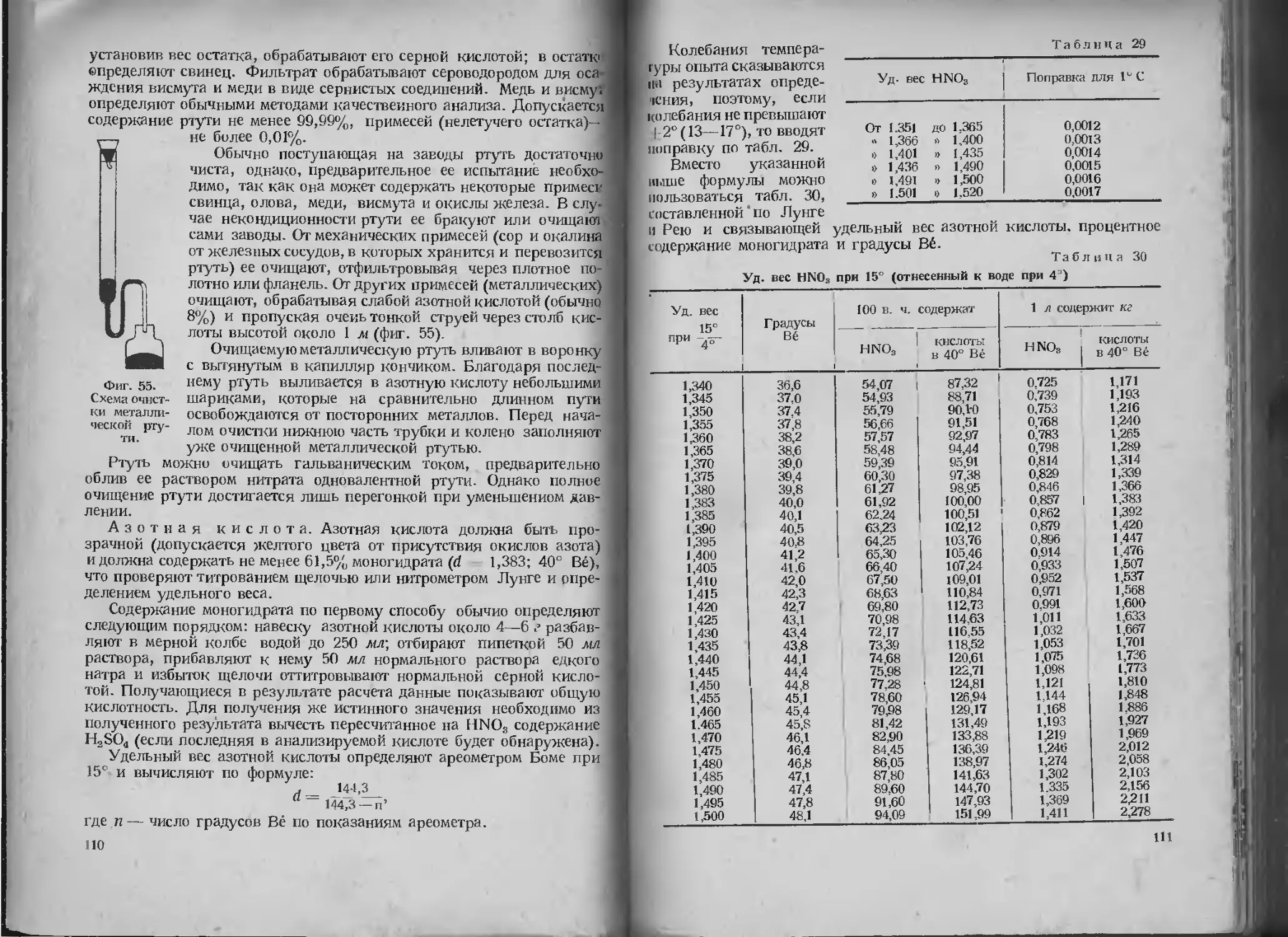
/




































































































































































Текст
пчд
11. Ф. БУБНОВ
ИНИЦИИРУЮЩИЕ
ВЗРЫВЧАТЫЕ ВЕЩЕСТВА
И СРЕДСТВА ИНИЦИИРОВАНИЯ
ЧАСТЬ 1
ГОСУДАРСТВЕННОЕ ИЗДАТЕЛЬСТВО ОБОРОННОЙ ПРОМЫШЛЕННОСТИ
МОСКВА 1940
Книга доцента Артиллерийской академии П. Ф. Бубнова
«Инициирующие взрывчатые вещества» содержит основные
теоретические положения и практические сведения из этой
области- Наиболее полно и подробно в ней изложены химия
и технология получения гремучей ртути, азида свинца и стиф-
ната свинца, широко применяемых в военном деле и в мир-
ной промышленности.
Книга предназначается в качестве учебника для слушате-
лей военных академий и для студентов втузов НКБ. Ее также
можно рекомендовать в качестве пособия для инженерно-тех-
нических работников заводов, производящих средства ини-
циирования и инициирующие взрывчатые вещества-
Редактор И. М. Хазанов Техн, редактор А. И. Савари
Сдано в набор 14/V 1940 г. Подписано к печ. 31/VIII 1940 г. Авт. дог. № 645.
Тираж 6000. Кол. печ. лист. 20т/4. Учетно-авт. лист. 22,25. Формат бумаги
60х92/гб. Инд. 5-2. А30767. Заказ № 92. Цена 9 руб. 4- 2 руб. пер.
Типография Оборонгиза. Киев, Крещатик, 42.
ПРЕДИСЛОВИЕ
Одним из самых ответственных элементов любого вида боепри-
пасов артиллерии, авиации и др. является капсюль, в который всегда
входят в качестве составной части инициирующие взрывчатые ве-
щества (ВВ). Капсюли, а стало быть, и инициирующие ВВ, имеют
laicoe же важное значение для мирной промышленности и для тех-
ники взрывного дела.
Между тем огромный, более чем столетний, опыт производства
инициирующих ВВ, а также многочисленные исследования, прово-
дившиеся в этой области, до сих пор не были систематизированы
в одном труде так, чтобы он мог служить в более или менее полной
мере учебным руководством для изучающих этот предмет и пособием
для работников заводов.
Стремясь восполнить этот пробел, автор собрал и обобщил основ-
ной материал, чтобы дать читателю в сжатой форме основные све-
юния по теории и технологии инициирующих ВВ, включая описа-
ния их физико-химических и взрывчатых свойств, способов полу-
чения и анализа, технологических процессов и пр. Наиболее полно
и подробно освещены главнейшие, широко применяемые, иници-
ирующие ВВ (гремучая ртуть, азид свинца и стифнат свинца).
В книге также уделено внимание вопросам, связанным с неко-
торыми мероприятиями по технике безопасности производства ини-
циирующих ВВ. Автор исходил из того, что техника безопасности
н данном случае является необходимейшим условием обеспечения
нормального хода производства, безаварийности и личной
безопасности работников заводов и набора-
г о р и й.
Книга составлена в основном применительно к учебным програм-
ма военных академий Красной Армии и втузов оборонной промыш-
ленности.
Пав. Бубнов
Москва
Артиллерийская ордена Ленина Акаде-
мия Красной Армии им. Дзержинского.
Март 1940 г.
ГЛАВА I
ВВЕДЕНИЕ
§ 1. ИСТОРИЧЕСКИЕ СВЕДЕНИЯ
I Iron ходимость в инициирующих В В и средствах инициирования
№ tinorjci. при переходе от так называемой баробалистической ар-
ni'1'И'рпп, г. с. артиллерийских машин, м^тателБное'-действме--кхщ^_
|м,|* основано на применении рычагов с противовесами, к артил-
рнн нпробалистической, метательное действие которой основано
н 1и н(н|1..10|й1нии пороха. В дальнейшем было установлено, что усо-
। рпн нс топание артиллерийских систем, систем ручного оружия и
...... рода снарядов, а также возможность применения в артил-
рии и промышленности бризантных ВВ вообще тесно связаны
ра пи п псм средств инициирования и инициирующих ВВ.
1 к горшо развития средств инициирования и инициирующих ВВ
-uno разделить на четыре периода:
Первый п е р и о д — с момента начала применения дымного
пороча в качестве источника метательной силы до появления хло-
। иных составов, взрывчатые свойства которых были открыты фран-
г к «им химиком Бертолле в 1786 г.
Второй период — со времени применения в средствах
ппнцппротпшя хлоратных составов с гремучей ртутью и без нее,
•о iHii/ । , когда инженером Нобелем был изобретен гремучертутный
пн юhi.-детонатор.
I р с т п и период — с 1867 г. до конца первой империали-
। ih’H’ckoU войны, т. е. до начала широкого применения в средствах
инициирования азида свинца и других новых инициирующих ВВ.
’ I спертый период-— с конца первой империалистиче-
. ной иойпы до настоящего времени.
I Irpiiuii период развития средств инициирования характерен при-
ш цепнем для воспламенения дымного пороха в артиллерийских ору-
иых, и ручном огнестрельном оружии, в горной промышленности
и в лр-iii плерийских снарядах того же дымного пороха в качестве
111ПЩ11.1|Ор<1.
У первых образцов артиллерийских орудий, заряжавшихся
I • ин, пороховой заряд воспламенялся весьма примитивно. Порох
г ни । реп пили тогда в виде мякоти, которая опудривала канал ору-
пн; воспламенялся порох от предварительно накаленных на жаровне
। нарядов (каменные и чугунные ядра).
5
Последующие образцы артиллерийских орудий и ручного оружия
имели запальные отверстия, расширяющиеся наружу и заканчиваю-
щиеся полкой для затравочного пороха. Для сообщения огня заряду
употреблялись раскаленные на жаровнях железные прутья или лу-
чинки, предварительно вываренные в селитре.
Таким образом первым инициирующим веществом был тот же
дымный порох (пороховая мякоть), употреблявшийся в качестве
метательного вещества. Этот порох вследствие разнообразия состава
и ряда других неудовлетворительных качеств не всегда был при-
годен в качестве инициатора. Поэтому уже в XV веке для иници-
ирования стали употреблять более сильный и более дорогой (с боль-
шим содержанием селитры) зерненый порох (в виде небольших
комков).
В последней четверти XIV века для воспламенения порохового
заряда ручного оружия начали применять фитиль, свитый из пень-
ковой веревки, пропитанной раствором свинцового сахара. Затем,
в начале XV века для зажигания зарядов артиллерийских орудий
применяли фитиль из орехового дерева, вываренного в селитрен-
ном растворе, или затравку, которую зажигали издали при помощи
дорожки пороха. В середине XV века стали применять ручное
оружие с фитильными замками. Однако замки эти были очень
неудобны: во-первых, стрелявший всегда должен был иметь при
себе зажженный фитиль, из-за чего порох часто преждевременно
воспламенялся; во-вторых, в дождливое время фитиль гаснул, что
приводило к осечкам; в-третьих, темп стрельбы замедлялся, ибо для
безопасности стрелок после выстрела обычно вынимал и гасил фи-
тиль, чтобы перед следующим выстрелом вновь его разжечь; в-чет-
вертых, зажженный фитиль, так же как раньше жаровня, ночью
был виден издали.
Эти недостатки фитильного замка привели к мысли высекать искру
для воспламенения пороха по образцу распространенного тогда ог-
нива !. В начале XV] века появился немецкий колесцовый замок.
Замок этот состоял из колеса, соединенного цепочкой с нижней ветвью боевой
пружины. Колесо заводили особым ключом. Цепочка наматывалась на ось колеса и
натягивала боевую пружину. Взведенное колесо удерживалось особой задержкой.
Наружную поверхность колеса зазубривали. Часть поверхности колеса, спрятан-
ного в замке, выдавалась наружу около полки, на которую насыпали порох.
Снаружи замка располагали на особой пружине курок, в лапках которого зажи-
мался кремень. Для того чтобы выстрелить, отодвигали задвижку, и боевая пру-
жина начинала быстро вращать колесо; кремень терся о зубья боковой поверх-
ности колеса, высекая искры, которые и воспламеняли затравочный порох. Ко-
лесцовые замки хотя и были удобнее фитильных, но все же имели недостатки:
кремни в них скоро сбивались, требовали частой замены, а конструкция замка
для того времени была слишком сложна и дорога в производстве.
В том же столетии наряду с фрикционным колесцовым замком
было изобретено так называемое ударное огниво (кремневый замок).
1 Именно ц этом направлении до XIX века шли усовершенствование воспла-
менения .ырнда ручного оружия.
II».i. го ввели его в употребление только в XVII веке. В некоторых
Брлн IX (например во Франции) не доверяли кремневому оружию
0 hi тому применяли ружья-мушкеты, т. е. ручное огнестрельное
•«1*1 снабженное и кремневым и фитильным замками одновре-
ши». В первых образцах этих ружей в курке помещали серный
• П i'ic'i.ih. Спускаемый курок' ударял о шершавую заднюю грань,
цп родившуюся перед полкой с порохом, и высекал искру; но вскоре
< > |1ц|.п*| колчедан заменили более твердым и прочным кремнем или
Но шрованным агатом. Однако и кремень часто давал отказы. Co-
nic и пя французов были вполне основательными: произведенные в
• ргдипс XIX века в Саксонии специальные опыты показали, что
» ж’мнсвое оружие давало 31% осечек.
Ни все же в начале XIX века кремневые ружья были окоича-
ь н.п» введены во всех странах мира.
Воспламенение зарядов артиллерийских орудий оставалось почти
। ним, каким оно было в XVI веке, только вместо затравочного по-
I” л дополнительно к фитилю стали пользоваться палительными
• псымн, стопином и так называемыми скорострельными трубками
(вначале бумажными, а затем металлическими), наполненными мел-
ким юрценым порохом.
Попытки использовать порох ие только как метательную силу,
пи и к качестве разрывного заряда, производились еше в XVI веке,
। цы1«», особого успеха они тогда не имели. Только в XVII веке по-
iiitiixncb разрцвные снаряды, изготовлявшиеся из железа или из
г*рпц.1Ы. Они обстояли из двух половин, образующих как бы фут-
I i|> сферической или овальной формы для помещения разрывного
шрм щ дымного пороха. Обе половицы соединялись пропускаемыми
иш кпозь одним или двумя болтами. В особое очко снаряда вставляли
i niiiuyio железную трубку с отверстиями, набитую медленно горя-
щим пороховым составом (смоченным льняным маслом) и присоеди-
ненную к стопину. Стреляли таким снарядом вначале «двойным огнем»
(ш> йн| пли стопин трубки, а затем поджигали затравку порохового
пряди орудия), а впоследствии—«одиночным» (стопин зажигался
ы • 1ми порохового заряда), причем снаряд помещали в орудии труб-
• Н к стенке канала, чтобы обеспечить воспламенение порохового
«•«vi.nt.-i трубки от газов и огня заряда. Позднее стали применять
ipV”Mi, рассчитанные по времени горения.
Попупю следует отметить, что в горной промышленности, раз-
ни i не которой требовало применения ВВ, дымный порох стали упо-
ip'iniHTi. для подрывных работ в начале XVII века (г. Хемниц,
И» Ч г.). Воспламенение и здесь, как и в артиллерии, производилось
< IU1IIIIHIM (через соломенную трубочку, наполненную мелкозернистым
порочим), к которому присоединялась зажигательная нить для
«и шн.юного воспламенения на расстоянии. Только в середине
\1\ цгк.1 (1831 г.) был изобретен медленно горящий бикфордов
ннцр, что в соединении с фитилем сделало воспламенение порохового
• ряда и шпуре сравнительно безопасным.
7
Если в первый период развития средств инициирования исследо-
ван ели и мастера обращали особое внимание на оформление меха-
нической части, что не дало значительного улучшения качества ру-
жейного огня, то во второй период внимание было обращено также
и на изыскание новых инициирующих ВВ, так как попытки улуч-
шения состава дымного пороха 1 не увенчались успехом.
Дальнейшее улучшение действия огнестрельных средств могло
быть достигнуто только путем новых изобретений. Таким изобрете-
нием, сделавшим переворот, особенно в военном деле, было открытие
Говардом в 1799 г. гремучей ртути. В 1786 г. французский химик
Бертолле открыл, что хлораты в смеси с горючими веществами от
удара взрываются. Это навело на мысль применять вместо затравоч-
ного пороха смесь бертолетовой соли с серой и углем. Смесь смачи-
вали и зернили, как порох, или изготовляли из гранулей величиной
с горошину ударный состав, который для предохранения от сырости
заливали воском. Согласно Гельцу и Эскалесу изобретателем первого
ударного состава для огнестрельного оружия был Форсайт (1805—
1807 гг.) 2. Форсайт применял рыхлый или формованный хлоратный
состав, который, однако, был далеко не безопасен в употреблении
и, кроме того, вредно действовал на металл оружия. Тем не менее,
даже это не вполне удачное изобретение оказало значительную услугу
развитию техники воспламенения, ибо, во-первых, порох, применяв-
шийся для запала кремневого ружья, быстро засорял продуктами
своего сгорания запальные отверстия, что зачастую приводило к осеч-
кам, применение же состава Форсайта почти совершенно уничтожило
засорение; во-вторых, сберегалось время, необходимое для насыпания
пороха на полку, что увеличило скорострельность ружья.
Изменение систем ручного оружия заставило изменить ударные.
составы и конструкцию капсюля. В России первые оболочки для кап-
сюлей вырубали из листовой красной меди в виде крестообразной
пластинки, в центральной части которой на лаке располагали ле-
пешку из прессованного ударного состава. Затем, концы креста за-
гибали под прямым углом, а сверху лепешку покрывали тем же ла-
ком. Только с 1858 г. оболочки стали изготовлять цельнотянутыми
(для ружей Крика). Кроме того, оказалось необходимым разработать
конструкцию оружия, заряжаемого с казенной части. Все это при-
вело к мысли о целесообразности соединить в один патрон пулю,
порох и капсюль.
Решающую роль в развитии средств инициирования сыграла гре-
мучая ртуть. Уже Лепаж, давший особую конструкцию воспламене-
ния, пользовался ударными составами с гремучей ртутью. Затем
появилось прусское игольчатое ружье, сконструированное Дрейзе
в 1820 г., а в 1841 г. принятое на вооружение в прусской армии ипока-
1 В 1756 г. Лебланд делал опыты приготовления пороха без серы; в 1766 г.
Делаваль взял патент на замену угля в порохе каменноугольным порошком;
в 1788 г. Бертолле и Лавуазье пробовали прибавлять в порох бертолетову соль.
2R. Escales u. A. Stettbacher, Initialexplosivstoffe. стр. 3, 19l7~
8
лип свои хорошие качества в кампании 1864 г. против Дании и
oil । против Австрии. В стреляющем механизме этого оружия
111 . । пг.и.1, которая при спуске курка пронизывала пороховой заряд.
Buipiiini п накалывала ударный состав, помещавшийся между поро-
В нулей.
(» ui.iko гремучая ртуть приобрела особое значение только после
> I о, как был изобретен капсюль в медной оболочке (Эгг, 1815 г.).
П ibpcH'nHC капсюля возвратило исследователей к мысли о целесо-
iipaiiiocni применять инициатор, расположенный вне заряда по-
г I, и иб использовании силы удара для воспламенения инициирую-
кц| состава. Первоначально гремучая ртуть не входила в ударные
1.1ЮЫ, так как ее высокая чувствительность вызывала опасения:.
><»ц|>му пользовались следующим (старым) ударным составом:
/ бертолетовой соли . 70,6% $
1 серы.................. 17,6’ь I
I угля.......... .11,8% | Q *« 1
Нит состав впрессовывали в медйК1Гткозтяачки, а для предозфа-
н ним от сырости и высыпания покрывали лаком или оловянной
|ш n.iiih. Капсюли надевали на запальный стержень; они воспламе-
nnici. от удара курка и сообщали огонь пороховому заряду.
II ю время порох, пуля и капсюль заряжались отдельно друг от
i| уы. Несмотря на это, а также и на то, что капсюли сами по себе
• hi.un несовершенными, капсюльный способ воспламенения был при-
и и во всех странах, и благодаря этому осечки ружей снизились до 1%.
1 IH.il г. гремучую ртуть начали применять в капсюльных составах.
На изыскание новых рецептур составов было затрачено очень
мини» времени, так как применявшиеся в капсюлях ударные составы
I"- к* сгорания оставляли на пистонах ружей значительное коли-
чн ню нагара, вследствие чего было трудно надевать новые капсюли.
I h правильно же расположенный на пистоне капсюль часто давал
"I гчкп.
В ряде стран (в Бельгии, России, Голландии и Испании) в
in х годах прошлого столетия были созданы специальные «кап-
i ниц.hi,к заведения». Бельгийская пиротехническая школа обучала
' 1иму искусству не только отечественных специалистов, но и коман-
дируемых для обучения специалистов других стран. В России про-
|| чшдсгво капсюлей началось в 1843 г. иа Охтенском пороховом
ншоде. Капсюльными заведениями был разработан целый ряд рецеп-
тур ударных составов, содержавших в основном гремучую ртуть,
«и рюлстову соль, антимоний или серу и уголь, стекло и склеивающие
вещества (шеллак, гумми-арабик и др.).
К середине прошлого столетия (1866 г.) уже имелись унитарные
и ироны (Флобер, Лефоше, затем Бердан) ручного оружия с капсю-
i iMii, причем последние в разных системах располагались различно.
I Li фиг. 1 показан патрон со штифтиком. Он имеет выдающийся
и i края подвижной штифтик, нижний заостренный конец которого
и р/ипгся на ударном составе капсюля- Удар курка по штифтику
&
-воспламенял ударный состав, луч огня которого передавался поро-
ховому заряду. Такого типа патроны охотники применяли около
30 - 40 лет. Военные же ведомства от применения патронов с по-
движными штифтиками отказывались вследствие недостаточной безо-
пасности их в обращении.
На фиг. 2 показан патрон центрального боя. Ударный состав
в нем насыпали на середину дна гильзы и воспламеняли ударом бойка.
Фиг. 1- Схема
патрона со
штифтиком.
Фиг. 2. Схема
патрона цен-
трального боя.
Фиг. 3. Схема
патрона боко-
вого огня.
На фиг. 3 показан введенный позже патрон, в котором ударный
состав располагали по краям и взрывали ударом или сжатием между
срезом ствола и головкой курка. Эти патроны особенно рекомендо-
вались потому, что при осечке достаточно было только повернуть
их другим боком, чтобы произвести выстрел.
Наконец, во второй половине XIX века во всех странах были
приняты металлические патроны с центральным воспламенением.
Некоторые из разработанных в это время составов сведены
в табл. 1.
Таблица 1
Ударные составы для ружейных капсюлей
Компоненты удар- ный составов Процентный состав
I 1 2 3 4 5 6 7 8 9
Гремучая ртуть . 10 10 13 27 18 30 30 40 48,8
Хлорат калия . 37 42 43 37 35,5 25 30 24 24,4
Антимоний . . . 40 35 34 29 28 40 16 26.2
Стекло .... 13 13 Ю 7 8.5 5 20 31 -
Связывающие ве-
щества .... 0.6 0,6 0,6 0,5 0,5 4 5 0,6
Попытки воспламенения пороховых зарядов артиллерийских ору-
дий ударными составами начались впервые в 1809 г. Так как кап-
сюли невозможно было ставить непосредственно за зарядом в канале
орудия, то пришлось устроить вместо запального отверстия для сто-
10
I । и ini iдлительных свечей запальный стержень с отверстием,
| w как и у ружей. Однако такой способ воспламенения орудий-
।рядов не обеспечивал безотказности действия.
iini.il ы применения ударного состава в «скорострельных» труб-
\ венчались успехом. Ударный состав из бертолетовой соли (20 ч.),
| нимопия (18 ч.) и стекла (3 ч.) помещали в скорострельной трубке
н р\ пороховой забивки и воспламеняли ударом молотка, закреп-
"||"<ип па шарнире.
11нц,ко в середине XIX века был применен фрикционный способ
• ч 1П1.1МС11СННЯ зарядов орудий. Во второй половине XIX века были
I нч1ы почти всюду вытяжные трубки, фрикционный состав кото-
ри> идтпял из бертолетовой соли, антимония и серы или из берто-
iiuioh соли, гремучей ртути и антимония.
Наряду с фрикционным способом воспламенения пороховых заря-
। »м артиллерийских орудий применялся в этот период и фитильный
। in .tub воспламенения. Воспламенение оформлялось фитильными
। i.n| и к-1 рольными трубками. Трубки изготовляли из листовой латуни
и । наряжали зерненым порохом, который воспламеняли фитилем
и in пилитсльноЙ свечой.
В России фрикционные трубки были окончательно введены в по-
|''|Ц1о артиллерию «кабинетным ордером» от 9 августа 1851 г. Затем
мни ныли введены и в крепостной и в осадной артиллерии по мере
со одования запасов фитильных трубок. Одна из первых фрикцион-
ных трубок, применявшихся в русской артиллерии, состояла из
i'iiviiiioh цельнотянутой гильзы и терки. Верхний конец гильзы
расширен в виде воронки и затянут шейкой. Терка состояла
I' । in лы и латунной фольговой коробочки, снаряженной сухим соста-
вим п i равных количеств бертолетовой соли и антимония. Игла пред-
। i.iiHiMjia собой кусок латунной проволоки со скрученными концами
и \ Ишим, за которое крючком можно было вытащить терку из гильзы.
I наряженную терку помещали в гильзе непосредственно под шейкой,
। \ unco иглы выводили наружу выше воронки. Затем гильзу снаря-
4<||»н1 артиллерийским порохом и оба конца гильзы закрывали масти-
кой, содержавшей равные количества воска, терпентина и асфальта.
Заряды разрывных Гранат того времени воспламеняли при помощи
> исциальных трубок, представлявших собой закрытый с одного конца
Деревянный цилиндр, снаряженный внутри составом, рассчитанным
па определенное время горения. Обычно применяли для этой цели
«ан называемый серый состав, содержавший калиевой селитры 75% и
। еры 25% в смеси с 7 в. ч. пороховой мякоти. Поверх столбика серого
।'и<ава в русских трубках располагали слой пороховой мякоти
uni обеспечения воспламенения, а в прусских—еще и переходный
И if та в.
В результате изобретения шрапнели в 1803 г. и повышения тре-
бований к разрывным гранатам появилась необходимость-в улучше-
нии воспламенения пороховых зарядов шрапнелей и гранат. При-
мерно в 1850 г. уже существовало несколько различных способов
использования для этого гремучей ртути. Первоначально капсюль
11
прикрепляли к снаряду. При падении капсюль взрывался и переда-
вал взрыв пороховому заряду. Однако такое приспособление оказа-
лось ненадежным, ибо снаряд мог упасть и не на капсюль. Затем
появилась идея использования инерции снаряда, претворенная
в конце 50-х годов в конструкцию ударной трубки с жалом и капсюлем
из гремучей ртути или ударного состава.
Принятием на вооружение гремучей ртути, вернее, ударных и
фрикционных составов, основой которых являлась гремучая ртуть,
заканчивается второй период развития средств инициирования.
Развитие химии в XIX веке создало мощное основание для быст-
рого прогресса химии и технологии ВВ. Были изобретены новые
ВВ: пироксилин, нитроглицерин, пикриновая кислота, тротил, тет-
рил, а также бездымный порох. Однако применение ВВ, а иногда
даже обнаружение их взрывчатых свойств или вызов взрыва, оказа-
лось возможным только с 1867 г., когда инженером Нобелем был
изобретен капсюль-детонатор, положивший начало третьему периоду
развития средств инициирования.
В самом деле, пироксилин был получен еще в 1832 г. химиком
Браконо, а достаточно чистый продукт—в 1845 г. Шецбейном; нитро-
глицерин впервые был получен итальянским химиком Собреро в 1846 г.
Однако получение должного эффекта взрыва этих двух веществ и
достаточно безопасное с ними обращение оказались возможными только
при помощи изобретенного позднее гремучертутного капсюля-дето-
натора.
Точно так же пикриновая кислота была известна еще алхимикам
XV века, а с конца XVIII века она находила применение в промыш-
ленности в качестве красящего вещества для шелковых тканей. Только
в 1886 г. французский химик Е. Тюрпен, пользуясь капсюлем-де-
тонатором, открыл ее взрывчатые свойства и впервые предложил
применять ее в прессованном или плавленом состоянии для сна-
ряжения артиллерийских снарядов.
Если введение капсюля-воспламенителя с ударным составом спо-
собствовало развитию промышленности порохов и повысило безопас-
ность их применения, то изобретение капсюля-детонатора произвело
переворот во взрывной технике и ускорило пополнение арсенала
ВВ. В этом отношении весьма показательны опыты Абеля и Нобеля:
первого — с пироксилином, второго — с нитроглицерином и дина-
митами.
В конце XIX и начале XX века были предложены и приобрели
серьезное значение аммонийно-селитренные ВВ; появились оксили-
квиты (горючие вещества, наполненные жидким кислородом) и др.
Это привело к необходимости предъявить к капсюлям-детонаторам
более разнообразные и более высокие требования в отношении их
мощности и способности вызывать детонацию новых, менее чувстви-
тельных, но более мощных, суррогатных ВВ. Эти требования были
удовлетворены вначале введением гремучертутных комбинированных
капсюлей-детонаторов, в которых наряду с гремучей ртутью в ка-
честве первичного заряда стали применять и бризантные ВВ в ка-
12
Bhrm*' вторичного заряда (тротил, пикриновая кислота, тетрил,
«• ан..гдстнии тэн и гексоген).
•' и1-и«> коренным образом был разрешен вопрос о мощности кап-
о кин.ко в результате открытия в 1891 г. Курциусом азотистово-
кислоты, вернее, ее свинцовой соли. Кроме того, появи-
сь I. иычнюльное количество патентов на новые инициирующие ВВ
। |ип!< гый азот, бензоилсупероксид, тетразолы и т. п.).
П промышленности стали переходить от воспламенения капсюлей
.. ..|и'рдивым шнуром к воспламенению электрическим путем (элек-
1<|| i.iii.iftbi, электродетонаторы и т. п.).
I Ьшпление детонирующего шнура и опыты с ним показали, что
*<-iiuikicп, бризантного ВВ может быть повышена применением так
II. u.iii.icMoro кумулятивного 1 способа инициирования, когда ини-
iiiiipiiii.uiiie ВВ производится с двух концов.
В артиллерии развитие техники и появление капсюлей-
। к>па горов дало возможность применять бризантные ВВ в качестве
pa 1р1.|ипых зарядов, а также воспользоваться суррогатными ВВ
(имм'1111[й|!о-селитренными и прочими смесями).
Одновременно развивались и усовершенствовались средства вос-
н шмспспия пороха. В 1867 г. в России была введена на вооружение
•'•шишка Бердана, что привело к изменению снаряжения капсюлей-
'нц||4.1мснителей. В связи с последовавшим затем принятием бездым-
||*ч и пороха в качестве метательного вещества и введением в 1894 г.
in вооружение армии 3-линейной винтовки опять пришлось изменить
и конструкцию и снаряжение патронного капсюля.
С IK70 г. для артиллерийских систем начали постепенно вводить
мп подымный порох; это требовало переконструирования запальных
। рубок и замены их другими с тем же фрикционным, но более надеж-
ным способом воспламенения и с большей скорострельностью. Появи-
»шп. вытяжные трубки.
В ЧПП-х годах появились новые системы артиллерийских ору-
дий, были введены для зарядов гильзы, были усовершенствованы
по tuny ручного оружия стреляющие приспособления и в качестве
нис||п.1мснителя начали применять капсюль. Последний в орудиях
мл лого калибра вставляли в специальное гнездо в гильзе, а в ору-
диях среднего и крупного калибра — в так называемые капсюльные
тулки, которые ввертывали в дно гильзы.
11.1 протяжении всего этого периода производились изыскания
Пшцч‘ рационального ударного состава, а также был сделан ряд
1ШПЫГПК заменить дорогую и, кроме того, «оржавляющую» канал
। iito на оружия гремучую ртуть.
I .IKHM образом третий период характеризуется дальнейшим усо-
вершенствованием средств воспламенения пороховых зарядов, откры-
том явления детонации, изобретением капсюля-детонатора на основе
। рему чей ртути и появлением на сцену конкурента гремучей ртути—
। шдм свинца.
<>г лагпнского слова cutnulo— увеличиваю.
13
Четвертый период развития средств инициирования — послевоен-
ный период — характеризуется вытеснением из артиллерийских
капсюлей-детонаторов гремучей ртути и заменой ее более мощным
азндом свинца. В результате этого оказалось возможным создать
капсюли-детонаторы малого размера и тем самым значительно
увеличить безопасность в обращении и безопасность стрельбы бое-
припасами с большими начальными скоростями. Это, кроме того,
способствовало еще большему расширению круга применяемых
суррогатных ВВ.
В целях борьбы с ©ржавлением канала ствола винтовок на про-
тяжении последних лет проводились опыты по изысканию новых
окислителей взамен хлората калия. В литературе опубликован ряд
рецептов ударных составов, не оржавляющих канал ствола и содер-
жащих тетразен и стифнат свинца, в качестве горючих — антимоний,
или металлы, дающие при сгорании большое количество тепла, ак-
кумулируемого их окислами, а в качестве окислителя — бариевую
селитру.
§ 2. КЛАССИФИКАЦИЯ СРЕДСТВ ИНИЦИИРОВАНИЯ
Конструкции, предназначенные для безопасного и однообразного
возбуждения взрывчатого превращения ВВ (горение, взрыв, детона-
ция), принято называть средствами воспламенения. Термин этот имеет
исключительно историческое значение и не охватывает полностью
тех функций, которые на эти средства возлагаются. В самом деле,
почти до конца прошлого столетия средства воспламенения приме-
нялись .только для зажигания дымного пороха так или иначе полу-
ченным лучом огня- Ныне от средств воспламенения требуется не
только воспламенение пороховых зарядов, но и инициирование взрыв-
чатых веществ с целью вызова их детонации. Поэтому рационально
вместо термина «средства воспламенения» употреблять имеющий
более широкое значение термин «средства инициирования».
В зависимости от назначения средства инициирования подразде-
ляются на две группы:
1) основные средства,
2) вспомогательные средства.
К основным средствам инициирования относятся капсюли, пред-
ставляющие собой конструкции, заключающие небольшой заряд ини-
циирующего ВВ или инициирующей смеси и назначенные для того,
чтобы в первом случае вызвать детонацию бризантных ВВ, а во вто-
ром случае — или детонацию ВВ, или воспламенение порохов.
В связи с этим основные средства инициирования, или капсюли,
разбиваются на две категории:
1) капсюли-детонаторы,
2) капсюли-воспламенители.
Капсюли-детонаторы представляют собой заключенные в метал-
лическую оболочку заряды инициирующего ВВ, одного или совместно
с бризантными ВВ. На фиг. 4 представлен один из подрывных кап-
п
< тлей-детонаторов— азидотетриловый капсюль-детонатор, в алюми-
ниевой оболочке которого запрессованы 1 г тетрила, 0,20 г азида
пикша и 0,10—0,15 г тринитрорезорцината свинца (ТНРС), прикры-
тые шелковой сеткой и алюминиевой чашечкой.
Действие капсюля-детонатора состоит в том, что от
луча огня воспламеняется ТНРС: получаемый при этом
тепловой эффект вызывает взрыв азида свинца; последний
в свою очередь вызывает детонацию вторичного заряда —
тетрила, —взрыв которого служит импульсом для детона-
ции заряда бризантного ВВ.
Луч огня может быть сообщен капсюлю-детонатору
бикфордовым шнуром, электрозапалом
или капсюлем-воспламенцтелем; послед-
ние особенно часто применяются в
артиллерийских взрывателях наряду
с капсюлями-детонаторами, действую-
щими от накола жалом. Один из таких
капсюлей-детонаторов изображен на
фиг. 5.
Он представляет собой металличе-
скую оболочку, в которую запрессован
з#ряд тетрила, заряд азида свинца в ка-
честве инициатора и заряд ударного
Подрывной^с0сТава, содержащего ТНРС, тетразен,
детонатор, бариевую селитру и антимоний, при-
j крытый сверху никелированной медной
—фольгой и чашечкой.
Капсюли-воспламенители подразделяются по их боевому приме-
нению на следующие четыре подгруппы:
1. Капсюли-воспламенители, назначенные для воспламенения по-
Фиг. 5. Капсюль-
детонатор, дейст-
вующий от накола
жалом.
фиг. 4. I
роховых зарядов винтовочных патронов, а также для капсюльных
и запальных втулок и действую-
щие обычно от удара бойка. §ти
капсюли носят название пат-
ронных капсюлей-воспламените-
Фиг. 7- Капсюльная втулка.
Фиг. 6- Патронный капсюль-
воспламе цитель.
лей. Они, как правило, представляют собой металлическую оболочку
(фиг. 6), в которую впрессована навеска ударного состава, прикрытая
сверху металлической фольгой, пергагиентной бумагой или лаком.
15
2. Капсюльные втулки (фиг. 7), служащие для воспламенения по-
роховых зарядов в орудиях гильзового заряжания. Снаряженная
втулка является усиленным патронным капсюлем-воспламенителем.
Она состоит из латунного корпуса, капсюля, втулочки, наковаленки,
•заряда ружейного или охотничьего пороха, кружка из пропитанной
селитрой папиросной бумаги, пергаментного кружка и латунного
кружка с отверстием посредине, покрытого снаружи шеллачным
лаком с примесью киновари. Действие капсюльной втулки состоит
в том, что при выстреле боек ударника вминает
Фиг. 8- Трубочный
капсюль - воспламе-
нитель.
металл дна корпуса втулки против капсюля.
Последний, ударяясь о наковальню, взрывается,
дает луч огня и воспламеняет заряд и поро-
ховые лепешки, огонь которых передается поро-
ховому заряду гильзы.
3. Трубочные капсюли-воспламенители
(фиг. 8), действующие от накола жалом и пред-
назначенные для воспламенения дистанционных
составов дистанционных трубок или усилите-
лей, замедлителей и капсюлей-детонаторов
взрывателей.
Эти капсюли состоят из медной никелиро-
ванной оболочки, фольгового дна и фольговой чашечки, снаряжен-
ных (в одном из частных случаев) двумя слоями ударного состава, из
которых нижний состоит из 28% .гремучей ртути, 36% бертолетовой
соли и 36% антимония, а верхний — из 15% гремучей ртути и
85% бертолетовой соли.
Действие трубочного капсюля-воспламенителя состоит в том, что
дистанционный ударник трубки в момент выстрела, двигаясь вслед-
ствие инерции, накалывает капсюль на жало. Капсюль воспламе-
няется и через передаточные отверстия головки стебля и через окошко
верхней дистанционной части трубки зажигает дистанционный со-
став последней. Действие трубочного капсюля-воспламенителя во
взрывателе в общем аналогично описанному, но воспламенение его
происходит не в момент выстрела, а в момент удара о преграду, и луч
огня передается замедлителю, усилителю или непосредственно кап-
сюлю-детонатору в зависимости от конструкции взрывателя.
4. Вытяжные трубки, ранее имевшие широкое распространение,
но применяющиеся и поныне для воспламенения пороха в неко-
торых системах с раздельным картузным заряжанием.
На фиг. 9 приведена одна из таких вытяжных трубок. Состоит она
из латунной гильзы, фрикционного приспособления, малой прово-
лочной петли, или хомутика, и большой проволочной петли. Гильза
наполнена дымным порохом и у открытого конца замазана мастикой.
Фрикционное приспособление служит для воспламенения трубки
действием терки на фрикционный состав из 60 ч. бертолетовой соли,
10 ч. серы и 30 ч. антимония; оно состоит из латунной гильзы, напол-
ненной на половину своей длины запрессованным составом, и терки
из латунной проволоки с лопаточкой на конце и с зубцами. Действие
i рубки состоит в том, что при дерганье за вытяжной шнур ^лопаточка
терки обламывается о края гильзы, зубцы терки царапают фрикцион-
ный состав и воспламеняют его; огонь передается дымному пороху
г ильзы и от него — боевому заряду орудия.
Фиг. 9. Вытяжная трубка-
К группе вспомогательных средств инициирования относятся все
i рсдства, предназначенные для передачи горения или детонации на
|мсстояние или же только для воспламенения того или иного объекта,
h ‘.«той группе прежде всего
• I НОСИТСЯ фитиль, Представ-
ЩЮЩИЙ собой пучок пень-
• оных нитей, пропитанных
Г<1Ш'ВОрОМ селитры И заклю- фиг- IO- Бикфордов шнур.
чинных в плотную оплетку
и ( таких же нитей. Зажженный спичкой или искрой пеньковый
фитиль начинает медленно тлеть. Скорость горения (тления) пень-
1.оного фитиля — около 4 см/мин; при ветре скорость горения фитиля
немного увеличивается. Пеньковый фитиль обеспечивает быстрое и
1Г1дсжное воспламенение зажигательного шнура. Он, кроме того,
удобен в обращении, дешев и заслуживает поэтому самого широкого
। нсдрсния во все взрывные работы при огневом способе взрывания.
К этой же группе относятся огнепроводы и передатчики детонации,
Л именно: 1) медленно горящий (бикфордов) шнур, 2) быстро горя-
щим зажигательный шнур, 3) детонирующий шнур и др.
Медленно горящий (бикфордов) шнур представляет собой (фиг. 10)
iujiGhk дымного пороха, заключенный в нитяную оплетку, и в зави-
• ।(мости от условий применения покрытый с поверхности тем или
иным изолирующим составом. Дымный порох, применявшийся в Гер-
•«fiiiiiit для бикфордова шнура, содержал следующие компоненты:
Скорость горения такого шпура колеблется между 40 и 15(1 мм/сек.
Применяемый в СССР бикфордов шнур горит со скоростью 1 см1сек
и дает сильный луч огня.
Быстро горящий шнур, имеющий непрессованную сердцевину,
горит со скоростью 150—300 мм/сек. Быстро горящие шнуры были
распространены в германской армии, но в настоящее время их вытес-
няют так называемые детонирующие шнуры.
Фиг. 11. Схема электрозапала.
Детонирующие шнуры представляют собой столбики бризантных
или инициирующих ВВ, заключенные в металлическую (свинцовую)
или нитяную оболочку и служащие для передачи детонации на рас-
стояние с той или иной скоростью.
Фиг. 12. Схема электродетонатора.
К вспомогательным средствам инициирования следует отнести
также и целый ряд электрозапалов, позволяющих превращать элек-
троэнергию в тепловую и тем самым обеспечивать воспламенение кап-
сюлей.
Одна из схем электрозапалов дана на фиг. 11. Электрозапал со-
стоит из гильзы, внутри которой расположены пропущенные через
эбонитовую или мастичную колодочку тонкие изолированные мед-
ные проволоки, между концами которых напаяна платиновая, никели-
новая или константановая проволока диаметром 0.035—0,05 мм и
длиной 4—6 мм, образующая мостик накаливания. На колодке на-
дета бумажная гильза, внутри которой помещен воспламенительный
состав.
В практике взрывного или артиллерийского дела очень часто со-
четают вспомогательные и основные средства инициирования. Так,
например, сочетание электрозапала с капсюлем-детонатором дает
электродетонатор (фиг. 12). Последний находит применение в гор-
ной промышленности и в артиллерийской технике для одновремен-
ного взрыва ряда объектов. Для этой цели применяют также и дето-
нирующий шнур с капсюлем-детонатором.
Сочетание электрозапала и капсюля-воспламенителя может найти
применение в электрических дистанционных трубках. Сочетанием
бикфордова шнура, фитиля и капсюля-детонатора получаются так
называемые зажигательные или запальные трубки.
18
| Средства инициирования независимо от начального импульса
мною разбить на две категории.
К первой категории относятся средства, инициирующий эффект
рнорых основан на действии теплоты. Они служат или для воспла-
Дпц-ния пороховых зарядов, выполняя при этом самостоятельную
|Ю'П. (капсюльная втулка, вытяжная трубка, капсюль-воспламени-
'•ль, бикфордов шнур, электрозапал и пр.), Или для инициирова-
нв| других инициаторов, выполняя в этом случае вспомогательную
|if«ill, (бикфордов шнур, электрозапал, капсюль-воспламенитель).
Ко второй категории относятся средства, инициирующий эффект
нгпрых основан на действии мгновенно возрастающего местного
" 'i ni» высокого давления (удара) на небольшую часть ВВ. Эти сред-
। им имеют только самостоятельное значение, а именно, для иници-
•1|«чыния зарядов бризантных ВВ. Сюда относится капсюль-детона-
юр, действующий от луча огня, капсюль-детонатор, действующий
" накола жала, электродетонатор и т. п.
Отсюда и требования к этим средствам инициирования различны,
Н если от средств первой категории в основном требуется создание
|\ча огня возможно более высокой жгучести, то от средств второй
!• .к сгорим требуется высокое инициирующее действие по отношению
• бризантным ВВ.
S 3. КЛАССИФИКАЦИЯ ИНИЦИИРУЮЩИХ ВЗРЫВЧАТЫХ ВЕЩЕСТВ
Под взрывчатыми веществами вообще мы понимаем системы, спо-
шбцые под влиянием внешних воздействий освобождать большие
количества энергии, иначе говоря, склонные к быстрым экзотерми-
ческим превращениям, сопровождающимся газообразованием, а в
• пили с этим значительным повышением давления в месте взрыва.
Гамой характерной особенностью этих превращений является их
чрезвычайная кратковременность, измеряемая для практически
применяемых количеств ВВ промежутками от тысячных до стоты-
• *1чпых долей секунды.
Все взрывчатые системы в зависимости от характера их приме-
нения подразделяются на три большие группы:
I. Инициирующие ВВ, служащие для создания началь-
ною импульса и применяемые в различных средствах инициирова-
ны, главным образом, в капсюлях.
2. Бризантные ВВ, служащие для изготовления разрыв-
ных зарядов артиллерийских снарядов, авиабомб, ручных гранат,
мин, торпед и прочих боевых припасов, основным назначением
|'О1орых является фугасное или бризантное действие.
3. Метательные ВВ (пороха), служащие для выбрасыва-
ния боевых припасов из артиллерийских систем и ручного оружия.
Под инициирующими ВВ мы понимаем системы, способные в ре-
цльтате взрыва небольшого количества под влиянием простого на-
Ильного импульса вызвать воспламенение, взрыв или детонацию
ipyioro ВВ.
19
Все известные инициирующие вещества можно разделить на два
класса: инициирующие химические соединения и инициирующие
смеси.
Первый класс в зависимости от химического состава в свою
очередь подразделяется на следующие группы:
1. Металлические производные к а р Сил-
ок с и м а, отличительным признаком которых является наличие
группы атомов:
^С—N—О—
В эту группу входят производные карбилоксима с двухвалент-
ным углеродом (первая подгруппа) и производные карбилоксима,
углерод которого четырехвалентен (вторая подгруппа). Представи-
телями первой подгруппы являются соли гремучей кислоты, или фуль-
минаты: гремучая ртуть Hg(ONC)2, применяемая в качестве иниции-
рующего ВВво всех странах, и фульминат меди Си (ONC)2-Cu (ОН)3 —
весьма нежелательный и вполне возможный спутник гремучертут-
ных капсюлей в медных оболочках, хранившихся в ненадлежащих
условиях. Представителями второй подгруппы являются металли-
ческие производные амидкарбилоксима NH. CH = NOH, метил-
ураловой кислоты СзН2О2Н4, метилазооксиураловой кислоты
C2H4O3N4 и др.
2. Производные азотистоводородной кис-
лоты, или азиды, основной характеристикой которых является
наличие в них группы атомов:
N
— || или — N = N -z N
XN
Неорганические производные (соли тяжелых металлов) состав-
ляют первую подгруппу азидов, наиболее ярким примером которой
служит азид свинца Pb(Ns)2. Органические производные составляют
вторую подгруппу, многочисленную по количеству, однако, еще не.
вполне изученную. Подробно исследовались лишь немногие органи-
ческие азиды, но в военном деле их пока не применяют. Представи-
тели этой подгруппы — циануртриазид C3N3(N3)3, тринитротриазидо-
бензол Cc(NO2)s(N3)3, свинцовая соль азидотиоугольной кислоты
PbC2N6S4, гцдразиндикарбоназид N3—СО—NH—NH—СО—N3, свин-
цовая соль динитроазидофенола [CcH.2(NO2)2N3O]2Pb и др.
3. Органические перекиси, характерным призна-
ком которых служит группа атомов:
— о—о—
Основные представители этой группы — перекись ацетона
(СН3)2С = (О—О)а =С — (СН3)2, перекись ацетилацетона (С5Н8О4)Я,
перекись бензоила С6НБСО—О -О—СО—С6Н5, гексаметилентрипер-
оксиддиамин (сокращенно ГМТД) N = (СН2 — О — О — СН2)3 = N,
трициклоацетонпер оксид С8Н18О6 и др.
20
.па группа, несмотря на сравнительную дешевизну продуктов
। простоту их получения, ни в военном деле, ни в мирной промыш-
। внести пока не применяется.
* I. Нитродиазосоединения, вернее соли различных
in । родиазопроизводных бензола, характерным признаком которых
1111Я1ОТСЯ группы атомов:
zZ°
— N, н - N = N —
X)
К этой группе относятся: диазобензолнитрат C6H5N == N • NO3,
ипштродиазофенол C6H3(NO2)2 — N = N — ОН, нитродиазобензол-
। рхлорат CeH;J(NO2) — N /= N • СЮ4, нитробисдиазобензолперхло-
н CeH8(NO2) • (N= N • C10j2, динитродназобензолперхлорат
,II.XNO2)2— N = N • C1O4 и др. Из перечисленных веществ заслу-
। икаютf внимания только три последние.
а. Производные непредельного азотоводо-
» о д a N4H4, известные под общим названием «тетразены». Пред-
I «жители этой группы — гуанилнитрозоаминогуанилтетразен
1I.,—C(NH)—NH—NH—N...N—C(NH)—NH—NH—NO., диазотетра-
it п.1мидогуанидин HN4 = C—N2-—NH—NH—C(NH2) — NH • H3O,
пшзотетразолсемикарбазид HN4C—N2—NH—NH—C0~NH2 • H2O,
|и.|.<отетразолфенилгидразин HN4 = CNa—N(NHa)—CcH5 и диазоте-
гразолбенЗолоаминогуанцдин HN4e CN2—N(N—C7HG)—C(NH2)—NH2,
•i которых только первый в последнее время применяют в кап-
‘олях-воспламенителях.
(>. Производные тетразола, отличительным призна-
ем которых является наличие тетразолового кольца:
N — NH. N = CH.
]| >СН или | -/NH
I N —JN ? _______N = N<
К этой группе надлежит отнести соли тяжелых металлов самого
гразола,
соли азотетразола
N—NH. N =N
II >С —N = N—N< |
N — N CH = N
соли оксиазотетразола
Я—"NH. N = N
|| \C-N = N-N( \
N-N z XC(OH)=N
соли диазоаминотетразола
N —NH NH —N
II >C-N = N —NH —C< II
N — N N — N
21
диазотетразол
соли тетразилазида
бистетразол
N-N
II
N —NH
/С —N3
N —NH NH — N
11 11
N— N " N — N
соли и эфиры нитротетразола
N —NH
II >С —NO,
N— N 77
Эта группа веществ еще недостаточно изучена.
7. Соли тяжелых металлов оке и нитросое-
динений бензола, к которым относятся пикраты железа
и свинца [CgH2(NO2)sO]2Fe, [CeH2(NO2)3O]2Pb, тринитрорезорцинат
свинца (ТНРС) C6H(N02)30,Pb • Н2О,динитрорезорцинат свинца (Д НРС)
C0H2(NO2)2O2Pb, тринитрофлороглюЦинат свинца [Cc(N02)303]2Pb3
и т. п. Из указанных солей только TH PC применяется в военной
и в мирной технике.
8. Дммониакаты и г идцр_а^-и-н-а т ы хлоратор
и перхлоратов двухвалент цы х тяжелых ме-
таллов, отличительным признаком которых является комплекс-
ный характер соединении хлоратов и перхлоратов с аммиаком и
гидразином. Представители этой группы — аммониакат хлората
меди Си(С103)2 • 4NH8, аммониакат перхлората меди Си(С1О4)3 • 4NH3,
гидразинат хлората кадмия Cd(C103)2 • 2N,H^ гидразинат хло-
рата никеля Ni(C103)2 • 3N2H4, гидразинат перхлората кадмия
Cd(C104) - Cd(OH)2 • 3N2H4 • 2Н2О, гидразинат перхлората никеля
Ni(C104)3 - Ni(C104) (ОН) • 5N2H4 ЗН3О и др. Все они обладают
высокими инициирующими свойствами, но вследствие недостаточной
стойкости не находят распространения.
9. Хлораты и перхлораты тримеркуральде-
гидов:
Hg3CAH • CIO,
HgaC.OzH • С104
Эти соединения превосходят по инициирующему действию грему-
чую ртуть, но их стойкость весьма мала, и поэтому они не заслужи-
вают внимания.
22
10. Ацетилениды — металлические производные ацети-
лена. Их особенностью, как известно, является тройная связь между
углеродными атомами:
— С = С
Представители этой группы — ацетиленистая медь Сп2С2 и аце-
тил енистое серебро Ag2C2-
11. Галоидные и сернистые соединения
азота, к которым относится хлористый азот N = Cl3, NH — С12,
иодистый азот N = J3, NH = J2, сернистый азот N4S4.
Второй класс — инициирующие смеси. Каждая иницииру-
ющая смесь состоит из нескольких компонентов, из которых хотя бы
один является горючим, а' другой окислителем. Кроме того, к ним
обычно прибавляют некоторое количество других компонентов, по-
вышающих чувствительность смеси, ее вязкость и т. п. В зависимости
от того, входят ли в смесь взрывчатые компоненты или смесь состав-
лена исключительно из невзрывчатых материалов, этот класс разби-
вается на две группы:
1. Смеси, в состав которых входят один или несколько взрывча-
тых компонентов. К ним, например, относятся воспламенительные
составы, содержащие гремучую ртуть и применяемые для снаряже-
ния винтовочных, трубочных и других капсюлей и для снаряжения
капсюлей-детонаторов. Содержание отдельных компонентов составов
различно и обусловливается служебными требованиями к капсюлям.
В качестве примеров можно привести следующие составы, применяе-
мые для капсюлей-детонаторов и распространенные в западноевро-
пейских странах.
Компоненты % содержания
Гремучая ртуть - . 90 87.5
Бертолетова соль . 10 15 10
Калиевая селитра . . । — 2,5
К этой группе относятся также ударные составы, применяемые
для снаряжения капсюлей-воспламенителей для патронов Наган,
патронов к 3-линейной винтовке и для 22-секундной трубки.
Ка пасли -воспламенители Hg(ONC), КСЮз SbsS3
К патронам Наган К патронам 3-линейной винтовки . . - Трубочные (22 сек. трубка) 25,8 16,7 28 37,1 55 36 37.1 28,3 36
23
К той же группе относятся применяемые, например, в Германии
неоржавляющие ударные составы для капсюлей-воспламенителей:
тетразена . . 0,25— 5%
ТНРС . .25 —55%
Ba(NOs). 25 —55%
РЬО2 • - 5 —10%
Sb2S3......................... О —10%
силицида кальция.............. 3 —25%
стеклянного порошка 0 — 5%
2. Смеси, составленные из невзрывчатых компонентов и обычно
назначаемые для воспламенения порохов. Примером таких смесей
может служить состав первых капсюлей-воспламенителей:
бертолетовой соли . . 70,6%
серы . . . 17,0%
угля 11,8%
или применявшийся в Германии состав для снаряжения капсюльных
втулок:
бертолетовой соли . . 52,9%
антимония.......... 29,5%
пороховой мякоти . 17,5%
а также применявшийся в обыкновенных французских ружейных
капсюлях состав:
бертолетовой соли ... . 50%
роданистого свинца . . 10%
железисто-синеродистого
свинца............... 40%
Все инициирующие ВВ в зависимости от цели, для которой они '
применяются, разбиваются на две больших группы:
1) инициирующие вещества, применяемые для вызова детонации
бризантных ВВ,
2) инициирующие вещества, применяемые для целей воспламене-
ния порохов (метательных ВВ) и капсюлей - детонаторов.
ГЛАВА II
ОБЩАЯ ХАРАКТЕРИСТИКА ИНИЦИИРУЮЩИХ ВВ
Характерными особенностями инициирующих ВВ, отличающими
их от прочих ВВ, являются:
I) эндотермический характер большинства из них;
2) значительно более быстрое нарастание скорости взрывчатого
превращения и связанное с этим исключительное влияние на рас-
пространение реакции взрывчатого превращения в массе инициируе-
мого вещества;
3) чувствительность к простому начальному импульсу.
§ 1. ЭНДОТЕРМИЧЕСКИЙ ХАРАКТЕР ИНИЦИИРУЮЩИХ ВВ
Известно, что все вещества в термохимическом отношении можно
разделить на экзотермические и эндотермические. К первым отно-
сятся вещества, образование которых из химических элементов свя-
зано с выделением теплоты, ко вторым же относятся вещества, на обра-
зование которых из элементов затрачивается то или иное количество
теплоты.
Количество теплоты, которое выделяется или поглощается при
образовании молекулы вещества из химических элементов, назы-
вается теплотой образования. Экзотермические соединения обла-
дают положительной теплотой образования, а эндотермические —
отрицательной. Взрывчатые вещества также могут быть и эндотер-
мическими и экзотермическими соединениями.
Эндотермические В В характеризуются, вообще говоря, более
сильными экзотермическими взрывчатыми превращениями, так как
чем больше затрачено энергии на образование молекул, тем больше
энергии выделится при их разложении.
В отличие от прочих взрывчатых соединений значительное, почти
абсолютное, большинство инициирующих веществ является соеди-
нениями эндотермическими, что видно из табл. 2.
В табл. 2 приводятся данные об инициирующих веществах (гекса-
мстилентрипероксиддиамин и трициклоацетонпероксид), обладающих
положительной теплотой образования. Они, так же как и еще неко-
торые другие, являются исключением из общего правила, ибо эндо-
юрмичность вещества как безусловную (необходимую и достаточ-
ную) характеристику инициирующих ВВ рассматривать нельзя.
25
Таблица 2
Теплоты образования инициирующих В В
| <dou ou | Наименование вещества Брутто-формула Теплота образо- вания Источник
Кал]кг Кал/мол
1 Гремучая ртуть . Hg(ONC)2 —221,5 —62,8 Каст, ВВ и
2 Азотистоводород- ная кислота . . HN, —1558.0 —67.0 средства вос- пламенения, стр. 70 Рот иМюл-
3 Треххлористый азот NC13 —453,0 —54,7 лер Б Р у н с в и г.
4 Диазобензолнитрат C„H5(N- NjNO. —284,0 —47,6 Теория ВВ, стр- 14—16
5 Азид свинца . . . ИШ —378,0 —110,8
6 Циануртрмазид . —1073,0 —219,0 М ю р а у р,
7 Сернистый азот . N,6< —700,0 —129,0 Bull- Soc-
8 Гексаметалентри- пероксиддиамин N(CH2O-O-CH2)sN +417,0 +86,7 Chem-, т- LI— L11, стр. 1152—
9 Трицикл оацетои- пероксид CsHjbOb +123,0 +21,7 1166, 1932
В самом деле, если рассмотреть теплоты образования бризант-
ных веществ (в большинстве своем' соединений экзотермических), то
и среди них найдется несколько веществ с отрицательной теплотой
образования. Так, например, теплоты образования (по Ринкенбаху):
тринитро фенил мети л нитрамина (тетрил) ... —4,686 Кал)/лол
гексаиитродифени ламина (гексамии) . . —13,682 >>
Стало быть резкой границы между инициирующими и бризант-
ными веществами по теплотам образования провести нельзя. Однако,
если сравнить теплоты образования тетрила и гексамина с теплотами
образования большинства инициирующих веществ, то надлежит
признать, что последние обладают эндотермическим характером,
превышающим в несколько раз эндотермичность гексамина. Следо-
вательно, отрицательная теплота образования является фактором,
благоприятствующим приобретению ВВ инициирующих свойств.
§ 2. УСКОРЕНИЕ ВЗРЫВЧАТОГО ПРЕВРАЩЕНИЯ
Процесс взрыва, вызванный тем или иным начальным импульсом,
как и всякий процесс, протекает во времени и может быть характе-
ризован скоростью взрывчатого превращения. Величины скорости
превращения можно выражать или в единицах времени, в течение
26
которого взрывается единица объема или веса; или в единицах длины
образца ВВ, по которому распространяется реакция взрывчатого
превращения в единицу времени.
Скорость взрывчатого превращения прогрессивно возрастает и
при соответствующих условиях может достигнуть максимальной,
вполне определенной и свойственной данной взрывчатой системе
скорости детонации. На фиг. 13 изображен предположительный
ход нормального нарастания скорости взрывчатого превращения
до скорости детонации в зависи-
мости от длины столбика ВВ или
от времени. - —-—
Опытами установлено, что ус- f
корению взрывчатого превращения /
способствует прежде всего харак- /
гер начального импульса. Рассмот- /
рим это положение в применении
к инициированию лучом ОГНЯ. И ______________________________
Сокращению продолжительности
периода воспламенения ВВ способ- фш. (3 Граф[1К иарастания скорост„
ствуют Следующие факторы. взрывчатого Превращения.
1) высокая температура пламени
или увеличение количества теплоты, сообщаемой единице поверх-
ности инициируемого вещества;
2) хорошая воспламеняемость от пламени самого инициируемого
вещества;
3) большая поверхность воспламенения;
4) незначительная плотность воспламеняемого поверхностного
слоя вещества;
5) уменьшение величины зерна, в результате чего увеличивается
поверхность воспламенения.
Переход от воспламенения к детонации также зависит от следую-
щих факторов:
1) от свойств самого инициирующего вещества и прежде всего
от количества энергии, выделяющейся в единицу времени при раз-
ложении единицы объема инициатора; увеличение количества энер-
гии ускоряет процесс взрывчатого превращения инициируемого ве-
щества;
2) от оболочки, в которую заключено ВВ; сокращению времени
перехода к детонации способствует прочная наружная оболочка и
незначительная ее теплоотдача, а также незначительное сечение для
вытекания газообразных продуктов взрывчатого превращения;
3) от степени прессования (плотности) ВВ. При малой плотности
воздух, находящийся между кристаллами, препятствует передаче
энергии от слоя к слою.
Основное отличие инициирующих ВВ от бризантных в том и со-
стоит, что у первых условия для действия указанных выше факто-
ров имеются или мы легко их можем создать. Вследствие этого уско-
рение взрывчатого превращения инициирующих ВВ значительно
27
выше ускорения превращения бризантных веществ. Большую роль
в этом играет, конечно, чувствительность инициирующих веществ
к внешним воздействиям.
Явление взрыва, несмотря на свою кратковременность, может
быть расчленено на два самостоятельных последовательно протекаю-
щих процесса: инициирования начальным импульсом и распростра-
нения взрывчатого превращения в массе ВВ.
Самая ничтожная доля азида свинца при соприкосновении с на-
каленной проволокой детонирует по всей массе в течение кратчай-
шего промежутка времени. В этом случае процесс иницииро-
вания немедленно переходит в процесс распространения взрыва
Фиг. 14- Кривые нарастания ско-
рости взрывчатого превращения
бризантных и инициирующих
веществ.
Фиг. 15. Кривые нарастания ско-
рости взрывчатого превращения
Pb(Ns)s и Hg(ONC)2
в массе азида свинца, причем скорость взрывчатого превращения
становится равной или по крайней мере весьма близкой к скорости
детонации. Тринитрофенол или тринитротолуол в тех же условиях
только сгорают в течение определенного промежутка времени за-
долго до приобретения свойственной им скорости детонации. Даже
относящийся к числу весьма легко воспламеняющихся веществ гли-
церинтринитрат может спокойно сгорать в сравнительно больших
количествах, и только при накоплении тепла от реакции горения
происходит его детонация-
Отсюда ясно, что для достижения скорости детонации у иниции-
рующих веществ требуется значительно меньше времени, чем у бри-
зантных, что, стало быть, у первых ускорение взрывчатого превра-
щения больше. На фиг. 14 изображены для иллюстрации кривые
нарастания скорости взрывчатого превращения бризантных и ини-
циирующих веществ.
У разных инициирующих веществ ускорение взрывчатого превра-
щения различно. Этим, в частности, можно объяснить значительное
различие между инициирующей способностью азида свинца и грему-
чей ртути (фиг. 15).
В самом деле, навеска азида свинца, воспламененная от пламени
спички, пробивает картон, а такая же навеска гремучей ртути только
быстро сгорает.
28
Для того чтобы уяснить механизм инициирования вторичного за-
ряда взрывом инициатора, необходимо знать механизм детонации
инициатора и вторичного заряда. В настоящее время мы еще не распо-
ыгаем достаточным опытным материалом о том, что происходит на
। ранице взорвавшегося инициатора и вторичного заряда. Однако,
пользуясь опытами по вопросам, связанным со скоростью детонации,
можно сделать несколько заключений.
Сначала условимся, что в массе ВВ взрывчатые превращения на-
чинаются не с одинаковой скоростью и что скорость, с которой воз-
никает взрывчатое превращение, зависит от количества энергии,
сообщенного начальным импульсом в единицу времени. Эту ско-
рость условимся называть начальной скоростью взрывчатого превра-
щения. Количество энергии в единицу времени, сообщаемое вторич-
ному заряду инициирующим веществом, зависит от той скорости,
с которой эта энергия выделяется последним, т. е. от скорости
взрывчатого превращения инициатора.
Гремучая ртуть, запрессованная в металлическую оболочку, при
прочих равных условиях оказывает тем большее действие на вторич-
ный заряд, чем интенсивнее был сообщенный ей луч огня. Началь-
ная скорость процесса растет с увеличением интенсивности импульса,
так как при этом среднее количество энергии, сообщенное вторич-
ному заряду в единицу времени, повышается.
Следовательно, чем выше начальная скорость взрывчатого превра-
щения инициирующего вещества, тем сильнее его инициирующее
действие.
Опыты Каста по исследованию скоростей детонации тринитро-
толуола и тринитрофенола показали, что скорость распространения
взрывчатого превращения в массе ВВ в сильной степени зависит от
начального импульса, пока он не достигнет определенной величины,
вызывающей детонацию. Начальный импульс, дающий больше энергии
в единицу времени, только в непосредственной близости к капсюлю
создает временную повышенную скорость, которая затем (на рассто-
янии около 200 мм от капсюля) переходит в нормальную скорость.
При импульсе же меньшем вызывается взрывчатое превращение со
скоростью ниже скорости детонации. В случае применения еще более
слабого импульса наблюдается падение скорости, а иногда и отказ.
Опыты М. Сотеу и В. Holmes по исследованию скоростей дето-
нации глицеринтринитрата под действием капсюлей, содержащих
от 0,8 до 1,5 г инициатора (90% Hg(ONC)2 и 10% КСЮ3), показали
колеблющиеся в широких пределах скорости взрывчатого превраще-
ния от 1288 до 8410 м/сек. При применении же более сильного ини-
циатора они получили вполне согласные результаты, потому что
скорость взрывчатого превращения достигала скорости детонации.
К. К. Снитко в своей книге «Теория взрывчатых веществ» отме-
чает, что на возбуждение правильной и нормальной для данных усло-
вий скорости детонации весьма существенное влияние оказывает
интенсивность начального импульса, что можно иллюстрировать сле-
дующими данными Сотеу для скорости детонации динамитов (см.
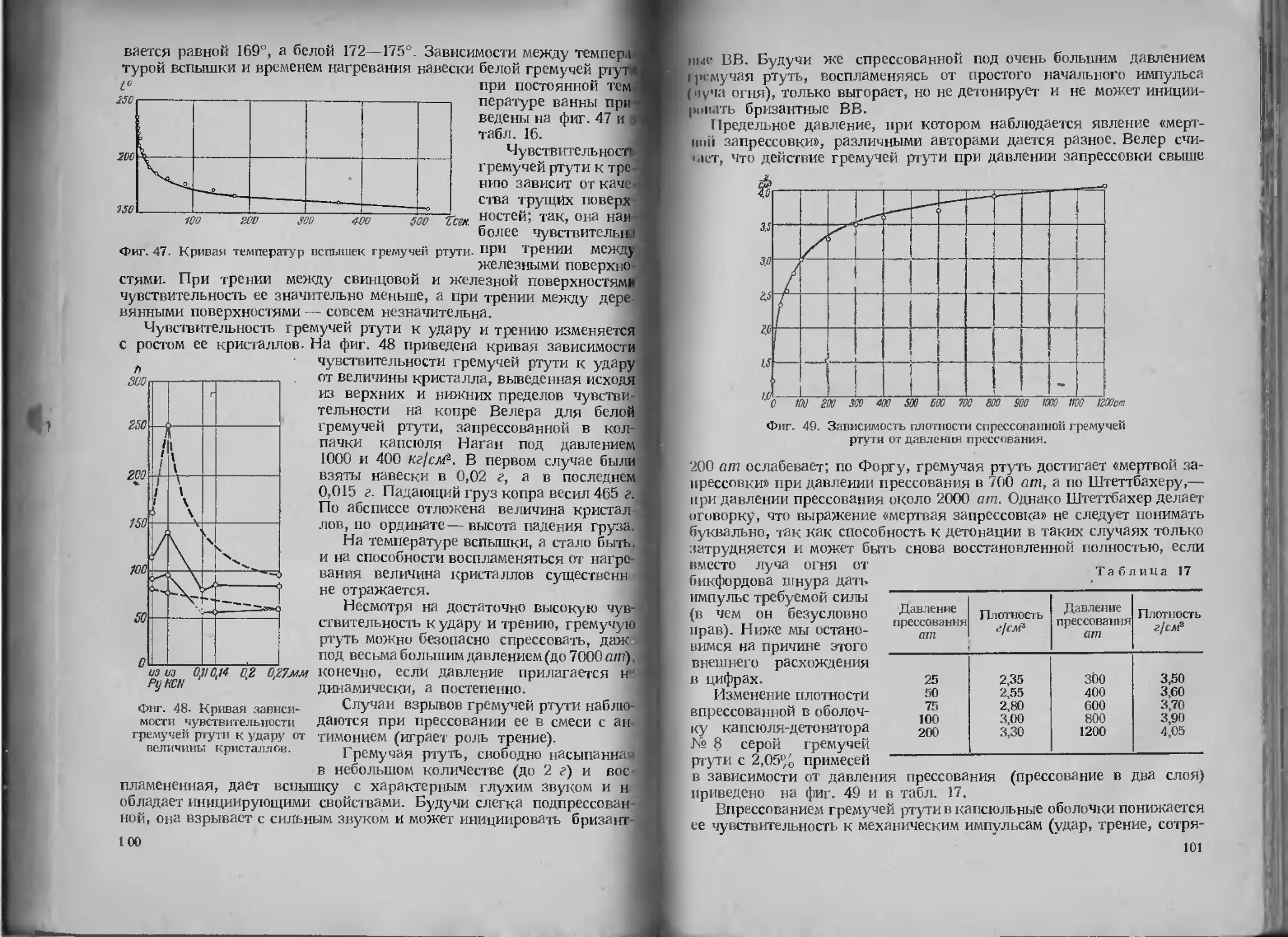
габл. на стр. 30). 2д
А.
Содержпнис
нитроглицери-
на в динами гс
Скорость дето-
iiatfui при шш-
цпмровапии I
капсюлем
Mjceii.
Скорость дето-
нации при ини-
циировании
детонатором
м:сек
30 2484 5122
40 2278 5544
50 2279 5862
60 2104 6606
75 2165 6999
Наконец, опыты Бертло с газовыми гремучими смесями показали,
что при скоростях, меньших 1000 м/сек, детонации не наблюдается
и что в газовых смесях также играет большую роль интенсивность
импульса.
Эти опыты позволяют высказать предположения, что при про-
чих равных условиях и безусловно при навесках инициатора, содер-
жащих равное количество энергии:
1) чем больше ускорение взрывчатого превращения инициатора,
тем больше скорость взрывчатого превращения вторичного заряда
(до предела, каковым является скорость детонации);
2) если скорость взрывчатого превращения инициирующего ве-
щества больше начальной скорости, необходимой для достижения
скорости детонации вторичного заряда, то детонация обеспечена;
наоборот, если первая меньше второй, то произойдет только взрыв
(неполная детонация) илн отказ.
3. ИНИЦИИРУЮЩАЯ СПОСОБНОСТЬ
Способность инициирующих веществ вызывать детонацию других
ВВ называется их инициирующей способностью. От инициирующего
В В требуется, чтобы после кратковременного периода нарастания
скорости его взрывчатого превращения детонировало инициируемое
бризантное ВВ. Рассмотрим факторы, от которых зависит инициирую-
щая способность.
В предыдущем параграфе была отмечена роль ускорения взрыв-
чатого превращения как фактора, отличающего инициирующие ве-
щества от бризантных. Оказывается, что чем выше ускорение взрыв-
чатого превращения инициатора, тем больше и инициирующая его спо-
собность. Самое быстрое нарастание скорости было найдено у фуль-
мината кадмия Cd(ONC)2,0,008 г которого достаточно для детонации
тетрила (в оболочке капсюля-детонатора № 8). Гремучая ртуть обла-
дает сравнительно медленным нарастанием скорости, и для возбуж-
дения взрыва тетрила ее необходимо 0,29 г. Другим примером влия-
ния ускорения взрывчатого превращения на инициирующую способ-
ность может служить опыт Велера, показавшего, что добавление к
гремучей ртути 0,5 мг азида свинца повышает инициирующую спо-
зо
собность первой примерно в два раза. Происходит это потому, что
ускорение взрывчатого превращения азида свинца весьма велико.
Инициируясь от начального импульса, он инициирует гремучую
ртуть, причем нарастание скорости взрывчатого превращения по-
следней происходит быстрее, чем без азида свинца.
Следовательно, нарастание скорости взрывчатого превращения
является одним из факторов, влияющих на инициирующую способ-
ность.
Для уяснения других факторов, влияющих на инициирующую
способность, заметим сперва, что детонация бризантного ВВ является
следствием работы удара газообразных продуктов взрывчатого пре-
вращения инициатора. Эти газообразные продукты, выделяющиеся
со все возрастающей скоростью, ударяют по поверхности бризант-
ного вещества, сообщая некоторый импульс /. Скорость движения
газообразных продуктов уменьшается в течение времени удара т от
величины 4-С до 0 и далее, если предположить эластичность бри-
зантного вещества, за то же время т — до величины—С. Вследствие
падения скорости возникает давление газообразных продуктов, рав-
ное некоторой величине Р.
Вычисление среднего давления Р во время удара по уравнению
импульсов и по формулам, вытекающим из кинетической теории,
приводит к следующему выражению:
р —----1----Д . Ck
г 3-081 ’
где Р — давление в г/см2;
А — плотность газообразных продуктов разложения в момент
их образования, в г/см9;
С — скорость газообразных продуктов превращения в см/сек;
2>Л> I.
Повышению давления Р, от которого зависит работа удара, спо-
собствует, как это видно из уравнения:
1) большая плотность газообразных продуктов взрывчатого пре-
вращения, а также, конечно, большая плотность самого инициатора;
2) большая скорость движения газообразных продуктов взрыв-
чатого превращения.
Развитию же большой скорости движения газообразных продук-
тов разложения благоприятствуют следующие свойства инициирую-
щего ВВ:
1) большая температура взрыва;
2) большая скорость детонации.
Работа удара зависит также от удельной энергии инициирующего-
ВВ. Капсюль, изготовленный из чистой гремучей ртути в качестве
инициатора и тетрила (1 г) в качестве вторичного заряда, обладает
меньшей пробивной способностью, чем капсюль, у которого инициа-
тор состоит из смеси гремучей ртути с хлоратом калия. Это объяс-
няется тем, что гремучая ртуть при взрыве разлагается по урав-
нению
Hg(0NC>2-> Hg + N2 + 2СО + 116 Кал,
31
т. е. мы имеем случай неполного сгорания углерода, а стало быть,
и меньшее выделение энергии. Для полного сгорания всего углерода
гремучей ртути необходим кислород, который и вводится примесью
бертолетовой соли. Если рассчитать весовой процент хлората калия,
необходимого для полного сгорания углерода гремучей ртути, т. е.
по реакции, при которой весь углерод превращается в углекислоту:!
3Hg(ONC)2 + 2КС1О3 ->3Hg 4- 2КС14- 3N2 + 6СО2 4- 757,8 Кал, I
то окажется, что потребуются следующие весовые количества:
гремУчей ртути....... 77,7%
бертолетовой соли . 22,3%
Однако опыты показывают, что примесь 22,3% хлората калия
к белой гремучей ртути не дает высшего инициирующего действия
Наибольший эффект дают капсюли, инициаторы которых состоят
из гремучей ртути с 10—15% хлората калия (при давлении прессо-
вания 200 ат). Таким образом этот пример показывает, что увеличение
содержания энергии, вообще говоря, повышает инициирующую спо-
собность, но что вместе с тем примесь к инициатору других веществ,
содержащих кислород, полезна только до известного предела, так как
последние в то же время, видимо, понижают скорость детонации.
Поэтому наивыгоднейшее количество примесей не может быть уста-
новлено только на основании уравнения взрывчатого превращения.
Плотность инициирующего ВВ также оказывает значительное
влияние на инициирующую способность, так как от нее зависит
плотность заряжания капсюля-детонатора, т. е. отношение веса ини-
циирующего заряда к объему капсюля. Чем большее количество ини-
циирующего ВВ поместится в меньшем объеме, тем большая работа
им будет совершена и тем больше будет инициирующее действие
вещества.
Плотность гремучей ртути, например. 4,307, а азида свинца 4,73.
Отчасти благодаря этому азид свинца обладает большей инициирую-
щей способностью.
Зависимость инициирующей способности от величины кристал-
лов может быть определена, например, количеством вещества, не-
обходимого для полной детонации тротила. Произведенные Велером
и Бертманом опыты показали, что между величиной кристаллов и
наименьшим количеством инициирующего ВВ, необходимого для
детонации тротила, существует зависимость, могущая быть выра-
женной гиперболической кривой (абсцисса — длина кристалла гре-
мучей ртути по большой диагонали, а ордината — вес предельного
заряда). € увеличением длины кристалла от 0,01 до 0,06 мм вели-
чина предельного заряда уменьшается от 1,0 до 0,22 г; дальнейшее
увеличение длины кристалла также уменьшает предельный заряд,
но уже значительно медленнее. Так, при увеличении длины кри-
сталла от 0,067 до 0,183 мм предельный заряд уменьшается только
от 0,22 до 0,20 г.
32
В теории ВВ все эти факторы, от которых зависит работа удара,
объединяются общим понятием — бризантность. Точной математи-
ческой формулировки бризантности еще не дано. Для практических
целей Каст предложил пользоваться формулой:
1 -де / — удельная энергия;
v — скорость взрывчатого превращения;
А — гравиметрическая плотность.
Согласно этой формуле бризантность является удельной энергией,
«гнесенной к единице объема и единице времени, т. е. мощностью
единицы объема ВВ.
Итак, инициирующая способность вещества зависит от его бри-
’ МНТНОСТИ и ускорения взрывчатого превращения.
§ 4. ПРЕДЕЛЬНЫЙ ИНИЦИИРУЮЩИЙ ЗАРЯД
На практике инициирующая способность инициирующих ВВ
характеризуется предельным
мильным количеством веще-
ства, способным вызвать
полную детонацию 0,5 г
ютрила или другого бризант-
ного ВВ, запрессованного
д медную оболочку капсюля-
детонатора № 8 под давле-
нием 500 ат.
Сравнительные величины
предельных зарядов по отно-
шению к какому-либо опре-
деленному бризантному ВВ
позволяют также классифи-
цировать инициирующие ве-
щества по их инициирующей
способности.
Полнота детонации опре-
деляется характером проби-
инициирующим зарядом, т. е. мини-
Фиг. 16.
Матрица для
снаряжения
капсюлей-дето-
наторов.
Фиг. 17. Камера для
подрыва капсюлей-
детонаторов.
ши взрываемым капсюлем-
детонатором свинцовой пла-
«гонки толщиной 4 мм.
Детонацию принято считать
полной, а инициирующий за-
ряд предельным, если в результате детонации капсюля-детонатора
и свинцовой пластинке пробивается отверстие, диаметр которого не
менее наружного диаметра капсюля-детонатора.
Капсюли-детонаторы снаряжают, прессуя ВВ в оболочку на прессе
тобой существующей системы, принимая при этом необходимые
меры безопасности (щиты).
Пубнов —92—3
Медную оболочку капсюля-детонатора № 8 вставляют в матрицу
(фиг. 16). Навешивают на технических весах 0,5 г тетрила, всы-
пают навеску через воронку в гильзу и осторожно подпрессовывают
пуансоном вручную. Затем навешивают на аналитических весах ори-
ентировочную навеску инициирующего ВВ, всыпают ее через воронку
в гильзу и, очистив закраины гильзы и ее стенки от возможной пыли
инициатора, осторожно вводят в гильзу сначала чашечку с сеткой,
а затем пуансон. Нив коем случае нельзя, после того как вставлена
чашечка и пуансон, подпрессовывать ВВ вручную.
Собранную матрицу ставят на подставку пресса, окруженную
с трех сторон щитами. Рукоятку рычага 1-го рода снимают с крюка
и плавно опускают до тех пор, пока не станет подниматься рычаг
2-го рода. Как только поднимется рычаг 2-го рода, система приходит
в равновесие, и в этом состоянии ее выдерживают в течение 5—10 сек.
Следует всегда выдержку давать одну и ту же — или 5 или 10 сек.,
так как она влияет на плотность прессуемого вещества. После этого
рукоятку рычага 1-го рода поднимают и вешают на крюк. Матрицу
снимают с подставки пресса и пуансон извлекают из матрицы на спе-
циальном станке за щитом.
После изъятия пуансона капсюль-детонатор вынимают из мат-
рицы рукой, если он свободно выходит. В противном случае из мат-
рицы извлекают поддон, ставят ее так, чтобы. капсюль-детонатор
был расположен вверх дном, на стальной полый цилиндр с ватой
в нижней его части и вводят вместо поддона пуансон. Все это ставят
на площадку пресса и, плавно опуская рычаг, выдавливают капсюль
из матрицы.
Снаряженные таким образом два-три капсюля подрывают на
4-миллиметровой свинцовой пластинке.
В качестве камеры для подрыва капсюлей-детонаторов приме-
няют чугунный или стальной, освинцованный внутри, муфель (фиг. 17),
на цилиндрическую, полую внутри, подставку которого наклады-
вают свинцовую пластинку. Затем извлекают из муфеля верхнюю
втулку и через отверстие в ней снизу вверх просовывают отрезок
бикфордова шнура такой длины, чтобы он доходил до свинцовой
пластинки и выступал над муфелем на 5—6 см.
Бикфордов шнур на деревянной подставке отрезают от бухты
острым чистым ножом. Один конец бикфордова шнура срезают на-
искось, другой конец срезают перпендикулярно оси шнура и загла-
живают о какую-нибудь гладкую поверхность (например лезвие
ножа). Этот конец отрезка предназначается для введения в капсюль-
детонатор; он должен быть чист и иметь открытый состав, в против-
ном случае могут быть отказы не по вине инициирующего ВВ. От-
казы могут быть и в случае косого среза, так как косо срезанный
конец шнура, ложась на отверстие чашечки капсюля-детонатора,
может закрыть его своей оплеткой. Заглаженный конец бикфордова
шнура вводится в капсюль-детонатор до упора. При этом нельзя
сильно нажимать и, в особенности, вращать бикфордов шнур ил1
капсюль, чтобы не создавать трения, от которого капсюль-детонатор
34
может взорваться. Легко нажимая на бикфордов шнур, убеждаются
i гом, что конец его касается металлической чашечки капсюля.
Капсюль-детонатор с бикфордовым шнуром устанавливают в цен-
। ре пластинки строго вертикально. Когда все операции произведены,
работающий заходит за закрытую стенку муфеля, пользуясь пенько-
вым тлеющим фитилем, воспламеняет бикфордов шнур и сейчас же,
но без особой спешки, уходит за щит. По сгорании бикфордова шнура
работающий слышит обычно или взрыв капсюля, или вспышку
инициатора без детонации самого капсюля, или слабозвучный взрыв.
В первом случае свинцовая пластинка оказывается пробитой, во вто-
ром случае наблюдается выгорание, а о третьем — образование жучка
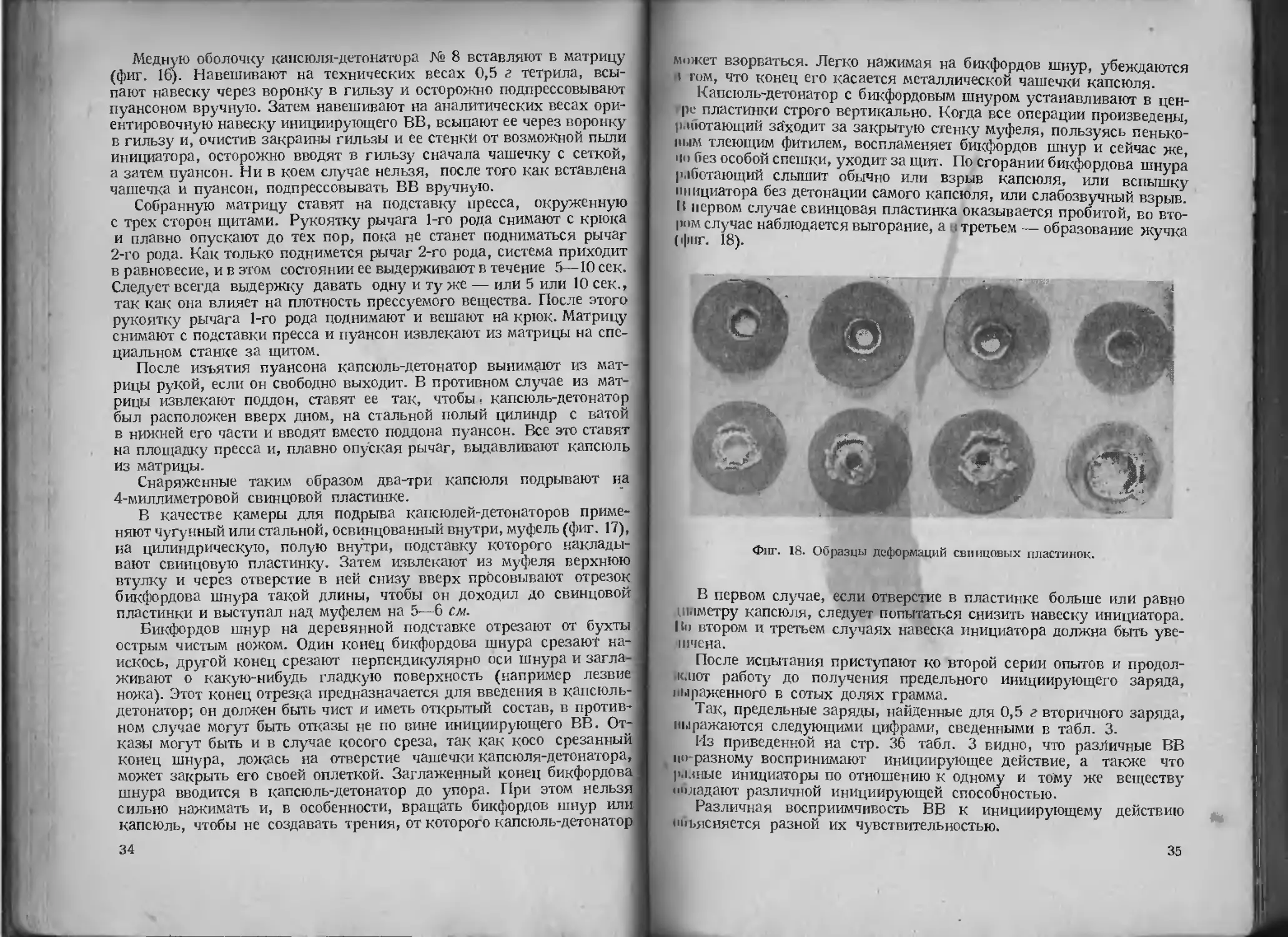
(фиг. 18).
Фиг. 18. Образцы деформаций свинцовых пластинок.
В первом случае, если отверстие в пластинке больше или равно
ишметру капсюля, следует попытаться снизить навеску инициатора.
Во втором и третьем случаях навеска инициатора должна быть уве-
шчена.
После испытания приступают ко второй серии опытов и продол-
Испот работу до получения предельного инициирующего заряда,
ныраженного в сотых долях грамма.
Так, предельные заряды, найденные для 0,5 г вторичного заряда,
иыражаются следующими цифрами, сведенными в табл. 3.
Из приведенной на стр. 36 табл. 3 видно, что различные ВВ
ц(|разному воспринимают инициирующее действие, а также что
разные инициаторы по отношению к одному и тому же веществу
ичладают различной инициирующей способностью.
Различная восприимчивость ВВ к инициирующему действию
объясняется разной их чувствительностью.
35
Ml Та блица 3
Предельные заряды инициирующих В В по отношению к 0,5 г детонатора
(по литературным данным)
о Е По пикри-
О 'Название инициирующих ВВ По тетрилу НОВОЙ ки- По тротилу
слоте
£
1 Гремучая ртуть 0,29 0,30 0,36
2 Гремучее серебро .... 0,02 0,05 0,095
3 Азид свинца .... 0,025 0.025 0,09
4 Азид ртути ... 0,045 0,075 0,145
5 Азид серебра 0,02 0,035 0,07
6 Хлораттримеркуральдегид . . — 0,40 ——
7 Перхлораттримеркуральдсгид . 0,10 —
8 Диазобензолнитрат ..... — >0,50 —
9 Нитродиазобензолисрхлорат • 0,015 —
10 Гексаметиле нтрипероксидднами 11. - 0,10 —
11 Сернистый азот ... -— >0,50 —
12 Циапуртриазид . 0,04 0,05 0.10
13 Трииитротриазидобензол 0,01 — 0.02
14 Ацетиленид серебра 0,29 -
Велер и Мартин определяли предельные инициирующие заряды
азидов и фульминатов по отношению к 0,5 г бризантных ВВ и полу-
чили следующие данные (табл. 4).
Таблица 4
Предельные инициирующие заряды по опытам Велера и Мартина, г
Инициатор Вторичный заряд
тетрил пикриновая кислота тротил тринитро- анизол тринитро- ксклол
Азид кадмия - 0,01 0,02 0,04 од
» серебра . 0,02 0,035 0,07 0,26 0,25
» свинца . 0,025 0,025 0,09 0,28 —
в меди . . 0,025 0,045 0,095 0,375 0,40
о ртути - 0,045 0,075 0,145 0,55 0.50
» таллия . 0,07 0,115 0,335 —
Фульминат се-
ребра . 0,02 0,05 0,095 0,23 0,30
» кадмия 0,008 0,05 0,11 0,26 0,35
» меди . 0,025 0,08 0,15 . 0,32 0,43
>> ртути 0,29 0,30 0,36 0,37 0,43
» таллия 0,30 0,43 — — —
36
Из этих данных можно сделать вывод, что последовательность
Мд вствительности бризантных ВВ к инициирующему действию раз-
личных инициаторов в общем одинакова.
J 1оэтому, если нанести в системе координат по оси абсцисс чув-
• । пительность различных бризантных ВВ по отношению к лучшему из
инициаторов [к Cd(ONC)2], выраженную предельным зарядом, распо-
южив бризантные вещества в виде ряда со все уменьшающейся чув-
i пиггельностью (тетрил, пикриновая кислота, тротил, тринитроанизол,
||шнитроксилол), а по оси ординат—предельные инициирующие за-
ряды, необходимые для их полной детонации, то получатся кривые,
Фиг. 19. Кривые предельных зарядов инициирующих ВВ.
характеризующие инициирующие ВВ с точки зрения их инициирую-
щей активности (фиг. 19). Количество необходимого для иницииро-
илнмя вещества возрастает с уменьшением чувствительности нитро-
соединений, и тем быстрее, чем меньше активность инициатора. Так,
V фульмината и азида таллия, обладающих относительно меньшей
инициирующей способностью, кривая быстро растет, в то время
кнк для лучших инициаторов — азида кадмия, фульминатов серебра
। меди она остается прямой.
Продолжая кривые до пересечения с осью ординат, мы получим
-к’личины предельных количеств инициаторов, которые необходимы
для полной детонации ВВ бесконечно большой чувствительности.
Продолжения кривых до оси ординат не направляются в начало
координат, и поэтому отрезок на оси ординат от начала до точки
пересечения с отдельными кривыми указывает ту массу инициатора,
которая каждый раз расходуется до наступления собственно иници-
ирующего действия.
Таким образом предельный инициирующий заряд q для всех
инициирующих ВВ представляет собой сумму двух величин:
1) количества инициатора, необходимого для достижения пол-
ноты детонации самого инициирующего ВВ;
37
Фиг. 20- Свинцовый
цилиндр для испыта-
ния ка пс юл ей-дето -
наторов.
2) количества qt инициатора, необходимого для получения такого!
количества газообразных продуктов взрывчатого превращения, чтобы
была совершена работа удара, достаточная для обеспечения начала
взрывчатого превращения бризантного ВВ.
§ 5. МЕТОДЫ ОПРЕДЕЛЕНИЯ ИНИЦИИРУЮЩЕЙ СПОСОБНОСТИ
Инициирующую способность инициирующих ВВ определяют, как
уже было указано, по отношению 0,5—1 г вторичного заряда (тро-
тила, тетрила, пикриновой кислоты и т. п.), запрессованного в мед-
ную оболочку капсюля-детонатора № 8.
Существующие методы регистрации инициирующего действия кроме
изложенного в предыдущем параграфе можно разделить на два класса.
К первому относятся методы, в основе которых лежит определение
механического эффекта и по которым можно лишь косвенно судить
об инициирующем действии. Ко второму — прямые методы, непо-
средственно характеризующие инициирующую
способность.
К косвенным методам относятся следующие:
I) проба в свинцовых цилиндрах, 2) проба
на свинцовых пластинках, 3) гвоздевая проба.
4) песочная проба, 5) водяная проба,6) действие
через влияние.
К прямым—все методы определения иниции-
рующей способности подрывом чистых и флег-
матизированных ВВ.
Косвенные методы
Проба в свинцовых цилинд-
рах. Эта проба основана на сравнении величин
расширения гнезда свинцового цилиндра от
подрыва в нем капсюля.
Пробу производят в свинцовом цилиндре
(фиг. 20) высотой 100 мм и диаметром 100 мм.
По оси цилиндра имеется углубление диаметром
7 мм и глубиной 55 леи. Изготовляются цилиндры
из возможно более чистого рафинированного
свинца одной и той же партии для каждой серии опытов. Свинцовые
цилиндры отливают в соответствующих формах и употребляют не
ранее 48 час. после их отливки. Все это необходимо для того, чтобы
создать вполне определенные тождественные условия в отношении
механических свойств металла цилиндров и в отношении испытуемых
капсюлей.
Снаряженную испытуемым инициирующим ВВ гильзу диаметром
6,85 мм и длиной 45 мм вводят в осевое гнездо и подрывают при
помощи бикфордова шнура. Вследствие давления взрыва происходит
расширение осевого гнезда цилиндра. Объем осевого углубления до
взрыва и после него измеряют объемом воды, которую приливают из
градуированной бюретки.
38
Эскалес приводит следующие цифры о величинах расширений
цилиндров в зависимости от инициирующего вещества:
цистой гремучей ртути .......................... 25,3 мл
80% гремучей ртути, 20% хлората калия . . 33,6 »
азида свинца..................................... 21,5 »
Необходимо отметить, что хотя гремучая ртуть с 20% хлората
калия дает большее расширение/чем чистая, вследствие более пол-
ного ее сгорания, однако, инициирующая способность этой смеси
меньше, чем смеси 93% Hg(ONC)2 -ф 7%КС1О3.
Велер на ряде опытов показал, что расширение цилиндров зави-
сит от теплоты взрыва. Из табл. 5 видно, что наибольшее расшире-
ние получается при взрыве диазо бензо лнитрата и сернистого азота,
выделяющих больше теплоты, чем гремучая ртуть и азид свинца,
хотя последние являются лучшими инициаторами.
Таблица 5
Расширения гнезда цилиндра после взрыва
Испытуемое вещество Расширение от 2 г МЛ Плотность заряжания г/см3 Теплота взрыв- чатого пре- вращения Кал! кг
Диазобензолиитрат ... 43.1 1,459 687
Сернистый азот . 39,2 2,112 700
Гремучая ртуть 25,6 3,368 403
Азид серебра . . 22,6 3,382 452
Как видно из изложенного, точно регистрировать инициирующее
действие этим способом невозможно. Им только до известной степени
определяют количество энергии, выделяемой веществом при взрыве.
Проба на свинцовых пластинках. Имеется не-
сколько разновидностей этой пробы. Первая и наиболее ранняя раз-
новидность состоит в том, что на большую квадратную свинцовую
пластинку определенной толщины вертикально устанавливают
гильзу, снаряженную испытуемым веществом, как указано ранее,
п производят взрыв его при помощи бикфордова шнура или электро-
напала. От удара взрыва происходи'! деформация пластинки, выра-
жающаяся в пробитии ее или в образовании воронки. Кроме того,
н результате взрыва на пластинке появляются лучеобразные чер-
точки от осколков оболочки (фиг. 18).
Для пробы применяют провальцОванные пластинки из мягкого
свинца, квадратные, размером 35—45 мм, или круглые того же диа-
метра. Свинцовые пластинки устанавливают так, чтобы они опира-
юсь краями на основание (фиг. 17). По характеру пробитого отвер-
стия судят об инициирующей способности испытуемого вещества.
Для некоторого уточнения пробы обмеряют отверстия или опреде-
ляют потерю веса пластинки.
39
Эта простая и сравнительно дешевая проба не имеет строго науч-
ного характера, но она все же дает достаточно наглядное представ-
ление о мощности капсюля-детонатора и вследствие этого пригодна
как приемочная проба.
Гвоздевая проба. Предложенная в Америке в 1913 г.
Холлом и Хоуеллом гвоздевая проба по технике выполнения является
наиболее простой и дешевой. Сущность ее заключается в том, что сна-
ряженный испытуемым веществом
капсюль взрывают под гвоздем
определенного размера и затем
измеряют угол изгиба последнего.
Этот угол и характеризует отно-
сительную инициирующую спо-
собность ВВ.
Фиг. 21. Гвоздевая проба.
Фиг. 22. Образцы дефор-
мированных гвоздей.
Для этого применяют круглые гвозди длиной 100 .им и толщиной
2,8 мм без головок. Само собой разумеется, что длина, диаметр и вес
гвоздей должны быть одинаковыми. Капсюль прикрепляют к гвоздю
'Проволокой так,чтобы заряд капсюля приходился по середине гвоздя
(фиг- 21)- Подрыв подвешенного гвоздя производят над чаном с во-
дой на расстоянии 25—30 см. Вода необходима для смягчения удара
падающего после взрыва гвоздя и предотвращения возможной его де-
формации в результате падения. Из чана гвоздь извлекают магнитом
м обмеривают (фиг. 22).
Велер указывает, что эта проба весьма не точна и что в руках
даже опытного экспериментатора получается не менее 20% несовпа-
дающих результатов. Сухаревский в 1926 г. получил еще больший
процент отклонений.
Таблица 6
Показатели Капсюли
гремучертут- ный «Кг 8 гремучертутно- толовый № 8 гремучертутно- тетрнловый № 8
Число взрывов Наибольший угол изгиба . . Наименьший угол изгиба . . Средний угол изгиба 30 55 27 36,2 50 71 36 48,4 50 110 66 85,1
40
Фиг. 23. Бомбочка для песоч-
ной пробы-
Результаты опытов Сухаревского сведены в табл. 6.
Па результатах этой пробы отражаются механические качества
। по.здей, положение капсюля на гвозде и ряд других более мелких
'условий опыта. Все это свидетельствует о весьма случайном харак-
гре результатов данной пробы-
Песочная проба. Эта проба, предложенная в 1910 г.
( юллингом в Америке, зафиксирована в некоторых технических
с ювиях США, например, в технических
условиях на прием тетрила, гремучей
ргуги и т. п.
Проба основана на дроблении кварце-
вого песка силой взрыва капсюля-дето-
н,пора. Для испытания применяют квар-
цеиый песок определенных качеств; одного
и 1<>го же месторождения, с одинаковым
держанием кварца (99,9%) и одинаковых
p.i шеров зерна. Обычно берут фракцию,
проходящую через сито с 8 отверстиями
ч линейном сантиметре и не проходящую
через сито с 12 отверстиями в линейном
с ;цггимстре.
Испытание производят в бомбочке
(фиг. 23), состоящей из основания 7, соб-
11 вснно бомбочки 2, крышки 3, ярма 4 и
"и ri гов 5 с гайками 6.
40 г кварцевого песку, отвешенного с
ирпюстьюдо 0,1г, засыпаютв бомбочку 2.
Несколькими легкими ударами деревян-
но! о молотка по боковым стенкам бомбочки
песок уплотняют и выравнивают. Испы-
|усмый капсюль-детонатор, обжатый с
отрезком вставленного
и пего бикфордова шнура длиной 20—25 см, пропускают сквозь от-
нирстие крышки до соприкосновения с песком. После этого в бом-
Гпчку всыпают еще 60 -I- 0,1 г кварцевого песку. Так же, как и после
п< рвой насыпки, песок в бомбочке уплотняют и выравнивают, посту-
кивая деревянным мойотком по корпусу. Для того чтобы избежать
|н>герь песка при взрыве, на бикфордов шнур плотно надевают рези-
понос кольцо длиной около 5 мм и диаметром около 3 мм так, чтобы
при установлении капсюля-детонатора в бомбочке оно находилось как
p.i.i в отверстии крышки. После этого крышку тщательно прикреп-
ипот к корпусу бомбочки, не сдвигая капсюля в песке. Бикфордов
шнур воспламеняют от тлеющего пенькового фитиля- После взрыва
Оимйочку разбирают, содержимое ее высыпают на глянцевую бумагу,
гщ.пельно снимают зерна песка, по тем или иным причинам при-
илншие к стенкам бомбочки. Из песка извлекают остаток сгорев-
iik'i <». бикфордова шнура и крупные осколки гильзы капсюля.
I.нем песок просеивают через сито с 12 отверстиями на линейный
н|1гиметр.
Количество кварцевого песка, прошедшего после взрыва чере-J
сито с 12 отверстиями на линейный сантиметр, и принимают за мер-J
инициирующей способности капсюля-детонатора.
По американским условиям, песок отсеивают через сита с 30, 40.
60, 80 и 100 отверстиями в дюйме или только через сито с 30 отвер-]
стиями в дюйме.
Таблица 7 В табл. 7 приведены результаты
испытания различных по величине 1
заряда капсюлей-детонаторов, пт I
данным Шторма и Копа.
По мнению Каста, результаты
песочной пробы должны быть ди|
статочно показательными, так как
действие взрыва капсюльного со]
става в условиях пробы фикси
руется достаточно полно.
Комиссия по взрывчатым вещ*
ствам при НТС горнорудной про-
мышленности 1 подвергла изучению
вопрос о целесообразности прим.
Средний вес кап- Средний вес раз-
сюлыюго заряда дробленного песка
г г
0,4933 22,03
0,6358 27,44
0,7220 30,90
0,9842 36,61
1,4377 47,16
1,8345 54,53
2,8691 68,74
нения песочной пробы и пришла к следующим выводам:
1) песочная проба нечувствительна к различиям в плотности з:н
ряда капсюля;
2) песочная проба дает достаточно однообразные показания для
каждого сорта капсюлей;
3) песочная проба с достаточной рельефностью выражает измь<
нение силы капсюлей разных размеров (по количеству заряда);
4) вариации в дозировке капсюлей данного сорта сказываются
ясно;
5) различие между разными капсюлями по снаряжению (напри-
мер, между гремучертутнотротиловым и азидотетриловыми) не нахо-
дит достаточно яркого выражения в пробе;
6) в качестве стандартного песка может быть принят: а) молотый
кварц, б) вельский стандартный песок № 1.
Определение инициирующей способности
капсюлей-детонаторов по воДяной пробе. ОП
инициирующей способности, определяемой данной пробой, судя!
по величине обжатия крешера при подрыве капсюлей-детонатороз
в замкнутом объеме, заполненном водой.
Испытание производят в бомбе (фиг. 24), изготовленной из корпуса
152-мм фугасной гранаты 1. Бомба снабжена тремя боковыми и одним
донным (изготовленными из стали) крешерными приборами, состоя-
щими из корпуса 2, в который вкладывается притертый к внутренней
поверхности корпуса поршень 3 и медный крешер 4 диаметром 7 мм
и высотой 10,5 мм, конец которого, обращенный к поршню, обточен
1 М. Сухаревский и Ф. Першаков, Курс теории взрывчатых
веществ, стр. 126, ГХТИ, 1932-
42
гы конус. Крешер должен быть хорошо центрован. Центрование про-
н ишдят отрезком резиновой трубки
высотой около 4 мм, надетым на
цилиндрическую часть крешера. В
корпус крешерного прибора вверты-
илот стальной упор 5 с хорошо от-
шлифованной поверхностью сопри-
косновения с крешером.
В крышке 6 бомбы имеется на-
нпнтованное центральное отверстие,
и которое ввертывают державку 7
и» сквозным отверстием по оси. На-
чинение державки — удерживать
лксюль-детонатор в бомбе на том
и мп ином уровне. Благодаря винто-
imli нарезке державка может быть
VI' гановлена в том или ином положе-
нии по высоте и после установки
>.|креплена контргайкой 8. Для опре-
•спения уровня расположения ниж-
него конца державки к крышке
прикреплен винтом 9 указатель 10.
Нижний конец державки имеет
приспособление для удерживания
к.шсюля-детонатора в строго верти-
кальном положении. На фиг. 25
л шбражен нижний конец державки
Фиг. 24- Бомба для водяной пробы.
7 с вкладышами 11 и 72, в которые вставляют
> лпсюль-детонатор 14. Вкладыши с капсюлем
поджимают к державке гайкой 13. Бомбу устанав-
пшают на стальной плите 15 с двумя болтами
(последние на чертеже не показаны). Перед опытом
пчеле подготовки бомбы на крышку надевают
прмо 16, притягиваемое гайками.
Подготовка бомбы к опыту состоит в следующем.
Испытуемый капсюль-детонатор с электрозапалом
шкрепляют на нижнем конце державки. Для
предохранения от проникновения воды в капсюль-
цтонатор место стыка его с электрозапалом по-
крывают водонепроницаемой замазкой или изоля-
ционной лентой. Державку ввертывают в крышку
1юмбы так, чтобы середина капсюльного состава
1"-щеголя-детонатора была расположена против
гннсовых крешеров. Корпус крешерного прибора
Фиг. 25. Державка
капсюля-детона-
। и вставленным в него поршнем устанавливают тора-
ПЛ стальной полированной плитке. В корпус
пипдят обмеренный крешер с надетым на него отрезком центрующей
резиновой трубки и ввертывают упор до соприкосновения с крешером.
43
Собранные таким образом крешерные приборы ввертывают в их
гнезда в бомбе.
* Бомбу заполняют водой, закрывают крышку с ввернутой в нее
державкой, причем избыток воды выходит через отверстие а в крышке.
На крышку бомбы накладывают ярмо и поджимают его гайками. По
указателю проверяют расположение капсюля-детонатора в бомбе.
В случае неправильного расположения державку поднимают или
опускают. В случае правильного расположения капсюля-детонатора
провода электрозапала присоединяют к проводникам подрывной
машинки и производят взрыв.
После взрыва извлекают державку, освобождают ее от остатков
электрозапала, выливают воду из бомбы, вывертывают крешерные
приборы, извлекают из них крешеры и обмеривают последние. Вели-
чину обжатия крешеров и принимают за меру инициирующей спо-
собности капсюля-детонатора.
Действие через влияние. Относительно этой пробы
достаточно указать, что ее производят для того, чтобы определить
расстояние, при котором получается детонация ВВ от взрыва
капсюля. Расстояние в данном случае в большей степени зависит
от положения капсюля-детонатора и направления детонационной
волны, а так как создание всегда одинаковых условий для пробы
затруднительно, то результаты ее не могут служить критерием для
определения инициирующей способности.
Все косвенные методы не характеризуют полностью инициирую-
щего действия, а дают понятие только об одном или нескольких фак-
торах, от которых оно зависит.
Прямые методы
Прямые методы определения инициирующего действия по отно-
шению кВВ дают несомненно лучшие результаты. В основе этих мето-
дов лежит подрыв капсюлей с испытуемым инициирующим ВВ в шаш-
ках, изготовленных из труднодетонируемых или флегматизированных
взрывчатых веществ. Выбор подходящего ВВ представлял и представ-
ляет некоторое затруднение; надо, чтобы ВВ регистрировало полностью
инициирующее действие и, кроме того, не изменялось при хранении.
Предложение об испытании капсюлей-детонаторов на флегмати-
зированных ВВ сделано было еще в 1899 г. К. Эзопом. Оно сводилось
к тому, чтобы при помощи примесей масел к нитросоединениям в раз-
личных количествах получать вещества с пониженной чувствитель-
ностью к детонации и на этих веществах определять инициирующее
действие.
Не менее трудным оказался и выбор флегматизатора. Эзоп поль-
зовался для этого хлопковым маслом и производил взрывы на свин-
цовых цилиндрах, обжатие которых затем обмеривал. Способ этот
применялся и в Англии, но без установленных правил. В. Гротта
флегматизировал ВВ окисью железа. Цибульский в Верхней Силезии
пользовался смесью поваренной соли с аммонийной селитрой, Каст
и Гайд пользовались шашками из литого тротила. Однако оказа-
44
юсь, что эти шашки вследствие своей легкой восприимчивости к дето-
в.щии слабо выражают различие в действии инициатора. Смеси тро-
шла с аммонийной селитрой и динитрото-
луола с мелинитом также оказались непри-
шдными для этой цели.
Исследования показывают, что лучше
пользоваться тротилом в качестве основного
вещества для испытания, так как тротил —
хорошо известное, вполне определенное, не
изменяющееся вещество высокой чистоты.
Кроме вышеупомянутых флегматизаторов
предлагались касторовое масло, парафин,
парафиновое масло, тальк и др.
Опыты Гайда с тринитротолуолом, флег-
мптизированным парафином, показали, что
можно легко приготовить пригодные для
опытов шашки с содержанием парафина от
I до 15%. Шашки изготовляли следующим
образом. Нагретый до 60° тротил вводили
и расплавленный парафин (50—55°) и пере-
мешивали вручную. Смесь прессовали под
давлением 400 кг/см* в цилиндрические
пышки (d = 25 мм, h = 40 мм, углубление
для капсюля dr = 7 мм, hx = 25 мм). При
з гом было установлено, что парафинирован-
ный тротил после нескольких дней хранения
становится менее чувствительным к детона-
ции, вероятно, вследствие изменения струк-
туры парафина, не представляющего собой
однородного в химическом отношении ве-
щества.
В 1925 и 1926 гг. Велер опубликовал свои
опыты с тринитротолуолом, флегматизиро-
ианным парафиновым маслом. Он считает,
чго парафиновое масло имеет достаточно
определенный химический состав и является
пеществом стойким (смесь тротила с флегма-
гнватором хранилась без изменений в течение
* мес.), но в то же время указывает, что при
очень большом содержании парафинового ма-
<• да смесь может стать чувствительной к лет-
пен температуре. Велер брал тротил,' выкристаллизованный из смеси
। пйрта с бензолом (90% спирта, 10% бензола). К высушенному тротилу
он прибавлял в фарфоровой чашке рассчитанное количество пара-
финового масла {d = 0,89 при 18,5°, вязкость 32,1° по Энглеру при
И|,4°) в эфирном растворе (объем эфира в миллилитрах был равен
числу граммов тротила), перемешивал, сушил 4 часа при 50° и
прессовал в медные гильзы (ft =55 мм, а —10 мм, толщина стенки
45
Фиг. 26. Схема подрыва
капсюля-детонатора по
пробе Велера:
а—подставка; b —свинцовая
пластинка; с—внешняя гиль-
за; d — капсюль-детонатор;
е •— чашечка; f — бикфордов
шнур; g— направляющая
шнура; h—пробковый клин.
вверху 0,15 мм, толщина диа 0,5 мм) [под давлением 100 кг}см?
пуансоном с шипом в зависимости от размеров капсюля.
Велер подрывал испытуемые патроны (гильзы) на свинцовой
пластинке 40 х 40 х 5 мм посредством воспламенения от бикфор-
дова шнура (фиг. 26) и при этом устанавливал предельный случай,
когда вследствие увеличения флегматизатора убыль веса свинцовой
пластинки резко умень-
шалась, что указывало
на отсутствие полной
детонации. На графике
(фиг. 27) показан пре-
дельный случай для
гремучертутных капсю-
лей. t
К недостаткам этой
пробы следует отнести
то, что вводить в тротил
более 9% парафинового
масла нельзя, так как
при большем содержа-
нии оно выжимается из
шашек при прессовании.
Осложняющим работу
условием является не-
обходимость строгого
соблюдения совершенно
одинаковых температург
ных условий во всей
Вес тротила, 2
Вынос свинца.2
Фиг. 27. График зависимости убыли веса
свинцовой пластинки от содержания флегма-
тизатора:
I—предел флегыатизации (процент парафина) в зави- Серии ОПЫТОВ, ТЭК КаК
симостиот инициирующей способности капсюля-детона- ВЯЗКОСТЬ ПарафиНОВОГО
тора;//—вынос свинца в г в зависимости от содержания . „ г; Г. . •
парафина. МЭСЛЭ (СТЭЛО быТЬ, И его
флегматизирующая спо-
собность) легко меняется с изменением температуры. Кроме того,
этот метод не применим для испытания мощных капсюлей-детонато-
ров с мощными инициирующими ВВ (например, для азидотетрило-
вых, азидотэновых и азидогексогеновых капсюлей-детонаторов).
В 1930 г. Гайд и Коенен опубликовали свои опыты с тротилом,
флегматизированным тальком. Тальк — более слабый флегматизатор,
чем парафиновое масло, и поэтому он позволяет фиксировать малей-
шее изменение инициирующего действия. Но, с другой стороны,
тальк — твердое тело, и вследствие этого передача детонации в
тротиле может оказаться разнородной в зависимости от степени
смешения.
Мерилом инициирующей способности капсюля-детонатора при
этой пробе служит максимальная степень флегматизации тротила,
при которой испытуемый капсюль-детонатор еще способен вызывать
его детонацию. О полноте детонации тротиловой шашки судят по де-
формации свинцовой пластинки или по обжатию медного крешера
46
Фел
Фиг. 28. Шашка
В В для пробы
Гайда.
ii.i бризантометре Каста. В зависимости от способа фиксирования
.механического действия взрыва шашки существуют два варианта
оформления этой пробы.
Прежде чем перейти к описанию отдельных вариантов, укажем
нищие для них правила производства работы.
Тротил, просеянный для отделения от пыли через сито с отвер-
I гнями диаметром 0,2 мм, и тальк в тонкораздробленном состоянии*
просеянный через сито с 36 отверстиями в линей-
ном сантиметре, тщательно перемешивают роговой
южкой в большой фарфоровой чашке. Так как
условия перемешивания влияют на характер флег-
млтизации тротила, то автором установлено за
правило готовить флегматизиров энный тротил
порциями по 200 г при продолжительности пере-
мешивания 15 мин. непосредственно перед прес-
сованием шашек.
Из смешанного с тальком тротила прессуют
цилиндрические шашки весом около 35 г, высотой
•I I —42 мм и диаметром 25 мм (фиг. 28) под дав-
нснием 1250 ат, в один прием, при помощи мат-
рицы, на поддоне которой имеется шип по разме-
рам испытуемого капсюля-детонатора. Так, напри-
мер, для азидотетрилового капсюля-детонатора
№ 8 диаметр гнезда шашки 6,9 мм, глубина 25,2 мм, а для гремуче-
ртутного капсюля-детонатора № 8 тот же диаметр, но глубина 32 мм.
Испытуемый капсюль-детонатор вставляют в назначенное для
него гнездо шашки и подрывают при помощи электрозапала, воспла-
меняемого тем или иным источником
тока (лучше всего пользоваться под-
рывными машинками).
Подрыв шашки производят в бро-
неяме, в изолированной от других
помещении специальной будке с кир-
пичными стенами или просто на воз-
духе за образующими замкнутое
пространство железными щитами.
Вариант I. Механическое
действие взрыва шашки в этом
случае определяют по действию его на свинцовую пластинку разме-
рим 100 х ЮО х 30 мм. Прежде всего на пол укладывают горизон-
тально железную или стальную плитку, имеющую крючки по углам.
I ki эту плитку накладывают свинцовую пластинку, на которой уста-
навливают шашку со вставленным капсюлем. Для того чтобы шашка
не сбивалась со своего места и не падала при случайном натяжении
проводов, ее укрепляют на месте, притягивая шпагатом и закрепляя
последний за крючки стальной плитки (фиг. 29). Затем присоеди-
няют провода к электрозапалу и производят взрыв, в результате
которого на свинцовой пластинке получается деформация большей
47
Риг. 29. Схема испытания по Гайду.
S3
Фиг. 30. Деформация свинцовых пластин при определении инициирующей
способности методом Гайда.
ii ini меньшей величины (фиг. 30). Так как мощность взрыва очного
и того же заряда ВВ зависит от мощности сообщен-
ного ему начального импульса, то полученную
деформацию при данной степени флегматизации ВВ
принимают за меру инициирующей способности ис-
пытуемого капсюля-детонатора.
Вариант И. Механическое действие взрыва
шашки определяют по обжатию медного крешера на
иризантометре Каста (фиг. 31). Медный крешерный
цилиндрик (8 х 13 мм или 7 х 10,5 мм) помещают
инд поршень бризантометра. Для того чтобы предо-
хранить поршень от действия взрыва, на него на*-
киадавают стальную накладку, а на нее —две свин-
цовых пластинки 4 мм толщины. На эти последние
устанавливают шашку и вставляют капсюль с элек-
трозапалом. Для большей устойчивости, как и в
первом случае, шашку притягивают шпагатом, поль- фиг 3J вризан-
луясь той же подкладкой. Далее производят подрыв, тометр Каста.
к.и< описано выше. Степень обжатия крешера после
нарыва, выраженная в милли-
метрах, и служит характерис-
шкой инициирующей способ-
ности капсюля-детонатора. 06-
Mtiij крешера до и после взрыва
производят при помощи паль-
мера.
Работы автора по методике
испытания капсюлей-детонато-
ров на их инициирующее дейст-
нне показывают, что метод Гайда
г некоторыми изменениями дает
пиолне удовлетворительные ре-
зультаты и позволяет классифи-
цировать капсюли-детонаторы
in» их мощности. На фиг. 32
Фиг. 32. Обжатие медных цилинд-
риков 10,5x7 мм-.
/—капсюль-детонатор № 8 азидогексогемсвый;
II — капсюль-детонатор № 8 азцдотеновый;
III—капсюль-детонатор № 8 азидотетриловый;
IV— капсюль-детонатор №8 гремучертутно-
толовый; V—капсюль-детонатор № 8 гремуче-
ртутный.
показаны графически резуль-
11 гы сравнительного испытания
р ыличных сортов подрывных
1ьн1сюлей“детонаторов № 8.
Техническое оформление ра-
Поты таково, что метод можно
применять только для научно-
in следовательских целей; применение же его в заводских условиях
п качестве приемочной пробы затруднительно.
§ 6. ЧУВСТВИТЕЛЬНОСТЬ ИНИЦИИРУЮЩИХ ВВ
В химии известно много взрывчатых веществ, которые по мощ-
|шсги превосходят, например, гремучую ртуть, но которые, однако,
с. Опив —92—4 49
непригодны в качестве инициирующих веществ. Это является след-
ствием или большей их чувствительности, повышающей опасность
непреднамеренного взрыва, или слишком медленного нарастания
скорости их взрывчатого превращения. Гремучая ртуть занимает
среднее положение: она достаточно чувствительна для воспламенения
от луча огня и от накола жалом и в то же время сравнительно без-
опасна при фабрикации и применении.
Эндотермические ВВ обладают требуемой чувствительностью
к лучу огня, но некоторые из них слишком чувствительны к трению
и удару, а следовательно, опасны в обращении.
Чувствительность инициирующих ВВ зависит от природы ВВ,
структуры и величины кристаллов, наличия примесей, степени их
прессования и температуры опыта.
О чувствительности инициирующих ВВ к начальному импульсу
в зависимости от природы вещества можно судить по следующим
примерам.
Соединения галоидов с азотом принадлежат к наиболее чувстви-
тельным веществам. При этом чувствительность их возрастает с ро-
стом атомного веса галоида. У фульминатов чувствительность к на-
греву и удару возрастает с увеличением атомного веса металлов.
Однако окончательного ‘суждения о том, как изменяется чувстви-
тельность инициирующих ВВ в зависимости от их химической при-
роды, еще нельзя сделать. Так, например, азид ртути Hg(N3)2 прево-
сходит по своей чувствительности гремучую ртуть: достаточно сломать
кристалл его, чтобы произошел взрыв. Между тем оба эти инициатора
обладают рядом одинаковых свойств: одинаковым молекулярным
весом, одинаковым содержанием ртути, одной и той же плотностью
и почти одинаковой теплотой взрыва. Судя по реакциям их взрыв-
чатого разложения:
Hg(N3)2->Hg + 3N2
Hg(ONC)2^Hg + N2 + 2CO
они выделяют одинаковый объем газообразных продуктов взрыва.
Не меньшее влияние на чувствительность инициирующих ВВ ока-j
зывает и их физическое состояние, в первую очередь, структура и
величина кристаллов. Так, при получении одной из двух кристалли-
ческих форм азида свинца получаются частые взрывы в момент обра-
зования кристаллов, что, повидимому, связано с разным содержа-,
нием энергии в азидах разных кристаллических форм.
С увеличением кристаллов ряда инициаторов обычно повышается
их чувствительность к удару. Например, кристаллы азида ртути
Hg(N3)2 длиной 3 мм и толщиной 0,25 мм взрывают от излома, между'
тем как кристаллы более мелкие (0,1 мм) значительно менее чув-
ствительны. Первые взрывают от прикосновения пером, вторые же—
только при падении на них груза 0,6 кг с высоты 65 мм. Еще более
мелкие кристаллы взрывают только при падении того же груза с
высоты 200 мм, т. е. с высоты, в три раза большей.
50
I 1римеси, более твердые, чем инициаторы, повышают чувствитель-
мпсть последних. Наоборот, примеси менее твердые понижают чув-
ПИИСЛЬНОСТЬ.
11рессование понижает чувствительность к удару и трению, и
whim пользуются с двоякой целью: для повышения безопасности
п обращении и для повышения гравиметрической плотности. Опыты
I рвиова показали, что действие азида свинца тем больше, чем плот-
нее он спрессован. Капсюль с
мод давлением 12 800 ат, про-
пинает свинцовый кружок ТОЛ-
ЩИНОЙ в б мм.
С другой стороны, повышение
и нот пости запрессовки влечет
• । собой ухудшение воспламе-
няемости. Так, при прессовании
и ища свинца под давлением,
превышающим 5000 ат, покры-
ли лаком поверхность азида
i пипца уже не воспламеняется
щ обычного луча огня. Особенно
но касается гремучей ртути,
не значительная перепрессовка
| порой вызывает отказы и вы-
। орание. Поэтому для иниции-
0,3 г азида свинца, спрессованного
Фиг. 33. Кривые чувствительности
C2H8ON10h Hg(ONC)s к удару в зави-
симости от температуры опыта.
рующих веществ довольствуются плотностями, получаемыми при
прессовании под давлением 200—800 ат.
С повышением температуры, как правило, повышается чувстви-
• ♦•и|,|юсть инициатора. По опытам Тейлора и Уиля, влияние темпе-
р.чуры на чувствительность к удару выражается следующими кри-
иимц (фиг- 33).
I пресечение кривых с осью абсцисс отвечает температуре вспышки
чих веществ.
Наряду с достаточной чувствительностью к простому началь-
ному импульсу не менее существенным требованием является и без-
.••ысность инициирующих веществ в обращении.
С химической точки зрения очень интересным является предло-
жение Штаудингера о применении в качестве инициаторов смеси
щелочных и щелочноземельных металлов с ВВ или с веществами,
Магыми хлором, например четыреххлористым углеродом (герм,
наг. № 391345, 391346 и 396209, 1922 г.). Одна капля жидкого
к пава металлических калия и натрия при соприкосновении с нитро-
। in церином или четыреххлористым углеродом вызывает взрыв даже
при незначительном сотрясении. Однако использование таких смесей
возможно, так как они опасны в обращении.
Г Для производства, и особенно для создания условий применения
Инициирующих ВВ, требуется определенная степень безопасности.
• Илсдствие высокой чувствительности значительное количество пред-
•н1авшихся ранее инициирующих ВВ, например азидов ртути (II)
51
и меди (II), ряда фульминатов и др., не может быть применено на
практике.
Производство больших количеств инициирующих ВВ, а также
применение их в значительных дозах всегда сопряжены с опасностью.
Поэтому вполне законно желание применять инициирующие вещества
в возможно малых количествах. Данное ВВ тем более подходит в ка-
честве инициатора, чем меньшее количество его требуется для дости-
жения полной детонации вторичного заряда. С этой точки зрения
введение более мощного азида свинца вместо гремучей ртути яв-
ляется уже значительным шагом вперед.
Из изложенного выше ясно, что чувствительность инициирующих
ВВ к удару, трению и лучу огня является одной из важнейших харак-
теристик их. В табл. 8 дана сравнительная оценка чувствительности
нескольких В В (по данным Брунсвига).
Таблица 8
Сравнительная оценка чувствительности некоторых ВВ
№ по пор. I Название ВВ Высота паде- ния груза в 2 кг, с кото- рой происхо- дит взрыв Температура вспышки Чувствитель- ность к треш- » В фарфоровой ступке
1 Гремучая ртуть . . . 2 160-165 Взрывает
2 Глицеринтринитрат 4 200—205
3 Пикриновая кислота . . 35—95 300—310 Слабый запах
4 Тринитротолуол 60—180 295—300 11икакого дей-
ствия 1
Определение чувствительности инициирующих ВВ к удару
Чувствительность инициирующих ВВ к удару определяют hj
копре Велера (фиг. 34). Он состоит из железного основания 7, к кч
торому прикреплена измерительная дуга 2 с делениями, представ
ляющими собой проекцию делений вертикальной миллиметровя
шкалы, стойка 3, направляющая 4 бойка 5 и стойка 6 рамы
с ударником 8.
Ударник имеет навинтованный сверху штырь, на который наде
вают по надобности свинцовый груз .0 того или иного веса и закреп
ляют при помощи гайки. Груз удерживается на желаемой высок
задержкой 10, привинченной к латунной дуге 2 при помощи винта 11
Полный вес падающего груза слагается из веса ударника 8, вес
рамы 7 и свинцового груза 0.
Бойки, употребляемые для испытания, изготовляют из стал
(серебрянка, Добрыня № 10) и должны быть длиной 50 мм. Коне,
бойка, обращенный к испытуемому ВВ, должен иметь вид усеченное
конуса (фиг. 35) диаметром ударной поверхности в пределах 1,5-1
1,6 мм. Обмер ударных площадок производят на компараторе п•
52
двум взаимно перпендикулярным диаметрам, причем берут среднее
«качение. Ударные площадки должны быть строго перпендикулярны
к оси бойков и хорошо отшлифованы (отсутствие трещин, заусенцев
Фиг. 34. Копер Велера.
и т. п.).
Испытуемое инициирующее
ВВ навешивают по 0,02 г и при
помощи воронки засыпают в
находящийся в матрице (фиг. 36)
для прессования латунный кол-
пачок капсюля к револьверным
патронам системы Наган. Лег-
кими ударами рукой по стенке
матрицы ВВ подравнивают,
после чего в матрицу вставляют
пуансон, матрицу переносят под
Фиг. 35- Боек копра Велера.
пресс и инициирующее ВВ запрессовывают под давлением 1000 или
>00 кг}см2.
Эту операцию можно производить на любом из прессов сущест-
вующих систем с соблюдением необходимых мер безопасности (щиты).
Для лабораторных опытов удобно пользоваться небольшими на-
стольными ручными прессами. Схема одного из них
приведена на фиг. 37. Этот пресс представляет собой
сочетание двух рычагов: рычага первого рода 7,
служащего для приложения силы руки прессующего,
и рычага второго рода 2, с навешенным на него
Крузом, служащим для сообщения ВВ заданного
давления прессования. Ось 3 является осью опоры
рычага 7. Короткий конец этого рычага вставлен в
вырез в нижней подвижной колонке 4 пресса. На
столик нижней колонки, являющейся нижней подуш-
кой пресса, ставят подготовленную к прессованию
матрицу с пуансоном. Когда матрица поставлена,
рычаг 7 опускают вниз. Тогда нижняя колонка
Фиг. 36. Матри-
поднимается вверх и прижимает головку пуансона ца для прессова-
к верхней подвижной колонке 5, являющейся верх- паградамТисте^
пей подушкой пресса. Рычаг 2 с подвешенным грузом ^Ь1 наган,
имеет опору на оси 6. Когда приходит в действие
первый рычаг и пуансон прижимается к верхней колонке, создается
давление на рычаг 2 в точке соприкосновения его с расположенным
па верхней колонке роликом 7. Как только при нажиме рукой на пер-
ши рычаг достигнуто требуемое давление, второй рычаг с грузом
53
приподнимается, после чего следует прекратить прессование. Давле
ние регулируют весом груза, находящегося на свободном конце ры
чага 2.
Фиг. 37- Схема ручного пресса.
На фиг. 38 приведена схема рычага 2 с приложенными к нему силами. Обо
значим вес груза, приложенного к рычагу р, вес рычага р1г давление, развиваем
прессом q, длинное плечо рычага L, короткое плечо I и расстояние от опоры
центра тяжести рычага' L,
Исходя из уравнения мо
меитов для этого рычага
мы можем папнсать-
pL Р ptLt -ql = 0,
откуда
ИЛИ L
Р
Задаваясь определенным давлением прессования q3aa и определенным вес on
груза р и зная вес линейки рычага pt, плечо его центра тяжести L, и малое плечо I
мы .можем установить плечо L.
Величины plt I и являются постоянными для каждого пресса и служат ег.
характеристиками.
При расчете следует учесть, что обычно давление задают в килограммах нг
квадратный сантиметр, а прессование производят в капсюли площадью попереч-
ного сечения, меньшей 1 см*' поэтому силы и должны быть выражены в килограм-
мах на площадь поперечного сечения пуансона путем соответствующего пересчет*
заданного давления:
jcd3
54
Рассмотрим пример- Необходимо запрессовать инициирующее ВВ в капсюль
in i давлением 1000 кг]см*. Пусть в нашем распоряжении имеется пресс со следую-
щими характеристиками:
вес линейки пресса 10 кг
Длина....................... НО см
короткое плечо....... 6 »
Диаметр применяемого пуансона равен 0,4 см, а груз — 10 кг. Требуется
ii.iIith длину плеча L.
Найдем сначала давление на площадь пуансона q. Оно будет равно:
1000 • 3,14 • 0,16 t0o
q =--------5-----— = 128 кг.
4
Подставляя известные нам величины в выведенную выше формулу, вычисляем
| uiuy плеча L-
128-6 — 10- 55 _1Q __
L ------ --------- = 21,8 — 22 см.
После расчета пресса на 1000 кг[см~ и установки на рычаге груза приступают
К снаряжению капсюлей.
В каждый снаряженный колпачок вкладывают кружок медной
фольги толщиной 0,10—0,12 мм, диаметром 3,7 мм так, чтобы кру-
жок не прикасался к стенкам капсюля (фиг. 39). Фольговые кружки
вырубают на специально приспособленном для этого
ручном прессе. Всего для испытания одного образца
инициирующего ВВтребуется максимум 50—60 таких
капсюлей.
Для производства испытаний копер Велера
следует помещать в вытяжном шкафу или под зонтом
I тягой в целях удаления образующихся при взрыве Снаряженный
Ипсюлей газов. Перед испытанием необходимо капсюль,
осмотреть копер и тщательно вытереть его для уда-
ления пыли вообще, а особенно пыли ВВ, могущей остаться от преды-
|ущих испытаний. Далее должны быть проверены задержки зажимного
пинта и ход рамы. Нельзя допускать шатания рамы в стойке (шата-
ние устраняется подвинчиванием зажимов), но в то же время паде-
ние рамы с бойком должно происходить свободно. Место соприкос-
новения зажимов с рамой рекомендуется смазывать вазелином.
После этого на штырь ударника надевают выбранный груз и за-
крепляют его гайкой. Задержку устанавливают на какой-либо ориен-
। ировочной высоте, исходя из предполагаемой чувствительности
испытуемого ВВ. Раму с грузом отводят и подвешивают на крюч-
ке задержки. г
Испытуемый капсюль ставят фольговым кружком кверху и вво-
дят на свое место под направляющей при помощи центрующей вилки
гак, чтобы он находился точно под осью канала направляющей.
Далее через этот канал вставляют боек. Для защиты работающего
иг осколков капсюля поверх направляющей надевают стальную
покрышку-
Подготовленные к испытанию остальные капсюли помещают в
специальной кабинке на расстоянии 0,5—1 м от копра.
55
Когда копер таким образом подготовлен, то плавным движение!
руки, стараясь во всех случаях соблюдать однообразие движений
отдергивают задержку, освобождающийся груз падает и ударникм
производится удар по бойку. При этом в зависимости от величин
живой силы удара происходит взрыв или отказ капсюля.
Капсюль в случае отказа или остатки капсюля после взрыва в>.>
талкивают из-под направляющей, прибор обтирают, причем о собе ни,
обращают внимание на то, чтобы не осталось распыленного ВВ, чпл
обычно получается при отказе капсюля.
В результате испытаний определяют максимальную высоту, пр!
падении груза с которой получается 100% отказов (нижний пред!!
чувствительности), и минимальную высоту, при падении груза с ко«
торой получается 100% взрывов (верхний предел чувствительности),
По получении результата с какой-либо высоты последнюю из!
меняют в зависимости от полученных данных. Для нахождения пр*4«
дела всегда следует сначала захватить этот предел в вилку, т. е. по-
лучить с двух разных высот плюс и минус (взрыв и отказ). После
этого следует споловинить вилку и в зависимости от результата по-
лучить новую, суженную, вилку.
Для закрепления результатов с каждой предельной высоты прон
изводят не менее десяти опытов.
После каждого взрыва боек подвергают осмотру на отсутствие
трещин и сколов и наощупь проверяют отсутствие заусенцев.
Таблица 9 I
Чувствительность инициирующих ВВ к удару на копре Велера
о с Вес падаю- Верхний Нижний
' о Наименование ВВ щего груза предел предел
г мм мм
1 Гремучая ртуть серая . . 600 «0 50
2 Гремучая ртуть бе'лая . . 600 85 55
3 Азид ртути (1) HgN3 . . 600 165 65
4 Азид ртути (II) Hg(N3)3 . . . Азид меди (I) CuNa ..... 600 80 10
5 600 125 70
6 Азид меди (11) Cu(Ns)2 600 70 10
7 Гремучее серебро 600 125 70
8 Тетразен 600 65 45
9 Гексаметилентри пероксиддиамин . 600 210 140
10 Азид свинца 975 235 65—70
11 Трипитротрназидобеизол . 1215 >250 160
12 Азид серебра . ... 914 245 150
13 14 Тринитрорезорцннат свинца ... Пикрат свинца . ...... 1215 1215 >250 >250 140 >250
15 Сернистый азот . . 1215 >250 >250
При проведении ответственных испытаний необходимо сменять
• •оПки после каждого взрыва или в случае получения отказов после
нух-трех ударов. Бойки, бывшие в употреблении, могут быть исполъ-
1 шпаны при последующих работах после обмера их компаратором при
цчюпии удовлетворения указанным выше требованиям.
Капсюли, давшие отказ, сметки ВВ и прочие остатки уничто-
iK.iiot растворением в азотной кислоте.
Чувствительность разных инициирующих ВВ в удару приведена
о габл. 9.
Определение чувствительности к трению
Если определенная чувствительность инициирующих ВВ к удару
•шляется положительным свойством вещества, ибо мы имеем немало
Йчучаев применения удара в качестве начального импульса, то чув-
11 кнтельносгь к трению очень часто не может быть отнесена к поло-
«чптельным свойствам.
Чувствительность к трению — свойство, отличное от чувствитель-
ности к удару. Это видно хотя бы из того, что вещества, обладающие
• "знаковой чувствительностью к удару, неодинаково относятся
- прению. В США испытание на чувствительность к трению введено
о ряд стандартов на ВВ. Вопрос об отношении к трению инициирую-
щих ВВ чрезвычайно важен как для производственников, так и для
артиллеристов. В самом деле, одной из основных операций ироиз-
подства капсюлей является прессование инициатора в металличес-'
। ую оболочку. Прессование производится в стальных матрицах,
причем неизбежно трение стального пуансона об инициирующее ВВ,
пли трение инициирующего ВВ между пуансоном и оболочкой кап-
। гоня. С другой стороны, при артиллерийской стрельбе не исключена
пи чможность трения инициирующего вещества о стенки капсюля
••следствие инерции инициатора при смещении снаряда в канале
орудия, особенно при современных начальных скоростях.
Обычное испытание на трение, производимое для других не столь
чувствительных веществ в фарфоровой ступке с шероховатым дном,
•ля инициирующих ВВ неприменимо. Для испытания иницииру-
ющих веществ на трение применяют приборы двух типов: 1) маят-
никовый (американский) и 2) вращающийся (германский)..
В США определяют чувствительность к трению маятником типа
Питтсбургской станции. Прибор состоит (фиг. 40) из стальной нако-
вальни и из подвешенного над нею маятника длиной 2 м со стальным
башмаком на конце. При помощи съемных грузов вес башмака можно
менять от 1 до 20 кг. Маятник, отклоняемый для первоначального
размаха, можно закреплять в различных положениях по высоте..
Подошва башмака съемная и может быть изготовлена из различных
материалов. При качаниях маятника она проходит над наковальней
пилотную, но не касается ее. На наковальне имеются три углубления,
к которых и помещается испытуемая проба.
57
Чувствительность к трению характеризуется высотой (наимень
шей), при которой в десяти опытах подряд не получится взрыва.
Тейлор и Ринкенбах для определения чувствительности к тре
нию также применяли маятниковый прибор, сконструированны<
ими по образцу описанного выше прибора, но в четыре раза мень
ших размеров. Башмак и наковаленка были сделаны из мягкой стали
Фиг. 40. Схема маятникового прибора-
Наковаленка располагалась слегка наклонно, и ее центральная
часть была прорезана поперечными бороздами, чтобы препятствовать
сметанию кристаллического вещества башмаком. Остальные детали
прибора были изготовлены из бронзы. Башмак весил 7,4 г, а стер-
жень маятника имел длину 50 см и весил 310 г. При помощи съем-
ных грузов вес башмака можно менять до 4,35 кг, а для изменения
степени трения наковаленке можно придавать различные положения.'
Работа на приборе производится следующим образом: напрессо-
вывают небольшое количество инициатора на центральную, покры-
тую бороздами часть наковаленки. Свободно опускают с высоты
'25, 33 или 37,5 мм башмак (с дополнительным грузом или без него),
который при своем прямом и обратном движении вызывает трение
вещества, и отмечают число качаний маятника до момента взрыва.
Если взрыва не происходит, то на наковаленку напрессовывают
новую навеску вещества и увеличивают вес добавочного груза или
высоту падения маятника. Опыты продолжают до тех пор, пока не
будет найдена наименьшая комбинация веса груза и высоты падения
маятника, при которых происходит взрыв. Полученные результаты
закрепляют десятью опытами.
Данные, полученные в результате испытаний на этом приборе
инициирующих ВВ по их чувствительности к трению, приведены
в табл. 10. Следует иметь в виду, что описанный метод показывает,
58
I ущности, чувствительность не к простому трению, а к наиболее
in встречающейся в практике его разновидности — к трущему
В'РУ
Таблица 10
Чувствительность к трению по опытам Тейлора и Ринкенбаха
Наименование В В Добавоч- ный груз кг Высота, падения см Количе- ство ка- чаний до взрыва Примечание
Ансшленид серебра Цп.шуртриазид 1 рему чая ртуть 10% КС 103 . 1 ргмучая ртуть+20% KC1OS . 1 рему чая ртуть Грнпитрорезорцинат серебра • 11 ксаметилентрипероксиддиа- F МИН «ид свинца .... А нт ртути (окисный) Пикрат свинца \ ии( серебра . 0.0 0,0 0,0 0,0 0,0 0.0 0,2 0,45 1,0 1,0 4.35 12.5 12,5 25,0 25.0 25,0 33.0 37.5 37,5 50,0 50,0 33,0 2 3 3-5 3-10 3-10 7 5—7 12 16—17 23—37 39 Чувствительность ацетиленида сереб- ра зависит от спо- соба его получе- ния Здесь приведена самая чувствитель- ная форма из всех примененных при исследовании Тей- лора и Ринкенбаха
I I ацетиленидов
В 1929 г. вопрос о чувствительности к трению исследовал Ратс-
* ург, предложивший методику испытания, резко отличающуюся от
Американской. Прибор был построен на принципе трения ВВ между
1»умя вращающимися поверхностями. Нижняя трущая поверхность,
и.i которую насыпают определенный объем ВВ, была сделана непо-
дпижной и лишь поднимаемой и опускаемой особой передачей вруч-
ную. Верхнюю трущую поверхность приводят во вращение со ско-
ростью 20 об/мин. На особую тарель, связанную с верхней трущей
поверхностью, накладывают груз различной величины, давление кото-
рого передается на ВВ. Верхнюю трущую поверхность прибора изго-
। овляют из фарфора или из закаленной стали. В первом случае ини-
циирующие ВВ испытывают в чистом виде, во втором случае для уси-
ления трения примешивают 10% кварцевого песку весьма высокой
испени измельчения.
Мерилом чувствительности инициирующих ВВ к трению при
данной конструкции прибора является величина груза, положенного
ид тарель. Данные, полученные при испытании инициирующих ВВ
по их чувствительности к трению наэтом приборе, приведены в табл. 11.
Другим, еще не разработанным окончательно, прибором для испы-
тания чувствительности ВВ к трению может служить прибор, пока-
чанный на фиг. 41.
Конструкция этого прибора такова: небольшого диаметра сталь-
ной круглый стержень, закрепленный в центре металлического диска,
59
между двумя стоиками, прикрепле]
ними к металлическому основании
Для того чтобы обеспечить плат
устанавливается вертикально
Фиг. 41. Прибор для определения
чувствительности инициирующих ВВ
к трению.
ное вращательное движение стер)
ня, последний в средней свосЦ
части пропущен через стальну!
муфту, а внизу—через латунны
вкладыш стальной планки, приче:
конец стержня выступает из вк:
дыша; к нижнему концу стержня
привинчен конический усечённый
пуансон с диаметром нижнего ос1
нования 10 мм. Это—верхняя тру
щая поверхность. Нижняя труща:
поверхность представляет co6oi
дно выточенного в стальном щ
линдре углубления- Цилиндр систе
мой рычагов связан с рычагом, на
который вешается тот или иноз
груз, поднимающий цилиндр и
тем самым увеличивающий усилие
трения по нашему усмотрению.
Навеску вещества в 0,05 г поме-
щают на дне углубления цилиндра,,
который вводят под пуансон. Па
рычаг навешивают груз 1 кг. Пуан{
сон приводят во вращательное
Чувствительность к трению по опытам Ратсб,
I Степень нагрузки, кг
Наименование ини- циирующих ВВ Поверх- ность—фар- фор Поверх- ность—ста
Азид ртути (окисный) 2 V
Азотетразолсвинец . 12—13 3—4
Гремучая ртуть. . . Тринитрофл орогл у- 15 12—13
цииат свинца . . . Гексанитродирезор- 20 15
цинат свинца . . . Гуанилнитрозоамимо- 22—23 22—23
гуанилтетразен . . 22—23 22—23
движение с определенной скоростью и засекают время пуска мотора
и взрыва. Время до
взрыва и служит мери-
лом чувствительности
вещества к трению.
При испытаниях на
этом приборе были по-
лучены следующие дан-
ные о чувствительности
к трению:
1) гремучей ртути—
от 34 сек. и более,
2) основного фуль-
мината меди — 3 — 5
сек.
Мерилом чувствитель-
ности к трению на по-*
добного типа приборах
может служить также
н нагрузка на пуансон
при постоянном времени и постоянной скорости вращения пуансона,
60
Чувствительность к трению зависит не только от конструкции
прибора и материала матрицы и пуансона, но также и от количества
и качества примесей. Установлено, что наличие примесей, обладаю-
щих большей твердостью, чем испытуемое инициирующее ВВ, повы-
Iилет чувствительность последнего к трению.
Определение чувствительности к лучу огня и нагреву
Очень часто по условиям применения, особенно в капсюлях-дсто-
ит горах, инициирующих ВВ в виде начального импульса сообщается
|уч огня. Поэтому при выборе инициатора приходится учитывать
го отношение к лучу огня. Нельзя применять инициирующие ВВ,
обладающие чрезмерно высокой нечувствительностью к обычным
л левым средствам инициирования (бикфордов шнур, капсюль-вос-
II заменитель или электрозапал). Это можно делать только как исклю-
чение в том случае, если остальные свойства выгодно отличают трудно
воспламеняемый инициатор от других.
С другой стороны, при слишком низкой температуре воспламене-
ния вещества оно является небезопасным в обращении, производстве
(сушка) и хранении.
Обычный способ определения чувствительности к лучу огня со-
стоит в воспламенении инициирующего вещества, запрессованного
о капсюль, от бикфордова шнура. При этом отмечают, в какой именно
<|и>рме проба реагировала на воспламенение: не воспламенилась,
загорелась и дала вспышку или детонировала.
Показательной величиной чувствительности к нагреву служит
температура вспышки. Температурой вспышки называется та наи-
меньшая температура, до которой должна быть нагрета часть вещества,
1ля того чтобы вызвать в нем реакцию взрывчатого превращения со
скоростью, достаточной для получения по крайней мере звукового
аффекта.
Эта температура для одного и того же ВВ может довольно значи-
тельно колебаться в зависимости от устройства прибора, величины
навески испытуемого ВВ и способов ее нагревания.
На практике температурой вспышки считают ту минимальную
1смпературу, при которой происходит воспламенение определенной
навески ВВ в строго определенных условиях опыта.
Для определения температур вспышки применяют прибор, изо-
браженный на фиг. 42. Он состоит из железной цилиндрической
бани (диаметр 60 мм, высота 85 мм), наполненной сплавом Вуда
(13% олова, 25% свинца, 12% кадмия, 50% висмута), уровень кото-
рого не доходит до верхнего края на 15 мм. Баню обогревают элек-
трическим током и регулируют температуру реостатом. Для удобства
работ и уменьшения теплоотдачи баню укрепляют в латунном фут-
ляре, причем между нею и футляром должен быть воздушный зазор.
Сверху баню накрывают железной крышкой с отверстиями; в цен-
тральное отверстие (диаметр 10 мм) вставляют термометр, погружае-
мый в сплав на 30 мм и защищаемый от взрыва металлической
61
Фиг. 42- Схема прибора
для определения темпе-
ратуры вспышки.
гильзой, другие четыре отверстия (диаметр 9 мм) служат для опуска-
ния в баню металлических (медных, луженых) гильзочек для наве
сок ВВ (диаметр 7,6 мм, высота 90 мм).
Методика определения. Через
отверстия в сплав погружают четыре гильзы
для навесок ВВ, баню нагревают до опре-
деленной температуры, близкой к ожидае-
мой температуре вспышки, гильзы опускают
на глубину около 30 мм и выдерживают
в течение не менее 1 мин. для нагрева их
до температуры сплава. Затем в одну из них
опускают навеску испытуемого инициирую-
щего ВВ в 0,01 г, спрессованную в таблетку
диаметром 3 мм под давлением 500 или
1000 ат.
Сохраняя температуру постоянной, одно-
временно с опусканием навески в гильзу
± засекают секундомером время и регист-
рируют продолжительность выдерживания
пробы до наступления вспышки. Рядом
опытов находят ту наименьшую темпера-
туру, ниже которой при выдерживании
навески в течение 5 мин. вспышки не про-
исходит. Эту температуру и принимают условно за температуру
вспышки ВВ.
Для большей точности определения каждый опыт повторяют
три-четыре раза и находят среднее значение температуры вспышки..
При этом способе определения температура вспышки зависит,
главным образом, от времени выдерживания вещества в ванне. По-
этому в целях более полной характеристики отношения ВВ к тепло-
вому импульсу рекомендуется дополнительное построение графика
зависимости температур вспышки от времени нагревания. Для постро-
ения графика (фиг. 43) производят ряд определений при различных
температурах, регистрируя каждый раз время до вспышки при помощи
62
Ькундомера. При этом наряду с промежуточными точками находят
I -in ическую температуру, при которой вспышка наступает сразу же
нпсче опускания пробы ВВ в нагретую гильзу, а также температуру,
под действием которой в течение 5 мин. вспышки не происходит.
f[n»i каждой точки производят три-четыре определения. Ради практи-
ческих удобств надо на-
чни.гы работу при тем-
li* р.| гурах, несколько
превышающих крити-
’нт кую температуру
in пышки взятого ве-
щества.
Температуры вспыш-
ки различных иниции-
рующих ВВ приведены
и гибл. 12.
I 7. ОСНОВНЫЕ ТРЕБОВА-
НИЯ, ПРЕДЪЯВЛЯЕМЫЕ
И ИНИЦИИРУЮЩИМ ВВ
Для того чтобы
ны.шать взрывчатое пре-
кращение зарядов бри-
мятных ВВ в подрыв-
ных патронах, артилле-
рийских снарядах, ми-
нах. торпедах, авиабом-
бах и др., а также для
uno, чтобы обеспечить
Таблица 12
Температуры вспышки инициирующих ВВ
& Е С Е £ Наименование вещества Темпе- ратура вспышки °C
1 Гремучая ртуть . 160—165
2 Гремучее серебро 170
3 Азид свинца . 305
4 Азид серебра . . 297
5 Азид ртути (I) 281
6 Хлораттримеркуральдегид 130
7 Диазобензолнитрат 90
8 Нитродиазобепзолперхлорат .... 150
9 Гексаметилентрипероксиддиамин . . 200
10 Триметиленамидоозонид 140
11 Сернистый азот ... ... 190
12 Ацетиленид серебра . ... 170
13 Цнануртриазид 207
14 Тринитрорезорцииат свинца .... 265
цозможность использования в боеприпасах, особенно в военное время,
разного рода суррогатов, обладающих пониженной чувствитель-
ностью, требуется возможно б 6 л ыи а я инициирую-
щая способность инициирующих ВВ. В самом деле, чем
пыше инициирующая их способность, тем большую работу совер-
шает бризантное ВВ и тем меньшее количество инициатора необхо-
димо для возбуждения взрыва. Кроме того, с уменьшением количества
инициатора уменьшается опасность в обращении и при стрельбе
цепкого артиллерийского снаряда. Требование повышения иници-
ирующей способности не может быть ограничено никакими преде-
лами, в том числе и техническими возможностями изготовления кап-
сюлей-детонаторов весьма малых размеров.
Возрастание начальных скоростей артиллерийских снарядов, вы-
зывающее увеличение сил инерции при выстреле и при ударе о пре-
।раду (броню) выдвигает второе требование —безопасность
инициирующих ВВ при практическом исполь-
। о в а н и и. Кроме того, требуется безопасность их
обращении для обеспечения нормального производства отно-
сительно больших количеств.
63
Из необходимости создания запаса боеприпасов в мирное время
с одной стороны, применения и хранения капсюлей-детонаторов в ус-
ловиях, отличных от нормальных, с другой, и, наконец, примене-
ния капсюлей в разных климатических условиях вытекает требова-
ние высокой химической стойкости инициирующихВВ.
При этом под химической стойкостью понимают неизменяемость фи-
зико-химических и взрывчатых свойств вещества под влиянием раз-
личных факторов: тепла, света, влаги, углекислоты воздуха, ме-
талла оболочки и др.
Максимальной естественной температурой, в которой приходится
применять средства инициирования, является температура ± 50—
60°. Инициирующие ВВ, не выдерживающие длительного хранения
при этих условиях, практического значения не имеют.
Инициирующие ВВ, разлагающиеся под влиянием лучей солнеч-
ного света, должны быть изолированы от последних при применении
и в производстве. Такие инициаторы вообще могут применяться,
так как необходимые условия их использования всегда могут быть
созданы.
Некоторые инициирующие ВВ при наличии влаги в значительной
степени флегматизируются, как, например, гремучая ртуть, или же
подвергаются разложению, особенно при повышенных температурах,
как, например, тетразен. Помимо этого присутствие влаги может
способствовать взаимодействию инициирующих веществ с металлом.
Углекислота также оказывает влияние на некоторые инициирую-
щие ВВ, изменяя их взрывчатые свойства. Поэтому всякое новое
инициирующее ВВ перед принятием на вооружение подвергают тща-
тельному испытанию на взрывчатые свойства и на химическую стой-
кость.
Так как инициирующие ВВ применяют всегда запрессованными
в ту или иную оболочку, причем прессованию предшествует насыпка
в оболочку навесок механическими мерками, то необходимо, чтобы
инициирующие ВВ обладали хорошей сыпучестью и
хорошей прессуемостью.
Вследствие жесткости всех этих требований приходится сильно
ограничить выбор инициирующего ВВ. До сих пор ни одно из вновь
предложенных (довольно ценных по ряду свойств) инициирующих
веществ не может конкурировать с гремучей ртутью и азидом свинца,
являющимися основными применяемыми в настоящее время иниции-
рующими ВВ.
ГРЕМУЧАЯ РТУТЬ
§ 1. ОТКРЫТИЕ ГРЕМУЧЕЙ РТУТИ И ЕЕ ПРИМЕНЕНИЕ
С достаточной достоверностью установлено, что некоторые соли
। ремучей кислоты были известны алхимикам еще в средние века.
Так, например, в рукописи так называемого Базилиуса Валенти-
пуса, относящейся к первой половине XVII века, упоминается
1 ремучее золото, открытие которого принадлежит, невидимому, гол-
мндцу Корнелиусу Ван-Дроббелю. Во всяком случае в первой поло-
вине XVII века уже знали характер действия этого очень бризант-
ного ВВ. В эфемеридах Германской академии естественных наук
в Нюрнберге за 1678 г. упоминается о двух взрывах во время работы
с гремучим золотом. По мнению Ромоцкого, Ван-Дроббелю была из-
вестна и гремучая ртуть (около 1628 г.). Однако первое указание
о приготовлении гремучей ртути принадлежит Кункелю (1690 г.).
В конце XVII века и в XVIII веке все эти работы над солями гре-
мучей кислоты были, видимо, основательно забыты. Вторичное откры-
тке гремучей ртути Говардом (1799 г.), а затем гремучего серебра
Бруньятелли (1802 г.) произошло в период, когда химия как наука
была очень молода и постепенно выходила нз тьмы схоластики. По-
грому понятны те совершенно случайные обстоятельства, при кото-
рых гремучая ртуть была приготовлена Говардом.
Согласно представлению того времени при воздействии спирта—
носителя водорода — на азотную кислоту, являющуюся носителем
кислорода, должна была получиться соляная кислота, которая в при-
сутствии окиси ртути должна была соединиться со ртутью и дать
хлорную ртуть. Говард действовал азотной кислотой на красную
окись ртути, растертую со спиртом, и в результате бурной реакции
вместо ожидаемой хлорной ртути получил гремучую ртуть. Пере-
кристаллизованная белая соль, которую Говард принял вначале
<а сулему, не показала реакции на связанную соляную кислоту.
Когда же Говард облил соль концентрированной серной кислотой,
го она с громадной силой взорвалась. Таковы обстоятельства получе-
ния Говардом гремучей ртути.
Гремучая ртуть является одним из весьма важных и распростра-
ненных ВВ, и несмотря на то, что в каждом отдельном случае в объек-
тах она применяется в очень небольших количествах (от 0,01 до 2 г),
Ьубнов—92—5
65
нередко можно встретить заводы производительностью в 2000—2500 кг
гремучей ртути в месяц. У Штеттбахера и Наума имеются указания
о производстве даже 7000 кг в неделю одним заводом.
По данным Каста, производство гремучей ртути в Германии
с 1910 по 1918 г. выражалось следующими цифрами (см. таблицу).
Области применения гремучей ртути
г Количество__обширны. В самом недалеком прошлом ею
° 1,1 кг снаряжались все капсюли-детонаторы. В
нио
1912
1914
1916
1917
1918
104 800
124 400
108 900
285 500
357 600
17 800
последнее же время она уступает свое ме-
сто в капсюлях-детонаторах азиду свинца.
Ударные составы всех капсюлей-воспла-
менителей в качестве обязательного ком-
понента содержат гремучую ртуть. Правда,
и здесь, особенно для капсюлей ручного
----------------------- оружия, делаются попытки заменить ее
стифнатом свинца и тетразеном.
Гремучая ртуть применяется также в некоторых фрикционных
составах и в качестве одного из компонентов сердцевины детонирую-
щих шнуров.
§ 2. ЭЛЕМЕНТАРНЫЙ СОСТАВ И СТРОЕНИЕ ГРЕМУЧЕЙ РТУТИ
Открытие Говарда вызвало большой теоретический интерес в на-
учном мире. Исследование гремучей ртути производилось и произ-
водится с момента ее открытия до настоящего времени. Ею серьезно
занимались такие великие химики, как Бертолле, Либих, Ге-Люссак,
Берцеллиус, Тенард, Дюма, Е. Деви, Велер, Жерар, Кекуле и др.
Во всех их исследованиях рассматривался прежде всего элементар-
ный состав гремучей ртути и только позже было обращено внимание
на изучение строения интересной во многих отношениях гремучей
кислоты.
Сам Говард на основании обширных исследований природы откры-
того им нового замечательного вещества считал гремучую ртуть со-
единением «эфирного селитренного газа» (nitrous etherized gas, ныне—
этилнитрит), щавелевой кислоты, ртути и кислорода в следующих
весовых отношениях:
щавелевой кислоты . 21,28%
ртути.................... . 64,72%
этилнитрата и кислорода . ... 14,00%
Интерес, проявленный по отношению к вновь открытому соединению, был,
невидимому, настолько велик, что уже в 1801 г. Бертолле опубликовал свою ра-
боту о гремучей ртути. Он обрабатывал гремучую ртуть разбавленной серной
кислотой, но вместо ожидавшейся им щавелевокислой соли ртути он получил
сернокислую закись ртути- При действии соляной кислоты ему также не удалось
получить щавелевую кислоту. Таким образом Бертолле показал, что щавелевая
кислота jHe входит в состав молекулы гремучей ртути. При энергичной обработке
гремучей ртути едким кали Бертолле обнаружил выделение аммиака. Между про-
чим он так же, как раньше Говард, заметил, что гремучая ртуть не получается
всегда одинакового состава, что иногда оиа полностью растворяется в соляной
кислоте, а в других случаях остается нерастворимый остаток. Из своей работы
Бертолле сделал вывод, что гремучая ртуть состоит нз «сильно окисленной ртути»,
аммиака и неизвестного органического вещества, родственного спирту.
66
I Позднейшие работы Фуркруа и Теннара не дали никаких существенных ре-
ни пин вопроса о составе гремучей ртути, однако, они внесли некоторую ясность
и противоречивые выводы Говарда н Бертолле, доказав, что состав гремучей ртути
iiiiuicHT от способа ее получения. Так, если растворы исходных веществ нагревать
hi ii»jiro (несколько мгновений и не до кипения), то гремучей ртути образуется
п< м нш'о, причем она будет в смеси с азотнокислой ртутью. Если кипятить несколько
минут, то получаемая гремучая ртуть достаточно чиста и не содержит щавелевой
V in лоты. И, наконец, если кипятить полчаса, то образуется, главным образом,
иыислевокнслая ртуть с небольшим количеством гремучей ртути.
Дескостиль исследовал в 1807 г. гремучее серебро, полученное так же, как
•11 рсмучая ртуть, и пришел к выводу, что оно представляет собой соединение окиси
|i'|M‘Rpa с аммиаком и «растительным веществом», т. е. является родственным гре-
мучей ртути. Надо отметить, что Дескостилю удалось произвести очень точное
И» тому времени определение содержания серебра в гремучем серебре (он нашел
11"„, в то время как по теории должно было быть 72%).
Развитие точных методов органического анализа дало возмож-
ность выяснить окончательно состав гремучей ртути и гремучего се-
ребра. Но для этого все же понадобилось около 35 лет (с 1811 г. по
IK48 г.).
Из работ этого периода прежде всего необходимо отметить работы
Чпбиха. Правда, первая его работа содержала много недостатков
и не могла способствовать разрешению вопроса. Например, из тех
фнктов, что выделяющийся при кипячении гремучего серебра с едким
iftuiM газ окрашивал красную лакмусовую бумажку в синий цвет
и что насыщенный раствор гремучего серебра в едком кали давал
I кальциевыми солями осадок, обладавший свойствами щавелево-
кислого кальция, Либих сделал заключение, что гремучее серебро
i «стоит из окиси серебра, аммиака и щавелевой кислоты. Этим своим
шключением Либих вновь запутал вопрос о составе. Впоследствии,
при применении более точной методики, Либих убедился в отсутствии
«ммиака и щавелевой кислоты в составе гремучего серебра, а также
и юм, что осадок, получающийся при действии кислот на раствор
। рсмучего серебра в едком кали, обладает свойствами какой-то особой
кислоты, не напоминающей щавелевую и обладающей взрывчатыми
। пойствами.
1 Следует отметить, что, исследуя осадок, получающийся при ней-
• рялизации щелочного раствора гремучего серебра, и наблюдая вы-
I'',пение синильной кислоты при разложении гремучекислых солей,
Либих сделал вывод, что гремучие соли можно рассматривать как
металлические соли кислоты, содержащей группу циана, подобно
железисто-синеродистым солям.
Первые анализы Либиха гремучей ртути и гремучего серебра
Пили очень неточны, однако, Берцеллиус на их основании вывел сле-
дующие формулы:
для гремучего серебра
Ag + 2N4C4HMO7 4-4Н2О
для гремучей ртути
Hg 4- N4C4H12O7 -Ь 2Н2О
67
Исходя из последующих данных Либиха и Ге-Люссака, была вы-
ведена для гремучего серебра формула: Ag.2O4C4N2. В современны]
атомных весах эта формула получает следующий вид:
Ag2O2^2^2-
В 1882 г. Велер получил циановосеребряную соль, и анализ по
казал, что она обладает тем же процентным составом, что и грему
чее серебро. Ге-Люссак объяснял еще тогда отличие в свойства
этих соединений при одной и той же эмпирической формуле различие]
структуры молекул. Однако Берцеллиус и его школа вначале строп
придерживались взгляда, что соединения одного и того же состав?
должны быть идентичными и не могут отличаться Друг от друга даже
по своему строению. Берцеллиус даже сомневался в правильности
анализа гремучего серебра, произведенного Либихом и Ге~Люсса
ком. Он считал, что гремучее серебро отличается и по составу от циа
новосеребряной соли и что оно является «циановокислой окисью
серебра», занимающей промежуточное положение между серебряным
солями синильной и циановой кислот.
Либих пытался выяснить строение молекулы гремучей ртути
причем особое внимание он обращал на то, как связаны между co6oi
отдельные части молекулы. Он пришел к выводу, что углерод'и азот
в гремучей кислоте находятся в том же положении, как и в циане,
но окончательно разрешить вопрос о строении гремучей ртути ему н1
удалось, несмотря на большое разнообразие опытов. Другие ученые
того времени (Кюи) также не сумели объяснить изомерию гремучей
и циановой кислот.
После этих неудачных попыток Берцеллиус в (844 г. отверг даже
возможность существования изомерии и возвратился к старому по-
ложению Либиха, что гремучее серебро является серебряной сольд
кислоты, содержащей металл, т. е. соединением комплексного порядка
Либих к этому времени считал мнение Берцеллнуса неверным
и остановился на формуле гремучей кислоты 2HgC4N2O2; он счита|
ее двухосновной и полимерной циановой кислоте.
Когда же в 1848 г. Гладстон получил мочевину и сульфоциановуЧ
кислоту взаимодействием сернистого водорода и медноаммонийног
фульмината, чем была доказана тесная связь между циановой и гре
мучей кислотами, Берцеллиус высказался за существование изоме
рии и вскоре стал настолько серьезным защитником этой теории, чт
и ныне за Берцеллиусом считается первенство введения в химик
понятия об изомерии.
Однако выяснение состава гремучей ртути и введение в хими
понятия об изомерии все же долгое время не привели к решени
вопроса о строении гремучей кислоты.
В 40-х годах прошлого столетия в связи с открытием нитросоед
нений многоатомных спиртов и углеводородов, с получением глицс
ринтринитрата, азотных эфиров маннита и клетчатки и други
взрывчатых соединений выработался новый взгляд на состав соле
гремучей кислоты. Носителем взрывчатых свойств стали считав
68
Rynny, а потому и в гремучей кислоте пытались обнаружить
этой группы. Жерар в своем учебнике органической химии
ривал гремучую ртуть как нитросоединение. В новом изда-
2го учебника (1854 г.) он отказался от своего мнения и выска-
дпсложение о том, что гремучая кислота является эфиром
кислоты.
ив этих утверждений и предположений Жерара возражали
Ч1й>их, Поггендорф и Велер, вполне справедливо указывавшие, что
|ущсствуют ВВ и без кислорода и что азот в гремучей кислоте не
может быть связан с кислородом, так как при действии сероводорода
ц.| медноаммонийный фульминат последний разлагается с образова-
нием мочевины и роданистого водорода.
Однако Кекуле, идя по стопам Жерара, пытался приписать грему-
чий кислоте формулу нитроацетонитрила: CH2NO2CN.
Интересно привести опыты, которыми Кекуле доказывал правоту своей фор-
»лы. Растворяя гремучую ртуть в соляной кислоте и сравнивая ее элементарный
. <н*г.-|в с гремучим серебром, Кекуле показал, что гремучая ртуть представляет
снГ)(»й соль закиси, а не окиси ртути. Он повторил опыты Либиха по изучению
ЬеЙствия хлора на гремучую ртуть. Продуктом реакции кроме хлорциана и сулемы
»к .дались и другие хлорзамещенные, среди которых был обнаружен хлорпикрин.
* нт эти продукта (хлорциан и хлорпикрин), казалось, доказывали существование
и гремучей кислоте нитрогруппы наряду с группой циаиа. Все же Кекуле не счи-
ны, что сообщенные им факты окончательно подтверждают данвую им формулу.
За два года до опубликования работы Кекуле Шишков открыл одноосновную
фульминуровую кислоту состава C3H3N3O3, названную им изоциануровой кисло-
•|»ц. Почти одновременно это открытие сделал и Либих, фульминуровзя кислота
была получена действием щелочных растворов галоидов на гремучую ртуть. Сво-
1шдный переход гремучей кислоты в фульмпнуровую Шишков объяснял аналогич-
ной группировкой атомов в обеих кислотах. Он пришел к заключению, что грему-
чая кислота не может содержать в себе иитрогруппы, так как полученная им новая
кислота оказалась стойкой по отношению к восстановителям, например, к сер-
нистому аммонию. Он также считал, что гремучая кислота — сложное соединение,
и солях которого металл непосредственно связан с азотом. Неправильное пред-
положение о неизменяемости гремучей ртути при обыкновенной температуре под
пипянием растворов йодистого калия и едкого кали навело его на мысль о том,
то гремучая кислота стоит ближе к амидам, чемккислотам. В дальнейшем Шишков
mказался от этой гипотезы в пользу предположения, что гремучая кислота содер-
жит нитрогруппу. В то же время он утверждал, что гремучая кислота не может
|ц.1гь простейшим нитроацетонитрилом, а должна иметь формулу.
(CONH)2CH2NO2CN.
Впоследствии Шишковым была предложена еще одна формула гремучей
кислоты:
С — HNOS
II + 2HCN
С ~ HNO2
и.। том основании, что фульминаты способны при разложении выделять синильну ю
кислоту и образовать муравьиную кислоту прн действии кислот на фульминаты
или бромистого циана при действии брома.
Теории Шишкова, частично опиравшиеся на блестящие экспериментальные
||.1боты, не внесли изменения в формулу Жерара—Кекуле и до 80-х годов не
могли поколебать взгляда на гремучую кислоту, как на нитроанётонитрил.
69
Соли гремучей кислоты по своей взрывчатости имеют сходство и с дназа •:
единениями- Основываясь на этом, Г рис в 1860 г. пытался присвоить гремуче
кислоте строение диазоуксусной кислоты:
/N
“(||
I \n
соон
Однако диазоуксусная кислота была получена в 1888 г. Ку роду сом, и таким обрг
зом была доказана неправильность предположения Гриса.
Попытка Вюрстера дать экспериментально не обоснованную опытом формул
гремучей кислоты:
CHNOe
II
С == NH
также не увенчалась успехом.
Следует привести еще формулу Армстронга, появившуюся в 1875 г.
С= NOH
II
НС —NO
Правда, серьезного обоснования этой формулы Армстронг не дал.
В 80-х годах Карстаньен и Эренберг установили, что при действи
крепкой соляной кислоты на гремучую ртуть выделяется гидроксил
амин- К этому же выводу пришел и Штейнер.
В это же время Ф. Мейер и его ученики доказали, что при дей-
ствии на гидроксиламин альдегидов и кетонов получаются изонитро-
зосоединения (оксимы), содержащие группу С = NOH, и что при
действии на них концентрированной соляной кислоты гидроксиламин
выделяется вновь. На основании сходства фульминатов с металличе-
скими производными оксимов Штейнер предложил приписать
гремучей кислоте формулу диизонитрозозтилена:
С = NOH
II
С = NOH
Штейнер установил, что весь азот гремучей кислоты выделяется
в виде гидроксиламина. Эренберг подробно исследовал реакцию
между фульминатами и соляной кислотой и пришел к тому же вы-
воду. Этими количественными опытами они установили необоснован-
ность нитроацетонитрильной формулы Жерара — Кекуле.
Дивере, основываясь на ошибочном наблюдении Либиха, что азо-
тистый ангидрид со спиртовым раствором солей серебра или ртути
дает фульминаты, предложил формулу оксифуразана:
HOC = N.
I >0
HC = NZ
70
Образование гремучей кислоты, по Диверсу, происходило по урав-
нению:
СН2ОН ON. НОС == N
1 + / О I /0-1-21^0
СН3 ON7 HC=N'
В то же время Армстронг, отказываясь от своей прежней фор-
мулы, данной им в 1845 г., и возражая Диверсу, предложил формулы:
х-он
N< | и
ХС = NOH
N =СН
I I
О —С= NOH
Точно так же в результате ошибочных наблюдений появились фор-
мулы:
С = NH НС = N
II ; I -
OaN — CH OjN — СН
Большинством этих формул авторы представляли гремучую кис-
лоту как производную этана — системы из двух углеродных атомов,—
базируясь на том, что исходным веществом для нее служит этиловый
спирт и что гремучая кислота является двухосновной.
Только Шолль и Голлеман в начале 90-х годов прошлого века
направили химию гремучей кислоты на новые рельсы и на основании
новых реакций доказали легкий переход гремучей кислоты в произ-
водные циановой кислоты.
Голлеман в 1890 г. для объяснения своих наблюдений над фуль-
минатами высказал предположение, что гремучая кислота реагирует
как таутомерное вещество в трех видах:
„О
ch2-n<
I
CN
CH = N — О С = NOH
I I Z “
CH = N -о C = NOH
I. е. как нитроацетонитрил, перекись глиоксима и диизонитрозоэти-
лен. В том же году Шолль предложил формулу гремучей кислоты
HON< >
NOH как производную формоксима.
Впоследствии Шолль сам доказал на основании ряда обстоятель-
ных исследований, что гремучую кислоту нельзя рассматривать ни
как нитро ацето нитрил, ни как перекись глиоксима, что производные
питроацетонитрила дают совершенно иные продукты превращения,
чем производные гремучей кислоты, и что, следовательно, последняя
не идентична с нитроацетонитрилом.
Окончательное подтверждение неправильности нитроацетони-
грильной формулы строения гремучей кислоты экспериментально
71
ch.no2
I +IV)
CN
было доказано только после получения самого нитроацетонитрила
Штейнкопфом в 1908 г. действием хлорангидрида сернистой кислоты
на метазон овую кислоту:
ch2no2
ch = NOH
Рядом исследований (реакции на CN и NO2, определение молекуляр-
ного веса, реакции восстановления ит. п.) было установлено, что это
вещество и есть нитро ацетонитрил.
Далее Шолль, получив монофенилглиоксимпероксид
С6Н5 — с = N — О
I I
H-C-N—О
пытался заменить в нем водород металлом. Это ему не удалось, так же
как не удалось получить фульминаты окислением глиоксима и его со-
лей. Этим была доказана невозможность существования гремучей
кислоты в форме перекиси глиоксима.
Исследуя процесс получения формоксима Н2С = NOH и его
свойства, Шолль пытался получить формоксим и из гремучей кислоты
путем восстановления, что могло бы иметь место, если принять строе-
ние последней как производной формоксима. Однако попытка не при-
вела к успешным результатам.. Далее Шолль остановился на изу-
чении перехода производных гремучей кислоты в производные циано-
вой кислоты. Он получил из гремучей кислоты ацстилизоциановую
кислоту действием, хлористого ацетила на гремучую ртуть и пришел
к заключению, что основным ядром в гремучей кислоте должно быть
изонитрозосоединение, т. е. карбилоксим CNOH, изомерный циано-
вой кислоте. Шолль показал параллельность явления Бекмачовского
превращения в отношении гремучей и изоциановой кислот.
По Бекману, оксимы переходят в изомерные амиды:
^С —СН3 -> / ^nh—со—сн3
Z || \___z
NOH
(ацетофеноноксим) (ацетанилид)
По Шоллю, то же самое происходит и в случае гремучей и изоциа-
новой кислот:
z°
C = N —ОН -> Cf
4 Nil
(карбилоксим) (изоциановая кислота)
Таким образом Шолль первым разрешил в принципе вопрос о
строении гремучей кислоты, но он не мог довести до конца своих рас-
суждений, а тем более заявить категорически о гремучей кислоте,
как о простейшем карбилоксиме, ибо по тем временам мысль о двух-
валентном углероде была бы слишком смелой.
72
Наконец, работы и выводы американского химика Нефа (1894 г.)
щхли ясность в последний вопрос; он окончательно установил для
рсмучсй кислоты формулу простейшего карбилоксима: CNOH.
В пользу этого строения говорят следующие факты:
L Гремучая ртуть образуется из Gci-нитрометаннатрия действием
норной ртути. Этим синтезом Неф объяснил казавшееся непонятным
йризование гремучей кислоты из этилового спирта и азотной кис-
j>^2 C= :H2
C .0 ^4-
N= 0 .
X \
0 Na ‘ Ci x Ox
+ i >Hg - * >Hg
0: Na Cl / oz
C„ X) <NC
\H 4 IL 6
С = N —
с = N — о/
Нитрометаннатрий и хлорная ртуть в водном растворе дают хло-
ристый натрий и желтую tzci-нитрометанртуть, которая при нагре-
вании частично переходит в гремучую ртуть с отщеплением воды.
2. Еще легче образуется гремучее серебро из серебряной соли ме-
нлнитроловой кислоты с отщеплением азотистой кислоты:
NOAg
НС( -> C = N — О -Ag + HNO2
4NO3
3. Так же легко образуется гремучее серебро из амидометилни-
розоловой кислоты в присутствии азотнокислого серебра в азотиокис-
ЮМ растворе:
ON ч
рС = NOH N2 4-Н2ОС = N — ОН
H2Nz
Виланд наблюдал изомеризацию формонитрилоксида в гремучую
кислоту:
О
/\ С —N —ОН.
HC=N
Собственно этим явлением правильнее было бы объяснить реак-
цию, показанную в п. 2. Метилнитро левая кислота расщепляется на
иотистую кислоту и на некоторое весьма реакционноспособное со-
динение (HONC)*, занимающее по своим свойствам промежуточное
положение между гремучей кислотой и изоциановой:
J4OH
ЗНС^
xno2
-> 3HNO2 + 3HONC
7a
Это соединение принадлежит к полимерному ряду, но показыв]
все реакции простого форме нитрилоксида, так Же как триоксимео
лен и формальдегид. Виланд назвал его трифульмином и дал ei
фор мулу.
О —N
нс ,/ХСН
/°
о-Хсн
Тр^ифульмин, отличающийся сильными взрывчатыми свойствами, га
действием минеральных кислот переходит в гремучую кислоту.
4. Образование формилхлоридоксима из свободной гремучей ки
лоты и хлористого водорода. Неф показал, что при действии на фул
минаты разбавленной соляной кислотой свободная гремучая кисла
не выделяется, но что, будучи вытеснена из ее солей, она присоёд:
няет к себе молекулу хлористого водорода и переходит в кристалл:
ческий хлорид:
С = N - ОН + НС1
н\
-> >C = N- ОН
СК
То же самое получил и Шолль независимо от Нефа. Ему такие
удалось выделить формилхлоридоксим, но он рассматривал это со-
единение, как солеобразное производное гремучей кислоты и не при-1
дал ему никакого значения- В чистом виде формилхлоридоксим был
получен из раствора фульмината натрия при приливании соляной
кислоты уд. веса 1,183 при 15°. Смесь сильно охлаждали, выделив^
шийся хлористый натрий быстро отфильтровывали, а раствор обра-
батывали эфиром. По испарении высушенного хлористым кальцием
эфира выделялись длинные иглы формилхлоридоксима.
Формилхлоридоксим, или гидр окса лова я кислота, легко гидро-
лизует с образованием муравьиной кислоты и хлористоводородного
гидроксиламина:
Н.
)С = N — ОН 4- 2Н2О -►
СК
HCOOH4-NH2OHHC1
Одновременно Неф открыл обратное превращение формилхлорид-
«оксима в гремучее серебро и хлористое серебро при действии азотно-1
кислого серебра:
Н\
>С = N — ОН 4- 2AgNO3
СК
C = N - OAg 4- AgCl + 2HNO3
74
Шолль получил в таких же условиях гремучую ртуть.
5. Целый ряд реакций присоединения аналогичен реакциям при-
осдинения изонитрилов, например:
Н .
С = NOH 4- H2S УС= NOH
HSZ
Вгч
С = NOH 4- Вг« >С = NOH
ВГ
СН3СООЧ
С = NOH + СН3СООС1 -> >С = NOH
С1
Н х
С = NOH 4- HONO -> >С = NOH и т. п.
NO/
Попытки поколебать уверенность в формуле строения гремучей
кислоты, данной Нефом, производились неоднократно.
Так, в 1899 г. Лей и Киссель, определяя электропроводность растворов раз-
нимых ртутных соединений, в том числе и гремучей ртути, по способу Кольрауша,
н.1мененному Оствальдом, установили, что молярная электропроводность раствора
। ремучей ртути оказалась р = 0,7 • 10—7, т. е. что она мало отличается от электро-
проводности чистой воды, равной 1,3 -IO—10, и, следовательно, подобна электро-
проводности цианистой ртути Hg(CN)2. Отсюда Лей и Киссель сделали вывод,
что в гремучей ртути ртуть соединена не с кислородом, а с углеродом или азотом,
и что поэтому формулу Нефа следует заменить одной из следующих формул:
С —N —Hg—N —С или N —С —Hg —C = N
'cf' ''cf о
Основываясь на этих исследованиях, Солонина также считал формулу Нефа
мало вероятной.
Следует отметить, что так как растворимость гремучей ртути при обыкновен-
ной температуре очень мала, то Лей и Кнссель работали при незначительных кон-
центрациях.
В то же время Лей и Киссель установили, что приведенные ими формулы не
могут быть приписаны натриевой соли гремучей кислоты. Строение фульмината
натрия, по их мнению, иное. Так как эта соль в воде растворима и диссоциирована
и растворе, то образование нз нее формилхлоридоксима еще не может служить
предпосылкой для решения вопроса о строении гремучей кислоты.
Григорович для исследования вопроса о применимости формулы строения
Нефа действовал на гремучую ртуть меркурдиэтилом, атак как последний к гре-
мучей ртути не присоединяется, то он решип, что гремучей кислоте нельзя при-
писать формулу строения Нефа, и счел возможным рекомендовать следующие
формулы:
Н — С=-М нлиН —CSN-0
V
Jowitschitsch на основании опытных данных пытался доказать правильность
формулы Голлемана. Он действовал на гипероксидэфнр диизонитрозоянтарной
75
кислоты сначала крепкой серной кислотой, а затем раствором едкого натра
(1 .10) и получил гипероксидкарбоновую кислоту:
C2HS—СО2—С — С—СО2С2НВ НС — С—СООН
в н -> II й
NO —ON NO —ON
При действии более крепкого раствора едкого натра (1 -5) он получил после
нейтрализации азотной кислотой, извлечения эфиром и вторичной нейтрализации
щелочью продукт, который после взаимодействия с азотнокислым серебром давал
светложелтый взрывчатый осадок, чувствительный к пламени и трению и дающий
при нагревании с азотной кислотой гидроксиламин. Jowitschitsch принял его вна-
чале за глиоксимгипероксид серебра
Ag-C — С — Ag
II II
NO—ON
• и счел аналогичным гремучему серебру.
Солонина, критикуя предложенные в разное время формулы строения грему-
чейкислоты, делает вывод, что едва ли можно выразить строение гремучей кислоты
одной формулой для объяснения всех свойств гремучекислых солей и что оно
должно быть выражено несколькими формулами, вероятнее всего, следующими
тремя:
N = CH С = NH
\ \/ Н —C = N-0
О О
Все эти попытки опровергнуть формулу Нефа базируются на не-
достаточно точных опытах или только на экспериментально недока-
занных предположениях.
Хотя Нефом и его последователями и был решен вопрос о гремучей
кислоте как карбилоксиме, однако, следует заметить, что на осно-
вании только химических реакций еще нельзя утверждать, мономер-
ный или димерный карбилоксим является гремучей кислотой. Окон- t
чательно решил этот вопрос Велер, который определил молекулярный
вес полученного им безводного фульмината натрия и установил для
него формулу:
CNONa (мономерный карбилоксим).
Таким образом гремучей ртути должна
быть приписана структурная формула по
Нефу:
ONC
Hg<
XONC
Свободная гремучая кислота была получена только в водных
растворах при низкой температуре. Раствор свободной гремучей кис-
лоты был получен Шольвиндом, обработкой фульмината натрия раз-
бавленной серной кислотой. Изолировать свободную гремучую кис-
лоту не удалось, так как она оказалась неустойчивой. Даже в эфир-
ном растворе она сохраняется не более 10 мин., а затем превращается
(полимеризуясь) в /n-фульминуровую кислоту (оксимизонитрозоизо-
76
салолом): г NOH I
11
3C=NOH -> |_“С —С —C = NOHJ ->
II I I
NOH
NOH
II
-> НС С— С = NOH.
II ।
N — О
Раствор гремучей кислоты обладает резким запахом. Пары ее
весьма ядовиты.
§ 3. ХИМИЗМ ПОЛУЧЕНИЯ ГРЕМУЧЕЙ РТУТИ
Гремучая ртуть получается действием азотнокислого раствора
ртути на этиловый алкоголь. Реакция образования гремучей ртути
настолько своеобразна и протекает столь бурно и быстро, что иссле-
щватели долгое время (более 100 лет) не могли познать ее истинного
хода, так какие удавалось установить ни одной промежуточной стадии
этого процесса. Шишков первым попытался показать механизм обра-
зования гремучей ртути. Однако ни эта попытка, ни последовавшие
<атсм попытки Голлемана, Шолля и Штейнера не привели к положи-
тельным результатам. Лишь после того, когда был обнаружен распад
метилнитриловой кислоты иа гремучую и азотистую кислоты, а также
после обнаружения, что для реакции пригоден именно этиловый
спирт, а не метиловый, и что ацетальдегид дает при реакции грему-
чую ртуть легче, чем этиловый спирт, — были найдены основные
звенья и появилась возможность объяснить ход процесса образова-
ния гремучей ртути. В 1909 г. Виланд на основании своих много-
численных работ объяснил весь сложный процесс реакции по отдель-
ным стадиям. Ход реакций представляется ныне в следующем виде:
1. Вначале происходит окисление зтилового алкоголя в ацеталь-
дегид (его запах всегда чувствуется при получении гремучей ртути):
СНэСН20Н + 0 -> CH3Cf 4-Н.О
хн
2. Далее вдет реакция нитрозирования ацетальдегида в изони-
грозоацетальдегид:
>° Д’
СН„С< 4-HONO СН2 — С< |--Н2О->
ХН | ХН
NO
Х>
-> сн—<У + нао
п Чн
NOH
77
Кстати, существование этих двух фаз реакции предполагал не-
сколько ранее и Велер.
3. Затем происходит окисление изонитрозоацетальдегида в изони-1
трозоуксусную кислоту:
CH — Cf + 0->СН -С<
II Хн || чон
NOH NOH
4. Изонитрозоуксусная кислота нитруется азотной кислотой
в нитроизонитрозоуксусную кислоту:
сн—,,о
II 0H-|-H0N02 -» C(N02) — С< 4-Н.О
NOH || ХОН
NOH
5. Нитроизонитрозоуксусная кислота — соединение весьма нестой-
кое и переходит с выделением углекислоты в метилнитроловую кис-
лоту:
,ZO
C(NO2) — С< t
II ОН -» CII(NO2) +СО2
NOH ||
NOH
6. Метилнитроловая кислота диссоциирует на гремучую кислоту
и азотистую кислоту»:
CH(NO2)
II -> c = noh + hno2
NOH
Если последнюю фазу вести в присутствии ртутной или серебря-
ной азотнокислых солей, то вместо легко разлагающейся гремучей
кислоты образуется стойкая гремучая ртуть или гремучее серебро:
CH(NO.)
2 11 -|-Hg(NO3)2 Hg(ONC)a+2HNO2-'-2HNO3
NOH
По этой схеме азотистая кислота оказывается весьма активным
агентом в процессе образования фульмината. Образуется же она во
время реакции (фазы I и III) непрерывно из имеющейся в избытке
азотной кислоты. Необходимость присутствия азотистой кислоты (те-
оретически даже в незначительных количествах), для того чтобы
вызвать реакцию, подтверждается тем, что прокипяченная азотная
кислота, свободная от окислов азота, ни со спиртом, ни с уксусным
альдегидом гремучей кислоты не образует. Это положение в свое
время высказывали еще Зельми, Собреро и Жерар, а опыты Велера
и Теодоровича его окончательно подтвердили.
78
еакция образования гремучей ртути протекает весьма бурно
(делением тепла, вполне достаточного для разогрева и кипения
|иирта и азотной кислоты. Выделение кристаллов гремучей ртути
1<»пр»вождается выбрасыванием густых, белых взрывчатых паров.
|'н гоящих, главным образом, из этилнитрита и этилнитрата с при-
№ । i.io большого количества ацетальдегида и угольной кислоты. Кроме
Ич<», выделяются глиоксаль, глиоксалевая кислота, щавелевая кис-
пил и другие продукты окисления спирта.
Главнейшие побочные реакции следующие:
1) образование этилнитрита:
СН3СН2ОН + HONO -> CHgCbLONO + Н20
2) образование этилнитрата:
CH3CH2OH4-HONO2 -> ch3ch2ono2+н.2о
3) образование щавелевой кислоты:
соон
СН3СН2ОН 4- 50 -> I 4- 2Н2О
СООН
4) образование муравьиной кислоты:
СООН
| -> НСООН4-СО2
СООН
5) образование этилового эфира муравьиной кислоты:
НСООН-|-С2Н5ОН -> НСООС3Н34-Н2О
6) образование окиси углерода:
НСООН С0Ч-Н20
II др.
Для получения гремучей ртути пользуются в лабораториях и.
№ технике старым способом, установленным еще Либихом и лишь,
несколько измененным другими исследователями.
По Либиху, 1 в. ч. металлической ртути растворяют в 12 в. ч.
Ьотной кислоты уд. веса 1,36—1,40. Затем в два приема к раствору
приливают II—12 в. ч. обыкновенного спирта. Вторую половину-
снирта приливают постепенно только после начала кипения раствора.
В 1886 г. Бекман несколько изменил способ Либиха. Он предло-
жил растворять 50 г (1 в. ч.) металлической ртути в 600 г (12 в. ч.)
плотной кислоты уд. веса 1,40 и приливать 550 г (И в. ч.) спирта
крепостью 98,5% также в два приема. Спирт и раствор ртути в азот-
ной кислоте он рекомендовал подогревать до температуры 25—30°.
Вторую половину спирта Бекман постепенно вводил после перехода
цвета раствора из светложелтого в красноватый. Лобри де-Бри
не без основания находит способы Либиха и Бекмана довольно Я
ними, так как после приливания первой половины спирта происз
дит очень бурная реакция с выделением бурых паров (окислы азота]
Вследствие этого смесь может разбрызгиваться, а температура подни
.мается настолько высоко, что если во-время не подливать шоре
половины спирта, может получиться взрыв.
Лобри де-Брюен рекомендует приливать раствор ртути к спирт
а не наоборот. В этом случае реакция идет спокойнее, и можно вест
ее с 300—400 г металлической ртути сразу. Он также расходова
на 1 в. ч. металлической ртути 12 в. ч. азотной кислоты (уд. веек
1,34). Раствор ртути он нагревал до 70° и приливал его к 10 в. ч
90%-ного спирта.
Шанделон предлагает следующий режим: I в. ч. металлически
ртути растворить в 10 в. ч. азотной кислоты (уд. вес 1,4). Раствс
нагреть до 54° и выливать тонкой струей в 8,3 в. ч. 91%-hoi
спирта.
Кроме этих способов получения металлических производных rpl
мучей кислоты из спирта и азотной кислоты следует отметить ул
описанные нами выше синтезы Нефа (из нитрометаннатрия) и Шелл
(из формилхлоридоксима под действием азотнокислой ртути). Следу
методу Нефа, Джонс получил количественно гремучую ртуть, дей
ствуя разбавленной соляной кислотой на основную ртутную сол
нитрометана, а Бцддль показал, что хлорид ацетилформи/Токсима пр
длительном стоянии в водном растворе азотнокислого серебра пре
вращается в гремучее серебро, хлористое серебро и уксусную кислоту
Н .
>С1 -= NO—СО — СН3 + 2AgNOs + КО
Cl z
-> С — NOAg J- AgCl 4- СНдСООН + 2HNOS
В 1901 г. Ф. Анжелико доказал, что можно очень легко получит
гремучую ртуть из малоновой кислоты, если к раствору ртути в раз
бавленной азотной кислоте приливать концентрированный водны
раствор малоновой кислоты и несколько капель азотистонатриево^
соли. Осаждается гремучая ртуть при бурном выделении двуокис
углерода и повышении температуры. Анжелико объясняет эт
реакцию тем, что из малоновой кислоты СН.2(СООН)2 получает!
вначале изонитрозомалоновая кислота C(NOH)(COOH)2, котор.
переходит в изонитромалоновую C(NOOH)(COOH)2, а последня
•отщепляя угольный ангидрид, дает изонитроуксусную кисло-
CH(NOOH)(COOH), которая, выделяя вторую молекулу угольно!
уО
ангидрида, образует изонитрометан СН2= N^ и далее гремучу
кислоту.
80
Однако Г. Понцио указывает, что этот процесс протекает иначе,
I» именно, через изо нитрозомалоновую кислоту, изонитроуксусную
кислоту и метилнитроловую кислоту:
,/NOH ,NOH -NOH
Z Z
С— СООН С —Н -> С —Н C = NOH
. ч HNOi '
ХСООН ' СООН 4NO2
Следует привести также предложенный Г. Виландом способ,
основанный на разложении амидометилнитрозоловой кислоты. Схема
получения самой кислоты следующая:
NH —ОН
2H0N = С nh2 {диоксигуанидин) -> HON — С — N = N — С — NOH 1 NH» NH3 ,NH2 ZNO > C = NOH + C = NOH xnh!, xnh2 (оксигуанидин)
Метиловый спирт с азотной кислотой не образует гремучей кис-
ноты. Это было замечено еще Дюма и Пелиготом, а затем Шталь-
шмидтом, получившим, кстати сказать, гремучую ртуть из лигнона.
Все попытки многих исследователей получить гремучую ртуть из
монокарбоновых соединений не привели к цели.
Велер и Теодорович показали, что можно получить гремучую
ртуть окислением азотной кислотой целого ряда органических ве-
ществ, содержащих метильную группу — СН3 в соединении с одной
из следующих четырех групп:
СН.ОН: —СН< —с/ ; = Щ
хо- Ч> хо
в присутствии окислов азота .и азотнокислой ртути. Им удалось полу-
чить гремучую ртуть из диметилацсталя, из лигнона, содержащего
кроме диметилацеталя метиловый спирт, метилацетат и ацетон, а также
из диэтилацеталя, ацетальдегида, паральдегида и метальдегида.
Следует, конечно, отметить, что все новые синтетические методы,
предлагавшиеся Нефом, Велером, Виландом и др., очень дороги
или дают малый выход продукта.
§ 4. ПРИМЕСИ ГРЕМУЧЕЙ РТУТИ
Гремучая ртуть может образоваться в двух модификациях: белой
и'серой. Вопрос о разнице в цвете долгое время оставался и оста-
ется еще до сих пор спорным. По этому вопросу существуют различ-
ные точки зрения.
Бубнов—92—6
81
Каст пишет на основании своих исследований:
«Так называемая «серая» гремучая ртуть, приготовленная без до
бавления соляной кислоты, имеет слабокоричневую окраску и со
стоит из правильно образовавшихся прозрачных и светлых кристал-
лов. Углы, образуемые двумя смежными ребрами кристалла, были
измерены Риттером в 126° и 53,5°. Поэтому кристаллы относите;
к моноклинической системе. Продукт считается весьма чистым, если
он содержит 99,7—99,9% растворимой в соляной кислоте ртутно|
соли и растворяется в водном аммиаке со слабым помутнением. Не«
растворимый в соляной кислоте остаток на фильтре окрашиваете,
аммиаком в весьма слабый серый цвет и состоит, вероятно, из хло
ристой ртути от некондиционных сырых материалов».
«Так называемый белый продукт имеет почти чисто белый цве1
с весьма слабым серым оттенком и состоит не из отдельных кристал
лов, а из сросшихся в продольном направлении кристаллически!
систем. Вследствие своего сращивания кристаллы непрозрачны и
очень сильно отражают свет, почему они и кажутся белыми. Продук
менее чист, чем серый, содержит 99,3—99,4% растворимых в соляно
кислоте солей ртути и растворяется в водном аммиаке с образова
нием темного осадка. Нерастворимый в соляной кислоте остато!
(0,6—0,7%) на фильтре с аммиаком окрашивается в немного боле/
темный цвет, чем осадок серой гремучей ртути».
Таким образом Каст считает, что различная окраска гремуче^
ртути зависит, главным образом, от физических причин, т. е. объяс
няется более или менее сильным преломлением света, с одной сто
роны, и величиной кристаллов, с другой.
Такое объяснение цветности гремучей ртути не выдерживает
критики, тем более, что разница в величине кристаллов белой и се-
рой гремучей ртути невелика и что даже путем перекристаллизации
из аммиака можно получить еще большие кристаллы совершенно
белого цвета. Лучепреломление кристаллов белого и серого цвета,
как установил Бертман, одинаково.
Другие исследователи объясняют причину серого цвета приме
сями, но и здесь отсутствует единое мнение, так как даже вопрос о ко-
личестве примесей еще является спорным. О том, что белая гремуча*
ртуть загрязнена более, чем серая, наряду с Кастом указывает в свои-я
исследованиях и Филипп. Однако микроскопические исследование
кристаллов гремучей ртути показывают, что такое утверждение неч
верно: чистота того и другого продукта примерно одинакова.
Часть исследователей (И. Чельцов, Бертло и Вьель, Велер и др.
видят причину образования серой гремучей ртути в наличии при-
меси металлической ртути, которая в коллоидальном состоянии про-
низывает кристаллы и легко распознается под микроскопом, особенна
если гремучую ртуть ввести в бензол или в канадский бальзам, так
как тогда кристаллы становятся прозрачными с высоким показате
лем преломления. Точно так же при растворении гремучей ртут»
в тиосульфате натрия остаются нерастворимыми шарики металличе
ской ртути.
82
Это показывает следующая таблица, составленная по опытам Со-
лонина, Велера и Теодоровича.
Примеси Цвет гремучей ртути
к спирту к азотнокислому раствору ртути
1 3 «1 в 7 к 10 п 12 13 И 15 CuCL—0,8 г СиЕС12—I Си2С12—1,5 г С^СЦ-2 г Cu(NO2)2—3,8 г CuSO4-5H2O—5 г НС1(15° Её)—1 мл НС1( 15° Её)—1 мл Без CitgCla—0,8 г CugCla-1.3 г 0.5 г НС 1(15° Вё)+0,5гСи 1 мл НС1 (15° Её) NaCl—1 г. ВаС12-Н2О—1 примесей Белый » с незначительным сероватым оттенком Белый Светлее, чем в опыте 15 (без примесей) Цвет, как без добавки в опыте 15 Темнее, чем в опыте 15 Белый Чисто белый » » » » Серый, ио несколько светлее, чем в опыте 15 Белый, как в опыте 2 Серый
Примечание. Опыты производились со следующей рецептурой:
металлической ртути................... 50 г
азотной кислоты (d -.= 1,38) - 450 »
спирта (96%-ного) .... 500 »
Если тщательно присмотреться к условиям реакции, то можно
заметить, что серая гремучая ртуть с содержанием металлической
ргута получается в том случае, если процесс ее изготовления был не-
нормален. Если реакция образования протекает слишком медленно
вследствие большой теплоотдачи или применения холодных исход-
ных материалов, то получается серая гремучая ртуть с содержанием
металлической ртути. Объяснять цветность, базируясь на испыта-
ниях такой гремучей ртути, конечно, нельзя. Если же процесс обра-
ювания гремучей ртути протекает достаточно бурно, то образующийся
серый продукт всегда свободен от металлической ртути.
Пробой на амальгамирование золотой пластинки можно устано-
вить даже в так называемой чистой гремучей ртути независимо от ее
цветности наличие следов ртути. Однако это, как увидим ниже, не
металлическая ртуть, а ртутные соединения, отличные от гремучей
ртути.
Опыт показывает, что можно получить как серую, так и белую
гремучую ртуть с одним и тем же содержанием металлической ртути.
83
Больше того, можно получить серую гремучую ртуть и с меньшим
содержанием металлической ртути. Следовательно, наличие металли-
ческой ртути не является причиной окраски гремучей ртути.
Другая часть исследователей утверждает, что в процессе получе-
ния гремучей ртути образуется много распыленных органических
примесей, которые, попадая в кристаллы гремучей ртути, придают
ей серый цвет. При введении же даже ничтожного количества связан-
ного хлора (на 400 г металлической ртути 5 мл НС1 и 5 г меди или
соответствующее количество полухдористой меди) гремучая ртуть по-
лучается белой. Его действие сводится, видимо, к отбеливанию этих
органических примесей.
Данси 1 показал, что белая гремучая ртуть может быть получена
в присутствии малых количеств других медных солей и при полном
отсутствии галоидов. Так, при прибавлении при 50° к смеси, состоя-
щей из 25 ч. 95°-ного спирта и раствора 2,5 ч. ртути в 25 ч. HN03
(уд. вес 1,4) и 0,05 ч. меди (в виде нитрата) получается белая
гремучая ртуть. Выходы изменяются в зависимости от количества
нитрата меди. Приведенная ниже таблица показывает, что вначале
выходы остаются постоянными, затем при прибавлении нитрата
меди более 0,7 г они уменьшаются; дальнейшее прибавление нитрата,
например до 3,5 г, приводит к повышению выхода гремучей ртути,
но последняя становится загрязненной оксалатом меди.
Количество меди в виде нитрата г Выход фульми- ната г Цвет Содержание фульмината %
0,00 0,05 0.50 0,65 0,70 0,75 0.80 2.00 3,50 3,05 3,00 3,03 3,06 2,98 2.60 2,55 2,70 3,40 Серый Белый » »> » Белый, загрязнен- ный оксалатом Си 98—98,8 98,4—99,5
При замене нитрата меди нитратами никеля, цинка или кобальта
получается не чисто белый продукт, а более или менее темный.
Предполагают, что медь не образует промежуточного соединения
в виде фульмината меди, а только участвует в предшествующей ста-
дии образования гремучей кислоты. В самом деле, если подейство-
вать 0,4 г фульмината меди на 1 г ртути, растворенной в минималь-
ном количестве азотной кислоты, то получается 0,074 г серой
нечистой (93%-ной) гремучей ртути.
1 Dan si, Annali di chimica applicata, 23, 29 (1933).
84
Повидимому, наиболее верным является широко распространен-
ный взгляд, что серая окраска получается вследствие частичного обра-
зования смолистых полимеризованных продуктов бурого цвета, по-
хожих на азульмовую кислоту. В пользу этого говорит и то, что
условия реакции вполне подходящи для осмо ления. Например, раствор
5 г азотнокислой ртути в 25 мл азотной кислоты (уд. веса 1,4) окраши-
вается в бурый цвет при нагревании до 70—80° с 20 мл ацетальдегида,
а через некоторое время на стенках сосуда оседает бурое смолистое
вещество.
Продукты осмоления также образуются в процессе получения гре-
мучей ртути из ацетальдегида или изонитрозоацетальдегида, являю-
щихся, как известно, промежуточными продуктами.
Кроме этих продуктов полимеризации и металлической ртути
в технической гремучей ртути находятся и другие примеси. Если на
гремучую ртуть подействовать цианистым калием, аммиаком или
пиридином, то главная масса перейдет в раствор и при этом всегда
останется темный остаток, содержащий ртуть. Нельзя, однако, утвер-
ждать, что этот остаток состоит только из металлической ртути. Ка-
чественные исследования, произведенные А. А. Солонина, показали,
что в нем содержится и азот и углерод. Отсюда следует, что, повиди-
мому, это какое-то непрочное органическое соединение, легко разла-
гающееся при нагревании с выделением металлической ртути.
Известно также, что аммиак и цианистый калий действуют на за-
кисные соединения ртути с образованием металлической ртути. Хло-
ристая закисная ртуть дает с аммиаком хлористый мерку раммоний
с выделением металлической ртути:
Hg-Cl ,NH2
[ -|-2NH3 -* NH4C1 + Hg< J-Hg
Hg —Cl XC1
Аммиак при действии на азотнокислую закисную ртуть дает чер-
ный осадок меркураммониевой соли и металлической ртути:
Hg-NO3
2 | 4- 4NH3 -|- Н2О 3(NHi)NO3 +
Hg- NO,
Hg\
+ 0 >NH2-NO34-2Hg
Цианистый калий при действии на закисную азотнокислую ртуть
осаждает металлическую ртуть с образованием растворимой циани-
стой ртути:
Hg2(NO3l2 + 2KCN 2KNOS + Hg(CN)2 4- Hg
Повидимому, остаток от гремучей ртути после растворения ее
в пиридине, аммиаке и цианистом калии является следствием разло-
жения каких-то закисных соединений ртути, являющихся спутни-
ками гремучей ртути.
85
Если получать гремучую ртуть, пользуясь вместо спирта параль-
дегидом, то, прежде чем начинают образовываться кристаллы грему-
чей ртути, возникает осадок бесцветных листочков. Анализ этих
листочков, отфильтрованных тотчас после их образования, показал
наличие основного нитрата ртути:
Hgs(NO3)3OH
Вполне вероятно, что незначительное количество этой основной
соли получается и при изготовлении гремучей ртути обычным путем,
так как гремучая ртуть всегда содержит следы ионов N0'3.
Если растворить 5 г ртути в 36 мл очищенной от окислов азотной
кислоты (уд. веса 1,4) и затем короткое время нагревать при 70°
в 50 лит спирта, то образуется соль Купера:
NO3- Hgx 0
ОН- Hg С—
NO3-Hg/ Н
представляющая собой продукт [реакции основного нитрата ртути
с ацетальдегидом.
Не исключена возможность наличия и этого соединения в числе
примесей гремучей ртути, тем более, что концентрация отдельных
компонентов, при которых происходит образование соли Купера,
примерно соответствует (за исключением окислов азота) концентра-
ции тех же компонентов при получении гремучей ртути.
Применение для получения белого продукта полухлористой меди
или меди и соляной кислоты влечет за собой, конечно, образование,
хотя и в незначительном количестве, хлорида окисной ртути.
Дальнейшим источником загрязнений является маточный раствор,
остающийся в кристаллах гремучей ртути. Исследования кристал-
лов под микроскопом в бромбензоле или канадском бальзаме показы-
вают, что внутри кристаллов параллельно краям расположены ряды
маленьких пузырьков, в которых находится жидкость. Ратсбург
определил потерю в весе гремучей ртути при продолжительном выле-
живании. Он в течение полугода нагревал серую и перекристаллизо-
ванную из аммиака гремучую ртуть при 50—60° и нашел, что убыль
в весе серой гремучей ртути составила 3,7%, а перекристаллизован-
ного продукта — только 0,2%.
Бертман наблюдал процесс растворения гремучей ртути в аммиаке
и тиосульфате натрия под микроскопом. В результате он установил,
что те места кристаллов, в которых находятся пузырьки раствора,
растворяются быстрее.
Наконец, примесью гремучей ртути является также оксалат ртути.
Правда, если процесс образования гремучей ртути протекает вполне
нормально, то примеси оксалата не будет (об этом см. ниже). Оксалат
ртути образуется при неправильно ведомом процессе и в результате
долгого стояния готового продукта в маточном растворе.
86
[ Побочные и промежуточные {продукты процесса образования
I рсмучей ртути остаются только в незначительных количествах. Ос-
новными примесями являются, невидимому, маточные включения и
продукты их взаимодействия с гремучей ртутью в период хранения.
Содержание чистой гремучей ртути, получаемой как в завод-
mix, так и в лабораторных условиях, колеблется в пределах 98—99%.
Наиболее нежелательными примесями гремучей ртути являются
следующие:
1) металлическая ртуть и закисные ее соединения, которые, на-
ходясь в соприкосновении с медью в снаряженных капсюлях, взаимо-
действуют с металлом, об-
разуют амальгаму меди и
делают металл хрупким;
2) маточные включения
(кислотность), которые
могут разлагать составы,
изготовленные с гремучей
ртутью, и разъедать обо-
цочку;
3) случайно попавшие
нерастворимые посторон-
ние примеси (песок ит. п.),
повышающие чувствитель-
ность к внешним воздейст-
виям и вызывающие опас-
ность в обращении с сухой
гремучей ртутью при сна-
ряжении.
£ 5. ФИЗИКО-ХИМИЧЕСКИЕ
СВОЙСТВА ГРЕМУЧЕЙ
РТУТИ
Содержание фульмината Плотность
99.7 4,32
99,0 4,36
98,0 4,38
97,0 4,40
Гремучая ртуть полу- фиг' 44' Белая П>е™учая ртуть.
чается в виде белых или
серых блестящих шелковистых безводных кристаллов орто-ромби-
ческой (по Майльсу) системы (фиг. 44). Уд. вес (по Бертло) 4,42.
По Солонина, уд. вес очищенной в растворе цианистого калия белой
гремучей ртути 4,394. Уд. вес гремучей ртути, очищенной перекри-
сталлизацией из аммиака, по определе-
нию Майльса, равен 4,307 при 20°.
Причиной значительных расхождений
между результатами определения уд.
весов разными исследователями является
неодинаковая чистота испытываемых
образцов гремучей ртути.
Определение плотности гремучей
ртути различной чистоты привело Патри
к следующим результатам (см. таблицу).
87
Таким образом, чем менее чиста гремучая ртуть, тем больше ес.
удельный вес.
Форма кристалла гремучей ртути приведена на фиг. 45. Отноше-1
ние осей кристаллов гремучей ртути а : Ь: с ~ 0,712 : 1 : 1,353;
элементарная ячейка содержит 4 молекулы и характеризуется следую!
щими параметрами: а = 5,87 А, b =7,71 А, с -- 10,43 А; объем
о „
ячейки равен 440,7 А ; гравиметрическая плотность 1,22—1,25. I
Гремучая ртуть имеет сладкий металлический вкус и, как почти
все соединения, содержащие ртуть (за исключением киновари и
каломели), ядовита.
Действие гремучей ртути на организм
в основном сходно с действием металлиле!
ской ртути и вызывает более или мене!
аналогичные симптомы отравления. Пыль
Фиг. 45. Форма кристалла гремучей ртути вызывает раздражение
гпемччем dtvtii. г j у г г i
1 1 слизистых оболочек носа, гортани и глаз.
При длительном и систематическом дейст-
вии влажной гремучей ртути кожа раздражается. В тяжелых слу-
чаях заболевание напоминает мокнущую экзему.
Одним из основных условий предохранения от заболевания при
работе с гремучей ртутью является строжайшее соблюдение условий
производственной и личной гигиены и большая аккуратность в ра-
боте.
Гремучая ртуть практически не гигроскопична. Опыты определе-
ния гигроскопичности при различной относительной влажности дали
следующие результаты:
Относительная влажность Время храие- Гигроскопическая
НИЯ дни влага
50 60 0,02
80 80 0,02
100 80 0,16
Смеси гремучей ртути с ее обычными примесями обладают боль-
шой гигроскопичностью. Так, смесь ее с 20% каломели при 100%
относительной влажности обладает гигроскопичностью 4,03%. При
50 и 80% относительной влажности эта смесь не гигроскопична. Гиг-
роскопичность смеси гремучей ртути с хлоратом калия при 100%
относительной влажности равна 30,11%. При 50 и 80% относитель-
ной влажности гигроскопичность соответственно равна 0,015—0,02%.
Гумми-арабик также повышает гигроскопичность гремучей ртути. При
наличии 8% гумми-арабика гигроскопичность возрастает и становится
равной:
при 50% относительной влажности . 0,51%
»> 80% » » .... 0,81%
» 100% » « ..... 36,05%
Оксалат ртути, сопровождающий техническую гремучую ртуть.,
повышает ее гигроскопичность только при 100% относительной влаж-
ности.
Гремучая ртуть несколько растворима в воде. По Голлеману,
и 100 ч. воды растворяется:
при 12° . 0,07 г
» 49 . 0,175 г
» 100*............. 0.77 г
При длительном пребывании в водном растворе гремучая ртуть
теряет свои взрывчатые свойства. При кипячении с водой она частично
разлагается с образованием (по Либиху) окиси ртути, а затем (по
Кекуле) — фульминуровокислой ртути и углекислоты.
Растворимостью в воде одно время пользовались для очистки гре-
мучей ртути. Так, судя по отчету Яковлева, этот способ очистки при-
менялся на заводе в Карлсруэ (Deutsche Waffen und Munitions Fabri-
ken). Серую гремучую ртуть просеивали и не проходящие через сито
крупные кристаллы растворяли в кипящей воде; при охлаждении
раствора осаждались мелкие шелковистые кристаллы желтоватого
цвета. Опыты Шишкова и Гойтзема показали, что гремучая ртуть
из воды кристаллизуется с 0,5 молекулы воды.
Применять воду в качестве растворителя для очистки гремучей
ртути невыгодно. При очень медленном охлаждении раствора полу-
чают тонкие листочки разных форм, однако, это кристаллы одной си-
стемы. При быстром охлаждении водного раствора выпадают очень
мелкие кристаллы той же системы.
Несколько лучше гремучая ртуть растворяется в спирте, значи-
тельно лучше — в водном аммиаке. По Штейнеру, водный раствор
аммиака, подогретый до 30—35°, растворяет четырехкратное весовое
количество ее. Однако указание Штейнера нам кажется несколько
сомнительным. Растворимость гремучей ртути в аммиасе, по нашим
опытам, значительно ниже. Для растворения 1 ч. гремучей ртути
требуется около 30 ч. водного аммиака при 30°.
Аммиак с гремучей ртутью, невидимому, образует двойные соеди-
нения. Так, если быстро, при помешивании, внести чистую, мелко-
кристаллическую гремучую ртуть в раствор аммиака, то вначале про-
исходит полное растворение продукта, а затем большая часть фуль-
мината выделяется в виде соединения, теряющего на воздухе аммиак.
Раствор в водном аммиаке бесцветен, но не долговечен. Уже через
5 дней замечается разложение, раствор темнеет и на стенке колбы
появляется налет. Далее раствор разлагается окончательно с выделе-
нием закиси и окиси азота и металлической ртути. При нагревании
до 60° раствор разлагается с образованием мочевины, гуанидина
и т. п. Кристаллы, образующиеся посредством осаждения гремучей
ртути выливанием аммиачного раствора в разведенную уксусную кис-
лоту, большие, чисто белые, с содержанием фульмината 99,1—99,75%.
89
Способностью гремучей ртути растворяться в аммиаке иногд
пользуются в лабораториях для ее очистки, принимая, конечно, меЯ
против разложения. Приведем некоторые способы очистки гремуче,
ртути, предложенные Филиппом:
1. Белую гремучую ртуть растворяют в тридцатикратном коли-
честве аммиака и оставляют на ночь. Выпавший желтый или красно-
ватый осадок отфильтровывают, а фильтрат нейтрализуют уксусной
кислотой. Через 2 часа гремучую ртуть отфильтровывают и высушю
вают. Выход 52,5%; чистота 98,70—99,93%.
2. Условия работы те же, только нейтрализацию производя!
концентрированной уксусной кислотой. При этом происходит значи-
тельное выделение тепла (до температуры кипения). Выход 72%;
чистота несколько меньше (98,35—99,34%).
3. Белую гремучую ртуть растворяют, как описано выше, и быстро
фильтруют. Затем нейтрализуют 30%-ной уксусной кислотой при
охлаждении (температура не выше 35°). Выход 80%. Содержание
фульмината 98,60—100,02%.
Стойкость гремучей ртути после перекристаллизации из аммиака
значительно повышается. Так, помимо аналитических данных, при-
веденных выше, следует отметить, что при нагревании в течение
6 мес. при температуре 50—60° технический продукт потерял в весе
3,7%, в то время как потеря в весе очищенной кристаллизацией из
аммиака гремучей ртути оказалась равной 0.2%.
Хорошей растворимостью гремучей ртути в ацетоне, насыщенном
аммиаком, пользуются при анализе ударных составов, содержащих
гремучую ртуть.
Лангханс исследовал другие растворители '(метиламин, хино-
лин и анилин), но их растворяющая способность оказалась незначи-
тельной.
А. Майрих обнаружил, что неплохими растворителями гремучей
ртути являются моно-, ди- и триэтаноламины, причем лучшим раство-
рителем является монопродукт. Так, 1 в. ч. чистого моноэтаноламина
растворяет при 25° 2 в. ч. гремучей ртути. Растворимость падает с воз-
растанием гликолевых остатков в молекуле. Уже в 1 в. ч. диэтанола-
мина растворяется при 30° только 0,4 в. ч. гремучей ртути, а в 1 в. ч.
триэтаноламина растворяется 0,27 в. ч. гремучей ртути.
Моноэтаноламин представляет собой бесцветную, несколько вяз-
кую, уд. веса 1,22 при 16° жидкость с запахом аммиака, смешиваю-
щуюся с водой во всех отношениях. Безводный амин при стоянии
на воздухе легко притягивает влагу, несколько летуч, легко испа-
ряется при 100°. Примеси гремучей ртути в моноэтаноламине не раст-
воряются. Насыщенный и отфильтрованный раствор гремучей ртути
в моноэтаноламине (уд. вес 2,05) представляет собой густую жид-
кость, склонную при температурах выше 30° к очень бурному само-
разложению, сопровождающемуся обильным выделением светло-
серого дыма и вспученного темного продукта. Моноэтаноламин, как
аммиак и другие амины, разлагает гремучую ртуть. Через 24 часа
из раствора уже не удается выделить фульминат, так как образуются
90
Ikhc-to высокомолекулярные соединения. При разбавлении разло-
। шюго раствора водой он через некоторое время превращается
। ель.
Смесь моноэтаноламина с концентрированным раствором аммиака
юворяет гремучую ртуть в тем большей степени, чем больше амина
। ходится в смеси. 1 ч. смеси из одинаковых объемов аммиака и моно-
ыколамина растворяет 1,6 ч. гремучей ртути. Уд. вес раствора 1,7.
Хнгя растворимость гремучей ртути в этой смеси несколько меньше,
•им в чистом амине, однако, применение ее более удобно, так как
р.кгвор лучше фильтруется и не склонен к внезапному разложению.
Чистота фульмината, высаженного немного разбавленной уксус-
ной кислотой, 99,64; выход 85%.
Растворимость гремучей ртути в смеси моноэтаноламина и ацетона
Яше меньше: 1 в. ч. смеси из одинаковых объемов растворяет 0,85 в. ч.
I рсмучей ртути.
Также хорошим растворителем для гремучей ртути является пи-
ридин C5H5N, предложенный для этой цели А. А. Солонина. В 100 мл
пиридина растворяется 14,5 г гремучей ртути. При выпаривании раст-
tmpa выпадают большие кристаллы соединения пиридина с гремучей
р|утью, теряющие пиридин при обработке водой или при высушива-
нии и распадающиеся. Пиридиновый раствор более стоек, чем аммиач-
ный, но и он с течением времени разлагается. При длительном же
нагревании гремучей ртути с пиридином происходит полное разло-
жение с сильным окрашиванием раствора. Следует отметить, что
растворение гремучей ртути в пиридине сопровождается разогрева-
нием раствора; например, при размешивании 2,6 г сухой Hg(ONC)3
и 18 мл пиридина температура раствора поднимается на 6,5°.
Гремучую ртуть можно осадить из пиридинового раствора и во-
юй. Изготовленный в указанной выше пропорции светложелтый
раствор гремучей ртути в пиридине отфильтровывают и приливают
к воде, взятой в отношении 100 мл на 1 ггремучей ртути. Выделяющиеся
кристаллы промывают после фильтрования водой из расчета 200 мл
на 1 г гремучей ртути. Выход гремучей ртути около 80%, содержание
фульмината 99,7%.
Водный раствор цианида калия хорошо растворяет гремучую ртуть
С образованием двойной соли: Hg(ONC)2KCN. В концентрированном
видном растворе цианида калия растворяется приблизительно двой-
ное количество гремучей ртути. Последняя кристаллизуется вместе
с KCN; поэтому выделение из раствора цианида калия затруднительно
без дополнительной промывки разбавленной HN03. Методика пере-
кристаллизации гремучей ртути из водного раствора цианида калия
была впервые разработана Штейнером еще в 1876 г. Из раствора‘гре-
мучую ртуть можно также выделить кислотами, лучше всего разбав-
ленной азотной кислотой (Григорович, Броунсдон и Солонина).
Однако гремучая ртуть, кристаллизованная из кислот, менее чиста,
чем кристаллизованная из аммиачного раствора. Лей и Киссель поль-
зовались для выделения гремучей ртути слабой серной кислотой, что,
конечно, небезопасно. При пользовании разведенной уксусной кисло-
91
той фульминат получается в виде достаточно крупных кристалл
без наростов и после сушки обладает хорошей сыпучестью.
Водный раствор роданида калия также растворяет гремучую рту|
Хорошо растворяет гремучую ртуть водный раствор иодида кал*
но он почти моментально ее разлагает. Водный раствор тиосульф!
натрия растворяет ее с более медленным разложением. Оба эти pal
вора становятся щелочными, и это используется для объемного j
тода определения фульмината в гремучей ртути.
Таким образом ни один из описанных выше растворителей к
пригоден для целей промышленной кристаллизации гремучей рт}
а стало быть, для очистки ее: либо растворитель разлагает грему
ртуть или образует с нею комплексные соединения, либо практичес
осуществление совершенно невыгодно (вода).
Разбавленные кислоты не разлагают гремучую ртуть. Разбав;
ная азотная кислота растворяет ее, причем растворимость рас
с увеличением концентрации кислоты. Из азотнокислого раство
гремучая ртуть может быть осаждена выливанием раствора в холе
ную воду. При этом кристаллы получаются очень мелкими и такой i
чистоты, как осажденные из раствора. При нагревании же азота
кислого раствора гремучей ртути последняя разлагается с образ
ванисм нитрата ртути и углекислоты.
Концентрированные кислоты разлагают гремучую ртуть. Та
дымящаяся азотная кислота разлагает ее с образованием углекислот]
окислов азота, уксусной кислоты и азотнокислой ртути. Концентр
рованная серная кислота вызывает взрыв. При действии концентр
рованной соляной кислоты получается вначале, как установил Шелл
формилхло ридокс им:
CNO.
) Hg 4- 4НС1 -> 2 ) CNOH 4- HgCl2
CNOZ СК
который в свою очередь разлагается с образованием хлоргидра:
гидроксиламина и муравьиной кислоты (Карстаньен и Эренберг):
)CNOH + 2Н2О -> NH2OH • НС1 + нсоон
СК
Результативную реакцию можно написать следующим образр]
Hg(ONC)2 -J- 4HCI + 4Н2О -> 2NHoOH • НС1 4- 2НСООН 4- HgCi2
Сероводород и сернистые щелочи разлагают гремучую ртут
с образованием сернистой ртути; в первом случае, по данным Камб*
кроме того, образуется формотиогидроксаловая кислота:
Н V
JCNOH
HSZ
92
Вюкислота иа гремучую ртуть практически не действует. Хлор
ает влажную гремучую ртуть, образуя хлорциан, хлорпикрин
эистую ртуть.
юными щелочами гремучая ртуть разлагается легко, а слабые
и действуют на нее медленно. Для уничтожения небольших
:ств фульмината ртути можно рекомендовать пользование креп-
целочами. Анилин взаимодействует с гремучей ртутью с обра-
г~ «нмнием металлической ртути, фенилмочевины и дифенилгуаиидина.
С т омологами анилина реакция идет лишь при нагревании.
Интересная реакция, указывающая на родство гремучей кислоты
I синильной кислотой, происходит при действии гремучей ртути на
I бензол в присутствии хлористого алюминия, содержащего следы воды.
[ Реакция идет с образованием бензальдоксима:
С6Нв 4- С = NOH -> CflHsCH(NOl I)
При применении безводного хлористого алюминия образуется
нитрил бензойной кислоты, — несомненно, как продукт ангидриза-
ции первоначально образовавшегося бензальдоксима:
CfiH6C = N C6H6CN 4- Н2О
I I
н он
К различным металлам гремучая ртуть относится различно. Этот
вопрос является чрезвычайно важным, так как гремучая ртуть всегда
применяется впрессованной в металлическую оболочку.
Впервые упоминают о взаимодействии гремучей ртути с метал-
лами Гладстон, Уаррен и Чельцов. Наиболее полно этот вопрос ис-
следовали Солонина, Гедымин и др.
Упоминания первых исследователей о взаимодействии гремучей
ртути с медью не были подтверждены достаточными экспериментами.
Чельцов, например, указывает, что порошкообразная медь при на-
гревании с водой и гремучей ртутью замещает ртуть, которая выде-
ляется в свободном состоянии по уравнению:
Hg(ONC)2 4- Си -> Cu(ONC)2 4- Hg
В начале 90-х годов прошлого столетия Уаррен, действуя на мед-
1 ные стружки горячим водным раствором гремучего серебра, получил
один из фульминатов меди. Раствор последнего он разлагал электро-
литическим методом и выделял медь. Однако новых фульминатов он
в чистом виде, невидимому, не получал и их свойств не исследовал.
При взаимодействии гремучей ртути с медью в присутствии воды
(по Солонина) образуется смесь гремучей меди, гидрата окиси меди
и углекислой меди:
nCu(ONC)2 - mCu(OH)2 • pCuCO3
I где n — 98 (приблизительно), a m и P вначале не более 2—3, а затем
увеличиваются. Это соединение нестойкое и разлагается даже при
Комнатной температуре, особенно в присутствии воды. Образованию
93
его благоприятствуют металлическая ртуть и вода, обычно naxoj
щисся в качестве примеси в техническом продукте. Сухая гремуч
ртуть при обыкновенной температуре почти не реагирует с метг
лами.
Исследования Паникова и Сухова показали, что при взаим—
действии меди с гремучей ртутью образуется химически стойки!
основной фульминат меди состава Cu(ONC)2Cu(OH)2, обладающ^
пониженной чувствительностью к удару и нагреву, но зато рези
повышенной чувствительностью к трению. Для образования осно|
ного фульмината необходимо обязательное присутствие влаги. В бе
водной среде (толуол) он не образуется.
Если внести 3 г гремучей ртути и 6 г медных опилок, очищеннь
от железа магнитом и обезжиренных эфиром, в колбочку с 30 ж
дестиллированной воды и оставить в термостате при 30°, то уже чер<
сутки вода в колбочках окрашивается в зеленый цвет, а на повер>
ности ее появляется тонкий слой зеленого вещества. Если эти koj
бочки выдержать в термостате ври 30° до 30 суток без взбалтываний
затем снять образовавшуюся на поверхности зеленую корочку, вы-
сушить ее при 45° в продолжение 1—2 час. в эксикаторе над хло|
ристым кальцием, то окажется, что полученное вещество содержит;
через 5 суток
• Юм
• 15 о
• 20 •
fCu(ONC)8Cu(OH), .
|CuCO3 • Cu(OH)„ . . .
lCu(ONC)» - Cu(OH), .
tCuCO3 • Cu(OH).,
1Cu(ONC)3Cu(OH)2 . . .
|CtiCO3 - Cu(OH).,“ . .
KT:(C)NC)2 - Cu(OH)2 . .
(CuCO3 • Cu(OH)0 .
77,3%
22,7%
83,5%
16,5%
81.4%
18,6%
74,8%
25,2%
Если снарядить смоченные внутри водой оболочки капсюлей-дето-
наторов сухой гремучей ртутью и хранить их при обыкновенной тем-
пературе, то уже через 96 час. повысится чувствительность гремучей
ртути к трению, что было установлено опытами на массете.
Исследование капсюлей-детонаторов, хранившихся в течении
16—18 лет в обычных условиях (изготовления 1914—1916 гг.), под-
твердило возможность образования основного фульмината меди. Так,
например, в одном из капсюлей, снаряженном в 1916 г. и находив-
шемся до 1928 г. во взрывателе, ввернутом в снаряд, а затем в не-
отапливаемом помещении до 1932 г., гремучая ртуть по наружному
виду в изломе оказалась зеленого цвета с падающей интенсивностью
позеленения от стенок латунной гильзы к центру. При разрядке
капсюля было обнаружено, что гремучая ртуть на всем протяжении
гильзы имеет зеленый цвет, а латунная оболочка в месте соири-1
косновения ее с гремучей ртутью значительно амальгамирована;
и настолько разрушена, что при разрядке взрывателя капсюль дал
излом в месте нахождения гремучертутного заряда. Исследование
извлеченной из капсюля гремучей ртути показало содержание в ней
меди около 6—6,1%; чувствительность к трению оказалась значительно
выше, чем у гремучей ртути.
94
Все эти опыты позволяют сделать следующие выводы:
1) в условиях негерметического хранения гремучертутного капсю-
пя-детонатора в медной или латунной оболочке гремучая ртуть вза-
имодействует в присутствии влаги с оболочкой, образуется основной
фульминат меди и происходит обильное выделение металлической
’ртути;
2) гремучая ртуть благодаря образованию основного фульмината
меди становится значительно более чувствительной к трению.
Так как гремучая ртуть применяется исключительно в оболочках
и;। меди или ее сплавов (латунь или мельхиор), то естественно, что
вопросу изучения взаимодействия меди и гремучей ртути уделяется
Гюлыпое внимание. Не меньшее внимание уделяется изучению воз-
можности замены меди и ее сплавов в капсюлях-детонаторах или за-
щите тем или иным способом оболочки от действия ртути.
С магнием гремучая ртуть вступает в бурное взаимодействие со
значительным выделением тепла даже при небольшом количестве
поды. При этом выделяется аммиак и образуется невзрывчатое, раст-
воримое в воде, магниевое соединение.
С алюминием влажная гремучая ртуть реагирует очень бурно,
с образованием объемистой массы, состоящей, главным образом, из
окиси алюминия, гремучей ртути и металлической ртути. При реак-
ции, в начале ее, обычно появляется запах синильной кислоты.
С цинком влажная гремучая ртуть реагирует медленнее. Причем
образующийся при этом фульминат цинка взрывает не хуже грему-
чей ртути. Е. Деви показал, что при действии 1 ч. Hg(ONC)2 на 2 ч.
цинка на холоду в присутствии воды образуется гремучий цинк,
кристаллизующийся в виде ромбических таблеток. Со временем под
действием воды фульминат цинка разлагается и переходит в гидрат
окиси цинка.
На никель ни влажная, ни сухая гремучая ртуть не действуют.
На олово сухая гремучая ртуть не действует.
Техническая же гремучая ртуть в присутствии влаги действует
на олово, поверхность которого покрывается матовыми серыми пят-
нами, невидимому, представляющими собой амальгаму олова. Дейст-
вие это незначительно и увеличивается с увеличением в технической
гремучей ртути примеси металлической ртути и ее закисных соеди-
нений.
Со свинцом гремучая ртуть взаимодействует слабо; это нисколько
не сказывается на технических свойствах металла. Повидимому,
поверхностный слой свинца образует с имеющейся во влажной тех-
нической гремучей ртути металлической ртутью амальгаму. Затем
свинец вытесняет из гремучей ртути металлическую ртуть и
образует фульминат свинца, который гидролизуется водой с обра-
зованием нерастворимых основных солей свинца, покрывающих по-
верхностной защитной пленкой основную массу свинца, вследствие
чего прекращается дальнейшее взаимодействие металла с гремучей
ртутью.
95
1 ремучая ртуть при обыкновенной (комнатной) температуре весы
устойчива. При нагревании гремучей ртути до температуры вспыш!
разложение ее ускоряется по мере возрастания температуры. ПродО|
жительное нагревание гремучей ртути при температуре 50° и выи
вызывает ее разложение. При этом следует отметить, что разложен]
подвергаются в первую очередь примеси гремучей ртути. НаибоЯ
быстрое разложение без взрыва наблюдали Гесс и Дитль при нагр
вании до 90—95°, причем через 35—50 час. гремучая ртуть значнтелы
потеряла свои взрывчатые свойства, а после 75—100 час- преврат
Фиг. 46. Зависимость между давлением газа и временем
выдержки.
Фармер исследовал превращения чистой гремучей ртути при
нагревании в интервале 60—100°. Он показал, что разложение про-
исходит тем быстрее, чем выше температура, и что выделяющиеся]
газообразные продукты разложения состоят почти исключительно из
двуокиси углерода.
При разложении гремучей ртути наблюдается отчетливый индук-
ционный период, при котором выделение газа очень мало. Продолжив
дельность индукционного периода зависит от качества гремучей
ртути. Так, например, обыкновенная серая гремучая ртуть при 80°
имеет индукционный период продолжительностью около 80 час. Бе-
лая гремучая ртуть имеет больший индукционный период. В конце
индукционного периода кристаллы Hg(ONC)2 разламываются и окра-
шиваются, далее наступает период заметного ускорения разложения,
кристаллы становятся совершенно коричневыми. Затем скорость
разложения остается постоянной.
Поведение белой и серой гремучей ртути и в послеиндукционном
периоде различно. Разложение белой гремучей ртути происходит
с большим ускорением и конечная скорость ее разложения больше,
чем у серой гремучей ртути.
96
1 la фиг. 46 изображены кривые разложения по данным измерений
кишения выделяющихся при разложении 0,002 г гремучей ртути
гнзов (по данным В. Е. Гарнера и X. Р. Хильса) при температу-
рах 100°, 105° и 110°.
Лангханс исследовал коричнево-желтый трудно воспламеняю-
щийся порошок, получающийся в результате нагревания гремучей
р1ути в течение 72 час. при температуре 82°, и нашел в нем содержа-
ние ртути 76,4% вместо 70,4%. Он установил, что увеличение содер-
*|(.'1ния ртути в порошке пропорционально времени нагревания.
При температурах выше 100° гремучая ртуть может взрывать.
Однако, по данным Гойтзема, можно разлагать гремучую ртуть при
осторожном и медленном нагревании до 132°.
Опыты Солонина показали, что при нагревании гремучей ртути
при постоянных температурах получение вспышки в значительной
степени зависит от качества (в первую очередь от чистоты) продукта.
Нагревание при 134—135° белой гремучей ртути, очищенной кри-
сталлизацией из раствора цианистого калия, вызывает вспышку обычно
через 36—-40мин., а серая гремучая ртуть с содержанием 0,4% металли-
ческой ртути и 0,7% щавелевой кислоты дает вспышку через 20—
22 мин. При нагревании при 126,5° очищенный кристаллизацией из
раствора цианистого калия белый продукт взрывает через 1 ч. 14 м.—
I ч. 16 м. Серая гремучая ртуть при этих условиях не взрывает, даже
при нагревании ее в течение 12 час.
Патри и Лаффит показали, что при температурах ниже 139,5°
не происходит воспламенения, но процесс выделения газов значи-
тельно ускоряется.
Осторожно нагреваемая и разлагающаяся вследствие этого грему-
чая ртуть становится менее чувствительной к удару. Так, например,
при нагревании гремучей ртути при 133°:
а) от 0 до 20 мин. — при ударе еще происходит детонация;
б) от 20 до 50 мин. — характер взрыва указывает на уменьшение
силы гремучей ртути;
в) от 50 до 80 мин. — продукт горит, но не детонирует от удара;
г) свыше 80 мин. — образуется продукт нечувствительный к удару;
д) свыше 100 мин. — продукт даже не воспламеняется от луча
огня.
Таблица 13
Давление Время выделения газов в часах при 60° [Hg(ONC)B = f г]
1 CJlf 2 см- 3 см* 4 см3 5 см3 6 см-
В вакууме . ... 868 925 970 1006 1039 1069
» » ... 842 900 944 983 1020 1053
» » .... 945 1008 1052 1094 1135 —
500 мм рт. ст. ... 960 1026 1073 1115 1156 1193
Бубнов —92—7
97
Фармер, разлагая гремучую ртуть при 60° в вакууме (остаточное I
давление 5 мм рт. ст.), доказал, что давление мало влияет на ско-1
рость разложения.
Выделение газов происходит в вакууме несколько быстрее, чем I
под давлением 500 мм рт. ст., что видно из табл. 13.
Патри и Лаффит установили, что при нагревании между 139’ и
172° обнаруживается критическое давление Рг, ниже которого взрыв
уже более невозможен. Это давление всегда ниже 45 мм рт. ст. и умень-1
шается при повышении темпера-1
туры, как это видно из табл. 14. 1
Время задержки взрыва гре-1
мучей ртути при нагревании не
зависит от давления только в том
случае, если сравниваемое давле-1
ние выше критического.
При температуре выше 172° I
взрывы происходят вне зависимости от давления.
При обычной температуре гремучая ртуть может храниться в сухом
состоянии в течение длительного времени. Считают, что она практик I
чески не претерпевает при этом никаких изменений в своих свойствах. I
Свет также оказывает некоторое влияние на гремучую ртуть, ]
вызывая отчетливое выделение газа. Фармер установил, что белый
и серый продукты, хранившиеся на свету, выделили следующее ко-
личество газа в миллилитрах (табл. 15):
Таблица 15
Таблица 14
ГС 145 160 167 172
Рс мм 40 28 13 0.2
Время хранения в часах 80 160 240 320
Белая гремучая ртуть (За). 0,11 0,23 0,30 0,37
Серая » » (3 г) . . . 0.04 0.08 0,11 0,14
Как только препараты помещались в темноту, выделение газа |
прекращалось. У образцов, подвергнутых действию дневного света I
в продолжение пяти недель в летнюю ясную погоду, скорость выделе-
ния газа при последующем нагрейании при 80° повысилась.
Гремучая ртуть, освещаемая в течение продолжительного времени
солнечным светом, окрашивается в слегка желтоватый цвет без види-1
мого изменения остальных свойств. Будучи подвергнута действию
ультрафиолетовых лучей, она по прошествии нескольких часов при-
нимает коричнево-черную окраску (независимо от начального цвета ?
продукта).
Патри и Лаффит после месячной экспозиции с ежедневным тща-
тельным перемешиванием массы гремучей ртути получили почти
черный продукт, в котором анализом было установлено содержание I
ртути, равное содержанию ее в исходном продукте. Повидимому,
под влиянием ультрафиолетовых лучей происходит изомеризация
или полимеризация с сохранением той же кристаллической формы.
98
Полученный продукт растворим в водном растворе гипосульфита
«и щелочной реакцией и в водном растворе цианистого калия, но
в противоположность гремучей ртути не вполне растворяется в ам-
миаке. Этот продукт легче воспламеняется, чем чистый фульминат.
Чувствительность к удару облучаемой ультрафиолетовыми лучами
। ремучей ртути убывает. При облучении свыше месяца гремучая
|иуть становится совершенно нечувствительной к удару.
§ 6. ВЗРЫВЧАТЫЕ СВОЙСТВА ГРЕМУЧЕЙ РТУТИ
Гремучая ртуть при взрыве разлагается по уравнению:
Hg(ONC)3 > Hg-J- 2СО + N3 J- 116 Кал
Не исключена также возможность разложения ее частично и по
v равнению:
Hg(0NCj2 HgJ-CO2 + C + N2
На основании же анализа продуктов взрывчатого разложения гре-
мучей ртути в калориметрической бомбе можно выразить уравнение
се разложения следующим образом:
Hg(ONC)2 - - Hgnap + 0,05C03+ 1,9CO + 0,05C+N2+ 105,2 Кал
Температура взрыва гремучей ртути, вычисленная на основании
лого уравнения, равна 4450°С, объем газов Vo=311 л/лг, уд. энергия
/ = 5350 л - ат/кг; скорость детонации гремучей ртути около
4500 м/сек.
Гремучая ртуть — соединение эндотермическое и его молярная
теплота образования равна 62,9 Кал.
Импульсом для взрыва гремучей ртути обычно служит накол жа-
лом, а также удар бойка или луч огня. Гремучая ртуть чрезвычайно
чувствительна к пла-
мени и легко воспламе-
няется как от луча огня
бикфордова шнура, так
и ot электрической иск-
ры; только ацетиленид
серебра обладает более
легкой воспламеняемо-
стью. Гремучая ртуть
легко взрывает от незна-
чительного удара (0,08
кг/см2). На копре Велера
чувствительность ее к
удару определяется пре-
Таблица 16
Зависимость между температурой вспышки
гремучей ртути и временем нагревания
Г "сек Г ’сек
235 0 205 8,2 175
230 1.5 200 12,2 170
225 2,1 195 18.0 165
220 2.9 190 23,1 160
215 4,0 185 34,5 155
210 5Д 180 52,8
‘сек
85,0
135,0
194,0
339.0
510,0
Отказ
делами 55—85 мм. Температура вспышки лежит между 160—165° при
обычном способе ее определения. При определении же температуры
вспышки бросанием в сосуд с постоянной температурой и продолжи-
тельностью действия 5 сек. Велер получил температуру вспышки 215°.
Температура вспышки серой гремучей ртути, определенная при
возрастающей со скоростью 20° в минуту температуре ванны, оказы-
99
вается равной 169°, а белой 172—175°. Зависимости между темпер,
турой вспышки и временем нагревания навески белой гремучей ртут
tc при постоянной тел
пературе ванны при
ведены на фиг. 47 и |
табл. 16.
Чувствите ль носи
гремучей ртути к тре
нию зависит от каче
ства трущих поверх
ностей; так, она нам
более чуветвительц
Фнг. 48. Кривая зависи-
мости чувствительности
гремучей ртути к удару от
величины кристаллов.
Фиг. 47. Кривая температур вспышек гремучей ртути- при Трении межД¥
железными поверхно
стями. При трении между свинцовой и железной поверхностям!
чувствительность ее значительно меньше, а при трении между дере
вянными поверхностями —- совсем незначительна.
Чувствительность гремучей ртути к удару и трению изменяется
с ростом ее кристаллов. На фиг. 48 приведена кривая зависимости
чувствительности гремучей ртути к удару
от величины кристалла, выведенная исходя
из верхних и нижних пределов чувстви-J
тельносги на копре Велера для белой
гремучей ртути, запрессованной в кол-
пачки капсюля Наган под давлением
1000 и 400 кг/слГ2. В первом случае были
взяты навески в 0,02 г, а в последнем
0,015 г. Падающий груз копра весил 465 г.
По абсписсе отложена величина кристал
лов, по ординате—высота падения груза..
На температуре вспышки, а стало быть.'
и на способности воспламеняться от нагре-
вания величина кристаллов существенны
не отражается.
Несмотря на достаточно высокую чув-1
ствительность к удару и трению, гремучую
ртуть можно безопасно спрессовать, даже!
под весьма большим давлением (до 7000 ат)
конечно, если давление прилагается не<
динамически, а постепенно.
Случаи взрывов гремучей ртути наблю-
даются при прессовании ее в смеси с ан-
тимонием (играет роль трение).
Гремучая ртуть, свободно насыпанная
в небольшом количестве (до 2 г) и вое
пламененная, дает вспышку с характерным глухим звуком и н
обладает инициирующими свойствами. Будучи слегка подпрессован-
ной, она взрывает с сильным звуком и может инициировать б риза нт
100
|n.t<* ВВ. Будучи же спрессованной под очень большим давлением
। рсмучая ртуть, воспламеняясь от простого начального импульса
(пуча огня), только выгорает, но не детонирует и не может иниции-
ровать бризантные ВВ.
Предельное давление, при котором наблюдается явление «мерт-
запрессовки», различными авторами дается разное. Велер счи-
। ют, что действие гремучей ртути при давлении запрессовки свыше
Фиг. 49. Зависимость плотности спрессованной гремучей
ртути от давления прессования.
Табл ина 17
Давление прессования ат Плотность е/см3 Давление прессования ат Плотность г/см3
25 2,35 300 3,50
50 2,55 400 3,60
75 2,80 600 3,70
100 3,00 800 3,90
200 3,30 1200 4,05
200 ат ослабевает; по Форгу, гремучая ртуть достигает «мертвой за-
прессовки» при давлении прессования в 700 ат, а по Штеттбахеру,—
при давлении прессования около 2000 ат. Однако Штеттбахер делает
оговорку, что выражение «мертвая запрессовка» не следует понимать
буквально, так как способность к детонации в таких случаях только
затрудняется и может быть снова восстановленной полностью, если
вместо луча огня от
бикфордова шнура дать
импульс требуемой силы
(в чем он безусловно
прав). Ниже мы остано-
вимся на причине этого
внешнего расхождения
в цифрах.
Изменение плотности
впрессованной в оболоч-
ку капсюля-детонатора
№ 8 серой гремучей
ртути с 2,05% примесей
в зависимости от давления прессования (прессование в два слоя)
приведено на фиг. 49 и в табл. 17.
Впрессованием гремучей ртути в капсюльные оболочки понижается
ее чувствительность к механическим импульсам (удар, трение, сотря-
101
сение и т. п.), благодаря чему в значительной степени уменьшается
опасность ее воспламенения от инерционного смещения чашечки
капсюля. Но так как значительных давлений применять нельзя
не рискуя получить отказ в действии капсюля-детонатора, то при-
ходится на практике ограничиваться давлениями запрессовки no-j
рядка 250—350 ат.
Возможность перепрессовки является одним из существенных не-1
достатков гремучей ртути. При малых давлениях прессования нет
гарантии от преждевременных разрывов снарядов при ударе о броню,
а в случае высоких давлений форсирования—и при выстреле в канале
ствола орудия.
Велер и Матгер исследовали влияние прессования под давлением
от 100 до 2000 кг [ел? в оболочке капсюля-детонатора № 8 и реги-
стрировали взрывы этих
капсюлей в свинцовом
цилиндре диаметром’80
мм по методу Трауцля.
При заряде капсюля в 2 г
гремучей ртути они по|
лучили следующие ре-
зультаты (табл. 18).
Диаграмма расшиб
рения в зависимости от
давления, приведенная
на фиг. 50, показывает]
небольшое уменьшение
Таблица 18
Давление прес- сования кг/сл!2 Плотность г/см9 Расширение мл
100 1,923 30,6
400 2,564 28,6
800 2,984 26,1
1200 3,211 25.6
1600 3.228 25,4
2000 3,358 25.6
расширения с возра-
станием плотности гремучей ртути отJ 2,98 до 3,35, т. е. до давле-
ния в 800 кг(см2. Дальнейшее увеличение давления до 2000 кг/см2
Фиг. 50. Диаграмма расширения в
свинцовом цилиндре в зависимости от
давления прессования.
не оказывает влияния на
величину расширения гне-
зда свинцового цилиндра.
Таблица 19
Вес заряда г Расширение мл
0,5 4,95
1.0 11,15
1.5 16,35
2.0 25.60
Изменяя заряды гремучей ртути, Велер получил следующие ве-
личины расширения камеры свинцового цилиндра (табл. 19).
Он пришел в результате этого к заключению, что расширение
камеры почти пропорционально весу заряда гремучей ртути (фиг. 51).
102
Фиг. 51. Зависимость расширения в свин-
цовом цилиндре от веса заряда гремучей
ртути.
Бертло и Вьель взрывали в стальной бомбе при разных плотностях
.лряжания гремучую ртуть при помощи накаленной электрическим
гоком проволоки и опре-
делили давления, разви-
ваемые в моменты взры-
ва. Они установили, что
давление при взрыве
гремучей ртути меньше,
чем при взрыве пиро-
ксилина (табл. 20).
Литературные данные
о предельных иниции-
рующих зарядах гре-
мучей ртути различны:
некоторые из них сведе-
ны в табл. 21 (стр. 104).
Наиболее достовер-
ными и вполне соот-
ветствующими нашим
исследованиям являются
данные Мартина. Разно-
образие в полученных
величинах предельных
зарядов объясняется
следующим:
1. Величина кристал-
лов гремучей ртути, как
показали исследования
Велера и Бертмана, от-
ражается на инициирую-
щей способности: с уве-
личением размеров кри-
сталлов инициирующая
способность повышает-
ся- В своей работе они
пользовались образцами
гремучей ртути, кри-
сталлизованными из азотной кислоты, водного аммиака, водного
раствора цианистого калия и пиридина, и определяли предельный
инициирующий заряд по отношению к 0,85 г кристаллизованного
тротила, запрессованного в медной оболочке капсюля-детонатора №8
под давлением 250 кг/сж2. Ими было установлено, что предельный
заряд технической гремучей ртути равен 0,23 г и что изменение пре-
дельного заряда в зависимости от величины кристаллов подчиняется
закону гиперболы. Данное положение иллюстрируется на фиг. 52,
где на оси абсцисс нанесены размеры кристаллов гремучей ртути,
определенные по большой диагонали кристалла, а по оси ординат —
предельные заряды.
Таблица 20
Давления, кг
I при взрыве
1 гремучей ртути
Плотность
заряжания
при взрыве
пироксилина
0,1 477 1085
0.2 1730 3120
0,3 2697 5575
0,4 4272 8745
103
Таблица 21
Предельные инициирующие заряды гремучей ртути
Предел ьный инициирующий
заряд гремучей ртути для
тетрила г пикриновой кислоты г тринитро- толуола г три нитро- анизол а г тринитро- ксилола г Автор Примечание
0.29 0.30 0.36 0,37 0,43 Мартин, Велер и Мартин Давление прес- сования 1100 ат
—- —. 0,25 — — Велер .
0,40 — 0,35 — — Рейнско-Вестфальское акционерное общество, Тройсдорф
0,24 0 21 0,26 — — Тейлор и Ринкенбах —
0,24 — 0,26 — — Тейлор, Копе Давление прес- сования 200 ат
0.25 — 0,30 — — Л » Давление прес- сования 400 ат
0,42 » 0 С укороченной чашечкой, давле- ление прессова- ния 200 ат
0.22— 2,7 0 25 — — Нейгофф В различных условиях
— — — Брунсвнг —
— —‘ 0,21 — Нансен С усиленной ча- шечкой
0,36 0,36 0,38 — 0,62 Кестер Давление прес- сования 200 ат
в зависимости от величины кристаллов.
Причина изложенного, видимо, кроется в незначительной плот-
ности заряжания мелких кристаллов, окруженных при незначитель-
ных давлениях сравнительно большими воздушными прослойками-
I 04
!!<гному, если мы увеличим давление прессования даже весьма
шч|кокристаллических продуктов, полученных кристаллизацией
И । азотной кислоты и цианистого калия, до 600 кг} см2, то предель-
ный инициирующий заряд уменьшится с 1 до 0,27 г.
' К аналогичным выводам пришел и Кестер. Он исследовал только
1ри сорта более или менее крупной серой гремучей ртути (больше
И. 13 мм, от 0,13 до 0,10 мм и меньше 0,10 мм) по их действию в отно-
шении I г тротила (плотность 1,35), запрессованного в медной гильзе
капсюля-детонатора № 8. Гремучая ртуть им прессовалась отдельно
И медную чашечку под давлением 200 кг/см2 слоями по 0,2 г. В ре-
iy цитате опытов он по-
мучил следующие дан-
ные (см. таблицу).
2. Следующий фак-
lop, влияющий на ини-
циирующую способность
I рсмучей ртути, — это
II «отность оболочки и
ш'обенно покрытия (ча-
шечки), в которые она
и к лючена. Установлено,
Величина кристаллов мм Предельный ини- циирующий заряд
Больше 0,13 . . | 0,34
От 0,13 до 0,10 0,38
Меньше 0,10 0,41
•по гремучая ртуть хорошо и безотказно инициирует бризантные ВВ,
йплько будучи заключена в прочную металлическую гильзу под опре-
деленным давлением. Если оболочка недостаточно прочна или если от-
сутствует чашечка, прикрывающая гремучую ртуть в капсюле сверху,
п» количество гремучей ртути, необходимое для вызова детонации
иризантного вещества, значительно выше. Если предельный иници-
ирующий заряд гремучей ртути по тротилу равен при обычном спо-
ите его определения около 0,36 г, то, находясь в капсюле без чашечки,
предельный заряд возрастает до величины более 1,2 г. Из опытов же
Пейгоффа вытекает еще большая разница предельных зарядов гре-
мучей ртути по тетрилу, а именно:
в капсюле с чашечкой . . 0,50 г
» » без чашечки . . 2,7 г
Роль металла оболочки, как это видно из табл. 22, также велика-
□то было установлено испытанием серой гремучей ртути с содержа-
нием 2,05% примесей по тротилу (плотность 1,35), запрессованному
и капсюли-детонаторы №8. Чашечка снаряжалась отдельно: грему-
чая ртуть запрессовывалась слоями по 0,2 г в каждом слое под дав-
нснием 200 ат.
То же самое следует и из данных предельных инициирующих
ырядов гремучей ртути по тетрилу, определенных Нейгоффом
(табл. 23).
[ 3. Величина предельных инициирующих зарядов серой гремучей
ртути по тротилу в изложенных выше условиях показывает, что
К увеличением давления прессования предельный заряд также уве-
личивается. В табл. 24 и на фиг. 53 дано наглядное представление
об этом.
105
Таблица 22
Материал Предельный инициирую- щий заряд Примечание
гильзы чашечки
Медь, заключенная в стальную трубку, тол- щиной 3,5 мм Медь » » Алюминий » Медь » вязкая Латунь Железо Алюминий Медь АЛЮМИНИЙ 0,33 0,38 0,27 0.43 0,24 0.38 0,40 _>0,75 Стальная трубка плотно обхватывал гильзу Особого изготовле НИЯ
Таблица 23
Материал Предельный иницииру ющи й заряд
гильзы чашечки
Железо Железо 0.22
Латунь Латунь 0,24
Медь Медь 0,50
Алюминий Алюминий 0,70
Медь Латунь 0,25
Таблица 24
Давление прессования кг/см2 Плотность гремучей ртути г/см& Предельный инициирую- щий заряд 2
25 2.37 0.28
75 2,78 0,29
100 2.97 0,31
200 3,30 0,38
265 3,40 0,40
400 3.61 0.47
800 3,85 0.76
1600 Выгорания
106
Фиг. 53. График зависимости предельного за-
ряда гремучей ртути от давления прессования.
4. |Способ прессования гремучей ртути в капсюль также играет
немалую роль. Так, если
iy же серую гремучую
ргуть прессовать не
слоями по 0,2 г отдельно
и чашечку» а прессовать
навеску вместе с чашеч-
кой непосредственно в
гильзу, то предельный
заряд уменьшится с 0,38
до 0,29 г. Причина столь
значительного повыше-
ния инициирующей спо-
собности кроется в том,
что при прессовании
в чашечку слоями по-
являются плоскости раз-
дела, неблагоприятно отражающиеся на распространении детонации.
5. Кроме того, величина предельного заряда зависит от длины
чашечки, погружающейся во вторичный заряд капсюля-детонатора.
Опыты Кестера с серой гремучей ртутью показали следующие изме-
нения предельного заряда с увеличением длины чашечки при том
же способе снаряжения (табл. 25).
Таблица 25 Таблица 26
Погружение ча- шечки во вторич- ный заряд мм Предельный ини- циирующий заряд Высота чашечки мм Предельный ини- циирующий заряд г
0.0 015 1,6 4,0 6,0 0,38 0.38 0,36 0,32 0,30 3,5 4,5 5,0 5,5 6.0 0,80 0,85 0,70 0,65 0.65
То же самое следует и из опытов Сухова, повторившего опыты
Кестера и определившего предельные заряды белой гремучей
ртути, спрессованной под давлением 400 ат, по 1 г тетрила, спрес-
сованного под давлением 500 ат.
Результаты этих опытов представлены в табл. 26 и на фиг. 54.
Стало быть, чем длиннее чашечка (до предела в 5,5 мм), тем
меньше предельный инициирующий заряд гремучей ртути.
6. Степень чистоты гремучей ртути отражается незначительно
на величине предельного заряда, что видно из данных табл. 27.
Примесь воды сильно ослабляет взрывчатые свойства и умень-
шает чувствительность гремучей ртути, в особенности в непрессо-
ванном состоянии. Это становится заметным при 5% влаги. При со-
держании 10% влаги гремучая ртуть в небольшом количестве может
107
сгореть на открытом воздухе без взрыва, при содержании же 30"В
влаги гремучая ртуть даже не загорается. Капсюлем-детонатором и
такая гремучая ртуть может быть взорвана, что подтверждается
данными О. Гуттмана. Чувствительность гремучей ртути также умснь-1
шается в результате добавки жиров, глицерина или парафина, чем
пользуются при снаряжении детонирующих шнуров.
_________________________________________________т а с л н ц а 27 I
Цвет гремучей ртути Количество при- месей в гремучей ртути Предельный мни- | циирующий заряд г 1 Примечание
Белый . . 0.05 0.27
» . . 0,05 0,24 Круппокристалли- 1 ческпй продукт
Серый - . . . 0,75 0,26
»> . . 1,40 0,29
2.05 0,29
3,05 0.29
» 3,44 0,30
Понижение температуры гремучей ртути также отрицательно ска- j
зывается на взрывчатых ее свойствах. При температуре жидкого!
воздуха инициирующая ее
способность становится не-
значительной, и поэтому кап- |
сюли, снаряженные гремучей
ртутью, непригодны для
взрыва оксиликвитов.
Гремучую ртуть обычно
хранят под водой. Однако!
следует отметить, что в воде
она подвергается некоторому
изменению, что видно из табл.
28, в которой собран опыт-
ный материал по исследо-
ванию гремучей ртути ди !
хранения под водой, а также
после двухнедельного и ме-
сячного хранения под водой. 1
Из таблицы видно, что
Фиг. 54. Изменение предельного заряда ПР 1 хРанении П0Д водой в
гремучей ртути в зависимости от высоты течение даже сравнительно
чашечки. небольшого промежутка вре-
мени качество гремучей ртути
ухудшается. С этим приходится мириться, так как хранение гремучей
ртути в сухом виде, несмотря на то, что при этом не наблюдается прак-
тически заметных изменений ее качества, является чрезвычайно
опасным и недопустимым с точки зрения техники безопасности.
108
Таблица 28
Аналитические определения До хране- ния После хранения
через 15 дней через месяц
Содержание фульмината - . - 99,34% 99,17% 99,05%
Содержание щавелевой кислоты .... 0,06% 0,12% 0,12%
Содержание нерастворимых примесей . 0,39 К олз»„ 0,64%
Образование амальгамы ........ 3 мин- 1 м. 45 с- 1 м. 30 с-
Температура вспышки 167° 165° 161°
Перевозка гремучей ртути на большие расстояния не практикуется
из-за ее значительной чувствительности, поэтому она перерабаты-
вается на месте производства.
§ 7. ИСХОДНЫЕ ПРОДУКТЫ ДЛЯ ФАБРИКАЦИИ ГРЕМУЧЕЙ РТУТИ
Зав.одские способы получения гремучей ртути лишь незначительно
отличаются от лабораторных, так как выработка значительных коли-
честв ее в один прием не допускается и в заводском масштабе.
Исходными материалами для производства гремучей ртути слу-
жат металлическая ртуть, азотная кислота и этиловый спирт, а для
получения белого продукта *, кроме того, — соляная кислота и медь.
Все исходные материалы должны быть в достаточной степени чи-
стыми.
Ртуть. Применяемая для производства металлическая ртуть
марки Р-2 должна быть по наружному виду блестящего серебристо-
белого цвета, с зеркальной поверхностью, уд. веса 13,6; она не должна
приставать к стенкам стеклянного сосуда при сильном взбалтывании,
при разливании на чистую гладкую поверхность (бумаги или глазу-
рованного фарфора) должна образовывать совершенно круглые, бле-
стящие, легко подвижные и сливающиеся шарики; не должна иметь
никаких следов механических примесей. Металлическая ртуть должна
полностью растворяться в азотной кислоте (d — 1,2). При нагрева-
нии 5 г металлической ртути после испарения не должно оставаться
никакого остатка. Ртуть не должна содержать амальгам других ме-
таллов, что проверяется анализом раствора 20 г ртути в азотной
кислоте.
Для определения примесей, находящихся в ртути, обычно помещают
навеску в 50 г в небольшую стеклянную колбу или реторту и пары
ртути отгоняют на умеренном пламени, просасывая воздух до тех
пор, пока в реторте не останется около 1—2 г остатка; остаток раство-
ряют в нескольких миллилитрах крепкой химически чистой азотной
кислоты, фильтруют и выпаривают во взвешенном фарфоровом тигле
емкостью 15 мл, сушат и осторожно прокаливают при 500°. Затем,
1 В настоящее время готовят только белую гремучую ртуть.
109
»
установив вес остатка, обрабатывают его серной кислотой; в остатк
определяют свинец. Фильтрат обрабатывают сероводородом для оса
ждения висмута и меди в виде сернистых соединений. Медь и висму
определяют обычными методами качественного анализа. Допускается
содержание ртути не менее 99,99%, примесей (нелетучего остатка)—
не более 0,01%.
Обычно поступающая на заводы ртуть достаточна
чиста, однако, предварительное ее испытание необхо
димо, так как она может содержать некоторые пример
свинца, олова, меди, висмута и окисли железа. В слу
чае некондиционности ртути ее бракуют или очищаю
сами заводы. От механических примесей (сор и окалин
от железных сосудов, в которых хранится и перевозите?
ртуть) ее очищают, отфильтровывая через плотное по
лотно или фланель. От других примесей (металлических'
очищают, обрабатывая слабой азотной кислотой (обычне
8%) и пропуская очень тонкой струей через столб кис-
лоты высотой около 1 м (фиг. 55).
Очищаемую металлическую ртуть вливают в воронку
с вытянутым в капилляр кончиком. Благодаря послед-
нему ртуть выливается в азотную кислоту небольшим*
шариками, которые на сравнительно длинном пут*
освобождаются от посторонних металлов. Перед нача-
лом очистки нижнюю часть трубки и колено заполняю!
уже очищенной металлической ртутью.
можно очищать гальваническим током, предварительнс
раствором нитрата одновалентной ртути. Однако полное
ртути достигается лишь перегонкой при уменьшенном дав-
Фиг. 55.
Схема очист-
ки металли-
ческой рту-
ти.
Ртуть
облив ее
очищение
леями.
Азотная кислота. Азотная кислота должна быть про-
зрачной (допускается желтого цвета от присутствия окислов азота^
и должна содержать не менее 61,5% моногидрата (d 1,383; 40° Be),
что проверяют титрованием щелочью или нитрометром Лунге и опре-
делением удельного веса.
Содержание моногидрата по первому способу обычно определяю)
следующим порядком: навеску азотной кислоты около 4—б г разбав-
ляют в мерной колбе водой до 250 мл\ отбирают пипеткой 50 ли
раствора, прибавляют к нему 50 мл нормального раствора едкогс
натра и избыток щелочи оттитровывают нормальной серной кисло-
той. Получающиеся в результате расчета данные показывают общук
кислотность. Для получения же истинного значения необходимо и:
полученного результата вычесть пересчитанное на HNOS содержание
H2SO4 (если последняя в анализируемой кислоте будет обнаружена)
Удельный вес азотной кислоты определяют ареометром Боме пр*
15° и вычисляют по формуле:
я _
а 144,3 —п’
где п — число градусов Вё по показаниям ареометра.
110
[ Колебания темпе pa- I гуры опыта сказываются Таблица 29
1 ii>i результатах опреде- Уд- вес HNO, Поправка для 1° С
I колебания не превышают |-2° (13—17°), то вводят поправку по табл, 29. Вместо указанной | выше формулы можно I пользоваться табл. 30, I составленной ‘ по Лунге и Рею и связывающей содержание моногидрата Уд. вес HNOa От 1.351 до 1,365 “ 1,366 » 1,400 •> 1,401 » 1,435 » 1,436 » 1.490 и 1,491 » 1,500 » 1.501 о 1.520 0,0012 0.0013 0,0014 0,0015 0,0016 0,0017
удельный вес азотной кислоты, процентное и градусы Вё. Таблица 30 при 15° (отнесенный к воде при 43)
Уд. вес 1 15° при 4- Градусы Вё 100 в. ч. содержат 1 л содержит кг
HNO3 кислоты в 40° Вё HNOS кислоты в 40° Вё
1,340 1,345 1,350 1,355 1,360 1,365 1,370 1,375 1,380 1,383 1,385 1,390 1,395 1,400 1,405 1,410 1,415 1,420 1,425 1,430 1,435 1,440 1,445 1,450 1,455 1,460 1.465 1,470 1,475 1,480 1,485 1,490 1,495 1,500 36,6 37,0 37,4 37,8 38,2 38,6 39,0 39,4 39,8 40,0 40,1 40,5 40,8 41,2 41.6 42,0 42,3 42.7 43,1 43,4 43,8 44,1 44,4 44,8 45,1 45,4 45,8 46,1 46.4 46,8 47,1 47,4 47,8 48,1 54,07 54,93 55,79 56,66 57,57 58,48 59,39 60,30 61,27 61,92 62.24 63,23 64,25 65,30 66,40 67,50 68,63 69,80 70,98 72,17 73,39 74,68 75,98 77.28 78,60 79,98 81,42 82,90 84.45 86,05 87,80 89.60 91,60 94,09 87,32 88,71 90,1-0 91,51 92,97 94,44 95,91 97,38 98,95 100,00 100,51 102,12 103,76 105.46 107,24 109,01 110,84 112,73 114.63 116.55 118,52 120,61 122,71 124,81 126,94 129,17 131,49 133,88 136,39 138,97 141,63 144,70 147,93 151.99 0.725 0,739 0,753 0,768 0,783 0,798 0,814 0,829 0,846 0,857 0,862 0,879 0,896 0.914 0,933 0,952 0,971 0.991 1,011 1,032 1,053 1,075 1,098 1,121 1.144 1,168 1,193 1,219 1,246 1,274 1,302 1.335 1,369 1,411 1,171 1,193 1,216 1,240 1,265 1,289 1,314 1,339 1,366 1,383 1,392 1,420 1,447 1,476 1,507 1,537 1,568 1,600 1,633 1,667 1,701 1,736 1,773 1,810 1,848 1,886 1,927 1,969 2,012 2,058 2,103 2,156 2,211 2,278
Ill
Таблица 31
Содержание в азотной кислоте посторонних веществ должно укдад
дыватъся в следующие нормы:
I) содержание хлористых соединений в пересчете на НС1 — не более 0,1 4
2) содержание сернокислых соединений в пересчете на .HsSO4— не более 0,зЯ
3) содержание железа — не более 0,07%;
4) содержание окислов азота — не более 0,5%;
5) содержание твердого остатка — не более 0.1%.
На некоторых заводах руководствуются следующими нормами:
1) содержание хлора не более 0,08%;
2) содержание серной кислоты — не более 0,05%;
3) содержание железа — не более 0,01%;
4) содержание твердого остатка - не более 0,9%.
Количественно определяют примеси к азотной кислоте следующим I
образом:
1. Для определения содержания хлора навеску кислоты растворякя
в 10 ч. дистиллированной воды, нейтрализуют содой ититруютдецн- I
нормальным раствором азотнокислого серебра. Индикатор—хромовн
кислый калий К2СгО4.
2. Содержание серной кислоты определяют в виде сернокислой
бария BaSO4. Для этого навеску кислоты 50—100 г выпаривают
на водяной бане почти досуха, разбавляют кипящей водой и BaSO,
осаждают кипящим раствором хлористого бария ВаС12.
3. Для определения содержания железа навеску азотной кис
лоты около 100 г разбавляют водой в четыре-пять раз и, нейтрали
зуя аммиаком при нагревании, осаждают гидраты железа и алюми
ния. Фильтруют осадок после получасового стояния, промываю!
горячей водой и на фильтре растворяют слабой серной кислотой
(1 : 5). Раствор собирают в колбу и восстанавливают окись желез.
в закись металлическим цинком (около 3—5 г) при нагревании. Поел'
охлаждения содержимое колбы разбавляют водой и титруют децинор-
мальным раствором марганцовокислого калия КМпО4.
4. Окислы азота определяют титрованием навески азотной кис-W
лоты КМпО4.
5. Твердый осадок определяют выпариванием навески кислотыЯ
около 50 г и прокаливанием остатка до постоянного веса в платино-
вом или фарфоровом тигле.
Если на завод поступает азотная кислота большей крепости, то е(
разбавляют (в керамиковых горшках) соответствующим количеством
воды до определенного содержания моногидрата.
Спирт. Требования к этиловому спирту заключаются в еле-
дующем: ректифицированный этиловый спирт должен представ- ш
лять собой прозрачную бесцветную жидкость, без примесей и бе
неприятного запаха. Крепость принимаемого спирта должна бытьЯ
не ниже 96%, что определяют при помощи спиртометра Траллеса. I
Спиртометр (ареометр) Траллеса показывает, сколько объемных
процентов абсолютного спирта содержится в испытуемом образчике
В табл. 31 дано соотношение между удельным весом и содержанием I
объемных процентов абсолютного спирта в водном спирте при 15,56°.
С 1 CJ Э процент спирта Еес единицы объема Объемный процент спирта Вес единицы объема Объемный процент спирта Вес । единицы I объема Объемный процент спирта Вес единицы объема
I 0,9976 26 0,9689 51 0,9315* 76 08739
2 0,9961 27 0,9679 52 0.9ЭД5 77 0.8712
з 0,9947 28 09668 53 0,9275 78 0,8685
4 0,9933 29 0.9657 54 0.9254 79 0,8658
5 0.9919 30 0.9646 55 0,9234 1 80 0.8631
6 0,9906 31 0,9634 56 0.9213 81 0,8603
7 0,9893 32 0,9622 57 0.9192 82 0,8575
8 0,9881 33 0,9609 58 0,9170 I 83 0.8547
9 0.9869 34 0.9596 59 0.9148 84 08518
10 0,9857 35 0,9583 60 0.9126 85 0.8488
11 0,9845 36 0.9570 61 0,9104 86 08458
12 0,9834 37 0.9559 62 0.9082 87 0.8428
13 0,9823 38 0,9541 63 0,9059 88 0.8397
14 0.9812 39 0,9526 64 0.9036 89 0.8365
15 0.9802 40 0.9510 65 0.9013 1 90 0,8332
16 0,9791 41 0,9494 66 0,8989 01 0,8299
17 0 9781 42 0.9478 67 0,8965 92 0.8265
18 0.9771 43 0.9461 68 0,8941 93 0.8230
19 0.9761 44 0.9444 69 08917 94 0.8194
20 0,9751 45 0,9427 70 0.8892 95 0,8157
21 0,9741 46 0,9409 71 0,8867 96 0 8118
22 0.9731 47 0.9391 72 0,8842 97 0 8077
23 0.9720 48 0,9373 73 0,8817 98 0.8034
24 0,9710 49 0,9354 74 0.8791 99 0.7988
25 0,9700 50 0,9335 75 0.8765 100 0.7937
Поправку на удельный вес спирта при различных температурах
вносят по табл. 32.
Таблица 32
Удельный вес водного спирта при различных температурах (по Менделееву)
К Е 8 Вес единицы объема водного спйрта (вода при 4°) при
0° 10° 20“ 30
0 0.98988 0,99975 0.99831 0,99579
5 0.99135 0,99113 0,98945 0,98680
10 0,98493 0,98409 0,98195 0,97892
15 0.97995 0.97816 0.97527 0,97142
20 0.97566 0,97263 0.96877 0,96413
25 0,97115 0.96662 0.96185 0.95628
.U) 0,96540 0.95998 0,95403 0.94751
35 0,95784 0,95174 0,94514 0.93813
Ю 0,94939 0,94255 0.93511 0.92787
45 0,93977 0,93254 0.92493 0.91710
ЩСнов—92—8
113
112
род о л ж- таил.
Вес единицы объема водного спирта (вода при 4°) при
О 5 8Ё m S оэ 10° 20° 30°
50 0.92940 0,92182 0.91400 0,90577
55 0,91848 0,91074 0,90275 0,89456
60 0.90742 0,89944 0,89129 0,88304
65 0.89595 0,88790 0,87961 0,87125
70 0,88420 0,87613 0,86781 0.85925
75 0.87245 0,86427 0.85580 0,84719
80 0,86035 0,85215 0,84366 0,83483
85 0,84789 0,83967 0,83115 0,82232
90 0,83482 0,82665 0.81801 0,80918
95 0.82119 0.91291 0.80433 0,79553
100 0.80625 0.79788 0.78945 0,78096
При пользовании таблицей надо помнить, что весовые проценты
спирта (р %) рассчитываются из объемных процентов (V%) по фор-
муле:
0J|37 v = p%j
где 0,7937 — уд. вес абсолютного спирта;
а — уД. вес испытуемого спирта.
Спирт должен выдерживать пробы Саваля и Ланга (в течение
не менее чем 20 мии.). Содержание посторонних веществ не должно]
превышать следующих норм:
альдегидов не более . . .
сивушных масел не более .
фурфурола.........
сложных эфиров
0,002%
0,003%
недопустимо
не более 50 мг в I
Спирт подвергают анализам по следующим пробам:
1 Проба Сав ал я. К Юлм спирта, помещенного в колбочку, прибав-
ляют при взбалтывании 10 мл химически чистой серной кислоты (уд. вес 1,84)
я нагревают до выделения пузырьков. По охлаждении жидкость наливают в склянку
Саваля и ставят на лист белой бумаги между такими же двумя склянками, из ко
торых одна наполнена спиртом, а другая — химически чистой серной кислотой.
Цвет должен быть средним между спиртом и кислотой. Если пробная смесь по
окраске заметно отличается от спирта и кислоты, то спирт считается не выдер?
жавшим испытания и бракуется. Проба Саваля обнаруживает содержание в спирте
сивушных масел сверх нормы.
2. Определение сивушных масел. Содержание сивушных ма-
сел определяют сравнением с типовыми растворами изоамилового спирта (0,001%.
0,003%, 0,005%) в присутствии различных количеств уксусного альдегида. Ре-
гентами являются растворы салицилового альдегида и серной кислоты.
3. Определение содержания альдегидов. Содержание
альдегидов также определяют сравнением с типовыми растворами уксусного аль-
дегида и спирта при действии на них фуксино-сериой кислоты, которая дает ха раю
терную розовую окраску.
114
4- Проба Ланга. Цилиндр емкостью 50 мл наполняют спиртом, затем
[Жывляют 1 AUjpaCTBOpa, содержащего 0,2 г марганцовокислое окалин в 1л дистил-
। прованной воды, хорошо перемешивают и дают стоять при комнатной температуре;
1ри этом происходит в течение определенного промежутка времени изменение
окраски раствора.
5.
• канне
Определение содержания
сложных эфиров определяют омылением
сложных эфиров. Содер-
их щелочами, фурфурол же может
1ть легко обнаружен вследствие розового окрашивания спирта при прибавлении
ксуснокцелого или солянокислого анилина (на 10 мл спирта 10 капель анилина
। 2 капли соляной кислоты).
Соляная кислота, применяемая в Производстве гремучей
ртути, должна соответствовать требованиям по ОСТ для химически
чистых кислот, а металлическая медь должна быть достаточно чистой
: содержанием Си не менее 99,5% (обычно употребляют отходы мед-
ных колпачков капсюльного производства).
Количество перерабатываемой одновременно металлической ртути
колеблется от 50 до 600 г. Большинство заводов одновременно перера-
батывает 400—500 г металлической ртути. Увеличение навески не
представляется возможным при ручном способе работы вследствие
возрастания объема реакционных сосудов.
В основе всех рецептур, применяющихся в производстве гремучей
ртути, лежит рецептура Либиха: ртуть, азотная кислота, спирт.
Со времени появления первого рецепта Либиха было предложено
большое количество других способов получения гремучей ртути,
каждый из которых претендовал на оригинальность, выражавшуюся
а изменении удельного веса азотной кислоты или спирта, начальной
температуры или других деталей процесса.
Фабрикация гремучей ртути точно по методам Либиха и Бекмана
ныне нигде уже не производится- Количества исходных материалов,
применяемых для производства гремучей ртути, на различных заво-
дах и в разных странах, различно.
Гак, например, Н. Оландер для заводского получения гремучей
ртути рекомендовал пользоваться следующим соотношением исход-
ных материалов:
ртути................................ I в- ч.
азотной кислоты (40° Вё) . 11»»
спирта (91°) ....................... 11 » »
И. Чельцов немного позже предлагал:
ртути............................ . 450 2
азотной кислоты (d •= 1,4) 4500 »
спирта (90) . ... 5000 »
Велер дает следующий рецепт для получения серой гремучей
ртути:
ртути............................................... 500 г
азотной кислоты (d 1,40) - . 4200 »
подогретого спирта................ . 5000 »
а для получения белого продукта, кроме того, 15 г полухлОристой
меди (Си3С12), или 5 г меди и 5 мл соляной кислоты.
115
По данным Солонина, для приготовления белого продукта па за-
воде ВВ акционерного общества в Рейнсдорфе (Германия) и на Охтен-
ском заводе (Россия) применяли:
ртути..................... 50о г
азотной кислоты (d 1,383) . 4500 •
Спирта (92—95%)........... 5000 мл
соляной кислоты (d - 1,115) . 5 ?
меди................... 5 »
а для получения серой гремучей ртути:
ртути............................................ 400 ?
азотной кислоты (d = 1,383) . 4200 »
спирта (96%) . . . 4000 »>
По литературным данным, на германских государственных заво-
дах пользовались следующим соотношением:
ртути.............................. 150 г
азотной кислоты (d - 1,4) . - 1072 мл
спирта (79,5%).................... 1500 »
соляной кислоты (d - 1,185) . 0,064%
В Австрии употребляли:
ртути......................................... 50о г
гремучей азотной кислоты (d =1,383) 4500 »
спирта (96%)...................... 5000 мл
410 г
2600 »
1630 »
5 »
5 мл
На наших заводах применялось следующее соотношение исход-
ных материалов:
ртути .............
азотной кислоты (61,5%) .
спирта (96°)....
меди............
соляной кислоты
Таким образом на разных заводах применяют на 1 ч. ртути 8,4—
11 ч. азотной кислоты уд. веса 1,38—1,40 и 6,5—10,6 ч. 90—
96%-ного спирта. Технологический процесс, включая количествен-
ное соотношение реагентов, только в деталях разнится от процесса
времен Говарда, Либиха и Шанделона, и определяется в основном
размерами аппаратуры, качеством вентиляции и другими техническими
и экономическими причинами. Велер пытался доказать, что в про-
цессе производства не имеют значения ни совершенно определенная
концентрация азотной кислоты, ни определенный удельный вес
спирта, ни постоянная температура реакции и что основным требо-
ванием должно быть соотношение между металлической ртутью и спир-
том 1 : 8.15 (спирт 95%-ный). Однако это, вообще говоря, верное ука-
зание не исключает необходимости учета конкретных условий на каж-
дом заводе.
§ 8. ТЕХНОЛОГИЧЕСКИЙ ПРОЦЕСС ПОЛУЧЕНИЯ ГРЕМУЧЕЙ РТУТИ
Технологический процесс получения гремучей ртути состоит из
следующих операций:
1) подготовки азотной кислоты;
2) получения раствора азотнокислой ртути в азотиой кислоте;
3) по мучения сырой гремучей ртути;
4) промывки и очистки гремучей ртути;
5) сушки гремучей ртути;
6) сортировки гремучей ртути.
В некоторых случаях добавляют еще одну*операцию по флегма-
гизации и грануляции пыли и шишек с последующей сушкой и сорти-
ровкой.
Поступающая на завод азотная кислота обычно имеет несколько
большую концентрацию, чем это требуется для производства. Для
получения кислоты с содержанием 61,5% моногидрата, ее смешивают
с рассчитанным количеством воды.
Расчет производят по формуле:
Воздух
ВозЪихк
Фиг. 56- Баллон для
смешивания кислот.
где w — количество добавляемой воды в кг;
N — требуемая концентрация;
п — концентрация взятой -кислоты;
А — количество взятой кислоты в кг.
Подготовкаазотнойкислоты.
Подлежащую смешению азотную кислоту зали-
вают в керамиковые баллоны (фиг. 56) емкостью
около 300 л с тремя отверстиями сверху — для
заливки кислоты, для подачи воздуха при
перемешивании и для выхода газов при про-
дувке. В низу баллона имеется кран для спуска
готовой кислоты. Перемешивают вдуванием воздуха при помощи
компрессора в течение 10—15 мин., после чего берут пробу на анализ.
По небрежности рассчитывающего кислота может оказаться не
соответствующей требуемой концентрации. В таком случае ее исправ-
ляют. Если кислота крепче, то рассчитывают вновь по приведенной
выше формуле; если же концентрация кислоты меньше, то прибавляют
более концентрированную кислоту, рассчитывая по формуле:
х _ (N ~
<n-N) ’
где X — количество добавляемой крепкой кислоты в кг;
7V — требуемая концентрация;
п — концентрация крепкой кислоты;
пг — концентрация исправляемой кислоты;
А — количество исправляемой кислоты.
После вторичного перемешивания вновь производят анализ кис-
лоты. Только по получении удовлетворительных результатов кислоту
117
отправляют непосредственно на растворение металлической ртут.
или сливают в запасные керамиковые баллоны.
Установка для смешения должна быть помещена в отдельно!
здании или в отдельном, имеющем самостоятельный ход наружу
помещении мастерской приготовления гремучей ртути, или же, в край
нем случае, на воздухе под навесом. Если установка расположен
в помещении, то последнее должно!
быть оборудовано приточно-вытяжной 1
вентиляцией.
В целях предупреждения разбрыз- ]
гивания при сливе азотной кислоты
в баллон-смеситель вручную следует 1
выливать азотную кислоту из буты-
лей медленно и через керамиковую с
широкими краями воронку. В горло-
вину бутыли полезно вставлять изо-
гнутую кверху алюминиевую трубку
для ввода воздуха внутрь бутыли.
Получение раствора
азотнокислой ртути в
азотной кислоте. Металли-
Фиг. 57. Фиг. 58. Пони-
жу pa вл ь. ческая колба
ческую ртуть растворяют в азотной кислоте в плоскодонных стеклян-
ных колбах (фиг. 57) с длинным горлом (журавли) или в конических
колбах (фиг. 58). Вытянутое горло журавля затрудняет удаление
выделяющихся при реакции окислов азота и является удобным для
заливки полученного раствора в спирт. Плоское, сравнительно боль-
шого диаметра, дно колбы способствует распределению ртути тонким |
слоем, благодаря чему увеличивается поверхность соприкосновения
ее с кислотой и ускоряется процесс растворения.
Поступающую в мастерскую очищенную металлическую ртуть
вновь фильтруют через фланель или полотно и хранят в закрытых
сосудах. Работа с ртутью требует некоторых мер предосторожности, так как ее пары ядовиты. В воз- духе помещения при на- Таблица 33
личии открытой поверх- ности металлической Температура ртути содержится опре- °с деленное количество па- Давление паров ртути мм Концентрация ртути мг/л
ров ртути (табл. 33) в зависимости от темпера- 20 туры. 30 Безусловно опасными 59 для здоровья являются 0,0013 0,0029 0,006 0,030 0,0152 0.0339 0,1000 0.3500
количества около 0,1 мг
на 1 л£3 воздуха, т. е. примерно той концентрации, которая со-
здается за счет испарения ртути уже при обыкновенной темпера-
туре. При этом вначале появляется легкая головная боль, со време-
118
разрушению
Фиг. 59. Кув-
шин для кис-
лоты.
нем усиливающаяся и локализующаяся в висках, появляется нерв-
ность, переходящая в тяжелое самочувствие, сопровождающееся
тяжестью в голове и плохой способностью соображать. Почти исче-
зает обонятельная способность. Дальнейшее развитие отравления
ведет к воспалению слизистой оболочки рта, быстрому
зубов, ухудшению работы желудка, появлению боли в
суставах и поражению нервной системы.
Поэтому в качестве мероприятии по технике без-
опасности рекомендуется хранить металлическую
ртуть всегда в закрытых сосудах, при операциях
с нею стремиться как можно меньшее время держать
открытой поверхность ртути и не прикасаться к ней
голыми руками, работать в тонких резиновых перчат-
ках и защищать дыхательные органы ватной повяз-
кой. Периодически (особенно летом) следует произ-
водить анализ воздуха мастерской.
По мере необходимости металлическую ртуть
развешивают или отмеривают автоматическими мер-
никами по 410 г в картонные, лакированные внутри,
коробки.
Очищенную спиртом красную медь навешивают по 5 г на робер-
валевских весах в картонные коробочки.
Азотную кислоту чаще всего навешивают по 3600 г на робервалев-
ских весах в специальные кубшины (фиг. 59). Эта операция должна
производиться при действии вентиля-
ционной системы.
В журавль загружают последова-
тельно навеску хорошо промытой
спиртом и высушенной меди, отмерен-
ное мензуркой количество соляной
кислоты (rf= 1,2), навеску металличе-
ской ртути и навеску азотной кислоты
(61,5%). Горло журавля закрывают
стеклянной пластинкой размером
100 х 100 мм для предохранения от
удаления окислов азота. Серию журав-
лей ставят на деревянный рамочный
поддон в шкаф-ванну (фиг. 60) при тем-
пературе 40—45°, которую контро-
лируют вставленным в один из журав-
лей термометром. Шкаф-ванна пред-
ставляет собой обычный вытяжной
шкаф, выложенный листовым свинцом
и содержащий бассейн с водой. Под деревянной решеткой проложен
паропровод. На некоторых- заводах эту операцию ведут в воздушных
шкафах, обогреваемых паром, что менее удобно.
Уравнение реакции растворения ртути:
3Hg + 8HNOs -> 3Hg(NOs)24-4H2O 4-2140 + 28,9 Кал
фиг. 60. Шкаф-ванна.
119
Процесс длится около 2 час., после чего азотнокислый раствор
ртути охлаждают до 35° тоже в течение 2 час. Последующие операции
занимают меньшее время, поэтому обычно загружают поочередно
две серии журавлей, так что в одной серии происходит растворение,
а в другой — в то же время охлаждение раствора. Количество одно-
временно загружаемых колб определяется производственным заданием.
При этой операции надлежит обращать особое внимание на точ-
ность отвешивания
Фиг. 61. Шарообраз-
ная колба-
и отмеривания исходных продуктов, потому что
малейшая неточность сказывается на выходе
готового продукта и его качества. Например,
увеличение навески соляной кислоты влечет за
собой увеличение загрязненности гремучей
ртути примесями. Установлено, что при двой-
ном количестве соляной кислоты в готовом про-
дукте находится 1,4% хлористой ртути, а выход
уменьшается на 1,5—2%. При применении шести-
кратного и десятикратного количеств соляной
кислоты содержание хлористой ртути увеличи-
вается соответственно до 5,2% и 7,6%.
Сырую гремучую ртуть полу-
чают в круглых, шарообразных колбах (фиг.61),
емкостью 70—80 лис горловиной диаметром
около 10 см. В совершенно чистые колбы вливают
навеску этилового спирта крепости 96°. Зимой спирт подогревают в
специальных нагревателях до 20—25°. Готовый и охлажденный
до 35" темнозеленый раствор ртути в азотной кислоте вливают в
колбы с залитым в них спиртом. При этом журавль приводят в движе-
ние так, чтобы жидкость в нем пришла во вращательное движение.
После слива в одну нз колб помещают термометр и раз в 5—10 мин-
регистрируют температуру.
Колбы помещают в шкаф, закрываемый тонкими деревянными щи-
тами с застекленными окнами для наблюдения, иа поддоны, обитые
листовой резиной по сукну или войлоку- К горлу каждой колбы
подводят керамиковый патрубок вентиляции. Спирт и азотнокислый
раствор ртути заливают в колбы в указанной последовательности,
в противном случае может происходить быстрое разогревание содержи-
мого колб, сопровождающееся иногда выбрасыванием его из колбы
и ожогом работающих. Взаимодействие между спиртом и азотнокис-
лым раствором ртути начинается сразу же после слива. Имеющиеся
в азотнокислом растворе ртути окислы азота вступают в реакцию
и почти мгновенно исчезают.
Реакция продолжается около 2 час. и по своему характеру может
быть разбита на четыре периода. В течение первого периода (сред-
ней продолжительностью 10—20 мин.) жидкость прозрачна, осадка
еще нет и температура постепенно возрастает от 25—30° до 40—50°.
В этот период происходит в основном взаимодействие этилового спирта
и азотной кислоты и появляются первые пузырьки бесцветных газов.
Второй период (продолжительностью 15—20 мин.) характеризуется
120
«начале слабым выделением белых паров, что наблюдается при темпе-
ратурах 50—60° (в течение 3—5 мин.), а затем обильным выделением
белых паров с образованием белых кристаллов гремучей ртути. Тем-
пература возрастает чрезвычайно быстро от 60 до 82—85°. Пузырьки
газов становятся все меньше и меньше. Реакция протекает бурно.
Жидкость сильно кипит. Третий период (15—20 мин.) характери-
зуется выделением бурых паров окислов азота. Температура несколько
повышается (0,5—1°), а затем начинает постепенно падать. В течение
четвертого периода происходит постепенное падение температуры.
Фиг. 62- Диаграмма температурного режима реакции получения
гремучей ртути-
Образовавшаяся гремучая ртуть в большей своей массе (к моменту
спада температуры до 60—65°) оседает на дно колбы, а часть ее в
виде пены плавает на поверхности маточного раствора. Динамика
температуры выражается диаграммой, изображенной на фиг. 62.
Если температура в момент бурной реакции во втором периоде
не достигает нормальной температуры 82°, то это свидетельствует о том,
что взятая кислота оказалась слабее требуемой и что гремучая ртуть
может содержать так называемую «металлическую ртуть» в тем боль-
шей степени, чем ниже оказалась максимальная температура. Если же
температура превышает 82°, то гремучая ртуть кристаллизуется
в более мелких кристаллах и может содержать значительное количе-
ство щавелевой кислоты. Поэтому необходимо вести реакцию осаж-
дения гремучей ртути при средней температуре 82°, регулируя
режим крепостью кислоты и соотношением компонентов.
Эта операция в прошлом не всегда удавалась. Иногда получа-
лась белая порошкообразная масса с незначительным количеством
гремучей ртути или состоящая полностью из щавелевокислой ртути
и других ртутных солей. Это так называемая пустышка, которую
приходилось выбрасывать. Бывали и такие случаи, когда совсем не
образовывалось никакого осадка. Все это происходило в основном
из-за неправильного соотношения спирта и азотнокислого раствора
ртути. Например, если спирта взято слишком мало, то реакция вслед-
ствие большой кислотности раствора протекает весьма быстро и при
высокой температуре, причем гремучая ртуть, образуясь, частично
121
Фиг. 63- Приспособление для слива
гремучей ртути.
тотчас же разлагается, и получается конечный продукт окисления'
щавелевая кислота. Если, наоборот, спирта взято слишком mhoi
то реакции вовсе не происходит, так как раствор очень разбавлв
или же она скоро останавливается, и в этом случае выход гремуч^
ртути мал. При ошибках меньшего порядка гремучая ртуть образуете]
но выходы ее сравнительно меньше нормальных.
В случае явных ошибок в количестве приливаемого азотнокислог
раствора ртути наблюдается чрезвычайно быстрое разогревание р;м
твора, а иногда и выбрасывание его из колбы, что небезопасно для ра-
ботающих. При таких ошибках п
исключена возможность и восплаЗ
менения спирта.
Для получения доброкачествен^
ной гремучей ртути никоим образом
нельзя применять азотную кис-
лоту удельного веса, выходящего
за пределы 1,38—1,40, а спирт —
крепости ниже 95%. При меньших
концентрациях реакция не может <
начаться без подогрева, что в усло-
виях заводского процесса трудно
осуществимо.
Стало быть, вторая операция, как и первая, требует строжайшего
соблюдения рецептуры и режима работы (время заливки и т. д.). По
окончании реакции надлежит осмотреть раствор и осадок в каждой
колбе отдельно.
На некоторых заводах в начале третьего периода, т. е. с появле-
нием бурых паров, реакцию прерывают — «тушат» — добавкой эти-
лового спирта в два приема (по Велеру — 30 мл на 100 г гремучей
ртути, на ряде французских заводов 130 мл на 100 г гремучей ртути,
а на германских заводах 166 мл на 100 г гремучей ртути). По опы-
там Каста, той же цели можно достигнуть прибавлением вместо спирта
1 л воды. В Англии же поступали при ведении этой операции иначе:
к раствору 3 ч. ртути в 36 ч. азотной кислоты (d = 1,34) прибавляли
сначала 17 ч. 90%-ного спирта и только после начала реакции для ее
замедления понемногу вливали еще 17 ч. спирта.
В свое время считали, что при «тушении» реакции, а стало быть, и
регулировании хода процесса во времени может быть получена более
чистая гремучая ртуть. Однако на практике при «тушении» иногда выде-
ляется металлическая-ртуть, которая окрашивает продукт в серый цвет.
Процесс получения сырой гремучей ртути считают законченным,
когда температура в колбах понижается по истечении определенного
промежутка времени до 55—60°.
Промывка и очистка гремучей ртути. По
окончании реакции в колбах и достаточном охлаждении жидкости
осадок гремучей ртути вместе с маточным раствором сливают через
воронку в керамиковые горшки. Остатки кристаллов гремучей ртути
со стенок колбы смывают сильной струей воды (фиг. 63).
122
Промывка имеет целью удалить из гремучей ртути кислоту и про-
чие растворимые в воде вещества, а также, насколько возможно.
Фиг. 64. Аппарат для
промывки гремучей
ртути.
уменьшить содержание сопровождающей
продукт щавелевокислой ртути. Даже са-
мое ничтожное количество кислоты при
последующей сушке может стать весьма
концентрированным, а затем при хранении
снаряженных капсюлей — взаимодейство-
вать с оболочкой, а в некоторых случаях
и дать повод к взрывам.
Гремучую ртуть отделяют от маточного
раствора декантацией и переносят в стек-
лянные цилиндрические сосуды, нижняя
часть которых имеет конусообразную фор-
му. Вершина конуса срезана. На срез
натянута шелковая плотная сетка № 150.
Сосуд вставляют в гнездо и соединяют с
водопроводом. Вода поступает снизу, взму-
чивает гремучую ртуть в сосуде и стекает
через отверстие в верхней части сосуда
в деревянный ящик (фиг. 64).
Отмытую таким образом в течение 40—60 мин. от кислоты грему-
чую ртуть (что определяется лакмусовой бумажкой) выгружают из
Фиг. 65. Глазурованная чаша.
стеклянных промывных цилиндров
в глиняную глазурованную чашку
и несколько раз промывают дистил-
лированной водой, перемешивая ру-
кой, одетой в резиновую перчатку.
Воду сливают каждый раз вместе
с легкими плавающими на ней части-
цами. Хорошо промытую гремучую ртуть пропускают через шелковое
сито № 50 для удаления могущих попасть посторонних механических
примесей и загружают в банки для хране-
иия, по 5 кг в каждой. Образцы посылают
на испытание в лабораторию.
Значительно ранее промывку произво- _______
дили исключительно ручным способом в . ij
глиняных глазурованных чашах (фиг. 65)
глубиной 150—175 мм и диаметром около
500 мм. В такую чашу выливали содер- Ф,1Г- 66. Роговые ножи,
жимое трех—пята колб. Гремучая ртуть,
обладая большим удельным весом, быстро оседала на дно, .что давало
возможность сливать маточный раствор. Затем чашу наполняли водой и
массу перемешивали роговыми загнутыми ножами (фиг. 66) или рукой
в длинной резиновой перчатке. После отстаивания гремучей ртути в
течение некоторого промежутка времени воду снова декантировали. Эту
операцию повторяли до нейтральной реакции промывных вод. Что
касается щавелевой кислоты, которая тогда образовывалась в боль-
123
том количестве, то ее отделяли в тех же чашах, пользуясь разницей
в удельных весах. Для этого массу гремучей ртути в чаше обливали
водой и слегка утряхивали, более легкая щавелевокислая ртуть соби-
ралась наверх в виде пленки, затем осторожно сливали воду так,'
что пленка уходила вместе с нею. Эту операцию повторяли несколько
раз. Для того чтобы мелкие частицы гремучей ртути не попадали
в сточные колодцы, воду
сливали на сетку с натянутым
полотном.
Фиг. 67. Схема промывки гремучей ртути.
Несколько позднее, а на
настоя-
некоторых заводах и в
щее время, для промывки гре-
мучей ртути применяли сле-
дующее устройство (фиг. 67).
Гремучую ртуть, освобож-
денную от маточного раствора,
перекладывают в стеклянные
воронки а емкостью около
4—5 кг. На дно такой во-
ронки помещают разъемный деревянный круг б с шелковой сеткой в,
на которую и перекладывают гремучую ртуть. Воронку устанав-
ливают в отверстие деревянной подставки г. В заполненную поверх
гремучей ртути водой воронку опрокидывают колбу с водой 0,
установленную в верхнем гнезде той же деревянной подставки г.
По мере вытекания воды из воронки через гремучую ртуть и шелковую
сетку в колбу начинает поступать толчками воздух, следствием чего
является взмучивание верхнего слоя гремучей'ртути. Этот способ
промывки значительно более продолжителен, чем описанный выше
(иногда до 3—4 час.).
По литературным данным, на германских заводах с целью отмывки
от кислот, мелких частнц и загрязнений белый продую’ также под-
вергали процессу отмучивания до 20 раз, а серый — 2 раза. На одном
из германских заводов сырую гремучую ртуть отсасывают на нутч-
фильтре несколько раз, промывая кристаллический осадок холодной
водой. Под конец содержимое нутч-фильтра суспензируется один или
два раза в чистой воде и отсасывается. Такая гремучая ртуть содер-
жит, как правило, 1—2% маточного раствора и имеет характерный
запах. На некоторых заводах содержимое колбы фильтруют через
полотно и гремучую ртуть промывают на том же самом фильтре.
После этого фульминат ртути взмучивают в воде, а полученную су-
спензию пропускают через волосяное сито для отделения посторонних
веществ.
На одном из итальянских заводов гремучую ртуть после промывки
водой промывают еще спиртом. Штеттбахер отмечает, что при этом
получается особенно чистый красивый продукт. Гаген также счи-
тает, что для удаления трудно растворимых в воде органических
примесей из готового продукта его надлежит предварительно промы-
вать спиртом.
124
Основные меры безопасности на операции промывки — это абсо-
люгная чистота помещения, хорошая освещаемость рассеянным, све-
том, безукоризненная чистота аппаратуры и поддержание полов во
время работы во влажном состоянии.
Выход гремучей ртути разнообразен. По описанному процессу
на 100 кг ртути получается 125—127 кг вместо теоретически вычислен-
ных 142 кг. На других заводах он колеб-
лется от 100 до 130 кг.
Промытую и пропущенную через фильтр
гремучую ртуть выкладывают в чистые
стеклянные баночки емкостью около 4—5
кг деревянной или эбонитовой ложкой. Во
время выкладки, так же как и при других
операциях в промывочном отделении,
всегда наблюдают за тем, чтобы гремучая
• ртуть не подсыхала; для этого все при-
боры, столы и пол помещения постоянно
смачиваются водой.
В стеклянных банках си слоем воды
сверху гремучую ртуть отправляют на
хранение в специально оборудованные,
окруженные валами, погребки. При хране-
нии строго наблюдают за тем, чтобы вода
из банок не испарялась до обнажения
гремучей ртути. В погребке категорически
запрещается производить какие-либо ра-
боты, не связанные с хранением гремучей
ртути (перекладывание продукта из банки
в банку, смена воды и т. п.).
Температуру хранилища все время поддерживают в таких преде-
лах, чтобы вода в зимнее время не замерзала. Чаще всего рекомен-
дуется поддерживать температуру 15—20°. Отопление погребка
должно быть исключительно водяное.
Во Франции для того, чтобы предотвратить замерзание гремучей
ргути в зимнее время, после промывки се помещают в керамиковые
горшки, в которые затем заливают воду, содержащую спирт.
В погребках необходимо соблюдать безусловную чистоту. Столы,
на которых устанавливают банки с гремучей ртутью, должны быть
обиты войлоком и сверху накрыты клеенкой. Пол помещения покры-
вают сплошным куском линолеума или листовой резиной и проти-
рают каждый раз после посещения погребка влажной тряпкой.
В тех случаях, когда по небрежности работающих влажная грему-
чая ртуть замерзает, не следует производить с нею никаких операций
до полного ее отогрева.
Сушка гремучей ртути. Гремучую ртуть по мере на-
добности отправляют на сушку. Перед сушкой избыток воды (около
33%) отсасывают на нутч-фильтре (фиг. 68) до содержания влаги
•6-10%.
125
Фиг. 69. Вакуум-шкаф системы Пассбург.
126
Сырую гремучую ртуть выкладывают из банки на шелковое сито,
помещаемое в фарфоровую чашку с водой, и пропускают через него
для удаления могущих случайно попасть механических примесей.
Затем гремучую ртуть в количестве 15—20 кг перекладывают из фар-
форовой чашки на фильтр-полотно и переносят на нутч-фильтр.
Нутч-фильтр представляет собой деревянную кадку или фарфоро-
вый сосуд с закрепленной в верхней части деревянной или металли-
ческой решеткой, покрытой фильтром. Влажную гремучую ртуть
размещают равномерным слоем по всему фильтру, затем открывают
кран трубопровода, соединяющего нутч-фильтр с вакуумнасосом.
Разрежение в нижней части фильтра около 400 мм рт. ст.; длитель-
ность операции 15—20 мин.
При работе по обезвоживанию гремучей ртути следует наблюдать,
чтобы гремучая ртуть не подсыхала на фильтре. Чистота рабочего
места и своевременное уничтожение случайно упавших отдельных
кристаллов гремучей ртути являются одним из условий безопасности.
Фильтрат из нутч-фильтра пропускают через отстойники-ловушки
для улавливания прошедших через фильтр мелких частичек грему-
чей ртути.
Отсосанную гремучую ртуть выкладывают роговой ложкой в фар-
форовую чашку или эбонитовую банку, навешивают по 350 г девять
навесок и раскладывают на покрытые двухсторонней клеенкой нике-
лированные подносы (лотки), равномерно по ним распределяют рези-
новой или роговой гребенкой и отправляют в сушильный шкаф.
Для сушки гремучей ртути начали применять с середины 80-х го-
дов прошлого столетия вакуумсушильные шкафы. Ныне применяют,
главным образом, вакуумсушильные шкафы типа Пассбурга (фиг. 69).
Сушка в этих аппаратах, как и вообще сушка с применением вакуума,
не только значительно ускоряет технологический процесс и повышает
производительность завода, но и делает операцию более безопасной,
конечно, при условии соблюдения установленного температурного
режима.
Гаген еще в 1865 г. показал, что зажигание гремучей ртути в ва-
кууме при помощи платиновой проволоки вызывает взрыв лишь тех
частиц, которые непосредственно прилегают к проволоке, а остальные
частицы только разбрасываются. Дальнейшие опыты подтвердили
возможность применением вакуума ограничить разрушительное дей-
ствие ВВ в процессе их изготовления. Исходя из этого, Нейман даже
рекомендовал производить всю работу в помещении с разреженным
воздухом. Пиротехническая лаборатория в Шпандау поставила
в 1888 г. серию опытов по воспламенению гремучертутных составов
в вакуумсосудах. Оказалось, что при взрыве больших количеств этих
составов происходит разрушение сосудов даже в случае применения
высокого вакуума, но действие взрыва в этих условиях значительно
меньше. По данным этой лаборатории можно сушить 4 г гремучей
ртути в объеме 1 л безвоздушного пространства.
Опытным путем было установлено, что взрыв 150 г гремучей ртути
изменяет аппарат Пассбурга лишь в очень незначительной степени.
127
Поэтому загрузка первых образцов аппаратов была сразу доведем
до максимально возможного для них количества в 540 г гремучей
ртути.
Вакуумаппарат Пассбурга представляет собой горизонтальный
клепаный цилиндр с расположенной спереди чугунной, закрывающейся
герметически, крышкой на петлях. По всей окружности крышки
(по месту соприкосновения с корпусом) вделано резиновое кольцо-
прокладка, прн помощи которого и достигается герметизация. Внутри
аппарата расположены одна над другой 3 полые полки, соединенные
трубопроводами с паровой и водяными (горячей и холодной) сетями
для нагревания горячей водой и для охлаждения. Сверху и на задней
стенке аппарата имеется до десяти отверстий, закрытых крышками на
резиновых прокладках. Задняя стенка аппарата подвижна и прижи-
мается к корпусу достаточно сильными спиральными пружинами; гер-
метизация обеспечивается также резиновым кольцом-прокладкой. От-
верстия и подвижная задняя стенка служат для смягчения удара при
взрыве: крышки с отверстий просто сбрасываются под влиянием воз-
никшего давления, и для газообразных продуктов взрыва образуется
выход сразу через значительное поперечное сечение. Наличие же рези-
новых колец под крышками при нормальной работе аппарата обеспечи-
вает вакуум, для создания которого аппарат соединен с вакуумнасосом.
Испаряющаяся влага после выхода из вакуумаппарата Пассбурга
конденсируется в колонках, охлаждаемых водой. Аппарат оборудован
параллельными контрольно-измерительными приборами, позволяю-
щими наблюдать за степенью разрежения, давлением греющего пара
и температурой аппарата, находясь не только в помещении, но и вне
его. Точно также и управление кранами может быть осуществлено как
в помещении сушки, так и вне его.
Гремучую ртуть на никелированных подносах, покрытых клеен-
кой, ставят на полки аппарата Пассбурга, на которых расположены
асбестовые листы. Создают вакуум (720 мм) и подают пар в полки
на 10 мин. в начале сушки и на 5 мин. за 20 мин. до ее окончания.
Затем охлаждают полки в течение 15 мин. холодной водой, после
чего в аппарат постепенно подают воздух. Продолжительность сушки
1 час. По установлении нормального давления аппарат разгружают,
а гремучую ртуть с содержанием не более 0,02—0.03% влаги пересы-
пают в пайковые коробки и направляют на следующую операцию или
на хранение в расходные погребки. Внутри аппарат Пассбурга тща-
тельно обтирают влажной ветошью. Для безопасности работы это
мероприятие обязательно, ибо в противном случае скопление пыли
может привести к несчастному случаю.
На некоторых заводах, по литературным данным, сушку произво-
дят следующим образом. Гремучую ртуть, освобожденную от воды
декантацией, навешивают по 260 г в резиновые конусообразные
фильтры, которые затем вставляют в стеклянные или эбонитовые
воронки, соединенные с трубопроводом вакуумнасоса (фиг. 70).
В каждый из фильтров поверх гремучей ртути заливают по 50 мл
спирта крепостью 96°. Отсасывают при разрежении 150—200 мм
128
рт. ст. до тех пор, пока гремучая ртуть на фильтрах не начнет рас-
сыпаться на отдельные комочки.
фиг. 70. Воронки для обезвоживания
гремучей ртути.
системой
которым,
низкого
ПрОДОЛ-
Сушку производят или в неподвижных или вращающихся сушил-
ках. В первом случае фульминат ртути насыпают на лоток, изготов-
ленный из молескина (1,5 кг
на каждый); лотки помещают
на этажерки, обогреваемые
воздухом, предварительно
пропущенным над
трубопроводов, по
циркулирует пар
давления. Сушка
жается 8 час.
Во втором случае грему-
чую ртуть помещают в эбони-
товые коробочки (фиг. 71),
на дне которых имеется
несколько выступов, а в се-
редине — углубление, куда вставляется латунная муфта (фиг. 72).
Коробочки укрепляют на конце бронзового вала, наклоненного под
углом 45°, на котором имеется поверхность, снабженная углубле-
Фиг. 71. Коробочки для сушки.
ниями, в которые и входят выступы эбони-
товых коробочек. К центру этого сосуда
подведена трубка, по которой поступает
нагретый воздух (35—40°). Поверхность
гремучей ртути непрерывно обновляется
вследствие вращения аппарата, а сушка
заканчивается через 25—30 мин.
Следует отметить, что если первый
способ очень продолжителен, то второй,
Фиг. 72- Аппарат
для сушки грему-
чей ртути:
А — нагретый воздух;
D — латунная втулка;
/V— поперечные реб-
рышки; Р — шкив;
К — эбонитовя коро-
бочка.
вообще говоря, требует
принятия особых мер предосторожности. Отсутствие вакуума во
время сушки гремучей ртути приводит к неполному испарению почти
всегда сопровождающей гремучую ртуть так называемой металли-
ческой ртути.
Бубнов—92—9
129
Помещение, в котором производят сушку, обогревают батареямиj
водяного отопления- Коробочки загружают и разгружают через от-
верстия в капитальной стене, во время сушки плотно закрываемые!
щитами из котельного железа.
Сортировка гремучей ртути. Как уже было отме-
чено, величина кристаллов гремучей ртути оказывает влияние на
однообразие ее действия. Поэтому следующей операцией и является
сортировка продукта (фиг. 73). Операцию производят на двух ситах
а, а, поставленных одно
Фиг. 73- Ручная сорти-
ровка гремучей ртути-
над другим на деревянной рогатке б над,
большим поддоном в, имеющим пергамент-
ное дно. Рукоятка рогатки г пропущена
в щель большого железного полукруглого
щита ж. Сортировщик, находясь за щитом!
покачивает рукоятку вправо и влево, наб-
людая за ситами при помощи зеркала?. Для
сортировки употребляют сита № 50 (верх-
нее) и № 130 (нижнее). На верхнем сите
остаются крупные кристаллы и комки, так
называемая «шишка», на нижнем сите —
нормальный продукт, так называемое
«зерно», а на поддоне — пыль.
Отсортированное зерно с нижнего сита.
высыпают на клеенку и с последней пере-
сыпают в папковые коробочки. Наполнен-
ные коробочки тотчас же уносят в поме-
щение для хранения- Пыль и шишку
собирают отдельно и замачивают в воде.
Операция сортировки сухой гремучей
из самых опасных. Поэтому от работающего
внимание и осторожность при съемке сит с
ртути является одной
требуется наибольшее
деревянной опоры и при ссыпке рассортированного продукта. После
каждой разгрузки сортировочного «грохота» следует всю наружную
поверхность сит, стола и шита осторожно обтирать слегка влажной
губкой.
Наряду с ручной сортировкой в последние годы применяют и
механическую сортировку- Прибор для механической сортировки
представляет собой легкую деревянную коробку с двумя ситами,
могущую передвигаться в горизонтальной плоскости при помощи спе-
циальной штанги. Управление осуществляется из соседнего поме-
щения, а просеянный продукт ссыпается в процессе работы (без
участия человека) в соответствующие коробки-приемники.
Флегматизация и грануляция. Так как пыли и
шишки обычно получается от 4 до 6% (при неналаженности процесса
их количество может доходить до 15%), то с целью утилизации их
и уменьшения производственных потерь производится их флегмати-
зация и грануляция (зернение).
Для флегматизации гремучей ртути применяют разные флегмати-
заторы в зависимости от того, для какой цели предназначается гото-
130
вый продукт. В большинстве случаев гремучую ртуть флегматизи-
руют парафином; в некоторых случаях применяют аравийскую ка-
медь (гумми-арабик), трагокант, желатину и т. п.
Парафин вводят в гремучую ртуть, как правило, для снижения
ее чувствительности к внешним воздействиям; гумми-арабик или
желатину — для цементации пыли и шишки.
Пыль и шишку после отсеивания в сортировочной замачивают,
отправляют на нутч-фнльтры и отсасывают до содержания 9—10%
влаги. Затем загружают 3 к? влажной гремучей ртути и 3 г аравий-
ской камеди в водном растворе в фарфоре-
вую чашку, перемешивают и вручную пере- • й*
гирают около 40 мин. пальцами в рези- i
нивой перчатке. I
Деревянная Шелковая сетка
Фиг. 75. Схема грану-
ляции.
Фиг. 74- Станок для грануляции-
Фиг- 76- Ваза.
аппаратов Пассбурга на
Грануляцию флегматизированной гремучей ртути производят
в отдельных кабинках на деревянных станках (фиг. 74). Флегмати-
знрованную гремучую ртуть загружают на шелковое сито № 28 и
протирают через него при помощи рези-
новой пластины, укрепленной на дере-
вянной рукоятке, пропущенной через
прорез щита (фиг. 75). Во время работы
наблюдают через глазок из корабель-
ного стекла, вставленный в щит. Гра-
нулированный продукт раньше сушили
на так называемых вазах (до введения
них очень часто сушили и обычную гремучую ртуть).
Ваза представляет собой медный сосуд с двойным дном с горячей
(нагретой до кипения) водой (фиг. 76)/400 г гремучей ртути загру-
жают на плотную двухстороннюю клеенку, которую подвешивают
па четырех веревочках над вазой. Веревочки поочередно подерги-
вают, для того чтобы гремучая ртуть не приставала к клеенке. Кроме
гого, гремучую ртуть время от времени перемешивают и разравни-
вают опушкой гусиного пера. На вазе одновременно можно сушить
около 200—400 г гремучей ртути. Операция продолжается около
Ю—60 мин. при температуре воды около 90°. При понижении темпе-
131
Таблица 34
Время от
начала ре-
акции
мин.
Убыль веса
реакцион-
ной колбы
О
10
15
20
25
30
35
40
45
50
55
60
65
70
90
О
О
700
1600
2200
2600
2900
3150
3300
3400
3450
3500
3550
3600
3600
ратуры воды приходится через 25—30 мин. менять воду. Наблюде-
ние осуществляется из-за щита через специальное отверстие в нем,
закрытое толстым стеклом. Высушенную гремучую ртуть вновь сор-
тируют. Во время сушки необходимо следить за тем, чтобы гремучая
ртуть не прилипала к клеенке, и если это случилось, то следует при-
ставшую гремучую ртуть осторожно замачивать мокрой губкой.
Основным недостатком сушки на вазах является необходимость
постоянного перемешивания гремучей ртути в процессе сушки, что
сопряжено с опасностью взрыва.
Отходы производства. В про-
цессе производства гремучей ртути получается
значительное количество побочных и отход-1
ных продуктов. В третьей операции остается
маточный раетвор, и значительное количество
паров побочных продуктов выделяется во
время реакции. В четвертой операции появ-
ляются остатки отмучивания, а в четвертой—
седьмой операциях—гремучертутные отбросы.
Удаление и рациональное использование от-
бросов и побочных продуктов представляют
значительные затруднения. Паро- и газообраз-
ные вещества, обильно выделяющиеся при
реакции образования гремучей ртути, состоят, •
главным образом, из этилнитрита, этилни-
трата, спирта, уксусного альдегида и азот-
ной кислоты; кроме этих продуктов в них
содержатся частично синильная, муравьиная
и уксусная кислоты. Опыт показывает, что
количество выделяющихся газообразных
продуктов значительно и для каждой колбы
обычно превышает 3 кг.
В табл. 34 приводится среднее уменьшение веса колбы с реаги-
рующими веществами по мере хода реакции.
Газообразные продукты огнеопасны, чрезвычайно ядовиты и при
вдыхании вызывают рвоту и обморочное состояние. Выбрасывание
этих продуктов в воздух недопустимо.
На некоторых заводах конденсируют выделяющиеся пары, для
чего применяют очень громоздкие и сложные установки, одна из ко-
торых приведена на фиг. 77. Другие заводы пользуются менее громозд-
кими конденсационными системами, состоящими из нескольких по-
следовательно соединенных флорентинских сосудов (фиг. 78), или цел-
ляриусов, орошаемых водой, или содержащих некоторое, периодически
освежаемое, количество воды. Целляриусы с одной стороны соединены
с реакционными колбами керамиковыми трубопроводами, а с другой—
с вентиляционной системой того или иного типа. Для улучшения дей-
ствия системы обычно избегают примеси воздуха к выделяющимся
газообразным и парообразным продуктам, для чего реакционные колбы
соединяют с вентиляционной системок при помощи резиновых трубок
132
или место соединения плотно обвязывают тяжелой плотной тканью.
Некоторые заводы применяют для конденсации паров холодильники,
соединенные с двумя туриллами.
Фиг. 77. Конденсационная установка.
Состав газообразных продуктов не позволяет рассчитывать венти-
ляционную систему на полную конденсацию, ибо крылья обычных
вентиляторов весьма быстро разъедаются газами, применение же
керамиковых вентиляторов очень затруднительно. Поэтому поль-
зуются инжекторной системой, работающей воздухом или паром,
правда, требующей более значительного расхода энергии.
133
На части заводов совершенно не конденсируют пары и газы,
а просто выбрасывают их в воздух.
Исследованию конденсата и разработке методов его использования
было посвящено значительное количество работ.
Фридерики впервые тщательно исследовал побочные продукты и
определил общее количество кислот, количество связанной азотной
кислоты, этилнитрата и этилнитрита. Опыты его показали, что нейтра-
Фпг. 78. Флорентийский сосуд.
лизованный и перегнанный
конденсат может быть без-
опасно использован для фаб-
рикации гремучей ртути,
так как он содержит не
более 0,5% этилнитрата, и
гремучая ртуть на этом
конденсате получается хо-
рошего качества. Перегоняя
нейтрализованный конден-
сат с водяным паром и
экстрагируя затем эфиром,
он получил светложелтую
жидкость, нерастворимую в
воде, кислотах и щелочах,’
с температурой кипения
178 — 188°. По Солонина,
это глиоксальацеталь:
СН(0С,Н5)—СН(ОСоН5)
Солонина исследовал верхний и нижний слои конденсата. Согласно
его данным, верхний слой желтоватый, уд. веса 0,8923, легко воспла-
меняется от огня и начинает кипеть при 25°; верхний слой относи-
тельно больше нижнего (соответственно 203 и 95 г); прн разгонке
верхнего слоя получаются следующие фракции (из 32,6 г):
с 25 до 53° перегоняется 14,6 г
» 53 » 75° » 11,5 »
» 75 » 100° 6,4 »
Выше 100° остается лишь незначительное количество вещества,
разлагающегося с выделением окислов азота. Качественные исследо-
вания показали, что все фракции состоят из спирта, уксусного альде-
гида, уксусно-этилового эфира, этилнитрата, этилнитрита, азотной
и азотистой кислот, ацеталя, гликолевой кислоты; главная масса
состоит из спирта и альдегида (около 78%).
Нижний слой прозрачный, уд. веса 0,948. Жидкость сильно кис-
лая, хорошо растворимая в воде, содержит следы хлора, дает реак-
ции на азотную, азотистую и уксусную кислоты, не горит, содержа-
ние спирта и альдегида — только около 3%.
Этих данных достаточно, чтобы установить экономичность реге-
нерации. Регенерированный спирт и альдегид можно обратить снова
134
для получения гремучей ртути вместо спирта или в смеси с чистым
спиртом.
Чельцов рекомендовал обрабатывать конденсат гашеной известью
до исчезновения кислой реакции при постоянном перемешивании:
после отстоя перегонять конденсат на водяной ванне и в это время
постоянно небольшими порциями вносить в куб гашеную известь,
а пары охлаждать змеевиками. Таким образом в приемнике
получается смесь спирта и альдегида.
Он же полагал возможным вместо чистого спирта брать смесь
из 4 л спирта и 1 л перегнанного конденсата. По данным Годи, приме-
нялся следующий способ получения гремучей ртути с использованием
конденсата. К 5 л 90%-ного спирта и смеси 4 л спирта и 1 л альде-
гида, полученного из конденсата, прибавляли сначала 3,5 кг нагре-
той до 80° азотной кислоты, а затем раствор 450 г ртути в 1 кг азот-
ной кислоты (d = 1,333). Солонина прямо, без всякой разгонки,
примешивал верхний слой конденсата к 90%-ному спирту и. поль-
зуясь этой смесью, получал гремучую ртуть с хорошим выходом,
«совершенно белую и очень хорошую по своим взрывчатым свой-
ствам».
Гаген предложил следующий технический способ регенерации
спирта из конденсата: 400 л конденсата смешивают в нейтрализацион-
ном чане с известковой кашицей до щелочной реакции, нейтрализован-
ную жидкость перекачивают затем в дистилляционную реторту н
перегоняют. При этом первая фракция (15 л) уд. веса 0,910 обладает
кислой реакцией и содержит 34—35% нерастворимых в воде эфиров.
При дальнейшей перегонке получают 180 л спирта, растворенного
в воде и обладающего нейтральной реакцией.
Следующими отходами производства являются маточный раствор,
содержащий в себе азотную кислоту, растворенную в ней гремучую
ртуть и другие соли ртути, а также промывные воды, уносящие с собой
некоторое количество мелких кристаллов гремучей ртути и других
солей ртути.
Из приведенных данных явствует, что практический выход грему-
чей ртути составляет только 88—89% от теоретического. Таким
образом свыше 10% металлической ртути остается в маточном рас-
творе и в промывных водах, что, конечно, нерационально.
Раньше как маточный раствор, так и промывные воды спускали
в колодец под зданием производства, где гремучая ртуть отстаивалась.
Колодец периодически очищали, содержимое переносили в ящики
и заливали соляной кислотой с цинковой пылью. Время от времени
массу перемешивали деревянным веслом. Через месяц она приобре-
тала серо-черную окраску, что считали концом реакции. Содержи-
мое ящиков осторожно переносили в колодец вне здания и заливали
дополнительным количеством соляной кислоты. Чистку таких колод-
цев производили раз в 5—10 лет.
Подобный способ разложения отбросов гремучей ртути ие является
вполне надежным и не гарантирует безопасности. Были случаи само-
произвольных взрывов колодцев.
135
Чистка колодцев на одном из заводов показала, что в них содер-
жится значительное количество неразложившейся гремучей ртути.
Так, 20 кг землистого состава из колодца легко детонировало от кап-
сюля-детонатора № 8, образовав воронку глубиной до 1 м и диамет-
ром в 2—2,5 м. Анализ нескольких образцов изъятой из колодца
массы показал наличие около 25% фульмината и температуру
вспышки 166—170°.
Наряду с опасностью взрыва при чистке колодцев наблюдается
и сильное токсическое действие газов, выделяющихся из них. При
чистке колодцев на одном из заводов работающие испытывали затруд-
нения в дыхании и сильную головную боль. На третий день чистки
почти у всех имевших соприкос-
новение с отбросами или долгое
время находившихся у сруба ко-
лодца появилось кровотечение из
носа, нарывы на руках и на губах,
порезы не затягивались и все время
Фиг. 79. Отстойник. КРОВОТОЧИЛИ.
Скопление больших масс отбро-
сов гремучертутного производства вблизи производственных зданий
недопустимо, а колодцы, в которых из года в год скопляются
запасы опасных материалов, являются очагами, могущими в любое
время нарушить нормальную жизнь завода.
Существует и другой метод ликвидации отбросов. Промывные
воды спускают не в колодцы, а в отстойные ступенчатые ящики
(4мг. 79). Ящики очищают раз в 5—б дней, а отбросы уничтожают
подрывом. Этот способ значительно безопаснее первого.
Однако приведенные способы невыгодны тем, что значительные
количества сравнительно дорогой ртути уничтожаются нерацио-
нально. Поэтому вопрос о регенерации ртути из отбросов гремуче-
ртутного производства заслуживает серьезного внимания. Регене-
рировать ртуть можно только из растворов, ибо в данном случае
ни один пнрометаллургический метод не осуществим.
Исследования Каста показали, что при выпаривании маточного
раствора остается около 3% твердого остатка, содержащего 3—6%
ртути и 90—96% щавелевой кислоты.
Гаген рекомендовал нейтрализовать маточный раствор известью,
затем отгонять спирт, содержащий альдегид и нитрит, остаток же ре-
генерировать в воде в виде каломели или металлической ртути. Спирт,
содержащий альдегид и нитрит, по Делиону, можно употреблять для
получения гремучей ртути.
§ 9. ТЕХНИЧЕСКИЕ ТРЕБОВАНИЯ К ГРЕМУЧЕЙ РТУТИ
1. Гремучая ртуть должна иметь вид мелкокристаллического по-
рошка белого цвета. Допускается слегка сероватое окрашивание,
но ни в коем случае не допускается зеленое или желтое.
136
Гремучая ртуть должна быть промыта до нейтральной реакции
и не содержать видимых на-глаз примесей.
2. Допускаются следующие примеси:
Примеси 1 сорт % II сорт о/ /о
Нерастворимого остатка в аммиаке не более .... Металлической ртути и ее закисных соединений не 0,5 1,0
более 0,3 0.6
Щавелевой кислоты не более 0,3 0,5
Влажность не более - . . . . 0,02 0,03
Кремнезем и стекло . . Не допускается
3. Содержание фульмината должно быть не менее:
для I сорта . 99%
» II » 98%
4. Температура вспышки обоих сортов должна быть не ниже 150°.
По Касту и Метцу, допускается содержание в гремучей ртути не
более 2% примесей, нерастворимого в пиридине или в водном растворе
цианистого калия остатка не должно быть более 2%, металлической
ртути — не свыше 1%, а щавелевокислых солей и хлоридов не должно
содержаться вовсе. Цвет технической гремучей ртути допускается
от желтоватого до темнобурого.
Для качественного определения гремучей ртути может служить
ряд реакций.
По Лангхансу, при прибавлении к раствору гремучей ртути в пи-
ридине раствора 1 мл брома в 100 мл хлороформа появляется красно-
желтое окрашивание.
При смешивании 2%-ного раствора нитрита натрия с раствором
гремучей ртути в цианиде калия после подкисления разбавленной
соляной кислотой появляется постоянное окрашивание от сине-зеле-
ного до изумрудного цвета.
Если встряхивать гремучую ртуть с несколькими миллилитрами
раствора 5—6 мл брома и 10 г едкого кали в 100 мл воды, то полу-
чается небесно-голубое или васильковое окрашивание от образующе-
гося при этом бромнитрозометана.
При обливании фенилгидразином гремучая ртуть окрашивается
сначала в оливково-зеленый, затем в серый и, наконец, в красно-бурый
цвет с выделением ртути. Если после нескольких часов разбавить
фенилгидразин спиртом и смешать с разбавленной серной кислотой,
то получается красно-фиолетовая окраска. Реакция идет быстрее
при нагревании, причем последнее можно безопасно производить
и на голом огне, так как при обливании гремучей ртути фенилгид-
разином и встряхивании вся гремучая ртуть растворяется уже на
холоду.
137
Для определения гремучей ртути в составе берут 0,3 г последнего
обливают 0,5 мл фенилгидразина, встряхивают и нагревают. Когд
начинается бурная реакция с изменением цвета раствора и выделе!
нием азота, пробирку отставляют от пламени- По окончании реакци
содержимое пробирки разбавляют спиртом, фильтруют и подкисляю
серной кислотой, в результате чего появляется резкое красно-фиоле
товое окрашивание. Эта реакция настолько чувствительна, что даже
отдельный кристалл гремучей ртути, будучи смочен двумя-тремя
каплями фенилгидразина, дает соответствующую окраску. Она обна-
руживает гремучую ртуть и в растворах, но не во всех. Так, реакцию
можно наблюдать при действии фенилгидразином на раствор гремучей
ртути в пиридине, на свежеприготовленные растворы ее в тиосульфате
и соляной кислоте, однако, аммиачный раствор этой реакции не
дает.
Окраска раствора, невидимому, обусловливается появлением пара-
розанилина:
NHgCeH.s xCeH4NH2
/С\
NH2C6H/ хОН
Качественный и количественный анализ гремучей ртути сводится
к определению в ней фульмината ртути и тех вредных и ненужных
примесей, которые обычно ее сопровождают.
1. Определение кислотности производится перед
пуском гремучей ртути на сушку. Синюю лакмусовую бумажку при-
жимают к влажному продукту или вносят ее в промывные воды.
В том и другом случае не должно происходить изменения цвета бу-
мажки.
2. Определение влажности. В тарированный стакан-
чик помещают точную навеску гремучей ртути около 1 г. Стаканчик
с навеской вместе с другим стаканчиком с металлической ртутью
сушат в эксикаторе при температуре 40° и вакууме не менее 15 мм
в течение 5—6 час.; вследствие испарения металлической ртути ат-
мосфера в эксикаторе становится насыщенной парами ртути, что пред-
охраняет от улетучивания металлической ртути из взятой навески.
Эксикатор снабжен электрической плиткой с закрытой нагрева-
тельной обмоткой. Степень нагревания регулируют реостатом. Для
выкачивания воздуха эксикатор снабжен тубусом. Перед эвакуацией
воздуха эксикатор предварительно нагревают до 40°. Первый раз
взвешивают через 4 часа. По истечении еще одного часа взвешивают
второй раз. Если второе взвешивание не покажет уменьшения веса,
то можно считать сушку законченной; в противном случае сушку
продолжают до постоянного веса. Разница в весе стаканчика с грему-
чей ртутью до и после сушки дает количество влаги, содержащейся
в исследуемом продукте.
Проще и достаточно точно можно определить влажность высушива-
нием гремучей ртути в термостате при 45° до постоянного веса в при-
сутствии металлической ртути.
3. Определение нерастворимого остатка.
Сперва подготовляют фильтр, промывая его последовательно насы-
щенным раствором аммиака в воде (d = 0,885), дистиллированной
водой, 96%-ным спиртом и безводным эфиром. Для высушивания
фильтр помещают на сутки в эксикатор, в котором находится стакан-
чик с крепкой серной кислотой для поглощения паров эфира. После
этого фильтр взвешивают и сохраняют в банке с притертой пробкой.
Для определения берут такое количество влажной гремучей ртути,
чтобы после сушки получилось 0,7—1 г сухой, и сушат одни сутки
в эксикаторе над хлористым кальцием вместе со стаканчиком с метал-
лической ртутью. Высушенную гремучую ртуть растворяют в 30 мл
насыщенного раствора аммиака в воде (d == 0,885) и быстро фильт-
руют. Остаток на фильтре промывают 30 мл того же аммиака, затем
30 мл аммиака, наполовину разбавленного водой, затем дистилли-
рованной водой до удаления аммиачного запаха, затем 96%-ным
спиртом и, наконец, безводным эфиром. Фильтр сушат в присутствии
металлической ртути и крепкой серной кислоты в эксикаторе в про-
должение суток и взвешивают. Вместо бумажного фильтра можно
пользоваться фильтром Шотта; при этом устраняется необходимость
длительной подготовки фильтра. Разница в весе фильтра показывает
количество нерастворимого остатка.
4. Определение общего содержания метал-
лической ртути и ее закисных соединений
можно производить, пользуясь нерастворимым остатком, полученным
при предыдущем анализе, или, пользуясь навеской гремучей ртути»
оставшейся после определения влажности. В первом случае нераство-
римый остаток в тигле Шотта помещают в термостат, выдерживают при
температуре 150—160° в течение часа и взвешивают. Во втором случае
стаканчик с навеской гремучей ртути после сушки вновь помещают
в тот же эксикатор, предварительно удалив из него стаканчик с метал-
лической ртутью, поднимают температуру до 60° и осторожно выка-
чивают воздух. Эвакуацию воздуха повторяют через каждый час. По
истечении 4 час. пробу взвешивают и спустя 2 часа взвешивают по-
вторно. Если в этом случае вес не изменяется, то по разности вычис-
ляют процентное содержание металлической ртути и ее закисных
соединений; если будет обнаружена убыль веса, то высушивание
продолжают еще 2 часа.
5. Определение щавелевой кислоты можно про-
изводить, пользуясь новой навеской или фильтратом, оставшимся
при производстве анализа на нерастворимый остаток.
В первом случае навёску 0,7—1 г гремучей ртути в стакане
емкостью 250 мл заливают 150 мл дистиллированной воды и обраба-
тывают в течение 30—40 мин. сероводородом. Затем, не прерывая
пропускания сероводорода, к раствору прибавляют 10—15 мл соля-
ной кислоты (уд. веса 1,12), стакан помещают на кипящую водяную
баню и нагревают до тех пор, пока осадок не станет хорошо отстояв-
шимся, а раствор прозрачным. Полученную таким образом, хорошо
фильтрующуюся сернистую ртуть отфильтровывают и промывают
139
сероводородной водой, слабо подкисленной соляной кислотой. Ия
фильтрата кипячением удаляют сероводород. После удаления серо-,
водорода к раствору прибавляют несколько капель перекиси водорода
и аммиака до нейтральной реакции. Нейтральный раствор подкис-^
ляют несколькими каплями уксусной кислоты, нагревают до кипе-
ния и осаждают щавелевую кислоту 10 мл раствора хлористого каль-
ция (Ю%). Выпавший щавелевокислый кальций отстаивают в течение
12 час., после чего отфильтровывают через беззольный фильтр, промы-
вают горячей водой, сушат, сжигают фильтр над фарфоровым тиглем,
прокаливают последний и взвешивают в виде окиси кальция.
Формула расчета:
%С2Н2О4 =
где р — навеска гремучей ртути;
а — вес окиси кальция.
Во втором случае к аммиачному раствору, получившемуся при
определении нерастворимого остатка, приливают 10—15 мл сер-
нистого аммония и выпавшую сернистую ртуть отфильтровывают, j
Фильтр нагревают на водяной бане до удаления свободного аммиака |
(от 30 мин. до 1 часа). После этого прибавляют 3—5 мл крепкой
соляной кислоты и выпаривают иа водяной бане до удаления запаха
сероводорода, прибавляют аммиак до остающегося запаха и затем
вновь удаляют кипячением на бане. Выпавшую при этом серу отфиль-
тровывают и промывают горячей водой. В горячий фильтрат с про-
мывными водами приливают 10 мл 10%-ного фильтрованного рас-
твора хлористого кальция; выпавший при этом щавелевокислый каль-
ций отстаивают в течение 12—18 час. в теплом месте (около 25°),
после чего переносят на фильтр, промывают водой до полного удале-
ния иона хлора (проверка нитратом серебра), осадок на фильтре
растворяют в 10%-ной серной кислоте, кипятят и титруют 0,1N рас-
твором марганцовокислого калия.
6. Определение кремнезема и стекла. 2 г гре-
мучей ртутн растворяют в водном растворе аммиака. Раствор сливают,
а остаток смывают на фильтр и промывают 10—15% NH4OH и водой.
Фильтр подсушивают и остаток рассматривают под микроскопом. При
обнаружении стекловидных веществ остаток обрабатывают царской
водкой и вновь рассматривают под микроскопом.
7. Определение содержания фульмината
ртути основано на разложении гремучей ртути водными раство-
рами некоторых солей и образовании при этом определенного коли-
чества щелочи. Так, водные растворы тиосульфата натрия и йодистого
калия при растворении в них гремучей ртути дают щелочные рас-
творы с содержанием четырех ионов ОН на 1 моль гремучей ртути. При
пользовании в качестве растворителя тиосульфатом натрия анализ
можно сделать алкалиметрическим методом, оттитровывая образую-
щуюся щелочь кислотой, или йодометрическим методом, оттитровы-
вая не вступивший в реакцию с гремучей ртутью тиосульфат натрия
раствором иода в иодистом калии.
140
Алкалиметричесойметод был предложен Броуне-
доном в 1904 г. и несколько уточнен в 1912 г. Р. Филиппом. При поль-
зовании этим методом требуется соблюдение строгого количествен-
ного соотношения применяемых при анализе реактивов и точного
времени производства анализа. Объясняется это тем, что в результате
реакции гремучей ртути с тиосульфатом натрия получается нестой-
кий раствор, в котором щелочность изменяется в зависимости от ко-
личества тиосульфата натрия и времени растворения и титрования.
При растворении гремучей ртути в водном растворе тиосульфата
натрия образуется растворимое комплексное соединение ртути по
уравнению:
Hg (0NC)2 + 2Na2S2O3 -> [Hg(S2O3)2] Na2 + 2NaONC
Фульминат натрия, как соль слабой кислоты и сильного основания,
гидролитически распадается:
2NaONC + 2Н2О -> 2NaOH + 2HONC
Таким образом в результате реакции тиосульфата натрия с грему-
чей ртутью образуются только 2 иона НО' на 1 моль гремучей ртути.
Щелочной же раствор при титровании обнаруживает большее коли-
чество ионов ОН' на 1 мол, потому что в процессе реакции в рас-
творе возникают еще 2 дополнительных иона ОН' вследствие распада
избыточного тиосульфата натрия (образование других сильных осно-
ваний и аммиака в растворе исключено). Уравнение реакции возник-
новения двух дополнительных ионов ОН' еще не изучено и стехио-
метрически точно не может быть написано. Однако целый ряд
побочных показателей позволяет предположительно установить ход
их возникновения.
В самом деле в процессе титрования гремучей ртути, растворенной
в водном растворе тиосульфата натрия, зачастую совершенно ясно
ощущается запах цианистого водорода, в то время как при растворе-
нии ее никакого запаха не чувствуется- Поэтому вполне вероятно,
что вначале, как и указывает Филипп, образуется комплексное со-
единение тиосульфата натрия и гремучей кислоты, углерод которой
имеет ненасыщенные связи:
,Na
1) 2HONC + 2Na2S2O3 -> 2HONC<
xS2OsNa
или
zNa
IIONC(
/S2O3 • S2O8Na2
HONCf
xNa
/Н • HONa
2) 2HONC + 2Na2S2O3 + 2H2O -> 2HONC<
XS2O3Na
141
При титровании раствора происходит распад образовавшегося
комплексного соединения, конечными продуктами которого являются
цианистая, циановая и изоциановая кислоты:
/Na
honcz ,н
)S2O3 • S2O3Na2 4- H2SO4 4 КО -> HONCZ -f-
HONCZ “
(формогидроксамовая кислота)
,н
+ HONC( + Na^Oe + Na2SO4
1 (формоксим)
Формогидроксамовая кислота и формоксим распадаются на воду,
синильную и циановые кислоты.
Таким образом 1 моль гремучей ртути соответствует 4 ионам ОН'.
Опыт показывает, что щелочность раствора убывает с увеличе-
нием времени работы с ним. Установлено, что при объемной цене
капли дсцинормального раствора серной кислоты, равной 0,04’ мл,
понижение щелочности раствора 0,3 г гремучей ртути в 30 мл 20%-ного
водного раствора тиосульфата натрия соответствует 0,12 мл децинор-
мального раствора серной кислоты в 1 мин. в первые 2 мин. после
начала растворения. Результаты титрования, следовательно, нуж-
даются в положительной поправке.
Раствор титруют в присутствии метилоранжа. Вследствие того,
что переход цвета гремучертутного раствора при титровании заметить
очень трудно, приходится сознательно перетитровывать раствор до
достижения эталонного цвета. Эталоном является цвет 150 мл дистил-
лированной воды с 5 каплями метилоранжа (1 : 5000) и с 2 каплями
децинормальной серной кислоты. Действительная точка нейтрали-
зации, стало быть, на 2 капли ниже той, которую мы наблюдаем
при титровании, и полученный результат нуждается в поправке,
равной удвоенному объему капли, т. е.
0,04 • 2 = 0,08 мл.
Раствор тиосульфата натрия в воде обладает щелочной реакцией,
что также должно быть учтено при вычислении результатов титрова-
ния. Чтобы при нейтрализации 30 мл 20%-ного раствора тиосуль-
фата натрия, разбавленного 120 мл воды, получить эталонный цвет,
приходится добавить 8 капель децинормальной серной кислоты. Сле-
довательно, отрицательная поправка на эталонный цвет и щелочность
тиосульфата натрия будет равна:
(8 —2)-0,04 = 0,24 мл.
Как было указано выше, щелочность гремучертутного раствора
убывает со скоростью, соответствующей 0,12 мл 0,1 N серной кис-
142
лоты в минуту. Поэтому наиболее рациональным временем для про-
изводства работы является:
Следовательно, работа по растворению гремучей ртути в водном
растворе тиосульфата натрия и титрованию раствора должна быть
выполнена в течение 2 мин. При этом условии никаких поправок
вводить не нужно. Исходя из этих соображений, алкалиметрический
метод анализа осуществляют следующим образом.
Навеску гремучей ртути около 0,3 г (не менее 0,284 г и не более
0,355 г) помещают в совершенно сухую колбочку емкостью 300 л/л.
В случае применения влажной колбы или, еще хуже, колбы с каплями
воды происходит комкование кристаллов гремучей ртути и связанное
с этим замедление процесса растворения. Навеску гремучей ртути за-
ливают 30 мл 20%-ного раствора тиосульфата натрия (раствор тио-
сульфата натрия должен быть приготовлен за 40—48 час. до анализа
и отфильтрован от выпавшей серы). Отмечают время приливания
раствора тиосульфата натрия и навеску гремучей ртути растворяют
при встряхивании. Растворение в этих условиях практически зани-
мает 0,5—1 мин. Количество жидкости в колбе доводят до 100 мл,
приливая 70 мл воды. Прибавляют 5 капель метилоранжа (раствор
I : 5000) и титруют 0,1JV серной кислотой до получения эталонного
цвета.
Титрование следует всегда начинать в одно и то же время — че-
рез 1 мин. после начала растворения. Для того чтобы закончить тит-
рование в I мин., необходимо предварительно определить расход
серной кислоты на отдельно взятую иавеску гремучей ртути. Для
титрования раствора 0,3 г чистой гремучей ртути требуется 42,25 мл
0,17V серной кислоты. Рассчитывают по формуле:
%Hg(ONC)2= 145.1 А- %50-,
где 145,1 — отношение молекулярного веса гремучей ртути к
удвоенному молекулярному весу серной кислоты, ум-
ноженному на 100;
А — число миллилитров серной кислоты, пошедшей на тит-
рование;
Тн^о. — титр серной кислоты:
р — навеска гремучей ртути.
Титрование требует некоторой привычки, так как работу необхо-
димо производить быстро и перемена цвета вначале недостаточно
резка. Для получения надежных результатов следует производить
несколько опытов титрования (обычно достаточно трех-четырех).
Алкалиметрический метод дает несколько повышенные результаты.
Йодометрический метод. Лучшим методом, свобод-
ным от недочетов алкалиметрического, является йодометрический
метод.
143
Гремучая ртуть, растворяясь в водном растворе тиосульфата нат-
рия, разлагается, вступая в реакцию с последним:
mHg(ONC)a + n(Na2S2O3) -> m lHg(0NC)s 1 -f- (n — m) Na2S2O3
lNa3S2O3 J
Избыток тиосульфата натрия (л—т) определяют йодометрическим
путем. Однако титрование раствора гремучей ртути в растворе тио-
сульфата натрия без соблюдения особых условий сохранило бы все
погрешности алкалиметрического способа анализа, ибо, как мы ви-
дели, при последнем играет значительную роль время анализа. Кроме
того, влияние изменения щелочности, а стало быть, и количества
израсходованного тиосульфата натрия, не дало бы возможности
сделать результат более достоверным, чем при алкалиметрическом
методе. Поэтому применяют в качестве растворителя более сильный
реагент по отношению к гремучей ртути —• иодистый калий.
Для работы берут 0,1N раствор тиосульфата натрия и 0,1N рас-
твор иода. Раствор тиосульфата натрия приготовляют за 10—14 дней
до установления титра. Необходимость заблаговременной подготовки
раствора вытекает из того, что тиосульфат натрия вступает во
взаимо-действие с угольной кислотой, обычно находящейся в дистил-
лированной воде:
Na2S2O3 + 2Н3СО3 2NaHCO3 + H2S2O3
H2S2O3 -> H2SO3 + S
Сернистая же кислота, вступая в реакцию с иодом
H2SO3 + 2J -f- Н2О -> 2HJ + H2SO4
повышает титр тиосульфата натрия. Иными словами, титр меняется
до тех пор, пока вся угольная кислота не войдет в обменное разложе-
ние с тиосульфатом натрия. После удаления из раствора всей угле-
кислоты, что происходит через 10—14 дней, титр остается неизмен-
ным ряд месяцев.
В колбе емкостью около 300 мл с 50 мл О, IN водного раствора
тиосульфата натрия растворяют 3 г йодистого калия и быстро всы-
пают навеску гремучей ртути около 0,3 г. Содержимое колбы взбал-
тывают до растворения гремучей ртути.
В колбе протекают следующие реакции:
1) гремучая ртуть реагирует с иодистым калием, давая раствори-
мую комплексную соль и фульминат калия:
Hg (0NC)2 + 4КJ [HgJ4jK2+2K0NC
2) фульминат калия, как соль слабой кислоты и сильного осно-
вания, гидролизует:
2KONC-f-2H2O 2HONC+2KOH
144
3) гремучая кислота по приведенной выше схеме вступает во
взаимодействие с тиосульфатом натрия, конечными продуктами
которого являются тетратионат натрия, едкий натр, синильная
и изоциановая кислоты:
2HONC + 2NasS2O3 + Н2О -> Na£S4O6 + 2NaOH + HCN + HNCO
После растворения гремучей ртути к раствору прибавляют 50 мл
воды и 4—5 капель метилоранжа (1 : 5000) и титруют О, IN серной
кислотой до остающегося розового окрашивания. Нейтральный рас-
твор титруют О, IN раствором иода в присутствии крахмального рас-
твора в качестве индикатора до неисчезающего синего окрашивания;
2Na2S2O3 4- J2 = 2NaJ + Na2S4O6
и производят расчет, исходя из количества тиосульфата натрия, всту-
пившего в реакцию с гремучей кислотой.
8. Определение содержания ртутнпроизводится
осаждением ртути сероводородом из раствора гремучей ртути в слабо
разбавленной соляной кислоте в виде сульфата ртути.
Можно сделать анализ и электролитическим путем: 0,4—0,5 г высу-
шенной при 75° гремучей ртути вначале осторожно нагревают с 2,5—
3,5 мл азотной кислоты (d = 1,4), затем доводят раствор до кипения
и кипятят до исчезновения окислов азота. Разбавляют раствор водой
и ведут электролиз на холоду, вначале при-0,4 А и 1,9—2,0 V, затем
при 0,25 А и 2,6 V. Ртуть выделяется полностью через 6 час. Уско-
рения работы можно добиться, применяя вращающиеся электроды
и повышая силы тока до 0,8—1,2 А при напряжении 2V.
9. Определение азота производят методом гидриро-
вания при температуре около 250° в струе водорода в присутствии
катализатора.
Сущность этого метода заключается в следующем: навеску грему-
чей ртути от 0,015 до 0,03 г осторожно измельчают при помощи ре-
зиновой пробки, зажатой деревянной ручкой. После измельчения
гремучую ртуть тщательно перемешивают со свежевосстановленным
никелем, служащим катализатором. Недостаточно измельченная гре-
мучая ртуть может при последующем нагревании привести к вспышке.
Смешанную с никелем навеску помещают в фарфоровую лодочку,
которую вставляют в тугоплавкую стеклянную или лучше кварце-
вую трубку длиной 50 см, наполненную в средней своей части асбе-
стированным никелем. Стеклянную трубку помещают в электри-
ческую печь, причем тот конец трубки, в который вставлена лодочка,
печью не обогревается, а нагревается отдельной горелкой; другой же
конец трубки, выступающий на 4—-5 см из печи, соединяют изогнутой
трубкой с небольшим стаканчиком (с водой), где поглощается образу-
ющийся аммиак. Над стаканчиком устанавливают закрепленную на
штативе бюретку, наполненную 1/4oN раствором H2SO4. Трубку со
стороны лодочки соединяют с прибором Киппа, служащим для полу-
чения водорода. Последний пропускают для очищения от могущего
бубнов—92-10 145
образоваться сероводорода через раствор сулемы и для высуши-
вания—через крепкую серную кислоту.
Вначале для вытеснения воздуха пропускают водород, затем вклю-
чают печь в электрическую сеть и поддерживают температуру трубки
около 250° при помощи реостата. По достижении этой температуры,
на что с начала пропускания водорода требуется 1—Р/2 часа времени,
приступают к нагреванию лодочки. Перед этим прибавляют в стакан-
чик с водой 2 капли метилоранжа и 2—3 капли 1/40TV раствора H2SO4.
При этом появляется оранжевая окраска, свидетельствующая о том,
что воздух до нагрева печи был полностью вытеснен водородом. На-
гревание лодочки следует вести очень осторожно, для чего горелкой
обводят в течение короткого времени часть трубки, где помещена ло-
дочка, и отставляют горелку в сторону, многократно повторяя эту
операцию, постепенно увеличивая время обогрева. Пожелтение рас-
твора в стаканчике свидетельствует о наступлении момента гидриро-
вания. Затем прибавляют из бюретки несколько капель Н28О4до кис-
лой реакции, продолжая обогревать лодочку. Через некоторое время
раствор опять желтеет, вновь прибавляют H2SO4 и нагревают лодочку
до тех пор, пока избыточная капля серной кислоты не будет окраши-
вать раствор в оранжевый цвет, несмотря на длительное и сильное
нагревание лодочки. Такой способ титрования дает возможность сле-
дить за скоростью разложения гремучей ртути и регулировать ее.
Лодочку нагревают от Р/2 до 2 час.
Асбестированный никель следует после нескольких определений
прокалить на воздухе, чтобы сжечь накопившиеся в нем углеродистые
вещества и вновь восстановить в струе водорода.
ГЛАВА IV
АЗИД СВИНЦА
§ 1. ОТКРЫТИЕ АЗИДА СВИНЦА
Азид свинца был получен впервые Курциусом еще в 1891г. Сразу же
после появления первой статьи о его получении им занялись военно-
технические учреждения главнейших стран мира. Опыты Билля и
Ленца уже в 1892—1893 гг. показали ряд его преимуществ по сравне-
нию с прочими инициирующими ВВ. в частности, по сравнению с
гремучей ртутью. Независимо от этих исследований к тем же ре-
зультатам пришли в 1907 г. Велер и Маттер.
Азид свинца был предложен в качестве самостоятельного ини-
циирующего ВВ Гиронимусомв 1907 г. сначала во Франции, а позднее
в Германии, США и Австрии. В том же 1907 г. Велер получил патент
(сначала в Германии, а несколько позднее и в других странах) на при-
менение азидов тяжелых металлов вместо гремучей ртути в капсюлях-
детонаторах, а Рейнско-Вестфальское акционерное общество взрыв-
чатых веществ в 1910 г. патентовало применение азида свинца в
смесях с сернистым азотом или диазо бензо лнитратом в качестве ини-
циирующего заряда для капсюлей-детонаторов.
В 1909—1910 гг. азидом свинца заинтересовалось и военное ведом-
ство России. В конце 1910 г. вышла в свет монография преподавателя
Артиллерийской академии А. А. Солонина, посвященная азидам
тяжелых металлов и явившаяся результатом работы автора в лабо-
ратории проф. Велера в Карлсруэ, а в Артиллерийском журнале
была напечатана его статья об азидах.
Однако до войны 1914—1918 гг. азид свинца не нашел широкого
применения, несмотря на то, что он обладает, как показал своими
исследованиями Велер, значительно большим постоянством при хра-
нении, даже под действием повышенной температуры, чем гремучая
ртуть. Необходима была серьезная практическая проверка.
Во время первой империалистической войны к производству азида
свинца и ег,о применению стали постепенно переходить Швейцария,
США, Россия и другие страны. Недостаток в металлической ртути
для производства гремучей ртути вынудил и Германию, хотя и в огра-
ниченных количествах, организовать производство азида свинца и
начать применять его в подрывных капсюлях-детонаторах. В Англии
же так до конца войны и не могли использовать патента на азид
147
свинца в производстве капсюлей-детонаторов, а в Австрии только
тогда занялись серьезным исследованием вопроса о применении
азида свинца, когда вследствие наступления итальянцев на Goriz
оказались под обстрелом австрийские ртутные разработки.
В России, по инициативе проф. С. П. Вуколова и Р. В. Мусселиуса,
приступили к снаряжению азидных капсюлей-детонаторов для ряда
морских боеприпасов (в частности, капсюля-детонатора образца
1913 г.). Взрыватели, снабженные азидными капсюлями-детонато-
рами, ни разу за время войны не вызывали нареканий. Сухопутное же
ведомство не решилось на применение азида свинца, несмотря на про-
веденные большие полигонные работы; только в 1917 г. Артиллерий-
ский комитет, разбирая вопрос о применении азида свинца в капсю-
лях-детонаторах для ручных гранат и для 9-сл* бомбомета, отметил,
что желательно начать изготовление азидных капсюлей-детонаторов:
1) к трубкам образца 1916 г.; 2) к трубкам ружейных гранат; 3) к труб-
кам для нарезных ружейных мортирок; 4) к трубкам образца 1915 г.;
5) для взрывателей 10-ДТ.
Серьезными аргументами против азида свинца были общеизвестные
его недостатки: действие лучей солнечного света, возможность само-
произвольных взрывов отдельных кристаллов, недостаточная чув-
ствительность к лучу огня, пониженная чувствительность к наколу
жала, а также плохая сыпучесть и плохая прессуемость. По мере
того как улучшалось производство азида свинца,— устранялись от-
дельные недостатки и появлялись способы их локализации. Необхо-
димость же повышения начального импульса, а также стремление
уменьшить размеры капсюлей-детонаторов ныне привели к тому, что
азид свинца повсеместно стал вытеснять гремучую ртуть.
В настоящее время во многих странах применяют азид свинца для
снаряжения комбинированных капсюлей-детонаторов для военных
целей и для горной промышленности. В Германии, например, азид
свинца в больших количествах изготовлялся на заводе Рейнско-Вест-
фальского общества взрывчатых веществ в Тройсдорфе. Одним этим
заводом до 1932 г. было выработано 750 000 кг азида свинца. Точно
так же в больших количествах азид свинца производили во Франции.
По неполным данным Герца, общее количество азида свинца, вырабо-
танное заводами важнейших стран мира к 1932 г., составляло
1 500 000 кг.
§ 2. АЗОТИСТОВОДОРОДНАЯ КИСЛОТА, ЕЕ СТРОЕНИЕ И СВОЙСТВА
Органические соединения, содержащие группу N3, были известны
давно. Первое такое соединение было синтезировано Петером Гриссом
еще в 1866 г.; он получил фенилазид (диазобензолимид), обрабаты-
вая пербромидбензолдиазоний водным раствором аммиака. Двена-
дцать лет спустя Эмиль Фишер получил тот же фенилазид, действуя
гидратом окиси калия на фенилнитрозогидразин. Еще позднее был
получен целый ряд соединений с группой N3. Природа этих соеди-
нений не была точно известна до открытия азотистово до родной
кислоты.
148
Азотистоводородную кислоту — это интересное неорганическое со-
единение, характеризующееся чрезвычайной взрывчатостью,—открыл
в 1890 г. Курциус. Она была впервые получена в водном растворе
омылением азидов органических кислот RCON3.
«К числу блестящих химических открытий текущего года,— писал
в том же году великий русский химик Д. И. Менделеев, —бесспорно
относится открытие Курциуса, показавшего существование HNS,
путь ее получения и аналогию этой азотистоводородной кислоты
(азоимида) с галоидными кислотами. Я считаю нелишним высказать
свои соображения, потому что они, по моему мнению, помогут предви-
деть реакцию и найти пути для новых способов получения этой
примечательной кислоты».
Методы получения азотистоводородной кислоты и ее производ-
ных, применявшиеся и применяемые разными исследователями, можно
разбить на шесть групп.
К первой группе принадлежат синтезы, в основе кото-
рых лежит действие азотистой кислоты и ее органических или неорга-
нических производных на гидразин и его производные. Простейший
вид реакции этой группы:
NHa — NH2 + HNO2 -> HN3 + 2H2O
был осуществлен Курциусом в 1893 г. обработкой охлажденного
льдом раствора гидразингидрата конденсированными бурыми парами
окислов азота, выделяющихся из смеси азотной кислоты и трехокиси
мышьяка.
К синтезам этой группы следует отнести:
1. Классический синтез азотисговодородной кислоты, произве-
денный Курциусом и сводившийся к следующему:
а) получение из этилового эфира бензоилгликолевой кислоты дей-
ствием гидразина, вернее гидразингидрата. бензоилгидразина:
CGH5CO . ОСН2 • СОО • С2Н5 + 2N2H4 -> С6Н5СО • NH • NH2 +
+ NH2NH • СН2СОО - С2Н5 + Н2О
б) получение из бензоилгидразина действием азотистой кислоты
азида бензойной кислоты или бензоилазоимида:
CcH5CONHNH2+HONO -> C6H5CON3 + 2Н2О
в) получение из бензоилазоимида действием щелочи азида щелоч-
ного металла:
C6H5CON3 + 2NaOH -> NaN3 + C6H5COONa + H20
т) наконец, получение самой азотистоводородной кислоты из ще-
лочного азида действием минеральной кислоты:
2NaN3 + H2S04 > 2HN3 + Na2SO4
2. Получение солей азотистоводородной кислоты действием ни-
тритов металлов на гидразинсульфат или гидразиннитрат.
149
Например, Анжели получил азид серебра при взаимодействии
раствора гидразинсульфата на насыщенный раствор нитрита се-
ребра. Он объясняет образование азоимида следующими реакциями:
NH2 — NH24-HONO -> NH2—N =N —OH + H2O
NH2 - N = N OH HN3 + H2O
При взаимодействии гидразингидрата и нитрита натрия в запаян-
ной трубке при 150° Годкинсон также получил некоторое количество
азида натрия.
Деннштедт и Голинг получили азотистоводородную кислоту обра-
боткой гидразинсульфата раствором нитрита калия в кислой среде:
3N2H4 HsSO4 + 6KNO2 + 3H2SO4 ->
2HN3 + 8H3O + 6KHSO4 + N3 + O2 + 2N3O
3. Способ Сабанеева и Деньгина, заключающийся в нагревании
кислого азотнокислого гидразина, распадающегося по уравнению:
6N2H4 HNO3)2 -> 2HN3 + NH4NO3 + X,
где X — продукты распада (Н2О, КО, HN03 и др.).
4. Получение азотистоводородной кислоты способом Тиле из
гуанидиннитрата или нитрата мочевины, путем их амидирования,
диазотирования (азидирования) с последующим разложением азидо-
соединений едкими щелочами или нитратом серебра.
5. Получение азо имида действием спирта и алкилнитрита на сер-
нокислый гидразин:
NH2 — NH2 + RO - NO 4- RONa -> NaN3 + 2ROH + H2O
Штолль для этого применял амилнитрит, а Тиле рекомендовал приме-
нение этилнитрита, дающего, однако, меньший выход продукта.
6. Синтез Штаудингера, предложившего получать азотистоводо-
родную кислоту действием гидразина на вторичный нитрозоамин,
как, например, на дифенилнитрозоамин в щелочном растворе. При
обыкновенной температуре реакция идет медленно, при нагрева-
нии же — быстро, по уравнению:
(C6H5)2N — NO + N3H4 + NaOH ->
NaN3 + (CcH5)2 NH + 2H2O
Ко второй группе относятся реакции окисления гидра-
зина. В 1902 г. Танатар нашел, что при окислении молекулярной смеси
гидразина и гидроксиламина бромной водой, перманганатом, дву-
окисью свинца или свинцовым суриком в кислом растворе всегда обра-
зуется некоторое количество азотистоводородной кислоты. Выход
при применении перекиси водорода 24,3%, а при применении
хромовой кислоты 29,27%.
150
Реакция окисления представляется в следующем виде:
NH2— NH2 + NH20H + 20 HN3 + ЗН2О
Браун и затем Мартин получали взаимодействием перекиси водо-
рода с гидразинсульфатом очень чистую азотистоводородную кислоту.
К этой группе синтезов примыкает и реакция Танатара, по кото-
рой азоимид получается действием гидразина на хлористый азот;
NH2 - NH2 + NC13 HN3 + ЗНС1
К третьей группе относятся реакции взаимодействия
амидов металлов с закисью азота, в основе которого лежит реакция;
NH3 + N30 -> HN3 + Н20
В 1892 г. В. Вислиценус нашел, что при взаимодействии расплавлен-
ного амида натрия с сухой газообразной закисью азота образуется
азид натрия:
2NaNH2 + N3O NaN3 + NaOH + NH3
Подобная реакция происходит, если применять вместо амида натрия
амид калия или цинка.
Четвертая группа охватывает реакции окисления три-
азенов и разложения других азотоводородов.
Курциус показал, что диазосоединения и гидразин или его про-
изводные реагируют, образуя в качестве промежуточного продукта
изотетразены — гипотетические азотистоводородные соединения, на-
званные им буциленами:
RN = N OH+H2N —NH-R ->
-> R — N = N — NH — NH — R + Н20
которые затем разлагаются, образуя амины и производные азотисто-
водородной кислоты:
r~N = N —NH-NH-R RNH2+RN3
Соли гидроксиламииа и диазония в присутствии карбоната натрия
образуют азиды:
(C6H5N2)2SO4 + 2NHoOH • НС1 -> 2C6H5N2 - NH(OH) ->
Na-CO3
C6HBNS+ Hs0
Бомбергер получил фенилазид, обрабатывая p-фенилгидроксил-
амин гвдроксиламином в присутствии окислителя:
CcH,NHOH + О C6H6NO + Н20
CeH6NO 4- NH2OH CoH6N2OH + Н.0
С6Н5М2ОН + NH2OH C6H5N(OH)N2H + 0
CcH6N(0H)N,H -> CcH5N3+H.O
151
Димрот наблюдал, что при окислении фенилтриазена образуется
фепилазид:
СЛМД + О -> C«H,N3 + Н2О
Дарапский окислял гидразиндикарбамид и получал производные
азотисговодородной кислоты:
NH —CONH2 N—CONH2
I 4-0 -^ || + Н20
NH - CONH2 N - CONH,
N — CONH2
II + 0 CH2N —N = N-CONH.
N — CONH2
NSCONH2 +H2o + co2
N3CONH24-NaOH NaNg + NH3 + C02
Фреунд и Шацдер по лучили' азотистоводородную кислоту дей-
ствием щелочи на амидотриазосульфоль:
N—N
II II
H.N —С С — NIL,
К п я т о й группе относятся методы получения азидов не-
посредственно из металла и азота.
В. Мольденгауер и Г. Меттиг непосредственным действием активи-
рованного азота при электрическом разряде под уменьшенным давле-
нием на металлический натрий, калий, цезий и рубидий получали
азиды соответствующих металлов. При этом продукты соединения
(азиды) содержали незначительные количества нитридов, получаю-
щихся в результате последующего термического распада азидов.
X. Баттенберг считает, что при этом синтезе вообще происходит
одновременное образование и нитридов и азидов, т. е. что нитриды
в данном случае могут образоваться не только вследствие терми-
ческого распада азидов, но и непосредственно.
К шестой группе следует отнести методы получения
азотистоводородной кислоты, в основе которых лежит гидролиз аро-
матических азидов. Методы эти не являются вполне самостоятельными,
так как азидогруппа в соединениях образуется по одному из рас-
смотренных выше методов. Мы их выделяем в особую группу, так
как они показывают, что азотистоводородная кислота аналогична
галоидным кислотам.
В 1891 г- Нольтинг и Гранд-Мужен получили азид калия действием
спиртового раствора гидрата окиси калия на динитрофенилазид.
В следующем году Нольтинг, Гранд-Мужен и Мишель нашли, что азид
калия получается также действием спиртового раствора калия на
152
орто- и паранитрофенилазиды и ряд других орто- и парапроизводных,
фенилазвда. Метапроизводные подобным образом не реагируют, вслед-
ствие чего выход HNS при гидролизе метапроизводных невелик (не
более 40%).
Азотистоводородная кислота (безводная) представляет собой бес-
цветную, прозрачную, легкоподвижную жидкость, обладающую чрез-
вычайно резким, неприятным запахом.
Впервые безводная азотистоводородная кислота была получена
Курциусом и Раденгаузеном. Получение сопровождалось несчаст-
ным случаем — Раденгаузен был ранен.
Безводная азотистоводородная кислота может быть получена по-
одному из следующих двух методов:
I. Фракционной дистилляцией водного раствора азотистоводород-
ной кислоты и дальнейшей сушкой первой фракции хлоридом каль-
ция- При перегонке водного раствора кислоты при 45° первая фракция
второй перегонки содержит около 91% HN3 и не является гидратом.
Деннис и Бенедикт применяли перегонку под уменьшенным давле-
нием.
2. Путем воздействия на сухой азид калия разбавленной серной
кислоты (2 : I), отгонки образующейся азотистоводородной кислоты
не содержащим углекислоты воздухом через сушильные трубки
с хлористым кальцием и конденсацией кислоты в охлажденном
жидким воздухом приемнике.
Безводная азотистоводородная кислота тяжелее воды. Она кипит
при температуре 37°. При обычных (комнатных) температурных
условиях стойка, но обладает значительной летучестью. Температура
плавления —80°. Д парообразном состоянии азотистоводородная
кислота мономолекулярна.
Работа с безводной кислотой очень опасна вследствие чувстви-
тельности к малейшим внешним воздействиям. Наблюдались случаи
взрыва даже в результате трения об острые края кусочков стекла
пузырьков пара кислоты при температуре ее кипения.
В парообразном состоянии азотистоводородная кислота также чув-
ствительна к внешним импульсам и взрывает. Нижний предел взрыв-
чатости парообразной HN3 до сих пор не установлен.
Д. В. Алексеев нашел, что разложение HNS при воспламенении
распространяется по всей массе даже при общем давлении в 1 мм рт. ст.
Данные о взрывчатых характеристиках азотистоводородной кис-
лоты, рассчитанные нами по вычисленной Ротом и Мюллером теп-
лоте образования, для сравнения с некоторыми другими наиболее
мощными ВВ сведены в табл. 35.
Табл. 35 совершенно ясно показывает, что азотистоводородная
кислота является самым мощным из всех до сих пор известных ВВ.
Достаточно взрыва небольшого количества азотистоводо родной
кислоты, чтобы вызвать относительно большие разрушения- Так,
например, опыт по определению плотности пара, по Гофману, закон-
чился печально: как только трубочка с 0,05 г HN3 достигла поверх-
ности ртути, произошел сильный взрыв, и аппарат был превращен
153
Таблица 35
Взрывчатые вещества Характеристика Азотисто- | к М X 4 rt 0.0 §5 = £ 1 иг ' Нитрогли- коль Нитрогли- церин S Гексоген
Теплота образования, Кал/мол Объем газообразных продуктов, Уо л/кг . -67 - -
1 042 737 715 800 900
Теплота взрыва, Qu Кал/кг .... 1592 1 580 1490 1400 1 300
Потенциальная энергия, Р т-м/кг . . Температура взрыва. Т°К - . .’ 669 - - —
5160 4 750 4 200 390С 3400
Сила, / л-ат/кг 19680 12 800 - 11 430 11 240
Скорость детонации. D м/сек ...... 8 300 8200 8400 8 380
в пыль. Такой же взрыв произошел и у Курциуса, когда он совместно
с Раденгаузеном впервые получал безводную HN3. Взрыв произошел
при извлечении трубочки с 0,07 г HN3 из охладительной смеси. Сила
взрыва была настолько велика, что осколками были пробиты далеко
стоявшие толстостенные сосуды.
Азотистоводородная кислота ядовита. Водяные растворы ее разъ-
едают эпидерму. Газообразная HN3 даже при слабых концентрациях
вызывает головокружение, головную боль и раздражение слизистых
•оболочек, особенно глаз и носа, а при более высоких концентрациях—
и остановку дыхания. В условиях производства пары азотистоводо-
родной кислоты очень часто приводят к обморокам. При попадании
внутрь азотистоводородная кислота вызывает спазмы, симптомы па-
ралича сердца и легких. Кролики умирают после впрыскивания под
кожу 0.03 г азида натрия. Зарегистрировано несколько случаев
отравлений азотистоводородной кислотой.
Азотистоводородная кислота растворима в воде и спирте в любых
•соотношениях. Водные растворы постоянны в процессе хранения.
При кипячении их азотистоводородная кислота улетучивается без раз-
ложения, однако, кипячением нельзя „удалить ее полностью. При
перегонке водных растворов кислоты первая фракция дистиллята
содержит 91% HN3. После же отгона основной массы кислоты продол-
жает перегоняться до конца ее водный раствор с содержанием 0,005%
HN3. При кипячении с умеренно разбавленными кислотами HNS мед-
ленно разлагается. Так, нагревание с соляной кислотой приводит
к образованию хлорида аммония:
3HN3+ НС1 > 4N2 + NH,C1
а при взаимодействии с серной кислотой образуется комплекс
HN3 • 3H3SO4. 7%-ный водный раствор HN3 растворяет металлическое
железо, цинк, медь, алюминий и магний. Продуктами реакции в данном
случае являются азиды металлов, азот, аммиак и небольшое количество
гидразина. Магний может вытеснить небольшое количество водорода,
так же как из очень разбавленной азотной кислоты. Азид двухвалент-
154
Л-105
7
5,38
15.98
45.97
1,98
1,80
1,66
него железа при избытке HN3 превращается в азид трехвалентного
железа. Более концентрированные растворы действуют на золото и
серебро. Платиновая чернь, губчатая платина разлагают водные
растворы HNg с выделением NH3 и N2. Таким образом действие HNS
на металлы аналогично действию на металлы азотной кислоты.
Азоимвд—одноосновная кислота, несколько превосходящая по силе
уксусную кислоту. Константа диссоциации HN3 при 25° выражается
следующими цифрами (см. таблицу). -----
HN3 — плохой проводник элек-
тричества. При электролизе обра-
зуются только азот и водород, но ------
возможно и образование в процессе J()
вторичных реакций небольшого 100
количества аммиака. юоо
Ультрафиолетовые лучи вызы-
вают разложение HN3 с образова- -------
нием азота, водорода и азида аммония.
Восстанавливающие вещества (например амальгама натрия) пере-
водят азотистоводородную кислоту в некоторых случаях в гидразин.
Другие вызывают количественное разложение ее на азот и аммиак.
Так. например, при действии йодистого водорода происходит следую-
щая реакция:
HN3 + 2HJ -> N2 + NH3 -I- 2J
Равным образом при действии иода в присутствии небольшого коли-
чества тиосульфата можно полностью разложить эту кислоту до
азота:
2HN3 + J2 3N2-|-2HJ
Окислители, например, КМпО4, дают при взаимодействии с HN3
азот и воду.
Строением азотистоводородной кислоты занимались и занимаются
многие исследователи, однако, в настоящее время еще нет оснований
принять за истину какую-нибудь совершенно определенную формулу.
Существует ряд экспериментальных доказательств как в пользу ци-
клической формулы:
,N
Н —N( ||
XN
так и в пользу формулы с открытой цепью:
И N = N=N
Курциус, Фишер и Кекуле предполагали, что азотистоводородная кис-
лота имеет циклическую форму строения, исходя из реакции образо-
вания азидов из нитрозогидразинов с отщеплением воды:
/NO zN
R N( R — N( И +Н2О
Чн, XN
155
откуда вытекает, что и при омылении азидов растворами щелочей
должны получаться азиды щелочных металлов строения:
zN
• K-N( ||
XN
Деннис показал, что азид таллия в токе водорода дает аммиак, что
можно выразить только следующим уравнением:
N Н
H)N-H+ N2+NH3
Nz Н
Эрдманн утверждает, что образование азотистоводородной кислоты
по методу Вислиценуса может служить доказательством такого ее
строения:
N N
|| >0 + H2NH -> Н20 + И Н
N/!....... Nz
Но так как строение закиси азота к настоящему времени достаточно
точно неизвестно, то последняя реакция может быть выражена также
и следующим уравнением:
N = N = ?б+”Н2;НН -> Н20 + N= N = N—Н
На последнем, между прочим, настаивает и Тиле. Он основывает
свои рассуждения на аналогии между азотистоводородной и родановой
кислотами, а именно, что кроваво-красное окрашивание раствора азида
железа аналогично окрашиванию раствора роданида железа, образо-
вание азида натрия аналогично образованию роданида натрия:
NaN’Hs‘ + S = С = S -> Na -- N = С = S + H2S
Тиле это подтверждает также расщеплением азидов магний-органи-
ческими соединениями с образованием диазо аминов:
RN3 + R,MgJ RN(MgJ)N = NR,
RN(MgJ)N = NR, 4- H20 -> RNH = NR, + MgJ(OH)
Менделеев рассматривал HN3 как производную вторичного орто-
нитрата аммония:
z°
N=(ONH4)a -> 4Н2О + Н - N = N 5 N
х ОН
156
Франклин рассматривает HN3 как продукт десольватации аммоно-
оргоазотной кислоты
NH2
|/NH2
N—NHj—3NHS HN = N = N
|xnh2
nh2
а азиды — как аммоннитраты. С этой точки зрения азотистоводород-
ную кислоту можно было бы получить из HNO8 и NH3.
Непосредственный нагрев NH4NO3 и KNO3 в растворе жидкого
аммиака к цели не ведет, однако, при взаимодействии в этом раство-
рителе нитрата калия с амидом калия при 80—140° и выше протекает
реакция:
KONO2 + 3KNH2 -> KN = N = N + 3KOH + NH3
При более высоких температурах выход KN3 понижается за счет
выделения значительных количеств Na. Этим путем можно получить
и Pb(N3)2 с выходом в 80%.
При взаимодействии водной HNS с магнием, цинком, железом,
марганцем, никелем и медью образуются соответствующие азиды и
небольшие количества гидразина и происходит выделение N2 и NH3,
а с магнием выделяется еще и водород.
Предполагается следующий механизм реакции:
Zn + 2HN = N = N Zn(N = N = N)2 + 2Н
HN = N Е N + 2Н -> HN = N — NH2
HN - N — NH2 -> NH3 + N2
Соединение HN = N — NH2 можно представить как аммоноазо-
тистую кислоту. Таким образом имеется полная аналогия с раство-
рением металлов в азотной кислоте.
Смесь водной HN3 и НС1 обладает свойствами царской водки
в смысле растворения золота и платины. При кипячении водной HN3
и НС1 или НВг происходит выделение хлора или брома; HJ восста-
навливает HN3 до N2 и NH3.
HN3 и азвды окисляют целый ряд неорганических соединений
(и органических): Fe" в Fe'“,H2S до серы, аммоностанит натрия
(из азида натрия и SnCl2) в аммоностанат, этанол до ацетальдегида,
уксусной кислоты, метиламмония и аммиака, бензиловый спирт до
бензальдегида, бензойной кислоты и анилина.
Лангмюир указал, что группы цианата и азида изостеничны и что
многие азиды обладают сходными свойствами с цианатами. Так,
157
азид калия и цианат калия кристаллизуются в тетрагональной системе
и имеют приблизительно одинаковые соотношения осей:
у KNCO
а :с= 1:0,5766,
У KN3
g:c = 1:0,5798,
что предполагает наличие изоморфизма и наличие цепочечной формы
строения HN3. Кранстон и Ливингстон изучали коэфициенты прелом-
ления, растворимость и проводимость водных растворов пар:
KNS —KCNO и NaN3— NaCNO. Результаты показали, что структуры
ионов цианата и азида должны быть сходными. Эрленмейер и Лео на
основании своих кристаллографических и физических исследований
также установили сходство структуры ионов в KN3 и KCNO.
С. Б. Гендерикс и Л. Паулинг на основании рентгенографических
исследований заключили, что в цепочках азида все три атома азота
расположены линейно. Бергманн и Щютц, определявшие дипольные
моменты для фенилазида, р-хлорфенилазида и других азидов, также
считают, что азидогруппа имеет цепочечную структуру.
Г. Линдеманн и Г. Тиле путем определения парахоров эфиров
азотистоводородной кислоты пытались доказать их циклическую
структуру. Они показали, что парахоры ряда азидов принимают
величины, которые следует ожидать от кольцевой структуры. Однако
Сиджвик считает это доказательство недостаточно убедительным, так
как разница между установленными значениями, вычисленными для
кольца (38,6) и для открытой цепи (44,8), составляет только около 2%
действительного общего парахора, и хотя наблюденные величины
(в среднем 40,3) ближе к кольцевому строению, Сиджвик настаивает
на возможности влияния ошибок опыта.
Сиджвик высказал предположение, что если азиды имеют кольце-
вую структуру, то они должны кипеть примерно при тех же темпера-
турах, что и соответствующие им хлориды или бромиды, и наоборот,
если они имеют структуру с открытой цепью, то точки их кипения
должны быть много выше и соответствовать точкам кипения нитро-
соединений. Исследование около 80 азидо-, галоидо- и нитросоедине-
ний показало, что точки кипения азидов лежат близко к точкам кипе-
ния бромидов, что еще раньше было отмечено Форстером и Нейма-
ном. Этими опытами Сиджвик подтверждает кольцевую структуру.
Сэттон, исследуя дипольные моменты азидов, показал, что в фенил-
азиде азидогруппа имеет кольцевое строение, а в азиде калия — ли-
нейное. Берто высказал предположение, что азидогруппа, обладая
кольцевой структурой, может реагировать, как кольцевая и как ли-
нейная, т. е. HN3 может существовать в двух таутомерных формах.
Затем Сэттон, Сиджвик и Томас в результате тщательного исследова-
ния дипольных моментов вновь пришли к выводу о кольцевой струк-
туре иона N3'.
158
Ганч путем определения абсороционных спектров азотистоводо-
родной кислоты и ее сложных эфиров установил кольцевую струк-
^Однако в своей новой работе Сиджвик, разбирая строение органи-
ческих азидов, приходит к противоположному выводу о цепочечном
строении и считает, что органические азиды представляют собой смесь
двух форм с открытой цепью:
R - N = N N
R —N <- N = N
Сэттон из рентгенографических и других исследований цианур-
гриазида выводит молекулярную решетку, в которой азидогруппа
образует короткую цепь. Брегг показал, что расположение атомов
в молекуле циануртриазида соответствует следующей формуле:
N
Ганч, базируясь на принципах, положенных в основу понятий
о псевдокислотах и псевдосолях, приходит к заключению, что в ще-
лочных азидах металл как катион одинаково связан со всеми
атомами аниона, и дает им структуру:
' О]
В тех же солях, которые содержат слабо положительный ион, т. е.
в псевдосолях, металл связан с одним атомом аниона, как, например-
в азвде серебра:
Ag - N< 11
Наконец, следует остановиться еще на одной работе, выполнен-
ной Г. Герцбергом, Ф. Пататом и Г. Ферлегером, которые для выяс-
нения структуры HNS исследовали спектр колебаний газообразной
HN3 в инфракрасном свете. Структура спектра, по их мнению, опре-
деленно показывает, что три атома азота находятся очень близко
15£>
друг к другу на прямой, и что у большей части молекул атом водо-
рода лежит на той же прямой. Отсюда они заключают, что возможно
^существование двух видов геометрической структуры:
N —N — N — Н и N-N —N
I
Н
Приведенный выше краткий обзор работ по вопросу о строении
HN8 показывает, насколько трудно остановиться окончательно на
той или иной формуле строения. Следует отметить только, что в по-
следнее время склоняются к признанию цепочечной формы строения
азотистоводородной кислоты:
Азотистоводородная кислота,, как и всякая кислота, может обра-
зовать два обширных класса химических соединений: неорганических
и органических.
Металлические производные азотистоводородной кислоты, т. е.
ее соли (азиды), по ряду своих физико-химических свойств аналогичны
солям галогеноводородных кислот.
Если, например, сравнить растворимость в воде щелочных солей
азотистоводородной кислоты с растворимостью щелочных же солей
галогеноводородных кислот, то окажется, что растворимость первых
занимает среднее место между растворимостями хлоридов и броми-
дов. Так, в 100 мл чистой воды при 16е растворяется г:
Из этой таблицы
можно вывести следую-
щие закономерности:
1) растворимость ази-
дов Na', К', Rb' и Cs
увеличивается так же,
как и галогенидов — от
Na‘ и Cs';
2) кислотный остаток
Ионы cr N3 Вг'
Na- 36 41 86
к- 35,5 49 62,5
Rb- 97 107 105
Cs- 176 307 —
N3' в ряде галоидов должен быть расположен между СГ и Вг', что и
отвечает его групповому весу (3 х 14 — 42).
Если сравнить растворимость азидов и галогенидов щелочноземель-
ных металлов, то ока-
жется, что в 100 мл воды
при 16° растворяется г
{см. таблицу):
Из этой таблицы выте-
кает, что растворимость
азидов Са-, Sr”, Ва--
увеличивается, так же
как у галогенидов, —
от Ва-- к Са”, и что
растворимость азидов щелочноземельных металлов лежит между
растворимостью фторидов и хлоридов.
Ионы F' n; Cl'
Са- 3,7-Ю~3 46,0 68,0
Sr- Очень мало 46,5 51,5
Ва- 0.16 17,0 34.0
160
Азиды щелочных и щелочноземельных металлов не разлагаются
в воде при кипячении, азиды же тяжелых металлов при нагревании
в воде медленно распадаются на гидрат окисла соответствующего ме-
талла и азотистоводородную кислоту.
Из всех азидов щелочных металлов только азид лития детонирует
при нагревании, азиды же калия, рубидия, цезия плавятся и затем
разлагаются на азот и металл. Азиды щелочноземельных металлов
при нагревании, так же как и азид натрия, не плавятся, а разлагаются
без взрыва на свободный металл и азот. Разложение их можно регу-
лировать так, чтобы оно происходило лишь постепенно. Равномер-
ное выделение азота при термическом разложении азидов получается,
как показано в табл. 36.
Таблица 36
Азиды Na* К* Rb- св* Са- «г- Ва-
Температуры термического разложения с равномерным выделением азота 230° 360° 310° 350е 100° 110° 120
Следует отметить, что в тех случаях, когда необходимо получить
особо чистые щелочные и щелочноземельные металлы, можно реко-
мендовать разложение азидов нагреванием. Если же подвергать на-
греванию азиды тяжелых металлов, то они вспыхивают со взрывом.
Наибольший интерес представляют соли тяжелых металлов, обла-
дающие чрезвычайно большой чувствительностью. Чувствительность
некоторых из этих солей, особенно азидов окисной ртути и окисной
меди, настолько велика, что они иногда взрывают при малейшем при-
косновении.
Что касается инициирующих свойств, то ими обладают азиды эле-
ментов побочных подгрупп периодической системы химических эле-
ментов Менделеева. Из солей тяжелых металлов лучше всех других
удовлетворяет требованиям, предъявляемым инициирующим ВВ, азид
свинца.
Азотистоводородная кислота помимо солей металлов образует
значительное количество других неорганических и органических про-
изводных, отличающихся значительной взрывчатостью и обладающих
высокой инициирующей способностью. К числу их между прочим
относятся:
I. Азвд иода, обладающий большей взрывчатой силой, чем другие
взрывчатые соединения иода и азота (иодистый азот). Азид иода
N3 J окрашен в белый или слегка желтоватый цвет и отличается острым,
напоминающим иодистый циан, запахом. Он хорошо растворим в воде
и многих органических растворителях. Водные растворы азида иода
гидролизуют по уравнению:
М + Н20 -> HN3+HJO
Бубнов—92—11
161
В присутствии основании гидролиз происходит мгновенно.
2. Азид хлора N3C1 аналогичен азиду иода. Он отличается от неп
тем, что если в присутствии щелочи в водном растворе он разлагаете)
с образованием азотистоводородной кислоты (азида щелочного ме
талла), то в кислом растворе реакция протекает нацело обрати®
HNS + HOC! -+ N3C1+H2O
На этом основано получение азида хлора. Подкисляя смесь азид«
натрия и гипохлорита натрия слабой кислотой, например борной,
получают газообразный азид хлора — бесцветный газ, по запаху
аналогичный хлорноватистой кислоте.
3. Азидотиоугольная кислота
ZNS
C=S
\sh
соли тяжелых металлов которой являются инициирующими ВВ
(герм. пат. № 376448; англ. пат. № 188302), получается простым при-
соединением азотистоводородной кислоты к сероуглероду:
ZS /Ns
С + HNS -> С = S
^SH
Реакция совершенно аналогична присоединению к сероуглероду
вторичных аминов, например:
(сероуглерод)
N = (СзН^
I
Н
ZN = (С.НД,
C = S
XSH
(ды»,илксантогеновая
кислота)
С
S
+
Азидотиоугольная кислота с окисными солями меди дает осадок
великолепного кадмиево-желтого цвета и может поэтому считаться
реактивом на медь подобно ксантогеновой кислоте. Взрывчатость
солей азидотиоугольной кислоты различна. Если соли щелочных ме-
таллов, содержащие кристаллизационную воду, устойчивы по отно-
шению к начальному импульсу, то соли тяжелых металлов легко
воспламеняются, чувствительны к механическим воздействиям и обла-
дают большой энергией. Так, например, творожистая белая серебря-
ная соль азидотиоугольной кислоты в сухом виде взрывает уже при
самом легком прикосновении. Лучшей цз солей азидотиоугольной
кислоты по своим инициирующим свойствам является свинцовая соль:
N3— C-S-Pb — S — С - N3
162
Несколько милиграммов азидотиоугольного свинца лучше вызывают
детонацию тэна, чем азид свинца.
Органические производные азотистоводородной кислоты много-
численны. Более или менее изученные их представители будут рас-
смотрены в особой главе.
§ 3. АЗИД НАТРИЯ» ЕГО ПОЛУЧЕНИЕ И СВОЙСТВА
Азид натрия в чистом виде получается нейтрализацией азотисто-
нодородной кислотой водного раствора гидрата окиси натрия:
NaOH + HN3 NaNs + Н2О
последующей концентрацией раствора и кристаллизацией из него.
Химически чистый азид натрия обычно получают путем разложе-
ния дважды перекристаллизованного технического азида разбавлен-
ной серной кислотой и отгонки образующейся азотистоводородной
кислоты в водный раствор гидрата окиси бария. Получаемый в прием-
нике азид бария отфильтровывают и вновь разлагают той же кисло-
той, а азотистоводородную кислоту улавливают водным раствором
1 идрата окиси натрия.
Такой длинный путь получения химически чистого азида натрия
применяется для тщательного освобождения его от углекислых солей.
Можно также получать раствор азотистоводородной кислоты,
обрабатывая азид натрия такой кислотой, натриевая соль которой
трудно растворима в воде, например кремнефтористоводородной
(растворимость силикофторида натрия в воде при 17,5°—0,65 г
в 100 мм), или щавелевой (растворимость кислого оксалата натрия
вводе 1,4 а в 100 мм). Далее, обрабатывая отфильтрованный раствор
гидратом окиси бария, получают азид бария.
Однако этот способ не всегда применим и дает азид натрия не очень
высокой чистоты.
Азотистоводо родная кислота как более сильная вытесняет из
раствора угольную кислоту. Если через раствор химически чистого
карбоната натрия пропускать газообразную азотистоводородную кис-
лоту, то получающийся азид натрия обычно содержит 99,75—-99,89%
NaN3 и отпадает надобность в получении азида натрия через азид
бария. Необходимо только брать такой избыток азида натрия, чтобы
свободной азотистоводородной кислоты было достаточно не только
для связывания свободной щелочи, но и для вытеснения угольной
кислоты.
Химически чистый азид натрия получают следующим образом.
В колбу Вюрца емкостью 300 мл всыпают 10 а перекристаллизован-
ного один раз из воды технического азида натрия и заливают 125 мл
освобожденной от углекислоты и других примесей воды. Колбу сое-
диняют с длинным холодильником Либиха, который в свою очередь
соединяют с приемником, содержащим 125 мл 4%-ного раствора
химически чистого гидрата окиси натрия. К приемной колбе присое-
диняют последовательно две контрольных склянки Дрекселя: первая—
163
с раствором гидрата окиси бария, вторая — с раствором нитрата се-
ребра (концентрация контрольных растворов безразлична). Послед-
нюю склянку соединяют с водяным инжектором для создания разре-
жения во всей системе.
Затем к раствору азида натрия в колбе Вюрца приливают 10%-ный
раствор серной кислоты в количестве 96 мл, колбу закрывают пробкой
с термометром и подогревают на бане с раствором хлористого каль-
ция, имеющим температуру кипения около 112°. Азотистоводородную
кислоту отгоняют с паром и поглощают щелочью. Так как в системе
находится небольшое количество углекислоты, то ее вытесняют азо-
тистоводородной кислотой и просасывают в первую контрольную
склянку Дрекселя, где она поглощается водным раствором гидроксида
бария (раствор мутнеет от образования ВаСО8).
К концу реакции, т. е. к моменту полного превращения гидроксида
натрия в приемнике в азвд натрия, избыток азотистоводо род ной кис-
лоты окрашивает раствор в приемной колбе в светлорозовый цвет,
переходит в первую склянку Дрекселя, растворяет муть карбоната
бария и, замещая в нем углекислоту, осветляет раствор. Концом
реакции считается осветление раствора в первой склянке Дрекселя
и образование мути AgN3 во второй.
Указанное количество исходных продуктов взято из расчета по-
лучения растворов, содержащих 7—8% свободной НМ3. Большой
избыток ее (при нормальном или близком к нормальному давлению)
представляет опасность вследствие ее взрывчатости и ядовитости.
Однако и при описанных условиях следует вести всю работу, прини-
мая должные меры предосторожности (щиты из толстого стекла
(20—25 мм), очки со слюдяными стеклами и предохранительной сет-
кой, постоянно работающая тяга).
По окончании перегонки раствор азида натрия, содержащий избы-
ток азотистоводородной кислоты, переливают в фарфоровую чашку
после получасового отстаивания в открытой колбе. Затем осторожно
(особенно вначале) выпаривают на водяной электробане под тягой
до концентрации сиропа, после чего охлаждают. Выпавшие кристаллы
азида натрия промывают водой и спиртом и сушат при температуре
около 80°.
Этим способом получается продукт с содержанием до 99,97%
NaN3, не более 0,03% нерастворимых в воде примесей и только следы
щелочи.
Менее чистый продукт можно получить обычной перекристаллиза-
цией технического заводского продукта. Молесс и Глеер получали
химически чистый азид натрия многократной перекристаллизацией
его из водного раствора над крепкой серной кислотой. При этом
потери равны примерно 30%.
В лабораториях, если не требуется особо высокой чистоты про-
дукта, перекристаллизацию ведут обычно два раза, упаривая водные
растворы азида натрия или осаждая NaN3 из водного раствор'а спир-
том. Чистота получаемого при этом продукта в зависимости от числа
перекристаллизаций дана в табл. 37.
164
Таблица 37
Чистота азида натрия в зависимости от числа перекристаллизаций
Содержание После 1-й пере- кристаллизации После 2-й пере- кристаллизации о/ /о После осаждения спиртом из вод- ного раствора %
NaNa • - ... 92,4 98,8 99,91
NaBCOs . . . . NaOH ... 7,2 1,2 Нет
0,3 Следы »
Н2О . . 0,1 0,01 0.05
Азид натрия является исходным продуктом для большинства дру-
гих азидов.
Азид натрия представляет собой прозрачные бесцветные кристаллы
гексагональной системы, плавящиеся при нагревании с разложением
на составные части. Уд. вес азида натрия 1,846. Он хорошо раство-
рим в воде; в 100 г воды растворяется технического азида натрия:
при 10' . . . 40,16 г
» 15,2° - 40,70 »
» 17° . . . . 41,70 »>
Растворимость химически чистого азида натрия в воде несколько
меныпая:
мри 18,6° - 35,3 г
»> 32,2° . 36,244 »
» 64,4‘ . 38,17 »
»> 104е . . 42,92 »
Свежеприготовленный водный раствор азида натрия дает ней- тральную реакцию; при стоянии же появляется щелочная реакция
вследствие гидролиза. Температура кипения насыщенного водного Таблица 38 Растворимость NaNs в водных растворах NaOH
раствора азида натрия 110,5°. Растворимость азида натрия в воде в присут- Температура ГС Концентрация NaOH % Растворимость NaN3 %
ствии растворенных в ней других солей или гидратов окислов щелоч- ных металлов уменьша- ется. Падение раствори- мости с увеличением концентрации гидрата 18,3 19,6 19,0 21,0 21,0 19,4 17,0 1 10- 20 30 33 41 50 29,5 25,0 14,9 9,5 7,7 3,7 2.0
окиси натрия видно из -----------------------------------------
табл. 38. Этим пользуются при производстве азида натрия на
заводах.
165
Значительно меньше растворимость азида натрия в спирте. 100 -•
спирта растворяет:
при 0° - - - 0.22 г
» 16° . • 0,3153 •
В абсолютном эфире азид натрия нерастворим.
Азид натрия не летуч и практически негигроскопичен. При выпа-
ривании из водного раствора он не изменяется. Несмотря на то, что
азид натрия является солью очень чувствительной кислоты, сам он
стоек и нечувствителен к механическим воздействиям — трению и
удару. При воспламенении азид натрия вспыхивает, давая при этом
блестящее желтое пламя. При быстром непосредственном нагреве до
очень высокой температуры (t > 330°) он может взрывать со значи-
тельной силой, а при нагревании в вакууме начинает разлагаться
при t = 330° на металлический натрий и азот. Начавшееся разложе-
ние протекает равномерно и при снижении температуры до 280°,
очевидно за счет каталитического действия натрия.
Азид натрия, так же как и HN3, представляет собой очень силь-
ный яд. Кайзер описывает случай действия NaN3 на человеческий орга-
низм. Введение в желудок таблетки NaN3 весом 0,005 г вызвало вна-
чале сильное сердцебиение, затем несколько следующих друг за дру-
гом глубоких обмороков. Вопрос о профилактике и противоядиях не
является еще окончательно разработанным. На основании некоторых
опытов считают, что противоядиями могут служить нитрат кобальта
или тиосульфат иатрия.
Технический азид натрия обычно слегка окрашен, что зависит от
качества очистки. Очень часто такой азид натрия при растворении
в воде окрашивает раствор в слабый желтоватый цвет. Технический
азид натрия сопровождается обычно карбонатами натрия, гидратом
окиси железа и нерастворимыми в воде примесями.
Азид натрия на практике получают несколькими методами. Наи-
более распространенным является метод Вислиценуса. Он состоит
из трех операций:
а) получения амида натрия,
б) получения азида натрия,
в) освобождения азида натрия от примесей.
Исходными продуктами для первой операции служат металли-
ческий натрий и аммиак. На сухой металлический натрий, расплавлен-
ный в закрытом сосуде, действуют сухим аммиаком при температуре
около 300°:
2Na + 2NH3 - > 2NaNHs+ На
Получившийся натрийамвд подвергают действию закиси азота при
температуре 210—220°:
NaNH2+N2O NaN3 + Н2О
Выделяющаяся вода разлагает натрийамид:
NaNH3+H2O NaOH+NH3
166
Таким образом суммарная реакция может быть; выражена следую-
щим уравнением:
2NaNH2 + N2O -+ NaN8 -J- NaOH + NH3
Отсюда видно, что только 50% металлического натрия расходуется
на изготовление азида натрия. Этот недостаток, а также длительность
технологического процесса заставляют искать другие более простые
и экономичные методы его получения.
Другим методом, бывшим одно время очень распространенным
(особенно в Германии), является метод Штолле, заключающийся
в том, что гцдразингидрат в спиртовом растворе кипятят (с обратно по-
ставленным холодильником) с изоамилнитритом и метилатом натрия,
или гидразинсульфат в водном растворе смешивают с водным раство-
ром гидрата окиси натрия и встряхивают с этиловым спиртом и изо-
амилнитритом. Часто изоамилнитриту предпочитали этилнитрит,
потому что удаление образующегося в реакции изоамилового спирта
затруднительно вследствие высокой температуры его кипения (136°).
Заслуживают внимания опробованные в свое время Тиле реак-
ции получения азида натрия из мочевины и гуанвдиннитрата. Реак-
ции эти были повторены рядом других исследователей и сведены ими
в более или менее стройную систему.
Получение азида натрия из мочевины схематически сводится
к нитрации мочевины, амидированию нитрогдрчевины, азидированию
получающегося семикарбазида и, наконец, омылению раствором гид-
рата окиси натрия:
znh2
С = О
xnh2
/NH - NO2
с=о
/NH NH2
c=o
Xnh2
С - О -+ NaN3
xnh2
Весь синтез слагается из:
1) получения нитрата мочевины действием на водный раствор
мочевины азотной кислотой уд. веса 1,4:
/NH2 /NH2. HNOS
С = О + HNO3 -* С = О
4NH2 xnh2
2) превращения нитрата мочевины в нитромочевину под действием
концентрированной серной кислоты:
zNH2.HN0? zNH-N02
/ 2 n2so< /
С = О ----------->С = О +Н2О
\ <7=1,84 X
xnh2 xnh3
3) восстановления нитромочевины электролитическим путем с при-
менением в качестве католита 20%-ной серной кислоты в семикарб-
167
азидсульфат:
/NН • NO., / NH • NH2 H,SO4
C=O +H2SO4->C=O
xnh2 xnh2
4) получения азида карбаминовой кислоты действием на семи-
карбазидсульфат нитритом натрия:
ZNH • NH2 • H2SOa
С = О + 2NaNO2 ->
xnh2
/NH — N = NOH
С = О + Na2S04 + Н2О
XNH2
/NH - N = NOH N3
с — о -> c = o-i- H2O
XNH2 XNHa
5) получения азида натрия омылением азида карбаминовой кис-
лоты раствором гидрата окиси натрия:
С = О + 3NaOH -> NaN3 + NH3 Na2CO34- Н2О
XNHE
Сущность одного из вариантов получения азвда натрия из гуанн-
диннитрата состоит в том, что вначале гуанидиннитрат обычной
дегидратацией переводят в нитрогуанвдии:
/NH2 /NH-NO2
С = NH HNO3 -> С = NH
XNH2 xnh2
взмученную в серной кислоте суспензию последнего электролити-
чески или другим путем восстанавливают до аминосоединения,
которое затем осаждают бикарбонатом натрия в виде аминогуани-
дннбикарбоната:
nhno2
C = NH
xnh2
/NH NH2
C = NH
XNH2
/NH-NH2
-> C = NH • H2CO3
xnh2
IBS
Далее действием теоретического количества азотной кислоты,
необходимой для вытеснения углекислого газа и последующего диа-
зотирования, получают аминогуанидиинитрат:
zNH-NH2 ^NH. NH2
С = NH • Н2СО3 + HNOS С = NH • HNO3 + H2O + CO2
xnh2 xnh2
который при нагревании до 40—50° диазотируют нитритом натрия
в присутствии азотной кислоты и получают нитрат гуанилазвда:
yNH NH2
С = NH - HNO3 + NaNO2 + HN03 ->
XNH2
/N3
-> C = NH • HNO3 + NaNO, + 2H2O
xnh2
Сульфатным методом при тех же условиях получают аминогуани-
Динсульфат, а из последнего сульфатгуанилазида:
zNH-NH2 zNH-NH2
С = NH • Н2СО3 + H2SO4 > С = NH • H2SO4 + Н2О +СО2
-NH2 ' XNH2
ZNH NH2
C = NH • H2S04 4- NaNO2 + H2SO4 ->
XNH2
/N,
-> C .= NH • H2SO4 + NaHSO4 + 2H2O
XNH„
Растворы нитрата или сульфата гуанила зида расщепляют на азид
и цианамид натрия действием гидрата окиси натрия при охлаждении:
/N,
С = NH H2SO4 + 5NaOH -э- NaN3 + NaHCN2 +
4nh2
+ NaOH + Na2SO4 + 5H2O
/N,
C = NH . HNO3 + 4NaOH -» NaN3 + NaHCN3+
XNH3
+ NaOH + NaNOs + 4H2O
169
В целях освобождения от цианамида всю смесь подвергают дейст-
вию высокой температуры и давления в автоклаве, вследствие чего
цианамид гидролизует и, присоединяя воду и избыток щелочи, пере-
ходит в Карбонат натрия и аммиак:
NaHCN2 + NaOH + ЗН2О -► NH3 + Na2CO3 + Н2О
Добавлением серной или азотной кислоты вытесняют углекислоту:
NagCOg + H2SO4 -> Na2SO4 + СО2 + Н2О
и, наконец, путем фракционной кристаллизации выделяют часть
сульфата натрия, а остальной сульфат осаждают азидом бария в виде
сульфата бария. В растворе остается один азид натрия:
Na2SO4 4- Ba(N3)2 -► 2NaN3 + BaSO4
Получающийся по всем способам азид натрия кристаллизуется из
воды. Разбавленными минеральными кислотами он разлагается, как
и все азиды, с выделением азотистоводородной кислоты.
§ 4. УСЛОВИЯ ПОЛУЧЕНИЯ АЗИДА СВИНЦА И ЕГО ФИЗИКО-
ХИМИЧЕСКИЕ СВОЙСТВА
Азид свинца получается действием водного раствора ацетата или
нитрата свинца на видный
раствор азида натрия:
2NaN3 + Pb(NO3)3
-> Pb(N3)2 + 2NaNO3
Азид свинца осаждается
в виде белого тонкокри-
сталлического порошка
(фиг. 80) уд. веса 4,73.
Впервые азид свинца был
получен Курциусом прили-
ванием к раствору азида
натрия или аммония раст-
вора уксусносвинцовой
соли:
2NaNs + (СН3СОО)2РЬ ->
Pb(N3)2 + 2CH3COONa
Фиг. 80. Азид свинца. Внимательное исследо-
вание процесса получения
азида свинца показывает, что при его осаждении для обеспечения
хорошего выхода необходимо всегда брать соль свинца с небольшим
избытком. При этом условии отфильтрованный маточный раствор
всегда прозрачен и не мутнеет при долгом стоянии. Если же взяты
количества реагирующих веществ в точных стехиометрических отно-
170
тениях, то фильтруемый раствор всегда содержит тончайшую муть,
исчезающую при длительном промывании. Наоборот, если будет пре-
обладать избыток азцда натрия, то отфильтрованная прозрачная
жидкость при дальнейшем промывании азида свинца мутнеет.
В лабораториях и на заводах азид свинца осаждают при обыч-
ных комнатных температурах (около 20°). Лабораторный процесс
осаждения весьма прост и состоит в медленном приливании раствора
азида натрия к раствору нитрата свинца при постоянном помешивании
реагирующей жидкости. Когда осадок азида свинца собирается в комки,
то на некоторое время прерывают осаждение и перемешивают массу.
Тяжелый осадок азида свинца быстро садится на дно. Если исходные
растворы взяты в строго стехиометрических отношениях, то конец
реакции можно легко определить по опалесценции жидкости. Обычно
и в лабораторных условиях употребляют избыток азотнокислого
свинца. После осаждения осадок фильтруют,промывают водой, рассти-
лают фильтр на многократно сложенную фильтровальную бумагу,
отделяют роговым шпателем азид свинца и сушат при температуре 50°.
Осаждение азида свинца из горячих растворов и при медленном
охлаждении влечет за собой наряду с большой его чистотой увеличе-
ние размеров кристаллов. Таким образом осаждение из горячих
растворов увеличивает опасность работы, что подтверждается проис-
ходящими иногда взрывами. Это объясняется, невидимому, тем, что
с повышением температуры растворимость азида свинца возрастает
и часть его осаждается при охлаждении в кристаллизованном виде.
Кристаллизованный же азид свинца чувствительнее обычного, полу-
ченного на холоду, продукта. Осаждение из горячих растворов должно
сопровождаться мерами предупреждения охлаждения продукта в ма-
точном растворе, что может быть достигнуто быстрым фильтрованием
теплого раствора, удалением фильтрата и последующей холодной
промывкой отфильтрованного азида свинца.
При получении азида свинца смешением исходных растворов раз-
меры кристаллов зависят от температурного режима и концентра-
ций, но не зависят от самых исходных веществ (табл. 39).
Таблица 39
Влияние концентрации растворов на размер кристаллов Pb (N3)2
Температура опыта °C Концентрации Внешний вид кри- сталлов Размер кристал- лов мм
NaN3 рь<мад„
21—23 1 1 Игольчатан форма 0,060-0,075
21—23 1 15 » » 0,035—0,053
21-23 10 1 » » 0,052—0,067
21-23 5 5 » А 0,022—0.035
21 23 10 15 1 Комки СЛИПШИХСЯ
21—23 30 30 | кристаллов; нх аг- j регаты
171
При концентрациях 1 : 1 образуются кристаллы игольчатой формы
длиной 0,06—0,075 мм. При более концентрированных растворах
образуются такие же кристаллы, но меньших размеров. При приме-
нении концентрации Ю : 15 уже замечается образование комков из
агрегатов кристаллов, почти неразличимых под микроскопом. В этом
случае могут быть обнаружены только отдельные иглы длиной 0,002—
0,003. При концентрациях порядка 30 : 30 нельзя различить отдель-
ных кристаллов.
При постоянной концентрации, но переменных температурах во
всех случаях также получаются кристаллы игольчатой формы. При
температуре 0—1° и концентрации 1 : 1 образуются иглы длиной
0,003—0,008 мм. С повышением температуры происходит рост разме-
ров кристаллов. Уже при температуре 21—23° и той же концентрации
длина кристаллов достигает 0,06—0,075 мм, а при температуре 49—50°
до 0,145—0,168 леи. Таким образом с уменьшением концентра-
ции и с повышением температуры размеры кристаллов увеличиваются.
Выдержка кристаллов в маточном растворе способствует повыше-
нию их размеров. Автор изучал этот вопрос при концентрациях раст-
воров 5 : 5 и времени выдержки 3—4, 60 и 120 мин. Результаты
ясно показали, что с увеличением выдержки размеры кристаллов
увеличиваются.
Избыток или недостаток той или иной соли для стехиометрической
реакции не оказывает никакого влияния на 4»рму кристалла. Во всех
случаях образуются игольчатые кристаллы или смесь из длинных
призм и игольчатых пластинок причем наблюдается очень большое
количество неправильно образованных кристаллов (сростков, двой-
ников, дендритов).
В 1909 г. на заводе в Тройсдорфе, где готовили азид свинца по
патенту Велера, т. е. с применением ацетата свинца, произошел силь-
ный взрыв на операции осаждения. Во Франции тогда же вполне
успешно готовили азид свинца по патенту Геронимуса, т. е. с приме-
нением нитрата свинца.
Солонина в результате своих исследований также пришел к
выводу о необходимости применить нитрат свинца.
Мартином был поставлен ряд опытов по изучению условий кри-
сталлизации азида свинца, причем во время кристаллизации из горя-
чего насыщенного раствора ацетата натрия при извлечении кристал-
лов произошел взрыв. Причиной взрыва предполагали образование
крупных кристаллов, обладающих большей чувствительностью, чем
мелкие, или образование нестойких соединений, вследствие способ-
ности свинца быть не только двух-, но и четырехвалентным, а именно,
соединений типа:
PbNJ2; PbN6O; PbN6(OH)2; PbNc • fflHN3
Однако опытами было установлено, что крупные кристаллы азида
свинца все же менее чувствительны, чем гремучая ртугь. Поэтому
едва ли можно объяснить самопроизвольные взрывы образованием
172
больших кристаллов. Доказать образование нестойких соединений
также не удалось.
В одной из лабораторий при медленном охлаждении маточного
раствора с азидом свинца, полученным* прибавлением к раствору
ацетата свинца раствора азида натрия при температуре 95°, через
2г/2 часа произошел взрыв.
Приведенные работы по изучению горячего и холодного осаждения
азида свинца показывают, что последний надлежит получать, поль-
зуясь нитратом свинца, но не решают вопроса о причинах взрывов
при кристаллизации и осаждении азида свинца. Автор заметил, что
все они происходят при наличии маточного раствора и при отсут-
ствии его перемешивания. На этом основании он пришел к выводу,
что самопроизвольные взрывы азида свинца в маточном растворе
являются следствием растворения получившегося мелкокристалли-
ческого порошка и последующей перекристаллизации его в виде кри-
сталлов соли иной формы или иной модификации, нестойкой и склон-
ной к взрывам при определенных условиях.
а-аз ид ₽-ашд
Фиг. 81. Форма кристаллов азида свинца по Майльсу.
Опыты показали, что азид свинца может существовать в двух
кристаллических формах— игольчатой и короткостолбчатой, — отно-
сящихся к одной и той же кристаллической сингонии по нашим
исследованиям, или к двум сингониям по исследованиям Майльса
(фиг. 81).
Обыкновенный азид свинца короткостолбчатой формы, или о.-азид,
получается вполне безопасно путем двойного обмена между нитратом
свинца и азидом’ натрия при обыкновенной (комнатной) температуре.
Опасность получения увеличивается с повышением температуры вслед-
ствие образования больших кристаллов при медленном охлаждении
горячих насыщенных растворов азида свинца в воде.
Чистые и удовлетворительные кристаллы «-формы также могут
быть получены перекристаллизацией азида свинца из водных раство-
ров ацетата аммония или натрия.
Кристаллы игольчатой формы, иначе говоря, 0-азид свинца, обра-
зуются при диффузии между растворами ацетата или нитрата свинца
и азида натрия. Такие условия, например, можно создать следую-
щим образом..
Стаканчик диаметром около 2,5 см наполняют раствором азида
натрия и помещают в кристаллизатор диаметром 100 мм с раствором
нитрата свинца. Затем очень медленно по внутренней стейке прили-
вают дистиллированную воду до уровня на 10—12 мм выше верхнего
173
среза внутреннего стаканчика. Все оставляют стоять в покое. Через
несколько часов (в зависимости от концентрации растворов) кристал-
лизатор наполняется кристаллами в виде тонких игл, длина которых
зависит от радиуса кристаллизатора.
При получении 0-формы методом диффузии можно пользоваться
и сухими солями, вводимыми в кристаллизатор и стаканчик, уже
заполненные водой.
а
б е
Фиг. 82. Габитусы игольчатых кристаллов азида свинца:
а—зарисовки кончиков кристаллов; б—кристалл короткостолбчатой формы;
в—кристалл игольчатой формы
Исследование образования кристаллов азида свинца в случае
диффузии позволяет сделать следующие выводы:
1. В результате диффузии растворов азида натрия и нитрата или
ацетата свинца происходит образование азида свинца нескольких
габитусов, могущих быть сведенными к двум формам: короткостолб-
чатой и игольчатой. Игольчатая форма может иметь вид пластинок
или призм (фиг. 82, о, б, в), склонных срастаться в дендриты. Иголь-
чато-пластинчатые кристаллы обычно покрыты вицина ло идами,
возникающими вследствие расщепления граней и искажения кри-
сталлической решетки.
2. Количественно соотношение между короткостолбчатой и иголь-
чатой формами различно для разных исходных материалов.
3. Образование той или иной формы зависит от времени пребыва-
ния кристаллов в маточном растворе, причем определенно можно
сказать, что игольчатая форма в виде призматических игл образуется
позднее из других форм путем роста короткостолбчатой формы в
длину или игольчатой формы в виде пластинок в толщину, или путем
перекристаллизации последней.
4. Образование игольчатой формы азида свинца зачастую сопро-
вождается взрывами при любых условиях (при применении ацетата
и нитрата свинца, при высоких и низких температурах, на свету и
174
в темноте, при любых концентрациях растворов: разбавленных и
концентрированных). Следует отметить, что чем концентрированнее
раствор, тем вероятнее получение взрыва при диффузии. Поэтому
иа производстве необходимо учесть, что при применении 10—15%-ных
концентраций исходных веществ особое внимание должно быть обра-
щено на работу мешалки, чтобы предотвратить возможность длитель-
ной диффузии между растворами.
Удельный вес азида свинца по данным различных авторов раз-
личен:
d = 4,7969
d =4,9
• = 4,71
dp = 4,93
d =4,73
в чашечку капсюля техни-
по Велеру и Крупко
» Бруисвигу - - -
» Майльсу .
Таблица 40
Сорт азида свинца Давление ат Плотность г/см2
Технически!^ мелкий . 100 2,4
» » 200 2,7
» » 400 2,9
» » 800 3,1
Крупнокристаллический 200 3,2
» 800 3,5
» исследованиям автора
Изменение плотности спрессованного
ческого азида свинца, содержащего около 11,9% примесей, и крупно-
кристаллического, содержащего 5,6% примесей, от давления прес-
сования приведено в
табл. 40.
Азид свинца практи-
чески нерастворим в
холодной воде, в кипя-
щей несколько лучше,
но менее, чем хлорид
свинца. Так, в 100 мл
воды растворяется:
при 18° . . . 0,023 г
» 70° . . • 0,090 »
Растворение в кипя-
щей воде сопровождает-
ся разрушением части азида свинца с образованием нерастворимого
гидрата окиси свинца.
По охлаждении из горячего водного раствора азид свинца осаж-
дается в виде весьма чувствительных сантиметровых блестящих бес-
цветных игл.
Азид свинца лучше растворяется в растворах солей, так:
100 мл 4N водного раствора нитрата натрия рас-
творяют при 18° ..............................
То же при 80° ..........................
100 мл 4N водного раствора ацетата натрия рас-
творяют при 18° ........................
То же при 80° .... .
Хорошо растворяется азид свинца в моноэтаноламине:
этаноламина растворяют 14,6 г азида. Из концентрированного раст-
вора выделяется белый, мелкокристаллический продукт, и весь раст-
вор затвердевает. Азид свинца из раствора может быть осажден водой,
однако, регенерированный азид нечист, очень трудно воспламеняется
и взрывает без обычного для азида свинца звука.
0,125
0,487
г
1,542
2,020
10 г моно-
175
При длительном нагревании азида свинца с водой происходит
постепенное разложение его с выделением азотистоводородной кис-
лоты. Азид свинца легко растворяется в теплой уксусной кислоте и
при этом он также постепенно выделяет азотистоводородную кислоту.1
Минеральные кислоты на него действуют значительно энергичнее.
Едкие щелочи также разлагают азид свинца с образованием ще-
лочных азидов, однако, действие их очень медленное вследствие обра-
зования оксидной пленки на поверхности кристаллов азида свинца.
Нагревание и перемешивание ускоряет действие щелочей. Гидро-
ксид аммония действует на азид свинца аналогично щелочам.
В связи с тем, что азид свинца обычно применяется запрессованным
в металлическую оболочку, немаловажное значение имеет отношение
его к разным металлам.
Во влажной среде в присутствии углекислого газа азид свинца
частично с поверхности расщепляется, а образующаяся свободная
азотисговодородная кислота легко вступает в соединение с медью,
образуя азиды окисной или закисной меди. В случае образования
азида окисной меди несчастные случаи неизбежны вследствие весьма
высокой чувствительности этого соединения к механическому воздей-
ствию.
Действие азида свинца на медь изучал в свое время Солонина.
Его опыты сводились в основном к следующему:
1. Пластинки из красной меди нагревались на водяной бане
в дистиллированной воде с азидом свинца. В результате нагревания
в течение 3 дней по 9 час. ежедневно на медных пластинках и на дне
сосуда появлялся зеленоватый налет, состоящий из гидрата окиси
меди, гидрата окиси свинца и следов азида свинца.
2. Восстановленная металлическая медь в запаянной трубке на-
гревалась без доступа воздуха с азидом свинца в среде дистиллиро-
ванной воды. В результате непрерывного нагревания в течение 7 дней
при 65—66° анализом был обнаружен в осадке свободный металли-
ческий свинец.
3. Красная медь, вырезанная из оболочек капсюлей, хранилась в
дистиллированной воде с азидом свинца в течение 3 мес. при 15—18°.
Через месяц было обнаружено, что все медные листочки покрылись
зеленым и отчасти буроватым налетом. Осторожно снятый с меди
зеленоватый порошок сильно взрывал при накаливании, но оказался
мало чувствительным к удару. Качественный анализ зеленого осадка
показал наличие меди, свинца и азота.
Можно предполагать, что здесь образовались смеси основных ази-
дов свинца и меди наряду с гидратами окисей свинца и меди. Однако
высушенный осадок оказался мало чувствительным к механическим
воздействиям и не взрывающим при падении груза 600 г с высоты
225 леи, в то время как окисный азид меди взрывает уже при падении
того же груза с высоты 15 мм, а закисный — с высоты 190—210 мм.
В результате упомянутых исследований Солонина приходит к вы-
воду, что при нагревании азида свинца с медью в дистиллированной
воде можно предполагать образование свободной азотистоводородной
176
кислоты благодаря расщеплению азида свинца. Эта кислота действует
на медь, образуя окисный азид меди, который при дальнейшем нагре-
вании разлагается водой на гидрат окиси меди и свободную азоти-
стоводо родную кислоту, постепенно удаляющуюся из нагреваемой
системы.
Если медь нагревать с азидом свинца в дистиллированной воде без
доступа кислорода воздуха в сфере угольной кислоты, то азид свинца
разлагается с образованием окисного азида меди и свободного метал-
лического свинца. В практике подобных обстоятельств при хранении
капсюлей почти не бывает, поэтому этот случай можно опустить.
Что же касается хранения красной меди в воде с азидбм свинца, то
следует отметить вслед за Солониной, что вряд ли при таком хранении
образуется азид окисной меди, ибо чувствительность получающегося
зеленого вещества заметно отличается от чувствительности окисного
азида меди.
Ряд испытаний, произведенных позднее, также показал, что
между негранулированным азидом свинца и медью в присутствии
влажности, или в воде происходит медленное взаимодействие, причем
поверхность медных пластинок от соприкосновения с азидом свинца
покрывается налетом зеленого цвета. Анализ этого налета не показал
наличия чистого азида окисной меди.
Тот же азид свинца был применен для опытного снаряжения кап-
сюлей-детонаторов 24/31 (медный). Последние хранились в течение
7 и 9 мес. соответственно:
1) в увлажнителе при комнатной температуре,
2) в негерметической укупорке в атмосферных условиях.
В первом случае после 7-месячиого хранения при разрядке капсю-
лей-детонаторов было обнаружено, что азид свинца остался белого
цвета, но содержал много включений — корочек желто-коричневого
цвета. Внутренняя поверхность гильз, соприкасавшаяся с азидом
свинца, также имела налет желто-коричневого цвета. Чувствитель-
ность к удару, определенная на копре Велера, уменьшилась, верхний
и нижний пределы после хранения повысились с 225 и 110 до 255 и 125.
Инициирующая способность не изменилась (0,05 г азида свинца по
тетрилу). Покрытый желто-зеленой корочкой, азид свинца содержал
от 2,74 до 5,68% меди.
Во втором случае, т. е. после 9 мес. хранения таких же капсюлей-
детонаторов в атмосферных условиях, после разрядки было замечено,
что внутренняя поверхность гильзы покрыта слабым налетом желто-
зеленого цвета. Температура вспышки и инициирующая способность
азида свинца не изменились.
Гранулированный азид свинца после снаряжения им медных обо-
лочек капсюля-детонатора № 8 хранился в негерметической укупорке
в ’ неотапливаемом помещении. Снаряженные капсюли по своему
наружному виду оказались вполне удовлетворительными. После их
хранения как в увлажнителе при 50°, так и при комнатной темпера-
туре, инициирующая способность не изменилась после 7-месячного
хранения.
Бубнов—02—12
177
То же можно сказать и о медных азидотетриловых капсюлях-дето-
наторах 24/31, хранившихся в негерметической укупорке, но в отап-
ливаемом помещении в течение 4 лет, а после этого в течение 6 мес.—
в увлажнителе при комнатной температуре. Их инициирующая спо-
собность и чувствительность не изменились.
Кроме того, железные омедненные капсюли-детонаторы № 8 были
снаряжены негранулированпым азидом свинца. После 8г/2 мес. хра-
нения при температуре 50° они были разряжены. Осмотр показал,
что на внутренней поверхности сквозных поражений защитного (мед-
ного) слоя нет. но покрытие, соприкасавшееся с азидом свинца, стало
желтым. Азид свинца остался белого цвета, но содержал от 0,032 до
0,14% меди. Чувствительность к удару по нижнему пределу почти не
изменилась (вместо 100—110), а по верхнему пределу повысилась
(вместо 225—165).
Те же капсюли-детонаторы № 8 (омедненные) хранились в течение
6 мес. в увлажнителе при комнатной температуре. Разрядка показала,
что азид свинца значительно загрязнился включениями желто-бурого
цвета. Защитное покрытие в месте соприкосновения с азидом свинца
не сохранилось: оно было разрушено полностью. Инициирующая
способность свинца не изменилась. То же было обнаружено при раз-
рядке, произведенной после хранения в тех же условиях в течение
9 мес. Полученный в этом случае азид свинца содержал от 0,61 до
3,13% меди. Чувствительность к удару понизилась (верхний предел
повысился от 225 до 260; иижний — со 100 до 165).
Те же капсюли-детонаторы № 8 (омедненные) хранились в течение
81/2 мес. в негерметической укупорке в атмосферных условиях. После
разрядки было обнаружено, что азид свинца имел значительное количе-
ство включений желто-бурого цвета. Защитное покрытие полностью
было обнажено и азид свинца содержал 0,49% меди. Инициирующая
способность не изменилась. Нижний предел чувствительности к уда-
ру почти не изменился (110 вместо 100). а верхний—снизился с 225
до 170.
В 1914 г. Солонина исследовал взаимодействие азида свинца с ла-
тунью, содержащей 66,61% меди и отожженной при 650°. Исследо-
вание было весьма элементарным и свелось к определению привеса
латунных пластинок после хранения в течение 6—7 дней в условиях:
1) сухой азид свинца + латунная пластинка;
2) азид свинца + латунная пластинка в воде;
3) латунная пластинка в воде.
Вес пластинок, хранившихся с сухим азидом свинца, не изменился,
поверхность же оказалась покрытой едва заметным налетом. В резуль-
тате хранения азида свинца с латунью в воде произошел привес пла-
стинки на 0,33%, а принимая во внимание убыль веса самой латуни
при хранении, привес оказался равным 0,34%. То же самое показала
другая серия опытов Солонина. На основании их можно заключить,
что происходит какое-то взаимодействие, но нельзя иайти ответа на
вопрос о характере взаимодействия.
178
Позднее были снаряжены негранулированным азидом свинца ла-
тунные капсюли-детонаторы № 8 и после хранения их в разных усло-
виях были произведены некоторые испытания.
Разрядка этих капсюлей-детонаторов после 5 мес. хранения в увлаж-
нителе при комнатной температуре показала, что азид свинца имеет
«чень немного включений (корочек) серовато-зеленого и коричне-
вого цветов. На внутренней поверхности гильзы появились отдельные
небольшие пятна коричневого цвета. Инициирующая способность
азида свинца не изменилась. Чувствительность к удару понизилась
(верхний предел 255, нижний 150). После разрядки, произведенной
через 7х/2 мес., оказалось, что азид свинца сильно засорился короч-
ками желтого и коричневого цвета и содержал от 0,12 до 1,2% меди.
Гранулированный азид свинца был исследован в капсюлях-детона-
торах, снаряженных Охтенским заводом в 1916 г. Эти капсюли-детона-
торы на протяжении 12 лет (до 1928 г.) находились во взрывателях,
ввернутых в снаряды, а с 1928 г. по момент разрядки в 1932 г. — в не-
герметической укупорке в неотапливаемом помещении. Наружная
поверхность этих капсюлей-детонаторов потемнела, фольга большин-
ства из них покрылась зеленью. Пять таких капсюлей-детонато-
ров было сброшено с 23-го зуба Массета, и взрывов не последовало.
После разрядки оказалось, то азид свинца стал серовато-желтым и
содержал много включений от стенокгильзы зеленоватого цвета.
Фольга между зарядом азида свинца и тетрилом покрылась налетом
коричневого цвета и имела значительное позеленение. Инициирующая
способность азида свинца оказалась ниже нормальной для техничес-
кого продукта. Анализ азида свинца с применением уксуснокислого
аммония, в качестве растворителя показал содержание 75% Pb(N3)g.
Два года спустя были исследованы подобные же капсюли, снаря-
женные еще в 1914 г. Наружный осмотр дал Примерно те же резуль-
таты. Анализ показал содержание в азиде свЦнца меди. Испытание
на Массете показало повышенную чувствительность: первый же бро-
шенный с 23-го зуба капсюль-детонатор дал вЗрыв, четвертый — дал
взрыв с 17-го зуба,
В 1914 г. Солонина также определил, что привес мельхиоровой пла-
стинки (79,24% меди, отожженной при 750°) пбсле годичного хранения
ее в воде с азидом свинца равен 0,14%. При хранении же с сухим ази-
дом свинца вес не изменился, но пластинка покрылась едва
заметным налетом.
Произведенные в 1932 г. испытания показали, что после 10 мес.
хранения в тех же условиях поверхность мельхиоровой пластинки
становится матовой, появляются пятна желто-зеленого цвета. Привес
незначителен.
Капсюли-детонаторы в мельхиоровой оболочке УГГ, снаряженные
азидом свинца, были поставлены на хранение в увлажнителе при 50°.
Через 4 мес. после разрядки было обнаружено,что азид свинца имеет
включения от зеленого до серо-черного цвета. На внутренней поверх-
ности гильз появились кристаллические образования желто-коричне-
вого цвета. Однако исследовать эти образования не удалось,
179
так как при распиловке гильз после их разрядки происходили
вспышки.
Сухой иегранулированный азид свинца с мельхиором не взаимо-
действует. При разрядке тех же капсюлей-детонаторов У ГТ после
4-мес. хранения никаких изменений азида свинца и внутренней по-
верхности гильз не было обнаружено.
Результаты испытаний после хранения этих же капсюлей-детона-
торов в увлажнителе при комнатной температуре через 4 мес. пока-
зали, что азид свинца имеет только отдельные включения — комочки
темного цвета, а внутренняя поверхность гильз — следы зеленых пя-
тен. Анализ извлеченного азида свинца на медь и никель показал их
отсутствие. После разрядки этих же капсюлей, произведенной через
7V2 мес. хранения, не было обнаружено нового изменения. Иници-
ирующая способность азида свинца не изменилась. Чувствительность
к удару также почти не изменилась (240—НО). Анализ на медь дал
отрицательное показание.
Поведение гранулированного азида свинца было испытано в мель-
хиоровых оболочках при хранении:
1) в увлажнителе при комнатной температуре,
2) в термостате при 50°.
Во всех случаях изменения инициирующей способности не про-
изошло. По 5 шт. было сброшено с 23-го зуба Массета, и взрывов не
произошло.
В том же 1914 г. Солонина установил, что привес дюралюминиевой
пластинки после годичного хранения ее в воде с азидом свинца значи-
тельно меньше привеса чистой алюминиевой пластинки и составляет
только 0,27% и что сухой азид свинца на дюралюминий не действует
(вес пластинки и поверхность совершенно не изменились).
Снаряженные в 1932 г. капсюли-детонаторы в дюралюминиевой обо-
лочке были - поставлены на 6-месячное хранение в увлажнителе при
50°. Изменения инициирующей способности не произошло. При испы-
тании на Массете с 23-го зуба 7 капсюлей-детонаторов взрывов
не последовало.
Другая партия таких же капсюлей-детонаторов была поставлена
на хранение при комнатной температуре и периодически увлажнялась
в течение 6 мес. При испытании на Массете 7 таких капсюлей-детона-
торов взрывов не было. При испытании на воспламенение от луча
огня бикфордова шнура во всех случаях были получены отказы.
Испытания после разрядки показали, что азид свинца побурел
и не воспламенялся от луча огня. Внутренняя поверхность гильзы
значительно корродировала и стала медно-желтой.
Изложенные результаты работ по исследованию взаимодействия
азида свинца с медью и ее сплавами не являются полностью исчер-
пывающими и не дают совершенно отчетливого представления о ходе
взаимодействия. Все же они позволяют сделать следующие
заключения:
1. Сухой азид свинца не взаимодействует ни с медью, ни с ее
сплавами (латунь, мельхиор, дюралюмин).
1S0
2. При продолжительном хранении взаимодействие технического
азида свинца с медью и ее сплавами возможно (а стало быть, воз-
можно и образование азида окисной меди, опасного в обращении
и практическом использовании), но только в условиях, при которых
от азида свинца может отщепляться свободная азотистоводородная
кислота, легко вступающая во взаимодействие с медью.
3. Образованию свободной азотистоводородной кислоты в тех или
иных количествах способствует, главным образом, следующее: 1) про-
должительное хранение азида свинца в воде, особенно при нагрева-
нии; 2) наличие кислых продуктов в среде, окружающей азид свинца;
3) хранение азида свинца во влажной атмосфере, насыщенной угле-
кислотой.
Если первый фактор при практическом хранении по техническим
условиям невозможен, то второй и третий в практике вполне возможны.
4. Отношение меди и ее сплавов к азиду свинца при неблагопри-
ятных условиях различно. Наибольшее взаимодействие происходит
между азидом свинца и медными оболочками капсюлей и оболочками
других металлов, покрытых медью. Образование вдоль стенок гильзы
корочки азида свинца, содержащего медь в относительно большом
проценте, уже само по себе нарушает механическую стойкость капсю-
ля-детонатора и после продолжительного хранения может повлечь
за собой преждевременный взрыв при применении.
Примерно таково же взаимодействие между латунью и азидом
свинца.
Азид свинца не взаимодействует с алюминием и свинцом. Взаимо-
действие с железом возможно.
По отношению к нагреванию азид свинца является веществом
стойким. Позднейшие опыты показали, что даже после нагревания
азида свинца в течение 3-—5 лет при температуре 50° его свойства
сколько-нибудь заметно не изменяются. Исследование азида свинца
после нагревания в темноте при 115° показало неизменяемость его
в течение 24 час., а при 170° в то же самое время нагрева наблюдалась
заметная потеря в весе. Выше 200° разложение происходит за сравни-
тельно короткий промежуток времени, причем взрывчатые свойства
исчезают, и азид свинца превращается в невзрывчатый порошок.
Удалось полностью разложить азид свинца без взрыва при осторож-
ном нагревании его при температуре 245—250°. При температуре
выше 250° в вакууме азид свинца взрывает.
Гигроскопичность азида свинца больше, чем у стифната свинца.
Каст и Гайд приводят следующие данные о гигроскопичности, полу-
ченные ими в результате хранения в насыщенном влагой воздухе
образцов азида свинца и стифната свинца (табл. 41).
Опытами Велера и Крупно, с одной стороны, и Форга, с другой,
было показано, что взрывчатые свойства азида свинца при хранении
на воздухе или под водой изменяются; происходит процесс окисления
азида свинца с образованием азотистоводородной кислоты и аммиака.
Поэтому совершенно понятно, что первые опыты применения азида
свинца в медных оболочках, как отмечает Каст, «сопровождались
181
многочисленными несчастными случаями, возникавшими благодаря
отщеплению HN3 углекислым газом в присутствии влаги с образова-
нием азида меди на стенках капсюля».
Таблица 41
Г игроскопичность
Количество дней хра- нения азвда свинца стифната свинца
1 обр. 2 обр. I обр. 2 обр.
10 20 30 40 0,88 1,58 1 90 1,90 0,42 1.40 1,56 1.60 0,02 0,36 1 0,46 0.46 , 0.60 0,60 0.60
Аналитические исследования образцов азида свинца, хранивше-
гося в разных условиях (нагревание, влага, углекислота и их комби-
нации), показывают, что азид свинца только с поверхности покры-
вается желтой пленкой, в своей же массе под пленкой он сохраняет
белый цвет. После тщательного перемешивания через некоторое время
появляется новая пленка. Таким образом разложение азида свинца
происходит только на поверхности и весьма медленно проникает
в толщу вещества. Если предотвратить возможность распространения
процесса вглубь вещества, например, прессованием, то взрывчатые
свойства азида свинца не изменяются, а уменьшается лишь чувстви-
тельность его к воспламенению.
Опыт показал, что прессованный азид свинца флегматнзированный
и не флегматизированный парафином, будучи подвергнут длитель-
ному нагреванию при 50°, действию углекислоты или влаги, значи-
тельно изменяется только с поверхности: уменьшается содержание
азота в поверхностном слое и образуется пленка из основных солей
свинца.
Наименьшее влияние оказывает нагревание при 503, несколько
большим является действие влаги, влаги и углекислоты, и, наконец,
наибольшее влияние на изменение качества азида свинца оказывает
совместное действие всех трех факторов.
Изменение качества азида свинца в процессе хранения тем больше,
чем меньше чистота продукта.
Поверхность азида свинца на свету окрашивается через незначи-
тельное время в желтый цвет. Раньше предполагали, что вследствие
действия света происходит саморазложение больших кристаллов и
что свет влияет на инициирующую способность азида. Однако в даль-
нейшем было установлено, что потемнение кристаллов азида свинца
происходит только с поверхности, что внутренние слои, даже при дли-
тельном освещении, защищаются тонким поверхностным слоем, и что
изменения взрывчатых свойств при этом не происходит. Поверхност-
ные слои коисталлов разлагаются на свободный металл и азот.
182
Еще позднее было установлено, что разложение распространяется
очень глубоко лишь в том случае, если разложившийся слой постоянно
во время освещения удаляется встряхиванием. В этих условиях
азид свинца постепенно теряет свою инициирующую способность и
чувствительность. Так, азид свинца, освещавшийся в течение 300 дней,
по данным анализа, содержал: 83,60% свинца, 12,48% азота и дал из
10 подрывов 1 отказ, а при определении чувствительности к удару
на копре Велера при грузе 500 г с высоты 300 мм в оболочках 3-ли-
нейного капсюля дал 0% взрывов.
Особый интерес представляет действие света на азид свинца под
водой. При этом получается разложение азида свинца с образованием
основного азида свинца, азотистоводородной кислоты, азота и аммиака.
Процесс этот можно представить следующим образом:
Pb(N3)3 + 2H2O Pb(OH)2 + 2HN3
Pb(Ns)2 Pb + 3N2 на свету
Pb + HN3 -f- 2H2O + PbfOHk + N2 + NH3
Pb(OH)2 + Pb(N3)2 -> PbO • Pb(Ns)2 4- H2O
Существует несколько методов получения основных азидов свинца.
Наиболее характерными из них. являются следующие:
1. Нагревание рассчитанных количеств гидрата окиси свинца и
азида свинца под водой в запаянной трубке при 140° в течение 12 час.
Этот метод небезопасен.
2. Пропускание лишенного углекислоты воздуха через кипящую
водную суспензию азида свинца до тех пор, пока образующаяся азо-
тистоводородная кислота не нейтрализует рассчитанного количества
баритовой воды (индикатор фенолфталеин).
3. Нагревание азида свинца на кипящей водяной бане в течение
20 час. с рассчитанным количеством свежеосажденного гидрата окиси
свинца под водой при сильном встряхивании в длинногорлой колбе.
§ 5. ВЗРЫВЧАТЫЕ СВОЙСТВА АЗИДА СВИНЦА
Азид свинца при взрыве разлагается по уравнению:
Pb(Ns)2 -> Pb4-3N24-106 Кал
Этому уравнению соответствуют следующие характеристики: теплота
взрывчатого разложения Qv = 364 кал/кг', объем газов Уо = 308 л/«г;
температура взрыва Т = 3050°; удельная энергия / == 3440 л • ат/кг',
скорость детонации азида свинца около 4500 м/сек.
Таким образом азид свинца по сравнению с гремучей ртутью
обладает несколько пониженными взрывчатыми характеристиками.
Тем не менее 'азид свинца обладает более высокой инициирующей
способностью вследствие значительно большего ускорения его взрыв-
чатого разложения, а стало быть, вследствие большего выделения
энергии в единицу времени.
183
Ускорение взрывчатого превращения, вплоть до скорости детона-
ции, у азида свинца происходит в чрезвычайно короткий промежуток
времени (почти мгновенно). Если поджечь спичкой на картоне грему-
чую ртуть и азид свинца, то последний мгновенно взрывает с резким
звуком и пробивает картон, в то время как гремучая ртуть дает лишь
вспышку, и картон остается целым.
Практически применяемым импульсом для взрыва азида свинца
обычно является луч огня, так как к механическим видам возбужде-
ния взрыва азид свинца менее чувствителен, чем гремучая ртуть.
Таблица 42
Чувствительность к удару
Взрывчатые вещества Верхний предел мм Нижний предел мм Вес груза
Азид свинца, химически чистый, по- лученный обычным смешением исходных растворов (размеры кристаллов нераз- личимы в микроскопе) 234 65-70 975
Азид свинца химически чистый, пе- рекристаллизованный (игольчатой или короткостолбчатой формы) 175 60—65 975
Гремучая ртуть белая ... 85 55 478
В табл. 42 приведены для сравнения величины чувствительности
к удару обоих веществ на копре Велера.
Данные, приведенные в таблице, показывают, что чувствитель-
ность азида свинца к удару зависит от размеров кристаллов, но не
зависит от исходных продуктов (ацетат или нитрат свинца).
Сопровождающие технический азид свинца примеси, главным
образом, основной карбонат свинца, несколько понижают чувствитель-
ность мелкокристаллического продукта к удару.
Чувствительность к трению азида свинца игольчатой и коротко-
столбчатой формы различна. Наиболее чувствительным является
азид свинца игольчатой формы, а наименее чувствительными — азид
короткостолбчатой формы и обычный некристаллизованный азид
свинца, полученный осаждением из растворов азида натрия и нитрата
свинца при тщательном перемешивании. Среднее положение занимает
азид свинца, полученный осаждением из растворов азида натрия и
ацетата свинца и дающий в результате трения только частичные
вспышки. •
Добавки инертных веществ, по своей твердости превосходящих
твердость азида свинца, значительно увеличивают чувствительность
азида свинца, особенно к трению.
Чувствительность азида свинца к трущему удару несколько мень-
шая, чем чувствительность гремучей ртути, и характеризуется следую-
щими цифрами, полученными на маятниковом приборе (табл. 43).
184
Таблица 43
Инициирующие В В Добавочный груз кг Высота паде- ния см Количество качаний до взрыва
Hg(ONC)t 0,0 25 3-10
Pb(N8)a 0.45 37,5 12
От накола жалом азид свинца не взрывает, и это является суще-
ственным его недостатком, из-за которого его нельзя применять в на-
польных капсюлях-детонаторах без дополнительных составов, более
чувствительных к наколу (в накольных капсюлях-детонаторах можно
также применять смешанные кристаллы азида свинца с другими более
чувствительными к трению инициаторами или продукт осаждения
азида свинца в присутствии тонко дисперсированного вещества, обла-
дающего также большой чувствительностью к трению).
Температура вспышки у азида свинца, как и вообще у всех азидов
тяжелых металлов, значительно выше, чем у гремучей ртути и дру-
гих фульминатов. Температуры вспышки у различных азидов и фуль-
минатов, по данным Солонина и Мартина, сведены в табл. 44.
Таблица 44
Температуры вспышки азидов и фульминатов
•dou QU Металл Температура вспышки °C
азида фульмината
1 Свинец . ... 327
2 Серебро . . 297 170
3 Медь (11) . 215 205
4 Ртуть (II) . 225 186
5 Кадмий . 291 210
Температура вспышки азида свинца уменьшается с увеличением,
количества взятого для пробы вещества, что видно из табл. 45.
Так как температура вспышки в значительной степени зависит от
метода ее определения, то другие исследователи указывают иные
цифры, так, например, Велер — 345°, Гич — 360°. Наши исследова-
ния показали, что температура вспышки химически чистого азида
свинца равна 305°.
Зависимость температуры вспышки от времени нагревания хими-
чески чистого азвда свинца, полученного осаждением из растворов
нитрата свинца и азида натрия, показана в табл. 46 и на фиг. 83.
185
Таблица 45
Зависимости температуры вспышки азида свинца от его количества
Навеска 0,005 г 0,01 г 0,02 г
t° тсек тсек ‘° теек
342 76 340 80 339 108
330 90 337 125 331 225
338 97 331 183 329 240
337 — 329 — 327 —
Таблица 46
Зависимости температуры вспышки от среднего времени нагревания навески
азида свинца 0,01 г
1° тсек Тсек t° ’сек
390 ссО 350 2,9 315 33.4
380 0,4 340 6,2 310 53,9
370 0.8 330 10.9 305 123,6
360 1,5 320 20:3 303 Отказ
Температура вспышки азида свинца зависит также от чистоты его-
С увеличением количества неактивных примесей температура вспышки
азида свинца увеличивается, что видно из табл. 47.
Та б ли на 47
Зависимость температуры вспышки от чистоты азида свинца
Чистого про- дукта о' /о Температура вспышки °C Примечание
99,65 313 Определялась минимальная температура,
98,09 337 при которой вспышка происходит не позднее
94,78 363 5 мии. после начала нагревания
Весьма крупным недостатком азида свинца является то, что он
•от луча огня воспламеняется значительно труднее, чем гремучая ртуть.
Это объясняется прежде всего его сравнительно большей теплоемкостью
в спрессованном виде, а также сравнительно высокой температурой
воспламенения.
486
Солонина и Крупно, разрабатывая в 1917 г. азидотетриловый
капсюль-детонатор для 9-сантиметрового германского бомбомета и для
ручных гранат, для улучшения воспламенения ввели поверх чашечки
с азидом свинца около 0,1 г черного ружейного пороха, спрессо-
ванного под давлением 100 а т.
Так пришли к мысли о том, что для улучшения воспламенения азида
свинца необходимо применять специальные воспламенительные на-
кладки. Дымный порох оказался непригодным для этой цели вслед-
ствие ряда его неудовлетворительных качеств (медленность про-
цесса сгорания, гигроскопич- Е-
ность и т. п.).
Лучшим воспламенителем
для азида свинца оказался три-
нитрорезорцинат свинца, хотя
он и обладает незначительным
по сравнению с азидом свинца № л
ускорением взрывчатого пре-
вращения. Вначале При ИЗГОТОВ- Фиг. 83. График температур вспышки
лении капсюлей применяли азида свинца,
смесь азида свинца с ТНРС и
только позднее перешли на расположение ТНРС в виде осо-
бой накладки над азидом свинца. Такое расположение устра-
няет одновременно и действие влажной углекислоты на азид
свинца.
ТНРС относительно дорог, чувствителен к трению и способен
электризоваться в сравнительно высокой степени. Однако попытки
замены его другими воспламеняющими средствами оказались до сих
пор безуспешными. Так, по опыту одной из американских фирм,
воспламенительный слой, состоящий из бертолетовой соли, роданида
свинца и нитроцеллюлозы, приводит к 3% отказов в воспламенении,
не говоря уже о том, что эта смесь сгорает еще более медленно, чем
ТНРС, а стало быть, не может обеспечить мгновенность действия там,
где это требуется.
Маттер утверждает, что плохое воспламенение азида свинца за-
висит, главным образом, от присутствия в нем углекислых солей.
Это утверждение опровергается Штеттбахером, который ссылается
на опыт Тюрингенской фабрики взрывчатых веществ в Германии.
На этой фабрике в течение долгого времени при изготовлении азида
свинца очищали раствор азида натрия прибавкой раствора уксусно-
кислого бария и отфильтровывали образовавшийся углекислый ба-
рий. Но эта очистка не давала никакого улучшения качества азида
свинца по сравнению с тем, который получался из азида натрия с
содержанием 1,9% углекислых солей.
Е. Герц, отрицая возможность применения азида свинца без ТНРС,
основывается на опыте завода в Тройсдорфе. Технический азид свинца,
получаемый на этом заводе, не представляет собой, как правило,
чистого продукта,-он содержит только 90—95% Pb(N8)2, остальное—
основные соли и карбонаты. Е. Герц, повидимому, стоит даже на той
187
точке зрения, что применять высокопроцентный азид свинца опасно
вследствие его большой чувствительности.
Наши опыты показывают, что свежеприготовленный, химически
чистый азид свинца, содержащий 99,9% Pb(N3)2, воспламеняется от
лучей огня безотказно. Увеличение содержания примесей влечет за
собой получение отказов в воспламенении в процессе хранения сна-
ряженных без ТНРС капсюлей-детонаторов. При этом, чем загрязнен-
нее азид свинца, тем большего числа отказов можно ожидать.
Причина плохой восприимчивости азида свинца к лучу огня со-
стоит, кроме того, в том,что, вследствие влияния влажности и углекис-
лоты, на поверхности кристаллов азида свинца любой чистоты обра-
зуется тонкий слой основного карбоната свинца, нечувствительного
к лучу огня.
Таблица 48
Предельные инициирующие заряды азида свинца
Предельные заряды в г по отноше- нию к Автор Примечание
тетрилу пикриновой кислоте тротилу
0,025 0.025 0,09 Мартин н Велер С медной чашечкой,, давление прессования 1100 кг/сл?
0,01 — 0,08 Рейнско-Вест- фальское обще- ство взрывча- тых веществ С алюминиевой чашеч- кой
0,04 - 0,14 Тейлор и Копе С медной чашечкой, давление прессования 400 кг[см-
0.03 о;о2 - 0.15 0,10 Кестер С чашечкой. Давление прессования 200 кг/см** В первом случае азид технический; во втором— лабораторного приготов- ления
- 0,05 005 Брунсвиг (Нансен -
188
Прессование не изменяет сколько-нибудь значительно чувстви-
тельности азвда свинца к начальному импульсу, как это бывает у гре-
мучей ртути. Азид свинца, по опытам Велера, воспламеняется даже,
если он запрессован под давлением в 2000 кг/см2, а по Форгу, — даже
при 6000 кг[слР. Опыты Громова показали, что капсюли-детонаторы,
снаряженные 0,3 г азида свинца, спрессованного под давлением
12 800 от, пробивают свинцовую пластинку в 6 мм толщиной и могут
сыть воспламенены лучом огня. При одинаковой степени запрессовки
азид свинца детонирует легче, чем гремучая ртуть.
Инициирующая способность азида свинца значительно выше ини-
циирующей способности гремучей ртути. Литературные данные о
предельных инициирующих зарядах различны. Некоторые из них
сведены в табл. 48.
Исследованиями автора установлено, что предельный заряд азида
свинца зависит как от степени чистоты и формы кристаллов, так и от
качества исходных продуктов (табл. 49).
Таблица 49
Предельные инициирующие заряды азида свинца ио тетрилу в зависимости
от чистоты продукта и формы кристаллов
№ по пор. 1 Образец азида свинца Чистота % Предельный заряд г
I Азид свинца, полученный осаждением из 10%-иого раствора NaN3 и 15%-ного раствора Pb(N03)e Химически 0,02
2 Азид свинца, полученный осаждением цз 10%-ного раствора NaN3 и 15%-иого раствора (СН3СОО)ЙРЬ ..... чистый То же 0,04
3 Азад свинца кристаллизованный, игольчатой формы - - * 0,02
4 Азид свинца кристаллизованный, коротко- столбчатой формы . . . ... » 0,03
5 Азид свинца, полученный осаждением из 10%-ного раствора NaN3 и 15-иого раствора гнход 98,16% 0,03
6 Азид свинца, полученный осаждением из 10%-ного раствора NaNs и 15%-ного раствора PtXNO,), .... 87,45% 0.035
Следует отметить, что между азидом свинца, полученным из аце-
тата свинца или в среде растворов уксуснокислых солей, и азидом
свинца, полученным из нитрата свинца или в среде растворов азотно-
кислых солей, наблюдается значительная разница. При одинаковой
чистоте продуктов азид свинца, полученный в присутствии иона NOS',
обладает в Р/3—2 раза большей инициирующей способностью, чем
азид свинца, полученный в присутствии иона СН3С00'. Величина
189
кристаллов сказывается только на азиде свинца, полученном в при-
сутствии иона СН3СОО'. С увеличением размеров кристаллов увели-1
чивается и их инициирующая способность, однако, не настолько,
чтобы достигнуть инициирующей способности продукта, полученного!
в присутствии иона N0'3.
Примеси (сами не являющиеся инициаторами) снижают иниции-
рующую способность азида свинца.
Азид свинца обычно получается в виде тонкого микрокристалли-
ческого порошка, собирающегося в комки. В таком виде он чрезвы-
чайно неудобен для применения в капсюльном производстве вслед-
ствие плохой сыпучести, а также сравнительно высокой чувствитель-
ности к скользящему удару, в результате чего могут происходить
вспышки при прессовании, насыпке и других операциях. Оба эти
недостатка устраняют путем флегматизации (обычно парафином) с
последующей грануляцией.
Количество флегматизатора влияет, конечно, не только на чув-
ствительность к механическим воздействиям, но и на инициирующую
способность азида свинца.
Предельный инициирующий заряд чистого, не флегматизирован-
ного, азида свинца не зависит от давления прессования. По нашим
опытам, он остается не-
изменным и при давле-
ниях порядка 3000 —
4000 кг1см*.
Иная картина полу-
чается при применении
азида свинца, флегма-
тизированного парафи-
ном. При повышении
давления прессования
на азид свинца, содер-
жащий 1,5 — 2% пара-
фина, предельный ини-
Таблица 50
Давление прессования ат Предельный заряд г
азида свинца тетрила
500 500 0,02
1000 1000 0,02
2000 2000 0,08
3000 3000 >0,25
4000 4000 >0,25
циирующий заряд за-
метно увеличивается, что видно из табл. 50.
Применение азида свинца, флегматизированного значительным
количеством парафина, влечет за собой уменьшение числа полных
детонаций капсюлей-детонаторов. Увеличение давления прессова-
ния на флегматизированный азид свинца также приводит к уменьше-
нию числа полных детонаций (табл. 51).
Поэтому оптимальным количеством парафина во флегматизиро-
ванном азиде свинца следует считать 1—2%.
Для изготовления капсюлей-детонаторов, обладающих нормаль-
ной чувствительностью к сотрясению при выстреле, можно было бы
пользоваться азидом свинца с содержанием флегматизатора меньше 1%
(до 0,4%), однако, еще не вполне ясно, как подобные капсюли будут
действовать в боеприпасах, предназначенных для стрельбы по брони-
рованным объектам.
190
Таблица 51
Давление прес- сования кг/см- Число полных детонаций при процентном содержании парафина
0.40 1.68 7,52 12,36
200 10 10 е 6 0
500 10 10 0 0 0
1100 2 1 0 0 0
1500 0 ° 0 0 0
При испытании ряда образцов азидотетриловых капсюлей-детона- торов, снаряженных азидом свинца, флегматизированным 1% пара-
фина или 1% гумми- арабика, путем подрыва Таблица 52
тротила в бомбах Тра- уцля были получены следующие расширения (табл. 52). Некоторые колебания между отдельными про- бами должны быть от- несены за счет неодно- родности отдельных бомб и ошибки опыта. Таким образом увели- чение количества азида свинца в капсюле-детона- Количество азида свинца в азидотетр и- ловом капсюле-дето- Нетто расширения бомбы Трауцля мл
наторе г с гумми- арабиком с парафином
0,05 0,10 0,15 020 025 0 30 297 293 301 309 299 294 302 295 305 286 305 297
юре от и,ио до и,о г не .. .. сказывается на мощности последнего (при запрессовке тетрила под давлением 450 кг[слр). Примесь воды к чистому азиду свинца не уменьшает сколько-ни- будь значительно взрывчатых свойств его. В противоположность гремучей ртути он также чувствителен в присутствии 30% влаги, как и сухой. На флегматизированном парафином азиде свинца влия- ние влаги сказывается и зачастую может приводить к неполным
взрывам.
К положительным свойствам азида свинца относится большая стой-
кость к высоким температурам, отсутствие способности перепрессо-
вываться и высокая инициирующая способность.
§ 6. ИСХОДНЫЕ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ ДЛЯ ФАБРИКАЦИИ
АЗИДА СВИНЦА
Исходными продуктами для фабрикации азида свинца служат
азид натрия и нитрат или ацетат свинца. Азид натрия изготовляется
самими заводами инициирующих ВВ, в последнее время преимуще-
ственно по способу Вислиценуса из амида натрия и закиси азота. Оба
191
продукт также производятся на заводах, изготовляющих азид
свинца. Кроме того, на практике применяются еще флегматизаторы,
растворители для них и прочие вещества, например, для увеличения
чистоты продукта.
Остановимся на характеристике указанных исходных продуктов
и полуфабрикатов:
1. Закись азота, бесцветный газ, сладковатого вкуса, со
слабым приятным запахом. При вдыхании его в незначительных ко-1
личествах он вызывает состояние легкого опьянения; в смеси с 7—20% I
кислорода закись азота обладает наркотическим действием. Вдыха-1
кие чистой закиси азота быстро вызывает грозящее смертью прекра-
щение дыхания.
Уд. вес закиси азота, при точке кипения 1,2257; вес 1 л при 0° и
760 лем давления 1,9777 г; точка кипения—89,6°; точка плавления
—102,4°; критическая температура 4- 36,5°; критическое давление
71,7 ат.
В воде растворимость закиси азота довольно значительна. Один
объем воды поглощает:
при 0 1,3052 объема NoO
» 5° . • 1,0954 »> N„O
!01 . 0,9196 » n;o
» 15° . . 0.7778 »> NaO
20° . 0,6700 N2O
25е . 0,5962 ► n2o
Водный раствор имеет сладковатый вкус и при кипячении выде-
ляет закись азота полностью.
Эфир и жирные масла хорошо растворяют закись азота. Так же
хорошо закись азота растворима в этиловом спирте: 1 объем спирта
при 18° растворяет 3 объема N2O, при 0° — 4,178 объема N2O.
При нагревании до температуры 500° 1,5% закиси азота находится
в состоянии равновесия:
2N2O 2N2 4- Оа
При нагревании до 900° происходит полное ее разложение на азот
и кислород.
Вещества, обладающие сильным сродством к кислороду, напри-
мер, уголь и некоторые металлы, сгорают в закиси азота почти так
же энергично, как в кислороде.
Смесь разных объемов закиси азота и водорода при соприкоснове-
нии с пламенем дает энергичную вспышку:
N2O -Ь Hg -> N2 + Н2О 4- 86,4 Кал
Смесь закиси азота с аммиаком при воспламенении взрывает с боль-
шой силой
3N2O4-2NH8 4N2 + 3H2O
Строение закиси азота до сих пор является спорным.
192
Предположения высказывались о следующих четырех формулах
строения:
N —О —N; N = N = О;
N - N = О; /°Х
N = N
В сжиженном состоянии закись азота представляет собой подвиж-
ную бесцветную жидкость уд. веса 0,937. Жидкая закись азота дей-
ствует на кожу, вызывая ожоги.
Закись азота образуется из нитратов при кипячении их в безвод-
ной муравьиной кислоте:
2KNO3 4- 7НСООН - N2O + 5COS + 5Н2О + 2НСООК
или нагреванием растворов гидразиннитрита или гидроксиламинни-
трита:
N2H4 • HNO2 -* N20 + NH3 + H2O
NHjOH • HNO3-> NgO + 2HgO
Однако наиболее дешевым является способ получения ее путем
разложения аммонийной селитры нагреванием:
NH4NOS - > NgO + 2Н2О
Аммонийная селитра. Аммонийная селитра, или нит-
рат аммония, представляет собой белую, почти бесцветную, кристал-
лическую массу, расплывающуюся на влажном воздухе, и обычно
кристаллизующуюся в ромбическую систему. Кристаллы аммонийной
селитры в нормальных условиях обладают уд. весом 1,725. Аммоний-
ная селитра может существовать в пяти различных кристаллических
модификациях, вернее кристаллических формах, а именно: одной
тетрагональной, двух ромбических, одной гексагональной и одной
правильной. Точки превращения следующие:
- 16° 32,3°
тетрагональная а-ромбическая 3 0-ромби-
85° 125°
ческая гексагональная правильная
Аммонийная селитра хорошо растворима в воде:
в 100 г воды при 0° растворяется 118,3 г
» 100*» » »> 25° » 214,2 »
» 100 » » » 50° » . . 371 »
» 100 » • » 100° ... 871 »
Растворение в воде сопровождается сильным понижением темпе-
ратуры раствора; поглощение теплоты равно 6,2 Кел/мол.
Растворимость в этиловом спирте значительно меньшая: 100 г
абсолютного этилового спирта растворяют при 20° только 3,8 а се-
литры. *
кубнов—92—13
193
Гигроскопичность аммонийной селитры очень велика; при хра-
нении во влажной атмосфере она способна расплываться, а под влия-
нием переменных температур — слеживается в сплошную камени-
стую массу.
При длительном нагревании аммонийной селитры при 90—110°
наблюдается потеря в весе. Температура плавления сухой селитры
169,6°. При температуре, немного превышающей температуру плав-
ления, происходит диссоциация:
NH4N03 NH3+HNO3 41,3 Кал (1)
протекающая весьма медленно.
При осторожном нагревании выше 200° NHtNO.t наряду с диссо-
циацией на аммиак и азотную кислоту разлагается по уравнению:
NH4NO3 -> N2O 4- 2Н2О + 10,2 Кал (2)
При температуре выше 220° разложение селитры в основном со-
ответствует уравнению (2). Повышение температуры до 250—260°
влечет за собой частичное разложение селитры по уравнению:
NH4NO3 0,5N2 -Ь NO J- 2Н->0 4- 9,2 Кал (3)
Дальнейшее повышение температуры до 400—450° уже приводит
к реакциям взрывчатого превращения селитры:
NHjNO3 -> 0,75N2 + 0,5N02 + 2Н3О 4- 29,5 Кал ( >
или
NH4NOs N2 4- 0,5О2 4- 2Н,0 4- 30,7 Кал (">
Быстрое нагревание селитры до 300° также приводит к взрыву.
Содержание в аммонийной селитре хлорида аммония ускоряет ее раз-
ложение.
Теплота взрыва аммонийной селитры до сих пор точно не установ-
лена, так:
но Бертл о Qv 421 кал/кг
» Гейзе- - Qp 381 >>
*' Касту ... . Qv 347 »
» Ауфшлегеру - . 375
Совершенно понятны поэтому и колебания в данных о вычисленных
температурах взрыва аммонийной селитры от 1121 до 2120°.
Аммонийная селитра поступает на заводы в деревянных сухотар-
ных бочках по 200 кг в каждой или в плотных (крафтцеллюлозных,
бумажных, битумированных пятислойных) мешках по 40—45 кг и
в таком виде хранится обязательно в сухом помещении.
К аммонийной селитре, идущей на производство закиси азота,
предъявляют следующие требования- По внешнему виду она должна
представлять собой мелкие кристаллы белого цвета; допускается
желтоватая окраска при условии удовлетворения прочим требова-
ниям, изложенным в табл. 53.
194
Таблица 53
Содержание 1 % | Примечание
____________________________________________I_____________________________________
I
Нитрата аммония NHjNOg Влаги в мелкокристаллическом продукте . Влаги в крупнокристаллическом продукте Не менее 99,4 Не более 0,3 Прп пересчете на сухой продукт азота должно быть 34,8%
Остатка после прокаливания . Нерастворимых в воде примесей . Кислотности в пересчете на азотную ки- слоту ... Сернокислых соединений . Хлористых соединений ... Окисляемых веществ . Не более 0,2 Не более 0.1 Не более 0,02 Не более 0,1 Следы »
Методика испытания принимаемой аммонийной селитры состоит в следующем:
1. Определение содержания нитрата аммония производят при помощи нитро-
метра Луиге, т. е. определяют азот нитрата (по теории 17,5%). Навеску около
0,15 0,30 г. высушенной и хорошо измельченной селитры осторожно всыпают в
воронку нитрометра н туда же вливают 1 мл дистиллированной воды. После ра-
створения селитры раствор вводят в нитрометр. После двукратного смывания
стенок воронки нитрометра 0,5—1 мл дистиллированной воды, которая после
смывания каждый раз вводится в нитрометр, туда же добавляют 5—6 мл химиче-
ски чистой серной кислоты уд- веса 1,84-
Раствор в нитрометре разлагают энергичным встряхиванием в продолжение
не менее 10 мин. Выделяющуюся окись азота собирают в измерительной трубке.
Отсчет количества выделившейся окиси азота производят после полного уравнения
температур жидкости в нитрометре и окружающей среды, на что требуется 15—
20 мин- Объем окиси азота, приведенный к 0° и 760 мм давления, должен быть
не менее 275 мл на 1 а селитры.
На некоторых французских заводах азот нитрата определяют и методом Шле-
зиига, основанным на восстановлении азотной кислоты хлористым железом FeCl3
и соляной кислотой и на последующем определении количества образующейся при
этом окиси азота.
Иногда для определения чистоты селитры определяют содержание аммиака
в ней (по теории 21,28%) путем перегонки с окисью магния.
Общее содержание азота в селитре (по теории 35%) может быть определено
метод ом Деварда или методом Арнда:
а) метод Деварда: к 50 мл раствора 10 г селитры в 1000 мл воды, не содер-
жащей углекислоты, в колбу для перегонки прибавляют 60—80 мл воды и около
4 г сплава Деварда в порошке (45% AI, 50% Си и 5% 2/1) и 10 мл этилового спирта-
Соединяют колбу с дистилляционным аппаратом, прибавляют 50 мл чистого, не
содержащего азота, 35%-ного раствора едкого натра и оставляют Стоять. Спустя
полчаса реакцию заканчивают и начинают перегонку аммиака. Дистиллят улав-
ливают в 45 мл 0,5 N серной кислоты, разбавленной приблизительно 150 мл воды,
и затем титруют;
195
б) метод Арнда отличается от метода Дсварда только тем, что вместо сплава
Деварда применяют 10 г сплава меди и магния и вместо раствора едкого натра —
25 мл 45%-ного раствора хлорида магния.
ОСТ 2465 рекомендует следующий метод определения содержания NH4NO3
в продукте:
навеску NH4NO3 около 4 г взвешивают на аналитических весах и растворяют
в мерной колбе иа 500 мл. Затем 40%-ный формалин нейтрализуют по фенолфта-
леину до слаборозовой окраски- К 25 мл примерно 0,1 А' раствора NH4NO3 при-
бавляют 10 мл нейтрализованного раствора формалина и оставляют стоять 1 мин.
Затем титруют раствором едкого натра (свободного от карбонатов) до появления
розовой окраски.
Содержание NH4NO3 вычисляют по формуле:
«/NH NO - “ ’ “ ' °-008 ’ 100'500 ‘ 100
АГОущ------25 (100— и>)~
где а— число миллилитров 0,1N раствора NaOH:
к — коэфициепт нормальности NaOH;
с — навеска;
w— процент влаги в NH4NO3.
2. Определение влажности производят сушкой навески селитры- Около 5 г
•селитры помещают в предварительно высушенный и взвешенный стаканчик диа-
метром 5 см, снабженный притертой пробкой. Стаканчик с навеской помещают
в термостат и сушат при температуре 100° в течение 2 час. По охлаждении в экси-
каторе стаканчик с навеской взвешивают. Разность в весе дает содержание втаж-
ности в селитре.
Это рекомендуемое ОСТ определение влажности, конечно, неточно, так как
при высокой температуре сушки улетучивается некоторое количество селитры
Можно было бы пользоваться трехдневным высушиванием в вакууме над
свежеконцеитрированной серной кислотой при комнатной температуре, но такое
определение занимало бы много времени.
Для сокращения времени сушки в вакууме применяют повышенную темпе-
ратуру, ио не более 70°.
3. Для определения остатка после прокаливания навеску селитры около
5—7 г помещают в предварительно прокаленный до постоянного веса фарфоровый
тигель и прокаливают на горелке Бунзена. Вначале во избежание потерь от раз-
брызгивания селгггры нагревание ведут медленно и осторожно. По окончании уле-
тучивания селитры пламя горелки увеличивают до возможного предела. После
прокаливания тигель охлаждают в эксикаторе и взвешивают. Привес тигля дает
количество остатка после просаливания.
4- Для определения нерастворимых в воде примесей навеску селитры около
50 г растворяют в 100 мл дистиллированной воды и фильтруют через высушенный
и взвешенный фильтр. Фильтр несколько раз промывают горячей дистиллирован-
ной водой и высушивают при 100° в термостате до постоянного веса. Привес фильтра
после высушивания дает количество нерастворимых в воде примесей.
5- Для определения кислотности 50 г аммонийной селитры растворяют в 100 мл
дистиллированной воды. Качественно кислотность может быть установлена лак-
мусовой бумажкой. При наличии кислотности раствор титруют 0, IN раствором
едкого натра в присутствии метилрота в качестве индикатора.
б. Содержание сернокислых солей определяют осаждением сульфата бария
из кипящего, подкисленного соляной кислотой, раствора 5 г селитры в 100 мл
дистиллированной воды действием однопроцентного раствора хлорида бария-
После 12-часового стояния в теплом месте осадок отфильтровывают на беззольный
фильтр, промывают до тех пор, пока промывные воды перестанут давать осадок
с нитратом серебра. Затем фильтр высушивают, сжигают и прокаливают до по-
стоянного веса в предварительно прокаленном и взвешенном тигле. Количество
полученного сульфата бария пересчитывают на сульфат аммония-
По ОСТ 2465 рекомендуется несколько иной метод, а именно:
5 г NH4N03 растворяют в 30 мл воды в колбе емкостью 200—250 лл и слегка
нагревают на водяной бане. К горячему раствору приливают 8 мл НС! (cl — 1,19),
196
свободной от иона SO4, н 10 мл 40%-ного формалина, после чего колбу снимают
с бани- Через 1—3 мин. начинается медленное, затем постояине усиливающееся,
выделение окислен азота. Во избежание потерь колбу накрывают часовым стеклом.
Если реакция не начинается, то через 5 мин. колбу осторожно подогревают на
бане. По окончании реакции раствор переносят в фарфоровую чашку на 200 мл,
выпаривают досуха на водяной бане и остаток сушат при 100' в течение получаса-
Далее осадок растворяют в 50 мл воды, фильтруют и промывают 50 мл воды.
Фильтрат с промывными водами подкисляют 1 мл НС1 (<! 1,10), нагревают до
кипения, ион SOj осаждают 10%-ным раствором ВаС1г и оставляют стоять |2 час-
Затем осадок фильтруют через фарфоровый пористый тигель, промывают горя-
чей водой до исчезновения реакции на ион С!', прокаливают и взвешивают.
7- Содержание хлористых солей устанавливают, прибавляя капли, раствора
нитрата серебра к подкисленному химически чистой азотной кислотой раствору
5 г селитры в дистиллированной воде- Допускается лишь слабая опалесценция
раствора.
8. Для определения окисляемых веществ к 100 мл дистиллированной воды,
подкисленной химически чистой серной кислотой, при температуре 40—50° при-
ливают по каплям 0.1N раствор перманганата калия до прекращения обесцвечи-
вания. Затем добавляют еще одну каплю этого раствора. При растворении в по-
лученной жидкости около 5 г селитры не должно происходить обесцвечивания
раствора-
2. Амид натрия- Амид натрия в чистом виде представляет
собой бесцветную, лучистую кристаллическую массу, уд. веса 1,39
при 25°. Технический амид натрия большей частью окрашен в зеле-
новато-серый цвет. Температура плавления амида натрия 208°. На-
гревание при более высоких температурах ведет к возгонке амида
натрия и к частичному его разложению. Так, при нагревании в ва-
кууме он начинает разлагаться уже при достижении 300°, а при 450°
разлагается полностью.
Расплавленный амид натрия растворяет металлический натрий,
но не вступает с ним в соединение. Раствор металлического натрия
в расплавленном амиде натрия имеет темноголубой цвет.
На воздухе амид натрия сравнительно быстро разлагается- Вода
также разлагает амид натрия с образованием гидрата окиси натрия
и аммиака. В расплавленном виде он действует как сильный восста-
новитель. При накаливании с углем амид натрия образует цианид
натрия. С окйслами азота в присутствии влаги амид натрия образует
соли азотной и азотистой кислот. При получении амида натрия необ-
ходимо следить за тем, чтобы применяемый для этой цели аммиак
был свободен от влаги и воздуха.
Если при температуре 100—110° пропускать через порошок амида
натрия струю сухого, свободного от СОо, воздуха, то через 170 час.
9,5% амида натрия превращается в окисленный продукт желтого
цвета состава NaNH3 • О2. Кислород воздуха действует на расплав-
ленный амид значительно быстрее.
Наряду с применением амида натрия в технике для получения
азида и цианида натрия он используется также в промышленности
органически^ веществ, например, при производстве индиго из фенил-
глицина.
Металлический натрий. Металлический натрий пред-
ставляет собой мягкий, серебристо-белый металл, легко режущийся
1S7
обыкновенным ножом. Удельный вес натрия 0,971. При нагревании
до температуры 97,7° плавится, при температуре 880° кипит. Пары
натрия сильно разъедают стекло. Плотность пара натрия была опре-
делена лишь в 1930 г. Родебушем и Волтерсом, причем оказалось,
что она отвечает смЬси одноатомных с небольшим количеством двух-
атомных молекул натрия. В жидком аммиаке натрий растворим, что
облегчает реакции аммонолиза. Натрий — сильный восстановитель,
очень легко теряет электрон и превращается в катион. Азотная кис-
лота под действием натрия восстанавливается до аммиака.
Натрий очень энергично реагирует с кислородом воздуха, свеже-
срезанная поверхность металла почти мгновенно тускнеет и покры-
вается слоем окиси. С водой бурно реагирует, образуя гидрат окиси
натрия и водород с большим выделением тепла (44,1 Нал/мол). Выде-
ление такого большого количества теплоты может вызывать вспышки
и серьезные взрывы, особенно если взяты большие куски металла.
Попадая на влажную кожу, металлический натрий вызывает болез-
ненные ожоги. Совершенно ясно поэтому, что работать с натрием
необходимо в спецодежде и защитных очках.
С аммиаком при повышенной температуре натрий также энер-
гично реагирует, образуя натрийамид и водород. При пониженных
температурах натрий растворяется в жидком аммиаке, образуя раст-
вор темноголубого цвета.
Металлический натрий хранится в запаянных жестяных короб-
ках, в жидкостях, которые с ним не только не реагируют, но и пре-
дохраняют от воздействия кислорода воздуха и влаги. Чаще всего
для этой цели применяют керосин, иногда парафин.
На заводы металлический натрий поступает в кусках до 1—2 кг
весом в жестяной укупорке, или залитым в железные барабаны
весом 24—28 кг, обязательно смоченные керосином, и в таком виде
хранится до употребления.
Применяемый в производстве металлический натрий должен иметь
уд. вес в пределах 0,97—0,98, что определяют пикнометрическим
методом в керосине с последующим пересчетом на воду. Содержание
натрия должно быть не менее 99,6%. Определение содержания натрия
производят следующим образом: 2—3 г металлического натрия раст-
воряют в 50 мл 96%-ного этилового спирта, доливают 150 мл воды и
титруют O,1N раствором соляной кислоты. Ни в коем случае недо-
пустимо наличие в нем примесей меди, ртути и свинца. Реактивом
на катион меди является NH4OH (не должно появляться посинения
нейтрализованного раствора натрия в воде), на ртуть — иодистый
калий (не должен появляться красный осадок HgJa), а на свинец —
хромат калия (не должен появляться желтый осадок РЬСгО4).
Аммиак. Аммиак представляет собой бесцветный газ с едким
запахом и вкусом. Он значительно легче воздуха, уд. вес, отнесенный
к воздуху, равен 0,5983. 1 л аммиака при 0° и 760 мм рт. ст. весит
0,7709 г. Легко сжижается при давлении 8.46 ат и 20°, образуя
легкоподвижную бесцветную жидкость с температурой кипения 33,4°;
удельный вес жидкого аммиака при 0° 0,6382; критическая темпера-
198
тура аммиака 132,4': критическое давление 112 ат; температура плав-
ления твердого аммиака 77,7°; теплота плавления 81 кал{г; теплота
испарения при температуре кипения 327 кал(г, а при 0° 302 кал[г.
Аммиак легко растворим в воде. Растворимость аммиака в воде
следующая:
Температура °C В ТОО г воды растворяется NH3 г Температура ‘С В 100 г воды растворяется NHS г
0 89.5 16 58.7
8 72.0 20 53.1
° 65.1 24 48.2
При кипячении из водных растворов аммиак улетучивается пол-
ностью. Водные растворы аммиака ведут себя, как растворы основа-
ний.
На воздухе аммиак в обычных условиях не горит, однако, некото-
рые смеси аммиака с воздухом от "луча огня воспламеняются и дают
сильный взрыв.
Лейбольд описывает случай, когда при поломке компрессора для
аммиака большое количество последнего проникло в машинное отде-
ление, следствием чего были взрыв и пожар. Опыты показали, что
смеси воздуха с аммиаком при содержании последнего от 17 до 27%
(по объему) взрывают от пламени при обыкновенном давлении, вызы-
вая повышение давления до 6 ат, т. е. то же, что и при взрыве смесей
водорода с воздухом.
Аммиак разлагается при пропускании его над многими металлами
или окислами металлов. В некоторых случаях азот аммиака при этом
соединяется с металлом, образуя нитриды. Щелочные и щелочнозе-
мельные металлы замещают в аммиаке 1 атом водорода, образуя
амиды, например:
2NH3 + 2Na 2NaNH2 + Н2.
Аммиак раздражающе действует преимущественно на верхние
дыхательные пути. Большие количества его действуют возбуждающе
на центральную нервную систему. Отравления аммиаком возможны,
главным образом, при авариях с аппаратурой и баллонами и в общем
носят острый характер. Человек ощущает раздражение уже при кон-
центрации 0,01 мг/л; концентрацию 0,25 мг{л человек переносит
с трудом в течение 1 часа, более высокие концентрации аммиака вы-
зывают резкое слезотечение, кашель, удушье.
В СССР предельная допустимая концентрация аммиака в воздухе
рабочих помещений установлена в 0,02 мг/л.
На заводы аммиак поступает в виде жидкости в специальных сталь-
ных баллонах, рассчитанных на давление 40 ат и выше. Емкость
таких баллонов 20—30 л. Баллоны при хранении' необходимо обере-
199
гать от резких толчков и ударов во избежание
так как железные и стальные предметы легко
руют при взаимодействии с водным раствором
Применяемый для производства амида натрия аммиак должен
удовлетворять следующим техническим условиям:
1) отсутствие механических примесей,
2) содержание влаги не более 0,2%.
3. Нитрат свинца представляет собой прозрачные кри
сталлы, в зависимости от условий кристаллизации принадлежащие
к правильной или к моноклинной сингонии. Растворимость нитрата
свинца в воде хорошая; в 100 мл воды растворяется:
при
взрыва и от сырости
и энергично корроди
аммиака.
0е - ... 38,8 г
20е’ - 56,5 »
40 75 »>
60 . 95 «
80' . 115 •
100е . 139 .
Так же как и многие другие соли свинца, нитрат свинца легко обра-
зует продукты присоединения, главным образом, с окисью свинца
(основные нитраты).
Водные растворы нитрата свинца вызывают сильную коррозии
(это надо иметь в виду!) металлического свинца. Согласно Тиле, ни-
трат свинца, переходя в нитрит, окисляет металлический свинец,
разъедая, главным образом, загрязненные поверхности свинца, та!
называемые «промежуточные вещества» (по Тамману), находящиеся
между отдельными кристаллами.
К применяемому в производстве нитрату свинца предъявляются
следующие технические требования:
1) по внешнему виду — белый кристаллический порошок;
2) содержание нерастворимых в воде примесей не более 0,3%;
3) содержание меди, ртути и иона СН3СОО' не допускается;
4) железа допускаются только следы;
5) кислотность в пересчете на HNO3 не более 0,5%;
6) содержание Pb(NO3)2 не менее 99,2%.
Для определения нерастворимых 10 г Pb(NO3)2 растворяют в 100 мл воды
фильтруют, промывают до исчезновения реакции на ион NOg - Остаток на фильтре
высушивают до постоянного веса-
Для определения иона СН3СОО' к 5 мл насыщенного раствора Pb(NO8)2 при
ливают 2 мл 10%~ного раствора FeCI3. При этом не должно происходить потем
нения раствора сравнительно с «пустой» пробой.
Для определения примеси меди стальную или железную, предварительно про
мытую спиртом и эфиром, пластинку опускают- в насыщенный раствор Pb(NO2)
на 1 час. Затем пластинку переносят в водный раствор аммиака. Посинения
раствора аммиака происходить не должно.
Для определения примеси ртутных солей в насыщенный раствор Pb(NOs)
погружают обезжиренную пластинку красной меди на 2 часа- После этого на
пластинке не должно быть даже следов побеления.
Наличие железа устанавливают обычной реакцией с роданидом аммония.
Для определения кислотности 5 г Pb(NO3)2 растворяют в 50 мл воды. К рас-
твору добавляют 50 мл нейтрального раствора" Na2SO4 (7 г Na2SO4 + 50 мл Н2О)
200
от полученной смеси отфильтровывают 50 мл, прибавляют 3 капли спиртового
раствора фенолфталеина и титруют 0,11V раствором щелочи.
Pb(NO3)3 в исходном продукте определяют по свинцу сульфатным методом
осаждением свинца в виде PbSO4.
4. Из вспомогательных материалов, применяемых в производстве,
следует остановиться только на бензоле, бензине и парафине, идущих
на изготовление флегматизирующего раствора.
Нитраты бария и кальция, применяемые для очистки азида нат-
рия, должны отвечать нашим ОСТ на чистые реактивы.
Что касается применяемого для промывки азидов натрия и свинца
спирта, то требования к нему ничем не отличаются от требований
к спирту, идущему на производство гремучей ртути.
Остальные вспомогательные материалы: 1) раствор гидроксида
натрия для очистки закиси азота от высших окислов азота; 2) хло-
ристый кальций для осушки газа; 3) азотная кислота для уничтожения
остатков азидов; 4) серная кислота для поглощения аммиака и др.
не требуют особой чистоты; могут применяться технические продукты.
Бензол — каменноугольный, чистый — представляет собой
бесцветную, прозрачную, сильно преломляющую свет жидкость, со
своеобразным запахом и жгучим вкусом; температура кипения 80, 18°;
уд. вес при 15° 0,8786; точка замерзания 5,4°. С воздухом чистый
бензол образует взрывчатые смеси.
Следует иметь в виду, что бензол относится к ядам с преимуще-
ственно наркотическим действием при остром отравлении.
Повышенная температура (летняя жара, теплый комнатный воз-
дух) способствует испарению бензола; при отсутствии вентиляции
на рабочем месте возможны отравления. Иногда несколько вдыханий
уже может вызвать отравление.
Идущему на растворение флегматизаторов бензолу’ предъявляются
следующие технические требования:
1) продукт должен быть бесцветен и прозрачен;
2) удельный вес его при 15° должен находиться в пределах
0,880—0,885;
3) при нагревании он должен испаряться без остатка;
4) пределы его кипения при давлении 760 мм рт. ст. должны быть
равными 78,5—80,6°; в пределах 1° должно перегоняться не менее
95% всего бензола;
5) окраска бензола в смеси с серной кислотой по шкале Кремера
и Шпилькера должна быть не выше 0,5;
6) бромное число должно быть не выше 0,6 брома на 100 мл
бензола.
Б е н з и н, идущий на производство азида свинца, должен удов-'
летворять следующим требованиям:
1) уд. вес 0,725—0,750;
2) дробная перегонка по Энглеру:
а) начало перегонки 50—75°,
б) погонов до 100°—не менее 30%,
в) конец перегонки не выше 175°,
201
г) остатка в колбе не более 1,5%;
3) кислотность (в мг КОН в 100 мм) не более 2%;
4) серы не более 0,1%.
Парафин представляет собой смесь твердых предельных угле-
водородов вида СпН2п + 2* Эмпирическая формула чистого парафи-
на С24Н50, уд. вес находится в пределах 0,907—0,915. Идущему в
производство азида свинца парафину предъявляются следующие
требования:
1) цвет белый;
2) механические примеси не допускаются;
3) температура затвердевания, по Жукову, не менее 54 .
4) содержание масла, по Гольцу, не более 1%;
5) полное отсутствие кислот и щелочей;
6) при освещении в течение 7 суток рассеянным светом не должен
желтеть;
7) при нагревании с серной кислотой не должен изменяться;
8) должен быть полностью растворим в ацетоне и сероуглероде.
§ 7. ТЕХНОЛОГИЧЕСКИЙ ПРОЦЕСС ПРОИЗВОДСТВА АЗИДА СВИНЦА
Технологический процесс производства азида свинца состоит из
.следующих операций:
1) получение к очистка закиси азота;
2) получение амида натрия;
3) получение азида натрия;
4) очистка азида натрия.
5) приготовление растворов исходных продуктов;
б) осаждение азида свинца;
7) фильтрация, промывка и флегматизация;
8) грануляция азида свинца;
9) сушка азида свинца;
10) сортировка азида свинца.
Получение и очистка закиси азота. Аммиач-
ную селитру подают в мастерскую отдельными партиями, примерно
по 40—50 кг, чтобы избежать перегрузки здания мастерской.
Аммонийную селитру для получения закиси азота, по Уль-
ману, разлагают в длинногорлых литых чугунных ретортах емкостью
около 30 л. Максимально допустимая загрузка реторты 16—18 кг
NH4NO3. Подвергать одновременной обработке большие массы не при-
нято, так как разложение вследствие экзотермичности реакции может
принять взрывной характер. При одновременном разложении таких
масс рекомендуется для торможения реакции применять песок, а еще
лучше — керамиковые осколки.
Чаще применяют алюминиевые реторты (фиг. 84), имеющие вид
цилиндрических сосудов емкостью около 30—35 л. Сверху реторты
.закрываются алюминиевыми же крышками на болтах. В крышке
имеются отверстия для загрузки, для пирометра или ртутного термо-
метра и для выхода газов. Для обогрева на реторты навивается электро-
202
нагревательная спираль. Реторта вместе со спиралью помещается
в железном кожухе. При разложении более или менее больших масс
NH4N03 перед пуском реторты в работу загружают около половины
объема битой керамики. Однако при небольших загрузках (5к г и менее)
можно легко регулировать процесс разложения и без керамики. При
наличии последней невозможно простыми способами определять свое-
временно конец разложения
загруженной порции селитры-
Измельченную селитру за-
гружают в реторту периодиче-
ски, порциями, величина кото-
рых зависит от объема реторты,
и только после того, как рацее
загруженная порция полностью
разложится. Контроль осущест-
вляют металлическим прутом,
опускаемым время от времени
через загрузочное отверстие в
реторту. При наличии в реторте
расплавленной, но не разложив-
шейся до конца, селитры конец
прута покрывается белой плен-
кой затвердевшей селитры.
Селитру загружают вручную, как можно быстрее, через воронку,
по возможности в один прием, не пропуская в реторту воздуха (кисло-
род воздуха — вредная примесь). Температура реторты во время за-
грузки не должна превышать 240°. После загрузки загрузочное от-
верстие закрывают асбестовой пробкой с дополнительным грузом
1—2 кг\
Селитру разлагают при температуре 240—245°. Наблюдение за
температурой и ее регулирование являются самой важной частью
работы мастера-аппаратчика, ибо от того, при какой температуре
ведется разложение селитры, зависит состав газов. Ниже 200° в со-
ставе газов может находиться значительный процент аммиака, при тем-
пературах выше 260° появляются в большом количестве высшие окислы
азота. Дальнейшее повышение температуры, принимая во внимание
все возрастающую экзотермичность процесса, становится опасным
и может привести к взрыву реторты. Ни в коем случае не допускается
повышение температуры до 285°. Если температура в реторте почему-
либо превысит 285°, то немедленно должен быть создан во всей системе
вакуум и начата откачка газа во избежание взрывчатого разложения
селитры.
Не менее существенным является содержание влажности в се-
литре. Чем влажнее селитра, тем ниже температура ее плавления
1 Весьма желательным было бы непрерывное питание аппарата аммонийной
селитрой. Задача эта, однако, пока еще конструктивно не разрешена. Попытки
решения, предложенные разными авторами, неудовлетворительны.
203
п тем скачкообразнее идет процесс плавления п разложения. При на-
личии влаги наблюдается сильное вспенивание и выпучивание селитры
до тех пор, пока вся влага не улетучится- На производстве это может
приводить к забиванию газоотводной трубы реторты, к выделению
части газов в помещение и к вынужденной остановке реторты.
При большой влажности (выше 2,5%) разложение селитры с выделе-
нием газов начинается уже при температурах 170—180°, но оно скоро
прекращается (говорят «реторта киснет»). Тогда требуется поднять
температуру в реторте дополнительным нагреванием ее при помощи
электрического тока, к чему приходится подходить очень осторожно,
так как несвоевременным выключением обогрева можно вызвать очень
бурную реакцию, что отразится на состоянии аппаратуры. Сухая
селитра разлагается спокойнее при температуре около 220°, и управ-
лять ее разложением довольно легко.
Для получения газа с максимально возможным содержанием за-
киси азота необходимо соблюдение еще одного условия— отсутствия
вакуума или давления в реторте.
При благоприятных условиях работы (температура разложения
240—245°, сухая аммиачная селитра, отсутствие притока воздуха
извне и т. п.) состав сырого газа после освобождения его от паров воды
примерно следующий:
N2O 80%
NH3 .
Ns - 18%
Nb,No03 н др. I
o2. :......... 2%
Повышение температуры до 280° влечет за собой понижение содер-
жания закиси азота:
NoO . 71,2%
О2................... 2,5%
прочих газов . . - 26,3%
При неправильной работе реторты, а в особенности при наличии
в аммонийной селитре большой влажности, анализ сырого газа дает:
14,0......... 45%
окнслов азота . 13% (иногда 17—23%)
NHa 10%
О.. . 6-^-12% (иногда 14—15%)
N; 28—35%
Закись азота поступает на производство азида натрия. Примеси
вредно влияют на ход последующей реакции азцдирования и на аппа-
ратуру. Окислы азота, например, разъедают аппаратуру (трубопро-
воды, компрессор, азидный аппарат) и загрязняют железными солями
азид натрия. Обладая кислыми свойствами, они, кроме того, дей-
ствуют разрушающе на амид натрия, обладающий щелочными свой-
ствами.
Не менее вредными примесями являются пары воды и кислород.
Как видно из изложенного, пары воды разрушают NaNH2 с образова-
204
кием NaOH и NH3, вследствие чего значительно уменьшается выход
NaNg. Кислород при высоких температурах окисляет NaNH2 и NaNs,
что также влечет за собой снижение выхода азида натрия. Поэтому
очистке закиси азота уделяется большое внимание.
Для освобождения от большого количества паров воды горячие
газы из реторты пропускают через змеевиковый холодильник, где
пары воды конденсируются, а с конденсированной водой частично
удаляются и растворяющиеся в ней окислы азота и аммиак. Образую-
щийся раствор (конденсат) собирается в сифоне холодильника и по
мере его заполнения поступает в керамиковый горшок или непосред-
ственно в канализацию.
Анализ конденсата показывает наличие в нем около 4% аммиака
и около 5% азотной и азотистой кислот (4,5% HN03 и 0,5% HN02).
Охлажденные газы из холодильника поступают в скрубберы, оро-
шаемые 8—30%-ным раствором гидроксида натрия. При этом проис-
ходит полное освобождение закиси азота от высших окислов азота.
Промывка газа осуществляется по принципу противотока: снизу
в скрубберы поступает газ, сверху сливается раствор щелочи. Скруб-
бер представляет собой железную башню емкостью около 550 л, на-
полненную для увеличения поверхности поглощения щелочи насадкой
из колец Рашига, представляющих собой керамиковые кольца и по-
лые цилиндры разных размеров. В верхней части башни расположен
цилиндрический бачок емкостью около 250 л со щелочью и ороситель-
ным приспособлением. Нижняя часть башни покоится на втором бачке
той же емкости, представляющем собой сборник отработанной щелочи.
По мере освобождения верхнего бака и загрузки сборника щелочь
при помощи монжуса перекачивают обратно в верхний бак. Циркуля-
ция щелочи производится до содержания в промывающем растворе
7—9% NaOH.
Анализ промывающего раствора, уже непригодного для использо-
вания, следующий:
NaOH 7 9%
NaNO3 13—15%
NaNCh, . 0,4-0,7%
NH3 . 0,11—0,13%
Отработанную щелочь удаляют и заменяют свежей.
Очищенный газ из скрубберов вновь проходит через холодиль-
ник, затем — через колонки с хлористым кальцием для осушки,
вернее, для освобождения от частиц щелочи, увлекаемой газом из
скрубберов, в железный газгольдер, где и собирается над водой.
Во всей системе реторта разложения NH4NO8 — газгольдер дав-
ление должно быть равным атмосферному, что контролируют рядом
манометров, расположенных у каждого аппарата, и регулируют гру-
зом, уравновешивающим колокол газгольдера.
Контроль наполнения газгольдера, располагаемого, как правило,
вне мастерской получения и очистки закиси азота, осуществляют
При помощи световой электрической сигнализации.
205
По мере наполнения газгольдера закись азота перекачивают ком-
прессором через сушильные колонки в аккумулятор, где она хра-
нится под давлением 4—8 ат (в зависимости от прочности аккумуля-1
тора). Схема очистки показана на фиг. 85.
Фиг. 85. Схема очистки закиси азота.
Получение амида натрия- Амид натрия получают по
методу Кастнера пропусканием чистого сухого газообразного аммиака
через расплавленный металлический натрий:
2Na + 2NHS -> 2NaNH2 + Н2.
Для этого служит специальный стальной цилиндрический аппарат,
снабженный нагревающей электро спиралью (фиг. 86). В крышке
аппарата вмонтированы: 1) штуцер с трубкой, проходящей почти до
дна (10 мм от дна) и заканчивающейся спиралью барбатера для ввода
аммиака, 2) штуцер для отвода газообразных продуктов реакции,
3) штуцер с трубкой, служащей футляром термопары. Загрузочное
отверстие закрывается герметически. У дна на боковой поверхности
аппарата имеется отводная трубка с краном для спуска готового рас-
плавленного амида натрия.
Перед загрузкой аппарата необходимо проверять исправность
газопроводов, кранов электрообогрева; все краны необходимо тща-
тельно протирать и смазывать графитом, аппарат — продуть и высу-
шить при 150° в слабом токе аммиака (около 100 г в час).
Металлический натрий перед загрузкой разрезают тонким сталь-
ным ножом на куски по 100—150 г весом. Количество загружаемого
натрия зависит от размеров аппарата. Обычно натрий в расплавлен-
ном состоянии занимает */3 объема амидного аппарата.
При резке натрия необходимо соблюдать элементарные правила
безопасности (отсутствие воды на столе и ноже, резиновые перчатки
на руках и предохранительные очки на глазах). Если металлический
натрий загорается, его следует тушить сухим песком (но не водой!)
206
Натрий загружают в аппарат также при слабом токе аммиака.
Аммиак поступает из баллона через сушилку и уловитель (предохра-
нительную колонку). Во время загрузки натрия при неосторожной
работе (особенно при наличии влаги) могут иметь место вспышки;
к этом случае загрузка должна быть приостановлена и загрузочное
нгверстие спешно закрыто асбестом и крышкой.
Фиг. 86. Амидный аппарат.
После загрузки аппарата необходимым количеством натрия гер-
метически закрывают загрузочное отверстие и поднимают темпера-
туру вначале до 380°, усиливая при этом подачу аммиака в аппарат
до 500—600 г в час. Затем подачу аммиака доводят до 1,0—1,5 кг
в час. Нагревание при 380е продолжают 6—8 час., т. е. до тех пор,
пока реакция амидирования не достигнет достаточной скорости. Ско-
рость процесса амидирования определяют путем анализа отходящих
газов. Надлежащая скорость процесса характеризуется содержанием
в отходящих газах 70—80% водорода при подаче 1—1,5 кг аммиака
в час. По достижении наивыгоднейшей скорости реакции температуру
аппарата следует снизить до 340—360° и дальнейший процесс вести
в пределах этой температуры. Скорость процесса при этом не умень-
шается, так как, невидимому, образовавшийся амид натрия дейст-
вует каталитически. Проведение всего процесса при температуре 380°
и выше нежелательно, потому что амид натрия может сублимироваться
и засорять газопровод.
Ток аммиака во время всего процесса должен быть ровным, давле-
ние в азидном аппарате не должно превышать атмосферного. В слу-
чае обнаружения в аппарате давления необходимо прекратить пуск
аммиака и найти место закупорки газопроводов — сначала выход-
ных, а затем входных (при закрытых кранах самого аппарата).
В результате процесса в аппарате образуются два слоя: нижний—
амид натрия (d ~ 1,3) и верхний — металлический натрий (d = 0,97).
По мере хода реакции верхний слой уменьшается.
207
Выделяющийся при реакции водород и изоыток аммиака отводя
через пылсотделитель и водоуловитель с водомерным стеклом в бутыл!
с водой, где аммиак поглощается, а водород выпускается в атмосфере
вне мастерской. Назначение пылеуловителя — улавливание уносимых
газами частичек амида натрия, назначение водоуловителя-буфера
состоит в том, чтобы вода из бутылей при внезапной закупорке вход!
ных газопроводов для аммиака не могла проникнуть в амидный аппа-
рат и вызвать катастрофу.
Процесс амидирования длится 20—25 час. Скорость амидирова-
ния зависит от скорости пропускания аммиака и от температуры,
причем избыток аммиака (20—30%) и повы-
Фиг. 87. Кривые ами-
дирования.
шение температуры способствуют ускорению
процесса. Однако повышение температуры
возможно только до известного предела
(350—360°); при дальнейшем повышении тем-
пературы начинается возгонка NaNHs.
Очень вредно сказывается на выходе и
качестве конечного продукта так называемое
«переамидирование», т. е. действие аммиака
на получившийся амид натрия. Переами-
дирование ведет к разрушению амида натрия,
образованию примесей, стало быть, к умець-1
шению выходов. О готовности продукта
можно судить только на основании того, что I
в конце процесса увеличивается содержание [
водорода в отходящих газах.
Аналитическому обслуживанию процесса
амидирования необходимо уделять весьма
серьезное внимание. Контроль содержания
водорода в газах в начале процесса должен осуществляться через
каждые 4—5 час., затем через 2 часа, а в конце процесса через 30 мин.
Кривые изменения процентного содержания водорода для ряда
процессов амидирования изображены на фиг- 87. Пропускание ам-
миака следует прекратить по достижении на кривой минимума, за-
крепленного двумя последующими анализами, указывающими иа
дальнейшее повышение кривой. Кривые также показывают, что мини-
мальное содержание водорода в отходящих газах должно быть в пре-
делах 5—7%.
Получение амида натрия по методу Кастнера нельзя признать экономически I
.выгодным и достаточно эффективным для полного удовлетворения потребностей
производства (азид натрия, цианамид натрия и пр-), так как в качестве исходного
сырья требуется металлический натрий.
Исследования Крауса о динамическом равновесии системы (жидкий раствор):
Na -J- NHS NaNH„ + 0,5Н2
показали, что стабильность растворов в системе Na—NH3 сильно сдвигается в сто-
рону образования амида натрия катализаторами. Различными авторами были ука-
заны катализаторы для получения амида натрия из раствора металлического натрия
в аммиаке. Оказалось, что окисям железа, щелочи, углеродистое железо, наконец,
самый амид натрия сильно ускоряют реакцию амидирования.
208
Эванс1 предлагал получать амид натрия из растворов натрия в аммиаке, упо-
| ребляя углеродистое железо в качестве катализатора. Однако это предложение
не решает вопроса об удешевлении стоимости производства амида натрия и эконо-
мически является мало эффективным.
Г. И- Войиилович, Б- Б. Васильев и Е. И.
Лхутов2 (ГИПХ) разработали новый метод полу-
чения амида натрия электролизом хлористого
натрия в жидком аммиаке. В качестве аппарата
для электролиза они пользовались рассчитанным
па давление в 25 ат автоклавом (фиг. 88), кор-
пус которого служил катодом. В центре авто-
клава помещали графитовый электрод, окру-
женный диафрагмой из асбестового картона,
разделяющей анодное и катодное пространства.
В зависимости от условий работы аиод приводили
ио вращение, чем достигали перемешивания элек-
тролита- К аппарату были приделаны вентили
в анодном и катодном пространстве для анализов
к отвода газов и карман для термометра (темпе-
ратура при электролизе 10—0°). Давление в
автоклаве измерялось манометром. Средние плот-
ности тока 0,82 Щсм'2- на аноде и 0,0175 к[см* на
катоде. Напряжение на зажимах аппарата под-
держивалось в пределах 4—5 V.
В результате электролиза в катодном про-
странстве получается амид натрия,который оседает
на дно автоклава в виде белого кристаллического
порошка. Газообразная фаза обогащается водо-
родом, выделяющимся в результате реакции-
В анодном пространстве выделяется хлор, кото-
рый, реагируя с газообразным аммиаком, дает
хлористый аммоний и свободный азот. Давление
в катодном пространстве должно превышать давление газов в анодном простран-
стве па 0,2—0,5ат во избежание проникновения хлора в катодное пространство.
Процесс образования побочных продуктов при электролитическом получении
амида натрия еще не изучен, но можно предполагать, что он идет по следующей
схеме:
Фиг- 88. Автоклав для
аммонолиза.
3CI24-4NHs -> NCIS + 3NH4CI
NCl3 + 4NH3 > N2 3NH4CI
или суммарно:
ЗС lz 4- 8NH3 -> N2 + 6NH4C I.
Процесс электролиза протекает спокойно. Поэтому, казалось бы, нет основания
предполагать возможность образования в этих условиях хлористого азота, являю-
щегося, как мы уже знаем, взрывчатым веществом н к тому же весьма чувстви-
тельным. Однако этот вопрос подлежит еще изучению, так как у нас нет доказа-
тельств о течении реакции именно по первым двум уравнениям.
Рассматривая электролитический процесс амидирования в целом, как состоя-
щий из двух фаз:
6NaCl + 6NHS 6NaNH2 4- 3CI2 4- ЗН8 (первичная реакция)
ЗС1я 4- 8NH3 -> N2 4- 6NH4C1 (вторичная реакция)
6NaCl 4- 14NH3 -> 6NaNHB 4- 6NH4C14- 3H2 4- N£
следует отметить, что в результате вторичной реакции теряется 14,3% азота-
1 Т. Evans, герм. пат. № 411732, 1924. .
2 Получение азида натрия по способу ГИПХ, журн. «Хим. пром.», № 10,
стр. 54—59, 1934.
Бубнов—92—14 209
Технологическая схема электролиза (фиг. 89) авторами метода представлена
непрерывно действующей с регенерацией аммиака и состоящей из ряда автокла-
вов с охлаждающими рубашками (промыватели, осушители, колонки и т. п-).
Жидкий аммиак из сборника 7 поступает в осушитель 2, содержащий окись
кальция, и направляется в солерастворитель 3, где растворяет осушенный хлори-
стый натрий, подвергающийся далее электролизу в температурном интервале от
10до0° в ваннах 4, рас-
фиг. 89. Технологическая схема получения NaNH„.
положенных каскадом.
Образующийся на катоде
амид натрия оседает на
коническом днище элек-
тролизера, откуда он
периодически выпускает-
ся на фильтры 5. На
фильтрах остается амид
натрия, который промы-
вается безводным аммиа-
ком из осушителя 2. По-
сле промывки амид нат-
рия расплавляется н по-
ступает в расплавленном
состоянии в тару или
непосредственно в азид-
ный аппарат.
Анодные и катодные
газы через каплеотдели-
тели 7 'и 9, теряя в них
хлористый аммоний, про-
ходят в сжижающие ап-
параты 8
и
10,
где аммиак
конденсируется и возвращается в производство, а водород улавливается в отдель-
ные сборники-
Солеотделитель 3 представляет собой колонный аппарат с охладительной
внешней рубашкой, наполненный хлористым натрием, расположенным на не-
скольких сетчатых полках.
Аппарат 5 представляет собой комбинированное устройство для фильтрования
отработанного электролита, для промывания осадка амида натрия жидким аммиа-
ком и для плавления амида натрия- Аппарат-автоклав имеет внешнее охлади-
тельное и внутреннее нагревающее устройства, а также специальное фильтрующее
приспособление. Расплавленный и очищенный амид натрия выливается через
выпускное отверстие.
По мнению авторов метода, аппаратура может быть изготовлена из железа,
которое является достаточно стойким по отношению к жидкому аммиаку. Жидкий
аммиак благодаря малой константе диссоциации (порядка 10-33) является веще-
ством инертным. Аммонийный же ион (NH4) следует изолировать от железа.
Выход продуктов при настоящем способе производства авторы определяют
следующим образом:
NaCl 4- 2NHa-> NaNH, 4- NHjCl
1 tn 0,9 m i m 1,5 m
Получение
рия спускают из амидного аппарата в азидный. Азидный аппарат
(фиг. 90) представляет собой стальной расположенный горизонтально
цилиндр, снабженный валом-мешалкой с насаженными стальными
зубьями- Корпус аппарата имеет внутри, по стенкам, насаженные
зубья, расположенные так, что при вращении мешалки ее зубья про--
ходят между зубьями корпуса. Такое перемешивание необходимо
азида натрия. Расплавленный амид нат-
210
потому, что в процессе амидирования происходит реакция между
жидкой (NaNHs) и газообразной (N2O) фазами, в результате чего
'образуется твердый NaN8. Здесь необходимо перемешивание, сопро-
рождаемое растиранием, так как твердая фаза NaN3 может окружать
Жидкую фазу (NaNH3) и комковать ее, что затруднит непосредственное
соприкосновение последней с газообразной закисью азота и приведет
к замедлению или, что еще хуже, к приостановлению реакции.
Фиг. 90. Азидный аппарат.
Азидный аппарат оборудован электро обмоткой для нагревания
и тремя колонками. Средняя колонка имеет штуцер для водопровода,
крайние же имеют: 1) штуцер для спуска NaNH2, 2) штуцер для впуска
п выхода газа, 3) штуцер для впуска воды, 4) штуцер для трубопро-
вода, назначенного для выхода паров воды и впуска аммиака, и, на-
конец, 5) отверстие для трубки, являющейся оболочкой пирометра.
В нижней части аппарата имеется отверстие, соединенное с трубопро-
водом, для удаления готового продукта.
Перед началом работы на азидном аппарате его тщательно подго-
товляют. Аппарат нагревают до 170—200°, сушат при этой темпера-
туре и одновременно продувают аммиаком (слабый ток 90—100 г в час)
при открытых кранах к холодильникам и при закрытых к пылеотде-
лителям. Это необходимо для удаления влаги, которая может вызвать
разложение поступающего в аппарат расплавленного амида натрия,
особенно чувствительного к влаге. В присутствии значительного ко-
личества воды расплавленный амид натрия может дать взрыв.
В таким образом подготовленный аппарат после проверки исправ-
ности трубопроводов спускают расплавленный амид натрия с темпе-
ратурой около 350° при работающей на небольшой скорости мешалке
(около 60 об/мин.). При установлении в аппарате температуры
210—220° пропускают 68—75%-ную закись азота и увеличивают
скорость мешалки примерно до 90 об/мин-
В процессе азиднрования периодически производят анализ отхо-
дящих газов (смесь NH3 н N3O). Как только содержание аммиака
в отходящих газах достигает примерно 15%, можно заменить высоко-
211
процентную закись азота низкопроцентной отработанной закисью
азота, содержащей около 35—40% N2O, с тем чтобы в конце процесса
вновь дать свежую закись азота.Одновременно увеличивают скорость
перемешивания до 200—300 об/мин.
Закись азота для реакции поступает из аккумуляторов, проходит
через цилиндрическую сушильную колонку с безводным хлористым
кальцием, редуктор давления газа и пылеотделители. Закись азота
для азидирования берется в избытке, чтобы обеспечить полностью
образование азида натрия и сократить время процесса.
Закись азота пропускают попеременно через правую и левую
колонки в таком количестве, которое обеспечивает поддержание тем-
пературы 210—215° без подогрева аппарата и содержание в отработан-
ном газе около 40% NaO и около 25% NH3.
Отработанный газ из азидного аппарата через пылеотделитель и
водоуловитель поступает в три последовательно соединенных металли-
ческих бачка с водой, где происходит поглощение аммиака, и в склянку
с H2SO4 (1 : 5). Отсюда освобожденная от аммиака отработанная за-
кись азота через предохранительные и сушильные колонки поступает
в резиновый газгольдер. Отработанная закись азота содержит в сред-
нем 35—40% NaO.
В процессе азидирования надлежит наряду с анализами поступаю-
щей и отработанной закиси азота внимательно наблюдать за тем,
чтобы в азидном аппарате давление было не выше атмосферного.
Конец азидирования определяют по содержанию в отработанном
газе не более 2—3% аммиака (анализ), по падению температуры
аппарата (пирометр) и по прекращению разогрева склянки с серной
Кислотой.
На скорость азидирования и выход продукта влияют следующие
факторы:
1. Интенсивность перемешивания. Ее необхо-
димо непрерывно увеличивать от 60 до 200—300 об/мин. во избежание .
образования твердого и неплавкого NaN3, могущего забить аппарат
комками NaNH2, окруженными коркой из NaN3.
2. Чистота поступающей на азидирование
закиси азота. Большое количество примесей, особенно кисло-
рода и окислов азота, недопустимо. Содержание N2O в закиси азота I
в начале и конце процесса должно быть не менее 70%.
3. Скорость пропускания закисиазот а. В каж-
дом отдельном случае для каждого отдельного аппарата наилучшая
скорость определяется опытом.
4. Температура азидирования- При температуре
170° реакция идет слишком медленно, а при температуре 250° ско-
рость ее настолько велика, что начинают появляться местные очаги!
разложения азида натрия и вспышки. Последнее надо иметь в виду I
всегда и ни в коем случае не допускать повышения температуры. В слу-
чае вспышки или повышения температуры до 250° необходимо немед-
ленно выключить подачу закиси азота и выждать понижения темпера-
туры до нормы.
212
Продолжительность азидирования колеблется в пределах от 25 до
37 час.; при правильно поставленной работе нормальное время азиди-
рования 27—30 час.
Как видно из реакций образования азида натрия:
NaNH2 -}- NqO —> NaN3 -J- HZO
NaNHg-K H2O -> NaOH + NH3
гсоретически возможным выходом, рассчитанным на взятый NaNH2,
является выход в 50%. Однако и этот выход обычно уменьшается за
счет недостаточной чистоты закиси азота. Употребление для реакции
чистой сжиженной закиси азота сокращает время процесса и увели-
чивает выход.
По окончании реакции азидирования выщелачивают образовав-
шиеся продукты: азид натрия и гидроксид натрия. Для этого прекра-
щают подачу закиси азота, аппарат охлаждают до 100—120°, откры-
вают краны, соединяющие его с холодильниками, и при малом числе
оборотов -мешалки в аппарат начинают постепенно подавать воду
через соответствующий штуцер. Эта операция требует особой осторож-
ности, так как в случае неправильного или недостаточного переме-
шивания в процессе азидирования могут остаться частицы NaNH2,
еще не прореагировавшие с N2O. Большое количество воды, бурно
реагируя с амидом натрия, может вызвать значительное повышение
давления в аппарате и его разрыв. В случае весьма бурного выделе-
ния аммиака следует временно прекратить пуск воды и возобновить
его только спустя некоторое время, лучше после совершенного прекра-
щения выделения аммиака.
Полученный в аппарате раствор азида и гидроксида натрия через
расположенный внизу трубопровод спускают в бак-отстойник.
Остаток продуктов реакции в азидном аппарате обрабатывают
в два приема таким же количеством воды и полученный раствор заса-
сывают в специальный сборник, где он сохраняется для использования
при выщелачивании следующей партии.
Поступивший в бак-отстойник раствор направляют на нутч-фильтр,
где он фильтруется от механических примесей и гидратов железа
через два слоя фланели с проложенным между ними листом фильтро-
вальной бумаги.
Отфильтрованный раствор, обычно содержащий:
азида натрия 15—16%
едкого иатра . - - 14—15%
карбоната натрия ... 3— 4%
подают в вакуумвыпариватель, представляющий собой цилиндриче-
ский стальной сосуд емкостью около 250 л, окруженный паровой ру-
башкой и снабженный мешалкой. Раствор упаривают под вакуумом
(500—700 мм рт. ст. до плотности 1,57, соответствующей примерно
50%-ной концентрации NaOH, при которой растворимость азида
натрия является минимальной и при которой он частично выпадает
в осадок.
213
По достижении необходимой плотности упариваемого раствора
подачу пара прекращают и дают водяное охлаждение. При этом пу-
скают в ход мешалку. Азид натрия выпадает из раствора, после чего
маточный раствор вместе с осажденным азидом натрия спускают на
нутч-фильтр. Фильтрующим материалом служит фланель; вакуум
при фильтровании и отсосе от 200 до 600 мм рт. ст. На нутч-фильтре
азид натрия промывают сначала дистиллированной водой, затем спир-
том. Промытый и отсосанный азид натрия сушат в сушильных камерах
на лотках по 2—3 кг при температуре 90—100°.
Высушенный азид натрия хранят в жестяных оцинкованных бан-
ках по 10 кг. Размер партии 100—400 кг.
Получающийся азид натрия имеет вид белых, достаточно крупных,
кристаллов и их агрегатов. Содержание NaN3 и примесей обычно ко-
леблется в следующих пределах:
азида натрия . . . 92—97%
гидроксида натрия.... 1%
карбоната натрия........... 3— 6%
нерастворимых в воде примесей... 0,01—0,3%
влажности . . ........ 1— 1,5%
Содержание примесей в азиде натрия предопределяет количество
и качество примесей в азиде свинца. Для полного анализа азида нат-
рия определяют содержание в нем едкого натра, карбоната натрия,
бикарбоната натрия и нерастворимых в воде примесей.
Определение нераствор им ы х примесей. Точ-
ную навеску около 2 г азида натрия растворяют в мерной колбе в
200 мл воды. Раствор фильтруют через высушенный при 100° и взвешен-
ный фильтр. Осадок промывают водой, ставят в термостат при 100°
и сушат до постоянного веса. Привес фильтра дает содержание нерас-
творимых в воде примесей.
Формула расчета:
р 100
процент нерастворимых = ——.
где р — привес фильтра;
а — навеска азида натрия.
Определение содержания едкого натра.
К 25 мл фильтрата, полученного при определении содержания нерас-
творимых примесей, приливают 10%-ный водный раствор хлористого
бария до тех пор, пока следующая капля его не вызовет нового осадка.
При этом карбонат натрия переходит в осадок в виде углекислого
бария, а в растворе вместо него появляется хлористый натрий. Затем
к раствору прибавляют 2—3 капли фенолфталеина и оттитровывают
0,1N раствором азотиой кислоты, отсчитывают количество милли-
литров кислоты, пошедшей на титрование, и вычисляют содержание
едкого натра по формуле:
% NaOH = 507,9 Thn°3 ’ " .
где Т hno3— титр азотной кислоты;
b — число миллилитров азотной кислоты, пошедшей на тит-
рование;
а — навеска азида натрия;
„ Mn-oh -200-100
507,9 = -----5=- -
MHNOj ‘ 25
Определение содержания карбоната н а т-
р и я. 25 мл фильтрата, полученного при определении содержания
нерастворимых примесей, оттитровывают 0, UV раствором азотной
кислоты с фенолфталеином. В результате титрования происходит
реакция:
Na2CO3 + HNO3 -> NaHCO3 + NaNO3
NaOH + HNO3-> NaNO3 4- H2O.
Отсчитывают число миллилитров азотной кислоты, пошедшей на тит-
рование, и отнимают от него число миллилитров азотной кислоты,
пошедшей в предыдущем анализе на нейтрализацию едкого натра.
Разность показывает расход азотной кислоты на превращение карбо-
ната натрия в бикарбонат натрия. Расчет производится по формуле:
% Na2CO8 = 1346 TlW±'b ,
где 7hno2 — титр азотной кислоты;
b — число миллилитров азотной кислоты, пошедшей на
превращение карбоната натрия в бикарбонат;
а — навеска азида натрия:
1346 =
AWo,-200- 100
A^HNOs ‘ 25
Определение содержания азида натрия.
Нейтрализованный при определении карбоната натрия раствор азида
натрия оттитровывают 0, \N раствором нитрата серебра в присутствии
нискольких капель хромовокислого калия в качестве индикатора:
NaN3 + AgNO3 -> AgN3 + NaNO3.
По мере того как азидогруппа будет полностью осаждена в виде азида
серебра, последующая капля нитрата серебра окрасит жидкость в не-
исчезающий красноватый цвет, который останется и при сильном
взбалтывании. Появление неисчейающего красноватого окрашивания
и считают концом анализа.
Расчетная формула:
% NaN3 = 306,2 ,
где TAgNOs — титр азотнокислого серебра;
215
b — число миллилитров азотнокислого серебра, пошедшего
на титрование;
а — навеска азида натрия;
;Ж2=^-2ОО.-’«’
MAgNOs ‘ 25
Следует помнить, что азид серебра—в сухом состоянии очень чув-
ствительное инициирующее ВВ. Поэтому сразу же после анализа содер-
жимое реакционного сосуда необходимо переносить в разбавленную
азотную кислоту для разложения азида серебра (в вытяжном шкафу).
Определение содержания бикарбоната
натрия- Это определение производится только в случае отсут-
ствия едкого натра.
К 25 мл фильтрата, полученного при определении нерастворимых
примесей, приливают несколько миллилитров титрованного 0, UV
раствора едкого натра, взбалтывают и оставляют в покое около
30 мин.:
NaHCO3 + NaOH -> Na2CO3 + H2O.
Затем осаждают описанным выше порядком карбонат натрия
10%-ным раствором хлористого бария:
Na^COg + ВаС12 -> ВаСО3 + 2NaCJ.
После этого прибавляют 2—3 капли фенолфталеина и оттитровывают
избыток щелочи 0,1N раствором азотной кислоты. Отсчитывают
число миллилитров азотной кислоты, пошедшей на титрование, и пере-
считывают их на число миллилитров прибавленного титрованного
раствора едкого натра по формуле:
NaOH^ = 0.635 ,
1 NaOH
где с — число миллилитров азотной кислоты, пошедшей на нейтра-
лизацию;
Тhno3— соответственно титры азотной кислоты и едкого натра;
0,635 = .
MHNO3
Из первоначально прибавленного количества 0,17V раствора едкого
натра вычитают количество едкого натра, нейтрализованного азотной
кислотой, и по разности d рассчитывают процентное содержание
^карбоната натрия, пользуясь формулой:
% NaHCO3 = ,
где а — навеска азида натрия;
1680= ‘200 “100
MNaOH ‘ 25
216
Очистка и приготовление 10%-н ого рас-
твора азида натрия. Азид натрия растворяют в фарфоровом
баке при комнатной температуре и при энергичном размешивании
мешалкой из луженого железа (и = 50—60).
В бак загружают технический азид натрия и дистиллированную
воду, исходя из расчета получения 10%-ного раствора. Расчет по-
гребного количества азида натрия производят по формуле
1000- ь
1де р —навеска технического азида, необходимого для образования
10%-лого раствора;
b — число литров приготовленного раствора;
а — процентное содержание NaN3 в техническом азиде натрия.
Различные методы очистки NaN3 влияют на выход и чистоту азида
свинца, особенно, если в техническом азиде натрия содержится зна-
чительное количество примесей.
Существующие методы очистки азида натрия можно разделить
на три вида;
1) кислотный метод очистки;
2) бескислотный метод очистки;
3) смешанный метод очистки.
Наиболее старый, рекомендованный герм. пат. № 224669, кислот-
ный метод очистки состоит в нейтрализации щелочности раствора
(от NaOH и Na2CO3) 7—8%-ной азотной кислотой, которую прили-
вают тонкой струей в размешиваемый раствор до отсутствия реакции
на щелочь, с применением в качестве индикатора фенолфталеина.
Под действием азотной кислоты NaOH переходит в NaNO3, a NasCO3
в NaHCOs. Затем добавляют избыток азотной кислоты до полной ней-
трализации, так как, оставляя в растворе NaHCOs, мы рискуем при
действии Pb(NOs), получить вместе с азидом свинца осадок бикарбо-
ната свинца, который уменьшит воспламеняемость и инициирующую
способность азида свинца. Вследствие того, что NaN3 есть соль силь-
ного основания и недостаточно сильной кислоты, при добавлении
к раствору NaN3 (технического или химически чистого) азотной
кислоты происходит частичное разложение азида натрия с образо-
ванием NaNO3 и HN3, значительный запах которой всегда ощу-
щается при этом методе очистки.
Таким образом кислотный метод всегда ведет к понижению выхода
азида свинпа (разложение части NaN3) и его качества (содержание
карбонатов). Кроме того, нежелательным является выделение азо-
тистоводородной кислоты.
Описание бескислотного способа очистки азида натрия изложено
в общих чертах в герм. пат. № 513933, по которому можно получить
чистый, легко воспламеняемый азид свинца, если водный раствор
азида натрия предварительно обработать раствором каких-либо солей
щелочноземельных металлов (предпочтительнее ацетат или нитрат
бария или кальция), ведя обработку до тех пор, пока отфильтрован-
217
ная проба при дальнейшем прибавлении осаждающего вещества не
перестанет выделять осадок. Этим способом все щелочные карбонаты
осаждаются в виде нерастворимых щелочноземельных карбонатов]
В целях повышения содержания азида свинца в готовом про
дукте и повышения его чувствительности изложенный выше кислот-
ный метод был заменен на некоторых .заводах бескислотным методом,
Вместо нейтрализации щелочи в азиде натрия стали обрабатывать
водный раствор азида натрия рассчитанным количеством ацетата
бария или нитрата бария или кальция-
Существует несколько вариантов способа бескислотной очистки!
раствора технического азида натрия, но следующие два из них наи-
более часто применяются.
По растворении навески технического азида натрия вне завися-1
мости от первоначального анализа отбирают пробу раствора для опре-
деления содержания в нем NaOH. В зависимости от содержания
NaOH приливают двойное количество бикарбоната натрия, потребного
для перевода NaOH в Na2C03.
Раствор бикарбоната приготовляют в отдельном сосуде и направ-
ляют в бак-растворитель азида натрия- После тщательного переме-
шивания раствору азида натрия дают отстояться 30—40 мин. и берут
вновь пробу на содержание NaOH. При получении отрицательного
результата на NaOH и после определения процентного содержания
в растворе карбоната натрия приливают рассчитанное количество
40%-ного раствора нитрата кальция.
Растворимость карбоната кальция в воде очень мала и характери-
зуется следующими цифрами:
при 25° 1,445 • 10-3%
» 50° 1,515 10-3%
» 75°.........1,816 • 10 %
Вследствие такой невысокой растворимости карбоната кальция
достигается почти полное освобождение раствора от углекислых солей.
Проверка полноты очистки азида натрия осуществляется добавлением
к фильтрованной пробе раствора небольшого количества раствора
Ca(NO3)2. При этом не должен появиться осадок.
Очищенный от карбонатов раствор фильтруется через бязевый
фильтр на нутч-фильтре и поступает на осаждение азида свинца.
При применении этого способа очистки увеличивается содержа-
ние азида свинца в готовом продукте с 86—90 до 97—98%, что, ко-
нечно, сказывается на чувствительности к начальному импульсу и
в первую очередь к удару и трению.
Другой бескислотный метод сводится к применению вместо нитрата
кальция нитрата бария. В результате обработки раствора технического
азида натрия концентрированным раствором нитрата бария выпадает
карбонат бария, растворимость которого также ничтожна и характе-
ризуется следующими цифрами:
при 16° 1,65-10-3 %
» 25° . 3.5 • IO"3 %
лишь несколько превышающими растворимость карбоната кальция.
218
Удобнее, дешевле и рациональнее применять для очистки не
Ba(NO8)2, a Ca(NO8)2, во-первых, потому, что растворимость нитрата
кальция значительно больше растворимости нитрата бария, благо-
даря чему в раствор вводится меньшее количество воды:
Растворимость Ba(NOs)2 и Ca(N03)3
Ba(NO,), Ca(NOs)2
/° % 1° °/ /о
0 4,8 0 48,2
20 8,1 18 54,8
40 12.4 42,7 69,5
Растворимость Ва(0Н)2 и Са(0Н)2
Г Ва(О.Н)2 Са(ОН)г
0 1,5% 0,131 %
20 3,84% 0,126%
50 11.75% 0,098%
80 90.8% 0,060%
этом приходится вводить двойной избыток
Во-вторых, потому, что расход Ca(NO3)2 меньше, вследствие меньшего
его молекулярного веса.
Растворимость гидроксидов бария и кальция в воде показывает
приведенная ниже таб-
лица.
Отсюда явствует, что
без особого ущерба для
чистоты продукта можно
опустить предваритель-
ную обработку раствора
технического азида нат-
рия раствором бикарбо-
ната при применении
Са(ЬЮ8)2. Кроме того,
следует учесть, что при r__________ „
бикарбоната натрия и заведомо образовать углекислые соли свинца.
Что касается способа осаждения карбонатов нитратом бария, то
в этом случае полностью осадить NaOH в виде Ва(ОН)2 не удается,
так как вся имеющаяся в техническом азиде натрия щелочь остается
в растворе.
Для устранения последнего недостатка и применяется так назы-
ваемый смешанный способ очистки, который состоит в том, что по осаж-
дении нитратом бария карбонатов и отфильтровании последних
в раствор вводят рассчитанное количество азотной кислоты для нейтра-
лизации NaOH иЬ1аНСО8. Азотную кислоту вводят прямо в мерники.
Приготовление 15% - него раствора нитрата
свинца производят в аналогичном баке-растворителе (но ни в коем
случае не в том же). Мешалка—алюминиевая. Полученный раствор
фильтруют через фланелевый фильтр.
Полученные и отфильтрованные растворы NaN3 и Pb(NO3)2 при
помощи вакуума засасывают в стеклянные мерники и уже оттуда
направляют для осаждения азида свинца.
219
Фиг. 01. Нутч-фильтр.
Основные меры предосторожности во время приготовления раство I
ров следующие: чистота помещения и баков-растворителей, хорошем
проветривание помещения от выделяющейся при нейтрализации азо
тистоводородной кислоты, обязательное переливание растворов в мер
ники через разные трубки во избежание образования азида свинца
и периодическая промывка слабой азотной кислотой и водой все,1
аппаратуры.
Осаждение азида свинца производится в баке-oca J
дителе. Последний представляет собой железный каркас, освинцо-1
ванный со всех сторон; он снабжен деревянной мешалкой и имеет
отверстие в дне для спуска маточного рас-
твора и готового продукта.
В бак-осадитель из мерника самотеком
сливают 15%-ный раствор нитрата свинца,
пускают в ход мешалку (п= 40) и из другого
мерника тонкой струей спускают раствор
азида натрия. Отношение исходных реагентов
Pb(NO3)2 и NaN33: 1. т. е. нитрата свинца
берут на 18% больше теоретически необходи-
мого количества. Температура осаждения
18—22°. Реакция образования азида свинца
происходит мгновенно. Время слива раствора
азида натрия обычно равно 5 мин.
После слива азида натрия образовавше-
муся азиду свинца дают выдержку в маточном растворе 10—15мин.
при неработающей мешалке.
При нормальном процессе осаждения (концентрация, температура,
характер перемешивания и время выдержки) получается мелкокри-
сталлический азид свинца. Размеры кристаллов неразличимы на-глаз
и трудно поддаются измерению под микроскопом. Понижение кон-
центраций влечет за собой образование кристаллов более крупного
размера, а увеличение времени выдержки и температуры наряду
с этим приводит к образованию (в результате все время продолжаю-
щегося растворения и перекристаллизации) больших кристаллов
игольчатой формы, склонных к самопроизвольным взрывам.
Так как операция осаждения азида свинца является взрывоопас-
ной, то она производится в хорошо изолированной кабинке в отсут-
ствии людей. Спуск растворов из мерников, пуск и остановка мешалки,
наблюдение за работой мешалки при помощи зеркал и выпуск готового
продукта с маточным раствором производятся извне кабины. До
и после каждой операции осаждения необходимо тщательно про-
верять исправность аппаратуры и строго соблюдать чистоту
помещения.
Фильтрация, промывка и флегматизация
азида свинца производятся на нутч-фильтре, расположенном
также в изолированной кабине. Нутч-фильтр (фиг. 91) изготовляется
из фарфора или представляет собой, как и бак-осадитель, освинцо-
ванный со всех сторон железный каркас, разделенный фильтрующей
220
перегородкой на две части. Лучше пользоваться более прочным же-
ненным освинцованным нутч-фильтром.
Из соседней кабины осаждения от бака-осадителя к нутч-фильтру
протягивают резиновый шланг, который во время осаждения зажи-
мают у выходного отверстия специальным зажимом.
Фильтрующий материал обычно представляет собой кружок филь-
тровальной бумаги, располагаемый на поверхности нутч-фильтра,
«а гем кружок льняного полотна и, наконец, льняной мешок с несколь-
к ими прикрепленными к нему бечевками или тесемками.
После подготовки нутч-фильтра взмучивают мешалкой выдержан-
ный в баке-осадителе азид свинца и вместе с маточным раствором сли-
пают на нутч-фильтр. Маточный раствор отсасывают при вакууме
около 600 мм, азид свинца промывают последовательно водой, спир-
том и бензолом при выключенном вакуумнасосе и при обязательном
управлении заливкой извне кабины-
По мере наполнения нижней части нутч-фильтра маточным раство-
ром и промывными жидкостями последние спускают из него на другой
нутч-фильтр, на котором улавливают случайно прошедшие через пер-
вый фильтр кристаллы азида свинца. Затем спускают промывные
жидкости с маточным раствором в канализацию.
Промытый азид свинца флегматизируют. Поверх лепешки отсосан-
ного азида свинца наливают 7,5%-ный раствор парафина в бензоле,
подогретый до 30—35°. Некоторые заводы применяют раствор пара-
фина в авиационном бензине, но тогда последняя промывка должна
производиться не бензолом, а бензином.
Лучше, конечно, пользоваться при промывке бензолом, так как
с бензином вода и спирт не смешиваются и, следовательно, при помощи
бензина невозможно окончательно удалить воду и спирт.
Залитый раствор флегматизатора через 5 мин- отсасывают при ва-
кууме около 300 мм рт. ст. в течение 1 мин. Для достижения одно-
родной флегматизации большое значение имеют концентрация и тем-
пература раствора флегматизатора, степень выдержки, время и вакуум
отсоса. Введение большого количества флегматизатора приводит
к уменьшению взрывчатых свойств азида. При недостаточном введе-
нии флегматизатора, наоборот, чувствительность выше нормы, и,
кроме того, получаются нестойкие грану ли, легко распадающиеся даже
от пересыпания, а это может вызвать несчастные случаи при изготов-
лении капсюлей.
Флегматизированный азид свинца извлекают из нутч-фильтра
вместе с фильтровальным мешком при помощи подъемного крана (при-
водимого в вертикальное и горизонтальное движение извне кабины),
переносят на рядом расположенный стол и выкладывают на алюминие-
вый, лакированный со всех сторон, лоток.
Таким образом обслуживающий персонал посещает кабину во
время нахождения в ней азида свинца только два раза: первый — для
того, чтобы надеть тесемки фильтровального мешка на крюк подъем-
ного крана, и второй—для снятия освобожденного мешка-фильтра с
крючка и передачи готового азида свинца на раскладку для грануляции.
221
Промывка и флегматизация азида свинца — операции взрыво-|
опасные, а потому требуют тех же мер предосторожности, что и при
осаждении. Особое внимание следует обратить иа отходы азида свинца)
Своевременное улавливание отходов, сбор и ликвидация их подрывом
или разложением, должны находиться под непосредственным контря
лем начальника мастерской. С дополнительного нутч-фильтра необ-
ходимо регулярно снимать и подрывать отходы; периодически следует
очищать ловушки. Ни в коем случае нельзя допускать накопления
азида свинца на дне нижней части нутч-фильтра. Всю систему (бак-
осадитель, спускные шланги, нутч-фильтры и систему ловушек) необ-
ходимо промывать не реже одного раза в шестидневку разбавленной
азотной кислотой для разложения оседающего в разных местах азида
свинца.
Применение в практике всякого рода отстойников в виде распо-
ложенных возле здания мастерской колодцев недопустимо, так как
опыт показывает, что, несмотря на периодическую обработку отхо-1
дов в этих колодцах азотной кислотой, разложение азида свинца |
идет неравномерно и остающаяся масса представляет собой неодно-
родную смесь азида свинца и флегматизатора с содержанием в неко-
торых участках до 90% азида свинца. Чистка таких колодцев может
приводить к несчастным случаям.
Фильтрующий материал (льняные мешки) необходимо очищать
после каждой операции.
Пребывание в кабинах осаждения и флегматизации (кроме огово-
ренных здесь случаев) недопустимо; наблюдение должно произво-
диться только посредством зеркал.
Грануляция азида свинца, т. е. превращение’ плохо
сыпучего азида свинца в однородные сыпучие гранули (зерна), имеет
целью сделать продукт удобным для снаряжения капсюлей.
Перед грануляцией азид свинца раскладывают на порции, для
чего лоток с флегматизированным продуктом устанавливают наклонно
в специальной кабине и из-за стены, наблюдая через глазок из кора-
бельного стекла, при помощи специального приспособления, на-глаз
ссыпают необходимое количество азида свинца (около 250 г). Назна-
ченные порции переносят в кабину грануляции. Операция состоит
в протирке флегматизированного азида свинца через шелковое си-
то № 28.
Очень часто бывает, что азид свинца после отсоса флегма тизирую-
щего раствора слишком сух для грануляции, в результате чего гра-
нули могут получиться слишком мелкими и во время сортировки
могут дать повышенный процент пыли. Вследствие этого азид свинца
перед грануляцией часто смачивают бензолом.
Грануляция производится на деревянном протирочном станке
(с натянутой шелковой сеткой), укрепленном в специальном столе,
обитом по войлоку черной или красной глянцевой клеенкой.
Протирка через сито производится из-за капитальной стены при
помощи резиновой пластины, укрепленной на деревянной рукоятке,
пропущенной через имеющуюся в стене прорезь. Протертые гранули
222
падают на лоток из плотной глянцевой бумаги, установленный внизу
сгинка под шелковой сеткой.
В процессе грануляции в кабине не должно быть никого. По окон-
чании работы лоток с гранулированным продуктом относят на сушку,
и аппаратуру и инструмент тщательно протирают губкой, смоченной
бензолом или бензином.
Сушка азида свинца. Гранулированный азид свинца
переносят на бумажных листах в камерную сушилку, изолированную
от прочих кабин мастерской, или, еще лучше, в соседнее здание,
соединенное с мастерской коленчатым коридором. В сушилке распола-
гают деревянные стеллажи с мягкой обивкой для установки на них лот-
ков. Азид свинца сушат при температуре 40° в течение 17—18 час.
Температуру контролируют термографом.
Сушка в камерных Сушилках, вообще говоря, мало производи-
гельна; требуются большие рабочие площади. Основные мероприятия
но технике безопасности: чистота помещения, отсутствие пыли азида
свинца, соблюдение температурного режима сушки и отсутствие пере-
грузки.
В последнее время применяют и метод сушки в пассбургах (одно-
временно до 3 кг азида свинца), аналогично гремучей ртути. Следует
отметить, что сушка в пассбурге производительнее сушки в камерных
сушилках, но при этом следует учесть, что взрыв гремучей ртути не
разрушает пассбурга, а взрыв азида свинца его разрушает. Это про-
исходит потому, что в вакууме гремучая ртуть при воспламенении
только вспыхивает, а азид свинца взрывает так же, как иа воздухе.
Лотки с высушенным азидом свинца переносят из кабины сушилки
в отдельную кабину, где азид свинца пересыпают в пайковые коробки.
Сортировка азнда с в и и ц а.Флегматизированный и вы-
сушенный азид свинца содержит довольно большое количество пылн
и крупных склеившихся друг с другом грануль. Применение несорти-
рованного продукта недопустимо, во-первых, потому, что его не-
удобно дозировать насыпными приборами и колебания в навесках
выходят за допускаемые по стандарту пределы; во-вторых, потому,
что гранулированный азид свинца в зависимости от размеров грануль
очень часто содержит различное количество парафина.
Сортировка производится на механических сортировочных аппара-
тах, описанных в главе о гремучей ртути. Порядок работы тот же.
Верхнее сито имеет 7 отверстий на линейный сантиметр, нижнее — 25
отверстий на линейный сантиметр.
Сортировочный аппарат располагают изолированно в железо-
бетонной кабине и приводят в движение извне кабины.
Пыль и шишка, отделяющиеся в процессе сортировки, замачивают
в спирте и направляют на переработку.
Непрерывный метод производства азида
свинца. Описанный выше общепринятый прерывный метод произ-
водства азида свинца разрабатывался в течение ряда лет. Техника
безопасности постепенно улучшалась, но все же возможность аварий
в производстве не устранена, главным образом, вследствие примене-
223
Фиг, 92. Колон-
ка для осажде-
ния азида
свинца.
пня ручного груда. Попытками введения непрерывного и механизи-
рованного процесса производства кроме достижения безопасностиI
производства преследуют также цель — увеличить производительное®
труда. Однако в настоящее время еще не разработан такой непрерыв-1
ный метод, который охватывал бы весь процесс от осаждения д-
сушки.
Предложение о непрерывном осаждении было!
сделано Акционерным обществом Лигноза в Берлине!
(герм. пат. № 440568, 15 сентября 1921 г.). Сущность
его состоит в том, что эквивалентные количества
растворов азида натрия и ацетата свинца спускают из
запасных сосудов в воронку, откуда они при помощи I
поставленной под углом к горизонтали трубки подво-
дятся к фиЛьтру". При этом требуется установить!
такие концентрации жидкостей и такую скорость их |
истечения, чтобы все время приходили в соприкосно-!
вение эквивалентные количества.
Однако этот метод неприемлем вследствие недоста-1
точного перемешивания растворов.
Другим, уже осуществленным в заводском мас-
штабе, является непрерывный метод осаждения азида 1
свинца, предложенный Мейснером. Он заключается
в том, что исходные продукты пропускают через
аппарат в строго определенных количествах, а про-
дукт их реакции непрерывно удаляют. Аппарат пред-
ставляет собой более или менее высокую колонку ]
(фиг. 92), через верхнюю часть которой проходит
мешалка. Автоматически отмериваемые количества
растворов поступают сверху, а образующийся азид
свинца спускается вниз. Приток растворов и удале-
ние продуктов реакции регулируют таким образом,
чтобы реакционная жидкость вместе с маточным раствором оставалась
в колонке в течение времени, достаточного для того,чтобы кристаллы
азида свинца могли вырасти до определенной, необходимой по тех-
ническим условиям, величины. Полученный продукт вместе с частью
маточного раствора выливается через специальный прерыватель на
промывной фильтр. Главная масса маточного раствора остается в ко-
лонке и через сифонную трубку, служащую одновременно регулято-
ром уровня Жидкости, вытекает наружу с верхней части колонки.
Выход азвда свинца — 97%.
Штеттбахер считает, что так как маточный раствор и кристаллы
при пользовании этим методом удаляются из колонки непрерывно,
то всякая ручная работа и транспорт становятся излишними. Без-
опасность в значительной степени увеличивается еще и благодаря тому,
что в каждый данный момент в аппарате находится небольшое количе-
ство азида свинца (около 300 г). Выделяющийся азид свинца поступает
на один 143 нутч-фильтров, где он при помощи вакуума отсасывается
от маточного раствора и промывается. В то время как один фильтр
224
медленно наполняется, другой, наполненный 1—2 кг азида свинца,
переносят в сушильню или в погреб. Штеттбахер рекомендует этот
метод также потому, что при пользовании им степень чистоты азвда
свинца достигает 98%, в то время как при пользовании старыми
методами выход чистого продукта равен 91—95%.
§ 8. ТЕХНИЧЕСКИЕ ТРЕБОВАНИЯ К АЗИДУ СВИНЦА
Нефлегматизированный азид свинца должен представлять собой
кристаллический порошок без видимых на-глаз посторонних приме-
сей. Цвет его может меняться от белого до светлорозового или
сероватого. Флегматизированный парафином азид свинца должен
представлять собий однородные сыпучие и несклеивающиеся гранули.
Различают два сорта азида свинца, для каждого из которых дей-
ствующими ОСТ устанавливается максимально допустимое содержа-
ние примесей (влаги, нерастворимых в кислоте, флегматизатора),
и минимально допустимое содержание Pb(N3)2.
1. Определение влажности и летучих ве-
ществ. Навеску около 3—4 г азида свинца помещают в стаканчик
для взвешивания и нагревают при 60° примерно в течение 4 час.
до постоянного веса. Затем стаканчик помещают на 15—20 мин. в
эксикатор над хлористым кальцием для охлаждения- Убыль в весе
показывает содержание влаги и летучих.
2. Определение нерастворимого остатка. На-
веску азида свинца около 3 г растворяют в стакане в 120 мл 25%-ной
азотной кислоты. Раствор должен остаться прозрачным и не содержать
остатка или мути. Если обнаружен остаток, то его рассматривают
под микроскопом. Присутствие минеральных частиц совершенно не
допускается.
3. Содержание свинца. Если азид свинца свободен от
посторонних металлов, то для определения содержания свинца обра-
батывают навеску его серной кислотой в большом платиновом тигле
я выпаривают-досуха. Свинец остается в виде сульфата.
Практически эта операция проводится несколько иначе.
Навеску около 0,5 г высушенного азвда обрабатывают в конической
колбочке разведенной азотной кислотой при слабом нагревании.
Затем добавляют 4—5 мл крепкой серной кйслоты и нагревают на
песчаной бане до появления паров серной кислоты.
По охлаждении переносят содержимое колбочки в стакан с дистил-
лированной водой, туда же тщательно смывают колбочку. К жидкости
прибавляют равный объем ректификованного спирта и оставляют
отстояться в течение ночи. По отстаивании сливают жидкость через
фильтр, вес золы которого известен, осадок промывают несколько раз
спиртом посредством декантации, затем переносят на фильтр и окон-
чательно промывают спиртом. Фильтр с осадком подсушивают и оса-
док переносят на черную или красную глянцевитую бумагу, а фильтр
сжигают на платиновой проволоке над предварительно прокаленным
взвешенным фарфоровым тиглем. Золу фильтра смачивают азотной
Бубнов—92—15 225
кислотой, выпаривают, затем смачивают 1—2 каплями серной кислоты
и тоже выпаривают, после чего слабо прокаливают и только после
этого переносят в тигель осадок с глянцевитой бумаги и слабо прока-
ливают его до постоянного веса. По полученному сернокислому свинцу
вычисляют процентное содержание свинца в азиде свинца по формуле:
v 0,9632 • а
где X — процентное содержание свинца в азиде свинца;
а — вес полученного сульфата свинца;
р — навеска азида свинца:
0,9632 — отношение молекулярных весов азида свинца и сульфата
свинца.
Содержание свинца можно установить также и электролитическим
путем.
Навеску азида свинца около 0,3 г растворяют в слабой азотной кис-
лоте (3 мл HNO3 в 40° Be и 15 см'3 воды) в фарфоровой чашке и
выпаривают на водяной бане досуха. Сухой нитрат свинца смывают
15%-ным раствором азотной кислоты в платиновую чашку, которую
доливают 15%-ным раствором азотной кислоты до 120 мл. Раствор под-
вергают электролизу, причем катодом служит обычная платиновая
спираль. Сила тока 0,5—1,0 А, разность потенциалов 12 V.
Перед началом электролиза раствор подогревают до 60—65°.
Через Р/2—2 часа проверяют на полноту осаждения и затем под током
содержимое чашки промывают водой и спиртом, после чего чашку
сушат в течение 30 мин. при 100° в шкафу, охлаждают в эксикаторе
и взвешивают. Количество свинца определяют по формуле:
Х= 8'5-62 'и
где X — процент содержания свинца;
Н — разность веса платиновой чашки пустой и с PbOs;
с — навеска азида свинца.
4. Определение азота. Существующие методы анализа
азида на азот можно разделить на две группы:
а) определение азота методами объемного анализа:
б) определение азота газометрическим путем.
Методы объемного анализа. Классическим методом
анализа свинца до последнего времени был метод, предложенный Кур-
циусом и Риссом. Он основан на расщеплении азида разбавленной
серной кислотой при кипячении по реакции:
Pb(N3)2 + HaSO4-> PbSOj + 2HN3
Азотистоводородную кислоту ОТГОНЯЮ!’ с водяным паром и погло-
щают титрованным раствором щелочи:
2HN3 + Ва(ОН)2 ->Ba(N3)2 + 2Н2О.
Избыток щелочи оттитровываюг.
226
Навеску около 1 г тщательно высушенного азида свинца поме-
щают в круглодонную полулитровую колбу с газоотводной трубкой.
В шейку колбы вставляют пробку с капельной воронкой. Газоотвод-
ную трубку при помощи каучуковой пробки вставляют в длинный
холодильник Либиха, форштос которого входит в коническую колбу,
«крытую пробкой с двумя отверстиями.
В коническую колбу наливают 0,11V раствор едкого бария с избытком
и 20%. Предварительно через всю систему, начиная с капельной во-
ронки, пропускают воздух, лишенный углекислого газа, в течение
I часа.
В круглодонную колбу приливают (принимая при этом меры пре-
досторожности против попадания в систему углекислого газа) 50 мл
свежепрокипяченной воды и затем медленно из капельной воронки —
300 мл слабого раствора серной кислоты (кислоты берут на 10% больше,
чем требуется по теории). Затем закрывают кран капельной воронки
н нагревают круглодонную колбу в ванне, наполненной насыщенным
раствором хлористого кальция- Перегонку заканчивают, когда в колбе
остается около 50 мл жидкости. После охлаждения жидкость в колбе
не должна давать кроваво-красного окрашивания с хлорным желе-
зом.
Избыток едкого бария титруют 0,1 N раствором азотной кислоты.
Количество азота определяют по формуле:
о/ N - (A-£/QT-42- 100
/о 85,69 р ’
где А — количество миллилитров 0,11V раствора едкого бария, на-
литого в коническую колбу;
Т титр едкого бария;
g—отношение титра азотной кислоты к титру едкого бария;
й — количество миллилитров 0,11V раствора азотной кислоты,
пошедшей на обратное титрование;
р — навеска азида свинца.
Однако при определении азота этим методом получаются неточные,
всегда пониженные, результаты, так как азид свинца при этом расщеп-
ляется очень медленно, а при длительном кипячении он разлагается
с отщеплением азота и образованием аммиачной соли.
Маркгейроль и Лоритти несколько улучшили этот метод, употре-
бив вместо серной кислоты уксусную. В качестве поглотителя HN3
они применяли раствор азотнокислого серебра, а выпавший азид
серебра после сушки взвешивали. Однако их метод является небез-
опасным вследствие большой чувствительности сухого азида серебра.
Рейнско-Вестфальская компания взрывчатых веществ предлагала
следующий способ, по которому навеску околи 0,5 г азида свинца
помещают в мерную колбу емкостью в 250 мл, прибавляют 150 мл
дистиллированной воды и 5 мл 2N азотной кислоты. Колбу закры-
вают резиновой пробкой и сильно взбалтывают для растворения азида
свинца. Затем приливают из бюретки точно 50 мл титрованного 0,11V
раствора AgN03. Для нейтрализации азотной кислоты прибавляют
227
еще 10 мл 2N ацетата натрия, доливают колбу до метки и оставля
стоять на ’2 часа в темном месте. Затем фильтруют через высушена
фильтр и в 100 мл фильтрата определяют’ избыток AgNO3. Пред]
рительно прибавляют в раствор несколько капель железоаммон»
ных квасцов и HNO3 до обесцвечивания появляющегося при это
окрашивания и оттитровывают 0,1N титрованным раствором родаш
стого аммония.
Определяют процент Pb(N3)2 но формуле:
%Pb(N3)2 =
Г/Т NHjCNS^ - 250 rNHiCNS MAgNOa
= V AgNO3 ’ ---- —-j(M) . M --
L eIUU MNH,CNS
A4pbN„• 100
2NAgN03 ’ P ’
Видоизменением метода Рейнско-Вестфальской компании служв
следующий: навеску азида свинца около 0,3 г помещают' в мерну
колбу емкостью 250 мл и растворяют в 10 мл 10N раствора уксуснс^
кислого аммония. По растворении азида свинца добавляют 100 мл
воды и затем 50 мл О, IN раствора нитрата серебра при встряхивании.|
Колбу доливают водой до метки и полученный азид серебра отстаи-!
вают в течение 60 мин. После этого фильтруют через фильтр в сухую
колбу. Берут 100 мл фильтрата и избыток нитрата серебра оттитро-
вывают О, IN раствором роданистого аммония с несколькими каплями
приготовленного, по Тредвеллу, азотнокислого раствора железоаммо-
нийных квасцов, употребляемых в качестве индикатора. Конец титро-
вания определяют по розовой окраске раствора. L
Расчет производят по формуле
%Pb (N3)2 = (50 Tabno. — 2,2323 2,5 . а Tnucns) —J°°,
где TAgNo3'—титр нитрата серебра;
TwHiCNs — титр роданистого аммония;
а— число миллилитров роданистого аммония, употреб-
ленного на обратное титрование;
р— навеска азида свинца;
9 23 9 3 молекулярный вес AgN03
** молекулярный вес NH4CNS
д 8571 молекулярный вес Pb(N8)2
’ 2 молекулярных веса Ag(NOs)j'
Газометрические методы. Для анализа азида свинца
наиболее подходящим является метод определения азота по способу
Зоммера и Пинкаса, основанному на окислительном действии нейтраль-
ного или слабокислого раствора церийаммонийнитрата. При наличии
в азиде хлоридов раствор должен быть, безусловно, нейтрален, ибо
в этом случае может выделяться наряду с азотом и хлор.
Окислительное действие церийаммонийнитрата сводится К разру-
шению молекулы азида свинца:
Pb (N3)2-»[Pb]'‘-f-3N2
228
Выделившийся азот собирают в измерительную трубку.
Анализ азида свинца производят в приборе, изображенном на
фиг. 93. В хорошо высушенный реакционный стаканчик прибора по-
мещают стеклянное ведерко с точ-
ной навеской сухого азида. Навеску
илида около 0,3 г.берут из расчета,
чюбы выделившийся после разло-
жения азот занимал объем не менее
70—80 мл (в этом случае ошибки
шгыта сведутся к минимуму). В тот
же стаканчик помещают навеску
церийаммонийнитрата из расчета
2 г цериевой соли на 0,1 г азида
свинца. В градуированную воронку
прибора наливают 3—4 воды.
Реакционный стаканчик соединяют
с измерительной частью прибора,
наполненной 50%-ным раствором
КОН, и открывают соединительный
кран. Из градуированной воронки
иоду спускают в реакционный ста-
канчик. При этом необходимо на-
блюдать за тем, чтобы ведерко с ази-
дом свинца не упало, прежде чем
закроется кран градуированной во-
ронки. Встряхивая прибор, опро-
кидывают ведерко. При этом азид Ф,1Г* 93’ ази а<^,ределения
свинца приходит в соприкоснове-
ние с раствором церийаммонийни-
трата. Реакция наступает мгновенно и продолжается около 2—3 мин.
Окисление идет по схеме
2HN3 + 2СеО2 -> 3N2 + Се2О3+Н2О
Азот выделяется количественно и собирается в газометрической
трубке. По истечении 1 часа, необходимого для установления посто-
янства температуры, производят отсчет объема, отмечают температуру
и атмосферное давление н делают обычный расчет.
Метод этот чрезвычайно прост, точен и не требует значительного
расхода времени.
5. Определение флегматизатора. Навеску в 3 г
азида свинца обрабатывают 100 мл петролейного эфира, дают отсто-
яться и фильтруют через двойной фильтр, промывают два раза по 25 мл
эфиром и выпаривают на водяной бане. Полученный осадок оконча-
тельно высушивают до постоянного веса в термостате при 100° в тече-
ние 2 час. По охлаждении в эксикаторе над хлористым кальцием
взвешивают.
ГЛАВА V
СТИФНАТ СВИНЦА (ТНРС)
§ 1. СТИФНИНОВАЯ КИСЛОТА И ЕЕ СВОЙСТВА
Стифниновая кислота впервые была получена в 1808 г. Шеврелем,
который, обрабатывая азотной кислотой фернамбуковый экстракт, |
получил вещество (по его описанию «комбинацию азотной кислоты
и маслянистого вещества»), обладающее взрывчатыми свойствами,
очень похожее на пикриновую кислоту, но несомненно не являющееся
пикриновой кислотой.
Затем в 1846 г. Боттгер и Виль, обрабатывая целый ряд различных
экстрактов и эфирных масел азотной кислотой (уд. веса от 1,2 до 1,5)
при нагревании, также получили вещество Шевреля и по аналогии
с пикриновой кислотой дали ему название «стифниновая кислота».
После исследований Боттгера и Виля были найдены и другие
экстракты и смолы, из которых нитрацией получается та же стифнино-
вая кислота. Что стифниновая кислота получается из резорцина, было
высказано впервые Шредером, сначала в виде предположения, так как
во всех экстрактах, эфирных маслах и смолах, нитрацией которых
можно получить стифниновую кислоту, был найден резорцин. Затем,
когда Штенхузу в 1871 г. удалось получить тринитрорезорцин из ре-
зорцина СсН.(ОН)>, Шредер доказал идентичность стифниновой кис-
лоты и тринитро резорцина C6H(NO2)3(OH).2.
Долгое время стифниновую кислоту получали в лабораториях
обработкой азотной кислотой разного рода смол и экстрактов. Однако
выходы были незначительны. Поэтому, опустив подробности о спосо-
бах получения стифниновой кислоты из смол и экстрактов, так как эти
способы ныне не представляют ни теоретического, ни практического
интереса, перейдем прямо к описанию современных способов получе-
ния стифниновой кислоты.
Наиболее рентабельно получение, стифниновой кислоты из ре-
зорцина.
Первым таким способом был способ Штенхуза, нитровавшего
резорцин в твердом виде и в водном растворе. При нитрации ре-
зорцина в твердом виде тонко измельченный резорцин при очень
хорошем размешивании и охлаждении до 5 10° присыпали в азот-
ную кислоту уд. веса 1,38—1,40. Когда резорцин растворялся, то
230
получившийся раствор по каплям прибавляли к серной кислоте
уд. веса 1,84, при охлаждении и хорошем перемешивании. Через
час по окончании слива реакционную смесь осторожно нагревали,
благодаря чему заканчивалось образование и выпадение в осадок
стифниновой кислоты.
Этот способ оказался нерентабельным, так как при пользовании
им относительно большое количество резорцина окисляется и тем
самым снижается выход. Поэтому Штенхуз стал искать другие спо-
собы получения стифниновой кислоты. Однако и при помощи другого
предложенного им способа нитрации резорцина в водном растворе не
удалось получить достаточно хороших выходов.
Далее следует отметить способы Бантлина и Бланкзама, предло-
живших получить стифниновую кислоту кипячением динитрофенола
или метамононитрофенола с дымящейся азотной кислотой. Получение
стифниновой кислоты из метанитрофенола происходит количественно и
может быть рекомендовано при отсутствии резорцина. Однако дорого-
визна исходных продуктов не позволяет осуществить этот метод в за-
водской практике.
Наконец, рассмотрим способ получения стифниновой кислоты по
Цеттеру и Мерцу, который с незначительными изменениями широко
применяется в настоящее время во многих странах.
Собственно способов Цеттера и Мерца два. В первом из иих про-
межуточным продуктом является диацетилрезорцин, а во втором —
д и Сул ьфо р е зорцин.
Первый способ. Тонко измельченный резорцин растворяют
в ледяной уксусной кислоте или в хлорангиДриде уксусной кислоты;
при этом образуется раствор диацетилрезорцина ц-Н^СНзСО^ОН^,
который при тщательном перемешивании и охлаждении приливают
к дымящейся азотной кислоте, взятой в тройном количестве против
теоретически необходимого для полной нитрации взятого резорцина.
После слива реагирующую смесь оставляют стоять 3—4 часа, про-
должая перемешивание, и затем ‘начинают осторожно нагревать,
наблюдая за тем, чтобы не было газообразования. При появлении
пузырьков газа в реагирующей смеси нагревание прекращают и смесь
охлаждают. Нагревание возобновляют через 1—Р/г часа.
Затем продукт, представляющий собой кашицеобразную желтую
массу, выливают постепенно в 5—6 ч. серной кислоты уд. веса 1,84
при тщательном перемешивании и охлаждении. После этого остав-
ляют всю массу стоять при комнатной температуре 1—Р/2 часа. За-
тем осторожно нагревают на водяной бане до 60° в течение 1—Ч час.,
внимательно наблюдая за тем, чтобы реагирующая масса не на-
чала вспениваться. В этом случае немедленно дают охлаждение,
иначе, упустив начало вспенивания (газообразования), остановить
его трудно. Нагревание поддерживают 1—2 часа. Получившуюся
красно-бурую массу выливают в избыток воды, где и выделяются
светложелтые кристаллы стифниновой кислоты. Осадок отделяют,
промывают водой, затем нагревают с гидратом окиси натрия, вслед-
ствие чего образуется раствор стифната натрия C6H(NO2)B(ONa)2, из
231
которого чистую стифниновую кислоту осаждают соляной кислотой.
Выход около 70% от теоретического.
Второй способ получения стифниновой кислоты, более
простой и удобный, состоит в следующем.
В нагретую до 40° серную кислоту уд. веса 1,84, взятую в
количестве 5—б ч., вносят небольшими порциями 1 ч. хорошо из-
мельченного резорцина. В растворе образуется дисульфорезорцин
Реакцию ведут при тщательном перемешивании.
Следующую порцию резорцина присыпают только тогда, когда первая
растворилась полностью, так как в противном случае образуются
очень трудно растворимые красные комки. Растворение тонко измель-
ченного резорцина в серной кислоте протекает быстро и со столь
большим выделением тепла, что температура без дальнейшего нагре-
вания остается равной 40°, а иногда поднимается и выше. По раство-
рении резорцина раствор охлаждают до 10—12°, после чего начинают
приливать дымящуюся азотную кислоту (небольшой избыток). Полу-
чающаяся масса имеет вид кашицы, состоящей из блестящих желтых
кристаллов стифниновой кислоты и красноватой жидкости. Массу
оставляют стоять для полной денитрации на ночь при комнатной темпе-
ратуре. На следующий день ее выливают в двойное по объему коли-
чество воды, декантируют, отсасывают на воронке Бюхнера, про-
мывают водой до удаления кислот и высушивают. Выход 91—95% от
теоретического.
Н. Н. Ефремов несколько изменил этот способ. Он производил
нитрацию крепкой азотной кислотой уд. веса 1,52 при температуре
1—2° и затем, после выливания реакционной массы в воду, нагревал
смесь на водяной бане около 8—10 час. до полного удаления окислов
азота. Этот способ выгоднее потому, что при нагревании образовав-
шегося продукта с разведенной азотной кислотой донитровываются
продукты неполной нитрации — нитрорезорцинсульфоновая кислота,
2,4-динитрорезорцин и 4,6-динитрорезорцин. Продукты неполной ни-
трации могут образоваться при получении стифниновой кислоты по
второму способу Цеттера и Мерца, так как при приливании азотной
кислоты к раствору дисульфорезорцина образуется очень вязкая,
слипающаяся в комки масса, которую не всегда удается растереть
даже при самом тщательном перемешивании.
Некоторый интерес представляют также попытки получения
стифниновой кислоты из изомеров пикриновой кислоты:
ОН ОН он
IZ 'jNOo Л'Ч|Н02 No/^jNOg
4Jno2 zOH \/0Н
NO„ NO2 NO.
Осуществлению этого метода пока мешает то, что конечный про-
дукт обычно засорен пикриновой кислотой, освободиться от которой
чрезвычайно трудно.
232
Стифниновая кислота представляет собой тринптрорезорцин, точ-
нее 2,4, б-тринитро-1,3-диоксибензол 1
ОН
NoZX.NO2
\/0Н
NO2
Она кристаллизуется из воды и спирта в виде светложелтых кристал-
лов гексагональной системы уд. веса 1,83, вяжущего вкуса.
Стифниновая кислота — довольно сильная кислота и по своим
свойствам приближается к пикриновой кислоте. Являясь двухоснов-
ной кислотой, она может давать средние и кислые соли. Средние соли
стифниновой кислоты так же стойки, как солн пикриновой кислоты.
Растворимость стифниновой кислоты в воде очень мала (меньше,
чем пикриновой кислоты). 1 ч. стифниновой кислоты при 14е раство-
ряется в 156 ч. воды. В горячей воде растворимость ее несколько по-
вышается и при 62° 1 ч. стифниновой кислоты растворяется в 88 ч.
воды. В спирте, эфире, уксусно этилов ом эфире она растворима значи-
тельно лучше. Растворимость в спирте может быть характеризована
следующими цифрами:
при 0° 100 г спирта растворяют CeH(NO2)3(OH)s . . 5,10 «г
" 17° 100 г спирта растворяют CeH(NO2)8(OH)3 . . 6.22 »
»> 68" 100 г спирта растворяют CcH(NO2)3(OH)» . 14.65 й
В бензоле и толуоле растворимость стифниновой кислоты меньше,
чем в спирте, так, например, в 10 мл бензола при 5° растворяется
только 0,45 г, а при 68° —4,7 г стифниновой кислоты: в 10 мл
толуола растворяется при 10° 0,51 г.
Стифниновая кислота очень хорошо растворяется в ацетоне: в
Юли ацетона при 18° растворяется 31,31 г.
Водный раствор стифниновой кислоты окрашен в яркожелтый
цвет, так как в водном растворе она диссоциирована на ионы. Кон-
центрированный водный раствор стифниновой кислоты растворяет
железо и цинк с выделением водорода; особенно легко протекает
реакция при нагревании. На медь, серебро, свинец, олово и кадмий
стифниновая кислота не действует. Карбонаты растворяются стиф-
ниновой кислотой с выделением двуокиси углерода.
Азотная и соляная кислоты (разбавленные и концентрированные)
не разлагают стифниновую кислоту даже при кипячении- В азотной
кислоте стифниновая кислота растворима несколько больше, чем в со-
ляной. При разбавлении азотнокислого раствора водой стифниновая
кислота выпадает в виде желтого порошка.
Стифниновая кислота разрушается при нагревании с царской
водкой (с образованием щавелевой кислоты), а также при кипячении
концентрированной (d = 1,84) серной кислотой.
1 В литературе может встретиться также название «оксипикрнновая кислота».
233
Зажженная стифниновая кислота горит ярким пламенем, но без
взрыва; от более мощного теплового импульса или от капсюля-дето-
натора она детонирует.
Температура плавления чистой стифниновой кислоты лежит между
174,5 и 175,5°. Температура плавления технической кислоты обычно
несколько ниже (173—174°). Однако наличие в техническом продукте
недонитровацного резорцина почти всегда повышает температур],
плавления, потому что температура плавления динитро резорцина
?12,5°, т. е. па 37° выше. инн
§ 2. ПОЛУЧЕНИЕ СТИФНАТА СВИНЦА И ЕГО ФИЗИКО-ХИМИЧЕСКИЕ
СВОЙСТВА
Стифнат свинца получается осаждением его из водного раствора
стифната натрия водным раствором нитрата свинца:
CcH(NO2)3(ONa)2 + Pb(NO3)2 + Н2О -
2NaNOs + CcH(NO2)3O2Pb • H2O.
f г стифниновой кислоты взмучивают в 125 мл дистиллированной
воды и к полученной суспензии небольшими порциями присыпают
3,5 а бикарбоната или 2,5 г карбоната натрия. При этом происходит
бурное выделение углекислого газа и образование стифната натрия,
который растворяется в воде вначале быстро, а затем все медленнее!
Процесс растворения ускоряют нагреванием на водной бане при тем-
пературе 50—60 и перемешиванием стеклянной палочкой образовав-
шихся на дне комочков. В результате получается раствор, окрашен-
ный в темнооранжевый цвет.
По растворении соли раствор фильтруют от механических приме-
сей и к нему приливают 0,5 мл 98—99%-ной уксусной кислоты или
соответствующее количество менее концентрированной кислоты. Рас-
твор нагревают до 70—80°.
К нагретому раствору стифната натрия быстро приливают (если
можно, то в один прием) 100 мл 7%-ного фильтрованного раствора ни-
трата свинца, нагретого почти до кипения. При сливании раствора непре-
рывно взбалтывают содержимое стаканчика. Сразу же начинается
выделение стифната свинца в виде песочного, легко осаждающего^
осадка, В процессе охлаждения периодически перемешивают рас-
твор с осадком. После охлаждения до комнатной температуры
жидкость проясняется до пикриново-желтого цвета, а осевший на дно
стифнат свинца получает темнооранжевую окраску.
После отделения осадка от маточного раствора его промывают
спиртом, эфиром и сушат. Выход при тщательной работе составляет
около 93% от теоретического.
Применение несколько более концентрированных растворов с
целью достижения больших выходов ведет к образованию студе-
нистых осадков неравномерного состава. В этом случае, если процесс
получения ведется при комнатной температуре и без предваритель-
ного подогрева растворов исходных веществ, образование кристаллов
234
стифната свинца происходит после некоторой выдержки не непосред-
ственно из раствора, а из студенистых осадков темнооранжевого цвета.
Чистый стифнат свинца представляет собой золотисто-желтые, тем-
неющие на воздухе (особенно при нагревании) кристаллы монокли-
нической системы. Хорошие кристаллы можно получить по методу
диффузии из растворов стифната магния и нитрата свинца.
Кристаллы стифната свинца-соответствуют формуле
C6H(NO2)sO2Pb • ИД
т. е. содержат 1 мол кристаллизационной воды. Эта молекула воды
прочно удерживается даже при 100° и может быть удалена посред-
ством нагревания в вакууме при 120° в течение сравнительно неболь-
шого промежутка времени. Без при-
менения вакуума она удаляется через
12 час. при температуре 110° или
через 5 час. при температуре 130°.
После оставления обезвоженной соли
во влажной атмосфере она вновь
притягивает количество влаги, рав-
ное 1 мол воды 4-1% гигроскопиче-
ской влаги на 1 мол соли.
Дегидратация не производит ни-
каких изменений в форме, очертаниях
и прозрачности кристаллов. Форма кристаллов изображена на фиг. 94.
Элементарная ячейка кристалла стифната свинца характеризуется
следующими параметрами:
Фиг. 94- Форма кристалла
стифната свинца.
а = 10,02 А,
b = 12,54 А,
«
с = 8,00 А.
О
Объем элементарной ячейки 1003,9 А3. Ячейка содержит 4 молекулы.
Ул. вес стифната свинца 3,085 при 20 .
Мы описали получение средней свинцовой соли. В нейтральной
(не подкисленной слабой органической кислотой) среде образуется
основной стифнат свинца следующего состава:
С6Н (NO2)s (ОН)2 • РЬО • Н20 ,
или
[CcH(NO2)3OH • О].РЬ н»о.
В слабощелочной среде образуется основная соль тринитрорезор-
Цината свинца следующего состава:
С0Н (NOJj 02РЬ • РЬ (ОН)2.
Основные свинцовые соли стифниновой кислоты пока в практике
не распространены вследствие их пониженных взрывчатых свойств.
Однако патентная литература рекомендует применять их в воспламе-
нительных (ударных) составах.
2.35
Растворимость средней соли в воде очень мала: при 17° в 100 мл
воды растворяется только 0,07 а. Растворимость в хлороформе, бен-
золе, метаноле, азотной и уксусной кислотах также ничтожна. Пере-
кристаллизация соли из этих растворителей практически невозможна.
Несколько лучше растворяется стифнат свинца в концентрированном
водном растворе ацетата аммония, чем и пользуются при анализах
ударных составов, содержащих ТНРС.
Гигроскопичность стифниновой кислоты ничтожна; поставленная
в атмосферу, насыщенную влагой, она через 40 дней дала привес
от 0,4 до 0,5%, в то время как технический азид свинца дает привес
1,6—1,9%; технический циануртриазид 4,6—6,3%.
С металлами стифниновая кислота не взаимодействует и поэтому
может применяться в любых оболочках.
§ 3. ВЗРЫВЧАТЫЕ СВОЙСТВА СТИФНАТА СВИНЦА
Стифнат свинца при взрыве распадается согласно следующему
уравнению:
40С6Н (NO3)3 О2РЬ - Н3О -► 40РЬпар + 86СО2 + 117СО + 7HCN +
+ 1э,5Н2 + 41HSO + 56,5N2 + 38,6 Кал.
Исходя из этого уравнения, взрывчатые характеристики стифната
свинца выражают следую-
щими цифрами: объем газо-
образных продуктов Vo =
=470 л/кг, температура взры-
• ва 100°; удельная энергия
I /- 423 л-amiкг, скорость дето-
J нации D 5200 м/сек.
™ Стифнат свинца чувстви-
«к v .. „ телен ко всем видам простого
Фиг. 95. Кривая температур вспышки , „ „
стифната свинца- начального импульса. Темпе-
ратура вспышки обычно при-
нимается равной 275°, т. е. несколько ниже, чем у азида свинца;
зависимость между температурой вспышки и временем нагревания
при постоянной температуре ванны нами приводится на фиг. 95
и в табл. 54.
Таблица 54
Зависимость между температурой вспышки ТНРС и временем нагревания
г сек г тсек '° тсек .
350 0,0 315 5,0 275 62,0
345 1,0 305 8,3 265 143,0
335 2,2 295 18,0 260 Отказ
325 3,5 285 32,0 —
236
Длительное нагревание стифната свинца может привести к вспышке
и при температурах ниже указанных температур; так, например,
нагревание при температуре 200° приводит к вспышке через 25 час.
Кроме того, следует отметить, что температура вспышки стифната
свинца зависит от чистоты продукта и может возрасти с 275 до 305°.
Чувствительность стифната свинца к удару меньше, чем чувстви-
тельность гремучей ртути, примерно в шесть раз, и меньше, чем чув-
ствительность азида свинца, примерно в два раза. Опыты показы-
вают, что при весе падающего груза в 600 г для взрыва необходима
высота:
для гремучей ртути ... 85 лш
» азида свинца - . 250 Л
» стифната свинца - - 500 »
По другим опытам чувствительность к удару запрессованных в
латунный колпачок веществ при весе груза в 5 кг выражается сле-
дующими высотами:
для циануртриазида 40 aw
» азида свинца . - - 120 »
» стифната свинца . . . 140 »
Чувствительность стифната свинца к трению, определение кото-
рой производилось растиранием навесок в фарфоровой ступке пести-
ком, находится между чувствительностью азида свинца и чувстви-
тельностью гремучей ртути.
Испытание капсюлей-детонаторов, снаряженных 2 г стифната
свинца, в свинцовых цилиндрах высотой 100 мм и диаметром 80 мм
с центральным цилиндрическим углублением (d == 7 мм) по сравне-
нию с другими инициаторами показывает у стифната несколько мень-
шую «силу», чем у гремучей ртути и у азида свинца (табл. 55).
Таблица 55
Сравнительная оценка «силы» инициирующих В В
Инициирующие ВВ Приблизи- тельная плотность Глубина отверстия мм Расширение бомбы мл
Стифнат свинца . . 1.8 30 29,1—29.0
Гремучая ртуть . 1.7 31 38.1 -28,7
Азид свинца . . - - 2,0 26 32,0- 26.6
Циаиуртриазид - . 1,2 43 131,3
Таким образом по силе ТНРС стоит ниже всех испытанных ини-
циирующих ВВ.
Впервые ТНРС был предложен Герцем (герм. пат. № 285902,
1914 г.). ИЛ совершенно правильно было указано и направление при-
менения его в ударных составах капсюлей-воспламенителей. Проверка
показала, что применять ТНРС в качестве самостоятельного первич-
ного заряда капсюлей-детонаторов нецелесообразно вследствие слиш-
ком незначительного ускорения его взрывчатого превращения.
237
Рейнско-Вестфальское общество взрывчатых веществ (герм,
пат. № 309210, 1922 г.) рекомендовало увеличить ускорение взрывча-
того превращения ТНРС в капсюлях-детонаторах путем увеличения
прочности оболочки капсюля; в патенте указано, что предельный
инициирующий заряд ТНРС по отношению к бризантным ВВ может
быть снижен до 0,5 г и что капсюлем-детонатором, содержащим такое
количество его, можно безотказно инициировать все нитросоединения
и аммонийиоселитрепные ВВ. Однако исследования автора показали,
что даже 2 г ТНРС недостаточно, для того чтобы вызвать полную
детонацию тетрила.
Применять же его в ударных составах капсюлей-воспламенителей
вместо гремучей ртути вполне возможно, и ТНРС действительно ис-
пользуют в ударных составах, изготовляемых в Германии, прежде
всего для мелкокалиберного оружия- При этом следует отметить одно
весьма ценное качество ТНРС — его способность увеличивать началь-
ную скорость пули и ее меткость. Герц приводит следующие сравни-
тельные данные результатов стрельбы 6-миллиметровыми флобертов-
скими патронами с круглой пулей:
Состав V(, м}сек Рассеивание на расстоянии 50 м
Гремучертутный состав . . ... 225 50 пуль в прямоугольник 85x105 мм
Состав с ТНРС 261 50 пуль в прямоугольник 65x65 мм
Еще характернее становится разница в начальных скоростях при
повышении калибра и величины заряда. Вследствие меньшей бризант-
ности ТНРС можно достичь, не опасаясь разрыва гильзы, начальных
скоростей, недоступных при этих условиях в случае применения гре-
мучей ртути.
Особое значение ТНРС приобрел в последние годы в качестве глав-
ной составной части некорродирующих и неэродирующих ударных
составов капсюлей-воспламенителей. Так, например, по данным ли-
тературы, новый немецкий неэродирующий состав Sinoxid содержит
в большем количестве ТНРС с примесью нескольких процентов
тетразена.
В ряде патентов ТНРС также рекомендуется для изготовления
детонирующих шнуров взамен гремучей ртути. Шнур с ТНРС дейст-
вует в воде безотказно и имеет большую, чем у гремучертутного шнура,
скорость детонации. При этом предлагается следующий способ приго-
товления детонирующего шнура: ТНРС смешивают с раствором флег-
матизатора в кашицу, которую в виде нитей сейчас же покрывают
предохранительной оплеткой. Рекомендуется также применять со-
вместно с ТНРС в шнурах бризантные ВВ.
Однако получать ТНРС и обрабатывать его в больших массах
опасно вследствие относительно высокой чувствительности его к тре-
238
цпю и большой склонности к электризации при трении кристалла
<» кристалл или о стенки аппаратуры- Электризация при этом может
достигать напряжения в несколько тысяч вольт. В производстве стиф-
п.гга свинца часто случаются взрывы вследствие отсутствия должного
внимания к этому вопросу. Так, например, 23 мая 1932 г- на заводе
Dynamite Actien Gesellschaft в Германии имел место взрыв 3 кг ТНРС
о г «неизвестной» причины в предварительном помещении сушильного
лиания-
Наиболее опасные операции в отношении ’ электризации %-это
пересыпка сухого продукта и сортировка его.
§ 4. ИСХОДНЫЕ ПРОДУКТЫ ДЛЯ ФАБРИКАЦИИ СТИФНАТА СВИНЦА
Для получения стифната свинца необходимы следующие исходные
материалы: а) резорцин1, б) серная кислота, в) азотная кислота,
г) бикарбонат натрия, д) уксусная кислота, е) нитрат свинца, ж) спирт,
.<) бензол, и) воск.
Свойства и требования, предъявляемые к нитрату свинца, спирту
и бензолу, те же, которые описаны в главе «Азид свинца».
Резорцин — метадиоксибензол С6Н4(ОН)2 представляет собой
кристаллическое вещество белого цвета, уд. веса 1,283. Кристаллы его
относятся к ромбической сингонии. Температура плавления 110°,
температура кипения 277° при нормальном давлении и 210° при
давлении 7 мм рт. ст.
В воде резорцин растворим хорошо;
в 100 ч. воды при 0° растворяется ч.
» 100 » » » 12,Б° » 147 •>
» 100 » " 30е » 228,6 »
Почти так же хорошо резорцин растворяется в спирте: в 10О ч.
90%-ного спирта при 15° растворяется 127,5 ч. В углеводородах
резорцин растворим значительно труднее. Так, в 100 мл бензола при
24° растворяется только 2 г резорцина. В хлороформе и сероуглероде
резорцин практически нерастворим. _
В присутствии аммиака резорцин краснеет. При действии хлора
или брома на водные растворы его образуется трихлор- или трибром-
резорцин.
С водными растворами хлорного железа резорцин дает синее окра-
шивание. В аммиачном растворе нитрата серебра резорцин восста-
навливает серебро. Резорцин не дает осадка с ацетатом свинца, в то
время как его изомер — пирокатехин — дает осадок.
Для качественного определения резорцина может служить реак-
ция образования флуоресцина.
1 Резорцин является одним из важных исходных продуктов для получения
целого ряда красителей из группы флуоресцинов, азокрасителей, сернистых и т. п.
Находит также применение и непосредственно на текстильных фабриках для обра-
зования красок на волокне. Благодаря своему антисептическому действию приме-
няется и в медицине.
239
Небольшое количество резорцина нагревают с фталевым ангидри
дом почти до кипения; полученный продукт буроватого цвета раствв
ряют в водном растворе гидроксида натрия. Если влить нескольн
капель этого раствора в пробирку с водой, то образовавшаяся в пре
бирке буроватая жидкость в проходящем свете будет окрашива!
раствор в зеленый цвет.
Резорцин обычно производится сплавлением натриевой соли
метадисульфо бензола с едкими щелочами:
CcH4(SO3Na)2 + 4NaOH CeH4(ONa)2 -J- 2Na2SO3 + 2H2O
с последующей обработкой резорцината натрия разведенной соляном
кислотой:
CcH4(ONa)3 + 2НС1 -> CfiH4(OH)2 + 2NaCl
и экстрагированием эфиром или амиловым спиртом.
Натриевую соль метадисульфобензола получают сульфированием]
бензола 20%-ным олеумом при температуре порядка 200° в один или
несколько приемов. Полученную массу разбавляют водой, нейтрали-
зуют мелом, направляют на фильтрацию от образовавшегося сульфата |
кальция, затем при кипячении обрабатывают содой, отфильтровывают
далее от карбоната кальция, выпаривают сначала в выпаривателе до
26° Вё и, наконец, в вакуумсушильном аппарате досуха.
Технический резорцин получают одним из следующих методов. |
250 кг каустической соды разбивают на куски и загружают в пла-
вильный котел, где она расплавляется до тех пор, пока не перестанут
•образовываться корки па внутренней стенке котла. В расплавленный
каустик при механическом перемешивании прибавляют в течение
получаса 125 кг натриевой соли метадисульфобензола, наблюдая затем,
чтобы не происходило обильного вспенивания. Затем вспенивание
исчезает и продукт принимает жидкую консистенцию; плав сперва
желтеет, потом становится коричневым; температура достигает 270°.
На этом заканчивают сплавление; массу выливают на железные про-
тивни и охлаждают.
Твердым плав разбивают на куски и вносят в железный футеро-
ванный чан с 500 л воды, куда затем приливают серной кислоты до пре-
кращения выделения сернистого ангидрида и до слабой реакции на
конго-бумажку. Подкисленный плав подают в экстрактор, где его под-
вергают четырехкратной экстракции амиловым спиртом (по 100 л
последнего на каждую экстракцию) при перемешивании в течение
получаса. После каждой экстракции содержимое экстрактора переда-
вливают в отстойник. Отстаивают в течение 1 часа, после чего водный
раствор снова направляют в экстрактор, а спиртовой раствор — !
в особый резервуар.
Четырехкратной экстракции вполне достаточно, ибо водный рас-
твор бывает к этому времени почти полностью лишен содержания
резорцина, и последний экстракт редко бывает окрашен.
Все четыре экстракта соединяют вместе, дают им отстояться в те-
чение 12 час., сливают отстоявшуюся воду и направляют спиртовой
240
Фиг. 96- Схема непрерывного экстра-
гирования резорцина эфиром.
1—экстрактор; 2—смотровые банки; 3—аппа-
рат для отгонки эфира; 4—холодильник;
5—приемник для эфира; 6—сборник отходя-
щего раствора.
раствор в дистиллятор, где и приступают к отгонке амилового спирта.
При этом вначале нагревают раствор глухим паром до 100°, а затем
пропускают острый пар внутрь дистиллятора до полной отгонки
амилового спирта.
Оставшийся водный раствор резорцина направляют е выпарные
эмалированные чаши для удаления воды.
Другой метод, более совершенный, отличается от описанного
тем, что работа производится не с сухой солью метадиоксибензола,
а с водным раствором.
В этом случае приблизительно
50%-ный раствор натриевой соли
метадисульфобензола сливают в
плавильный чан с раствором каусти-
ческой соды в 45° Вё. Массу на-
гревают при температуре 170—185
до густой консистенции, затем
разливают черпаком на противни.
Противни задвигают в особую на-
гревательную печь с температурой
около 400° на 5—6 час., после чего
сплавление считается оконченным.
Противни механически извлекают-
ся из печи, содержимое их переме-
щается в особый аппарат, орошае-
мый водой. Через дырчатое дно
аппарата раствор резорцината
натрия стекает в расположенный
ниже котел, откуда он подается
на нутч-фильтр для отделения от
сульфата.
Отфильтрованный раствор резорцината натрия обрабатывают со-
ляной кислотой до превращения темнокоричневой окраски раствора
в желтую и, наконец, после вторичной фильтрации подают на экстрак-
цию, проводимую непрерывным процессом по схеме, указанной на
фиг. 96.
Экстрактор представляет собой вертикальный цилиндрический
аппарат, снабженный мешалкой. Кислый раствор резорцина загру-
жают сверху, в то время как эфир по свинцовой трубе непрерывно
поступает в нижнюю часть аппарата и, поднимаясь вверх, экстраги-
рует резорцин. Из верхней части аппарата эфир непрерывно отводят
на дистилляцию, при этом через смотровое стекло наблюдают за цве-
том эфирного раствора. Когда эфир начинает проходить бесцвет-
ным, экстракцию прекращают. Из дистиллятора пары эфира поступают
в холодильник, где они конденсируются, и через особый приемник
вновь поступают на экстракцию.
Оставшийся в экстракторе водный раствор спускают в специальный
резервуар, фильтруют и направляют в обогреваемые паром аппараты
для отгонки остатков' эфира и направляют для выщелачивания плава.
Бубнов—92—16 241
Полученный сырой резорцин, так же как и по первому способу,
поступает для очистки на перегонку в вакууме.
При нагревании перегонного куба вначале отгоняется немного
воды и фенола. Когда температура достигает 190е, дают вакуум до
130 мм остаточного давления. Резорцин закипает и, конденсируясь,
собирается в приемнике.
Охлажденный резорцин размалывают на медной луженой мель-
нице.
В зависимости от качества очистки технический резорцин может
содержать в виде примесей свои гомологи и изомеры: фенол, пирока-
техин, гидрохинон, флорглюцин, дирезорцин. Наряду с ними ветре
чаются тиорезорцин, органические кислоты (салициловая, фталевая),
смолы и нерастворимые в воде примеси. Поэтому технический резорцин
имеет обычно сероватый оттенок и запах фенола. Недостаточно очи-
щенный резорцин имеет коричневую окраску.
После сравнительно тщательной очистки возгонкой, дистилляцией
с водяным паром и кристаллизацией технический резорцин все же
может содержать фенол и пирокатехин. Следует иметь в виду, что
при нитрации технического резорцина примеси изомеров и гомоло-
гов менее вредны, чем примеси всякого рода смолистых веществ.
Примесь к резорцину смолистых веществ резко снижает его качество
и ухудшает процесс нитрации (сильное вспенивание, понижение вы-
хода, значительное понижение температуры плавления стифниновой
кислоты).
Поэтому, ограничивая содержание в резорцине его гомологов и
изомеров, не следует допускать наличия смолистых веществ (кроме
следов).
Применяемому в производстве стифниновой кислоты резерпину
предъявляются следующие технические требования:
№ пс п. р. Наименование характеристик Сорт I Сорт 11
1 Гвешний вид Кристаллы бе- Кристаллы могут
лого цвета с ро- быть окрашены до
зоватым оттенком коричневого цвета
2 Влажность не более 0,3% 2,0%
3 Нерастворимость в воде не более 0.05% 0,5%
4 Зольность не более 0.01% 0.3%
5 Фенола и других бронирующихся
веществ не более 0,1% 1,0%
6 Пирокатехина нс более . . 0.3%, ол%
7 Температура плавления 109—111° Не менее 101
8 Резорцина не менее 09% 91 ° о
Методика анализа резорцина следующая:
1. Для определения влажности 5 г резорцина в тарированном бюксе высу-
шивают в термостате при 80—00° в течение 2 час. Убыль веса, выраженную в
процентах, принимают за влажность.
42
2. Для определения нерастворимых в воде примесей 10 г резорцина раство-
ряют в 100 мл воды, затем раствор фильтруют через высушенный и взвешенный
гигель Шотта и остаток на тигле Шотта сушат в термостате до постоянного веса
при 100—105°. Привес тигля Шотта, выраженный в процентах, н выражает коли-
чество нерастворимых.
3- Для определения зольности 5 г резорцина сжигают в фарфоровом тигле
и золу прокаливают. Привес фарфорового тигля показывает зольность продукта.
4. Для определения пирокатехина в фильтрат после определения нераствори-
мых в воде примесей прибавляют 50 мл 4%-ного раствора ацетата свинца и остав-
ляют на сутки, затем фильтруют через тигель Шотта, промывают осадок водой и
сушат его при 70° в течение 4 час.-
5. Для определения содержания резорцина 1,5—2 г резорцина растворяют
в 500 мл воды. Берут из приготовленного раствора в колбу Эрленмейера 25 мл
раствора, к которому добавляют 25 мл концентрированной соляной кислоты
уд. веса 1,19 и 200 мл воды. Колбу с содержимым ставят на лед н титруют при
тщательном перемешивании 0,1N раствором нитрита натрия. Индикатор-— нодо-
крахмальйая бумажка. О конце реакции судят по не исчезающему в течение 30 мин.
синему окрашиванию бумажки при нанесении на нее капли титруемого раствора.
Температура титрования не выше 10°.
6- Для определения фенола и других бронирующихся веществ из приготовлен-
ного выше общего раствора берут 50 мл в мерную колбу емкостью 250 мл н доводят
водой до метки. Для бромирования из этого вновь разбавленного раствора отби-
рают 25 мл в банку с пришлифованной пробкой и 25 мл водного раствора бромида
калия и бромата калия (раствор 2,7819 КВгО3 и 60 г КВг в 1 л воды), затем добав-
ляют 5 мл концентрированной соляной кислоты (d = 1,19), оставляют стоять на
холоду 5 мин., добавляют 20 мл 10%-ного раствора иодида калия, вновь дают по-
стоять раствору на холоду 5 мин. и, наконец, титруют 0, IN раствором тиосульфата
натрия.
Разница в количестве тиосульфата натрия, пошедшего на титрование пробы
и на контрольный опыт, и есть количество NasSsOa, израсходованное на брони-
рующиеся вещества.
Серная и азотная кислоты, а также применяе-
мый иногда для нитрации меланж, должны удовлетворять тем же
требованиям, которые ставятся промышленностью бризантных ВВ.
Бикарбонат натрия — белый кристаллический поро-
шок со щелочным вкусом, уд. веса 2,2. Получается при введении
двуокиси углерода в холодный насыщенный раствор NaaCO3. Бикар-
бонат натрия в водном растворе, как и в сухом виде, уже при комнат-
ной температуре медленно выделяет СОа. Выше 65° из раствора соли
происходит энергичное выделение СО3.
Растворимость бикарбоната в воде, насыщенной СОа, по Федо-
тьеву, составляет;
при 0° в 100 мл воды
» 15° » 100 » »
» Зо°» юо » »
» 45° » 100 »> »
6,9 г NaHCO3
8,8 » NaHCO3
11,02 »> NaHCOs
13,86 » NaHCO3
Вследствие гидролиза водные растворы обнаруживают щелоч-
ную реакцию, хотя и очень слабую. Щелочная реакция слабо обнару-
живается лакмусом и метилоранжем и вовсе не обнаруживается
фенолфталеином.
Бикарбонату натрия предъявляются следующие требования;
I) по внешнему виду он должен представлять собой белый кри-
сталлический порошок;
243
2) нацело должен растворяться в воде;
3) содержание бикарбоната натрия в продукте должно быть не
ниже 98%;
4) содержание карбоната натрия не более 0,5%;
5) содержание хлористого натрия не более 0,05%;
6) содержание влаги не более 1%;
7) совершенно недопустимо содержание солей тяжелых металлов
и аммиака.
Воск пчелиный по своим физическим свойствам представь
ляет собой пластичную массу с зернистым изломом, легко раствори-
мую при нагребании в бензоле, сероуглероде, хлороформе и других
растворителях.
Пчелиный воск с химической стороны является смесью сложных
эфиров жирных кислот и высокомолекулярных спиртов. Кроме того,
он обязательно содержит некоторое количество неомыляемых веществ
и углеводородов парафинового ряда. К жирным кислотам, входящим
в состав воска и выделяемым из него кипячением в спирте, относится,
главным образом, церотиновая кислота С26Нб2О2. Из эфиров, входя-
щих в воск, надо отметить эфир пальмитиновой кислоты и мирицило-
вого спирта С31Н64О2, из углеводородов — C27H5G,C31H64 и СзсН74.
Натуральный пчелиный воск желтого цвета. Под действием солнеч-
ных лучей он отбеливается и становится совершенно белым.
Следует иметь в виду, что пчелиный воск очень часто фальсифици-
руют церезином, парафином, канифолью и другими веществами.
Применяемый для флегматизации стифната свинца воск должен
удовлетворять следующим техническим требованиям:
1) цвет в пределах от белого до желтого;
2) содержание видимых на-глаз механических примесей недопу-
стимо; 1
3) нерастворимого в бензоле остатка должно быть не более 0,2%:
4) температура плавления должна быть в пределах 62—64°;
5) уд. вес при температуре 98—100° — в пределах 0,961—0,968:
6) содержание золы не более 0,8%, в том числе кремнезема не
более 0,02%;
7) кислотное число в пределах 17—21 (кислотное число — коли]
чество миллиграммов КОН, необходимое для нейтрализации жир-
ных кислот, содержащихся в I г воска);
8) число омыления — в пределах 88—96;
9) эфирное число 69—70;
10) содержание минеральных и стеариновой кислот недопустимо;
11) содержание фальсификаторов (церезин, парафин, канифоль)
также недопустимо.
Нерастворимые в бензоле примеси определяют обычным способом. Навеску
воска растворяют в бензоле, раствор фильтруют через тарированный фильтр, про-
мывают бензолом и сушат до постоянного веса. Привес фильтра равен количеству
нерастворимых.
Температуру плавления определяют капиллярным методом. Берут открытый
с Обоих концов капилляр, опускают в расплавленный воск, для того чтобы в ка-
пилляре задерживалось минимальное количество воска. После этого капилляр
244
охлаждают (очень часто оставляют для охлаждения на ночь). Охлажденный ка-
пилляр с воском прикрепляют к термометру резиновым кольцом и опускают в ста-
кан с дистиллированной водой. Воду в стакане нагревают. Та температура, при
которой вода своим давлением на нижний открытый конец капилляра поднимает
воск в капилляре, и отмечается как температура плавления воска.
Удельный вес определяют на весах Мора-Вестфаля при температуре
98—100° (температуру поддерживают нагреванием водяным паром) посредством
уравнения удельных весов смеси спирта с водой и испытуемого расплавленного
образца. Для этого помещают навеску воска на дно стакана, приливают в стакан
воды и затем, добавляя спирт, добиваются уравнения удельных весов, т. е. свобод-
ного плавания воска в смеси спирта и воды. После этого ареометром определяю?
удельный вес спиртоводвой смеси, а стало быть, устанавливают удельный вес
воска.
При этом методе определения неизбежны ошибки. Поэтому очень часто, и со-
вершенно правильно, определяют при комнатной температуре удельный вес воска
в виде твердых, свободных от воздуха, высушенных шариков. Удельный вес по
воде устанавливают при комнатной температуре. Точность при этом методе вполне
достаточная и работа менее сложна.
Зольность определяют, так же как в резорцине, только прокаливанием на
платиновом тигле.
Содержание кремнезема устанавливают обработкой золы плавиковой кислотой.
Для определения кислотного числа 5 г воска растворяют в спирто-эфирной
смеси (I ч. спирта 4- 4 ч. эфира). К полученному прозрачному раствору приливают
1 мл 1%-ного спиртового раствора фенолфталеина и титруют 0,17V спиртовым
раствором гидроксида калия до появления розовой окраски:
ткон'°
кислотное число —-------,
Р
где а — число миллилитров раствора КОН, пошедших на титрование;
р — навеска.
Для определения числа омыления в колбу емко.стью 150—200 мл отвешивают
1 г воска и приливают 30 мл 0.5N спиртового раствора гидроксида калия н нагре-
вают на кипящей водяной бане с обратно поставленным холодильником. Затем
оттитровывают избыточную щелочь в присутствии фенолфталеина 0,57V водным
раствором соляной кислоты.
Параллельно в другую колбу отмеривают 30 мл применявшегося раствора
едкого кали и титруют применявшимся раствором НС1:
(Й —• G)T hcj
число омыления = ______'____,
р
где а — число миллилитров НС), пошедших на первый Опыт;
b - число миллилитров НС), пошедших на параллельный опыт;
р — навеска.
Эфирное число может быть определено или по разности число омыления —
кислотное число, или непосредственно. В последнем случае к нейтрализованному
после определения кислотного числа раствору приливают 25—50 мл полунормаль-
ного спиртового раствора КОН и дальше ведут анализ так же, как при опреде-
лении числа омыления.
Для определения стеариновой кислоты 1 г воска нагревают до кипения с 15 мА
80%-ного спирта. После охлаждения до 18—20е раствор фильтруют в мерный ци-
линдр емкостью 200 мл и разбавляют водой примерно до 200 мл. Если в воске
имеется стеариновая кислота, то она выделяется при этом в виде хлопьев ня
поверхности раствора.
Для определения наличия фальсификаторов 5 г воска обрабатывают 25 мл
спиртового раствора щелочи при кипячении с обратно поставленным холодиль-
ником. Затем спирт выпаривают и остаток нагревают на водяной бане с 20 мл
чистого глицерина до растворения. К раствору прибавляют Ю мл кипящей воды.
Чистый воск дает при этом прозрачный раствор, в присутствии же парафина или
церезина раствор становится мутным.
245
Далее растворяют 0,1—0,2 г воска в уксусном ангидриде и осто-
рожно по стенке спускают в раствор 1—2 капли серной кислоты
(d = 1,84). В присутствии незначительного количества канифоли'
появляется интенсивная фиолетовая окраска, переходящая в буро-
желтую.
§ 5. ТЕХНОЛОГИЧЕСКИЙ ПРОЦЕСС ПОЛУЧЕНИЯ СТИФНАТА СВИНЦА
Тринитрорезорцин очень часто изготовляется на тех же заводах,
что и стифнат свинца. Поэтому весь технологический процесс про из-1
водства стифната свинца делится на две самостоятельных части:
I) получение тринитрорезорцииа или стифниновой кислоты;
2) получение TH PC (стифната свинца).
Производство стифниновой к и г л о т ы- Посту-
пающий в мастерскую резорцин тонко измельчается в железных бара-
банах с чугунными шарами или на мельницах типа краскотерок.
Нитрации предшествует сульфирование резорцина, которое про-
изводится в нитраторе с обогревательной рубашкой и крышкой,
прикрепленной болтами к корпусу нитратора. В крышке расположено
смотровое (оно же загрузочное) окно, штуцер для термометра, штуцер
для спуска кислот и отверстие для удаления окислов азота. Нитратор
снабжен мешалкой пропеллерного типа.
В предварительно подготовленный и высушенный нитратор зали-
вают из мерника концентрированную серную кислоту (rf = 1,84),
пускают в ход мешалку и через загрузочное окно в крышке нитратора
постепенно малыми порциями присыпают тонко измельченный резор-
цин. Серной кислоты необходимо брать пятикратное весовое количество
против теоретического расчета. Начальная температура реакции
должна находиться в пределах 20—25°. При этом происходит раство-
рение резорцина в серной кислоте и образование дисульфорезорцина:
С6Н4 (ОН)2 + 2H2SO4 С6Н2 (HSO3)2 (ОН)2 + 2Н2О.
Реакция образования дисульфо резорцина экзотермична, и поэтому
содержимое нитратора необходимо тщательно перемешивать и все
время охлаждать водой, для того чтобы температура реагирующей
смеси не поднималась выше 35°.
В случае недостаточности охлаждения и при больших скоростях
засыпки резорцина температура реагирующей массы может подняться
до 90°, и реакция закончится выгоранием. Очень часто подъем темпе-
ратуры выше 35° может сопровождаться загустеванием реагирующей
смеси, вследствие чего дальнейшее ведение процесса сульфирования
затрудняется. Надо иметь в виду, что прибавление новых порций ре-
зорцина надлежит производить только после того, как растворится
предыдущая порция, иначе образуются красные комья, которые
также затруднят дальнейший процесс сульфирования. Ликвидация
комкования и загустевания массы должна производиться выдержкой
ее при температуре не выше 35° и хорошим перемешиванием. При
правильно ведомой операции получается фиолетово-красный про-
зрачный раствор дисульфорезорцина в серной кислоте.
246
После того как весь резорцин внесен в серную кислоту и нацело
растворился в ней, прекращают водяное охлаждение нитратора.
аппарат нагревают паром и поднимают температуру реагирующей
массы до 60—70°.
Для полного завершения реакции выдерживают при этой темпе-
ратуре в течение I—Р/2 час » после чего охлаждают раствор дисуль-
форезорцина до 30° и приступают к нитрации.
Нитрация проводится в том же аппарате, что и сульфирование.
Она может быть осуществлена тремя методами:
I. Нитрация азотной кислотой разной крепости (сначала слабой,
затем крепкой).
2. Нитрация одной концентрированной азотной кислотой (d =
- 1,52).
3. Нитрация смесью азотной и серной кислот.
При нитрации следует учесть необходимость ведения процесса
при низкой температуре, так как при температурах выше 38° нитрую-
щаяся масса приобретает бурый оттенок и загустевает в вязкую
массу, весьма трудно поддающуюся размешиванию. В таких случаях
требуется дополнительное количество азотной кислоты или значи-
тельный подъем температуры, чтобы масса вновь растворилась. Даль-
нейший нагрев до 60—70° в этом случае может вызвать значительное
увеличение скорости реакции недонитрованного продукта, что при-
водит к самопроизвольному подъему температуры до 100°, бурному
процессу и частичному окислению. Масса при этом сильно вспучивается,
вспенивается, увеличиваясь в объеме до четырех-пяти раз против пер-
воначального, и не исключена возможность выбрасывания ее из ни-
тратора. Только значительной выдержкой при 40—50° удается отре-
гулировать процесс реакции.
При нитрации крепкой кислотой имеет большое значение и харак-
тер смешения. Требуется одновременно хорошее охлаждение и хоро-
шее перемешивание. Остановка мешалки может закончиться значи-
тельным разогревом и порчей продукта.
При нитрации на кислотах разной крепости, как уже сказано,
раствор дисульфорезорцина охлаждают до 30° и начинают слив азот-
ной кислоты (d = 1,38). На 1 в. ч. резорцина берут 1 в. ч. HNO3
(d = 1,38). Температуру во время слива поддерживают около 38°
и ни в коем случае не выше 40°. Температуру регулируют охлаждением
и временем спуска HNOS.
После слива слабой кислоты нитруемая масса обогащается кри-
сталлами и дальнейшую нитрацию можно вести при более высокой
температуре. Поэтому крепкую азотную кислоту (d =1,5) сливают
при температуре 40 — 45°. На I в. ч. резорцина берут 1,6 в. ч.
крепкой азотной кислоты. После слива кислот повышают температуру
до 50° и выдерживают около I часа, затем вновь повышают темпера-
туру до 65—75° и выдерживают до осветления массы. При выдержке
нитруемая масса сначала густеет, приобретая темнобурый оттенок,
в конце же она становится менее густой и более светлой.
247
Значительно более экономичным и эффективным является третий
путь нитрации, т. е. с применением кислотной смеси.
К охлажденному до 30° раствору дисульфорезорцина в серной
кислоте приливают постепенно через алюминиевую сливную трубу
кислотную смесь (HNO3, H2SO4, Н20) в 1,3 раза больше теоретиче-
ского расчета. Кислотную смесь сливают так, чтобы максимальная
температура реагирующей массы не превышала 37°. Особое внимание
следует обратить на состояние реагирующем массы к концу слива
кислотной смеси, когда наступает переход моно- и динитропродук-
тов в тринитрорезорцин и выпадание основной массы кристаллов
последнего.
Процесс перехода моно- и динитрорезорцина в тринитропродукт
сопровождается обильным выделением окислов азота, а иногда и за-
густеванием массы. При недостаточно внимательном отношении рабо-
тающих к температурному режиму и скорости слива кислотной смеси
пенящаяся и увеличивающаяся при этом в объеме в несколько раз
реагирующая масса может быть выброшена из нитратера.
Причина этого явления заключается в том, что реагирующая масса
становится густой и трудноперемешиваемой, а при недостатке пере-
мешивания и несвоевременном охлаждении происходит местный
разогрев и вспенивание массы.
Иногда в этом случае достаточно дополнительного перемешивания,
чтобы процесс нитрации вновь протекал спокойно. Однако в случае
резких скачков температуры следует прекратить слив кислотной
смеси и давать максимальное охлаждение при перемешивании. Если
после принятия всех этих мер все же температура будет возрастать
и вспучивание массы с выделением окислов азота будет продолжаться,
то при достижении температуры реагирующей массы до 70° следует
открыть аварийный кран нитратора и всю массу спустить в располо-
женный ниже бак-разбавитель.
При нормальном течении процесса последние литры кислотной
смеси следует приливать быстро с тем, чтобы довести температуру
до 50°. Затем дают часовую выдержку, температуру поднимают до
60—65° и выдерживают реагирующую массу еще I час.
К концу выдержки реагирующая масса приобретает желтую
окраску. Если же она будет иметь красноватый оттенок, что говорит
о наличии больших количеств недонитрованного продукта, то следует
продлить последнюю выдержку до изменения цвета.
В результате нитрации образуется:
C6H2(SO8H)2(OH)2 + 3HNO3 -> CeHCNOj.yOHb + 2H2SO4 + Hp
кристаллическая стифниновая кислота.
Нитрованную массу охлаждают в нитраторе до температуры
25—35° и затем опускают в керамиковый бак-разбавитель, где маточ-
ный раствор разбавляется водой. Эта операция сопровождается вспе-
ниванием и повышением температуры, поэтому ее необходимо произ-
водить постепенно, небольшими порциями, чтобы избежать резкого
выделения окислов азота и возможного разбрызгивания раствора.
248
В результате разбавления растворенная в кислотной смеси стиф-
ниновая кислота выпадает в осадок, так как ее растворимость в воде
меньше, чем в отработанной кислоте. Затем содержимому бака-разба-
вителя дают отстояться в течение 30 мин.—1 часа, после чего кри-
сталлы стифниновой кислоты отфильтровывают на керамиковом нутч-
фильтре при вакууме 400—700 мм рт. ст.
В качестве фильтрующего материала обычно применяют шинель-
ное сукно. Разбавленный маточный раствор по мере накопления
спускают из бака нутч-фильтра через деревянные ловушки в кана-
лизацию. Ловушки периодически очищают от стифниновой кислоты.
Продукт на нутч-фильтре промывают водой порциями по 7—10 л,
сначала теплой (/ 50°), а затем холодной (/ 20°). После этого
продукт отсасывают до достижения хорошей его сыпучести.
При применении резорцина первого сорта можно ограничиться
одной промывкой, при этом получается выход продукта около 84%.
В случае применения для производства резорцина второго сорта
недостаточно одной промывки водой для отделения серной кислоты,,
а приходится прибегать к специальной очистке стифниновой кислоты.
Очистка в случае необходимости осуществляется путем обработки
стифниновой кислоты бикарбонатом натрия:
CcH(NO2)3(OH)2 + 2NaHCO3 -► C6H(NO2)3(ONa)2 + 2СО2+2Н2О
Одновременно с этой реакцией происходит и нейтрализация серной
кислоты до Na2SO4. Дальнейшая обработка полученного раствора
азотной кислотой приводит к выделению чистой стифниновой кислоты.
Следует отметить, что при очистке стифниновой кислоты часть ее
теряется (около 5%).
Порядок очистки стифниновой кислоты следующий.
К загруженной в деревянный (снабженный мешалкой) бак стифии-
новой кислоте приливают воду в отношении 1:2, включают мешалку
и дают обогрев до 85—90°. Затем тонкой струей приливают 12,5%-ный
раствор бикарбоната натрия с небольшим избытком против теорети-
ческого расчета.
Полученный примерно 15%-ный раствор стифната натрия охлаж-
дают до 40—45° и обрабатывают крепкой азотной кислотой. Осажден-
ную стифниновую кислоту вновь отфильтровывают и промывают
водой.
Выход стифниновой кислоты при применении резорцина второго
сорта --до 78%.
Очищенную стифниновую кислоту отправляют на сушку в сушилки
стеллажного типа при температуре 50—60°. Стифниновую кислоту'рас-
сыпают ровным слоем (толщиной 2—3 см) на алюминиевых листах.
Сушка в камерных сушилках требует около полутора суток.
Очень часто, чтобы избежать непроизводительной траты времени
на сушку стифниновой кислоты, сырой продукт после его анализа
отправляют непосредственно для приготовления раствора стифната
натрия или оставляют на хранение в складе, а содержание влаги
учитывают перед пуском его в производство.
249'
Технический продукт обычно содержит в большей или меньше
степени различные посторонние примеси. Из химических примесе
обычными спутниками стифниновой кислоты являются изомеры и пре
дукты низшей нитрации (моно- и динитро резорцины), неблагоприятп
отражающиеся на кондиционности продукта. Технический продуц
может содержать и некоторое количество серной кислоты. Неизбея
ными примесями также являются гидраты окислов железа, получае
мые вследствие корродирующего действия кислот на аппаратуру.
Идущей на производство ТНРС стифниновой кислоте предъявляю
следующие требования (все цифры в пересчете наtсухой продую)
1) температура плавления — не ниже 174°;
2) нерастворимых в воде примесей — не более 0,5%;
3) зольность — не более 1%;
4) содержание серной кислоты — не более 0,2%;
5) содержание азота — в пределах 16,2—17,4%.
Производство стифната свинца. Соответствую
щий указанным техническим условиям продукт направляют на при
готовление раствора стифната натрия. Для осаждения стифната свинц
применяют обычно 3- или 4%-ный раствор стифната натрия. При рас
чете концентраций раствора учитывают влагу, содержащуюся в исход
ном продукте. Количество воды, необходимой для растворения стифни
новой кислоты, подсчитывают по формуле
ТНР, — g *100
''г ~ 100 — %Н2О’
где а — количество сухой стифниновой кислоты, необходимой дл
получения 3- или 4%-ного раствора.
В фарфоровый бак, снабженный мешалкой, заливают рассчитанно
количество воды и при работающей мешалке (п = 50—60) засыпаю
порциями рассчитанное количество стифниновой кислоты. К получен
ной суспензии добавляют необходимое количество бикарбоната натри
небольшими порциями во избежание вспенивания. При этом стифни
новая кислота растворяется полностью с образованием стифнат:
натрия. Полученный раствор подкисляют 75 — 80%-ной уксусно!
кислотой, которой на I кг тринитрорезорцина берут'250 мл.
После перемешивания полученный раствор фильтруют через плот
ный фланелевый фильтр при разрежении 400—500 мм рт. ст., подогре
вают до 45—50е и засасывают в мерники.
Приготовление 15%-ного раствора нитрата свинца ничем не отли
чается от описанного в главе об азиде свинца. Раствор после фильтра
ции и подогрева до 45—50° также засасывают в мерники.
Оборудование, применяемое для осаждения стифната свинца, ничеь
не отличается от оборудования для осаждения азида свица. Опера
ция ведется в изолированной кабине, вход в которую во время оса-
ждения должен быть запрещен, все управление процессом осажденш
и наблюдение за ним совершается извне кабины.
В бак осаждения заливают из мерников подогретый раствор ни-
трата свинца, затем при работающей мешалке (п = 35—40) сливакл
250
из мерника подогретый раствор стифната натрия. Для полноты осажде-
ния дают избыток нитрата свинца около 18—20%.
При применении подогретых растворов быстро выпадают желтые
кристаллики ТНРС.
Остальные операции (фильтрация, промывка, флегматизация, гра-
нуляция, сушка и сортировка) ничем не отличаются от соответствую-
щих операций при производстве азида свинца.
Флегматизатором стифната свинца является воск, раствор кото-
рого в бензоле вносят на нутч-фильтр после промывки отсосанного про-
дукта. Меры по технике безопасности производства аналогичны мерам
по технике безопасности при получении азида свинца.
ТНРС сушат в камерных сушилках при 40—50° или в вакуумшка-
фах Пассбурга. При сушке в последних необходимо соблюдать те же
правила работы, что и при сушке гремучей ртути и азида свинца с
одном лишь небольшим, ио существенным изменением: в конце
сушки полки аппарата не охлаждают холодной водой, а медленно
охлаждают сухой продукт, чтобы избежать спекания его в один кусок
при быстром охлаждении.
§ 6. ТЕХНИЧЕСКИЕ ТРЕБОВАНИЯ К СТИФНАТУ СВИНЦА
1. По внешнему виду нефлегматизированный стифнат должен
представлять собой кристаллический порошок без видимых на-глаз
посторонних примесей. Цвет — от светложелтого до темнокоричневого-
Флегматизированный воском стифнат свинца должен представлять
собой однородные сыпучие и не склеивающиеся между собой гранули.
2. Анализ стифната свинца должен давать результаты, уклады-
вающиеся в допуски, приводимые в ОСТ. Ограничивается содержание
влаги и летучих примесей, содержание нерастворимых примесей, со-
держание воска и регламентируется содержание свинца.
Технические условия обычно не регламентируют содержания
в стифнате свинца азота, хотя это было бы наиболее надежным способом
контроля качества.
Контрольные лаборатории применяют следующие методы анализа
стифната свинца:
I. Определение влажности илетучих. Точную
навеску около 3 г нефлегматизированного стифната свинца, помещен-
ную в тарированный бюкс, сушат при температуре 60° до постоянного
веса. Затем бюкс, закрытый пришлифованной пробкой, охлаждают
в эксикаторе над хлоридом кальция и взвешивают. Разница в весе
вещества до и после сушки, выраженная в процентах, дает общее коли-
чество влаги и летучих. В случае испытания флегматизированного
стифната свинца сушку производят при 45° также до постоянного веса.
2. Определение свинца. Во взвешенную платиновую
чашку с матовой внутренней поверхностью помещают навеску ТНРС
•около 0,2 г и растворяют в 120 см315%-иой химически чистой азотной
кислоты при нагревании на водяной бане. По растворении ТНРС рас-
твор еше горячим подвергают электролизу; чашка служит анодом,
251
а катодом — электрод Каблукова. Сила тока 0,3—0,35 А и напря-
жение 4—4,5 V. Электролиз ведут при перемешивании мешалкой
(100 об/мин-). При этом свинец осаждается на платиновую чашку'
в виде двуокиси. Чтобы избежать осаждения свинца в виде металла,]
в электролит добавляют Ю мл 10%-ного раствора сульфата меди. По
истечении Р/г—2 час. содержимое чашки проверяют на полноту осаж-
дения, для чего чашку слегка нагибают и раствору дают соприка- i
саться со стенкой платиновой чашки, еще не покрытой двуокисью
свинца. Если через 20 мин- покрытая раствором поверхность платины
не покроется осадком двуокиси свинца, то осаждение можно считать
законченным. Раствор из чашки сифонируют под током при одновре-
менном и непрерывном прибавлении дистиллированной воды до тех
пор, пока стрелка амперметра не покажет отсутствия тока. Затем
чашку споласкивают дистиллированной водой и несколько раз спир-
том и эфиром. Высушенную при 80° в течение 30 мин. и охлажденную
в эксикаторе над хлористым кальцием чашку взвешивают.
Содержание свинца вычисляют по формуле:
/о™ мРЬО1 - р ’
где а — разность весов чашки с осадком и пустой;
р — навеска ТНРС.
Содержание свинца можно с успехом определять и сульфатным ме-
тодом, для чего в стакан емкостью 150 мл помещают точную навеску
0,6 г ТНРС и обрабатывают 15 мл крепкой серной кислоты. После
этого стакан ставят на песочную баню и выпаривают до выделения
паров серной кислоты. Затем содержимое стакана охлаждают, прили-
вают 75 мл этилового спирта и 75 мл воды и оставляют стоять 12 час.
Раствор фильтруют через беззольный фильтр, последний промывают
холодной водой и несколько раз этиловым спиртом, сушат в течение
1 часа и осадок на фильтре высыпают во взвешенный фарфоровый
тигель. Фильтр сжигают на платиновой проволоке над тиглем, золу
его присоединяют к осадку и все вместе, смочив несколькими каплями
серной кислоты, прокаливают на слабом пламени до постоянного веса.
Тигель с остатком охлаждают в эксикаторе над хлористым кальцием
и взвешивают.
3. Содержание нерастворимых примесей.Точ-
ную навеску okojjo 0,5 г растворяют в 75 мл 25%-ной уксусной кислоты
при нагревании на водяной бане. При этом навеска должна раство-
риться полностью и не должно быть мути или нерастворимого остатка.
При наличии нерастворимого остатка следует проверить его природу
под микроскопом: содержание минеральных примесей не допускается-
В гранулированном воском ТНРС допускается только наличие
следов аморфного остатка воска и резины.
4. Определение флегматизатора (воска). 3 г
стифната свинца обрабатывают в 50 мл безводного бензола и фильтруют
через тройной фильтр в тарированный стаканчик. Фильтр промывают
два раза по 25 мл бензола и ставят для выпаривания Досуха на водя-
252
ную баню. Затем стаканчик с остатком сушат до постоянного веса в
термостате при температуре не выше 60°, охлаждают в эксикаторе и
взвешивают.
Это же определение можно производить и несколько иным мето-
дом. Точную навеску флегматизированного тринитрорезорцината
свинца около 2—3 г помещают в аппарат Сокслета. Воск экстрагируют
безводными бензолом или спиртом. После шести-семи экстракций аппа-
рат разбирают, содержимое колбы выпаривают на водяной бане,
сушат при 80° в течение 3 час. и взвешивают. Процентное содержание
воска определяют по формуле:
lOOjjz
Р ’
где а — разность весов колбы с флегматизатором и пустой;
р — навеска флегматизированного стифната свинца.
5. Определение содержания азота. В кониче-
скую колбу емкостью около 200 мл навешивают 0,15—0.20 г тринитро-
резорцината свинца. Навеску растворяют в смеси спирта и соляной
кислоты, взятых в количестве 20 мл спирта (96°) и 30 мл соляной кис-
лоты (уд. веса 1,19). После растворения тринитрорезорцината свинца
над раствором в течение 15 мин. пропускают углекислый газ. Затем,
не прерывая тока, в колбочку с раствором спускают 50 мл титрован-
ного раствора хлористого олова. Все последующие операции ведут
обязательно в токе углекислого газа. Колбочку с раствором нагре-
вают на водяной бане до полного исчезновения зеленой скраски, после
чего ее снимают с бани и дают охладиться. В охлажденный раствор
приливают I—2 мл приготовленного обычным способом крахмала,
и избыток хлористого олова оттитровывают раствором иода до появ-
ления фиолетовой окраски.
Содержание азота рассчитывают по формуле:
o/N - (5О-У)7Х.Л-^
/о 12 3SnCle • р 9
в которой
SnCJ2 - /fiT,,
Т I . т 1
Jz 1 X.O
где SnCl2 и J-2 — молекулярные веса хлористого олова и иода;
N - атомный вес азота;
Т*.о и Тн — титры растворов хлористого олова и иода;
к{ — число миллилитров раствора иода, пошедшего на
титрование;
р — навеска тринитрорезорцината свинца.
Титрованный раствор хлористого олова приготовляют примерно
1,4%-ный (по олову) с содержанием соляной кислоты 5%. Титр устана-
вливают 0, IN раствором иода в токе углекислого газа,для чего в кониче-
скую колбочку емкостью 150—200 мл спускают 40 мл О,IN раствора
иода и 25 мл соляной кислоты уд. веса 1,19. Над смесью в течение
15 мин. пропускают углекислый газ и затем иод титруют раствором
253
хлористого олова до слабожелтой окраски, после чего к раствору-
прибавляют крахмал и титруют до обесцвечивания- После обесцвечи-
вания титруют обратно по каплям иодом до появления фиолетовой
окраски.
Титр хлористого олова рассчитывают по формуле:
_8пС12-^.Ги
J2 • ’
где |SnCl2, J2 — молекулярные веса;
— число миллилитров раствора иода;
— число миллилитров раствора хлористого олова;
Тх,о, Ти — титры растворов хлористого олова и иода.
Титр раствора хлористого олова устанавливают непосредственно
перед анализом стифната свинца.
Следует отметить, что изложенный метод требует весьма тщатель-
ной и кропотливой работы, а также хорошего навыка в его выпол-
нении.
ГЛАВА VI
ТЕТРАЗЕН
§ 1. ОБЩЕЕ ПОНЯТИЕ О ТЕТРАЗЕНАХ
Под общим названием «тетразеньр> известен целый ряд веществ—
производных непредельного азотоводорода N4H4.
Большинство этих веществ является веществами взрывчатыми и
обладающими в той или иной степени инициирующими (вернее вос-
пламеняющими) свойствами.
К тетразенам относятся, например, следующие соединения'-
1. Гуанилнитрозоаминогуанилтетразен
NH2 /NH —NH —N = O
С — NH С = NH
NH —NH —N — N
2, Гуанилтетразилтетразен
/NH,
N-N C=NH-HjO
II ><4 \
N — NHZ XN = N —NH—NH
3. Диазотетразолсемикарбазид
N - N . x NHa
II >C C = О H.,0
N — NH \
N=N—NH—NH
4. Диазотетразолбензолоаминогуанцдин
/NH3
N — N x C — NH
11 \
N—NH' XN = N —N(NC$H,)
5. Диазотетразолфенилгвдразин
N — N .
H / — N = N — N — NH2
N — NH' |
CeH5
255
Наибольшего внимания заслуживает первый из них, а именно,
гуанилнитрозоаминогуанилтетразен, обычно называемый просто
•«тетразен»-
Впервые это вещество было получено Гофманом и Ротом в 1910 г.
На возможность его практического использования в качестве ини-
циирующего ВВ указал в 1922 г. Ротербург в Германии.
§ 2. ПОЛУЧЕНИЕ ТЕТРАЗЕНА И ЕГО ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Открытие тетразена Гофманом и Ротом произошло, можно cKasaTbJ
случайно. Ими была предпринята попытка диазотировать амино*
гуанидин в присутствии хлорной кислоты. Вместо ожидавшегося
диазогуанидина образовался взрывчатый перхлораттетразен. Гид-
ролиз образовавшегося перхлораттетразена в щелочной среде дал
тетразен.
Они же выяснили, что тетразен может быть получен и при взаимо-1
действии аминогуанидиннитрата с нитритом натрия в нейтральной
среде:
^NH -NH.
2С — NH • HNO3 + 2NaNO2 -+ CcH8ON10 + ЗН2О + 2NaNO3
XNH3
Тетразен также может быть получен при пропускании нитратных
газов через аммиачный раствор нитрата аминогуанидина.
Первые синтезы тетразена были очень длительными. Например,
синтез Гофмана состоял в том, что 5 г аминогуанидиннитрата раство-
ряли в 30 мл воды и при охлаждении до 0° смешивали с раствором
2,5 г нитрита натрия в 15 мл воды. Полученную смесь выдерживали
при температуре около 10° в течение 6—7 дней и при этом получали
выход в 3,2 г.
Позднее методика была значительно усовершенствована и было
установлено, что умеренный избыток нитрита ускоряет реакцию без
понижения выхода.
Ринкенбах и Бертон получали тетразен из солей аминогуанидина
при взаимодействии растворов аминогуанидинсульфата с нитритом
натрия при 30° с прибавлением уксусной кислоты. Продолжитель-
ность реакции сократилась, но тетразен получался загрязненный
желтоватыми примесями. Далее, Ратсбург показал, что реакцию
можно вести и при повышенной температуре в специальной аппара-
туре, снабженной термоизоляцией, чтобы использовать теплоту реак-
ции для ускорения образования тетразена. Он рекомендовал раство-
рить 3 кг аминогуанидинсульфата в 10—20 л воды, нагреть до 25—35°
и смешать с нагретым до той же температуры раствором 1,8—2 кг
нитрита натрия в 6 л воды. По описанию Ратсбурга процесс происхо-
дит без механического перемешивания и выделения газов и закан-
чивается через 3—4 часа. Выход продукта зависит от температуры
реакции и при 35—55° составляет 80% от теоретического. В настоя-
щее время реакция образования тетразена требует не более 2—3 час.
256
Механизм реакции образования тетразена до сих пор не ясен.
Написанное выше уравнение этой реакции является, конечно, пред-
положительным .
Тетразен по внешнему виду представляет собой кристаллический
порошок с желтоватым оттенком в толстом слое. Кристаллы мелкие,
стекловидные, клинообразные. Уд. вес его равен 1,641. Перекристал-
лизованный же из азотной кислоты, т. е. освобожденный от желтого,
сопровождающего технический тетразен, продукта, обладает уд. ве-
сом 1,635. Гравиметрическая плотность 0,45. При прессовании плот-
ность тетразена изменяется так, как показано на фиг. 97, и при дав-
лении 2000 ат становится равной 1,47.
Фиг. 97. Плотность тетразена в зависимости от давления прессования.
Тетразен в обычных условиях хранения является весьма стой-
ким веществом; он негигроскопичен и практически нерастворим
в воде. Гигроскопичность его может быть охарактеризована следую-
щими цифрами: при хранении тетразена при температуре 30° и 90%
относительной влажности происходит увеличение веса только на
0,77%. Растворимость в 100 мл воды составляет только 0,02 г.
^Тетразен также практически нерастворим и в органических рас-
творителях (этиловом и амиловом спиртах, этиловом эфире, ацетоне,
толуоле, бензоле, лигроине, четыреххлористом углероде, хлороформе,
нитробензоле, хлорбензоле, дихлорбензоле, бромбензоле и дихлор-
этилене).
Воздушно-сухой тетразен содержит около 0,4% удаляемых суш-
кой летучих. Нагревание при 50° он выдерживает, но при этом уда-
ляются сопутствующие тетразену летучие. Повышение же темпера-
туры выше э0° влечет за собой дальнейшую потерю в весе. Так, при
нагревании выше 75° тетразен теряет в весе через 10 суток около 8%
и превращается в желтый, но еще взрывчатый порошок. Нагрева-
ние же выше 85° влечет за собой побурение продукта, потерю веса
его в течение 20 суток около 20%. При еще более длительном иагре-
Бубяов—92—17 257
вании при этой температуре он превращается в невзрывчатое ве~ I
щество.
Углекислота в сухом состоянии на тетразен практически не дей- I
ствует. Влажная же углекислота действует на тетразен (не столько I
углекислота, сколько сопутствующая ей влага).
Вода при комнатной температуре на тетразен также не действует. 1
При нагревании в воде разложение тетразена становится заметным 1
только при 50°; при 60° происходит энергичное разложение с виде- |
лением газообразных продуктов.
Гофман установил, что в кипящей воде тетразен разлагается ‘
так же, как и гуанилтетразилтетразен. При этом параллельно про-
текают два процесса: гидролиз и расщепление. Если структурную
формулу тетразена изобразить так:
H2N — С — NH — NH —N = N —С — NH- NH-NO
II II
NH NH
то схематически можно ее переписать следующим образом:
Ri — NH — NH—N—N— R2
и тогда гидролиз можно представить по схеме:
R, -NH - NH — N — N — R2 + Н2ОRj^NHNHg + R2N = NOH
т. е. образуются аминогуанидин и диазооснование нитрозоамино-
гуанидина, которые при нагревании дают гидразин, мочевину, циан,
азот и воду:
/NH - NH2 zNH2
С —NH +Н2О->С = О +NH3 — NH2;
~nh2 \nh2
/NH — NH— NO //N — N
2C = NH ->2HO N = N—C |+2HsO
XN = N - OH XNH-N
2 С II + 4H2O -* 4H2O + C2N2 + 5N2
N
Происходящее в меньшей степени расщепление тетразена можно
представить по схеме:
RiNH— NH - N = N — R2-> RxNH2 + R2N3
258
При этом образуются гидразин и нитрозоаминогуанилазцд:
yNH2
С = NH-J-N3 — С — NH —NH — N — О
которые превращаются в цианамид, аммиак, тетразилазцд и воду:
/NH2 N
С = NH -> С + NH3
XNH2 XNH2
zN - N
N3 — C—NH — NH — N = О -+ Ns —C< || 4-H2O
II NH — N
NH
Реакция образования тетразена из амииогуанидиннитрата сопро-
вождается частичным разрушением образующегося тетразена, уве-
личивающимся по мере повышения температуры. Так как этот про-
цесс обычно проводится в нейтральной (или близкой к нейтральной)
среде, то следует предположить, что здесь . происходят реакции,
наблюдающиеся и при нагревании тетразена с водой, т. е. гидролиз
и расщепление.
С металлами и солями тяжелых металлов тетразен не взаимо-
действует. Однако с хлористым оловом в присутствии соляной кис-
лоты он может образовать тетразилгцдразин.
Тетразен растворяется в умеренно концентрированных водных
растворах щелочей и кислот. Щелочные растворы не стойки. Наобо-
рот, кислотные растворы тетразена в нормальных условиях стойки.
Предполагается, что тетразен в зависимости от условий способен
изомеризоваться и переходить из нитрозосоединения (в нейтральной
среде) в диазосоединение (в щелочной или кислой среде) по схеме:
Rx — NH —NH — N= О Rj-NH — N = N — ОН
Установлено, что тетразен на холоду реагирует с минеральными
кислотами как кислородное основание C2H7N10OH и дает соли типа
СгНДЧщХ, где X — кислотный остаток.
Соли эти обладают свойствами инициирующих ВВ, из них неко-
торые по мощности превосходят самый тетразен, но обладают весьма
крупным недостатком: они способны гидролизоваться при наличии
влаги. Однако замечено, что способность солей тетразена к гидро-
лизу не одинакова и, невидимому, уменьшается в случае образо-
вания двойных солей.
Известны, например, следующие соли тетразена, обладающие инициирующими
свойствами:
I- Перхлорат тетразена C2H7NJOC104, получающийся обработкой тетразена
холодной смесью хлорной кислоты и эфира, кристаллизующийся в бесцветные,
блестящие призмы или остроконечные пирамиды. Он растворим в абсолютном
спирте. Водой легко гидролизуется при комнатной температуре. По взрывчатым
259
свойствам он слабее тетразена. При резком нагревании взрывает; от луча огня
вспыхивает.
2. Нитрат тетразена CEH7NI0NO8> получающийся обработкой тетразена холод-
ной смесью азотной кислоты и эфира, кристаллизующийся в виде игл. Он также
растворим в абсолютном спирте. Водой легко гидролизуется при комнатной тем-
пературе. По взрывчатым свойствам ои слабее перхлората тетразена. При резком
нагревании взрывает; от луча огня вспыхивает.
3. Хлорид тетразена C2H7NIOC1, получающийся растворением тетразена в хо-
лодной дымящейся соляной кислоте с последующим осаждением эфиром. Кри-
сталлизуется в бесцветные блестящие призмы или иглы. Немного растворим в аб-
солютном эфире. Водой легко гидролизуется при комнатной температуре- Взрыв-
чатые свойства меиыие, чем у нитрата.
4- Сульфат тетразена (C2H7N10).,SO4, получающийся встряхиванием тетразена
с 20%-ной серной кислотой. Кристаллизуется в длинные бесцветные иглы. По
взрывчатым свойствам аналогичен нитрату тетразена.
5- Йодид тетразена C2H7NI(J, получающийся растворением тетразена в бес-
цветной иодистоводородной кислоте в атмосфере углекислоты и выделяющийся
эфиром. Кристаллизуется в виде слабожелтых плоских игл. Водой гидролизуется. |
По взрывчатым свойствам превосходит перхлорат тетразена.
6- Периодид тетразена C2H7N1OJS, получающийся из раствора иодида тетра-
зена в концентрированной HJ при стоянии на воздухе. Кристаллизуется в черные
ромбоэдрические кристаллы. Растворим в спирте. Водой не гидролизуется. Обла-
дает слабыми взрывчатыми свойствами.
7. Хлористый периодид тетразена C2H7N1OJ2C1, получающийся растворением
тетразена в дымящейся НС1, с последующей обработкой раствора KJ или дымя-
щейся HJ. Кристаллизуется в зеленые кристаллы, обладающие металлическим
блеском. В спирте растворим, в воде слабо гидролизует. Обладает взрывчатыми
свойствами.
Известны также следующие двойные соли тетразена:
1. Двойная соль кислого сульфата тетразена и сульфата серебра
C2H7N10HSO4 • Ag2SO4, получающаяся при смешивании растворов тетразена
в 50% H2SO4 и сульфата серебра в 20% H2SO4- Соль кристаллизуется в блестящие
иглы, водой не гидролизуется.
2. Двойная серебряная соль тетразена и нитрата серебра
C2HsNJ0OAg- AgNOs • ЗН2О, получающаяся осаждением из азотнокислого раствора
нитрата тетразена избытком водного раствора нитрата серебра. Кристаллизуется в
светложелгые, почти бесцветные, блестящие иглы. Водой почти не гидролизуете»
При разбавлении кислотных растворов водой тетразен выделяется
вновь. На этом свойстве тетразена основана очистка его от приме-
сей, окрашивающих технический продукт в желтый цвет. Очистка
тетразена может быть произведена следующим способом: 6 г техни-
ческого тетразена растворяют при перемешивании в 40 мл азотной
кислоты (68%), полученный раствор выливают в 1500 мл воды, через
некоторое время осаждают совершенно чистый продукт. Аморфно»
желтое вещество, сопровождающее технический продукт, остается
при этом в растворе.
Возможно также осаждение тетразена из кислотных растворов
нейтрализацией их аммиаком, ацетатом натрия или бикарбонатом
натрия.
Нагревание кислотных растворов влечет за собою разложение
тетразена, которое происходит по следующему уравнению:
zNH-NH2 n_n
C2HsON10 -> С = NH + || V — N = N — ОН
\мы N — NH
260
с образован почти коли уравнению: ием аминогуанидина и диазотетразола, причем последний чественно распадается дальше на циан, воду и азот по N — N , 2 || 7C-N =N— ОН -> N —NHZ
N N. * 2 || >С + 2Н,0 -> C2N2 + 5N2 4- 2Н2О N —Nz \ \ N
Таким с кислого ра< уравнением )бразом процесс распада тетразена при нагревании его створа может быть выражен приблизительно следующим /NH - NH2
2С2 H8ONw -+ 2С = NH + СЛ 4- 5N2 4- 2Ц>О ^nh2
Нагрева тетразена, ние щелочных растворов также влечет за собой распад протекающий по реакции:
( /NH2 N .. N :2H«ON„ -> С= NH + || )С — N3 + Н20 \nh2 n-nh/
с образоваь ной среде р шем гуанидина, воды и тетразилазида. Гуанидин в щелоч- >азлатается далее на цианамид и аммиак по реакции: /NH2 С = NH -> NH2—CN4-NHs ^NHo
Вопрос чательного считают: о строении тетразена до сих пор еще не получил окон- ответа. Наиболее вероятной структурной формулой его NH NH —N = N сС NH С = NH XNH2 4NH — NH — N = o
Этой формуле соответствует и его название гуанилнитрозоамиио-
гуанилтетразен. Это соединение рассматривается как производное
гипотетического непредельного азотоводорода—«тетразена»:
NH2 —NH~N —NH
у которого по одному водороду у крайних атомов азотной цепи заме-
щено радикалами — гуанилом и нитрозоаминогуанилом.
261
§ 3. ВЗРЫВЧАТЫЕ СВОЙСТВА ТЕТРАЗЕНА
Реакция взрывчатого превращения тетразена точно неизвестна.
Теплота взрывчатого превращения его Qv порядка 550 кал/кг. Про-
дукты взрывчатого превращения обладают щелочными свойствами.
Объем газов Уо порядка 400—450 л/кг. В насыпанном состоянии в не-
больших количествах тетразен под действием луча огня вспыхивает
без ощутительного звука; количества же его порядка 1—2 г при
воспламенении детонируют. По чувствительности к удару тетразен
несколько превосходит гремучую ртуть. Так, при испытании на
копре в приборчиках Каста при падающем грузе 100 г были установ-
лены следующие минимальные высоты:
для гремучей ртути 12 см
» тетразена - - - ....... 10 »
Пределы чувствительности к удару на копре Велера соответственно—
45 и 65 мм.
Чувствительность тетразена к трению значительно меньше чув-
ствительности гремучей ртути; примесь твердых инертных веществ
(песок) повышает чувствительность тетразена, однако, в меньшей
степени, чем у гремучей ртути.
Чувствительность тетразена к наколу жалом характеризуется
пределами 10 и 15 см при стандартном жале и грузе весом 200 г.
Температура вспышки тетразена 135—140°.
По фугасному действию тетразен несколько превосходит грему-
чую ртуть, а по бризантности уступает ей. Бризантность и ини-
циирующая способность тетразена зависят от давления прессования.
Таблица 56 Зависимость бризантности тетразена от давления Испытания по песочной пробе показывают, как это видно из табл. 56, что с повышением дав- ления прессования бри- зантность его умень- шается. Тетразен обладает
прессования
Навеска тет- разена г Давление прес- сования' ат Вес раздроб- ленного песка г
0,4 0,4 0,4 0,4 0 250 500 3000 13.1 9.2 7,5 2,0 весьма слабой 'иниции- рующей способностью. Для увеличения его инициирующей способ- ности требуется добавка
небольшого количества
стифната свинца, или, лучше всего, гремучей ртути (около 0,2 г при
давлении прессования около 225 кг/см2). Примесь к тетразену окис-
лителей (чтобы повысить содержание кислорода) почти не меняет его
мощности.
Вследствие этого тетразен применяется не в качестве самостоя-
тельного инициирующего ВВ, а только преимущественно в качестве
сенсибилизирующей добавки в неоржавляющих, содержащих стифнат
свинца, ударных составах.
262
§ 4. ОСНОВНЫЕ ИСХОДНЫЕ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ
ДЛЯ ПОЛУЧЕНИЯ ТЕТРАЗЕНА
Из предыдущего ясно, что для производства тетразена необхо-
димы: нитрат аминогуанидина и нитрит натрия. Последний изготов-
ляется на стороне; первый же производится на самом заводе из
аминогуа нидинбикарбоната.
Аминогуанидинбикарбонат может быть получен различными спо-
собами;
1, Химическим или электролитическим восстановлением нитрогуа-
нидина:
/NH • NOa /NH • NH2
С = NH 4- 6Н -> С = NH 4- 2Н2О
XNH2 ^NHa
2. Конденсацией гидразина и цианамида
.„N /NHNH2
С 4- HoN - NH> -> С = NH
XNHa XNH2
3. Конденсацией метилизотиомочевины с гидразином
/S —СН8 /NH-NH3
С — NH 4- HaN - NH2 -> С = NH 4- HSCH8
xnh2 xnh2
Таким образом в зависимости от способа синтеза аминогуанидин-
бикарбоната применяются разные исходные материалы. Из приве-
денных выше способов синтеза, конечно, заслуживает внимания вто-
рой, т. е. синтез из гидразина и цианамида, как наиболее простой,
продуктивный и требующий наличия только гидразинсульфата и
кальцийцианамида. Однако в литературе мы не встретили вполне
достоверных данных об этом процессе производства.
Химическое восстановление нитрогуанидина осуществляют в кис-
лой среде цинковой пылью или в щелочной среде алюминиевой пуд-
рой. Электролитическое восстановление нитрогуанидина требует не
только соответствующего католита (обычно серная кислота), но и
хороню подобранных по материалу электродов.
Рассмотрим теперь свойства основных исходных продуктов и
полуфабрикатов тетразена.
Нитрогуанидин получается обычной дегидратацией нит-
рата гуанидина концентрированной серной кислотой
/NH2 • HNO3 /NH - N02
С = NH -> H2O + С = NH
XNH2 'no2
On представляет собой иглообразные кристаллы белого цвета,
плавящиеся при температуре 230“ с разложением. Нитрогуанцдин
263
негигроскопичен и весьма мало растворим в холодной воде (0,3%
при 20°) и в этиловом спирте; в эфире же совершенно нерастворим.
Таблица 57 В в0дн0м Растворе гад-
рата окиси калия рас-
творяется на холоду
Растворимость нитрогуанидина в HeS04
Нитрогуанидин в г на
100 мл раствора
rf % при 0° при 25°
1,12 17,0 0,447 0,700
I,l3 18,3 0,495 0,920
1,15 20,9 0,664 1,280
1,18 24,8 0,755 1,800
1,22 29,8 1,506 2,714
1.26 34,6 5,421
1,349 44,7 6,956 11,129
Z—вриО°; II—при 25е.
рогуанидина дает слабо кислую реакцию,
чить, что соединение это содержит ациформу:
с поглощением тепла
и может быть вновь
осажден действием СО2.
В концентрированных
минеральных кислотах
растворяется легко и
из них может быть осаж-
ден водой.
Растворимость нит-
рогуанидина в серной
кислоте была определе-
на Иве ном и Юнгом в
1921 г. и Девисом в
1922 г. Результаты ра-
боты последнего даны в
табл. 57 и на фиг. 98.
Горячей серной кис-
лотой нитрогуанидин
разлагается по реакции
NH2-C(NH) —
— nhno2+ Н3О ->
N2O + СО2 + 2NHS
Качественной реакцией
на нитрогуанидин слу-
жит цинковая пыль и
FeSO4, прибавляемые к
щелочному раствору
нитрогуанидина и вызы-
вающие интенсивно-
кровавое окрашивание
вследствие образования
железистого соедине-
ния нитрозогуанидина.
Водный раствор нит-
из чего можно заклю-
,N = N^°
С = NH 0Н
'Nli2
264
Щелочи медленно разлагают нитрогуанидин даже на холоду.
Нитрогуанидин взрывчат; реакция его взрывчатого превращения
CeH4N4O2 -> СО + Н2О + Н3 + 2N2.
При инициировании капсюлем-детонатором, снаряженным 1,5 г гре-
мучей ртути, дает полную детонацию. Взрыв 40 г нитрогуанидина
дает обжатие свинцовых столбиков диаметром 67 мм, равное 7 лои;
мелинит в тех же условиях дает 10,5 мм. Температура взрыва нитро-
гуанидина близка к 1900°. Однако самостоятельным ВВ он служить
не может; рекомендуется его использование лишь в качестве добавки
к суррогатным ВВ и нитроглицериновым порохам для понижения
температуры взрыва. Следует упомянуть, что во Франции нитрогуа-
нидин в смеси с тетрилом применяют также для снаряжения детони-
рующих шнуров.
Нитрогуанидин получается непосредственной нитрацией сырого
роданистого гуанидина и из гуанидин нитрата. Получение его по
методу Тиле заключается в том, что сырой роданистый гуанидин
обрабатывают серной кислотой (d — 1,84) и этим самым переводят
его в гуанидинсульфат, после чего к раствору прибавляют некоторое
количество олеума с содержанием 10% SO3. Затем, к охлажденному
раствору приливают азотную кислоту (d = 1,5), и как только на
поверхности раствора появляются пузырьки газа (начало полного
разложения нитрогуанидина), раствор выливают в холодную воду,
из которой сразу же выпадает нитрогуанидин. Метод нерентабелен
и требует большого расхода крепких кислот. Ивен и Юнг получали
нитрогуанидин тремя методами:
1) действием серной кислоты на гуанидйннитрат;
2) действием азотной кислоты на гуанидиннитрат;
3) действием смеси H2SO4 и HNO3 на гуанидиннитрат.
Лучшим методом является первый, т. е. метод дегидратации
гуанидиннитрата серной кислотой; сущность его состоит в сле-
дующем.
Высушенный и хорошо измельченный гуанидиннитрат присыпают
при перемешивании (обязательно небольшими порциями) к концен-
трированной серной кислоте. После введения всего гуанидиннитрата
раствор оставляют стоять в течение некоторого времени при ком-
натной температуре, а затем выливают в ледяную воду. Воды надо
брать столько, чтобы в конце слива раствора образовалась не более,
чем 20%-ная серная кислота. Нитрогуанидин выпадает в виде кри-
сталлов белого цвета. Выход около 80—85%.
ZNH - NH2
Аминогуанидин. Аминогуанидин С—NH в свобод-
XNH2
ном состоянии представляет собой сильное основание, однако, не-
прочное, легко разлагающееся. Соли же аминогуанидина — прочные
вещества, обладающие ядовитыми свойствами.
265
При гидролизе аминогуанидин расщепляется на гидразин, аммиак
и угольную кислоту:
/NH — NH2
С — NH -> со2 + NHa - NH2 4- 2NH3
4NH2
Действие азотистой кислоты на аминогуанидин различно и в зна-
чительной степени зависит от той среды, в которой происходит диа-
зотирование.
Так, азотистая кислота в присутствии минеральной кислоты пере-
водит его в соль гуанилазида (азидоформамидина):
/NH — NH2 /N3
C = NH с = NH
'4NH., ^NIL,
•с последующим, возможным, превращением в аминотетразол:
yN- N
h2n - с ||
'4NH—N
а при действии щелочей — в азид щелочного металла.
Та же азотистая кислота в отсутствии минеральных кислот обра-
зует тетразен, причем здесь протекает реакция не диазотирования,
а только нитрозирования:
/NH — NH2
2 С = NH2 + 2HNO2 -> C2H5ON10 + Н2О
XNH=
Наконец, в уксуснокислом растворе действие азотистой кислоты
приводит к иным результатам. В этом случае образуется диазоамидо-
тетразол, что может быть объяснено следующим образом. Вначале
реакция протекает, как и при наличии минеральных кислот, с обра-
зованием уксуснокислой соли гуанилазида. Эта соль гуанилазида
неустойчива и подобно свободному основанию превращается в амино-
тетразол. Затем происходит взаимодействие со второй молекулой
азотистой кислоты, образуется диазосоединение, которое с неизме-
нившейся молекулой аминотетразола дает диазоамидосоединение:
.NH—N мн— N
H2N — С< || -> НО — N = N — II -+
х N — N <N — N
N-NH NH — N
-ь II >C-NH — N = N-C< II
N —N ? — N
266
Нитрат аминогуанидина может подвергаться окислению в азотно-
кислом растворе, особенно в присутствии перманганата калия, что
ведет к образованию азодикарбондиамидина:
HN ,NH
- N = N - C<f
H2NZ 'NIL,
Реакцию образования азодикарбондиамидина, очевидно, можно представить
следующим образом: вначале образуется производное дигидротетразена, которое
отдает азот с образованием гидразодикарбондиамидина, а последний под влия-
нием окислителей переходит в азосоединение:
HN. HN х
2 ^С —NH-NH2-> ^С —NH —N = N-NH-C<
иж h2nz XNH2
HN .NH
-> >C —NH —NH-C^ ->
H2NZ xNH2
HN. ,NH
-> 7-CN = NC<
ILJV XNH2
Бикарбонат аминогуанидина весьма устойчив в
обычных условиях хранения, легко превращается в любую соль
аминогуанидина непосредственной обработкой соответствующей кис-
лотой. Он представляет собой белый порошок с температурой плав-
ления 166—167°. Применяющийся в производстве бикарбонат должен
обладать именно этой температурой плавления, не содержать в себе
комков и не иметь запаха аммиака.
Нитрит натрия, идущий на производство тетразена, должен
по внешнему!виду представлять собой кристаллы белого цвета с желто-
ватым оттенком. По чистоте он должен соответствовать ОСТ 8880/2323
и содержать:
азотистокислого натрия, в расчете
иа сухое вещество, не меиее............ 98%
влаги не более......................... Зуо
нитрата натрия не более 1,5%
нерастворимого в воде остатка на более 0,1%
§ 5. ФАБРИКАЦИЯ ТЕТРАЗЕНА
Производство тетразена слагается из двух отдельных операций:
1) получение аминогуанидинбикарбоната,
2) получение тетразена.
П олучение аминогуанидинбикарбоната. Ныне
применяемые способы получения аминогуанидина сводятся на прак-
тике к восстановлению нитрогуанидина химическим или электро-
литическим путем.
267
Следует иметь в виду, что нитрогуанидин является алифатиче-
ским читроамином, восстановление которого связано с возникнове-
нием непрочных промежуточных соединений.
Из продуктов его восстановления были изолированы и изучены
нитрозогуанидин, аминогуанидин и гуанидин:
/NH-NO2 /NH-NO /NH-NH2 /NH3
C = NH -> C----NH -> С = NH ->C = NH
\NH2 ^NHg \чнз ^NHj
Смиту и Сабетти удалось, видимо, кроме этих хорошо изученных
продуктов выделить и промежуточное соединение состава:
/NHNO /NHNO2
2С - NH • ЗС = NH
xnh2 xnh2
Наряду с этими продуктами известен еще из работ Тиле продукт
взаимодействия нитрозогуанидина с аминогуанидином:
/NH-NO
С = NH
^nh2
zNH-NH2 zNH — N = N--zNH
+ С = NH -> C NH C = NH 4- H20
'4nh2 \nh2 \nh2
Этот так называемый бигуанилтетразен нестоек и легко переходит
в прочный гидразобикарбонамидин:
/NH —N = N —/NH /NH-/NH
C = NH C = NH -> C = NH C=NH + NS
'XNII2 'NIL, "^NHj \mH2
Гидразобикарбонамидин дает с минеральными кислотами хорошо
кристаллизующиеся соли. Он может быть получен также взаимо-
действием 2 мол нитрозогуанидина с 1 мол гидразина, из чего можно
заключить, что слабо щелочная восстановительная среда благоприят-
ствует возникновению этого вещества.
Нитрозогуанидин — первый промежуточный продукт восстанов-
ления — имеет амфотерную природу и в зависимости от среды спо-
собен изомеризоваться по схеме
NH • NO (щелочная
среда)
C = NH
^NHa
(кислая
среда)
/NH2
C = NH
\n =n —он
Переход от нитро- к нитрозогуанидину также обратим.
268
Как нитрогуанидин, так и продукты его восстановления являются
мало прочными веществами по отношению к гидролизующим агентам
и легко разлагаются по схемам:
/NH —NO2
C = NH +Н2О -> N2O4-СО2 + 2NH3
4 NH2
/NH • NO /NHg
C — NH + H2O -> HNO2 + C = NH
^NH2 ^NHa
/NH—NH2
C = NH 4-2H2O -> CO2 4- H3N — NH2 4- 2NH3
XNH2
Таким образом продолжительное пребывание этих продуктов
в кислых и щелочных растворах (особенно при повышенных темпера-
турах) неизбежно ведет к частичному их разрушению.
Восстановление нитрогуанидина в кислой среде осуществляется
в уксуснокислом растворе, металлическим пылевидным цинком или
активированной медноцинковой пылью. Сущность этого процесса
состоит в том, что 150 г нитрогуанидина и 500 г цинковой пыли сме-
шивают с 1000 г льда, а затем обрабатывают 180 г 50%-ной уксусной
кислоты. Водород in statu nascendi восстанавливает нитрогуанидин.
Продолжительность реакции восстановления около 40—50 мин.,
конечная температура 45°. От прореагировавшей смеси отфильтро-
вывают осадок, фильтрат упаривают и обрабатывают бикарбонатом
натрия, а осадок аминогуанидинбикарбоната отделяют от раствора
и высушивают.
Сущность восстановления в щелочной среде состоит в том, что
к суспензии нитрогуанидина в воде приливают 25%-ный раствор
аммиака, присыпают хлористый аммоний и, наконец, медленно при-
бавляют смоченную спиртом алюминиевую пудру при энергичном
перемешивании реагирующей смеси. Температура реакции не должна
превышать 45°; регулирование производят льдом, прибавляемым при
подъеме температуры выше 45°. Время реакции 2—4 часа.
Конец реакции устанавливают пробой на содержание нитрозо-
соединения при помощи сульфата железа (II). Отсутствие зеленого
окрашивания показывает конец реакции.
Прореагировавшую смесь охлаждают до 30°, затем отфильтро-
вывают осадок гидроксида алюминия и избыток алюминиевой пудры,
а фильтрат упаривают до V4—-1/5 объема. Далее теплый фильтрат
обрабатывают бикарбонатом натрия и образовавшийся аминогуани-
динбикарбонат после промывки водой и спиртом сушат.
Теория и практика электролитического восстановления органи-
ческих нитросоединений изложена в общеизвестных руководствах
по химии. Совершенно ясно, что решающим фактором для хорошего
269
выхода при электролитическом восстановлении является католит и
материал катода. Кроме этого фактора в практике следует учитывать,
конечно, и расход тока и энергии, которые обычно выражаются от-
ношением теоретически необходимых количеств тока и энергии для
получения данного количества вещества к практически затрачивае-
мым току и энергии.
Восстановление алифатических нитроаминов до соответствующих
гидразинов удачно протекает обычно при помощи оловянных и свин-
цовых катодов. В качестве кислых католитов пользуются разбавлен-
ной H2SO4 или смесью H2SO4 и СН3СООН. При наличии нейтральных
католитов (например смесь СН8СООН и CH3COONa) или щелочных
(Na2COs) восстановление протекает также удачно.
Что же касается нитрогуанидина, то его восстановление в 2%-ном
спиртовом или водном растворе H2SO4 с свинцовым катодом при
температуре не выше 20° удавалось с выходом аминогуанидина до
30—40%. При восстановлении с ртутным, никелевым или платиновым
катодами в H2SO4 и СН3СООН и свинцовым катодом в €H3COONa
получаются конечные продукты восстановления — гуанидин и ам-
миак.
По Берингеру, при электролизе с оловянным катодом в H2SO4
при t = 8—10° и плотности тока 250 A/jw2 выход аминогуанидина
достигает 81%. Хорошие выходы были также получены Брокма-
ном при: применении катода, покрытого оловянной чернью (выход
около 74%).
Недостаток электролиза с оловянным катодом состоит в том,
что олово значительно корродирует.
Так как вопрос о выборе материала катода окончательно не ре-
шен, то, конечно, заслуживают внимания и требуют проверки све-
дения, касающиеся электролитического восстановления аналога нитро-
гуанидина — нитромочевины. Судя по литературным данным, вос-
становление последней возможно при помощи железного катода в рас-
творе хлорида натрия, оловянного катода в растворе серной кислоты,
свинцового катода в соляной кислоте и медных, никелевых, свин-
цовых и ртутных катодов — в растворах соляной и серной кислот.
Получение тетразена. Для получения тетразена необ-
ходимо нерастворимый в воде аминогуанидинбикарбонат перевести
в растворимую соль аминогуанидина. Обычно переводят его в амино-
гуанидиннитрат. Для этого к рассчитанному количеству азотной
кислоты уд. веса 1,35 небольшими порциями присыпают эквива-
лентное количество аминогуанидинбикарбоната при нагревании. В ре-
зультате реакции:
/NH — NH2 /NH- NH2
С = NH . H2COS + HNO3 -> С = NH • HNO3 dr H2O 4-CO2
'xnh2 4'h.
образуется раствор аминогуанидиннитрата, насыщенный двуокисью
углерода. Для удаления последней раствор нагревают до 85—90°.
270
Освобожденный от двуокиси углерода раствор нейтрализуют гидро-
ксидом аммония (d=0,91), фильтруют и охлаждают до 50—55°. За-
тем к раствору приливают несколько капель азотной кислоты.
Одновременно с этим готовят раствор 1,5 мол нитрита натрия
на 1 мол аминогуанидиннитрата (исходя из расчета 100 г на 160 мл
воды). Растворение ведут при повышенной температуре (50—55°),
а затем раствор фильтруют.
Нагретые до 50—55° растворы быстро сливают вместе в алюми-
ниевый бак осаждения с работающей мешалкой. Через несколько
минут начинается реакция образования тетразена:
ZNH— NHZ
2С = NH HNO3 + 2NaNO3 C2H8ON10 + 2NaNO3 + 3H2O
XNH2
Реакция экзотермична. Допустимая максимальная температура
реакционной массы 55—58°. Температуру регулируют льдом или
ледяной водой. Время реакции 80—90 мин., после чего реакционную
массу охлаждают, тетразен отфильтровывают на нутч-фильтре, про-
мывают несколько раз водой, по одному разу спиртом и эфиром и
сушат (провяливают) при комнатной температуре.
На выход и чистот продукта влияет продолжительность реакции,
причем решающее значение имеет первый час реакции, в течение
которого должно выпасть не менее 85—90% тетразена. Дальнейший
ход процесса протекает медленно.
Наилучшие результаты, как правило, получаются тогда, когда
удается провести реакцию получения исключительно за счет теплоты
реакции, без дополнительного подогрева. Стало быть, температурный
режим является вторым решающим фактором.
Маточный раствор содержит кроме NaNO3 и NaNO2 целый ряд
органических соединений, являющихся продуктами побочных реак-
ций, зависящих от условий ведения процесса диазотирования амино-
гуанидина. Если выпарить досуха маточный раствор, то остается
желтоватый осадок, слабо вспыхивающий от луча огня.
Спиртовая вытяжка сухого остатка при удалении спирта дает
кристаллы двух видов: призматические желтоватого цвета и бесцвет-
ные иглы. При нагревании этот остаток сильно взрывает.
ГЛАВА VI I
ДРУГИЕ ИНИЦИИРУЮЩИЕ ВЗРЫВЧАТЫЕ ВЕЩЕСТВА
Применяемые в практике инициирующие ВВ обладают некоторыми
недостатками технического или экономического порядка. Так, напри-
мер, гремучая ртуть обладает следующими недостатками:
1. Незначительная инициирующая способность (для детонации
1 г тротила необходимо 0,36 г гремучей ртути).
2. Недостаточная стойкость (гремучая ртуть начинает разлагаться
при температурах порядка 60°).
3. Способность подвергаться перепрессовке при давлениях выше
-500 кг на 1 см2.
4. Неоднородность кристаллов по величине.
5. Относительная дороговизна.
Азид свинца наряду с положительными его качествами по срав-
нению с гремучей ртутью [как то: большая стойкость (разлагается I
при температурах много выше 100°), отсутствие способности пере-
прессовываться и высокая инициирующая способность] обладает
следующими недостатками:
L Пониженная чувствительность к воспламенению, вследствие |
чего необходимо добавлять к нему дополнительный воспламенитель.
2. Пониженная чувствительность к наколу жала, что затрудняет 1
применение его в капсюлях-детонаторах, действующих от накола,
-без дополнительных ударных составов.
3. Возможность образования слишком чувствительных больших
кристаллов игольчатой формы.
4. Сложность технологического процесса.
Эти недостатки заставляют продолжать и далее работу по изы-
•сканию новых инициирующих ВВ. Так как инициирующие ВВ в на-
стоящее время употребляются, главным образом, в качестве пер-
вичных зарядов для капсюлей и, следовательно, требуются в относи-
тельно небольшом количестве, то экономические соображения не
имеют такого значения, как в случае бризантных веществ и порохов.
Поэтому, хотя стоимость большинства предложенных в последние
годы инициирующих веществ сравнительно высока, все же остано-
вимся вкратце на некоторых из них.
:272
§ 1. НЕОРГАНИЧЕСКИЕ ПРОИЗВОДНЫЕ ГРЕМУЧЕЙ КИСЛОТЫ
Из солей гремучей кислоты в настоящее время известны и в той
или иной степени изучены только немногие. Обычный алкогольно-
азотнокислый способ получения фульминатов применим только для
так называемых благородных металлов (Hg, Ag, Au). Этим способом
не могут быть получены фульминаты неблагородных металлов.
Поэтому долгое время были известны только соли ртути и серебра.
Простым замещением ртути в гремучей ртути металлом с более
высоким потенциалом также не удается получить новых фульминатов
[исключение составляет образование основного фульмината меди
Cu(ONC>, • Си(ОН)3]. Также не удается замещение ртути при обмен-
ном разложении гремучей ртути с солями вследствие большой склон-
ности гремучей кислоты к образованию комплексных соединений.
Гремучее серебро (фульминат серебра) впервые было
получено Бруньятелли в 1802 г. и аналогично гремучей ртути пред-
ставляет собой серебряную соль гремучей кислоты:
Ag — О — N - С
Надо иметь в виду, что под гремучим серебром понимают иногда и другое сое-
динение, получающееся в виде черного порошка при растворении свежеосажденной
окиси серебра в концентрированном водном растворе аммиака. Это соединение
было получено впервые Бертолле в 1788 г. выпариванием аммиачного раствора
нитрата серебра- Препарат отличается чрезвычайной чувствительностью к нагреву,
удару и трению, так что при выпаривании при 80- 90° аммиачного раствора окиси
серебра могут происходить взрывы из-за образования кристаллов бертолетова
«•гремучего серебра».
Никакого отношения к фульминату серебра это соединение не имеет; Рашит
приписывает ему формулу нитрида серебра NAg3-
Фульминат серебра одно время применялся наряду с гремучей
ртутью для инициирования вторичных зарядов; однако вследствие
большой чувствительности, особенно к трению, применение его было
приостановлено. В 1906 г. в Англии (англ. пат. № 13983) предлагали
применять его в количестве 10% в качестве добавки к гремучей ртути
в капсюлях для увеличения их инициирующей способности. Однако
и это предложение не было осуществлено вследствие высокой стои-
мости гремучего серебра. Зато в малых количествах оно служит
иногда для пиротехнических целей (хлопушки, Knallbombons и т. п.).
Получение гремучего серебра аналогично получению гремучей
ртути. Исходным продуктом служит чистопробное серебро или ни-
трат серебра. Вследствие меньшей растворимости нитрата серебра
в азотной кислоте при получении гремучего серебра приходится
брать относительно большее количество азотной кислоты и спирта,
чем при получении гремучей ртути.
Говард получал гремучее серебро, исходя из следующей рецеп-
туры: он растворял 40 гран (1 гран 0,6 г) серебра в 2 унциях
(1 унция ^ 30 ?) концентрированной, разбавленной равным объемом
воды азотной кислоты, и приливал 2 унции спирта.
По Нефу, для получения гремучего серебра растворяют 5 г се-
ребра в 100 мл азотной кислоты (d = 1,34), выливают горячий рас-
твор в 150 мл 90%-ного спирта и нагревают в течение 5—10 мин.
Бубнов—92—18 273
По Ге-Люссаку и Либиху, растворяют 1 ч. серебра в 20 ч. азот
ной кислоты (d = 1,36— 1,38), приливают полученный раствор
к 27 ч. 85—90%-ного спирта и осторожно нагревают до кипения
Из раствора выделяются иглы гремучего серебра.
Получение гремучего серебра в лаборатории рекомендуется одни»
из следующих методов:
1. В колбочке емкостью 100 мл растворяют при слабом нагре-
вании на водяной бане 5 г чистопробного серебра в 50 мл азотно!
кислоты 1,38—1,40, причем горло колбочки прикрывают часовыд
стеклом, чтобы из колбочки по возможности не уходили наружу
бурые окислы азота. Серебро растворяется в азотной кислоте до-
вольно быстро; однако к концу реакции происходит частичное вы-
деление нитрата серебра, для растворения которого добавляют
в колбу 10 мл воды и при встряхивании нагревают на водяной бан(
до исчезновения осадка. Еще теплый раствор приливают быстро
к 70 мл 96%-ного этилового спирта, помещенного в эрленмейеров-
скую колбу емкостью около 600 мл.
Реакция в большинстве случаев начинается самопроизвольно
Если, однако, реакция не идет вовсе или идет медленно, то реакцион-
ную смесь нагревают на водяной бане до начала появления первых
пузырьков газа.
Жидкость вскоре начинает бурно кипеть, сильно пенится, и гре-
мучее серебри выделяется в виде блестящих белых, очень мелкиз
тяжелых кристаллов. Для прекращения дальнейшего вспенивания
жидкости, протекающего толчками, колбу ставят в воду. Химизм
процесса образования гремучего серебра аналогичен химизму обра-
зования гремучей ртути.
2. Вместо чистого металлического серебра берут более доступный
нитрат серебра в количестве 5 г, растворяют в 5 мл горячей воды,
добавляют 35 мл азотной кислоты уд. веса 1,4, нагревают на водяной
бане до полного растворения выделившегося нитрата серебра и теп-
лый раствор выливают в 50 мл спирта. Так как при этом способе
получения гремучего серебра не образуется окислов азота, то для
возбуждения реакции в раствор добавляют 0,5 г нитрита натрия
и подогревают раствор на водяной бане.
При обоих методах получения содержимое колбы после охла-
ждения отсасывают на небольшом нутч-фильтре или на простой во-
ронке, гремучее серебро хорошо промывают холодной водой и вы-
сушивают в термостате при температуре 50° в течение 6—7 час.
При работе с гремучим серебром следует помнить, что оно чув-
ствительнее -гремучей ртути. Поэтому осторожное обращение с гре-
мучим серебром есть одно из основных условий безопасности работы.
При фильтровании на нутч-фильтре целесообразно пользоваться
двойным или тройным круглым фильтром, который в виде мешочка
вкладывается в нутч-фильтр, чем предотвращается возможность со-
прикосновения осадка со стенками фарфоровой воронки.
Изложенные способы получения гремучего серебра являются по
сути дела вариантами одного и того же способа. Кроме них суще-
274
ствует ряд совершенно иных методов получения гремучего серебра,
аналогичных методам синтеза гремучей ртути (например из хло-
ридформилоксима С1НС = NOH, из метилнитроловой кислоты
HC(NO2)=^NOH и др.). Оригинальным способом получения гремучего
серебра является его синтез действием окислов азота (образующихся
восстановлением азотной кислоты, мышьяковистым ангидридом
As2Os или крахмалом) на спиртовой раствор нитрата серебра.
Гремучее серебро имеет вид белых блестящих шелковистых игл
или их сростков, уд. веса 4,09. Гремучее серебро плохо растворимо
в воде. Так, в 1 л воды растворяется:
при 13° 0,075 г
» 30° • 0,18 »
В кипящей воде растворимость заметно повышается (до 2,8%). Из
водных растворов выкристаллизовывается совершенно чистый про'
дукт.
Растворимость гремучего серебра в гликокольаминах значительно
больше. Оно легко растворяется в моно-, ди- и триэтаноламинах,
из которых может быть осаждено только кислотами в виде мелко-
кристаллического порошка, чистотой 98,7—98,9%.
В азотной кислоте гремучее серебро нерастворимо. К химическим
реагентам гремучее серебро относится так же, как гремучая ртуть.
Только по отношению к концентрированной серной кислоте оно
обладает повышенной чувствительностью — малейшая капля кислоты
вызывает взрыв гремучего серебра.
Гремучее серебро отличается значительной стойкостью. Оно мало
гигроскопично. При обыкновенной температуре не изменяется (при
нагревании в течение 60 дней при температуре 50° никаких призна-
ков разложения не было замечено). Его можно хранить под водою
весьма долго. По опытам Петера, гремучее серебро, хранившееся
под водой в течение 37 лет, нисколько не изменилось.
Как и все соли серебра, гремучее серебро на воздухе и на свету
темнеет с поверхности, однако, признаков разложения при этом не
замечается. *
Температура вспышки гремучего серебра около 170°. Чувстви-
тельность к удару зависит от величины кристаллов. В литературе
по вопросу о чувствительности к удару гремучего серебра приво-
дятся противоречивые данные. Одни утверждают, что, находясь
между двумя твердыми телами, гремучее серебро взрывает от самого
слабого удара, даже под водой, другие, наоборот, отмечают, что
гремучее серебро менее чувствительно, чем гремучая ртуть. Так,
Тейлор приводит следующие данные: запрессованные под давлением
500 ат навески гремучей ртути и гремучего серебра взрывают при
падении груза с высоты:
для Hg(ONC)z - .127 мм
» AgONC . . . - 327 »
Сухое гремучее серебро рекомендуется размешивать деревянными
шпателями на мягкой бумаге, так как оно очень чувствительно и
к трению.
275
В отличие от гремучей ртути гремучее серебро с содержанием
больших количеств влаги не теряет своей восприимчивости к дето-
Таблица 58
Предельные инициирующие заряды
Инициатор Детонатор
тнт ТНФ тетрил
Pb(N3)3 . 0,09 0.025 0,025
AgONC . 0,095 0.05 0,02
Hg(ONC)s 0.36 0,30 0,29
лочке капсюля-детонатора № 8 показан в
нации и, так же как и
азид свинца, взрывает
и под водой.
По инициирующей
способности гремучее
серебро значительно
превосходит гремучую
ртуть и равно примерно
азиду свинца. Так, пре-
дельный инициирующий
заряд их по 0,5 г вто-
ричного заряда в обо-
табл. 58.
Тейлор при подрыве 0,4 г тротила в оболочках капсюля-дето-
натора № 6 определил следующие предельные инициирующие заряды:
для Hg(ONC)s без чашечки . 0,30 г
то же с чашечкой ................. 0,24 »
для AgONC без чашечки 0,14 >'
то же с чашечкой ....... 0,07 »
Гремучее серебро образует с рядом других металлов двойные
соли, как, например: NH4[AgONC], Na[AgONC], K[AgONC] и т. п.
Так же как н гремучая ртуть, AgONC образует с пиридином легко
разлагающееся соединение типа AgONC • C6HSN.
Фульминат натрия был получен Эренбергом в 1886 г.
действием натриевой амальгамы на гремучую ртуть в присутствии
Н2О. Его состав соответствовал формуле: CNONa - Н2О. Безводный
фульминат получался при долгом стоянии над серной кислотой.
Такой способ получения безводного фульмината небезопасен, так как
при испарении в эксикаторе водного раствора фульмината натрия
могут происходить взрывы. Кроме того, водный раствор фульмината
натрия нестоек и уже по истечении I часа желтеет.
Велер, имея целью более удобное и безопасное получение безвод-
ного фульмината натрия, действовал на гремучую ртуть 8%-ной
амальгамой натрия в присутствии абсолютного спирта. Через 30 мин.—
1 час выделялся кристаллический безводный фульминат натрия,
который Велер промывал абсолютным спиртом для удаления алко-
голята натрия, отделял от тяжелой металлической ртути, промывал
эфиром и высушивал.
При растворении в воде фульминат натрия гидролизует, но
меньше, чем цианистый калий.
Фульминат натрия представляет собой тонкие бесцветные иглы.
С сернокислым железом дает железистогремучий натрий:
NaJFe(CNO)c]. 18Н2О
соль формально соответствует железистосинеродистому натрию. Тем-
пература вспышки фульмината натрия 215°.
276
Фульминат таллия T10NC впервые был получен дей-
ствием гремучей ртути на стружки металлического таллия. Однако
вследствие гидролиза его извлечение водой трудно выполнимо- Полу-
чают фульминат таллия поэтому следующим способом: встряхивают
в темном сосуде в продолжение 24 час. в атмосфере водорода и в при-
сутствии абсолютного спирта 100 г 10%-ной амальгамы таллия
с 1—2 г гремучей ртути. Фульминат таллия представляет собой белые
кристаллы в виде призматических игл, на свету окрашивающихся
в желтый цвет. На воздухе продукт буреет и постепенно лишается
взрывчатых свойств; чувствительность к удару у него больше, чем
у гремучей ртути; температура вспышки 120°; при длительном на-
гревании он превращается в невзрывчатый порошок; недостаточно
стоек также и к химическим реагентам: даже азотная кислота способна
вызвать взрыв его. Инициирующая способность его меньше, чем
у гремучей ртути.
Фульминат кадмия Cd(ONC)3 получается взаимодей-
ствием 2 г гремучей ртути с 30 мл безводного метанола при встряхи-
вании и ледяном охлаждении в продолжение 25 мин. с 20%-ной амаль-
гамой кадмия. Спиртовой раствор фульмината фильтруют в токе
водорода и высушивают при 55° над фосфорным ангидридом. Фуль-
минат кадмия представляет собой белый кристаллический порошок,
хорошо растворимый в этиловом и метиловом спиртах. В воде он
также хорошо растворим, однако при этом гидролизует и абсорби-
рует углекислоту. Чувствительность его к удару аналогична чув-
ствительности гремучей ртути. Температура вспышки 215°. Ини-
циирующая способность очень высока: предельный заряд его:
по тетрилу 0,008 <
по пикриновой кислоте - 0,05 >
по тротилу...................... . 0,11 *>
по тринитроанизолу л,26 »
Фульминат закисной меди Cu2(ONC)2 получается
продолжительным встряхиванием под водой в атмосфере водорода
2 г гремучего серебра с 12—20 мл пластичной медной амальгамы.
Фульминат закисной меди представляет собой светлосерый с зелено-
ватым оттенком кристаллический порошок, легко окисляющийся на
воздухе, но стойкий в сухом состоянии. В воде нерастворим и почти
не подвержен гидролитическому расщеплению. Температура вспышки
205°. К удару он несколько менее чувствителен, чем гремучая ртуть.
Инициирующая способность фульмината закисной меди выражается
следующими предельными зарядами:
по тетрилу............ 0,025 г
по пикриновой кис/уэте 0,08 »
по тротилу......... 0,15
по трмнигроанмзолу . . 0,32
217
§ 2. НЕОРГАНИЧЕСКИЕ ПРОИЗВОДНЫЕ АЗОТИСТОВОДОРОДНОЙ КИСЛОТЫ
Неорганических производных азотистоводородной кислоты из-
вестно значительно больше, чем производных гремучей кислоты.
Сравнение их взрывчатых свойств позволяет сделать предположение
о том, что из азидов инициирующими веществами могут быть только
азиды побочных подгрупп периодической системы элементов Мен-
делеева. Поэтому мы остановимся, главным образом, на описании
свойств последних.
Азид серебра N3Ag. Азид серебра впервые был получен
Курциусом в 1890 г.
Германское военное министерство сразу же после первой публикации о полу-
чении азида серебра пыталось применить его в качестве инициатора для военных
целей. Первая серьезная попытка этого рода была сделана еще в 1893 г., когда
в германском военно-исследовательском институте в Шпандау были произведены
Виллем и Ленцем, по предложению Курциуса, очень обстоятельные- Опыты по
применению азида серебра. Однако эта попытка не увенчалась тогда успехом,
и вследствие большого взрыва, при котором был убит один химик, работы временно
были прекращены. Только тогда, когда Л- Велер и О- Маттер показали практи-
ческую целесообразность применения азидов тяжелых металлов в качестве ини-
циирующих В В вообще, научно-исследовательские работы возобновились, и опыты
применения азида серебра были продолжены.
В дальнейшем азид серебра изучал в 1909—1910 гг. А. А. Солонина, в 1918 г.
Hitch и затем Тейлор и Ринкеибах в 1925 г.
Азид серебра,, как это достоверно известно из литературных дан-
ных, производился в заводском масштабе в Италии на заводе -в Taino,
в Испании и в других странах, причем в некоторых из них азид
серебра не только вытеснил из снаряжения капсюлей-детонаторов
гремучую ртуть, но, повидимому, успешно вытесняет и азид свинца.
Получение азида серебра. Курциус получал азид
серебра перегонкой азотистоводородной кислоты и поглощением ее
нейтральным раствором нитрата серебра; впоследствии он указал
на возможность получения нерастворимых в воде азидов путем двой-
ного обмена растворимой соли металла и растворимого азида в вод-
ной среде.
Велер и Маттер получали азид серебра смешением водных раство-
ров нитрата серебра и азида аммония*.
AgNO3 -Ь NH4N3 = AgN3 4- NHflNO3
Штеттбахер описывает методику получения азида серебра путем
двойного обмена в водной среде азида натрия и нитрата серебра:
AgNO3 4- NaN3 = AgN3 4- NaNO3
При применении неконцентрированных растворов (не более 5%)
происходит мгновенное образование кристаллов азида серебра, по
они получаются настолько мелкими, что осадок представляется
хлопьевидным, творожистым. Раствор нитрата серебра вливают в рас-
твор азида натрия при перемешивании до тех пор, пока не перестанет
образовываться осадок азида серебра. Раствору дают отстояться,
сливают прозрачную, находящуюся над осадком, жидкость и про-
278
мывают азид серебра на фильтре дистиллированной водой до тех
пор, пока в промывных водах исчезнет ион серебра (проба хлоридом
натрия). Осадок содержит большое количество адсорбированного
маточного раствора, который очень трудно удаляется при промы-
вании. Промытый азид серебра сушат в течение 24—48 час. при
температуре 50°.
В сущности этим же методом пользовались Тейлор и Ринкенбах.
Однако они, опасаясь образования крупных кристаллов азида се-
ребра при осаждении из разбавленных растворов, пользовались
концентрированными растворами. 34 г нитрата серебра они раство-
ряли в 50 мл воды и полученный раствор медленно приливали к рас-
твору 13,5 г азида натрия в 70 мл воды при непрерывном перемеши-
вании. Кристаллы отфильтровывали и промывали водой, спиртом
и эфиром. Сушка производилась в струе сухого воздуха.
А. А. Солонина готовил азид серебра приливанием раствора 2,6 г
азида натрия в 20 мл воды к 20 мл 2N раствора нитрата серебра.
Азид натрия, видимо, содержал неопределенное количество карбо-
ната или гидроокиси натрия, и поэтому необходимо было предвари-
тельно нейтрализовать его разбавленной азотной кислотой. Осадок
он промывал водой, спиртом и эфиром и сушил при температуре 98°
в течение получаса, а затем над хлористым кальцием в эксикаторе
с разреженной атмосферой.
Браун показал, что азид серебра можно получить количественно
электролизом азотистоводородной кислоты с применением серебря-
ных электродов.
Физико-химические свойства азида серебра.
Азид серебра представляет собой белое кристаллическое вещество,
уд. веса 4,81 + 0,05 (азид свинца имеет уд. вес 4,71, а гремучая
ртуть 4,3—4,42). Кристаллическая решетка азида серебра относится
к орторомбической сингонии и имеет следующие параметры:
0 = 5,58 + 0,03 А, 6 = 5,93 + О,ОЗА, с = 6,04 ±0,03 А
Элементарная ячейка содержит 4 молекулы, что дает вычисленную
плотность 4,94 ± 0,07, хорошо согласующуюся с непосредственно
измеренной при помощи пикнометра (4,81 ±0,05). Рентгенограммы
вращения показывают, что структура кристаллов ионная.
Азид серебра, осажденный из концентрированных растворов ни-
трата серебра и азида натрия, имеет вид чрезвычайно мелких, не
поддающихся измерению под микроскопом, белых кристаллов, при
сушке превращающихся в очень прочные комки. Величина кристал-
лов возрастает по мере уменьшения концентрации растворов исход-
ных веществ.
Из водных растворов азид серебра кристаллизуется в небольшие,
игольчатого типа кристаллы. Из раствора в водном аммиаке он кри-
сталлизуется в виде крупных почти бесцветных кристаллов иголь-
чатой формы, достигающих 1 см в длину и обладающих повышенной
чувствительностью.
279
Таблица 59
Растворимость азида серебра
Растворитель В 100 рас- творителя рас- творяется азида серебра ?
Вода 95%-ный этиловый 0.006
спирт 0.007
Эфир . . 0.017
Ацетон . . 0 015
Растворимость азида серебра в
воде ничтожна. В спирте и эфире j
она только несколько больше. В
табл. 59 приведены данные о рас- ’
творимости азида серебра в обыч-
ных растворителях.
В водном растворе аммиака
азид серебра растворяется легко,
причем раствор этот стоек даже
при кипячении.
Азид серебра практически неги-
гроскопичен. Увеличение его веса
после 2-недельного храпения при
комнатной температуре во влажной
атмосфере составляет всего 0,4%.
Разбавленные кислоты растворяют и действуют на азид серебра
медленно при комнатной температуре. При кипячении же, а также
в случае применения концентрированных кислот действие значительно
ускоряется. В обоих случаях образуется соль действующей кислоты
и отщепляется азотистоводородная кислота.
Водные растворы щелочей действуют на азид серебра значительно
медленнее, чем на азид свинца.
Азид серебра, так же как и азид свинца, изменяется под дей-
ствием лучей солнечного света. Установлено, что под влиянием пря-
мых лучей солнечного света изменение окраски азида серебра про-
текает относительно быстро, на рассеянном же свету — медленно.
Цвет азида серебра меняется постепенно: из белого порошка он
превращается сначала в светлофиолетовый, затем серо-фиолетовый,
темносерый и, наконец, в черно-серый. Бекк показал, что азид
серебра, подобно хлористому или бромистому серебру, можно при-
менять в фотографии.
При освещении азид серебра разлагается с выделением тонко-
раздробленного (до размера коллоидальных частиц) и способного
окрашивать металлического серебра и азота; 2 г вещества, по-
мещенные в кварцевый сосуд, соединенный с манометром (из
системы перед опытом эвакуировали воздух), под действием света
повысили давление в течение 27 дней при 22" на 78 лш рт. ст. При
встряхивании освещаемой навески азида серебра разложение идет
несколько быстрее. Без встряхивания разлагается только поверх-
ностный слой азида и разложение вглубь кристаллов нс идет, пока
с них встряхиванием не будет снят слой разложившегося ве-
щества.
В то время как у азида свинца взрывчатые свойства в резуль-
тате освещения изменяются достаточно резко (падает чувствитель-
ность к удару и повышается температура вспышки), у азида серебра
после освещения температура вспышки и чувствительность к удару
остаются неизменными. У азида свинца в результате освещения
замечается снижение инициирующей способности; у азида же се-
ребра, по данным Штсттбахера, фотохимические изменения не ока-
зывают никакого влияния на инициирующую его способность.
При освещении азида серебра под водой (как, впрочем, и других
азидов тяжелых металлов) наблюдается разложение.
По отношению к нагреванию азид серебра не менее стоек, чем
азид свинца; будучи подвержен нагреванию при 75° в течение 24 час.,
азид серебра не изменяется в весе.
Азид серебра, в противоположность азиду свинца, может пла-
виться. Курциус нашел, что температура его плавления 250°, Тейлор
и Ринкенбах определили температуру плавления равной 251°, a Hitch
251,5°. При нагревании до температуры, несколько превышающей
температуру плавления, азид серебра быстро разлагается на азот и
металлическое серебро. Температура вспышки азида серебра не-
сколько ниже, чем температура вспышки азида свинца, но значи-
тельно выше, чем температура вспышки гремучей ртути. По данным
Велера и Мартина, температура вспышки:
азида свинца 327'
азида серебра 297*
гремучей ртути 186'
Взрывчатые свойства азида серебра. Азид,
серебра при взрыве распадается по следующему уравнению:
AgN3 -> Ag-j- l,5N2+65,5 Кал
Чувствительность азида серебра к удару, как показывают опыты,
зависит от величины кристаллов: чем больше размеры кристаллов,
тем выше их чувствительность к удару. Солонина для навесок тон-
кого порошкообразного азида серебра, равных 0,05 г, не смог уста-
новить на копре Велера при весе груза 600 г и ударной поверхности
бойка диаметром 1,7 мм верхнего предела чувствительности, в то
время как для кристаллов азида серебра размером 0,1—20 мм верх-
ний предел оказался равным 315 мм. Тейлор и Ринкенбах устано-
вили при весе груза в 500 г высоту, при падении с которой проис-
ходит' 100% взрывов:
для мелкокристаллического AgN3 . 777 лш
для крупнокристаллического AgNs 287 »
Те же авторы несколько позднее произвели сравнительную оценку
чувствительности к удару азидов свинца, серебра и гремучей ртути
при весе падающего груза 500 г и навеске вещества в 0,02 г и полу-
чили следующие данные о высотах падения груза, обеспечивающих
100% взрывов:
Hg(ONC)3 • 240 мм
AgN.t ... . - 410 "
Pb(N3)2 .... 430 »
Из этих опытов следует, что чувствительность к удар)/ азидов
свинца и серебра примерно одинакова, вернее, азид серебра только
несколько чувствительнее азида свинца. Результаты наблюдений
других авторов противоречат результатам работы Тейлора и Ринкен-
баха.
28t
Велер и Мартин приводят следующие сравнительные данные
с чувствительности азидов серебра и свинца к удару в зависимости
от величины навесок.
Испытание производилось на копре Велера. Азиды прессовались
в латунные колпачки капсюля-воспламенителя под давлением
1100 кг/см- (табл. 60).
Таблица 60
Чувствительность к удару азидов свинца и серебра
Наименование, вещества Ударная поверхность] бойка мм'2 Вес груза кг Высота падения груза Работа кгем
0.01 г 0,02 г 0,05 г
AfiNs - 1 2,14 0.964 310/13,87 24541,04 180 8,11
Pb(Ns). . . 2.14 ! 0,599 170/4.76 170'4,76 145/4,06
Из табл. 60 видно, что азид серебра обладает меньшей чувстви-
тельностью к удару, чем азид свинца. То же вытекает и из опытов
А. А. Солонина.
Чувствительность к трению была определена Тейлором и Рин-
кенбахом при помощи маятникового прибора. Результаты сведены
в табл. 61.
Таблица 61
Чувствительность к трению
Наименование вещества Вес добавоч- ного груза кг Высота па- дения ма- ятника мм Количество качаний до взрыва
Гремучая ртуть . 0,0 25,0 3-10
Азид свинца 0,45 37,5 12
Азид серебра 4,35 33,0 39
Этот метод показывает, в сущности, чувствительность не к про-
стому трению, а к наиболее часто встречающейся в практике его
разновидности — к трущему удару. Результаты опытов показывают,
что чувствительность азида серебра к трущему удару значительно
ниже, чем чувствительность азида свинца и, особенно, гремучей
ртути.
Инициирующее действие азидов серебра, свинца и гремучей ртути
по отношению к различным бризантным ВВ, выраженное предель-
ными зарядами инициатора, приведено в табл. 62, составленной по
опытам Велера и Мартина.
Таким образом результаты опытов показывают, что инициирую-
щая способность азида серебра в большинстве случаев несколько
282
превосходит инициирующую способность азида свинца и значи-
тельно — гремучей ртути.
Таблица 62
Предельные заряды инициаторов ио различным вторичным зарядам, г
Инициирующее В В Тетрил Пикри- новая кислота Тримм- трокси- лоя
Азид серебра . 0,02 0.035 0 07 0,26 0,25
Азид свинца . . 0 025 0.025 0.09 0.28
Гремучая ртуть . . 029 0,30 0.36 037 0.40
Некоторые другие характеристики азида серебра были опреде-
лены Тейлором и Монро, а затем Тейлором и Ринкенбахом. Резуль-
таты их опытов сведены
в табл. 63. Условия
опыта: капсюли-детона-
торы № б с различными
навесками инициатора,
запрессованными под
давлением около 72
•кг[см-, подрывались в
песочной бомбе; песок
перед опытом пропуска-
ли через сита с 30 отвер-
стиями на 1 линейный
дюйм.
Результаты подрыва
в песочной бомбе, по
существу, характеризу-
ют дробящее, бризант-
ное действие. И по этой
пробе азиду серебра по
Таблица 63
Результаты испытания инициатороа по песочной
пробе
Вес заряда Вес раздробленного песка, г
Hg(ONC)2 | Pb(N,)2
0,1 1 3.3
02 3.8 5,9 6.8
0,3 8.0 10,4
0.4 12,2 12.7 —
0.5 16.0 16,1 189
0.6 20.1 20.9 —
0.75 — 30 0
0.8 28.2 28.5
1.0 30.8 33.6 41.1
сравнению с прочими инициаторами принадлежит первое место.
Азид меди (1) CuN3. Впервые был получен Солонина в 1910г.
восстановлением сернокислой меди кислым сульфитом натрия с по-
следующим осаждением раствором азида натрия. Осажденный азид
меди (I) представляет собой кристаллический порошок серого цвета,
очень плохо растворимый в воде: 1 л воды растворяет лишь 0,044 г
CuNs при 20°. Кислоты разлагают азид меди (1) при нагревании.
Щелочи не растворяют азида меди (I), но медленно разлагают его
при кипячении с образованием кирпично-красного осадка гидрида
окиси меди. CuNs растворим в аммиаке, еще лучше в ЮЛ/ растворе
ацетата нЯтрия. Температура вспышки азида меди (I) 215° (по данным
Солонина). К удару азид меди (I) несколько более чувствителен,
чем азид свинца.
283
Азид меди (II) Cu(Ng)2 был получен впервые Курциусом
в 1890 г., затем Курциусом и Риссом в 1898 г. Они получали азид
меди (II) действием раствора азида натрия на раствор сульфата меди
или действием меди на водный раствор азотистоводородной кислоты.
Азид меди (11) кристаллизуется при этом с 0,5 молекулы воды. Он пред-
ставляет собой буро-желтое вещество, трудно растворимое в воде.
В зависимости от среды, в которой осаждается азид меди (II), могут
быть получены как средние, так и основные соли. Из кислой
среды осаждается средняя соль, из нейтральной или слабо щелочной
среды—основная соль.
Кислоты разлагают азид меди; щелочи также разлагают его, но
значительно медленнее. В водном аммиаке азид меди хорошо рас-
творим и образует комплексное соединение, выпадающее после одно-
дневного стояния в виде темнозеленых игл, нерастворимых в воде
и обладающих слабо взрывчатыми свойствами. Исходя из анализа
кристаллов, можно предполагать образование комплексного медно-
аммиачного азида:
• Н2О
Деннис и Ишам в почти аналогичных условиях получили аммо-
ниакаты и пиридинаты азида меди (II):
Cu(N3)2 • 2NH3
Cu(N3)3 • 2C5H6N
При кипячении с водой Cu(N3)3 медленно разлагается с выделение.м
HN3. Азид меди (II) легко взрывает от трения, удара и нагрева. Он
является одним из наиболее чувствительных азидов.
Так, если азид меди (I) взрывает при падении на него груза весом
600 г с высоты 190—210 мм, то в тех же условиях для азида меди (II)
достаточна высота в 15 мм. Температура вспышки азида меди (II)
205—210°.
Азид ртути (I) HgN3 был получен впервые Курциусом в
1891 г., а затем Бертло и Вьслем действием растворов азида натрия
или аммония на нитрат ртути (I). Он представляет собой белый кри-
сталлический порошок, быстро желтеющий на свету. Под действием
аммиака, как и все соли ртути (I), чернеет. Растворимость в воде
очень невелика (в 1 л воды при 18° растворяется 0,2320 г азида
ртути). Действует на металлы. Температура вспышки 295'. Чувстви-
тельность к удару при весе груза 600 г в пределах 225—240 мм.
Азид ртути (II) Hg(N3)3 впервые был получен Бертло и
Вьелем растворением свежеосажденной окиси ртути в азотистоводо-
родной кислоте. Затем Солонина получал его взаимодействием рас-
творов сулемы и азида натрия. Азид ртути (II) представляет собой
бесцветные кристаллы игольчатой формы, плохо растворимые в хо-
лодной [при 20° в 100 мл растворяется 0,527 г Hg(N3)3] и легко в горя-
чей воде. По охлаждении горячего водного раствора выпадают боль-
шие иглы, очень чувствительные к механическому воздействию.
284
Фиг. 99. Форма
кристаллов азида
ртути (II).
Интересно сравнить Hg(Ns)fe с Hg(ONC)3. Обе соли имеют одина-
ковый молекулярный вес, почти одинаковую плотность, одинаковое
содержание ртути, продукты их взрыва имеют одинаковые объемы
и состоят из недиссоциирующих молекул газов, что видно из следую-
щих уравнений:
Hg(Ns)2 -> Hg + 3N2
Hg(ONC)2 - Hg + N2 + 2CO
Однако между этими двумя веществами наблюдается большая раз-
ница по чувствительности к .механическому воздействию. Чувстви-
тельнее азид ртути, особенно в виде больших
кристаллов.
Азид ртути (II) может существовать в двух
модификациях. a-Hg(N3)2 может быть приготовлен
смешением насыщенного раствора сулемы с экви-
валентным раствором азида натрия, подкисленным
слегка азотистоводородной кислотой. Кристаллы
при этом имеют вид тонких, правильно развитых
призмочек длиной 2—4 .мм (фиг. 99). a-Hg(N3)3
не является анормально чувствительным, что
было доказано при опытах многократной сушки
и грубом измельчении продукта.
При получении азида ртути перегонкой азо-
имида в водную суспензию окиси ртути получается
большое количество переплетающихся игольчатых
кристаллов, образующихся при охлаждении рас-
твора и часто детонирующих во время кристаллизации или в момент
приведения сосуда в движение. Оказалось, что эти игольчатые кри-
сталлы (фиг. 99) являются р-азидом ртути (II), необыкновенно чув-
ствительным к трению и способным иногда самопроизвольно дето-
нировать во время сушки. При этом способе получения образуется
обычно смесь а- и p-модификаций; то же наблюдается при получении
азида ртути по методу диффузии в воде или (более быстро) в
растворе нитрата ртути (II). p-форма нестабильна и легко переходит
в а-форму.
Данные о чувствительности известны только для a-формы. Чув-
ствительность ее к удару несколько меньше, чем у азида меди (II),
и может быть выражена высотой в 65 мм при весе груза 600 г. Тем-
пература вспышки 220—230°.
Азид цинка Zn(N3)2 был получен впервые Велером и Мар-
тином в 1913 г. из карбоната цинка и эфирного раствора азотисто-
водородной кислоты с последующей тщательной просушкой получен-
ного продукта.
Азид цинка представляет собой белый порошок, состоящий из
столбикообразных кристаллов, гигроскопичный и легко поддающийся
гидролизу с образованием основной соли Zn(OH)N3. - Азид цинка
вспыхивает при воспламенении, как и азиды щелочных металлов;
от сильного удара детонирует.
285
Основной азид цинка Zn(0H)N3 был получен Кур-
циусом растворением свежеосажденного гидроксида цинка в азоимиде
или при взаимодействии сульфата цинка с азидом бария в разбав-
ленных растворах. Он представляет собой белый, плохо растворимый
в воде (0,123 г в 1 л при 20°) кристаллический порошок. При кипя-
чении с водой легко выделяет HNS. Кислоты и щелочи легко разла-
гают Zn(OH)Ns. Температура вспышки около 290°. Чувствительность
к удару аналогична чувствительности азида серебра.
Азид никеля Ni(N3)2 может быть получен действием кар-
боната никеля на эфирный раствор азоимида. Он представляет собой
зеленый порошок, легко растворяющийся в воде и гидролизующийся.
Азид никеля особенно чувствителен к трению. Слабое давление или
трение между стеклом и металлом приводят к сильным взрывам.
Легко переходит в основной азид Ni(OH)N3, значительно менее рас-
творимый в воде (0,1709 г в 1 л при 20°).
Азид кобальта Co(N3)g получается взаимодействием кар-
боната кобальта с эфирным раствором азоимида в виде красно-
коричневого порошка, склонного к гидролизу в воде, аналогично
азиду никеля. Он еще более чувствителен к трению, чем азид никеля.
§ 3. ОРГАНИЧЕСКИЕ ПРОИЗВОДНЫЕ А30ТИСТ0ВОДОРОДНОЙ КИСЛОТЫ
Органические азиды (алкилазиды и азиды кислот) составляют
обширный класс органических соединений. Однако, несмотря на
большое количество уже известных азидов, до сих пор еще не пред-
ставилось возможности в достаточной степени точно оценить зави-
симость не только инициирующих, ио даже просто взрывчатых свойств
от строения молекулы и количества азидогрупн. Мы останавливаемся
поэтому на тех органических азидах, которые являются, как пока-
зали исследования, инициирующими ВВ, в достаточной степени изу-
чены и в той или иной степени при необходимости могут быть исполь-
зованы в практике.
Циануртриазид был получен впервые Фингером в 1907 г.
путем взаимодействия цианурхлорида с гидразингидратом с после-
дующей обработкой образующегося при этом циануртригидразида
нитритом натрия:
C3N3C13 + 3NH2-NH2 -> C3N3(NHNH2)3 + ЗНС1
C3N3(NHNH2)3 + 3NaNO3 -> C3NS(N3)3 + 3NaOH + ЗН2О
Получающийся при этом продукт содержит некоторые примеси.
В чистом виде циануртриазид получен Оттом и Озе, которые
предложили в качестве исходных продуктов цианурхлорид и азид
натрия.
Цианурхлорид получается действием хлора на цианистый водород:
3HCN + ЗС13 -> 3CNC1 + 3HCI
3CNCI -> C3N3C13
286
Метод получения состоит в обработке хлором цианистоводородной
кислоты в присутствии какого-либо недеятельного растворителя.
Хлор можно пропускать через эфирный раствор цианистого водорода,
или через раствор цианистого водорода в хлороформе. Наконец,
можно пропускать одновременно хлор и цианистый водород через
насыщенный хлором хлороформ.
При указанных методах получения цианурхлорида полимеризация
хлористого циана в цианурхлорид до конца не доходит, и обычно
окончательный продукт содержит различные количества хлорциана.
Добавлением к реагирующей смеси 1% этилового спирта несколько
улучшается выход, однако он остается неравномерным. Попытка
заменить хлороформ ацетоном не дала положительных результатов.
Позднее Джемс усовершенствовал метод получения цианурхло-
рида, применив вместо синильной кислоты метилтио цианат. По его*
методу цианурхлорид получают следующим образом: 100 г метил-
тиоцианата отфильтровывают в эрленмейеровскую колбу, погружен-
ную в ледяную воду. Через охлажденный метилтиоцианат в течение
5 час. пропускают струю хлора. Примерно через Р/3 часа жидкость
в колбе ‘ становится теплой и окрашивается сначала в желтый, а
затем в оранжевый цвет. Приблизительно через 2 часа происходит
значительное выделение хлористого водорода, а затем начинается
образование кристаллов. После пропускания хлора колбы неплотно
закрывают и оставляют на несколько часов в покое. Кристаллы
отфильтровывают, сушат на фильтровальной бумаге и очищают
перекристаллизацией из горячего хлороформа.
Реакция протекает по уравнению:
3CH3SCN + 11 С12 -> C3N3C13 + 2CSC14 + CSC12 + 9НС1
Кристаллы цианурхлорида моноклинической системы, чрезвычайно
мягки и не могут быть растерты в порошок- Температура плавления.
147°. Выход около 95% от теоретического.
Для получения циануртриазида чистый азид натрия растворяют
в небольшом избытке воды и в полученный раствор всыпают неболь-
шими порциями эквимолекулярное количество цианурхлорида. Реак-
ция экзотермична и температура поднимается до 45°. Смесь тщательно
перемешивают и оставляют стоять в течение 12 час.
В результате реакции:
C3N3Cl3+3NaN3 -> C3N3(N3)3 + 3NaCl
получается твердое вещество, которое отфильтровывают, промывают
водой и сушат. Выход около 90%.
Вещество представляет собой кристаллы чисто белого цветаг
среднего размера в 0,1 мм. Самые крупные кристаллы, получающиеся
при этом методе, имеют длину не более 0,3 мм.
Циануртриазид может быть очищен однократной перекристалли-
зацией из горячего этилового спирта. Однако следует работать с чи-
стыми исходными материалами, чтобы избежать перекристаллизации,
так как при последней образуются крупные, чрезвычайно чувствитель-
287
ные кристаллы циануртриазида. Крупные кристаллы иоиучаюгсм
также при выливании спиртового раствора в холатную иоду. Полтому
следует применять вместо воды ацетон, при выливании спирюнснп
раствора в который образуются кристаллы значительно мепышп
размеров.
Ввиду того, что циануртриазид представляет собой удачно»
-сочетание радикалов гремучей кислоты и азоимида
N
z\
Ns С С —N3
U I
С
I
N3
-было высказано предположение, что он по своим взрывчатым свой-
ствам превосходит азид свинца. В связи с этим были проведены
весьма тщательные исследования циануртриазида. Кристаллы цианур-
триазида отличаются сильным блеском и, подобно всем азотоугле-
родам, — большой светопреломляемостыо. Циануртриазид нераство-
рим в воде, немного растворим в холодном этиловом спирте, хорошо
растворяется в бензоле, хлороформе, ацетоне, эфире и кипящем
этиловом спирте. Из растворов кристаллизуется в форме игл.
Влага не изменяет взрывчатых свойств циануртриазида. Он мало
гигроскопичен и стоек также по отношению к разбавленным минераль-
ным кислотам.
При нагревании до 100° не разлагается, однако уже при темпера-
туре 50° начинает возгоняться; плавится при 94° и в плавленном
виде имеет плотность, равную 1,54. При продолжительном нагре-
вании несколько выше 100° циануртриазид начинает разлагаться
и может быть разложен нацело без детонации. Продолжительное
нагревание при температурах порядка 150—160° приводит к взрыву,
причем часть вещества, конечно, сублимируется.
Температура вспышки циануртриазида порядка 200°. Однако
точно установить ее затруднительно, так как циануртриазид может
давать взрыв и при 170—180° после сравнительно короткого нагре-
вания.
Чувствительность циануртриазида к трению больше, чем чув-
ствительность гремучей ртути. Наблюдались случаи взрыва мелко-
кристаллического циануртриазида при прессовании капсюлей-дето-
наторов. Большие кристаллы зачастую детонируют даже при расти-
рании их резиновой палочкой. Чувствительность к удару на копре
Велера порядка 150 мм.
Следует также отметить, что в расплавленном состоянии цианур-
триазид несколько более чувствителен к трению, чем в твердом.
288
ПнИЦИИруКИЦ.В! СНСНЧИиИК II» Ц11<И!у111'р11П.111Д>1 CpillllllMCJIl.llO ПЫСО
кая — предельный инициирующий «и ряд его равен:
ЦП гстрилу 0.0(» .•
и<» пикриновой кислоте 0.06 й
ио тротилу ...................... . 0,13 »
по тетранитроанилину 0,09 >
по пикрату аммония - 0,15 »
Циануртриазид являлся очень хорошим инициатором, но в то же
время он весьма чувствителен и небезопасен в обращении. Не ядовит,
но материалы, применяемые для его приготовления, ядовиты и дей-
ствуют раздражающим образом на слизистые оболочки. Вследствие
этого циануртриазид пока еще не нашел распространения во взрыв-
ной технике.
Т ринитро триазидобензол впервые был получен в
1924 г. О. Туреком по следующей схеме:
C.HfOII
nh2 nh2 N = N
j C1- CI,Z X,CI H.SO, C,H.OH
/ "™- \Z
Cl Cl
no2 no,
С1|Х ^C1 HNO. C]/ 'S:C1 NaNe
[ h,so,+so, NO„X JnO2 NoJ^ Jno2
'ci Cl N,
Трихлоранилин получается хлорированием взмученного хлористо-
водородного анилина в индиферентной жидкости (CHCIB, СС1„ CSa,
СвНв), взятых в соотношении 15—30 в. ч. первого на 100 объемн. ч.
второй. Более высокое содержание хлористоводородного анилина
в значительной степени затрудняет перемешивание тестообразной
массы в конце реакции.
Газообразный сухой хлор вводят в реакционную смесь при не-
прерывном перемешивании, причем смесь защищают от попадания
в нее влаги. Для ускорения реакции прибавляют спирт в количестве
0,5—2%. Трихлоранилин выделяют промывкой полученного осадка
водой с воздушным перемешиванием. Продукт получается чистый.
Выход'- 100 в. ч. анилина дают 175 в. ч. чистого трихлоранилина,
т. е. 80% от теоретического.
Трихлорбензол получается удалением NH2 обычным путем. Три-
хлоранилии растворяют в спирте с прибавлением H,SO4(y,'i, вес 1,84)
и мелкими порциями всыпают Na.NO,, диазосоединение разрушают
на водяной бане при 50°, отгоняют спирт и, наконец, с водяным
паром отгоняют трихлорбензол. Выход: из 100 в. ч. трихлоранилина
получается 83 в. ч. трихлорбензола, т. е. 90% от теоретического.
Бубнов—92—19 289
Трихлортринитробензол получают в две фазы. Сначала сульфи-
руют трихлорбензол олеумом с содержанием 65% SO8 и затем нитруют
азотной кислотой (d=l,52). HNO3 приливают при температуре 70—90°,
после чего продукт пвдерживают при температуре 150° в течение
30 час. Выход: из 1и0 в. ч. трихлорбензола 140 в. ч. тринитротри-
хлорбензола.
Тринитротриазидобензол получается действием спиртового рас-
твора азида натрия на тринитротрихлорбепзол при 25е при соотно-
шении 5 г азида натрия и 10 г тринитротрихлорбензола. Время
реакции 1—1г/2 часа. Выход: из 10 г тринитротрихлорбензола 7 г
тринитротриазидобензола, т. е. 70% от теоретического.
Фиг. 100. Зависимость разложения трииитротриазвдобензола в зависимости
от температуры и времени нагревания.
Тринитротриазидобензол представляет собой желтые с зеленова-
тым оттенком кристаллы. Он очень хорошо растворяется в ацетоне,
довольно хорошо в хлороформе, плохо в спирте и почти совершенно
нерастворим в воде. Тринитротриазидо бензо л очень нестойкое соеди-
нение по отношению к температуре: уже при 20° при продолжитель-
ном хранении наблюдается потеря в весе вследствие разложения
с выделением азота по уравнению:
Cg(NO3)3(N3)3 -> c6(N0)g+3N2
Разложение ускоряется с повышением температуры и при 100°
заканчивается примерно через 16 час. Нафиг- 100 приведены графики
230
разложения тринитротриазидобензола при 20, 35, 50 и 100°. Тем-
пература плавления трииитротриазидобензола 131°. При этой тем-
пературе выделяется свободный азот и тринитротриазидобензол пре-
вращается почти в совершенно чистый гексанитрозобензол. Вода
и двуокись углерода на тринитротриазидобензол не действуют:
с металлами ои не реагирует. Плотность равна 1,8054. На свету,
как и все азиды, меняет цвет из светложелтого в темнозеленый.
Чувствительность к удару и трению меныпая, чем у гремучей ртути
и азида свинца.
Испытания в бомбах Трауцля 100 х 100 мм показали, чти сила
трииитротриазидобензола больше целого ряда бризантных и ини-
циирующих ВВ (табл. 64).
Значительное превосходство силы трииитротриазидобензола обес-
печивает и более высокое инициирующее действие его. Так, предель-
ный инициирующий заряд равен:
но тетрилу
по тротилу
0,01 г
0,02 »
Недостатки триннтротриазидобензола следующие: пониженная
стойкость к повышенным температурам, способность перепрессовы-
ваться, недостаточная
чувствительность к уда-
ру и плохая сыпучесть.
Т ринитроазидобензол
запатентован в ряде
стран и предлагается
для снаряжения кап-
сюлей- в осп ламен ите лей,
капсюлей - детонаторов
и детонирующих шну-
ров. Применяться он
может и в комбинации
с тротилом, тетрилом и
другими бризантными
ВВ в качестве первич-
Таблица 64
Относительные величины расширений но пробе
Трауцля для ряда ВВ
ВВ
Относительное
расширение
I
Пентаэрьггриттетранитрат .
Тринитротриазидобснэол
Тетрил ...
Тротил ............
Гремучая ртуть .
Азид свинца . .
100
90
70
60
23
16
него заряда.
Динитротриазидобензол C6H(NO2)2(N3)3 был пред-
ложен впервые в 1931 г. В. Фредерики (герм. пат. № 531253). Это
более дешевый продукт по сравнению с тринитротриазидо бензолом.
Он получается при взаимодействии азида натрия с динитротрихлор-
бензолом. В отличие от трииитротриазидобензола он может быть
сравнительно легко перекристаллизован из органических раствори-
телей (например ацетон), причем он кристаллизуется из них в виде
порошка или очень мелких кристаллов. От луча огня вспыхивает
с ярко светящимся пламенем. Чувствительность его к удару высока
и, судя по патенту, вполне достаточна для применения его в капсю-
лях-воспламенителях и патронах бокового огня-
291
§ 4. АЦЕТИЛЕНИДЫ (ЭТИНИДЫ)
Ацетилен СНеСН, представляя собой эндотермическое соединение
(теплота образования его—61,4 Кал/мол), сам является в определенных
условиях взрывчатым веществом. Особый интерес для нас представ-
ляют его металлические производные, из которых инициирующими
свойствами обладают производные тяжелых металлов (серебряные,
ртутные, медные).
С водными и кислыми растворами солей серебра ацетилен реаги-
рует с образованием двойных соединений. Такие же двойные соеди-
нения образуются при действии ацетилена на аммиачные растворы
ацетата, хлорида или бромида серебра.
Соединение ацетилена с серебром выпадает из разбавленного
аммиачного раствора нитрата серебра в виде светложелтого осадка.
Выпадающее из более крепких растворов желтое творожистое веще-
ство имеет тенденцию становиться белым и уменьшаться в объеме.
В обоих случаях осадок легко увлекает за собой окись серебра. Он
получается более чистым при введении ацетилена в раствор ацетата
серебра, который при этом полностью разлагается. Анализы полу-
ченного таким образом и высушенного над серной кислотой ацетиле-
нида серебра приводят к цифрам, лежащим между вычисленными
для формул:
2C2Ag2 - Н2О и 3Ag2C2 • Н2О
Из водных децинормальных растворов нитрата серебра ацетилен
освобождает только 4/б содержащейся в веществе азотной кислоты,
причем и выпадающий осадок содержит азотную кислоту. Еще в боль-
шей степени это относится к осадку, выделяемому ацетиленом из
спиртового раствора нитрата серебра.
Найденный для осадков, образовавшихся из растворов нитрата
серебра, состав отвечал значениям между
3Ag2C2 • 2AgNO3 • Н2О и Ag2C2 • 2AgNQ, • Н2О
Последняя формула соответствует осадку, полученному из спир-
тового раствора. Из раствора сернокислого серебра, содержащего
0,2 г AgoS04 в 100 мл, при действии ацетилена осаждается соединение
2Ag2C3 Ag2SO4 • Н2О.
Могущая образоваться в процессе окисления при хранении сереб-
ряная соль диацетилена СН = С—Се СН также чрезвычайно взрывчата.
Ацетиленид серебра можно получить пропусканием ацетилена
как в аммиачный, так и в нейтральный и слабокислый (подкисленный
азотной кислотой) раствор нитрата серебра.
Действие медных солей на ацетилен столь характерно, что этой
реакцией пользуются для доказательства присутствия ацетилена и
для количественного отделения меди от других тяжелых металлов
путем осаждения ацетиленом из аммиачных растворов медных солей.
При этом получается ацетиленистая медь, известная или в безводной
форме Си2С2 или с кристаллизационной водой Сп8С2 • Н2О.
Ацетиленид медн следует рассматривать, так же как и ацетиленил
серебра, как конечный продукт действия ацетилена на аммиачные
292
растворы медных солей; промежуточными являются комплексные
соединения меди.
Ацетиленид меди в сухом состоянии при хранении на воздухе
взрывчат. Полагают, что при его окислении образуются небольшие
количества нестойкого диацетиленида меди: CuCeeC—С = ССи, являю-
щегося причиной разложения.
При действии ацетилена на медные соли в кислых растворах
образуются двойные соединения ацетилена с медными солями. Ацети-
лен соединяется также с солями окиси меди, но при этом одновременно
идет процесс полимеризации. Равным образом ацетилен может образо-
вать ацетиленид меди и при соприкосновении с металлической медью
(конечно, в том случае, если металл влажен или загрязнен аммиаком).
В патентной литературе имеются указания о попытке применять
ацетиленид меди в воспламенительных смесях.
Из раствора двойной соли тиосульфата натрия и золота при дей-
ствии ацетилена выпадает ацетиленид золота.
По данным Штеттбахера, в качестве инициатора наиболее под-
ходящим из ацетиленидов является ацетиленид серебра. По своим
взрывчатым свойствам он должен находиться между гремучей ртутью и
азидом свинца. Получение чистого ацетиленида серебра сложно; обычно
получаются, как мы уже видели ранее, продукты различной чистоты.
Лучшие результаты получаются при осаждении ацетиленида сере-
бра из слабоподкисленного раствора. Белый или желтоватый осадок
ацетиленида серебра на свету приобретает красно-фиолетовую окраску.
Температура вспышки ацетиленида серебра лежит в пределах
170—225°. Предельный инициирующий заряд по тетрилу составляет
0.29 г.
Чувствительность ацетиленида серебра к внешним воздействиям
зависит от способов его приготовления. Тейлор и Ринкенбах исследо-
вали ацетилениды серебра, полученные пятью различными способами:
1. Ацетиленид серебра А-коллоидные агрегаты в форме хлопьев.
Получены пропусканием ацетилена через нейтральный раствор ни-
трата серебра.
2. Ацетиленид серебра В получен пропусканием ацетилена в рас-
твор 5 г нитрата серебра в 200 мл воды и 5,3 мл концентрированного
водного раствора аммиака.
Таблица 65
Наименование веществ Маятник трения Удар грузом 500 г с высоты см Темпе- ратура вспышки °C
добавоч- ный груз кг I высота падения см количе- ство ка- саний до взрыва
Ацетиленид серебра А - 1,0 33,0 12 43 225
» » В . 0.0 33.0 4 15 177
С . 0,0 12.5 2 66 171
. D . 0.2 50.0 8 79 200
• 1 Е . 2.0 50.0 16 64 200
293
3. Ацетиленид серебра С получен пропусканием ацетилена в рас-
твор 5 г нитрата серебра в 200 мл воды и 20 мл концентрированного
водного раствора аммиака.
4. Ацетиленид серебра D получен пропусканием ацетилена в рас-
твор 5 г нитрата серебра в 200 мл воды и 5 мл концентрированной
азотной кислоты.
5. Ацетиленид серебра Е получен пропусканием ацетилена в рас-
твор 5 г нитрата серебра в 200 мл воды и 25 мл концентрированной
азотной кислоты.
Чувствительность этих пяти ацетиленидов серебра сведена в
табл. 65.
Высокая стоимость серебра препятствует применению ацетиленида
серебра; ацетиленид меди применяется для воспламенительных смесей
электрозапалов.
§ 5. НИТРОДИАЗОСОЕДИНЕНИЯ
Хотя в свое время в качестве инициирующих ВВ был рекомендо-
ван целый ряд диазосоединений, в том числе и их перхлораты, од-
нако, практика показывает, что одной группы—N=N—недостаточно,
чтобы вещество было инициирующим. Нитраты диазосоединений,
являясь веществами взрывчатыми и в достаточной степени чувстви-
тельными, в то же время являются веществами недостаточно стой-
кими и обладающими значительно меньшими инициирующими свой-
ствами, чем, например, перхлораты. Сочетание в молекуле групп
—N=N—, —NO2, —ОН и —С1О4 может все же привести к соеди-
нениям, практически применимым в качестве инициирующих ВВ.
Рассмотрим поэтому некоторые из диазосоединений.
Диазобензолнитрат CeH6N = N * N03 получается вве-
дением N2O3 в ледяную воду, содержащую нитрат анилина. После
вливания полученной жидкости в спирт и прибавления эфира обра-
зуется белый объемистый осадок кристаллов в виде игл. Продукт
очень нестоек. На свету приобретает коричневый цвет, а после месяч-
ного хранения над безводным хлоридом кальция разлагается даже
в затемненном эксикаторе. Вследствие своей рыхлости диазобензол-
нитрат очень трудно прессуется в капсюли-детонаторы. Он обладает
весьма слабыми инициирующими свойствами.
Диазобензолперхлорат C6HBN—N—С1О4 впервые был
получен Форлендером следующим путем: в 125 мл 20%-ной хлорной
кислоты растворяли 9 г анилина и затем полученный раствор диазо-
тировали при 0° раствором 7,5 г нитрита натрия в 10 мл воды. Диазо-
бензо лперхлорат выпадал в виде игольчатых призм белого цвета.
Диазобензолперхлорат можно получить также, смешивая 10%-ный
раствор диазобензола с разбавленным раствором хлорной кислоты.
Продукт, получаемый в результате описанных реакций, является
настолько чувствительным, что он способен взрываться даже во влаж-
ном состоянии при трении и ударе. В сухом состоянии его чувстви-
тельность к механическим воздействиям еще выше.
294
Растворимость диазо бензо л перхлората в воде при 0° равна 70%.
Температура вспышки лежит около 84°.
Перхлораты орто- и пара диазотолуолов получаются путем ди-
азотирования соответствующих орто- и паратолуидинов. Продукты
диазотирования
СН3
СН3 /\
N = N ClOj и .
I I и
\/ N = N • С1О4
менее мощны, чем перхлорат диазобензола.
Перхлораты нитродиазобензолов были рекомендованы в качестве
инициирующих ВВ в 1911 г. Э. Герцем (герм. пат. № 258679). Их
относительная дешевизна, простота, безвредность, относительная безо-
пасность изготовления и, в особенности, их весьма сильное инициирую-
щее действие, почти полное отсутствие гигроскопичности и стойкость
во влажной атмосфере доказывают, что они имеют серьезные преиму-
щества перед гремучей ртутью и азидом свинца. Остановимся на
некоторых представителях этой группы.
М е т а н и т р о д и а з о б е н з о л п е р х л орат CcH4(NO2)N =
==N • С1О4 получается из метанитранилина диазотированием в раство-
ре хлорной кислоты. 5 г чистого свежеперекристаллизованного из
воды метанитроанилина растворяют в разбавленной до уд. веса 1,12
хлорной кислоте. Последнюю берут в небольшом избытке, в данном
случае, 12 г хлорной кислоты. К -полученному раствору, охлажден-
ному льдом, приливают по каплям концентрированный водный рас-
твор 3 г азотистокислого натрия- Выпавший осадок отфильтровывают,
промывают два раза водой, подкисленной хлорной кислотой, затем
последовательно спиртом и эфиром и сушат. Из 5 г метанитранилина
получается 7—7,5 г нитро диазобензолперхлората. По Штеттбахеру, 5 г
метанитранилина в эрленмейеровской колбе смешивают с 200 г воды;
причем вначале прибавляют только четверть необходимого коли-
чества воды, 5 мл концентрированной соляной кислоты и 22 мл
20%-ной хлорной кислоты. При встряхивании нитранилин раство-
ряется, после чего прибавляют остальные три четверти воды и по-
лучают раствор желтого цвета. Прибавив в колбу несколько кусков
тонко измельченного льда, осторожно приливают концентрирован-
ный раствор 2,5 г нитрита натрия. Осадившиеся кристаллы отсасы-
вают, промывают холодной водой или разбавленным раствором
хлорной кислоты, затем спиртом и, наконец, эфиром.
Нитродиазобензолперхлорат (НДБП) представляет собой массу
тонких белых кристаллов, трудно растворимых в воде, спирте и
эфире. При хранении в нормальных условиях стоек, однако длитель-
ного хранения при 60° не выдерживает и уже на 43-и сутки теряет
свои взрывчатые свойства. Температура его вспышки, по данным
Штеттбахера, 154—155°, по данным других исследователей, 141°.
295
Чувствительность его к удару больше, чем у гремучей ртути, что
видно из следующего
(см. таблицу). Нитроди-
азобензолперхлорат не
теряет своих взрывча-
тых свойств во влаж-
ном состоянии, од-
нако, при хранении
влажный продукт раз-
лагается с поверхности.
Взрывчатое разложе-
ние нитродиазобензол-
Наименование веществ Высота паде- ния груза см
Диазобензолперхлорат . . 3—4
Нитродиазобензолперхлорат. . 10
Гремучая ртуть 40-50
перхлората протекает по следующему уравнению:
CcH,j(NO2)N = NCIO4 — 3N2 + 9СО J- ЗН2О + ЗНС1 + ЗС
Объем газообразных продуктов при разложении его значительно
больше, чем у гремучей ртути и у азида свинца, вследствие чего и
расширение в бомбе Трауцля несколько больше (около 25 см3).
Из этого уравнения следует также, что здесь происходит далеко
не полное сгорание. Если же принять во внимание ту энергию, кото-
рую приобретает молекула нитробензола вследствие диазотирования,
то нитродиазобензолперхлорат должен быть признан веществом,
богатым энергией. Инициирующая способность его больше, чем
у азида свинца. Так, на 1,5 г тротила, запрессованного в оболочке
минного капсюля под давлением 1000 ат, требуется:
азида свинца .
гремучей ртути
НДБП .
0,15 г
0,30 »
0.02 »
Метабисдиазобензол перхлорат может быть по-
лучен диазотированием метафснилендиамина:
ЬШ2
+ 2НС1 + 2НС1О4 + 2NaNO2
\Jnh2
N = NC104
I | +2NaCl + 4H2O
= NCIO4
2 г мета фенилендиамина растворяют в небольшом количестве воды
с б мл дымящейся соляной кислоты. Отфильтровав нерастворимый
осадок, к фильтрату прибавляют 2,6 г азотистокислого натрия.
Осадок обрабатывают обычным способом. Полученное соединение,
представляющее собой кирпично-красную массу, обладает большей
чувствительностью к простому начальному импульсу, чем нитродиазо-
296
бензолиерхлорат, и иногда взрывает при высушивании на фильтро-
вальной бумаге.
N = N • СЮ*
Нитробисди азобензол перхлорат
N = N • СЮ*,
получается диазотированием нитропарафенилендиамииа. Инициирую-
щая способность этого вещества очень высока. Для детонации пикри-
новой кислоты требуется его около 0,015—0,020 г. При применении
в капсюлях только одного нитробисдиазобензолперхлората для дето-
нации гурдинамита, желатиндинамита, шедитта, пикриновой кис-
лоты и тринитротолуола необходимо от 0,02 до 0,05 а.
Диазодинитрофенол, представляющий собой 4,6-динн-
тробензол-2-диазо-1-оксид, был впервые получен Петером Гриссом,
положившим начало исследованию диазосоединений, при действии
на аминодинитрофенол (пикраминовую кислоту) азотистой кислотой
в спиртовом растворе.
Диазодинитрофенол принадлежит к особой группе диазосоедине-
ний, получающихся из замещенных аминофенолов путем диазотиро-
вания- Обычно амины диазотируются по схеме:
QNH..HC,
В данном же случае
ОН
NO2j/'\ NH,
'мо2
=N-HC1
+ HNO2 + HSO
мы имеем несколько иной процесс:
+ HNO2 -> 1 + 2НЮ
\о2
при котором образуется соединение, относящееся к ряду внутренних
ангидридов свободных диазофенолов и известных в литературе под.
названием «диазоксидов».
Ганч рассматривает их, как хинондиазиды:
О
il n
NO./V II
I | N
N02
(4,6-динитрохинон-2-диазид)
Отличительная черта подобных соединении — стойкость их при
кипячении с водой, чего обычно не выдерживают простые диазоси-
единения.
Диазодинитрофенол используется в мирной промышленности как
диазокомпонент при производстве некоторых протравных красителей.
Вследствие того, что пикраминовая кислота трудно растворима
в воде (0,14 г в 100 мл воды при 22°), Абель рекомендует перед диазо-
тированием растворять ее в крепкой соляной кислоте уд. веса 1,16,
в которой растворим и сам диазодинитрофенол. Это важно для
производства красителей, где процесс купелирования азокомпонентов
ведется в гомогенной среде:
ОН о /у*
NO./\n
I + NaNO2 + HC1 -> I + Naci + HSO
Xno2 no2
Клерк считает наилучшим методом приготовления диазодинитро-
фенола диазотирование натриевой соли пикраминовой кислоты ни-
гритом натрия в присутствии 5,5%-ной соляной кислоты.
1 в. ч. пикрамида натрия взбалтывают при 15° с 12,5 ч. 5,5%-ной
соляной кислоты. К этой смеси прибавляют небольшой избыток
азотистокислого натрия, растворенного в 150—200 мл воды. Тем-
пература поднимается до 18—20°. Темнокоричневый, характерный
для пикрамвда, цвет переходит в светложелтый. Взбалтывают в те-
чение одного часа при температуре 15°, затем отфильтровывают и про-
мывают дистиллированной водой, насыщенной диазодинитрофенолом.
Применяющаяся для этой реакции пикраминовая кислота является
Инициирующие ВВ Максимальная высота па- дения груза, при которой не происходит взрыва (в пяти опытах при навеске 0.02 г и падающем грузе 500 г), см
Гремучая ртуть 15
Азид свиица . . 17.5
Стифнат свинца 20
Диазодинитрофенол . 22.5
продуктом восстановления пикриновой кислоты при помощи железа
или его солей или посредством сернистых щелочей.
Диазодинитрофенол — порошок яркожелтого цвета, состоящий из
микроскопических кристаллов игольчатой формы. Плотность техни-
ческого продукта при 25° равна 1,634, перекристаллизованного из
ацетона — 1,71. Г рави-
метрическая плотность
порошкообразного ди-
азодинитрофенола 0,27,
спрессованного же под
давлением 239 ат—0,86.
Диазодинитрофенол
менее чувствителен к
удару и трению, чем
гремучая ртуть и азид
свинца. Чувств иге ль-
ность его к удару, опре-
деленная Клерком, по сравнению с чувствительностью основных
инициаторов, видна из таблицы.
298
Чувствительность к трению, определенная на маятниковом при-
боре, примерно аналогична чувствительности к трению стифната
свинца, что видно из следующих данных:
Инициирующие В В Навеска Высота падения маятника см Максимальный добавочный груз, при котором не происходит взры- ва (в десяти по- следовательных опытах), г Число качаний маятника
Гремучая ртуть Азид свинца . . Стифнат свинца .... Диазодииитрофенол 0,02 0,02 0,02 0.02 50 50 50 50 25 1000 5000 5000 8 13—15 30-33 28—32
Давление прессования, повидимому, не изменяет его взрывча-
тых свойств; он не спрессовывается намертво даже при давлении
9139 kzIcaP. Под водой не взрывает даже от действия капсюля-дето-
натора № 8.
Относительная сила диазодинитрофенола, измеренная расшире-
нием в бомбе Трауцля, примерно в три раза выше, чем у гремучей
ртути и азида свинца. 1 г вещества, спрессованный под давлением
239 кг}см\ дал расширение:
при диазодииитрофеноле 25,0 сма
" гремучей ртути 8,1 »
, >: азиде свинца 7,8 »
Повышенную силу диазо динитрофенола показала и песочная проба,
что видно из следующей таблицы:
Вес вещества г Давление прес- сования кг/см2 Вес измельченного песка, г
диазодииитро- фенол гремучая ртуть азид свинца
0,10 239,0 9,1 3.1 3.5
0.20 239,0 19,3 6,5 72
0,40 239,0 36,2 17,0 14.2
0.50 239,0 45,6 25,0 20.0
0 60 239 0 54,3 27,5 21.5
0,80 239.0 72,1 38,0 28.7
1,00 239.0 90,6 48,4 36 0
Следует отметить, что диазо динитрофенол, являясь инициирую-
щим ВВ, обладает относительной силой, свойственной бризантным ВВ.
В самом деле, его относительная сила, определенная методом
песчаной пробы, по сравнению с другими нитро производным и арома-
тического ряда, оказалась равной относительной силе тетрила и
299
тринитробензальдегида, и несколько больше относительной силы
тротила и пикриновой кислоты, что видно из следующих данных
(инициирующий заряд гремучей ртути во всех случаях 0,30 г).
Название бризантного вещества Давление Вес измельченного песка г
Kzjcjf- всем заря- дом ннтропро- изводным
Тринитрорезорцин 239,0 52,9 41.2
Тринитротолуол - . 239,0 55,3 43 6
Тетранитроанилин . . 239.0 56,3 44-6
Пикриновая кислота 239,0 57,0 45-3
Гексанитроднфениламгш . . 239.0 60.2 49.5
Тринитробензальдегид . 239,0 62,8 51,1
Диазодинитрофенол . . 239,0 63,9 52.2
Тетрил 239.0 65 9 54,2
Диазо динитрофенол при взрыве разлагается по уравнению:
CcH2N4O6 4СО + 2С + Н2О + 2N2
При прибавлении к нему окислителей относительная сила взрыва
смеси не ниже силы взрыва чистого диазо динитрофено ла.
Смесь диазодинитрофенола с аммонийной селитрой при содержа-
нии последней 43,5% при взрыве разлагается по уравнению:
CcH3N4O5 + 2NH4NO3 -> 4СО + 5Н2О + СО2 + 4NS + С
а при содержании NH4NO3 75%:
CGHaN4O5 4- SNHjNOg -> 6СО.2+ 17H2O4-10N8
Инициирующая способность диазодипитрофепола, по данным
Кларка, несколько ниже инициирующей способности азида свинца
и значительно выше, чем гремучей ртути, что видно из следующих
величин предельного заряда:
Инициирующие ВВ Предельный заряд
по тротилу по тетрилу | по тетранн- троанилину
Гремучая ртуть . Диазодннитрофенол . Азид свинца ... 0,240 0,163 0,160 0,165 0 075 0,030 0175 0,083 0,050
Опыты же инж. Беньковского не согласуются с данными Кларка!
и показывают меньшую инициирующую способность диазодинитро-
фенола по сравнению с гремучей ртутью.
300
Динитродиазофенол нечувствителен к рассеянному свету, однако
при действии прямых солнечных лучей он быстро темнеет, причем
при экспозиции более 5 час. его взрывчатые свойства несколько
убывают. Хорошо выдерживает нагревание до 60°, но при более
высокой температуре наблюдается незначительный распад его. Так,
потеря в весе после нагревания при 75° в течение 48 час. равна 0,3%,
а после нагревания при 100° в течение 96 час. 1,25%. Температура
вспышки технического продукта 170—193°, перекристаллизованного
из ацетона 173,5°. Диазодинитрофенол растворим в нитробензоле,
ацетоне, пиридине, анилине, уксусной кислоте, крепкой соляной
кислоте, ацетонхлориде, глицеринтринитрате и ряде других обычных
органических растворителей. Лучшими же растворителями его яв-
ляются этилацетат, этиловый и метиловый спирты (соответственно
2,45, 2,43 и 1,25 г в 100 г растворителя).
Диазодинитрофенол можно перекристаллизовать из нитробензоль-
ного или ацетонного раствора, вливая последние в холодный эфир.
Осажденный из ацетонного раствора диазодинитрофенол имеет вид
пластинок (прямоугольники и квадраты). Его можно также очистить
прибавлением большого избытка ледяной воды к быстро перемеши-
ваемому горячему ацетонному раствору; при этом оседает яркожелтый
аморфный порошок. Разлагается он водными растворами щелочей
(и даже аммиака) на холоду с выделением азота, динитрофенола
и других продуктов. Горячая концентрированная серная кислота
также разлагает диазодинитрофспол.
Диазодинигрофенол может применяться как в качестве самостоя-
тельного инициатора, так и в смеси с другими веществами для вос-
пламенительных составов в капсюлях-воспламенителях.
Haules в 1916 г. взял патент на применение диазодинитрофенола
в качестве инициирующего ВВ. Затем Герц в 1923 г. предложил
применить его для ударных составов капсюлей-воспламенителей.
Также предлагали использовать диазодинитрофеиол в смеси с нитро-
соединениями в качестве инициатора для торпед, мин, гранат и т. п.
В 1932 г. были рекомендованы следующие составы с диазодинитро-
фенолом (ам. пат. № 1862295):
Наименование веществ Сорт 1 Сорт П
Диазодинитрофенол . 37 4
Тетразен 3 1
ТНРС . . — 40
Pb(NO„)2 30 29
Pb(CNS)s . 7 7
Стекло . . 19 19
Попутно следует отметить, что смеси эти в отличие от гремуче-
ртутных смесей не вызывают коррозии ружейных стволов.
301
В to^j же 1932 г. Кайзер рекомендовал применение диазодинитро-
фенола в следующем составе капсюлей-воспламенителей:
дназод! нитрофенола.... 0,45 г
дппнкратпарафепилевдиамин 0,15 »
Ba(NO3)2 ................... 0,20 »
стекла 0,20 »
Входящий в данный состав дипнкратпарафенилендиамин представ-
ляет собой
NH^O-C^NO^
О I
NH2.O.C6H2(NO2)3
и может быть получен растворением при нагревании диаминобензола
в спиртовом растворе пикрата и кипячением полученного раствора
в течение нескольких минут.
§ 6. ОРГАНИЧЕСКИЕ ПЕРЕКИСИ
Органические перекиси следует рассматривать как производные
перекиси водорода, в которой либо один атом, либо оба атома водо-
рода замещены одновалентной или двухвалентной группой.
Все органические перекиси в той или иной степени представляют
собой более или менее сильные ВВ, чувствительные как к быстрому
нагреванию, так и к механическим воздействиям (удару, трению).
Однако не все перекиси являются веществами инициирующими.
Следует отметить, что усложнение радикала в перекисях влечет за
собой падение как инициирующих свойств, так и взрывчатых свойств
вещества вообще. Например, гидроперекись уксусной кислоты
.О
ХО —О —Н
крайне взрывчата; при медленном нагревании до 110° она взрывает
с большой силой. В то же время гидроперекись пропионовой кис-
О
лоты СН3—СНо—С/ при нагревании лишь вспыхивает, а гид-
4 О—ОН
У°
роперекись масляной кислоты СН8—СН,—СНЧ—Cf вспыхи-
ХО—ОН
вает еще слабее, причем происходит частичное ее обугливание.
То же можно заметить и при рассмотрении гидроперекисей алки-
лов. Так, например, гидроперекись метила СН3—О—ОН настолько
взрывчата, что ее изучение затруднено вследствие этого, в то время
как гидроперекись этила СН3—СН2—О—ОН является взрывчатым
веществом, поддающимся изучению.
302
Таковыми являются и перекиси — продукта замещения двух
атомов водорода.
Наряду с этим увеличение количества пероксидных групп
- О—О— влечет за собой повышение взрывчатых свойств вещества.
По своим физико-химическим свойствам все перекиси являются
веществами нейтральными. Исключение составляют гидроперекиси,
обладающие очень сильными кислотными свойствами и могущие
образовывать с гидроксидами щелочных и щелочноземельных метал-
лов растворимые в воде кристаллические соли.
Общий метод получения органических перекисей состоит в дей-
ствии на органические вещества (алкилы, ацилы, кетоны, альдегиды)
перекиси водорода, гидрата перекиси натрия или озона. Кроме того,
возможно образование перекисей при расщеплении озонидов.
Качественно органические перекиси могут быть обнаружены сле-
дующими реакциями:
1. Вытеснением иода из йодистого калия. Трудно растворимые
в воде перекиси реагируют с KJ крайне медленно, но процесс можно
ускорить прибавлением ледяной уксусной кислота или спирта, повы-
шающих растворимость перекисей.
2. Обесцвечиванием индиго в присутствии концентрированной
серной кислоты.
3. Переводом ацетата марганца в двуокись марганца.'
4. Окислением закисных соединений в окисные.
5. Получением хлора из соляной кислоты.
О чистоте продукта можно судить по количеству содержащегося
в нем активного кислорода. Методы определения активного кислорода
основаны на измерении:
1. Количества иода, вытесненного перекисью из KJ.
2. Количества хлорида олова, необходимого для расщепления,
перекиси.
3. Количества водорода, необходимого для восстановления пере-
киси.
4. Количества индикотиисульфокислоты, окисляемой перекисью.
В качестве примеров перекисей, являющихся инициирующими ВВ,
рассмотрим перекись ацетона, перекись трициклоацетона, перекись
бензоила и гексаметилентрипероксиддиамин.
Перекись ацетона легко и с хорошим выходим полу-
чается при действии ацетона на концентрированную перекись во-
дорода в кислом растворе. Обычно ее получают при прибавлении кон-
центрированной соляной кислоты к смеси ацетона с концентриро-
ванной перекисью водорода. Строение перекиси ацетона следующее-
,0-0 ,сн3
с<
хо ох си.,
СН3х
сн/
Перекись ацетона представляет собой белые мелкие кристаллы-
обладающие достаточно высокой химической стойкостью, плавящиеся
без разложения при 95—95,5° и выдерживающие продолжительное
ЗОВ
нагревание при 100°. Температура вспышки около 180°. К удару
и трению перекись ацетона чувствительна, однако, ее можно без-
опасно прессовать.
По repaid пат.. № 423176, 192.5 п—рекомендуется снаряжать кап-
сюли I г тринитротолуола и<Х),3\г перекиси ацётонй.
-П е-р-е-ктг с ь—г р и ц"1Гк ло'а'ц е тона? Это вещество, откры-
тое Вольфенштейном, имеет формулу (С8О2Н0)3:
(СН8)2 = С- О — О — С = (СН3)2
о о
°\ /° 8 г. ацетона- 10.1 и
с-(сн3)3 13г. КВД.ИСЫ8.1 и
Получается перекись трициклоацетона, по Байеру и Веллигеру,
следующим образом.
8 г ацетона смешивают при охлаждении ледяной водой с 16 г
30%-ной перекиси водорода. Затем при продолжающемся хорошем
охлаждении и перемешивании прибавляют 19 г концентрированной
соляной кислоты. Полученный продукт отфильтровывают, промы-
вают водой до нейтральной реакции и перекристаллизовывают из
ацетона.
Перекись трицикло ацето на представляет собой небольшого раз-
мера белые кристаллы, очень летучие и обладающие запахом, напо-
минающим запах ацетона. Температура плавления точно не опре-
делена (указывают пределы 90—97°).
Патри, изучая трициклоацетон, пришел к выводу, что характер
взрыва его при нагревании не зависит от температуры, до которой
нагревается вещество. Повышение температуры нагревания не во
всех случаях приводит к детонации. Опыты Патри показали, что
трициклоацетон:
1. При нагревании до 97° медленно возгоняется.
2. При нагревании между 97 и 245° вначале плавится, а затем
улетучивается.
3. При нагревании между 245 и 250° претерпевает неясно выра-
женное разложение, в течение которого в зависимости от ряда обстоя-
тельств может происходить: а) горение, сопровождающееся коптящим
пламенем; б) детонация; в) разложение без пламени, представляющее
собой нечто промежуточное между горением и детонацией.
4. При нагревании в интервале температур 250—285° дает дето-
нацию.
5. При нагревании до 285—305° ведет себя так же, как и при
температурах 245—250°.
6. При 305° и выше горит с коптящим пламенем.
Изменение плотности запрессованной в оболочку перекиси три-
циклоацетона видно из табл. 66.
304
Предельный инициирующий заряд пере-
киси трициклоацетона по тетрилу равен
0,09 г, а по тротилу 0,18 г.
Г ексаметилентрипероксид-
диамин (ГМТД) впервые был получен
Лангером в 1885 г.; предложен в качестве
инициирующего ВВ в 1912 г. Гирзевальдом
(герм. пат. №274522, 1912 г.). Получается он
внесением мелкораздробленного уротропина
(гексаметилентетрамина) в перекись водорода.
Наилучший выход, по Гирзевальду (герм,
пат. № 283459, 1912 г.), обеспечивается при
следующих условиях.
28 г гексаметилентетрамина и 42 г лимон-
ной кислоты растворяют в 140 г 30%-ной
перекиси водорода. Роль лимонной кислоты
сводится к связыванию выделяющегося при
реакции аммиака. Реакция протекает спо-
койно в течение 10—20 час. При этом наблю-
дают, чтобы температура не поднималась выше 30°. В результате
реакции из раствора выпадает белый мелкокристаллический осадок
ГМТД. Кристаллы продукта—ромбической системы. Осадок промы-
вают водой и спиртом и сушат. Выход около 65—70% от теоретического
в расчете на гексаметилентетрамин.
Тейлор и Ринкенбах несколько изменили соотношения исходных
продуктов. Их опыты показали, что хороший выход обеспечивается
при следующей пропорции:
Таблица 66
Плотность запрессовки
перекиси трициклоацето-
ва в зависимости от дав-
ления прессования
Давление прессования ат Плотность г/см9
25 0,70
50 0,80
75 0,85
100 0,90
200 1,00
500 1.15
750 1,20
1000 1,20
гексаметилентетрамина . . 50 г
лймоннбйТ<ислоты Г'т.................... 84 »
"перекиси водорода (30%-noii)........... 158 »
Реакция, как и в первом случае, идет по уравнению
CeH18N4 + ЗН2О2 -> C6H12N2O6 + 2NH3
Выход 68—71% от теоретического при расчете на гексаметилен-
тетрамин.
Опыты автора показали, что при уменьшении перекиси водорода
до количества, рекомендуемого Тейлором и Ринкенбахом, уротропин
не окисляется полностью, а частично остается в продукте реакции.
Об этом свидетельствует то, что содержание азота в нем, определен-
ное методом гидрирования, обычно больше теоретического.
По Байеру и Веллигеру, гексаметилентрипероксиддиамин может
* быть получен следующим образом: 50 г сернокислого аммония рас-
творяют при нагревании в 50 г 30%-ной перекиси водорода. Раствор
фильтруют и при 55° смешивают с 40%-ным раствором формалина.
В результате реакции выпадает белый кристаллический осадок,
который отделяют от раствора и промывают водой.
Бубнов-92—20 305
г
По Легору, ГМТД может быть получен также действием аммиака
„а перекись формальдегида:
/0-0.
2СН20 + 2Н2О2 -> СНУ )СН2 4- 2Н,0
хо—си
,0 0
ЗСН2" )CHZ + 2NH3 -> С„Н12ОЛ + 6Н2О
oz
Строение гексаметилентрипероксиддиамина по одним представ-
лениям:
О —СН2Х ,сн2— о
I >n-ch2 — о — О — CH2-Nz I
О—сн2 О
по другим:
ZCH2 — О — О — СП2\
N—СН2 - О - О - СН2 -N
ЧСН2—О 0-СН2/
Давление прессования кг/слР Плот- ность
100 1,64
200 1,15
«00 1,30
Последнюю формулу считают более близкой к истине.
Удельный вес ГМТД d=l,57. Плотность
запрессовки в зависимости от давления прес-
сования может быть выражена следующими
цифрами (см. таблицу).
Он весьма чувствителен к удару; при испы-
тании на копре Велера показывает чувстви-
тельность по высоте падения груза порядка
100 мм. Температура вспышки около 139°.
При поджигании легко вспыхивает и дето-
нирует без отказа. При растирании в фарфо-
ровой ступке взрывает. Его инициирующая способность характери-
зуется следующими цифрами:
по тротилу ........................ 0,08 г
по пикриновой кислоте - 0,00 »
по тетрилу - • • • 0,00 »
т. е. превосходит гремучую ртуть более чем в четыре раза.
Стойкость по отношению к нагреванию небольшая. При нагре-
вании в присутствии влаги он разлагается уже через 40 час. При
нормальной же темпе-
ратуре в закрытом со-
суде его можно хранить
в течение продолжитель-
ного времени. Тейлор
и Ринке нбах приводят
следующие данные об
отношении ГМТД к по-
вышенной температуре:
306
Время на- гревания «ас. Потеря веса, %
60° 75° 100°
2 . 0,1 0.25 3,25
8 . 0,35 0.60 39.0
24 0.50 1,30 68,0
48 . 0,50 2,25 - 1
Растворимость ГМТД в различных растворителях относительно
невелика:
в 100 г воды .
» 100 » спирта - .
»> 100 » эфира
» 100 » ацетона - .
» 100 » хлороформа -
0,01 г
0,01 »
0,017 »
0,33 »
0,64 »
В воде гидролизует. При кипячении с водой разлагается:
CeH12OeN2 4- 6Н2О -> 2NH3+3C2H4(OH)2 + 3O2
С2Н4(ОН)2 + О2 -> Н2О 4- НСНО + нсоон
То -же происходит в кислой и щелочной средах. ГМТД — ней-
тральное вещество. При обычных условиях совершенно негигроско-
пичен, на' металлы, повидимому, не производит никакого действия.
Перекись бензоила СвНб—СО—О—О—ОС—СвНв. 100 г
10%-ной перекиси водорода взбалтывают с соответствующим коли-
чеством хлористого бензоила и раствора гидроксида натрия до тех
пор, пока не прекратится выделение осадка и не пропадет запах
хлористого бензоила. Отфильтрованную перекись перекристаллизо-
вывают из кипящего спирта с добавкой небольшого количества воды.
Так как перекись бензоила растворима и в хлороформе, то можно
перекристаллизовывать ее из раствора в хлороформе осаждением
метиловым спиртом. Температура плавления перекиси 106—108°.
Впервые она была предложена в качестве инициатора по англ. пат.
№ 23450. Однако перекись бензоила — худший инициатор, чем пере-
кись ацетона.
§ 7. ПРОИЗВОДНЫЕ ТЕТРАЗОЛА
Тетразол представляет собой органическое соединение, принад-
лежащее к пятичленной гетероциклической системе, состоящей из
четырех атомов азота, замкнутых одним атомом углерода:
3 4
N — N 5
II >СН
N—NHZ
2 1
4 5
N = CH i
или | /NH
N — N /
3 2
Впервые тетразол был получен в 1885 г. при действии азотистой
кислоты на дицианфенилгидразин. Впоследствии было установлено,
что тетразол является родоначальником целого класса органических
соединений.
Тетразол образуется:
1) при восстановлении диазотетразола двухлористым оловом;
2) при окислении формазиловых соединений в азотнокислой среде
перманганатом калия;
307
3) при постепенном окислении дифенилтетразола по следующей
схеме:
zN — N — ceH5 ,;N - N - С6Н5
НвСсС< | -> НООС—С< | ->
XN = N XN = N
НС
.N — NH
f H
XN = N
NH - NH2
4) путем обработки метилтиосемикарбазида С — SCH3 азота-
XNH2
стой кислотой с последовательным окислением полученного таким
образом метилмеркаптотетразола:
N— N
II >CSCHa
N —NHZ
азотистой кислотой и затем перманганатом калия;
5) при окислении бензтетразолка рборундовой кислоты в щелочной
среде перманганатом калия;
6) конденсацией формимидхлора (из HNC и НС1):
Н,
ХС = NH
СИ
с азотистоводородной кислотой.
Этот способ аналогичен способу Тиле, получившего еще в 1892 г.
аминотетразол перегруппировкой карбамидимидазида:
,NH N — N
H2NC< -> HaN —С< II
XN3 XNH — N
Оливерий Мандола, вводя циан в конденсированный водный рас-
твор азотистоводородной кислоты, получил циантетразол, который
после омыления щелочью и отщепления СО2 от образовавшейся
тетразо лкарбазоловой кислоты дает тетразол по схеме:
N—N х N —N N — N к
|| >С • CN -> || >С • СООН -> || >СН
N —N— NHZ N —NHZ
В литературе описывается также процесс распада производных
тетразола и нафталина при окислении их перманганатом калия
в уксуснокислой среде с количественным выходом тетразола.
Из приведенного обзора можно заключить, что для синтеза тетра-
зола исходными продуктами являются вещества, богатые азотом:
308
диазосоединения, производные гидразина, азотистоводородная кис-
лота и ее органические производные. С другой стороны, тетразол
может быть получен в результате распада его производных. Тетразол
является слабой одноосновной кислотой. Кристаллизуется из уксусно-
этилового эфира в виде бесцветных иголочек или листочков, имею-
щих температуру плавления 155—156°. Легко растворим в воде и
спирте, трудно — в эфире и бензоле. Способен возгоняться. Обра-
зует легко растворимую в воде натриевую соль, кристаллизующуюся
в призмы с 1 мол воды. Тетразол отличается большой стойкостью
к химическим реагентам и к механическим воздействиям. Что ка-
сается производных тетразола, то надлежит отметить, что и в них
тетразольное кольцо удерживается достаточно прочно. Ярким
примером может служить производное тетразола — фениЛтетразол.
то вещество очень стойко по отношению к кислотам и щелочам;
при обработке дымящейся азотной кислотой дает нитрофенилтетра-
зол, причем нитрогруппа может восстанавливаться. После же окис-
ления перманганатом калия в кислом растворе вновь образуется
тетразол по следующей схеме:
N — N-x hno3 N — Н н.
II YH —> II СН -4
N N( N —
\ссн5 VcHaNO2
н2 N — N-a kmho4 N •— N
-> II zCH II 'CH
N - - N—nhz
\CcH4NH2
Будучи связан боковой цепью с несколькими атомами азота,
тетразол становится крайне чувствительным к механическим воз-
действиям, приобретая при этом сильно взрывчатые свойства. До-
статочно указать на диазотетразол, который самопроизвольно взры-
вает в водных растворах при концентрации выше 6—7% даже при
температуре 0°.
Тетразол, являясь «родоначальником» целого класса соединений,
в противоположность обычному правилу не может служить непосред-
ственным исходным продуктом для их синтеза.
Причина этого явления кроется в сравнительной трудности и
сложности процесса получения самого тетразола, с одной стороны,
и в легкости и доступности методов получения производных тетра-
зола из соединений, иногда совершенно отличных от тетразола и
принадлежащих к другим классам.
Рассмотрим некоторые производные тетразола.
N—N
А м и н о т е т р а з о л || ^>С—NHg может быть получен по
N—NHZ
способу Штоле, который заключается в конденсации азотистоводо-
309
znh8
родной кислоты HN3 с дициандиамидом NH==C<
XNH • CN
или
из
аминогуамидинбикарбоната по способу Тиле посредством диазиро-
вания его в азотнокислой среде в карбамидимидазид (гуамилазид)
и перегруппировки при кипячении с раствором соды.
68 г аминогуанидинбикарбоната постепенным прибавлением рас-
творяют в 200 мл азотной кислоты (32%-ной) при помешивании:
yNH2 /NH2
C=NH • Н2СО3 + HNO3 -> С NH. HNO3 + H.2O + C02
^NH2 \nH NH,
Полученный азотнокислый аминогуанидин диазотируют в азотно"
кислой среде при температуре не выше 25—30° постепенным при-
бавлением 35%-ного раствора нитрита натрия (90—100 мл) в течение
10—15 мин. при охлаждении реакционного сосуда водой:
/NH„
C=NH HNO3 + NaNO2 + HNO3 ->
^NH NHS
/NH„ ,NH,
- C=NH -> C=NH42H3O + NaNOs
XNH-N = N —NOS \n8
Конец диазотирования определяют реакцией на иодокрахмаль-
ную бумажку. Раствор до конца диазотирования должен быть кислым
(проба на конго-красную бумажку).
Затем раствор переносят в круглодонную колбу, куда при пере-
мешивании небольшими порциями прибавляют 58 г соды. Колбу
соединяют с обратным холодильником и нагревают в течение 4 час.
при температуре кипения Жидкости:
/NH2
C=NH 4- Na2CO3 ->
^NH —N = N —NO3
/N — N
-> HaNC || + NaNOs + NaHCO,
VNH—N
После нейтрализации 30%-ной серной кислотой содержимое колбы
переливают в кристаллизатор. Выпавшие кристаллы отжимают на
воронке Бюхнера и промывают два-три раза ледяной водой. Темпера-
тура плавления получаемого продукта 199—20Г.
310
Аминотетразол является слабой одноосновной кислотой. С метал-
лами образует соли типа:
N — N
н ;c-nh2
N —
XR
Свободный аминотетразол кристаллизуется из воды в виде бесцвет-
ных призм или блестящих листочков с 1 мол воды. При нагревании
до 100° теряет воду,.а при 199—203° — плавится. По Тиле, 1 ч. воды
растворяет при 18,4° 85,25 ч. амииотетразола.
Натриевая соль аминотетразола кристаллизуется в виде больших
желтых призм с 3 мол воды, легко растворяясь в воде. Раствор вслед-
ствие гидролиза обладает щелочной реакцией. Несмотря на наличие
ацидифирующей группы ^СН, аминотетразол обладает частично и
щелочными свойствами, что видно из его способности давать с кис-
лотами соли типа —NH„ • НС1.
Последние в водном растворе легко подвергаются гидролизу и
при кристаллизации из воды выделяют свободный аминотетразол.
Характерной реакцией на свободный аминотетразол является
образование аморфного осадка зеленого цвета при взаимодействии
с раствором сульфата меди в присутствии ацетата натрия. Осадок
этот нерастворим в уксусной кислоте; растворяется в НС1.
Как аминотетразол, так и его натриевая соль к удару нечувстви-
тельны. При нагревании слабо вспыхивают.
N—ЬК
Диазотетразол || —N =N • Cl получается по спо-
N—NHZ
собу Тиле диазотированием аминотетразола в солянокислой среде.
5 г аминотетразола растворяют в 30 мл воды, куда прибавляют
сначала 6 мл 25%-ного раствора едкого натра, а затем 3,4 г нитрита
натрия. По растворении жидкость охлаждают ледяной водой и из
капельной воронки медленно в течение 10—12 мин. при перемеши-
вании прибавляют к смеси, состоящей из 170 г льда и 16 мл соляной
кислоты уд. веса 1,15 (30%-ной). Смесь охлаждают все время ледя-
ной водой, с тем чтобы температура не превышала 0°.
Жидкость в конце реакции принимает зеленоватую окраску.
Реакция протекает по следующему уравнению:
N — N .
|| >С — NH2 4- NaOH + NaNO2 + ЗНС1 ->
N —
N—N .
-> II >C —N = NCl-|-2NaC14-2H2O
N —NHZ
311
Диазотетразол представляет собой чрезвычайно чуне ши ic.ni.нос В|.
водные растворы его при концентрациях выше б—' 7%, даже при п
самопроизвольно взрывают, дробя стеклянную посуду, в которой
ведется процесс диазотирования.
Растворы диазотетразола после их нейтрализации щелочью дани
вполне устойчивые, легко растворимые в воде, изодиазотаты:
N-Nx
|| \С—N = N — ONa
N —N<
^Na
Поэтому обычно получают 2,5—3%-ный раствор диазотетразоло
с применением надлежащих мер предосторожности (щиты) и немед-
ленно же, если он не нужен для дальнейших синтезов, переводят
его добавлением 25 мл 25%-ного раствора едкого натра (до щелоч-
ной реакции) в натриевую соль диазотата. Если же диазотетразол
необходим для дальнейших синтезов, раствор изодиазотата натрия
охлаждают до 0° или ниже и к нему прибавляют соответствующее
количество кислоты.
Йзодиазотат натрия представляет собой белые иглы. При отсут-
ствии углекислого газа не разлагается при кипячении в водном
растворе. Натриевая соль диазотетразола при нагревании лишь
слабо вспыхивает. В нейтральном или слабощелочном растворе
натриевая соль диазотетразола дает с солями свинца свинцовую соль
оксиазотетразола, нерастворимую в воде, спирте и эфире, но легко
растворимую в соляной кислоте, едком натре и аммиаке. Свинцовая
соль взрывает при нагревании до 360°.
П. Атти описал процесс получения окси- и метилтетразолов
в- результате конденсации азотистоводородной и гремучей кислот
с метилкарбиламином:
N — N. N —N-
|| \СН и II ,'СН
N —N< N-N<
\он xCHj
Он же исследовал азоокситетразол
] \СН
NH — iC
!О
Сложным путем, исходя из тиосемикарбазида
C—SH
XNHNH3
312
посредством мстилпровипня, конденсации, омыления, окисления и
сплавления со щелочью получается 5-иксигстр.иол.
Штоле, вводя при 60° раствор диазоте гразола в суспензию Сп(ОН)2
в водной среде, получил тот же 5-окситетразол с температурой плав-
ления 254°, растворимый в воде.
5-5-бистетразол, Энергичным действием азотистоводородной
кислоты на циантетразол получается 5-5-бистетразол, в котором
два тетразо льных кольца связаны между собой:
N — N . — N
11 ;с—с/ и
N-— NHZ ;'NH —N
При комнатной температуре это вещество оказалось стойким
к действию кислот и щелочей. Кристаллы его (призмы) плавятся
при температуре 254°. Вещество обладает кислым характером, легко
растворимо в воде, растворяется в спирте и ацетоне, трудно в эфире
и нерастворимо в бензоле.
Виланд еще раньше синтезировал бисоксибистетразол:
N — N. N — N
и >с - а у
N - N< >N — N
хон oi-и
действуя азотистой кислотой на оксалдигидразидоксим
H2N — NHX zNHNH2
_____________£/
НО —№ \N —ОН
5-5-бисдиазотетразолгидразин
N —N ч <N — N
К >С — N = N — NH • - NH — N = N —Ci 11
N — NHZ XNH —N
Это вещество получается путем купелирования диазотетразола
с солянокислым гадразином в присутствии ацетата натрия-
Так, например, 0,75 г гидразинхлорида и 1,5 г ацетата натрия
растворяют в 3 л/л воды. К раствору прибавляют 3 г льда. К охла-
жденному извне льдом с поваренной солью раствору постепенно
при помешивании приливают охлажденный раствор диазотетразола.
Реакция происходит по следующему уравнению:
N —N
2 11 )С —N = NC1 4- H2N — NH2 • 2НС1 ->
N—NHZ
N — Nv zN - N
-» || ЭС —N = N—NH-NH N = N —C< li-J-4HC1
N—NHZ XNH— N
313
Выпавший аморфный осадок желтоватого цвета промывают ледяной
водой, спиртом и эфиром и сушат при комнатной температуре. Работа
эта требует чрезвычайной осторожности, так как соединение в силу
накопления в его молекуле 14 атомов азота обладает сильными взрыв-
чатыми свойствами. При 90° оно может взрывать. Это соединение
трудно растворимо в воде и в органических растворителях. Чистый
продукт, хорошо промытый спиртом, плавится при 120—123°. Кри-
сталлизуется в виде желтовато-окрашенных табличек с характерными
звездообразно заостренными углами. При комнатной температуре
оно достаточно- стойко, но очень чувствительно к удару и трению.
Щелочами быстро разлагается с образованием тетразилазида, амино-
тетразола, азота, аммиака и циана. При хранении под водой проис-
ходит медленное его разложение вследствие гидролиза.
Если в водную суспензию бисдиазотетразолгидразина при 0°
прибавить 20%-ный раствор сульфата меди, то выпадает зеленоватый
исадок, имеющий температуру вспышки 185° и взрывающийся от
трения, удара, накола и луча огня-
5-5'- азотетразол. Азотетразол получается окислением
аминотетразола перманганатом калия в щелочной среде (избыток
NaOH). Свободный азотетразол вследствие его нестойкости получен
быть не может; обычно получают натриевую соль азотетразола.
51г аминотетразола растворяют в круглодонной колбе в 500 мл
15%-ного раствора едкого натра при нагревании до температуры 100°.
Затем при помешивании постепенно прибавляют 70 г порошкообраз-
ного перманганата калия до неисчезающей окраски жидкости. При
этом наблюдается сильное вскипание жидкости, очевидно вследствие
термического эффекта реакции окисления:
N — N,
2 || УС - NHS + 4КМпО4 4- 2NaOH ->
N — Nz f
NN zN —N
-> [| X— N = N — Cx || 4-4KOH + 4MnO2 + 4H2O + O2.
N —Nz )N — N
xNa Naz
По окончании окисления аминотетразола избыток перманганата
калия удаляют прибавлением 1-—2 мл спирта и горячий раствор
быстро отфильтровывают от обильного осадка двуокиси марганца.
Осадок промывают горячей (около 100°) водой. Из фильтрата и про-
мывных вод после их испарения до 1/з первоначального объема вы-
кристаллизовывается натриевая соль азотетразола в виде желтых
призм. Выход около 7.0%.
Азотетразол является двухосновной кислотой. Двунатриевая соль,
кристаллизующаяся из кипящей воды, содержит 5 молекулы воды. В рас-
творе она нейтральна. Будучи обезвожена, натриевая соль от удара
и нагрева сильно взрывает. Температура вспышки 245—250°. При
подкислении раствора натриевой соли выделяется азотетразол, кото-
314
рый немедленно разлагается с образованием тетразилгидразина,
муравьиной кислоты и азота по следующей схеме:
N — N N — N
II >С —N = N —С< ] ->
N —NH' XNH— N
N — N
-> 2N£+ II %C —NH — N = C
N — NHZ
Последнее гипотетическое вещество гидролизует:
N —N x
И )C — NH -N = C+ H2O
N — NH'
N — N
-> II Sc —NH —NH2 + HCOOH
N - NHZ
Свинцовая соль азотетразола, по исследованиям Лосева, может
быть получена смешением нагретых до 90—95° растворов натриевой
соли азотетразола (15%-ного) и нитрата свинца (20%-ного) и отстаи-
вания на остывающей водяной бане в течение 3 час. При этом проис-
ходит реакция:
WoNaa-SHsO^-P^NO^ -> CaN10Pb. 5Н2О + 2NaNO3
Свинцовая соль нерастворима в воде и органических растворите-
лях. Растворяется в слабых кислотах и щелочах. Температура вспышки
199—202°. При хранении в течение месяца при 40° не изменяет своих
свойств. От луча огня вспыхивает, от трения и удара взрывает.
Железная соль азотетразола получается смешением 30%-ных рас-
творов натриевой соли азотетразола и сульфата железа при 25°.
Выпадает черный коллоидальный осадок. После часового продувания
через раствор воздуха и трехчасового отстаивания отфильтровывают
мелкие темнокоричневые кристаллы и промывают водой, спиртом и
эфиром. Вещество плохо растворимо в воде и спирте, нерастворимо
в ацетоне; слабые щелочи и растворы солей растворяют эту соль.
От трения и удара вещество не взрывает. Температура его вспышки
164—165°.
Цинковая соль азотетразола получается смешением 30%-ных рас-
творов натриевой соли азотетразола и сернокислого цинка при 25°.
Выпадающие после часового продувания воздуха и трехчасового
отстаивания желтые блестящие кристаллы в виде ромбов промывают
на фильтре водой, спиртом и эфиром. Соль в воде растворима. От
трения и удара не взрывает. Температура вспышки 166—176°. При
температуре 40° не изменяется, однако, при повышении темпера-
туры до 60° разлагается.
315
При смешении 10%-ного раствора натриевой соли азотетразола
и сульфата меди после часовой продувки воздухом образуется голубо-
ватый осадок медной соли. Ее фильтруют, промывают водой, спиртом
и эфиром и сушат при 40°. Температура вспышки 155°. В воде она
мало растворима. Соль очень чувствительна к трению, удару, наколу
и лучу огня-
Тетразилазид. В литературе это соединение известно также
под названием тетразилазоимида, диазотетразол имида или азидо-
тетразола. Его структурная формула:
N — N v ZN
II )С— N< II
N — NHZ
Это соединение тетразола получается диазотированием тетразол-
гидразина, образующегося, как выше было изложено, при подкисле-
нии натриевой соли азотетразола.
По Тиле, тетразилазид можно получить следующим образом.
К нагретому до 60—70° раствору натриевой соли азотетразола при-
бавляют соответствующее количество соляной кислоты и образую-
щийся тетразилгидразин диазотируют обычным порядком при охла-
ждении.
Тиле получал тетразилазид, восстанавливая диазотетразол SnCl2.
Тетразилазид является самым богатым по содержанию азота
углеродным соединением. От малейшего трения он взрывает. Из
бензола он кристаллизуется в виде белых иголочек. Легко растворим
в воде, ацетоне и метиловом спирте. В эфире нерастворим. Реакция
его кислая. Аммонийная соль CN7H • NH3 может быть получена
пропусканием аммиака через раствор тетразилазида в бензоле. Оиа
представляет собой белый порошок, при комнатной температуре
достаточно постоянный. При прибавлении к водному раствору тетра-
зилазида раствора сульфата меди выпадает ^еленая медная соль его,
очень чувствительная к трению, удару и лучу огня.- Температура
вспышки 170°.
5 -5'-диазоаминотетр азо л. Согласно указанию Гоф-
мана это соединение легко получается диазотированием аминогуани-
диннитрата в уксуснокислой среде в присутствии ацетата натрия.
По Лосеву, 25 г аминогуанидиннитрата растворяют при нагре-
вании в 125 мл 12—13%-ной уксусной кислоты, добавляют 12,5 г
уксуснокислого натрия и по охлаждении жидкости до 8—10° мед-
ленно (при помешивании) в течение 30—40 мин. приливают раствор
нитрита (17,5 г в 75 мл Н2О). Температура реагирующей массы не
должна превышать 12—15°. Затем жидкость оставляют стоять при
комнатной температуре. При этом иногда наблюдается повышение
температуры до 25°. Выпавшие кристаллы отфильтровывают, про-
мывают ледяной водой, подкисленной уксусной кислотой, и -сушат
при комнатной температуре.
Другой путь его получения, экономически менее выгодный, со-
стоит в диазотировании аминотетразона.
316
Диазоаминотетразол является трехосновной кислотой строения:
N —N ч N — N
|| >С — N=N—NH— Ct ||
N- NHZ XNH —N
Как самый диазоаминотетразол, так и его соли отличаются по-
стоянством. Мононатриевая соль легко растворима в воде и кристал-
лизуется в виде желтых игл.
Медная соль диазоамииотетразола получается следующим спосо-
бом: 43 г натриевой соЯи диазоаминотетразола растворяют в 350 мл
воды, к которой прибавлено 23 мл 25%-ного раствора едкого натра.
К полученному раствору при комнатной температуре при помеши-
вании приливают 350 мл 10%-ного раствора сульфата меди. Выпав-
ший осадок зеленовато-оливкового цвета отфильтровывают, промы-
вают водой, спиртом и эфиром и сушат при комнатной температуре.
Медная соль диазоаминотетразола трудно растворима в воде, не-
растворима в органических растворителях. К удару и трению почти
не чувствительна. При усиленном растирании в фарфоровой ступке
взрывает только часть продукта, находящегося непосредственно под
пестиком. От луча огня взрывает. Температура вспышки 195—220°.
Состав соли (C2Nu)2Cu3.
Двойная медноаммиачная соль диазоаминотетразола получается
при обработке осадка медной соли аммиаком. Осадок вначале рас-
творяется, а затем, через непродолжительное время, выпадает в виде
длинных призм, темнозеленого цвета. К трению и удару двойная соль
более чувствительна. Температура вспышки 220—230°. Состав соли
(C2Nn)3Cu3(NH3)2.
Свинцовая соль диазоаминотетразола получается смешением при
комнатной температуре 9—10%-ного раствора натриевой соли диазо-
аминотетразола с 10%-ным раствором ацетата свинца до полноты
осаждения. Если к выпавшему желтому аморфному осадку приба-
вить 5 мл азотной кислоты уд. веса 1,4 и разбавить равным количе-
ством воды, то! коллоидный характер осадка пропадает, и осадок
принимает мелкокристаллический вид. Чувствительность свинцовой
соли равна чувствительности медной соли.
Соли нитротетразола. Эти вещества в качестве ини-
циирующих предложены для снаряжения капсюлей-детонаторов и
капсюлей-воспламенителей (герм. пат. № 547685, 1931 г.). Нитро-
тетразол, имеющий строение
N — N х
II )с- no,
N — NI-K
может быть получен, по Гессу, в виде медной соли обработкой азо-
тистой кислотой диазотетразола в присутствии сульфата меди или
металлической меди.
317
Для получения медной соли нитротетр азола приливают тонкой
струей диазотетразол к кипящему водному раствору нитрита натрия
и медной соли. Реакция протекает с выделением азота и его окислов
по следующей схеме:
N-N
2 II /С — N N • Cl + 2NaNO2-|-Си" —•
N —NHZ
ДЛ - Гк
>Cu +2N3+2NaCl
N — N<
II >C —NO2
N — №
При этом выпадает зеленовато-голубоватый осадок медной соли
нитротетразола, который затем отфильтровывают, промывают холод-
ной водой и сушат при комнатной температуре. Она является хорошо
кристаллизующимся веществом, очень гигроскопичным и разлагаю-
щимся на воздухе.
Уже по одному тому, что образующийся при реакции нитротетра-
зол способен разлагать соли минеральных кислот, можно заключить,
что он является веществом с весьма сильными кислотными свой-
ствами и что нитрогруппа увеличивает кислотные свойства самого
тетразола. Хотя нитротетразол и его медная соль и обладают взрыв-
чатыми свойствами, однако вследствие своей гигроскопичности в прак-
тике они неприменимы.
Техническое значение могут иметь только соли тяжелых металлов:
серебра, ртути, свинца. Соли серебра и ртути могут быть получены
осаждением (при нагревании) концентрированных азотнокислых рас-
творов нитратов этих металлов водным раствором натриевой соли
нитротетразола. Применяемая для этой реакции натриевая соль
нитротетразола получается обработкой медной соли тётразола кипя-
щим раствором едкого натра. По охлаждении раствора натриевая соль
кристаллизуется в виде длинных призм с содержанием 4 молекул
воды. Основная свинцовая соль нитротетразола получается обработ-
кой горячего водного раствора нитротетразола избытком окиси свинца
или его гидрата:
N-N
II V-NOs
N — Nx N - N<
2 || V - NOB + PbO )Pb + HaQ.
N — Nz N-N<
II >C- NO2
N— Ж
Упомянутые выше соли нитротетразола обладают значительными
инициирующими свойствами, в четыре раза превосходящими азид
318
свинца. Эти соли обладают хорошей воспламеняемостью; так, напри-
мер, медная соль имеет температуру вспышки в пределах 210—216°.
При обычных условиях хранения они химически стойки, влажность
и углекислота на них не оказывают заметного действия.
Соли нитротетразола могут быть использованы не только в ка-
честве самостоятельных инициаторов, но и в качестве добавок к ини-
циирующим ВВ, обладающим относительно незначительным ускоре-
нием взрывчатого превращения, а вследствие этого и невысоким
инициирующим действием. Так, при добавке 10% ртутной соли нитро-
тетразола к гремучей ртути ее предельный инициирующий заряд
уменьшается в 5—6 раз, а при добавке 20% — в 40 раз. Предельный
заряд тринитрорезорцината свинца по тетрилу равен 0,03 г при
добавке 20% ртутной соли нитротетразола.
Эфиры нитротетразола также обладают хорошей инициирующей
способностью, но несколько меньшей, чем его соли. Так, например,
метиловый эфир нитротетразола дает в три раза лучшее инициирую-
щее действие по сравнению с гремучей ртутью.
Несмотря на все изложенные выше положительные свойства
нитротетразолов, промышленного значения они еще не имеют, так
как применяемый для их получения исходный продукт — диазотетра-
зол — вследствие своей неустойчивости весьма опасен в обращении..
§ 8. ГАЛОИДНЫЕ СОЕДИНЕНИЯ АЗОТА
Галоидные соединения азота имеют чисто научное значение, прак-
тического же применения в качестве инициаторов они не имеют.
Хлористый азот был открыт в 1812 г. Дюлонгом. Приготовляется
он действием хлора на концентрированный водный раствор хлори-
стого аммония. Реакция протекает по уравнению:
NH.C1 + ЗС12 -> NC13 + 4НС1,
Хлористый азот представляет собой маслянистую жидкость темно-
желтого цвета с сильным запахом хлора, уд. веса 1,653. Хлористый
азот растворяется в бензоле и других органических растворителях.
Это — эндотермическое соединение, взрывающееся не только под
влиянием удара,'но и от действия различных веществ: фосфора,
мышьяка, селена, различных смол, жиров и других органических
веществ, вернее всех веществ, на которые он может оказать хлори-
рующее действие. При температуре 93° пары хлористого азота легко
взрывают. Дюлонг при открытии хлористого азота из-за взрыва этого
вещества потерял глаз и три пальца. Деви и Фарадей были серьезно
ранены при исследовании NC13. Наряду с этим NC13 является ядови-
тым веществом.
Согласно Миллону при обработке хлористого азота раствором
бромида натрия или калия получается соединение типа NBr3—бро-
мистый азот, представляющий собой коричневое взрывчатое масло.
Данные эти, однако, еще не проверены окончательно.
319
Иодистый азот приготовляется посредством обработки иода или
водного раствора его в иодистом калии разбавленным раствором
соли аммония.
В сухом состоянии он взрывает от малейшего толчка, разлагаясь
на азот и иод. Медленно разлагается и в воде, даже на холоду. Отно-
сительно состава йодистого азота мы не знаем ничего определенного.
Только по аналогии с хлористым азотом иодистому азоту приписы-
вается формула NJS. Согласно результатам исследований Миллона,
Маршака и Вино, можно предполагать о существовании соединений
NHJ и NHJ2.
При разложении иодистый азот, так же как и хлористый азот, вы-
деляет небольшое количество тепла, поэтому полезная работа их при
взрыве ничтожна.
Согласно Г. В. Уаррену, электролизом концентрированного рас-
твора фтористого аммония может быть получен фтористый азот—
желтое масло, взрывающее или разлагающееся даже от контакта
со стеклом, салициловой кислотой и другими органическими веще-
ствами.
В 1934 г. G. Cady дал описание триоксифтористого азота, пред-
полагаемое строение которого следующее:
ч
J4-OF
о/
Способ получения его такой: фтор, для большей безопасности
смешанный с сухим воздухом, пропускают со скоростью 3 л/ч через
никелевую трубку, снабженную платиновым наконечником, в 3/V рас-
твор азотной кислоты, протекающей со скоростью 1 л/ч. Аппарат
закрывают металлической крышкой, посаженной на мастике из воска,
смолы и окиси железа. Такая легкая посадка сделана из соображений
сохранности аппарата в случае непредвиденного взрыва.
Триоксифтористый азот представляет собой газообразное веще-
ство. По выходе из аппарата газ направляют в сосуд, охлаждаемый
жидким кислородом, где он затвердевает в виде белых кристалликов,
плавящихся при—175°; жидкий триоксифтористый азот кипит при —42°.
Другие исследователи указывают, что образование триоксифто-
ристого азота происходит и при взаимодействии фтора с твердым
нитратом калия.
Триоксифтористый азот в запаянной стеклянной ампуле может
храниться не разлагаясь в течение многих дней. При сильном ударе
и при нагревании до 200—300° он взрывает. При соприкосновении
со спиртом, эфиром и другими органическими растворителями три-
оксифтористый азот также взрывает. С металлами (медь, латунь,
сталь V2A и железо) не взаимодействует.
Пары вещества имеют запах гнилого картофеля и уже в небольшом
количестве раздражают дыхательные пути и вызывают кашель, а при
больших концентрациях — головную боль и удушье.
320
§ 9. СЕРНИСТЫЙ АЗОТ
Сернистый азот N4Se сам по себе является сравнительно слабым
инициирующим веществом.
Из всех предложенных структурных формул его следует считать
наиболее отвечающей экспериментальным данным формулу, предло-
женную Руффом:
N=S S=N
N = S —N
Эта формула подтверждается следующим:
1) реакцией между сернистым азотом и хлористым водородом:
N4S4+12HC1 -> 4NH34-4S4-6C12
из которой можно заключить, что сера связана с азотом 12 валент-
ностями и, следовательно, атомы азота между собой не связаны;
2) составом металлсодержащих соединений, полученных из ам-
миачного раствора N4S4:
ZN\ ZN\
S = S< >Pb и S = Sq j:Hg
\N/ XnZ
3) данными, полученными С. А. Вознесенским при изучении
гидролиза.
Получение сернистого-азота может быть осуществлено двумя ме-
тодами. Первый из них основан на реакции между серой и жидким
безводным аммиаком. По Руффу и Гайселю, в этом случае проис-
ходит обратимая реакция:
I0S4-4NH3 61^8 +N4S4
Поэтому, если раствор серы в аммиаке обработать иодидом се-
ребра, то равновесие нарушается, и реакция идет вправо до конца.
После отделения выпавшего сульфида серебра фильтрат испаряют
и получают кристаллы сернистого азота. Метод этот весьма прост,
но обладает всеми неудобствами работы с жидким аммиаком.
Другой метод, предложенный Грегори и Субейраном, основан
на реакции между двухлористой серой в сероуглероде и газообраз-
ным аммиаком (в избытке). В результате реакции получаются хлорид
аммония, сера и сернистый азот:
6SC12 + 16NH3 -> N4S4 + 12NH4C1 + 2S
С. А. Вознесенский, пользуясь этой реакцией, получил выход
20% от теоретического. Он применял раствор двухлористой серы
в бензоле и через него в течение 4 час. пропускал газообразный ам-
миак, предварительно смешанный с воздухом, при охлаждении реак-
БубнОв—92—21 l.’l
ционного сосуда. По окончании процесса он фильтровал полученную
смесь через фланель; сернистый азот он экстрагировал бензолом,
по испарении бензола выкристаллизовывал и перекристаллизовывал.
По мнению Микаэлиса, сернистый азот образуется также при
действии газообразного аммиака на хлористый тиснил.
Сернистый азот получается в форме оранжево-желтых кристаллов
ромбической системы со слабым характерным запахом. Температура
плавления 179°, однако, еще до начала плавления сернистый азот
возгоняется и частично разлагается. При температурах выше точки
плавления он взрывает.
Температура плавления сернистого азота находится вблизи тем-
пературы вспышки его, поэтому определение первой температуры
должно сопровождаться особыми мерами предосторожности. Опре-
деление температуры следует вести в приборе, наполненном не серной
кислотой, а парафиновым маслом, за стеклянным щитом.
Удельный вес сернистого азота 2,22. Он незначительно раство-
ряется в этиловом и метиловом спиртах и эфире, несколько более
в бензоле и сероуглероде. Сернистый азот гигроскопичен, во влажном
виде непостоянен и в воде гидролизуется. При обычной температуре
разложение идет медленно; нагреванием значительно ускоряют про-
цесс. При 110° он относительно легко летуч; температура вспышки
190°. К трению и удару чувствителен. Он считается слабым инициа-
тором, потому что ускорение его взрывчатого разложения весьма
мало. Предельный инициирующий заряд его по пикриновой кислоте
почти в два раза больше, чем предельный заряд гремучей ртути, и ра-
вен 0,5г. При добавлении к сернистому азоту даже одного кристаллика
или зернышка азида свинца настолько повышается ускорение его
взрывчатого превращения, что он становится полноценным ини-
циатором. Точно так же накладка из 0,05 г ТНРС вызывает значи-
тельное снижение предельного инициирующего заряда: достаточно
0,15 г сернистого азота, чтобы при этих условиях детонировать тротил.
ПЕРЕЧЕНЬ ЛИТЕРАТУРЫ
R. Escales u. Stettbacher, Initiaiexplosivstoffe, Leipzig, (917.
O. Guttman, Die Industrie der EXplosivstoffe, Braunschweig, 1895.
А. А- Солонина, Гремучая ртуть, СПБ, 1909.
А- А- Солонина. Азиды свинца, меди, ртути и серебра, СПБ. 1900.
Г. Каст, Взрывчатые вещества и средства воспламенения, ГХТИ, 1932.
Н. Koester, Untersuchungen liber die sogenannte Grenzladung von Initial-
explosivstoffen, Berlin, 1929.
Г. К а с т н Л- M e ц. Химические исследования взрывчатых и воспламени-
тельных веществ, ГХТИ, 1934-
А. Штеттбахер, Пороха и взрывчатые вещества, ОНТИ, 1936-
Н- А. Соколов, Курс теории взрывчатых веществ, ОНТИ, 1937.
М. Патри, Горение и детонация взрывчатых веществ, Оборонгиз, 1938-
С. Зорич, Инициирующие взрывчатые вещества, ОНТИ, 1937.
П. М- К у к с и н с к и й, Инициирующие взрывчатые вещества, ГХТИ,
1934.
Л. Бенен, Э. Бюрло и А. Лекорше, Пороха и взрывчатые вещества,
ОНТИ, 1936.
Г. Брунсвиг, Теория взрывчатых веществ, ГХТИ, 1932.
П. Паскаль, Взрывчатые вещества, пороха, боевые газы, ГХТИ, 1932.
К- К. Снитко, Теория взрывчатых веществ, Артакадемия, 1936.
М. Сухаревский и Ф. Пермаков. Курс теории ВВ, ГХТН,
1932.
Журнальная и периодическая техническая литература по 1939 г. включительно.
ОГЛАВЛЕНИЕ
Стр.
Предисловие ......................................... 3
Глава 1. Введение.................................... 5
§ 1. Исторические сведения ........................
§ 2. Классификация средств инициирования...........
§ 3. Классификация инициирующих взрывчатых веществ - -
Глава II. Общая характеристика инициирующих ВВ .............
§ 1. Эндотермический характер инициирующих ВВ
§ 2. Ускорение взрывчатого превращения............ . .
§ 3. Инициирующая способность.............. ..................
§ 4. Предельный инициирующий заряд...............
§ 5. Методы определения инициирующей способности -
§ 6- Чувствительность инициирующих ВВ ......................
§ 7. Основные требования, предъявляемые инициирующим ВВ . - .
Глава III. Гремучая ртуть .......................................
§ 1. Открытий гремучей ртутн и ее применение................
§ 2. Элементарный состав и строение гремучей ртути .
§ 3. Химизм получения гремучей ртути ............
§ 4. Примеси гремучей ртути ................
£ 5. Физико-химические свойства гремучей ртути
§ 6. Взрывчатые свойства гремучей ртути . . . ......
§ 7. Исходные продукты для фабрикации гремучей ртути
§ 8. Технологический процесс получения гремучей ртути........
§ 9. Технические требования К гремучей ртути .........
Глава IV. Азид свинца....................................
§ 1. Открытие азида свинца..........................
§ 2. Азотистоводородная кислота, ее строение и свойства ...
§ 3. Азид натрия, его получение и свойства...................
§ 4. Условия получения азида свинца него физико-химические свойства
14
19
25
26
30
33
38
49
63
65
66
77
81
87
99
109
117
136
147
148
163
170
§ 5. Взрывчатые свойства азида свинца .... -................. 183
§ 6. Исходные и промежуточные продукты для фабрикации азвда свинца 191
§ 7. Технологический процесс производства азида свинца........202
§ 8. Технические требования к азиду свинца ...................225
Глава V. Стифнат свинца (ТНРС)..................................... 230
§ I. Стифниновая кислота и ее свойства ........................ —
§ 2. Получение стифната свинца и его физико-химические свойства 234
§ 3. Взрывчатые свойства стифната свинца...................... 236
§ 4- Исходные продукты для фабрикации стифната свинца . 239
§ 5. Технологический процесс получения стифната свинца . . 246
§ 6. Технические требования к стифнату свинца . . 251
Глава VI. Тетразен ................................................ 255
§ 1-. Общее понятие о тетразенах ............................... —
§ 2. Получение тетразена и его физико-химические свойства . . 256
§ 3. Взрывчатые свойства тетразена .......................... 262
§ 4. Основные исходные и промежуточные продукты для получения
тетразена............................................... 263
§ 5. Фабрикация тегразена.................................. 267
Глава VII. Другие инициирующие ВВ ................................. 272
§ 1. Неорганические производные гремучей кислоты ............ 273
§ 2. Неорганические производные азотистоводородной кислоты 278
§ 3. Органические производные азотистоводородной кислоты . 286
§ 4. Ацетилениды (этиииды) . . 292
§ 5. Нитродиазосоединения . . . 294
§ 6. Органические перекиси .... 302
§ 7. Производные тетразола .................................. 307
§ 8. Галоидные соединения азота 319
§ 9. Сернистый азот.......................................... 321
Перечень литературы - . .
323
ОПЕЧАТКИ
Стр. Строка Напечатано Должно быть По чьей вине
н\ н\
80 15 снизу >CI --NO- СО—СН. >C- NO- СО—СН3 считчик
сК СИ
236 8 сверху стифниновой кислоты стифната свинца редактор
236 12 ь стифниновая кислота стифнат свинца
262 3 »> 550 кал'кг 550 Кал [кг автор
Бубнов П. Ф. Инициирующие ВВ № 92.