведомственном уровне.17.3. ГХ методы анализа природных газов, газовых конденсатов,
нефтепродуктов и нефтей.В таблице 17.2 приведены методы хроматофафии, применяемые в га¬
зовой, нефтяной промыЕшенности.Таблица 17.2Хроматография нефтей и нефтепродуктовМетодыОбласти примененияГазовая хроматографияАнализ газовых конденсатов, бензиновых, кероси¬
новых фракций, масел, групповой анализ ПИОНА,
имитированная разгонка нефтепродуктов и нефтейВысоюэффеюгивная
жидкостная хроматографияАнализ полиядерных ароматических соединений,
анализ порфиринов, групповое разделение сырых
нефтей, определение степени отработаннности ма¬
сел, селеютвное определение присадок к маслам,
анализ асфальтенов, селективное определение irre-
роциклических соединенийСверхкритическая флюид¬
ная хроматографияАнализ тяжелых фракций, разделение сырых нефтей
и нефтепродуктовИонная хроматографияОпределение в исходной нефти общего содержания
S, N, тяжелых металлов, кислотГельфильтрационная
хроматографияРазделение тяжелых фракций по молекулярным мас¬
сам, разделение асфальтеновАнализы проводятсяна всех этапах: при шологоразведке газонос-ных и нефтеносных районов, в процессе добычи газа и нефти, в процессе
транспортировки, на разных стадиях технологической переработки и, на¬
конец, для контроля готовой продукции. Кроме того, следует указать, что
контроль загрязнений окружающей среды (почвы, воды, воздуха, донных
отложений и др.) нефтепродуктами проводится преимущественно хрома¬
тографическими методами.Развитие газовой хроматографии с момента ее открытия в значитель¬
ной мере стимулировалось аналитическими задачами в нефтехимии. Вы¬
сокие разделительные способности капиллярной хроматографии впервые
были продемонстрированы на смесях бензиновых фракций сотрудниками
Института нефти (Англия) в шестидесятые годы.В настоящее время на современных высокоэффективных капиллярных
колонках возможно полное покомпонентное разделение бензиновых фрак¬
ций (200 - 1000 компонентов из одной пробы). Ни один другой метод не
дает подобных результатов.464
ГЛАВА 17 Области применения газовой хроматографииГХ применяется для контроля природных и попутных нефтяных газов,
газовых конденсатов, бензиновых и керосиновых фракций, дизельного то¬
плива, смазочных масел, тяжелых фракций нефтей.Методами ГХ определяют как индивидуальный (покомпонентный),
так и групповой состав нефтепродуктов, проводят имитированную разгон¬
ку нефти и нефтепродуктов по температурам кипения, оценивают экологи¬
ческие характеристики нефтепродуктов, в частности содержание бензола,
общей серы и азота.Групповое разделение (парафины - нафтены - ароматика) бензинов
проводят на колонках с цеолитами 13Х с последующим разделением на
капиллярных колонках. Хроматографическое определение оксигенатов в
бензине проводят с помощью О-ПИД (пламенно-ионизационного детекто¬
ра, селективного к кислородсодержащим соединениям). Принцип действия
детектора основан на переводе кислородсодержащих соединений в метан,
что позволяет избежать влияния углеводородного фона и определять окси¬
генаты на следовом уровне.Фирма «Перкин-Эльмер» совместно с фирмой «Ариел» выпускает бо¬
лее 16 анализаторов природных и нефтяных газов, в которых используются
сочетания детекторов ДТП-ДТП, ДТП-ПИД, 2 ДТП-ПИД. Кроме того, та¬
кие анализаторы могут включать обратную продувку, два крана-дозатора,
в том числе и дозатор жидких проб.Подобные схемы есть на отечественных хроматографах Цвет-800,
Кристаллюкс-4000, Кристалл-2000М и Кристалл-5000.Для анализа ароматических углеводородов в бензинах можно успещно
использовать фотоионизационный детектор. Интересные возможности от¬
крываются при одновременном детектировании пламенно-ионизационным
и фотоионизационным детекторами.Многомерная газовая хроматография применяется для разделения
сложных многокомпонентных смесей в нефтехимии, а также для анализа
компонентного углеводородного состава природного, попутного газов, га¬
зовых конденсатов и сжиженного газа.Разработаны следующие общие схемы хроматографического анализа
сырой нефти.От сырой нефти отделяют асфальтены, которые анализируются жид¬
костной хроматографией. Оставшуюся часть разгоняют дистилляцией на
две части: часть, выкипающую при температуре ниже 350“С, и часть, вы¬
кипающую при температуре выше 350“С. Отгон, выкипающий при тем¬
пературе менее 350“С, анализируется газовой хроматофафией, а более
высококипящая часть разделяется и анализируется жидкостной хромато¬
фафией. Жидкостная хроматофафия позволяет разделять эту смесь на
насыщенные, моноароматические, диароматические, полиароматические
углеводороды, полярные соединения и соединения основного характера.46530 Газовая хроматография
Яшин Я.И.. Яшин Е.Я. Яшин А.Я. ГазоваяхраштюграфшОсновные области применения газовой хроматографии в нефтехимии:• анализ природного газа;• анализ попутных природных газов;• анализ газовых конденсатов;• анализ бензиновых фракций;• анализ керосиновых фракций;• анализ масел;• групповой анализ ПИОНА;• имитированная разгонка нефтепродуктов и нефтей;• анализ азот-, кислород- и серосодержапщх органиче¬
ских примесей;• групповой анализ на цеолитах;• определение октанового числа бензинов;• анализ тяжелых фракций нефтей пиролизной газовойхроматографией;• ГХ-МС фракций нефти;• ГХ-ИКС фракций нефти.Важность надежного анализа природного газа сильно возросла с ро¬
стом цены на него для добывающих, транспортирующих и потребляющих
предприятий при введении взаиморасчетов.В таблице 17.3 приведены отечественные ГОСТы по определению
природного газа, бензинов и нефти.В таблице 17.4 приведены сорбенты, применяемые для разделения
природного газа, в таблице 17.5 указаны детектирующие системы, описан¬
ные для анализа природного газа, газовых шнденсатов.Специализированные газохроматографические системы для природ¬
ного газа приведены в таблице 17.6, которые позволяют реализовать стан¬
дартный метод анализа природного газа Nj, Oj, COj, С,-С, и сумма С^.Таблица 17.3Перечень ГОСТовп/п.ГОСТ1.«Газы углеводородные сжиженные. Метод определения
углеводородного состава» (определение фракции С, - С,
и их смеси, находящихся под избыточным давлением соб¬
ственных паров при их содержании 0,01 % масс, и выше).гост 10679-762.«Газы горючие природные. Расчетный метод опреде¬
ления теплоты сгорания, относительной плотности и
числа Воббе».ГОСТ 22667-823.«Газы углеводородные сжиженные. Расчетный метод
определения плотности и давления насыщенных паров».ГОСТ 28656-90466
ГЛАВА 17Области применения газовой хроматографиип/п.ГОСТ4.«Газы горючие природные. Хроматографический метод
определения юмпонентного состава» (метод А - опреде¬
ление Nj ,0j, Hj, COj, с, - Cg при содержании не более
20% об., углеводородов С, и выше не более 1 % об., метод
Б - определение угаеводородов С^- при содержании от
0,001 до 0,5 % об.).гост 23781-875.«Газ сухой. Метод определения юмпонентного состава»
(определение С, - а также N^, 0^ СО, СО^ и H^S при
содержании 0,1 % масс, и выше).ГОСТ 14920-796.«Нефть. Метод определения сероводорода, метил- и этил-
меркаптанов».ГОСТ Р 50802-957.«Бензины. Метод определения бензола и суммарного со¬
держания ароматических угаеводородов».ГОСТ 29040-918.«Нефть. Определение углеводородов С, - С^ методом га¬
зовой хроматографии».ГОСТ 13379-829.«Газы угаеводородные сжиженные. Методы отбора
проб».ГОСТ 14921-7810.«Хроматография газовая. Термины и определения».ГОСТ 17567-81Таблица 17.4Сорбенты для разделения компонентов природного газа№№СорбентыРазделяемые компоненты1.Пористые полимеры:
Порапак Q
Хромосорб 102Углеводороды С,-С^, СО^2.Цеолит СаХHj,Oj,Nj,CH,,CO3.PoraPlot с AljOjУглеводороды С,-С,„4.PoraPlot с карбоситомC,-C,„,0„N„C0,5.PoraPlot с полимероммеркаптаны6.Капиллярные колонки широкого ди¬
аметра с разными жидкими фазамис.-с,„467
Яшин Я.И., Яшин ЕЯ., Яшин А.Я.Газовая хроматографияТаблица 17.5Детекторы для газохроматографического анализа природного газаJVa№Тип детектораАнализируемые соединения1.Пламенно-ионизационныйУглеводороды С,-С^,метанол,диэтиленгликоль2.КатарометрH„0„N„C0, СО,3.МикрокатарометрC,-q,0„N„C0,4.ФотоионизационныйС|-С,|,, HjS, меркаптаны5.Пламенно-фотометрическийHjS, меркаптаны6.Пульсирующийпламенно-фотометрическийHjS, меркаптаны7.ХемилюминисцентныйHjS, меркаптаны, сульфиды, дисульфиды8.Электрохимический:- окислительный режим- восстановительный режимС|-С|д, меркаптанынр,о,, СО,9.Гелиевый разрядныйуниверсальный10.АтомноэмиссионныйуниверсальныйИ.МасспектрометрическийуниверсальныйТаблица 17.6Переченьгазохроматографической аппаратуры для анализа природного газа№№Название прибораПроизводителиХарактеристики системы1.Цвет-800ОАО «Цвет»,
г. ДзержинскДТП + ПИД + коп + КД2.АХК-05 (06)(на основе Цвет-800)«БАКС»,
г. СамараАХК-05: 2 ДТП + ДИП
АХК-06: 2 ДТП + КОП.
БДГ-1153.Кристалл-5000«Хроматэк»,
г. Йошкар-ОлаДТП + ДИП, КД, КОП4.Кристаллюкс-4000НПЛ «Химприбор»,
г. Йошкар-ОлаДТП + ДИП, КД, коп5.Яуза-200НПО «Химавтоматика»,
г. МоскваДТП + ДИП, КД, копПримечание; ДТП - детектор по теплопроводности, ПИД - пламенно-ио¬
низационный детектор, КОП - крап обратной продувки, КД - кран дозирующий,
БДГ-115 - газовый блок.468
ГЛАВА 17 Области применения газовой хроматографии17.4. Применение ГХ для контроля загрязнений окружающей среды.По применению ГХ в контроле загрязнений окружающей среды больше
всего публикаций и больше всего гостированных и аттестованных методик.Перечень основных загрязнений 01д>ужающей среды приведен в таблице 17.7.ГХ применяется для контроля питьевой воды в США (таблица 17.8) и
в странах ЕС (таблица 17.9).В таблице 17.10 приведены основные аттестованные методики для
анализа воды в нашей стране.Перечень ASTM загрязнителей в разных средах приведен в таблице 17.11.По анализу загрязнителей окружающей среды каждый год публикуют¬
ся сотни-тысячи работ.Наиболее интересные статьи и обзоры приведены в таблицах 17.12 (по
общим вопросам), 17.13 (по анализу атмосферы городов, сел, воздуха поме¬
щений), 17.14 (по анализу водных сред), 17.15 (по анализу почв и осадков).В этих таблицах следует выделить новые работы по анализу воздуха
помещений и компонентов химического оружия в окружающей среде.Таблица 17.7Перечень основных загрязнителей окружающей среды,
анализируемых методами газовой хроматографииОсновные загрязнителиДетекторыМетодыконцентрированияОрганические примеси в атмосфере, воде и почвеПИДТФЭ; ЖЖЭВредные газы в атмосфере (NO^, SO^, SO^, NHj
и др.)ДТПНефтепродукты в водеПИДЖЖЭ; ТФЭПестициды в воздухе, воде, почвеПИД; ЭЗД;
ТИДЖЖЭ; ТФЭХлорорганические соединения в воздухе и водеЭЗДТФЭАроматические соединения (бензол, толуол,
этилбензол, ксилол) в воздухе и водеФИД; ПИДАРП; ТФЭПолиядерные ароматические соединенияПИД; ФИДТФЭФенол и его производныеПИДТФЭФреоны в стратосфереЭЗД-ПолихлорбифенилыЭЗДТФЭПолихлорированные диоксиныМС; ЭЗДЖЖЭ; ТФЭЛетучие хлорорганические соединения в
питьевой водеЭЗДАРППримечание: МС - масс-спектрометрия; АРП - анализ равновесного пара;
ФИД - фотоионизационный детектор; ПИД - пламенноионизационный детек¬
тор; ЭЗД ~ электронозахватный детектор; ДТП - детектор по теплопро¬
водности; ТИД - термоионный детектор; ТФЭ - твердофазная экстракция:
ЖЖЭ - жидкостножидкостная экстракция.469
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я. Газовая храчатографииТаблица 17.8Методы ЕРА 500 для идентификации и определения органических
соединений в питьевой воде методом газовой хроматографииНомерметодаАнализируемыесоединенияМетодыхроматографииДетекторМетод кон¬
центрирова¬
ния502.2Летучие органические сое¬
диненияКГХФИДIhirge and Trap504.11 ,2-дибромэтан,
1,2-дибромо-З-хлорпропан,
1,2,3-трихлорпропанГХ-Микроэк¬стракция505Галоидоорганические пе¬
стициды и полихлорирован¬
ные бифенилыГХ-Микроэк¬стракция506Эфиры фталатов и адипатовГХФИДЖЖЭ, ТФЭ507Азот- и фосфорсодержащие
пестицидыГХТИД-508Хлорированные пестицидыГХЭЗД-508.1Хлорированные пестициды,
гербициды и галоидоорга¬
нические соединенияГХЭЗДТФЭ508.АПерхлорированные бифйш-ГХ--515.1515.2Хлорированные кислотыГХЭЗДТФЭ524.2Выдуваемые органические
соединениякгх/мсМС-525.1525.2Органические соединениякгх/мсМСТФЭ548548.1Эндоталлгх/мсЭЗДТФЭ552552.1552.2Галоуксусные кислотыГХЭЗДТФЭПримечание: КХГ - капиллярная газовая хроматография, ТФЭ - твердофаз¬
ная экстракция, ЖЖЭ - жидкостно-жидкостная экстракция.470
ПАВА 17 Области применения газовой хроматографииТаблица 17.9Список приоритетных загрязнителей питьевой воды в странах ЕЭС,
альтернативные методы их анализа и пробоподготовкиСоединенияМетоды анализа1МетодыпробоподготовкI1. Летучие галоидоорганиче¬ские соединения (СС1,. хлоро¬
форм, хлоропрен, 3-хлоропрен,1.2-дибромэтан, 1,1-дихлорэтан,1.2-дихлорэтан, 1,1-дихлор>
тилен, дихлорметан, 1,2-дих-
лорпропан, 1,3-дихлорпропан,2.3-дихлорпропан, 1,1,2,2-те-
трахлорэтан, 1,1,1-трихлорэтан,
1,1,2-трихлорэтан, трихлорэти-
лен, 1,1,2-трихлорфтор-этан, ви-
нилхлоридКГХ/МСКГХ/ЭЗДЖЖЭПФАМТФЭнения (бензол, толуол, этилбен-
зол, ксилолы, изопропилбензол)КГХ/МСКГХ/ФИДКГХ/ПИДЖЖЭПФАМТФЭческие соединения (хлор&нзол,
1,2,4-трихлорбензол, 1,1,1-трих-
лорэтан, 1,1,2-трихл(Ч)этан)КГХ/МСКГХ/ФИДКГХ/ПИДЖЖЭМТФЭ4. Хлорфенояы (4-хлор-З-
мегалфеноя, 2-хлорфенол, З-хл^)-
фенол, 4-хлорфенол, 2,4-дихлор-
феноя, 1,2-дихлорфенол, пентах-
лорфенол, трихлор^нолКГХ/МСЖЖЭТФЭдер.(эльдрин, хлордан, ДДТ, дизяь-
дрнн, эндосульфон, эвдфин, геп-
тахлор, гексахлорбензол, линдан)КГХ/ЭЗДКГХ/МСКГХ/ЭМДЖЖЭТФЭ6. ПАХВЭЖХ/ФЛУВЭЖХ/СПФКГХ/МСЖЖЭТФЭды (азинфос-зтил, азинфос-метил,
кумафос, диметон, дихлофос, дис-
ульфотон, фентион, фенитротион,
метамидофос, мевинфос, ометоат,
метилоксидеметон, 4к>ксим, триа-
з(ф)с, трихлорофон)ВЭЖХ/СПФКГХ/МСЖЖЭТФЭПримечание: Кроме приведенных, необходимо определять: хлорнитробензо-
лы, хлорнафталины, хпоранилины, оловоорганические соединения, хлорбензидины,
полихлорбифенилы и др.471
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я.Газовая хроматографияОбозначения: ПФА - парофазный анализ, МТФЭ - микротвердофаз-
ная экстракция, ТФЭ - твфдофазная экстракция, ФЛУ - флуориметриче-
ский детектор, СПФ - спектрофотометрический детектор.Таблица 17.10Анализ примесей в воде ГХп/пШифр документаНазвание методикиПринципДиапазонмг/дм’1.ПНДФ 14.1:4.201-03МВИ ацетона и мета¬
нола в природных и
сточных водахПрямой ввод. ПИД,
насадочная или ка¬
пиллярная колонка0,3-6,02.ПНДФ 14.1:2.57-96
(2004)МВИ бензола, толуо¬
ла, 0,- М-, п-ксилолов,
стирола, этилбензола
в питьевых, природ¬
ных, сточных водахHead Space ПИД,
насадочная и капил¬
лярная колонки0,0025-3,03.ПНДФ 14.1:2:4.191МВИ гидразина в пи¬
тьевых, природных и
сточных водахПИД. Определение в
виде бензалазина, ка-
пиллфная колонка0,005-0,24.ПНДФ 14.1:2.141-98МВИ жиров в при¬
родных и сточных
водахЖидкостная экстрак¬
ция; гидфолиз; мети¬
лирование: ПИД; на¬
садочная колонка0,5-2505.ПНДФ 14.1:2:4.211-05МВИ капролактама
в природных и сточ¬
ных водахТвердофазная экс¬
тракция; ПИД, на¬
садочная колонка0,1-5,06.ПНДФ 14,1:2:4.71-96
(издание 2004 г)МВИ летучих га-
логенорганических
соединений в питье¬
вых, природных и
сточных водахHead Space, ДЭЗ, ка¬
пиллярная колонка0,(Ю005-107.НДП 30.1:2:3.72-02МВИ летучих органи¬
ческих соединений в
питьевых, природных
и сточных водахГХ-МС; Puige & Trap
(выдувание и улавли¬
вание)0,000005-0,38.НДП 30.1:2:3.74-01МВИ монохлорфено-
лов и нитрофенолов в
питьевых, природных
и сточных водахЖидкостная экс¬
тракция; ацетили-
рование, ГХ-МС0,0001-1,09.НДП 30.1:2:3.68-00МВИ органических
соединений в питье¬
вых, природных и
сточных водахГХ-МС;Жидкостно-
жидкостная экс¬
тракция0,000002-1,0472
ГЛАВА 17Области применент газовой хроматографиип/пШифр документаНазвание методикиПринципДиапазонмг/дм’10.ПНДФ 14.1:2:4.225-06МВИ фенола и фе-
нолпроизводных в
питьевых, природных
и сточных водахЖидкостная экс¬
тракция; ПИД, ка¬
пиллярная колонка0,0005-5.0И.1ШДФ 14.1:2:4.205-04МВИ фосфороргани-
ческих и симм-триа-
зиновых гербицидов
в питьевых, природ¬
ных и сточных водахЖидкостная экс¬
тракция; термион-
ный детектор; ка¬
пиллярная колонка0,00005-3.012.ПНДФ 14.1:2:4.204-04МВИ хлорорганиче-
ских пестицидов и
полихлорированных
бифенилов в питье¬
вых, природных и
сточных водахЖидкостная экс¬
тракция, ДЭЗ, ка¬
пиллярная колонка0,00001-0,113.ПНДФ 14.1:2:4.212-05МВИ 2,4-Д в питье¬
вых, природных и
сточных водахЖидкостная экс¬
тракция, ДЭЗ, ка¬
пиллярная колонка,
ГХ-МС0,0005-0,1Таблица 17.11ASTM для контроля загрязнений окружающей среды методом
газовой хроматографии№№Номер ASTMНазвание ASTM1.D6160-98Стандартный метод определения (СМО) полихлориро¬
ванных бифенилов в сбросах2.D5812-96 (2002)СМО хлорорганических пестицидов в воде капиллярной
ГХ3.D2360-00СМО следовых примесей моноциклических ароматиче¬
ских соединений4.D2306-00СМО ароматических углеводородов5.05175-91(1996)СМО галоидоорганических пестицидов и полихлори¬
рованных бифенилов в воде микроэкстракцией и ГХ6.D5739-00Стандартная практика идентификации разлива нефтей
ГХ-МС по положительным ионам с хемиионизацией7.D5830-95 (2001)СМ анализа растворителей в опасных выбросах473
Яшин Я.И., Яшин Е.Я., Яшин А.Я.Газовая хроматографияНомер ASTMНазвание ASTM8.D3695-95 (2001)СМО летучих спиртов в воде прямым вводом водных проб9.D6142-97СМ анализа фенола капиллярной газоюй хроматографи¬
ей10.D5317-98СМО хлорированных органических кислот в воде ГХ с
элекгронозахватным детектором11.D5316-98СМО 1,2-дибромметана и 1,2-дибромо-З-хлорпропана в
воде микроэкстракцией и ГХ12.D5790-95 (2001)СМ измерений выдуваемых органических соединений
в воде капиллярной ГХ-МС13.D5475-93 (2002)СМО азот- фосфорсодержащих пестицидов в воде ГХ с
азот-фосфорным детектором14.D5466-01СМО летучих органических соединений в атмосфере
(забор пробы в канистры)15.D6420-99СМО газообразных органических соединений ГХ-МС16.05241-92(1998)Стандартная практика микроэкстракции из воды для ана¬
лиза летучих и полулетучих органических соединений17.05075-01СМО ншютина и 3-этинилпиридина в юздухе помеще¬
ний18.03973-85 (1995)СМО низкомолекулярных галоидоорганических соеди¬
нений в воде19.05836-95СМО 2,4-диизоцианита толуола (2,4-TDI) и 2,6-диизо-
цианита толуола (2,6-Т01)20.06520-00Стандартная практика твердофазной микроэкстракции
и headspace для анализа летучих и полулетучих органи¬
ческих соединений в воде21.06209-98СМО полициклических ароматических соединений в
окружающем воздухе (сбор на фильтр с сорбентом и
анализ ГХ-МС)Таблица 17.12Публикации по разным применениям ГХ для контроля загрязнителейАнализируемые соединенияУсловия: метод,
детектор, методы
концентрированияСсылки1.Остатки пентахлорфенола Гканиероген"» в
текстиле, коже, и кожаных изделияхГХ-МС52.Летучие нитрозоамины в окружающей среде63.Определение метилртути в кровиГХ - ЛАС; АРП74.Определение метилолова в антарктических
водахГХ-ПФД; выдувание
и улавливание8474
ГЛАВА 17O&iacmu применения газовой хроматографии№№Анализируемые соединенияУсловия; метод,
детектор, ме1иды
концентрированияСсылки5.Определение следов люизита в воде и почве96.Контроль загрязнений окружающей средыГХ-АЭС. Обзор-59
ссылок107.Новые тенденции анализа и мониторинга за¬
грязнений окружающей средыОбзор - 260 ссылок118.Канцерогенные и мутагенные нитро ПАУ в
окружающей среде129.Доямердаванные бнф^ишш в Антарктике1310.Бромированные пламягасители в окружаю¬Обзор - 68 ссылок14щей среде11.Примеси нитрофенолов в антарктической
воде и снеге1512.Скрининг зарина в воздухе и волеГХ - МС; МТФЭ16Сокращения: ААС - атомно-адсорбционный спектрометрТаблица 17.13Перечень работ по анализу летучих органических примесей в
атмосфере городов и сел, в помещениях и офисах, в воздухе
производственных помещений методом ГХАнализируемые соединенияУсловия: метод, детектор,
способ концентрированияСсылкаАтмосферный ВОЗДУХ1.Определение полиароматических
соединений (ПАУ) в выхлопах ди¬
зельных двигателейГХ-МС172.Летучие органические соединения
(ЛОС) С^ - С, в атмосфере городов
и селГХ - термодесорбер183.ЛОС в атмосфереАвтоматический анализ ГХ -
адсорбционная ловушга - кри¬
офокусирование194.ЛОС в атмосфере г ИокогамаАвтоматическая ГХ система205.Мониторинг ЛОС в окружающем
воздухеПостоянный отбор проб216.Полихлорированные бифенилы в
окружающем воздухе22475
Яшин Я.И., Яшин Е.Я., Яшин А.Я.Газовая хроматография№№Анализируемые соединенияУсловия: метод, детектор,
способ концентрированияСсылка7.ЛОС в воздухеОбзор - 280 ссылок238.ИзоцианатыОбзор - 138 ссылок24Воздух жилых и пооизволственных помещений9.ЛОС - выделения из строительных
материаловМежлабораторное сравнение
в Европе2510.ЛОС микробиологического проис¬
хождения в помещенияхГХ-ГХ-ПИД26И.Анализ воздуха в чистых комнатах2712.Бутиловый спирт, акриловая кисло¬
та, бутиловый эфир акриловой кис¬
лоты в воздухе рабочей зоны2813.Оксид углерода в атмосфере2914.ЛОС, адсорбированные на части¬
цах пыли внутри помещенийГХ-УФ, ГХ-МС
Термическая десорбция30Примечание: ПИД - пламенно-ионизационный детектор; УФ - ультрафио¬
летовый детектор.Таблица 17.14Перечень работ по анализу примесей в водных средах№№Анализируемые соединенияУсловия: метод,
дете1сгор, способ
концентрирования
и пр.СсылкиВода1.Летучие амины в сточных водахГХ-ТИД, МТФЭ312.Органические примеси в водеЭкспрессная ГХ323.Определение 27 ЛОС в морской водеГХ, определение на
уровне ppt334.Примеси этанола, метилтретбутилового
эфира и других оксигенатовГХ - МС; МТФЭ345.Загрязнения в снеге городов и деревень в
России и Финляндии356.ЛОС в сточных водах нефтехимических
производствГХ - ИКС367.Обнаружение в питьевой воде продуктов де¬
зинфекцииГХ-МС; ЖХ-МС37476
ШАВЛ17Области применения газовой хроматографии№№Анализируемые соединенияУсловия: метод,
детектор, способ
концентрирования
и пр.Ссылки8.Летучие жирные кислоты в сточных водахГХ-АРП-МТФЭ389.Определение бензола, толуола, этилбензола,
ксилолов3910.Полихлорбифенилы40И.Полихлордибензодиоксины, полихлорди-
бензофураныГХ-МС4112.Хлорированные пестициды4213.Следы ПАУ в поверхностных водахГХ - МС; ТФЭ4314.ПАУ в сточных водах44Примечание; ТИД - термоионный детектор; МТФЭ - микротвердофазная
экстракция; ppt - одна триллионная часть (1-1(У‘^) или 1-10'"'%; АРП - анализ
равновесного пара; ТФЭ - твердофазная экстракция; ПАУ - полиароматические
углеводороды.Таблица 17.15Перечень работ по анализу загрязнителей в почве, осадках и твердыхотходах№№Анализируемые соединенияУсловия: меюд,
детектор, способ
концентрированияСсылкиДочщ, Осадки, Отходы1.Фенольные соединения в почве и осадкахГХ + ТФЭ452.Органические соединения в осадкахГХ-АРП-МТФЭ463.ПАУ, гетеро ПАУ (N, S, О) в креозотзагряз-
ненных почвах474.Нефтяные углеводороды в твердых отходахГХ485.Летучие моноароматические углеводороды
в почвеГХ - МТФЭ496.Полибромированные дифениловые эфиры в
осадках и биоте507.Полихлорированные нафталины в почвах
Англии518.Нефтяные углеводороды в почвах52477
Яшин Я.И., ЯштЖЯ., Яшин А.Я.Газовая хроматография№№Анализируемые соединенияУсловия: метод,
детектор, способ
концентрированияСсылки9,Изомеры полихлорированных дибензофура-
новГХ^МС5310.Бутилолово в осадкахГХ - АРП - МТФЭ5411. Примеси лекарств в водных средахОбзор - 58 ссылок55Примечание; ТФЭ - твердофазная экстракция; АРП - анализ равновесного
пара; МТФЭ - микротвердофазная экстракция.17.5 Контроль качества и безопасности пищевых продуктов и
напитков.Анализ пищевых продуктов - в настоящее время наиболее актуальная
аналитическая задача. Она важнее проблемы загрязнений окружающей сре¬
ды, так как по данным немещсих специалистов, более 70 % вредных загряз¬
нителей в организм человека попадают через пищу, 20 % с водой и 10 % с
вдыхаемым воздухом (Эйхлер В. Яды в нащей пище. Москва, Мир, 1993).
Несомненно, техногенные загрязнения окружающей среды через почву,
воду и воздух непосредственно попадают в пищевые продукты. Однако
пищевые продукты загрязняются природными вредными веществами, по¬
являющимися при неправильном хранении, при нарушениях технологий
пищевых обработок. В пищевые продо'кты вводятся многочисленные пи¬
щевые добавки, пища загрязняется через упаковку и т. д. В связи с этим
в последние годы, к сожалению, безопасность человека в наибольшей
степени определяется чистотой и доброкачественностью пищевых про¬
дуктов, алкогольных и безалкогольных напитков, так как многие вредные
загрязнители обладают канцерогенными, мутагенными, тератогенными и
иммуноугнетающими действиями. Кроме прямых отравлений недобро¬
качественными продуктами и напитками, иногда с летальным исходом
(более 40 тыс. человек в год), загрязненные пищевые продукты ухудшают
иммунитет, приводят к изменениям наследственности и непосредственно
становятся причиной болезней. Во многих случаях аллергические, онко¬
логические, сердечно-сосудистые и другие опасные заболевания являются
результатом нарушения биохимических реакций в организме, вызванных
главным образом некачественной пищей. Очень большое распространение
получила фальсификация пищевых продуктов. В последние годы во всем
мире широко используются продукты длительного хранения при темпе¬
ратуре окружающей среды в связи с распространением мелкорозничной
торговли без холодильного оборудования. Хроматографические методы
широко применяются для контроля пищевых продуктов. Можно выделить478
ГЛАВА 17 Области применения газовой хроматографииследующие основные цели хроматографических анализов: установление
пищевой ценности продуктов, в частности, определение белков (состава
аминокислот), жиров, сахаров, витаминов, микроэлементов; определе¬
ние доброкачественности, свежести пищевых продуктов, стадии порчи
продуктов; установление фальсификации пищевых продуктов, шнтроль
техногенных загрязнителей (пестицидов, ПАУ, диоксинов, полихлорби-
фенилов и др.); контроль природных загрязнителей (биогенных аминов,
миштоксинов: афлатоксинов, охратоксинов, зеараленона, фуманизинов,
патулина и других); определение пищевых искусственных добавок (анти¬
оксидантов, синтетических красителей, подслащивающих соединений,
ароматизаторов, горечей и др.); контроль ароматов пищевых продуктов;
определение трансгенных продуктов; шнтроль загрязнений от пищевых
упаковок; контроль специальной обработки пищевых продуктов, в част¬
ности, радиацией или нагреванием; анализ ветеринарных средств (анти¬
биотиков, сульфамидных препаратов, гормонов, анаболических стероидов,
цитостатина). По хроматографическим методам анализа пищевых продук¬
тов опубликованы десятки книг и сборников, сотни обзоров. Опубликова¬
ны как книги общего характера, так и специальные по анализам отдельных
классов соединений (Ящин Я.И., Яшин А.Я. Пищевая промышленность
2005, т.6, с. 14). Хроматографическими методами, в том числе и ГХ, в на¬
стоящее время анализируется большинство основных пищевых продо'ктов:
мясо и мясные изделия, рыба и морепродукты, растительные и животные
масла, молочные продукты, сыр, зерновые 1д'льтуры, хлеб, яйца, овощи,
фрукты, ягоды, джемы, соки, напитки, сахара, мед, орехи, вино, спирто¬
водочные изделия, пиво, чай, шфе, какао, приправы, специи и многие
другие. Сегодня хроматография - наиболее универсальный метод анализа
пищевых продуктов и напитков. Хроматографические методы позволяют
определять летучие и нелетучие компоненты пищи, основные ингредиен¬
ты и примеси. Благодаря универсальности, чувствительности, экспресс-
ности, доступности аппаратуры хроматографические методы применяют
для анализа исходного пищевого сырья, на всех стадиях технологичесшго
процесса, для контроля качества на выходе продукции.В современных условиях упашвочные матфиалы становятся в боль¬
шинстве случаев обязательным элементом в производстве пищи. Упашвка
необходима для защиты пищевых продуктов от микроорганизмов, химиче¬
ских и биологических изменений в процессе хранения. Упашвка продлевает
сохранность пищевых продуктов. В качестве упашвочных материалов в на¬
стоящее время применяются более 30 пластишв. Загрязнения от упашвки
появляются при их шнтакте с пищей за счет выделения мономеров и олиго¬
меров, таких как хлорвинил, изоцианат, стирол, капролакгам, полиэтиленте-479
Яшин Я.И.. Яшин Е.Я, Яшин А.Я. Гтошам .храчатографиирефталат и др.; технологических добавок в полимерных упаковках: пласти¬
фикаторов, термических стабилизаторов, антиоксидантов и др.; продуктов
разложения, термодеструкции полимеров и других факторов, влияющих на
миграцию вышеуказанных соединений в пищевые продукты - это диффузия
в полимерах, растворение при контактах полимер - пища. Некоторые из них
токчичные соединения: хлорвинил, фталаты (пластификаторы) - канцеро¬
генные соединения, изоцианат - сильное токсичное соединение. Загрязне¬
ния в пище от упаковки определяются ГХ-МС, ВЭЖХ, ГХ с устройством
равновесного пара. Полимерные упаковочные материалы влияют на аро¬
мат пищевых продуктов. Загрязнение пищевых продуктов винилхлоридом
выявляется в полихлорвиниловых бутылках, стиролом из полистирольных
контейнеров, ароматическими углеводородами из джутовых мешков. На¬
питки в пластиковых бутылках загрязняются за счет фотоокисления, осо¬
бенно при длительном хранении на солнце. В странах ЕС имеются дирек¬
тивные требования к упаковочным материалам для пищевых продуктов.Отдельная область применения ГХ -анализ состава аромата пище¬
вых продуктов. Обнаружены тысячи летучих компонентов, из которых
несколько десятков являются основными, определяющими характер за¬
паха, остальные могут придавать запаху и вкусу продукта его индивиду¬
альность. В последние годы возникло новое направление - энантиоселек-
тивный анализ компонентов пищи. По соотношению оптических изомеров
определенных аминокислот, оксикислот и некоторых иных соединений
можно однозначно определить, является ли данный продукт натуральным
или он содержит синтетические имитаторы и добавки. Энантиомерный ана¬
лиз показал, что микроволновая обработка пищевых продуктов, в отличие
от жесткой термической, не приводит к рацемизации аминокислот. Однако
все молочные продукты, подвергнутые процессам сбраживания, содержат
немало (нетоксичных) D-аланина и D-аспарагиновой кислоты - продуктов
жизнедеятельности молочнокислых бактерий. В природных жирах преоб¬
ладают цис-изомеры жирных кислот. Недавно обнаружено, что транс-изо-
меры повышают содержание липопротеинов низкой плотности и умень¬
шают концентрацию липопротеинов высокой плотности в крови, что
может способствовать развитию атеросклероза. Разработанная методика
газо-хроматографического разделения и анализа всех изомеров жирных
кислот заставила производителей в несколько раз уменьшить содержание
транс-изомеров ненасыщенных кислот в маргарине.С помощью ГХ бьио выявлено, что в некоторых сырах содержится мно¬
го нежелательных физиологически активных биогенных аминов и эти сорта
бьши запрещены. Из-за использования в Японии в пищевых продуктах L-
триптофана, полученного с помощью генной инженерии и биотехнологии.480
ГЛАВА 17Области применения газовой хроматографииТЫСЯЧИ людей получили неизвестное ранее заболевание и десятки умерли.
С помощью хроматографии выявлено, что эти трагические последствия вы¬
званы наличием токсичных загрязнений в триптофане (было обнаружено 60
примесей). Очень много публикаций имеется по газово-хроматографическо¬
му анализу вин, шньяков и другой спиртсодержащей продукции. В 1997 г.
в России вышел ГОСТ по определению микропримесей в водке и пищевом
этиловом спирте с помощью метода газовой хроматофафии.Таблица 17.16Анализ пищевых продуктов ГХ№№Анализируемые соединения или
продуктыУсловия: метод,
детектор, способ
концентрированияСсылки1.Жирные кислоты в растительных маслахКапиллярная ГХ562.Токоферолы и стеролы в растителы1ых маслахГХ ^ ТФЭ573.Определение подлинности растительных
маселГХ и ВЭЖХ; Обзор ^
152 ссылки584.Малые примеси в растительных маслахГХ595.Оливковое маслоГХ и ВЭЖХ; Обзор -
162 ссылки606.Фенольные соединения в олившвом маслеГХ-МС617.Стеролы в растительном маслеГХ и ВЭЖХ628.Масло какаоГХ и ВЭЖХ639.Стероидные липидыГХ и ВЭЖХ6410.Холестерин и а-токоферол в яйцахГХ и ВЭЖХ6511.Мониторинг щюдукгов разложения липидовГХ-МТФЭ6612.Углеводороды в растительных маслахГХ и ВЭЖХ; Обзор ^
103 ссылки6713.Анализ липидовГХ-ИКС6814.Окись холестерина в пищеГХ и ВЭЖХ6915.Профиль андрогенов, прогестогенов и гпюю-
кортикоидов в мясе крупного рогатого скотаГХ^МС7016.Моно- и олигосахариды в медеГХ7117.Летучие амины в рыбеГХ7218.Хлорорганические пестициды в томатахгх-эзд7319.Высокомолекулярные органические соеди¬
нения в дыме мясаГХ7448131 Газовая xp(»mron^iu
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я.Гтовая хроматография№№Анализируемые соединения или
продуктыУсловия: метод,
детектор, способ
концентрированияСсылки20.Пестициды в медеГХ7521.ПАУ в жирах и маслахГХ; Обзор-51
ссылка7622.Ртутьорганические соединения в пищеГХ7723.Загрязнения пищевых продуичв от упако¬
вочных материаловГХ7824.Пестициды и их метаболиты в пищеГХ + ЭЗД + ТИД7925.Гетероциклические ароматические амины в
пищеГХ и ВЭЖХ8026.Транс-резвератрол в винеГХ-МС + ТМФЭ +
дериватизация8127.Диэтилстилбистрол, тестостерон, 17р-эстра-
диол в мясе и мясных продуктахГХ и ВЭЖХ8228.Анаболические стероиды в пище и БАДахГХ и ВЭЖХ8329.Экстракты цитрусовыхГХ-МС84Таблица 17.17Анализ алкогольных напитковАнализируемые соединенияУсловия: метод,
детектор,способ
концентрированияСсылкиАлкогольные напитки1.Диметилсульфил в пивеГХ - АРП852.Альдегиды в алкогольных напиткахГХ863.Анализ водки и белого ромаГХ-МС-МТФЭ874.Диамины, полиамины и ароматические
амины в портвейне и виноградном сокеГХ-МС885.Энантиомеры аминокислот в пиве896.Хараетеристика вискиГХ-МС-МТФЭ907.Транс-резвератрол в винахГХ-МС-МТФЭ918.4-этилфенол и 4-этилгваякол в красных
винахГХ929.Сернистые соединения в пивеГХ-ПФД-АРП-МТФЭ9310.Основные компоненты ромаГХ9411.Идентификация бренди и коньяковГХ-МС95482
тЛВА 17 Области применения газовой хроматографииТаблица 17.18
Анализ ароматов и запахов пищевых продуктов№№Анализируемые соединенияУсловия: метод, детектор,
способ концентрированияСсылкиАроматы, запахи и летучие соединения пишевых продуктов и напитковНовая концепция определения лету¬
чих соединений в пище и напитках962.Летучие соединения в пивеКГХ-ПИД; АРП973.Аромат коммерческих винГХ-АРП-МТФЭ984.Летучие соединения сыраГХ - МС; динамический
АРП995.Достижения в исследовании запаха1006.Аромат винГХ-МС; АРП + МТФЭ1017.Летучие соединения в пищевых про¬
ектах Разные методы концентри-
рования 102Пробоподготовка для анализа арома¬
тов1039.Летучие компоненты аромата моло¬
дого токаяГХ-МС10410.Летучие компоненты картофеляГХ - АРП10511.Анализ ароматов пищиГХ - олфактометрия10612.Анализ запахов и ароматовГХ - МСВП10713.Анализ летучих компонентов пищиГХ-АРП10814.Дифференциация кофе по анализу
летучих соединений ГХ-МТФЭ10915.Профиль аромата (букета) высокока¬
чественных испанских винГХ11017.6 Применение ГХ в медицинеУспех лечения наиболее опасных болезней в значительной степени за¬
висит от их ранней диагностики. В последние десятилетия в медицине для
клинических анализов внедряются современные методы, среди них одно
из наиболее важных считается хроматография. Раннюю диагностику про¬
водят на основании определения специальных биохимических маркеров
или метаболитов, появляющихся в биологических жидкостях человека при
конкретных заболеваниях.Анализ биохимических маркеров используется:- для скрининга населения и выявления опасных заболеваний при про¬
филактических осмотрах;483зг
шин Я.И., Яшин Е.Я., Яшин А.Я. Газовая хроматография- ДЛЯ подтверждения заболеваний;- для мониторинга эффективности терапии;- для предсказания прогноза лечения;Во многих случаях достаточно определение только маркеров для диа-
ностики заболеваний, однако в некоторых случаях необходимо опреде-
ять метаболический профиль уровня многих компонентов.По зарубежным данным методом хроматографии можно диагностиро-
ать сотни заболеваний среди них наиболее опасные социально значимые
олезни, как сердечно сосудистые, онкологические, диабет и другие.Современные хроматографические методы позволяют определять
иохимические маркеры на уровне 10 ’ - 10 '^ г (в зависимости от методов
онцентрирования и применяемых детекторов), что и позволяет проводить
иагностику заболеваний на самых ранних стадиях, при которых в боль-
1инстве случаев возможно полное излечение.Маркерами чаще всего являются биологически активные сравнитель-о низкомолекулярные соединения; нуклеозиды, нуклеотиды, катехолами-
:ы, углеводороды, производные аминокислот (в частности, гомоцистеин и
р.), стероиды, сахара, индолы, органические кислоты, витамины и др.Для определения биохимических маркеров чаще всего применяют
(етоды газовой и высокоэффективной жидкостной хроматографии. Это
вязано с тем, что хроматографические методы имеют преимущества по
равнению с другими аналитическими методами, среди которых, можно
казать следующие;- высокая информативность метода за счет разделения и анализа де-
ятков ~ сотен соединений за один цикл;- как уже указывалось выше низкие пределы обнаружения;- экспрессность анализа (в основном время анализа от 1 до 15 мин.);- хорошая воспроизводимость и правильность анализа (СКО в преде-
ах 1-5% в зависимости от концентрации определяемых маркеров);- надежное аппаратурное оформление;- автоматизация процесса анализа с выдачей полного протокола, со-
ранение данных анализа в архиве ПЭВМ;- универсальность применения, один и тот же хроматограф можно ис-
юльзовать для многих задач;- за счет низкого предела обнаружения необходимы небольшие коли-
[ества биологических проб для анализа (несколько микролитров);- использование в основном неинвазивных методов отбора проб (моча,
люна, слезы, пот, волосы, выдыхаемый воздух и др.).Естественно, наибольший интерес представляют маркеры наиболее
шасных заболеваний, в частности онкологических.484
ГЛАВА 17Области применения газовой хроматографииВ таблице 17.19 приведены примеры разных применений ГХ в меди¬
цине. В последние годы большой интерес вызывает выдыхаемый воздух
человека для ранней диагностики заболеваний (таблица 17.20).Таблица 17.19
Применение газовой хроматографии в медицине№№Анализируемые соединенияУсловия: метод,
детектор,
пробоподготовкаСсылки1.Желчные кислоты и стеролыГХ1112.Стероиды, жирные кислоты, органические
кислоты и катехоламины для диагноза мета¬
болических нарушенийГХ; микроволновая
ускоренная дерива¬
тизация1123.Фитостеролы, станолы и метаболиты холе¬
стерина в сыворотке человекаГХ1134.Высшие жирные кислоты в плазме крови
человекаГХ1145.Липиды в биологических пробахГХ - ИКС1156.Измерение содержания жирных кислот в
сыворотке и состав жирных кислот у паци¬
ентов с гиперлипидемиейГХ1167.Простагландины в биологических жидко¬
стяхГХ1178.Определение Р,-изопростана как индикатора
оксидантного стрессаГХ-МС1189.Липиды крови и кожного жира у пациентов
с болезнью ACNEГХ11910.Клинический анализ стероидовГХ-МС12011.Стероиды в моче женщин с раком грудиГХ-МС12112.Быстрый диагноз фенилкетонурии по ана¬
лизу аминокислот в кровиГХ-МС12213.Ашхантоин в сыворотке человекаГХ-МС12314.Значение измерения рибозы крови у пациен¬
тов с коронарной болезньюГХ с дериватизацией12415.Липиды крови (плазма, сыворотка) у паци¬
ентов с псориазомГХ12516.Жирные кислоты, липопротеины, роль ли-
попротеинов низкой плотностиГХ; ВЭЖХ126485
Яшин Я.И., Яшин Е.Я.. Яшин А.Я.Гтовая хроматография№№Анализируемые соединенияУсловия: метод,
детеет-ор,
пробоподготовкаСсылки17.Цистеин и индольиые производные маркеры
злокачественной меланомыГХ; ВЭЖХ12718.Мониторинг оксидантного стресса по про¬
дуктам разложения ДНКГХ; ВЭЖХ12819.Анализ стеровдальных липидовГХ; ВЭЖХ: обзор -
275 с.12920.Нехолестернновые стеролы плазмыГХ; ВЭЖХ: обзор-
137 с.13021.Природные кортиюстероиды в моче спор¬
тсменовГХ; ВЭЖХ13122.Природные стероидыГХ; ВЭЖХ13223.Анализ стероидов для диагностикиГХ; ВЭЖХ: обзор -
74 с.13324.Анализ аминов в вагинальной жидюсти при
бактериальном вагнносисеГХ-МС13425.Определение в плазме и моче в-изо-РОР^а-
биомаркера липидного перокислениягх-мс-мс13526.Холин и ацетилхолинГХ13627.Мелатонин и его предшественникиГХ13728.Гомоцистеин, цистеин и метионин в плазме
человекаГХ-ЭЗД13829.Определение сахаров в моче для определения
кишечной проницаемостиГХ с дериватизацией13930.Нитриты в крови и желудочном сокеГХ14031.Алкилфосфорхолины в SK-BR-3 при раке
грудигх-мс14132.Сульфид в крови как маркер ферментатив¬
ных процессов в кишечникеГХ-МС14233.Определение низкомолекулярных силико¬
нов в плазме крови у женщин после имплан¬
тации в грудь силиконаГХ-МС14334.Биохимические маркеры хронического ал¬
коголизмаГХ144486
ГЛАВА 17Области применент газовой хроматографии№№Анализируемые соединенияУсловия: метод,
детектор,
пробоподготовкаСсылки35.Определение ”N0 для диагностики и тера¬
пии заболеваний человекаГХ14536.Вариации летучих органических соедине¬
ний в вьщыхаемом воздухе здоровых людейГХ-МС14637.Сахара и спирты в моче во время беремен¬
ностиГХ-МС14738.Изопрен в выдыхаемом воздухе человекаГХ-МС-МТФЭ14839.Экдистерол в мочеГХ14940.а-токоферол и а-токоферолхинон в плазме
человекаГХ; ВЭЖХ15041.Этиловые эфиры жирных кислот в воло¬
сах - возможный маркер алкоголизмаГХ-МС-МТФЭ-АРП15142.Новый прибор для исследования плохого
дыханияГХ-ИСПМС15243.Летучие сернистые соединения в выдыхае¬
мом воздухеГХ-МС15344.Определение анестетиков и их метаболитовГХ-МС15445.Сравнение стероидного профиля у трех¬
дневных новорожденныхГХ; ВЭЖХ15546.Скрининг ошибок метаболизма у новорож¬
денныхГХ-МС15647.Скрининг уреазы у новорожденныхГХ-МС157Таблица 17.20Анализ выдыхаемого воздуха человека№№Анализируемые соединения/задачиСсылка1.Проект ранней диагностики рака легких1582.Летучие маркфы рака груди в выдыхаемом воздухе159,1603.Диагностика рака легких по летучим маркерам в выдыхаемом воз¬
духе1614.Тестирование выдыхаемого воздуха: потенциальный неинвазив¬
ный метод диагностики рака и других болезней1625.Биомаркеры оксидантного стресса при диабете в выдыхаемом воз¬
духе. Твердофазная экстракция для анализа выдыхаемого воздуха1636.Маркеры оксидантного стресса в выдыхаемом воздухе1647.Оксиданты и антиоксиданты при раке груди1658.Свобоные радикалы: их история и их роль в старении и болезнях166487
Яшин Я.И., Яшин Е.Я., Яишн А Я. Гззовая хроматографт№№Анализируемые соединения/задачиСсылка9.Углеводороды в выдыхаемом воздухе в исследовании липидного
пероксиления: принципы и практика 16710.Потенциальная роль углеводородов в вьщыхаемом воздухе для из-
мерения липидного перокисления 168И.Влияние возраста на спектр метилированных алканов как новых
маркеров оксидантного стресса 16912.Пентан в выдыхаемом воздух как маркер ревматоидного стресса17013.Высокая концентрация пентана во время инфаркта миокарда17114.Повышенное содержание пентана и дисульфвда угаерода в выды-
хаемом воздухе у пациентов с шизофренией 17215.Патологии по выдыхаемому воздуху17316. Твердофазная экстракция для анализа выдыхаемого воздуха17417.7 Применение ГХ в судебной медицине и в криминалистике.Аналитические задачи в судебной экспертизе весьма разнообразны: от
точных количественных анализов до простого сравнения или установле¬
ния идентичности разных проб или предметов. Аналитический контроль
важен при расследовании таких преступлений, как употребление нарко¬
тиков и спиртных напитков, неумышленных и умышленных отравлени¬
ях, при злоупотреблениях ле1арствами, при убийствах, пожарах, кражах,
взрывах, авариях и проч.Объектами хроматофафического анализа чаще всего являются по по¬
следним публикациям наркотики (морфин и его производные, кокаин, кан-
набиноиды, ЛСД и др.), амфетамины, барбитураты, различные лекарства,
и яды, этанол, метанол, ацетон, изопронанол, толуол, хлороформ, дихлорэ¬
тан, этилацетат и др. растворители.В судебных экспертизах методом гаювой хроматофафии анализируют
также нефтепродукты и горюче-смазочные магериалы, а также вещества,
применяемые в качестве горючих средств при поджогах; выявляют поддел¬
ки и фальсификации горюче-смазочных материалов. Анализируют также ла¬
кокрасочные материалы и покрытия, в том числе частицы окраски автомо¬
билей, краски в чернилах для выявления особенностей письма или давности
документов, древесину, взрывчатые вещества, продукты выстрела и др.Сотни работ опубликованы по хроматофафическим анализам биоло¬
гических объектов для судебной экспертизы, в частности крови, сыворот¬
ки, мочи, слюны, пота, выдыхаемого воздуха, волос человека и др. Только
за последние два года опубликованы сотни статей по применению газовой
хроматофафии в судебной и судебно-медицинской экспертизе. Для бы-
сфого скрининга разработаны систематические токсикологические ана¬
лизы лекарств и их метаболитов при злоупофеблениях.488
ГЛАВА 17 Области применения газовой хрсматографииГазохроматографические индексы удерживания токсикологически
значимых веществ на насадочных и капиллярных колонках с полиметил-
силиконовыми неподвижными фазами приведен в специальных обзорах,
созданы банки масс-спектрометрических и газо-хроматографических дан¬
ных для лекарств, ядов, пестицидов, загрязнителей и их метаболитов.Из других достижений представляют интерес следующие: газо-хрома-
тографический анализ продуктов взрыва, определение лекарств в крови,
слюне и поте, в волосах при злоупотреблениях, определение амфетаминов
в крови и моче, бензодиазепинов в биологических жидкостях. Роль газовой
хроматографии в различных областях судебной химии хорошо отражена в
периодических обзорах.В таблицах 17.21 и 17.22 приведены обзоры типичных применений ГХ
в судебной медицине и криминалистике.Таблица 17.21
Некоторые применения ГХ в судебной медицине.№Анализируемое соединениеУсловия: метод,
детектор, условия
концентрирования
и прочее.Ссылка1Системный токсикологический анализ на
основе ГХ и ЖХ.Обзор - 123 ссылки(с)1752Системный токсикологический анализ кислых
лекарств и их метаболитов для клинической и
судебной медицины и допинг-контроля.Обзор - 132с1763Системный токсикологический анализ ле¬
карств и ядов в биопробах.Комбинация хро¬
матографических и
спектрометрических
методов1774Лекарства в ногтях.ГХ-МС1785Лекарства в волосах как при терапевтических
приемах, так и при злоупотреблениях.ГХ-МС1796Анаболические соединения в волосах.ГХ-МС1807Хроматографические и электрофоретические
методы для анализа чернил.Обзор - 76с1818Автомобильные краски (определение проис¬
хождения).ГХ-МС182489
Яишн Я.И., Яшт Е.Я.. Яшт А.Я.Гтомя хроматография№Анализируемое соединениеУсловия: метод,
детектор, условия
концентрирования
и прочее.Ссылка9Определение лекарств при злоупотреблениях.Общий обзор18310ГХ-МС в клинической и судебно-медицинской
токсикологии с целью контроля.ГХ-МС18411Допинг-анализ.18512Эфедрин в волосах при употреблении с целью
допинга.18613Скрининг лекарств при злоупотреблении.ГХ-ИКС18714Взрывчатые нитросоединения в воде.Микроэкстракция188Таблица 17.22Применение ГХ в судебной медицине (анализ наркотиков и
наркотических средств).№Анализируемое соединениеУсловия: метод,
детектор, условия
концентрирования
и прочее.Ссылка1Сравнительный анализ героина и кокаина
после конфискации.Обзор-116 ссылок (с)1892Кокаин, кодеин и их метаболиты в поту че¬
ловека.ГХ-МС. Обзор 54с1903Героин и морфин в биологических материалах.1914Кодеин, морфин, 6-ацетилморфин, гидроко¬
дон, гидроморфон, оксикодон и оксиморфон
в моче.ГХ-МС1925Опиаты и кокаин в волосах.ГХ-МС триметил-
силильных произво¬
дных ГХ-МС.1936Опиаты в слюне и моче.ГХ-МС1947Морфин и кодеин в сыворотке и слюне.ГХ-МС1958Амфетамины, кокаин и опиаты в волосах.ГХ-МС1969Каннабиноиды в пробах слюны.ГХ - МТФЭ19710Каннабиноиды в биологических жидкостях.198490
ГЛАВА 17Области применения газовой хроматографии№Анализируемое соединениеУсловия; метод,
детектор, условия
концентрирования
и прочее.Ссылка11Каннабиноиды в биологаческих жидкостях
(кровь, слюна, пот, кал).Обзор - 50с19912«Экстази» в волосах.20013«Экстази» в биологических жидкостях.Обзор - 29с20114Профиль «Экстази» и амфетаминов.ГХ - МТФЭ20215Лизергиновая кислота (LSD) и ее метаболи¬
ты в физиологических жидкостях.ГХ-МС20316Лизергиновая кислота и диэтиламид ЛК в
моче.ГХ-МС20417Метадон и его метаболиты в плазме.ГХ-МС - МТФЭ20518 Метадон в слюне.ГХ-МС20617.8. Применение ГХ в фармацевтике и фармакологии.В таблице 17.23 приведены примеры основных применений ГХ в фар¬
мацевтике и фармакологии.В фармацевтике, в основном, определяется чистота лекарств по фар¬
макопейным статьям. В новых фармакопеях США, Великобритании, стран
ЕС, Японии, Украины имеются как общая статья о газовой хроматографии,
так и многие конкретные фармакопейные статьи по определению приме¬
сей в лекарствах. Особенно часто газовая хроматография применяется для
контроля остаточных количеств растворителя в лекарствах методом анали¬
за равновесного пара (head-space).К 2008 г. в Российской Федерации зарегистрировано более 17000 ле¬
карств и лекарственных средств (см. Государственный реестр лекарствен¬
ных средств, т. 1 и т. 2), из них наиболее востребованы 1000 - 1500. Многие
малолетучие лекарства и лекарственные средства анализируются методом
высокоэффективной жидкостной и тонкослойной хроматографии.В фармакологии анализируют лекарства в биологических жидкостях
для изучения их воздействия на организм человека. Эти работы прово¬
дятся на основе «Руководства по экспериментальному (доклиническому)
изучению новых фармакологических веществ». Отдельный раздел в Ру¬
ководстве посвящен фитопрепаратам, обладающим свойствами антиокси¬
дантов и хелаторов.В таблице 17.24 приведены последние статьи и обзоры по примене¬
нию ГХ в фармацевтике и фармакологии.491
ЯшинЯ.И., Яшт Е.Я., Яшин А.Я.Газовая хроматографияТаблица 17.23Основные применения методов газовой хроматографии в
фармацевтике и фармакологиип/пОсновные применения1.Фармакопейный контроль качества лекарств2.Мониторинг примесей в лекарствах3.Исследования стабильности лекарств4.Анализ экстрактов лекарственных трав5.Анализ лекарств из растений традиционной Китайской медицины6.ГХ-МС в разработке новых лекарств7.Определение энантиомеров лекарств8.Технологический контроль в фармацевтической промышленности9.Определение природных фенольных соединений в растительных материалах
(флавоноидов, оксикислот, природных красителей и др.)10.Анализ лекарств при допинг-контроле11.Клиническая фармация12.Фармакокинетика13.Анализ лекарств при злоупотреблениях (токсикологический анализ)14.Исследования взаимодействия лекарств в организмеТаблица 17.24Некоторые применения ГХ в фармацевтике и фармакологии№№Анализируемые соединенияУсловия: метод,
детектор,
пробоподготовкаСсылки1.Экспертная система фармацевтических ана¬
лизовГХ2072.Лекарства и токсичные веществаГХ-МС2083.Лекарства в биологических жидкостяхГХ - МС; автомати¬
ческая пробоподго¬
товка2094.Лекарства в биологических пробахГХ - МТФЭ2105.Остаточные количества растворителей в ле¬
карствахГХ-МС АРП2116.Летучие примеси в водорастворимых лекар¬
ствахГХ-АРП2127.Полный анализ лекарств и токсикантов в
биологических пробахГХ-МС-МТФЭ213492
ГЛАВА 17Области применения газовой хроматографии№№Анализируемые соединенияУсловия: метод,
детектор,
пробоподготовкаСсылкиМикроэкстракция лекарствГХ - МТФЕ; Обзор
131 ссылка2149.Мелатонин и его производныеГХ21510.Одновременное определение мелатонина и
пиридоксина в таблетках ГХ-МС21611.Антиканцерогенные соединения в биологи¬
ческих жидкостяхГХ и ВЭЖХ; Обзор
99 ссылок21712.Антинеопластические соединенияГХ; Обзор-167
ссылок21813.Лекарства традиционной китайской меди¬
цины, показывающие неопластическую ак¬
тивностьГХ21914.Циклофосфамиды и близкие антиканцеро¬
генные лекарстваГХ22015.Трициклические антивирусные лекарстваГХ22116.Антивирусные фосфорсодержащие лекарстваГХ22217.Бензодиазепины (15) в моче человекаГХ-МС-ТФЭ22318.Растительные сапониныГХ22419.Лидокаин в плазмеГХ - МТФЭ22520.Скрининг бензодиазепинов в кровиГХ22617.9 Газовая хроматография для космических исследованийПортативные газовые хроматографы для космических исследований
(анализ атмосферы и грунта планет, анализ воздуха космических кораблей)
начали разрабатываться с 1962 г. Опубликованы обзоры по этим вопросам
[227-229]. Специальные требования к газовым хроматографам для косми¬
ческих исследований подробно описаны в работах [227-230]. Эти требо¬
вания определяют конструкции автоматических систем пробоподготовки
(доведение пробы до давления в колонке, сжатие или разряжение и др.) и
введения проб, применение капиллярных или микронасадочных колонок с
небольшими расходами газа-носителя и с очень стабильными сорбентами,
применение чаще всего программирования температуры, автоматическое
переключение разных колонок и др.В течение 1962-1970 гг. было разработано несколько проектов газовых
хроматографов для космических исследований. В табл. 17.25 приведены
основные технические характеристики этих приборов.493
Яшин MM. Яшиным. Яшин А.Я.Газовая хроматографияТаблица 17.25.
Проекты портативных газовых хроматографов для
космических исследований.НазначениеДетекторыКолонкиТемпера¬турныйрежимГа>носи¬тельВес,кгМощ¬ность,ВтГодразработкиАнализ газов, воды,
углеводородов и орга¬
нических кислот(лун-
ный проект) Три разряд¬
ных детек¬
тораТрирал.сад.лонкиИзотер¬мический,105°СНе5.6721962-1963Анализ продуктов
пиролиза почвы на
МарсеМасс-спек¬трометрМНКИзотерми¬ческийНе7(ГX-^MC)0,1(ГХ)101966Анализ газового соста-
ва атмосферы МарсаМикроката¬рометрДве МНКИзотерми¬ческийНе1.21966Анализ атмосферы
шсмических кораблей
(постоянные газы, вода,
угаеводороды и ;ф.)Детектор
поперечно¬
го сечения
ионизацииТри па-
рал. ко¬
лонкиИзотер¬мический,65°СНе1966Анализ газов и лету¬
чих органических со¬
единений в атмосфере
МарсаМикроката¬
рометрДве ко¬
лонки с
порапа-
ком Q и RПрограм¬
мирован¬
ный, 25-
150°СНе1968Анализ атмосферы
Марса (миниатюрный
газовый хроматограф)МСМНКИзотер¬
мический,
23°С, про-
граммирН,0,07-0,15
(без элек¬
тронного
блока)2.5-51970Сокращения: МНК - микронасадочная колонка
Начиная с 1982 г. в СССР и США стали запускаться ракеты к Венере и
Марсу с комплексом научной аппаратуры, включая и портативные газовые
хроматофафы. Впервые газовые хроматофафы и газовые хроматофафы-
масс-спекфомефы проанализировали атмосферы этих планет [232-235].
В СССР эти исследования проводились по профаммам Венера 11-14 и
Вега, а в США по профаммам «Viking» и «The Pioneer-Venus».В Табл. 17.26 приведены основные технические характеристики газо¬
вых хроматофафов для этих исследований. Кроме вышеперечисленных
требований, приборы обладали высокой виброустойчивостью и ударо¬
прочностью, требовалось также, чтобы характеристики прибора не изме¬
нялись в течение 10 лет (срок полета до некоторых планет).Миниатюрный газовый хроматофаф по профамме «Viking» имел раз¬
меры 9x9x6 см, бьш соединен с дозирующей системой и с источником
газа-носителя (Не). В приборе было две колонки и катаромеф. Дозирующее
усфойство представляло собой пять миниатюрных соленоидных кранов и
позюляло дозировать пробы объемом 0,1 мл. Одинаковые колонки размером494
ГЛАВА 17Области применения газовой хроматографии7.6 м X 1.1 мм, одна из которых фавнительная, выполнены из нержавеющей
стали и заполнены Порапаком Q (размер фраищи 100-120 меш). Разделение
проходило при 24°С, скорости газа-носителя 13.5 мл/мин. Смесь HLj, N^, О^,
СН^ и COj разделялась полностью, а Аг и СО разделялись частично.Эти приборы впервые проанализировали состав атмосферы на плане¬
тах. Чувствительность определения достигала 0.1 нмоль. Миниатюрный
автоматический газовый хроматограф работал хорошо и полностью вы¬
полнил программу.Таблица 17.26.Технические характеристики портативных газовых хроматографов
для космических кораблей.НазначениеПрограммаполетаДетекторыКолонкиТермо¬статГазноси-тельДозаторАнализ аимосферн
и грунта Марса«Viking» 1975-
1976 гг. (США)ДТП, мс7.6 X !.! мм с Порапа¬
ком Q, 2 м X 0,75 мм с
тенаксом24°СНе, 13.5
мл/мйнКраныАнализ атмосферы
Венеры«Венера 11-14»
ГХ «Сигма»
1978-1982 гг.Неоновыйионизацион¬ныйТри юлонки: с По-
лисорбом, цеолитом и
оксидом магния 2 м;
2.5 м70°СNeКраныАгализ атмосферы
Венеры и кометы
Галлея«Вега» 1984-
1985 гг. ГХ
«Сигма-3»Гелиевый
ионизаци¬
онный ДТП,
Два ДЭЗТри колонки с Пора-
паками QS, N и Е70°СNe, И,КраныАнализ атмосферы
Венеры«Pioneer-Venus»
1978 г.ДТП15.85 м X 1.1 мм с По-
рапаюм N; 2.1 м х .,1
мм с poly-DVB18°СНе, 35 мл/
минКраныАнализ атмосферы
Титана {спутник
Сатурна)«Cassini-
Huygens» 1997-
2(Ю4ггМСТри парал. колонки;
рога PLOTQ, PLOT
А1,0^, цеолит Про-грам-мир.Газовый хроматограф-масс-спектрометр (также программа «Vi¬
king»), кроме анализа атмосферы, должен был определить наличие органи¬
ческих примесей в почве Марса после ее пиролиза [236], те. определить,
есть ли жизнь на этой планете.Весь комплекс включал дозирующее устройство, колонки, систему
обеспечения газом-носителем и масс-спектрометр.При заборе газа для анализа предварительно удалялись СО, СО^ и Н^О
до масс-спектрометра для улучшения предела детектирования атмосфер¬
ных постоянных газов. СО и СО^ поглощались гидрооксидом лития и окси¬
дом серебра, воду поглощали перхлоратом магния. Проба почвы подаерга-
лась измельчению и просеиванию до размеров частиц <300 мкм, вводилась
в печь пиролизера и нахревалась в пределах 50-500°С.Колонка размером 2 м х 0.75 мм из нержавеющей стали была на¬
полнена 2,6-дифенил-и-фениленоксидом (Тепах GC), покрытым поли-и-495
Яшт Я.И.. Яшт Е.Я., Яшт А.Я. Газовая хроматографияфеноксиленом (Ро1у-МРЕ). В начале анализа температура колонки была
50°С в течение 10 мин, затем повышалась со скоростью 8 град/мин до
200°С и сохранялась около 1 ч. Скорость газа-носителя 2 мл/мин. В масс-
спектрометре ионизация проводилась методом электронного удара, диапа¬
зон определения масс 12-215, газ-носитель водород перед входом в ион¬
ную ловушку поглощался палладиевым сепаратором.В результате этих исследований был определен состав атмосферы
Марса (COj - 95.3%, -2.7%, «Аг - 1.6%, О, 0.13%, СО - 0.08-0.27%,
HjO - 0-0.3%). На уровне млн ‘(1 х 10^% об.) установлено присутствие
Ne - 2.5, ’‘Аг - 0.5, Кг - 0.3, Хе - 0.08, и О, - 0.003) (таблица 17.27).Программа «Венера И, 12». Аналитический прибор «Сигма» состоял
из газового хроматографа и масс-спектрометра [234, 235]. Газовый хрома¬
тограф включал три колонки и три детектора. Первая шлонка длиной 2 м
была заполнена Полисорбом-1 для разделения H^S, COS, SO^, Н^О и СО^.
Вторая колонка длиной 2.5 м была заполнена молекулярными ситами и
предназначалась для разделения постоянных и благородных газов (Н^, О^,
Nj, со. Не, Аг, Кг и СН,). Аргон определялся на третьей колонке с оксидом
марганца. Температура колонок была 70°С. Неон использовался как газ-
носитель, детектор - неоновый ионизационный с р-радиоактивным источ¬
ником. Предел детектирования составлял менее 1 млн ' при объеме пробы
1 мл. Газовые хроматографы работали в атмосфере Венеры 4 ч, выполнено
8 заборов проб; последняя проба была отобрана на поверхности, несмотря
на очень жесткие условия (давление 90 атм и температура 730°К).В программа «Венера 13» и «Венера 14» [235] были использованы
приборы «Сигма-2». Основные технические характеристики этих газовых
хроматографов не отличались от описанных выше.Программа «Вега». По программе «Вега» определяли состав атмос¬
феры Венеры в июне 1985 г. и газовый состав кометы Галлея в марте
1986 г. [237, 238]. В состав газового хроматографа «Сигма-3» входили
три дозирующие системы, три колонки и три детектора, соединенные
параллельно. Колонки были наполнены Порапаком QS, Порапаком N и
Порапаком Т для разделения и анализа газов и паров серной кислоты.
Первая колонка соединена с гелиевым ионизационным детектором и с ка¬
тарометром (газ-носитель - гелий). Вторая и третья колонки соединены с
электронозахватным детектором; газом-носителем служил особо чистый
азот. Пределы детектирования были от 0.1-1 млн ‘ (100 млн ‘для воды).Анализ атмосферы Венеры (программа «The Pioneer-Venus»,
США). Газовый хроматограф включал дозирующий кран, два детектора
и две колонки [239]. Газовый хроматограф близок к газовому хромато-
фафу программы «Viking» с некоторыми улучшениями. Объем пробы496
ГЛАВА 17Области применения газовой хроматографии0.35 мл, использовался катарометр на термисторах. Колонки размером
15.85 м X 1.1 мм были наполнены Порапаком N и могли разделять смеси
Не, Hj, Nj, Oj, Ar, СО, СН^, Кг при изотермических условиях (18°С) и
при скорости потока 35 мл/мин. Вторая колонка 2.1 м х 1.1 мм с пори¬
стым полимером из полидивинилбензола (poly-DVB) с размером фрак¬
ции 180-220 меш служила для разделения СО^, NHj, Н^О, H^S, СО,, С^Н^,
SOj, НС1 и HF. Этот сорбент был специально разработан для разделения
этой смеси [240]. Температура этой колонки 60°С, скорость газа-носи-
теля 40 мл/мин. Предел детектирования 1-60 млн'. Колонка работала
30 мин; за это время проведено три анализа. Прибор работал хорошо и
обеспечил анализы на высотах 51.6, 41.7 и 21.6 км над поверхностью
Венеры [241].Измерены концентрации COj, Nj, Н^О, Аг, СО, Ne и SOj. На верхнем
пределе обнаружения определены Н^, СН^, Кг, Nj, NOj, С^Н^, С^Н^, H^S,
COS, С3Н,.В результате газохроматографических измерений по программам «Ве¬
нера 11-14» и «Pioneer-Venus» определен следующий состав атмосферы
Венеры: COj- 96%, Nj ~ 4%. На уровне млн ' определены: Н^О - 1 - 1000,
СО - 20 - 2000, SOj - 0/1 - 200, COS - 0/35 - 40, Н^ - 25, Аг - 70, Ne - 7,
НС1 - 0.4, HF - 5x10-5 (.^абл. 17.27).В дальнейшем планируется использование миниатюрных газовых
хроматографов для исследования газового состава атмосферы Титана, наи¬
большего спутника Сатурна [242, 243], комет, в повторной экспедиции на
Марс и в других экспедициях.В июле 2008 г была обнаружена вода на Марсе нвой системой США.Таблица 17.27Состав атмосферы Марса и Венеры, определенный
хроматографическими и масс-спектрометрическими
методами (СССР и США).Атмосфера МарсаАтмосфера ВенерыКомпонентыМол. долиКомпонентыМол. долиОсновные юмпоненты (%):Основные компоненты (%)СО,95,3СО,96N,2,7N23,4-«Аг1,6Оа0,13СО0,08-0,27нр0,0349732 Газовая хроматография
Яишн ЯМ.. Яшин ЕЖ. Яишн А.Я.Газовая хроШвфтАтмосфера МарсаАтмосфера ВенерыКомпонентыМол. доляКомпонентыМол. долиПримесные номпоиентыПримесные компоненты(ppm)(ppm)Ne2,5н,о1-1000“Аг0,5СО20-2TOКг0,3Ne2,5Хе0,08SO,0,1-2000,0,003-0,03H,S1-80COS0,35-40н,25Аг70Ne7HCl0,4HF510-’S,41fr»498
ГЛАВА 17 ЛитератураЛитература1. Яшин Я.И., Яшин А.Я. Ж.анлит. химия 2001, т.56, с.231-245.2. Даванков В.А., Яшин Я.И. Вестаик Российской академии наук. 2003,
т.73, №7, с.637-646.3. Яшин Я.И, Яшин А.Я. Химическая техника, 2003, №6, с.30.4. Яшин Я.И., Яшин АЛ Нефтяная промышленность, 2004, т.8, №3, с.50.5. Мои J. et al. Sepu, 1999, v 17, р 386.6. Protois J. С., Ошу В. Arm. Falsif. Expert. Chim. Toxicol. 1999, v 92, p 17.7. Brunetto M. R. et al. Analyst (Cambridge), 1999, v 124, p 1493.8. Liu J. et ol. Fenxi Shizanshi. 2001, v 20, p 76.9. Станьков И. Н. Сергеева А. А., Тарасов С. Н. Ж. анал. химии. 2000,
т 55, с 66.10. Bogheri Н. Saraji М., Brinkman LF. А. Т. Tech. Instrum. Anal. Chem.2000, V 21, p 221.11. Namiesnik J. Crit. Rev. Anal. Chem. 2000, v 21, p 211.12. lamiceli A. L. et al. Food Sci Technol (N. Y.) 2000, V 102, P 673.13. Fuoco R., Ceccarini A. Environ Contam. Antarct. 2001, p 237 - 273.14. Kuosmanen K. et al. J. Chromat. 2002, v 943, p 113.15. Vanni A. et al. Int J. Environ Anal. Chem. 2001, v 79, p 349.16. Schneider J. F, Boparoi A. S., Reed L. L. J. Chromat. Sci 2001, v 39, p 420.17. Fleurat - Lassard P., Pointet K., Renou - Gounord M.-F. J. Chem. Educ
1999, V 76, p 962.18. Fernandez - Martinez G et al. Anal. Lett 1999, v 32, p 22851.19. Wang }. - L., Chen S. - N., Chew G. J. Chromat 1999, v 863, p 183.20. Yamamoto N. et al. Atmos. Environ, 2000, v 34, p 4441.21. Uchiyama S., Hosegawa S. Environ. Sci. Technol. 2000, v 34, p 4656.22. Garcia - Alonso S. et al. Anal. Chim. Acto. 2001, v 440, p 223.23. Aragon P., Atienza J., Climent M. D Chrit. Rev. Anal. Chem. 2000,V 30, p 121.24. Гугля E. B. Ж. анал. Химии, 2000, v 55, p 508.25. De Bortoli M.et al. Indoor Air, 1999, v 9, p 103.26. Elike K. et al. J. Environ. Monit. 1999, v 1, p 455.27. Shiramizu V. Jpn Kokai Tokkyo Koho Jp 11218526 A2 lOAug 1999
Appl. Jp 1998 23146 4 Feb. 1998 4pp.28. Зайцева H. B. и др. Гигиена и санитария. 1999, №2, с 74.29. Gros V., Bosnany В., Esteve R. S. Chemosphere: Global Changa Sci.1999, v l,p 153.30. Lagesson H. V, Nilsstm A., Т^жот С. Chromatographia 2000, v 52, p 621.31. Abalos М., Bayona J. U., VentxuB F. Anal. Chem. 1999, v 71, p 3531.499
Яшин Я.И., Яшин Е.Я., Яшин А.Я. Гтовая хроматографим32. Current R. W., Borgerding А. J. Anal, Chem. 1999, v 71, p 3513.33. Huybrechts Т., Moerman O., Everaert М., Meded. - Fac. Land boumkd
Toegepaste Biol Wet, 1999,v 64, p 133.34. Cassada D. A. et al. Anal. Chem. 2000, v 72, p 4654.35. Poliakova O. V., Lebedev A. Т., Haimien O. Toxicol. Environ Chem.2000, V 75, p 181.36. Zhang R., Sun G. Fenxi Huaxue 2000, v 28, p 915.37. Richardson S. D., J. Environ. Monit, 2002, v 4, p 1.38. Abalos M. Bayona J. М., Powliszyn J. J. Chromat. 2000, v 873, p 107.39. Valor L et al. Food Sci. Technol. (N. Y.) 2000, v 102, p 721.40. Fuoco R., Ceccarini A. Food Sci Technol (N. Y.) 2000, v 102, p 655.41. lamiceH A. L. et al Food Sci Technol. (N. Y.) 2000, v 102, p 673.42. Mangani E, Maione М., Palma P. Food Sci Technol. 2000, v 102, p 577.43. Crozier P. W., Plomley J. B., Matchuk L. Analyst (Cambridge) 2001,V 126, p 1974.44. Peraz S. et al. J. Chromat. 2001, v 938, p 57.45. Helaleh M. L H. et al. Anal. Sci. 2001, vl7, p 1225.46. Dermietzel J., Strenge G. Fres. J. Anal. Chem 1999, v 364, p 645.47. Meyer S., Cartellieri S., Steinhart M. Anal. Chem. 2000, v 71, p 4023.48. Donan R., Marten S., Wronski B. Fres. J. Anal. Chem. 2СХЮ, v 367, p 220.49. Zgymunt B., Namiesnik J., Anal. Chem. 2001, v 370, p 1091.50. De Boer J. et al. TrAC 2001, v 20, p 591.51. Meijer S. H. et. al. Environ. Sci. Technol. 2001, v 35, p 4205.52. Cam D., Cagni S. J. Chromat Sci, 2001, v 39, p 481.53. Pehlivan M, Pehlivan E, Turk. J. Chem. 2001, v 25, p 459.54. Liu J. et al. J. Sep Sci. 2001, v 24, p 459.55. Terkes T. A. TrAC 2001, v 20, p 419.56. Aitzetmuller K. GIT Lab. J. 1999, v. 3, n. 2.57. Lechner М., Reiter B., Lorbeer E. J. Chromat. 1999, v. 857, p. 231.58. Aparicio R., Aparicio-Ruiz R. J. Chromat. 2000, v. 881, p. 93.59. Cert A., Moreda W., Perra-Camino M. C. J. Chromat. 2000, v. 881, p. 131.60. Lercker G., Rodrignez-Estrada M. T. J. Chromat. 20СЮ, v. 881, p. 105-129.61. Tasiola-Margani М., Okogeri O. J. Food Sci. 2001, v. 66, p. 530.62. Adibi S. L. J. Chromat 2001, v. 935, p. 173-201.63. Barcarolo R., Akklam E. J. Assoc, off Anal. Chem. Int 2001, v. 84, p. 1485.64. Volin P. J. Chromat. 2001, v. 935, p. 125-140.65. Jin М., Wang L., Fu Z. Sepu 2001, v. 19, p. 478.66. Doleschall F. et. al. J. Microcolumn Separ 2001, v. 13, p. 215.67. Moreda W. et. al. J. Chromat. 2001, v. 936, p. 159.68. Mossoba M. M. et. al. Eur. J. Lipid. Sci. Technol. 2001, v. 103, p. 826.500
ГЛАВА 17 Литература69. Dutta P. С. et. al. Spec. Publ. - R. Soc. Chem. 1999, v. 240, p. 309.70. Fritsche S., Scmidt G., Steinhert H. Z. Lebensim.-Uaters Forsch A.
1999, V. 209, p. 393.71. Barez J. A. G. et. al. Chromatographia, 1999, v. 95, p. 403.72. Oetjen K., Karl H. Dtsch. hebensm.-Rundsch 199, v. 95, p. 403.73. Morelli-Cardoso M. H. W. et. al. JHRC 1999, v. 22, p. 619/74. Nolte C. G. et. al. Environ. Sci. Technol. 1999, v. 33, p. 3313.75. Menkissoglu-Spiroudi U. et. al. J. Assoc off. Anal. Chem. Int. 2000,V 83, p. 178.76. Moret S., Conte L. S. J. Chromat. 2000, v. 882, p. 245.77. Carro A. М., Mejuto M. C. J. Chronat. 2000, v. 882, p. 283-307.78. Lan O.-W., Wong S.-K. J. Chromat. 2000, v. 882,p. 255-270.79. Ahmed F. E. TrAC 2001, v. 20, p. 649.80. Pais P., Knize M. G. J. Chromat. B. 2000, v. 747, p. 139.81. Luan Т., Li G., Zhang Z. Anal. Chim. Acta. 2000, v. 424, p. 1982. Баркатина E. H. и др. Ж. аналит химии 2001, т. 56, с. 740.83. De Cock К. J. S. et. al. J. Pharm. Biomed Anal. 2001, v. 25, p. 843.84. Ruperez F. J. et. al. J. Chromat. 2001, v. 935, p. 45-69.85. Wu Z. V. Anal. hett. 1999, v 32, p. 2691.86. Lau M. N., Ebeler J. D., Ebeler S. E. Am. J. Ekil. Vitic 1999. v. 50, p. 324.87. Ng L.-K. In: Pawliszyn J. (Editor) Application Solid Phase Microextrac¬
tion. Royal Society of Chemistry. Cambridge 1999, p. 393-406.88. Fernandes J. O., Ferreara M. A. J. Chromat. 2000, v. 886, p. 183.89. Erbe T. Bruckner H. J. Chromat. 2000, v. 881, p. 81.90. Fitzgerald G., James K. J. J. Chromat. 2000. v. 896, p. 351.91. Luan Т., Li G., Zhang Z. Anal. Chim. Acta 2000, v. 424, p. 19.92. Pollnitz A. P. et. al. J. Chromat. 2000, v. 874, p. 101.93. Hill R G., Smith R. M. J. Chromat. 2000, v. 872, p. 203.94. Martinez A., Ribas М., Porto O., Rev. Deriv. Cana. Aziucar 1999, v. 33,
p. 1-12.95. Власов В. Н., Маручешюв Д. С. Analusis, 1999, v. 27, p. 663.96. Azodanlou R. et al. Z. Ltbensm.-Unters. Forech. A 1999, v. 208, p. 254.97. Miao Y. Zhongguo Niangzoo 2000, v. 4, p. 26.98. Vas G. et al. Acta Aliment. 1999, v. 28, p. 133.99. Valero E. et al. Chromatographia 2000, v. 52, p. 340.100. Grosch W. Lebensmittelchemie 1993, v. 47, p. 129.101. Francioli S. et al. Am. J. Enol. Vitic. 1999, v. 50, p. 404.102. Sides A., Robards K., Helliwell S. Tr. Ac. 2000, v. 19, p. 322.103. Wilkes J.G. et al. J. Chromat. 2000, v. 880, p. 3.104. Miklosy E. et al. Chromatographia 2000, v. 51, p. 305.501
Яшин Я.И.. Яшин Е.Я., Яишн А.Я. ГшsaяJmmmsmФш105. Arkes J.M., Duckham S.C., Ваккег J. Adv. Exp. Med. Biol. 2001,
V. 488, p. 133.106. Blank I. Food Sci. Technol. (N.Y.) 2002, v. 115, p. 297.107. Holland J.F., Gardner B.D. Food Sci. Technol. (N.Y.) 2002, v. 115,
p. 107-138.108. Wampler T.R Food Sci. Technol. (N.Y.) 2002, v. 115, p. 25.109. Costa Freitaz A.M. et al. Chromatographia 2001, v. 54, p. 647.110. Ferreira V. et al. J. Agric. Food Chem. 2001, v. 818, p. 4824.111. Batta A.K. et al. i. Lipid Res 1999, v. 40, p. 1148.112. Agatha G., Kauf E. Clin. Lab. (Heidelberg) 1999, v. 45, p. 387.113. PhilUps K.M., Ru^o D.M., Bailey J.A. J. Chroimt В 19^, v. 732, p. 17.114. Лобачев А.Л., Лобачева И.В., Ревинская E.B. и Колосюва С.В.
Клин. лаб. Диагностика, 2001, № 7. с. 13.115. Mossoba М.М. et al. Eur. J. Lipid Sci. Technol. 2001, v. 103, p. 826.116. Ueda K. et al. Rinsko Kagaka 2002, v. 31, p. 58.117. Baranowski R., Pacha K. Med. Chem. 2002, v. 2, p. 135.118. Morrow J.D., Roberts L.J. Methods Mol. Biol. (Totowa) 2002, v. 186,
p. 57.119. Коляденко В.Г., Брузгина T.C., Усенш Г.Д. Лабораг. диагностика2001, №4, с. 13.120. Wudy S.A., Homoki J., TeUer W.M. Chromat Sci. Ser. 2001, v. 86, p. 309.121. Poor V. etal. Chromatograhpia 2002, v. 56, p. S 145.122. Deng C. et al. J. Chromat. В 2002, v. 775, p. 115.123. Pavitt D.V. et al. Clin. Chem. Acta 2002, v. 318, p. 63.124. Строев Ю.А., Бухов B.A., Оюроков В.Г., Чуриюв Г.И. Клин. лаб.
диагностика 2001, № 5, с. 23.125. Андрашио Ю.В., Коляденко В.Г. и jq). Мед. хим. 2001, т. 3, № 2, с. 49.126. Blaha V. et al. Microchimica Acta 2001, v. 136, p. 23.127. Hortleb J, Arndt R. J. Chromat. В 2001, v. 764, p. 409.128. Dizdaroglu M. Nato Sci. Ser. A 2000, v. 313, p. 76.129. Volm R J. Chromat. 2001 v. 495, p. 12.130. Kuksis A. J.Chromat 2001, v. 935, p. 203.131. Rivero-Morabe J.J, et al. J. Chromat. В 2001, v. 761, p. 77.132. Shimade K., Mitamura K., Higashi T. J. Chromat 2001, v. 935, p. 142.133. Nozaki O. J. Chromat. 2001, v. 935, p. 267.134. Wolrach H., Forsum V. et al. J. Clin. Microbiol. 2001, v. 39, p. 4026.135. Richelle M. et al. Eur. J. Mass Spectrom. 2001, v. 7, p. 427.136. Tsai T.-H. J. Chromat. В 2000, v. 747, p. 111.137. Horumi Т., Matsushima S. J. Chromat. В 2000, v. 747, p. 95.138. Myung S.W. et al. Anal. Sci. Technol. 1999, v. 12, p. 408.502
ГЛАВА 17 Литература139. Ghaly S.M., Al-Sebai А.А. Egypt J. Biomed. Sci. 1998, v. 1, p. 139.140. Яблочкин В.Д., Симонова H.C., Кирнел В.В. Судеб.-мед. экспер¬
тиза 1999, т. 42, с. 26.141. Brohez V. etal. Lipids 1999, v. 34, p. 511.142. Hyspler R. et al. J. Chromat. В 2002, v. 770, p. 255.143. Flossbeck D. et al. Anal. Chem. 2001, v. 73, p. 606.144. Jeszenszky E. et al. Z. Zagadnich Nauk Sadowych 2000, v. 43, p. 118.145. Plath C. et al. Ger DE 19719098 (cl. AG1K49/00) 12 May 1999,
Appl. 19.719098 6 May 1997.146. Phillips M. et al. J. Chromat. В 1999, v. 729, p. 75.147. Tetsuo М., Zhang C. et al. J. Chromat. В 1999, v. 73, p. 111.148. Hyspler R. et al. J. Chromat. В 2(КЮ, v. 739, p. 183.149. Tsitsimpikon C. et al. Rapid Commun. Mass Spectr 2СЮ1, v.l5,
p. 1796.150. Mottier P. et al. Anal. Biochem. 2002, v. 301, p. 128.151. Pragst F. et al. Forensic Sci. Int. 2(Ю1, v. 121, p. 76.152. Rodriguez-Femandez L. et al. Anal. At. Spectrom. 2001, v. 16, p. 1051.153. Ochiai N. et al. J. Chromat. В 2001, v. 762, p. 67.154. Desaga М., Guitton J. J. Chromatog. Sci. Ser. 2001, v. 86, p. 267.155. Веккет L.C., B^ MJ., Hinler R J. Microcolumn Sq>. 2001, v. 13, p. 280.156. Suss J.O., Sewell A.C. Chromat. Sci. Ser. 2001, v. 86, p. 341.157. Kuhera T. et al. J. Chromat. В 1999, v. 731, p. 141.158. Henschke C.L, McCauley D.I., Yankelevitz D.F., et al. Lancet 1999,
V. 354, p. 99-105.159. Phillips M„ Cataneo PW., DitkoflFB.A. Breast J. 2СЮЗ, v. 9 (4), p. 334.160. Phillips М., Cataneo P.W., Ditkoff B.A. et al. Breast J. 2003, v. 9, №3,
p. 184-191.161. Phillips М., Cataneo P.W., Cummin A.R.C. et al. Chest. 2003, v. 123,
№6, p. 2116-2123.162. Fireman J. Am. Clin. Lab. 1998, v. 17, №5, p. 12-13.163. Phillips М., Cataneo P.W., Cheema Т., Greenberg J. Clin. Chim. Acta.
2004, V. 344, №1-2, p. 189-194.164. Phillips М., Cataneo P.W., Greenberg J. et al. Heart. Dis. 2003, v. 5 (2),
p. 95-99.165. Ambrosone C.B. Antioxid Redox Signal 2000, v. 2, p. 903-917.166. Knight J.A. Ann. Clm. Lab. Sci. 1998, v. 28, p. 331-346.167. Kneepkens C.M., Ferreira C., Lepage G., Roy C.C. Clin. Invest. Med.
1992, v. 15, p. 163-186.168. Kneepkens C.M., Lepage G., Roy C.C. Free Radic. Biol. Med. 1994,
V. 17, p. 127-160.169. Phillips М., Cataneo P.W., Greenberg J., Gunawardena R., Naidu A.,
Rahbari-Oskoui F. J. Lab. Clin. Med. 2000, v. 136, p. 243-249.503
Яшин Я.И; Яишн Е.Я.. Яшин АЯ. Гаювая хроматография170. Humad S., Zarling Е., Clapper М., Skosey J.L. Free Radic. Res. 1988,
v.5, p. 101-106.171. Weitz Z.W., Bimbaum A.J., Sobotka P.A., Zarling E.J., Skosey J.L.
Lancet 1991, v 337, p. 933-935.172. Pliillips М., Sabas М., Greenberg J. J. Clin. Pathol. 1993, v. 46, p. 861-864.173. Rosen P.P. Philadelphia: Lippincott-Raven, 1997.174. Grote C., Pawliszyn J. Anal. Chem. 1997, v. 69 (4), p. 587-596.175. Drummer O.N. J. Chromat. B. 1999, v. 733, p. 27.176. Meatherall R. J. Anal. Toxicol. 1999, v. 23, p. 177.177. Polletini A. J. Chromat. B. 1999, v. 733, p. 47.178. Lomos N.P., Anderson R.A., Robertson J.R. J. Anal. Toxicol. 1999,
V. 23, p. 147.179. Nakahora Y. J. Chromat. B. 1999, v. 733, p. 161.180. Cirimele V. et al. J. Chromat. B. 2000, v. 740, p. 265.181.Zlotnik J.A., Smith F.P. J. Chromat. B. 1999, v. 733, p. 265.182. Kochanowski B.K., Morgan J. Chromat. Sci. 2000, v. 38, p. 100.183. Brodbelt J.S. et al. Chromatog. Sci. Set. 2001, v. 86, p. 369.184. Maurer H.H. Chromatog. Sci. Ser. 2001, v. 86, p. 355.185. Schaenzer W. GIT Lab-Fach. 2001, v. 45, p. 1096.186.Dumestre-Toulet Y, Kintz P. J. Anal. Toxicol. 2000, v. 24, p. 381.187. Proisler M. et al. Talanta 2000, v. 53, p. 177.188.Psillanis E., Kologerakis N. J. Chromat. 2001, v. 909, p. 211.189.Chiarotti М., Fucci N. J. Chromat. B. 1999, v 733, p. 127.190. Huestis M.A. et al. J. Chromat. B. 1999, v. 733, p. 247.191. Karawya M.S., Selim M.A., Zayhloul S.S. Egypt. J. Pharm. Sci. 1998,
v. 38, p. 339.192. Meatherall R. J. Anal. Toxicol. 1999, v. 23, p. 177.193. Pichoni S. et al. J. Anal. Toxicol. 1999, v. 33, p. 348.194. Specki I.M. et al. J. Toxicol. Clin. Toxicol. 1999, v. 37, p. 441.195. Janowska E. Chem. Anal. (Warsaw) 2001, v. 46, p. 857.196. Skender L. et al. Forensic Sci. Int. 2002, v. 125, p. 120.197. Fucci N. etal. Forensic Sci. Int. 2001, v. 119, p. 318.198. Teske J. et al. J. Chromat. В 2002, v. 772, p. 229.199. Stoub C. J. Chromat. В 1999, v 733, p. 119.200. Cooper G.A.A. J. Forensic Sci. 2000, v. 45, p. 400.201. Kintz R, Samyn N. J. Chromat. В 1999, v 733, p. 137.202. Kongshang K.E. et al. Chromatographia 1999, v. 50, p. 247.203. Reuschel S.A., Eades D., Foltz R.L. J. Chromat. В 1999, v. 733, p. 145.204. Sklerov J.H., Kalasinsky K.S., Ehoen C.A. J. Anal. Toxicol. 1999, v.23, p. 474.504
ГЛАВА 17 Литература205. Bermejo A.M. et al. J. Anal. Toxicol. 2000, v. 24, p. 66.206. Dos Santos Lucax A.C. et al. J. Anal. Toxicol. 2000, v. 24, p. 93.207. Xiao Y. Fenxi Kexue Xuebao, 2001, v. 17, p. 279.208. Ishii A., Kurihara R., Katsumata Y. Rinsho Kenza 2000, v. 44, p. 1507.209. Valeariel M. et al. Chrom. Sci. Ser. 2001, v. 86, p. 247.210. Snow N.H. J. Chromat. 2000, v. 885, p. 445.211. Urakami K. et al. Chem. Pharm. Bull. 2000, v. 48, p. 1894.212. Hong L., Altorfer H. Chromatographia 2001, v. 13, p. 76.213. Li L. et al. Yaoxue Xuebao 2000, v. 35, p. 521.214. Lord H., Pawliszyn J. J. Chromat. 2000, v. 902, p. 17.215. Harumi Т., Matsushima S. J. Chromat. B. 2000, v. 747, p. 95.216. Nunez-Vergara L.G. J. Pharm. Biomed. Anal. 2001, v. 26, p. 929.217. Oliveira E.J., Watson D.G. J. Chromat. B. 2001, v. 764, p. 3.218. Paci A. et al. J. Chromat. B. 2001, v. 764, p. 255.219. Tsoi T.-H. J. Chromat. B. 2000, v 747, p. 27.220. Bauman £., Preiss R. J. Chromat. B. 2001, v. 764, p. 173.221. Suckow R.F. J. Chromat. B. 2001, v. 764, p. 313.222. Ba B.B., Saux M.-C. J. Chromat. B. 2001, v. 764, p. 349.223. Borrey D. et al. J. Chromat. B. 2001, v. 765, p. 187.224. Oleszek W.A. J. Chromat. B. 2002, v. 967, p. 147.225. Koster E.N.M. et al. J. Chromat. B. 2000, v. 739, p. 175.226. Rasaken I. et al. J. Anal. Toxicol. 2000, v. 24, p. 46.227. Raulin F., de Vanssay E., Do L. Gas Ghromatography in Space, LC-
GC Intl. 1992, V. 5, № 7, p. 22-31.228. Akapo S. O., Dimadjia J.-M. D., Kojiro D. R., Valentin J. R., Carle G. C.,
Gas Ghromatography in Space, J. Chromatogr. 1999, v. 843, p. 147-162.229. Brazhnikov V. V, Mukhin L. M. Chromatogr. Rev. 1971, v. 15, p. 151.230. Schmidt D. Proc. Symp. on exploration of Moon and Space. Munchen
Apr 22. 1966.231. Wilthie W. F. Gas Chromatogr 1966., v. 4, p. 47.232. Brown F. S., Adelson H. E., Chapman M. C. et. ol. Rew. Sci Instrum.
1978, v. 49, p. 139.233. Oyama V. I., Berdhal B. J. J. Geophys. Res 1977, v. 82, p. 4669.234. Gehnan B. G., Zolotukhin V. G., Muchin L. M. et. al. Spase Res. 1980,
v.20, p. 219.235:Мухин Jl. М., Гельман Б. Г., Ламонов Н. И. и др. Космические ис¬
следования 1983, V. 21, р. 225.236.Rushneck D. R., Diaz А. V., Nowazth D. W. et. al. Res. Sci. Instrum.
1978, V 49, p. 817.237. Venus-Holley mission, Balebanov V. М., Skuridin C. A. Vorontzova E.
V, Bassolo V. S. International Scientific and Technical Committee (MNTK)
Report 1985, p. 181.505
Яишн ЯМ. Ятт ЕЖ. ЯшинАЯ. Гтотя зттштгтФым238. Mukhin L. М., Nenarokov D. R, Porschnev N. V. et. ol. Adv. Spase
Res. 1987, V. 7, n. 12, p. 329.239. Oyama V. I., Carle G. C., Woeller F. et. al. ШЕ Trans. Geoshi. Remote
Sensing, 1980, GE-18, p. 85.240. Woeller F. H„ Pollock G. E. J. Chromat. Sci. 1978, v. 16, p. 137.241. Oyama V. I., Carle G. C., Woeller F. et. al. J. Geophys. Res. 1980, v. 85,
p. 7891.242. Announcement of opportunity Cassini-Huygens Probe, European
Space Agency (ESA) Sci. 1989, v. 2, p. 89.243. Niemann H. B. Gas Chromatograph-Mass Spectrometer (proposal to
BSACassini+mission-Haygens probe, 1990).506
СодержаниеСОДЕРЖАНИЕПредисловие 4Глава 1 Введение 51.1 Определение хроматографии 51.2 Классификация методов хроматографии 61.3 История открытия хроматографии 101.4 Основные достижения хроматографии 171.5 Наукометрия газовой хроматографии 191.6 Преимущества газовой хроматографии 201.7 Высказывания о хроматографии 23Глава 2. Основы теории газовой хроматографии 282.1. Параметры удерживания 282.1.1. Время удерживания 282.1.2. Объем удерживания 292.1.3. Относительные параметры удерживания 302.1.4. Параметры хроматографического пика 312.2. Основные процессы в хроматографической колонке 322.2.1. Теория теоретических тарелок 342.2.2. Основные виды размывания 352.3. Понятие об эффективности, селективностии степени разделения 472.3.1. Основные критерии оценки степени разделения 472.3.2. Связь критериев разделения с параметрами опыта 492.4. Влияние температуры на хроматографическое разделение 522.4.1. Зависимость удерживаемых обьемов от температуры 522.4.2. Хроматографическое разделение с программированием
температуры колонки во времени 552.4.3. Хроматографическое разделение с программированием
температуры по длине слоя (хроматермография).. 592.5. Влияние параметров на хроматографическое разделение 612.5.1. Основные группы параметров 612.5.2. Выбор оптимального режима разделения взависимости от характера аналитической задачи 622.5.3. Влияние основных параметров на степень разделения 63Глава 3. Газо-жидкостная хроматография 773.1. Преимущества и недостатки газо-жидкостной
хроматографии 773.2. Основные требования к жидким неподвижным фазам 77507
Яшин Я.И.. Яшин Е.Я.. Яшин А.Я. Гтовая хроматография3.3. Классификация жидких фаз 793.4. Межмолекулярные взаимодействия, определяющие
удерживание соединений в газо-жидкостной хроматографии 883.5. Селективность жидких фаз в ГЖХ 903.6. Стандартизация жидких фаз 953.7. Бинарные жидкие фазы 963.8. Полярность жидких фаз по Роршнайдеру и
МакРейнольдсу. 983.9. Новые жидкие фазы для газовой хроматографиии новые технологии их производства 1073.10. Химичес1ая прививка (иммобилизация) жидких фазк внутренней поверхности и к поверхности твердых носителей 1113.11. Твердые носители для жидких фаз ИЗ3.12. Способы нанесения жидких фаз на твердые носители 1223.13. Заполнение и тренировка колонки 123Глава 4 Газо-адсорбционная хроматография 1284.1 Преимущества и недостатки газо-адсорбционной
хроматографии 1284.2 Влияние химии поверхности на удерживаемые обьемы
и селективность разделения. Неспецифические и
специфические межмолекулярные взаимодействия 1304.3 Классификация молекул и адсорбентов по характеру
межмолекулярных взаимодействий 1304.4 Влияние геометрической структуры адсорбентовна удерживание объема 1364.5 Геометрическое модифицирование адсорбентовна примере силикагеля 1384.6 Структура пор адсорбента и удерживаниеразличных веществ 1394.7 Структура пор адсорбента и размывание полос 1424.8 Изменение химической природы поверхностиадсорбентов (модифицирование адсорбентов) 1474.9 Основные типы адсорбентов и их применения.Силикагели. Пористые стекла. Пористые полимеры.Оксид алюминия. Цеолиты (молекулярные сита).Углеродные адсорбенты. Углеродные нанотрубки, фуллерены 150Глава 5 Газовая адсорбционно-адсорбционная хроматография 2465.1 Зависимость удерживаемого объема от количестважидкой фазы в адсорбционно-адсорбционной хроматографии 246508
Содержание5.2 Макропористые кремнеземы в качествеадсорбентов-носителей 2485.2.1. Макропористые кремнеземы с пленками жидких фаз 2485.2.2. Макропористые силикагели с полимерными пленками 2605.2.3. Макропористые силикагели с пленкамижидких кристаллов 2625.3 Пористые полимеры как носители жидких фаз 2655.4 Углеродные адсорбенты в качестве носителейжидких фаз 2695.5. Силикагели с привитыми алкильными группами 2745.6. Определение коэффициентов распределения методом
адсорбционно-абсорбционной хроматографии 275Глава 6 Капиллярная хроматография. 2806.1 Ведение. Основные этапы развитиякапиллярной хроматографии 2806.2 Размывание в капиллярных колонках 2856.3 Размер пробы для капиллярных колонок 2876.4 Особенности определения числа теоретическихтарелок для капиллярных колонок 2876.5 Материал капиллярных колонок 2886.6 Приготовление капиллярных колонок 2896.7 Влияние толщины пленки жидкой фазы 2906.8 Колонки PLOT. 2916.9 Поликапиллярные колонки 2956.10 Колонки PoraPLOT. 2966.11 Циркуляционная капиллярная хроматография 3026.12 Монолитные капиллярные колонки 3026.13 Основные области применения капиллярнойгазовой хроматографии 303Глава 7 Методы реакционной газовой хроматографии.. 3087.1. Введение 3087.2. Аналитическая реакционная хроматография 3087.3. Пиролизная газовая хроматография 3097.4. Химическая дериватизация анализируемых проб 3167.5. Реакционная газовая экстракция в анализеобъектов окружающей среды 320Глава 8 Двумерная хроматография 325Глава 9. Высокоскоростная газовая хроматография 3309.1. Отличие высокоскоростной хроматографииот «обычной» хроматографии 330509
Яшт ЯМ, Яшин ЕЛ. Яшин А.Я. Гтотя хроматография9.2. Преимущества высокоскоростной хроматофафиии возможные области ее применения 3319.3. История развития методов высокоскоростнойгазовой хроматофафии 3319.4. Основные способы сокращения времени анализа 332Глава 10 Высокотемпературная газовая хроматография 33710.1. Краткий исторический обзор 33710.2. Основные области применения высокотемпературной
газовой хроматофафии 33810.3. Высокотемпературная газо-адсорбвдюнная
хроматофафия паров металлов и солей 338Глава 11 Хиральная газовая хроматография,разделение энантиомеров 34211.1. Краткая история 34211.2. Молекулярная структура и хиральность 34211.3. Природа энантиомерной селективности 34311.4. Области применения газовой хиральной хроматофафии 344Глава 12. Аппаратура для газовой хроматографии 34612.1. Введение 34612.2. Функциональная схема газового хроматофафа 34612.3. Источники сжатых газов 34812.3.1. Генераторы чистого водорода 34912.3.2. Генераторы чистого азота 35012.3.3. Генераторы особо чистого воздуха 35112.4. Блок подготовки газа 35112.5. Дозирующие устройства 35312.6. Колонки для газовых хроматофафов 35612.7. Термостаты 35712.8. Детекторы для газовой хроматофафии 35812.8.1. Пламенно-ионизационный детектор (ПИД) 36112.8.2. Детектор по теплопроводности (ДТП) 37012.8.3. Элекфонозахватный детектор (ЭЗД) 37412.8.4. Термоионный детектор (ТИД) 37512.8.5. Пламеннофотомефический детектор (ПФД) 37612.8.6. Фотоионизационный детектор (ФИД) 37712.8.7. Масспекфомефический детектор (МСД) 38012.8.8 Другие детекторы 38012.8.9. Комбинации детекторов 38112.9. Дополнительные устройства для газовой хроматофафии 381510
Се^ержание12.10. Краткая история развития газохроматографического
приборостроения 38412.11. Основные типы газовых хроматографов 38512.11.1. Лабораторные газовые хроматографы 38712.11.2. Портативные газовые хроматографы 39112.11.3. Промышленные автоматические газовые хроматографы....395
Глава 13 Комбинированные методы................. ... .......40113.1. Основные комбинированные методы ГХ 40113.2 ГХ-МС 40113.3. Газовая хроматография - ИКС Фурьеспектросшпия (ГХ-ИКС) 40513.4. Газовая хроматография - атомно-эмиссионное
детектирование 407Глава 14. Качественный анализ в газовой хроматографин...............41014.1. Введение 41014.2. Методы идентификации, основанные наизмерении параметров удерживания 41014.3. Методы идентификации с использованием
селективных химических реакций 14.4. Методы идентификации с применениемспециальных детектирующих устройств 41214.5. Методы идентификации с использованиемкачественных химических реакций на выходе колонки 41414.6. Методы идентификации другимифизико-химическими методами 41514.7. Методы идентификации, основанные наудалении некоторых компонентов смеси 416Глава 15. Методы количественного анализа ............ .41815.1. Введение 41815.2. Метод абсолютной калибровки 41815.3. Метод внутреннего стандарта 41915.4. Метод внутренней нормализации 41915.5. Источники ошибок в газохроматографическихизмерениях 42015.6. Определение сходимости показаний 421Глава 16. Методы концентрирования в газовой хроматографии. 42516.1. Введение 42516.2. Концентрирование из воздуха и газовых сред 427511
Яшт Я.И.. Яшин Е.Я.. Яшин АЛ Гтовая хттатогпафш16.3. Твердофазная экстракция (ТФЭ) загрязнителей из воды 43416.4. Микротвердофазная экстракция 43616.5. Ион-парная твердофазная экстракция 44216.6. Иммуноаффинная ТФЭ 44216.7. ТФЭ на полимерах с порами молекулярныхразмеров (molecular imprinted polymers) 44316.8. Жидкостно-жидкостная экстракция (ЖЖЭ) 44316.9. Жидкостная микроволновая экстракция 44616.10. Сверхкритическая флюидная экстракция (СФЭ) 44716.11. Мембранная экстракция 44816.12. Газовая экстракция (метод анализаравновесного пара, head-space) 44916.13. Динамическая газовая экстракция - методвыдувания и улавливания 452Глава 17. Области применения газовой хроматографии 45917.1. Введение 45917.2. Технологический анализ 46017.3. Анализ природного газа, газовых конденсатови нефтепродуктов 46417.4. Применение ГХ для контроля загрязненийокружающей среды 46917.5. Контроль качества и безопасности пищевых продуктов 47817.6. Применение газовой хроматографии в медицине 48317.7. Применение газовой хроматографии в судебноймедицине и криминалистике 48817.8. Применение газовой хроматографии вфармацевтике и фармакологии 49117.9. Газовая хроматография в космических исследованиях 493512
Научно-производственное объединение
«Химавтоматика»Газовый хроматограф «Яуза-100»НОВЫЙ ЛАБОРАТОРНЫЙ ГАЗОВЫЙ ХРОМАТОГРАФ
С ВЫСОКОЙ НАДЁЖНОСТЬЮПредназначен для анализа широкого круга смесей органических и неорганических сое¬
динений.ОСНОВНЫЕ ОБЛАСТИ ПРИМЕНЕНИЯ- технологический контроль производственных процессов в химической, нефтехимичесюй
и газовой промыишенности;- контроль загрязнений окружающей среды (возцух, вояа, почва);- контроль пищевых продуктов и напитков;- контроль летучих примесей лекарств и лекарственных форм;- анализ загрязнений воздуха жилых, офисных и производствен¬
ных помещений с предварительным концентрированием на пред-
колонке;- анализ загрязнений атмосферного воздуха в составе передвиж¬
ных лабораторий;- анализ смесей постоянных и благородных газов.ОСНОВНЫЕ ОСОБЕННОСТИ ХРОМАТОГРАФА- компьютерное управление параметрами режима;- надёжная MHiqwnpoueccopHafl профамма обработки результатов t
ного протокша;- лёгкость обслуживания и доступность основных рабочих элементов;- высокая надёжность конструкции термостата;- возможность применения насадочных, микронасадочных, кашшлярных и поликапил¬
лярных юшонок;- ввод проб как михрощприцем, так и газовым краном;- обогатительное устройство для концентрирования соединений из газовых и воздушных
сред оригинальной конструкции;- надёжный электронный блок управления, стабилизации и измерения газовых потоков;- может комплектоваться дополнительно по желанию заказчика 6-ходовым краном для
дозирования газовых проб, термодесорбером оригинальной конструкции.ТЕХНИЧЕСКИЕ ХАРАКТЕРИСТИКИ:и вьмачи пол-Темпе^шу^тый диапазон колонокег50аоЗ<ЮЧ:Темпй^»тжый шашкзк» жшцкитш!«л50ао350Ч:3 10-"по тепдонрсао^остм по нонану, в г/см‘5Т0'"Ii0'>Поя(»мий штостата2дПиш1в«220 В. 50 ГцМшснмаяью1 потоеблясмая мшшюогь700 Вт4Мх332х417ммВес. не ботеНС бояее 15 кгСертификат об утверждеини типа средств измерений № 17188 от 05.04.2(Ю4 г.
Зарегистрирован в Госреестре под № 26625-04.
РАЗРАБОТЧИК И ИЗГОТОВИТЕЛЬОАО «НПО Химавтоматика»129226, г. Москва, ул. Сельскохозяйственная, д. 12А,
www.chimavtonmtika.ruНг^но-технический центр «Хроматография»,
тел.: (499) 181-53-27, 181-14-02,
факс: (499) 181-80-58, 181-14-02
e-mail: ya.shinchrom@mail.ru
http://chromatograph.narod.ruОтдел научно-технической информации
(ОНТИ):телефон/факс: (499) 181-34-04
e-mail: info@chimavtomatika.ru33 Газовая хроматография
SchelTecTotal l„3boratory РвГк1пЕ1гПвГ'Хроматографическое оборудование PerkinElmerКомпания Scheltec AG является авторизованным дистрибьютором фирмыPerkinElmer - одного из лидеров мирового аналитического приборостро¬
ения, которая первой в мире представила коммерческий газовый хрома-
тофаф - “Vapor Fractometer 154” в 1955 году. В настоящее время фирма
производит газовые хроматографы серии Clarus 400/500/600 - от простых
приборов до больших аналитических систем для комплексного решения
аналитических задач.Газовый хроматограф Clarus 600 оснащен наиболее совершеннымтермостатом колонок с макси¬
мально возможной скоростью
нагрева и охлаждения, тех¬
ническое решение которого за¬
патентовано. Имеется функция
активации промывки шприца ав¬
тодозатора и отбора пробы до
готовности хроматографа к вводу
пробы. Все это резко повышает
производительность за счет су¬
щественного сокращения вре¬
мени аналитического цикла. ГХ
Clarus 600 легко адаптируется к
любому сколь угодно сложному
анализу и позволяет добиться выдающейся продуктивности лаборатории с
максимально быстрым возвратом инвестиций.Газовые хроматографы Clarus 500 и 400, сочетающие простоту исполь¬
зования с выс№4айшей чувствительностью и гибкостью, используются для
решения, как рутинных, так и исследовательских задач. На базе Clarus
500/600 изготавливается более 100 моделей анализаторов для решения
задач «Под ключ».Наличие уникальных технологических и профаммных особенностей та¬
ких, как PSS, РРС, Prevent, ProTect, MSVent, Soft-Cooling, GC-Tune и
встроенный автодозатор, делает хроматографы PerkinElmer непревзой¬
денными по производительности, гибкости, точности, простоте и удобству
использования.Clarus 560/600 MS - квадрупольные масс-спектрометры с префильтром,
уменьшающим загрязнения аналитического квадруполя. Использование
современных материалов и решений продиктовано требованиями вы¬
сокой инертности к анализируемым веществам и стабильности системы.
Компоненты системы и новейшая электроника обеспечивают работу с мак¬
симальной скоростью и чувствительностью. Приборы легко обслуживают¬
ся, быстро выходят на режим, наращиваются до химической ионизации.
MSVent - первое в мире универсальное устройство для автоматическо¬
го манипулирования газовыми потоками при работе с масс-спектрометром- позволяет упростить работу с масс-спектрометром до уровня обычных
хроматофафических детекторов, дает возможность менять колонку без на¬
рушения вакуума масс-спектрометра. А возможность лепсо менять потоки
газаноситепя при разделении без изменения вакуума трудно переоценить.Turbomatrix HSАвтоматические парофазные дозаторы Turbornatrlx HSI основаны на уникальной разработанной и запатентован¬ной PerkiElmer технологии, не требующей использования
шприцев и газовых кранов - ввод пробы перепадом дав¬
ления (pressure balance). Эта технология позволяет про¬
водить анализ летучих компонентов пробы с высочайшей
чувствительностью, скоростью и без искажения состава.
Использование системы Trap существенно увеличивает
чувствительность анлиза.Turbornatrlx TD, ATDТермодесорберы Turbornatrlx основаны на разра¬
ботанной PerkiElmer технологии двухстадийной тер¬
мической десорбции, позволяющей анализировать
образцы в широком диапазоне концентраций и влаж¬
ности с фокусировкой в охлаждаемой эффектом Пель¬
тье ловушке. Эти устройства разработаны для анализа
загрязнения воздуха, однако нашли широкое примене¬
ние в криминалистике, парфюмерии, фармацевтике,
материаловедении. Продуманная конструкция, сенсор¬
ное управление и полная автоматизация превращают
сложный, требующий искуалва анализ, в простой и обычный.TotalChrom WSuCSПрограммное обеспечение TotalChrom служит для управления газовыми
и жидкостными хроматофафами и обработки данных. Встроенный редактор
отчетов TotalChrom Publisher позволяет создавать отчеты любой сложности
в любых форматах. В варианте WS (рабочей станции) ПО дает возможность
работать одновременно с 8 хроматографами, а в варианте CS - клиент сер¬
вера - число приборов и пользователей практически не ограничено.Удобство в работе в сочетании с высокими техническими и ана¬
литическими характеристиками делает приборы PerklnElmer по-
настоящему современными.Все приборы сертифицированы по ISO 2001 и Федеральным агенством
по техническому регулированию и метрологии РФПодробная информация о приборах размещена на сайте www.scheltec.ruМы ответим на все Ваши вопросы, если Вы обратитесь на Scheltec AG119334 Москва, ул. Косыгина 19.Тел/495 935 8888, факс 495 564 8787,lnfo@scheltec.ru www.scheltec.ruScheltec AG Авторизованный дистрибьютор PerkinElmer LAS в странах СНГ
:!0!; Agilent Technologies-AeHeriudOiaraiiterAgilent 7000A QQQ
ГХ/МС/МСПовседневный фемтофаммовый анализ
ПревосхоАная селективность
Сверхнизкий уровень шума
Уникальная скорость сбора данныхАнализ целевых соединений
на самом низком пределе определения.Новейший тандемный квадрупольный газовый хромато-масс-спектрометр
Agilent 7000А - самые низкие пределы детектирования и очень быстрый
ГХ/МС/МС анализ, даже при работе с загрязненными и сложными по
составу пробами. Простой в использовании прибор специально разработан
для рутинных высокоточных анализов с очень высокой
производительностью. Agilent 7000А - идеальная система для
количественного определения следовых количеств целевых соединений
при анализе: пестицидов в воде и пищевых продуктах, лекарственных
препаратов и наркотиков в биологических матрицах, экологических
загрязнителей. Такие результаты обеспечиваклся сочетанием
патентованного инертного источника ионов, кварцевых квадруполей,
инновационной ячейки соударений, нового трех-осевого детектора с
флагманом газовой хроматографии Agilent 7890.За дополнительной информацией обращайтесь;127055, Моаша, Тихвинский пер., д.11 стр.2
т. (495) 788-09-82/83, ф. (495) 755-77-61
www.interlab.ru; e-mail: lnterlab@interlab.ruЕкатеринбург; т. (343) 379-5733
Новосибирск: т. (383) 335-64-16
Санкт Петербург; т. (812) 605-07-39
Agilent 7820Просто устанавливать
Просто обучиться
Просто использовать
Просто обслуживатьКомпания Interiab представляет новый газовый хроматофаф
MaestroGC (Agilent 7820) разработан совместно со
специалистами Agilent Technologies специально для российского
рынка. В основе прибора лежит флагманская платформа Agilent
7890, отлично зарекомендовавшая себя во всем мире.MaestroGC - это прибор российского производства идеально
подходящий для массовых рутинных анализов в заводских и
экспертных лабораториях, а также для обучения газовой
хроматофафии на самом современном уровне.istroШ Все, что Вам нужно, и ничего лишнего!
За дополнительной информацией обращайтесь:1270S5, Москва, Тихвинский пер., д.11 стр.2
т. (495) 788-09-^2/83, ф. (495) 755-77-81
www.interlab.ru; e-mail: Inter1ab@interiab.ruЕкатерин^; т. (343) 379-5733
Новосибирск: т. (383) 335-64-16
Санкт Петербург: т (812) 605-07-39
Access to your successSHIMADZUОборудование SHIMADZU для газовой хроматографии0Газовый хроматограф
GC-2014 предназначен для
работы как с капиллярньШи,
так и с набивными колонками
и идеально подходит для вы¬
полнения рутинных анализов.
GC-2014 - единственный хро¬
матограф на мировом рынке,
обеспечивающий цифровой
контроль газовых потоков
при работе с набивными ко¬
лонками.Особенности модели:• одновременная установка капиллярных и набивных колонок• установка до 3-х инжекторов различного типа и до 4-х независи¬
мо температурно-шнтролируемых детекторов• цифровое электронное управление потоками газа-носителя до
давления 970 кПа• обеспечение постоянной линейной скорости потока газа-носителя
при работе с капиллярными колонками• высокие скорости нагрева и охлаждения термостата• встроенные функции самодиагностики и валидации для обе¬
спечения качества измерений в соответствии с требованиями
GLP/GMP• компактный дизайн при большом объеме термостата (13,7 л)• большой встроенный 16 строчный ЖК-дисплей, демонстрирую¬
щий текущую хроматограмму и режим работы хроматографа• контроль, сбор и управление данными при помощи программы
GCSolution.
Газовый хроматофаф
гС-2010 идеально при-
пособлен для проведения
нализа на капиллярных
олонках. Благодаря со-
ершенной электронной
истеме контроля потоков
аза-носителя и детектор-
ых газов прибор позво-
яет проводить анализы
ри высоких значениях
явления газа-носителя на
ходе в колонку (до 970 кПа), что позволяет использовать метод быстрой
роматографии, на порядок сокращая время анализа.Особенности модели:• установка до 3-х инжекторов для капиллярных и/или набивных
колонок и до 4-х детекторов; температуры инжекторов и детекто¬
ров могут контролироваться независимо друг от друга• максимальная частота сбора данных со всех детекторов - 250 Гц• режим поддержания постоянной линейной скорости газа-носителя
обеспечивает эффективную работу капиллярной колонки на про¬
тяжении всего времени анализа вне зависимости от использован¬
ной температурной программы• высокие скорости нагрева и охлаждения термостата• встроенные функции самодиагностики и валидации для обе¬
спечения качества измерений в соответствии с требованиями
GLP/GMPНа базе газового хроматографа модели GC-2010 разработаны ком-
лексы для работы в режиме многомерной хроматографии, ш также
роматомасс-спектрометры GCMS QP-2010Plus и GCMS QP-2010S.Более подробная информация на сайтах www.shimadzu.com,
www.shimadzu.eu,www.shimadzu-sng.ruПредставительство Shimadzu Europe GmbH в Москве:121059, пл. Европы, 2, гост. «Рэдиссон-Славянская»,
Бизнесс-центр, оф. 716
Тел.: (495) 941-8290/8108, факс: (495) 941-8109
E-mail: shimadzu@shimadzu-sng.ru
Все это - МультиХром!Сомпания АМПЕРСЕНД предлагает модульное программное обеспечение для компьютерной
втоматизации хроматографии МультиХром.Начните с простого - сложность вашей системы
всегда будет на уровне сложности ваших задач!Цифровое управление оборудованием; более 100 приборов российских и зарубежных
трошводителей: Детекторы - Насосы - Автосамплеры - Термостаты - Краны-переключатели
10T0K0B - Коллекторы фракций.Управление несколькими хроматографическими системами.Подключение любых хроматографов с помощью 24-битного аналого-щ|фрового
преобразователя (гарантия до 24 месяцев).: детвкто^ж
;насосы
: с*юс:
i термостспы
i (жолого-чифровывUMS,
мини-UMS
^кстнзт данных
и реэ^ьштов
Веб пу&т1в(ации
печать отчетов
электронные документыОБЕСПЕЧИВАЕТСЯ РАБОТА В УСЛОВИЯХ ПОВЫШЕННОЙ ОТВЕТСТВЕННОСТИ:
соответствие стандчяу "надлежащая лабораторная практика” (GLP и GALP), электронные
подписи по 21CFR part 11.ПЕРЕКЛЮЧЕНИЕ ЯЗЫКА ИНТЕРФЕЙСА С РУССКОГО НА АНГЛИЙСКИЙ.ДЛЯ РОССИЙСКОГО ПОТРЕБИТЕЛЯПофобная техническая документация на русском языке.«Горячая линия» поддержки.Регулярное проведение школ-семинаров с целью лучшего освоения системы.
Низкие цены.Метрологическое обеспечение системы.
Регистрация в Госреестре РФ средств измерений.
полный НАБОР СОВРЕМЕННЫХ ВОЗМОЖНОСТЕЙ ОБРАБОТКИ ХРОМАТОГРАММ
Автоматизированное вьшолнение интегрирования, идентификации юмпонентов, градуировки,
количественного анализа.РАЗДЕЛЕНИЕ ПЕРЕКРЬШАЮЩИХСЯ ПИКОВ
Разделение пиков по форме.Использование факторного анализа для многоканальных хроматограмм.ГРУППОВАЯ ОБРАБОТКА ХРОМАТОГРАММАвтоматизированное исполнение и обработка серии анализов (очереди).
Пакетный пересчет хроматограмм.Статистические отчеты для серий хроматограмм.ЭКСПОРТ И ИМПОРТ ХРОМАТОГРАММ РАЗЛИЧНЫХ ФОРМАТОВ,
ВКЛЮЧАЯ AIA-CDF, XMLHTXTОТЧЕТЫШирокие возможности настройки внешнего вида: форматирование текста, таблицы, графика.
Расчет параметров пиков по формулам пользователя.Программируемые формулы, условные шрифты и цвета - для специальных отчетов.
Одновременная подготовка для одного анализа нескольких отчетов по разным шаблонам.
Поддержка электронного документооборота; отчеты в форматах PDF, RTF, TXT, HTML.
Формирование отчетов по стандартизованным бланкам.Настраиваемые отчеты поверки методик и приборов.СПЕКТРАЛЬНЫЙ АНАЛИЗ И МНОГОКАНАЛЬНАЯ ХРОМАТОГРАФИЯ
Обработка хроматограмм, полученных с помощью фотодиодных линеек и других
многоканальных детекторов.Идентификация и расчет пиков; библиотеки спектров.Анализ образцов неизвестного состава "одной кнопкой".2D и 3D представления спектров.ПОДДЕРЖКА КАПИЛЛЯРНОГО ЭЛЕКТРОФОРЕЗА
Вычисляемый канал Отклик/Время.Расчеты подвижности.Специальные методы расчета для пиков с выраженной асимметрией.ПОДДЕРЖКА ЭКСКЛЮЗИОННОЙ (ГЕЛЬ-ПРОНИКАЮЩЕЙ) ХРОМАТОГРАФИИПОДДЕРЖКА ПЛАНАРНОЙ ХРОМАТОГРАФИИ И ГЕЛЬ-ЭЛЕКТРОФОРЕЗАПОДДЕРЖКА ПРЕПАРАТИВНОЙ ХРОМАТОГРАФИИСбор фракций по программируемым условиям для сигнала детектора.Повторное использование элюента.Работа в условиях повышенных требований к надежности систем.МУЛЬТИХР0М-МИНИЕ1М8: система сбора, учета, хранения и обработки хроматографических
данных масштаба лаборатории или предприятия. Обеспечивает централизованное хранение и
доступ к информации по локальной сети.ЗАО Амперсенд123060 г. Москва, а/я 80+7-916-675-25-92, +7-499-196-18-57, +7-499-196-52-90
support@ampersand.ru http://www.ampersand.ru
Научно-производственная фирма «Мета-хром» -
разработчик и изготовитель хроматографов серии «Кристаллюкс»Уже почти 15 лет Научно-производственная фирма «Мета-хром» занимается разработкой,
производством и внедрением газовых хроматографов и комплектующих к ним. Костяк фирмы
составляют разработчики газовых хроматографов с 30-летним стажем. Последней разработ¬
кой фирмы является газовый хроматограф «Кристаллюкс-4000М», который превосходит по
многим показателям не только своих предшественников хроматографы «Кристаллюкс-4000»
и «Кристалл-2000», но и современные отечественные и зарубежные хроматографы. В хрома¬
тографе применены наиболее современные технологии, комплектующие материалы и изде¬
лия, технические решения. О квалификации специалистов фирмы говорит тот факт, что при
разработке приборов ими создано свыше 50 изобретений, многие из которых были внедрены
в хроматографы «Кристалл-2000», «Кристаллюкс-4000М» и в другие разработки. Фирма име¬
ет лицензию на выпуск хроматографического оборудования, новый производственный кор¬
пус, оснащенный оборудованием и станками для выпуска сложной аналитической техники.Основным направлением совершенствования хроматографа является оснащение его
сервисными устройствами, повышающими качество, удобство и скорость работы. Для этого
хроматограф оснащен:1) возможностью независимой и одновременной работы с двумя аналитическими канала¬
ми хроматографа («два хроматографа в одном») при одинаковых режимах термостатируемых
объектов для увеличения производительности, причем оба канала разделения могут быть
оснащены капиллярными колонками с независимым управлением по газу-носителю;2) автоматическим дозатором жидких проб НТЗООА (производство фирмы LabHat США),
который является одним из лучших дозаторов в мире, и способен вводить как жидкие пробы
для ГХ и ВЭЖХ, так и равновесный пар (НТ200Н). Дозатор может быть установлен на любой
отечественный хроматограф. Особенностью дозатора является возможность ввода пробы по¬
следовательно в оба испарителя хроматографа, что в сочетании с п.1 очень актуально;3) дозатором равновесного пара ДПР-10 на базе продуваемого газом-носителем газо¬
плотного термостатируемого шприца и электромагнитным перемешиванием пробы в термо¬
статируемых отдельно от шприца контейнерах (4 шт);4) термодесорбером, который служит для ввода в хроматограф летучих веществ из воз¬
духа и твердых веществ, например, пластмасс и др. Простота и надежность термодесорбера
позволяют применить его на любом хроматографе. Особенностью термодесорбера является
минимальный мертвый объем между пробой и хроматографической колонкой;5) пиролизером для анализа состава каучуков, красок и др. Пиролиз проб осуществляется в
кварцевой капсуле при нагреве до фиксированной температуры со скоростью более 500 °С/мин.;6) автоматическими кранами для дозирования пробы, обратной продувки, причем краны
могут быть как горячими (до 250 “С), так и в обычном исполнении и устанавливаться по два
на прибор, подвод анализируемых газов также может быть выполнен обогреваемым;7) устройством для автоматической полуобратной продувки капиллярных колонок с
предколонкой. Применение его при анализах проб с тяжелыми юмпонентами, например,
нефти позволяет значительно сократить время анализа;8) гелиевым детектором Pulsed Discharge Detector Models D2 (фирма VICI), обладающим
уникальной чувствительностью к постоянным и редким газам;9) фильтром для каталитической очистки газов, в т.ч. азота от кислорода и воздуха от
органических примесей, возможна одновременная очистка азота и воздуха;10) выносной панелью управления на базе микрокомпьютера Pocket PC;11) генераторами чистого водорода САМ-1-30 и САМ-1-90 производительностью до 30 и
90 л/ч соответственно, «Мета-хром-10» производительностью 10 л/ч;12) масс-спектрометрическим квадрупольным детектором «Маска».Постоянно совершенствуется программа управления и обработки сигналов детекторов
хроматографа «NetChrom V2.0». В программу введены дополнительные функции:
- втможносгь выб^я и переключения рабочих языков программы (русский, английский ихф.);- поддфжка требований GLP, в т.ч. многоуровневый доступ к данным программы, устра¬
няющий возможность случайного или несанкционированного изменения параметров управ¬
ления и обработки данных, встроенная система безопасности на основе пароля позволяет
ограничить уровень доступа пользователя, в соответствии с его квалификацией, исходные
данные (хроматофамма), параметры обработки и конфигурация хроматографа хранятся в
одном файле;- запись всех событий, произошедших с хроматографом и операциях над хромагофамма-
ми и методами в электронный журнал, регистрация изменений, произведенных пользовате¬
лем в хроматофамме,- построение градуировочной характеристики с помощью специальных математических
формул, использование технологаи RTL (фиксация времен уцерживания), ведение журнала
регламентных работ, обеспечение передачи данных в заводские системы АСУТП и многоедругое;- расчет концентрации примесей в спиртных напитках с учетом отклика пика этанола,
таким образом повышается точность измерения концентрации токсичных примесей;- построение зависимости фадуировочных коэффициентов (фафика) для методов рас¬
чета «внутренняя нормализация» и «внутренний стандарт» в зависимости от концентрации
компонентов в пробе;- статистический расчет одновременно по всем компонентам метода для 15 анализов;- разработана программа диагностики развивающихся дефектов трансформаторного
оборудования по результатам хроматофафического анализа газов, растворенных в транс¬
форматорном масле;- разработана профамма расчета контрольных карт Шухарта по ГОСТ Р 50779.42-99
(ИСО 8258-91). Профамма позволяет вводить данные из хроматофамм, полученных на хро-
матофафе с помощью профаммы «NetChrom V2.0», или вручную. Профамма позволяет
строить карты средних размахов, выборочных стандартных отклонений, выводить фафиче-
ские карты на монитор и печать. Реализован также как отдельный расчет карт пределов по
ГОСТ Р 51698-2000 «Водка и спирт этиловый. Изменение Ns 1»;- разработана профамма идентификации многокомпонентных смесей, например, рас¬
тительных масел по соотношению интенсивностей пиков отдельных компонентов;- разработана профамма визуализации параметров технологических процессов в ре¬
жиме реального времени;- разработана профамма и устройство для поверки хроматографов и хроматофафических
профамм - генератор хроматограмм.Кроме основного направления деятельности, фирма занимается автоматизацией старых
хроматографов, например: «ЛХМ», модель 3700, «Цвет», «Хром» и др. с помощью:- профаммно-аппаратного комплекса на базе аналого-цифрового восьмиканального пре¬
образователя АЦП, логарифмического цифрового измерителя тока (ИТЛЦ), персонального
компьютера и профаммы обработки хроматофафической информа¬
ции «NetChrom V2.0»,- формирователя газовых потоков от 1 до 5 пяти каналов регули¬
рования расхода и давления газов.Фирма «Мета-хром» занимается кроме хроматофафии разработкой
испытательного и измерительного оборудования по заданию различ¬
ных отраслей промышленности.НПФ «Мета-хром» тел./факс (836-2)42-49-97, 42-22-66, 43-04-40,41-14-10,41-10-63424000 г Йошкар-Ола, ул. Баумана, 100E-mail; m_chrom(gmari.ruhttp;//www.meta-chrom.ru
Разработка и производство аналитического оборудования в СКБ “Хроматэк"СКБ «Хроматэк» - признанный лидер в разработке и производстве газохроматогра¬
фического оборудования в России.Лабораторные газовые хроматографыКомпания работает иа рынке аналитического приборостроения с 1991 года. СКБ «Хрома¬
тэк» разрабатывает и производит газохроматографические ашшратно-профаммные шмплек-
сы, иггорые включают в себя:• газовые хроматографы• устройства ввода пробы и пробоподготовки - дозатор автсмагический жидюстный, доза¬
тор равновесного пара, дозатор автоматический газовый, краны поворотные для ввода газовых
проб, инжектор бесшприцевого ввода проб, находящихся под высоким давлением, кран-дозатор
сжиженных газов, дозатор твердых проб• вспомогатеяыюе оборудование - генерагсф водорода, кшшрессор воздуха, фильтр катадши-
чесюй очистки газов, формирователь газовых погонов, устройства АЦП• программный пакет “Хроматэк Аналитик” и специализированное программное обеспе¬
чение• методическое обеспечение по проведению анализов• хроматофафические колонки и расходные материалыВ настоящее время выпускаются три модели лабораторных газовых хроматографов:• Кристалл 2000М• Хроматэк-Кристалл 5000 исполнение 1• Хроматэк-Кристалл 5000 исполнение 2В сравнении с другими отечественными производителями компания предлагает наиболее
широкий выбор детекторов, устройств ввода пробы и автоматических внешних устройств.В состш газовых хроматофафов могут вхсздить как универсальные детекторы (ПИД, ДТП,
ДТХ), так и селективные дегекг(Ч)ы (ЭЗД, ТИД, ПФД, ФИД). Хроматографические анализы более
сложного уровня выполняются с применением масс-спектрометрического детектора DSQ П.Хромато-маес-спектрометрыС 2005 года СКБ «Хроматэк» поставляет на российский рынок хромато-масс-спектроме-
тры, в состав которых входит газовый хроматофаф «Хроматэк-Кристалл 5000» и квадруполь¬
ный масс-спектрометрический детектор DSQ II (производства Thermo Fisher Scientific Inc.,
США). Сегодня эти комплексы успешно конкурируют с зарубежными производителями.Промышленные хроматографыВ 2008 году начинается серийное производство взрывозащищенного промышленного хро-
матофафа “Хроматэк-Кристалл 7000”, Хроматофаф обеспечивает автоматическое проведе¬
ние анализа и обработку результатов за счет применения встроенной микро-ЭВМ, Прибор
удобен в экаиуатации и имеет высокую надежность за счет модульного исполнения и встро¬
енной электронной системы диагностики, Хроматофаф по заказу оснащается детекторами
ДТП и ПИД, кранами для ввода проб и переключения колонок при анализе газообразных и
жидких проб.Конструктивные особенностиГазовые хроматофафы и другие устройства, производимые в СКБ «Хроматэк», разрабо¬
таны с учетом принципов максимальной надежности в работе, удобства в эксплуатации и
обеспечения правильности результатов проводимых анализов.Современные хроматофафы представляют собой автоматизированные юмплексы со
встроенной системой самодиагностики, контроля и управления, а также передачи и хранения
данных. Благодаря электронным регуляторам расхода и давления достигается высокая вос¬
производимость времен удерживания и удобство управления газовыми потоками.
Взаимозаменяемость узлов, простота обслуживания, унифицированность конструктивных
решений делают хроматографы компании СКБ «Хроматэк» надежным инструментом в лю¬
бой лаборатории.Хроматографические комплексы изготавливаются по заказу в соответствии с конкретными
задачами потребителя. Тестирование комплексов и дополнительного оборудования, постав¬
ляемого в их составе, производится согласно заявленным методикам анализа.Программное обеспечениеПрограммное обеспечение «Хроматэк Аналитик» предназначено для управления хрома¬
тографом и автоматическими устройствами ввода и пробоподготовки в составе прибора, а
также для сбора и обработки хроматографических данных. Гибкий интерфейс программы
позволяет настроить ее индивидуально для каждой методики анализа и сделать работу опе¬
ратора максимально эффективной и удобной. На выбор предлагаются две версии профаммы:
версия 1,5 - модификация прежней версии 1,21, а также версия 2,5 - современный пакет с
широкими возмсмшостями, по достоинству оцевенпый многими пользователями.Полбор оптимальной комплектацииДополнительное оборудование, поставляемое с хроматофафами, в том числе устройства
ввода, источники питания и системы очистки газов, хроматофафические колонки и расхо¬
дные материалы для хроматофафии индивидуально подбираются специалистами техниче¬
ского отдела для решения конкретной аналитической задачи. В лабораториях СКБ «Хрома¬
тэк» производится тестирование капиллярных и насадочных колонок в соответствии с мето-
диками анализаВиеаренне и сервисная поддержкаМонтаж оборудования и пусконаладочные работы, стартовое обучение персонала на ре¬
альных методиках в течение 3-4 дней в лаборатории заказчика гарантируют эффеетивное ис¬
пользование приборов с первых дней после запуска оборудования. Внедрение и сервисную
поддержку на площадках заказчика обеспечивают более 25 квалифицированных специали¬
стов компании, а также представители в регионах России и ближнем зарубежье. По любым
вопросам, связанным с работой оборудования, оказываются консультации в режиме “горячей
линии". Кроме того, ежемесячно в специально оборудованной лаборатории СКБ “Хроматэк”
проводятся бесплатные учебные курсы для лаборантов и специалистов КИПиА, которые по¬
лучают практические навыки по работе с хроматофафом, техническому обслуживанию и
ремонту.Вся продукция, выпускаемая СКБ «Хроматэк», сертифицирована.Федеральным агентстюм по техническому регулированию и метрологии выданы сертифи¬
каты об утверждении типа средств измерений:• RU,C. 31,004, А № 30878 от 14.04.2СЮ8 г на хроматофаф газовый “Кристалл 20(ЮМ”, За¬
регистрирован в Госреестре средств измерений под Xs 14516 -08• RU.C. 39.004,А № 31051 от 14.04.2008 г на комплекс аппаратно-профаммный для меди¬
цинских исследований на базе хроматофафа “Хроматэк-Кристалл 5000”.Зарегисфирован в Госреестре средств измерений под № 18482-08• RU.C. 31.004.А X» 26797 от 27.02.2007 г. на промышленный хроматофаф “Хроматэк-Кри¬
сталл 7000”. Зарегистрирован в Госреестре средств измерений под Ха 33907-07Система менеджмента качества ЗАО СКБ “Хроматэк” сертифицирована по ГОСТ Р ИСО
9001-2001424000, г, Йошкар-Ола,ул. Строителей, 94Главпочтамт, а/я 84e-mail: salesCoichromatec.ru www.chromatec.ni
Яков Иванович Яшин,Евгений Яковлевич Яшин,
Александр Яковлевич ЯшинГАЗОВАЯ ХРОМАТОГРАФИЯГлавный редаетор В.В. Бондалетов
Компьютерная верстка Н.Г Щитнев
Дизайн обложки Н.Г. ЩитневПодписано в печать 15.10.2008 г.Формат 60x90 1/16. Гарнитура «Times New Roman»
Печать офсетная. Уел. печ. л. 33.00
Тираж 1000 экз. Заказ № 66.Издательство «ТрансЛит»115432, Москва, 2-ой Южнопортовый пр. д. 5 корп. 2
(495) 960-72-13Отпечатано в полном соответствии с качеством
предоставленных материалов в ОАО «Дом печати — ВЯТКА».
610033, г. Киров, ул. Московская, 122.
^ Яшин Яков Иванович - директор
научно - технического центра «Хромато¬
графия» НПО «Химавтоматика», доктор
химических наук, профессор, Л§^еат
Государственных премий СССр и Россий¬
ской Федерации., Автор более 600 науч¬
ных работ, 20 книг и сборников, 42 патен¬
тов’1Й Ti5o6peTeH и й.Яшин Евгений Яковлевич - замести¬
тель директора научно - технического
центра «Хроматография» НПО «Химавто-
ма]щ'НЗучные интересы в газовой хроматогра¬
фии связаны С анализом нефтепродуктов.
Учась в аспирантуре Всесоюзного научно -
и ссл е до вател ьс ко го геол ощразведоч н ого
нефтяного инститрЗ/ опубликовал более
десяти науч^^ых раббт; 'Яшин Александр Яковлевич --началь¬
ник отдела хроматограф^ НТЦ «Хромато¬
графия» НПО «Химавтоматика», кандидат
химически)$^ук.''Автор более 110 публи¬
каций по газовой, жидкостной и ионной
хроматографии, 3 патентов, является
соавтором Международной энциклопедии
по аналитической химии и Нового Россий¬
ского справочника химика и технолога.