/
















































































































































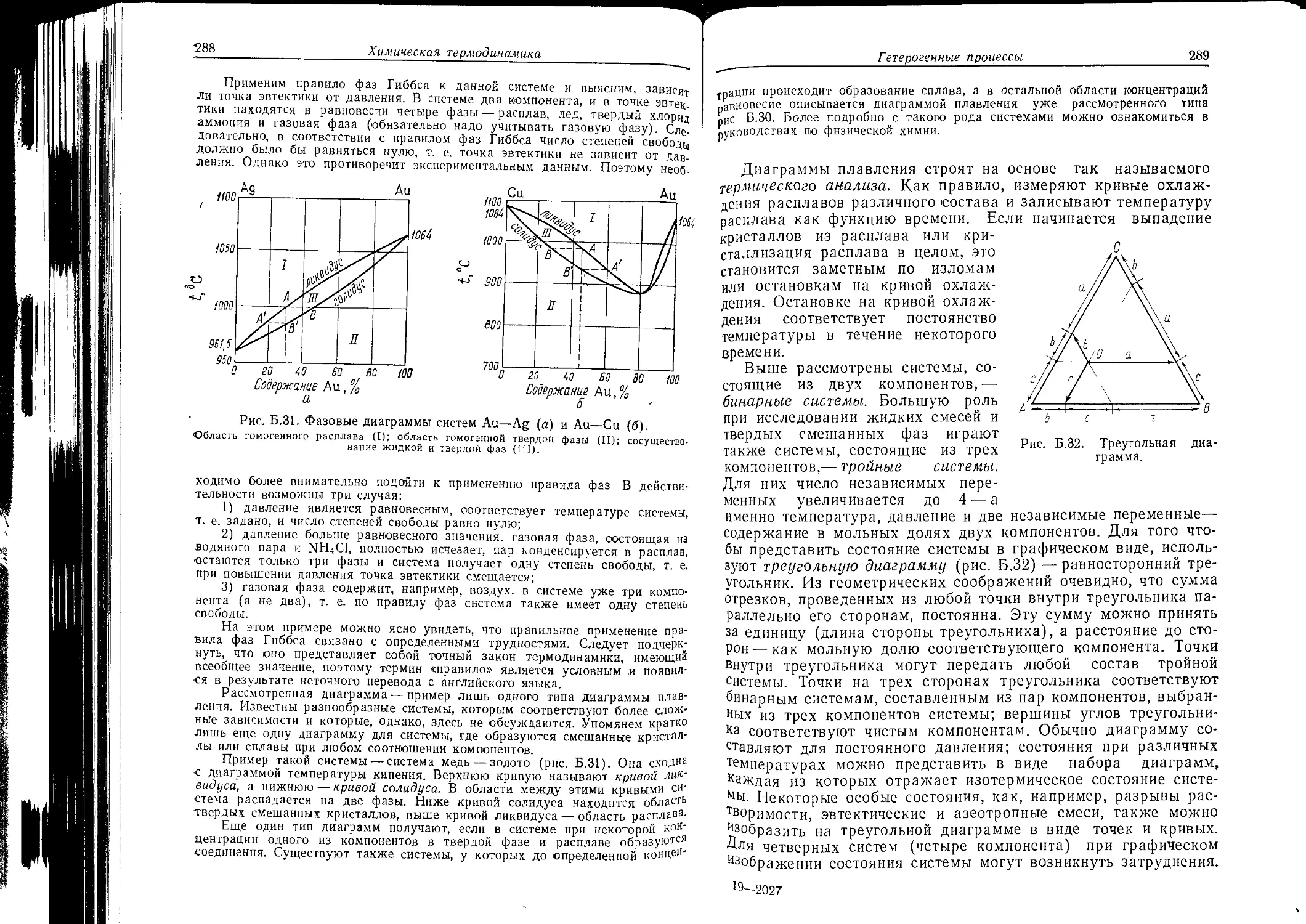































































































































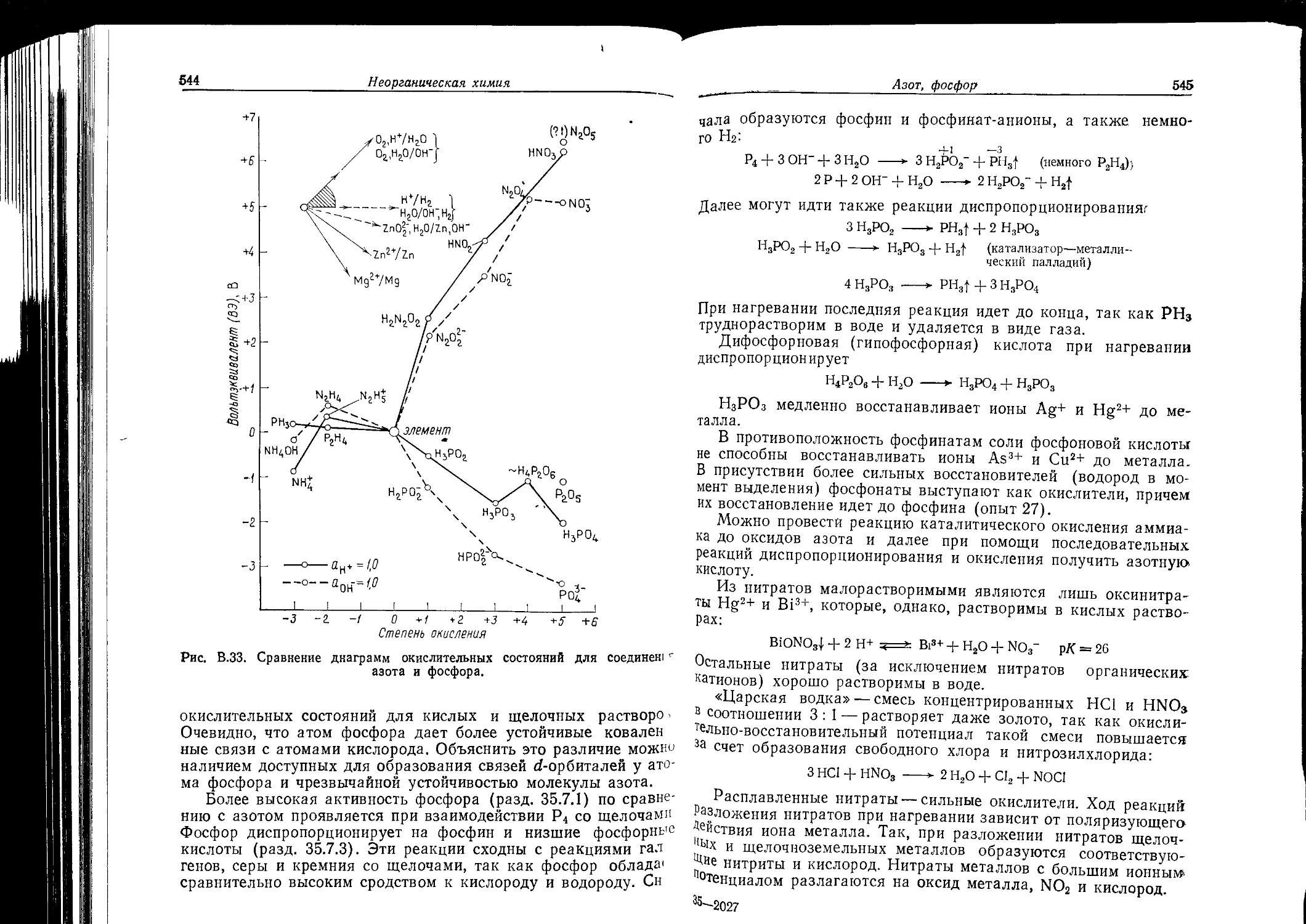




































































Текст
ANOR6ANIKUM
Lehr- und Praktikumsbuch der anorganischen Chemie mit einer Einfiihrung in die physikalische Chemie
Von Autorenkollektiv:
G. Blumenthal, S. Engels, I. Fitz, W. Haberditzl, K.-H. Heckner, G. Henrion, R. Landsberg, W. Schmidt, G. Scholz, P. Starke, I. Wilke, К.-Th. Wilke
Herausgegeben von LOTHAR KOLDITZ
10., uberarbeitete Auflage in 2 Teilen
Teil 1
VEB Deutscher Verlag der Wissenschaften Berlin 1984
АНОРГАН И ИА
В двух томах Том 1
СТРОЕНИЕ МАТЕРИИ ХИМИЧЕСКАЯ КИНЕТИКА ТЕРМОДИНАМИКА ЭЛЕКТРОХИМИЯ НЕОРГАНИЧЕСКАЯ ХИМИЯ: СОЕДИНЕНИЯ И РЕАКЦИИ
Редактор Л. Кольдиц
Перевод с немецкого канд. хим. наук В. В. Соболя и канд. хим. наук С. И. Троянова под редакцией д-ра хим. наук А. Ф. Воробьева
МОСКВА «МИР» 1984
ББК 24.4
А69
УДК 543+542
Блументаль Г., Энгельс 3., Фиц И., Хабердитцль В., Хекнер К.-Х-, Хенрион Г., Ландсберг Р., Шмидт В., Шольц Г., Штарке П., Вильке И., Вильке К--Т.
Анорганикум: В 2-х т. Т. 1. Пер. с нем./Под ред. А69 Л. Кольдица. — М.: Мир, 1984.1— 672 с., ил.
В учебном пособии, составленном авторами из ГДР и выдержавшем таи 10 изданий, излагается курс неорганической химии с основами физической и аналитической ХИМИН.
Том I посвящен основам строения материи, физической химии (термодинами ка, кинетика и электрохимия) и неорганической химии (свойства элементов и соединений с описанием соответствующих опытов)
Для студентов и преподавателей высших я средних химических и химикотехнологических учебных заведений
. 1802000000-428
А~04ЦЬГ)-84 95~84’ 41
ББК 24.4
543
Редакция литературы по химии
© VEB Deutscher Verlag der Wis-senschaften, Berlin, 1984
© Перевод на русский язык, «Мир», 1984
ПРЕДИСЛОВИЕ РЕДАКТОРА ПЕРЕВОДА
Рекомендуемое читателю издание является фундаментаЛ ным учебным пособием по неорганической химии. Основной 0 личительной особенностью книги является современный поД*° к отбору и подаче материала, необходимого при изучении не°ре ганической химии в высших учебных заведениях. В настой^ время совершенно необходимым элементом преподавания на°Р ганической химии в вузах является предварительное ознак0 ление студентов с теоретическими основами химии, после че’а свойства элементов и их соединений могут быть рассмотрены н современном уровне.
Авторы «Анорганикума»— коллектив преподавателей линского университета имени В. Гумбольдта под общей реД jj. цией профессора Л. Кольдица — весьма основательно и мет0^ чно следуют этим соображениям при написании книги. О*1 санию свойств элементов и их соединений («собственно неор1^ нической химии») предшествует изложение основ физичесК® химии. Серьезное внимание уделено изложению элемента?111’ д основ строения вещества; дан материал по основам химичек термодинамики (в том числе элементам статистической тер*1 динамики) и химической кинетики; рассмотрены основы эЛеК рохимии. Отбор материала для этих глав книги, что всегда я ляется не тривиальной задачей, выполнен на очень хоро^1 „ уровне и весьма последовательно. Наличие этого матери позволяет рассматривать свойства химических веществ на с л временном уровне, с привлечением всех необходимых сведен11 из теоретической химии. д
Безусловный интерес представляет оригинальный поД*^, авторов «Анорганикума» к изложению и логике подачи матер* ала в ряде разделов книги
Об успехе этого большого труда свидетельствует тот Фа^р что за сравнительно короткий период книга выдержала в 9 изданий. Настоящий перевод осуществляется с 10-го изданй , которое выйдет в свет одновременно и в ГДР, и в нашей стр^ не. Последнее обстоятельство оказалось возможным благодаря, активной помощи со стороны наших коллег из «Дойчер фер-113 дер виссеншафтен».
6 Предисловие редактора перевода
Содержание данного тома делится на три части. Часть А посвящена основам строения вещества (атомно-молекулярное учение, элементарные частицы, строение атомного ядра); в части Б рассмотрены основы физической химии; часть В объединяет данные по соединениям и реакциям неорганической химии.
Естественно, в таком большом труде неизбежны некоторые неточности и нестрогости. Переводчики и редактор считали своей обязанностью принять меры к их устранению, или внося необходимые уточнения в текст книги, или обращая внимание читателя на неточности посредством примечаний. При этом, конечно, были приложены усилия для сохранения авторской манеры и нюансов изложения.
Редактор посчитал возможным уточнить в ряде случаев терминологию, сделав ее более современной, а также заменить устаревший числовой иллюстративный материал на более надежный. И то, и другое, в первую очередь, относится к некоторым разделам, в которых излагаются вопросы термодинамики.
Мы надеемся, что книга окажется полезной и интересной советскому читателю и в первую очередь студентам, начинающим изучение химии в высших учебных заведениях. Несомненно, что аспиранты, научные сотрудники и инженерно-технические работники различных областей химической науки также будут в числе ее активных читателей.
А. Воробьев
ПРЕДИСЛОВИЕ К 10-МУ ИЗДАНИЮ
После значительной переработки «Анорганикума», предпринятого при подготовке 7-го издания, эта книга была выпущена еще в двух мало отличающихся изданиях. Для удобства пользования книга в 8-м издании разделена на два тома. При подготовке 10-го издания «Анорганикума» потребовалась более существенная переработка в связи с введением единиц СИ и приведением названий элементов в соответствие с требованиями IUPAC. Мы воспользовались этой возможностью для повторной переработки некоторых глав, сохранив при этом основной замысел книги.
В части Б, написанной доц. К.-Х. Хекнером (термодинамика и кинетика) и проф. Р. Ландсбергом (электрохимия), мы ввели безразмерные переменные и пояснили преимущества их употребления. Кроме того, в главу, посвященную электрохимии, включен раздел, посвященный уравнению Лютера. Д-р В. Шмидт —автор первых разделов части В — написал дополнительный раздел по химии твердого тела, а в целом часть В сокращена. Некоторые дополнения внесены в главу «Количественный анализ» части Г. В эту часть внесены существенные изменения в соответствии с современными воззрениями. Главы «Методы и проблемы анализа следовых количеств веществ» и «Теория измерений в аналитической химии» написаны заново.
Мы надеемся, что, как и предыдущие, новое издание «Анорганикума» (также в двух томах) будет принято с неослабевающим интересом.
Мы хотим сердечно поблагодарить сотрудников отдела химии «Дойчер ферлаг дер виссеншафтен», особенно И. Вениг и М. Мидлих, которые на протяжении нашего многолетнего сотрудничества всегда оказывали нам большую помощь.
Берлин, осень 1980 г. Лотар Кольдиц
ПРЕДИСЛОВИЕ К 1-МУ ИЗДАНИЮ
Во многих университетах и вузах делаются попытки найти новый подход к проведению практических занятий студентов. В основу обычно кладется классический подход, успешно применявшийся в течение многих лет, но теперь уже требующий внесения изменений. В секции химии университета имени В. Гумбольдта эти вопросы обсуждаются сравнительно давно и предпринимаются попытки их решения. Имеется много различных перспективных путей подхода к проблеме, и трудность состоит в том, чтобы сделать определенный выбор. Из многочисленных предложений можно выделить прежде всего два главных требования: 1) широкое приложение теории при проведении практических занятий как по неорганической, так и по органической химии и 2) важность привлечения студентов к исследовательской работе уже в первые годы их обучения.
Издавна принято, что основам общей химии студент обучается в первом семестре при прохождении курса неорганической химии. При этом часто расходятся во мнении, что же считать относящимся к области общей химии. В настоящее время вместо курса общей химии принято давать студентам как можно раньше введение в физическую химию как прочный фундамент для дальнейшего обучения. Лучше всего это осуществлять в тесном взаимодействии с физикохимиками. В результате такой совместной работы и возникла эта книга. При обучении следует больше уделять внимания взаимному перекрыванию областей неорганической и физической химии. В случае органической химии это не так актуально, поскольку она изучается значительно позднее, когда основы физической химии студентами уже усвоены. В связи с этим совершенно ясно, что книга для практических занятий по неорганической химии не должна быть узкоспециальной и в нее следует включать гораздо более широкий круг вопросов.
Наша книга состоит из восьми крупных разделов, порядок работы с которыми совсем не обязательно соответствует их расположению в учебнике. Большей частью эти разделы целесообразно прорабатывать в тесной связи один с другим. В первом разделе, написанном И. Вильке и К.-Т. Вильке, рассмотрены наиболее важные приборы и методы работы, а также пра
Предисловие к 1-му изданию
9
вила техники безопасности. Этот материал прост для усвоения и, несомненно, должен быть пройден в начале обучения. Порядок работы с остальными разделами может в значительной степени зависеть от преподавателя. Большое значение для глубокого понимания химических процессов имеет большая глава «Основы строения материи», составленная проф. В. Хабердитц-лем. Несмотря на то что наш учебник предназначен для студентов, лишь начинающих обучение, в нем сделана попытка отойти от модели атома по Бору и ввести элементы квантовой механики. При этом предполагается наличие у студентов умения дифференцировать в объеме школьной программы. Большое место уделено разделам «Химическая кинетика и механизмы реакций», «Равновесия и химическая термодинамика» и «Электрохимия». Начало первых двух разделов написано П. Штанге, который, к сожалению, скоропостижно скончался; дальнейшую работу проводили К.-Х. Хекнер и проф. Р. Ландсберг («Электрохимия»).
В разделе «Неорганическая химия: соединения и реакции» (В. Шмидт) дается обзор предмета неорганической химии с рассмотрением классов веществ и типов реакций.
Разделы «Неметаллы» (И. Фиц) и «Металлы» (3. Энгельс) носят более специальный характер и предназначены для изучения химии элементов периодической системы по группам. Проработку этих разделов необходимо вести с использованием обзора препаративных методов разделения, составленного И- Вильке и К.-Т, Вильке (обзор частично оформлен в виде таблиц).
При изложении раздела «Качественный анализ» (Г. Шольц и П. Штарке) основное внимание уделено используемым методам работы. Несколько меньшее, чем обычно, внимание уделено «классическому» ходу анализа. Все же, учитывая его большое методическое значение, ход анализа рассмотрен полностью. Глава «Количественный анализ» подразделяется на «Основы количественного анализа» (Г. Блументаль) и «Электрохимические методы количественного анализа» (Г. Хенрион). В этих разделах повышенное внимание было уделено основательному теоретическому обоснованию используемых методов.
В соответствии с учебным планом по завершению 3-летнего обучения (т. е. после прохождения практикума по органической химии) студенты проводят работу, имеющую исследовательский характер. Как правило, темы работ связаны с промышленностью, но они могут также даваться в рамках научной тематики института. Наш опыт показывает, что в обучении студентов эти задания очень важны. Они стимулируют комплексное использование знаний, полученных в предшествующий период обучения, оценки по этим заданиям влияют на оценку заключительного экзамена, предшествующего практическому выполнению дип-
10
Предисловие к 1-му изданию
домной работы. Небольшая глава, посвященная этим заданиям, написана Д. Хайнцем.
Этот учебник нельзя рассматривать как строгое руководство для изучения неорганической химии. Эта книга должна побудить студента к самостоятельному мышлению, а не к механическому выполнению ряда последовательных заданий. «Анор-ганикум» — это прежде всего пособие для обучения, требующее в значительной степени направляющего руководства преподавателей. Преподаватель получает широкие возможности варьирования процесса обучения за счет различных комбинаций отдельных разделов учебника.
Мы приносим глубокую благодарность народному предприятию «Дойчер ферлаг дер виссеншафтен», особенно сотрудникам химического отдела, а также сотрудникам типографии «Прогресс» (г. Эрфурт) за прекрасное оформление книги.
Берлин, лето 1967 г.
Л. Кольдиц
А. СТРОЕНИЕ МАТЕРИИ
1. АТОМИСТИЧЕСКИЕ ПРЕДСТАВЛЕНИЯ О СТРУКТУРЕ
МАТЕРИИ
Основы современных представлений о структуре материи были заложены в те далекие времена, когда люди только еще пытались вникнуть в сущность окружающих их вещей. Такие неотделимые от материи понятия, как движение и прерывность (дискретность), были уже предметом дискуссий древнегреческих натурфилософов. Понятие «атом» (от греческого атород — неделимый) восходит к Демокриту (V в. до н. э.). Изучающим химию полезно проследить историю развития атомистических представлений, а также основы кинетической теории. Ниже весьма кратко изложены наиболее важные экспериментальные доказательства, которые послужили краеугольным камнем атомно-молекулярной теории строения материи и так называемой «теоретической химии» (именно так Нернст назвал одну из своих классических работ, снабдив ее подзаголовком «Теоретическая химия с точки зрения правила Авогадро и термодинамики») .
1.1. Закон сохранения массы
Закон сохранения массы при химических превращениях был открыт Ломоносовым в 1748 г. и экспериментально подтвержден им же в 1756 г. (окисление свинца воздухом в запаянной стеклянной реторте). Независимо от Ломоносова в 1774 г. Лавуазье доказал справедливость этого закона с помощью некоторых других опытов.
В 1908 и 1909 гг. Ландольт и Этвеш также подтвердили закон сохранения массы, причем их эксперимент до настоящего времени не превзойден по точности (погрешность < 10-6 %).
На основе достижений современной атомной физики и теории относительности было установлено, что закон сохранения массы тесно связан с законом сохранения энергии (Ломоносов, 1748 г., Мейер, 1842 г.). Соотношение между массой и энергией было установлено Эйнштейном, который показал, что изменение энергии системы прямо пропорционально изменению массы:
ЬЕ — с* кт
Коэффициент пропорциональности здесь — квадрат скорости света, т. е. это огромное число по сравнению с измеряемым зна-
12
Строение материи
чением Дт. Поэтому изменения энергии, сопровождающие обычные химические реакции, соответствуют настолько малым изменениям массы, что последние нельзя определить с помощью современных экспериментальных методов (при сгорании 1 г водорода до воды изменение массы составляет 1,576-10~В 9 г). Уравнение Эйнштейна получило подтверждение при исследованиях ядерных реакций, происходящих с выделением колоссального количества энергии.
Сохранение массы при химических реакциях (без учета тех случаев, когда нельзя не обращать внимания на соотношение .между энергией и массой) логически следует из гипотезы об атомарной структуре материи: при химическом превращении мельчайшие элементарные частицы вещества (атомы) не изменяются, а только соединяются между собой в иных сочетаниях, так что их суммарная масса остается постоянной.
1.2. Законы постоянства состава и кратных отношений
Рихтер (1791г.), Пруст (1799г.) и Дальтон (1799 и 1802гг.) сформулировали основные законы химической стехиометрии (экспериментальное подтверждение получено путем измерения массы веществ, вступающих в химические реакции):
отношение массы элементов или масс группы элементов, входящих в состав химического соединения, всегда постоянно;
если два элемента образуют несколько химических соединений, то массы этих элементов в соединениях, как правило, относятся как небольшие целые числа.
Используя атомистические представления, Дальтон (1803 г.) раскрыл смысл этих законов и окончательно ввел представление об атоме в химическую науку. Согласно Дальтону, атомам каждого элемента можно приписать некоторую относительную массу. На основе обычных стехиометрических закономерностей можно произвольно считать атомный «вес» одного из элементов равным единице (Дальтон выбрал в качестве такого элемента водород), а затем найти атомные «веса» других элементов.
1.3. Газовые законы. Закон Авогадро.
Химические уравнения
В 1805 г. Гей-Люссак и Гумбольдт экспериментально установили, что объемы газов, вступающих в реакцию, относятся как целые числа, например
1 объем кислорода-Ь2 объема водорода=2 объема водяного пара
1 объем хлора+1 объем водорода=2 объема хлороводорода
1 объем азота+ 3 объема водорода=2 объема аммиака
Атомистические представления
13
Столь простой эмпирический закон теоретически очень просто объяснить, если предположить, что равные объемы газов содержат (при одинаковых давлении и температуре) равное количество реагирующих частиц-молекул. Это утверждение было выдвинуто Авогадро в 1811 г. как гипотеза (известно сейчас как закон Авогадро). Авогадро считал, что частицы, участвующие в рассмотренных выше реакциях газообразных элементов, могут включать несколько (группу) атомов, т. е. представляют собой молекулы. Гипотеза Авогадро имела выдающееся значение для дальнейшего развития химической науки, в частности, потому, что на ее основе стало возможным составлять уравнения химических реакций.
Запишем уравнение реакции образования воды с учетом гипотезы Авогадро
1ОХ 2Ну = 2HyO0sx
где х^2. Относительно у можно сделать какие-либо выводы, лишь если привлечь к рассмотрению другие газовые реакции с участием водорода, например
1Нр 4" 1С1г = 2Н0 5уС10>5г
ИЛИ
ЗН^ -J- INa, ==
Получается, что y—z=w^2. Однако неизвестны реакции, в которых х>2 и г/>2. Следовательно, х—у=2, и формула воды запишется так: НгО. Прямое доказательство числа атомов в молекуле можно получить из данных по теплоемкости газов (гл. 2).
1.4. Идеальные газы. Уравнение состояния.
Экспериментальные методы определения молекулярной массы
Для химических реакций в газовой фазе закон Гей-Люссака точно выполняется только в том случае, если реакция происходит при низком давлении и относительно высокой температуре. Соотношения, разбираемые ниже, также относятся к газам, находящимся при низком давлении и повышенной температуре, т. е. в состоянии, близком к идеальному.
Основное уравнение состояния идеального газа предложено Клапейроном:
= C (1)
где р — давление, v— объем, Т — абсолютная температура, С — постоянная (для некоторого определенного количества данного газа). Уравнение состояния выведено из эмпирических закономерностей.
Закон Бойля-Мариотта (для 7’=const): давление обратно пропорционально объему (графически зависимость давления от объема при постоянной температуре изображается гиперболой; Pv = const.
14
Строение материи
Закон Гей-Люссака (для р = const): v = °0 ( 1 + 273,15 где t — температура в градусах Цельсия, v0 — объем газа при температуре О °C. Принимая во внимание, что абсолютная температура (в кельвинах) T = To-\-tt где Т = 273,15 К принята за нуль в шкале Цельсия, получим
= -12- = const
Т 273,15 То 1
В соответствии с законом Авогадро в объеме любого газа при одинаковых температуре и давлении содержится одинаковое число молекул, поэтому
C = kN (2)
где N— число молекул в объеме и.
Сколько же молекул соответствует единице количества вещества, вступающего в химические реакции? Вначале это число Na определяли как число атомов, которое содержится в одном грамме водорода («грамм-атоме»), или число молекул в 2 г водорода («грамм-моле»), или просто моле*. Несколько позднее за Na принимали число молекул в 32 г кислорода (однако, как подробно изложено далее, кислород содержит в небольшом количестве изотопы ,7О и 18О, поэтому надо различать «изотопный атомную массу» и «химическую атомную массу»), В настоящее время принято, что 1 моль любого вещества содержит столько молекул, сколько атомов в 12 г изотопа углерода 12С (подробнее в разд. 4.4).
Истинное число молекул в одном моле вещества {число Авогадро) было установлено позднее различными методами [первое такое определение выполнено Лошмидтом (1865 г.)]:
Ул = (6,02252 ± 0,00009)-1023 моль'1
Насколько велико это число? Если 1 л воды, каждая молекула которой содержит изотопную метку, вылить в Мировой океан, то после полного перемешивания в 1 л морской воды будут находиться 20 000 меченых молекул воды.
Система массой m содержит N{ — nNA} молекул (где п — число молей; А4 = Адтм, где М — масса 1 моля вещества, тм — масса одной молекулы; п = т/М). Из уравнений (1) и (2) следует
* Количество вещества — одна из семи основных физических величин, на которых основана СИ. [Номенклатурные правила ИЮПАК по химии. Т. 1. Полутом 2. — М.: ВИНИТИ АН СССР, 1979, с. 295]. — Прим, перев.
Атомистические представления
15
Это — уравнение состояния идеального газа (уравнение Менделеева— Клапейрона). R = kNA— универсальная газовая постоянная. Величину k называют постоянной Больцмана. Числовые значения k n R меняются в зависимости от размерности переменных в уравнении (3) (см., например, табл. A.l, k~ = 1,381 «10-23 Дж/К).
Таблица А.1. Значения универсальной газовой постоянной в различных шкалах
Размерность Численное значение
л-атм-К-1-моль-1 мм. рт. ст.-см3-К-1-моль-1 эрг-К-1-моль'1 Дж-К_1-моль-‘ КГС-М-К-1-МОЛЬ-1 кал-К-1-моль-1 0,08205 62396 8,314- 10’ 8,314 0,8477 1,987
С помощью уравнения (3) можно определить массу, соответствующую I молю различных веществ. Сделаем некоторые преобразования с уравнением состояния (3). Пусть мольный объем К=-^-=-^- (р — плотность), тогда pV~RT. Мольный объем идеального газа одинаков для всех газов и равен 22,415 л/моль.
Введем в уравнение (3) молярную концентрацию c = nlv, тогда p = cRT. При экспериментальном определении молекулярной массы (массы 1 моля) пользуются уравнением (3): обеспечивают постоянство двух или трех параметров, входящих в это уравнение, и измеряют остальные. Определение выполняют по методам Дюма (Г, р и v постоянны, m— измеряемый параметр), Гей-Люссака — Гофмана (Т и m постоянны, р и v— измеряемые параметры) и Мейера (Г, р и m постоянны, и — измеряемый параметр).
Метод Мейера (рис. А 1). Точную навеску исследуемого вещества помещают в разогретую испарительную трубку. Вещество испаряется, переходит в газообразную форму и вытесняет из трубки горячий воздух, равный по объему образовавшемуся пару. Количество вытесненного воздуха измеряется в пробирке (с делениями) с водяным затвором. Таким образом можно сразу найти объем газа, вытесненного из разогретой трубки в результате испарения исследуемого вещества, при комнатной температуре.
Пример. Исследуется вещество, формула которого определена элементным анализом, например (СгНЮ)*. Таким образом, М=44х, т. е. надо най-Ти х. Такую задачу можно решить, используя метод Мейера, хотя определяе
16
Строение материи
мое вещество в газообразной форме ни в коей мере не является идеальным газом Если путем соответствующих измерений получено, например, значение М=88,2, то можно считать, что, несмотря иа малую точность метода, надежно найдена формула С4Н3О2.
Более точные определения молекулярной массы проводят при нескольких различных давлениях, а М находят путем экстраполяции к р=0.
Рис. А.1. Прибор для определения молекулярной массы (по Мейеру).
/ — трубка для испарения вещества, помещенная в толстостенную пробирку для обеспечения равномерного подогрева; 2 — градуированная пробирка, 3 —
1.5. Идеальные твердые тела
Идеальный газ можно представить себе как некоторое предельное состояние материи. Наряду с таким состоянием, пользуясь гипотезой об атомарной структуре вещества, необходимо рассмотреть противоположное предельное состояние материи, представление о котором позволяет сделать полезные, хотя и предварительные, выводы о строении вещества. В таком состоянии находятся так называемые «идеальные» твердые тела, аналогом которых можно с некоторым приближением считать кристаллы при очень низких температурах. Атомы (например, алмаза) или электрически заряженные ионы (например, хлорида натрия) в «идеальных» твер
дых телах расположены в упорядоченных решетках с определенной снимет-рией. Структура таких пространственных решеток может быть исследована рентгеновскими методами (разд. 6.4.1). Наиболее простые модели таких
исследуемое вещество. Структур МОЖНО ПОЛУЧИТЬ, раССМаТрИ-вая возможные способы образования плотнейшей сферической упаковки. На рис. А.2 показаны два типа (плотнейшей сферической упаковки с различной симметрией. Незаштрихованные кружки изображают сферы А нижнего слоя. На нем размещен слой сфер В (заштрихованные кружки), посаженные на «пустоты». Во втором слое можно заметить
два типа пустот: совпадающих с пустотами слоя А и не совпадающих с ними. При размещении третьего слоя сферы можно
уложить в пустоты над совпадающими пустотами первого и второго слоев. Тогда четвертый слой займет такое же положение, как первый,— такую структуру называют «плотнейшей кубической упаковкой» (рис. А.З). В другом случае получается «плотнейшая гексагональная упаковка» (рис. А.4). Для обоих типов
А томистические представления ___________,
Рис. А 2. Схема расположения сфер при образовании двух слоев плотнейшей упаковки.
рйС, А.З. Плотнейшая веская упаковка.
плотнейшей упаковки вокруг каждой сферы расположено 12 й седних шаров*.
На основе простых геометрических соображений и зная мольный объем вещества, для которого известен тип упаковки, можно сделать выводы о размерах частиц; например, простой расчет показывает, что при гексагональной упаковке пространство заполнено на 74%:
4л
r3NA = 0,74V
О
откуда радиус сферы равен
г = 0,56 у
Приияв мольный объем равным V=14,8 см3/моль, получим г=0,163 нм.
В то же время, считая известным радиус сферической частицы, можно приближенно определить число Авогадро ЛАд. Атомарная структура твердых тел может быть выявлена во всех подробностях из измерений рассеяния рентгеновских лучей и пучков электронов в веществе. Электронные
Рис А.4. плотнели [ексагональнаЯ Упаь°’‘
микроскопы позволяют визуально наблюдать структуру твердого тела. Между предельными состояниями идеального газа и идеального твердого вещей11
тела существует наиболее важное для химика реаль?ое состояние д Некоторые виды таких состояний, например жидкости8 «РеаЛьные суждаются более подробно несколько ниже.
„_ * Совпадающие пустоты слоев А и В называют октаздрическ ими, а* ге“аДаЮ1ЦИе ~ тетРаэДРическими; последовательное» слоев «плотней №лХ^НаЛЬп0И Упак°вкн»- 12121..., а «кубнческой»-1231231... [Коттон МнГЙ ДдаСгт СовРемениая неорганическая химия * 1- Пер. е англ.^>
ИР> 19о9]. — Прим, перед.
2—2027
18
Строение материи
2.
КИНЕТИЧЕСКАЯ ТЕОРИЯ МАТЕРИИ
Выше уже говорилось, что дискретная структура материи, понятие об атоме и молекуле лежат в основе научных представлений современной химии. Важнейшее свойство материи — движение — рассматривается кинетической теорией, развитой во второй половине XIX в. Клаузиусом, Максвеллом и Больцманом, главным образом кинетической теорией газов. Было постулировано, что элементарные частицы материи — атомы и молекулы— находятся в постоянном движении. Рассмотрим сначала поступательное движение молекул в идеальном газе, подчиняющееся законам классической механики.
Вследствие упругих соударений молекул газа между собой, а также о стенку сосуда они постоянно меняют скорость и направление движения. В соответствии с теоремой Максвелла в течение некоторого промежутка времени все молекулы иезависнмо от нх массы имеют кинетическую энергию, мало отличающуюся от среднего значения (закон равномерного распределения по энергиям). Суммарное воздействие всех молекул на стенку проявляется как давление газа.
Пусть молекула массой т, движущаяся со скоростью w перпендикулярно стенке, упруго ударится о стенку и отразится обратно. Стенка получит импульс 2inw (удвоенный первоначальный нмпульс, так как после удара о стенку направление движения меняется на обратное). В 1 смз газа, находящегося вблизи стенки, имеется Mi молекул, из которых '/« часть движется по направлению к стенке. (Мы полагаем ради простоты, что по */з всех молекул движется в направлении осей координат, причем в равиых количествах в прямом и обратном направлениях). Тогда каждую секунду о стенку ударяется (Ni/Q)-w молекул (т е. те молекулы, которые находятся от стенки на расстоянии ш-1с). Импульс этих молекул, переданный стенке, составляет
М
2mw —g— w (4)
Таким образом, полученный 1 см2 стенкн за 1 с импульс, т. е. давление р.
р = mmfl == -у- да2
(5)
где к.'2 — среднее значение квадрата скорости молекулы (более строгое доказательство можно найти в специальной литературе). Перейдем к любому объему v, Ni= (гМа)/и, так как МЛ = М1-V~Ninfv) и
nNд — nMw- . n.Mw2
P = ~^Tmw = (6)
Эти уравнения лежат в основе кинетической теории идеального газа. Из уравнений (6) и (3) получим _
1 — 2 Mw*
ЯТ=—= -к-
О о
т е кинетическая энергия молекул газа и температура связаны между собой соотношением
3 Мш2
2 RT= 2
ИЛИ
3 mui2
2 kT = ~2~
(7)
Кинетическая теория
19
Таким образом, температура газа—макроскопический параметр, соответствующий кинетической энергии молекул.
К^[см/с] = ]/^= 1,5840* (8)
Следует различать среднюю скорость w и корень из среднего квадрата скорости Vw2. Из уравнений (6) и (7) непосредственно следует закон Авогадро: при равенстве р, и и Т независимо от природы газа совпадает и п. Из теоремы Максвелла следует (см. ниже) гй = 0,92 V w2.
Как следует из уравнения (7), скорость движения молекулы, а следовательно, скорость диффузии и скорость истечения газов из узких отверстий зависят от молекулярной массы. Эта закономерность, между прочим, используется при разделении газовых смесей и изотопов.
Истинное распределение скоростей молекул газа можно установить теоретически и на основании опытных данных. Теоретический вывод закона распределения молекул по скоростям
Рис. А.5. Принципиальная схема опыта Штерна.
(распределение Максвелла) можно найти в учебниках по физической химии. Ниже кратко рассмотрен эмпирический метод установления максвелловского распределения путем экспериментального определения скорости движения частиц в газе (рис. А.5).
Этот метод был разработан Штерном на основе использования стробоскопического эффекта. С разогретой до высокой температуры посеребренной проволоки А в высоком вакууме испаряются атомы серебра. «Атомные лучи» проходят через щелн Bt и В2 и осаждаются на латунном барабане С Все устройство (щели и латунный барабан) приводятся во вращение вокруг оси — проволоки А (скорость —2000 об/мнн). Поэтому траектория атомов серебра относительно всего устройства изгибается, и в зависимости от своей скорости они попадают на различные участки барабана в области СС' (аналогично дрейфу на запад или восток воздушных потоков, направляющихся от полюсов Земли к экватору, — пассатов). Получаемый при этом «спектр» (распределение) скоростей можно измерить; Максвелл предложил аналитическую формулу для доли молекул dN, имеющих скорость в интервале ш + +dw:
dN f М as/2 / Mw2 \ ехр^-2^-^Л®
Для двух газов распределение молекул по скоростям при различных температурах представлено на рнс. А.6.
2*
Кинетическая теория
21
20
Строение материи
Поскольку Л1ш<2/2=£'кин (Емп—кинетическая энергия газа), то приведенное выше выражение можно записать в следующей приближенной форме:
ЛУ [ ЕКИН \
N ~ехД~ RT )
Следовательно, для доли молекул NE, имеющих (кинетическую или потенциальную) энергию >Е, справедливо
Не / _£_\
N ~ exp — RT )
Рис. А.6. Распределение молекул легкого (Н2) и тяжелого (Ог) газа по скоростям для двух различных температур (по Максвеллу — Больцману).
2.1. Молярная теплоемкость идеальных газов
Внутренняя энергия идеального газа U представляет собой сумму кинетической энергии отдельных молекул. Для одноатомного газа (например, инертного) кинетическая энергия определяется только поступательным движением. Поэтому для одной молекулы газа из уравнения (7) получим
ma)2 3RT 3 . гр ~=-2^ = -TkT
а на 1 моль газа
U = s/2RT
(8а)
Таким образом, внутренняя энергия идеального газа не зависит от объема. Изменение U при изменении температуры газа на 1 К для одного моля при постоянном объеме называют молярной теплоемкостью Cv.
Со=-%- R « 12,48 Дж/(К • моль)
У многоатомных молекул имеются и другие возможности накопления энергии — за счет вращения молекулы и колебаний атомов в молекуле.
Из кинетической теории газов следует, что закон равномерного распределения энергии относится к различным видам движения молекулы. Например, для описания поступательного движения необходимо знать, как изменились координаты молекулы в трехмерном пространстве; поэтому говорят, что поступательное движение молекулы имеет три степени свободы*. Аналогично вращение двухатомной молекулы имеет только две степени
Таблица А.2. Молярная теплоемкость С„ некоторых газов (25 °C, 10s Па)
с Газ п V ?’ 1 Дж-К-'-моль-1 Газ С°’ 1 3 Дж К-‘ моль-'
Одноатомные газы Трехатомные газы (линейные
Не 12,48 молекулы )
Аг 12,48 СО2 28,97
N2O 30,48
Двухатомные газы
Трехатомные газы (угловые
Нг 20,56 молекулы)
n2 20,81 Н2О 25,16
СО 20,91 H2S 25,71
О2 21,06 SO2 31,61
НС1 20,81 Другие многоатомные газы
NO 21,56 NH3 27,21
С12 25,46 СН4 27,47
свободы, так как поворот какого-нибудь тела однозначно определяется двумя заданными углами относительно выбранного направления. Число степеней свободы при колебательном движении в молекуле растет с увеличением числа входящих в ее состав атомов. В соответствии с законом равномерного распределения энергии в среднем на каждую степень свободы должно приходиться одинаковое количество энергии, которое, согласно Уравнению (8а), составляет RTf2 (на моль на степень свободы).
В табл. А.2 и А.З приведены молярные теплоемкости для молекул различной природы. Интересно отметить, что молекула водорода имеет настолько малый момент инерции, что при низких температурах не проявляются вращательные степени сво-
* Число степеней свободы определяется числом независимых перемен-ных, однозначно определяющих положение системы в пространстве (в данном случае молекулы) [Ландау Л. Д., Лифшиц Е. М. Механика. — М.: Нау-ка> 1965, с. 9].—Прим, перев.
22
Строение материи
Таблица А.З. Поступательные и вращательные степени свободы
Газы Степени свободы Ср (без учета колебаний)
поступательные вращательные
Одноатомные 3 0 3/гк
н2 3 0 или 2 3/2к-5/2к
Двухатомные 3 2 ’/2/г
Трехатомные и многоатомные (линейные молекулы) 3 2 Ь/2К
Трехатомные и многоатомные (угловые молекулы) 3 3 %/?
боды. Более полное понимание причин этого явления можно получить после изучения гл. 3.
Таким образом, становится ясным, что из данных о молярной теплоемкости газов можно сделать некоторые выводы о строении молекул. В гл. 3 мы познакомимся с важными квантовомеханическими уточнениями этих представлений.
2.2. Число столкновений и средняя длина свободного
пробега. Диффузия и теплопроводность
На основе кинетической теории газов можно получить важные характеристики процессов переноса в газах, эти характеристики имеют важное прикладное значение. Пусть Z — число столкновений молекулы с другими молекулами за 1 с в 1 см3, Л — средняя длина свободного пробега молекулы:
г— nN л — Z = /2 —no2ai
w V__________
2 j/Д пЛ/дла2
где а — диаметр молекулы (вывод соответствующих соотношений можно найти в учебниках физической химии). Можно видеть, что средняя длина свободного пробега при постоянной плотности не зависит от температуры. Для воздуха Z«s6-109 с_|; Л~ 1 • 10_5 см. Таким образом, для газа в нормальных условиях* число столкновений очень велико. Если при постоянной температуре давление снизится до 10~' Па, то длина свободного пробега увеличится до ~10 см. Приведенный выше цифровой материал показывает, что явления, связанные с диффузией молекул газа, протекают сравнительно медленно (перемешивание газов, промывка и высушивание газов, газовая хроматография).
Коэффициент внутреннего трения газов (вязкость) также связан с Z и Л; этот фактор надо учитывать при изучении протекания газов в трубопроводах. Можно показать, что «коэффициент внутреннего трения» идеального газа при постоянной температуре не зависит от давления. Физический смысл
* Нормальные условия: давление 101 325 Па, температура 273,15 К- — /Трим, перед.
Основы квантовой теории
23
этого вывода можно объяснить следующим образом- при повышении давления увеличивается число столкновений, связанное с внутренним трением обратно пропорциональной зависимостью (при уменьшении числа столкновений молекул параллельных «слоев» газа увеличивается их взаимопроникновение, т. е. усиливается сцепление слоев), но в то же время и увеличивается число частиц (концентрация) в данном объеме, а внутреннее трение прямо пропорционально концентрации. Таким образом, оба эффекта компенсируются, в результате чего коэффициент внутреннего трения оказывается независимым от давления.
Теплопроводность идеальных газов связана с вязкостью, поэтому зависимости теплопроводности от температуры и давления аналогичны соответст вующим зависимостям для вязкости. В газовых смесях при перепаде температуры происходит незначительное расслаивание. Для этого явления, которое называют термодиффузией, характерно обогащение более легким газом той части объема, в котором поддерживается более высокая температура. Используя это явление, Клузиус предложил метод разделительных трубок и разработал соответствующую аппаратуру, с помощью которой оказалось возможным разделять изотопы элементов (гл. 4). Термодиффузия в жидкостях известна как эффект Соре.
2.3. Молярная теплоемкость твердых тел
Свойства идеальных твердых тел не должны зависеть от температуры в предположении, что частицы этого твердого тела не испытывают колебаний, не могут вращаться или перемещаться в пространстве. Иначе говоря, идеальное твердое тело соответствует состоянию реального кристалла вблизи температуры абсолютного нуля. Для алмаза уже при 50 К молярная теплоемкость близка к нулю. При более высокой температуре, в частности при комнатной, значения, наблюдаемые для молярной теплоемкости, вполне объясняются с помощью представлений о колебательных степенях свободы элементов решетки твердого тела. Таким образом, в первом приближении молярная теплоемкость всех твердых тел должна быть одинакова и составлять С„ = 3/? = 24,95 Дж-моль-'-К-'.
Уже в 1819 г. Дюлонг и Пти установили, что молярная теплоемкость многих твердых веществ равна ~26 Дж-моль-'-К-1. Впрочем, известно и немало веществ, теплоемкость которых существенно отклоняется от указанного значения, что можно объяснить только с привлечением квантовой механики (гл. 3). Изменение теплоемкости происходит также вблизи температур перехода вещества из одной модификации в другую (например, вблизи точки Кюри железа 1045 К, когда ферромагнитное железо переходит в парамагнитное) .
3. ОСНОВЫ КВАНТОВОЙ ТЕОРИИ
С самого начала бурного развития атомной физики, т. е. с конца прошлого столетия, многое указывало на то, что атомы, из которых построена материя, в свою очередь также имеют Дискретную структуру и состоят из «элементарных частиц». Большую роль при этом сыграло открытие дискретной природы электричества и доказательство существования свободных электронов. Уже Гельмгольц, основываясь на законах электролиза Фарадея, высказал предположение о том, что частицы обладает зарядом, кратным некоторому «элементарному заряду», электрон был первой элементарной электрически заряженной частицей, для которой определены заряд и масса, а также ис-
24
Строение материи_____________________
следованье некоторые магнитные и электрические свойства. Развитие методов ядерной физики и первые исследования взаимодействия радиоактивного излучения и вещества (гл. 4) способствовали созданию модели атома в виде положительно заряженного атомного ядра сравнительно большой массы и электронов вокруг него (Резерфорд, 1911 г.}. Более полное понимание строения и свойств атома, а также «частиц», входящих в его состав, стало возможным только с позиций квантовой механики. Наиболее важные в химии представления о строении атомного ядра и свойствах «элементарных частиц» даны в гл. 4.
3.1. Классическая физика и атомарная структура
материи
В первых двух главах дискретная структура материи обсуждалась с привлечением «классической» физики, которая на исходе прошлого века составляла фундамент теоретической химии. Однако на пороге следующего столетия произошел грандиозный переворот в основах физики, обусловленный появлением квантовой теории и теории относительности. Эти события повлияли и на развитие химии, хотя и получили должную оценку только в последнее время, после создания квантовой химии. Ушло время, когда химику достаточно было иметь общее представление о строении атома, ограничивая себя моделью Бора. В гл. 2 были сделаны два предположения, которые, как теперь известно, принципиально неверны:
движение частиц, из которых состоит материя, подчиняется законам классической механики,
изменение энергии этих частиц происходит непрерывно.
Замена этих положений на правильные, подтвержденные экспериментом было одним из первых шагов на пути создания современной «неклассической» физики.
3.2. Квант действия. Квантовая теория и теплоемкость
В 1900 г. при исследовании теплового излучения тел Планк установил некоторые закономерности, постулируя, что энергия дискретна. Вскоре после этого на основе анализа отклонений удельной теплоемкости, в частности для твердых тел, от правила Дюлонга — Пти (гл. 2) Эйнштейн пришел к аналогичным выводам (1907 г.). Так, при понижении температуры тела его молярная теплоемкость начинает уменьшаться. Такое изменение теплоемкости может происходить при сравнительно высокой температуре, например для твердых тел, характеризующихся сильным взаимодействием между частицами, располо-женными в узлах кристаллической решетки, т. е. имеющими до- статочно большую частоту колебаний v. Таким образом, только> I
Основы квантовой теории__________________25
в том случае, когда v/T достаточно малая величина, кинетическая теория приводит к правильным выводам. Установлено простое соотношение, связывающее температуру Т и частоту колебаний -v:
hv - kT (9)
где h—константа (имеет размерность энергия-время). Экспериментально найдено, что при температуре ~1000 К колебательные степени свободы осуществляются при частотах ~ 1014 с-1, т. е.
10-23- 103 _ 1П_31 п h ~ ~ 10 Дж- С
Таким образом, частота колебаний определяет «порцию энергии» hy^kT, которую может принимать частица, находящаяся в узле кристаллической решетки («осциллятор»), на каждую степень свободы и которую она принимает при нагревании. Эти порции энергии названы квантами, a h —постоянной Планка. С помощью введенных понятий можно объяснить температурную зависимость молярной теплоемкости. Введем характеристическую температуру 0, соответствующую температуре «пробуждения» колебательной степени свободы:
-?- = 0 k
«Собственные колебания» осциллятора (частота vo) можно при соответствующих условиях определить спектроскопическими методами. Реальные соотношения в отдельных деталях несколько сложнее, так как собственные колебания решетки происходят не с одной частотой, а в более или менее широком диапазоне частот (Дебай, 1912 г.). Однако <в целом квантовая теория молярной теплоемкости блестяще подтверждает квантовые представления Планка.
3.3. Фотоэлектрический эффект. Фотоны
Фотоэлектрический эффект (Гальвакс, Столетов, 1888 г.) также связан с квантовыми явлениями. Облучение некоторых металлов светом в коротковолновом диапазоне освобождает элементарные частицы, которые можно идентифицировать как электроны. Оказывается, что кинетическая энергия освободившегося электрона зависит не от интенсивности излучения, а от частоты света, а именно
то2 , .
- = hv-A
где А — работа выхода электрона, т. е. количество энергии, необходимое для того, чтобы разорвать связь электрона с кри-
26
Строение материи
сталлической решеткой металла. Как показал в 1905 г. Эйнштейн, подобно тепловой энергии свет поглощается и излучается только квантами с энергией hv. Скорость электрона v можно определить, измерив потенциал V при и—>-0, т. е. при mv2/2 = = eV. Тогда, зная заряд электрона е (см. ниже), можно найти hz
h--= (6,62559 ± 0,00016). 10-34 Дж-с
В соответствии с уравнением Эйнштейна световые кванты, или-фотоны, характеризуются определенной массой:
Е = hv = тс2
Существование инерционной массы света (давление света) и тяжелой массы (отклонение света при прохождении вблизи Солнца) доказано экспериментально.
В этом и предыдущем разделах для энергии использовались, различные размерности. Суммируя вышесказанное, энергия одного кванта выражается следующими соотношениями:
hv — kT = v2 = eV
Приведем некоторые постоянные, которые могут понадобиться при дальнейшем изложении основ квантовой химии:
k= 1,38-10'23 Дж-К-1 /и=9,1-10'31 кг
е =1,60-10"13 Кл 1 эВ= 1,60210-10~19 Дж
3.4. Электроны
Электроны в «связанной форме» являются частицами, поведение которых в значительной мере определяет химические-свойства вещества. Говорят даже, что химия — это физика «электронных оболочек». При исследовании именно этих элементарных частиц был установлен так называемый «корпускулярно-волновой дуализм» материи. Рассмотрим сначала некоторые свойства электронов, в которых проявляется их корпускулярная природа. Прежде всего отметим, что можно определить-заряд и массу электрона; интересны в этом отношении и методы получения электронов. К последним относятся термоэмиссия (при высокой температуре электроны сравнительно легко покидают решетку некоторых металлов, в особенности щелочных) и ударная ионизация.
Под ионизацией понимают образование электрически заряженных частиц (электронов и ионов) из нейтральных частиц. Если энергия столкновения таких нейтральных частиц между собой достаточно велика, то от них могут оторваться электроны. Такая термическая ионизация происходит, например, при электрическом разряде в газах при пониженном давлении. В катодной трубке образовавшиеся в результате термоэмиссии электроны ускоряются электрическим полем настолько, что становится возможной ударная иониза
Основы квантовой теории
27
ция. Минимальная энергия, необходимая для ионизации, и кинетическая энергия частицы связаны соотношением
При достаточно высоком напряжении и давлении ~10-6 атм появляются •«катодные лучи», которые после выхода из трубки через алюминиевое окошко могут быть исследованы (вне трубки). В основе простого метода определения Отношения е/m лежит указанное выше соотношение. При этом и определяется методом, с которым мы познакомились в гл. 2.
Другие методы определения отношения elm основаны на отклонении электронов в электрическом и магнитном полях.
Зная ejm и заряд электрона, можно вычислить массу электрона. Прямое определение заряда электрона впервые было выполнено Милликеном, который измерял силу, воздействующую в электрическом поле на парящую в пространстве между электродами заряженную капельку масла. Сила, действующая на одинаковые капельки, зависела от заряда капельки; отношение сил для разных капелек выражалось целыми числами, т. е. было кратным элементарному заряду. Согласно последним данным, заряд и масса электрона равны
е = (1,60210 нь 0,00007) • 10~19 Кл
(9,1091 ± 0,004)-10'31 кг
Пользуясь «классическими» закономерностями для процессов столкновений частиц, можно вычислить «классический» радиус электрона: гл 10-13 см.
Выше уже говорилось, что свет, который в классической физике рассматривается с позиций волновой механики, проявляет и корпускулярные свойства. В то же время, можно показать, что электроны также обладают волновыми свойствами. Так, Дэвиссон и Джермер (1927 г.) установили, что электроны рассеиваются на кристаллической решетке подобно рентгеновским лучам (разд. 6.4.1). Еще до этого де Бройль (1925 г.) обобщил уравнение Эйнштейна
V 1
тс=й • — = й • — (так как с — Kv)
в том смысле, что для каждой элементарной частицы с массой m и скоростью v справедливо соотношение
й mcv = Р — ~
Это фундаментальное соотношение — формула де Бройля — сопоставляет каждой частице некоторую «материальную волну». чем меньше масса и скорость частицы, тем больше длина вол
28
Строение материи
ны. Можно рассчитать, например, длину волны электрона для некоторого ускоряющего напряжения:
У m У 2mU
Если напряжение измерено в вольтах, то
Таким образом, длина волны де Бройля в области напряжений 102—103 В имеет порядок ~ 0,1 нм, т. е. близкий длине волны рентгеновского излучения. Волновые свойства других элементарных частиц также получили экспериментальное подтверждение.
Из классической волновой механики известно, что волновой процесс (пакет волн протяженностью х) можно охарактеризовать длиной волны X только в том случае, если протяженность пакета х стремится к бесконечности. Этот факт отражается так называемым «соотношением неопределенностей»
Дх •Д£ = 1 ( k = ~ = J-')
Воспользовавшись формулой де Бройля, можно получить соотношение неопределенностей Гейзенберга для импульса р и координаты частицы х
&x-kp = h
Оно дает нам границы возможной точности измерения параметров элементарной частицы, поскольку они интерпретируются в рамках классической физики.
3.5. Квантовая теория и классическая физика
Рассмотренные выше теоретические представления и экспериментальные данные убедительно свидетельствуют о том, что с помощью классической физики нельзя полностью интерпретировать свойства элементарных частиц. Раздельное рассмотрение «волны» и «частицы» не позволяет проникнуть в сущность микромира. Электрон, например, — это и не частица и не волна, тем не менее это вполне реальный объект, во многом определяющий свойства химических веществ. Заслугой Гейзенберга, Борна, Шрёдингера и Дирака является то, что они заложили основы такой «механики», которая правильно описывает свойства электронов и позволяет более глубоко понять сущность материи. Чтобы более ясно представить себе основы квантовой механики, необходимо отойти от привычных понятий, которые от долгого употребления стали слишком «наглядными». Физика
Основы квантовой теории
2»
микромира качественно отличается от физики макромира. Однако между ними нет четкой границы, поскольку, согласно принципу соответствия, для всех процессов, в которых ft значительно меньше аналогичных коэффициентов в уравнениях энергии (например, kT/v), закономерности классической физики совпадают с квантовомеханическими. В связи с этим классическая физика является предельным случаем более общей неклассической физики.
3.6. Некоторые основные принципы квантовой механики
Ниже приводятся некоторые основные представления квантовой механики, с которыми необходимо познакомиться, чтобы в дальнейшем понять ту роль, которую играют в химических свойствах веществ электроны, «связанные» с атомами, молекулами и кристаллическими решетками веществ.
Два основных положения являются как бы мостом в классическую физику, так как имеют всеобщее значение:
Во-первых, закон сохранения энергии, а также законы сохранения массы, заряда, импульса и вращательного момента действительны и в квантовой механике. Например, полная энергия равна
£ = £кин -]-(/ = const
где £Кин — кинетическая энергия ЕКИн=шп2/2=р2/2т, (/ — потенциальная энергия. Тогда закон сохранения энергии запишется так:
£-----(£-17) = О
2пг 4 7
Это выражение после некоторых преобразований в дальнейшем примет вид так называемого уравнения Шрёдингера.
Во-вторых, хотя движение электронов происходит не по законам классической физики, можно задать функцию, квадрат которой определяет вероятность нахождения электрона в точке С координатами q. Эту функцию f(q) обозначают как ^-функцию (в нашем изложении зависимость от времени опущена). Для химических систем эта функция выражается с помощью тригонометрических, экспоненциальных и сферических функций.
Рассмотрим основные характерные особенности квантовой механики. Чем раньше читатель освоит эти основные понятия, тем ему легче будет в дальнейшем применять на практике достижения квантовой химии.
1) Известным в классической механике параметрам—динамическим переменным (например, импульсу или вращательному моменту), которые могут быть точно измерены, — в квантовой
30
Строение материи
механике соответствуют операторы, т. е. некоторые математические операции (например, дифференцирование), которые следует производить с волновой функцией. В гл. 5 введен соответствующий оператор импульса и с его помощью из закона сохранения энергии получено уравнение Шрёдингера.
Оператор импульса
А д i dq означает следующие операции: дифференцирование по координате, умножение на й = й/2л и деление на мнимую единицу
Более подробно об операторах можно прочесть в учебниках по квантовой механике. Здесь заметим лишь, что применение операторов в границах используемых приближенных методов дает правильные, согласующиеся с экспериментом результаты.
2) Знание ф-функции само по себе недостаточно для описания состояния элементарной частицы. Последняя характеризуется еще одним параметром, не имеющим аналогии в классической физике, —так называемым спиновым вращательным моментом, который определяет особые свойства элементарной частицы, открытые Гаудсмитом и Уленбеком (1925 г.) и подробнее рассмотренные в гл. 5. Эти ученые установили, что спиновая функция о, соответствующая волновой функции ф, может быть записана в ад- и p-формах. Для а проекция механического момента вращения частицы на ось вращения равна +’/2^, а для Р она равна —’/гй. Функция состояния системы определяется как Чг=фст. [Функции перемножаются при условии независимости поступательного движения частицы и спина (отсутствует «спинорбитальное взаимодействие»).] (Подробнее об умножении вероятностных функций см. также разд. 6.2.1.)
3) В многоэлектронных системах не могут существовать электроны, находящиеся в одинаковом состоянии,— по крайней мере ф и ст должны различаться (принцип Паули). Этот принцип является выражением одной из основополагающих закономерностей химической науки, связанной с математической структурой Т-функции. Эта функция для данной элементарной частицы может быть либо «симметричной», либо «антисимметричной».
Симметричная функция (например, с координатами ла и лаг) при изменении знака переменных не меняет знака, антисимметричная в таком же случае меняет знак на обратный, например, симметричная функция •sin Xi cos х2 + cos Xi sin х2 и антисимметричная sin Xi cos х2—cos Xi sin x2.
Легко видеть, что принцип Паули идентичен требованию, чтобы функция электронов Чг=фст была антисимметричной (нечетной) . Действительно, для системы из двух электронов при
Элементарные частицы и атомное ядро 31
одинаковых ф и о Т-функция становится равной нулю, т. е. вероятность нахождения двух электронов в одинаковом состоянии равна нулю. Этого как раз и требует принцип Паули.
Рассмотрим в качестве примера состояние двух электронов с одинаковыми о-функциями и разными ф-функциями. Тогда получится следующая антисимметричная функция Чг:
= Фх(1) а(1) ф2(2) а(2)—ф,(2) а(2) фй(1) а(1) =
= а(1) а(2) №(1) Ш-Ф^) Ф2(1) I
(можно убедиться, что эта функция антисимметрична Чгл = = озфл) •
Подставим теперь ф1=ф2, что не разрешается принципом Паули: 4^ = 0. Соответствующая симметричная функция (можно получить ее, если заменить в Т знак — на +) при ф1 = =ф2 не обращается в нуль, что также запрещено принципом Паули. Такое же положение имеет место при неодинаковых функциях о и одинаковых ф (проверьте это самостоятельно).
Все эти соотношения играют важную роль в теории химической связи.
4. ЭЛЕМЕНТАРНЫЕ ЧАСТИЦЫ И АТОМНОЕ ЯДРО
Выше рассмотрены свойства таких важных элементарных частиц, как электроны. Остановимся вкратце на характеристиках некоторых других элементарных частиц, особенно тех, представление о которых необходимо для понимания строения атомного ядра. Попутно коснемся и некоторых закономерностей в строении атомного ядра, имеющих большое значение в химии. Помещаемый здесь материал можно рассматривать лишь как краткий очерк по ядерной физике и ядерной химии. С основной аппаратурой, устройствами, методами анализа, применяемыми в ядерной физике и химии, можно ознакомиться по специальной литературе (ускорители, реакторы, масс-спектрографы, камеры Вильсона и пузырьковые камеры и т. д.).
Элементарные частицы
В теории и методах современной химии знание свойств элементарных частиц имеет большое значение. Ниже приведен краткий обзор известных в настоящее время элементарных ча-
Прежде всего познакомимся с важнейшими характеристиками элементарных частиц.
Масса покоя то. Для движущихся со скоростью v т^то1У 1—J-
32
Строение материи
Элементарные частицы и атомное ядро
33
Особую роль это уравнение играет в ядерной технике, в частности при рассмотрении процессов, происходящих в мощных ускорителях, m измеряют в атомных единицах массы ('/ы массы изотопа углерода 12С).
Заряд. Указывают обычно относительный заряд частицы, приняв за единицу заряд электрона е. Все элементарные частицы имеют заряд 1, —1 или 0. Каждой частице с полуцелым спином соответствует античастица с такой же массой и противоположным знаком заряда. (О понятии «античастица» см. ниже.)
Время жизни. Для нестабильных частиц составляет 10 6— 10-15 с.
Спин. Частицы с полуцелым спином называются фермионами, а с целым спином — бозонами.
Зарядовая независимость, изотопический спин, гиперонный заряд, странность — свойства элементарных частиц, ставшие известными в последнее время, — связаны с взаимодействием между частицами (рассмотрение этих вопросов выходит за рамки этой книги).
Во всех реакциях между частицами, в том числе и при распаде частиц, обязательно соблюдаются законы сохранения (энергии, заряда, массы, импульса, вращательного момента). Существует правило, что фермионы либо образуются парами при поглощении излучения с высокой энергией, либо такая пара аннигилирует с излучением энергии. Поскольку для незаряженных фермионов, например нейтронов, доказана возможность их аннигиляции, таким частицам также соответствует античастица.
4.1.1. Некоторые важнейшие элементарные частицы
Лептоны и мезоны
а) Фотон у (разд, 3.3).
б) Электрон е~ (разд. 3.4).
в) Позитрон е+. Открыт Андерсоном (1932 г.) при исследовании космического излучения в камере Вильсона. Образуется в паре с электроном.
г) Нейтрино V. Существование нейтрино постулировано Паули исходя из закона сохранения вращательного момента при 0-распаде протона (см. ниже). Прямое доказательство было получено значительно позднее. Так как нейтрино имеет спин 7г, ему соответствует античастица.
д) Антинейтрино v*. Частица открыта также недавно (Райне и Коуэн, 1959 г.). Наблюдается при ядерных реакциях с высокими энергиями.
е) ц-Мезоны (ц~ и ц+). «Средние» по массе частицы (между барионами и электронами). Были предсказаны Юкава
(1935 г.) и позднее открыты в «жестком» излучении высокой энергии.
ж) л-Мезоны (яг, л+,л). Это—«кванты» ядерного поля подобно тому, как фотоны — кванты электромагнитного поля. В противоположность фотонам, образующимся при изменении электронного состояния системы, л-мезоны обнаруживаются при переходах из одного стационарного состояния ядра в другое. Изучение л-мезонов сильно затруднено по сравнению с ц-мезонами вследствие малого времени жизни и сильного взаимодействия. и л-Мезоны могут входить в состав атомов вместо электронов, образуя мезонные атомы (найдены при исследованиях в пузырьковой камере).
Барионы (тяжелые частицы)
а) Протон р+. Положительный ион водорода впервые в качестве свободной частицы стал объектом изучения ядерной физики в лучах газоразрядной трубки. При так называемом 0-распаде протона (когда образуются е^ или е+) возникает нейтрино п:
р+ --->- п + е+ + v
что подтверждает закон сохранения вращательного момента, так как р+, п и е+ имеют спины, равные V2 (спины могут быть только параллельными или антипараллельными). Масса покоя протона 1,0078 а. е. м.
б) Нейтрон п. О его существовании догадывался еще Резерфорд (1920 г.). Однако, поскольку нейтральную частицу труднее обнаружить, например с помощью камеры Вильсона, только в 1930 г. Боте и Беккер открыли новое излучение, возникающее при взаимодействии а-лучей (разд. 4.3) с бериллием и способное отрывать от других атомов протоны. В 1932 г. Чедвик установил, что это излучение представляет собой поток нейтронов. Эти нейтральные частицы сыграли большую роль в развитии ядерной физики, так как, имея существенную массу при отсутствии заряда, они легко проникают в другие атомы и вызывают многие ядерные реакции. Нейтрон нестабилен и распадается на р+ и е~. Поразительное открытие того, что у незаряженного нейтрона имеется магнитный момент, привело к развитию теории строения нуклонов (нейтроны и протоны объединяют понятием «нуклоны») в новом направлении. Масса покоя нейтрона 1,0087 а.е. м.
в) Антипротон р~. Открыт Сегре в космическом излучении, гииД(6 2 °io9aPgMeH ПРИ °^стРеле яДеР протонами высокой энер-
Р+~Уядро --> ядро* + р+ 4- р~ (ядро*—возбужденное ядро)
пн Г) Антинейтрон п*. Открыт в 1956 г., при реакции аннигиляции п-^п* выделяется энергия 2 ГэВ.
3—2027
34
Строение материи
Элементарные частицы и атомное ядро
35
д) Гипероны и К-мезоны («сверхтяжелые» частицы). Открыты в 1947 г. в космическом излучении и при ядерных реакциях, сопровождающихся излучением высокой энергии. Представляют большой интерес при исследованиях в области теоретической ядерной физики.
4.2. Атомные ядра
Атомные ядра включают N нейтронов и Z протонов. Параметры и свойства атомных ядер влияют на протекание химических процессов, так как масса, заряд, энергия связи, устойчивость и ядерный спин ядра в значительной мере определяют свойства атома в целом. Отметим прежде всего, что с помощью масс-спектроскопических методов можно обнаружить разность между массой ядра и массой, найденной простым суммированием масс составляющих его нуклонов, — так называемый дефект массы Ат. Энергетический эквивалент дефекта массы представляет собой энергию связи нуклонов в ядре. Дт = = 1,0078 Z+1,0087 N —т. Для ядра гелия Ат = 0,03 а. е. м., что соответствует 27,9 МэВ. Энергия связи ядра химического элемента приблизительно линейно зависит от массового числа A = Z-\-N. Если построить график зависимости средней энергии связи на один нуклон от массового числа, наблюдается максимум при средних значениях массового числа. Таким образом, ядра со средним массовым числом более устойчивы, чем тяжелые или легкие. Следует отметить, что тяжелые ядра богаче нейтронами, чем легкие. При Z>84 уже не существует стабильных ядер. Различают следующие виды ядер: изотопы (равные Z, неравные N), изотоны (неравные Z, равные N), изобары (неравные Z, неравные N, равные А), изомеры (равные Z и N, однако внутренняя энергия неодинакова). Для нечетных А имеется лишь одно стабильное ядро, а для четных — несколько стабильных ядер изобаров (правило изобар Маттауха)
О ядерных силах известно, что они распространяются на рас стояние ~5-10'13 см. Плотность нуклонов во всех ядрах примерно одинакова; между величиной А и радиусом ядра соблюдается зависимость
г = 1,4-10-1’(см)
Нуклоны распределены в ядре относительно равномерно; и' расположение близко к плотнейшей кубической упаковке (разд. 1.5), подобно молекулам в капле жидкости. Другая аналогия основана на равномерном увеличении энергии связи по мере увеличения числа частиц (капельная модель ядра-" Бор, Гамов). Ядра с четными Z и N наиболее часто встречают' ся и более устойчивы, причем особенно выделяются ядра с чй«'
лами 2, 8, 14, 20, 28, 50, 82, 126 (эти числа поэтому называются «магическими»), Йенсен и Гопперт-Майер с помощью квантовомеханических методов показали, что для атомного ядра применим принцип, аналогичный правилу «заполненной оболочки» инертных газов (гл. 5), в соответствии с которым наиболее стабильными являются ядра с определенным набором протонов и нейтронов. Исследования в этой области — изучение «сильных взаимодействий» между частицами в ядре и роль мезонов как квантов ядерного поля — продолжаются.
Наряду с энергией связи и стабильностью ядер большое значение в химических процессах имеют также магнитный и электрический моменты ядра. Спин ядра складывается из спинов нуклонов ('/г^) таким образом, что составляет четное или нечетное число, кратное исходному спину */2й. Поэтому спин ядра может для разных элементов меняться от 0 до 4,5. Он проявляется в сверхтонкой структуре атомных спектров и является основой метода ядерного магнитного резонанса. Так называемый квадрупольный момент ядра Q отражает асимметрию распределения заряда в ядре. Он особенно важен при взаимодействии между неполярными молекулами (например, молекулами СО2 в газовой фазе). Q дает также информацию об отклонении ядра от сферической формы.
4.3. Радиоактивность. Ядерные реакции
Уже при первых исследованиях стабильности атомных ядер и ядерного распада на объектах, проявляющих естественную радиоактивность (Беккерель, 1896 г.; М. и П. Кюри, 1898 г.), получены существенные результаты. При распаде радиоактивных веществ наблюдаются следующие виды излучения:
а-излучение — ядра гелия (идентифицированы путем измерения отношения elm и химическими методами);
Р-излучение — быстрые электроны;
у-излучение — электромагнитное излучение с длиной волны меньшей, чем у рентгеновского (не отклоняется в электрическом и магнитном поле).
В результате испускания атомными ядрами а-лучей массовое число А уменьшается на 4 а.е. м., а заряд — на 2; при иссекании р-лучей Z увеличивается на 1, а массовое число не меняется (правила смещения Фаянса и Содди). Кинетика (скорость реакции) ядерного распада подчиняется уравнению пер-кюп П01РЯдка- Активность радиоактивных веществ выражают в вкп 1 Кн — это такое количество радиоактивного вещества, ___тором_за 1 с происходит 3,7-1010 расп*.
Керелях Гт?и?1И11'ах СИ активность радиоактивных веществ выражают в бек-нздат Ю7К1К'* * Ки = 3,7-1010 Бк. [Справочник физических величин —М.: Атом-
’ 1У/ч|. — Прим, перев.
3*
36
Строение материи
Элементарные частицы и атомное ядро
37
В 1919 г. Резерфорд при бомбардировке ядер азота а-лу. чами открыл явление искусственной радиоактивности (в верх-нем индексе A = N-[-Z, в нижнем Z):
14,N ф %Не--> i’8O + \Н
Эту реакцию можно записать в упрощенном виде (указать исходное ядро и конечный продукт с соответствующими А и Z, а в скобках между ними — последовательно бомбардирующую частицу и частицу — продукт ядерной реакции):
и7Ы (а,р)178О
Наибольший интерес представляют ядерные реакции с участием следующих частиц (d— дейтрон):
(п, у), (п, 2п), (п, р), (п, а)
(d, р), (d, п), (d, а), (d, 2л)
(р,п), (p,d), (р, а)
(а, п), (а, р)
(?,п)
Тяжелые ядра при бомбардировке медленными нейтронами могут распадаться на два больших осколка (ядерный распад, Ган и Штрассман, 1938 г.). При распаде 23592U находят ядра-осколки с зарядом Z от 30 до 63. Не будем останавливаться подробнее на теории и практике ядерного распада, которые рассматриваются в специальных руководствах, а также на реакциях синтеза ядер, например,
i3H + /Н----► 25Не
i2H + ?H ---<- /Не
при которых освобождается колоссальное количество энергии Химия искусственных трансурановых элементов, получаемых в результате ядерных реакций, хорошо описана в специальных разделах учебников неорганической химии.
4.4. Стабильные и нестабильные нуклиды*
Некоторые методы разделения изотопов были упомянута ранее; сведения о других можно найти в руководствах по физической химии.
Юри (1932 г.) на основе изучения спектров водорода сдела-11 вывод о том, что в природном водороде содержится изотоп с массой 2 а. е. м. (['Н] : [2Н] = 1 :7000). Этот изотоп получил на звание «дейтерий» (D), а его ядро — «дейтрон» (d). Большов
* Нуклид — общее название всех типов ядер; изотоп, например, являет ся одним из типов ядер какого-либо элемента.
различие в массе между Н и D облегчает их разделение. Н2О и ргО, например, можно сравнительно легко разделить с помощью фракционной перегонки или путем электролиза обычной воды (табл. А.4).
Таблица А.4. Свойства тяжелой (D2O) и легкой (Н2О) воды
Свойство d2o нго
Температура плавления, °C 3,82 0,00
Температура кипения, °C 101,42 100,0
Максимальная плотность, г/см3 1,1071 1,000
Температура максимальной плотности, °C Н,6 4,0
Вязкость (20°C), П-с 0,00126 0,001009
Первоначально для выражения атомных масс в химии была предложена и использовалась кислородная единица, равная Vie средней атомной массы кислорода естественного происхождения, который, как считалось, состоит практически из постоянной смеси изотопов О’6 (99,76%), ,7О (0,04%), 18О (0,2%). Однако масс-спектрометрические исследования показали, что состав природного кислорода зависит от происхождения пробы. Поэтому кислород не может быть принят в качестве основы для введения относительной единицы атомной массы. По международному соглашению с 1961 г. в качестве атомной единицы массы (а. е. м) принята масса изотопа углерода-12:
1 а.е.м. равна х/12 массы изотопа 12С.
Тогда для протонов, нейтронов и электронов были приняты следующие приближенные значения массовых чисел:
тп= 1,0087 а.е.м.; тр= 1,0078 а.е.м.; те= 0,0006 а.е.м.
Массовые числа можно определить с точностью до 10-7 а. е .м.
Определение атомных масс элементов имеет исключительно важное значение для всех разделов химической науки. Атомная масса—-это среднее значение относительных атомных масс изотопов элемента с учетом их процентного содержания в данном образце. При протекании химических реакций соотношение изотопов не меняется, поэтому атомная масса остается практически постоянной. Исключение составляет только свинец, который в различных соединениях имеет неодинаковый изотопный состав: это зависит от месторождения. Свинец из урансодержа-ших руд имеет атомную массу 206. В минералах, в которых сви-На\пяРа3овался ПРИ Распаде тория, атомная масса свинца рав-влп™ В наиболее распространенном минерале свинца — свин-
ом блеске PbS — атомная масса РЬ равна 207,21. Таким об
Электронные оболочки атомов 39
38 _____________Строение материи______________________
разом, свинец состоит из смеси изотопов с массовыми числами 204, 206, 207 и 208.
Для определения атомной массы элемента необходимо с высокой точностью провести аналитическое определение стехиометрического соотношения атомов элементов, входящих в состав тщательно очищенного вещества. Выдающихся результатов в этой области добились школы Ричардса (США) и Хо-нигшмидта (Германия). Публикации в этой области показывают, насколько тщательно и продуманно должны проводиться подобные исследования.
В настоящее время для точного определения атомной массы в первую очередь используются методы масс-спектрометрии. В масс-спектрометре газообразный ионизированный поток вещества попадает в электрическое и магнитное поля, расщепляется в зависимости от соотношения заряда и массы и регистрируется (см. руководства по физике). Расшифровка спектра дает также возможность установить относительное содержание изотопов. Для хлора, например, установлено следующее процентное содержание изотопов: 75% 35 *С1 и 25% 37С1. Отсюда атомная масса хлора
. _ 75-35 + 25-37 _ .
z'ci =----iooo------35’5 а-е'м-
В этом расчете использованы округленные величины; точное значение атомной массы хлора равно 35,453 а. е.м. Атомные массы элементов, рассчитанные по новой системе, лишь незначительно отличаются от принятых ранее. Например, атомная масса кислорода в настоящее время принята равной 15,9994 а. е.м. Только в трех случаях имеются существенные отличия:
Массовое число, а. е. м.
по кислородной
Элемент по углеродной шкале шкале
Бром 79,909 79,916
Хлор 35,453 35,457
Серебро 107,870 107,880
Новое базисное значение единицы массы привело к некоторому изменению числа Авогадро Na и связанных с ним величин, таких, как число Фарадея, универсальная газовая постоянная и молярный объем идеального газа. Если раньше число Авогадро определялось как число атомов в 16 г кислорода, то в настоящее время —в 15,9994 г кислорода, так как за базис принят изотоп углерода 12С. Согласно последним данным (сМ-также разд. 1.4), Na= (6,022045+0,000031) • 1023 моль-1.
Во многих аналитических и физико-химических методах ис-
следования применяются как радиоактивные, так и стабильные нуклиды. Здесь можно упомянуть лишь некоторые из них: активационный анализ (определение неактивного элемента в пробе путем превращения его в радиоактивный изотоп с последующим измерением активности), применение так называемых меченых атомов в химических и биохимических исследованиях, определение возраста геологических и биологических объектов с помощью изотопа 14С.
5. ЭЛЕКТРОННЫЕ ОБОЛОЧКИ АТОМОВ
Во многих учебниках химии понятия и термины вводятся на основе представлений теории строения атома Бора; такое положение затрудняет изучение основ квантовой химии. Поэтому в дальнейшем не применяются такие, например, термины, как круговые (эллиптические) орбиты электронов. В то же время представления об электронном облаке и электронных оболочках находят применение при квантовомеханическом описании строения атомов.
Рассмотрим сначала состояния одного электрона в центральном поле со сферической симметрией, иначе говоря, атом водорода в стационарном и возбужденном состоянии. Прежде всего сделаем краткий обзор экспериментальных данных, лежащих в основе теории, в особенности полученных методами атомной спектроскопии.
5-1- Стационарные состояния атома: экспериментальные данные
Наиболее важный экспериментальный материал об энергетических состояниях и электронных оболочках атомов получен при исследовании бомбардировки вещества электронами, а также из рентгеновских и оптических спектров.
5-1-1- Бомбардировка вещества электронами
Опыты Франка и Герца (1912 г.) наглядно показали, что, одобно частицам, колеблющимся в узлах кристаллической ре-етки твердых тел (разд. 2.3), атомы не могут принимать лю-е количество энергии. Если атомы вещества (например, ртути рост3°В°й $азе) подвергнуть бомбардировке электронами, ско-ряюь КОт°рых постепенно возрастает по мере увеличения уско-(рНсЩед°7?апРяжения> то можно снять вольт-амперную кривую (~5 ' °ЧеВИДН0’ что д0 определенного напряжения
электпо происходят упругие соударения между атомами и нейщр Нами и с Удлинением напряжения ток растет. При даль-
м увеличении напряжения ток внезапно падает. Затем при
Электронные оболочки атомов
41
40
Строение материи
увеличении напряжения подобная зависимость для тока периодически повторяется.
Из этого опыта можно сделать вывод, что электроны могут передавать энергию атомам при соударении до тех пор, пока их энергия не достигнет некоторого минимального значения. Это минимальное значение энергии соответствует разности энергий атома в различных состояниях.
5.1.2. Рентгеновские спектры
Наличие дискретных энергетических состояний атома объясняет также особенности рентгеновских спектров. Рентгеновские лучи возникают при воздействии катодных лучей на атомную решетку анода. Как установил Мозли (1913 г.), рентгеновские спектры для различных видов атомов имеют много общего. Они состоят из нескольких групп линий, расположенных на небольшом расстоянии друг от друга; с увеличением длины волны группы линий обозначают как К-, L-,
Рис. А.7. Зависимость силы тока от ....серии (рис. А.8). Объ-
фТаОнРкТиТерцТсР"и мнение строения рентгеновских
спектров предложено Косселем (1914 г.), который предположил, что существует закономерное соотношение между энергией электронов в атоме и их расстоянием от ядра. Рентгеновское излучение возникает в том случае, если ближайший к ядру электрон выбивается из атома, а электрон, находившийся на более далеком расстоянии от ядра, занимает его место. При
этом возникает излучение с частотой v, которое соответствует разности энергий этих двух электронных состояний. Очень важно то, что число рентгеновских серий значительно меньше суммарного числа электронов в атоме. Вероятнее всего, что в ато-
ме всегда имеется несколько электронов, находящихся в приблизительно одинаковом энергетическом состоянии. Поэтому и
Рис. А 9. Схема, позволяющая объяснить появление серий в рентгеновских спектрах.
возникло представление о том, что электроны в атоме распределены по «оболочкам», причем все электроны каждой такой оболочки имеют одну и ту же энергию. Чем меньше энергия электрона, тем больше вероятность найти его на более далеком расстоянии от ядра, т. е. тем «слабее» он связан с ядром. Происхождение рентгеновских серий можно легко понять из рис. А.9
Длина
Мозли
волны для каждой серии определяется уравнением
= cor st-(Z—s)
42
Строение материи
Электронные оболочки атомов
43
где/ — порядковый номер в периодической системе (число электронов в атоме), Л — длина волны, s — коэффициент экранирования. Согласно этому уравнению, по мере увеличения заряда ядра электроны должны более прочно связываться, хотя при этом необходимо учитывать экранирование ядра расположенными вблизи него электронами. С помощью уравнения Мозли оказалось возможным точно установить присутствие новых элементов. В последнее время рентгеновская спектроскопия успешно используется для исследования химической связи.
5.1.3. Оптические спектры
Наиболее полное представление об энергетическом состоянии атома дает атомная спектроскопия — прежде всего вследствие возможности с высокой точностью определять длину волны излучения.
а /? / it st, SG
ООО 500 400 000™
Рис. А.10. Спектр атомарного водорода по Бальмеру.
Первая важная закономерность в строении спектров атомарного водорода была установлена Бальмером (1885 г.). Он выделил «серию» линий (рис. А.10) и предложил формулу для соответствующей частоты излучения:
„ ( 1 1 \
V (22 п2 )
где Ry называется постоянной Ридберга (Ry = 3,2869• 101S Гц)-Позднее в спектре атомарного водорода наряду с серией Бальмера обнаружены серии в других областях длин волн (серии Лаймана, Пашена, Брэккетта, Пфунда). Частоту излучения для каждой серии можно рассчитать также по формуле Бальмера: ’=^(-у-у) (1°)
Если т=\ (серия Лаймана), 2 (серия Бальмера), 3 (серия Пашена), 4 (серия Брэккетта), 5 (серия Пфунда), соответствующие значения п находят следующим образом: n = mJr^ m+2, m+......
Непреходящая заслуга Бора (1913 г.) состоит в том, что оЯ сумел объяснить наблюдаемые закономерности наиболее про' стым (и правильным) способом. Если умножить уравнение (10)
Рис. А.11. Энергетические уровни и схема термов атома водорода.
на постоянную Планка h, то выражение в правой части уравнения можно представить в виде разности энергий, отвечающих двум различным состояниям атома («термам»). Эти состояния можно охарактеризовать с помощью квадратов целых h
чисел; если записать выражение Ку Для чисел х, равных
1- 2, 3, ..., то получится ряд возможных уровней энергии атома водорода (в электронвольтах):
х 12 3
Ry-^T 13,59 3,4 1,55 ...
На рис. А.11 схематически представлены энергетические Уровни (термы), соответствующие трем (из пяти известных) сериям, длины волн которых находятся в диапазоне 100— •ООО нм. По ординате слева за нуль принято основное состоя-иие атома водорода (с наибольшей энергией), а справа — минимальное значение энергии (при котором электрон еще связан в чюме), иными словами, — граница серии. Максимальная энер
44
Строение материи
Электронные оболочки атомов
45
гия связи (13,59 эВ) эквивалентна энергии ионизации атома водорода, которая может быть найдена и с помощью других методов.
Спектры более тяжелых атомов, вообще говоря, значительно сложнее, чем спектры водорода. Излучение (эмиссия) и поглощение (абсорбция) света обусловлены очень многими энергетическими переходами внешних, так называемых «световых» электронов.
Рис А 12. Спектр щелочного металла: три серии атома натрия.
Рассмотрим относительно простой спектр атома щелочного металла (три серии атома натрия представлены на рис. А.12). Следует иметь в виду, что показанные штрихами линии на самом деле в отдельных случаях представляют собой «дублеты» (двойные линии), как, например, известная D-линия натрия, соответствующая переходу из основного состояния «главной серии» в основное состояние «второй побочной серии». Здесь разрешены не все переходы из одного состояния в другое: существуют «правила отбора». Одновременное появление в спектре нескольких серий с различными основными состояниями, а также удваивание, утраивание и т. д. линий рассмотрено в последующих главах.
5.2. Атом водорода: основное состояние
Пусть нашей первой «химической» электронной системой, изучаемой с позиции квантовой механики, будет атом водорода. При этом будем руководствоваться в основном законом сохранения энергии (и его математическим выражением — уравнением Шрёдингера), а также принципом Паули.
Запишем закон сохранения энергии (разд. 3.6);
4-(£-(/) = О (11)
Умножим уравнение (11) на ф и заменим импульс р на оператор Т&/ КОТ°РЫЙ 03начает дифференцирование по координате,
а затем умножение на й/i. Тогда уравнение (11) принимает форму уравнения Шрёдингера:
. , , 2m rr. . „ . , д2ф /д2 , дг , д2 \ , Аф+ (£ ^)Ф —0, Аф — ^.2+ ду2 + az2)1*’
Уравнение Шрёдингера лежит в основе всех квантовомеханических расчетов. Его можно получить на основе «волновых» представлений, если связать классическое уравнение волны с сравнением де Бройля. Углубленное изучение возможных методов решения уравнения Шрёдингера показывает, что его можно свести к известным дифференциальным уравнениям теории колебаний, имеющим ряд решений, каждое из которых отвечает «собственным колебаниям» натянутой струны. Ниже затрагиваются лишь некоторые решения уравнения Шрёдингера для частных случаев и более глубоко его свойства не исследуются. Заметим лишь, что изучение классических представлений о «волне», соответственно «поле», открывает путь в квантовою теорию химической связи и дает возможность понять природу химической связи уже в классическом приближении.
Потенциал электрического поля протона — центрально-симметричная функция, —Тогда из уравнения (11)
М’+>(е+4)1>-о
(13)
Из всех возможных решений рассмотрим только такие, для которых ф зависит от г. Исследуем сначала выражение
Аф = /^-+-Д-+^1-')ф (14)
\ дх2 1 ду2 1 dz2 ) т 4 7
Принимая во внимание, что
r= ’Kx2-|-z/24'Z2;
dr _ 1 2х _______ х
~д7 ~~2 у “ 7
Получим первую и вторую производную ф по х:
дф дф дг х дф дх дг дх г дг
д^ф _ 1 дф Л"2 дф ( х2 д2ф
дх2 г дг г3 ’ дг г2 дг2
Вторую производную находим следующим образом. Введем функцию g==f(r'i д [ х дф \
/1 ) вместо выражения в скобках —-------. Перейдем к сферическим
дх\ г дг !
Электронные оболочки атомов
47
46 _______________Строение материи________________________
де да dr „ dg х
координатам ~. Для определения принимаем —и. Затем
' дх дг дх дг г
дифференцируем произведение и частное.
Другие члены уравнения (14) находим аналогичным образом:
д2ф 1 дф у2 дф ( у2 д2ф
ду2 ~~ дг ~~ г3 дг “г г2 дг2
д2ф 1 дф z2 дф z2 д2ф
dz2 = г dr г3 дг г2 дг2
Рассчитаем теперь для атома водорода значение энергии и соответствующую ф-функцию, иначе говоря, собственное значение энергии и собственное значение ф-функции системы. Да-
При суммировании следует учитывать, что x2+y2+z2 = r2. Тогда
3 Л|) 1 дф д2ф д2ф 2 дф
Дф = — — г дг + дг2 — дг2 + г дг
Теперь уравнение Шрёдингера представляет собой функцию только г:
Рис. А.13. Вероятность нахождения электрона вблизи ядра для осиовиого состояния атома водорода.
д2ф дг2
(15)
Наиболее простое решение последнего уравнения запишется в виде функции (убедитесь подстановкой) ф (Г) = е~га
(15а)
для которой соответствующие производные по г равны
дф
— = — ае-га
дг
д2ф дг2
= а2е~га
Таким образом, множитель е~га присутствует во всех слагаемых уравнения (15). Разделим уравнение (15) на этот множитель
, 2 . 2m / р . е2 \ п
а2--а+-^(£+—) = 0
Приравняем нулю попарно первый и третий, а затем второй и четвертый члены этого уравнения:
(16)
так как уравнение (15) должно удовлетворяться при всех с Объединим уравнения (16):
р a2 ft2 me1
'fc= 2m 2Й2"
лее мы увидим, что при решении уравнения Шрёдингера получается набор собственных значений энергии и ф-функций. Прежде всего обратим внимание на Е. Поскольку при присоединении электрона энергия выделяется, Е — отрицательная величина. Полагая m, е ati известными, рассчитаем энергию основного состояния атома водорода: Е = —13,5 эВ. Это хорошо согласуется с экспериментально определенной энергией ионизации атома водорода. Собственному значению энергии соответствует экспоненциальная вероятностная Функция. При этом необходимо помнить, что вероятность нахождения электрона в некотором объеме dx Равна ТС^ф2^. Определим теперь объем сферического слоя «толщиной» dr, находящегося на расстоянии г От центра (для наглядно-
сти представим себе кожуру апельсина). Очевидно, объем тако-0 слоя dx=^nr2dr. Таким образом, К7~г2ф2, а приняв во вни-
мание уравнение (15а), W~r2e~2ra.
Распределение вероятности нахождения электрона для ато-а водорода показано на рис. А.13. Найдем г, при котором
Функция вероятности имеет максимум (приравнивая нулю пер-
выше выражение для
полученное
г
3
Рис. А.14. Симметрия s и р-функций.
48
Строение материи
Электронные оболочки атомов
49-
вую производную функции вероятности):
__ 1 _ ft2 rmax а ~ fngZ
Здесь мы также находим хорошее согласие с экспериментальными данными; прежде всего отметим, что распределение заряда описывается сферической функцией, что уточнило атомную модель Бора, согласно которой заряд распределен в плоском кольце. Электроны, функция вероятности которых имеет сферическую симметрию, называют s-электронами (рис. А.14, а).
5.3. Атом водорода: возбужденное состояние
Существуют и другие ф-функции со сферической симметрией, представляющие собой решения уравнения Шрёдингера для атома водорода [уравнение (13)], например ф=(2—ra)e~rali. Максимум функции, описывающей вероятность распределения заряда, рассчитанный в соответствии с таким решением уравнения Шрёдингера, отвечает большему расстоянию г от ядра. Значения энергии для электронов на таком расстоянии можно выразить в следующем виде (вывод этих уравнений не рассматривается) :
Еп=-^ «=1,2,3,...
Ех — Е для п = 1 (основное состояние)
п называется главным квантовым числом. Оно в первую очередь определяет энергию данного состояния атома. Разность этих энергий представляет собой не что иное, как уравнение частоты для атомных спектров, экспериментально установленное ранее (разд. 5.1.3):
, / 1 1 \
v = const • —=---5-
I m2 n2 I
Наряду с рассмотренными выше основным и возбужденными s-состояниями для электронов атома водорода известны возбужденные состояния, волновые функции которых ф не имеют сферической симметрии. Решение уравнения Шрёдингера, которое приводит к этим функциям, значительно сложнее, так как требует учета наряду с г также и других полярных координат и <р (см. учебники по атомной физике). Здесь эти выводы не приводятся; ограничимся тремя возможными решениями уравнения Шрёдингера, полученными для одного и того же значения энергии:
фж=^7(0; Фг = ?-Ж
[Если читатель имеет хорошую математическую подготовку, то он может самостоятельно получить эти функции как решения уравнения (13).] На рис. А.14, б—г наглядно показана симметрия функции вероятности нахожде-
ния электрона для этих так называемых p-состояний электрона, имеющих одинаковую энергию. Состояния, имеющие одинаковую энергию, называют вырожденными. Для атома имеется еще так называемое «случайное» вырождение (разумеется, оно не случайно, а основано на особой сферической симметрии кулоновского потенциала в Н-атоме), так как собственное значение энергии указанных трех p-состояний совпадает со значением энергии 5-состояния при п=2. Кроме функций, описывающих p-состояния, известны и Другие решения уравнения (13) в виде функций, не имеющих сферической симметрии. Функции вероятности для них имеют более сложную зависимость (РиХ’А15)2 (напРиМеР* х2’ ХУ' хг и т. д.). Это так называемые d-функции
Некоторые другие проблемы решения уравнения Шрёдингера, например, то обстоятельство, что для каждого решения ф величина с-ф также является Решением (с — любая константа), что требует так называемого нормирова-ия> здесь не рассматривается.
Орбитальный момент. Атом водорода в магнитном поле
Важным параметром, характеризующим состояние атома, вляется орбитальный момент электронов. В классической ме-анике орбитальный момент частицы в прямоугольных коорди-4-2027
50
Строение материи
натах можно разложить на три компоненты:
Мх=ург—гру; Му = грх~хр2, Mz--=xp,—ypz
Применив уже известный прием, заменим импульс р наопе-ратор импульса — Тогда орбитальный момент электрона описывается некоторой волновой ф-функцией. Для s-функции ф = ф(г) получим
м- h A.Д. \ = о
~[х дх у дх i (Д дг г у дг г )
(аналогично можно записать для Мх и Му)
Итак, мы видим, что орбитальный момент электрона в s-состоянии равен нулю. Расчет орбитального момента р-электро-на, который здесь не приводится, дает замечательный результат: только одна пространственная компонента (проекция на одну координату) орбитального момента значима (имеет определенное значение или несколько значений), две другие всегда .не определены. Какая это компонента, зависит от выбора ф-функции. Для р-электрона оказывается, что компоненты орбитального момента могут принимать значения +Й, 0, —й.
Орбитальный момент определяет также магнитные свойства атома. Классическая электродинамика дает следующее соотношение между орбитальным моментом и магнитным моментом ц заряженной частицы (для проекции на одну из осей координат) :
= Ч (17)
г 2 2шс 2 4 ’
В магнитном поле с напряженностью Н частица обладает дополнительной энергией
W = p2H=-^- MZH
г 2 2тс 2
Если атом водорода в возбужденном p-состоянии поместить в магнитное поле, то окажется, что вырождение трех р-функций снимается, p-состояние расщепляется на три энергетических уровня
Это важное явление называют эффектом Зеемана. Можно доказать, что d-состояния в магнитном поле распадаются на
Электронные оболочки атомов 51
пять уровней, причем проекции орбитального момента определяются следующими значениями: 2Ъ, 1г, 0, —h, —2Й.
В соответствии с вышеизложенным следует ожидать, что s-электрон, не имеющий орбитального момента, не обладает также и магнитными свойствами. Кроме того, в магнитном поле можно наблюдать расщепление на 3, 5 и более уровней, поскольку существуют состояния с еще большим орбитальным моментом. Однако экспериментальные данные свидетельствуют об ином: для s-электронов в магнитном поле наблюдается расщепление на два уровня. Это объясняется существованием спина электрона (разд. 3.6), так что орбитальный момент характеризуется двумя проекциями на одну ось координат: !/2Й и —72й.
Между спиновым магнитным моментом и орбитальным моментом имеется связь, иная по сравнению с уравнением (17):
е ,, eh u,=----М.= -я---
'"s me ,s 2mc
Поскольку электрон имеет как орбитальный момент, так и спиновый момент, то общий момент частицы определяется обеими величинами, т. е. частицу можно характеризовать также суммарным вращательным моментом (см. далее).
5.5. Электронные конфигурации. Атомные состояния.
Периодическая система элементов
На основе развитых ранее представлений можно уже при некоторых упрощающих предположениях установить строение также и других атомов периодической системы. Наиболее существенное упрощение теории состоит в том, что в ней не учитывается взаимодействие между электронами, т. е. предполагается, что каждый электрон движется в поле атомного ядра не-зависимо от других электронов и его потенциал равен U= —— (Z — зарядовое число ядра). Таким образом, в данном случае Речь идет о так называемой электронной модели строения атома. Позднее мы увидим, что эта модель играет важную роль в квантовой химии, в частности в так называемом методе молекулярных орбиталей (МО).
Расположение элементов в периодической таблице определяется целиком и полностью принципом Паули (разд. 3.6), а также некоторыми правилами вычисления орбитальных моментов, которые рассмотрены ниже. Отметим некоторые соотношения Между так называемыми квантовыми числами. Понятие квантового числа п уже введено ранее; установлено, что это число-непосредственно связано с энергетическим состоянием электро-ов. Орбитальные моменты характеризуются квантовым числом 4»
52
Строение материи
Электронные оболочки атомов
53
I: 1 = 0 (s-электроны), 1=1 (р-электропы), 1 = 2 (d-электроны), | 1 = 3 (/-электроны).
Таким образом, квантовые числа п и I связаны с энергией и симметрией электронных состояний. Другие квантовые числа характеризуют уровни энергии, которые возникают при расщеплении вырожденного состояния, относящегося к-квантовому
Рис. А. 16. Основные состояния Н, Не, Li, Be и В.
числу I, отличному от нуля. С этой целью используется магнитное орбитальное квантовое число т = 4-1, 0, —1. Вообще, m может принимать 2/+1 значений, а именно все целые числа между +Z и —I. Наконец, последнее квантовое число, необходимое для полной характеристики состояния электрона, — это спин электрона. Магнитное спиновое число может принимать значения Д-'/г и —'/2. То, каким образом в многоэлектронных атомах проявляются вращательные моменты и как суммарный орбитальный момент обозначается через квантовые числа, проще всего понять, если построить электронные модели нескольких атомов.
В разд. 5.3 говорилось о «случайном» вырождении в атоме водорода. Оно снимается в многоэлектронных атомах вследствие того, что поле, в котором движутся электроны, не является, строго говоря, чисто кулоновским. Поэтому схема энергетических уровней электронов для таких систем выглядит несколько иначе, чем в атоме водорода; например, уровни 2s и 2р не совпадают, хотя, впрочем, разность энергий между 2s- и 2р-уров-нями, обусловленная «эффективным полем ядра», значительно меньше, чем для уровней с разными п.
При заполнении уровней электронами будем учитывать принцип Паули*. Первый многоэлектронный атом в периодической системе-—гелий.
* Принцип Паули в применении к строению атома формулируется следующим образом: в одном атоме не существует электронов, имеющих одинаковые наборы квантовых чисел. [Яворский Б. М., Детлаф А. А. Справочник по физике. — М.: Наука, 1979, с. 770].—Прим, перев.
Два электрона гелия занимают ls-уровень, имеют одинаковое главное квантовое число, один и тот же орбитальный момент, а следовательно, могут отличаться только спином. Для антипар аллельных спинов + 1/2 и —’/г суммарный спин равен нулю. Суммарный орбитальный момент (два электрона с 1 = 0), также равен нулю. Для обозначения суммарного орбитального момента L и суммарного спинового момента 5 принято исполь-
м -г. +г -/ о+/ -io +1
Рис. А.17. Атомные состояния углерода.
зовать так называемые термы. С этой целью для каждого L ввели буквенные обозначения: 5 (L = 0), Р (L=l), D (L = 2), F (L = 3) и т. д. Суммарный спиновой момент учитывают с помощью мультиплетности* 25+1, которую указывают при соответствующей букве в виде верхнего левого индекса. Например, основному состоянию атома гелия соответствует терм '5.
В атоме лития имеется еще один, третий, электрон, который занимает уровень 2s; основному состоянию атома лития соответствует терм 2S (суммарный спиновый момент равен */2). На рис. А.16 приведены основные состояния атомов нескольких
элементов (от водорода до бора). Сложнее картина строения атома углерода. Здесь имеются два электрона на уровне 2р. Этот уровень, как мы видели ранее, может расщепляться на три уровня: с т = 1, 0 или —1. На рис. А.17 показано, как, согласно принципу Паули, можно расположить электроны на этих Уровнях. Для каждого случая приведены также суммарные спиновые и магнитные квантовые числа. Если 5 = 1, М =— 1, 0, +1. Всем значениям М соответствует -суммарный орбитальный момент L=l, т. е. состояние такого атома 3Р. Если 5 = 0, М =
0, 1, 2 и еще раз 0. Этому наб’ору М отвечают различные L, а именно L = 2 и L = 0. Таким образом, получили еще Два возможных состояния атома углерода: lD и '5. Какое же стояние из этих трех состояний 3Р, ’£> и ’5 — основное? На —2l_gPnP°c нельзя получить правильный ответ, если исходить
со * Число уровней энергии, на которое расщепляется энергия некоторого Ник-Т0ЯНИя атома в магнитном поле. [Яворский Б. М„ Детлаф А. А. Справоч-к по физике.— М.: Наука, 1979, с. 760]. — Прим, перев.
Таблица А.5. Основные состояния элементов
к L M N 0 p Q
Z Элемент ЗТерм"5
Is 2s 2p 3s Зр 3d 4s iP id 41 5s 5p 5d 6s 6p 64 7s
1 н 2«l/2 1
2 Не xSo 2
3 Li 251/2 2 1
4 Be ^0 2 2
5 В *?1/2 2 2 1
6 С 3P0 2 2 2
7 N 4«3/2 2 2 3
8 О 2 2 4
9 F ^3/2 2 2 5
10 Ne 4s„ 2 2 6
11 Na 2s1/2 2 2 6 1
12 Mg x50 2 2 6 2
13 Al aPr/2 2 2 6 2 1
14 Si 3Po 2 2 6 2 2
15 P 4S3/2 2 2 6 2 3
16 S 3P2 2 2 6 2 4
17 Cl 2 2 6 2 5
18 Ar LSo 2 2 6 2 6
19 К 2S1/2 2 2 6 2 6 1
20 Ca xSo 2 2 6 2 6 2 )
IJ 1
21 1 Sc 1 2P>3/i 2 2 6 2 6 1 2
22 Ti 3P2 2 2 6 Г £ 2 2
23 V ^3/2 2 2 6 г € 3 2
24 Cr 7S3 2 2 6 2 6 5 1
25 Mn 6S5/2 2 2 6 с € 5 2
26 Fe 2 2 6 2 6 6 2
27 Co ^9/2 2 2 6 2 6 7 2
28 Ni ^4 2 2 6 2 6 8 2
29 Cu 2«l/2 2 2 6 2 6 10 1
30 Zn 2 2 6 2 6 10 2
31 Ga 2-Pl/2 2 2 6 2 6 10 2 1
32 Ge 3P0 2 2 6 2 6 10 2 2
33 As 4S3/2 2 2 6 2 6 10 2 3
34 Se 3P% 2 2 6 2 6 10 2 4
35 Br 3P3l2 2 2 6 2 6 10 2 5
36 Kr XSO 2 2 6 2 6 10 2 6
37 Rb 2«l/2 2 2 6 2 6 10 2 6 1
38 Sr xSo 2 2 6 2 6 10 2 6 2
39 Y 2 2 6 2 6 10 2 6 1 2
40 Zr 3P2 2 2 6 2 6 10 2 6 2 2
41 Nb 6Dl/2 2 2 6 2 6 10 2 6 4 1
42 Mo 753 2 2 6 2 6 10 2 6 L5 1
43 Tc 6S5/2 2 2 6 2 6 10 2 6 5 2
44 Ru ^5 2 2 6 2 6 10 2 6 7 1
45 Rh 4T9/2 2 2 6 2 6 10 2 6 8 1
Продолжение
К L M N 0 p Q
z Элемент Терм
Is 2s 2p 3s 3p 3d 4s id if 5s 5p 5d 6s 6p Gd 7s
46 Pd '«о 2 2 6 2 6 10 2 6 10
47 Ag 2s1/2 2 2 6 2 6 10 2 6 10 1
48 Cd 'So 2 2 6 2 6 10 2 6 10 2
49 In 2^/2 2 2 6 2 6 10 2 6 10 2 1
50 Sn JPo 2 2 6 2 6 10 2 6 10 2 2
51 Sb 'S1/2 2 2 6 2 6 10 2 6 10 2 3
52 Те 3R2 2 2 6 2 6 10 2 6 10 2 4
53 1 2^/2 2 2 6 2 6 10 2 6 10 2 5
54 Xe 'So 2 2 6 2 6 10 2 6 10 2 6
55 Cs 2s1/2 2 2 6 2 6 10 2 6 10 2 6 1
56 Ba 'So 2 2 6 2 6 10 2 6 10 2 6 2
57 La 2D3/2 2 2 6 2 6 10 2 6 10 2 6 1 2
58 Ce 3Ht 2 2 6 2 6 10 2 6 10 1 2 6 1 2
59 Pr V3/2 2 9 6 2 6 10 2 6 10 3 2 6 2
60 Nd 5Л 2 2 6 2 6 10 2 6 10 4 2 6 2
61 Pm 6W5/2 2 2 6 2 6 10 2 6 10 5 2 6 2
62 Sm 7Л> 2 2 6 2 6 10 2 6 10 6 2 6 2
63 Eu 8s7/2 2 2 6 2 6 10 2 6 10 7 2 6 2
64 Gd 9O2 2 2 6 2 6 10 2 6 10 7 2 6 1 2
65 Tb *^7/2 2 2 6 9 6 10 2 6 10 9 2 6 2
к \ 1
66 / Dy v8 \ 2 l 2 6 2 6 10 2 6 10 10 2 6 2 \
67 / Ho %. 5/2 2 2 6 2 6 10 2 6 10 11 2 6 2
68 Er 2 2 6 2 6 10 2 6 10 12 2 6 2
69 Tu 2 2 6 2 6 10 2 6 10 13 2 6 2
70 Yb 'So 2 2 6 2 6 10 2 6 10 14 2 6 2
71 Lu 2-^3/2 2 2 6 2 6 10 2 6 10 14 2 6 1 2
72 Hf 3^2 2 2 6 2 6 10 2 6 10 14 2 6 2 2
73 Ta 'A3/2 2 2 6 2 6 10 2 6 10 14 2 6 3 2
74 W 6 2 2 6 2 6 10 2 6 10 14 2 6 4 2
75 Re eS6/2 2 2 6 2 6 10 2 6 10 14 2 6 5 2
76 Os 2 2 6 2 6 10 2 6 10 14 2 6 6 2
77 Ir V9/2 2 2 6 2 6 10 2 6 10 14 2 6 7 2
78 Pt (3D3) 2 2 6 2 6 10 2 6 10 14 2 6 9 1
79 Au 2s1/2 2 2 6 2 6 10 2 6 10 14 2 6 10 1
80 Hg 'So 2 2 6 2 6 10 2 6 10 14 2 6 10 2
81 T1 2Л/2 2 2 6 2 6 10 2 6 10 14 2 6 10 2 1
82 Pb 3Л> 2 2 6 2 6 10 2 6 10 14 2 6 10 2 2
83 Bi 4S3/2 2 2 6 2 6 10 2 6 10 14 2 6 10 2 3
84 Po 3r2 2 2 6 2 6 10 2 6 10 14 2 6 10 2 4
85 At 3P3/2 2 2 6 2 6 10 2 6 10 14 2 6 10 2 5
86 Rn 'So 2 2 6 2 6 10 2 6 10 14 2 6 10 2 6
87 Fr 2Sl/2 2 2 6 2 6 10 2 6 10 14 2 6 10 2 6 1
88 Ra 'So 2 2 6 2 6 10 2 6 10 14 2 6 10 2 6 2
89 Ac ^з/г) 2 2 6 2 6 10 2 6 10 14 2 6 10 2 6 1 2 ?
90 Th (3^2) 2 2 6 2 6 10 2 6 10 14 2 6 10 2 6 2 2 ?
Электронные оболочки атомов
59
только из нашей упрощенной электронной модели атома. Необходимо учитывать взаимодействия электронов в атоме. Такое взаимодействие обсуждается немного далее; здесь же уместно сформулировать так называемое правило Хунда: наиболее стабильно состояние атома, в котором спины электронов параллельны. Поэтому стабильное (основное) состояние углерода соответствует терму 3Р, что подтверждается результатами спектроскопических исследований.
Таким же образом, и даже, может быть, еще проще, можно найти основные состояния ближайших, следующих за углеродом атомов N, О, F, Ne. У неона s- и р-уровни слоя п = 2 полностью заполнены, т. е. электроны не могут появиться на этих оболочках, не нарушив принципа Паули. Поэтому для следующего элемента начинается заселение уровней слоя « = 3. Это происходит точно так же, как и для слоя « = 2; в результате образуется электронная оболочка инертного газа аргона. Термы этого периода также одинаковы, т. е. электронные оболочки атомов элементов первых двух «коротких» периодов периодической системы имеют аналогичное строение. Опустим подробности построения электронных моделей остальных элементов периодической системы. С последовательностью заполнения энергетических уровней электронов в слоях и особенностями заполнения, например появлением побочных групп и лантаноидов, можно ознакомиться с помощью табл. А.5. В термы включен также индекс справа внизу, который указывает на суммарный орбитальный и спиновый моменты.
5-6. Границы применимости электронной модели атома.
Атомы и молекулы
Из вышеизложенного очевидно, что периодичность заполнения электронных оболочек можно довольно хорошо представить себе, не рассматривая взаимодействия электронов между собой. Правда, не удается объяснить некоторые важные явления, которые лежат в основе, например, правила Хунда; кроме того, нельзя определить строение даже такого простого атома, как гелий, в возбужденном состоянии. При изучении электронного взаимодействия прежде всего следует учитывать некоторые особенности рассмотренной в разд. 3.6 симметричной и антисимметричной волновой ф-функции. Однако сначала рассмотрим эти чрезвычайно важные особенности (хотя они проявляются и в атоме гелия) взаимодействия на примере молекулы в°дорода — системы с двумя электронами. В следующей главе Рассмотрены некоторые теоретические представления по проблеме образования химической связи. Следует лишь принять во внимание, что причины образования такого прочного атома, как гелий, те же, что и для молекулы водорода, как стабильной си
60
Строение материи
стемы из нескольких частиц. Вообще говоря, не существует принципиального физического различия между атомами и молекулами. Тем не менее принято считать, что при объединении частиц в стабильную систему с несколькими атомными ядрами между ними появляется химическая связь.
6.
ПРЕДСТАВЛЕНИЯ О ХИМИЧЕСКОЙ СВЯЗИ
6.1. Уровни энергии и структура молекул
Наиболее простые системы с химической связью — двухатомные молекулы газов (N2, Н2, О2), состав которых установил еще Авогадро. Ион Н2+, содержащий два протона и электрон,— вот самая простая система из трех частиц с одной химической связью. Для того чтобы понять, что же такое химическая связь в самом простом ее проявлении, выясним причины устойчивости этих простых молекул. Однако прежде всего познакомимся с экспериментальными данными об энергетических уровнях молекул. Они значительно более разнообразны, чем в атомах, так как в молекулах наряду с электронными энергетическими переходами происходят также изменения колебательной и вращательной энергии. Поскольку все эти изменения энергии накладываются друг на друга, молекулярные спектры по большей части имеют очень сложное строение. Можно различать три ти-Таблица А.6. Характеристика спектров электромагнитного излучения
Излучение Длина волны, м Волновое число, м—1 Энергия кванта излучения Процессы, сопровождающиеся излучением
эВ КДж/МОЛЬ
Гамма-излуче-иие Рентгеновское излучение УФ в вакууме УФ и ВИДИМЫЙ свет Ближнее ИК Дальнее ИК Микроволновое излучение Радиоволновое излучение Ю-12 ю-11 10-10 io-9 10-8 ю-7 10-6 10~5 10-4 10—5 10~2 ю-1 I 10 100 1000 10е 106 104 1,24-10® 1,24-105 1,24-104 1240 124 12,4 1,24 0,124 0,0124 0,00124 1200 1200 120 12 1,2 Ядерные процессы Электронные переходы вблизи ядра Электронные переходы в валентных оболочках Электронные переходы в валентных оболочках Колебания молекул Вращение молекул Зеемановские переходы Спиновые переходы в атомных ядрах
Химическая связь
61
па спектров: вращательные спектры в длинноволновой инфракрасной области (от 500 до 50 мкм), вращательно-колебательные спектры в коротковолновой инфракрасной области (от 10 до 1 мкм) и электронно-вращательно-колебательные спектры в видимой и ультрафиолетовой областях.
Наиболее характерные свойства спектров электромагнитного излучения представлены в табл. А.6.
6.1.1. Вращательные спектры
Вращательные спектры двухатомных молекул в самом простом случае представляют собой полосы поглощения (абсорбции) излучения; они, впрочем, характерны только для таких молекул, у которых имеется дипольный момент. Вращательные термы можно получить довольно просто из уравнения Шрёдингера для «жесткого ротатора» с моментом инерции <Э — тг2. Подставим в уравнение Шрёдингера в качестве координаты путь по круговой орбите q~r&, т. е. рассмотрим частный случай вращения в одной плоскости. Так как d(r(o) =rd&, то
д2гр . 2т /с, Т, д2ф . 2т „ ,
(У/2 ' Й.2 ' ' о (гео)2 ' Й2 вр т
д2ф । 2тгг- г, , д2ф , 20 г, , п = Н-------• Ф = ЛчЧЧ—*2” • Чп • Ф = о
ОСО2 1 Й.2 ВР т 1 Й2 вр г
Решение этого уравнения приводит к следующим собственным: волновым функциям:
ф — cos у —со и ф = sin у ----------СО
В качестве граничного условия примем, что ф-функция после-полного оборота ротатора вокруг оси принимает прежнее значение: ф(со) =ф(со+2л) = ф(<вф-4л). Таким образом, вращательная энергия квантуется следующим образом:
/^^=0,1,2,3,...; EBP = ^--fe2 (fe = 0,1,2,3)
В трехмерном решении вместо тригонометрических волновых Функций появляются более сложные сферические функции, а в. собственных значениях k2 изменяется на Вращатель-
ные квантовые числа подчиняются правилу отбора А&=±1.
Положение терма в инфракрасном диапазоне можно получить из численного значения момента инерции соответствующей Молекулы. Порядок величины <Э~10~40 г-см2. Вращательные спектры позволяют вычислить моменты инерции различных мо-ЛекУл. Однако для тяжелых атомов с большими моментами Инерции частота вращения настолько мала, что попадает уже-
<62
Строение материи
и микроволновую область. Благодаря развитию микроволновой 1ехники достигнуты заметные успехи и в микроволновой спектроскопии. Так, в последние годы этим методом получены новые ценные данные о геометрических размерах молекул.
Рис. А 18. Модель (типа гантели) вращающейся двухатомной молекулы.
Познакомимся на примере вращательного спектра молекулы 'СО, каким образом выполняется расчет размеров молекулы. Вращение двухатомной молекулы можно моделировать (рис. А. 18). Воспользовавшись законом сохранения момента количества движения m1r1 = m2r2, момент инерции системы 0 = /Н1Г12+ 4-m2^22 можно выразить в следующем виде:
О nVn2Г2 (18)
4- m.2 v ’
где r = ri+r2> т. е. г — расстояние между атомами. Таким образом, вращение обоих масс можно представить как вращение -одной (усредненной) массы:
находящейся на расстоянии г от оси вращения, перпендикулярной прямой, соединяющей ядра атомов в молекуле. Итак, момент инерции равен
0 = рг2 (20
Выведенное ранее уравнение
£вр = Т^Ш+1) (2D
связывает уровни энергии вращения с моментом инерции молекулы и вращательным квантовым числом k. Из рис. А. 19 очевидно, что разность энергий соседних состояний увеличивается по мере увеличения k.
Химическая связь
63
Энергию возбуждения для случая поглощения излучения можно вычислить, применяя правило отбора Д& = ±1:
^E^EвPk+-EвPk = ^[(k+lЦk+2)-k(k+\)]
А£ = 1Йб^+1) <22>
Соответствующие частоты поглощения равны
vw = ^-=-4^-(^+l) для й = 0,1, 2,... (23)
Таким образом, приняв за основу рассмотренную выше модель
ротатора, можно получить вращательный спектр, состоящий из ряда эквидистантных линий, часто-
ты поглощения которых отличаются л
на величину
(24)
Изучение экспериментально полученных спектров показывает, что это условие в общем хорошо выполняется. На рис. А.20 схематически представлен спектр поглощения моноксида углерода. Линии поглощения на нем появляются при частотах (длинах волны), соответствующих энергии кванта излучения, возникающего при переходах с ближайшего вращательного состояния. Первая частота поглощения vi (переход от k = 0 к k=l) приблизи-
Рис. А.19. Уровни вращательной энергии двухатомной молекулы.
тельно равна разности частот сосед-
них линий. Точное измерение дает значение vi = 1,152706-•10й с-1 (настолько высока точность измерения в микроволновом диапазоне частот!) Для дальнейших вычислений достаточно принять vi = 1,153-1011 с-1. Соответствующая длина волны равна
- _ с _ 2,998-1010
Л1~ vt — 1,153-Ю11
Поглощенный квант энергии
= 0,26
см
ХЕ = hv-L = 6,625 • 10’3) • 1,153 • 1011 = 7,64 -10'23 Дж Рассчитаем теперь расстояние г между ядрами атомов молекулы СО. Из уравнения (20) получим
где
п\т2 mi + tns
«4
Строение материи
Из уравнения (24), подставив в него частоту vb получим момент инерции молекулы СО
®СО 4n2vt
1,67-10-28
“ 1,15-1011
== 1,45-10'39 г-см2
Усредненную массу молекулы можно получить, воспользовавшись атомными массами углерода и кислорода и числом Аво-тадро Уд/
и —___________16-J2__________ । 14.1 п-23 г
(12+ 16)-6,023-1023 ’
••Отсюда получим 1 / 1,45-10-»» , 1П_8
гсо у 1,14-Ю-23 1,13-10 см
Определенное из спектроскопических данных значение частоты позволяет вычислить величину гео с большим числом
Дем--*-
0,260 0,130 0,0877 0,065
к=0—! к=1^2 к=2-~3 к=3-^
JJJ_________________________
2,30 3,56 5,6f
D IO'1,'с'--
Рис. A.20. Спектр поглощения молекулы моиооксида углерода.
значащих цифр, однако такой расчет не оправдан из-за ограниченной точности определения постоянной Планка и массы атомов. Расчет с более высокой точностью также не имеет смысла потому, что при этом пренебрегали собственными колебаниями атомных ядер.
Для того чтобы выполнялось условие антисимметричности суммарной волновой функции, при расчете вращательных состояний молекулы с ядрами, имеющими полуцелый спин, например для водорода, необходимо учитывать симметрию ядерного спина. Поэтому без вывода примем, что, например, водород существует в двух модификациях с четным и нечетным вращательным квантовым числом. Эти модификации называют пара-и ортоводородом.
6.1.2. Вращательно-колебательные спектры
Прежде чем рассматривать структуру этих спектров, необ-ходимо в общих чертах описать способ квантования колебательной энергии. В упрощенном виде можно предположить, что
Химическая связь
65
периодические колебания атомных ядер в молекуле протекают по «гармоническому закону», т. е. между ними существует упругое взаимодействие, подчиняющееся закону Гука. Тогда
F=— k(r—r0)
где г —расстояние между ядрами в данный момент, г0 — равновесное расстояние между ядрами. Колебания такого гармонического осциллятора происходят с частотой
’4/4
Коэффициент упругости k является характеристической константой молекулы. Частота колебаний увеличивается при уменьшении массы. Потенциальная энергия гармонического осциллятора зависит от расстояния между ядрами:
П =: Г°)2
Рис. А.21. Функция Морзе для потенциальной энергии с уровнями колебательной энергии.
откуда функция J7=f(r) графически изображается параболой. Однако модель гармонического осциллятора «работает» только при Дг—>0. Понятно, что моле-
куда не может принять любое количество энергии; при некотором расстоянии между атомами, соответствующем определенному значению поглощенной энергии, следует говорить о диссоциации молекулы на атомы. Более правильно зависимость U от расстояния г дает функция Морзе (рис. А.21):
U = D (1 — exp [—const (г—r0)]2
где D — так называемая энергия диссоциации. Если подставить потенциальную энергию колебаний как функцию от г в уравнение Шрёдингера, то можно получить следующее выражение Для собственных значений колебательной энергии:
£Кол=( ы+4") hv (« = 0, I, 2,3,...)
Таким образом, для гармонического осциллятора уровни энергии находятся на одинаковом расстоянии друг от друга, в то время как для функции Морзе уровни энергии уже не ЭК' 5—2027
x_jfgTep»»____________________
I). Ва5Нц
*ует эН% 0 Отметить, что наиболее низкой КР^5И11 большей, чем минимальное °и (на 1/aftv); эту величину на--.IBafoT «нулевой» колебательной ьНеРРией. Она играет важную л°Ль при определении энергии ^Иссоциации молекулы на основе ^Кспериментально полученных °Твнциальных кривых.
~ Теперь можно разобраться а Ол^е сложной структуре поло-vbI вращательно-колебательного ПеКтра (например, для НС1 по-OcU поглощения при 3,5 мкм): *аРактерно изменение как коле-ательных, так и вращательных ^Вантовых чисел. На рис. А.22 ^°Лебательные состояния обозна-^ены как А и А'. Здесь же отме-Чецы разрешенные переходы **е}вду двумя вращательными состояниями.
и*\трь,
I'У|вл’,е1^ 15>ЛС1женнЬ1е в диапазоне удимого к'\х,к°л х^.?’ возникают в результате взаимо-1 <0 егруК'. ба^ельных и электронных перехода тФ ^УРУ. На рис. А.23 и А.24 приведе-^адовн ^ов двухатомной молекулы. На ЭЧ2Г° состояния с колебательными и vm пр ^ергии. Диссоциированная молеку-У5г'-ТцД1Ицмать лГОбое количество кинети-ДргиРТ^Ует заштрихованным областям у1касштх льные термы приведены в другом, psbix х^бе. на рИС_ д.24 показаны анало-rt^o^x^Pвходов возбужденной молекулы. иНокб лов состоит из ряда полос, соответ № стру .^тельным переходам, а те в свою рйадииС^туру. связанную с вращением М°
Мс>леКуЛЬ1 можно определить, уста-'31051 Полосатый спектр переходит ггеМ<х^ледует учитывать энергию возбу РеХ(хХ)в. Положение колебательных ур°® \Да>: в молекуле определяется приа Ри электронных переходах рассто
Химическая связь
67
нце между атомами не изменяется, т. е. молекулы в электронновозбужденном состоянии имеют такое же колебательное состояние, как и в основном.
Рис. А 23. Электронное основное со- Рис. А.24. Возбужденное электронное стояние и вращательно-колебатель- состояние.
ные термы двухатомной молекулы.
6.1.4. Спектры растворов
Для многих веществ, например для большинства комплексных соединений, электронные переходы можно исследовать методами абсорбционной спектроскопии в растворах соответствующих веществ в видимом и ультрафиолетовом свете. Метод основан на законах поглощения света. Согласно закону Ламберта, понижение интенсивности I световой волны в абсорбирующей (поглощающей) среде обусловлено толщиной слоя ds: dl~ —aids или -/-= e~as
Бер установил, что коэффициент а пропорционален концентра-вещества в растворе /Д0 = ехр (—sncs) или Е~—\qIIIq —
Е называют экстинкцией (поглощением) среды, коэффициентом экстинкции*. Абсорбционный спектр
Ды Р аналитической практике 1g/До часто называют «поглощением» ере-’ а Щ /о// — «пропусканием». — Прим, перев.
•5*
66
Строение материи
видистантны (рис. А.21). Важно отметить, что наиболее низкий уровень соответствует энергии большей, чем минимальное значение на потенциальной
А
А'
7 S И 2 ! 13-1 -2 -3-4 -5 -6 -7
кривой (на ll2hv); эту величину называют «нулевой» колебательной энергией. Она играет важную роль при определении энергии диссоциации молекулы на основе экспериментально полученных потенциальных кривых.
Теперь можно разобраться в более сложной структуре полосы вращательно-колебательного спектра (например, для НС1 полосы поглощения при 3,5 мкм): характерно изменение как колебательных, так и вращательных квантовых чисел. На рис. А.22 колебательные состояния обозначены как А и А'. Здесь же отмечены разрешенные переходы между двумя вращательными состояниями.
Рис. А.22 Вращательные и колебательные переходы
6.1.3. Электронные спектры
Полосы на спектрах, расположенные в диапазоне видимого и ультрафиолетового излучения, возникают в результате взаимодействия вращательных, колебательных и электронных переходов и имеют сложную структуру. На рис. А.23 и А.24 приведена упрощенная схема термов двухатомной молекулы. На рис. А.23 дана схема основного состояния с колебательными и вращательными уровнями энергии. Диссоциированная молекула, атомы которой могут принимать любое количество кинетической энергии, соответствует заштрихованным областям (рис. А.23 и А..24). Вращательные термы приведены в другом, значительно меньшем масштабе. На рис. А.24 показаны аналогичные термы электронных переходов возбужденной молекулы. Полоса электронных переходов состоит из ряда полос, соответствующих различным колебательным переходам, а те в свою очередь имеют тонкую структуру, связанную с вращением молекул. Энергию диссоциации молекулы можно определить, установив частоту, при которой полосатый спектр переходит в сплошной, однако при этом следует учитывать энергию возбуждения образовавшихся атомов. Положение колебательных уровней при электронных переходах в молекуле определяется принципом Франка — Кондона: при электронных переходах расстоЯ-
Химическая связь
67
иле между атомами не изменяется, т. е. молекулы в электронно-,возбужденном состоянии имеют такое же колебательное состояние, как и в основном.
Рис. А 24. Возбужденное электронное состояние.
Рис. А 23 Электронное основное состояние и вращательно-колебательные термы двухатомной молекулы.
6.1.4. Спектры растворов
Для многих веществ, например для большинства комплексных соединений, электронные переходы можно исследовать методами абсорбционной спектроскопии в растворах соответствующих веществ в видимом и ультрафиолетовом свете. Метод основан на законах поглощения света. Согласно закону Ламберта, понижение интенсивности / световой волны в абсорбирующей (поглощающей) среде обусловлено толщиной слоя ds:
di = —aids или ~ — e~as io
Sep установил, что коэффициент а пропорционален концентрации вещества в растворе //20=ехр (—encs) или Е ——[gl[lo= ^^ncs/2,303 Е называют экстинкцией (поглощением) среды, Ц еп-—коэффициентом экстинкции*. Абсорбционный спектр
* В аналитической практике 1g I/Io часто называют «поглощением» сре-a \glo/l — «пропусканием».— Прим, перев.
5*
68
Строение материи
(спектр поглощения) обычно снимают как зависимость Е, еп или 1g еп от длины волны падающего света (или частоты). При этом получают так называемую «типичную» кривую цветности. С анализом абсорбционных спектров и основными методами спектрофотометрии можно ознакомиться в специальных руководствах.
Следует лишь упомянуть, что в последнее время благодаря успехам «отражательной спектроскопии» (эллипсометрии) стало возможным спектроскопическое исследование нерастворимых (или изменяющихся при растворении) веществ.
6.1.5. Спектры комбинационного рассеяния (спектры КР)
Возможность непосредственно наблюдать вращательные и колебательные переходы в области видимого света основывается на открытии Раманом и Мандельштамом явления комбинационного рассеяния света. При прохождении монохроматического света через вещество в спектре рассеянного света наряду с линией излучения источника света появляются также линии с более высокими и более низкими частотами. Эта разность частот относительно основной частоты источника света соответствует изменению энергии при колебательных переходах. Основное достоинство спектроскопии комбинационного рассеяния (КР) состоит в том, что с ее помощью можно точно и просто определять собственные частоты колебаний молекулы. При этом можно различить «валентные» и «деформационные» колебания. Последние возможны у многоатомных нелинейных молекул. Так, например, молекула воды Н2О имеет два валентных колебания. О—Н и одно деформационное (угла между связями О^^). ;
В табл. А.7 приведены частоты валентных колебаний для некоторых типов связи. Легко заметить, что для различных типов связи валентные колебания попадают в разные частотные диапазоны. На основе колебательных спектров, полученных методами инфракрасной спектроскопии и спектроскопии комбинационного рассеяния, можно сделать важные выводы о симметрии молекул. Число возможных собственных колебаний lV-атом-ной молекулы (А'>2) равно 3N—6, из них N—1 валентных и 2.V—5 деформационных колебаний. В зависимости от симметрии молекулы некоторые колебательные частоты совпадают (вырожденные колебания).
Рассмотрим, например, четырехатомную молекулу с условной формулой ZA3. В общем случае такая молекула должна проявлять 6 собственных колебаний, однако для конфигураций равностороннего треугольника это число уменьшается до 3, а для конфигурации типа пирамиды — до 4. При изучений строения молекулы инфракрасные спектры и спектры КР ча'
Химическая связь
69
Таблица А.7. Валентные колебания и энергия связи
Связь Частота колебаний, см-1 Энергия диссоциации, кДж/моль Расстояние между ядрами, им
n=n 2331 926,8 0,110
С=о 2168 942,4 0,113
(НС1)№ 2089 ~687 0,116
0=0 1556 490,7 0,121
F—F 892 150,7 0,143
С1-С1 557 239,5 0,200
Вг—Вг 321 190 0,228
I-I 213 149 0,267
H-F 4141 561 0,093
Н-С1 2989 427,5 0,128
Н—Вг 2650 359,6 0,142
сто дополняют друг друга, так как они основаны на различных принципах поглощения энергии. Молекулы с одним центром симметрии, у которых, как правило, колебания вызваны изменением дипольного момента, не имеют спектров комбинационного рассеяния. Это объясняется тем, что у них не меняется поляризуемость в направлении, совпадающем с вектором поля излучения. Поэтому линейные трехатомные молекулы имеют две интенсивные полосы в инфракрасном спектре и одну интенсивную полосу в спектре КР. У нелинейных трехатомных молекул, не имеющих центра симметрии, как в одном, так и в другом спектре имеются три линии.
Методы инфракрасной, КР- и микроволновой спектроскопии представляют собой ценное средство исследования структуры молекул. Прежде всего это касается упомянутых в разд. 5.4 пер’еходов зеемановских уровней. Расстояния между этими «магнитными» энергетическими уровнями в магнитных полях, создаваемых обычными лабораторными установками, соответствуют микроволновому диапазону для спина электронов, дециметровому и метровому диапазонам для ядерного спина.
6-1-6. Спектры ядерного магнитного резонанса (спектры ЯМР)
Ядерный магнитный резонанс представляет собой явление поглощения энергии, сопровождаемое изменением спинового состояния атомного ядра, которое так же, как и электрон, имеет ‘ агнитный момент. Магнитный дипольный момент у, — вектор-ая величина и измеряется в ядерных магнетонах ця. Анало-чно магнетону Бора (разд. 6.5.3) ядерный магнетон опре-д ляется с помощью следующего выражения:
Ря = -2^Г-/г = 4,019°.10-22 А-м2 (25)
70
Строение материи
где nip — масса протона, с —скорость света. Так как отношение массы электрона к массе протона равно 1 : 1836, ядерный магнетон во столько же раз меньше магнетона Бора. Количественно различаются и магнитные характеристики, связанные с электронной оболочкой атома и спином ядра.
Во внешнем магнитном поле с напряженностью Но вектор магнитного момента может устанавливаться в 27-}-1 положени-
Рис. А 25. Возможные ориентации вектора ядерного магнитного момента с ядерным спином 1=11г.
ях относительно вектора внешнего поля (I— ядерный спин). Следовательно, в некоторых часто встречающихся ядрах с /=’/2 (Н, 19F, 3!Р) существуют два состояния с соответствующими магнитными ядерными квантовыми числами — и =
В том случае, если вектор магнитного момента параллелен полю,
Ия0 = ( т; = +4- или-------------(26)
где g — гиромагнитное отношение, которое зависит от строения ядра и поэтому имеет некоторое характерное значение для любого типа ядра.
При повороте магнитного диполя во внешнем магнитном поле его потенциальная энергия меняется. Из курса физики известно, что эта энергия равна
Е — —|л7У0созф (27)
где ф — угол между векторами ц и Но (см. рис. А.25). При пи^ = ’/2 со5ф = цц0/ц, и из уравнений (26) и (27) следует
^(+1/2) = — ^я77° (28)
Химическая связь
71
Следовательно, при такой ориентации магнитного момента выделяется энергия. При mi =—’A cos(n—ср) = coscp = — и
E(_i/2) =-^-§|ляЯ0 (29)
Таким образом, диполь поглощает энергию магнитного поля. Разность уровней энергии составляет
\E = g^H0 (30)
Отсюда можно сделать важный вывод о том, что в случае проявления ядерного магнитного резонанса энергия квантов пропорциональна напряженности внешнего магнитного поля.
Необходимая энергия AE=7rv подводится к ядру высокочастотным излучением. При этом изменение энергии в обоих направлениях, т. е. излучение и поглощение, индуцируется при постоянной переориентации от магнитного момента ядра.
При любом взаимодействии излучения с веществом поглощение энергии происходит только в том случае, если число уровней N- с более низким энергетическим состоянием (незаселенных) оказывается меньше, чем число уровней N+ с более высоким энергетическим состоянием (заселенных).
Отношение числа заселенных и незаселенных уровней при тепловом равновесии определяется распределением Больцмана (k— константа Больцмана, Т — абсолютная температура):
М / ХЕ \ ,о..
-jy— = ехр kT j (31)
При постоянной температуре с ростом Д£ отношение N+/N-^величивается.
Поэтому оно растет с увеличением напряженности магнитного поля Но. Следует, однако, отметить, что при использовании Других спектроскопических методов это отношение имеет значительно более высокое значение. Поглощение энергии высокочастотного поля соответствующей частоты способствует быстрому заполнению всех незаселенных уровней. Так называемые релаксационные процессы обеспечивают преобразование поглощенной энергии в тепловую с помощью механизмов, природа которых здесь не обсуждается. Время, необходимое для установления теплового равновесия (время релаксации), должно быть как можно меньшим, в противном случае будет проявляться эффект насыщения, который препятствует наблюдению резонансного сигнала.
Во внешнем магнитном поле с Яо = 7,9-1О5 А/м резонансные астоты находятся в диапазоне 1—50 МГц, что соответствует етР°вому диапазону радиоволн. Поэтому в методе ядерного агнитного резонанса используется аппаратура, аналогичная
72
Строение материи
Химическая связь
73
применяемой в радиосвязи. Основная проблема при постановке эксперимента состоит в создании постоянного магнитного поля. Обычно при измерении спектров ядерного магнитного резонанса частоту высокочастотного поля поддерживают постоянной, а меняют Но. Поэтому, как правило, ширину линий и расстояние между ними измеряют в единицах напряженности магнитного поля.
Ядра различных элементов отличаются своим ядерным спином. Все ядра с четным числом протонов и нейтронов имеют ядерный спин /=0 и поэтому не обнаруживают ядерного парамагнитного резонанса. В наибольшей степени резонанс проявляется в ядрах с / = 7г и большим магнитным моментом, а именно в нуклидах 'Н, 19F, 31Р, имеющих сравнительно большое значение гиромагнитного отношения g.
Резонансная частота зависит как от g, так и от внешнею магнитного поля
gjMA /ось
Рис. А.26. Резонансный спектр протона в молекуле этанола.
Из-за экранирующего действия электронного облака атома магнитное поле вблизи ядра Яэфф несколько ослабляется:
Я9фф«Я0-аЯ0 (33)
где о называют константой ядерного экранирования. Поэтому если в веществе (или различных веществах) содержатся одинаковые резонансные атомы, но в различном окружении, то резонансный эффект для этих атомов наблюдается при различных значениях напряженности магнитного поля.
Классическим примером может служить спектр ядерного магнитного резонанса этанола, на котором резонансные линии атомов водорода, имеющих различное химическое окружение, находятся на расстояш и,
соответствующем напряженности магнитного поля ~0,1 А/м. По мере увеличения напряженности поля можно заметить три линии, интенсивности которых относятся как 1:2:3 (рис. А.26). Это явление, полезное для выявления структуры молекул, называют химическим смещением.
На практике проводят сравнение исследуемой пробы с образцовым препаратом, в котором содержится тот же резонансный атом, как и в пробе; молекула вещества образцового препарата имеет симметричное строение и поэтому на спектр6
ЯМР проявляется лишь одна четкая резонансная линия (например, Si(CH3)4 для резонанса протона). При постоянной частоте v высокочастотного поля по мере увеличения напряженности магнитного поля Но получают резонансное значение Н для вещества-пробы и ЯобР для образцового препарата. Для оценки величины химического смещения при постоянной частоте используют отношение
6 = -Н ~ Нобр (34)
"обр
где 6 — безразмерная величина, как правило выраженная в миллионных долях (м. д.).
С увеличением разрешающей способности аппаратуры можно достигнуть дальнейшего расщепления резонансных линий,
Рис. А 27. Сверхтонкая структура спектра ядерного магнитного резонанса PF3. причем в отличие от химического смещения расстояние между линиями не зависит от напряженности внешнего поля. Это расщепление связано с взаимодействием магнитных моментов соседних ядер через связывающие пары электронов. Поэтому это явление называют спин-спиновым взаимодействием.
В связи с различными возможностями ориентации ядра А под влиянием магнитного момента ядра В со спином I линия Wa А расщепляется на мультиплет (2/-J-1). В присутствии п эквивалентных соседних ядер с ядерным спином I число состояний становится равным 2п/+1. Распределение интенсивности линий зависит от статистического распределения ядерных спиновых состояний и для ядер с /=72 соответствует последовательности биномиальных коэффициентов. В качестве примера рассмотрим сверхтонкую структуру спектра молекулы PF3. Резонансная линия ядра 19F под влиянием соседного ядра 31Р со спином >/2 расщепляется на две линии (рис. А.27, а). Резонансная линия ядра фосфора под действием трех одинаковых ядер „ „ 0 спином I=ll2 дает «квартет» с отношением интенсивности 1:3:3 : 1 (рис. А.27, б).
6 1 7
Дифракционные методы
Дифракционные методы основаны на применении рентгенов-их лучей, быстрых электронов и нейтронов с массой m и ско-стью v, т. е. частиц, соответствующих длине волны X = h!mv,
74
Строение материи
Если длина волны близка по порядку величины размерам молекул и расстояниям между ними, то наблюдается известная интерференционная картина, изучение которой позволяет получить ценные сведения о структуре вещества. Рентгеновские лучи и электроны рассеиваются на электронных оболочках атомов, причем в первом случае (рентгеновские лучи) главную роль играют максимумы электронной плотности, а во втором случае (пучки электронов) — неоднородность электрического поля вблизи атомных ядер. Рентгеновский метод наиболее ценен при определении структуры кристаллических соединений (его основы рассматриваются в разд. 6.4.1). Здесь обсуждают только наиболее существенные аспекты определения строения отдельных молекул с помощью дифракционных методов. Строение молекулы можно установить вполне однозначно, если получить дифракционную картину вещества в газовой фазе (пар). Однако из-за низкой плотности рассеивающей среды для получения дифракционной картины в рентгеновских лучах необходима экспозиция в течение многих часов, а для получения элект-ронограммы — в течение нескольких секунд. Поэтому для исследования молекул в газовой фазе применяется преимущественно метод электронографии.
По сравнению с простыми ионными решетками расшифровка структуры молекул представляет собой значительно более сложную задачу, в особенности для несимметричных молекул. Наряду с определением углов рассеяния необходимо измерять интенсивность рассеянного излучения. Так как из дифракционной картины нельзя получить фазовые характеристики волнового излучения, рассеянного в веществе, прямое определение структуры во всех подробностях становится невозможным. Поэтому рассчитывают интенсивность рассеянного излучения исходя из нескольких возможных модельных структур молекулы и сравнивают результаты расчета с экспериментальными данными. При таком методе «проб и ошибок» параметры модельной структуры меняют до тех пор, пока не получат полного совпадения между теорией и экспериментом.
Интенсивность рассеянного излучения в первую очередь зависит от количества электронов в электронной оболочке и увеличивается по мере повышения порядка симметрии. Тем самым становится возможным локализовать положение отдельных атомов в молекуле. Однако положение атомов водорода можно установить лишь косвенно — как областей с минимальной электронной плотностью.
В отдельных случаях важные сведения о строении вещества могут дать дифракционные картины, полученные с помошьЮ нейтронного излучения, когда дифракция происходит на атомных ядрах, а интенсивность рассеяния определяется спином
Химическая связь
75
лдра. Потоки нейтронов необходимой интенсивности получают в ядерных реакторах.
Длина волны де Бройля нейтрона близка по порядку величины размерам молекул, поэтому нейтроны тормозятся веществом (например, тяжелой водой D2O). Измерение дифракции потока нейтронов (обычно для твердых препаратов) проводится, как правило, с помощью «борфторидных счетчиков» (в результате ядерной реакции 'QsB + 'on—^73Li + 42He образуются «-частицы, которые можно обнаружить обычными методами).
Несмотря на необходимость применения сложной аппаратуры, результаты измерения дифракции нейтронов оправдывают затраты и имеют большую ценность. Так, например, этим методом удалось точно установить расположение некоторых легких атомов относительно более тяжелых. Были также исследованы структура льда и строение ионов HF2“, благодаря чему выявлена природа водородной связи в этих соединениях. Дифракция нейтронов позволяет также различать атомы с одинаковым порядковым номером, так как сечение захвата нейтронов сильно зависит от природы элемента.
6.2. Химическая связь
Перейдем теперь к коренной проблеме теоретической химии. Что такое химическая связь? Что связывает атомы в молекуле? Почему одни комбинации атомов стабильны, а другие — нет? Почему, например, устойчива молекула Н2, а не Н3 (насыщение химической связи)? Почему инертные газы не образуют двухатомных молекул? На все эти вопросы может дать ответ только квантовая химия. Поскольку все эти проблемы представляют собой в известной мере сущность химической науки, каждому начинающему химику необходимо уяснить себе основные подходы к решению этих проблем.
Ранее уже упоминалось, что нет принципиального различия между природой межатомной химической связи и природой устойчивости самих атомов. «Силы», которые удерживают систему— атом гелия (ядро и два электрона), те же, что и в молекуле водорода Н2 (два ядра, два электрона) или в молекулярном ионе водорода Н2+ (два ядра, один электрон). Рассмотрим образование химической связи на примере Н2+-иона и молекулы Н2, так как на этих примерах удобнее всего познакомиться с методами квантовой механики.
6’2.1. Молекулярный ион водорода Н2+
Продолжим ту же линию, которая была намечена при построении периодической системы на основе электронной модели атома. Будем исходить не из полностью построенных атомов,
76
Строение материи
а из ядерного остова молекулы (например, из двух протонов) и исследуем, в каких состояниях может находиться электрон в потенциальном поле двухъядерного остова. Последовательно заселим эти молекулярные состояния («молекулярные орбитали») электронами и тем самым построим систему двухатомных молекул, учитывая принцип Паули и последовательность энергетических уровней, подобно тому как это было сделано при построении моделей элементов периодической системы. Возьмем за исходную модель молекулярный ион Нг+, который играет в теории химической связи такую же роль, как атом водорода в теории строения атомов. Этот метод можно затем улучшить, полагая, что электрон движется в эффективном усредненном поле остальных частиц.
Рассмотрим, что собой представляет в общем виде волновая функция электрона ф в двухъядерном поле. Примем, что срд — функция состояния электрона в поле ядра А, а фв— аналогичная функция в поле ядра В. Естественно предположить, что в нулевом приближении (не рассматривая условий нормирования) волновая функция ф является линейной комбинацией функций Фа и фв, т. е. фа±фв. При этом состояние, соответствующее сумме волновых функций и учитывающее взаимодействие электронов, оказывается более стабильным:
Ф2 = Фа2_ЬФв2 ± 2фдФв
Значение члена 2фАфв становится тем более весомым, чем больше вероятность нахождения электрона между ядрами, т. е. (как увидим ниже) состояния, «благоприятного» в энергетическом отношении. С помощью приближенных методов решения «одноэлектронного» уравнения Шрёдингера
д , , 2m (г-, е2 , е2 , е2 \ , п
ДФ+~ь2~ £-----dH-----------Ф = °
Y 1 Й2 \ R 1 г а 1 гв }
(где R — расстояние между ядрами, гА — расстояние электрон — ядро А, Гв — расстояние электрон — ядро В) можно определить параметры молекулы, хорошо согласующиеся с экспериментом.
Познакомимся с помощью приближенных методов вариационного исчисления с основами количественного расчета этой системы. За основу примем приближение Борна — Оппенгеймера: движение ядер и электронов происходит независимо друг от друга; каждому заданному состоянию ядра соответствует определенная энергия электронов. Вследствие сравнительно большой массы ядра погрешность расчета очень мала (например, по ван Флеку для Н2+ составляет <0,0075 эВ). Энергетические переходы можно грубо оценить следующим образом: электронные переходы — от 1 до 10 эВ; колебательные—10~' эВ, крутиль
Химическая связь
77
ные— Ю-2 эВ, вращательные— Ю-3 эВ. Переведем рассмотренное выше уравнение Шрёдингера
(35>
в принятую в квантовой химии форму
//ф — £ф (36)
Особенность уравнения (35) состоит в том, что в нем использован оператор Гамильтона
Я= — ----------— (37)
2т 1 R гл гв v ’
Умножим уравнение (36) на ф (напомним, что с левой стороны уравнения стоит оператор) и после интегрирования по всему объему получим следующее выражение для энергии:
I ibfftydv
£ = Л------ (38)
1 фМи
Для того чтобы использовать методы вариационного исчисления, введем в качестве вариационного параметра линейную комбинацию коэффициентов:
ф = слФаЧ-свФв (39)
Энергия минимальна, если
_££_==_5£_ = 0 дсА ~ дсв
Подставим сначала уравнение (39) в уравнение (38):
I (слФа + свфв) Н (сдфА + св<рв) dv
Е — ----------------------------
J (Сафа + свФв)2 dv
Вынесем постоянные за знак интеграла:
Е сА2 J <pA//<pAdu + 2сасв J фл^фв^с + св2 J фцЯфв* СА2 У Фа2^ + 2сАСВ у ФаФв* + св2 У фв2*)
и введем следующие обозначения:
[ ФА//<рв(й) = /7АВ—рЛВ —резонансный интеграл (ковалентный)
J tpA//cpAdo = ЯАА — аА —кулоновский интеграл
J ФаФв<^ = 3Ав —интеграл перекрывания
78
Строение материи
Тогда Р____________________ са2"аа 2сдсв//дв Ч~ св2//вв f40\
ca2Saa + 2сд<?в5дв + Св2-Ь’вв '
Продифференцируем последнее выражение по сА:
1 ЭЕ __ (сдНдь + сасв//ав) (са25аа + 2сасв5аб + cb2Sbb)_
2 дед (ca2Saa + 2cacbSab + cb2SBb)2
__ (са2#аа + 2сдсвЯдВ 4- св2Явв) (СД^АА + CB'Sab) __ о
(ca2Saa + 2cbcaSab + св25вв)2
Умножим это уравнение на выражение в знаменателе, сделаем сокращения и, учитывая уравнение (40), получим
са(^аа—^Аа)4*СВ (^АВ ^ЛВ)=0 (41)
Аналогичный расчет для дЕ/дсъ = 0 дает
са(^ав £Зав)-}--с’в (Явв- £Sbb) = 0
Эта система линейных однородных («вековых») уравнений имеет нетривиальное решение для сЛ и св только в том случае, ес-
Рис. А.28. Связывающая (ips) и разрыхляющая (1рд) линейные комбинаций ls-орбиталей.
ли детерминант системы уравнений равен нулю. Проведя нормирование (£Аа = £вв:=1) и учитывая, что ал=«в^а, получиА а—£ Рав~£3 ав =Q I
Рав — £^ав а I
Отсюда, решая квадратное уравнение относительно Е, получим’ два корня:
£s = -”£oAB и £As= ?--LAb (42)
5 1 + Sab 5 1 — Sab
Химическая связь
79
Коэффициенты сА и св можно получить после подстановки Es и Eas, в уравнение (41). Кроме того, следует учесть, что функции фэ и фл должны быть нормированы. Тогда для симметричного решения получим
са~~ св
1
/2(1 +SAB)
а для антисимметричного
1
£ л — ~ 1 - —~ — С д
А у 2(1- SAB)’
На рис. А.28 показана зависимость функций Фэ и фА от R для ls-орбитали.
Таким образом, для расчета энергии необходимо знать следующие три интеграла: а, рАв и Sab. С расчетом этих интегралов можно ознакомиться в специальной литературе. Зависимость энергии от расстояния между ядрами, полученная
Рис. А.29 Энергия иона IM как функ ция расстояния до ядра.
на основе этих интегралов для симметричного и антисимметричного состояний,
представлены на рис. А.29.
Такой расчет дает ~60% экспериментально найденного значения для энергии связи Е—Е^ (268,4 Дж/моль). Уточнение нашей весьма простой модели (увеличение числа вариационных параметров, введение эффективного ядерного заряда) приводит
к хорошему согласию между теорией и опытными данными.
6.2.2. Природа химической связи
Попробуем внести ясность в проблему определения энергетического баланса молекулярного иона водорода Н2+. Часто обсуждают вопрос о том, за счет какой энергии электронов образуется химическая связь: кинетической или потенциальной? Однако такой альтернативы не существует, так как в квантовой механике, так же как и в классической, справедлива так называемая вириальная теорема, которая утверждает, что при Равновесии между средней кинетической энергией Т и средней кулоновской потенциальной энергией V имеет место следующее соотношение:
Г=-1/2У
80
Строение материи
Химическая связь
81
Нулевая точка отсчета.
и+ энергии
о г-ион и ядро совершенно
Н-атом
Кинетическая энергия
-30 Н
Потенциальная (кулоновская) энергия
Суммарная энергия
Энергия химической связи
удалены друг от друга)
Рис. А.30. Кинетическая н потенциальная энергия атома Н и нона На+.
Общую энергию Е = Г+ V можно выразить через кинетическую или потенциальную:
Е = —Т; E = V/2
Таким образом, при образовании химической связи, когда общая энергия системы понижается (по сравнению с энергией отдельных атомов), уменьшается уровень потенциальной энергии и (до некоторой степени автоматически) происходит увеличение кинетической энергии. Схематически это показано на рис. А.30.
Согласно принятой (хотя и неверной) точке зрения, изменения V и Т чаще всего объясняют следующим образом: для связывающей молекулярной орбитали волновая функция соответствует повышению плотности заряда между ядрами, т. е. в нее входит член + 2фАфв, который добавляется к «классической» плотности заряда фд2+фв2 и соответствует «интерференции» атомных волновых функций или так называемой «плотности заряда перекрывания». Это все в известной мере верно. Однако во многих книгах такая интерференция рассматривается как непосредственная причина понижения V («компенсация отталкивания ядер») и увеличения Т. Вот этот вывод и является ошибочным! «Интерференция» в большей степени приводит к понижению кинетической энергии. Вернемся к схеме, изображенной на рис. А.28. Если учесть, что кинетическая энергия Т в направлении R пропорциональна d2ty/dR2, то, вследствие то
го что минимум функции ф2 между ядрами становится менее-резким, она понижается. Из равновесных значений Е, V и Т, соответствующих «вириальному состоянию», Е имеет более низкое значение вследствие «сжатия» всей молекулы (с более значительным понижением V по сравнению с увеличением Т). Такое сжатие «электронного облака» согласуется с теорией, если «уточнить» расчет, сделанный в разд. 6.2.1 на основе вариационного исчисления путем введения второго вариационного параметра (наряду с линейной комбинацией коэффициентов с). Таким параметром служит коэффициент в показателе степени экспоненциальной волновой функции исходных атомов*. Минимум энергии наблюдается при значении параметра, соответствующем сокращению электронного облака. Итак, природу химической связи можно представить себе следующим образом: при «перекрывании» исходных электронных оболочек атомов возникает «выгодный» в энергетическом отношении эффект «интерференции», сущность которого может быть раскрыта только-методами квантовой механики. Такая «интерференция» вызывает увеличение заряда в пространстве между ядрами за счет заряда, находившегося вблизи них. Таким образом, «провал» плотности заряда между ядрами «выравнивается», что приводит к сильному понижению кинетической энергии (при небольшом увеличении потенциальной). Это вполне соответствует балансу энергии, но противоречит вириальной теореме. Последняя удовлетворяется за счет того, что при образовании молекулы идет и другой энергетически выгодный процесс — сжатие электронного облака всей молекулы. Оба процесса протекают таким образом, что вириальная теорема выполняется: устойчивое состояние молекулы достигается на более низком уровне энергии.
6.2.3. Обменное вырождение. Молекула Н2. Метод валентных связей (ВС)
Прежде чем перейти к двухэлектронным системам, рассмотрим еще один из важнейших подходов к решению проблемы химической связи, который, впрочем, не касается общего энергетического баланса (разд. 6.2.2).
Он распространяется не только на такую двухэлектронную систему, как молекула водорода, но и на атом гелия, и на любую другую двухэлектронную систему. Оба электрона 1 и 2 характеризуются координатами Xi и х2 и волновыми функциями 'I’m и фп. Отвлечемся сначала от взаимодействия между части-
* п л I 1 V/»
Волновая функция записывается, например, в виде ф= — I е-аг/яо
1Аей К., Селбин Д. Теоретическая неорганическая химия. Пер. с англ. — М.: иМня, 1969, с. 165]. — Прим, перев.
6-2027
82
Строение материи
цами и представим себе, что мы изучаем нейтральные частицы, например нейтроны. В этом случае общая энергия представляет, собой сумму энергий обоих электронов
£ = £14-£2
а волновая функция системы — произведение соответствующих волновых функций
Ф(1, 2) = фт(1)-фп(2)
Это следует из того, что вероятность независимого («несв} ванного») совместного появления двух независимых событи пропорциональна произведению их вероятностей (разд. 3.6)*.
Мысленно поменяем местами оба электрона. Очевидно, эне] гия системы останется прежней, в то время как волновые фун! ции электронов изменяются. (Пример: <pm = sinx, <pn = cosx, Xi = 0, х2=л/2. Тогда -ф (1, 2)=0 и ф (2, 1) =фт(2)-фп(1) = 1.)
Таким образом, одному и тому же значению энергии соответствуют две волновые функции ф(1, 2) и ф(2, 1), поэтому эти состояния являются вырожденными, иначе говоря, наблюдается обменное вырождение. В действительности никакого «обмена» не происходит, поскольку нельзя различить, «имеет» ли данный электрон волновую функцию <ра или ф&. Поэтому все электроны неразличимы, и функции ф(1,2) и ф(2, 1) не совсем точно отражают состояние двухэлектронной системы. Более удачно приближение, согласно которому, если волновые функции относятся к системам с одной и той же энергией, решение уравнения Шрёдингера можно представить в виде так называемой линейной комбинации волновых функций:
ф = аф0+₽фь (а, ₽—некоторые константы)
Таким образом, общее решение уравнения Шрёдингера для данной системы при условии отсутствия электростатического взаимодействия между частицами представляет собой линейную комбинацию функций:
ф = аф(1,2)+₽ф(2, I)
Вероятности состояния системы при обмене электронов равны, т. е.
[аф(1,2)4-рф(2, 1)]2 = [аф(2,1) + ₽ф(1,2)]2
Отсюда а = ±|3, а в наиболее простом случае «=1, р = ±1-Таким образом, для данной системы существуют только две
* Если между частицами наблюдается взаимодействие, правило произведения вероятностей соблюдается лишь приближенно. При использовании это-ю правила в расчетах говорят о «модели независимых частиц».
Химическая связь
S3
волновые функции, имеющие физический смысл:
ф<. = ф(1,2)+ф(2, 1)
фА = ф(1,2)—ф(2, 1)
Их называют симметричной и антисимметричной собственными волновыми функциями (разд. 3.6).
При более подробном изучении этих волновых функций, если представить их графически на координатной плоскости, можно заметить примечательный факт. Несмотря на то что было исключено всякое взаимодействие между частицами, волновые функции фз и фд отличаются тем, что в фА-состоянии частицы движутся так, как будто они избегают друг друга. Для выбранного примера фт=зшх, фп = созх это легко показать:
ф3 = sin Xi cos х2 + sin х2 cos
фА = sin cos x2—sin x2 cos xt
Если координаты частиц совпадают, т. е. Xi=*2, после подстановки в вышеприведенные уравнения получим, что фА = 0> a <ps имеет некоторое конечное значение. Напрашивается один из вариантов трактовки: несмотря на принятое допущение об исключении взаимодействия, между частицами действует какая-то «сила», которую можно было бы назвать «обменной силой». В природе известен другой пример того, что в системе, состоящей из большого числа частиц, некоторое состояние предпочтительнее по сравнению с другими возможными состояниями системы. При этом оказывается ненужным привлекать к рассмотрению никакие силы; для объяснений достаточно понятие энтропии, введенного термодинамикой. Таким образом, легко видеть, что если учесть взаимодействие частиц, т. е. их электростатическое притяжение или отталкивание, то из-за различий в характере движения электронов в состояниях фА и фз вырождение снимается. Оба состояния характеризуются различными энергиями. Какое состояние при этом «устойчиво» — симметричное или антисимметричное, — зависит от значения потенциала, под действием которого находятся частицы. Если последний равен нулю, то принимается во внимание только электростатическое взаимодействие электронов между собой и состояние, характеризующееся волновой функцией фА, «устойчивее», чем для функции ф§. Как было показано в разд. 3.6, функция описывает состояние электронов с одинаковым спином. В этом случае обменное взаимодействие коррелирует с кулоновским взаимодействием. Такое обменное взаимодействие для антисимметричной функции фА называют также «корреляцией по ФеРми». В фэ-состоянии такой корреляции с кулоновским взаимодействием не существует.
6*
Химическая связь
85
84________________ Строение материи________________________
В уравнении Шрёдингера для нашей системы
(Е-и^=о
(Д1 и Д2 относятся к координатам Xi, yi и z1 и х2, Уг и z2 электронов 1 и 2) потенциальная энергия £ представляет собой довольно сложную функциональную зависимость, в которой учитываются не только взаимодействия ядро — ядро и ядро — электрон, но и взаимодействие между электронами.
Строгое решение этой задачи для четырех частиц невозможно, поэтому применяют приближенные методы, например метод возмущений. Последний был развит в классической теории движения небесных тел, в особенности для солнечной системы. Примем, что молекула водорода Н2 в нулевом приближении состоит из двух невзаимодействующих атомов водорода а и b в основном состоянии (1s). Тогда общая энергия равна сумме энергий отдельных атомов:
Если учесть межатомное взаимодействие, возрастающее по мере сближения атомов, то вырождение двух возможных состояний системы фз и Фа снимается. Расчет изменения энергии за счет возмущающего воздействия здесь опущен; остановимся только на конечных результатах такого расчета и дадим качественное описание поведения системы. Устойчивое состояние в противоположность «невозмущенному состоянию» имеет более низкую энергию и соответствует симметричной волновой функции
Фэ = Фо(1) Ф6(2)+<Ра(2) Ф6(1)
Это объясняется тем, что вследствие уже названных причин устойчивым является состояние, имеющее более высокую вероятность нахождения электронов между ядрами. Даже в случае отсутствия взаимодействия электроны в этом состоянии «не избегают друг друга», т. е. орбиты электронов симметричны без корреляции по Ферми.
Найдем вероятность нахождения электронов для молекулы водорода (для чего возведем в квадрат волновые функции как симметричного, так и антисимметричного состояний):
симметричная функция
ФЛО фЛ2) + фв2(2) <р62(1) + 2Фа(1) <р6(1) Фа(2) Фб(2)
антисимметричная функция
Фа2(1) Фь2(2)+фа2(2) ФЬ2(1)- 2фа(1) ф6(1) фа(2) ф6(2)
Из сравнения этих выражений (а именно третьего члена) следует, что вероятность нахождения частицы между ядрами вы-
ще для фз-состояния, чем для фд. Третий член этих выражений, имеющий для симметричной функции положительный знак, указывает на то, что как электрон 1, так и электрон 2 принадлежат обоим ядрам. На рис. А.31 показаны вероятность нахождения электронов для обоих состояний в виде так называемых «геодезических» линий, т. е. линий одинаковой плотности за-
Рис А.31. Линии одинаковой электронной плотности для симметричного (а) и антисимметричного (б) состояний молекулы Н2.
Следует также выяснить, не противоречит ли волновая функция фз принципу Паули. Согласование наблюдается, если при одинаковых значениях п, I и т электроны отличаются спином, вследствие чего общие волновые функции электронов од становятся антисимметричными. Тогда выполняется требование (разд. 3.6) об антисимметричности волновой функции молекулы водорода
Фа = Фз°а
Решение методом возмущений дает следующие значения энергии ез и ед для состояний фз и фд (вывод опущен): es = 2£0-}-£s (/?); £s(fl)=-£±£
8A=2£0+£aCR); £aW = 4e4
Энергии С, А и S представляют собой интегральные функции Расстояния между ядрами R (эти функции называют кулоновским интегралом, обменным интегралом и интегралом перекрывания). Вычисление значений этих интегралов, как правило, очень сложно. А и С в определенной области R отрицательны, причем |А| > |С|. С соответствует классическому, а А — неклассическому электростатическому взаимодействию. Тогда для ^s-состояния молекулы водорода общая энергия оказывается больше по абсолютной величине, чем энергия 2£о для двух изолированных атомов водорода. Следовательно, состояние, соот-еетствующее связанным в молекулу атомам, более устойчиво
86
Строение материи
(Ео =—1320 кДж/моль — энергия, которая выделяется при образовании */г моль молекул водорода из атомов). Разность энергии для фз- и фл-состояний зависит от значения обменного интеграла. На рис. А.32
Теоретическая и эксперимен-зависимость потенциальной
Рис А 32. тальная энергии молекулы Н2 от расстояния между атомами (Ра — расстояние в единицах воровского радиуса 1/а).
представлены графики функций £а(Д), Es(R) и C(R). В Фа-состоянии атомы только отталкиваются, для ф3-состояния получается потенциальная кривая, хорошо согласующаяся с экспериментальными данными. Из нее можно определить и равновесное расстояние между атомами, и энергию связи.
Энергия Es молекулы водорода значительно меньше энергии образования связи Ео. Для Н2 2Ео = —2640 кДж/ /моль, a Es = —452 кДж/ /моль. Для более крупных молекул Es еще меньше; например, для связи О—Н в молекуле воды энергия Es составляет лишь 0,4% энергии, которую необходимо затратить на диссоциацию воды на составляющие ее атомы.
Со времени опубликования (1927 г.) предложенного Гейтлером и Лондоном квантовомеханического расчета строения молекулы водорода приближенные методы были существенно улучшены, и в настоящее
время экспериментальные и расчетные данные хорошо согласуются между собой.
Следует лишь еще раз отметить, что именно характер изменения потенциала вблизи ядра — причина наибольшей предпочтительности в энергетическом отношении той или иной (симметричной или антисимметричной) многоэлектронной волновой функции Для двухъядерной системы (молекулы водорода Н2) это симметричная волновая функция. Однако если обратиться к системе с электронами, имеющими различные волновые функции и одинаковую энергию (например, р- или d-электроны атома), то наиболее выгодна в энергетическом отношении антисимметричная функция: электроны при такой «геометрии» потенциала как бы «избегают» друг друга Это и послужило причиной введения правила Хунда (разд. 55) Так как функция Ф
Химическая связь
87
симметрична, то в соответствии с принципом Паули о должна быть антисимметрична, т. е. спины должны быть параллельны (разд 3 6).
Надо совершенно ясно представлять себе, что приближенный метод, с которым мы здесь познакомились, является лишь одним из многих возможных Другой подобный метод описан в следующем разделе. Однако рассмотренный метод и полученные результаты полезны, поскольку соответствуют привычной картине так называемой «атомной» или «ковалентной» связи (метод валентных связей). Для наглядности такие связи изображаются с помощью пары точек (пара электронов) (Н : Н) или черточки (Н—Н). Понятие валентности и явление насыщения химической связи хорошо объясняются с помощью метода валентных связей (разд. 6.3).
6.2.4. Молекулярные состояния (метод молекулярных орбиталей, МО)
Итак, мы познакомились с двумя приближенными решениями уравнения Шредингера для молекул. Ранее (разд. 6.2.1) было показано, как, исходя из одноэлектронной модели молекулярного иона водорода Н2~, можно построить в некотором роде «периодическую систему» двухатомных молекул. Для применяемого при этом метода «молекулярных орбиталей» (МО) характерно «заполнение» молекулярной (а не атомной) орбитали ф последовательно одним, а затем и двумя электронами В методе валентных связей (ВС) Гейтлера — Лондона исходят из атомных орбиталей, занятых одним электроном, а далее переходят к двухэлектронной системе (Не или Н2) путем линейной комбинации занятых атомных орбиталей, в которой учитывается неразличимость электронов.
Теперь, используя метод молекулярных орбиталей, составим волновую функцию молекулы водорода. Для связывающей молекулярной орбитали двух электронов (коэффициент нормирования при этом опускается) запишем функцию ф5 и по тем же соображениям, что и при решении по Гейтлеру — Лондону, составим произведение
^н2м0= [(Фа+Фв) (1)1 [(Фа+Фв) (2)1
Поскольку эта двухэлектронная функция симметрична, она Должна быть, так же как и функция по Гейтлеру — Лондону, Умножена на антисимметричную спиновую функцию суА, которая, разумеется, соответствует состоянию двух электронов с противоположными спинами.
Определим, чем отличаются волновые функции молекулы водорода, рассчитанные методом МО и ВС. Для этого упростим
88
Строение материи
полученное выше выражение
7JrH2MO = Фа(0 Фв(2)+Фа(2)<Рв(1)+Фа(1) Фа(2)+Фв0) Фв(2)
Сравним полученную сумму с функцией, соответствующей методу ВС:
^н2ВС = Фа(1)Фв(2) + Фа(2)Фв(1)
Мы видим, что в этих уравнениях первые два члена одинаковы, однако в методе МО появляются два дополнительных члена. Член <ра(1)фа(2) дает вероятность того, что оба электрона находятся вблизи ядра А, а фв(1)фв(2)—вероятность, что оба электрона находятся у ядра В. Эти члены отвечают в ходе приближенных расчетов за ионное состояние, которое схематически можно записать так:
[НА:ГНв+ и НА+[НВ:]-
При подходе Гейтлера—Лондона такие состояния не рассматриваются. Расчет методом МО дает лучшее согласие с экспериментом, если «ионные члены» подставить в функцию МО с «весом» меньше 1.
¥ = Фа(П Фв(2)+Фа(2) Фв(1) + 0,2 [Фа(1) Фа(2)+фв(1) Фв(2)1
Из дальнейшего будет легко понять, почему гипотетическая молекула Не2 значительно менее устойчива, чем молекула Н2. В последней имеются два связывающих электрона, а в гипотетической молекуле Не2 кроме двух связывающих имеются и два разрыхляющих электрона. С молекулярными орбиталями двухатомных молекул, образованных следующими элементами периодической системы, мы познакомимся позднее.
6.3. Молекулы с преимущественно ковалентными
связями
Чтобы убедиться в больших возможностях квантовомеханических методов, рассмотрим молекулы более сложного состава и строения, а также соединения с некоторыми особыми свойствами. При этом надо всегда помнить, что многие свойства веществ вообще становятся понятными лишь на основе представлений квантовой механики (например, валентный угол, магнитные свойства кислорода).
Прежде всего воспользуемся методом валентных связей (ВС), иначе говоря, когда связь образуется парой электронов, а затем познакомимся с несколькими примерами применения метода молекулярных орбиталей (МО) в неорганической химии.
Химическая связь
89
зб.ЗЛ. Валентность, валентный угол. Гибридизация
Попытаемся присоединением атома водорода получить из молекулы Н2 молекулу Н3. В Н2 спины антипараллельны, в атоме водорода спин может принимать любое из двух возможных значений
Н-Н н АВС Ф I t 12 3
При перестановке электронов 2<->3 получится следующая структура:
Н-Н н t t I
Однако этому состоянию, как мы видели, соответствует антисимметричная функция, т. е. состояние является неустойчивым, и ядра атомов отталкиваются друг от друга. При перестановке электронов с симметричным спином 1*-> 3 также получается антисимметричная волновая функция
Фа(1)Фс(3)—<Ра(3) ФсО)
г. е. и в этом случае происходит отталкивание ядер. (Этот вывод, впрочем, верен только для сравнительно больших расстояний.) Итак, мы видели, что молекула Н3 неустойчива, так как в молекуле Н2 химическая связь насыщена. В этом смысле водород обладает валентностью 1. Таким же образом можно показать, что молекулы НеН и Не2 также неустойчивы, так как в атоме гелия в основном состоянии 1s2 спины электронов антипараллельны. Это верно также и для остальных инертных газов, валентность которых равна нулю. В настоящее время получены соединения инертных газов, однако характер связи в этих соединениях является предметом дискуссии.
Вообще, понятие «валентность» можно без всяких оговорок применять только к ковалентным, или, как иногда говорят, «го-Таблица А.8. Валентность элементов первых периодов
Характеристики н Не ы Be в с N 0 F Ne
S Валентность основного состояния простейшие соединения с водородом 1/ /2 1 н2 0 0 1 LiH 0 0 (ВеН2) V2 1 вн8 1 2 СН4 з/ / 2 3 NH3 1 2 ОН2 ’! FH 0 0
90
Строение материи
меополярным», соединениям. В табл. А.8 приведены валентности первых десяти элементов периодической системы.
Для определения валентности элемента недостаточно рассматривать только основное состояние атома. Это видно на примере атома углерода. Он имеет два нескомпенсированных 2р-электрона. Энергия, необходимая для того, чтобы возбудить электроны из состояния 2s до состояния 2р, довольно велика и
Рис. А.34. Образование двух s—р-связей.
Рис. А.ЗЗ. Образование s—s-связи.
составляет 436,2 кДж/моль. Однако эта энергия возвращается, если происходит насыщение четырех нескомпенсированных валентных электронов за счет связи с атомами водорода. Расчет показывает, что при образовании метана СН4 выделяется энергия 102,6 кДж/моль. Таким образом, sp3 является истинным валентным состоянием углерода. Эти соображения, однако, неверны в том случае, когда необходим переход электрона на уровень с большим главным квантовым числом, так как на это затрачивалась бы слишком большая энергия. Поэтому, например, азот не образует пяти, а кислород — четырех ковалентных связей. Напротив, существует ион
г н Т
I H-N-H
I н
в котором атом азота имеет на один электрон меньше, чем в несвязанном виде, — положительно заряжен — и валентное состояние которого аналогично состоянию углерода в метане.
При обсуждении строения молекулы водорода было показано, что связь между ядрами организована таким образом, что плотность электрического заряда становится наибольшей вдоль оси, соединяющей оба ядра. В этом случае «электронные облака» атомов перекрываются в наибольшей степени. Такая s—s-связь показана на рис. А.ЗЗ. Вследствие сферической симметрии
Химическая связь
91
s-состояния все направления в пространстве относительно связи s—s равноценны. Однако это не так, если атом с р-электрона-ми вступает во взаимодействие с двумя атомами, имеющими s-электроны. На рис. А.34 показаны такого рода связи в молекуле воды. Видно, что при максимальном перекрывании валентный угол между связями должен быть равен 90°. На рис. А.35
изображена такая же схема для пирамидальной структуры молекулы NH3. Однако экспериментально определенный валентный угол в этих молекулах, как правило, несколько больше. Это можно объяснить взаимным отталкиванием не связанных непосредственно между собой атомов, а также гибридизацией атомных волновых функций кислорода или азота (табл. А.9).
Для определения валентного угла недостаточно исходить только из волновых функций, соот-
ветствующих основному Рис А 35 Образование трех з—р-связей. состоянию атомов. На
пример, обратимся вновь
к атому углерода. Судя по валентному состоянию, в молекуле СН4 должны быть три связи s—р и одна связь s—s. Однако опытные данные показывают, что все четыре связи С—Н полностью равноценны. По-видимому, «самые лучшие» функции состояния, т. е. с наивысшей энергией связи, соответствуют некоторым линейным комбинациям s- и p-связей, т. е. «гибридным функциям», а не самим исходным s-или р-волновым функциям. В СН4 имеются четыре полностью равноценные гибридные орбитали, направленные по углам тетраэдра (табл. А. 10).
Было показано, что в некоторых случаях гибридизация возможна и в двухатомных молекулах. Например, в НС1 нет просто связи 1s—Зр; связь между атомами Н и С1 осуществляется с участием Зз-электронов хлора.
Возможность образования различных гибридных сочетаний из волновых функций одного атома имеет большое значение для объяснения существования соединений с кратными связями.
В последнее время получила значительное распространение внешне простая теория строения молекулы, которая пытается объяснить расположение и углы связи в молекуле, не прибегая
92
Строение материи
Таблица А.9. Валентные углы
Молекула Связи, образующие валентный угол Экспериментальное значение валентного угла
Н2О н—о—н 105°
С12О С1—О—С1 116°
f2o F—О—F 100°
SC12 Cl— S—С1 102°
H2S H-S-H 92°20'
NH3 H-N-H 108°
PF3 F—Р—F 104°
PC13 Cl-Р—С1 101°
PBr3 Вг— Р—Вг 100°
PI3 I—Р—I 98°
AsCl3 Cl—As—Cl 103°
SbCl3 Cl—Sb—Cl 104°
к представлениям об одноэлектронных орбиталях с определенной симметрией (так называемая модель отталкивания пар электронов). Здесь эта теория не рассматривается, так как попытки ее вывода и интерпретации (с помощью принципа Паули) внесли бы известное замешательство в наши представления. Следует, однако, указать на то, что уменьшение угла связи по сравнению с теоретическим значением в ряду соединений
н. /И >С< щ хн
109,5°
.Н
:N°—Н ХН
107,3°
104,5°
действительно связано с наличием свободной пары электронов, которая оказывает «отталкивающее» действие, сокращающее валентный угол. Это влияние можно рассчитать в рамках квантовомеханической теории.
6.3.2. Соединения с кратными связями
В табл. А.10 для атома углерода наряду с «тетраэдрической» гибридизацией приведены и другие линейные гибридные комбинации. Прежде всего обратимся к случаю тригональных функций, которые отражают строение радикала СН3. Оси трех сг-связей находятся в плоскости ху под углом 120° друг другу (рис. А.36). В молекуле этилена углерод-углеродная и углеродводородные связи также расходятся к вершинам треугольника (рис. А.37). В этой молекуле существует обычная (однократная) связь, описываемая двумя cr-функциями. В то же время происходит образование кратной связи путем перекрывания
Химическая связь
93
Таблица АЛО. Гибридные функции атома углерода
1 . 5р3-Гибридизация (тетраэдрическая)
01= ll2(s+px + py + pz) cr2=l/2(s+px—py—pz)
1/2 (s—Рх-\~Ру-pz)
O4=ll2(s—px+py+pz)
2 $р2-Гибридизация (тригональная)
Р?
1 l/”
ai==/rs+^ 3 Рх
1 1 1
°2 ~ у'У5 ~ УУ Рх~г уУ Ру
____i_ 1 1
°з _ у уs ~ уу рх~ у гру
3 sp-Гибридизация (дигональная)
Ру
Р*
1
= ^=(s + ft)
<?2= y==(s~Px)
волновых рг—Рг-функций в плоскости, перпендикулярной плоскости ху. Эта связь уже не имеет вращательной симметрии относительно оси С—С, ее называют л-связью в отличие от
имеющей такую вращательную»
a-связи, рассмотренной ранее и симметрию. В молекуле этилена мы впервые встретились с двойной связью, которая включает о-ил-связи. Поскольку перекрывание л-связи меньше, чем сг-связи, соответственно меньше и энергия л-связи. Поэтому совершенно неверно, если говорят, что двойная связь в два раза прочнее, чем одна сг-связь.
Из табл. А.10 можно видеть, что имеется еще одна
возможность связи С атомом Рис. А 36. Электронная структура Углерода — образование двух радикала СН3.
^-связей, т. е. в сумме трех
связей между атомами углерода. Электронная структура тройной связи наглядно изображена на рис. А.38.
Атомы третьего и последующих периодов периодической таблицы образуют также связи с d-электронами. При этом осуществляется квадратное или октаэдрическое расположение связей, 0 котором речь пойдет в связи с изучением комплексных соединений.
Рис. А.38. Электронная структура ацетилена.
'б.З.З. Мезомерия
Ранее уже говорилось о том, что атом азота может образовать, самое большее, четыре связи, получая при этом положительный заряд. Аналогично атому углерода это соответствует ар3-валентному состоянию, для которого возможны различные случаи гибридизации и образования двойных связей. Например, следует рассмотреть следующие формулы нитросоединений, образованные с помощью пары электронов — одновалентный радикал)
В—N+
/°-
/О R-N+/
O'-
Обе валентные структуры полностью эквивалентны. Поэтому двойная связь, как и отрицательный заряд, не может быть' локализована в молекуле. Такой вывод подтверждают и неко-
Химическая связь
95
торые физические свойства этих соединений. Таким образом, их строение нельзя правильно передать с помощью какой-либо одной валентной структуры. Однако следует особенно остерегаться представлений о том, что молекула как бы непрерывно переходит из одного состояния в другое, т. е. имеет место «резонанс». Иногда приходят к подобным неверным выводам, так как энергия связи такой «мезомерной» молекулы больше, чем рассчитанная на основе валентной структуры. Эту добавочную энергию называют «резонансной энергией». Мы будем избегать-таких вводящих в заблуждение выражений и применять термины «особая часть энергии» (по Хюккелю) или «энергия делокализации». Ниже изображены мезомерные структуры для карбоната и нитрата.
О(-)
О
II
(У хо
(-) (-)
0<->
Следует всегда помнить, что таким образом отражается лишь, то обстоятельство, что метод валентных связей является приближенным и что валентные структуры, которые составляются с помощью «черточек», следует понимать лишь как схему, условно отображающую действительное строение молекулы. В противном случае может сложиться опасное заблуждение, например, о том, что мезомерия.— реальное явление. Однако следует также иметь в виду, что валентные структуры дают очень полезные указания о физико-химическом поведении молекул. Если строение молекулы можно представить в виде нескольких валентных структур, то говорят, что связи «делокализованы». В рамках теории МО, которая не рассматривает локализованных связей, образованных парой электронов, а следовательно, и валентных связей, такого рода недоразумения невозможны, в чем можно убедиться ниже.
6-3.4. Применение метода МО
На примере таких молекул, как N2, 02 и т. п., проще всего Уяснить подход, принятый в теории молекулярных орбиталей пРи рассмотрении кратных связей. Познакомимся с энергетиче-ской последовательностью молекулярных состояний. В линейных комбинациях орбиталей не нужно учитывать ls-состояния-атомов, так как у атомов, имеющих достаточно большой номер»
Химическая связь
97
96
Строение материи
Тнс. А 39 Линейные комбинации атомных состояний (справа приведены другие, также принятые в литературе обозначения этих состояний).
периодической таблице, из-за большого заряда ядра электронные оболочки, находящиеся вблизи ядра, испытывают сильное «сокращение» и не принимают участия в «перекрывании» при обычных расстояниях между ядрами. Поэтому наиболее прочные в энергетическом отношении линейные комбинации — связывающая симметричная и разрыхляющая антисимметричная функции двух 2$-состояний. Обозначим их 2sog и 2$<уи* (разрыхляющая молекулярная орбиталь отмечена звездочкой). Из 2р-состояний можно образовать две сравнительно сильно перекрывающиеся линейные комбинации, если ось х p-состояния совпадает с прямой, соединяющей ядра атомов, связанных в молекулу. Поэтому следующим на шкале энергии будет молекулярное состояние 2pag, в то время как после него располагается сильно разрыхляющее состояние 2раи*, составленное как комбинация атомных состояний 2ру и 2рг (при линейной комбинации этих функций наблюдается меньшее перекрывание)-Получающиеся при такой комбинации молекулярные орбитали не имеют вращательной симметрии относительно прямой, соединяющей ядра, и обозначаются как 2рпи и 2png*. Из пространственных соображений, как легко видеть, можно иметь две схемы перекрывания, разумеется обладающих одинаковой энер'
гией, т. е. две вырожденные орбитали. Таким образом, получается следующий ряд молекулярных состояний с увеличивающейся энергией (рис. А.39): 2scrg, 2scru*, 2pcrg, 2рли (два), 2png* (два), 2рои*- Все эти уровни заполняются парами электронов; в результате получим следующие схемы заполнения молекулярных орбиталей молекул С2, NO, N2, СО, О2 и F2:
Число связей
с2 (2scf) 2 (2so*)2 (2po)2 (2ря)2 2
NO (2so)2 (2scr)2 (2pa)2 (2ря)4 (2ря*) 2,5
n2, со (2so)2 (2sa)2 (2po)2 (2ря)4 3
02 (2so)2 (2s<r)2 (2pa)2 (2ря)4 (2ря*)2 2
f2 (2so)2 (2sa*)2 (2po)2 (2ря)4 (2ря*)4 1
Оправдывает себя следующее правило определения числа связей в молекулах: вычитаем из числа электронов на связывающих орбиталях число электронов на разрыхляющих орбиталях и делим разность на два. Рассчитанное таким образом
Рис. АДО.
Схема
молекулярных орбиталей молекулы О2
число связей приведено выше в последней колонке. Валентность в этом смысле является иным понятием по сравнению с тем, которым мы пользовались при изучении теории валентных свя-Зеи- На примере NO видно, что полуцелые валентности естест-иенным образом появляются в теории МО, а не как «резонанс» Различных валентных структур.
7—2027
98
Строение материи
Остановимся на схеме заполнения электронами молекуляр. ных орбиталей кислорода (рис. А.40). Мы видим, что оба электрона на орбитали 2png* в соответствии с правилом Хунда имеют параллельные спины. Это является причиной парамагнетизма кислорода (магнитные свойства веществ см. в разд. 6.5 3), который с трудом поддается объяснению с помощью других теорий строения. Парамагнетизм NO также легко понять, если рассмотреть заполнение его молекулярных орбиталей электронами. Естественно, для молекулы, составленной из разных ато-мов, атомные волновые функции вступают в линейную комбинацию с различным «весом». Весовые коэффициенты ci и с2 в линейной комбинации
Чг = с1ф1+с2ф2
находят с помощью приближенных (вариационных) методов. «Правильными» коэффициентами будем считать такие, при ко
торых решение уравнения Шрёдингера дает минимальное зна
Рис А 41. Пространственная структура молекулы СН4 (в соответствии с методом МО)
чение энергии молекулы (наиболее устойчиво, как здесь, так и в других аналогичных физических системах, состояние молекулы с минимальной энергией). Различие коэффициентов ci и с2 при функциях ф указывает на то, что вероятность нахождения электронов у одного из атомов оказывается большей, чем у другого. Таким образом, здесь намечается переход к ионной связи, которая до некоторой степени является предельным случаем ковалентной связи.
В качестве второго примера применения метода МО об
судим в общих чертах молекулу СН4, которая уже упоминалась в связи с изучением мето-
да валентных связей, где с привлечением понятия «гибридизация» для нее получена тетраэдрическая структура. Четыре атомные ls-орбитали фч, ф2, фз, и ф4 четырех атомов водорода можно комбинировать различными способами (табл. А. 11). Ес' ли атомы водорода расположены по углам тетраэдра, то полУ' ченные четыре комбинации (рис. A.4I) имеют такую же симметрию, как и четыре атомных состояния s, рх, ру, pz центрального атома углерода. Поэтому при линейной комбинации эти* четырех волновых функций углерода с волновыми функциям# четырех атомов водорода получается очень хорошее перекрыва'
Химическая связь 99
Таблица А.11. Линейные комбинации волновых функций водородных атомов в метане
Комбинация волновых функций Симметрия функций атома углерода
Ф1+Ф2+ФЗ+Ф4 S
Ф1+Ф2—Фз—Ф'4
Ф1—Фг+Фз—фЧ Р«
Ф1—Ф2—Фз + Ф< Рг
ние. Можно показать, что все другие способы взаимного расположения атомов С и Н и все другие комбинации атомных волновых функций дают значительно меньшее перекрывание. Таким образом, тетраэдрическое строение СН4 хорошо объясняется с помощью метода МО. Из этого примера ясно видно, что молекулярные орбитали в методе МО не имеют ничего общего с атомными связями. Это волновые функции, относящиеся ко всей молекуле в целом.
6.3.5. Полярные молекулы. Дипольные моменты
Согласно методу МО, переход к ионным связям сопряжен с тем, что в зависимости от коэффициентов Ci и сг вероятность нахождения электронов у одного из ядер оказывается выше, чем у другого. Вследствие этого в молекулах электрический заряд распределен неравномерно, и в них появляется так называемый дипольный момент (произведение расстояния между центрами зарядов на заряд [i = el). При измерении дипольного момента всегда надо иметь в виду, что существует различие между постоянным и индуцированным (наведенным) дипольным моментом
Прежде всего остановимся на индуцированной поляризации. Если молекулу с симметричным распределением зарядов внести в электрическое поле с напряженностью Е, то на ядра и электронную оболочку будут действовать противоположно направленные силы, вследствие чего центры тяжести зарядов Раздвинутся и возникнет диполь Oi = (iE. Коэффициент пропорциональности а называют поляризуемостью. Она характеризует Размер и «подвижность» электронной оболочки, поэтому ее из-еряют как степень ослабления электрического поля, т. е. как диэлектрическую проницаемость е. Между е и средним моментом всех диполей ц. имеется связь, которая выражается урав-нием Клаузиуса — Мосотти
е+ 2 3 А Е
7*
100 Строение материи
где V — молярный объем, Р — молярная поляризация. Если имеются только индуцированные диполи, то
Р=—МАа
Как известно, преломление света на границе двух сред объясняется его поляризацией. Диэлектрическая проницаемость е и показатель преломления п связаны между собой:
е = п2
Таким образом, поляризация определяет молярную рефракцию Р:
п г?—1 ,7 4л
т. е. из измерений показателей преломления можно сделать выводы о поляризуемости электронных оболочек.
Индуцированная поляризация проявляется и для веществ с постоянным дипольным моментом. Для последних надо, однако, принять во внимание, что макроскопическая поляризация постоянных диполей зависит от температуры, так как из-за теплового движения диполи отклоняются от направления, заданного электрическим полем. Для среднего момента постоянных диполей справедливо следующее выражение:
и — ^--Е
3kT
Если подставить это выражение в уравнение Клаузиуса — Мо-сотти, можно получить уравнение Дебая
р__ s — 1 у__ 4л кг („ ; Нп2 \
64-2 V ~ 3 3kT J
Таким образом, молярная поляризация веществ с постоянными диполями складывается из двух членов, один из которых зависит, а другой не зависит от температуры.
Если поляризующее поле колеблется с высокой частотой, ю из-за инерции постоянных диполей они не успевают следовать за колебаниями поляризующего поля. Поэтому постоянные диполи не оказывают никакого влияния на молярную рефракцию (свет представляет собой высокочастотное электромагнитное поле). При частотах 10е Гц (длина волны 10—100 см, т. е. область дециметровых волн) возбуждается также и «ориентационная поляризация». Такое возбуждение зависит от внутреннего трения среды и в твердых телах вообще не наблюдается. Дипольные моменты молекул газа можно непосредственно определить из уравнения Дебая, измерив температурную зависимость диэлектрической проницаемости. Значения « и ц нахО'
Химическая связь
101
Таблица А.12. Дипольные моменты и ионный характер галогеноводородов
Галогеново-дорОДЫ Го, НМ ег§, 10-29 Кл м м, ю-2у Кл м ц/его
HF 0,092 1,47 0,66 0,45
НС1 0,128 2,02 0,34 0,17
НВг 0,143 2,27 0,26 0,12
HI 0,162 2,58 0,13 0,05
дяг либо графическим, либо численным методом. Дипольный момент молекул в растворе (общая поляризация раствора аддитивно складывается из поляризаций отдельных компонентов) определяют следующим образом. Сначала измеряют на рефрактометре индуцированную поляризацию, затем находят общую поляризацию, а ориентационную поляризацию вычисляют, как разность молярной поляризацииРи молярной рефракции Р. В настоящее время определение дипольных моментов проводят также на основе анализа влияния электрического поля на микроволновые спектры вещества.
В табл. А.12 представлены данные измерений дипольных моментов ц, галогеноводородов. Указаны также значения межъядерного расстояния Го и дипольный момент, рассчитанный как произведение ег0. Тогда можно попытаться выбрать соотношение Цэкс/Ционов в качестве меры перехода от ковалентной связи к ионной. Например, связь в молекуле HF в соответствии с табл. А.12 была бы на 45% «ионной», в НС1 — на 17%, в НВт— на 12% и в HI —только на 5%. Эти данные о доли «ионного характера» непосредственно подтверждают реальность мезомер-ных состояний и согласуются с другими характеристиками химической связи (см. далее). Прн определении дипольных моментов многоатомных молекул при любых сравнительных расчетах необходимо учитывать валентный угол. В молекуле Н2О Для расстояния Н—О 0,101 нм и валентного угла 104° центр тяжести Н-атомов лежит на расстоянии 0,101-cos 52° = 0,062 нм От ядра атома кислорода. Теоретическое значение дипольного момента в случае ионной связи составляет 1,97-lCF29 Кл-м. Измеренное значение дипольного момента воды равно 0,61-•Ю-29Кл-м.
Электроотрицательность. Доля ионной связи
Пол Оц.енки Менять
инг предложил другой полезный метод количественной полярности межатомных связей, который можно при-независимо от не всегда наглядных приближенных ме-
102
Строение материи
годов квантовой механики. Полярную связь между атомами представляют как простую мезомерную систему трех валентных структур
А-В, [А;-][В+], [а+][В;~]
Оказалось, что основной вклад вносит лишь одна из приведенных структур. Согласно Полингу, энергию связи гипотетического ковалентного соединения А—В можно вычислить из энергий связи в молекуле А—А и В—В. Разность между истинной (экспериментальной) энергией связи и гипотетической (вычисленной) может рассматриваться как мера ионного характера валентной структуры. Одновременно повышается устойчивость системы, так как для ионных валентных структур всегда имеет место понижение энергии. Если энергии связи А—А и В—В мало отличаются, то энергию связи 1ЕА-в можно найти как среднее арифметическое IEa-a и 1ЕВ-в; в противном случае №д-в — среднее геометрическое этих величин yiFA-A-IFb-b- Полинг предложил эмпирическое уравнение, в соответствии с которым уменьшение энергии связи (заметим, что здесь не употребляется термин «резонансная энергия»)
Дав = ^А-в~ 1 /2 (ГА_А+1ЕВ_В) выражается как квадрат разности некоторых характерных для атомов А и В величин х — так называемых «электроотрицательностей»:
дав= 23 (хА хв)2
Если одному из элементов произвольно приписать определенное значение электроотрицательности (для атома углерода С х = 2,5), то можно рассчитать эти значения для других элементов (табл. А.13, рис. А.42). Чем объясняются различия вэлект-роотрицательности элементов, явно имеющие периодический характер (рис. А.42)? Возьмем первый период периодической системы: от Li до F; х регулярно увеличивается на 0,5 единицы для каждого последующего элемента; очевидно, это объясняется увеличением зарядового числа ядра и вследствие этого усилением электростатического притяжения 2s- и 2р-валентных электронов. При переходе от F-->-(Ne)->Na это притяжение резко падает, так как валентные электроны этих элементов находятся на более удаленных от ядра уровнях 3s (соответственно Зр). Действительно, электроотрицательность проявляет такдо же периодичность, как и энергия ионизации элементов. Согласно’ Малликену, мерой притяжения валентного электрона в нейтральном атоме, иначе говоря, электроотрицательностью, является среднее арифметическое между энергией ионизации (за-
О О оо lO oj
Рн’Ф О со СО О? *-<04 <СоГ
0 3,5 LO CO 04 0) CO 04 <D ”"1 H 04 Po 2,0
о « o CD CD
Z co" Ct 04 < CM uo - m-.'
Ж cm" UO О 04" Si l..8 Ge 1,8 Sn 1,8 Pb 1,8
00
та O-" c
c Ь0°1
N Z -
=°1 s'*
u- < ~ 04
00 n-t 01 04
z~ Q< cm" Q< cm
о °0. J==T 04
u- CX 04 — 04
00 s*! »=1
ь —• DC cm О 04
C L0 Те 1.9 Re 1,9 Np 1,3
o qo
—г □ O
CD lO
Z-H H —
UP 00 л00.
H «—« X-."
04
=3 5s 1
о Ю 00 co г Д та 0 *1
CQ 04 < — GO > — >—2 —< <-
ra°. 0 rr, CD та
1 CQ^ —< c~ (Л —< mo Ko
О Л 05 qo -Q 00
i—J —< Zo «0" a 0 60 Ёо"
аДаииые таблицы относятся к обычным степеням окисления Идя некоторых элементов в зависимости от степени окисления наблюдя ются другие значения электроотрицательности, например Fe2^- 1,8, Fe3+ 1,9, Cu+ 2Д Cu2+ 1,8, Sn2+ 1,8, Sn4- h9 Данные по- другим элементам см Gordy W, Thomas W J О J Chem Physic 24, 439 (1956).
104
Строение материи
трата энергии на реакцию X—>-Х++е_) и сродством к электрону Е (выигрыш энергии при реакции Х-|-е~=>Х~) (см. табл. А.14). Приближенные квантовомеханические расчеты показывают, что общую энергию связи можно выразить через сумму энергии отталкивания Ei в положительно заряженном атомном остове, энергии притяжения Ег между атомным остовом и двумя валентными электронами, а также энергии отталкивания Ез между валентными электронами. Е2 и Ез можно объединить в связывающий терм С. Тогда энергия связи определяется следу-Таблица А.14. Сравнение электроотрицательности со средними значениями ионизации и сродством к электрону (25 °C) (по Полингу)
Элемент Энергия ионизации Ei, кДж/моль Сродство к элек трону Ез, кДж/моль £i+£a 523 X
F 1688,5 349,6 3,9 4,0
С1 1257,3 365,5 3,10 3,0
В 1149,7 343,3 2,86 2,8
I 1014,0 316,9 2,54 2,5
Н 1318,8 74,5 2,66 2,1
Li 526,7 0 1,01 1,0
Na 502,4 0 0,96 0,9
К 425,0 0 0,81 0,8
Rb 409,5 0 0,78 0,8
Cs 382,3 0 0,73 0,7
-
105
Химическая связь
ющими выражениями (Z — эффективный заряд атомного остова молекулы):
7*2
^в = < + Свв ГДВ = ^ + СЙВ
Далее предположим (что с определенной степенью точности может быть подтверждено экспериментально), что Сав = = */2(Саа+Свв), а также
ГЛВ = (ГЛЛ' ГВ1з)1' 2
Тогда получается следующее уравнение, аналогичйое уравнению Полинга, связывающему электроотрицательность и выигрыш энергии:
Дав = ^ав-1/2(Гаа+^вв) = -1/2
\ гаа1/2
Полинг предложил также выражение для зависимости между разностью электроотрицательностей хА—хв и степенью ионности (долей ионного характера связи (табл. А.15, рис. А.43):
Степень ионности связи =1—е'~0,25 Иа-хв)2
Zb У
Лвв1/2 /
Кроме того, на рис. А.43 нанесены рассчитанные из дипольных моментов степени ионности для некоторых соединений. Рассчитанные таким образом значения хорошо аппроксимируются кривой, построенной по вышеприведенному уравнению.
Электроотрицательность по Полингу, хотя она и зависит от многих молекулярных параметров (гибридизации орбиталей, заряда атомов и т. д.), хорошо помогает при ориентации в обширной области химии, занимающейся изучением химической связи. Поэтому полезно познакомиться с этим понятием как можно раньше.
Таблица А.15. Разность электроотрицательностей и степень ионности связи в некоторых простых соединениях
*А~ ХВ Степень ионности связи, % ХА ~ *В Степень ионности связи, %
№>
0,2 1 1,8 55
0,4 4 2,0 63
0,6 9 2,2 70
0,8 15 2,4 76
1,0 22 2,6 82
1,2 30 2,8 86
1,4 39 3,0 89
-— 1,6 47 3,2 92
106
Строение материи
6.3.7. Атомные и молекулярные решетки. Межмолекулярные силы
Среди систем с ковалентными связями встречаются вещества, которые по своим свойствам в значительной степени приближаются к описанным ранее «идеальным» предельным состоя-
Рис. А 43. Зависимость степени ионности от разности электроотрицательностей.
ниям материи. Например, водород в газообразном состоянии при низком давлении и высокой температуре является моделью идеального газа. Другим предельным состоянием является так называемая атомная решетка, например алмаза (или другого подобного вещества, например SiC). В алмазе четыре зр'А-гнб-ридизованные валентные связи атома углерода насыщаются другими атомами углерода, в результате чего образуется регулярная кристаллическая структура, в которой каждый атом углерода тетраэдрически окружен четырьмя другими. В данном случае нельзя уже применять понятие «молекула»; можно даже сказать, что кристалл алмаза представляет собой одну «гигантскую молекулу». Прочные межатомные связи обусловливают большую твердость и малую испаряемость веществ с атомными кристаллическими решетками.
Переходная система от регулярно построенных атомных решеток к обычным ковалентным молекулам (с молекулярной массой ~ 102—103 — это неорганические «макромолекулы», которые включают много атомов, связанных преимущественно ковалентными связями; в таких молекулах часто осуществляются цепочечные структуры (например, PNC12, «неорганический каучук», некоторые конденсированные фосфаты).
Большинство веществ с ковалентными связями находится Б агрегатном состоянии, промежуточном между идеальным газо^
Химическая связь
107
Н2 и алмазом. Обычно пользуются следующей классификацией: почти идеальные газы (например, N2, СН4), реальные газы (например, СО2), жидкости (например, Вг2, Н2О), молекулярные решетки (например, 12, лед). В такой же последовательности увеличиваются межмолекулярные силы, за счет которых происходит образование молекулярных, а в отдельных 'случаях и слоистых кристаллических решеток.
Силы притяжения между молекулами с насыщенными валентными связями могут иметь различную природу (природа этих связей еще мало изучена). Наиболее просто поддается количественному описанию сила взаимодействия (притяжение) между молекулами-диполями
А__
/г---—
где pi и ц2— дипольные моменты i кул, г—расстояние между диполями одной прямой). Сила взаимодействия между диполями уменьшается в большей степени, чем для точечных зарядов. С большим трудом поддаются интерпретации так называемые дисперсионные силы между молекулами, квантовомеханическая теория которых здесь не излагается. Заметим лишь, что по Лондону энергия взаимодействия пропорциональна 1/г6. Рассмотрим также межмолекулярные силы, известные как вандерваальсовы. Они приводят к образованию связей между молекулами с энергией до 20 кДж/моль, т. е. бы молекулярных соединений (энергия так ной валентной связи составляет "
реальных газов так называемое
взаимодействующих моле-i (диполи расположены на
0
А 44.
Рис
Тетраэдрическая структура воды.
1°
н+
к образованию как называемой основ-400 кДж/моль). Поведение приближенно описывает уравнение состояния — уравнение Ван-дер-Ваальса:
(p+-£-yv~b) = RT
Оно отличается от уравнения состояния идеального газа двумя поправочными членами: a/V2 и Ь. Поправка a/V2 связана с ^ежмолекулярным притяжением в газе; это взаимодействие создает как бы «внутреннее давление» (которое добавляется к внешнему давлению), увеличивающееся при уменьшении объема V газа, т. е. с уменьшением расстояния между молекулами газа. Поправка к объему учитывает то обстоятельство, что мо
108
Строение материи
лекулы не геометрические точки, как это постулирует кинетическая теория; они имеют некоторый объем V. Теория показывает, что b^4v. Наличие вандерваальсовых сил объясняет ряд свойств жидкостей и твердых тел (свойства адсорбционных слоев, хемосорбцию, образование мицелл, коллоиды).
Особым случаем межмолекулярных взаимодействий, имеющим важное значение в химии, является водородная связь (энергия, как правило, 20—40 кДж/моль). Она осуществляется прежде всего в соединениях с группой ОН; образование водородной связи приводит к выигрышу энергии, поскольку протон взаимодействует одновременно с двумя электронными облаками. Например, две молекулы уксусной кислоты образуют димер
О--Н-О\
CH3-C<f >С-СН3
Хо-Н-О^
Подобные ассоциаты образуют также HF, спирты и вода. Водородные связи определяют возникновение характерной структуры воды (рис. А.44).
6.4. Ионы в кристаллических структурах
В химической практике, в особенности при аналитических определениях, часто имеют дело с веществами, имеющими ионное строение. Правда, лишь в чрезвычайно редких случаях свободные ионы существуют как таковые (что и отмечалось в начале изложения атомистической теории строения материи). Ионные соединения по строению можно разделить на две основные группы:
соединения с регулярным расположением анионов и катионов в кристаллической решетке, определяемым в основном их геометрическими размерами («идеальный» ионный кристалл): химическая связь имеет чисто электростатическую природу;
соединения, имеющие вокруг ионов оболочку из «лигандов» (ионов, дипольных молекул и т. п.) в виде изолированного «координационного полиэдра» (многогранника), причем при некоторых условиях связь между центральным ионом и лигандами может быть частично или полностью ковалентной. С такими координационными системами имеют дело при изучении комплексных ионов и соединений, а также ионов в растворе («сольватированные» ионы), в химическом анализе, особенно часто в водных растворах.
В данной главе рассматриваются преимущественно соединения, относящиеся к первой группе, включая переходные формы к кристаллическим системам, в которых координационные полиэдры выступают как более или менее изолированные составные части кристаллической решетки. Здесь же намечен переход к
Химическая связь
109
уже известным нам атомным и молекулярным кристаллическим решеткам. Соединениям второй группы посвящен специальный раздел (разд. 6.5); состояние и свойства ионов в растворах обсуждаются при изложении физической химии электролитов, термодинамические и кинетические закономерности которых изучает электрохимия.
6.4.1.
Применение рентгеновского излучения для исследований структуры веществ
Рассмотрим сначала основные принципы исследования строения кристаллических решеток вещества. Мы уже познако
мились с некоторыми типами нейшей сферической упа-
ковкой» (гл. 1). (При изучении металлической связи мы еще раз вернемся к этой проблеме.) Для того чтобы правильно охарактеризовать кристаллическую решетку, необходимо иметь в виду следующее: вследствие периодичности строения кристалла для каждой кристаллической решетки можно выделить некоторый минимальный объем — элементарную ячейку, при последовательном повторении которой во всех направлениях
решеток,^, например «плот-
можно получить кристалл в
целом. В качестве простого примера на рис. А.45 изображена элементарная ячейка кубического кристалла NaCl. Кристаллическая решетка NaCl построена из гранецентрированных «кубиков», образованных ионами Na+ и СК.
Мысленно построим в кристалле прямоугольную систему координат, оси которой расположены параллельно поверхностям, на которых лежат узлы решетки. Тогда каждая такая узловая
плоскость отсекает на осях координат отрезки, которые можно использовать в качестве характеристики строения кристалла. При этом используют обратные величины отношений этих отрезков к «постоянной решетки», выражаемые как наименьшие Целые числа. Эти числа называют индексами Миллера. Например, плоскость (123) отсекает на осях координат отрезки I, ‘/2 и '/з длины ребра элементарной ячейки. Узловая плоскость (100) параллельна плоскости yz и смещена в направлении оси х Иа расстояние постоянной решетки. Плоскость (110) проходит
по
Строение материи
через передний правый и задний левый узлы элементарной ячейки (рис. А.45) и параллельна оси z.
При структурных исследованиях кристаллических веществ используется взаимодействие рентгеновского излучения с кристаллом. При этом проникающие в кристалл рентгеновские лучи (с длиной волны 7) всегда отражаются от атомов (ионов) кристаллической решетки под углом а в соответствии с формулой Вульфа — Брэгга:
2dsina = nZ (и= 1,2, 3,...)
(d — расстояние между узловыми плоскостями, на которых происходит отражение рентгеновских лучей; рис. А.46). Разрабо-
Рис. А.46. К выводу уравнения Вульфа — Брэгга.
таны методы, использующие как монохроматические рентгеновские лучи, так и рентгеновское излучение с широким спектром. Первые рентгенограммы были получены Лауэ (совместно с Фридрихом и Книппингом) на монокристалле с применением рентгеновского излучения широкого спектра Рефлексы на такой «лауэграмме» относятся к различным узловым поверхностям и разным длинам волн, так что расчет такой диаграммы довольно сложен. Тем не менее из нее сразу можно сделать выводы о симметрии кристалла относительно пучка рентгеновских лучей (например, для лауэграммы, приведенной на рис. А.47, кристалл имеет ось симметрии третьего порядка). «Индексация» поверхностей кристалла удается только в том случае, если сделано несколько снимков в направлениях, параллельных различным осям в кристалле, а также измерено расстояние между узловыми плоскостями на основе рентгенограмм, полученных в монохроматическом излучении. Для расшифровки структуры кристалла часто применяется метод Дебая — Шерера (порошковая рентгенография, метод дебаеграмм). Он использует монохроматическое излучение и часто проводится на порошках кристаллических веществ, не требуя монокристалла. Принцип метода помогает понять рис. А.48.
Химическая связь
Ш
Поскольку в пробе вещества микрокристаллы ориентировали в самых разнообразных направлениях, на рентгенограмме наблюдаются многочисленные резкие кольцевые линии, из которых можно рассчитать угол рассеяния а и расстояние между плоскостями d. Разработанные в последнее время методы рент
геноструктурного анализа в нашем курсе не рассматриваются. Важную информацию о связи в кристаллах можно получить
с помощью так называемого фурье-преобразования рассеяния
рентгеновских лучей на электронах кристаллической решетки монокристалла. Так как в кристалле электронная плотность периодически изменяется, снимают диаграммы электронной плотности, иа которых проводят линии одинаковой электронной плотности. Диаграммы, полученные этим методом Бриллем, Гриммом и Петерсом, показаны на рис. А 63, а и б, а также А.64 (при заключительном обсуждении различных типов связи в твердых телах).
Наиболее важным методом рентгенографии монокристаллов
с осью симметрии третьего поряд-
ка
в монохроматическом излучении
является метод вращающегося
кристалла Зеемана Однозначная расшифровка рефлексов от кристалла по Брэггу и определение узловых плоскостей облегчается, если производится регистрация угла поворота рефлек-
сов при синхронном движении пленки и повороте кристалла (рентгеновский гониометр).
Уже в 1929 г Брэгг показал, что функцию распределения электронной плотности р для некоторого объема v можно записать через коэффициенты Сны трехмерного ряда Фурье:
р=4" 2См sin [2п (Ях+^+/г)+фм^ hkl
Коэффициенты Сны определяются из измерений интенсивности Рефлексов. Для расчета распределения электронной плотности необходимо знать фазовую константу Фо?, которую нельзя непосредственно измерить Поэтому применяется «приближение тяжелых атомов»- на первой итерационной ступени рассчитывают (х, у, z) с приближенными значениями фаз, соответствующих структуре, состоящей только из сильно рассеивающих тяжелых атомов (например, Вг) В этом приближении появляют-
112
Строение материи
Химическая связь
113
ся дополнительные максимумы, которые можно идентифицировать, относя к легким атомам. Для них получаются фазовые константы второй итерационной ступени, для которой снова проводят расчет электронной плотности р и т. д. В результате достаточно громоздких вычислений можно определить распределение электронной плотности сложных молекул.
Наряду с рентгенографическим анализом следует упомянуть методы дифракции электронов и нейтронов. Однако электроно-
Рис. А.48. Метод Дебая —Шерера (порошковая рентгенография).
графическим методом можно исследовать лишь тонкие пленки, так как электроны значительно сильнее поглощаются веществом, чем рентгеновское излучение. С помощью потоков нейтронов можно изучать сравнительно толстые металлические пленки. Нейтроны рассеиваются на атомных ядрах, причем рассеивание зависит не от заряда ядра, а прежде всего от его спина. Поэтому изотопы одного и того же элемента по-разному рассеивают нейтроны. Ядра водорода и дейтерия рассеивают нейтроны лишь немногим слабее, чем ядра более тяжелых элементов. Тем самым открывается возможность установить расположение атомов водорода в кристалле, что не так просто сделать рентгенографическим методом.
6.4.2. Энергия решетки
Рассмотрим теперь энергетические соотношения при образовании ионной связи. Энергия взаимодействия катиона и аниона (заряд Z, расстояние между ионами г) равна
—ZW
ррафик зависимости энергии от расстояния г приведен на рис. А.49 (кривая 2). При достаточно малых г необходимо учитывать силы отталкивания между ионами, вызванные взаимодействием электронных оболочек. Энергия отталкивания очень быстро растет по мере сближения ионов (рис. А.49, кривая примерно пропорционально а/гп (где пЗ>1). Общая энергия (рис. А.49, штриховая кривая) оценивается по уравнению
г 1
равновесное расстояние го можно найти, продифференцировав функцию Е по г и приравняв нулю полученную производную:
1 (па \ п—1 )
Отсюда
a = Z2e2 п
Таким образом, для равновесного состояния энергия взаимодействия равна
(в большинстве случаев п^9). Условно полагая п = оо, получим, что в первом приближении кривая 1 (рис. А.49) преобразуется в прямую, параллельную оси Е. Это означает, что ионы представляют собой жесткие сферы, а расстояние между ними не может быть меньше, чем сумма соответствующих радиусов. Действительно, эксперимент показывает, что в разных соеди-ениях радиусы одних и тех же ионов остаются неизменными.
табл. А. 16 приведены радиусы некоторых ионов.
нергию ионных кристаллов можно получить, сложив энер-и взаимодействия всех пар ионов (для п = оо):
ноНяг--3арядовое числ0 Z’ro иона’ Zi~ зарядовое число /-го ное пГ,/ Расстояние между t-м и /-м ионом в кристалле, крат-
Расстоянию между соседними ионами г0 (если кристалл &-2027
114
Строение материи
Таблица А.16. Радиусы (нм) ионов
н- 0,154 L1+ 0,068 Ве2+ 0,030
О2~ 0,145 F-0,133 Na+ 0,098 Mg2+ 0,065 А13+ 0,045
S2- а- К+ Са2 + Sc3+
0,190 0,181 0,133 0,094 0,068
Se2~ Вг- Rb+ Sr«+ Y3+
0,202 0,196 0,148 0,110 0,090
Те2- I- Cs+ Ва2+ La3+
0,222 0,219 0,167 0,129 0,104
увеличить, не изменив его геометрической формы, гч меняются пропорционально), г Ct,
£0=-УУ-?^-=-ф
го г0
Ео пропорциональна квадрату минимального элементарного заряда Z, а также числу Авогадро (в I моле «кристалла» содержится 6,023-1023 «формульных единиц»). Итак,
Ф = — ANaZ2
Коэффициент пропорциональности А зависит от типа кристаллической решетки и называется числом Маделунга. Определе-
Na(me) + ^CL^acu) _ 1/п Г) Na (газ) + Cl (газ)
'‘-а, At/NaCl F ^Na. ^Cl
, , NaCl Na.CL(me) * Ма+(газ)+ Cl'(газ)
Рис. А.50. Цикл Габера — Борна.
ние числа Маделунга — довольно сложная математическая задача. Числа Маделунга решеток важнейших типов для кристаллов стехиометрического состава АВ, АВ2 и А2В3 приведены
Химическая связь
115
Таблица А.17. Числа Маделунга
Тип решетки
Число Маделунга
Хлорид натрия М+Х-Хлорид цезия М+Х-Цинковая обманка М2+Х2-Вюрцит М2+Х2~ Флюорит М2+Х2-
Куприт М2+Х2~
Рутил М2+Х2-
Анатаз Мг+Х2~
Иодид кадмия М2+Х2~
Кварц М4+Х22~ Корунд М23+Х3г-
1,74756 1,76267
1,63806
1,64132 5,03878 4,11552 4,816
4,800
4,71
4,4394
25,0312
табл. А.17. Подставив число Маделунга, получим следующее выражение для энергии решетки Е:
~~E — ANaZ2 — г0
Борн показал, как можно сопоставить теоретические (рассчитанные) значения энергии решетки с экспериментальными данными. На рис. А.50 приведена схема так называемого термодинамического цикла Габера — Борна. 1 моль NaCl в виде кристалла можно получить при образовании кристаллической решетки соли из ионов Na+ и С1~, при этом высвобождается энергия решетки Е. В то же время мысленно можно осуществить процесс в несколько стадий: перевести Na+ и С1~ в атомарные Na и С1, при этом нужно затратить энергию на преодоление сродства к электрону иона С1~ ЕА, а выделится энергия ионизации иона натрия /. Далее атомарные Na и С1 можно перевести в металлический натрий и газообразный С12, при этом выделится энергия сублимации натрия L и энергия диссоциации хлора 1/2Z7ci2. Наконец, при образовании хлорида натрия из металлического натрия и газообразного хлора выделится энергия, Равная теплоте образования 1 моля хлорида натрия. Таким образом, на основе закона сохранения энергии можно записать
E—AU—L— ^D—/4-£Л = 0
В табл. А.18 приведены энергии решетки некоторых галогенидов щелочных металлов, рассчитанные теоретически и определенные по экспериментальным данным с помощью цикла ь°рна. Рассчитанная этими способами энергия кристаллической Решетки относится к ионам или атомам, окруженным такими е частицами, и представляет собой энергию, которая высво-8*
116
Строение материи
Таблица АЛ8. Энергия решетки некоторых галогенидов щелочных металлов
Галогенид щелочного металла Энергия, кДж/моль
экспериментальные данные (цикл Борна) рассчитанные значения
NaCl 766 762
NaBr 712 716
Nal 666 662
КС1 691 678
KBr 645 649
KI 603 603
RbCl 674 649
RbBr 632 620
Rbl 590 578
бождается в объеме кристалла при его образовании из моля частиц — ионов или атомов. Однако рост кристалла в растворе, расплаве или газовой фазе происходит на поверхности кристалла. Выделяющаяся при этом энергия присоединения частиц всегда меньше, чем энергия решетки, и зависит от места присоединения частиц к поверхности (по Косселю). Выделяющаяся при кристаллизации энергия максимальна при так называемом регулярном росте кристалла, т. е. присоединении ионов (атомов) к недостроенным участкам решетки. Рост кристалла на сформированной плоскости кристалла, например для NaCl, составляет лишь 7,6% максимальной энергии связи, ввиду того что присоединение частицы может происходить лишь на одной чз шести граней куба. В термически равновесных условиях всегда осуществляются только наиболее благоприятные в энергетическом отношении возможности присоединения частицы, поэтому в основном кристаллы растут регулярно.
6.4.3. Радиусы ионов и типы решеток
Попробуем ответить на вопрос, почему данное ионное соединение образует кристаллическую решетку определенного типа. Поначалу ответ кажется очень простым. Наиболее стабильной будет кристаллическая решетка, имеющая минимальную энергию, а следовательно, наибольшее число Маделунга. Из табл. А. 18 следует, что ионные соединения стехиометрического состава (АВ) должны кристаллизоваться по типу CsCl. Однако это не так: известна и решетка NaCl, число Маделунга для которой меньше, чем для CsCl. Другие бинарные соединения кристаллизуются по типу решеток с еще меньшим числом Маделунга (например, решетка вюрцита — цинковой обманки)-Кроме того, упущено еще одно важное обстоятельство. Рассмот
Химическая связь 47
рим рис. А.51. Очевидно, что
г+Ц-г_ = 1/2Я И—пространственная диагональ куба
Я = ]/За а—длина ребра, a = D-{-2t
W г+4-г_ = уз_
a D + 2л_ 2
О + 2Г-=^-(Г+ + Г-)
Пусть в решетке CsCl при постоянном радиусе аниона г_ радиус катиона г+ уменьшается, так что в конце кондов анио-
ны соприкоснутся между собой. Тогда
У3г_ = г++г_, т. е. ^=]/3—1 = 0,732
При г+/г_<0,732 соединение кристаллизуется по типу решетки NaCl. При дальнейшем уменьшении г+1т- до критического значения
r+/r-=V2— 1 = 0,414
соединение кристаллизуется по типу вюрцита. Таким образом, очевидно, что ионные радиусы играют определяющую роль при образовании кристаллической решетки того или иного типа. ° табл. А. 19 включены соединения АВ2 с решетками типа рути-Ла и флюорита (критическое отношение радусов 0,732).
118
Строение материи
Таблица А.19. Отношение радиусов атомов (решетка рутила и флюорита)
Решетка рутила r+/r- Решетка флюорита r+/r-
MgF2 0,6 CaF2 0,87
ZnF2 0,65 SrF2 0,97
TiO2 0,55 BaF2 1,12
GeO2 0,43 CdF2 0,84
SnO2 0,55 HgFa 0,92
PbO2 0,60 SrCl2 0,73
6.4.4. Переходные типы связи в кристаллах
Важнейшие факторы, вызывающие отклонения кристаллических решеток от «идеального» строения, — это поляризация ионов, размеры электронной оболочки и разность электроотрицательностей ионов, входящих в состав кристалла. Если вернуться к индуцированным диполям (разд. 6.3.5, табл. А.16), то можно увидеть, что анионы поляризуются значительно сильнее, чем катионы. Это особенно заметно, если многозарядный катион небольшого размера вступает во взаимодействие с большими анионами, например I", О2-, S2~; в этом случае ионная связь становится частично атомной. Поэтому, например, в оксидах и сульфидах тяжелых металлов связь между ионами сильно отличается от обычной ионной связи в «идеальном» кристалле Кроме того, следует обратить внимание на следующее. Представим себе в газовой фазе молекулы iNaO, MgCU, А1С1з-SiCl4, образованные из соответствующих ионов; для данного ряда соединений электронная плотность вблизи катиона увеличивается за счет анионов, следовательно, увеличивается экранирование центрального катиона. При конденсации в жидкость или образовании кристалла в этом ряду увеличивается тенденция к образованию элементарной ячейки в виде координационного полиэдра. Таким образом, происходит переход к образованию менее прочной молекулярной кристаллической решетки, для которой характерна более низкая температура плавления, вследствие того что частицы связаны между собой лишь ела быми вандерваальсовыми силами. Одновременно обнаруживается существенное различие в электропроводности в этом ряд' соединений. Электронная плотность вблизи катиона, разумеется, зависит от отношения радиусов катиона и аниона: напри мер, ионы хлора С1~ могут экранировать В3+ и А13+, но нс La3+, имеющий большие размеры. В то же время ионы F~ нс могут образовать оболочку вокруг А13+, поэтому AIF3— чистс ионное соединение. В табл. А.20 и А.21 приведены некоторые физические свойства некоторых галогенидов, которые позволь ют сделать соответствующие выводы о типе связи.
Химическая связь
119
Таблица А.20. Температура плавления (и кипения или сублимации) некоторых галогенидов
—- — Галогенид Температура, °C
С1 Вг I
MgX2 1266(2260) 716(1418) 700(?) 650(?)
А1Х3 (1291) 193(180) 97(255) 191(381)
S1X4 —90(—95) —69(57) 5(155) 120(290)
Таблица А.21. Эквивалентная электропроводность X (Ом-1-г-экв.-1-см2) хлоридов при температуре плавления_______________________________________
1аС1 166 | ВеС12 0,086 ВС13 0 CC14 0
NaCl 133,5 MgCl2 28,8 А1С13 15 • 10-’ SiClt 0
КС! 103,5 СаС12 51,9 8сС13 15. T1C14 0
RbC! 78,2 SrCl2 55,7 YC13 9,5 ZrCl4 ?
CsCl 66,7. ВаС12 64,6 LaCl3 29,0 HfCl4
*1 . — — — ThCl4 16
Между ионными и молекулярными решетками находятся так называемые слоистые кристаллические решетки. Например, Cd2+ недостаточно экранируется электронными оболочками ионов I-, для того чтобы образовалась молекулярная решетка; в то же время ионы I- слишком слабо поляризуются, чтобы могла образоваться трехмерная кристаллическая решетка. В результате образуются двумерные слои
I- Г I-
Cd2+ Cd2+ Cd2+ Cd2+
I- I- Г
Связь между слоями осуществляется благодаря слабым ван-Дерваальсовым силам. Поэтому такие кристаллы легко раскалываются.
Обзор всех типов связи, встречающихся в твердых телах, будет дан после того, как мы познакомимся также с атомной связью в металлах.
6-5. Ионы в поле лигандов
Обратимся вновь к координационным системам, о которых Уже упоминалось при обсуждении различных типов элементарных ячеек кристаллов. Координационные системы всегда возникают вокруг ионов при растворении электролитов (ионные с°льваты). В подобных системах, называемых комплексными соединениями, встречаются настолько разнообразные и измен
120
Строение материи
чивые формы связи и структуры, что исследования в этом направлении способствуют развитию и теоретической, и прикладной химии (теория поля лигандов, комплексонометрия). Для того чтобы упорядочить дальнейшее обсуждение свойств комплексных соединений, сольватов, комплексных ионов и т. д., остановимся прежде всего на одной из важнейших проблем структурной молекулярной химии — симметрии молекул.
6.5.1.
Симметрия молекул
При изучении симметрии молекулы или любой другой координационной системы всегда будем принимать, что данная система построена из точечных атомов. Операцией симметрии называют любое перемещение точек системы, при котором точ-
ки-атомы занимают
Рис. А.52. Элементы симметрии молекулы Н2О.
первоначальное положение, т. е. одинаковые атомы совмещаются. Такой операцией является, например, зеркальное отражение о атомов в молекуле Н2О в двух плоскостях симметрии (рис. А.52). В одной из этих плоскостей лежит сама молекула, другая плоскость расположена перпендикулярно к ней и делит угол Н—О—Н молекулы воды пополам. Плоскость симметриии обозначают <т. Кроме того, Н2О имеет еще ось симметрии второго порядка. Порядок п означает, что поворот относительно оси симметрии на угол
360° _ 360"-2 . 360°-(Л — 1)
п ’ п ’''' п
меняет местами одинаковые атомы, — остов молекулы совмещается. Ось симметрии /г-го порядка обозначают Сп. Симметричные операции с молекулой воды можно отнести к типу С2,-Индекс v означает, что плоскости симметрии направлены вертикально и параллельно оси С2. Если «оставление системы на месте» тоже считать операцией симметрии, так называемой идентичной операцией Е, то группа симметрии* молекулы Н2О содержит следующие операции симметрии: Е, С2-~, , (То2-
Для операций одной группы выполняется правило: последовательное проведение нескольких операций симметрии в группе всегда идентично одной операции симметрии этой группы. Это свойство (сформулированное более абстрактно) в теории групп
* Группа симметрии — все операции симметрии, относящиеся к данной точечной конфигурации [Физическая химия./Под ред. К. С. Краснова, —М-Высшая школа, 1982, с. 46]. — Прим, перев.
Химическая связь
121
имеет важное значение для многих областей физики и химии.
Рассмотрим некоторые примеры. Молекула NH? имеет ось Сз, совпадающую с высотой равносторонней пирамиды. Операциями симметрии здесь являются также повороты на 360°: 3=120° и 360°-2 : 3=240°. Через каждую связь N—Н и ось Сз проходит плоскость симметрии о„. Молекула бензола имеет ось Се и одну плоскость симметрии от, (индекс h означает, что эта плоскость симметрии перпендикулярна оси Се); в плоскости лежит сама молекула бензола. Кроме того, можно убедиться, что молекула бензола имеет шесть осей второго порядка С2, лежащих в плоскости молекулы, и шесть плоскостей симметрии, перпендикулярных к о>>. Бензол имеет центр симметрии — это точка, через которую происходит отражение точек системы (такое отражение называют также инверсией I). Молекулы NH3 и Н2О ие имеют точки инверсии.
Молекулы могут иметь оси 2, 3, 4, 5 и 6-го порядков. Кроме того, введена ось симметрии бесконечно большого порядка (п = оо), когда возможно любое вращение вокруг оси, например для линейных молекул Н2, СО2, HCN и т. д. Сводная система символики групп симметрии Шенфлиса приведена в табл. А.22 и на рис. А.53 (в кристаллографии применяется символика Германа — Могена).
Симметрия молекулы, или, что то же самое, расположение лигандов вблизи центрального атома, имеет существенное значение при решении уравнения Шрёдингера для соответствующей системы, так как этот фактор определяет потенциальную энергию U системы. По соображениям симметрии часто можно значительно упростить методы решения уравнения Шрёдингера, а также провести качественную оценку последовательности энергетических уровней, что и будет сделано в разд. 6.5.6 для вырожденных d-состояний в поле лигандов.
Появление определенной симметрии в молекулах было уже объяснено (разд. 6.3.2) на основе метода валентных связей при образовании ковалентной связи (гибридизация). Однако как чисто электростатические, так и геометрические соотношения могут привести к определенной симметрии в координационных соединениях, если исходить из ионной модели строения молекулы. Рассмотрим, например, координационный полиэдр Ап+Вр, в котором центральный ион с зарядом -фп окружен р однозарядными лигандами. Потенциальная энергия комплекса складывается из отдельных членов, учитывающих кулоновское взаимодействие ионных пар. Сумма отрицательных (связывающих) членов тем больше, чем меньше расстояние между ионом и лигандом. Минимальное расстояние между ионом и лигандом Равно ги+г (гп — радиус центрального иона, г — радиус лиган-4а). Для октаэдрического комплекса с симметрией Oh
jj______бгсе2 , Зе2 , 12г2
_ ГП + г "Т” 2(г„ + г)'4 р2(гл+ г)
122
Строение материи
Рис. А.53. Важнейшие типы симметрии
Таблица А.22. Группы симметрии молекул
^Обозначение групп Элементы симметрии Примеры
— .Ci Ось первого порядка, симметрия от- Вторичный бутиловый
/=Сг сутствует Центр симметрии i^S2 Н спирт Н3С—С—ОН 1 С2Н8 Мезовинная кислота
Сг Ось симметрии второго порядка С2 Пероксид водорода Н2О2
Сз Плоскость симметрии а НОС1 (нелинейная молекула) гранс-Дихлорэтилен
Сгъ Ось второго порядка Сг, горизонталь-
D^V ная плоскость, перпендикулярная оси ffh, центр симметрии i Три взаимно перпендикулярные оси (плоская молекула) Частично повернутая
C2v С2 Ось Сг и две плоскости с.., проходя- форма этилена Н2О, фосген СОС12,
J)2h = Г’ь гцие через ось Три взаимно перпендикулярные пло- гщс-дихлорэтилен С2Н2С12 Н\ /Н
D2ds Va скости а, пересекающиеся по трем осям второго порядка С2, центр симметрии 1 Зеркально-поворотная ось S4, две Этилен уС=С\ , Нх \н тетрахлорэтилен (плоские молекулы) Аллен Н2С = С = СН2
Csv перпендикулярные к ней осн С2 и две плоскости, пересекающиеся по s4 Ось Сз, три эквивалентные плоскости NH3, СНС13 (тригональ-
^3h симметрии Оу Ось Сз, три эквивалентные перпенди- ные пирамиды) ВС13, NO3-, СОз2-, i,3,5,-трихлорбензол, затененная (цис-) форма этана, циклопропан Шахматная (транс-) фор-
Z?3(l кулярные к ней оси Сг, три эквивалентные плоскости ffv, одна плоскость (Th Ось Se, три оси Сг, три эквивалент-
Bui ные плоскости ot, проходящие между осями Сг через ось S6, центр i Ось С4, две эквивалентные перпенди- ма этана, циклогексан (кресло) Циклобутан (ато-мы С в
Dib кулярные к С4 оси С2, еще две такие же оси, четыре попарно эквивалентные вертикальные плоскости (ov, Cd), горизонтальная плоскость Oh, центр i Ось Сэ, пять эквивалентных перпен- одной плоскости) Циклопентан (атомы С
^6h дикулярных к ней осей Сг, пять эквивалентных плоскостей сг., плоскость Oh Ось Се, три эквивалентные оси С2, в одной плоскости) Бензол, гексахлорбензол
Тл еще три эквивалентные оси С2, три эквивалентные плоскости ov, еще три эквивалентные плоскости оа, плоскость Оь, центр 1 Четыре эквивалентные оси третьего Тетраэдрические молеку-
порядка, три эквивалентные оси S4, шесть эквивалентных плоскостей о лы СН4, SiCl4, C(GH3)4, ионы SO42-, С1О4~
124
Строение материи
Продолжение
Обозначение групп Элементы симметрии Примеры
оь Четыре эквивалентные оси Se, три эквивалентные оси С4т шесть эквивалентных плоскостей, еще три эквивалентные плоскости, центр i SF6> ион PtCl62“ и другие комплексы
Coov Ось симметрии С™, бесконечное множество плоскостей av Несимметричные линейные молекулы НС1, HCN, HC = CCR
Z^ooh Ось симметрии С^, бесконечное множество ПЛОСКОСТеЙ СТч, ПЛОСКОСТЬ Oh, бесконечное множество осей С2, центр с Симметричные линейные молекулы Н2, С12, СО2, С2Н2, С3О2
В общем случае энергия связи между центральным ионом и лигандом равна —рп/(гп-\-г).
При данной симметрии расстояние между лигандами пропорционально расстоянию центральный ион — лиганд. Поэтому несвязывающая (определяемая взаимным отталкиванием лигандов) энергия пропорциональна 1/(гте-|-г). Таким образом,, оказывается, что суммарная энергия электростатических взаимодействий в комплексе равна (sp называется коэффициентом экранирования):
t/= Р (» — «р) е2
'"л + г
В табл. А.23 представлены коэффициенты экранирования sp, рассчитанные Магнусом для различных видов симметрии. Видно, например, что для комплекса с координационным числом 4 наиболее благоприятна тетраэдрическая конфигурация, а для комплекса с р = 6— октаэдрическая. Для одного и того же цент-
Таблица А.23. Константы экранирования по Магнусу
Число лигандов Расположение лигандов SP
2 Диагональное 0,25
3 Равносторонний треугольник 0,58
4 Тетраэдр 0,92
4 КвадраТ 0,96
5 Правильный пятиугольник 1,38
6 Октаэдр 1,66
6 Правильный шестиугольник 1,83
7 Правильный семиугольник 2,30
8 Куб 2,47
8 Правильный восьмиугольник 2,80
Химическая связь
125
Таблица А.24. p(n—sp) по Магнусу
pin—sp) для
п р=1 р=2 р=3 р=4 р=5 р=б Р=7 р=8
1 1,00 1,50 1,26 0,32
2 3,50 4,26 4,32 3,12 2,04
3 7,26 8,32 8,12 8,04 4,90 4,24
4 12,32 13,12 14,04 11,90 12,24
5 18,12 20,04 18,90 20,24
6 26,04 25,90 28,24
7 32,90 36,24
8 44,24
рального иона и одинаковых лигандов величина р(п—sp) является мерой энергии, освобождающейся при образовании комплексного соединения. Значения р(п—sp) для различных зарядовых чисел центрального иона и координационных чисел приведены в табл. А.24. Из таблицы видно, что для каждого зарядового числа центрального иона имеется предпочтительное координационное число. С увеличением п максимальное значение р(п—sp) смещается к большим координационным числам, что подтверждается экспериментальными данными.
6.5.2. Основы магнетохимии
Если поместить образец вещества, атомы, ионы или молекулы которого обладают магнитным моментом (разд. 5.4), в магнитное поле, и образец намагнитится по полю, то такое вещество называют парамагнитным. Ранее уже указывалось на то, что атом имеет механический орбитальный вращательный момент Мг, спиновый вращательный момент Ms> а следовательно, и орбитальный магнитный момент
и. М,
‘г 2тс г
и магнитный спиновый момент
ц = 2Иа
•s тс s
Единица измерения магнитных моментов атомов магнетон Бо-Ра цв= “2^-В СИ цв = 9,273,10-24 Дж/м. Значения Mz и Ms определяются квантовыми числами L и S. У многих атомов и иОнов (в особенности с электронной конфигурацией инертных газов, т. е. в основном состоянии S, см. разд. 5.5) L и S равны нУлю, а следовательно, орбитальный магнитный и спиновый'
126
Строение материи
магнитный моменты также равны нулю. Такие вещества уже не являются парамагнитными. В данном случае проявляется только магнетизм, индуцированный внешним магнитным полем, так как он не перекрывается собственным магнитным моментом— парамагнетизмом. Такое индуцированное намагничивание (в противоположность индуцированному дипольному моменту) направлено против внешнего поля, т. е. имеет отрицательный знак, и называется диамагнетизмом. Образец диамагнитного вещества намагничивается перпендикулярно («диагонально») линиям магнитного поля. Такое поведение диамагнетиков объясняется тем, что индукция кольцевых токов в атомах происходит таким образом, что индуцированные магнитные моменты направлены навстречу постоянному магнитному полю. Классическая теория показывает, что диамагнитный момент пропорционален усредненному квадрату радиуса орбиты. Как и индуцированная электрическая поляризация, диамагнитный момент не зависит от температуры. Исследование температурной зависимости парамагнитных свойств веществ (Ланжевен) приводит к результату, аналогичному полученному для постоянного дипольного момента. Средний магнитный момент моля парамагнитного вещества в магнитном поле Н равен
Р2 гт
I-1 — 3>kT 'Н
Мерой макроскопического намагничивания материалов является магнитная восприимчивость
J
Ъ— н
где J — магнитный момент 1 см3 вещества. Для моля вещества — Хм’Н
где хм — молярная магнитная восприимчивость. Хм и 7я (отнесенной к 1 г вещества) связаны между собой (где р — плотность):
Хм= Mxg = Мрх0
Для диамагнитных веществ xg«10~6 ед. СГС, а для парамагнитных х^~ 10-54-10-3 ед. СГС. (О ферромагнетизме см. разд. 6.6.1.)
Измерение магнитной восприимчивости диа- или парамагнитных веществ обычно проводят в постоянном магнитном поле. Образцы парамагнитного вещества втягиваются в магнитное поле, а диамагнитного — выталкиваются из него. Возникающую при этом силу, пропорциональную магнитной восприимчивости, можно измерить с помощью весов или каким-либо дру гим методом. Если концентрация парамагнитных частиц в ве
Химическая связь 127
ществе незначительна, ее можно определить с помощью метода электронного парамагнитного резонанса, основанного на эффекте Зеемана (разд. 5.4). Разность энергий между расщепленными уровнями (уровнями Зеемана) близка по порядку величины к энергии электромагнитного излучения в области сантиметровых длин волн. Таким образом, за счет поглощения энергии электромагнитного излучения в диапазоне сантиметровых волн индуцируется электронный переход от низких к более высоким зеемановским уровням. Измерение такого рода поглощения требует сложной радиотехнической аппаратуры, однако с помощью этого метода можно получить ценные данные о локализации электронов в молекуле вещества.
Обратимся снова к комплексным соединениям. В качестве грубого приближения можно пренебречь диамагнитными свойствами, а также магнитными свойствами, определяемыми орбитальным магнитным моментом, и учитывать только магнитные свойства, связанные со спином электронов. Эффективный магнитный момент атома или иона зависит от спинового квантового числа, т. е. от числа электронов с неспаренными спинами: ______________________
Иэфф = («+2) |тв
Ниже приведены значения магнитных моментов в зависимости от числа электронов с неспаренными спинами:
п 1 2 3 4 5
Цэфф 1J2 2,83 3,87 4,90 5,91
В табл. А.25 представлено число электронов на d-оболочках ионов переходных металлов. Оказывается, что для большого Таблица А.25. Магнитные моменты ионов элементов первого большого периода в водном растворе
Ион Число З^.элект-ронов Число неспаренных электронов Спиновый момент, магнетоны Бора
рассчитанный экспериментальный
К+, Са2+, Sc3+, Ti4+ 0 0 0,00 0,00
Ti3+ v4+ 1 1 1,73 1,78
V3+ 2 2 2,83 2,80
V2+, Cr3+, Mn4+ 3 3 3,88 3,7—4,0
Cr2+, Mn3+ 4 4 4,90 4,8-5,0
Mn2+ Fe3+ 5 5 5,92 5,9
Fe$+ 6 4 4,9 5,2
Co2+ 7 3 3,88 5,0
Np+ 8 2 2,83 3,2
Cu2+ 9 1 1,73 1,9
Cu+, Zn2+ 10 0 0,00 0,00
Химическая связь
129
128
Строение материи
числа комплексов, и прежде всего (но не всегда) для непрочных комплексов присоединения, рЭфф согласуется с данными табл. А.25. Однако многие прочные комплексы внедрения проявляют значительно меньший магнетизм — некоторые из них даже диамагнитны.
Как можно объяснить эти экспериментальные данные? Впервые это по-пытался сделать Полинг Он считал, что существуют два резко отличающихся вида комплексов, с ионной связью (нормальный ионный магнетизм) и комплексы с ковалентной связью; для последних причины уменьшения магнитной восприимчивости можно представить себе, рассмотрев диамагнитные свойства комплексного иона [Fe(CN)6]-4. Шесть свободных электронных пар лигандов переходят к иону Fe2+ и занимают его незанятые атомные орбитали— две Зг/-орбитали, одну 45-орбиталь и три 4р-орбитали.
Поэтому шесть электронов попарно занимают оставшиеся 5</-орбитали, вследствие чего неспаренных электронов больше не остается, что и обуслов ливает диамагнетизм системы. Кроме того, теория показывает, что возможная здесь гибридизация типа d3sp3 обладает октаэдрической симметрией, что полностью подтверждается исследованиями структуры (Fe(CN)6]4-. Однако эти представления недостаточны для того, чтобы объяснить все свойства комплексов. На основе представлений Полинга, например, невозможно пра вильное истолкование спектров большинства комплексов металлов.
Уменьшение магнитной восприимчивости можно также объяснить иным образом (разд. 6.5.5) на основе исследований Ван Флека. Преимущество предложенной им теории состоит в том, что она правильно учитывает особенности спектров поглощения комплексных соединений.
6.5.3. Ядерная гамма-резонансная спектроскопия (мёссбауэровская спектроскопия)
Новый метод исследования поля лигандов использует явление поглощения (или, наоборот, эмиссии) атомными ядрами у-квантов. Наиболее существенное отличие этого метода от электронной спектроскопии состоит в проявлении очень резкого резонансного максимума, соответствующего энергетическим переходам при излучении. Уже относительное изменение энергии на 10'2 у-кванта достаточно для того, чтобы подавить резонанс. Однако это означает, что энергия отдачи ядра при поглощении у-кванта изменяет условия резонанса и подавляет его В 1958 г. Мёссбауэр при исследовании ядер 1911г нашел условия ядерного резонанса с отдачей на весь кристалл. Энергия отдачи в условиях проявления эффекта Мёссбауэра вследствие прочной связи всех атомов в кристалле достаточно мала для того, "чтобы обеспечить возможность резонансного поглощения у-лу чей. Тем самым становится возможной у-спектроскопия с высокой разрешающей способностью. Даже эффект Допплера, обусловленный перемещением источника у-излучения со скоростью
несколько миллиметров в секунду, достаточен для того, чтобы вызвать изменение частоты, которое сравнительно легко можно измерить спектроскопическими методами.
Этот новый вид спектроскопии твердых тел может дать химику полезную информацию о непосредственном окружении ядра, т. е. об его электронных оболочках. Однако этим методом можно исследовать не слишком легкие ядра (в настоящее время ядра тяжелее, чем 19К). Смещение резонансных линий, связанное с различными видами химической связи между атомами излучателя (или, наоборот, поглощающего излучения вещества), называют «изомерным смещением», соответственно «химическим смещением» (открыто на атомах железа). Это смещение происходит в результате взаимодействия с s-электронами. Расщепление спектральных линий, связанное с взаимодействием между электрическим ядерным квадрупольным моментом (разд. 4.2) и орбитальным моментом р- и d-электронов, называют квадрупольным расщеплением. Тем самым становится возможным отдельно исследовать распределение s-, р- и d-электронов. Большие успехи были достигнуты, например, при исследовании соединений железа и олова методом мёссбауэровской спектроскопии.
6.5.4. Вращательная дисперсия света и круговой дихроизм
За последние годы при изучении стереохимии оптически активных веществ получили развитие различные спектрофотометрические методы исследования, основанные на явлениях, связанных с поляризацией света. Оптическая активность комплексных соединений проявляется в том случае, когда расположение лигандов в координационной системе «хирально», т. е. в ней отсутствует зеркально-поворотная ось, вращение вокруг которой переводит молекулу в соответствующий стереоизомер. Линейно-поляризованный свет можно представить себе как совокупность Двух циркулярно-поляризованных волн с одинаковыми частотами и амплитудами. Тогда «оптическая активность» обусловлена тем, что право- и левополяризованный свет распространяется в веществе с разной скоростью. Угол поворота плоскости поляризации а пропорционален разности коэффициентов преломления право- и левополяризованного света:
® ®уд ХваК (^пр1 ^лев)
где I—.длина образца, ^вак — длина волны света в вакууме; п показатель преломления. Коэффициент пропорционально-СТи ауд называют «удельным вращением».. Если измерить за-М027
130
Строение материи
висимость угла вращения от длины волны — вращательную дисперсию (ВД),— го можно отметить, что она может быть нормальной и аномальной. Нормальная вращательная дисперсия характеризуется монотонным убыванием угла вращения по мере увеличения длины волны. Для объяснения более сложного случая аномальной вращательной дисперсии необходимо рассмотреть явление кругового дихроизма (КД). Последнее состоит в том, что в оптически активных средах в области длин волн, соответствующих полосе поглощения света, право- и левополя-ризованного поглощение света происходит по-разному. Для характеристики кругового дихроизма используют разность десятичного молярного коэффициента поглощения
апр ~ ^лев
Круговой дихроизм является причиной того, что в области полосы поглощения на нормальную вращательную дисперсию
Рис. А. 54. Положительный и отрицательный круговой дихроизм и эффею Коттона.
Химическая связь
131
нитном поле. Таким скую вращательную хроизм.
образом, можно измерить дисперсию и магнитный
магнитооптиче-
круговой ДИ-
нЭиЛйеКТР0СТаТИЧеская те°РИя комплексных соедине.
ранственноерасположение пяти й-орбитаТе/^нтрал^ного^о-
аХу аналогично dIZ, dyZ
Рис. А.55. d-Состояиия в октаэдрическом поле лигандов.
накладывается S-образный участок. Такую аномальную враша тельную дисперсию называют также эффектом Коттона. Каь показано на рис. А.54, в зависимости от знака Ае можно гово рить о положительном и отрицательном эффекте Коттона. В по следние годы проводятся интенсивные исследования вращатеп ной дисперсии и дихроизма, результаты которых могут бы11 использованы для исследований структуры молекул оптичеЛ' активных веществ. Однако совсем недавно появился мет^ который позволяет применить эти методы к оптически нсаь тивным веществам. Он основан на так называемом эффеьт‘ Фарадея, т. е. появлении оптической активности веществ в УаГ
ма. На рис. А.55 показаны два типа орбиталей (с/г2 и dX2_ua— ^-состояния), направленные вдоль осей, на которых, как можно себе представить, находятся шесть лигандов октаэдрическо-го комплекса с симметрией Ок и три другие орбитали (dxy-, dyz-, dxz—^-состояния). Последние направлены к середине ребер куба, оси которого расположены параллельно осям координат. Легко понять, что электроны в е§-состоянии сильнее отталкивается лигандами, чем в ^-состоянии. В результате взаимодействия с полем лигандов снимается вырождение пяти d-состоя-иий. Энергия расщепления уровней обозначается 10 Dq. На Рис. А.56 показано расщепление как в октаэдрическом, так и в Тетраэдрическом поле лигандов (кристаллическом поле) с сим-9*
132
Строение материи
метрией Т-типа. Пространственные представления можно получить с помощью рис. А.55. Если энергия расщепления достигнет некоторого предела, d-электроны, например, в поле с Ол-сим-метрией занимают не все пять d-орбиталей, а лишь более выгодные в энергетическом отношении орбитали tig. Вообще говоря, заполнение орбиталей электронами зависит от трех факторов: энергии расщепления уровней, «обменного взаимодействия» и электростатического взаимодействия, которые далее будут рассмотрены в указанной последовательности.
Если разместить электроны по всем d-орбиталям, то условно можно считать, что каждый из них на 3/s находится в ^-состоянии, а на 2/э — в ^-состоянии. В этом случае электроны имели бы среднюю энергию 4 Dq. Если принять эту среднюю энергию за уровень отсчета, то электрон в /^-состоянии бу-
октаэдрическое поле тетраздричесые поле
Рис. А.56. Расщепление d-состояний в октаэдрическом и тетраэдрическом поле лигандов.
дет иметь энергию —4 Dq, в ^-состоянии +6 Dq. Выраженную в единицах Dq разность энергий между возмущенным и невоз мущенным состояниями называют энергией стабилизации в кри сталлическом поле (ЭСКП). Значения этой энергии приведень в табл. А.26 для различных электронных состояний (4—7 элект ронов можно разместить различными способами в зависимости от того, заполняют ли электроны все пять орбиталей или тон ко /2г-орбитали). Зависимость ЭСКП от числа электронов \о рошо согласуется с теплотой гидратации двухзарядных ионог элементов первого переходного периода (рис. А.57). Велг'11' ну Dq можно вычислить из спектроскопических данных комплексных соединений. Например, полоса поглощен!1 видимого света для водного раствора комплексного i011 [Ti(H2O6)]3+ соответствует переходу d-электрона с ^-орбита' на eg-орбиталь, т. е. энергии расщепления 10 Dq. Исследован1! показали, что Dq сильно зависит от природы лиганда. Поле 11 ганда, а следовательно, и величина Dq увеличиваются в ду 1-< Вг-<С1- <F~ <Н2О <С2О42~ <C5H5N <NIV <h2n-ch2—nh2<no2-<cn-. j
«Обменное» и электростатическое взаимодействие уд о® I
Химическая связь
1 35
134
Строение материи
всего рассмотреть совместно. Вспомним свойства симметричных и антисимметричных собственных волновых функций (разд. 6.2.1), а именно соответствие наиболее устойчивому состоянию. Если учесть также и электростатическое взаимодействие между двумя d-электронами, то наиболее устойчивому состоянию отвечает антисимметричная функция, так как даже в том слу-чае, если исключить электростатическое взаимодействие между электронами, они избегают друг друга. По понятным причинам
Рис. А.57. Энтальпия гидратации двухвалентных ионов первого переходного периода.
/ — теоретические значения; 2 — экспериментальные данные
энергию стабилизации можно определить в некоторых произвольных единицах измерения. Например, пусть для с?5-конфи-гурации электронов с параллельными спинами (г 2f;-e g) i~2~ Ь/ десять пар электронов вступают в стабилизирующее взаимодействие. В то же время для конфигурации t52s (2) 4" (2) имеются четыре взаимодействующие пары, т. е. на шесть меньше. Следовательно, конфигурация t52g на шесть единиц энергии менее стабильна, чем рассмотренная выше (назовем единицы энер-гии взаимодействия за счет симметрии S-единицами). Можно тая же убедиться, что ^-конфигурация с параллельными сгчг нами на 4 S-единицы стабильнее, чем конфигурация со спаренными электронами, что и определяет в действительноеП1 такую «стабилизацию за счет симметрии». Последняя хараь-теризуется также другим параметром, а именно отталкивате Ц' ной энергией пары электронов с антипараллельным спин >v
(напомним, что в таком состоянии электроны сближаются в большей степени). Каждая пара вносит одну единицу энергии (кулоновской, С-единицу).
Итак, мы познакомились со всеми параметрами, которые определяют распределение электронов в комплексе, и после этого рассмотрим на нескольких примерах их взаимосвязь. Почему, например, [Fe(H2O)e]3+ проявляет обычные магнитные свойства, обусловленные спином, a [Fe(QN)6]3- не проявляет их? Это объясняется тем, что в первом случае поле лигандов значительно слабее [Z?e(H2O) <Dq(CN)] и S-стабилизации оказывается недостаточно, чтобы компенсировать их влияние. Далее становится ясным, что ^-конфигурация скорее всего будет иметь «высокий спин», так как разность энергии в S-еди-ницах между таким состоянием и конфигурацией с «низким спином» значительно больше, чем с любой другой конфигурацией. Кроме того, понятно, что при равных Dq для ^-конфигурации более характерен «низкий спин», чем для d5, так как величина С в обоих случаях одинакова, а разность энергии в S-единицах по отношению к (^-конфигурации равна 4, а по отношению к (^-конфигурации — 6. Если учесть также случай тетраэдрической симметрии (в табл. А.27 сопоставлены энергии в /^-единицах для октаэдрических и тетраэдрических комплексов), то можно сделать еще одни вывод: ионы Zn2+, Fe3+ и Tls+, которые имеют либо 5, либо 10 d-электронов, образуют менее прочные тетраэдрические комплексы, чем другие ионы,—для них всегда характерна октаэдрическая симметрия. ЭСКП для тетраэдрической симметрии максимальна для двух (соответственно семи) d-электронов в случае высокого спина и для 4d-3neKTpoHOB в случае низкого спина (табл. А.27). Поэтому TF+, уз+, со2+ при высоком спине и Сг2+ при низком спине одинаково склонны образовывать тетраэдрические комплексы. Таким образом, электростатическая теория комплексных соединений, или теория поля лигандов, позволяет хорошо объяснить многие закономерности, наблюдаемые в химии комплексных соединений.
Таблица А.27. Сравнение энергии стабилизации в октаэдрическом и тетраэдрическом кристаллическом поле лигандов
Число электронов
Энергия (в тетраэдрическом поле в /^-единицах) высокий спин низкий спин
Энергия (в О,-единицах) высокий спин (октаэдр) высокий спин (тетраэдр)
Разность энергии (энергия стабилизации в /^-единицах) для системы октаэдр— тетраэдр
Ковалентные связи в комплексах
Сведения, полученные при изучении электростатической теории комплексных соединений, могут быть уточнены и расширены, если принять во ®нимание также наличие в комплексных соединениях ковалентной связи. То, Что такие связи образуются с некоторыми лигандами (например, с пиридином,
136
Строение материи
Таблица А.28. Линейные комбинации функций лигандов в пространстве
Металл
Линейная комбинация
Oig <p4s 7Ag — yg (°1 + °2 + CT3 + °4 + + ffe)
<p3dz2 <fidx2_y2 Xeg,z“ — g + 2ae — ох o2 as a4) Xgg,*2,s/2 — 2 ""b °2 — °s °4)
rfiu.x = -yy (CTi — стг)
Ф4Ру /Au,?/ — (°3 °4)
(р4рг фЗ^ЗД ^2g фЗ<Лгг <p3cf^z /Au,г — y^~ (°® ffe) Могут образовывать л-связи
CN- или СО),, т. е. имеет место делокализация электронов, можно показать с помощью спинрезонансной спектроскопии. Необходимо построить молекулярные орбитали комплексных соединений подобно тому, как это было показано при рассмотрении молекулярных орбиталей СН4 (разд. 6.3.4). Для этого берутся определенные линейные комбинации молекулярных орбиталей лигандов, которые имеют такую же симметрию, как и атомные d-орбитали центрального иона Линейные комбинации для октаэдрических комплексов приведены в табл. А.28, а в более наглядном виде—на рис. А.58. (Индексы симметрии ale, еЙ, tiu и т. д. взяты из системы обозначений, принятых в теории групп, и здесь не обсуждаются.) Молекулярные орбитали комплексных соединений образуются линейной комбинацией таких атомных орбиталей ме талла и орбиталей лиганда, которые имеют одинаковую симметрию, так как в этом случае наблюдается максимальное перекрывание. Результаты энергетических расчетов молекулярных орбиталей представлены на рис. А.59, Разрыхляющие орбитали отмечены звездочкой. Заполнение электронами происходит, как обычно, попарно. Если в образовании связи принимают участие-12 электронов от шести октаэдрических лигандов и п d-электронов металла, то первые заполняют связывающие а>е-, tiu- и ег-орбитали, а d-электроны несвязывающие tag- и разрыхляющие ег*-орбитали. Последние две молекулярные орбитали играют ту же роль, как и в теории поля лигандов. Их расщепление также обозначают 10 Dq, хотя на энергию расщепления влияет перекрывание при образовании ковалентных связей.
Дальнейшие исследования показывают также, что атомные ^е-орбиталЯ металла имеют симметрию, согласующуюся с линейной комбинацией лигандов л-типа, т. е. между атомом металла и некоторыми лигандами (например, CN~) могут образоваться л-связи (двойная связь). Это приводит к тому, что расщепление 10 О, еще больше возрастает. Однако едва ли возможна
Химическая связь
137
Рис. А.58. Линейные комбинации s- и p-функций лигандов.
уровни энергии уровни мргш' уровни энергии свободного металось комплекса лигандов
Рис. А.59. Молекулярные состояния октаэдрического комплекса.
Химическая связь
139
138
Строение материи
количественные расчеты положения термов для простой модели молекуляр. пых орбиталей, так как рассматривается «электронная модель», не учиты-вающая взаимодействия электронов.
6.6. Металлическая связь
Металлы отличаются от других твердых тел некоторыми особыми свойствами — высокой электрической проводимостью и теплопроводностью. Изучая эти свойства, а также зависимость от температуры, можно сделать ряд важных выводов о природе металлической связи.
6.6.1. Металлическая связь и решетка металлов. Ферро- и антиферромагнетизм
Уже сравнительно давно стало известно, что в металлах валентные электроны покидают свои атомы, образуя как бы электронный газ, пронизывающий кристаллическою решетку металла. Наличие электронного газа является основной причиной высокой электропроводности (ст) и теплопроводности (X) металлов. С помощью таких представлений можно объяснить, хотя и не очень легко, закон Видемана — Франца
Однако применение законов кинетической теории газа к электронному газу приводит к значению а, отличающемуся от эксперимента. Делокализация валентных электронов в кристаллической решетке металла, а следовательно, отсутствие в ней направленных валентных связей объясняет тот факт, что металлы имеют большое координационное число Д, плотнейшую сферическую упаковку, а также чаще всего кубическую объемно-центрированную элементарную ячейку решетки. Некоторые металлы могут кристаллизоваться в различных типах ре" шеток; например, при температуре <768 °C магнитное а-желе-зо имеет Д=8, а при температуре >906 °C устойчивым является немагнитное у-железо с Д=12. Впрочем, для некоторых тяжелых металлов наряду с металлической связью, образованной Зй-электронами, реализуются слабые ковалентные связи ме»'-ду атомами, в то время как 4з-электроны образуют электроИ" ный газ. Для такой смешанной металлической и межатомной связи характерно образование пар электронов как с пара-11' лельными, так и с антипараллельными спинами (для марганца— антипараллельные, для железа — параллельные). Эти'1 объясняется различие в магнитных свойствах металлов; пара-1' лельные спины обусловливают ферромагнетизм, т. е. поло#11' тельная магнитная восприимчивость на два или три порЯД1"'
более высокая, чем у обычных парамагнитных веществ. При повышении температуры слабые межатомные связи рвутся и ферромагнетизм исчезает (для железа так называемая точка Кюри соответствует 1045 К). Межатомные связи, образованные электронами с антипараллельными спинами, являются причиной появления антиферромагнетизма. Температура, при которой разрываются такие связи, называется точкой Нееля. Такие магнитные явления определяются отношением межатомного расстояния к радиусу атома в различных кристаллических структурах.
6.6.2. Электронный газ
Термодинамические свойства металлов, например тот факт, что электронный газ не дает никакого вклада в теплоемкость, можно объяснить лишь в том случае, если вместо статистики
Рис А 60 Распределение по энергиям N электронов, содержащихся в 1 см3 вещества при трех значениях температуры [Lehrbuch der Physikalischen Chemie/ /Eggert, Hock, Schwab. — S. Hirzel-Verlag, Stuttgart, 1968].
a — распределение, подчиняющееся статистике Ферми — Дирака, вблизи уровня энергии S Для Г=0, 700 и 7000 К для меди, б — схематическое сравнение статистики Максвелла (без нулевой энергии) со статистикой Ферми (нулевая энергия £о); при температуре TF обе кривые сливаются.
Больцмана, основанной на максвелловском распределении ча-стиц в газе по скоростям, использовать статистику Ферми, учитывающую принцип Паули. Тогда при температуре абсолютно-г° нуля электронный газ обладает некоторой энергией, так как &се электроны должны обладать различной энергией, т. е.толь-0 один электрон может иметь энергию, равную нулю. На р • А.60 показано распределение энергии N электронов в объ-MeJ см3 для трех значений температуры. Верхний энергетиче-Ии Уровень, занятый электронами при абсолютном нуле тем-
140
Строение материи
пературы, называется уровнем Ферми. Этот уровень может быть выражен так:
_ Л2 ( зк уй
8m у л J
где m — масса электрона. Если приравнять £ и классическую кинетическую энергию ^МгТ, то можно получить так называемую температуру вырождения
т — k2 13N У/3 р 12mfe л j
Состояние электронного газа ниже этой температуры называют вырожденным, так как энергия электронного газа в этом случае настолько больше тепловой, что изменения температуры
Рис. А.61. К зонной теории металлов [Lehrbuch der Physikalischen Chemie/Eg-gert, Hock Schwab. — S. Hirzel-Verlag, Stuttgart, 1968].
Воронкообразные кривые схематически изображают изменение потенциальной эиер ин вблизи атома медн в свободном состоянии (а) и в кристаллической решетке (б). £ "я свободного состояния термы четкие, а для кристаллической решетки расщепляются ча зоны, которые относятся ко всему кристаллу, зоны расширяются по мере увеличен ня квантового числа, а для 3d и 4s перекрываются. Запрещенные зоны ие заштрихова ы, заштрихованные области — зоны, заполненные электронами. Область, отмеченная пре* рывнстой штриховкой, — разрешенная зона, не заполненная электронами (зона «прово* днмости»), Ее иижняя граница совпадает с энергией Ферми £
не оказывают существенного влияния на распределение энер' гии электронов. Например, для электронного газа в металлической меди 7’к=53 000 К.
6.6.3. Зонная теория (электронная модель металла)
Свободные атомы металлов и атомы в кристаллической ре' шетке имеют характерные отличия. Как показано на рис. А.бЬ
Химическая связь
141
Рис. А.62. Модели основных вариантов взаимного расположения зон [Lehrbuch der Physikalischen Chemie/Eggert, Hock, Schwab. — S. Hirzel-Verlag, Stuttgart, 1968].
a~ изолятор; б — одновалентный металл; в — двухвалентный металл; г — собственный полупроводник; д — примесный полупроводник n-типа, е — прнмесиый полупроводник p-типа Е — энергия; L — зона проводимости; S — термы прнмеси (донора или акцептора), U7 — вероятность заселения прн данной температуре, G — основная зона; s — энергия активации; 5 — энергия Ферми.
отдельные атомы имеют однозначные энергетические термы электронов, в то время как в атомах, входящих в кристаллическую решетку металла, существует взаимодействие электронов, находящихся на внешних электронных орбитах, с электрическим полем решетки. В результате этого энергетические уровни электронов расщепляются и образуют более или менее широкую энергетическую зону. В соответствии со статистикой Ферми наиболее высокая энергетическая зона заселяется свободными электронами и может быть почти полностью заполнена электронами, в особенности если энергетическим термам отдельного атома соответствуют два электрона с антипарал-лельными спинами (основная или валентная зона). Однако эта зона может быть заполнена лишь частично, что дает возмож-н°сть электронам переходить на более высокие энергетические Уровни. Тогда эта зона называется зоной проводимости. В од-новалентных металлах (например, щелочных металлах и меди)
142
Строение материи
число возможных уровней в зоне ровно в два раза больше, чем число электронов, вследствие чего она является зоной проводимости. Этим объясняется также высокая электрическая проводимость этих металлов. Существует несколько основных типов взаимного расположения энергетических зон (рис. А.62), соот-ветствующих изолятору, одновалентному металлу, двухвалентному металлу, полупроводнику с собственной проводимостью, примесному полупроводнику и-типа и примесному полупроводнику p-типа. Соотношение энергетических зон (рис. А.62) определяет также тип проводимости твердого тела.
Изолятор. Незаполненная зона проводимости L расположена значительно выше заполненной валентной зоны (для алмаза, например, расстояние между зонами соответствует ~5 эВ). Перенос электронов в зону проводимости при увеличении энергии Ферми за счет подвода тепловой энергии возможен только при очень высокой температуре (рис. А.62, а).
Металлические проводники. У одновалентных металлов (рис. А.62, б) L наполовину заполнена валентными электронами; при увеличении энергии электронов за счет внешнего источника по проводнику может протекать ток. У двухвалентных металлов валентная зона полностью заселена электронами, однако из-за сильного взаимодействия с полем кристаллической решетки металла происходит перекрывание валентной зоны с более высокими незаполненными зонами, так что электроны могут заполнять более высокие энергетические уровни, принимая энергию (рис. А.62, в).
Полупроводник с собственной проводимостью (рис. А.62, г). Разность энергии (Еа) между заполненной валентной зоной G и зоной проводимости L настолько мала, что становится сопоставимой с уровнями тепловой энергии. Отсюда
1g о = const-
Приведем значения Еа для некоторых полупроводников: Ge Mg3Sb2 a-Sn
Еа, эВ 0,72 0,82 0,08
В "результате перехода электронов в зону проводимости в валентной зоне образуются «положительные дырки» («дефекты электронов»), которые также могут изменять энергетический уровень и обеспечивать перенос электрического заряда.
Примесные полупроводники п-типа (рис. А.62, д). При внесении в собственный полупроводник посторонних атомов, способных быть донорами электронов, возникающие примесные энергетические термы находятся вблизи зоны проводимости Е Переход электронов в зону проводимости L требует лишь незначительных затрат энергии.
Химическая связь
143
Примесные полупроводники p-типа (рис. А.62, е). Внесение примесных атомов, способных быть акцепторами электронов, приводит к тому, что примесные энергетические термы находятся несколько выше валентной зоны G. Вследствие переноса электронов из валентной зоны на уровни атомов-акцепторов в валентной зоне становится возможной «дырочная проводимость». (Комбинации германиевых и кремниевых полупроводников с р- и «-проводимостью применяются в транзисторах.)
6,6.4. Смешанные кристаллы (твердые растворы)
и интерметаллические фазы
Для лучшего понимания свойств металлических сплавов и соединений с металлической связью напомним некоторые определения химии и физики металлов. Образование смешанных кристаллов происходит в том случае, если размеры атомов различных металлов близки между собой. При этом различают смешанные кристаллы замещения и внедрения.
Смешанные кристаллы замещения. При высокой температуре распределение атомов неупорядоченное, при низкой температуре атомы одного и другого металла размещаются в определенном порядке и образуют «упорядоченные» кристаллы или «сверхструктуры», строение которых можно установить рентгенографическим методом (например, медь с золотом).
Смешанные кристаллы внедрения образуются, если один из атомов может занять пустоты в решетке, образованной атомами другого металла. Например, углерод может внедряться в структуру у-железа, в результате чего получается смешанный кристалл (твердый раствор) аустенит.
Ближе всего к настоящим химическим соединениям с определенным стехиометрическим составом находятся так называемые интерметаллические фазы. Так, например, металлы литий и магний образуют с металлами IV—VII групп периодической системы соединения следующего состава: Mg3Sb2, Li3As, Li3Bi, Li4Pb, Mg3As2, Mg2Ge, Mg2Sn, Mg5Pb и т. д. Эти соединения кристаллизуются, как правило, в структурах, сходных со структурой ионных кристаллов, и имеют значительную энтальпию образования. Однако другие интерметаллические соединения не имеют такого определенного стехиометрического соста-ва> а только довольно широкую область гомогенности (напри-мср, система Си—Zn). Для такого рода интерметаллических Фаз Юм-Розери вывел следующее правило. Если установить отношение v/a (v — число имеющихся валентных электронов, а — исло участвующих в металлической связи атомов), то наблю-аются главным образом следующие четыре отношения: 21 :21 ля о-фаз, 21 ; 14 для 0-фаз, 21 : 13 для у-фаз и 21 : 12 для 8-фаз.
5
Рис. А.63. Диаграмма электронной плотности NaCl (а) и алмаза (б).
Химическая связь
145
Интересно отметить, что металлы группы железа и платины имеют в данном случае нулевую валентность. Это проявляется также в их магнитных свойствах. В то время как в свободном виде они парамагнитны, в сплавах они становятся диамагнитными, так как их свободные электроны заполняют вакантные ^-уровни.
6.6.5. Виды связи в твердых телах
Воспользовавшись диаграммами электронной плотности, рассмотрим еще раз различные типы связи в твердых телах. На рис. А.63, а и б приведены диаграммы электронной плотности
Рис. А.64. Схематическое изменение электронной плотности между соседними атомами [Lehrbuch der Physikalischen Chemie/Eggert, Hock, Schwab. — S. Hir-zel-Verlag, Stuttgart, 1968].
хлорида натрия и алмаза, а на рис. А.64 схематически показано изменение электронной плотности в зависимости от расстояния между соседними атомами (ионами) в решетках хлорида натрия, алмаза, магния и аргона. На этих примерах можно наглядно и убедительно показать различия в распределении электронов четырех основных видов связи — ионной, атомной, металлической и вандерваальсовой. В случае ионной связи между ионами имеются области, в которых электронная плотность падает до нуля; напротив, в алмазе вследствие делокализации электронных пар между атомами электронная плотность никогда не равна нулю. Такое же состояние характерно и для металлической связи, хотя в данном случае электроны не локализуются, а распределяются по всему кристаллу. Диаграмма электронной плотности магния показывает, что между соседними атомами средняя плотность соответствует двум электронам. Поскольку при вандерваальсовой взаимодействии не обра-УЮтся электронные валентные связи, электронная плотность ежду атомами, например аргона, равна нулю.
Гримм условно показал эти четыре типа связи с помощью тРаэдра, расположив вещества, рассмотренные выше в каче-1о--2027
146
Строение материи
Рис. A.G5. Типы связи в твердых телах [Lehrbuch der Physikalischen Che-mie/Eggert, Hock, Schwab. — S. Hirzel-Verlag, Stuttgart, 1968].
стве примеров данного типа связи (рис. А.65). Ребра тетраэдра изображают переходные типы связи в твердых телах. Например, для висмута характерно сочетание атомной и металлической связей. Интерметаллическое соединение Mg3Sb2 соответствует переходному типу связи — между металлической и ионной. В графите его слои обладают металлическими свойствами, а связь между слоями осуществляется вандерваальсовы-ми силами. В силикатах наблюдается типичная переходная форма связи — промежуточная между ионной и атомной (ионный характер SiO связи составляет ~5О°/о). Восьмичленные кольца молекулы серы образуются за счет атомных связей. Между собой эти кольца связываются вандерваальсовыми силами.
Таким образом, с помощью тетраэдра химических связей Гримма можно ориентироваться при изучении многообразия связей в химических веществах. Всегда следует попытаться при знакомстве с новым химическим веществом найти для него место на этом тетраэдре.
Б.1. ХИМИЧЕСКАЯ КИНЕТИКА
Б.1. ХИМИЧЕСКАЯ КИНЕТИКА И МЕХАНИЗМЫ РЕАКЦИИ
7. ПРЕДМЕТ И МЕТОД ХИМИЧЕСКОЙ КИНЕТИКИ
Существуют два основных подхода к изучению химической реакции: 1) термодинамический, с помощью которого определяют возможность и направление протекания реакции и 2) кинетический,— рассматривающий механизм реакции и ее скорость. Рассмотрим обобщенное уравнение химической реакции v4A + vBB vcC 4- vdD (1)
где va, vb, vc и vD-—стехиометрические коэффициенты, А и В —исходные вещества, С и D — продукты реакции.
Термодинамика рассматривает эту реакцию в состоянии равновесия, т. е. при условии, что концентрации всех участвующих в ней веществ не меняются. Термодинамический подход позволяет определить константу равновесия реакции, равновесные концентрации всех реагирующих веществ, тепловой эффект, а также условия смещения равновесия в ту или другую сторону. Термодинамические соотношения ничего не скажут нам о механизме и скорости реакции, а также о возможных промежуточных стадиях реакции.
Химическая кинетика исследует протекание реакции во времени. С помощью химической кинетики можно выяснить закономерности, в соответствии с которыми устанавливается состояние химического равновесия, а теоретический анализ зависимости скорости реакции от времени может дать полезную информацию о промежуточных стадиях реакции.
При этом важно отметить, что и в состоянии термодинамического равновесия молекулы веществ, участвующих в реакции, могут взаимодействовать между собой. Однако вследствие того, что скорости прямой и обратной реакций равны между собой, мы не замечаем изменения концентрации реагирующих веществ. Такое состояние называют динамическим равновесием в химической системе. Для расчета константы равновесия Нз термодинамических данных вполне достаточно знать обычае Уравнение химической реакции со стехиометрическими ко-эФфициентами, например
Н2 + 12 «=* 2HI (2)
Или
Н2+Вг, ^=±: 2НВг (3)
Ю*
148
Химическая кинетика и механизмы реакций
Предмет и метод
149
Напротив, из этих уравнений нельзя сделать никаких выводов о скорости этих реакций и их механизме. Учитывая, что эти реакции однотипны с точки зрения стехиометрических соотношений, можно было бы предположить, что они имеют аналогичные механизмы реакции и параметры ее скорости. Однако, как будет подробно показано (разд. 12.1), элементарная реакция образования HI согласуется с уравнением реакции (соударение молекул), в то же время образование НВг происходит в результате сложной цепной реакции, при которой сначала предполагается диссоциация молекулы Вг2, а затем реакции между атомами и молекулами:
Вг2 -< — > 2Вг (За)
Вг + Н2 ч=±: НВгН (36)
Н + Вг2 з НВг + Вг (Зв)
Характер изменения скорости реакции образования НВг (разд. 12.4) соответствует такому сложному механизму реакции.
Термодинамика представляет собой более законченную и теоретически обоснованную дисциплину, чем химическая кинетика. Однако за последние годы был достигнут значительный прогресс в развитии последней, особенно в истолковании механизма элементарного акта химической реакции.
До настоящего времени нельзя сделать определенных выводов, например, о механизме наиболее часто применяемых в аналитической химии окислительно-восстановительных реакций Остановимся на реакции между перманганатом калия и пероксидом водорода в щелочной среде:
2МпО4“+НаО2+2ОН- 2МпО42-+ О2 + 2 Н2О (4)
Согласно современным кинетическим данным, реакция протекает через следующие стадии:
НОО“ + МпО4- ---> [ООМпОзГ + ОН" (5)
[ООМпО3]- --> МпО3- + О2 (6)
МпОд- + Н2О --> Н2МпО4“ (7)
МпО4” + Н2МпО4" -J- 2 ОН" -> 2МпО42~ + 2 Н2О
При объяснении этой последовательности реакций следует вспомнить, что слабая кислота Н2О2 существует в щелочных растворах (при рН>12) в виде иона ООН-. В результате первой стадии реакции возникает пероксидный комплекс, распадающийся затем на МпО-3 и О2 (вторая стадия). МпО3- — весьма неустойчивая частица, еще не обнаруженная препаративными методами. Mn(V) имеет координационное число 4, поэтому по реакции (7) тотчас образуется Н2МпО4-, в свою очередь реагирующий реакция синпропорционирования) с МпО4~ с образованием МпО42~. Еще более сложно протекает реакция при из
бытке Н2О2, когда образуется диоксид марганца (разд. 36.14.1); в кислой среде реакция идет еще дальше до Мп2+.
Аналогичные затруднения возникают при рассмотрении большинства неорганических окислительно-восстановительных реакций. Несмотря на это, оказывается возможным составить такие стехиометрические уравнения, чтобы суммарная реакция представляла собой сложную последовательность нескольких стадий. На основе исследования механизма реакции можно сделать вывод об истинной последовательности этих стадий (или их параллельном протекании). В рассмотренном выше примере можно было бы предположить, что образование О2 обусловлено двумя электронными переходами, например
ООН" -J- МпО4- -> МпО42- + НО2
(8)
НО2 -J- МпО4- -4- ОН- -> МпО42- + О2 -J- Н2О
Однако исследования кинетики процесса показывают, что скорее всего реакция протекает в соответствии с уравнениями (5)-(7).
В большинстве случаев определение скорости реакции само по себе не позволяет сделать выводов о каком-либо конкретном механизме реакции. Здесь для выяснения механизма реакции может быть очень полезным применение стабильных или радиоактивных изотопов. В качестве примера рассмотрим взаимодействие сульфитов с перманганат-ионом в щелочной среде*:
2МпО4" + SO32~ + Н2О -> SO42- -J- 2НМпО4~ (9>
Вопрос о том, происходит ли здесь прямой перенос кислорода от МпО4~ к SO32- или имеет место двухстадийный электронный переход с образованием промежуточной частицы ионного радикала SO32- может быть решен, если провести реакцию с перманганатом, меченным 18О. Было установлено, что содержание 18О в продукте реакции SO42- точно такое же, как и в перманганате. Это доказывает, что реакция осуществляется по первому механизму, что соответствует следующему уравнению:
so32- 4- 18ОМпО3- ---► [O3S—l8O-MnO3]3- -> OgS^O2- MnO3- (10>
Ион MnO3“ реагирует в соответствии с уравнением (7).
Другими методами, которые можно привлечь к исследованию Механизмов реакции, являются изучение короткоживущих промежуточных продуктов реакции с помощью масс-спектрометрии и электронного парамагнитного резонанса.
* В сильнощелочной среде (сон >10 моль/л) образуется устойчивый продукт реакции МпО43~ (разд. 38.14.1).
150
Химическая кинетика и механизмы реакций
Наряду с исследованием механизма химических реакций методы химической кинетики используются также для решения такой важной задачи, как определение основных параметров процессов химической технологии (выбор оптимального времени реакции, температуры, концентрации реагирующих веществ, катализаторов и т. п.).
8. ОСНОВНЫЕ ПОНЯТИЯ ХИМИЧЕСКОЙ КИНЕТИКИ
(ПОРЯДОК РЕАКЦИИ, МОЛЕКУЛЯРНОСТЬ, КОНСТАНТА СКОРОСТИ)
8.1. Скорость реакции
Рассмотрим снова реакцию (1) и предположим теперь, что все стехиометрические коэффициенты равны 1, а реакция идет <лева направо (реакция справа налево заторможена). Тогда
Тис. Б.1. Схема изменения во времени концентрации исходных веществ и продуктов простой химической реакции. Индексом 0 отмечены начальные скорости ( de \ ( dx \ реакции —— .
\ dt /о\ dt /о
скорость реакции г определяется как производная концентрации исходных веществ
dC\_____dev ,,
dt ~ ~dt~ '
или продуктов реакции по времени (рис. Б.1)
rfee _ dco м2)
dt ~ dt
Основные понятия
151
Достаточно знать изменение во времени концентрации одного вещества, так как концентрация других веществ определяется стехиометрическими коэффициентами уравнения реакции Для случая реакции (1) при va = vb=1 уравнение скорости реакции определяется следующим образом:
г=—^-=^саСв (13)
т. е. скорость реакции пропорциональна концентрациям исходных веществ, k называют константой скорости химической реакции (она зависит от температуры) и равна скорости реакции при концентрации исходных веществ, равной единице (например, при Сд=1 моль/л и <?в = 1 моль/л). В данном случае k имеет размерность (моль • л-1)-1-с-1.
8.2. Порядок и молекулярность реакции
Порядком реакции называют сумму показателей степени концентраций, входящих в уравнение скорости реакции. Следовательно, реакция, скорость которой описывается уравнением (13), имеет второй порядок п = 1 + 1 = 2. Часто более целесообразно определять порядок реакции по каждому компоненту; для уравнения скорости реакции (13) порядок реакции по компонентам А и В равен 1. Для общего случая (1), если стехиометрические коэффициенты va и vb отличны от единицы, уравнение скорости реакции записывается в следующем виде:
г=-----=------------(14)
va di vb at ad \ /
Одна из задач химической кинетики состоит в определении порядка реакции (tn и п) по отдельным компонентам. Лишь редко случается, что порядок реакции по данному компоненту совпадает со стехиометрическим коэффициентом уравнения реакции. Одной из таких редких реакций является образование иодоводорода (2):
г
dcH. ~dT
(15)
Рассмотрим также более сложную реакцию, например окисление метанола хромовой кислотой в кислом растворе:
ЗСН3ОН + 2Н2СгО4 + 6Н3О+ ► ЗСН2О + 2Cr3+ + 14Н2О (16)
Для этой реакции стехиометрические коэффициенты не имеют никакого отношения к порядку реакции, и уравнение скорости Реакции можно записать в следующем виде:
Г = kc^Q^ ССго42- • С2н3О+ (1 Ъ
152
Химическая кинетика и механизмы реакций
Порядок этой реакции по каждому компоненту определяется ее механизмом, т. е. элементарными промежуточными стадиями и их последовательностью. Из уравнения скорости реакции нельзя сделать никаких выводов о том, как происходит восстановление Cr(VI). Оно может быть результатом одноэлектронных переходов Cr(VI)->-Cr(V)->Cr(IV)->Cr(III), а также обусловлено двухэлектронным переходом при переносе О или И. Нельзя также выяснить, в какой стадии реакции участвуют ионы НзО+ и появляется ли промежуточный радикал (СН2ОН).
Таким образом, порядок реакции следует рассматривать лишь в связи с механизмом реакции в целом, помня, что этот механизм складывается из отдельных элементарных стадий. В то время как порядок реакции определяется для реакции в целом, понятие молекулярность реакции относится к ее отдельным стадиям. Молекулярность реакции равна числу молекул, которые сталкиваются в элементарном акте химического превращения (на некоторой промежуточной стадии процесса). Очевидно, что чаще всего происходят двойные столкновения (двух частиц) между реагирующими молекулами, а следовательно, в большинстве случаев элементарные стадии (или элементарные реакции) бимолекулярны. Вероятность тройных соударений (соответствующая тримолекулярным реакциям) уже значительно меньше, а реакции с молекулярностью более трех практически не наблюдаются. Настоящие мономолекулярные реакции, в которых молекулы распадаются сами без какого-либо внешнего воздействия, также встречаются очень редко. Наиболее известный пример мономолекулярного процесса, протекающего по первому порядку, — это радиоактивный распад. Он происходит спонтанно, и на него практически не оказывают влияния внешние воздействия. Скорость распада в любой момент времени t пропорциональна числу имеющихся атомов У:
-4г
В большинстве случаев молекулярность реакции не совпадает с ее порядком. Уравнение скорости бимолекулярной реакции может, например, соответствовать реакции первого порядка, если одно из реагирующих веществ имеется в избытке, так что его концентрация в ходе реакции почти не меняется. Примером такой реакции может служить кислотный гидролиз эфиров:
нчо+
СН3СООС2Н5 + Н2О —СН3СООН+С2Н5ОН (19)
Эта реакция протекает по бимолекулярному механизму в том •смысле, что она осуществляется при взаимодействии молекул
Основные понятия
153
эфира и воды, однако она может быть описана уравнением скорости реакции первого порядка:
г k • • сэфир k сэфир (20)
Поскольку реакция протекает в большом избытке воды по сравнению с эфиром, ее концентрация почти не меняется и может быть принята постоянной, а следовательно, сн2о можно ввести в константу реакции. Таким образом, эта реакция имеет кажущийся первый порядок.
9. ПРОСТЫЕ КИНЕТИЧЕСКИЕ ЗАКОНОМЕРНОСТИ.
АНАЛИЗ РЕЗУЛЬТАТОВ ИССЛЕДОВАНИЯ КИНЕТИКИ ХИМИЧЕСКИХ РЕАКЦИИ
При исследовании кинетики химической реакции прежде всего надо определить изменения концентрации реагирующих веществ во времени (рис. Б.1). Используя эти данные, необходимо найти порядок химической реакции, иначе говоря, записать уравнение скорости реакции, а затем определить и константу скорости. Из зависимости константы скорости от температуры можно получить значение для энергии активации.
Если стехиометрия реакции позволяет ожидать достаточнопростую закономерность, часто оказывается целесообразно привлечь для исследования кинетических закономерностей интегральные уравнения скорости реакции, имеющие весьма простой вид для реакции первого, второго и третьего порядков.
9.1. Уравнение скорости реакции первого порядка
Итак, как уже упоминалось, радиоактивный распад может служить идеальным примером реакции первого порядка. Предположим сначала, что исходный радиоактивный нуклид А превращается в стабильный нуклид В в результате радиоактивного излучения (а-, р- или у-излучение)
А ---> В (21 >
Этому процессу соответствует уравнение скорости реакции (18); после разделения переменных получим
-~ = kdt (22)
Интегрирование дает
—1пА = &4-С (23)
Константу интегрирования С находим из начальных условий; при t—Q и (V=2Vo C=lnA0- Тогда из уравнения (22) следует
In ЛЖ0 = —kt; А = Ао • е-« (24)
154
Химическая кинетика и механизмы реакций
Результаты исследования такой реакции могут быть представлены графически. Очевидно (рис. Б.2, а), что радиоактивность N экспоненциально снижается во времени в соответствии с уравнением (24), потому удобно пользоваться полулогарифмическими координатами (рис. Б.2, б). Из наклона прямой можно определить константу скорости k.
Рис. Б 2. Изменение активности или концентрации вещества во времени для реакции первого порядка.
а — в обычных координатах, б —в логарифмических координатах
Важный параметр в химической кинетике — время полупревращения Д/2 (для случая радиоактивного превращения — период полураспада). Это время, в течение которого начальная концентрация исходного вещества (или количество молекул исходного вещества) снизится в два раза. Для случая радиоактивного распада это означает, что N=N0/2, т. е.
1п2 = Ц/2 или £1/2 = 0,693/& (25)
Часто первый продукт распада радиоактивного нуклида не является стабильным, а распадается далее. За немногими исключениями, так ведут себя почти все естественные радиоактивные вещества, входящие в три основных семейства (ряда) радиоактивных элементов (ряд уран —радия, ряд тория и ряд актиния). В этих радиоактивных семействах имеется один наиболее долгоживущий материнский элемент, распадающийся на дочерние и внучатные короткоживущие радиоактивные элементы. В общем случае превращения можно представить в виде схемы
(26)
Простые кинетические закономерности
155-
В качестве примера приведем основные стадии радиоактивного распада для семейства уран — тория:
4,51 109 лет 24,5 сут 1,14 мин
23892U-----------► 231eoTh ----------> 234пРа------------>
1590 лет
_____Ь. 234 Т I _____*, ... 226 Рд ----------->. 4 . . 206 DK
-----► 92U * 88Л<1 82ги
(стабильный изотоп)
(27}
Система такого рода, поскольку на нее не оказывается внешнего воздействия, находится в радиоактивном равновесии. Оно соответствует стационарному состоянию, при котором скорости образования и распада отдельных компонентов равны между собой:
dNpJdt — dNn!dt = dNc/dt = dNG/dt
(28)
Используя уравнение (18), получим
^(VA = ^B = ^VC (29)
Подставим вместо k период полураспада h/2 [уравнение (25)]:
ВД/2 (А) = ВД/2 (В) = ад/2 (С) (30}
Согласно уравнению (29), активность компонентов радиоактивного равновесия обратно пропорциональна константам скорости.
Аналогичные соотношения наблюдаются и при химических реакциях, скорость которых подчиняется уравнению первого порядка. Гомогенные химические реакции, протекающие строго в соответствии с уравнением скорости первого порядка, встречаются не часто. К ним относятся в первую очередь реакции разложения, например дегидрирование этана до этилена
С2Н6 » С2Н|УН2 (31}
или циклопентена до циклопентадиена
(32)
Активация молекул, т. е. подвод к ним энергии, необходимой Для осуществления химического превращения, здесь также обеспечивается за счет соударений с молекулами. Эта реакция подчиняется закономерностям мономолекулярной реакции преж-До всего потому, что возбуждение реагирующих молекул происходит при столкновении с любыми молекулами, даже не уча-
156
Химическая кинетика и механизмы реакции
у ПодвоЗ энергии соударением
Рис. Б.З. Молекулярный механизм мономолекулярного дегидрирования циклопентена.
Дезактивация соударением
. Распределение н колебательной 11 энергии
в возбужденной молекуле
ствующими в реакции, например с молекулами инертного газа. Поскольку такие соударения относятся к бимолекулярным процессам, реакции термического разложения часто называют псевдомономолекулярными; можно даже сказать, что, кроме радиоактивного распада, не существует реакций, протекающих строго по мономолекулярному механизму.
Для наглядности на рис. Б.З схематически изображены процесс возбуждения (активирования) молекулы, а также дальнейшие стадии реакции на примере дегидрирования циклопентена. Энергия активации в виде колебательной энергии распределяется на возбужденной молекуле А*; последняя может либо дезактивироваться при соударении с другой исходной молекулой А, либо при благоприятной локализации энергии перейти в В с отщеплением Н2.
Известные реакции, лишь формально протекающие пс уравнению реакции первого порядка, как это было показан! для омыления эфира в кислой среде [уравнение (19)]. Вооб ще, если один из компонентов имеется в большом избытке 1 вследствие этого его концентрация почти не снижается в ход! реакции, то, хотя эта реакция протекает со скоростью, опреДе ляемой уравнением второго порядка, расчет кинетики такойре акции можно проводить на основе уравнений скорости реакпи1
Простые кинетические закономерности 157
первого порядка. Кроме того, различные каталитические реакции (как гомогенные, так и гетерогенные) идут в соответствии с закономерностями уравнения первого порядка [разд. 14.2, уравнение (122)]. Гетерогенная реакция, скорость которой определяется диффузией реагирующего вещества к границе раздела фаз, также описывается уравнением скорости реакции первого порядка (разд. 14.1).
Часто рассматривают также скорость образования продукта реакции во времени; тогда по аналогии с уравнением (18) можно записать
~ = k(a~x) (33)
где а — начальная концентрация исходного вещества, ах — концентрация продукта реакции в момент времени t. Используя начальные условия (/ = 0, х = 0), после разделения переменных и интегрирования получим
In a-^x- = kt (34)
х = а(1—e~kt) (35)
k можно определить графическим методом (рис. Б.2, а), построив график зависимости изменения концентрации от времени в координатах lg(a—х)—t-, полученная прямая пересекает ось координат в точке 1g а, а ее коэффициент наклона равен —k/2,3. В соответствии с уравнением (33), размерность константы скорости реакции первого порядка с-1.
9.2. Реакции второго и более высокого порядков
Для многих химических реакций кинетические закономерности подчиняются уравнению скорости реакции второго порядка. К таким реакциям относятся образование Ш и его разложение на исходные вещества. В качестве классического примера реакции второго порядка часто рассматривают омыление эфира в Щелочной среде:
СН3СООС2Н6 + ОН" --*- СН,СОО- + С2Н6ОН (36)
Многие реакции, сопровождающиеся переносом электрона, также являются реакциями второго порядка, например
Fe2+4-Mn3+ >- Fe3 + + Мп2+ (37)
Се4+ + Fe2+ -> Се3+ + Fe3+ (38)
В наиболее простой форме уравнение скорости реакции второго порядка получается в том случае, если концентрации исход
158
Химическая кинетика и механизмы реакций
ных веществ равны между собой’
-ДГ=ЙС (39)
где с—концентрация исходного вещества. После разделения переменных и интегрирования получаем
<4°>
Для начальных условий 1 = 0 и с=с0 находим константу интегрирования С= 1/с0, тогда
= (41>
Обработку кинетических данных по уравнению (41) удобнее всего проводить графически, построив график в координатах 1/с—t. Полученная прямая отсекает на оси ординат отрезок 1/с0; графически определяют и коэффициент k.
Время полупревращения /1/2 можно найти, подставив с = — с012 в уравнение (41).
k имеет размерность (моль • л-1)-1 • с-1.
При равенстве концентраций реагирующих веществ кинетические уравнения более высокого порядка, соответствующие соотношения между концентрацией Со и периодом полупревращения Zi/2 находят так же, как и уравнения скорости реакции второго порядка (39)—(42). Уравнение скорости реакции п-го порядка можно записать в следующем виде:
de
~^ = kcn ИЗ)
Разделив переменные и проинтегрировав уравнение (43), получим
При начальных условиях 1 = 0 и с = Со получим константу С = = 1/(п—1) соп~Тогда
(я — 1) ('с”7! Со"-1 )= (44)
Уравнение (44) справедливо для всех п, кроме п=1. При подстановке п=\ в (44) получается неопределенность (кинетическое уравнение первого порядка рассмотрено ранее). Время по
Простые кинетические закономерности.
лупревращения реакции первого порядка можно выразить через уравнение (44), если подставить с = Со/2:
2«-i _ 1 ^/2= (Л—
Известны также уравнения нулевого порядка (разд. 14.2). После подстановки п = 0 в уравнение (44) получим
с0—с = kt (46)
Очевидно, в этом случае количество образовавшегося вещества пропорционально времени; зависимость концентрации от времени представляет собой прямую, отсекающую на оси координат отрезок Со и имеющую тангенс угла наклона k. Такого типа кинетические закономерности встречаются главным образом в гетерогенных реакциях.
Кинетические уравнения принимают несколько более сложную форму, если концентрации исходных веществ не равны между собой. Рассмотрим снова реакцию второго порядка. Пусть а—'начальная концентрация вещества А, b — концентрация вещества В, х — концентрация одного из продуктов реакции Тогда
—- = k(a—x)(b—x) (47)
После разделения переменных интегрируем
f 7---Йь----Г = f kdt (47а)
J (а — х) (Ь — х) J
Далее разложим подынтегральное выражение на сумму дробей: определение коэффициентов М и
N дает А=1/(а—Ь) и М=1/(6—а). Таким образом, интеграл складывается из двух легко решаемых интегралов. Несложные вычисления дают
-Л-1 - In = W+C (Ь — а) а —х 1
Так же как и ранее, из начальных условий (/ = 0; х = 0) получим С= —- in В результате получается выражение зависимости концентрации конечного продукта от времени:
= (476)
b —а Ь (а —х) ’
Обработка результатов опыта проводится довольно просто в координатах lg(A—х)1(а—х)—<t. При этом экспериментальные Данные по кинетике реакции укладываются на прямую, тангенс
160 Химическая кинетика и механизмы реакций
угла наклона которой равен k(b—а)/2,3; прямая отсекает на оси ординат отрезок lg a/b. В данном случае понятие «время полупревращения» для суммарной реакции теряет свой смысл, так как необходимо уточнить, относительно какого компонента следует рассчитать значение ti/2.
9.3. Кинетические уравнения как функции безразмерной
переменной
Для решения ряда задач химической кинетики оказывается целесообразным использовать вместо концентрации безразмерную переменную. Для этого относят текущую концентрацию реагирующего вещества к исходной концентрации:
C'red = C/C0 (48)
Переменную с' удобно применять для обработки экспериментальных данных, в особенности, когда масса вещества прямо пропорциональна концентрации:
М const с
Мо const -с0 С red
(48а)
Для математического описания кинетической кривой как функции времени, например с помощью аналоговой ЭВМ, также необходимо введение безразмерной переменной. Подстановка (48) в (44) и соответствующие преобразования дают
-z- ~ тс ((£')1-"— 1) = £O"-1^ = T (44а)
\!1, ч
Как можно видеть из уравнения (43), константа скорости имеет размерность (моль-л-1)1-71-с-1, следовательно, x = con~i kt— безразмерный параметр, пропорциональный времени. Подстановкой в (44а) целочисленных значений п от 0 до 4 получим следующие выражения зависимости безразмерной концентра-
ции от времени: п= 0 с' = 1 —т (446)
п= 1 с' — е-т (44в)
п = 2 с' = 1/(1 +т) (44г)
п = 3 с'=1/|/1-|-2т ' (44д)
п = 4 с' = 1/р/ 1 -|-Зт (44е)
Подстановка п—\ в (44а) приводит к неопределенности (типа 0/0). В соответствии с правилом Лопиталя (правило предельных переходов)
, (с')1-"—1 (с')1-71 In С 1 , ,
Iim-------;---= Iim —---------j----- = —In c
1 J можно получить уравнение (44в).
Простые кинетические закономерности
Зависимости с' — т, соответствующие уравнениям (446) — (44е), представлены на рис. Б.4. Можно заметить, что при прочих равных условиях скорость реакции увеличивается при понижении порядка реакции.
9,4. Методы определения
порядка реакции
Рассмотренные выше соотношения пригодны для расчета кинематики многих химических реакций. Однако для некоторых реакций наблюдаются более сложные зависимости изменения концентрации во времени (кинетические кривые), ин
Рис. Б.4. Сравнение реакций различного порядка в безразмерных переменных.
теграл которых невозможно выразить через элементарные функции. Для решения таких задач приходится применять приближенные мето-
ды (разложение в ряд, использование вычислительной техники). Определение порядка реакции таких более сложных хими-
ческих реакций проводят главным образом методом начальных скоростей и методом времени полупревращения.
9.4.1, Определение порядка реакции из зависимости начальной скорости реакции от концентрации
Для любой сложной реакции, порядок которой по каждому компоненту следует найти, можно записать следующее дифференциальное уравнение для скорости реакции:
de
= kcAm-cBn-ccP....
(49)
гДе m, п, р,... — порядок реакции, который необходимо определить. Проведем следующий кинетический эксперимент. Для определения порядка реакции m вещества В и С берутся в таком избытке, что в течение некоторого времени их концентрация не меняется. Начальную скорость реакции определяют Для нескольких исходных концентраций со вещества А. Для них Можно использовать графический метод: провести касательные к кинетической кривой в начальный момент времени и определить тангенс угла наклона (рис. Б.5). Для т>1 следует ожидать, чТ0 с увеличением начальной концентрации вещества А НаЧальная скорость реакции также увеличивается. Подстанов-11 —2027
162
Химическая кинетика и механизмы, реакций
ка начальной скорости в уравнение (49) и логарифмирование дают
/ de \
lg = lgA:4-mlgcA+nlgcB4-plgcc (50)
Если все концентрации, кроме сА, постоянны, то построенный по экспериментальным данным график в координатах lg (dcA/dt)0~~ —1gсА представляет собой прямую, тангенс угла наклона ко-
Рис. Б 5. Уменьшение концентрации исходных веществ во времени (порядок реакции можно найти по начальной скорости реакции).
торой равен т. Подобным же образом можно найти порядок реакции п и р остальных веществ, участвующих в реакции, при изменении соответствующих начальных концентраций. Разумеется, расчет соответствующего порядка реакции можно проводить по уравнению прямой.
9.4.2. Метод времени полупревращения
Этот метод основан на использовании зависимости между порядком реакции, временем полупревращения и начальной концентрацией Со [уравнение (45)]. В ходе эксперимента варьируют также концентрацию компонента, по которому определяется порядок реакции (основного компонента), поддерживая концентрацию остальных веществ постоянной. Кроме того, измеряют время полупревращения ti/2 как функцию начальных концентраций Cq основного компонента. Концентрации остальных реагирующих веществ, как и ранее, поддерживают посто-
Простые кинетические закономерности 163
" г
явными. После логарифмирования уравнения (45) получаем
[ОН —1_ 1 1
(П~ 1}fe ] —(^~ l)lg^o (51)
Если представить полученные результаты графически в координатах 1g/1/2—1g са, то в соответствии с уравнением (51) из наклона прямой можно определить (п— 1) или же рассчитать это значение из соотношения
lg = (n_ 1) lg (52)
Г1/2 (2) C01
Следует, однако, отметить, что значения порядка реакции, полученные с помощью двух рассмотренных методов, не всегда согласуются между собой. Это объясняется тем, что на начальную скорость реакции не оказывают влияния промежуточные продукты реакции, существенно влияющие на скорость ее протекания. Напротив, метод времени полупревращения учитывает влияние промежуточных веществ.
Пример 1. В присутствии оснований нитроамид NH2NO2 разлагается: NH2NO2->N2O (газ) +Н2О (жидк.). В раствор ацетатного буфера при 15°С поместили 50,0 мг нитроамида. Через 70 мин выделилось 6,19 мл газа (N2O) (объем отнесен к 15 °C и 101,325 кПа). Рассчитайте константу скорости, время полупревращения П/2 и время, за которое разложится 90% нитроамида. Разложение нитроамида является реакцией первого порядка.
Решение. Используем уравнения (24) или (34)
kt = 2,303 1g—
& п.
Рассчитаем исходную концентрацию NH2NO2 • По = 50,0-10-3 г/62,032 г= =0,806-Ю-3 моль (A4nh2no2 = 62,032 г). Воспользовавшись законами идеальных газов, найдем количество выделившегося за 70 мин N2O в молях:
6,19-10-в м3-101 325 Па
= 8,314 Па-м-моль-1-к-1.288,2К = 0,261‘° 3 моль
Тогда через 70 мин останется п= (0,806—0,261) • 10-3 моль=0,545-10-3 моль
NH2NO2. Отсюда
2,303-1g (0,806/0,545) 60-70 с
= 9,33-10-4 с-1
Время полупревращения /,/2 [уравнение (25)] равно 7440 с. Время, необходимое для разложения 90% исходного вещества, равно
2,303-lg (1/0,9) °.9~ 9,33-10-4с-1
= 1,137-105 с = 31,6 ч
Данная реакция катализируется основаниями, поэтому в более щелочных средах, чем ацетатный буфер, константа скорости увеличивается.
.„ Пример 2. Диметиловый эфир при высокой температуре разлагается: ”з)2О^СП4-|-Н2Ч-СО. В эвакуированную колбу при температуре 504 °C
И*
164 Химическая кинетика и механизмы реакций
вводился диметиловый эфир и измерялось давление через определенные промежутки времени:
t, °C 390 777 1587 3155 сю
р, кПа 54,4 65,06 83,19 103,86 124,12
Рассчитайте константу скорости, время полупревращения, а также давление в сосуде через 30 мии после начала опыта.
(/24 /23-р), Пф
Рис. Б.6. Кинетический график для реакции разложения диметилового эфира.
Решение. В уравнение (24) вместо концентраций (или числа частиц) подставим парциальные давления эфира в момент времени 4=0 (ро) и 4(р):
Рп
kt = 2,303 1g —
Р
Выясним прежде всего, как зависит парциальное давление эфира от общего давления в сосуде. В соответствии с уравиеиием реакции р«—р— =ран =Рн —рао; Робщ = р+рсн +Рн2+рсо = 3ро—2р. Для 4=<ю,р=0, или Роо=ЗРо, т. е. для данного опыта ро=рсо/3=41,37 кПа. Тогда р= = 1/2(Зро—робщ) ='/2(124,12—робщ). Подставим полученное выражение для давления эфира в исходное уравнение:
£4=lg ро — 1g р= 1g 82,74 — 1g (124,12 — рОбщ)
Обработку результатов опытов целесообразнее всего проводить графически в полулогарифмических координатах. График зависимости lg(124,12—р0бш) от 4 на рис. Б.6 представляет собой прямую, а следовательно, реакция разложения эфира является реакцией первого порядка. По графику можно определить время полупревращения 41з=1550 с. Зная Н/г можно с помощью уравнения (25) вычислить константу скорости k—0,693/1550 с=4-10-4 с"1-Графически можно иайти также общее давление через 30 мии=1800 с, р= = 86,93 кПа.
Пример 3. Пусть реакцию 1- + ОС1~-ИЭ1- + С1- проводят при 25 С в 1,0 М растворе NaOH, начальная концентрация C]-=eoci-=2-10~3 моль/Л
Простые кинетические закономерности
165
концентрация реагирующих веществ во времени изменяется следующим образом (спектрофотометрический метод индикации):
t, с 2 3 5 6 8 14
= СОС1-, моль/л 0,00169 0,00158 0,00135 0,00123 0,00101 0,00073
Постройте кинетическую кривую и определите константу скорости реакции.
Решение. Для исследуемой реакции неизвестен порядок. Поэтому прежде всего необходимо попробовать представить экспериментальные результаты в
координатах, удобных для расчетов при различных порядках реакции (например, 1g с—t или 1/с—£), а именно для первого и второго порядков. Исходя из уравнения реакции, можно ожидать, что рассматрнваемая реакция является реакцией второго порядка. Поэтому целесообразно провести обработку результатов в соответствии с уравнением (41). Отложим на оси ординат \/с= = 1/Coci" как функцию времени I. Из наклона прямой найдем k= = 61 (моль/л)’1 -с-1.
Точки на рис. Б.7 имеют некоторый разброс относительно прямой. Однако, учитывая, что данные получены для довольно быстрой реакции, можно считать сходимость результатов достаточно хорошей. Для обработки результатов можно использовать метод наименьших квадратов.
Пример 4. Эйгеиом и Мейером для реакции нейтрализации Н3О+ + ОН~—>-—>2Н2О найдена константа скорости k= (1,3±0,2) • 1011 (моль/л)1^'1 при
25 °C.
Рис. Б.7. Кинетический график для реакции между иодидом и гипохлоритом (реакция второго порядка).
Для реакции нейтрализации сильной кислоты сильным основанием рассчитайте /jy (концентрации кислоты и основания одинаковы и равны 10-4 моль/л); найдите также время, за которое реакция пройдет на 90%.
Решение. С помощью уравнения (42) рассчитаем время полупревращения реакции нейтрализации при заданных условиях:
Zi/2 — 1,3- 10й-10~4 - 7,7-10 « с
Если концентрация веществ уменьшается на 9/10, с=0,1 с0; подставив эти величины в> уравнение (41), получим время, необходимое для такого снижения концентрации:
9
Z=^r=9/V2=6’9-10’5 с
Этот пример показывает, с какой большой скоростью идет реакция нейтрализации; разработка техники эксперимента и аппаратуры для исследования таких быстрых реакций относятся к важнейшим достижениям современной химии за последние 15 лет.
Вопросы и задания
1. Для определения степени гидролиза метилацетата СН3СООСН3 в 1 М Растворе НС1 при 25 °C из исследуемого раствора через определенные про
166
Химическая кинетика и механизмы реакций
межутки времени отбирали пробы по 0,5 мл, которые титровали 0,1852 моль/л NaOH.
t, с 339 1242 2745 4546 оо
V, мл NaOH 26,34 27,8 29,7 31,81 39,81
а) Определите константу скорости реакции и время полупревращения, а также формальный порядок реакции, б) Рассчитайте, сколько процентов эфира гидролизуется через 60 мин.
2. Для реакции CH3CH2NO2+OH_—► H20 + CH3CH = N02_, протекающей при 0°С, /г=38,1 (моль/л)~‘-мин-1. а) Пусть водный раствор с концентрацией 0,004 моль/л нитроэтана омыляется раствором NaOH (скаон= = 0,005 моль/л). Сколько времени потребуется, чтобы реакция прошла На 90% ? б) Для тех же условий рассчитайте время, через которое расходуется половина нитроэтана? щелочи?
3. Вещество А смешивают с эквимолярным количеством вещества В. Через 1 ч прореагировало 75% вещества А. а) Рассчитайте константу скорости реакции, полагая порядок реакции равным 0, 1 и 2. б) Какая часть вещества А прореагирует за 2 ч, если порядок реакции равен 0, 1, 2?
4. В слабокислом растворе пероксид водорода Н2О2 и тиосульфат-иоч ЗгОз2~ реагируют по уравнению Н2О2 + 2 S2O32~ + 2H+->-2H2O + S4Oe2_. В области pH от 4 до 6 скорость реакции не зависит от pH. При температуре 25 °C, pH 5 и начальных концентрациях сн2О2=0,0368 моль/л и с§2о,2_ == = 0,0204 моль/л была обнаружена следующая зависимость концентрации С32О32'~ ОТ вреМенИ:
/, мин 16 36 43 52
с52Оз2-’'03, моль/л 10,3 5,18 4,16 3,13
Определите: а) порядок реакции; б) константу скорости реакции; в) количество S2O32-, оставшегося в растворе через 1 ч, если при сохранении прочих условий концентрацию Н2О2 увеличить в два раза.
10. ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДЫ В ХИМИЧЕСКОЙ
КИНЕТИКЕ
Как уже неоднократно упоминалось, первичными исходными данными, которые получают при проведении кинетичесю к экспериментов, является зависимость концентрации реагирующих веществ от времени. Поэтому исследование кинетики реа ции выливается прежде всего в определение концентрации в ществ с помощью химических или физико-химических метод( анализа. Критерием при выборе того или иного аналитическо метода является время, необходимое для проведения анализа, оно должно быть намного меньше, чем общее время кинетического эксперимента. Для исследования достаточно длительных реакций (0,5 ч</1/2< 100 ч) через определенные промежутки времени из реакционной смеси отбирают пробы; в пробе приостанавливают реакцию (для этой цели применяют достаточно сильное ее охлаждение, поскольку, согласно эмпирическому пра
Экспериментальные методы
167
вилу Вант-Гоффа, скорость реакции уменьшается в 2—3 раза при понижении температуры на 10°C, либо удаляют из реакционной смеси одно из реагирующих веществ, например путем перевода его в труднорастворимое вещество). Однако этим методом (отбор проб) можно найти лишь отдельные точки на кинетической кривой. Значительно удобнее непрерывные методы контроля протекания реакции во времени, единственно пригодные для исследования кинетики быстрых химических реакций.
Чаще всего для исследования процесса протекания химических реакций во времени применяются следующие методы анализа:
Прямое определение концентрации проводится гравиметрическим или масс-аналитическим методами. В современной химической кинетике эти методы почти полностью заменены физико-химическими методами, с помощью которых измеряют некоторый физический параметр, пропорциональный концентрации вещества. Например, в газовых реакциях прослеживают изменение давления газа в системе в ходе протекания реакции. Впрочем, предпосылкой использования последнего метода является разность в количестве молей исходных веществ и продуктов реакции; тогда протекание реакции сопровождается заметным изменением давления. В последнее время для изучения газовых реакций используют масс-спектрометрический анализ, а также методы, основанные, в частности, на измерениях теплопроводности газов (например, газовая хроматография).
Реакции в растворах также прослеживаются с помощью физических или физико-химических методов, например из измерений электропроводности (применяются для исследования кинетики ионных реакций, в результате меняется общее число ионов), из измерений поглощения света (в соответствии с законом Ламберта — Бера поглощение света пропорционально концентрации вещества). Различие в оптической активности исходных веществ и продуктов реакции также может быть использовано для определения концентрации (например, при исследовании инверсии сахарозы). Применение полярографических методов анализа основано на том, что предельный ток Диффузии пропорционален концентрации.
Преимущество физико-химических методов анализа состоит в том, что с их помощью можно осуществить постоянное наблюдение за изменением концентрации во времени, не прерывая ход исследуемой реакции.
Одна из насущных задач химической кинетики состоит в получении кинетических кривых для быстрых реакций; с помощью современных методов исследования можно решить эту задачу для многих реакций. Из подходящих для этой цели методов можно назвать осциллографический и полярографиче-
Константа скорости реакции 169
168 _______Химическая кинетика и механизмы реакций_____
ский*. Методы изотопного обмена также позволяют изучай очень быстрые реакции, хотя следует отметить, что этот метод можно применять лишь к таким реакциям, механизм которых связан с обменными процессами. Сама по себе реакция может протекать исключительно быстро, однако оказывается возможным измерить ее скорость, так как перераспределение нуклидов в ходе реакции происходит за счет многократного обмена в прямом и обратном направлениях, и поэтому оно происходит значительно медленнее, чем происходит сама реакция обмена.
Для изучения очень быстрых химических реакций, а также для установления короткоживущих промежуточных продуктов применяется метод парамагнитного электронного резонанса. К наиболее быстрым химическим реакциям, для которых константа скорости практически идентична числу столкновений (hx 1012 ш1), относятся реакции переноса протона, а также различные реакции с электронными переходами. Совсем недавно для определения констант скорости с большим успехом применяют релаксационные методы. В самом общем виде сущность этих методов состоит в том, что на систему, находящуюся в состоянии термодинамического равновесия, оказывают кратковременное воздействие, выводящее ее из равновесия (например, воздействуют ультразвуком). Скорость установления нового равновесного состояния регистрируется, например, на осциллографе. Время, необходимое для перехода к новому состоянию, называют временем релаксации; оно количественно связано с константой скорости реакции. Для нарушения равновесия используют также кратковременное повышение температуры.
График зависимости lgk от 1/Г представляет собой прямую с коэффициентом наклона А; эта прямая пересекает ординату в точке В. Аррениус и Вант-Гофф конкретизировали константы в уравнении (53):
1п£=1п/г0—(54)
или
_ Е
k — kQQ~ RT (55)
где Е — энергия активации, R — универсальная газовая постоянная, a <k0 — фактор частоты или фактор столкновений (разд. 11.2). Из уравнения (55) следует, что не каждое столкновение реагирующих частиц приводит к химическому превращению, а лишь те столкновения, энергия которых больше энергии активации Е. Уравнение Аррениуса (55) можно также записать в дифференциальной форме
din k Е
~dT~ = ~Rf*
Интегрирование в пределах температур от Л до Г2 дает
С помощью уравнения можно рассчитать энергию активации, если найдены константы скорости при двух различных температурах.
11. КОНСТАНТА СКОРОСТИ РЕАКЦИИ
Зависимость константы скорости от температуры
прошлом столетии было установлено, что для многих -------------------------------------------~ ~ — . т т Т
в
11.1.
Еще _ реакций скорость увеличивается при повышении температуры. Вант-Гофф показал, что при повышении температуры на 10°С скорость реакции увеличивается примерно в 2—3 раза. Для температурной зависимости константы скорости было найдено эмпирическое соотношение
\gk=B~ 4-
(53)
* Для исследования быстрых стадий реакций в растворах применяют мс тод осциллографической полярографии [Дамаскин Б. Б., Петрий А. О. Введение в электрохимическую кинетику. — М.: Высшая школа, 1975]. — При1,1 перев.
11.2. Физический смысл фактора столкновений
Первоначальное теоретическое истолкование уравнения Аррениуса (55) было дано на основе кинетической теории газов. Для того чтобы произошла реакция, должны быть выполнены следующие два условия:
молекулы должны столкнуться между собой;
в момент столкновения полученная энергия должна быть достаточна для образования активированного состояния.
Что такое активированное состояние, будет пояснено несколько позднее. Однако первое представление об активированном состоянии можно дать уже здесь, обратясь к рис. Б.8. Исходному состоянию молекулы соответствует энтальпия Hi, а конечному состоянию — энтальпия Н2 (Hi>H2). В процессе реакции исходные вещества с уровня энергии Hi переходят в конечные с уровнем энергии Н2, при этом происходит изменение энтальпии АЯ (разд. 20.1). Для того чтобы могла произойти реакция, необходимо, чтобы во время соударения взаимодействую-
170
Химическая кинетика и механизмы реакций
Константа скорости реакции______ 171
щие частицы получили энергию, достаточную для образования активированного комплекса; эта энергия как раз и есть энергия активации. Из рис. Б.8 также следует, что разность энергий активации реакций, протекающих в прямом и обратном направлениях, равна энтальпии реакции &Н=Е—Е'. Пусть q — число (активных) молекул, которые вступают в реакцию, Z — число столкновений в 1 см3, k— константа скорости реакции. Тогда
k — Zq (58)
Активированные комплекс
Исходное вещество ;
-АН
Продукт реакции
Рис. Б.8. Схема, показывающая взаимосвязь энергий активации прямой и обратной реакций и энтальпии суммарной реакции.
следующее выражение для и = 0,921уз/?77М. Подставим это
Z и q можно рассчитать на основе кинетической теории газов. Из простых соотношений (разд. 2.2) можно найти число столкновений Z в газе, содержащем молекулы одного и того же вещества:
Z = ]/2 ло2ш2
(соударений-см"3-с'1) (59)
где v — средняя скорость, о — эффективный диаметр столкновений; п — число молекул в 1 см3.
Кинетическая теория дает средней скорости частицы: выражение в уравнение (59):
(60)
Z = 6,5-10Vn2]/~
Если газ состоит из различных молекул, вступающих в реакцию, то получается несколько иное выражение.
Отношение количества молекул, вступающих в реакцию, к общему числу молекул также можно получить по кинетической теории газов. Из закона распределения по энергиям Максвелла (разд. 2.2) следует, что если при температуре Т в 1 см3 газ содержит п молекул, то число молекул п, обладающих энергией Е, определяется следующим соотношением:
n'=ne~El'RT (61)
9 = е-Е/дт (62)
С учетом уравнения (58) получим
k=Ze-ElRT (63)
Таким образом, мы установили, что множитель ko в уравнени11
Аррениуса (55) действительно характеризует число столкновС'
нИй молекул в зоне реакции. На примере распада HI на Н2 и j2 при температуре 556 К проверим, насколько согласуется с экспериментом константа реакции, вычисленная из теории столкновений. Экспериментальное определение k при этой температуре привело к значению k = 3,5-10“7 (моль/л)-1-с-1; энергия активации равна 184,16 кДж/моль. Эффективный диаметр о для HI равен 3,5-10-8 см; для концентрации HI, равной 1 моль/л, в 1 см3 содержится ~6-1020 молекул; Мш = 127,9. Подставив эти значения в уравнение (60), получим Z=6,0-1031 столкновений -см-3-с-1. С учетом энергии активации [уравнение (62)] получим q = 5,2• 10"18. Подставив вычисленные значения Z и q в уравнение (58), получим
k = Zq = 3,1 • 1014 Зкул- ~= 5>2 • 10’7 (моль/л)-1 с’1
п ’ см3-с 6,02-1033 '
Итак, можно считать, учитывая приближенный характер расчета, что экспериментальное значение константы скорости хорошо согласуется с рассчитанным из теории столкновений.
Однако реакция разложения HI — это один из немногих примеров реакций, для которых теория столкновений дает удовлетворительное истолкование экспериментально найденным константам скорости. Для многих других газовых реакций, а также для реакций в растворах теория столкновений зачастую дает неудовлетворительные результаты, иногда отличающиеся от экспериментальных данных чуть ли не в 109 раз. Некоторое уточнение теории столкновений достигается введением так называемого стерического фактора Р (фактора вероятности). Фактор Р отражает то обстоятельство, что столкновение является эффективным, только если оно происходит при некотором критическом взаимном расположении молекул (например, для простых молекул перпендикулярно оси связи). Значение Р особенно мало для больших молекул, так как вероятность попадания в реакционный центр тем меньше, чем больше размеры молекулы. В рамках теории столкновений точный количественный расчет стерического фактора Р невозможен.
Линдеманн (1922 г.) применил теорию столкновений к моно-Молекулярным реакциям. Он предположил, что такие реакции также осуществляются по бимолекулярному механизму, т. е. элементарному акту реакции предшествует активация молеку-лы в результате столкновения. Активные молекулы имеют только Две возможности дальнейшего превращения: либо дезактивацию при следующем столкновении, либо превращение в проект реакции
активация, реакция
А + A t А + А* > продукты реакции (64)
дезактивация, k% 1
172 Химическая кинетика и механизмы реакций
При протекании мономолекулярной реакции в равновесии на. ходится активные и дезактивированные молекулы, а скорость суммарной реакции определяется превращением активной мо лекулы в продукты реакции. Таким образом, скорость реакции пропорциональна концентрации активных молекул.
r = kiCv (65^
Концентрацию активных молекул сд* можно определить с по мощью закона действующих масс
= (66)
^А ЬА
Тогда
г=/г1'КсА=/г1сл (67)
11.3. Теория абсолютных скоростей реакции Эйринга
Теория абсолютных скоростей основана на предположении о том, что между исходными веществами и активированным
комплексом в ходе реакции устанавливается термодинамическое равновесие. При этом дела-Исходные Активированный Продукты ется попытка вычислить кон
КмрОинам рещии.
Рис Б 9. Потенциальные кривые, позволяющие объяснить происхождение энергии активации в бимолекулярной реакции
станту равновесия методами статистической термодинамики на основе спектроскопических данных. Кроме того, энергия активации рассчитывается теоретически методами квантовой ме ханики.
Рассмотрим некоторые качественные представления теории Эйринга, имеющие важное значение для более глубокого понимания сущности химической реакции Согласно теории Эйринга реакция идет через следую щие стадии:
А + ВС ч=* (А . В С)* -*
АВ + С
(б8>
Энергия системы изменяется в ходе химической реакции в соо ветствии с потенциальной кривой, схематически изображенн на рис. Б.9. По оси ординат откладывают энергию, по оси ас*
__________________Константа скорости реакции__________ 173
цисс — так называемую координату реакции, связанную с изменением расстояния между реагирующими частицами в процессе реакции. Для того чтобы произошла реакция, необходимо преодолеть потенциальный барьер между энергетическим состоянием исходных веществ и энергетическим состоянием активированного комплекса; требующаяся для этого энергия и есть как раз энергия активации. Возможность иного (квантовомеханического) варианта преодоления этого энергетического барьера (туннельный эффект) обсуждается позже. Энергию активации можно также связать с изменением длины связей молекул, участвующих в реакции
А- + ВС (А-В---С)* АВ •••-» С (69)
Если молекула А приближается к молекуле ВС, связь в которой должна быть разорвана в ходе реакции, то сначала ослабляется связь В—С, причем тем заметнее, чем больше сближаются А и В. Энергетическая характеристика этого процесса находит свое выражение в энергии активации. В активированном комплексе (А---В---С)* связи между тремя частицами ослаблены приблизительно в равной мере, и, наконец, при дальнейшем сближении А с В частица С удаляется из активированного комплекса. На основе теории Эйринга можно также дать качественное объяснение фактора столкновения k0. Скорость реакции определяется распадом активированного комплекса на продукты реакции и пропорциональна частоте колебаний активированного комплекса вдоль координаты реакции:
r = v[ABC]* (70)
Константа равновесия между исходными веществами и активированным комплексом равна
„ [АВС]*
= [А] [ВС] ; [АВС1* = [ВС] (71)
Тогда
г = vA* [А] [ВС] (72)
ЧТО соответствует уже известному выражению для скорости реакции второго порядка, если
k' = vK* (73)
Теперь необходимо пояснить смысл величин v и К*. Колебательная энергия на одну степень свободы (разд. 2,3) (колебание вдоль координаты реакции) равна
hv = kT; v = (74)
Сложные реакции 175
174 Химическая кинетика и механизмы реакций
Отсюда
ьт
k' = ^-K* (75)
Запишем выражение константы равновесия К* из уравнения изотермы реакции Вант-Гоффа (разд. 24.2):
К* = едз*/л е-дн*/яг (76)
После подстановки в уравнение (75) получим
k' = (77)
Из этого уравнения следует, что энтальпия активации АН* с некоторым приближением идентична энергии активации (эти величины отличаются на ~2,1 кДж/моль, что не превышает точности измерения энергии активации). Сравнение уравнения (77) с уравнением (55) показывает, что фактор столкновений в уравнении Аррениуса соответствует множителю в уравнении константы скорости из теории Эйринга:
° h
(78
Для условий комнатной температуры 1012-с-1; по свое-
му физическому смыслу этот коэффициент соответствует максимально возможной скорости реакции, а экспонента eAS*^— сте-рическому фактору Р, введенному в теории столкновений (разд. 11.2). Величина AS* (энтропия активации) зависит от строения и свойств активированного комплекса; ее можно рассматривать как выражение степени разупорядоченности активированного комплекса. В большинстве случаев AS* отрицательна; в соответствии с вероятностным истолкованием функции энтропии (разд. 22.1.2) она уменьшается при повышении упорядоченности структуры, т. е. при образовании активированного комплекса образуется более упорядоченная структура, чем у системы исходных веществ. Теория Эйринга — значительный шаг вперед в теоретическом исследовании химической кинетики по сравнению с теорией столкновений; она дает, например, количественное представление о стерическом (или вероятностном) факторе Р, позволяет рассчитать энергию активации и константу скорости реакции. Однако ее значение и возможности ограничены постольку, поскольку до настоящего времени еще мало известно об истинной структуре и свойствах активированного комплекса.
12. БОЛЕЕ СЛОЖНЫЕ РЕАКЦИИ
12.1 - Протекание обратной реакции
Зачастую химические реакции не идут до конца, т. е. эквимолярная смесь исходных веществ не полностью превращается в конечные продукты реакции. В таких случаях реакция приближается к некоторому равновесному состоянию, которое может быть заранее рассчитано на основе термодинамических данных (разд. 4.2). Примерами таких реакций являются образование аммиака, иодоводорода, многие окислительно-восстановительные реакции и т. д. Обратимость реакции особенно заметна в тех случаях, когда константа равновесия близка к 1. Если необходимо довести реакцию до конца, то следует удалять продукты реакции из равновесной смеси. На скорость таких реакций оказывает большое влияние обратная реакция, как только в достаточной степени повысится концентрация продуктов реакции. Скорость обратной реакции становится тем больше, чем ближе находится система к равновесному состоянию. При учете обратной реакции скорость образования, например, HI для реакции (2) вблизи состояния равновесия равна
'' = ^iPh2Pi2—V?2hi (79)
где ki и ki —константы скорости прямой и обратной реакции соответственно. Уравнение (79) показывает, что из-за вычитания скорости обратной реакции из скорости образования продуктов (прямой) реакции скорость реакции падает быстрее, чем скорость (односторонней) реакции второго порядка. По мере протекания реакции постепенно достигается состояние, при котором скорость обратной реакции становится равной скорости прямой реакции, т. е. суммарная скорость реакции г в уравнении (79) становится равной нулю. В этом состоянии концентрации веществ в системе не меняются. Такое состояние называют химическим равновесием, для которого
^iPh2Pi2 = ^i>2hi (79а)
Если выделить из этого выражения соотношение парциальных Давлений и обозначить отношение констант прямой и обратной Реакций как константу равновесия К, то можно получить закон действующих масс
(796) ki Ph2Pi2 ’
С более общим выражением закона действующих масс мы познакомимся в дальнейшем (разд. 24.2). Если сравнить уравнение изобары Вант-Гоффа (разд. 24.1) с уравнением Аррениу-Са (56), то можно заметить справедливость выдвинутого ранее
,1
176
Химическая кинетика и механизмы реакций
утверждения о том, что энтальпия реакции равна разности энергий активации прямой и обратной реакций:
d. In К. d In (kjJkj1) Ег — E±' dT~ ~ dT RT2 RT2
Вклад обратной реакции необходимо учитывать при обработке данных, полученных при исследовании реакций такого типа. В большинстве случаев константу скорости kp обратной реак ции заменяют отношением константы скорости ki к константе равновесия:
^ = -Фг; R
Можно также непосредственно ввести равновесные концентра ции в уравнение скорости реакции и проинтегрировать его. Рассмотрим, например, простой случай, когда реакции, протекаю-шие в прямом и обратном направлениях, имеют первый порядок:
А В (81)
Уравнение скорости реакции в этом случае имеет вид
-^- = R1(a—x)~k1'x (82)
В равновесии концентрации не меняются (dxldt = Q), и уравнение (82) переходит в
kx' = (83)
Хр
(хр — равновесная концентрация вещества В), или после подстановки этого выражения в уравнение (82) получим
^=^(хР-х) (84)
После интегрирования и подстановки £ = 0, х = 0 получим
-^-•1п [ - —= kJ a ( %p — х 1
(85)
Из уравнения следует, что в данном случае, так же как и ранее, можно определить константу скорости реакции, воспользовавшись логарифмическими координатами.
12.2. Параллельные реакции
Реакции называют параллельными в том случае, если пр1* их протекании вещество реагирует одновременно с нескольк11' ми компонентами реакционной смеси или если химическое взаИ'
Сложные реакции
177
модействие двух веществ приводит к различным продуктам (например, для окислительно-восстановительных реакций образуются соединения различной степени окисления данного элемента). Такие реакции встречаются довольно часто. Если в водном растворе содержатся одновременно различные способные к окислению ионы, например БОз2-, NO2" и Fe2+, и в этот раствор добавить сильный окислитель, например Се4+ в кислой среде, то в растворе одновременно протекают три реакции:
SO32- + 2Се4+ + Н2О -> S04a- -+ 2Се3+ + 2Н+
NO2- + 2Се4+ + Н2О --► NO3- + 2Се3+ + 2Н+
Fe2+ -|- Се4+ -- Fe3+ 4- Се3+
Примером образования двух продуктов реакции при взаимодействии двух веществ является нитрование фенола. В этой реакции можно получить одновременно о- и /г-нитрофенол
ОН
NOZ
Скорости параллельных реакций могут настолько различаться, что одна из них практически не оказывает влияния на протекание процесса и тем самым выпадает из наблюдения. Даже растворитель может играть определенную роль как участник реакции. Его влияние заметно, например, при реакциях окисления перманганатом в щелочной среде (наряду с основной реакцией окисления метильной группы органического соединения вода окисляется до кислорода вследствие каталитического действия гидроксид-иона).
В том случае, если параллельные реакции протекают со сравнимыми скоростями, еще возможно точное решение кинетического уравнения. Рассмотрим, например, реакцию нитрования (86). Пусть xi — количество образовавшегося о-нитрофенола (в молях), а х2 — n-нитрофенола, а исходное их количество, как обычно, а я Ь. Тогда уравнение скорости образования для о-нитрофенола и п-нитрофенола
-^ = ^(«-х)(й-х); ^ = ^2(«-х)(й-х) (87)
12-2027
Сложные реакции
179
178
Химическая кинетика и механизмы реакций
а для суммарного процесса
4- = (88)
Решение этого уравнения совпадает с уравнением (48) для реакции второго порядка с тем отличием, что константа скорости в уравнении (48) здесь меняется на сумму констант ki и kz. Из отношения скоростей параллельных реакций получим
fej kj (89)
Из уравнения (89) можно найти отношение констант скорости параллельных реакций, пользуясь отношением концентраций продуктов реакции х/9 и x2<z) в момент времени t.
12.3. Последовательные реакции
Во многих случаях устойчивые конечные продукты химической реакции или радиоактивного превращения образуются не непосредственно из исходных веществ, а через промежуточные вещества, которые можно выделить из реакционной смеси; если последние не удалены из системы, то идет дальнейшее превращение в конечные продукты. В качестве примера рассмотрим снова процесс радиоактивного распада [уравнение (26)]. В общем случае
А ---> В----> С (90)
Такого типа реакции встречаются довольно часто, однако протекание промежуточной стадии не всегда заметно. Если то скорость реакции зависит только от второй стадии процесса, напротив, если то скорость определяется константой
скорости kp, в обоих случаях кинетические уравнения принимают простую форму. Наметим в общих чертах математическое решение этой задачи.
Скорости уменьшения концентрации А, увеличения концентрации В и образования конечного продукта подчиняются следующим уравнениям:
—dcpJdt — kiC^ (91)
dc^dt = k^— k2eB (92)
dcjdt — k2cB (93)
После интегрирования уравнения (91) получим уравнение типа (24). Выражение (92) является неоднородным дифференциальным уравнением первого порядка, решение которого можно провести, например, методом Лагранжа.
В результате интегрирования уравнений (91), (92) и (93) можно получить временные зависимости для концентраций отдельных компонентов:
сА = сАое-^ (91а)
= (е"*1'—е"*2/) (92а)
На рис. Б. 10 схематически показан ход изменения концентрации веществ — участников реакции во времени. Пока концентрация вещества А снижается по экспоненте, концентрация веще-
Рис. Б 10. Зависимость концентрации реагентов от времени для последовательной реакции типа А->-В->-С.
ства В увеличивается и достигает через некоторое время максимума, а затем начинает уменьшаться. [Упражнение. Найдите из уравнения (92а) выражение для времени (тах, при котором концентрация компонента В становится максимальной]. Концентрация продукта реакции С постепенно увеличивается и постепенно достигает предельного значения (максимум концентрации В соответствует перегибу на кривой зависимости концентрации С от времени).
Рассмотрим еще несколько примеров последовательных реакций. Диспропорционирование гипохлорита в щелочной среде.
ЗОС1- ----► 2С1" + СЮ3- (94)
В соответствии со схемой (94) можно предположить, что это реакция третье-Го порядка, в действительности она имеет две последовательные стадии, обе второго порядка:
2ОС1" ----> СГ + С1О2~ (скоростьопределяющая стадия)
(95) С1О2- + ОС1- ---> Cl" + сю3-
, Другой весьма интересный пример реакций такого типа—обмен кисло-между перманганатом и водой в щелочной среде в присутствии МпОД-. омен кислорода между перманганатом и водой заметно катализируется 12*
180
Химическая кинетика и механизмы реакций
манганатом (в отсутствие манганата эта реакция обмена происходит очець медленно, кислородный обмен манганата и воды—очень быстрый процесс) Кроме того, происходит быстрый электронный обмен между МпО4~ ч МпО42~. В раствор, содержащий немеченый МпО42-, в щелочной среде бц., введен МпО4_, меченный 18О, тогда
Мп*О4 + МпО42 < -• >: МпО4 + Мп*О42- (электронный обмен)
(96)
Т2
Мп*О42- + Н2О <=> МпО42- + Н2*О
Уменьшение содержания 18О в перманганате происходит в соответствии с уравнением (91а), увеличение содержания 18О в МпО42~— по уравнению (92а)
Очень часто последовательные реакции типа (90) встречаются при распаде не только естественных, но и искусственных радиоактивных нуклидов. Рассмотрим процесс распада li0Ba, образующегося при ядерном делении 23SJJ
₽ ₽
140Ва ------->- i«La-------->- 11"С1
12,8 сут 40 ч
(97)
Характер изменения во времени радиоактивности нуклидов, участвующих в реакции (97), соответствует кривым рис. Б 10.
12.4. Цепные реакции
Реакции, при протекании которых возникают промежуточные вещества с высокой энергией (радикалы), часто имеют механизм цепных реакций. Обычно в момент элементарного акта взаимодействия между активными молекулами появляются реакционноспособные промежуточные вещества — активные центры,— которые в свою очередь реагируют с компонентами реакционной системы, воспроизводят подобные себе частицы, в результате чего происходит циклическое повторение стадий реакции. Таким образом, возникает цепь реакций, так как после первичного акта цепной реакции появляется активная частица с высокой энергией (например, при воздействии излучения), которая продолжает последовательность стадий реакции. Такого рода процессы характерны прежде всего для реакций в газовой фазе (взрыв гремучего газа, реакция водорода с хлором), а также для некоторых реакций в растворах (фотохимические реакции, реакции полимеризации и т. д.). Возникновение реакционноспособной частицы часто называют реакцией зарождения цепи, например реакция (За) при образовании НВг (гл. 7). Под развитием цепи понимают последовательное продолжение элементарных стадий с постоянным образованием активных центров, продолжающих цепь радикалов. К реакциям обрыва цепи относится рекомбинация, т. е. реакция, обратная (За). Еще раз обратимся к уже описанному выше процессу образования бромоводорода (гл. 7). Для него найдена следую
Сложные реакции
181
щая экспериментальная зависимость скорости реакции от концентрации:
Предлагаемый механизм реакции [уравнения (За) — (Зв)] согласуется с уравнением (98), что можно показать следующим образом. Рассмотрим как прямые, так и обратные реакции (За) —• (Зв). Для скорости образования НВг выражение
Г = ^бСВгСц2 ~г4,С11СВг2 ^-бСНСНВг (99)
выведено на основании предположения, что молекулы НВг образуются в результате прямых реакций (36) и (Зв), а исчезают по реакции (36). Реакцию, обратную (Зв), можно не учитывать, так как атом Вг не имеет достаточно энергии для того, чтобы разорвать связь Н—Вг. Концентрации участвующих в реакции промежуточных веществ (атомов Н и Вг) неизвестны, поэтому нельзя рассчитать скорость реакции из уравнения (99). Для того чтобы преодолеть эту трудность, Боденштейн (1916 г.) предложил «принцип стационарности». Согласно этому принципу, в ходе реакции, в которой участвуют короткоживущие промежуточные частицы, устанавливается стационарное состояние, т. е. скорость образования таких частиц равна скорости их исчезновения (в результате распада или вступления в следующий цикл реакции). Этот принцип имеет исключительно важное значение и до настоящего времени часто привлекается для объяснения экспериментально найденных кинетических данных. Принцип Боденштейна позволяет рассчитать концентрации промежуточных частиц. Покажем, как проводится этот расчет на примере реакций (За) — (Зв). Атомы Н образуются в реакции (36) и исчезают в реакции, обратной (36), и прямой реакции (Зв). Тогда
^бСВгсН2 — &вСНСВг2 4" ^-бСНСНВг (100)
Сн = т-----г-г-1-- (101)
«вСвг2 + К-бСнВг ’
Концентрация атомов Вг рассчитывается из равновесия диссоциации (За) [предполагается, что прямая реакция (36)—ско-Ростьопределяющая]:
cBr = V kcBr2 (102)
Если подставить выражения (101) и (102) в уравнение (99), то Действительно можно получить экспериментально найденную зависимость (98) для образования НВг.
182 Химическая кинетика и механизмы реакций
• —— - ,
Напрашивается вопрос: почему образование НВг и HI, сте-хиометрические уравнения которых совпадают, имеют разный механизм протекания реакции? Чтобы ответить на этот вопрос необходимо учитывать, что, вообще говоря, из возможных механизмов реакции осуществляется лишь тот, который обеспечивает наибольшую скорость реакции. Очевидно, для образования НВт также возможен механизм типа реакции образования HI, однако мы не можем его наблюдать, так как цепная реакция обеспечивает более высокую скорость образования молекул
Ранее (разд. 2.1) уже выведен закон действующих масс для элементарного акта реакции. Для сложных реакций, состоящих из нескольких стадий, приведенное доказательство не является очевидным (хотя иногда это доказательство представляют в литературе как строгое). Выведем закон действующих масс для реакции образования бромоводорода. Эта реакция имеет цепной механизм в соответствии с уравнениями (За) — (Зв). Запишем уравнения скорости образования продуктов реакции, учитывая прямые и обратные реакции и все возможные механизмы протекания процесса (включая обратную реакцию (Зв), которая не оказывает существенного влияния на кинетику образования НВг (разд. 12.1)):
dCy^/dt = ^бСВгСН2 ^-бсНВгСн4”^вСНСВг ^-вСНВгСВг dcn/dt = k5cBTcH2—&_6cHBrcH — йиснсВг2Ц- &_всНВгсВг
В состоянии равновесия можно приравнять нулю скорости изменения концентрации, тогда получим
^бсВгСН2— ^-бСНВгсн + ^вСНСВг2—^-вСНВг“Вг = О
^бСВгСН2— ^-бсНВгсН ^вСНСВг2 + &-вСНВгСЕг = О
Сложим эти уравнения:
&бСВгСН2 &-бсНВгСН — О а затем вычтем из первого уравнения второе:
^вСНСВг2 ^-uCHBrCBl = О
Из последних двух уравнений можно получить условия равновесия обеих стадий реакции в цепи (36) и (Зв):
Снвгсн _ IСнВгСВг _1
СВгСН2 k-б СНСВГ2 А-в
Перемножив эти уравнения, получим наконец
C2HBr _ Mb
сН2сВг, ’ &-б^-в
Это и есть уравнение закона действующих масс для реакций образования бромоводорода. Вывод оказался более сложным,
Сложные реакции
183
чем для реакции образования иодоводорода. Однако формаль-но уравнение закона действующих масс имеет тот же вид, что й прежде: в числителе стоят концентрации продуктов реакции, а в знаменателе — исходных веществ. Если предположить, что любая сложная реакция состоит из элементарных стадий и суммирование уравнений реакций элементарных стадий после сокращения промежуточных веществ дает суммарное уравнение реакции, то аналогичным путем можно получить уравнение закона действующих масс для любой многостадийной реакции из кинетических данных. Однако это предположение верно только в том случае, если выполняется принцип микроскопической обратимости, согласно которому в равновесном состоянии многостадийной реакции каждая ее стадия также должна быть в равновесии. Этот принцип учитывался при выводе рассмотренных выше уравнений.
13. ГОМОГЕННЫЕ РЕАКЦИИ В РАСТВОРАХ
13.1. Общие представления о влиянии растворителя
на механизм реакции
Теоретическое исследование кинетики и механизма химических реакций в растворах — намного более сложная задача по сравнению с исследованием газовых реакций, поскольку в растворах реагирующие вещества могут взаимодействовать с растворителем (следует учитывать влияние диэлектрической проницаемости растворителя, степень гидратации, присутствие посторонних компонентов и т. д.). Существует много различных типов реакций в растворах; для некоторых из них влиянием растворителя можно пренебречь (особенно в тех случаях, когда используются неполярные растворители). При некоторых условиях участники реакции взаимодействуют с такой же скоростью, как и в газах, как, например, при разложении N2O5. Существенным фактором является число столкновений между молекулами реагирующих веществ в растворе (включая растворитель). Дебай и Рабинович провели оценку числа столкновений в растворе, согласно которой оно примерно в три раза больше, чем в газовой фазе. Это согласуется с экспериментальными данными, также подтверждающими, что фактор столкновений для реакций в растворах увеличивается примерно в три раза. Так как энергия активации практически не меняется, скорость реакций в Растворе также увеличивается в три раза по сравнению с газовыми реакциями. Для реакций в растворе характерна также Небольшая подвижность реагирующих частиц (по сравнению с реакциями в газовой фазе). Для цепных и других реакций, в Которых появляются в качестве промежуточных частиц радика
184
Химическая кинетика и механизмы реакций
лы, это приводит к тому, что в растворах происходит преимущественно первичная рекомбинация двух радикалов (например, Вг + Вг—^Вг2), а не продолжение или разветвление цепи. Эю явление называют эффектом клетки (растворитель играет роль стенки сосуда). Особенность реакций в растворах заключается также в том, что в них часто принимают участие ионы. При этом необходимо учитывать электростатические (кулоновские) взаимодействия. Например, реакция нейтрализации между противоположно заряженными ионами (НзО+ + ОН_—>-2Н2О) протекает с максимально возможной скоростью (~1012 с-1) при минимальной энергии активации. Существенную роль играет заряд ионов (реакции между ионами с большим зарядом протекают с более высокой скоростью). Более подробное рассмотрение влияния растворителя здесь не приводится.
13.2. Влияние ионной силы на химические реакции в растворах (первичный солевой эффект)
Что такое первичный солевой эффект? Самые общие представления о нем позволяют более глубоко понять сущность элементарных процессов химических реакций, протекающих в растворах. Рассмотрим один из аспектов теории сильных электролитов Дебая — Хюккеля (разд. 31.4), а именно зависимость коэффициента активности от ионной силы раствора. [Теория влияния нейтральных солей на скорость химических реакций в растворах была развита Бренстедом и Бьеррумом.]
Пусть происходит ионная реакция
А А + В в < :fc X х ----> продукты реакции (ЮЗ)
В основе теории Бренстеда и Бьеррума также лежат представления о том, что реакция идет через стадию образования активированного комплекса X, который находится в равновесии с исходными веществами. Заряд активированного комплекса складывается из зарядов вступающих в реакцию ионов (zx=Z4 + zb). Скорость реакции определяется скоростью превращения активированного комплекса в продукты реакции (медленная стадия)
г = k'cyi (104)
В присутствии нейтральной соли в уравнении закона действующих масс для реакции (ЮЗ) необходимо принять во внимание коэффициенты активности исходных веществ и активированного комплекса
1 огда
_ сх/х сдсв ^А'^в
г = k'K*c^ св- -
(105)
(106)
Произведение k'K* можно обозначить k°. т. е. константой скорости при условии, что член, включающий коэффициенты активности, равен 1.
(107)
/х
Гомогенные реакции в растворах
185
Константа скорости реакции в присутствии любой концентрации нейтральной соли, активность которой можно вычислить на основании теории Дебая — Хюккеля (разд. 31.4), определяется уравнением (107). Логарифмированием (107) получим
lg А = lg А" + lg (108)
Между коэффициентом активности и ионной силой раствора в первом приближении существует следующая зависимость (разд. 31.4):
-lg ft = 0,509z£-2 VT (109)
где I — ионная сила (7=1/22cIz,2). Принимая во внимание, что zx = za+zb, получим
lg А = 1g А» - 0,509 /7[(zA2 + zb2) - (zA + zB)2] (ПО)
lg А= lg Ао-ф 1 ,02zazb V1
С помощью последнего уравнения легко оценить влияние нейтральной соли на скорость реакции. При взаимодействии одноименно заряженных частиц (обе либо положительные, либо отрицательные) можно ожидать увеличения
Рис. Б. 11 Зависимость скорости различных ионных реакций от ионной силы раствора:
Г — 2а2в = 4,
2[Co(NH3)sBr]2++Hg2+--»2H2O+2[Co(NH3)sH2O]3++HgBr2
2 — zazb = 2,
S2O82-+2I-^I2+2SC>42-
3 ~ zazb = ^
O2N=N-COOC2Hs-+OH—>N2O t- СОза-+С2Н5ОН
zazb=°-
CH3COOC2Hs+OH—>ch3coo-+ C2H6OH + H2O
$ ~ zazb= i;
Н2Ог+2Н++2Вг—>2H2O + Br2
6~zazb=-2;
[Co(NH3)5Brp++OH—> [Со(ЫНз)5ОН]2++Вг~
скорости реакции при повышении концентрации соли. Напротив, для реагирующих ионов с разноименным зарядом следует ожидать уменьшения скорости реакции при увеличении концентрации соли (отрицательный катализ). При участии в реакции нейтральной молекулы (z = 0) не наблюдается никакого влияния ионной силы па скорость реакции (рис. Б.11). Эти подтвержденные па опыте положения можно рассматривать как доказательство существования активированного комплекса в реакциях типа (103).
14. ГЕТЕРОГЕННЫЕ РЕАКЦИИ
До настоящего времени теория гетерогенных реакций разработана значительно слабее, чем теория гомогенных реакций, Хотя гетерогенные реакции имеют очень большое практическое значение, особенно в химической промышленности.
186
Химическая кинетика и механизмы реакций
Гетерогенными называют реакции, при которых реагирую-щие вещества находятся в разных фазах, т. е. сама реакция протекает на границе раздела фаз. Выделяют следующие стадии гетерогенной реакции: 1) перенос одного или нескольких реагирующих веществ из глубины фазы в зону реакции (часто совпадающую с поверхностью раздела); 2) собственно химичес-
Рис. Б.12. Схема изменения концентрации в диффузионном слое при растворе нни магния в соляной кислоте.
кую реакцию и 3) перенос (транспорт) продуктов реакции из зоны реакции в глубь фазы.
К гетерогенным реакциям относятся, например, процессы растворения (соли водой или металла кислотой); кристаллизацию соли из раствора также можно рассматривать как гетерогенную реакцию. Следует отметить, что на поверхности раздела фаз газовые реакции протекают с большей скоростью, чем в объеме (гетерогенный катализ). Здесь граница раздела фаз играет роль катализатора (например, в различных реакциях гидрирования) .
Как уже упоминалось ранее, в общем случае суммарная скорость химической реакции определяется замедленной стадией процесса. Для большинства гетерогенных реакций ско-ростьопределяющей стадией является подвод (транспорт) реагирующих частиц к активному центру реакции. Транспорт реагирующих веществ может осуществляться конвекцией или диффузией (коэффициент диффузии ~ 10~6—10~4 см2/с).
Рассмотрим в качестве примера процесс растворения металла кислотой. Если путем интенсивного перемешивания обеспе чить некоторую постоянную конвекцию, то растворение металл^ определяется скоростью диффузии ионов водорода в диффузионном слое и может быть количественно рассчитано на основа законов диффузии. Обратимся к рис. Б. 12 (где сг — концентрация ионов водорода в глубине раствора, Со — концентрация ионов водорода на поверхности металла, 6 — толщина диффузионного слоя). Согласно Нернсту, градиент концентрации (dc/cw
Гетерогенные реакции
187
на границе раствор/поверхность металла можно считать постоянным, так что скорость процесса подчиняется закону фика
(1Н)
где п — число молей, D — коэффициент диффузии, q — площадь поверхности металла; так как (дс/дх) = const, можно переписать уравнение (111) в следующем виде:
-^ = -Dq^-^- (112)
или, поделив выражение (112) на объем V и полагая, что концентрация кислоты на поверхности металла Со = О, получим
de Dq
dF "" Vb Ci
(ИЗ)
т. e. уравнение скорости реакции первого порядка, где константа скорости равна k = Dq!V&. Рассмотренные выше закономерности справедливы также и для процесса растворения солей. Проводя эксперименты при различных точно измеренных скоростях перемешивания, можно получить подробные сведения о кинетике растворения различных веществ. Отклонения от простых кинетических закономерностей наблюдаются только в том случае, когда на поверхности растворяющегося вещества имеет место адсорбция.
14.1. Адсорбция и гетерогенные реакции
Адсорбционные явления, происходящие на поверхности раздела между твердой, жидкой и газообразной фазами, оказывают большое влияние на кинетику гетерогенных реакций. Прежде всего наблюдаются отклонения от порядка реакции соответствующей гомогенной реакции (получают порядок реакции от О До 1). Скорость реакции в таких случаях зависит от площади поверхности раздела. Ввиду важности явлений адсорбции для гетерогенных реакций рассмотрим сначала более подробно адсорбционное равновесие.
Изотерма адсорбции Ленгмюра. Адсорбция атомов или мо-лекул осуществляется либо под действием межмолекулярных сил (физическая адсорбция), либо с образованием химической связи (хемосорбция или активированная адсорбция). Адсорбция зависит не только от площади поверхности, но и от концентрации (парциального давления), а также от температуры. ° случае физической адсорбции количество адсорбированного Вещества уменьшается с ростом температуры, а при хемосорбции оно, как правило, увеличивается. Следует, однако, помнить,
Химическая кинетика и механизмы реакций
что нельзя провести резкой границы между этими двумя видами адсорбции.
При исследовании адсорбции часто используют изотерму адсорбции Ленгмюра; соответствующее математическое выражение выводится из кинетических закономерностей. Для вывода
уравнения Ленгмюра необходимо применить к процессам адсорбции и десорбции те же методы, которые уже нашли применение в разд. 12.1 при исследовании образования HI, т. е. следует рассматривать адсорбцию как прямую, а десорбцию как обратную реакцию. Пусть 0 — степень заполнения поверхности (0 =
Рис. Б.13. Изотерма адсорбции рованного к моменту времени t
Ленгмюра.
количества вещества а к количеству адсорбированного вещества а«>, соответствующего полному
заполнению поверхности адсорбированным веществом). При адсорбции газа скорость адсорбции пропорциональна парциальному давлению и доле свободной поверхности (1—0):
Мг) = V(1-©) \ /1
(П4)
Скорость десорбции пропорциональна заполненной поверхности
(П5)
При адсорбционном равновесии скорости адсорбции и десорбции равны, откуда
Используя приведенное выше определение степени заполнения, получим выражение для количества адсорбированного вещества
Изотерма Ленгмюра представляет собой типичную кривую с на сыщением (рис. Б. 13). При малых давлениях можно пренебречь величиной р в знаменателе, и адсорбированное количество ста новится пропорциональным давлению р. Напротив, при доста точно больших давлениях газа отношение k2/ki становится малым по сравнению с р и а—>-ах.
Гетерогенные реакции
189
При математической обработке экспериментальных данных изотерму Ленгмюра обычно записывают в следующем виде:
(118) а ах 1 ах 7
Построив график в координатах p/а, получим прямую, коэффициент наклона которой дает предельное заполнение ах, а величина отрезка на оси ординат позволяет вычислить константу адсорбционного равновесия.
При исследовании реакций в растворах вместо давлений следует использовать концентрации адсорбирующихся веществ.
14.2. Применение изотермы адсорбции
к гетерогенным реакциям
Скорость реакции на поверхности твердого тела (например, для дегидрирования этана до этилена) пропорциональна количеству адсорбированного вещества а, следовательно,
^T = ka (119)
Подставив а из уравнения (117), получим
= - (120)
&2/&1 + р v ’
Уравнение (120) показывает, что порядок реакции зависит от давления (или концентрации) участвующих в ней веществ. Следует рассмотреть несколько случаев.
Высокое давление (или большая концентрация),
JL^kax.--_k" (121)
т. е. реакция протекает по уравнению нулевого порядка, когда поверхность полностью покрыта реагирующими частицами. Поэтому скорость реакции определяется только реакцией на поверхности. Концентрация продуктов реакции пропорциональна времени [уравнение (46)].
Низкое давление, p<k2/ki. Можно пренебречь величиной р в знаменателе [уравнение (120)]:
= k • j- а.„р — k'p (122)
т- е. скорость реакции подчиняется уравнению первого порядка, Что соответствует восходящей ветви изотермы адсорбции (скорость реакции определяется преимущественно диффузией исход-Нь1х веществ к активным центрам на поверхности). Поскольку Ге1«пературная зависимость диффузионных процессов незначи
190
Химическая кинетика и механизмы реакций
тельна, в обоих разобранных случаях следует ожидать сравнительно небольшую энергию активации (8—21 кДж/моль).
'Давление, промежуточное по сравнению со значениями, рассмотренными выше [т. е. когда нельзя пренебречь величиной р в знаменателе уравнения (120)]. Здесь наблюдается дробный порядок реакции. Скорость реакции записывается в следующем виде:
— (0<п<1) (12ч
(Ji
Примером реакций, имеющих при обычных давлениях дробны i порядок, является разложение АзНз, РН3, HI и NH3 и т. п. hi поверхности металлов (Au, Pt, W, Mo).
Следует иметь в виду, чт ।
/Д---------
Рис. Б.14. Зависимость константы скорости от температуры для гомогенного (/) и гетерогенного (2) механизмов протекания реакции, отличающихся разной энергией активации при высокой и низкой температуре
различные реакции, протеках щие в гомогенных условиях г бимолекулярному механизм (например, образование Н1) на поверхности металлов, име ют первый порядок. В то вре мя как в гомогенной системе предпосылкой осуществления реакции является столкнове ние двух молекул, на поверх ности возможен непосредственный распад молекулы (выигрыш энергии за счет образования адсорбционной связи с поверхностью). Поэтому энер-
акции оказывается значительно
гия активации гетерогенной ре' более низкой, чем для той же
реакции, протекающей в гомогенной системе. Часто на некото-
рых типах поверхности реакция идет через параллельные стадии гомогенного и гетерогенного механизма; при высокой температуре преобладает гомогенная реакция, при более низкой — гетерогенная. Скорость гомогенной реакции увеличивается с температурой быстрее, чем скорость гетерогенной, вследствие более высокой энергии активации гомогенной реакции; поэтому при повышении температуры преобладает гомогенная реакция (рис. Б.14).
15. КАТАЛИЗ
15.1. Общие представления
Константа скорости, вообще говоря, функция температурь1’ при некоторой заданной температуре это постоянная величина-Однако известны вещества, которые при определенных условия*
Катализ
191
способны ускорять реакцию, причем сами эти вещества практически не претерпевают изменений (положительный и отрицательный катализ). О некоторых явлениях такого рода уже шла речь ранее при изучении влияния ионной силы на скорость гомогенных реакций в растворах, а также при исследовании причин укоренил химических реакций на поверхности твердых тел.
Рис Б 15. Энергия активации реакции, протекающей без участия катализатора я в присутствии его (катализатор образует промежуточный продукт с исходными веществами)
Вещества, способные изменять скорость реакции, не участвуя в суммарной стехиометрической реакции, называют катализаторами. Эти вещества могут, правда, участвовать в отдельных элементарных стадиях процесса, образуя, например, реакционноспособные промежуточные вещества, однако на следующих стадиях процесса они выделяются снова. Отрицательный катализ называют также ингибированием.
Процесс катализа открывает возможность протекания реакции по пути с более низкой энергией активации (например, благодаря образованию более реакционноспособных промежуточных соединений), что связано с понижением энергии активации (рис. Б.15). Каталитическая реакция идет в две стадии, причем каждая стадия имеет меньшую энергию активации, чем та же реакция в отсутствии катализатора.
Составим схему протекания реакции по новому пути (К — катализатор):
А + В ---->- АВ
К + А КА (124)
«-1
*2
КА + В --->- АВ+К
Очевидно, что катализатор не оказывает влияния на положе-Ние равновесия реакции. В присутствии катализатора также не пРоисходит изменения энтальпии реакции. Отсюда однозначно
192
Химическая кинетика и механизмы реакций
следует, что катализатор воздействует как на прямую, так и ц, обратную реакцию, так как если Ki = kilk2 = const, то при увели чении ki в той же степени должна увеличиваться константа k-
Рассмотрим еще раз общую схему каталитического процес са (124) и применим к нему принцип стационарности (разд 12.4). Скорость образования продуктов реакции в этом случае описывается следующим уравнением:
-^- = fe2cKAcB (125)
Условие стационарности для активного промежуточного соединения КА можно записать в следующем виде:
^ickca = ^-1ск\+^2скасв! ска = 026)
Тогда
61сав ^1^2сКсАГв /197
dt — А^ + ^св ' 1
Возможны два крайних случая.
1) Можно пренебречь вторым членом в знаменате-
ле уравнения (127):
(128)
Уравнение (128) соответствует скорости реакции третьего порядка, однако, так как активность катализатора постоянна, расчет проводится по уравнению скорости реакции второго порядка. Энергия активации в соответствии с уравнением Аррениуса связана с энергиями активаций отдельных стадий соотношением Е = Еу-\-Е2—Е-1.
2) Можно пренебречь первым членом в знамена-
теле уравнения (127):
(1И)
Таким образом, константа скорости равна k\, а энергия активации каталитической реакции Е идентична энергии активации первой стадии Е = Е{.
Рассмотренные выше на примере каталитической реакции кинетические закономерности для реакций, проходящих через две стадии, одна из которых представляет собой обратимый процесс, а последующая протекает преимущественно в прямом направлении, — эти закономерности присущи многим химическим реакциям в растворах и поэтому имеют общее значение.
Катализ
193
15.2. Гомогенный катализ
Особенность гомогенного катализа состоит в том, что и катализатор, и все реагирующие вещества находятся в одной и той дее фазе. Наиболее разнообразные возможности имеет применение гомогенного катализа в растворах.
Широко распространен кислотно-основной катализ. К нему относится, например, катализируемое кислотой омыление эфиров [реакция (19)]. При реакциях окисления анионами различных оксокислот (призводные оксокислот галогенов, хрома, марганца и др.) зачастую заметно, что протонированный анион реагирует значительно быстрее, чем непротонированный. Например, многие органические соединения (спирты, альдегиды) очень медленно окисляются хроматом в нейтральном растворе, в то же время при уменьшении pH раствора скорость окисления резко возрастает. Очевидно, на реакцию окисления влияет протолитическое равновесие:
СгО42- + Н3О+ НСгО4’ + Н2О
^2
НСгО4“ + S 4-Н2О --► продукты реакции + Н3О+ (130)
(S — любое окисляющееся органическое вещество). В связи с тем что реакции в растворе, катализируемые кислотой, имеют большое значение в химии, рассмотрим подробнее их механизм. Последовательность стадий реакции можно записать в следующем виде:
S + ВН+ SH+ + в
м
SH+ + H2o Р+Н3О+ (131)
Здесь S обозначены реагирующие исходные вещества, Р — продукты реакции, ВН+ — любой кислотный катализатор. Одновременно с реакцией (131) осуществляется протолитическое равновесие
свсн3о+ /1оп.
ВН+ + Н2О В + Н3О+; К№СС = - 3. (132)
По аналогии с уравнением (127) для реакции (131)
г = Wscbh+ (133)
^-1СВ +
Для одного из возможных случаев, когда ^2<^-1Св, уравнение (133) упрощается:
г cscbh+ (134)
'св k ’
'3—2027
194
Химическая кинетика и механизмы реакций
Подставим концентрацию основной формы катализатора св, полученную из уравнения (132):
rz=='k'K'"' csch3o+ 035)
«-1Лдисс °
Описанный выше случай называют также специфическим кислотным катализом: уравнение скорости реакции не содержит концентрации кислоты ВН+, действующей, как катализатор, так как она входит в равновесие (132) и определяется концентрацией Сн о+- При постоянной концентрации исходных веществ скорость реакции зависит только от pH, т, е. от концентрации кислоты.
Для другого предельного случая, когда /г2>/г-щв, в соответствии с уравнением (133)
r = ^cscBH+ (136)
В данном случае предшествующая реакция (131) не является равновесной и в выражение скорости реакции входит непосредственно концентрация каталитически активного компонента ВН+. Здесь говорят об общем кислотном катализе; скороеи> реакции не зависит от концентрации ионов гидроксония.
15.2.1. Примеры гомогенного катализа
Очень часто следы тяжелых металлов играют роль катализаторов химических реакций в растворах. Рассмотрим разложение пероксида водорода. Механизм этой реакции можно записать в виде следующей последовательности реакций:
Н2О2 2 -ОН (скоростьопределяющая стадия) (137а)
• ОН + Н2О2 -о- Н2О + НОО - (1376)
НОО- + Н2О2 ----► О2 -у Н2О + -ОН (137в)
Реакция начинается с образования радикала -ОН—активного центра цепной реакции. Обрыв цепи становится возможным в результате следующих реакций:
2 .ОН ---> Н2О2 (138а)
• ОН+НОО- ----->Н,О4-О, (1386)
Образование радикалов -ОН [уравнение (137а)] идет очень медленно в растворах, не содержащих других компонентов (примесей). Однако в присутствии способных окисляться катионов тяжелых металлов, например Fe2+. образование -ОН ускоряется, а следовательно, облегчается протекание суммарного процесса. Участие Fe2+ в реакции можно представить себе следующим образом.
Fe2+ + Н2О2 --► Fe3+ + • ОН + ОН" (139)
По аналогии со схемой (139) действуют на процесс разложения Н2О2 я другие соединения, способные к окислению. Подобный каталитический механизм имеет место также при окислении сульфита кислородом в присутствии Си2+.
Катализ
195
Следует особо подчеркнуть возможность применения катализаторов для изменения направления реакции («управления реакцией»), например в физиологических процессах, а также в органических реакциях. Примером изменения направления реакции с помощью катализа может служить окисление тиосульфата пероксидом водорода в кислой среде. В присутствии иодид-иона реакция заканчивается образованием S4Os2 ', а в присутствии молибденовой кислоты — образованием SO42-.
Н2О2 -ф 2 S2O32- + Н3О+ ---> S4O62- + 4 Н2О (140а)
4 H2O2 + S2O32- ---► 2 SO42- + 2 Н3О+ + Н2О (1406)
Наиболее выгодной в термодинамическом отношении является реакция окисления БгОз2- пероксидом водорода до сульфат-иона. При протекании реакции по уравнению (1406) вначале образуется пероксомолибденовая кислота, которая передает кислород иону $20з2-. В случае реакции (140а) сначала из Н2О2 и Ш получается иод, способный окислить тиосульфат до тетратионата в соответствии с известной йодометрической реакцией.
Если в ходе реакции образуется или расходуется каталитически активное вещество, такая реакция называется автокаталитической. Изменение во времени концентрации исходных веществ в ходе автокаталитической реакции, в которой образуется одно каталитически активное вещество, показано на рис Б. 16. Видно, что в начале реакция протекает медленно, однако по мере накопления каталитически активного вещества реакция ускоряется, а в конце снова замедляется. Таким образом, кривая зависимости концентрации от времени имеет точку перегиба. Примером автокаталитической реакции может служить восстановление перманганата щавелевой кислотой, часто используемое в
ной точки эта реакция протекает очень медленно, однако после появления в растворе ионов Мп2+ реакция очень быстро проходит до конца*.
Так называемая реакция Ландольта (окисление сульфита йодатом в Растворе уксусной кислоты) также относится к автокаталитическим реакциям
Юз’ + З HSO3- + 3H2O ------> Г + 3 SO42~+3 Н2О+ (141)
® Данном случае автокатализ осуществляется иодом, образовавшимся в результате реакции йодата с иод-ионом, полученный молекулярный иод окисляет сульфит
Ю3- + 5 Г + 6 Н3О+ -----3 12 + 9 Н2О
3 (I2 + HSO3- + 4 Н2О ---► 2 Г + SO42- + 3 Н3О+) (142)
Рис Б 16 Изменение во времени концентрации исходного вещества при протекании автокаталитической реакции при условии, что один из продуктов реакции — катализатор.
аналитической практике. Вблизи началь-
* Время, в течение которого реакция протекает медленно, а в реакцион-°и смеси накапливается катализатор, называют временем индукции. [Эмма-М. Кнорре Д. Г Курс химической кинетики. — М.: Высшая школа, 969]—Прим перев
13*
196
Химическая кинетика и механизмы реакций
Изотопные методы
197
15.3. Гетерогенный катализ
Гетерогенным называют катализ на поверхности твердых тел, находящихся в контакте с реагирующими веществами в газовой фазе или в растворах. Основные теоретические положения, необходимые для понимания сущности гетерогенного катализа, уже изложены в гл. 14 в связи с обсуждением роли адсорбции в гетерогенных реакциях. При проведении реакции на поверхности твердых тел последняя играет вполне определенную роль: благодаря адсорбции на поверхности понижается энергия активации катализируемой реакции. До настоящего времени еще не существует удовлетворительной количественной теории катализа. В любой каталитической реакции важнейшее значение имеет структура поверхности. Катализ протекает не на всей поверхности твердого тела, а главным образом на активных центрах (дислокациях, ребрах кристаллов и других дефектах кристаллов). Кроме того, известно, что каталитическая активность зависит от кристаллографической плоскости, — кристаллы, ориентированные в некоторых определенных направлениях, обладают максимальной активностью. Большое значение в гетерогенном катализе имеют смешанные катализаторы. Примером могут служить почти все известные газовые реакции, используемые в химических технологических процессах (синтез аммиака, синтез SOs, гидрирование угля по Бергиусу или Фишеру— Тропшу, окисление аммиака по Оствальду и многие другие).
16. ИЗОТОПНЫЕ МЕТОДЫ В ХИМИЧЕСКОЙ КИНЕТИКЕ
16.1. Общие представления
Ранее было показано, что данные кинетических исследований позволяют установить закономерности изменения скорости химических реакций. На их основе можно проследить изменение концентрации реагирующих веществ во времени, однако нельзя надежно и достоверно установить механизм протекания реакции. Например, водород в щелочной среде окисляется МпО4“ до Н2О с образованием МпО42-. Скорость окисления зависит от концентрации:
Суммарная реакция протекает по уравнению
2 ОН- 4- Н2 + 2 МпО4- > 2 Л1пО42-+ 2Н2О (144)
Однако, как видно из уравнения (143), кинетика подчиняется уравнению второго порядка. В элементарном акте реакции реа'
гируют только Н2 и МпО4". Такой акт может осуществляться, например, так:
МпО4“ + Н2 --->- МпО3_ + Н2О (перенос кислорода) (145)
МпО3- + МпО4" + 2 ОН- ---> 2 МпО42- + Н2О (146)
Едва ли возможно установить обычными методами химической кинетики, какой из механизмов имеет место в действительности. В данном случае могут помочь изотопные методы, из которых в химической кинетике наиболее часто применяются измерение изотопного эффекта, исследования изотопного обмена и исследования с мечеными атомами.
16.2.
Кинетический изотопный эффект
Кинетическим изотопным эффектом а называют отношение констант скорости одной и той же реакции, которая проводится С реагирующим веществом, содержащим более легкий изотоп, (ki) и более тяжелый (fe2):
ь
а = А- (147)
Вследствие того что изотопы одного и того же элемента имеют одинаковую электронную структуру, которая в основном определяет химическое поведение атома, изотопный эффект зависит исключительно от массы изотопов, взятых для сравнения. Это различие в массе влияет на формы движения молекулы или атома (поступательное движение, вращение, колебания). Имеются два метода исследования изотопного эффекта. В одном из них проводится измерение изотопного состава исходных веществ и продуктов реакции, пока она еще не закончилась. Вследствие различия в скорости реакции веществ с легким и тяжелым изотопами (более легкий изотоп реагирует быстрее, что будет обосновано далее) обычно исходные вещества обогащаются тяжелым изотопом, а продукты реакции — легким. В другом методе проводится непосредственно измерение скорости реакции как с веществами, содержащими легкий изотоп, так 11 с веществами, содержащими тяжелый изотоп. Последний ^тод применяется, правда, только в случае, если изотопы сильно различаются по массе, и поэтому практически ограничен Реакциями с участием изотопов водорода. Основной областью Применения изотопного эффекта как раз и является исследование реакций веществ, содержащих атомы водорода. Отношение ^ассы дейтерия к протию (одно из названий легкого водорода) Равно 2, трития к протию — 3. Для более тяжелых изотопов это
198
Химическая кинетика и механизмы реакций
соотношение значительно меньше; например, для 18О и 16О сно составляет 1,125. При дальнейшем изложении ограничимся и ,(J топным эффектом для дейтерия.
С изотопным эффектом дейтерия приходится считаться в том случае, если скоростьопределяющей стадией реакции является разрыв связи водорода (дейтерия) с другим элементом (М—1).
активированное состояние
Рис. Б. 17. Объяснение изотопного эффекта дейтерия для реакции углеводорода, если скоростьопределяющей стадией является расщепление связи С—Н или С—D.
разрыв). Изотопный эффект дейтерия особенно важен для исследования химических реакций с участием органических соеди-
нений, так как из измерений изотопного эффекта можно уста-новить, является ли разрыв С—Н связи скоростьопределяющей
стадией. В настоящее время не имеется достаточно точного метода теоретического расчета изотопного эффекта главным образом потому, что отсутствует достаточное количество данных о структуре активированного комплекса. Ниже приводится грУ' бая оценка изотопного эффекта исходя из уравнения АррениУ'
са (разд. 11.2).
k = PZe~E/RT
(148)
где Р— стерический фактор, a' Z— число столкновений. МоЖ110 заранее сказать, что на стерический фактор Р не оказывает влияния изотопный состав вещества. Применим уравнение (148) к изотопному эффекту и получим следующее значение для эффициента а:
а = ^н. е(£о-сн)/«г (149)
Изотопные методы
199
рассмотрим сначала влияние изотопного эффекта на число столкновений. Для молекул, в которых атом М связан с Н (или D), отношение числа столкновений обратно пропорционально отношению корней квадратных из усредненной массы. Усредненная масса определяется выражением
= МмМн Iх Л4М + Мн
(150)
(Дм и Мн — молекулярные массы МиН).
Полагая Mm>MhMd, получим для изотопного эффекта числа столкновений:
(151>
Подставив это значение в уравнение (149), получим
а= 1,41е(£о-£н)^7' (152) -
Для того чтобы наглядно представить себе влияние энергии активации на изотопный эффект, обратимся вновь к потенциальной кривой элементарного акта химической реакции (рис. Б. 17). Как для исходных веществ, так и для активированного комплекса наиболее низким уровнем энергии является нулевая энергия Ео= '/2hv. Введение изотопа (например, в углеводороды) не меняет форму потенциальной кривой, но смещает уровень нулевой энергии. Это смещение связано с зависимостью частоты от усредненной массы:
где v — частота, k — константа упругого взаимодействия. Таким образом, частота обратно пропорциональна корню из усредненной массы. Кроме того, при дальнейших рассуждениях принято, что вследствие сильно разрыхленной связи в активированном комплексе по сравнению с исходными веществами частота \
колебаний в активированном комплексе почти не меняется, так 'Ке как и нулевая энергия. Следовательно, для расчета изотоп-кого эффекта необходимо принимать во внимание исключитель-Но разность нулевых энергий исходных соединений с легким и ТяЖелым изотопами. Разность нулевых энергий колебаний связи С—Н (соответственно С—D) составляет АД0 = 4,8 кДж/моль.
Согласно уравнению (149), при 25°C ct = 6,9.
Полученные на опыте значения изотопных эффектов часто °Казываются значительно более низкими, чем можно ожидать Из Расчета, так как не всегда выполняются допущения, положенные в основу его вывода. Тем не менее можно на основе измерений изотопного эффекта сделать заключение о том, являет-я ли распад связи С—Н или С—D скоростьопределяющей ста-
Изотопные методы
201
200
Химическая кинетика и механизмы реакций
дней. В качестве примера рассмотрим окисление альдегида хромовой кислотой:
ОН
К I 1/
RCHO + Н2СгО4 =₽=^» R—С—0Сг03Н rcO2H + Н2СгО3 (154)
li-'
[Н2СгОз — короткоживущее, быстро распадающееся соединение Cr(IV)]. В соответствии с предложенным здесь механизмом скоростьопределяющей (лимитирующей) стадией процесса является перенос гидрид-иона к окислителю (хромовой кислоте). Другим примером может служить окисление Н2 перманганатом [уравнение (145)]. Для этой реакции найден изотопный эффект а «2,5, что наряду с другими данными свидетельствует о том, что окисление молекулярного водорода происходит с участием атомарного водорода:
Н2 + МпО4“--->- НМпОГ + Н-
Н- + МпОГ + Н2О ---> МпО42~ + Н3О+ (155)
16.2.1 . Изотопный эффект растворителя
Важные выводы из измерений изотопного эффекта могут быть сделаны не только для веществ, содержащих различные изотопы, но и при сравнении скорости одной и той же реакции, проведенной в разных средах — в Н2О и D2O. Скорость реакции в D2O может быть как более медленной, так и более быстрой по сравнению с реакцией, проведенной в Н2О. Какая из этих возможностей осуществится, зависит от механизма реакции. В большинстве случаев реакции в D2O протекают медленнее, чем в Н2О. Это объясняется меньшей подвижностью реагирующих веществ в D2O (более высокая плотность и вязкость D2O по сравнению с Н2О).
Однако если в механизм реакции входит равновесие с участием слабых кислот или оснований, то можно наблюдать более высокую скорость реакции в D2O. Причина этого состоит в том, что константы диссоциации слабых кислот и оснований в DzO меньше, чем в Н2О (в 2—4 раза). Ионное произведение тяжелой воды Kd2o = 0,145-10-14 также более чем в пять раз меньше, чем ионное произведение Н2О (разд. 35.4.1.4). Этот фактор становится особенно существен при кислотно-основном катализе. Вернемся к уравнению (135); можно легко понять, что в случае специфического кислотного катализа, т. е. при наличии предшествующего протолитического равновесия ([реакция (131) ]> скорость катализируемой реакции в D2O оказывается больше-чем в Н2О. Константа скорости в уравнении (135) равна
а так как К в D2O меньше, чем К в Н2О, то константа скорости реакции в D2O оказывается больше, чем в Н2О. Численное значение изотопного эффекта растворителя а для специфического кислотного катализа находится в пределах от 1 до 4. В данном случае, говоря об изотопном эффекте растворителя, имеют в виду отношение констант скорости в D2O и Н2О: а = ^в0о/^н2о. Напротив, в случае общего кислотного катализа [уравнение (136)] речь идет об обычном изотопном эффекте растворителя (а=^о2о/^н2о< 1). Таким образом, становится ясным, что значение изотопного эффекта может служить критерием типа кислотного катализа, или, в более общем виде, определяет, имеется ли в реакции предшествующее равновесие, в котором участвует ион водорода.
16.3. Исследования методом меченых атомов
При исследованиях методом меченых атомов пользуются возможностью проследить путь атомов в ходе реакции, «пометив» реагирующее соединение, т. е. заменив один из его атомов стабильным или радиоактивным изотопом. Примером использования этого метода снова может служить реакция окисления альдегида хромовой кислотой [уравнение (154)]. Было установлено, что, если пометить хромовую кислоту изотопом 18О, то оказывается, что в продукте реакции — органической кислоте — содержание 18О примерно такое же, как и в исходной хромовой кислоте. Это является прямым доказательством того, что кислород от окислителя непосредственно переходит к окисляемому веществу. Другой пример прямого перехода кислорода был приведен в гл. 7 (в самом начале изучения химической кинетики) при рассмотрении реакции окисления сульфита перманганатом. В этой связи интересно отметить, что в большинстве реакций окисления анионами оксокислот наблюдается прямой перенос кислорода, сопровождающийся одновременным переносом двух электронов. Например:
3 Мп2+ + МпО4" + 6 Н2О -> 5 МпО2 + 4 Н3О+ (157)
При использовании МпО4_, меченного 18О, было установлено, что в случае проведения реакции в щелочной среде в присутствии ионов бария (МпО42- образует с ионами Ва2+ труднорас-чворимую соль ВаМпО4, сразу же выпадающую в осадок) содержание 18О в диоксиде марганца составляет половину его содержания в перманганате. На основании этих данных можно предложить следующий механизм реакции:
Мп2+ + Мп18ОО3~ -> Mn18O2+ + MnO3~ (158а)
Мп18О2++2ОН" ---> Mni«O(OH)2 -> Мп18ОО-Н2О (1586)
МпО3- + Мп18ОО3~ + 3 Н2О -► МпО42- + 2 Н3О+ + Mn18OO32- (158в)
202
Химическая кинетика и механизмы реакций
Реакция (1586) с участием промежуточного вещества Мп18О\ образовавшегося по уравнению (158а), объясняет, почему в МпО2 содержится только половина атомов 18О, введенных в МпО4~.
Окисление сульфита хлоратом также относится к этому же типу реакций, причем было показано, что эта реакция протекает через последовательные стадии образования СЮ2, С1О~ С1-.
16.4. Исследования изотопного обмена
Остановимся на некоторых аспектах исследований изотопного обмена. Эти исследования, во-первых, необходимы для правильной оценки результатов опытов с мечеными атомами, так как если требуется установить, какое количество кислорода переходит от аниона оксокислоты непосредственно к продукту реакции, то необходимо знать, в какой степени окислитель и продукт окисления обмениваются атомами кислорода с растворителем. Во-вторых, с помощью исследований изотопного обмена становится возможным изучение очень быстрых химических реакций (например, реакций электронного обмена).
Реакцией изотопного обмена называют химическую реакцию, при которой изотопы одного и того же элемента меняются местами в различных химических соединениях (ионах, молекулах и т. д.). В общем виде (*Х — обменивающийся изотоп)
АХ + В*Х ^=>- А*Х + ВХ (159)
Такой обмен, как и любая другая химическая реакция, протекает по какому-либо определенному механизму реакции.
Это означает, что обмен может идти через стадию образова ния промежуточных продуктов и имеет некоторую константу равновесия, которая, впрочем, почти всегда равна единице (что соответствует равномерному распределению изотопа межД} обменивающимися соединениями). Поскольку при изотопном обмене не происходит собственно химических превращений, энтальпия реакции обмена А// = 0, т. е. процесс идет адиабатически (разд. 19.2.3). Единственной движущей силой этой реакции является увеличение энтропии, происходящее при выравнивании концентрации изотопа в обменивающихся веществах (подобно энтропии смешения газов, разд. 22.1.2).
Важнейшими типами обменных реакций изотопов в неорг^ нической химии являются реакции электронного обмена и обм« на лигандами.
16.4.1. Реакции электронного обмена
К такого рода реакциям относятся прежде всего взаим( действия между веществами, содержащими один и тот же эЛ
Изотопные методы 203
------------------------- ----------------------------------------— мент в разной степени окисления, например
*МпО4“ + МпО42- Л111О.Г + *МпО42~ (160а)
[*Fe(H3O)6]2+ + [Fe(НаО)6)]3 + [Fe(H2O)6l2+ + [*Fe(H3O)6]3+ (1606)
[*Fe(CN)6]3~ + [Fe(CN)6]i- =₽==►; [Fe(CN)6p- + [*Fe(CN)6p- (160b)
В одно из обменивающихся веществ вводят в качестве метки радиоактивный или стабильный изотопна затем в ходе реакции измеряют изменение количества меченых атомов в другом веществе. Реакции электронного обмена особенно интересны тем, что константа скорости обмена электронов пропорциональна току обмена соответствующей электрохимической реакции (разд. 31.5.3). Примечательно, что все участники обменной реакции имеют одинаковый знак заряда, в результате чего между ними действуют значительные кулоновские силы отталкивания. Несмотря на это, реакции электронного обмена протекают с большой скоростью, период полупревращения составляет доли секунды. Высокая скорость этих реакций объясняется прежде всего тем, что мало различаются размеры координационных сфер участников реакции, что характерно как для анионов оксокислот марганца, так и для цианидных комплексов железа. В энергию активации такого рода реакций вносят вклад следующие компоненты: энергия, необходимая для преодоления кулоновского отталкивания, энергия «выравнивания» размеров координационной сферы и энергия, связанная с туннельным переходом электрона от одного участника реакции к другому. Энергия, связанная с различием размеров координационной сферы, качественно может быть оценена следующим образом. Прежде чем произойдет адиабатический электронный переход (т. е. переход с минимальной затратой энергии), должны стать почти одинаковыми расстояния между центральным атомом и лигандами; для реакции (1606), например, расстояние между Fe3 h и Н2О должно увеличиться настолько, чтобы сравняться с расстоянием между Fe2+ и Н2О. Для такого изменения расстояния необходима затрата некоторой энергии (энергии активации). Очевидно, реакции с электронными переходами протекают особенно быстро в том случае, если эти расстояния мало отличаются для соединений с различной степенью окисления.
Уже было упомянуто, что в этих реакциях перенос электронов происходит по туннельному механизму; это означает, что электрон не преодолевает энергетического барьера, а «просачивается» через него. Туннельный эффект объясняется корпускулярно-волновым дуализмом частиц на основе соотношения Неопределенности Гейзенберга, если рассматривать электрон как волну де Бройля (подробнее см. в учебниках атомной Физики). В данном случае возможность туннельного перехода
204
Химическая кинетика и механизмы реакций
электрона означает, что лиганды не должны подходить друг к другу слишком близко, так как иначе существенно увеличивается энергия отталкивания, а туннельный переход может уже осуществляться на некотором расстоянии между лигандами. Часто отмечается, что реакции переноса электрона сильно катализируются ионами противоположного знака. Реакции (160а) и (160в) катализируются катионами, а (1606)—анионами. Каталитическое воздействие на реакции с обменом электронов можно объяснить встраиванием катиона или аниона в активированный комплекс; противоположный по знаку заряда ион снижает кулоновскую долю энергии активации. Следует лишь отметить, что в этих реакциях можно различать два типа активированных комплексов — внешнесферные комплексы и мостиковые комплексы. В первом случае электронный переход происходит между полностью изолированными комплексами, т. е. не происходит перекрывание координационных сфер обменивающихся частиц, во втором случае перенос электронов связан с переносом лиганда от одной частицы к другой.
' о о у-
OMnO ОМпО О О
Комплекс внешнеарернцго 1(Н2О)5 Fe—Cl— Fe(H2O)5]4+ (161)
типа. Комплекс мостикового типа
Катализ электронного обмена катионами между обоими оксоанионами марганца можно, например, объяснить образованием следующего активированного комплекса:
о о у-
ОМпО К ОМпО о О
К — одновалентный катион
(162)
16.4.2.
Обмен лигандами
Обмен лигандами часто наблюдается при обмене кислорода между кислородсодержащими соединениями и водой (например, между оксоанионами и водой). Можно различить два типа обмена:
-Х=О + Н218О^
1«ОН —ХМО + Н2О
-хг'
\он = Х18ОН +ОН-
обратимое образо-
вание ангидрида /1 S3) нуклеофильное за- ' '
мещение
В качестве примера укажем реакцию обмена кислорода межДУ СО32- и Н2О:
-ох -ох ^ОН О2С18О2- + Н2О
ХС=О + НЛО
-о/ “ -0х хон О2С18ОН- + он-
обратимое образование ангидрида нуклеофильное замещение
Изотопные методы
205
В данном случае осуществляются два варианта одного и того же механизма. На первой стадии образуется ортоформа, распадающаяся в зависимости от pH среды на разные продукты.
Из измерений констант скорости этих обменных реакций можно найти константу скорости соответствующих химических реакций, что, как правило, нельзя сделать обычными методами химической кинетики. Проведенные до настоящего времени исследования обменных реакций кислорода позволяют сделать следующие выводы:
скорость обмена кислорода для элементов одной группы периодической таблицы увеличивается по мере увеличения заряда ядра
и снижается по мере уменьшения заряда центрального иона.
Например, ион С1О3- очень медленно обменивает кислород с водой (время полупревращения — несколько суток), а ион Юз- — очень быстро (период полупревращения — доли секунды) вследствие возможности проявления более высокого координационного числа у более тяжелого атома. Быстрый обмен кислорода воды с ионом Ю3~ по сравнению с С1О3~ можно объяснить следующим механизмом реакции:
НЮз + Н218О Н3118о4 (164)
Протекание такой же реакции с ионами С10з- очень затруднено.
Увеличение скорости обмена при уменьшении заряда центрального атома [например, в ряду СЮ4-, СЮз-, СЮ2-, СЮ-) также объясняется увеличением способности к проявлению более высокого координационного числа.
Другие возможности обмена кислорода здесь не рассматриваются.
Б.2. РАВНОВЕСИЯ И ХИМИЧЕСКАЯ ТЕРМОДИНАМИКА
17. ПРЕДМЕТ И ЗАДАЧИ ХИМИЧЕСКОЙ
ТЕРМОДИНАМИКИ
В отличие от химической кинетики, основной задачей кото рой, как мы видели в предыдущих главах, является исследова ние протекания химической реакции во времени и выяснение ее механизма, химическая термодинамика исследует поведение хи мических систем в состоянии равновесия. Как следует из кинетического рассмотрения химического равновесия, здесь речь идет о динамическом равновесии, т. е. отсутствии внешних изменений в системе и равенстве скоростей прямой и обратной реакций.
Методами химической термодинамики можно определить при каких условиях и в каком направлении протекает химическая реакция, ее энергетический баланс, выход конечных про дуктов и влияние температуры и давления на равновесие.
Прежде чем делать выводы о закономерностях протекания химической реакции во времени или ее равновесном состоянии необходимо условиться о форме записи уравнений химической реакции. Нельзя, например, изучать реакцию образования ам миака, не привлекая к рассмотрению уравнение
3 Н3 + N3 +=> 2 NH3 (165)
или
з/3Н3 + V2N3 «=* NH3 (166)
Оба уравнения описывают реакцию получения аммиака, однако численные значения теплоты реакции и константы равновесия реакции по уравнению (165) отличаются от соответствующих значений уравнения (166). Пока еще не известно, являются Л1 вещества, стоящие в левой части уравнения, исходными веще ствами, а с правой — продуктами реакции. Задача термодина мического исследования как раз состоит в том, чтобы опре делить направление протекания реакции. При записи урав нений реакции можно использовать и дробные коэффициент [уравнение (166)]. Однако, вообще говоря, в качестве коэффи циентов всегда стремятся использовать минимальные целые чис ла, и уравнение реакции записывают таким образом, чтобы оно соответствовало самопроизвольному протеканию реакции еле ва направо.
Предмет и задачи
207
Реакции можно разделить на гомогенные и гетерогенные. Гомогенными называют реакцию, все компоненты которой образуют одну фазу, например газовую или жидкую. Оба этих случая рассмотрены ниже. Реакция, протекающая между различными фазами, называется гетерогенной. Фазой называют объем, Бсе части которого имеют одинаковые физические (и химические) свойства. На границе двух фаз наблюдается скачкообразное изменение этих свойств (показателя преломления, плотности и т. д.).
Если в одной фазе содержится несколько веществ, концентрацией которых нельзя пренебречь, то ее называют смешанной (многокомпонентной). Существуют различные способы выражения концентрации компонентов в смешанной фазе, основанные на измерении количества вещества в молях (1 моль содержит Na = 6,023-1023 атомов, молекул, ионов или других частиц).
При теоретических исследованиях, особенно термодинамических, как для газообразных, так и тля твердых веществ наиболее удобно применение в качестве меры концентрации мольной доли х, (безразмерный параметр). Она определяется как отношение числа молей nt компонента i к общему числу молей всех компонентов Sn,:
i
ni
2^
I
Отсюда
2^=1
I
Концентрация в мольных долях определяется только составом фазы и не зависит от температуры и давления. Если умножить ее значение на 100, то получится концентрация компонента в мольных процентах (моль %).
В растворе концентрация растворителя обычно значительно превышает концентрацию растворенного вещества Если концентрацию выражают в молях на килограмм растворителя, се называют моляльностью (моль/кг, илч Мл)
В аналитической химии число молей растворенного вещества относят к 1 литру раствора, т. е. указывают молярность раствора (моль/л, или М). Концентрацию, выраженную в грамм-эквивалентах на литр раствора, называют нормальностью (г экв /л, или н ) Концентрация в молях на литр и грамм-эквивалентах на литр, так же как и объем, зависит от температуры и Давления, и в этом отношении такой способ выражения концентраций менее Удачен, чем если концентрация указана в мюльных долях или как отношение количества вещества к массе растворителя (более подробно и последовательно о способах выражения концентрации см. в гл. 21).
В термодинамической системе могут одновременно находиться несколько твердых и жидких фаз (если жидкости несмеши-вающиеся), однако система может состоять и из одной фазы, Например газовой. В связи с этим понятие «реакция» необходимо расширить. Любой процесс, записанный в виде уравнения Реакции, можно рассматривать как реакцию. Так, например, ис-
208 Химическая термодинамика
Термодинамические функции состояния 209
парение воды можно записать как особый тип гетерогенной реакции:
Н2О(жидк.) Н2О (газ) (167)
В зависимости от условий, в которых протекает реакция, различают изотермические (если во время реакции температура поддерживается постоянной) и адиабатические реакции (если невозможен тепловой обмен с окружающей средой). Несмотря на то что почти во всех химических реакциях происходит выделение или поглощение теплоты, условия их протекания чаще всего приближаются к предельному случаю изотермического процесса, если они идут настолько медленно, что скорость теплопередачи за счет теплопроводности, конвекции или излучения значительно превышает скорость протекания реакции. К адиабатическим можно отнести только очень быстрые реакции горения и взрыва. Кроме того, различают изобарные и изохорные реакции, т. е. реакции, протекающие при постоянных давлении и объеме соответственно. Изобарные реакции встречаются значительно чаще.
Уже говорилось о том, что часто протекание химических реакций сопровождается выделением или поглощением теплоты (тепловым эффектом). Экзотермической называют реакцию, протекающую с выделением теплоты, а эндотермической — с поглощением теплоты из окружающей среды. В экзотермических реакциях принято считать теплоту реакции отрицательной (энергия реагирующей системы уменьшается), а в эндотермических— положительной (энергия увеличивается).
18. ТЕРМОДИНАМИЧЕСКИЕ ФУНКЦИИ СОСТОЯНИЯ. I
ИХ СВОЙСТВА fl
18.1. .Математическое описание функций состояния Н
Как уже отмечалось, химическая термодинамика занимается j
в основном описанием состояний равновесия и изучением зако- I нов, в соответствии с которыми происходят изменения энергии системы в ходе физико-химических процессов. Всякий такой процесс в общем является изменением состояния системы, которое можно описать с помощью функций состояния (энергия энтропия и др.). Функция состояния определяется независимы ми переменными (или параметрами состояния системы), например давлением, температурой. Однако невозможно достаточно четко и однозначно разделить функции состояния и независимые переменные системы.
Функция состояния зависит от многих переменных. В общем Л виде можно записать
z=f(x, у, п1г п2,..., nit..., пь) (168) fl
где z— функция состояния, х и у — независимые переменные (например, давление и температура), nt-— еще одна независимая переменная — содержание (в мольных долях) данного компонента в химической многокомпонентной системе. В реальных условиях число независимых переменных не так велико, поскольку некоторые параметры системы поддерживаются постоянными. Если рассматривать равновесный химический процесс, например, при постоянных давлении и температуре, то независимые переменные —содержание (мольные доли компонентов в системе. Особенно простой вид принимает функция состояния для гомогенной однокомпонентной системы, так как она характеризуется постоянной концентрацией данного компонента. В этом случае z — функция двух независимых переменных:
z=f(x, У) (169)
Такими являются например, термическая функция состояния однокомпонентной системы v = f(p, Т) (разд. 4.1, уравнение (3)) и калорическая функция состояния для той же системы и = ~f(v, Т), описывающая зависимость внутренней энергии от объема и температуры.
При более глубоком изучении термодинамики (разд. 22.1) станет ясно, что не всякая функция независимых переменных z=f(x, У) удовлетворяет требованиям, предъявляемым к термодинамической функции состояния. Накопленный в ходе развития науки и техники опыт, сконцентрированный в основных законах термодинамики, показывает, что состояние системы может быть однозначно охарактеризовано только такими функциями, приращение которых не зависит от пути, по которому система переходит из одного состояния в другое. Зависимость функции состояния от пути означала бы, что если при переходе системы из состояния 1 в состояние 2 к ней подводилось бы некоторое (пусть небольшое) количество энергии, то при переходе из состояния 2 в состояние 1 от нее отбиралось бы большее количество энергии в виде работы. Тогда на основе такой системы можно было бы создать вечный двигатель (perpetuum mobile), что противоречит основным законам термодинамики (природы).
Введем основные математические понятия и соотношения, характеризующие функцию состояния. Функция z=f(x, у) списывает некоторую поверхность (функции трех и более неза-нпсимых переменных нельзя геометрически изобразить в трехмерных координатах). Например, на рис. Б.18 представлена п°верхность, построенная в соответствии с термодинамическим Уравнением состояния v=f(T, р). Изменение функции состояния системы (в данном случае объема и) в общем случае сопровождается одновременным изменением других параметров системы (давления р и температуры Т). Гёометрически это со-|4-2027
210
Химическая термодинамика
Термодинамические функции состояния
211
ответствует перемещению точки на поверхности (например, От точки Pi к точке Р2). Если изменяется только одна из перемен-них, а другая поддерживается постоянной, говорят о парциаль-ном изменении. Перемещение точки вдоль кривой CD, напри-мер, соответствует изотерме (уравнение Бойля — Мариотта разд. 1.4), вдоль прямой АВ — изобаре (закон Гей-Люссака’ разд. 1.4). Переход системы от pi к р2 может (кроме уже рас’
Рис. Б. 18. Поверхность в координатах v, р, Т, соответствующая термическому уравнению состояния идеального газа (пример функции трех переменных)
смотренного случая одновременного изменения переменных р и Т) происходить по пути Р1В{Р2 или P{CiP2. Первый случай вначале соответствует процессу при р = const и v меняется в зависимости от Т, а затем при 7'=const v меняется в зависимости от р. Второй случай сначала соответствует процессу при 7’=const и v меняется в зависимости от р, а затем при р = const v меняется в зависимости от Т. Суммарное изменение v не зависит от пути, т. е. функция v удовлетворяет требованиям, предъявляемым к термодинамическим функциям.
Можно убедиться в этом, сделав вычисления с помощью законов идеальных газов (разд. 1.4).
Основополагающее значение в термодинамике имеет матеМЯ' тическое выражение дифференциального приращения:
(170)
где (дг/дх)у— частная производная z по х при постоянно»* у, a (dz/dy)x — частная производная z по у при постоянном '
Запишем в виде дифференциального уравнения в частных производных функцию состояния v=f(p,T)i
dv = (-g^\ + dp (171)
\ 01 /р \ °? Jt
Вернемся вновь к рис. Б. 18. (dv/dT)o численно равна тангенсу угла наклона касательной к кривой, образованной пересечением поверхности v = f(p, Т) с плоскостью ABKL, с плоскостью р—Т в точке Pi, a (dvfdp)? — тангенсу угла наклона касательной к кривой, образованной пересечением поверхности v = f(p, Т) с плоскостью CEGHD, с плоскостью р—Т в точке Рь
Частные производные, вообще говоря, сами представляют собой функции независимых переменных х и у, обозначив
и (w)x==Q(y,y)
можно записать уравнение (170) в следующем виде:
dz--P(x, у) dx~rQ (х, у) dy (172)
Уравнение (171), например, после подстановки в него уравнения состояния идеального газа (у = nRT/p) преобразуется в
dv=d^-dT-----dp (173)
Установим, можно ли считать дифференциал в уравнении (170) [соответственно (172)] полным дифференциалом-, для этого воспользуемся правилом Шварца: если смешанные частные производные функции f(x, у) в некоторой области являются непрерывными функциями х и у, то в пределах этой области они равны между собой:
или (174)
дхду дудх \ ду )х \ дх )у v ’
Итак, дифференциалы термодинамических функций состояния являются полными дифференциалами, т. е. для них выполняется соотношение (174), и, напротив, при выполнении условия (174) Данная функция термодинамических независимых переменных является функцией состояния.
Проверим справедливость соотношения (174) для функции и (Уравнение (173)):
д (nR \ д ( nRT \ nR nR
др \ Р ) дТ ( р2 ) ~ р2 ~ Р2
В термодинамике соотношения (174) используются не только как критерий для определения того, является ли данная Функция функцией состояния; с помощью этих соотношений 14*
212
Химическая термодинамика
можно установить взаимосвязь между различными функциями состояния и параметрами системы (разд. 21.1 и 22.3).
Другую важную зависимость между функцией состояния и параметрами системы можно получить из уравнения (170). Пусть переменные х и у меняются таким образом, что функция состояния остается постоянной, т. е. dz=0. Тогда
dx4-(ЛМ dy=0
I дх Л Л дУ L
/ ду \ / дг \ / ду \ [ дх \ I дг \ ( ду \ ,
Д-Дч(175>
Уравнения (175) удобнее всего использовать при неявном диф. ференцировании функций с одной независимой переменной, т. е. в том случае, когда неявную функцию f(x, z/)=0 нельзя преобразовать в явную y = j(x), так как при дифференцировании явной функции y=f(x) необходимо проделать значительно более сложные вычисления. Применив соотношение (175) к уравнению состояния (171), получим следующее выражение для температурной зависимости объема ((dv/dT)p— термический коэффициент расширения):
\ дТ (др/дфт
Преимущество уравнения (176) становится очевидным, например, в том случае, когда надо рассчитать коэффициент термического расширения реального газа (уравнение Ван-дер-Ваальса), так как определение функции (dv/dT)p требует дифференцирования кубического уравнения.
При выводе термодинамических функций состояния на основе теоретических представлений и экспериментальных данных (выраженных в виде основных законов термодинамики) соотношения между дифференциалами обычно получают из уравнений типа (170) и (172). Вычисление приращения функции состояния для конечного изменения параметров системы эквивалентно определению значения функции z в точках (Xi, yi) и (х2, у2) ПУ’ тем интегрирования уравнения типа (172). Первоначально по-х-г и2
лучим интегралы J Р(х, y)dx и J* Q(x,y)dy.
•Ч У1
В общем случае такие интегралы берутся, если в подынтегральном выражении у можно представить в явном виде y=f(xY Это равноценно вычислению пути на плоскости х—у, вдоль которого проводится интегрирование, т. е. вычислению криволинейного интеграла. В общем случае существует множество ПУ' тей перехода из одного состояния в другое, и результат интегрирования зависит от пути. Решение сильно упрощается, есл" dz является полным дифференциалом, тогда для термодинЯ'
ч
Термодинамические функции состояния
213
^лческой задачи z — функция состояния, т. е. ее изменение не зависит от пути перехода. Примем, что
z=Jp(x,y)c?x4-K(y) (177)
Поскольку подынтегральная функция зависит от х и у, константа интегрирования по х может быть функцией от у, т. е. Х(у). Преобразуем уравнение (177):
<178>
Однако из уравнения (172) (dz/dy)x = Q(x, у), т. е.
Q(x,y)--^^P (х, у) dx) (179)
Предпосылкой применимости этого метода является выполнение правила Шварца для рассматриваемого дифференциала, поэтому соотношения (174) называют также условием интегрируемости функции.
Поскольку освоение методов интегрирования полных дифференциалов необходимо для глубокого понимания термодинамики, ниже предлагаются два примера его применения.
Пример 1. Является ли полным дифференциалом выражение dz= (2ах+ +2by+c)dxJr (bx+2cy + e')dy? Если это условие выполняется, определить функцию z.
Решение. Условие интегрируемости выполняется, так как д д
-g^-(2ax+by + c)= (bx + 2с у + с)
b = b
В соответствии с условием (177)
г— I (2ax-!rby-!rc)dx-\-K(y) = ax‘2-\-bxy-\-cx-1rK(y)
Теперь определим К. (у). Из уравнения (178) получим (дг/ду)х=Ьх+ д/\(у [ дг\
+ ), а из исходного уравнения для дифференциала dz I g^\=bx+2cy+
+е; сравним эти выражения и получим дК(у)/ду~2су+е; после интегрирования К. (у) =су2+ су+С. Итак, функция г равна
z — ах2 -|- Ьху -ф сх су2 су -J- С
Пример 2. Пусть дана функция состояния v=f(p, Т); полный дифферен-nR nRT
ЧИад этой функции равен —dT—-------- dp. Рассчитаем изменение функции
Р Р
состояния v, если параметры системы меняются От pi, Ti до рз, Т%.
Решение. Этот пример тривиален, поскольку функция v = f(p, Т) уже Введена как функция состояния; достаточно подставить начальные и конеч-Нь1е значения р и Т в уравнение идеального газа, чтобы вычислить v. Одна-к° при решении поставленной задачи необходимо выполнить такие же мате-
1 U
214
Химическая термодинамика
магические преобразования, как и при нахождении неизвестной функции состояния.
Воспользовавшись уравнением (177), получим
v=^dT+K(p) = -^-^K(p)
Описанным ранее способом найдем константу интегрирования К(р). Т огда
/ dv \ nRT , дК(р) __ nRT
др ) р2 “Г др р2
т. е. дК.(р)/др = 0 и 7<(р)=С. Таким образом, решение задачи имеет вид _ nRT
v ~—+С,а подстановка заданных значений р и Т приводит к выражению, которое можно было ожидать из уравнения (3) в разд. 1.4:
v = и2—I»! = nR (Г2/р2— Тi/pj)
Как показано в разд. 1.4, константа С не зависит от давления и температуры и в случае идеального газа равна нулю.
Из свойств полного дифференциала следует, что при интегрировании в тех же пределах, но в обратном направлении изменение функции состояния имеет ту же величину, но обратный знак. Это означает, что при интегрировании полного дифференциала по замкнутому контуру криволинейный интеграл становится равным нулю:
dz= О
(180)
Напротив, если dz не является полным дифференциалом, то изменение функции z зависит от пути перехода, а интегрирование по замкнутому контуру дает результат, отличный от нуля.
Таким образом, условие (180) также представляет собой критерий существования функции состояния, а следовательно, имеет важное значение в термодинамике (термодинамика круговых процессов).
Рассмотренные выше соотношения для функций двух переменных можно распространить также на функции с большим числом переменных. Так, например, дифференциал функции (168) равен
dz~l-^-\ + (181)
\ дх /УМ1 \ дУ )x,nt ^\dni )х,у,п^п} 1 v
Переменные, которые поддерживаются постоянными, указань индексами у скобок. Суммирование в третьем члене уравнения (181) проводится по всем щ (содержание в мольных долях) от п{ до Пу. Здесь правило Шварца также является критерием того, что данное уравнение является полным дифференциалом функции состояния.
Термодинамические функции состояния 215
48,2. Интенсивные и экстенсивные параметры системы и парциальные мольные величины
Интенсивными параметрами называют характеристики системы, не зависящие от количества рассматриваемой фазы (температура, давление, плотность, диэлектрическая проницаемость и т. д.). Параметры системы, значение которых зависит от количественной характеристики фазы, называют экстенсивными (масса, внутренняя энергия, энтропия и т. п.). В так называемых идеальных смешанных фазах (газах или жидкостях) большинство экстенсивных параметров аддитивно. К экстенсивным параметрам вещества относятся также все функции состояния г (в том числе те, которые определены далее). Можно записать
г = /7171+/г222 + --- + »Л + ---+«А = 2,l>Zi (182)
г = 1
Здесь и в дальнейшем приняты следующие обозначения: строчные буквы — для экстенсивных функций состояния, а прописные — для функций состояния, отнесенных к 1 молю вещества, — мольные функции состояния. Объем идеальной смешанной фазы можно записать через парциальные объемы ком-, понентов [уравнение (182)]:
(183)
i=i
где V/ — мольный объем одного из компонентов; «идеальные» свойства системы предполагают, что между молекулами компонентов нет никакого физического или химического взаимодействия. Ясно, что в этом смысле идеальное состояние является предельным состоянием, которое никогда не осуществляется полностью. Несмотря на это, уравнение (183) выполняется для некоторых жидких смесей (например, бензола с толуолом). Для большинства смесей, однако, уравнение (183) не оправдывается. (При смешении серной кислоты с водой или растворении солей происходит заметное уменьшение объема.) Существование специфических сил взаимодействия между молекулами можно отметить также и при исследовании других функций состояния смешанных фаз, так что в общем случае уравнение (182) является приближенным. Для того чтобы дать количественную характеристику смешанных фаз, вводят понятие о парциальной мольной величине-, по аналогии с уравнением (181) изменение функции состояния смешанной фазы представляет собой полный дифференциал, т. е. при постоянных давлении и температуре
+ . d«2+...+
\ 0п1 \ОН2 /ч;^/г2
dnh (184)
\ дп[ Jn.+tij ( дп^
Парциальными мольными величинами здесь являются частные производные рассматриваемой функции состояния по числу молей соответствующего компонента при постоянном содержании остальных компонентов:
дг \ д,г1
(185)
По определению парциальные мольные величины, как подчеркивается в самом названии, относятся к бдному молю, не зависят от количества вещества, т. е. являются интенсивными параметрами системы. Однако они зависят °т состава смешанной фазы, и, как будет показано ниже, с их помощью
216
Химическая термодинамика
можно количественно охарактеризовать движущую силу физико-химического процесса (превращения фаз) на каждой его стадии.
В отличие от экстенсивных параметров мы будем обозначать парциальные мольные величины прописными буквами с чертой, отличая их таким образом от обычных мольных величин. Далее будет введена парциальная моль-ная величина—химический потенциал (размерность энергия/моль).
Используя парциальные мольные величины, можно записать для реальной смешанной системы следующее выражение [аналогичное уравнению (182)]:
z=niZl-\-n2Z2-[-... -]~nkZk—^ niZi (186)
Откуда _ _
dz==nldZ1 —Z^dtiy —~~*• *Z^dn^ (187)
Если подставить уравнение (185) в уравнение (184) и приравнять dz из уравнений (184) и (187), получим важное соотношение, известное как урав-некие Гиббса — Дюгема:
nidZ1-]-n2dZ2-\-... -\~nkdZk — 0 (188)
С методами определения парциальных мольных величин и возможностями использования уравнения (188) можно познакомиться подробнее по учебникам физической химии.
18.3. Стандартное состояние
Применяемые на практике мольные или парциальные мольные функции состояния Z, характеризующие термодинамическую систему, как правило, зависят от температуры, давления, а при реакциях в растворах также и от концентрации. В связи с этим, для того чтобы можно быЛо табулировать данные о функциях состояния (что удобно для сравнения), оказалось целесообразным выбрать некоторое стандартное состояние, исходя из которого можно расчетным путем перейти к функции состояния для любого конкретного состояния системы. Примем и будем использовать в дальнейшем следующее определение: стандартное состояние* газа осуществляется при давлении р=101 325 Па= 101,325 кПа (величины, относящиеся к стандартному состоянию, обозначают соответствующим символом с индексом 0 вверху справа). Давление 101 325 Па будем в дальнейшем называть нормальным давлением. Стандартное состояние зависит от температуры и поэтому записывается с индексом Т (Z°T). В таблицах, как правило, приводят значения функций состояния при температуре 298,15 К (Z°2!>8,i5)**. Чтобы охарактеризовать стан
* Стандартным состоянием веществ, находящихся в конденсированной фазе, является реальное состояние веществ, находящихся при данной температуре под давлением в одну атмосферу (101,325 кПа).
Стандартным состоянием газообразного вещества при любой температуре является состояние гипотетического идеального газа, летучесть которого равна единице, а энтальпия равна энтальпии реального газа при той же температуре и давлении, стремящемся к нулю.
Стандартным состоянием раствора является состояние гипотетического идеального раствора, в котором парциальная мольная энтальпия и теплоемкость растворенного вещества те же, что и в реальном бесконечно разбавленном растворе, а энтропия и свободная энергия те же, что в растворе с моляльностью, равной единице [Воробьев А. Ф. Теоретич. и эксперим. химия 8, 705—708, 1972].'—Прим. ред.
** Точнее эту температуру называть стандартной. Принятая специальным соглашением, она к определению стандартного состояния отношения не имеет [Воробьев А. Ф. Теоретич. и эксперим. химия, 8, 705—708, 1972] " Прим. ред.
Первый закон термодинамики
217
дартное состояние какого-либо компонента в растворах, принимают его активность в растворе равной единице (а=1 моль/л), также указывая индекс сверху справа.
19. ПЕРВЫЙ ЗАКОН ТЕРМОДИНАМИКИ
19.1. Общие положения
Первый закон термодинамики, строго установленный Мейером (называемый в физике также законом сохранения энергии), утверждает, что энергия не исчезает и не создается, а переходит из одной формы в другую, другими словами, невозможно создать «вечный двигатель первого рода». Воспользовавшись представлениями, развитыми в гл. 18 о функциях состояния [уравнения (174) и (180)], можно сформулировать первый закон термодинамики следующим образом: внутренняя энергия системы есть функция состояния. Если бы внутренняя энергия не была функцией состояния, то при ее изменении в круговом процессе можно было бы получить дополнительное количество энергии, т. е. создать «вечный двигатель первого рода», что противоречит первому закону термодинамики (одному из основных законов природы).
Вопрос об изменении энергии в химических реакциях также относится к области приложений первого закона термодинамики, так как является особым случаем общего закона сохранения энергии, а именно возможности изменения внутренней энергии системы. Последнюю понимают как общую энергию системы, исключая кинетическую энергию системы в целом (не имеет значения, где идет газовая химическая реакция—-в лабораторном сосуде или в аэростате) и ее потенциальную энергию, связанную с внешним полем (не имеет значения, происходит реакция в подвале дома или на 4-м этаже дома, хотя в последнем случае воздействие гравитационного поля будет более сильным). Внутренняя энергия — это сумма энергий отдельных атомов и молекул, из которых состоит система, включая потенциальную энергию, связанную с межмолекулярным взаимодействием.
Первый закон дает также представление о путях преобразования внутренней энергии. Прежде всего следует отметить, что система может обмениваться энергией с внешней средой. Поток энергии может иметь место в виде теплового потока через поверхность раздела системы с внешней средой, если между Ними имеется разность температуры. Если эта разность температур представляет собой бесконечно малую величину, такой тепловой поток называют обратимым. Для обратимого теплового потока характерен бесконечно медленный перенос энергии. В любой момент времени поток энергии может быть как бы остановлен и при изменении знака ДЕ направлен в противополож
218
Химическая термодинамика
ном направлении. Поток энергии может быть также преобразован в работу, например в работу расширения газа. Рассмотрим цилиндр с поршнем, под которым расширяется газ, в результате чего поршень смещается вверх. В этом процессе энергия газа преобразуется в работу. Если разность давления газа и внешнего давления бесконечно мала, произведенную работу называют обратимой. Обратимая работа расширения равна
dtv=,—pdv (189)
При химических реакциях может совершиться работа расширения, а также электрическая работа (в химических источниках тока).
Рассмотрим подробнее работу объемного расширения. Уравнение (189) соответствует обычному определению механической работы. Пусть сила F действует вдоль бесконечно малого отрезка ds; совершается бесконечно малая работа dw. Давление р на поршень в цилиндре определяет силу F, действующую на площадь q. Отсюда dw = Fds=p-q-ds = pdv, нли, принимая
Рис. Б. 19. Схема к объяснению работы расширения.
термодинамическое правило знаков, произведенная системой работа отрицательна (—pdv).
Можно назвать и другие виды работы, например работа, связанная с увеличением поверхности, или работа перемещения зарядов в электрическом поле. Одиако и это не последняя возможность преобразования энергии в работу. Такая возможность осуществляется всегда, когда некоторая сила действует вдоль координаты пути.
Таким образом, первый закон термодинамики можно кратко записать в следующем виде* *:
аи=Ьит\-Ьс/ (190)
* Буква 6 вместо d показывает, что работа и теплота ие являются полными дифференциалами, а лишь бесконечно малыми приращениями.—ПриМ-перев.
Первый закон термодинамики 219
4-—"1 . — - - - - _ _ . .
или [уравнение (189)]
du=8q—pdv (191)
Одной из важных для термодинамики величин является мольная теплоемкость С, определяемая как количество теплоты, необходимое для того, чтобы нагреть 1 моль вещества на 1 градус. Тогда количество теплоты, необходимое для того, чтобы повысить температуру п моль вещества от 7\ до Т2, равно
q=--nC(7'2—7\) или, полагая Q=~~, (192)
В изохорных условиях (при o = const, dv = 0) (например, при разложения СаСО3 в закрытом сосуде) из уравнений (191) и (192) получим
(dQ)p=dU=CddT- =С0 (193)
\ < V
Индекс v означает, что процесс проводится при o = const; Cv — молярная теплоемкость при постоянном объеме.
Таким образом, для изохорного процесса изменение внутренней энергии равно принятому или отданному системой количеству теплоты, а для изохорной реакции выделившееся количество теплоты определяет изменение внутренней энергии. Однако, так как изобарные реакции встречаются на практике значительно чаще, было бы желательно найти функцию, изменение которой при постоянном давлении соответствовало бы принятому или отданному количеству теплоты. Такой функцией является энтальпия
h = u-^-pv
H~U^pV (194)
Выразим энергию через энтальпию и подставим полученное выражение в уравнение (191) при условии dp = 0:
d(H—pV') — 8Q~pdV
dH—pdV~Vdp = 8Q—pdV (195)
8Q=dH—Vdp
В изобарных условиях (p = const, dp = 0) получим
(8Q)p=dH-, Ж = Cp (196)
\ /p
Полезное в дальнейшем соотношение между Ср и Cv можно получить из определения энтальпии (194):
(st) =с.= |("+Л, \ /р
CP^CV+R ° (197)
220
Химическая термодинамика
Уравнение (197) справедливо только для идеальных газов. Для них молярная теплоемкость при постоянном давлении равна сумме молярной теплоемкости при постоянном объеме и работе R расширения идеального газа в количестве 1 моль при его нагревании на 1 градус.
При формулировке первого закона термодинамики предполагается, что энергия может преобразовываться только в теплоту или работу. Однако принципиально энергия системы может меняться также при изменении количества вещества: при удалении вещества из системы оно уносит часть внутренней энергии этой системы, а при поступлении вещества в систему последняя получает дополнительное количество энергии. Системы, в которых возможно изменение количества вещества за счет его притока или выноса из системы, называют открытыми. Если такой процесс невозможен, систему называют замкнутой. Следует отличать еще изолированную систему, в которой невозможен обмен с внешней средой не только веществом, но и энергией. В изолированных системах энергия всегда остается постоянной. Термодинамическое исследование открытых систем приобрело важное значение при переходе к живым организмам, которые находятся в обмене веществом с внешней средой. Эти системы также широко используются при моделировании непрерывных процессов в химической промышленности, где в химический реактор (систему реакторов) непрерывно поступают исходные вещества, а на выходе— конечные продукты. Теория открытых процессов (систем) достаточно хорошо разработана, поскольку исторически она возникла одновременно с термодинамикой необратимых процессов, однако при дальнейшем изложении теория открытых процессов не будет рассматриваться более глубоко.
19.2. Изотермические, изобарические и адиабатические процессы
Для лучшего понимания общего подхода к физико-химическим методам изучения термодинамических процессов рассмотрим подробнее изменение функции состояния и, а также параметров w и q для изотермических, изобарических и адиабатических процессов.
19.2.1. Изотермическое расширение и сжатие
Еще Гей-Люссак установил, что внутренняя энергия идеального газа не зависит от объема (второй закон Гей-Люссака). Для этого он провел следующий опыт: два сосуда соединены трубкой с краном (рис. Б.20); в одном из сосудов содержится газ, из другого сосуда газ откачивается; после открывания крана газ устремляется в вакуумированный сосуд, однако при этом не наблюдается изменения температуры, т. е.
(-в-)г=« <198>
Однако для реальных газов это соотношение не выполняется. В большинстве случаев при расширении сжатого газа при нормальной температуре происходит его охлаждение (эффект Джоуля—Томсона), так как при его расширв' нии преодолеваются силы притяжения между молекулами, аналогично тому как это происходит при испарении жидкостей. (Ознакомьтесь с применением эффекта Джоуля — Томсона для сжижения газов.)
Первый закон термодинамики
221
Рассмотрим изотермическое расширение идеального газа, находящегося в цилиндре с поршнем, от объема Vi до объема v2. Как указывалось в предыдущем разделе, этот процесс протекает обратимо в том случае, если внешнее давление, против которого совершается работа, в каждый момент времени бесконечно мало отличается (на dp) от давления в цилиндре. Согласно второму закону Гей-Люссака, u = const, du = 0; тогда первый закон термодинамики записывается в следующем виде:
—dq — dw=—pdv (199)
Увеличение объема от до v2 в процессе расширения в соответствии с законом Бойля — Мариотта вызывает понижение давления от pt до р2- Для обратимого процесса (так как dp^O) внешнее давление и давление в цилиндре почти равны, поэтому можно подставить р,
Рис. Б 20. Расширение газа в вакуум (опыт Гей-Люссака).
полученное из уравнения состояния идеального газа p = nRT)v в (199):
—dq~dw= —nRT
1 v
—q=w=~nRT\n^ (200)
Таким образом, в изотермическом процессе расширения газа внутренняя энергия системы, преобразованная в работу против внешнего давления, восполняется за счет притока теплоты. В рассмотренном здесь случае обратимого проведения процесса совершенная работа идентична максимальной полезной работе, которая, как показано ниже, равна изменению функции состояния. Минимальная обратимая работа сжатия, необходимая для перевода системы в исходное состояние, равна 7?7’1п(г'2/У1)-При необратимом проведении процесса (потери на трение, Ар>0) часть полезной работы теряется, переходя в теплоту. В предельном случае расширения газа в вакуум работа не совершается, однако для возвращения в исходное состояние необходима работа по крайней мере не меньшая, чем соответствующая уравнению (200).
Пример. Требуется вычислить минимальную работу изотермического расширения идеального газа в количестве 1 моль от давления 1 МПа до 0,1 МПа ПРИ температуре 0°С и количество теплоты, поступившей из внешней среды.
Решение.
w — —q =—8,314 Дж-моль-1-К-1-273 К-1 моль-2,303-lg 1/0,1 w = —q = —5,23 кДж/моль
Максимальная полезная работа равна 5,23 кДж, эквивалентное этой работе количество теплоты поступает из внешней среды.
Первый закон термодинамики
223
222
Химическая термодинамика
19.2.2. Изобарно-изотермические процессы
Многие процессы (фазовые превращения, некоторые химические реакции) протекают в изобарно-изотермических условиях в связи с чем необходимо выяснить, как изменяются в этих условиях w, q, и и h. Рассмотрим, например, испарение жид-кости при постоянном давлении, в процессе которого соверша-ется работа против внешнего давления-
dw=—pdv; w =—p(v2 — ux) (201)
При испарении жидкости всегда w2^>wi (обычно 1 моль жидкости занимает объем 20 мл, а 1 моль пара — около 20 л), так что можно пренебречь объемом жидкости щ по сравнению с объемом ее пара ц2. Предположив, что в газовой фазе, полученной при испарении жидкости, выполняются законы идеального газа, подставим в уравнение (201) вместо рц2 выражение nRT-
w= —nRT (202)
Точно так же можно рассчитать работу расширения для газовой реакции, при протекании которой меняется количество вещества (в молях), например для реакции разложения СаСО3.
Изменение энтальпии Д/г для таких процессов представляет собой теплоту испарения или теплоту реакции, а внутреннюю энергию можно определить из уравнения (194):
Au=A/i—рДц или Au=A/i— nRT (203)
Ди процесса испарения можно также назвать внутренней энергией испарения (затраченной на преодоление межмолекулярных сил), а энтальпия испарения включает также работу против внешнего давления.
Пример. Вычислить работу расширения при испарении воды в количестве 1 моль при 100 °C, а также внутреннюю энергию испарения. Энтальпия испарения Д//=40,66 кДж/моль.
Решение.
w = —1 моль-8,314 Дж• моль-1 К-1-373,3 К
w = —3103,6 Дж
u = h — nRT = 40,66 кДж — 3,1 кДж = 37,56 кДж
Работа расширения при испарении 1 моля воды равна 3,1 кДж/моль, внутренняя энергия испарения — 37,56 кДж/моль.
Адиабатические процессы
19.2.3.
Процессы, протекающие в изолированных системах, т. е. системах, которые не обмениваются теплотой и веществом с внешней средой, называю, адиабатическими. Такой процесс, например, можно себе представить прот<-кающим в сосуде Дьюара. Кроме того, процессы, в которых выделение теГт лоты происходит значительно быстрее, чем обмен с внешней средой за сче теплопередачи и излучения, также можно приближенно рассматр
аТь как адиабатические. В соответствии с принятым определением при 0диабатическом процессе AQ = 0; используя параметры состояния системы, из Равнения (190) следует, что d.U=d.W, а с учетом уравнений (189) и (193) p(ff=—pdV. p=RT[V, тогда Cv(dTIT) =—R(dV/V) и после интегрирования
“пределах от до Тг и от У] до У2 Со ln(T2/Ti) =— R ln(y2/yt). В соот-„еГствин с уравнением (176) R = CP—Cv; обозначим Cp/Cv = x, тогда ]n(7’2/7’i) = (1—х)1п(У2/У1). После интегрирования получим
т / У,
-TJT- = —п~ ИЛИ = const
(204)
Подставим в уравнение (204) T=pV)R
рУ’к const (205)
Уравнения (204) и (205) называют также уравнениями Пуассона. Как следует из кинетической теории газов (разд 2.1), значение х зависит от числа атомов в молекуле газа. Для одноатомного газа х= 1,66; для двухатомных —
Рис. Б 21. Идеальный газ в адиабатическом и изотермическом процессах. а — адиабата, б — взаимное расположение адиабаты и изотермы.
1,4, а для линейных и угловых трахатомных молекул — около 1, 33. Очевидно, в любом случае х больше единицы, что объясняет тот факт, что в координатах р—у при У<1 адиабата расположена выше изотермы, а при У>1— ниже ее, при У=1 изотерма и адиабата пересекаются (рис. Б.21).
Процессы, протекающие в природе, а также в аппаратах химических производств, никогда не бывают строго изотермическими или адиабатическими. Такие реальные процессы называют политропическими. Уравнения Пуассона (204) и (205) для них выполняются, однако вместо х показателем степени является константа политропы m (которая может принимать значения
Работа и равное ей изменение внутренней энергии адиабатического процесса для идеального газа (1 моль) равно
Д{/ = w = Cv (Т2 — TJ (206)
Из х=Ср/С„ и R = CP—Cv следует Cv = R/%—1. Подставив это выражение в ^Равнение (206), получим
A R (Т„ — Tt) P2V„ — р.У,
Д{/ = W = —х _ t = х _ t (207)
н Пример. Водород в количестве 2 моль при температуре 0°С и нормаль-м давлении адиабатически сжимают до объема 10 л. При каких давлении
224
Химическая термодинамика
и температуре находится газ после сжатия? Какая работа требуется для этого и каково изменение внутренней энергии? Хн2 = 1,41; Со=20,51 Дж. • моль-’-К-1.
Решение. 2 моля Н2 в заданных условиях занимают объем 44,8 л.
Рг = Pi = Ю1 325 Па- (44,8/10)1,41 = 841 000 Па
р2г2__________841 000 Па-Ю~2 м3________
2~ пД ~ 2 моль-8,314 м3-Па-моль-1-К-1'~ 505 К
Ди = Дш = 20,51 Дж-моль-1-К-1-2 моль-(505 — 273,3) К = 9,50 кДж
В результате адиабатического сжатия (совершения работы над системой) при заданных условиях давление увеличивается до 841 кПа, температура повышается до 232,3 °C, внутренняя энергия увеличивается на 9,71 кДж.
Вопросы и задания
1. При растворении металла в кислоте выделилось 35 л водорода (атмосферное давление). Какую работу совершает газ против внешнего давления?
2. 100 г жидкого бензола испаряется при температуре кипения (т. кпп.= =80,2 °C). Удельная энтальпия испарения при постоянном давлении составляет 395,1 Дж-г-1. Рассчитайте ш, q, ДИ и Ди. Является ли w максимальной работой?
3. Рассчитайте минимальную работу, необходимую для сжатия 20 г О2, занимающего объем 10 л, до объема 5 л при 0 °C. Потребляется ли системой теплота (энергия) или выделяется? В каком количестве?
4. Какова работа обратимого изотермического расширения, которая совершается при изменении объема 10 г СО2 от 5 до 10 л при 27 °C? Найти q, Ди и ДА для этой системы, если предположить, что СО2 ведет себя как идеальный газ.
5. 1 моль аргона при 25 °C и нормальном давлении расширяется до объема 50 л. Найти конечное давление для а) изотермического и б) адиабатического процессов.
6. 8 г О2 при 27 °C и при давлении 1 МПа обратимо адиабатически расширяются до давления 0,1 МПа. Какую температуру будет иметь газ по окончании процесса, какую работу совершит газ, если молярная теплоемкость О2 СР = ’/2Д?
7. 10 г N2 при температуре 17 °C в объеме 8 л обратимо и адиабатически сжимают до объема 5 л. Найти температуру газа по окончании процесса, затраченную работу, а также Ди и ДА (Ср=7/2Р).
20. ТЕРМОХИМИЯ 1
20.1. Энтальпия реакции, энтальпия образования *
химического соединения
Рассмотрим с точки зрения первого закона термодинамики химическую реакцию
v,\A vBB = vcC vdD (2C8)
Мольная внутренняя энергия исходных веществ равна tAicx^ = Ua+Ub, а мольная внутренняя энергия продуктов реакции Ukoh—Uc+Ud. Изменение внутренней энергии, связанное с пр0’ теканием химической реакцищ равно разности внутренней энер'
Термохимия
225
гИи конечных и исходных веществ. В соответствии с первым законом она равна тепловому эффекту реакции и работе расширения:
^U = UKOn-Un^Q-\-W (209)
Экспериментальное определение теплового эффекта реакции проводится в калориметрической бомбе* (ознакомьтесь с термохимическими методами по учебникам и практическим руководствам по физической химии). Поскольку объем калориметрической бомбы заранее известен и не меняется, можно измерить QV = AU, т. е. тепловой эффект при постоянном объеме, расчет энтальпии реакции при известном AU проводится при помощи уравнения (203):
\H=MJ±p\V= MJ+bvRT (210)
где Av — разность количества молей газообразных продуктов реакции и количества молей исходных веществ.
Рассмотрим реакцию
2Н2 (газ) + О2 (газ) — 2Н2О (жидк.) (211)
Энтальпия реакции при 298 К ДЯ298 =—571,88 кДж/моль. Внутреннюю энергию реакции можно вычислить по уравнению (189):
А Я298 = ДЯ298—&vRT = —571,66 кДж/моль -J-
-|-3-298К-8,314 Дж• моль-1 • К"1 =—564,23 кДж/моль
Поскольку U и Н —• функции состояния, изменения АЯ и ДЯ однозначно определяются лишь конечным и начальным состояниями системы, т. е. не зависят от пути процесса и способа осуществления реакции. Для реакции с точно известными исходными и конечными веществами не имеет значения, сразу ли получаются из исходных веществ конечные продукты или через промежуточные стадии (например, СО2 может образоваться непосредственно по реакции С + О2 = СО2 или через промежуточное соединение СО, которое затем окисляется до СОг). Эта закономерность играет чрезвычайно важную роль в термохимии и позволяет проводить расчет теплоты образования и теплового эффекта реакции из экспериментальных данных. Еще в 1840 г. Гессом был установлен закон, два следствия из которого наиболее важны для термохимии:
* Экспериментальное определение теплового эффекта в калориметрах Иожет проводиться как при v = const, так и при р—const; таким образом, Пожет непосредственно измеряться как Д17, так и ДЯ [Скуратов С. М., Кобсов В. И., Воробьев А. Ф. Термохимия. Т. 1. — М: Изд-во МГУ, 1964].— "Рюц. ред
•5—2027
226
Химическая термодинамика
а) Энтальпия реакции АНР равна сумме энтальпий образования продуктов реакции за вычетом суммы энтальпий образования исходных веществ
'\’^-Л/7^одр (212)
i
Стехиометрические коэффициенты конечных продуктов считаются положительными, а исходных — отрицательными. (Такое правило знаков используется в термодинамике и далее.)
б) Энтальпия любой химической реакции равна сумме энтальпий сгорания исходных веществ за вычетом суммы энтальпий сгорания продуктов реакции:
ДЯобр = -2^-ДЯ/сг (213)
i
Для практического применения закона Гесса необходимо знать стандартную энтальпию образования 1ХН°Т соединений и элементов. Очевидно, невозможно определить абсолютное значение энтальпии и внутренней энергии, поэтому оказалось необходимым выработать специальное соглашение* о правилах вычисления стандартной энтальпии образования АЯ°29з обр. Стандартная энтальпия элементов в стабильной модификации при 298,15 К принята равной нулю. Так, например, стандартная энтальпия образования Н2, О2, N2 и аналогичных двухатомных молекул принята равной нулю, в то же время энтальпия образования атомов Н, О и N не равна нулю, так как, для образования атомов из молекул необходимо затратить энергию. Стандартная энтальпия образования углерода также принята равной нулю для модификации углерода — графита при температуре 25°C** и нормальном давлении, а стандартная энтальпия образования алмаза равна 0,92 кДж/моль. На основе закона Гесса из энтальпий сгорания, энтальпий реакции или энтальпий растворения можно рассчитать и свести в таблицы стандартные энтальпии образования химических соединений. В таблицах также указано агрегатное состояние, в котором находятся эти соединения в стандартном состоянии (индекс внизу справа).
* Точнее, речь идет не о соглашении, а О четких правилах расчета энтальпии образования соединений из простых веществ. Естественно, энтальпия образования простого вещества «самого из себя» равна нулю [Скура" тов С. М., Колесов В. И., Воробьев А. Ф. Термохимия. Т. II. — М.: Изд-в0 МГУ, 1966]. — Прим. ред. .
** Стандартная энтальпия образования графита (как и любого дРУгоГ простого вещества, являющегося «нулем отсчета») равна в стандартном с стоянии нулю при любой температуре [Воробьев А. Ф. Теоретическая и эксп рименгальная химия, 8, 705—708, 1972]. — Прим. ред.
Термохимия
227
Пример. Расчет (по табличным данным) энтальпии реакции из стандартной энтальпии образования. Рассчитаем стандартную энтальпию реакции
3 С (тв.) 4-2 Fe2O3 (ТВ.) ч=£ 4 Fe (тв.) 4-3 С02 (газ) (214)
g таблицах находим следующие значения стандартной энтальпии образования веществ, участвующих в реакции:
АЯ’гэз (СО2) = —393,15 кДж/моль (Fe2O3) = —817,0 кДж/моль
Энтальпия образования С и Fe, как указано выше, равна нулю. Уравнения реакций образования СО2 и Fe2O3 необходимо записать с такими коэффициентами, чтобы они соответствовали коэффициентам при СО2 и Fe2O3 в уравнении (214):
С (тв.) + О2 (газ) —- СО2 (газ)
AH°W8 (СО2) = -393,43 кДж/моль (215)
2 Fe (тв ) -j- 1,5 О2 (газ) Fe2O3 (тв.)
А^°298 (Fe2O3) = —817,0 кДж/моль (216)
Эти уравнения реакций можно умножать и складывать, как любые алгебраические выражения. Умножим уравнения (215) на 3, а (216) на 2 и вычтем из второго первое. Легко убедиться, что в этом случае получим уравнение (214). Проведя те же математические операции со значениями энтальпии образования соответствующих веществ, получим энтальпию реакции
А^%э8Р — ЗА№298 (СО2) 2А№298 (Fe2O3) =
= —1180,29 кДж/моль + 1634,0 кДж/моль = 453,71 кДж/моль
Полученный результат хорошо согласуется с экспериментом. Уравнения реакций образования реагирующих веществ записаны здесь лишь для контроля; поскольку доказана справедливость закона Гесса, от этого можно отказаться и для вычислений использовать сразу уравнение (212).
Пример. Расчет энтальпии образования из энтальпии сгорания [уравнение (213)]. Часто невозможно определить энтальпию образования соединения непосредственно путем измерений этой величины, так как эта реакция либо вообще не может протекать, либо имеет очень небольшую скорость. В то же время теплоту сгорания соединения, в особенности органического, сравнительно легко измерить. Попробуем вычислить энтальпию образования уксусной кислоты из известных значений энтальпии сгорания элементов, из которых Она состоит:
2 С (тв ) 4- 2 Н2 (газ) 4- О2 (газ) ч—=^^СН3СООН (жидк.)
АД»298 (СН3СООН) = ? (217)
Известны
С (тв ) 4- О2 (газ) ч * СО2 (газ) АД°298 (СО2) = —393,45 кДж/моль (218) Н2 (газ) 4- 0,5 О2 (газ) ч~*** Н2О (жидк.)]
АД°298 (Н2О) =—285,28 кДж/моль (219)
СН3СООН (жидк.) 4- 2 О2 (газ) ч —> 2 СО2 (газ) 4- Н2О (жидк.)
А№298 сг (CHgCQQH) = —870,14 кДж/моль (220)
Умножим уравнения (218) и (219) на 2, сложим результат и из полученной УМмы вычтем уравнение (220), в результате чего получим уравнение (217).
15*
228
Химическая термодинамика
Такие же действия проведем с энтальпиями сгорания:
ДЯ°298 (СН3СООН) = 2Д//о298 (СО2) + 2А/7°298 (Н2О) - ДЯ»298 (СНзСООН) = = —786,86 кДж/моль — 571,90 кДж/моль—(—870,14 кДж/моль) =
= —488,62 кДж/моль
В этом случае также можно отказаться от написания отдельных уравнений реакций сгорания и использовать уравнение (213).
Наряду с только что рассмотренными примерами известны и другие возможности комбинирования уравнений реакции с использованием закона Гесса. В этом читатель может убедиться, решая предлагаемые в конце главы задачи. В растворах энтальпия образования соединений и энтальпия реакций также определяются на основе первого закона термодинамики. При этом энтальпия реакции характеризует только взаимодействие компонентов раствора, вступающих в химическую реакцию. Этим объясняется тот факт, что энтальпия нейтрализации всех сильных кислот сильными основаниями одинакова* и равнг —57,8 кДж/моль, так как в любых реакциях нейтрализации всегда идет одна и та же реакция:
Н+ (р-р) + ОН' (р-р) = Н2О (жида.)
20.2. Температурная зависимость энтальпии реакции [закон Кирхгофа]
Зависимость энтальпии реакции и энтальпии образования химических соединений можно получить из уравнений, определяющих теплоемкость веществ [уравнение (193) или (194)]. Мольную теплоемкость газов можно оценить на основе данного в разд. 2.1 молекулярно-кинетического истолкования сущность мольной теплоемкости. По мере приближения температуры к абсолютному нулю мольная теплоемкость стремится к нулю пропорционально температуре в третьей степени (Г3) в отличие от мольной энтальпии, которая имеет конечное значение. Рассмотрим снова обобщенное уравнение химической реакции (208). Согласно уравнению (212), энтальпия реакции следующим образом складывается из энтальпий образования ингредиентов реакции:
АЯр° = АЯс° + ДЯо°— ДЯА°-ДЯВ° (221'
Температурная зависимость энтальпии каждого вещества, уча ствующего в реакции, в соответствии с уравнением (196), равн-
ЛД/УР __с z222
dT ~^pW
* Это верно только при бесконечном разбавлении растворов [Воро^' ев А. Ф. В сб. Современные проблемы физической химии. Т. 6, 165—192. М.: Изд-во МГУ, 1972]. — Прим. ред.
Термохимия
229
Отсюда температурная зависимость энтальпии реакции складывается из температурных зависимостей энтальпий отдельных веществ [аналогично уравнению (212)]:
•^'-=^„=2 v,Cp,„ (223)
i
Уравнение (223) представляет собой закон Кирхгофа в дифференциальной форме. Для практического расчета энтальпии реакции при любой температуре Т необходимо вывести закон Кирхгофа в интегральной форме, а также знать энтальпию при некоторой заданной температуре. Обычно известна стандартная энтальпия образования при стандартной температуре, а требуется определить ее значение при любой другой температуре. Из уравнения (223) следует
т
J = ^bCpdT (224)
//«298 298
Если предположить, что Ср в данном температурном интервале постоянна, то
ДД0Гр = ДЯ°298р + ДСр (Т - 298) (225)
Уравнение (225) представляет собой закон Кирхгофа в интегральной форме. Очевидно, что температурная зависимость энтальпии для газовых реакций, в уравнении реакции которых по обе стороны знака равенства находятся одинаковые молярные количества газов, очень невелика, так как молярная теплоемкость газов почти не зависит от природы газа. В то же время для реакций, в которых образуется или расходуется газообразное вещество, можно ожидать существенную зависимость энтальпии от температуры.
Пример. Рассчитать стандартную энтальпию образования аммиака при 500 °C, не учитывая зависимость теплоемкости от температуры, CP(N2) = =29,13 Дж-моль-1-К-1, СР(Н2) =28,88 Дж-моль-1-К-1, CP(NH3) = =35,58 Дж-моль-1-К-1, ДЯ°2эз(NH3) ——46,04 кДж/моль.
Решение. N2 + 3H2 = 2NH3.
С помощью уравнения (212) можно найти стандартную энтальпию реакции при нормальных условиях
А#°298Р = —92,08 кДж/моль
ДСр = (2-35,58 — 3-38,88 — 1-29,13) = —44,62 Дж/(К-моль) Подставив полученные значения в уравнение (225), получим
ДЯ°77з(МН3) = — ( 92,08— (773—298)J = — 113,42 кДж/моль
1аким образом, энтальпия реакции образования аммиака при 500 ®С на £’84 кДж меньше, чем при 25 °C.
230
Химическая термодинамика
Способы выражения концентрации
231
Для более точного расчета энтальпии необходимо принимать во внима. иие зависимость молярной теплоемкости от температуры. Обычно ее пред, ставляют в виде степенного ряда
Ср = а+ЬГ + сГ (226)
Если необходимо учесть температурную зависимость энтальпии реакции, то в уравнение (224) следует подставить ДСр = Да+Д67'+Дс7'2. Тогда после интегрирования получим
ДЯ°г = ДЯ°298Р+\а (Т— 298)+(Г2— 2982) +
^_А^(Гз_ 2983)
(227)
Если необходимо провести интегрирование в более широком температурном интервале, то может оказаться, что в этой области температуры происходят фазовые превращения. С последними связано соответствующее изменение энтальпии, которое не учитывается при интегрировании [уравнение (227)]. В таких случаях интегрирование надо проводить в интервале температур, в которых не происходит фазового превращения.
гф Т
АН°т = ДД°2э8 + (ж) dT + Д№ф + j ХСр (Газ) dT (228)
298 Лф
здесь Гф—температура фазового превращения, например температура кипения, а ДДф° — энтальпия фазового превращения.
Вопросы и задания
1. Стандартная энтальпия сгорания нафталина (Л4= 128,16) составляет 5155,6 кДж/моль. Рассчитайте теплоемкость калориметра, если при сгорании 0,55 г нафталина температура повысилась на 2,05 °C.
2. Рассчитайте энтальпию сгорания органического соединения, если при сгорании 1,52 г этого соединения температура калориметра с общей теплоемкостью, найденной в задаче I, поднимается на 1,845 °C.
3. Энтальпия сгорания н-бутана ДА+э^сДн-С'.Нц) =—2879,1 кДж/моль Определите стандартную энтальпию и энергию образования н-бутана в нормальных условиях; ДД°298(Н2О)——242,08 кДж/моль, ДЯ°298 (СО2) == ——393,15 кДж/моль.
4. Рассчитайте стандартную энтальпию образования AgCl в нормальны' условиях, принимая во внимание следующие данные:
Ag2Q (тв ) + НС/ (газ) :₽=£ 2 AgCl (та.) 4- Н2О (жидк.)
ДЛ*»298Р = —326,88 кДж/моль
2 Ag (тв ) + 72О2 (газ) <=> Ag2O (тв.) А№298Р = —28,88 кДж/моль
1/2Н2 (газ) + 1/2С12 (газ) ч-- т» НС1 (газ) Д/7°298Р = —92,00 кДж/моль
Н2 (газ) + Х/2О2 (газ) Н2О (жидк.)
ДДо298Р = —285,28 кДж/моЛ
5. Рассчитайте, пользуясь таблицами стандартных ния, стандартные энтальпии следующих реакций:
a) Fe2O3 (тв.) + СО (газ) *= СО2 (газ) + 2 FeO (тв )
б) 2 NO2 (газ) 2 NO (газ) + О2 (газ)
энтальпий образов
в) ЗС2Н2(газ) > С6Н„ (жидк.)
г) HgO (тв.) + 2 НС1 (газ) Н2О (жидк.) + HgCI2 (тв.)
6. В каких случаях можно ожидать наиболее значительный тепловой эффект при смешивании разбавленных водных растворов ряда соединений в следующих сочетаниях:
a) CH3COONa + HCl
б) KCl + MgSO4
в) NI14С1 + Ва(ОН)2
г) СаС12 + Д2СО3
150 мл 0,4 н. раствора (0,4 моль/л) НС1 нейтрализуют избытком рас-аммиака в сосуде Дьюара. При этом температура повысилась на
Ряггиитайто ткгчттт- --- ---«•
«ГВОра ---Г“. “8“ -- ---
2,36 °C. Рассчитайте мольную энтальпию нейтрализации, если теплоемкость сосуда Дьюара равна 1318 Дж/Д.
8. Безводный Na2CO3 растворен в большом объеме воды. При этом выделилось 23,0 кДж/моль теплоты. Стандартная энтальпия образования Na2CO3 равна —1134,2 кДж/моль. Найдите стандартную энтальпию образования Na2CO3 в разбавленном растворе.
9. Из энтальпии сгорания графита до СО2 при 25 °C (—393,15 кДж/моль) и энтальпии сгорания Н2 до воды при 25 °C (—285,83 кДж/моль) рассчитайте соответствующие энтальпии сгорания при температурах 18 и 40 °C. Даны средние молярные теплоемкости в данном температурном интервале: Ср(СО2) =38,38 Дж-моль-1-К-1, СР(О2)=29,51 Дж-моль-*-Д-*, СДН2О) = =75,42 Дж • моль-*-Д-1, СР(Н2) =28,96 Дж-моль-2-Д-*, СР(С)=9,54 Дж-• моль"1-К-1.
10. Энтальпия гидрирования этилена до этана при 83°C составляет —138,2 ДДж-моль-1-Д_|. Рассчитайте энтальпию гидрирования при 25°С, используя следующие значения теплоемкости: СР(Н2) =28,96 Дж-моль-*-Д-1, Ср(С2Н6) =56,46 Дж-моль-*-Д-*, СР(С2Н4) =46,71 Дж-моль-*-Д-‘.
11. Рассчитайте энтальпию сгорания моноксида углерода при температуре 1000°С 2СО(газ)+О2(газ) =2СО2(газ), если ДАГ0298 р=—558,38 кДж/моль, а зависимость Ср (Дж-моль-*-Д-*) описывается следующими уравнениями:
Ср (СО) = (27,62+ 5,02-10-зТ)]
Ср (СО2) = (29,3 + 29,7- 10-3Т — 7,78- 10~5 672) Ср (О2) = (28,29+ 2,54- 10-3Т + 0,54- 10-вГ2)
2’- СПОСОБЫ ВЫРАЖЕНИЯ КОНЦЕНТРАЦИИ
До сих пор обсуждалась зависимость физико-химических функций состояния от объема и температуры или от давления и температуры. Еще одной переменной, необходимой для изучения физико-химических свойств смешанных /аз> является концентрация. Рассмотрим способы выражения концентрации (единицы измерения концентрации) (см. также гл. 17). Целесообразно разделить единицы концентрации в смешанных фазах на две группы, из которых ем°Дной рассматриваются соотношения масс, а в другой — соотношения объ-
К первой группе относятся часто встречающиеся массовые доли и мас-вые проценты, удобные для применения во многих химических расчетах ПоЛьные доли и мольные проценты, а также часто используемую моляль-которая указывает число молей растворенного вещества на 1 кг рас-па°РИтеля. До второй группе относятся выражения концентрации в виде РЦиального давления, а также применяемая для разбавленных растворов
232
Химическая термодинамика
молярность, которая указывает число молей растворенного вещества в 1 -раствора.
Как легко показать, единицы концентрации второй группы в отличие первой зависят от давления и температуры. Для пересчета одних едиг концентрации в другие полезно располагать готовыми формулами. Пусть д мольная доля, с' — моляльность, с — молярность, ш— масса (в грамма । М — молярная масса, Р (%) — содержание в процентах (массовые процент;о Величины, относящиеся к растворенному веществу, будут иметь индо с 2, а величины, относящиеся к растворителю, — индекс 1.
Массовые проценты (процентная концентрация):
Мольные доли:
«2 tn2/M2 (рЩу.
Ъ = П1 _|_ П2 - mj/Mi + m2/M2
ni
Xi~
(231)
(232)
Для бинарной смеси, как следует из уравнения (230), xi+xj 1.
В соответствии с законом Дальтона общий объем смеси идеальных га <ов равен сумме парциальных объемов Vi, а общее давление равно сумме пар циальных давлений рр
f = P = JjPl
Применяя уравнение состояния идеальных газов, получим
Vi Pi
Xl~ ^i ~ % Pi
Если содержание в мольных долях умножить на 100, то получим содер жание в мольных процентах, а при умножении на 100 выражения (232) — со держание в объемных процентах.
Моляльность:
<*’ = M2.(m?+^j'1000 (МОЛЬ/КГ) <233)
Если р — плотность смеси, то смесь (раствор) массой /щ+тг-Ь— нимает объем mi + тг + .../р мл.
Молярность:
с=-тг~/—-----------;• 1000 моль/л (23
М2 (mi + m2 + ...)
Так как в уравнение (234) входит плотность, эта единица концентрации висит от давления и температуры. , м
Пример. 24%-ный (по массе) раствор карбоната калия (K2CU3) и при 20°C плотность р= 1,232 г/мл (или кг/л). Вычислить концентрацию F твора в молях на литр (молярность), в молях на килограмм раствор (моляльность) и в мольных долях.
Решение. Из формул (229) и (234) следует
С_АТ.1ЮО= =2,14 моль/л
/И 2 1ОО,.£
_______________________Второй закон термодинамики 233
Из формулы (229) и (233) следует
_Р2-1000 0,24-1000
С ~ М2’(1 — Р2) ~ 138,2-0,76 = 2,284 моль/кг
Из формул (229) и (230) следует
240/138,2
<К2СО3 — 240/138,2 + 760/18,20 ~ ° ’0396
Вопросы и задания
1. Плотность водного раствора, содержащего 8 г NaCl в 100 г раствора, составляет 1,0541 г/мл при 25 °C. Рассчитайте молярность, моляльность Й мольную долю NaCl.
2. Растворением 22,5 г Na2CO3-10H2O в воде получен раствор Na2CO3 в объеме 200 мл. Плотность полученного раствора равна 1,04 г/мл. Рассчитайте молярность, моляльность, а также содержание Na2CO3 в массовых и молярных процентах.
3. Газовая смесь состоит из 15% (об.) Н2, 10% (об.) СО и 75% (об.) N2. Рассчитайте мольную долю газов, а также их содержание в массовых процентах.
4. Плотность, 60%-ного раствора фосфорной кислоты равна 1,426 г/мл. а) Сколько молей Н3РО4 содержит 1 л раствора? б) Сколько молей Н3РО4 приходится на 1000 г растворителя? в) Вычислите мольную долю кислоты в растворе.
22. ВТОРОЙ ЗАКОН ТЕРМОДИНАМИКИ
22.1. Энтропия как функция состояния
Второй закон термодинамики рассматривает вопрос о движущей силе всех совершающихся в природе самопроизвольных процессов. Первый закон термодинамики не затрагивает этого вопроса. В прошлом веке за меру движущей силы реакции принимали тепловой эффект реакции. Томсон и Бертло считали, что самопроизвольно протекают только экзотермические реакции, а эндотермические, как правило, не являются самопроизвольными. Однако этому противоречило существование самопроизвольно протекающих, но в то же время эндотермических процессов Растворения многих веществ, а также многих равновесных про-Чессов, степень превращения в которых соизмеримы в прямом 11 обратном направлениях (если в прямом направлении идет экзотермическая реакция, то при установлении равновесия должен протекать и обратный процесс и в соответствии с первым законом термодинамики обратная реакция должна быть эндотермической) .
В своей первоначальной формулировке второй закон термодинамики утверждает, что имеются некоторые мыслимые провесы, не противоречащие первому закону термодинамики, ко-т°рые не могут происходить в природе. Так, например, невоз-°исно, чтобы теплота самопроизвольно переходила от менее
234
Химическая термодинамика
нагретого тела к более нагретому телу без каких-либо измене-ний в других телах. Точно так же невозможно, чтобы при охлаждении тела за счет его теплоты совершалась работа против внешних сил (например, поднимался какой-либо груз) без ю-менений во внешних телах. Эти, а также некоторые другие формулировки второго закона термодинамики были предложены Клаузиусом, Томсоном и Планком. Второй закон термодинамики можно выразить в математической форме в виде уравнений. (Подробное описание метода, с помощью которого из приве. денных выше формулировок (постулатов Клаузиуса и Томсона) можно вывести выражения для энтропии и ее соотношения с другими термодинамическими функциями, можно найти в курсах физики и физической химии). Одно из следствий второю закона заключается в том, что смешение двух различных газов, О2 и N2, протекает самопроизвольно, а их разделение те является самопроизвольным процессом.
Второй закон термодинамики вводит новую функцию состояния— энтропию. Это экстенсивная величина; она обозначается буквой 5 для 1-го моля вещества, и s — для любого количества вещества (разд. 18.2). Второй закон термодинамики дает количественное выражение изменения энтропии AS. В замкнутых системах (разд. 19.1) энтропия может меняться двояким образом. Энтропия системы уменьшается, если поток энтропии направлен из системы, и, наоборот, увеличивается при поступлении энтропии в систему извне. Такой тип изменения энтропии называют потоком энтропии. Не касаясь математической формулировки энтропии, полученной из постулатов второго закона термодинамики, можно сделать вывод о том, что поток энтропии пропорционален потоку теплоты dQ, а именно dQ]T. Другой тип изменения энтропии наблюдается, если в системе происходят необратимые процессы. В этом случае энтропия может только увеличиваться (возникновение энтропии). Запишем возникновение энтропии в виде dl/T-, di всегда положительно. Тогда можно записать второй закон термодинамики в следующем виде:
(235)
Иногда дают неверную формулировку второго закона, учитывая лишь первое слагаемое уравнения (235), т. е. dS~dQ’T-Это выражение справедливо лишь для обратимых процессов. В то же время известно утверждение, что энтропия может лишь увеличиваться; это верно только для адиабатических процессов, когда dQ = O. Часто без всякой необходимости сужают формулировку второго закона, полагая, что в изолированных системах dS^>0. Это является неоправданным ограничением, так как преобразование энергии в работу не влияет на энтропию: по
Второй закон термодинамики
235
ток энтропии определяется только потоком теплоты. Из уравнения (235) следует, что различные процессы (рассмотрим шесть случаев) сопряжены со следующими изменениями энтропии:
1) адиабатический обратимый dS = 0
2) адиабатический необратимый dS^>0
3) эндотермический обратимый dS>0
4) эндотермический необратимый dS>0
5) экзотермический обратимый t/S<0
6) экзотермический необратимый dSSO
Для последнего случая нельзя сказать заранее, что будет наблюдаться — увеличение или уменьшение энтропии. Примером такого процесса может служить кристаллизация переохлажденной жидкости. В этом процессе (поскольку выделяется теплота плавления) поток энтропии направлен из системы, вследствие чего энтропия понижается. В то же время процесс идет необратимо, т. е. возникает энтропия. Как правило, преобладает поток энтропии во внешнюю среду.)
Таким образом, изменение энтропии в системе является критерием обратимости протекающего процесса. В основном все процессы в природе протекают необратимо, т. е. с возникновением энтропии. Обратимые процессы являются предельным случаем реальных процессов, если представить их как протекающие бесконечно медленно. Несмотря на это, как мы увидим в дальнейшем, имеется возможность исследования необратимых процессов методами равновесной термодинамики, если мысленно представить необратимый процесс как последовательность обратимых процессов. z
С помощью введенных понятий и представлений можно подойти к некоторым весьма важным выводам о направлении изменения энтропии в конкретных процессах. При этом необходимо подчеркнуть, что здесь не было дано развернутого определения энтропии. Постепенно, по мере знакомства с ее свойствами, станет ясна и сущность энтропии. Рассмотрим сначала изменение энтропии в изотермических процессах, а также зависимость энтропии от температуры.
22.1.1 , Изменение энтропии в изотермических процессах
Рассмотрим процесс изобарно-изотермического превращения, а также изотермическое расширение и сжатие, которые Уже обсуждались в гл. 19. Примером такого фазового превращения может служить испарение жидкости, которое протекает практически обратимо. При испарении энтропия увеличивается, так как в систему поступает теплота (с. 235, случай 3). Теплота, которая необходима для испарения одного моля жидкости При постоянных давлении . и температуре, называется мольной теплотой испарения. Она равна разности энтальпий жидкости и
236
Химическая термодинамика
ее пара. Если сравнить теплоты испарения различных соединений (воспользуемся таблицами), то можно заметить, что они сильно отличаются друг от друга. Однако если вычислить энтропию испарения этих соединений
Л5ИСП = ^=- (236)
'(где Т — температура испарения при нормальном давлении), • оказывается, что энтропия испарения большинства химичесю соединений равна ~88 Дж• моль-1 К-1 (правило Пикте — Тр тона). Точно так же при конденсации соединений энтропия для температуры кипения уменьшается на те же 88 Дж• моль-1 • К-1 (с. 235, случай 5). Ассоциированные жидкости, такие, как вода (имеющие большой дипольный момент), отклоняются от правила Пикте — Трутона; энтропия испарения для них имеет более высокое значение (для воды ~110 Дж-моль-1-К-1). Энтропия плавления, т. е. теплота плавления, деленная на температуру плавления, также приближенно постоянна (правило Ричарда), хотя интервал, соответствующий изменениям энтропии плавления, несколько шире, чем интервал изменений энтропий испарения. Энтропия плавления сильно зависит от структуры молекулы: для сферических молекул она может меняться от 12 до 88 Дж-моль-1 К-1-
Теперь рассмотрим изменение энтропии при изотермическом расширении или сжатии газа. В гл. 19 показано, что уменьшение внутренней энергии при ее превращении в работу компенсируется притоком теплоты извне [уравнение (200)]. Поэтому д = =n/?Tln(v2/Vi) и
As = nR In (vjvi) или AS = 7?ln(V2/V1) (237)
При расширении идеального газа в количестве 1 моль до двукратного объема энтропия увеличивается на 5,8 Дж-моль-1-•К-1. При испарении объем возрастает в ~ 1000 раз, что соответствует увеличению энтропии на 57,5 Дж-моль-1-К-1. Однако последнее меньше, чем требует правило Пикте — Трутона; причины такого отклонения будут выяснены далее. То обстоятельство, что энтропия плавления значительно ниже, чем энтропия испарения, связано с тем, что при плавлении почти не происходит изменения объема.
22.1.2 . Зависимость энтропии от параметров состояния системы (р, v и Т)
Для того чтобы установить соотношения между энтропией, введенной на основе второго закона термодинамики, и параметрами состояния системы, необходимо, сделав некоторые допу
Второй закон термодинамики 237
щения, исследовать математические свойства функций
S = /(V,T) (a) S — f(p,T) (б) (238)
Запишем выражение полного дифференциала энтропии [уравнение (170)]:
dS=(-w)TdV + ^)vdT
(239)
(б)
Для того чтобы найти частные производные в уравнении (239а), обратимся к первому закону термодинамики [уравнение (191)]:
dQ = dU±pdV (240)
Для идеального газа [согласно второму закону Гей-Люссака, уравнение (198)] dU=CvdT и p=RTIV, тогда
dQ = CvdT-]r~-dV (241)
Если применить правило Шварца для полного дифференциала dQ [уравнение (174)], то окажется, что dQ не удовлетворяет условиям полного дифференциала
т. е. теплота dQ не является функцией состояния. Дифференциалы, не являющиеся полными дифференциалами, часто можно перевести в полный дифференциал умножением на соответствующий коэффициент — интегрирующий множитель (см. руководства по математике). Можно показать, что интегрирующим множителем для dQ является величина 1/Т:
-^-=-^-dT+-^dV = dS (242)
Легко показать, что для этого выражения выполняется правило Шварца
д / Cv \ _ д / R \ л____«
dV \ Т дТ \V )т’ и
Таким образом, dQIT представляет собой дифференциал функции состояния, идентичный дифференциалу энтропии [уравне-Ние (235)] обратимого процесса, введенной на основе второго закона термодинамики (т. е. для dI/T=0).
238
Химическая термодинамика
Точно таким же образом, используя уравнения (1У0) и (196), можно получить дифференциальные уравнения зависимости энтропии от давления:
dS=-^-dT+-^-dp
(243)
Сопоставляя уравнения (242) и (239а), соответственно (243) и (2396) и приравняв коэффициенты при dp и dT, получим следующие выражения для производных энтропии по температуре, давлению и объему:
( dS \ Cv [ dS \ _ Ср
\ W )v~ Т ’ \дТ /р Т
/ dS \ _/ др \ / dS \ ___/ 0V \
(243а)
(2436)
Следовательно, при повышении температуры и постоянном объеме или давлении энтропия увеличивается. При повышении дав-
ления, как правило, энтропия уменьшается, а при
увеличении
объема — увеличивается (этому правилу не подчиняется изменение энтропии воды в интервале 0 — 4 °C).
Аналитические выражения функций (238а) и (2386) можно получить интегрированием уравнений (242) и (243) [см. разд. 18.1, уравнения (177) — (179) и примеры 1 и 2]. Интегрируя в пределах от исходного состояния 1 до конечного 2, получим
AS = Cv In (TJTJ + R In (V2/Vj)
(244а)
AS = Cp In (T2/7\) - R In (p2/P1) (2446)
Уже известное нам уравнение (237) оказывается частным случаем более общего уравнения (244а) при Т = const. Кроме того, из уравнений (244а) и (2446) можно определить зависимость энтропии от температуры для изохорного и изобарного процесса (n = oonst, P = const):
Ta
As ~ S2 S1 = Ct,(p)-ln (245)
Пример. 1 моль переохлажденной воды изотермически замерзает при температуре —10 °C. Рассчитать изменение энтропии системы, изменение энтропии внешней среды, а также составить суммарный баланс изменений энтропии в системе и внешней среде.
Решение. В данном случае происходит необратимый процесс (лед при —10°C не может самопроизвольно растаять). Как уже упоминалось, изменение функции состояния для необратимого процесса можно вычислить, разделив его на ряд обратимых стадий. Рассмотрим следующие стадии и с помощью уравнения (245) найдем изменения энтропии:
Н2О (жидк. при —10°C) ----->- Н2О (жидк. при 0°С)
Н2О (жидк. при 0°C) ----► Н2О (тв. при 0°С)
Ло _г , 273
‘'Л — ьжндк. ш 2gg
Ь2С (тв. при О °C) ------------»- Н2О (тв. при —10 °C)
* о „ , 263
AS3 — Ств. In 273
AS = -£ 2 7
Второй закон термодинамики
239
Теплота плавления при О °C равна 334,8 Дж/г;
удельная теплоемкость воды 4,185 Дж-г-1-К-1;
удельная теплоемкость льда 2,093 Дж-г-1-К"1.
Суммарное изменение энтропии системы складывается из изменений энтропии отдельных обратимых стадий:
273
Д5== 18 г/моль-4,185 Дж-г-1 -К-1 -2,303 1g- +
Zoo
18 г/моль-(—334,8 Дж/г) 263
-J- 273К г/моль-2,093 Дж-г 1 -2,303 1g 273 =
273К
= 2,8 —22,1 — 1,42 = —20,72 Дж моль-1-К’1
Д5=—20,72 Дж-моль-1-К-1 (такое изменение энтропии соответствует случаю 6 в гл. 22.1).
Для расчета изменения энтропии внешней среды следует предположить, что вода находится в тепловом контакте с большим резервуаром, имеющим температуру —10 °C. При замерзании воды происходит выделение теплоты (теплота плавления при —10°С равна 313,9 Дж/г), которая поглощается резервуаром без заметного изменения его температуры. Поэтому изменение энтропии внешней среды составляет
АЗвн.среда —
18 г/моль-313,9 Дж-г-1-К-1 263К
= 21,5 Дж • моль-1 К-1
Если рассматривать воду и внешнюю среду как единую систему, то суммарное изменение энтропии в этой системе составляет
ASo6m == “1“ ASbh.среда = 6,78 Дж • моль 1 * К 1
С помощью уравнения (235) можно вычислить лишь изменение энтропии, и нельзя сделать никаких выводов о ее абсолютном значении. На основе измерений теплового эффекта реакций при постепенном понижении температуры Нернст установил так называемый тепловой закон (который рассматривают также как третий закон термодинамики); по мере приближения температуры к абсолютному нулю изменение энтропии стремится к нулю. Справедливость теплового закона достоверно подтверждена на опыте. Планк предложил считать энтропию любого вещества при абсолютном нуле равной нулю. Тем самым открывается возможность точно рассчитать энтропию любого вещества при любых температуре и давлении, воспользовавшись Уравнениями (244а) и (2446). Например, рассмотрим изменение энтропии воды в зависимости от температуры при постоянном Давлении (рис. Б.22). При абсолютном нуле энтропия льда в соответствии с тепловым законом Нернста равна нулю. При возрастании температуры энтропия изменяется пропорционально Т3, при дальнейшем повышении температуры обнаруживается более сложная зависимость от Т. В точке плавления энтропия скачкообразно увеличивается на величину энтропии плавления. В интервале 0—100°С энтропия снова непрерывно увеличивается, а при 100 °C обнаруживает скачок, равный энтропии испарения. При температуре ~100°С энтропия пара постелен-
240
Химическая термодинамика
но увеличивается. Такой ход изменения энтропии характерен для большинства веществ. В физико-химических справочниках можно найти значения энтропии для различных веществ при 25 °C и нормальном давлении.
Статистическая термодинамика дает более глубокое истолкование понятия энтропии. Методами статистической термодинамики было выведено уравнение Больцмана, которое связывает энтропию с термодинамической вероятностью W состояния (разд. 27.1)
S = 61nIF (246)
где k — константа Больцмана, W— число микросостояний, соответствующих одному макросостоянию. Под макросостоянием
в
Рис. Б.22. Изменения энтропии воды зависимости от температуры.
следует понимать обычное термодинамическое состояние системы, определяемое параметрами Т, р, v, п. Число микросостояний — это число возможных распределений частиц по группам, имеющим различные значения скорости и энергии. Очевидно, что одно и то же макросостояние системы соответствует огромному числу микросостояний. Статистическая механика разработала методы расчета этого числа W и тем самым в конечном счете методы расчета энтропии вещества при заданном состоянии. В соответствии со вторым законом термодинамики в изолированных адиабатических системах при протекании необратимых процессов энтропия увеличивается и стремится к максимальному значению. Если, на
пример, молекулы газа занимают лишь половину предназначенного для них пространства, то в таком состоянии W не достигает своего максимального значения даже в том случае, если молекулы равномерно распределятся в пространстве. Это можно сформулировать следующим образом: все термодинамические системы стремятся достигнуть состояния наибольшего разупоря-дочения. Это стремление проявляется во все большей степени по мере увеличения температуры. При достаточно высокой температуре газа его молекулы начинают ионизироваться и раз-упорядочение в системе увеличивается. При еще большем повышении температуры сложные молекулы распадаются на бол< простые, наконец, газ превращается в плазму — смесь ионов > электронов. При температуре, господствующей внутри звез^ (~107К), вообще не существует таких частиц, как молекул^
Второй закон термодинамики 241
й атомы, есть только электроны и ядра, причем последние начинают уже распадаться на протоны и нейтроны. Все это является одним из проявлений второго закона термодинамики в смысле увеличения числа микросостояний W и снижения упорядоченности системы при распаде каждой структурной единицы материи на атомные и элементарные частицы. Таким образом, становится понятным различие между энтропией испарения, рассчитанной по уравнению (236) и равной 88 Дж-моль-1-• К-1, и энтропией объемного расширения, возникающей при увеличении объема жидкости при ее испарении [рассчитанной по уравнению (237) и равной 59,0 Дж-моль-1-К-1]. Разность этих величин составляет 29 Дж-моль-1 -К"1- Испарение жидкости соответствует переходу от квазикристаллической структуры жидкости к полностью разупорядоченному состоянию газа. Эти представления согласуются и с тем, что энтропия плавления составляет лишь примерно 21 Дж-моль-1-К-1, что соответствует переходу кристаллического вещества в жидкое состояние. То, что энтропия плавления меньше, чем указанное выше значение 29 Дж-моль-1 • К-1, является доказательством того, что жидкость по своей структуре ближе к твердому телу, чем к газу.
Все рассмотренные выше термодинамические соотношения, раскрывающие смысл второго закона термодинамики, относятся к замкнутым системам. В открытых системах энтропия может изменяться в результате обмена вещества с внешней средой. Тогда в уравнении (235) появится дополнительный член, учитывающий изменение количества вещества (числа молей) в системе. Более подробно этот вопрос не будет здесь обсуждаться; следует лишь упомянуть о том, что изучение открытых систем открывает возможность для применения второго закона термодинамики к живым организмам. Ранее вызывала сомнение сама возможность применения второго закона термодинамики к живым организмам, поскольку такие системы характеризуются сложными процессами (из почти бесструктурной клетки развивается сложно организованная система), связанными с понижением энтропии. В то же время в организме постоянно происходят необратимые процессы, вызывающие увеличение энтропии. Частично энтропия может передаваться во внешнюю среду в процессе теплообмена, в большей степени она переходит во внешнюю среду при обмене веществ.
Если принять нижний предел интегрирования уравнения (243) постоянным, а верхний — переменным (что равносильно постоянному исходному состоянию системы), то зависимость энтропии S от р и Т можно выразить следующим образом:
S = S0-i-Cpln-^-—Д1п — (247)
•о Ро
16—2027
242
Химическая термодинамика
rjpe. So — энтропия при некоторой температуре (обычно Т = = 298 К) и при давлении р0. В справочниках приведены значения энтропии для давления р0 = Ю1 325 Па. Зависимость энтропии, а также и других термодинамических функций (разд. 22.3 и 23.2) от давления целесообразно представить, используя относительное давление р° = р/(101325 Па), т. е. безразмерный параметр. Если ввести относительное давление р°, энтропию идеального газа можно представить в виде суммы двух членов, один из которых зависит только от температуры, а другой — от относительного давления (где S°T — стандартная энтропия):
S = Sf>T—R\nf/> (248)
s = s°r(i) —/?1пр°г (248а)
Рис. Б.23. к объяснению возникновения энтропии смешения N2 и О2.
В последнем уравнении вместо относительного давления р° всей системы стоит относительное парциальное давление pi0, pi° = =хгр° (где Хг — содержание i-ro компонента в мольных долях), следовательно,
S. = S°T(i) — /?1пр° — /?1пх£ (249)
Последнее выражение дает зависимость энтропии от температуры, давления и состава системы. Уравнения (247) и (249) записаны для идеального газа в количестве 1 моль. Для п молей следует энтропию обозначить буквой s и ввести в правую часть уравнения множитель п. С помощью уравнения (249) можно рассчитать изменение энтропии для процесса самопроизвольного смешения двух разных газов. Пусть имеется сосуд, в котором находятся два газа, имеющие одинаковые температуру и давление, каждый в количестве 1 моль (рис. Б.23). Смешение газов происходит после удаления разделяющей их перегородки. Общее давление и температура при этом не меняются, однако парциальное давление каждого газа уменьшается в два раза. Энтропия при этом уве личивается на 27?-1п2 (энтропия смешения). Энтропия смешения не зависит заметным образом от природы смешивающихся газов. Однако, если по обе стороны перегородки находился один и тот же газ, то энтропия не меняется (парадокс Гиббса)-Причиной увеличения энтропии при смешении в адиабатических условиях может быть только возникновение энтропии в результате необратимого процесса смешения газов.
Второй закон термодинамики
243
22.2. Другие функции состояния, введенные на основе второго закона термодинамики (максимальная полезная работа)
Утверждение о том, что в самопроизвольных процессах возникает энтропия, а следовательно энтропия системы постепенно увеличивается, справедливо лишь для изолированных систем. Однако в большинстве случаев химические реакции протекают при заданной температуре и в условиях теплообмена с окружающей средой. Чтобы найти функции состояния для таких процессов, необходимо объединить первый и второй законы термодинамики. Запишем первый и второй законы термодинамики:
dU — dQ-j-dlF (первый закон термодинамики) (250)
TdS = dQ-]-dI (второй закон термодинамики) (251)
Совместное решение этих уравнений дает нам выражение для dW, из которого исключена теплота dQ:
dW = dU-- TdS-^dl (252)
Одно из следствий первого закона состоит в том, что dU представляет собой полный дифференциал функции состояния, a dQ и dW зависят от пути перехода, di также зависит от пути протекания процесса, однако, кроме работы, теплоты, в термодинамике не существует других функций, которые зависят от пути процесса. В связи с этим одной из наиболее важных проблем химической термодинамики является нахождение условий, при которых работа равна изменению функции состояния, т. е. работа химического процесса не зависит от его пути. Из уравнения (252) следует, что при обратимом процессе
&W — d(U~ TS) (253)
Это означает, что работа, совершенная при .обратимом изотермическом процессе, не зависит от пути, так как представляет собой полный дифференциал функции состояния. Эта функция состояния по определению равна
F=U— TS (254)
й называется свободной энергией. Обратимая изотермическая работа не зависит от пути процесса и равна изменению свободной энергии
Ц7обр = AF= А£7— T&S (255)
В соответствии с вторым законом термодинамики TAS можно называть обратимой теплотой*. В первом приближении смысл
* T&S (в общем случае TS) называют также связанной энергией, так Как она не может быть переведена в работу. — Прим, перев.
16*
244
Химическая термодинамика
выражения (255) заключается в следующем: внутренняя энергия системы меняется на At7, причем часть этого изменения, т. е. А/7, может быть преобразована в работу. Член T’AS в любом случае проявляется как тепловой эффект. Это утверждение не совсем точно, так как поначалу неясно, какой знак имеют величины А£7, А/7 и FAS. В некоторых процессах можно было бы получить работу в большем количестве, чем изменение внутренней энергии системы, если к системе будет подводиться необходимое количество теплоты. Указанные представления строго верны для обратимых процессов; для необратимого процесса работа, совершаемая над системой, больше, а работа, получаемая от системы,— меньше, чем для обратимого процесса [уравнение (252)]. Функция F, которая введена с целью решения, казалось бы, второстепенного вопроса об обратимости процесса получения работы от системы, имеет первостепенное значение для определения условий равновесия термодинамических систем (см. далее).
При исследовании физико-химических процессов методами термодинамики оказалось целесообразно разделить совершаемую системой работу на работу расширения и полезную работу:
dW=-pdV-[-dW11 (256)
Такое разделение связано с тем, что работа расширения, совершаемая при изменении объема реагирующих веществ в химической реакции, не является «полезной» в термодинамическом смысле. «Полезная работа» может быть получена в гальванических элементах*. Если подставить уравнение (256) в (252) и решить его относительно d lFn, получим
dWn = dU—TdS-[-pdV-\-dI (257)
В случае обратимого изобарно-изотермического процесса
dWu = d(U-TS+pV) (258)
т. е. полезная работа изобарно-изотермического процесса также является функцией состояния. По определению
G=U—TS+pV =
= H—TS= (259)
— F-\-pV (260)
Функцию G называют свободной энтальпией** [термин, хорошо понятный из уравнения (259)]. По аналогии с уравнением (255)
AG=A/f—TAS (261)
* Полезная работа может проявляться также в виде энергии химической реакции, механической, световой и иного рода энергии. — Прим, пер ев. _
"* Функцию G называют также энергией Гиббса или свободной энергией при постоянном давлении, а функцию F—энергией Гельмгольца или свободной энергией при постоянном объеме. [Рекомендации ИЮПАК]. — Прим. ред.
Второй закон термодинамики
245
В то время как свободная энергия F является функцией V и Т, для функции G в качестве независимых переменных приняты параметры р и Т. Так как на практике такое сочетание параметров встречается значительно чаще, чем пара V, Т, функция G применяется значительно более широко, чем свободная энергия F. Зависимость свободной энтальпии от параметров системы дается следующими уравнениями [см. уравнения (279)
(262)
Из уравнений (262) и (259) следует*
(263)
выво-
Второе из последних уравнений играет важную роль при де температурной зависимости константы равновесия.
Пример. Один моль водяного пара обратимо конденсируется при 100 °C (энтальпия испарения 2258,9 Дж/г). Рассчитать W и Q, а также изменение функций состояния BU, ВН, ВО и Д8.
Решение. Г=р(1/ЖИДк— 14аз) = 1 •/?7'=8,314 Дж-моль-1-К”1-373 К= =3103,3 Дж/моль
Q — ВН = 18 г/моль-2258,9 Дж/г = 40,66 кДж/моль
BU — ВН — pBV = (—40,66 -f- 3,1) Дж/моль = —37,56 кДж/моль
Д5 = Q06p/T = —40 660 Дж/моль- 373 К = —109 Дж-моль-1- К-1
BF = BU — TBS = (—37,56—(40,66)) кДж/моль = 3,1 кДж/моль
BG = ВН — TBS = (—40,66 — (—40,66)) кДж/моль = 0
Интерпретируйте эти результаты с точки зрения рассмотренных в этом разделе понятий, «максимальная полезная работа», «связанная и свободная энергия».
Вопросы и задания
1. Рассчитайте изменение энтропии для следующих процессов: а) плавление алюминия в количестве 3 моль в точке плавления (660°C), ВНПл = =7,99 кДж/моль; б) испарение 1 моля жидкого кислорода в точке кипения (—182,97 °C), Д//исп=6,82 кДж/моль; в) нагревание 20 г H2S от 50 до 100°С, Ср = 29,93+0,0139 Т.
2. 1 моль льда плавится при температуре 0 °C (теплота плавления 333,6 Дж/г); образовавшаяся вода нагревается до температуры 100°C (Удельная теплоемкость 4,185 Дж-г-^Д-1) и при температуре 100 °C испаряется в водяной пар (энтальпия испарения 2,26 кДж/г). Рассчитайте суммарное изменение энтропии.
3. 1 г идеального газа при температуре 300 К и начальном давлении 1 МПа обратимо изотермически расширяется до объема 10 л. Рассчитайте Максимальную работу и теплоту, а также Ди, Bh, Bf, Bg, Bs.
* Уравнения (263) называют также уравнениями Гиббса — Гельмголь-- Прим, перев.
246
Химическая термодинамика
4. 10 г гелия при 100 °C обратимо изотермически сжимают от 2-Ю5 до 10s Па. Рассчитайте w, q, kf, &g, Ыг, Au и As.
5. 1 моль толуола испаряется при температуре кипения 111 °C (энтальпия испарения 362 Дж/г). Рассчитайте W, Q, &Н, &U, AG, АД и AS.
6. Рассчитайте общее изменение энтропии при смешении 350 г воды при температуре 5 °C с 500 г воды при температуре 70 °C (удельная теплоемкость воды 4,185 Дж-г-'-К-1)-
7. Рассчитайте суммарное изменение функций состояния Ди, Д/г, Af, Ag-и As при испарении 2 молей воды в точке кипения и последующем (изотермическом) расширении пара, находящегося при температуре 100 °C и нормальном давлении, до конечного объема 50 л.
8. 150 г ртути при температуре 100 °C вносят в калориметр, содержа щий 80 г воды при 20 °C. Теплоемкость калориметра равна 83,7 Дж/К. Рас считайте изменение энтропии а) ртути; б) воды в калориметре; в) ртути, воды и калориметра вместе. Темплоемкость воды принята равной 4,185 Дж/(г-К) ртути — 0,143 Дж/(г-К).
22.3. Термодинамические функции состояния, введенные на основе объединения первого и второго законов термодинамики. Фундаментальные уравнения Гиббса
В результате совместного рассмотрения основных законов термодинамики получили четыре основные функции состояния имеющие размерность энергии. Ниже эти функции исследуютсг более подробно, так как с их помощью можно количественнс описать химические равновесия. Для того чтобы связать первый и второй законы термодинамики, введем в параметры системы наряду с р, Т и v также энтропию s. Для четырех функций состояния получим
u=f(s, v, п(,. (264)
h = f(s, р, nit. (265)
f=f(v, Т, (266)
g=f(p, Т, (267)
Изменение функций состояния определяется с помощью полных дифференциалов:
, / ди \ , , / ди du = -3—) as -4- -j— \ ds /Vi ( V+Ж 1 dn. (268)
л / dh \ , , I dh dh = I —5— I ds I —Д— (, ds Jp„. ‘ ( dp L,dp+^ ) dnt h’P>nj (269)
‘НЯ.-Л+Ш vtn^ j 1 dn; 'v,T,nj (270)
p,nl | dn; )p,T,nj (271)
Индекс п3 означает, что содержание (в мольных долях) всех компонентов, кроме i-го, поддерживается постоянным. Все рас*
Второй закон термодинамики
247
смотренные выше уравнения справедливы только для однофазной системы. Если в системе имеется несколько фаз, то для каждой из них необходимо составить соответствующую систему уравнений.
Значения частных производных можно найти, написав уравнения для полных дифференциалов и сравнивая их с выражениями полных дифференциалов (разд. 22.2). Ограничиваясь обратимыми процессами, можно считать диссипативный член в уравнении (235) равным нулю (d/=0).
При выполнении этих условий, подставив выражение для теплоты, полученное из (235), в уравнение (191) (первый закон термодинамики), получим полный дифференциал внутренней энергии:
du—Tds—pdv-^-^f^dni (272)
где цг — частные производные внутренней энергии по содержанию i-го компонента (пг-):
= (273)
\ д/li ]s,Vtnj
Подставив уравнение (272) в уравнение (194), получим полный дифференциал функции dh:
dh=Tds-{-vdp-st-'2i\kidni (274)
Выразив внутреннюю энергию и через свободную энергию f (u=f4-Ts) и подставив полученное выражение в (272), получим после преобразований
df~ —sdT—pdv-j-^ Pidrij (275)
Наконец, подстановка в уравнение (275) уравнения, определяющего функцию g, дает нам дифференциал dg:
dg=—sdT(276)
Уравнения (272), (274), (275) и (276) имеют важное значение Для количественного расчета термодинамических равновесий и называются фундаментальными термодинамическими уравнениями Гиббса.
Сопоставление уравнений (268) — (276) дает ряд важных соотношений:
(277)
(278)
248
Химическая термодинамика
В уравнениях (272) — (276) присутствует один и тот же член ^ihdrii Гиббса; после сопоставления с коэффициентами уравнений (268)— (271) получим
ц, называют химическим потенциалом. Это парциальная мольная величина [некоторое представление о таких величинах уже вводилось в разд. 18 2, уравнение (185)]. Химический потенциал— это интенсивный параметр системы; он не зависит от количества вещества в фазе. Величина выражаемая как производная (ди/дпг)в, г,, п; или (dhldnt)s,P,ni, несколько абстрактна, поскольку ее изменение происходит при постоянной энтропии, т. е. при условиях, которые редко встречаются в обычных физико-химических процессах. В то же время химический потенциал, определяемый двумя последними выражениями, т. е. как парциальная свободная энергия и парциальная свободная энтальпия вещества в смеси нескольких компонентов, имеет важнейшее значение для расчета фазовых химических равновесий.
Далее показано, что как раз разность химических потенциалов отдельных компонентов в смеси реагирующих веществ и есть движущая сила химического превращения, а разность химических потенциалов соответствующих компонентов в разных фазах — движущая сила фазового превращения.
23. ХИМИЧЕСКИЕ И ФАЗОВЫЕ РАВНОВЕСИЯ
23.1. Общие условия равновесия
Ранее уже упоминалось об одном из условий равновесия. При установлении равновесия в адиабатическом процессе энтропия принимает максимальное значение и dS = Q. Однако такой путь протекания процесса сравнительно редко встречается в химии, большинство равновесий, которые наблюдаются в химических реакциях, устанавливаются в условиях изобарно-изо термического или изохорно-изотермического процесса. Укажем без вывода условия равновесия термодинамических процессов (экстремальные значения соответствующих функций; полный дифференциал функции равен нулю):
Химические и фазовые равновесия
249
Процесс адиабатический изохорно-изотермический изобарно-изотермический
Исследуемая функция
S
F
G
Экстремум максимум минимум минимум
Дифференциал dS=O
dF=O
dG=Q
Второе условие равновесия о том, что полный дифференциал функции состояния должен быть равен нулю, менее важно, чем условие максимума или минимума функции. Однако для дальнейшего изложения этого условия вполне достаточно, и мы будем пользоваться исключительно этим условием.
Из рассмотренных условий равновесия непосредственно следуют также условия равновесия, которые будут выведены ниже.
Пусть в системе осуществляется равновесие
А <—^ в
(282)
причем не имеет значения, фазовое это превращение или химическая реакция. Из основного условия равновесия (dS = 0) получим
dSA+dSB=0
—dQJT k-\-dQJT в = О
ТВ = ТА (283)
Таким образом, температура начального и конечного состояний должна быть одинаковой, т. е. должно иметь место термическое равновесие.
Аналогичный подход к изохорно-изотермическому равновесию дает
dFA+dFB = О
-Ра^+Рв^=0
Ра = Рв (284)
Следовательно, давление в начальном и конечном состояниях должно быть одинаковым, т. е. должно осуществляться механическое равновесие.
Введем еще одно важное условие равновесия, связанное с химическим потенциалом цг; далее химический потенциал используется только в виде парциальной мольной свободной энтальпии:
=рг (285)
По определению химический потенциал вещества i равен изменению свободной энтальпии системы при изменении числа молей dn-i этого компонента при постоянном давлении и температуре и при постоянстве числа молей остальных компонентов си
250
Химическая термодинамика
стемы пр Таким образом, третье условие равновесия, выраженное через химический потенциал из уравнения (285), записывается следующим образом:
У М". = 0
i
Применяя это условие равновесия к процессу (282), получим — = О
Поскольку увеличение числа молей пв приводит к уменьшении) числа молей пА, то при равновесии
М-а= Ив
В зависимости от того, у какого вещества химический потенциал больше (ца>цв или наоборот), процесс (282) идет в прямом или обратном направлении, т. е. химический потенциал является движущей силой всех химических реакций и фазовых превращений.
Более общая формулировка этого условия равновесия для процесса с любым количеством компонентов i и стехиометрическими коэффициентами Vi имеет вид
= 0 (285а)
L
23.2. Химическое равновесие и закон действующих масс
При протекании химической реакции изменяется давление, соответственно концентрация компонентов, участвующих в химической реакции. Отсюда следует, что необходимо учитывать зависимость химического потенциала от давления (концентрации). Зависимость свободной энтальпии от давления задается уравнением (279). Если применить правило Шварца для полных дифференциалов к фундаментальному уравнению (276), то можно получить следующее выражение для зависимости химического потенциала от давления:
<286>
где Vi — парциальный молярный объем. Разделение переменных и интегрирование в пределах от нормального давления 101 325 Па до парциального давления Pi компонента i дает
Иг= W + RT In-10^25 = (i) +RT In p°t (287)
Использованное здесь парциальное давление рг° введено уравнением (248). ц°т(г) представляет собой стандартный химиче-
Химические и фазовые равновесия
251
ский потенциал компонента i, т. е. химический потенциал при относительном давлении р°г=1. ц°т(о зависит только от температуры. RT In р°г — второй член уравнения (287)—называют иногда также остаточным потенциалом.
Используем выведенное выше уравнение (285а) для расчета равновесия гомогенной химической реакции, например реакции образования аммиака в газовой фазе:
3H2 + N2 =8=3= 2NH3 (288)
2r-nh3—Зр-н2— P-n2 = 2p-°NH3— 3[х°Н2 P-°n2+
+ 2RT In p°NH3~ ЗЯТ In p°H2- RT In р\ (289)
Так как
2V^ = AGP (290)
определяет работу химической реакции или изменение свободной энтальпии, можно записать уравнение (287) также в виде
AGP = AG°P -4-7?Т1п /о- - (291)
р Р Г ' (Р°Н2) P°n2 '
или в более общей форме
AGp= AGp°-{-/?74n П (РФ (291а)
Первый член с правой стороны уравнения (291) называют иногда также основной работой реакции, а второй — остаточной работой реакции. В состоянии равновесия в соответствии с уравнением (285а) AGp = 0, и в уравнении (291) должны стоять равновесные парциальные давления компонентов. Тогда из уравнения (291) получим
AGp° = — RT\n
(P°NH3(PH))2
(P<’h2(Ph))®P<’N2(Ph)
= -RT In П (р°г(рн)Р = ~RT 1П Kp
(292)
Здесь Kp — константа равновесия, рассчитанная для относительных давлений компонентов. Таким образом, используя термодинамические соотношения, получили закон действующих масс для конкретной реакции. Этот вывод может быть обобщен на любую химическую реакцию. Общая форма уравнения (292) называется уравнением изотермы химической реакции Вант-Г оф фа.
Пример. Рассчитаем константу равновесия реакции образования аммиака (288) при 25 °C из стандартной энтальпии и стандартной энтропии образования компонентов, взятых также при нормальных условиях.
Решение. Свободную энтальпию можно определить из уравнения (261)
AG°nh3 = д#°21)8 (NH3) — 7’д5°298 (NH3) (293)
252
Химическая термодинамика
Найдем в таблицах стандартные значения термодинамических величин:
Д^°2В8 (NHS) = —46,04 кДж/моль; S°298 (NH > = 192,65 Дж • моль'1 • К-1
5°2Э8(Н2) = 130,71 Дж-МОЛЬ-1-Л-1; S298(Na) == 191,65 Дж-моль'1-Ю1
Учитывая знаки стехиометрических коэффициентов, получим для стандартной энтальпии реакции в нормальных условиях
Д^°298Р = 2 (—46,04) = —92,08 кДж/моль а для стандартной энтропии в нормальных условиях
Д5°2в8Р = (2 -192,63 — 3-130,71 — 191,65) - 199,31 Дж/(К-моль)
Отсюда получим для стандартного изменения свободной энтальпии
ДО°298Р (NHS) = —92,08 — ^—199,31 - = —33,07 кДж/моль
Тогда из уравнения (292) получим
33 070
1g Кр = 2,303-8,317298 = 5,8; ^р = 6’3 105
Полученная нами константа равновесия содержит равновесные относительные „парциальные давления отдельных компонентов, поэтому представляет собой безразмерную величину. Рассмотренный выше расчет показывает также, что реакция синтеза аммиака полностью смещена в сторону его образования; то, что эта реакция практически не происходит в нормальных условиях, объясняется кинетическим торможением.
Для того чтобы обобщить рассмотренный выше вывод закона действующих масс на любую реакцию, надо более подробно исследовать понятие химического потенциала компонента в смешанной фазе. Для идеальных газов химический потенциал всегда может быть определен при помощи уравнения (287). Если имеется лишь один идеальный газ, то вместо парциальных давлений в уравнение подставляется общее давление в системе.
Другим важным для практических приложений случаем являются растворы. Предположим сначала, что раствор является идеально разбавленным; следует также знать, какой компонент раствора принимает участие в реакции — растворитель или растворенное вещество. Если в уравнении (287) ио аналогии с уравнением (249) разделить парциальное давление на содержание в.мольных долях и общее давление и перенести член, зависящий от давления, в стандартный потенциал, то из уравнения (287) получим
Hi = Н°г (о (P°)+RT In Xi (294)
таких конденсированных системах, как растворы, зависи-РТЯ TJ ТТО ----------
в
мость стандартного потенциала от давления значительно меньше, чем в газах. Принимая во внимание уравнение (286), следует ожидать, что эта зависимость для конденсированных систем в '-—1000 раз меньше, чем для газов, поэтому в большинстве случаев ее можно не учитывать. На основе более сложных статистических расчетов для идеально разбавленных растворов можно показать, что уравнение (294) точно определяет химиче-
Химические и фазовые равновесия
253
ский потенциал как для растворителя, так и для растворенного вещества. Сами расчеты здесь не приводятся, и далее используются только результаты расчетов. Мольная доля редко применяется в качестве меры концентрации в химии растворов. Если раствор достаточно разбавлен, то [уравнения (230) и (234)]*
Y _ _______^1С2______ /9Q52
2 1 ОООр + (Л^ - М2) сг ~ Тобф С2 '
т. е. концентрация в мольных долях приблизительно пропорциональна молярной (моль/л) концентрации, р — плотность раствора. Не будет большой ошибки, если химический потенциал растворенного вещества в разбавленном идеальном растворе связать с молярной концентрацией с,-:
И4=Нг(Лр0)+ЯПпс/> (296)
По аналогии с относительным давлением [см. уравнение (248)] введем относительную концентрацию сг° = сг/(1 моль-л-1) и условно назовем концентрацию 1 моль-л-1 нормальной. Член, который содержит логарифм коэффициента пропорциональности между молярной концентрацией и концентрацией в мольных долях, можно внести в стандартный химический потенциал. Однако такой способ расчета невозможен, если он относится к растворителю. Для растворителя
Н1= (297)
Неверное использование такого рода соотношений встречается довольно часто, что приводит к типичным ошибкам, например при расчете процесса диссоциации воды по закону действующих масс.
Необходимо иметь в виду, что, как показано выше, для чистых газов имеется остаточный химический потенциал, отсутствующий в конденсированных системах. Это является определенным преимуществом такого рода систем, так как стандартный химический потенциал газовых систем по определению не зависит от давления, а стандартный химический потенциал в разбавленных растворах и чистых конденсированных фазах почти не зависит от давления. Так как константа равновесия в соответствии с уравнениями (289) — (292) определяется лишь стандартной свободной энтальпией, это означает, что константа равновесия практически не зависит от давления для всех химических реакций. Имеются лишь немногие случаи, когда необходимо учитывать ее зависимость от давления вследствие того, что оказывается невозможным пренебречь молярным объемом конденсированной фазы (пример такого процесса приведен позднее). Выделим четыре случая:
* Индекс 1 относится к растворителю, а индекс 2 — к растворенному веществу.
254
Химическая термодинамика
идеальные газы и смеси газов
^(win^
растворенное вещество в разбавленном идеальном растворе ^ = ^(7, рО)4-/?71пС.о
растворитель в разбавленном идеальном растворе ;
индивидуальные конденсированные фазы I
и< = И/0(Т,р«)4-/?Пп1
или в обобщенной форме
Цг = Ц;°(ЛЛ+^1птг (298)
где вместо т, в зависимости от физического состояния вещества можно подставить величины рг> сг, Xt и 1. Таким образом, имеются все исходные данные для вывода закона действующих масс в случае идеальных смешанных фаз. Аналогично выводу закона действующих масс для равновесия в газах из уравнений (289)—-(292), учитывая условие равновесия 2vigi = 0 и уравнения для химических потенциалов конденсированных фаз [уравнения (294) и (296)], можно получить уравнение константы равновесия, где содержание компонентов выражено в мольных долях (Кх) или относительных единицах (М). Тогда получаем следующие соотношения между константами равновесия и равновесным составом системы:
кр=П(рЛрн)Р (а); П (*hph))v' (б);
Кс = ГШрн)Р (в) (2")
Можно связать между собой константы равновесия, определяемые уравнениями (299). Соотношения для взаимного пересчета констант равновесия получают после подстановки зависимости pi от сг и pi от хг [используют законы идеальных газов, уравнение (299)]. Введем обозначения ро=1О1 325 Па и с0 = 1 моль/л. Рг^СгКТ, рг — Хгр И
KP = Kc(^)2V‘(Co/Po)2Vi
„ / RT \2v.
(300а) (3006) (ЗООв)
~ = VM— средний молярный объем газовой смеси [уравнение (300 в)]. Для разбавленного идеального раствора
Кх=Кс(УмС0)^‘
(300г)
Химические и фазовые равновесия 255
_______________________________
где Ум-—молярный объем чистого растворителя.
Следует еще раз подчеркнуть, что рассчитанные из термодинамических данных на основе изотермы химической реакции рант-Гоффа константы равновесия являются безразмерными величинами. При расчете равновесного состава системы пользуются также константами равновесия, имеющими определенную размерность. В отличие от константы равновесия, заданной уравнением (299), такую константу обозначим Кррз:
v=[W=mb‘ <301>
Связь между этими константами равновесия можно получить, подставив в уравнение (299) Pi°=pt/Po (где р0= 101 325 Па). Для реакций, в которых 2v(t^0, Кр и Дррз отличаются по своему значению.
Пример. Рассчитаем из полученной в предыдущем примере константы равновесия образования аммиака Кр=6,3-105 константы равновесия Кс и К/3.
Решение. Из уравнения реакции (288) получим
^v£ = 2- 3- 1 =-2
Из уравнения (300а) следует
Кс = КР (RT)~^ (cQ/pQ)~^ = Кр (RT)2 (с0/рй)2 „ / 1000 моль/м3 \2
Кс= 6,3-104(8,314 Па-М®.моль-1 К-1-29» К)2 lnl q9e Д-) =Кс = 3,8-10»
\ 1U1 0^0 lid /
Из уравнения (3006) получим 7G=Kp(p0)_2v> =КР(р0)2; таким образом, Кх пропорциональна относительному давлению в квадрате. Для р°= = 101 325 Па р°=1, а следовательно, К*=ДР=6,3> 105.
Константа равновесия Кррз получается из уравнения (301):
КрРз = /СрРо-2 = 6,3-105-(101 325 Па)-2 = 6,14-10~5 Па-2 = 61,4 кПа"2
До сих пор речь шла о идеальных системах — идеальных газовых смесях и идеальных разбавленных растворах. Эти системы являются основной областью применения закона действующих масс. Для газовых смесей обычно не учитываются отклонения от законов идеального газа, если только не рассматриваются газовые реакции, протекающие при высоких давлениях. Для реакций в растворах дело обстоит не так просто. Уже при обычных небольших концентрациях растворенного вещества (10~3<-10-2 моль/л) наблюдаются отклонения от идеального состояния. Эти отклонения учитываются умножением значений концентраций в законе действующих масс на поправочные коэффициенты ; /г — сложная функция концентрации компонен-Та сг, а также концентраций остальных компонентов (в некоторый момент времени), температуры и давления (слабая зависимость) ; Д называют коэффициентом активности. Произведение
256 Химическая термодинамика
Химические и фазовые равновесия 257
коэффициента активности и концентрации называют активностью аг. Произведение Д- и парциального давления Pi называют летучестью. Коэффициент активности в газовых системах можно рассчитать из уравнения состояния реального газа или реальной газовой смеси. Важное значение имеют также коэффициенты активности растворенных компонентов. Теория Дебая-— Хюккеля дает возможность рассчитать коэффициенты активности в области концентрации примерно до 10~2 моль/л (гл. 31.4). Их можно также определить из данных о равновесии между раствором и растворенным веществом и измерений э. д. с. гальванической цепи (гл. 30.4).
23.3. Применение закона действующих масс
к гомогенным и гетерогенным процессам
23.3.1. Гомогенная газовая реакция
2Н2О ?—t 2Н2 + О2
При достаточно высокой температуре эта реакция происходит в газовой фазе и сопровождается изменением числа молей. Если реакция протекает слева направо, то вступают в реакцию 2 моля, а образуются 3 моля. При постоянном давлении объем системы увеличивается на RT/p на каждый моль прореагировавшего вещества. Закон действующих масс для этой системы запишется следующим образом:
Крр3 = (Р2н2Ро2/Р2н2о) (302)
Выразим парциальное давление через общее давление и мольные доли:
Крр3 = р (х2Н2 • Хо2/х2Н2о) (303)
Таким образом, уравнение (303) определяет зависимость равновесия от давления. Если давление увеличивается, то дробь, в которую входят мольные доли газов, должна уменьшаться, чтобы КрР3 осталась постоянной. Поэтому знаменатель в дроби должен увеличиться, т. е. равновесие сместится в сторону образования водяного пара. Это можно пояснить еще следующим образом: при повышении давления равновесие смещается таким образом, чтобы реагирующая система заняла меньший объем, а давление снизилось. Иначе говоря, система стремится скомпенсировать внешнее воздействие (в данном случае давление). Из такого поведения системы следует правило Ле Шателье — Брауна, представляющее собой одну из первых качественных форму' лировок, предшествовавших второму закону термодинамики.
23.3.2. Гетерогенная химическая реакция
СаСО3 = СаО + СО2 (304)
Это реакция обжига известняка; она является примером гетерогенной реакции — присутствуют две твердые фазы. Поскольку в газовой фазе содержание в мольных долях твердых компонентов равно 1, то
V = Pco2 (305)
Поэтому над двухфазной системой из СаО и СаСОз устанавливается постоянное давление СО3. С увеличением давления диоксида углерода равновесие смещается влево. Зависимостью константы Кррз от давления, которая определяется слабой зависимостью химических потенциалов СаО и СаСО3 от давления, практически можно пренебречь.
23.3.3. Гетерогенная реакция в жидкой фазе
2Ag + Hg2Cl2 ^=> 2AgCl + 2Hg (306)
В этой реакции принимают участие только конденсированные фазы — три твердые и одна жидкая. Реакция может протекать в гальванической ячейке, если соединить между собой каломельный и хлорсеребряный электроды (разд. 30.5). Закон действующих масс для этой реакции запишется следующим образом* *:
1 (307)
Однако рассчитанная из экспериментальных данных для 25°C константа равновесия оказывается равной 5,8. Это значение не согласуется с теорией [уравнение (307)]. Очевидно, система не может прийти к равновесию и всегда находится вдали от него. Реакция идет в одном направлении до тех пор, пока не израсходуется одно из веществ, стоящих слева в уравнении реакции. Такое поведение системы часто наблюдается для гетерогенных реакций, которые не доходят до состояния равновесия, а заканчиваются, как только израсходуется одно из исходных веществ. В случае реакции (304) также может оказаться, что при небольшом количестве СаСО3 это вещество израсходуется раньше, чем будет достигнуто состояние равновесия.
23.3.4. Равновесная реакция фазового превращения
Рассмотрим, например, равновесный процесс испарения воды
Н2О (жидк.) Н2О (газ) (308)
* В уравнение закона действующих масс можно подставлять только Равновесные концентрации веществ. Для реакции (306) в равновесном состоянии концентрация по крайней мере одного нз исходных веществ должна °ыть значительно меньше 1, что и соответствует указанному опытному значению константы равновесия, т. е. тому, что реакция идет практически до Конца слева направо. Соотношение (307) соответствует условию ДО°Р<0. —• ‘‘Рим. перев.
17.
2027
528
Химическая термодинамика
Химические и фазовые равновесия
259
В данном случае как жидкость, так и газ представляют собой индивидуальное вещество. Тогда закон действующих масс можно записать в следующем виде:
КРрз = Рн2о (309)
В системе присутствует жидкая фаза, поэтому КРрз зависит практически только от температуры и пренебрежимо мало зависит от давления. Уравнение (309) является уравнением давления пара. В равновесном состоянии давление пара над жидкостью— индивидуальным веществом — является функцией только температуры. Что собой представляет эта функция, разбирается далее при знакомстве с другими параметрами, влияющими на константу Кррз.
23.3.5. Первый закон Рауля
Вернемся вновь к равновесию (308). Однако пусть в данном случае испарение воды происходит из раствора, причем предполагается, что испаряется только вода. Согласно введенным ранее правилам, концентрацию растворителя следует выражать в мольных долях. Тогда закон действующих масс запишется следующим образом:
К=р!Х1=рй (ЗЮ)
где ро — давление пара чистого растворителя,^ если х=1. Уравнение (310) показывает, что при одной и той же температуре (т. е. при постоянстве К) давление р над раствором ниже, чем над чистым растворителем р0- Уравнение (310) называют первым законом Рауля. Его можно записать в несколько иной форме, введя мольную долю х2=1—Xi растворенного вещества:
(ро—р)/Ро = Ьр/Ро = х2 <31
Если рассматривать реальный раствор, то вместо х2 следует подставить f0x2 (fo — осмотический коэффициент; он связан с коэффициентом активности растворителя, соответственно растворенного вещества). Если растворенное вещество полностью или частично диссоциировано, то уравнение (311) видоизменяется.
23.3.6. Равновесие растворимости
AgCl Ag+H-СГ (312)
Подобное равновесие наблюдается в том случае, если в рас* творе имеется осадок нерастворившейся соли AgCl. Закон Дей ствующих масс для этой системы можно записать следуют11 образом:
К = сАе+-ссГ (313)
Нерастворившаяся соль AgCl находится в твердой фазе и поэтому не входит в уравнение закона действующих масс. Константу называют произведением растворимости или константой растворимости. Поскольку в равновесии находятся ионы, более правильно было бы подставить в уравнение (313) вместо концентраций активности, например если речь идет о растворе AgCl в ‘присутствии КС1.
23.3.7. Закон распределения Нернста
Пусть имеются две несмешивающиеся жидкости (например, вода и бензол) и третье вещество, растворимое в обеих жидкостях. Если растворить некоторое количество этого вещества в одной из жидкостей, а затем энергично перемешать их, то растворенное вещество распределится между обеими несмешиваю-щимися жидкостями. Отношение количества вещества, растворившегося в этих жидкостях, задается законом распределения Нернста, который представляет собой частный случай закона действующих масс. Рассмотрим несколько более сложный случай распределения бензойной кислоты между водой (в которой она находится в виде недиссоциированных молекул В) и бензолом (в виде ассоциатов В2):
2В «=> В2 (314)
К = сВ2/св2 (315)
В более простом случае, когда растворенное вещество не образует ассоциатов и не распадается на ионы, уравнение (315) (закон распределения) означает, что отношение концентраций (точнее, активностей) вещества в обоих растворителях постоянно и не зависит от общего количества растворенного вещества. Так как константа К в уравнении (315) является константой равновесия, она подчиняется рассмотренным выше зависимостям от температуры и давления и может быть рассчитана так же, как и для всякого другого равновесия.
23.3.8. Растворимость газов
Пусть газ или смесь газов под некоторым давлением находится в контакте с растворителем, в котором возможно растворение газа. Рассмотрим, например, систему диоксид углерода— иода, где осуществляется равновесие
СО2 (раствор) ч=±: СО2(газ) (316)
Закон действующих масс определяет константу равновесия
К — РсоУссоъ (317)
соответствии с уравнением (317) концентрация газа в раство-Ре пропорциональна его давлению (закон Генри).
17*
260
Химическая термодинамика
Если в жидкости растворяется смесь газов, то в уравнение закона действующих масс входит парциальное давление растворенного газа, а константа К меняется в зависимости от природы газа (она как раз соответствует стандартному (химическому) потенциалу данного газа). Например, растворимость кислорода в воде в два раза выше, чем растворимость азота; это име. ет большое значение для процесса обмена веществ у рыб. В применении к газовым смесям рассмотренная выше закономерность называется законом Генри — Дальтона Константа К может иметь различные размерности. Для применяемых чаще всего размерностей эта константа называется коэффициентом поглощения Бунзена. Он представляет собой отношение объема газа (приведенного к 0°С и нормальному давлению) к единице объема растворителя при парциальном давлении газа р=101 325 Па.
23.3.9. Электролитическая диссоциация растворенных газов
н2о + 4- н2 + С12 =F=h н3о+ + С1- (318)
Эта реакция в направлении слева направо используется в гальванической ячейке, состоящей из двух полуэлементов, в один из которых пропускается газообразный хлор, а в другой—-газообразный водород (разд. 30.1). Реакция (318) является реакцией, обратной электролизу раствора НС1. Закон действующих масс для этой реакции можно записать следующим образом:
= ^нзО+’^сг/Т^Рнз'Рсц (319)
Содержание (в мольных долях) воды близко к единице, и поэтому этот параметр не влияет на константу равновесия. Такая запись константы равновесия, в которую входят как концентрации, так и парциальные давления реагирующих веществ, обычно применяется в электрохимии для выражения э. д. с. электрохимической ячейки, в которой протекает реакция (318), в соответствии с уравнением Нернста (разд. 30.1).
23.3.10. Ионное произведение воды
2Н2О 3==* Н3О++ОН- (320)
Это равновесие имеет очень важное значение в химии водных растворов. В соответствии с законом действующих масс
К ~ ан3о" • йон“ (321)
Х2н2о здесь сразу принимается равным единице по тем же причинам, что и при рассмотрении реакции (318). Константу равновесия (321), как правило, называют ионным произведением воды или константой автопротолиза. При комнатной температуре она равна ~10~14 моль2/л2; эта константа растет при повышении температуры: например, при 65 °C К=10-13. Закон действующих масс для равновесия диссоциации воды можно выра
Химические и фазовые равновесия
261
зить также другим способом, подставив в знаменатель «концентрацию воды» 55,5 (вернее даже, 55,52); в этом случае получим так называемую константу диссоциации воды, которая отличается от ионного произведения воды по своему численному значению. Константа диссоциации воды применяется главным образом в теории кислот и оснований для сравнения с константами диссоциации других соединений (разд. 35.4.1, табл. В.10).
Примеры
1. Составить уравнение закона действующих масс для реакции разложения карбамината аммония
NH2COONH4 (тв.) s=h 2NH3 (газ) + СО2 (газ)
Решение. Довольно большое количество (избыток) карбамината аммония поместим в вакуумированный сосуд. В соответствии со стехиометрией реакции парциальное давление NH3 оказывается в два раза больше, чем парциальное давление СО2; давлением пара карбамината аммония можно пренебречь. Следовательно,
/2 \2 / 1 \ 4
Кр?3 = (Pnh3)2 (Рсо2) = (-g-pj "27“ р3
где р — общее давление в системе.
2. Применение закона Рауля. Рассчитать давление пара воды над 10%-иым раствором глицерина (Л1=92) в воде при 25 °C; давление пара чистой воды при 25 °C равно 3,16 кПа.
Решение. Рассчитаем содержание (в мольных долях) глицерина в растворе [уравнение (230)]:
0,10/92
Xi ~ 0,90/18 4- 0,10/92 — °>0216
С помощью закона Рауля (311) найдем давление пара над раствором: р° — р 3,16—р
= з, jg = 0,0216
р= (3,16 —0,0216-3,16) = 3,08 кПа
Разумеется, если известны давление пара над чистым растворителем и раствором, то можно решить обратную задачу, т. е. найти концентрацию растворенного вещества.
Пусть в воде растворено вещество, распадающееся в растворе на ионы (а не глицерин — вещество, не диссоциирующее на ионы). В уравнении, Выражающем закон Рауля, необходимо учитывать дополнительный множитель Др/р° = х21. Этот множитель зависит от степени диссоциации а и числа ионов v, на которые распадается растворяющееся в растворе вещество:
i=14-a(v— 1) (322)
Из этого уравнения, зная понижение давления пара над раствором, можно вычислить степень диссоциации а.
3. Применение закона распределения Нернста 0,35 г серы в 100 мл бензольного раствора находятся в равновесии при 25 °C с 0,65 г серы, растворенной в 250 мл тетрахлорида углерода, а) Рассчитать коэффициент распределения серы в системе тетрахлорид углерода — бензол, б) Вычислить, Колько серы необходимо растворить в 20 мл СС14, для того чтобы этот аствор находился в равновесии с 1,00 г серы, растворенной в 100 мл бен
262
Химическая термодинамика
зольного раствора в) Рассчитать растворимость серы в CCU при 25 °C, если растворимость серы в бензоле при тех же условиях составляет 17,93 г/л В указанных растворителях сера находится в виде молекул Sg.
Решение. Уравнение закона действующих масс для равновесия распределения имеет вид K=c'/crt, где с1 и с11 — концентрации серы в СС14 и бензоле соответственно.
„ 0,655/250
0,350/100 -°-749
б) Таким образом, найдена константа К- Тогда
ws/20
1,00/100
= 0,749
откуда получим ms = 0,1498 г.
в) В первом приближении можно предположить, что закон распределения Нернста справедлив также для насыщенных растворов
растворимость S в СС14
растворимость S в СвНв 91
Откуда найдем cs в СС14; cs—0,749-17,93= 13,43 г/л.
4. Применение закона Генри. Рассчитать растворимость СО2 в воде и коэффициент поглощения Бунзена при 24 °C и давлении 101325 Па. СО2 имеет небольшую растворимость в воде, поэтому примем, что 1 л раствора содержит 1000 г воды.
Решение. Найдем константу для процесса растворения СО2 в воде при 25 °C:
РСО„
К = -~-= 1,67- Ю8 Па
*со2
101 325 / Ю00 \
К = 1,67-10» Па = псо* упсо2 + 18,02 )
Первым членом в скобке можно пренебречь по сравнению со вторым; тогда 101325-55,5 „ „о ,
пСо2 = —Й67Й08-------= 3,38• 10-2 моль/л
Вычислим коэффициент поглощения Бунзена (разд. 23.3.8)
аБ = 3,38-10-2 моль/л-22,41 л/моль = 0,747
Вопросы и задания
1. Напишите уравнения для констант равновесия Кр следующих реакций:
С2Нв (газ) С2Н4 (газ) + Н2 (газ)
2NO (газ) + О2 (газ) 2NOa (газ)
NOa (газ) 4- SO2 (газ) SO3 (газ) + NO (газ)
ЗО2 (газ) 2О3 (газ)
2. Для реакций, указанных в задании 1, определите отношение Кр к при 25 °C.
3. Изменение стандартной свободной энтальпии образования НС1 из элементных газов в нормальных условиях составляет 95,01 кДж/моль. Рассчи тайте термодинамическую константу равновесия диссоциации НС1 на Нг С12 при 25 °C.
Химические и фазовые равновесия
263
4. Напишите уравнения для констант равновесия следующих реакций: О2 (газ) + 2Hg (жидк ) ч=ь 2HgO (тв )
НСО3" (водн ) + Н+ (водн.) :<.СО2 (газ) + Н2О (жидк.)
NH3 (газ) 4- Н2О (жидк.) NH4OH (водн.)
MgO (тв.) + 2НС1 (газ) MgCl2 (тв.) -|- Н2О (газ)
5. Соединение NH4HS разлагается по уравнению
NH4HS (тв.) т—» NH3 (газ) + H2S (газ)
При 25 °C давление над твердым соединением равно 66,7 кПа. Рассчитайте: а) константу равновесия и б) общее давление в условиях равновесия, если в систему введен аммиак под давлением 40,0 кПа.
6. Константа равновесия для реакции
2CaSO4 (тв ) <=£ 2СаО (тв ) + 2SOa (газ) + О2 (газ)
при 1625 К равна КР—1,45-10-5. Рассчитайте общее давление в системе при разложении CaSO4 при температуре 1625 К.
7. Пусть SO2 распределяется при 20 °C между 200 мл СНС13 и 75 мл Н2О; в результате в СНС13 содержатся 0,14 моля SO2, а в воде — 0,05 моля SO2. Рассчитайте коэффициент распределения SO2 между водой и хлороформом при 20 °C.
8. Коэффициент распределения Н3ВО3 между водой и амиловым спиртом при 25 °C К=3,35. Сколько молей Н3ВО3 можно экстрагировать из 50 мл 0,2 М водного раствора борной кислоты при а) однократной экстракции с помощью 150 мл амилового спирта и б) трехкратной экстракции с помощью 50 мл амилового спирта.
9. Из приведенных ниже данных о распределении бензойной кислоты между водой и бензолом при 20 °C а) показать, что бензойная кислота в бензоле находится в виде ассоциатов, включающих две молекулы, и, учитывая это обстоятельство, б) рассчитайте коэффициент распределения (диссоциацией кислоты в воде пренебречь).
Равновесная фаза Содержание бензойной кислоты, моль/л
Вода 0,015 0,0195 0,0289
Бензол 0,242 0,412 0,970
10. Давление паров воды при 40 °C равно 7375 Па. Рассчитайте давление пара над раствором 9,206 г глицерина в 360 г воды при той же температуре.
11. В 100 г эфира растворено 10 г нелетучего вещества. Давление пара над раствором при 20 °C равно 56,8 кПа, давление пара чистого растворителя при тех же условиях — 59,0 кПа Вычислите молекулярную массу растворенного вещества.
12. Растворимость СО2 и NO2, выраженная в виде коэффициентов поглощения Бунзена, равна 0,757 и 0,539. Рассчитайте, сколько молей этих соединений растворится в 10 кг воды.
Константа равновесия гомогенных реакций 265
264 Химическая термодинамика
24. ЗАВИСИМОСТЬ КОНСТАНТЫ РАВНОВЕСИЯ
ОТ ТЕМПЕРАТУРЫ И ДАВЛЕНИЯ (ГОМОГЕННЫЕ РЕАКЦИИ)
24.1. Зависимость константы равновесия от температуры и давления (основные уравнения)
Если объединить уравнение (2636) с уравнением изотермы Вант-Гоффа (292), можно получить уравнение изобары Вант-Гоффа
венно уравнением для константы равновесия этих реакций) Из уравнений (286), (290) и (292) следует
din К ДЕ
—др—= ~ ~RT' (дЛЯ конденсированных фаз) (326)
Входящая в уравнение (326) разность объемов веществ, участвующих в реакции, настолько мала в случае конденсированных фаз, что ею можно пренебречь в практических расчетах, т е не учитывать зависимость константы от давления.
24.2. Расчет констант равновесия для данной температуры с различной степенью приближения
Это уравнение показывает, что кочстанты равновесия для экзотермических реакций уменьшаются при увеличении температуры, а для эндотермических — увеличиваются. Если в первом приближении пренебречь температурной зависимостью энтальпии, то интегрирование уравнения (323) дает
к2 _L\
ln = R \ Л - Т2 )
(324)
Полученное уравнение аналогично уравнению Аррениуса в логарифмической форме Этого следовало ожидать, так как константа равновесия представляет собой отношение констант скорости прямой и обратной реакций, а энтальпия реакции — это разность энергий активации тех же реакций, входящих в химическое равновесие Если измерить константы равновесия при двух температурах, то из уравнения (324) можно рассчитать энтальпию реакции Если же известны энтальпия реакции и константа равновесия при одной температуре, то при помощи уравнения (324) можно вычислить константу равновесия при любой температуре. Зная константы равновесия при двух температурах, можно вычислить константу равновесия при любой температуре Если измерены константы равновесия в некотором диапазоне температур, то в соответствии с уравнением (324) можно предложить следующий графине ский метод расчета равновесия результаты измерений наносят на график зависимости 1g Л' от 1/Г Если диапазон температур не слишком велик, то результаты измерений укладываются на прямую, которая имеет положител^ ный или отрицательный наклон в зависимости от того, является реакция экзотермической или эндотермической, из коэффициента наклона этой прямой можно определить энтальпию реакции.
Пренебречь температурной зависимостью энтальпии можно только для не больших интервалов температуры.
Пример Рассчитать среднюю энтальпию реакции Na+Ch^^NO в диапа зоне температур от 2000 до 2500 К Kpi (2000 К) =4,08-10“4, Кр2 (2500К)я = 36 ю-4
Решение Преобразуем уравнение (323)
> 2 _ (^а ~ Л) /325)
lg Кр1 ~ 2,ЗЪЗРТ\Т2 (
0,0036 AН° (2500 — 2000) g 0,000408 = 2,303 8,314 2500 2000
А//0 = 181,2 кДж/моль
Рассмотрим в общих чертах зависимость константы К от давления Д1' конденсированных фаз (для 1аэовых фаз давление учитывается непосрет
Как уже было показано, уравнение (324) пригодно для расчета констант равновесия для любой температуры лишь в ограниченной степени (в узком диапазоне температур). Однако на практике часто сталкиваются с необходимостью определить константу равновесия для любой, значительно удаленкой от нормальной температуры из взятых в таблицах значений стандартных энтальпии и энтропии для нормальных условий, а также теплоемкости реагирующих веществ Иногда говорят об абсолютном расчете константы равно весия с помощью выведенного ранее уравнения изотермы Вант-Гоффа Аб°р
[уравнение (292)] 1пАР=——у~. Изменение свободной энтальпии [уравнение
(259)] в свою очередь включает энтальпию реакции ДЯР° и энтропию реакции Д8р0
AGP° = ДЯр» — TASpO (327)
Из уравнения (327) с учетом уравнения (292) следует
ДЯр» Д8п°
1п^р— RT + (328)
Это уравнение уже использовалось в ра>д 23 2 для расчета константы равновесия при нормальных условиях Следует подчеркнуть, что с помощью этого уравнения можно вычислить константу равновесия для любой температуры, если известна зависимость молярной теплоемкости от температуры [уравнение (226)] Тогда по аналогии с уравнением (227) из температурной зависимости молярной теплоемкости прежде всего находим по закону Гесса (212) энта 1ьпию реакции для заданной температуры Т
Определим зависимость энтропии реакции от температуры при постоянном давлении [уравнение (245)]
т
AS01 p = AS°2i)SP viCp (г) ~yi (329)
298
H после подстановки 2у,Ср(г ) = Аа1+А&1Г+Дс,Г’ получим 29! Ас,
ASOj-р = AS°248P + А«! 1п-г~ i А6, (Г - 298) + —2^-(Л - 2982) (330)
Рассчитав энтальпию и энтропию реакции, можно ле] ко найти константу Реакции для заданной температуры.
Часто для расчета константы равновесия с известной степенью приближения используют более простые соотношения, также полученные из урав-нения (328), а именно приближенные уравнения Улиха.
266
Химическая термодинамика
24.2.1. Первое приближение Улиха
Положим SvjCp(i)=O. Тогда в уравнение (328) можно подставить стандартные значения энтальпии и энтропии н найти константу равновесия
1п*„ =
Д^°298Р RT
AS°298p R
(331)
Подставив в это уравнение заданную температуру, получим константу равновесия исследуемой реакции.
24.2.2. Второе приближение Улиха
Пусть 2viCp(i) = a не меняется во всем заданном интервале температур, т. е. будем считать теплоемкость независимой от температуры. Тогда, объединив уравнение (328) с уравнениями (225) и (329), получим
1пКр =
Д#°298Р . Д5°298Р , Я / Т
~ RT + R R [ 298
(332)
Выражение в скобках в этом уравнении содержит только константы (во многих учебниках физической химии оно табулировано). В диапазоне температур до 1600 К можно принять, что 1п?/298—298/Т—1=0,0007?—0,2.
Пример, Рассчитать в первом и втором приближении Улиха, а также с учетом зависимости теплоемкости от температуры константу равновесия Кр реакции 2Н2 + О2Ч±2Н2О при 2500 °C, если известны следующие термодинамические данные:
Д//°298, кДж/моль Д5°298, Дж/(моль-К) СР298, Дж/(моль-К) Ср=а+ьт
н2 0 130,71 28,88 27,21+0,0038 Т
о2 0 205,21 29,38 27,21+0,0042 Т
Н2О (газ) —242,08 188,93 33,48 34,11+0,0021 Т
Решение. Стандартная энтальпия реакции и стандартная энтропия реакции при нормальных условиях равны
Д//°298Р — 2(—242,08) = —484,16 кДж/моль
AS°288P = 2S°298 Н2О 2S°298 Н2 '5°298 О2 ~
= 2-188,93 — 2-130,71 — 205,21 = —88,77 Дж/(К-моль)
Согласно первому приближению Улиха, подставим ДЯ+ар и AS°2esp в уравнение (331):
л ол 1А оо 77
lg = 19,148-2773 — 19,148’ = 9>118 — 4.636 = 4,482
Кр = 3,03-104
Согласно второму приближению Улиха, 1
°= 2чСр (г, = 2-33,48— 2-28,88 — 29,38 = —20,17 Дж/(моль-К) |
Расчет Кр проводится по уравнению (332); два его первых члена идентичны с рассчитанными в первом приближении Улиха; требуется рассчитать лишь
Константа равновесия гомогенных реакций
267
третий член уравнения (332) и прибавить его к выражению, рассчитанному в первом приближении:
а / Т 298 \
2,3/? П 298 + Т ~ J =
20,17 / , 2773 298 \
-----19,148 \i2,303 1g 298 + 2773 —1) = ~М13
Учитывая результат, полученный при расчете первого приближения, получим \gKp = 4,482— 1,418 = 3,064
Кр = 1,16-Юз
Запишем третье приближение, учитывающее зависимость теплоемкости от температуры, SviCp(i)=—13,39—0,0075 Т. Применив уравнение (227) для зависимости энтальпии от температуры, получим
Д //»277 зр = —496 160 — 13,39 (2773 — 298) — 0,0075
— ---(27732 — 2982) = —557 803 Дж/моль
Стандартная энтропия при температуре 2500 °C в соответствии с уравнением (330) равна
2773
Д5°2773р = — 88,77 — 13,38 2,303 • -ggg- —
— 0,0075 (2773 — 298) = —137,3 Дж/(К • моль)
Наконец, подставив полученные значения Д//°277зР и Д502773р в уравнение (328), получим
557 803 137,3
1g Др - 19,148-2773 — 19,148 = 3>И
КР= 1,3-103
Сравнение констант равновесия, рассчитанных в трех приближениях, показывает, что результат, полученный с помощью первого приближения Улиха, в ~20 раз преувеличивает истинную константу равновесия. В то же время результаты вычислений по второму и третьему приближению мало отличают, ся друг от друга. Таким образом, применение второго приближения Улиха вполне оправдано при расчетах большинства химических реакций можно не Принимать во внимание зависимость молярной теплоемкости от температуры), Расчет по первому приближению дает лишь грубую оценку равновесия. .
24.3.
Интерпретация температурной зависимости свободной энтальпии реакции
Сущность температурной зависимости химического равновесия можно себе наглядно представить, обратясь к уравнению (327).
В зависимости от знака энтропии н энтальпии могут осуществляться четыре типа химических реакций:
1) Д/7р»>0 и ASp“<0,
2) Д//рр<0 и Д£р°>0,
при любой Т равновесие смещено влево, так как
ЛОО и к<1
при любой Т равновесие смещено вправо
268
Химическая термодинамика
3) Д//р»>0 и ASpo>O,
при высокой Т равновесие смещено вправо
4) Д//р°<0 и Д$р«<0,
при низкой Т равновесие смещено вправо
Проведение реакций в промышленных масштабах при низкой температуре оказывается невыгодным из-за низкой скорости реакции^ поэтому в таких случаях применяют катализаторы. Примером реакции, протекающей по типу 4, является реакция синтеза аммиака — одна из наиболее известных каталитических реакций: при увеличении давления равновесие смещается вправо.
Здесь, по-видимому, полезно сделать еще некоторые замечания о значении свободной энтальпии (максимально полезной работы, или меры сродства). В соответствии с уравнением (291) свободная энтальпия включает две составляющие — основную и остаточную работу. Уравнение (291) целесообразно записать, введя обозначение
П = p2nh3/P3h2’Pn2
п
Дбр0 = RT In (333)
С помощью уравнения (333) можно вычислить работу реакции Дб% для любого состояния. Если П = КР, то система находится в состоянии равновесия, и для нее соблюдается закон действующих масс. Если П<КР, то система находится слева от равновесия и реакция самопроизвольно протекает слева направо; наконец, если П>КР, то система находится справа от равновесия и реакция самопроизвольно идет справа налево.
Самопроизвольному протеканию реакции слева направо соответствует отрицательное значение Дб°р, и из уравнения (327) следует, чтр отрицательное значение Дб°р тем больше по абсолютной величине, чем более отрицательна энтальпия реакции и чем выше положительное значение энтропии реакции. Тем самым подтверждаются две закономерности, характерные для процессов, протекающих в природе: каждая система стремится занять состояние с минимальной энергией (электроны в атомах, например, расположены на самых низких энергетических уровнях); одновременно каждая система стремится к состоянию с максимальной энтропией. Совместное действие этих закономерностей, регулирующих направление процессов в природе, приводит к требованию минимума свободной энтальпии в состоянии равновесия. Выразим энтальпию реакции через свободную энтальпию и энтропию [уравнение (327)]:
ДЯрО = Дбр° + TASpO (334)
Получим уравнение первого закона термодинамики. Д//°р в данном случае является мерой общего изменения энергии при превращении 1 моля вещества в соответствии с уравнением химической реакции. Дб°р — работа, совершаемая реакцией, a TASOP — обратимая теплота. Найдем из последнего уравнения Д5°р:
. <•, „ а^р° АСР°
ASp — уч гр (335)
По форме это уравнение совпадает с уравнением второго закона термодинамики (235). ЛН°Р/Т в данном случае представляет собой поток энтропии, а Дб°Р/Г—возникновение энтропии. Таким образом, величину Дб°р можно
Закон действующих масс для газовых реакций
269
представить себе не только как меру максимальной работы в обратимом изобарно-изотермическом процессе, но также как меру энтропии, возникающей при образовании 1 моля реагирующих веществ в ходе необратимого процесса.
25. ПРИМЕНЕНИЕ ЗАКОНА ДЕЙСТВУЮЩИХ МАСС
К ГАЗОВЫМ РЕАКЦИЯМ (ОПРЕДЕЛЕНИЕ ВЫХОДА РЕАКЦИИ)
Ранее уже показано, что из закона действующих масс, зная константу равновесия и исходный состав системы, можно вычислить равновесный состав реагирующих веществ. Такие расчеты называют определением выхода реакции. Читатель должен приобрести достаточный навык в технике таких расчетов, поэтому ниже приведено несколько подробно разобранных примеров расчета газовых реакций (гомогенных и гетерогенных). Реакции в растворах будут обсуждаться несколько позднее (гл. 35 и разд. 40.4).
Пусть имеется газ, который может диссоциировать, давая два продукта реакции. В исходном состоянии в системе присутствуют только молекулы газа и нет конечных продуктов. Долю газа, распавшегося на продукты реакции к данному моменту времени, называют степенью диссоциации а. Степень диссоциации— безразмерная величина, значение которой может меняться от 0 до 1. Если а=0, то распад газа не происходит, если а= = 1, то газ полностью диссоциирован.
Рассмотрим, например, реакцию диссоциации пентахлорида фосфора
РС15 РС13 + С12 (336)
В начальный момент имелось п0 молей PCI5. Закон действующих масс для этой реакции, если выразить его через мольные доли (p=Xip°), можно записать следующим образом:
(337)
Очевидно, что с увеличением давления равновесие смещается влево. Определим молярный состав в системе для начального состояния и для состояния, соответствующего степени диссоциации а, а также суммарное число молей в системе:
PCls РС1з
Исходное состояние п0 —
Равновесное состоя- По(1—и) апо
йие
С12
— Ло
ап» ло(1 + а)
270
Химическая термодинамика
Теперь можно рассчитать содержание (в мольных долях) веществ в реакционной смеси:
1 — а а а
xpci5 1 > ХРС13 1 _|_ а ’ хс1з 1 а
Подставим эти выражения в уравнение (337), после чего получим уравнение для расчета степени диссоциации а из константы равновесия реакции диссоциации Кр'
W-Rir <338>
В это уравнение не входит число молей по вещества в исходном состоянии, и его легко решить относительно а:
<339>
Из последнего уравнения легко вычислить а для заданного значения константы /Ср (последняя зависит от температуры). Для р° = 1 получим
Кр=10-* а я s 10~2
а й до-1
*₽ = ! а г к 0,707
КР=Ю2 а я - 1 10~2 ' 1 2
а л 1 _ 10~4 ' 1 2
Полученные результаты можно нанести на прафик зависимости а от 1g Кр (рис. Б.24). Приведенная на рис. Б.24 кривая рассчитана для частного случая, однако она типична для большинства подобных реакций.
При малых Кр происходит постепенное увеличение а, при /СР=1 наблюдается сильный рост а, и, наконец, при достаточно больших Кр а постепенено приближается к 1. Практически это означает, что если константа равновесия на несколько порядков меньше единицы, то выход реакции близок к нулю, а степень диссоциации значительно меньше единицы. Если же константа равновесия на 2 или 3 порядка больше 1, то равновесие реакции полностью смещено направо и реакция протекает практически до конца. При очень больших или очень малых КР можно упростить уравнение (338). При малых а можно принять 1—а«1; ири а, близком к 1, можно считать числитель-а=1 и учитывать только знаменатель (1—а). Такие допущения особенно важны для расчетов более сложных газовых реакций!, описываемых уравнениями третьего или четвертого порядков.
Закон действующих масс для газовых реакций
271
Рассмотрим другой пример газовой реакции:
О2 ч—» 20 (340)
Эта реакция отличается от рассмотренной ранее только тем, что исходная молекула распадается на два одинаковых атома. Это
LgX
Рис Б 24. Зависимость степени диссоциации от логарифма константы равновесия
приводит к несколько иному соотношению между Кр и а. Вновь определим состав реакционной смеси
^=Р°^ (341)
О, О s«(.
Исходное состояние По — По
Равновесное состояние л0(1—а) 2п«а По(1 + а)
Содержание веществ в реакционной смеси (в 1—а 2а
14-а 14-а
мольных долях)
Согласно закону действующих масс,
4а2
1 —а2
=
(342)
Особенность этого уравнения — появление коэффициента 4, что можно объяснить кинетическими закономерностями. При одинаковых константах реакции и давлении р° величина а, вычисленная из уравнения (338), оказывается больше, чем полученная из уравнения (342). Это объясняется тем, что реакция образования Ог идет быстрее, чем реакция образование пента-хлорида фосфора (336), в которой для осуществления элементарного акта реакции должны столкнуться разные молекулы.
272
Xимическая термодинамика
Закон действующих масс для газовых реакций
275
Рассмотрим реакцию, в которой принимают участие два исходных вещества:
СО2 + Н2 со + Н2о (343)
Здесь уже нельзя говорить о степени диссоциации. Вместо нее введем также безразмерную степень превращения у. Однако эта величина имеет смысл только в том случае, если в исходной смеси содержится одинаковое количество молей СОг и Н2. Предположим также, что в исходной смеси нет продуктов реакции (речь идет о так называемой стехиометрической смеси). По закону действующих масс
Р°со-Р°н2о Р°со2-Р°н2 хС0'хИ2О — *СО2-*Н, = КХ (344)
Равновесие не зависит от давления, так число молей газа в системе не меняется. как в ходе реакции Определим состав ре-
акционной смеси: со 2 На со H2Oj
Исходное состояние п0 «о — 2л»
Равновесное состояние «о(1 -Y) л0(1 — Y) ПоУ п0У 2п„
Содержание веществ в реакционной смеси (в мольных долях) 1 —Y 2 (1-Y) 2 У 2 У 2
кР- У2 (1-Y)2 (345)
После извлечения корня из уравнения (345) получим линейное уравнение для степени превращения.
Для реакции синтеза аммиака закон действующих масс, выраженный через мольные доли, можно записать в следующем виде:
Это уравнение имеет довольно сложный вид, однако после извлечения квадратного корня из выражений, стоящих по обе-стороны знака равенства, получается обычное квадратное уравнение относительно у.
Если исходные вещества не находятся в стехиометрическом соотношении,, то величинами а и -у пользоваться нельзя. Введем новый параметр — число прореагировавших молей Оно характеризует число молей исходных веществ, прореагировавших в эквивалентном соотношении до равновесного состояния. Параметр £ измеряется в молях; он показывает, сколько молей продуктов реакции в эквивалентном соотношении находится в системе в данный момент времени. Если £ увеличивается, то реакция идет слева направо, если уменьшается, то реакция протекает в обратном направлении. Предположим, что перед тем, как началась реакция (343), было смешано и°со, и п°н2 молей. Тогда закон действующих масс, выраженный через £, имеет следующий вид:
р («0со2-?)(«°н2-^)
(348)
Здесь весьма уместен вопрос- в каком стехиометрическом соотношении должны быть исходные вещества, для того чтобы выход был наибольшим, иначе говоря, величина £ достигала максимума’ Расчеты показывают, что-максимальный выход достигается как раз при стехиометрическом составе системы, поскольку рассматриваются смеси, в которых сумма молей исходных веществ постоянна. Для реакции (343), например, для смеси 5 молей СО? и 5 молей Н2 выход больше, чем если, например, смесь содержит 4 моля СОг и 6 молей Н2 или 3 моля СО3 и 7 молей Н2 Однако выход реакции будет еще выше, если в смеси содержится 5 молей СО2 и 6 молей Н2 *
Вопросы и задания
1. Константа диссоциации диоксида углерода па монооксид углерода и кислород при 1200 К равна 3,276 10~16. Если энтальпия этой реакции в заданной области температур равна 565,45 кДж/моль, рассчитайте константу-равновесия а) по уравнению (324); б) по уравнению (324), учитывая температурную зависимость молярной теплоемкости (Ср Дж/(моль-К)):
Ср(сО) = Ср(о2) =(27,21+0,0042 Г)
и Ср(со2, = (32,23+0,0222 7—3,48 10~6 Г);
в) в соответствии со вторым приближением Улика из следующих данных:
Найдем состав реакционной смеси: NH3
Н2 n2
Исходное состояние Зи0 «0 — 4и0
Равновесное состояние Зп„( 1 — у) М1 — у) 2и07 2л0(2 —7)
Содержание веществ 3(1-7) 1 —у 27
в реакционной смеси (в мольных долях) 2(2-7) 2(2-т) 2-7
Д//°298) Обр, кДж/моль —393,51 —110,45
0
S°298, Дж/(моль-К)
213,83
198,05
205,21
298 ’ Дл</(МЪЛЬ • К)
37,29
29,13
29,38
СО2 СО О2
2. Рассчитайте константу равновесия реакции С2Н...+ 112О,+ДН5ОП при 25 °C в газовой фазе из следующих данных:
Подставим ети данные в уравнение закона действующих масс:
С2Н4
Н2О (газ)
С2Н3ОН (газ)
ДД^298, обр> КДж/МОЛЬ
52,57
—242,08
-235,18
1 64?2(2-т)2
лр— (р°)2 ' 27(1 - у)*
(347)
S°29s, Дж/(моль К)
219,61 188,93 277,91
, -г------—„..„..а константу равновесия
;2Н2О+2С12 при 400 °C и нормальном давлении. Равно-
РеакцииЭ4НС11Т+ОТе приближению Улиха
ls-2027
274
Химическая термодинамика
весный состав укажите в мол.% Определите состав при давлении 5-10® Па.
обр кДж/моль S°298. Дж/(МОЛЬ-К) с Р 298’ Дж/(молъ- К)
НС1 —91,62 186,92 29,13
О2 0 205,21 29,39
Н2О (газ) —242,08 188,93 33,48
С12 0 223,17 33,78
4. Коистаита равновесия реакции CO + H2Os±CO2+H2 при 800 К равна Лр=4,12. Смесь, содержащая 20% СО и 80% Н2О (массовые %), нагревается до 800 К. Рассчитайте равновесный состав и выход водорода, если в реакцию вступит 1 кг воды.
5. Константа равновесия реакции H2 + I2s±2HI при 444°С равна KrJ = = 46,7. Рассчитайте, сколько молей HI распадется на водород и иод, если 1 моль HI нагреть до 444 °C. Найдите степень диссоциации HI.
6. 1,518 ммоль 12 при 800 °C занимает объем 249,3 мл при давлении 58,075 Па, Рассчитайте константы равновесия Кр и Кс реакции диссоциации
7. При 49,7 °C и давлении р=34,85 кПа N2O4 диссоциирует: N204=f±2N02 Определите степень диссоциации при той же температуре и давлении 12,506 кПа.
8. При каком давлении в замкнутом объеме должен находиться РС15, для того чтобы произошло его разложение на 30%? Кр Для реакции РСЬ** ч±РСЫ-С12 при 250°C равно 1,78.
9. Для реакции А+В+*АВ при 27 °C AG% = 8,4 кДж/моль. При каком общем давлении газовой смеси произойдет превращение в АВ иа 40% ?
10. Изменение стандартной свободной энтальпии реакции (Дж/моль) СО(газ)+С12(газ) ч±СОС12(газ) можно записать в виде следующего уравнения Дб°р=(—105+16,74 Т In Т + 14,65 Т). Рассчитайте парциальное давление хлора в равновесии с фосгеном при 250 °C и нормальном общем давлении, предполагая, что для этой системы выполняются законы идеальных газов.
11. Даны две реакции с соответствующими константами равновесия:
2Н2 + О2 ^=4: 2Н2О
24900К
Кс =---1,335 lg Т + 9,65- IO"5 Т — 1,37-10-7 Т2 4- 1,08
W + VsCl, 4=^ НС1
lgKc =
9586К
Т ~
0,44 lgT4- 2,16
'Определите константу равновесия реакции 4НС1+О2^2Н2О+С12 при 450 и 600 °C, а также внутреннюю энергию и энтальпию этой реакции при Т= -=800 К.
26. ГЕТЕРОГЕННЫЕ ПРОЦЕССЫ
При выводе закона действующих масс не делалось никаких особых предположений о фазовом составе системы, поэтому его можно применять как к гомогенным, так и к гетерогенным системам. В гл. 23 уже были разобраны некоторые частные случаи равновесия в гетерогенных системах, которые, однако, нельзя назвать химическими реакциями — они подчиняются более общим физическим закономерностям. В этой главе будут более подробно рассмотрены такие гетерогенные процессы. Все коли
Гетерогенные процессы 275-
чественные соотношения для гетерогенных равновесий, которые-обсуждаются далее, можно легко получить из фундаменталь-ных уравнений (276) и общего условия равновесия dg = 0. В следующем разделе используются и другие уравнения, которые, впрочем, также тесно связаны с уравнением (276) 'и условием равновесия dg = Q.
26.1. Фазовые равновесия
26.1.1. Фазовые равновесия в однокомпонентных системах
С частным случаем такого равновесия мы уже познакомились на примере равновесия вода — пар [уравнение (310)]. К равновесиям такого же рода можно отнести системы твердая1 фаза — расплав, твердая фаза — пар (сублимация), а также-равновесие между модификациями одного и того же соединения, например фазовый переход между ромбической и моноклинной серой. Равновесие между жидкостью и паром в координатах р— Т можно изобразить графически, исследуя зависимость равновесного давления пара над жидкостью от температуры. Если диаграмму р — Т расширить и поместить там зависимость температуры плавления от давления и давления пара от температуры сублимации, то получим диаграмму состояния рассматриваемого вещества (рис. Б.25). Ход всех этих кривых на р — Т-диаграмме определяется общим термодинамическим уравнением, известным как уравнение Клаузиуса — Клапейрона [его можно вывести из уравнения (276) и условия равновесия dg=O; вывод здесь не приводится]:
др ЛНф ~dT~~ ТДУ
Здесь АНф — теплота фазового превращения, т. е. разность молярных энтальпий вещества в различных агрегатных состояниях, ДУ — разность молярных объемов равновесных фаз, ДТ/ф/Т— энтропия фазового превращения. Если использовать это уравнение для равновесия жидкость — пар, то можно пренебречь молярным объемом жидкости по сравнению с молярным объемом пара и при не слишком больших давлениях считать ДУ как объем газовой фазы, вычисленный с помощью законов идеальных газов:
din р &Нф / о к
ат ~ rt2
Это уравнение похоже на уравнение (323); давление при фазовом превращении р, как показано ранее, имеет смысл константы равновесия. По аналогии с уравнением (323) уравнение (350) можно интегрировать. Зная давление pi при температуре 18*
276
Химическая термодинамика
Ti, можно определить значение р2 при температуре Т2 по уравнению
1пА
Pi
ДЯф / 1 R I Т\
(351)
Таким образом, из данных о давлении пара при различных температурах можно определить теплоту испарения или субли-_____________________________ мации. Напротив, зная теп
Рис. Б.25. Диаграмма состояния воды.
лоту фазового перехода, можно вычислить давление пара при любой температуре, если оно известно для какой-либо определенной температуры. При исследовании более широких диапазонов температур необходимо учитывать зависимость энтальпии испарения от температуры [по аналогии с уравнениями (224) или (227)].
Кривая зависимости давления пара от температуры имеет физический смысл лишь в определенной области давления и температуры. При увеличении температуры и давления плотности жидкости и пара начинают сближаться между собой по своему значению, и наконец достигается такое
состояние, при котором жидкость и пар становятся неразличимыми по всем термодинамическим параметрам (например, по плотности и молярному объему), исчезает граница жидкость — пар, соответственно поверхностное натяжение становится равным нулю. Это состояние называется критическим. Энтальпия испарения с увеличением температуры понижается и в критическом состоянии также становится равной нулю. Соответствующая температура называется критической Ткрит (аналогично ркРИТ, УКРит, ркрит). В критическом состоянии пар и жидкость неразличимы. Нельзя счи
тать, как это иногда делают, что критическое состояние отличается тем, что вышё критических температуры и давления невозможно превратить газ (пар) в жидкость. Критическая плотность составляет примерно треть плотности жидкости в нормальных условиях, соответственно она в 300 раз больше плот-
Гетерогенные процессы
277
дести пара этой жидкости. Вещество в критическом состоянии более сходно с жидкостью, чем с газом. В качестве примера приведем критические параметры воды:
Лрйт:=б47К, Ркрит = 22,14 МПа, Екрит==55,6см3/моль;
Ркрит = 0,324 г/см3
Если написать уравнение Клаузиуса — Клапейрона для процесса плавления, то в знаменателе окажется разность молярных объемов конденсированных фаз. Поэтому знаменатель — очень малая величина, а следовательно, тангенс угла наклона кривой зависимости р—Т очень велик; при не очень больших давлениях эта ветвь диаграммы представляет собой практически вертикальную прямую (рис. Б.25). Если молярный объем жидкой фазы больше, чем твердой, то температура плавления увеличивается при повышении давления; это и наблюдается почти для всех твердых веществ. Исключением является вода: температура плавления льда уменьшается с повышением давления, так как молярный объем льда больше, чем жидкой воды (поэтому айсберги плавают, а не тонут).
Точка на диаграмме р—• Т, в которой сходятся кривые зависимости давления от температуры для равновесий жидкость-— пар, жидкость — твердая фаза и твердая фаза — пар, называется тройной точкой. При термодинамических параметрах тройной точки в системе находятся в равновесии одновременно три фазы: твердая, жидкая и газообразная. Кривая сублимации твердой фазы идет от тройной точки до температуры абсолютного нуля, при которой давление в соответствии с тепловым законом Нернста приближается к нулю по касательной, параллельной оси температуры. Кривые равновесий жидкость — пар, жидкость — твердая фаза и твердая фаза — пар делят диаграмму состояния на три области: области существования пара, жидкости и твердой фазы (рис. Б.25). Видно, что при температуре тройной точки кончается область жидкости. Твердая фаза и пар могут существовать вплоть до абсолютного нуля температуры (даже вблизи абсолютного нуля над 'твердой фазой имеется некоторое давление пара данного вещества). Особую диаграмму состояния имеет гелий: на ней нет тройной точки; гелий находится в жидком состоянии при температуре, максимально близкой к абсолютному нулю; для того чтобы перевести его в твердое состояние, необходимо увеличить давление До 2 МПа.
Для веществ, которые имеют несколько твердых модификаций, диаграмма состояний усложняется, так как появляются кривые, соответствующие равновесию между этими модификациями. Так, например, при повышенном давлении существует Несколько модификаций льда, равновесие между которыми ^ожно показать при помощи кривых на диаграмме состояния.
278
Химическая термодинамика
Для таких веществ можно было бы полагать, что существует состояние, при котором одновременно в равновесии находятся четыре фазы («четверная точка»). Однако из общих термодинамических соображений для однокомпонентной системы это невозможно; согласно правилу фаз Гиббса (приводим без вывода)
С = К + 2-Ф (352)
Здесь Ф — число фаз, К — число компонентов, т. е. различных по химическому составу Веществ, С — число степеней свободы, т. е. число интенсивных термодинамических параметров, которые могут меняться в системе при условии, что число фаз остается неизменным. В качестве примера рассмотрим однокомпонентную систему, К=1. Если имеется лишь одна фаза, то, согласно уравнению (352), число степеней свободы равно 2. Это может быть температура и давление либо жидкости, либо газа, либо твердой фазы. При равновесии двух фаз С=1. Если, например, задано давление пара, то температура кипения есть функция давления пара. Если одновременно сосуществуют три фазы (тройная точка), то С = 0. Следовательно, тройная точка одного вещества характеризуется единственным набором значений температуры и давления. В четверной же точке (четыре фазы) для однокомпонентной системы число степеней свободы было бы равно —1, следовательно, равновесие четырех фаз в такой системе невозможно. Для серы, например, не существует состояния, при котором одновременно находились бы в равновесии две твердые фазы (ромбическая и моноклинная сера) — жидкость и пар. Четверная точка наблюдается только на диаграммах состояния двухкомпонентных систем.
Особого внимания заслуживает случай, когда в многокомпонентной системе протекают одна или несколько гомогенных реакций. Тогда К в уравнении (352) правила фаз равно числу имеющихся в системе видов молекул минус число гомогенных реакций между этими молекулами. Число К называют числом независимых составляющих термодинамической системы. На примере воды можно показать, насколько полезно это уточнение. Как известно, в жидкой воде существуют ассоциаты, т. е. кроме молекул Н2О также молекулы (Н2О)2, (Н2О)4 и (Н2О)в-Однако даже если принять это во внимание, число независимых составляющих должно быть равным единице. Для молекул (Н2О)2, например, осуществляется равновесие
2Н2О (Н2О)2 (353)
Два вида молекул, связанные одним уравнением реакции, Да' ют одну независимую составляющую, то же самое получается для ассоциатов с большим числом молекул Н2О. Следователь*
Гетерогенные процессы
279
но, в системе имеется одна независимая составляющая независимо от того, сколько равновесий ассоциации типа (353) могут осуществляться в воде.
26.1.2. Равновесие между раствором и растворителем
Один из примеров такого равновесия уже разобран в разд. 23.3.5 (понижение давления пара раствора по сравнению с давлением пара чистого растворителя при той же температуре). При этом предполагалось, что из раствора может испаряться только растворитель. Далее будем полагать растворы идеально разбавленными, т. е. считать осмотический коэффициент и коэффициент активности равным 1. Нанесем на диаграмму состояния чистого вещества изменение давления пара над раствором (понижение относительно чистого растворителя) некоторой концентрации от температуры (рис. Б.26). Из сопоставления кривых сразу же становится ясным, что понижение давления пара при постоянной температуре соответствует повышению температуры кипения при постоянном давлении. Отношение &р/\Т приблизительно равно тангенсу угла наклона кривой р — Т для раствора, и с хорошим приближением можно заменить его наклоном кривой р—Т чистого растворителя. Если подставить значение Ар из первого закона Рауля (311) d\npxp\plp = x2, то с учетом уравнения (350) получим
Таким образом, повышение температуры кипения пропорционально мольной доле растворенного вещества. Коэффициент пропорциональности зависит только от свойств чистого растворителя. Можно преобразовать уравнение (354) таким образом, чтобы вместо мольной доли в него входила моляльность m растворенного вещества [используя уравнение (233)]:
ДТ = £эт (355)
Константу Еэ называют молярным повышением точки кипения или эбулиоскопической константой. Если продолжить кривую Давления пара над раствором до пересечения с кривой равновесия -между твердой фазой растворителя и паром (с кривой сублимации), то получится тройная точка раствора. Кривая сублимации для раствора совпадает с кривой сублимации для растворителя, так как с полным правом можно принять, что при Постепенном охлаждении раствора прежде всего переходит в Твердое состояние растворитель.
Кривую зависимости температуры замерзания раствора от Давления получим, если проведем из тройной точки раствора Линию, параллельную аналогичной кривой для чистого раство
280
Химическая термодинамика
рителя. В соответствии с уравнением Клаузиуса — Клапейрона кривая замерзания раствора также почти вертикальна. Из всех этих соображений становится ясно, что точка замерзания раствора находится ниже, чем точка замерзания растворителя. Используя теплоту замерзания растворителя, можно вывести фор.
Рис Б 26. Повышение температуры кипения и понижение давления пара
мулу для определения понижения температуры замерзания (здесь также удобнее всего использовать моляльную концентрацию растворенного вещества):
DT2
АТ==-Х77---х2=Ект (356)
Ек зависит только от природы растворителя и называется криоскопической константой. Уравнения (355) и (356) называют также вторым законом Рауля. Из уравнений (355) и (356) следует, что повышение температуры кипения и понижение температуры замерзания зависит только от моляльности раствора (от мольной доли или числа растворенных частиц), т. е. от концентрации раствора, а не от их природы.
Такие свойства (еще пример — мольный объем идеального газа), которые зависят только от числа частиц, а не от их химического состава, называют коллигативными свойствами-
Гетерогенные процессы
281
Коллигативные свойства можно использовать для определения молекулярной массы вещества. Например, если, зная массу пг растворенного вещест-ва, определить температуру замерзания (кипения) раствора, то, найдя понижение, повышение) температуры замерзания (кипения) раствора, можно вычислить число молей п растворенного вещества, а затем и саму молекуляр-ную массу вещества М=т/п Таким образом можно определить степень диссоциации или ассоциации вещества в растворе. В этом случае следует умножить правую часть уравнений (355) и (356) на введенный Вант-Гоффом в соответствии с уравнением (322) коэффициент I. Понижение температуры замерзания раствора повареной соли примерно в два раза больше, чем для раствора сахарозы той же моляльной концентрации. На практике чаще используют криоскопический метод, так как он более прост в экспериментальном исполнении, а кроме того, как правило, криоскопическая константа для одного и того же растворителя больше, чем эбулиоскопическая. Для растворителя камфары, например, /Д = 40 К-кг/моль.
Пример. Найдите молекулярную массу динитробензола, если при растворении 1 г этого вещества в 50 мл бензола температура кипения повышается на 0,3°C Для бензола £э==2,53 К-кг/моль.
Решение. В соответствии с определением моляльности [уравнение (233)] уравнение (355) можно записать следующим образом:
Отсюда г, и2-1000 л гп 1,00-1000 ,
= -ТОГ = 2,53 "0,3-50,б-= 170 Г/МШ1Ь
С коллигативными свойствами растворов связано и осмотическое давление. Рассмотрим так называемую ячейку Пфеффе-ра. Она представляет собой трубку, заполненную раствором и закрытую снизу полупроницаемой мембраной (перегородкой); последняя проницаема для молекул растворителя и не пропускает растворенное вещество. Трубка погружена в сосуд с растворителем (рис. Б.27). В начальный момент времени система неравновесна, и растворитель начинает переходить через мембрану в трубку. Теоретически равновесие должно характеризоваться равенством химических потенциалов чистого растворителя и раствора. Разумеется, такое положение недостижимо, так как химический потенциал раствора включает остаточный потенциал ЯТЧплц, а следовательно, химический потенциал раствора всегда меньше, чем химический потенциал растворителя.
Движущая сила, обусловленная разностью химических потенциалов, «переносит» растворитель через мембрану в трубку. Проникновение растворителя можно объяснить как диффузию под действием градиента концентрации: в результате проникновения растворителя в раствор содержание растворителя Xi увеличивается и постепенно приближается к 1, хотя не может быть ей равным, сколько бы растворителя ни прошло в трубку, так как в растворе остается исходное количество растворенного вещества. Поэтому, для того чтобы система находилась в
28:
Химическая термодинамика
равновесии, стандартный потенциал растворителя в растворе должен увеличиться, чтобы компенсировать остаточный потен-циал (концентрационный член). Напомним, что стандартный химический потенциал зависит от температуры и давления. В результате поступления растворителя в трубку уровень раствора в ней повышается и давление в растворе (за счет гидростатического давления) оказывается на л больше, чем в растворителе, находящемся в сосуде. Увеличение давления над раствором соответствует увеличению стандартного потенциала растворителя в соответствии с уравнением (287). Равновесие в системе установится после того, как л достигнет такого значения, при котором химические потенциалы растворителя в сосуде и растворителя в растворе станут равными.
Условие термодинамического равновесия между растворителем с Н1°(Т, Ро) и раствором с химическим
пар
h-h0
растворитель у
раствор
полупроницаемая меморана
Рис. Б.27. Схема опыта для наблюдения осмотических явлений.
химическим потенциалом Ц1°(7, ро) и раствором с химическим потенциалом gi° (Т, p)+RT\nxt записывается следующим образом:
Н1°(Л Ро)=Н1°Г. рНЯПпх,
Далее зависимость химического потенциала раствора от давления с учетом гидростатического давления л [уравнение (286)] состоит в следующем:
Р1° (Т, р) = И1о (Т, р0)+J Vxdp=(Т, р0) +^л (358)
Ро
Подставив это выражение в указанное выше уравнение условия равновесия, получим
— RTlnx^ — ДПп(1—х2) (359;
Мольная доля растворенного компонента очень мала по сравнению с единицей, поэтому после разложения в ряд In(I—Хг) и отбрасывания членов более высокого порядка, получим 1п(1—х2) да—Х2- Тогда из уравнения (359) следует
ViJt=RTx2 (360)
Так как для разбавленного раствора то X2~n2/tii. ЕсЛЯ
подставить это выражение в уравнение (360) и принять во PHli'
Гетерогенные процессы
283
мание, что tiiVi равно с достаточной степенью точности 1000 см3, то получим
л — cRT (361)
Это уравнение называют законом Вант-Гоффа. В него входит молярная (моль/л) концентрация раствора (равновесная!). Осмотическое давление пропорционально количеству частиц в растворе, т. е. это — коллигативное свойство раствора. Если в уравнении (361) вместо с подставить п/V, то оно примет форму уравнения состояния идеального газа. Таким образом, можно сказать, что осмотическое давление равно давлению, при котором находились бы частицы растворенного вещества, если бы они заполняли весь объем раствора в виде идеального газа. Однако в действительности имеют дело не с идеальным газом, а с реальными молекулами вещества, взаимодействующими с молекулами растворителя.
Такие упрощенные представления приводят иногда к широко распространенным неверным выводам о том, что осмотическое давление аналогично парциальному давлению растворенного вещества. Подобные представления господствовали в начале развития химической термодинамики и использовались, например, для того, чтобы установить зависимость химического потенциала от концентрации для идеального разбавленного раствора. При этом в термодинамике растворов придавали излишне большое значение исследованиям осмотического давления.
Осмотическое давление также можно использовать для определения молекулярной массы. Согласно уравнению (361), осмотическое давление раствора сахарозы с концентрацией 1 моль/л при 25°C составляет ~2,5 МПа. Это соответствует гидростатическому давлению столба воды высотой 240 м (для раствора поваренной соли такой же концентрации осмотическое давле ние равно 5,0 МПа). Таким образом, осмотические явления проявляются в количественном отношении в значительно большей степени, чем криоскопические и эбулиоскопические измерения. Поэтому измерения осмотического Давления для определения молекулярной массы можно проводить со значительно меньшей концентрацией растворенного вещества.
Концентрация насыщенных растворов различных веществ, выраженные в граммах на литр, близки между собой по порядку величин. Поэтому соответствующие молярные концентрации оказываются тем меньше, чем больше молекулярная масса растворенного вещества. Поскольку с помощью измерений осмотического давления можно определить молекулярную массу в весьма Разбавленных растворах, этот метод особенно удобен для исследования высокомолекулярных соединений. Он обладает большими преимуществами также по той причине, что изготовление подходящей мембраны оказывается тем проще, чем больше различаются размеры молекул растворенного вещества и Растворителя.
26.1.3. Равновесие между раствором и растворенным веществом в твердой фазе и между двумя растворами
К рассмотренным ранее равновесиям такого типа, как растворимость газов (закон Генри — Дальтона), растворимость трудиорастворимых твердых °€Ществ и закон распределения Нернста (разд. 23.3.6—23.3.8), мы уже не УДем больше возвращаться.
284
Химическая термодинамика
26.1.3.1. Диаграммы давления пара и диаграммы плавлени-
До сих пор обсуждались термодинамические свойства силь но разбавленных растворов. Обратимся теперь к таким жидкостям, которые смешиваются между собой в любом соотношении. Основными параметрами системы здесь являются температура, давление и состав смеси, который будет выражаться в мольных долях Xi и (1—%i) (для компонента 1 и компонента 2 со-
Рис Б 28 Диаграмма давления пара над смесью двух жидкостей
/ — давление пара в зависимости от мольной доли жидких компонентов жидкой фазыс 2 —давление пара в зависимости от мольной доли компонентов в газовой фазе
ответственно). Довольно сложно изобразить на плоскости взаимосвязь всех трех параметров (плоская координатная система связывает только два параметра). Известны три варианта диаграмм состояния таких систем. Во-первых, можно составить р — T-диаграмму для системы с постоянной мольной долей Xi, как это было показано на рис. Б.26. Во-вторых, для того чтобы более наглядно выявить зависимость состояния системы от состава, пользуются диаграммой р — Xi при постоянной температуре Т — диаграммой давления пара. Наконец, можно построить диаграмму Т — х1 при постоянном давлении (обычно при атмосферном давлении). Такие диаграммы наиболее важны, так как чаще всего применяются на практике; их называют обычно диаграммами кипения.
Рассмотрим сначала идеальные смеси жидкостей. Они характеризуются прежде всего тем, что для них во всем диапазоне концентраций выполняется первый закон Рауля:
PilPi=xt (362)
Гетерогенные процессы
285
где pi — парциальное давление компонента 1 над смесью, р<°— давление пара чистого компонента 1. Точно так же для компонента 2 получим
р2/р2°=х2 (363)-
Очевидно, что каждое из этих уравнений в случае сильно разбавленного раствора переходит в рассмотренное ранее уравнение первого закона Рауля (310); Xi и х2— мольные доли соответствующих веществ в растворе. Для неидеальной смеси форма уравнений не меняется, если вместо мольных долей подставить значения активностей щ и а2.
Уравнениям (362) и (363) соответствуют прямые на диаграмме зависимости парциального давления от мольной доли раствора (на рис. Б.28 изображена также зависимость общего-давления пара от состава жидкой смеси, представляющая собой прямую). Пар над жидкой смесью, как правило, имеет иной состав, чем состав жидкой фазы. Пусть У и х'2 — мольные доли каждого компонента в газовой фазе (состав пара), полагаем ^ = ^[4-^2. В соответствии с законом Дальтона парциальное-давление в газовой смеси равно произведению мольной доли на общее давление:
Р1 = х/р, р2 = х2'р (364)
Учтем, что p = Pi-\-p2, и подставим парциальные давления из уравнений (362) и (363):
Таким образом получили состав газовой фазы, выраженный через состав жидкой смеси. Уравнение (365) нелинейно. Зависимость давления пара р от мольной доли Xi компонентов в газовой фазе на диаграмме р — х изобразится кривыми (рис. Б.28). Из уравнения (365) следует, что Xi совпадает с x'i только при P°i = p°2- В других случаях пар обогащается компонентом с большим давлением пара. С помощью диаграммы, изображенной на рис. Б 28, можно определить состав жидкой фазы, если известен состав пара. Однако, так как парциальное давление компонентов не представляет большого интереса для практики, более удобно использовать график другого типа, а именно зависимость общего давления пара от Xi и х/, т. е. от состава Жидкости и пара. Представим себе, что для некоторого температурного интервала имеются необходимые данные для до-Строения таких диаграмм. Тогда для некоторого давления р Можно найти соответствующую температуру кипения и построить диаграмму температуры кипения (рис. Б.29), которая для Идеальной смеси представляет собой две кривые (на диаграмме давления пара для идеальных смесей имеется только кри
•286
Химическая термодинамика
вая, отвечающая составу пара). Кривые на диаграмме темпера-туры кипения называются кривой кипения и кривой конденса-.ции (выпадения росы). Кривая конденсации соответствует сс
Рис. Б.29. Диаграмма кипения смеси О2 и Nj.
ставу пара, а кривая кипе ния — составу жидкой фа зы. Область на диаграмме расположенная выше кри вой конденсации, является областью пара, а ниже кривой кипения — областью жидкого состояния. Область, расположенная между кривыми конденсации и кипения, отвечает составу смеси, при котором не может существовать одна фа за — смесь распадается на жидкость и пар. Для реальных растворов на таких ди аграммах всегда имеются обе кривые, однако их фор ма может заметно меняться На некоторых диаграммах температуры кипения могут наблюдаться, например, максимумы или минимумы, в которых кривые конденсации и кипения совпадают, причем касательная в точке экстремума параллельна оси абсцисс. Смесь имеющая та-
кой состав, называется азеотропной.
Диаграммы температуры кипения имеют важное значение прежде всего для разделения жидкостей методом перегонки и ректификации. Как происходит разделение жидкостей, можно наглядно пояснить при помощи рис. Б 29. Нагреем смесь жидкостей данного состава до температуры, соответствующей температуре кипения, и удалим образовавшийся пар. Очевидно, что остающаяся жидкость обогащается компонентом 1 (О2). При неоднократном повторении этого процесса жидкая фаза обогащается компонентом 1 (правда, этим способом нельзя получить чистый компонент 1). Подробно с методами перегонки и ректификации, имеющими большое значение в химической практике, можно ©знакомиться по обширной специальной литературе.
Рассмотрим в общих чертах еще одну систему. Пусть имеются две жидкости, которые не могут смешиваться в любых отношениях. В некоторой средней области концентраций система распадается на две жидкие, фазы, одна из которых обогащена компонентом 1, а другая — 2. В такой системе имеет место расслаивание. Состав обеих жидких фаз зависит от температуры При некоторой достаточно высокой температуре разрыв растворимости исчезает (верхняя критическая температура растворения) Верхняя критическая температура может проявиться, если до этого момента смесь еще не испЯ'
Гетерогенные процессы
287
Рис. Б.ЗО. Система без смешанных кристаллов.
ится. Точно так же, если смесь еще не закристаллизовалась (не замерзла), Еаблюдается нижняя критическая температура растворения — достаточно ииз-^аЯ температура, при которой также исчезает разрыв растворимости.
26'1,3.2. Диаграммы плавления
Равновесие между твердой двухкомпонентной системой и ее расплавом также можно описать при пом'ощи диаграммы. Наиболее часто используют диаграмму плавления в координатах температура — мольная доля одного из компонентов бинарной системы при постоянном давлении. На рис. Б.ЗО приведена схема диаграммы плавления системы вода — NH4C1. Мольная доля NH4CI обозначена х\. Левая сторона диаграммы соответствует разбавленным растворам NH4C1 в воде. При понижении температуры раствор начинает замерзать при более низкой температуре, чем чистая вода (понижение точки замерзания). В соответствии со вторым законом Рауля при малых концентрациях NH4C1 наблюдается близкая к прямолинейной зависимость
температуры замерзания
раствора от состава, далее
отклонения от прямолинейности увеличиваются. То же самое наблюдается, если исходить из чистого хлорида аммония, постепенно добавляя к нему воду и понижая температуру. В этом случае также происходит постепенное понижение температуры замерзания. Обе кривые наконец пересекаются в некоторой точке, называемой эвтектической. Составы твердой фазы и расплава в этом случае называют эвтектическими. Следует подчеркнуть, что в нашем примере при замерзании водного раствора хлорида аммония прежде всего выпадают кристаллы льда и, Наоборот, при понижении температуры расплава хлорида аммония с небольшим содержанием воды прежде всего кристаллизуется хлорид аммония. Эвтектика (твердая фаза) представляет собой твердую смесь кристаллов льда и хлорида аммония, а не °Дну фазу. В других двухфазных системах эвтектика может представлять собой одну фазу. На рис. Б.ЗО показаны область Расплава и области существования расплава+кристаллы 1 (лед) и расплава + кристаллы 2 (хлорид аммония).
288
Химическая термодинамика
Применим правило фаз Гиббса к данной системе и выясним, зависит ли точка эвтектики от давления. В системе два компонента, и в точке эвтек. тики находятся в равновесии четыре фазы — расплав, лед, твердый хлорид аммония и газовая фаза (обязательно надо учитывать газовую фазу). Следовательно, в соответствии с правилом фаз Гиббса число степеней свободы должно было бы равняться нулю, т. е. точка эвтектики не зависит от давления. Однако это противоречит экспериментальным данным. Поэтому необ-
Рис. Б.31. Фазовые диаграммы систем Au—Ag (а) и Au—Си (б).
Область гомогенного расплава (I); область гомогенной твердой фазы (II); сосуществование жидкой и твердой фаз (III).
ходимо более внимательно подойти к применению правила фаз В действительности возможны три случая:
1) давление является равновесным, соответствует температуре системы, т. е. задано, и число степеней свободы равно нулю;
2) давление больше равновесного значения, газовая фаза, состоящая из водяного пара и NH4C1, полностью исчезает, пар конденсируется в расплав, остаются только три фазы и система получает одну степень свободы, т. е. при повышении давления точка эвтектики смещается;
3) газовая фаза содержит, например, воздух, в системе уже три компонента (а не два), т. е. по правилу фаз система также имеет одну степень свободы.
На этом примере можно ясно увидеть, что правильное применение правила фаз Гиббса связано с определенными трудностями. Следует подчеркнуть, что оно представляет собой точный закон термодинамики, имеющий всеобщее значение, поэтому термин «правило» является условным и появился в результате неточного перевода с английского языка.
Рассмотренная диаграмма-—пример лишь одного типа диаграммы плавления. Известны разнообразные системы, которым соответствуют более сложные зависимости и которые, однако, здесь не обсуждаются. Упомянем кратко лишь еще одну диаграмму для системы, где образуются смешанные кристаллы или сплавы при любом соотношении компонентов.
Пример такой системы — система медь — золото (рис. Б.31). Она сходна с диаграммой температуры кипения. Верхнюю кривую называют кривой ликвидуса, а нижнюю — кривой солидуса. В области между этими кривыми система распадается на две фазы. Ниже кривой солидуса находится область твердых смешанных кристаллов, выше кривой ликвидуса — область расплава.
Еще один тип диаграмм получают, если в системе при некоторой концентрации одного из компонентов в твердой фазе и расплаве образуются соединения. Существуют также системы, у которых до определенной конце11'
Гетерогенные процессы
289
трацпи происходит образование сплава, а в остальной области концентраций равновесие описывается диаграммой плавления уже рассмотренного типа рис Б.30. Более подробно с такого рода системами можно ознакомиться в руководствах по физической химии.
Диаграммы плавления строят на основе так называемого термического анализа. Как правило, измеряют кривые охлаж-
дения расплавов различного состава и записывают температуру расплава как функцию времени. Если начинается выпадение
кристаллов из расплава или кристаллизация расплава в целом, это становится заметным по изломам или остановкам на кривой охлаждения. Остановке на кривой охлаждения соответствует постоянство температуры в течение некоторого времени.
Выше рассмотрены системы, состоящие из двух компонентов,— бинарные системы. Большую роль при исследовании жидких смесей и твердых смешанных фаз играют также системы, состоящие из трех компонентов,— тройные системы. Для них число независимых переменных увеличивается до 4 — а
Рис. Б.32. Треугольная диаграмма.
именно температура, давление и две независимые переменные— содержание в мольных долях двух компонентов. Для того чтобы представить состояние системы в графическом виде, используют треугольную диаграмму (рис. Б.32) —равносторонний треугольник. Из геометрических соображений очевидно, что сумма отрезков, проведенных из любой точки внутри треугольника параллельно его сторонам, постоянна. Эту сумму можно принять за единицу (длина стороны треугольника), а расстояние до сторон— как мольную долю соответствующего компонента. Точки внутри треугольника могут передать любой состав тройной системы. Точки на трех сторонах треугольника соответствуют бинарным системам, составленным из пар компонентов, выбранных из трех компонентов системы; вершины углов треугольника соответствуют чистым компонентам. Обычно диаграмму составляют для постоянного давления; состояния при различных температурах можно представить в виде набора диаграмм, Каждая из которых отражает изотермическое состояние системы. Некоторые особые состояния, как, например, разрывы растворимости, эвтектические и азеотропные смеси, также можно Изобразить на треугольной диаграмме в виде точек и кривых. Для четверных систем (четыре компонента) при графическом Изображении состояния системы могут возникнуть затруднения.
19—2027
290
Химическая термодинамика
Вопросы и задания
1. Углеводород СпН2л+2 был растворен в 1,2-дибромэтане (7’пл= 10,0 °C). Раствор, содержащий 0,81 г углеводорода в 190 г 1,2-дибрамэтана, замерзает при температуре 9,47 °C. Определите число п.
2. Белок альбумин имеет Л1=69ООО. Рассчитайте осмотическое давление раствора, содержащего 2 г альбумина в 100 мл его водного раствора, при 25 °C.
3. Понижение температуры замерзания раствора, содержащего 2 г вещества в 1000 г бензола, составляет 1,28 °C. При растворении того же количества вещества в 100 г воды понижение температуры замерзания равно 1,395 °C. Полагая, что вещество ие диссоциирует в бензоле и полностью диссоциировано в воде, определите, на сколько ионов оно» распадается в воде. Криоскопическая константа воды—1,86 К-кг/моль, бензола — 5,12 К-кг/моль.
4. 68,4 г сахара (’41=342) растворяют в 1000 г воды. Рассчитайте а) давление пара при 20 °C; б) температуру замерзания; в) осмотическое давление при 20°C; г) температуру кипения раствора. Давление паров воды при 20°C равно 2,315 кПа, теплота испарения при температуре кипения — 2,256 кДж/г, теплота плавления при температуре плавления — 333,2 Дж/г.
5. Давление пара над водным раствором мальтозы при 25 °C равно 3,13 кПа, для чистой воды при той же температуре — 3,167 кПа. Рассчитайте осмотическое давление раствора.
6. 0,401 г салициловой кислоты содержится в 10,6 г этанольного раство ра. Раствор кипит при температуре на 0,337 °C более высокой, чем этанол Рассчитайте молекулярную массу салициловой кислоты.
7. Водный раствор сахарозы замерзает при температуре —0,989 °C. Чис тая вода имеет при этой температуре давление пара 567,3 Па. Рассчитайте давление пара над раствором. £/=1,86 К-кг/моль.
8, Рассчитайте процентный состав и молекулярную массу углеводород, по следующим данным: из 0,2 г вещества образуются 0,687 г СО2 и 0,1125 г Н2О. Температура замерзания раствора 0,0925 г этого вещества в 10 г бен зола на 0,354°C ниже, чем чистого бензола. Для бензола £к=5,12 К-кг/моль
9. Давление пара воды при 20 °C равно 2,338 кПа, давление пара на, раствором при той же температуре— 2,296 кПа. Определите осмотически давление раствора при 40 °C, если его плотность при этой температуре 1,01 г/мл, а молекулярная масса растворенного вещества 60.
10, При 30°C давление пара бензола составляет 16,08 кПа, а толуола 4,89 кПа. Рассматривая систему бензол — толуол как идеальную, составьте диаграмму парциальных давлений бензола и толуола в зависимости от состава, выраженного в мольных долях. Определите из этой диаграммы давление пара над смесью, содержащей 30% бензола. Определите парциальное давление и общее давление над смесью, содержащей по 100 г каждого компонента.
11. Пусть давление пара веществ А и В при 50 °C составляет 46,663 и 101,325 кПа соответственно. Предполагая, что они образуют идеальный раствор, рассчитайте состав пара, находящегося в равновесии со смесью состава 0,5 моля А и 0,7 моля В.
27.
ВВЕДЕНИЕ В СТАТИСТИЧЕСКУЮ ТЕРМОДИНАМИКУ
В предыдущих главах при изложении термодинамики всегда рассматривался ансамбль многих молекул, т. е. была принята макроскопическая точка зрения. В основу изучения систем были положены основные законы термодн иамики, которые характеризуют энергетический баланс (I закон) н определи1’ направление протекания химической реакции (II закон). Существенное зна чение имели и представления о функциях состояния системы. В° ' никает вопрос: как термодинамические функции состояния связаны с энерг
Статистическая термодинамика
291
-рическими характеристиками отдельной молекулы, т. е. как свойства системы в целом выявляются из свойств отдельных молекул? Очевидно, свойства всей системы не могут быть просто суммой свойств составляющих ее частиц, -рак как взаимодействие между ними обусловливает появление качественно рровых свойств. В то время как энергия в различных видах, характерных для отдельной молекулы, вносит вклад во внутреннюю энергию системы, понятие «энтропия» не имеет смысла для отдельной молекулы, так как она появляется как результат статистического поведения системы, состоящей из огромного количества молекул.
Задача статистики и разработанных на ее основе методов состоит в том, чтобы из свойств составляющих систему молекул сделать выводы о свойствах системы в целом. Она представляет собой как бы мост между микроскопическими и макроскопическими методами изучения природы. В классической механике свойства, иначе говоря параметры, тела характеризуются его начальными координатами и их изменением во времени. В статистической механике вследствие большого числа частиц и их беспорядочного движения начальные условия неизвестны, а число уравнений для описания системы таким способом слишком велико. Поэтому в статистике используются методы теории вероятности.
Среди статистических теорий в химии наиболее широко используется классическая статистика Больцмана. Лишь поведение электронного газа в твердых телах нельзя описать с помощью этой статистической теории. Тем не менее при обсуждении свойств систем, содержащих множество молекул, используются уже введенные ранее представления (гл. 6) квантовой механики, так как в первую очередь наша цель состоит в том, чтобы показать, как через параметры, определяющие энергию молекулы (поступательного, вращательного, колебательного движения), можно выразить термодинамические свойства всей системы (причем энергетические характеристики задаются как решения уравнения Шрёдингера).
27.1. Термодинамическая вероятность и энтропия
Макросостояние системы, состоящей из многих частиц, однозначно определяется приведенными в предыдущих главах уравнениями термодинамики, в которые входят параметры состояния, поддающиеся непосредственному измерению (например, если речь идет о газе — это объем, давление н температура). Однако, как уже было отмечено, невозможно установить значения этих параметров, зная скорость и координаты молекулы. Координаты молекулы в пространстве и ее скорость (импульс) постоянно меняются. Если было бы возможно в некоторый момент времени зафиксировать эти координаты, то Удалось бы установить микросостояние системы. Разумеется, это практически невозможно. Поэтому следует сделать вывод о том, что макросостояние системы реализуется как огромное количество микросостояний, которые отличаются координатами и скоростями движения отдельных молекул. Число микросостояний, соответствующее макросостоянию, называется термодинамической вероятностью W. Очевидно, речь идет о термодинамической вероятности, которая представляет собой большую величину, в отличие от математической вероятности, которая по определению равна отношению числа благоприятных событий ко всему количеству возможных событий и может Меняться от 0 до 1. Однако методы теории вероятности применимы и к термодинамической вероятности. Так, например, общая вероятность некоторого числа независимых отдельных событий, происходящих с вероятностью Равна*
V = (366)
ных
* Уравнение (366) можно пояснить на примере бросания двух играль-костей. Вероятность выбросить какое-либо определенное число при бро
19*
292
Химическая термодинамика
Известно, что физико-химические системы стремятся прийти в состояние с максимальной вероятностью его осуществления, что эквивалентно равновесию системы. Вернемся к уже рассмотренному примеру о самопроизвольном перемешивании двух газов (разд. 22.12, рис. Б 23). Такой самопроизвольный процесс сопровождается увеличением энтропии системы В состоянии равновесия энтропия достигает максимального значения (разд. 22.1.2). Связь между вероятностью и энтропией системы была установлена Больцманом [уравнение (246)]*:
з = /?1п1Г (367)
Уравнение (367) связывает вероятность с термодинамической функцией состояния — энтропией; далее прежде всего следует установить, каким образом в наиболее доступной форме можно выразить вероятность системы многих молекул, учитывая их свойства и параметры Для этой цели имеет смысл все так называемые фазовые пространства, включающие все молекулы системы, разделить на некоторые подпространства. Так как молекулы отличаются значениями координат пространства и импульсов, оказалось целесообразным представить фазовое пространство (соответственно подпространство) как математическое пространство с практически бесконечным числом указанных координат. Представим себе, что это большое число молекул разделено на группы, причем в каждой группе значения независимых переменных находятся в некоторых узких границах, например пространственные координаты от х до x+dx, от у до y+dy, от z до z+rfz, а в координатах импульсов от рх до px+dpx, от до py+dp,,, от рг до p^+dp?, где рх, ру и р- — проекции импульса молекулы на оси координат. Фазовый пространственный элементарный объем, который приписывается отдельной молекуле (называемый также ^-пространством) равен
dp — dxdydzdpxdp,jdpz (368)
сании кости, например 3 или 5, равна 1/6. Вероятность выбросить это же число на другой кости также равна '/в- Однако общая вероятность, т. е. вероятность одновременного выпадения на двух костях одного и того же числа, является произведением этих вероятностей, т. е. равна И7=1/б-1/б = 1/зб-
* Общий ход рассуждений, который привел к выводу этого уравнения, можно продемонстрировать на следующем примере. Предположим, что система состоит из двух независимых подсистем, характеризуемых вероятностью IFj и IF2. Общая вероятность в соответствии с уравнением (366) равна W'=W7i-W’2. Энтропия системы складывается из значений энтропии подсистем s=s1+s2. Таким образом, необходимо найти функцию f, связывающую энтропию и вероятность s=f(W). Из свойств вероятности и энтропии системы следует
s = S1 + S2 = /(F1) + /(F2) = /(F1F2) = /(F)
Дифференцируем это выражение по IT] и IF2:
/' (^1) = VW 1 /' = й7!/' (Г1Г2)
Отношение производных дает
/' (Гг)//' (Г2) = W2IW,
или в общем виде W-f'(W)=k. После разделения переменных = и интегрирования
s = f (IF) = k In IF + const
Константа интегрирования равна нулю, что можно показать в этом же вЫ воде.
Статистическая термодинамика
293
Из уравнения (368) и соотношения неопределенностей Гейзенберга ЬрЬхязЬ. (которое представляет собой произведение изменения импульса — на изменение координаты и соответствует кванту действия Планка) следует, что величина фазового пространственного элементарного объема в общем случае равна h2’ (f — число степеней свободы; для одноатомной молекулы f=3, двухатомной )=6 и т. д.). Фазовое пространство для всех молекул системы называют Г-пространством, и его размер, разумеется, значительно больше, чем для ц-пространства. Объем фазового пространства по аналогии с уравнением (368) можно записать в следующем виде:
= П dqcdpi i
(369)
где dqi и dpt — дифференциалы обобщенных пространственных и импульсных координат, а индекс i относится ко всем молекулам данного подпространства. Поэтому размер фазовой ячейки в Г-пространстве можно представить себе
ячейка ( ячейка 2 &
Рис. Б.33. Возможности распределения трех частиц в двух ячейках (а) и, согласно этой схеме, микрооостояния для распределения частиц (б).
как h!Nt. При таком методе изучения системы микросостояние является точкой в Г-пространстве. Основная задача статистики состоит в том, чтобы найти распределение, реализующееся наибольшим число микросостояний, т. е. наиболее вероятное распределение, отвечающее равновесию.
Для решения этой задачи Больцман сделал несколько гипотетических допущений.
1) Частицы (например, молекулы), распределение которых необходимо определить, по своей природе неразличимы, однако для статистических исследований и расчетов их можно рассматривать как различимые (например, отмеченные цифрами). Напротив, в квантовой статистике частицы считают принципиально неразличимыми.
2) Изучаемые объекты независимы друг от друга.
3) Объем фазового элемента в координатах пространства и импульсов, в котором исследуется распределение, остается постоянным. Это следует из теоремы Лиувилля, согласно которой изменение во времени объема элемента фазового пространства dil/dt—Q, а следовательно, и сами элементы остаются постоянными (они перемещаются практически, как несжимаемая жидкость).
4) Все элементы фазового пространства равновероятны относительно Распределения по энергиям, т. е. вместо начальных условий, необходимых Для решения задач методами классической механики, здесь выдвигается статистическая гипотеза о равновероятности элементов фазового пространства. “ квантовой статистике гипотеза о равновероятности ограничивается запретом Паули.
Итак, надо распределить N молекул в М различных фазовых ячейках (элементов), принимая во внимание допущения Больцмана, а также найти вероятность такого распределения. Из математики известно, что для N
294
Химическая термодинамика
объектов возможно N различных перестановок; однако это число перестановок уменьшается, если в какой-то области, например в фазовой ячейке, содержится более одной молекулы. Тогда вероятность равна* *
№ = (370)
где N,— число молекул в фазовой ячейке I.
Из уравнений (367) и (370) следует
ДП
s=feln——------- (371)
И
I
Далее будем пользоваться приближенной формулой Стирлинга**
M = (W/e)tf (372)
Подставим (372) в (370) и прологарифмируем:
In W=N (InW — 1)— [2M(lnM— 1)4-1].
После дальнейшего упрощения (SW,=W)
ln№ = WlnW—^Л'( InW, (373)
Из уравнений (367) и (373) получим
s= k (w In W — InWfj (374)
Предпосылкой справедливости формулы Стирлинга является достаточно большое число N, а также W), так как только в этом случае оправдывается переход от суммирования к интегрированию при вычислении InW. Это условие выполняется при статистической обработке данных для достаточно больших систем (например, 1 моль газа).
Выясним физический смысл константы Больцмана А, сравнив уравнение (374) с уравнением (237). Предположим, что N частиц необходимо разместить в М ячейках фазового пространства, предполагая равновероятность размещения, а также приняв условие Считаем, что в каждой от-
дельной ячейке содержатся NjM частиц. С учетом этих предположений из уравнения (370) следует
W! W/eW , „
W1~~ ~ ~MN 37 >
* Поясним уравнение (370), играющее важную роль в статистике. Пусть необходимо разместить три частицы в двух ячейках. Очевидно, существуют три возможности их размещения (рис. Б 33), т. е. трн различных микросо-стояння. Перестановка частиц в ячейке не меняет микросостояння; так, например, мнкросостояния на рис. Б 33, б не отличаются друг от друга. Таким образом, для того чтобы найти число различных мнкросостояннй, соответствующих одному макросостоянню, следует найтн общее число перестановок (здесь 3!) и поделить его на число перестановок к каждой ячейке, т. е. Г=3!/2!1!=3.
** Формулу Стирлинга можно вывести следующим образом. Поскольку N=l -2-3 ..., то
N
In W! = In 1 + In 2 4- • • + In N — 2 1° x ~ I 1° xd-x = x=i X
= [x (In x — 1)]^ = N (InN — 1) + 1 ss Win (W/e)
следовательно, A/! = (N/e)N.
Статистическая термодинамика 295
Индекс 1 обозначает вероятность начального состояния; переход от М к оправдывается только в предположении, что в каждой из М ячеек находится одинаковое количество молекул. Представим себе, что общий объем в начальном состоянии с вероятностью Wt увеличится в х раз. Тогда увеличится также объем ячеек фазового пространства, н для нового состояния (обозначенного индексом 2)
Г2 = (xM)N (376)
Связанное с изменением состояния увеличение энтропии можно найтн, подставив W2 и W-L в уравнение (367), а затем, найдя разность энтропии конечного и начального состояний:
xN-MN
As = k In (W2/WJ = k In -MN = k In XN (377)
Если число частиц равно NA, т. е. имеется 1 моль, то
AS = k In xNa = kNA In x (378)
Поскольку x=V2/Vi, т. e. представляет собой коэффициент, характеризующий увеличение объема при переходе от одного состояния к другому, то уравнение (378) можно сравнить с соответствующим уравнением феноменологической термодинамики [разд. 22.11, уравнение (237)]:
AS = R In (Va/VO (379)
Сравнив эти уравнения, можно легко установить, что kNA=R„ т. е введенная Больцманом константа играет универсальную роль в физической химии.
27.2. Функция распределения Больцмана
Возникает вопрос, как распределить N молекул,_ или, в более общем смысле, частиц, по фазовым ячейкам пространства координат н импульсов так, чтобы вероятность и связанная с ней уравнением (367) энтеропия приняли равновесное, т. е. максимальное, значение. Для того чтобы выведенными соотношениями можно было пользоваться в дальнейшем, будем рассматривать фазовые ячейки также и как квантовые состояния. Запишем основное условие следующим образом:
In W /\ max, а следовательно, б In W = 0 (380)
Кроме основного условия, согласно которому вероятность должна быть максимальной, т. е. ее вариация 61nWz=0, необходимо учитывать, что имеются Дополнительные условия равновесия:
Л7; = N, следовательно, б N[ = 0 (381)
и = jVt8t-, следовательно, б = ° (382)
Эти условия показывают, что число частиц в системе постоянно, а следовательно, вариация этого числа равна нулю. Условие (382) указывает на то, ‘то внутренняя энергия и системы постоянна, что соответствует равенству ее °ариацин нулю, е, — энергия частицы в t-й ячейке фазового пространства (энергия i-го квантового состояния молекулы).
Поставленная задача представляет собой типичную задачу вариационно
296
Химическая термодинамика
го исчисления, т. е. нахождения решения, для которого одновременно выполняются все три условия*.
Задача, которую предстоит решить, аналогична по своей форме задаче, рассмотренной выше. Три уравнения, которые должны одновременно выполняться,— это основное условие (380) и два дополнительных условия (381) и (382). Используя точное выражение для In W [уравнение (373)], можно получить следующее выражение для условия (380):
б In W = б pV In N — W(- In JV£) = 6 (Nt In Ni) = 0 (383)
Здесь принято, что Л/ In JV= const, и поэтому вариация этого члена равна нулю. Найдем производную правой части уравнения (383) и приравняем ее нулю:
б (Ni In Ni) = 6N‘ (ln N‘ + ’) ~ 2 ln Ni = 0
По аналогии с рассмотренным выше примером введем множители а и р и просуммируем все трн уравнения:
t>Ni (а + ре(- + In Ni) = 0 (384)
Очевидно, что это уравнение выполняется только в том случае, когда выражение в скобках равно нулю при любом значении ег и Л\ т. е.
а₽ei + In tVj = 0 (385)
Из этого уравнения, а также уравнений (381) и (382) можно сделать следующие важные выводы:
1) число частиц с энергией в фазовой ячейке равно
Ni = е~“ е~рЕ‘ (386)
2) общее число частиц равно
(387)
3) при делении (386) на (387) е~“ сокращается; получим функцию распределения Больцмана
(388)
Эта функция определяет долю частиц с энергией е; относительно общего числа частиц. Выражение в знаменателе суммируется по всем фазовым ячейкам (квантовым состояниям) и поэтому называется суммой по состояниям.
м
5=2 е~^‘ (389)
___________ i=l
* Продемонстрируем общий принцип вариационного исчисления. Пусть имеются три независимых уравнения: х=0; г/=0; г=0. Необходимо найти условие, при котором выполняются одновременно все три уравнения. Это можно сделать, умножив каждое уравнение на соответствующий коэффициент и скомбинировав их так, чтобы одновременно выполнялись все три уравнения:
Ах + By + С г — 0
Характер этого уравнения дает возможность обойтись лишь двумя коэффициентами:
а-х fiy + z = 0, где а=А/С и $ — В/С.
Статистическая термодинамика
297
Если рассматривать энергию как функцию координат пространства и импульсов s(q,p), то можно определить число частиц в интервале q, q+dq и р, p+dp в виде больцмаиовской функции распределения
dN/N kT dqdp | J е dqdp (390)
Форма функции распределения Больцмана [уравнение (388) или (390)] выбирается в зависимости от поставленной задачи. Физический смысл множителя Р в уравнении (388), который в уравнении (390) предполагается уже известным, требует дополнительного объяснения. Для этого обратимся к уравнению (374) для энтропии и заменим Nt под логарифмом выражением (388), предварительно преобразовав его:
Hi = Ne^i/Q (391)
Тогда получим
s = k [win N — 2 Ni(\nN — рег — InQ)j (392)
После умножения скобки под знаком суммы на N; и обозначения сумм 2W,- In N=N In N и SA/,- ре,-= р« получим наконец
s = fe(Pu + WlnQ) (393)
Продифференцируем энтропию по энергии и при постоянном объеме:
/ ds \
("аг =*₽ <394)
\ /у
В то же время в феноменологической термодинамике выведено соотношение [уравнение (278)]
/ ds \ 1
(^гХ = т- <395>
Из уравнений (394) и (395) можно установить физический смысл 0:
0 = 1/67 (396)
Подставив уравнение (396) в уравнение (393), получим после некоторых преобразований
и - Ts = —kTN InQ = f (397)
Итак, получено выражение, связывающее сумму по состояниям н свободную энергию, которое имеет фундаментальное значение для вывода других соотношений между термодинамическими функциями состояния и суммой по состояниям. Так как в Q входит энергия молекулы, уравнение (397) связывает также различные формы энергии с функцией состояния.
27.3. Некоторые свойства суммы по состояниям
и физический смысл множителя а
Используя вместо 0 полученное для него выражение (396), запишем сУмму по состояниям в следующем виде:
Q = 2 (398)
298
Химическая термодинамика
В этой форме сумму по состояниям часто называют суммой по квантовым состояниям, т. е. суммирование проводят по всем квантовым состояниям Так как в это число входят также и вырожденные состояния, т. е. квантовые состояния, для которых энергия одинакова, некоторые члены суммы могут многократно повторяться*.
Из вышеизложенного следует, что для решения отдельных задач целесообразно записать выражение для суммы по энергетическим состояниям, т. е. провести суммирование по всем энергетическим состояниям, а вырожденные энергетические состояния умножить па коэффициент вырождения g,:
Q^'2igl^i/kT (399)
Суммы по состояниям (398) и (399) относятся к одной молекуле. Для решения большого числа задач, особенно термодинамических, применяется понятие суммы по состояниям ансамбля частиц. Смысл этого понятия ясен из уравнения (397). Если ввести множитель W под знак логарифма, то получим
f = —kT In QN —kT In Z (400)
Z = (W (401)
Здесь Z обозначает сумму no состояниям ансамбля. Выражение для Z в соответствии с уравнением (401) справедливо только для ансамбля не обменивающихся местами, т. е. локализованных, частиц, например для идеального твердого тела. Если частицы могут меняться местами, как это имеет место в газе, то нужно учесть при суммировании, что состояния, которые получились в результате перестановки, неразличимы. В этом случае число различных состояний уменьшается в М раз, т. е.
Z = QV/M (402)
Если подставить это выражение для суммы по состояниям газа в уравнение (400), то получим зависимость между свободной энергией и суммой по состояниям молекулы:
/= — In (QAT/at!) = — kTN in Q + kT in N\ (403)
и, полагая lit ,V1«/V In N—N, получим
/ = —kTN In (Q/N) - kTN = -nRT In (Q/nNA) — nRT (404)
В уравнении (404) сделана замена N=nNA (где п — число молей).
Из уравнения (404) и определения химического потенциала как частной производной свободной энергии по числу молей при постоянном объеме и температуре (280) получим уравнение, связывающее химический потенциал с молекулярной суммой по состояниям:
И = (dfldn)ViT = -RT In (405)
* Пусть состояние молекулы описывается различными волновыми функциями, причем некоторые из них имеют одинаковую энергию:
fe fe. .fe fe fe fe fe,
f'u
Здесь волновая функция фо имеет однократное вырождение, ф[ — трехкратное, а ф2— пятикратное. Сумма по состояниям в данном случае равна
Q = 3e-ei/fer 5е-82/АГ
________________Статистическая термодинамика 299
Если найти число молей п из этого уравнения и из уравнения (387), то, учитывая, что N=nNA, а также уравнение (389), получим
<«>
Сравним показатели степени при экспонентах '
ц
а = -£Г (407)
Важное свойство суммы по состояниям состоит в том, что общая сумма по состояниям для нескольких независимых видов энергии представляет собой произведение сумм по состояниям этих видов энергии. Это следует из аддитивности независимых форм энергии. Если предположить, что общая энергия складывается из нулевой энергии е0, энергии поступательного движения елосд. вращательной энергии еВр и колебательной энергии еКол
8 = ео + ^ПОСТ + евр + екол (408)
то для суммы по состояниям получим
<2 = 2 kT =
—е0 “епост,г “евр,г “*еколД
kT~ V kT ЛТ kT
—е е ее
откуда следует наконец
—ео kT
Q — <2о<2пост?вр<7кол ~ е Спост?вр?кол (409)
Множители </вр и qK0^ являются суммами по состояниям вращения п колебаний молекулы без соответствующей нулевой энергии, т. е. суммирование производится начиная с квантового числа, равного 1. В поступательную сумму состояний не входит нулевая энергия, т е. при температуре 7=0 всякое поступательное движение прекращается В то же время колебательная энергия, как это видно из рис. А 21, равна e0=1/2/zv (разд. 22.1.2). Остальные составляющие нулевой энергии с трудом поддаются точному определению, поэтому при вычислении термодинамических функций из сумм по состояниям нулевая энергия и соответствующая ей часть термодинамической функции не выражается в явной форме [например, уравнение (410)].
27.4. Связь между термодинамическими функциями газов и молекулярными суммами по состояниям
В этом разделе показано, как можно найти связь между термодинамическими функциями и свойствами ансамбля из большого числа молекул и энергий молекул, т. е характеристиками отдельной молекулы. Эту связь мы продемонстрируем на примере термодинамических функций газов. В соответствии с определением суммы по состояниям ее получают суммированием по всем энергетическим состояниям. Выводы, которые будут проведены ниже» основаны на уравнениях (400), (402) и (409):
—ео_ ~ Е°
аг „ kT
/ —kT In Z; Z == QN/N', Q e <2пост?вр<7кол — e 4
300
Химическая термодинамика
Сумма по состояниям q в последнем уравнении представляет собой произведение всех сумм по состояниям начиная с квантового числа 1, т. е. без нулевой энергии. Далее в выводе используются соотношения между различными термодинамическими функциями (разд. 22 2 и 22 3).
Связь между свободной энергией f и суммами по состояниям уже найдена [уравнение (404)]. Сделаем подстановку Q=e—q:
f = —kTN In €-^lkT — kTN In — kTN
После логарифмирования первого члена этого уравнения, считая, что Ne!l=E(l, получим
f — Ео = — kTN In — kTN (410)
В то время как е0 — нулевая энергия молекулы, Ео—нулевая энергия всего ансамбля из N молекул. Выразим из уравнения (260) свободную энтальпию g
g = f+pv=f+kTN
и подставим f из уравнения (410) , ''''
g-Ea = -kTNln-fr (411)
Используя уравнение (280), найдем выражение для энтропии
з= -(-^Д =kNln^- + kNT( ) (412а)
\ и /р \ /р
или
/ df \ о / 0 In о \
з= — — kN In + kNT + &V (4126)
Сравнение уравнений (412а) и (4126) показывает, что суммы по состояниям также связаны:
Из уравнений (254), (410) и (4126) получим соотношение между внутренней энергией и суммой по состояниям:
I д In q \ ,
и— E0=f+Ts = NkT^—gj~- j (414)
а также для энтальпии из уравнений (259), (411) и (412а):
h-Ea=g+Ts = NkTt (415)
\ ! р
Из двух последних уравнений можно также получить молярную теплоемкость при постоянном объеме (постоянном давлении), например из (414), если привлечь уравнение (193):
(ди \ /<Э21по\ / 0 In о \
-лГ) =NkT^ +%NkT (416)
/ V \ f V ' / V
Статистическая термодинамика 301
Таким образом, с помощью этих соотношений становится возможным, зная энергию молекул, вычислить термодинамические функции состояния системы Из сумм по состояниям.
Подобным методом можно вывести аналогичные уравнения для идеального твердого тела, если вместо величины Z из уравнения (402) подставить величину Z для локализованной системы гармонических осцилляторов из уравнения (401).
Сравнение уравнений, полученных для различных функций, показывает, что для всех термодинамических функций нулевая энергия одинакова, т. е. становится совершенно ясно, что при абсолютном нуле не существует различий между термодинамическими функциями, определенными при постоянном давлении н постоянном объеме:
St=o ~ h'=o = ит=о ~ ^т=о ~ &о (417)
Кроме того, мы установили, что все термодинамические функции, для которых характерны свойства потенциала, т. е. имеющие размерность энергии, могут быть рассчитаны из спектроскопических данных с точностью до величины Ео; из этих данных в лучшем случае можно рассчитать Ео для двухатомных молекул. Напротив, все термодинамические функции, имеющие размерность энергия/температура, не содержат нулевой энергии; поэтому из спектроскопических данных можно рассчитать, например, энтропию s, а также Cv и Ср.
Константа равновесия также связана с суммой по состояниям Соотношение между ними можно вывести из общего условия равновесия (разд. 23 1):
(418)
Вспомним уравнение для химического потенциала (405) и подставим его в уравнение условия равновесия
2VZRTln ^7 (419>
Индексом i отмечен каждый компонент системы. В случае изотермического процесса можно разделить это уравнение на RT и после логарифмирования получим
П П J* = « (420)
•ч
i t
IB соответствии с принятым ранее соглашением стехиометрические коэффициенты считаются положительными для продуктов реакции и отрицательными для исходных веществ.] Получили выражение для константы равновесия химической реакции. Если вынести за скобку член с нулевой энергией, т. е. полагая Q1 = e— *otJkT qlt вместо уравнения (420) получим
к П,,’< (421)
i
По аналогии с законом Гесса [уравнение (212)]
Ае° = S Vi&oi (422)
27.5. Суммы по состояниям для важнейших видов движения молекул
Для того чтобы можно было проводить расчеты по уравнениям, выведенным в предыдущей главе, в которых термодинамические функции выражены нерез суммы по состояниям, необходимо знать достаточно точные выражения
302
Химическая термодинамика
сумм по состояниям для каждого вида движения молекул. Детальный вывод таких уравнений выходит за рамки настоящей книги, поэтому будем пользоваться ими как готовым результатом и будем принимать во внимание только суммы по состояниям для поступательного, вращательного и колебательного движения. Как правило, они в наибольшей степени определяют значение термодинамической функции. Мы не будем учитывать возбужденное состояние электронов и ядерный спин. Возбужденное состояние электронов проявляется в энергетическом отношении лишь при очень высокой температуре — для большинства молекул в основном состоянии коэффициент вырождения энергии электронов равен 1.
При решении уравнения Шрёдингера [уравнение (12) в разд. 5.2] было получено уравнение для энергии поступательного движения частицы
h2
Ёпост = 8а2т • («? + ПУ2 + «г2) (423)
Здесь h—квант действия Планка, m — масса частицы, а — линейный размер пространства, в котором движется частица, пх, пу, п2—квантовые числа для поступательного движения вдоль осей координат в пространстве. Подстановка уравнения (423) в уравнение суммы по состояниям (399) и переход от суммирования к интегрированию по квантовым числам, как переменным интегрирования, дает
/ 2nmkT \3/2
Споет= дг j v (424)
Уравнение (424) можно вывести из уравнения (423) следующим образом. Подстановка (423) в общее уравнение суммы по состояниям (399) (учитывая, что поступательное движение не имеет вырождения) дает
оо оо оо-------"--- , 2_(_ 2_ьл 2\
V V V Sma^kT (пх^пу+пг)
Фпост = Z Z Z е <425>
п =0 п =0 п = 0 Л с/ X
Так как при свободном движении частицы нельзя выбрать какое-то предпочтительное направление, вместо тройной суммы можно записать
Knici'-kT
— (1Фпост)8
(426)
'фиоет обозначает сумму по состояниям поступательного движения на одну степень свободы. Так как различие между значениями квантов энергии поступательного движения очень невелико, а суммирование проводится практически в области от нуля до бесконечности, то с полным основанием (вспомнив правила нахождения определенного интеграла) можно перейти от суммирования к интегрированию. Обозначим
h2
с ~ 8ma2kT
(427)
Тогда из уравнения (426) получим
ОО
^посг = ) е-с" dn = ~ (-- j о
(428)
Статистическая термодинамика
303
Этот интеграл представляет собой известный интеграл ошибок Гаусса. Подставив в него выражение для с, получим наконец
/ 2nmkT к1/2
3QnOCT = ( ^2 j а (429)
Таким образом, полная сумма по состояниям поступательного движения записывается следующим образом:
Л / 2nmkT \3/2
QnocT == ( ^2 j V (430)
Вращательная энергия двухатомной молекулы равна
й2
евр — gn2j ^1(^+0 (431)
Это уравнение для квантованного вращательного движения двухатомной молекулы уже приводилось в разд. 6.1.1 [уравнение (21)].-Л—вращательное квантовое число, J —момент инерции.
J = рг2 (432)
и— усредненная масса mi •/n2/(m1+/n2), а г — расстояние между атомами в молекуле. Подстановка уравнения (431) в уравнение (399) дает, таким образом, сумму по состояниям вращения молекулы:
СО _ Л2 (Д-1-1) К
Qbp = 2 W + ’)е 8ЯШГ (433)
Л=о
При не очень низких температурах (T>ft2/8n2/fe) вполне оправдан переход от суммирования к интегрированию по вращательному квантовому числу
8n2JkT 1 h2 о
— число симметрии, которое не является результатом интегрирования. Оно вводится нз общих представлений о симметрии молекулы для расчета вращательных сумм по состояниям. Это число представляет собой число неразличимых положений при полном обороте молекулы; для двухатомной молекулы с одинаковыми атомами а = 2, а для двухатомной молекулы (например, НС1) с разными атомами а=1.
Значительно более сложное выражение получается для суммы по состояниям вращательного движения многоатомной молекулы, так как в общем случае она может иметь различные главные оси вращения.
Энергия колебательного движения для гармонического осциллятора равна
еКол = (п 4- 1/2) hv, (п — 0, 1, 2,. ..) (435)
При учете нулевой колебательной энергии
еКОл = е®кол + п^, (п = 1,2,...) (436)
Подставив уравнение (436) в уравнение для суммы по состояниям, получим
ОО Ну
п ______е°кол^7 . _ __""V р kT п (437)
Мкол — е <?кол > </кол С )
п=1
Так как в общем случае при частотах, для которых необходимо принимать во внимание колебательное возбуждение, hv^-kT, а следовательно,
304
Химическая термодинамика
е hv/kTдля расчета СуММЫ (437) можно использовать известную форму, лу суммы геометрической прогрессии
?кол = (1 — е hvlkT) 1 (438)
27.6. Расчет поступательной составляющей термодинамических функций
Уравнения, выведенные в предыдущем разделе для сумм по состояниям для различных видов энергии молекулы, дают возможность конкретно рассчитать термодинамические функции. Этот раздел знакомит с методикой такого расчета на примере поступательной составляющей термодинамической функции. В разд. 27.3 было показано, что общая сумма по состояниям представляет собой произведение сумм по состояниям для отдельных видов движения [уравнение (409)], в то же время термодинамические функции определяются, как логарифмы сумм по состояниям [уравнение (410)]. Отсюда можно сразу же сделать вывод о том, что составляющие термодинамических функций для соответствующих видов энергии имеют аддитивные свойства. Это означает, что, например, при подстановке уравнения (409) в уравнение (404)
f = Ео + fПОСТ 4*/вр + /кол (439)
Между [пост и <2пост, f»p и QBP, а также /кол и <2Кол имеются соотношения, аналогичные уравнению (410). При расчете поступательной составляющей термодинамических функций, как уже отмечалось, не учитывается нулевая энергия.
Подстановкой <2Пост из уравнения (424) в уравнение (404) получим
/ 2nmkT \з/2
/пост — kTN In ) v kTN (440)
Для поступательной составляющей свободной энтальпии [подстановка (424) в (411)]
/ 2nmkT 8/2
£пост---£7Wln^ /{Здг j v (441)
или (полагая pv=NkT)
[ (2nmkT)3!2 1
£ПОст = ~kTN In Г hSp! - • kT (442)
Для вычисления внутренней энергии необходимо в уравнение (414) подставить частную производную (d In QnoCtldT)„. Из уравнения (424) следует
1п Споет = 3/2 in ( j + 3/2 ,п Т + 1П и (443)
Так как первые члены в уравнении (443) не зависят от температуры, то
Подстановка этого выражения в уравнение (414) дает для поступательной составляющей внутренней энергии
Щ.ост = 3/2Л?«- (445)
Таким образом, если принимать во внимание только поступательное движение, получается простое соотношение между внутренней энергией и темпера-
•дтистическая термодинамика
305.
турой (ср. разд. 2.1, уравнение (8а)). Аналогичным методом из выражения для (д In Q ПОСТ !дТ)р, после замены v в уравнении (443) на v=NkTfp, полу-
чим
д In Qnocr \ ___5 . 1
дТ 1 ~ 'г' Т ' р
(446).
й после подстановки в (415), полагая нулевую энергию поступательного движения равной нулю
7^ пост = 5/%NkT (447)>
Важное значение имеет поступательная составляющая энтропии, так как из нее вычисляется абсолютное значение энтропии молекулярного газа. Подстановкой (443) и (444) в (4126) получим
«пост = W In — о + kN (448}
соответственно молярная энтропия при условии N—Na и m=MINA и сведении всех постоянных в один член равна
SnocT=3/2Rln7’ + RlnV + 3/2R In41 — 46,33 Дж/(моль-К) (449>
Выразив объем через давление при помощи уравнений идеального газа, получим
5ПОст = 6/2^1пТ — 7? In р + 8/27? 1п 41 — 9,68 Дж/(моль-К) (450>
Это уравнение называют также уравнением Сакюра — Тетроде-, на его основе можно рассчитать энтропию атомарного газа в зависимости от параметров системы р и Т.
Пример. Рассчитать стандартную энтропию гелия, аргона, паров ртути и кадмия, а также поступательную составляющую энтропии Н2 и НС1 при-298 К- Сравнить полученные результаты с экспериментальными данными (приведены в таблице).
Решение. Для вычисления стандартной энтропии при 298 К подставим в уравнение (450) 7'=298 К и р=1. Получим уравнение для расчета
S°nocT = 47,877 lg298-|- 28,727 lg41 — 9,681 Дж/(моль-К) (451>
После подстановки М соответствующих газов в уравнение (451) получим следующие значения для стандартной энтропии при 298 К:
Вещество 5°П0ст’ рассчитанное 1 по уравнению (451), Дж /(моль-К) Экспериментальное значение 5°ПОСТ(298 Ю. Дж_/(моль-К)
Не 126,02 122,21
Аг 154,69 152,35
Hg 174,82 172,86
Cd 174,95 172,86
н2 117,57 130,63
НС1 153,48 186,92
Экспериментальные данные для атомарных веществ получены из измерений теплопроводности, а для Н2 и НС1 взяты из таблиц.
Очевидно, что для одноатомных молекул наблюдается хорошее согласие ^жду рассчитанными [уравнение (451)] и экспериментальными данными, .'схождение между результатами расчета и табличными данными для модулярных газов можно объяснить тем, что в уравнении (451) учтена толь-20—2027
306
Химическая термодинамика
ко поступательная составляющая энтропии. Сравнение этих данных показывает также, что поступательная составляющая дает наибольший вклад в общую величину энтропии
27.7. Расчет вращательной и колебательной составляющей энтропии газов
В предыдущем разделе установлено, что термодинамические функции, относящиеся к различным формам движения, аддитивны. Поэтому методом’ аналогичным рассмотренному выше, можно рассчитать также вращательною и колебательную составляющие термодинамических функций. Это имеет особо важное значение для расчета энтропии, так как она не содержит нулевой энергии [уравнение (412)]. Поэтому становится возможным вычислить абсолютные значения энтропии молекулярных газов. Если подставить вращательную сумму по состояниям для двухатомного газа [уравнение (434)] в уравнение, связывающее энтропию с суммой по состояниям, и определить
8л2Л
In QBp = In h2(y - 4- In 71 (452)
’И
/ d In QBp \
I dT ) ~ T '
\ z p
то после подстановки последних в уравнение (412а) получим молярною энтропию (для N=Na)
tScf-JkT
SBP = R In + R (454)
и после соответствующих преобразований
SBp = # + R In —+R ln J + R In T — R Inc (455)
•Соотношение для колебательной составляющей энтропии молекулярного двухатомного газа можно найги из колебательной суммы по состояниям (438) н уравнения (412а). Из уравнения (438) следует
1п ?кОл = —In (1 - e-AvH (456)
~ I)-1 (457)
V /р
Постановка этого выражения в (412а) дает для молярной энтропии следующее уравнение (для N=Na)'-
5кол = —R In (1 — +R-^ (eftv/^ _ 1)’1 (458)
При помощи этого уравнения можно найти колебательную составляющую энтропии двухатомных газов. Применение этих уравнений для расчета поступательной, вращательной и колебательной составляющих энтропии рассмотрено ниже на примере двухатомного газа СО.
Пример. Рассчитать энтропию СО прн 600 К через составляющие ЭНТР пии для поступательной, вращательной и колебательной форм движени^ Сравнить результат с экспериментальными данными: = 222,89 Дж/(моль-К). Длина связи в молекуле СО равна г0=1,128-Ю .волновое число основного колебания v=2167 см-1.
Статистическая термодинамика 307
Решение. Рассчитаем с помощью уравнения (450) поступательную составляющую энтропии; после подстановки в него Л4со=48 и Г=600 К оказалось, что SnocT=184,91 Дж/(моль-К). Вращательную составляющую рассчитаем по уравнению (455) после подстановки в него значений универсальных постоянных:
SBP= 19,14 lg J 4-19,14 1g Г — 19,14 lg с + 744,16 Дж/(моль-К)
Число симметрии с для СО равно 1, так что этот член уравнения исчезает, усредненная масса была найдена в разд. 6 1.1 и равна рсо= 1,14-10-23 г. Таким образом, получим вращательную составляющую энтропия 5вр = =32.94 Дж/(моль-К).
Колебательная составляющая энтропии может быть получена после подстановки в уравнение (458) волнового числа v = 2167 схг1 [учесть, что v = c (разд. 6.1.1)]. 5Кол = 0,293 Дж/(моль-К). Тогда общая энтропия газа равна
S = SnocT + SBP + З’кол = 184,91 + 32,94+ 0,293 — 218,14 Дж/(моль-К)
Полученное значение энтропии хорошо согласуется с экспериментально найденным: 5ЭКс = 222,89 Дж/(моль-К). Таким образом, методы статистической термодинамики применены для расчета энергетических характеристик молекул и термодинамических функций соответствующего вещества из спектроскопических данных. Кроме юго, этот пример наглядно показывает, что энтропию газа определяет в основном поступательная составляющая, а колебательная составляющая энтропии почти не сказывается на ее общей величине.
Вопросы и задания
1. Рассчитайте термодинамические функции U, F и S, а также молярную теплоемкость Cv идеального твердого тела Твердое тело следует рассматривать как систему из Na локализованных осцилляторов, т е следует рассчитать только колебательную составляющую термодинамических функций. Общий ход вычислений обсуждался в разд. 27 4. Поскольку речь идет о локализованных частицах, Z=QN.
2. В табл. А.7 приведены расстояния между ядрами и колебательные волновые числа двухатомных газов. Рассчитайте для этих газов поступательную, вращательную и колебательную составляющие энтропии, а также общую энтропию как сумму составляющих для различных форм движения. Сравните полученные результаты с табличными данными, приведенными в руководствах по физической химии.
20*
Б.З. ЭЛЕКТРОХИМИЯ
з 28. ТЕОРИЯ ЭЛЕКТРОЛИТИЧЕСКОЙ ДИССОЦИАЦИИ '
Рассмотрим подробнее условия появления в растворе ионов. До создания Аррениусом теории электролитической диссоциации (1883 г.) господствовало мнение, что диссоциация молекул на ионы происходит только под действием электрического поля. В настоящее время достоверно известно, что процесс образования ионов идет самопроизвольно, так как при протекании соответствующей реакции, например
НС1 + Н2О <=> Н3О+ + Cl-
изменение свободной энтальпии AG<0.
Теория электролитической диссоциации Аррениуса опирается на ряд экспериментально установленных фактов.
Во-первых, описанные ранее (разд. 26.1.3) коллигативные характеристики растворов определяются количеством находящихся в нем частиц. Для бинарных электролитов, т. е. веществ, диссоциирующих на два иона, в разбавленном растворе такая коллигативная характеристика, как, например, понижение температуры замерзания, увеличивается примерно в два раза по сравнению с раствором вещества той же молярной концентрации, если оно распадается на ионы.
Во-вторых, измерения электропроводности растворов, например раствора серной кислоты двумя медными электродами, показало, что в растворах некоторых веществ соблюдается закон Ома; этот факт согласуется с теорией электролитической диссоциации. Между тем ранее полагали, что ток в растворах электролитов может протекать только после того, как приложенное напряжение достигнет некоторого минимального значения, необходимого для разложения молекулы на ионы.
В-третьих, ионные реакции протекают с необычно высокими скоростями. Часто скорость реакции настолько велика, что ее измерение оказалось возможным лишь с помощью новейших методов. Высокая скорость некоторых реакций, например реакций нейтрализации или осаждения, может быть объяснена только на основе теории электролитической диссоциации исходя из представлений о том, что образование ионов происходит непосредственно в процессе растворения вещества.
Закон Фарадея
309
Закон Фарадея
Как известно, в растворах электролитов протекание тока обеспечивается движением ионов, а в металлических проводниках— движением электронов. Поэтому на границе металлический электрод — раствор протекает электрохимическая реакция, в которой ионы принимают от электрода или отдают ему электроны (восстанавливаются или окисляются). Количественное соотношение между массой прореагировавшего вещества и количеством электричества известно как объединенный закон Фарадея. Количество прореагировавшего вещества т так относится к его молекулярной массе М, как количество электричества it, протекшего в цепи, относится к количеству электричества nF, необходимому для превращения одного моля вещества:
т __ it
М nF
где i — сила тока, t — время (протекания тока в цепи), F— число Фарадея.
Обычно на электродах имеют место одновременно несколько электрохимических реакций, поэтому лишь некоторые электрохимические системы можно использовать для измерения количества электричества с помощью ^специальных приборов — кулонометров, принцип действия которых основан на применении закона Фарадея'5. Уже Гельмгольц высоко оценил значение открытия Фарадеем закона электролиза, поскольку благодаря этому открытию и используя атомно-молекулярные представления были сделаны выводы о «корпускулярных свойствах электричества».
Вопросы и задания
Сопоставьте закон Фарадея с законом кратных отношений. Какие выводы можно сделать из такого сопоставления’
30. ЭЛЕКТРОХИМИЧЕСКАЯ ТЕРМОДИНАМИКА
30.1. Э. д. с. ячейки
Сосуд, в который помещены два электрода и раствор электролита, называют гальванической ячейкой. В краткой записи вертикали обозначают межфазную границу металл — электро-
* В электрохимическом кулонометре должны быть обеспечены условия протекания только одной электрохимической реакции — той, для которой проводится расчет количества электричества по массе выделившегося вещества. [Антропов Л И Теоретическая электрохимия —М Высшая школа, 1982] — Прим перев.
310
Электрохимия
лит. В гальванической ячейке
pt pt
н2 | НС11 С1а (459)
две платиновые пластинки помещены в раствор соляной кислоты, причем к одной из них подводится газообразный водород, а к другой — хлор. В такой системе может происходить следую, щая Химическая реакция:
Н2 + С12 ч=±: 2НС1 (460)
газ газ раствор
На катоде восстанавливается хлор:
4“ е~ < СГ (461)
а на аноде — окисляется водород*:
1/2Н2 Н++<Г (462)
Реакция (460) протекает самопроизвольно, что соответствует понижению свободной энтальпии. При протекании электродных реакций (461) и (462) между электродами измеряется разность потенциалов 8. При этом потенциал водородного электрода оказывается отрицательнее хлорного. При образовании 1 моля соляной кислоты ток совершает работу Fe, где F— число Фарадея, равное количеству электричества, необходимому для выделения при электролизе 1 г-экв. вещества (F=96491 А-с/г-экв.). Если в реакции принимает участие п электронов, то суммарная работа равна nFe, например, для реакции (460) 2Ёе. В том случае, если процесс проводится обратимо (при бесконечно малом токе), работа системы равна изменению свободной энтальпии химической реакции. Согласно первому закону термодинамики,
AG°=.- —nFe° (463)
В то же время Sv,p, = AG [уравнение (290)]. Поэтому, объединив стандартные химические потенциалы реагирующих веществ в член 8°, получим для реакций (461) и (462)
e^£o£Llu <W-Ca-
nF PH ]/2-РС1 1/2
(464)
* Реакции (461) и (462) протекают слева направо в электрохимической ячейке (гальваническом элементе), подключенном к внешнему (нагрузочному) сопротивлению. Электрод, на котором происходит восстановление вещества, называют анодом; он имеет положительную полярность по отношению к другому электроду — катоду. Те же реакции протекают в ячейке при электролизе, т. е. при подключении ее к внешнему источнику тока; при этом реакция (461)—восстановление С12— происходит на электроде, соединенном с отрицательным полюсом внешнего источника, т. е. на катоде, а реакция (462)—окисление Н2— протекает на электроде, соединенном с положительным полюсом источника тока, т. е. на аноде. [Антропов Л. И. Теоретическая электрохимия. — М.: Высшая школа, 1982]. — Прим, перев.
Электрохимическая термодинамика 311
Уравнение (464) дает выражение для э. д. с. электрохимической ячейки. При включении в электрическую цепь нагрузки (например, сопротивления) разность потенциалов на выводах ячейки понижается (в данном случае измеряется «напряжение на клеммах»). Реакции (461) и (462) называют потенциалопре-деляющими. Из уравнения (464) можно сделать следующие выводы. Во-первых, напряжение ячейки понижается при: а) увеличении концентрации продукта реакции соляной ,кислоты; б) уменьшении парциального давления исходных веществ, т. е. водорода или хлора. В предельном случае, когда парциальное давление водорода и хлора равно их равновесному значению в реакции (460), напряжение на ячейке становится равным нулю. Во-вторых, при увеличении в два раза количеств реагирующих веществ [уравнения (461) и (462), п = 2] одновременно удваиваются показатели степени при концентрациях и парциальных давлениях в уравнении (464).
30.2. Схема измерения э.д. с. ячейки компенсационным методом
Для того чтобы измеренные значения э. д. с. ячейки совпадали с вычисленными по термодинамическому уравнению (463), а также по уравнению Гиббса — Гельмгольца, необходимо проводить измерения при бесконечно малом токе. Только при этом услов1ии процесс идет обратимо и система остается в равновесии. Так как в уравнение (464) входят концентрации веществ вблизи поверхности электрода, то они должны быть одинаковы как на поверхности электрода, так и в объеме раствора. Если концентрации вблизи электрода отклоняются от среднего значения, то возникает концентрационное перенапряжение и процесс становится термодинамически необратимым. Другие виды перенапряжения будут рассмотрены позднее.
Компенсационный метод Погтендорфа наиболее удобен для измерения э. д. с. ячейки, так как измерительная схема работает с минимальным потреблением тока (рис. Б.34). Источником напряжения является аккумулятор А. Сопротивление /?1 регулирует падение напряжения на высокоомной проволоке (реохорде) ВС; это падение напряжения всегда должно быть больше, чем неизвестное напряжение X или известное напряжение нормального элемента N. Источники X или X включены навстречу аккумулятору А, поэтому движок на проволоке ВС можно установить таким образом, чтобы через гальванометр G
312
Электрохимия
не протекал ток (стрелка гальванометра указывает нуль) В этом случае между точками В /или S) и соответствующими электродами ячейки X (или N) ток отсутствует и разность по-тенциалов равна нулю. В то же время между точками В и 5 существует разность потенциалов, создаваемая аккумулятором А. Работа, совершаемая при переносе едицичного заряда по проволоке от В к S точно такая же, как и при переносе заряда через ячейку X. Иными словами, разность потенциалов на от-
Д
------II----ПГ~1---------
Рис. Б.34. Измерительная схема компенсационного метода Поггендорфа.
резке BS равна напряжению на выводах ячейки X. Для измерения неизвестного напряжения ячейки X можно либо провести градуировку проволоки BS с помощью элемента с извес!-ной э. д. с. (метод Дольрауша— Вальце), либо использовать вместо проволоки магазины сопротивления (компенсационный метод). В любом варианте схемы если напряжение на X и на отрезке BS равны, то через ячейку X (соответственно N) ток не проходит. Чтобы снизить ток через ячейку X (У) во время уравновешивания компенсационной схемы, в нее включают добавочное сопротивление R2.
Вопросы и задания
Почему нельзя использовать компенсационный метод, если в цепь включено большое сопротивление?
30.3. Активность
Уравнение Нернста (464) дает «идеализированную» зависимость напряжения на ячейке (э. д. с.) от концентрации и парциального давления веществ, принимающих участие в реакции, поскольку в этом уравнении не учитываются силы взаимодействия между реагирующими частицами. В общем виде следует вместо концентраций подставлять в уравнение (464) значения
Электрохимическая термодинамика 313
активности (разд. 23.2). Последние можно вычислить для разбавленных растворов на основе теории, разработанной Дебаем й Хюккелем, в которой рассматриваются электростатическое взаимодействие ионов и их собственный объем. Активность ложно также определить, измерив э. д. с. соответствующих электрохимических ячеек и воспользовавшись затем уравнениями типа (464). Значения активности, вычисленные с помощью этого уравнения, учитывают силы взаимодействия между ионами в объеме раствора. Такой расчет дает возможность определить активность растворенного вещества в растворе, а не катионов или анионов в отдельности. В связи с этим вводится понятие средней активности. Средняя активность а± бинарного электролита выражается через активности отдельных ионов (а+ и а-) следующим образом:
а± = ]/а+ • а_ (465)
Измерения э. д. с. ячейки позволяют рассчитать только средние активности. Численное значение коэффициента активности (разд. 23.2), очевидно, зависит от выбранного способа выражения концентрации (гл. 21).
30.4. Ряд напряжений. Стандартные потенциалы
Для элемента Даниеля
Си|CuSO4 ZnSO4 | Zn (466>
в котором протекает реакция Cu2+ + Zn Си + Zn2+ (467)
если не учитывать диффузионного потенциала, э. д. с. ячейки выражается уравнением
e=8°cuZn+4^W—(468) cuzn I 2F xCu aZna+ / ’
Полагая мольные доли индивидуальных конденсированных фаз равными единице, получим
* = s’+#'n(-g£) <469)
Если измерения проводят при 25 °C и активность обоих ионов Равна единице (т. е. для стандартного состояния), уравнение (469) упрощается, а именно е = е°.
Значения е° связаны с изменением стандартной свободной энтальпии химической реакции AG0:
8 — nF
(470)
314
Электрохимия
т. е. с работой, необходимой для переноса соответствующего ко* личества электричества. Вспомнив первый закон термодинамики, можно говорить об аддитивности 8°. Уравнение для э. д. с. элемента Даниеля можно преобразовать:
® 8^ат' 8ан Cu/Cu2+H 2р ^Си2
— f8°Zn/Zn2+4—) (471)
I Zn2+J
Значения 8° можно определить на опыте, если выбрать некоторый универсальный электрод сравнения. По предложению Нернста в качестве такого электрода выбран водородный электрод. Он представляет собой платинированную платиновую пластинку, погруженную в раствор кислоты, через который пропускается газообразный водород. Активность ионов гидроксо-ния в растворе должна быть равна 1; стандартный потенциал водородного электрода по определению равен нулю. Э. д. с. ячейки, составленной из стандартного водородного электрода и электрода, на котором идет окислительно-восстановительная реакция между веществами, активность которых одинакова и равна 1, дает нам стандартный потенциал е° соответствующего окислительно-восстановительного электрода (редокс-электро-да). Измеренные таким образом значения стандартных потенциалов сведены в таблицы.
Оба электрода в элементе Даниеля можно рассматривать как окислительно-восстановительные для гетерогенной системы. Если же восстановленная форма содержится в растворе, то для соответствующего полуэлемента можно записать уравнение Нернста в следующем виде:
где п—число электронов, принимающих участие ,в окислительно-восстановительной реакции, аОх и агеа — активности вещества в окисленной и восстановленной форме. Уравнение (472} также определяет э. ,д. с. ячейки, составленной из электрода, на котором протекает^ соответствующая окислительно-восстановительная реакция, и стандартного водородного электрода. Уравнение (472) можно понимать только в этом смысле, так как потенциал полуэлемента измерить невозможно.
С .помощью таблицы стандартных потенциалов (табл. В. 15) и уравнения Нернста, пользуясь аддитивностью потенциалов полуэлементов, можно рассчитать э. д. с. практически любой комбинации электродов в электрохимической ячейке.
Вклад концентрационного члена в э. д. с. ячейки можно оце-RT
нить, имея в виду, что при 25°С-р In 10 = 59 мВ. Таким обра
Электрохимическая термодинамика
315
Зом, для п=1 при 10-кратном изменении концентрации потен-циалопределяющего вещества измеряемое значение э. д. с. смещается на 59 мВ. Для п = 2 этот член равен 29,5 мВ. Металлам, которые в стандартном состоянии являются восстановителями по отношению к системе Н2/НзО+ в стандартном состоянии, принято приписывать отрицательный стандартный потенциал; эти металлы считаются менее благородными, чем металлы, обладающие окислительными свойствами и имеющие положительный знак 8° по отношению к стандартному водородному электроду. Ряд металлов, расположенных в порядке увеличения стандартного потенциала системы металл — ион металла, называют рядом напряжений металлов.
Если реакция в элементе Даниеля (467) пройдет до конца, то э. д. с. элемента станет равной нулю, тогда из уравнения (468) получим
8° = -^-Jn I (473)
2F \ аси2+ ) ’
где azn2+ и tzcu2 + — равновесные концентрации соответствующих ионов; их значения можно вычислить из стандартного потенциала е°. Для химического равновесия имеет место аналогичное уравнение AG° = —Воспользовавшись табличными дан-)ыми для 8°, получим
' 17.Ю« (474)
• «Си2+ ’
т. е. ионы меди полностью вытесняются из раствора металлическим цинком и осаждаются в виде металлической меди. При замене цинкового электрода водородным также осаждается медь, а раствор подкисляется. Добавлением сульфидов или селенидов можно настолько снизить ничтожную концентрацию ионов Cu2+-aq, находящихся в равновесии с осадком, что для сохранения условий равновесия (474) необходимо понизить также и концентрацию Zn2J_ или Н3О+.
30.5.
Электроды второго рода. Произведение растворимости
Если потенциалопределяющий катион образует с анионом, находящимся в растворе, труднорастворимую соль, то потенци-алопределяющим становится анион, а электрод в такой системе называют электродом второго рода.
Произведение растворимости Кь для насыщенного раствора Хлорида серебра можно записать следующим образом:
aAg+ 'ac\~ — ^L
316
Электрохимия
Потенциал такого электрода относительно стандартного водо-родного электрода равен
е ео . I R? in / \ „о in д, _
8—8 Ag/Ag+ I р ш [ дс1_ I —8 Ag/AgCl р 1П МС1
Хлорсеребряный электрод обратим относительно ионов хлора, при акс1= 1 его стандартный потенциал равен 0,2225 В (для сравнения приведем потенциал серебряного электрода: Ag—>Ag+4-e~; 8°Ag/Ag+=0,799 В).
На хлорсеребряном электроде протекает электродная реакция
AgCl + е~ <=* Ag + ci-
Электроды второго рода имеют более воспроизводимый потенциал, поэтому такие электроды чаще применяют в качестве электродов сравнения
В разд. 416 3 приведены стандартные потенциалы некоторых электродов сравнения. В компенсационной схеме для измерения э. д. с методом Поггендорфа используется нормальный элемент Вестона. Он состоит из двух электродов второго рода’ один из них — это ртутьсульфатный электрод, второй — насыщенная амальгама кадмия в насыщенном растворе сульфата кадмия:
CdxHg | CdSO4 : Hg2SO4 | Hg
При 20 °C э. д. с. нормального элемента составляет 1,0183 В.
Вопросы, и задания
Пользуясь приведенными в этом разделе данными, рассчитайте произведение растворимости хлорида серебра
30.6. Концентрационные цепи
Пусть имеется электрохимическая ячейка, в которую залиты два раствора с различной концентрацией AgNO3 и одинаковой, достаточно большой концентрацией KNO3; опыт проводится таким образом, чтобы исключить перемешивание растворов (по крайней мере в начальный момент); в растворы погружены серебряные электроды.
Ионы серебра осаждаются на электроде с большей скоростью из более концентрированного раствора азотнокислого серебра (химический потенциал ионов серебра имеет более высокое значение в более концентрированном растворе AgNO3 с активностью а2 и электрод в таком растворе заряжается положительно). Э. д. с. ячейки можно выразить через активности серебра:
е=-^-1п — (475)
г ах
Электрохимическая термодинамика
317
для обеспечения условия электронейтральности системы нит-рат-ионы переносятся из более концентрированного раствора в более разбавленный. Однако, вследствие того что концентрация нитрат-ионов в обоих растворах достаточно велика, изменение активности ионов NO3_ практически незаметно.
Вопросы и задания
Каким соотношением связаны напряженность поля и разность потенциалов’
30.7. Подвижности ионов и числа переноса.
Цепи с переносом
В том случае, когда в концентрационной цепи раствор AgNO3 не содержит избытка нитрата калия, необходимо считаться с переносом ионов нитрата из раствора с большей концентрацией AgNO3 (1) в более разбавленный раствор (2). При подключении ячейки к нагрузке в цепи протекает ток, одинаковый по величине как во внешней цепи, так и в растворе элек-
Катод
Катодное пространство
+5 -5 +J
Катионы
Анионы
Анод
Объем I Анодное раствора. । пространство +j|-5 +5
-2+2 0 0
Рис Б 35 Изменение концентрации в результате протекания тока в при-электродных пространствах в ячейке с двумя серебряными электродами, погруженными в раствор нитрата серебра
На катоде выделилось 5 г экв катионов и столько же катионов растворилось на аноде, rI+/n_=d/2 Поступление (потеря) катионов и анионов условно показано иа границах ка-
тодного и анодного пространства
тролита, однако в растворе электрический заряд переносится ионами. Из-за того что гидратированные ионы имеют разные размеры, в одном и том же электрическом поле скорость их движения неодинакова, иначе говоря, подвижности ионов различны. Вклад ионов в электрический ток также неодинаков. Доля тока, соответствующая переносу электрического заряда ионами одного вида, называется числом переноса п ‘Сумма чисел переноса катионов п+ и анионов п- равна 1: «++«_=!.
318
Электрохимия
Пусть через замкнутую электрическую цепь проходит количество электричества, равное 1 фарадею. Тогда на катоде (рис. Б.35) выделится 1 г-экв. серебра, а на аноде такое же количество перейдет в раствор в виде ионов.
На рис. Б.35 приведена схема изменения концентрации в различных частях электрохимической ячейки с двумя серебряными электродами, заполненной раствором AgNO3. Граница раздела анодного и катодного пространства, т. е. «середина» раствора, выбирается произвольно; необходимо только выполнение условия постоянства концентрации нитрата серебра в середине раствора.
Пусть в результате протекания тока через ячейку на катоде выделится 1 г-экв. катионов, а на аноде точно такое же количество катионов перейдет в раствор; примем также «+/«-=3/2. Тогда из середины раствора в катодное пространство будет перенесено п+ г-экв. катионов серебра, и из катодного пространства исчезают (1—ц+)=п_ г-экв. серебра, а также п- г-экв анионов. Перенос анионов происходит, во-первых, в соответствии с введенным выше представлением о числах переноса, во-вторых, ввиду необходимости обеспечения электронейтральности раствора. Аналогичные соображения приводят к заключению о том, что в анодном пространстве появляются дополнительно г-экв. азотнокислого серебра (ср. рис. Б.35). [Измерения концентрации в катодном и анодном пространстве используются для определения чисел переноса по методу Гитторфа.] Таким образом, изменение свободной энтальпии равно
AG = [(р"д§++h"no3“) (и'Ag+ Ч~Нко3-)] —
= n_RT In a;Ag+,a,N-°-^ 2п_RT \n^-=Fe (476)
a Ag+ a no.-t a ±
Э. д. с. ячейки с двумя водородными электродами в растворе соляной кислоты и э. д. с. ячейки с двумя хлорными электродами равны соответственно
ен==^Г1п^Х (477)
ес1 = 2п+ In (478)
'Следовательно, из измерений разности потенциалов такого рода электрохимических ячеек можно получить значения чисел переноса, однако следует заметить, что метод Гитторфа дает ‘более точные результаты.
Вопросы и задания
Подумайте, одинакова ли полярность водородного и хлорного электрода, погруженного в раствор концентрированной соляной кислоты?
Электрохимическая термодинамика
319*
30.8. Диффузионный потенциал
Число переноса ионов водорода в растворе соляной кислоты оказывается большим, чем число переноса хлорид-ионов, поэто-чу в соответствии с уравнениями (477) и (478) э. д. с. ячейки с двумя хлорными электродами имеет более высокое значение, чем э. д. с. ячейки с водородными электродами.
Это можно представить себе более наглядно, введя понятие диффузионного потенциала. Рассмотрим границу растворов соляной кислоты различной концентрации, залитых в полуэлементы электрохимической ячейки. Химический потенциал разбавленного раствора ниже, чем потенциал граничащего с ним концентрированного раствора. Поэтому ионы водорода и хлора из концентрированного раствора под действием разности химических потенциалов диффундируют в более разбавленный раствор (химический потенциал этих ионов примерно одинаков). Однако подвижность ионов водорода более высокая, чем подвижность ионов хлора. Ионы водорода как бы «спешат» и создают в более разбавленном растворе избыток положительных зарядов, т. е. электрическое поле. Это поле выравнивает скорость переноса ионов, ионы водорода «тормозятся» этим полем, а движение хлорид-ионов ускоряется.
Вопросы, и задания
Пусть раствор содержит избыток постороннего электролита Какое влияние это оказывает на движение ионов?
Рассмотренное выше незначительное разделение ионов вызывает появление разности потенциалов на границе между растворами, находящимися в полуэлементах,— диффузионного потенциала. Для ячейки с хлорными электродами полярность диффузионного потенциала совпадает со знаком э. д. с. ячейки. Для ячейки с водородными электродами диффузионный потенциал имеет противоположный знак, поэтому э. д. с. такой ячейки с переносом меньше, чем э. д. с. ячейки с хлорными электродами. Можно в значительной степени подавить диффузионный потенциал добавлением солей, для которых подвижности катионов и анионов практически одинаковы, напримёр КС1, KNO3, NH4CI. Такой прием часто используют на практике, так как при электрохимических измерениях диффузионный потенциал желательно исключить.
В отсутствие диффузионного потенциала э. д. с. ячейки определяется уравнением (475). Хлорный электрод в более концентрированном растворе соляной кислоты имеет отрицательный знак относительно второго хлорного электрода.
Можно представить э. д. с. цепи с переносом (епер) как сумму э. д. с. концентрационной цепи 8К и диффузионного потенциала 8d:
епер = 8к~Ь eD (479)}
320
Электрохимия
Числа переноса можно выразить через подвижности ионов и т. е. скорость движения иона при напряженности поля, равной 1:
п_ = —v— ; w. = —щ— (480)
U+ + U- ’ + и+ 4- U— V V)
Воспользовавшись этими соотношениями, получим из (475), (478) и (479) уравнение Нернста для диффузионного потенциала 1,1-зарядного электролита:
( 2и+ »+ +«- RT 1t, а"+
О ГЛ 1 1 1 1 I I I ГУ 111 /
и \ и+ + и- и+ + j г а ±
= “+~“~ ^-1пД±- (481)
и^. —Р F а '
.Для многозарядного иона (заряд гг) диффузионный потенциал равен
2, z, RT , а"± ,.О1 .
Bn = In —(481 а)
и Ui + Ua F а'± v 1
.30.9. Ионообменники. Равновесие Доннана. Электрохимический потенциал
на границе фаз. Двойной электрический слой
Пусть растворы электролита различной концентрации разделены пористой перегородкой. Тогда диффузионный потенциал между растворами по обе стороны перегородки определяется уравнением (481), где «+ и «_ представляют собой эффективные значения подвижности ионов в объеме перегородки. Предположим теперь, что эта перегородка является полупроницаемой (мембрана), т. е. через нее могут проходить ионы только одного вида (катионы или анионы). Тогда разность потенциалов равна
где п — заряд проходящего через мембрану иона. Эта разность потенциалов возникает при переносе некоторого количества ионов через мембрану и препятствует их дальнейшему движению; иными словам'и, более разбавленный раствор имеет тот же знак заряда, что и перемещающийся в мембране ион.
Полупроницаемая мембрана препятствует диффузии в растворах бинарных электролитов и тем самым их перемешиванию-Катионо- или анионообменные мембраны представляют собой систе'мы, в которых анионные (соответственно катионные) труп-
Электрохимическая термодинамика
321
цы включены в неподвижную структуру (связанные ионы), в то время как катионы (соответственно анионы) обмениваются с раствором (противоионы). Это дает возможность удалять посторонние или мешающие ионы из раствора. Ионы малого размера того же знака что и связанные в мембране ионы, могут до некоторой степени проникать в мембрану (см. ниже).
Аналогичное положение имеет место на фазовой границе мембрана — раствор: мембрана заряжается относительно раствора, причем знак заряда соответствует знаку связанных ионов. Разность потенциалов равна
RT , а"
8 = —рг In —
nF а
где а" и а' — активности обменивающихся с мембраной ионов. Эта разность потенциалов соответствует электрической части работы, которая совершается при переносе единичного заряда через границу фаз. Для полупроницаемой мембраны его называют потенциалом Доннана.
При изучении самопроизвольно протекающих процессов необходимо наряду с химическими потенциалами учитывать разность электрических потенциалов, поэтому для ионных систем часто используют понятие электрохимический потенциал, который включает в себя обе составляющие термодинамического потенциала. Для случая мембраны, по обе стороны которой находятся растворы электролита, отличающиеся только концентрацией, электрический и химический потенциалы равны и противоположны по знаку:
/ а , nFe=RT In
\ а ±
(482)
В случае контакта двух различных фаз разделение электрического и химического потенциалов может быть только произвольным, так как все носители зарядов имеют различные химические и электрические потенциалы. При равновесии разность потенциалов также равна и противоположна по знаку разности химических потенциалов контактирующих фаз, между которыми происходит перенос зарядов.
Для полупроницаемой мембраны уравнение (482) является условием равновесия для ионов, обменивающихся с мембраной. Ионы того же знака, что и противоионы, накапливаются в мембране, так как она имеет небольшой избыточный заряд того же знака, что и связанные ионы, и принимает соответствующий потенциал относительно раствора. Если противоионы являются Катионами (мембрана заряжена отрицательно), то
„ RT . ( а"кат \I/n a"Na+ а"к2+ / а"са+ \1/2 1
е = —— In , т- , например —Д2—=——Л
F I а кат I Г г a'Na+ а>^2+ I а Са+ /
21—2027
322
Электрохимия
Из этих соотношений можно сделать вывод о том, что многозарядные катионы накапливаются в мембране в большей степени, чем однозарядные. X называют доннановским коэффициентом распределения.
Для анионов также справедливо соотношение
а градиент концентрации имеет противоположный знак. Итак,, из этих соотношений следует условие равновесия Доннана
а"а~— а Na+ • (fl,,Na+)2 ’ fl,,SO42- = (^Na*)2 ‘ а SO42-
30.10. Э. д. с. ячейки и составляющие скачка потенциала на границе фаз. Двойной электрический спой
Все фазовые границы в электрохимической ячейке проницаемы для носителей заряда одного типа (например, электронов между контактирующими фазами металлов) и непроницаемы для носителей другого типа. На границе фаз, через которую происходит обмен электрических зарядов, устанавливается равновесие, при котором электрохимические потенциалы по обе стороны границы равны между собой. В процессе установления
Pt(Cla) HClfy) Hctto2) Pt(Cl2)
£d
Рис. Б.36. Изменение потенпиала в ячейке, если ai>ai.
Отдельные скачки потенциала на границе фаз не измеряются, однако известно, что 1 1 а"+
ei+e2= In ~ , если
F а'±
равновесия, вообще говоря, необходим перенос зарядов через межфазную границу. В результате на границе фаз образуется двойной электрический слой, скачок потенциала в двойном электрическом слое препятствует дальнейшему переносу зарядов через межфазную границу.
Образование двойного электрического слоя происходит на поверхности раздела разнородных фаз. Сумма всех скачков потенциала в электрической цепи электрохимической ячейки (включая скачки потенциала между разными металлами) равна э. д. с. ячейки. В отличие от скачка потенциала между различными .фазами э. д. с. ячейки может быть измерена, напри
Электрохимическая термодинамика
323
мер, компенсационным методом (рис. Б.36). В двойном электрическом слое можно выделить плотную и диффузную части. Между поверхностью металла и слоем ионов, находящихся на минимальном расстоянии от металла, находится плотная часть двойного слоя (слой Гельмгольца). В этом слое напряженность электрического поля может достигать значений до 107 В/см. Плотная часть двойного слоя оказывает значительное влияние
Рис. Б.37. Изменение потенциала в двойном слое в отсутствие (а) и при наличии (б) адсорбции.
€о — суммарный скачок потенциала, ej — скачок потенциала в диффузном слое.
на протекание электродного процесса. К плотной части двойного слоя примыкает диффузный слой, в котором электрическое поле (нелинейное) и температурный градиент (тепловое движение), обеспечивающие равномерное распределение ионов, действуют в противоположных направлениях, что приводит к установлению равновесного распределения ионов вблизи поверхности металла. Чем выше концентрация ионов, тем меньше толщина диффузного слоя (ср. «радиус ионной атмосферы» в теории Дебая— Хюккеля, рис. Б.37).
30.11. Основы потенциометрического титрования
Логарифмическая зависимость уравнения Нернста, связывающая потенциал электрода и концентрацию (активность) по-тенциалопределяющего вещества (чаще всего ионов) в растворе, очень удобна для определения конечной точки титрования при аналитическом определении концентрации вещества. Метод титрования, при котором конечная точка определяется по разности потенциалов соответствующей электрохимической ячейки, Называется потенциометрическим титрованием.
Предположим, что при титровании потенциалопределяющее вещество удаляется из раствора в результате реакции с тит-Рзнтом; при этом могут происходить окислительно-восстановительная реакция (редокс-титрование), реакция осаждения (осадительное титрование), нейтрализация (кислотно-основное тит-21*
324
Электрохимия
Электрохимическая термодинамика
325
рование) и комплексообразование. Вначале потенциал электрода меняется медленно, так как концентрация в уравнении Нернста входит под знак логарифма (потенциал меняется только на 59 мВ при снижении концентрации в 10 раз). В конечной точке титрования исходное потенциалопределяющее вещество исчезает не полностью —его концентрация определяется константой равновесия реакции взаимодействия титранта с анализируемым веществом. Чем больше константа равновесия этой реакции, тем меньше концентрация исходного вещества в точке эквивалентности. Соответственно, чем больше десятичный логарифм отношения начальной .концентрации реагирующего вещества к его концентрации в точке эквивалентности, тем резче выражен скачок потенциала, указывающий конечную точку титрования. В качестве титрантов более всего подходят осадители, образующие осадки с низким значением произведения растворимости, комплексообразователи, образующие комплексные соединения с большой константой устойчивости и редокс-системы, стандартный потенциал которых в достаточной степени отличается от стандартного потенциала титруемой системы Точность определения возрастает с увеличением соответствующей константы равновесия.
Пример. Рассмотрим связь между концентрацией титранта и потенциалом в конечной точке титрования. Какие факторы влияют иа точность определения?
Если в титровании принимают участие две редокс-системы, то измеряемая разность потенциалов определяется взаимодействием четырех веществ, участвующих в реакции. При этом в большинстве случаев равновесное состояние устанавливается очень быстро.
При титровании железа(П) церием(1У) (см. рис. Д.133) протекают следующие реакции:
Fe2+ -<—> Fe3+ + е~
Се4+ + е~ Се3+
8° = 0,77 В
8» = 1,61 В
Fe2++Ce4+ Fe3++Ce3+
е° = 0,84 В; tf = 2,5-1014
RT , 8 = 1,61 + р 1П
Ссе4+
ССе3+
RT , = 0,77+ -j?-In
cFe3+
CFe2+
Поскольку титрование в данном случае проводят в сильиокислых растворах с постоянной концентрацией кислоты, коэффициенты активности можи< сократить или перенести их в константу е° и все расчеты проводить со зна чениями концентрации. После достижения в ходе титрования стандартной потенциала e°Fe3+/Fe2+ половина исходного количества Fe(II) переходит i окисленное состояние. В данном случае изменение потенциала при добавлс нии титранта (Да/AV) имеет минимальное значение. Такая же картина на блюдается прн кислотно-основном титровании. Функциональная зависимое!) между значением pH и отношением концентрации соли к концентрации кис лоты точно такая же, как между разностью потенциалов и отношением кон центраций компонентов редокс-системы. Как только половина слабой кислс ты будет оттитрована, концентрация слабой кислоты и концентрация ее сол ’ соавниваются. Это означает, что значение pH примерно равно значению
слабой кислоты, а буферная емкость достигает максимума В точке эквивалентности
CFe3+ = Cfe3+ и CFe2+ = сСе4+
Если ввести эти соотношения в константу равновесия
„ cFe3+ •сСе3+
Сре2+'Ссе4+
то получим
/ cFe3+ \ __ / сСе3+ \ =
\ CFe2* /еэкв \ сСе4+ Аэкв У *
RT RT
еэкв = e°Fe3+/Fe2+ + ~2р~ % = е°Се4+/Се3+ + ~^р~ 1П К
ИЛН
р — e°Fe3+/Fe2+ + /Се3+ —lion
ьэкв — -----------2 ------------ ““ 1 > 1У Ь
Для симметричной кривой титрования Де/ДК имеет максимальное значение в точке эквивалентности.
Вопросы и задания
1. Как изменится кривая титрования, если в раствор были заранее введены железо(Ш) или церий(П)?
2. Как из кривой титрования определить стандартный потенциал редокс-системы, а также число электронов п, принимающих участие в реакции?
Предположим, что число электронов п, принимающих участие в реакции, неодинаково для редокс-систем, использованных в потенциометрическом титроваиии. Тогда точка эквивалентности расположена ближе к потенциалу редокс-системы с большим значением п. Кривая титрования становится несимметричной, а производная de/dV при 8ЭКв ие достигает максимума. Если в реакции принимают участие ионы гидроксония, то положение точки эквивалентности зависит и от pH.
Рассмотрим в качестве примера потенциометрическое титрование с осаждением галогенида серебра. Оно проводится с серебряным индикаторным электродом. Перед достижением точки эквивалентности в растворе имеется избыток ионов серебра, в точке эквивалентности концентрация ионов серебра равна концентрации галогенид-ионов, продолжение процесса титрования создает избыток последних. Концентрация ионов серебра в растворе определяется произведением растворимости:
3KBcAg+ ~ эквсСП = J/ Пр; ~ 1
и
RT RT
&экв — e°Ag+/Ag "Г -р 1П sKBcAg+ = B°Ag+/Ag 4“ OF
Поскольку иоиы Ag+ и С1~ имеют единичный заряд (п=1), кривая титрования симметрична и производная d&jdV максимальна в точке эквивалентности. При осаждении каломели Hg2Cl2 дело обстоит иначе, так как для катиона Hgs2+ п=2. Однако в точке эквивалентности концентрации ионов Hg22+ и '-I- соответствуют стехиометрии реакции нх взаимодействия.
326
Электрохимия
Вопросы и задания
1. Выведите формулу для потенциала в точке эквивалентности при осаждении каломели.
2. Выведите формулу для потенциала в точке эквивалентности при титровании комплексообразователем.
3. Почему редокс-система при потенциале е° ведет себя так же, как буферная система, в которой рН=рК?
30.12. Уравнение Лютера
Если элемент образует ионы, соответствующие различной степени окисления, то между стандартными потенциалами
6 м*1+/м’ 6 6 mZ1+/mZz+
имеется известное термодинамическое соотношение — уравнение Лютера.
Рассмотрим вывод этого уравнения на примере меди, которая в водном растворе может образовывать однозарядные и двухзарядные ионы. Стандартный потенциал электрода измеряется в ячейке относительно водородного электрода. В ячейке протекают следующие химические реакции:
Си + Н+ Си+ + V8H8 (I)
Си+4-Н+ ч==* Cua++VH2 (И)
Си + 2Н+Си2++Н2 (Ш)
Изменение свободной энтальпии ие зависит от пути реакции, поэтому сумма изменений свободной энтальпии реакции (I) и (II) равна изменению свободной энтальпии реакции (III):
ДО»! + ДО0п = AG’in (483)
Используя уравнение (470), получим
1-е0! + 1 е% = 2-е0э или
е% = 2е°3 — е»!
В обобщенной форме уравнение Лютера имеет следующий вид:
*2 V2+/m “ ^м^/м /ЛЯЛ,
‘ 1
По таблице В. 19 найдем
e°Cu2+/Cu-'= 0,345 В и е°Си2+/С1)+ = 0,15 В
Отсюда
e°Cu+/Cu — 2-0,345 — (2— 1)0,15 = 0,54 В
Равновесие диспропорционирования (разд. 33.5.2.2) устанавливается при погружении меди в раствор, содержащий Си(II) в результате протекания ре-акции
Си2+ + Си 2 Си+ (485)
Концентрацию образовавшейся Си+ можно вычислить из условия равновесия
0,345 + ~^Г 1п (Дси2+) = °>54 + ~7Г' 1паСи+
Электрохимическая термодинамика
327
т_ е. при 25 °C
Дси2+ а2Си+
0,195
= 10 0,0296 = 106>688 = 3,87-106
Это и есть константа равновесия, определенная из стандартных потенциалов. Определим активность ионов меди(1), если, например,
сСи2+ = 0,1 моль/л
—7,588
10-1 ~2----
nW = Т06>888~ = 10~7’688; аСи+ = Ю = Ю-3>794 = 1,6Ь10-4 моль/л
Очевидно, что во многих случаях приходится учитывать концентрацию ионов Си+.
Реакция диспропорционирования в растворах меди(1) протекает с большой скоростью с образованием ионов Си2+ и меди до достижения равновесного состояния и в отсутствие металлической меди [уравнение (485)], т. е. растворы меди(1) неустойчивы (разд. 35.5.2.2).
31. ПРОТЕКАНИЕ ЭЛЕКТРИЧЕСКОГО ТОКА
в электрохимической ячейке
Протекание электрического тока через гальванический элемент или электрохимическую ячейку нарушает состояние равновесия, в результате чего термодинамические условия равновесия более не выполняются. Протекание тока подчиняется кинетическим закономерностям, наблюдающимся, во-первых, при переносе ионов в растворе электролита и, во-вторых, при переносе электрона через границу металл — электролит.
3.1.1. Электропроводность растворов электролитов
Электропроводность растворов электролитов подчиняется закону Ома (ток / пропорционален напряжению 77), следует лишь обеспечить такие условия измерений, при которых вблизи электродов не происходят изменения концентрации в результате протекания тока. Чаще йсего измерения электропроводности растворов электролитов проводят при помощи моста Уитстона, подключенного к источнику переменного тока (частота переменного тока обычно равна 1 кГц).
Подчинение электропроводности растворов электролитов закону Ома означает, что количество носителей заряда в системе остается неизменным и при постоянной напряженности электрического поля скорость движения носителей заряда также постоянна. Таким образом, можно считать, что скорость движения ионов пропорциональна напряженности поля Е (если не Учитывать переходное время, в течение которого ионы получают ускорение). Скорость движения иона w при напряженности электрического поля £=1 называют подвижностью иона и.
It
wJ. — u+E=—u+dU/дх
(486)
328
Электрохимия
Ток через объем электролита с площадью поперечного сечения равен
i=.qN+z+ew+-\-qN_z.ew_=N+z+e(w+-\-w.) q (487)
где z+e — зарядовое число катиона, z_e— зарядовое число аниона, е— заряд электрона. Из соображений электронейтральности N+z+ = N-Z-, где N+, N-— соответственно число положительных и отрицательных носителей заряда в единице объема раствора электролита.
Сопротивление объема раствора электролита между электродами с площадью q, удельным сопротивлением р и расстоянием между электродами I равно R = pl/q. Учитывая закон Ома i=El/R и уравнения (486) и (487), получим
FI
i=N+z+e (и++«_) Eq=-у
или
JL =:l.=Ji^N+Z+e(U++u_} (488
Таким образом, удельная электропроводность н пропорциональ на концентрации электролита в растворе. Однако на опыте наблюдаются отклонения от пропорциональности, которые связаны с взаимодействием между ионами в растворе. В растворах слабых электролитов химическое взаимодействие приводит к неполной диссоциации молекул на ионы; в растворах сильных (полностью диссоциированных) электролитов наблюдается электростатическое взаимодействие между ионами. Для того, чтобы провести оценку данных по электропроводности независимо от концентрации носителей заряда и их взаимодействия введем понятие эквивалентной электропроводности X; это элек тропроводность, отнесенная к постоянному числу носителей за ряда Х=и/с. В зависимости от способа выражения концентрации (г-экв./мл или моль/мл) ее называют эквивалентной или молярной электропроводностью.
Концентрацию (в грамм-эквивалентах на литр) можно выразить в виде отношения общего заряда частиц к объему, воспользовавшись числом Фарадея: c = N+-z+-e!F. Отсюда, учитывая (488), получаем
^=-^ = К(Н+ + «_)
K+=Fu+ называют ионной подвижностью; величины и+ и и-связаны с числом переноса соотношением (разд. 30.8):
+ X X.J. 4* X— и* -j- и—
Электрохимическая термодинамика
329
Значения подвижности можно найти из измерений электропроводности, определив числа переноса, например, методом Гитторфа.
Вопросы и задания
1. Подвижность можно определить из вполне доступных измерений электропроводности х, а также чисел переноса. Если х задана в обычных единицах, в каких единицах получатся значения: а) ионной электропроводности, б) ионной подвижности, в) эквивалентной электропроводности? Проверьте, в каких единицах (по определению) задается подвижность. В таблицах обычно приводятся значения ионной электропроводности.
2. Выведите уравнение для измерения сопротивления при помощи моста Уитстона, аналогично тому как это было сделано для компенсационной схемы измерения э. д. с. ячейки.
31.2. Коэффициент диффузии и подвижность
Движение ионов в растворе происходит как под действием электрического поля (градиента электрического потенциала), так и в результате различия химических потенциалов в отдельных частях системы. Перенос ионов в электрическом поле называют миграцией, а перенос ионов под действием градиента химического потенциала — диффузией. Установим связь между этими явлениями. Движущая сила, определяемая градиентом химического потенциала и отнесенная к 1 молю вещества, равна /=—ду.г/дх. После введения коэффициента пропорциональности Vi, связывающего / и скорость ионов w, получим ш =—Vi-•дщ/дх. Через единицу поверхности в единицу времени проходит поток ионов / =—ViCi-dy,t/dxi, где с,— концентрация в единице объема. Полагая = In (что справедливо для разбавленных растворов), получим для градиента химического потенциала (дуц'дх) = (RT/Ci) (dct/dx), а для потока ионов ji = -RTVi-dcildx.
В соответствии с первым законом диффузии Фика (1885 г.) количество вещества, диффундирующего в единицу времени через единицу поверхности, пропорционально градиенту концентрации ji — —Di-dc^dx, где Di — коэффициент диффузии ионов i. Из сравнения двух последних уравнений для /, видно, что Di= — RTVi. В то же время движение ионов со скоростью в электрическом поле с напряженностью Е определяется подвижностью ионов Ui = Wi)E. Таким образом, движущая сила / для 1 моля ионов с зарядом г, в поле Е равна I~ZiFE. Отсюда ^iiZiFE = UilziF. Сравнение этих выражений показывает, что Vi — UifziF, т. е. между подвижностью и коэффициентом диффузии существует следующее соотношение (В. Нернст, 1888 г.):
гл _ RTui
~ Z(F
330
Электрохимия
Вопросы и задания
Почему в последнем уравнении появляется температура? Как опытным путем измерить коэффициенты диффузии и подвижность?
31.3. Слабые электролиты
Зависимость эквивалентной электропроводности от концентрации в растворах слабых электролитов объясняется прежде всего их неполной диссоциацией. Предельную эквивалентную электропроводность Хо, соответствующую полной диссоциации вещества, получают экстраполяцией экспериментальных данных, полученных при постепенном разбавлении раствора, на бесконечное разбавление. Если не принимать во внимание электростатическое взаимодействие, то можно, как это предложено Аррениусом, определить степень диссоциации как а = ХДо-
Пользуясь законом действующих масс, получим закон разбавления Оствальда
(«9)
который преобразуется в форму, удобную для графического определения Кс и X:
Вопросы и задания
Какие величины следует откладывать на осях координат? Как из измеренных значений X и с установить графически неизвестные Кс и Хо?
31.4. Сильные электролиты
В растворах сильных электролитов при уменьшении концентрации эквивалентная электропроводность меняется не так резко, как для слабых электролитов. Чем больше заряд иона, тем значительнее изменения эквивалентной электропроводности с концентрацией. Отношение электропроводности при концентрации с к предельной эквивалентной электропроводности называется коэффициентом электропроводности: 'Kcl‘Ko=‘f‘K (табл. Б.1). Величины ионной электропроводности аддитивны как для сильных, так и для слабых электролитов [уравнение (489)]. Для сильных электролитов это справедливо также и в разбавленных растворах, если учитывать ионную силу.
Зависимость эквивалентной электропроводности от концентрации установлена Кольраушем (1900 г.):
\.=Х0—А /с
Константа А для одного и того же растворителя зависит в основном от заряда ионов. В сильных электролитах снижение
Электрохимическая термодинамика
331
Таблица Б.1. Коэффициент электропроводности электролитов различных типов
Тип электролита f для с, г-экв./л
10-J io-2 10~‘
1,1-зарядиый 0,98 0,93 0,83
1,2-зарядный 1 0,95 0,87 0,75
2,1-зарядный J
2,2-зарядный 0,85 0,65 0,4
электропроводности при увеличении концентрации, очевидно, объясняется электростатическим взаимодействием ионов. На этих представлениях строится теория сильных электролитов Дебая и Хюккеля (1923 г.).
В растворе электролита вблизи каждого иона сосредоточивается больше ионов противоположного знака, чем ионов одноименного. Образованию такой ионной атмосферы благоприятствуют более высокий потенциал иона ф и увеличение его заряда; размыванию (разрушению) ионной атмосферы благоприятствует увеличение температуры.
Количественная картина распределения заряженных частиц в объеме электролита дается законом распределения Больцмана
N+dV=exp ( - ) dV (490)
ЛГ_(/К=Угехр(-!-^^рК (491)
где AZ+, N-—число положительных и отрицательных носителей заряда в элементарном объеме dV, z+ (соответственно z_) —• зарядовое число катиона (аниона); е — заряд электрона; А/,— средняя концентрация ионов в единице объема. Плотность заряда р и электрический потенциал ф связаны дифференциальным уравнением Пуассона, которое записывается в сферических координатах в следующем виде:
1 d / 2 t/ф \ 4лр г2 dr у dr j е
где г — расстояние от центрального иона, 8 — диэлектрическая проницаемость.
По сравнению с энергией теплового движения частиц электрическая энергия иона очень невелика (геф^йТ’), поэтому экспоненциальный множитель в уравнениях (490) и (491) можно разложить в ряд
21 31
332
Электрохимия
и ограничиться первым членом ряда. Просуммировав число заряженных частиц из уравнений (490) и (491) (первые члены) и умножив их на соответствующие заряды, получим плотность заряда
Р== Iг-1 е) -\-z+e — | г_ | е j =
= -^[АМЛе2+1г-М1
Подставим выражение для плотности заряда р= I iSA/jZi2 в уравнение Пуассона. Его решение относительно потенциала представляет собой функцию от расстояния:
-у ,р р"Л^ .£-------er-w (492)
1 er 1 -j- %а ' '
где а — минимальное расстояние, на которое ион может подойти к центральному иону. Коэффициент х2 определяется как
I
где Na — число Авогадро, сг — молярная концентрация, 1/х имеет размерность длины и часто называется дебаевской длиной или радиусом ионной атмосферы. Ионная атмосфера, состоящая из ионов, окружающих центральный ион, эквивалентна шарику диаметром 1/х с зарядом, компенсирующим заряд центрального иона.
Снижение эквивалентной электропроводности электролита при увеличении концентрации можно представить себе наглядно. Пусть при движении центрального иона в электрическом поле ионная атмосфера возникает перед ним и исчезает позади него. Появление ионной атмосферы происходит с некоторой задержкой времени (релаксацией). Время релаксации обратно пропорционально концентрации и заряду ионов, а также электропроводности. В результате движения иона равнодействующая всех зарядов ионной атмосферы смещается назад по движению ионов, иначе говоря, ионная атмосфера деформируется, становится асимметричной и поэтому тормозит движение центрального иона из-за электростатического взаимодействия (эффект релаксации). Кроме эффекта релаксации возникает также электрофоретическая сила. Она создается вследствие того, что ионная атмосфера состоит преимущественно из ионов противоположного знака и при движении в направлении, противоположном центральному иону, увлекает за собой молекулы растворителя; в результате возникают как бы дополнительные силы трения. Обе эти силы обратно пропорциональны радиусу
Электрохимическая термодинамика 333
ионной атмосферы, что соответствует установленной Кольрау-щем зависимости эквивалентной электропроводности от концентрации.
Теория Дебая — Хюккеля позволяет рассчитать коэффициент активности ионов в электролите. Коэффициент активности соответствует работе, которая была бы совершена, если 1 моль вещества из некоторого воображаемого раствора без электростатического взаимодействия перенести в раствор, в котором он имеет место.
Электрический потенциал, который создает ионная атмосфера в точке, соответствующей центральному иону, легко вычислить с помощью уравнения (492). Он как раз соответствует той работе, которую совершает единичный заряд при рассмотренном выше воображаемом переносе. Эта работа, умноженная на заряд центрального иона гге, дает коэффициент активности, достоверный в тех пределах, в которых остается справедливым предельный закон Дебая — Хюккеля. Для одно-однозарядных электролитов в водных растворах область применимости предельного закона ограничивается концентрациями 10-3 моль/л (для бинарного электролита f+=f_=f±). Средний коэффициент активности определяется формулой
1 + DCL у 1
где А и В — коэффициенты, зависящие от температуры и природы растворителя; здесь вместо концентрации введена ионная сила раствора /=1/2Хсггг2. Коэффициент 1/2 указывает, что для одно-однозарядных электролитов ионная сила равна концентрации. В электрохимии и теории растворов вместо концентрации часто используется ионная сила, так как электростатическое взаимодействие ионов играет важную роль в электрохимических процессах.
В настоящее время можно считать уже установленным фактом, что на графике зависимости коэффициента активности от концентрации наблюдается минимум, а в концентрированных растворах коэффициент активности зачастую превышает единицу, в то время как, согласно теории межионного взаимодействия, коэффициент активности должен монотонно уменьшаться с увеличением концентрации. Ниже приводится эмпирическое Уравнение для зависимости коэффициента активности от концентрации, которое хорошо соответствует экспериментальным Данным вплоть до концентрированных растворов:
Л | 2_ | 2+ // 1 + Ba V1
lg
VCI
334 Электрохимия
где С—некоторый коэффициент при втором слагаемом. Член CI учитывает собственный объем ионов в ионной атмосфере, который не рассматривается в теории Дебая — Хюккеля (ср. зависимость pv от v для реальных газов).
Вопросы и задания
1. Влияет ли присутствие избытка соляной кислоты на ионную электропроводность К+ в растворе КС1?
2. Какие параметры следует измерить, чтобы получить данные о силах взаимодействия между ионами? Предложите методы измерения. Какие параметры имеют наиболее важное значение?
3. Какое влияние оказывают сильное электрическое поле и высокочастотный переменный ток на экспериментально определяемые значения электропроводности?
31.5. Кинетика электродных процессов
В отличие от химической кинетики для кинетики электрод ных процессов важнейшее значение имеет зависимость скоро сти процесса от потенциала электрода. Скорость электрохимической реакции определяется электрическим током, протекающим через электроды ячейки. Количество вещества, прореагировавшего на единице поверхности электрода за единицу времени (dN/dt), эквивалентно плотности тока; эти величины связаны уравнением
t dN \ _ t
\ dt )х-^о~ nF
где N — число прореагировавших частиц (ионов, отдавших или принявших электроны), х — расстояние от поверхности электрода. Суммарный электрохимический процесс превращения вещества протекает в несколько последовательных стадий:
1) перенос исходных веществ к электроду (осуществляется в основном путем конвекции и диффузии);
2) реакция перехода электронов через фазовую границу;
3) отвод продуктов реакции от электрода (как правило, происходит по механизму конвективной диффузии).
Если не рассматривать начального периода времени после включения тока, то в стационарном состоянии скорость всех последовательных стадий одинакова, в противном случае на одной из этих стадий будет происходить накопление вещества и не будут выполняться условия стационарности.
До тех пор пока на фазовой границе не образуется насыщенного раствора продуктов реакции (непосредственно вблизи поверхности электрода преобладают процессы диффузии), отвод продуктов реакции точно соответствует подводу исходных веществ, т. е. не является скоростьопределяющей стадией. Ско-ростьопределяющая стадия вызывает наибольшее отклонение от равновесия. Иногда обе стадии могут лимитировать проте-
Электрохимическая термодинамика
335
какие процесса (случай смешанной кинетики). Если только одна из ст'цдий определяет скорость всего процесса, то характеристики суммарного процесса зависят только от параметров этой стадии.
В некоторых случаях перед стадией перехода электрона через межфазную границу или после нее вблизи поверхности электрода протекает химическая реакция; если такая гомогенная химическая реакция имеет не очень большую скорость, то она накладывается на процессы переноса вещества к электроду или от него. Теория таких процессов достаточно сложна и здесь не рассматривается.
Если система находится в равновесии, то ток во внешней цепи не протекает. Протекание тока в электрохимической ячейке вызывает смещение потенциала ф от его равновесного значения <рр. Отклонение потенциала от равновесного при протекании тока называют перенапряжением т] = <р—фр. Электроды, на которых наблюдается высокое перенапряжение уже при небольших плотностях тока, называют поляризуемыми; электроды же, перенапряжение которых невелико даже при больших плотностях тока, называют неполяризуемыми. Вещество, которое окисляется или восстанавливается на электроде (причем в отсутствие этого вещества на электроде должен протекать процесс с более высоким перенапряжением), иногда называют деполяризатором.
В том случае, если лимитирующей стадией процесса является перенос вещества к электроду (первая стадия), концентрация на поверхности электрода существенно отличается от концентрации в объеме раствора и на электроде возникает перенапряжение диффузии. Однако, как известно, потенциал электрода определяется концентрацией потенциалопределяющего вещества вблизи электрода, поэтому при протекании тока потенциал электрода отличается от равновесного значения. Перенапряжение диффузии часто проявляется в полярографии.
Если лимитирующей является вторая стадия, то на электроде наблюдается перенапряжение перехода электрона. В этом случае концентрация вещества одинакова как вблизи поверхности электрода, так и в объеме раствора, однако появляются существенные отклонения от функциональной зависимости, характерной для равновесия (уравнение Нернста), поскольку нарушено равновесие между переходом электронов в прямом и обратном направлениях.
Рассмотрим более подробно оба вида перенапряжения.
31.5.1. Перенапряжение диффузии
Перенапряжение диффузии тесио связано с изменениями коицеитрации в приэлектродиом слое. Это в свою очередь приводит к изменению плотности вблизи поверхности электрода; в результате происходит стекание рас
336
Электрохимия
твора, имеющего более высокую плотность, вниз и вытеснение раствора меньшей плотности к верхней части электрода —происходит естественная конвекция. Если принять меры к уменьшению естественной конвекции, то фронт диффузии медленно отодвигается в глубь объема раствора и стационарное состояние не достигается. Оно наблюдается при равномерном перемешивании раствора (вынужденная конвекция)-, при не очень малой скорости перемешивания естественной конвекцией можно пренебречь.
Известно, что вблизи твердого тела наблюдается градиент скорости течения жидкости (обусловленный вязкостью жидкости). Скорость потока равна нулю на поверхности тела, а затем почти линейно увеличивается по мере удаления от поверхности.
Для того чтобы установить закономерности диффузии в стационарных условиях, Нернст (1904 г.) сделал упрощающее предположение о том, что на поверхности твердого тела имеется неподвижный слой (толщиной 5), в котором справедливы только законы диффузии. На расстоянии б от поверхности электрода происходит резкий переход к объему раствора, в котором перенос вещества происходит только при помощи конвекции.
Рис. Б.38. Зависимость концентрации с и скорость потока v от расстояния до поверхности электрода при ламинарном течении жидкости.
Sjjp — толщина пограничного слоя Праидтля; 6Н — толщина диффузионного слоя Нернста, эквивалентная истинному диффузионному слою,
Однако на основе законов гидродинамики было установлено, что в водных растворах толщина пограничного слоя, в котором скорость потока меняется от нуля до максимальной, примерно в 10 раз больше, чем толщина диффузионного слоя, в котором концентрация меняется от максимального значения на поверхности электрода до некоторого среднего значения в объеме раствора. В отличие от представлений Нернста в действительности наблюдается плавный переход концентрации и скорости от нулевого значения на поверхности до максимального в объеме раствора. Можно представить себе эквивалентный реальному слой Нернста толщиной бн, через который к поверхности электрода проходит за единицу времени столько же вещества, сколько и через реальный диффузионный слой (рис. Б.38).
К слою Нернста применимы законы диффузии Фика. На поверхности электрода поток диффузии эквивалентен электрическому току i (условие непрерывности, первый закон Фика):
i nF
/ дс \
D -д-\ дх jx=o
(494)
ч Электрохимическая термодинамика 337 g стационарных условиях вблизи поверхности электрода устанавливается постоянный градиент концентрации (в любой точке объема электролита концентрация постоянна), поэтому, согласно второму закону Фика,
а после интегрирования
дс
~дГ = const
т. е. концентрация меняется линейно, поэтому уравнение (494) можно записать в следующем виде:
i = nFD.-^- (495)
где с — концентрация в объеме раствора, со=концентрация на поверхности электрода. Если со=0, то ток представляет собой предельный ток диффузии id. Это означает, что ток, протекающий через ячейку, не зависит от потенциала
id = (496>
Для ионных растворов, вообще говоря, следовало бы добавить член, учитывающий перенос ионов в электрическом поле (ток миграции). В зависимости от знака заряда иона ток id (или i) увеличивается или уменьшается. Однако, как правило, измерения проводят при значительном избытке постороннего, хорошо проводящего электролита (основного электролита). В этом случае перенос потенциалопределяющих иоиов в результате миграции становится пренебрежимо малым, и можно пользоваться уравнениями для диффузионного тока.
Из уравнения (496) с учетом уравнения (495) следует
• nFD (id—i)b . .
i = td — —g— Со или с0 = —— (49/а)
о
Поскольку c=id—получим для перенапряжения диффузии nr D
»14 = фр — ,ф= (фо+ ^Нпс) —(<Ро + ~^-1ПСо) =
RT , с RT id = <497б>
Уравнения (497а) и (4976) показывают, что предельный ток диффузии пропорционален концентрации потенциалопределяющих ионов в объеме раствора, поток диффузии соответствует электрическому току; для продуктов реакции все меняется: градиент концентрации определяется током с учетом некоторого коэффициента пропорциональности [см. ниже, уравнение (498)].
31.5.2. Основы полярографии
Пропорциональность между предельным током диффузии и концентрацией Деполяризатора используют в полярографии для количественного анализа катионов металлов и других веществ, способных электрохимически восстанавливаться на электроде.
22—2027
Электрохимическая термодинамика
3391
338
Электрохимия
Полярографические измерения проводят в присутствии избытка постороннего (основного) электролита. В качестве катода используют капельный ртутный электрод (время жизни капли ~3 с). Преимущество капельного электрода состоит в том, что его поверхность постоянно обновляется, так что загрязнения, присутствующие в растворе, значительно меньше мешают определению, чем на твердом электроде.
Потенциал капельного ртутного электрода определяется, во-первых, концентрацией разряжающегося осаждаемого катиона на поверхности электрода (с0) и, во-вторых, концентрацией осажденного металла, растворенного в ртутной капле (hrCs). Hgc0 пропорциональна протекающему току В капле ртути также идет конвекция, а следовательно, образуется диффузионный слой с толщиной 6'; если коэффициент диффузии в ртути обозначить D', то
(б' HgCo - nFD, а с учетом (497а)
. RT . СО RT
<P = <Po + ^FIn-^7 = <Po + ^FIn
(498)
, RT , (id — i Ф = Ф1/2 + In —-t---------------
(499)
Последнее уравнение представляет собой уравнение поляризационной кривой (вольт-амперной кривой, или полярографической волны), <Pi/2 называют потенциалом полуволны, так как при i=i<il2 ф=ф1/2. (От использования коэффициентов активности можно отказаться, так как они входят в ф!/2.) Потенциал полуволны тесно связан со стандартным потенциалом редокс-систе-мы, а также однозначно характеризует компоненты раствора, реагирующие на электроде.
Если в системе имеет место только перенапряжение диффузии, то она считается «обратимой» (в полярографическом отношении). Критерии обратимости можно найти в специальной литературе.
Предельный ток диффузии с трудом поддается строгому теоретическому анализу, так как, во-первых, вытекающие из капилляра капли не имеют строго сферической симметрии, во-вторых, устье капилляра экранирует часть капли, вследствие чего плотность тока вблизи устья капилляра отличается от среднего значения, и, в-третьих, вторая капля по сравнению с первой попадает в раствор, уже обедненный разряжающимся веществом.
Капельный ртутный электрод применяется для исследования реакций, протекающих в области потенциалов от +0,3 до —1,8 В (относительно н.к. э). Этот диапазон потенциалов ограничивается при положительных потенциалах растворением ртути, а при отрицательных — электровосстановлением катионов щелочных металлов из основного электролита или выделением газообразного водорода.
Было показано, что на вращающемся дисковом электроде толщина диффузионного слоя б постоянна на всей поверхности диска:
где v — кинематическая вязкость (динамическая вязкость т]> отнесеииая К плотности), п — скорость вращения диска (в оборотах в секунду).
Дисковый электрод наиболее удобен для применения в исследованиях окислительных процессов и при полярографии в окислительных средах; оН может использоваться при более положительных потенциалах па сравнений с ртутным электродом.
Ячейка, в которой проводятся измерения с вращающимся дисковым электродом, должна быть достаточно большой для того, чтобы не мешать движению потоков раствора вблизи электрода. При больших скоростях вращения точность измерений на вращающемся дисковом электроде значительновыше, чем на капельном ртутном электроде. Критерий того, что процесс является чисто диффузионным, заключается в выполнении прямой пропорциональной зависимости между предельным током и корнем квадратным из скорости диска. Дисковый электрод применяется также для выяснения вопроса о лимитирующей стадии электрохимического процесса на твердых электродах, так как из измерений на вращающемся дисковом электроде можно-разделить диффузионную, электрохимическую и другие составляющие перенапряжения процесса, протекающего по сложному смешанному механизму, хотя здесь мы и не рассматриваем такие методы исследования механизма реакции подробно.
Вопросы и задания
Составьте уравнение полярографической волны для редокс-системы для случая вращающегося дискового электрода; концентрация продукта реакции в растворе принимается равной нулю и не входит в выражение градиента концентрации В какой степени входит коэффициент диффузии в выражение потенциала полуволны?
31.5.3. Перенапряжение перехода электрона
Уравнение Нернста справедливо в том случае, если на электроде имеет место равновесие:
Ох + е < ,т-Ь Red (переход электрона) (500)
i_
Здесь Ох обозначает окисленное, a Red — восстановленное вещество; i+ и t_ — токи, соответствующие окислению вещества Red (справа налево) и восстановлению вещества Ох (слева направо). В электрохимии реакции перехода электрона имеет очень большое значение, так как ее скорость определяется потенциалом электрода, т. е. электрическим полем в двойном электрическом слое. В плотной части двойного слоя напряженность электрического поля настолько велика, что под ее действием скорость обычной химической реакции сильно изменяется
Функциональная зависимость между плотностью тока, определяемой стадией перехода электрона, и перенапряжением установлена эмпирически Тафе-лем еще в 1905 г. (где а и b — постоянные):
Т] = а + b In i
Однако то, что именно замедленная стадия перехода электрона характеризуется экспоненциальной зависимостью тока от перенапряжения, было показано теоретически значительно позднее Батлером (1924 г.), Фольмером и Эрдей-Грузом (1930 г.). Появление экспоненциальной зависимости можно представить себе следующим образом. При протекании реакции с замедленным переходом электрона электрический заряд должен преодолеть разность потенциалов между электродом и раствором, на что необходимо затратить определенную энергию. В соответствии с законами химической кинетики такая энергия необходима для достижения переходного состояния (энергия активации). Для электрохимической реакции переходное состояние локализуется в плотной части двойного слоя. Поскольку плотная часть двойного слоя ограничена поверхностью металла и плоскостью, отстоящей .от нее на расстояние радиуса иона, то в одной области плотной части двойного слоя потенциал ускоряет прямую реакцию, а в другой — замедляет обратную реакцию (рис. Б.39).
22*
340
Электрохимия
Рис. Б.39. Влияние изменения потенциала в двойном слое на катодный и анодный процессы.
Изменение потенциала в плотной части двойного слоя Aij>s, а также энергия активации в отсутствие электрического поля не могут быть измерены
Следует отметить, что невозможно измерить разность потенциалов в плотной части двойного слоя Если поддерживать ионную силу раствора, а тем самым падение потенциала в диффузионном слое постоянным, то изменение потенциала электрода оказывается равным изменению потенциала в плотной части двойного слоя. Влияние такого изменения потенциала на скорость реакции уже можно измерить. То, что было верно для недоступного измерению скачка потенциала в двойном слое, оказывается справедливым и для измеренной разности потенциалов: в одной области ускоряется, например, катодная реакция, в то время, как в другой замедляется обратная анодная реакция. Это происходит потому, что изменения разности потенциалов относятся ко всему двойному слою, в то время как переходное состояние локализовано в пределах его плотной части. Ранее полагали, что переходное состояние симметрично относительно начального и конечного положений на координате реакции (a=Vs)- Для анодного тока перехода заряда
<+ = zFA+c*rRed exp ( (501)
Здесь z — число единичных зарядов, обмениваемых в окислительно-восстановительной реакции (в уравнении (501) z=l). В большинстве случаев это соответствует экспериментальным данным. са цеа — концентрация восстановленного вещества на поверхности электрода, Дфз — падение потенциала в плотной части двойного слоя, а — коэффициент перехода (обычно принимает^ ся равным '/s в предположении, что прямая и обратная реакции в одинаковой степени зависят от потенциала). Вообще говоря, 0<и<1; k+— константа, которую можно сравнить с константой скорости гетерогенной химической
Электрохимическая термодинамика
341
реакции (размерность длина/время, например, см/с). Для катодного процесса .имеем
(l-f-a) zFAi|;s
i_ = zF^eOxexp
RT
(502)
Здесь все обозначения те же, что и для уравнения (501). Показатели степени Zr и Zo указывают на порядок электрохимической реакции и имеют такое же значение для установления механизма электрохимической реакции (перехода электрона), как порядок химической реакции для выяснения ее механизма. Как будет показано ниже, в большинстве случаев порядок электрохимической реакции равен единице
Как уже указывалось, измерению доступны только изменения потенциала в плотной части двойного слоя, найденные из измерений потенциала электрода при постоянной ионной силе. Поэтому целесообразно принять в качестве точки отсчета равновесный потенциал системы <рр. Это удобно в связи с тем, что при <р₽ прямая и обратная (сопряженная) реакции протекают с одинаковой скоростью. Ток обмена определяется следующим выражением:
/ анКфр \ / (1— а)г?фр
Ц — 2г«.j. Cq exp I I — zFk - Cy Ox exp 1
(503)
Полагая т] = <р—<pP и учитывая, что измеряемый ток i во внешней цепи представляет собой разность частных токов прямой и обратной реакций, получим
.[ azFr\ IZ-1 = exp
(1 — a) zFrj RT
(504)
Разложим экспоненты уравнения (504) в ряд и при условии r\<^RT[zF ограничимся первым членом разложения. Тогда получим
. . । агРц (1—oQzFt] iqZ/д
1 — lo — RT \uUoj
1 di \ zF _ л
U )n=o = RT l° ( }
Таким образом, из наклона поляризационной кривой при равновесном потенциале можно найти плотность тока обмена io. Наклон не зависит от коэффициента перехода и в соответствии с уравнением (506) определяет поляризационную проводимость и обратную ей величину — сопротивление перехода заряда (поляризационное сопротивление).
При достаточно большом удалении от равновесия (RTfzF) можно Це учитывать обратного процесса, поэтому из уравнения (504) получим
azFt]
In t+ = In ia 4- = *n 1
Это выражение представляет собой преобразованное уравнение Тафеля, хотя сам он объяснял полученную зависимость совсем иначе. Наклон прямой в координатах т]—Ini дает коэффициент перехода а, а экстраполяция на г| = 0 Позволяет получить ток обмена i0 иным методом по сравнению с указанным выше.
Электрохимические системы с высоким током обмена наиболее удобны Аля применения в потенциометрическом анализе. Диапазон концентраций, в котором остается справедливым закон Нернста, расширяется в сторону низ-Ких концентраций в тем большей степени, чем выше плотность тока обмена, отнесенная к некоторой концентрации другого участка реакции (например,
342
Электрохимия
стандартная плотность тока обмена относится к раствору с концентрацией 1 моль/л). Уравнение (503) устанавливает зависимость плотности тока обме-на от концентрации, причем всегда следует принимать во внимание зависимость срр от концентрации потенцналопределяющего вещества. Таким образом оказывается, что, чем выше плотность тока обмена, тем выше полярографий ческая обратимость системы.
Вопросы и задания
1. Выведите из основного уравнения электрохимической кинетики уравнение Нернста для равновесного процесса.
2. Как зависит плотность тока обмена от концентрации потенциалопре-деляющего вещества? (Указание. Прологарифмируйте уравнение (503) и возьмите производную по логарифму концентрации, от которой зависит также и равновесный потенциал. Используйте соотношение ду'дх= (ду!дг}У Х(дг/дх).
3. Ответив на предыдущий вопрос, подумайте, как найти порядок электрохимической реакции относительно вещества, принимающего участие в реакции перехода электрона. Каков порядок реакции по компоненту, не участвующему в реакции?
4. Как определить порядок реакции из плотности тока перехода? Какие экспериментальные трудности при этом возникают?
5. Говорят, что лимитирующая стадия дает наибольший вклад в отклонение от равновесия. Какие величины необходимо измерить для количественного определения таких отклонений? Как провести сравнение этих отклонений для случая смешанной кинетики (перенапряжение диффузии и перехода) ?
В. НЕОРГАНИЧЕСКАЯ ХИМИЯ: СОЕДИНЕНИЯ И РЕАКЦИИ
32. ВАЖНЕЙШИЕ ТИПЫ НЕОРГАНИЧЕСКИХ
СОЕДИНЕНИЙ
32.1. Простые вещества и соединения
При классификации веществ по их составу важнейшая роль отводится понятию элемента. Первая научно обоснованная формулировка этого понятия восходит к английскому исследователю Бойлю. В изданном в 1661 г. сочинении «Химик-скептик» он называет элементами простые вещества, на которые могут быть разложены все «смешанные тела». Лишь столетие спустя многим исследователям удалось, применяя химические, термические и электрохимические методы, выделить важнейшие простые вещества и экспериментально доказать их химическую неразложимость. Лавуазье в 1789 г. в своем выдающемся классическом труде «Начальный курс химии» дал определение химических элементов как веществ, которые не могут быть дальше разложены химическим путем; это определение сыграло большую роль для развития экспериментальной химии.
Существенное углубление понятия «элемент» принесло открытие периодической системы элементов в 1869 г. Менделеевым и Мейером*, что сделало возможным обнаружение многочисленных связей между элементами. Выдающиеся естественнонаучные открытия на пороге XX в. побудили к уточнению понятия «элемент». Так, открытие явления радиоактивности указывало на возможность превращения элементов. Почти одновременно было обнаружено существование изотопов элементов, вследствие чего стали различать атомы различной массы одного и того же элемента. Большое значение имела открытая Мозли (1913 г.) закономерность, согласно которой существует линейная зависимость между квадратным корнем из волнового числа
* Таблица химических элементов была составлена Мейером в 1870 г. (вслед за Менделеевым). По словам самого Мейера, эта таблица «в существенном идентична данной Менделеевым» [Семинин В. И. Периодическая система химических элементов Д. И. Менделеева. — М.: Наука, 1972, с. 40; Кемпбелл Дж. Современная общая химия. Т. 1. — М.: Мир. 1975]. —~Прим. перев.
344
Неорганическая химия
характеристического рентгеновского излучения определенной серии и зарядом ядра элемента (разд. 5.1.2). Это послужило надежной теоретической основой для нового метода идентификации элементов и вместе с тем позволило обосновать понятие «элемент» с позиций ядерной физики. В рамках модели атома Резерфорда-—Бора порядковый номер элемента соответствует заряду ядра (т. е. числу протонов в ядре). Поэтому однозначное и точное определение понятия «элемент» может быть дано только на «атомном» уровне.
Химические элементы — это различные виды атомов, характеризующиеся определенным зарядом ядра.
Это определение элемента абстрагируется от всех других свойств атома, например массы, заряда, характера его связей в рассматриваемом соединении. Так, символ элемента углерода С объединяет все виды атомов с зарядом ядра, равным шести, независимо от того, сколько нейтронов содержится в ядре, ион это или нейтральная частица, находящаяся в решетках графита или алмаза.
Из определения элемента, данного на атомном уровне, следует более раннее определение этого понятия: элементы — это простейшие части химических соединений, комбинирующиеся различным образом, но остающиеся практически неизменными, не считая небольших изменений некоторых «второстепенных свойств (например, заряда частиц). Это позволяет для химических явлений постулировать закон сохранения элементов, используемый при (Символическом описании химических реакций в каждом химическом уравнении количество символов всех элементов с обеих сторон должно быть одинаковым. Этот закон верен, если исключены ядерные,процессы, при протекании которых меняется число протонов в ядре. Происходящее при этом превращение элементов относится к области ядерной химии
При сопоставлении свойств микросоставляющих материи (атомов, молекул, ионов) и свойств макроскопических объектов (газов, жидкостей, твердых тел) заметны существенные различия. Они зависят от пространственного расположения и взаимодействия микросоставляющих между собой. Многие свойства вещества, такие, как температура плавления, давление пара, плотность, электропроводность и др., обусловлены свойствами системы в целом, а для микросоставляющих не имеют смысла. Поэтому следует четко различать свойства микро-и макрообъектов.
В зависимости от числа содержащихся элементов все вещества можно разделить на две группы, а именно
простые вещества, содержащие атомы лишь одного элемента, и соединения, в которых присутствуют атомы или ионы различных элементов.
Простые вещества и соединения
345
Можно предложить следующую классификацию химических систем;
гетерогенные тс те мы
гомогенные системы
(растворы)
гомогенные смеси
индивидуальные вещества.
/ соединения
простые вещества,
Один из тот же элемент может образовывать несколько простых веществ. Так, элемент кислород при нормальных условиях образует два газообразных вещества — кислород и озон, сильно различающиеся по своим физическим и химическим свойствам. Причина различия заключается в неодинаковом числе и неодинаковом способе соединения атомов в молекулах этих веществ. ,Обычный газообразный кислород состоит из двухатомных молекул, тогда как молекулы озона — трехатомные, угловые. Эти же виды молекул присутствуют в жидком и твердом кислороде и озоне соответственно и, таким образом, характерны для данных простых веществ. В других случаях, например в модификациях серы, различные способы соединения атомов могут и не сопровождаться столь сильными отличиями в свойствах.
Простые вещества в зависимости от преимущественного типа связи могут проявлять металлические или неметаллические свойства. Металлические свойства имеют, как правило, вещества, состоящие из электроположительных элементов, а неметаллические— из электроотрицательных элементов. Некоторые элементы, имеющие промежуточные величины электроотрицательности, например As и Sb, в зависимости от условий их получения могут образовывать простые вещества как с металлическими, так и с неметаллическими свойствами. Термодинамическая устойчивость этих модификаций элемента различна.
У соединений многообразие свойств гораздо больше. Комбинация различных элементов приводит к разнообразию состава соединений. При этом оказывается, что связь между атомами имеет полярный характер, причем степень полярности может сильно варьировать. В предельном случае происходит передача электронов одного атома другому, т. е. образование ионов. Поэтому в отличие от простых веществ соединения мо-гУт быть построены из катионов и анионов.
346
Неорганическая химия
32.2. Классификация веществ по строению и типу связей
Сведения о природе частиц, входящих в состав вещества, ц о характере взаимодействия между ними позволяют свести все многообразие веществ к нескольким основным типам. Однако такая классификация соответствует крайним случаям, и веще-ства, полностью отвечающие параметрам данного типа, встречаются редко. Для большинства веществ их принадлежность к определенному типу выражена нечетко, и по своим свойствам они могут занять промежуточное место между основными типами. Несмотря на это, классификация позволяет упорядочить вещества по их строению и объяснить их важнейшие физические и химические свойства. Итак, различают несколько основных типов соединений.
Ионные соединения (гетерополярные соединения, солеобразные вещества) состоят из противоположно заряженных ионов (простых или комплексных). Связь осуществляется преимущественно за счет электростатических сил.
Молекулярные вещества (мономерные ковалентные соединения) построены из молекул, атомы которых связаны более или менее полярными ковалентными связями. Объединение молекул в жидком или твердом состоянии осуществляется за счет межмолекулярных сил.
Полимерные соединения (высокомолекулярные ковалентные, координационные соединения) построены из частиц больших размеров, состоящих из ковалентно связанных между собой мономерных частиц. Иногда осуществляется преимущественное связывание в определенных направлениях. Этот тип соединений в полной мере реализуется в решетке алмаза, в которой все атомы углерода связаны тетраэдрически и образуют бесконечный трехмерный каркас.
Металлы. Строение металлов характеризуется наличием положительно заряженного атомного остова, связанного за счет взаимодействия с делокализованными электронами. Такое строение обусловливает осуществление связей металлического типа.
Следует отметить, что в кристаллической структуре молекулярных веществ могут одновременно реализоваться несколько типов связи. Такие структуры называются гетеродесмическими. В алмазе, так же как в простых солях, преобладает один тип связи. В этих случаях речь идет о гомодесмических структурах.
32.3. Характерные свойства соединений различных типов
32.3.1. Ионные соединения
Ионные соединения образуются чаще всего при взаимодействии элементов с металлическими и неметаллическими свойствами. Вследствие большой разницы электроотрицательностей
Свойства соединений
347
партнеров происходит передача валентных электронов атомов металла атомам неметалла. Образовавшиеся катионы и анионы рассматриваются в рамках ионной модели как несжимаемые (неупругие) шары определенного радиуса. Сила притяжения между противоположно заряженными ионами определяется законом Кулона. В единицах СИ справедливо следующее уравнение:
Р__ ?1'?2
4ле (ту + г2)2
где F— сила притяжения (в ньютонах), qlt q2— заряды ионов (в кулонах), Г1, Г2 — ионные радиусы катиона и аниона (в метрах), е — диэлектрическая проницаемость среды (Кл-В-1-м-1)*.
Электростатические силы тем больше, чем выше заряд ионов и чем меньше ионные радиусы. Поле кулоновских сил имеет сферическую симметрию, что приводит к ненаправленно-сти ионной связи. Можно выделить три особенности строения ионных соединений.
1) Каждый ион стремится окружить себя как можно большим числом ионов противоположного знака. Вследствие этого следует ожидать (и это действительно имеет место) проявления весьма высоких координационных чисел. При координации одного иона не происходит насыщения связи. Поэтому в идеальном случае должна образоваться бесконечная трехмерная решетка, построенная по способу плотнейшей шаровой упаковки (разд. 1.5).
2) Отталкивание одноименно заряженных ионов обусловливает расположение их на возможно большем расстоянии друг от друга. Это приводит к осуществлению наиболее симметричного распределения ионов вокруг центрального иона. Поэтому в решетках ионных солей чаще всего встречается высокая локальная симметрия окружения ионов.
3) Для всей системы должен выполняться закон электронейтральности: общее число положительных зарядов в системе должно быть равно общему числу отрицательных зарядов. Поэтому справедливо следующее равенство: число катионовХза-ряд катиона = число анионовХзаряд аниона. Это соотношение определяет стехиометрический состав ионного соединения:
Общая формула Примеры соединений
м+х-М2+Х2-М2+Х2-М2+Х2-м2з+х32- КС1, NaOH, NH4CIO4 СаО, MgSO4 CaF2, Pb(NO3)2 Li2O, К2СгО4 A12O3, La2(C2O4)3
* Диэлектрическая проницаемость е равна произведению относительной Диэлектрической проницаемости вещества ег (безразмерная величина) на Диэлектрическую проницаемость вакуума ео = 8,854-10~12 [Кл.В-1-м-1].
348
Неорганическая химия
Совершенно очевидно, что для ионных соединений справед. лив основной закон стехиометрии: атомы различных элементов соединяются друг с другом в соотношении простых целых чисел.
Для структуры соли определяющим является не столько тип формулы, сколько координационные числа катиона и аниона и соотношение их ионных радиусов (разд. 6.4.3). В структуре хлорида цезия каждый ион Cs+ окружен восемью ионами Cl-соответственно каждый ион С1~ — восемью ионами Cs+. В структуре хлорида натрия координационные числа катиона и аниона равны шести. В структуре фторида кальция вокруг иона Са2+ расположено восемь ионов F-; по принципу электронейтральности координационное число иона F~ должно быть равно четырем. Координационные числа катиона и аниона можно указывать при написании формулы соединения (по Ниг-гли), например для хлорида цезия CsCl8/8, для хлорида натрия! NaCl6/6, для хлорида кальция CaF8/4. Электростатическая модель объясняет в первом приближении ряд физических свойств, ионных соединений — твердость, температуры плавления и кипения.
Кристаллы ионных солей обычно довольно твердые, поскольку при их разрушении разрывается значительное число связей со сравнительно высокой прочностью, обусловленной электростатическим взаимодействием между ионами. При плавлении надо затратить значительную энергию для того, чтобы сделать подвижными ионы, занимающие в твердом теле строго фиксированные положения. То же справедливо и для процесса испарения. Вследствие этого ионные соединения имеют сравнительно высокие температуры плавления и кипения. При испарении из расплава в газовую фазу переходят небольшие осколки, которые следует рассматривать как мономерные или полимерные ионные пары. Например, в парах LiF при 1100°C присутствуют молекулы LiF, IJ2F2 и Ы3Рз в соотношении 1:0,73: : 0,32. При еще более высоких температурах происходит диссоциация на атомы.
Обычно температуры плавления и кипения растут (в соответствии с законом Кулона) при увеличении заряда ионов и уменьшении расстояния между ними. Исключения из этого правила, часто наблюдающиеся у ионных соединений с большими размерами ионов, простых или комплексных, могут быть объяснены увеличением ковалентной составляющей связи из-за поляризационных эффектов.
Еще одной особенностью ионных кристаллов является их малая деформируемость, т. е. высокая хрупкость. Это свойство наглядно демонстрируется на рис. В.1. Смещение отдельных слоев ионов относительно друг друга требует приложения значительных сил, так как при этом происходит смещение многих
Свойства соединений
349
атомов из положений, соответствующих минимальной потенциальной энергии.
Мерой силы электростатического взаимодействия ионов в кристалле служит энергия решетки (разд. 6.4.2), которая растет с увеличением заряда ионов и с уменьшением расстояния между ними (суммы ионных радиусов). В табл. В.1 на примере галогенидов щелочных металлов показана взаимосвязь энергии решетки, механических и термических свойств веществ.
Ионные соединения обладают также характерными электрическими свойствами. В твердом состоянии ионные кристаллы, как правило, являются диэлектриками. Небольшая ионная проводимость может наблюдаться в ионных кристаллах с дефекта-
е©е©е ©0©е© о©е©е ©е©е©
е©е©е ©е©е©
Рис. В.1. Действие деформирующей силы на слои решетки ионного кристалла.
ми (разд. 33.9.1). В расплавах солей электропроводность скачкообразно возрастает. Это свойство весьма типично для ионных соединений.
Таблица В.1. Некоторые характеристики ионных соединений и их механические и термические свойства (на примере галогенидов щелочных металлов)
Соединение Сумма ионных радиусов, нм Энтальпия решетки, кДж/моль Твердость по Бринеллю, Н/мм2 К ^"кнп’ К
NaF 0,229 —909 1265 1968
NaCl 0,276 —767 122 1073 1714
NaBr 0,290 —737 90 1020 1666
Nal 0,314 -687 82 935 1573
KF 0,266 —808 — ИЗО 1778
KC1 0,314 —703 57 1043 1690
КВг 0,329 —674 53 1015 1654
Ki 0,352 —632 31 955 1604
32.3.2. Молекулярные вещества
Представление о молекулярных веществах может относиться к любому из трех агрегатных состояний, хотя использование Понятия молекулы для твердых веществ встречает некоторые 3атРУДнения.
В противоположность силам кулоновского взаимодействия Ковалентная связь имеет свойства насыщаемости. Обычно это-
Свойства соединений
351
350
Неорганическая химия
приводит к соединению ограниченного числа атомов с образованием молекулы. Кроме того, для ковалентной связи характерна ее направленность, так что связи между атомами в молекулах расположены под определенными углами.
В противоположность прочным ковалентным связям между атомами в молекуле вандерваальсовы силы, действующие между молекулами, относительно слабые (разд. 6.3.7). В зависимости от полярности молекул можно подразделить молекулярные вещества на неполярные и полярные.
•32.3.2.1. Неполярные ковалентные соединения
К ним относятся прежде всего простые вещества, образованные неметаллами. Ввиду того что друг с другом соединены атомы одного и того же элемента, не может возникнуть несимметричное распределение заряда между атомами. Примерами могут служить двухатомные газы — водород, кислород, азот, фтор и хлор. В газообразных благородных газах присутствуют сво-
бодные атомы, конденсирующиеся при низких температурах под действием вандерваальсовых сил в бесконечные атомные агрегаты. Другие простые вещества, например бром, иод, фосфор и сера, представляют собой при нормальных условиях жщдкие или твердые вещества. В молекулярных решетках конденсированных фаз присутствие молекул в качестве структурных единиц может быть однозначно доказано путем рентгеноструктур-ного анализа. При этом оказывается, что внутримолекулярные расстояния значительно короче, чем межмолекулярные. В струк" туре кристаллического иода, которая, как и многие другие моле
кулярные решетки, является слоистой, межатомное расстояние в молекуле 12 составляет 0,268 нм, тогда как расстояние между атомами иода соседних молекул 12 в слое равно 0,354 нм (рис. В.2). При сублимации молекулы иода за счет энергии тепловых колебаний отщепляются от кристаллического остова и переходят в газовую фазу.
Структурными единицами неполярных ковалентных веществ могут быть также неполярные молекулы, состоящие из атомов различных элементов. Диполи отдельных связей в таких молекулах полностью компенсируют друг друга благодаря симметричному расположению связей. Примерами подобных веществ могут служить СС14, СО2, SiF4, SFe, WC16, Ст (СОД, HgCl2, а также многие симметрично построенные алифатические углеводороды.
Свойства неполярных молекулярных веществ в полной мере соответствуют их строению. Это в большинстве случаев легколетучие или сублимирующие вещества с низкими температурами плавления и кипения. В твердом состоянии такие вещества построены из молекул, слои которых под действием механических сил легко сдвигаются друг относительно друга. Эти вещества— типичные диэлектрики. Жидкие фазы представляют собой растворители с низкой диэлектрической проницаемостью.
32.3.2.2. Полярные ковалентные соединения
Молекулы этих веществ полярны. Наряду со всегда действующими вандерваальсовыми силами отдельные молекулы в кристалле или в жидкой фазе оказывают друг на друга более сильное электростатическое влияние, обусловленное взаимодействием диполей. Наличие постоянного и индуцированного дипольного момента приводит к взаимной ориентации молекул в твердом или жидком состоянии (рис. В.З). За счет дипольного взаимодействия межмолекулярные силы, а следовательно, и энергии решетки увеличиваются. Это является при
чиной того, что температуры плавления и кипения полярных Молекулярных веществ, как правило, выше, чем сравнимых с Ними по молекулярной массе неполярных соединений. В случаепростых молекул можно, используя значения дипольного момен-Та g и поляризуемости а, рассчитать обусловленную межмоле-кулярным взаимодействием энергию решетки. Соответствующие Данные для некоторых неполярных и полярных молекулярных неществ приведены в табл. В. 2.
Рис. В.З. Ориентация дипольных молекул в твердом теле и в жидкости.
352
Неорганическая химия
Таблица В.2. Связь между свойствами молекул, межмолекулярными силами и температурами плавления и кипения неполярных и полярных молекулярных веществ
Соединение Молекулярная масса Дипольный момент 1030, Кл м Полярнзуе мость 1030, ма Энтальпия решетки, кДж/моль т пл’ К т КИП’ К
n2 28,02 0,0 1,76 7,79 63 77
о2 32,00 0,0 1,60 8,63 54 90
сн4 16,04 0,0 2,60 11,31 90 112
со 28,01 0,40 1,99 8,75 69 81
НС1 36,47 3,45 2,63 21,16 159 188
NH3 17,03 4,88 2,21 29,60 196 240
32.3 2 3. Соединения с водородной связью
Дополнительные силы притяжения между молекулами полярных ковалентных веществ могут возникать за счет образования так называемой мостиковой водородной связи. При этом через атом водорода происходит связывание двух атомов с высокой электроотрицательностью, таких, как, например, F, О и N. Связь атома водорода этого водородного «мостика» с одним из двух атомов (которые могут быть одинаковыми или различными) обычно прочнее. Схематически это обозначается следующим образом; X—Н--Х. В некоторых случаях, например для гидродифторида калия KHF2, доказана симметричность водородных связей в водородных мостиках.
Следует различать внутри- и межмолекулярные водородные мостиковые связи. Последние осуществляются значительно чаще, чем первые. Энергия межмолекулярной водородной связи составляет в среднем 25 кДж/моль. Ее наличие приводит к более или менее выраженной ассоциации молекул, возможной в любом из трех агрегатных состояний. Особенно часто происходит ассоциация молекул, содержащих ОН-группы, например Н2О, Н2О2, спирты, фенолы, кислородсодержащие неорганические и органические кислоты (HNO3, H2SO4, Н3РО4 и НСООН) Ассоциация имеет место в структурах некоторых кислых солей, например у NaHCO3 и КН2РО4, а также в жидких фтороводороде HF, аммиаке NH3 и циановодороде HCN. В результате ассоциации могут образовываться димеры, например, уксусной кислоты, линейные или цепочечные полимерные частицы. Установлено, что в газообразном HF существует несколько видов молекул (HF)n с п= 1 ч-6. Среди них молекула (HF)6 особенно устойчива, по-видимому, благодаря образованию дополнительной водородной связи при замыкании кольца. В структурах жидкой воды и льда имеется трехмерный каркас из молекул воды, связанных водородными мостиками (рис. В.5).
Свойства соединений
353
Внутримолекулярные водородные связи соединяют электроотрицательные атомы одной молекулы. Эти связи часто образуются в хелатных комплексах, как, например, в бис(диметил-глиоксимато) никеле (II):
h3c-c/N'
Н3С-С,
о н ...о
I I
он "С— Ctij
,с—сн.
I I 0....H-..-0
Расстояние между атомами кислорода укорочено за счет образования симметричных мостиков О---Н---О, причем благодаря скр2-гибридизации орбиталей атома никеля комплекс имеет плоское строение.
“2ГО
3 4
Номер переда,
Рис В.4. Влияние образования водородных мостиковых связей на температуры кипения бинарных гидридов элементов главных подгрупп четвертой — седьмой групп.
Наличие водородных связей можно обнаружить с помощью расчетных методов. Однозначные выводы позволяют сделать инфракрасные спектры поглощения. Довольно узкая полоса при 3600—3700 см-1 соответствует валентному колебанию свободной гидроксильной группы. Если же атом водорода выступает в качестве мостикового, появляется широкая полоса в области 23—2027
354
Неорганическая химия
больших длин, причем величина смещения может служить мерой прочности водородной связи.
Методом ядерного магнитного резонанса на протонах (ПМР) можно обнаружить водородную связь по изменению химического сдвига резонансных линий.
Наличие водородных связей в веществе вызывает появление ряда характерных свойств. Прежде всего ассоциация молекул ведет к повышению температур плавления и кипения по сравне нию со значениями, ожидаемыми из хода соответствующих величин для однотипных соединений. Это видно из сравнения тем ператур кипения бинарных гидридов элементов главных подгрупп IV—VII групп (рис. В.4). Ход зависимости температур кипения гидридов элементов четвертой группы показывает, что у них водородных связей не образуется.
Увеличение полярности агрегатов молекул проявляется также в повышении диэлектрической проницаемости жидкостей. Так, для воды при 25 °C ег=78,5, а для серной кислоты ег = 100 (другие примеры см. в табл. В.17). Свойства жидкостей как растворителей и сольватирующих агентов также в значительной мере определяются способностью их молекул выступать донором или акцептором при образовании водородных связей (разд. 34.2.1).
Способность к образованию водородных мостиков у воды выражена осо бенно сильно благодаря тому, что в каждой молекуле имеются два атома
водорода и две неподеленные элект ронные пары, которые могут был акцепторами атомов водорода. Эт< приводит к тому, что в кристаллической структуре льда каждый ато» кислорода окружен четырьмя атома ми водорода: двумя связанными ковалентно и двумя мостиковыми В результате образуется тетраэдрическая структура с необычно рыхлой упаковкой частиц (рис. В.5). При плавлении льда часть водородиы? связей разрушается, и в жидкости находятся в динамическом равновесии молекулярные ассоциаты, содер жащие до 100 молекул воды, представляющие собой осколки структу ры льда. Отдельные молекулы воды могут путем быстрого обмена (~10-н с) присоединяться к ассоциату или отщепляться от него В этих кластерах имеются и иетет-
Рис. В.5. Тетраэдрическая структура раэдрические (изогнутые) водород-льда, ные мостики, в которых атомы во-
дорода несколько смещены в сторону от линии, соединяющей атомы кислорода. Таким образом упаковка молекул воды становится более плотной что проявляется в аномальном увеличении плотности при плавлении льда. При нагревании воды от 0 до 4 °C происходит дальнейшее разрушение тетраэдри
Свойства соединений
355
ческой структуры, так что плотность воды увеличивается. Лишь при температуре выше 4 °C этот эффект перекрывается обычным термическим расширением благодаря усилению теплового движения молекул. Выше 4 °C плотность воды с ростом температуры убывает.
Кластерной структурой объясняются и другие аномалии воды — высокие значения теплоты испарения, поверхностного натяжения и удельной теплоемкости, так же как влияние на’ свойства воды растворенных в ней веществ.
Для объяснения строения жидкостей часто используются кривые радиального распределения электронной плотности, получаемые методом рентгеноструктурного анализа. Эти кривые дают зависимость вероятности нахождения окружающих частиц в элементе объема в зависимости от расстояния от центральной частицы. По положению максимумов на кривой можно судить о среднем расстоянии до различных координационных сфер, а по площади под кривой — о числе частиц в данной координационной
сфере. На рис. В.6 приведена кривая радиального распределения для воды при 1,5 °C. Вследствие незначительной рассеивающей способности атомов водорода рентегеновские методы обычно дают сведения лишь о положении атомов кислорода. Максимум на кривой при 0,29—0,30 нм отвечает среднему расстоянию до первой гидратной оболочки. Площадь под этим максимумом соответствует наличию в этой гидратной оболочке от четырех до пяти молекул воды. При расстояниях 0,5 нм и 0,7 обнаруживаются другие максимумы, соответствующие более удаленным гидратным оболочкам. В последнее время для исследования структуры воды успешно использовались методы микроволновой спектроскопии н спектроскопии ЯМР.
Рис. В 6. Кривая радиального распределения для воды при 1,5 °C.
Тенденция к образованию водородных связей обусловливает также строение гидратированных протона и гидрооксид-иона. Основываясь на результатах многих исследований, можно считать, что в воде протон присутствует как Н9О4+, а гидроксидион— как Н7О4-. Строение этих частиц схематически показано на рис. В.7. Поэтому часто используемые схемы для процесса диассоциации воды
Н2О Н+ + ОН~ или Н2О + Н2О я—>: Н3О+4- ОГГ
могут рассматриваться лишь как чисто символические.
Ион Н9О4+ имеет пирамидальное строение. Центральный атом кислорода связан тремя сильными водородными связями с тремя молекулами воды. Прочность водородных связей может быть количественно охарактеризована высокой энтальпией реакции гидратации протона (Д№ =—1185 кДж/моль). По данным ИК-спектроскопических исследований в кристаллическом тетрагидрате бромоводорода присутствуют ионы Н9О4+ и Вг_. 23*
356
Неорганическая химия
При экстракции сильных кислот из их водных растворов с помощью основных растворителей, например экстракция в три-я-бутилфосфат, Н9О4+-ИОНЫ вместе с анионами кислоты переходят в органическую фазу. В ионизованном водяном паре ион Н9О4+ также был обнаружен масс-спектрометрически. Анало
Рис. В.7. Строение ионов Н9О4+ и Н7О4-.
гичный характер гидратации протона (так же как гидроксид иона в виде Н?О4_) был установлен и в водных растворах при исследовании их различными физико-химическими методами.
32.3.3. Полимерные соединения
Соединения с водородной связью по своему строению занимают промежуточное положение между низкомолекулярными i полимерными соединениями. Представление о полимерных со единениях в дальнейшем используется лишь для соединений । ковалентным типом связи, хотя кристалл соли или решетку ме талла можно было бы рассматривать тоже как полимерное образование. Кроме того, термин «полимерные» не отражает механизма образования соединения. Ведь для описания типичных свойств таких соединений совершенно несущественно, происходило ли его образование из мономерных единиц путем конденсации или полимеризации (разд. 33.7).
В полимерных соединениях атомы могут соединяться кова лентными связями с образованием пространственной решетк. (координационные структуры), сеток (слоистые структуры) или цепей (волокнистые структуры).
32.3.3.1. Координационные структуры
К типичным представителям этой группы относятся структуры алмаза и цинковой обманки (рис. В.8). Обе структуры отличаются лишь тем, что в одной из них все позиции заняты атомами одного сорта, а в другой — атомами двух сортов. Для этих структур характерна тетраэдрическая координация всех атомов, что приводит к высокосимметричной и довольно плот-
Свойства соединений
357
Рис. В.8. Структура цинковой обманки.
ной упаковке. По такому структурному типу кристаллизуются также карбид кремния, кремний, германий и серое олово. Прочность связей в структурах может сильно различаться. В алмазе неполярные ковалентные связи очень прочны. В других веществах заметными становятся металлические свойства, проявляющиеся в увеличении электропроводности. В структурном типе цинковой обманки кристаллизуются многие полупроводниковые соединения.
Трехмерный каркас имеется также в различных модификациях диоксида кремния SiOa — кварце, тридимите, кристобалите. Во всех этих структурах каждый атом кремния соединяется с четырьмя атомами кислорода, а атом кислорода — с двумя атомами кремния, причем ковалентные связи имеют полярный характер. Таким образом, структурная формула вещества — SiOi/2.
Наличие трехмерного каркаса в структуре соединения с ковалентными связями легко объясняет их важнейшие свойства: высокую твердость, очень высокие температуры
плавления и кипения, нерастворимость как в неполярных, так и в полярных растворителях. Растворить такое вещество можно лишь путем проведения химической реакции, т. е. при далеко идущих разрушении и перестройке связей между атомами.
32.3.3.2. Слоистые структуры
Вторая важная модификация углерода — графит. Здесь реализуется типичная слоистая структура (рис. В.9). В каждом слое плоские шестиугольники из атомов углерода объединены в плоские сетки, напоминающие соты. В а-графите атомы третьего слоя находятся точно над атомами первого слоя, так Что последовательность чередования слоев может быть обозначена как АВАВ. 10-Графит имеет другое чередование слоев. Расстояние между атомами углерода в слое составляет 0,141 нм, расстояние между слоями значительно больше (0,335 нм). Это указывает на существование лишь слабой связи Между слоями, тогда как в плоскости в пределах слоя наблюдается даже некоторое укорочение расстояния углерод-углерод До сравнению с длиной однократной С—С-связи (0,154 нм), Происходящее за счет дополнительного л-связывания.
Итак, расположение атомов углерода в графите характеризуется менее плотной упаковкой, чем в алмазе. Слабое связыва-
358
Неорганическая химия
Свойства соединений
359
ние между соседними слоями объясняет хорошую спайность параллельно плоскости слоев и мягкость вещества. Различия в состоянии электронов в обеих модификациях углерода приводит к появлению сильных различий в электрических и оптических свойствах.
Слоистую структуру имеют также солеобразные соединения типа АВ2 с большими легкополяризуемыми анионами, например
Cdl2, РЫ2- В их структурах можно выделить слоевые пакеты, состоящие из одного слоя катионов и двух слоев анионов. Последовательность слоев можно представить как BAB ВАВ. Расстояния между ближайшими слоями анионов из соседних пакетов довольно велико, что приводит к появлению хорошей спайности по этим плоскостям. Напротив, внутри слоевого пакета рас стояния ВАВ благодаря эффектам поляризации несколько укорочены по сравнению с сум-
мой ионных радиусов. Значение валентного угла Cd—I—Cd ~90° указывает на перекрывание р-орбиталей аниона с орби талями катиона.
Со слоистой структурой некоторых силикатных минералов связано наличие у них ряда весьма ценных свойств. Так, способность глин к набуханию и их пластичность обусловлены тем, что между двойными слоями кремнекислородных колец могут внедряться молекулы воды. В слюдах отдельные слои связываются через катионы. Способность ионов К+ вытеснять из слюд другие катионы (Na+, Са2+) приводит к накоплению калия, который может извлекаться растениями и использоваться при п\ росте, что немаловажно для сельского хозяйства.
в
Рис. В 9. Структура графита.
32.3.3.3. Цепочечные или волокнистые структуры
Соединения этого типа построены из длинных цепочек молекул, связанных между собой вандерваальсовыми силами Эти соединения по своим свойствам в значительной степени приближаются к молекулярным веществам. К этой группе соедп нений относятся, например, изополикислоты цепочечного строения и их анионы, образующиеся при конденсации мономерньл кислот. Сходное строение имеют также асбестовидная модификация триоксида серы и шелковистые волокна сульфида крем ния (неорганическое волокно). В SiS2 цепи состоят из тетраэдров
SiSi, соединенных друг с другом через ребра:
Напротив, тетраэдры SiO4 в полисиликатах связаны через вершины:
ООО
I I I
...О—Si—О—Si—О—Si—О—
ООО
Высокомолекулярные цепи содержатся в структурах многих органических природных или синтетических веществ, которые часто обладают весьма ценными свойствами (например, в целлюлозе, белках, каучуке, полиэтилене и перлоне). Параллельно расположенные или скрученные цепи образуют гибкую нить. Эластичность резиноподобных веществ обусловлена нерегулярным сшиванием макромолекул между собой.
32.3.4. Металлы
32.3.4.1. Общие свойства
Металлы можно разделить на:
1) простые вещества,
2) интерметаллические соединения (фазы),
3) металлические сплавы.
Все металлические вещества образуются из элементов, атомы которых характеризуются небольшим значением электроотрицательности и вследствие этого легко образуют положительные ионы. Кроме того, число валентных электронов у атомов этих элементов невелико, что обусловливает многие особенности металлического состояния. Так, оказываются не полностью заселенными электронами многие энергетические состояния верхней энергетической зоны, что приводит к типичной электронной ненасыщенности металлических веществ. В противоположность этому энергетические зоны неметаллических веществ полностью заняты.
Большинство простых веществ является типичными металлами. У ряда элементов металлическими свойствами обладают лишь некоторые их модификации. К металлам относятся элементы главных подгрупп первых четырех групп периодической системы, все элементы с внешними d- и f-оболочками электронов. Несколько модификаций, как с металлическими, так и с неметаллическими свойствами, образуют, например, С, Р, As, Sb, Se. Устойчивость отдельных модификаций сильно зависит °т внешних условий. В последнее время подробно исследовано 8лияние давления на фазовые превращения. Установлены общие
360
Неорганическая химия
Свойства соединений
361
закономерности, справедливые для любого вещества, согласно которым при повышении давления соединение с гетерополярны-ми связями сначала превращается в другую модификацию с ковалентными связями, а при дальнейшем повышении давления вещество переходит в металлическое состояние. По оценкам, такое превращение для водорода должно происходить при давлении 1800 ГПа.
Характерные свойства металлических веществ также можно предсказать путем рассмотрения типа связей и структуры.
Электропроводность служит важнейшей физической характеристикой металлического состояния. Металлы принадлежат к проводникам 1-го рода, в которых электропроводность осуществляется электронами. У проводников 2-го рода, например расплавов солей или растворов электролитов, ионный механизм проводимости.
Металлическая проводимость возникает при наличии частично занятых электронами энергетических зон, в пределах которых электроны обладают высокой подвижностью. В непроводящих веществах (изоляторах) имеются полностью заполненные энергетические зоны, отделенные от свободных энергетических зон широкой запрещенной зоной (рис. А.26). У полупроводников ширина запрещенной зоны мала, так что уже при подводе тепловой энергии электроны могут переходить в более высоколежащие зоны. Поэтому в противоположность веществам с металлической проводимостью у полупроводников повышение температуры вызывает увеличение электропроводности. Тот же эффект может наблюдаться при воздействии световой энергии. Это объясняет фотопроводимость у селена.
С наличием металлической проводимости тесно связаны высокая теплопроводность и оптические свойства металлических веществ. Так, электроны могут вследствие их высокой подвижности осуществлять отвод тепла путем переноса энергии из областей с более высокой температурой в области с более низкой температурой. Высокие коэффициенты поглощения и отражения излучения у металлов объясняются наличием в энергетических зонах очень тесно расположенных чередующихся занятых и свободных состояний. Этим обусловлены металлический блеск и непрозрачность. В тонкодисперсном состоянии все метал ты имеют черный цвет.
Другие важные свойства металлов связаны с особенностями их структуры. В трех главных структурных типах металлов реализуются весьма высокие координационные числа (разд. 1.5)-В кристаллических решетках кубической и гексагональной плотнейших упаковок каждый атом металла окружен 12 ближайшими соседями. В то же время в кубической объемно-центрированной решетке число ближайших соседей равно восьми, н° можно считать дополнительно координированными еще шесть
атомов, расположенных по вершинам октаэдра на несколько большем расстоянии, так что общее число атомов ближайшего окружения равно 14. Уже из этих высоких координационных чисел можно заключить, что при наличии лишь небольшого числа валентных электронов атомы металла должны быть соединены многими делокализованными связями.
Отдавая свои валентные электроны, атомы металла в значительной мере теряют свою химическую индивидуальность. По-
Рис. В. 10. Действие деформирующей силы на слои решетки металла.
этому атомы или ионы других элементов, особенно если они имеют те же размер и заряд, могут легко замещать атомы основного элемента. Это дает возможность получения разнообразных сплавов, часто обладающих исключительно ценными свойствами. С этим же связаны, однако, и трудности очистки металлов от металлических примесей.
Из-за равноценности всех связей в кристаллической решетке металлы могут сравнительно легко деформироваться под действием механической обработки. Особенно мягки чистые металлы, в которых слои атомов могут беспрепятственно смещаться друг относительно друга (рис. В.10). При внедрении в пустоты структуры металла других небольших по размеру атомов твердость материалов может существенно возрасти. Эта особенность находит широкое применение при промышленном производстве различных сплавов.
32.3.4.2. Структура и связь в интерметаллических соединениях и фазах
Взаимодействие металлов между собой может приводить к различным продуктам в зависимости от природы металлов.
Если оба металла бинарной системы схожи как по размерам их атомов, так и по их электронной конфигурации (например, серебро и золото), в решетке металла возможно полное взаимное замещение атомов разных сортов без изменения структуры. При этом образуются твердые растворы замещения во всей области составов (рис. В.11,б). В подобных системах и в жидком состоянии наблюдается полная смешиваемость во всех отношениях.
Если размеры и электронные структуры атомов металлов Различны, образуются интерметаллические фазы, структуры кобрах отличаются от структур исходных металлов. Например,
362
Неорганическая химия
Свойства соединений 363
в системе Си—Zn устойчивы фазы CuZn, CusZns, CuZn3, структуры которых (кубическая объемно-центрированная, сложная кубическая, гексагональная плотнейшая упаковка соответственно) определяются правилом Юма — Розери в зависимости от величины отношения общего числа валентных электронов к числу атомов (разд. 6.6.4).
• • • О О • ООО*«о 0*0*00 • о • о о • • о • • о о • • О • • о 6
• о • о • о о • о • о • • о • о • о о • о • о • • о • о • о о • о • о •
б
Рис. В.11. Строение интерметаллических фаз.
а — решетка чистого металла, б — твердый раствор замещения, в — свсрхструктура, г — твердый раствор внедрения
Обычно интерметаллические фазы не имеют постоянного стехиометрического состава. Их состав может меняться в определенных пределах без изменения структуры. Иногда удается наблюдать переход из состояния со статистическим распределением в упорядоченное состояние. Так, в кубической объемно-центрированной p-фазе CuZn атомы Си и Zn статистически распределены по позициям; при отжиге этой фазы при низких температурах возникает сверхструктура типа CsCl (рис. В.11,в).
Если при протекании реакции в решетку металла внедряются атомы других элементов, имеющие небольшие размеры, происходит образований твердых растворов внедрения, сопровождающееся лишь незначительными изменениями исходной структуры (рис. В.11,г). Особенно часто такие фазы образуют d-элементы IV, V и VI групп, атомы которых достаточно велики, чтобы в октаэдрических или тетраэдрических пустотах решетки металла могли поместиться атомы меньших размеров, например углерода или азота. По типу твердых растворов внедрения построены карбиды (ZrC, ТаС, W2C) и нитриды (ZrN, Nb3N, U2N3), которые получаются при нагревании порошкообразных металлов в атмосфере паров углеводородов, N2 или NH3. Эти фазы также не являются дальтонидами. Например, в фазе УгСо,74-1,о атомы углерода могут занимать 3/8—‘/2 всех октаэдрических пустот; при большем содержании углерода образу* ется новая фаза. Хотя в этих фазах присутствуют атомы неметаллов, металлический тип связи сохраняется. Подобные соединения обладают металлической электропроводностью, отличаются чрезвычайно высокой твердостью и инертностью. Из всех
известных веществ они имеют самые высокие температуры плавления. Например, температура плавления соединения состава Ta4ZrCs составляет 4215 К.
Для ряда интерметаллических соединений структура определяется в первую очередь геометрическими факторами. Особенно наглядно это проявляется для фаз Лавеса, которые имеют постоянный стехиометрический состав АВ2 и характеризуются примерно постоянным отношением атомных радиусов элементов А и В (гА : 1,225). Структуру фаз Лавеса можно
представить как вставленные одна в другую решетки двух металлов с весьма плотной упаковкой координационных полиэдров. Примеры фаз Лавеса — MgZn2, MgCu2, MgCa2, СаА12, NaAu2.
Некоторые интерметаллические соединения, связь в которых заметно ковалентна, обладают полупроводниковыми свойствами. Наиболее важны соединения типа AinBv (например, GaAs, InSb), которые изоэлектронны с кремнием или германием. Эти соединения кристаллизуются, как правило, в структурном типе цинковой обманки.
При взаимодействии металлов, еще более отличающихся по электроотрицательности, обычно образуются соединения определенного стехиометрического состава с гетерополярным характером связей, например Li4Pb, Li3Bi, Mg2Pb, Mg3Bi. Реакции образования этих соединений из простых веществ сопровождаются заметным выделением тепла; происходит частичный переход электрона к более электроотрицательному элементу и образование анионной подрешетки. Такому характеру связи в полной мере соответствуют плохая электропроводность, относительно высокие температуры плавления, хрупкость, свидетельствующие о переходе от металлических веществ к ионным соединениям.
Интерметаллические фазы и соединения с частично полярным типом связи известны как цинтл-фазы. Это прежде всего соединения, впервые исследованные Цинтлом, типа NaTl (Naln, LiGa и Liln). По данным рентгеноструктурного анализа, в этих соединениях радиус атомов менее активного металла имеет практически постоянную величину, в то время как радиус атома более активного металла значительно уменьшен. Атомы менее активного металла образуют основную решетку, в пустотах которой находятся атомы более активного металла.
Существование полиатомных анионов металлов доказано в соединениях типа Na4Pb7, Na4Pb9, Na3As7, синтезированных при проведении реакции в жидком аммиаке. Анионы [РЬ7]4- и lPb9]4— можно представить себе как [РЬ4~-РЬ6] и [РЬ4“-РЬ8] соответственно, т. е. с октаэдрическим или кубическим окружением малоустойчивого иона РЬ4~ атомами свинца, что ведет к стабилизации полиатомного аниона.
364
Неорганическая химия
33. ВАЖНЕЙШИЕ ТИПЫ РЕАКЦИЙ В НЕОРГАНИЧЕСКОЙ
ХИМИИ
33.1. Химические уравнения
При протекании химической реакции происходит переход исходной системы в новую, в которой связи и структура претерпевают существенные изменения по сравнению с исходной. При этом сначала возбуждаются или расщепляются определенные связи в исходных веществах. Часто затем происходит постадий-ное образование новых групп ,частиц и структур. С-точки зрения термодинамики каждая химическая реакция сопровождается выравниванием химических потенциалов компонентов системы (разд. 23.1).
При схематическом изображении химических реакций, т. е. при написании уравнений реакций, следует соблюдать общие правила.
1) В левой части уравнения реакции пишутся формулы исходных веществ, а в правой части — продуктов реакции. Обе части уравнения соединяются знаком равенства или стрелкой, направленной в сторону конечных веществ. Химическое равновесие между исходными и конечными продуктами обозначается с помощью двух противоположно направленных стрелок. При этом подразумевается, что все вещества находятся в равновес-.ных концентрациях. Если в результате прохождения реакции из раствора выпадает осадок или образуется газообразное вещество, это обозначается с помощью стрелок, направленных вниз .и вверх соответственно, например
СО2 + С ч=ь 2СО
NaCl + AgNO3 -► AgClj + NaNO3
CaCO3 --► CaO + CO2f
2) В обеих частях (левой и правой) уравнения химической реакции должно находиться одинаковое число символов каждого элемента (закон сохранения элементов). Для этого перед формулами исходных и конечных веществ ставятся соответствующие числовые коэффициенты (определяющие число молей вступающих в реакцию и получающихся в результате реакции веществ), например
Ni + 4 СО Ni(CO)4
3) Уравнения могут быть записаны в ионной или молекулярной форме. Обычно при этом твердые фазы (в том числе соли) .записываются в виде брутто-формул, неэлектролиты и слабые электролиты — в виде молекулярных формул, а сильные элект
Типы реакций
365
ролиты — в виде ионных формул, например
С12 + 2Н2О ч—> Н3О++ Cl" + НОС1
Cu2+ + H2S + 2 Н2О CuS + 2 Н3О+
Если нужно указать агрегатное состояние соединения, это делают либо с помощью индексов, либо подписывая соответствующее обозначение под формулой или рядом с формулой в скобках (обычно приняты обозначения: крист.— кристаллическое вещество, тв.— твердое вещество, жидк.— жидкость, газ— газ). Например,
NaTB_ < х. Nara3
4) Если в уравнение входят ионы или электроны, сумма зарядов в обеих частях уравнения должна быть одинаковой, например
Ca2+ + 2F" --> CaF2
Fe2+ --► Fe3+ + e“
5) С формулами химического уравнения связывают определенные количества вещества; обычно формуле вещества отвечает его количество, равное 1 молю, например
AgNO3 + NaCl --► AgClj + NaNO3
169,89 г-f-58,45 г = 143,34 г+ 85,00г
Вследствие незначительности энергетических эффектов химических реакций изменение массы в результате реакции практически равно нулю. Поэтому сумма масс веществ с обеих сторон уравнения должна быть одной и той же (закон сохранения массы при химических реакциях).
6) Значения энтальпий реакции ДЯ° (стандартные величины*) для количеств веществ, указанных в уравнении реакции, записываются отдельно. При этом обязательно нужно указывать агрегатное состояние, а для твердых фаз — модификацию, например
Н2 (газ) + VaOa (газ) -----► Н2О (газ)
Н2 (газ) + !/2О2 (газ) ----► Н2О (жидк.)
С (а-графит) + О2 (газ) ----->- СО2 (газ)
Д№ = —242,1 кДж/моль
Д№ =—285,8 кДж/моль
Д№ = —393,2 кДж/моль
* Стандартными энтальпиями реакций называют величины, относящиеся К реакциям, все ингредиенты которых находятся в стандартных состояниях (при любых температурах), поэтому для строгости нижним правым индексом следует указывать температуру, к которой относится ДЯ° этой реакции. Говорить, что реакция протекает при стандартных условиях (25 °C и нормальном давлении), ошибочно. — Прим. ред.
366
Неорганическая химия
33.2. Превращения без изменения состава
33.2.1. Изменения агрегатного состояния индивидуальных
веществ
При изменениях агрегатного состояния происходит сильное изменение сил взаимодействия, определяющих строение вещества. При повышении температуры твердого вещества частицам, его слагающим, сообщается все более сильное колебательное движение относительно положений равновесия. При определенной температуре в структуре разрывается часть связей, тем самым возникает состояние с более низким структурным порядком (более высокой энтропией), в котором частицы обладают большей подвижностью. Различают переходы твердое тело — жидкость (плавление), твердое тело — газ (сублимация) и жидкость— газ (испарение). При охлаждении, т. е. при обратной последовательности процессов, возникают состояния с более высоким порядком.
При изменении агрегатного состояния скачкообразно изменяются такие свойства вещества, как плотность, теплосодержание, оптические свойства и т. д. Для воды, например, изменение энтропии при испарении составляет ~ 109 Дж/(К-моль). Эта величина значительно выше, чем для «нормальных» жидкостей (~88 Дж/(К-моль)) (разд. 22.1.1), так как при испарении воды разрушаются водородные связи между молекулами.
Изменения агрегатного состояния подчиняются тем же закономерностям, что и химические реакции. Так, равновесие между двумя фазами устанавливается лишь при равенстве химических потенциалов вещества в обеих фазах.
33.2.2. Фазовые превращения в твердом состоянии
Фазовые переходы в простых веществах или в соединениях могут осуществляться при определенных температурах и давлениях. Обычно при этом происходят заметные изменения в расположении частиц в кристаллической структуре, сопровождающиеся даже изменениями характера связей между ними.
О чем же идет речь, если рассмотреть фазовые превращения с позиций термодинамики? Пусть имеется однокомпонентная система, состоящая из двух твердых фаз. Точка перехода отвечает равновесию между двумя фазами. На диаграмме состояния это соответствует температуре, при которой обе модификации имеют одинаковое давление пара (излом на кривой давления пара). Ниже температуры перехода устойчива модификация с меньшими величинами энтальпии и энтропии; выше температуры перехода устойчива модификация с большими величинами энтальпии и энтропии.
Превращения без изменения состава
Если фазовый переход между модификациями может осуществляться в обоих направлениях, такие модификации гтазы-ваются энантиотропными. Такому условию отвечают модиф^ика-ции серы. Устойчивая при комнатной температуре ромбическая сера переходит^ при 95,5 °C в серу моноклинную с поглощением теплоты (~37/ Дж/моль) и небольшим увеличением объема (0,42 мл/моль). При температуре ниже 95,5 °C происходит' обратное превращение моноклинной серы в ромбическую, однако часто этот процесс замедлен из-за уменьшения скорости превращения при понижении температуры.
У модификаций фосфора наблюдается явление монотропии, при котором превращение модификаций может осуществляться лишь в одном направлении, часто через расплавленное состояние. Равновесие между двумя модификациями не может установиться, поскольку метастабильная модификация плавится до достижения температуры превращения твердых фаз. Так, белый фосфор, содержащий в молекулярной решетке тетраэдрические молекулы Р4 (7’пл = 44 °C) может быть переведен в черный фосфор либо при нагревании до 220 °C и одновременном действии ударной волны (1,2 ГПа), либо при длительном отжиге при 220 370 °C в присутствии катализатора — металлической ртути. Черный фосфор — термодинамически наиболее устойчивая модификация фосфора вплоть до 550°C. Он построен из полимерных гофрированных сеток, причем каждый атом фосфора связан с другими тремя ковалентными связями.
Превращение черного фосфора в белый возможно лишь через состояние газа. При конденсации паров, состоящих из молекул Р4, образуется более энергоемкая и менее плотная белая модификация фосфора. Это соответствует правилу Оствальда, согласно которому сначала образуется наименее устойчивая модификация,переходящая через промежуточные ступени в наиболее устойчивую. Из этого правила имеются и исключения. Например, при термическом разложении карбида кремния выделяется графит, хотя энтальпия образования алмаза больше на 2,8 кДж/моль (при нормальных условиях).
Красный и фиолетовый фосфор не могут считаться определенными полиморфными модификациями. Их структуры состоят из неупорядоченного полимерного каркаса, в котором также могут быть статистически распределены атомы примесей.
При рассмотрении фазовых переходов важны еще и кинетические факторы, поэтому различают:
1) Переход с изменением первой координационной сферы. Происходят существенные изменения в расположении ближайших соседей рассматриваемого центрального атома. Например, У металлов относительно часто возможен переход кубической объемно-центрированной модификации с координационным числом 8 в кубическую гранецентрированную модификацию с ко-
368
Неорганическая химия
Образование и разрушение ионных решеток
369
ординационным числом 12. Положения и связи атомов изменяются незначительно, так что при соответствующей температуре переход происходит быстро. В противоположность этому для перехода арагонита в кальцит, сопровождающегося полной перестройкой структуры, необходима значительная энергия активации. Поэтому это превращение протекает относительно медленно.
2) Переход с изменением второй координационной сферы. Координация ближайшими соседями остается неизменной. В структуре могут меняться расстояние до более далеких частиц и их число. Такой тип перехода наиболее распространен; он сопровождается лишь небольшим изменением энтальпии.
3) Превращения с одновременным изменением характера связи. Переход графита в алмаз, белого фосфора — в черный. В то время как белый фосфор — типичное молекулярное вещество, связи в черном фосфоре носят частично металлический характер.
33.2.3. Превращения изомерных веществ с различным типом связей
Изомерами могут быть вещества одинакового химического состава, но с различным характером связей между частицами. Особенно часто это явление наблюдается в галогенидах фосфора (V), мышьяка (V) и сурьмы (V), причем в одной из модификаций этих соединений связи носят ковалентный характер, а в другой преимущественно ионный. Например, для соединения состава Р : F: С1 = 1 :3:2 известны молекулярная форма PF3CI2 в ионная форма [PCUj^EPFg]-, сильно отличающиеся по свойствам. Так, PF3CI2 — газ, конденсирующийся при температуре +7,1 °C. Изомерное гетерополярное соединение [РС14]+[PF6]^— твердая соль с температурой сублимации +135°C, растворяю щаяся в безводных полярных растворителях, например ацето нитриле. Оба вещества могут сосуществовать в отсутствие вла ги, вызывающей мгновенное гидролитическое разложение (В этом случае можно говорить об истинной изомерии связей.
Другим примером может служить соединение PC14F (ТКИп = = 67°С), образующееся наряду с PF5 (ТКип — —75°С) при тер мическом разложении [РС14]+[PF6]_:
[PC141+[PF6]- PC14F + PF5
Получающиеся вещества могут быть легко разделены благодаря сильному различию в температурах кипения. Соединение PCl^F не проводит электрического тока, что указывает на присутствие в жидкости либо отдельных молекул, либо молекулярных агрегатов. При комнатной температуре это соединение по реакции первого порядка переходит (с периодом полупревра
щения 35 ч) в солеобразную форму. Эта последняя, соответствующая формулам [PC14]+F~ или [PC14]+[PC14F2]~, плавится при +175°С; ее растворы в полярных растворителях проводят электрический ток. Переход PC14F в [PC14]+F~ можно представить как превращение изомера с ковалентным типом связей в энергетически более выгодную гетерополярную форму.
Подобного типа превращения довольно часто происходят и при изменении агрегатного состояния. Так, пентахлорид фосфора, построенный (по данным КР-спектроскопии и рентгеноструктурного анализа) из катионов [РС14]+ и анионов [РС16]_, в газовой фазе состоит из молекул PCU. В жидком PCI5, который можно получить при нагревании вещества под давлением,, присутствуют агрегаты молекул PCU, соединенных посредством хлорных мостиков. Подобные явления наблюдаются и в других жидкостях. В безводной азотной кислоте HNO3 присутствуют ионы Н2ЫОз+ и NO3~, обусловливающие сравнительно высокую электропроводность. В газовой фазе азотная кислота состоит только из одних молекул HONO2.
В некоторых случаях превращения изомерных веществ происходят при взаимодействии с растворителем. Так, ряд молекулярных веществ, в чистом виде практически не диссоциирующих на ионы, под влиянием растворителя переходят в гетерополяр-ные формы. В качестве примера можно указать на пентахлорид сурьмы, построенный во всех трех агрегатных состояниях из молекул SbCU. При взаимодействии с ацетонитрилом сначала образуется аддукт SbCU-CHsCN, который при дальнейшем добавлении растворителя обратимо диссоциирует на комплексные ионы:
2SbCl6-CH3CN [SbCl4(CH3CN)2]++ [SbCl6]~
Положение равновесия подобных реакций сильно зависит от природы растворителя, концентрации и температуры (разд. 34.2.4).
По данным ЯМР, в некоторых смешанных галогенидах под действием полярных растворителей также устанавливается равновесие между изомерами в ионной и молекулярной формах, часто сопровождающееся процессом диспропорционирования. Так ведет себя, например, дихлоротрифторид ниобия(V) в ацетонитриле:
2 NbCl2F3 + 2 CH3CN [NbCl4(CH3CN)2]+ + NbFy
33.3. Образование и разрушение ионных решеток
33.3.1. Процесс растворения
При растворении соли происходит разрушение ионной решетки под действием растворителя. При этом дипольные молекулы растворителя в соответствующей ориентации присоединя-24—2027
370
Неорганическая химия
ются к катионам и анионам, находящимся на поверхности кристалла, что сопровождается переходом этих ионов в раствор. В дальнейшем молекулы растворителя окружают ионы со всех сторон. По существу этот процесс присоединения дипольных молекул является реакцией комплексообразования. Число сольватных молекул зависит от различных факторов. Например, растворение хлорида лития в воде можно описать с помощью следующего уравнения:
Li+Cl- (тв.) + (х + у) Н2О -► [Li(H2O)x]+ + [С1(Н2О)у]~
Растворение солей может сопровождаться протеканием и других процессов, например протеканием реакций протолиза или окислительно-восстановительных реакций (ВаСО3+кислота, LiH+H2O).
Для решения вопроса о том, растворяется ли данное вещество в определенном растворителе, можно провести термодинамическую оценку. При постоянных температуре и давлении решающим будет изменение свободной энергии Гиббса AG=AH— —TAS, которое учитывает как изменение энтальпии (разрыв и образование связей), так и энтропийные факторы (изменение степени упорядоченности).
Изменение энтальпии при растворении (энтальпия растворения) может быть экспериментально определено. Оно равно разности энтальпии решетки и энтальпии сольватации Растворение безводных солей обычно сопровождается повышением температуры, тогда как соответствующие гидраты, имеющие в кристаллической структуре уже частично сольватированные ионы, растворяются часто с поглощением теплоты.
Растворимость большинства веществ при повышении температуры увеличивается, что связано как с дополнительным подводом тепловой энергии, компенсирующим энергию решетки, так и с увеличением энтропийного фактора благодаря увеличению величины TAS. В некоторых случаях наблюдается уменьшение растворимости при повышении температуры, что связано с разрушением стабилизирующих сольватных оболочек.
Скорость растворения зависит и от других факторов, например степени измельчения и скорости перемешивания.
Если при растворении часть соли остается нерастворенной, устанавливается равновесие между твердой фазой и раствором Это равновесие носит динамический характер, т е. происходит непрерывный обмен между ионами кристаллической решетки и ионами, находящимися в растворе, что было установлено с помощью радиоактивных изотопов.
Пусть имеется насыщенный раствор твердой фазы состава АтВп, т. е. гетерогенное равновесие
АтВ„ ч—>-. тАп+-j-пВт~ (О
Образование и разрушение ионных решеток
371
Согласно закону действующих масс,
= (2>
где Яап+ и авт----активности катиона с зарядом п+ и аниона
с зарядом m~\ Kl называется произведением активностей. Оно является для данной системы величиной постоянной и зависит только от температуры (табл. ВЗ). Активность твердой фазы не входит в уравнение (2), поскольку активности всех индивидуальных веществ в конденсированном состоянии приняты равными единице. Следует, однако, иметь в виду, что это уравнение верно лишь в присутствии твердой фазы.
Таблица В.З. рКс некоторых труднорастворнмых неорганических
соединений при 25 °Са
Соединение Соединение PKL
Бромиды AgBr 12,4 Йодиды Agl 16,0
CuBr 7,4 Cui 11,3
Карбонаты ВаСОз 8,5 Оксалаты CaC2O4 8,1
СаСОз (кальцит) 8,3 MgC2O4 4,1
Сс1С0з 13,6 Фосфаты Ag3PO4 19,9
L12CO3 2,4 MgNH4PO4-6H2O 12,6
Si-СОз 9,5 Сульфаты BaSO4 10,0
Хлориды AgCl 9,96 CaSO4 2H2O 4,6
CuCl 6 PbSO4 8,0
Hg2Cl2 17,5 SrSO4 6,6-
РЬС12 4,9 Сульфиды Ag2S 49
Хроматы Ag2CrO4 11,7 AS2S3 25
BaCrO4 9,7 B12S3 96
PbCrO4 13,8 CdS 27
Фториды CaF2 10,5 CoS 24
MgF2 8,2 CuS 41
Гидроксиды A1(OH)3 32,7 HgS 52
Ca(OH)2 5,1 MnS 15
Cr(OH)3 30,2 NtS 24
Fe(OH)2 16,0 PbS 28
Fe(OH)3 37,4 ЬЬгЗз 93
Mg(OH)2 10,9 SnS 28
SnS2 70
ZnS 25
а По аналогии с pH pKL=—lg KL=—1g Пр Вследствие Данных в ряде случаев приведены усредненные значения.
расхождения литературных
Если произведение активностей ионов оказывается меньше чем /(т, осадок растворяется, пока не будут достигнуты значения равновесных активностей. Если же, наоборот, произведение активностей ионов больше, чем Ад (состояние пересыщенного Раствора), осадок выпадает до тех пор, пока не будет достигнуто равновесное состояние системы.
24*
372
Неорганическая химия
В разбавленных растворах можно в первом приближении заменить активности молярными концентрациями, а произведение активностей — произведением растворимости:
• с\т- = = Пр (3)
Соотношение (3) позволяет в хорошем приближении рассчитать молярную (моль/л) концентрацию соли в ее насыщенном растворе; при этом не учитывается, в каком виде находятся растворенные частицы. Простая зависимость между растворимостью и произведением растворимости может быть установлена лишь в том случае, если ионы не участвуют в каких-либо других равновесиях. В этом случае для растворимости (моль/л) вещества состава АтВ« справедлива следующая формула*:
2+у^Г f4.
ЬАгиВл У тт-Пп 'Ч
Пример. Рассчитайте молярную концентрацию ионов серебра в насыщенных растворах хлорида и хромата серебра. Произведения растворимости веществ при 25 °C равны для AgCl Пр=1-10-10 моль2/л2 и для AgjCrO, Пр=2-10-12 моль3/л3.
Решение. Сравнение абсолютных значений Kl еще не свидетельствует о том, что растворимость хромата серебра меньше, чем хлорида серебра. Действительно, размерность произведений растворимости для двух солей различна. Для AgCl
AgCl (тв.) Ag+ + Cl-
CAg+ 'ссГ = 1 ^-1° моль2/л2
Так как оба вида ионов в насыщенном растворе находятся в одинаковых концентрациях, cAg+ = сс1_. Тогда
CAg+=y Ю"1о= 10*5 моль/л
Для хромата серебра
AgjCrOj (тв.) 5=t’2Ag++СгО42“
clg+ • ссго42~ = 2-1 О*12 моль3/л3
Согласно уравнению реакции диссоциации, cAg+=2сСгО42-- Тогда
CAg+ • 1/aCAg+ = 2-10'12 моль3/л3
Сд8+ = ^4-10"12= 1,6-10-4 моль/л
Таким образом, в насыщенном растворе Ag2CrO4 концентрация ионов серебра больше, чем в насыщенном растворе AgCl.
Использование формулы (4) дает те же результаты. Молярная концентрация Ag2CrC>4 равна концентрации СгС>42_-ионов:
3/ 9,1 п-12
C'Ag2CrO4 = ccrO42- = )7 b4' =0,8-10' моль/л
* Этой формулой можно пользоваться, если в растворе нет избытка оД ного из ионов: Ап+ или В"1-, например за счет других электролитов, содеР жащих эти ионы. — Прим, перев.
Образование и разрушение ионных решеток 373
для концентрации ионов серебра получим:
cAg+= 2cCi.q42-= 1,6-10-4 моль/л
Строго говоря, произведение растворимости Klc не является константой, а зависит от коэффициентов активности ft, р свою очередь зависящих от ионной силы, раствора. Справедливо следующее соотношение:
Пр=Лд7—Й77—-п (5)
'An+
По теории Дебая — Хюккеля, коэффициенты активности ионов в водных растворах при 20 °C приблизительно вплоть до значения ионной силы 7 = 0,1 могут быть рассчитаны по формуле {zi — заряд иона) _
(6)
Ионную силу раствора можно вычислить по формуле 7=0,5^^ (7)
При точных расчетах растворимости солей следует учитывать вышеописанные закономерности. Обычно при возрастании ионной силы коэффициенты активности уменьшаются. Поэтому растворимость растет при увеличении концентрации посторонних ионов и сильно зависит от величины их заряда (табл. В.4). Этот солевой эффект может заметно сказываться Таблица В.4. Коэффициенты активности3 ионов в водных растворах в зависимости от ионной силы I и от заряда иона z.
I f
г.=1 г/=2 г.=з
0,001 0,97 0,87 0,73
0,005 0,93 0,74 0,51
0,01 0,90 0,66 0,40
0,05 0,81 0,43 0,15
0,1 0,76 0,33 0,1
а Рассчитаны по формуле (6).
Даже при небольших значениях ионной силы (7=0,001). Для соли, состоящей из двухзарядных ионов, например ВаЭОд, при Ионной силе 0,1 происходит трехкратное увеличение растворимости. Для соли, состоящей из однозарядных ионов, увеличение растворимости при той же ионной силе значительно Меньше.
374
Неорганическая химия
Пример. Дан насыщенный раствор AgCl, находящийся в равновесии с твердой солью, в 0,1 М растворе NaNO3 Какова растворимость AgCl в этом растворе?
Решение. Найдем ионную силу раствора [формула (7)]:
I = 0,5-(I2-0,1 +Р.0,1)=0,1
Вследствие того что концентрация ионов Ag+ и С1_ незначительна, их влиянием на ионную силу можно пренебречь. В соответствии с данными табл. В.4 коэффициенты активности равны fAg<-=[ci_=0,76.
Kl Ю-10
пр ~ /Ag+7ci~ ~ о,7б2 = 1>73'10 10 моль2/л2
Растворимость AgCl оказывается теперь равной
CAg+ = сщ- = VКсl = 1,31 -10-5 моль/л
Таким образом, за счет добавления посторонних ионов растворимость хлорида серебра увеличилась на 30%.
33.3.2. Осаждение труднорастворимых солей
Труднорастворимая соль выпадает в осадок в том случае, если превышено произведение активностей ионов [формула (2)]. Напишем в ионной форме уравнение осаждения BaSO4 (обратное процессу растворения):
Ва2+ + SO42- --► BaSO4|
Согласно закону действующих масс,
/<=------!-----или p/о—р/С (8)
аВа+-aso42- Kl н 1 L '
где /С—константа равновесия реакции осаждения сульфата бария, численно равная обратной величине произведения активностей (значения рК и р/Сд для процессов осаждения и растворения соответственно различаются лишь знаком).
Влияние на осаждение одноименных и посторонних ионов, а также кинетика образования кристаллов соли подробно описаны в разд. 38.3.4.2. Все эти факторы имеют большое значение в количественном анализе.
Рассмотрим теперь процесс сопряженного растворения и сопряженного осаждения соли. Эти термины означают превращение труднорастворимой соли в еще менее растворимую. Если, например, осадок CaSO4 обрабатывать раствором карбоната | натрия, происходит взаимодействие с образованием менее рас- 1 творимого СаСОз.
Константа равновесия этой реакции может быть легко рассчитана, если весь процесс рассматривать состоящим из двух стадий, причем р/< для суммарной реакции вычисляется как сумма рКд или р/< для отдельных стадий:
Теория кислот и оснований
375
CaSO4 (тв.) zi==z Са2+ + SO42- рКл (CaSO4) = 4,6
Са2+ + СО32- СаСО3 (тв.) рК (СаСО3) = -рКл (СаСО3) = -8,3 CaSO4 (тв.) + СО32- :₽=t СаСО3 (тв.) + SO42~ р7< = 4,6 — 8,3 = —3,7
Следовательно, К = 10’’7 «5000, т. е. сs0(12-: сСОз2- = 5000 : 1. Равновесие сильно сдвинуто в сторону образования осадка карбоната кальция.
Перевод труднорастворимых слоей в карбонаты весьма важен в аналитическом химии, поскольку исследуемая проба в такой форме гораздо легче подвергается дальнейшей обработке (разд. 37.2.1 4). Если процесс проводить не с раствором, а с карбонатным расплавом, степень превращения в карбонаты значительно увеличивается. Так, при обработке раствором соды сульфата бария (рЛь=10,0) происходит лишь частичное превращение в несколько более растворимый карбонат бария р/(ь = 8,5. Однако при сплавлении BaSO4 с карбонатами переход в карбонат бария количественный. Расчет равновесия с использованием р/( здесь невозможен, поскольку определение соответствующих значений для расплавов затруднено.
Направление реакций обмена с участием солей в растворах может быть определено также на основе рассмотрения энергии решетки. Как правило, в системе с двумя ионными парами наиболее прочную решетку образуют ионы с более высоким значением ионного потенциала, который вычисляется как отношение заряда иона к его радиусу (в какой-то степени это мера «концентрации» заряда). Ионы с небольшими ионными потенциалами взаимодействуют слабо и остаются в растворе и лишь при упаривании растворителя дают кристаллическую фазу, например
RbF + Lil --► LiF^ + Rbl
СаС12 4- 2 NaF -► CaF2| + 2 NaCl
CaCO3 + Na2S ---► CaS| -f- Na2CO3 (получение соды по Леблану)
33.4. Теория кислот и оснований
33.4.1. Основы теории кислот и оснований
33.4,1.1. Границы применимости классической теории кис-
лот и оснований
В соответствии с классическим определением Аррениуса U887 г.) кислотами называют вещества, которые в водном растворе диссоциируют с образованием ионов водорода, а основаниями — вещества, диссоциирующие с образованием ионов гидроксила. Это определение было большим шагом вперед по сравнению с эмпирическими критериями, которые, правда, и До настоящего времени используются на практике для оценки кислотно-основных свойств веществ. В соответствии с классической теорией для кислот и оснований характерна реакция нейтрализации, в результате которой образуется вода, а типичные свойства прореагировавших компонентов исчезают. При выпаривании раствора получается соль, катионы которой остались от основания, а анионы — от кислоты. Теория объясняет также электропроводность образовавшегося раствора соли, понижение температуры замерзания и осмотические явления.
376
Неорганическая химиЯ~
Однако в дальнейшем эта теория оказалась недостаточной по следующим причинам:
не оправдывается предположение о существовании в водном растворе свободного протона, как эТ° требует классическая теория; высокий ионный потенциал этой чрезвычайно маленькой частицы (радиус — 10~15 м) способствует очень прочной связи между протоном и другими часТиЦами> например молекулами растворителя.
теория не позволяет распространить определение кислот и оснований на обширную область апротонных растворителей.
33.4.1.2. Понятие кислоты и основания по Бренстеду
В 1923 г. независимо друг от друга Бренстед в Дании и Лаури в Великобритании предложили следующее более широкое определение кислоты и основания:
кислоты — вещества, отдающие протон;
основания — вещества, принимающие протон-
Таким образом, постулируется, что кИСЛ0ТЬ1 и основания вступают в следующую реакцию:
кислота ч > основание прОТ0Н
A у—ВЧ-Н'*' (9)
Каждой кислоте соответствует основание, которое возникает при потере кислотой протона. Кислоту и основание одной и той же протолитической системы называют сопряженной парой, например: 1 .
A В + Н+
нс1 сг + н+
H2SO4 HSO4- + Н'1'
HSO4- ч—SO42- + н+ NH4+ ч-^- nh3 + Н+ N2H62+ N2H5++H+
N2H5+ =р=±: N2H4 + Н+
Понятие кислоты по Бренстеду относился не только к нейтральным молекулам, таким, как НС1 или H2SO4, но и к заряженным частицам (HSO4_ или NH4+). Поэтому можно различать нейтральные (молекулярные), катионные и анионные кислоты. То же самое относится и к основаниям. Так, например, ион гидразиния(1+ ) N2Hs+ представляет собой основание в протолитической реакции с ионом гидр^зиния (2+) N2H4 • Существуют также частицы, которые могут нести себя и ка кислоты, и как основания (амфолиты).
Теория кислот и оснований
377
33.4.1-3. Протолитические реакции
Представляемая уравнением (9) реакция имеет только гипотетический характер. Вследствие того что свободные протоны не могут существовать отдельно в химических системах, в реальных условиях кислотно-основные реакции протекают между двумя сопряженными парами кислот и оснований, причем одна из них поставляет протоны, а другая их принимает. Рассмотрим этот процесс на нескольких примерах.
При пропускании газообразного хлороводорода через воду образуется соляная кислота и раствор нагревается. Протон хлороводорода присоединяется к молекуле воды:
НС1 ч—CJ- + Н+
Н+ + НгО ч=* Н3О+
НС1 + Н3О 4=5= Нзо+ + С1-
В данном случае НС1 выступает как донор протонов (кислота), а НгО—как акцептор протонов (основание). Выделение теплоты в этой реакции происходит как на стадии присоединения протонов, так и на стадии гидратации образовавшихся ионов гидроксония и хлора.
Аналогично протекает реакция аммиака с водой:
Н2О 4=5= ОН- + Н+
H++NH3 4—^ NH4+
Н2О + NH3 4=* NH4+ + ОН~
Здесь донором протонов является вода (кислота), а их акцептором— NH3 (основание). Эта реакция также экзотермическая.
Запишем в общем виде протолитический процесс между Двумя сопряженными парами:
Ах 4=* Bx-f-H+
Н+ + В2 4=5- А2
Ах + В2 4=fc Аа -f- Вх
Переход протона не всегда происходит в жидкой фазе; этот процесс может происходить как в твердой, так и в газообразной фазе, например, выделение циановодорода при растирании Цианидов с KHSO4
KHSO4 (тв.) + KCN (тв.) -> HCN) (газ) + K2SO4 (тв.)
нли образование твердого иодида фосфония из фосфина и иодоводорода.
РН3 (газ) + HI (газ) ^=±= [РН4]1 (тв.)
378
Неорганическая химия
33.4.1.4. Автопротолиз воды
Вода характеризуется амфотерными свойствами, т. е. в ней протекает протолитическая реакция с очень небольшой константой равновесия:
НаО+ Н2О > Н3О+4- ОН"
Перенос протона между одинаковыми частицами называют автопротолизом или кислотно-основным диспропорционированием. Существование ионов доказывается наличием небольшой электропроводности воды (при 18 °C х = 4,41-10-10 См/м). В чистой воде концентрация ионов НзО+ и ОН- одинакова. Для этой протолитической реакции принята еще одна характеристика, кроме обычной константы равновесия, — ионное произведение воды (разд. 23.3.10):
^аЧ — аН30^'аОН~ (10)
Эту величину можно также назвать константой автопротолиза ВОДЫ. Kaq СИЛЬНО ЗЭВИСИТ ОТ Температуры; при 22 °C K.aq — = 1,00-10-14.
Уравнение (10) справедливо для всех водных систем, в которых находятся и другие протолиты, при условии замены концентрации на активности. Коэффициенты активности и в этом случае определяются через ионную силу раствора. Уже при ионной силе />10~3 значения pH сильно изменяются, что необходимо учитывать при сравнении данных, полученных расчетом и из потенциометрических измерений.
В чистой воде и в сильно разбавленных водных растворах активности можно заменить на концентрации:
^%==сн3о+-Сон- [моль2/л2] (П)
В такой форме ионное произведение воды часто применяется для оценки значений pH и при более высоких значениях ионной силы.
33.4.1.5. Сила кислот и оснований в водных растворах
Кислоты и основания могут сильно отличаться друг от друга по степени протолиза в воде. Сильные протолиты почти нацело реагируют с растворителем. Слабые электролиты реагируют только частично, т. е. значительная часть исходного вещества находится в равновесной смеси в неизмененном виде-Так, например, в растворе соляной кислоты практически все молекулы НС1 диссоциированы на гидратированные ионы Н3О+ и СК. В 1 М уксусной кислоте только ~ 1 % исходного количества кислоты распадается на ацетат-ионы и ионы гидроксо-ния, а преобладающая часть вещества находится в неизменен-
Теория кислот и оснований
379
цом состоянии. Количественной мерой силы кислоты является константа равновесия, которую получают, применив к протолитическому равновесию закон действующих масс. Для протолиза кислоты
а + н2о у—* н3о++в
= аВ'ан3о+ ~ ав,ан3о<~
а «А-ан2О ~ аА ' >
Здесь Ка — константа кислоты-, приближенное значение константы кислоты можно получить, принимая активность воды в разбавленных растворах равным 1.
Аналогично для протолиза основания
Н2О + В А + ОН~
д- __ ад-йон- аА'аон~
ь “В-“Н2О ~ «в ' '
где Кь — константа основания.
Для упрощения вычислений вместо констант Ка и Кь часто используют их логарифмы с обратным знаком (рК). рК важнейших кислотно-основных пар приведены в табл. В.5, причем все данные относятся к стандартному растворителю — воде.
Константы кислоты Ка(Н3О+) и основания Кь(ОН-) требуют особого обсуждения. Если принять некоторые упрощающие предположения, из уравнений (12) и (13) получим для них следующие значения:
аН2О'аН3О+
Ка (Н3О+) = —---------= ан2о = /н2о-сн2о ~ Сн2О = 55,3 моль/л
чн2о-аон- „ „
Кь (ОН ) = —---------= ан2о = /н2о-сн2о ~ сн2о = 55,3 моль/л
Отсюда при 25 °C
рКа (Н3О+) = ?Кь (ОН-) =-1,74
На практике обычно принимают /н2о=1, хотя вследствие большой концентрации воды (Сн2о = 55,3 моль/л) и сильного взаимодействия между молекулами (водородная связь) это предположение не совсем оправданно. Истинный коэффициент активности значительно меньше 1, однако его очень трудно измерить.
Для термодинамического определения констант надо выразить активность воды, участвующей в протолитической реакции, так же как и активность воды — растворителя, в мольных долях. Тогда получим
Ка' (Н3О+) = Кь' (ОН-) = 1
?Ка' (Н3О+) = рКь' (ОН-) = О
Впрочем, при этом получаются безразмерные константы кислот и оснований, Которые нельзя сравнить с соответствующими константами других протолинов. Поэтому в большинстве случаев применяют константы, выраженные через концентрации (табл. В.5).
Таблица В.5. Распределение сопряженных пар кислота — основание в зависимости от силы кислоты и основания
рка
Кислоты
Основания
р*ь
10 НС1О4 4=t H+ + C1O4-
« 10 HI ?=ЗН++Г
g ~-9 НВг 4=3 Н++Вг-
5 ~-7 НС1 4=з Н++СГ
л /'4Z—3 H2SO4 4=fc H+ + HSO4-
2 -1,74 н3о+ 4=t H+ + H2O
— 1,32 HNO3 4=± H++NO3-
1,42 Н2С20а 4=3: h++hc2o4-
1,92 H2SO3 4=3 H+ + HSO3-
1,92 HSO4- 4=3 H+ + SO42-
„ 1.96 Н3РО4 4=3: H+ + H2PO4-
я 2,0 НС1О2 4=3 H++C1O2-
§ 2,22 [Fe(H2O)e]’+ 4=3: H+ + [Fe(H2O)sOH]2+
2,32 H3AsO4 4=3: H+ + H2AsO4-
3,14 HF 4=3 H++F-
3,35 hno2 4=3: H++NO2-
очень слабые слабые средней силы
4,75 4,85 CH3COOH [Al(H2O)e]«+
6,52 H2COS
6,92 H2S
7,12 h2po4-
7,25 HC1O te
9,24 H3BOS
9,25 nh4+
9,4 HCN
9,5 H4SiO4
10,4 HCO3"
11,62 H2O2
12,32 HPO42-
4=3: Н++СН3СО2-4=з Н++[А1(Н2О)6ОН]2+ 4=* Н++НСО3-4=3: H++HS-
4=t Н+ + НРО42-4=3: Н+ + С1О-
Ч=* Н++Н2ВО3-4=^ H++NH3 4=* Н++ CN-4=3: н+ + H3SiO4-4=3 Н++СО32-4=3 Н+ + НО2-4=t Н++ РО43"
15,74 H2O
23 NHS
24 OH-
34 CH4
38,6 H2
H++OH’i H++NH2-Н++О2-н++сн3-Н+ + Н-
~24
~24
О)
з ~23 ю
§ ~21
й ~17
| 15,74
15,32
12,58
12,08
12,08
12,04
3 12,0
ю
§ 11,78
11,68
10,86
10,65
9,25
ч 9,15
5 7,48
>s
н 7,08
2. 6,88
6,75
4,76
4,75
<D 4,8
3 X 4,5
J3
4 g 3,6
3,38
1,68
<u —1,74
9
g , ~--10
20
s
S' о —24,6
Теория кислот и оснований
381
В зависимости от силы (соответствующие значения рКД кислоты можно разделить на:
1) очень сильные (рК-<0);
2) сильные (0<рК<4,5);
3) средней силы (4,5<рЛ<9);
4) слабые (9,0<рК< 14);
5) очень слабые (рК>14).
В водных системах константы кислоты и основания связаны между собой:
т. т. ав-«н3о+ «А-аон-
-----аХ '—~ анзО+' аон- 0 4)
р/<а+рДь = рДа(?=14 (при 25 °C) (15)
При помощи уравнения (14) можно из известного значения рК. кислоты или основания определить рК сопряженного основания или кислоты. Например, из значения рКь аммиака (4,75) получим рКа катиона аммония (9,25). Уравнение (14) показывает, что сила кислоты обратно пропорциональна силе сопряженного основания. Например, очень сильной кислоте НСЮ4 соответствует очень слабое основание CIO4-. Для протолитов средней силы значения рК сопряженных кислоты и основания примерно равны, т. е. скорости протолитических реакций в прямом и обратном направлениях примерно одинаковы, например
H2S 4- Н2О HS" + Н3О+ рКа = 6,92; рКь = 7,08
Термодинамическое исследование протолитических систем привело к следующему выводу: сильная кислота (основание) протолитической системы сопряжена со слабым основанием-кислотой). Эта закономерность подтверждается для всех приведенных ранее примеров.
Рассмотрим еще одну протолитическую реакцию — взаимодействие этилата натрия с водой:
н2о + С2н6о- —► с2н6он + он-Ai В2 Ай Bi
В этой системе вода является более сильной кислотой, чем этиловый спирт, а этокси-анион — более сильным основанием^ чем ОН-, поэтому равновесие реакции сильно смещено направо.
С помощью этого примера можно понять механизм нивелирующего действия, которое оказывает вода на сильные кисло-TbI- Если в растворе присутствуют сильные кислоты с более вЫсокой константой кислоты, чем для Н3О+, то в результате-протолитической реакции в системе устанавливается равнове-
382
Неорганическая химия
Теория кислот и оснований
383
сие, при котором образуется эквимолярное количество ионов гидроксония:
НХ + Н2О----► н3о+ + х- (Х = СЮ4", Г, Cl’)
Тем самым уровень кислотности всех кислот снижается д0 уровня кислотности Н3О+-иона.
Подобным же образом протолиз сильного основания, такого, как, например, С2Н5О_, NH2~, Н~ с Кь>К.он~, приводит в водных растворах к выравниванию уровня основности. При растворении солеподобного соединения одновременно с разрушением кристаллической решетки происходит протолитическая реакция
H2O-(-NH2- ->- NH3 + ОН"
В результате энергичного протекания реакции выделяется газообразный аммиак, и образуется эквимолярное количество едкого натра в качестве конечного продукта. В этом случае основность амид-иона снижается до уровня основности ОН~-иона.
Можно легко показать, что константа равновесия К реакций между кислотой и основанием Ai + B2^Bi+A2 может быть получена из констант сопряженных пар кислота — основание:
К = Ка1!Ка„ или рЛ=рЛа1—рКа2 (16)
Например,
Н2РО4- + NH3 <—h НРО42- + NH4+ Ai Bg Bi A2
рЛ = 6,88—9,25=—2,37
Подобно тому как диссоциацию соли определяют степенью диссоциации, в качестве меры протолиза вводят степень протолиза. Степень протолиза — это доля исходной (аналитической) концентрации кислоты с0, которая в результате взаимодействия с водой переходит в сопряженное основание:
(Ч) ^0
Исходная концентрация — это сумма равновесных концентраций образовавшегося основания и непрореагировавшей кислоты:
Со = Св+с> 1
Отсюда степень протолиза кислоты
Аналогично для степени протолиза основания
Для сильных кислот Св = С0, так как протолиз идет до конца, й ца=1. В слабых кислотах, напротив, Св<С0 и аа<1-
Степень протолиза слабых электролитов зависит от исходной концентрации кислоты и описывается законом разбавления Оствальда:
= (2»
Так как %а (Ль) — некоторые постоянные, степень протолиза увеличивается по мере снижения исходной концентрации кислоты и становится равной единице при бесконечном разбавлении. Поэтому в сильно разбавленных растворах вода почти полностью протолизирует слабые протолиты. В отличие от К а и Кь степень протолиза зависит от разведения и поэтому малопригодна для количественного сравнения силы кислот и оснований.
33.4.2. Особые случаи протолитических реакций
33.4.2.1. Реакции между сильными кислотами и основа-
ниями
Рассмотрим реакцию соляной кислоты с едким натром. Как уже указывалось, кислотные свойства любого водного раствора сильной кислоты объясняются исключительно тем, что в нем содержатся ионы гидроксония. В то же время в растворе натриевой щелочи содержится сильное основание — ионы гидроксила, которые образуются при взаимодействии кристаллической решетки NaOH с молекулами воды как гидратирующими частицами. Если смешать эквивалентные количества кислоты и основания, то вследствие низкой константы автопротолиза воды из ионов Н3О+ и ОН- образуются недиссоциированные молекулы воды:
Н3О++ ОН- 2 Н2О
Имеющиеся в растворе ионы Na+ и С1_ не участвуют в реакции. Образуется нейтральный раствор соли с низкой концентрацией ионов Н3О+ и ОН~:
сНзо+ = сон- = 10-7 моль/л (при 25 °C)
Во всех реакциях нейтрализации сильных кислот и оснований в водных растворах стандартное изменение энтальпии Д№ = = —-57,4 кДж/моль. Это также является доказательством того, что независимо от выбранной пары кислота — основание в растворе идет всегда одна и та же реакция.
Несколько иначе идет реакция между сильными основаниями — анионами, такими, как CN_, СО32-, S2~, и растворами-
384 Неорганическая химия
Теория кислот и оснований 285
сильных кислот. При подкислении раствора цианидов в резуль тате протолиза в соответствии с уравнением реакции
CN- + Н3О+ » HCNf + Н2О образуется циановодород, который вследствие низкой раствори, мости в воде удаляется из раствора. Точно таким же образом ведут себя и другие названные анионы.
33.4.2.2. Реакция между сильными и слабыми (средней силы) протолитами
Примером реакции между сильной кислотой и основанием средней силы может служить взаимодействие растворов соляной кислоты и аммиака:
Н3О+ + NH3 NH4+ + Н2О
Образовавшийся раствор хлорида аммония имеет слабоки лую реакцию, так как содержит катионную кислоту NH4 Раствор с [NH4+] = 1 моль/л имеет pH 4,6. Аналогично прог кает реакция между относительно сильным основанием — аце тат-ионами(Ас_) и слабой кислотой — водой;
Н2О + Ac- НАс + ОН"
В результате реакции устанавливается равновесие, для ко торого характерно наличие в растворе всех этих частиц. Раствор с [NaAc] = 1 моль/л имеет pH 9,4.
Классическая теория кислот и оснований рассматривает эту реакцию как обратную реакции нейтрализации, причем для растворения в воде соль частично разлагается на исходные ук сусную кислоту и основание. Поэтому такой процесс назвали гидролизом. Однако в рамках теории Бренстеда в данном случае происходит протолиз основания в ионной форме, протекаю щий по тем же законам, что и обычный протолиз, поэтому из лишне вводить новый термин. Понятие «гидролиза» следует применять только в том случае, если разрываются связи меж ду атомами в результате взаимодействия вещества с водоп (разд. 35.8).
Гидратированные многозарядные катионы металлов реаги руют с водой как катионные кислоты средней силы. Водный раствор соли алюминия содержит, например, ион гексаакваалю-миния(Ш) [А1(Н2О)6]3+. Вследствие высокого заряда катион3 между ионом А13+ и атомами водорода гидратной оболочки возникает сильное отталкивание. Это приводит к переносу про тона от молекулы воды гидратной оболочки к молекуле во ды — растворителя с образованием иона гидроксония. Реакцш11 протолиза можно записать в виде следующего уравнения:
[А1(Н2О)6]з++ н2О Н3О++[А1(Н2О)5ОН]2+
Координационное число катиона остается равным шести. Даль-нейшее отщепление протонов может привести к образованию соединений типа [А1(Н2О)4(ОН)2]+, [А1(Н2О)3(ОН)3] и т. д.
Появляющиеся вокруг катиона гидроксильные группы способны к конденсации, что может приводить к образованию цепей
но Н20 но н2о но н2о
•••НО—А1 -о н" НО — А1—О Н-НО — AL—ОН ••
Н20 Нго Н20 Н2О HjO Н2О
При добавлении в раствор слабых оснований, например аммиака, число гидроксильных групп, способных к конденсации, увеличивается, цепи распространяются во всем трехмерном пространстве и возникают гелеобразные осадки. При старении таких осадков происходит дальнейшая конденсация. Такого рода процессы можно назвать протолитическим осаждением.
Амфотерный характер гидроксида алюминия проявляется в том, что при добавлении к нему раствора щелочи снова происходит растворение осадка с образованием гидроксокомплек-сов. Можно предположить, что сначала происходит гидролитическое расщепление связей А1—О—А1 и образуются аквагидрок-сокомплексы А1(Ш). При дальнейшем протолизе ОН~-ионы становятся акцепторами протонов:
[А1(Н2О)8(ОН)3] + ОН- H2O + [A1(H2O)2(OH)J-
Вообще, почти все гидратированные ионы металлов можно рассматривать как аквакислоты. Впрочем, ионы щелочных и щелочноземельных металлов практически не проявляют свойств кислоты, так как эти ионы имеют сравнительно большие размеры, малый заряд, а следовательно, не могут отщеплять протон от молекул гидратной оболочки. Совсем иначе обстоит дело с ионами элементов побочных групп периодической системы, имеющих малый размер и высокий электрический заряд. Например, ион {Ре(Н2О)б]3+ имеет рАа = 2,2 и относится к сильным кислотам.
Реакции кислотно-основных индикаторов также являются Реакциями между слабыми (средней силы) и сильными протолитами. Окрашенные индикаторы представляют собой, как правило, органические кислоты и основания, изменяющие свой Цвет при обратимом переносе протона. Рассмотрим механизм Действия индикаторов на примере п-нитрофенола.
Индикатор и-нитрофенол — кислота средней силы (рАа= = 7,2)
02N-<>-0H + Н20 =F=* 02n4”jH’ + Нз°* бесцветный желтый
25—2027
386
Неорганическая химия
Теория кислот и оснований
387
Молекула кислоты бесцветна, в то время как анион имеет ин тенсивно желтый цвет вследствие существенного изменения электронного состояния (делокализации электронов). В разбав ленном 0,02 М растворе присутствуют обе формы сопряженно! пары — кислота и основание. Присутствие анионов можно оп ределить по слабому желтому окрашиванию раствора. Пр!, добавлении нескольких капель 2 М НС1 анион-основание прак тически полностью переходит в бесцветный «-нитрофенол. Наоборот, при добавлении нескольких капель 2 М NaOH идет следующая протолитическая реакция:
02N-£>0H + ОН'=^= 02N^>-0- + Н2О
В результате образования нитрофенолят-иона раствор окрашивается в ярко-желтый цвет.
Для определения pH растворов применяют, как правило, двухцветные индикаторы. Изменение окраски индикатора происходит при pH, при котором концентрации сопряженных кислоты и основания равны. Применение закона действующи^ масс к протолитической реакции А + Н2О*^В + Н3О+ без учет, коэффициентов активности дает следующую приближенную формулу для вычисления pH:
PH = pKa-|-lg-|y- (22)
В точке изменения окраски <?в=са, следовательно,
рН = рКа (23)
Таким образом, показатель степени в константе индикатора-кислоты соответствует pH, при котором происходит изменение окраски индикатора. На практике цвет индикатора изменяется не сразу, а постепенно в некоторой области pH, которая определяется соотношением концентрации обеих форм индикатора Область изменения окраски распространяется на ~2 единиць pH. Применение набора индикаторов, отличающихся значения ми рКа, дает возможность определять pH любых растворов
В табл. В.6 указаны наиболее часто применяемые в лабора торной практике индикаторы. Чтобы найти приближенное зна чение pH раствора, применяют универсальную индикаторную бумагу, пропитанную смесью нескольких индикаторов, изменение окраски которых происходит в различных областях pH, благодаря чему можно охватить достаточно большой диапазон pH. Более точное определение можно провести при помощи бумаги, рассчитанной на более узкий диапазон pH. Здесь можно определить pH с точностью до 0,5 единицы. Еще более точные определения pH производят с помощью электрохимических ме тодов (разд. 39.6.6).
Таблица В.6. Области перехода pH наиболее употребительных индикаторов
Индикатор Область изменения окраски, pH Цвет
иидикатор-кислота индикатор основание
Тимоловый голубой Диметиловый желтый Детиловый оранжевый Метиловый красный Бромтимоловый голубой Фенолфталеин Тимолфталеин Ализариновый желтый R 1,2—2,8 2,9—4,1 3,1—4,4 4,4—6,2 6,2—7,6 8,0—9,8 9,3—10,5 10,0—12,1 Красный » » » Желтый Бесцветный » Светло-желтый Желтый » Оранжевый Желтый Голубой Красно-фиолетовый Голубой Коричневый
33.4.2.3. Реакции между протолитами средней силы
В том случае, если реагируют между собой кислота и основание средней силы, то не происходит полного перехода протонов от одного компонента к другому. Если, например, смешать эквивалентные количества уксусной кислоты и аммиака, то устанавливается протолитическое равновесие в соответствии с уравнением
СН3СООН + NH3 СН3СОО- + NH4+
и в полученной смеси остается по ~0,5% уксусной кислоты и аммиака. pH такого раствора ~7, так как сила обоих протолитов, как кислоты, так и основания, примерно одинакова.
33.4.2.4. Буферные растворы
Буферные растворы (или просто буферы) представляют собой такие растворы, которые содержат в определенном отношении слабую (или средней силы) кислоту и сопряженное основание. Эти растворы обладают очень важным свойством: в некотором интервале поддерживать постоянным pH раствора при его разбавлении или добавлении небольших количеств кислоты или основания. Каким же образом «работает» буфер? Пусть например, имеется аммиачный буфер, который состоит из эквивалентных количеств соли аммония и аммиака. В водном растворе для отдельных компонентов буфера устанавливаются равновесия:
NH3 + Н2О <—>: NH4+ + ОН~
NH4++H,0 NH3+H3O+
Обмен протонами как бы приостанавливается из-за наличия в Растворе сопряженных протолитов, так что концентрация ела-25»
388
Неорганическая химия
бой кислоты NH4+ и слабого основания NH3 почти не меняется. Если добавить в буферный раствор постороннюю сильную кислоту, то появившиеся в растворе протоны будут взаимодействовать с основанием NH3, вследствие чего их влияние на pH раствора окажется сравнительно небольшим. Подобным же образом при добавлении сильного основания с ним взаимодействует кислота NH4+. Таким образом, добавленные в раствор сильные кислоты и основания не могут реагировать с водой, образуя ионы Н3О+ или ОН~. Разумеется, изменение pH остается небольшим лишь до определенного предела, если количество добавленной кислоты или основания не слишком велико
Для pH буферной смеси в середине области буферного действия можно записать следующее приближенное выражение:
pH=p^+lg-^- (24)
где рЛа — относится к буферной кислоте, св — концентраци? буферного основания, с\—буферной кислоты. Для аммиачно го буфера NH4+/NH3 величина са соответствует количеству со ли аммония, отнесенному к объему раствора. Так как в отно шение концентраций в уравнение (24) входит один и тот же объем, при вычислении pH буферных растворов следует принимать во внимание только отношение количества кислоты и основания в растворе, выраженное в молях.
Оптимальное буферное действие имеет раствор, в котором кислота и основание находятся в эквимолярном отношении 1:1, так как при этом изменение pH минимально при добавлении как сильной кислоты, так и сильного основания. pH такой буферной смеси равен рКа буферной кислоты. Например, для эквимолярной смеси NH4+/NH3, исходя из рЛа кислоты NH4+, получим pH 9,25. Для приготовления других важных буферных смесей применяются следующие сопряженные пары кислота — основание: НАс/Ас- (pH 4,7), НгРС^/НРС^2- (pH 7,1) к НСО3_/СО32~- (pH 10,4). Важное значение имеют буферные свойства белков, содержащихся в крови; последние являются слабыми амфолитами и поддерживают pH крови в постоянных узких пределах 7,3—7,5.
33.4.2.5. Протолитические реакции в неводных протогенные растворителях
Так же как и вода, некоторые другие протогенные растворители образуют заряженные частицы в результате протеканиг реакций автопротолиза (разд. 34.1 и 34.3). На основе измере ний электропроводности и с помощью некоторых других физико-химических методов доказано существование следующи'
Теория кислот и оснований
389
равновесий в жидком аммиаке, жидком сероводороде и в безводной серной кислоте:
NH3 + NH3 <=> NH4+4-NH2-
H2S + H2S H3S+ 4- HS~
H2SO4 + H2SO4 H3SO4+ + HSO4~
Аналогично равновесию автопротолиза воды протонированные катионы являются кислотами, а анионы — основаниями.
Константа автопротолиза 100%-ной H2SO4 Kh,so4 =2,7-10~4; это довольно высокая величина. В этом растворителе НС1О4 ведет себя как слабая кислота:
H2SO4+HC1O4 h3so4+ + cio4-
На этом примере можно ясно представить себе, что кислотноосновные свойства вещества не являются его индивидуальным свойством, а определяются свойствами других участников протолитической реакции и растворителя. Теория Бренстеда далеко продвинула наши представления о кислотах и основаниях; она показала относительность понятий «нейтрализация», «кислота», «основание» и ввела количественные соотношения, в основном правильно отражающие реальное поведение рассмотренных выше систем.
33.4.3. Дальнейшее развитие теории кислот и оснований
33.4.3.1. Представления о ионотропии в кислотно-основных равновесиях
В более широком смысле обмен протонами представляет собой частный случай реакций с переносом ионов. Многие процессы в апротонных растворителях можно объяснить переносом ионов от одного участника реакции к другому. Поэтому полезно расширить понятия «кислота» и «основание» и на такие системы.
Соответствующие теоретические представления были развиты Гутманом и Линдквистом, а также Эбертом и Конопиком. Согласно их теории, все процессы переноса ионов между ингредиентами реакции можно объединить в понятие «ионотро-пия». Растворители, в которых происходит перенос катионов и перенос анионов, различаются между собой. В качестве ка-тчонотропных сольвосистем в настоящее время известны только системы, в которых происходит перенос протонов и поведение которых можно достаточно ясно представить себе с помощью теории Бренстеда. В химии апротонных растворителей, таких, например, как N2O4, SO2, BF3, РОС13, БЬВгз и ICI, можно отметить много общих свойств, которые можно объяснить обменом анионов, соответственно О2~, F-, С1~, Вг- или
390
Неорганическая химия
Теория кислот и оснований
391
I- . Поэтому эти растворители относятся к классу анионотроп.. ных сольвосистем.
Для ионотропных систем можно дать следующее определение кислоты и основания: кислота — это вещество, которое увеличивает концентрацию кислотного катиона в растворителе; основание — это вещество, которое увеличивает в растворителе концентрацию основного аниона. Эти определения составляют основу концепции сольвосистем (разд. 36.1). Рассмотрим под-робнее некоторые ионотропные реакции.
К оксидотропным системам относятся растворители N2O4 и SO2. Незначительная собственная электропроводность этих соединений связана с их диссоциацией в соответствии с уравнениями
N2O4 no++no3-
2 SO2 SO*++SO32-
Эти реакции сравнимы с равновесием автопротолиза воды и аммиака: 2Н3О Н3О++ОН“
2NH3 =f=fc NH4++NH2-
Как и в этих системах, образующиеся в N2O4 и SO2 ионы имеют сольватную оболочку из молекул соответствующего растворителя.
Ионам Н3О+ и NH4+ соответствуют катионы-кислоты NO+ н SO2+, а ионы NO3- и SO32~ следует рассматривать как основания. Точно так же как в системах Н2О и NH3) кислотные и основные частицы нейтрализуются в процессе обмена протонами, в системах N2O4 и SO2 кислотные и основные частицы обмениваются ионами кислорода.
Если, например, раствор NOC1 в жидком N2O4 смешать с твердым AgNO3, то ионы NO+ и NO3- реагируют с образованием N2O4:
NOC1 + [AgNO3] (тв.) --> [AgCl] (тв.) + N2O4
После отгонки растворителя остается соль — хлорид серебра.
Аналогичные реакции протекают также н в системах с растворителем SO2. Так, например, после внесения соединений тионила SOX2 или сульфитов M2SO3 в жидкий SO2 полученные растворы имеют очень высокую ^электро-проводность. Можно предположить, что в результате их взаимодействия с растворителем появляются катионы SO2+ и анионы SO32~. При смешении этих растворов тионил-катионы реагируют с анионами SO32- с образованием недиссоциированного SO2. За ходом реакции можно проследить, воспользовавшись методами кондуктометрии. Точка эквивалентности реакции
SOC12 + Cs2SO3 ---► 2 CsCl + 2 SO2
легко определяется по четкому излому на кривой зависимости электропроводности от количества добавленного вещества.
В качестве примера растворителя с переносом галогена можно назват трифторид брома. Это чрезвычайно реакционноспособное вещество (Ткип== = 126°C), которое имеет небольшую собственную электропроводность. Мо»' но предположить существование следующих равновесий:
BrF3 BrF2+ + F"
F" + BrF3 BrF4~_________
2BrF3 BrF2+ + BrF4~
кислота основание
При растворении KF или AgF в BrF3 получается хорошо проводящий раствор, в котором в избытке имеются анионы (основание):
KF+BrF3 K+[BrF4]-
При растворении SbF3 или SnF4 в BrF3 образуются кислотные катионы растворителя
SbF6 + BrF3 [BrF2]+[SbF6J-
а при смешении этих растворов образуется недиссоциированный растворитель
K+[BrF4J- + [BrF2]+[SbF6J- ---> K+[SbF6J- + 2 BrF3
После отгонки растворителя можно получить солеобразный продукт реакции высокой чистоты.
33.4.3.2. Реакции кислот и оснований в расплавах
Реакции ионного обмена происходят также в кислых и основных расплавах солей. По Люксу, у солей с анионами, содержащими кислород (Na2CO3, KNO3, K2S2O7), ионы кислорода основных частиц переносятся на кислотные, т. е. основания являются донорами О2-, а кислоты — их акцепторами. Для того чтобы подчеркнуть отличие от протонных кислот, у которых ярко выражена их способность отдавать ионы Н+, Бьеррум предложил для кислот — акцепторов О2- — название антиоснование.
Так же как и для других донорно-акцепторных систем, можно представить себе сопряженные пары основание — антиоснование, которые переходят друг в друга в результате присоединения и отщепления ионов О2-.
Основание Антиоснование.
2SiO44- Si2O/-+ О2-
2РО4з- ь Р2О/-+О2-
2SO42- S2O72-+O2-
2NOS- N2O5 + O2- (?==* 2КОг + 1/2О2)
CO32- CO2 + o2-
При дальнейшем отщеплении ионов О2“ от дисиликат- и пирофосфат-ионов происходит их дальнейшая конденсация. Разумеется, этот процесс возможен лишь при наличии в расплаве акцепторов ионов О2“.
Например, при плавлении соли NaNH4HPO4 кислород удаляется (Н2О): n[NH4HPO4]~---------> nNH3 + (« - I) Н2О + [PzAn-i (ОН)2р-
Образовавшиеся полифосфат-анионы вследствие высокого заряда присоединяют катионы — частично с образованием ковалентной связи. При этом расплав так называемой фосфорной пробы окрашивается в цвета, характерные Для присоединившегося катиона (разд. 37.2.1.2). Такого рода реакции протекают также в реакции образования перла буры.
Прн сплавлении кристаллических оксидов (например, TiO2, Fe2O3, AI2O3 и Подобных нм соединений с прочной кристаллической решеткой) с веществами типа персульфатов и карбонатов схему взаимодействия можно себе предъявить следующим образом. В решетке оксидов положительные ионы раскатаются в тетраэдрических или октаэдрических пустотах плотноупакован-Ь’х ионов кислорода. Атомы или катионы освобождаются от этих оболочек, ли сильные антиоснования связывают ионы кислорода, входящие в их со-
(А12О3) (ТВ.) + 3 S2o,2- Ч=± 2 А13+ + 6 SO42-
392 Неорганическая химия
Теория кислот и оснований 393
Водный раствор плава содержит гидратированные ионы А13+ и сульфат-ионь’ Разрушение оксидов амфотерных металлов происходит также при Ц' сплавлении с основаниями, причем после частичного разрыва связи металл--кислород в кристаллической решетке происходит образование комплексно, 3 оксо-иона:
(А12О3) (тв.) + СО32“ =ё=> 2 А1О2- + CO2f
При растворении плава в воде образуются гидроксоалюминий-ионы.
При сплавлении полевого шпата с карбонатами происходит следующая реакция:
[AlSi3O8]- (тв.) + 6 СО32“ А1О2- + 3 SiO44- + 6 CO2f
Пространственная решетка силиката, в которой каждый четвертый атом кремния изоморфно замещен на атом алюминия, разрушается в результате этой реакции с образованием диоксоалюминат- и ортосиликат-ионов.
Сила оснований и антиоснований в реакциях, протекающих в расплавах, увеличивается в том случае, если вещество, входящее в сопряженную пару основание — антиоснование, — газ. Так, например, СО32~ является особенно сильным основанием, поскольку антиоснование СО3 удаляется из система в виде газа.
33.4.3.3. Теория кислот и оснований Льюиса
Одновременно с Бренстедом и Лаури со своей концепцией кислот и оснований выступил Льюис. Он предположил, что поведение кислот и оснований определяется следующими четырьмя многократно проверенными на практике критериями:
образование продуктов нейтрализации кислоты и основания происходит очень быстро;
кислота (основание) вытесняет более слабую кислоту (основание) из их соединений;
кислоты титруются основаниями с применением цветных индикаторов;
кислоты и основания катализируют многие химические реакции.
Льюис связал эти характерные свойства кислот и оснований с их электронной структурой, в особенности с парой электронов, образующих координационную ковалентную связь, и предложил следующее определение кислоты и основания:
кислоты — это вещества, которые при образовании ковалентной связи принимают пару электронов (являются акцептором пары электронов);
основания — это вещества, которые при образовании ковалентной связи отдают пару электронов (являются донорами пары электронов).
Выбор электронной конфигурации в качестве фундаментального критерия для обоснования понятий «кислота» и «основание» дает возможность применить их для более широкого класса веществ. По типу кислот Льюиса реагируют, например молекулы с незаполненной восьмиэлектронной конфигура цией (BF3, SO3);
катионы — центральные атомы комплексных соединений (Fe3+, Со2+);
галогениды с ненасыщенными координационными связями (TiCl4, SbF5);
молекулы с поляризованными двойными связями, в которых более положительный атом обусловливает проявление веществом кислотных свойств (СО2, SO2).
По типу оснований Льюиса реагируют, например,
молекулы с вытянутыми электронными орбиталями (NH3, Н2О);
анионы — лиганды в комплексных соединениях;
органические соединения с двойными и тройными связями (СН2=СН2); олефины и ароматические соединения, например, вступают в кислотно-основные реакции с кислотами Льюиса BF3, А1Вгз и Ag+ с образованием л-комплексов.
Таким образом, в соответствии с представлениями Льюиса можно рассматривать следующие реакции:
Кислота Основание Продукты реакции
BF3 + NH3 --► F3BNH3
Ag+ + 2NH3 --► [Ag(NH3)2]+
TiCl4 + 2 Cl- --► [TiCla]2-
o=c=o + OH---»- O=C—O-
4 I
1 OH
более гидрокарбоиат
положительный
атом
Несмотря на тот вклад, который теория Льюиса внесла в представления о кислотах и основаниях благодаря единому подходу к явлениям кислотно-основного равновесия, надо отметить и некоторые ее недостатки.
Во-первых, важнейшие протонные кислоты, такие, например, как НС1 и H2SO4, не находят себе места в теории Льюиса, так как не содержат ковалентных связей, хотя их химическое поведение является основой для построения всех теорий кислот и оснований. Поэтому они занимают совершенно особое место в теории. Другим недостатком теории Льюиса является трудность количественного измерения относительной силы кислот и оснований, что легко доступно с помощью теории Брен-стеда. Поэтому теория Льюиса не позволяет предсказать положение равновесия реакции и ее направление. Это можно объяснить тем, что более широкое обобщение, предложенное Льюисом, не позволяет количественно оценить некоторые важ-нЫе факторы поведения кислот и оснований.
Имеются также возражения против предложенных Льюисом экспериментальных критериев. Высокая скорость реакций кислот и оснований наблюдается в основном для реакций об
394
Неорганическая химия
Теория кислот и оснований
395
мена протона в отличие от реакций образования комплексных соединений, имеющих значительно более сложный кинетический механизм. Каталитическая активность кислот Льюиса также оспаривается. В отдельных случаях каталитическая активность связана с наличием в реакционной системе незначительного ко-личества протонных кислот. В других случаях надежно доказано, что ускорение реакции происходит благодаря присутствию кислот Льюиса. Вообще говоря, каталитическое действие кислот обоих типов имеет различный механизм.
33.4.3.4. Представления о жестких и мягких кислотах Пирсона
В 1963 г. теория кислот и оснований, основанная на представлениях об электронном взаимодействии, была существенно развита и дополнена Пирсоном. Как и Льюис, Пирсон рассматривает в качестве основного процесса кислотно-основного равновесия взаимодействия акцептора пары электронов А (кислоты) с донором пары электронов В (основанием) с образованием стабильного кислотно-основного комплекса АВ:
А+ :В <—> А—В
Однако, в то время как Льюис считал самым важным при образовании комплекса появление ковалентной связи, Пирсон включил в рассмотрение и другие типы взаимодействия между электрофильными и нуклеофильными частицами, в том числе те, которые приводят частично или полностью к электростатической (ионной) связи. Таким образом, к кислотно-основным реакциям относятся, например, реакции образования комплексных катионов и анионов, а также формирование кристаллической решетки солей. Примеры, приведенные в табл. В.7, поясняют возможности применения представлений Пирсона.
В теории Пирсона рассматривается вопрос о том, какие свойства кислоты А и основания В обеспечивают термодинами-Таблица В.7. Важнейшие кислотно-основные реакции по модели Пирсона
Тип реакции Кислота Основание Кислотно-основный комплекс
Перенос протона Образование АПЭ-комплекса Образование соли Образование катионного комплекса Образование анионного комплекса Образование л-акцепторного комплекса НС1 BF3 Ag+ Cu2+ SnCl4 {Ni} NH3 or2 I-4NH3 2C1- 4CO {NH4+C1-} F3B : OR2 {Ag+I-} [Cu(NH3)4]2+ [SnClep- Ni(CO)4
ческую стабильность образовавшегося комплекса АВ. Теория предполагает, что в качественном отношении эта стабильность определяется жесткостью и мягкостью участников реакции. При этом постулируется правило:
жесткие кислоты комбинируются преимущественно с жесткими основаниями;
мягкие кислоты комбинируются преимущественно с мягкими основаниями.
Классификация кислот и оснований. Жесткими частицами называют молекулы с реакционноспособными центрами (атомами или группами атомов), обладающими прочной малоде-формируемой электронной структурой. Это могут быть атомы элементов с высокой электроотрицательностью (F, О, N) или катионы с большим зарядом. Напротив, мягкие частицы имеют подвижную деформируемую электронную структуру и высокую поляризуемость.
Таблица В.8. Распределение кислот и оснований по Пирсону
Кислоты
Жесткие Средние Мягкие
H+, Li+, Na+, K+, Mg2+, AJ3+, Las+, Si4+, Ti4+, As3+, Mn2+, Crs+, Cr6+, Cos+, Fe3+, Cl’+, I’+, BF3, BC13, A1H3, Be(CH3)2, RCO+, SO3, CO2 Sn2+, Pb2+, Sb3+, Bis+, Fe3+, Co2+, Ni2+, Cu2+, Zn2+, Ru2\ Rh3+, Ir»+, B(CH3)3, NO+, R3C+ Cs+, Cu+, Ag+, Au+, T1+, Hg+, Hg2+, CH3Hg+, Pt2+, Cd2+, Aus+, Tl3+, Cr®, Fe®, Mn1-, Cr1-, I+, I2, ICN, BH3, cbo бодные радикалы (Cl, Вг, I, 0)
Основания
Жесткие Мягкие
F~, ОН’, О2-, Н20, R20, ROH, OR", NH3, N2H4, RNH2, CH3COO-, CO32-, PO43-, SO42-, N03", C104- ВГ, Г, CN", SCN", H-, R", RS", RSH, R2S, R3P, R3As, (RO)3P, CO, RNC, олефины, ароматические соединения
Частицы — доноры и акцепторы электронов — можно распределить по степени жесткости и мягкости в зависимости от свойств их электронной системы и реакционной способности.
396
Неорганическая химия
Теория кислот и оснований
397
(табл. В.8). Некоторые частицы занимают промежуточное положение. Поясним общие принципы классификации на отдельных примерах.
Жесткие основания. К ним относятся прежде всего анионы сильноэлектроотрицательных элементов, такие, как F-, О2~, ОН-. Вследствие прочной и устойчивой электронной оболочки, а также соответствующего строения электронных орбиталей эти ионы не имеют склонности к образованию ковалентных связей с катионами. Рассматривая реакционную способность воды как донора пары электронов, можно отметить, что, например, при гидратации катионов, кислород молекулы воды как раз является жестким центром. Относительно высокая электроотрицательность атомов азота — причина того, что азотные основания (NH3, N2H4 и их замещенные производные) являются жесткими основаниями. Анионы кислородсодержащих кислот, таких, как С1О4~, SO42-, РО43-, СО32-, также имеют малодеформируемую структуру.
Мягкие основания. Среди наиболее поляризуемых частиц можно назвать анионы с большими ионными радиусами: Р3-, S2-, Se2-, I-, Вг~. Ион С1~ занимает промежуточное положение между мягкими и жесткими основаниями. Вследствие того что гидрид-ион Н- также имеет большую легко деформируемую электронную оболочку, его также можно отнести к мягким основаниям. Малая электроотрицательность углерода — причина того, что его производные типа СН3_ и CN~ относятся к мягким основаниям. Часто в донорных атомах мягких оснований имеются незанятые орбитали с низкой энергией, которые могут быть использованы для образования дативной связи (обратного связывания). Вследствие высокой поляризуемости л-элект-ронных систем моноксид углерода, а также ароматические и непредельные соединения также можно отнести к классу мягких оснований.
Жесткие кислоты. Электронная оболочка жестких кислот характеризуется высокой стабильностью относительно внешних электрических полей. Наиболее жесткой кислотой является протон, который из-за отсутствия электронной оболочки и чрезвычайно малого радиуса прочно связывается с активным^ центром молекулы основания. Недеформируемой электронной обо^ лочкой обладают также катионы с электронной конфигурацией инертного газа, такие как Li+, Са2+, Al3+, Ti4+, в которых электрические и магнитные моменты всех электронов полностью скомпенсированы. Эти катионы образованы в основном элементами главных подгрупп периодической системы. К последним близки по свойствам некоторые катионы переходных металлов с не полностью занятой, d-оболочкой, например Мп и Fe3+. Способность к присоединению оснований возрастает по мере увеличения ионного потенциала. Кроме того, к жестким
кислотам относится большая группа соединений с электрофильными центрами, как, например, тригалогениды и триалкилы бора и алюминия (это соединения с дефицитом электронов*). Относительно жесткий характер имеют также не содержащие металла группы, как, например, ацил-катион сн3со+.
Мягкие кислоты — большие катионы с деформируемой электронной оболочкой (элементы главных подгрупп периодической системы, например Cs+, Т1+), а также катионы переходных металлов, в электронной оболочке которых имеются d-электроны (например, Cu+, Hg2+). Электронные оболочки d-электронов имеют сравнительно большой радиус, в результате чего становится возможным их взаимодействие с электронными оболочками лигандов-оснований. Мягкость соединений увеличивается по мере уменьшения положительного заряда иона. Поэтому наиболее мягкими являются соединения, в которых атом металла не имеет заряда или даже заряжен отрицательно. К мягким кислотам относятся также катионы неметаллов (I+, Вг+), электрофильные молекулы с деформируемой электронной оболочкой (Ь, IC1), а также реакционноспособные атомы и свободные радикалы (О, CI, Вг).
Ионы с конфигурацией электронных оболочек ns2, как, например, Pb2+, Sn2+ и Sb3+, занимают промежуточное положение между жесткими и мягкими кислотами.
Применение теории жестких и мягких кислот и оснований. Теория жестких и мягких кислот и оснований оказалась во многих отношениях полезной. Она позволяет качественно предсказать наиболее стабильные продукты реакции между электрофильными и нуклеофильными соединениями, т. е. оценить положение равновесия реакций, для которых не имеется достаточно точных термодинамических характеристик ввиду сложности их определения. Основную роль в теории играет уже рассмотренное выше правило о том, что предпочтительными являются комбинации жесткая кислота — жесткое основание и мягкая кислота — мягкое основание. Этот эффект упрочнения связи между участниками реакции одинаковой степени жесткости назван Йоргенсеном «симбиозом».
Поясним эти соображения на примере. При взаимодействии мягкой кислоты ВН3 (радикал, образовавшийся при термическом разложении диборана) с мягким основанием СО возникает устойчивое соединение ВН3-СО, в то время как при взаимодействии ВН3 с жесткими основаниями (аминами, эфирами) стабильный аддукт не образуется. В то же время жесткая Кислота BF3 легко соединяется с жестким основанием с обра-
* В соединениях с дефицитом электронов количество электронов в молекуле меньше, чем можно было бы ожидать, исходя из числа валентных связей.
398
Неорганическая химия
зованием таких соединений, как ВРз-ОИг или BF3-NR3. Аддукт BF3-CO (комбинация жесткая — мягкое) неустойчив.
Пользуясь теми же соображениями, можно предсказать продукты обменных реакций между солями. Например,
Lil + AgF <—*: LiF+ Agl
В результате реакции, протекающей в растворе или в твердой фазе, образуются более стабильные соли (жесткая кислота — жесткое основание LiF и мягкое основание — мягкая кислота Agl).
Теория жестких и мягких кислот и оснований объясняет также различия в способности галогенид-ионов образовывать координационные соединения с катионами различной степени жесткости. С катионом А13+ (жесткая кислота) последовательность имеет следующий вид (в порядке убывания стабильности): F-, С1_>Вг~>1_; с катионом Hg2+ (мягкой кислотой) более стабильные соединения получаются в обратной последовательности.
Становится также понятным, почему происходит стабилизация металлов с высокой степенью окисления жесткими основаниями типа F-, ОН-, О2-. Это объясняется тем, что центральные ионы металла с большим зарядом представляют собой жесткие кислоты. Напротив, мягкие кислоты — ионы с малым зарядом — стабилизируются мягкими основаниями, например CN-, I- и СО (разд. 36.14.2).
Теория кислот и оснований Пирсона хорошо интерпретирует также реакции между акцепторами л-электронов (СО, R = N = C, NO, замещенные фосфины) и переходными металлами (соответствующими ионами). Типичным примером является образование карбонильного соединения никеля:
{Ni} + 4СО <—> Ni(CO)4
Атомы никеля в кристаллической решетке не заряжены и являются мягкими кислотными центрами, к которым присоединяются мягкие основные молекулы СО, имеющие свободную пару электронов. Четыре о-связи между центральными атомом никеля и атомами углерода молекул СО усиливаются л-датив-ными связями, причем электроны заселенных d-орбиталей никеля принимают участие в образовании этой связи за счет незаселенных л-орбиталей атома углерода. Тем самым высокая электронная плотность нейтрального атома металла, которая особенно велика для атомов с низкой степенью окисления, перераспределяется на лиганды, в результате чего достигается более равномерное распределение электронной плотности по всей молекуле.
Стабильными соединениями в комбинации мягкая кислота — мягкое основание являются также различные л-комплеК-
Теория кислот и оснований
399
сы В этих соединениях донором электронов являются л-элект-ронные системы ароматических или непредельных соединений, а в результате реакции с веществом-кислотой также образуется л-связь. Типичные представители таких соединений — бис (бензол) хром (С6Н6)2Сг, а также бис(л-циклопентадие-нил)железо ((л-С5Н5)2Ре — ферроцен), которые имеют структуру сандвича или двойной кегли; к комплексам непредельных соединений относится и соль Цейзе К[С2Н4Р1С1з]1. Структура этих соединений изображена на рис. В.12.
Рис В 12. Структура л-комплексов и диметилсульфоксида.
В перечисленных выше л-комплексах мягкими основными компонентами являются ароматические или непредельные лиганды, а мягкой кислотой — атом d-металла в низкой степени окисления. За счет занятой л-орбитали лиганда и свободной гибридной орбитали d-металла образуется донорная связь о-типа. В этих комплексах выравнивание электронной плотности и упрочнение связи также достигается за счет образования дативной связи.
Некоторые молекулы имеют как жесткие, так и мягкие центры. Так, в диметил сульф оксиде (рис. В.12) атом кислорода вследствие высокой электронной плотности проявляет жесткие свойства, а атом серы вследствие особенностей его электронной структуры является мягким основным центром. Соответственно жесткие кислоты, как, например, протон, прочно связываются с атомом кислорода. Мягкие кислоты, например соли платины и палладия, образуют прочные координационные соединения с серой.
Попытки теоретической интерпретации теории жестких и мягких кислот « оснований. С помощью этой теории возможны качественные предсказания и Объяснения для реакций между нуклеофильными и электрофильными соединениями, а также оценка стабильности образовавшихся веществ. Поскольку ход реакции и стабильность связей зависят от целого ряда факторов, количественная трактовка всех этих факторов возможна только с определенной степенью приближения. Однако, несмотря на эти ограничения, можно представить себе основные принципы теории Пирсона с помощью известных Моделей химической связи.
400
Неорганическая химия
Сильное взаимодействие между жесткими кислотами и жесткими основаниями приводит преимущественно к образованию ионной связи. Она оказывается тем прочнее, чем меньше радиус ионов и выше их заряд. Напротив мягкие соединения образуют в основном ковалентные связи. Комбинаций между жесткими н мягкими соединениями неустойчивы прежде всего потому, что они имеют тенденцию образовывать связи различного типа.
Упрочнение связи при комбинации мягких соединений во многих случаях объясняется образованием дативной связи. После образования донорной связи за счет пары электронов основания л-электроны кислоты-акцептора заселяют незаполненные орбитали основания-донора. Такое поведение наиболее характерно для типичных мягких кислот и оснований.
Другая возможность стабилизации мягких кислот участников реакции осуществляется за счет дисперсионного взаимодействия, которое пропорционально произведению поляризуемости участников реакции.
Количественное сравнение жесткости и мягкости кислот и оснований. С момента создания теории Пирсона предпринимались многочисленные попытки количественного определения «жесткости» и «мягкости» соединений, а также составление последовательности соединений по мере увеличения (уменьшения) их количественных характеристик. Примером количественного подхода является нахождение константы равновесия обменной реакции типа
А' + АВ А'В + А
В' + АВ т=->: АВ' + В
относительно принятого за основу вещества сравнения АВ для различных кислот А' и оснований В'. Реакции идут слева направо, если А' — более сильная кислота, чем А, а В' — более сильное основание, чем В.
При сравнении оснований в качестве вещества сравнения наиболее пригоден гидроксид метилртути(1) CH3HgOH или соответствующий аквакомплекс [CH3Hg(H2O)]+. Эти вещества представляют собой реакционноспособные комплексы очень мягкой кислоты CH3Hg+ и жестких оснований ОН~ и Н2О. Чтобы сравнивать экспериментальные данные для различных обменных реакций в воде с указанными веществами, запишем уравнение реакции в обобщенном виде:
CH3Hg(H2O)++ НВ+ CH3HgB++ Н3О+
Константа равновесия этой реакции К=Кобр-Ка, где Кобр — константа обра зования соединения CH3HgB+ по реакции CH3Hg+ + B^=CH3HgB+, а Ка~ константа кислоты НВ+, сопряженной с основанием В. Равновесие реакцш тем сильнее смещается направо, чем мягче основание В. В качестве мерь жесткости (мягкости) основания В можно использовать логарифм константы равновесия К (табл. В.9).
Кроме того, Пирсон предложил следующее выражение для константы кислотно-основного равновесия образования комплекса А+В^В:
lgtf = SASB + aAaB (25)
где Sa и Sb характеризуют силу кислоты А и основания В в реакциях обме на протона (например, логарифм констант кислоты и основания). Жесткость и мягкость соединений А и В учитывают члены Оа и ав- У жестких кисло, и оснований а имеет положительное значение, а у мягких — отрицательное Таким образом, комбинации «жесткая — жесткое» и «мягкая — мягкое» соответствуют увеличению Оа и <тв, а следовательно, и константы равновесия
Однако, несмотря на то что были проведены исследования многих кис лотно-основных реакций, оказалось невозможным установить однозначнук последовательность изменения жесткости (мягкости) кислот и оснований. Прь изменении вещества сравнения константы равновесия менялись, а иногда происходило изменение даже направления найденной ранее последовательности. Вообще говоря, оказалось, что результаты измерений сравнимы только
Теория кислот и оснований
401
Таблица В.9. Константа равновесия (рК) обменной реакции
СНзНё(Н2О) ++НВ+ч^СНзН2В++НзО+ с основаниями различной жесткости (мягкости) в водном растворе при 25 °C
Основание В 1g К Основание В 1g К
он- —6,3 Р (С2Нг)з 6,2
NH3 —1,8 SCN- 6,7
НРО42- —1,8 s2- 7,0
НРОз2- —1,6 а- 12,3
F- —1,4 Вг- 15,6
SO32- 1,3 I- 18,1
CN- 5.0 СНз- 30
при условии проведения реакции в одном и том же растворителе. Оценка всех имеющихся экспериментальных материалов показывает, что жесткость донорных атомом уменьшается в ряду F>O>N>Cl>Br>I>S~P~C; в этом же ряду увеличивается мягкость соответствующих атомов.
Клопманом была предпринята попытка при помощи квантовой механики рассчитать жесткость и мягкость ионов. Исходными данными для расчетов послужили энергии внешних электронных орбиталей. Для оснований в качестве внешней орбитали донорного атома была принята заселенная орбиталь с наибольшей энергией, для кислот — незаселенная орбиталь атома-акцептора с минимальной энергией. В том случае, если разность энергий этих орбита-лей достаточно велика, при образовании комплекса кислота — основание электронный переход не происходит, что соответствует случаю «жесткая кислота — жесткое основание». Взаимодействие атомов осуществляется только посредством взаимодействия их зарядов — возникает ионная связь. Наоборот, если энергии внешних орбиталей примерно одного порядка, то становится возможным электронный обмен с образованием ковалентной связи, что соответствует комбинации мягкая кислота — мягкое основание.
Используя данные по энергии ионизации, сродства к электрону, ионные радиусы и энергию гидратации, Клопман рассчитал для ряда катионов и анионов энергии внешних орбиталей; распределение этих иоиов по мере убывания энергии поразительно хорошо совпадает с ходом изменения степени Жесткости (мягкости) ионов в водной среде (табл. В 10). Приведенные в таблице данные следует сравнивать отдельно в ряду катионов и анионов. Для катионов жесткие кислоты имеют положительное значение энергии Е, мягкие кислоты — отрицательное. Это распределение в основном согласуется с активностью соответствующих соединений в реакциях. Единственным исключением является протон, который представляет собой более жесткую кислоту, чем это следует из данных табл. В. 10. В то же время теория верно предсказывает, что Т13+ — более мягкий ион, чем Т1+. Причиной этого является /^-конфигурация электронов Т1+ (наличие инертной пары электронов). В последовательности анионов энергия Е имеет только отрицательное значение (около —10 эВ). Область энергии около 10 эВ является границей между Жесткими и мягкими соединениями.
Термодинамический подход к теории Пирсона. Особый интерес представляют результаты, полученные Арландом при изучении равновесий, соответствующих образованию галогенидиых и цианидных комплексов с катионами главных и побочных групп периодической системы. В соответствии с законами термодинамики отрицательное значение свободной энтальпии реакции ДО = ДЯ—TAS отвечает образованию устойчивого комплекса. Изучение экспериментальных данных показывает, что при образовании соединений в комбинации жесткая кислота — жесткое основание ДО<0 вследствие увеличения энтропии при образовании комплекса. Так, например, образование фторид-
26-2027
-402
Неорганическая химия.
Таблица В.10. Энергия внешней орбитали акцепторов и доноров электрона как характеристика жесткости и мягкости катионов и анионов
Катион йак,ц эВ Анион £дон' эВ
А13+ 6,01 F- —12,18
Са2+ 2,33 он- — 10,45
Fe2+ 0,69 а- — 9,94
Н+ 0,42 Вг- — 9,22
Na+ 0,0 CN- — 8,78
Т1+ . —1,88 SH- — 8,59
Т13+ —3,37 I- — 8,31
Hg2+ —4,64 н- — 7,37
ных комплексов с жесткими трехзарядными катионами (AF+, Fe3+ и др.) в воде связано с увеличением энтальпии (ДЯ>0), так как разрушение гидратной оболочки катионов и анионов требует больших затрат энергии, чем -ее выделяется при образовании ионной связи. В то же время освободившиеся из гидратной оболочки молекулы получают дополнительные вращательные и колебательные степени свободы, т, е. получают возможность дополнительного накопления энергии, что приводит к увеличению энтропии AS>0. Отрицательный член 7AS оказывается по абсолютной величине больше, чем АЯ, вследствие чего изменение свободной энтальпии становится отрицательным: AG<0.
При взаимодействии мягких анионов (например, Вг-, I-, CN-) с типично мягкими катионами (например, Cd2+, Hg2+, Pd2+) термодинамические соотношения становятся совсем другими. Распад сравнительно рыхлой гидратной оболочки вокруг ионов не требует больших затрат энергии; наоборот, образование ковалентных связей сопровождается значительным выделением энергии, поэтому АЯ<0. Изменение энтропии, напротив, невелико. Увеличение энтропии в результате отщепления гидратной воды почти полностью компенсируется уменьшением количества частиц в системе при образовании комплекса катион—анион.
33.4.3.5. Теория кислот и оснований Усановича
Наиболее общая теория кислот и оснований, включающая в себя и другие теории, предложена Усановичем. Согласно его определению,
кислоты — это вещества, отдающие катионы или принимающие анионы (соответственно электроны);
основания — это вещества, отдающие анионы (или электроны) и принимающие катионы.
На основе такого определения можно предсказать и интерпретировать протекание многих реакций. В качестве примера рассмотрим реакцию взаимодействия между триметиламином и метилиодидом:
N(CH3)3 + CH3I -> [N(CH3)4]++ I-
Здесь метилиодид — кислота, отдающая катион СНз+, а три-метиламин — основание, его присоединяющее. В результате реакции образуется соль тетраметиламмонийиодид.
Теория кислот и оснований
40»
Согласно теории Усановича, кислотно-основные процессы протекают также между ангидридами кислот и оснований, например
NaaO + SO3 --► 2Na+ + SO4a"
и при образовании комплексных соединений, например Sb2S5 + 3Sa----------------------->- 2SbS4s-
Теория Усановича включает также реакции с электронным переносом и поэтому рассматривает окислительно-восстановительные реакции как частный случай реакций между кислотами и основаниями. Действительно, внутреннюю связь между реакциями этих типов можно увидеть при сравнении процесса нейтрализации основания кислотой и растворения металла сильной кислотой. Уравнения соответствующих реакций можно записать следующим образом:
Са(ОН)2 + 2 НС1 --► СаС12 + 2Н2О
Са (тв.) + 2 НС1 -»- СаС12 + Н2
Как в одной, так и в другой реакции кислота исчезает и образуется раствор соли.
Большое значение Усанович придает координационно-ненасыщенному характеру центрального атома. Для положительно' заряженного иона его кислотные свойства проявляются в присоединении анионов. Для отрицательно заряженных ионов характерны основные свойства, выражающиеся в присоединении катионов. Для значительного числа соединений, в которых имеются как положительные, так и отрицательные координационно-ненасыщенные ионы, характерны амфотерные свойства.
Взяв за основу электростатическую модель строения вещества, теория предложила количественный метод оценки силы кислоты и основания как функции ионного потенциала. Если в каком-либо соединении, в состав которого входит водород, Центральный ион имеет положительный заряд, то кислотность увеличивается по мере повышения заряда и уменьшения радиуса иона. При отрицательном заряде иона с увеличением потенциала иона кислотность снижается, а основность увеличивается. В зависимости от того, потенциал какого иона преобладает в амфотерном соединении, вещество обладает либо кислотными, либо основными свойствами.
Теории кислот и оснований Льюиса и Усановича отличаются выбором модели связи между участниками реакции. Кислотно-основные реакции по Льюису осуществляются преимущественно за счет образования и разрыва ковалентной связи, что Дает возможность с успехом применять ее в органической химии. Кислотно-основное равновесие гетерополярных соединений, составляющих, как правило, предмет неорганической хи
26*
404
Неорганическая химия
мии, удобнее интерпретировать с помощью теории Усановича. -Однако большинство соединений имеет промежуточный характер, поэтому можно надеяться, что в будущем появится теория, в которой удастся объединить обе точки зрения на поведение кислот и оснований.
33.4.4. Связь между структурой и силой кислот
Для соединений с гидроксильными группами и аквакомплексов металлов теория Усановича устанавливает четкую связь между потенциалом центрального и положительного ионов и кислотностью. Сила кислоты увеличивается по мере увеличения силы, отталкивающей атом водорода в группе ОН от положительного заряда. При этом наблюдаются следующие закономерности изменения силы кислоты в зависимости от места, которое центральный атом занимает в периодической системе элементов:
в пределах одного периода периодической системы элементов сила кислоты растет с увеличением заряда и уменьшением радиуса центрального иона, например
H4SiO4 < Н3РО4 < H2SO4 < НС1О4
в пределах одной группы периодической системы элементов сила кислоты уменьшается по мере увеличения радиуса центрального иона, например
HNO3 > Н3РО4 > H3AsO4
[Mg(H2O)e]2+ > [Ba(H2O)xl2+
Сила различных кислот одного и того же элемента увеличивается по мере увеличения степени окисления центрального иона, что согласуется с использованной в теории моделью и объясняется повышением заряда и уменьшением радиуса центрального иона, например
НСЮ < НС1О2 < НС1О3 < НС1О4
Сила многоосновных кислот уменьшается по мере отщепления ионов водорода. Это легко понять из рассмотрения электростатического взаимодействия в молекуле кислоты. Энергия отщепления протона становится тем больше, чем более отрицателен заряд частицы — аниона. В то же время все гидроксильные группы в молекуле кислоты проявляют одинаковую способность к отщеплению протона.
С оценками на основе упрощенной модели следует обращаться осторожно; это можно показать на примере бинарных соединений с водородом. В табл. В.11 представлены округленные значения рКо этих соединении. рКа для НС1, НВг, HI ие могут быть измерены в водных растворах вследствие взаимодействия с растворителем —водой. Приведенные данные относятся
Теория кислот и оснований
405
Таблица В.11. рК.а бинарных гидридов
Гидрид PKa Гидрид PKa Гидрид
NH3 23 H2O 16 HF 3
H2S 7 HC1 — 7
H2Se 4 HBr — 9
H2Te 3 HI —10
к менее основному растворителю, в котором возможна дифференциация кислотности этих трех кислот (разд 34.3).
Изучение данных в табл. В 11 позволяет выявить ряд закономерностей. Распределение кислот по их силе в ряду NH3-•-НгО-•-HF следует общему принципу, согласно которому с увеличением электроотрицательности полярность связи Н-Х возрастает, а следовательно, облегчается отщепление протона. Однако в ряду Н2О-- Н2Те и HF---HI наблюдается противоположная закономерность: если бы главную роль играла электроотрицательность, то наиболее сильными кислотами были бы первые члены ряда: Н2О и HF.
Более точный анализ причин различного кислотно-основного поведения галогеноводородов основывается на термодинамической трактовке протолитического процесса. Термодинамически кислотно-основная реакция галогеноиодорода в водной среде соответствует изменению состояния системы при переходе гидратированных молекул галогеиоводорода НХ в гидратированные иоиы:
НХ (aq) ---► H+(aq) + X'(aq)
Непосредственно измерить изменение энтальпии этого процесса невозможно, одиако ее можно вычислить, воспользовавшись термодинамическим циклом, для которого известны изменения энтальпии для каждой стадии. Как уже указано ранее, изменение термодинамического потенциала (в том числе энтальпии) не зависит от пути протекания процесса, а только от начального и конечного состояний системы.
Проведем следующий мысленный эксперимент: переведем молекулы HX(aq) в безводную форму и разделим ее на атомы. Далее проведем ионизацию атомов галогена и водорода, наконец переведем ионы в гидратирован-яую форму:
НХ (aq)
НХ (газ)
Н (газ)
X (газ) + е~
Н+ (газ)
X" (газ)
—энтальпия дегидратации
D—энтальпия диссоциации
/—энтальпия ионизации
Е—сродство к электрону
ДЯ2—энтальпия гидратации протона
Д//3—энтальпия гидратации аниона
НХ (газ)
Н (газ) + X (газ) Н+ (газ) + е~ X* (газ) H+(aq) X- (aq)
НХ (aq) ----► H+(aq) + X-(aq)
Изменение энтальпии протолитической реакции в стандартных условиях:
Д/Д = д#! + D + 1 - Е + Д//2 + ДЯ3
Изменения энтальпии стадий указанного процесса для некоторых галогеноводородов представлены в табл. В. 12. В ней же даны изменения энтропии AS0 суммарного процесса:
AS0 == Sh+ (aq) + Sx- (aq) — ^HX (aq)
406
Неорганическая химия
Таблица В.12, Энтальпия и энтропия протолиза галогеиоводородов в водных растворах
Галогеио-водород ДНа. кДж/моль D, кДж/моль I, кДж/моль Е, кДж/моль ДЯ2+ДЯ3, кДж/моль ДЗО, кДж/моль
HF 48,1 563,5 1318,8 344,2 —1598,9 —87,1
НС1 17,6 432,0 1318,8 365,5 —1460,4 -56,1
НВг 20,9 366,3 1318,8 343,3 —1426,4 —38,1
HI 23,0 299,4 1318,8 316,9 —1382,9 — 13,4
Изменение свободной энтальпии
ДО» = ДЯ» — TAS0 (Т = 298 К) (26)
а константу равновесия можно вычислить из уравнения
ДО» = —RT In На = 5,711 -рКа (ДО0 в кДж/моль) (27)
Данные по ДО», ДЯ», ТД8» и вычисленные из них значения рКа приведены в табл. В.13. Как видно из таблицы, рассчитанные и экспериментально найденные величины рКа близки между собой.
Таблица В.13. Рассчитанные и экспериментальные значения рКа галогеиоводородов в водных растворах
Галогено-водород А Я» ГАЗ0 ДО» рК (расч.) РК (экс.)
HF — 12,6 —26,0 13,4 2,3 3
НС1 —57,4 —16,8 —40,6 —7,1 — 7
НВг —63,6 — 11,3 —52,3 —9,2 — 9
HI —58,6 — 4,2 —54,4 -9,5 —10
Выяснение роли каждой стадии процесса протолиза позволяет дать ответ на причины необычного поведения галогеиоводородов. Особое место HF в последовательности этих соединений объясняется относительно низким значением энтальпии ДЯ» и относительно высоким отрицательным значением энтропии Д8» процесса протолиза. Небольшая энтальпия процесса протолиза ДЯ» обусловливается относительно высоким значением энтальпии диссоциации молекулы HF и сравнительно небольшой электроотрицательностью атома фтора. Этот эффект несколько снижается за счет высокой энтальпии гидратации иона F~, имеющего малый радиус. Разность изменений энтальпии ДЯ° протолиза НС1 и HF (44,8) показывает, что протолиз HF происходит в значительно меньшей степени, чем НС1. Причиной отрицательного изменения энтропии протолиза HF является более сильная связь иона F- с водородом, что приводит к меиьшей вероятности его существования в виде гидратированного иона. Это также является причиной относительно небольшой протолитической активности фтороводорода.
Окислительно-восстановительные реакции
407
J3.5. Окислительно-восстановительные реакции
(редокс-процессы)
33.5.1. Основные положения теории окислительно-восста-
новительных процессов
33.5.1.1. Современные представления об окислении и вос-
становлении
Так же как и в теории кислот и оснований, представления об окислении и восстановлении веществ постепенно развивались, с тем чтобы выработать единый подход к широкому кругу родственных явлений. Первоначально под окислением понимали реакцию присоединения кислорода. Вообще говоря, при такой реакции неметаллических элементов образуются типичные ангидриды кислот (СО2, SO2, Р4О10), относящиеся к классу полярных молекулярных соединений. Вследствие того что кислород энергично притягивает электроны, электронная плотность связывающей пары электронов увеличивается вблизи атома кислорода.
При реакции кислорода с металлами, т. е. с электроположительными элементами, образуются оксиды металлов, которые представляют собой солеобразные соединения и имеют в узлах кристаллической решетки катионы металла и ионы кислорода, например
2Hg + О2 ---> 2 {Hg2+O2-} (тв.)
В данном случае электроны металла переходят к кислороду, в результате чего образуются ионы О2-.
Общим для окисления как металлов, так и неметаллов является переход электронов от электроположительного элемента к электроотрицательному.
Обратный процесс отщепления кислорода назвали восстановлением. Например, при нагревании оксида ртути из-за смещения положения равновесия электроны переходят от О2- к Hg2+, вследствие чего снова образуются исходные вещества — ртуть и кислород.
Известны аналогичные реакции, в которых кислород не принимает участия, однако также происходит переход электронов от одного вещества к другому:
2Fe + ЗС12 -> 2FeCl3
Н2 + F2 ► 2HF
Так же как при реакции горения с участием кислорода, эти процессы сопровождаются выделением тепла и световым излучением.
Окислительно-восстановительные реакции
409
408 Неорганическая химия
Таким образом, можно предложить более широкое опреде ление:
окислением называется отдача электронов молекулами ее щества;
восстановлением называется присоединение электронов молекулами вещества.
Вещество, принимающее электроны от другого вещества, называют окислителем.
Присоединение и отдача электронов — сопряженные процессы, так как свободные электроны могут существовать в химических системах в исключительно малых концентрациях. Поэтому окисление всегда происходит одновременно с восстановлением, и наоборот. Например, при образовании HgO из Hg и О2 ртуть окисляется до Hg2+, а кислород восстанавливается до О2-. Вообще, все процессы, в которых происходит обмен электронами, называют окислительно-восстановительными или редокс-процессами.
33.5.1.2. Сопряженные окислительно-восстановительные пары
Вещество-окислитель (ох) и соответствующее вещество-восстановитель (red) называют сопряженной окислительно-восстановительной парой (системой). Реагирующие вещества отличаются количеством электронов, содержащихся в соответствующей частице. Приведем несколько примеров важнейших сопряженных окислительно-восстановительных систем:
red > ох + пе~
Ка(тв.) у z-h Na+4-e~
Zn (тв.) Zn2+ + 2е-
Fe2+ ч > Fe3+ -j- е~
Sn2+ Sn4+4-2e-
H2 + 2HaO 4=^ 2H3O+ + 2e“
2C1- 4=t Cl2 + 2e~
B любой окислительно-восстановительной реакции всегда принимают участие две сопряженные пары — системы, отдающие электроны, и системы, принимающие их. Аналогичное положение наблюдается и для сопряженных пар кислота — основание в теории Бренстеда, от которой уравнения окислительно-восстановительных реакций отличаются только тем, что в них вместо обмена протонов происходит обмен электронами.
Число стадий окислительно-восстановительного процесса эквивалентного превращения веществ определяется на основе общего принципа, в соответствии с которым число отданных электронов должно быть равно числу принятых. В общем виде
окислительно-восстановительную реакцию можно записать следующим образом:
redj ч -fc nte~ 4- охх х«2 охз + пгв- ч=> red2 ХЩ
^redj -f- п1ох2 ч n2°xi + nired2
Пример: Sn2+ t -±. Sn4+ 4- 2e_ xi
Fe3+ 4- e~ ч—> Fe2+ X2
Sn2+ + 2Fe3+ 4=t Sn4+ + 2Fe2+
33.5.1.3. Правила определения степени окисления
При записи уравнений окислительно-восстановительных реакций целесообразно указывать состояние атомов элементов или ионов в составе соединений с помощью некоторых чисел — степени окисления. Для атомарных ионов степень окисления равна заряду иона. У молекул и комплексных ионов определение степени окисления не всегда однозначно, так как атомы в
их составе могут быть не только ионами, но и частицами с нецелочисленным относительным зарядом. Можно лишь гипотетически представить себе, что эти частицы состоят из ионизированных атомов, и, исходя из этого, считать заряд ионов рав-
ным степени окисления атома соответствующего элемента в соединении. Вообще говоря, такой подход не совсем соответствует действительности, однако вполне оправдан при составлении стехиометрического баланса окислительно-восстановительных реакций. Для определения степени окисления следует
пользоваться следующими правилами:
сумма степеней окисления всех атомов в молекуле равна нулю, а в комплексном ионе — общему заряду частицы;
в простых веществах атомы имеют степень окисления, равную нулю; так, например, углерод в графите и алмазе, кислород в озоне и в О2, металлы во всех кристаллических модификациях имеют степень окисления, равную нулю;
заряд атома в соединении приписывается ему в соответствии с его степенью окисления. Степень окисления некоторых
наиболее часто встречающихся элементов подчиняется следующим закономерностям: металлы имеют положительную степень окисления, фтор — всегда степень окисления, равную —1, водород — обычно +1, а кислород —2, галогены в галогеноводородных соединениях —1, например
LiH: Li (+1)Н(-1) OF2: О (4-2) F (-1)
Н2О: Н (4-1) О (-2) Н2О2: Н (4-1) О (-1)
NH2OH: N(-l)H(4-l)O(-2) NH4C1: N (—3) Н (4-1) Cl (-1)
Na2S2O3: Na (4-1) S (4-2) О (-2) K2Cr2O7: К (4-1) Cr (4-6) О (-2)
№
410
Неорганическая химия
После введения понятия «степень окисления» можно дать на его основе следующее определение для процессов окисления и восстановления:
повышение степени окисления соответствует окислению;
понижение степени окисления соответствует восстановлению.
33.5.1.4. Составление уравнений окислительно-восстановительных реакций
Для того чтобы записать уравнение окислительно-восстановительной реакции, прежде всего надо знать исходные вещества и конечные продукты реакции. В отдельных случаях однозначный ответ можно получить из расчета, основанного на данных об окислительно-восстановительных потенциалах соответствующих редокс-пар (разд. 33.5.1.5). Однако часто приходится устанавливать полученные в реакции вещества с помощью химического анализа. Особое внимание следует обращать на возможность выделения в ходе реакции газов. Например, при реакции пиролюзита МпО2 с соляной кислотой цвет и запах выделяющегося газа указывает на образование хлора, а цвет и другие свойства раствора — на образование Мп2+. Зная компоненты системы, можно установить состав сопряженных окислительно-восстановительных пар, взаимодействующих в данной реакции. В нашем примере такими парами являются МпО2/Мп2+ и С1_/С12. Сначала запишем полуреакции для обеих сопряженных пар. Начнем с определения степени окисления, которую атомы элементов имеют в окисленном и восстановленном состоянии. Далее найдем число электронов, которые участвуют в каждой полуреакции:
(+4) +2е_ (+2)
МпО2 ---► Мп2+ (восстановление)
2 (—1) _2е_ (0)
2С1" -->- С12 (окисление)
В данном случае меняется только степень окисления марганца, ионы О2- не принимают участия в обмене электронов. Однако освободившиеся в результате реакции ионы О2- являются сильными основаниями и вступают в протолитическую реакцию с ионами Н+ соляной кислоты:
О2- + 2Н+----> Н2О
Для ионов О2- из МпО2 требуются 4 иона Н+:
МпО2 + 2е' + 4Н+ ---»- Мп2+ + 2Н2О
2d" ---->- С12 + 2е~
МпО2 + 2СГ + 4Н+ ---»- Мп2+ + С12 + 2НаО
Окислительно-восстановительные реакции
411
Суммарное уравнение реакции появляется в результате сложения двух уравнений для полуреакций, так как число электронов, принятых и отданных в частных реакциях, одинаково.
Рассмотрим другой пример:
раствор перманганата реагирует с Н2О2 в щелочном растворе. В такой среде молекулы Н2О2 вступают в протолитическую реакцию с ионами ОН- и образуют ионы НО2“. Продуктами реакции являются кислород и МпО2. Таким образом, в системе имеются окислительно-восстановительные пары MnO4_/MnO2 и НО2~/О2. В ходе реакции степень окисления веществ меняется:
(+7) _|_3е- (+4)
МпО4~ ---► МпО2 (восстановление)
2(-1) _2е- (0)
НО2“ --► О2 (окисление)
В щелочной среде образовавшиеся из МпО4- ионы О2- и протоны диссоциированного пероксида водорода в присутствии избытка воды (или ионов ОН-) реагируют с образованием ОН- и воды:
О2- + Н2О---► 2ОН-
Н++ОН- -----<- Н2О
Учитывая эти реакции, можно записать
МпО4" + 2Н2О + Зе' ---► MnO2 + 4О1-Г I Х2
НО/ + ОН' ---► О2 + Н2О + 2е" I хЗ
После выравнивания баланса электронов умножением уравнений частных реакций на соответствующие множители и суммирования этих уравнений получим
2МпО4" + ЗНО2" + НгО ->- 2МпО2 + ЗО2 + 5ОН“
Имея некоторый навык, можно без особого труда составлять уравнения окислительно-восстановительных реакций, следуя схеме составления уравнения реакции, которая рассмотрена ниже на примере окисления хлор-ионов перманганатом в кислой среде:
а) исходные вещества и продукты реакции разделяют только стрелкой и расставляют условные стехиометрические коэффициенты а, Ь, ..., f:
а МпО4" + Ь СГ + сН+-->- d Mn2+ + е С12 + f Н2О
б) указывают соответствующие значения степени окисления над соединениями
(+7) (-1) (+2) (0)
а МпО4 + b С1 + с Н+ -► d Mn2+ + е Cl2 f Н2О
412
Неорганическая химия
в) по разности степени окисления сопряженных окислительно-восстановительных пар находят стехиометрические коэффициенты окислителя и восстановителя:
2МпО4- + ЮС1- + с Н+ ->- 2Мп2++ 5С12 + / Н2О
г) введением необходимого количества ионов Н+ (в кислой среде), ОН- (в щелочной среде) или О2~ (в оксидных солевых расплавах) уравнивают заряд ионов с обеих сторон уравнения реакции. При этом становится ясным, сколько молекул воды (в водном растворе) образовалось или, наоборот, прореагировало:
2МпО4" + 10С1- + 16Н+ ->- 2Мп2++ 5С12 + 8Н2О
Наконец, следует проверить полученное уравнение реакции на равенство заряда каждого знака и число атомов каждого элемента с обеих сторон знака равенства.
33.5.1.5. Определение направления реакции по редокс-по-тенциалам ингредиентов реакции
Как показано в разд. 30.4, каждой сопряженной окислительно-восстановительной паре (редокс-паре) red^*ne_+ox соответствует потенциал, определяемый уравнением Нернста
е=ео| (28)
1 nF ared v '
Этот потенциал при данной температуре зависит только' от активности веществ ох и red. При 25 °C и после пересчета в десятичные логарифмы
е=е°+ 0,059 1g(29) 1 п ь ared
где е — стандартный потенциал при температуре 25°C при активности всех реагирующих веществ, равной единице; п — число электронов, обменивающихся в редокс-реакции; аох и и aired—произведение активностей всех компонентов окислительно-восстановительной реакции, находящихся в уравнении реакции на стороне окислителя (аох) и на стороне восстановителя (ared). Для определения активности приняты следующие правила:
активность химически однородной конденсированной фазы равна 1;
активность газа равна его парциальному давлению;
активность растворителя в разбавленном растворе равна ~1;
активность веществ, участвующих в окислительно-восстановительной реакции, в весьма разбавленных растворах равна их
Окислительно-восстановительные реакции 413
молярной концентрации, для средних значений ионной силы a^fc (f вычисляется из теории Дебая — Хюккеля).
Пример. Для окислительно-восстановительного равновесия металл — ион металла
М (тв.) <..± пе 4- Мп+
уравнение Нернста при 25 °C записывается следующим образом: „ , 0,059 . . J_
е = е°Н---~ lgaMn+
Для системы О2+4Н++4е-^2Н2О
е = ео_j_ А059. ig рог. атн+ = 1,23- 0,059рН+1g р02
Оказывается, что окислительная способность кислорода в водных растворах зависит от pH среды и парциального давления О2.
Стандартные потенциалы ряда редокс-систем, расположенные в порядке увеличения потенциала, приведены в табл. В. 14. Потенциалы определены относительно стандартного водородного электрода, потенциал которого принято считать равным нулю. Следовательно, стандартный потенциал системы Fe/Fe2+ (е°=—0,44 В) равен э. д. с. гальванического элемента, составленного из водородного электрода и полуэлемента Fe/Fe2+ в стандартном состоянии. Знак «—» означает, что железный электрод является отрицательным полюсом рассмотренного элемента. Положение металлов в табл. В. 14 соответствует их способности переходить в раствор в виде гидратированных ионов. В стандартном потенциале отражается не только энергия решетки металла и энергия ионизации атома металла, но также и энтальпия и энтропия гидратации ионов. Гидратацией ионов объясняется, в частности, высокое отрицательное значение стандартного потенциала лития.
Термодинамическое равновесие между двумя редокс-пара-ми определяется прежде всего разностью их стандартных потенциалов. При этом выполняется следующее правило: сопряженная пара с относительно высоким (более положительным) потенциалом (окислитель) присоединяет электроны от сопряженной пары с относительно низким (более отрицательным} Потенциалом (восстановитель). В соответствии с этим правилом происходит осаждение более благородных металлов (имеющих более положительный стандартный потенциал) из раствора их солей при внесении в него менее благородного металла. Этот процесс используется в технологическом процессе обогащения бедных руд благородных металлов методом цементации (вытеснения).
Знание стандартных потенциалов дает возможность рассчитать константу равновесия окислительно-восстановительного» Процесса.
Таблица В.14. Стандартные потенциалы сопряженных окислительно-восстановительных пар
Восстановленная форма Окисленная форма Е», В
Li ч= * Li 4- е- —3,02
Cs ч= ± Cs+ 4- е~ —3,02
Са » Са2+ + 2е" —2,82
Na 4= ±: Na+ 4- е~ —2,712
Mg 4= ±: Mg2+ 4- 2е" —2,34
н- ч= * 1/2Н24-е- —2,24
А1 AF+ 4- Зе“ —1,67
Мп .< > Мп2+ 4- 2е~ —1,05
Zn 4= Zn2+ 4- 2е~ —0,762
Н2С2О4 ч=* 2СО2 4- 2Н+ + 2е" —0,47
Те Fe2+ 4- 2е~ —0,44
Сг2+ ч= =fc Сг3+ 4- е' —0,41
Sn ч > Sn2+ + 2е~ —0,136
Ti3+ —- т;4+ _|_ е- 0,0
Н2 4= 2Н++2е" 0,00
Си+ ч= Cu2+ 4- е~ 0,15
Си ч -А Си2+ + 2е" 0,345
-21' ч= => 124-2е" 0,535
Н2О2 =₽= =t О2+2Н+4-2е" 0,68
Fe2+ ч= рез+ _±_ е- 0,771
Ag ч= =fc Ag+ 4- е' 0,800
2Вг- ч-- Btg -J- 1,065
2Н2О =₽= =t О2 4- 4Н+ 4- 4е- 1,23
Мп2+ 4- 2Н2О =₽= =h МпО2 4-4Н+4-2е' 1,28
2С1" ч~=* С12 4- 2е" 1,358
2Сг3+ 4- 7Н2О ч=з± Сг2О72" 4- 14Н+ + бе" 1,36
Мп2+ 4- 4Н2О ч= МпО4- 4- 8Н+ 4- 5е- 1,52
МпО2 4- 2Н2О ч= ± МпО4- 4- 4Н+ 4- Зе" 1,68
2Н2О Н2О2 4- 2Н+ 4- 2е" 1,78
Со2+ ч -* Со3+4-е" 1,84
2SO42- S2O82- 4- 2е- 2,06
Ю2 + Н2О ч=^ О3 4- 2Н+ 4- 2е~ 2,07
2F- F2 4- 2е' 2,82
Окислительно-восстановительные реакции
415.
Пример. Для реакции между металлическим железом и ионами меди (II) Fe (тв.) + Си2+ д.-т»: Си (тв.) 4- Fe2+
константа равновесия равна
_ aFe2+
Яси2+
(30>
В ходе окислительно-восстановительной реакции концентрация (активность) ионов Fe2+ постепенно возрастает, а активность ионов Си2+ падает вследствие уменьшения их концентрации в растворе. Поэтому потенциалы женных редокс-пар также изменяются: 0,059 ,
sFe = s°Fe +---g— lgaFe2+ (gFe увеличивается)
сопря-
(31)
0,059 ,
ecu = S°Cu + —2— g aCu2+ (SCu Уменьшается)
(32>
После того как ере = вси, установится равновесное состояние, и из (31) и (32) получим
уравнения
0,059 S°Fe — S°Cu =
_____,а аСи2+
2 lg aFe2+
Из уравнения (30) следует
ё aFe2+
= -lgK
Поэтому
, 2 (s°Fe — s°Cu)
~IgA~ 0,059
2 (-0,44 — 0,35) 0,059
—27
Таким образом, константа равновесия К=10~27, т. е. реакция практически1
полностью смещена слева направо.
Подобным же образом, исходя из соответствующих стандартных потенциалов, можно вычислить произведение растворимости труднорастворимых солей при стандартной температуре 25 °C.
Пример. Пусть происходит осаждение хлорида серебра:
Ag (тв.) ч_.-> Ag+ + е~ е2° = 0,8 В
AgCl (тв.) 4-е’ <=* Ag(TB.) + Cr е2° = 0,21В
AgCl (тв.) > Ag+ 4- Cl-
Потенциалы соответствующих полуреакций равны е1 = 0,8 4- 0,059 1g o\g+
в, = 0,21 4- 0,059 1g-—
2 ь вещ
В состоянии равновесия e.i = &2, откуда
0,21—0,8
1g Пр = 1g (ауь • асг) =-оТо59--= —1 а
II
416
Неорганическая химия
Таким образом, произведение растворимости хлорида серебра
Пр = 10-10 моль2/л2
Потенциал окислительно-восстановительных процессов, связанных с протолитическими реакциями, зависит от pH.
Пример. Рассмотрим систему Сг3+/Сг2О72-. Для реакции
Сг2О72- + 14Н+ + бе" 2СгЗ+ + 7Н2О
запишем уравнение Нернста
0,059 асг2о,г--а14н+
е = е» 4- —4— 1g----------------
~ 6 6 а2сг3+
аСг,О7г“
е = 1,36 — 0,14рН + 0,01 1g- 2
а Сг’>+
Отсюда для pH 0 (например, 1 М НС!)
«Сг2о7г-
е = 1,36 + 0,01 1g
для pH 3 (например, 0,1 М НАс)
. аСггО,г~
е = 0,94+ 0,01 1g
“ Сг*
Полученные соотношения показывают, что рассмотренная окислительно-восстановительная пара при pH 0 способна окислять хлориды, бромиды и иоди-ды, а при pH 3 — только иодиды.
В отличие от быстрых протолитических реакций окислительно-восстановительные реакции часто протекают очень медленно, вследствие чего ожидаемая в соответствии с расчетом константы равновесия реакция не наблюдается. Такое поведение характерно, например, для явлений пассивации металлов в кислотах.
33.5.2 . Особые случаи окислительно-восстановительных
реакций
33.5.2.1 . Окислительно-восстановительные реакции в гомо-
генных и гетерогенных системах
Переход электронов в окислительно-восстановительной реакции может происходить как в объеме раствора между находящимися в нем частицами, так и на границе раздела твердая фаза — раствор. Примерами гомогенных реакций могут служить взаимодействия между SnCb и FeCl3 или между FeSC>4 и К2СГ2О7 в водном растворе. Подобные реакции часто используются в химическом анализе для определения окислителен или восстановителей. При потенциометрическом титровании (разд. 39.6) точка эквивалентности совпадает со скачкообразным изменением потенциала.
Длительно-восстановительные реакции
417
акцииТвТто^ Р^
жащими Н30+ Этот nS? металлов с растворами, содер-образом: ’ Т прОцесс может быть записан следующим
М(тв.)+пн+------ м'г+ + «/ н
с^гидратирова^ные^плтл И°НаМ в0Д0Р0Да’ т- е- восстанавлива-дорода со степенью ДгТ’' С° степенью окисления +1 до воду атомами ВЮ окисления, равной 0. Прочная связь меж-ронного газа» с обусловленная взаимодействием «элект-^ичХй решетки o°cZTbH° 3аРЯЖеввыми атомами кристал-рушается Ри мртяп °С 6яется’ кристаллическая решетка раз-вУраствор’. ЛЛ В ВВДе гидРатированных ионов переходит растворщИТеЛЬНаЯ способн°еть системы Н+/Н2 зависит от pH
2 Н+ + 2е~ -у > м
е_ 0.059 . д2н+ а
2 8 Рн2 0,059 pH (е°=0, нормальное давление) (33)
В воде (pH 7) потенциал водородного электрода равен
е = —0,059-7 = —0,413 В
риалов <н^од°ятсяМеТаЛЛЬ1’ К0Т0РЬ1е в ряду стандартных потен-ионами водорода изТдыТТ ЭТ°Г° потенРиаЛа> окисляются Де всего шаггапы И3 воды- таким металлам относятся преж-энергично пря Ые И щел°чноземельные металлы, которые примет ZРУЮТ С ВОД0Й- Нек°торые другие металлы, на-ются вР воле иГЛ’ МаГНИЙ И ЦИНК’ весьма медленно растворя-Ния водорода ня А высокого перенапряжения процесса выделе-пассивапияДинтЛ 3JHX ме?аллах- Важную роль при этом играет кого слоя ня ьзванная образованием тонкого и прочного оксидного слоя на поверхности металла.
Ор РИ Р потенциал сн+/н2 =0. Окислительная способность отрицательным1 В03растает’ так что в принципе все металлы с твопах с ятгттл тандаРтным потенциалом растворяются в рас-Вос™ стадии вХСТЬЮ И°Н0В В0Д°Р°ла, равной 1- Замедленных металлахьделения водорода, которая имеет место на чи-вотопых Ля а’ снимается Добавлением следовых количеств не-тво я™ блаðаДНЫХ металлов. При этом на поверхности растре гяпнпяиЯ металла образуются локальные короткозамкну-^еталпа 1кятАпС1\Ие элементы —иа включении благородного Ренаппяжрни1 Де ПРОИСХОЛИТ выделение водорода, так как пе-РоваХ н я а НеМ невелик°: одновременно начинает ионизи-Вый металл13 гидРатиР°ванных ионов основной неблагород-
С 1 d л ,/ц ЗНОД.
2?-~-2027
418
Неорганическая химия
Некоторые металлы также растворяются в щелочном растворе с выделением водорода, хотя в этих условиях концентрация ионов водорода очень невелика: в 1 н. едком натре (pH 14) она составляет 10-14 моль/л. Если не учитывать влияния коэффициента активности, ен + /н2 = —0,83 В. Однако этот потенциал достаточно положителен для того, чтобы окислить такие металлы, как алюминий и цинк. Окислительно-восстановительной реакции благоприятствует образование гидроксо-комплекса:
Zn (тв.) + 2 ОН' + 2 Н2О-> [Zn(OH)4l2- + Н2
В результате этой реакции концентрация, а следовательно, и активность ионов Zn2+ уменьшается, и понижается потенциал:
e==_0,76+-^-lgaZn2+ (34)
Точно таким же образом на потенциал системы влияет образование комплексных соединений (разд. 33.6.3).
33.5.2.2 . Диспропорционирование и контрдиспропорционирование
Диспропорционирование — это особый случай окислительно-восстановительной реакции, в ходе которой происходит переход одного и того же вещества средней степени окисления на более низкую и более высокую степень окисления. Вещество как бы само себя окисляет и восстанавливает. Это явление называют также редокс-амфотерностью. Реакции такого типа часто встречаются в химии галогенов. Например, при растворении иода в растворе едкого натра молекулы иода сначала диспро-порционируют на гипоиодид- и иодид-ионы. Нестабильный ги-поиодид быстро диспропорционирует с образованием иодида и йодата:
(0) (—1> (+о
Ц + 2ОН- Г+Ю’ + НаО
В результате реакции раствор обесцвечивается.
При подкислении раствора в результате связывания ионов ОН- вновь образуется иод. Обратный процесс — образование вещества средней степени окисления из веществ с более высокой и более низкой степенью окисления — называют контрдиспропорционированием..
Для элементов побочных групп периодической системы, таких, как марганец, ртуть и медь, также можно наблюдать подобные реакции. Металлическая медь образует с гидратированным ионами Си+ и Си2+ две редокс-пары со стандартными по
Окислительно-восстановительные реакции
419
тенциалами: e°cu/cu+ =4-0,52 В и e°cu/cu2+=4"0,35В, т. е. система Cu/Cu+ имеет более высокий стандартный потенциал. Поэтому при высокой концентрации ионов Сп+ в водном растворе Cu(I) окисляется с образованием Сп2+ и меди. Это является причиной того, что в водных растворах нельзя получить соли CuNO3 или CU2SO4. В растворах труднорастворимых соединений Си2О или Cui или в прочном комплексном соединении [Cu(CN)4]3- концентрация гидратированного иона Си+ очень мала. В соответствии с уравнением Нернста потенциал системы снижается до значения, соответствующего потенциалу системы Cu/Cu2+. Поэтому из металлической меди и солей меди(II) в избытке хлор-ионов происходит реакция контрпропорциониро-вания в образованием дихлорокупрат(1)-иона:
Си (тв.) + Си2+ + 4 СГ ч—> 2 [СиС12]~
Интересно также поведение окислительно-восстановительной системы Н2О2 в присутствии солей Мп (II) и МпО2; для нее ха-
рактерны следующие редокс-потенциалы:
2Н2О НА4-2Н+4-2Г
;Мп2++2Н2О з—h МпО2+4;н++2е-
Н2О2 О2+2 Н++ге-
ев = 1,78В ев = 1,28 В
ев = 0,68 В
Таким образом, пероксид водорода является восстановителем в реакции МпО2—>-М.п2+ и окислителем, обеспечивающим протекание реакции в обратном направлении. Это объясняет сильное каталитическое действие небольших добавок МпО2 на процесс разложения пероксида водорода:
МпО2
НО, /Мп2+\ Н2о, , МпО2 н о2/Мпг+
\о2 \нго хо2
Н2О2 диспропорционирует в термодинамически более стабильные соединения со степенью окисления кислорода 0 и —2.
(-1) (-2) (0)
2Н2О2 ---- 2Н2О + О2
Диспропорционирование и контрдиспропорционирование часто протекают при нагревании веществ
(+5) 400 °C (—1) <+7>
4 КС1О3 --> КС1 + ЗКС1О4
(-3) (+3) (0)
NH4NO2 ----- N2 + 2 Н2О
33.5.2.3 . Окислительно-восстановительные процессы в расплавах солей
Окислительно-восстановительные реакции особого типа протекают в расплавах солей, если в них присутствуют окислители (например, KNO3, КСЮз, Na2O2) или восстановители (KCN, углерод). В этом случае одновременно с обменом электронов происходит также перенос анионов кислорода.
27*
420
Неорганическая химия
Реакции окислителей:
NO3- + 2е“ ---- NO2- + О«-
С1О3~ + бе" —* Cl" + 3 О2-
О2~ + Зе~ -► 2 О2-
Реакции восстановителей:
С + О’- ---и СО + 2е"
CN- 4- О2- --> CNO- + 2е“
В окисляющих расплавах ионы О2- используются атомами окисляющегося элемента. Более высокий заряд частицы, возникший в результате ее окисления, компенсируется координированными с ней ионами О2-. Поэтому окислительное сплавление облегчается при добавлении веществ со свойствами оснований, таких, как Na2CO3, которые также могут поставлять ионы О2-. При окислительном сплавлении оксида хрома (III) в нитрат-карбонатном расплаве идет реакция
Сг2О3 (тв.) + 3 NO3- + 2 СО32- -> 2 СгО„2- + 3 NO2* -j- 2 СО2
Для превращения в растворимую форму касситерита SnO2, который так же, как и SiO2, с трудом поддается растворению, в отличие от рассмотренного ранее примера необходимо разорвать связи —О—Sn—О—. Эта задача решается сплавлением с цианидами, которые в результате этого процесса переходят в цианат-иоиы. При этом происходит восстановление SnO2 до металлического олова:
SnO2 (тв ) + 2 CN- -► Sn (тв ) + 2 CNO~
33.6. Реакции комплексных соединений
33.6.1. Образование и диссоциация комплексов
При добавлении аммиака в водный раствор соли серебра связанная с ионом серебра вода последовательно замещается аммиаком:
[Ag(H2O)2]+ + NH3 [Ag(NH3) (H2O)]+ + H2O
(Ag(NH3) (Н2О)]++ NH3 =₽=& [Ag(NH3)2]++ H2O
Образование комплексного иона из гидратированного в результате обмена воды на соответствующие лиганды обычно называют комплексообразованием, хотя более точно этот процесс можно было бы назвать обменом лигандов.
К этому обратимому последовательному процессу можно применить закон действующих масс. Поскольку речь идет ° разбавленных растворах, активность воды будем считать раВ' ной 1. Тогда, воспользовавшись экспериментальными данными, можно записать для первой и второй констант реакции образе вания иона [А§(ЫНз)2] +
к .fl[Ag(ML==6 оз
Реакции комплексных соединений
421
к _ arAg(NH3)2]+
2 a[Ag(NH3)]+‘ “NH3
«= 2,0-103 л/моль
(связанная в комплексе вода обычно не записывается в формуле соединения).
Константа равновесия, соответствующая суммарной реакции
Ag++2NH3 [Ag(NH3)2]+
называется суммарной константой образования или константой устойчивости КуСт комплекса
X _ a[Ag(NH3)2]+ уст aAg+-a2NH3 Можно легко показать, что константа Куст является произведением констант Ki и К2, и для иона [Ag(NH3)2] +
Ауст = Ах-А2 = 1,3-10’ моль~2-л2
Рассмотренные выше соотношения справедливы вообще для процессов комплексообразования. Для комплекса с п лигандами суммарная константа устойчивости-—произведение п констант отдельных стадий:
КУСТ=К1К2...КП (35)
Константа КуСт — термодинамическая мера устойчивости комплекса. В табл. В. 15 приведены рКУст некоторых важнейших комплексных соединений. Реакцией, обратной комплексообразованию, является реакция диссоциации. Диссоциация также протекает ступенчато, причем при обмене лигандов с водой координационное число обычно остается неизменным. Между константой равновесия и рА прямой и обратной реакций имеет место следующее соотношение:
^уст 1/^НСТ’ Р^уст-----' Р^НСТ (36)
[Кнст — константа нестойкости (диссоциации)]. Знание констант нестойкости необходимо при рассмотрении более сложных равновесий в растворах комплексных соединений. Наглядный пример этому показан на рис. В.13, где представлена зависимость содержания (в мольных процентах) комплексов с Различным числом лигандов от логарифма концентрации комп-тексообразователя для системы Cu2+/H2O/NH3. В области концентрации cNHs Ю~6—10-1 моль/л происходит ступенчатое образование комплекса [Cu(NH3)4]2+. Вообще говоря, при некоторых значениях концентрации аммиака одновременно существуют комплексы с различным содержанием лигандов NH3. Например, при cnhs = 10 2 моль/л молярные концентрации ди-, три- и тетраминных комплексов находятся в следующем соотношении: [СиА2] 2+ = 3°/о ; [CuA3] 2+ = 32%; [СиА4] 2+=65%. 11РИ концентрации аммиака 10-2—1 моль/л в растворе имеет-
422
Неорганическая химия
Таблица В.15. Коистаиты образования рК некоторых комплексных соединений
Реакция образования комплекса P^yCT
Ag+ + 2NH3 4==^ [AgtNHa),]* —7,1
Ag+ + 2S2O32 - [Ag(S2O3)2]2- —13,6
Ag+ + 2CN- [Ag(CN)2]- -21,0
Au+ + 2CN- [Au(CN)2J- —28
Cd2++4NH3 =e=* [Cd(NH3)4]2+ —7,0
Cd2+ + 4CN- [Cd(CN)4l2~ —16,9
Co2++6NH3 [Co(NH3)e]2+ —5,1
Co3++6NH3 [Co(NH3)e]3+ —35,2
Cu2++4NH3 [Cu(NH3)J2+ —13,3
Cu++4CN- [Cu(CN)4]3- —27,3
Fe2++6CN- =?=* [Fe(CN).]‘- —44
I2+I- ,=4 I8" —2,9
Ni2+ + 4CN- [Ni(CN)4]2- —15,3
Hg2+4-4CNT [Hg(CN)4]2- —40,5
Hg2+ + 4I' 4 [Hgl,]2- —30,3
Zn2+4-4NH3 ,=> [Zn(NH3)4]2+ —9,0
ся в основном [Cu(NH3)4]2+. При более высокой концентрации аммиака доказано существование [Си(ЫН3)5]2+. При этом координационное число меняется от 4 до 5. Это объясняет также
относительно широкую область существования ионо
[Cu(NH3)4]2+.
Для многих практических целей достаточно знать сумма? ную константу равновесия. Тогда для устойчивых комплекса® и при большом избытке комплексообразователя можно вычи^ лить концентрацию свободного (гидратированного) иона в воД
Реакции комплексных соединений 423
ном растворе. Эта концентрация имеет очень важное значение для решения многих препаративных и аналитических задач. При названных ранее упрощающих предположениях можно считать, что практически все центральные ионы связаны в комплекс (чаще всего с наибольшим координационным числом) и концентрация свободных лигандов равна суммарной аналитической концентрации комплексообразователя.
Пример. Рассчитать равновесную концентрацию гидратированных ионов меди, если сси2+ = 10-3 моль/л и Cnh8 = Ю'1 моль/л.
Решение. Из рис. В 13 следует, что при этой концентрации в растворе имеются исключительно ионы [Cu(NH3)4]2+, концентрация которых приблизительно равна суммарной концентрации ионов Си2+ (~10-3 моль/л-1).
Концентрация аммиака при комплексообразовании меняется мало: c4H3«cNH3—4сси2<-=0,1—0,004»0,1 моль/л. Тогда из константы устойчивости тетраммиииого комплекса КУст=1013-3 л2>моль-2 получим
_ _ 5. „„ь/я
Луст'СТЧНя 10хл>а-10~4 '
33.6.2. Комбинированные равновесия комплексообразования и осаждения
В аналитической химии часто проводится растворение труднорастворимых солей путем перевода в комплексное соединение. Равновесие между насыщенным раствором твердой соли и твердой фазой смещается при изменении концентрации одного из ионов. Равновесие восстанавливается только за счет растворения труднорастворимой соли. При избытке комплексообразователя можно растворить весь осадок. Направление течения реакции в сторону осаждения или растворения соли зависит от произведения растворимости и константы устойчивости комплексного соединения.
Пример. Можно ли добавлением аммиака перевести Agl в комплексное соединение [Ag(NH3)2] + ?
Решение. Реакция включает следующие стадии:
Agl (тв.) ч =* * Ag++ I- —1gПр = 16,0
Ag++ 2NH3 ?==£ [Ag(NH3)2]->- pK = -7,1
Agl (тв) + 2NH3 =₽=* [Ag(NH3)2]+ +1“ pK = 8,9
Константа равновесия суммарной реакции равна
К = ^AgWHah]*- °Г. = 10_8(9 ~ ! 3.10-9 а%1н3
* е Добавление аммиака даже в больших концентрациях не может привести Растворению Agl.
р В то же время обработка Agl раствором цианида приводит к полному Раствореиию соли, так как константы устойчивости иоиов [Ag(CN)2]~
424
Неорганическая химия
рК>ст=~21 Константа равновесия соответствующей суммарной реакции равна
а С№
Следовательно, реакция идет в сторону комплексообразования.
33.6.3. Комплексообразование и окислительно-восстановительный потенциал
В случае комплексообразования потенциал сопряженной окислительно-восстановительной системы может сильно понизиться. Это явление используется, например, в технологическом процессе обогащения золота методом выщелачивания в цианидных растворах.
Известно, что в обычных условиях золото не окисляется кислородом воздуха, что соответствует значениям стандартного потенциала следующих редокс-пар:
Au Au+ + e~ e° =-- 1,46 В
2 HSO O2 + 4 H+ + 4e~ e° = 1,23 В
Однако обработка металлического золота раствором цианида, содержащим растворенный кислород, переводит золото в раствор в виде комплексного иона [Ag(CN)2]~:
4 Au + О2 + 8 CN' + 2 Н2О -> 4 [Au(CN)2f + 4 01 Г
Здесь .кислород выступает как окислитель, который переводит золото в ионы со степенью окисления +1. Причиной значительного смещения потенциала пары Au/Au+ является существенное уменьшение концентрации ионов Аи+ за счет комплексообразования. Константа устойчивости комплекса равна
К = -CWcN2)r. = 1028 моль-2. Л2
ycl CAu+-C2CN-
Полагая ccn_ = 1 моль/л и qau(cn)2] =10-2 моль/л получим
1 П-2
САи+ = 10287]—= 10’30 МОЛЬ/Л
e=e°+0,0591gcAu+==l,46—30-0,059=—0,31В
Таким образом, потенциал системы О2/Н2О становится достаточно положительным для того, чтобы обеспечить окислени. золота даже при небольшой концентрации растворенного кис лорода.
33.6.4. Обмен лигандов в комплексных соединениях
Из данных о термодинамической устойчивости комплексно го соединения нельзя сделать выводы о скорости, с которой ганды одного вида замещаются (обмениваются) другими
Реакции комплексных соединений
425
гандами. Кинетика реакции обмена в большей степени определяется возможными механизмами реакции и соответствующей энергией активации. Экспериментальные данные можно интерпретировать на основе рассмотрения двух предельных типов реакций обмена в октаэдрических комплексных соединениях (эта конфигурация наиболее характерна для многих комплексных соединений).
В случае механизма SN1* первой стадией является отщепление одного лиганда, в результате чего возникает неустойчивый комплекс с конфигурацией тригональной бипирамиды или квадратной пирамиды. Координационно-ненасыщенное состояние снимается за счет присоединения нового лиганда:
[МХ6р+ - ~Х > [МХ5]("+1’+ [MX5Y]"+
медленная быстрая ° J
Энергия активации первой стадии значительно больше, чем второй, соответственно первая стадия протекает медленнее, чем вторая. Следует однако отметить, что скорость реакции зависит только от концентрации исходного комплекса и не зависит от концентрации замещающего лиганда Y-.
В случае механизма SN2 аналогичная реакция является бимолекулярной. Сначала происходит присоединение лиганда к октаэдрическому комплексу. Условием для этого является наличие трех свободных орбиталей, энергия которых немного выше уровня остальных э/73сГ-гибридных орбиталей. При этом возникает активированный комплекс с координационным числом 7 (пентагональная бипирамида). Реакция заканчивается отщеплением одного лиганда:
[МХв]«+ ——— + [MXeY]<«-n+ —[MXsYr-t-медленная ° быстрая
В этом случае первая стадия также является медленной и определяет скорость всего процесса. Скорость реакции зависит как от концентрации лиганда Y~, так и от концентрации неходкого комплекса [МХ6]П+, т. е. реакция является бимолекулярной.
В исследованных до настоящего времени системах не всегда возможно однозначно решить, какой механизм осуществляется при обмене лигандов. Можно предположить, что во многих случаях происходит постепенный обмен лигандами с одновременной дисссоциацией и присоединением лигандов:
Y- 4- (MX,,]'1*- -► [Y.. .МХ5... Х](л-1)+ -► [МХ5У]Л+4-X"
*5?? 1- и SK2-механизмы — это мономолекулярное и бимолекулярное нук-Рильное замещение соответственно.
426 Неорганическая химия
Кроме того, можно предположить образование ионной пары [MXg]n+Y~. Часто на механизм обменной реакции влияют также молекулы растворителя.
33.7. Полимеризация и конденсация
Полимеризацией называют реакцию присоединения едина-ковых молекул или атомов друг к другу. Молекулярная масса полимера является кратной молекулярной массе составляющих его молекул (мономеров).
Одним из частных случаев полимеризации является связывание координационно-ненасыщенных молекул. Так, свободная орбиталь у атома серы в молекуле SO3 дает возможность образования тримерного кольцеобразного у-БОз, в котором сера имеет координационное число 4:
О. /О ,SZ 0х \О 3S°3 - о J Го
>S Sz
о/\о/ 0
Особенно часто реакции полимеризации протекают через стадию образования свободных радикалов. Свободные радикалы, имеющие неспаренные электроны, образуются при гомолитическом разрыве связей между атомами, особенно часто при пиролизе, а также в некоторых окислительно-восстановительных реакциях. Так, например, восстановление пероксида водорода ионами Fe(II) приводит к образованию радикала гидроксила:
Н2О2 + Fe2+ -► Fe3+ + - ОН + ОН"
Одно из доказательств существования радикала «ОН состоит в том, что полимеризация акрилонитрила СН2 = СН—CN в полиакрилонитрил происходит только в присутствии смеси Fe(U) и Н2О2 как катализатора, хотя по отдельности эти вещества не оказывают влияния на реакцию полимеризации.
Конденсацией (поликонденсацией) называют присоединение одинаковых или разнородных молекул, при котором выделяются такие вещества, как Н2О, NH3, H2S, галогеноводороды и спирты. Поэтому молекулярная масса продуктов конденсаций меньше, чем сумма молекулярных масс исходных компонентов В неорганической химии известно много примеров денсации:
2 KHSO4 ---► K2S2O7 + Н2О
2 MgNH4PO4 --► MgsP2O7 4- 2 NH3 + H2O
реакций кон-
ci-s-s-a
-| г
H-S-H
Cl—S-S—Cl Cl-S-S-S-S-S-Cl +J$cl
_________________Полимеризация и конденсация_______________Ш
Конденсация оксо- и гидроксокомплексов с удалением в раствор молекул воды приводит к образованию поликатионов, как это было показано на примере гидроксоаквакомплекса алюминия (разд. 33.4.2.2). Подобным же образом идет образование основных солей при нагревании кристаллогидратов солей:
[Mg(H2O)e]Cl2 -► Mg(OH)Cl (тв.) + НС1 + 5 Н2О
Другим возможным примером конденсации является образование полианионов, которое, как и при полимеризации, ведет к соединениям самых разнообразных состава и структуры. К таким системам относятся, например, изополикислоты бора, кремния, фосфора, молибдена и вольфрама, поверхностные и объемные структуры силикатов и многие гетерополикислоты.
Особенность внутримолекулярной конденсации состоит в том, что отщепление воды происходит от одной и той же молекулы, способной к конденсации. Примером такой реакции является замыкание кольца трифосфорной кислоты конденсацией конечных гидроксильных групп и образование тримета-фосфорной кислоты.
н н ООО । и ।
но-р-о-р-о-р-он
II I II
—-НО I I о +Н2° \р рг о/ \0/ \он
33.8.
Термическая диссоциация и сольволитическое расщепление
Термической диссоциацией называют реакции, в которых при воздействии тепловой энергии происходит разрыв межатомных связей, в результате чего происходит распад молекулы. В более узком смысле это реакции, обратные полимеризации.
Поскольку процесс полимеризации можно считать обратимым, константа равновесия зависит от температуры, и при повышении температуры полимерные молекулы распадаются на низкомолекулярные блоки и молекулы мономера:
N2O4 2NO2 (SO3)3 3SO3 S8 4S2
При термической диссоциации молекулы могут также распадаться на отдельные атомы. Процесс диссоциации зависит Не только от энтальпии диссоциации, являющейся мерой энер-гии связи в молекуле. Он в значительной мере определяется Изменением энтропии реакции. В процессе диссоциации, вооб
428
Неорганическая химия
Термическая диссоциация и сольволиз
429
ще говоря, увеличивается число возможных энергетических состояний, а следовательно, и энтропия. Число энергетических состояний молекулы определяется числом возможных колебательных и вращательных состояний. Кроме того, происходит накопление энергии за счет поступательного движения. Уровни энергии поступательного движения расположены значительно более плотно, чем уровни энергии колебательного и вращательного движения. При диссоциации молекулы на атомы колебательные и вращательные термы исчезают, однако за счет увеличения общего числа частиц значительно увеличивается возможность поглощения энергии поступательного движения, так что в целом энтропия системы возрастает: AS>0.
С увеличением температуры свободная энтальпия AG = = АН—TAS уменьшается, поскольку положительная энтальпия диссоциации перекрывается энтропийным членом TAS.
Сравнение процесса диссоциации молекул различных галогенов провели Вике и Франк; они показали, что при диссоциации фтора изменение энтропии оказывается больше, чем для остальных галогенов. Значительное изменение энтропии при диссоциации фтора F2^=2 F объясняется особой устойчивостью электронной структуры молекулы фтора. Вследствие этого число вращательных и колебательных уровней молекулы фтора сравнительно невелико, а следовательно, остается очень небольшим и соответствующее значение энтропии молекулярного фтора. Поэтому при появлении возможности поглощения энергии за счет поступательной энергии образовавшихся атомов энтропия увеличивается в большей степени. Таким образом, при одной и той же температуре степень диссоциации фтора оказывается больше, чем, например, степень диссоциации иода, хотя энтальпия диссоциации фтора (ДЯ° = 156,6 кДж/моль) примерно на 8 кДж больше, чем энтальпия диссоциации иода (АЯ°=149 кДж/моль). Эта относительно более сильная диссоциация фтора — одна из причин его большей реакционной способности.
В более широком смысле как реакции термической диссоциации можно рассматривать также и другие реакции, сопровождающиеся изменением степени окисления, например РС155=ьРС1з + С12. или реакции, сопровождающиеся выделением газов при нагревании твердых веществ
СаСО3 (тв.) -► СаО (тв.) -|- СО2
Термическое разложение карбонатов щелочноземельных металлов также является равновесной реакцией, подчиняющейся закону действующих масс (разд. 23.3.2). Температура разложения достигается при условии равенства парциального давления СО2 внешнему давлению. В ряду MgCO3—СаСОз—ВаСОз
термоустойчивость повышается, так как с увеличением радиуса катиона способность оксида отдавать ион О2- увеличивается, а распад карбоната замедляется:
О2’ + СО3 «=* СОд2'
Другой реакцией разложения вещества является сольволиз. В этой реакции под влиянием растворителя разрываются полярные и поляризованные межатомные связи, причем, как правило, растворитель образует соединения с продуктами реакции. При добавлении в пиросерную кислоту воды происходит гидролитическое расщепление с образованием двух молекул серной
кислоты:
II HO-S-0
о нюн о
II I II
О : О
о о
II II
-ОН -> HO-S-OH + HO-S-OH
II II
О О '
прочных связей между ________
(разд. 33.4.3.2 и 33.5.2.3)
конденсированных атомами
В более мостиковых сплавлении ____________,, .
«Омыление» синильной кислоты теплой серной кислотой также является процессом распада. Можно предположить, что наиболее ре: собными центрами в полярной молекуле синильной кис ляется азот, несущий некоторый отрицательный заряд род, несущий некоторый положительный заряд последовательно присоединяются водород и гидроксил молекулы воды. С каждой молекулой HCN реагируют три молекулы воды. Присоединение последней молекулы ппл^т ,„— разрыву связи С—N:
(—) н2о HC=N ----> HC(OH)=NH
соединениях распад возможен только при
теплой концентрированной _____________________ .......процессом гидролитического распада. Можно предположить, что наиболее реакционноспо-
— ----- - г кислоты яв-
. [, и угле-; к этим атомам
последней молекулы воды приводит к
Н,0 Н2О
---> НС(ОН)2—NH2 ----> НС(ОН)3 + NH3
Возникающая в реакции ортомуравьиная кислота неустойчива и после отщепления воды образует муравьиную кислоту, которая под действием серной кислоты отдает еще одну молекулу воды, превращаясь в ангидрид СО. Таким образом, конечными продуктами реакции являются моноксид углерода и сульфат аммония. В теплой концентрированной щелочи NaOH, в Которой также имеет место гидролитическое расщепление, из NaCN образуется соль муравьиной кислоты — формиат натрия и аммиак.
В неорганической химии часто применяются также сольво-Литические реакции в неводных растворителях (разд. 34.1).
430
Неорганическая химия
33.9. Реакции веществ в твердой фазе
33.9.1. Общие предпосылки реакционной способности твердой фазы
Химия веществ в твердом состоянии охватывает все химические процессы, которые протекают с участием веществ в твердой фазе. Частным случаем таких реакций являются реакции твердых веществ с жидкостями и газами.
Реакции твердых веществ имеют некоторые особенности по сравнению с реакциями в растворах. В жидкой фазе, как правило, диффузионные процессы протекают довольно быстро, что способствует сильному сближению ингредиентов реакции, находящихся в растворе в виде сольватированных ионов, молекул или комплексных частиц. Протекание суммарной реакции,
• •••••• Ад+ Вг" Ад+ Вг- АЭ+ ВГ
• □ • Ф Ф • • @+
• Ф^Ф •’Ф □ • ВГ □ ВГ Agv ВГ Ад+
ф • 9 □ ф ф ф Ад+ ВГ Ад+ ВГ □ ВГ
• □ • • •
Ф Ф Ф ф ф ф ф ВГ Ад+ ВГ Ад4* ВГ Ад4"
а 6
Рис. В.14. Схема атомной решетки с дефектами по Френкелю (а) и дефекты по Френкелю в решетке AgBr (б).
как правило, лимитирует стадия образования активированного комплекса; последняя зависит от энергетических и стерических факторов. Например, при нагревании смеси порошков двух твердых веществ протекание химической реакции определяется как образованием зародышей и реакциями на поверхности раздела фаз, так и относительно медленной диффузией вглубь кристаллической решетки. Для переноса вещества к местам взаимодействия частиц и отвода продуктов реакции необходимо преодолеть длинный диффузионный путь в потенциальном поле кристаллической решетки.
Поэтому, как правило, для ускорения переноса вещества твердофазные реакции проводят при повышенной температуре. Реакцию можно проводить и при относительно низкой температуре, если структурные элементы исходных веществ остаются неизменными (разд. 33.9.2). В процессе присоединения частицы к исходной структуре энергия должна подводиться в таком количестве, чтобы, с одной стороны, обеспечить достаточную подвижность присоединяющейся частицы, с другой стороны — не нарушить структуру исходной решетки. Исследование кинетики реакций в твердой фазе показывает, что химическая активность твердых тел в первую очередь зависит от степени совершенства структуры реального кристалла. Под несовершенством структуры понимают общее число дефектов решетки, причем можно различать макродефекты кристалла (границы зерен, смещения (дислокации), примесные атомы) и микродефекты (точечные) однородного кристалла.
Существуют два вида точечных дефектов, проявляющиеся в зависимости от размеров частиц, образующих решетку. В структурах с достаточно большими незаполненными объемами, например в металлах с объемными октаэдрическими и тетраэдрическими пустотами в ионных решетках, состоящих
Реакции в твердой фазе
431
из небольших катионов и больших поляризуемых анионов, преобладает заполнение ионами междуузлий с образованием пустот (вакансий) (рис. В.14, а и б). Точечные дефекты такого рода называются дефектами по Френкелю.
В структурах с плотнейшей сферической упаковкой, например в кубических объемно-центрированных металлических решетках, а также в ионных решетках с катионами и анионами, мало отличающимися по своим размерам (например, NaCl), ион не может занять междуузлие. Точечные дефекты в этом случае образуются при перемещении элементов ячейки на поверхность, в результате чего также образуются пустые места в решетке
• ••••• е сг
••••□• Nq+
• •□••••
□•••••
•••••• Na+
а СГ
Na СГ Na
□ Na+ СГ Na+ СГ
Na+ СГ □ СГ Na+
СГ Na+ СГ Na+ СГ
□ СГ Na+ □ Na*
Na+ СГ
Na+ СГ б
Na+ СГ
Рис. В. 15. Схема атомной решетки с дефектами по Шоттки (а) и дефекты по Шоттки в решетке NaCl (б).
(рис. В.15,а и б). Такого рода точечные дефекты называют дефектами по Шоттки. Интересным примером является вюстит состава от Fe0,gtO до Feo.ssO, образующийся при термическом разложении FeC2O4 и остающийся в метастабильном состоянии после быстрого охлаждения. Его кристаллическая •структура аналогична структуре NaCl, причем до 5% катионных мест оказываются незанятыми. Электронейтральность системы достигается за счет того, что часть катионов железа в решетке имеет заряд +3, а не +2.
Оба вида точечных дефектов имеют решающее значение для процесса перемещения ионов частиц в решетке; они обеспечивают возможность переноса вещества при химической реакции и электрическую проводимость ионной решетки. Концентрация дефектов, устанавливающаяся в обратимом тепловом равновесии, однозначно определяется температурой (экспоненциально растет с температурой); соответственно при охлаждении устанавливается более низкое равновесное значение концентрации дефектов. Например, в решетке AgBr при 300 °C 0,4% всех ионов Ag+ занимают места вне узлов решетки. Причиной появления несовершенств в кристаллической решетке чистого вещества являются колебания элементов кристаллической решетки, энергия которых зависит от температуры.
Кроме точечных дефектов несовершенство кристалла в значительной степени определяется смещениями и перестановками элементов решетки (линейными дефектами), поворотом слоев в кристалле (плоскостные дефекты), образованием пор, пустот и включений (объемные, трехмерные дефекты). Наиболее заметные отклонения от идеальной структуры наблюдаются на поверх
432
Неорганическая химия
ности, вследствие чего именно здесь в наибольшей степени протекают процессы адсорбции. Ход реакции на поверхности раздела фаз в основном определяется находящимися на ней активными центрами. Частицы, которые удерживаются поверхностью, в общем не очень прочно связаны с поверхностью и имеют относительно высокую подвижность. В связи с этим поверхностная диффузия имеет более высокую скорость по сравнению со скоростью диффузии в объеме твердой фазы и сравнима по величине с диффузией в растворах.
33.9.2. Виды реакций в твердой фазе
33.9.2.1. Однокомпонентные реакции без изменения аналитического состава
При изменении параметров состояния температуры и давления твердые вещества индивидуального состава могут переходить из одной структурной формы в другую без изменения стехиометрического состава. Примеры таких переходов — обратимые (энантиотропные) и необратимые (монотропные) превращения модификаций ряда простых веществ и соединений (разд. 33.2.2). Предпосылкой таких процессов является подвижность элементов решетки и перенос вещества, вызванный несовершенством строения твердой фазы. Некоторые свойства твердых веществ определяются не только их структурой и характером дефектов, но и строением микрокристаллитов, в том числе их формой, размерами и составом. Особенно большое влияние строение микрокристаллитов оказывает на механические свойства твердого тела, такие, как твердость, пределы пластической деформации. Проведением специально подобранной твердофазной реакции можно добиться направленного изменения структуры. В результате повышения температуры и достаточно длительного нагревания при постоянной температуре (отжига) можно ускорить рост отдельных кристаллических зерен до больших кристаллов и рекристаллизацию, что обеспечивает улучшение некоторых свойств материала. В отдельных случаях рекристаллизация играет отрицательную роль, например приводит к понижению активности некоторых катализаторов.
Изменение структуры происходит также при спекании порошкообразных твердых веществ. Температурой спекания обычно считают температуру, равную '/г—3А абсолютной температуры плавления. При длительном нагревании при такой температуре кристаллиты спекаются между собой, при этом происходит обмен частицами через границы между зернами. Одновременно уменьшаются число и объем пор, в результате чего происходит значительное уменьшение объема вещества. Процессы спекания имеют особенно большое значение, например, в технологических процессах керамического производства.
Реакции в твердой фазе
433
33.9.2.2. Однокомпонентные реакции с изменением аналитического состава
При подводе энергии, особенно в виде тепла или излучения, твердые вещества часто разлагаются; общий признак такого процесса — выделение газа и образование новых твердых веществ с иным стехиометрическим составом.
Многие такие реакции имеют важное прикладное значение, например получение негашеной извести СаО термическим разложением известняка СаСОа при 900—1000 °C и термическое, разложение сульфата кальция
1200 °с
2 CaSO4 ---> 2 СаО + 2 SO2-[- О2
ГДР располагает месторождениями гипса, и поэтому диоксид серы для производства серной кислоты получают в основном по этой реакции. С целью экономии энергетических ресурсов температуру разложения понижают введением добавок кокса и глины. При этом образующийся СО уносит избыток кислорода, а СаО в ходе твердофазной реакции с глиной образует силикат и алюминат кальция (основную составную часть цемента).
Рассмотрим некоторые типичные примеры такого рода реакций разложения.
Дегидратация кристаллогидратов. При обезвоживании пентагидрата сульфата меди установлена следующая последовательность отщепления молекул воды:
100 °C >100 °C >200 °с
CuSO4-5H2O ---> CuSO4-3H2O ----> CuSO4-H2O ---> CuSO4
При изотермическом нагревании кристаллогидрата при 100 °C отщепляются только четыре молекулы воды, связанные дипольными взаимодействиями. Последняя молекула воды связана непосредственно с сульфат-анионом водородной связью и поэтому удаляется из кристалла только при температуре выше 200 °C.
Так же как и вода, другие растворители, например жидкий аммиак и жидкий диоксид серы, образуют сольваты, которые разлагаются при повышении температуры:
80 °с
A1C13-SO2 —> aici3 + so2
Дегидратирование с одновременным отщеплением элементов кристаллической решетки. Гидраты некоторых галогенидов, например MgCl2-6H2O, А1С13-6Н2О, FeCl3-xH2O, при нагревании не только отдают воду, но и реагируют с образовавшейся водой, образуя галогеноводород в результате реакции сольволи-тического расщепления гетерополярной или полярной ковалентной связи металл — галоген
MgCl2 - 6Н2О -> Mg(OH)Cl + НС1 + 5НаО
28—2027
434
Неорганическая химия
В твердой фазе остается эквивалентное количество ОН-групп, которые могут конденсироваться при дальнейшем нагревании системы.
Термическая дегидратация и конденсация. Многие кристаллические фазы, содержащие в решетке гидроксильные группы, при нагревании соединяются в новые структурные элементы, образуя мостики из атомов кислорода и отщепляя воду. Реакция конденсации осуществляется за счет перемещения протона по водородной связи соседних групп ОН. Реакционноспособные группы ОН есть, например, в кристаллических кисл тах, гидроксидах металлов, кислых и основных солях, а таю во многих силикатных структурах. Примерами таких реакц могут служить дегидратация борной кислоты, дегидратация гидроксида магния, конденсация гидрофосфата натрия (в результате реакции образуется дифосфат, структурные единицы которого состоят из двойных тетраэдров фосфата с мостиковым атомом кислорода), конденсация силикатов [в результате более сложной твердофазной реакции из серпентина (слоистой структуры присоединения); при отщеплении воды образуются ортосиликат магния (островковая структура) и диоксид кремния (объемная структура)]:
400 °C
2В(ОН)3 ---> В2О3+ЗН2О
>200 °C
Mg(OH)2 ---> MgO + H2O
>250 °C
2 Na2HPO4 ---> Na4P2O7 ф- H2O
800 °C
Mge[Si4O10](OH)8 -> 3Mg2SiO4+SiO2H-H2O
Окислительно-восстановительные реакции в твердой фазе. В связи с изменением строения решеток при воздействии тепловой энергии в твердых телах часто происходят электронные переходы, вследствие чего исходное вещество распадается на соединения с различными степенями окисления. Так, например, при нагревании кристаллического хлорида германия(II) до температуры 100 °C в результате диспропорционирования образуются металлический германий и газообразный хлорид германия (IV) :
2 GeCl2 --► Ge 4- GeCl4
Для получения каталитически активных форм различных металлов, таких, как Ni, Fe или Pt, большое значение имеет реакция пиролитического разложения их формиатов и оксалатов. Здесь также идет довольно сложная перестройка структуры, связанная с окислительно-восстановительной реакцией. В связи с существенной перестройкой структуры, которая пр0'
реакции в твердой фазе
435
исходит при переходе от исходного вещества к продукту реакции, возникают твердые вещества с развитой поверхностью, имеющие высокую степень несовершенства.
Путем воздействия световой энергии или других видов излучения можно вызвать в твердых веществах окислительно-восстановительные реакции, например фотохимическое разложение галогенидов серебра или таллия(I):
hv
AgBr --->- Ag Вг
Такой же механизм имеет и индуцируемое излучением освобождение валентного электрона из анионной решетки галогенидов щелочных металлов, что является причиной образования окрашенных центров (голубая каменная соль).
Получение активных промежуточных состояний. В ходе перестройки кристаллической решетки при увеличении подводимой к месту перестройки энергии образуются реакционноспособные промежуточные состояния, имеющие более высокую' энергию [эффект Гедвалля). Это имеет место как при превращениях модификаций простых веществ, так и при всех других изменениях структуры, обнаруженных при твердофазных реакциях. При дегидрировании гипса CaSO4-2H2O при 100°С образуется активная промежуточная форма CaSO4-i0,5H2O (жженый гипс), способный к активному взаимодействию с водой. Рост образующихся при этом кристаллов гипса приводит к его затвердеванию.
33.9.2.3. Многокомпонентные реакции в системе твердая.
фаза — газ
Многие химические процессы, применяемые в промышленности, и главным образом в основном химическом синтезе, основаны на реакциях твердой фазы с газом. К таким процессам относятся, например, получение металлов восстановлением газами, обжиг сульфидных руд, получение основных полу-продуктов неорганического синтеза *— аммиака, серной кислоты и многих органических соединений методами гетерогенного катализа, а также очистка веществ и выращивание монокристаллов (полупроводниковая промышленность). Очень важно здесь то, что в таких гетерогенных системах концентрация дефектов зависит не только от температуры, ио и от равновесия между соответствующими компонентами твердой и газовой фаз. Так, например, состав решетки NiO меняется при увеличении парциального давления кислорода, причем в результате окислительно-восстановительной реакции увеличивается количество ионов О2- в решетке и одновременно образуется эквивалентное количество ионов Ni3+. В соответствии с требованиями об электро-
нейтральности системы в целом, в решетке появляются катионные вакансии.
Наиболее подробно было исследовано взаимодействие металлов с газами, в результате которого на их поверхности образуются твердые соединения, например „ , . „ „
2 Си ....* Си2О
3Mg+N2 --->- Mg3N2
Ag+V2Cl2 ---- AgCl
28*
436
Неорганическая химия
Образование тонких слоев этих соединений на поверхности металла вызывает появление цветов побежалости, увеличение толщины слоя продуктов реакции приводит к окалине. Стадии этого довольно сложного процесса включают адсорбцию газа на поверхности, реакции на поверхности раздела, фаз, образование зародышей кристаллов, образование поверхностного слоя и процессы диффузии подвижных частиц сквозь этот слой в обоих направлениях. Это движение обусловлено уменьшением концентрации реагирующих частиц на поверхности и возникшим вследствие этого градиентом концентрации диффундирующих по ионным вакансиям катионов металла (например, Си+) и одновременным движением дефектов электронов (дырок) (например, Си2+) к поверхности раздела твердых фаз. На поверхности протекает окислительно-восстановительная реакция с образованием нового твердого вещества. Для системы Си/О2 происходит, например, образование оксида меди(1)-:
2 Си+ + !/2О2 + 2е~ -► Си2О
При гетерогенном катализе в качестве катализаторов чаще всего используются смеси твердых веществ, каждое из которых играет определенную роль в стадиях каталитического процесса. Нескомпенсированное потенциальное поле и большое число дефектов кристаллической структуры приводят к тому, что на поверхности возникают особые активные центры адсорбции, а также донорные и акцепторные участки (центры), на которых происходит присоединение или отщепление нуклеофильных и электрофильных частиц, протонов и электронов. Чаще всего используемый в настоящее время катализатор синтеза аммиака имеет состав Fe/K2O/Al2O3. Первой стадией реакции синтеза аммиака является адсорбция N2 иа (1,1,1)-поверхности кубической объемно-центрированной решетки железа. На поверхности катализатора происходит также расщепление Н2 на атомы Адсорбированная и активированная молекула N2 постепенно гидрируется атомарным водородом до промежуточного образования N2Hb. При последующем присоединении атома водорода связь N—X разрывается и образуется молекула аммиака NH3. Другие компоненты катализатора оказывают активирующее и стабилизирующее воздействие на отдельные стадии этого химического процесса.
При реакциях твердых веществ с газами большое значение имеют процессы переноса в газовой фазе. Такого рода химические реакции с переносом всегда возможны в том случае, если в результате химического взаимодействия образуется новое газообразное вещество, а реакция происходит в замкнутом объеме с заранее установленным перепадом температуры. При эндотермической реакции возгонки образовавшееся газообразное вещество разлагается в зоне с более низкой температурой. Экзотермическая реакция возгонки, напротив, приводит к выделению исходных веществ в зоне с более высокой температурой (разд. 36.2.1.4). Этим методом можно получить вещества очень высокой степени чистоты.
33.9.2.4. Многокомпонентные реакции в системе твердое вещество — жидкость
Растворение твердого вещества в растворителе и кристаллизация твердой фазы из раствора являются одними из основных операций препаративной химии, необходимых как в начальных, так и в заключительных стадиях химического синтеза. Особым случаем является разрушение и образование ионного соединения в присутствии полярного растворителя (разд. 33.3). Растворение и кристаллизация твердого вещества в соответствующем растворителе также можно рассматривать как химическую реакцию с переносом вещества. Этим методом можно добиться очистки твердого вещества, а также получать монокристаллы. Процессы образования зародыша, а также особенности его роста рассматриваются в разд. 38.3.4.2. Знание закономерностей процессов кристаллизации позволяет проводить направленную кристаллизацию. Кинетика растворения металлов рассмотрена в гл. 14.
Реакции в твердой фазе
437
Более сложной реакцией, в которой принимают участие одновременно три фазы — твердая, жидкая и газообразная, является коррозия. В процессе коррозии в результате окисления поверхности металла постепенно происходит полное разрушение металла в твердой фазе. Коррозия ускоряется при наличии пленки жидкости (воды), так как продукты реакции могут в ней частично или полностью растворяться. Вследствие этого затрудняется образование слоев, препятствующих диффузии. Образовавшийся раствор электролита способствует протеканию различных окислительно-восстановительных реакций на поверхности. При коррозии железа н его сплава в качестве конечного продукта реакции образуется Fe2+. Согласно одной из схем, обсуждавшихся в литературе (механизм Бокриса), процессы, происходящие при коррозии в упрошенном виде, можно записать следующим образом:
Fe + Н2О -----> FeOH (аде.) + Н+ + е"
FeOH (аде.) ---> FeOH+ + е~
FeOH+ + Н+ ------ Fe2+ + Н2О
Процесс коррозии можно замедлить, если удается создать на поверхности плотные (беспористые) слои оксида. В качестве примера можно привести пассивацию алюминия — метод анодного окисления (анодирования).
33.9.2.5. Многокомпонентные реакции в системе твердое вещество — твердое вещество
При реакциях между твердыми веществами наряду с процессами, протекающими на поверхности раздела фаз, и процессами образования зародышей кристаллов при образовании новой фазы большое значение имеют также процессы переноса в кристаллах. Для ускорения относительно медленной объемной диффузии необходим подвод тепловой энергии. Поэтому все реакции между твердыми веществами, как правило, проводятся при повышенных температурах. Поскольку химическая активность твердых веществ в значительной мере определяется их структурой и величиной поверхности, исходные вещества перед проведением реакции размалывают в тонкий порошок или измельчают каким-либо иным способом, т. е. переводят вещества в состояние с сильно развитой поверхностью. Тем самым осуществляется активация за счет механической энергии (разд. 33.9.2.6). Для проведения реакций между твердыми соединениями чаще всего используют смеси порошков или прессованные таблетки. Для установления равновесия обычно требуется постепенное нагревание до довольно высокой температуры. Для исследования конечных продуктов и кинетических измерений особенно удобны структурно-аналитические и физические методы анализа. При определении механизмов реакции было установлено, что в некоторых твердофазных реакциях перенос компонентов реакции происходит через газовую фазу.
Реакции между твердыми веществами можно разделить на аддитивные и двойные. В качестве примеров аддитивных реакций, в которых новое соединение образуется в результате взаимодействия двух исходных веществ, можно назвать следующие:
Получение шпинели Mg + А12О3 -----► MgAl2O4
Синтез комплексных галогенидов 2 КС1 Ц- SrCl2 --► K2[SrCl4]
Образование силикатов 2 СаО -|- SiO2 -»- Ca2[SiO4]
Во всех этих реакциях перенос вещества происходит главным образом в результате движения ионов в глубь кристаллической решетки. Например, при получении шпинели ионы Mg2+ в решетке MgO и ионы Als+ в решетке А1гО3 движутся в противоположных направлениях к поверхности раздела фаз, гДе и происходит образование шпинели. В последнее время исследование
438
Неорганическая химия
различных типов шпинелей показало, что эта сложная реакция может протекать по-разному. Особые механизмы наблюдаются при получении шпинели в атмосфере кислорода или азота.
Можно показать, что при образовании ортоснликатов M2SiC>4 (M=Mg, С a, Pb, Zn) движущимися частицами являются ионы М2+ и О2~ или ионы М2+ и электроны, причем в последнем случае к фазовой границе переносится кислород из газовой фазы. Иоиы (электроны) в этих системах движутся в одном и том же направлении к поверхности раздела фаз. В системе оксид металла/диоксид кремния в качестве последующих реакций могут протекать также и другие твердофазные превращения, приводящие к образованию пространственной структуры силикатов.
Проведенные в последнее время исследования показывают, что перенос вещества происходит не только в результате перемещения атомов и электронов. При определенных условиях, в особенности при высокой температуре, через газовую фазу могут двигаться также более сложные частицы, например группы МО.
При бинарных твердофазных реакциях из двух исходных веществ образуются также два новых вещества:
Си AgCl ------►* CuCl -ф- Ag
ВаО + СаСО3 —* ВаСО3 + СаО
В случае реакций ионных соединений катионные и анионные составляющие решетки обмениваются, причем движение ионов обеспечивается в основном за счет более подвижного катиона. Менее подвижные комплексные ионы оксокислот, например СО32-, уже при относительно небольшой температуре начинают разлагаться.
GO32~ ----► СО2 4- Со-
образовавшиеся промежуточные частицы СО2 и О2- обеспечивают перенос вещества и используются затем для образования новой решетки.
33.9.2.6. Трибохимические реакции
Трибохимия—раздел механохимии — изучает влияние механической энергии на реакции между твердыми веществами и их структуру. Под влиянием энергии, выделяющейся при трении или ударе, элементы неупорядоченности кристаллической структуры, возникающие за счет теплового движения, увеличиваются, в результате чего возникает активное состояние. За счет ме-ханохимического активирования наблюдаются значительные адсорбционные эффекты, при этом адсорбированные компоненты заполняют субмикроскопи-чсские поры и пустоты более глубоко лежащих слоев твердой фазы. При импульсном торможении струи песка из пескоструйного аппарата на короткое время (10~5—10~6 с) достигается высокоэнергетическое состояние, соответствующее короткоживущей твердотельной плазме. Оно характеризуется электронным и световым излучением (триболюминесценцией), переносом заряда, а также высокой химической активностью.
Трибохимические реакции несколько отличаются от реакций, активируемых тепловой энергией. Целый ряд синтезов, как, например, получение метана из углерода и водорода и образование карбонилов металлов из металла и оксида углерода, вследствие того что они являются эндотермическими, обычно проводят при повышенной температуре Однако если эти синтезы проводить при постоянном подводе механической энергии, то они проходят и при нормальных условиях. Другой реакцией, которая не идет самопроизвольно даже при высокой температуре, является, например,
SIC + 2Н2 ----► Si + СН4
В то же время она, как трибохимическая реакция, идет при комнатной температуре и нормальном давлении. При достаточно интенсивном механохимиче-
*
Реакции в твердой фазе 439
ском активировании многие твердофазные реакции можно провести при низких отрицательных температурах, например при температуре кипения жидкого кислорода.
Трибохимическое активирование химического процесса находит самое разнообразное применение, например для ускорения спекания, повышения растворимости твердых веществ и увеличения активности катализаторов. Напротив, необходимо замедлить трибохимические процессы разложения смазочных веществ в зазорах между трущимися поверхностями, а также вызываемую трением коррозию.
33.9.2.7. Топохимические реакции
Наряду с прочностью связи в твердых соединениях большую роль в процессе протекания твердофазных химических реакций играют кристаллическая структура и строение поверхности реагирующих веществ, а также соответствующие дефекты строения этих веществ. В некоторых реакциях этот эффект настолько усиливается, что направление реакции начинает меняться в зависимости от участков кристаллической структуры. Такие реакции называют топохимическими. При кинетических исследованиях газофазных реакций на медном монокристалле было, например, установлено, что различные кристаллические плоскости имеют разную каталитическую активность и избирательность, что может служить методом избирательного получения продуктов реакции.
Топохимические реакции часто встречаются в химии твердых веществ и имеют большое значение, в частности для понимания процессов гетерогенного катализа, коррозии и полимеризации в твердой фазе.
Во многих случаях структура исходных веществ воспроизводится в структуре продуктов реакции. При некоторых условиях, например при не очень большой температуре, основные структурные элементы не меняют своего положения. Примером могут служить многочисленные соединения включения, возникающие при заполнении атомами, ионами или молекулами структурных пустот или слоев кристаллического соединения. Способность к набуханию и обмену катионов некоторых силикатов, имеющих слоистую структуру, указывает необратимость этих процессов.
Закономерная морфологическая связь между исходным веществом и продуктом реакции наблюдается в реакциях, протекающих на твердых гидроксиде меди, гидроксиде цинка и гидроксиде магния, причем кристаллическая структура исходного соединения в большой степени определяет образование и рост зародыша твердого продукта реакции. Это явление называют топо-таксией.
Примечательны некоторые реакции, в которых влияние ориентации исходного вещества можно проследить на протяжении нескольких стадий реакции. Так, например, рентгенографическая Т-модификация Nb2O5 при дальнейшем нагревании образует различные продукты реакции в зависимости от того, была ли оиа получена окислением NbO2 кислородом воздуха, хлором или нагреванием «ниобиевой кислоты». Это указывает на то, что возможный механизм реакции определяется не идеальной структурой реагирующего вещества, а несовершенствами реальной структуры.
Интересное топохимическое явление открыто при термическом разложении гексафторсиликатов состава M2SiFe (М=К, Rb, Cs, Т1). Все представители этой группы кристаллизуются в кубической кристаллической решетке антифлюорита*. При нагревании происходит частичное превращение, вещества; промежуточно образуется соединение состава M3S1F7 с ионами SiFe н F-в качестве анионных структурных элементов решетки, причем в зависимости от катиона проявляется различие в строении решетки и создаются высокоактив-
* Иначе говоря, в решетке флюорита место Са2+ занимает иои SiFe2 s а место F- — ион М+. — Прим, перев.
440
Неорганическая химия
ные промежуточные состояния. При дальнейшем нагревании отщепляется газообразный SiF4 и остается соответствующая твердая бинарная соль — фторид MF в активной форме. Уравнение реакции можно записать в следующем виде:
M2SiFe --► 2 MF + SiF4
Рентгеноструктурное исследование показывает, что между строением промежуточного вещества M3SiF7 и структурой продукта реакции MF существует тесная топохимическая связь. Например, в случае калиевого соединения оказывается, что кубическая структура KF уже была заложена в структуре решетки K3SiF7. Точно так же положения ионов Т1 и F в Tl3SiF7 способствуют образованию ромбического T1F. В известной мере механизм этой реакции разложения заранее предопределен структурой продукта реакции.
34. ТЕОРИИ РАСТВОРИТЕЛЕЙ В НЕОРГАНИЧЕСКОЙ
ХИМИИ
Как правило, обменные реакции проводят не путем непосредственного взаимодействия исходных веществ, особенно если они находятся в твердом состоянии, а предварительно растворив их в соответствующем растворителе. При растворении твердого вещества связи в кристаллической решетке разрываются и вступающие в реакцию частицы становятся более реакционноспособными. Часто выбор подходящего растворителя позволяет воздействовать в нужном направлении на термодинамику и кинетику химической системы.
Развитые до настоящего времени теории кислот и оснований позволили многое понять в свойствах растворителей и растворов. И наоборот, исследования свойств растворителей в значительной мере способствовали развитию теорий кислот и оснований. Однако еще не создана всеобъемлющая теория растворителей, которая на основе единой концепции строения системы растворитель — растворенное вещество могла бы количественно описать все ее важнейшие свойства. В то же время для различных классов растворителей разработаны теории, которые могут качественно объяснить и предсказать результат влияния природы растворителя на процесс растворения и поведение растворенного вещества в различных реакциях. Среди этих теорий можно назвать теорию сольвосистем, которая разработана для ионизирующихся растворителей, координационную теорию, рассматривающую по большей части растворители с донорно-акцепторными свойствами, протонную теорию, пригодную для растворителей, в которых происходит перенос протонов.
34.1. Теория сольвосистем
Теория сольвосистем, разработанная в основном Яндером, считает важнейшим свойством растворителя его способность к диссоциации. В процессе диссоциации у некоторых молекул
Теории растворителей
441
растворителя происходит гетеролитический разрыв связи. Отделившийся ион соединяется с молекулой растворителя, так что в результате диссоциации растворителя в нем появляются катионы и анионы. Образовавшиеся ионы, особенно если они ко-ординационно-ненасыщенны, сольватируются растворителем. Процесс диссоциации растворителя рассматривают как ионо-тропию. Соответственно можно различать катионотропные и анионотропные растворители (разд. 33.4.3.1). При исследовании различных растворителей было установлено, что чаще всего переносятся ионы Н+, О2~, S2-, F~, СП, Вг-, I-. Схемы диссоциации некоторых ионизирующихся растворителей приведены в табл. В.16.
Интенсивность диссоциации определяется произведением активностей (концентраций) ионов, образовавшихся из растворителя (ионное произведение). Наиболее сильная диссоциация наблюдается в безводной серной кислоте. K=Ch3so+4‘chso-4= = 2,7-10~4 моль2-л-2. Для некоторых других растворителей ионное произведение можно найти в табл. В.18. В слабодиссо-циирующих растворителях экспериментальное определение ионного произведения (например, при помощи потенциометрических п кондуктометрических измерений) связано с определенными экспериментальными трудностями из-за сильного влияния загрязнений.
В соответствии с теорией сольвосистем поведение растворенного вещества определяется его взаимодействием с ионами растворителя. Сольвокислотами называют соединения, которые после растворения и диссоциации повышают концентрацию катиона растворителя. Соответственно сольвооснованиями называют вещества, которые при их растворении и взаимодействии с растворителем повышают концентрацию аниона растворителя. Вещества, которые при диссоциации в растворителе распадаются на ионы, не взаимодействующие с растворителем, называют сольвосолями.
Наряду с описанными выше веществами существуют многочисленные соединения, которые в результате реакции с растворителем смещают равновесие диссоциации растворителя и тем самым вызывают повышение концентрации одного из ионов. Такие вещества называют ансолъвосоединениями. Например, пентафторид сурьмы в трифториде брома является ансольво-соединением:
2 BrF3 BrF2+ + BrF4-
SbF6 + BrF4" SbFe~ + BrF3
SbF6 + BrF3 4=^ BrF2+ + SbFe-
Аммиак в жидком диоксиде серы также проявляет свойства знсольвооснования:
2 NH3 + 2 SO2 (H3N)2SO2+ + SO32-
=CH3, C2HS
Теории растворителей
443
Бромиды, растворенные в расплаве бромида ртути(II), также ведут себя как ансольвооснования. В результате присоединения ионов Вг~ к HgBr2 образуются анионы (HgBr3~).
На основе представлений теории сольвосистем можно описать и использовать для синтеза новых соединений целый ряд реакций, протекающих в ионизирующихся растворителях. Теория является также основой для систематического исследования неводных растворителей, использование которых существенно расширяют возможности препаративной и аналитической химии.
Примеры. Проведение в жидком аммиаке кислотно-основной реакции
Mg2Si + 4 NH4+ ---> 2 Mg2+ + SiH4 + 4 NH3
позволило получить силан высокой чистоты со значительно более высоким выходом продукта реакции, чем в водном растворе. В растворителе трифториде брома, обладающем высокой ионизирующей и фторирующей способностью оказалось возможным синтезировать гексафторовисмутаты и тетра-фтороаураты. В основе синтеза — реакция нейтрализации сольвокислоты и сольвооснования, например
BrF2AuF4 + AgBrF4 ---»- AgAuF4 2 BrF3
Очень часто наблюдается аналогия между химическими реакциями в ионизирующихся растворителях и в водных растворах. Так, многие соединения, имеющие в своем составе анионы растворителя, проявляют амфотерные свойства, т. е. как и слабоосновные гидроксиды в водных растворах, по-разному взаимодействуют с кислотами и основаниями. Так, выпадающий при добавлении в жидкий аммиак раствора амида калия осадок Al (NH3)3 растворяется в избытке KNH2 в результате комплексообразования
A1(NH2)3 + nh2- --► HHNH^r
При добавлении сольвокислоты, например NH4C1, растворение объясняется нейтрализацией амид-ионов с образованием сольватированных ионов AF+:
A1(NH2)3 + 3NH4+ A13+4-6NH3
Поскольку амид-ионы в жидком аммиаке являются более сильными основаниями, чем гидроксил-иони в воде, амидные комплексы образуются даже с элементами, для которых неизвестны одноядерные гидроксокомплексы, например Ks[Zr(NH2)4].
Особое значение для реакций синтеза имеют сольволитические взаимодействия в ионизирующихся растворителях. Под действием растворителя происходит распад полярных связей с одновременным присоединением иона растворителя (разд 33 8), например синтез диамида сернистой кислоты аммонолизом тионилхлорида в жидком аммиаке:
SOC12 + 4 NH3----- OS(NH2)2 + 2 NH4+ + 2 Cl-
Аналогичным образом протекает сольволитическое расщепление перхлоратов ® Жидком диоксиде азота, в результате которого образуется нитрозилпер-хлорат:
Mg(C104)2 + 2 N2O4 -->- Mg(NO3)2 + 2 NOC1O4
34.2. Координационная теория растворителей
Теория сольвосистем, основанная на представлениях о диссоциации молекул растворителя, несмотря на свои большие возможности, не может объяснить многие явления, так как не
444
Неорганическая химия
учитывает донорно-акцепторных свойств растворителей. В то же время эти свойства в значительной мере определяют растворяющую способность ионизирующихся ковалентных соединений, сольватирование растворенных частиц и, наконец, термодинамику и кинетику протекающих в растворе реакций.
34.2.1. Растворители — доноры и акцепторы пары электронов
Для понимания многих химических процессов в растворах оказалось целесообразным разделить растворители на донорные (ДПЭ — донор пары электронов) и акцепторные (АПЭ — акцептор пары электронов) в зависимости от доминирующего в молекуле реакционного центра. Однако нельзя упускать из виду, что в молекулах растворителя могут одновременно существовать нуклеофильные и электрофильные центры, которые могут вступать в реакцию в зависимости от свойств взаимодействующей с ними частицы.
Важнейшей группой ДПЭ-растворителей. являются л-доно-ры, обладающие одной или несколькими несвязывающими парами электронов. Нуклеофильные центры часто сосредоточиваются на О-, N- и S-атомах молекулы растворителя. Самые известные представители этой группы — вода, спирты, эфиры, кетоны, амины, амиды кислот, тиоэфиры и тиокетоны. В л-донор-ных растворителях, к которым относятся среди прочих также ароматические и непредельные углеводороды, координационное присоединение основано на взаимодействии с л-электронной системой.
В акцепторных растворителях преобладают центры, имеющие координационно-ненасыщенный характер и способные к присоединению нуклеофильных частиц, так как их молекулы имеют незаполненные орбитали. К таким соединениям относятся ковалентные галогениды, обладающие? свойствами кислоты Льюиса, такие, как SbF5, SbCls, TiCl4, AsCU и BrF3, которые в нормальных условиях находятся в жидком состоянии.
Важную группу АПЭ-растворителей составляют протонные растворители. Общим признаком этих растворителей является наличие у них кислого атома водорода, который обеспечивает возможность образования водородных мостиков с основными электроотрицательными центрами. За счет такого механизма образования связи протонные растворители обладают свойствами акцепторов пары электронов.
34.2.2. Донорные и акцепторные числа
Растворяющая и сольватирующая способности растворителя — результат комбинированного действия многих факторов. Наряду с электростатическим взаимодействием постоянных и
Теории растворителей
445.
индуцированных диполей для поляризующихся молекул — участников реакции характерно также наличие дисперсионных сил, а для протонных растворителей — жесткой связи через водородные мостики. Кроме того, в процессе растворения необходимо преодолеть силы взаимодействия между молекулами растворителя, при этом структура жидкости, особенно ярко выраженная в растворителях с водородными мостиковыми связями, должна сильно меняться. Такое сложное поведение растворителя нельзя описать при помощи только физических характеристик (дипольного момента и диэлектрической проницаемости). Поэтому для оценки координирующих свойств растворителя пытались найти эмпирический параметр, при помощи которого можно выразить как бы результирующую всех взаимодействий. Для этого измеряют наиболее характерные параметры некоторой стандартной реакции (энтальпию реакции, константу равновесия, константу скорости, максимум поглощения света сольватохромного красителя) в различных растворителях. Затем на основе этих данных распределяют изученные растворители по их растворяющей способности.
Для количественной оценки координирующего действия растворителей наиболее удобны определяемые из эксперимента донорные числа, как мера донорной способности, и акцепторные числа, как мера акцепторной способности растворителя.
Определение донорного числа (DN) (Гутман, 1966 г.) основано на координационном взаимодействии между сильным акцептором SbCU и нуклеофильными центрами молекулы ДПЭ-растворителя в разбавленном растворе 1.2-дихлорэтана. Этот растворитель выбран в качестве среды сравнения вследствие’ его инертности в соответствующих реакциях. Стандартная реакция протекает следующим образом:
1,2-дихлорэтан
ДПЭ + SbClj —....— ДПЭ-SbClg
Донорное число определяется как установленная калориметрическим методом энтальпия реакции с обратным знаком:
Dn = — кН (З7)-
Донорное число Dn учитывает все, в том числе электростатические, взаимодействия в этой системе.
Выбранное вещество сравнения SbCU оказалось очень Удобным, так как оно образует только аддукты состава 1:1, а его акцепторная способность дает возможность измерить достаточно заметный эффект даже в слабых ДПЭ-растворителях.
Удалось также найти подходящую единицу измерения и воспроизводимый стандартный способ определения акцепторной способности растворителя (Гутман, 1975 г.). Акцепторное-число An оценивается экспериментально путем измерения хи
446
Неорганическая химия
мического смещения сигнала ядерного магнитного резонанса атомов 31Р, которое наблюдается при растворении вещества сравнения триэтилфосфиноксида в соответствующем растворителе. Образование координационной связи между основным атомом О в соединении фосфора и электрофильным центром растворителя вызывает поляризацию Р—О-связи:
R
I Г+
R—Р=О —>АПЭ
R (*=СгН5)
Уменьшение электронной плотности вблизи атома Р можно измерить в виде смещения сигнала ядерного магнитного резонанса, что позволяет достаточно ясно установить различие в акцепторных свойствах растворителя.
(СаНУзРО оказалось практически идеальным веществом для определения акцепторного числа. Оно обладает лишь одним основным центром в стереохимически фиксированном положении. Центральный атом Р прикрыт инертными этильными группами. Сильно основные свойства соединения позволяют также обнаружить взаимодействие со слабоэлектрофильными частицами. Для практического применения важно также, что соединение с этильными группами растворимо почти во всех растворителях и очень устойчиво к сольволитическому расщеплению даже в сильиокислых растворах.
Измеренные значения химического смещения экстраполируют на бесконечное разведение. Наиболее целесообразная шкала установлена по соглашению. Значения акцепторного числа Адг в отличие от донорного числа являются безразмерными. Следует также иметь в виду, что эти шкалы не коррелируют между собой.
Поскольку каждый растворитель в зависимости от свойств системы может проявлять как нуклеофильные, так и электро--ф ильные свойства, его координационное поведение следует характеризовать парой чисел: DN и An. В табл. В.17 приведены донорные и акцепторные числа растворителей, а также их диэлектрические проницаемости. Неполные данные по некоторым растворителям указывают на современное состояние экспериментальных исследований этой проблемы.
Из рассмотрения табл. В.17 можно сделать некоторые выводы. Как и следует ожидать, небольшие значения DN имеют углеводороды и галогеноводороды, а также соединения, в которых основность центра понижена за счет индуктивных факторов (например, РОС13). Растворители с DN~>20 характеризуются сильными донорными свойствами. Наибольшие значения донорного числа наблюдаются у замещенных аминов.
Самые низкие значения акцепторного числа имеют апротонные растворители. Соединения с кислой группой СН—, такие, как нитрометан и хлороформ, уже имеют заметные акцепторные свойства. Далее следуют растворители с группами ОН, на'
Таблица В.17. Донорные и акцепторные числа, а также диэлектрическая проницаемость наиболее распространенных растворителей
Растворитель Формула dn an er (25 °C)
«- Гексан СНз- (СН2)4- СНз — 0,0 1,9
1,2-Дихлорэтан СН2С1-СН2С1 0,0 16,7 10,1
Тетрахлорид углерода СС14 — 8,6 2,2
Хлороформ СНС1з — 23,1 4,7
Бензол С6Нб 0,1 8,2 2,3
Нитрометан СНз—NO2 2,7 20,5 36,7
Нитробензол c6h6no2 4,4 14,8 34,8
Оксихлорид фосфора ОРС13 11,7 — 14,0
Бензонитрил CgHgCN 11,9 15,5 25,2
Ацетонитрил CH3CN 14,1 18,9 36,0
1,4-Диоксан °\ /° 14,8 10,8 2,2
Ацетон СН3СОСН3 17,0 12,5 20,7
Метанол СНзОН 30,0 41,3 32,6
Этанол С2Н5ОН — 37,1 24,3
Диэтиловый эфир С2Н5ОС2Н5 19,2 3,9 4,2
Тетрагидрофуран (СН2)4О 20,0 8,0 7,4
н-Трибутилфосфат («-С4Н9)зРО 23,7 — 6,8
М,Ы-Диметилформамид НСОЩСНзЪ 26,6 16,0 36,7
N,N-Диметилацетамид »CH3CON(CH3)2 27,8 13,6 37,8
Диметилсульфоксид (CH3)2SO 29,8 19,8 46,7
Вода н2о 33,0 54,8 78,5
Пиридин c5h5n 33,1 14,2 12,3
Гексаметилфосфортри-амид [ЩСНзЫзРО 38,8 10,6 29,6
Аммиак NH3 59,0 — 22,0 (-33°C>
Триэтиламин N(CH3)3 61,0 — 2,4
Уксусная кислота СН3СООН — 52,9 8,2 (20 °C)
Трифторуксусная кислота CF3COOH — 105,3 —
Трифторметилсерная кислота CF3SO3H — 129,1
448
Неорганическая химия
пример вода и спирты. Самые большие акцепторные числа имеют протонные кислоты.
Между донорными и акцепторными числами и диэлектрической проницаемостью растворителей нельзя найти какой-либо закономерной связи.
Из табл. В.17 также видно, что некоторые растворители, как, например, вода и метанол, проявляют одновременно сильные донорные и акцепторные свойства.
Оценка имеющегося экспериментального материала показывает, что координационные свойства растворителя можно количественно описать и предсказать с определенной степенью точности на основе донорных и акцепторных чисел. Это касается прежде всего ряда свойств, связанных с сольватацией растворенных частиц. Если доминируют нуклеофильные свойства растворителя (большое Dn, малое Ау), то достаточно учитывать донорные числа. Так, при полярографическом осаждении катионов из таких растворителей установлена связь между потенциалом полуволны окислительно-восстановительной системы, па-пример Na++e‘'4^Na, и донорным числом ДПЭ-растворителя, что позволяет заранее оценить неизвестное значение потенциала полуволны при заданном донорном числе. Потенциал полуволны оказывается тем более отрицательным, чем прочнее сольватная оболочка, т. е. чем больше донорное число DN В то же время в случае преобладания электрофильных свойств растворителя можно ограничиться рассмотрением акцепторных чисел. Они особенно удобны для выявления различий сольвати-рующей способности растворителей при взаимодействии с анионами. Если же одновременно проявляются ДПЭ- и АПЭ-свой-ства растворителя, то необходимо привлекать оба числа—донорное и акцепторное, так как наиболее полная характеристика координационной способности растворителя становится возможной лишь в рамках модели двух параметров.
34.2.3.
Сольватация в донорных и акцепторных растворителях
Сольватация вносит значительный вклад в свободную энтальпию процесса растворения. Наблюдаются существенные различия в специфическом взаимодействии растворителя и растворенной частицы. Электрофильные частицы, например катионы, сольватируются преимущественно ДПЭ-растворителями. Вследствие присоединения молекул растворителя значительно увеличивается эффективный ионный радиус. Так, например, в диметилсульфоксиде размеры сольватированного иона лития достигают размеров иона тетрабутиламмония. Основные ненТ; ры молекул растворителя (атомы О, N или S) в сольватной оболочке ориентированы к иону металла. Связь имеет хараьте!
Теории растворителей
449
полярной межатомной связи, так называемого ион-дипольного взаимодействия. Сольватация оказывается, вообще говоря, тем сильнее, чем больше донорное число растворителя. Растворители можно расположить в следующем порядке по мере уменьшения свободной энтальпии сольватации катионов:
[(CH3)2N]3PO > CH3CON(CH3)2,
(CH3)2SO > HCON(CH3)2 > CH3CN > CH3NO2
Аналогичным образом степень сольватации нуклеофильных частиц, например анионов, в АПЭ-растворителях пропорциональна акцепторному числу Ац. Частицы, имеющие в своем составе сильноотрицательные атомы, например F, О, N, в большей степени сольватируются протонными АПЭ-растворителями. Эти растворители являются донорами протонов и на основе механизма образования водородных мостиков создают стабильную структурированную сольватную оболочку, в которой протон находится в потенциальном поле двух электроотрицательных атомов. Особенно прочные водородные мостики образуются с группами О—H---F, О—Н---О, О—H---N и N—Н---О. Впрочем, способность к образованию водородных мостиков, а следовательно, и способность к сольватации резко снижается при увеличении размеров и поляризуемости анионов.
В ДПЭ-растворителях, напротив, сольватация анионов выражена очень слабо. Причиной этого является отталкивание отрицательных основных центров аниона и молекул растворителя. В соответствии с теорией жестких и мягких кислот образование сольватной оболочки около больших поляризуемых анионов (I-, SCN~, S2") возможно только под действием дисперсионных сил (разд. 33.4.3.4). Жесткие же анионы (F-, ОН", NR2~) в таких средах совершенно «обнажены» и поэтому проявляют высокую активность в реакциях с нуклеофильными заместителями. Предпочтительная сольватация катионов, вследствие чего образуются сольватные комплексы большого размера, снижает электростатическое притяжение между сольватированными катионами и анионами, у которых практически не имеется сольватной оболочки. Такое состояние ионов в растворе способствует увеличению реакционной способности анионов, которая увеличивается еще и за счет высокой диэлектрической проницаемости растворителя.
Вода, метанол, этанол и другие подобные растворители, обладающие одновременно донорными и акцепторными свойствами, способны к сольватации как электрофильных, так и нуклеофильных частиц. Это объясняет способность этих растворителей растворять самые разнообразные вещества.
При растворении ионного соединения или соединения, состоящего из полярных молекул, в смеси ДПЭ- и АПЭ-раство-Рйтелей катионы сольватируются преимущественно молекулами 29~2027
450
Неорганическая химия
ДПЭ-растворителя, а анионы — молекулами АПЭ-растворите-ля. Это явление называют селективной сольватацией. Так, например, в водном ацетонитриле AgNO3 образует катионы состава [Ag(CH3CN)4-n]+ (n=0, 1, 2) и анионы [МОзСНгО)*]-. Однако часто молекулы обоих растворителей входят в состав сольватной оболочки.
34.2.4. Образование ионов и диссоциация
Первой стадией процесса растворения вещества, состоящего из полярных молекул, является поляризация ковалентной связи растворителем, что, вообще говоря, приводит к гетеролити-ческому расщеплению на положительную и отрицательную частицы. Многочисленными примерами можно доказать, что способность растворителя расщеплять вещество на ионы в первую очередь определяется его донорным и акцепторным числами, а не диэлектрической проницаемостью ег. Даже растворитель с большой диэлектрической проницаемостью не способен гете-ролитически расщепить связи растворенной частицы, если он не имеет достаточной координирующей способности. Так, например, хлорная кислота в серной кислоте (ег = 80) не образует ионов, в то время как в водном растворе (ег = 78,5) О—Н-связь в молекуле НС1О4 полностью разрывается.
При растворении какого-либо вещества механизм образования ионов и вид полученной частицы в значительной мере зависят от специфической химической координирующей способности растворителя. В ДПЭ-растворителе полярная ковалент-6-|- 6—
ная связь М—X разрывается под действием нуклеофильно ’ атаки на положительный центр М, причем большая часть нс обходимой для разрыва связи энергии выделяется при сольва тации образовавшегося катиона, например
FeCl3 + 6 (CH3)2SO -> [Fe((CH3)2SO)e]3+-t-3 Cl-
Стабилизация хлор-ионов происходит благодаря образованию аквакомплекса.
При растворении в АПЭ-растворителе расщепление полярной связи происходит в результате электрофильной атаки на более отрицательный центр X, причем гетеролизу способствует образование комплексных или сольватированных анионов, на пример
FeCls + NOCl --> [NO]++[FeCl4]-
У растворителей с более высокими значениями донорного или акцепторного числа основная часть энергии, необходимой для разрыва связи, выделяется за счет координационной стабилизации возникающей частицы. Поэтому на процесс растворения влияет также диэлектрическая проницаемость средЫ-
Теории растворителей
451
В растворителях с высокой диэлектрической проницаемостью участие растворителя в образовании ионов увеличивается за счет влияния диэлектрических свойств. В зависимости от значения диэлектрической проницаемости ионы, образовавшиеся в результате разрушения ионной решетки или гетеролиза полярной связи, либо ассоциированы, либо находятся в растворе в виде отдельных ионов, окруженных сольватной оболочкой. При использовании растворителей с низкой диэлектрической проницаемостью возникают преимущественно ионные ассоциаты и ионные пары, в которых два или более иона связываются электростатическими силами. Ассоциированные ионы образуют самостоятельные частицы и вследствие взаимного насыщения электрических зарядов не дают вклада в электрическую проводимость раствора. При переходе к среде с более высокой диэлектрической проницаемостью электростатическое притяжение между катионами и анионами в соответствии с законом Кулона (разд. 32.3.1) ослабляется и образуются отдельные, большей частью сольватированные ионы. При растворении полярных соединений в растворителе с высокой диэлектрической проницаемостью это состояние достигается без каких-либо промежуточных состояний. Процесс перехода ионных ассоциатов в свободные ионы называют диссоциацией. Весь процесс можно записать с помощью следующей схемы последовательных реакций образование ионных ассоциатов диссоциация
1МХ]5 ZL • —---------* [М+Х-] ----------- М+Соль + X СОЛЬВ
твердое ионная отдельные сольва-
вещество пара тированные иоиы
В растворителях с ег<20 существуют исключительно ионные пары; при ez>40 преобладают свободные ионы. В растворителях 20<8г<40 имеется равновесие между обеими формами, причем соотношение между содержанием ассоциированных и свободных ионов зависит также от заряда и размеров ионов.
Как можно показать на примере хлорида алюминия А1С13, зная донорное и акцепторное число растворителя, можно сделать выводы о составе комплекса в растворе, а также о механизме образования ионов.
При растворении безводного хлорида алюминия в растворителе с небольшими значениями донорного и акцепторного чисел (например, в дихлорэтане (DN = 0, 4и=16,7)) в результате разрушения решетки образуются частицы состава AlsCU, в которых две молекулы А1С13 координационно-насыщенны за счет образования «хлорных мостиков». Вследствие слабой координационной способности растворителя дальнейший распад оказывается невозможным.
По-иному ведет себя А1С13 в апротонных растворителях, которые имеют высокую донорную и низкую акцепторную спо-29*
452
Неорганическая химия
собность [например, для тетрагидрофурана (ТГФ) £>№=20, Лд/ = 8]. При растворении вокруг катиона возникает стабильная сольвоструктура, однако сольватация аниона С1_ в этом растворителе невозможна. Поэтому стабилизация аниона происходит в результате его реакции с A1CU с образованием иона [A1CU]-. Этот особый механизм образования ионов, при котором центральный атом исходного соединения при его расщеплении входит в состав как катиона, так и аниона, называют аутокомплексообразованием.
4 А1С13 + 4 ТГФ -► [А1 (ТГФ)4]3+ + 3 [А1С14]“
Если растворитель обладает одновременно высокой донорной и акцепторной способностью (например, метанол ZAi = 30, A,v = 4l,3), то происходит гетеролиз связи А1—С1 с образованием сольватированных ионов А13+ и С1_:
А1С13 + (х + 4)СН3ОН -» [А1(СН3ОН)4]3+-ЬЗ[С1(СН3ОН)х]-
34.2.5. Влияние растворителя на термодинамику и кинетику реакций
Как правило, при смене растворителя, в котором проводится реакция, ее равновесие смещается. Причина этого в основном состоит в том, что сольватация исходных веществ и продуктов реакции в различных растворителях происходит по-разному. Для проведения реакции наиболее удобно использовать такой растворитель, в котором равновесный выход продуктов реакции будет наибольшим. Это особенно выгодно, если изменение свободной энтальпии сольватации AZ<0 достаточно велико по абсолютному значению, т. е. сольватация обеспечивает устойчивость продуктов реакции.
Вследствие сочетания различного рода взаимодействий между растворителем и участниками реакции положение равновесия зависит от многих факторов. Так, протолитическое равновесие между кислотой и основанием при изменении растворителя зависит не только от кислотности (основности) растворителя, но и от его способности к образованию координационных соединений. Поэтому, например, константы диссоциации карбоновых кислот в воде в 105—106 раз больше, чем в безводном этаноле.
Изменение состава растворителя может оказать влияние на скорость и порядок гомогенной реакции.
В реакциях обмена лигандов комплексного соединения (5л-1-механизм) полярная молекула ‘RX в переходном состоянии распадается на катион R+ и анион Х~. В протонных растворителях с большим значением А.у этот процесс сильно ускоряется, так как они способны сольватировать анионы Х~ с образованием водородных мостиков. Одновременно снижается
Теории, растворителей
453
энергия образования активированного комплекса (энергия активации) . Реакция ускоряется по мере увеличения кислотности растворителя, поэтому реакции, протекающие в этаноле по бимолекулярному механизму, в безводной муравьиной кислоте становятся мономолекулярными.
Реакции типа SN2, напротив, протекают с большой скоростью в апротонных растворителях с высоким донорным числом Dn и малым акцепторным числом Я#. В данном случае не происходит отщепления аниона на лимитирующей стадии процесса (образовании активированного комплекса). Исследования кинетики процесса показывают, что константы скорости в апротонных растворителях, как правило, в 105 раз больше, чем в протонных. Это объясняется тем, что присоединяющийся анион (нуклеофильная частица) находится в исключительно реакционноспособном состоянии ввиду отсутствия сольватной оболочки.
Нуклеофильная реакционная способность анионов зависит не только от степени их сольватации, но и от степени ассоциации с соответствующим катионом. Связанный в ионную пару анион имеет значительно меньшую реакционную способность, чем свободный. С увеличением размеров ионов ионные пары становятся неустойчивыми, поэтому в апротонных растворителях галогениды тетраалкиламмония, имеющие сравнительно большие размеры, более активно обменивают галогены в комплексных соединениях, чем галогениды лития и натрия.
34.2.6. Представления о химических функциях
В своих исследованиях кислотно-основных равновесий и теории растворов Усанович доказал, что различия в реакционной способности веществ при обмене электронами, ионами или молекулами основываются на различиях координационных свойств участников реакции (разд. 33 4.3.5). Гутман подробно изучил проблемы взаимодействия между частицами, различающимися электронной донорно-акцепторной активностью и. на этой основе изложил представления о химических функциях.
Отправной пункт этих представлений — реакционная способность не есть свойство молекул вещества, а есть свойство, проявляющееся только во взаимодействии с участниками химической реакции. Электронное взаимодействие в процессе химической реакции, для которого характерны координационное взаимодействие донора и акцептора электронов или обмен электронов в окислительно-восстановительной реакции, имеет место только в том случае, если Участники реакции — ионы, атомы или молекулы — обладают различными Функциями электронной плотности. Только с этой точки зрения можно представить себе в целом электронное состояние системы, состоящей из реагирующих между собой частиц.
Определяющей тенденцией в ходе реакции является выравнивание электронной плотности в системе. В процессе осуществления донорной функции Частицы ее электронная плотность понижается, а электронная плотность акцепторной функции — увеличивается. Электронные взаимодействия различного типа соответствует следующим химическим функциям:
Функция донора пары электронов (ДПЭ) — основание Льюиса;
функция акцептора пары электронов (АПЭ) — кислота Льюиса;
454
Неорганическая химия
функция донора электрона (ДЭ) — восстановитель;
функция акцептора электрона (АЭ) — окислитель.
Для коррелирующих функций характерны донорные и акцепторные свой ства одного типа, например пара ДПЭ — АПЭ или ДЭ — АЭ.
При анализе отдельных стадий процесса может оказаться, что последе-вательно проявляются функции электронного взаимодействия различного типа; так, например, редокс-процесс мысленно можно разделить на две стадии: собственно электронный переход (ДЭ — АЭ-взаимодействие) и стабилизация образовавшейся частицы в ходе химической реакции с образованием
обращение
Рис. В. 16. Схема обращения функций для реакции Li + 1/2F2->Li+ + F-.
координационного соединения (ДПЭ — АПЭ-взаимодействие). В этом случае наблюдается об ращение функций, когда образовавшиеся частицы проявляют противоположные некоррелирующие функции. Восстановитель (ДЭ) после перехода в окисен ную форму действует как кислота Льюиса (АПЭ), а окислитель (АЭ) после перехода в восстановленную форму — как основание Льюиса Химические функции, связанньь с обращением функций, назы вают сопряженными, паприме ДЭ —АПЭ или АЭ —ДПЭ. Эти общие положения можно пояснить на примерах.
1. Реакция металлического лития (ДЭ), как сильного вое становителя, приводит к образованию Li—>Li++e~, который практически не проявляет спо собности к обратной реакции при соединения электрона. Поэтому он
компенсирует низкую электронную плотность за счет присоединения частиц со свободными парами электронов, например F-, Н2О, т. е. ион лития является уже акцептором пары электронов (АПЭ).
2. Напротив, при реакции сильного окислителя фтора (АЭ) (УгРег+е-—>-F_) образуются ионы фтора, которые могут выравнивать повышенную электронную плотность за счет присоединения к свободной паре электронов акцепторов пары электронов, имеющихся в системе, например ионов Li+. В данном случае ионы фтора имеют функцию доноров пары электронов (ДПЭ).
На рис. В 16 наглядно представлены соотношения функций при протекании реакции Li+1/2 F2—>-Li++F-. Сопряженные функции изображены в одному ряду, а коррелирующие — по диагонали.
В процессе выравнивания электронной плотности слабых окислителей И восстановителей после окончания первой стадии процесса участвуют различные функции. Так, например, ион иода I- в воде проявляет слабую функцию донора электронов. В процессе восстановления какого-либо вещества иона’ ми иода появляется окисленная форма 12, которая имеет слабую АПЭ-фуию цию. Низкая электронная плотность повышается в результате реакции иоДа либо как акцептора пары электронов (кислоты Льюиса), либо как акцептора
электронов — окислителя
Подобным же образом в реакцию образования координационных соеДИ' нений типа ДПЭ — АПЭ включается окислительно-восстановительная реаК' ция, в ходе которой происходит обращение функций и компенсация элекТ' ронной плотности. Так, усиление окислительных свойств КМпО4 при подкДС' лении раствора объясняется присоединением к основным атомам О коми'
Теории растворителей
455
лексного иона протонов из кислоты (АПЭ). Тем самым облегчается отрыв ионов О2~, и центральный ион Мп7+ имеет возможность интенсивнее проявлять функции акцептора электронов (АЭ).
34.3. Протонная теория растворителей
Многие растворители обладают способностью переносить протоны к соответствующим акцепторам (кислотные свойства) или присоединять протоны к молекуле растворителя (основные свойства). К этому классу растворителей можно применить развитую Бренстедом теорию кислот и оснований, которую можно также назвать протонной теорией растворителей.
34.3.1. Классификация растворителей в зависимости от их кислотно-основных свойств
Оценку кислотно-основных свойств растворителей наиболее удобно проводить на основе констант автопротолиза растворителя, а также констант диссоциации кислот и оснований (табл. В.18). Значения рК относят к стандартной системе, в качестве которой выбрана вода, т. е. для получения сравнимых данных рассматривают протолиз молекул растворителя в двух направлениях:
Растворители-кислоты HL + Н2О < —> Н3О+ + L”
Растворители-основания HL + Н2О < —> H2L+ + ОН-
Этим протолитическим разновесиям соответствуют:
константа кислотного растворителя Ка (HL) = -Д-Нз^ L - (38)
константа основного растворителя Kb (HL) = (39)
Активность присутствующей в избытке воды принята равной единице (aHgo=l).
В протонных растворителях идет процесс автопротолиза, в Результате которого образуются катионы лиония и лиат-ионы:
HLH-HL ч=ь H2L++ L-
Константа равновесия диссоциации является константой авто-вротолиза, или, что то же самое, ионным произведением растворителя:
Ащь=йн2ь+' яь~ (4 0)
3 зависимости от значения константы кислоты и основания Растворители можно разделить на классы. Растворители, способные к диссоциации и проявляющие как кислотные, так и Основные свойства и для которых можно найти определенные
456
Неорганическая химия
Таблица В.18. Распределение растворителей по их кислотно-основным свойствам3
растворитель РДа (HL) РКЬ (HL) РКщ "г Классификация
Метиламин Аммиак 33 3,36 4,75 29 22 Амфипротные,
9,42 (-50 °C) (—33 °C) основные
Анилин 27 —. 6,8
Вода Метанол 15,74 15,2 15,74 16 14 16,9 78,5 33 Амфипротные,
Этанол 16 16 19,5 25 нейтральные
Уксусная кисло- 4,75 20,2 14,45 6,3
та 3,75 6,2 (19 °C)
Муравьиная 16 58,5 Амфипротные,
кислота Фтороводород 3,14 27 9,7 84 кислые
Серная кислота —7 28 3,85 100
Пиридин — 8,85 — 12,3
Диметилформа-мид Диметилсуль- — 14 — 36,7 Апротонные,
— 14 48,9 основные
фоксид Ацетон Ацетонитрил 21,2 24 — 20,7 37,5 Апротонные,
Бензол — — — 2,3 нейтральные
Нитрометан 10,2 — — 35,9 1 Апротонные,
Нитроэтан 8,4 — — 29,5 ' кислые
а рХо, а также и рД автопротолиза относятся к стандартной системе — водным растворам; здесь же приведены данные по диэлектрической проницаемости ег Если это не отмечено особо, все данные приведены для 25 °C
значения, относят к классу амфипротных растворителей. Остальные растворители, у которых диссоциация не доказана, называют апротонными. Впрочем, не существует однозначного деления на эти два класса растворителей, так как определение малых значений степеней диссоциации связано с большими экспериментальными затруднениями.
Оба класса растворителей можно в свою очередь разделить на основные, нейтральные и кислотные растворители. У нейтральных амфипротных растворителей, к которым можно отнести воду и спирты, сила кислоты и основания одинакова, хотя их диэлектрическая проницаемость может сильно различаться. Большое значение Ка и малое — Къ характерно для кислотных амфипротных растворителей, таких, например, как безводная серная и уксусная кислоты, в то же время основные амфипротные растворители, такие, как жидкий аммиак и метиламин, имеют невысокое значение Ка и большое — Кь-
Апротонные растворители не способны отщеплять протоны, поэтому для них невозможно определить константу кислоты-
Теории, растворителей
457
Исключением являются немногие апротонные растворители, которые в особых условиях проявляют кислотные свойства. К ним относятся первичные нитроалканы RCH2—NO2, которые в сильно основной среде переходят в ациформу RCH — NO (ОН).
Основная функция апротонных растворителей проявляется значительно сильнее, так как молекулы растворителя часто содержат атомы О, N или S, к которым, как к основным центрам, могут присоединяться протоны. Апротонные растворители, у которых основная функция выражена наиболее сильно, называют основными апротонными растворителями. К ним относятся, например, такие широко распространенные растворители, как диметилформамид и диметилсульфоксид.
К нейтральным апротонным растворителям относится большая группа соединений, у которых нет кислотных и основных свойств (например, бензол и тетрахлорид углерода) или у которых чрезвычайно малы Къ (например, ацетонитрил, ацетон).
Важной характеристикой растворителя, влияющей на механизм реакции, является диэлектрическая проницаемость, от которой в первую очередь зависит состав частиц, на которые распадаются вещества-электролиты в растворе (разд. 34.2.4). Кроме того, диэлектрическая проницаемость растворителя влияет на процесс диссоциации, а также кислотно-основное равновесие. Так, рекомбинация ионов в нейтральные молекулы происходит преимущественно в растворителях с низким значением диэлектрической проницаемости е, а увеличение & способствует их диссоциации. Выбор подходящего растворителя или их смеси позволяет получить любое значение е среды, в которой протекает реакция. Этим широко пользуются при титровании в неводных растворителях (разд. 39.9).
34.3.2. Применение суперкислот и сильноосновных сред в неорганическом синтезе
При растворении пентафторида сурьмы в жидком фтороводороде или во фторосерной кислоте образуются среды с исключительно высокой активностью протона — суперкислоты.
В системе HF — SbFs идет реакция
SbF6 + 2 HF H2F+ + SbFe-т- e. образование ионов дигидрофторония, которые отличаются °чень высокими кислотными свойствами (рКа« —17). Возникшие в результате реакции диссоциации HF ионы F~ связывается SbFs в координационно-насыщенные ионы SbF6~. Супер-кислотные свойства этой смеси проявляются вследствие того, 'iT° ионы SbF6' практически не способны присоединять прото-ftbI- Поэтому освободившиеся протоны вынуждены присоеди
458
Неорганическая химия
няться к слабоосновному центру F в молекуле HF и находятся в исключительно реакционноспособном состоянии в ионах H2F+.
Совсем иначе идет реакция в системе HSO3F — SbFs:
+HSO3F
SbF6 + HSO3F H[SbF6(SO3F)] --------> H2SO3F+ Д [SbF6(SO3F)F
Вследствие устойчивости связи сера — фтор во фторосерной кислоте не происходит отщепления фторид-ионов и образования комплексного соединения — кислоты. Протоны присоединяются к находящейся в избытке фторосерной кислоте с образованием иона лиония H2SO3F+ (рКа~—20), который считается самым сильным протонирующим соединением.
Кислотность может быть увеличена также добавлением триоксида серы, образующего комплексный анион дифторотетра (фторосульфато)сурьмы (V), который почти не проявляет основных свойств, т. е. не может присоединять протонов:
HSO3F
H[SbF5(SO3F)] + 3SO3 H[SbF2(SO3F)4] --> H2SO3F+ + [SbF2(SO3F)4]'
Такие кислоты могут протонировать даже насыщенные углеводороды. Например, если в смесь HSO3F—SbFs (1:1) ввести метан в десятикратном избытке при 140°C под давлением, то сначала образуется промежуточный продукт СН5+, распадающийся на СН3+ и Н2, а далее получаются ион карбония углеводородов с большим числом атомов углерода и непредельные соединения. Существование иона СН5+ доказано масс-спектрометрическими методами.
Для решения некоторых задач неорганического синтеза большое значение имеют среды с сильноосновными свойствами В водной среде невозможно создать основность большую, чем та, которую имеют гидратированные ионы ОН-, —Ль = 55,3 (разд. 33.4.1.5). Гидратированные ионы ОН- сильно отличаются по степени основности от свободных ионов ОН-. Стабилизированная водородными мостиками гидратная оболочка экранирует свободную пару электронов гидроксид-иона, в то же время для свободного иона ОН- (Кь~ 1020) способность к присоединению протона возрастает на несколько порядков Применение в качестве среды дипольных апротонных растворителей, в которых невозможна сольватация анионов, позволяет проявиться сильноосновным свойствам свободного иона ОН'
В соответствии с методикой Джолли гидроксид калия в виде тонкого порошка суспендируют при перемешивании в диметил сульфоксиде или диметоксиэтане. При этом часть КОН растворяется. Затем добавляют кислоту, которую необходимо депр° тонировать. Суммарная реакция записывается следующий образом:
2КОН (тв ) + НА ► к+ + А- + КОН • Н2О (тв.)
Водород
459
В данном случае имеет место равновесие между двумя твердыми фазами. Растворенные в дипольном апротонном растворителе свободные ионы ОН- отнимают от кислоты протоны с образованием аниона. Вода связывается с нерастворившимся КОН в виде твердого гидрата и тем самым выводится из реакции. Таким образом, КОН является также и осушителем.
Значение рК константы равновесия реакции составляет рК=рКа—31. Следовательно, в таких средах можно депрото-нировать очень слабые кислоты, например GeH4 (рКа = 25) и РНз (рКа = 27), и получать необходимые продукты реакции. Этим методом можно также с хорошим выходом получить из циклопентадиена CsH6 ион пентадиенил С5Н5-— важный промежуточный продукт в синтезе ферроцена.
35. НЕМЕТАЛЛЫ
Неметаллические свойства элемента выражены тем сильнее, чем легче его атомы принимают электроны. Связь электрона с ядром определяется средним расстоянием электрона на данной орбитали от ядра и эффективным зарядом ядра. Последний зависит прежде всего от степени экранирования заряда ядра внутренними электронами, а также от перекрывания орбита-лей внутренних и внешних электронов. Поэтому неметаллы занимают правую верхнюю часть периодической системы элементов. Легко также понять, что в соединениях одного и того же элемента его неметаллические свойства усиливаются с ростом положительного заряда иона. Неметаллы отличаются еще и тем, что у их атомов заселенность валентных орбиталей близка к максимально возможной согласно принципу Паули. Поэтому атомы неметаллов проявляют тенденцию путем присоединения электронов приобретать электронную конфигурацию ближайшего инертного газа. Неметаллы называют также электроотрицательными элементами.
Лишь немногие из неметаллов встречаются в природе в виде простых веществ. Это — инертные одноатомные газы, газообразные кислород и азот, состоящие из неполярных молекул, сера в виде твердого вещества, структурные единицы которой— кольцевые молекулы Ss, углерод — в виде графита и алмаза (полимерные ковалентные структуры). Преобладающая часть неметаллов встречается в природе в виде соединений с другими элементами.
35.1. Водород
Весь комплекс физических и химических свойств водорода определяется особенностями строения его атома, на которых следует остановиться особо.
460
Неорганическая химия
Емкость электронной оболочки атома водорода — два электрона, так как при п=1 квантовые числа I и т равны нулю.
Водород образует, как правило, ковалентную двухэлектронную связь, причем его валентная оболочка оказывается полностью заполненной. Однако, как показывает существование иона Н2+ (разд. 6.2.1), спаривание электронов не является необходимым для образования связи.
Отсутствие внутренних оболочек и, следовательно, эффекта экранирования приводит к тому, что электроны сильно взаимодействуют с ядром.
К особенностям химического поведения водорода следует отнести способность к образованию гидридов различных типов, в которых возможно образование как «протонных» (например (HF)X), так и «гидридных» водородных мостиковых связей , электронодефицитных соединениях (В2Нв). В некоторых комн лексах переходных металлов атом водорода непосредственна связан с атомом металла.
Необходимо отметить, что реакции водорода в различны, соединениях можно в одних случаях трактовать как окисли тельно-восстановительные либо как кислотно-основное взаимодействие (разд. 33.4), в других случаях — как кислотно-основное взаимодействие либо как реакции осаждения (разд. 33.3-.2), а в третьих — как окислительно-восстановительные либо ка < реакции обмена лигандами (разд. 33.6.4).
Во всех трех агрегатных состояниях водород состоит из молекул Н2. Это неполярное молекулярное вещество, в твердом и жидком состоянии между молекулами обнаруживаются чрезвычайно слабые вандерваальсовы силы. Поэтому водород имеет очень низкие температуры плавления и кипения.
Для протекания реакции очень прочная молекула водорода должна диссоциировать, что требует большой затраты энергии:
Н2 ---► 2Н АД0 ==432,1 кДж/моль
Диссоциация молекулярного водорода может протекать по трем механизмам.
1. ) Гомолитический распад, когда образуются водородные радикалы, т. е. атомарный водород
2) Образование иона Н~ за счет присоединения одного электрона. Это возможно лишь в реакциях с сильными донорами электронов, например щелочными металлами. Реакция образования Н~ из молекулярного водорода — эндотермический процесс:
^^(газ) ----► Н (газ) А/7 = -(-216,0 кДж/моль
Н (газ) -j- е~ -*- Н" (газ) АД =—67,0 кДж/моль
1/2Н2 (газ)е~ ----->- Н_ (газ) АД = 4-149,0 кДж/моль |
Гидрид-ионы могут присутствовать в солеобразных соединениях (например! гидриды щелочных металлов). Если центральный атом металла не обладаем
Водород
461
столь явно выраженными электроположительными свойствами, то связь металл — водород содержит ковалентную составляющую.
3) Отщепление электрона с образованием Н+-иона (протона):
Н (газ) — е~ --»- Н+ (газ)
Водород в химических реакциях может выступать как восстановитель или как окислитель. В этом проявляется промежуточное положение водорода в окислительно-восстановительном ряду.
Н Рис. В. 17. Типы гидридов в периодической системе элементов.
Связь водорода с другими элементами в зависимости от их электроотрицательности носит более или менее полярный характер (рис. В. 17), что может служить основой для классификации бинарных гидридов. Вследствие того что водород находится примерно в середине шкалы электроотрицательности, он образует как ковалентные, так и ионные соединения (рис. В.17), а также соединения промежуточных типов. Особый класс составляют соединения включения водорода с металлами (разд. 36.16.1).
В зависимости от положения элемента в периодической системе атом водорода в соединении с ним приобретает либо отрицательный, либо положительный заряд (табл. В. 19). Величина и направление дипольного момента связи Н—X в значительной степени определяют физические и химические свойства гидридов.
Гидриды щелочных и щелочноземельных металлов гетеро-полярны. Менее полярные гидриды других элементов, например (ВеНг)х, (MgH2)x, (А1Н3)х, (1пН3)л; и др., благодаря наличию «гидридных» водородных мостиков образуют полимерные структуры; эти гидриды труднолетучи и термически малоустой-
462
Неорганическая химия
LIF
1172,3
§1 1088,6
£ 1004,8
586,2
502,4
418,7
334,9^
251,2 §
О
-83,7
-167,5
1675 -
[ и Ш W V W ' I И Ш IV V VI VH
Номер группы периодической, системы элементов
Рис. В. 18. Энтальпии образования гидридов.
537 >
чивы. Известны также легколетучие молекулярные ассоциаты типа В2Н6 (разд. 32.3.2), SiH4, Si2H6 (рис. В. 17, табл. В. 19). Чем больше отрицательный заряд на атоме водорода, тем более сильным восстановителем может быть соединение. Однако восстановительная активность зависит также от многих других факторов (разд. 33.5.1).
Чем больший положительный заряд сосредоточен на атоме водорода в соединении, тем более кислые свойства он проявляет. И наоборот, основные свойства атома водорода усиливаются с ростом отрицательного заряда на нем. Протон является жесткой кислотой вследствие отсутствия электронной оболочки
Водород
463
f Таблица В.19. Парциальный заряд на атоме водорода в бинарных гидридах
Соединения с положительным зарядом Заряд , усл. ед. Соединения с зарядом, близким нулю Заряд, усл. ед. Соединения с отрицательным зарядом Заряд, усл. ед.
HF +0,25 AsH3 +0,02 SiH4 —0,05
НС1 +0,16 Н2Те --0,01 1пНз —0,05
Н2О -0,12 СН< —0,01 в2нв —0,05
НВг +0,12 SbH3 —0,01 ZnH2 —0,06
NH, +0,06 РН3 —0,01 СаН2 —0,09
H2Se +0,06 GeH4 —0,01 А1Н3 —0,12
H2S HI +0,05 4-0,04 SnH4 —0,02 ВеН2 MgH2 СаН2 SrH2 ВаН2 LiH NaH КН RbH CsH —0,13 —0,18 —0,29 —0,31 —0,33 —0,50 —0,50 —0,58 —0,58 —0,59
весьма малого радиуса, а также из-за его устойчивости к воздействию внешних электрических полей.
В разд. 33.3.2.3 было дано объяснение закономерностям изменения температур кипения для большого числа ковалентных полярных соединений, в том числе для соединений с водородными мостиковыми связями.
Гидриды можно разделить на экзотермические и эндотермические в зависимости от знака энтальпий их образования (рис. В.18). Экзотермичны реакции водорода с самыми электроположительными металлами, а также с некоторыми сильно электроотрицательными неметаллами. Если же разность электроотрицательностей невелика, реакции образования гидридов эндотермичны. Положение равновесия реакций элементов с молекулярным водородом определяется изменением свободной энергии Дб°. Для расчета равновесия необходимо знать изменение энтропии в этой реакции, например
2 Li (тв.) 4- Н2 (газ) -► 2 LiH (тв.)
AS° =—56,0 —130,44-49,3 = —137,1 Дж/(моль-К)
Изменение энтропии в этом случае относительно велико, так как при образовании кристаллического продукта степень упорядоченности повышается. Напротив, образование большинства летучих гидридов не сопровождается значительным изменением энтропии.
464
Неорганическая химия
35.1.1. Реакции водорода
Молекулярный водород не очень реакционноспособен. С галогенами водород реагирует после инициирования по радикально-цепному механизму. Обычно при нагревании молекула Н2 гомолитически расщепляется. Образующийся атомарный водород восстанавливает, к примеру, многие оксиды до низших оксидов или до металлов (разд. 36.2.1). В присутствии платинового, никелевого или палладиевого катализаторов водород вступает в реакции уже при комнатной температуре. Каталитическое действие оказывают также соединения некоторых тяжелых металлов или их ионы. Например, ионы Ag+ и МпО4~ восстанавливаются молекулярным водородом. Реакции водорода при низких температурах протекают вследствие образования реакционноспособной связи с металлом-катализатором (переходным металлом). При этом происходит поляризация молекулы водорода.
В воде водород плохо растворим. Раствор его практически не обнаруживает кислых свойств рКа = —38,6.
Молекулярный водород реагирует с некоторыми солями и комплексами переходных металлов. Протекающие при этом реакции можно систематизировать по их механизмам.
Реакции присоединения
Среди реакций этого типа достойны внимания окислитель ные реакции с присоединением водорода. В качестве примера можно привести реакцию образования IrH2CICO(PR3)2 и: IrClCO(PR3)2 путем окислительного гидрирования при ком натной температуре и нормальном давлении.
Реакции замещения
Примером реакции этого типа может служить замещение i соединении [ (CH3)4N]3 [Pt(SnCl3)5] группы SnCl3 — на водо род. Реакцию проводят в ацетоне. При 30 °C и 4,9-107 Па обра зуется соль состава [(CH3)4N]3 [HPt(SnCl3)4].
Комбинированные реакции
В комбинированных реакциях гидрируются органические лиганды. Далее происходит отщепление органического лиганда от центрального иона. Металл, с которым был связан лиганд, сам катализирует реакцию гидрирования и использование специального катализатора не обязательно.
При реакциях с водородом алкильных или арильных производных металлов образуются соответствующие углеводороды. Так, реакция водорода с раствором (n-C5H5)2Ti (СН3)2 в алифатическом углеводороде при комнатной температуре приводит к образованию метана и дипиклопентадиенилтитана.
Водород
465
^5.1.1.1. Гидрид-ион
Образование гидрид-иона при реакциях водорода с сильно электроположительными металлами можно доказать, проводя электролиз солеобразных гидридов в расплавленных галогенидах щелочных металлов. При этом водород выделяется на аноде. Химическое поведение солеобразных гидридов можно объяснить присутствием в них гидрид-иона.
Свободный Н~-ион имеет неожиданно большой радиус 0,208 нм. В кристаллической решетке соединений радиус Нойона значительно меньше (~ 0,153 нм). Энергии решеток гидридов сравнимы с энергией решеток фторидов (рис. В. 18) и хлоридов. Гидрид-ион — сильный восстановитель. Стандартный потенциал пары Н2/Н~ составляет £° = —2,24 В. По отношению к воде и многим органическим соединениям ридрид-ион проявляет восстановительные свойства. Протекающую при этом реакцию синпропорционирования Н~-}-Н+—И12 в то же время можно рассматривать как кислотно-основное взаимодействие. При взаимодействии с водой гидрид связывает ионы II" и образуется щелочной раствор: II ф-Н2О—ЯДф-ОН.
Гидрид-ион—настолько сильное основание, что действие на него воды проявляется как окислительно-восстановительная реакция (разд. 33.4.1.5). С точки зрения теории жестких и мягких кислот и оснований (разд. 33.4.3.4) гидрид-ион — чрезвычайно мягкое основание.
Гидрид-ион выступает в качестве донорного лиганда при образовании гидридных комплексов. Различают комплексы элементов главных подгрупп (Х = В, Al, Ga) и переходных элементов (М.п(СО)3Н, Та(СбН5)Н3, [Р1(РН3)гС1Н]). Гидрид-ион реагирует с акцепторами электронов ХН3, имеющими свободную орбиталь. Устойчивость получающихся соединений падает в ряду B>Al>Ga, что соответствует изменению кислотности по Льюису соединений ХН3 (разд. 33.4.3.3)
Доказательством того, что в комплексном ионе ХН4~ лигандом является отрицательно заряженный ион водорода, может быть ход реакции:
4 Н2О + ХН4---> 4 Н2 4- Х(ОН)3 + ОН-
Происходящая окислительно-восстановительная реакция Между протонами воды и Н"-лигандами сопровождается реакцией кислотно-основного типа.
Гидрид-ионы реагируют с комплексами металлов так же, как Молекулярный водород (разд. 35.1.1).
В реакциях замещения происходит нуклеофильный обмен ^Держащихся в комплексах галогенид-ионов на гидрид-ионы. ^та реакция может сопровождаться восстановлением центрального иона. В качестве источника гидрид-ионов могут выступать 3°—2027
466
Неорганическая химия
как солеобразные гидриды, так и гидридные комплексы, на-
пример NaBHj С5Н5Те(СО)2С1 С5Н5Те(СО)2Н NaBH4 CoBr2(PR3)2 CoH(PR3)2
Во второй реакции помимо реакции нуклеофильного замещения происходит еще восстановление центрального иона гидрид-ионами.
Реакция присоединения происходит, например, между сульфатом меди(II) в этанольном растворе и NaBH4 в присутствии трифенилфосфина PR3, при этом образуется соединение
R3P\ „ЛК н
R5PZ
Могут протекать также комбинированные реакции, т. е. одновременно с процессом восстановления центрального иона происходит гидрирование органического лиганда, например
н
Мп+(СО)3
NaBHz, или UAIH4
Мп(С0)3
н
35.1.1.2. Ион водорода Н+
Процессы, приводящие к образованию положительно заряженного иона водорода, представляют особый интерес (разд. 35.1). Для осуществления этого процесса необходима значительно большая энергия, чем для образования других однозарядных положительных ионов, например ионов щелочных металлов. Те соединения, в которых атом водорода имеет хотя бы небольшой положительный заряд (при электролизе водород выделяется на катоде), ведут себя в определенных растворителях как протонные кислоты. При этом образуются сольватированные ионы водорода. Состояние протона в воде подробно рас-смотрено в разд. 33.4.
Важно отметить, что у соединений с частично положительным атомом водорода наблюдается повышенная склонность к образованию ассоциатов. Соединения этого типа (HF, Н2О, NH3) обладают пониженной летучестью. Водородные мостиковые связи в них отличаются от таковых в боранах (разд-32.3.2.3) тем, что в первом случае имеет место положительная поляризация атома водорода (по терминологии Сандерсона это «протонные мостики»).
Водород
467
Кислотные свойства бинарных гидридов, кроме прочего, зависят от величины положительного заряда на атоме водорода (табл. В.19). Определенную роль играют также электронодонорные свойства атома, связанного с протоном. Так, фторид-ион—.сильный донор (разд. 35.3), поскольку его электронные пары сосредоточены в небольшом пространстве (для них невозможно нахождение на ^-орбиталях). Вследствие этого, несмотря на сильную полярность связи Н—F, плавиковая кислота в водном растворе —кислота слабая (разд. 33.4.4).
Водородные соединения с положительным зарядом на атоме водорода не всегда кислоты (например, NHs). Если в соединении, содержащем водород, имеются орбитали, занятые свободной электронной парой, возможно даже присоединение протона. Подобные соединения в воде ведут себя как основания.
Из-за чрезвычайно малого размера протона напряженность поля вблизи этой частицы весьма велика, поэтому Н+ обладает очень большим поляризующим действием.
Поведение протона в реакциях определяется, помимо кислотно-основных равновесий, также его окислительно-восстановительными свойствами. Протон — это чрезвычайно жесткая кислота.
В воде (ан+=Ю-7 моль/л) окислительно-восстановительный потенциал системы Н+/Н2 меньше, чем стандартный потенциал (Е°=0):
Н+ (aq) (10-7 М) + е~ -► 1/2Н2 Е<> = —0,414 В
Некоторые металлы, такие, как натрий и кальций, окисляются даже в этих условиях. Большинство же металлов или ионов металлов способно окисляться протоном лишь при больших его концентрациях (растворение металлов в кислотах).
В реакциях обмена лигандами с образованием комплексов ион Н+ часто выступает как «кислотный катализатор».
Взаимодействие иона Н+ с комплексными соединениями со смешанными лигандами рассматривались ранее (разд. 35.1.1).
Реакции присоединения. Твердый дициклопентадиенилрений-гидрид адсорбирует газообразный хлороводород с образованием [ (ft'CsHs) 2ReH2] Cl.
Реакции замещения. Простейшая реакция этого типа состоит в замещении катиона на протон. Например, из соединения Na [Со (СО) 4] образуется соединение Н[Со(СО)4]. Примером реакции замещения с участием протонов может служить следующая реакция:
2НВг IrH3(PR3)3 -> IrHBr2(PR3)3 + 2H2f
Комбинированные реакции. Из множества возможных реакций можно выделить
Зо*
468
Неорганическая химия
реакции протонов с лигандами, связанными с центральным атомом о-связью;
реакции протонов с лигандами, связанными с центральным атомом л-связью.
Рассмотрим, например, взаимодействие аллильного комплекса вольфрама с HCI. При этом образуется комплексный «про-неновый катион», в котором пропиленовый лиганд, первоначально присоединявшийся о-связью, образует л-связь с атомом вольфрама.
HCL
Л-связывание
Можно выделить соль «пропенового катиона» с анионом гексафторофосфата.
Электрофильная атака с участием Н+ протекает через образование «неклассического» карбониевого иона:
сн2
СНз
I сн3
© сн, М "*=“— II сн I сн3
На этих примерах можно убедиться в том, что неорганические и органические реакции полезно рассматривать с единой точки зрения.
Рекомендуемые опыты
1. Каталитические реакции водорода. В три последовательно соединенные промывные склянки наливают коричневый раствор К1з (раствор К1з получают растворением металлического иода в растворе иодида калия). В течение 15 мин через склянки пропускают ток чистого водорода. Восстановления не происходит. В растворы К1з добавляют 2 мл золя платины и снова пропускают водород. В этом случае каталитически активированный водород восстанавливает коричневый раствор К1з в бесцветный иодид.
2. Реакция водорода при комнатной температуре. Собирают прибор, описанный в опыте 1. В среднюю промывную склянку наливают 30 мл 0,1 М раствора AgMnO4. Сильный ток водорода пропускают до тех пор, пока на стенках склянки не образуется серебряное зеркало. Ион МпО42- также восстанавливается до бесцветного иона Мп2+.
3. Реакция симметричного синпропорционирования, Н~ и Н+. Небольшое (на кончике шпателя) количество гидрида кальция или лития вносят в фарфоровую чашку с водой. В результате бурной реакции образуется водород, который иногда самовоспламеняется. Сразу после окончания реакции опре' деляют pH раствора.
Кислород
469
4. Зависимость реакции окисления Н~ от парциального отрицательного заряда на атоме водорода. В воду вносят немного полимерного (А1Н3)Х. Реакция окисления Н- хотя и идет, но не так бурно, как в случае СаН2.
5. Донорные свойства гидридного лиганда. 20 мг борогидрида натрия NaBHi вносят в стакан с водой (100 мл). Борогидрид натрия до тех пор реагирует с водой, пока реакция раствора не станет щелочной (индикаторная бумага). При подкислении раствора 0,1 М раствором НС1 (добавляют до каплям) выделение водорода возобновляется (почему?).
6. Кислотно-основные взаимодействия. С помощью индикаторной бумаги определяют pH нескольких растворов: 0,01 М НС1, 0,01 М HF и раствор NH3. Результаты определения объясняют с помощью представлений теории кислот и оснований (разд. 33.4).
7. Реакция осаждения протона. К небольшому количеству (несколько капель) разбавленной НС1 добавляют 1,5%-ный раствор бензоата калия до выпадения белого осадка труднорастворнмой бензойной кислоты. Эта реакция может рассматриваться также как реакция кислотно-основного взаимодействия:
С6Н6СОО~ 4- Н3О+ ----> С6Н5СООН + Н2О
8. Окислительные свойства положительно поляризованных атомов водорода. Кусочек ленты или стружки магния помещают в 1 М раствор NH4C1. Наблюдают выделение газа. Ионы водорода, образующиеся при протолизе [NH4]+-hohob, действуют как окислитель, принимая электроны атомов магния.
Вопросы, и задания
1. В чем заключается сходство и различие электронных конфигураций атома водорода, с одной стороны, и атомов элементов главных подгрупп первой, четвертой и седьмой групп — с другой?
2. Могут ли существовать следующие соединения: АгН, SH6, НС13 и TiH4? Ответ обоснуйте.
3. Как можно используя ионную модель, объяснить различия энтальпий образования ионных гидридов и соответствующих фторидов (рис. В. 18)?
4. Обсудите термодинамические и кинетические аспекты процессов синтеза из элементов LiH, (А1Н3)Х, AsH3 и HI.
5. Объясните, почему HI в большей степени подвергается протолизу в воде и является более сильным восстановителем, чем HF.
6. В гидридных комплексах переходных металлов водород выступает в виде простого лиганда Н~. а) В какие реакции обмена лигандами он способен вступать? б) Как доказать присутствие иона Н_? в) В каких случаях гидридные комплексы термодинамически и кинетически особенно устойчивы? (Учтите влияние других лигандов, связанных с центральным атомом.)
7. Объясните поведение водорода и его ионов в рамках теории жестких и мягких кислот и оснований.
35.2. Кислород
Как это характерно для всех элементов второго периода, кислород по своим химическим и физическим свойствам заметно отличается от более тяжелых элементов той же группы (главной подгруппы). (Этот вопрос объясняется в разд. 35.3, посвященном фтору.) Химическое поведение кислорода в значительной степени определяется электронной конфигурацией его атома ls22s22p4.
Такая электронная конфигурация обусловливает осуществление устойчивой рп—рл-связи в парамагнитной молекуле кис-
Неорганическая химия
& (РазЛ- 6.3.4). Атом кислорода может образовывать лорох^ двойные связи за счет рп—рпили ря—йл-связывания. такя< соответствии с электронной конфигурацией атом кислоро-о?кет образовать две о-связи (координационное число ато-Да М-^слорода в этом случае равно двум), либо иметь координа-ма к .ое число три или четыре за счет образования одинарных ЦИ011 гтвНТНЬ1Х связей.
К0ВгТо правилу октета атом кислорода может присоединить два 1л.рона, давая ион О2-.
элек кислорода может получить законченную, устойчивую кронную конфигурацию, присоединяя один электрон и об-элек *двая одну одинарную ковалентную связь (ОН~-ион). Раз°₽я ектронные оболочки атома или иона кислорода характе-дся гораздо меньшей электронной плотностью, чем в ато-ризуи-2 ионе фтора, поэтому их поляризуемость значительна. ме ^гоянь1е соединения, включающие О2-ионы, имеют высокие решеток. При взаимодействии с кислородом образуют-энер1 сТабилизируются соединения элементов в высоких степе-ся 11 ^целения (разд. 33.4.3.4).
няХ 1Сокая электроотрицательность атома кислорода и наличие го несвязывающих электронных пар (которые не могут де-У не п1<зоваться с использованием d-орбиталей) приводит к об-лока0анию мостиков О—Н-0 (разд. 32.3.2.3).
ра3ПрОсТОе вещество — кислород — относится к классу непо-1 Лх молекулярных веществ. Он реагирует со всеми другими ЛЯРн Ь1МИ веществами*, давая оксиды. При этом молекула кис-ПР°С да сначала должна диссоциировать на атомы — процесс, лороА одЯщий с затратой энергии. Для образования оксид-иона ТмоЛеКУляРного кислорода требуется затрата энергии о|() кД*/МОЛЬ’
-Постараемся по возможности классифицировать реакции 1 кУлЯРного кислорода.
мол?о^олитическое расщепление на атомы
^гОДгаз) --> О (газ) Д//° — 246,8 кДж/моль
раСщепление связи между атомами кислорода и присоедине-электР0Н°в с образованием оксид-иона
Ние О (газ) + 2е~ -> О2- (газ) и 700 кДж/моль
Присоединение электронов без расщепления связи кисло-
^3-кислород с образованием пероксид- или надпероксид-ио-нов
О2 + 2е- —> О22~ О2 + е~ -----► О2_
Т~Кр°ме гелия> неона к аргона, Ахметов Н. С. Неорганическая химия.
М рь1сц1ая школа> 1975. — Прим, перев.
Кислород
471
Потеря одного электрона и образование диоксигенил-катио-на
О2 — е~ --► О2+
Присоединение одного атома кислорода с образованием полиморфной модификации — озона
О2 + О ---► О3 ДН° ==—144,4 кДж/моль
Путем присоединения электрона к молекуле озона образуется озонид-ион
О3 -)- в” 1 ► О3“
Перечисленные выше ионы в виде изолированных частиц могут присутствовать в ионных соединениях.
35.2.1. Классификация оксидов
Оксиды элементов можно классифицировать по различным признакам, в том числе по энтальпиям их образования.
Несмотря на высокую положительную энтальпию образования О2~-иона, энтальпия образования оксидов большинства металлов и оксидов некоторых неметаллов отрицательны (табл. В.20).
Можно было бы проводить классификацию и по величинам AG°o6p. В первом приближении изменением энтропии реакции можно пренебречь* и заменить AG°06p на ДЯ°Обр. Таким обра-Таблица В.20. Парциальный заряд на атоме кислорода и свойства некоторых оксидов
Оксид Заряд на атоме кислорода А"°обр-кДж/г-экв. Координационное число атома кислорода Кислотно-основные свойства
Na2O -0,81 208,1 8 Сильное основание
СаО —0,57 317,8 6 То же
MgO —0,42 301,0 6 Слабое основание
A12O3 -0,31 273,7 4 Амфотерные свойства
ZnO —0,29 174,2 4 То же
B2O3 —0,24 21,6 2 Слабая кислота
SiOs —0,23 211,3 То же
SnO2 —0,17 145,3 Слабые амфотерные
свойства
Р«О10 —0,13 150,7 Кислота средней силы
co2 —0,11 98,7 Слабая кислота
so3 —0,06 66,6 Сильная кислота
SeO3 —0,06 — То же
N2O3 —0,05 4,2 » »
СЦО7 —0,01 18,8 » »
* Вклад энтропийного фактора в Дб°обр Для различных оксидов пример-но одинаков (см. напр.,, разд. 21.Ц2). — Прим, перев.
472
Неорганическая химия
зом, энтальпия образования служит мерой прочности связи в соединении.
Прочность связи находится в прямой зависимости от ее полярности. Полярность рассматриваемых связей элемент — кислород зависит от разности соответствующих электроотрицательностей. На этом основан принцип классификации оксидов
Рис. В.19. Зависимость свойств некоторых оксидов непереходных элементов от величины парциального заряда на атоме кислорода.
по величине парциального заряда на атоме кислорода в данном соединении (ср. разд. 35.1).
Разность электроотрицательностей определяет принадлежность соединения к тому или иному типу (табл. В.20).
Оксиды элементов главных подгрупп со значительным парциальным зарядом на атоме кислорода являются ионными соединениями (Na2O, СаО). Соединения с немного меньшим парциальным зарядом на атоме кислорода имеют полимерное строение, причем связь элемент — кислород в них приобретает в значительной степени ковалентный характер (В2О3, SiO2). И наконец, оксиды с атомами кислорода, на которых сосредоточен очень небольшой отрицательный заряд, представляют собой молекулярные вещества (Р4О10, СО2, оксиды азота, серы и некоторые оксиды галогенов).
Классификация оксидов непереходных элементов в зависимости от их строения и типа связи представлена на рис. В.19.
Кислород
473
Координационное число центрального атома в оксидах понижается с уменьшением отрицательного заряда на атоме кислорода (табл. В.20).
При увеличении разности электроотрицательностей в оксидах неметаллов происходит изменение типа соединений:
Увеличение отрицательного заряда на атоме кислорода, увеличение разности электроотрицательностей xq—х^
Мономерные Полимерные Полимерные соедиие- Иоиные соеди-
молекуляр- ——> молекуляр » ния с высокой сте- — > нения
ные вещества ные вещества пенью агрегации
(имеют сходство с интерметаллическими соединениями, характеризуются металлическом электропровод-
L НОСТЬЮ)
I Важный отличительный признак полимерных соединений с высокой степенью агрегации (такой тип соединений часто встречается также у сульфидов, селенидов, нитридов и фосфидов) — металлический тип проводимости. Остальные же свойства оксидов (температуры кипения, плавления, летучесть, растворимость и др.) определяются их принадлежностью к соединениям определенного типа.
Другая схема классификации оксидов основана на проявляемых ими кислотно-основных свойствах при реакциях с водой. Чтобы и здесь как критерий можно было использовать величину отрицательного заряда на атоме кислорода, будем придерживаться определения кислот и оснований по Усановичу (разд. 33.4.3.5): кислота или кислотный оксид —это акцепторы электронов.
Оксиды с большим отрицательным зарядом на атоме кислорода—'доноры электронов, т. е. основные оксиды. При уменьшении разности электроотрицательностей кислорода и его партнера в оксиде этот оксид проявляет как акцепторные, так и донорные свойства, т. е. речь идет об оксиде с амфотерными свойствами. При незначительной разности электроотрицательностей, т. е. при еще меньшем отрицательном заряде на атоме кислорода, оксиды ведут себя как акцепторы электронов и проявляют кислотные свойства (табл. В.20).
Действие воды на оксид можно рассматривать как реакцию между кислотным и основным оксидами. Результат реакции зависит от того, больший или меньший отрицательный заряд имеет атом кислорода в оксиде по сравнению с зарядом атома кислорода в молекуле воды (—0,25).
С позиций термодинамики имеется взаимосвязь между энтальпией гидратации оксида и его кислотно-основными свойствами. На рис. В.20 сделана попытка показать это для элементов третьего периода. Оказывается, что амфотерные оксиды ха-
474
Неорганическая химия
Рис. В.20. Энтальпия гидратации высших оксидов элементов третьего периода.
рактеризуются также наименьшей энтальпией гидратации (рис. В.21, а и б).
Кислотно-основные свойства большинства амфотерных оксидов, труднорастворимых в воде, можно выразить с помощью следующей схемы:
ОН" + Е+ (БОН) (ТВ.) Н++ ЕСТ
В зависимости от партнера по реакции образуются гидратированные катионы E+(aq) или анионы EO~(aq). Процесс установления равновесия протекает через множество стадий (гидратация, гидролиз, реакции конденсации, см. разд. 33.7). Применительно к реакциям оксида алюминия возможные равновесия можно условно* выразить следующими уравнениями:
Д1О2--|-Н2О+Н+ ч—> А1(ОН)3(тв.) :₽=> АР++ЗОН-
Линия «амфотерности» на рис. В.21,а не совсем строго следует определенной разности электроотрицательностей. Во мно-
* В действительности в растворе присутствуют гидроксокомплексы типа [АКОНК]-, [А1(ОН)]2+ (см. разд. 36.8). — Прим, перев.
Кислород
457
Sc
Ti
Сг
Мп Fe Со Ni Си Zn
V
г
г
2
г г г г /. г= г
з
3
3
3
3 3 3=3 3
4 == = 4 =~~=. 4=4 = 4'
5
5
е
6 6
7
5
Рис. В.21. Положение линии амфотерных оксидов.
— степень окисления элемента в оксиде соответствует номеру группы; амфотерные ок-ды со степенью окисления N— 2 соединены тонкими линиями; 6 — оксиды элементов в различных степенях окисления.
х случаях положение линии лишь приблизительно описывает кислотно-основные свойства соответствующего оксида. Так, ок-
сиды цинка и галлия действительно амфотерны, тогда как оксиды кадмия, ртути(II), индия(III) и таллия(III) имеют слабо основной характер (разд. 36.3 и 36.17.1).
Если элемент образует несколько оксидов, то всегда оксид элемента в более высокой степени окисления проявляет более кислотные свойства (разд. 33.4.4). Разность электроотрицатель-
В
476
Неорганическая химия
ностей при повышении степени окисления становится меньше, поскольку электроотрицательность тем больше, чем выше положительный заряд на атоме. В качестве примера можно рассмотреть оксиды марганца в различной степени окисления (разд. 36.14). Линия на рис. В.21, б показывает положение амфотерных оксидов ^-элементов первого ряда в различных степенях окисления.
Вблизи «линии амфотерности» происходит резкое изменение некоторых свойств многих других соединений; это замечание касается следующего:
гидролиза солей (соли металлов, образующих основные оксиды, гидролизуются быстро; склонность солей к гидролизу, как и кислотно-основной характер оксида металла, зависит от степени окисления элемента);
растворимости галогенидов в апротонных полярных и неполярных растворителях (элементы, образующие кислые оксиды, дают растворимые полярные ковалентные галогениды — молекулярные вещества);
летучести галогенидов (летучесть галогенидов меняется по-разному в зависимости от того, кислый, амфотерный или основной оксид образует данный элемент; приведем ряды изменения летучести: для кислотных оксидов F>Cl>Br>I ; для амфотерных оксидов F<§cCl>'Br>-I; для основных оксидов F< <С1<Вг<1);
кислотных свойств бинарных гидридов (разд. 33.4.4);
растворимости сульфидов в сульфидах щелочных металлов в результате образования комплексных тиоанионов (разд. 38.5);
проявления в основном «металлических» типов соединений основных оксидов (величина электронной проводимости может быть мерой основных свойств);
образования стекол и основных солей (слабокислотные оксиды склонны благодаря образованию полимерных анионов давать стекла. Слабоосновные оксиды образуют основные соли, часто содержащие полимерные катионы).
35.2.2. Реакции кислорода
В виде молекул кислород находится в воздухе, а в связанном состоянии-—во многих оксидах и солях кислородных кислот. Кислород получают из воздуха (ректификацией жидкого воздуха — процесс Линде) и из его соединений (путем термического разложения последних). При нагревании соединений — оксидов или солей — происходит отщепление кислорода. Реакции разложения смещаются вправо при повышении температу
Кислород
477
ры, так как энтальпия реакций положительна (разд. 33.8), например:
2 HgO----->2Hg + O2f
2 ВаО2 ---► 2 ВаО 4- ОД
2 KNO3 ---> 2 KNO2 + ОД
2 KC1OS --► 2 КС! + 3 O2f
Под действием коротковолнового ультрафиолетового излучения или электрического разряда из обычного кислорода можно получить его полиморфную форму (разд. 33.2.2)—озои — газ с диамагнитными свойствами.
При образовании молекулы озона из молекулы кислорода и атома кислорода происходит выделение энергии за счет образования связи (разд. 35.2). Образование же озона только из молекулярного кислорода — эндотермический процесс, так как для расщепления молекулы кислорода на атомы требуется значительная энергия (494 кДж/моль). «Эндотермические» молекулы озона способны легко разрушаться и вследствие этого проявляют сильные окислительные свойства. Так, озон окисляет серебро до сине-черного Ag2O2 и иодиды до иода (молекулярный кислород в такие реакции не вступает).
Наличие неспаренного электрона на разрыхляющей ^-молекулярной орбитали О2 ведет лишь к весьма незначительной склонности к ассоциации. Упоминаемая в литературе молекула О4, которая могла бы образоваться в результате спаривания электронов двух молекул О2, в действительности не существует даже при очень низких температурах.
Для молекулярного кислорода характерны реакции окисления— восстановления. Почти все реакции окисления кислородом протекают с выделением энергии. Однако при комнатной температуре молекулярный кислород довольно инертен. Например, он менее сильный окислитель, чем иодат, бром, азотная кислота или ионы [Fe(H2O)e]3+.
При подведении к системе энергии из молекулярного кислорода получается атомарный, окислительный потенциал которого выше, чем озона или дифторида кислорода, и ненамного меньше, чем потенциал молекулярного фтора.
При использовании катализаторов окисление молекулярным кислородом происходит при комнатной температуре и нормальном давлении. В апротонных растворителях окисление идет по «одноэлектронному» механизму. На промежуточной стадии образуется надпероксид-ион:
О2 4- е~ -► О2_
Напротив, в водных растворах окисление осуществляется по «двухэлектронному» механизму через образование НО2_-ио-на:
О2 4- 2е- 4- Н2О -► НО2' 4- ОН-
478
Неорганическая химия
Некоторые ионы и комплексы переходных металлов (Си, Fe Мп, V и Со) часто выступают в качестве «обратимых» переносчиков молекулярного кислорода, т. е. служат катализаторами окисления. При этом молекулярный кислород и ионы металла соединяются в комплексы, строение которых пока точно не вы-яснено. Наиболее вероятно образование комплексов следующего типа:
о Fe || или Fe-~®O=o
°
а также образование двухъядерных комплексов, как это, по-ви димому, имеет место при использовании соединений кобальЛ как катализаторов
NH3X /NH3 0 NH3X /NH3 1
NH3-Co ч-----II-----► Co—NH3 I
NH3/ \nH3 0 K’H3/ \nh4 I
Каталитические свойства ионов металлов в реакциях моля
кулярного кислорода играют особо важную роль в биологических системах. Именно каталитический эффект позволяет объяснить окисление ионов Сг2+ и Fe2+ в воде, насыщенной кислородом (в воде, свободной от кислорода, они устойчивы).
Реакционная способность молекул О3 и О2 очень сильно различается. Озон окисляет многие соединения при таких условиях, когда кислород еще не реагирует. В кислых растворах окислительные свойства озона усиливаются. По окислительному действию его превосходят лишь фтор, атомарный кислород, ОН-радикалы и перксенат-ионы. Приведем окислительно-восстановительные потенциалы пары О3/О2 для некоторых полуре-акций в водных растворах:
О34-2Н+4-2е- -----► О24-Н2О
О3 + Н2О 4- 2 е~ -► О2 + 2 ОН-
О34-2Н+ + 2е- ----> О2 + Н2О
(с=10—’ моль/л)
£0 = 4-2,07 В £0 = 4-1,24 В £»= 4-1,65 В
Озон способен медленно окислять воду до кислорода:
2О3 4- 4 Н3О+ 4- 4е~ -► 2О24-6Н2О
6Н2О — 4е~ -<- О24-4Н3О+
2 О3 ---► ЗО2
Вода может быстро окисляться до озона лишь под действием фтора и ионов Ag2+.
Кислород
479
35.2-2-1- Оксид-ион
Как показывают рентгеноструктурные исследования, в ионных соединениях могут присутствовать различные ионы кислорода. Отрицательные ионы кислорода, такие, как оксид-ион Q2-( пероксид-ион О22- и надпероксид-ион О2_, в водных растворах существовать не могут. Реагируя с водой, они дают основания (разд. 33.4.1.5):
О2" 4- Н2О --> 2 ОН~ (aq) К > Ю22
о22- + н2о —> ног + он-
2 О2- + Н2О -► О2 4- НО2- 4- ОН-
Реакцию протолиза этих ионов в воде можно считать одной из наиболее для них характерных.
Благодаря сильно выраженному свойству оксид-иона быть донором электронов, с акцепторами он может давать оксоние-вые ионы.
Менее характерны для оксид-иона окислительно-восстановительные реакции. В связи с тем что большинство реакций протекает в водных растворах, особенно важно знать условия, при которых вода окисляется до О2, либо восстанавливается до Н2:
2 Н2О 4- 2е~ у- Н2 4- 2 ОН~
6 Н2О --> 4е- 4- О2 4- 4 Н3О+
Для этих процессов можно написать уравнение Нернста (разд. 33.5.1), причем давление выделяющихся газов принимается равным одной атмосфере. Тогда потенциалы £н2/н+и £о2-/о2 (в вольтах) равны
£Н2/н+=—0,82 4-0,059 lg = — 0,82-)-0,059 рОН= —0,059 pH
ЕО2~ /Ог = 1,23 + 0,0591g аНз0+ = 1,23—0,059 pH
Можно рассчитать потенциалы для различных значений pH:
₽н £'н2/н+> в ео2-/о2> в
0 0 1,23
7 —0,41 0,81
14 —0,83 0,40
Все окислительно-восстановительные пары, потенциал которых £<£н2/н+, должны восстанавливать воду до водорода; если же потенциалы £>£о2-/о2, вода будет окисляться до молекулярного кислорода. Однако многие восстановители или окислители, удовлетворяющие одному из этих условий, все же устойчивы в воде вследствие возникающих кинетических затруднений.
480
Неорганическая химия
35.2.2.2. Гидроксид-ион
Гидроксид-ион образуется при протолизе оксид-ионов. В ви. де самостоятельной частицы он существует лишь в гидрооксщ дах наиболее электроположительных элементов. Как и оксид, ион гидроксид-ион — очень жесткое основание. При растворе-нии гидроксидов в воде происходит не протолиз, а гидратация гидроксид-иона с образованием Н7О4_-иона. В присутствии гидроксид-иона водный раствор дает основную реакцию. Основные свойства гидроксид-иона полностью нейтрализуются при взаимодействии с протоном:
ОН- + Н+ Н2О
Если ковалентный характер связи Е—О возрастает, гидро-ксид-группа может отщеплять протон, т. е. проявлять кислотные свойства:
ЕОН + Н2О --► ЕО" (aq) + Н3О+ (аф
Амфотерные гидроксиды с сильными кислотами реагируют как основания, а с сильными основаниями — как кислоты. Для гидроксидов различных элементов периодической системы применима та же система классификации, что и для оксидов (разд. 35.2.1).
Гидроксид-ионы образуются также при реакции протолиза гидратированных катионов тяжелых металлов (разд. 33.4.2).
Соединения, содержащие гидроксид-ионы, могут полимеризоваться, причем гидроксид-ионы выступают как мостиковые лиганды различного типа. В полимерных гидроксидах группы ОН могут образовывать структуры с кислородными или водородными мостиками
н
н
ЕО—Н---ОЕ
Образование полимерных структур может происходить также в результате процессов «конденсации — полимеризации» между молекулами, содержащими гидроксид — группы:
2 ЕОН > Е—О—Е + Н2О
Однако не все гидроксиды полимеризуются в растворах или в твердом состоянии. Гидроксиды с особенно сильно выраженными кислотными или основными свойствами не полимеризуются (рис. В.22).
Кислород
481
По своим комплексообразующим свойствам, как и в других отношениях, ОН~-ион близок к F'-иону. Известно большое число устойчивых гидроксокомплексов. При протекании реакции замещения лигандов в комплексных соединениях гидроксид-ион выступает в качестве нуклеофильного реагента. Его особое положение среди нуклеофильных реагентов выражается в том, что скорости реакции замещения у него в 104—108 раз выше, чем
Рис. В.22. Элементы, образующие полимерные оксогидроксо-анионы и оксо-гидроксо-катионы.
при замещении протонами. Согласно кинетическим данным, реакцию комплекса кобальта с ОН~-ионами можно рассматривать как идущую по 5ы2-механизму (разд. 11.3 и 33.6.4), поскольку скорость реакции подчиняется уравнению о = £скомпл’Сон_. В соответствии с этим ОН_-ионы как «нуклеофильные агенты» должны непосредственно взаимодействовать с центральным атомом комплекса.
Другая возможность — осуществление реакции по Sn 1-механизму через стадию сопряженного основания; при этом реакция замещения протекает в три стадии:
очень быстро
[Co(NH3)3Cl]2+ 4- ОН- ------>- [Co(NH3)4NH2Cl]+ + Н2О
[Co(NH3)4NH2cii+ —ленн^- [Co(NH3)4NH2P+4-C1-
[Co(NH3)4NH2]2+4-H20 [Co(NH3)3OH]2+
На первой стадии происходит депротонирование содержащего активный протон лиганда, остающегося в комплексе (скорость в 105 раз превышает скорость других стадий). На второй стадии образующееся сопряженное основание диссоциирует; эта стадия определяет скорость всего процесса. В активированном 31—2027
482
Неорганическая химия
комплексе координационное число равно пяти. На третьей стадии происходит быстрый процесс гидратации.
Различие механизмов SN2 и SnI (через сопряженное основание) достаточно велико для того, чтобы их можно было надежно отличить друг от друга.
Пирсон и Эдингтон (1962 г.) исследовали реакционную спо-собность НО2- как основного агента. Группа НО2-— более слабое основание, чем ОН-, но более сильный нуклеофил. Основной гидролиз с участием НО2- должен идти медленнее, чем с участием ОН-, в том случае, если он связан с образованием в результате удаления протона сопряженного основания. Если же реакция представляет собой прямое взаимодействие с ионом металла, скорость гидролиза с участием ООН- должна быть больше. Экспериментальные данные согласуются с протеканием реакции по механизму SnI через сопряженное основание.
35.2.2.3. Пероксид-, надпероксид- и озонид-ионы
Ионные пероксиды, содержащие ионы О22-, образуют щелочные металлы, Ba, Sr и Са. Ион О22- вступает в следующие реакции:
1) с водой он взаимодействует, давая щелочную реакцию (разд. 35.2.2.1); возникающий ион О2Н- ведет себя как нуклеофильный реагент; с протоном в кислых растворах реакция происходит мгновенно, при этом НО2--ион переходит в Н2О2 (пероксид водорода);
2)пероксид-ион может выступать и как лиганд при комплексообразовании. Важнейшие реакции О22--иона — реакции окисления — восстановления — рассмотрены на примере пероксида водорода.
Вследствие того что в пероксиде водорода кислород формально имеет промежуточную степень окисления (—1), Н2О2 может вести себя как окислитель или как восстановитель. Н2О2 неустойчив. За счет принятия или отдачи электронов он переходит в более устойчивые соединения Н2О и О2:
Н2О2 + 2 Н+ + Че~ ---► Н2О Е° = 1,77
Ог4-2Н++2е- ----► Н2О2 £» = 0,68;В
О2Н“ -Ь Н2О + Че~ ---> 3 ОН" Ео = 0,87 В
Такие восстановители, как I-, Fe2+, N2H4, в присутствии ка-
тализаторов— шестивалентного молибдена или восьмивалентного осмия отдают электроны иону О22-. Два образовавшихся оксид-иона О2- мгновенно реагируют с водой, давая гидроксидионы, которые в случае кислой среды реагируют дальше с образованием воды. В щелочных растворах пероксид водорода окисляет Мп(ОН)2 и СгО2- до МпО2 и СгО42- соответственно.
Такие окислители, как перманганат, гипохлорит и оксид се'
Кислород
483
ребра(II) в нейтральных, а также РЬОг и иодат в кислых растворах, переводят ион О22- в молекулярный кислород.
Разложение пероксида водорода сопровождается значительным выделением энергии. При этом происходит реакция диспропорционирования:
Н2О ---» 2е- + О2 + 2 Н+
Н2О2 + 2Н+ + 2е“ > 2Н3О
—1 —2 о
2Н2О2 ---► 2Н2О+О2 ЛЯ = —98,8 кДж/моль
Системы МпО2/МпО и СоОг/СогОз, как и порошок платины, катализируют реакции диспропорционирования. Ацетанилид и тидрофосфат калия действуют как ингибиторы и препятствуют разложению (почему?).
Кислотные свойства пероксида водорода в протолитических реакциях выражены очень слабо
Н2О2 НО2- + Н+ РКХ = 11,7
Н2О2 о22- + 2Н+ рК = 25
В зависимости от pH в растворе могут присутствовать частицы: Н2О2, НО2“ и О22-. Они способны образовывать более или менее устойчивые комплексы. Четырехвалентный титан в кислом растворе образует комплексные катионы, придающие раствору оранжевый цвет: [Ti(H2O2)]4+; [Ti(HO2)]3+; [Т1(Ог)]2+ и [Ti(O2) (ОН)]+. Соединение TiO2(OH)2 содержит кислород в различных степенях окисления: О22- и 2О2-. Образование окрашенных растворов используется для качественной реакции на Н2О2.
Шестивалентный уран в щелочном растворе с Н2О2 дает оранжевое окрашивание благодаря образованию комплексных анионов, а в нейтральном растворе — светло-желтый осадок UOraq. При добавлении кислоты (сильная кислота вытесняет более слабую Н2Ог) снова образуется Н2О2.
Надпероксиды (супероксиды), содержащие ионы Ог-, известны лишь для наиболее электроположительных металлов, придем устойчивость надпероксидов находится в прямой зависимости от степени электроположительности соответствующего металла.
Путем действия озона на КОН с последующей перекристаллизацией продукта реакции из жидкого аммиака получают красный кристаллический озонид калия. Магнитные измерения показали, что в КО3, NaO3 и CsO3 присутствует ион О3~ с неспаренным электроном (угловое строение). Соединений, содержащих ионы Оз2-, получено не было.
Для надпероксидных соединений характерны свойства сильных окислителей. Они медленно окисляют воду до молекуляр-31*
484
Неорганическая химия
ного кислорода, в качестве промежуточной частицы образуется ион НО2_:
2 Ог- + Н20 --> 02 + Н02- + ОН"
2 Н02" ---► 2 ОН" 4- 02 (медленно)
35.2.2.4. Катион диоксигенила
Как молекула, так и атом кислорода могут давать положительные ионы. Катион диоксигенила (разд. 35.4.1) существует в виде изолированной частицы в ряде соединений. Катион 02+ похож на катион N0+. В отличие от соединений с азотом (например, оксид азота (II)) кислород со фтором при различных условиях образует фториды кислорода состава 0F2, О2Рг, O3F2 и O4F2, в которых атомы кислорода имеют положительный заряд.
В присутствии сильных акцепторов атомов фтора из O2F2 может образоваться катион 02+. Так AsFs, PF5 и SbFs взаимодействуют с O2F2 и дают «ионные» соединения диоксигенила:
O2F2 + MF5 ---> O2+[MFe]" + V2F2
Фториды кислорода действуют как сильные окислители и фторирующие агенты: ОН~-ионы окисляются быстро, а Н20 — несколько медленнее. Формально происходят реакции синпро>-порционирования:
OF2+2OH" -----► O2 + 2F" + H2O
0F2 + Н20 ----> 02 + 2 HF
Рекомендуемые опыты
1. Каталитическое окисление кислородом.
В две пробирки помещают по 2 мл 5%-ного раствора МпС12. Через раствор в одной из пробирок пропускают ток аргона, не содержащего кислорода. После этого в обе пробирки добавляют немного 2 М раствора NaOH и плотно закрывают их резиновыми пробками. В пробирке, через которую пропускался аргои, внешний вид раствора не меняется, тогда как в другой происходит окисление с образованием МпО(ОН)2 (коричневое окрашивание). Если открыть резиновые пробки (в пробирки попадает воздух), Мп2+ и в другой пробирке окисляется. Опыт повторяют с тем лишь отличием, что вместо щелочи добавляют 2 М соляную кислоту. Как объяснить различную скорость окисления в щелочном и кислом растворе?
2. Окислительное действие озона. В большую пробирку помещают немного (——100 мг) ВаО2 и добавляют 15 капель конц. H2SO4. Пробирку закрывают резиновой пробкой с отводящей трубкой, согнутой под прямым углом. При осторожном (!) нагревании пробирки выделяется газ, содержащий озон (запах!), который окрашивает иодкрахмальную бумагу (напишите уравнение реакции). На проволоке из серебра, помещенной внутрь отводной трубки, образуется Ag2O2 (напишите уравнение реакции).
3. Н2О2 как окислитель. Немного соли Сг(Ш) растворяют в 1 мл воды и добавляют конц. NaOH до тех пор, пока выпавший сначала осадок ие растворится. При добавлении нескольких капель 2,5 М Н2О2 и при последующем нагревании зеленый хромит окисляется до желтого хромата:
2 Сг(ОН)в3" + 3 Н2О2 -> 2 СгО42" + 8 Н20 + 2 ОН"
Фтор 485
Вследствие того что соли тяжелых металлов каталитически ускоряют реакцию диспропорционирования пероксида водорода, происходит также- частичное разложение Н2О2 с выделением кислорода.
4. Н2О2 как восстановитель. К 1 мл 2,5 М Н2О2, подкисленного двумя каплями 2,5 М H2SO4, добавляют по каплям 0,02 М раствор КМпО4. Перманганат обесцвечивается и одновременно выделяется кислород:
2 МпО4- + 5 Н2О2 + 6 Н+---> 2Мп2++5О2 + 8Н2О
5. Диспропорционирование Н2О2. К *. мл 2,5 М Н2О2 добавляют несколько капель коллоидного раствора платины. Можно также к 1 мл 2,5 М Н2О2 добавить немного MnSO4, подкислив раствор несколькими каплями 2,5 М H2SO4. В обоих случаях выделяющийся газ идентифицируют с помощью тлеющей лучинки.
6. Обнаружение Н2О2. К 1 мл 0,1 М Н2О2, подкисленного несколькими каплями 2,5 М H2SO4, добавляют несколько капель 0,1 М сульфата титаиила T1OSO4 (желтое окрашивание).
Вопросы и задания
1. Назовите соединения, в которых координационное число атома кислорода больше двух.
2. Чем определяется величина угла, образованного двумя ковалентными одинарными a-связями при атоме кислорода?
3. Присоединение двух электронов к атому кислорода — процесс эндотермический. Объясните, почему, несмотря на это, известно большое число «ионных» оксидов. Составьте диаграмму энтальпий образования оксидов элементов главных подгрупп.
4. Как можно объяснить, почему энтальпия образования ZnO больше, чем Cs2O, хотя цезий — один из наиболее электроположительных элементов.
5. Почему окисление ионов Fe2+ кислородом воздуха в кислых растворах протекает медленнее, чем в щелочных? (следует учесть кинетику реакции окисления).
6. Какое влияние на реакции соединений кислорода оказывает наличие нли отсутствие воды?
7. Каким путем можно установить наличие в соединении мостиковых связей следующих типов: Е—ОН, Е—О—Н---О, Е—О—Е?
I
Е
8. Через некоторое время после растворения [Со(МН3)в]С!з в D2O в молекулах аммиака может быть обнаружен дейтерий. Обсудите возможный механизм реакции обмена.
9. На примере нескольких соединений объясните, почему оксид- и гидроксид-ион относятся к жестким основаниям. Какие соединения по теории жестких и мягких кислот и оснований термодинамически наиболее устойчивы?
10. Проследите зависимость окислительных и восстановительных свойств оксидов элементов главных подгрупп от величины отрицательного заряда на атоме кислорода.
11. Составьте таблицу для иллюстрации зависимости типа оксида от положения элемента в периодической системе и от его степени окисления.
35-3. Фтор
При рассмотрении химического поведения фтора и фторид-иона необходимо учитывать, что:
атом и ион фтора характеризуются весьма высокой плотностью электронных оболочек (рис. В.23);
высокая плотность заряда проявляется в сравнительно высо-
486
Неорганическая химия
кой энергии ионизации атома, в относительно небольших величинах атомного и ионного радиусов и в незначительной поляри
зуемости иона фтора;
причиной высокой реакционной жит как небольшая величина
Рис. В.23. Электронная плотность U(г) как функция расстояния от центра в атоме фтора (прерывистая линия) и во фторид-ионе (сплошная линия). 1а.е.= = 0,05284 нм.
способности молекул F2 слу. энергии ее диссоциации (154кДж/моль), так и большая энергия связи в образующихся соединениях фтора;
низкая энергия связи в молекуле F2 обусловлена отталкиванием несвязывающих электронов; в молекулах других галогенов межэлектронное отталкивание меньше из-за большего расстояния между атомами галогена и более компактного расположения внутренних электронов; кроме того,
связь в молекулах других галогенов может усиливаться благодаря участию d-op-биталей; ।
из всех элементов у атомов фтора наибольшее зна-
чение электроотрицательности, и поэтому в соединениях фтора могут осуществляться максимальные значения положительной
валентности других элементов;
в полярных ковалентных соединениях отрицательно поляризованный атом фтора вследствие своего небольшого размера и связанной с этим повышенной электронной плотностью (невозможна делокализация электронов на d-орбитали) может частично предоставлять в общее пользование свою электронную пару;
по этой же причине соединения фтора (особенно с элементами второго периода) очень устойчивы (повышение кратности связи);
из-за жесткости электронной оболочки фторид-ион обладает особенно сильным полем;
по величине электроотрицательности и размерам сходство со фтором в наибольшей степени проявляют не более тяжелые галогениды, а кислород и гидроксид-радикал.
35.3.1. Реакции фтора
До сих пор известны лишь такие реакции, в которых молекула фтора выступает как окислитель, причем атом фтора присоединяет электрон. При окислительном действии фтора может
Фтор
487
образоваться большое число разнообразных соединений. В этой связи интересно отметить, что взаимодействие фтора с сухим K2SO4 не идет. Напротив, с KHSO4 как в водном растворе, так и в твердом состоянии, фтор реагирует, давая пероксодисульфат. Этот процесс окисления фтором сходен с анодным окислением гидросульфата до пероксосульфата. Таким образом, для протекания реакции важно присутствие водорода в гидросульфатах. Реакция идет по радикальному механизму, т. е. через образование НЭО4-радикалов и затем H2S20s:
2 KHSO4 + F2 --> K2S2O8 + 2 HF
В действительности ход реакции несколько более сложен, поскольку получаются еще KSO3F и KHSO5.
35.3.1.1. Фтороводород
Фтороводород — это полярное ковалентное соединение, которое даже в газообразном состоянии ассоциировано за счет образования «протонных мостиков». Эту ассоциацию можно
a
6 .
''H--F" 'н—V' ''H—F'"
Рис. В.24. Различные варианты строения ассоциатов (HF)n.
описать в рамках днполь-дипольного взаимодействия. Ассоциаты с формулой (HF)n представляют собой цепи, для описания строения которых предложены различные варианты (рис. В.24). Часть цепей замыкается в шестичленные кольца. Ассоциация обусловливает относительно высокую температуру кипения фтороводорода + 19,5 °C.
Для фтороводорода характерно образование устойчивых фторгидрогенатов. Во фторгидрогенат-ионе [FHF]- водород протонного мостика занимает симметричное положение (пока единственный пример симметричного мостика). Известно много Устойчивых кислых солей фтороводорода, в то время как другие галогеноводороды вообще не дают кислых солей.
Рентгенографически найдено, что в анионе HF2~ расстояние F—F составляет 0,226 нм, что лишь на 0,04 нм больше, чем Удвоенная длина ковалентной связи Н—F (0,092 нм). На особый характер взаимодействия в этом ионе указывает также
488
Неорганическая химия
очень большая для притонного мостика энергия связи •(113 кДж/моль). По-видимому, симметричное строение иона можно объяснить отсутствием остаточной энтропии (имеющейся, например, в ассоциатах воды) и возникновением лишь одного минимума потенциальной энергии с «центральной» симметрией.
Энтальпии процессов растворения HF при 18 °C составляют
HF (газ) в 200 моль Н2О АЯ=—48,4 кДж/моль
HF (жидк.) в 200 моль Н2О АЯ=—19,3 кДж/моль
Растворение HF в воде нельзя рассматривать только как физический процесс; при этом происходит химическая реакция и устанавливается равновесное состояние. В образовавшейся плавиковой кислоте присутствуют ионы Н3О+, F~ и HF2~. Плавиковая кислота — довольно слабая кислота*. Частично это можно объяснить аномальной реакцией протолиза:
2 F- + НаО -»- HFa- + ОН-
Сильное увеличение кислотных свойств в концентрированных растворах плавиковой кислоты происходит за счет аутокомплексообразования:
2Н2О
3HF «=* 2Н3О+ + F” + HF2-
н2о
2 HF HF2- + Н3О+
Ион HF2~ представляет собой сильную анионную кислоту.
35.3.1.2. Фторид-ион
Фторид-ион во многом отличается от других галогенид-ио-нов. Его особые свойства проявляются в растворимости фторидов.
При осаждении смешанных галогенидов, например PbBrF, фторид-ион выпадает в виде труднорастворимой соли. Разумеется, растворимость выпавшей соли сильно зависит от pH раствора. Следует помнить, что фторид-ион в водных системах присутствует в виде ионов F~ и HF2~. Понижение pH раствора в результате осаждения объясняется освобождением протонов из имеющегося «анионного амфолита» HF2_. При достаточном избытке протонов смешанный галогенид снова растворяется.
Фторид-ион может давать очень устойчивые фторокомплек-сы: BF4~, PF6~, SiF62-, AsF6_, SbF6~, A1F63- и др. Следует от
* Обычно плавиковую кислоту считают кислотой средней силы К=7Х ХЮ-4. [Ахметов Н. С. Неорганическая химия. — М.; Высшая школа, 1975].—' Прим, перев.
Фтор
489
метить, что путем комплексообразования с фторид-ионом удается стабилизировать необычные степени окисления. Обычно у переходных металлов стабилизируются степени окисления вплоть до -|-5 (разд. 36.14.2), причем координационное число не превышает шести: Кз[№Рб]; RbzfNiFg]. У атомов больших размеров возможны координационные числа 7 и 8; [NH4]3ZrF7; СэгТеРв. В соответствии с теорией жестких и мягких кислот и оснований вполне объяснимо, что ион F-, как жесткое основание, стабилизирует высокие степени окисления элементов. Центральный атом в этих соединениях благодаря высокому заряду представляет собой жесткий кислотный центр, т. е. жесткую кислоту (разд. 33.4.3.4). Способность к образованию комплексов используется в различных качественных реакциях на фторид-ион.
Рекомендуемые опыты
1. Окислительное действие фтора. В холодный насыщенный раствор1 KHSO4, находящийся в платиновой или медной колбе (Эрленмейера), в течение нескольких часов пропускают газообразный фтор. При этом выделяются газообразные фториды кислорода с характерным запахом и твердый пероксодисульфат, который можно отфильтровать через полиэтиленовый фильтр и затем проделать характерные для него реакции (разд. 35.6.4.2).
Немного фильтрата, содержащего HF, добавляют в раствор KI. Тотчас же происходит выделение иодида (коричневое окрашивание).
2. Получение HF. В тигель из свинца, никеля, платины или серебра помещают 1 г KHF2. Тигель накрывают часовым или предметным стеклом, покрытым воском, причем на нижней стороне стекла часть воска удаляют. Осторожно нагревают тигель на песчаной бане. Выделяющийся газообразный фтороводород разъедает стекло. При этом HF взаимодействует с SiO2 стекла, давая SiF<.
3. Кислотно-основные реакции Р~-иона. Растворяют немного NaF в воде и определяют pH раствора. Раствор сохраняют для других опытов.
За. Осаждение фторид-иона. К раствору NaF в двух пробирках добавляют по несколько капель растворов ВаС12 и СаС12. Свежевыпавшие осадк» растворяют, если необходимо при нагревании, в разб HNO3 или разб. НС1. Свежевыпавшие осадки легко растворимы в кислотах. Повторяют опыты по осаждению, но растворы с осадками в течение некоторого времени нагревают до кипения, затем центрифугируют, отделяют осадки и недолго прокаливают. Теперь осадки нерастворимы в разб. НО и разб. HNO3. Концентрированные кислоты медленно растворяют осадки, благодаря протеканию гетерогенной кислотно-основной реакции. Происходит вытеснение легколетучега фтороводорода.
36. Изучение растворимости AgF и TIF. К раствору NaF, подкисленному Разб. HNO3, добавляют растворы AgNO3 или TINO3. Выпадения осадков не Наблюдается (другие галогенид-ионы образуют осадки с ионами Ag+ и Т1+).
4) Изучение растворимости галогенидов. В четырех пробирках к небольшому количеству слабокислого (pH 3,5—4,0) 0,1 М Pb(NOs) добавляют по каплям 0,1 М KF, КО, КВт и KI. После добавления каждой капли пробирки энергично встряхивают. Отмечают и число капель, необходимое для получения скоагулированных осадков PbF2, РЬС12, РЬВг2 и РЫ2. В этом ряду Происходит постепенное уменьшение растворимости солей (что объясняется Различной поляризуемостью анионов). Кроме того, меняется и тип структуры от ионной (PbF2) до более ковалентной молекулярного типа (РЫ2).
5. Смешанные галогениды: осаждение PbBrF. К раствору 0,1 М NaF
490
Неорганическая химия
добавляют несколько капель конц. раствора Na2CO3 и затем индикатор — бромфеноловый синий, который окрашивается в синий цвет. Добавлением 1 М HNO3 доводят pH раствора до 3,2—3,4 ((изменение окраски в зеленую). К этому раствору прибавляют немного 0,8 М КВг и после этого по каплям несколько миллилитров 1,6 М Pb(NO3)2. Через минуту после выпадения осадка при сильном перемешивании добавляют по каплям еще несколько миллилитров 0,8 М КВг. Осадок PbBrF становится крупнокристаллическим, а желтое окрашивание индикатора указывает на понижение pH. С помощью 15%-ного раствора ацетата натрия доводят pH до 3,5—3,7 (цвет индикатора зеленый). Осадок центрифугируют, растворяют в разб HNO3 и к раствору добавляют Ag'NO3: выпадает AgBr. Этот опыт можно про водить и с раствором КС1.
6. Свойства фторид-иона как лиганда. В пробирку с двумя каплям! 1%-ного раствора ZrOCl2 добавляют несколько капель 0,1%-ного раствор; ализарина S и подкисляют 5 М соляной кислотой. Образуется ярко-красны; лак — комплекс цнрконнлхлорида с ализарином S. Если теперь добавлять пс каплям раствор NaF, происходит переход окраски в желтую, что соответст вует цвету свободного ализарина в кислом растворе.
Ализарин S образует комплексы, в которых связь с цент ральным атомом осуществляется через атомы кислорода. Из-з; стерических затруднений ионы металла связываются лишь с частью атомов кислорода в молекуле лиганда. Затем образуется малорастворимый многоядерный комплекс с мостиковыми ОН-группами.
101 O-ZrO^
101
Фторид-ион — более прочно связываемый лиганд, и поэтому цутем обмена лигандами образуется устойчивый комплекс [ZrFd*-.
Вопросы и задания
1. Систематизируйте (удобно составить таблицу) данные о взаимодействии Ег с водой. Какие продукты при этом образуются? Напишите уравнени! реакций взаимодействия фтора с водными растворами карбоната, титаната ванадата, молибдата и хромата натрия.
2. Какие соединения образуются: при пропускании HF в сильно охлажденные конц. растворы НСЮ4 и H2SO4; при реакции HF (жидк.) с Н3ВОз, <НРО3)П, КМпО4 и К2СгО4?
3. К каком типе структуры кристаллизуется CaF2?
4. Систематизируйте (составьте таблицу) данные о растворимости галогенидов свинца в кислотах; укажите также цвет галогенида и его произведение растворимости. Дайте теоретическое объяснение изменению свойств для различных галогенидов.
5. Дайте объяснение факту устойчивости и процессу осаждения PbBrF С позиции теории жестких и мягких кислот и основании.
6. Покажите, что, согласно теории жестких и мягких кислот и основании, ионы F~ и О2- являются жесткими реагентами.
Инертные газы
491
35.4. Инертные (благородные) газы
Гелий и другие инертные газы занимают в периодической системе положение промежуточное между весьма реакционноспособными элементами — галогенами и щелочными металлами. В соответствии с их физическими свойствами (летучесть, растворимость) инертные газы относятся к неметаллическим элементам.
Неон, аргон, криптон и ксенон кристаллизуются в кубической плотнейшей упаковке (гелий — в гексагональной плотнейшей упаковке). В твердых телах между атомами действуют лишь вандерваальсовы силы.
Полностью завершенная электронная оболочка атомов; [s2p6, а у гелия s2) приводит к чрезвычайной инертности этих веществ. В газообразном состоянии инертные газы состоят из атомов. В гипотетических молекулах (Не2, Ne2,...) число заполненных связывающих молекулярных орбиталей было бы равным числу заполненных разрыхляющих молекулярных орбита-лей, так что такие молекулы не существуют (разд. 6.2.4).
С различными веществами, в кристаллах которых имеются относительно большие пустоты, инертные газы образуют соединения включения (клатраты). Так, например, за счет наличия пустот в кристаллическом гидрохиноне в нем удерживается три атома инертного газа на каждую молекулу гидрохинона. Соединения подобного рода преимущественно с тяжелыми инертными газами образует и лед. Связь в таких соединениях осуществляется силами Ван-дер-ваальса.
35.4.1. Реакции инертных газов :
Вплоть до 1962 г. не удавалось получить «валентные» соединения инертных газов. Если рассмотреть величины энергий ионизации неметаллов (табл. В.21), оказывается, что теоретически тяжелые инертные газы могут соединяться с другими наиболее электроотрицательными элементами. Энергия ионизации криптона практически равна энергии ионизации кислородам Энергия ионизации ксенона ниже таковой для кислорода и хло-Ра, и ненамного выше, чем для брома.
Еще в 1951 г. на основании теоретических расчетов связей в ионах Юг и 1з~ было высказано предположение, что можно> получить соединения инертных газов. Бартлетт и Ломанн показали, что при комнатной температуре гексафторид платины Реагирует с кислородом с образованием диоксигенилгексафто-Роплатината(У) O2+[PtFe]_. Гексафторид платины является одним из наиболее сильных окислителей; его сродство к электрону составляет 712 кДж/моль (339 кДж/моль для фтора). PtF®
492
Неорганическая химия
Таблица В.21. Первые потенциалы ионизации (в электронвольтах) атомов неметаллов
В 8,28 С 11,24 Si 8,14 N 14,46 Р 10,43 О 13,57 S 10,43 F 17,46 С1 13,01 Вт 11,82 I 10,43 Не 24,48 Ne 21,41 Аг 15,68 Кг 13,94 Хе 12,08 Rn 10,69
легко взаимодействует с ксеноном, давая Xe+[PtF6]~. Тем самым было получено первое «валентное» соединение ксенона. Аналогично ксенон реагирует с RhF6 с образованием Xe[RhF6],
35.4.2. Фториды и кислородные соединения
После 1962 г. путем прямого синтеза из элементов при различных условиях (давление, температура, электрический разряд) удалось синтезировать ряд фторидов инертных газов (табл. В.22).
Таблица В.22. Некоторые соединения ксенона и криптона
Фториды XeF2, (XeF3), XeF4, XeFe. XeFg, (Xe2F10) KrF2, KrF4
Оксифториды (XeOF2), XeOF4, (XeO2F2)
Оксиды XeO3, XeO4
Кислоты HXeO-4, H4XeOe
Соли Na4XeOe-8 или 6 H2O, Na4XeOe, K4XeOe-9 H2O, Ba2XeOe-l,5 H2O, (Ba3XeOe)
Помимо фторидов должны существовать и соединения инертных газов с сильно электроотрицательным кислородом. Такие соединения образуются при довольно сложно протекающем гидролизе фторидов (полярных молекулярных веществ). Путем гидролиза и диспропорционирования можно получить — сами инертные газы, их оксиды, кислородные кислоты и при нейтрализации последних — соли. Гидролиз XeF4 идет с образованием Хе, Оя и ХеОз:
6XeF4 + 12Н2О ► 2ХеО3 + 4Хе + ЗО2 + 24HF
XeF6 гидролизуется в водном растворе до ХеО3:
XeFe + ЗН2О --► ХеО3 + 6HF
Инертные газы
493
В щелочном растворе гидролиз сопровождается диспропорционированием с образованием ХеО64~, Хе и Ог:
ХеО3 + ОН" ---»- ХеОв4- + Хе -f- О2 + Н2О
Ионы ХеОб4- и Ю65~ дают сходные спектры. Перксенат серебра в щелочном растворе неустойчив. Анион окисляет катион, восстанавливаясь при этом до Xe(VI). При пропускании в раствор озона снова получают степень окисления ксенона +8.
В табл. В.22 приведены некоторые из известных к настоящему времени соединений инертных газов. Сведения о их физических свойствах и подробности, касающиеся химического поведения, можно почерпнуть в обзорной статье [Hoppe R. Fort-schr. Chem. Forsch. 5, S.214—346 (1965)*].
Для объяснения характера связи в соединениях инертных газов и типов возможных реакций с их участием не требуется новых теоретических представлений. Так, XeF4 образует кристаллы молекулярного типа, в которых атом ксенона имеет плоскую квадратную координацию четырьмя атомами фтора. Такая геометрия часто реализуется также в структурах межгалогенных соединений; XeF4 соответствует ионам 1С14~ и BrF4-**. При качественном рассмотрении по методу М.0 (разд. €.3.4) предполагается образование делокализованных трехцентровых четырехэлектронных связей при участии только р-орби-талей валентных оболочек.
Рекомендуемые опыты
1. Получение XeFt. При наличии фтора и ксенона можно легко получить тетрафторид ксенона по методике Голловей и Пикок. Реакцию проводят в аппаратуре, изображенной на рис. В.25.
Ксенон (~500 мл) в течение 24 ч диффундирует из колбы и смешивается с фтором, который проходит затем через никелевую трубку, нагреваемую тремя горелками Бунзена. Тетрафторид ксенона в виде бесцветных кристаллов может быть сконденсирован в расположенных далее охлаждаемых ло-
* См. также обзоры: Нейдинг А. Б. Усп. химии 34, 1965, с. 969; Нейрине А. Б., Соколов В. Б. Усп. химии 43, 1974, № 12, с. 2147. — Прим, перев.
** Такая геометрия возникает при зр3сР-гибридизации центрального атома с неподеленными парами, занимающими транс-положения в октаэдре. [См., напр., Коттон Ф„ Уилкинсон Дж. Современная неорганическая химия. Пер. с англ. — М.: Мир, 1969, т. 2J
494
Неорганическая химия
вушках. Выход составляет ~30%. Реакция проводится при полном отсутствии воздуха и влаги.
2. Гидролиз XeFt; окислительно-восстановительное диспропорционирование. Небольшое количество XeF«, полученного в опыте 1, вносят в полиэтиленовый сосуд с 5 М NaOH. В результате энергичной реакции выделяется, кислород (тлеющая лучника).
В полиэтиленовую бутылку с 3 М H2SO< добавляют немного XeF«. В этом случае также происходит выделение газа — смеси ксенона и кислорода. К раствору добавляют раствор иодида и раствор крахмала, чтобы убедиться в окислительном действии продуктов гидролиза (синее окрашивание)^
Вопросы и задания
1. Почему инертные газы имеют низкие температуры плавления и кипения?
2. Возможно ли получения хлоридов тяжелых инертных газов?
35.5. Хлор, бром, иод
Элементы второго периода в валентном состоянии не имеют d-орбиталей, и поэтому их химическое поведение существенно отличается от поведения более тяжелых гомологов в той же группе. По этой причине химия остальных галогенов рассматривается отдельно.
Все атомы галогенов имеют сходную электронную конфигурацию— семь электронов во внешнем слое (s2p5). Эти элементы— наиболее электроотрицательные члены соответствующих периодов. Атомы и ионы галогенов характеризуются также довольно высокой компактностью расположения орбиталей.
Существенные различия в химии галогенов обусловлены различиями в строении предпоследнего электронного слоя их атомов (скачок электроотрицательности, кислородные соединения). В предвнешнем слое у атома хлора содержится 8, а у атомов брома и иода—18 электронов. Для образования связей атомы этих элементов могут использовать также свободные d-орбитали.
В свободном состоянии галогены образуют двухатомные молекулы; при этом каждый из атомов приобретает устойчивую октетную конфигурацию внешних электронов.
Активность галогенов в реакциях высока, поскольку из-за довольно низких энергий диссоциации может осуществляться распад двухатомных молекул.
35.5.1. Неполярные молекулярные вещества
Физические свойства галогенов определяются в значительной степени величиной вандерваальсовых сил, действующих между молекулами (разд. 32.2.2.1). Эти силы растут с увеличением размеров и поляризуемости атомов; так, фтор и хлор при нормальных условиях представляют собой газы, бром — жидкость, а иод — твердое вещество. Другие свойства галогенов, такие,
Хлор, бром, иод
495
Таблица В.23. Связь между окраской соединения и положением полос поглощения
Положение максимума поглощенного света Окраска соединения
нм цветовая область спектра
400—440 Фиолетовая Желто-зеленая
440—480 Синяя Желтая
480—490 Зеленовато-синяя Оранжевая
490—500 Сине-зеленая Красная
500—560 Зеленая Пурпурная
560—580 Желто-зеленая Фиолетовая
580—595 Желтая Синяя
595-605 Оранжевая Зеленовато-синяя
605—750 Красная Сине-зеленая
как растворимость, летучесть, способность к сублимации, также весьма типичны для неполярных молекулярных веществ (более подробно см. в других учебниках).
Окраска галогенов возникает благодаря поглощению видимого света. Остановимся кратко на связи между цветом соединения и положением полос поглощения (табл. В.23). Например, иод имеет полосу поглощения при 570 нм, т. е. он поглощает желто-зеленый цвет и его окраска — фиолетовая.
Положение полос поглощения в общем зависит от разности энергий стационарного и возбужденных электронных состояний в молекуле.
На примерах молекул галогенов можно убедиться в том, что при повышении числа электронов соответствующие электронные переходы требуют меньшей затраты энергии. Так, для возбуждения молекулы хлора необходимо более жесткое, фиолетовое излучение, и цвет хлора поэтому зеленый, тогда как в случае иода соотношения обращены, т. е. поглощенный цвет — зеленый, а окраска паров— фиолетовая.
Поглощение света в ультрафиолетовой и инфракрасной частях спектра обсуждалось в разд. 6.1.
Неполярный ковалентный тип связей в молекулах галогенов соответствует их хорошей растворимости в неполярных растворителях, причем растворы не проводят электрический ток и молекулы в них не ассоциированы. Растворимость галогенов в ноде незначительна. Растворы в неполярных растворителях скрашены так же, как галогены в свободном состоянии. При наличии у молекул растворителя донорных свойств (основание) окраска растворов меняется.
В сильно полярных растворителях благодаря гетеролитиче-скому расщеплению молекулы иода и при дальнейшем диспропорционировании образуются иодид- и иодат- ионы (уравнение реакций).
496
Неорганическая химия
Так, в растворах иода в различных растворителях может осуществляться в различной степени взаимодействие неполярного молекулярного вещества с растворителем. Взаимодействие может протекать с образованием комплексов с переносом заряда и даже приводить к гетеролитическому расщеплению молекулы иода. Например, комплекс иода с бензолом относится к комплексам с переносом заряда, в которых при возбуждении происходит переход электронов с занятой орбитали одного атома на свободную орбиталь другого атома. Возбуждение молекулы приводит, таким образом, к переносу заряда от одного атома к другому.
В технике галогены получают иначе, чем в лаборатории, но принцип их получения — путем окисления галогенид-ионов — остается неизменным в различных вариантах (электролиз, окисление воздухом, диоксидом марганца).
При окислении НС! с помощью МпО2 реакция идет через: промежуточное образование МпСЦ, распад которого, согласна кинетическим исследованиям, и определяет скорость всей реакции.
При лабораторном получении брома сначала происходит кислотно-основное взаимодействие КВг с H2SO4 с выделением бромоводорода, который далее подвергается действию окислителя МпО2.
35.5.2. Галогеноводороды — полярные молекулярные ве-
щества
Галогеноводороды представляют собой полярные молекулярные вещества. Благодаря своей летучести они выделяются при действии труднолетучих кислот на галогениды
NaCl + H2SO4 -► NaHSO4 + HClf
Протеканию реакции способствует также незначительная растворимость НС! в конц. H2SO4. При нагревании растворимость НС! уменьшается в равновесии протолиза смещается вправо:
HSO4- + СГ HClf + so42-
При проведении реакции с фосфорной кислотой, обычно замещается только один из протонов Н3РО4.
В табл. В.24 приведены некоторые свойства галогеноводородов. Хорошая растворимость полярных молекулярных веществ НХ в полярном растворителе — воде связана с протеканием при растворении химического процесса. При этом проис-
Хлор, бром, иод
497
Таблица В.24. Свойства галогеноводородов
Галогеиоводород Длина ковалентной связи, нм Дипольный момент D Ионность связи» % <9 00 о> Устойчивость Эо ‘VUJ Гкип- °с Растворимость, г/100 г Н2О Энергия связи X—Н, кДж/моль
(HF) ж 0,092 1,9 43 10-ез Очень устойчив —92 19,4 00 при 0°С 563,5
на 0,127 1,0 14 Ю-31 Очень устойчив -112 -84,0 82,3 при 0°С 432,1
НВг 0,141 0,8 12 Ю-ш Устойчив при комнатной темпера- —89 -67,0 221 при 0°С 366,3.
HI 0,160 0,4 5 3 туре Не устойчив —50,8 —35,5 224 при 10 °C 298,9*
а Константа реакции 2НХт^Н2+Х3.
ходит протолитическая реакция, которую следует рассматривать как сопряженное кислотно-основное взаимодействие:
НХ + Н2О Н3О+ + X-
кислота 1 основание 2 кислота 2 основание^! очень слабое сильная очень сильная слабое
Степень протолиза газообразных галогеноводородов в воде увеличивается в ряду НР<НСКНВг<Н1.
35.5.3. Реакции галогенов
Окислительно-восстановительные свойства галогенов зависят от электроотрицательности, энтальпии диссоциации и других факторов. С увеличением радиуса атомов окислительные свойства галогенов ослабевают. Поэтому легче всего окисляется ио-Дид-ион, тогда как фторид-ион не может быть окислен обычными окислителями.
Опыты 7—9, описанные в конце раздела, демонстрируют окислительное действие галогенов. Образуются полярные молекулярные вещества:
25Ь---> 2Sb3++ 6е"
ЗС12 + 6е~ -► 6С1-
2Sb + ЗС12 -► 2SbCl3
Sn + 2Вг2 ► SnBr4
Hg+I2 —► Hgia
32—2027
498
Неорганическая химия
35.5.3.1. Галогенид-, полигалогенид-ионы
Галогенид-ионы образуются при восстановлении галогенов. -Возможность их присутствия в виде ионов, например в водном растворе, зависит от величины парциального отрицательного заряда. Присоединение электрона к атому галогена (с образованием конфигурации s2p6) сопровождается выделением энергии, и поэтому галогенид-ионы весьма устойчивы.
Галогенид-ионы с однократно заряженными катионами дают соли, т. е. вещества с гетерополярной связью (NaCl, NaBr, Nal). Большинство галогенидов металлов растворимы в полярных растворителях.
Небольшие катионы с большим зарядом оказывают на галогенид-ионы поляризующее действие. В наибольшей степени деформируется и поляризуется иодид-ион, вследствие чего ио-диды металлов в высоких степенях окисления оказываются окрашенными. Гетерополярный характер веществ выражен у иодидов в наименьшей степени. В табл. В.25 приведены некоторые данные для галогенид-ионов.
Таблица В.25. Некоторые физические характеристики галогенид-ионов
Радиус, нм Стандартный потенциал, В 2Х-*±Хг+2е Поляризуе-MOCTbXlO24, см3/атом Энтальпия гид-ратации, кДж/моль
F- / 7 0,133 2,82 1,04 510,8
Cl- 0,181 1,36 3,66 372,6
Br- 0,196 1,06 4,77 339,1
1- 0,219 0,53 7,10 301,5
Как было показано ранее, растворимость солей тесно связана с энергиями их решеток и энергией сольватации ионов (разд. 33.3.1). NaF хуже растворим в воде, чем Nal, так как энергия решетки NaF больше. Высокая энергия сольватации NaF по сравнению с энергией сольватации Nal в данном случае недостаточна для компенсации вклада энергии решетки.
Обычно энергия решетки тем больше, чем выше поляризуемость анионов (исключение: фториды). Плохая растворимость -соли определяется, конечно, не только поляризуемостью аниона. Так, например, хлориды, бромиды и иодиды одновалентных меди, серебра, золота плохорастворимы. Электронные конфигУ' рации ионов Cu+, Ag+ и Аи+ сходны — у всех полностью занят ^-уровень:
Cu+ Is2 2s22p6 3s23p83d10
Ag+ Is2 2s22pe 3s23pe3d10 4s24pW>
Au+ Is2 2s22p8 3s23p83d10 4s24pe4d1« 5s25p85dw
Хлор, бром, иод
499»
Более того, с этими ионами имеют формальное сходство ионы. Hg22+, Т1+ и РЬ2+, у которых d-уровень также заполнен полностью. Имеющиеся во внешнем s-уровне электроны ионов таллия и свинца при образовании труднорастворимых галогенидов ведут себя как «инертная электронная пара», которая, находясь на периферии электронной оболочки, не оказывает заметного влияния на связь.
Основываясь на значениях стандартных потенциалов (табл, В.25), можно найти системы, селективно окисляющие галогениды. Это оказывается важным для аналитической химии галогенид-ионов.
В области pH от 1 до 7 окислительно-восстановительное’ равновесие в системах галоген/галогенид почти не зависит от pH; только в очень сильнокислых средах (pH 0 или —I) стандартный потенциал системы 21~/12 несколько понижается (pH 1„ £0=0,53 В; pH —1, £'° = 0,44 В). Уменьшение потенциала в этих условиях можно объяснить тем, что при высокой кислотности; происходит увеличение коэффициента активности иодид-иона вследствие дегидратирующего действия протонов. Иодид-иош восстанавливает ионы Fe3+, а также Т13+ и Си2+, а бромид-ион — лишь Си2+.
Реакция с ионами Fe3+ протекает не количественно, так как в противоположность реакциям с Т13+ и Сн2+ взаимодействие не-сопровождается образованием твердых продуктов.
Применение уравнения Нернста для окислительно-восстановительных равновесий, зависящих от величины pH, рассматривалось в разд. 33.5.1.5. Справедливо уравнение
£=£o-O,O59 — pH-4—^Llg-^ п ‘ 1 п ь ared
где п — число электронов, т— число протонов. Для системы: Мп2+/МпО4~ получим
Е = Е°~ 0,059-1,6pH4-]g а.Мп2С.
Влияние величины pH для этой системы особенно заметно, так Как отношение mln велико. Если положить активности всех участвующих в реакции ионов равными единице и изменять лишь Р“, легко видеть, что можно осуществить селективное окисле-Нйе галогенид-ионов. Побочные реакции в системе Мп2+/МпО4_ Исключаются. На рис. В.26 приведены зависимости стандартах потенциалов систем 2 С1~/С12; 2 Вг~/Вг2; 2 1-/12 и Мп2+/МпО4_
Годных растворах от pH. Зная потенциал соответствующей Лислительно-восстановительной системы, можно определить, Ри каком pH будет идти реакция окисления. В области рН>7" Реакции протекают более сложно: ион МпО4~ восстанавливает-32*
-500 Неорганическая химия
ся уже не до иона Мп2+, а образующиеся галогены в щелочной среде подвергаются диспропорционированию.
Галогенид-ионы можно рассматривать как корреспондирующие основания галогеноводородных кислот. Образование ковалентной связи между основанием и протоном соответствует реакции нейтрализации. Этот тип реакции представляет частный случай более общих реакций. Если катион не является протоном, то реакцию такого типа называют реакцией координирования (или упорядочивания). Известны «нормальные» реакции координирования, в которых осуществляется комбинация двух
/ Рис. В.26. Влияние pH на окисление ионами МпО4~.
частиц различной электроотрицательности. При этом образуется «нормальная» связь, например в Agl. Если катион за счет так называемой координационной связи присоединяет большее число отрицательных частиц, происходит образование комплексного иона. В этом случае слабые основания выступают как относительно сильные лиганды, способные давать большое число комплексных ионов. Иодид- и бромид-ионы представляют собою типичные мягкие основания.
В лабораторной практике часто используют растворы иода. Растворимость иода в воде -мала (опыт 3) и для ее повышения к раствору добавляют хорошо растворимые иодиды. Причина увеличения растворимости иода состоит в образовании полиио-дидных ионов. Изучение этого равновесия показало, что в основном присутствуют ионы 1з_ и Is-:
KI + nI2 К12П+1
При л=1 константа равновесия этой реакции в CCI4 равна 7,1-10-2. В твердом состоянии известны полигалогенид-ионы с л^4. Большие по размерам ионы такого типа стабилизирУ'
Хлор, бром, иод
501
ются при координации с более крупными катионами, например [(С2Н5)4N]"''ly- и НЬ+19~-2СбНб (эффект сольватации). Помимо этого, известны полигалогенидные ионы, содержащие два или даже три вида атомов галогена (1С12~, 1С14~, 1С1Вг~, IBrF~, IFC13~). Эти ионы можно рассматривать как производные межгалогенных соединений.
Для некоторых равновесий ассоциации типа
АВ + В’ АВ2-
были определены значения констант равновесия при 25 °C: Cig— Вг3~ IClg- 1Вгз— 13~
К, л/моль 0,01 17,8 167,0 370,0 725,0
35.5.3.2. Межгалогенные соединения
Соединения IC1 и 1С13 играют определенную роль в количественном анализе. Эти полярные молекулярные вещества построены таким образом, что более тяжелый атом координирует вокруг себя более легкие атомы. Всегда нечетное число атомов в молекуле увеличивается при увеличении соотношения радиусов ^бол/^мал. Так, атом иода может соединяться с семью атомами фтора, но лишь с одним атомом брома. Бром может координировать самое большое пять атомов другого галогена. Данные по устойчивости межгалогенных соединений, представленные на рис. В.27, дают информацию и о прочности связей в их молекулах. Геометрию молекул можно предсказать исходя из ее электронной конфигурации и типа связей.
Молекулы IF и BrF неустойчивы, хотя изменения свободной энергии Гиббса AG0 реакций их образования отрицательны (—117,7 и —76,6 кДж/моль соответственно). Они диспропор-ционируют с образованием продуктов с большей степенью фторирования:
5IF =e==t IF64-2I2
3 BrF BrF3 + Br2
Полярные ковалентные связи в молекулах IF5 и BrF3 очень прочные, а свободные электронные пары расположены достаточно симметрично. С термодинамической точки зрения диспропорционирование, несмотря на отрицательные значения AG°o6p для IF и BrF, объясняется тем, что значения AG°06P для IF5 и BrF3 еще более отрицательны. Аналогичные соотношения наполняются и для энергий диссоциации (рис. В.27). Молекула BrF устойчивее, чем молекула IF, поскольку в первой возможно участие рп—с?л-связывания. Сравнение устойчивости различных межгалогенных соединений типа АВ, АВ3, АВ5 и IF7 между собой, а также с неполярными молекулярными веществами типа А—А и В—В можно провести, используя рис. В.27.
502
Неорганическая химия
Рис. В 27. Свободная энтальпия образования газообразных межгалогенных соединений из газообразных галогенов и их энергии диссоциации (шкала-справа).
Межгалогенные соединения — сильные окислители. Почти все элементы периодической системы окисляются ими до смешанных галогенидов*.
35.5.4. Реакции кислородных соединений галогенов
Наиболее заметны различия галогенов между собой в соединениях, где они проявляют положительные степени окисления. В основном это соединения галогенов с наиболее электроотрицательными элементами — фтором и кислородом, которые
* Или до смеси галогенидов. — Прим, перев.
Хлор, бром, иод
503
могут частично переводить s- и р-электроны атомов хлора, брома и иода на d-орбитали.
При указании на различные, иногда очень высокие, степени окисления речь идет лишь о смещении электронов, и в действительности, например, ион С17+ не существует. Перемещение s- или р-электронов на d-орбиталь приводит к появлению новой, частично заполненной d-орбитали, причем s- или р-орбитали становятся также лишь частично заполненными. Тем самым оказывается возможным образование еще двух ковалентных связей и поэтому у галогенов следует ожидать преимущественной устойчивости нечетных степеней окисления: —1, +1, +3, 4-5 и 4-7. Известно лишь немного соединений, в которых галогены имеют степени окисления 4-4 и 4-6.
Степень окисления 4-1 неустойчива. До сих пор не удалось получить свободные гипогалогенные кислоты, так как они мгновенно диспропорционируют.
Степень окисления 4-3 также неустойчива. Оксиды галогенов в этой степени окисления неизвестны. Хлор образует малоустойчивую хлористую кислоту НС1О2, тогда как для брома и иода подобные соединения не известны.
Устойчивее соединения галогенов в степени окисления 4-5. Иод образует 12О5 и устойчивую иодную кислоту НОЮ2.
Соединения галогенов в степени окисления 4-7 вследствие симметричного строения молекул и максимального координационного числа устойчивы (исключение НВгО4).
Причиной неустойчивости бромной кислоты можно считать плохое перекрывание 4й-орбиталей атома брома и 2р-орбиталей кислорода, так что не происходит стабилизации связей за счет дополнительного л-связывания. В третьем периоде периодической системы ослабление л-связывания при переходе от SiO44- к С1О4- компенсируется наличием достаточно прочных а-свя-зей В четвертом периоде устойчивость ионов такого типа (GeO44-, AsO43~ « SeO42-) вообще меньше. Ослабление в этом ряду л-связывания приводит к тому, что ион ВгО4- оказывается неустойчивым.
Оксиды галогенов — полярные молекулярные вещества, в которых атомы галогенов (кроме фтора) имеют положительные степени окисления.
Кислотные свойства в наибольшей степени выражены у оксидов хлора, так как разность электроотрицательностей хлора и кислорода наименьшая (разд. 35.2.1). В соответствии с общими закономерностями С12О7 дает наиболее сильную кислоту (табл. В.26). Следует учитывать также свойства воды как растворителя. В таблице В.26 указаны продукты, образующиеся при взаимодействии оксидов галогенов с водой. Свойство оксидов, а следовательно, и кислородных кислот образовывать соединения полимерного типа в соответствии с общими правилами (разд. 35.2.1) наиболее типично для иода. Перечень извест-
Таблица В.26. Кислородные соединения галогенов
Хлор Бром Иод
Оксиды С12о Вг2О 12О4
сю2 ВГдОд
С120в С120? ВгО2 (Вг2О;) 12О6 (экзотерм.)-
Теплоты гидратации Д//, кДж/моль
Х2О (газ) + Н2О ► НХО (aq) 21 а —а
ХО (газ) + ХО2 (газ) + Н2О ► * НХО2 (aq) 75® (?)а Неизв.
ХО2 (газ) + ХО, (газ) + Н«О ► > НХО3 (aq) 168а а а
Х2О7 (газ) + Н2О »- НХО4 (aq) 243а Неизв. н6ю6, Н412О3 в ЮМ р-ре НС1О4 также-в виде 1(0Н)6
Константы равновесия
Х2 (газ,жидк., тв.) ► Х2 (aq) 0,062 0,21 0,0013
Х2 (aq) Н+ + X- + НОХ 4,2-10-9 7,2-10-» 2,0-10-1®
Х/ + 2ОН- ► Х-+ХО" + Н2О 7,5-1С1» 2-Ю8 ~30
3X0" ► 2Х" + Х03- 10ь7 10*5 1020
2НХ0 >- X- + Н+ + НХО2 ~10-» — —
2X0- ► Х~+ХО2“ ~10* — —
4Х03" > Х"+ЗХО4" 1029 — Ю-53
Равновесная концентрация в водно» растворе при 25 °C (моль/л)
Растворимость 0,091 0,21 0,0013
«н+ = сх~ = «нох 3-Ю"2 1,15-10-а 6,4-10-а
Стандартные потенциалы, В
Н+4-НОХ + е- ч=* Ч' 1/2Х2(газ, жидк., тв.) + Н20 4-1,63 +1,59 +1.4S
ЗН+ + НХО2 + Зе_ ч=> ч=~* VjXjjraa, жидк., тв.) 4 2Н2О 4-1,64 — —
6Н+ 4- Х03 4- 5е“ <77^» ч ’ > V2X2 (газ, жидк., тв.) + ЗН2О 4-1,47 +1,52 +1,20
8Н+ 4- Х04- 4- 7е~ ч=* ч • > VaXa (газ, жидк., тв.) + 4Н2О 4-1,42 — +1,34
1/зХ2(газ> ЖИДК., ТВ.) 4-С- 4 —Ь X- + 1,36 +1,07 +0,54б
Хлор, бром, иод
505
Продолжение
Хлор Бром Иод
ХО- + Н2О + 2е“ =s==t X- + 2ОН- +0,89 +0,76 +0,49
ХО2_ + 2Н2О + 4е“ X" + 4ОН' +0,78 — —
ХОз- + ЗН2О + 6е‘ ч=ь X- + 6ОН- +0,63 +0,61 +0,26
ХО4- + 4Н2О + 8е“ X- + 8ОН~ +0,56 — +0,39
2Х- + 2Н+ + V2O2 Х2 + Н2О —0,13 0,16 0,69
Константы диссоциации КИСЛОТ, рКа
(см. также табл. В.6)
HXO »- Н+ + ХО- 7,25 8,7 И
нхо2 —► н+ + ХО2- 2,0 — —
НХО3 —> н+ + ХО3- 0 0,7 0,8
НХО4 »- Н++ХО4- 10 — —
Н6ХО„ —<- н+ + н4хо„- — — 3,2
н4хо„- —► н++н,хов2- — — 6,7
я Соединение неустойчиво.
® Это значение показывает, что I” в водном растворе может окисляться кислородом
вых оксидов галогенов приведен в табл. В.26. Их химические реакции не имеют особого практического значения.
Реакции кислородных соединений галогенов лучше изучать на примере кислородных кислот галогенов.
35.5.4.1. Сопряженные кислотно-основные и окислительно-восстановительные реакции
Большинство известных кислородных кислот можно получить (хотя бы формально) путем взаимодействия соответствующих оксидов или их смесей с водой (табл. В.26).
В то же время гетеролитическое расщепление галогенов как Неполярных молекулярных веществ в воде (полярном растворителе) или в растворах, содержащих ОН_-ионы, также приводит к образованию кислородных кислот галогенов или их анионов. Сначала происходит сольватация растворенных галогенов. Б результате взаимодействия с диполями воды неполярная связь в молекулах галогенов подвергается индуцированной поляризации, а затем протекает быстро идущее окислительно-восстановительное диспропорционирование. 'При этом рассматри-®аемый формально катион галогена оказывает поляризующее
506
Неорганическая химия
действие на ион ОН- и благодаря этому стабилизируется. Степень протекания этих реакций для различных галогенов характеризуется константами равновесия (табл. В.26). Сравнивая эти величины, можно заключить, что в присутствии ОН~-ионов константы равновесия значительно увеличиваются. За счет связывания протонов равновесие смещается в сторону образования кислородных соединений галогенов.
С термодинамической точки зрения можно предполагать, что на устойчивость гипогалогенит-ионов в присутствии ОН_-ионов должно оказывать существенное влияние их способность к дальнейшему диспропорционированию. При этом термодинамически наиболее вероятно диспропорционирование до галогенат-ионоа (табл. В.26). Следует также отметить, что на равновесие реакций диспропорционирования галогенов и гипогалогенитов сильно влияет изменение температуры. Несмотря на то что константа равновесия реакций диспропорционирования хлората на перхлорат и хлорид достаточно велика (табл. В.26), в растворах при 100 °C реакция идет очень медленно. Это еще один пример* того, что при рассмотрении хода реакций следует учитывать как термодинамические, так и кинетические факторы. Броматы и йодаты в водных растворах при нормальных условиях не дис-пропорционируют.
Для характеристики изменения свойств в ряду галогенов и их кислородных соединений помимо констант равновесия можно использовать также величины окислительно-восстановительных потенциалов (табл. В.26). Эти данные позволяют определить, в каких условиях и при каких pH данная группа будет окисляться или восстанавливаться, т. е. какие из конкурирующих реакций в действительности пойдут.
Все кислородные кислоты галогенов представляют собой сильные окислители. Их окислительные потенциалы зависят от pH, что показано на примерах хлорноватистой и бромновати-стой кислот на рис. В.28, а и б. Сила окислительного действия возрастает с увеличением относительной доли недиссоциирован-ной кислоты, т. е. с ростом концентрации протонов (разд. 35.7.3.1). Для протекания данной окислительно-восстановительной реакции необходимо присутствие недиссоциированной кислоты в какой-то минимальной концентрации. Это означает, что чем сильнее кислота, тем более низкое значение pH должно поддерживаться (см. опыт 19).
В разбавленных растворах НСЮ4 не является окислителем, поскольку она полностью диссоциирована, а окислительное действие иона С1О4~ невелико. Присутствие протонов в большой концентрации, а также повышение температуры вызывают искажения высокосимметричной тетраэдрической координации иона С1О4~, так что концентрированная НСЮ4 действует как окислитель.
Хлор, бром, иод
507
Рис. В.28. Диаграмма окислительно-восстановительной системы в зависимости от pH.
а - С1(1)/С1»/С1-; б — Вг(1)/Вг°/Вг~.
Осторожно! В концентрированном виде НС1О4 взрывоопасна.
При повышенных температурах НС1О4 окисляет иодид в йодат
135 °C +1 200 °C +5
1 нею* 10 нсюГ 10а
Периодные кислоты также обладают сильным окислительным действием. Они реагируют быстро, т. е. без кинетических затруднений, в соответствии с величинами их потенциалов.
Некоторые из галогенат-ионов в нейтральных водных раство-₽ах не проявляют сильных окислительных свойств, которые **ожно было бы ожидать на основании величин их потенциалов. При комнатной температуре нейтральные водные растворы Хлоратов и йодатов не диспропорционируют с образованием
508
Неорганическая химия
пергалогенатов. Нейтральный водный раствор NaClCU не проявляет окислительных свойств.
Вследствие сложного механизма окислительно-восстановительных реакций в водных растворах — протекания большого числа последовательных реакций — часто наблюдаются кинетические затруднения. При повышении температуры и в присутствии катализаторов реакции начинают идти без кинетических затруднений.
Отсутствуют кинетические затруднения и при реакциях расплавленных солей галогенкислородных кислот. По окислительному действию расплавы этих солей приближаются к атомарному кислороду. В сильнощелочных расплавах (присутствие иона О2-) центральный атом (на промежуточной стадии) окисляет ионы О2- до атомарного кислорода, который далее и оказывает окисляющее действие. Этот механизм сходен с окислительным действием ионов МпО4“ в сильнощелочных средах.
При нагревании солей галогенкислородных кислот могут происходить различные окислительно-восстановительные реакции: диспропорционирование, окислительно-восстановительное разложение с выделением кислорода и галогенида или с образованием свободного галогена и оксида металла:
Диспропорционирование 4 КС1О3 --► 3 КС1О4 + КС1
Окислительно-восстано- 2 КВгО3 -► 2 КВг -г 3 О2
вительное разложение
85.5.4.2. Реакции осаждения
Некоторые ионные соединения галогенкислородных кислот представляют интерес как труднорастворимые вещества и вследствие особенностей протекания окислительно-восстановительных реакций с их участием.
Интересные особенности были обнаружены в структурах перйодатов, принадлежащих к различным типам соединений. Так, могут быть получены кислые гексаоксоиодаты (табл. В.26) Na2H3IO6, NaH4IO6-6H2O, Na3H2IO6 и тетраоксоидаты(VII) (ме-тапериодаты), в структурах которых имеются тетраэдрические ионы Ю4_. В соединении КДгОэ-НгО содержатся ионы [Оз(ОН)Ю2Юз(ОН)]4~, в которых октаэдрические группы Ю& соединены общим ребром. Максимальное координационное число атомов хлора и брома по кислороду равно четырем.
Растворимость солей кислородных кислот галогенов уменьшается при увеличении порядкового номера галогена и его положительной степени окисления. Так, все гипохлориты и хлораты хорошо растворимы в воде. Напротив, йодаты четырехвалентных металлов — Се, Zr, Hf и Th — труднорастворимы Да; же в 6 н. HNO3, а йодаты серебра и бария — в разбавленной HNO3.
Хлор, бром, иод
50>
Перхлорат-ион образует труднорастворимые соли с крупны-:и или комплексными катионами, из которых следует назвать <+, Rb+, Cs+, Т1+, а также (C2H5)4N+ и (C6H5)4As+.
Перхлорат тетрафениларсония используется в количествен-ом анализе для определения С104~-иона. Благодаря своей ма-1ОЙ поляризуемости ион СЮ4~ стабилизирует высокие степени жисления, давая простые соли. Согласно теории жестких и мягких кислот и оснований, СЮ4~ относится к жестким основапи-ш. В водных растворах он не образует анионных комплексов, рак что в перхлоратных растворах можно, например, проводить Точные измерения стандартных потенциалов катионных окислительно-восстановительных систем. Окислительный потенциал кислого раствора сульфата Ce(IV) в присутствии ионов СЮг больше, чем в присутствии ионов NO3_, SO42- или С1_.
Рекомендуемые опыты
1. Неполярное молекулярное вещество /2. По нескольку кристаллов нода добавляют в пробирки с тетрахлорндом углерода, бензолом, пиридином и разб. раствором NaOH; отмечают окраслу растворов.
2. Экстракция — доказательство неполярности молекул галогенов. Приготавливают насыщенные водные растворы хлора, брома н нода (осторожно, галогены ядовиты!). Хлор пропускают до насыщения в дистиллированную воду. Несколько капель брома встряхивают с небольшим количеством воды. Иод встряхивают с водой более продолжительное время, лучше с использованием «трясучки». По 1 мл приготовленных таким образом растворов галогенов разбавляют 4 мл воды, добавляют к каждому нз растворов по 3 мл ’ ССЦ илн СНС1з и встряхивают.
3. Сублимация иода. В широкую пробирку с боковым штуцером для откачивания вставляют на резиновой пробке пробирку меньшего размера. Внутреннюю пробирку охлаждают холодной водой. Для проведения сублимации в широкую пробирку загружают немного нода и осторожно нагревают ее небольшим пламенем горелки. Из темно-фиолетовых паров иода на охлаждаемом «пальце» осаждаются черно-фиолетовые кристаллы с металлическим блеском. При быстром нагревании иод плавится (Тпл= 113,6 °C; 7'КИп= = 184,5 °C).
4. Получение хлора. В пробирке нагревают немного диоксида марганца, смоченного несколькими каплями конц. НС1. Через отводную трубку, имеющуюся у пробирки, выделяющийся газ пропускают в пробирки с водой н с 0,1 М KI. В пробирке с водой наблюдается слабо-зеленое окрашивание; из раствора в другой пробирке нод можно экстрагировать тетрахлоридом углерода.
5. Получение брома. В прибор, описанный выше, вносят несколько кристаллов КВг, немного МпО2, добавляют 50%-ный раствор H2SO4 и осторожно нагревают. Выделяющийся бром собирают в другую, охлаждаемую льдом, пробирку (осторожно! резиновые перчатки! защитные очки!).
6. Получение галогеноводородов. В прибор для получения газов помещают в отдельных опытах следующие количества веществ:
0,5 г NaCl н 2 мл конц. H2SO4;
0,5 г КВг и 2 мл конц. H2SO4;
0,5 г KI н 2 мл конц. H2SO4;
те же количества солей обрабатываются конц. фосфорной кислотой.
Образовавшиеся в опытах с конц. фосфорной кислотой бесцветные газообразные галогеноводороды пропускают в пробирки, наполненные водой. *ззц легко поглощаются водой (внимание! Следите за тем, чтобы вода не-
ДЮ Неорганическая химия
затягивалась в отводящую трубку!). С помощью индикаторной бумаги про-веряют pH полученных водных растворов и затем добавляют в каждую про. Днрку немного порошка цинка.
7. Окислительное действие С12. В литровую колбу, снабженную трубками для входа н выхода газов, помещают тонкий порошок сурьмы и затем про-пускают ток сухого хлора. Реакция начинается без нагревания и идет с воспламенением.
8. Вг2 менее сильный окислитель, чем С12. В литровую колбу помещают тонкую оловянную фольгу, добавляют каплю воды и затем 3—4 капли брома. Если реакция не начинается, немного нагревают.
9. /2 — слабый окислитель. Небольшую капельку ртутн и немного иода помещают в пробирку. Смесь осторожно нагревают; при этом на холодных частях пробирки появляется красный или желтый налет Hgl2.
10. Окислительно-восстановительные реакции галогенов. К нескольким каплям 0,1 М КВг и 0,1 М KI по каплям добавляют хлорную воду. Выделяющиеся свободные бром и иод экстрагируют встряхиванием с ССЦ (опыт 2). Затем к раствору KI прибавляют избыток хлорной воды. После встряхивания смеси фиолетовое окрашивание тетрахлорнда углерода исчезает. Окраска становится желтоватой нз-за присутствия 1С13 При еще большем избытке хлорной воды образуется иодат-ион и раствор обесцвечивается.
11. Различие окислительно-восстановительных потенциалов. 0,01 М KI подкисляют 2 М H2SO4 н затем добавляют немного раствора нитрита натрия. Иод экстрагируют тетрахлоридом углерода
Проводят те же операции с 0,1 М раствором КВг. Окрашивания не наблюдается.
12. Влияние pH на окислительно-восстановительные свойства. К 0,1 М КВг, подкисленному 0,1 М уксусной кислотой, прибавляют несколько капель 0,1 М ацетата натрия. Затем приливают несколько миллилитров 0,1 н. КМпОд. При нагревании выделяется свободный бром (запах). Доказать присутствие брома можно с помощью полоски иодкрахмальной бумаги (синее окрашивание).
Опыт повторяют с КС1 (ч д. а.); посинения иодкрахмальной бумаги наблюдаться не должно.
13. Иодид-ион окисляется легче, чем другие галогенид-ионы. К небольшому количеству (несколько капель) 0,1 М КС1, КВг и KI добавляют несколько кристаллов NH4Fe(SO4)2 и кипятят, а затем встряхивают с ССЦ. -Лишь в пробирке с раствором KI наблюдается красно-фнолетовое окрашивание.
2 Fe3+ 4-2Г 4—2 Fe’-+ -|- I2f
14. Ион Hg2+ и его комплексы с галогенид-ионами. 0,1 М Hg(NO3)2 подкисляют 2,5 М HNO3, после чего приливают растворы КС1, КВг и KI-
15 Соединение включения полииодида. а) К сильноразбавлениому раствору К1з (раствор очень слабо окрашен в желтый цвет) добавляют немного раствора крахмала. Исследуют поведение образовавшегося соединения включения при нагревании. При повышении температуры синяя окраска исчезает, при охлаждении —• вновь появляется.
б) Приготавливают слабый раствор иода в воде и разбавляют его до тех пор, пока он не перестанет давать синее окрашивание с раствором крах--мала В другой пробирке сливают свежеприготовленный раствор KI с раствором крахмала. Синяя окраска не должна появляться, если случайно не произошло окисления иодида Оба бесцветных раствора смешивают н наблюдают интенсивное синее окрашивание. . .
16 Диоксид хлора — полярное молекулярное вещество. Немного кончике шпателя) КС1О3 помещают в пробирку из тугоплавкого стекла и д° бавляют немного конц. H2SO4. Затем укрепленную в держателе пробирку очень осторожно нагревают. Смесь днспропорционнрует на СЮ2 н Н1-! *-Газообразный диоксид хлора иногда сильно взрывает.
Хлор, бром, иод 51
Осторожно! Оксиды галогенов (за некоторыми исключениями) весьма реакциоииоспособиы и склонны к разложению со взрывом. Некоторый интерес представляет реакция образования С1О2.
17. Окислительно-восстановительное диспропорционирование. К хлорной» бромной и иодной воде добавляют ССЦ. Затем по каплям прибавляют несколько миллилитров 1 М раствора NaOH. По изменению окрашивания растворов судят о различной степени диспропорционирования галогенов.
Те же опыты повторяют при нагревании растворов на водяной бане к при добавлении горячего 1 М NaOH. Объясните наблюдаемые явления.
18. Диспропорционирование 1О~. Иод (на кончнке шпателя) растворяют в горячем растворе NaOH средней концентрации. В этих условиях в растворе присутствуют только нодид- и нодат-ионы. Берут ~ 1 мл этого раствора» добавляют одну каплю раствора бромтнмолового синего и нейтрализуют несколькими каплями 2 М HNO3 до перехода окраски индикатора от синей в зеленую. При этом образующийся в месте падения капли HNO3 элементарный иод каждый раз снова растворяют, встряхивая раствор. Затем, добавля® по каплям раствор AgNO3, наблюдают последовательное осаждения Agl и AgIO3. После полного осаждения осадки центрифугируют, промывают и обрабатывают разбавленным раствором аммиака. AgIO3 растворяется с обра-зеванием комплексного соединения [Ag(NH3)2]IO3. Путем центрифугирования отделяют осадок нодида от раствора. К последнему добавляют по каплям избыток раствора H2SO3, что приводит к выпадению Agl (нерастворимого в аммиачном растворе). Сернистая кислота восстанавливает иодат до иодида (уравнение реакции).
19. Зависимость окисления иодида галогенатами от pH раствора. В три пробирки наливают 0,1 М КЮ3, в другие трн пробирки — 0,1 М КВгО3 и еще в трн — 0,1 М КС1О3. Расставляют эти пробирки по три штуки в трех сериях: одна — раствор йодата, вторая — бромата и третья — хлората. Получают трн серин пробирок. К растворам первой серии добавляют буферную смесь (для ее приготовления смешивают 15 мл 2 М ацетата натрия н 2 мл 2 М уксусной кислоты), растворы второй серии подкисляют разбавленной уксусной кислотой, а растворы третьей серии — разбавленной соляной кислотой. После этого в каждую из пробирок добавляют по 1 мл 5 %-кого раствора КЦ содержащего раствор крахмала. В пробирках первой серии синее окрашивание появляется прн реакции с йодатом, во второй серин — в пробирках с йодатом и броматом, тогда как в растворах, подкисленных соляной кислотой, выделение иода наблюдается во всех трех случаях (иногда раствор хлората необходимо еще сильнее подкислить несколькими каплями конц. НС1) (уравнения реакции).
20. Кинетическая устойчивость иона ClOt~. К раствору NaC104 добавляют разб HNO3. цинк н кипятят.
Проводят такой же опыт, но вместо цинка добавляют H2SO3. Восстановления не происходит. Прн прнлнванни раствора AgNO3 осадка не образуется.
21. Каталитическое окисление в щелочной среде. К раствору NaClO*. приливают избыток раствора FeSO4, к которому добавлен NaOH в количестве недостаточном для полного осаждения Fe(OH)2. Некоторое время смесь кипятят, затем центрифугируют; раствор подкисляют конц. HNO3. При добавлении раствора AgNO3 наблюдается выпадение AgCl.
22. Каталитическое восстановление в кислой среде. Немного разбавленного раствора NaClO4 наливают в стакан и добавляют ~ 1/4 объема конц. H2SO4. Затем прикапывают немного раствора сульфата титана (IV) и нагревают до кнпення. Не прекращая кипятить, раствор, небольшими порциями вносят маленькие кусочки цинка. Раствор отфильтровывают и добавляют по несколько капель конц. HNO3 и раствора AgNO3, убеждаясь в присутствии хЛорид-иона.
п 23. Окислительно- восстановительные реакции в солевых расплавах. ° трех тугоплавких пробирках нагревают по 50 мг КС1О3, КВгО3 и КСЮ4.
512
Неорганическая химия
Сера, селен, теллур
513
В двух последних с помощью тлеющей лучинки можно доказать выделение кислорода. После охлаждения расплавов содержимое пробирок растворяют в воде и доказывают присутствие галогенид-ионов реакцией с раствором AgNO3, подкисленным азотной кислотой. Из расплава КС1О3 после его растворения в горячей воде можно выделить при охлаждении раствора кристал-.лический КС1О4.
Внимание! Следует избегать смешивания КС1О3 с легкоокисляющимися веществами, например серой, красным фосфором. Такие смеси сильно взры-<вают!
24. Реакции осаждения. К нескольким каплям 0,1 М растворов КС1О3, КВгО3 и КЮ3 приливают растворы AgNO3 и ВаС12. При этом осаждаются лишь йодаты, причем осадок AgIO3 быстро темнеет.
Готовят концентрированный раствор КВгО3 и, добавляя растворы AgNO3 и ВаСЬ, осаждают белые AgBrO3 и Ва(ВгО3)2. Отделяют осадки центрифу-гированием, к ним приливают холодную разб. HNO3. Убеждаются в том, что осадки плохорастворимы.
25. Труднорастворимые перхлораты. К разбавленному водному раствору NaC104 приливают растворы КС1, TINO3, RbCl и CsCl. Выпадают плохорастворимые осадки. КСЮ» хорошо растворяется в горячей воде; при охлаждении раствора КС1О4 выпадает в виде кристаллов.
Вопросы и задания
1. Почему для получения НВг и HI используют сиропообразную фосфорную кислоту?
2. Объясните, почему восстановительные свойства усиливаются в ряду F-—>С1~—»-Вг-—И-.
3. Какова роль плотности заряда катиона, его координационного числа и наличия у него свободных орбиталей при образовании комплексов?
4. Дайте объяснение устойчивости галогенных комплексов ртути в рампах теории жестких и мягких кислот и оснований.
5. Как построены полигалогенид-ионы? Почему неизвестен ион F3_?
6. Как можно получить RbF из RbCl?
7. Как можно доказать, какие из ионов I3~, Is-, I?- и 19- присутствуют <в растворе?
8. Составьте таблицу свойств межгалогенных соединений.
9. Какие продукты образуются при взаимодействии с водой различных <межгалогенных соединений?
10. Сравните между собой строение хлорной, иодной, теллуровой и сурь-гмяной кислот. Дайте объяснение сходству и различиям в поведении этих
КИСЛОТ.
35.6. Сера, селен, теллур
Электронная конфигурация валентной оболочки атомов этих элементов д2р4 в значительной степени определяет их химическое поведение. В соответствии с правилом Хунда на двух из трех р-орбиталей находится по одному неспаренному электрону-'Тем самым у атома возникает возможность образовать две ковалентные связи путем соединения с двумя атомами того же -или иного вида. С использованием свободных d-орбиталей атомы серы, селена и теллура в зависимости от типа лиганда могут давать от шести до восьми связей. Гибридная зр3й2-конфИ' гурация соответствует октаэдрическому расположению, напрй' мер, в SF6. С ростом радиуса атомов и, следовательно, увеличением способности к предоставлению орбиталей усиливается
(к теллуру) металлический характер элементов. Наиболее типичные степени окисления этих элементов в соединениях —2, 4-4 и +6. Вследствие того что связи носят преимущественно ковалентный характер, происходит выравнивание зарядов, так что они становятся гораздо меньше, чем степени окисления. Соединения с высокими степенями окисления для тяжелых элементов менее устойчивы.
35.6.1. Неполярные молекулярные вещества
Сера, селен и теллур при комнатной температуре — твердые вещества. Они образуют многочисленные полиморфные модификации (разд. 33.2.2), отличающиеся друг от друга кристаллическим строением, в частности размерами молекул в кристалле (табл. В.27).
Таблица В.27. Свойства халькогенов
Сера Селен Теллур
Модификация Ромбическая: S8 Моноклинная: S8 Ромбическая: Se8 Моноклинная: Se8 Ромбоэдрическая: SCoo Ромбоэдрическая
Превращение 96 °C SpOM6<= “^МОНОКЛ 50°С ЭеМонокл-« -Зеромб
Температура плавления SpoM6 114°С $монокл И9°С ^ромбоэдр 220,2°С $емонокл 180,0°С IsW*
Температура кипения 444,6°С 688,0°С 1390°С
Металлическая форма Нет ^ромбоэдр Те
Газообразное состояние *— $8 $2 Se8—Se2 Те2
Электроотрицательность 2,5 2,4 2,1
Две основные формы селена отличаются друг от друга гораздо сильнее, чем а- и (3-модификации серы. У теллура — одна м°дификация с четко выраженными металлическими свойствами (разд. 32.3.4 и 36.1). В полиморфных формах этих элемен-33—2027
514
Неорганическая химия
тов можно наблюдать довольно ясные переходы между неметаллами и металлами.
Устойчивый при комнатной температуре серый селен проводит электрический ток при освещении. Это указывает на то, что характер связей в нем близок к металлическому. В сером селене присутствуют восьмиатомные молекулы*. Они образую! кольца, а вообще структура построена по спиральному типу. В паровой фазе молекулы S2, Se2 и Тег — парамагнитны.
35.6.2. Полярные молекулярные вещества
Реакции образования полярных ковалентных или ионных соединений из неполярных ковалентных могут быть поняты на основе рассмотрения электронной конфигурации атомов (шесть валентных электронов). При использовании двух наружных наполовину занятых р-орбиталей атомы могут образовать две простые ковалентные связи, причем атом халькогена обычно заряжен отрицательно.
На примерах соединений этих элементов особенно хорошо прослеживаются переходы между соединениями различных типов. Это связано с тем, что у самих элементов наблюдается переход от неметаллов к металлам.
Из халькогенидов путем проведения гетерогенной кислотноосновной реакции (присоединение протонов) можно получить халькогеноводороды. Они более летучи, чем кислоты, вступающие в реакцию, и поэтому выделяются в виде газов. Гидриды халькогенов имеют весьма характерный запах и чрезвычайно ядовиты. В табл. В.28 приведены некоторые свойства газаообраз-ных гидридов халькогенов — строение, характеристика связей Таблица В.28. Свойства халькогеноводородов
Соединение Энергия связи Х-Н, кДж/моль Энтальпия образования кДж/моль Угол связи н-х-н, градус Степень ионности связи Х-Н, % °с т кип, °C Термическая устойчивость
Н2О 461,4 —242,0 104,45 33 0 100 Очень устойчив
H2s 345,8 - 22,2 92,2 ~4 —85,6 —60,8 Устойчив
H2Se 305,6 + 77,5 91,0 — 2,5 —60,4 —41,5 Мало устойчив
Н2Те 240,7 +143,2 89,5 ~0 —51,0 —1,8 Мало устойчив
* В основе структуры серого селена — бесконечные цепи Se«>. ^отгок Уилкинсон Дж. Современная неорганическая химия: Пер. с англ. — М.: МИР’ 1969 Т. 2, с. 381]. — Прим, перев.
Сера, селен, теллур
515
в молекуле, термическая устойчивость. Приведенные значения для степени ионности связи имеют оценочный характер. Они вычислялись с помощью соотношения Полинга из разности электроотрицательностей и дают верное представление лишь о ходе изменения степени ионности связи. Следует также учитывать, что в молекулах с числом связей больше двух степень ионности несколько меньше значения, вычисленного для простой связи X—Н (гл. 6).
Растворимость газообразных халькогеноводородов в воде невелика. Как и в случае галогеноводородов, при пропускании в воду Нц5, HaSe и НгТе идет протолитическая реакция (в две стадии):
Н2Х 4- Н2О Н3О+ 4- НХ-
НХ- +Н2О Н3О+-|- х2-
Вторая стадия протолитической реакции сдвинута вправо в гораздо меньшей степени, чем первая. Это видно из значений констант диссоциации кислот:
Н2О HaS H2Se Н2Те
pKi 15,74 6,92 3,89 2,64
рХг ~13,0 10,0 5,0
Ионы HS~, HSe~ и НТе- представляют собой анионные амфолиты, так что они могут реагировать как кислоты и как основания.
35.6.3. Реакции халькогенов
В виде простых веществ сера, селен и теллур довольно инертны. Для прохождения реакции требуется за счет подвода энергии разорвать связи между атомами в простых веществах.
Расщепление связей S—S катализируют амины. Реакциям кольцевых молекул Ss обычно предшествует реакция «размыкания» кольца. При этом сначала происходит нуклеофильная атака связи S—S, которая в большинстве случаев осуществляется по 5к2-механизму (разд. 11.3). Энергия активации находится в обратной зависимости от длины связи S—S. Акцепторные свойства атомов серы по отношению к присоединяющимся нуклеофилам (CN~, SO32~) выражены слабо. Следующие реакции идут по Эк2-механизму с разрывом связей S—S:
N=C“ 4- X—S-S-Y ► NCS4- SY-
I
X
so32- 4- X-S-S-Y ► X—S-SO3- 4- SY"
Расщепление связей S—S происходит при взаимодействии и Другими нуклеофильными реагентами, например (СбНз)зР.
Зз*
5J6
Неорганическая химия
35.6.3.1. Халькогенид- и полихалькогенид-ионы
В противоположность оксид-иону О2- сульфид-ион S2- может существовать в водном растворе, хотя возможно равновесие протолиза сильно смещено вправо
S2- + Н2О HS- + ОН-
В количественном и качественном анализе при изменении pH осуществляют фракционированное осаждение сульфидов.
Гидратированный ион S2- не поглощает в видимой области спектра. При образовании сульфидов за счет эффектов поляризации происходят сильные изменения в связывающей электронной системе, и поэтому сульфиды тяжелых металлов, как правило, интенсивно окрашены. Сульфид-, селенид- и теллурид-ионы выступают в реакциях как мягкие основания.
Сера окисляет целый ряд простых веществ, давая сульфиды. В зависимости от степени полярности связи образуются либо ионные, либо полярные молекулярные (S4N4) вещества, либо вещества промежуточного типа. Последний случай реализуется наиболее часто, например полимерные ковалентные соединения SiS2, Sb2S3 и Bi2S3, а также нестехиометрические фазы многих сульфидов металлов, обладающие сходством с металлическими веществами. Расстояния между атомами металла в структурах FeS, CoS и NiS составляют от 0,260 до 0,268 нм, что указывает на наличие взаимодействия металл — металл (полуметаллический характер).
В сульфидах металлов связь более ковалентная, чем в соответствующих оксидах. Поэтому часто не наблюдается аналогии в стехиометрии сульфидов и оксидов одного и того же элемента.
Плохую растворимость многих сульфидов, например PbS, обычно объясняют исходя из ионной модели (ионы РЬ2+ и ионы S2~). Однако SrS тоже соль. Почему же первая из них труднорастворима, имеет черный цвет, является полупроводником, тогда как другая этими свойствами не обладает? Объяснение, основанное на различии величин электростатических сил притяжения, явно недостаточно. Оба сульфида кристаллизуются в структурном типе NaCl, но связи имеют значительную долю ковалентной составляющей. В SrS это приводит к тому, что одна из р-электронных пар иона S2~ частично снова занимает квантовое состояние, освободившееся в результате ионизации атома стронция. Сила этой связи будет определяться энергией ионизации атома стронция и степенью перекрывания р-орбиталей атома серы с s-орбиталью атома стронция. Имеющая сферическую симметрию s-орбиталь плохо перекрывается (разд. 5.3) с другими орбиталями. По принципу Паули лишь один из шести ионов S2-, окружающих ион Sr2+, может образовать с ним ковалентную связь. s-Орбиталь может быть лишь однократно за'
Сера, селен, теллур
517
нятой, и поэтому мезомерная (резонансная) часть ковалентной связи мала.
Энергия ионизации атома свинца на 2164—1607=557кДж/ /моль больше, чем у атома стронция, что ведет к более сильному притяжению электронов. р-Орбитали атомов свинца имеют значительную плотность в направлении двух соседних атомов серы и это приводит к хорошему перекрыванию р-орбиталей ионов серы и свинца. Всего у атома серы имеются три р-электронные пары, а у атома свинца три свободные р-орби-тали. Тем самым осуществляется связывание в трех направлениях. Мезомерная (резонансная) гомеополярная составляющая связи в сульфиде свинца больше, чем в сульфиде стронция. Вследствие двустороннего перекрывания р-орбиталей образуются резонансные цепи, в которых при удалении, например, одного электрона из иона S2~ возникает дефект, распределяющийся по всей цепи. Этим объясняется черный цвет и полупроводниковые свойства сульфида свинца.
Селен и теллур в еще большей степени склонны к образованию ионных соединений с ковалентной составляющей связи с переходом к металлическому типу связи (разд. (6.6), поскольку они располагают еще большими возможностями предоставления орбиталей для образования связей.
Теллурид-ион в пределах группы — наиболее сильный восстановитель и наиболее мягкое основание. Ионы халькогенов могут окисляться даже воздухом. Так, при долгом стоянии из водного раствора H2S выпадает сера. Используя стандартные потенциалы реакций:
X2-—>Х+2е-
X-________О________ S Se Те
£°, В 0,41 —0,50 —0,76 • —0,91
можно выбрать подходящие окислители халькогенид-ионов.
Склонность халькогенид-ионов выступать в качестве лиганда при образовании анионных комплексов уменьшается при увеличении порядкового номера халькогена. Даже такое сильное основание, как ион S2~, представляет собой слабый лиганд. На образование соответствующих анионных комплексов сильное влияние оказывает pH раствора.
В щелочной среде концентрация ионов S2~ повышается и становится возможным образование тиокомплексиых соединений (тиосолей). В присутствии сильных щелочей ионы ОН- и конкурируют между собой, как лиганды, причем происходит также обмен лигандами. Гидр оксо комп лексы могут подвергаться далее конденсации с отщеплением Н2О и образованием сложных структур.
518
Неорганическая химия
За счет окисления воздухом, которое усиливается в щелочной среде {почему?), образуется сера, реагирующая далее с Sb2S3, давая Sb2S5:
Sb2S3 + 2 S -> Sb2S5 Sb2S5 -h 3 S2- -► 2 [SbSJs-
Эти уравнения можно рассматривать лишь как формальные.
Атомы серы и селена способны к образованию цепей или колец (табл. В.27). По этой причине у этих элементов возможно существование полихалькогенид-ионов. Для серы выделены цепочечные анионы вплоть до S92-, а для селена — до Se52^. По аналогии с пероксид-ионом S/~ может выступать и окислителем, и восстановителем. Полисульфиды с более тяжелыми анионами представляют собой слабые окислители.
Многосернистые водороды (полисульфаны) получают в виде желтого маслянистого вещества при вливании раствора полисульфида в избыток сильно охлажденной конц. соляной кислоты. Эти соединения весьма неустойчивы и быстро разлагаются на серу и сероводород. Полисульфаны обладают более сильными кислотными свойствами по сравнению с H2S (ср. Н2О и Н2О2). ,
35.6.3.2. Галогениды халькогенов
В молекулах галогенидов халькогенов атомы халькогенов присутствуют всегда в виде частиц, поляризованных положительно. С наиболее электроотрицательным элементом — фтором они дают SF6, SeF6, TeF6. В соответствии с общими закономерностями периодической системы SI4 не известен, тогда как Те14— вполне устойчивое соединение.
В октаэдрической молекуле SF6 должна осуществляться 5р3с?2-гибридизация. Однако расчеты интегралов перекрывания орбиталей в соединениях серы с другими элементами показывают, что в предположении гибридизации с участием Sd-орбита-лей не должно образовываться устойчивой связи. Как же можно сделать возможным участие d-орбиталей в образовании связей?
При достаточно полярном характере связи Z—X, обусловленном большой разностью электроотрицательностей, на центральном атоме возникает поле положительного заряда. Это поле несколько сжимает электронные облака центрального атома, причем орбитали фги благодаря своей большей поля-
ризуемости сжимаются сильнее, чем другие d-орбитали. Таким образом, сильная полярность связей в молекуле из-за большой разности электроотрицательностей может сказываться на ра мерах 3s-, Зр- и Sd-орбиталей, что в свою очередь делает во можным осуществление вышеуказанной гибридизации.
Сера, селен, теллур
519
Этот подход справедлив и для d-орбиталей фосфора (PF5, PCI5) и других элементов.
SF6 отличается весьма малой реакционной способностью (газообразный изолятор). Особенно устойчива молекула SF6 по отношению к гидролизу. Причины этого могут быть следующие: значительная сила связи S—F;
атом серы имеет максимальное координационное число и хорошо экранирован;
молекула SF6 неполярна.
Соединения TeF6 и SeF6 более реакционноспособны, чем SF6. В воде они полностью гидролизуются. TeF6— координационно ненасыщенная молекула и поэтому представляет собой слабую кислоту Льюиса. Например, присоединение CsF приводит к образованию Cs2TeFs. В противоположность SF4 соединения SeF4 и TeF4 являются сильными кислотами Льюиса, поскольку у них для образования связи могут быть предоставлены энергетически более выгодные орбитали.
Гидролиз всех галогенидов халькогенов идет в соответствии с полярностью связи халькоген—галоген. Часто гидролиз сопровождается реакцией окислительно-восстановительного диспропорционирования. Например, гидролиз S2CI2 идет через промежуточное образование тиосернистой кислоты (НО—S—S— —ОН), которая подвергается диспропорционированию. При этом выделяются НС1, SO2 и H2S. Побочными продуктами могут быть также сера, тиосерная кислота (H2S20s) и политионовые кислоты (H2SzOs).
S2C12— первый представитель гомологического ряда дихлор-замещенных многосернистого водорода.
35.6.4. Реакции кислородных соединений халькогенов
В кислородных соединениях халькогенов наблюдаются переходы между соединениями различных типов. Диоксиды, триоксиды и кислородсодержащие кислоты могут быть веществами молекулярного типа, полимерными или даже ионными соединениями.
Так, SO2 — газ, SeO2 — легколетучее твердое вещество, тогда как ТеО2 — малолетучее твердое вещество. Простая ковалентная о-связь в SO2 и SeO2 усиливается за счет рл—рл- и Рл—dn-связывания соответственно. SO2 — мономерное полярное молекулярное вещество. Напротив, SeO2 — уже полимерное соединение, построенное из длинных цепей, изогнутых в разных плоскостях. По две полиморфные модификации имеют ТеО2 и ^0С>2, причем второе соединение можно уже считать ионным.
SO3 и SeO3 — типичные кислотные оксиды, поскольку раз-н°сть электроотрицательностей образующих их элементов очень мала (разд. 35.2.1). Кислоты, соответствующие другим оксидам,
520
Неорганическая химия
Таблица В.29. Кислородные соединения халькогенов
Реакции Сера Селен Теллур
Стандартные потенциалы, В
X + ЗН2О ч—>- Н2ХО3 + 4Н+ + 4е~ 0,45 0,74 0,559
ХО2 + 4Н2О у—Н,ХО, + 2Н+ + 2е~ — — 1,02
Н2ХО3 + Н2О ХО42- + 4Н+ + 2е~ 0,17 1,15 1,04
Х + 6ОН- -<=t ХО32- + ЗН2О + 4е" — —0,366 —0,577
ХО32- + 2ОН- ХО42- + Н2О + 2е~ —0,93 0,05 0,40
S2O82- + 2е~ 2SO42- 2,06
S4Oe2- + 6Н2О 4H2SO3 + 4Н+ + бе" 0,51
S2Oe2" + 2Н2О 2SO42- + 4Н+ + 4е“ —0,22
HS2O4- + 2Н2О 2H2SO3 + Н+ + 2е~ —0,08
S2O42- + 4ОН- 4==t 2SO32- + 2Н2О + 2е~ —1,12
S2O32- + 6ОН- 2SO32- + ЗН2О + 4е- —0,584
Константы кислотной дис-
социации (рКа)
Н2ХО3 н+ + нхо2- 1,92 2,6 2,7
НХО3" н+ + хо32- 7,7 8,3 8,0
h2xo4 =«=* н++нхо4- -3,0 —1,0 7,68
НХО4- Н+ + ХО42- 1,92 2,0 11,19
также довольно сильные (табл. В.29). Будучи двухосновными, эти кислоты образуют анионные амфолиты.
Окислительно-восстановительные потенциалы кислородных кислот халькогенов (табл. В.29) зависят от pH. Для протекания окислительно-восстановительной реакции необходимо присутствие кислоты в недиссоциированной форме, хотя бы в минимальной концентрации. Чем сильнее кислота, тем ниже должно быть значение pH, при котором идет окислительно-восстановительная реакция. Для кислородных соединений селена и теллура наиболее характерна степень окисления +4.
35.6.4.1. Сопряженные реакции кислотно-основного взаимодействия и окисления — восстановления
В SC14 атом серы поляризован положительно. При взаимодействии с водой в результате ряда последовательных стадий: гидролиза, образования аквакомплекса, отщепления молекул воды— образуется сернистая кислота. Формально аквакомплекс
Сера, селен, теллур
521
четырехвалентной серы можно рассматривать как катионную кислоту (разд. 33.4.2.2), дающую сульфит-ион:
6Н2О
[S(H2O)4]4+ -> SO32- + 6Н3О+ + Н2О
или
4Н2О
[S(H2O)4]*+ -> O=S(OH)2 4- 4 Н3О+ + Н2о
Кислород взаимодействует с серой с образованием SO2. Триоксид серы (SO3) в заметных количествах не образуется, так как равновесие
SO2 ‘/2О2 <—SO3
в широком интервале температур смещено в сторону исходных продуктов*. Молекула SO2 нелинейна; она может выступать как кислота Льюиса (разд. 33.4.3.3) или как основание, быть окислителем или восстановителем:
основание восстановление Льюиса,
:O=S
плота' окисление
Льюиса,
В качестве основания SO2 взаимодействует с BF3, давая O2S : BF3. Как кислота SO2 выступает в реакциях с водой и гид-роксид-ионом.
Халькогениты и соответствующие им кислоты — довольно сильные восстановители. При взаимодействии со слабыми окислителями сульфиты переходят в дитионаты, а с сильными — в сульфаты:
2SO32- --> S2O,2-+2e-
SO32- + 3 Н2О-► SO42- + 2 Н3О++ 2е-
Так, в кислых растворах MnO2, IO3~, Ag+ и Fe3+ окисляют сульфиты и дитионаты.
Халькогениты проявляют также окислительные свойства. Например, сероводород окисляется всеми тремя халькогенита-ми до серы. При окислении сульфидов путем несимметричного
* При температурах ниже 500 °C равновесие этой реакции смещено в сторону образования SO3, но вследствие кинетических затруднений реакция идет лишь в присутствии катализатора (Pt) и при повышенных температурах JSP-, с. 522). [Щукарев С. А. Неорганическая химия.—М.: Высшая школа, ‘974. Т. 2, с. 215]. — Прим, перев.
522
Неорганическая химия
синпропорционирования и последующей конденсации образуются политионовые кислоты.
Селениты в присутствии конц. НС1 и теллуриты в среде разб. НС1 восстанавливаются при действии SO2.
Как и для соединений других элементов, находящихся в состоянии окисления, промежуточном между высшим и низшим (см. также кислородные соединения галогенов), для соединений селена(IV) и теллура(IV) можно ожидать протекания реакций диспропорционирования. Такая реакция для сульфит-иона сопровождается окислением до устойчивого оксокомплекса — сульфат-иона и восстановлением до сульфид-иона с законченной октетной электронной конфигурацией атома серы. Диспропорционирование катализируется платиновой чернью.
Реакции разложения твердых сульфитов тяжелых металлов протекают иначе. При термическом разложении образуются либо оксид металла и диоксид серы, либо, если оксид металла термически нестабилен, металл, кислород и диоксид серы.
Селеновая и теллуровая кислоты являются окислителями уже в разбавленных растворах, тогда как разбавленная серная кислота не проявляет окислительных свойств. Концентрированная H2SO4 окисляет иодиды. Такие металлы, как медь и серебро, восстанавливают ее даже до H2S*.
С окислительным действием концентрированных кислот сходно окислительное действиехалькогенат-ионов (SO42~ SeO42-и ТеО66-) в расплавах. При этом окислительное действие усиливается благодаря высокой концентрации ионов кислорода (часто добавляется Ыа2СОз), так как это способствует координационному насыщению образующихся ионов в высокой степени окисления.
35.6.4.2. Кислородные кислоты халькогенов. Халькогенаты
У оксидов халькогенов наблюдается переход от молекулярных веществ к полимерным ковалентным соединениям. SO3 — легко летуч, но склонен к полимеризации; SeO3 — труднополучаемое полимерное вещество, сильный окислитель («эффект инертной пары»), ТеО3 — полимерное соединение, устойчивое до 700 °C.
Триоксид серы получают каталитическим окислением SO2 кислородом воздуха. Без катализатора окисление идет слишком медленно.
В молекуле SO3 связи сера — кислород имеют повышенную кратность. Поскольку, однако, сера в треугольной координации не может образовать устойчивые двойные связи, молекула БОз довольно реакционноспособна. С водой идет энергичная реак-
* Более характерно восстановление конц. H2SO4 до SO2.— Прим, перев.
Сера, селен, теллур
523
дня; при этом достигается устойчивое состояние благодаря образованию тетраэдрического иона SO42-
|о|
I
jo —s + |о—н—►
JOI н
101
|О — s — О| “ I -
+ 2,Н +
|О|
Водоотнимающее действие серной кислоты связано с образованием устойчивых гидратов, имеющих в своем составе ион гидроксония. Протоны гидратируются водой с выигрышем энергии.
Благодаря таким свойствам серной кислоты, как водоотнимающее действие, способность быть донором протонов и малая летучесть (азеотропная смесь кипит при 338 °C), серная кислота находит широкое применение при проведении многих химических реакций.
Существует класс соединений серы с общей формулой HO3S—Sm.—SO3H, так называемые политионовые или полисуль-фандисульфоновые кислоты*, причем известны члены ряда с msg 10. Некоторые из этих кислот входят в состав жидкости Вакенродера**. Они могут быть получены также при взаимодействии полисульфанов (Нг5п) с SO3:
Ю| |О|
JO—S + Н-?„-Н + S-O| 121
Это кислоты средней силы. Например, диоксид углерода не вытесняет дитионовую кислоту из ее солей. В аналитической и препаративной практике иногда находят применение тетратионовая и тритионовая кислоты (их молекулы содержат четыре и три атома серы соответственно).
Тиосерную кислоту можно назвать также моносульфанмоно-сульфоновой кислотой. Это соответствует одному из способов ее получения:
[О—S + H-S-H H-O-S-S-H |i| 101
Тиосерная кислота — начальный член ряда полисульфанмо-носульфоновых кислот, которые, за исключением тиосерной, одноосновны. Строение тиосульфат-иона подсказывает путь син
* При т=0, кислота называется дитионовой. — Прим, перев.
** Раствор, образующийся при пропускании сероводорода в водный раствор SO2. — Прим, перев.
524
Неорганическая химия
теза тиосульфатов из сульфитов и серы, причем сера, восстанавливаясь до S2~, окисляет центральный атом в ионе SO32~* (опыт 4).
Свободная тиосерная кислота — кислота сильная, но очень неустойчивая. Соответствующее ей основание — хороший комп-лексообразователь, используемый в фотографии. Устойчивые комплексы с тиосульфат-ионом, кроме иона Ag+, дают также ионы Fe3+, Сг3+, Bi3+, Hg2+, Cu2+, Pb2+, Sb3+ и As3+. В сильнокислой среде эти комплексы разрушаются: образуются сера и соответствующий сульфит.
В противоположность сульфитам щелочных металлов тиосульфаты при термическом разложении дают сульфид, сульфат и серу, что отвечает их строению. В кислых растворах H2S2O3 разлагается (со скоростью, сильно зависящей от концентрации) на диоксид серы и серу. В щелочных же растворах идет распад на анионы со степенями окисления серы, уже имевшимися в тиосульфате:
Кислая среда HS2O3" Н3О+ ► SO2f -|- 2Н2О + S|
Щелочная среда S2O32- + 2ОН" ► SO42- S2~ 4- Н2О
Ион тиосульфата проявляет восстановительные свойства:
2 S2O3-2 -> S4Oe"2 + 2е- £° = 0,1 В
Слабые окислители, такие, как иод, Fe3+ и Си2+, окисляют ион ЗгОз2- до тетратионата. Хлор, бром, МпО4~ и СггО72~ окисляют тиосульфат до сульфата.
Благодаря более слабому окислительному действию иод не разрушает анион ЗгОз2-, а дает лишь радикал
|О U-S-А I— —
соединяющийся с другим таким же радикалом с образованием иона тетратионата
Пероксодисерную и пероксомоносерную кислоты можно рас-
* Вопрос о степенях окисления атомов серы в тиосульфате неоднозначен. — Прим, перев.
Сера, селен, теллур
525
сматривать как производные Н2О. Это показывает и и ход гидролиза:
|О|
_ _ I _
Н—О—О—S—О|
|О| |О|
_ I — I _
[O-S-O-O-S-OI
I I “
|О| |О|
н.. ,н н _ _
+ 2 ЧИ х0-0
Растворы довольно сильной пероксодисерной кислоты устойчивы на холоду. При нагревании происходит разложение на серную и пероксомоносерную кислоты, причем последняя лишь одноосновна и в свою очередь способна разлагаться на Н2О2 и сульфат-ион. Интересный путь синтеза мононадсерной кислоты— взаимодействие хлоросерной кислоты с Н2О2. Обе пероксокислоты не образуют труднорастворимых солей. Для них типичны реакции окисления — восстановления.
Пероксокислоты селена и теллура не описаны.
Окислительный потенциал пероксодисульфата довольно высок:
S2O82- + 2е" <=-> 2 SO42~ Е° = 2,06 В
Однако вследствие кинетических затруднений эффективный потенциал составляет ^1,1 В. В присутствии катализатора Ag+ эти затруднения снимаются.
От Н2О2 ион S2Os2~ отличается тем, что он с солями тита-на(1У) не образует окрашенных пероксокомплексов, аоткисло-ты Каро H2SO5 — тем, что лишь медленно окисляет раствор KI до иода.
Рекомендуемые опыты
1. Сублимация селена. Немного (на кончике шпателя) селена осторожно нагревают в тугоплавкой пробирке (селен ядовит!). Вначале появляются красные пары, из которых образуется черный сублимат. Часто иа верхнем крае пробирки образуется белый сублимат. (Какие реакции имели место!)
2. Получение халькогеноводородов. Небольшие кусочки FeS и Ag2Te обрабатывают 5 М. НС1 (несколько капель) (тяга!). Селеноводород получают гидролитическим разложением селенида алюминия водой (осторожно! халькогеиоводороды ядовиты). Испытывают реакцию выделяющихся газов с помощью влажной индикаторной бумаги.
3. Разрыв связей S — S под действием нуклеофилов. На часовом стекле смешивают несколько капель раствора цианида с несколькими каплями раствора полисульфида аммония. Раствор упаривают досуха иа водяной баие. Остаток увлажняют несколькими каплями соляной кислоты и добавляют сильно разбавленный раствор FeCl3. Наблюдается красное окрашивание (уравнение реакции):
NaCN + (NH4)2Sx --> NaSCN + (NH4)2SX_1
526
Неорганическая химия
Сера, селен, теллур
527
4. Тиосульфат натрия. В небольшой колбе со шлифом готовят раствор 8 г NajSOs-SHaO в 50 мл воды. В раствор вносят 1 г тонкоизмельченного порошка серы, смоченного небольшим количеством этанола. Смесь кипятят с обратным холодильником до тех пор, пока почти вся сера не перейдет в раствор- Непрореагировавшую серу отфильтровывают, а раствор осторожно упаривают и оставляют для кристаллизации Na2S2O3-5H2O.
5. Окислительное действие серы. Энергично растирают в спирте немного серы с каплей ртути. Образуется HgS.
В тугоплавкой пробирке нагревают смесь эквивалентных количеств порошков железа или меди с серой. Реакция идет с воспламенением смеси. Таким же образом получают белый ZnS.
6. Осаждение ионов металлов в виде сульфидов. В растворы сульфатов меди, цинка и железа (II), подкисленные разб. НС1, в течение 60 с пропускают сероводород. Осаждается лишь CuS. К обоим прозрачным растворам добавляют разб. раствор ацетата натрия (зачем?), что приводит к выпадению ZnS. Значение pH повышают, прибавляя NH3, после чего осаждается чернокоричневый FeS.
В основе этих качественных реакций на ионы металлов лежит сочетание
кислотно-основной реакции и реакции осаждения.
7. Гетерогенная кислотно-основная реакция осаждения сульфидов. В прибор для получения газов (разд. 37.1.5.5) помещают немного Na2S и добавляют 1 мл 5 М. НС1. Выделяющийся газ испытывают свинцовоацетатной бумагой, которая окрашивается в черный цвет (PbS). Немного Na2S помещают на пластинку из серебра. Образуется коричневый Ag2S.
8. Восстановительное действие сульфид-иона. Несколько миллилитров 0,5 М К2СЮ4 подкисляют 3—4 каплями 2,5 М. HNO3 и затем в раствор пропускают ток газообразного H2S. Наблюдают выделение серы и изменение оранжевой окраски раствора (СггО?2-) в сине-фиолетовую, соответствующую образованию гидратированных ионов Сг(Ш) (уравнение реакции!).
д Сопряженная реакция кислотно-основного взаимодействия и комплексообразования. В слабокислый 0,1 М. раствор соли Sb (III) пропускают сероводород. Оранжево-красный осадок Sb2S3 отделяют, добавляют к нему несколько мияилитров 5 М NH3 и снова пропускают H2S. При этом осадок SbzS3 растворяется. При подкислении раствора 5 М. соляной кислотой осадок
сульфида выпадает снова.
10. Получение полисульфидов. Немного серы смачивают этиловым спиртом и в закрытом сосуде продолжительное время встряхивают с 10%-иым раствором Na2S. После фильтрования подкисляют раствор соляной кислотой. Образуется H2S и выделяется сера.
И. Реакция гидролиза в сочетании с диспропорционированием. Немного S2C12 вносят в воду. Выделяющиеся SO2 и H2S узнают по запаху и с помощью свинцовоацетатной бумаги. Наличие хлорид-ионов доказывают, добавляя к раствору AgNO3 и обрабатывая осадок (Ag2S и AgCl) коиц. раствором NHs- Аммиачный раствор испытывают на присутствие ионов С1~ (уравнения реакции).
12 Халькогениты как восстановители. Восстановительные свойства ти-пичинЫ лишь для сульфитов; в случае селенитов и теллуритов они выражены
слабо.
Проводят реакции подкисленного 5 М уксусной кислотой раствора сульфита натрия с 2,5 М. раствором НгО2, 0,02 М. раствором КМпО4 и 0,2 М. раствором КгСгО4 (уравнения реакций!).
Образование сульфата доказывают его осаждением ионами Ва2+.
13. Окислительное действие халькогенитов. Свойства селенитов и теллуритов как окислителей выражено гораздо сильнее, чем у сульфитов, причем окислительное действие зависит от pH (рис. В.29).
а) К 0,5 М. раствору SnCl2 в 12 М НС1, находящемуся в трех пробирках, приливают 0,2 М растворы, содержащие SO32~, SeO32~ и ТеО32-. Во всех трех пробирках наблюдается выпадение осадков, которые представляют собой S11S2, Se и Те соответственно.
б) 0,5 М раствор SnCl2 обрабатывают 5 М. NaOH до растворения первоначально выпавшего осадка Sn(OH)2. С этим раствором, содержащим Sn(OH)42~, повторяют опыты, описанные выше. Восстанавливается только раствор, содержащий ионы ТеО32-.
в) Оставшиеся прозрачными растворы из предыдущего опыта, содержащие ионы SO32- и SeO32-, подкисляют 5 М уксусной кислотой. Произойдет
ис. В.29. Зависимость окислительно-восстановительного потенциала сульфита
от pH.
восстановление лишь SeO32~ до красного селена (напишите все уравнения реакций!).
14. Качественные реакции на Se и Те. Очень небольшие количества Se и Те смачивают 10 каплями конц. H2SO4 и осторожно нагревают. В пробирке с селеном появляется зеленое окрашивание; теллур растворяется в конц. H2SO4, давая красное окрашивание. При разбавлении полученных растворов водой снова выпадают красный селен и черный теллур.
15. Реакции диспропорционирования и разложения. В двух тугоплавких пробирках нагревают безводные соли K2SO3 и A12(SO3)3. Газообразный SO2, выделяющийся при разложении сульфита алюминия, определяют по запаху. Часть темно-окрашенного расплава, образовавшегося при иагреваиии K2SO3, наносят на полированную серебряную пластинку, увлажняют водой и наблюдают почернение (Ag2S). Другую часть расплава обрабатывают соляной кислотой (запах H2S), отфильтровывают и в фильтрате осаждают сульфат раствором хлорида бария.
16. Различная сила окислителей. В три пробирки помещают солянокислый раствор хлорида олова (II) и при нагревании добавляют разбавленные растворы H2SO4, H2SeO4 и H4TeOe. H2SeO4 и Н4ТеО4 окисляют [SnCl4]2- до [SnCle]2-, причем они сами восстанавливаются до красного селена и черного теллура. Разбавленная H2SO4 ие проявляет окислительных свойств.
17. Водоотнимающее действие H2SO4. Смешивают (под тягой!) 2—3 мл конц. муравьиной кислоты с коиц. H2SO4. Моноксид углерода (осторожно! Угарный газ!), образующийся сразу или при легком нагревании, горит яркосиним пламенем
HCOOH-|-H2SO4 -----> CO + H3O++HSO4-
18. Реакции осаждения, комплексообразования и разложения тиосульфа-?ов. Смешивают равные объемы 0,5 М AgNO3 и 1 М Na2S2O3. Выпадает белый осадок, растворяющийся в присутствии избытка ионов S2O32-.
528
Неорганическая химия
Немного труднорастворимого AgBr обрабатывают раствором Ка25гО3; наблюдают растворение AgBr.
К другой порции раствора AgNOg добавляют такое количество раствора Na2S2O3, чтобы выпал белый осадок. Спустя значительный промежуток времени, можно наблюдать изменение вида осадка в результате его разложения, которое может также происходить и при нагревании раствора (уравнения реакций!).
19. Термическое разложение тиосульфата. Тиосульфат натрия, содержащий кристаллизационную воду (опыт 4), нагревают в тугоплавкой пробирке. В холодной зоне осаждается сера; выделение H2S обнаруживается по запаху^ Остаток от прокаливания растворяют в разб. НС1 и осаждают BaSO4 раствором ВаС12.
20. Зависимость скорости распада тиосульфата от его концентрации. К сильно разбавленному раствору тиосульфата натрия приливают немного разбавленной H2SO4 н раствор перемешивают. Прозрачный вначале раствор постепенно мутнеет за счет выделения мельчайших частиц серы; при этом из раствора выделяется сернистый газ (запах). Разложение наступает сразу, если опыт проводить с концентрированным раствором Na2S2O3.
21. Восстановительные свойства тиосульфата, образование различных продуктов окисления. Немного разбавленного раствора Na2S2O3 встряхивают с раствором иода. Окраска иода быстро исчезает. Раствор не дает реакций сульфат-иона. К разбавленному раствору Na2S2O3 приливают бромную воду до тех пор, пока выпавшая вначале сера не окислится далее:
Na2S2O3 Вг2 Н2О --------► 2 NaBr H2SO4 -|- S
S + 3 Вг2 + 4Н2О ---- 6 НВг + H2SO4
22. Окислительно-восстановительные реакции, сопровождаемые реакциями осаждения.
Свежеприготовленный раствор (NH4)2S2O3 подкисляют НС1 и добавляют к нему раствор ВаС12. Осадка не образуется. При кипячении раствора выпадает BaSO4.
К другой части раствора (NH4)2S20s, подкисленной конц. HNO3 (1), добавляют раствор AgNO3. Осаждения не наблюдается. После непродолжительного кипячения выпадает коричневый AgO.
23. Различия окислительно-восстановительных реакций НАО^, HqSOs и Н2О2. Подкисленный раствор (NH4)2SiO3 смешивают с раствором KI. Выделение иода происходит медленно и постепенно.
Кислоту Каро получают следующим образом: 0,5 г K2S2Os растирают с 2 мл конц. H2SO4 и оставляют на 15 мин. Затем смесь выливают на растолченный лед и разбавляют 20 мл воды. К части полученного таким образом раствора приливают немного раствора KI. Сразу наблюдается выделение иода.
В пробирку наливают разбавленный раствор Н2О2, а в другую пробирку-раствор H2SO5. В обе пробирки добавляют 10%-иый раствор КВг. Взаимодействие КВг с кислотой Каро приводит к выделению свободного брома.
Вопросы и задания
1. На основании рассмотрения равновесия протолиза H2S обоснуйте возможность осаждения сульфидов при различных значениях pH.
2. Напишите уравнения идущей в присутствии воздуха реакции Na2S с серебром. Обоснуйте возможность ее протекания, используя уравнение Нерн-ста и величину произведения растворимости Ag2S.
3. Объясните причину постепенного пожелтения растворов сульфид0 щелочных металлов на воздухе.
4. Сведите в таблицу свойства (включая реакции с водой) всех извест ных галогенидов халькогенов.
Азот, фосфор
529
5. На основании электронного строения атомов обоснуйте возможность осуществления рл— йл-связывания в SO2 и ТеО2.
6. Как меняются свойства оксидов халькогенов в степени окисления +6? Почему изменения менее отчетливо выражены, чем в соединениях со степенью окисления +4?
7. Каково пространственное строение молекулы SO2, ионов SO32- и SO42-?
8. С привлечением теории жестких и мягких кислот и оснований объясните, почему соединения селена и теллура в низших степенях окисления действуют как яды по отношению к поверхности металлов — катализаторов.
35.7. Азот, фосфор
В основном состоянии атомы этих элементов имеют «^-электронную конфигурацию. В соответствии с правилом Хунда три р-орбитали заняты тремя неспаренными электронами, а s-орбиталь— парой электронов. Атомы могут путем комбинации с атомами того же или иного вида образовать три ковалентные связи: N=N, РС13, AsH3.
Максимальное число связей атома азота в его соединениях
равно четырем, как и для других элементов второго периода. Это реализуется, например, в катионе NH4+, где осуществляется «р3-гибридизация.
Более тяжелые элементы пятой группы могут благодаря наличию свободных d-орбиталей образовывать пять и шесть связей. Состояние 8р3с?-гибридизации соответствует тригонально-бипирамидальному окружению центрального атома (PF5, SbCU), а состояние sp^d2-гибридизации — октаэдрическому окружению (PF6~, SbCls-).
Помимо о-связей, элементы главной подгруппы пятой группы способны к образованию л,-связей. В случае азота — это главным образом рл—рл-связи. Особые свойства молекулы азота частично обусловлены ротационной симметрией обеих л-свя-зей. У других элементов, особенно у фосфора, возможно образование рп—dn-связей. Это обычно имеет место в соединениях со связями Р—F и Р—О.
Химическое поведение элементов характеризуется наличием большого числа степеней окисления (от —3 до +5). Однако в большинстве случаев эти степени окисления не соответствуют величине заряда ионов, так как при образовании ковалентных связей в значительной степени происходит выравнивание зарядов на атомах.
В соответствии с их электронной конфигурацией атомы рассматриваемых элементов могут давать ионы X3-, которые присутствуют в структурах твердых нитридов, фосфидов и т. д. Вследствие больших размеров поляризуемость этих ионов высо-Ка- С другой стороны, сами эти ионы благодаря их высокому заряду должны оказывать поляризующее действие на катионы больших размеров. Поэтому в этих соединениях можно наблю-
34—2027
530
Неорганическая химия
Таблица В.ЗО. Полиморфные модификации фосфора
Свойства Белый фосфор Красный фосфор Черный фосфор
Температура плавления, °C Температура кипения, °C Плотность, г/см3 Температура воспламенения, °C Токсичность 44 287 620 587
1,83 60 2,36 от 300 до 400 2,67
Ядовит Не ядовит Не ядовит
Самоокисление На воздухе в темноте светится Не светится Не светится
Растворимость Растворим в CS2, CCU Не растворим В CS2, СС14 Не растворим в CSs
дать переход от ионного типа связи к атомному и металлическому типу.
В ряду оснований Льюиса типа ХН3 (X = N, Р, As, Sb, Bi) при увеличении размера атома X присутствие свободной электронной пары сказывается в меньшей степени и тем самым уменьшается сила оснований. Причиной этого можно считать наличие у атомов более тяжелых элементов d-орбиталей, что препятствует осуществлению зр3-гибридизации. В соответствии с этим величина угла между связями в группе НХН уменьшается от 107 (NH3) до 93° (РН3).
При увеличении атомного радиуса и одновременно с возрастанием числа участвующих в связях орбиталей закономерно меняются свойства простых веществ. Ясно прослеживается переход от неполярных молекулярных веществ (N2 и Рбел) к слоистым структурам (Рчери, As) и к металлическим веществам (Bi).
Химия гидроксо- и оксокомплексов сурьмы и висмута характеризуется сильной склонностью к конденсации, приводящей к образованию сложных каркасных структур. Это свойство начинает проявляться уже у кислородных соединений фосфора.
35.7.1. Неполярные молекулярный вещества
Азот образует двухатомные молекулы с кратной и очень прочной связью NssN и с очень коротким расстоянием между атомами (0,109 нм). Белый фосфор построен из тетраэдрических молекул (Р4), в которых отсутствуют связи повышенной кратности за счет рп—рл-связывания. Фосфор имеет три основные полиморфные модификации. Сведения о структурах и свойствах этих модификаций фосфора приведены в табл. В.ЗО. Белый фосфор переходит в красный при 400°C. У мышьяка и сурьмы из-j вестны также металлоподобные модификации.
Азот, фосфор
531
Таблица В.31. Энергии связей углерод-—углерод и азот — азот
Энергия связи, кДж/моль Энергия связи, кДж/моль Д(С-C)-(N-N)
С-С Д(С=С) - (С-с, 347,5 268,0 N—N A(N=N) — (N—N) 159,1 259,6 188,4
с=с С) (С С) 615,5 196,8 N=N A(№N) — (N=N) 418,7 (196,8) 196,8
СэС 812,3 NsN (615,5 по оценке) 946,2 найдено (196,8) по аналогии
В большинстве случаев для вступления в реакцию простых веществ необходимо их расщепление на атомы.
Очень высокая энергия связи в молекуле азота делает его чрезвычайно инертным. Сравнение энергий связей между атомами углерода (в полимерных соединениях) и между атомами азота (табл. В.31) показывает, что оцениваемая по аналогии величина энергии тройной связи в N2 лежит значительно ниже, чем истинное значение 946 кДж/моль. Большинство простых соединений азота эндотермично и может разлагаться с образованием устойчивых молекул N2 (см. опыт 2).
Тройная связь в N2 обладает повышенной устойчивостью также к реакциям присоединения; это связано с неполярностью молекулы N2 и ее высоким потенциалом ионизации. Напротив, группы —С=С— и —C^N проявляют л-электронодонорные свойства. В последние годы было показано, что и молекула N2 может выступать в качестве л-донорного лиганда, образуя комплексы при нормальных условиях. Были получены комплексы типа [Ru(NH3)5N2]Х2, (PR3)2IrN2Cl, (РКз)2СоЫ2 и др.
На рис. В.ЗО схематически показан механизм образования связи атома металла с молекулой азота. Он предусматривает активирование молекулы N2 и затем ее связывание в мягких условиях. Аналогичным образом объясняется действие соединений d-элементов как катализаторов в процессах синтеза аммиака или при связывании азота микроорганизмами. Сначала происходит образование донорно-акцепторной связи о-типа. Это требует переноса электронной плотности за счет перекрывания занятой орбитали атома азота (свободная пара электронов) с незанятой орбиталью атома металла. Далее происходит перекрывание заполненных dn- или dn — рл-орбиталей атома метал-34*
532
Неорганическая химия
Азот, фосфор
533
ла со свободными разрыхляющими р*л(л*)-орбиталями молекулы азота. Эта дативная связь обусловливает устойчивость комплекса (связи металл — азот). Одновременно за счет ослабления связи NsN происходит активация молекулы N2.
При комнатной температуре с азотом взаимодействует также литий.
донорно-акцепторная
-связь
М
:N = N:
электронная пара дативная связь участвующая дп—зт,* , в о-связи
(Nj)
Рис. В.ЗО. Связь металл—N2 в комплексах молекулярного азота.
При повышенных температурах, особенно в присутствии катализаторов, азот становится более реакционноспособным. Типичные реакции:
N2+3H2 ------> 2NHS
N2 + О2 -----> 2 NO*
N2-f-3Mg-----> Mg3N2
N2 -f- CaC2 ->• CaCN2 C
Присутствие молекулы P4 в белом фосфоре проявляется в его повышенной химической активности. $р3-гибридные орбитали, обычно располагающиеся под углом друг к другу от 90 до 120°, в тетраэдре Р4 сильно деформированы (угол 60°). В то же время осуществление зрс?2-гибридизации, которой и соответствует угол 60°, требует большой энергии возбуждения. Отсюда понятна высокая реакционная способность белого фосфора. Как видно из табл. В.ЗО, полиморфные модификации фосфора обнаруживают существенные различия в своем химическом поведении.
Здесь следует еще раз обратить внимание на различий свойств атомов и молекул. Электроотрицательность атомов азо
* Равновесие этой реакции вплоть до температур 3000 К сильно смещено влево [Коттон Ф., Уилкинсон Дж. Современная неорганическая химия. — №•• Мир, 1969, т. 2, с. 176]. — Прим, перев.
та больше, чем атомов фосфора, и они более активны химически. Напротив, в двухатомных молекулах N2 связи прочнее, так что молекулы Р4 оказываются химически активнее.
35.7.2. Различные типы соединений азота и фосфора
При прямом взаимодействии азота и фосфора со многими металлами и неметаллами образуются нитриды и фосфиды. В зависимости от полярности связи Е—X можно наблюдать переходы от связей ионного типа к ковалентным или к металлическому типу связи (X = N, Р). При этом происходят переходы между тремя основными типами соединений; меняются также химические свойства соединений.
Ионные соединения. ЫазХ, СазХ2 и ЬаХз, так же как и нитриды и фосфиды некоторых других сильно электроположительных металлов, построены по типу солей. В них доказано присутствие ионов X3-. Типичная реакция этих соединений — полный гидролиз до NH3 и РН3 и соответствующего гидроксида металла.
Молекулярные вещества. Атомы азота и фосфора образуют ковалентные связи как в соединениях между собой, так и со многими другими элементами. Их соединения с S, Se, Те представляют собой летучие молекулярные вещества. Примерами полимерных веществ могут служить BN, AIN, P3N5 и др. Кристаллы BN и A1N можно рассматривать как огромные трехмерные молекулы (аналогия с алмазом).
Металлические вещества, нестехиометрические соединения. Переходные металлы склонны к образованию соединений включения, в которых атомы X занимают пустоты в плотнейшей упаковке металла. Часто эти соединения имеют нестехиометрический состав. Их отличительные свойства — металлический блеск, высокая твердость и хорошая электропроводность, что связано с сохранением зонной структуры металла. У некоторых нитридов обнаружена даже сверхпроводимость. Сами металлы и их соединения включения (а также карбиды и бориды) по величине проводимости можно расположить в следующий ряд: металл карбиды > фосфиды >нитриды> бориды.
Нитриды титана, циркония, тантала и ванадия плавятся при 3220, 3255, 3360 и 2570 °C соответственно, а их твердость составляет 8—9 по шкале Мооса. Как нитриды, так и фосфиды — весьма инертные химические вещества.
Фосфиды обычно характеризуются повышенной термической Устойчивостью, так как для большинства фосфидов металлов (за исключением благородных металлов) равновесие реакций образования при температурах, используемых при синтезе, сменены в сторону фосфида.
534
Неорганическая химия
35.7.2.1. Гидриды азота и фосфора и их производные
Аммиак и фосфин можно получить при гидролизе ионных нитридов и фосфидов. Есть и другие способы получения. Эти соединения— при обычных температурах газы. В жидком состоянии аммиак ассоциирован за счет возникновения водородных мостиковых связей (сходство с водой). Вследствие высокого значения диэлектрической проницаемости (при — 34 °C ег=22) он хороший растворитель. В жидком аммиаке устанавливается следующее равновесие аутопротолиза:
2 NH3 ч=+ NH4+ + NH2- Х223 ~ IO-3»
NH3 — чрезвычайно жесткое основание.
Сильноядовитый монофосфин (фосфин) в жидком состоянии не ассоциирован и не подвергается аутопротолизу.
Многие электроположительные металлы способны растворяться в жидком аммиаке. Образующиеся синие растворы проявляют сильные восстановительные свойства вследствие присутствия сольватированных электронов (поляронов).
Ионы X3- — очень сильные, но одновременно и мягкие основания. Растворяясь в воде, они подвергаются ее нивелирующему действию, в результате чего образуются ионы ОН- (опыт 6):
X3- + 3 Н2О ->- ХН3 + 3 он-
Растворимость в воде NH3 гораздо больше, чем для РН3, так как NH3 способен к образованию с молекулами воды водородных мостиковых связей. Растворы в воде имеют не кислую реакцию (за счет гипотетического отщепления протона), а основную благодаря протолитическому взаимодействию:
ХН3 + Н2О ХН4+ + ОН"
Нейтральное основание NH3 характеризуется следующими значениями констант;
рКа = 9,25 NH4+ NH3 + Н+ рКь = 4,75
Раствор РН3 в воде имеет практически нейтральную реакцию:
рКа ~ 29 РН4+ ---- РН3 + Н+ рКь ~ 26
Молекулярные основания ХН3 взаимодействуют с молекулярными кислотами, давая аммонийные и фосфониевые соли:
NH3 + НС1 ---»- NH4CI
PHg + HI > РН41
Устойчивость тетраэдрических ионов зависит от донорных свойств свободной электронной пары у атома X и от вида аниона. Соли аммония устойчивее солей фосфония. В свою очередь РН41 устойчивее, чем PH4F (не существует). Устойчивость воз
Азот, фосфор
535
растает при увеличении сродства к протону исходных гидридов й при усилении кислотных свойств участвующего в реакции га-догеноводорода.
Многие соли аммония и фосфония сублимируют с одновременной диссоциацией:
NH4C1 ч. NH3 + НС1 АН = 177,1 кДж/моль
NH4NO3 4 >• NH3+ HNO3 ДЯ = 171,2 кДж/моль
Растворы солей аммония и фосфония в воде имеют слабокислую реакцию, так как эти соли полностью диссоциируют и катионная кислота ХН4+ подвергается протолизу водой
ХН4У ХН4+4-У" К. ~ 8 (Y—анион сильной кислоты)
|Н2О
ХН4+ Н3О++ ХН3
Известны производные аммиака, дающие в водных растворах в отличие от аммонийных солей, сильнощелочную реакцию. Эти соединения имеют ионное строение (соли) и включают анионы NH2~; последние в воде дают щелочную реакцию вследствие быстро идущей протолитической реакции с образованием NH3 (разд. 33.4.2.5). Поэтому такие солеобразные амиды можно получать лишь в неводных средах (например, в жидком NH3). При этом окислительная способность молекулы NH3 достаточна для ее реакции с металлическим натрием, приводящей к образованию амида натрия и выделению водорода:
NH3 + Na -->- NaNH2 + V2H2
Ион NH2~ почти такое же сильное основание, как ион О2-, т. е. его основные свойства выражены гораздо сильнее, чем У иона ОН-, что видно из сравнения значений ^К,а и рД& (табл. В.5).
Некоторое формальное сопоставление свойств соединений элементов второго периода можно провести с помощью следующих реакций:
2 СН4 --► СН3—СН3 + Н2
2NH3 ---► NH2—NH2 + H2
2 ОН2 --► НО—ОН-|-Н2
(2 FH --► F—F + Н2)
Гидразин можно рассматривать как гомолог аммиака. Свойства соответствующих водородных соединений меняются в зависимости от строения атомов элементов. В гидразине, пероксиде водорода и фторе порядок связи и ее энергия существенно ни-Же, чем во многих других соединениях (табл. В.32). Это связа-Во с сильным ослаблением связи вследствие отталкивания электронных пар. Если эти электронные пары оказываются во-
536
Неорганическая химия
Таблица В.32. Свойства аналогов гидразина (второй период периодической системы)
Характеристика соединения НзС-СН3 hsn-nh2 но-он F-F
Порядок связи 1,00 0,68 0,55 0,49
Энергия связи, кДж 347,5 159,1 139,0 153,2
Окислительно-восстановительные свойства Не окислитель (не восстановитель) Восстановитель Окислитель или восстановитель Окислитель
влеченными в химическую связь, порядок связи и ее энерпи возрастают:
[H2N—NH3]+ Д'= 0,83
[HSN—NHS]2+ N=l,01
В сульфате гидразиния (2 + ) связь не ослаблена.
Еще больше уменьшается порядок связи, если ранее связанная пара электронов становится свободной; например, в результате аутопротолиза Н2О2 порядок связи кислород-кислород по нижается, ^=0,49:
Н-О-О-Н—- [н-О-О|]~+Н +
Как следует из табл. В.32, восстановительные свойстве соединений ослабляются с убыванием порядка связи. Так, гидразин в присутствии ионов Мо5+ восстанавливает пероксид водорода до воды, окисляясь при этом до N2. Окислительно-восстановительные свойства гидразина сильно зависят от pH:
N2H5+ 5Н++ N2 + 4е_ Е»=0,23В
N2H4 + 4 ОН" N2 + 4 Н2О + 4е" Е° = 1,16 В
При уменьшении порядка связи усиливаются кислотные свойства аналогичных соединений. Присутствие свободной электронной пары, играющей определяющую роль в химии азот; существенно также и в случае гидразина. В водных раствора' гидразин, так же как NH3, дает щелочную реакцию; с сильными кислотами он дает «ониевые» соединения. В рамках теорш жестких и мягких кислот и оснований гидразин — жесткое о< нование.
Р2Н4, образующийся наряду с фосфином, на воздухе може1 самовоспламениться. От N2H4 дифосфин отличается отсутстви
Азот, фосфор
537
еМ основных свойств. Так же как и гидразин, дифосфин — восстановитель.
Соли гидразиния — это катионные кислоты:
[H3N—NH3]2+ + Н2О ?=* [H2N-NH3]+ + Н3О+ рК = —0,3
[H2N-NH3]+ + H2o;^=t H2N-NH2 + Н3О+ рХ = 7,9
Если в предыдущих главах обсуждались примеры анионных амфолитов, то ион [H2N—NH3] +— пример катионного амфолита.
Еще одним производным аммиака можно считать гидроксиламин H2N—ОН. Его окислительно-восстановительные свойства также зависят от pH, причем при его окислении не всегда получается азот; могут образовываться также N2O и HNO2:
2 [NH3OH]+ =₽=* 6 Н+ + N2O-f + Н2О + 4е“ Е° = —0,05 В
2NH2OH ?==i: N2OH-H2O + 4H+H-4e- Е«=—1,05В
В уксуснокислом растворе происходит окисление гидроксиламина иодом до азотистой кислоты, образование которой можно доказать, используя реакции с сульфаниловой кислотой и а-нафтиламином:
NH2OH -j- 2I2 + Н2О -► NO2" + 41- + 5Н+
При работе на холоду присутствие гидразина не мешает ходу реакции.
В воде гидроксиламин реагирует как основание. Он образует «ониевые» соли, проявляющие свойства катионных кислот. Действуя на эти соли более сильными основаниями, можно вытеснить более слабое основание — гидроксиламин: [HONH3]+—>NH2OH+H+ (рК=6,0).
35.7.2.2. Соединения с галогенами
Состав галогенидов азота и фосфора в основном соответствует формулам ЕХ3 и ЕХ3, где E=N, Р и X —галоген. Существуют также смешанные (по галогену) три- и пентагалогениды. Из-за наличия свободной электронной пары молекулы тригалогенидов имеют пирамидальное строение. Соединения состава ЕХ3 в газовой фазе имеют тригонально-бипирамидальное строение.
Пентагалогениды азота не известны; в этих соединениях атом азота должен был бы образовывать пять связей. Энергия, Необходимая для распаривания 2х2-электронной пары и перевода одного электрона на орбиталь с более высоким главным квантовым числом (п=3), слишком высока и не может быть Получена за счет прохождения химической реакции. Поэтому Для азота максимальное число ковалентных связей — четыре.
Результаты последних исследований показывают, что, по-ви
538
Неорганическая химия
димому, распаривание «2-электронной пары атома азота все-та-ки происходит при взаимодействии NH3, AsF5 и F2. При этом образуется катион тетрафторазота (V) NF4+, обнаруженный в продуктах реакции (NF4+AsF6~).
Реакции гидролиза тригалогенидов азота дают представление об обращении распределения парциальных зарядов в связях N—Hal. Гидролиз газообразного весьма инертного неполярного молекулярного вещества NF3 идет в жестких условиях до HNO2 и HF:
«+«—
2NF3 + 4H2O ---» HF + 2HNO2
Соединение C13N, которое вследствие обращения в полярности связи правильнее было бы называть нитридом хлора, а не трихлоридом азота, представляет собой светло-желтое масло. C13N медленно гидролизуется уже при комнатной температуре:
б-t- в—
C13N + Н2О --> NH3 + 3 нею
Соединения C13N (А//°ОбР = +232 кДж/моль) и I3N эндотер-мичны и поэтому неустойчивы. I3N даже при комнатной температуре разлагается со взрывом.
В соответствии с полярностью связей в тригалогенидах фосфора они гидролизуются с образованием кислородных соединений фосфора и соответствующих галогеиоводородов. Пентагалогениды дают при гидролизе фосфорилгалогениды. Экзотермич-ные реакции гидролиза РВг3 и РС13 дают фосфоновую кислоту, а в определенных условиях и другие кислоты фосфора в низших степенях окисления. Тригалогениды фосфора используются как апротонные растворители с хорошей сольватирующей способностью.
NF3, PF3 и РС13 не способны присоединять галогенид-ионы и образовывать комплексные анионы. В то же время PF? и РС13, подобно СО, могут выступать как лиганды в комплексах с переходными металлами. Сначала за счет их присоединения в функции сильного основания Льюиса (свободная электронная пара) образуется о-связь, затем благодаря наличию у них свободных dn-орбиталей — дативная связь с участием d-электронов атома металла.
PF3 и РС15 ведут себя как очень сильные кислоты Льюиса. PF3 присоединяет ион F- и образует устойчивый комплексный анион PFe~. Это происходит благодаря тому, что электроотрицательный фтор делает возможным использование d22 и dxz~^ орбиталей центрального атома в виде гибридных d2sp3-op6HTa-лей (разд. 35.6.3.1). Тем самым одна из орбиталей оказывается незанятой, что приводит к проявлению кислотных свойств и° Льюису.
Азот, фосфор
539
У некоторых галогенидов главной подгруппы пятой группы (в том числе у смешанных, например, PCUF) могут существовать «связевые» изомеры (разд. 33 2.3):
твердое состояние, полярный растворитель
(pci5)2 . - •: —рс14++рс1,-
газообразное состояние
неполярный растворитель
35.7.3. Реакции кислородных соединений азота и фосфора
Известны следующие оксиды азота и фосфора: N2O, NO, N2O3, NO2, N2O4, N2O5(NO3, ЫгОб), Р4О10, Р4О6 и РО2. Некоторые из оксидов азота (полярные молекулярные вещества) мономерны,
другие представляют собой димерные молекулы. Оксиды фосфора даже в газовой фазе по крайней мере димерны, но, как правило, образуют более крупные агрегаты. Свойство образовывать полимерные оксиды возрастает с увеличением разности электроотрицательностей элементов (разд.
35.2.1).
При растворении в воде оксиды азота и фосфора дают кислую реакцию*. В пределах всей подгруппы кислотные свойства проявляют оксиды N(III), Р(Ш), As(III); Sb2O3 уже основание.
Рис. В 31. Структура Р4О10.
амфотерен, а В120з—
Устойчивость высших степеней окисления убывает с ростом порядкового номера элемента.
Молекулы NO и NO2 имеют нечетное число электронов, т. е. они содержат неспаренный электрон. Однако NO не ассоциирован и бесцветен. Напротив, NO2 обнаруживает типичное для этою вида молекул свойство — ассоциацию, степень которой зависит от температуры
2NO2 gp->-- N2O4
бурый
пара- бесцветный
магнитные диамагнитные
свойства свойства
Р2О5 существует в нескольких полиморфных модификациях. В них присутствуют группы Р4О]0 (рис. В.31). Атомы фосфора находятся в вершинах тетраэдра, шесть атомов кислорода — Ча его ребрах, а остальные четыре атома кислорода — на продолжении осей третьего порядка тетраэдра. Связи Р—Окоицевой Имеют характер двойных благодаря рл.— с?я-связыванию.
* Некоторые оксиды, например N2O, NO, с водой не реагируют — "рич перев
540
Неорганическая химия
Помимо реакций ассоциации — диссоциации надо также рассмотреть диссоциацию N2O4 на ионы. N2O4 — хороший ионизиру. ющий растворитель-
N2O4 (жидк.) ---* NO+ + NO3"
нитрозил-катион
Оксиды азота в водных растворах — окислители, сравнимые с бромом:
N2O4 + 2 Н+ + 2е- -» 2 HNO2 Е° = +1,07 В
Р4Ою — прекрасный осушитель для веществ, содержащих воду. Он отнимает воду даже от HNO3 с образованием N^j. Взаимодействие Р4Ою с водой приводит к образованию фосфорных кислот различного состава. Р4Ою взаимодействует не только с водой, а также со спиртами и многими другими веществами. Вначале образуется «активированный комплекс», затем следует расщепление связей Р—О. ,
Аналогичным образом идут реакции P4Si0, а также взаимоде] ствие Р4 со щелочами до гипофосфита и РН3.
35.7.3 1. Оксокислоты и оксоанионы
Окислительно-восстановительные реакции весьма типичны дзя кислородных соединений азота и фосфора. Для определен тя направления протекания окислительно-восстановительных реакций можно использовать значения стандартных электродных потенциалов или свободной энтальпии AG. Наглядное представление о положении равновесия или о направлении хода реакций (без учета кинетических факторов) можно получить аз диаграммы окислительных состояний элемента в водном растворе. Для ее построения необходимо найти степень окислений элемента в простом соединении или ионе (если атомы элемента связаны только с атомами кислорода или водорода), которая численно равна формальному заряду на атоме элемента, если принять для атома кислорода заряд —2, а для атома водоро-
Азот, фосфор
541
Рис. В.32. Диаграмма окислительных состояний для соединений азота в кислом и щелочном растворе.
Да +1, а также вольтэквивалент (ВЭ) соединения или иона,, определяемый как произведение степени окисления на стандартный потенциал сопряженной окислительно-восстановительной, пары. Тогда для реакции
HNO2 + 3 Н+ + Зе- -► V2N2 + 2 Н2О Е° = +1,45 В
вольтэквивалент азотистой кислоты составляет 3-1,45 = 4-4,35 В. При построении диаграммы окислительных состояний (рис. В.32) Для соединений азота в кислом растворе ак+=I значения ВЭ откладываются по оси ординат, а степени окисления — по оси абсцисс. Так называемый градиент — это прямая, соединяющая Две точки диаграммы, каждая из которых соответствует одному Из участников редокс-пары. Величина градиента (тангенса уг-
542
Неорганическая химия
Азот, фосфор
543
ла наклона прямой) численно равна «формальному»* стандарт, ному потенциалу редокс-пары. Чем больше положительное значение градиента, тем сильнее окислительные свойства данной редокс-пары. Чем больше отрицательное значение градиента, тем сильнее выражены свойства редокс-пары как восстановителя.
Для сравнения на диаграммы окислительных состояний часто наносят градиенты редокс-пар Н+/Н2 или О2/Н2О. Можно указывать градиенты других редокс-пар (рис. В.32).
• Как указывалось ранее, равновесие окислительно-восстановительной реакции часто зависит от pH раствора. Построение диаграммы окислительных состояний в щелочном растворе производится таким же образом, но с использованием величин стандартных потенциалов в щелочном растворе. Соответствующие величины вольтэквивалентов в щелочном растворе обозначаются ВЭ(ОН). Для соединений, которые не являются ни кислотой, ни основанием (например, N2, N2O, NO), вольтэквива-ленты в щелочном и кислом растворах равны. Напротив, если соединение в кислом и щелочном растворах присутствует в различных формах (например, азотная кислота HNO3 и ионЫОз-), эти значения вольтэквивалентов могут существенно отличаться.
Между величинами обратимого окислительно-восстановительного потенциала и изменением свободной энтальпии химической реакции имеется прямая взаимосвязь, поэтому величины последних также можно наносить на диаграммы окислительных состояний. Шкала значений iAGO6p 298 находится с правой стороны диаграммы. Значение ВЭ азотной кислоты ( + 6,23 В) для реакции
V2N2 + ЗН2О --► HNO3 + 6/2Н2
соответствует изменению свободной энтальпии +96,6-6,23 = = +601,5 кДж/моль.
Известный метод получения N2O из NH4NO3 (опыт 17) можно пояснить с использованием диаграммы окислительных состояний: редокс-пара HNO3/N2O обладает более сильным окисляющим действием, чем пара N2O/NH4+. Поэтому HNO3 окисляет ионы аммония до N2O (в водном растворе реакция кинетически затруднена), соответственно при нагревании нитрата аммония образуется N2O. Происходит реакция несимметричного синпропорционирования**.
3 4-5 +1
NH4NO3 ---► N2O + 2Н2О
* Данная редокс-пара может оказаться практически нереализуемой. — Прим, перев.
Qntyky&r напомнить, что строение молекулы N2O соответствует различ-ной степени окисления атомов азота. — Прим, перев.
Из рисунка В.32 видно, что (Zn2+/Zn) более сильный восста-ловитель, чем, например, редокс-пары N2/[NH3OH]+, HN3/[NH3OH]+ или N2/NH3*. Нитраты и нитриты количественно восстанавливаются металлическим цинком (а также сплавом Деварда) до аммиака. Эти реакции используются при количественном определении азота по Кьельдалю.
Окислительное действие азотной кислоты сильно зависит от ее концентрации, как это видно из значений окислительно-восстановительных потенциалов для реакции
NO3- + 3Н+ + 2е" HNO2 + Н2О = 0,93 В
Концентрация HNO3, 1 4 12
моль/л
Е°, В 0,93 1,00 1,10
Разбавленная азотная кислота или водные растворы нитратов не могут быть сильными окислителями.
Зависимость окислительного потенциала от концентрации азотной кислоты используется при открытии нитратов с помощью качественной реакции «образования бурого кольца». При осторожном добавлении раствора, содержащего нитрат, к конц. H2SO4 в месте соприкосновения растворов возникает высокая концентрация азотной кислоты. В этом месте происходит окисление ионов Fe2+, причем сама азотная кислота восстанавливается до NO. При этом образуется нитрозокомплекс бурого цвета в виде кольца (разд. 37.2.1.6).
Окислительное действие нитрита на ионы Fe2+ и связывание образующейся молекулы NO в комплексе — пример сочетания двух различных типов реакций в качественной реакции на нит-рит-ион (окислительное действие зависит от pH; опыт 21).
При взаимодействии производных аммиака (CO(NH2)2; NH2SO3H) с азотистой кислотой по реакции симметричного синпропорционирования образуется азот:
—з Ч~з
NH2SO3H+HNO2 ----► H2SO4 + N2 + H2O
Если хотят таким путем удалить нитрит из его смеси с нитратом, следует учесть, что при подкислении раствора образуется азотистая кислота, склонная к диспропорционированию. По этой причине при несоблюдении указанной методики нитрат в аналитической пробе может быть «переоткрыт».
На рис. В.33 для сравнения окислительно-восстановительных свойств соединений азота и фосфора приведены две диаграммы
* На диаграмме (рис. В.32) градиенты для редокс-пары N2/[NH3OH]+ и
НКг/[МН3ОН]+более отрицательны, чем для пары Zn2+/Zn, а положение NH> соответствует положению NH4OH. — Прим, перев.
544
Неорганическая химия
Азот, фосфор
545
Рис. В.ЗЗ. Сравнение диаграмм окислительных состояний для соединен! азота и фосфора.
окислительных состояний для кислых и щелочных растворо > Очевидно, что атом фосфора дает более устойчивые ковален ные связи с атомами кислорода. Объяснить это различие можно наличием доступных для образования связей d-орбиталей у атома фосфора и чрезвычайной устойчивостью молекулы азота.
Более высокая активность фосфора (разд. 35.7.1) по сравнению с азотом проявляется при взаимодействии Р4 со щелочами Фосфор диспропорционирует на фосфин и низшие фосфорные кислоты (разд. 35.7.3). Эти реакции сходны с реакциями гал генов, серы и кремния со щелочами, так как фосфор облада1 сравнительно высоким сродством к кислороду и водороду. Сн
чала образуются фосфин и фосфинат-анионы, а также немного Н2* __2
Р4 + 3 ОН- + 3 Н2О ->- 3 Н2РО2- + РН3) (немного Р2Н4);
2 Р + 2 ОН" + Н2О -- 2 Н2РО2" + H2f
Далее могут идти также реакции диспропорционированияг
3 Н3РО2---<- РН3| + 2 Н3РО3
Н3РО2 + Н2О ---> Н3РО3 + НД (катализатор—металли-
ческий палладий)
4Н3РО3 ---> РН3| + ЗН3РО4
При нагревании последняя реакция идет до конца, так как РН3 труднорастворим в воде и удаляется в виде газа.
Дифосфорновая (гипофосфорная) кислота при нагревании диспропорционирует
Н4Р2О6 + Н2о---► Н3ро4 + Н3РО3
Н3РОз медленно восстанавливает ионы Ag+ и Hg2+ до металла.
В противоположность фосфинатам соли фосфоновой кислоты не способны восстанавливать ионы As3+ и Си2+ до металла. В присутствии более сильных восстановителей (водород в момент выделения) фосфонаты выступают как окислители, причем их восстановление идет до фосфина (опыт 27).
Можно провести реакцию каталитического окисления аммиака до оксидов азота и далее при помощи последовательных реакций диспропорционирования и окисления получить азотную кислоту.
Из нитратов малорастворимыми являются лишь оксинитраты Hg2+ и Bi3+, которые, однако, растворимы в кислых растворах:
BiONO3| 4- 2 Н+ Bi3+ + Н2О + NO3- рК = 26
Остальные нитраты (за исключением нитратов органических катионов) хорошо растворимы в воде.
«Царская водка» — смесь концентрированных НС1 и HN03 в соотношении 3:1 — растворяет даже золото, так как окислительно-восстановительный потенциал такой смеси повышается За счет образования свободного хлора и нитрозилхлорида:
3 НС1 + HNO3 --> 2 Н2О + С12 + NOC1
Расплавленные нитраты — сильные окислители. Ход реакций Разложения нитратов при нагревании зависит от поляризующего действия иона металла. Так, при разложении нитратов щелочных и щелочноземельных металлов образуются соответствующие нитриты и кислород. Нитраты металлов с большим ионным* Потенциалом разлагаются на оксид металла, NO2 и кислород. 35—2027
546
Неорганическая химия
Рекомендуемые опыты
1. Превращение фосфора. Небольшой кусочек белого фосфора помещают в пробирку, отверстие которой закрывают ватным тампоном. При осторож-ном нагревании фосфор сначала плавится, затем испаряется и конденсируется на холодных стенках пробирки в виде красноватого налета. В небольших ко-личествах образуется также Р4О10.
2. Получение при симметричном синпропорционировании. В приборе для получения газов нагревают до 70 °C концентрированный водный раствор нитрита аммония или смесь насыщенных растворов NH4C1 и NaNO2. По схеме NH4NO2—>-N2+2H2O образуется азот. Для поглощения оксидов азота и паров воды выделяющийся газ пропускают через «хромовую смесь», затем его собирают в колбе.
3. Реакции азота. В тугоплавкой пробирке осторожно нагревают 1 г NaN3. Выделяющийся азот собирают в колбе объемом 0,5 л, вытесняя из нее воздух. После этого в колбу помещают очищенный кусочек лития и колбу плотно закрывают. Через несколько дней в колбе образуется продукт, в основном нитрид лития (сохранить!).
4. Различие реакционной способности белого и красного фосфора. Небольшие количества красного и белого фосфора помещают в открытые пробирки и наблюдают в темноте (свечение белого фосфора).
Кусочек белого фосфора и такое же количество красного помещают на концы полоски из жести, которую затем нагревают посередине пламенем горелки.
5. Получение Са3Р2. В тугоплавкой пробирке осторожно нагревают до температуры красного каления 1,2 г кальция и 0,6 г красного фосфора. Чтобы избежать частичного окисления фосфора до оксида, пробирку можно заполнить аргоном. Образующийся первоначально расплав застывает по мере протекания реакции, поскольку фосфид кальция — тугоплавок.
6. Различия в прочности ковалентной связи. В приборах для получения газов проводят взаимодействие Mg3N2 и Са3Р2 с водой (осторожно! тяга! Следует помнить о возможности самовоспламенения фосфина). Выделяющиеся газы испытывают с помощью влажной иидикаториой бумаги. Убеждаются в различной степени проявления основных свойств NH3 и РН3 в воде.
7. NH3 — молекулярное основание. Две пробирки наполняют концентрированными растворами НС1 и NH3. Держа обе пробирки рядом, дают смешиваться парам НС1 и NH3. Поместив на пути движения паров большое часовое стекло, можно получить на нем белый налет хлорида аммония.
8. Вытеснение слабого основания более сильным. В небольшой ступке смешивают немного NH4C1 с NaOH или КОН; смесь растирают пестиком. Проверяют действие выделяющегося газа иа индикаторную бумагу и идентифицируют этот газ по запаху.
К 20 мг РН41 добавляют 5 мл воды. Вследствие незначительной растворимости РН3 в воде, он выделяется в виде газа. В данном случае нейтральный амфолит Н2О выступает как более сильное основание, вытесняя более слабое молекулярное основание из соли (разд. 35.8.4).
9. Термическая диссоциация (гл. 33.8) в качестве транспортной реакции. В большую пробирку помещают немного NH4C1. Внутри пробирки приклеивают иа разной высоте полоски влажной индикаторной бумаги. Держа пробирку в наклонном положении, осторожно нагревают NH4CI. Нижняя полоска индикаторной бумаги показывает кислую реакцию, а верхняя — щелочную-Спустя некоторое время и верхняя полоска указывает на кислую среМ (образование NH4C1). Этот опыт демонстрирует различную скорость дифФУ' зии газов — НС1 и NH3.
10. Окислительное действие аммиака. В железную лодочку помещают 1 г > очищенного от корки металлического натрия. Лодочку быстро вдвигают в трубку из тугоплавкого стекла, через которую пропускают ток газообразного аммиака. После вытеснения воздуха трубку с лодочкой нагревают до температуры 350°C (трубчатая печь). Реакцию можно считать оконченной, когда
Азот, фосфор
547
выходящий газ будет полностью поглощаться водой (осторожно! водород!).
11. NaNH% в качестве основания. В небольшое количество воды вносят немного NaNH2 (полученного в опыте 10). Испытывают действие выделяющегося газа и образовавшегося раствора на индикаторную бумагу.
12. Восстановительные свойства N^Ih, и Н%0. В пробирку наливают 2 мл разб. раствора Н2О2, добавляют 2 капли раствора молибдата и затем 2 мл разб. раствора гидразина. Если выделение газа сразу не происходит, пробирку немного нагревают. Получающийся газ не поддерживает горения (лучинка1}
13. Кислотные свойства Nrfit и Н^О^. Определяют pH разбавленных растворов гидразингидрата и пероксида водорода. Водные растворы гидразина имеют отчетливую щелочную реакцию, тогда как раствор Н2О2— слабокислую. Обратите также внимание на различные способы образования солей.
14. Восстановительные свойства гидразина и гидроксиламина. Для проведения опыта необходимо приготовить следующие растворы:
1) растворяют 50 мг сульфаниловой кислоты в 5 мл 5 М СН3СООН, содержащей 500 мг CH3COONa;
2) 500 мг а-нафтиламина кипятят с 20 мл воды; раствор отделяют от осадка декантацией, к раствору добавляют 5 мл ледяной уксусной кислоты;
3) 500 мг иода растворяют в 10 мл ледяной уксусной кислоты;
4) 500 мг тиосульфата натрия растворяют в 20 мл воды.
В две пробирки наливают по 1 мл уксуснокислых растворов сульфата гидразина и солянокислого гидроксиламина, в которые затем добавляют CH3COONa (образуется «буферный раствор»), К этим растворам при охлаждении льдом приливают 1 мл раствора сульфаниловой кислоты, 0,5 мл раствора иода и энергично встряхивают. Через две-три минуты удаляют избыток иода, добавляя по каплям раствор тиосульфата, затем добавляют раствор а-нафтиламина (запишите результаты наблюдений).
15. Вытеснение слабого и летучего основания более сильным основанием. Проводят опыт с твердыми [NH3OH]C1 и КОН также, как в опыте 8. Испытывают действие выделяющегося газа на индикаторную бумагу (основная реакция).
16. Гидролиз PCI-.. В пробирке сливают 5 мл Н2О и 1 мл РС13, содержимое пробирки встряхивают; 2 мл этого раствора нейтрализуют разбавленным раствором NaOH и затем, добавляя раствор, содержащий ионы Ва2+, осаждают труднорастворимый ВаНРО3. Осадок отделяют центрифугированием и высушивают в сушильном шкафу при 100 °C.
17. Получение и реакции N^O. В сухую тугоплавкую пробирку помещают немного NH4NO3 и осторожно нагревают. Через отводную трубку газ собирают в три пробирки и
1) убеждаются в том, что газ не окрашивается на воздухе в бурый цвет (отличие от NO);
2) во вторую пробирку вводят тлеющую лучинку. N2O легко отдает кислород, так что лучинка загорается;
3) газ встряхивают с разбавленным раствором перманганата калия, подкисленного серной кислотой, что приводит к обесцвечиванию раствора. Если же раствор перманганата оказывается слишком концентрированным, в него пропускают N2O до обесцвечивания.
18. Свойства PtCK0. 2 г Р4Ою, очищенного сублимацией, вносят небольшими порциями в 10 мл воды. Наблюдается энергичная реакция. Испытывают действие полученного раствора на индикаторную бумагу.
19. Восстановление NO$~ до КН3. В щелочной раствор нитрата вносят Цинковую пыль или сплав Деварда (45% А1, 50% Си, 5% Zn) и нагревают. Выделяющийся газ состоит из водорода и аммиака (запах!).
20. Окислительно-восстановительные реакции HNO3. В конц. HNO3 вносят Несколько кусочков цинка. Выделяются бурые оксиды азота* (NO, NO2).
* NO — бесцветный газ, окисляющийся на воздухе до бурого NO2—-Прим. мрев.
35*
548
Неорганическая химия
Азот, фосфор
549
Несколько капель раствора иодида калия добавляют в конц. HNO3 и в разбавленную HNO3. Выделение иода происходит лишь в первом случае. К нескольким кусочкам цинка приливают разбавленной HNO3 и слегка нагревают. Выделяется водород (осторожно!).
21. Сочетание реакций окисления — восстановления и комплексообразования. Несколько капель разбавленного раствора нитрита подкисляют разбавленной серной кислотой (отличие от реакции на нитрат-ион) и добавляют несколько кристаллов FeSO4. Появление темно-бурого окрашивания кристаллов FeSO4 указывает на образование нитрозокомплекса железа.
Fe*+ + NO2- + 2 Н+------> Fe’+ + NO + Н2О
Fe2+4-NO ------► [Fe(NO)]a+
22. Реакция симметричного синпропорционирования. Свежеприготовлен->ный раствор нитрита вливают в холодный раствор амидосерной кислоты. Немедленно происходит выделение газа (тлеющая лучинка гаснет). При кратковременном кипячении происходит разрушение взятого нитрита.
23. Устойчивость связей Р—О и N—O. Реакция диспропорционирования (тяга!). В колбу Эрленмейера помещают 2 г Ва(ОН)2 и 30 мл воды. Колбу закрывают пробкой с двумя трубками. Трубка для подачи газа должна доходить до дна колбы, а конец газоотводной трубки лишь немного выступает за пробку. Другой конец отводной трубки согнут и погружен в фарфоровую чашку с водой, нагретой до 50 °C. В колбу вносят 0,5 г белого фосфора (резиновые перчатки!) и для вытеснения воздуха пропускают сильный ток •азота. После того как воздух из колбы вытеснен, ток азота уменьшают и ,раствор нагревают до кипения. Пробулькивающий через водяной затвор фосфин сгорает (Р2Н4 самовоспламеняется, при недостаточном доступе воздуха на поверхности воды образуются пленки из желтого и красного фосфора). После того как весь фосфор растворится, в колбу с кипящим раствором пропускают вместо азота ток СО2, что ведет к осаждению избыточного Ва(ОН)2 в виде ВаСО3. Осадок отфильтровывают, а раствор Ва(Н2РО2)2, из которого можно методом ионного обмена получить свободную кислоту, сохраняют для дальнейшей работы.
24. Диспропорционирование иона [ДгРОг]-. Часть раствора Ba(H2PO2)2 упаривают до начала кристаллизации. Через некоторое время выпадают бесцветные кристаллы Ва(Н2РО2)2. Эти кристаллы нагревают в небольшой колбочке. При этом выделяется фосфин (запах!)-, твердый остаток представляет собой пирофосфат бария, нерастворимый в уксусной кислоте.
25. Диспропорционирование ВаНРО$. Немного ВаНРО3 нагревают в тигле (под тягой!). По запаху идентифицируют выделяющийся фосфин. В остатке содержатся Ва3(РО4)2 и Ba2P2O7.
26. Восстановительное действие ВаЦЦРСЦ);,. Остаток раствора Ba(H2PO2)2 добавляют к разбавленному раствору CuSO4. Выпадает краснокоричневый осадок, состав которого приблизительно соответствует формуле СиН. Ионы Ag+, Hgs+ и As3+ в кислом растворе также восстанавливаются до металла.
27. Восстановительное действие W[PH2O2] и Н2[РНО3]. Растворяют при нагревании немного As2O3 в разбавленной соляной кислоте. К одной части раствора добавляют раствор Ва(Н2РО2)2, а к другой — раствор Н3РО3. В первом случае происходит выпадение серого металлического мышьяка, а во втором реакция не идет
28. Царская водка. Нагревают серебряную проволоку и листовое золото с азотной кислотой средней концентрации. Серебро растворяется с выделением оксидов азота. Золото удается перевести в раствор лишь после добавления трехкратного количества конц. НС1. Растворению золота благоприятствует образование комплексной кислоты.
29. Термическое разложение нитратов. Немного KNO3 и Pb(NO3)2 помещают в две тугоплавкие пробирки. Обе пробирки нагревают. В пробирке с KNO3 выделяется бесцветный газ—кислород. Остаток в пробирке про
веряют на присутствие нитритов (см. ранее). В пробирке с Pb(NO3)2 выделяется бурый NO2 и кислород. В остатке от нагревания (РЬО) можно доказать отсутствие нитрата.
35.7.3.2. Полимерные оксокислоты и оксоанионы
При растворении Р4О10 в воде наряду с ортофосфорной кислотой образуется смесь различных конденсированных фосфорных кислот. Эти кислоты и их соли, называемые конденсированными фосфатами, можно обнаружить и разделить методами хроматографии на бумаге и электрофореза на бумаге (разд. 38.3.6).
Ион РО43~ — это комплексный анион, в котором центральный атом фосфора тетраэдрически окружен четырьмя атомами кислорода. В конденсированных фосфатах часть атомов кислорода принадлежит более чем одному центральному атому. Образуются состоящие из тетраэдров многоядерные комплексы, в которых фосфор-кислородные тетраэдры вследствие электростатического отталкивания высокозаряженных центральных ионов соединяются между собой только через вершины. Соединение через ребра и тем более через грани возможно лишь при комбинации октаэдрических групп, так как в этом случае расстояние между центральными ионами не так мало, как при соединении тетраэдров.
В ряду HC1O4>H2SO4> (HNO3) >H2SiO3 сила кислот уменьшается. Склонность к образованию полимерных кислот или анионов возрастает с уменьшением силы кислот или с усилением основных свойств соответствующих анионов.
Для фосфатов известно три гомологических ряда: изополифосфаты (с цепочечными анионами); метафосфаты (с кольцеобразными анионами) и каркасные фосфаты (в структурах которых присутствуют разветвленные цепи и кольца, причем тетраэдры РО4 имеют две и более общие вершины с другими тетраэдрами) .
Пирофосфорная кислота, пирофосфаты. Пирофосфорная кислота Н4Р2О? — первый член ряда кислот с цепочечными анионами, образуется как один из продуктов при растворении Р4О10 в воде. Это сильная четырехосновная кислота. Отдельные ступени протолиза многоосновных кислот обычно существенно отличаются. В случае пирофосфорной кислоты первая и вторая ступени мало отличаются друг от друга, так же как третья и чет-8еРтая. Этот факт связан с особенностями ее строения. Различие энергий отщепления одного протона от одного тетраэдра и Второго протона от другого тетраэдра невелико. Третий протон значительно менее «кислый», так как его отщепление идет от °трицательно заряженного тетраэдра. Пирофосфорная кислота Имеет следующие константы диссоциации: p/G = l,O, р/(2=1,9, Раз=6,6 и рД4 = 9,6. За исключением пирофосфатов щелочных баллов, все остальные пирофосфаты труднорастворимы в во
550
Неорганическая химия
Азот, фосфор
551
де. Эти соли отличаются от средних фосфатов по растворимости в кислотах, по цвету и форме кристаллов.
Пирофосфаты, которые можно также получить путем нагревания гидрофосфатов (например, Na2HPO4), при нагревании в водных растворах гидролизуются до монофосфатов (РО43^),
Конденсированные фосфаты. Олигомеры циклофосфатов можно разделить хроматографически вплоть до октаметафосфата. Один из гексаметафосфатов был выделен в препаративных количествах. Полимерные метафосфорные кислоты — это сильные или средней силы кислоты. Например, для тетраметафосфорной кислоты (НРОз)4 p/Ci = 2,6, рК2 = 6,4, р/Сз = 8,8, р/<4= 11,4. Солг этих кислот хорошо растворимы в воде. Осаждение происходит лишь после их гидролиза до монофосфатов, который ускоряете?, в присутствии разбавленных кислот и при нагревании. При образовании перлов фосфорных солей связь Р—О—Р разрывается благодаря протеканию реакций с оксидами металлов. В предельном случае процесс приводит к образованию монофосфатов, причем получающиеся смешанные соли имеют характерную окраску. Считавшаяся ранее специфичной для метафосфатов реакция свертывания яичного белка идет лишь в присутствии растворов изополифосфатов с большим размером цепи. Олигофосфатами с п^15 раствор яичного белка не сворачивается
35.7.3.3. Сочетание кислотно-основных реакций и реакцш осаждения
Оксокислоты азота в основном представлены «метая-формой содержащей меньше воды, а оксокислоты фосфора — «орто» формой. Сила оксокислот меняется в соответствии с общим! закономерностями (разд. 33.4.4).
Во всех оксокислотах имеются связи X—ОН, причем атом водорода способен к диссоциации. Атомы водорода связей X—Н не диссоциируют (X = N, Р). Ниже приведены оксокислоты азота и фосфора и соответствующие значения рК-
Н О
н о
нитриламид е,5
азотистая кислота
5,213,35)
он
^N = N//
но
гипоазотистая кислота
рК,: 7,7
рКг: И,7
фос<ринова.я кислота
рк( U
рК3 '
фос(роновая кислота,
*.8
6,2
азотная кислота,
-/.32
просторная кислота.
2,0
1,1
v 12.3
Раствор Р4Ою в воде имеет кислую реакцию. Наряду с другими фосфорными кислотами он содержит трехосновную орто-фосфорную кислоту. Ион РО43-— довольно сильное основание. Сама фосфорная кислота в присутствии очень сильных кислот может играть роль основания, например Р(ОН)4С1О4. Протекание отдельных стадий протолиза фосфорной кислоты тесно связано с возможностью осаждения ионов металлов в виде фосфатов. Одно-, двух- или трехзамещенные (средние) фосфаты можно получить только при поддержании точного pH. Для получения гидрофосфатов следует поддерживать pH=(pKi + -|-рК2)/2 = 4,5. В таком растворе 90% фосфата находится в виде недиссоциированного «анионного амфолита» Н2РО4_. При pH 9,72 в основном присутствует «анионный амфолит» НРО42~. Лишь при больших pH достигается более высокая концентрация ионов РО43~.
Средние фосфаты всех металлов (за исключением щелочных металлов) труднорастворимы в воде. Однако для осаждения средних фосфатов необходима согласно произведению растворимости определенная минимальная концентрация ионов РО43-. Поэтому на процесс осаждения фосфатов металлов можно влиять изменением концентрации протонов и связанной с ней концентрации ионов РО43-. Кислые фосфаты водорода, как правило, растворимы гораздо лучше, чем средние. В сильнокислой среде все фосфаты (кроме фосфата циркония) легкорастворимы. Сведения о труднорастворимых фосфатах содержатся в соответствующих разделах, посвященных химии металлов. Поэтому ниже приведены Пр лишь для некоторых фосфатов.
—1g Пр
В1РО4 ВР+ + РО43- 20
РЬ3(РО4)2 3 РЬ2+ + 2 РО43- 32
РЬНРО4 ^7—> РЬ2+ + НРО42- 10—11,4 Са3(РО4)2 3 Са2+ + 2 РО43~ 25
СаНРО4 ч=±: Са2+4-НРО42- 5,3-6,6
Li3PO4 <=* 3 L1++ РО43~ 12,6
Методы количественного определения фосфатов и магния основаны на осаждении MgNH4PO4-6H2O (разд. 37.2.1.8.6).
Рекомендуемые опыты (продолжение)
30. Осаждение пирофосфатов. В фарфоровом тигле прокаливают Na2HPO4; после охлаждения продукт растирают в ступке. Часть полученного Порошка растворяют в холодной воде и проводят с раствором следующие °Пыты, используя каждый раз несколько миллилитров раствора:
1) добавляют раствор ВаС12: образуется белый Ва2Р2О7, нерастворимый 8 Уксусной кислоте;
, 2) добавляют раствор AgNO3: образуется белый Ag4P2O7, растворимый 8 HNOs и NH3;
3) добавляют раствор ZnSO4: образуется белый Zn2P2O7, нерастворимый
552
Неорганическая химия
в уксусной кислоте при кипячении; осадок нерастворим вплоть до pH 3,4;
4) добавляют магнезиальную смесь: образуется белый аморфный (п^ внешнему виду) осадок.
31. Получение триметафосфата. При получении метафосфатов исходят из дигидрофосфатов. NaH2PO4 нагревают несколько часов в сушильном шкафу при 60 °C для удаления кристаллизационной воды. В иеглазурованном тигле медленно нагревают обезвоженную соль до температуры красного каления причем в результате должен образоваться прозрачный расплав соли Грэма’ По охлаждении тигель разбивают, а его содержимое переносят в другой фар.' форовый тигель. Стеклообразный пек отжигают в течение 24 ч при 480 °C в электрической печи и получают кристаллический трициклофосфат.
32. Реакции идентификации. В четыре пробирки, содержащие свежеприготовленный раствор яичного белка, добавляют
1) соль Грэма: белок коагулирует;
2) кристаллический триметафосфат: 1
3) раствор монофосфата: ! коагуляции нет.
4) раствор пирофосфата: J
33. Хроматографическое разделение монофосфата, дифосфата и трициклофосфата. При охлаждении в смеси поваренной соли со льдом готовят растворы следующих веществ (по ~40 мг) в 5 мл воды: монофосфат, пирофосфат и триметафосфат. Часть этих растворов смешивают. Следует познакомиться с методикой работы методом хроматографии на бумаге (разд. 38.3.6). При помощи стеклянной палочки или микропипетки отбирают по капле каждого из четырех полученных таким образом растворов и наносят их на бумагу. Используют метод восходящей хроматографии. Для приготовления элюента смешивают 120 мл метанола, 10 мл разбавленной уксусной кислоты (2 мл ледяной уксусной кислоты и 8 мл воды) и 30 мл буферного раствора. Готовят в колбе на 1 л смеси из 133,3 г трихлоруксусной кислоты и 30 м.т 25%-ного раствора NH3 и разбавляют дистиллированной водой до метки.
Примерно через 5—6 ч бумагу вынимают, отмечают на ней линию фронта растворителя и сушат при 60 °C. Для проявления хроматограммы готовят раствор, содержащий 4 г молибдата натрия и 5 г нитрата аммония; раствор разбавляют дистиллированной водой до 100 мл и вливают в 10 мл HN<X (d=l,4). Затем хроматограмму обрабатывают восстанавливающим раствором: 30 г дисульфита натрия, 1 г сульфита натрия и 0,2 г метола растворяют в 10 мл дистиллированной воды и, если необходимо, фильтруют. Фосфаты дают синие пятна, так как гетерополикислоты легко восстанавливаются до молибденовой сини. Не следует брать слишком много восстанавливающего раствора, так как иначе может произойти восстановление молибдата натрия, что приведет к слабому голубому окрашиванию всей хроматограммы.
34. Реакция осаждения иона РО^~. Используют 0,1 раствор Na2HPO4 В различных пробирках проводят следующие реакции:
1) добавляют раствор ZrOCl2: несколько минут раствор кипятят и образовавшийся осадок далее пытаются растворить в разбавленной НС1;
2) после добавления буфера (2 мл 0,1 М раствора ацетата натрия и 2 мл 0,1 М уксусной кислоты) приливают раствор FeCU: осаждается бледно-желтый FePO4 (который может быть и коричневым из-за образования Fe(OH)s); при большей концентрации ацетат-иона образуется многоядерньп комплекс железа(III), который при нагревании разрушается вследствие гидро лиза с осаждением основного фосфата железа:
г ас ас П +
/ \ / \ (+) (ас—)
HO-Fe-ac-Fe—ас-Fe-OH ОН-
^ас'
В отличие от пирофосфатов фосфат-ион дает желтый осадок AgsP^4’ растворимый как р кислотах, так и в аммиаке.
Углерод, кремний
553
35. Осаждение Ag3POt. При приливании раствора AgNO3 к раствору фосфата, подкисленному азотной кислотой, осаждения не происходит. Затем осторожно приливают имеющий меньшую плотность разбавленный раствор .аммиака. На границе фаз устанавливается перепад pH и образуется желтое кольцо, свидетельствующее об образовании Ag3PO4 (разд. 35.7.3.1).
Вопросы и задания.
1. Приведите примеры, показывающие близость химии фосфора и мышьяка в отличие от химии азота и фосфора. В чем причины сходства и различия в химии этих элементов?
2. Дайте объяснение тому факту, что соединения типа P(OR)3 существуют, a N(OR)3— нет. Сравните между собой связи в R3NO и R3PO.
3. В каких соединениях фосфор имеет максимальную валентность?
4. Как меняются основные свойства следующих аминов: N(SiR3)3, N(GeR3)3 и N(SnR3)3 (R=CH3), если известно, что тенденция к образованию Рх—Ля -связей падает от Si к Sn?
5. Азот при комнатной температуре — газ, а фосфор — твердое вещество. Температура кипения СС14 выше, чем для SiCl4. Дайте объяснение этим фактам.
6. Предложите возможный механизм гидролиза NC13 и NF3. Что образуется при гидролизе PF5?
7. При помощи каких реакций можно получить фосфорилгалогениды? К какому типу соединений их следует отнести?
8. Напишите реакции взаимодействия различных оксидов азота с водой. Как можно получить оксиды азота в низшей и в высшей степенях окисления?
9. Обсудите пространственное строение нитрат- и фосфат-ионов с позиций теории химической связи.
10. Рассчитайте положение равновесия реакции
NH3 + Н2О NH4+ + ОН-
йспользуя величины ионного произведения воды и Ка иона аммония.
11. Объясните факт возрастания электропроводности некоторых модификаций неметаллов в ряду: иод, серый селен, черный фосфор, графит и бор.
12. Покажите, что Р3- — мягкое основание, a NsFK— жесткое.
13. Объясните причину парамагнетизма NO. Химическую связь в NO обычно рассматривают в рамках теории МО (см. также N3). Каковы следствия того, что в молекуле NO неспарениый электрон занимает разрыхляющую л*-молекулярную орбиталь?
14. Сульфиды имеют сходство с оксидами. Каковы различия и сходство в типе соединений и в реакциях сульфидов и оксидов фосфора?
15. Обсудите наиболее важные окислительно-восстановительные реакции «ксокислот азота и соответствующих им анионов с привлечением диаграммы окислительных состояний.
35.8. Углерод и кремний
Атомы элементов главной подгруппы четвертой группы в основном состоянии имеют электронную конфигурацию s2p2. При образовании ковалентной связи электронная пара s2 распадается и один s-электрон занимает свободную р-орбиталь.
Р ЕПП
I 'I чч
Углерод, кремний
555
554
Неорганическая химия
Электронная конфигурация определяет основные химические свойства этих элементов.
Их потенциалы ионизации, ковалентные радиусы и радиусы ионов закономерно меняются при переходе от одного элемента к другому. Небольшое отклонение наблюдается лишь в отношении электроотрицательностей, по величине которой эти элементы располагаются в следующий ряд: C>Ge>Si»Sn>Pb. Это отклонение, как и для элементов главной подгруппы третьей группы, обусловлено влиянием переходных элементов и лантаноидов, заполнение в атомах которых d- и f- подуровней приводит к значительному экранированию валентных электронов, у следующих за ними элементов.
Углерод и кремний относятся к неметаллическим элементам, олово и свинец—металлы, а германий — полуметалл. Максимальное число ковалентных связей (координационное число), у атома углерода — четыре, у атомов остальных элементов — шесть.
„Атом углерода способен к образованию кратных рп—рл-свя-зей, в то время как атом кремния в ограниченной степени может образовывать кратные связи за счет использования d-орбиталей при dn—рд-связывании.
Склонность к образованию цепей —Э—Э—Э— убывает в ряду C»Si>Ge«Sn»Pb, поскольку прочности связи Э—Э уменьшаются, например для С—С она равна 347,5 кДж/моль, а для Si—Si — 175,9 кДж/моль.
Углерод и кремний в большинстве своих соединений имеют формальную степень окисления +4. В карбидах и силицидах на атомах С и Si сосредоточен частично отрицательный заряд. Главные типы реакций соединений углерода и кремния — это кислотно-основные, гидролиз и реакции образования комплексов. Окислительно-восстановительные реакции для них не характерны. Напротив, для соединений элементов с более высокими порядковыми номерами характерны окислительно-восстановительные реакции. Эти элементы могут также образовывать-устойчивые двухзарядные ионы.
Углерод и, в меньшей степени, кремний образуют ряд неполярных молекул (СН4, СС14, СО2, SiF4).
Для кремния характерна высокая энергия связи с некоторыми элементами, например
Si—F 540,1 кДж/моль
Si—О 368,4 кДж/моль
Si—С! 360,0 кДж/моль
Для соединений углерода и кремния характерны переходы от одного типа соединений к другому. Например, у карбидов и силицидов можно наблюдать переходы между полимерным,-интерметаллическим и солеобразным типами соединений.
В виде простых веществ углерод и кремний при комнатной температуре — твердые вещества. Структура и связи в модификациях углерода обсуждались в разд. 32.2.3. По кристаллическому строению кремний аналогичен алмазу. Особый интерес представляют свойства кремния как полупроводника. Температуры плавления простых веществ в группе понижаются с уменьшением энергии связи X—X.
Относительно малая активность углерода и кремния при обычной температуре —следствие их полимерного строения и большой энергии связи между атомами. Более высокая реак-ционноспособность кремния, чем углерода, обусловлена устойчивостью образующихся соединений, а также другими причинами (размер частиц, строение поверхности, содержание примесей). При обычной температуре углерод и кремний не изменяются (не реагируют) на воздухе. При более высокой температуре образуются диоксиды. Вода при повышенной температуре восстанавливается ими до Н2:
С + Н2О —> CO + H2f
Si4-2H2O ---► SiO2 + 2H2f
Из кислот с кремнием реагирует только плавиковая кислота. Протекание этой реакции объясняется высокой устойчивостью образующихся ионов SiF62—
Si + 6 HF --» H2SiFe + 2 H2f
Благодаря склонности кремния к образованию силикат-ио-яов, он легко реагирует со щелочами:
Si + 4 ОН" ->- SiO44- + 2 Н2(
Для растворения кремния достаточно даже незначительной концентрации ионов ОН-, так как благодаря сильным основным свойствам ионов SiO44~ в растворе вновь образуются гидроксид-ионы по реакции протолиза:
SiO44~ + 4 Н2О ->- H4SiO4 + 4 ОН"
35.8.1. Карбиды и силициды ;
В зависимости от типа связи выделяют
ковалентные карбиды, интерметаллические карбиды и солеобразные карбиды.
Разумеется, существуют также карбиды, занимающие промежуточное положение между указанными тремя типами. Карби-
556
Неорганическая химия
ды различного строения ведут себя в реакциях по-разному (см. опыты).
Атомы углерода и бора могут образовывать прочную ковалентную связь, давая соединения, весьма сходные с алмазом. Так, карбидом В4С можно шлифовать алмазы. Карбиды бора очень устойчивы и к химическим воздействиям.
Для интерметаллических карбидов характерны высокие температуры плавления (от 3000 до 4200°C), большая твердость (9—10 по шкале Мооса) и металлический тип проводимости. Электронная структура и другие характерные свойства металлов в основном сохраняются при внедрении атомов углерода в кристаллическую решетку. Атомы металлов, образующие интерметаллические соединения, имеют радиус ^0,13 нм. Это — более тяжелые элементы побочных подгрупп четвертой, пятой и шестой групп. Здесь следует назвать ТаС (4150 °C), ZrC (3800°С), и в особенности смешанный карбид 4TaC + ZrC с самой высокой известной в настоящее время температурой плавления (4215°C).
Металлы Сг, Мп, Со и Ni, атомные радиусы которых меньше, чем 0,13 нм, не образуют интерметаллических карбидов. Их карбиды гидролизуются водой и разбавленными кислотами, причем образуются водород и жидкие или (реже) твердые углеводороды. Видимо, в этих карбидах уже имеются цепочечные группы из атомов углерода, сходные с углеводородами, в которых водород замещен металлом. Расстояния С—С равны 0,161 нм, т. е. несколько больше, чем в парафиновых углеводородах*.
Солеобразные карбиды образуют ионную кристаллическую решетку, состоящую из катионов металла и разнообразных анионов углерода, производных этина (С22“, аллилена (С34") и метана (С4-). Ковалентная составляющая связи в этих соединениях относительно мала. Солеобразные карбиды — бесцветные, прозрачные кристаллические вещества. При их взаимодействии с водой образуются различные продукты; соответственно с этим принята классификация солеобразных карбидов.
Метаниды — карбиды, дающие при реакции с водой метан и содержащие в структуре ионы С4~ (например, Ве2С, АЦСз)-
Этиниды — карбиды, дающие при реакции с водой этин (ацетилен) и содержащие в структуре ионы С22- (например, Na2C2, К2С2, СаС2, SrC2, ВаС2, Cu2C2, Ag2C2).
Другие карбиды, при взаимодействии с водой дающие смесь ацетилена и ненасыщенных углеводородов и также содержащие в структуре ионы С22- (например, U2C2, LaC2). Катионы металлов в этих карбидах легко могут изменять степень окисления,
* В карбидах указанных элементов имеются изолированные атомы углерода, окруженные шестью атомами металла. — Прим, перев.
Углерод, кремний
557
что сопровождается протеканием вторичных реакций, например:
ХаС2 + 2Н2О -> Ъа(ОН)2 + HfeCH
Ъа(ОН)2 + Н2О La(OH)3 + 1/?Н2
=1-> Н2С=СН2
HfcCH-X^ 2СН4 знГ^ 4
MgzCe, в структуре которого содержатся ионы Сз4-, с водой дает главным образом аллилен СН3—С^СН.
Большинство силицидов относится к интерметаллическому типу соединений. Разлагаться водой могут только силициды щелочноземельных металлов и лития. При этом образуются кремневодороды, сгорающие на воздухе.
Для получения силанов, силициды разлагают эфирным раствором бромоводорода. Поскольку у кремния склонность к образованию двойных связей невелика, в силицидах анионы имеют обычно слоистую или сетчатую структуру, схожую со строением кремния.
35.8.2. Углеводороды и силаны
Насыщенные углеводороды — неполярные молекулярные вещества. Этот класс соединений изучает органическая химия.
Метан не вступает с водой в протолитическую реакцию. СН3_, как и О2-, NH2-, а также другие подобные ионы,— очень сильное основание. Согласно концепции жестких и мягких кислот и оснований, все они считаются мягкими основаниями.
В водных растворах эти ионы подвергаются нивелирующему действию и тотчас же образуют СН4, ОН-, NH3, переводя молекулы Н2О в ОН--ионы.
рКа -34,0 СН4 +=t Н+ + СН3- рЯ6 —20
Силаны вступают в другие реакции, поскольку электроотри-е+ 6-цательность кремния меньше, чем углерода. Связи Si—Н отли-е— б+
чаются по полярности от связей С—Н, что сказывается на ходе реакций.
Силицид магния, реагируя с разбавленными кислотами, не образует моносилана. В этой реакции Mg2Si скорее напоминает сплав. Наряду с водородом образуется несколько низших олигосиланов от SiH4 до SigHi4
Mg2Si + 4НС1 -► 2MgCl2 + SiH4 (идет в незначительной степени)
Mg2Si + 4 НС1 + 2Н2О -► 2MgCl2 + SiO2 4- 4Н2 (основное направ.
ление реакции)
Соединения кремния только по физическим свойствам имеют некоторую аналогию с соответствующими углеродными соединениями; по химическим реакциям, так же как по термической
55°Неорганическая химия
устойчивости, они сильно различаются (в этом можно убедиться, выполняя опыты). Силаны легко окисляются и на воздухе самовоспламеняются. Они склонны к гидролизу под действием воды, особенно в щелочном растворе, причем связи Si—I-] и Si—Si расщепляются и образуется молекулярный водород. Вода участвует сначала в реакции гидролиза, азатем как окислитель. Окислительно-восстановительная реакция с участием Sri, идет потому, что благодаря меньшей электроотрицательное и кремния атомы водорода в SiH4 заряжены более отрицательно, чем атомы водорода в связи О—Н*.
Формальные аналоги этана и этина (SiFUK [полиси-лан(П] и (SiH)n [полисилан(1)] ведут себя в реакциях совершенно по-другому. Эти различия связаны с полимерным характером полисиланов, что в свою очередь объясняется отличиямч в строении атомов С и Si. Так, атом кремния имеет больший радиус и меньшую электроотрицательность; уровень 3d в основном состоянии не занят.
35.8.3. Галогениды углерода и кремния
Соединение СС14 принадлежит к группе неполярных молекулярных веществ; молекула СС14, несмотря на большую полярность каждой связи вследствие симметричного строения (тетраэдр), не имеет дипольного момента.
SiCI4 тоже неполярное молекулярное вещество. Но полярная связь Si—CI разрушается при действии воды (гидролиз). С помощью этой реакции можно определить направление поляризации связи:
Углерод, кремний 559
—— —— •
Гидролиз SiCl4 происходит по механизму SN1 (внутремоле-кулярное нуклеофильное замещение). Полярная молекула Н2О атакует тетраэдрически координированный атом кремния по механизму замещения. При этом d-орбитали атома кремния могут быть использованы для образования промежуточного соединения. Таким образом, в «активированном» комплексе осуществляется переходное состояние с координационным числом 5. Затем происходит обозначаемое как «внутренний механизм» отщепление НС1. Атомы С1 последовательно замещаются на ОН. Одновременно идут процессы конденсации, приводящие в конечном счете к трехмерному полимеру.
Первая стадия конденсации
CljSiO |н + он| SiCl3-—Cl3si — О — SIC13
Суммарное уравнение можно записать следующим образом: SiCl4 + 4H2O --------------->- Si(OH)4 + 4HCl
Si(OH)4 --> SiO2 + 2 H2O
Следует указать, что кремний может образовывать и устойчивые соединения с координационным числом 5. Так, гидролиз SiF4 проходит через анион SiFs~, который может быть выделен, и далее до аниона SiF62~. В производных силила — аддуктах соединений типа R3SiX и различных оснований Льюиса, имеется пятикоординированный комплексный катион кремния:
f CHoClo
(C6H5)3SiI + 2,2,-Dtpy-----
СБН5\ |
Si—N
+ Г
С6Н= с6н5
Предполагаемый механизм реакции:
+ НС1
активированный комплекс
* Атомы водорода в молекуле воды имеют небольшой положительный заряд. [Мартыненко Л. И., Опицын В. И. Методические аспекты курса неорганической химии. — М.: Изд-во МГУ, 1983, с. 26] — Прим, перев.
В SiF4 гетерополярная составляющая связи больше, и можно было бы ожидать, что это соединение будет более чувствительно к гидролизу. Однако это не так (опыт 9). В тетрафтори-Де кремния порядок связи Si—F равен 1,41. В соответствии с этим надо принять либо возможность образования кратных связей, либо использовать представления, развиваемые ниже, которые помогают объяснению ряда реакций SiF4.
Как было показано на примере SF6, элементы второго периода могут использовать для образования связей d-орбитали. Вследствие высокой электроотрицательности фтора на центральном атоме кремния, который значительно менее электроотрицателен, создается положительное поле (разд. 35.6.3.2). Благодаря этому d-орбитали сжимаются и становятся способными принимать участие в с125р3-гибриц.изац.ии. Число связывающих
560
Неорганическая химия
орбиталей становится больше числа связывающих пар электронов, т. е. молекула SiF4 имеет как бы дефицит электронов. Поэтому SiF4 реагирует с донорами электронов, например с фторидами, давая ион SiFg2-. Частичное образование двойных связей происходит за счет того, что из-за дефицита электронов на центральном атоме свободные электронные пары атомов фтор, могут участвовать в связывании. Кратность связи при этом по вышается.
H2SiF6 — это сильная двухосновная кислота. В сильнокисло! среде, например в конц. H2SO4, она реагирует следующим обра зом:
SiFe2~ + H2SO4 ->- SO42-+ SiF4 + 2 HF
Ее соли, фторосиликаты, можно отличить от фторидов и О' «силикатов по тому, что они с Н2О дают реакцию без добавления SiCh или CaF2 (опыт 9).
35.8.4. Оксиды, оксокислоты и сульфиды
При сгорании углерода и кремния образуются оксиды, приче! оксиды углерода и кремния имеют весьма различные свойства Углекислый газ СО2, реагируя с водой, образует угольную кис лоту Н2СОз, в то время как кварц SiO2 в воде практически не растворим. Кварц плавится выше 1500 °C, а СО2 — газ. Связи в обоих соединениях полярные ковалентные, но атом углерода способен к образованию двойных связей, а атом кремния к этому не очень склонен. Вместо этого образуются четыре прочные одинарные связи Si—О. Октетная конфигурация атома кремния ц SiO2 достигается за счет образования полимерной структуры
|О| |О|
£- 2<У+ £- _ | _ | _
О=С = О; —О —Si —О—Si—О—
|О| |О|
1 I
Различным типом ковалентных связей объясняется переход от неполярного молекулярного вещества к полимерному соединению.
При пропускании СО2 в воду часть его в соответствии с законом Генри физически растворяется, а другая часть, реагируя с водой, образует двухосновную угольную кислоту Н2СО3. При этом поляризованная двойная связь гидролитически расщепляется и образуется точно так же, как в случае иона NO3~, мезо-
Углерод, кремний
561
мерный карбонатный ион плоского строения:
<У- 5-
о=с=о +
Н — он-*
2— '
+ 2Н+
к = сн3о+-снсо3- = j 0_3)3 = сНзо^Снсо3- = j 0_м
1 СН2СО3 1 СН2со3+ссо2 раств
Значение Ка = Ю~3’3 соответствует истинной константе кислотной диссоциации Н2СО3. Если же учитывать и растворенный СО2, получают важную для практики «кажущуюся» константу Kt' (Kt'<Ki). На кислотно-основное равновесие, устанавливающееся при растворении СО2, можно влиять, например сдвигать его в сторону образования ионов СО32- путем добавления ионов ОН-. Следует заметить, что образование кислоты при растворении СО2 в воде происходит медленно (опыт 10), так как присоединение молекулы воды к двойной связи С = О идет не по ионному механизму.
Равновесие СО2—НгО имеет значение в биологии. В крови его установление ускоряется ферментом карбоангидразой.
Ион СО32- — более сильное основание, чем анионный амфолит НСОз-.
Как видно из сопоставления произведений растворимости, большинство карбонатов, кроме карбонатов щелочных металлов и таллия, труднорастворимо. Напротив, все бикарбонаты (кроме NaHCO3) хорошо растворимы в воде (табл. В.З).
В водном растворе СО2 концентрация ионов СО32- так мала, что при добавлении ВаС'12 произведение растворимости ВаСО3 не достигается.
Карбонаты растворяются в слабых кислотах со вспениванием (выделение СО2, качественная реакция). Даже вода может Действовать как слабая кислота:
ВаСО3 + Н—ОН у—>• ОН" + НСО3- 4- Ва2+
Так, суспензия ВаСО3 в воде имеет щелочную реакцию. Анион СО32- — жесткое основание.
В биологических системах, в частности в крови, система Н2СО3 — НСОз-ИгРает очень важную роль буфера (опыт 15) (разд. 33.4.2.4):
нсо3- 4- н3о+ <—>• н2со3 4- н2о —> co2f 4- 2 н2о
Н2СО3 -|- ОН- * НСО3- 4- Н2О
При гидролизе SiCT4 образуется ортокремневая кислота: ;
SiCl4 4- 8 Н2О -► Si(OH)4 4-4 Н3О+4~ 4 Q-
36-
2027
562
Неорганическая химия
Это очень слабая кислота (pKi=9,5; р/Сг— Н,8); она сразу конденсируется в олигомерную поликремневую кислоту:
Si(OH)4 + (HO)4Si -> HeSi2O7 + Н2О и т. д.
В случае низких значений pH образуются высокомолекулярные изополикислоты, нерастворимые в воде. При высокой степени полимеризации состав осадка отвечает формуле (H2SiO3)n-•Н2О, где п — большое число. Выпадающий при pH 0 SiO2-aq находится в состоянии геля, а после высушивания благодаря большой поверхности пор представляет собой прекрасный адсорбент (силикагель).
При выполнении количественных анализов силикатов следует обращать внимание на процессы адсорбции, происходящие при осаждении и промывании осадков, имеющих сильно развитую поверхность.
Сероуглерод CS2 — мономерное неполярное молекулярное вещество (в чистом виде — бесцветная жидкость), в то время как дисульфид кремния S1S2 — полимер с волокнистой структурой (разд. 32.3.3.3).
CS2 гидролизуется водой при 150°C; гидролиз S1S2 идет очень быстро уже в холодной воде:
XS2 4- 2 Н2О -► ХО2 + 2 H2S
При взаимодействии CS2 с сульфидами щелочных и щелочноземельных металлов, а также с сульфидом аммония образуются растворимые тиосоли:
CS2 + Na2S ---»- Na2CS3
тиокарбонат натрия
Эта реакция аналогична реакции диоксида углерода:
СО2 + 2 NaOH---► Na3CO3 + HjO
35.8.5. Моноксиды углерода и кремния
В водных растворах Н2СО3, СОг и СО32- не могут быть восстановлены до НСООН, СО, НСОО- или СгОд2-*. Кислородные соединения кремния в водных растворах также не восстанавливаются обычными восстановителями.
Моноксиды получают взаимодействием диоксидов с соответствующими элементами:
СО2 + С ч=> 2СО
SiO3 + Si 2 SiO
Первое равновесие имеет большое значение в технике. Моноксид углерода применяется как восстановитель во многих
* Восстановление углекислоты при фотосинтезе протекает в водных системах.— Прим, перев.
Углерод, кремний^
563
кр\ тих производствах. Он является составной частью светиль-ноп>, генераторного, колошникового, водяного и коксового газов.
При рассмотрении диаграммы окислительных состояний соединений углерода при pH 7 (рис. В.34) можно сделать следующие выводы:
высокая стабильность элементарного углерода обусловливает стремление (по термодинамическим причинам) соединений углерода* к разложению на уголь и воду;
Степень окисления
Рис. В.34. Диаграмма окислительных состояний для соединений углерода.
небольшая величина градиента меЖДУ СН4 и СО2 обусловливает то, что большинство соединений диспропорционирует на метан и диоксид углерода.
СвН12Ов --г 2 СО2 + 2 С2Н6ОН (диспропорционирование)
Последнее особенно важно для химико-биологических процессов, протекающих чаще всего за счет солнечной энергии. В промышленности особое значение имеют реакции С, СО и СН4 с кислородом.
Моноксид углерода можно рассматривать как ангидрид муравьиной кислоты, хотя в отличие от настоящих ангидридов он в обычных условиях не способен к протолитической реакции с водой или щелочью**. Муравьиная кислота — кислота средней силы, в растворе диссоциирует:
НСООН + Н2О Н3О+ + НСОСГ рК = 3,7
* Содержащих водород и кислород. —Прим, перев.
** При повышенных давлениях и температурах СО взаимодействует со Щелочами с образованием формиатов. — Прим, перев.
36*
564
Неорганическая химия
Муравьиная кислота — простейшая монокарбоновая кисло-та, щавелевая — простейшая дикарбоновая кислота. В последней углерод формально находится в степени окисления 3 (фор. мальные правила для определения степени окисления см. в разд. 33.5.1.3). Структурная формула щавелевой кислоты:
о. .он
>с-с< но/
щавелевая кислота
Соли щавелевой кислоты — оксалаты — часто труднорастворимы, например для СаС2О4 —1§Пр = 8,1; в то же время для MgC2O4—1g Пр = 4,1 и оксалат магния растворим значительно лучше. Особенно плохо растворимы оксалаты лантаноидов:
—1g Пр —1g Пр
Се2(С2О4)з 28,6 Sm2(C2O4)3 28,5
Ьа2(С2О4)з 27,7 Y2(C2O4)3 26,3
Рг2(С2О4)з 27,3
Молекула моноксида углерода благодаря наличию свободной пары электронов — отличный лиганд в реакциях комплексообразования. В карбонилах металлов атомы металла и углерода связаны ковалентно; металл в них имеет формальную степень окисления «нуль». На примере гексакарбонила хрома удобно подробно рассмотреть механизм образования дативной связи.
Наряду с этим особым классом соединений СО дает комплексы также с ионами в водных растворах. Образование продуктов присоединения при пропускании СО в аммиачный раствор CuCl используется в промышленности, а также и в аналитической химии.
Рекомендуемые опыты
1. Реакционная способность кремния. 50 мг тонкого порошка кремния растворяют в 10 мл 40%-ной HF. Реакцию проводят в платиновом тигле. Затем раствор разбавляют до объема 50 мл и осаждают H2SiFe в виде ее труднорастворимой соли BaSiFe. Такое же количество кремния растворяют в 10 мл 2 М раствора NaOH и нагревают. Выделяющийся водород собирают и сжигают на воздухе (осторожно!).
2. ВцС — инертное вещество. Немного карбида бора кипятят в кона. HNO3. Никаких изменений не наблюдается. Попытайтесь растворить В4С в смеси КСЮз и НС1. Устанавливают, что В4С не подвержен действию хлора (в момент выделения).
3. Карбиды переходных элементов. Небольшое количество ТаС обрабатывают в пробирке водой, разбавленной и концентрированной НС1 и другим» кислотами. Реакции не идут.
Карбид хрома и немного цементита (Fe3C) кипятят с водой, при этом происходит реакция: выделяется водород; образуется также жидкий углеводород или иногда уголь.
4. Гидролиз солеобразных карбидов. В приборе для выделения газов проводят реакцию А13С4 с водой.
Углерод, кремний
565’
1) Образующийся бесцветный газ пропускают в воду и испытывают его действие на индикаторную бумагу: кислой реакции нет. Затем пропускают газ в сильноразбавленный раствор NaOH. pH раствора проверяют перед пропусканием и через каждые пять минут во время пропускания газа: нейтрализации не происходит.
2) Выделяющийся бесцветный газ поджигают и в пламя вносят глазурованный фарфоровый тигель. Благодаря недостаточному поступлению воздуха происходит неполное сгорание метана и на тигле оседает сажа.
3) Другую часть газа собирают в цилиндре, вытесняя из него воду. Цилиндр обертывают полотенцем (защитные очки!) и собранный метан сжигают. После этого в цилиндр наливают несколько миллилитров известкрвой воды и встряхивают.
кароиа кальция tu^-ный NaOH 50"/а нг5Щ и К^Сгг07
Рис. В.35. Схема прибора для получения этина (ацетилена).
5. Получение этина (ацетилена). Собирают прибор (рис. В.35). Из капельной воронки на карбид кальция капают водой. Образуется ацетилен, который для очистки пропускают через две промывные склянки—одна с 10%-ным раствором NaOH и другая — с раствором К2Сг2О7 в разбавленной-H2SO4.
1) Собирают немного газа под водой в пробирку и проверяют его горючесть (осторожно!).
2) Ток газа пропускают в раствор небольшого количества брома в уксусной кислоте, раствор обесцвечивается.
3) Ток газа пропускают в раствор, полученный смешением 2 мл 0,1 М. AgNO3 с 1 мл разбавленной аммиачной воды. Выпадает бесцветный этинид серебра.
6. Силаны. Немного MgaSi обрабатывают в пробирке разбавленной соля-Чой кислотой. Выделяющийся газ сгорает на воздухе. В приборе для получения газов Mg2Si обрабатывают эфирным раствором НС1 при осторожном Нагревании. Образующийся газ пропускают в разбавленный раствор NaOH. Затем к этому щелочному раствору приливают НС1 (1:1), при этом выпадает осадок SiO2-aq (определите тип реакции).
7. Неполярное молекулярное вещество ССЦ. Немного ССЦ встряхивают в пробирке с водой и конц. NaOH. Опыт показывает, что ССЦ не смешивает-Ся с полярным растворителем — водой. При встряхивании с конц. NaOH не-происходит гидролитического расщепления.
, 8. Гидролиз 31СЦ. В пробирке встряхивают немного SiCl4 с водой-Пяга!). Определяют pH раствора.
666
Неорганическая химия
9. Устойчивость молекул SiF4 и S1CI4. Приготавливают две смеси:
1) из трех частей SiO2 и одной части CaF2;
2) из трех частей SiO2 и одной части ВаС12.
Обе смеси хорошо высушивают и помещают в два свинцовых тигля. В каждую смесь добавляют немного конц, H2SO4 и хорошо размешивают, а затем покрывают крышками с просверленными отверстиями и нагревают на песчаной бане. На отверстия в крышках кладут смоченную черную фильтровальную бумагу.
На черной бумаге, покрывающей тигель, содержащий CaF2, появляется белое пятно вследствие образования и гидролиза SiF4. При гидролизе SiF образуется гексафторкремиевая кислота (почему?).
2SiF44-2H2O -----► SiO2-aq + 2 H2SiFe
, 10. Равновесие протолиза СО2. В пробирку помещают 5 мл охлаждение
го льдом раствора СО2 и каплю фенолфталеина, затем прибавляют раствор полученный добавлением 3 капель разбавленного раствора NaOH к 1 м-воды. Раствор окрашивается в красный цвет, окраска бледнеет лишь чере несколько часов.
СО2 + Н2О ч=>: Н2СО3 Н2СО3 + ОН- НСО3- + Н2О
11. Равновесия протолиза. В водные растворы NaHCO3 и Na2CO3 добавляют по одной капле фенолфталеина. Раствор NaHCO3 окрашен слабо, а раствор Na2CO3 — интенсивно, так как концентрация иоиов ОН- увеличивается вследствие протолиза:
НСО3- + н—он —>- н2со3 + ОН-
со32- + Н-ОН------->- НСО3- + ОН-
12. Реакция осаждения иона СОз2~. Несколько капель раствора Na2CO3 разбавляют водой и по каплям добавляют раствор ВаС12. Выпадает белый осадок.
13. Гетерогенная кислотно-основная реакция (растворение соли). Кусочек мрамора в пробирке обливают разбавленной соляной кислотой. У отверстия пробирки держат стеклянную палочку с висящей на ней каплей баритовой воды. В капле образуется муть от выпадающего ВаСО3. Если у отверстия пробирки будет находиться капля раствора ВаС12, помутнения не произойдет (почему?).
14. Кислотно-основное равновесие с участием СО2. В воде взмучивают СаСО3, пропускают в эту суспензию СО2 и добавляют каплю фенолфталеина. После обесцвечивания индикатора осадок растворяется. Одну часть этого раствора нагревают, к другой добавляют раствор, содержащий ионы ОН". В обеих пробирках вновь выпадает СаСО3.
СаСО3 + Н2О + СО2 ------- > Са2+ + 2 НСО3_
нагревание
15. Буферное действие. В пробирках разбавляют водой (1 : 4) по нескольку миллилитров 2 М. НС1 и 2 М NaOH. Немного NaHCO3 растворяют 8 10 мл охлажденной льдом воды, насыщенной СО2, добавляют 4 капли тйМ°' левого синего; этот раствор разливают в две пробирки. В одну из них Д0.' бавляют по каплям разбавленный раствор НС1, в другую — разбавлений» раствор NaOH. Изменение цвета от добавления кислоты наступает толь»8 после добавления 20—30 капель. При добавлении же 2—3 капель щелочу цвет индикатора меняется иа синий, ио прежняя окраска вскоре восстаИаВ' ливается, так как из растворенного СО2 снова образуется Н2СО3. Только после добавления 8—10 капель щелочи окраска индикатора меняется окончатель ио, т. е. когда весь буфер израсходован.
Псевдогалогены
567
16. Сильное ^анионное основание». К раствору силиката натрия («растворимое стекло») добавляют несколько капель фенолфталеина и убеждаются в сильнощелочиой реакции раствора. Раствор приливают в две пробирки, в одной из которых находится разбавленная НС1, в другой — раствор NH4C1 (катионная кислота). Пробирки встряхивают. Оба раствора застывают в «студень» раньше, чем меняется цвет индикатора. Силикаты щелочных металлов могут оставаться в растворе только при pH 12—14.
17. Реакция осаждения SiO^-. К раствору CuSO4 понемногу добавляют аммиачную воду, до тех пор пока образовавшийся осадок снова не растворится. К полученному раствору добавляют раствор силиката натрия. Выпадает силикат меди бирюзового цвета. С AgNO3 жидкое стекло дает осадок, состоящий из силиката серебра и оксида серебра.
18. Реакция BaCS3. Из желтого BaCSs готовят 10 мл 0,1 М. раствора, определяют его pH индикаторной бумагой и проводят реакцию с уксуснокислым свинцом.
19. СО как восстановитель. Полученный, как указано в опыте 17, СО или светильный газ пропускают для очистки через промывную скляику с КОН, далее через пустую скляику и, наконец, через скляику с конц. H2SO4. Очищенный таким образом газ пропускают через:
1) раствор, содержащий [Ag(NH3)2]+: выпадает черный осадок серебра;
2) смесь I2O5 с разбавленной H2SO4: уже иа холоду СО окисляется; иод можно экстрагировать растворителем, например СС14;
3) раствор PdCl2: Pd2+ восстанавливается до металлического палладия;
4) раствор КМпО4 в присутствии иоиов Ag+ : СО окисляется до СО2 и наблюдается обесцвечивание раствора.
Вопросы и задания.
1. Почему в щелочных растворах ие идут реакции диспропорционирования углерода и кремния?
2. Составьте таблицу пяти главных типов структур силикатов. Изобразите для каждого типа схему его строения и назовите для каждой структуры пример встречающегося в природе силикатного минерала.
3. Подсчитайте число атомов в элементарной ячейке 0-кристобалита и покажите, что результат соответствует эмпирической формуле диоксида кремния.
4. К какому типу соединений принадлежат силанолы и силоксаны? Как объясняется их химическое поведение?
5. В некоторых реакциях в качестве промежуточных образуются «карбеновые» частицы: :СН2, :SF2; :SiF2. Как объяснить большую реакциоииоспо-собность этих промежуточных состояний? Какие вещества могут образоваться, например, из :CF2 или :SiF2?
6. Объясните, почему СО, олефины и ароматические соединения реагируют как мягкие основания.
7. Объясните ход измеиеиия относительной устойчивости различных степеней окисления элементов главной подгруппы четвертой группы.
Псевдогалогены
Как показывает название, псевдогалогены по своему поведению °Чень напоминают группу галогенов. Они образуют димерные ^одекулы. Наиболее известные молекулы этого рода — дициан ^N)2, дитиоциан (диродан) (SCN)2, диселеноциан (SeCN)2 и Диазидодитиодисероуглерод (SCSNsh-
Псевдогалогеноводороды протолизуются в воде и образуют одноосновные кислоты. Ионы псевдогалогенидов дают трудно-Рэетворимые соли с ионами свинца, серебра и ртути(1).
£68
Неорганическая химия
Псевдогалогены можно получать в общем случае окислением соответствующих ионов: 2Х_—>-Х24-2е~. Молекулы Х2 диспро-порционируют в щелочи. Сильно поляризуемые ионы псевдогалогенидов являются лигандами во многих комплексных соединениях.
35.9.1. Дициан, циановодород, цианид-ион
Как все соединения, содержащие углерод в степени окисления + 2. псевдогалогениды и псевдогалогены чрезвычайно ядовиты.
Дициан получают по внутримолекулярной окислительно-восстановительной реакции (опыт 1). Для его получения требуется менее сильный окислитель, чем для получения галогенов по аналогичной реакции:
2 HCN --► (CN)2 + 2 Н+ + 2е" Е° = 0,80 В (при pH = 0)
2 CN- --> (CN)24-2e- Е» = 0,27В
Ионы Си2+ способны окислять ионы CN", так как величина потенциала Cu2+/Cu меняется благодаря образованию комплекса [Cu(CN)4]3- (разд. 33.6.3).
При пропускании (CN)2 в раствор NaOH (рН>13) он дис-пропорционирует на ионы цианида и цианата:
(CN)2 + 2 ОН' -► CN- + OCN- + Н2О
Синильная кислота — очень слабая кислота (рК=9,1) и сильно летуча. СО2 вытесняет HCN из цианидов. Поэтому растворы цианидов щелочных и щелочноземельных металлов име-пот сильно щелочную реакцию. Образующаяся при протолизе синильная кислота частично улетучивается.
Синильная кислота гидролизуется до NH3 и муравьиной кислоты медленно на холоду и быстро при нагревании. В присутствии сильных кислот или оснований гидролиз ускоряется (см. также гл. 33.8).
HCN4-2H2O ------ HCOOH4-NH3
Цианид-ион дает труднорастворимые соли и, будучи сильным основанием, является прекрасным комплексообразовате-лем. По поляризуемости он близок к ионам галогенидов. Приведем произведения растворимости некоторых цианидов:
Ag[ Ag(CN) 2]2 Ag+ + 2 Ag(CN)2 -1g Пр = 11,4
Hg2(CN)2 Hg22+ + 2 CN- -lg Пр = 39,0
Ион CN- изоэлектронен с молекулой CsO и катионом нИ--трозила NO+ и поэтому может ими замещаться в устойчивых жомплексах, причем, разумеется, происходит изменение заряДа ^комплекса.
Псевдогалогены
569
«Амбидентная» (двойственная) природа группы C = N~ служит причиной полимерного строения многих цианидов неметаллов и металлов.
Схемы строения:
Цепи—линейные:
спиральные:
Плоская сетка
Трехмерное строение
-Ag-C=N-Ag-C=N-
—Си—C=N—Си—С—
AgCN
KCu(CN)2
Тетраэдрическая координация центрального атома
CuCN-NHs
Zn(CN), Cd(CN)2
35.9.2. Дитиоциан, родановодород, тиоцианат-ион
В кислых растворах тиоцианаты действуют как восстановители, окисляясь сами при этом до дитиоциана (диродана):
2SCN- «=> (SCN)2 + 2е" Е° = 0,77 В
В водном растворе диродан диспропорционирует:
3 (SCN)2 + 4 Н2О -> 5 SCN- + SO42- + HCN + 7 Н+
Родановодородная кислота (р/(=0,85) сильнее, чем синильная. Роданид-ион образуется путем присоединения серы к циа-нид-иону. Вследствие сильной поляризации роданид-иона многие роданиды окрашены. Роданиды Ag+, Hg22+, Cu+, Pb2+ труднорастворимы в воде, например для CuSCN и Hg2(SCN)2 —1g Пр =13,4 и 19,5 соответственно. В разбавленной H2SO4 (1:1) они разлагаются на (NH4)2SO4 и газообразный карбо-нилсульфид COS.
Рекомендуемые опыты
1. Дициан. В сухой пробирке (под тягой!) нагревают немного цианида Ртути(Н) (уравнение реакции!). Образуются газообразный дициан и немного Парациана (CN) п. Выделяющийся в этом опыте газообразный дициан мож-Но поджечь, его пламя имеет пурпурную кайму.
2. Легколетучая синильная кислота. В прибор для получения газа помечают немного раствора KON и добавляют твердый NaHCO3 до насыщения. ° приемник помещают раствор AgNO3, подкисленный азотной кислотой, затем через систему пропускают равномерный ток СО2. При этом происходит Улетучивание HCN и выпадение AgCN в приемнике.
3. Гидролиз HCN. Немного K4[Fe(CN)6] нагревают с конц. H2SO4. Выделяющийся газ (СО) поджигают. Остаток разбавляют водой и доказывают Рисутствие в растворе аммонийной соли.
570
Неорганическая химия
4. Образование комплекса. К небольшому количеству раствора цианида калия добавляют по каплям раствор AgNO3. В месте падения капель обр i-зуется осадок AgON. При встряхивании в присутствии избытка ионов СК-осадок растворяется (образование комплекса). Когда соотношение [Ag+] j [CN-] становится больше, чем 1 :2, начинает выпадать белый AgCN; он м жет быть растворен в NH3.
5. Совокупность реакций восстановления, комплексообразования и осел дения. Несколько кристаллов сульфаниловой кислоты HzNCeHtSOjH и и большой кусочек металлического натрия помещают в сухую пробирку; про-бирку медленно нагревают на горелке до температуры красного каления (осторожно!). Когда реакция закончится, еще горячую пробирку опускают в более широкую пробирку, в которую налито ~2 мл воды. Горячая пробирка лопается и ее содержимое растворяется в воде, полученный раствор фильтруют и добавляют к нему свежеприготовленный раствор FeSO4. Смесь некоторое время нагревают до кипения, добавляют раствор FeCl3 и подкисляют разбавленной H2SO4. Раствор окрашивается в ярко-синий цвет или образуется синий осадок «берлинской лазури» Fe4[Fe(CN)6]3.
Углерод органического соединения в присутствии азота восстанавливг ется натрием до иона CN~. Последний реагирует с Fe2+, образу-Ге! (CN) е] 4~, который в кислой среде с Fcs+ дает «берлинскую лазурь».
6. Получение (SCN)2. 2 г AgSCN высушивают в сушильном шкафу при 70 °C. В конической колбе объемом 50 мл к 5 мл перегнанного над Р4О|0 .сероуглерода (осторожно!) прибавляют 1 г AgSCN. При энергичном перемешивании путем вращения колбы к этой смеси по каплям добавляют ~0,2 мл брома. Реакция заканчивается через несколько минут. Раствор отфильтровывают от выпавшего AgBr и охлаждают до —70 °C (СО2+ацетон). При этом образовавшийся диродан выпадает в осадок.
7. Полярное молекулярное вещество COS. К раствору роданида прибавляют растворы H2SO3 и CuSO4, выпадает белый осадок CuSCN. Его отфильтровывают и высушивают в сушильном шкафу при 110 °C. К этому веществу прибавляют H2SO4 (1:1) и слегка нагревают. Выделяется COS; он горит синим пламенем (осторожно, COS очень ядовит!).
35.10. Бор
Бор в третьей главной подгруппе — единственный неметалл. Скачок свойств между ним и более тяжелыми гомологами очень резкий. Здесь следует упомянуть о правиле «диагонального сходства» в периодической системе. Согласно этому правилу, первый элемент главной подгруппы по своему химическому поведению имеет сходство с вторым элементом следующей главной подгруппы, а этот второй — с элементом побочной подгруппы той же группы. Ниже, в качестве примера, обсуждается лишь сходство в химии бора и кремния.
Гидриды бора и кремния летучи и самовоспламеняются йа воздухе. Водой гидролизуются.
Галогениды бора и кремния легко гидролизуются, давая соответствующие кислородные кислоты. BF3 и S1F4 одинаков0 ведут себя в реакции гидролиза, отличаясь от остальных гаЛ°‘ генидов этих элементов.
Важнейшие соли бора и кремния — производные изополиКИС' лот. Оксокислоты этих элементов очень слабые.
Оксиды с максимальной валентностью — полимерные веШе' ства с каркасной структурой или стёкла.
Бор
571
Бориды и силициды при взаимодействии с кислотами дают соответствующие водородные соединения.
У элементов главной подгруппы третьей группы особенно ярко проявляется тенденция к уменьшению способности образовывать ковалентные связи по мере увеличения атомной массы. Бор образует преимущественно ковалентные соединения.
Согласно спектральным данным, атомы бора и других элементов главной подгруппы третьей группы в своем основном состоянии имеют только один неспаренный электрон. В соответствии с этим могла бы образоваться только одна ковалентная связь. Для энтальпий образования соединений этого типа расчетом получены очень большие положительные значения, например ДДв-н = 364,3 кДж/моль и A//ai-h= 146,5 кДж/моль. Оказывается, однако, что для перевода одного из s-электронов из спинспаренного 52-состояния в p-состояние с тем же главным квантовым числом требуется очень небольшая энергия (для В 0,17; для А1 1,34 кДж/моль). В возбужденном 5р2-состоянии атомы этих элементов имеют три неспаренных электрона и могут образовать три ковалентные связи. 5р2-Гибридные орбитали расположены в одной плоскости под углом 120°. Такие молекулы, как ВС13, В(СНз)з, имеют форму плоских равносторонних треугольников.
В решетке элементного бора связи между атомами неполярные, ковалентные, что обусловливает такие его свойства, как большая твердость и химическая устойчивость к процессам окисления — восстановления, растворения и образования комплексов. В образовании сильных ковалентных связей участвуют делокализованные электроны, о чем свидетельствуют цвет и наличие небольшой электропроводности.
35.10.1. Бориды и бораны
В солеобразных боридах не содержится ионов В3-. Против ионного строения этих соединений говорят большая твердость, металлическая проводимость и высокая химическая устойчивость.
В боридах атомы бора хотя и несут отрицательный заряд, их структуры сходны с силицидами, где нет 514--ионов, а отрицательно поляризованные атомы кремния расположены в основном в виде цепей, слоев и трехмерных каркасов. Структуры боридов нельзя классифицировать, пользуясь обычными представлениями о валентности. В большинстве своем это «нестехиомет-Рические» соединения. В зависимости от их строения бориды Чожно разделить на следующие группы:
Бориды с изолированными атомами бора. В них атомы бора Размещаются в тригонально-призматических пустотах между атомами металлов (~EaB2, Е2В).
572
Неорганическая химия
Бориды, в которых имеются простые или двойные цепи атомов бора. Так, в Ni4B3 две трети атомов бора образуют бесконечные зигзагообразные цепи.
Бориды с двумерными сетками из атомов бора (соединения состава ~ЕВ2 и Е2В5). Бориды этого типа лучше других проводят ток, обладают наибольшей твердостью и тугоплавкостью.
Рис. В.36 Схема взаимного превращения боранов
Бориды с трехмерным каркасом из атомов бора (соединения состава ~ЕВ4, ЕВв, ЕВ12). В СаВв атомы бора образуют октаэдр, состоящий из шести расположенных рядом атомов бора.
Бораны делятся на две группы: термически устойчивые В„Нл+4 и неустойчивые ВлНл+б. Те и другие гидролизуются водой и еще легче щелочами, подобно кремневодородам, с образованием боратов и водорода. Однако они не столь легко самовоспламеняются, как силаны. На рис. В.36 схематически показаны условия взаимного превращения боранов. С другими реакциями боранов, особенно многочисленными у диборана, можно ознакомиться по специальной литературе.
Большое значение имеют соединения, содержащие ион боро-гидрида ВН4_, которые вследствие отрицательного парциального заряда на атоме водорода являются восстановителями и источником гидридных ионов.
35.10.2. Галогениды бора
Тригалогениды бора по своему поведению напоминают галогениды кремния. Так, например, в молекуле BF3 связи тоже имеют повышенную кратность. Свободная орбиталь у бора используется для связывания электронных пар атомов галогена. Сво-
Бор
573
родная орбиталь, имеющаяся в молекулах тригалогенидов бора, и малый размер атома бора определяют их поведение в реакциях. Важнейшие реакции для них — это гидролиз и образование донорноакцепторных комплексов с основаниями Льюиса.
BF3 летуч и, как все летучие соединения бора, окрашивает пламя в зеленый цвет (способ обнаружения бора). Он медленно гидролизуется в воде; гидролиз протекает иначе, чем у ВОз, который тотчас же дает В(ОН)3 и НС1 (см. разд. 35.8, гидролиз SiCl4). Быстрый гидролиз ВС13, ВВгз, В13 указывает на то, что эти галогениды — более сильные кислоты Льюиса, чем BF3. Ход изменения энтальпий реакций взаимодействия галогенидов бора с пиридином в нитробензоле показывает, что в этих условиях электронно-акцепторное действие (а следовательно, и сила кислот Льюиса) возрастает в ряду ВР3<ВС1з< <ВВг3<В13.
Соединение C6H6N—>-BF3 C5H6N—>-ВС13 C6H6N—>-BI3
Энтальпия реакции, —132,7 —165,4 —186,3
кДж/моль
Гидролиз BF3 (ср. SiF4):
4BF3 + 6Н2О ЗН3О+ + 3BF4- + В(ОН)3
Сходство ионов F~ и ОН- проявляется в том, что ион BF4~ в водных растворах может участвовать в следующих равнове-
H[BF3OH] + Н2О H[BF2(OH)2] + HF рХ = 2,0
BF4- + Н2О [BF3(OH)J- -J- HF p/f = 2,6
Первая стадия реакции гидролиза — присоединение молекулы воды благодаря наличию в молекуле BF3 (кислота Льюиса) орбитали, не занятой электронами:
•р
\з- 3+ /Н F-В-О< р/ И
В соответствии с этим теоретическим рассмотрением сущест-вУют два твердых «кристаллогидрата» трифторида бора; вВз-Н2О (Тпл= 10,18 °C) и BF3-2H2O (7™ = 6,36°C).
H[BF4] — сильная кислота. Во многих случаях благодаря комплексообразованию кислотные свойства соединений бора Усиливаются.
35.10.3. Борная кислота и бораты
Наряду с сродством к фтору бор обладает также очень сильным сродством к кислороду (см. получение элементов из их оксидов алюмотермией в разд. 36.2). Соединения бора с кислородом обычно можно получить путем окисления бора кислородом °здуха. Гидролиз галогенидов бора (ВС13 и др.), в которых
574
Неорганическая химия
бор поляризован положительно, приводит к образованию бор. ной кислоты:
ВС13 + ЗН2О --► В(ОН)з4-ЗНС1
Борная кислота — слабая одноосновная кислота. Она не протолизуется обычным образом в водном растворе. Вследствие координационно-ненасыщенного характера атома бора, она реагирует как кислота Льюиса с присоединением иона ОН-:
В(ОН)3 + Н2О [В(ОН)4]~ + Н+ рК = 9,00
При концентрации борной кислоты 0,02 моль/л в растворе присутствуют группы В(ОН)3 и [В(ОН)4]-. Измерения pH показывают, что при более высоких концентрациях борной кислоты кислотность раствора возрастает. Это связывают с образованием полимерных групп
ЗВ(ОН)3 --► [В3О3(ОН)4]- + Н+ + 2 Н2О p^ = 6,84
Борная кислота столь слаба, что ее водные растворы нельзя с достаточной точностью титровать сильными основаниями. Напротив, соответствующее анионное основание Na2B4O7 (бура) можно титровать сильными кислотами:
В4О72-4-2Н++ 5Н2О ---»- 4Н3ВО3
При титровании одновременно происходит гидролитическое расщепление полимерного аниона В4О72-.
Чтобы иметь возможность оттитровать борную кислоту щелочью, предварительно проводят реакцию комплексообразования с многоатомными спиртами, например с глицерином СН2ОН—СНОН—СН2ОН или маннитом СН2ОН—(СНОН)4— СН2ОН. При этом борная кислота образует комплексную одноосновную алкоксиборную кислоту, р7(==5:
Н2С-ОН НО/ /ОН н^-он + но/ H,i-OH
но-сн2
но-(!:н ->
I
но-сн2
Н2С-ОХ ,0-СН/
I /в\ I НС-о/ \о-сн
_Н2С-ОН но-сн2
+ НаО+ + 2HjO
Увеличение силы кислоты вызывается не образованием эфира, а присоединением кислорода с его несвязанной парой электронов к секстету электронов атомов бора. При взаимодействии борной кислоты с диолом в мольном соотношении 1 : 1 увеличения кислотности не происходит:
- zR о-с< H-o-B^ IR
- I R
О-С\
Катионы тяжелых металлов осаждают из растворов борн°-кислоты нерастворимые соединения, производные не исходи
Бор
575
ортоформы борной кислоты, а ее обезвоженной формы НВОг. Однако ионов BO2_ в этих солях нет. Они образуют два типа кристаллов. В кристаллах орторомбических метаборатов содержатся кольца, включающие три атома бора. В структурах метаборатов имеются также водородные мостиковые связи, так что координационное число бора может быть 3 или 4. Моноклинная форма имеет цепочечную структуру.
Если буру медленно нагревать до 350—400 °C, получается безводный тетраборат натрия, который при дальнейшем нагревании застывает при 878 °C в стеклообразную массу. Расплав буры может растворять ряд оксидов металлов, приобретая характерную окраску (перлы буры).
Рекомендуемые опыты
1. Летучесть и устойчивость BF3. Смешивают В2О3 и CaF2 в мольном отношении 3; 1, помещают шихту в свинцовый тигель и замешивают с конц. H2SO4 (см. разд. 35.8, опыт 9).
1) Немного полученной смеси помещают на платиновую проволоку и держат ее на краю некоптящего пламени газовой горелки: пламя на некоторое время становится зеленым.
2) Остальное количество смеси в свинцовом тигле нагревают до 200 °C, положив на отверстие в крышке смоченную черную фильтровальную бумагу. На нижней стороне бумаги через некоторое время появляется белый осадок.
2. Равновесие фтороборат — гидроксофтороборат. В аппарате для получения газов получают метиловый эфир борной кислоты и его пропускают в приемник с 1 мл раствора, содержащего Mrj(NO3)2, AgNO3 и KF. [Этот раствор готовят так: 2,9 г Mn(NO3)2 и 1,7 г AgNO3 растворяют в 100 Мл воды После добавления 1—2 капель 0,1 М раствора NaOH образуется темный осадок MnO2+Ag, который отфильтровывают К прозрачному нейтральному фильтрату добавляют раствор 3,5 г KF в 50 мл воды, нагревают до кипения и фильтруют.] При пропускании газа в этот раствор происходит гидролиз эфира, а образовавшаяся борная кислота реагирует по уравнению H3BO3-|-4F_—>-BF4“+3 ОН~. Выделяющиеся ионы ОН~ тотчас же осаждают МпО2 и Ag:
Мп2+ + 2 Ag+ + 4 ОН" --> МпО2| + AgJ. 4- 2 Н2О
Осадок будет содержать также KBF4; эта соль изоморфна с КС1О4 и трудно-растворима.
3. Повышение силы кислоты при комплексообразовании. Готовят насыщенный раствор борной кислоты и определяют pH с помощью смешанного индикатора (метиловый красный+метиловый синий). Затем прибавляют равный объем концентрированного нейтрального раствора маннита. Если добавлять такой же объем концентрированного раствора двухатомного спирта, то заметного изменения pH не происходит.
4. Осаждение и гидролиз полиборатов. К раствору Na2B40z прибавляют Раствор AgNO3, выпадает белый осадок. При кипячении осадок становится коричневым, так как белый полиборат серебра гидролизуется до Н3ВО3 с од-°Временным образованием Ag2O.
В4О72-+ 4 Ag++ Н2О ----► 4]AgBO2 + 2H+
2 AgBO2 + 3 Н2О -->• 2 Н3ВО3 + Ag2O|
Вопросы и задания
к I- При реакции ацетилена с борогидридами образуются карбораны. Кано их строение, к какому типу соединений они принадлежат?
Металлы
577
576
Неорганическая химия
2. Объясните изменение акцепторных свойств молекул BF3, ВС13, ВВг3 и В1з как следствие различной доли л-связывания в связях бор — галоген.
3. При каких условиях происходит образование ионов ВС14- и ВВг«-и какова их устойчивость по сравнению с BF4~?
4. В каких реакциях BF3 применяется в качестве катализатора? Объясните механизм действия катализаторов на основе BF3.
5. Сделайте обзор важнейших реакций BF3, ВС13 и ВВг3 с Н2О, С2Н5ОН, R3N, NH3 и ОН'. Рассмотрите соответствующий каждому случаю тип ре’ акции.
6. На основе знаний о типах соединений классифицируйте по типам различные кислородные соединения бора.
7. Постройте диаграммы pH/lg с и отметьте на них возможные точки эквивалентности при кислотно-основном титровании 0,02 М В(ОН)3 и 0,02 М НагВдО?. Какой индикатор надо выбрать, чтобы погрешность титрования не превышала 0,1%.
8. Сделайте обзор различных реакций диборана. Как следует классифицировать диборан в рамках теории жестких и мягких кислот и оснований?
36. МЕТАЛЛЫ
36.1. Переход от неметаллов к металлам
Простые вещества с сильно выраженным металлическим характером— за некоторыми исключениями — кристаллизуются в одном из трех структурных типов (разд. 1.5, 32.3.4.1):
кубическая гранецентрированная решетка, кубическая объемноцентрированная решетка, гексагональная плотнейшая упаковка.
смен мышьяк acvai
Рис. В.37. Структуры селена
(гексагон), мышьяка и алмаза.
Для этих структур характерны высокие координационные числа: 12 — для первого и третьего типов, 14 — для второго типа (с учетом 6 атомов второй координационной сферы, расстояния до которых лишь на 15% больше, чем до ближайших соседей). Значительно меньшие координационные числа встречаются в структурах некоторых элементов IV—VII главных подгрупп (рис. В.37): j
Таблица В.ЗЗ. „Расположение структур металлов и неметаллов
в периодической системе элементов3. Цифры под символами элементов обозначают число ближайших соседей (а также вторую координационную сферу) и межатомное расстояние (в А) в решетках. Числа в рамочках означают отношение двух расстояний.
1д 8:3,04 Be | Ва |С(алма 6:2,14 4:1,54 12:2,281 112:2,52 | | С (граф 3:1,42 » I 6:2,46 3) 1,63 urn) 1,73 N 0 F
Na 8: 3,72 Mg 12:3,20 Al 12:2,86 Si 4:2,35 12:3,84 1,63 P (белы P(чернь 2:2,17 1:2,20 2:3,68 0 1G) 1,68 S 2:2,04 1:3,69 1,81 Cl 1:2,02 2:3,34 1,65
Си 12: 2,56 Zn 6:2,66 6:2,91 Ga 1:2,44 2:?,71 2:2,74 2:2,80 Ge 4:2,45 12:4,00 1,63 As 3:2,51 3:3,15 1,25! Se 2:2,32 4:3,46 1,49 Br 1:2,27 2:3,30 1,45
Ag 12: 2,89 Cd 6:2,97 6:3,29 In 4:3,24 8:3,37 Sn (ce pc 4:2,81 12:4,59 Sn(бело 4:3,02 2:3,17 4:3,76 e) 1,63 e) Sb 2:2,90 3:3,36 1,16! Те 2:2,87 4:3,74 1,31 I 1:2,68 2:3,55 1,32
Аи !2: 2,88 Hg 6:2,99 6:3,45 «-Т1 6:3,40 6:3,45 Д-Т1 8:3,36 Pb 12: 3,50 Bi 3:3,11 3:3,55 1,14! Po 6:3,29 12:4,64
данным работы [Klemm W., Spitzer Н„ Niermann Н. Angew. Chem. 72, S. 985;
(I960)]По
_ & Бор
относят к неметаллам с некоторой оговоркой.
координационное КУЛЫ);
число 1: галогены (двухатомные моле-
координационное число 2: Sp, Se (гексаг.), Те (гексаг.) (це~ ф ’. Свернутые в спирали, параллельные одной из кристаллогра-веских осей, заканчивающиеся кольцами);
37-2027
578
Неорганическая химия
координационное число 3: Р (черный), As, Sb, Bi (слоистые структуры);
координационное число 4: С (алмаз), Si, Ge, Sn (серое).
Во всех перечисленных случаях между соседними атомами 'существуют локализованные гомеополярные связи. Поэтому максимальное количество соседей у одного атома равно числ\ его валентных электронов (см. структуру алмаза). Если число валентных электронов меньше четырех, они не способны к обра зованию локализованных связей*. Стремление к проявлению высоких координационных чисел характерно для структур металлов. Как видно из табл. В.ЗЗ, граница между металлами о высокими координационными числами и «полуметаллами;. с низкими координационными числами проходит через клетк\ «олово». На примере двух его форм («серого» и «белого») мож но проследить переход от неметаллических к металлическим структурам. В то время как серое олово кристаллизуется в ре тетке алмаза (к.ч. = 4), структуру белой модификации можи< рассматривать как тетрагонально искаженную алмазную; к. возрастает до 6 (приближается к металлическому состоянию!! С другой стороны, дрЗ-гибридизация, свойственная структур серого олова, сохраняется даже при значительной деформащ (тенденция к проявлению направленных связей, свойственна; структурам неметаллов). Результаты ряда исследований влияния температуры на структуру полуметаллов позволяют наме тить следующую картину:
а) При повышении температуры происходит переход от неме таллических структур к расплаву, структура которого носит уже более металлический характер. Точка перехода совпадает с Тпл (исключение Sn). Об этом свидетельствуют и болышк значения энтропий плавления. Например, для мышьяка, сурьм-и висмута: 19,7; 22,2 и 21,4 Дж/(К-моль). Поэтому Sb и Bi i жидком состоянии обладают более высокой электропроводке стью, чем в твердом.
б) Уже в твердом состоянии могут подготавливаться измене ния структуры, о чем свидетельствует, например, возрастаю электропроводности (сурьма).
в) Изменения структуры часто происходят при темпераТ рах даже выше ТпЛ- Так, у расплава мышьяка удельная элек' ропроводность растет с температурой (происходит переход металлическим структурам).
При сравнении строения металлических элементов мо»н' сделать следующие выводы: начиная с III периода, металлич^
* Согласно правилу Юм — Розери, в атомных или молекулярных сТР;,'[к турах простых веществ координационное число=8—п, где п — номер ГР' пы. — Прим, перед.
Металлы
579
екая структура образуется только в тех случаях, когда у атомов на внешних s- и р-орбитах находятся один, два или три электрона. Это объясняет также существование большого числа соединений металлических элементов. В случае присутствия четырех или пяти электронов на внешних s- и р-уровнях металлический характер структуры усиливается с возрастанием атомной массы (переход от Sn и РЬ сопровождается появлением кубической гранецентрированной решетки!).
36.1-1- Электронная теория переходных металлов
Атомы переходных металлов характеризуются существованием внутренних незаполненных электронных уровней. Энергия электрона зависит не только от главного квантового числа п, но и от побочного (азимутального); орбитали (n+l)s и (n+1) р оказываются энергетически более предпочтительными, чем nd или nf. Однако не исключено, что для всех элементов свободные атомы имеют в основном состоянии на внешней s-op-
битали два электрона. Один или оба этих электрона могут переходить на d-уровень; например, хром 3d54s, медь 3dI04s, палладий 4d10, платина 5d96s1.
Таким образом, наблюдается «конкуренция» между d- и 5'°рбиталями. Эти переходы объясняются незначительными энергетическими различиями между отдельными состояниями, ак, на переход 4d10—>-4d95s в атоме палладия затрачивается ‘°>2 кДж/моль.
При образовании кристаллической решетки из атомов ме-аЛла квантовые состояния дискретных уровней, наиболее уда-5<ейнЫх от ядра, испытывают заметные изменения. Внешние ’ Р- и d-электроны ведут себя не так, как в изолированных °Мах, Они образуют в первом приближении единую электрон-УЮ систему, обладающую квантовыми состояниями с постояи-37» ___
Металлы
581
580
Неорганическая химия_______________________
ними энергетическими зонами (ср. разд. 6.6.3). Схема располо-жения энергетических зон по Мотту и Джонсу изображена на рис. В.38.
Для ряда переходных металлов и значительного числа сплавов трактовка электронных структур на основе «жесткой» зонной модели приводит к довольно простой картине. Она базиру. >ется на предположении о перекрывании зон, возникших при
Ер Энергия
Рис. В.39. «Жесткая» зонная модель: (п—1) d- и из-зоиы.
— переходный металл (например Ni, Pd, Pt); б — металл с полностью занятой d-зоной (например Си, Ag, Au); EF — энергия уровня Ферми.
расщеплении (п—l)d-, ns- и пр-уровней атома, при этом остальные орбиты не меняются. Схема перекрывания (п—1) d- и rzs-op-биталей приведена на рис. В.39. Из-за частичного заполнения (п—1) d-уровней у переходных металлов граница Ферми проходит в областях (п—l)d- и ns-зон (рис. В.39,а), а у непереходных (Си, Ag, Аи и т. д.) —выше заполненной (п—1) d-зоны в области ns-зоны проводимости (рис. В.39,б). К ним относятся все металлы, у которых не достигается максимальное заполнение d-зон (10 электронов/атом). Согласно такой классификации, металлы I и II побочных подгрупп периодической системы элементов не принадлежат к переходным. Обычно их, как и переходные металлы, называют металлами побочных подгрупп. В табл. В.34 сопоставлены степени заселенности зон некоторых переходных металлов с электронными конфигурациями изолированных атомов. Как же можно объяснить «дробное» число электронов в коэффициенте заселенности зоны? Рассмот-ренная модель предполагает, что атомы в металле обладают различными электронными конфигурациями. У никеля вклад 3d10-, 3d9- и 3d8-cocTOHHHfi равен примерно 55, 35 и 10%.
Появление незаселенных уровней («d-дырок») во внешних d-зонах приводит к ряду особенностей поведения переходных металлов. Часто размеры d-дырок играют при этом решаюшУ1^ роль (ср. способность некоторых переходных металлов абсор' бировать водород, разд. 36.16.1).
• Дальнейшими примерами глубоких изменений квантовых со-
Таблица В.34. Электронные конфигурации и заселенность зон
Элемент Электронная конфигурация Заселенность зоны
Сг ...3d5 4s 3d5>344s°,68
Мп ...3d5 4s2 3d5,S54s1-15
Fe ...3d» 4s2 3d’>0B4s°>8B
Со ...3d7 4s2 3d8,2B4s°,78
Ni ...3d8 4s2 3d9,454s°,BB
Pd 4d9,«45s°,3e
Pt ...5ds 6s 5d9>7 6s°,3
стояний исходных d-оболочек при переходе от свободного атома к твердой фазе могут служить нецелочисленные магнитные моменты атомов, различия в величине моментов изолированных атомов у ферро- и антиферромагнитных d-металлов (Fe, Со, Ni, Сг, Мп), а также аномально высокие значения удельной теплоемкости электронов у d-металлов. В то же время, если как у лантаноидов происходит экранирование внутренних 4 f-орбиталей электронами с более высокими энергиями (5s25p6), часто можно наблюдать «идеальное поведение» атомов. Например, лантаноиды в степени окисления +3 ведут себя и в кристалле как свободные ионы.
Наряду с изучением магнитных свойств важную информацию о деталях электронного строения переходных металлов можно получить с помощью и других методов:
низкотемпературные измерения удельной теплоемкости электронов;
рентгеновские эмиссионные и абсорбционные спектры;
рентгенографическое и нейтронографическое исследование структур;
измерение электропроводности.
Наряду с «жесткой» зонной моделью для чистых металлов и сплавов получила дальнейшее развитие модель электронных структур, отличающаяся разнообразными подходами к соответствующей проблеме.
а) Согласно методу валентных структур (валентных связей), свойства металлов описываются на основе ковалентного связывания двух атомов (разд. 6.2.3).
б) В ряде случаев применение к структуре металлов теории П°ЛЯ лигандов (разд. 6.5.6) приводит к наглядным представлениям об их свойствах. Принцип этого метода можно объяснить На примере металлов, кристаллизующихся в кубической гране-^нтрированной структуре (например, Ni) (рис. В.40).
Расположение атомов металла на грани куба неизбежно приводит к перекрыванию соответствующих dxy-, dxz- и dyz-орби-алей данного центрального атома с теми же орбиталями ато-
582
Неорганическая химия
мов, находящихся в ближайшем окружении на соответствующей грани. Это значит, что три d-орбитали центрального атома,
направленные по диагонали между осями декартовых координат (dXJ/, dxz, dyz), приводят к оптимальному перекрыванию тех же орбиталей 12 ближайших соседей (координационное число атомов в кубической гранецентрированной решетке равно 12). Это V обстоятельство обусловли
Рис. В 40. Кубическая гранецентрированная решетка плоскость (001), перекрывание б/ху-орбиталей.
вает выделение одной энер. гетической зоны (^g-зона), делокализованной у всех атомов в структуре металла.
Иные соотношения существуют для двух орбиталей рассматриваемого атома, ориентированных вдоль
осей координат (dX2-yi, dZ2). Эти орбитали относятся к атомам второй координационной сферы, находящимся в октаэдрическом окружении центрального атома. Вследствие больших рас-
стояний от них до центрального атома — не происходит перекрывания соответствующих орбиталей.
в) К теориям электронного строения сплавов относятся моде-
ли «минимальной полярности» и «виртуально связанных состояний». Согласно первой, каждый компонент сплава обладает той же электронной структурой, что и в свободном состоянии. На этой теории основывается, например, метод определения состава поверхности сплава с помощью селективной адсорбции газообразного субстрата («титрование» поверхности металла). Согласно теории виртуально связанных состояний, d-электроны атомов растворенного металла в матрице (например, Мп в Си) локализуются на них самих. Вследствие резонансного взаимодействия соответствующие d-электроны имеют локализованные, но
расширенные уровни энергии.
Для дальнейшего изучения электронного строения кристаллической решетки металлов необходимо обратиться к специальной литературе.
36.2. Методы получения металлов. Значение «редких» металлов
В основе методов получения металлов (за исключением неко торых специальных случаев) лежат несколько основных типе химических и физических процессов. Поскольку достижению о
Металлы
S83
ределенных, заданных свойств препятствует присутствие разй0' образных примесей, в разделе, посвященном методам получения металлов, рассмотрены некоторые методы их очистки. При этом количественная характеристика степени чистоты не всегда является достаточно простой. Для полупроводниковой техники» ракетостроения и т. д. представляется особенно важным отсутствие определенных примесей (целевая очистка). Например, при получении полупроводникового германия особое вниманИе обращают на удаление элементов, обладающих донорным иЛи акцепторным действием (разд. 6.6.3); металлические рабочИе
I жидкости атомных реакторов, материалы оболочек ядерного гО" ' рючего, графита и т. д. также должны быть свободны от элементов, имеющих высокое сечение захвата тепловых нейтронов (ядерная чистота) и т. д. Было показано, что нарушения струК" туры решетки (например, замещения в ней) вызывают разнообразные эффекты, эквивалентные определенным химическим нарушениям («физические примеси», см. также разд. 33.3.1).
Хотя нельзя провести четкую границу между «химическими» я «физическими» методами, для получения металлов и их соединений используются в основном первые, применение вторых предпочтительно в процессах очистки.
36.2.1. Получение металлов
Для получения металлов и их соединений практическое значение имеют в основном следующие методы:
восстановление оксидов углеродом или водородом;
восстановление оксидов металлами (металлотермия);
восстановление галогенидов или сульфидов водородом ил** металлами;
термическое разложение подходящих соединений (например, галогенидов и карбонилов);
электрохимические методы.
Три последних метода используются также при получений высокочистых металлов.
Наряду с оксидными рудами часто встречаются сульфидный Месторождения металлов. Кроме того, следует упомянуть кар' еоиаты, фосфаты, вольфраматы, молибдаты, а также — особен' н° в случае щелочных и щелочноземельных металлов — их со' Ляиые месторождения (главным образом хлориды).
^2-1.1. Восстановление оксидов углеродом и водородом Сульфидные руды перед восстановлением подвергают тща' льному обжигу, так как присутствие серы исключительно не' агоприятно влияет на металлургические свойства металлов-Л ’ 2MS-f-3|Oj -->- 2 МО + 2 SO2f
Б84 Неорганическая химия
Металлы 585
Затем оксиды восстанавливают углеродом при высоких темпе, ратурах:
С + о2- --► СО + 2е~
Из температурной зависимости изменения энтальпии образов?, ния (разд. 20.2) оксида металла и моноксида углерода определяют область температур, в которой возможно восстановление. Подобные сопоставления можно применять и при восстановде. нии оксидов водородом (см., например, получение германия, мс. либдена и вольфрама) или металлами.
В некоторых случаях сульфиды подвергаются лишь частид. ному обжигу, а затем смесь оксидов и сульфидов нагревав-без доступа воздуха:
2MO + MS ----> ЗМ + SOjf
В лаборатории в качестве восстановителя удобны также цщ-ниды щелочных металлов (см. разд. 33.5), например:
CN* + О2* ---► OCN* + 2е“
As8+ + 3 е~ --► As
Sn1+ + 4e“ --► Sn
36.2.1.2. Восстановление оксидов металлами (металлотермия)
При металлотермии в качестве восстановителей используютс? в первую очередь металлы, оксиды которых даже при высоки' температурах обладают достаточно большой энтальпией образования. Обычно в качестве восстановителей используются магний, алюминий и кальций. Кальций сохраняет свою восстано вительную активность вплоть до самых высоких температур поэтому может восстанавливать оксиды практически всех Mi таллов.
Вопросы и задания
1. Примеси каких элементов не допускаются в германии и кремнии <®>" лупроводниковой» чистоты?
2. Почему нз графита для реакторов полное удаление таких элементов, как бор, кадмий, марганец и кобальт, более важно, чем, например, натрия» магния, алюминия и олова?
3. Что такое «монокристалл»?
4. Путем рассмотрения равновесия С+СО2ч±2СО объясните пренМУ® ства выделения СО нлн СО2 (в качестве продуктов окисления) при во®1 новлеиии оксидов металлов углеродом.
5. Чем ограничено более широкое применение углерода в качестве в стаиовителя (например, при получении молибдена и вольфрама)? р0.
6. Как достигнуть полного восстановления оксида германия (IV) воДОР дом GeO2+2 Н2_»Ge+2 Н2О (AG°=67 кДж/моль)? яе.
7. В каких случаях применение металлов в качестве восстановителей^ возможно, несмотря на то что значения теплот образования позволяют ществить реакцию?
$.2.1.3. Восстановление галогенидов и сульфидов водородом или металлами
faлoгeниды также можно восстановить водородом или металлами:
МХ„ + п/2Нг -> М + иНХ
(Х=С1, Вг, I; М— например, Та, Mo, W)
MlX„ + Мп ---> Mi + МПХ„
(Mj — например, U, Zr, Ti, Si; 'Мп — например, Zn, Са, Mg, К, Na).
Восстановление сульфидов неблагородными металлами находит ограниченное применение (метод осаждения), например:
HgS + Fe ► Hg + FeS
36.2.1.4. Термическое разложение соединений
Следующий «сухой» метод получения металлов — восстановление в процессе термического разложения некоторых их соединений, главным образом галогенидов и наиболее часто иодидов (почему?):
МХп М + п/2Хг
Если процесс осуществляется непрерывно (синтез и разложение происходят в замкнутом сосуде), возможна очистка металла че-Таблица В.35. Равновесные транспортные реакции
Класс веществ Примеры3 Градиенты температур
Простые веще- Zr + 2ГЙ(4Т) Zrl4 (газ) 280 — 1450 °C
ятва (элементы) W + 3C12(6C1) WCle (газ) 400 — 1400 °C
Al + ViAlXg 1,5А1Х(газ) (X = F, Cl.Br.I) 1000 — - 600 °C
Оксиды Fe2O3 + 6HC1 ц—2FeCl3 (газ) 3H2O (газ) 1000 — 800 °C
SiO2 + Sil4 (газ) 4 -b 2SiO (газ) + 41 (газ) 1270 — - 1000 °C
Галогениды VC13 + i/2Cl2 VC14 (газ) 300 — - 250 °C
MoCl3 + MoCls (газ) л—* 2MoC14 (газ) 300 — ► 250 °C
Сульфиды FeS + I3 ч=±: Felj (газ) + VA (газ) 900 — ► 700 °C
^аРбонилы Ni + 4CO Ni(CO)4 (газ) 70 — » 200°C
Полужирным шрифтом обозначено вещество, которое подвергается транспорту.
586
Неорганическая химия
рез газовую фазу (метод Ван Аркеля — Де Бура). Здесь речь идет о «химической транспортной реакции».
К «химическим транспортным реакциям» относятся процес сы, в которых твердое или жидкое вещество А реагирует с газом с обязательным образованием только газообразных продуктов. Последние в другой части системы разлагаются с выделе-нием А:
nA (тв., жидк.) + иВ (газ) + ... qp==t хС (газ) + ...
Таким образом, осуществляется транспорт одного вещества через газовую фазу. В табл. В.35 приведены уравнения некоторых транспортных реакций, протекающих в пространстве с определенным градиентом температур.
Вопросы и задания
1. Что такое коэффициенты переноса при химическом транспорте?
2. Как отделить титан (цирконий) от избытка магния и образовавшегося хлорида магния при получении Ti(Zr) по методу Кролля?
3. Что вы знаете о «карбонильном методе» получения чистых металлов?
36.2.1.5. Электрохимические методы
К электрохимическим методам относятся: простые процессы электролиза (из водных или неводных электролитов), электролиз жидких расплавов и электролиз с образованием амальгам (обменные реакции на границе фаз).
Простой электролиз. Осаждение («цементация») менее активного металла из раствора его соли при действии более активного металла позволяет рассматривать электрохимический процесс как реакцию восстановления (разд. 33.5.1).
Как наиболее целесообразно очистить раствор соли металла от примесей других металлов, менее активных, чем основной компонент?
Электролиз расплавов. Получение ряда металлов осуществляется при электролизе расплавов. Такие методы разработаны не только для натрия, магния и алюминия, но и для «редких» металлов — бериллия, ниобия, тантала, урана, тория и т. д.
Электролиз с образованием амальгам. К методам амальгамной металлургии относятся наряду с другими обменные реакции «на границе раздела фаз», а также электролиз с образованием амальгам. Сущность обмена на границе фаз очень напоминает реакции цементации в водных растворах: если амальгама активного металла входит в соприкосновение с водным рас' твором менее активного металла, первый переходит в раствор^ в то же время эквивалентная часть второго превращается 5 амальгаму (обменная реакция между фазами). 'Докажите обобщение! См. учебники электрохимии!
Применение электролиза с образованием амальгам основы* вается, во-первых, на высоком перенапряжении водорода на ртути (см., например, получение едкого натра, свободного °
Металлы
587
хлора), а во-вторых, на использовании обменной реакции на границе фаз. В последнем случае на катоде выделяется наряду с соответствующим металлом и менее активный. Активные металлы тотчас переходят в раствор в результате обменной реакции между фазами. При анодной поляризации переходят в раствор только активные компоненты, в то же время менее активные остаются в амальгаме.
Рекомендуемые опыты
1. Смешивают очень тщательно — в соответствии с приведенным уравнением реакции — несколько граммов РЬО и PbS в эквивалентных количествах, переносят эту смесь в маленький тигель, иа дне которого находится слой флюса, состоящего из равных частей безводных Na2CO3 и К2СО3. Тем же флюсом засыпают сверху реакционную смесь. Затем тигель нагревают в вытяжном шкафу примерно 45 мни при темнокрасном калении (тяга/). Рекомендуется часто перемешивать реакционную смесь и добавлять флюс. Затем тигель охлаждают, разбивают и обрабатывают водой.
2. В сухую трубочку из термостойкого стекла загружают тесную смесь AsjOj, NaCN и безводной соды, смесь нагревают. Образующийся мышьяк испаряется и осаждается в виде металлического зеркала иа холодных частях трубочки.
3. Проведите реакцию алюмотермии (разд. 50.1.1.3).
4. В тугоплавком реакционном сосуде нагревают эквивалентные количества HgS и порошка железа (по возможности свободного от оксидов) в количествах, соответствующих уравнению реакции. Верхнюю часть сосуда охлаждают влажной тканью. Ртуть отгоняется и частично оседает иа Охлаждаемой стейке сосуда. \Опыт проводится обязательно под тягой, так как пары ртути очень ядовиты!}
5. Проведите «простой электролиз», как описано в «препаративной части» разд. 50.1.2.
Если «простой» электролиз в большинстве случаев проводится в водных растворах, для получения некоторых металлов развиваются методы, связанные с применением неводных растворителей (например, пиридин, жидкий аммиак).
6. Получение лития как пример электролиза расплава (разд. 50.1.2).
Вопросы и задания
1. Используя уравиеиие Нернста, опишите условия проведения электролиза с образованием амальгамы для получения едкого иатра, свободного от хлора!
2. Опишите схему получения высокочистого индия иа основе разделенного катодного и анодного процессов по амальгамному методу!
3. Как можно осуществить коицеитрироваиие разбавленного раствора соли с помощью обмена иа границе фаз?
36.2.2 . Операции очистки
Электрохимические методы позволяют осуществить процессы очистки металлов или получения их соединений. Обычно используется лишь несколько методов. При решении комплексной задачи здесь, как и в случае прямых методов получения, речь ’1Дет главным образом об оптимальной комбинации отдельных ’Иетодов при создании стандартной методики и, в значительно "’еньщей степени, о разработке новых принципов. «Физические» Методы сводятся в основном к следующим.
588
Неорганическая химия
Использование различной летучести отдельных металлов для их разделения
Фракционная перегонка — испарение металлических примесей из матрицы (см. также электронно-лучевая плавка высокоплавких металлов).
Разделение веществ на несколько (обычно две) фаз
Зонная плавка и экстракция растворителем (экстракция жидкость — жидкость, см. также в разд. 38.3.5).
Вопросы и задания
I. Рассмотрите процессы, происходящие прн зонной плавке, с точки зрения диаграммы состояния соответствующей двухкомпонентной снстемк (разд. 26.1).
2. Что такое «коэффициент распределения»?
3. Какие преимущества имеет метод бестнгельной зонной плавки?
4. Как можно проследить за очисткой германия в процессе зонной плавки без отбора проб?
5. Возможно лн разделение водорода и дейтерия методом зонной плавки? Каков вероятный эффект разделения у нуклидов тяжелых элементов?
6. Какие преимущества н недостатки имеет метод плавки во взвешенно» состоянии?
Методы ионного обмена (см. разд. 38.3.7)
Разделение отдельных соединений металлов на основе различия их растворимости (фракционная кристаллизация)
Ввиду неэкономичности этих методов — особенно при малых различиях в растворимости — во многих случаях их применение лишено смысла. Поэтому, например, разделение редкоземельных металлов, ниобия и тантала или циркония и германия наиболее эффективно осуществляется с помощью жидкостной экстракции или ионного обмена.
Вопросы и задания
1. Какая количественная закономерность лежит в основе процесса экстракции растворителем?
2. Каким требованиям должен удовлетворять растворитель, используемый в качестве экстрагента?
36.2.3 . Значение «редких» металлов и их сплавов
Успехи естественных наук, а также развитие технологии прн-водят к непрерывному возрастанию потребностей в таких металлах, которые до последнего времени представляли лишь-
«академический» интерес (рис. В.41).
При рассмотрении периодической системы становится ясныМг что понятие «редкий» металл не следует принимать буквально-К этой группе относятся и металлы, не образующие собственных месторождений, однако содержание которых в земной кор® достаточно велико; и металлы, которые трудно восстановить Д, элементного состояния. Физические и химические свойства П°'
следних часто весьма затрудняют их использование.
Металлы
589
Период 1 Главная подгруппа Подочная подгруппа. Главная подгруппа.
Ф гНе
' 2 6P> 7N Bo 9F IO№
3 t)Na qm9 cAL 15P 16^ 17ci isAr
IA /7А ШБ IV5 V5 VIB VIB VllIB IB ПБ IlIA IVA VA VIA VUA VIHA
--—— 4 )9К 20Ccl 25V 24Cr 25Mn 26Fe 27Co 28Ni 29Cu зо^п 3> 33As 35Br 36Kr
5 38s r 39'И 4tNb wMo «Тс 44Ru i5Rh 47A9 48Cd 49^ 505n 51Sb 52Te 531 54Xe
6 55Ss 56ЙО- 7jTq 74W 75^ 79Au 00^9 yt 82Pb 6jBi B6Rn
7 Ж ..
58-71 Ламикоиды
HCe Pp 59 60Nd 6lpm 62Sm 63^ b4 65Tb 66°У 67H° 68^Г 69 70Vb 71LU
90-103 Актиноиды
90Th|9f₽(1 9ZU 93NP 94Ри|д5Ат)дбСгп 97Bk 98Cf |э9Ез iooFm lolMdlo/i°|l03Lr
Рис. В.41. Периодическая система элементов (редкие металлы и полуметаллы отмечены штриховкой).
Несмотря на исключительно многообразные возможности применения редких металлов и их сплавов, выделим здесь лишь некоторые основные области их применения. Это прежде всего> ядерная техника, где необходимы такие металлы, как бериллий,, ниобий и цирконий и др., в качестве материалов оболочки-ядерного горючего в различных типах реакторов. Эти металлы отличаются малым сечением захвата тепловых нейтронов, высокой твердостью при рабочих температурах, хорошей теплопроводностью, устойчивостью к коррозии и т. д. Галлий и литий предложены, кроме того, в качестве рабочих жидкостей {последний— при условии его отделения от изотопа 63Li (почему?)]. Благодаря свойству значительно поглощать нейтроны гафний, индий и европий используют для изготовления регулирующих стержней. Значительное количество редких металлов потребляет производство стали. Наряду с чистыми легирующими компонентами (например, Mo, V, W, Y) ряд редких и др. металлов Используется в качестве раскислителей (например, редкоземельные элементы, кремний). Для современной авиационной Промышленности и космической техники необходимы жаростой
590
Неорганическая химия
кие конструкционные материалы. В этой области объектами интенсивного исследования и применения являются бериллий, ниобий, молибден, рений, тантал, титан и вольфрам, а также их сплавы. Среди потребителей редких металлов необходимо упомянуть и промышленность полупроводников, использующую галлий, индий и др. — наряду с уже ставшими «классическими» полупроводниковыми материалами — кремцием и германием. Успехи в области электротехнической промышленности, а также радиотехники и электроники были бы невозможны без применения таких металлов, как молибден, рений, платиновые металл селен, цезий и др. С развитием электроники тесно связаны ; пехи в области высоковакуумной техники. Там в качестве сет в электронных лампах используются цезий, ниобий, тантал, г рий, цирконий и др. Затем следует упомянуть еще о широких возможностях применения многочисленных сплавов цветных металлов, а также радиоактивных редких металлов в процессах технологического контроля, в качестве источника нейтронов, в медицине и т. д.
В предлагаемом обзоре приводится лишь краткая сводка важнейших областей применения редких металлов. Ввиду большого значения этих элементов в дальнейшем будет необходимо еще вернуться к этой теме.
36.3. Галлий, индий и таллий
Галлий, индий и таллий относятся к главной подгруппе III группы периодической системы элементов (разд. 35.10). В соответствии с номером группы в своих соединениях они проявляют степень окисления +3. Возрастание устойчивости низших степеней окисления с ростом атомного номера элемента иллюстрируется на примерах соединений индия(III) (легко восстанавливающихся до металла), а также большей прочности соединений таллия(1) по сравнению с производными таллия(Ш). Ввиду того что между алюминием и галлием находится скандий — элемент первого переходного периода — вполне можно ожидать, что изменение физических и даже химических свойств этих элементов будет происходить не вполне закономерно. Действительно, обращает на себя внимание очень низкая температура плавления галлия (29,78°C). Это обусловливает, в частности, его применение в качестве запорной жидкости при измерениях объема газа, а также в качестве теплообменника в ядерных реакторах. Высокая температура кипения (2344 °C) позволяет использовать галлий для наполнения высокотемпературных термометров. Свойства галлия и индия часто рассматривают совместно с алюминием. Так, их гидрооксиды растворяются с образованием гидроксокомплексов (опыт 1) при более высоких значениях pH, чем остальные М(ОН)3. Гидратированные ионы М3+ этой
Галлий, индий, таллий
591
группы (М3+=А13+, Ga3+, 1п3+, Т13+)—кислоты средней силы. Например, даже у соединений галлия(III) и индия(III) в водных растворах легко происходит «протолиз» (опыт 2). Гал-лий(Ш) и индий(Ш) образуют квасцы. Однако химические свойства этих элементов обнаруживают известное сходство с их соседями слева в периодической системе (цинком и кадмием). Свойства соединений таллия близки, с одной стороны, щелочным металлам (ТЮН, Т^СОз и Т13РО4 легкорастворимы, их водные растворы имеют сильнощелочную реакцию), а с другой— серебру [труднорастворимы некоторые галогениды (опыт 3) и сульфид таллия (I) — (опыт 2)].
Ионы Al3+, Ga3+ и 1п3+ относятся к «жестким» кислотам (разд. 33.4.3.4), а Т13+ и TI+ — к группе «мягких» кислот. Такая классификация обусловлена в основном различными размерами ионов (ионные радиусы, нм): А13+ 0,057; Ga3+ 0,062; In3+ 0,092; Tl3+ 0,105; Т1+ 0,149).
Соли таллия (III) являются окислителями:
Т13+ + 31----> ТП + 12
Процесс Т13+-{-2е_^:Т1+ происходит только в сильнокислых растворах (рН<0,3 при концентрации солей таллия 10~2 моль/л). При рН>0,3 осаждается гидроксид таллия(Ш)’
Т1(0Н)3ф + 2е- + ЗН+ «=> Т1+ + 3 Н2О
Е=1,25-|—^-1g-^-. — = 1,25-|~^^-lg -----------------0,09pH
1 2 & а3он~ «Т1+ 1 2 ь ат1+ г
Возрастание устойчивости низших степеней окисления с ростом порядкового номера элемента (почему?) означает, в частности, возможность восстановления цинком ионов индия(Ш) до металла в сильнокислых растворах. (Проведите этот опыт.)
In ч—to>+ + Зе Е° = —0,34 В
Сравните Ga ^... >. Ga3+-f-3e' Е° = —0,53 В
Соли таллия также восстанавливаются до металла цинком, алюминием и магнием.
Почему в бокситах галлий замещает алюминий?
Рекомендуемые опыты
Для опытов используют растворы GaCl3, 1п(НОз)з и T12SO4 или T1NO3. (Внимание! соединения таллия ядовиты!)
1. К растворам солей Ga3+, 1п3+ и Т1+ добавляют по каплям щелочь: выпадают белые осадки гидрооксидов: Ga(OH)3— при pH 3,5* * (Пр=10-35-3,)> 1п(ОН)з — при pH 3,4 (Пр«10“34). Осадки растворимы в избытке осадителя (Ga(OH)3 —при pH 9,74-13, 1п(ОН)3—при pH 14). Очень упрощенно (почему? см. разд. 33.4.2.2) это можно записать с помощью следующих уравнений;
Ga(OH)3 + ОН’ ₽=± Ga(OH)4'
* Данные относятся к осаждению из 0,01 М растворов солей.
•592
Неорганическая химия
-или
Ga(OH)3 + 2HaO 4=* Ga(OH)4-+ Н30+
2. В аммиачный раствор солен галлия пропускают H2S: осаждается ‘Ga(OH)3:
2Ga3+ + 3 S2~ + 6НОН ----> 2 Ga(OH)3| + ЗН2
Пропускают H2S в уксуснокислые растворы соединений индия(Ш) и таллия (I): осаждаются желтый In2S3 и черный T12S (Пр = 10“22).
Оба сульфида легко растворимы в разбавленных кислотах:
2 Т1+ + H2S «=> T12S + 2 Н+ (рД = -1)
3. К растворам солей таллия(1) добавляют растворы NaCl, NaBr и Nal. Труднораствори^ые галогениды очень похожи на аналогичные соли серебра (разд. 36.17.1.3 и 35.5.3.1): Прт1ш = 10~3'8, ПрТ1Вг=10-’-7, ПрТц = 10-Ч Т1С1 нерастворим в NH3, так как ион таллия(I) в водном растворе не образует аммиачных комплексов. С иодом Т1(1) реагирует с образованием поли-лодида таллия (I) Т1 (13).
4. Сильные окислители окисляют таллий до степени окисления +3:
TI Т13+ + 3е- 5° = 0,72 В
Т1+ч=ь Т13++2е- £°=1,25В
Поэтому Т1С1 растворим в конц. HNO3 [с образованием Tl(III)]. Аналогичным образом соли таллия(1) окисляются, например, бромом:
Т1+ + Вга ---► Т13+ + 2 Вг"
Из такого раствора (избыток брома удаляют кипячением) щелочь осаждает (при pH 0,3!) черно-коричневый осадок Т1(ОН)3 (Пр«10-43).
5. Спектральное определение (разд. 37.2.1.2, см. также гл. 40). Легколетучие соединения галлия и индии окрашивают плами бунзеновской горелки в фиолетовый цвет [Лоа=417,2 нм (403,3 нм); %in=451,l нм], а соединения таллия — в зеленый. У таллия наблюдается интенсивная зеленая линия при Ji= 535,1 нм.
Вопросы и задания
1. Предпочтение одной из двух возможных степеней окисления, свойственных элементам данной группы, мы впервые встречаем у тяжелого элемента III главной подгруппы. Обсудите это положение в связи с изменением прочности связей у соединений элементов одной группы. Дайте при этом критическую оценку часто встречающемуся «объяснению», согласно которому по группе с ростом атомного номера увеличивается устойчивость «^электронного состояния («эффект инертной пары»),
2. Известны ли у галлия, индия и таллия соединения в степени окисления +2?
3. В какой мере осаждение Ga(OH)3 в опыте 2 является «протолитическим»? Сравните с гидроксидами алюминия (разд. 36.8), хрома (разд. 36.12.1) и железа (разд. 36.15.1).
4. Почему Т1(ОН)3 осаждается при значительно более низких pH, чем Ga(OH)3 и 1п(ОН)3?
5. С чем связана желтая окраска иодида таллия(I)?
36.4. Германий, олово, свинец
Эти элементы относятся к главной подгруппе четвертой группы периодической системы (см. также разд. 35.8). Способность проявлять степень окисления, соответствующую номеру группы
Германий, олово, свинец
593
здесь тоже падает с ростом номера элемента. У германия, близкого по некоторым свойствам кремнию и олову, наиболее устойчива степень окисления +4; в то же время соединения олова (II) уже вполне устойчивы. Легкость перехода к степени окисления +4 позволяет использовать их в качестве сильных восстановителей. Наконец, у свинца соединения в степени окисления + 2 наиболее устойчивы, а производные свинца (IV) — уже сильные окислители. В гидридах типа МН4 (M = Ge, Sn, Pb) степень окисления элементов 4.
Ионы Sn2+ и РЬ2+ относятся к группе кислот, занимающих промежуточное положение между жесткими и мягкими (разд. 33.4.3.4).
Рекомендуемые опыты
1. Переходы между различными степенями окисления
а) Немного олова обрабатывают разбавленной соляной кислотой:
Sn =₽=> Sn2+ + 2е- £°==—0,136В
Этот раствор по каплям (!) приливают к раствору HgCl2: наблюдается вое* становление ртути (II) (см. разд. 36.17.2.1).
б) К тому же раствору хлорида олова (II) прибавляют по каплям бромную воду. Окраска исчезает:
Sn2+ =?=> Sn4+ + 2е~ Е* = 0,15 В
или
[SnCl3]~ + 3 СИ т—[SnClJ2- + 2е- £° ~ 0
2Вг" г Вг2 + 2е“ Ей — 1,065
Бромную воду следует добавлять только до слабо-желтой окраски.
в) В полученный раствор опускают железный гвоздь с очищенной поверхностью. Олово в степени окисления +4 медленно восстанавливается до Sn2+, что можно установить по восстановительному действию раствора (см. опыт 1.а).
Таким же образом восстанавливают цинком растворы солей олова(II) и олова (IV). Проводят ту же операцию с раствором Pb(NO3)2.
г) Растворяют небольшое количество олова илн свинца в концентрированной азотной кислоте. Образуются SnO2-aq (Пр,,5п(он)4”«Ю-66) или Pb(NO3)2.
д) На раствор РЬ(СН3СОО)2 действуют бромной водой; выпадает черный осадок PbO2-aq (почему?)
е) Немного SnO2 (оловянного камня) нли РЬО2 обрабатывают НС1 (РЬО2 — сильный окислитель):
РЬ2+ + 2Н2О РЬО2+4Н++2е- Ей — 1,47В
Из-за высокого заряда и относительно малого ионного радиуса гидратированные ионы олова(1У) и свинца(IV) — сильные кислоты. Они очень легко •образуют оксокомплексы, которые затем переходят в труднорастворнмые SnO2-aq (pH 0,5) или PbO2-aq (рН<0). (См. опыты 1,д н 1.е.)
ж) Раствор соли олова (IV), полученный в опыте 1.6, нагревают в течение длительного времени, а затем добавляют немного Na2SO< (зачем?)
2. К растворам солей олова(Н) и свинца(Н) по каплям добавляют NaOH. Образующиеся белые осадки «Sn(OH)2> (5SnO-2H2O; Пр» 10“ PH 1,5) и «РЬ(ОН)2» (гидратированный оксид нестехиометрического соста
38—2027
594
Неорганическая химия
ва; ПраНО-15; pH 7,2) растворимы при рН~13 в избытке NaOH (см. также разд. 33.4.2.2):
Sn(OH)2 + ОН" [Sn(OH)3]_
РЬ(ОН)2 + 2 ОН" [РЬ(ОН)4]2-
При кипячении раствора гидроксостаината (II) происходит диспропорционирование (почему?):
2[Sn(OH)3]- ---> P-Sn + [Sn(OH)ep-
Свежеосаждениые SnO2-aq или PbO2 — как аитиосиоваиия (разд. 35.4.3.2)—растворимы в сильных щелочах с образованием соответствующих гексагидроксокомплексов.
3. Из слабосолянокислых растворов соединений олова (II) и олова (IV), а также раствора Pb(NO3)2 осаждают сероводородом сульфиды:
, SnS (коричневый) Пр » 10-28
SnS2 (желтый) Пр ~ 10-70
PbS (черный) Пр « 10~28
Если раствор свинцовой соли содержит галогенид-ионы, из него осаждается галогенсульфид свинца (по аналогии, например, Hg3S2Cl2).
Олово в степени окисления +4 образует тиостаинаты—[SnS3]2-. Поэтому SnS2 растворим в (NH4)2S, a SnS — только в (NH4)2SX (реакция окисления) :
SnS + 2 S2- [SnS]32- + 2е~
GeSs (белый) осаждается H2S только из очень сильиокислых растворов (как и мышьяк, в степени окисления +5). С (NH4)2S он образует тиосоль [GeS3]2-, из которой сульфид может быть осажден только в сильнокислой среде.
4. К раствору Pb(NO3)2 по каплям добавляют НС1, KI и конц. H2SO4. Осадки РЬС12 (белый, Пр«10~4-8) и РЬ12 (желтый, Пр« 10~7>9—8Л) легко растворимы при нагревании. Поэтому осаждение следует проводить из холодных растворов, а в случае РЬС12—из-за его значительной растворимости— только из концентрированных растворов.
РЬС12 и РЫ2 растворимы при добавлении галогенидов с образованием галогенокомплексов: [РЬС13]~ (pAj = l,4), [РЫ3]_ (p/G=5,4)*.
PbSO4 (белый, Пр= 10-7>7~8’0) образует с конц. H2SO4 кислоту H2[Pb(SO4)2]; сульфат свинца заметно растворим и в горячей соляной кислоте.
Вопросы и задания
1. Рассмотрите изменение химических и физических свойств по этой группе элементов. Сравните энергии ионизации для переходов М—>-Мп и Мп—->M,V.
2. Почему соли олова (II) в солянокислом растворе являются более сильными восстановителями, чем в сернокислом?
3. Какие ионы находятся в растворе GeCl4 в конц. НС1?
4. Что такое кассиев золотой пурпур?**
5. Объясните различия в свойствах железа и цинка в качестве восстановителей олова (IV).
* Если не оговорено специально (рЛ), рКа и т. д.), в таких случаях речь идет о суммарной константе диссоциации — рКо.
** Имеется в виду качественная реакция открытия золота по интенсивной окраске его коллоидного раствора, образующегося при восстановлении Au(I) хлоридом олова(II). — Прим, перев.
Мышьяк, сурьма, висмут
595
6. Как ведут себя соединения германия в (серно)кислом растворе по отношению к магнию, алюминию и цинку?
7. Из какого сырья получают сурик?
8. Объясните, почему не существует РЬВг4 и РЫ4.
9. Почему водный раствор желтого иодида свинца бесцветен?
10. Обсудите образование [SnSb2- из SnSs+Ss-, используя концепцию основание—антиоснование (фрайбергский способ вскрытия-, разд. 37.2.1.4).
35.5. Мышьяк, сурьма, висмут
В отличие от азота и фосфора (разд. 35.7) мышьяк, сурьма и висмут входят главным образом в состав катионов.
Относительно необычные катионы Sbg2+ и Sbi2+ образуются при окислении раствора сурьмы фторосерной кислотой и персульфатом калия или пероксодисульфурилдифторидом S2O6F2.
По аналогии с предшествующими элементами главных подгрупп у рассматриваемых элементов с ростом атомного номера падает устойчивость высшей ( + 5) степени окисления. Так, соединения мышьяка и сурьмы в степени окисления +5 проявляют окислительную активность в кислых растворах (опыт 1.е); соединения висмута в той же степени окисления [например, BiP5, Li3BiC>4, NagBiOs, Ba7(BiOe)2] —особенно сильные окислители (опыт 1.з).
Ионы Bi+ были обнаружены с помощью рентгеноструктурного анализа впервые в гексагональных кристаллах соединения BiioHf3Cli8. Наряду с катионами висмута в степени окисления + 1 в его структуре существуют ионы Big5+ и [HfCl6]2~. Из гидридов этих элементов (AsH3, As2H2, As2H(?), SbH3, BiH3, B12H2) получили применение AsH3 и SbH3 благодаря своей доступности, значительной летучести, а также малой термической устойчивости. Последнее качество лежит в основе метода определения малых количеств мышьяка и сурьмы («проба Марша», опыт 1.а).
Аналитические реакции элементов определяются возможностью осаждения сульфидов, склонностью мышьяка и сурьмы к изменению степени окисления, а также изоморфизмом между фосфатами и арсенатами.
Рекомендуемые опыты
Растворимые соединения мышьяка и сурьмы ядовиты!
1.а) Проба Марша. В большой реакционный сосуд помещают несколько гранул цинка (свободного от мышьяка, слепой опыт!) и несколько капель раствора CuSO4 (зачем?); затем вводят вещество, испытываемое иа присутствие мышьяка или сурьмы, и разбавленную серную кислоту (реакция?). Сосуд закрывают пробкой, в которую вставлена стеклянная трубка, оттянутая в Капилляр. После вытеснения воздуха из реакционного сосуда (проба на гремучий газ) выделяющийся водород поджигают. В присутствии мышьяка (или сурьмы) он горит бледно-голубым пламенем (реакция?). Если внести в пламя фарфоровую чашку с холодной водой, мышьяк и (или) сурьма осаждаются в виде черного зеркала. (Гидриды очень ядовиты, поэтому опыт проводят под тягой!)
38*
596
Неорганическая химия
Чтобы отличить зеркало мышьяка от сурьмы, к нему надо прикоснуться раствором NaOH — Н2О2 (реакция?).
Аналогичными мышьяку свойствами обладает и германиевое зеркало, которое может быть получено таким же образом.
б) В концентрированной соляной кислоте растворяют немного As2Os или NasAsO4 и добавляют раствор хлорида олова (И). Происходит восстановление до металла («проба Беттендорфа», при очень слабом нагревании):
2 As3+ + 3 Sn2+ --> 2As + 3Sn4+
5п4+ + 6СГ ----► [SnCl6]2-
в) Аналогичная реакция происходит в растворе соли сурьмы (Ш) при действии очищенной железной проволоки:
2Sb3++3Fe -----> 2Sb| + 3Fe2+
г) Висмут в степени окисления +3 в щелочном растворе тоже можно восстановить раствором соли олова(II) (раствор не нагревать! см. разд. 36.4, опыт 2):
[Sn(OH)3]“ + 3 ОН" [Sn(OH)6]2- + 2е~ Е° = —0,96 В
3[Sn(OH)3r + 2Bi(OH)3 + 3OH- ------- 3 [Sn(OH)6]2" + 2 ВЦ
д) В щелочном растворе мышьяк и сурьма в степени окисления +3 — сильные восстановители:
AsO2* + 4 ОН" AsO43- + 2 Н2О + 2е~ Е° = —0,7 В
SbO2- + 2ОН- -и—>- [SbO3]- + Н2О + 2е~ Е° = — 0,59 В
Окислительно-восстановительный переход следует представлять следующим образом (ионы SbO3_ в растворе практически отсутствуют):
[SbO2J- + 5 ОН- [H3SbO6]4“ + Н2О + 2е- £°восст = -0,4 В
К щелочному раствору арсената или антимоната натрия прибавляют аммиачный раствор соли серебра и нагревают: выделяется серебро.
е) В кислой среде арсенаты и антимонаты, напротив, сильные окислители:
! H3AsO3 + Н2О H3AsO4 + 2 Н+ + 2е~ Е° = 0,56 В
2SbO+ + 3H2O Sb2O3-aq+6Н++4е“ F«=0,58B
[ср. Н3РО3 + Н2О Н3РО4 + 2 Н+ + 2е~ Е° = -0,28 В]
К кислому раствору соли мышьяка(Ш) или сурьмы(Ш) прибавляют немно-го раствора иода. Окраска не меняется. После добавления твердого NaHCOj трехвалентные мышьяк и сурьма окисляются (исчезновение окраски! почему нельзя добавлять Na2COs?) При подкислении этого раствора иод опять выделяется в свободном состоянии:
[AsO3]3- + I2 + Н2О [AsO4]3- + 2 Г 4- 2 Н+
Sb3+ + 12 + 6 Н2О [Sb(OH)6]" + 2 Г + 6Н+
ж) При окислении арсенита меняется pH: к раствору арсенита добавля-ют несколько капель фенолфталеина (табл. В.6) и разбавленный раствор серной кислоты (чтобы уменьшить интенсивность малиновой окраски). Затем добавляют по каплям раствор иода. При этом окраска индикатора исчезает.
з) К нескольким каплям раствора MnSO4 добавляют 1 мл конц. HNO* и на кончике шпателя висмутат натрия NaBiOa; смесь встряхивают. Мп на холоду окисляется до МпО4~ (разд. 36.14 1). В случае необходимости добавляют небольшое количество воды, а затем центрифугируют:
Bi3++3H2O НВ1О3 + 5Н+4-2е Е3«1,7В
Натрий, калий, литий
597
2. Растворяют немного As2O3, Na3AsO4, Sb2O3, K[Sb(OH)3] или Bi(NO3)s в соляной кислоте средней концентрации и пропускают H2S. Выпадают осадки желтого сульфида мышьяка As2S3 (Пр аг 1 б-25), As2Ss (или As2S3+S; почему?), оранжево-красные сульфиды сурьмы Sb2S3 (Пр«10-93), Sb2Ss (или Sb2S3+S), а также коричнево-черный Bi2S3 (Пр«10-96) (испытайте растворимость сульфидов в конц. соляной кислоте!).
Мышьяк и сурьма образуют в водных растворах тиосоли типа [MlnS3]s“‘ и [MVS4]3-. Поэтому рассматриваемые сульфиды растворимы в [NH4]2S. Соответствующие тиокислоты H3[MinS3] и H3[MVS4] в свободном состоянии неустойчивы, поэтому при подкислении растворов тиоарсенитов (антимонитов) и арсенатов (антимонатов) сульфиды снова выпадают (см. также разд. 35.6.3.1).
Аналогичным образом испытайте отношение отдельных сульфидов к ще-лочгм и NH3! Какие реакции происходят?
3. Близость свойств ионов [AsO4]3~ и [РО4]3* (см. разд. 35.7.3.3 и 36.7,. опыт 2) демонстрирует образование осадков состава: Ag3AsO4 (шоколаднокоричневый; Пр= 10-22), Mg[NH4]AsO4-6 Н2О (белый), [NH4]3[AsO4(Mo3Og)4] (желтый).
К кислому (но не солянокислому!) раствору Na3AsO4 прибавляют раствор AgNO3 и нейтрализуют его NH3 (по каплям); выпадает осадок Ag3AsO4. Испытайте его растворимость в щелочах и аммиаке (реакции?).
Вопросы и задания
1 . Что происходит при открытии мышьяка методом Гутцайта*?
2 В какой степени элементы мышьяк, сурьма и висмут способны к образованию свободных (гидратированных) ионов М111?
3 Приведите примеры соединений, в которых мышьяк, сурьма и висмут имеют координационное число 6.
4 . Как получить достаточно чистые препараты As2S5 или SbjSj? Что такое сурьмяный блеск (антимонит)?
5 Какое соединение образуется при растворении As2S3 (или Sb2S3) в. [NH4]2Sx (окислительно-восстановительный переход, реакция основание — антиоснование!).
36.6. Главная подгруппа первой группы
В главной подгруппе первой группы периодической системы находятся литий, натрий, калий, рубидий, цезий и франций. В соответствии с номером группы в своих соединениях (в большинстве случаев ионных) они проявляют всегда степень окисления + 1. Чисто ковалентное о—о-связывание имеет место в газообразных молекулах Na2, К2 и т. д. Эти элементы — самые «неблагородные». Их стандартные потенциалы порядка от —2,7 До —3,0 В (ср. табл. В. 14). Ионные радиусы сопоставлены в табл. А. 16. Обращает на себя внимание тот факт, что при переходе от натрия к калию изменение радиусов оказывается большим, чем в следующем за ними ряду элементов К—Rb—Cs (почему?). Это обстоятельство является главной причиной отличия свойств натрия от его более тяжелых аналогов. С учетом этого становится понятной аналогия в свойствах соответствующих соединений калия, рубидия и цезия. Особо следует под
* После восстановления его в реакции Марша. — Прим, перев.
.'598
Неорганическая химия
черкнуть небольшой радиус лития — атомный и ионный, что приводит к аналогии свойств этого элемента с магнием (диагональное сходство).
Совершенно естественно, что ион Li+ (наряду с ионами Na+ и К") относится к группе жестких кислот (разд. 33.4.3.4).
36.6.1. Натрий
Большинство солей натрия легко растворимы. Для его открытия, используются микроаналитические реакции осаждения в виде натрийуранилацетата NaUO2(CH3COO) 3, натриймагний-. уранилацетата NaMg(UO2)3(CH3COO)9-9H2O и натрийвисмут-сульфата 3Na2SO4-2Bi2(SO4)3-2H2O.
36.6.2. Калий (рубидий и цезий)
Химические свойства рубидия и цезия совершенно аналогичны калию; то же относится и к соответствующим - их соединениям. Растворимость солей калия, рубидия и цезия меняется незначительно и постепенно. NH4+-hoh часто мешает аналитическим реакциям осаждения ионов К+, Rb+ и Cs+ (почему?). Из труднорастворимых солей калия — КС1О4, K2Na[Co(NO2)6], KsfPtCle] (гл. 36.16, опыт 3), тетрафенилборат калг ' К[В(С6Н5)4] и гидротартрат калия КН (С4Н40б)первые дг годятся для аналитического определения. Применение H2[PtCl( жак осадителя для ионов калия ограничивает высокая стоимосг. платины; осаждение калия в виде К[В(С6Н5)4] служит преимущественно для количественного определения; КН[С4Н40б] — часто образует пересыщенные растворы и легко растворим как в кислотах, так и в щелочах (осаждение проводят при pH 3,4—3,6).
О специфических реакциях осаждения ионов рубидия и цезия см. специальную литературу. Лучше всего рубидий и цезий открывать методом спектрального анализа.
.36.6.3. Литий
Образование труднорастворимых фосфатов, фторидов и кар-'бонатов позволяет рассматривать литий как металл, свойства которого промежуточны между свойствами щелочных и щелочноземельных (диагональное сходство) элементов.
Растворимость ряда соединений лития в органических (преимущественно кислородсодержащих) растворителях также аналогична солям некоторых щелочноземельных металлов.
Рекомендуемые опыты
1. а) На предметном стекле соединяют каплю не очень разбавленного -раствора соли натрия и каплю раствора ураиилацетата (насыщенный раствор .иО2(СНзСОО)2 в ~ 10%-ной уксусной кислоте). Через короткое время поД
Натрий, калий, литий
599»
микроскопом наблюдается кристаллизация светло-желтых тетраэдров натрий-ураннлацетата. Определению мешают фосфаты, ферроцианйды, а также мио* гне катионы металлов. Значительные количества солей аммония, а также из* быток кислоты должны быть удалены прн упаривании. Ионы калия мешают только в значительных количествах.
б) Хлоридсодержащий раствор соли натрия осторожно упаривают досуха, добавляют каплю разбавленной H2SO4 н еще раз упаривают. Затем к препарату прибавляют каплю 1 М H2SO4 и 1%-иый раствор Bi(NO3)3. При этом образуются гексагональные столбики гидрата натрийвисмутсульфата.
Ионы К+ в тех же условиях дают шестиугольные таблички двойной соли аналогичного состава. Открытию натрия этим методом не мешает присутствие ионов Li+ и Mg2+.
Раствор нитрата висмута, используемый в этой реакции, получают при растворении 1 г Bi(NO3)3 в минимальном количестве 2 М HNO3 (при нагревании) и дальнейшем разбавлении дистиллироваииой водой до 100 мл.
2. Спектральный метод качественного определения отличается большой чувствительностью, причем не существенно, является ли натрий основным компонентом пробы (разд. 37.2.1.2).
3. а) К раствору соли калия добавляют несколько капель 60—70%-ной хлорной кислоты НС1О4: выпадает белый осадок КС1О4. Под микроскопом видны ромбические кристаллы с сильным двулучепреломлением. 1ЧН4+-ноны мешают только в значительном избытке, так как NH4CIO4 растворим значительно лучше Осадок нагревают с достаточным количеством воды (?).
б) Несколько миллилитров раствора соли калия слегка подкисляют слабой уксусной кислотой, затем прибавляют свежеприготовленный раствор гек-санитрокобальтата(Ш) натрия (разд. 36.15.2, опыт 11): образуется желтый осадок K2Na[Co(NO2)e]. Соли аммония должны быть предварительно удалены; соответствующие соли рубидия и цезия тоже плохо растворимы.
4. Соли калия окрашивают пламя бунзеиовской горелкн в фиолетовый цвет; аналогичное окрашивание вызывают летучие соли рубидия и цезия. О спектральном определении элементов см. разд. 37.2.1.2.
Полоса поглощения, им
К 768,2 (красная) и 404,5 (фиолетовая)
Rb 780 (красная) и 421,5 (фиолетовая)
Cs 456—459 (красио-фиолетовая)
5. На раствор L1C1 действуют
a) Na2HPO4 — NaOH (иагреваиие);
б) NaF;
в) Na2CO3.
Образуются трудиорастворимые: Li3PO4, LiF и Li2CO3:
3Lt+ + НРО42- LI3PO4} + Н+ Пр = Ю-12.6- '
Li+ + F" ----- LiF|
2Li+ + CO32~ ---->- Li2CO3| Пр =10-°,5
6. Летучие соли лития вызывают интенсивное карминово-красное окрашивание пламени бунзеиовской горелки (линии: 670,8 нм, красная н 610,3 нм. Желто-оранжевая).
Вопросы и задания
1. Напишите уравнение реакции гидрида лития с водой (разд. 35.1.1.1).
2. Объясните, к какому типу соединений относятся гидроксиды щелочных металлов.
3. Почему большинство солей щелочных металлов легко, растворимы в> воде? В этой связи оцените также величину радиуса иона [Li-aq]+.
ФОО
Неорганическая химия
4. Почему ионы элементов первой главной подгруппы имеют слабую тенденцию к комплексообразованию?
5. Какие продукты образуются после сгорания всех металлов от лития до цезия? В чем причина того, что содержание кислорода в иих возрастает?
6. Какое влияние на тип структуры хлоридов MCI оказывает возрастание радиуса соответствующего иона М.+?
36.7. Главная подгруппа второй группы
Во второй главной подгруппе находятся элементы от бериллия до радия. Во всех своих устойчивых соединениях они проявляют степень окисления +2, причем образуют только бесцветные ионы [M-aq]2+. Ионы Ве2+, Mg2+, Са +, а также Sr относятся, по классификации Пирсона, к жестким кислотам (разд. 33.4.3.4); поэтому с жестким основанием НгО они образуют устойчивые кислотно-основные комплексы типа [М(Н2О)„р+.
Реакции, связанные с растворением щелочноземельных ме-таллов в их галогенидах, ни в коем случае не приводят к образованию ионов М+.
Весьма интересен в этой группе скачок от малых атомных и ионных радиусов первых членов (Be и Mg) к более тяжелым аналогам (Са, Sr, Ва, ср. табл. А.16). В этом заключается одна из существенных причин различий свойств бериллия и магния по сравнению с кальцием, стронцием и барием. Характер изменения физических констант свидетельствует об особом положении кальция. Он обладает более высокими температурами плавления и кипения, а также более высокой энтальпией испарения, чем его аналоги — магний и стронций. Это объясняется возрастанием энергии связи в решетке металла, так как у кальция впервые становятся вакантными Зс?-орбитали. В результате происходит перекрывание эффекта обычного уменьшения этих величин с ростом атомного радиуса. Барий плавится ниже, а кипит при более высокой температуре, чем стронции. Вследствие большей атомной массы бария для перехода его атомов в расплав требуется более высокая энергия, чем в случае стронция (несмотря на то что в расплаве они, вероятно, связаны менее прочно, чем атомы стронция).
Химия бериллия, соединения которого в основном ковалентны (разд. 36.7.2), очень напоминает химию алюминия (диагональное сходство). С другой стороны, меньшие различия ионных радиусов кальция, стронция и бария очень часто обусловливают общность реакций этих элементов. Меньший радиус иона Mg служит, например, причиной значительной растворимости суль фата (большая энергия гидратации иона Mg2+), малой римости гидроксида (деформация поляризуемого иона Оп ) и низкой температуры разложения карбоната магния по сРа лению с карбонатами кальция, стронция и бария (сильная Д
Магний, кальций, стронций, барий, бериллий
601
формация карбонат-иона, приводящая к легкому распаду его на: СО2 и О2-; ср. также разд. 33.8).
Образование иона ![Mg(H2O)6]2+ объясняет очень слабокислую реакцию водного раствора соли магния:
[Mg(HaO)„]2+ ^=±: [Mg(HaO)„_1OH]++H+
Ход растворимости соединений кальция, стронция и бария часто демонстрирует четкую зависимость от размера иона (примеры!). С другой стороны, существуют и «исключения», так как. причины растворимости вещества весьма многообразны.
36.7.1. Магний, кальций, стронций н барий
Рекомендуемые опыты
Для опытов используют растворы хлоридов.
1. а) Кипятят растворы хлоридов с несколькими миллилитрами раствора. (NH^sCOs (зачем?). Выпадают карбонаты МСО3 (М=Са, Sr, Ва), а также-основной карбонат магния (все белые).
б) Опыт повторяют в присутствии NH4CI. Магний в противоположность Са2+, Sr2+ и Ва2+ не осаждается (почему?) и может быть отделен от этих элементов.
MgCO3 СаСО3 SrCO3 ВаСО3
(тригидрат) кальцит арагонит
Пр~10-5 10-8'3 10-8’2 10—9.0—10,0 10-8,3-8,8
2. Осаждение фосфатов имеет аналитическое значение главным образом» для иона Mg2+ (разд. 37.2.1.8.6). К слабоуксуснокислому раствору Mg2+ прибавляют немного NH4C1 и NaHPO4, а затем по каплям (!) разбавленный раствор NH3 — до слабого помутнения, предшествующего выпадению кристаллического осадка MgNH4PO4-6H2O; Пр=10-12'6. Затем добавляют избыток NH3. (Почему не проводят осаждение сразу в сильно аммиачной среде?) Если образуется некристаллический (слипшийся) осадок, его растворяют в разбавленной уксусной кислоте и повторяют осаждение. Осадок рассматривают под микроскопом.
3. Осаждение кальция, стронция и бария [при добавлении (NH4)2C2O4] в виде оксалатов МС2О4 находит аналитическое применение только у кальция,, так как СаС2О4 (наряду с CaF2) относится к наименее растворимым солям., кальция.
MgC2O4 СаС2О4 SrC2O4-H2O ВаС2О4-2Н2О ВаС2О4 Пр 10-4-1 10-8'1 10-’.2 10-’-8 10-6-8
Проводят осаждение и изучают растворимость осадков в разб. НС1 и уксусной кислоте!
Так как щавелевая кислота по своей второй константе является слабой: кислотой (pKi=l,42, рК2=4,21), все оксалаты легко растворимы в разбавленной НС1; СаС2О4 нерастворим в уксусной кислоте (SrC2C>4 и ВаС2СЦ в-зависимости от концентрации уксусной кислоты частично или полностью растворимы в ней).
Различия в растворимости сульфатов и хроматов иллюстрируют опыты 4 и 5.
4. а) К 0,1 М растворам хлоридов добавляют по несколько капель разб. H2SO4. Тотчас выпадают осадки SrSO4 (Пр=10_6-6) и BaSO4 (Пр = 10-1(,>0)-(оба белые; у кальция осаждение происходит только из концентрированных растворов (Прсазо4*2н2О =10-4'6, см. также разд. 33.3.2).
«02
Неорганическая химия
б) Готовят насыщенный раствор CaSO4 (свежеосажденного и промытого), т. е. «гипсовую воду» ~0,25%-ный раствор CaSO4. К нескольким миллилитрам этого раствора прибавляют раствор SrCl2 или ВаС12. Выпадают суль->фаты бария (тотчас) и стронция (медленно!), и наблюдается сильное помутнение. Из щелочноземельных хроматов ВаСгО4 наименее растворим (Пр$гСго4 = 10-М; ПрваСгС4 = Ю-’.7).
Равновесие (разд. 36.12, опыт 7)
Сг2О72~ + Н2О 2СгО42- + 2Н+
•зависит от pH, поэтому осаждение StCtO4 не происходит, а барий осаждается в виде ВаСгО4. И даже из кислого раствора происходит лишь частичное соосаждение SrCrO4 вследствие сокристаллизации.
5. К раствору ВаС12, содержащему ацетатный буфер, добавляют при нагревании раствор К2СгО4; выпадает желтый осадок ВаСгО4. Он легко растворим, например, в НС1, так как хромовая кислота, устойчивая только в водном растворе, — кислота средней силы (рК, = 0,7; рК2=6,4). Осаждение SrCrO4 происходит только в слабоаммиачном растворе.
6 Открытие методом спектрального анализа: для качественного определения Са, Sr и Ва хорошо подходит спектральный анализ (разд. 37.2,1.2). Легколетучие соли этих элементов вызывают характерное окрашивание пламени бунзеиовской горелки: Са — кирпично-красное, Sr — карминово-красное, Ва — от светло-зеленого до зеленого (синяя линия Sr 460,7 нм проявляется очень редко).
36.7.2. Бериллий
Малый атомный радиус бериллия (в сравнении с радиусом элементов-аналогов и лития), а также его более высокий потенциал ионизации придают ему слабо электроположительный характер. Так, практически во всех соединениях бериллия связи имеют в большей или меньшей степени ковалентный характер. На химические свойства бериллия значительно большее влияние, чем в случае магния, оказывает малый ионный радиус Ве2+, который оценивается примерно в 0,03 нм. Так, соли бериллия имеют значительно более кислую реакцию, так как гидратированный катион бериллия является кислотой (разд. 33.4.4):
[Ве(Н2О)4]2+ [Ве(Н2О)3(ОН)]+ + Н+
Промежуточное осаждение Ве(ОН)2 демонстрирует опыт 7.
Рекомендуемые опыты
1. К раствору ВеС12 приливают растворы аммиака (или NH3/NH4C1), NaOH (по каплям) и (NH4)2S. При pH 5,8 выпадает белый осадок Ве(ОН)» (Пр«10“м~26), растворимый в избытке едкого натра (при pH 5= 13,5); очень схематично можно представить образование растворимых ионов [Ве(ОН)+]л при незначительном избытке гидроксид-ионов:
Ве(ОН)2 + 2 ОН’ [Ве(ОН)4]2-или
Ве(ОН)а + 2 Н2О [Ве(ОН)4]2- + 2 Н+
При малых концентрациях Ве2+ речь идет об образовании оксо- и гидро-ксокомплексов с мостиками Be—О—Be и Be—ОН—Be (разд. 33.4.2.2). В присутствии тартрата натрия-калия комплексообразование препятствует •осаждению гидроксида.
Алюминий
60S
Вопросы и задания
1. р/( кислотной диссоциации ионов [М(Н2О)„]2+ (разд. 33 4) в соответствии с уравнением
[М(НаО)л]2+ =F=t [М(Н2О)„_1(ОН)]+ + Н+
составляет
Mg Сз Sr Ва
р/С 11,5 12,6 13,2 13,3
Сравните диссоциацию, например, Sr(OH)2 и Ва(ОН)2 с Mg(OH)2.
2. Объясните промежуточное положение Mg2+ по сравнению с элементами-аналогами, привлекая отношение заряд/радиус для ряда Be—Ra.
3. Какими простейшими реакциями можно различить MgNH4PO4-6 Н2О и MgNH4AsO4-6H2O?
4. Объясните осаждение СаНРО4 при рН~7, исходя из значений кон-стант диссоциации фосфорной кислоты!
5. Как отделить радий от элементов, сопутствующих ему в урановой смоляной руде (уранините) ?
6. Можно ли осадить сульфат бария насыщенным раствором SrSO4?
7. Объясните изменение растворимости по ряду соединений элементов-аналогов из хода соответствующих энергий гидратации и энергии решетки!
36.8. Алюминий
В соответствии с положением в периодической системе основная степень окисления алюминия +3. В так называемых «субсоединениях» алюминий проявляет степень окисления +1 (заполненный Зз2-уровень). Эти соединения (А1Х, X=F, Cl, Вг, I; АЬУ, Y=O, S, Se, а также A1O) образуются при высоких температурах при реакциях, обратных диспропорционированию:
2 А1 + А1Х3 (газ) ц.. .А 3 А1Х (газ)
Или
4 Al + А12У3 (газ) 3 A12Y (газ)
При выделении из газовой фазы «субсоединения» диспропорцио-нируют на алюминий и соответствующее соединение А1 (1П> (малая стабильность «2-электронного состояния!). «Химический транспорт» этих «субсоединений» через газовую фазу (разд. 36.2.1.4) открывает возможность термического получения алюминия, удобного для технологии.
Несмотря на то что стандартный потенциал (А1«±А13++Зе-) составляет —1,67 В, алюминий очень устойчив к коррозии, так как на воздухе и в воде моментально покрывается очень прочной и плотной пленкой оксида алюминия AI2O3. Если эта пленка покрывает не всю поверхность, происходит окисление алюминия (опыт 1). Для повышения коррозионно- и износоустойчивости алюминия методом анодного окисления на нем создают Плотный слой А12О3 толщиной 10—30 мкм. В декоративных целях этот слой можно окрасить органическими красителями.
<04
Неорганическая, химия
Гидратированный катион алюминия (координационное число 6), как и катионы более тяжелых элементов-аналогов в степени окисления +3 (разд. 36.3), — кислота средней силы:
[А1 (Н2О)в]3+ [А1(Н2О)5(ОН)]2+ + Н+ рА = 4,85
А1(ОН)3 рассматривать как ангидрид кислоты, так как при действии сильных оснований он может присоединять ОН- с образованием комплексного иона; упрощенно это можно выразить уравнением:
А1(ОН)3 + ОН" [А1(ОН)4Г
Более правильно говорить об образовании [А1(Н2О)2(ОН)4]-. «Формально А1(ОН)3 реагирует с водой как ангидрид кислоты {обсудите величину рК):
А1(ОН)3 + Н2О [А1(ОН)4]- + Н+ рА = 12,4
Кроме того, существует и другое равновесие:
А1(ОН)з + 6Н3О ч-=> [А1(Н2О)е]3+ + 3 ОН-
При добавлении минеральных кислот свежеосажденный гидроксид алюминия растворяется, так как равновесие сдвигается вправо в результате нейтрализации ионов гидроксила. Если осадок гидроксида оставить на длительное время или проводить осаждение при нагревании, он хуже растворяется в щелочах и кислотах (?).
Поскольку А1(ОН)3 — значительно более слабое основание, чем NH3, последний осаждает гидроксид из растворов солей алюминия. Ионы алюминия(III) не образуют аммиачных комплексов, поэтому некоторая растворимость А1(ОН)3 в большом избытке концентрированного NH3 обусловлена образованием «алюмината».
В присутствии солей аммония не происходит растворения •гидроксида алюминия, так как [NH4] +— гораздо более сильная кислота, чем [А1(ОН)4]р (для NH4+ рКа=12,4):
[NH4]+ + [А1(ОН)4]- А1(ОН)з( + NH3 + Н2О
Гидроксид алюминия в зависимости от условий образования может выделяться в виде гидраргиллита, байерита или нордст-рандита. Кристаллическая структура этих модификаций отличается последовательностью расположения слоев ОН. Решетка гидраргиллита, по-видимому, стабилизуется только в присутствии незначительных количеств Na+.
На рис. В.42 показаны стадии разложения трех фора* А1(ОН)3 и диаспора на основе многочисленных исследовании процесса дегидратации как функции размера частиц, скорости нагревания образцов, а также парциального давления атмо* сферной влаги. Все процессы завершаются образованием
Алюминий
605
А1(0Н)3 м^цеЗоватепность 1 -групп ОН
АЮОН
А1203 pnffXOitilMliep^m/рше
мотемтратдрныг формы)
Рис. В.42. Последовательность превращений при термической обработке гидраргиллита, байерита, нордстрандита и диаспора (прерывистые линии показывают гидротермальные переходы).
а-А12О3 (корунда). Этот оксид практически нерастворим в кислотах и водных щелочах. Его удается перевести в растворимое состояние только с помощью сплавления со щелочами. Вследствие амфотерного характера А12О3 возможно сплавление его с основаниями и антиоснованиями (разд. 33.4.3.2).
Как низко-, так и высокотемпературные формы оксида алюминия в твердом состоянии реагируют с рядом оксидов двухвалентных металлов с образованием шпинелей, например:
MgO + А13О3 ---► MgAl2o4
Рис. В.43, Механизм образования MgAbOt.
Двойной оксид магния и алюминия, образующийся при этой твердофазной реакции присоединения, относится к группе шпинелей типа AIIX2IIIO4 (например ZnAl2O4, FeCr2O4, СО3О4, т. е. СопСо2П104). Известны также шпинели типа AIVX2nO4 (например, TiCo2O4, TiZn2O4, SnCo2O4).
При твердофазных реакциях (разд. 33.9.2.5), приводящих к образованию алюмомагниевой шпинели, происходит встречная ДиффуЗИя катионов, т. е. ионы Mg2+ и А13+ проникают через образующийся «промежуточный слой» MgAl2O4 (рис. В.43). При таком механизме реакции неизбежно сохраняется условие электронейтральности в ионной решетке. Реакции, протекающие
606
Неорганическая химия
на границе твердых фаз, можно описать следующими уравнениями:
4 MgO + 2 А13+ («-) -> MgAl2O4 4- 3 Mg2+ (—)
иа границе фаз MgO/MgAl2O4
4 А12О3 + 3 Mg2+ (—) -> 3 MgAl2O4 + 2 Al3+ (*-)
на границе Al2O3/MgAl2O4
Аналогично образуются Mg—АЬ или Zn—AI-шпинели и многие другие (например, NiAl2O4, СоА12О4, MgCr2O4, CuFe2O4 и т. д.)( хотя количественные данные о механизме их образования часто отсутствуют. Так, кобальтовая шпинель СоА12О4 образуется в атмосфере азота со значительно большей скоростью, чем в присутствии кислорода. При образовании титанокобальтовой шпинели TiCo2O4 встречную диффузию катионов удается наблюдать только в атмосфере азота. На воздухе образование шпинели имеет место при диффузии ионов Со2+ и электронов через реакционный слой (при одновременном транспорте кислорода через газовую фазу):
2 Со2+ (—) + 4е~ (—) + Т1О3 + О2 (газ) -► TiCo204
Рекомендуемые опыты
1. На алюминиевой пластинке растирают несколько капель раствора HgCl2 и после промывания водой оставляют на воздухе. В результате окислительно-восстановительной реакции
2;А1 + 3 Hg2+ ->- 2 АР+ + 3 Hg(
на поверхности алюминия образуется амальгама (?), препятствующая образованию сплошной оксидной пленки. Через некоторое время образуются «выцветы» А1(ОН)3:
2А1 + ЗН2О + 1,5О2 ---->- 2А1(ОН)3
2. Определяют pH раствора сульфата алюминия. Затем по каплям прибавляют к нему щелочь. При pH 3,8 выпадает белый осадок Al(OH)s (Пр ~ 10“33), который снова переходит в раствор при дальнейшем прибавлении щелочи (pH 11—1.3) с образованием многоядерных комплексов (разд. 35.4.2.2).
3. К нескольким каплям раствора «алюмината» добавляют иа конце шпателя твердый NH4CI. В другую порцию раствора «алюмината» пропускают СО2 нли добавляют твердый NaHCO3. Обсудите сдвиг равновесия [A1(OH).J-^A1(OH)3+OH- в терминах реакции основание — антиоснова-иие?
4. К раствору соли алюминия прибавляют несколько миллилитров насыщенного раствора уротропина (гексаметилентетрамина); раствор нагревают. Уротропин — продукт конденсации аммиака и формальдегида, и при нагревании его водного раствора происходит обратная реакция:
CgHlaN4 + 6 Н2О --► 4 NH3 + 6 НСНО
А13+4-ЗН2О+ 3NH3 -----► A1(OH)3| + 3NH4+
Аналогичное осаждение из слабокислых растворов (pH 5—6) в результат® протолиза наблюдается для многих элементов (например, Fe, Cr, Ga, *+’ редкоземельные металлы, Th); это позволяет отделять их, например, от Zn • Ni2+ и Со2+.
Редкоземельные металлы,
607
5. В фарфоровом тигле сплавляют прокаленный А12О3 (на кончике шпателя) с пятикратным количеством смеси Na2CO3— К2СО3:
А12О3 + СО32- ---->- 2 А1О3~ + CO3f
При выщелачивании плава «алюминат»-ионы гидратируются: >’
А1О2- + 2Н2О -----» [А1(ОН)4]-
Аналогичным образом сплавляют А1гО3 с пятикратным количеством K2S20t (анион — антиосноваиие S2O72-). Расплав нагревают при возможно более низкой температуре, чтобы избежать потерь SO3. Реакция протекает количественно, если образуется прозрачный расплав:
А12О3 + 3 S2O72- --► 2A1»++6SO42-
При температурах >500 °C реакция становится обратимой (температурная зависимость реакции основание — антиосиование!).
6, Небольшое количество свежеосаждениого А1(ОН)3 тщательно промывают и осторожно обезвоживают в углублении пластинки из MgO. Затем смачивают несколькими каплями очень разбавленного (!) раствора Co(N03)2 и прокаливают в окислительном пламени (разд. 46.1.1). Кобальтовая шпинель окрашена в голубой цвет.
Квасцы. Сульфат алюминия, как и ряд других сульфатов трехвалентных катионов (например, Ti, V, Сг, Мп, Fe, Ga, In, Rh, Ir), из водных растворов может кристаллизоваться с сульфатами однозарядных катионов (М+ = К+» Rb+, Cs+, NH4+, Т1+) в виде двойных солей состава (квасцы):
М1МШ [SO4]3-I2H3O = [Ml (Н2О)е]+[МШ (H2O)6]3+[SO4122-
Растворимость алюминиевых квасцов падает в ряду щелочных металлов. Растворимость в 100 г Н2О при 20 °C составляет: для Na-квасцов 39,7, NHLG-квасцов — 6,59, К-квасцов — 6,0, Rb-квасцов — 1,52, Cs-квасцов — 0,46 г (в расчете на безводную соль).
Вопросы и задания
1. Ионные радиусы Ве2+ и В**-, Mg2+ и А13+ различаются на 0,01— 0,02 нм соответственно. Разность радиусов Са2+ и Ga3+ или Sr2+ и 1п®+ составляет уже более 0,03 нм. В чем причина такого резкого возрастания радиусов при переходе от элемента II группы к III в V ряду?
2. Обсудите с точки зрения координационной химии процессы осаждения Щелочью и растворения в ней А1(ОН)3.
3. Что происходит при действии (NH4)2S, KCN или CH3COONa на нейтрализованный раствор сульфата алюминия (ср. разд. 36.12.1, опыт 2)?
4. Опишите кристаллическую структуру шпинели. Что такое обратные Шпинели?
5. Относятся ли шпинели типа МА12О4 (например, M=Mg, Zn, Ni, Со) к алюминатам?
36.9. Редкоземельные металлы
К редкоземельным металлам относятся элементы скандий, иттрий и лантан, а также элементы от церия до лютеция. Последние называют «лантаноидами». Главная степень окисления Всех редкоземельных металлов +3. Церий, празеодим и тербий относительно легко приобретают степень окисления +4, а европий, иттербий и самарий +2.
608
Неорганическая химия
В соединениях Lal2 и Се12, как следует из их хорошей электропроводности, существуют катионы трехвалентных металлов и свободные электроны: La3+e_(I~)2 и Се3+е_(1~)2.
Поскольку у лантаноидов электроны заполняют только 4/-уровень, с ростом заряда ядра происходит сжатие электронной оболочки («лантаноидное сжатие»). В связи с большой близостью ионных радиусов лантаноиды обнаруживают глубокую аналогию в химических свойствах (экранирование 4f-op6n-талей электронами 5s- и 5р-орбиталей). Несколько большее различие в свойствах проявляют скандий, иттрий и лантан.
Щавелевая кислота Н2С2О4 служит групповым реактивом на редкоземельные металлы (которые она осаждает даже из слабокислого раствора).
Аналогия с алюминием проявляется в осаждении гидроксидов, которые, однако, нерастворимы в избытке щелочи (опытЗ).
Рекомендуемые опыты
Для опытов воспользуйтесь растворами продажных солей лантаноидов (обратите виимаиие на их квалификацию} или смесями различных солей редкоземельных металлов.
1. К слабокислому раствору прибавляют по каплям щавелевую кислоту: кристаллический осадок нерастворим в избытке осадителя, 11Рсе2(С2О4)з‘1 =
= 10~28-6.
В связи с тем что ряд элементов соосаждается в этих условиях и внедряется в кристаллическую решетку редкоземельных оксалатов, редкоземельные металлы часто отделяют в виде трудиорастворимых фторидов.
2. К слабокислому раствору прибавляют по каплям плавиковую кислоту: выпадает белый осадок соответствующих фторидов, растворимый в избытке плавиковой кислоты.
3. При добавлении к испытуемому раствору по каплям NaOH или NHj гидроксиды осаждаются на холоду в виде студенистых осадков; при нагревании они образуют творожистую массу. Значения Пр гидроксидов лантаноидов имеют порядок 10~2“~24.
4. Переход в производные других степеней окисления хорошо виден на примере церия. Их окисление до степени окисления +4 может происходить как в кислой, так и в щелочной среде.
К раствору соли церия(Ш), подкисленному разбавленной H2SO4, добавляют (NH4)2S2O8 и кипятят; появляется желтая окраска, свидетельствующая об окислении иона Се(III). В щелочном растворе происходит, например, окисление с помощью КМпО4 или гипохлоритов:
ЗСеЗ+ + МпО4~ + 8ОН- + ЗН2О --->• ССе(ОН)4 + МпО(ОН)2
Вопросы и задания
1. Перечислите возможные степени окисления лаитаиоидов. В этой связи дайте критическую оценку часто встречающемуся утверждению, что «стремление» к f°-, f7- и ^-конфигурациям является единственной причиной проявления степеней окисления, отличных от +3.
2. Объясните различия в основности редкоземельных металлов на основ свойств соответствующих катионных кислот.
3. Какие выводы можно сделать об изменении термической устойчивост • например, нитратов и карбонатов по группе редкоземельных металлов (и менении силы основания Льюиса, донорной активности оксидов М20з)-
Титан, цирконий
609
4. Рассмотрите окраску иоиов лантаноидов и объясните появление узких полос поглощения. Их можно наблюдать при рассмотрении лантансодержащих минералов при дневном свете в ручной спектроскоп.
5. Сравните атомные объемы Ей и Yb с другими лантаноидами. Наблюдается ли сходство со щелочноземельными металлами?
36.10. Титан, цирконий (гафний и курчатовий)
В соответствии с положением в периодической системе основная степень окисления этих элементов +4. Только титан относительно легко проявляет низшие степени окисления ( + 3 — ^'-конфигурация; +2 — ^-конфигурация), причем соединений с двухвалентным титаном известно лишь незначительное количество (опыт 5). Формальные степени окисления —1 и 0 реализуются лишь в исключительных случаях (табл. В.39). Из соединений циркония низших степеней окисления можно указать только ZrCI3 и ZrCI2. Первый образуется при восстановлении ZrCl4 цинковой пылью при 3,4—6,0 МПа и температурах 460—500 °C:
3ZrCl4 (газ) + Zr (тв ) <- > 4[ZrCl3J (тв.)
Хлорид курчатовия (IV) КиС14 образуется при 300—350 °C при хлорировании атомов Ku (ср. разд. 36.13).
Химические свойства ионов титана (IV), циркония(IV) и гафния (IV) напоминают свойства ионов урана, церия, олова, свинца, германия и кремния той же степени окисления; свойства ионов титана (III) обнаруживают общность с ионами V(III), Fe(III) и Al (III). Имея почти одинаковые атомные и ионные радиусы вследствие лантаноидного сжатия (Zr 0,145 нм; Hf 0,144 нм; Zr4+ 0,074 нм; Hf+ 0,075 нм), цирконий и гафний очень похожи друг на друга по химическим свойствам. Цирконий и гафний образуют всегда общие минералы. Наиболее Удобными технологическими методами разделения циркония и гафния являются ионный обмен или жидкостная экстракция.
Ионы Ti3+, Zr+ и НР+ относятся к группе жестких кислот (Разд. 33.4.3.4).
Труднолетучие оксиды МО2 элементов этой группы амфотерны. В кислотах и водных щелочах они после прокаливания растворяются с трудом. Кислоты и щелочи реагируют с МО2 в процессе «окислительного плавления», т. е. при сплавлении с соединениями основного (например, Ыа2СОз) или антиоснов-Нрго («кислого») характера (например, K2S2O7) (разд. 33.4.3.2). *ак, TiO2 лучше всего сплавлять с дисульфатом калия (анион ^2О72- — антиоснование). Поскольку TiO2 является еще и сла-Ым основанием, в расплаве диоксида титана существует ТЮ2* 'Титанил-ион) t
TiOj ---► TiOas+-|- Ог~; S2O,2- + O2- --► 2SO42~
39-2Q27
610
Неорганическая химия
В растворе аквакомплексы Ti(IV) не существуют, так как они являются сильными катионными кислотами. Но и простой «титанил»-ион TiO22+, по-видимому, не существует ни в растворах, ни в многочисленных кристаллических соединениях. В структуре TiOSO4-H2O, например, обнаружены цепи (TiO)п2п+. То же справедливо для понятий «цирконил»- и «гаф. нил»-ионов. Так, в решетке ZrOC12-8H2O существует ион '[2г4(ОН)8(Н2О)16]8+; а для соединений ZrO(NO3)2-2H20, HfOF2 и т. д. также до сих пор нет данных о существовании островных ZrO2+- или НЮ2+-ионов.
При сплавлении TiO2 со щелочными катионитами образуются «щелочные титанаты» формального состава Мг'ТЮз*. То же справедливо и для оксидов циркония и гафния:
СО32- ---- CO2f + О2-; ZrO2 + О2- ---► ZrO32~
Оксосоединения устойчивы только в сильнощелочных растворах (образование гидроксокомплексов). При разбавлении водой происходит полный протолиз до соответствующих гидратов оксидов, которые сравнительно легко осаждаются при добавлении раствора кислоты (почему?) .Для реакции TiO(ОН)2—>-TiO2++ +2 ОН- значение рК вычислено ~29. Для продукта, названного выше гидратом диоксида титана, методами ИК- и ПМР-спектроскопии установлено, что связанная вода, занимающая фиксированное положение, существует в виде ОН-групп.
Легкое протолитическое осаждение некоторых гидратов оксидов демонстрирует опыт 3.
Цирконий очень склонен к образованию ацидокомплексов (например, карбонатных, оксалатных, сульфатных (опыт 4))’.
Соли титана (III)—очень сильные восстановители (например, восстановление перхлората, титанометрия (разд. 38.3.2.4))-Исключительно чувствительна качественная реакция на титан с образованием пероксотитанил-иона, имеющего окраску от желтой до оранжево-красной:
(TiO)2+ + H2O2 —► (Т.О2)2+ + н2О
Рекомендуемые опыты
1. В фарфоровом тигле в течение 5—10 мин сплавляют TiO2 (на кончике шпателя) с пятикратным количеством K2S2O7. Нагревают до образования прозрачного расплава, но стараются избежать сильного выделения серного ангидрида (реакция основание — антиоснование обратима). Плав раствори» в разбавленной серной кислоте. При правильно проведенном сплавлении н должен оставаться нерастворимый осадок.
2. Часть раствора оксисульфата титана разбавляют небольшим количес вом воды и кипятят: образуется осадок TiO2-aq. 0
3 К части раствора оксисульфата титана (приготовлеииого, как опнса»
* Соединения такого типа часто предпочитают рассматривать в внЛ® двойных оксидов, так как нет доказательств существования определенна^, оксоаниоиов и оксокатиоиов в их структуре [Selbin J., Angew. Chemie, 78, (1968)]. — Прим, перев.
Титан, цирконий
611
выше), оксинитрата циркония ZrO(NO3)2-2 Н2О (или ZrOCl2-8H2O) прибавляют водные растворы NaOH, NH3 (NH^zS и уротропина. Конечными продуктами осаждения являются TiO2-aq (рН<0) или ZrO2aq (pH 1). TiO2-aq и ZrO2-aq легко растворимы в сильных кислотах. При нагревании осадков происходит их быстрое старение (?).
4. К разбавленному раствору оксинитрата или оксихлорида циркония добавляют по каплям раствор (NH4)2CO3. Образующийся осадок (?) растворим в избытке осадителя. При нагревании он снова выпадает (почему?). В качестве предварительной пробы на титан рекомендуется сплавление вещества с натрием.
5. В маленькой пробирке нагревают пробу TiO2 с кусочком натрия до размягчения стекла (защитные очки! опустить тягу!). Затем горячую пробирку бросают в конический сосуд с холодной водой и подкисляют раствор. Появляется красно-фиолетовая окраска иона [Ti(H2O)6]3+ (d'-конфигурация).
Соединения титана в степени окисления +3 образуются также при действии на раствор соли титана(IV) водорода «в момент образования» (например, цинк +НС1). Проделайте этот опыт!
6. Раствор оксисульфата титана разбавляют и прибавляют несколько капель 3%-ного раствора Н2О2: появляется интенсивное окрашивание, исчезающее в избытке фторид-ионов (образуется [TiF6]2-). Этой реакции мешают соли урана, ванадия н молибдена, а также ионы железа(П) и СгО42-.
Цирконий дает с Н2О2 в нейтральном растворе белый осадок пероксоцир-конисвой кислоты.
Наиболее важной является качественная реакция на цирконий с Na2HPO4.
7. Раствор соли циркония подкисляют 12 М НС1 (почему не H)SOt?), к нему добавляют несколько капель раствора Na2HPO4i выпадает бесцветный осадок примерного состава Zr3(PO4)2.
Малой растворимостью фосфата даже в сильиокислых растворах цирконий и гафний отличаются от всех других металлов.
Вопросы и задания
1. К какому типу относится реакция:
ZrOa + СО32~ ----> ZrO32- + СО2
2. Почему осаждение ZrO2-aq (или HfO2-aq) происходит при более высоких pH, чем TiO2-aq (опыт 2)?
3. Происходит ли простое осаждение в реакциях, проводимых в опыте 3?
4. Что происходит, если слабокислый раствор оксисоли титана прокипятить с Na2S2O3 или CH3COON (уравнение реакции) ?
5. Какие ионы существуют в растворах TiO2-aq в различных кислотах (и соответственно ZrO2-aq)-
6. Расскажите об устойчивости ацидокомплексов циркония и отношении к сильным кислотам — с точки зрения их аналитического применения (например, разделения Zr — Til). Какие комплексы титана имеют аналитическое значение (см. также опыт 6) ?
7. Каково основное состояние иона Ti3 4 5 6 7 8 9 10 11+ в октаэдрическом поле лигандов?
8. Какому электронному переходу обязана своим происхождением крас-н°-фиолетовая окраска иона [Т1(Н2О)б]3+?
9. Каково положение, интенсивность и ширина полосы поглощения в спектре иона [Ti (Н2О)6]3+ ?
10. В чем состоит аналогия в интерпретации спектров иоиов с конфигурациями d' и d2?
11. Что Вы думаете о применении сиропообразной фосфорной кислоты, часто рекомендуемой для осаждения фосфатов?
39»
612
Неорганическая химия
36.11. Ванадий, ниобий, тантал (и элемент 105)
В соответствии с номером группы основная степень окисления этих элементов +5, однако при нормальных условиях для ванадия стабильной является +4. В то время как у ванадия легко достигаются низшие степени окисления ( + 4, +3, +2; конфигурации d1, d2 и d3), ниобий обычным путем можно восстановить только до степени окисления +3 (опыт 2). Восстановление тантала в водном растворе вообще невозможно. Известны соединения с формальной степенью окисления —1 ([М(СО)б]~, где M=V, Nb, Та) и +1 ([VDipy3] , л-С5Н5М(СО)4, где M = Nb, Та) (табл. В.39). Низшие и «дробные» степени окисления этих элементов встречаются в соединениях, содержащих группы Мп (разд. 36.11.1). Химические свойства соединений нанадия(П) весьма напоминают свойства соединений цинка, а ванадия(Ш)—титана(Ш), железа(Ш) и алюминия. Донорные основные свойства оксидов ванадия ослабляются с увеличением формальной степени окисления.
Вследствие лантаноидного сжатия ниобий и тантал проявляют большое сходство в химических свойствах. Хлорид элемента 105 (разд. 36.13) в высшей степени окисления получают, например, при замещении тионилхлоридом при 300°C «атомов отдачи» реакции 24395Am + 22wNe. Он летуч менее хлорида ниобия (V) NbClg, но более, чем хлорид гафния (IV) HfCl4.
Элементы этой группы в высших степенях окисления легко образуют изополикислоты. Последние представляют собой многоядерные комплексы и образуются из одинаковых одноядерных оксоанионов (например, SO42-, РО43~, SiO44-, СгО42~, МоО42-, WO42-, причем формально отщепляются ионы О2-):
2VO43~ -->- [V2O;F“ + о2-
Координационное число центрального иона сохраняется неизменным (опыт 1). «Конденсация» различных одноядерных оксокомплексов приводит к анионам соответствующих гетерополикислот. О строении атома элемента 105 см. разд. 36.13.
Рекомендуемые опыты
1. К водному раствору ванадата натрия, Na3VO4, по каплям добавляют разб. H2SO4. Первоначально бесцветный раствор приобретает желтую, а затем оранжево-желтую окраску (образование декаванадат-ионов [VioOzs]6*)-Из концентрированных растворов на последнем этапе происходит осаждение гидрата оксида V2Os-H2O (разд. 36.12, опыт 7), который при дальнейшем Д°’ бавлении кислоты растворяется с образованием светло-желтого (мономерного) ванадил(У)-иона VO2~. Раствор ванадата в зависимости от pH содер' жит частицы различного размера. При подкислении растворов ванадата уста-навливаются следующие равновесия:
vo43- + Н+ [HVO4I2-
2[HVO4p- 4=fc |V2O7b-+HtO
Ванадий, ниобий, тантал
613
В 2[VaO7]4- + 4Н+ «=* [V4O12]<- + 2Н2О
1 5[V4O12p- + 8Н+ =г=± 2[V10O28]e- + 4Н2О
При дальнейшем добавлении кислоты образуются [HVio02s]s—, [H2Vio02s]4” й, наконец, свободная декаванадиевая кислота. Последняя находится в равновесии с высокомолекулярным V2O5-xH2O, который в свою очередь под действием Н+ образует УО2+-ионы:
HaV10O28 -> 5V20j-aq 4* хН2О
V2O5- aq + 2Н+ 2VO2+ + *Н2О
Необходимо отметить, что существование ионов VO2+ до сих пор достоверно не доказано ни в растворе, ни в твердом состоянии. Из оксокатионов ванадия (V) в вышеприведенных уравнениях необходимо еще упомянуть ионы VO3~, чаще других встречающиеся в твердых и жидких соединениях (например, VOX3, где X=F, Cl, Вг, OR).
Как VO3+-, так и ¥02+-ионы (см. ниже) относятся к группе жестких кислот (разд. 33.4.3.4).
Различие действия отдельных восстановителей демонстрирует опыт 2.
2. H2S или SO2 пропускают в подкисленный раствор ванадата: происходит восстановление ванадия до степени окисления +4 (VO2+ окрашен в си-нии цвет). То же происходит при кипячении раствора ванадата в присутствии концентрированной соляной кислоты:
VO2+ -Ь 2Н+ + СП ----- vo2+-h/2ci2 + h2o
Иодоводород восстанавливает ванадий в кислом растворе до сине-зеленого иона ванадця(Ш), окраска которого становится различимой при удалении иода кипячением:
V3++H2O VO2+ + 2Н+ + е~ £° = 0,36В
При взаимодействии ванадатов с натрием в расплаве образуются также ионы ванадпя(Ш). Восстановление водородом в момент образования (при действии цинка на раствор ванадата, слабоподкисленный серной кислотой) заканчивается образованием фиолетового иона ванадия(П) [V(H2O3]2+, чувствительного к действию кислорода воздуха. Переходы между отдельными степенями окисления легко различимы по изменению окраски раствора. Соли ва-надия(П) —сильные восстановители:
V2+V3+ + e' £° = —0,25В
Так, в кислом растворе он восстанавливает NO3~ до NH4+, С1О4~ до С1~ и Н+ до Н2.
Для восстановления ниобия в степени окисления +5 к кислому раствору СОЛИ ниобия добавляют небольшое количество цинка или олова; реакционную смесь можно слегка нагреть. Наблюдается восстановление с появлением синей или коричневой окраски:
NbO3+ + 2Н+ Н- 2е~ Nb3+ + Н2О
Аналогия свойств ванадатов с вольфраматами, молибдатами, хроматами, ман-ганатами(У), арсенатами, фосфатами и сульфатами проявляется также в воз-можности их осаждения при действии ряда ионов тяжелых и щелочноземель-НЫХ[ металлов (см. также разделы, посвященные соответствующим элемен-. 3. К раствору ванадата натрия прибавляют растворы ВаС12, FeC!3, AgNO3, Hg2(NO3)2 или Pb(NO3)2. Ванадаты железа, серебра (Прдеуо3== J=10~9.9) и свинца имеют окраску от желтой до красной, а ртути и бария — °елые.
614
Неорганическая химия
4. Для аналитической химии весьма важно отношение нейтральных или аммиачных растворов ванадатов к H2S и (NH4)2S.
H2S пропускают в аммиачный раствор ванадата. Раствор приобретает интенсивную красно-фиолетовую окраску ([VS4]3-). При добавлении (NH4)2S раствор, в котором образуются тиованадаты, окрашивается от коричневого до красно-фиолетового цвета в зависимости от содержания серы в (NH4)2S. При подкислении раствора тиованадата осаждается коричневый V2S5. Раствор, образующийся после центрифугирования, имеет голубоватую окраску (почему?).
5. Для открытия ванадия в степени окисления +5 можно использовать, также образование иона пероксоваиадия(У).
Катионы VO3*, присутствующие в растворах солей ванадия(У) наряду с ионами УО2+, реагируют с Н2О2 с образованием красновато-коричневых ионов УО23+. При дальнейшем действии Н2О2 последние образуют светло-желтую пероксованадиевую кислоту Нз[УО2(О2)2]; поэтому по возможности следует избегать избытка Н2О2. Этот опыт проводят в возможно более кислом растворе (-—15—20%-ная H2SO4 или HNO3) (почему?). Об открытии ванадия с помощью Н2О2 в присутствии хрома см. разд. 36.12, опыт 12.
6. Реакции ниобия и тантала целесообразно проводить с растворами щелочных ниобатов или танталатов, которые можно получить, например, при сплавлении с К2СО3 соответствующих пентаоксидов Nb2Os или Та2О5:
ЗСО32- ---> ЗСО2| + зо2-
Nb2O5 + ЗО2- ---► 2NbO43“
Плав растворяют в воде.
Кислоты, соответствующие ниобатам и танталатам, неизвестны в свободном состоянии. Наблюдаемая у ионов ванадия в степени окисления +5 тенденция к конденсации и в этом случае выражена весьма сильно.
Растворы ниобатов и танталатов подкисляют минеральной кислотой. Даже при pH 0 (почему?) выпадают гелеобразные осадки соответствующих гидратированных оксидов M2Os-aq (M=Nb, Та). При непродолжительном нагревании они практически нерастворимы в большинстве кислот (почему?).
7. Для получения кислых растворов ниобия (опыт 2) лучше всего растворить свежеосаждеиные гидратированные оксиды в минеральных кислотах или пентаоксиды — в плавиковой кислоте (реакции?). Раствор в HF в платиновом тигле упаривают с конц. H2SO4 и затем растворяют в воде.
8. Кроме открытия ниобия с помощью восстановления (опыт 2), для идентификации его в присутствии тантала используется образование пероксонио-биевой кислоты H[NbO4]-aq.
К свежеосаждеиному Nb^s-aq приливают на холоду небольшое колИШЯ чество пергидроля и разбавленной H2SO4: образуется желтая пероксоииобвд^И вая кислота. Соответсвующая пероксокислота тантала бесцветна.
Вопросы и задания
1. Как можно объяснить явление уменьшения донорной активности осн<^^ вания с возрастанием степени окисления (наблюдаемое и у других элементов, например хрома, марганца и т. д.)?
2. VO2—амфотерный оксид. Дайте качественное объяснение существования соединений VSO4-7H2O и Na3VO4.
3. Справедливо ли тетраванадаты, например Па4[Н2У4О1з], называть «метаванадатами» [(МаУОз)4-Н2О]*? Сравните ситуацию в случае полифос-фатов.
4. Почему в водном растворе ие существуют гидратированные иоиы в^ надия (V) и ванадия (IV)?
* Данные о существовании тетраванадат-ионов в водном растворе поД вергаются сомнению. [Коттон Ф., Уилкинсон Дж. Современная неоргани ская химия. Ч. 3. Пер. с англ. — М.: Мир, 1969, с. 222]—Прим, перев.
Ванадий, ниобий, тантал
615
5. Почему в опыте 2 равновесие смещено полностью вправо, хотя стандартный потенциал реакции С1-=^У2 С12+е- (£°= 1,358 В) оказывается выше, чем реакции VO2++H2O=^VO2++2 Н++е~ (£°= 1,00 В)?
6. Возможны ли для октаэдрически координированного иона V2+ различия в расположении орбиталей в случаях лигандов сильного или слабого поля?
7. Насколько радиус иона V2+, полученный на основе несферической модели, оказывается меньше, чем у (гипотетического) сферического иона? Обсудите это важное обстоятельство в связи с изменением относительных ионных радиусов для других двухзарядных 3</-ионов1
36.11.1. Связь метал л — металл
Почти для всех переходных ^-элементов известны соединения, в которых осуществляется связь двух или более атомов металла друг с другом. Были исследованы соединения с груп-
[W^lg]3' NbOCla Nb02 NbCl4; a-Nbl;
NbSaClj, MoCl3 Nb3Cl0 ERe3CL(a]3'
Рис. B.44. Координационные полиэдры металла (черные кружки).
Связи М—М показаны жирными линиями. Дополнительные связи анионов обозначены стрелками. Анионы — пустые или заштрихованные кружки.
пами Мп (га = 2, 3 и 6), состоящими из атомов одного и того же металла (табл. В.36). При классификации этих соединений ока. зывается весьма целесообразным различать соединения с двух-Центровыми связями металл — металл (группы М2) и соединения с многоцентровыми связями (группы М3 и М6). В последнем случае каждый из атомов металла находится в обменном взаимодействии со многими или со всеми остальными атомами группы (соединения с кластерами из атомов металла) (рис. В.44 Й в.45).
Особую склонность к образованию подобных соединений Проявляют ниобий, тантал, молибден, вольфрам и рений. Как правило, эти элементы образуют соединения, в которых атомы
616
Неорганическая химия
металлов находятся в низших нях окисления, например
Степень окисления
2,33
2,50
2,67
(иногда нецелочисленных) степе-
Соединение нли нон
Nb6Cl14, [Та6С112]2+
NbgFis, TasClis
Nb3Cl8
При рассмотрении связей металл — металл следует учитывать, что прочность связи может изменяться в широких преде-
° Q
Рис. В.45. Расположение атомов в структурах, содержащих группы М.п.
Металл — черные кружки; анионы — пустые кружки, X1 —анноны «внутренней сферы» Ха — анноны «внешней сферы».
лах: от слабого спин-спинового взаимодействия до очень прочных кратных связей.
Указанием на наличие связей М—М могут служить относительно более короткие расстояния между атомами металла*. Возможность образования связей М—М в основном определяется такими факторами, как степень окисления** металла, вид лигандов и др. Магнитные свойства многих этих .соединений также не могут непосредственно свидетельствовать о простом спаривании электронных спинов атомов металла и образования связи М—М. На магнитных свойствах могут сказываться не только М—М-взаимодействия, но и сильное перекрывание орбиталей атомов металла и лигандов. Например, в RuO2 молекулярные орбитали имеют значительную протяженность. Это соответствует образованию энергетических зон, что сильно влияет на магнитные свойства соединения.
Факторы, определяющие возможность образования связей М—М, удалось выявить, привлекая обширный экспериментальный материал. При этом, помимо препаративных методов, широко использовались термохимические и подробные структур-
* Обычно расстояния М—М в соединении сравниваются с расстояниям» в металле, хотя этот критерий не всегда справедлив.—Прим, перев.
** Число внешних (валентных) электронов атома М, не участвующих » связях с лигавдамв.— Прим, перев.
Хром, молибден, вольфрам
617
Таблица В.36. Соединения с группами М„
Число атомов п в группе Мл Примеры
2 Двухъядерные карбонилы, например Мп2(СО)10, Fe2(CO)9a, Со2(СО)8а, (ОС)5Мп— Re(CO)5, R3Sn—Мп(СО)5« NbX4 или Nb2X8 (X = С1, Вг, 1в); ТаХ4 или Та,Х8 (X =С1, Вг, 1) NbOX2 (Х = С1, Вг, I); МоОС12; NbO2; NbS2Cl2 [Re2Br8]2~; a-ReO2
3 Трехъядерные карбонилы, например Fe3(CO)12, Os3(CO)12 NbCl2j67 или Nb3Cl8 CsReCl4 или Cs3[Re3Cl12] Zn[Mo308]
4 Co4(CO)12, [Ni4(CO)9]2- r
6 Rh6(CO)16 NbF2 6 или Nb6F15; (Nb6Cl12]2+; TaCl2 33-7/6H2O или Ta6Cl14-7H2O TaBr2S или Ta6Br15 [MoeCls]4+; МоВг2 или Мо6Вг12 аналогично WeCl12 WeBr16 wi<[WeBrgq Br4a[Br4]2/2a-a
J Наряду с СО мостиками осуществляется связь М—М.
R— алкильная или арильная группа
a-NbI4
Структура полностью не решена.
ные данные. Для теоретического объяснения образования связей в группах М3 и Мб применялись методы валентных связей (ВС) и молекулярных орбиталей (МО), причем метод МО дает возможность предсказывать многие из свойств соединений (например, магнитные свойства групп Мб, содержащих 15 или 16 d-электронов).'
36.12. Хром, молибден, вольфрам (и элемент 106)
В соответствии с номером группы во многих своих соединениях эти элементы находятся в степени окисления +6, которая особенно типична для молибдена и вольфрама. Для хрома наи
618
Неорганическая химия
более устойчивая степень окисления +3 (й3-система; заполнение наполовину ^-орбиталей при октаэдрической координации). Помимо этого, могут быть получены (в некоторых случаях достаточно легко) соединения этих элементов в более низких степенях окисления (разд. 36.14.2).
Соли хрома (II), окрашенные в водных растворах, как правило, в голубой цвет, весьма чувствительны к кислороду и обладают сильным восстановительным действием:
Сг2+ ч—h Сг3* + е~ Е» = —0,41 В
Для всех трех элементов известны также соединения с формальной степенью окисления металла —2, например [М(СО)5]2~ (табл. В.39).
По своему химическому поведению молибден и вольфрам гораздо сильнее отличаются от хрома, чем между собой. Например, в отличие от хрома степень окисления +3 для молибдена и вольфрама реализуется лишь в небольшом числе катионных комплексов. Реакции хрома (III) во многом сходны с реакциями железа(III) и алюминия. В степени окисления +6 хром несколько напоминает ванадий ( + 5).
Сходство химического поведения ионов Сг3+, Fe3+ и А1+ может быть качественно объяснено в рамках теории жестких и мягких кислот и оснований (разд. 33.4.3.4). Все эти ионы — жесткие кислоты. О строении атома элемента 106 см. разд. 36.13.
36.12.1. Химические свойства хрома
Сг(ОН)3 (правильнее, Cr2O3-aq) труднорастворим (—1§Пр« »30); это —амфотерный гидроксид. В упрощенном виде можно записать:
Сг(ОН)3 <=> Сг3* + ЗОН“
Сг(ОН)3 + Н2О 4=t [Сг(ОН)4Г + Н+ рК « 16
Так же как это было сделано для А1(ОН)3, следует и для Сг(ОН)3 обсудить значение этой величины рА (разд. 36.8, опыт 2).
Будучи слабым основанием, Сг(ОН)3 не дает растворимых солей со слабыми кислотами.
При взаимодействии солей хрома (III) с (NH4)2S осаждается Сг(ОН)3; так же ведут себя ионы циркония(ГУ), титана(ГУ) и алюминия.
Из раствора с концентрацией соли хрома (III) 0,01 моль/л осаждение начинается при pH 5,0. При дальнейшем добавлении гидроксида щелочного металла при pH 13—14 начинается растворение осадка Cr2O3-aq с образованием [Сг(ОН)4]' и [Сг(ОН)6]3-. В избытке аммиака Cr2O3-aq также растворяется, причем хром частично переходит в комплексный ио» {Cr(NH3)6l3+.
Хром, молибден, вольфрам 619
Можно было бы предполагать, что при добавлении к раствору соли хрома (III) ацетата натрия основание ацетат-ион будет связываться с ионами гидроксония в малодиссоциированную уксусную кислоту СН3СОО+Н3О+—>-НСН3СОО+Н2О. Далее должно было бы происходить образование буферной системы СНзСООН/СН3СОО_ и осаждение хрома в виде Сг(ОН)з. В действительности однако, образуется, так же как в случае реакции с ионами железа (III) и алюминия, ацетатный комплекс вероятного состава [Сг3(ОН)2Ас6]+. В отличие от комплексов алюминия и железа, разлагающихся при нагревании до труднорастворимых основных ацетатов, комплекс хрома в этих условиях не гидролизуется.
Если же в растворе присутствовали ионы хрома (III), алюминия и железа(III), то хром частично переходит в осадок, а алюминий и железо(III) частично остаются в растворе. Это происходит потому, что ионы хрома (III), алюминия и железа (III)’ могут замещать друг друга в растворимых и труднорастворимых комплексах.
Соединения хрома (III) могут окисляться до степени окисления хрома +6 как при действии окисляющих расплавов, так и в водных растворах. При этом всегда образуются оксосоединения. В щелочном растворе также можно легко осуществить окисление до хроматов.
В кислой среде окисление до хрома (VI) идет только под действием наиболее сильных окислителей.
Хром, так же как молибден и вольфрам, может образовывать изополикислоты. В кислой среде простая конденсация оксокомплекса СгО42~ дает ион дихромата Сг2О?2-.
При добавлении основания равновесие смещается в сторону хромата. То же самое происходит при разбавлении водой (закон действующих масс):
Сг2О72- + Н2О ч=± 2 HCrO4- рК = 1,6
НСгО4' ч=±= Н+ + CrO42- рК = 6,5
Образование многоядерных анионов изополикислот можно наблюдать по изменению окраски раствора при медленном (постепенном) добавлении конц. H2SO4 к почти насыщенному раствору Na2CrO4. В присутствии большого избытка кислоты большая часть хрома осаждается в виде полимерного (СгО3)х. Одновременно образуется CrO2SO4 (можно проверить это на опыте).
В кислых растворах хром в степени окисления +6 — сильный окислитель. При этом происходит восстановление до хро-ма(Ш), так что наблюдается изменение окраски раствора из °ранжевой в зеленую (разд. 33.5.1.5):
2Сг3++7Н2О Сг2О,2-+14Н++6е- £<>== 1,36 В
620
Неорганическая химия
Для аналитической химии имеет значение образование ряд труднорастворимых хроматов тяжелых металлов. В этом прояв ляется сходство хроматов с ванадатами, вольфраматами, мо либдатами, арсенатами, фосфатами и сульфатами.
С некоторыми особенностями осаждения хроматов можн познакомиться, выполнив опыт 10.
В аналитической химии при открытии хрома используется реакция образования хромилхлорида СгО2С12.
Весьма специфичной реакцией может служить открытие хрома в виде пероксида хрома CrOs, т. е. СгО(О2)2, который может быть стабилизирован некоторыми органическими растворителями (например, эфиром, метилизобутилкетоном).
Пероксосоединения титана (разд. 36.10, опыт 6) и ванадия (гл. 36.11, опыт 5) нерастворимы в органических растворителях. Благодаря этому хром может быть открыт в присутствии этих элементов. При наличии ванадия не следует работать в слишком кислой среде (разложение CrOs), хотя в этих условиях чувствительность реакции на ванадий также понижается.
36.12.2. Химические свойства молибдена и вольфрама
Для проведения опытов используют растворы парамолибдата аммония (NH4)6Mo7O24-4H2O. (Упрощая, часто указывают следующий состав соли молибдена (NH4)2MoO4. Можно ли это делать в данном и в некоторых других случаях, например для Li2WO4-4/7 Н2О или вольфрамата натрия Na2WO4?)
Осаждение гидратов оксидов молибдена и вольфрама протекает по сложному (еще не полностью изученному) механизму через промежуточные стадии образования полианионов. Конденсация молибдат- и вольфрамат-анионов при добавлении кислоты идет путем связывания через ОН-группы и при отщеплении молекул Н2О (разд. 36.11, опыт И и разд. 36.12, опыт 7). В растворах молибдатов уже при добавлении небольших количеств кислоты образуются полимерные ионы. В этой системе доказано, например, присутствие ионов [Мо7024]6-:
7 Мо042- + 8 Н+ [Мо7О24]в- 4- 4 Н2О
В растворах вольфраматов при подкислении сначала обра зуется анион, содержащий шесть атомов вольфрама:
6 WO42- + 7 Н+ [HWeO21-x aqp- + (3 - х) Н2О
В дальнейшем этот анион находится в равновесии с другивд полианионом (включающим 10 атомов вольфрама).
Неудивительно поэтому, что соединения состава H2MoO4-H2^’ Н2МоО4, H2WO4-H2O, H2WO4, а также HVO3 (с ионами НзО или ОН~ в решетке) в твердом состоянии не существуют, устойчивыми являются лишь гидраты оксидов. Эти положена
Хром, молибден, вольфрам
621
были подтверждены исследованием гидратов оксидов состава МоО3-2Н2О, МоО3-Н2О, WO3-2H2O, WO3-H2O и V2O5-H2O методами ЯМР- и ИК-спектроскопии.
Взаимодействие некоторых одноядерных оксокомплексов (AsO43-, РО43~, GeO44-, SiO44- и т. д.) с оксокомплексами молибдена и вольфрама можно условно изобразить с помощью уравнения, например:
РО43-+ 12МоО42- -► [Р(ОМо3О0)4]3-+ 12 О2-
Для аналитической химии некоторое значение имеет отношение растворов молибдатов и вольфраматов к H2S (опыт 15).
36.12.3. Молибденовая и вольфрамовая синь
Восстановление соединений молибдена и вольфрама в степени окисления +6 дает соединения с более низкими степенями окисления. В веществах, известных под названием молибденовой или вольфрамовой сини, молибден и вольфрам нельзя считать входящими в состав одного определенного соединения или имеющими определенную степень окисления. В зависимости от выбора исходных соединений (например, МоО3, Мо03-Н20, молибдаты; то же самое для вольфрама), используемого восстановителя (например, Zn, SnCl2 или Pb в солянокислом растворе; нагревание Мо03-2Н20 в ампуле при 110°С с порошкообразным молибденом и т. д.) и продолжительности процесса могут быть получены различные соединения, содержащие оксидные или гидроксидные группы (табл. В.37). В аналитической практике при открытии вольфрама в виде вольфрамовой сини име-
Таблица В.37. Различные формы молибденовой и вольфрамовой сини
Степень окисления Кристаллические соединения Рентгеиоаморфные соединения
5,76 МоО2 яз’^О МоОд о8 * хН2О Н[Мо3О9]
5,66 —
5,50 МоО2>6(ОН)015 МоО2 в0-хН2О Мо2>75-хН2О
5,20
5,00 моо2;0(он)1>0 —
Степень окисления Соединения, полученные ИЗ WO3 Соединения, полученные из WO3 Н2О
5,90 W02t9o(OH)0_i -
5,67 ——
5,64 — WO2 82-xH2O
5,50 WO2i5(OH)0|5
622
Неорганическая химия
ют дело исключительно с «синими гидроксидами». Для соединений, приведенных в табл. В.37, постоянство состава подтверждено методами химического и термического анализов. Убедительные аргументы в пользу «гидроксидной природы» этих веществ были получены методом протонного магнитного резонанса.
Весьма чувствительной качественной реакцией на молибденсодержащие вещества может служить проводимое в открытом тигле «отдымливание» пробы с несколькими каплями конц. H2SO4 почти досуха. В результате восстановления, например при попадании частичек пыли из воздуха и т. д., также появляется синее окрашивание. В присутствии металлического вольфрама эта реакции неоднозначна, поскольку при окислении металла конц. H2SO4 образуются соединения, также окрашенные в синий цвет.
Для открытия молибдена в присутствии вольфрама используют реакцию образования в растворе ионов [Mo(SCN)6]3~ (красное окрашивание).
Рекомендуемые опыты
Для следующих опытов используют раствор соли хрома (III).
1. Немного фиолетовой соли хрома (III) растворяют на холоду в небольшом количестве воды. В фиолетовом растворе присутствует «кислотноосновной комплекс» Сг[(НгО)б]3+. При нагревании раствор становится темнозеленым вследствие образования ацидоаквакомплекса (гидратная изомерия).
2. К раствору соли хрома(Ш) добавляют раствор (NH4)2S; происходит «протолитическое осаждение» Cr(OH)3 (Cr2O3-aq):
6НОН 6Н+ + 6ОН"
3 S* 2~ 4- 6 Н+ 3 H2S
2&3++6ОН- 2Сг(ОН)3|
2Cr3++3S2-4-6HOH 2 Сг(ОН)3| + 3 H2S р/< = —36,4
3. Совершенно так же идет осаждение Cr2O3-aq при добавлении NaOH, NH3, Na2CO3 и уротропина (кипячение)
Сг»++ ЗН2О ч—* Cr(OH)3| + ЗН+
4. В лодочке из оксида магния немного Сг2О3 обрабатывают расплавленной смесью безводных iNa2CO3 и KNO3. Расплав окрашивается ионами СгО42- в желтый цвет (окислительные смеси, разд. 33.5.2.3, см. также разд. 36.14 1).
Сг2О3 (тв.) + 2СО32- -► 2 СгО2~ + 2 CO2f 4- О2-
2 СгО2" + 3 NO3- + О2- -> 2 СгО42- + 3 NO2"
Сг2О3 (тв.) 4- 2 СО32- + 3 NO3" ->- 2 СгО42- + 2 СО2 4- 3 NO3"
5. К раствору соли хрома (III) добавляют смесь растворов NaOH и Н2О2. Наблюдается желтое окрашивание:
Сг(ОН)3 4- 5 ОН‘ Sr—* СгО42- 4- 4 Н2О 4- Зе' Е° == — 0,13 В
2 ОН’ 5Р=±: Н2О2 4- 2е~
Напишите суммарное уравнение.
Хром, молибден, вольфрам 623
6. К раствору соли хрома(III), подкисленному разб. H2SO4, добавляют немного твердого пероксодисульфата калия и раствор короткое время кипятят. Вследствие образования иоиа дихромата Сг2О72- раствор окрашивается в оранжевый цвет:
2Сг3+ + 3S2O82- + 7Н2О ---> Сг2О72- + 6SO42- + 14Н+
7. Немного К2СгО4 растворяют в воде; раствор подкисляют разб. HNO3 или НС1О4. Происходит переход окраски раствора в оранжевую:
2СгО42- + 2Н+ <- г 2СгО3ОН_ (протонирование)
2СгО3ОН“ Сг2О72“ + Н2О (димеризация)
' 2СгО42- + 2Н+ ^=±: Сг2О72- + Н2О
8. К2СгО4 нагревают с конц. HCI (тяга/). К раствору хромата в нескольких пробирках, подкисленного конц. H2SO4, прибавляют сероводородную воду (разд. 35.6.3.1), раствор H2SO3 (разд. 35,6, опыт 12) и раствор K.I. Напишите ионные уравнения проведенных реакций.
9. К раствору КгСгО4 добавляют раствор ВаС12, AgNO3, Hg2(NO3)2 И Pb(NO3)2. Все осадки интенсивно окрашены.
BaCrO4 Ag2CrO4 Hg2CrO4 PbCrO4
желтый темио-кирпичио» при осаждении на хо- желтый
красный лоду ярко-оранжевый;
при нагревании красный
-ignp 9,7 11,7 8,7 13,8
10. К 0,01 М раствору Иа2СггО7 (Сг2О72-+Н2Оч*2НСгО4~) добавляют каплю индикатора бромкрезолового зеленого. Цвет индикатора в растворе Na2Cr2O7 — зеленый (синий при pH 5,4; желтый при pH 3,8). При добавлении нейтрального раствора соли-осадителя, например РЬ(МО3)2, наблюдается переход цвета индикатора в желтый (почему?). Более четко можно видеть изменение окраски раствора, отфильтровав осадок РЬСгО4.
11. Опыт по получению хромилхлорида, описанный в разд. 49,2.3.1, используют как метод открытия хрома. Для этого образующийся хромилхло-рид гидролизуют раствором NaOH (уравнение реакции/). Открытию мешает присутствие фторид-ионов (образование хромилфторида) и значительных количеств иодидов (окисление до иода).
12. Несколько капель раствора хромата подкисляют разбавленной H2SO4 или HNO3, приливают диэтиловый эфир или метилизобутилкетон. Затем добавляют немного Н2О2 и тотчас встряхивают. Органическая фаза окрашивается в ярко-синий цвет:
HCrO4- + Н+ + 2Н2О2 ----->- СгО(О2)2 + ЗН2О
Через некоторое время окраска бледнеет, а водная фаза приобретает зеленую окраску за счет образования аквакомплекса хрома (III):
4СгО(О2)2 + 12Н+ ----> 40-3++ 6О2 + 2Н2О2 + 4Н2О
Образовавшаяся при этой реакции Н2О2 в дальнейшем может разлагаться.
13. К растворам молибдата и вольфрамата по каплям добавляют разбавленную HNO3. В растворе молибдата при этом сначала образуется белый осадок состава МоО3-2Н2О, растворяющийся при дальнейшем добавлении Кислоты вследствие образования катионов МоО22+. Аналогичным образом в Растворах солей Mo(V) при концентрации НС1 10—12 моль/л молибден присутствует исключительно в виде мономерного катиона МоО3+, представляющего собой жесткую кислоту (разд. 33.4.3.4).
Из раствора вольфрамата при добавлении кислоты выпадает белый садок гидрата оксида WO3-2H3O, желтеющий при нагревании (почему?).
624
Неорганическая химия
14. Раствор молибдата или вольфрамата сильно подкисляют азотной кислотой; к нему добавляют немного NH4CI или КС1 и несколько капель разбавленного раствора Na2HPO4. При слабом нагревании из молибдатного раствора выпадают желтые, а из вольфрамового — бесцветные кристаллы (фосфоромолибдата, соответственно фосфоровольфрамата). Проверяют, растворимы ли осадки в NaOH (уравнение реакции!).
Арсенаты также дают желтый осадок, а германаты и силикаты—желтые растворимые соединения аналогичного состава.
15. В подкисленный раствор молибдата пропускают H2S. Синее окрашивание раствора указывает на частичное восстановление молибдена. При дальнейшем пропускании H2S часть молибдена осаждается в виде темпокоричневого MoS3. Его количественное осаждение достигается лишь под давлением H2S. MoS3 практически нерастворим в конц. НС1. С (NH4)2S образуется растворимый тиомолибдат красного цвета
MoS34-S2- -----> [MoS4]2-
При подкислении происходит его разложение.
16. В солянокислый раствор молибдена или вольфрама вносят немного Zn или SnCl2. Происходит восстановление до молибденовой или вольфрамовой сини.
Постепенное изменение окраски можно наблюдать при восстановлении суспензии WO3 (ио ие гидрата оксида) гранулированным цинком в соляно кислом растворе. Окраска раствора постепенно меняется от синей к фиолетовой и далее до коричневой. При длительном восстановлении па кусочках цинка, находящихся на дне сосуда, появляются оливково-зеленые пятна, исчезающие при встряхивании. В результате восстановления раствор ста новится зеленым.
17. На фильтровальную бумагу, предварительно смоченную разбавленной (1:1) НС1 (конц. НС1 разрушает комплекс), наносят по капле раствор мо либдата и 10%-иого раствора KSCN. При добавлении на то же место капли раствора SnCl2 (~5%-ный раствор в 3 М НС1) образуется пятио или коль цо, окрашенное в розовый цвет комплексом молибдена (III). Мешают ли проведению этой реакции соли железа(Ш)?
При одновременном присутствии вольфраматов при восстановлении обра зуется пятно синего цвета, окаймленное розовым кольцом комплекса молпб дена (капиллярная диффузия).
18. Из нейтральных (почему?) растворов молибдатов и вольфраматов можно осадить труднорастворимые соли (аналогия с хромом, см. опыт 91, например;
Ag2WO4 Ag-jMoOj РЬМо04
—Ignp 9,3 10,6 5,4
Вопросы и задания
1. Происходит ли изменение окраски при нагревании сильноразбавлеп-ного раствора, содержащего ионы [Cr(H2O)oJJ+? Учтите закон действующих масс
2 Аква- и ацидогруппы в комплексах хрома связаны весьма прочно При помощи каких реакций можно различить два гидратных изомера [Сг(Н2О)6]С13 и [СгС12(Н2О)4]С1-2Н2О-
3 Влияет ли сила поля лигандов на электронное строение октаэдрических комплексов хрома (III)?
4. Познакомьтесь с принципом действия рубинового лазера.
5 К какому типу относится реакция, соответствующая первому уравнению опыта 4? Назовите примеры аналогичных реакций.
6. Напишите в общем виде уравнение для процессов окисления нитратными расплавами.
Торий, уран
625
Будет ли образовываться ион СгО42- также в отсутствие Na2CO3 (реакции окисления в расплавах)?
7. Используя уравнение Нернста для реакции Сг3+ + 4Н2Очэ=СгО42~ + 4-8Н+ + Зе~, объясните, почему окисление хрома(Ш) идет легче в щелочной среде.
8. Почему при использовании нейтрального раствора дихромата не происходит количественного осаждения хроматов (например, хромата бария)? Какое влияние на степень осаждения оказывает добавление ацетата натрия?
9. К какому классу соединений относится хромилхлорид?
10. Почему при подкислении растворов молибдатов или вольфраматов не образуется гидратированных ионов M(VI)? Ознакомьтесь с составом и свойствами ацидокислот молибдена.
И. Что такое вольфрамовые бронзы?
36.13. Элементы семейства актиния (торий, уран)
Элементы от актиния (Z=89) до лоуренсия (Z==103) образуют семейство актшия, тогда как, собственно, к актиноидам относятся элементы, у которых идет заполнение 5/-уровпя. В отношении размещения электронов на 5f- или бй-уровпях для некоторых из этих элементов нет полной ясности (таб I. А 5). Следует надеяться, что в дальнейшем для трансурановых (транс-актчнневых) элементов можно будет надежно различать энергетические урс ни, соответствующие 5/-, 6d-, 7s- и 7р-орбиталям. Уже благодаря совет» 7 иому радиоактивному излучению происходит постоянное возбуждение ронов и тем самым образование «гибридоподобиых» состояний.
кктиний, торий, протактиний и уран с учетом особенностей их свойств । помещали в побочные подгруппы соответственно третьей, четвертой,
1 I и шестой групп периодической системы элементов. Оказалось, однако,
оТп элементы вместе с трансурановыми элементами (включая эле-' 103) образуют группу, аналогичную семейству лантаноидов. Все же
‘нты от тория до лоуренсия ие обнаруживают того сходства болыпин-( своих свойств, какое наблюдается в группе лантаноидов. В качестве 1 jpa можно указать на большое разнообразие степеней окисления акти-। щ (габл. В 38).
)ткрытый по реакции 242g4Pu (22ioNe, 4n)2e0i04Ku в конце 1964 г. эле-1 сурчатовий (Z=104) уже не принадлежит к актиноидному семейству, ; ляется аналогом гафння. В дальнейшем были получены по реакции ‘"..т (188О, 5н)261ю4Ки атомы более долгоживущего изотопа, Ku-261 (период полураспада 65 с). Теперь известны изотопы курчатовия с массовыми числами 254, 256, 258 и 259. По реакции 243g5Am (24i0Ne, 4 или 5л)261 юз или 260105 был получен еще один трансактиноидный элемент с порядковым номером 105, который можно рассматривать как аналог тантала. Синтез атомов элемента 105 удалось осуществить также при бомбардировке мишени с Cf-249 ядрами N-15; 24998Cf (157N, 4ii)2«’io5.
Атомы элементов 106 (экавольфрама) и 107 (экареиия) образуются при бомбардировке свинцовой или висмутовой мишеней ионами хрома большой анергии.
2о882РЬ 4- 5124Сг ->- 25910в -► спонтанный распад
2o%3Bi + ^24Cr __>2ei10e + 2n
Атомы экарения с массовым числом 261 на ~20% подвергаются спонтанному делению, а на 80% испытывают а-распад; в последнем случае образуются атомы элемента 105 (257105),
Химическое поведение актиноидов определяется тем, что 5f-, 6d-, 7s-_ 7р-орбитали перекрываются в пространстве, а их энергии оказываются лизкими Вследствие этого (в противоположность лантаноидам) для образо-
^0—-2027
626
Неорганическая химия
Таблица В.38. Степени окисления элементов с порядковыми номерами от 89 до 107
Элемент Порядковый (номер Степени окисления3
Ас 89 3
Th 90 (3), 4
Ра 91 (3), 4, 5
и 92 3, 4, 5, 6
Np 93 3, 4, 5, 6, 7
Pu 94 3, 4, 5, 6, 7
Am 95 (2), 3, 4, 5, 6, 7
Cm 96 3, 4, 5, 6
Bk 97 3, 4
Cf 98 (2), 3, 4
Es 99 2, 3
Fm 100 2, 3
Md 101 2, 2, 3
No 102 2, 3
Lr 103 3
Ku 104 4
Элемент 105 5
Элемент 106 (6)
Элемент 107 (7)
аНаиболее типичные степени окисления выделены полужирным шрифтом.
вания связей могут использоваться практически все из вышеперечисленных типов орбиталей. Отличия в химических свойствах лантаноидов от актиноидов в значительной степени определяются способностью атомов последних к образованию гибридных связей с вовлечением б^орбиталей.
Ниже будут рассмотрены лишь реакции и аналитическая химия тория и урана.
Ионы Th‘+, U4+ и иОа2+ — жесткие кислоты.
36.13.1. Торий
В своих соединениях торий проявляет почти исключительно степень окисления +4.
Рекомендуемые опыты
Для опытов используют водный раствор Th(NO3)4-5H2O.
1. К раствору соли тория добавляют по каплям раствор щавелевой кислоты или оксалата аммония и нагревают. Осадок оксалата тория при дальнейшем добавлении (NH4)2C2O4 растворяется (уравнение реакции). Прй подкислении этого раствора конц. НС1 и нагревании снова осаждается Th(C2O4)2 (уравнение реакции).
2. По аналогии с редкоземельными элементами торий дает трудиорас-творимый, гидратированный фторид; однако, он не дает фторидных комплексных анионов — фтороторатов. Поэтому осадок ThF4 не растворяется в избытке плавиковой кислоты. Проводят соответствующий опыт.
Торий, уран
627
36.13.2. Уран
В наиболее устойчивых соединениях уран имеет степени окисления +4 и +6. Соединения урана (VI) в кислой среде присутствуют в виде производных иона уравнила UO22+, а в щелочной среде — в виде производных иона диураната U2O72-. Ионы UO22+ и U2O72- могут быть осаждены в виде труднорастворимых солей.
Об открытии урана в виде дипероксоуранат-иона см. разд. 37.2.1.2.
Перевод урана из степени окисления +6 в степень окисления + 4 удается провести лишь под действием сильных восстановителей:
U4++2H2O 4=i UO22++ 4Н++ 2е* £° = О,ЗЗВ
Рекомендуемые опыты
Для проведения опытов готовят раствор UO2(CH3COO)2 или UO2(NO3)2.
1. При добавлении к раствору соли уранила растворов NaOH, КОН или NH3 выпадает желтый осадок диураната, например:
2UOg2+ + 2Na++ 6ОН" -----► Na2U2O7| + ЗНаО
Испытывают действие на осадок раствора (NH4)2CO3 (уравнение реакции'.).
2. К раствору соли уранила прибавляют раствор (NH4)2S. Образуется коричневый осадок состава (UO2)S. Испытывают отношение осадка к раствору (NH4)2CO3.
3. При добавлении к нейтральному или слабокислому раствору соли уранила раствора K4[Fe(CN)3] образуется коричневый осадок (иО2)г[Ре(СМ)б]. При обработке осадка раствором NaOH его цвет становится желтым (уравнение реакции'.). Как ведет себя в аналогичных условиях соответствующее соединение меди Cti2[Fe(CN)e] (разд. 36.15, опыты 12 и 13)?
Так же как и в опытах 1 и 2, проверяют действие на осадок раствора (NH4)2CO3.
4. Для отделения урана от хрома и ванадия оказывается существенным то, что ионы UO22+ в солянокислом растворе в присутствии избытка ионов SCN- образуют желтый UO2(SCN)2, который можно экстрагировать эфиром или метилизобутилкетоном. Проводят соответствующий опыт.
5. Раствор соли уранила подкисляют серной кислотой и вносят металлический цинк. Постепенно раствор окрашивается в зеленый цвет. При взаимодействии раствора соли урана (IV) с гидроксидом щелочного металла выпадает коричневый U(OH)4(—lgEIp=45).
Вопросы и задания
1. В чем состоит отличие в поведении тория от а) редкоземельных металлов и циркония при осаждении оксалатов? б) от титана и циркония при осаждении плавиковой кислотой?
2. Каковы возможности осаждения тория растворами КаСО3, КЮ3 и ^а2Н2Р2Об-6Н2О? В чем состоит отличие в поведении по отношению к этим Реагентам тория от поведения титана, циркония и редкоземельных метал-
s. Как объяснить способность атома тория к комплексообразованию’
4. Почему при растворении оксида урана (VI) в минеральных кислотах °s. бразуется ион UO22+?
40*
628
Неорганическая химия
36.14. Марганец (технеций, рений и элемент 107]
Для марганца известны все степени окисления от —3 д< 4-7, причем наиболее устойчивы степени окисленщ + 2(d5-), + 4 (d3-), +6 (d1-) и +7 (^-конфигурация). Соединс ния марганца(II) в значительной мере сходны с соединениям!-железа, кобальта и никеля в той же степени окисления.
Мп2+ относятся по Пирсону (разд. 33.4.3.4) к группе жестких кислот, тогда как Fe2^ Со2+ и NF4 причисляют к переход ной группе между жесткими и мягкими кислотами.
Наблюдается сходство марганца и его левого соседа в периодической системе — хрома. В подгруппе марганца также происходит повышение устойчивости высших степеней окисления при увеличении порядкового номера элемента. Так, например, пер-ренаты (KReO4)—менее сильные окислители, чем перманганаты (КМпО4):
МО2 + 2Н2О МО4" 4- 4Н+ 4- Зе~
Mn Re
Ео, В 1,68 0,51
Ионы ТсО4- занимают по окислительным свойствам промежуточное место между марганцем и рением. Для технеция известны лишь искусственно синтезируемые радиоактивные изотопы. Его максимальная, соответствующая номеру группы степень окисления реализуется в оксиде ТС2О7, сульфиде Tc2S? и фтороксиде ТсОзЕ. По своему химическому поведению технеций стоит ближе к рению, чем к марганцу.
Рений проявляет некоторое сходство со своими соседями по периодической системе элементов (слева — вольфрам, справа — осмий). Как и марганец, рений образует соединения различных степеней окисления от —1 до +7. Из солей перрениевой кислоты HReO4 наиболее известны калиевая и аммониевая соли. В ряде соединений рения осуществляются связи М.—М (разд. 36.11.1).
Для химии марганца наряду с реакциями осаждения окрашенного в розовый цвет иона М.п(Н2О)б2+ (опыты 1—3) важную роль играют реакции, идущие с изменением степени окисления (опыты 4—6).
Если лаборатория располагает соединениями рения, опыты проводят также с ними и сравнивают химические свойства ре' ния и марганца. О строении атома элемента 107 см. разд. 36.13-
36.14.1. Соединения марганца в различных степенях окисления
Исходя из соединений марганца(II), можно получить соедИ" нения марганца в более высоких степенях окисления; эти пре0' ращения могут быть осуществлены как в растворе, так и 0 расплавах солей.
Марганец
62»
Рекомендуемые опыты
1. Проводят реакции раствора соли марганца(II) с растворами NaOH, а2СО3 или (NH4)2CO3 и со смесью NH4C1—NH4OH.
В первых двух случаях осаждаются соединения Мп (ОН) а (белый, -Пр=12,7, pH начала осаждния 8,3) и МпСО3 (белый, — lgI7p=10,l). При стоянии на воздухе происходит частичное окисление марганца (II) до марганца (IV) и осадок быстро приобретает коричневый цвет. Образуются соединения диоксида марганца. Полное окисление солей марганца(II) до MnO2-aq можно осуществить в щелочном растворе при действии брома, Н2О2 и т. д.
Можно осадить марганец(П) в виде сульфида MnS (розовый, —Пр» 15) и аммонийфосфата Mn[NH4]PO4-H2O (слабая розовая окраска лучше заметна при больших количествах осадка).
2. При взаимодействии нейтрального или аммиачного раствора соли марганца (II) с (NH4)2S выпадает гидратированная форма MnS. Из кислых растворов осаждение не идет, так как концентрация ионов S2- оказывается недостаточной для достижения произведения растворимости MnS:
H2S 4- Н2О <=* HS- + Н3О+ рКа = 6,92
HS- + Н2О S2- + Н3О+ рКа ~ 13
Часто осадок MnS окрашен в грязные тона, так как вследствие окисления ионов Мп2+ до соединений Мп (IV) и ионов S2- до серы он представляет собой смесь MnS, МпО(ОН)2 и серы.
3. К раствору соли марганца(II) прибавляют раствор двухзамещенного фосфата натрия или аммония и раствор аммиака. Кристаллический осадок состава Mn(NH4)PO4-H2O при добавлении нескольких капель Н2О2 окрашивается в коричневый цвет (почедц/?) в отличие от соответствующих бесцветных солей магния и цинка (разд. 36.7, опыт 2).
4. а) Окисление ионов Мп?+ до МпОц~ в кислом растворе. В качестве окислителей могут быть использованы (NH4)2S2O3 в сернокислом растворе (ионы Ag+ в качестве катализатора; по-видимому, происходит их окисление до ионов Ag2+), а также РЬО2 и NaBiO3 (разд. 36.5, опыт 1) в азотнокислом растворе.
К нескольким каплям раствора MnSO4 добавляют немного РЬО2 (несодержащего марганца; провести холостой опыт), а затем несколько миллилитров конц. HNO3 и кипятят в течение нескольких минут. После разбавления водой и отделения избыточного РЬО2 путем центрифугирования можно наблюдать ярко-фиолетовое окрашивание вследствие образования МпО4_:
2Мп2+ + 5РЬО2 + 4Н+----->• 2МпО4“ + 5РЬ2+ + 2НаО
Окислению марганца в кислом растворе мешает присутствие CI", Вг~, I-, а также Н2О2 и др. (опыт 6).
б) Окисление марганца(П) до МпО^~ в щелочном растворе. Окисление Марганца (II) в щелочном растворе удается провести при действии гипобро-мита в присутствии катализатора (Си(II)):
2Мп2++ 5ОВг~ + 6ОН" -----> 2МпО4~ + 5Вг" + ЗН2О
Проводят соответствующий опыт.
в) Окисление в солевом расплаве (разд. 37 2.1.2). В лодочке из оксида Магния сплавляют немного соли марганца (II) или пиролюзита с трехкратным количеством тонкоизмельченной смеси равных частей безводной соды и Нитрата калия (ср. разд. 36.12, опыт 4). Расплав окрашивается в зеленый Ивет вследствие образования манганата (VI) МпО42~:
MnO2 + 2О2- ----> МпО43~ + 2е~
Б небольшом количестве образуется также синий манганат(V) МпО43-. Последний является основной составной частью «расплава манганата натрия»
«30
Неорганическая химия
(влияние катиона на устойчивость комплекса). Манганат(V) образуется также в присутствии значительного избытка щелочи или нитратов (почему?),
5. При растворении расплава манганата(VI) (опыт 4. в) в небольшом количестве воды и подкислении его разбавленной уксусной кислотой происходит изменение окраски раствора из зеленой в красно-фиолетовую (МпО4~). Через некоторое время осаждается коричневый MnO2-aq. Ион МпО42- может диспропорционировать:
2МпО4* 2~ --► 2МпО4~ 2е~
МпО42- + 4Н+ + 2е- ----> МпО2; + 2Н2О
ЗМпО42- + 4Н+ -----► 2МпО4" + МпО2( + 2Н2О
Свободный ион марганца (III) также весьма неустойчив. В кислом растворе
он диспропорционирует по схеме:
Мп2О3 + 2Н+ -----► МпО2 + Мп2+ + Н2О
Следует отметить, что MnOs-aq (—1g Пр»56) менее растворим, чем Мп(ОН)3 (—1g Пр»36).
6. Перманганаты — очень сильные окислители. В пробирках проводят соответствующие реакции. При проведении опытов 6. а подкисление следует производить добавлением разбавленной H2SO4, так как хлорид-ион окисляется перманганатом в сильнокислых растворах. С разбавленной НС1 реакция не идет, что связано, по-видимому, с кинетическими затруднениями процессов разрушения иона МпО4_ и образованием молекулы хлора из атомов. В соответствии со значениями стандартных потенциалов Мп2+/МпО4~ (1,52 В) и .2 С1—/С12 (1,358 В) должно идти окисление хлорид-иона перманганат-ионом.
а) В кислой среде
|Мп2+-|-4Н2О МпО4- + 8Н++5е" Е° = 1,52В
бледно- красно-
розовый фиолетовый
Сравните, например:
Fe2+ Fe®+ + е~ Е° = 0,771 В
21- =г=±= 1а + 2е~ Е° = 0,535 В
Н2С2О4 =s=±: 2CO2f + 2Н+ + 2е~ Е« = 0,47 В
Н2О2 ч=* O2f + 2Н+ + 2е' Ео = о, 68 В
б) В щелочной среде:
МпО2 + 4ОН" ч=* МпО4" + 2Н2О + Зе’ Е° = 0,59 В
Сравните, например:
SO32- + 2ОН- SO42- + Н2О + 2е- Е° = 0,93 В
Mn(OH)2 + 2ОН" МпО2( + 2Н2О + 2е~ Е° = 0,05 В
7. В сильнощелочном растворе МпО4~ восстанавливается под действие'1 иона С2О42~ лишь до (зеленого) иона МпО42~. Восстановление сульфитом идет до манганата(V), который в сильнощелочном растворе днспропорционирует-
/-НН0Н )
2МпО43- ----> МпО42-+ МпО44 * 6 7- (--> MnOJ + 2Н2О + 4ОН'^
К нескольким миллилитрам 0,01 н. раствора перманганата добавляю'1' приблизительно равный объем 0,5 н. раствора NaOH и 1—2 капли 10%-ног° раствора сульфита. Происходит изменение окраски раствора в зеленую.
Марганец
63L
Вопросы и задания
1. Как можно объяснить розовую окраску иона Мп2+, находящегося под действием слабого поля лигандов, например молекул воды?
2. Объясните, исходя из ^^-конфигурации иона [Mn(CN)e]4_ (октаэдрическая координация; сильное поле лигандов), его магнитные свойства.
3. На примере различных комплексов марганца (III) (слабое н сильное поле лигандов) поясните зависимость их магнитных свойств от различной заселенности и ее-орбитален.
4. Ознакомьтесь с магнитными свойствами соединений (в том числе комплексных) технеция и рения. Имеются ли отличия по сравнению с магнитными свойствами соединений марганца?
5. При добавлении NH3 к раствору соли марганца (II) в аммонийном буфере осадка не образуется (в этом случае также может идти быстрое окисление до марганца (IV)). На основе теории кислот и оснований Бренстеда поясните роль ионов NH4+. Кроме того, следует учитывать, что ион марганца (II) образует гексаминный комплекс, благодаря чему концентрация свободных (точнее, гидратированных) ионов марганца (II) уменьшается.
6. Что происходит при прокаливании соли состава Mn(NH4)PO4-H2O?
7. Почему марганец в высшей степени окисления находится в водных растворах в виде одноядерных оксокомплексов, а не в виде гидратированного иона, например [Мп(Н2О)4]6+?
8. Можно ли свойства комплексов марганца(VI) (^'-состояние) рассматривать в рамках теории поля лигандов?
9. Для процесса
Мп2+ + 4Н2О «=* МпО4- + 8Н+ + Бе-
йз зависимости окислительно-восстановительного потенциала от pH следует, что окисление Мп (II) до МпО4~ должно идти гораздо легче в щелочной среде. Это предположение не подтверждается (если не считать каталитического ускорения реакции в опыте 4.6). Дайте объяснение этому.
10. Напишите уравнение реакции перевода Мп2+ в МпО4- в окисляющем расплаве. Какую роль в этом процессе играют ионы СОз2-?
11. Почему прн восстановлении иоиа МпО4~ в присутствии фторид-ионов процесс восстановления может остановиться на степени окисления марганца + 3.
12. Что произойдет при пропускании Н2 в сильнокислый раствор перре-ната?
13. Какова степень окисления рения в ренидах?
36.14.2. Стабилизация степеней окисления
То обстоятельство, что у переходных элементов при образовании соединений в связях участвуют d-орбитали, энергетические уровни которых затем расщепляются при октаэдрическом, тетраэдрическом или квадратном расположении лигандов (ср. Разд. 6.5.6), способствует существованию большого числа степеней окисления у этих элементов.
В приведенной ниже обзорной таблице указаны известные к Настоящему времени (частью формальные) степени окисления элементов первого переходного периода:
£32 Неорганическая химия
-3 —2 —1 0 +1 +2 4-3 +4 +5 +6 +7
Sc + ? + +
Ti + + + + + +
V ? + + + 4- + + +
Сг + + + + + + + + +
Мп + + + + + + + + + , 1 +
Fe + 1 + + + + + + +
Со + + + + + ?
№ + + + + + +
Си + + -ф-
Zn +
Прежде всего обращают на себя внимание низкие степени окисления +1, 0, —1 и —2 (—3), встречающиеся в целом ряде комплексов. В табл. В.39 приведены лишь немногие из известных соединений, в которых лигандами являются нейтральные молекулы СО, NO, 2,2‘-дипиридил (Dipy), бензол СеН6, фталоцианин или NO+ и циклопентадиенил-иоп
Путем комплексообразования могут быть стабилизированы и высокие степени окисления, причем наиболее подходящие для этого лиганды — фтор и кислород (почему?). Если при комплексообразовании со фтором считать координационное число равным шести (что часто и наблюдается в действительности), т0 максимальная стабилизируемая степень окисления оказывается равной +5. При образовании оксокомплексов могут быть стабилизированы более высокие степени окисления. Так, уже при достаточно низких координационных числах (например, 4) стабилизируются высокие степени окисления (например, МпОГ> ReO4-).
В табл. В.40 приведены некоторые примеры фторо- и оксокомплексов с центральными атомами в различных степенях окисления, причем в меньшей степени уделено внимание теМ соединениям, в которых степень окисления центрального атома совпадает с номером группы.
Марганец
633
Таблица В.39. Стабилизация низших степеней окисления
Элемент Соединения
Титан -1 0 [Ti-DiPy3]“, TiDiPy3
Ванадий -1 -1 0 +1 [V(CO)6]-, [VDiPy3]-, V(C6H6)2, [VDiPy3]+
Хром -2 -1 0 +1 Na2[Cr(CO6)], Na2[Cr2(CO)10], C6H6Cr(CO)2NO, [Cr(C6He)2]I
Марганец Mn(NO)3CO, [Мп (фталоцианин)]2-, [Mn(CO)5]~
0 +1 [Mn(CO)5]2, C5H5Mn(CO)3
Железо oo+l [Fe2(CO)8]2-, [C6H6Fe(CO)2]-, Fe(CO)5, [FeNO(H2O)5]2+
Кобальт -1 0 [Co(CO)4]_, [Co(CO)4]3, [Co(NH3)CO]2-
Никель —1 -1/2 0 0 [Ni2(CO)6]2-, [Ni4(CO)9]2-, Ni(CO)4, [Ni(CN)4F-,
C5H5n‘i(CO)2
Теория жестких и мягких кислот и оснований (разд. 33.4.3.4) позволяет дать объяснение возможности стабилизации различных степеней окисления за счет комплексообразования с различными лигандами. Мягкие лиганды (например, СО, Р(СН3)3, C2H5NC) стабилизируют низкие степени окисления металлов (мягкие кислоты). Й наоборот, жесткие лиганды (такие, как ионы F- и О2-) способствуют стабилизации высоких степеней окисления металлов [Ni(CO)4 и KafNiFs], Na2FeO4].
Вполне объяснимо также существование соединений со связями металл — металл (например, Мп2(СО)ю, Fe2(CO)82~). Образование связи металл — металл формально можно рассматривать как кислотно-основное взаимодействие:
L— М+ :М'—L ---> L—М:М'—L
где L —мягкий лиганд. Как было показано выше, мягкие лиганды способствуют образованию соединений элементов в низких степенях окисления.
Вопросы и задания
Следует иметь в виду, что в ряде случаев указанные степени окисле-иия имеют формальный характер (например, у никеля).
1. Какова природа связи М—СО в карбонилах?
2. Почему такие соединения, как дипиридил и о-феиантролии являются
•634
Неорганическая химия
Таблица В.40. Фторо- и оксокомплексы
Фторокомплексы
Центральный атом в степени окисления +5
J»l{MVFe] Ml = Na, К, Rb, Cs
MV = Mo, W, Re
Ag[MVF6] MV = ru, os, Ir
K3[MVF8] MV = Mo, W
Центральный атом в степени окисления +4
MaI[MiVF6] MI = K, Rb, Cs
MIV = Y, Cr, Re, Ni, Rh, Pd, Os, Pt, Pr
•MII[MIVFe] МП = Ba
MIV =pt, Ir, Ru, Re, Mn
Центральный атом в степени окисления +3
Ag[AuniF4], Cs[AgIHF4], K3[NiiIIFe], K2[FeHlF5]-H2O, [NH^MnlilFg]
Оксокомплексы
Хроматы (IV) и хроматы (V) Манганаты (IV) и манганаты (V)
Na4CrO4 1ЛЗСЮ4 Li2MnO3 Na3MnO4
Ва2СгО4 Na4MnO4 Rb3MnO4
Ва3СгО5 Ba6(CrO4)3(OH) Ba3MnO5 Sr3[MnO4]2-Sr(OH)2
Ba5[MnO4]3(OH)
Другие простые оксокомплексы
SrFeO3 Na4CoO4 LiNiO2 NaCuO2
Sr2FeO4 Ba2CoO4 KNiO2 Na3CuO3
K3FeO4 B^gCoOg Ba2Ni2O6 Sr[CuO2]2-aq
M2lFeO4
-Ml = Na, K, Rb, Cs
Na3ReO5 KRuO4 Na5OsO8
Ba3(ReO6)2 KgRuOg K2OsO4
AyiReOj CaRuOs Li3IrO4
Li2RhO3 Na2PtO3
Li2PdO3
хорошими комплексообразователями? В этой связи следует ознакомиться с понятием о rt-акцепторных (я-кислотных) лигандах.
3. Рассмотрите электронную структуру ферроцена.
4. Как можно осуществить синтез фторокомплексов, приведенных в табл. В.40?
5. Какое влияние на стабилизацию определенных степеней окисления может оказывать размер ионов щелочного или щелочноземельного металла, не входящих в координационную сферу комплексного фторидного иона?
6. Как влияет относительное количество катионов иа возможность Д0' стижения определенной степени окисления? Сравните, например, соединения KstVFe] и K[VF6],
Железо, кобальт, никель
635.
7. Каковы возможности получения оксокомплексов с «аномальной» степенью окисления центрального атома?
8. Какие реакции могут происходить при внесении галогене- и оксокомплексов в воду? [О возможности осуществления ковалентных связей в комплексах, содержащих в качестве лигандов CN-, пиридин или СО, см. разд. 6.5 6].
36.15. Железо, кобальт и никель
У металлических железа, кобальта и никеля в возрастающей степени идет заполнение электронами 3 d-зоны, в то время как степень заполнения 4 s-зоны ниже, чем у изолированных атомов (ср. разд. 36.1.1): Fe Со NI
Число электронов в 4х-зоне, при- ~0,96 ~0,75 ~0,55
ходящихся на атом
Ни один из этих элементов в своих соединениях не достигает степени окисления, соответствующей номеру группы. Наиболее-устойчивы степени окисления +2 и +3, причем для никеля, за некоторыми исключениями (например, в K2[NiF6],> см. также-опыт 1), наиболее типична степень окисления +2 (конфигурация d8) (опыт 1). Во многих соединениях кобальта он также имеет степень окисления +2 (d7); степень окисления + 3 (d6) характерна главным образом для комплексных соединений кобальта, которые имеют сходство с комплексами хрома(III). Соединения железа в степени окисления +2 (d6) сходны с соединениями цинка; реакции иона железа (III) (d5) во многом похожи с реакциями ионов алюминия и хрома(III). Обладающие сильным окислительным действием ферраты(\71) (d2) FeO42~ напоминают хроматы (VI) и манганаты (VI); ферраты имеют тот же состав, что и сульфаты, и часто им изоморфны. Реакции соединений железа, кобальта и никеля в своем большинстве определяются склонностью этих металлов к изменению степени окисления и их способностью к комплексообразованию.
Причины различной относительной устойчивости многочисленных комплексов этих элементов можно понять с позиций теории жестких и мягких кислот и оснований. В качестве примера рассмотрим комплексные соединения кобальта в степени окисления +3: [Co(NH3)6]3+ (жесткая кислота), [Со(СМ)б]2~ (мягкая кислота). Если в кислотно-основном комплексе кислота — жесткая, то более устойчив комплекс с жестким основанием (например, с ионом F-) и менее устойчив комплекс с мягким основанием (например, с ионом I~): [Co(NH3)5F]2+ более устойчив, чем [Co(NH3)5l]2+. Подтверждением правила, согласно которому мягкие кислоты образуют более устойчивые комплексы с мягкими основаниями, служит сравнение различных сме
636
Неорганическая химия
шанных цианидных комплексов: [Co(CN)5I]3~ более устойчив чем [Со(CN)5F] 3~.
В то же время жесткий акцептор — ион Со3+ — сохраняет эти свойства лишь при комплексообразовании с жестким основанием NH3 ([Co(NH3)5]3+). Свойства пентацианидного комплекса как мягкой кислоты объясняются легкой поляризуемостью мягкого лиганда — иона CN-, благодаря чему на центральный атом переходит значительный отрицательный заряд, так что его степень окисления, а следовательно, и жесткость кислоты, понижаются* *.
Осадок Fe(OH)2 на воздухе меняет окраску от грязно-зеленой через черную (Fe3O4-aq) до коричневой (Fe2O3-aq). По-видимому, реакция идет с промежуточным образованием иона FeO22+ с последующим переходом в ион Fe(OOH)2+, который быстро разрушается. Формальное уравнение:
4Fe(OH)2 + О2 + 2Н2О 2Fe2O3-3H2O
Розовая окраска гидроксида кобальта также меняется** в результате аутоокисления и образования Со3О4.
Если при осаждении гидроксидом натрия заранее ввести окислитель (Н2О2, Вг2, МпО4~, Ю4~ и т. д.), получаются гидратированные оксиды металлов в более высоких степенях окисления. Из раствора соли железа сразу осаждается красно-коричневый Fe2O3-aq. Из растворов солей кобальта и никеля в зависимости от выбранного окислителя можно получить осадки различного состава: смеси Co2O3-aq и Co3O4-aq, Co2O3-aq и CoOa-aq, чистый Co2O3-aq; осадки сходного состава получаются из растворов солей никеля.
Можно написать следующие реакции (М = Со, Ni):
ЗМО + Н2О <—>• М3О4 + 2Н+ + 2е~
2М3О4 + Н2О 1—fc ЗМ2О3 + 2Н+ + 2е-
М2О3 + Н2О <—2МО2 + 2Н+ + 2е-
CoO2-aq и NiCh-aq сразу взаимодействуют с водой:
2МО2 + 2Н+ + 2е~ -> М2О3 + Н2О
Н2О --->- 2Н++1/2О2 +2е~
2МО2 --> М2О3 + 1/2О2 "
В отличие от Со2О3 Ni2O3 способен окислять воду, хотя реакция идет значительно медленнее, чем с NiO2-aq:
3Ni2O3 + 2Н+ + 2е~ -► 2Ni3O4-}-H2O
Н2О ---э- 2Н++ 1/2О24-2е-
3Ni2O3 ---» 2Ni3O4+ 1/2О2
* При фактическом переносе заряда, формальная степень окисления остается прежней--1-3. Речь идет об эффективном заряде.—Прим, перев.
** Переходит в коричневую.— Прим, перев.
Железо, кобальт, никель
637
При доступе воздуха в растворах, содержащих железо и кобальт, идет медленное окисление: осаждение Fe2O3-aq, окрашивание раствора соли кобальта в красный цвет вследствие образования аква- и ацидопентаминных комплексов кобальта(III).
Окисление соединений кобальта(II) в присутствии сильных лигиндов идет гораздо легче, чем в их отсутствие; это связано с тем, что Со3+ образует с этими лигандами прочные комплексные ионы:
[Со(Н2О)в]2+ [Со(Н2О)в]3+ + е~ £о = 1,84 В
[Co(NH3)e]2+ [Co(NH3)e]3+ + е~ = 0,1 В
В присутствии солей аммония гидроксиды раствором аммиака не осаждаются (почему?). На воздухе идет окисление солей железа и кобальта до соединений со степенью окисления металлов + 3.
Для аналитической химии имеют значение реакции осаждения кобальта и никеля сульфидом аммония.
36.15.1. Реакции железа(П) и (III)
Окисление соединений железа (II) в соединения железа (III) в щелочной среде идет очень легко (опыты 1 и 2), а в кислой среде требует применения сильных окислителей.
Гидратированные ионы Fe(III) (имеющие слабую малиновую окраску) представляют собой сильную кислоту:
[Fe(H2O)„]’+ [Fe(H2O)5(OH)]2++ Н+ ptfa = 2,22
Поэтому гексааква-ионы преобладают лишь в сильнокислых растворах (рН<1). Различные гидроксо-ионы [Fe(H2O)e-n (ОН) „](3~’1)+ окрашены в желтый цвет, так как их полосы переход >з с переносом заряда простираются и в видимую часть спектра.
При осторожном проведении реакции протолиза растворов солей железа (III) (нитрата, сульфата, аммонийсульфата) при pH 2,2 наблюдается появление красно-коричневого окрашивания вследствие образования коллоидных растворов, содержащих изополиоснования [FeO(OH)]x. Эти частицы образуются Путем конденсации одноядерных гидроксокомплексов. При дальнейшем повышении pH раствора происходит полное осаждение Железа в виде Fe2O3-aq. Исследование этих осадков методами ИК-спектроскопии и ЯМР указывают на присутствие в них ОН-групп, что дает основание называть их конденсированными гидроксидами. При старении осадков и при их нагревании процессы конденсации приводят к продуктам с меньшим содержанием воды и в конце концов к безводному оксиду а-Ре20з (гематит).
Помимо комплексообразования с цианид-ионами (опыты 9, 12, 13 и 14) для железа(III) представляют интерес реакции « Cl- SCN-, F- и РО43-.
638
Неорганическая химия
При добавлении к растворам солей железа (III) роданида калия происходит окрашивание раствора в кроваво-красный цвет вследствие образования Fe(SCN)3 и комплексных роданидных анионов (вплоть до [Fe(SCN)6]3~). Это — чувствительная качественная реакция на ионы железа(III), если растворы достаточно концентрированы.
При разбавлении водой идет диссоциация комплексов и происходит ослабление красной окраски растворов:
[Fe(SCN)e]3- Fe(SCN)3 + 3SCN” рК = 3,5
Fe(SCN)3 «=t [FeSCNF+ 4- 2SCN рЛГ = 5,4
[Fe(SCN)F+ ч—> Fe3+ + SCN’ рК = 2,9
Если к раствору роданида железа (III) прибавить избыток фторида калия или несколько миллилитров сиропообразной фосфорной кислоты, можно наблюдать обесцвечивание раствора:
[FeFe]3- х—* Fe3++ 6F" рК ж 16
В растворе присутствуют также фторокомплексы с координационным числом по фтору меньшим, чем шесть
[FePO4H]+ 4==h Fe3+ 4. [НРО4]2~ рК = 9,4
Кроме этих ионов в растворе присутствуют фосфорные комплексы, например [Ре(РО4)г]3-.
36.15.2. Реакции комплексообразования железа, кобальта и никеля
Железо в степени окисления +2 и кобальт в степени окисления + 3 образуют чрезвычайно устойчивые комплексы; это приводит к тому, что для комплексных ионов обычные реакции железа(II) и кобальта(III) в большинстве случаев не наблюдаются.
Процесс окисления с образованием комплексных ионов железа (III) и кобальта(III) идет легко, несколько различаясь для этих двух металлов (опыт 9).
Большая константа устойчивости [Co(CN)e]3~ по сравнению с [Ni(CN)4]2- приводит к тому, что при действии на соответствующие растворы смеси NaOH—Brs кобальт не осаждается в виде Co2O3-aq (опыт 10).
На склонности солей кобальта к образованию комплексов основана важная в аналитической химии реакция ионов кобальта (II) с нитритами в уксуснокислом растворе (опыт 11).
В аналитической химии имеют значение также реакции ионов [Fe(CN)6]4~ и [Fe(CN)s]3~ с катионами тяжелых металлов. Высокую устойчивость ионов [Fe(CN)6]4- и [Fe(CN)s] (р7(«44 и р/С~35) иллюстрирует опыт 14. Все труднораствО" римые цианоферраты разрушаются при нагревании со Ш671 чами.
Железо, кобальт, никель
639
36.15.3. «Берлинская лазурь» и «турнбулева синь»
При взаимодействии раствора K4[Fe(CN)6] с избытком раствора ионов Fe(III) образуется «нерастворимая берлинская лазурь» Fe4[Fe(CN)6]3. Согласно исследованиям мёссбауэровских спектров (разд. 6.5.4) атомы железа в этом соединении имеют вполне определенные степени окисления, а именно +2 и +3. При этом возможность «резонанса» в пределах молекулы* между обоими состояниями окисления Fe2+/Fe3+-<->-Fe3+/Fe2+ исключается.
«Турнбулева синь» образуется при добавлении избытка соли железа(II) к раствору Кз[Ре(СМ)6]. Сходство мёссбауэровских спектров «турнбулевой сини» и «берлинской лазури» подтверждает ранее высказанные предположения о том, что при осаждении первой происходит взаимный обмен состояний окисления:
Fe2+ + [FeHl(CN)e]3- -► Fe3++[Fell(CN)e]4-
Эта реакция идет в соответствии со значениями стандартных потенциалов процессов:
Fe2+ 2: Fe3+-|- ё~ Еа = 0,771 В [Fe(CN)e]4~ [Fe(CN)e]3- + е~ Е° = 0,36 В
Если при реакции мольное соотношение между K4[Fe(CN)6] и ионами Fe(III) или Ka[Fe(CN)6] и ионами Fe(II) равно единице (1:1), то получают «растворимую берлинскую лазурь» состава KFeni[Fen(CN)6] Сравнение мёссбауэровских спектров этого соединения со спектрами «нерастворимой берлинской лазури» и «турнбулевой сини» показывает, что речь идет действительно о гексацианоферрате(П) калия-железа(III).
Рекомендуемые опыты
Для проведения опытов используют растворы солей железа (соль Мора), кобальта и никеля в степени окисления +2.
1. К растворам соответствующих солей прибавляют NaOH, нагревают (раствор, содержащий кобальт) до кипения и оставляют на воздухе, часто перемешивая. Выпадают осадки: Fe(OH)2 (—1g Пр» 15—19) при рН>5,8 (в отсутствие следов ионов Fe(III) чисто белый осадок); Со(ОН)2 (—1g Пр» 15—18) при pH3=7,5 (на холоду выпадает основная соль синего Цвета, которая при нагревании дает розовый Со(ОН)2); зеленый Ni(OH)2 (—1g Пр» 15—18) при >7,4.
2. Проводят реакции осаждения водным раствором аммиака в присутствии и в отсутствие NH4C1.
При добавлении лишь раствора аммиака (и без доступа воздуха) образуются труднорастворимые гидроксиды (см. опыт 1). При избытке осадителя «ни растворяются, переходя в соответствующие гексаминкомплексы lM(NH3)e]2+ (M=Fe бесцветный комплекс; М=Со желтый комплекс, РЛ=5,1; M=Ni синий комплекс, рК=8,8).
* Точнее, по-видимому, говорить о кристаллической решетке. — Прим, пе* Рев,
640
Неорганическая химия
3. При пропускании сероводорода в кислые растворы солей осаждения сульфидов ие происходит:
М2+ + HaS + 2Н3О2 5=t MS + 2Н3О+
Fe Со Ni
рК реакций +3 —1 ±0
—1g Пр MS 18-22 21—27 20—28
Черные сульфиды осаждаются при пропускании H2S в нейтральные растворы или в растворы, содержащие ацетатный буфер; осаждение также идет при действии раствора (NH4)2S. Если вместо (NH4)2S берут (NH4)2SX, Ni и Со часто образуют коллоидные растворы, коагуляции которых можно добиться кипячением с раствором ацетата аммония.
Испытывают действие холодной разбавленной НС1 на осадки сульфидов, находившиеся некоторое время на воздухе. Осадок FeS легко растворяется, в то время как сульфиды кобальта и никеля, переходящие под действием воздуха в сульфиды с большим содержанием серы, могут быть переведены в раствор лишь при действии конц. HNO3, смеси Н2О2—СН3СООН и др.
4. К раствору соли железа(II), подкисленному разбавленной H2SO4> прибавляют несколько капель конц. HNOs и нагревают. Наблюдается переход окраски раствора в желтую.
' 3Fe2+ + NO3~ + 4Н+ -----► 3Fe3+ + NO + 2H2O
Образование на промежуточной стадии комплексного нона [Fe(NO) (H2O)S]2+ сопровождается появлением бурого окрашивания раствора, обусловленного переходами с переносом заряда в системе Fe—N—О.
5. Железо(Ш) в кислой среде действует как мягкий окислитель:
Fe2+
Fe3+-j- е~
a) SO2 + 2Н2О HSO4" + ЗН+ + 2е~
б) Sn2+ Sn4+ + 2е~
=0,771 В £о = О,17В Е° = 0,15 В Е° = 0,44 В
в) Fe fc Fe2+-f-2e~
Последнюю из приведенных реакций используют, когда необходимо и раствора солн железа(II) удалить ионы железа(III):
2Fe3+ + Fe ----> 3Fe2+ рК = —41
6. Проводят на холоду реакции раствора соли железа (II) с растворам*: NaOH, NH3—NH4C1, Na2CO3; при кипячении проводят реакции с растворами NaCH3COO (разд. 36.12) и уротропина (разд. 36.8, опыт 4).
7. Немного FeCl3 растворяют в воде. Желто-коричневый раствор ° (см. выше) при добавлении НС1 светлеет (уравнение реакции)-, при дальнейшем прибавлении НС1 образуется желтый раствор хлороферрата(III), в котором главным образом присутствует тетраэдрически координированный ион [FeClJ- Из этого раствора железо может быть экстрагировано эфиром или метилизобутилкетоном. При концентрации ионов железа, превышающей концентрацию ионов водорода, происходит частичное образование ионов с октаэдрической координацией [FeCl4(H2O)2]~.
8. К нейтральным растворам солей железа (II), кобальта (II) и никеля (II) прибавляют по каплям раствор KCN (тяга!). Выпадают осадки Fe(CN)2 (коричневый), Co(CN)2 (красно-коричневый; структура цианида кобальта(П) соответствует формуле Co[Co(CN)3]2). Цианиды кобальта и никеля Ni(CN)2-aq, светло-зеленый; Ni[Ni(CN)<]) растворяются при дальнейшем добавлении KCN с образованием цианидных комплексов. Следует избе
Железо, кобальт, никель
64Г
гать избытка KCN (см. опыт 9):
Цвет рХ
[Fe(CN)ep- Бледно-желтый ~44
[Co(CN)6]4- Коричневый 19,1
[Ni(CN)4p- Желтый 15,3
Растворы комплексов сохраняют для проведения других опытов.
9. При нагревании раствора, содержащего ионы [Fe(CN)6]4~r с бромной* водой окраска раствора становится красновато-коричневой:
[Fe(CN)6]*- ч==Ь [Fe(CN)6]3- 4- е~ Е° = 0,36 В
Избыток цианида калия взаимодействует с бромом, давая очень ядовитый бромциан BrCN. Поэтому обязательно нужно работать под тягой.
Уже при соприкосновении раствора комплексной соли кобальта (II) с воздухом начинается окисление, которое значительно ускоряется при добавлении нескольких капель Н2О2:
[Co(CN)6]4- [Co(CN)e]s- -|- е- £'° = — 0,83 В
Раствор становится желтым. Даже в отсутствие кислорода воздуха происходит медленное окисление (уравнение реакций).
10. К растворам комплексных цианидов кобальта и никеля прибавляют окисляющую смесь NaOH—Вг2 и нагревают (тяга!). Из раствора соли никеля осаждается осадок состава Ni2O3-aq (опыт 1), в то время как в растворе соли кобальта изменений не наблюдается:
2 [Ni(CN)4]2~ + 6 ОН" + 9 Вг2 ---► 2 Ni(OH)3| + 10 ВГ + 8 BrCN
11. Раствор соли кобальта (II) почти насыщают твердым KNO2. При подкислении этого раствора уксусной кислотой образуется желтый осадок Кз[Со(И02)б]. Выделяющаяся при подкислении азотистая кислота окисляет Со (II) до Со (III), который с избытком нитрита дает комплексный гексанитрокобальт (III) :
Со2+ + NO/ + 2 Н+ ------► Со3+ -J- NO -J- Н2О
Co3+ + 6NOa- -----> [Co(NOa)e]3-
12. Проводят реакции растворов гексацианоферратов (II) и (III) с солями серебра(I) или меди (II). Образуются окрашенные осадки цианоферратов серебра и меди: Ag4[Fe(CN)6] (белый, — 1§Пр=40,8), Ag3[Fe(CN)6) (оранжево-красный), Cu2[Fe(CN)6] (красно-коричневый), Cu3[Fe(CN)e]2 (зеленый) .
Таким же образом проводят реакции гексацианокомплексов с ионами Железа (II) и железа(III). Так как эти реакции характеризуются очень низким пределом обнаружения, в качестве соли железа (II) используют свежеприготовленный раствор соли Мора (более подробно о «берлинской лазури» н «турнбулевой сини» см. выше).
13. Осадок цианоферрата нагревают с раствором гидроксида щелочного Металла:
Fe4[Fe(CN)e]3 + 12 ОН“ ----> 4 Fe(OH)3+ 3 [Fe(CN)6]«-
14. К растворам Ks[Fe(CN)e] и K4[Fe(CN)3] прибавляют растворы аммиака и (NH4)2S. Ни гидроксиды, ни сульфиды железа в осадок не выпадают.
Вопросы и задания
1. Используя уравнение Нернста объясните, почему в щелочной среде Железо (II)—чрезвычайно сильный восстановитель:
Fe(OH)a + ОН' Fe(OH)3 + е~ Е° = —0,56 В
2027
642
Неорганическая химия
Следует учесть, что
Fe(OH)2 ч=±= Fe2++2OH- —1g Пр « 16
Fe(OH)3 Fe3+ + 3 ОН" —1g Пр « 38
2. Используя значения стандартных потенциалов и уравнение Нернста рассчитайте константу равновесия реакции Sn2++2Fe3+^tSn‘l++2Fe2+, которая применяется при количественном определении железа в солянокислом растворе
3. Из уравнения Нернста следует, что в системе Fe3+/Fe2+ потенциал Е будет равен бесконечности, если активность ионов Fe2+ равна нулю: £= = 0,771 + 0,059 lg [Fe3+]/0 На практике, однако, значение £=1,5 В Почему это так? Сходные рассуждения можно распространить иа ряд аналогичных систем
4. Используя уравнение Нернста объясните, почему при добавлении фто-рид-ионов окислительное действие ионов Fe(III) ослабляется, а восстановительное действие ионов Fe(II) увеличивается.
5. Рассмотрите расщепление и заполнение электронами d-уровней центральных атомов для цианидных комплексов (сильное поле лигандов) железа и кобальта в степени окисления +2 (конфигурации d6 и d1). Объясните причину различной устойчивости этих комплексов
6. Сравните силу лигандов CN- и F~ и для случая октаэдрического комплекса железа(Ш) (^-конфигурация); объясните различия в заполнении tig- и ег-орбиталей в ионах [Fe(CN)e]3- и [FeFe]3-.
7. Какое влияние оказывает энергия стабилизации в поле лигандов иа образование тетраэдрических и октаэдрических комплексов кобальта (II) (сР-конфигурация)? Сравните устойчивость октаэдрических и тетраэдрических комплексов кобальта(II).
8. Дайте объяснение изменению окраски при подкислении водного раствора СоС12 соляной кислотой Для объяснения проведите рассмотрение спектров поглощения Используя спектрохимический ряд как характеристику силы поля лигандов, объясните изменение окраски, происходящее при добавлении избытка раствора аммиака к раствору NiSO4.
9. Какова должна быть бгег-коифигурация основного состояния иона Со2+ при октаэдрической координации в слабом и в сильном поле лигандов?
36.16. Платиновые металлы
Эти элементы подразделяются на группу «легких» (рутений, родий, палладий) и «тяжелых» платиновых металлов (осмий, иридий, платина). При сравнении с «группой железа» можно сразу отметить большое разнообразие степеней окисления (табл. В.41). Лишь в оксидах рутения и осмия эти элементы имеют степень окисления +8, соответствующую номеру группы периодической системы. Соединение дикарбонилоктафторид платины Pt(CO)2F8 следует, по-видимому, все же рассматривать как (FCO+)2[PtF6]2+
В соединениях OsF/hOsOFs, а также в LisOsOe, Ва5[ОзОб]2 и КзОэОб осмий имеет степень окисления +7. За исключением палладия для всех платиновых металлов известны соединения со степенью окисления +6. Примером могут служить гексафт0' риды, представляющие собой очень сильные окислители. Так PtF6 может окислять кислород и ксенон (разд. 35.4.2). Для этих элементов очень характерно образование комплексных ионов,
Платиновые металлы
643
Таблица В.41. Степени окисления платиновых металлов
Ru (В, (II), (III), (IV) (V). VI, (VII), VIII Rh (II), III, (IV), (VI) Pd П, (III), IV
Os Ir Pt
(II), (III), IV, VI, (VII), VIII I, (II), III, IV, VI II, (III), IV, (VI), (Vlliy
что часто используется при их аналитическом определении. В то же время комплексообразование делает затруднительным точное определение величин стандартных потенциалов.
Палладий растворим в конц. HNO3. Остальные платиновые металлы, за исключением рутения, родия и иридия, могут быть растворены в «царской водке». Окисление Ru, Rh и Ir удается провести при повышенных температурах, например, при их нагревании с кислородсодержащей соляной кислотой. Важное значение имеет способность некоторых платиновых металлов i (платины и, особенно, палладия, см. опыт 2) растворять значи-; тельные количества водорода.
Ниже рассматривается лишь химия палладия и платины; с*Для опытов используют раствор палладия в конц. HNO3 (ионы Pd(II), и, по-видимому, нитритные комплексы палладия) и раствор платины в «царской водке» (ионы [PtClg]2-). В обоих случаях избыток кислот осторожно удаляют упариванием и затем растворы разбавляют водой до нужной концентрации.
Особенность обоих металлов, как принадлежащих к группе благородных, состоит в их способности легко восстанавливаться в водных растворах:
Pd Pd2+ + 2е” £° = 0,99 В
Pt + 6СГ [PtCIe]2- + 4е- £о = о,73 В
В активации водорода, растворенного в палладии, можно убедиться, выполнив опыт 2.
Для аналитической химии платины в степени окисления +4 имеют значение реакции с КС1 и NH4C1. Осадки солей палладия образуются лишь в том случае, если и раствор палладия, и раствор осадителя достаточно концентрированы (для KzPdCLt р7<=5,2). При отделении осадков комплексных солей платины центрифугированием они, вследствие разложения и Выделения тонкораздробленной платины, становятся черными. В ходе систематического анализа палладий и платина должны, отделяться в процессе восстановления. Есть опасность, что, бла* годаря устойчивости иона [PtCle]2' (опыт 1), платина попадет в ходе разделения в «группу H2S».
В целях отделения иона Pd(II) от катионов других тяжелых металлов проводят осаждение диацетилдиоксимом (опыт 5). 41*
644
Неорганическая химия
Рекомендуемые опыты
1. К нейтральным или слабокислым растворам солей металлов добавляют восстановитель, например, муравьиную кислоту, и слабо нагревают (выделение газов, почему?). В противоположность палладию восстановление платины идет вяло и лишь при длительном нагревании через стадию коллоидного раствора приводит к постепенной коагуляции металла Также очень медленно идет восстановление платины формальдегидом НСОН в растворе NaOH.
2. Металлический палладий, полученный в опыте 1, осторожно отфильтровывают на небольшом стеклянном пористом фильтре. При этом наблюдается появление налета палладия на стенках, а осадок довольно сильно разогревается (каталитически активированный синтез гремучего газа). Особенно отчетливо эти явления можно наблюдать при больших количествах взятого металлического палладия (~ 100 мг и больше).
3. К слабокислым растворам солей палладия и платины добавляют растворы КС] или NH4C1. Выпадают осадки KiPdClJ (коричневый), KJPtCl6] (желтый, —1§Пр = 5,0), (NHj^PdClj (оливковый) и (NHJ^PtClsJ (желтый, —1g Пр «5).
4. В солянокислые растворы ионов Pd(II) и [PtCl6]2- пропускают H2S. Раствор, содержащий платину, подогревают. Выпадают осадки PdS (черный) и PtS2 (темно-коричневый), растворимые в царской водке. PtS2 растворяется также в (NH4)2SX с образованием [PtS3]2~.
5. К нейтральному или слабокислому раствору соли Pd(II) добавляют на холоду 1%-ный спиртовой раствор диацетилдиоксима. Выпадает желтый осадок диацетилдиоксимата палладия, свертывающегося в хлопья при кратковременном нагревании.
Вопросы и задания
1. Используя уравнения Нернста покажите возможность увеличения активности благородных металлов при комплексообразовании.
2. Что можно сказать о величине константы устойчивости иона [PtCl6]2-, зная, что процесс его восстановления протекает довольно медленно?
3. Что представляют собой палладированный и платинированный асбест?
4. Объясните принцип действия зажигалки Деберейнера.
36.16.1. Системы переходный металл — водород
Наряду с солеобразными и ковалентными гидридами (разд. 35.1.1.1) существуют соединения водорода, так называемые «металлические гидриды», образуемые переходными элементами. В них водород тем или иным образом внедрен в решетку металла. Часто при этом не образуется стехиометрических соединений и в системе М — Н имеют место весьма сложные фазовые соотношения. Ниже в качестве примера приведены данные для системы гафний — водород:
Состав Содержание водорода, ат. % Равновесные фазы
HfHfl,023-0,995 2,25—49,9 Hf+псевдокубическая фаза
HfHl.se-1,53 57,6—60,5 Псевдокубическая фаза+Hf
HfH1.7a-1.sa 63,0-64,8 Кубическая фаза
HfHt Д8— 1,8в 64,0—65,0 Кубическая и тетрагональная фаза
HfHi.sT-i.as 65,3—66,4 Тетрагональная фаза
Медь, серебро, золото, цинк, кадмий, ртуть 645
При теоретическом объяснении растворимости водорода в металлах используют модель «диссоциации абсорбированных атомов водорода на протоны и электроны». Этой диссоциации предшествует процесс диссоциативной хемисорбции молекул водорода на поверхности металла. Можно предположить, что в гидридах никеля, палладия и платины (в известной степени прототипах металлических гидридов вообще) освобождающиеся электроны переходят в d-зоны, у которых плотность электронных термов гораздо выше, чем в s-зонах (разд. 36.1.1). Внедренный водород находится преимущественно в виде протонов в октаэдрических пустотах кубической гранецентрированной решетки этих металлов (протонная модель растворения водорода).
Нахождение электронов водорода в электронном газе соответствующей решетки металла дает основание говорить в таких случаях о «металлическом» типе связи водорода. Этот тип химической связи полностью реализуется лишь в гидридах переходных металлов VI—VIII групп. У переходных металлов V, IV и у некоторых металлов III групп происходит постепенный переход к солеобразным гидридам, которые типичны для непереходных металлов I и II групп. Основной причиной этого перехода от металлического к ионному типу связи следует считать уменьшение электроотрицательности металлов при продвижении влево по периоду и, как следствие, оттягивание валентных электронов металлов к атому водорода. В то же время гидриды переходных металлов I и II групп, также как непереходных металлов III группы занимают промежуточное положение между солеобразными гидридами и летучими гидридами непереходных элементов V, VI и VII групп. В этом же направлении, начиная с типично металлических гидридов, наблюдается плавный переход и в типе связи — от металлической к атомной связи: валентные электроны атома водорода во все большей степени оттягиваются к его партнеру по связи вследствие возрастания электроотрицательности последнего. Таким образом, оказывается, что у гомеополярных гидридов элементов главной подгруппы VII группы атом водорода поляризован положительно.
36.17. Элементы побочных подгрупп первой и второй групп
36.17.1. Медь, серебро н золото
В металлических Си, Ag и Au имеются практически полностью заполненные d-зоны; число d-электронов, приходящихся на атом в металлах, соответствует электронной конфигурации изолированных атомов (и—1) d^ns1. Все же относительно легко идет возбуждение электронов из d-зон в ближайшие по энергии s-зоны. ,
646
Неорганическая химия
При образовании соединений в степенях окисления 4-2 и 4-3 также затрагиваются электроны из d-оболочки.
Для серебра наиболее типичны степени окисления +1 (4d10), для меди 4-2 (3d9) и для золота 4-3 (5d'8). Медь и золото образуют также соединения в степени окисления 4-1, которая соответствует номеру группы. В то же время медь и серебро могут иметь и более высокие степени окисления, например
CsjCuFJ, KCuOa, K7[Cu(IOe)2] • 20Н2О, Na9[Cu(TeOe)2] • 20Н2О
AgF2, [Ag(Py)4]S2O8, «AgO»=AgIAgI11O2, K[AgF4], BafAgFj, K3H4[Ag(IO6)2] • 4H2O, Na7H2[Ag(TeO6)2] • 24H2O
По теории жестких и мягких кислот и оснований (разд. 33.4.3.4) Cu+, Ag+, Au+ и Au3+ относятся к мягким кислотам Льюиса. Си2+ занимают промежуточное место между жесткими и мягкими кислотами.
Химические свойства меди, серебра и золота определяются малой химической активностью металлов (благородные металлы) :
Си •<=. Си+ 4- е"
Си Си2+ 4- 2е"
Е° = 0,52В
Е° = 0,35 В
Ag ч=* Ag+4-е"^, £о = 0,80 В
Аи ч=Ь Аи+4-е- £0= 1,46В
Аи «-=. > Аи3+ 4- Зе" £° = 1,50 В
Для химии этих элементов характерны их способность к изменению степени окисления и возможность образования ряда труднорастворимых солей. Особое значение для химического поведения этих элементов имеет комплексообразование. В противоположность элементам побочной подгруппы второй группы (разд. 36.17.2) медь, серебро и золото могут кроме электронов Л5-уровня (где я = 4, 5 или 6) отдавать один или два электрона (я—1) d-уровня. В последнем случае образуются соединения этих элементов в степени окисления 4-3, которая не известна для цинка, кадмия и ртути.
Образование труднорастворимых соединений меди и серебра и в то же время очень легкая способность к восстановлению соединений золота, определяют при проведении процесса разделения принадлежность этих элементов к различным аналитическим группам:
Медь «Группа H2S» CuS или Cu2S
Серебро «Группа соляной кислоты» AgCl
Золото Выделение при действии восстановителей Аи
Медь, серебро, золото, цинк, кадмий, ртуть
647
36.17.1.1. Свойства меди
Медь и серебро растворяются в HNO3 и в конц. H2SO4 (при нагревании). Золото растворимо только в царской водке.
NO + 2 Н2О NO3" + 4Н+ + Зе" £° = 0,96 В
SOa + 2H2O HSO4-4-3H++ 2е" £" = 0,17 В
(в качестве побочных продуктов могут образоваться сера и сероводород)
Си Си2+ + 2е‘
Ag у=* Ag+ 4- е~
Аи 4СГ y==t [AuCUl' + Зе' £° = 1,00 В
При количественном определении меди ее осаждают в виде труднорастворимого роданида CuSCN (—1g Пр =13,4) (опыт 5).
Ион Си (II) в водных растворах образует гидроксо- и аминокомплексы.
При осаждении раствором NH3 выпавший сначала осадок основного сульфата меди растворяется, давая темно-синий раствор, содержащий ионы [Cu(NH3)4(H2O)2]2+ (рХ=13,3, ср. также разд. 33.6.1), которые, как и множество подобных комплексов с жесткими основаниями Льюиса Н2О и NH3, можно считать «кислотно-основным комплексом».
Принадлежность меди к аналитической «группе сероводорода» доказывает опыт 7.
36.17.1.2. Реакции иона серебра(1)
Некоторое сходство серебра (I) и меди(1) обнаруживается в том, что их соли с одними и теми же анионами оказываются труднорастворимыми. Оба эти иона — мягкие кислоты. Кроме этого, для серебра (I) типичны реакции комплексообразования (разд. 33.6.1).
У иона Ag+ имеются разнообразные возможности для образования гибридных орбиталей и, соответственно, различных типов комплексных соединений. Наибольшее значение среди них имеют комплексы AgX2 (например, [Ag(NH3)2]+ с линейной координацией). Этот вид координации может осуществляться благодаря небольшой разности в энергиях заполненной 4d-op-битали и свободной внешней 55-орбитали и образованию гибридных ds-орбиталей. Аналогичные представления могут использоваться при описании большинства линейных комплексных ионов типа Си’Х2 и Ag‘X2.
При увеличении поляризуемости анионов катионом серебра (переход от ионной к атомной связи) происходит постепенное уменьшение растворимости и углубление окраски соответствующих соединений:
648
Неорганическая химия
AgF AgCl AgBr Agl Ag,S
—1g Пр Хорошо растворим 9,96 12,4 16,0 49
Цвет Бесцветен Белый Желтоватый Светло-желтый Черный
Ход изменения растворимости галогенидов серебра можно объяснить и в терминах теории жестких и мягких кислот и оснований. Фторид-ион — более жесткое основание, чем хлорид-ион; свойства бромид-иона занимают промежуточное положение при переходе к типично мягкому основанию — иодид-иону. Поскольку ион Ag+ представляет собой мягкую кислоту, силы взаимодействия катиона и аниона возрастают от AgF к Agl, что имеет следствием уменьшение растворимости галогенидов в том же направлении. Различие в растворимости труднорастворимых соединений серебра можно качественно наблюдать в опыте 8.
Соотношения между величинами констант устойчивости ряда комплексов серебра и произведений растворимости различных соединений иллюстрирует опыт 9.
36.17.1.3. Золото
Как и у серебра, у золота в степени окисления +1 труднорастворимы хлорид, бромид и иодид (—1g Пр =16). Соединения золота(I) очень легко диспропорционируют, и поэтому ион Аи+ (как и ион Си+) может присутствовать в растворе лишь в очень малой концентрации:
3 Аи+ --► 2 Au| + Аи3+ рА » 10
Осадок Aul образуется при добавлении KI к раствору золота (III) (сходство с Cui):
Au3+ + 31- --► Aul) + I2|
Золото в степенях окисления +1 и +3 дает ряд устойчивых комплексов, что в значительной степени определяет все химическое поведение этого элемента:
[Au(CN)a]- =«=t Au+ + 2 CN" pA w 28
[AuC14]- «=> Au3+ + 4 Cl’ pX « 21
[Au(SCN)4J- Au3+ + 4 SCN" pK w 42
Комплексный ион [Au(CN)2]~ образуется, например, при обработке металлического золота растворами цианидов щелочных металлов в присутствии воздуха или Н2О2.
Медь, серебро, золото, цинк, кадмий, ртуть
649
Все соединения золота могут легко восстанавливаться, т. е. ♦сами действуют как сильные окислители:
Au Аи+ + е~ Аи+ ч—Аи3+ + 2е‘ Au/AuCla_ Au[AuC14]_
[АиС14]-ДАиС14Г
Е°= 1,46 В £о= 1,41В £° = 1,148В £о = 0,995 В £0 = 0,921 В
Рекомендуемые опыты
1. Растворимость металлов в кислотах. Исследуют растворимость меди, серебра и золота в 12 М НС1, 2,5 М H2SO4, 14,5 М HNO3, 18 М H2SO4 и (только для золота) в царской водке.
2. Концентрированный раствор CuSO4 слабо подкисляют разбавленной H2SO4 и нагревают с порошком меди.
Голубая окраска ионов [Си(Н2О)4]2+ исчезает и одновременно часть порошка меди переходит в раствор с образованием ионов Cu(I):
Cu2+ -J- Си (тв.) 2 Си+
При охлаждении раствора голубая окраска ионов [Си(Н2О)4]2+ снова появляется.
3. К раствору CuSO4 добавляют раствор KI. Образуется белый осадок •иодида меди(1) (—1§Пр=11,3), цвет которого маскируется одновременно выделяющимся иодом:
Cu3++5I- ------> CuI| + VaIa|
Иод можно удалить прибавлением сернистой кислоты. Окисления Cui на воздухе не происходит вследствие очень малой величины произведения растворимости.
Совершенно аналогичным образом идет взаимодействие ионов Си2+ с цианид-ионами.
4. К раствору CuSO4 по каплям добавляют раствор KCN. Выпадающий сначала желтый Cu(CN)2 при нагревании переходит в труднорастворимый СиСМ (белый, —lgllp=19,5). Одновременно образуется дициан (CN)a. (Тяга! Дициан чрезвычайно ядовит!)
2Си2+ + 2е- ---► 2Си+
2CN" -----► (CN)a + 2e“
При дальнейшем добавлении KCN осадок CuCN растворяется с образованием «онов [Cu(CN)4]3-. Из этого раствора, благодаря большой величине константы устойчивости комплексного цианидного иона, сероводород не осаждает труднорастворимый сульфид меди (отличие от кадмия):
[Cu(CN)4]3- [Cu(CN)a]_ + 2 CN- рК = 11,3
[Cu(CN)aJ- Cu+ + 2 CN- pK = 16
5. При добавлении к раствору CuSO4 сернистой кислоты и раствора । KSCN образуется белый осадок CuSCN.
В отсутствие восстановителя процесс восстановления Cu(SCN)2 хотя и медленно, но все же идет:
6 Cu(SCN)a + 4 НаО ---► CuSCN + SO42~ + HCN + 5 SCN" 4- 7 H+
6 При добавлении NaOH из раствора CuSO4, начиная с pH 5,0, вьша< |дает голубой Cu(OH)2 (—1g Пр «18—20), при нагревании которого происхо-[Дит отщепление воды и образование черного СиО. При прибавлении избыт
650
Неорганическая химия
ка NaOH при рН^15 Си(ОН)2 растворяется, переходя в синие ионы гид-роксокупрата(П) [Сип(ОН)2л+2]2-:
Cu(OH)2 + 2 ОН” <—>r [Cu(OH)4]2- рК = 2,9
7. В раствор CuSO4, подкисленный соляной кислотой до ее концентрации 2 моль/л, на холоду пропускают H2S. Выпадают черные осадки CuS (—1g Пр ~ 36—44) и Cu2S (—1g Пр «49). Сульфиды растворимы в конц. НС1, а при нагревании —в разбавленной HNO3. Сульфид меди частичка растворяется при длительном нагревании с раствором (NH4)2SX с образованием тиосолей.
8. Из раствора AgNO3 осаждают хлорид серебра, промывают его несколько раз водой и взвесь встряхивают с раствором KI.
Аналогичным образом к суспензии AgCl добавляют несколько капель раствора Na2S
В обоих случаях идут реакции, представляющие собой сочетание процессов растворения и осаждения:
AgCl + Г 3p=t Agl + Cl-
2 AgCl + S2~ 4=fc Ag2S -J- 2 Cl-
О. К раствору AgNO3 по каплям добавляют водные растворы NH3, Na2S2O3 и KCN. Осадки Ag2O (коричневый), Ag2S2O3 (белый, —lg ITp= 13,0) и AgCN (белый, —lg Пр=13,8) при дальнейшем прибавлении осадителя растворяются с образованием комплексов (разд. 33.6.2):
рК
[Ag(NH3)2]+ 7,1
[Ag(S2O3)2]3— 13,6 (разд. 35.6, опыт 18)
[Ag(CN)2]_ 21,0 (разд. 35,9, опыт 4),
Все значения рК соответствуют полной диссоциации комплекса. Проводят опыт по осаждению AgCl, AgBr, Agl и Ag2S из растворов комплексных солей; осадки этих соединений серебра обрабатывают растворами NH3 (различной концентрации), Na2S2O3 и KCN.
Осадок AgCl растворяется даже в растворе (NH4)2CO3. При добавлении к такому раствору КВг выпадает осадок AgBr. Объясните эти явления, сравнивая величины соответствующих произведений растворимости и констант устойчивости.
При работе с аммиачными растворами солей серебра следует соблюдать предосторожности: эти растворы нельзя оставлять иа продолжительное время, поскольку возможно образование легко взрывающегося нитрида серебра Ag3N.
10. Из солянокислого раствора AuCl3 (Н[АцС14) может быть получек в виде желтых кристаллов состава Н3О+[АиС14]--ЗН2О) при прибавлении FeSO4, SnCl2, N2H4-2H2O, НСООН, НСОН и других восстановителей осаждается металлическое золото При работе с очень разбавленными растворами сначала образуются коллоидные растворы золота, окрашенные в цвета от красного до сине-фиолетового.
Вопросы и задания
1. Используя уравнения Нернста, покажите возможность изменения «благородного характера» металла при переводе его в раствор в присутствия комплексообразователя.
2. Несмотря на то, что серебро и золото имеют почти одинаковые радиусы атомов, золото окисляется гораздо труднее. Почему^
3. Соединения меди(1) устойчивы в водных растворах лишь при условии, что активность гидратированных ионов Cu(I) весьма мала (образование-
Медь, серебро, золото, цинк, кадмий, ртуть
651
комплексов или труднорастворимых соединений). Подтвердите это положение для процесса 2Си+—>Cu2 + + Cu|, используя закон действующих масс и зная рК полуреакций
РК
Си+ Cu2+ + е~ 2,98
Cu+4-e_ -<.-fe. Си
—8,83
Объясните склонность ионов Cu(I) к диспропорционированию, пользуясь величинами стандартных потенциалов реакций и уравнением Нернста
Си+ ч— — Си2+ + е~
Си ч fe Си+ + е~
Для этого следует рассчитать отношения [Cu2+]/[Cu+]2 при установлении равновесия, если в равновесном состоянии системы справедливо уравнение
ас..2+
Е = 0,15 + 0,059 lg-гЧ- =0,52+ 0,059 lg аС11+ “Cu*
4. Почему хлорид- и бромид-ионы не могут восстанавливать ионыСи(П)?
5. Дайте объяснение устойчивости иона тетрацианокупрата (I) принимая во внимание его электронную конфигурацию. Какой вид гибридизации осуществляется? Каковы магнитные свойства этого иона?
6. Рассмотрите реакции, в которых равновесие Cu24-Cu^*2Cu+ смещается в сторону образования Си2+.
7. Вытеснение даже одной, не говоря о следующей (второй), молекулы Н2О в комплексе [Cu(NH3)4(H2O)2]2+ молекулами NH3 в водных растворах практически не идет. Рассмотрите этот факт как следствие искажения октаэдрической координации комплекса вдоль оси z при стабилизации ег-элек-тронов Си2+ (конфигурация Z62ge3g).
8. Как связано изменение окраски комплексов меди(II) при замещении молекул Н2О на молекулы NH3 и этилендиамина с различиями в силе поля этих лигандов?
9. Какому типу перехода соответствуют полосы, наблюдаемые в спектрах октаэдрических комплексов Си2+?
10. К раствору, содержащему одновременно хлорид- и иодид-ионы, добавлен раствор AgNO3. Какова должна быть концентрация иодид-иона, чтобы началось осаждение хлорида серебра? Проведите аналогичный расчет для раствора, содержащего хлорид- и бромид-ионы. (Разделение хлорид- и бро-мид-ионов путем «фракционированного» осаждения на практике дает значительно худшие результаты, чем следует из расчета. Это происходит вследствие образования твердых растворов AgCl и AgBr )
И. Какие стадии включает процесс растворения AgCl в растворе NH3? Следует учитывать существование иона [Ag(H2O) (NH3)]+.
12. Полагая, что разность энергий для заполненных 4+орбиталей и свободных 5э-орбиталей ионов Ag+ невелика, поясните возможность существования линейной координации (координационное число 2), например в IAg(CN)2]-, [Ag(NH3)2]+ н AgSCN.
36.17.2. Цинк, кадмий и ртуть
Цинк в своих соединениях практически всегда проявляет степень окисления +2. То же самое, за немногими исключениями, справедливо и для кадмия. При добавлении А1С13 в расплав Cd—CdCl2 образуется и может быть выделено соединение CdAICU которому, основываясь на его магнитных свойствах
652
Неорганическая химия
(соединение диамагнитно) следует приписать формулу Cd22+(A1C14“)2. В воде и других донорных растворителях наступает мгновенное диспропорционирование:
Сф2+ Cd + Cd2+
Напротив, ион ртути (I) Hg22+, в котором имеется связь металл— металл (разд. 36.11.1), устойчив и в водных растворах. Все же соединения ртути (I) могут легко диспропорциониро-вать (опыт 2):
Hg22+ Hg; + Hg2+
Цинк, кадмий и ртуть по своему химическому поведению несколько напоминают переходные элементы первой группы (близкие значения электроотрицательности; сходство в растворимости и окраске ряда соединений). В то же время благодаря наличию полностью заполненных d-орбиталей, у этих элементов не может происходить стабилизации под действием поля лигандов. В связи с этим их стереохимия практически полностью определяется размерами ионов. Реакции Zn2+ и Cd2+ в значительной мере соответствуют реакциям Mg2+; Cd2+ проявляет также сходство с Си2+.
Си2+ и Hg2+ можно однозначно отнести к мягким кислотам (разд. 33.4.3.4), тогда как Zn2+ занимает промежуточное положение между явно жесткими и явно мягкими кислотами.
36.17.2.1. Свойства ртути
Ртуть и ее соединения сильно ядовиты!
Для химии ртути можно считать определяющими приведенное выше равновесие диспропорционирования, способность к комплексообразованию, также как отчетливо ковалентный характер ряда ее соединений.
В то время как цинк и кадмий следует отнести к сравнительно «неблагородным» металлам, восстановление соединений ртути(II) до соединений ртути(I) и до металла идет очень легко:
Zn Zn2++2e-
Е° = — 0,76 В
Cd Cd2+ + 2e'
Е° = —0,40 В
2 Hg Hg2+ + 2е-
Hg аг Hg2++ 2е"
Е° = +0,79 В
£° = +0,85 В
Склонность соединений ртути(I) к диспропорционированию объясняется близостью величин стандартных потенциалов двух последних реакций.
Медь, серебро, золото, цинк, кадмий, ртуть 653
Пользуясь значениями р/С для окислительно-восстановительных реакций
Hga2+ 2 Hg2+ + 2е“ рК0Х = 30,8
Hga2+ + 2е~ 2Hg| pXrej = —27,1
можно для процесса диспропорционирования
Hga2+ ^=±: Hg| + Hg2+
найти р/С, равную 1,9.
Понижение активности ионов ртути(II) путем осаждения труднорастворимой соли или при комплексообразовании спо-{ собствует протеканию реакции диспропорционирования (закон j действующих масс).
> Hgl2 (осадок красного цвета, —lg Пр «26—28) растворяется | в избытке KI, переходя в очень устойчивый комплекс [HglJ,2-:
Hgl2 + 2 Г ч=±: [Hgi4]2-
На примере галогенных и псевдогалогенных комплексов ртути(П) можно убедиться в справедливости эмпирической закономерности (верной и для многих других комплексов): устойчивость комплексов увеличивается при возрастании деформируемости анионов:
[HgX4]2- Hg2++4X-
X Cl- SCN- Br- I- CN-
pX 16,0 19,3 21,6 30,3 40,5
Ряд соединений ртути(П) (галогениды и псевдогалогениды) в растворах практически не диссоциируют и, вследствие этого, не дают качественных реакций на гидратированный ион рту-ти(П).
36.17.2.2. Реакции цинка и кадмия
Zn(OH)2 амфотерен и при избытке щелочи (рН>13,5) растворяется с образованием гидроксокомплексов:
Zn(OH)a + 2 ОН" «=> [Zn(OH)4]2- рКа = 14,5
Из-за низкой устойчивости кадмат-ионов Cd(OH)2 лишь незначительно растворим в щелочах.
Если осаждение гидроксидов проводить водным раствором аммиака, то оба осадка растворяются в избытке осадителя с образованием аммиачных комплексов:
Zn(OH)a + 4 NH3 --> [Zn(NH3)4]2++2OH-
Для реакции [Zn(NH3)4]2+5=tZn2+-|-4NH3 рК=9,0.
Для аналитического разделения Си и Cd важна реакция образования цианидных комплексов (разд. 37.2.1.8).
654
Неорганическая химия
Рекомендуемые опыты
1. а) В раствор HgCl2 на короткое время опускают медную проволоку. На проволоке появляется серый налет, который при растирании материей становится серебристым (амальгама меди):
Си + Hg2+ ----> Си2+ + Hg|
б) Восстановление ртути(П) до степени окисления +1 идет под действием SnC12:
Hg22+ <—> 2 Hg2+ + 2е' Е° = 0,92 В
В раствор HgCl2 прибавляют по каплям раствор SnCl2. Осаждается труднорастворимый Hg2Cl2 (белый, —lgllp=17,5), который при дальнейшем прибавлении SnCl2 восстанавливается до металлической ртути:
2 Hg2+ + Sn2+ ---► Hg22+ + Sn4+
Hg22+-f-2Cl- ---► Hg2Cl2|
Hg22+ + Sn2+ ----► 2Hg|-}-Sn4+
2. К раствору Hg2(NO3)2 прибавляют растворы NaOH, H2S и KCN; к Hg2Cl2--раствор NH3, a Hg2I2 — раствор KI-
Hga2+ + 2 OH' ---Hg| + HgO| + H2O
Hga2+ + S2- -----> Hg| + HgSj. (в кислой среде)
Hga2+ + 2CN- ----»- Hg| + Hg(CN)21
Hg2Cl2 + 2 NH3 --- Hg| + Hg(NH2)Cl + NH4+ + Cl'
(может также образоваться
Hg2NCl«H2O, качественная
реакция на каломель)
HgaI24-2I- ------> Hg| + [Hgl4l2-
Те же продукты, как в последней реакции, образуются при добавлении по каплям раствора KI к раствору Hg(NO3)2. Зеленовато-желтый осадок Hg2I2 (—1g Пр=28,4) легко разлагается при нагревании:
Hg2Ia ---> Hg + Hgla
3. а) К водному раствору Hg(SCN)2 добавляют раствор соли Fe(III). Красный Fe(SCN)3 образуется лишь после добавления бромида:
Hg(SCN)2 + 4Br [HgBr4]2-+ 2 SCN-
б) К растворам HgCl2 и Hg(CN)2 по каплям прибавляют растворы NaOH, KI и AgNO3. Напишите уравнения реакций.
в) Из раствора Hg(CN)2 при действии H2S осаждается черный HgS (—lgnp«51—54), не растворимый в соляной и в разбавленной азотной кислотах.
4. При добавлении по каплям раствора NaOH к растворам ZnSO4 и CdSO4 выпадают гидроксиды Zn(OH)2 (начиная с pH 6,8; —Ig Пр «16) и Cd(OH)2 (при pH8,3; —1g Пр«14).
5. Из растворов солей кадмия, слабо подкисленных минеральными кислотами, при действии сероводорода выпадает желтый или желто-коричневый CdS (—lgnp = 27); из раствора солей цинка, к которому добавлен ацетатный буфер (NaCH3COO + CH3COOH) выпадает белый ZnS (рентгеноаморфиый; —1g Пр (сфалерит или цинковая обманка) «25, —1g Пр (вюрцит) «23):
M2+ + Has MS + 2Н+ pKzns = —2
pKcds = —6
cp- P^ues = —31
Медь, серебро, золото, цинк, кадмий, ртуть
655
Как следует из величины рК для реакции осаждения ZnS, из нейтральных растворов количественное осаждение невозможно.
6. При добавлении по каплям раствора KCN к растворам солей цикла или кадмия сначала выпадают белые осадки Zn(CN)2 и Cd(CN)2 (—lgnp=10,6). При избытке цианид-ионов эти осадки растворяются:
[M(CN)4]2- M2++4CN-
Zn Cd
рК 19 16,9
К раствору комплексного цианида цинка прибавляют несколько капель раствора (NH4)2S (идет ли реакция?)-, в раствор комплексного цианида кадмия пропускают H2S (?).
Вопросы и задания
1. Каким образом можно доказать, что ион Hg22+—биядерная частица?
2. Почему при взаимодействии растворов Hg(NC>3)2 и HgCl2 с водным раствором аммиака получаются соединения ртути различного состава?
3. Что такое реактив Несслера?
4. Можно ли рассматривать основание Миллона как ионообменник?
5. Почему насыщенный раствор HgCl2 после добавления кристаллического NaCl показывает нейтральную реакцию?
6. Почему ртуть может быть растворена в конц. иодоводородной кислоте (с выделением водорода)?
7. Что происходит при нагревании HgCl2 с конц. растворами хлоридов?
8. Напишите уравнение реакции протолиза гидратированных ионов цинка [Zn(H2O)4]2+ с образованием ионов тетрагидроксоцинката.
9. Какую электронную конфигурацию имеет цинк в тетраминном комплекс-се? Какой тип гибридизации осуществляется в тетраэдрически координированных комплексах цинка и кадмии?
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Адсорбция 187
Азот 529
Азот, оксиды 539
— тригалогениды 538
Азотная кислота 543
Активированный комплекс 172
Активность 312
Актиноиды 625
Акцепторное число 445
Алюминий 603
Амиды 535
Аммиак 534
Антиферромагнетизм 138
Атомное ядро 34
Бериллий 602
Берлинская лазурь 638
Бор 570
Бораны 572
Бораты 573
Бориды 571
Борная кислота 573
Валентность 89
Ванадий 612
Взаимодействие спин спиновое 73
Висмут 595
Вода, диссоциация 355
— ионное произведение 260
— строение 354—356
— тяжелая 37
Водород 459
— диссоциация на атомы 460
— молекула 81
— реакции 464, 468
Водородная связь 108, 352, 356, 449,
487
Вольтэквивалент 541
Вольфрам 620
Время полупревращения 154
— релаксации 332
Галлий 590
Галогениды 498, 509
Галогеноводороды 496
Галогены 494
— кислородные соединения 502
Гафний 609
Германий 592
Гибридизация 91
Гидразин 535, 547
Гидрид ион 460, 465
Гидриды 461, 644 |
Гидроксиды 480
Гидроксиламин 537, 547
Графит, строение 357
Давление пара 258
Двойной электрический слой 322
Дейтерий 36
Дефект массы 34
Дефекты по Френкелю и по Шоттки 430-431
Диаграмма давления пара 284 — окислительных состояний 540 — плавления 257 Диоксигенил ион 484
Диоксид углерода 561
Дипольный момент 9, 351
Диспропорционирование 418
Диссоциация в растворе 451
— термическая 427
— электролитическая 260, 278
Дифосфин 536
Дифракция нейтронов 74, 112 — рентгеновских лучей 74, 110 Дициан 568
Диэлектрическая проницаемость 457
Предметный указатель
657
Длина свободного пробега 22
Донорное число 444
Единица массы атомная 37
Железо 635
Закон Авогадро 12
— Бойля — Мариотта 13
— Вант-Гоффа 283
— Гей-Люссака 14, 220
— Генри 259
> — Генри — Дальтона 260
— Гесса 225
— Дальтона 285
— действующих масс 175, 250
— Кирхгофа 228
— кратных отношений 12
— Нернста 259
— постоянства состава 12
— разбавления Оствальда 383
— сохранения массы 11
— Фарадея 309
— Фика 329
Законы термодинамики
--- второй 233
•--первый 217
---третий 239
Золото 648
Зона проводимости 141
Зонная теория 140
Идеальные газы 13
— теплоемкость 20
Изомерия 368
Изотопный обмен 202
Индексы Миллера 109
Индий 590
Индикаторы кислотно основные 385
Инертные газы 491
Иод, кристаллическое строение 350
Ион Н+ 466
Ионизация ударная 26
Ионная атмосфера 331
Ионная сила раствора 184, 373
Ионность связи 105
Ионный потенциал 375
42—2027
Ионотропия 387
Кадмий 651, 653
Калий 598
Кальций 600
Карбиды 555, 564
Карбонаты 561
Катализ 190
— гетерогенный 196, 436
— гомогенный 193
— кислотно основной 193
Квант действия 24
Квантовые числа 48, 52
Кислород 469
Кислота серная 523
— тиосерная 523
Кислоты Льюиса 392
— пероксосерные 525, 528
— политионовые 523
Кластеры 615
Кобальт 635
Колебания валентные 68
Комплексные соединения 420
Комплексы молекулярного азота 531
Константа равновесия 175
Концентрация 231
Коррозия 437
Коэффициент активности 255, 333,
373
Кремний 554
Криоскопия 280
Круговой дихроизм 13
Летучесть 256
Литий 598
Магний 600
Магнитный момент эффективный 127
Марганец 148, 628
Медь 645
Мезомерия 94
Металлическая связь 138
Металлы 359, 576
— методы получения 582
— очистка 587
— переходные 579
Метод валентных связей 81
— Дебая — Шерера 110
658
Предметный указатель
— меченых атомов 201
— молекулярных орбиталей 87, 95
Молекулярная масса 15
Молекулярность реакции 152
Молекулярные вещества 349
Молибден 620, 623
Молибденовая синь 621
Моль 14
Мольные доли 232
Моляльность 232
Молярность 207
Моноксид углерода 562
— молекула 64
Мультнплетность 53
Мышьяк 595
Надпероксиды 483
Натрий 597
Нейтрон 33
Неметаллы 459, 577
Нивелирующее действие 381
Никель 635
Ниобий 612
Нитраты 545, 548
Нитриды 533
Нормальность 207
Обмен лигандами 204, 420, 424
Озон 477
Оксиды 471
Оксокомплексы 634
Олово 592
Осаждение сопряженное 374
Осмотическое давление 281
Основания Льюиса 393
Палладий 643
Парадокс Гиббса 242
Первый закон Рауля 258
Перенапряжение 335
Период полураспада 154
Пероксид водорода 148, 194, 482
Перхлораты 509, 511
Пирофосфаты 549
Платина 643
Платиновые металлы 642
Плотнейшие упаковки 17
Подвижность ионов 317
Полигалогенид-ионы 500, 510
Полупроводники 142
Поляризация молекул 99
Полярография 338
Порядок реакции 151, 161
Постоянная Планка 25
Потенциал диффузионный 313, 319
— электрохимический 321
Правило Оствальда 367
— фаз 278, 288
— Хунда 59
— Паули 30, 52
Произведение активностей 371
— растворимости 259, 372
Простые вещества 344
Протон 33
Работа выхода электрона 25
Радиоактивность 35
Радиусы ионов 116
Расплавы 391, 419
Растворение 369
Растворители апротонные 456
— протонные 444
Растворы буферные 387
Реакции второго порядка 157, 165
— гетерогенные 185, 256
— гомогенные 183, 256
— дегидратации 433
— конденсации 426
— окислительно-восстановительные
407, 434
— осаждения 374, 423
— параллельные 176
— первого порядка 153, 164
— полимеризации 426
— последовательные 178
— протолитические 377
— твердых веществ 430
— топохимические 439
— трибохимические 438
— цепные 180
— экзотермические 208
— эндотермические 208
Редкие металлы 588
Редкоземельные элементы 601
Предметный указатель
659
Рений 625
Ртуть 652
Рубидий 598
Ряд напряжений металлов 315
Свинец 592
Связь кратная 92
— металл — металл 615
Селен 513
Сера 513
— оксиды 521—522
Серебро 647
Сероводород 514
Симметрия молекул 120
Системы бинарные 287
— однокомпонентные 275
— тройные 289
Скорость движения молекул 18
— реакции 150
Соединения 344
— интерметаллические 143, 361
— ионные 108, 346
— межгалогенные 501
— неполярные ковалентные 350
• — полимерные 356
— полярные ковалентные 351
Сольволиз 429
Соотношение неопределенности Гейзенберга 28
Спектры вращательно-колебательные 64
— вращательные 61
— комбинационного рассеяния 68
• — оптические 42
— растворов 67
— рентгеновские 40
— электронные 66
— ядерного магнитного резонанса 69
Спин ядра 34, 72
Сплавы 557, 589
Стабилизация степеней окисления 631
Стандартное состояние 216
Степень диссоциации 269
— окисления 409
— превращения 272
Стронций 600
Структура льда 354
Структуры координационные 356
— слоистые 357
— цепочечные 358
Суперкислоты 457
Сульфид кремния 358
Сульфиды 516
Сурьма 595
Таллий 590
Тантал 612
Твердые растворы замещения 143, 361
Твердые тела, идеальные 16
--- теплоемкость 24
Теллур 513
Температура характеристическая 25
Теория жестких и мягких кислот и оснований 394
— кислот и оснований 375
— сольвосистем 440
Тепловые эффекты при растворении 370
Теплоемкость мольная 219
Термический анализ 289
Термодинамика статистическая 290
Термодиффузия 23
Термохимия 224
Термы атомов 53
Тетратионаты 524
Технеций 628
Типы соединений 346
Тиосульфаты 524, 527
Титан 609
Титрование потенциометрическое 323
Торий 626
Турнбулева синь 638
Углеводороды 557
Уравнение Аррениуса-169
— Больцмана 240, 292
— Ван-дер-Ваальса 107
— Вант-Гоффа 251, 264
— Гиббса 246
— Клаузиуса — Клапейрона 275
— Ленгмюра 188
— Лютера 326
— Менделеева — Клапейрона 15
660
Предметный указатель
।— Мозли 41
— Нернста 314
— Шрёдингера 29, 45, 76
Уравнение реакций 364
Уран 627
Фаза 207
Фазовые переходы 366
Фазовые равновесия 275
Фазы Лавеса 363
Ферромагнетизм 138
Формула Вульфа — Брэгга 110
— де Бройля. 27
Фосфаты 549
Фосфин 534
Фосфор 367, 529
— галогениды 538
— оксиды 539
Фтор 485
Фториды 488
— кислорода 484
Фтороводород 487
Фторокомплексы 634
Функция состояния 209
Халькогены 512
— галогениды 518
— кислородные соединения 519
Химическая связь 75
Химическая термодинамика 206
Химические функции 453
Химический потенциал 249
Химическое равновесие 175, 250, 269
Хром 618, 622
Царская водка 545, 548
Цезий 598
Цианиды 568
Цикл Габера — Борна 114
Цинк 651, 653
Цинковая обманка, структура 357
Цинтл-фазы 363
Цирконий 609
Числа переноса 317
Число Авогадро 14, 38
— Маделунга 114
— Фарадея 310
Эбулиоскопия 279
Эвтектика 287
Электродвижущая сила 309
Электролитическая диссоциация 308
Электрон 26
Электронный газ 139
Электронный обмен 202
Электроотрицательность 101
Электропроводность удельная 328
— эквивалентная 328
Элемент 343
Элемент 105, 625
Элементарная ячейка 109
Элементарные частицы 32
Энергия решетки 112
— свободная 243
— стабилизации в кристаллическом поле 132
Энтальпия образования 226
— реакции 224
— свободная 244
Энтропия 234
Эффект Джоуля — Томсона 220
— Зеемана 50
— изотопный 197
— Мёссбауэра 128
СОДЕРЖАНИЕ
Предисловие редактора перевода . . , .............................. 5
Предисловие к 1-му изданию.......................................
Предисловие к 10-му изданию........................................ 8
А. СТРОЕНИЕ МАТЕРИИ..................................................И
1. Атомистические представления о структуре материи..................11
1.1. Закон сохранения массы..........................................11
1.2. Законы постоянства состава и кратных отношений .... 12
1.3. Газовые законы. Закон Авогадро. Химические уравнения ... 12
1.4. Идеальные газы. Уравнение состояния. Экспериментальные методы определения молекулярной массы..................................13
1.5. Идеальные твердые тела.........................................16
2. Кинетическая теория материи................................... 18
2.1. Молярная теплоемкость идеальных газов 20
2.2. Число столкновений и средняя длина свободного пробега. Диффузия и теплопроводность...........................................22
2.3. Молярная теплоемкость твердых тел..............................23
3. Основы квантовой теории.........................................23
3.1. Классическая физика и атомарная структура материи ... 24
3.2. Квант действия. Квантовая теория и теплоемкость .... 24
3.3. Фотоэлектрический эффект. Фотоны......................25
3.4. Электроны.............................................26
3.5. Квантовая теория и классическая физика 28
3.6. Некоторые основные принципы квантовой механики .... 29
4. Элементарные частицы и атомное ядро ........ 31
4.1. Элементарные частицы..................................31
4.1.1. Некоторые важнейшие элементарные частицы .... 32
4.2. Атомные ядра..........................................34
4.3. Радиоактивность. Ядерные реакции......................35
4.4. Стабильные и нестабильные нуклиды.....................36
5. Электронные оболочки атомов.....................................39
5.1. Стационарные состояния атома: экспериментальные данные . . 39
5.1.1. Бомбардировка вещества электронами....................39
5.1.2. Рентгеновские спектры..................................40
5.1.3. Оптические спектры.....................................42
5.2. Атом водорода: основное состояние.....................44
5.3. Атом водорода: возбужденное состояние.................48
5.4. Орбитальный момент. Атом водорода в магнитном поле ... 49
662
Содержание
5.5. Электронные конфигурации. Атомные состояния. Периодическая система элементов....................................................5;
5.6 Границы применимости электронной модели атома. Атомы и молекулы ............................................................59
6. Представления о химической связи................................6(>
6.1. Уровни энергии и структура молекул............................6о
6.1.1. Вращательные спектры...................................61
6.1.2. Вращательно-колебательные спектры......................6;
6.1.3. Электронные спектры....................................66
6.1.4. Спектры растворов......................................67
6.1.5. Спектры комбинационного рассеяния (спектры КР) . . 63
6.1.6. Спектры ядерного магнитного резонанса (спектры ЯМР) 69
6.1.7. Дифракционные методы...................................73
6.2. Химическая связь..............................................75
6.2.1. Молекулярный ион водорода Н2+..........................75
6.2.2. Природа химической связи...............................79
6.2.3. Обменное вырождение. Молекула Н2. Метод валентных
связей (ВС)............................................81
6.2.4. Молекулярные состояния (метод молекулярных орбита-
лей, МО) ..............................................87
6.3. Молекулы с преимущественно ковалентными связями .... 88
6.3.1. Валентность, валентный угол. Гибридизация .... 89
6.3.2. Соединения с кратными связями..........................92
6.3.3. Мезомерия..............................................94
6.3.4. Применение метода МО...................................95
6.3.5. Полярные молекулы. Дипольные моменты...................99
6.3.6. Электроотрицательность. Доля ионной связи .... 101
6.3.7. Атомные и молекулярные решетки. Межмолекулярные
силы..................................................106
6.4. Ионы в кристаллических структурах..........................108
6.4.1. Применение рентгеновского излучения для исследований
структуры веществ.....................................108
6.4.2. Энергия решетки.......................................112
6.4.3. Радиусы ионов и типы решеток..........................116
6.4.4. Переходные типы связи в кристаллах....................118
>6.5. Ионы в поле лигандов.......................................119
6.5.1. Симметрия молекул.....................................120
6.5.2. Основы магнетохимии...................................125
6.5.3. Ядерная гамма-резонансная спектроскопия (мёссбауэровская спектроскопия).........................................128
6.5.4. Вращательная дисперсия света и круговой дихроизм . . 129
6.5.5. Электростатическая теория комплексных соединений . . 131
6.5.6. Ковалентные связи в комплексах........................135
>6.6. Металлическая связь..........................................138
6.6.1. Металлическая связь и решетка металлов. Ферро- и антиферромагнетизм ............................................138
6.6.2. Электронный газ...................................139
6.6.3. Зонная теория (электронная модель металла) ... 140
6.6.4. Смешанные кристаллы (твердые растворы) и интерметаллические фазы 143
6 6.5. Виды связи в твердых телах....................145
Б. ОСНОВЫ ФИЗИЧЕСКОЙ ХИМИИ....................147
Б.1. ХИМИЧЕСКАЯ КИНЕТИКА И МЕХАНИЗМЫ РЕАКЦИЙ . . I47
7. Предмет и метод химической кинетики
147
Содержание
без-
s. Основные понятия химической кинетики (порядок реакции, молекулярность, константа скорости) .................................. 150
8.1. Скорость реакции...........................................150
8.2. Порядок и молекулярность реакции...........................151
9. Простые кинетические закономерности. Анализ результатов исследования кинетики химических реакций ............................... 153
9.1. Уравнение скорости реакции первого порядка.................153
9.2. Реакции второго и более высокого порядков..................157
9.3. Кинетические уравнения как функции безразмерной переменной 160
9.4. Методы определения порядка реакции......................161
9.4.1. Определение порядка реакции из зависимости начальной скорости реакции от концентрации .......................... 161
9.4.2. Метод времени полупревращения.......................162
10. Экспериментальные методы в химической кинетике...............166
11. Константа скорости реакции .................................. 168
11.1. Зависимость константы скорости от температуры..............168
11.2. Физический смысл фактора столкновений......................169
11.3. Теория абсолютных скоростей реакций Эйринга................172
12. Более сложные реакции........................................175
12.1. Протекание обратной реакции................................175
12.2. Параллельные реакции.......................................176
12.3. Последовательные реакции...................................178
12.4. Цепные реакции.............................................180
13 Гомогенные реакции в растворах...............................183
13.1. Общие представления о влиянии растворителя на механизм
реакции....................................................183
13.2. Влияние ионной силы на химические реакции в растворах
(первичный солевой эффект).................................184
14. Гетерогенные реакции.........................................185
14.1. Адсорбция и гетерогенные реакции...........................187
14.2. Применение изотермы адсорбции к гетерогенным реакциям . . 189
15. Катализ......................................................190*
15.1. Общие представления........................................190
15.2. Гомогенный катализ.........................................193
15.2.1. Примеры гомогенного катализа.......................194
15.3. Гетерогенный катализ.......................................196
16. Изотопные методы в химической кинетике.......................196
16.1. Общие представления........................................196
16.2. Кинетический изотопный эффект..............................197
16.2.1. Изотопный эффект растворителя......................200
16.3. Исследования методом меченых атомов........................201
16.4. Исследования изотопного обмена.......................... 202
16.4.1. Реакции электронного обмена........................202
16.4.2. Обмен лигандами....................................204
Б.2. РАВНОВЕСИЯ и ХИМИЧЕСКАЯ ТЕРМОДИНАМИКА . . . 206-
17. Предмет и задачи химической термодинамики....................20&
664
Содержание
18. Термодинамические функции состояния. Их свойства .... 208
18.1. Математическое описание функций состояния.....................208
18.2. Интенсивные и экстенсивные параметры системы и парциальные мольные величины...................................................215
18.3. Стандартное состояние.........................................216
19. Первый закон термодинамики......................................217
19.1. Общие положения...............................................217
19.2. Изотермические, изобарические и адиабатические процессы . . 220
19.2.1. Изотермическое расширение и сжатие....................220
19.2.2. Изобарно-изотермические процессы......................222
19.2.3. Адиабатические процессы...............................222
20. Термохимия.....................................................224
20.1. Энтальпия реакции, энтальпия образования химического соединения .............................................................224
20.2. Температурная зависимость энтальпии реакции (закон Кирхгофа) 228
21. Способы выражения концентрации..................................231
22. Второй закон термодинамики......................................233
22.1. Энтропия как функция состояния.............................. 233
22.1.1. Изменение энтропии в изотермических процессах . . . 235
22.1.2. Зависимость энтропии от параметров состояния систе-
мы (р, v и Т)..........................................236
22.2. Другие функции состояния, введенные на основе второго закона термодинамики (максимальная полезная работа).......................243
22.3. Термодинамические функции состояния, введенные на основе объединения первого и второго законов термодинамики. Фундаментальные уравнения Гиббса ...................................... 246
23. Химические и фазовые равновесия................................248
23.1. Общие условия равновесия......................................248
23.2. Химическое равновесие и закон действующих масс .... 250
23.3. Применение закона действующих масс к гомогенным и гетерогенным процессам . . ................................ 256
23.3.1. Гомогенная газовая реакция............................256
23.3.2. Гетерогенная химическая реакция.......................256
23.3.3. Гетерогенная реакция в жидкой фазе....................257
23.3.4. Равновесная реакция фазового превращения .... 257
23.3.5. Первый закон Рауля....................................258
23.3.6. Равновесие растворимости..............................258
23.3.7. Закон распределения Нернста...........................259
23.3.8. Растворимость газов...................................259
23.3.9. Электролитическая диссоциация растворенных газов . . 260
23.3.10. Ионное произведение воды.............................260
24. Зависимость константы равновесия от температуры и давления
(гомогенные реакции)................................................264
24.1. Зависимость константы равновесия от температуры и давления (основные уравнения)...............................................264
24.2. Расчет констант равновесия для данной температуры с различной степенью приближения...........................................265
24.2.1. Первое приближение Улиха.............................266
24.2.2. Второе приближение Улиха..............................266
Содержание 665
24.3. Интерпретация температурной зависимости свободной энтальпии реакции.............................................................267
। 25. Применение закона действующих масс к газовым реакциям (опре*
i деление выхода реакции) ......................................... 269
t 26. Гетерогенные процессы.........................................274
I 26.1. Фазовые равновесия..........................................275
| 26.1.1. Фазовые равновесия в однокомпонентных системах . . 275
| 261 2. Равновесие между раствором и растворителем . . . 279
I 26.1.3. Равновесие между раствором и растворенным веществом
в твердой фазе и между двумя растворами .... 283
; 27. Введение в статистическую термодинамику.......................290
27 1. Термодинамическая вероятность и энтропия.....................291
27.2. Функция распределения Больцмана...............................295
27.3. Некоторые свойства суммы по состояниям и физический смысл множителя а.........................................................297
27.4. Связь между термодинамическими функциями газов и молекулярными суммами по состояниям.......................................299
1 27.5. Суммы по состояниям для важнейших видов движения молекул 301
I 27 6. Расчет поступательной составляющей термодинамических функций 304
, 27 7. Расчет вращательной и колебательной составляющей энтропии газов...............................................................306
i Б.З. ЭЛЕКТРОХИМИЯ...............................................308
28. Теория электролитической диссоциации ........................ 308
29. Закон Фарадея.................................................309
30. Электрохимическая термодинамика...............................309
30.1. Э. д. с. ячейки.............................................309
30.2. Схема измерения э. д. с. ячейки компенсационным методом . . 311
30.3. Активность..................................................312
30.4. Ряд напряжений. Стандартные потенциалы......................313
30.5. Электроды второго рода. Произведение растворимости . . . 315
30.6. Концентрационные цепи.......................................316
30.7. Подвижности ионов и числа переноса. Цепи с переносом . . 317
30.8. Диффузионный потенциал......................................319
30.9. Ионообменники. Равновесие Доннана. Электрохимический потенциал ..........................................................320
30.10. Э. д. с. ячейки и составляющие скачка потенциала на границе фаз. Двойной электрический слой...................................322
30.11. Основы потенциометрического титрования.....................323
30.12. Уравнение Лютера...........................................326
31. Протекание электрического тока в электрохимической ячейке . . 327
31.1. Электропроводность растворов электролитов...................327
31.2. Коэффициент диффузии и подвижность..........................329
31.3. Слабые электролиты..........................................330
&1.4. Сильные электролиты.........................................330
'31 5. Кинетика электродных процессов.............................334
. 31.5 1. Перенапряжение диффузии.............................335
' 31.5 2. Основы полярографии.................................338
31.5.3. Перенапряжение перехода электрона..................339
>666
Содержание
В. НЕОРГАНИЧЕСКАЯ ХИМИЯ: СОЕДИНЕНИЯ И РЕАКЦИИ . .
32. Важнейшие типы неорганических соединений.....................
32.1. Простые вещества и соединения...............................
32.2. Классификация веществ по их строению и типу связей .
32.3. Характерные свойства соединений различных типов ....
32.3.1. Ионные соединения...................................
32.3.2. Молекулярные вещества...............................
32.3.3. Полимерные соединения...............................
32.3.4. Металлы.............................................
33. Важнейшие типы реакций в неорганической химии................
33.1. Химические уравнения........................................
33.2. Превращения без изменения состава...........................
33.2.1. Изменения агрегатного состояния индивидуальных веществ
33.2.2. Фазовые превращения в твердом состоянии ....
33.2.3. Превращения изомерных веществ с различным типом связей......................................................
-33.3. Образование и разрушение ионных решеток....................
33.3.1. Процесс растворения.....................
33.3.2. Осаждение труднорастворимых солей...................
-33.4. Теория кислот и оснований..................................
33.4.1 . Основы теории кислот и оснований...................
33.4.2 . Особые случаи протолитических реакций..............
33.4.3 . Дальнейшее развитие теории кислот и оснований .
33.4.4 . Связь между структурой и силой кислот..............
33.5. Окислительно-восстановительные реакции (редокс-процессы) .
33.5.1. Основные положения теории окислительно-восстановительных процессов ..........................................
33.5.2. Особые случаи окислительно-восстановительных реакций
33.6. Реакции комплексных соединений..............................
33.6.1. Образование и диссоциация комплексов................
33.6.2. Комбинированные равновесия комплексообразования и осаждения...................................................
33.6.3. Комплексообразование и окислительно-восстановительный потенциал ..................................................
33.6.4. Обмен лигандов в комплексных соединениях . . . .
.33.7. Полимеризация и конденсация................................
33.8 . Термическая диссоциация и сольволитическое расщепление .
33.9 . Реакции веществ в твердой фазе............................
33.9.1 . Общие предпосылки реакционной способности твердой фазы
33.9.2 . Виды реакций в твердой фазе........................
34. Теории растворителей в неорганической химии..................
34.1. Теория сольвосистем.........................................
34.2. Координационная теория растворителей........................
34.2.1. Растворители — доноры и акцепторы пары электронов
34.2.2. Донорные и акцепторные числа........................
34.2.3. Сольватация в донорных и акцепторных растворителях
34.2.4. Образование ионов и диссоциация.....................
34.2.5. Влияние растворителя на термодинамику н кинетику реакций ....................................................
34.2.6. Представления о химических функциях.................
34.3. Протонная теория растворителей..............................
34.3.1. Классификация растворителей в зависимости от их кислотно-основных свойств......................................
34.3.2. Применение суперкислот и снльноосновных сред в неорганическом синтезе .......................................
343
343
343
346
346
346
349
356
359
363
363
365
365
366
368
369
369
374
375
375
383
389
404
407
407
416
420
420
423
424
424
426
427
430
430
432
440
440
443
444
444
448
450
452
453
455
455
457
СодержаниеЁЁ1
35. Неметаллы .... ...............................459
35.1. Водород..................................................
35.1.1. Реакции водорода.................................
35.2. Кислород.................................................
35.2.1. Классификация оксидов............................
35.2.2. Реакции кислорода................................
35.3. Фтор.....................................................
35.3.1. Реакции фтора....................................
35.4. Инертные (благородные) газы..............................
35.4.1. Реакции инертных газов...........................
35.4.2. Фториды и кислородные соединения.................
35.5. Хлор, бром, иод..........................................
35.5.1. Неполярные молекулярные вещества.................
35.5.2. Галогеноводороды — полярные молекулярные вещества
35.5.3. Реакции галогенов................................
35.5.4. Реакции кислородных соединений галогенов .
35.6. Сера, селен, теллур......................................
35.6.1. Неполярные молекулярные вещества.................
35.6.2. Полярные молекулярные вещества...................
35.6.3. Реакции халькогенов..............................
35.6.4. Реакции кислородных соединений халькогенов .
35.7. Азот, фосфор.............................................
35.7.1. Неполярные молекулярные вещества .:....
35.7.2. Различные типы соединений азота и фосфора .
35.7.3. Реакции кислородных соединений азота и фосфора .
35.8. Углерод н кремний........................................
35.8.1. Карбиды и силициды...............................
35.8.2. Углеводороды и силаны............................
35.8.3. Галогениды углерода и кремния....................
35.8.4. Оксиды, оксокислоты и сульфиды...................
35.8.5. Моноксиды углерода и кремния.....................
35.9. Псевдогалогены...........................................
35.9.1. Дициан, циановодород, цианид-иои.................
35.9.2. Дитиоциан, родановодород, тиоцианат-ион
35.10. Бор......................................................
35.10.1. Бориды и бораны.................................
35.10.2. Галогениды бора.................................
35.10.3. Борная кислота и бораты.........................
459
464
469
471
476
485
486
491
491
492
494
494
496
497
502
512
513
514
515
519
529
530
533
539
553
555
557
558
560
562
567
568
569
570
571
572
573
36. Металлы .......................................................576
36.1. Переход от неметаллов к металлам..............................576
36.1.1. Электронная теория переходных металлов.............579
36.2. Методы получения металлов. Значение «редких» металлов . . 582
36.2.1. Получение металлов.....................................583
36.2.2. Операции очистки.......................................587
36.2.3. Значение «редких» металлов и их сплавов...............588
36.3. Галлий, индий и таллий........................................590
36.4. Германий, олово, свинец.......................................592
35.5. Мышьяк, сурьма, висмут........................................595
36.6. Главная подгруппа первой группы...............................597
36.6.1. Натрий.................................................598
36.6.2. Калий (рубидий и цезий)................................598
36.6.3. Литий..................................................598
36.7. Главная подгруппа второй группы...............................600
36.7.1. Магний, кальций, стронций и барий....................601
36.7 2. Бериллий...............................................602
36.8. Алюминий......................................................603
|36.9. Редкоземельные металлы.......................................607
668
Содержание
36.10. Титан, цирконий (гафний и курчатовий).........................609
36.11. Ванадий, ниобий, тантал (и элемент 105).......................612
36.111. Связь металл — металл.................................615
36.12. Хром, молибден, вольфрам (и элемент 106)......................617
36.12 1. Химические свойства хрома.............................618
36.12. 2. Химические свойства молибдена и вольфрама .... 620
36.12. 3. Молибденовая и вольфрамовая синь....................621
36.13 Элементы семейства актиния (торий, уран)......................625
36.13.1. Торий...............................................626
36.13.2. Уран..................................................627
36.14. Марганец (технеций, реннй и элемент 107)................... 628
36.14.1. Соединения марганца в различных степенях окисления 628
36.14.2. Стабилизация степеней окисления.......................631
36.15. Железо, кобальт и никель................................ ... 635
36.15.1. Реакции железа(Н) и (III).............................637
36.15 2. Реакции комплексообразования железа, кобальта и никеля 638
36.15.3. «Берлинская лазурь» и «турнбулева синь» .... 639
36.16. Платиновые металлы...................................... ... 642
36.16.1. Системы переходный металл — водород...................644
36.17. Элементы побочных подгрупп первой и второй групп . . . 645
36.17.1. Медь, серебро и золото................................645
36.17 2. Цинк, кадмий и ртуть................................651
Предметный указатель.................................................656
Уважаемый читатель!
Ваши замечания о содержании книги, ее оформлении, качестве перевода и другие просим присылать по адресу: 129278, Москва, И-110, ГСП, 1-й Рижский пер., д. 2, издательство «Мир».
Герд Блументаль, Зигфрид Энгельс, Ингомар Фиц н др,
АНОРГАНИКУМ
Редактор Л. Кольдиц
Т. 1
Научный редактор Т. И. Почкаева
Мл. научный редактор Н. Н. Устякова
Художник Б. П Кузнецов
Художественный редактор М. Н Кузьмина
Технический редактор Л. П. Бирюкова
Корректоры: С А. Денисова, В. И. Постнова
ИБ № 3693
Сдано в набор 17.02.84.
Подписано к печати 27.08.84.
Формат 60X90V16 Бумага типографская № 2.
Гарнитура литератур. Печать высокая. Объем 21,00 бум. л.
Усл. печ. л 42,00. Усл. кр.-отт. 42,25. Уч.-изД. л. 43,45.
Изд. № 3/2563. Тираж 8.400 экз. Зак. 2027. Цена 2 р. 50 к.
ИЗДАТЕЛЬСТВО «МИР»
129820, Москва, И-110, ГСП, 1-й Рижский пер., 2.
Московская типография № 11 Союзполиграфпрома при Государственном комитете СССР по делам издательств, полиграфии и книжной торговли.
Москва, 113105, Нагатинская ул., д. 1.
Издательство «Мир»
выпускает в свет в 1985 г. подписное издание
Руководство по неорганическому синтезу: В 6 томах. Пер. с нем /Под ред. Г Брауэра —Ориентировочная цена издания 13 р. 80 к. за комплект.
Перевод сделан с третьего издания широко известного во всем мире руководства, ставшего по существу энциклопедией неорганического синтеза (первое издание было переведено на русский язык издательством «Иностранная литература» в 1956 г.). В книге собран обширный материал, касающийся оптимальных путей синтеза большого числа (свыше 3000) неорганических и ме-таллорганических соединений (в виде высокочистых продуктов) и их основных свойств.
Издание предназначено для специалистов в самых различных областях науки и техники, а также для преподавателей и студентов химических вузов как ценное пособие для обучения современным методам препаративной химии, при выборе заданий для курсовых работ.
Из отзыва академика В. И. Спицына:
«Трудно переоценить необходимость подобного издания в практике обучения химиков современным методам препаративной химии. Обширность содержащегося в книге материала дает преподавателю возможность выбрать синтез, соответствующий поставленной задаче—либо отработке определенного метода работы, либо изучению химии элементов и т. д. Весьма полезным настоящий справочник может оказаться и при выборе заданий для курсовых работ студентов, изучающих химию».
Из отзыва академика А. В. Новоселовой:
«Изданное в 1956 году на русском языке «Руководство...» широко вошло в повседневную практику наших учебных заведений и научных учреждений. Им пользуются отнюдь не только неорганики, но и химики других специальностей, а также представители многих других профессий — технологи, биологи, геологи и другие. Сведения, содержащиеся в новом издании, касаются оптимальных путей синтеза и фундаментальных свойств неорганических соединений. Поэтому нх ценность и новизна сохранятся на протяжении еще многих лет».
Подписка принимается в 1984 г. магазинами, имеющими отделы подписных изданий.
Издательство «Мир» выпускает в свет в 1985 г. книги по химии
Киперт Д. Неорганическая стереохимия: Пер. с англ.— 19 л., — 2 р 20 к
В монографии собраны данные о структурах комплексов — информация, необходимая практически всем специалистам в области неорганической и физической химии. Автор — известный австралийский химик — обобщил свой многолетний труд в области стереохимии координационных соединений.
Книга предназначена для специалистов в области неорганического синтеза, спектроскопии, химии металлорганических соединений, а также как учебное пособие для преподавателей и студентов вузов.
Коттон Ф., Уолтон Р. Кратные связи металл — металл: Пер. с англ. — 35 л. — 5 р. 60 к.
Первая в мировой литературе монография, посвященная бурно развивающейся области химии — химии кластерных соединений металлов, — нашедшей к настоящему времени выход в практику (покрытия, композиционные материалы, катализ). Ф. Коттон широко известен советским химикам фундаментальными работами и книгами (Коттон Ф., Уилкинсон Дж., Современная неорганическая химия.—М.: Мир, 1969; Коттон Ф., Уилкинсон Дж. Основы неорганической химии. — М.: Мир, 1979).
Книга предназначена для научных работников — химиков-неоргаников, ка-талитиков, материаловедов, для аспирантов и студентов старших курсов химических вузов.
Заблаговременно оформляйте заказы на интересующие Вас книги. Заказы принимают магазины, распространяющие научно-техническую литературу. Своевременно сданный заказ гарантирует приобретение нужных Вам книг.