/



























































































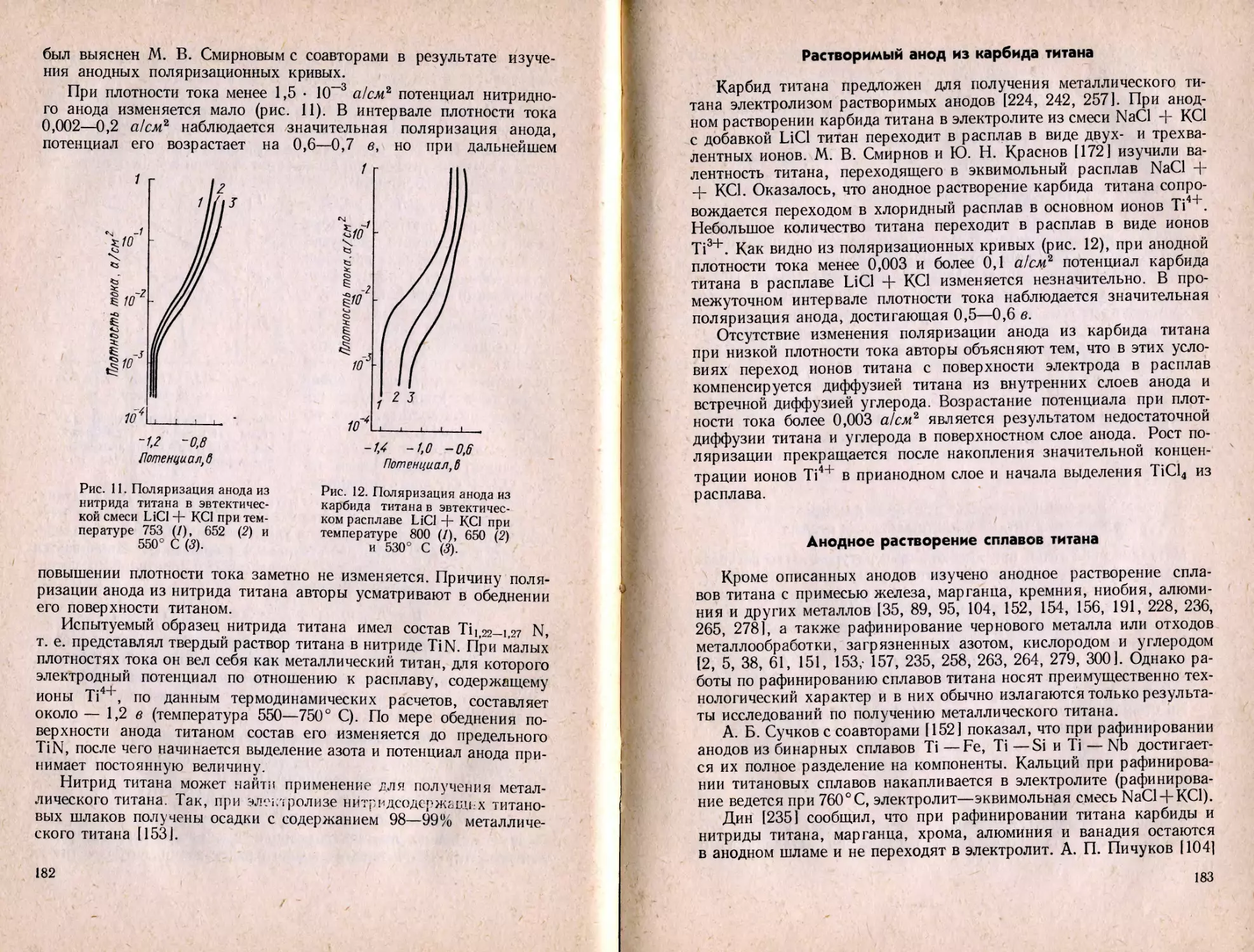

























































Текст
АКАДЕМИЯ НАУК УКРАИНСКОЙ ССР
ИНСТИТУТ ОБЩЕЙ И НЕОРГАНИЧЕСКОЙ ХИМИИ
Я. Г. ГОРОЩЕНКО
ХИМИЯ
ТИТАНА
ЧАСТЬ II
«НАУКОВА ДУМКА» КИЕВ — 1972
640
Г70
2—5—2
265—72М
УДК 546
В книге описаны комплексные и металлорганиче-
скиесоединения титана, его электрохимические свойства,
а также физико-химические основы производства тита-
новых шлаков, четыреххлористого титана, двуокиси
титана и металлического титана. В ней впервые изла-
гаются основы теории гидролиза сульфата титана, раз-
витой автором с сотрудниками. С помощью этой теории
стало возможным регулирование сложного процесса
гидролиза с целью получения продуктов с заданными
физико-техническими свойствами.
Представляет интерес для научных работников,
инженеров и техников, занятых в области химии и тех-
нологии титана, а также для студентов высших учебных
заведений, специализирующихся в области производства
металлического титана и его соединений.
Ответственный редактор
чл.-кор. АН УССР
И. А. Шека
Рецензенты:
докт. техн, наук А. К. Шарова,
канд. хим. наук И. С. Чаус
Редакция химической литературы
Зав. редакцией 3. С. Покровская
КИЕВСКИЙ ПОЛИГРАФИЧЕСКИЙ КОМБИНАТ
ПРЕДИСЛОВИЕ
Настоящая книга является второй частью монографии «Химия титана», вы-
пущенной в 1970 г. издательством «Наукова думка». В ней рассматриваются не во-
шедшие в первую книгу разделы химии титана, а также кратко освещается со-
стояние производства металлического титана и его важнейших соединений. В кон-
це книги приведены предметные указатели к обеим частям. При написании второй
4асти монографии использована литература, опубликованная до конца 1969 г.
включительно.
Автор испытывал большие затруднения при описании титанорганических
1 комплексных соединений титана из-за отсутствия резкой границы раздела меж-
iy этими классами соединений. В работах многих авторов комплексные соедине-
!ия титана с органическими лигандами, например аддукты типа TiCl4 • nL, где
- — эфиры, спирты, карбонильные соединения и другие органопроизводные
вносятся к титанорганическим соединениям. И наоборот, соединения типа титан-
фиров, например циклопентадиенильные производные, причисляются к комплекс-
ым соединениям титана. Кроме того, в первой части монографии, стремясь более
олно и всесторонне осветить свойства отдельных классов соединений титана,
втор вынужден был описать многие аддукты с органическими и неорганическими
игандами. Избегая повторений, мы не упоминаем их в соответствующих главах
юрой книги, к которым они относятся.
Таким образом, сведения о комплексных соединениях титана оказались рас-
'янными частично в отдельных главах первой и второй частей книги. От-
льные же представители титанорганических соединений попали в главу о комп-
жсных соединениях, и наоборот. При распределении материала между главами
*е соединения с валентной связью типа Ti — 9R (OR — органогруппы) относились
титанорганическим соединениям. К комплексным соединениям причислялись
юизводные с донорно-акцепторной связью между титаном и лигандами. Данный
>инцип нарушался только в том случае, когда природа связи между титаном и
ганопроизводными оставалась окончательно еще не выясненной.
До настоящего времени еще нет твердо установленных номенклатурных норм
я наименования титанорганических соединений. В тексте книги мы пользовались,
ычно, названиями соединений, которые приводились в цитируемых работах,
(нако при составлении предметных указателей с целью унификации была при-
Та единая система наименования соединений. Она распространена и на те со-
ления, которые в тексте книги обозначены только химическими формулами.
В основу наименования титанорганических соединений положены номенкла-
ые нормы, принятые в органической химии. Для наименования комплексных
соединений использована номенклатура, рекомендованная Н. Б. Некрасовым
в приложении к первому тому его книги «Основы общей химии», выпущенной изда-
тельством «Химия» в 1965 г. Описание соединений проводится нами «по ходу форму-
лы». Название комплексного иона, занимающего центральное положение, оканчи-
вается на «ал». Присоединенные к формуле через точку части ее в словесных
названиях оттеняются буквой -т- (точка). Наличие заряда у ионов обозначается
словами «анион» или «катион» с соответствующей числительной приставкой^
присоединенными через апостроф. Полимерное строение молекул обозначается
словом «полимер», присоединенным также через апостроф.
В четвертой главе книги мы попытались изложить вкратце основы теории гид-
ролиза сульфата титана, развитой нами совместно с сотрудниками. Разработка
ее еще не закончена, но мы полагаем, что приведенные сведения не являются
лишними. Знакомство с основами теории гидролиза сульфата титана может ока-
заться интересным для исследователей и специалистов, работающих в области
производства двуокиси титана.
Автор приносит большую благодарность чл.-кор. АН УССР И. А. Шеке,
проф. А. К. Шаровой, докт. хим. наук А. Л. Суворову и канд. хим. наук
И. С. Чаусу за ценные советы и замечания, сделанные ими при подготовке руко-
писи к печати.
ГЛАВА I
ТИТАНОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
К титанорганическим соединениям мы относим соединения, со-
держащие органические радикалы, атомы углерода которых свя-
заны с атомами титана непосредственно или через другие атомы.
Координационные соединения титана с органическими лигандами
рассмотрены нами в главе II и при описании отдельных классов
неорганических соединений.
Первые попытки получения титанорганических соединений от-
носятся еще к середине XIX ст., однако систематическое изучение
их началось только после второй мировой войны в связи с интересом,
который они представляют для синтеза различных полимерных ве-
ществ, обладающих, подобно кремнийорганическим соединениям,
повышенной термостойкостью, влагоотталкивающим действием и
другими важными техническими свойствами. Титанорганическим
соединениям посвящено много работ, в которых использована ли-
тература по 1958—1963 гг. [102, 221, 229, 249, 314, 316, 339, 402,
412, 419, 420, 439, 468, 497, 498, 554, 558, 585, 608].
С органическими производными титан образует соединения двух
типов, в которых связь между атомами титана и углерода осущест-
вляется либо непосредственно, либо с помощью мостиковых атомов
кислорода, азота, фосфора, серы и др.
Соединения со связью Ti—С нестойки и мало исследованы, за
исключением циклопентадиенильных производных с сэндвичной
структурой. Значительно лучше изучены органотитанаты с мости-
ковой связью. За последние 20 лет получено большое число предста-
вителей этого класса соединений: мономеров и полимеров. Синте-
зированы полимеры, содержащие в неорганических цепочках кроме
титана атомы кислорода, кремния, азота, фосфора и серы.
Титанорганические соединения получаются обычно замещением
атомов галогенов у галогенидов титана на органические радикалы.
ПРОИЗВОДНЫЕ СО СВЯЗЬЮ ТИТАН — УГЛЕРОД
ТИТАНАЛКИЛЫ И ТИТАН АР ИЛЫ
Попытки получения титаналкилов со связью Ti—С по реакциям
обменного разложения четыреххлористого титана с цинкорганиче-
скими [280, 318, 499, 5431, магнийорганическими [206, 309, 310, 398]
5
и ртутьорганическими соединениями [235, 280, 393, 422, 447], а
также взаимодействием с трибутоксихлортитаном и дибутоксиди-
хлортитаном [139] предпринимались неоднократно. Однако в чистом
виде из-за малой стойкости титаналкилы получить не удалось. Более
стойкими оказались смешанные алкилгалогениды титана.
Герман и Нельсон [399] изучили устойчивость соединений типа
R^TiG4_n в зависимости от природы углеводородного радикала R
и числа заместителей п в галогенидах титана.
Оказалось, что устойчивость соединений со связью титан — угле-
род возрастает в зависимости от природы радикала в следующем по-
рядке: бутил, метил, ацетил, n-анизил, фенил, а-нафтил, индил.
С увеличением п устойчивость соединений уменьшается. Хлорид-
ные производные менее стойки, чем фторпроизводные.
Из числа алкилтитанов к настоящему времени в чистом виде
получено только несколько соединений. Одним из первых синтези-
рован метилтрихлоридтитан CH3TiCl3 действием диметилалюминий-
хлорида на четыреххлористый титан [252]. Электронные спектры
этого соединения описаны в работе [361].
Леви [447] получил тетрафенилтитан (С6Н5)4 Ti. Данное соеди-
нение неустойчиво: при его распаде в эфире (0° С) в присутствии
дифенилацетилена образуется дифенил, дифенилтитан и гексафе-
нилбензол [213].
При взаимодействии четыреххлористого титана с фенилмагний-
бромидом [206, 399], фениллитием [393, 422], а-нафтилмагнийбро-
мидом, бензилмагнийхлоридом и смесью хлорбензола с металли-
ческим натрием [310] арилтитаны не были получены. Однако при
протекании этих реакций наблюдалось образование дифенила, что
может служить доказательством получения арилтитанов в качестве
нестойких продуктов, разлагающихся на дифенил и металлический
титан.
В английском патенте [13] описан синтез по реакции между тет-
раэтилоксититаном и фениллитием оранжевого кристаллического
осадка, содержащего титан, литий и хлор. Такое соединение в чис-
том виде не было получено, но в числе продуктов его разложения
обнаружен дифенил. Исходя из этого, можно предполагать, что оно
является нестойким арилтитаном.
Герман и Нельсон [398], действуя на тетраизопропоксити-
тан фениллитием, получили фенилтриизопропилтитан C6H5Ti -
• (OC3H7-rzso)3, в котором фенильная группа присоединена непо-
средственно к атому титана. Это соединение устойчиво в атмосфере
азота, плавится при температуре 88—90° С, но выше температуры
плавления разлагается. Выделено также неустойчивое, самопроиз-
вольно разлагающееся соединение, которому приписывают состав
[C6H5Ti(OC3H7-u3o)3J(uso-C3H7OLi)(LiBr)(C2H5OC2H5) [13, 3971. В
патентной литературе описано еще несколько арильных произ-
водных титана, например a-C10H7Ti (ОС4Н9)3, ft-CH3C6H4Ti (ОС4Н9)3
[200].
6
При взаимодействии тетрахлорида титана с метиллитием в аб- •
солютном бутиловом эфире при температуре от —50 до —80° С по-
лучен тетраметилтитан по реакции [191
TiCl4 + 4CH3Li = (СН3)4 Ti + LiCl.
Соединение неустойчиво и существует только в растворе.
Тетраметилтитан в этиловом эфире с пиридином, триметилами-
ном, триметилфосфином, дипиридилом, фенолом, N,N,N',N'-tct-
раметилендиамином, Р,Р,Р',Р'-тетраметилендифосфином и диокса-
ном образует желтые кристаллические аддукты состава (CH3)4Ti-L,
где L — лиганд [476, 575]. Пиридин и триметилфосфин дают
также красно-коричневые комплексы состава (CH3)4Ti • 2L [476].
Выпариванием раствора C6H5CH2MgCl и TiCl4 получено бензиль-
ное соединение титана (CeH5CH2)4Ti [384]. Продукт красного цвета,
точка плавления 70° С, устойчив при температуре 0° С, но в кипящем
гептане за несколько часов разлагается. С этанолом вступает в ре-
акцию обменного разложения, в результате которой получается
бис-(этокси)титан-бис-(бензил) с точкой плавления 102° С.
В нескольких патентах [62, 145, 578] описаны методы получения
алкилтитангалогенидов по реакции
Tif4 + М (Alk)„ -> (Alk)m Tir4_,n,
где Г = Cl, Br, I. Этим методом синтезированы алкилтитанхло-
риды CH3TiCl3, (CH3)2TiCl2, QHsTiClg и CD3TiCl3.
При взаимодействии Сб (СН3)6, А12С1б и алюминия с TiCl4 по-
лучен фиолетовый продукт {Ti3 [Св (СН3)6| С16}С1 [377].
бис-(Инденил)титангалогениды образуются при действии на тет-
рагалогениды индиллития в эфире при температуре 0° С:
Tif4 + 2RLi = R2Tir2 + 2Lif,
где Г — Cl, Вт, I, R — инденил
Получены также инденильные производные C5H5RTiCl2, RTiCl3 и
окись (RTiCl2)O [537].
Смесь триэтилсвинца и тетрахлорида титана в гептане при
температуре —80° С реагирует с образованием этилтитан-
7
трихлорида [259]:
(C2H5)4Pb + TiCl4
-> C2H5TiCl3 + (C2H6)3 PbCl.
При сравнении ИК-спектров поглощения смеси тетрахлорида
титана и диметилалюминийхлорида с ИК-спектрами индивидуаль-
ных веществ обнаружено присутствие в смеси этих компонентов
метилтитантрихлорида и метилалюминийдихлорида, образующихся
по реакции взаимного обмена [385].
Взаимодействие четыреххлористого титана с металлорганиче-
скими соединениями сопровождается не только реакциями взаим-
ного обмена, наблюдается также и комплексообразование. Впервые
образование комплексов титанорганических соединений, состав
которых не был установлен, отмечено Фридландом и Олта [381].
Натта и соавторы [493] при восстановлении тетрахлорида титана
металлическим алюминием и трихлоридом алюминия в бензоле
выделили кристаллический комплекс, состав которого выразили
формулой Al2TiCl8 • С6Н5. При перемешивании данного комплекса
с тетр а гидрофураном в бензоле они получили комплекс состава
AlTiCl5 . С6Н5 [487].
Мартин и Вогвинкель [480], однако, считают, что в этих комплек-
сах титан находится в двухвалентном состоянии и состав их следует
выражать формулой Al2TiCl8 • С6Нб. Продукт такого состава был
получен ими при перемешивании толуола и мезитилена с тетрахло-
ридом титана, тетрахлоридом алюминия и металлическим алюминие-
вым порошком при температуре 80° С в атмосфере аргона. Описаны
препараты (TiCl4)[А1(С2Н5)3] и (TiCl4)[(C2H5)2AlCU, являющиеся
катализаторами полимеризации непредельных углеводородов при
низком давлении, с помощью которых получены кристаллические
полимеры этилена, пропилена и изопрена [621]. Соединение (TiCl3)«
• [Al (С2Н5)3] предложено в качестве катализатора полимеризации
аллилсилана и аллилтриметилсилана [490].
Методами ядерного магнитного резонанса и ИК-спектроскопии
показано, что в растворе тетрагидрофурана титан в комплексах
с алкилхлоридами алюминия находится в трехвалентном состоя-
нии [556].
Т. С. Джабиев и соавторы [84] методом электропроводности и
спектроскопии доказали, что тетрабутоксититан Ti (OR)4 с три-
этил-алюминием образует два соединения, имеющие следующее
строение:
С2Н5 II
Комплекс II является каталитически активным.
Методом ЭПР [83] установлено, что при взаимодействии C5H6TiCl3
с А1(С2Н5)3, А1(СН3)3 и А1(С2Н5)2С1 происходит восстановление
титана до трехвалентного и образуются комплексы с мостиковой
связью:
где R = СН3, С2Н5; X = С1.
Синтезировано соединение (C2H5)TiAlCl5(C10H8N2)2 (C10H8N2 —
дипиридил) [141]. Изучено кристаллическое строение трш>(цик-
лооктатетраен)дититана. Кристаллы его имеют ромбическую ре-
шетку с параметрами а = 14,41, b = 35,99, с = 7,25 A, z = 8,
фед. гр. Fdd2.
Алкил- и арилтитаны обладают каталитической активность#) при
полимеризации непредельных углеводородов. Каталитические свой-
ства титанорганических соединений были открыты Циглером [64—
66], предложившим проводить низкотемпературную полимериза-
цию в присутствии смеси четыреххлористого титана и алюминий-
алкилов. В последующем катализаторы на основе соединений ти-
тана стали называть циглеровскими.
ЦИКЛОПЕНТАДИЕНИЛЬНЫЕ ПРОИЗВОДНЫЕ ТИТАНА
Циклопентадиенильные соединения титана получаются заме-
щением атомов галогена в галогенидах титана на циклопентадие-
нил С5Н5. В них осуществляется многоцентровая связь между ти-
таном и циклопентадиенилом посредством валентных электронов
металла и углеродных атомов. Исследование ИК-спектров погло-
щения [595, 597], химических и магнитных свойств циклопентадие-
нильных соединений титана показало, что они имеют сэндвичную
структуру, подобно ферроцену. - |
Общим методом получения циклопентадиенильных соединений
титана служит взаимодействие галогенидов титана с циклопента-
диенильными производными магния, натрия и лития [595, 597].
Очень удобным оказался метод, основанный на взаимодействии га-
логенидов титана с циклопентадиенилнатрием в растворе тетрагид-
рофурана [598] или диметилового эфира диэтилгликоля [597].
(C5H5)2Ti. Фишер и Уилкинсон [374] взаимодействием двуххло-
ристого титана с циклопентадиенилом натрия в тетрагидрофуране
получили бис-(циклопентадиенил)титан (C5H5)2Ti в виде темно-зе-
леных кристаллов, легко окисляемых на воздухе. Это соединение
имеет строение, аналогичное ферроцену. Его строение можно вы-
I
9
разить структурной формулой
Кристаллы (C5H5)2Ti возгоняются без плавления при темпера-
туре 120—180° С, диамагнитны, удельная магнитная восприимчи-
вость их х — —100 • Ю-6 (298° К). Они имеют моноклинную кри-
сталлическую решетку с параметрами а = 7,00, b = 12,26, с =
= 7,92А, Р = 114,85°, рреНтг = 1,378 г/см3, г = 2, фед. гр. РЬ. Мо-
лекула содержит одно плоское и второе гофрированное пентадие-
нильное кольцо [536].
Е. М. Шустарович и М. Е. Дяткина [233] произвели расчет ва-
лентных состояний Ti° в (C5H5)2Ti методом Слейтера — Кондона.
Расчет отвергает валентное состояние sd3, приводящее к парамагне-
тизму. Выбор валентных состояний, отвечающих диамагнитному
соединению (C5H5)2Ti, неоднозначен. Если энергия образования свя-
зей alg и e2g + e2g) может окупить большую затрату на возбужде-
ние конфигурации d4 (115 ккал), то бис-(циклопентадиенил)титану
следует приписать состояние dxzdyZd2X2_y2. Если же энергия связи
4-^ меньше 115 ккал, то исходным является валентное состоя-
ние s2dxzdyz с энергией 11 ккал (при этом связи alg и e2g не возникают)-
бш>(Циклопентадиенил)титан с тетрагидрофураном образует
этерат (C5H5)2TiC4H8O в виде зеленых парамагнитных кристаллов.
При стоянии кристаллы этерата принимают коричневую окраску
и становятся диамагнитными.
(C5H5)3Ti. Фишер и Лёхнер [376] получили т/шс-(циклопен-
тадиенил)титан действием на бис-(циклопентадиенил)дихлортитан
циклопентадиенилнатрия, взятого с избытком. По данным [327],
соединение имеет моноклинную решетку; магнитный момент его
(Нэфф ~ IJ1 рв) при температуре 100—320° К не изменяется.
Сигерт с соавторами [536] синтезировали (C5H5)3Ti разложением
(C5H5)4Ti в вакууме при температуре 125° С. Исследование ИК-спект-
ров и спектров ЯМР указывает на то, что т/шс-(циклопентадие-
нил)титан имеет строение (rc-C5H5)2Ti(o-C6H5).
(C5H5)4Ti. Получен по реакции (C5H5)TiCl2 с NaC5H5 в сухом
тетрагидрофуране при температуре 0° С [536]. Соединение имеет
строение. (л-С5Н5)2Т1(а-СбН5)2.
C5H5TiCl2. Циклопентадиенилдихлортитан получили Бартлетт
и Зейдел [269], действуя на бис-(циклопентадиенил)дихлортитан
в растворе смеси толуола с гептаном диизобутил хлор алюминием
10
(озо-С4Н9)2А1С1. Вещество имеет фиолетовый цвет, нерастворимо
в гептане, бензоле, дифениловом эфире и слабо растворимо в ацето-
нитриле. Кристаллы C5H5TiCl2 легко окисляются кислородом воз-
духа; при нагревании в вакууме до 150° С возгоняются, а при тем-
пературе около 300° С частично разлагаются. Основываясь на дан-
ных исследования спектров ЭПР и ЯМР, авторы считают, что связь
титана с циклопентадиенилом сэндвичного типа.
Циклопентадиенилтитандихлорид по реакции обменного раз-
ложения с LiSCN в ацетоновых растворах образует (C5H5)Ti (SCN)CI
и (C6H5)Ti(SCN)2 [354].
(C5H5)2TiCl. Бирмингем с соавторами [250], восстанавливая
бис-(циклопентадиенил)дихлортитан в растворе тетрагидрофурана
и метанола цинковой пылью, выделили зеленый продукт состава
(C5H6)2TiCl. Риид и Уайлз [523] получили бш>(циклопентадие-
нил)хлортитан взаимодействием TiCl3 с эквимольным или избыточ-
ным количеством (C5H5)2Mg в растворе тетрагидрофурана. бш?-(Цик-
лопентадиенил)хлортитан образуется также при электролитическом
восстановлении (C5H5)2TiCl2 в эфирном растворе [478] и при вос-
становлении (C5H5)2TiCl2 в тетрагидрофуране [256].
Кристаллы бис-(циклопентадиенил)хлортитана имеют фиоле-
тово-коричневый цвет, температура плавления их 282—283° С. Маг-
нитный момент при комнатной температуре равен 1,56 рв на 1 атом
титана, что свидетельствует о наличии у титана одного неспаренного
электрона. Методами ИК-спектроскопии и ЭПР показано, что при-
рода связи циклов С5Н5 с титаном одинакова как в твердом бис-(цик-
лопентадиенил)хлортитане, так и в его растворах. Согласно [327],
молекула (C5H5)2TiCl имеет димерную структуру.
При взаимодействии (C5H5)2TiCl с реактивом Гриньяра, по
данным ЭПР [256], образуются гидридные соединения состава
[(C5H5)2TiH2]~ и [(C5H5)2TiD2]_- При действии на (C6H5)2TiCI ме-
тил-, этил- и изопропиллития также наблюдается присоединение
алкильных групп к металлу с образованием [(C5H5)2Ti (СН3)2]~,
[(C5H5)2Ti (С2Н5)2]~ и [(С5Н5)2П(С3Н7)2Г. Аллиллитийбромид с
(C5H5)2TiCl дает соединение (C5H5)2Ti(C3H5).
Азиды типа RN3, где R = С6Н5, п - С1СбН4, п - O2NC6H4,
п - СН3С6Н4 и С2Н5, реагируют с бис-(циклопентадиенил)хлорти-
таном по схеме
(С5Н5) TiCl + R - N3
-> |(C5H5)2TiCl>NR + N2.
По этой реакции синтезировано соединение [(C5H5)2TiCl]2=NC6H5 —
твердое шоколадно-коричневое вещество, гидролизующееся водой
с образованием оксида (C5H5)2TiO, и аминохлорбензолпроизводное
[(C5H5)2TiCl]2=N (п - С1СбН4) [329].
По расчетам Е. М. Шустаровича и М. Е. Дяткиной [233], для
Ti+ в (C5H5)2TiCl наиболее вероятно валентное состояние dxzdyzdX2^yi
с энергией возбуждения валентной связи 19 ккал.
11
бис-(Циклопентадиенил)хлортитан может существовать в виде
димера. Натта [492] строение димера изображает формулой
/С1 ••
<w2Ti._ ;ткс6н3)2,
*С1
т. е. принимает для него мостиковую структуру. Мартин и Винтер
[482] считают, что в димере осуществляется ковалентная связь меж-
ду атомами титана, и предлагают строение его выражать формулой
[(C5H5)2TiCI2Ti (СВН5)2].
C5H5TiCl3. Циклопентадиенилтрихлортитан получен нагрева-
нием Ош>(циклопентадиенил)дихлортитана с избытком четырех-
хлористого титана в растворе ксилола при 120° С в течение 24 ч, а
также хлорированием (C5H5)2TiCl2 в растворе четырехлористого
углерода при 55—65° С с ультрафиолетовым облучением по реакции
[388]
(C5H5)2TiCl2 + С12 -> C5H5TiCl3 + С5Н5С15.
Слоан и Барбер [542] синтезировали C5H5TiCl3, нагревая TiCl4 с
(C5H5)2Mg в растворе ксилола.
В инфракрасных спектрах C6H5TiCl3 имеются только полосы,
отвечающие валентным колебаниям С—Н с длиной волны 3,3 мкм.
Это указывает на то, что связь пентадиенильного кольца с титаном
аналогична связи в (C5H5)2TiCl2.
Аллегро с соавторами [248, 389] изучил кристаллы C5H5TiCl3
методом рентгеноструктурного анализа и установил, что они имеют
моноклинную решетку с параметрами а — 6,66, b — 10,39, с =
= 6,65 А, Р == 115° 18', ризм = 1,76 г/см3, z = 2, фед. гр. р^/т.
Кристаллы C5H5TiCl3 плавятся, по данным [535], при темпера-
туре 210—212° С, по данным [520],— при 216—217,5° С и разлага-
ются при 186° С [388]. Частота валентных колебаний титан — ко-
льцо в моноциклопентадиенильных соединениях находится в пре-
делах 410—465 см~х [122]. На воздухе кристаллы относительно
устойчивы и с водой реагируют медленно. При действии водного
раствора едкого натра разлагаются с отщеплением всех трех ато-
мов хлора.
Из раствора в ароматических углеводородах в результате гид-
ролиза C5H5TiCl3 рассчитанным количеством воды получено соеди-
нение (TiCl2C5H5)2O. Кристаллы его плавятся при температуре
148—150° С [248], имеют моноклинную решетку с параметрами а =
= 7,47, b = 9,86, с = 12,58 А, Р = 127° 56', z = 2, фед. гр. р21/с
[247, 248]. При более глубоком гидролизе образуется желтое ве-
щество с температурой плавления 255—260° С, имеющее состав
С5Н5ТЮС1 [2481.
Атомы хлора в циклопентадиенилтрихлортитане более подвиж>
ны, чем пентадиенильная группа, ацильный и другие остатки.
Описаны следующие продукты замещения: C5H5Ti,(OCH3)3,
C5H5Ti(OC2H5)Cl2, C5H5Ti(OC2H5)2Cl [85], C5H5Ti(OC2H5)3 [85, 134],
12
C5H6Ti(OCOCH3)3 185]. А. Н. Несмеянов с соавторами [135] методом
масс-спектроскопии показал, что в соединении С5Н5Т1(ОС2Н5)з_лгС1п
степень ионной связи возрастает с увеличением п.
C5(CH3)5TiCl3. Рёгль с соавторами [522], нагревая бутен-1, бу-
тен-2, изобутен, изомерные пентаны, диизобутен, тетрапропилен
и другие олефины с четыреххлористым титаном в автоклаве под
давлением 30—60 атм при 300° С, получил пентаметилциклопен-
тадиенилтрихлортитан. Вещество оранжево-красного цвета, точка
плавления 200° С, устойчиво к действию влаги, щелочей и кислот,
однако при длительном нагревании с водными растворами ще-
лочи разлагается с образованием желтого комплекса состава
[C5(CH3)5TiO(OH)]rt, где п = 4—6. При частичном гидролизе
получается [C5(CH3)5TiOCl]n. Получены продукты замеще-
ния следующего состава: CH3C5H4TiCl3, CH3C5H4Ti(OC2H5)Cl2,
CH3C5H4Ti(OC2H5)2Cl, CH3C5H4Ti(OC2H5)3 и C2H5C5H4TiCl3 [85].
C5H5TiBr3, C5H5TiI3. При действии на TiBr4 и Til4 дипентадие-
нилмагния в растворе толуола при температуре около 20° С обра-
зуются кристаллы циклопентадиенилтрибромтитана C5H5TiBr3
(т. пл. 174,5—175,5°.С, разлагается) и циклопентадиенилтрииодтита-
на C5H5TiI3 (т. пл. 184—186° С, разлагается).
Изучение И К- и УФ-спектров и спектров ЯМР показало, что
соединения С5Н5ПГ3, где Г = С1, Вг и I, являются открытыми и
имеют сэндвичную структуру с л-связями между кольцом С5Н5 и
металлом. По данным измерений молекулярного веса и электропро-
водности, соединения типа С5Н5ПГ3 мономерны [542].
Электронографическими исследованиями C5H5TiBr3 в газовой
фазе установлено, что молекула этого соединения представляет со-
бой полусэндвич с равными по длине связями Ti—С и конфигура-
цией «рояльной табуретки» [210].
C5H5TiF2. Уилкинсон и Бирмингем [597] из четырехфтористого
титана в растворе тетрагидрофурана и циклопентадиенила натрия
получили бпс-(циклопентадиенил)дифтортитан в виде желтых кри-
сталлов. В ИК-спектре поглощения (C5H5)2TiF2 обнаружены полосы
с частотами 550 и 579 см~1, относящиеся к симметричным и асиммет-
ричным колебаниям связи Ti—F [367].
(C5H5)2TiCI2. Получен действием на четыреххлористый титан в
растворе тетрагидрофурана пентадйенила натрия [597]. С небольшим
выходом бнс-(циклопентадиенил)дихлортитан образуется при пря-
мом действии TiCl4 на C5H5Na в присутствии акцепторов НС1
(пиридин, аммиак) [251]. Слоан и Барбер [542] синтезировали
(С5Нб)2ПС12 кипячением раствора TiCl4 и (C5H5)2Mg в ксилоле (эк-
вимольные количества).
Кристаллы (C5H5)2TiCl2 ярко-красного цвета, температура плав-
ления 289° С [597]. Плавление кристаллов сопровождается их раз-
ложением [542].
Молекула (C5H6)2TiCl2 представляет собой клинообразный сэнд-
вич с равными расстояниями Ti—С 2,38 А и углом между
13
циклопентадиенильным кольцом 59°; расстояние Ti—Cl равно
2,24 А, угол С1—Ti—Cl составляет 100° [6, 209].
Хлор, бром и иод [22] при взаимодействии с (C5H5)2TiCl2 присое-
диняются к одному циклопентадиенильному кольцу. Получены про-
дукты присоединения хлора C5H5(C5H5Cl2)TiCl2 и С5Н5 (C5H5Cl4)TiCl2.
Изучен изотопный обмен лигандов в системе (C5H5)2TiCl2 — Li36Cl
1539].
Молекулы (C5H5)2TiCl2 в камере масс-спектрометра ионизиру-
ются, образуя ионы Ti(C5H5)2Cl“ и Ti(C5H5) С1Г [366]. При вос-
становлении (C5H5)2TiCl2 цинком получен комплекс [Ti (С5Н5)2С1]2 •
• ZnCl2 [538]. Магнитный момент его, равный 1,71 рв на 1 атом ти-
тана, указывает на наличие одного неспаренного электрона.
В водных растворах (C5H5)2TiCl2 вступает с солями в реакции
обменного разложения, образуя продукты общего состава (С5Н5)2Т1Г2,
где Г = F, OCN, SCN и другие [368]. По реакции обменного
разложения получены также продукты состава (C5H5)2TiS5,
[(C5H5)2TiNO3]2O-4H2O и [(С5Н5)2Т1Г]2О-ЗН2О, где Г = Вг или I,
а при кипячении (С5Н5)2Т1Г2(Г = С1, Вг) с избытком NaCNS — ро-
данидное производное (C5H5)2Ti (CNS)2 [455].
В патенте ФРГ [317] упоминается метильное производное —
бш?-(метилциклопентадиенил)дихлортитан (CH3C5H4)2TiCl2.
(С5Н5)2TiBr2. б&с-(Циклопентадиенил)дибромтитан является пер-
вым полученным производным циклопентадиенильных соединений
титана. Его синтезировали Уилкинсон с соавторами [595], действуя
избытком циклопентадиенилмагнийбромида на четыреххлористый
титан в растворе толуола. Это — темно-красное кристаллическое
вещество с точкой плавления 240—243° С, диамагнитное, X =
= —145 • 10'6 (25° С). бис- (Циклопентади енил)дибромтитан по-
лучен также взаимодействием TiBr4c C5H5Na в растворе тетр а гид-
рофурана и 1,2-диметоксиэтана; температура плавления его 314° С?
% = —186 • КГ6 (25° С) [597].
Слоан и Барбер [542] получили кристаллы (C5H5)2TiBr2 смеше-
нием растворов TiBr4 и (C5H5)2Mg в толуоле при комнатной темпера-
туре. По их данным, это вещество плавится при температуре 309—
310° С с разложением.
б«с-(Циклопентадиенил)дибромтитан частично гидролизуется
водой с образованием желтого раствора. В редукторе Джонса
(C5H5)2TiBr2 восстанавливается и образуется зеленый раствор, со-
держащий ион бш?-(циклопентадиенил)титана-3.
(C5H5)2TiI2. Получен действием на раствор четырехиодистого ти-
тана в гидрофуране пентадиенилом натрия в виде темно-красных
кристаллов с точкой плавления 319°С. Кристаллы (C5H5)2TiI2 обра-
зуются кипячением раствора Til4 и (C5H5)2Mg в ксилоле. По данным
[542],кристаллы (C5H5)2TiI2 плавятся при 317—318° С с разложением.
Дигалогениды бш?-(циклопентадиенил)титана имеют сэндвичное
строение типа ферроцена. Для Ti2+ валентное состояние имеет вид
14
dxi/li/z с энергией возбуждения связи 13 ккал [233]. Вследствие этого
возможно образование (C5H5)2Ti2+, несмотря на отсутствие связей e2g-
(C5H5)Ti (ОС2Н5)з, (C5H6)Ti(OC3H7)3, C5H5Ti(OC4H9)3. Синтези-
рованы действием циклопентадиенилнатрия на титанхлорэфиры со-
ответствующих спиртов по реакции [63, 120]:
C5H5Na + 2 (RO)2 TiCl2 -> C5H5Ti (OR)3 + NaCl.
(C5H5)2TiCl(OC2H5). Образуется при взаимодействии циклопен-
тадиенилнатрия с этоксититантрихлоридом [114]:
2C5H5Na + C2H5OTiCl3 -> (С5Н5)2 TiCl (ОС2Н5) + 2NaCl.
Получен также кипячением (C5H5)2TiCl2 в смеси бензола с триэтил-
амином [115].
C5H5Ti [OSi (СН3)3]3. Синтезирован по реакции обменного раз-
ложения триметилсилоксититантрихлорида с циклопентадиенил-
натрием:
(СН3)3 SiOTiCl3 + C5H5Na -> (СН3)3 SiOTi (С5Н5)2 Cl +
+ [(CH3)3SiO]3TiC5H5 + NaCl.
l(C5H5)TiC104]4. Кристаллы ромбической системы с параметрами
кристаллической решетки а = 15,32, b = 11,715, с = 14,515А,
Ризм = 1,66 г!см\ 2 = 4, фед. гр. СтС21 [540].
Реакции замещения
Характерным свойством бцс-(циклопентадиенил)титанов типа
(С5Н5)2Т1Г2 является подвижность атомов галогенов. На реакциях
замещения атомов галогенов основаны многочисленные синтезы
производных бис-(циклопентадиенил)титанов. За короткое время,
прошедшее после открытия этого класса соединений, синтезировано
уже около двух десятков различных производных.
Муррей [472, 473], нагревая бензольный раствор бис-(циклопен-
тадиенил)дихлортитана с циклопентадиенилом натрия и окисью
углерода в автоклаве при температуре 100° С под давлением 110 атм,
получил бш>(циклопентадиенил)дикарбонилтитан (C5H5)2Ti(CO)2.
Это соединение образуется также при нагревании в автоклаве
эфирного раствора TiCl4 и NaC5H5 или (C5H5)2TiCl2 и LiC4H9 с
окисью углерода [472]. бис-(Циклопентадиенил)дикарбонилтитан
кристаллизуется в виде темно-красных пирофорных игл, плавя-
щихся с разложением при температуре выше 90° С.
В растворе аммиака при температуре —37° С происходит заме-
щение одного атома хлора в £шс-(циклопентадиенил)дихлортитане
ца аминогруппу и образуется аммиакат (C5H5)2TiCl (NH2) • 2NH3
1245], который при нагревании разлагается по схеме
(С5НБ)а TiCl (NH2) . 2NH3 (С6НБ)2 TiCl (NH^ NH3 ->
130° C u .
------>(СБНБ)2Т1С12.
15
В растворе метиламина бш>(циклопентадиенил)дихлортитан при
температуре —37° С образует сольват состава (C5H5)2TiCl(NHCH3) •
•%CH3NH2, разлагающийся при нагревании аналогично аммиакату.
При действии на (C5H5)2TiCl2 триметилалюминия или диметил-
хлоралюминия образуется бис-(циклопентадиенил)метилхлортитан
(C5H5)2TiCH3Cl [317]. Это—кристаллическое вещество сточкой плав-
ления 168—170° С.
В патенте [317] описано замещение двух атомов галогенов в
бш>(циклопентадиенил)дихлор(бром, иод)титане на метильные груп-
пы действием CH3Li, CH3MgI и (CH3)2Zn.
Хендрик с соавторами [396], действуя аллилмагнийхлоридом
C3H5MgCl на раствор (C5H5)2TiCl2 или (C5H5)2TiCl в тетрагидрофу-
ране при комнатной температуре, получил фиолетовый раствор, из
которого выделен пурпурный осадок бис-(циклопентадиенил)аллил-
титана (C5H5)2TiC3H5. Кристаллы его парамагнитны и плавятся с
разложением при 118° С . В инфракрасном спектре этого соединения
имеется полоса поглощения при 1500 см~\ что подтверждает нали-
чие л-связи аллильной группы с титаном. На воздухе (C5H5)2TiC3H5
быстро окисляется.
Бритцингер [256] получил (C5H5)2TiC3H5 взаимодействием
(C5H5)2TiCl с аллилмагнийбромидом. А. Н. Несмеянов и соавторы
[134] синтезировали ацильное производное (C5H5)2Ti (ОСОСН3)2.
По реакции пентафторфениллития C6F5Li с бис-(циклопентадие-
нил )дихлортитаном получены кристаллы (C5H5)2Ti(C6F5)2 в виде
оранжевых игл с температурой плавления 228—230° С [326, 560].
В результате пиролиза этого соединения при 150° С выделено не-
большое количество желтого вещества состава (C5H5)2Ti (C6F5)F,
разлагающегося без плавления при 240° С. Синтезирован также про-
дукт замещения одного атома хлора (C5H5)2Ti(C6F5)Cl в виде блед-
но-оранжевого вещества с точкой плавления 201—203° С [326, 560].
Это вещество образуется при действии C6F5MgBr на (C5H5)2TiCl2
в эфире [515]; температура плавления его равна 187—188° С. Опи-
санным методом получен не вполне чистый бис-(циклопентадиенил)-
бромфенилтитан (C5H5)2Ti(C6H5)Br.
При щелочном гидролизе (C5H5)2Ti(C6F5)Cl образуется вещество
состава (C5H5)2Ti (C6F5)OH с точкой плавления 183—185° С [326].
Кипячением (C5H5)2Ti (C6F5)C1 с раствором NaOC2H5 в этаноле вы-
делены желтые кристаллы (C5H5)2Ti (C6F5)OC2H5 с точкой плавле-
ния 117° С, устойчивые в сухом воздухе [326].
Саммерс с соавторами [541, 544], действуя на бис-(циклопента-
диенил)дихлортитан литийариламином, получил бис-(циклопен-
тадиенил )дифенилтитан (C5H5)2Ti (СбН5)2, бис-(циклопентадиенил)-
-ди-н-толилтитан (C5H5)2Ti (н-С6Н4СН3)2, бис-(циклопентадиенил)-
-ди-лг-толилтитан (C5H5)2Ti(jw-C6H4CH3)2 и бис-(циклопентадиенил)-
-ди-лг-диметиламинофенилтитан (C5H5)2Ti • ри-С6Н3Н2 (СН3)2]2.
При термическом разложении дифенил -бис- (циклопентадиенил)-
титана происходит восстановление его до бис-(циклопентадиенил)-
16
титана с образованием свободных фенильных радикалов [212]. В эфи-
ре в присутствии дифенилацетилена дифенил-бис-(циклопентади-
енил)титан распадается с образованием продукта присоединения
(C5H5)2Ti • 2 (С6Н5)2С2 [457].
Г. Г. Дворянцева с соавторами [85] получила соединения
(C5H5)2Ti (ОСОС2Н5)2, (C5H5)2Ti (ОСОСН2С1)2, (C5H5)2Ti (OCOCF3)2,
(CH3C5H4)2TiCl2, (СН3С5Н4) (C5H5)TiCl2. В патенте [317] описано
метильное производное (CH3C5H4)2Ti (СН3)2.
В. И. Тельной с соавторами [226] измерил теплоты сгорания
C5H5TiCl3, (C5H5)2TiCl2, (С5Н5)2 Ti(CH3)2, (C5H5)2Ti(C2H5)2 и вы-
числил энергии диссоциации связей, равные для Ti—С5Н5 74,
Ti—СН3 60, Ti—С6Н5 84ц ккал/моль. Исследованы химические
сдвиги протонов циклопентадиенильного кольца ряда замещенных
бш>(циклопентадиенил)титановых соединений [263.] Приведены
данные о масс-спектрах циклопентадиенильных производных тита-
на [267, 567].
Методом спектрофотометрии изучен термический распад титан-
органических соединений [100]. При термическом распаде
(C5H5)2Ti (СН3)2 в тетрагидрофуране, я-гексане и хлороформе об-
разующиеся метильные радикалы превращаются в метан. Терми-
ческий распад (C5H5)2Ti (С6Н5)2 и C5H5Ti (С6Н5)3 происходит с отры-
вом фенильных радикалов, переходящих в бензол. В’изопропи-
ловом спирте кроме бензола получается ацетон.
В кипящей воде бш>(циклопентадиенил)дибромтитан дает
кристаллогидрат (C5H5)2Ti(OH)Br-H2O [597]. При нагревании
бш?-(циклопентадиенил)дибромтитана с раствором пикрата
калия образуется б^с-(циклопентадиенил)оксипикрат титана
(C5H5)2Ti (OH)C6H2N3O.
Уилкинсон и Бирмингем [597], нагревая (C5H5)2TiBr2 с 12-н.
плавиковой кислотой, получили фторзамещенный бис-(циклопен-
тадиенил)титан (C5H5)2TiF2, а при нагревании раствора (C5H5)2TiBr2
в ацетоне с иодистым калием в колбе с обратным холодильником
синтезировали иодзамещенное производное (C5H5)2TjI^<
бис-(Циклопентадиенил)титаны реагируют с-' р-дикетонами,
образуя хелатные комплексы с координационным числом титана
четыре [387]. Этим методом синтезированы соединения типа
l(C5H5)TiL+] X-где L — сопряженные основания: ацетилацетон,
бензо ил ацетон, дибензо ил метан, бш>(триметил)ацетилметан, тро-
полон; X = C10J", BFJ", PFjf, AsRT, SbFjf.
В водных растворах бш?-(циклопентадиенил)титанхлорид реа-
гирует с сульфатом и карбонатом натрия по реакции обменного раз-
ложения [334]. При этом получаются бледно-зеленые кристаллы
сульфата [(C5H5)2Ti]2SO4 (разлагаются при 248° С) и бледно-го-
лубые кристаллы карбоната [(C5H5)2Ti]2CO3 (разлагаются при 280° С)
бш?-(циклопентадиенил)титана.
бис-(Циклопентадиенил)дихлортитан в растворе ацетона при
нагревании вступает в реакции обменного разложения с цианатом,
2 1-1669 1 7
селеноцианатом и тиоцианатом калия, образуя титанаты с соответ-
ствующими ацидогруппами (C5H5)2Ti (NCO)2,(C5H5)2Ti (NCSe)2 и
(C5H5)2Ti (NCS)2 [266]. Цианидные и роданидные группы связаны
с титаном через азот и поэтому по химическому строению комплексы
являются изоцианатными и изороданидными производными [335].
Изоцианат (C5H5)2Ti (NCO)2 плавится при температуре 275—277° С
(разлагается), (C5H5)2Ti (NCS)2 — при 305—307° С (разлагается).
Кёпф и Шмидт [430] получили по реакции обменного разложе-
ния серопроизводные бш?-(циклопентадиенил)титана: темно-корич-
невые пластинки (C5H5)2Ti (SC6H5)2 с точкой плавления 201° С; чер-
но-красные кристаллы (C5H5)2Ti [S (п-СН3С6Н4)] сточкой плавления
198,5° С; темно-красные ромбические кристаллы (C5H5)2Ti [S (п-
С1СбН4)]2 с точкой плавления 178—182° С; рубиново-красные кристал-
лы (C5H5)2Ti [S (С6Н5СН2)]2 с точкой плавления 172° С; рубиново-
красные кристаллы (C5H5)2Ti [SC6H4(CH2)2]2 с точкой плавления
92° С и соединение (C5H5)2Ti(SC3H7)2, плавящееся при 107—110° С.
Гиддингс [386] синтезировал серопроизводные (C5H5)2Ti (SCH3)2
с точкой плавления 193—197° С и (C5H5)2Ti [S (п-С12Н25)]2, плавя-
щиеся при комнатной температуре.
Взаимодействием раствора Na2S2C2(CN)2 в метаноле с раствором
(C5H5)2TiCl2 в ацетоне получены темно-зеленые кристаллы
(C5H5)2TiS2C2(CN)2, плавящиеся при температуре выше 260° С; вы-
делены также кристаллы состава [(C2H5)4N] (С5Н5) Ti [S2C2 (CN)2]2
[454]. Смешением водных растворов NaB (С6Н5)4 и (C5H5)2TiCl в
атмосфере аргона получен светло-голубой осадок комплекса
[(C5H5)2Ti (Н2О)3]2[В (СбН5)4]2 с точкой плавления 92° С [336]. Из
раствора метанола выкристаллизован комплекс (C5H5)2Ti [N (СН3)2Ь
• В(СбН5)4, плавящийся при 234° С. Он, реагируя с пиридином, об-
разует кристаллы [(C5HB)2Ti (C5H5N)2] В (С6Н5)4 с точкой плавления
242° С. Продукты пиролиза [(C5H5)2Ti (Н2О)3]2 [В (СбН5)4]2 при тем-
пературе 150° С представляют из себя темно-голубой остаток, со-
став которого выражается формулой (C6H5)2TiOTi (С5Н5)2.
Добавляя к (C5H5)2Ti (NCS)2 н-бутиламин, Гидрингс [386] по-
лучил оранжево-красные кристаллы I(C5H5)2 Ti (NCS)2]2O.
Кланберг с соавторами [427] по реакции (C5H5)2Ti с CsB3H8 и
CsB3D8 получил борпроизводные (C5H5)2TiB3H8 и (C5H5)2TiBD4.
Восстановлением (C5H5)2TiCl2 метаборидом лития LiBH4 син-
тезирован бис-(циклопентадиенил)титанбор гидрид (C5H5)2TiBH4
[485], который дает б^с-(циклопентадиенил)титанбортетрафторид
(C5H5)2TiBF4, а с галогеноводородами — £шс-(циклопентадиенил)-
титангалогениды (С5Н5)2ПГ, где Г = Cl, Вг, I [485].
Взаимодействием (C5H5)2TiC3H7Cl с избытком ВС13 в присутствии
металлического натрия или калия получен-комплекс (C5H5)2TiBCl4
[408]. Спиновая плотность этого комплекса на один атом бора рав-
на 0,81. Она несколько выше, чем на атом алюминия (0,70), что
объясняется большей акцепторной активностью атома бора. Боро-
гидрид (C5H5)2TiBH4 синтезировали Джемс и соавторы [424].
18
Кипячением бш>(циклопентадиенил)титандихлорида с этанолом
получен циклопентадиенилтитанэтоксидихлорид C5H5Ti (ОС2Н5) С12
1115].
бш>(Циклопентадиенил)титандихлорид реагирует с трифенил-
силанолятом натрия, замещая атомы хлора на силоксигруппы:
(С5Н5)2 TiCl2 + (СбН5)3 SiONa -> (С5Н5)2 TiCl [OSi (С6Н5)3]+
+ (СбН5)3 SiONa -> (С5Н5)2 Ti [OSi (С6Н5)2]2.
Циклопентадиенилтитантрихлорид реагирует' с метиллитием,
образуя циклопентадиенилтитантриметил [3831:
C5H5TiCl3 + 3CH3Li -> С5Н5 Ti (СН3)3 + 3LiCl.
Коутс и Уелс [330] по реакции обменного разложения
(С5Нб)2 TiCl + RCOONa -> (C5H5)2TiOOCR + NaCl
синтезировали карбоксильные производные бис-(циклопентадиенил)-
титана (III). При действии HaC5H5TiCl3 в эфире фенил сульфгидридом
в присутствии триэтиламина при молекулярном отношении компо-
нентов 1 : 3 получаются оранжево-красные кристаллы комплекса
C5H5Ti (SC6H5)3 [431]. При отношении компонентов 1 : 1 выделяются
оранжево-желтые кристаллы C5H5Ti (SC6H5) С12 с точкой плавления
104° С, устойчивые в атмосфере азота.
При взаимодействии C5H5TiCl2 в ацетоне с KSCN образуются
красные кристаллы C5H5Ti (NCS)2 с точкой плавления 164° С [431].
По реакции (C5H5)2TiCl2 с Na2Sn, где п ~ 2—7, в водном или спир-
товом растворе ацетона получен серусодержащий комплекс
(C5H5)2TiS5 и димер [(C5H5)2SJ2, где п = 2 и 3 [4331. Мономерному
комплексу приписывается строение гексамера (C5H5)2Ti
где п = 1—3, реагирует с (C5H5)2TiCl2 с образованием поли-
Он имеет температуру плавления 201° С, в бензоле мономерен. Серусо-
держащие комплексы образуются также по реакции (C5H5)2TiCl2 с
(NH4)2S5 или (C5H1oNH2)2S7 в бензоле и ацетоне. По аналогичной
реакции синтезированы соединения (C5H5)2TiS3 с точкой плавления
205° С и (C5H5)2TiS4, плавящееся при 185° С [518].
При действии на (C5H5)2TiCl2 сероводорода в присутствии третич-
ных аминов получен комплекс (C5H5)2Ti (SH)2 [434]. Политиохлорид
S„C12, ‘ _ - - - -
сульфидов типа (C5H5)2TiSm (т = 3—5) [434, 518]. Они содержат
циклы из атомов серы и титана. Окислением (C5H5)2Ti (SH)2 иодом
в присутствии пиридина получается производное с шестичленным
циклом из четырех атомов серы и двух атомов титана в положении 1,4.
Действием на (C5H5)2Ti (SH)2 иода в растворе ацетона полу-
чен сульфид (C5H5Ti)2Sn с точкой плавления 190—200° С, имеющий
2*
19
S —s
I I
строение C5H5 — Ti Ti — C5H5 [518]. Фотолиз его в бензоле,
S—S
по данным исследований радиохимического и спектрофотометри-
ческого методов [577], приводит к образованию полимерного про-
дукта (C5H5TiS4)„, содержащего Ti3+. В диметилформамиде полимер
не получается.
По реакции обменного разложения (C5H5)2TiCl2 с калиевыми
производными пиррола, индола и фталимида (KL) получены про-
дукты общего состава (C5H5)2TiL2 [411]. Литиевое производное кар-
базола образует продукт аналогичного состава. Взаимодействие
(C5H5)2TiCl2 с NaN [Si (СН3)3]2, KN [Si (СН3)312, LiN (СН3)2 и
LiN (С2Н5)2 дает димер [(C5-H5)2TiCl]2. Этилацетат с (C5H5)2TiCl2 в
безводном AgC104 образует окрашенное соединение [(C5H5)2TiL] С1О4
[363]. Действуя на (C5H5)2TiC104 калиевой солью скваратной кис-
лоты
К—О—С—С-О
К—О—С—С-О
можно выделить оранжевый комплекс (C5H5)2Ti (С4О4). Получены
также комплексы состава (C5H5)2Ti(acac) X, гдеасас — ацетилацетон,
X = FeCl4, SnCl3, HgCl3, ZnCl3, CdCl4, SnCl6.
При действии на (C5H5)2TiCl натриевыми солями дитиокарбонатов
синтезированы производные состава
где R = NH2, N (СН3)2, N (С2Н5)2, N (С3Н7)2, N (C5Hn)2, NBr2, пи-
перидил [331]. Окислительно-восстановительные реакции циклопен-
тадиенилтитан (IV) галогенидов рассмотрены в [214].
Реакции присоединения
бш>(Циклопентадиенил)галогентитаны подобно галогенидам
титана вступают в реакции соединения со многими веществами
с акцепторными свойствами, давая координационные соедине-
ния. Натта с соавторами получил в чистом виде соединения
(C5H5)2TiCl2AlCl (C2H5),(C5H5)2TiCl2AlCl2[494j и (C5H5)2TiCl2Al (С2Н5)2
[4891.
При нагревании бш>(циклопентадиенил)титандихлорида с триэ-
тилалюминием в бензоле выделен комплекс[(С5Н5)2Т1С12] [А1 (С2Н5)2]2
[143, 488]. Это — кристаллическое вещество с точкой
20
плавления 170—171° С и параметрами кристаллической решетки
а = 15,77, b = 14,24, с = 7,54 А [338, 494]. По данным ИК-спект-
роскопического и рентгеноструктурного исследования [488], в нем
к каждому атому титана присоединены две циклопентадиенильные
группы. Связь между атомами титана и алюминия в циклопентадие-
нильных соединениях слабая. В масс-спектрах осколочных ионов час-
тицы, содержащие одновремен но титан и алюминий, отсутствуют [567].
В патенте Натта [143] описан диамагнитный димер
[(C5H5)2TiAl (С2Н5)2]2. Он получен нагреванием бис-(циклопента-
диенил)титандихлорида и триэтилалюминия в бензоле. Кристаллы
его имеют ромбическую решетку с параметрами а = 9,50, b = 14,60,
с = 19,40 А, фед. гр. РЬет [489]. Структура состоит их димерных
единиц, объединенных связью Ti—Ti.
Комплексы бис-(циклопентадиенил)дихлортитана с алюминий-
алкилами являются катализаторами полимеризации этилена и про-
пилена [283, 491] (так называемые наттавские катализаторы). Ис-
следование каталитических свойств системы (C5H5)2TiCl2Al (СН3)2С1
методом ЯМР позволило прийти к заключению, что взаимодействие
мономеров в ней происходит по связи титан — алкил [79]. Наиболее
активным инициатором полимеризации этилена служит комплекс
[(C5H5)2TiCl2] Al (С2Н5)2, причем группа С2Н5 входит в состав по-
лимера. Этот комплекс катализирует образование кристаллических
полимеров этилена, а в смеси с TiCl3 — и пропилена. Каталити-
ческая активность комплекса [(C5H5)2TiCl2] А1С12 мала, и в полимер
входит группа С5Н5.
Фурман с соавторами [228] изучил каталитическую активность
системы (C5H5)2TiCl2 — Al (С2Н5)3 в среде 1,2-дихлорэтана при по-
лимеризации этилена. Каталитическая активность системы в основ-
ном определяется мольным отношением (C2H5)2TiCl : Al (С2Н5)3.
Циклопентадиенильные производные титана при смешении с ди-
этилалюминийхлоридом в толуоле образуют синее кристаллическое
вещество. Бреслов и Невбург [283] состав его выразили формулой
(C5H5)2TiCl • 0,5 (С5Н5)2 А1С1 • 0,5С2Н5А1С12. Однако впоследствии
было показано, что оно является смесью трех комплексов со-
става (C5H5)2Ti (С5Н5) Cl . С2Н5А1С12, (C5H5)2TiCl • (С2Н5)2А1С1 и
(C5H5)2TiCl • (С2Н5) А1С12 [449].
Исследуя взаимодействие б^с-(циклопентадиенил)титандихлори-
да и А1С13 в толуоле спектрофотометрическим методом, Лонг [4481
установил образование комплексов состава (C5H5)2Ti (А1С14) и
(C5H5)2Ti(AlCl4)2. При замене А1С13 на СН3А1С12 получаются ком-
плексы (C5H5)2TiCl • СН3А1С13 и (C5H5)2Ti [(СН3)2А1С12]2.
При восстановлении б^с-(циклопентадиенил)титандихлорида
металлическим натрием в присутствии А1С13 в тетрагидрофуране
получен комплекс (C5H5)2TiH2AlCl3 [409]. По данным спектров ЭПР
сделано заключение, что в рамках модели донорно-акцепторного
комплекса один из атомов водорода связан с титаном, а другой —
с алюминием [409].
21
бш>(Циклопентадиенил)дихлортитандиэтилалюминий получили
А. К. Зефирова и А. Е. Шилов [88]. Ю. В. Киссин с соавторами [90]
изучили ИК-спектры поглощения комплексов [(C5H5)2TiCl2]-
• Al (С2Н5)2 и [(CbH5)2TiCl2] Al (С2Н5) Cl, обнаружив некоторое раз-
личие между ними в области частот 820 и 870 см~х.
Адема с соавторами [244] на кривых спектрофотометрическо-
го титрования (C5H5)2TiCl2 раствором хлористого алюминия в бен-
золе наблюдал изломы, отвечающие образованию соединений
(C5H5)2TiCl2 • AICI3 и (C5H5)2TiCl2 • 2А1С13. Натта и Массанти [491]
получили комплексы (C5H5TiCl3) А1С12 и (C5H5TiCl3) А1С12 • А1С13,
инициирующие образование кристаллических полимеров (этилена
и пропилена).
Диксон и Вест [348] изучили взаимодействие между дихлоридом
бш>(циклопентадиенил)титана и производными щелочных металлов
с алкилами свинца и олова Na [Pb (С2Н5)3] и Li [Sn (С2Н5)3]. Они
получили продукты присоединения (C5H5Ti)2 • NH2Pb (С2Н5)3 (реак-
ция проводилась в жидком аммиаке) и (C5H5)2TiClSn(C2H5)3.
Натта с соавторами [494] на основании рентгеноструктурных
исследований приписал титаналюминиевым комплексам следующее
строение:
С5Нз .. С1.. СаН5
х- Tl._ А1
C5Hs ' CV' 4 С2Н5
Были исследованы гомогенные растворы (C5H5)2TiCi2 с
Al (изо-С4Н9)3 в толуоле методом ЭПР [232]. При отношениях ком-
понентов в пределах 2:1 — 100 : 1 получены три спектра ЭПР.
Первые два сигнала, судя по величине ^-факторов, относятся за
счет электронов, локализованных на атомах титана или алюминия
на уровнях, обладающих значительной примесью орбитального
магнетизма. Третий спектр объясняется взаимодействием локали-
зованного на алюминии неспаренного электрона со спином ядра А127
и спинами двух эквивалентных протонов.
В другой работе [87] авторы изучили методом ЭПР взаимодей-
ствие алюминийалкилов и алюминийарилов Al (С2Н5)3, А1 (С6Н5)3,
Al (озо-С3Н7)3, Al (СН3)3, Al (шо-С4Н9) и Al (QH^Cl с дигалоге-
нидами био (циклопентадиенил )титана (C5H5)2TiCl2, (C5H5)2TiBr2
и (C5H5)2TiI2. При соотношении компонентов 1 : 1 получены сход-
ные сигналы ЭПР с g = 1,975. Соединения Al (С2Н5)3, Al (rzso-C3H7)3
и Al (rzso-C4H9)3, кроме того, дают еще два сигнала ЭПР при соотно-
шениях Ti : Al, отличающихся от 1 : 1, форма которых не зависит
от природы алкила и галогена. Ответственными за первый сигнал
авторы считают соединения типа
с5н5 х ...С2н5
Ti Al
С5Н5Х ЧС1’’ '’C2HS
22
с двумя мостиковыми атомами хлора, содержащими неспаренныи
электрон. За второй и третий сигналы, по мнению авторов, отве-
чают примеси алкилгидридов алюминия, которые с диамагнитными
дигалогенидами бис-(циклопентадиенил)титана образуют соедине-
ния типа
, г-Н-. .. Н.
(W2Ti„ Al (изо -С4н9)а И (C5H5)2Ti; А1(И30-С4Н9)2
CI * С1
с водородными мостиками.
Методами ЭПР и протонного магнитного резонанса изуче-
ны системы (С5Н5) TiCl2 — [Al (СН3)2С1]2. и (С5Н5) TiCH3Cl —
—[Al (СН3) С12]2 [253]. Оказалось, что в этих системах сразу же после
их приготовления метильная группа мигрирует от атома алюминия
к атому титана; число неспаренных электронов равно числу молекул
(C5H5)2TiCl2, т. е. неспаренные электроны принадлежат частицам,
содержащим ионы Ti3+, Сигналы ЭПР состоят из трех спектров,
форма которых обусловлена взаимодействием неспаренного электро-
на с протонами циклопентадиенильного кольца и с ядрами атомов,
находящимися в комплексе. Значения g-фактора в обеих системах на-
ходятся в пределах 1,97—1,98, что указывает на существование
комплексов, в которых либо один атом хлора непосредственно при-
соединен к титану, либо два атома хлора присоединены к титану
мостиковыми связями.
А. Н. Несмеянов с соавторами [136] исследовал методами ЭПР
и ИК-спектроскопии влияние присоединенных к титану групп на
характер связи его с циклопентадиенильными кольцами. С умень-
шением электроотрицательных свойств присоединенных групп связь
титана с л-комплексами становится более ионной и менее ковалент-
ной.
бис-(Циклопентадиенил)титан с перекисью бензоила образует
бис-(циклопентадиенил)титандибензоат — соединение желтого цве-
та, плавящееся при 188° С [211].
Кёпф и Блок [432] провели в растворе толуола реакции окисли-
тельного присоединения к титаноцену:
|(С5Н5)2 Ti]2 + 4Х = 2 (С5Н5)2 TiX2,
где X = (C6H5Se)3(CH3S)3I3,(C5H5S).
Смешением (C5H5)2TiCl с некоторыми бидентатными лигандами
в тетр а гидрофуране получены комплексы (C5H5)2TiCl • 1,5L, где
L = С6Н5, o-C6H4(NH2)2, дипиридил, а-пиколинамин [333]. Все эти
комплексы — кристаллические окрашенные вещества, нераствори-
мые в органических растворителях. Магнитные моменты их равны
соответственно 1,77; 1,76; 1,83 и 1,64 р,в, что отвечает одному не-
спаренному электрону в атоме титана.
23
Соединения со связью металл — металл
Котте и У еле [332] синтезировали циклопентадиенильные соеди-
нения титана с металлметаллической связью: (C5H5)2TiSn (С6Н5)3,
(C5H5)2Ge (С6Н5)3 — изумрудно-зеленые кристаллы с точками плав-
ления 80 и 100° С соответственно, (C5H5)2Ti[Sn(C6H5)3]2 — зеле-
ное вещество, плавящееся при 80° С, (C5H5)2ClTiSn (С6Н5)3 и
(C5H5)2CiTiGe (С6Н5)3 — темно-зеленые вещества с точками плавле-
ния 177—180 и 193—196° С.
Получен комплекс (C5H5)2TiCI2GaCl2 с мостиковой связью. При
исследовании его методом ЭПР обнаружено взаимодействие неспа-
ренного электрона Ti (III) с ядром Ga69 и Ga71 [407]. Образование
соединения (C5H5)2ClTiPb (С6Н5)3 отмечено только в растворе [332].
Фиксация азота
М. Е. Вольпин и В. Б. Шур [69, 70] показали, что тетрахлорид
титана в смеси с (^зо-С4Н9)3А1, C2H5MgBr, а также с арильными
производными ПС14 обладает способностью катализировать при ком-
натной температуре в растворе арильных соединений фиксацию
молекулярного азота. Фиксация азота объясняется способностью
Таблица 1
Фиксация азота при комнатной температуре системами на основе
титана [70]
Система Растворитель Давление азота, атм Выход NH3 на I моль ти- тановой соли, моль
TiCl4—C2H5MgBr Эфир 150 0,10
(CsHs^TiCh—C2H6MgBr 150 0,84
(C6H6)2TiCl2—CaHsMgBr 1 0,67
(СбНб)2Т iCl 2—C2HsMgBr Тетрагидрофуран 150 0,40
(СбН6)2Т!С12—w-C4H9Li Гептан 150 0,50
титана как переходного металла катализировать реакции вслед-
ствие образования л-комплексов с ненасыщенными соединениями
по схеме
N ГН1
R—M + N2 -> RM <- || > NH3.
N
Выход аммиака по этой каталитической реакции при комнат-
ной температуре зависит от давления азота и за 8—10 ч достигает
0,84 моля на 1 моль TiCl4 в смеси (табл. 1). Фиксация азота приоста-
навливается после образования менее 1 моля NH3 на 1 моль TiCl4.
Г. И. Нечипоренко с соавторами [106] высказали другую гипо-
тезу о механизме фиксации азота в системе C2H5MgBr—(C5H5)2TiCl2.
Они установили, что аммиак, выделенный после проведения реакции
24
фиксации азота в растворе (C2D5)2O, а также с использованием в ка-
честве восстановителя C2D5MgBr, не содержит дейтерия. Отсюда
напрашивается вывод, что азот фиксируется в виде нитрида, кото-
рый может образоваться при взаимодействии азота с атомами ме-
таллического титана или магния в момент их образования.
Бритцингер [255], основываясь на спектрах ЭПР, полагает, что
фиксация азота происходит через промежуточное соединение—димер
[(C5H5)2TiH]2. Этот же автор пытался установить строение про-
дукта реакции — димера [(C5H5)2TiH]2 с азотом [257]. Выдерживая
смесь (C5H5)2TiCl2 и C2H5MgCl в тетрагидрофуране под давлением
азота 150 атм в течение 15 дней, он наблюдал образование соедине-
ния, которое при подкислении выделяло 1 моль аммиака на 1 моль
титанового комплекса. Спектр ЭПР этого соединения содержит пять
линий, обусловленных взаимодействием неспаренного электрона
с двумя эквивалентными ядрами азота, на основании чего выска-
зано предположение, что поглощение молекулы азота происходит
за счет внедрения ее по двум связям металл — водород димера
[(C5H5)2TiHl2, а л-кОмплекс азота является промежуточным про-
дуктом.
Газообразный азот фиксируют растворы (C5H5)2Ti (СН3)2 и
(С5Н5)2 Ti (С6Н5)2 в эфире и алифатических углеводородах [71]. При
температуре 90—100° С и давлении азота 80 атм за 10 ч присоеди-
няется от 0,08 до 0,20 г-ат азота на 1 г-ат титана. В присутствии
водорода количество фиксированного азота увеличивается до
0,32—0,36 г-ат на 1 г-ат титана.
Молекулярный азот может связывать бпс-(циклопентадиенил)-
титан [328]. При гидролизе изопропиловым спиртом выделяется
аммиак. Образовавшийся т£тршшс-(изопропокси)титан можно вос-
становить до комплекса двухвалентного титана электролитически
и осуществить фиксацию молекулярного азота в замкнутом процессе.
При стоянии эфирных растворов титановых комплексов с фи-
ксированным азотом наблюдается восстановление азота до гидрозина
и его производных [231].
Кинетика связывания молекулярного азота раствором
(C5H5)2TiCl2 и C2H5MgBr в диэтиловом эфире при давлении 1 атм
подчиняется уравнению реакции второго порядка [469, 470]. Коли-
чество связанного азота пропорционально концентрации (C5H5)2TiCl2.
КАТАЛИЗАТОРЫ ЦИГЛЕРА — НАТТА И ДРУГИЕ
к КАТАЛИТИЧЕСКИЕ РЕАКЦИИ ‘
Титанорганические соединения оказались очень эффективными
катализаторами для низкотемпературной полимеризации непре-
дельных углеводородов и нашли широкое промышленное примене-
ние в производстве полиэтилена, полипропилена и других синте-
тических материалов. Получаемые при этом кристаллические
полимеры обладают высоким молекулярным весом и линейной стерео-
25
регулярной структурой. Большинство катализаторов Циглера —
Натта представляют собой системы, состоящие из тетрахлорида ти-
тана или низших его хлоридов и металлорганических соединений.
За короткое время запатентовано огромное число каталитических
систем. Они обычно рекомендуются для полимеризации олефинов,
но в девяти случаях из десяти, как отмечают Филд и Коув [229],
речь идет об этилене или пропилене.
Способы получения и использования катализаторов, описанные
в разных патентах, мало отличаются друг от друга [229]. Катализа-
торы Циглера — Натта обычно готовят смешением соединений ти-
тана и других металлов в присутствии инертного растворителя.
Катализаторы применяются в форме твердых веществ, взвешенных
в растворителе. Ниже приведен перечень типичных каталитических
систем [229]:
Каталитические системы
TiCl4—(alc)sAl
TiCl4—(С2Н6)2А1С1
TiCl4—(С2Нв)3А1
TiCl3—(С2НВ)3А1
Ti(OC4H9)4—(alc)3Al
CBHBTiCl3—C2HBA1C12
TiCl4—(uso-C4H9)3A1
TiCl4—Zn(CeHB)2 или Mg(CeHB)2
Полимеризуемые мономеры
Олефины
Этилен, 2,3-диметилбутадиен-1,3
Этилен, децен-1, п-метилстирол, Af-метилсти-
рол, ацетилен, аллилсилан, аллилтриметил-
силан, диаллилдиметилсилан, триметилви-
нилсилан, триметил-3-винилпропилсилан, 3-
бутилтриметилсилан, /г-триметилсилилстирол
Пропилен
Этилен, бутен, гексен
Этилен
Пропилен, 2-фенилбутадиен-1,3, дивинилаце-
тилен
Этилен
Механизм действия катализаторов Циглера — Натта при низ-
котемпературной полимеризации окончательно еще не выяснен.
Следует, однако, признать, что активность катализаторов обуслов-
лена наличием в каталитических системах соединений со связью
титан — углерод. Эти смеси хотя и готовятся из хлоридов титана,
но между последними и металлорганическими соединениями, вхо-
дящими в состав катализаторов, возможно протекание реакций
обмена, приводящих к образованию титанорганических соединений.
Протекание реакций обмена между TiCl4 и (СН3)3А1, TiCl3 и
(С2Н5)3А1 установлено экспериментально. При добавлении TiCl4
к (С2Н5)2А1С1 в растворе четыреххлористого углерода образование
C2H5TiCl3 фиксируется по исчезновению полосы в спектре погло-
щения с максимумом при 285 нм, характерной для TiCl4 [260].
Физико-химические исследования каталитических систем позво-
ляют констатировать ряд явлений, имеющих отношение к их актив-
ности. В этих системах обнаруживается существование непрочных
комплексов между титанорганическими соединениями и непредель-
ными углеводородами. Дьячковский и соавторы [81] при распаде
CH3TiCl3 под действием органических оснований наблюдали появле-
ние свободных радикалов СН3н.
26
При исследовании систем TiCl4 — (С2Н5)2А1С1 и TiCl4—
(СН3)2А1С12 замечено повышение электропроводности после смеше-
ния компонентов [80]. Методом электродиализа показано наличие
в растворах каталитических систем ионов [TiCl2R]+ [80] и
[(C5H5)2Ti (СН3)]+ [74]. Измерение электропроводности в системах
бензол + гептан, бензол в дихлорэтане + гептан в дихлорэтане ис-
пользовано для определения константы диссоциации комплекса
(C5H5)2TiCH3ClAlCH3Cl2 на ионы. Переход от смеси бензол + геп-
тан к дихлорэтану сопровождается увеличением константы диссоци-
ации в 1010 раз [61].
Во многих каталитических системах происходит восстановление
четырехвалентного титана до низших степеней валентности. Мето-
дом ЭПР в системе TiCl4 — (изо-С4Н9)3Al обнаруживается четыре ак-
тивных парамагнитных центра, обусловленных присутствием атомов
титана с неспаренными электронами [572]. Восстановление Ti (IV)
до Ti (III) происходит в каталитических системах (C5H5)2TiRCl •
. A1RC12, где R - СН3, С2Н5 [470]. Система Al (С2Н5)2С1—TiCl3—
ацетилен, катализирующая образование тримеров ацетилена с вы-
сокой селективностью, дает сильный сигнал ЭПР eg = 1,97 (АТ/ 11 гс)
даже в присутствии сильных оснований [566]. При добавлении эта-
нола сигнал ЭПР исчезает и активность катализатора теряется.
Согласно [451], твердая фаза катализатора обладает наибольшей
активностью полимеризации, когда титан находится в трехвалентном
состоянии.
Исследование кинетики восстановления Ti(IV) в смесях
TiCl4 + (С2Н5) А1С12 и CH3TiCl3 + (С2Н5) А1С12 в гептане показа-
ло, что концентрация неспаренных электронов, измеренная методом
ЭПР, точно соответствует содержанию Ti (III) [447]. Полимериза-
ция этилена протекает со скоростью, линейно зависящей от концен-
трации Ti (III). Ткач [574] полагает, что необходимым, но недоста-
точным условием, обеспечивающим каталитическую активность
к полимеризации, является наличие неспаренных З^-электронов в
атомах Ti (III), связанных с (изо-С4Н9)2А1С1 в форме биметалличе-
ского комплекса.
Однако имеются системы, каталитическая активность которых
определяется содержанием титана в четырехвалентной форме [449].
И. В. Калечиц с соавторами [91] показал, что восстановление ти-
тана в системе (C2H5)2TiCl2—А1(С2Н5)3, являющейся катализа-
тором при гидрировании гексена-1, гептена-1, гептена-3 и гексена-2,
до трех- и двухвалентного приводит к дезактивации катализатора.
Введение в реакционную смесь следовых примесей кислорода
(0,001—0,08%) увеличивает активность катализатора.
Исследование системы TiCl4 — (изо-С4Н9)3А1 в гептане методами
ЭПР и ИК-спектроскопии при отношении Al : Ti = 0,7—1,5 по-
казало, что механизм реакции, природа сигналов ЭПР и активность
катализатора при реакциях полимеризации зависят от избытка
TiCl4 или (г/зо-С4Н9)3А1, поддерживаемого в ходе синтеза [573].
27
Следы AlCIg, влаги или галогенсодержащих растворителей от-
щепляют от катализаторов Циглера — Натта одно циклопентадие-
нильное кольцо [410]. Образующийся при этом моноциклопента-
диенильный комплекс обнаружен методом ЭПР как комплекс с дву-
мя молекулами А1С13.
Активность катализатора зависит от природы галогена, входя-
щего в состав каталитической системы. Полимеризация циклодо-
декатриена-1,5,9 с катализатором ПГ4 (изо-С4Н9)3А1 (Г = Cl, Вг, I)
повышается при переходе от Til4 к TiBr4 и TiCl4 [67].
Наибольшее признание получили две гипотезы, объясняющие
механизм действия катализаторов Циглера — Натта при реакциях
полимеризации. Согласно одной из них [381, 528], в основе механиз-
ма каталитического действия лежит образование и распад промежу-
точных комплексов между олефином и атомом переходного металла.
Постулируется различный состав переходных комплексов и различ-
ный механизм их распада.
Исследуя взаимодействие этил-бис-(циклопентадиенил)титан-
хлорида с триэтил литием, содержащим радиоактивный углерод,
Синн и Петет [528] показали, что эти компоненты образуют комплекс,
содержащий хлорный и углеродный мостики:
В дальнейшем такой комплекс распадается с образованием этана
и этилена в зависимости от температуры по схеме
щс9т(
5 2 ।
0,5С2Н4
0.5С2Н6
Ti(C2H5),(C И,) +А1С1(С*Н,)2
X. и 4» Q О 4 Z о 4
.•С1
+ (С5Н5)2тГ * -А1
с н2-
сн3
zC*Hs
ХН5
Улзман [576] предположил ионный механизм полимеризации с
образованием частиц (TiCl3)+(R3AlCl)“ или (TiCI2)'H(R3AlCl)~.
Например, полимеризация этилена должна сопровождаться обра-
зованием комплекса типа (TiCl3)+(RCH2CH2AlR2Cl)~.
Э. А. Григорян и соавторы [74] полагают, что в системе
(C5H5)2TiCl2—(СН3)2А1С1 образование промежуточного комплек-
са сводится к внедрению олефина по связи Ti—С в ионе
(C5H5)2Ti (СН3)+. Хенрики-Олив [406] показал, что координация
олефина с атомом переходного металла ослабляет связь металл —
углерод. В системах типа (С5Н5) TiRCl • A1R'C12, где R и R' —
28
алкилы, скорость восстановления Ti4+ -> Ti3+, сопровождающегося
раскрытием связи Ti—С и извлечением R из комплекса, повыша-
ется при введении в систему этилена.
Бредли и соавторы [279] рассчитали энергетические уровни для
молекулярных орбиталей октаэдрического комплекса RTiCl4 —
олефин, постулируемого в качестве промежуточного соединения
при реакции полимеризации. Разница между энергетическими уров-
нями максимально заполненной и полузаполненной орбитали ока-
залась близка к энергии активации полимеризации (10—
14 ккал!моль).
По другой гипотезе [493], каталитическое действие титанорга-
нических соединений объясняется свободно-радикальным механиз-
мом [381, 493]. Фридлендер и Ойта [381] образование этана и этилена
при взаимодействии этиллития с тетрахлоридом титана представля-
ют в виде следующей схемы:
LiC2H5 + TiCl4 -> LiCl + TiCl3 (С2Н5),
TiCl3 (С2Н5) -> TiCl3 + с2н5,
2С2Н5 С2Н„ 4-С2Н,.
Каталитической активностью отличается система СН31 —
(C5H5)2Ti-С4Н8О— СН3АП2 при полимеризации этилена [109]. Ме-
тильные группы СН31 при протекании реакции входят в состав по-
лимерных молекул. При действии на (C5H5)2Ti • С4Н8О йодистого
метила происходит реакция окислительного присоединения с обра-
зованием связи Ti—СН3.
Изучена кинетика полимеризации децена и этилена под дейст-
вием каталитического комплекса [(C5H5)2TiCH3l+ [61, 73], а также
кинетика гидрогенизации олефинов с помощью титана на основе
(C5H5)2TiCl2 [532].
Окись этилена в присутствии дихлордиэтилата и дихлордиизо-
пропилата титана превращается в вязкую жидкость [60]. Тетраизо-
пропилтитан является эффективным катализатором этерификации
при 200° С [529].
Бутен-1 в присутствии каталитических комплексов Циглера —
Натга (смеси A1R3 и A1R2C1 с TiCl4, TiCl3 или смеси TiCl3 и TiCl2)
изомеризуется до цис- или транс-бутена-2 [456].
ПРОИЗВОДНЫЕ СО СВЯЗЬЮ Ti—Э—С
Связь титана с углеродом типа Ti—Э—С может осуществлять-
ся через посредство атомов кислорода, азота, кремния, фосфора,
серы и других. Соединения с этой связью относятся к числу более
стойких веществ, чем образованные на основе титан — углерод.
Среди них следует отметить многочисленные эфиры гипотетической
29
ортотитановой кислоты, галогенэфиры, амиды, сложные эфиры ор-
тотитановой кислоты с карбоновыми кислотами, соединения с си-
ланами, фосфорорганическими производными и пр.
ЭФИРЫ
К эфирам ортотитановой кислоты относятся соединения, состав
которых выражается общей формулой Ti (OR)4, где R — алкил,
алкенил или арил. Титанэфиры чаще всего получаются при дей-
ствии на тетрагалогениды титана спиртов и алкоголятов:
TiT4 + 4ROH - Ti (OR)4 + 4НГ,
TiT4 + 4RONa - Ti (OR)4 + 4NaT.
При этерификации спиртами выделяющийся галогеноводород
необходимо связывать аммиаком или органическими основаниями,
так как в противном случае реакция останавливается на стадий
диалкоксидигалогентитанов (RO)2Tir2 [2, 107].
Применение безводного аммиака для фиксации хлористого во-
дорода, выделяющегося при реакции этерификации, предложе-
но в патентах [164, 165]. В патенте [171] рекомендуется фиксиро-
вать хлористый водород формальдегидом и диметилформамидом.
А. Н. Несмеянов и соавторы [118] при синтезе алкилтитанатов ис-
пользовали пиридин. В патенте [166] описан метод получения тет-
раалкилтитанатов с высоким выходом, в котором связывание хло-
ристого водорода производится кальциево-натриевой солью.
Алкилтитанаты получаются при действии на TiCl4 алкилено-
ксидов [172]:
TiCl4 + 4R—СН—СН2 - Ti (OCH2CHC1R)4.
Этим методом синтезированы Р-хлоралкилтитанаты из эпоксидов
(окисей этилена, пропилена, циклогексена) [25, 172].
Замещение одних RO-групп в титанэфирах на другие может
производиться по реакции переэтерификации:
Ti (OR)2 + 4R'OH - Ti (OR')4 + 4ROH.
Алкил- и арилтитанаты получаются при взаимодействии суль-
фида титана со спиртами и фенолами [173], а также натрий- и
калийтитангексафторидс в с алкоксидами или арилоксидами магния,
кальция и алюминия [24].
Альдегиды с ортотитанатами образуют винилтитаны. Реакция
идет через энольную форму по схеме
(RO)4Ti + R'— С = R"
I
ОН
Ti + 4ROH.
4
30
Ацетальдегид по этой реакции с тетраизопропилтитаном дает ди-
изопропилдивинилтитанат [167]. Исходя из паральдегида получен
дивинилдибутилтитанат. Синтезированы титанэфиры, содержащие
пропокси- и гексадиенильные группы [474]. Все эти соединения
являются вязкими жидкостями красного цвета. А. Н. Несмеянов
и соавторы [119] по реакции переэтерификации синтезировали тет-
и тетршшс-(2-этокси)титанаты.
Алкилтитанаты
Ti (ОСН3)4. Тетраметилоксититан впервые получили Бишоф и
Адкинс [254] из TiCl4 и CH3ONa. Тетраметил оксититан можно полу-
чить, используя реакцию этерификации [86, 285, 561]. Для этого
к раствору TiCl4 в бензоле по каплям прибавляют абсолютный ме-
тиловый спирт при температуре 0° С и пропускают ток сухого ам-
миака, пока не поглотится 4 моля NH3 на 1 моль TiCl4. Образовав-
шийся хлористый аммоний экстрагируется охлажденным метано-
лом, а осадок эфира высушивается над пятиокисью фосфора.
Тетраметилоксититан — светло-желтый порошок с температурой
плавления 209° С. Образует кристаллы с триклинной симметрией.
Параметры решетки их составляют а — 9,09, b = 10,62, с = 10,62 А,
а = 101° 13', ₽= 123° 14', у-_102°06', ризм = 1,496 г/см3, ррентг =
= 1,476 г!см\ z = 4, фед. гр. Р1 [602]. Под действием водяных паров
гидролизуется с образованием аморфного гидрата двуокиси титана
состава TiO2 • 0,69Н2О.
Гут [391] изучил равновесие образования метилатов титана в
растворе метанола. Форина и Брессан [380] получили оптически
активный (—) тетраметилтитанат.
Ti (ОС2Н5)4. Титанэтиловый эфир пытались получить еще Демоли
[349] и 'Пфордтен [495] взаимодействием TiCl4 со спиртом, но без-
успешно. Впервые он был получен, как и тетраметил оксититан, Би-
шофом и Адкинсом [254] действием на TiCl4 алкоголята натрия. Дру-
гие исследователи синтезировали его действием спирта и аммиака
или металлического натрия на раствор TiCl4 в бензоле [1, 285, 304,
312, 319, 320, 550] или нагреванием спирта с аммиакатом TiCl4 • 6NH3
[445, 611]. Кулинане с соавторами [323] получил Ti (ОС2Н5)4 по ме-
тоду переэтерификации.
Тетраэтилоксититан образует моноклинные кристаллы с пара-
метрами решетки а = 19,50, b = 14,67, с = 17,14 А, ₽ = 106,Г,
Ризм = 1,23, Ррентг = 1,27 г/см3, z = 16, фед. гр. С2/с [413]. Точка
плавления его равна 40° С, точка кипения 236—237° С (760 мм рт. ст.)
[323] и 150° С (15 мм pm. ст.), показатель преломления 1,5049 (35° С),
магнитный момент, по данным [276], 1,41, по данным [323], 1,50 р,в.
Тетраэтил оксититан с бензоилацетоном С6Н5СОСН2СОСН3 в бен-
золе образует соединение Ti(OC2H5)3(C6H5CO=CHCOCH3) в виде
оранжевых кристаллов с точкой плавления 82° С и продукт
31
Ti(OC2H5)2(C6H5CO=CHCOCH3)2 в виде желтого порошка с точкой
плавления 119° С [511].
Эти вещества реагируют с н-бутаналом, фенолом и этиленгли-
колем по реакции переэтерификации, замещая группы ОС2Н5
на остаток соответствующего спирта. При действии на
Ti(OC2H5)2(C6H5COCH2COCH3)2 хлористого водорода в растворе бен-
зола получен галогенэфир TiCl2(C6H5COCH2COCH3)2. Реакция эта об-
ратима. В присутствии аммиака атомы хлора замещаются на это-
ксигруппу по реакции
TiCl2 (С6Н5СОСН2СОСН3)2 + 2С2Н5ОН + 2NH3 =
= Ti (ОС2Н5)2 (С6Н5СОСН2СОСН3)2 + 2NH4C1.
Бредли с соавторами [285] в системе Ti (ОС2Н5)4—СН3СОС1
наблюдал образование продуктов TiCl4 • СН3 СООС2Н5 и
TiCl3 (ОС2Н5) • СН3СООС2Н5 (по реакции обменного разложения).
При действии стехиометрических количеств 2-гидрокси-
этиламина на Ti (ОС2Н5)4 в бензоле получаются продукты переэте-
рификации и присоединения аминоспирта (C2H5O)3TiOCH2CH2NH2,
(C2H5O)2Ti (OCH2CH2NH2)2, (С5Н5О) Ti (OCH2CH2NH2)3,
Ti (OCH2CH2NH2)4 и Ti (OCH2CH2NH2)4 . HOCH2CH2NH2. Все эти
соединения — белые кристаллические вещества.
Тетраэтоксититан дает продукты присоединения с органическими
основаниями Ti (ОС2Н5)4 • C5H5N [562], Ti (ОС2Н5)4 • C2H5NH2
[421], Ti (ОС2Н5)4 • NH2CH2CH2NH2 [284], Ti (OC2H5)4 • NH2NH2
[284].
По данным рентгеноструктурного анализа, твердый триэтил-
титанат существует в форме тетрамера [594]. В жидком состоянии,
как и большинство алкилортотитанатов, за исключением вторичных
и третичных алкилтитанатов, ассоциирован [571]. Ассоциацией
тетраэтилтитаната объясняются большие значения энтальпии в
реакциях взаимного обмена в системах (C6H5O)4Ti—[(CH3)2N]4Ti,
l(C6H5O)]4Ti - [(CH3)2CHO]4Ti, (C6H5)4Ti - TiX (X = Cl, Br) [604].
Судя по величинам молекулярной поляризации и дипольного момента
[601], тетраэтилтитанат ассоциирован в растворе н-гептана. Ис-
следования методом ЯМР и криоскопии показали, что тетраэтил-
титанат в широком интервале концентраций и температур в растворе
существует в форме тримера [295]. Капур и Мехротра [435] синте-.
зировали титанэфир Ti [OCH2CF3]n.
В растворе диоксана Ti (ОС2Н5)4 полимеризуется. Степень по-
лимеризации титанэфира с повышением концентрации увеличива-
ется, стремясь к пределу.
Ti (ОС3Н7)4. Тетраизопропоксититан Ti (ОС3Н7-^зо)4 получен
действием алко гол ята натрия на тетрахлорид титана [254], действием
на четыреххлористый титан изопропилового спирта и металлического
натрия или безводного аммиака [321], а также [611] синтезирован
кипячением изопропилового спирта с TiCl4 • 6NH3.
32
i
Это — жидкость с точкой кипения 220° С (760 мм рт. ст.);
под вакуумом (13 мм рт. ст.) кипит при 130° С [323]; показатель
преломления 1,4568 (35° С) [611]. Инфракрасный спектр поглоще-
ния изучен Беллом с соавторами [270]. В жидком состоянии ассо-
циирован [571]. Ассоциация наблюдается даже в разбавленных раст-
ворах н-гептанаД601]. В растворе тетрапропилтитанат существует
в форме тримеров [295].
С бор гидридом В2Н2 тетраизопропилтитан в тетрагидрофуране
образует комплекс Ti (ОС3Н7-изо) (ВН4)2 • С4Н8О [423]. При дей-
ствии триметилацетоксисилана протекает реакция обменного раз-
ложения только лишь с одной изопропилоксигруппой [137].
С бензоилацетоном образует соединения Ti (ОС3Н7-цзо)3 •
• (С6Н5СОСН2СОСН3) в виде оранжевой массы сточкой плавления
86° С и Ti (ОС3Н7-изо)2 (С6Н5СОСН2СОСН3)2 в виде оранжево-жел-
того осадка с точкой плавления 89° С.
Тетра-н-пропоксититан Ti (ОС3Н7-н)4 получил Спир [550] дей-
ствием на четыреххлористый титан пропилового спирта и металли-
ческого натрия. Этот эфир синтезирован по реакции переэтери-
фикации [323] и обменного разложения [461]:
Ti (ОС2Н5)4 + 4н-С3Н7ОСОСН3 = Ti (ОС3Н7-н)4 + 4С2Н5ОСОСН3.
Он представляет собой жидкость с точкой кипения 265° С
(760 мм рт. ст.) [323]. Под вакуумом (0,2 мм рт. ст.) кипит при
130° С [461]. Дипольный момент его 1,2 р,в [311], показатель прелом-
ления 1,4950 (35° С), удельный вес d4° = 1,0309, вязкость 1,6135 спз
(25° С) [323].
При действии на Ti (ОС3Н7-4/зо)4 2-гидроксиэтиламина в бен-
золе образуются соединения (u3o-C3H7O)3Ti (OCH2CH2NH2),
(aso-C3H7O)2Ti (OCH2CH2NH2)2, (изо-С3Н7О) Ti (OCH2CH2NH2)3,
Ti (OCH2CH2NH2)4, Ti (OCH2CH2NH2)4HOCH2CH2NH2 [514]. Все они
белые кристаллические вещества.
Получены продукты присоединения к тетраизопропоксититану
органических оснований и гидразина Ti (изо-ОС3Н7)4 • C5H5N [89,
562], Ti (изо-ОС3Н7)4 • C2H5NH2 [89, 421], Ti (^o-OC3H7)4 •
.NH2CH2CH2NH2 [89, 284], Ti (uso-OC3H7)4 • NH2NH2 [89, 284].
Ti (OC4H9)4. Тетра-н-бутоксититан получен действием на четы-
реххлористый титан алкоголята натрия [254], а также действием
на четыреххлористый титан н-бутанола и металлического натрия.
Он также может быть синтезирован методом переэтерификации
при нагревании тетраэтоксититана с н-бутанолом [2, 323] или тет-
раизопропилтитаната с к-бутанолом [23]. Мехротра [461] получил
Ti (ОС4Н9-н)4 по реакции обменного разложения:
Ti (ОС2Н5)4 + 4н-С4Н9ОСОСН3 = Ti (ОС4Н9-н)4 + 4С2Н5ОСОСН3,
Ti (ОС3Н7-цзо)4 + 4н-С4Н9ОСОСН3 - Ti (ОС4Н9-н)4 +
+ 4 изо-С3Н7ОСОСН3,
з 1-1669
33
а Юшино с соавторами [611]—нагреванием аммиаката TiCl4 • 6NH3
с н-бутанолом.
Наиболее удобным методом получения тетра-н-бутоксититана
является взаимодействие четыреххлористого титана с н-бутиловым
спиртом в растворе бензола в присутствии аммиака [99, 321,
565] или в смеси бензола с пиридином с последующей нейтра-
лизацией образовавшегося хлористого водорода аммиаком [107,
320, 321].
Бутиловый ортотитановый эфир — вязкая жидкость бледно-
желтого цвета [320]. На воздухе под действием паров воды полностью
гидролизуется [99]. Точка кипения его 290° С (760 мм рт. ст.), под
вакуумом (5 мм рт. ст.) кипит при температуре 175° С [611]. Удель-
ный вес 0,9927 (18° С) [579], показатель преломления 1,4864 (35° С),
дипольный момент 1,15 р,в [311]. В жидком состоянии ассоцииро-
ван [571]. По данным измерения молекулярной рефракции и диполь-
ного момента, ассоциация наблюдается и в жидком н-гексане [601].
Разлагается при нагревании до температуры 290° С [475].
В бензольном растворе Ti (ОС4Н9-н)4 полимеризован, молекуляр-
ный вес его с увеличением концентрации приближается к тримеру
[295, 503, 594]. Присоединяет аммиак, образуя аммиакат
Ti (ОС4Н9-к)4 • NH3 [351]. Бензоилацетон с ортоэфиром образует
продукты замещения Ti (ОС4Н9-н)3 (С6Н5СОСН2СОСН3) в виде
оранжевой вязкой массы и Ti (ОС4Н9-н)2 (С6Н5СОСНСОСН3)2 в виде
оранжево-желтого клейкого вещества [511].
При нагревании до 200—250° С тетра-н-бутоксититан полиме-
ризуется с выделением бензола. Полученный в результате полиме-
ризации продукт (цри 180—215° С) содержит 65,4% TiO2
(в мономере содержится 23,52% TiO2 [565]). Пури и Мехротра
[514] по реакции переэтерификации Ti (OC4H9-w) с OHCH2CH2NH2
получили белые кристаллические вещества, имеющие состав
(H-C4H9O)3Ti(OCH2CH2NH2), (H-C4H9O)2Ti(OCH2CH2NH2)2, (н-С4Н9О) .
• Ti (OCH2CH2NH2)3, Ti (OCH2CH2NH2)4 и Ti (OCH2CH2NH2)4-
• (HOCH2CH2NH2).
В толуоловом растворе тетршшс-(бутилокси)титан реагирует
с триметилалюминием, образуя соединение с отношением компонен-
тов 1 : 4 [242]. Циклооктатетраен (H2L) стетршшс-(бутилокси)тита-
ном в присутствии (С2Н5)3А1 вступает в реакцию обменного разло-
жения, образуя фиолетово-розовые кристаллы состава TiL2 и сое-
динение желтого цвета Ti2L3 [268]. Смешением растворов Ti (ОС4Н9)4
с Ga (С2Н5)3 в бензоле и гептане получен триметаллический комплекс
Ga...Ti...Ga... [82]. Через четыре месяца он переходитв биметалли-
ческий.
Из раствора Ti (ОС4Н9)4 и В2Н2 в тетрагидрофуране после от-
гонки растворителя получены голубые кристаллы состава
Ti (ОС4Н9) (ВН4)2 • С4Н8О [423]. Трихлорид бора реагирует с тет-
ршшс-(бутокси)титаном, образуя тетрахлорид титана и бутил хло-
рид [325].
34
/петршшс-( Бутокси) титан предложено использовать в качестве
связующего для алюминиевой краски, устойчивой до температуры
500—550° С [99].
Методом переэтерификации получен тетра-г/зо-бутоксититан [324].
Он представляет собой жидкость, температура кипения которой
163Q С (13 мм рт. ст.), показатель преломления 1,4749 (54° С),
удельный вес 0,9750 (55° С).
Тетра-втор-бутоксититан Ti (OC4H9-emop)4 и тетракис-(трет-
бутокси)титан Ti (OC4H9-mpem)4 получены по тем же методам, ко-
торые применялись для синтеза тетроде-(я-бутокси) титан а [26,
170, 320, 323, 461, 550, 611]. Первый из них — жидкость с темпера-
турой кипения 137° С (13 мм рт. ст.) и показателем преломления
1,4554 (35° С). Третичный бутилтитановый эфир также жидкость
с температурой кипения 155° С (14 мм рт. ст.) и показателем пре-
ломления 1,4740 (55,5° С) [611]. В жидком состоянии тетракис-
(трет-бутокси)титан не ассоциирован [571]. В растворе н-гептана
находится в форме мономера [601].
С этиленгликолем и оксиэтиленгликолем тетракис-(трет-6у-
токси)тйтан образует соединения с отношением компонентов 2 : 3,
а с -бутадиенолом-2,3— 1 : 3 [516]. Ацетобромид СН3СОВг дает
продукты замещения и присоединения в зависимости от соотношения
взятых для реакции компонентов [459]. Продуктами реакции явля-
ются соединения TiBr (OC4H9-mpem)3, TiBn 3 (ООСН3)07 •
• (OC4H9-mpem)2, Ti2OBr1>6 (ООСН3)3,2 (OC4H9-mpem)12 ’0,6С4Н9ОССН3-
трет, Ti2OBr1>5 (ООСН3)4>5.
Ti (ОС5НИ)4. /7гетржяс-(н-Амилокси)титан получен действием
на раствор четыреххлористого титана в бензоле [321 ] или в смеси бен-
зола с пиридином [320] изоамилового спирта и аммиака, а также по
реакции переэтерификации [323]. Это — вязкая бледно-желтая жид-
кость, точка кипения которой 314° С (760 мм рт. ст.), удельный
вес 0,9735 (25° С) [323].
тетракис-(изо-Амилокси)титан синтезирован методом переэте-
рификации [3231 и действием изоамилового спирта на аммиакат
TiCl4 • 6NH3. Он представляет собой жидкость с точкой кипения
140° С (2 мм рт. ст.) и показателем преломления 1,4499 (35° С) [611 ].
Получены титановые эфиры и остальных амиловых спиртов:
Ti [(С2Н5) (СН3) СНСН2О]4 [297], Ti [(СН3)3ССН2О14 [26,170,246,
297], Ti [(С2Н5)2СНО]4 [287, 297], Ti [(СН3) (С3Н7-н) СНО]4 [287,
297], Ti[(CH3)(C3H7-uso)CHO]4 [287, 297], Ti[(CH3)2(C2H5)CO]4 [285,
286, 297, 584], Ti [(СН3) CH (СН3) СН2СН2О]4 [380]. Все они явля-
ются легкоплавкими твердыми веществами или вязкими жидкостя-
ми. тотрякяс-(2-Метилбутокси)титан оказался оптически активным
(+) [380].
Ti (С6Н13О)4. тотршшс-(н-Гексокси)титан получен по реакции
переэтерификации [2, 323] и кипячением аммиаката TiCl4 • 6NH3
с н-гексиловым спиртом. Это — жидкость с удельным весом
0,9573 (20° С), показатель преломления 1,4800 (35° С), кипит при
з*
35
температуре 218° С (5 мм рт, ст.) [611]. При нагревании до 280° С
за 48 ч превращается в воскообразное вещество [475].
Кроме т?тршшс-(н-гексокси)титана получены эфиры с изоме-
рами других гексиловых спиртов: Ti [(СН3) (С4Н9-н) СНО]4, [287],
Ti [(СН3) (С4Н9-трМСНО]4 [287], Ti [(СН3) (С2Н5)2СО]4 [286, 584],
Ti [(СН3)2 (С3Н7-к) СО]4 [286, 584] и Ti [(СН3)2(С3Н7-цзо)СО]4 [286].
Ti (С7Н15О)4. тетршшс-(«-Гептокси)титан впервые получили
В. А. Арбузов, Т. Г. Шавша [3] и Бредли с соавторами [298]. Синте-
зировано также несколько титановых эфиров с изомерами гептило-
вого спирта, а именно: Ti [(СН3Н7-н)2СНО]4, Ti [(C3H7-uso)2CHO]4,
Ti [(С2Н5)3СО]4, Ti [(СН3) (С2Н5) (С3Н7-цзо) СО]4 [286] и
Ti [(СН3)2 (C4H9-mpem) СО]4 [465].
Ti (С8Н17О)4. В. А. Арбузов и Т. Г. Шавша [2, 3], нагревая тет-
ракис-(эткт)ттт с н-октиловым спиртом получили тетракис-
(н-октаокси)титан. Этот эфир получил также Спич [550]. Он явля-
ется твердым веществом, плавится и кипит под вакуумом при темпе-
ратуре 255—258° С (2 мм рт. ст.).
Бредли с соавторами [287] синтезировал титановый эфир с изо-
октиловым спиртом Ti [(СН3) (C6H3-w) СНО]4. Юшино с соавторами
[611], действуя на аммиакат TiCl4 • 6NH3 метил гептанолом, получил
эфир Ti [СН3 (СН2)5С (СН3) НО]4. Это — жидкость с показателем
преломления 1,4601 и точкой кипения 216° С (11 мм рт. ст.).
Эти же авторы получили /пе/лрж^с-(2-эти л гексокси) титан
Ti [СН3СН(С2Н5) (СН2)3СН2О]4 в виде жидкости с показателем прелом-
ления 1,4750 (35° С) и точкой кипения 248—249° С (11 мм рт. ст.)<
Ti (С9Н19О)4. По методу переэтерификации синтезирован тет-
ршшс-(н-нонаокси)титан —твердое вещество с точкой кипения 264—
265° С (1,5 мм рт. ст.) [2, 3]. Получен эфир Ti [(С4Н9-н)2СНО]4 [287].
Ti (СН2=СНСН2О)4. Тетрааллоксититан можно получить дей-
ствием на раствор четыреххлористого титана в смеси бензола и пи-
ридина аллиловым спиртом и аммиакатом [107], а также действием
аллилового спирта на аммиакат TiCl4 • 6NH3 [611]. Это —жид-
кость, показатель преломления которой 1,4601 (35° С), точка кипе-
ния 174° С (11 мм рт. ст.), удельный вес 1,1138 (25° С), вязкость
0,6225 спз [107].
Ti(CH3CHOHCH2O)4. т£тршшс-(2-Гидроксипропокси)титан [123]—
жидкость с точкой кипения 182,5—183° С (3 мм рт. ст.), пока-
затель преломления 1,5077 (20° С), удельный вес 1,1910 (18° С).
Ti (С2Н5СНОНСН2О)4. тетрж^г-(3-Гидроксибутокси)титан [123] —
жидкость с точкой кипения 187—188° С (3 мм рт. ст.), по- '
казатель преломления 1,4920 (21° С), удельный вес 1,1106 (21° С).
Ti (С5Н9О-цд^о)4. Данный эфир получил Дамм [357]. При тем-
пературе 230° С эфир подвергается пиролизу.
Ti (C6HnO ^u^o)4. тетрякцс-(Циклогексанокси)титан синте-
зирован действием на раствор четыреххлористого титана в смеси
бензола с пиридином циклогексанола и аммиака [107]. Это — жид-
кость с точкой кипения 190,5—192° С (1 мм рт. ст.).
36
Арилтитанаты
v /
Арилтитанаты могут быть получены теми же методами, которые
применяются для синтеза алкилтитанатов. Удобным является ме-
тод, основанный на вытеснении из алкилтитанатов алифатических
спиртов фенолами и нафтолами [72, 264, 401].
Ti (С6Н5О)4. тетршшс-(Фенокси)титан [72, 246, 401, 437] —
твердое вещество, точка плавления 153—154° С, точка кипения
267° С (3 мм рт. ст.).
Синтезированы титановые эфиры производных бензола:
Ti (ОС6Н4СН3-л/)4, точка кипения 323—329° С (3 мм рт. ст.) [72,
Таблица 2
Энтальпия образования и энергия разрыва связи титанэфиров
Формула -ДНобр. ккал/молъ A//®) ккал/моль Энергия раз- рыва связи, ккал/моль 1
Т1(С2НбО)4 (тример) 325 349 50,5
Ti(C3H?O-H)4 354 372 55,0
Ti(C3H7O-H3o)4 360 377 53,0
Ti(C4H9O-w)4 377 399 56,0
Ti(C4H90-i/3o)4 381 403 54,0
Ti(C4H90-e/nop)4 382 404 53,0
T i (C4H9O-mpe/7i)4 395 411 51,0
Ti(CsHnO-H)4 403 428 58,0
Ti(C5HnO-mpm)4 438 457 57,0
264, 441], Ti [ОС6Н3(СН3)2-о]4, точка кипения 460° С ($мм рт. ст.),
Ti (3,4-СН3С1С6Н3О)4, точка кипения 284° С (1,5 мм рт. ст.) [72,
264], Ti (о-С1С6Н40)4 [372], Ti (n-NO2C6H4O)4, точка плавления 97° С
[264, 496], Ti (2,4-О2С6Н3О)4, точка плавления 110,6° С [496],
Ti [(NO2)3C6H2O]4, точка плавления 152° С [496], Ti (ОСН2С6Н5)4
[123], Ti (o-N02C6H40)4, Ti (/г-С1С6Н4О)4, а также производные то-
луола — Ti (о-СН3С6Н40)4 и Ti (/г-СН3С6Н4О)4 [264].
Ti (С10Н7О)4. Из производных нафтола известны твердые вещест-
ва Ti (а-С10Н7О)4 и Ti (Р-С10Н7О)4 [72, 401]. Прасад и Трипати [401]
синтезировали эфир динитронафтола Ti [(NO2)2C10H5O]4 с точкой
плавления 122° С.
Ti [(C6H5)2SnO]4. Получен добавлением к бензольному раствору
т£тршшс-(бутилокси)титана раствора гидроокиси трифенилолова
[341]. После удаления растворителя из фильтрата выпадает светло-
желтый осадок тетра/шс-(трифенилстаннокси)титана.
Бредли и Хильер [293, 296] определили энтальпии образования
и энергии разрыва связи некоторых алкилтитанатов (табл. 2).
Делузарх [365] измерил молекулярную рефракцию группы TiO4
в алкилтитанатах и нашел ее равной 20,63 мл/молъ. Такатани [563]
вычислил инкремент молекулярной рефракции связи Ti—О в ал-
килтитанатах (4,08 мл/молъ).
37
Титанэфиры со смешанными оксигруппами
Существует несколько методов синтеза титанэфиров со смешан-
ными оксигруппами. А. Н. Несмеянов и О. В. Ногина [126] полу-
чили алкоксититаны типа (RO)3TiOR' и (RO)2Ti (OR')2 действием
спиртов на хлористые алкоксититаны, а также действием смесей
спиртов на четыреххлористый титан в присутствии пири-
дина и аммиака. По первому методу синтезированы эфиры
(u30-C5HnO)3TiOR, где R = С2Н5, СН2-СНСН2, н-С3Н7, изо-С3Н7
и др. С ~ помощью второго метода получены смешанные
эфиры (u30-C5HnO)3Ti (ОСН2СН=СН3), (w-C3H7O)3Ti (ОС5НП),
(ш?о-С5НпО)2П (СН2=СНСН2О)2 и пр.
Уиттер и Кофлан [600] изучили структуру кристаллов
Ti (ОСН3) (ОС2Н5)3 и нашли, что они имеют триклинную решетку
с параметрами а = 12,13, b = 12,14, с — 16,93 А, а = 76° 56', р =
= 77°2', у = 74° 15', z = 8, рренТг = 1,244г/с3, фед. гр. Р1.
Получены смешанные титанэфиры постепенным замещением хло-
ра в TiCl4 на алкоксигруппы в присутствии аммиака [319, 350]. Этим
методом синтезированы метилбутиловые и метилэтилбутиловые ти-
танэфиры.
Ишино, Минами [416] и Ямамото, Камбара [613] использовали
для получения смешанных титанэфиров реакцию переэтерификации.
В реакции переэтерификации с эфирами типа Ti (OR)4, где R =
= С2Н5, н-С3Н7, н-С4Н9, вступают ацетилацетон и ацетоуксусные
эфиры в энольной форме:
Ti (OR)4+ 2СН3С (OH)-CHCOOR'-Ti (OR)3 [ОС (CH3)-CHCOOR'],
Ti (OR)4 + 2CH3C (OH)-CHCOOR-Ti (OR)2 [ОС (CH3)-CHCOOR']2.
По реакции между алкилортотитанатами и (ацето)ацетатами
и их производными синтезированы смешанные алкокси-бис-(аце-
токси)титанаты [531] общего состава
Ti2 (C2H5O)6L, Ti2 (С3Н70-изо)6 L, Ti2 (OC2H5)4 [(CH3CO) -
=CHCOCH3]2L, Ti (C2H5O)4 (OC-CHCOCH3)2 l,
C2H5
Ti2 (C2H5O)4 (O - C-CCOCH3)2 L,
C2H5 C2H6
I
39
40
Ti2 (С2Н5О)2 [(ОССН3)=СНСОСН3]4
(СН2)
/СОСН2СО'
7\сосн2со.
где
L = - (СН2)
СОСН2СО
СОСН2СО,
zcoch2co
- (СН2)</
\сосн2со,
- (СН2)
/СОСН.СО
7.
ХОСН2СО.
В табл. 3 приведены смешанные титанэфиры, описанные в пе-
чати. В работах [416, 438, 465, 564, 613, 616, 620] изложены данные
исследования инфракрасных спектров титановых эфиров. Имеются
данные и о дипольных моментах [311, 313].
Галогенэфиры
К титановым галогенэфирам относятся соединения, состав кото-
рых выражается общей формулой Ti (OR)nF4_n, где R — алкил,
алкенил или арил, Г — галоген. Титановые галогенэфиры можно
получить по нескольким методам.
Действие спиртов и фенолов на тетрагалогениды титана. Впервые
титановые галогенэфиры действием этилового спирта на четырех-
хлористый титан пытались получить Демарсе [353] и Верлинт, Ги-
рауд [596]. Взаимодействие четыреххлористого титана со спиртами,
согласно [417], протекает вначале по реакции
TiCl4 + 3ROH = TiCl2 (RO)2 • ROH + 2HC1,
т. e. с образованием продуктов присоединения спирта. При длитель-
ном нагревании до температуры кипения молекула спирта отщеп-
ляется и образуются диалкилдихлортитаны TiCl2 (OR)2. Более двух
атомов галогенов в тетрагалогенидах титана на алкоксигруппу не
замещается.
Этот метод (нагревание спиртов с избытком TiCl4 в растворе че-
тыреххлористого углерода и петролейного эфира) оказался удобным
для синтеза моноалкокситрихлортитанов TiCl3 (OR) [125]. В усло-
виях опыта на RO-группу замещается только один атом хлора.
Г. А. Разуваев и соавторы [207, 517] получили галоидэфиры типа
ROTiCl3 (R=w3o-C3H7,emop-C4H9, mpem-C4H9, C6H4-^uoo) непосред-
ственным нагреванием TiCl4 с соответствующими спиртами. Ана-
логичным путем синтезированы алкилтитанхлориды с аллиловым
и mpem-бутиловым спиртами [239].
Фенолы при действии на TiCl4 могут замещать от одного до че-
тырех атомов хлора. Количество замещающихся атомов хлора за-
висит от строения фенола. А. Н. Несмеянов с соавторами [113] пред-
ложил при синтезе алкилтитанхлоридов из тетря/шс-(алкокси)тита-
нов и четыреххлористого титана добавлять пиперидин.
41
Действие галогенопроизводных на тетраорганотитановые эфиры.
Тетраорганотитановые эфиры при действии на них галогенпроизвод-
ных вступают в реакции обменного разложения с образованием га-
логенэфиров:
Ti(OR)4 + RT -> Ti (OR)nr4_n + R'OR.
Реакции обменного разложения протекают с гало ген ан гидр идами
[322, 581, 414], галогенводородами [460, 462], с тетрахлоридом ти-
тана [129, 285] и др.
Действие на тетраорганоэфиры титана свободных галогенов.
А. Н. Несмеянов и соавторы [123] показали, что при действии хлора
и брома на тетраалкоксититаны образуются двойные соединения
галогенэфиров со спиртами по схеме
Ti (RCH2O)4 + Г2 => TiT2 (RCH2O)2 + 2RCH2O°,
2RCH2O° — RCH2OH + RCHO,
(RCH2O)2 TiT2 + RCH2OH => (RCH2O)2 TiT2RCH2OH,
RCHO0 — 4- RCOOCH2R
(RCH2O)4 Ti + r2 = (RCH2O)2 TiT2 • RCH2OH + ~ RCOOCH2R.
Действие на галсгенэфиры спиртов. О. В. Ногина с соавтора-
ми [108] показала, что галогенэфиры могут вступать в реакции пе-
реэтерификации. Этим методом из (C2H5O)3TiCl с соответствующим^
спиртами получены соединения (г/зо-С5НпО)зТ5С1, (ClCH2CH2O)3TiCI
И др.
Действие на теш/шл:&с-(алкскси)титаны солянокислого пири-
дина. А. Н. Несмеянов и соавторы [111] получили с хорошим выхо-
дом трг/с-(алкокси)титанхлориды по реакции между тетракис-
(этилокси)-, тетракис-(пропокси)-, тешр«тас-(бутокси)титаном и со-
лянокислой солью пиридина:
Ti (OR)4 + C5H5N • НС1 = Ti (OR)3C1 + C5H5N + ROH.
Действие тетрахлорида титана на эфиры. Алкоксититанхлориды
получены взаимодействием тетрахлорида титана с диэтиловым эфи-
ром [262], диизопропиловым эфиром [403] и диалкоксититаноксида-
ми [116]. Описанными методами получено значительное количество
титановых галогенэфиров, и число вновь синтезированных соедине-
ний продолжает расти.
Четырехфтористый титан реагирует со спиртами, образуя стек-
ловидные продукты неопределенного состава [610]. Алкоксититан-
фториды этим методом получить не удается.
TiF3 (С2Н5О). Синтезирован кипячением тетрафторида титана
с т?тршшс-(этокси)титаном [452]. Получается в смеси с продуктом
состава TiF4 • 2Ti (С2Н5О)4.
42
TiF2 (C2H5O)2. Синтезирован также по реакции обмена между
TiF4 и Ti (С2Н5О)4 [452].
TiF (С2Н5О)3. Получен Брукером с соавторами [58] но реакции
Ti (С2Н5О)4 + CH3COF = Ti F(C2H5O)3 + СН3СООС2Н5.
Латхам [452] трш?-(этокси)титанфторид синтезировал кипяче-
нием тетрафторида титана с тетршшс-(этокси)титаном в растворе
бензола в течение нескольких часов. Это — кристаллы с точ-
кой плавления 75—78° С и точкой кипения 162—163° С (2 мм
рт. ст.).
Триэтоксифтортитан получен действием на тетраэтоксититан
трехфтористой сурьмы. Реакция протекает с образованием двойного
соединения Sb (ОС2Н5)3 • 3Ti (OC2H5)3F (т. пл. 82° С), которое при
перегонке в вакууме разлагается с образованием Ti (OC2H5)3F.
TiF (язо-С3Н7О)3. Получен по реакции между Ti (цзо-С3Н7О)4
и SbF3 [58]. Первоначально в результате взаимодействия этих сое-
динений образуется двойная соль 5Ь(ОС3Н7-^зо)3-ЗП(цзо-С3Н7О)3Р,
которая при перегонке под вакуумом разлагается на сос-
тавные компоненты. Образующиеся при этом кристаллы
(U30-C3H7O)3TiF имеют точку плавления 83—85° С и точку ки-
пения 140—150° С (6 мм рт. ст.).
TiF(H-C4H9O)3. Получен по реакции взаимодействия
Ti (н-С4Н9О)4 с SbF3 или с CH3COF [58]. Представляет собой крис-
таллическое вещество, точка плавления 45—48° С, точка кипения
175—180° С (2 мм рт. ст.).
TiCI3 (СН3О). Метилокситрихлортитан получил Бредли с со-
авторами [285] по реакции обменного разложения TiCl4 с Ti (ОСН3)4.
TiCl3 (С2Н5О). Получен по реакции обменного разложения между
TiCl4 и Ti (С2Н5О)4 [125, 129, 285], которая проводится в растворе
инертных растворителей (бензол или петролейный эфир), а также
по реакциям обменного разложения TiCl4 с SiCl3 (С2Н5О),
SiCl2 (С2Н5О)2, SiCl (С2Н5О)3 и SiCl4 с Ti (С2Н5О)4 [290]. Твердое
вещество с точкой плавления 81—82° С и точкой кипения 185—
186° С (760 мм рт. ст.) [125]. Дипольный момент в бензоле равен
2,798 рв [313].
Этоксититантрихлорид с тетраэтилхлорамином в ацетонитриле
образует аддукт [(C2H5)4N] [TiCl4 (С2Н5О)] • CH3CN [382], а из смеси
TiCl3 (С2Н5О) и TiCl2 (С2Н5О)2 в диоксане и ацетонитриле — ад-
дукты TiCl3 (С2Н5О) . 2CH3CN, TiCl3 (С2Н5О) • 1,5С4Н8О2.
Бредли с соавторами [285] при взаимодействии Ti (С2Н5О)4 с
СН3СОС1 получил продукт присоединения к этокситрихлортитану
, TiCl3 (С2Н5О) • СН3СООС2Н5 с температурой кипения 90° С
(0,5 мм рт. ст.).
Известны продукты присоединения к этокситрихлортитану пи-
ридина, имеющие состав TiCl3 (ОС2Н5) • C5H5N, TiCl3 (ОС2Н5) •
•2C5H5N, TiCl3 (ОС2Н5) . C5H5N . НС1, TiCl3 (OC2H5) . 2C5H5N .
•HC1, TiCl3(OC2H5) • 3C5H5N • 2HC1 [89].
43
TiCI3 (C3H7O). A. H. Несмеянов с соавторами [125] получил к-про-
покситрихлортитан — вещество с точкой плавления 65—67° С и точ-
кой кипения 100—102° С (23 мм рт. ст.). С пиперидином этот гало-
генэфир образует двойное соединение (н-С3Н7О) TiCl3 • C5H10NH
[128].
Изопропокситрихлортитан синтезирован несколькими авторами
[125, 285, 342, 371]. Он представляет собой твердое вещество с точ-
кой плавления 78—79° С и точкой кипения 65° С (1 мм рт. ст.) [125].
С уксусноэтиловым эфиром образует продукт присоединения
(изо-С3Н7О) TiCl3 • СН3СООС2Н5 [285].
Амиды C6H5CONH2, C6H5NHCOCH3, NH2COCH2, CH3CONH2
и HCONH2 с изопропоксититантрихлоридом образуют аддукты типа
TiCl3 (ОС3Н7-изб>) • L, где L — амид [618].
Гутман и Меллер [390] получили трихлортитанэфир фторзаме-
щенного пропилового спирта (CHF2CF2CH2O) TiCl3. Это соединение
реагирует с аммиаком, образуя смешанный аминохлорэфир
(CHF2CF2CH2O) Ti (NH2)2C1.
TiCl3 (C4H9O). Нормальный бутокситрихлортитан [129, 285,
325, 371, 483, 593] — вещество с точкой плавления 45—55° С и точ-
кой кипения 65—80° С (0,5 мм рт. ст.). В растворе бензола суще-
ствует в виде мономера [483, 593]. Присоединяет пиридин, пипери-
дин и аммиак, образуя двойные соединения: (н-С4Н9О) TiCl3-
• C6H10NH [128], (н-С4Н9О) TiCl3 • 3C5H5N, (н-С4Н9О) TiCl3-
•NH3, (w-C4H9O) TiCl3 • 3NH3 и (w-C4H9O) TiCl3 • 5NH3 [351].
Г. А. Разуваев с соавторами [207, 517], нагревая четыреххло-
ристый титан со спиртами, получил в/тгор-бутокситрихлортитан (втор-
С4Н9О) TiCl3 и mpem-бутокситрихлортитан (mpem-C4H9O)TiCl3-
Из бутанола выделены сольваты состава TiCl3 (С4Н9О) •
• 6С4Н9ОН, TiCl3 • (С4Н9О) • 2С4Н9ОН и [TiCl3 (С4Н9О)
•С4Н9ОН]2 [241].
TiCl3 (С5НИО). Нормальный амилокситрихлортитан в чистом
виде еще не получен. Известно[414] двойное соединение нормального
амилокситрихлортитана со спиртом H-CgHnOTiClg • С5НПОН с тем-
пературой кипения 105—110° С (0,5 мм рт. ст.). А. Н. Несмеянов
с соавторами [125] синтезировал изоамилокситрихлортитан — твер-
дое вещество с температурой плавления 50—60° С и точкой кипения
НО—111° С (17 мм рт. ст.). По реакции этерификации четырех-
хлористого титана с третичным амиловым спиртом получен трет-
амилокситрихлортитан TiCl3 (трет-С5НиО) [285].
TiCl3 (СбН13О). Получен нормальный гексокситрихлортитан
[125] — твердое вещество, точка плавления 47,5—49° С, точка ки-
пения 120—122° С (8 мм рт. ст.).
TiCl3 (СН2=СНСН2О). В чистом виде не получен. При действии
пропиленового спирта на четыреххлористый титан в растворе пи-
перидина образуется двойное соединение TiCl3 (СН2=СНСН2О) •
•C5H10NH [128].
TiCl3 (СН3ОСН2СН2О). Ацетилацетокситрихлортитан — твердое ве-
44
V.
щество с точкой плавления 164—166° С, возгоняющееся при тем-
пературе 160—180° С под давлением 5 мм рт. ст. [125].
Т1С13ЮС (СН3)=СН2СОСН3]. Получил Пури с соавторами [510].
Ранее был синтезирован [526] сольват TiCl3 [ОС (СН3) =
-СН2СОСН3] • (С2Н5)2О.
TiCl3 (цияло-СвНцО). Циклогексокситрихлортитан получен на-
греванием четыреххлористого титана с цикло гексанолом [207].
Это — твердое вещество с точкой плавления 75—76° С.
TiCl3 (С6Н5О). Получен действием на TiCl4 фенола, который за-
мещает только один атом хлора [103, 453]. С диэтиловым эфиром
образует комплекс с отношением компонентов 1:1, отличающийся
наличием в спектре поглощения полосы с максимумом при 25 000 смГ\
вызванной переносом заряда. Спектр поглощения комплекса в н-
гексане имеет три полосы поглощения с максимумами при 25 000
переход п а*), 31 000 (переход л л*) и 37 000 см~~х (переход
1Л1-> гТ2) [362]. Дипольный момент TiCl3 (С6Н5О) равен 2,79 D [313].
Г. П. Лучинский [101] аналогичным методом синтезировал jw-нит-
рофенокситрихлортитан TiCl3 (jw-NO2C6H4O), а Функ с соавторами
[372] - получил двойные соединения TiCl3 (л«-СН3С6Н4О) •
. (НОС6Н4СН3-лОо 5 и TiCl3 [сшюи-С2Н5 (СН3) С6Н3О] НОС6Н3 -
. (СН3)С2Н5.
TiCI2 (СН3О)2. В чистом виде не выделен. Дженингс [417] синте-
зировал двойное соединение TiCl2 (СН3О)2 • СН3ОН.
TiCl2 (С2Н5О)2. бнс-(Этокси)дихлортитан [129, 417, 460, 596] —
твердое вещество с точкой плавления 40—50° С [129]. По данным
[605], кристаллы моноклинной системы с параметрами решетки
а = 9,821, b = 14,006, с = 9,836 А, ₽ - 94°54', ризм - 1,49 г/см3,
z ~ 4, фед. гр. PZjn. В кристалле молекулы находятся в форме ди-
меров, расположенных в,оцентре симметрии. Связи Ti—О короче,
чем в Ti3O5 (1,83—2,7 А). Связь Ti—Cl длиннее, чем у TiCl4
(2,185 А). Хаазе и Хоппе [404] приводят иные параметры решетки:
а - 5,91, b = 10,19, с = 13,90 А, ₽ = 94,7°, ризм = 1,69, z = 4,
фед. гр. Р^/с. Кипит с разложением при температуре 142° С
(18 мм рт. ст.) [417]. В ацетонитриле реагирует с тетраэтил хлор-
амином, образуя аддукт I(C2H5)4N] [TiCl4 (С2Н5О)] • CH3CN. Из сме-
си TiCl2 (С2Н5О)2 и TiCl3 (С2Н5О) в диоксане и ацетонитриле получе-
ны аддукты TiCl2 (С2Н5О)2- CH3CN и TiCl2 (С2Н5О)2 • С4Н8О2. При-
соединяет спирты, пиперидин и пиридин, образуя двойные соедине-
ния TiCl2 (С2Н5О)2 • СН3ОН [417]. TiCl2 (С2Н5О)2 • С2Н5ОН [123,
417], TiCl2 (С2Н5О)2 • C5H1oNH [128], TiCl2 (ОС2Н5)2 • 2C5H5N [89].
TiCl2 (С3Н7О)2. По реакции обменного разложения тетракис-
(н-пропокси)титана с хлорангидридом пропионовой кислоты полу-
чен н-дипропоксидихлортитан, вещество с температурой кипения
159° С (18 мм рт. ст.) [322]. При действии хлора на тетракис-(н-
пропокси)титан в растворе четыреххлористого углерода образуется
двойное соединение (С3Н7О)2 TiCl2 • С3Н7ОН [125]. Известно также
двойное соединение с пиперидином TiCl2 (С3Н7О)2 • C6H10NH [128].
45
При нагревании четыреххлористого титана с изопропиловым
спиртом получен [377, 417] диизопропоксидихлортитан, жидкость
с температурой кипения 160° С (18 лш рт. ст.) [417]. Этот титановый
хлорэфир можно получить нагреванием тетраэфира с газообразным
хлористым водородом [460]. Дженингс с соавторами [417] синтези-
ровал двойное соединение (r/3o-C3H7O)2TiCl2 • (г/зо-С3Н7ОН).
Амиды C6H5NHCOCH3, NH2CONH2, CH3CONH2, C5H5CONH2
и HCONH2 с бг/с-(изопропокси)титандихлоридом дают аддукты со-
става TiCl2 (C3H7O-t/so)2 • L, где L — амид [618].
Гутманн и Меллер [390] получили титановый хлорэфир фторза-
метенного пропилового спирта (CHF2CF2CH2O)2TiCl2, который с
аммиаком образует хлораминоэфир (CHF2CF2CH2O)2TiNH2Cl —
желтое аморфное вещество, конденсирующееся при нагревании.
• TiCl2 (С4Н9О)2. Камбара и Окамото [436] синтезировали бис-(н-бу-
токси)дихлортитан. Соединение это получено также и другими ав-
торами [285, 322, 467]. Это — вязкая жидкость с точкой кипения
182° С (17 мм рт. ст.) [322]. Известны двойные соединения
(н-С4Н9О)2Т1С12 • С4Н9ОН [123] и (H-C4H9O)2TiCl2 • C5H10NH [128].
н-Дибутоксидихлортитан в бензольном растворе существует в виде
димера [483, 493]. тетршшс-(зтор-Бутокси)титан и треххлори-
стый бор дает бцс-(втор-бутокси)титандихлорид [325]. Дженингс
с соавторами [417] синтезировал бис-(изо-бутокси)дихлортитан<—
жидкость с температурой кипения 184° С (18 мм рт. ст.) и двойные
соединения состава (i/so-C4H9O)2TiCl2 • СН3ОН, (дао-С4Н9О)2Т1С12 •
• С2Н5ОН и (изо-С4Н90)2Т[С12 • C4H9O-uac>.
TiCI2 (С5НПО)2. Синтезирован пока только н-диамилоксиди-
хлоотитан [414].
TiCl2 (С6Н5О)2. Получены следующие производные титано-
вого хлорфенольного эфира: (о-СН3С6Н4О)2ПС12 • НОС6Н4СН3-о,
(н-СН3С6Н4О)2ПС12 • 2НОС6Н4СН3-я, (o-ClC6H40)2TiCl2,(jw-ClC6H40)2 -
•TiCl2 • НОС6Н4С1-Л1 [372], (n-ClC6H4O)2TiCl2, (o-NO2C6H4O)2 •
• TiCl2, (n-NO2C6H4O)2TiCl2 1101], [(NO2)2CeH2O]2TiCl2 [354].
TiCI2 (C10H7O)2. Описан (₽ -C10H7O)2TiCl2 [373].
TiCl2 [ОС (CH3) CHOCH3I2. Диацетоацетилдихлортитаи обра-
зуется в результате взаимодействия ацетилацетона с четыреххло’
ристым титаном [356, 510, 512, 612], а также по реакции [501]
Ti (ОС2Н5)2[ОС (СН3)=СНОСН5]2 + 2НС1 TiС12 ОС(СН3) =
= СНОСН3]2 + 2С2Н5ОН.
Юнг [612] этому соединению приписал комплексное строение
{Ti [ОС (СН3=СНОСН3]3} [TiClJ. Присоединяет спирты, об-
разуя двойные соединения TiCl2 [ОС (СН3)=СНОСН3]2 • С2Н5ОН
и TiCl2 [ОС (СН3)=СНОСН3]2 • изо-С3Н7ОН. При взаимодействии
с FeCl3 дает соединение Ti [ОС (CH3)=CHOCH3]3FeCl4 в виде оран-
жевых или красно-коричневых кристаллов с точкой плавления
165° С [513].
46
____
1
TiCl2 ЮС (OCH3) CHCOCH312. Получено взаимодействием че-
тыреххлористого титана с метил ацетоацетатом [68, 512, 290].
TiCl2 [ОС (ОС2Н5)=СНСОСН3]2. Синтезирован нагреванием
СН3СОСН2СООС2Н5 • TiCl4 [681 и взаимодействием четыреххлори-
стого титана с этил ацетоацетатом [512]. Кристаллы с температурой
плавления 122—123° С.
TiCl2 (ОС6Н4СООСН3)2. Получен взаимодействием четыреххло-
ристого титана с метилсалицилатом [512, 527].
Смешанные дихлорэфиры. Пури с соавторами [510] получил сме-
шанный дихлорэфир TiCl2 [ОС (СН3)=СНСОСН3](ОС2Н5) •
•С2Н5ОН.
TiCl (С2Н5О)3. Титановый хлорэфир описан в работах [107, 129,
322, 417]. Это — жидкость с точкой кипения 187° С и удельным
весом 1,1348 (25° С) [322]. Дипольный момент в растворе гексана
при температуре 25° С равен 2,51 [311]. Получены двойные соеди-
нения (C2H5O)3TiCl • С2Н5ОН [417] и (C2H5O)3TiCl • C5H10NH [128].
Бюргер [271] синтезировал аммиакат состава (C2H5O)3TiCl • 3NH3.
При нагревании в вакууме наблюдается ступенчатое отщепление
аммиака, в результате чего образуются продукты (C2H5O)3TiCl •
•NH3 и 3(C2H5O)3TiCl • 2NH3. Моноаммиакат с органическими
основаниями в растворе бензола образует кристаллические продук-
ты замещения состава (C2H5O)3TiCl • R, где R — пиридин, анилин,
диметилсульфоксид.
TiCl (С3Н7О)3. Получен [123, 129, 322] н-трипропоксихлор-
титан—жидкость с точкой кипения 168° С (18 мм рт. ст.),
а также двойное соединение с пиперидином (н-С3Н7О)3Т1С1 •
• C5H10NH [128]. Синтезированы аммиакаты и продукты присое-
динения органических оснований (uso-C3H7O)3TiCl • 3NH3,
(u3o-C3H7O)3TiCl • NH3,3 (uso-C3H7O)3TiCl • 2NH3, (изо-С3Н7О)3Т1С1 •
•R, где R — пиридин, анилин, диметил сульфоксид [271]. т/шс-(Изо-
пропокси)хлортитан с амидами образует аддукты состава
Т1С12 (С3Н70-изо)2 • 2L и TiCl2 (С3Н7О-мзо)2 • 3L, где L —
амид [618].
По реакции переэтерификации получен трис-(1-хлорпро-
покси)хлортитан в виде жидкости с точкой кипения 194—196° С
(2 мм рт. ст.) [108]. Синтезирован титановый хлорэфир фторзаме-
щенного пропилового спирта (CHF2CF2CH2O)3TiCl [390]. В растворе
абсолютного хлороформа это вещество реагирует с аммиаком, об-
разуя аминоэфир (CHF2CF2CH2O)3TiNH2 в виде игловидных темных
кристаллов, плавящихся при температуре 110° С с выделе-
нием аммиака. При длительном нагревании титановый ами-
ноэфир затвердевает, конденсируясь в продукт состава
(CHF2CF2CH2O)3Ti NHTi (OCH2CF2CHF2)3.
TiCl (C4H9O)3. т/шс-(н-Бутокси)хлортитан получен многими ав-
торами [128, 130, 131, 322, 371, 394, 436]. Это — светло-желтая жид-
кость с точкой кипения 156,2° С (2,5 мм рт. ст.), показатель пре-
ломления 1,5192 [130, 131]. В растворе бензола существует в виде
47
тримера [483, 593]. Данн [351] получил аммиакаты данного хлор-
эфира, имеющие состав (н-С4Н9О)3ТЮ • 3NH3 и (H-C4H9O)3TiCl •
•NH3.
Синтезирован/прис-(изобутокси)хлортитан, который представля-
ет собой жидкость с точкой кипения 147—148° С (6 мм рт. ст.),
показателем преломления 1,5158 (20° С) и удельным весом 1,1043
(20° С) [108, 322]. Бредли с соавторами [304] получил тре/п-трихлор-
метандиметилметоксихлортитан [(СС13) (CH3)2CO]3TiCl.
TiCl (С5НИО)3. т/шс-(к-Амилокси)хлортитан — жидкость с точ-
кой кипения 214° С (2 мм рт. ст.) [322, 414]. Производное изоами-
лового спирта (tz3o-C5H11O)3TiCl представляет собой жидкость, точка
кипения которой 173,5—175,5° С (2 мм рт. ст.), показатель пре-
ломления 1,5092 (20° С), удельный вес 1,0600 (20° С).
Бредли с соавторами [285] получил /прш?-(трет-амилокси) хлор-
титан (/npem-C5HnO)oTiCl в виде жидкости с температурой кипения
115—120° С (0,3 мм рт. ст.).
TiCl [ОС (СН3) = СНСОСН3]3. В некоторых работах [444, 503,
580, 581] показано, что триацетилацетоноксихлортитан образуется
наряду с дихлор производными при действии ацетилацетона на че-
тыреххлористый титан. Получен аддукт состава {TiCl [ОС (СН3)=
—СНС0СН)]2}20 • СНС13 в виде моноклинных кристаллов с пара-
метрами решетки а = 15,744, b = 22,628, с = 8,888 А, ₽ =
= 100,30°, ризм = 1,47 г/см3, z = 4, фед. гр. Р211п [607].
TiCl (ОС6Н13)3. /тг/шс-(н-Гексаокси)хлортитан — жидкость с тем-
пературой кипения 182—182,5° С (1 мм рт. ст.), показателем пре-
ломления 1,5050 (20° С) и удельным весом 1,0039 (20° С) [108, 130—
132, 322].
TiCl (СбН5О)3. Трифеноксихлортитан получил Гениц [72, 401].
Это — жидкость с температурой кипения 265° С (4 мм рт. ст.).
Эти же авторы синтезировали титановые хлорэфиры с некоторыми
производными фенола, а именно: (л/-СН3С6Н4О)3ПС1 — жидкость
с точкой кипения 272°С (1,5 мм рт. cm.), (n-Cl -jw-CH3C6H3O)3TiCl —
жидкость с точкой кипения 267° С (2 мм рт. ст.) и 1,3-
(CH3)2C6H3O3TiCl кипит под вакуумом 2 мм рт. ст. при температу-
ре 480° С. .
TiCl (С2Н5ОСН2СН2О)3. Получен по реакции обменного разложе-
ния триэтоксихлортитана со спиртом [108]. Жидкость, температура
кипения 182—183° С (1 мм рт. ст.), показатель преломления
1,5178 (20° С), удельный вес 1,2035 (20° С).
Смешанные хлорэфиры. Синтезированы [394] следующие сме-
шанные монохлортитановые эфиры: (С2Н5О) (ft-C4H9O)2TiCl (т. кип.
140—160° С при 1,5 мм рт. ст.), (шо-С3Н7О) (ft-C4H9O)2TiCl (т. кип.
152—154° С при 0,8 мм рт. cm.), (СН2=СНСН2О) (H-C4H9O)2TiCl
(т. кип. 162—180° С при 1,5—2 мм рт. ст.).
TiBr3 (С2Н5О). Получен по реакции обменного разложения тет-
раэтоксититана с четырехбромистым титаном [131].
TiBr3 (С3Н7О). Получены трибромтитановый эфир изопропило-
48
вого спирта и продукт присоединения к нему уксусноизопропилово-
го эфира состава (изо-С3Н7О) TiBr3 • СН3СООС3Н7-изо [581].
TiBr2 (С2Н5О)2. бис-(н-Этокси)дибромтитан — легкоплавкое ве-
щество, точка плавления которого 47—50° С, точка кипения 96—
105° С (5 мм рт. ст.) [130, 131, 462]. Образует двойные соединения
(C2H5O)2TiBr2 • С2Н5ОН [123] и (C2H5O)2TiBr2 . C5H10NH [131].
TiBr2 (С3Н7О)2. Синтезировано также двойное соединение
(н-С3Н7О)2Т1Вг2 • С3Н7ОН-н [123]. По реакции обменного разложе-
ния между тетршшс-(изопропокси)титаном и бромуксусным ангид-
ридом или бромистым водородом получен [581] диизопропоксиди-
бромтитан — жидкость с точкой кипения 100—102° С (2 мм рт. ст.)
[462].
TiBr2 (С4Н9О)2. бис-(н-Бутокси)дибромтитан [123, 581] с бути-
ловым спиртом образует продукт присоединения (н-С4Н9О)2Т1Вг2 •
• С4Н9ОН-н [123].
TiBr (С2Н5О)3. В чистом виде не получен. Синтезировано двойное
соединение (C2H5O)3TiBr • C5H10NH [131].
TiBr2 (ОС6Н5СОН)2. Образуется при действии на TiBr4 оксибен-
зойного альдегида в растворе хлорбензола [545].
TiBr (С3Н7О)3. Получено только производное изоамилового спир-
та TiBr (С3Н7О-цзо)3 [581].
Состояние титангалогенэфиров в растворах
Галогенэфиры титана, как и алкил(арил)титанаты, в жидком
состоянии и в растворах находятся преимущественно в полимери-
зованном виде и реже в форме мономеров. трис-(Бутокси)титанхло-
рид и бцс-(бутокси)титандихлорид, судя по криоскопическим и ка-
лориметрическим изменениям, в жидком состоянии димеризованы,
бутоксититантрихлорид мономерен [593], этоксититантрихлорид в
растворах бензола и диоксана мономерен [382]. бцс-(Этокси)титан-
дихлорид в растворе диоксана находится в виде мономерных моле-
кул, а в бензоле — в виде димеров. Этоксититантрихлорид и бис-
(этокси)титандихлорид в ацетонитриле частично диссоциируют. Рас-
творы их обладают заметной электропроводностью.
Методами рентгеноструктурного анализа показано, что молеку-
лярные соединения TiCl2 (С4Н9О)2 • С4Н9ОН, TiBr2 (С4Н9О)2-
• С4Н9ОН и TiCl2 (С2Н5О)2- С2Н5ОН в растворе бензола димеризо-
ваны [261].
Спектральные и магнитные свойства
Дьякграаф и Руссеау [360] полагают, что структуры титангало-
генэфиров TiCl3 (С3Н7О-цзо), ПС12(С3Н7О-изо)2 и Ti С1(С3Н7О-цзо)3
близки к тетраэдрической. В ультрафиолетовой области спектров
этих соединений обнаруживается по две полосы, относимые к пере-
ходам (2^2)6 (1^)6-> (24)6 (1^)5 (2е) и (2/2)6 (14)6(2/2)6(Пх)5 (3/2).
4 1-1669
49
Магнитная восприимчивость фрагмента TiCl3O в соединениях
с TiCl3OR, где R= С2Н5, С3Н7, изо-С3Н7, н-С4Н9, я-С5Нп, изо-С5Ни,
равна —30,5 • 10~6 [340]. Численно она слагается из магнитных
восприимчивостей связей Ti—О и Ti—О.
Диспропорционирование алкоксититанхлоридов
Перегонка нормальных алкоксититанхлоридов сопровождается
их диспропорционированием. Алкоксититанхлориды, содержа-
щие вторичные и третичные группы, при хранении или нагревании
разлагаются, образуя оксихлорид титана, алкилхлорид и полиме-
ризованные олефины [517].
Двойные титанэфиры
Смешением тетраокси-(алкокси)титанов с алкоголятами щелоч-
ных, щелочноземельных металлов и соответствующими спиртами
получены двойные титанэфиры (алкоголяты). Из этих производных
описаны следующие соединения: К2 [Ti (С2Н5О)6], КН [Ti (СН3О)6],
Са [Ti (С2Н5О)6], MgH2 [Ti (С2Н5О)6]2 [20, 158], LiTi (ОДО-изо)5,
LiHTi (C3H7O-r/so)6, NaTi (C3H7O-i/so)5, LiTi2(C3H7-wso)9, LiTi2(C4H9O)9,
LiTi2 (C4H9O-emop)9 [458].
Эфиры трехвалентного титана
Эфиры трехвалентного титана образуются при восстановлении
тетраэфиров щелочными металлами [132, 238]. Действуя на
(C2H5O)4Ti в спиртовом растворе металлическим калием, А. Н. Нес-
меянов с соавторами [132] получил трш?-(этокси)титан (C2H5O)3Ti.
Аналогичным методом получены трш>(н-пропокси)титан и трис-
(н-бутокси)титан [127]. Шлефер и Гётц [546] синтезировали трис-
(ацетилацето)титан Ti (СН3СО=СНСОСН3)3.
Триалкоксититаны — твердые вещества с розово-сиреневым цве-
том. Они обладают большой реакционной способностью и устойчивы
только в атмосфере сухого инертного газа. Окисляются кислородом
воздуха с образованием бг/с-(алкокси)титаноксидов. В спиртовом
растворе трш?-(алкокси)титаны восстанавливают сулему и броми-
стую ртуть, присоединяют хлор, со спиртами образуют двой-
ные соединения (н-С3Н7О) Ti • 2 (н-С3Н7ОН) и (H-C4H9O)3Ti •
.(я- ОДОН) [127].
Гидролиз титанэфиров
При действии на титановые эфиры воды они подвергаются гид-
ролизу. При гидролизе изоамилового эфира Ti [ОСН2С (СН3)3]3
наблюдается образование изоамилового спирта (СН3)3ССН2ОН [246].
Из этого авторы заключили, что гидролиз титановых эфиров про-
50
исходит с разрывом связи по месту титан — кислород, а не угле-
род— кислород, так как в противном случае в качестве продукта
реакции должен был бы получиться (в результате перегруппировки)
изоамиловый спирт состава (СН3) СНСН2СН2ОН.
Устойчивость титановых эфиров к гидролизу и скорость гидро-
лиза зависят от природы и строения RO-rpynn [274, 586, 588]. Аро-
матические титановые эфиры более устойчивы к гидролизу, чем
алифатические [72, 274].
Скорость гидролиза титановых эфиров увеличивается с повыше-
нием температуры и с увеличением количества взятой для реакции
воды [466]. В случае недостаточного количества воды для полного
гидролиза титановых эфиров происходит межмолекулярная кон-
денсация с образованием полимерных соединений. Состав продуктов,
образующихся в результате гидролиза титановых эфиров, и степень
полимеризации зависят главным образом от количества введенной
в реакцию воды [464].
Механизм гидролиза титановых эфиров был предметом исследо-
вания многих авторов, но до сих пор окончательно еще не выяснен.
Бойд [307] и Винтер [587, 590] показали, что средняя степень по-
лимеризации при гидролизе титановых эфиров зависит от соотно-
шения взятой для реакции воды и титанового эфира. По мнению Бой-
да [307], если отношение Н2О : Ti (OR)4 меньше единицы, то в резуль-
тате гидролиза получаются линейные полимеры по уравнению
nTi (OR)4 + (п - 1) Н2О = (RO)2 (л+1) + 2 (п - 1) ROH. (1,1)
При расходе воды более 1 моля на 1 моль эфира должны полу-
чаться полимеры с поперечными связями:
Ti (OR)4 + мН2О - [TiOm (OR)4-2mlx + 2mROH. (1,2)
Однако эти предположения не нашли подтверждения в работах
других авторов. Согласно уравнению (I, 1), гидролиз тетраэтокси-
титана с расходом воды 0,5 моля должен протекать с образованием
гексаэтилоксидититаноксана (C2H5O)6Ti2O. А. Н. Несмеянов с соав-
торами [1241 показал, что на самом деле образуется октаэтокситри-
титаноксан (С2 Н5) 8Ti 3О2.
Гидролиз титановых эфиров по уравнению (I, 2) протекает толь-
ко при условии, если отношение Н2О : Ti (OR)4 не превышает 1,5
[283, 307, 415, 464, 466, 587, 590]. В противном случае вода не всту-
пает в реакцию полностью, и для исчерпывающего гидролиза тита-
новых эфиров приходится затрачивать 3—3,5 моля воды вместо двух,
согласно уравнению (I, 2).
Бредли и соавторы [299, 300] изучили гидролиз тетраэтил окси-
титана. Молекулярный вес образующихся продуктов они определя-
ли эбуллиоскопическим методом. Оказалось, что простейшим про-
дуктом реакции гидролиза является соединение, молекулярный вес
которого соответствует формуле (C2H5O)16Ti6O4. Так как тетраэти-
ловый эфир не является мономерным продуктом, а ассоциирован,
4*
51
авторы предположили, что в ортоэфире и в продуктах его гидролиза
титан имеет координационное число 6 и состав продуктов гидролиза
выражается формулой [Ti3(x+i)O4x (ОС2Н5)4(х+3)], где х = 0, 1,
31г
2, 3,... В свою очередь, х = от» где —отношение Н2О : Ti (OR)4.
Если данная формула состав продуктов гидролиза титановых эфи-
ров выражает правильно, то можно показать, что состав конечного
продукта полимеризации выражается формулой [Ti3O4 (CO2H5)4L,
Рис. 1. Схема циклического строе-
ния тримеров титановых эфиров ти-
па Ti (OR)4.
а исходный тетраэтоксититан яв-
ляется тримером.
Экспериментальные данные при
Л С 1,1 находятся в согласии с при-
веденной формулой, при /г > 1,1 и
очень малых значениях h опыт не
подтверждает ее. Неподчинение экс-
периментальных данных формуле
Бредли с соавторами [301] объяс-
няет изменением величины h при
малых концентрациях воды за счет
сольволиза продуктов гидролиза
спиртом:
8С2Н5ОН + [(С2Н5О)1в Ti6O4]
<>2[(C2H5O)8Ti3O2-4C2H6OH].
В этом случае возможно обра-
зование двух структур, а именно:
[(RO)2(X-j-4) Ti2(x+i)O3x (ROH)2(x+i)l и [(RO)4_2x Ti2(X-j_i)O3x (ROH)2(x-f-nJ-
Первая из них основана на сольватированном димере Ti2(OR)8 •
• (ROH)2, а вторая — на мономерном сольвате Ti2(OR)4(ROH)2.
Пользуясь этими тремя моделями, Бредли с соавторами [301]
объяснил механизм гидролиза титановых эфиров этилового, пропи-
лового, бутилового и изоамилового спиртов.
Мартин и Уинтер [484], в отличие от Бредли с соавторами [299—
301], предположивших цепочечное строение титановых эфиров, при-
писывают последним циклическое расположение атомов титана
(рис. 1). Окружение атомов титана атомами кислорода при цикли-
ческом строении тригонально-призматическое. Такая симметрия
лучше согласуется с замещением трех групп OR на атомы хлора и
позволяет объяснить гидролитическую полимеризацию тримеров.
По мнению авторов, гидролиз титановых эфиров начинается с кон-
цевых алкоксигрупп и затем происходит межмолекулярное эли-
минирование спирта и внутримолекулярное элиминирование воды
по схеме (м — мостиковые, к — концевые группы):
Ti3 (OR)6 (OR)? + 6Н2О = Ti3 (OR)? (ОН)? + 6ROH;
Ti3 (RO)? (ОН)? + Ti3 (OR)? (OR)? =
= (RO)? (RO)? Ti3O? Ti3 (RO)? (ОН)? + 2ROH;
52
(RO)? (R0$ Ti3O2M Tis (OR)” (0H)4 ->
-> (RO)? (RO)e TiAMTi3 (0R)£ (0)? + 2H2O.
При этом образуются концевые группы Ti=O с пятивалентным
титаном, как и в титанатах калия K2Ti2O5 (см. гл. V).
Минами и Ишино [464] и А. Н. Несмеянов с соавторами [ПО]
также считают, что при полимеризации титановых эфиров в резуль-
тате гидролиза образуются циклические соединения. При изучении
полимеризации mcmpu/шс-(бутокси)титана цепные соединения не
обнаружены [464].
Исследованию гидролиза титановых эфиров посвящены работы
[301, 350, 582].
Продукты неполного гидролиза титанэфиров содержат оксо-
группы. Кокас и Винтер [429] в продуктах гидролиза Ti (СН3О)4
и Ti (С2Н5О)4 методом рентгеноструктурного анализа обнаружили
присутствие оксогрупп Ti3O и Ti4O. В ИК-спектрах поглощения
существованию этих групп отвечают полосы с частотами 780 и
725 см~\ характерные для полимерных связей титан — кислород.
Некоторые алкоксититаноксиды получены в индивидуальном сос-
тоянии.
А. Н. Несмеянов и соавторы [132] окислением трис-(этокси)-
титана получили гексаэтоксидититаноксид (C2H5O)3TiO •
• Ti (С2Н5О)3. Переэтерификацией этого оксида с соответствующи-
ми спиртами синтезированы гексапропокси- и гексабутоксидити-
таноксиды. Гексабутокси- и гексаамилоксидититаноксиды образу-
ются при взаимодействии трис-(изопропокси)титана с масляной и
валериановой кислотами [500].
В индивидуальном виде получен твердый продукт гидролиза
тс/иршшс-(бутокси)титана состава (C2H5O)19Ti7O5 [606]. Кристаллы
его имеют моноклинную решетку с параметрами а = 27,99, b —
~ 22,42, с = 23,21 А, Р = 117,3°, ризм = 1,305 г/см\ z = 8, фед.
гр. PZ-Ja.
Гидролиз тетрикис-(алкил)титана с водой при молярном соот-
ношении 1 : 1 приводит к образованию соединений с оксогруппой
состава (RO)2Ti = O [300, 301].
На основе определения молекулярного веса титаноксоэфирам
приписано полимерное строение.
Окислением трис-(алкокси)титанов сухим кислородом синтези-
рованы бис-(пропокси)- и бис-(бутокси)титаноксиды [127]. бис-(Ал-
кокси)титаноксиды вступают в реакции переэтерификации [121].
По реакции переэтерификации бис-(пропокси)титаноксида и соот-
ветствующего спирта синтезированы бис-(этокси)-, бис-(изоамило-
кси)-, бис-(гексанокси)-, бис-(фенокси)- и бис-(триметилсилено-
кси)титаноксиды.
53
Титаноксиэфирам приписывается циклическое строение [1121:
RO Dri RO RO
। /°\ I \ /
R0—T’ Ti—OR RO. ° T‘ ? z0R
I । и \ I I /
О 'Q Ti Ti
yTi\ Ro/i------Ti---oX°R
RO OR RO^ ^)R
По реакции
TiCl4 + (RO)oTiO -> ROTiCl3 + RO — Ti=O
I
Cl
получены этоксихлортитаноксид (т. плавл. 92—96° С) и пропокси-
хлортитаноксид (т. плавл. 125—129° С) [116]. При нагревании
б&с-(пропокси)титаноксида образуются системы из двух жидких
слоев. Нижний слой состоит из бш>(пропокси)титандихлорида, по-»
л у чающегося по реакции
(С3Н7О)2 TiO + (СН3)2 SiCl2 -> (С3Н7О)2 TiCl2 + [(СН3)2 SiO]„.
Каталитические системы и применение титанэфиров
Системы Ti (C4H9O-emop)4 — Al (С2Н5)3 и Ti (C4H9O-mpem)4 —
Al (C2H5)3 проявляют себя как катализаторы реакции полимериза-
ции [568]. Каталитической активностью обладают три парамагнит-
ные частицы, включающие ион Ti (III).
Многие свойства титанорганических эфиров представляют ин-
терес для техники. В литературе имеются предложения по исполь-
зованию титанорганических эфиров для получения лакокрасочных
покрытий, в том числе теплостойких [18, 16, 99, 197, 282, 315, 342,
343, 344, 370, 400, 441—443, 501, 548, 549, 589—592], для сшивания
полимеров вследствие способности титанэфиров вступать в реакции
переэтерификации [11, 57, 97, 99, 150, 153, 155, 175, 179, 183, 186,
188—190, 192, 347, 395, 460, 575, 547, 552], для гидрофобизации ма-
териалов [12, 14, 154, 156, 157, 176, 184, 185, 193, 227, 275, 307,
308, 551, 553] и для модифицирования полимерных смол [177, 180,
187]. Они также могут найти применение в качестве пластифика-
торов [346, 347, 552], как поверхностно-активные вещества [156, 157,
345, 347, 552, 314], и для других целей.
54
Ацильные соединения
Ацильные производные титана содержат группу RCOO. Они
получаются в результате взаимодействия органических кислот с
титанэфирами или четыреххлористым титаном:
Ti (OR)4 + nR'COOH = Ti (OR)4_n (R'COO)n + nROH,
TiCl4 + nR'COOH = TiCl4+n (R'COO)„ + HC1.
Взаимодействие эфиров с кислотами — процесс обратимый [463].
Кроме карбоновых кислот титан образует ацильные производные
с неорганическими кислотами, содержащими органо группы, напри-
мер С6Н5В (ОН)2 и СбН5Р (-0) ОН [371].
Кипячением карбоновых кислот с эфирами типа Ti (OR)4, где
R = С2Н5, С4Н9, изо-С3Н7, получены ацильные соединения (мыла)
состава (CH3COO)3TiOR, (C2H5COO)3TiOR, (C3H7COO)3TiOR,
(C4H9COO)3TiOR, (CnH23COO)3TiOR, (C13H27COO)3TiOR,
(C15H31COO)2Ti (0R)2 [500]. Четвертую ацильную группу из-за
пространственных затруднений ввести не удается. Реакцию
ацилирования проводят в растворе бензола или толуола при
избытке карбоновых кислот. После отгонки избытка карбоно-
вых кислот под вакуумом в остатке образуются соединения
типа [(CH3COO)3Ti]2O. Описанными методами синтезированы
соединения (r/so-C3H7O)3TiOCOCH3, (uso-C3H7O)2Ti(OCOCH3)2,
C3H7OTi(OCOCH3)3, [^o-C3H7OTi (OCOCH3)2]2O [500].
При действии на алкилортотитаны уксусного ангидрида образу-
ются соединения (RO)Ti(OCOCH3)3 и (RO)2Ti(OCOCH3)2 [457,
502, 505, 506]. Изопропилтитан при действии на него ук-
сусного ангидрида дает соединения [(СН3СОО)2(изо-С3Н7О) Ti2O] и
t(CH3COO)3Ti]2O.
При нагревании смеси четыреххлористого титана и кар-
боновых кислот в бензоле получены соединения Ti (ОСОС17Н35)4,
Ti (ОСОС17Н33)4 и Ti (ОСОС17Н31)4 [418]. Синтезированы титановые
соли нафтеновых кислот [140].
Из TiBr4 и карбоновых кислот получены титановые соли состава
Ti (RCOO)4 [504]. Одноосновные органические кислоты реагируют
в количестве четырех молекул с одной молекулой тетрабромида
титана, а двухосновные — в количестве двух молекул. Авторы [504]
синтезировали следующие титановые соли карбоновых кислот:
Формула соли
Ti[CH3(CH2)10COO]4
Ti[CH3(CH2)i2COO]4
Ti[CH3(CH2)14COO]4
Ti[CH3(CH2)16COO]4
Ti(CeH6CH=CHCOO)4
Ti(CeH5COO)4
Ti[(CH2)4(COO)2]2
Z°
Ti[CeH4— COO]3
Цвет продукта
Г рязно-коричневый
Желтый
Грязно-белый
Светло -кор и чневый
Серый
т. пл.,
°C
57
79
97
77
145
115
152
55
Ti[(CH2)2(COO)2]2
Ti
гсн2---COO
I
CH(OH)COO
2
Ti[2,5(NO2)2C6CH2—COO]2
Ti(n-NO2C6H4COO)4
Ti[C8H14(COO)2]2
Белый
i
Желтый
Коричневый
Бледно-желтый
Темно-коричневый
166
158
126
163
В английском патенте [8] описан метод получения смешанных
ацильных производных действием карбоновых кислот или их ан-
гидридов на дисульфид титана при температуре 40—100° С в при-
сутствии безводных растворителей. Смешанные галогенацилаты
титана образуются при действии на галогениды титана карбоно-
вых кислот и их ангидридов.
Из четыреххлористого титана и уксусной кислоты получен ди-
хлордиацетоксититан Ti (ОСОСН3)2С12 [371, 375]. Этот продукт
в виде белого порошка образуется при взаимодействии TiCl4 и
СН3СООН в газовой фазе [168] иТЮ4с РЬ (СН3СОО)2 при темпера-
туре ниже 25° С [507].
При кипячении карбоновых кислот с раствором четыреххлори-
стого титана в бензоле получены хлорацилы Ti (ОСОСН3)2С12,
Ti (ОСОС2Н5)2С12 и Ti (ОСОС5Н1Х) С13 [418]. Реакция четыреххло-
ристого титана с уксусной кислотой на холоду приводит к образо-
ванию основных (частично гидролизованных) хлорацетатов титана
[392].
Обработка четыреххлористого титана уксусным ангидридом и
смесью уксусного ангидрида и уксусной кислоты на холоду дает
дихлордиацетоксититан [503]. В условиях повышенной темпера-
туры образуется гексацетоксидититаноксан [(CH3COO)3Ti]2O. Син-
тезированы в кристаллическом состоянии TiCl2(CH3COO)2 и
TiCl2(C2H5COO)2 [557]. При действии на TiCl4 муравьиной, н-мас-
ляной, н-валериановой и н-капроновой кислот получены тягучие
резинообразные продукты. При кипячении четыреххлористого
титана с уксусной кислотой в соотношении 1,25 : 1 образуется три-
хлорацетоксититан TiCl3(OCOCH3). Это соединение описано также
в работе [371].
В результате термического разложения эфиров четыреххлори-
стого титана наблюдается образование ацетилхлоридов CCl3COOTiCl3
и C6H5COOTiCl3.
При действии на TiCl4 масляной, изомасляной, валериановой,
ди- и трихлор уксусной кислот синтезированы ацилаты состава
TiCl3RCOO и TiCl2(RCOO)2, где RCOO — остаток соответствующей
кислоты [426]. Соли титана типа TiCl3RCOO с уксусной и пропио-
новой кислотами дают устойчивые продукты присоединения со-
става TiCl3PCOO • NOC1.
56
Действием на Ti (C4H9O-mpem)4 ацетобромида СН3СОВг получе-
ны основные ацетаты Т^ОВп 6(СН3СОО)4 4, Ti2OBr3 6 • (СН3СОО)2 4 •
•2 mpem-C4H9COCH3, Ti2O (СН3СОО)б [459].
В патенте США [151] сообщается, что при обработке четырех-
хлористого титана ацетатом калия образуются основные аце-
таты титана, например Ti (ОН)2 (СН3СОО)2. Пури с соавторами
[510] получил смесь TiO (СН3СОО)2 с [Ti (СН3СОО)3]2О.
Филд [371], действуя уксусной кислотой на титановые эфиры
Ti (С3Н7О-изо)4 и Ti (С4Н9О-н)4, синтезировал смешанные
соединения CH3COOTi (С3Н7О-изо)3, (CH3COO)2Ti (С3Н7О-изо)2,
(CH3COO)Ti (С4Н9О-н)3 и (CH3COO)2Ti (С4Н9О-н)2.
В работе [258] описан дихлор-бш>(пентан-2,4-дионат)титан
TiCl2 [СН3СН (ОН) СН2СН (ОН)СО]2.
Четыреххлористый и четырехбромистый титан реагирует с ки-
пящей трифторуксусной кислотой, образуя 3TiO (CF3COO)2 •
•2CF3COOH [533]. В высоком вакууме это соединение разлагается
на CF3COOH и [TiO (CF3COO)2]n. Аналогичным методом полу-
чены [TiO (C2F5COO)2]m и [TiO (C3H7COO)2]rt.
бис-(Циклопентадиенил)титанхлорид реагирует с натриевыми
солями карбоновых кислот по реакции
(С5Н5)2 TiCl + RCOONa = (С5Н5)2 Ti (RCOO) + NaCl.
Получаемые при этом карбоксилатные производные бш>(цикло-
пентадиенил)титан (III) — парамагнитные продукты зеленого или
голубого цвета. Синтезированы производные (С5Н5)2ТЮОСН (т.
пл. 92° С), (С5Н5)2ТЮОССН3 (т. пл. 110° С), (С5Н5)2 •
• TiOOC (СН2)8СН3, (С5Н5)2TiOOC (СН2)16СН3 (т. пл. 35° С),
(С5Н5)2ТЮОСС6Н5 (т. пл. 83—85° С), (C5H5)2TiOOCCH2 •
•CH2COOTi (С5Н5)2 (т. пл. 245—250° С), (C5H5)2TiOOC — СН -
=CHCOOTi (С5Н5)2 (т. пл. 290—300° С) и аналогичное транс-
производное (т. пл. 260—262° С).
Галогениды титана, за исключением фторидов, реагируют с эфи-
рами карбоновых кислот и с моно- или полными эфирами дикарбо-
новых кислот, образуя полимерные ацилтитанаты [169]. Соедине-
ния RnTiCl4_n, где R = СН3СОО, С6Н5СН2СОО, а-С10Н7СН2СОО,
п = 0, 1, 2, после восстановления водородом титана до трехвалент-
ного проявляют каталитическую активность к реакциям гидри-
рования двойных и тройных связей, карбонильной группы и нит-
рогруппы [5].
При взаимодействии алкилтитанов с кислотами и ангидридами
кислот образуются смешанные алкоксититанацилаты [457, 505, 506]:
Ti (ОС2Н5)4 + С15Н31СООН -> (С15Н31СОО)3 ТЮ+
+ (С15Н31СОО)2 Ti (ОС2Н5)2 + (С15Н31СОО)3 TiOTi (ООСС15Н31)3,
(С15Н31СОО)2 Ti (ОС2Н5)2 + С4Н9ОН + (С15Н31СОО)2 Ti (ОС4Н9)2,
(С15Н31СОО)2 Ti (ОС4Н9)2 + С15Н31СООН -> (С15Н31СОО)2 тю +
+ (С15Н31СОО)2 Ti (ОС4Н8)2 OTi (ООСС15Н81)2.
57
Титановые эфиры Ti (OR)4 и салициловый альдегид дают сме-
шанные соединения (RO)3Ti (ОС6Н4СНО), где R = С2Н5, CjH7 и
С4Н9 [617]. Тетраэтилтитан с метиловым эфиром салициловой кис-
лоты образует смешанные эфиры (C2H5O)3Ti (ОС6Н4СООСН3) и
(С2Н5О)2 • Ti (ОС6Н4СООСН3)2. Дермер и Фернелиус [354] получи-
ли Ti (ООСНСНС6Н5)4.
Показано, что между производными титана типа TiX4, где X —
= С1, г/зо-С3Н7О, н-С4Н9О, и замещенными органическими радика-
лами неорганическими кислотами типа ROH, где R = CH3C6H4SO2,
С8Н5Р (=0) Н, С6Н5В (0Н)2, протекает реакция обменного разло*
жения:
TiX4 + nROH = (RO)„ TiX4_„ + nHX.
Величина п при этом равна 1 или 2. В случае взаимодействия
Ti (ОС4Н9)4 с С6Н5В (0Н)2 образуется борпроизводное
[(С4Н9О)3ТЮ]2ВС6Н5 [371].
Судя по ИК-спектрам поглощения ацилатных производных, связь
между титаном и ацильными группами в алкилортотитанатах носит
ионный характер [215]. Эти производные по своим свойствам близки
к солям.
АМИДЫ ОРТОТИТАНОВОЙ кислоты
Амиды ортотитановой кислоты — соединения, состав которых
выражается общей формулой Ti (NRR')4, где R — водород, алкил
или арил, R' — алкил или арил. Простейшим производным соеди-
нений этого типа является тетрааминотитан Ti(NH2)4, полученный
нагреванием смеси мочевины с четыреххлористым титаном при тем-
пературе 150° С [418].
Синтезирован тетракис-(диметиламидо)титан Ti [N (СН3)2]4, ко-
торый реагирует с тетракарбонилом никеля, образуя желто-корич-
невый продукт присоединения Ti [N (СН3)2]4 • 2Ni (СО)4 [291]. При
взаимодействии его с диметил мал онатом (HL) и ]\Г,М'-тетраме-
тилмалонамидом (HL') получены комплексы [(CH3)2N]2TiL2 и
[(CH3)2N]2TiL'2 [606]. Первое соединение существует в растворе в
форме изомер а. Интенсивное поглощение в спектре комбина-
ционного рассеяния [TiN (СН3)2]4 при 102 смГ{ отнесено к колеба-
нию 6-TiN4(E4 — Е2) [273].
При действии на TiCl4 литиевого замещенного этиламида обра-
зуется тетракис-(диэтиламидо)титан Ti [N (С2Н5)2]4 в виде оранже-
вой жидкости, перегоняющейся при 112° С (0,1 мм рт. ст.).
Амидотитаны из низшего вторичного амина подвергаются ами-
нолизу на высшие вторичные амины:
Ti (NR)4 + 4R'NH -> Ti (NR')4 + 4R2NH.
58
Исключение составляет аминолиз тетршшс-(диметиламидо)титана
диэтиламином, приводящий к образованию трис-(диэтиламидо)ди-
метиламидотитана [288].
Энтальпия образования Ti[N(C2H5)2]4(>K) составляет
—116 ккал/моль, энергия разрыва связи Ti—N около 73 ккал/моль
[293].
Тетрахлорид титана с метиламином дает соединения тройного
состава TiCl2 (NHCH3)2 • XNH2CH3, где X зависит от условий про-
ведения опыта\[378]. В индивидуальном состоянии выделены со-
единения TiCl (NHCH3)3 и TiCl (NHCH3) (=NCH3).
Диметиламин и диэтил амин замещают в тетрахлориде титана толь-
ко один атом хлора, образуя соединение TiCl3 (NR2) • 2NHR2, где
R = СН3 и С2Н5 [378]. Тетрабромид титана реагирует с диалкил-
аминами, замещая два атома брома. Этилендиамин с тетрахлоридом
титана дает соединение TiCl2 (NHCH2CH2NH2)2 • 2 (NH2CH2CH2NH2)
[379].
По реакции между тетршшс-(диалкиламидо)титаном и четырех-
хлористым титаном в кипящем бензоле получены моно-, ди- и три-
хлортитандиалкиламиды [265]. При действии на mempaKuc-faxwi-
киламидо)титан алкилортоэфирами титана получены бис- (диалкил-
амидо)титаны.
Дисульфид титана реагирует с первичными и вторичными аминами
[163], на чем основан синтез тетранилид титана Ti (NHC6H5)4, тет-
ракис-(бутиламидо)титана Ti (NHC4H9)4 и те/пршшс-(дибутилами-
до)титана Ti IN (С4Н9)2]4.
Кипячением смеси бензольного раствора четыреххлористого ти-
тана с аминами получены амиды Ti (NHC8H17)4, Ti (NHC18H37)4 и
(C4H9O)2Ti (NHC8H17)2 [418].
Из четыреххлористого титана и натрийдифенил амида (C5H5)2NNa,
а также натрийдибензиламида (C6H5CN2)2NNa образуется тетракис-
(дибензиламин)титан Ti [N (СН2С6Н6)2]4 и тетршшс-(дифениламин)-
титан Ti[N(C6H5)2|4 [354].
В результате взаимодействия тетрагалогенидов титана TiT4
(Г = Cl, Br, I) с первичными аминами типа RNH2, где R = СН3,
С2Н5, н-С3Н7 и н-С4Н9, получены смешанные галогенамиды [324].
Эти вещества являются кристаллическими продуктами, состав ко-
торых выражается общей формулой TiT2 (NHR)2 • nRNH2. При
нагревании в вакууме кристаллизационные молекулы аминов от-
щепляются.
Из TiCl4, N-карбозила лития СбН4----С6Н4 и индолила лития
^NLi^
/СНХ
С6Н4< >СН получены частично или полностью замещенные
xNLiz
амиды титана [450]. Это—устойчивые кристаллические вещества,
разлагающиеся при нагревании и при действии водорода. С орга-
59
ническими соединениями лития, натрия, бериллия и алюминия
амиды титана образуют каталитические системы, вызывающие сте-
реоспецифическую полимеризацию пропилена, пентена-1 и стирола.
При действии на TiCl4 гексаметилдисил азана [(CH3)3Si]2NH в
лигроине синтезирован (CH3)3SiNHTiCl3 [272]. Кипячение натрие-
вого производного (CH3)3SiNNaTiCl3 с (цзо-С3Н7О)3Т1С1 и (изо-
C3H7O)2TiCl2 в бензоле дает (t/so-C3H70)3TiN [Si (СН3)2]2 и (изо-
C3H7O)2Ti{N [Si (СН3)3]2}2. Натриевое производное гексаизопро-
пилдисилазана [(t/so-C3H7)3Si]2NNa с (f/3o-C3H7O)3TiCl образует
Rz30-C3H7O)3TiN [Si (СН3)3]2.
Синтезирован также хлорамид [(CH3)3Si]2NTiCl3. Реагируя
с пиридином в петролейном эфире, он превращается в аддукт
(CH3)3SiNTiCl2 • 2C5H5N [272].
В ПК-спектрах поглощения амидов типа Ti (NR2)X, где R = СН3,
С2Н5, к-С3Н7, н-С4Н9 и г/зо-С4Н9, имеются полосы с частотами в
пределах 500—700 смГ1 , относящиеся к колебаниям связи Ti—N
[289].
Трихлорид титана с метил- и этиламинами дает твердые кристал-
лические вещества серого цвета TiCl2 (NHCH3) • 2NH2CH3 и
TiCl2(NHC2H5)-NH2C2H5 [378], с метил- и этиллитийамидом — ко-
ричнево-красные /лр&с-(диметил амидо)титан [TiN (СН3)2]3 и трис-
(диэтиламидо)титан Ti [N (С2Н5)2]3 [240]. Взаимодействие трис-
(диметиламидо)титана с вторичными аминами HN (С2Н5)2,
HN (цзо-С3Н7)2 и NH [Si (СН3)3]2 приводит к образованию смешан-
ных амидов (C2H5)2NTi [N (СН3)212, (изо-С3Н7) NTi [N (СН3)2]2 и
[Si (CH3)3]2NTi [N(CH3)2]2. Метиловый и этиловый спирты вступают
с Ti [N (С2Н5)2]3 в реакции обменного разложения, превращаясь в
соответствующие алкоксититанаты (III).
При действии на трихлорид титана N-карбозил- и N-индоллития
получаются, как и с тетрахлоридом титана, частично или полностью
замещенные амиды титана [450].
Установлено, что метиламин при действии на бцс-(циклопента-
диенил)дихлортитан замещает в последнем не атомы хлора, а пента-
диенильные кольца. Получаемые при этом продукты имеют
неопределенный состав [378]. В другой работе [240] при дейст-
вии на (C5H5)TiCl диметиллитийамида (CH3)2NLi в результате
замещения хлора на амидогруппу получено твердое коричневое
вещество состава {C6H5Ti [N (СН3)2]2}2. Известен также амид
{(С5Н5)2 Ti[N (СН3)2]2}2 [240].
СОЕДИНЕНИЯ С СЕРОПРОИЗВОДНЫМИ
Получены пока единичные представители титанорганических
соединений, в которых связь между атомами титана и углерода осу-
ществляется через атом серы. Руней [524] при добавлении 1%-ного
водного раствора диэтилдитиокарбамината натрия к забуференному
раствору титановых солей (pH 1—6) выделил желтый осадок ди-
60
этилдитиокарбомината титана, состав которого выражается формулой
TiO [(C2H5)2NCSS]2.
Синтезирован N, N-дибензилдитиокарбаминат титана
{[C6H5CH2l2NCSS}4Ti [354].
При действии на тетршшс-(диалкиламидо)титаны сероуглерода
образуются серопроизводные состава Ti (S2CNR2)4, где R = СН3,
С2Н5, С3Н7. Добавляя к Ti (N (СН3)2)4 или Ti [N (С2Н5)2]4 алкил- •
сульфиды калия KCH3SH, KC2H5SH и К (изо-С3Н7) SH, Бредли
и Хамерслей [294] синтезировали серию комплексов состава
Ti (SCH3)4 (CH3SH)0,2 [(CH3)2NH]0,6, Ti (SCH3)4 [(CH3)2NH]o,55,
Ti (SC2H5)4 (C2H5SH)r(RNH)„ Ti (SC2H5)4 [(CH3)2NH]0.67,
Ti (SC2H5)X [N (CH3)2]„ Ti (SC3H7-U3o)4 (^o-C3H7SH)x (R2NH)„
Ti (С3Н7-озо)х [N (CH3)2]^, где R = CH3 и C2H5 (x и у — величины
переменные).
СОЕДИНЕНИЯ С СИЛАНАМИ
Силаны образуют с титаном соединения, аналогичные алкил-
(арил)титановым эфирам, в которых связь кремния с титаном осу-
ществляется через кислород. Производные со связью титан — крем-
ний до настоящего времени не известны. Состав алкил(арил)си-
локсититанов выражается общими формулами Ti (R3SiO)4 и
TiT4_n (R3SiO)n, где R — органический радикал.t Г — галоген или
алкил(арил), п = 1,2.
тетракис-Триалкил(арил)силоксит;<тзш
Методы синтеза алкилсилоксититанов сходны с методами полу-
чения алкоксититанов. тетршшс-Триалкил (арил)силоксититаны
получаются взаимодействием четыреххлористого титана с триал-
кил (арил)силоксититанами, триалкил (арил )силанами в присут-
ствии аммиака или органических оснований и по реакции переэте-
рификации (силанолиза) алкилтитанов или силаноксититанов с три-
алкил (арил)силанолами. Реакцию переэтерификации катализирует
примесь небольшого количества (около 0,01%) металлического нат-
рия [76].
Ti [OSi (СН3)3]4. тетршшс-(Триметилсилокси)титан является
простейшим представителем соединений титана с силанолами. Он
был получен пропусканием аммиака через раствор триметилсила-
нола и четыреххлористого титана в безводном бензоле [369]. В по-
следующем данное производное синтезировали многие авторы [42,
46, 47, 49, 76, 138,302,303,305,3551. Это — желто-зеленая жидкость,
кипит при температуре 112° С (11 мм рт, ст.), удельный вес
0,9051 (0°С), показатель преломления 1,4277 (20° С).
Ti [OSi (С2Н5)3]4. тетршшс-(Триэтилсилокси)титан образует-
ся по реакции переэтерификации [76, 234]:
Ti (ОС2Н5)4 + 4 (С2Н5)3 SiOH = Ti [OSi (С2Н5)3]4 + 4С2Н5ОН;
по реакциям обменного разложения между четыреххлористым ти-
61
таном с триэтилсиланолятом натрия [49] и четыреххлористым ти-
таном с двойными соединениями триэтил сил анол ята свинца с гид-
роокисью свинца [50]:
TiCl4'+ 4NaOSi (С2Н5) - Ti [OSi (C2H5)3J4 + 4NaCl,
2 {2Pb [OSi (C2H5)3] • Pb (OH)2) + TiCl4 ->
-> 2Ti [OSi (C2HJ3]4 + 6PbCl2 + Ti (OH)4
и по др. [44, 46, 48, 76—78, 138]. Представляет собой кристаллы,
точка плавления 96° С, точка кипения 176—178° С (2,5 мм рт. ст.),
удельный вес 0,9809 (20° С), показатель преломления 1,4812 (20° С)
[48].
Ti [OSi (СН3)2С2Н5]4. Получен по реакции переэтерификации
[623]. Жидкость, точка кипения 140—142° С (5 мм рт. ст.), показа-
тель преломления 1,4461 (20° С), удельный вес 0,9237 (20° С).
Ti [OSi (СН3) (С2Н5)2]4. Образуется по реакции переэтерифи-
кации между тетраэтилоксититаном и метилдиэтил сил аксанолом
[76], а также при действии на четыреххлористый титан метилдиэ-
тилсилаксанола и пиридина [77, 78]. Жидкость, точка кипения
154° С (3 мм рт. ст.), показатель преломления 1,4545 (20° С), удель-
ный вес 0,9244 (20° С) [76].
Ti [OSi (С3Н7-н)3]4. Получен по реакции переэтерификации между
тетраэтил оксититаном и диметил-н-пропилсиланолом. Жидкость,
точка кипения 190° С (3,5 мм рт. ст.), показатель преломления
1,4582 (20° С), удельный вес 0,9056 (20° С) [76, 138].
Ti [OSi (С3Н7)2СН3]4. Получен по реакции переэтерификации
между тетраэтил оксититаном и метилди-н-пропилсилоксанолом [76].
Жидкость, точка кипения 186° С (3 мм рт. ст.), показатель прелом-
ления 1,4570 (20° С), удельный вес 0,9044 (20° С).
Ti [OSi (С6Н5) (СН3)2]4. Образуется по реакции переэтерифика-
ции тетраэтилоксититана с трифенилсиланолом и в результате
взаимодействия четыреххлористого титана с трифенилсиланолом
в присутствии пиридина [76, 78, 138]. Кристаллическое вещество,
точка плавления 501—505° С (разлагается), показатель преломле-
ния 1,6488 (114° С), удельный вес 1,215 (29° С).
Ti [OSi (С6Н5)2 (CH3).J4. Получен из диметилфенилсилаксолята
натрия и четыреххлористого титана [45]. Жидкость, точка кипения
370—374° С (6 мм рт. ст.), показатель преломления 1,5960 (20° С),
удельный вес 1,248 (20° С).
Ti [OSi (С6Н5) (С2Н5)2]4. Жидкость, точка кипения 300—305° С
(5 мм рт. ст.), показатель преломления 1,5455 (20° С), удельный
вес 1,04559 (20° С) [138].
Ti [O5Si4 (С6Н5)8]2. Получили Цейтлер и Браун [622] переэте-
рификацией тетрабутоксититана с дифенилсиландиолом. Кристал-
лический продукт с точкой плавления 214—214,5° С. Авторы при-
писывают продукту следующее строение:
62
Алкокситриалкилсилоксититаны
с
Смешанные алкокси- и силоксититаны получаются действием
на алкоксититаны триалкоксисиланолов, взятых в соответствующих
пропорциях [355]. Показано, что реакция между бш?-(триметилси-
локси)титанхлоридом и спиртами является общей для синтеза
т/шс-(диметилсилокси)титаналкоксидов [29]. Однако в присутствии
металлического натрия (катализатора) на бутоксигруппу замеща-
ются как атомы хлора, так и триметоксисилоксигруппы [28].
При добавлении к тетракис-(триалкилсилокси)титану хелатов
(ацетил ацетона, дибензил метана, у-оксихинолина) происходит за-
мещение двух триалкилсилоксигрупп [521]. ИК'-спектры поглоще-
ния хелатных комплексов титана имеют полосы с частотами 1375,
1520, 1575 смГ\ Частоты 1245, 980 и 845 см~х относятся к связи
—Ti—О—Si—.
При умеренном нагревании смеси диалкоксититаноксидов с тет-
раалкоксисиланом образуются смешанные алкоксисиланоксититаны
с одной связью Ti—О—Si по схеме
ROX •
Ti=O + (R'O)4 Si = RO—Ti—О—Si (OR')3.
RO/
Методом ЭПР показано, что TiCl4 с триметилоксисиланом в рас-
творах циклогексана и четыреххлористого углерода вступает в ре-
акцию обменного разложения, образуя метокситрихлор титан или
диметоксидихлорсилан [237].
(изо-С3Н7О)2Т1 [OSi (СН3)3]2. Жидкость, точка кипения 120° С
(14 мм рт. ст.), показатель преломления 1,4408 (22° С) [355].
(#3O-C3H7O)3TiOSi (СН3)3. Жидкость, точка кипения 114° С
(13 мм рт. ст.), показатель преломления 1,4490 (22° С) [355].
RO
63
C4H9OTi [OSi (CH8)3]3. Жидкость, точка кипения 76—77° С
(2 мм рт. ст.), показатель преломления 1,4317 (20° С), удельный
вес 0,9070 (20° С) [47].
(C3H7O)3TiOSi (С3Н7)3. Жидкость, точка кипения 125—126° С
(1 мм рт. ст.), показатель преломления 1,4647 (20° С) [580].
(C3H7O)3TiOSi (C3H7)2OTi (ОС3Н7)3. Жидкость, точка кипения
78—8Г С (10-5 мм рт. ст.), показатель преломления 1,4910 [133].
(#3O-C4H9O)3TiOSi (ОС4Н9-язо)3. Жидкость, температура кипе-
ния 75—78° С (10~5 мм рт. ст.), показатель преломления 1,4610
(20° С) [1331.
[С6Н5СОСН—(С6Н5) CO]2Ti [OSi (СН3)3]2. Кристаллическое ве-
щество с точкой плавления 159—170° С [521].
[СН3СОСН=(СН3) C012Ti [OSi (СН3)3]2. Кристаллическое ве-
щество с точкой плавления 56° С [521].
(C3H7O)3TiOSi (СН3)2ОС3Н7 и (C3H7O)3TiOSi (СН3) OTi (ОС3Н7)3.
Образуются при взаимодействии дипропоксититаноксида(С3Н7О)2ТЮ
с диметил-бш>(пропокси)силаном.
Триалкилсилоксигалогентитаны
Получаются триалкилсилоксигалогентитаны нагреванием че-
тыреххлористого или четырехбромистого титана с гексаалкилди-
силоксаном в присутствии хлористого или бромистого алюминия.
В результате реакции один или два атома галогена у тетрагалоге-
нида обмениваются на триалкилсилоксигруппы:
nR3SiOSiR3 + 2TiX4 (R3SiO)n TiX4_„ + nR3SiX.
Триметилсилоксихлортитаны [30, 138] образуются при действии
на четыреххлористый титан триметилсилаксолята натрия [47]:
п (CH3)3SiONa + TiCl4 [(CH3)3SiO]n TiCl4_rt + nNaCl.
TiCl3OSi (CH3)3. Получен К. H. Андриановым и В. Г. Дуловой
[47], а также Н. Ф. Орловым и соавторами [138]. Легкоплавкое вещест-
во, точка плавления 32—33° С, точка кипения 82—88° С (17 мм рт.
ст.), удельный вес 1,305 (20° С) [138].
TiCl3OSi (СН3)2С2Н5. Жидкость, точка кипения 94—98° С
(17 мм рт. ст.), удельный вес 1,305 (20° С) [75, 138].
TiCl3OSiCH3(C2H5)2. Жидкость, точка кипения 122—126° С
(9 мм рт. ст.), удельный вес 1,217 (20° С) [138].
TiCl2 [OSi (СН3)3]2. Жидкость с точкой кипения 72—73° С
(3 мм рт. ст.) [47, 138].
TiCl2 [OSi (СН3)2С2Н5]2. Жидкость, точка кипения 114—118° С
(9 мм рт. ст.), удельный вес 1,111 (20° С) [138].
TiCl2 [OSiCH3 (С2Н5)2]2. Жидкость, точка кипения 127—135° С
(2 мм рт. ст.), удельный вес 1,068 (20° С) [138].
Ti [OSi (СН3)3]3С1. Жидкость с точкой кипения 103—105° С
(10 мм рт. ст.) [47].
64
TiBr3OSi (CH3)3. Кристаллическое вещество, точка плавления
32—34° С, точка кипения 102—104° С (10 мм рт. ст.), удельный вес
1,807 (40° С) [138].
Другие титанорганические соединения
Нагреванием четыреххлористого титана с четырехкратным (мо-
лярным) количеством о-фталонитр ила o-C6H4(CN)2 получен фталоциа-
нин титана. Состав его выражается формулой C32H15N8ClTi (ОН)2 [2301.
Известны комплексные соединения четырехвалентного титана с
фталоцианином (C32H16N8) H2(H2L) состава TiLCl2, (TiLO)x,
[TiLOP (С6Н5) О2]х, TiL [OSi (C6H5)3]2 [281]. Первое и второе сое-
динения — порошкообразные вещества темно-синего цвета, третье
и четвертое — голубовато-зеленого и зеленого цвета соответственно.
При взаимодействии галогенидов Ti4+ в безводном бензоле или
четыреххлористом углероде с третичным арсином образуют-
ся комплексные соединения (TiF4)3 • CH3As [(CH2)3As (СН3)2]2,
(TiCl4)2 • CH3As [(CH2)3As (CH3)2]2, TiBr4 • CH3As [(CH2)3As (CH3)2]2
и Til3 • CH3As [(CH2)3As (CH3)2]2 [277]. В американском патен-
те [204] описаны • соединения состава
/О—А \
RO-Ti—О—A'—N,
\О—А"/
где А, А' и А" — СН2 СН2 или СН2СНСН3, R —низший алкил.
Вебер с соавторами [599], действуя гексаметилбензолом на ка-
тализатор Циглера с последующим воздействием воды, синтезиро-
вал л-комплекс синего цвета [С6 (СН3)5] TiCl2OH.
При взаимодействии TiCl3 с магнием и азотом в тетр а гидрофуране
получен диамагнитный комплекс [TiNMg2Cl2 • С4Н8О] [615]. В при-
сутствии пиридина он превращается в соединение [TiNMgCl •
• С5Н5], которое, реагируя с С6Н5СОС1, дает темно-зеленый поро-
шок [TiNCl (C6H5CO)i,5].
Получены комплексы состава CH3TiCl2 • (C5H5N)2 и C6H5TiCl2 •
. (C5H5N)2 [471], которые устойчивы при 20° С, но на воздухе
быстро разлагаются. В системе TiCl4 — Si (С2Н5О)4 обна-
ружены соединения с соотношением компонентов 2 : 3 и 1 : 1 [95].
При действии [PN (С2Н5О)2]2 на TiCl4 образуется соединение
[P3N3(C2H5O)4O2TiCl2] [59]. Методом ПМР установлено в нем нали-
чие связей Р=О, N=C2H5 и Р—N.
Барботированием хлорамина через разбавленный раствор три-
этаноламинотитаната в присутствии диметилформамида получено
соединение [160] со структурой
5 1-1669
65
В патентах [21, 159] описано получение водорастворимых тита-
натов при взаимодействии тет^7/шс-(алкилокси)титанов с ди- или
триэтаноламинами, метилдиэтанол амином и Р-аминоэтилэтанол-
амином.
Триалкилфосфаты РО (OR)3 (R=C2H5 и С4Н9) реагируют с
TiCl4, образуя белые кристаллы (РО3)3 [OTi (R2PO4)2]xOTi (РО3)3.
Описаны также соединения OTi [О (РО) (OR2)2j2, (Н4С6О2)-
•Ti [О (РО) (OR)2]2, (ОНСС6Н4О) [(RO) (РО) О2]-Ti [О (РО) (OR)21
[508].
Алкильные производные сурьмы с хлоридами титана дают не-
сколько продуктов, которым приписывается определенный состав.
Из растворов Sb (С2Н5)4С1 и TiCl4 или TiCl3 в бензоле получены бе-
лые порошковидные комплексы [Sb (C2H5)4]2TiCl6, Sb (C2H5)2TiCl5,
Sb (C2H6)4Ti2Cl9, [Sb (C2H5)4]2TiCl5, Sb (C2H5)4TiCl4 [569]. При
взаимодействии Sb (C2H5)4C1 c TiCl2 в стальном реакторе образуются
TiCl3 [Sb (C2H5)4C1]X, где x ~ 1,5—1,9. Взаимодействием TiCl4 и
Sb (C2H5)3C12 получены также кристаллы состава Sb (С2Нб)3С12 •
. (TiCl4)n, где п = 0,5—1,3. Описаны продукты и более сложного
состава, которые, по-видимому, не являются индивидуальными со-
единениями [569].
ПОЛИМЕРНЫЕ ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ ТИТАНА
Титанорганические полимеры содержат неорганические цепочки
или циклы из атомов титана, соединенных, обычно, через кислород
и обрамленных органогруппами. В состав неорганических цепей и
циклов кроме титана могут входить атомы других элементов [41,
94]. В настоящее время уже получены полимеры, содержащие в не-
органических цепях и циклах кремний и фосфор.
Полиорганотитаноксаны
К полиорганотитаноксанам относятся полимеры, содержащие не-
органические цепочки или циклы из атомов титана и кислорода,
обрамленные алкильными или арильными группами. Они получают-
ся при гидролизе титановых эфиров или с помощью конденсации.
Гидролиз титановых эфиров происходит при пропускании через
них струи влажного воздуха [400] или при смешении эфиров в без-
водных растворителях с водными растворами соответствующих спир-
тов [7, 124, 180, 307, 466, 587, 590]. Второй метод позволяет точно
контролировать расход воды на протекание реакции и потому яв-
ляется более удобным.
В результате гидролиза тетраэтоксититана с эквимольным рас-
ходом воды получен смолообразный продукт состава TiO (ОС2НБ)2
[300]. Если расход воды увеличить, то при гидролизе образует-
ся кристаллический продукт Ti2O3(OC2H5)2 или гелеобразный поли-
мер с малым содержанием этоксигрупп. А. Н. Несмеянов и соав-
66
7
торы [124] при гидролизе тетраэтоксититана получили продукт
Ti3O2(OC2H5)8. Окисление этилового эфира трехвалентного титана
дает полимер состава Ti2O (ОС2Н5)б [132]. По реакции переэте-
рификации синтезированы другие эфиры трехвалентного титана:
Ti20(OC3H7-z/so)6 и Т12О(ОС4Н9-шю)6 [127, 132].
По реакции переэтерификации получены полимеры состава
(RO)2Ti3O2, где R= я-С4Н9 и я-С6Н13 [131], которые являются
вязкими жидкостями. При нагревании до температуры 210—220° С
они полностью деполимеризуются до алкилортотитанатов; вступают
в реакции обменного разложения с хлорангидридами, например
с СН3СОС1. При обменном разложении полиорганотитаноксанов с
хлор ан гидридом уксусной кислоты образуются хлорполиокси-
титанаты (C2H5O)7Ti3O2Cl, (C2H5O)6Ti3O2Cl2, (H-C4H9O)7Ti3Cl и
(/i-2C6H13O)6Ti3O2Cl, которые при нагревании в вакууме превраща-
ются в хлорэфиры.
Значительное число работ посвящено исследованию бутилтита-
новых полимеров. Бутилполититанаты получают гидролизом [307,
590] и непосредственно из четыреххлористого титана и бутилового
спирта [342, 587, 590]. Синтез бутилполимеров из четыреххлористого
титана и бутанола сводится к обработке четыреххлористого титана
водным раствором бутилового спирта с последующей нейтрализацией
образовавшегося хлористого водорода аммиаком. Данн [350] в резуль-
тате гидролиза триметил-н-бутилтитанового эфира водой получил по-
лимер метоксибутоксиполититаноксан с молекулярным отношением
СН3О : TiO2 = 1,24.
Полибутилтитанаты — вязкие жидкости или твердые вещества.
Если содержание ТЮ2 в них менее 56—58%, то они хорошо раство-
ряются в органических растворителях.
При взаимодействии алкилтитанатов с дикетонами образуются
полимерные комплексы [93]. Один из них получается при гидролизе
продуктов реакции между алкилтитанатами и бис-(Р-дикетон)-1,4-
бис-(ацетоацетил)бензолом. Второй полимер синтезирован взаимо-
действием бутилтитаната с ацетилацетоном и последующим гидро-
лизом продуктов реакции. Он плавится при температуре выше
250° С. Авторы [93] выделили также осадок полимера неустановлен-
ного состава с молекулярным весом 12 000.
Согидролиз этилацетоната алюминия и бутилортотитаната при
комнатной температуре [146] приводит к образованию светло-жел-
того полимера. Описаны светлые вязкие полимеры, состав которых
выражается общей формулой Ti [OZr (OR2)]n [144].
Полимерные титанацилаты
Полиорганотитаноксаны при нагревании с жирными кислотами
вступают с ними в реакции обменного разложения, в результате
которых образуются полимеры с ацильными обрамляющими груп-
пами [307, 308]. Полимерные ацилаты титана получаются также при
нагревании алкилтитанатов с жирными кислотами [446] и при взаи-
5* 67
модействии четыреххлористого титана с алифатическими монокар-
боновыми кислотами и водой в присутствии органических оснований
[375, 500]. По внешнему виду эти соединения напоминают воско-
образные вещества, хорошо растворяются в углеводородных раство-
рителях. В их состав входит остаток OTi(OCOR)(OH).
Полимерные титанацилаты описаны преимущественно в патент-
ной литературе [15—17, 156, 157, 174, 183, 196, 201—203]. Иссле-
дованиям методов синтеза и свойств данных полимеров посвящены
только единичные статьи. Например, В. С. Киселев и Т. А. Ермола-
ева [97, 98, 559] изучили получение полимерных ацилатов титаната
из тетрабутоксититана и жирных кислот, в частности олеиновой
кислоты. Синтезированные ими бутокситриолеатитан и бг/с-(трио-
леил)титаноксан оказались пригодными при производстве лакокра-
сочных материалов в качестве пленкообразующих.
Полиорганосилоксититаноксаны
К полиорганосилоксититаноксанам относятся полимеры с кис-
лородно-титановой цепочкой и силоксановыми обрамляющими груп-
пами. Впервые эти полимеры были получены К. А. Андриановым с
соавторами [43, 49] гидролизом тетржш>(триметилсилокси)ти-
тана в ацетоне при 50° С. В результате добавления 1 моля воды на
1 моль эфира образуется полимер, растворимый в органических
растворителях. С большим расходом воды и при нагревании в ре-
зультате гидролиза получаются нерастворимые полимеры. Гидро-
лиз, по мнению авторов, протекает по следующей схеме:
Ti [OSi (СН3)3]4 + Н2о -> [(СН3)3 SiO]3 TiOH + (СН3)3 SiOH;
2 (СН3)з SiO3TiOH -> [(СН3)з О], TiOTi [OSi (СН3)3]3 + Н2О ->
-> [(СН3)з SiO3]3 Ti {OTi [OSi (СН3)3]2}П OTi [OSi (CH3)3]3.
Изучен гидролиз Ti [OSi (CH3)3]4 в диоксане при комнатной тем-
пературе [306]. Показано, что в результате гидролиза получается
продукт Ti2O [OSi (СН3)3]6, который подвергается диспропорциони-
рованию с образованием полимеров:
3Ti2O [OSi (СН3)3]6 -> 4Ti [OSi (СН3)3]4 +
+ Ti2O3 [OSi (CH3)3]2 (полимер).
При действии на бутилортотитанат триметилсилоксиацета-
та получен полимерный продукт состава [(СН3СОО)2ТЮ]3 •
. OTi (ОСОСН3) OSi (СН3)3 [32]. Тетрахлорид титана с силилаце-
татом дает твердый полимер СпН21О12С151Т1з.
Поликонденсация а, со -диоксиметилфенилсилоксановых оли-
гомеров с бш?-(метилалкоксифосфинокси)дибутоксититаном при-
водит к сополимерам, содержащим в главной цепи Si, Ti и О, а в
боковых цепях — Р и О [33]. По реакции между бг/о(триметилси-
локси)диизопропоксититаном и уксусным ангидридом получены
68
поли-(триметилсилокси)титановые полимеры [530]:
п [(CH3)S SiO]2 Ti (u30-CsH,O)2 + n (CH3CO)2O -+
Г OSi (CH3)3-|
изо-С3Н70—
—Ti—o—
I
L OSi (CH3)3J
-C-CH3
+ (2n — 1) CH3CO2C3H7-rz30.
Полиорганотитансилоксаны
Полиорганотитансилоксаны имеют цепочку из атомов титана,
кремния и кислорода. Они получаются в результате совместного
гидролиза алкил (арил)хлорсиланов с тетраалкоксититанами и при
взаимодействии продуктов гидролиза алкил(арил)хлорсиланов с
титановыми эфирами, амидами и ацилатными производными орто-
титановой кислоты.
К. А. Андрианов с соавторами [27, 40, 42) при совместном гид-
ролизе диалкилдихлорсиланов с тетрабутоксититаном получил про-
дукты, строение которых можно представить следующей схемой:
/ R\ ОС4Н9
I 1 I 1
—О—Si —О—Ti—О—
\ | / I
\ R/n ОС4Н0
Отношение между кремнием и титаном в этих полимерах может ме-
няться в зависимости от соотношения взятых для реакции компонен-
тов в пределах от 5 : 1 до 30 : 1.
При действии тетрахлорида титана на мононатриевые производ-
ные алкил- и арилоксисиландиолы, алкил- или арилметокси-,
этокси-, фенокси- и винилоксисиландиолы образуются полиоргано-
титансилоксаны высокой степени сшивки с одной метильной, этиль-
ной или фенильной группой у атома кремния [38, 391.
Согидролиз диалкил- или диарилхлорсиланов и бш?-(ацетилаце-
тонато)дихлортитана дает линейные полимеры с главной цепью
Ti—О—Si [37]. Однако молекулярный вес полимеров, осаждае-
мых при постепенном добавлении к бензольному раствору гептана,
оказался низким 1208].
Из бутил ортотитаната а,со-диоксиполидиметилсилоксана по-
лучен полимер с молекулярным весом около 27 000, растворимый в
бензоле и толуоле [361. Поликонденсацией полимеров с хелатными
комплексами титано-бпс-(8-оксихинолинат)дибутоксититаном или
бш?-(ацетилаце1онат)дибутоксититаном получены полихелатотитан-
диметилсилоксаны с различным соотношением Si : Ti [35].
Реакция полимеризации протекает по уравнению
СН3
Октаметилциклотетрасилоксан с бутилортатитанатом при тем-
пературе 180—200° С образует полимер и линейные олигомеры [34].
Конденсация тетрафункциональных олигомеров дает полимеры,
содержащие 18 и 26 диметил силоксановых радикалов [31].
Полиорганотитансилоксаны с линейным строением при нагре-
вании конденсируются с образованием полимеров сетчатого строе-
ния. К. А. Андрианов и соавторы [531 провели конденсацию полити-
танодиметилсилоксанов общей формулы
с п — 23, 34, 52 и 128 и частично конденсированных олигомеров
с п = 2,3. При температуре 200° С эти олигомеры конденсируются
в результате взаимодействия гидроксильных групп по схеме
70
Н. Б. Барановская с соавторами [56] наблюдала отверждение
полимеризованного диметилоксисилана в результате взаимодействия
его с тетраэтилокси-, тетрапропокси- и тетрабутилоксититанами
при комнатной температуре.
В патентной литературе имеются предложения по использованию
полимерных титанкремнийорганических соединений для производ-
ства термостойкой резины, гидрофобизирующих веществ, катали-
заторов, для приклеивания полифторэтиленовых полимеров к метал-
лам и др. [9, 142, 147, 152, 185, 186, 191, 194, 195, 198, 199, 205, 235,
236, 130].
Полиорганофосфонилоксититаноксаны
Полимеры этого типа имеют цепи из атомов титана, кислорода
и фосфора. Впервые они были получены К. А. Андриановым и
И. К. Кузнецовой [51] поликонденсацией «-тетрабутоксититана с
х л ор а н ги д р и дом метил фосфи новой кисл оты.
Поликонденсация w-тетрабутилоксититана с дибутиловым эфи-
ром метилфосфиновой кислоты, взятых в соотношении 1:1, проте-
кает с выделением дибутилового эфира и образованием олигомеров
[551:
n(«-C4H9O)4Ti+«(C4H9O)2 —РОСН3 ->
(2п - 1) С4Н9ОС4Н9 + С4Н9
- ОС4Н9
—отю
_ ОС4Н9
СН.1
о
—ОС4Н#.
«-Тетрабутилоксититан реагирует с двухосновными фосфиновыми
кислотами с образованием олигомеров или /петршаа>(оксиалкил-
71
г:
фосфонил)титана [54]:
nTi (ОС4Н9)4 + nR—-Р = (ОН)2 -> (2п—1)С4Н9ОН +
II
О
Ti (ОС4Н9)4 + 4R-P = (ОН)2
О
4С4Н9ОН + Ti
где
R = CH3 и СН2=С(С6Нб).
тетршшс-Монометилфосфонилтитан при действии воды отщеп-
ляет только две группы, а две другие остаются с продуктами кон-
денсации:
/ сн3 \
/ о \
О—Р—ОН
(избыток) + Н2О = 2СН3—Р = (ОН)2 +
4
3
ОН
он
3
п
При взаимодействии тетршшс-а-фенилвинилфосфонилтитана с
избытком триэтилбутоксисилана и «-тетрабутилоксититана проис-
ходит замещение водорода в фосфонильном остатке на триэтилси-
локси- и «-трибутилтитаноксигруппы:
/ОН \
2
72
mempa/шс-Монометилфосфонилтитан реагирует с триэтилбуто-
ксисиланом с выделением бутилового спирта и бш?-триэтилсилильного
эфира метафосфиновой кислоты:
ОН
+ 4C4H9OSi (С2Н5)3 + 2 [(С2Н5)3 SiO]2 РОСН3 + C2H5OP2Ti.
Таким образом, в тетракис- ал кил фосфонил титанах два алкилфос-
фонильных остатка легко отрываются, а два других, по-видимому,
координационно взаимодействуют с титаном и поэтому не отщепля-
ются.
При взаимодействии н-трибутилоксититана с монометил- и а-
фенилвинилфосфиновой кислотой получаются олигомерные про-
дукты со связью Ti—О—Р, имеющие молекулярный вес 1500—
2000, и тетракис-моноалкилфосфонилтитаны.
В ИК-спектрах бш?-диметилфосфонилдибутоксититана и поли-бис-
диметилтитаноксанов имеется полоса поглощения Р=О, что объяс-
няется координационным взаимодействием атома титана с кислоро-
дом группы РО [521:
(СН3)2 р - о
0 х :-
- О - Ti - 0 -
(СН3)2Р^ о
Даль и Блок [364] синтезировали серию фосфинатных полититанов.
СОПОЛИМЕРЫ НЕНАСЫЩЕННЫХ ТИТАНОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Сополимеризация тетраметаллилтитана с винилацетатом приво-
дит к образованию эмалеподобных веществ, устойчивых к гидроли-
зу [147]. Дибутоксититандиметакрилат (C4H9O)2Ti (ОСОС (СН3) =
= СН2)2 дает сополимер со стиролом. Полимеризуются дивинилди-
73
изопропилтитанат (w3o-C3H70)2Ti (ОСН=СН2)2 и тетрастирилти-
танат Ti (ОСН=СНС6Нр4 ПО, 182].
Эфиры ортотитановой кислоты образуют полимеры с производ-
ными целлюлозы, полиэфирами и поливиниловым спиртом, в ре-
зультате чего происходит сшивание цепей полигидроксиполимеров.
Полимеры при этом становятся нерастворимыми в обычных раство-
рителях [96, 161, 162, 179, 181, 183, 395, 428, 519, 555, 570].
Взаимодействием аллилового спирта с бш?-(циклопентадиенил)
дихлортитаном в присутствии аммиака синтезирован бис-(циклопен-
тадиенил)аллилоксидихлортитан, сополимеризующийся со стиролом
или метилметакрилатом [92]. Сополимеры являются твердыми ве-
ществами оранжевого цвета с молекулярным весом 22 100 (метакри-
лат) и 47 000 (стирол).
А. Л. Суворов и С. С. Спасский [219] получили прозрачный крас-
но-коричневый полимер из конденсированного циннамата титана и
стирола.
Литература
1. Андреев Ю. Н., Никольский В. А.— В кн.: Сборник статей по
общей химии, 2. Изд-во АН СССР, Л., 1953, 1428.
2. А р б у з о в В. А., Исаева 3. Г.— Ж- общ. хим., 1952, 22, 566.
3. Арбузов Б. А., Ш а в ш а Т. Г.— ДАН СССР, 1949, 68, 859.
4. А р б у з о в Б. А., Ш а в ш а Т. Г.— ДАН СССР, 1951, 79, 599.
5. Абелеева В. В., ХидекельМ. Л.— Изв. АН СССР. Сер. хим.,
1969, 2087.
6. Алексеев Н. В., Ронова И. А.— Ж- структ. хим., 1966, 7, 103.
7. Австралийский пат. № 160036; РЖХим., 1955, 47 599.
8. Англ. пат. Ns 719340 от 1.12.1954.
9. Англ. пат. Ns 728751, 1955; С. А., 1955, 49, 12878.
10. Англ. пат. Ns 7332224; С. А., 1955, 49, 16 464.
11. Англ. пат. № 765603; С. А., 1957, 51, 1557.
12. Англ. пат. Ns 766810; С. А., 1957, 51, 10950.
13. Англ. пат. Ns 779490, 1957; С. А., 1957, 51, 17992. Ns 771167; С. А., 1958, 52, 1204.
14. Англ. пат.
15. Англ. пат. Ns 793760, 1958; С. А., 1959, 53, 1148.
16. Англ. пат. Ns 797391; С. А. 1959, 53, 2094.
17. Англ. пат. Ns 798412, 1958; С. А., 1959, 53, 31501. Ns 479470; С. А., 1938, 32, 5003.
18. Англ. пат.
19. Англ. пат. Ns 858540; С. А., 54, 18546.
20. Англ. пат. № 777216; С. А., 51, 12 542.
21. Англ. пат. Ns 755728; С. А., 51, 8128.
22. Англ. пат. Ns 800528; С. А., 53, 4134.
23. Англ. пат. № 798412; С. А., 53, 3150.
24. Англ. пат. Ns 738708; С. А., 50, 16 825.
25. Англ. пат. № 716126; С. А., 49, 12 534.
26. Англ. пат. Ns 787180; С. А., 52, 10 143.
27. А н д р и а н о в К. А., Жданов А. А., Ганина Т. Н.— В кн.:
Сообщение о научных работах членов Всесоюзного химического о-ва
им. Д. И. Менделеева, 3. Изд. ЖВХО, М., 1955, 2.
28. А н д р и а н о в К. А., Астохин В. В.— ДАН СССР, 1959, 127, 1014.
29. Андрианов К- А., Делазари Н. В.— Изв. АН СССР. Отд. хим.
наук, 1960, 1712.
30. Андрианов К- А., Курашова Н. Я.— ДАН СССР, 1960, 131, 825.
74
31.
32.
33.
34.
35.
36.
37.
Андрианов К. А.,
Хим. пром., 1963, 1, 14.
Петрашко Ю. К., Аснович Э. 3.—
Андрианов К. А., Гашина Т. Н.— Ж- общ. хим., 1959, 29, 605.
Андрианов К. А., Варламова Н. В., Борисов М. Ф.,
Колчина А. Г., Гребенщикова Г. В.— Пласт, массы, 1966, 33.
Андрианов К- А., Пичхадзе Ш. В., Комарова В. В.—
Изв. АН СССР. Отд. хим. наук, 1962, 724.
Андрианов
наук, 1962, 261.
Андрианов
Андрианов
Высокомол. соед.,
К- А., П и ч х а д з е Ш. В.— Изв. АН СССР. Отд.
К. А., Жданов А. А.—ДАН СССР, 1961, 138,
К- А., Пичхадзе Ш. В., Бочкарева И.
1961, 3, 1321.
хим.
361.
В.—
38. А н д р и а н о в К- А., Аснович Э. 3.— Высокомол. соед., 1959, 1,
744.
39. А н д р и а н о в К. А., А с н о в и ч Э. 3.— Высокомол. соед., 1960, 2, 136.
40. Андрианов К- А., Хрусталева Е. Н., Ганина К. А.—
Изв. АН СССР. Отд. хим. наук, 1956, 817.
41. Андрианов К- А. Полимеры с неорганическими главными цепями
молекул. Изд-во АН СССР, М., 1959.
42. А н д р и а н о в К- А., Ганина Т. Н., Хрусталева Е. Н.—
Изв. АН СССР. Отд. хим. наук, 1956, 798.
43. А н д р и а н о в К. А., Жданов А. А., К у р а ш о в а Н. А., Д у -
лова В. Г.— ДАН СССР, 1957, 112, 1050.
44. Андрианов К- А., Жданов А. А.— Изв. АН СССР. Отд. хим. наук,
1958, 779.
45. Андрианов К- А., Д е л а з а р и Н. В.— ДАН СССР, 1958, 122, 393.
46. А н д р и а н о в К. А., Жданов А. А.— Polymer Sci., 1958, 30, 513.
47. Андрианов К. А., Дулова В. Г.— Изв. АН СССР. Отд. хим. наук,
1958, 644.
48. А н д р и а н о в К. А., Жданов А. А., К а ш у т и н а Е. А.—
ДАН СССР, 1959, 126, 1261.
49. А н д р и а н о в К- А., Жданов А. А., Казакова А. А.— Изв.
АН СССР. Отд. хим. наук, 1959, 466.
50. А н д р и а н о в К. А., Жданов А. А., Кашу тин а Э. А.—
Ж- прикл. хим., 1959, 32, 463.
51. Андрианов К- А., Кузнецова И. К-— Изв. АН СССР.
Сер. хим., 1964, 651.
52. А н д р и а н о в К- А., Г а е ш н и к о в а Н. П., Кузнецова И. К- —
Изв. АН СССР. Неорг. материалы, 1965, 1, 460.
53. Андрианов К. А., Ку р а шов а Н. А., Манучарова И. Ф.,
Берлинер Е. М.— Изв. АН СССР. Неорг. материалы, 1965, 1,
294.
54. Андрианов К- А., Кузнецов И. К-, Смирнов Ю. М.—
Изв. АН СССР. Неорг. материалы, 1965, 1, 301.
55. Андрианов К- А., Кузнецова И. К, Смирнов Ю. М.—
Изв. АН СССР. Неорг. материалы, 1965, 1, 289.
56. Барановская Н. Б., Захарова М. 3., Мизикин А. И.,
Берлин А. А.— ДАН СССР, 1958, 122, 603.
57. Бельгийский пат. № 500963; С. А., 1954, 48, 9080.
58. Брукер А. Б., Френкель Р. И., Соборовский Л. 3.—
Ж- общ. хим., 1958, 28, 2413.
59. Б у с л а е в Ю. А., Л е в и н Б. В., Румянцев 3. Г. и др.—
Ж- неорг. хим., 1969, 14, 3245.
60. Бобинова Л. М., Разуваев Г. А.— В кн.: Высокомолекулярные
соединения. Гетероциклические высокомолекулярные соединения. «Наука»,
М., 1963, 2б0.
61. Бабкина О. Н., Григорян Э. А. и др.— Ж- физ. хим., 1969,
43, 1759.
62. Бельгийский пат. № 533477, 1954.
75
63. Бельгийский пат. № 550573; С. А. 54, 15 402.
64. Бельгийский пат. № 533362, 1954.
65. Бельгийский пат. № 534792, 1955.
66. Бельгийский пат. № 534888, 1955.
67. В а х о н и н А. П., Дудов и к А. Н.— ДАН СССР, 1969, 184, 348.
68. В о л ь н о в Ю. Н., Глезер П. М., Райкина И. Я-— В кн.:
Сборник статей по общей химии, 2. Изд-во АН СССР, М., 1953, 976.
69. В о л ь п и н М. Е., Шур В. Б.— Изв. АН СССР. Сер. хим., 1964, 9,
1728.
70. Вольпин М. Е., Шур В. Б.— Вест. АН СССР, 1965, 1, 51.
71. Вольпин М. Е., Шур В. Б., Л а т я е в а В. Н., Вышинс-
кая Л. Ч., Шульгацер Л. А.— Изв. АН СССР. Сер. хим., 1966, 385.
72. Г а н и ц Ф. — Сб. чехословацких хим. работ, 1955, 20, 990.
73. Григорян Э. А., Дьячковский Ф. С., Семенова Н. М.,
Шилов А. Е. Kinetics and Mechanics Polyreacts, 2, Prers, Budapest, 1969, 267.
74. Григорян Э. А., Дьячковский Ф. С., Хвостик К- M.,
Шилов А. Е.— Высокомол. соед., 1967, А9, 1233.
75. Д о л г о в Б. Н., Орлов Н. Ф., Воронков М. Г.— Изв.
АН СССР. Отд. хим. наук, 1959, 1408.
76. Долгов Б. Н., Орлов Н. Ф.—ДАН СССР, 1957,117,617.
77. Д о л г о в Б. Н., Орлов Н. Ф.— Изв. АН СССР. Отд. хим. наук,
1957, 1414.
78. Долгов Б. Н., Орлов Н. Ф.— Изв. АН СССР. Отд. хим. наук, 1957,
1395.
79. Д ь я ч к о в с к и й Ф. С., Яровицкий П. Я., Б ы с т р о в В. Ф.—
Высокомол. соед., 1964, 6, 659.
80. Дьячковский Ф. С., Ерицян М. Л., КаширенковО. Е.,
Матиска Б., Max К-, Шветна М., Шилов А. Е.— Высоко-
мол. соед., 1969, А4, 543.
81. Д ь я ч к о в с к и й Ф. С., Хрущ Н. Е., Шилов А. Е.— Кин.
и кат., 1968, 9, 1006.
82. Д ж а б и е в Т. С., Шилов А. Е.— Ж- структ. хим., 1969, 10, 549.
83. Д ж а б и е в Т. С., ШиловА. Е.— Ж. структ. хим., 1965, 6, 302.
84. Д ж а б иев Т. С., Сабирова Р. Д., Шилов А. Е.— Кин. и кат.,
1964, 5, 441.
85. Дворянцева Г. Г., Шейнкер Ю. Н., Несмеянов А. Н.,
Ногина О. В., Лазарева Н. А., Дубовицкий В. А.—
ДАН СССР, 1965, 161, 603.
86. Е м е л ь я н о в а И. В., Суворов А. Л., Позднев А. А.—
Труды Ин-та химии, 15. Уральский филиал АН СССР, Свердловск, 1968, 53.
87. 3 е ф и р о в а А. К., Тихомирова Н. Н., Шилов А. Е.— ДАН
СССР, 1960, 132, 1082.
88. Зефирова А. К., Шилов А. Е.— ДАН СССР, 1961, 136, 599.
89. Ионова Е. И.— Изв. АН СССР. Неорг. материалы, 1965, 1, 183.
90. К и с с и н Ю. В., Толстых Э. В., Чирков Н. М.— ДАН СССР,
1962, 145, 104.
91. К а л е ч и ц И. В., Л и п о в и ч В. Г., Ш м и д т Ф. К-— Кин. и кат.,
1968, 9, 24. Q
92. Коршак В. В., Сл адков А. Н., Лунева Л. К., Г е р ш о-
в и ч А. С.— Высокомол. соед., 1963, 5, 1284.
93. К о р ш а к В. В., Сл адков А. М., Лунева Л. K.s Булга-
ков а И. А.— Высокомол. соед., 1963, 5, 1288.
94. К о р ш а к В. В. (редактор). Успехи в области синтеза элементоорганичес-
ких полимеров. «Наука», М., 1966.
95. К о л о д я ж н ы й Ю. В., КаширенковО. Е., ОсиповО. А.—
Ж- неорг. хим., 1966, 11, 348.
96. К и с е л е в В. С., Е р м о л а е в а Т. А.— Ж- прикл. хим., 1956,
29, 432.
97. Киселев В. С., Ермолаева Т. А. — Ж- прикл. хим., 1958, 31, 111.
76
98. К и се л е в В. С., Ермолаева Т. А.— Ж- прикл. хим., 1957, 30,
1810.
99. Киселев В. С., Ермолаева Т. А.— Ж- прикл. хим., 1956, 29, 288.
100. Латяева В. Н., Вышинская Л. И., Вышинский Н. Н.—
В кн.: Применение молекулярной спектроскопии в химии. «Наука», М., 1966,
248.
101. Л у ч и н с к и й Г. П.— Ж- общ. хим., 1937, 7, 2044.
102. Лучинский Г. П. Химия титана. Госхимиздат, М., 1941.
103. Лучинский Г. П.— Ж- общ. хим., 1936, 6, 481.
104. Л ы с е н к о Ю. А., О с и п о в О. А.— Ж. общ. хим., 1958, 28, 1724.
105. Маляренко А. В., Суворов А. Л. Труды Ин-та химии, 15.
Уральский филиал АН СССР, Свердловск, 1968, 43.
106. Нечипоренко Г. Н., Табрина Г. М., Ш и л о в а А. К-, Ш и -
лов А. Е.—ДАН СССР, 1965, 164, 1062.
107. Ногина О. В., Фрейдлина Р. X., Несмеянов А. Н.—
Изв. АН СССР. Отд. хим. наук, 1950, 327.
108. Ногина О. В., Фрейдлина Р. X., Несмеянов А. Н.—
Изв. АН СССР. Отд. хим. наук, 1952, 74.
109. Невельская Э. Я., Дьячковский Ф. С.— Высокомол. соед.,
1969, Б11, 797.
ПО. Несмеянов А. Н., Ногина О. В., Дубовицкий В. А.—
Изв. АН СССР. Сер. хим., 1968, 1045.
111. Несмеянов А. Н., Фрейдлина Р. X., Ногина О. В.—
Изв. АН СССР. Отд. хим. наук, 1951, 518.
112. Несмеянов А. Н., Ногина О. В., Дубовицкий В. А.—
Изв. АН СССР. Сер. хим., 1968, 1045.
113. Несмеянов А. Н., Брайнина Э. М., Фрейдлина Р. X.—
Изв. АН СССР. Отд. хим. наук, 1954, 987.
114. Несмеянов А. Н., Ногина О. В., Берлин А. М.— Изв.
АН СССР. Отд. хим. наук, 1961, 804.
115. Несмеянов А. Н., Ногина О. В., Берлин А. М., Гир-
шкович А. С., Шаталова Г. В.—Изв. АН СССР. Отд. хим. наук,
1961, 2146.
116. Несмеянов А. Н., Ногина О. В., Берлин А. М., Кудря-
вцев Р. В.— Изв. АН СССР. Отд. хим. наук, 1960, 1206.
117. Несмеянов А. Н., Ногина О. В., Дубовицкий В. А.—
Изв. АН СССР. Сер. хим., 1968, 1045.
118. Несмеянов А. Н., Ногина О. В., Фрейдлина Р. X.—
Изв. АН СССР. Отд. хим. наук, 1950, 327.
119. И есмеянов А. Н., Ногина О. В., Фрейдлина Р. X.—
Изв. АН СССР. Отд. хим. наук, 1951, 518.
120. Несмеянов А. Н., Ногина О. В., Берлин А. М.— ДАН
СССР, 1960, 134, 607.
121. Несмеянов А. Н., Ногина О. В., Дубовицкий В. А.—
Изв. АН СССР. Отд. хим. наук, 1961, 437.
122. Несмеянов А. Н., Ногина О. В.,
Локшин Б. В., Дубо-
вицкий В. А. — ДАН СССР, 1968, 844, 182.
123. Несмеянов А. Н., Фрейдлина Р. X., Ногина О. В.—
Изв. АН СССР. Отд. хим. наук, 1951, 518.
124. Несмеянов A. H.s Брайнина Е. М., Фрейдлина Р. X.—
ДАН СССР, 1952, 85, 571.
125. Несмеянов А. Н., Фрейдлина Р. X., Ногина О. В.,—
Изв. АН СССР. Отд. хим. наук, 1952, 1037.
126. Несмеянов А. Н., Ногина О. В.— Изв. АН СССР. Отд. хим.
наук, 1954, 41.
127. Несмеянов А. Н., Ногина О. В., Фрейдлина Р. X.—
ДАН СССР, 1954, 95, 813.
128. Несмеянов А. Н., Фрейдлина Р. X., Брайнина
Э. М.— Изв. АН СССР. Отд. хим. наук, 1954, 987.
77
129. Несмеянов А. Н.» БрайнинаЭ. М., ФрейдлинаР. X.—
ДАН СССР, 1954, 94, 249.
130. Несмеянов А. Н., Брайнина Э. М., Фрейдлина Р. X.—•
Изв. АН СССР. Отд. хим. наук, 1955, 755.
131. Несмеянов А. Н., Брайнина Э. М., Фрейдлина Р. X.—
Изв. АН СССР. Отд. хим. наук, 1955, 838.
132. Несмеянов А. Н., Ногина О. В., Фрейдлина Р. X.—
Изв. АН СССР. Отд. хим. наук, 1956, 373.
133. Несмеянов А. Н., Ногина О. В.— ДАН СССР, 1957, 117, 249.
134. Несмеянов А. Н., Ногина О. В., Дубовицкий В. А.—
Изв. АН СССР. Отд. хим. наук, 1962, 1481.
135. Несмеянов А. Н., Дубовицкий В. А., Ногина О. В.,
Бочкарев В. Н.— ДАН СССР, 1965, 165, 125.
136. Несмеянов А. Н., Федин Э. И., Петровский П. В. и
др.— ДАН СССР, 1965, 163, 659.
137. Нокайдо, Я с у а к и, Охаси Хоруо — J. Chem. Soc. Japan.
Indastr. Chem. Sec., 1968, 71, 1207.
138. Орлов H. Ф., Долгов Б. H., Воронков M. B.— Химия
и практическое применение кремнийорганических соединений, 1. Труды
конференции. Изд. ЦБТИ, Л., 1958, 161, 172.
139. Плец В. М.— Ж. общ. хим., 1938, 8, 1298.
140. Плаксин И. Н., Клименко Н. Т., Солнышкин В. И.,
Шапиро А. П.— Научн. сообщ. Ин-та горного дела им. А. Н. Ско-
чинского, 1968, 45, 8.
141. П л юснI 1 н А. Н., Уваров Б. А., Цветкова В. И., Чир-
ков Н. М.- — Изв. АН СССР. Сер. хим., 1969, 2324.
142. Пат. ФРГ № 940482, 1948.
143. Пат. ФРГ № 1105416 ; С. А., 56, 4794.
144. Пат ФРГ № 1019285; С. А., 54, 1299.
145. Пат. ФРГ № 1023766; С. А., 54, 19 486.
146. Пат. ЧССР № 91718;( " А., 55, 17 081.
147. Пат. США № 2258718 С. А., 36, 595.
148. Пат. США № 2448556 С. А., 42, 9238.
149. Пат. США № 2445794 С. А., 42, 7565.
150. Пат. США № 2453520 С. А., 43, 1796.
151. Пат. США № 2489651 С. А., 44, 1534.
152. Пат. США № 2512068 С. А., 44, 8698.
153. Пат. США № 2518193 С. А., 44, 10 376.
154. Пат. США № 2505259 С. А., 44, 7069.
155. Пат. США № 2570499 С. А., 46, 3783.
156. Пат. США № 2621193 С. А., 47, 10 549.
157. Пат. США № 2621194 С. А., 47, 10 559.
158. Пат. США № 2720502 С. А., 50, 2204.
159. Пат. США № 2824115 С. А., 52, 1827.
160. Пат. США № 2991299 С. А., 56, 324.
161. Пат. США № 2518195 С. А., 44, 10 378.
162. Пат. США № 2686133 С. А., 49, 2754.
163. Пат. США № 2579413 С. А., 46, 6139.
164. Пат. США № 2187821 С. А., 34, 3764. ! )
165. Пат. США № 2655523 С. А., 48, 12 167. к :
166. Пат. США № 2663720 С. А., 49, 1778.
167. Пат. США № 2708205 С. А., 52, 4211.
168. Пат. США № 2670363 С. А., 48, 11 734.
169. Пат. США № 2966505 С. А., 55, 9352.
170. Пат. США № 2684972 С. А., 49, 10 999.
171. Пат. США № 2654770 С. А., 48, 13 710.
172. Пат. США № 2709174 С. А., 50, 5730.
173. Пат. США № 2579414 С. А., 46, 6139.
174. Пат. США № 2621195, 1953; С. А., 1953,47, 10 550.
78
175. Пат. США № 2620318; С. А., 1953, 47, 2508.
176. Пат. США № 2628170, № 2628171; С. А., 1953, 47, 5131.
177. Пат. США № 2643984; С. А., 1953, 47, 9668.
178. Пат. США № 2672455; С. А., 1954, 48, 7924.
179. Пат. США № 268108; С. А., 1954, 48, 10 378.
180. Пат. США № 2689858; С. А., 1955, 49, 667.
181. Пат. США № 2686133; С. А., 1955, 49, 2754.
182. Пат. США № 881508: С. А., 1956, 50, 1168.
183. Пат. США № 2720468; 1955; С. А., 1956, 50, 2336.
184. Пат. США № 2710267; С. А., 1956, 50, 10 362.
185. Пат. США № 2716656, 1955; С. А., 1956, 50, 1372.
186. Пат. США № 2721855, 1955; С. А., 1956, 50, 6090.
187. Пат. США № 2727918, 1955; С. А., 1956, 50, 6834.
188. Пат. США № 2732320; С. А., 1956, 50, 9038.
189. Пат. США № 2732799; С. А., 1956, 50, 6070.
190. Пат. США № 2733222; С. А., 1956, 50, 6812.
191. Пат. США № 2736721, 1956; С. А., 1956, 50, 7502.
192. Пат. США № 2736666; С. А., 1956, 50, 11687.
193. Пат. США № 2750307; С. А., 1956, 50, 15046.
194. Пат. США № 2751314, 1956; С. А., 1956, 50, 17 517.
195. Пат. США № 2717246, 1955; С. А., 1956, 50, 2188.
196. Пат. США № 2750303, 1956; С. А., 1957 , 51, 1626.
197. Пат. США № 2768909; С. А., 1957, 51, 13419.
198. Пат. США Ks 2774690, 1956; С. А., 1957, 51, 4730.
199. Англ. пат. № 791991, 1958; С. А., 1958, 52, 16 745.
200. Пат. США № 2886579, 1959; С. А., 1959, 52, 17 057.
201. Пат. США № 2897966, 1959; С. А., 1953, 53, 19 881.
202. Пат. США № 2898356, 1959; С. А., 1959, 53, 17 905.
203. Пат. США № 2913369 от 17.11.1959; РЖХим, 1961, ЗЛ402
204. Пат. США № 2935522 от 3.05.1960.
205. Пат. ФРГ № 959329, 1957; С. А., 1960, 54, 16 587.
206. Разуваев Г. А., Богданов И. Ф.— Ж- общ. хим., 1933, 3, 367.
207. Разуваев Г. А., Бобинова Л. М., Этлис В. С.— ДАН
СССР, 1958, 122, 618.
208. Рафиков С. Р., Андрианов К. А., Павлова С. А., Т вер-
дохлебова И. И., Пичхадзе Ш. В.— Изв. АН СССР. Отд.
хим. наук, 1962, 1581.
209. Р о н о в а И. А., Алексеев Н. В.— ДАН СССР, 1967, 174, 614.
210. Ронова И. А., Алексеев Н. В.— ДАН СССР, 1967, 174, 614.
211. Разуваев Г. А.—ДАН СССР, 1961,138,1126.
212. Разуваев Г. Л., Латяева В. Н., Вышинская Л. И.—
Ж- общ. хим., 1961, 31, 2667.
213. Разуваев Г. А., Латяева В. Н., Кислякова Г. А., Ба-
талов А. П,— ДАН СССР, 1969, 185, 369.
214. Смирнова С. А., Губин С. П.— Изв. АН СССР. Сер. хим., 1969,
1890.
215. Семернева Г. А., Суваров А. Л., Самарина Л. А.,
Алексеева И. А., Спасский С. С.—Ж- прикл. спектр.,
1965, 3, 555.
216. С е м е р н е в а Г. А., Суворов А. Л., Самарина Л. А.,
Алексеева И. А., Спасский С. С.— Труды Ин-та химии, 13.
Уральский филиал АН СССР, Свердловск, 1966, 65.
217. С е м е р н е в а Г. А., Суворов А. Л., Спасский С. С.—
Труды ин-та химии, 13. Уральский филиал АН СССР, Свердловск, 1966,
49.
218. Суворов А. Л., Спасский С. С.— Высокомол. соед., 1961, 3.
219. Суворов А. Л., Спасский С. С.— ДАН СССР, 1959, 127, 805.
220. Суворов А. Л., Спасский С. С.— ДАН СССР, 1964, 157, 669.
221. Суворов А. Л., Спасский С. С.— Усп. хим., 1959, 28, 1267.
79
222. Суворов А. Л., Спасский С. С.— Труды Ин-та химии, 13.
Уральский филиал АН СССР, Свердловск, 1966, 31.
223. Суворов А. Л., Спасский С. С.— Труды Ин-та химии, 13.
Уральский филиал АН СССР, Свердловск, 1966, 39.
224. Суворов А. Л., Моляренко А. В., Кватер А. И., Спас-
ский С. С. International Symposium on Macromolecul. Chem., Budapest,
1969, V, 479.
225. Суворов А. Л., Моляренко А. В., Кватер Л. И., Спас-
ский С. С.— Тезисы докладов на IV Всесоюзной конференции по химии
и применению кремнийорганических соединений. Изд. НИИТЭХИМ, М.,
1968, 132.
226. Тельной В. И., Рабинович И. В., Тихонов В. Д., Л а -
т я е в а В. Н.,— ДАН СССР, 1967, 174, 1374.
227. Французский пат. № 1033737; РЖХим., 1955, 22391П.
228. Фурман Э. А., Цветкова В. И., Чирков Н. М.— Изв.
АН СССР. Сер. хим., 1965, 2075.
229. Филд Р., Коув П. Органическая химия титана. «Мир», М., 1969.
230. Шкловер Л. П., Плющев В. Е., Роздина И. А., Нови-
кова Н. А.— Ж- неорг. хим., 1964, 9, 478.
231. Шилов А. Е., Шилова А. К.— Ж- физ. хим., 1970, 44, 288.
232. Шилов А. Е., Зефирова А. К-, Тихомирова Н. Т.—
Ж- физ. хим., 1959, 33, 2113.
233. Шустарович Е. М., Дяткина М. Е.—Ж- неорг. хим., 1959,
3, 2721.
234. Яковлев В. И., Виноградова Н. В.— Ж- общ. хим., 1959,
29, 695.
235. Японский пат. № 3343, 1953.
236. Японский пат. № 7568; С. А., 51, 18396.
237. Ястребов В. В., Чернышева А. И.— Ж- общ. хим., 1970, 40,
604.
238. Adkins Н., McCorguodale D. W.— J. Am. Chem. Soc., 1928,
50, 1938.
239. Altmann В. A., Wertyporoch E.— Z. Phys. Chem., 1934,
168, 1.
240. A 1 у e a E. С., В r e d 1 e у D. C., L a p p e r t M. F., S a n g e r A. R.—
Chem. Communs, 1969, 1064.
241. Archambault J., Rivest R.— Canad. J. Chem., 1957, 35, 879.
242. Angelescu E., Moldoveanu S.— Rev. roumaine Chim., 1966,
11, 541.
243. A d e m a E. H.— J. Polymer Sci., 1968, C, 7, 3643.
244. A d e m a E. H., Bos H., Vrinssen С. H.— Rec. trav. chim.,
1960, 79, 1282.
245. Anagnostopoulos A., Nicholls D.— J. Inorg. Nucl. Chem.,
1965 27 339.
246. Adamo A. F. D., Kienle R. H.— J. Am. Chem. Soc., 1955, 77, 4408.
247. Allegra G., Gan is P., Porri L.— Atti Acad. naz. Lincei. Rend,
cl. sci., fis., mat. e natur, 1963, 33, 4, 38.
248. Allegra G., Porri L., Corradini P.— Atti Accad. naz. Lincei.
Rend. cl. sci., fis., mat. e natur, 1961, 30, 44.
249. В i 1 M.— Chem. prumysl, 1955, 5, 4, 42.
250. Birmingham J. M., Fischer A. K., Wilkinson G.— Na-
turwis., 1955, 42, 96.
251. Birmingham J. M., Seyferth D., Wilkinson G.— J. Am.
Chem. Soc., 1954, 76, 4179.
252. В e e r m a n C.— Ang. Chem., 1959, 71, 195.
253. BartlinkH. J., BosH., Raayen W., S m i d t J.— Ark. Sci.,
1961, 14, 158.
254. Bischoff F., Adkins H.— J. Am. Chem. Soc., 1924, 46, 256.
255. Brintzinger H.— J. Am. Chem. Soc., 1966, 88, 4305.
80
256. Brintzinger H. H.— J. Am. Chem., Soc., 1967, 89, 6871.
257. Brinzinger H.— J. Am. Chem. Soc., 1966, 88, 4307.
258. Bull J. M., Fraser M. J., Measure s.—J. Chem. Communs, 1968, 21,
1310.
259. В a w п С. E. H.^ G 1 a ds t о n e.— J. Proc. Chem. Sos., 1959, 227.
260. Behar D., Feilchenfeld H.—J. Organometallic Chem., 1965, 4,
278.
261. В г о v n R. N., Winter G.— J. Chem. Soc., 1963, 734.
262. В e d s о n P. P.— Ann. Phys., 1875, 180, 235.
263. В 1 a c h e 1 1 H. C., Butter S. A.— Inorg. Chem., 1965, 4, 1133.
264. Berny M. F., Berrest M. O.— Bui. Soc. Chim. France, 1968, 4709.
265. E. Berzin g, W. Kornicker — Ber.; 1961, 94, 2263.
266. Burmeister J. L., Deardorff E. A., Dyke С. E.— Inorg.
Chem., 1969, 8, 170.
267. BruckeM. I., Thomas M. A.— Org. Mass-spec trochem., 1968, 1, 835.
268. В r e i 1 H., Wilke G.— Ang. Chem., 1966, 78, 942.
269. Bartlett P. D., Seidel B.— J. Am. Chem., Soc., 1961, 83, 581.
270. Bell J. V., Heisler J., Tannenbat H., G о 1 d e n s о n.—J. Ana-
lyt. Chem., 1953, 25, 1710.
271. Burger H.— M о n a t s h., 1963, 94, 574.
272. В й r g e r H., Wannagat W.— Monatsh., 1963, 94, 761.
273. Burger H., Stammreich H., Teixeira T. S.— Monatsh.,
1966, 97, 1276.
274. В i s t a n E.— J. Gomory Chem. Zvesti, 1956, 10, 91.
275. Boyd T.— Chem. abstr., 1956, 6834.
276. Berradough C. G., Marting R. L., Winter G.— J. Chem.
Soc., 1964, 758.
277. В a c 1 a у G. A., Gregor I. K., Wild S. B.— Chem. Ind., 1964,
1710.
278. Breslow D. S., Newburg N. R.— J. Am. Chem. Soc., 1959, 81, 81.
279. Begley J. W., P e n n e 1 1 a F.— J. Catalisis, 1967, 8, 203.
280. В и с к t о n G. B.— J. Chem. Soc., 1863, 16, 17.
281. Block В. P., Mel on i E. G.—Inorg. Chem., 1965, 4, 111.
282. Bowes C. R., Winter G.— Australia, Dept. Munitions, Paint No-
tes, 1948, 3, 114; C. A., 1948, 7066.
283. Breslow D. S., Newburg N. R.— J. Am. Chem. Soc., 1957, 79,
5072.
284. Ba i n s M. S., Bradley D. C.— Canad. J. Chem., 1962, 40, 2218.
285. Bradley D. C., Hancock D. C., Wardlaw W.— J. Chem.
Soc., 1952, 2773.
286. Bradley D. C., Mehrotra R. C., Wardlaw W.— J. Chem.
Soc 1952 4204
287. Bradley D. C., Mehrotra, Wardlaw W.— J. Chem. Soc., 1952,
5020.
288. Bradley D. C., Thomas I. M.— J. Chem. Soc., 1960, 3857.
289. Bradley D. C., G i t 1 i t z M. H.— J. Chem. Soc., 1969, A. 980.
290. Bradley D. C., Hill D. A. W.—J. Chem. Sos., 1963, 2101.
291. Bradley D. C., Charalambous J., Jain S.— Chem. Ind.,
1965, 41, 1730.
292. Bradley D. C., G i t 1 i t z M. H.— Chem. Communs, 1965, 13, 289.
293. Bradley D. С., H i 1 1 у e r M. J.— Trans. Farad. Soc., 1966, 62, 2374.
294. Bradley D. C., Hammersley P. A.— J. Chem. Soc., 1967, A,
1894.
295. Bradley D. C., Holloway С. E.— J. Chem. Soc., 1968, A., 1316.
296. Bradley D. С., H i 1 1 у e r M. J.— Trans. Farad. Soc., 1966, 62, 2367.
297. Bradley D. С., M e h ro r t a R. J., Wardlow W.— J. Chem. Soc.,
1952, 2027.
298. Bradley D. C., Mehrotra R. C., Swanwick J. D., War-
dlaw W.— J. Chem. Soc., 1953, 2025.
6 1-1669
81
299. Bradley D. C., Gaze R., Wardlaw W.— J. Chem. Soc., 1955,
3977.
300. Bradley D. C., Gaze R., Wardlaw W.— J. Chem. Soc., 1955,
721.
301. Bradley D. C., Gaze R., Wardlaw W.— J. Chem. Soc., 1957,
469.
302. Bradley D. C., Thomas I. M.— Chem. Ind., 1958, 17.
303. Bradley D. C., Thomas I. M.— Chem. Ind., 1958, 1231.
304. Bradley D. C., Sinha R. N., W a r d 1 a w W.— J. Chem. Soc.,
1958, 4651.
305. Bradley D. C., Thomas I. M.— J. Chem. Soc., 1959, 3404.
306. Bradley D. C., Prevedarou-Demag C.— Canad. J. Chem.,
1963, 41, 629.
307. Boyd T.—J. Polimer Sci., 1951, 7, 591.
308. Boyd T. Пат. США № 2666772; С. A., 1954, 4881; nam. США № 2614112;
С. A. 1953 524.
309. Challenger F., Jinks J.— J. Chem. Soc., 1924, 125, 878.
310. Challenger F., Pritchard F.— J. Chem. Soc., 1924, 125, 864.
311. Coughlan C. N., Katz W., Hodgson W.— J. Am. Chem. Soc.,
1951, 73, 5654.
312. Coughlan C. N., Smith H. S. et al.— J. Am. Chem. Soc., 1951,
73 5652
313. Growe R. W., Coughlan C. N.— J. Am. Chem. Soc., 1950, 72,
1694.
314. Cotton F. A.— Chem. Rev., 1955, 55, 501.
315. C h a r r i n V.— Peintures, pigments, vernis, 1959, 35, 144.
316. Chem. Week, 1956, 78, 9, 64.
317. Clauss К., В e s t i а и H. Пат. ФРГ № 1037446, 1959; РЖХим.,
1961, ЗЛ75.
318. C a h о u r s A.— Ann. Chim. Phys., 1861, 62, 257.
319. C h о s h J. C., G h о s h - M a z u m d a r B. N., Bose A. K-, Sen-
Gupta R.— Research., 1954, 7, 326.
320. Cullinane N. M., Chard S. J.— Nature, 1949, 710.
321. Cullinane N. M., Chard S. J., Price G. F., M i 11 w a r d В. B.—
J. Ind. Chem. Soc., 1950, 69, 38.
322. C u 1 1 i n a n e N. M., Chard S. J., Price G. F., Mill-
ward В. B.— J. Appl. Chem., 1952, 2, 250.
323. Cullinane N. M., Char d S. J. at al — J. Appl. Chem., 1951, 1, 400.
324. С о w d e 1 1 R. T., Fowles G. W., Walton R. A.— J. Less.
Common Metals, 1963, 5, 386.
325. Cooper R. M., Garrard W. — Chem. Ind., 1961, 320.
326. Chaudhari M. A., T r e i c h e 1 P. M., Stone F. G. A.— J. Or-
ganometall. Chem., 1964, 2, 206.
327. Canty A. J., С о u t t s R. S. P., W a i 1 1 e s P. C.— Austral. J.
Chem., 1968, 21, 807.
328. Chim. actual, 1969, 1396, 36.
329. Coutts R. S. P., Surtees J. R.—Austrdl. J. Chem., 1966, 19, 387.
330. Coutts R. S. P., W a i 1 e s P. C.— Austral. J. Chem., 1967, 20, 1579.
331. Coutts R. S. P., W a i I e s P. C., Kingston I. V.— Chem. Com-
muns, 1968, 19, 1170.
332. Coutts R. S. P., W a i 1 e s P . C.— Chem. Communs, 1968, 5, 260.
333. Coutts R. S. P., W a i 1 e s P. C.— J. Chem., 1968, 2199.
334. Coutts R. S. P., Wai les P. C.—Austral. J. Chem., 1968, 21, 1181.
335. Coutts R. S., W a i 1 e s P. C.— Austral. J. Chem., 1966, 19, 2069.
336. Coutts R. S., Kautzner B., W a i 1 e s P. C.— Austral. J. Chem.,
1969 22 1137.
337. Corradine P., Sirigu A.— Inorg. Chepi., 1967, 6, 601.
338. Corrodini P., Bassi I. W., N a 11 a G.— Atti acad. naz. Lincei,
Rend. cl. sci. fis., mat. e natur., 1958, 24, 45.
82
339. Chem. Eng. News., 1955, 34, 4226.
340. Choukroun R., Gervais D.— Compt. Rend., 1969, C269, 1233.
341. Cohen H. J.— J. organ. Chem., 1960, 25, 154.
342. Cox A. B., Winter G.— Australis, Dept. Supplay, Defence Research
Labs., Circ., 1953, 13, 1; C. A., 1953, 47, 10241.
343. Cox A. B., Winter C.—Paint Manuf., 1955, 25, 146; C. A., 1955, 8611.
344. Cox A. B., W i n t e r G.— J. Inst. Automt. and Aeronaut. Eng., 1954, 14, 81.
345. Chatfield H. W.— Paint Manuf., 1958, 28, 17; C. A., 1958, 4997.
346. Chatfield H. W.— Paint Manuf., 1958, 28, 43, 60; C. A., 1958, 8581.
347. Chatfield H. W.— J. Appl. Chem., 1957, 7, 456.
348. Dickson R. S., West В. O.— Austral. J. Chem., 1961, 14, 555.
349zD e n о 1 у A.— Compt. Rend. Traw. Chim., 1849, 5, 325.
350. Dunn P.— Austral. J. Appl. Sci., 1959, 10, 458.
351. Dunn P.—Austral. J. Appl. Sci., 1960, 13, 225.
352. Dryden R. P., Winston A.— J. Phys. Chem., 1958, 62, 635.
353. Demarcay E.— Compt. Rend., 1875, 80, 51.
354. Dermer О. C., Fernelius W. C.— Z. anorg. Chem., 1934, 221 83.
355. Danforth J. D.— J. Am. Chem. Soc., 1958, 80, 2585.
356. D i 1 t h e у W.— Ber., 1904, 37, 588.
357. Damm F.— Bull. Soc. Chim. France, 1965, 3, 798.
358. Dietrich H., Dierks H.— Ang. Chem.) 1966, 78.
359. Dietrich H., S о 1 t w i s c h M.— Ang. Chem., 1969, 81, 785.
360. Dijkgraaf C., Rousseau J. P.— Spctrochim. Acta, 1968, A24,
1213.
361. Dijkagraaf C., Rousseau J. P. G.— Spectrochim. Acta, 1969,
A25, 1455.
362. Dijkgraaf C., Rousseau J. P. G.— Spectrochim. Acta, 1969,
A25, 1831.
363. D о у 1 1 G., Tobias S. R.— Inorg. Chem., 1968, 7, 2484.
364. Dahl G. H., Block В. P.— Inorg. Chem., 1967, 6, 1439.
365. Deluzarche A.— Compt. Rend., 1954, 239, 1489.
366. Dillard J. G.— Inorg. Chem., 1969, 8, 2148.
367. Druce P. M., Kingston В. M. at al.— J. Chem. Soc., 1969, A,
2812.
368. EdmondS.— Bull. Soc. Chim. France, 1966, 3548.
369. English W. D., S о m m e r L. H.— J. Am. Chem. Soc., 1955, 77, 170.
370. Fabian R. J.— Materials and Methods, 1956, 43, 120.
371. Feld R.— J. Chem. Soc., 1964. 3963.
372. Funk H., Schleger A., Zimmermann K-—J. prakt. Chem.,
1956,3, 320.
373. Funk H., Rogler E.— Z. anorg. Chem., 1944, 252, 323.
374. Fischer A- K-, Wilkinson G.— Inorg. Nucl. Chem., 1956, 2, 149.
375. Fischer F., Reichnert F.— Helv. Chim. Acta, 1925, 7, 1082.
376. Fischer A. K., L о c h n e r A.— Z. Naturwis., 1960, 15b, 266.
377. Fischer E. D., Rohrscheid F.— J. Organometallic Chem.,
1966, 6, 53.
378. F о w 1 e s G. W., C a r t m e 1 1 E.— U. S. Dept. Comm. Office Tech.
Serv., P. B. Rep., 1962, 149423.
379. F о w 1 e s G. W., McGregor W. R.— J. Chem. Soc., 1958, 139.
380. Farina M., Bressan G.— Rend. Inst, lombardo sci. eletters. Sci.
mat., fis., chim. 6 geol., I960, 94, 593; РЖХим., 1960, 31314.
381. Friedlander H. N., Oita K-— Ind. Eng. Chem., 1957, 49, 1885.
382. F e 1 t z A.— Z. anorg. Chem., 1964, 334, 186.
383. Giannini U., C e s c a S.— Tetrahedron Letters, 1960, 19.
384. Giannini U., Zucchini U.— Chem. Communs, 1968, 940.
385 Graenewege M. P.— Z. phys. Chem., 1958, 18, 147.
386. Giddings S. A.— Inorg. Chem.. 1967, 6, 849.
387. Gerald D., Tobias S. R.— Inorg. Chem., 1967, 6, 1111.
388. G о r s i c h R. D.— J. Am. Chem. Soc., 1958, 80, 4744.
6*
83
f
389. Gan is P., A 1 1 e g r a G.— Atti acad. naz. Lincei Rend., cl. sci fis., mat.
e natur., 1962/63, 5, 303.
390. Gutmann V., M e 1 1 e r A.— Monatsh. Chem., 1964, 95, 1001.
391. G u t R.— Helv. Chim. Acta, 1964, 47, 2262.
392. G i u a M., M о n a t h E.— Z. anorg. Chem., 1925, 143, 383.
393. Gilman H., Jones R. G.— J. Org. Chem., 1945, 10, 505.
394. Ghosh J. C., Mazumdar B. N., Bose A. K-, S e n G. R.—
J. Ind. Chem. Soc., 1954, 31, 683.
395. H i г t R. P., Bruxelles G. N.— Ind. Eng. Chem., 1956, 48, 1325.
396. Hendrik D., Martin A., J e 1 1 i n e к F.— Ang. Chem., 1964,
76, 274.
397. Herman F. D., Nelson W. K.— J. Am. Chem. Soc., 1952, 74, 2693.
398. Herman F. D., Nelson W. K.— J. Am. Chem. Soc., 1953, 75, 3877.
399. Herman F. D., Nelson W. K.— J. Am. Chem. Soc., 1953, 75, 3882.
400. Hancock A., Sidlow R.— J. Oil and Colour Chemists Assoc., 1952,
35, 28.
401. H a n i c F.— Chem. Listy, 1955, 49, 370.
402. Hershenson H. H. Infrared Absorption Spectra Index for 1945—1957.
Academ. Press Inc., N. Y., 1959.
403. Hamilton P. M., M с В e h t R., Bekebrede W., S i s -
1 e r H. H.— J. Am. Chem. Soc., 1953, 75, 2881.
404. Haase W., Hoppe H.— Acta crystallogr., 1968, B24, 281.
405. Henrici-Olive G., Olive S.—Ang. Chem., 1968, 80, 796.
406. Henrici-Olive G.— J. Organometall. Chem., 1969, 16, 339.
407. Henrici-Olive G., Olive S.— Chem. Commun., 1969, 113.
408. Henrici-Olive G., Olive S.— Ang. Chem., 1968, 80, 796.
409. Henrici-Olive G., Olive S.— J. Organometall Chem., 1969,
19 309
410. Henrici-Olive G., Olive S.— Makromol. Chem., 1969, 121, 70.
411. I s s 1 e i b K-, В a t z G.— Z. anorg. Chem., 1969, 369, 83.
412. I s h i n о T., M i n a m i S.— Kagoku no Byoiki, 1957, 11, 9, 657; C. A.,
1959, 192, 657.
413. I b e r s J. A.— Nature, 1963, 197, 686.
414. I s h i n о T., M i n a m i S., A s a d a S.— Technol. Rept. Osaka Univ.,
1955, 5, 195; C. A., 1956, 11940.
415. I s h i n о T., M i n a m i S.— Technol. Rept. Osaka Univ., 1953, 3, 357;
C. A 1955 702
416. I s h i n о T., M i n a m i S.— Technol. Rept. Osaka Univ., 1957, 7, 439;
C. A., 1959 962.
417. Jennings J. S., W a r d 1 a w W., Way W. J. R.— J. Chem. Soc.,
1936, 637.
418. J a c i n i G.— Olii miner., sgrasi e saponi, colori e vernici, 1953, 30, 193.
419. Jones R. G., Gilman H.— Chem. Rev., 1954, 54, 835.
420. J af fe H. H., D о a к G. O.—J. Chem. Phys., 1953, 21, 196.
421. J a n z G. J., D a n у 1 u к S. S.— J. Am. Chem. Soc., 1959, 81, 3848.
422. J о m u s R. G.— Iowa state Col. J. Sci., 1942, 17, 88; C. A., 1943, 3400.
423. James B. D., W о 1 1 b r i d g e M. G.— J. Inorg. Nucl. Chem., 1966,
28, 2456.
424. James B. D., N a n d a R. K., W a 1 1 b r i d g e M. G.— Inorg.
Chem., 1967, 6, 1979.
425. Jensen A., Jorgensen E.— J. Organometall. Chem., 1967, 7, 528.
426. J agues A., Claude D.— Compt. Rend., 1967, C264, 2156.
427. К 1 an b er g F., Muetterties E. L., Guggenberger L. J.—
Inorg. Chem., 1968, 7, 2272.
428. Kraitzer I., McTaggart M., Winter G.— Austral. Dept.
Munitions, Paint Notes, 1947, 2, 348.
429. Kakos G. A., Winter G.— Austral. J. Chem., 1968, 21, 793.
430. К 6 p f H., S c h n i d t M.— Z. anorg. Chem., 1965, 340, 139.
431. К 6 p f H., Block B.— Z. Naturwis., 1968, 23b, 1534.
84
432. К 6 p f Н., Block В.— Z. Naturwis., 1968, 23b, 1536.
433. К б р f Н., Block В.— Вег., 1969, 102, 1504.
434. Кб р f Н.— Вег., 1969, 102, 1509.
435. Kapoor R. N., Mehrotra R. С.— Chem. Ind., 1966, 1034.
436. Kambara S., Okamoto S.— J. Chem. Soc. Japan. Ind. Chem. Sec.,
1950 53 370
437. King — Bull. Soc. Chim., 1898, 19, 190.
438. Kr iegsmann H., Licht K.— Z. Elektrochem., 1958, 62, 1163.
439. Kronstein M.— Praint and Varnish Production, 1950, 30, 10, 20.
440. К r a i t z e r I., M cT ag g a г/t K-, Winter G.— J. Oil and Colour
Chemists Assoc., 1948, 31, 410.
441. Kraitzer I., McTaggart K-, Winter G.—J. Counsil Sci.
Ind. Research., 1948, 21, 328.
442. Kraitzer I., McTaggart K-, Winter G.— Australia, Dept,
Munitions, Paint Notes, 1947, 2, 304; C. A., 1948, 1433.
443. Kraitzer I., McTaggart K-, Winter G.— J. Oil and Colour
Chemists Assoc., 1948, 31, 405.
444. Kapoor R. N., P a n d e К- С., M e h г о t r a R. C. — J. Ind. Chem.
Soc., 1958, 35, 157.
445. Lengyel B., G a r a i T.— Maguar kemiai folyoirat, 1953, 50,
343.
446. Langkammerer С. M. Пат. CHIA № 2621193; C. A.. 1953, 10549.
447. Levi L.— Ann. Chim. Phys., 1892, 25, 433.
448. Long W. P.— J. Am. Chem. Soc., 1959, 81, 5312.
449. Long W. P., Br eslaw D. S.— J. Am. Chem. Soc., 1960, 82, 1953,
450. Long W. P., N о r d i о U.— Chim. Ind., 1965, 47, 593.
451. Laszlo K., Artur S., Jozsel O.— Maguar kem. folyoirat, 1968,
74, 281.
452. Latham R.— Diss. Abstr., 1960, 22, 3386; C. A., 1963, 57, 4775.
453. Luchinskii G. P., Altmann E. Z.— Z. anorg. Chem., 1935,
255, 321.
454. Locke J., McClevecty J. A.— Inorg. Chem., 1966, 5, 1157.
455. Langford С. H., Ap lington I. P.— J. Organometall. Chem., 1965,
4, 271.
456. L a p u t t e R., G а у о t A.— Makromol. Chem., 1969, 129, 250.
457. Mehrotra R. C.— J. Indian Chem. Soc., 1961, 38, 509.
458. Mehrotra R. C., Agrawal M. M.— J. Chem. Soc., 1967, A, 1026.
459. Mehrotra R. С., M i s г a R. A.— J. Indian Chem. Soc., 1968, 45, 656.
460. Mehrotra R. C.— J. Indian Chem. Soc., 1953, 30, 731.
461. Mehrotra R. C-— J. Am. Chem. Soc., 1954, 76, 2266.
462. Mehrotra R. C.— J. Indian Chem. Soc., 1955, 32, 759.
463. McHatton L. P., Sou lai M. J.—Chem. Ind., 1953, 1337.
464. M i n a m i S., I s h i n о T.— J. Chem. Soc. Japan. Ind. Chem. Sec., 1955,
58, 36.
^65. M i n a m i S., Takano H., I s h i n о T.— J. Chem. Soc. Japan. Ind.
Chem. Sec., 1957, 60, 1406.
466. M i n a m i S., A к i у a m a T., I shi no T.— J. Chem. Soc. Japan. Ind.
Chem. Sec., 1955, 58, 32.
467. M i n a m i S., I s h i n о T.— J. Chem. Soc. Japan. Ind. Chem. Sec., 1956,
61, 66.
468. Metalorganic Compouds. Am. Chem. Soc., N. Y., 1959.
469. Maskill R.. Pratt J. M.— Chem. Communs, 1967, 950.
470 Maskill R., Pratt J. M.— J. Chem. Soc., 1968, A, 1914.
471. Matsuzaki K. Yasukava T.—J. Organometall. Chem., 1967, 10, 9.
472. M u r r a u J. G.— J. Am. Chem. Soc., 1961, 83, 1287.
473. Murray G. G.— J. Am. Chem. Soc., 1959, 81, 752.
474 Majdik F., Pfeifer G.— Amgyar kem, folyoirat, 1963, 69, 280.
475. Majdik F., Pfeifer G.— Proc. Sympos. Coordinat. Chem. Tinany,
Hungary 1964, Budapest, 1965, 317.
85
476. Muller J., Thiele К.- H.— Z. anorg. Chem., 1968, 362, 120.
477. Marconi W., Santostasi M. L., Madle M.— Chim. Ind.,
1962, 44, 229, 235.
478. Malatesta A.— J. Polymer Sci., 1962, 51, 45.
479. M e j z 1 i к J., К v i z M., P r i 1 у m M., Vesely K.— Chem. pru-,
mysl, 1965, 15, 85.
480. Martin H., V о h w i n к e 1 F.— Ber., 1961, 94, 2416.
481. Martin A. H., J e 1 1 i и e к F.— J. Organometall. Chem., 1967, 6, 293.
482. Martin R. L., Winter G.— J. Chem. Soc., 1965, 4709.
483. Martin W., Gruehn R., Schafer H.— J. Solid State Chem.,
1970, 1, 425.
484. Martin R. L., Winter G.— Nature, 1960, 188, 313.
485. Noth H., Harlwimmer R.— Ber., 1960, 93, 2238.
486. Not les J. G., Ker к G. I. M.— Rec. Trav. Chim. Pays-Bas., 1962,
81, 39.
487. N a t t a G., M a z z a n t i G., P r e g a g 1 i a G.— Gazz. Chim. ital.,
1959, 89, 2065.
488. Natta G., M a z z a n t i G., Carrodini P., Giannini U.,
C e s c a S. — Atti. Acad. Lincei Rend. cl. sci. fis., mat., natur., 1959, 26, 150.
489. N a t t a G., Pino P., M a z z a n t i G., Giannini U.— J. Am.
Chem. Soc., 1957, 79, 2975.
490. Natta G., Mazzanti G., Longi P., Bernardini F.— J. Po-
limer, 1958, 31, 181.
491. Natta G., Mazzanti G.— Tetrahedron, 1960, 8, 86. r
492. Natta G.— Ang. Chem., 1959, 71, 205.
493. Natta G., Pino P., Mazzanti G., G i a n n i n i U.— J. Inorg.
Chem., 1958, 8, 612.
494. Natta G., Corrodini P., Bassi I.— J. Am. Chem. Soc., 1958, 80,
755.
495. Pfordten O.— Lieb. Ann., 1884, 237, 201.
496. Prasad S., Tripathi J. B.— J. Ind. Chem. Soc., 1958, 35, 177.
497. Pilcher G., Skinner H. A.— J. Inorg. Nucl. Chem., 1958, 7, 8.
498. Post H. W. The Chemistry of the Aliphatic Ortoesters. Reinhold Pabishing
Corp., N. Y., 1943.
499. Paterno E., Perqtoner — Ber., 1889, 22, 467.
500. Pande К. C., Mehrotra R. C.— Z anorg. Chem., 1957, 290, 87.
501. Pande К. C., Mehrotra R. C.— Chem. Ind., 1958, 1198.
502. Pande К- C., Mehrotra R. C.—Chem. Ind., 1957, 114.
503. Pande К. C., Mehrotra R. C.— J prakt. Chem., 1957, 5, 101.
504. Prasad S., Srivastava R. C.— J. Indian Chem. Soc., 1962, 39, 9.
505. Pande К- C., Mehrotra R. C.— Z. anorg. Chem., 1957, 291, 97.
506. Pande К. C., Mehrotra R. C.— Z. anorg. Chem., 1957, 290,95<
507. P a n i к I. M., Sullivan W. F., Jacobsen A. E.— Am.
Dyest. Rep., 1950, 39, 509; C. A., 1950, 44, 9157.
508. Pandit S. K-, N e r i f к a r P. G., Gupta I.— Indian J. Chem.,
1967, 5, 121.
509. Puri D. M., Pande К. C., Mechrotra R. S.— J. Less-Common
Metals, 1962, 4, 393.
510. Puri D. M., Pande К. C., Mechrotra R. S.— J. Less-Common
Metals, 1962, 4 481
511. Puri D. M., Mehrotra R. C.— J. Indian Chem. Soc., 1962, 39, 499.
512. Puri D. M., Mehrotra R. C.— J. Less-Common Metals, 1961, 3, 247.
513. Puri D. M., Mehrotra R. C.— J. Less-Common Metals, 1963, 5, 2.
514. Puri D. M., Mehrotra R. C.— J. Indian Chem. Soc., 1962, 39, 447.
515. Rauch M.— Inorg. Chem., 1964, 3, 300.
516. Reeves R. E., Mazzeno L. M.— J Am. Chem. Soc., 1954, 76,
2533.
517. R as u w a j w G. A., Bobinova L. M., V. S. E t 1 i s — Tetrahe-
dron, 1959, 6, 154.
86
518. R a 1 e a R., Un guren as u C.,C 1 h о d ar у St.— Rev. roum. chim.,
1967, 12, 861.
519. Rinse J., Bokhout B.— Chim. peint., 1949, 12, 170.
520. Reid A. F., W a 1 1 e s P. C.— J. Organometall. Chem., 1964, 2, 329.
521. Rust J. В., T a к i m о t о H. H.— J. Organ. Chem., 1961, 26, 2467.
522. Rohl H., Lange E., Gossl T., Roth G.— Ang. Chem., 1962,
74 155.
523. Reid A. F., W a 1 1 e s P. C.— Austral. J. Chem., 1965, 18, 9.
524. Rooney R. C.— Anal. Chim. Acta, 1958, 19, 428.
525. Rinse J., Bokhout B.— Chim. reintres, 1949, 12, 170; C. A., 1949,
7239.
526. Rosenheim A., Loewanstamm W., Singer L.— Ber., 1903,
36, 1833.
527. Rosenheim A., SchabelR., Bilecki R.—Ber., 1915, 48, 447.
528. Sinn H., P at a t F.— Ang. Chem., 1964, 3, 98.
529. Stahley R. H.— Catalys in Ind., 1965, 1, 29.
530. Segal C. L., T a к i m о t о H. H., Rust J. B.— Am. Chem. Soc.,
Div. Polymer Chem. Preprints, 1960, 1, 34.
531. Saxena U. B.— Indian J. Chem., 1967, 5, 368.
532. Stern R., H i 1 1 i о n G., S a j u s Z.— Tetrahedron Letters, 1969,
1561.
- 533. Sartori P., Weidenbruch M.— Ber., 1967,100, 2049.
534. S u к a t a K., Jo к a g a w a K., Nakao C., Adzuma K-— J. Chem.
Soc. Japan. Ind. Chem. Sec., 1965, 68, 1248.
535. SloanC. L., Barber W. A.— Chim. Ind., 1960, 42, 877.
536. S i e g e r t F. W., M e i j e r H. J., d e L i e f d e— J. Organometall. Chem.,
1969, 20, 141.
537. Samuel E.— J. Organometall. Chem., 1969, 19, 87.
538. Solzmann J. J.— Helv. Chim. Acta., 19€8, 51, 526.
539. S c h i a v a n G., P a r a d i s i C.— Ricerca Sci., 1967, 37, 337.
540. Skapski A. C., Troughton P. G. H., Sutherland H. H.—
Chem. Communs, 1968, 14, 18.
541. Summers L., Uloth R., Holmes A.— J. Am. Chem. Soc., 1955,
77, 3604.
542. SloanC. L., Barber W. A.— Am. Chem. Soc., 1959, 81, 1364.
543. Schumann A.— Ber., 1888, 21, 1079.
544. Summers L., Uloth R. H.— J. Am. Chem. Soc., 1954, 76, 2278.
545. S c a g 1 i a r i n i G., T a r t a r i n i G.— Atti Line.., 1926, 4, 318.
546 SchlaferH. L, Go tz R.— Z. phyc Chem., 1964, 41, 95.
547. Schmidt F.— Ang. Chem., 1952, 64, 536.
548. Sachs G.—Verfkaniek, 1956, 29, 140; C. A., 1956, 16134.
549. Sachs G., Werther F.— Farbe und Lack., 1955, 61, 60.
550. Speer R. J.— J. org. Chem., 1949, 14, 655.
551. Speer R. J., Carmody D. R.— Ind. Eng. Chem., 1950, 42,
251.
552. S i d 1 о w R.— J. Oil and Colour Chemists Assoc., 1958, 41, 577.
553. S i d 1 о w R.— Dyer, 1958, 120, 310; РЖХим., 1959, 3734.
554. Sidlow R. S.— Chem. Products., 1953, 16, 215.
555. Schmidt F.— Ang. Chem., 1952, 64, 536.
556. SakuradaY., Huggins M. L., Anderson W. R.— J. Phys.
Chem., 1964, 68, 1934.
557. Schwartz D., Johnson C., Ludwig J., Morris M. L.—
J. Inorg. Nucl. Chem., 1964, 26, 2025.
558. Shiihar a I., Schawartz W., Post H. W.— Chem. Pev., 1961,
61, 1.
559. SubbaraoT. V.— Oils and Oilseeds J., 1953, 5, 109.
560. Treichel P. M., Chaudharb M. A., Stone F. G. A.— J. Or-
ganometall. Chem., 1963, 1, 98.
561. Teichner S.— Compt. Rend., 1953, 238, 900.
87
562. Takeshi V., Ichiro K-, M i у о к а О.— Bull. Chem. Soc. Japan,
1961, 34, 459.
563. T a к a t a n i T.— Bull. Chem. Soc. Japan, 1957, 30, 7, 705.
564. Takatani T., YoshinoT., Mashiko Y.— J. Chem. Soc. Japan.
Ind. Chem. Sec., 1957, 60, 1382.
565. Thomas A.— Peinteres, pigments, vernis, 1952, 28, 457; C. A., 1953, 4102.
566. Такэтами И., Судзуки T., Фудзии М.— J. Chem. Soc. Japan.
Ind. Chem. Sec., 1968, 71, 1544.
567. T a k e g a m i Y., Suzuki T., Fuchizaki Y.— Bull. Chem. Soc.,
Japan, 1968, 41, 2637.
568. T а к э д a M., Ч им у p а К., К о й д э Н.— J. Chem. Soc. Japan. Ind.
Chem. Sec., 1968, 71, 563.
569. Takashi Y.— Bull. Chem. Soc. Japan, 1967, 40, 1194.
570. T акесита T., Мияути H.— J. Chem. Soc. Japan. Ind. Chem. Sec.,
1965, 68, 1721.
571. Thomas L. H., Davies G. H.— J. Chem. Soc., 1969, A, 1271.
572. T k a c A.— Coll. Czechosl. Chem. Communs, 1968, 33, 1629.
573. T k a c A.— Coll. Czechosl. Chem. Communs, 1968, 33, 2004.
574. T k as A.— Coll. Czechosl. Chem. Communs, 1968, 33, 3001.
575. T h i e 1 1 К.- H., Muller J. —Z. anorg. Chem., 1968, 362, 113.
576. U e 1 z m a n n H.— J. Polymer Sci., 1958, 32, 457.
577. Ungurenasu C., Cecal A. A.— J. Inorg. Nucl. Chem., 1969,31,
1735.
578. de Vries L.—Rec. Trav. Chim. Pays-Bac., 1961, 80, 866.
579. Varma I. D., Mehrotr a R. C.— J. Less-Common Metals., 1961,
3 321.
580. Varma I. D., Mehrotra R. C.— J. prakt. Chem., 1959, 8, 64, 235.
581. Varma I. D., Mehrotra R. C.— J. Less-Common Metals, 1959, 1,
263.
582. W a t e n a b 1 A., S h i о z a w a K., Y a g u r a F.— J. Chem. Soc.
Japan. Ind. Chem. Sec., 1959, 62, 1081.
583. W a r d 1 a w W., В r e d 1 e у D. C.— Endeavour, 1958, 14, 140.
584. W a r d 1 a w W.— J. Chem. Soc., 1956, 4004.
585. W a r d 1 a w W., В r a d 1 e у D. C.— Research., 1950, 3, 462.
586. Winter G.— Australia, Dept. Supply. Defence Standards Lab. Rept.,
1953, 191, 10; C. A., 1956, 11028.
587. Winter G. —Australia, Dept. Supply. Defence Standards Lab. Rept.,
1953, 191, 1; C. A., 1956, 11027.
588. Winter G.— J. Oil and Colour Chemists Assoc., 1953, 36, 694.
589. Winter G.— Australia, Dept. Supply, Paint Notes, 1950, 5, 285; C. A.,
1951 4059.
590. Winter G.— J. Oil and Colour Chemists Assoc., 1953, 36, 689.
591. Winter G.— J. Oil and Colour Chemists Assoc., 1953, 36, 701.
592. Winter G.— J. Oil and Colour Chemists Assoc., 1951, 34, 30.
593. Winter G., Martin R. L.— J. Chem. Soc., 1961, 2947.
594. Winter G., Martin R. L.— Nature, 1963, 687, 197.
595. Wilkinson G., P a u s о n P. L., Birmingham J. M., С о t-
t on F. A.— J; Am. Chem. Soc., 1953, 75, 1011.
596. W i h r 1 i n E., G i r a u d E.— Compt. Rend., 1877, 85, 288.
597. Wilkinson G., Birmingham J. M.— J. Am. Chem. Soc., 1954,
76, 4281.
598. Wilkinson G., Cotton F. A.— Chem. Ind., 1954, 307.
599. Weber H., Ring W., Hochmuth U., Franke W.— Lieb.
Ann. Chem., 1965, 681, 10.
600. Witters R. D., Caughlan C. N.—Nature, 1965, 205, 1312.
601. Witters R. D., C a u g h 1 a n C. N.— J. Inorg. Chem., 1965, 4,
689.
602. Wright D. A., Williams D. A.— Acta crystallogr., 1968, B24,
1107.
88
603. Weingarten H., Miles M. G., E d e 1 m a n n N. K.— Inorg.
Chem., 1968, 7, 879.
604. Weingarten H., Van Wazer J. R.— J. Am. Chem. Soc., 1965,
87, 724.
605. Watenpaugh K-, Caughlan C. N.—J. Inorg. Chem., 1966, 5, 1782.
606. Watenpaugh K., Caughlan C. N.—Chem. Communs, 1967, 76.
607. Watenpaugh K-, Caughlan C. N.— J. Inorg. Chem., 1967, 6, 963.
608. Yoshida I.— Kagaku no Ryoiki, 1951, 5, 414.
609. YoshinoT., К i z i m a I. et al. — J. Chem. Soc. Japan, 1957, 60, 1124.
610. YoshinoT., Kizimal., Sugiyamal., Kanazawa M.—
J.Chem. Soc. Japan, Ind. Chem. Sec., 1962, 64, 1182.
611. Yoshino T., Kijima I., M as u da Y., Sugiyama I.—J. Chem.
Soc. Japan, Ind. Chem. Sec., 1959, 62, 77.
612. Young R. C.— Inorg. Sinthesis, 1946, 20, 119.
613. Yamamoto A., К a mb ar a S.— J. Am. Chem. Soc., 1957, 79, 4344.
614. Yamamoto A., Kamb ar a S.— J. Am. Chem. Soc., 1959, 81, 2663.
615. Yamamoto A., Ookawa M., Ikeda S.— Chem. Communs, 1969,
841.
616. Yamamoto A., К a mb ar a S.— J. Chem. Soc. Japan Pure Chem.
Sec., 1959, 80, 1239.
617. Yamamoto A., Kambara S.— J. Inorg. Nucl. Chem.,pl961, 21, 58.
618. Yosino N., Yosino T.— J. Chem. Soc. Japan. Ind. Chem. Soc.,
1968, 71, 1025.
619. Zigler K-, Martin H., Stedefeder J.—Tetrahedron Letters, 1959,
12.
620. Z e i t 1 e г V. A., Brown C. A.— J. Phys. Chem., 1957, 61, 1174.
621. Ziegler K-, Martin H.— Makromol. Chem., 1956, 18—19, 186.
622. Zeitler V. А., В г о w n C. A.— J. Am. Chem. Soc., 1957, 79, 4618.
623. Zeitler V. A., Brown C. A.— J. Am. Chem. Soc., 1957, 79, 4616.
ГЛАВА II
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ ТИТАНА
Будучи переходным металлом, титан образует большое число ком-
плексных соединений. В качестве лигандов в комплексных соеди-
нениях титана могут быть молекулы воды, перекиси водорода, эфи-
ров, спиртов, ионы многих неорганических и органических кислот
и органических оснований, молекулы органических кислот, содер-
жащие полярные группы, и др.
Комплексы титана могут быть нейтральными и заряженными
частицами. Нейтральные комплексы обычно удается выделить из
растворов в виде индивидуальных веществ препаративными мето-
дами. Заряженные комплексы образуются в результате диссоциации
нейтральных частиц и могут существовать только в растворах. Со-
став их устанавливается физико-химическим анализом, а также в
результате спектрофотометрических, потенциометрических, кондук-
тометрических и прочих исследований. Важнейшими характеристи-
ками комплексных частиц, диссоциирующих в водных растворах,
являются константы нестойкости (диссоциации) или устойчивости
(образования). Комплексы, подвергающиеся ступенчатой диссоци-
ации, характеризуются константами ступенчатой диссоциации.
В водных растворах титан проявляет склонность к комплексо-
образованию не только с неметаллическими аддендами, но и с иона-
ми переходных металлов (например, с железом-П, железом-Ш,
хромом-Ш), вероятно, в результате непосредственного перекрыва-
ния d-орбиталей.
В качестве центрального иона в водных растворах фигурируют
ионы трех- и четырехвалентного титана. При образовании титано-
вых комплексов в водных растворах могут принимать участие мо-
лекулы воды и продукты ее диссоциации (ионы ОН- и О2~), явля-
ющиеся конкурирующими лигандами. Вследствие этого с помощью
обычных методов определения состава комплексов достоверно уста-
навливается не их истинный состав, а только соотношение между
металлом и лигандами, входящими в состав комплекса. Участие
воды и продуктов ее диссоциации в комплексообразовании приводит
к ионам титана в виде гидратов или оксигидратов, например
Ti (ОН)3+, Ti (ОН)!+, Ti (OH).t, TiO2+, TiO (OH)+.
Нейтральные комплексы титана, образующиеся при взаимодей-
ствии галогенидов, сульфатов и других соединений титана, рассмот-
рены нами в соответствующих главах первой части книги. В на-
90
стоящей главе рассматриваются преимущественно титановые ком-
плексы, существующие в растворах, образованные при взаимодей-
ствии ионов титана с лигандами.
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ С НЕОРГАНИЧЕСКИМИ
ЛИГАНДАМИ
АКВОКОМПЛЕКСЫ
г
Ионы титана в водных растворах гидратированы. Состав акво-
комплексов ионов четырехвалентного титана изучен еще недоста-
точно. О существовании их можно только предполагать, исходя из
существования кристаллогидратов.
Ионы трехвалентного титана в водных растворах удерживают
по шесть координированных молекул воды [Ti (Н2О)6]3+. Существо-
вание гексакомплексов доказывается спектрами поглощения [228,
234] и измерением упругости пара над растворами гексагидратов,
например TiCl3 • 6Н2О [64]. Молекулы воды во внутренней сфере
аквополикомплексов титана могут замещаться молекулами спирта
[228], ионами С1“, ОН- [227], F- [93] и др.
По результатам спектрофотометрических исследований [43],
в солянокислых растворах при концентрации НО 0,1—10 моль!л
ионы Ti3+ находятся преимущественно в форме аквокомплексов
[Ti (Н2О)6]3+ и смешанных аквохлоркомплексов [Ti (Н2О)5С1]2+ и
[Ti (Н2О)4С12]1+, при концентрации НО 12—19 молъ/л — в форме
ионов [Ti (Н2О) С15]2“.
Энергия 2£^->2£^ перехода в [Ti (Н2О)б]3+, поданным расчетов
В приближении теории кристаллического поля [373], равна произ-
ведению дипольных моментов молекул Н2О, связанных с централь-
ным атомом, и функции ф0, зависящей от длины связи Ti ОН2
и величины заряда на атоме титана.
РОДАНИДНЫЕ КОМПЛЕКСЫ
Роданидные комплексы образуют ионы трех- и четырехвалент-
ного титана. Комплексы титана, полученные препаративным мето-
дом, описаны в работах [312, 313, 336, 346]. В настоящей главе опи-
саны роданидные комплексы титана, существующие в водных рас-
творах.
Деляфосс [184] методом спектрофотометрии в разбавленных вод-
ных растворах роданида титана установил существование только
одного иона, имеющего состав [Ti (ОН) (CNS]2+. А. К. Бабко
и М. М. Тананайко [19] обнаружили комплекс с отношением
Ti : CNS =1:3, константа образования которого (температура
20° С, ионная сила 1)
Д = [Ti <CNS>31±_ = 7 8 . 10\
[Ti4+][CNS~]
91
В системе титан — роданид — диантипирилметан образуются
комплексы тройного состава с отношением компонентов 1:4:1.
Этанол является конкурирующим лигандом по отношению к комп-
лексам титана с роданидом и диантипирилметаном [21]. В присут-
ствии этанола окраска таких комплексов титана ослабляется.
В сернокислых водных и метанольных растворах с концентраци-
ей 0,5—5,0% H2SO4 спектрофотометрическим методом установлено
образование комплексов Ti (IV) и SCN~ с отношением компонен-
тов 1 : 1 и 1 : 2 [205]. При избытке Ti (IV) в воднометанольных рас-
творах обнаружено присутствие полиядерных комплексов.
Органические основания [7] и кетоны [95] в качестве внешне-
сферных ионов способствуют упрочнению гексароданидных комплек-
сов титана состава R2 [Ti (CNS)6], где R — органическое основание
[7]. Спирты препятствуют комплексообразованию титана с родани-
дом, вытесняя роданид-ион из внутренней сферы комплекса.
Гексароданидные комплексы титана хорошо экстрагируются
из водных растворов органическими экстрагентами. Экстракция
из сернокислых растворов этиловым эфиром вызывает заметное раз-
деление изотопов [44]. Найдены следующие коэффициенты изотоп-
ного разделения: а = Ti50/Ti48 = 1,006 ± 0,004; а = Ti50/Ti46 =
= 1,014 ± 0,007, а = Ti46/Ti48 = 1,008 ± 0,003.
ЦИАНИДНЫЕ КОМПЛЕКСЫ
Из водных растворов TiCl3 и KCNS при энергичном перемеши-
вании получен двойной цианид титана и калия K3[Ti (CN)6] в виде
синего творожистого осадка [229]. При избытке цианистого калия
образуется комплекс K3[Ti (CN)6] • KCN. Шлефер и Готц [344, 3451,
действуя цианистым калием на раствор TiBr3 в жидком аммиаке,
получили комплес K3[Ti (CN)6] • 2KCN.
Двойные цианиды титана и калия — парамагнитные вещества,
так как содержат один неспаренный электрон на Зб/-орбитале. При
длительном соприкосновении с воздухом или с водой цианиды трех-
валентного титана окисляются и образуется комплекс со смешан-
ной валентностью титана состава K3Ti (CN)6 • K2Ti (CN)6. Окислен-
ные цианиды титана легко гидролизуются водой до гидроокиси
титана.
ХЛОРИДНЫЕ КОМПЛЕКСЫ
Методами спектрофотометрии, потенциометрии и кондуктомет-
рического титрования изучено комплексообразование ионов СГ~
с Ti (BF4)3 в пропандиол-1,2-карбонате и триметил фосфате [202].
Обнаружено наличие координационных форм [TiCl]2'4" и [TiClJ3
в первом растворителе, [TiCl2]+—во втором, [TiCl3]° и [TiCl4]~ —-
в обоих растворителях.
92
।
СУЛЬФАТНЫЕ КОМПЛЕКСЫ
Состояние титана (IV) в сернокислых растворах исследовано еще
недостаточно, что затрудняет изучение комплексных соединений,
образуемых его ионами с сульфогруппами. Кроме того, исследование
комплексообразования осложняется также и тем, что сульфатные
комплексы, будучи малостойкими образованиями, упрочняются под
влиянием ионов — внешнесферных заместителей. Состояние ион-
ных равновесий сульфата титана в водных растворах описывается
сложной схемой. Из нее трудно выделить одно или несколько звень-
ев, которые можно было бы рассматривать как независимые про-
цессы. При изучении комплексообразования титана в сернокислых
растворах исходят,-обычно, из упрощенных представлений, и по-
этому полученные данные носят сугубо ориентировочный характер.
Методами ионного обмена и ионообменной экстракции раствором
три-^-октиламина показано существование в сернокислых растворах
с концентраций 0,1—8,0 моль/л H2SO4 сульфатных комплексов со-
става [TiOSO4]°, [TiO (SO4)2]2~ и [Ti (SO4)3]2“ [8]. Константы нестой-
кости их при 20° С и ионной силе 8 равны соответственно /С1об =
= 5,9 • 10~3, = 3,8 • 10“2, . = 3,8 . 1(Г12.
В сернокислых растворах образуются только два сульфатных
комплекса: [TiOSO4]° и [TiO (SO2)2]2—, что доказано спектрофотомет-
рическим методом [29]. Для электронейтрального комплекса найде-
ны константы нестойкости при ионной силеЗ, 4 и 5, равные 7,1 • 10~3,
5,5 • 10-3 и 3,4 • 10~3 соответственно. Значение термодинамической
константы равно (3,0 ± 0,4) • 10-3, константа нестойкости двух-
зарядного комплекса при ионной силе 4 составляет 3,3 • 10—2.
Синтезированы сульфатные комплексы титана со смешанными
лигандами [207]. Действием на TiCl4 диметилсульфата в отношении
1:2 получен желтый гигроскопичный порошок, растворимый в
спиртах, состава CITi (CH3SO4) (SO4). При действии на него бензола,
тимолового спирта, салицилового альдегида, ванилина, метилса-
лицилата, ацетилацетона и дибензоилметана происходит замещение
атома хлора на остаток RO с образованием комплексов состава
ROTi (SO4) (CH3SO4).
Действием диметилсульфата на титановые - дихлорэфиры
(RO)2TiCl2 получены окрашенные комплексы (RO)2Ti (CH3SO4)2
и (RO)3Ti (CH3SO4). По реакции диметилсульфата с (CH3CO)2TiCl2
синтезировано комплексное соединение TiO (CH3SO4)2 в виде белого
порошка, растворимого в спирте.
БОРНОКИСЛЫЕ КОМПЛЕКСЫ
Борнокислые комплексы специально еще не изучались. Однако
анализ спектров ЭПР перлов буры указывает на то, что ионы Ti3+
входят в состав октаэдрических комплексов с очень сильным ани-
онным искажением [147].
93
АЗИДОКОМПЛЕКСЫ
В неводных растворах (ацетонитриле, пропандиол- 1,2-карбонате
и триметил фосфате) обнаружены азидокомплексы титана состава
lTi(N3)2]+, [Ti (N3)a]°, [Ti(N3)e]3-, [Ti(Ns)J- [201 [.
МОЛИБДАТНЫЕ И ФОСФОРМОЛИБДАТНЫЕ КОМПЛЕКСЫ
В результате взаимодействия молибдата аммония (NH4)2MoO4
с гексафтортитанатом аммония (NH4)2TiFe в солянокислой среде
получены золотисто-желтые кристаллы состава (NH4)4TiO4(MoO3)12-
. пН2О [113, 114, 251].
При добавлении раствора (C5H5)2Ti (SC6H5)2 к суспензии
(CH3CN)2 Мо (СО)4 в хлороформе образуется титанмолибденовый
комплекс состава (C5H5)2Ti (SC6H5)2Mo (СО)4 [250]. Аналогичным
методом синтезированы комплексные соединения состава
(C5H5)2Ti (SeC6H5)2Mo (СО)4 и 4uc-[(C5H5)2Ti (ОСбН5)2] • Мо (СО)2.
В растворе бензола первые два комплекса имеют мономерное строе-
ние. Структура титанмолибденовых комплексов характеризуется
наличием двух мостиковых групп (между атомами титана и молиб-
дена) состава ЭС6Н5, где Э = S, Se и О.
По данным кислотно-основного титрования аммонийная соль
титанмолибденовой кислоты является четырехзамещенной средней
солью, титанмолибденовая кислота соответственно — четырех-
основной кислотой [112].
Смешение растворов, содержащих Ti (IV), МпО^ и POl- при
pH ~ 1,5, приводит к образованию устойчивой титанфосфорномо-
либденовой кислоты с отношением компонентов 1 : 1 : 12 [301].
Действием водных растворов аммиакатов и аминатов Со3+ на фос-
фор нотитанмолибденовую кислоту [TiPMo12] выделены комплексы
состава (CoL)rt (TiPMo12)m, где L, п и т соответственно (NH3)6, 3 и 2;
NH2CH2CH2NH2, 3 и 2; (NH3)5NO2, 2 и 1; [NH2CH2CH2NH2 • NO2]2,
4 и 1; (NH2CH2CH2NH2 • Cl2)2, 4 и 1. Основность комплекса равна
примерно 4. Константа нестойкости фосфорнотитанмолибденовой
кислоты составляет 1,2 • 10“5 [82].
Методом катионного обмена из аммонийной соли получена ге-
терополимолибденовая кислота.
ПЕРЕКИСНЫЕ КОМПЛЕКСЫ
В водных растворах с перекисью водорода титан дает перекисные
комплексы. Если наряду с перекисью водорода в растворе содержатся •
другие лиганды, образуются смешанные комплексы, содержащие не-
94
сколько лигандов. Многие перекисные комплексы титана получены
препаративными методами в чистом виде. Перекисные комплексы
титана, относящиеся к соответствующим классам соединений, рас-
смотрены нами ранее при описании отдельных классов соединений
в первой части книги. В настоящей главе рассматриваются перок-
сокомплексы титана, существующие в растворах, и смешанные
комплексы.
В водных растворах перекись водорода, будучи слабой кислотой,
подвергается ступенчатой диссоциации по реакциям:
н2о2 = ног + н+
ног = оГ + н+
Анионы НОГ и О2-, а также молекулы недиссоциированной
перекиси водорода могут быть лигандами при образовании пере-
кисных комплексов титана..
В большинстве работ, посвященных исследованию перекисных
комплексов титана, устанавливается только количественное соот-
ношение между ионами титана и молекулами перекиси водорода
в составе комплекса. Относительно строения комплексов и природы
лигандов высказываются только предположения.
Образование окрашенных комплексов титана с перекисью водо-
рода наблюдали многие авторы [200, 259, 295, 296, 334, 369, 370].
Веелер [369, 370] предложил использовать перекисные комплексы
для колориметрического определения титана. Этот аналитический
метод широко применяется и в настоящее время. Добавление пере-
киси водорода к кислым растворам солей титана вызывает появле-
ние в зависимости от их концентрации интенсивного окрашивания
от слабо-желтого до оранжевого цвета. В щелочных растворах по-
лучаются бесцветные комплексы титана с перекисью водорода.
Существование перекисных комплексов титана в сернокислых
растворах доказано изучением переноса ионов [332], а также изме-
рением электродвижущих сил [226].
Исследованию состава и строения перекисных комплексов титана
посвящено большое число работ, но выводы различных авторов
плохо согласуются между собой. Причина этого состоит, по-види-
мому, в том, что перекисные комплексы титана имеют сложный
состав. В образовании их кроме перекиси водорода принимают
участие вода, продукты ее диссоциации и кислотные остатки.
В кислых растворах перекисные комплексы титана имеют полосы
поглощения между 350 и 450 нм. Отношение титана к перекиси
водорода в этих комплексах составляет 1:1 [149, 306—308, 333];
константа нестойкости перекисного комплекса (К = [TiO2+] .
.[HAl/lTi (О2)2+1) равна 10~5— 1(Г4 [149, 307, 333].
А. К. Бабко и А. И. Волкова [10] в результате исследова-
ния спектров поглощения установили существование двух видов
95
перекисных комплексов титана: окрашенных в интервале от pH 0,3
до [Н+] = 6-н. и бесцветных при pH > 5. Соотношения между тита-
ном и перекисью водорода в обоих видах перекисных комплексов,
по данным метода изомолярных серий, равно 1:1. Изучая влияние
концентрации водородных ионов на интенсивность окраски, авторы
пришли к заключению, что окрашенные комплексы получаются в
результате присоединения к иону Ti4+ недиссоциированной моле-
кулы перекиси водорода, а в образовании бесцветных комплексов
принимают участие ионы НОГ и О|-. Константа диссоциации окра-
шенного комплекса, по данным авторов, составляет
t
[Ti4+][H2Q2]
[Ti (Н2О2)]4+
= 0,9 • 10“4.
Изучая скорость окисления тиосульфата перекисью водорода в
присутствии титана, А. К. Бабко и В. А. Литвиненко [5] установи-
ли, что последний существует в форме перекисного комплекса с за-
рядом 1+. Константа диссоциации его при pH 2—5
[ТЮ2+] [НОГ] 14
К = 2.......и . 2J_ = о,96 • 1СГ14.
[TiO (НО2)+]
Существование комплекса [TiO (НО2)]+, в состав которого входит
анион НОГ, подтверждается также методом потенциометрического
титрования четыреххлористого титана перекисью водорода в вод-
но-спиртовых растворах [6]. Спектр поглощения этого комплекса
имеет полосу с максимумом при X = 340 нм. Соотношение компонен-
тов в комплексе при pH 2, равное 1:1, установлено по кривым
изомолярных серий. Константа диссоциации комплекса равна
1,4 • 1(Г14.
Данные А. К. Бабко и В. А. Волковой не согласуются с резуль-
татами исследования смешанных пероксидно-оксалатных комплек-
сов титана Пателя и Джере [144], которые обнаружили в комплек-
сах, полученных из кислых растворов, наличие пероксо групп.
В другой работе А. К. Бабко и П. В. Улько [17] установили, что
бесцветные комплексы с отношением Ti : Н2О2 =1:1 существуют
до pH 10—13. В растворах с pH 14 образуются комплексы с отно-
шением Ti : Н2О2 =1:2. Константа диссоциации этого комплекса
по схеме
[TiO (О2)2]2~ = TiO2+ + 2ОГ
составляет 10~.
Методами спектрофотометрии и ионного обмена установлено,
что при pH 2 титан с перекисью водорода образует оранжевый ком-
плекс [Ti (ОН)2(Н2О) (Н2О2)]2+ с максимумом светопоглощения при
Л = 400 нм [261]. Этот комплекс устойчив при комнатной темпера-
туре. Препаративным методом (см. ч. I) получена двойная соль со-
става K2SO4 • [Ti (ОН)2(Н2О) (H2O2)]SO4, содержащая этот комплекс.
96
В интервале pH 3—6 устойчив желтый комплекс
[Ti (ОН)3(Н2О2)]+ с максимумом светопоглощения при X = 330 нм.
Он также получен препаративным методом в виде двойной соли
4K2SO4 • [Ti (OH)3(H2O2)]2SO4. Желтый комплекс менее стоек, чем
оранжевый, и при стоянии разлагается с образованием осадка
TiO (ОН) (ООН).
При pH 7—9 и большом избытке перекиси водорода образуется
светло-желтый комплекс состава [Ti (ОН)3(ООН)] с максимумом
светопоглощения при X = 313 нм. Он также выделен препаративным
методом в чистом виде.
В щелочных растворах с pH > 10 образуется перекисный ком-
плекс [Ti (ОН)2(О2)]2- с максимумом светопоглощения в ультрафио-
летовой области. Препаративным методом получена соль
К2 [Ti (ОН)2(О2)], содержащая этот комплекс.
Комплексы [Ti (ОН)3(ООН)1 и [Ti (ОН)2(О2)2]2“ неустойчивы.
Они при стоянии растворов разлагаются с образованием светло-
желтого осадка состава (HO)2Ti (О2) OTi (ОН)2.
Оптическим методом определено, что желтый перекисный ком-
плекс образуется с соотношением TiO2+ : Н2О2 =1:1 [211]. Рас-
творы солей титана в серной, хлорной, соляной и азотной кислотах
с концентрацией кислот от 0,1 до 1 моль/л и титана 0,0005 г-экв/л
при X = 435 нм обладают одинаковыми коэффициентами молярного
погашения, равными 594. Так как хлорная кислота относится к числу
слабых комплексообразующих агентов, то из этого наблюдения мож-
но сделать вывод, что перекисные комплексы титана в этих условиях
во всех перечисленных средах имеют одинаковое строение и не со-
держат в качестве лигандов кислотных остатков. Опыты по электро-
миграции показали, что перекисные комплексы титана в этих рас-
творах имеют положительный заряд [277].
В разбавленной хлорной кислоте перекисные комплексы титана
имеют две полосы поглощения с максимумами при Л =230 и 410 нм
[215, 289, 371], отношение ТЮ2+ : Н2О2 =1:1. Константа образо-
вания перекисного комплекса титана в 3-н. хлорной кислоте со-
ставляет 103’51 [152]. При pH < 2 константа нестойкости, по дан-
ным [367], равна 0,49 • 10-4.
В 1-мол. сернокислом растворе при концентрации титана
0,00047—0,0012 г-экв/л максимум светопоглощения приходится на
длину волны 410 нм, как и в разбавленной хлорной кислоте [305].
При, большей концентрации титана закон Бера — Ламберна в сер-
нокислых растворах не выполняется. Максимум светопоглощения
с увеличением концентрации титана сдвигается в сторону коротких
волн, а коэффициент молярного погашения перекисного комплекса
уменьшается, так как взаимодействие перекисного комплекса ти-
тана с ионами SO2" в этих растворах отсутствует, то смещение мак-
симума светопоглощения с увеличением концентрации титана
в растворе объясняется полимеризацией перекисных комплексов
7 1-1669 97
титана [305]. Льюис [254] при спектрофотометрическом исследова-
нии перекисных комплексов титана не наблюдал смещения макси-
мума светопоглощения. Поэтому вопрос о полимеризации перекис-
ных титановых комплексов нуждается в дополнительном изучении.
Исследованы спектры светопоглощения перекисных комплексов
титана в 96%-ной серной кислоте [285] и в смеси серной и фосфор-
ной кислот (56% H2SO4 + 10% Н3РО4) [153].
С повышением температуры от 5 до 40° С устойчивость пероксо-
титанового комплекса в хлорной, соляной и серной кислотах умень-
шается, однако коэффициент молярного погашения остается посто-
янным, равным 590 [28]. По приближенной оценке, тепловой эффект
реакции присоединения Н2О2 к Ti4+ составляет 10 ккал/моль.
В сернокислых растворах с концентрацией 16—90% H2SO4 ти-
тан и ниобий совместных пероксидных комплексов не образуют
[104]. При pH 2—5 титан (IV) проявляет каталитическое действие
при окислении иона 1“ перекисью водорода [59], причем скорость
окисления линейно зависит от концентрации титана [60]. Соотно-
шение Ti : Н2О2 в каталитическом комплексе равно 1:1.
СМЕШАННЫЕ ПЕРЕКИСНЫЕ КОМПЛЕКСЫ
Смешанные перекисные комплексы титана образуются в резуль-
тате присоединения к комплексам простого состава многих неорга-
нических и органических лигандов.
Перекисный комплекс титана в водном растворе вступает во вза-
имодействие с фтор-ионом, из-за чего окраска его ослабевает [149].
К- Е. Клейн [46] методом изомолярных серий установила, что в
сернокислых и азотнокислых растворах титан образует с фтор-
ионом соединение, в котором отношение Ti : F = 1 : 1 [46]. Она
полагает, что состав этого иона выражается формулой TiOF+, кон-
станта нестойкости его
[TiO2+] [F~]
[TiOF-]
= 3,6 • IO-7.
В водных растворах титанилфторидный комплекс взаимодействует
с перекисью водорода по реакции
TiOF- + Н2О2 = [TiOH2O2]2+ + F-.
Таким образом, фтор не входит в состав перекисного титанового
комплекса, а разрушает его. Константа равновесия, по определению
К. Е. Клейн, составляет
д' ___ [TiOHgO^] [F ] _ з g К)-3
[TiOF+] [H2OJ
Согласно [152], перекисный комплекс титана с фтор-ионом дает
несколько соединений тройного состава. В растворах с малой кон-
98
центрацией фтор-иона образуется тройной комплекс состава [TiO-
• F • Н2О2]. Он имеет максимум светопоглощения при X = 380 нм,
константа образования его равна 104’22. При избытке фтор-иона
образуется тройной комплекс состава [TiO • (F)2 • Н2О21° с макси-
мумом светопоглощения при X = 365 нм. В растворах с высокой
концентрацией фтор-ионов получаются бесцветные комплексы, со-
став которых авторами [152] не изучен.
По мнению [220], при большом избытке перекиси водорода
устойчивость перекисных комплексов титана не изменяется в при-
сутствии фтор-ионов.
Нихольсон и Рейтер [276] при добавлении к раствору четырех-
хлористого титана в этилацетоне безводной перекиси водорода
получили окрашенный комплекс с отношением Ti : Cl : Н2О2 =
= 1:1:1. Массужели и Пантанелли [262, 263] смещением раствора
гидроокиси титана в щавелевой кислоте с перекисью водорода син-
тезировали смешанный перекисный комплекс, которому приписыва-
ют состав Ti2O3(C2O4)2. Харкер и Патель [242], повторившие опыты
Массужелли и Пантанелли, получили, однако, продукт иного со-
става, выражаемого формулой ТЮ2С2О4 • 3,5Н2О. Авторы полагают,
что препарат Массужелли и Пантанелли состоял из смеси двух про-
дуктов: TiO2C2O4 и TiOC2O4. Харкер и Патель строение тройного
комплекса выражают формулой
Оч /ООС
| >Ti(H2O)2< I . 1,5Н2О.
0х \(ЭОС
Л. И. Дубовенко и Е. И. Герасименко [24] в системе TiCl4—
Н2О2—Н2С2О4 при pH 1—2 установили существование комплекса
с отношением компонентов 1:1:1.
Джери и Патель [238, 290] изучили строение комплекса титана
с помощью ИК-спектров и нашли, что препарат содержал три моле-
кулы воды. Он имеет две полосы поглощения при частоте 1050 и
880 см~\ характерные для пероксогруппы титана. Наличие этих
полос указывает на неионный характер связи Ti—О2“ и нелинейное
строение пероксотитановой группы. Неустойчивость группы О2~,
оранжево-красный цвет комплекса, положение полос поглощения
в ИК-спектре и термическая неустойчивость, по мнению авторов,
свидетельствуют в пользу бидентатного присоединения пероксо групп,
образующих в комплексах трехчленные циклы. В спектре поглоще-
ния оксалатно-перекисного комплекса титана имеется широкая по-
лоса поглощения в области X = 370—410 нм, которая вызвана пе-
реходом несвязующих электронов группы О2“ на неразрыхляющие
6-орбитали.
Аналогичные комплексы тройного состава образуются с малеи-
новой и молоновой кислотами. Судя по ИК-спектрам поглощения,
молекулы воды в пероксокомплексах титана со щавелевой, малеи-
новой и молоновой кислотами непосредственно связаны с атомами
7* 99
титана (III) очень прочно и не удаляются даже после сушки в ва-
кууме. Аморфное строение пероксокомплексов титана авторы объяс-
няют беспорядочным характером ‘межмолекулярных водородных
связей.
Джери и Патель [235] изучили термическую устойчивость пере-
кисных комплексов титана со щавелевой, молоновой и малеиновой
кислотами, имеющими состав TiO2C2O4 • ЗН2О, TiO2CH^C2O4 •
• ЗН2О и ТЮ2С2Н4С2О4 • ЗН2О. Исходя из максимумов на термо-
граммах нагревания, перекисные группы в них разлагаются соответ-
ственно при 100, 100 и 88° С.
Растворением титаната натрия в смеси щавелевой кислоты и пе-
рекиси водорода получена натрйевая соль тройного титанового
комплекса с перекисью водорода и щавелевой кислотой состава
[Na2C2O4 • TiO3)2C2O3 • 4Н2О [265]. Аналогичным методом были по-
лучены калиевая и бариевая соли (КгС2О4 • ТЮ3) С2О3 • 2Н2О,
(ВаС2О4 • TiO3)2G2O3 • 4Н2О [264, 2651.
При смешении растворов калийтитаноксалата и перекиси водо-
Г°\ ]
рода образуется перекисный комплекс Кг I (С2О4)2 [309].
Он имеет максимум светопоглощения при X = 380 нм, а по составу
аналогичен соли Массужели. В присутствии уксуснокислого натрия
(pH 5,8—6,0) отношение между титаном и щавелевой кислотой
в перекисном комплексе становится равным 1 :2 и максимум све-
топоглощения сдвигается в коротковолновую область спектра.
Пропуская раствор соли через колонку с ионообменной смолой
зеокарб-225, Ричардсон [3091 получил свободную кислоту
Н2 [O2Ti(C2O4)2l почти с количественным выходом.
Оксалат-ион в смешанных перекисных комплексах титана может
замещаться на другие ионы, например SO4. В присутствии этилен-
диаминтетрауксусной кислоты (ЭДТА) и нитрилтриуксусной кис-
лоты при pH 1—6 образуются перекисные тройные комплексы ти-
тана [267, 294].
Перекисный комплекс титана с ЭДТА имеет состав TiO (H2O2)Y2y
где Y — остаток ЭДТА [2671. Тройной комплекс имеет максимум
светопоглощения при 365 нм. Константа устойчивости его при из-
бытке перекиси водорода равна 2,67 • 1020, а при избытке ЭДТА
2,33 • 10е.
Виаллет [367] состав тройного титанового комплекса с пере-
кисью водорода и ЭДТА выражает формулой Ti (H2O2) Y, константа
диссоциации его
К = [Tj (Н2О2) (ОН8)1 [Y4-] [Н+12 = 1>50 . 1025ф
[Ti (Н2О2) Y[
Джери и Патель [2371 получили пероксомолоновый комплекс
титана ТЮ2СН2(СОО)2 • ЗН2О. В водном растворе он диссоциирует:
ТЮ2СН2 (СОО)2 =г ТЮ2+ + СН2 (СОО)Г.
100
Константа диссоциации его равна 2,34 • КГ*3.
Пероксомолоновый титановый комплекс можег диссоциировать
и по другой реакции:
TiO2CH2 (СОО)2 + Н2О = TiOCH2 (СОО)2 + Н2О2.
Константа диссоциации по этому уравнению составляет 2,8 • КГ5.
Известны перекисный комплекс титана с винной кислотой и ка-
лиевая соль этого комплекса состава TiO3 • С4Н4О6 • 2КНС4Н4Об •
• 9Н2О [262, 2631. Соль представляет собой белый негигроскопич-
ный порошок, частично гидролизующийся при растворении в воде.
В системах Ti44— Н2О— X, где Х-НС1, НС1О4и НСООН, спек-
трофотометрическим методом установлено образование тройных ком-
плексов состава [TiX6(H2O)l2“ [3721. Цезиевая и рубидиевая соли
выделены в кристаллическом состоянии и им приписывается состав
Rb [TiO2Cl4 • Н2О1 и Cs [TiO2Cl4 • Н2О]. Пероксосоли кристалли-
зуются в моноклинной системе, параметры решеток их а = 14,32,
Ъ = 13,74, с = 10,053 А, ₽ = 101°53' и а = 14,65, b = 14,282,
с = 10,29 А, ₽ = 97е 27'.
Гриффит [2101 в ИК-спектрах некоторых тройных пероксидных
комплексов титана обнаружил следующие характерные полосы
поглощения:
Комплекс K2[TiO2(C2O4)2] K2[TiO2(SO4)2] [K3TiO2F5] K2[Ti(O2)3H2O]
и, см-1 840; 615 862; 667 901; 597 825
Полосы с частотами 800—930 см—1, ранее относимые к асиммет-
ричным колебаниям связей М—О, автор приписывает деформаци-
онным колебаниям связей О—О или О—М—О, а полосы с частота-
ми 500—600см-1 — симметричным колебаниям связи М—О. Комплек-
сы ТЮ3 • ЗН2О и ТЮ3 • 3D2O он рассматривает как полимерные
соединения состава [Ti (О2)О1П • aq с частотами колебания связей
О—О 856 см~х.
Спектрометрическим методом установлено образование тройного
комплекса [TiA (НО2)]“, где Н4А — этилендиаминтетрауксусная
кислота [97].
КОМПЛЕКСЫ С ОРГАНИЧЕСКИМИ
И СМЕШАННЫМИ ЛИГАНДАМИ
В комплексообразовании с титаном принимают участие раз-
личные классы органических соединений, содержащих энольные,
карбонильные, карбоксильные, аминные и другие функциональные
группы. В водных растворах прочные комплексы с титаном образу-
ют оксикислоты. Большинство изученных комплексных соединений
титана представляют собой продукты присоединения к галогенидам
недиссоциированных молекул органических лигандов. По хими-
ческому составу они являются смешанными комплексами с органи-
ческими и неорганическими лигандами.
101
МУРАВЬИНОКИСЛЫЕ КОМПЛЕКСЫ
Штелер и Бахрам [347] наблюдали образование комплексов с му-
равьиной кислотой и двойных формиатов титана с калием, аммони-
ем и барием. Эти соединения окрашены в зеленый цвет, легко гид-
ролизуются водой.
Смешением раствора TiCl3 с раствором муравьинокислого калия
получен осадок двойной соли Ti (НСО2)з • 3Ti (ОН) (НСО2)2 •
• 2К (НСО2). Аналогичным методом получена аммонийная соль
состава Ti (НСО2)3 • 3Ti (ОН) (НСО2) (НСО2) • 2NH4(HCO2)2 • Н2О.
Кристаллы двойных формиатов титана окрашены в оливково-зеле-
ный цвет.
Формиаты титана находят применение в качестве протравы при
крашении тканей.
УКСУСНОКИСЛЫЕ КОМПЛЕКСЫ
Уксусная кислота дает комплексы с титатом в безводной среде
и в водных растворах. При добавлении к безводной уксусной кис-
лоте и уксусному ангидриду четыреххлористого титана в результате
замещения двух атомов хлора в TiCl4 на остатки уксусной кислоты
образуется соединение TiCl2(CH3COO)2 [199, 217, 251, 2931.
Капур с соавторами [2511 получил трихлорацетат титана
СН3СОО • TiCl3, основной ацетат титана переменного состава
(СН3СОО)Х • TiO (ОН)4_х и полимер [(СН3СОО)2ТЮ1Х.
С. Я. Шнайдерман и И. Е. Калиниченко 1125] установили суще-
ствование в водных растворах двух уксуснокислых комплексов ти-
тана, имеющих состав ТЮСН3СОО+(pH 2—3) и ТЮ(СН3СОО)2
(pH > 3). Константы нестойкости этих комплексов равны соответ-
ственно 1 • КГ"6 и 4 • 10“3, т. е. они относятся к числу малостой-
ких соединений.
В растворах TiCl3 в уксусной кислоте и уксусном ангидриде про-
исходит медленное замещение хлора на карбоксильную группу и
образование соединений со смешанными анионами состава
TiCl2(CH3COO) и TiCl (СН3СОО)2 [272]. Из свежеполученпых рас-
творов TiCl3 в уксусной кислоте при добавлении к ним хлористого
цезия выделен комплекс Cs2TiCl5 • СН3СООН.
ЩАВЕЛЕВОКИСЛЫЕ КОМПЛЕКСЫ
Известны щавелевокислые комплексы с трех- и четырехвалент-
ным титаном. Так, при смешении раствора TiCl3 с раствором ща-
велевой кислоты получены желтые призматические кристаллы со-
става Ti2(C2O4)3 • 10Н2О [345, 346].
Оксалат трехвалентного титана с оксалатами аммония, калия и
рубидия образует двойные соли типа MTi3+(C2O4)2 • 2Н2О, где М —
102
щелочной металл и аммоний [346]. Получена также натриевая соль
этого состава [345]. Все эти двойные оксалаты — кристаллические
вещества золотисто-желтого цвета, хорошо растворимые в воде,
но нерастворимые в этиловом спирте.
С помощью ИК-спектров поглощения и спектров отражения по-
казано, что в соединениях Ti2(C2O4)3 • Н2О и MTi (С2О4)2 • 2Н2О
(М = NH4,K) молекулы воды не координированы [186]. Аюмы ти-
тана в них находятся в состоянии октаэдрической координации, что
объясняется полимерной структурой иона, выражаемой схемой
Величина магнитного момента (рв = 1,7) указывает на наличие
у атома титана одного неспаренного электрона.
Дрю и соавторы [174] исследовали рентгеноструктуру
Ti2(C2O4)3 • 10Н2О. Кристаллы этой соли имеют триклинную ре-
шетку с параметрами а = 8,242, Ь = 7,161, с = 9,934 А, а =
= 95,30, р = 122,54,_у = 102,60°, ризм = 1,92, ррентг = 1,922 г/см3,
z=l, фед. гр. Р1. Строение комплекса отвечает формуле
[C2O4Ti (H2O)3C2O4Ti(H2O)3C2O4l.
Магнитная восприимчивость оксалатных комплексов Ti (III) изу-
чена в работе [451.
Суббанна с соавторами [337] в результате исследования электро-
проводности и спектров светопоглощения в растворах TiOCl и Н2С2О4
обнаружил только одно соединение с отношением Ti3+ : С2О42' =
= 1:1, которое в растворе неустойчиво и подвергается гидролизу.
Константа его устойчивости равна 2,161 • 10“3. На этом основании
авторы выражают сомнение о возможности существования двойных
солей оксалатов титана со щелочными металлами.
Методами ЭПР и ИК-спектроскопии в водном растворе трехва-
лентного титана с этилендиаминтетрауксусной и щавелевой кисло-
тами обнаружены тройные комплексы с отношением компонентов
1:1:1 [156].
Четырехвалентный титан в водных растворах образует оксалат-
ные комплексы, по-видимому, только в форме титанила. Методом
физико-химического анализа в растворах установлено существова-
ние нескольких оксалатных комплексов титана.
Методами изомолярных серий и электромиграции показано, что
в 0,1—0,5-н. соляной кислоте и в 0,2-н. хлорной кислоте титан со
щавелевой кислотой образует нейтральный комплекс [TiOC2O4]°
[14]. При повышении pH титан движется к аноду, что указывает
на образование отрицательного заряженного комплекса состава
[TiO (С2О4)2]2~. Константы нестойкости нейтрального и анионного
комплексов равны 2,5 • 10~7 и 5 • 10 4 соответственно. Близкие
103
величины (Ki — 3 -10“7 и Кг = 3 • 10“6) получены Е. А. Мазу-
ренко и Б. И. Набиванием [62].
На кривой электропроводности водных растворов гидроокиси
титана и щавелевой кислоты обнаружены изломы, соответствующие
отношениям TiO2 : Н2С2О4 = 1: 2; 1 : 1; 1 : 3 и 1 : 4 [2431, а на кри-
вой потенциометрического титрования раствора тетрахлорида в
соляной кислоте щавелевой кислотой имеются максимумы при от-
ношениях TiO2 : Н2С2О4 = 1 : 1; 1 : 2; 1 : 3 и 1 : 4 1297]. Появле-
ние максимумов авторы [297] объясняют ступенчатой диссоциацией
четыреххлористого титана:
TiCI4 = TiCl|+ + 2СГ,
TiCl2+ = Ti4+ + 2СГ
с последующим образованием оксалатных комплексов титана со-
става TiCl2C2O4Ti (С2О4)2, Н2 [Ti (С2О4)з1 и [Ti (С2О4)2] • 2Н2С2О4.
Однако предположение о существовании в водных растворах окса-
латных комплексов с ионом титана Ti4+ нуждается в подтверждении.
Этот ион в водных растворах неустойчив, легко присоединяет груп-
пу О2~ или ОН~, с которой образует прочные ионы титанила и
гидроксокомплексов. Препаративным методом оксалаты гитана
приведенного состава не получены [243].
Бринцингер и Эккард [151 методом диализа установили суще-
ствование в оксалатных растворах иона [TiO (С2О4)2]2~. А. А. Грин-
берг и Л. В. Шихеева [35] с помощью потенциометрического ти-
трования определили первую константу нестойкости комплекса
[TiO (С2О4)2]2~, оказавшуюся равной 2 • 1О~10. Вторая константа
нестойкости составляет 0,77 • 10~5. М. И. Штокало [111] по ослаб-
лению окраски титанового комплекса с ксиленоловым оранжевым
определила первую константу нестойкости оксалатного комплекса,
равную 0,3 • 10-7-
Виалет [366] методом ионного обмена, полярографии, спектро-
фотометрии и рефрактометрии установил, что в концентрированных
растворах существуют комплексы с отношением Ti : С2О4 = 1:2,
а в разбавленных =1:1. Автор определил константы нестойкос-
ти комплексов Ti (ОН)2 (НС2О4)2 (0,5- 1СГ6) и Ti (ОН)2(С2О4)2
(1,02 • 10~13).
В твердом виде получено небольшое число оксалатных титановых
комплексов. Комплексная природа некоторых из полученных со-
единений нуждается еще в подтверждении.
Упариванием оксалатного раствора выделены белые кристаллы
Н2[ТЮ(С2О4)2] • 2Н2О, хорошо растворимые в воде и спирте [310].
Растворением гидроокиси титана в щавелевой кислоте получен
оксалат титана TiOC2O4, а из спиртового раствора — комплекс
TiOC2O4 • С2Н5ОН [312]. При смешении раствора оксихлорида ти-
ки
тана в соляной кислоте с концентрированным раствором щаве-
левой кислоты образуется аморфный осадок состава (ТЮ)2ОС2О4-
• пН2О.
Харкер и Патель [2431 из водного раствора получили оксалат
титана TiOC2O4 • 3,5Н2О, строение которого они выражают форму-
лой [TiO (Н2О)2С2О41 • 1,5Н2О.
Оксалатные комплексы титана с оксалатами щелочных и щелоч-
ноземельных металлов образуют двойные соли. Прасад и Трипати
[297] на кривых потенциометрического титрования солянокислых
растворов тетрахлорида титана оксалатами натрия, калия и аммония
наблюдали максимумы, отвечающие образованию соединений типа
М2 [Ti (С2О4)з1 и М4 [Ti (С2О4)4], а для оксалатов калия и аммония —
еще и соединений типа Ti (С2О4)4 • ЗМ2С2О4 и Ti (С2О4)2 • 4М2С2О4.
Авторы, однако, не приводят других доказательств, подтверждающих
существование этих соединений. Описана натриевая соль состава
Na2 [ТЮ (С2О4)21 [150].
При нагревании гидроокиси титана с водным раствором кислого
оксалата калия КНС2О4 образуется двойной оксалат титанила
и калия Кг!ТЮ (С2О4)2] • 2Н2О в виде бесцветных мелких игольча-
тых кристаллов [312]. Кристаллы этой соли имеют триклинную
симметрию. Отношение осевых параметров а : Ь; с= 1,7067 : 1 : 0,8180;
а = 103°12'36", 0 = 97° 25'47", у = 101° 3'59" [178, 179].
По данным дифференциального термического анализа, [146], мо-
лекула КгТЮ (С2О4)3 • 2Н2О при 70—200° С теряет кристаллиза-
ционную воду, а при 335° С безводная соль начинает разлагаться.
В остатке от разложения при 1100° С получается титанат K2TiO3.
При нагревании свежеосажденной гидроокиси титана с раство-
ром кислого оксалата аммония образуется двойной оксалат титанила
и аммония (NH4)2TiO (С2О4)2 • 2Н2О [312], а по реакции обменного
разложения между Кг [TiO (С2О4)2] • 2Н2О и-ВаС12 — бариевая соль
Ba [TiO (С2О4)] • 2Н2О в виде белых, плохо растворимых в воде
кристаллов [284].
Б. В.Стрижков с соавторами [89],добавляя к оксалатному раствору
титана водные растворы СаС12, ВаС12, SrCl2 и РЬС12, получил осад-
ки двойных оксалатов состава СаТЮ (С2О4)2 • 5Н2О, BaTiO (С2О4)2 •
. 4Н2О, SrTiO (С2О4)2 • 5,5Н2О, PbTiO (С2О4)2 • 4Н2О. Строение
оксалатных титановых комплексов изучено еще не достаточно.
Л. Г. Власов с соавторами [26], сопоставляя ИК-спектры по-
глощения оксалатов натрия, калия, аммония, кальция и бария с
двойными оксалатами титанила и щелочноземельных металлов
СаТЮ (С2О4)2 • 4Н2О, SrTiO (С2О4)2 • 5Н2О и BaTiO (С2О4)2 • 4Н2О,
обнаружили, что в области 900—750 и 1000—1100 см~1 двойные
соли содержат больше полос, чем простые. Это, по мнению авторов,
служит доказательством комплексного строения двойных оксала-
тов титана. Авторы предлагают состав двойных оксалатов титана и
щелочноземельных металлов выражать формулой М [ТЮ (С2О4)2] •
• пН2О.
105
Исследование Раман-спектров [2161 и ИК-спектров [176, 177]
свидетельствует о том, что в двойных оксалатах титана группа С2О4
координирована около иона ТЮ2+. Дин с соавторами [171] комплекс-
ному оксалатному иону приписывают строение [Ti (С2О4)3]2“.
И. П. Беляев и С. В. Бондарева [4] изучили систему Ва, К ||
| [TiO (С2О4)2]2~, О, Н2О методами растворимости, вязкости и др.
Л. В. Новикова и И. Н. Беляев [65, 661 получили двойные окса-
латы титана K2TiO (С2О4)2 • 2Н2О, Na2TiO (С2О4)2 • 2Н2О,
BaTiO (С2О4)2 • 4Н2О, PbTiO (С2О4)2 • 4Н2О, PbTiO(C2O4)2 •
• Н2С2О4 • 4Н2О и изучили растворимость их в интервале темпе-
ратур 8—60° С. Устойчивым оказался только двойной оксалат ти-
танила и калия. Растворимость его изменяется от 1,64 до 48,31 вес. %.
Остальные двойные оксалаты титана относятся к числу малораство-
римых и гидролизующихся при нагревании соединений.
А. В. Лапицкий с соавторами [52, 87, 88] исследовали термиче-
скую устойчивость двойных оксалатов титана и кальция, стронция,
бария, свинца. При нагревании они обезвоживаются, а затем рас-
падаются на карбонаты двухвалентных металлов и двуокись ти-
тана. Выше 700° С двуокись титана реагирует с карбонатами
щелочноземельных металлов, в результате чего получаются метати-
танаты. Барийлитанилоксалат, например, при нагревании разла-
гается по следующей схеме:
ВаТ1(С2О4)2 • 4Н2О------> BaTiO (QO^--------
-> BaTiO (С2О4)2 -520°С > BaTiO3(CO2) 670° - -+ ВаТЮ3 ->
(I) '
(П)
Галахер и Томсон [208] приводят иной механизм разложения
BaTiO (С2О4)2 • 4Н2О. По их данным, безводная соль при 300—400° С
разлагается на ВаСО3, TiO2, СО и СО2. В интервале температур 600—
700° С образуется титанат бария ВаТЮ3. Аналогично протекает
разложение SrTi (С2О4)2 • 4Н2О.
Синтезированы двойные оксалаты Со [Ti(C2O4)2] • Н2О [3] и
FeC2O4 • ЗТЮС2О4 • яН2О [163].
При взаимодействии растворимых оксалатных комплексов
титана с [CoCi (NH3)5] С12 получены соединения [Со (NH3)6]2 •
. [Ti3O2Cl2 (С2О4)61 - 5Н2О и [Со (NH3)5 Н2О12 [CoCi (NH3)5]
• [Ti (ОН)С1 (С2О4)2]4 [270]. Термическое разложение первого соеди-
нения [274] начинается при 170° С и протекает с образованием без-
водной соли [Co(NH3)6]2 • [TiO (С2О4)2]3. В интервале температур
170—300° С выделяется аммиак. Твердый остаток, получаемый при
170—300° С, состоит только из окислов титана и кобальта. Заканчи-
вается разложение оксалата образованием при 540° С титаната ко-
бальта 2СоО • 3TiO2.
106
ВИННОКИСЛЫЕ КОМПЛЕКСЫ
Существование виннокислых комплексов титана в водных рас-
творах отмечено в работах [36, 151, 223, 358]. Розенгейм и Шютте
[312] получили виннокислый комплекс титана препаративным ме-
тодом в виде белого порошкообразного продукта, хорошо раствори-
мого в воде. Состав его отвечает формуле Ti(C4H4O6)2-4H2O. Эти же
авторы из раствора хлорида титана в смеси соляной и винной кислот
в результате высаливания спиртом выделили второй комплекс со-
става (ТЮ)2С4Н4О6 • 7Н2О.
Этому комплексу они приписывают следующее строение:
О=С—СНОН—СНОН —С=О
I I
О-----Ti----Ti -----О • 7Н,О.
Ci
II II
о о
Методом потенциометрического титрования установлено суще-
ствование в водных растворах двух комплексов титана с винной кис-
лотой с отношением компонентов 1 : 1 и 1 : 2 [266]. В кислых вин-
нокислых растворах четыреххлористого титана по изменению угла
вращения поляризованного луча /-винной кислоты обнаружен вин-
нокислый комплекс титана с отношением металл : лиганд =1:2;
в аммиачных растворах существует комплекс с отношением ме-
талл : лиганд = 1 : 1. В растворах сульфата титана в винной кис-
лоте образуется виннокислый титановый комплекс с отношением
металл : лиганд =1:3.
И. В. <Пятницкий [76] с помощью метода распределения титана
между водными и неводными растворителями доказал существо-
вание следующих виннокислых комплексов:
Концентрация
винной кисло- Состав виннокислого
ты, моль/л Рн комплекса титана
0,05—0,15 4,6 TiOC4H4O6 • НС4Н4Ов“
0,05—0,15 5,5—6,2 ТЮС4Н3Об-
0,2—1,0 9,0—9,5 TiO(C4H3O6)2“
Вследствие неустойчивости иона Ti41’ в водных растворах вин-
нокислые титановые комплексы образуются, вероятно, вследствие
координации лигандов вокруг иона ТЮ2+. В кислых растворах
винная кислота, по данным Пятницкого, при образовании титано-
вых комплексов ведет себя как двухосновная кислота. В щелоч-
ных растворах титан реагирует только с одной карбоксильной
группой.
Методами ионного обмена, полярографии, спектрофотометрии
и рефрактометрии показано образование комплексов титана с
1
107
винной кислотой (H2L) состава Ti (OH)2(HL)2 и Ti (OH)2L2, константы
нестойкости которых равны 1,30 • 10“7 и 1,45 • 10~14 соответствен-
но [366]. Согласно [33], Ti (IV) с лимонной кислотой образует ка-
тионный комплекс, константа диссоциации которого составляет
6,9 • 10~5.
Ю. Д. Долматов [37] полагает, что в сернокислых растворах
виннокислый комплекс титана существует в форме иона
[Ti(OH)3(C4H4O6)3]5_, а в солянокислой среде образуются комплек-
сы [Ti (ОН)2(С4Н4О6)2]2“ и [Ti (ОН)з(С4Н4О6)3]5“. В щелочных рас-
творах устойчив титановый комплекс [Ti (ОН)4С4Н4О6]2-. Методами
спектрофотометрии и ионного обмена установлено, что в зависимо-
сти от pH растворов в них существует три виннокислых комплекса
титана [38]. При pH < 1 доминирует катионный комплекс состава
[Ti (ОН)2(С4Н5О6) (С4Н6О6)]1+, при pH 2—7 образуется анионный
комплекс [Ti (ОН)2С4Н4О6]2“, а при pH >7 — гидроксокомплексы,
включающие только один ион винной кислоты.
Со щелочными металлами и аммонием виннокислые комплексы
титана существуют в форме двойных солей. Нагреванием гидрооки-
си титана с раствором кислого виннокислого натрия получена нат-
риевая сопь виннокислого титанового комплекса TiO (C4H4O6Na)2 •
• 8Н2О [221]. Она образует белые призматические кристаллы, лег-
корастворимые в воде, но почти нерастворимые в этиловом спирте.
Известны натриевая, калиевая и аммонийная соли винноки-
слого титанового комплекса 2Na2O • 2TiO2 • ЗС4Н6О6 • ЮН2О,
(С4Н4О6К • ТЮ2)2С4Н4О5 • 6Н2О, 2 (NH4)2 С4Н4О6 • Ti2O3C4H4O6 •
. 10Н2О [312]. Нагреванием гидроокиси титана с раствором кислого
виннокислого калия синтезирована калиевая соль TiO (С4Н4О6К)2 •
•4уН,0 [222].
Описаны также двойные виннокислые соли титана с аммонием
и барием TiO(C4H4OeNH4)2 • 3-^-Н2О и ВаТЮ(С4Н4Ов)2 • 5Н2О
[2211.
ЛИМОННОКИСЛЫЕ КОМПЛЕКСЫ
В растворах с pH 5—6 образуется титановый комплекс с лимон-
ной кислотой состава TiOHCeH5O7 • СвН5О7~ 179]. Константа' не-
стойкости его
[TiO2+] [НС6Н5О72-] [С6Н5О?~] 12
А =--------------------------— 1,2 • 1U
[ТЮНСбНбО7 . СбН5О^“]
И. В. Пятницкий и Р. С. Харченко [80] изучили зависимость
экстракции трибутиламинцитратного комплекса титана «-амиловым
спиртом от pH раствора. Методом сдвига равновесия авторы уста-
новили, что молекулярное отношение амина к титану в экстраги-
108
руемом комплексе равно 4, а перенос ионов показал, что титан на-
ходится в анионной части соединения. В состав лимоннокислого
комплекса титана, экстрагируемого «-амиловым спиртом, входит
анион Н2СвН5О7".
t
КОМПЛЕКСЫ С ЯБЛОЧНОЙ И ЯНТАРНОЙ КИСЛОТАМИ
- ь
Методом потенциометрического титрования установлено суще-
ствование в водных растворах комплексов титана с яблочной и ян-
тарной кислотами с отношением металл : лиганд =1:1 и 1:2
12661.
Исследован состав и устойчивость яблочных комплексов титана
в кислой и щелочной средах по распределению оксихинолята титана
между хлороформным и водным растворами [34]. При pH 3,8—5,8
в интервале концентраций винной кислоты 0,01—0,4 моль/л в рас-
творе преобладает комплекс [TiOC4H4O5]. Константа диссоциации его
[Titf+i |С,н,о>т
Л “ [ТЮС4Н4О5] -1и •
В щелочном растворе (pH 10,5—12,5) в комплексообразовании
участвует оксигруппа и одновременно идет гидролиз комплекса.
Вероятный состав яблочного комплекса в щелочной среде выража-
ется формулой [TiO (ОН)С4Н3О5]2“.
МОЛОЧНОКИСЛЫЕ КОМПЛЕКСЫ
В патенте [69] описан метод получения молочнокислого титана
из сульфата титана и лактатов щелочноземельных металлов. Состав
молочнокислых комплексов титана не определен.
КОМПЛЕКСЫ С ТРИОКСИГЛУТАРОВОЙ КИСЛОТОЙ
1 I •' *.
Триоксиглутаровая кислота СООН—СНОН—СНОН—СНОН—
—СООН с трех- и четырехвалентным титаном образует прочные
комплексы, окраска которых зависит от pH раствора [252]. Состав
их еще не установлен.
< ' ♦
САЛИЦИЛАТНЫЕ КОМПЛЕКСЫ
Салициловая (о-оксибензойная) кислота с титаном образует не-
сколько комплексов. Одни из них получены в чистом виде препара-
тивным методом, а другие существуют только в растворах. Салици-
ловые комплексы окрашены и поэтому находят применение для ко-
лориметрического определения титана.
Получен комплекс (TiSal3)2TiCl6, где Sal — салициловая кис-
лота, и ее эфиры [180]. Из раствора нитробромида титана и
109
салициловой кислоты в абсолютном эфире выделен комплекс
TiBr (ОСвН4СО2Н)3 • НВг [3161. В эфирном растворе салициловая
кислота с тетрахлоридом титана образует пурпурно-красные кри-
сталлы состава [Ti (OC6H4CO2H)3C1] • НО, а в бензольном раство-
ре— Ti (ОС6Н4СО2)2 [315—317].
Из раствора тетрабромида титана и салицилового альдегида
в хлороформе выделены кристаллы титанового комплекса
TiBr2 (ОСвН5СНО2) • НВг, а смешением четыреххлористого титана
Таблица 4
Внутрикомплексные соединения титана с шиффовыми основаниями
Название
Формула
T. пл., °C
Салицилалметанитроанилат
Салицилаламинопиридинат
Салицилалпараиоданилинат
2-(4-Метил -2 -оксибеизолами -
но)пиридинат
3,5-Дибром-2-салицилалами-
нопиридинат
5-Бром-2-оксибензоланилинат
3,5-Д и хлор -2 -са л ици л а л ами -
нопиридинат
5-Хлор-2-(5-бром-2-оксибенз-
аламино)пиридинат
5-Х лор-2-салицилаламинопи-
ридинат
5-Бром-2-оксибензалметанит-
роанилинат
Дисалицилалэтилендииминат
2,6-Дисалицилаламинопири-
динат
Дисалицилал-о-дианизидинат
Ti(C13H9O3N2)2Cl2 Ti(C12H9ON2)2Cl2 Ti(C13H9ONI)2Cl2 Ti(C13H41ON2)2Cl2 Ti(C12H7ON2Br2)2Cl2 Красный Оранжевый Алый Светло- красный Оранжевый 168 175—176 176—177 133—134 202
Ti(C13H9ONBr)2Cl2 Красно-ко- ричневый 176—177
Ti(CuH7ON2Cl2)2Cl2 Красный 181—183
Ti(C12H7ON2BrCl)2Cl2 Малиновый 189—200
Ti(C12H8ON2Cl)2Cl2 Алый • 170—172
Ti(C13H8O3NBr)2Cl2 Коричневый 198—200
Ti(CleH14O2N2)Cl2 Темно-ко- ричневый —
Ti(C19H13N3O2)Cl2 Малиновый 218
Ti3(C28H22O4N2)2Cl4 Темно-ко- ричневый 198—199
с салициловой кислотой и ее метиловым и феноловым эфира-
ми — красные кристаллы состава Ti (OCeH4CO2H)2TiCle,
Ti (OC6H4CO2CH3)2TiCle и Ti (OC6H4CHO2)2 • HC1 [326]. Салицило-
вый альдегид с четыреххлористым титаном в хлороформном растворе
образует фиолетовое соединение TiCl2 (ОС6Н4СНО2)2 • НС1. В пири-
диновом растворе салициловый альдегид и четыреххлористый титан
реагируют с шиффовыми основаниями с образованием внутриком-
плексных соединений [292].
А. С. Кудрявцев и И. А. Савич [47], нагревая нитрат, хлорид и
бромид титана в абсолютном спирте или эфире с салицилал-о-ами-
нофенолом, синтезировали комплекс Ti (Ci3H9O2N)2, а Розенгейм
НО
>Ti (OCeH4COONa)2.
и Зорге [317] — золотисто-желтые кристаллы тройного комплекса
О
О
А. В. Лапицкий с соавторами [106] синтезировал серию внутри-
комплексных соединений титана с шиффовыми основаниями (табл. 4).
Все они получены по реакции между TiCl4 и шиффовыми основания-
ми в растворе диоксана. Строение их на примере дисалицилалэтилен-
диимината титана [54] можно представить формулой
Получены также следующие титановые комплексы с шиффовым
основанием, производным салицилового альдегида и 2-оксиэтил-
амина (H2L) : Ti (изо-С3Н7О)2Ь, точка плавления 129—13Г С; TiL2,
точка плавления 212—215° С; TiL2 • H2L, точка длавления 132—
134° С; Ti (OC4H9)2L, Ti (OC5Hh)2L, Ti (C5H10O2)L, Ti (C6H12O2)L,
Ti (изо-С3Н7О) OC6H5 • L и Ti (OC6H5)2L [65].
Бредли с соавторами [142] синтезировал тетракис-(R, N-сали-
цилалиминаты)титана, где R = C2H5, CH3, mpem-C4H9. Этильное
производное образует ромбические кристаллы с параметрами ре-
шетки а = 10,15, b = 17,01, с = 39,26 А, Ризм — 1,22, Ррентг —
= 1,26 г/см2, z = 8, фед. гр. РЬса.
В. А. Коган и соавторы [51] получили комплексы с шиффовыми
основаниями (ЬС12) состава TiCl2L2.
При добавлении к раствору сульфата титана салицилатов нат-
рия и аммония образуются кристаллы желтых титановых комплек-
сов, состав которых выражается формулами NaTiSal3 • ЗН2О и
NH4TiSal3 • 4Н2О [901. При нагревании аммонийной соли до 180° С
она отщепляет воду. В интервале температур 240—250° С отщепля-
ется аммиак и получаются ромбоэдрические кристаллы состава
TiO[CeH4COO (ОН)12.
Циглер с соавторами [377] экстрагировал титан из водных рас-
творов хлороформом в виде салицилатнотрибутилфосфатного комп-
лекса.
А. К. Бабко и соавторы [15, 18] установили, что в водных рас-
творах титан образует тройные комплексы с салициловой кислотой
и органическими основаниями (пиридином, пирамидоном, хиноли-
ном и др.). Комплексы эти получаются в виде труднорастворимых
осадков. При осаждении на холоду соотношение компонентов в по-
лученных продуктах Ti : Sal : Ру = 1 : 3 : 1 (Ру — органическое
основание). Из нагретых растворов осаждаются продукты с отноше-
нием компонентов 1:3:2.
111
Тройные титановые комплексы хорошо растворимы в органи-
ческих жидкостях. Они окрашены в оранжевый цвет. Строение
их авторы выражают формулами PyH[TiO (ОСвН4СООН)314 и
О
(РуН)2 Ti
. Ранее
тройные комплексы титана с
салициловой кислотой и пиридином получали только из неводных
сред.
Леви [258] описал комплекс темно-желтого цвета, состав которо-
го TiO (ОС6Н4СООН)2 • 2C5H6N. Розенгейм и Зорге [317] синтези-
ровали оранжевый комплекс С6Н4/ ^^Ti (OCeH4COOC5H6N)2.
'СО/
А. К. Бабко и соавторы [18] изучили препаративным методом
салицитатные комплексы титана, осаждаемые из растворов ед-
ким натром и аммиаком при различных pH:
pH Ti : Sal: Na Цвет осадка
2 1:2:1 Оранжевый
2—6 1:3:1 Желтый
6 1:2:2 Оранжевый
1,5—1,6 1:1:1 Красно-оранжевый
2,2 1:2:1 Т емно -ор анжевый
4,1 1:3:2 Желтый
7,1 1:2:2 Оранжевый
Авторы считают,
pH > 6 с отношением компонентов
что оранжевые комплексы, образующиеся при
1:2:2, имеют строение
М2
TiO
, где М = Na и NH4.
4 I
2 ' Ъ
Оранжевые комплексы, образующиеся при pH —> 2, имеют один
незамещенный водород, и строение их выражается формулой
, а у комплексов с отношением компо-
МН ТЮ
4
2
\ ХСО2
& _
нентов 1 : 3 : 2 и 1 : 3 : 1 следующее строение:
О
NaH Ti
со/
Ti
3
2
з
Методом спектрофотометрии установлено, что салициловая кис-
лота, взятая в избытке, при pH 2,13—4,93 образует с титаном ком-
плексы желто-оранжевого цвета с максимумом светопоглощения
112
в интервале 362—365 нм [353]. Методом изомолярных серий показа-
но, что титан с салициловой кислотой в водных растворах дает ком-
плекс с отношением Ti : Sal =1:3. Константа диссоциации его
равна 0,82 • 1СГ6 (35° С) [181].
А. К. Бабко с соавторами [22] этим же методом нашли, что в кис-
лых растворах с pH 2 титан образует с салициловой кислотой
окрашенный комплекс с отношением Ti : Sal = 1:1. При избыт-
ке салициловой кислоты получаются комплексы с отношением
Ti : Sal = 1 : 2 и 1 : 3. Авторы определили константы нестойкости
комплексов:
к _ [TiO2+] [Sal2-] _ 9 П_16
Л1 [TiOSal] ~ ‘ Ш ’
_ |Tltf+| iy-r _ . _5
2 [TiOSal^-]
В щелочных растворах титан с салициловой кислотой образует
бесцветные комплексы состава [Ti (OH)2Sal2]2~ или [Ti(OH)3Sal]3~.
Согласно [348], в концентрированной серной кислоте и
N,N- диметил формами де титан (IV) с салициловой кислотой дает
комплексы состава TiSal и TiSal2.
В результате изучения электропроводности, магнитной воспри-
имчивости и кажущегося молекулярного веса А. В. Лапицкий и
соавторы [107] решили, что дисалицилалэтилендииминат титана
TiRCl2 имеет комплексное строение. Во внешней сфере комплекса
находятся два хлор-иона. Молекула лиганда во внутренней сфере
комплекса занимает четыре координационных мерта, будучи свя-
зана с титаном двумя валентными связями и двумя связями через
атомы азота. Растворимость этого комплекса в четыреххлористом
углероде при температуре 20° С равна 1,0 • 10~6 моль!л [531.
В водных растворах титан реагируете карбоксильной и энольной
группами салициловой кислоты. В кислых растворах центральным
ионом комплекса является ион титанила TiO2+. В щелочных раст-
ворах и при избытке салициловой кислоты органогруппа замещает
кислород титанила [18].
В неводных растворах салициловая кислота может реагировать
с титатом как с помощью карбоксильной, так и энольной группы.
Салицилатный комплекс титана в свою очередь образует комплекс
с маннитом и глицерином в отношении 1 : 1 [77].
Из производных салициловой кислоты изучено комплексообразо-
вание титана с 2-сульфосалициловой кислотой. Четырехвалентный
титан с 5-сульфосалициловой кислотой дает комплексы состава TiR,
TiR2, TiR3 и TiR4 [280, 353, 356].
Описаны комплексные соединения титана с алкиламинами са-
лицилового альдегида состава TiCl4 • 2L, где L — салицилал-я-гек-
самин, салицилал-я-додециламин и салицилал-я-октадециламин [84].
8 1-1669 113
1
Связь титана с лигандами в этих комплексах осуществляется через
атом азота.
Нагреванием смеси L2TiCl2 [L == о-С6Н4(ОН)СОО], 8-оксихино-
лина (HL') и триэтиламина в бензоле получен смешанный салици-
латнохинилинатный комплекс титана L2L'Ti [167]. Салицилальдок-
сим (H2L) с титаном образует комплексы с отношением компонентов
1 : 1 и 1 : 2. Константы диссоциации этих комплексов в выражении
—1g К равны соответственно 18,50 и 17,20. Свободная энергия ком-
плексообразования составляет —25,39 и —23,67 ккал/моль [268].
Тиосульфосалициловая кислота (Н2Ь) с те/прякис-(изо-пропок-
си)титаном дает окрашенные растворимые в бензоле комплексы
состава (w30-C3H7)3TiL и (u3o-C3H7)2TiL2 [218].
В работах [136, 137] изучено комплексообразование титана с
салициловой и сульфосалициловой кислотами методом физико-
химического анализа.
В индивидуальном состоянии препаративным путем выделены
комплексные соединения титана с салициловой кислотой с отноше-
нием компонентов 1 : 1, 1 : 2 и 1 : 3 [118]. В этиловом, пропиловом
и изопропиловом спиртах титан образует с салициловой кислотой
комплексное соединение с отношением компонентов 1 : 1 [120].
Зоммер [355] полагает, что сульфосалицилатные комплексы ти-
тана образуются по схеме:
TiO2+ + 2RH2 = TiO (RH)2 4- 2Н+ (pH 1,5—2),
TiO (RH)2 + RH2 = TiRl~ + H2O + 2H+ (pH 2—5).
Хабаши [225] с помощью полярографии обнаружил два салици-
латных и три сульфосалицилатных комплекса титана, в состав ко-
торых входят ионы ОН“. Сульфосалициловая кислота образует
комплексы также и с трехвалентным титаном [81].
КОМПЛЕКСЫ С АСКОРБИНОВОЙ КИСЛОТОЙ
Аскорбиновая кислота с титаном (IV) образует желтое окраши-
вание с максимумом светопоглощения при 360 нм [224]. Известны
два комплекса титана с аскорбиновой кислотой с отношением
Ti : R = 1:2 [213, 355] и 1:3 [355] (R — аскорбиновая кислота).
Константы диссоциации этих комплексов равны 9,58 • 10~4 и
5,5 • 10“3 соответственно [212].
Согласно [327], один из аскорбинатных комплексов титана яв-
ляется катионом, а второй анионом. Анионный комплекс устойчив
в широком интервале pH, прочнее катионного, адсорбируется силь-
но щелочными анионитами и экстрагируется аминами. Катионный
комплекс адсорбируется на сильно кислых анионитах.
114
КОМПЛЕКСЫ С ХРОМОТРОПОВОЙ КИСЛОТОЙ
Хромотроповая (1,8-диоксинафталиндисульфокислота) образует
с титаном комплексы, отличающиеся высокой интенсивностью
окраски. Состав и строение хромотроповых комплексов титана
в водных растворах изучались Окачом и Зоммером.
В водных растворах обнаружены комплексы с отношением
Ti : H4Chr = 1 : 1; 1 : 2; 1 : 3 и 1 : 4 (H4Chr — хромотроповая кис-
лота). В образовании титановых комплексов принимают участие
энольные и сульфогруппы хромотроповой кислоты, подвергающейся
ступенчатой диссоциации с отщеплением одного, двух, трех и че-
тырех ионов водорода [354]. Состав титановых комплексов с хромо-
троповой кислотой зависит главным образом от кислотности раствора.
В опубликованных работах указываются следующие условия обра-
зования титановых комплексов.
Комплекс красного цвета с максимумом светопоглощения в ин-
тервале 470—480 нм с отношением Ti : H4Chr =1:1 образуется
при pH 1,52—1,62 [119, 280, 281]; по данным [283], он устойчив так-
же при pH 1,87—1,96. Комплекс фиолетового цвета также с отно-
шением Ti : H4Chr =1:1 обнаружен в растворах с pH ~ 0,1 [354]
и в 75—98%-ной серной кислоте [121]. Очевидно, что красный и
фиолетовый комплексы имеют различное строение, хотя отношение
в них металла и лиганда одинаково. Вероятно, в образовании крас-
ного комплекса принимают участие ионы титанила ТЮ2+, а в фио-
летовом комплексе лиганды координированы вокруг Ti4r.
Комплекс с отношением Ti : H4Chr =1:2, по данным различ-
ных авторов, красного (максимум светопоглощения 470 нм) и оран-
жевого цвета (максимум светопоглощения 420 нм). Красный ком-
плекс с отношением Ti : H4Chr =1:2 существует наряду с комплек-
сом с отношением металла к лиганду 1 : 1 в интервале pH 0—2 [280,
281, 283, 350, 351]. При pH 2,39—2,44 он существует наряду с ком-
плексом с отношением Ti : H4Chr =1:3 [2831. В интервале
pH 0,4—0,9 наряду с комплексом 1 : 1 существуют комплексы с
отношением 1 : 2 красного и оранжевого цвета [354].
А. К. Бабко и О. И. Попова [131 нашли, что красный хромотро-
повый комплекс титана с отношением Ti : H4Chr =1:2 существу-
ет при pH < 3, а С. Я. Шнайдерман [121, 122] считает, что при
pH 3—4 вначале образуется красный комплекс с отношением ком-
понентов 1:2, который через некоторое время переходит в комплекс
с отношением 1:4.
Относительно окраски комплекса с отношением Ti : H4Chr =
= 1:3 данные различных авторов расходятся. Согласно [280, 2831,
он имеет красный цвет и существует в интервале pH 3,32—4,69.
В интервале pH 2,39—2,44 наряду с ним находится комплекс
с отношением компонентов 1 : 2 [283]. По мнению [350, 3541,
комплекс 1 : 3 имеет оранжевый цвет и существует в интервале
pH 3,2—7, а поданным [131 — желтый цвет и существует при pH > 5.
8*
115
Комплекс с отношением Ti : H4Chr =1:4 имеет винно-красный
цвет [279, 280]. Он образуется в разбавленных сернокислых раство-
рах и существует в интервале pH 2—3 [280, 350]. С. Я- Шнайдерман
[121, 122], как уже отмечалось, наблюдал образование комплекса
с отношением компонентов 1 : 4 при pH 3—4 из комплекса с от-
ношением компонентов 1 : 2 через несколько суток после смешения
растворов.
Строение хромотроповых комплексов титана изучалось Зомме-
ром, однако данные его работ не являются еще достаточно убеди-
тельными. В одной из своих работ [3491 он считает, что хромотро-
повая кислота с титаном в водных растворах образует комплексы
следующего состава: [TiOChr]2“, [TiOChr2]6—, [TiOChr3]I0“ и
[TiChrJ12-. В другой работе [3501 автор пишет, что в зависимости
от концентрации компонентов и pH раствора между титаном и хро-
мотроповой кислотой происходит ступенчатое комплексообразова-
ние по следующим реакциям:
pH 0—2, ТЮ2++ 2Н2Спг2-= TiO(H Chr)2~ + 2Н+,
pH 1—3,5, TiO2++ 2H2Chr2-= TiOChr®" + 4Н+,
pH 3,2-7, TiOChit" + 2H2Chr|~ = TiOChr|°“ + 2H+,
pH 1,7—2,4, TiOChr^ + 2H2ChrI~ = TiChr'2-+ 2H+4-H2O.
Комплекс ТЮСЬгз0- окрашен в оранжевый цвет (максимум светопо-
глощения при 420 нм), а остальные — в красный (максимум све-
топоглощения при 470 нм).
Зоммер [3501 вычислил константы устойчивости комплексов
TiOChr2~, TiOChr8- и TiOChr|°“, оказавшиеся равными 1О23’40,
Ю40’96 и Ю56’80 соответственно (pi = 0,1).
В последней из опубликованных работ Зоммер [354] состав ти-
тановых комплексов в растворе хромотроповой кислоты выражает
формулами [TiH3Chr]3+, [TiOH3Chr]+, [TiO (HChr)2]4~, [TiOHChrf,
[TiOH2Chr]4~, [TiOChr2]6~, [TiChr3l8“, [TiOChr2]10~. Комплексу c
отношением Ti: H4Chr= 1: 1 приписываются три различные формулы.
Для определения титана колориметрическим методом Зоммер
[354] рекомендует использовать комплекс с хромотроповой кислотой
состава TiOChrf^ с максимумом светопоглощения при 470 нм в
интервале pH 1,8—2,5.
Хромотроповые красители состава (HO3S)2(OH)2Ci0H3N=NR,
где R = НО (Cl), С6Н5, HO3S (ОН)С6Н? и HO3As С6Н?, образуют
с титаном окрашенные растворимые и нерастворимые комплексы
[48]. В уксуснокислых и лимоннокислых растворах хромотроповая
кислота с титаном образует комплексы сложного состава.
Спектры поглощения указывают на то, что в присутствии щаве-
левой и малеиновой кислот состав титановых комплексов с хромо-
116
троповой кислотой изменяется в зависимости от кислотности среды
[135]. В диметилформамиде титан с хромотроповой кислотой обра-
зует комплексы с отношением компонентов 1 : 1 и 1 : 2 [102].
Препаративным методом выделен комплекс титана с хромотроп-
ной кислотой с отношением компонентов 1 : 3 [102].
В трехвалентном состоянии титан также образует комплексы
с хромотроповой кислотой. Методом спектрофотометрии установлено
существование комплексных соединений титана (III) с и-нитробен-
зохромотроповой кислотой светло-розового цвета с отношением ком-
понентов 1 : 2 [275]. Комплекс устойчив в интервале pH 2—9. Мак-
симум светопоглощения приходится на А = 520 нм, С хромотропо-
вой кислотой титан (III) образует также комплекс красного цвета
с отношением компонентов 1 : 1 [248]. Он устойчив при pH 7. Кон-
станта устойчивости его в логарифмическом выражении равна 3.
Максимум светопоглощения находится при А = 440 нм,
КОМПЛЕКСЫ С ТИОГИДРОКСАМОВОЙ
И ГИДРОКСАМОВОЙ КИСЛОТАМИ
При действии на растворы Ti (IV) п-метоксибензотиогидрокса-
мовой кислоты образуется желтый осадок, растворимый в хлоро-
форме и бутаноле [269]. Из 2-н. солянокислых растворов этот ком-
плекс экстрагируется хлороформом и может быть использован для
определения титана. Максимум светопоглощения комплекса при-
ходится на А = 420 нм,
n-Метоксигидробензогидроксамовая кислота (HR) в слабокислых
растворах дает с титаном комплекс состава TiOR2 [322], экстраги-
руемый хлороформом. Из концентрированных солянокислых рас-
творов хлороформ извлекает в органическую фазу соединение
TiCl3R2(HR)2. Константа экстракции этого комплекса 1g Кэкстр =
= 7,55, коэффициент распределения составляет 117.
Спектрофотометрическим методом установлено комплексообра-
зование титана (IV) с N-фенилсалицилгидроксамовой кислотой
(Н2Ь) [209]. В водно-спиртовой смеси (1:1) при pH 0,5 соотношение
компонентов в комплексе равно 1 : 2. При добавлении ЭДТА вы-
делены кристаллы комплекса состава Ti (HL)3OH.
КОМПЛЕКСЫ С КОРИЧНОЙ КИСЛОТОЙ
Коричная кислота не дает с титаном растворимых комплексов.
При нагревании из растворов с pH 2 она осаждает титан количест-
венно в виде основной коричнокислой соли [27].
КОМПЛЕКСЫ С СУЛЬФОКИСЛОТАМИ
Титан (IV) с 1-нитрозо-2-нафтол-3,6-дисульфокислотой образует
растворимые комплексы с отношением компонентов 1:1; 1 : 2 и
1 : 3 [271].
117
КОМПЛЕКСЫ С ПИКОЛИНОВОЙ КИСЛОТОЙ
Состав продуктов взаимодействия титана с пиколиновой кисло-
той еще не установлен. Методом спектрофотометрии в системе титан
(IV) — пирокатехин — пиколиновая кислота обнаружено сущест-
вование тройного комплекса с отношением компонентов 1:2:2
[117]. Он хорошо экстрагируется из водных растворов органиче-
скими растворителями.
КОМПЛЕКСЫ С МИНДАЛЬНОЙ КИСЛОТОЙ
И. В. Пятницкий и Г. И. Кравченко [75] изучили комплексо-
образование титана (IV) и миндальной кислоты, пользуясь методом
распределения оксихонолятного комплекса между хлороформным
раствором и раствором миндальной кислоты. В интервале pH 3—11
миндальная кислота реагирует с титаном как двухзарядный радикал
состава RCI ЮСОО2~, а соотношение компонентов в образующемся
комплексе равно 1 : 2. Константа нестойкости комплекса
= [TiO2+] [RCHOHCOQ-]2 [ОН]2 _ 4 4 . ] О~26
[TiO (RCHO“COO-)2]
По устойчивости в водных растворах миндальный комплекс ти-
тана аналогичен салицилатным.
КОМПЛЕКСЫ С ГЛИЦЕРИНОВОЙ, ГЛИКОЛЕВОЙ
И ДИОКСИТАРТРОНОВОЙ КИСЛОТАМИ
Титан образует комплекс с глицериновой кислотой состава 1:1,
а с гликолевой и диоксатартроновой — с отношением компонентов
1 : 2 [73].
КОМПЛЕКСЫ С ДИОКСИБЕНЗОЙНОЙ КИСЛОТОЙ
Методом спектрофотометрии исследовано комплексообразование
титана с 2,4-диоксибензойной кислотой [1]. При pH 3,3 титановый
комплекс образуется по реакции
TiO2+ + 2H3L = TiO (H2L)2 + Н+,
а в интервале pH 3,3—6,5 по реакции
TiO2+ + 2H2L- = TiO (H2L)2.
Константа нестойкости его равна 5,93 • 10—7, коэффициент моляр-
ного погашения при X = 353 нм составляет 1,2 • 104.
КОМПЛЕКСЫ С ВИОЛУРОВЫМИ КИСЛОТАМИ
При смешении концентрированных водных растворов солей ти-
тана и аммонийной соли дифенилвиолуровой кислоты получен про-
дукт серого цвета с отношением металл : лиганд =1:4 [339]. Рас-
118
твор этого комплекса имеет максимум светопоглощения при X =
= 420 нм.
Известно соединение титана с ди-п-толилтиовиолуровой кисло-
той (HL) в виде твердого вещества коричнево-оранжевого цвета со-
става TiOL2. Комплекс устойчив до температуры 139° С.
КОМПЛЕКСЫ СО СПИРТАМИ, ФЕНОЛАМИ И НАФТОЛАМИ
Одноатомные спирты с титаном в водных растворах не образуют
прочных комплексов. Они, обычно, замещают молекулы воды в ак-
вокомплексах титана, не подвергаясь протонизации. Многоатомные
спирты, фенолы и нафтолы с титаном подобны слабым кислотам, и
связь их с титаном осуществляется через кислороды оловых групп.
Комплексообразование с одноатомными спиртами
Метанол вызывает сольволиз четыреххлористого титана по ре-
акции
TiCl4 + nCH3OH = TiCl4_n (OCH3)n + пНС1.
Ф. Д. Шевченко и Л. А. Кузина 1108] изучили это равновесие
и определили константы диссоциации сольватов (Ki = 0,3, К2 —
= 5 • 10~3, К3 = 10-5 и = 2 • Ю-10).
При взаимодействии четыреххлористого титана с этиловым спир-
том и пиридином получаются комплексные соли хлоралкокситита-
натов [41]. Исследование растворов четырехфтористого титана в
этиловом спирте методом ЯМР указывает на существование в них
фторэтанольного комплекса [TiF5(C2H5OH)]- [300].
Методом рефрактометрического анализа показано, что в раство-
рах тетракис-(бутокси)-титана в бензоле образуются комплексы
состава NaH [Ti(C4H9O)6] и Na2[Ti (С4Н9О)61 [2911.
Комплексы с одноатомными фенолами и нафтолами
Методом изомолярных серий и сдвига равновесий установлено
существование комплекса с отношением Ti4+ : фенол : антипирин =
= 1:2:2 [127]. P-Нафтол с четыреххлористым титаном в растворе
сероуглерода реагирует с образованием комплекса TiCl2(Ci0H7O)2
[55].
А. С. Кудрявцев и И. А. Савич [47] наблюдали образование не-
растворимых в воде осадков титана с 2-окси-1-нафтол-о-аминофено-
лом. В патенте [70] описан синтез водорастворимых титановых ком-
плексов на основе р- и у-аминоспиртов и о-аминофенолов.
Фенол реагирует с хлор- и бензокарбоновыми производными
титана состава Y2TiCl2 (HY — салициловый альдегид, метилсали-
цилат или дибензоилметан) с образованием комплекса Y2Ti (С6Н5ОН)2
[2071.
119
При смешении растворов фенолов и четыреххлористого титана
в неводных растворителях получаются комплексные соединения
титана, окрашенные от красного до фиолетового цвета [56, 103, 126,
131, 255]. Шуман [238] из бензольного раствора выделил темно-крас-
ные кристаллы состава Ti (ОС6Н5)4 • НО, а Г. П. Лучинский [56,
255] — соединение TiCl3OC6H5. Функ и Роглер [198] нагреванием
четыреххлористого титана с фенолом получили комплекс
Ti (ОС6Н5)4 . С6Н5ОН.
Известны комплексы с n-хлорфенолом, о- и n-нитрофенолом со-
става Т1С12(ОС6Н4С1)2и TiCl2(OC6H4NO2)2, а также комплекс с ^-нит-
рофенолом TiCl3OC6H4NO2 [55].
Тимол (HL) и ванилин (HL') при взаимодействии с TiCl4 в бен-
зольном растворе дают комплексные соединения состава LTiCl3,
L4Ti, L'TiCl3, L2TiCl2 и L3TiCl [207]. Тимоловые комплексы окра-
шены, максимум светопоглощения находится при к = 440 нм [214].
Комплексы с пирогаллолом и его производными
Пирогаллол (H3G) в водных растворах образует с титаном ком-
плексы состава TiG2, TiG3~, TiOG и TiOGf- [124]. Константы об-
разования TiOG и TiOG2“ равны 0,91 и 1,8 • 10“5 соответственно.
С. Я. Шнайдерман [122] методом изомолярных серий показал,
что в сернокислых растворах титан с пирогаллолом образует ком-
плексы с отношением Ti : H3G = 1 : 4 и 1 : 2. Комплекс с отноше-
нием 1 : 2 существует в концентрированной серной кислоте. В рас-
творе метанола и в водно-метанольных растворах при pH 1,2—5,5
титан образует с пирогаллолом комплекс с отношением компонен-
тов 1 : 1 [128].
Препаративным методом выделена аммонийная соль пирогал-
лолтитанового комплекса HOTi [ОС6Н3(ОН) ONH4]3 • ЗН2О [317].
С производным пирогаллола — галловой кислотой С6Н2(ОН)3СООН
(H4R) титан дает комплексы состава TiR, TiR2 и TiR3 [280, 282].
Комплекс TiR существует при pH 1,7—1,8 в присутствии избытка
лиганда. При повышении pH образуется комплекс TiR2, существу-
ющий совместно с комплексом TiR. При pH 2 в растворе находится
только один комплекс TiR2, который в интервале pH 3,5—3,8 пол-
ностью превращается в комплекс TiR3.
Максимум светопоглощения растворов титановых комплексов
галловой кислоты TiR2 приходится на к = 375—390 нм, а растворы
TiR3 имеют максимум светопоглощения при к = 365 нм,
Мемжэм изомолярных серий показано, что в разбавленных сер-
нокислых растворах титан с галловой кислотой образует комплексы
с отношением Ti : H4R = 1 : 2, а в концентрированной серной кис.
лоте — ярко-красный комплекс с отношением Ti4+ : H4R = 1:4
[1221.
В метаноле, этаноле, пропиловом и изопропиловом спиртах при
pH 1,5—7,5 титан дает с галловой и пирогалкарбоновой кислотами
120
комплексные соединения с отношением металл : лиганд = 1: 1 [119].
Препаративным методом из метанольных, ацетоновых и диоксано-
вых растворов получены комплексы титана с пирогаллолом, галловой
и пирогалкарбоновой кислотами с отношением компонентов 1 : 3
[118]. В диметилформамиде титан образует титангаллатные комп-
лексы TiO(HR)+ и TiO(HR)2 (H2R — галловая кислота) [101].
Строение титановых комплексов с пирогаллолом и его производ-
ными еще не изучено. Приводимые химические формулы комплексов
отражают только соотношение между металлом и лигандами, но
не строение их.
С пирогалкарбоновой (С6Н2(ОН)3СООН) кислотой при pH 1,5—2
титан образует комплексы TiR и TiR2. В менее кислых растворах
(pH 3,5—5,8) существуют только комплексы TiR2. На кривой све-
топоглощения растворов титановых комплексов с пирогалкарбоно-
вой кислотой имеется полоса с максимумом при 370—372 нм [280,
282].
Е. И. Князева и С. Я- Шнайдерман [50] изучили спектрофото-
метрическим методом систему титан — пирогалкарбоновая кислота
(RH2) — пирамидон (Руг). Они обнаружили в ней признаки суще-
ствования тройного соединения состава (Руг H)2[TiOR2].
В системах титан (IV) — галловая кислота — антипирин и
титан (IV) — пирогалкарбоновая кислота — антипирин образуются
окрашенные комплексы тройного состава. Из слабокислых раство-
ров (pH 2,5) они экстрагируются хлороформом. С. Я. Шнайдерман
и Е. И. Князева [129] состав этих комплексов выражают формулой
(Ant H)4[TiOR2], где Ant — антипирин [129].
Комплексы с пирокатехином и его производными
Пирокатехин в эфирном растворе реагирует с четыреххлористым
титаном с выделением хлористого водорода и с образованием ком-
/°\
плексной соли СвН4<^ ^>Ti (OC6H4ONH4) • Н2О [314, 317]. В вод-
ных растворах титан с пирокатехином дает окрашенные комплексы
оранжево-желтого цвета [280, 348, 352]. В растворах, содержащих
эквимольные количества металла и лиганда при pH 3—4, существует
смесь двух комплексов с отношением Ti : Н2Р = 1 : 1 и 1 : 2. При
избытке лиганда получается комплекс с отношением Ti : Н2Р =
= 1:3 желтого цвета.
Существование комплекса с отношением Ti : Н2Р = 1 : 1 в раст-
ворах с pH 4, экстрагируемого бутанолом, подтвердили А. К. Бабко
и О. И. Попова [12]. С. Я. Шнайдерман и И. Е. Калиниченко [123]
установили существование в интервале pH 2—4 титановых комплек-
сов с отношением Ti : Н2Р = 1 : 1 и 1 : 2. Сате и Венкатесварлу
[335] наблюдали образование всех трех комплексов титана с пиро-
катехином. В слабокислых и сильнокислых сернокислых растворах
121
обнаружено существование еще одного титанпирокатехинового
комплекса с отношением Ti : Н2Р =1:4 [122]. Титанпирокатехи-
новый комплекс с отношением Ti : Н2Р = 1:1 при pH 3 имеет по-
лосу светопоглощения с максимумом при X = 330—400 нм [20].
В комплексообразовании с пирокатехином в водных растворах
принимают участие ионы титана TiO2+ [12, 123, 355, 357] и Ti4+
[12, 355, 357]. Зоммер [357] показал, что пирокатехин вступает в
реакцию с ионом титана с отщеплением водорода, т. е. ведет себя
как кислота. На основании спектрофотометрии и зонального
электрофореза он пришел к выводу, что при избытке лиганда
и pH < 1 образуются пирокатехиновые комплексы титана состава
TiO (НР)+ и Ti (НР)3+. В интервале pH 1,5—2,5 существует комплекс
ТЮ(НР)2. Таким образом, водород от второй оксигруппы пиро-
катехина при формировании титановых комплексов в кислых
растворах не отщепляется.
В растворах с pH > 2 титан реагирует с пирокатехином как с
двухосновной кислотой, отщепляющей водород от двух оксигрупп.
При этом образуется комплекс TiH~* Области существования ком-
плексов титана с пирокатехином с отношением компонентов 1 : 2
и 1 : 3 резко разграничены.
Согласно [1231, пирокатехин реагирует с ионами титана в водных
растворах с отщеплением двух водородных атомов. Константы дис-
социации пирокатехиновых комплексов титана при ионной силе
растворов, равной 0,5, составляют:
[TiO24"] [Р2“]
[ТЮР]
= 3,3 • 1(Г23,
[ТЮР] [Р2~~]
[ТЮР2- J
= 1,1 • 10~16.
Зоммер [348] определил константы образования титановых ком-
плексов с пирокатехином с отношением металла к лиганду =1:1
и 1 : 2 в концентрированной серной кислоте и в растворе N, N-ди-
метилформамида. В диметилформамиде пирокатехин дает с титаном
комплексы состава TiO(HP)+, TiO(HP)2 и [TiOP3]2~“ (Н2Р — пиро-
катехин) [101]. Из метанольных, ацетоновых и диоксановых раство-
ров выделены титанпирокатехинатные комплексы с отношением ком-
понентов 1 : 1; 1 : 2 и 1 : 3 [118].
В петролейном эфире тетрахлорид титана образует с пирокате-
хином коричневый комплекс состава PTiCl2. При избытке пирока-
техина из бензола кристаллизуется комплекс PTi (НР)2 [207]. По-
лучен также окрашенный комплекс состава Y2Ti(HP)2, где Y —
салициловый альдегид, метилсалицилат или дибензоилметан.
В этиловом, пропиловом и изопропиловом спиртах титан с пи-
рокатехином и пирогаллолом образует комплексы с отношением
компонентов 1 : 1 [120]. Реакция протекает с вытеснением одного
атома водорода из гидроксильной группы фенола.
122
Пирокатехиновые комплексы титана реагируют с органическими
основаниями, давая при pH 3 труднорастворимые в воде тройные
соединения, которые из водных растворов экстрагируются невод-
ными растворителями, например хлороформом [16].
А. К. Бабко и М. М. Тананайко [201 установили существование
тройных комплексов в системе титан (IV) — пирокатехин — дианти-
пирилметан. Экстракт тройного комплекса из водного раствора
1—2-н. соляной кислоты хлороформом и дихлорэтаном имеет мак-
симум светопоглощения при 330 нм. Из растворов с низкой кислот-
ностью экстрагируются комплексы с максимумом светопоглоще-
ния, сдвинутым в длинноволновую часть спектра (X = 320—420 нм).
В системе титан — пирокатехин (Н2Р) — пирамидон (Руг) ме-
тодом спектрофотометрии установлено существование комплекса
с отношением Ti : Н2Р : Руг =1:2:1, подвергающегося в водном
растворе ступенчатой диссоциации [50]. В системе титан — пирока-
техин — пиридин (Pyd) при pH 2,3 обнаружен комплекс ТЮР •
. Pyd, который при повышении pH и концентрации пирокатехина
превращается в комплекс TiP2 • Pyd с вытеснением кислорода из
координационной сферы [130]. В системах титан — пирокатехин —
пиколиновая кислота (N-окись пиридина) существует комплекс
с отношением титан : пирокатехин : пиколиновая кислота (N-окись
пиридина), равным 1:2:2.
Титан (IV) с пирокатехиновым фиолетовым в кислой среде дает
комплекс, окрашенный в синий цвет [303]. С производным пирока-
техина (тироном) в водных растворах титана получаются комплексы
с отношением металла к лиганду 1:1; 1 : 2 и 1 : 3, аналогичные
пирокатехиновым [280, 3521.
Титан также образует комплексы с протокатехиновой (3,4-ди-
оксибензойной) кислотой (H3L) [184]. Они имеют состав TiL+, TiL^T
и TiL|~ [335]. Константы образования их составляют:
[TiL+J [Н+]2
[Ti4+j[H2L“]
= 3,55 • 10~5,
2 —
к3 =
fTiL22~] [Н+Р _
[Til+j [H2L-J
[TiL|~] [Н+]2
[TiL2-] [H2L~] "
Зоммер [357] считает, что при pH 1,5—2,5 и избытке протокате-
хиновой кислоты получаются титановые комплексы TiO(H2L)2.
В менее кислых растворах (pH 2) существуют комплексы Ti (НЬ)з-
и частично TiL|“.
Пирокатехин-3,5-дисульфокислота (Н4К) с титаном в водных
растворах образует комплексы с отношением Ti : Н4К — 1 : 1 и
123
.1:3 [355, 357]. Как и пирокатехин, пирокатехин-3,5-дисульфокис-
лота реагирует с ионами титана с отщеплением водорода не только
от сульфогруппы, но и от оксигрупп. При pH < 1 и избытке лиганда
в водных растворах образуются комплексы TiO [НКП и Ti [НК1+
без отщепления водорода от второй оксигруппы лиганда. При pH 2
получается комплекс [TiKsl8 , в котором титан связан с лигандом
с помощью кислородных атомов сульфогрупп и обеих оксигрупп.
Комплексы с резорцином
Добавлением резорцина (H2R) к бензольному раствору TiCl4
получены комплексы RTiCl2 и RTi (HR)C1 [207].
Комплексы с диоксинафталином
1,8-Диоксинафталин при pH 1—4 с титаном образует соедине-
ние красного цвета с отношением металл : лиганд =1:2 (макси-
мум светопоглощения при 490 нм), а при pH 6 — желтый комплекс
с отношением металл : лиганд =1:3 (максимум светопоглощения
при 430 нм). Оба комплекса нерастворимы в воде, но растворяются
в бутиловом спирте и других органических растворителях [11, 13].
Комплексы с глицерином и маннитом
С глицерином и маннитом при pH 10,5 титан дает комплексы с
отношением компонентов 1 : 2 [78]. Соединения эти очень прочные,
константы нестойкости их равны 3,7 • 10~27 и 6,9 • 10-2' соответ-
ственно.
Комплексы с тироном
1,2-Дигидроксибензен-3,5-дисульфонат (тирон) образует с тита-
ном комплексные соединения желтого цвета [232]. Методами потенцио-
метрии и спектрофотометрии установлено, что при pH 2—9 образу-
ется два комплекса тирона (H4L) с титаном состава [TiO (HL)3]7"“
и [TiO (HL)2]8~ [260]. Константы устойчивости их равны 2,69 • 1025
и 9,24 • 1034.
В присутствии кальция из растворов выделяется кальциевоти-
тановый комплекс с тироном [144] желтого цвета. В интервале
pH 4—10 цвет переходит в красно-коричневый.
Комплекс с фенилфлуороном
2,2,7-Триокси-9-фенилфлуорон в солянокислом растворе обра-
зует с титаном окрашенный комплекс, имеющий максимум свето-
поглощения при 525 нм [340, 341]. Окрашенные комплексы с тита-
124
ном образуют и некоторые производные фенилфлуорона [342). Они
могут найти применение для колориметрического определения
титана.
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ ТИТАНА (III) СО СПИРТАМИ,
ФЕНОЛАМИ, НАФТОЛАМИ И ИХ ПРОИЗВОДНЫМИ
Известны комплексные соединения со спиртами, фенолами и наф-
толами и для трехвалентного титана. Гигенбах и Брубакер [204,
203] синтезировали комплексы трехвалентного титана со спиртами
следующего состава: TiCl3 • (СН3ОН)2, TiCl3 • (СН3ОН)4, TiCl3 •
. (СН3ОН)5, (C5H5N) TiCl4 • (СН3ОН)2, (C5H5N)2TiCl5 • (СН3ОН),
Н [TiCl4(C4H9OH)l.
Действуя на твердый TiCl3 спиртами в разных растворителях,
Зикмунд с соавторами [376] получил комплексы TiCl3 • (СН3ОН)4,
TiCl3 . (С2Н5ОН)4, TiCl3 • (изо-С3Н7ОН)4, TiCl3 • (С3Н7ОН)3, TiCl3 •
• (мзо-С3Н7ОН)3 и TiCl3 • (^зо-С4Н9ОН).
При действии на Ti (С2Н5О)4 и Ti [(СН3)2СНО]4 оксипроизвод-
ными (СН3)2С(ОН) СН2СН (ОН) СН3, о-С6Н4(ОН)2 и (НО) С6Н4СООН
выделены комплексы состава Ti(C2H5O)2L и Ti [(CH3)2CHO12L, где
L — бидентатный лиганд [170], а также Ti 1(СН3)2С (О)—
—СН2СН (О)—СН3] (С2Н5О)2 — вязкая жидкость, Ti (о-С6Н402) •
•(С2Н5О)2 — желто-красное вещество с точкой плавления 85—90° С,
Ti (о-С0Н4О2) [(СН3)2СНО]2 — желто-оранжевые кристаллы с
точкой плавления 112—114° С, Ti(OC0H4COO)(C2H5O)2 — жел-
тое вещество с точкой плавления 166—168° С, Ti(OC0H4COO) •
• [(СН3)2СНО]2 — оранжево-желтое вещество с точкой плавления
166—173° С. При взаимодействии Ti [(СН3)2СНО]4с СН2(ОН)СН2(ОН)
получен 1ермостойкий комплекс Ti [О(СН2)3О12.
Выделен бромидный комплекс (TiBr3)2[CH2(OH) СН(ОН) СН2СН3]3
и исследована его структура [1961. Кристаллы этого комплекса име-
ют моноклинную решетку с параметрами а = 15,61, b = 13,55, с =
= 14,05 А, Р = 113°10', ризм = 2,0, рреНТ-г = 2,08 г!см\ z = 4, фед.
гр. P2ilc. Структура состоит из дискретных комплексных анионов
[TiBr4(C4H40O2]~ и катионов [^c-TiBr2(C4Hi0O2)2]+. Атом титана
октаэдрически координирован с четырьмя атомами брома и двумя
атомами кислорода хелатного цикла.
Методом спектрофотометрии установлено, что TiCl3 в растворе
образует красно-коричневый комплекс с какотелином, красный с
ализарином и интенсивно зеленый с дитизоном [1381. Методом изо-
молярных серий показано, что во всех этих комплексах отношение
металла к лиганду равно 1 : 3. Какотелин и ализарин образуют
с титаном также комплексы с отношением компонентов 1:1.
Полярографическим методом констатировано существование в
растворе при pH 3—4,5 комплекса титана (III) с протокатехиновой
кислотой состава 1 : 1 [175].
125
КОМПЛЕКСЫ С Р-ДИКЕТОНАМИ
Р-Дикетонатные комплексы титана относятся к внутрикомплекс-
ным соединениям с хелатной структурой типа
В них атомы титана связаны двукратно с каждым остатком р-ди-
кетона. Получаются р-дикетонатные комплексы титана по реакции
взаимного обмена между солями титана и р-дикетонами в энольной
форме
TiX4 + п С-СНСО—R2\ =
\ ОН )
= TiX4-n
Rx — С-СНСО—R2\ +пНХ
I
о
По этой реакции получены комплексы состава Ti (асас)2Х2, где
X = F, С1, Вг, асас—(СН3—С=СНСОСН3) [191]. Исследование
О
их методом ПК-спектроскопии показало, что атомы фтора находятся
в транс-, а хлора и брома — в цис-положении [158]. Днс-строение
Ti (асас)2С12 подтверждено также методом ЯМР [325]. Кристаллы
этого комплекса имеют триклинную симметрию с параметрами ре-
шетки а = 11,315, b = 8,074, с = 9,04 А, а = 96,4, р = 99,25,
у = 87,0°, z = 2, фед. гр. Р1 или Р1 [158].
Из раствора хлората титана и ацетилацетона в хлороформе по-
лучено соединение Ti (асас)3С1О4 [316]. Это — гексагональные ко-
ричневые кристаллы с точкой плавления 184° С. Ацетилацетоновые
комплексы титана описаны Каррикье [156].
Синтезированы комплексы титана с ацетоацетанилидом (HL)
состава TiCl4 • 3HL, TiBr4 • 3HL, TiCl3L, TiClL3, TiBrL3, TiIL3
[321]. Получен также комплекс Til2(acac)2.
Согласно спектрофотометрическим измерениям [1821, титан (IV)
с р-метил-, Р-изопропил- и а-карбоксил-Р-метилтрополоном образу-
ет желтые комплексы с отношением компонентов 1 : 2; 1 : 3; 1 : 4.
В водных растворах титан (III) с ацетил ацетоном дает комплексы
с отношением компонентов 1 : 1; 1 : 2 и 1 : 3 [2531. Общие константы
устойчивости их в логарифмическом выражении таковы: 1g pt =
= 10,43, 1g р2 = 18,81 и 1g р3 = 24,92 соответственно.
При взаимодействии трихлорида титана с ацетилацетоном вы-
делен комплекс Ti (асас)3 в виде темно-синего вещества, возгоняю-
щегося в вакууме [164]. Исследование электронного строения
126
Ti (acac)3 указывает на отсутствие в нем чистых с^-орбиталей, а есть
смешанные л — ск-орбитали [132]. Поэтому наблюдаемые малоин-
тенсивные полосы в спектрах поглощения этого комплекса не отно-
сятся к d—d-переходам.
Добавлением TiCl3 в концентрированной НС1 к смеси ацетил -
ацетона (HL), Na2CO3, СбН6 и Н2О получен темно-синий раствор,
из которого окислением воздухом выделен кристаллический ком-
плекс [TiOL]2 [166].
Известен синий кристаллический комплекс Ti (C5HF6O2)3, рас-
творимый в эфире, хлороформе, метаноле, диметил карбониле и
нерастворимый в нитрофеноле [192]. Из раствора в бензоле выделен
желтый продукт состава [TiO (C5HF6O2)]n. Исследуя окисление
Ti (асас)3 кислородом воздуха методом ЭПР, Гарвей и Таппер [273]
установили образование ионов TiO2^. Синтезирован нерастворимый
в избытке ацетилацетона полимер [OTi (асас)2]л. [188].
Кокс с соавторами [157] получил комплексы Ti3+ с Р-дикетона-
ми (3-ацетиленацетоном, 3-цианоацетилацетоном, бензоилацетоном,
дибензоил метаном и трифтортеноил ацетоном). Комплексы образу-
ются при смешении растворов Ti2(SO4)3 и р-дикетонов (HL) и экс-
трагируются бензолом. Состав комплексов выражается формулой
TiL3. На воздухе они быстро окисляются, превращаясь в диамаг-
нитные соединения четырехвалентного титана состава TiOL2.
КОМПЛЕКСЫ С КАРБОНИЛЬНЫМИ СОЕДИНЕНИЯМИ
Карбонильные комплексы титана известны только как продукты
присоединения к галогенидам титана.
Известны комплексы TiF4 с ацетальдегидом TiF4 • СН3СНО,
диэтилкетоном TiF4 • (С2Н5)2СО, бензальдегидом TiF4 • С6Н5СНО
[187]. Т. Н. Сумарокова и Д. С. Сакенова [83] выделили аддукт с
фурфуролом TiCl4 • 2С4Н3ОСНО. Четыреххлористый титан с 5-Ы-ди-
алкиламинофурфуролами (L) образует продукты присоединения со-
става TiCl4 • L [115]. В случае S-N-диметил-, 5-М-диэтил-, S-N-пипери-
дино-, 5-М-марфолинофурфурола связь лиганда с титаном осуще-
ствляется через атом азота, а в соединениях 5-М-метил-М-фенилами-
нофурфурола в комплексообразовании участвует карбонильный
кислород [71].
При взаимодействии с тетрагалогенидами титана в бензольном
растворе бензатрона (L), антрона (L') и нитроантрона (L") образу-
ются кристаллические окрашенные соединения состава TiX4 • 2L,
TiX4 • 2L'(X = Cl, Br,I) и TiX4 • 2L"(X - Cl, Br) [288].
Получена серия комплексов TiCl4 с кетонами: TiCl4 •
• (СН3СбН4СОСН3-п), TiCl4 • (п-СН3СОСбН4СОСН3) (кристаллы
желтого цвета с т. пл. 133° С), TiCl4 • СН3С6Н4СОСН3 (кристаллы
желтого цвета, разлагаются при 110° С) [162]. Бензофенон образует
желтое кристаллическое вещество, плавящееся при 150° С, соста-
ва TiCl4 • 2(С6Н5)2СО [162]. В 2-н. соляной кислоте бензофеноновый
127
комплекс Ti 1(С6Н5)2СО]4 гидролизуется с восстановлением ти-
тана до Ti3+. При аминолизе получается тетрафенилэтилен (С6Н5)2С=
-С(С6Н5)2 [141].
В водных растворах титан (IV) с 2-(а-М-пиперидино)изопропил-4-
окситетрагидрофураном-3 дает устойчивый комплекс, состав кото-
рого еще не определен [30]. Он экстрагируется в органическую фазу
хлороформом, дихлорэтаном и ^-амиловым спиртом. Взаимодей-
ствие TiCl4 с некоторыми кетонами изучалось методом ИК-спектро-
скопии [31].
2,5-Диокси-З-ундецил-п-бензохинон (эмберлин) в кислой среде
с титаном образует комплексы переменного состава в виде осадков
грязно-зеленого цвета [368].
КОМПЛЕКСЫ С АМИНАМИ, АМИДАМИ И ИХ ПРОИЗВОДНЫМИ
Амины и амиды образуют комплексы присоединения к галогени-
дам титана. В этом разделе описаны преимущественно новые со-
единения, полученные в последние годы. Антипирин- и диантипирил-
фенилметан образуют с титаном в 80%-ном метаноле комплексы
с отношением компонентов 1 : 1 [96]. Константы образования их
равны 15,58, и 11,03 соответственно.
Смешением TiCl4 с ацетамидом (Am), N, N-диметилацетамидом
(Dm), ацетанилидом (Ап), бензамидом (Bad) и оксамидом (Od) в
растворе СН2С12 получены комплексы желтого цвета (TiCl4 • Am)2,
(TiCl4 • Dm)2, (TiCl4 • Bad)2, 2TiCl4 • Od с точками плавления 133—
134, 201—204, 215, 225° С (разлагается) соответственно и красный
комплекс TiCl4 • Ап с точкой плавления 117—118° С [331]. Коорди-
национная связь в комплексах желтого цвета, по данным ИК-спек-
тров, осуществляется через атом кислорода карбонильной группы.
В молекуле красного комплекса координация осуществляется через
атом кислорода и атом азота.
В концентрированных солянокислых растворах титан (IV) с N-
фураилдифенилгидроксиламином (HL) образует окрашенный ком-
плекс [Ti (HL)2C12]C12, при высушивании переходящий в TiL2Cl2
[681. Из сернокислых растворов (3—12-н. H2SO4) выпадает осадок
оранжевого комплекса TiL2SO4. В более концентрированных раство-
рах серной кислоты, по данным спектрофотометрии, образуются рас-
творимые комплексы H2[TiL2(SO4)2], H3[Ti(HL) (SO4)3] (HSO4)
и H5[Ti (HL) (SO4)3] (HSO4)3.
Ю. я. Харитонов с соавторами [99] из водных растворов TiCl4
с пиридином выделил комплексы (C5H5N)2HTiOCl3 • Н2О,
(C5H5NH)2TiOCl4 • Н2О, (C5H5NH)4Ti4O3Cl14 • 2C5H5N • ЗН2О,
(C5H5NH)2Ti2O3Cl4 • ЗН2О и (C5H5NH)2Ti4O7Cl4 • 5Н2О.
Добавлением раствора N-оксипиридина (L) в безводном спир-
те к спиртовому раствору TiO (С1О4)2 синтезирован комплекс
TiOL (С1О4)2 [242] в виде белого вещества, разлагающегося со взры-
вом при 215° С.
128
По реакции между комплексами Ti2OCl6 • 4CH3CN и Ti4O4Cl8 •
• 7С4Н8О2 [N (С2Н5)4]С1 в ацетонитриле получено соединение
[N (C2H5)4]2TiCl4O [194]. Судя по ИК-спектрам поглощения, в этом
комплексе присутствуют изолированные ионы TiCl4O2-.
При нагревании аддукта TiCl4 • CH3CN с хлорамином N (С2Н5)4С1
в растворе ацетонитрила образуются чешуйчатые кристаллы ком-
плекса N (C2H5)4TiCl5 • CH3CN [1931. Нагревание под вакуумом
(70° С) сопровождается отщеплением ацетонитрила и образованием
комплекса [(C2H5)4N]TiCl5.
Смешением раствора TiCl3(C2H5O) в ацетонитриле с хлорамином
N (С2Н5)4С1 получен желтый осадок состава [(C2H5)4N]TiCl4(C2H5O) •
• CH3CN. Он также отщепляет ацетонитрил при нагревании в ва-
кууме, образуя желтые кристаллы [(C2H5)4N]TiCl4(C2H5O). Ана-
логичным методом синтезированы комплексы (C5H5NH)2TiCl5 •
• [TiCl4(C2H5O) (СН3С)1 и (C5H5NH)2TiCl5(C2H5O) [193].
Действием на TiCl4 солянокислых третичных аминов
синтезированы легкоплавкие соединения [N (C2H5)3H]2TiCl6,
[C6H5N(CH3)2H]2TiCl6, [(u3o-C3H7)3NH]2TiCl6 и [C6H5N(CH3)2H]2TiCI6.
43301.
Препаративным путем получены продукты присоединения к
TiCl4 4-(Р-диэтиламиноэтил)-, 4-(Р-пиперидиноэтил)-, -2-(Р-диэтил-
аминоэтил)хинолина и 1,3-дипиперидино-2-(4'-хинолин)пропана
с отношением компонентов 1 : 1 [72]. К TiCl4 присоединяются две
молекулы 2-(Р-диэтиламиноэтил)-5-винил пиридина.
Нагреванием TiCl4 с бензоил ацетанилидом (HL) получен комп-
лекс коричневого цвета TiCl2L2 [320]. 1-Фенил-З-метил-бензолпи-
разолон образует с титаном (IV) комплекс, растворимый в бензо-
ле [58].
Изучены ИК-спектры поглощения комплексов титана с этилен-
диамином (Ей), 2,4-бутадиамином (BDM) и гексаметилендиамином
(GMD) состава TiEnCl4, TiEnBr4, Ti (BDM)C14, Ti (GMD) Cl4,
Ti (GMD)Br4 [219]. В комплексах TiEnCl4 и TiEnBr4 лиганд—хелат
с гош-конфигурацией. Исходя из предположения о аош-конфигу-
рации, произведено отнесение частот в ИК-спектрах поглощения
в комплексах TiEnCl4 и TiEnBr4 [1401.
В слабокислых растворах титан (IV) с папаверином и пирокате-
хином образует тройной оранжевый комплекс с отношением компо-
нентов 1:3:1 [32]. Он устойчив при pH 2,5—3,5. Молярный коэф-
фициент поглощения его 32000.
Методом ЯМР изучены комплексы N, N-диметилформамида
с TiCl4 [190].
Ароматические амины с тетрабромидом титана образуют амино-
производные состава [Ti{R (NH2)2)2]Br4 (с диаминами) и
[Ti (RNH2)4]Br4 (с моноаминами) [298]. Известны соединения с бен-
зидином, о-толлидином, о-фенилдиамином, дианизидином, о-нафтил-
диамином, анилином, о-толуидином, ^-толуидином, о-анизиди-
ном, о-фенетидином, n-фенетидином, бензоламином, п-толуидином,
9 1-1669 129
Р-нафтиламином, а-нафтиламином, и-анизидином [2981. Эти соедине-
ния имеют точки плавления выше 180° С, устойчивы при комнатной
температуре, окрашены в различные цвета.
При действии замещенными аминами на растворы тетраметилти-
тана в эфире при температуре —20° С образуются оранжево-красные
кристаллы тетраметиламиновых комплексов [Ti (Тк)41, где Тк-пири-
дин, 2,2'-дипиридил, 1,10-фенантралин, N, N, N', N''-пипераме-
тилендиамин [361].
Амиды кислот R—CONH2, где R = СН3, С3Н7, С4Н9, СбНи и
С6Н13, по реакции с TiS2 образуют послойные комплексные соеди-
нения [374]. Они располагаются в соединениях состава TiS2 •
. x(R—CONH2) между слоями TiS2, раздвигая их на расстоя-
ние, в 2—3 раза большее, чем в кристаллах чистого дисульфида
титана.
Смешением галогенидов титана с фталимидом [L], фталамидом
(L'), 1,2-циклогексафталимидом (L") и сукцинимидом (Lz//) получе-
ны комплексы состава 2TiCl4 • L, TiBr4 • 2L, 2TiCl4 • L, TiCl4 • L',
TiBr4. L', TiCl4 • L'",TiBr4 • L"',TiCl4 • L", TiBr4 • L" [233]. Это —
кристаллические вещества с точками плавления около 200° С.
Дипиколиновая кислота в солянокислых растворах (1-н. НС1)
образует с титаном (III) комплекс с максимумом светопоглощения
при 500 нм [161]. При действии на TiCl3 имидозолидина (этиленмо-
чевины) и антипирина (2,3-диметил-1-фенил-5-пиразолона) получены
комплексы состава [Ti (C3H6N2O)61C13 • 2С2Н5ОН, [Ti (C3H6N2O)e]I3,
[Ti (СиН12Ы2О)6]Вг3. 2C2H5OH и [Ti (ChH12N2O)61I3 [143]. Никоти-
намид c TiCl3 в растворе ацетонитрила дает комплексы с отношением
компонентов 2:1 и 3:1 [272], а диэтилентетрамин — комплекс
состава 2:1 [168].
Синтезированы комплексы титана (III) с пиридином
и тетраэтил амином состава (C5HN)3TiCl6, (C5H5N)3TiBr6,
(C6H5N)3TiCl4Br2, [(C2H5)4N1 TiCl4-2CH3CN, [(C2H5)4N] TiBr42CH3CN,
I(C2H5)4N] TiCl3Br • 2CH3CN [1951. При термическом разложении
аддуктов с ацетонитрилом получены комплексы (C2H5)4NTiCl4 и
(C2H5)4NTiBr4. Сплавлением диэтиламмонийхлорида с TiCl3 выделены
светло-желтые кристаллы [(C2H5)2NH2]3TiCl6 и светло-коричневый
продукт [(C2H5)2NH2]3Ti2Cl9. Аналогичным методом полу-
чены желто-коричневые комплексы состава [(C3H7)4N]3Ti2Cl9
и l(C3H7)4N]iдеПСЦдб П68].
Изучены магнитные свойства и определены параметры расщепле-
ния молекулярных энергетических уровней в поле лигандов следу-
ющих аминокомплексов титана (III): (C5H5N)3TiX6(X = Cl, Br),
[Ti [CO(NH2)2]6} X3(X = Cl, I), [Ti (H2NCH2COCH2NH2)6IC13 ,
• 2C2H5OH, [Ti (H2NCH2COCH2NH2)6]I3, Ti[C6H5C5H2(NH2)2OH]eBr8.
. 2C2H5OH, {Ti [CeHBC5H2(NH2)2OH]6)I3 [343].
130
Комплексы с этилендиаминтетрауксусной кислотой
Комплексообразование титана (IV) с ЭДТА еще не изучено. Ти-
тан (III) в кислых растворах с ЭДТА (Н4Ь) образует комплекс
(HTiL), диссоциирующий с отщеплением протона [286].
[HTiL] = Н+ + [TiL“].
Константа диссоциации в логарифмическом выражении 1g =
== —2,02. В щелочном растворе этилендиаминатный комплекс
гидролизуется по реакциям:
[TiL-] + Н2О = [TiLOH-2] + Н+,
[HTiLOH2-] + Н2О = [TiL (ОН)Г[ + Н+.
Константы гидролиза
, __ [Н+] [TiLOH2~~] b _ [Н+] [TiL (ОН)^]
2~ [TiL"”] И 3~ [TiLOH2“]
составляют lg k2 = — 8,64, lg ks = — 11,64. Этилендиаминатные
комплексы титана (III) образуют соединения типа двойных солей.
Синтезированы следующие двойные комплексы этого типа:
BaTiLCl • 8Н2О, LiTiL • 4Н2О, HTiL . ЗН2О, Ba (TiL)2 • ЮН2О
и MTiL • мН2О, где М = Na, К, Rb, Cs, NH4 [286].
Карбамидный комплекс
Мочевина в разбавленных кислых растворах с треххлористым'
титаном образует комплекс [TiCO (NH2)2]3+ [133]. Константа не-
стойкости его при 18—20° С и ионной силе раствора 0,261 равна
2,69. С трехиодистым титаном получен комплекс Til3 • 6СО (NH2)2
[172]. Поляризованная дублетная полоса в спектре поглощения это-
го комплекса с интервалом между максимумами 2500 см~У отнесена
к переходу 2л, + 2£ -> 22 (D3) центрального иона. Слабая зависи-
мость интенсивности данной полосы от температуры объясняется
действием эффекта Яна — Теллера.
Комплекс с карбоксилгалланилидом
А. Т. Слюсарев и А. Л. Гершунс [911 получили комплекс титана
с n-карбоксилгалланилидом (H2R) [TiR4]”4. Константа диссоциации
его
[TiO2+] [H+j2[R2—= 4>3 . 10-43,
4
Комплексы с диантипирилметаном
В солянокислых растворах с концентрацией 0,1—5-н. дианти-
лирилметан (Dp) с титаном образует комплекс с отношением
Ti : Dp = 1 : 3 [40,631. Константа устойчивости его при температуре
9*
131
20° С и ионной силе раствора, равной 1, составляет [191:
[TiDps]
[Ti4+l[Dp]3
= 7,8 • 107.
В присутствии роданид-ионов образуется несколько комплексов
тройного состава, которые окрашены и используются для колори-
метрического определения титана. В 2-н. соляной кислоте домини-
рует комплекс с отношением Ti : Dp : CNS =1:1:4 [191.
Окрашенная комплексная соль (DpH)2Ti (CNS)6 выделена в
чистом виде [94]. При растворении ее в хлороформе она распадается
на малорастворимые комплексы DpTi (CNS)4 и Dp [(HCNS)21.
Добавка к хлороформному раствору двукислого роданида
диантипирилметана сдвигает равновесие в сторону образования
(DpH)2Ti (CNS)6 [9]. Роданидные соли диантипирилметана и рада-
нистоводородной кислоты повышают растворимость окрашенного
титанового комплекса.
В растворе метанола (80%-ный водный раствор) диантипирил-
метан вступает в соединение с титаном в отношении 1 : 1 [96]. Кон-
станта образования комплекса равна 186,0.
Диантипирил-о-оксифенилметан в нейтральном и слабощелочном
растворах образует с титаном плохо растворимые светло-желтые
комплексы, состав которых выражается общей формулой 2DpTiOX2,
где X = NO3, С ЮГ, С1ОГ, CNS“, 1“ [25]. Из растворов хлоридов
и сульфатов осадки не выпадают, но растворы окрашиваются
в оранжевый цвет.
Комплексы с дипиридилом
Действием на TiCl3 2,2'-дипиридила (D) получено комплексное
соединение [TiCl2D2]Cl в виде бесцветных кристаллов [360]. При
восстановлении смеси дипиридила с четыреххлористым титаном в
тетрагидрофуране (ТГФ) металлическим литием получены кристал-
лические осадки состава TiD3 и LiTiD3 • 3,7ТГФ [230, 231]. Пер-
вый комплекс диамагнитен. Игольчатые кристаллы его имеют фио-
летовый цвет и быстро окисляются кислородом воздуха. Кристаллы
второго соединения в виде листочков черного цвета также легко
окисляются кислородом воздуха и даже водой.
Фоульс и Лестер [197] изучили комплексообразование
титана (III) с дипиридилом (Dp) методами спектрофотометрии,
ИК-спектроскопии, магнитных измерений и электропроводно-
сти. Они установили существование следующих комплек-
сов: {[TiBr2(DpC5H5N)2l+Br-}, {TiBr3(DpC5H5N)^Br'_ • CH3CN},
{[TiBr2(DpC5H5N)2]+Br- • CHC13}, {[TiBr2(DpC5H5N)2]+TiBr4
(DpC5H6N)-}, {[TiCl2(DpC6H6N)2]+TiCl4(DpC5H5N)-}.
132
Комплексы с фенантралином
Фенантралин вступает в реакции комплексообразования с трех-
и четырехвалентным титаном. М. И. Штокало [НО] металлиндика-
торным методом показала, что титан (IV) с фенантралином (Ph)
образует комплекс состава (TiOPh)2+. Константа диссоциации его
при pH 4 и 5 равна 1,1 • 10~"8 и 2,1 • 10“9 соответственно. С титаном
(IV) известны трифенантролиновых комплекса состава TiPh4(H2O)43+
[TiPh2(H2O)2]3+ и [TiPh3]3+, константы диссоциации которых со-
ставляют 6,56 • 10~5, 5,1 • 10-9 и 3,16 • 10~13 [100]. Фенантрали-
новые комплексы предложены для химико-аналитического опреде-
ления титана (III) в присутствии титана (IV) и для разделения их.
КОМПЛЕКСЫ С ЭФИРАМИ
Комплексные соединения с простыми и сложными эфирами из-
вестны для галогенидов титана. Они описаны нами в соответствую-
щей главе первой части книги. Приводим некоторые дополнитель-
ные сведения.
Исследованы магнитные и магнитооптические свойства комплек-
сов TiCl4 с алифатическими эфирами состава 1 : 1 и показано, что
в основе комплексообразования между этими компонентами лежит
донорно-акцепторная связь О+—Ti”.
Получены комплексы TiCl4 с эфирами циклогександиолов и трио-
лов, а также с моно- и олигосахаридами 1165]. Тетрагидрофурано-
вый комплекс TiCl3 • ЗТГФ при нагревании разлагается с образо-
ванием соединений состава TiCl3 • 2ТГФ и TiCl3 • ТГФ [375]. Ком-
плекс TiCl3 • 2ТГФ зеленого цвета получается при смешении
TiCl3 • ЗТГФ с гексаном. В присутствии бензола образуются кри-
сталлы TiCl3 • 2ТГФ • СвН6 зеленого цвета.
Эскулатин (лактон триоксикоричной кислоты) и 4-метилэскула-
тин в водных растворах образуют с титаном комплексы с отношени-
ем металла к лиганду 1 : 3 [239]. Комплексы имеют максимумы све-
топоглощения при 420 и 400 нм соответственно.
КОМПЛЕКСЫ С НЕПРЕДЕЛЬНЫМИ УГЛЕВОДОРОДАМИ
Методом спектрофотометрии показано, что TiCl4 в растворе че-
тыреххлористого углерода дает комплексы типа 1:1с пропиленом,
транс- 2-бутеном, изобутеном, триметилэтиленом, тетраметил эти-
леном, стиролом, /пранс-2-стильбеном, 1,1-дифенилэтиленом, три-
фенилэтил еном, тетрафенилэтиленом, 1 -бутеном, транс-транс-
транс- и ^ис-траяс-гпрбшс-циклододекатриеном и циклогексеном
1247].
Тетрахлорид титана образует комплексные соединения с карбо-
ксилкаротиноидами [313]. Синтезированы комплексы TiCl4 • L,
133
где L — кантаксантин, родоксантин или (3-апа-8'-каротиналь, и
комплексы 2TiCl4 • L, где L — родоксантин или р-апа-8'-кароти-
наль.
Додекопрено-|3-каротин с TiCl4 в растворе бензола образует ком-
плекс [TiCl3L]Cl [362]. Раствор его в диметилформамиде имеет по-
лосу светопоглощения при 560 нм.
КОМПЛЕКСЫ С ОКСИХИНОЛИНОМ
Титан с 8-оксихинолином (НА) образует комплексы, будучи
в трех- и четырехвалентном состоянии. Связь между ионами титана,
как впрочем и ионами других металлов, и 8-оксихинолином осуще-
ствляется через кислород оксигруппы после отщепления водорода.
Стивенс [3281 методом хроматографии на бумаге наблюдал обра-
зование между TiCl3 и 8-оксихинолином в этанольном растворе ком-
плексов зеленого цвета. Эти комплексы устойчивы только в атмо-
сфере инертного газа. Спиртовые растворы имеют максимум свето-
поглощения при 660 нм. Из хлорнокислых растворов титана
хлороформ экстрагирует оксихинолиновый комплекс состава
Ti (ОН)2А2 [256]. Комплекс подвергается ступенчатой диссоциации
с образованием ионов Ti (ОН)2А+. Из растворов солей титана, со-
держащих перекись водорода, хлороформ экстрагирует оксихи-
нол инпероксидные комплексы [39]. Растворы их в хлороформе имеют
полосы светопоглощения с максимумами при 360 и 430 нм.
Титан (IV) с оксихинолином в системе хлороформ — вода обра-
зует комплексы переменного состава [Ti (OH)„Am,4~("+,n) 1851. Пре-
паративным методом выделены галогенхинолятные и алкоксихино-
лятные комплексы титана в индивидуальном состоянии.
Взаимодействием TiCl4 с 8-оксихинолином получен бш>(8-окси-
хинолинато)титандихлорид Ti (OC9H6N)2C12 [323, 319]. Это моно-
клинные кристаллы с параметрами а = 14,06, b = 8,54, с = 14,97 А,
Р = 111°, ррентг = 1,16, г!см3у рИзм = 1,64 г/см\ г = 4, фед. гр. С2/с.
Аналогичным методом получены комплексы Ti (OC9H6N)2F2
и Ti (OC9H6N)2Br2 [189].
Нагреванием бис-(8-оксихинолинат)титандихлорида с триэти-
ламином в бензоле выделен красный кристаллический комплекс
тетракис-(8-оксихинолинато)титана TiA4 [167]. Нагреванием TiCl4
и 8-оксихинолина [3651 в бензоле с последующим гидролизом про-
дуктов реакции получены твердые комплексы (TiOA2)2 • Н2О
и(ТЮА2)4 • Н2О. По реакции 8-оксихинолина с ортоэфирами тита-
на в бензоле выделены твердые окрашенные комплексы состава
(RO)2TiA2, где R = С2Н5, пзо-С3Н7, С4Н9.
При действии тетрагалогенидов на 8-оксихинолин в хлороформе
синтезированы аддукты TiF4 • п (HOC9H6N), TiBr4 • п (HOC9H6N),
Til4 • п (HOC9H6N) [1891, где п = 1, 2 или 4. Нагреванием бис-(8-
оксихинолинато)титан дифторида с 8-оксихинолином при 160° С
с последующей сублимацией в вакууме получен фторэфир с тремя
134
хинолинатными остатками Ti(OC9H6N)3F. Кипячением тетрахло-
ридов титана с 8-оксихинолином в хлороформе получены галоген-
эфиропроизводные Ti (OC9H6N)3Br и Ti (OC9H6N)3I. Пиридин и
аддукт TiCl4 • HOC9H6N при кипячении дают коричневый смешан-
ный аддукт TiCl4 • (HOC9H6N) • (C5H5N). Аддукт TiCl3OC9H6N •
• (C5H5N) образуется при смешении растворов пиридина и тетрахло-
рида титана в хлороформе. Смешением 8-оксихинолинатотитантри-
хлорида с 8-оксихинолином в хлороформе выделен аддукт
TiCl3OC9H6N • (HOC9H6N). Цирконий и вольфрам соосаждаются
с титаном в виде оксихинолинатов [2991.
КОМПЛЕКСЫ С КУПФЕРРОНОМ, НЕОКУПФЕРРОНОМ
И П-ФЕНИЛКУПФЕРРОНОМ
N-Нитрозофенилгидроксиламин (купферрон), N-a-нафтил гид-
роксиламин (неокупферрон) и N-n-ксилилгидроксиамин в кислых
растворах образуют с титаном комплексы состава TiX4 [185], где
X — остаток лиганда [185]. Связь лигандов с титаном в комплексах
осуществляется посредством кислорода гидроксильной группы пос-
ле отщепления атома водорода. Эти соединения плохо растворимы
в воде и разбавленных кислотах. При температуре 25° С раствори-
мость в воде титановых комплексов с купферроном, неокупферро-
ном и n-фенилкупферроном равна соответственно 2,3 • 10~7,
3,2-10~7 и 2,5- 10~8люлб/л. Купферроновые комплексы используют-
ся для осаждения титана из водных растворов для отделения его от
щелочно-земельных металлов при химическом анализе руд и мине-
ралов.
Состав купферронатных комплексов титана зависит от среды,
из которой производится их осаждение [42]. Из солянокислых раст-
воров титан осаждается в виде нормального купферроната состава
TiX4. Из разбавленных сернокислых растворов в осадок выпадают
основные купферронаты титана переменного состава. При концен-
трации H2SO4 более 2-н. образуется нормальный купферронат.
Изучена диссоциация титанкупферронатного комплекса TiX4
[57]. Последовательные константы устойчивости титановых ком-
плексов составляют 1g = 10,39, 1g р2 = 10,07, 1g рз = 9,75,
Р4 = 9,43 (температура 10° С, pH 1,5, среда СНС13 + Н2О +
+ НС1О4, ионная сила 3,0). Константа распределения купферронат-
ного комплекса титана между водной фазой и хлороформом равна
|а2,26
АЛИЗАРИНОВЫЕ КОМПЛЕКСЫ
Ализарин S с трех- и четырехвалентным титаном в отдельности
комплексных соединений не образует. Смесь же солей трех- и четы-
рехвалентного титана с ализарином S образует соединение зеленого
цвета с максимумом светопоглощения при 754 нм [252]. Эти ком-
плексы аналогичны крапп-лакам, получающимся при действии на
135
ализарин смеси солей двух- и трехвалентных металлов, например
железа и алюминия. Определена константа диссоциации ализари-
нового лака, равная 2,7 • 10~5 [183].
КОМПЛЕКСЫ С АЗОКРАСИТЕЛЯМИ
Окрашенные комплексы с титаном образуют азокрасители-
производные нафталина и бензола: HO3S (NH2) (ОН)С4оН4—N=
=N—(С6Н4)2—N=N—С6Н3(ОН)СООН, HO3S (NH)2(OH)C10H4-N=
=N—C3H4—N=N—C6H3(OH)COOH и O2N (Cl) (OH)C6H2— N=
=N—Ci0H5(OH)SO3Na [98]. Последний краситель с титаном дает
комплекс розового цвета, в котором отношение металла к лиган-
ду равно 1 :2.
3,4-Диокси-4-хлоразобензол с титаном образует окрашенные
комплексы с отношением металл : лиганд =1:1 и 1:3 [318].
Нагреванием тетршшс-(изопропокси)титана в бутаноле с азо-
красителем (4-нитро-2-аминофенол и 1-фенол-3-метил-5-пиразолон)
получен комплекс с отношением титан: краситель =1:2 [134].
КОМПЛЕКСЫ С ФТАЛОЦИАНИНОМ
Известен темно-фиолетовый комплекс титана с фталоцианином,
имеющий состав TiClC32H16N8 [359]. Он образуется при кипячении
раствора TiCl3 и диметилфталоцианина с хинолином. Из раствора
темно-фиолетового комплекса в кипящем хинолине при доступе
воздуха выделены кристаллы фенолятного вещества состава TiO •
• O32Hi6N8.
Нагреванием при температуре 180° С четыреххлористого тита-
на и фталоцианина в хинолине получен фталоцианинтитандихлорид
TiCl2C32H16N8. Соединение идентифицировано по ИК-спектрам по-
глощения [302].
КОМПЛЕКСЫ С КУМАРИНОМ
Четырехвалентный титан с 3-бензил-4,5-диоксикумарином об-
разует комплексное соединение с отношением компонентов 1 : 3
[2401. Комплекс устойчив до pH 2,7, а в более щелочных растворах
претерпевает гидролиз.
7,8-Дигидроксикумарин реагирует с титаном (IV), образуя со-
единение с отношением компонентов 3 : 1 [241]. Строение комплекса
изображается формулой
136
КОМПЛЕКСЫ С КВЕРЦЕТИНОМ
Впервые на комплексообразование титана с кверцетином обратил
внимание И. П. Алимарин с соавторами [2] при изучении спектров
поглощения растворов. В. А. Назаренко и соавторы [67] показали
спектрофотометрическим методом, что титан (IV) с кверцетином (H2L)
в 40%-ном водно-спиртовом растворе образует комплекс состава
Ti (OH)2(HR)2. Максимум светопоглощения комплекса находится
при 440—460 нм.
Изучена экстракция кверцетинового комплекса титана бутано-
лом и показано, что в органическую фазу извлекается соединение
с отношением компонентов 1 : 1 [1731. Константа нестойкости ком-
плекса равна 5 • 10-5. Предложен метод экстракционно-фотометри-
ческого определения титана с помощью кверцетина.
КОМПЛЕКСЫ с ЭРИОХРОМЧЕРНЫМ
Спектрофотометрическим методом показано, что титан (III) с
эриохромчерным (H3L) в водном растворе образует комплек-
сы Ti (H2L)3 (pH 4, 2), Ti (HL)2~(pH7—8) и TiL (pH 12) [139]. Добав-
ление раствора TiCl3 к раствору эриохромчерного приводит к обра-
зованию красно-коричневого осадка состава Ti(H2L)3.
В 98%-ном спирте титан (IV) с эриохромчерным дает комплекс
с отношением компонентов 1:2.
КОМПЛЕКСЫ С ПИРИДИЛАЗОРЕЗОРЦИНОМ
4-(2-Пиридилазо)резорцин с титаном (IV) в кислой среде обра-
зует комплекс с отношением компонентов 1 : 1 [109]. Ему приписы-
вается строение
N=N-----/ОН
N......TiO 0
СН3СОО
В щелочной среде при pH 8 обнаружено существование второго ком-
плекса с отношением титан : лиганд =1:2.
КОМПЛЕКС С КРИПТОКСАНТИНОМ
Получен комплекс TiCl4 с криптоксантином или криптоксантин-
пальмитином (L) состава [TiCl3L]Cl [364]. Строение его установлено
по данным спектрофотометрических измерений.
137
ГЛИОКСИМАТНЫЕ КОМПЛЕКСЫ
Синтезированы комплексы титана (IV) и титана (III) с диметил-
(H2L), дифенил глиоксимом (H2L'), а- и fj-бензилмонооксимами
[169]. Последний состоит из двух геометрических изомеров. У «-изо-
мера (a-HL") ОН-группы расположены в транс-, а у р-изомера
(P-HL") - в цис-положении относительно карбонильной группы.
Суспензированные в хлороформе или петролейном эфире лиганды
образуют с титаном окрашенные комплексы состава TiCl4 • H2L,
TiCl4 • 2H2L, TiCl3 • HL, TiCl3 • HL', TiCl4 • a-HL", TiCl4 • ₽-HL"
и TiCl3 • HL' • HL". Первые четыре соединения при нагревании до
100° С взрываются. Получены также аддукты с тетрагидрофураном
TiCl2L" • ТГФ и хлороформом TiCl4 • 2 (a-HL") • 0,5СНС13 и TiCl4 •
• 2 (P-HL") • 0,5 СНС13.
КОМПЛЕКСЫ С ЭЛЕМЕНТООРГАНИЧЕСКИМИ СОЕДИНЕНИЯМИ
КОМПЛЕКС С БУТИЛЛИТИЕМ
При добавлении бутиллития к раствору четырех хлор истого ти-
тана в гексане титан восстанавливается до трехвалентного и с избыт-
ком реактива образует тройной комплекс [49]. Состав его еще не
выяснен.
КОМПЛЕКСЫ С БОРПРОИЗВОДНЫМИ
Кикер и Шрам [244] по реакции между TiCl4 и [(CH3)2N]4B2 или
[(CH3)2N]2 ВС1 получили комплекс {[(CH3)2N]2BC1 }2 • 3TiCl4.
бш?-(Циклопентадиенил)титан реагирует с (С6Н5)2ВС1, ВВг3, или
ВС13 в эфире, давая черно-фиолетовые соединения [324].
Синтезированы борпроизводные состава (C5H5)2TiClB(C6H5)2,
(C5H5)2TiQBCl2 и (C5H5)2TiBrBBr2. При взаимодействии [(CH3)2N]4B2
с избытком TiCl4 получено мелкокристаллическое коричневое веще-
ство ЦСН3)2М]2ВС1 • 2TiCl3 [245].
Действуя на [(CH3)2N]2BC1 • 2TiCl3 хлористым водородом под
давлением, авторы [246] синтезировали продукт присоединения,
которому приписывают строение {[(CH3)2NH]2BC12} • Ti2Cl7, рас-
сматривая его как двуядерный со связью Ti—Ti. На существование
в составе этого комплекса катиона и аниона указывают ПК- и УФ-
спектры поглощения. Вероятной структурой кластера [Ti2Cl^~]
является следующая: атомы титана имеют тригонально-бипирами-
дальное окружение из атомов хлора, причем три атома хлора из
семи являются мостиковыми.
Нагреванием комплекса {[(CH3)2NH]2BC12} • Ti2Cl7 получен про-
дукт пиролиза [(CH3)2NH2] • Ti2Cl7 и летучий бораминохлорид
[CH3]2NBC12.
138
КОМПЛЕКСЫ С СЕЛЕНПРОИЗВОДНЫМИ
В кислой среде титан образует осадки с бензол- и нафталинсе-
ленитами состава TiO (C6H5Se2O)2 и TiO (C10H7Se2O)2 [92]. Они
• малорастворимы в кислотах и предложены для осаждения титана
из растворов при химических анализах.
КОМПЛЕКСЫ С АРСИНПРОИЗВОДНЫМИ
Получены осадки титанарсиновых комплексов состава
[CH3As(CH2)3As(CH3)2]2 • 2TiCl4 (белый), [CH3As (CH2)3As (СН3)2]2 •
• 3TiF4 (с малиновой окраской), [CH3As (CH2)3As(CH3)2]2 . TiBr4
(желтый) и [CH3As(CH2)3As(CH3)2]2 • Til4.
При смешении раствора TiBr4 и смеси изомеров цис- и транс-
1, 2-бпс-(диметил)арсинэтилена (L) выделены комплексы состава
TiBr4- L [159]. Вначале при наличии семикратного избытка лиган-
да выпадает осадок светло-красных кристаллов ф/с-изомера, а за-
тем темно-красные кристаллы транс-изомера. Оба комплекса легко
сублимируются без разложения.
о-Фенилен-бпс-(диэтил)арсин с TiCl4 и TiBr4 образует комплексы
с отношением компонентов 1 : 1 [160]. При избытке лиганда полу-
чаются комплексы с отношением титан : лиганд =1:2.
КОМПЛЕКСЫ С ФОСФОРПРОИЗВОДНЫМИ
В системе TiCl4 — НО — Н2О — трибутилфосфат (ТБФ) обнару-
жен комплекс состава [Н3О • ТБФ]^ [Ti (ОН)2С14]2~. Константа
образования его равна 0,1 [86].
Л. К. Чучалин и соавторы [105], смешивая растворы ТБФ в СС14
и TiCl3 в воде, получили комплекс TiCl3 • 2 (С4Н9О)3РО. Молекула
ТБФ и ионы хлора в этом соединении, согласно спектральным дан-
ным, связаны с титаном координационной связью.
Четыреххлористый титан образует непрочные комплексы с ди-
ал кил фосфорными кислотами [61]. Координационная связь титана
с лигандами в этих комплексах осуществляется через атом кислоро-
да фосфорильной группы. Комплексы TiCl4 с диалкилфосфорными
кислотами легко претерпевают превращения с образованием внут-
рикомплексных полимерных продуктов. Изучена экстракция три-
бутилфосфатных производных титана из водных растворов орга-
ническими растворителями [74, 148].
Даль и Блок [170], действуя на Ti [(СН3)2СНО]4 фосфорпроиз-
водным С6Н5РО (ОН)СН2РО(ОН)С6Н5, получили комплек-
сные соединения Ti [С6Н5РО (О)СН2РО (О)С6Н5]2[(СН3)2СНО]2 и
Ti [С6Н5РО (О)СН2РО (О)С6Н514. Галогениды титана с Р-фосфон-
карбонильными соединениями (ФС) образуют аддукты TiT4 • ФС
и TiT4 • 2ФС (Г — галоген)! 1161. В соединениях первого типа связь
титана с лигандом осуществляется за счет карбонильной и фосфо-
рильной групп.Комплексообразование в соединениях второго типа
происходит с участием только фосфорильной группы.
139
Литература
1. Астахов А. И., Князев Е. Н., Блейхер Я- И., Ш н а й-
д е р м а н С. Я-— Ж. общ. хим., 1970, 40, 347.
2. Алимарин И. П., Головина А. П., Кутейников А. Ф.,
Степанов Н. Ф.— Вести. МГУ. Сер. мат. мех., астрон., физ., хим.,
1958, 203.
3. Б е л я е в И. Н., Бондарева С. В., Скибина Н. Д.— Ж-
неорг. хим., 1968, 13, 2620.
4. Б е л я е в И. Н., Бондарева С. В.— Ж- неорг. хим., 1969, 14, 219.
5. Бабко А. К., Литвиненко В. А.— Ж- неорг. хим., 1965, 10, 2075.
6. Бабко А. К., Волкова А. И., ЛисиченокС. Л.— Ж- неорг.
хим., 1966, 11, 478.
7. Бабко А. К., Тананайко М. М.— Ж- неорг, хим., 1969, 14, 1618.
8. Б а б к о А. К., Мазуренко Е. А., Набиванец Б. И.— Ж-
неорг. хим., 1969, 14, 2079.
9. Б а б к о А. К-, Тананайко М. М., Лозовик А. С.— Укр. хим.
ж., 1969, 35, 1267.
10. Б а б к о А. К-, Волкова А. И.— Ж- общ. хим., 1951, 19, 1949.
11. Бабко А. К-, Попова О. И.— Укр. хим. ж., 1957, 23, 376.
12. Б а б к о А. К-, Попова О. И.— Ж- неорг. хим., 1957, 2, 147.
13. Бабко А. К-, Попова О. И.— Ж- неорг. хим., 1957, 2, 138.
14. Б а б к о А. К-, Д у б о в е н к о А. Л.— Ж- неорг. хим., 1959, 4, 372.
15. Б а б к о А. К., Волкова О. И.— ДАН УРСР, 1959, 12, 1336.
16. Бабко А. К., Гордеева Л. М.— Укр. хим. ж., 1960, 26, 762.
17. Бабко А. К-, У л ько Н. В.— Укр. хим. ж., 1961, 27, 101.
18. Б а б к о А. К-, Волкова А. И., Гетьман Т. Е.— Ж- неорг.
хим., 1961, 6, 354.
19. Бабко А. К-, Тананайко М. М.— Ж- неорг. хим., 1962, 7, 562.
20. Б а б к о А. К-, Т а н а н а й к о М. М.— Ж. неорг. хим., 1962, 7, 2549.
21. Бабко А. К-, Тананайко М. М.— Ж- неорг. хим., 1962, 7, 571.
22. Б а б к о А. К., Волкова А. И., Гетьман Т. Е.— Ж- неорг.
хим., 1962, 7, 284.
23. Б а б к о А. К., Волкова А. И., Гетьман Т. Е.— Ж- неорг.
хим., 1962, 7, 2167.
24. Д у б о в е н к о Л. И., Герасименко Е. И.— Укр. хим. ж., 1963,
29 255.
25. Б е й л е с Р. Г.— Зав. лабор., 1956, 22, 1296.
26. В л а с о в Л. Г., С т р и ж к о в Б. В., Л а п и ц к и й А. В., С а л и-
м о в М. А.— ДАН СССР, 1962, 145, 1055.
27. Волков И. М., Остроумов Э. А.— Труды Ин-та океанологии
АН СССР, 1965, 79. 81.
28. В а с и л ь е в В. П., Воробьев П. Н.— Изв. высш. уч. зав. Хим.
и хим. технол., 1968, 11, 971.
29. В а с и л ь е в В. П., Воробьева П. Н., Белякова А. Ф.—
Изв. высш. уч. зав. Хим. и хим. технол., 1969, 12, 115.
30. Г у л е н к о в Р. Г., Скороход О. Р.— Вести. Белорусск. ун-та.
Сер. 2, 1969, 15.
31. Гайворонский В. И.— Материалы IV Научной конференции аспи-
рантов. Изд. Ростовского ун-та, Ростов-на-Дону, 1962, 146.
32. Горьковская Г. П., Джнянбаева Р. X., Тали-
пов Ш. Т.— Узб. хим. ж., 1967, 3, 26.
33. Григорьева В. В., Кекендорф Л. П.— Bi’ch. КДУ. Сер.
ф1*з. та Х1*м., 1967, 7, 105.
34. Григалашвили К. И., Пятницкий И. В.— Укр. хим. ж.,
1968, 34, 402.
35. Гринберг А. А., Шихеева Л. В.— Ж- неорг. хим., 1960, 5, 599.
36. Д у м а н с к и й А. В., К ни га А. Г.~ ЖРФХО, 1928, 60, 229.
140
37. Долматов Ю. Д.— Лакокрасочные материалы и их применение, 1966,
5, 29.
38. Ж од н и н А. В., Долматов Ю. Д.— Ж« неорг. хим., 1969, 14, 1212.
39. ЖаровськийФ. Г., П i н а е в а С. Г.— BicH. КДУ. Сер. астрой.,
ф!з., Х1*м., 1962, 1, 120.
40. Живописцев В. П., Минин А. А.— Зав. лабор., 1960, 26, 1346.
41. Ионова Е. А.— Изв. АН СССР. Неорг. материалы, 1965, 1, 1853.
42. К а р л ы ш е в а К- Ф., М а л и н к о Л. А., Шека И. А.— Ж*
неорг. хим., 1966, 11, 540.
43. Карпинская Н. М., Андреев С. Н.— Ж- неорг. хим., 1968,
13, 50.
44. Кузнецова Е. М., Закурин Н. В., Никитин О. Т.— Ж-
неорг. хим., 1962, 7, 676.
45. К а л и н н и к о в В. Т., Зеленцов В. В., Жарких А. А.,
Алимов Г. Г.— Изв. АН СССР. Сер. хим., 1969, 2837.
46. Клейн К- Е.— Ж- общ. хим., 1952, 22, 17.
47. Кудрявцев А. С., Савич И. А.— ЖВХО, 1962, 7, 591.
48. К о р е н м а н И. М., Фрум Ф. С., Рыжков а Л. В.— Уч. зап.
Горьк. ун-та, 1958, 32, 113.
49. Коротков А. А., Артамонова И. Л.— Высокомол. соед.,
1962, 4, 145.
50. Князева Е. Н., Шнайдерман С. Я.— Ж- неорг. хим., 1965, 10,
1848.
51. К о г а н В. А., Соколова В. П., Осипов О. А.— Ж- общ.
хим., 1970, 40, 322.
52. Л а п и ц к и й А. В., С т р и ж к о в Б. В.— В кн.: Физико-химический
анализ. Изд-во СО АН СССР, Новосибирск, 1963, 232.
53. Л а п и ц к и й А. В., Ч ж у а н Я-у й, Савич И. А.— Вести. МГУ.
Хим., 1960, 5,78.
54. Л а п и ц к и й А. В., Ч ж у а н Я-у й, СавичИ. А.— Ж- неорг.
хим., 1961, 6, 653.
55. Л у ч и н с к и й Г. П.— Ж- общ. хим., 1937, 7, 2045.
56. Л у ч и н с к и й Г. П.— Зав. лабор., 1936, 5, 233.
57. Л о б а н о в Ф. И., Савостина В. М., Пешкова В. М.—
Вести. МГУ. Хим., 1969, 95.
58. Лобанов Ф. И., Савостина В. М., Пешкова В. М.—
Гестн. МГУ. Хим., 1969, 121.
59. Литвиненко В. А.— Укр. хим. ж., 1966, 32, 1115.
60. Литвиненко В. А.— Укр. хим. ж., 1966, 32, 1160.
61. М у р а т о в а А. А., Яркова Э. Г., П р и бы л ов а А. П., П у до-
в и к А. Н.— Ж- общ. хим., 1968, 38, 2772.
62. Мазуренко Е. А., Набиванец Б. И.— Укр. хим. ж., 1967, 33,
98.
63. М и н и н А. А.— Уч. зап. Пермского ун-та, 1956, 9, 177.
64. М о р о з о в И. С., Топтыгина Г. М.— Ж. неорг. хим., 1957, 2,
1629.
65. Н о в и к о в а И. В., Беляев И. Н.— Ж- неорг. хим., 1965, 10, 294.
66. Новикова Л. В., Беляев И. Н.— Ж- неорг. хим., 1966, 11, 940.
67. Н а з а р е н к о В. А., Бирюк Е. А., Р а в и ц к а я Р. В., Анто-
нович В. П.— Укр. хим. ж., 1968, 34, 115.
68. П и л и п е н к о А. Т., Шпак Э. А.— Укр. хим. ж., 1966, 32, 383.
69. Пат. США 2926183; РЖХим., 1961, 10П 667.
70. Пат. США 2824114; С. А., 52, 7743.
71. Пустоваров Б. С., Шелепина В. Л., Шелепин О. Е.,
Назарова 3. Н., Осипов О. А.— Ж- общ. хим., 1968, 38, 2763.
72. Панюшкин В. Т., Каган Е. Ш., Горновский А. Д.,
Осипов О. А., Семенова И. М.— Ж- общ. хим., 1967, 37, 1566.
73. Пятницкий И. В., Григалашвили К- И.— Сообщ.
АН Груз.ССР, 1970, 57, 53.
141
74. Пятницкий И. В., Харченко Р. С.— Укр. хим. ж., 1967, 33,
734.
75. Пятницкий И. В., Кравченко Т. И.— Укр. хим. ж., 1969,
35, 478.
76. П я т н и ц к и й И. В.— Изв. высш. уч. зав. Хим. и хим. технол., 1958,
6, 20.
77. П я т н и ц к и й И. В., К л и б у с А. X.— Укр. хим. ж., 1963, 29, 440.
78. П я т н и ц к и й И. В., К л и б у с А. X.—Укр. хим. ж., 1964, 30, 151.
79. П я т н и ц к и й И. В., К о с т ы н и н а А. П.— Изв. высш. уч. зав.
Хим. и хим. технол., 1960, 3, 794.
80. Пятницкий И. В., Харченко Р. С.— Укр. хим. ж., 1965, 31,
714.
81. Пешкова В. М., Ким Хен Ра к.— Вести. МГУ. Сер. мат., мех.,
астроном., физ., хим., 1958, 2, 187.
82. Резник Б. Е., Вороятягина В. Д.— Ж- неорг. хим., 1968,
13, 3080.
83. Сумарокова Т. H.f Сакенова Д. С.— Ж- общ. хим., 1962,
32, 3.
84. С о к о л о в В. П., Коган В. А., Осипов О. А., Румян-
цева М. И.— Ж- общ. хим., 1968, 38, 2489.
85. С а в о с т и н а В. М., Лобанов Ф. И., Пешкова В. М.— Ж-
неорг. хим., 1967, 12, 2162.
86. С т а р ц е в В. Н., Санников Ю. И., Беньян Г. Н., Кры-
лов Е. И.— Ж- неорг. хим., 1968, 13, 1122.
87. С т р и ж к о в Б. В., Л а п и ц к и й А. В., С и м а н о в Ю. П., В л а-
с о в Л. Г.— Ж- неорг. хим., 1962, 7, 2181.
88. С т р и ж к о в Б. В., Лапицкий А. В., Власов Л. Г.— Ж*
прикл. хим., 1960, 33, 2009.
89. С т р и ж к о в Б. В., Лапицкий А. В., Ц в е т к о в А. И., В л а -
сов Л. Г.—ДАН СССР, 1960, 133, 1347.
90. С у д а р и к о в Б. Н., Смирнов Л. М.— Ж- неорг. хим., 1956, 1,
2327.
91. С л ю с а р е в А. Т., Г е р ш у н с А. Л.— Укр. хим. ж., 1962, 28, 458.
92. С о т н и к о в В. С., А л и м а р и н И. П.— Научи, докл. Высшей
школы. Хим. и хим. технол. 1959, 2, 296.
93. С е м е н о в а Е. И.— ДАН СССР, 1962, 143, 1369.
94. Т а н а н а й к о М. М., Царенко Г. В.— Укр. хим. ж., 1965, 31, 530.
95. Т а н а н а й к о М. М., ЛозовикА. С.— Ж- неорг. хим., 1970, 15, 1070.
96. Т а н а н а й к о М. М., Царенко Г. В.— BicH. КДУ. Сер. ф1’з. та
х1м., 1967, 7, 146.
97. Т о р о п о в а В. Ф., Заббарова Р. С.— Изв. высш. уч. зав. Хим.
и хим. технол., 1969, 12, 1487. >
98. Фрум Ф. С., Тишина Е. П.— Уч. зап. Горьк. ун-та, 1958, 32, 119.
99. X а р и т о н о в Ю. Я., И о н о в а Е. А., Буслаев Ю. А.— Изв.
АН СССР. Неорг. материалы, 1968, 4, 720.
100. Церковицкая И. А., Новикова Е. И.— Ж- анал. хим; 1969,
24, 1160.
101. Черная Н. В., Шнайдерман С. Я-— Изв. высш. уч. зав. Цвет-
ная металлургия, 1966, 3, 370.
102. Черная Н. В., Шнайдерман С. Я-— Ж- общ. хим., 1966, 36,
1165.
103. Черная Н. В., Шнайдерман С. Я-— Вести. Киевского политехи,
ин-та. Сер. хим. машиностроения и технол., 1965, 1, 33.
104. Черняк А. С., Хамутников В. А., Мишнев Г. Г., Б о-
е з у е в А. А.— Ж- неорг. хим., 1969, 14, 1569.
105. Чучалин Л. К., Пецевицкий Б. И., К из ин И. А., Гран-
кина 3. А., Дулепов Е. В.— Ж- структ. хим., 1968, 9, 213.
106. Чжун Я-у й, Савич И. А., Лапицкий А. В., Самору-
ков В. Р., Титов Л. Г.— Вести. МГУ. Хим., 1960, 3, 40. ’
142
107. Ч ж у а н Я-уй, Лапицкий А. В., Савич И. А.— Вести. МГУ.
Хим., 1960, 4, 43.
108. Шевченко Ф. Д., Кузина Л. А.— Укр. хим. ж., 1965, 31, 347.
109. Штокало М. И.— Укр. хим. ж., 1967, 33, 319.
ПО. Штокало М. И.— Укр. хим. ж., 1968, 34, 630.
111. Штокало М. И.— Укр. хим. ж., 1962, 28, 555.
112. Шахова 3. Ф., Семеновская Е. Н.— Ж- неорг. хим., 1968,
13 1887
113. Шахова 3. Ф., Семеновская Е. Н.— Вести. МГУ. Хим., 1968,
2, 122.
114. Шахова 3. Ф., Семеновская Е. Н.— Ж. неорг. хим., 1962, 7, 1084.
115. Шелепина В. Л., Пустоваров В. С., Осипов О. А., На-
зарова 3. Н.—Ж- общ. хим., 1968, 38, 666.
116. Швец А. А., Осипов О. А., Шакиров А. М.— Ж. общ. хим.,
1967, 37, 2717.
117. Шнайдерман С. Я., Князева Е. Н.— Ж- неорг. хим., 1967,
12, 438.
118. Ш н а й д е р м а н С. Я-, Черная Н. В.— Вести Киевского по-
литехи. ин-та. Сер. хим. машиностроения и технол., 1968, 178.
119. Шнайдерман С. Я-, Черная Н. В.— Ж- неорг. хим., 1966,
11, 1292.
120. Шнайдерман С. Я., Черная Н. В.— Ж- неорг. хим., 1966,
11, 1297.
121. Шнайдерман С. Я-—Ж- неорг. хим., 1957, 2, 2122.
122. Шнайдерман С. Я-— Укр. хим. ж., 1957, 23, 92.
123. Шнайдерман С. Я-, Калиниченко И. Е.— Ж* неорг. хим.,
1961, 6, 1843.
124. Шнайдерман С. Я-, Калиниченко И. Е.— Изв. высш. уч.
зав. Хим. и хим. технол., 1961, 4, 897.
125. Шнайдерман С. Я-, Калиниченко И. Е.— ДАН СССР,
1961, 139, 910.
126. Шнайдерман С. Я-— Изв. высш. уч. зав. Хим. и хим. технол., 1963,
6, 364.
127. Шнайдерман С. Я-, Князева Е. Н.— Укр. хим. ж., 1964, 30,
1136.
128. Шнайдерман С. Я., Черная Н. В.— Ж- неорг. хим., 1965, 10, 224.
129. Шнайдерман С. Я., Князева Е. Н.— Укр. хим. ж., 1965, 31,
27.
130. Шнайдерман С. Я-, Князева Е. Н.— Укр. хим. ж., 1966, 32, 119.
131. Шнайдерман С. Я., Черная Н. В.— Ж- неорг. хим., 1966,
11, 134.
132. Юрченко Э. Н., Авдеев В. И., Щ у г а м Е. А.— ТЭХ, 1970,
6, 84.
133. Я син скене Э. И., Я цимирск и й К- Б.— Изв. высш. уч. зав.
Хим. и хим. технол., 1958, 2, 31.
134. Ямамото М., Маси о Ф.— J. Chem. Soc. Japan. Ind. Chem. Sec.,
1965, 68, 688.
135. Athavale V. T., Mathur P. K., S a t h e R. M.— Indian J. Chem.,
1968, 6, 323.
136. Athavale V. T., Krishnan С. V., Ramanathan P. S.,
Ven k a t ens war 1 u C.— Indian J. Chem., 1969, 7, 1040.
137. Athavale V. T., Krishnamoorthy T. S., Krishnan С. V.,
Ramanathan P. S., Ven ka t es war 1 u C.— Indian J. Chem.,
1968, 6, 528.
138. Akhtar S. M. S., Ahmad N., Rehman S. M. F.— Indian J.
Chem., 1966, 3, 253.
139. Akhtar S. M. S., Ahmad N., Rahman S. M. F.— J. Ind. Chem.
Soc. 1965 42 875.
140. Ashley P. J., T о г г i b 1 e E. G.— Canad. J. Chem., 1969, 47, 167.
143
141. В 1 a n с к E.— Liebbigs Ann. Chem., 1969, 724, 56.
142. Bradley D. C., Hursthouse M. B., Rendall I. F.— Chem.
Communs, 1969, 672.
143. Blazekova M., ScheaferH. L.— Z. anorg. Chem., 1968, 360, 169.
144. Barzily I., Yaalon D. H., A v i n u r P.— Israel J. Chem., 1967,
5, 289.
145. Barclay G. A., Gregor I. K., Lambert M. J., Wild S. B.—
Austral. J. Chem., 1967, 20, 1571.
146. Broadbent D., D о 1 1 i m о r e D., D о 1 1 i m о г e J.— Analyst,
1969 94 543.
147. Burley S. P.—Austral. J. Phys., 1964, 17, 543.
148. В 1 а к e С. A., В a e s C. F., Brown К. B.— Ind. Eng. Chem., 1958,
50, 1763.
149. Bending M., Hirschmiiller H.— Z. anal. Chem., 1940, 120, 385.
150. Barnes J.— J. Soc. Chem. Ind., 1896, 15, 420.
151. Brintzinger H., Eckardt W.— Z anorg. Chem., 1935, 224, 93.-
152. Ciavatta L., Liberti A.— Ricerca Sci., 1960, 30, 1186.
153. Chariot G.— Chim. analyt., 1953, 35, 51.
154. C a g 1 i о t i V., S a r t a r i G.— Gazz., 1936, 66, 741.
155. Sudsuo F., Kodso N., Maurice C.— J. Chem. Soc. Japan, Pure
Chem. Sec., 1964, 85, 305.
156. Carriques C. J. Contribution a la chimie des solutions aqueuses de ti-
tane tetravalent. Pubis scient. et techn. Ministere air, 1960, NN. T 93, V;
РЖХим., 1963, 15 683.
157. Cox M., Lewis J., N i h о 1 m R. S.— J. Chem. Soc., 1965, 2840.
158. Cox M., Clark R. J. H., M zi 1 1 e d g e H. J.— Nature, 1966, 212, 1357.
159. Clark R. J. H., Negrotti R. H. U.— Chem. and Ind., 1968, 5, 154.
160. Clark R. J. H.— J. Chem. Soc., 1965, 5699.
161. С о h e n-N о r d m a n n M.-E.— Bull. Soc. chim. France, 1967, 1820.
162. C u 1 i n a n e N. M., Chard S., Leyshon D. M.— J. Chem. Soc.,
1952, 4106.
163. Choundury P. C.— Ind. J. Chem., 1967, 5, 121.
164. Chakravarti B. N.— Naturwis., 1958, 45, 286.
165. Csaros Z., Deak G. et al.—Acta chim. Sci. Acad. Hung., 1968, 55, 347.
166. Collins R. E.— J. Chem. Soc., 1969, A, 1895.
167. Cuningham D. et al.—J. Chem. Soc., 1969, A, 2133.
1C8. Crouch P. C., F о w 1 e s G. W. A., Walton R. A.— J. Chem.
Soc., 1969, A, 972.
169. Charalambous J., Fraser M. J.— J. Chem. Soc., 1968, A, 2361.
170. Dahl G. H., Block В. P.— Inorg. Chem., 1966, 5, 1394.
171. Dean P. A., Evans D. F., Phillips R. F.— J. Chem. Soc.,
1969 A 363.
172. Dingle R.— J. Chem. Phys., 1969, 50, 545.
173. Dragulescu C., Simonescu T., Shiff H.— Rev. roumaine
chim., 1969, 14, 583.
174. D r e w M. G., F о w 1 e s G. W. A., Lewis D. F.— Chem. Communs,
1969, 876.
175. Dhaneschwar R. S.— Current Sci., 1969, 38, 61.
176. Douville F., Duval C., Lecomte J.— Compt. Rend., 1941, 212,
697.
177. Douville F., Duval C., Lecomte J.— Bull. Soc. Chim. France,
1942 9 548.
178. Dufet H.— Z. Kristallogr., 1897 , 27, 633.
179. D u f e г t H.— Bull. Soc. Min., 1895, 18 421.
180. D i 1 t h e n W.— Ber., 1904, 37, 588.
181. Dutt N. К., С о s w a m i N.— Z. anorg. Chem., 1959, 298, 258.
182. Dutt Y., Singh R. P.— Indian. J. Chem., 1966, 4, 424.
183. D о r t a-S c h a e p p i Y., Hiirzeler H., Treadwell W. D.—
Helv. Chim. Acta, 1951, 34, 797.
144
184. Delafosse D.— Compt. Rend., 1953, 236, 2313.
185. E 1 v i n g P. J., Olson E. C.— J. Am. Chem. Soc., 1956, 78 , 4206.
186. Eve D. J., Fowles G. W.— J. Chem. Soc., 1966, A, 1186.
187. Emeleus H. J., С a о G. S.— J. Chem. Sos., 1958, 4245.
188. Fernelius W. C. et al.— Wright Air Development Corp., Techn.,
1957 2 56____203.
189. Frazer M. J., Rimmer B.— J. Chem. Soc., 1968, A, 69.
190. F r a t i e 1 1 о A., Miller P., Schuster R.— Molecul. Phys., 1967,
12, 111.
191. Fag R. C., Lowry R. N. — Inorg. Chem., 1967, 6, 1512.
192. Fry F. H., Walt W. R.—J. Inorg. Nucl. Chem., 1968, 30, 3115.
193. F e I t z A.— Z. anorg. Chem., 1965, 338, 147.
194. F e 1 t z A.— Z. anorg. Chem., 1965, 334, 242.
195. Fowles G. W., Russ B. J.— J. Chem. Soc., 1967, A, 517.
196. Fowles G. W., Lester T. E., Wood G. S.— J. Inorg. Nucl.
Chem., 1969, 31, 657.
197. F о w 1 e s G. W. A., Lester T. E.— J. Chem. Soc., 1968, A. 1180.
198. Funk H., Rogler E.— Z. anorg. Chem., 1944, 252, 323.
199. Fichter F., Reichert F.— Helv. Chim. Acta., 1925, 7, 1078.
200. Faber P.—Z. analyt. Chem., 1907, 46, 277.
201. Gutmann V., L e i t m a n n O., Scherhaufer A., Czu-
ba H.— Monatsh., 1967, 98, 188.
202. Gutmann V., Scherhauser A., C z u b a H.— Monatsh., 1967,
98, 619.
203. Giggenbach W., Brubaker H.— Inorg. Chem., 1969, 8, 1131.
204. Giggenbach W., Brubaker С. H.— Inorg. Chem., 1968, 7, 129.
205. Gan a pa t hy I. S., Ven к a t es w a 1 u C.— Indian J. Chem., 1968,
6, 578.
206. Gervais D., Basso-Bert M., Lab ar re J.-F., G a 1 a I s F.—
Compt. Rend., 1968, C266, 1183.
207. Gopinathan C., Gupta J.— Indian J. Chem., 1966, 4, 374.
208. Galagher P. K-, Thomson J.—J. Am. Ceram. Soc., 1965, 48, 644.
209. Ghosh N. N., Bhattachoryya A.— J. Indian Chem. Soc., 1967,
44, 972.
210. Griffith W. P.— J. Chem. Soc., 1964, 5248.
211. Gastinger E.— Z. anorg. Chem., 1954, 275, 331.
212. Gregorozy K- Z.— Acta polon-pharmac. 1958, 15, 333.
213. Gregorozik Z.— Acta polon-pharmac., 1958, 15, 129.
214. G r i e 1 J. V., R о b i n s о n R. J.— Anal. Chem., 1951, 23, 1871.
215. Ginsberg H.— Z. anorg. Chem., 1931, 198, 162; 1933, 211, 401.
216. Gupta J.— Indian J. Phys., 1936, 10, 465.
217. G i u a M., M о n a t h E.— Z. anorg. Chem., 1925, 143, 383.
218. Hasan M., Sankhla B. S., Kapoor R. N.— Austral. J. Chem.,
1968, 21, 1651.
219. H о r 1 e у D., Terrible E. G.— Canad. J. Chem., 1965, 43, 3201.
220. Herrington K-, Kinsburg D. E.— J. Am. Chem. Soc., 1957,
79 5893
221. Hen d er son G. G., Orr T. W., Whit head R. J.—J. Am.
Chem Soc., 1899, 75, 54, 2.
222. Henderson G. G., Schutte O.— J. Am. Chem. Soc., 1899, 75, 542.
223. Haissinsky M., Emmanuel-Zaviziano H.— J. Chem.
Phys., 1937, 34, 641.
224. Hines E., Boltz D. F.— Anal. Chem., 1952, 24, 947.
225. H a b a s h у G. M.— J. Electroanal. Chem., 1964, 8, 237.
226. H а к о m о r i S.— Technol. Rep. Tohoku Univ., 1931, 9, 567, 607.
227. Hartmann H., Schlafer H. L., Hansen К- H.— Z. anorg.
Chem., 1957, 289, 40.
228. Hartmann H., Schlafer H. L., Hansen К. H.— Z. anorg.
Chem., 1956, 284, 153.
10 1-1669
145
229. Heintz E.
230. Herzog s.,
231. Herzog s.,
232. Harvey A.
4744.
233. Jain F. E.,
234. L e s e F. E.,
235. J e r e G. V.,
236. J e r e G. V.,
237. J e r e G. V.,
238. J e r e G. V.,
239. Jain B. D.,
240. Jain B. D-,
241. Jain B. D„
242. К h a r k a r D
243. К h a r k a г D
A.— Nature, 1963, 197, 690.
Taube R.— Z. anorg. Chem., 1960, 306, 159.
Taube R.— Angew. Chem., 1958, 70, 469.
E., Manning D. L.— J. Am. Chem. Soc.,
1952, 74,
R i v е s t R.— J. Inorg. Nucl. Chem., 1969, 31, 399.
Hartmann H.— Z. phys. Chem., 1951, 197, 239.
Patel С. C.— J. Inorg. Nucl. Chem., 1961, 20, 343.
Patel С. C.— Nature, 1962, 194, 470.
Patel С. C.— Proc. Indian Acad. Sci., 1963, A57, 69.
Patel С. C.— Indian J. Chem., 1964, 2, 383.
Singh H. B.— J. Indian Chem. Soc., 1964, 41, 29.
, Kumar R.— Proc. Indian Acad. Sci., 1964,A60, 265.
Singh H. B.— Indian J. Chem., 1964, 2, 245.
. P.,, Patel
С. C.— Current Sci., 1956, 25, 56.
С. C.— J. Indian Inst. Sci., 1957, AB39, A41.
E. P.— J. Am. Chem. Soc., 1968, 90, 3672.
E. P.— Inorg. Chem., 1969, 8, 2306.
E. P.— Inorg. Chem., 1969, 8, 2319.
.— Z. Naturforsch., 1965, 20b, 630.
N.— J. Inorg. Nucl. Chem., 1967, 29, 2961.
249. Krishnan V., Patel С. C.— Canad. J. Chem., 1965, 43, 2685.
250. К 6 p f H., R a t h 1 e i n К- H.— Angew. Chem., 1969, 81, 1000.
251. Kapoor R. N., Pandl
Chem., Soc., 1958, 35, 157.
252. Ким Хен Рак — Хвахак ка хвахак конон, 1958, 2, 285; РЖХим.,
1959, 53134.
244. К у k e r G. S., Schram
245. К у k e r G. S., Schram
246. К у k e r G. S., Schram
247. Kraus s H.-L., N i c kl J
248. Khan M. M., Ahmad
Mehrotra R. C.— J. Indian
253. Linden W. E., В о e f G., Den A.— Anal. Chim. Acta., 1967, 37,
179.
254. Lewis D.— J. Phys. Chem., 1958, 1145.
255. Lutschinsky G. P.— Z. anorg. Chem., 1935, 225, 321.
256. Liberti A., Chi an tel 1 a V.— Ann. Chim., 1962, 52, 495.
257. Liberti A., Giombini G., С e r v о n e E.— Ricerca Sci., 1955,
25 883
258. Levy L.— Ann. Chim. Phys., 1892, 6, 25, 433.
259. Levy L.— Compt. Rend., 1889, 108, 294.
260. L a n d i M. F.— Ricerca Sci., Parte 2, Sez A, 1964, 7, 165.
261. Mori M., S h i b a t a M., К у uno E., Ito S.— Bull. Chem. Soc.
Japan, 1956, 29, 904.
262. Mazzucchel 1 i A., Pantanelli E.— Gazz., 1910, 40, 661.
263. Mazzucchelli A., Pantanelli E.— Atti Line., 1909, 18, 518.
264. Mazzucchelli A.— Atti Line., 1907, 16, 265.
265. Mazzucchelli A.— Gazz., 1907, 37, 545.
266. Mattock G.— J.Chem. Soc., 1954, 989.
267. Musha S., Ogawa K.— J. Chem. Soc. Japan. Pure Chem. Sec., 1957,
78, 1689.
268. M a n d a 1 S., Dey A. K-— J. Inorg. Nucl. Chem., 1968, 30, 1221.
269. Minczewski J., Skork o-T г у b u 1 a Z.— Chem. anal., 1964,
9, 397.
270. Macaravici C. G., Strajescu M.— Rev. roumaine Chim., 1968,
13, 1477.
271. M a n d a 1 S., Dey A. K-— Rev. chim. miner., 1968, 5, 783.
272. McDonald G. D., Thompson M., Larsen E. M.— Inorg.
Chem., 1968, 7, 648.
273. McGarvey B. R., Tapper E. L.— Inorg. Chem., 1969, 8, 498.
274. Macaravici C. G., Strajescu M.— Rev. zoumaine Chim., 1968,
13, 160.
275. Mahfooz К. M., Nasser A.— Proc. Nat. Acad. Sci., India, 1968,
A36, 326.
146
276. Nicholson D. Y., Reiter M. A.— J. Am. Chem. Soc., 1937*
59, 151.
277. N e r n s t W.— Z. Elektrochem., 1897, 3, 308.
278. О t t J. B., G о a t e s J. R., Jensen R. J., Ma ngels on N. F.—
J. Inorg. Nucl. Chem., 1965, 27, 2005.
279. Okac A., Sommer L.— Chem. Listy, 1953, 47, 659.
280. Okac A., Sommer L.— Chem. Listy, 1955, 49, 1093.
281. Okac A., Sommer L.— Chem. Listy, 1956, 50, 1711.
282. Okac A., Sommer L.— Anal. Chim. Acta., 1956, 15, 345.
283. Okac A., Sommer L.— Spicy Vyd. prirodoved. fak. Masarykovy univ.,
1956 E13 213.
284. P e c h a г d E.— Compt. Rend., 1893, 116, 1513.
285. P a 1 i 1 1 a F. C., Adler N., H i s к e у C. F.— Anal. Chem., 1953,
25 926.
286. Podlahova J., Podlaha J.— J. Inorg. Nucl. Chem., 1966, 28,
2267.
287. Prashar P., T andon J. P.— J. Less-Common, Metals, 1967, 13, 541.
288. Paul Ram Chand, Parkash Ram, Sandhu Sajit
Sing h.— Indian J. Chem., 1966, 4, 426.
289. Patel С. C., Krishnan V.— J. Sci. Ind. Res., 1961, B20, 604.
290. Patel С. C., J e r e G. V.— J. Inorg. Nucl. Chem., 1963, 25, 1155.
291. Pfeifer G.— Magyar kem. flyoirat, 1966, 72, 93.
292. Pfeiffer P., Thiebert H.— Ber., 1938, 71, 119.
293. Pande К. C., Mehrotra R. C.— J. prakt. Chem., 1957, 5, 101.
294. P u s c h e 1 R., L a s s n e r E.— Z. anorg. Chem., 1964, 326, 317.
295. P i с c i n i A.— Gazz., 1882, 12, 151.
296. P i с c i n i A.— Gazz., 1883, 13, 57.
297. Prasad S., T r i p a t h i J. P.— Proc. Nat. Acad. Sci. India, 1961,
A30 24.
298. Prasad S., Tripathi J.— J. Indian Chem. Soc., 1956, 33, 838.
299. Q a i m S. M.— J. Inorg. Nucl. Chem., 1969, 31, 1737.
300. Ragsdale R. O., St ewort В. B.— Inorg. Chem., 1963, 2, 1002.
301. R i p a n R., В u d i u T.— Rev. raumaine Chim., 1968, 13, 77.
302. R a f a e 1 a f f R., D j e r a s s i J.—Israel Atomic energy commiss. (Rpt),
1968, 1168, 87.
303. Ruba О., C i f к a J. et al.— Chem. Listy, 1957, 51, 1462; Collect.
Czechosl. Chem.Communs, 1958, 23, 71.
304. Reeves R. E., Blouin F. A.— J. Am. Chem. Soc., 1954, 76, 5233.
305. Reeves R. E., JonassenH. B.— J. Am. Chem. Soc., 1954, 76, 5354.
306. R u m p f M. E.— Ann. Chim., 1937, 8, 456.
307. R u m p f M. E.— Compt. Rend., 1935, 200, 317.
308. R i v e n g F.— Bull. Soc. Chim., 1945, 12, 283.
309. Richardson E.— J. Less-Common Metals, 1960, 2, 458.
310. R e c h a r d E.— Compt. Rend., 1893, 116, 1513.
311. Reynolds M. L. — J. Inorg. Nucl. Chem., 1964, 26, 667.
312. R о s e n h e i m A., Schutte O.— Z. anorg. Chem., 1901, 26, 238.
313. R о s e n h e i m A., Cohn R.— Z. anorg. Chem., 1901, 28, 167.
314. R о s e n h e i m A.— Ber., 1905, 38, 2770.
315. R о s e n h e i m A., Schnabel R.— Ber., 1905, 38, 2776.
316. R о s e n h e i m A., Schnabel R., Bilecki R.— Ber., 1915, 48,
447.
317. Rosenheim A., Sorge O.— Ber., 1920, 53, 932.
318. S h i m a M., S a n о H., L i m u г a F.— J. Chem. Soc. Japan. Pure Chem.
Sec., 1958, 79, 1032.
319. S w a 1 1 о w A. G., S t u d d B. F.— Chem. Communs, 1967, 1197.
320. S у a n с о 1 A.— J. Inorg. Nucl. Chem., 1969, 31, 1850.
321. Sen D. N., Umapathy P.— J. Indian Chem. Soc., 1968, 45, 810.
322. S к о г к o-T rybula Z., Boguszewska Z.— Chem. anal., 1969,
14, 549.
10*
147
323. S t u d d B. F., Swallow A. G.— J. Chem. Soc., 1968, A, 1961.
324. Schmid G., P e t z W., А г 1 о t h W., Noth H.— Ang. Chem.,
1967, 79, 683.
325. Smith J. A., Wilkins E. J.— J. Chem. Soc., 1966, A, 1749.
326. Scagliarini G., Tar tar ini G.— Atti Line., 1926, 4, 318.
327. Susie M.— Bull. B. Kidtic Inst. Nucl. Sci., 1963, 14, 125.
328. Stevens H. M.— Anal. Chim. Acta., 1958, 18, 359.
329. Singh R. P.— J. Indian Chem. Soc., 1960, 37, 569.
330. Schwartz D., N a e g 1 i D.— Inorg. Nucl. Chem. Letters, 1966, 2,
149.
331. Schwartz D., Heyer R.— J. Inorg. Nucl. Chem., 1967, 29, 1384.
332. Schwarz R.— Z. anorg. Chem., 1933, 210, 303.
333. S c h a e p p i Y., Treadwell U. D.— Helv. Chim. Acta., 1948, 31,
577.
334. Schon n— Z. anal. Chem., 1870, 9, 41.
335. S a t h e R. M., Venkateswarlu C.— Coll. Czech. Chem. Communs,
1962, 27, 701.
336. Schmit z-D u M о n t O., Simons P., В г о j a G.— Z. anorg. Chem.,
1949, 258, 307.
337. Subbanna V. V., RaoC. S., Bhattacharya A. K. — J. Sci.
Ind. Res., 1959, BC18, В127.
338. Schumann A.— Ber., 1888, 21, 1079.
339. Singh R. P.— Current. Sci., 1955, 24, 208.
340. S a n о H.— Japan Analyst, 1958, 7, 235.
341. S a n о H.— Bull. Chem. Soc. Japan, 1957, 30, 790.
342. S a n о H.— Bull. Chem. Soc. Japan, 1958, 31, 974.
343. Schlafer H. L., Lenz W., Staab J.— Z. Phys. Chem., 1968,
62, 290.
344. Schlafer H. L._, G d t z R.— Z. anorg. Chem., 1961, 309, 104.
345. Schlafer H. L., Go t z R.— Z. phys. Chem., 1964, 41, 93.
346. Stabler A., Wirthwein H.— Ber., 1905, 38, 2619.
347. Stabler A., Bachran F.— Ber., 1911, 44, 2906.
348. Sommer L.— Coll. Czech. Chem. Communs, 1962, 27, 439.
349. Sommer L.— Spisy Vyd. prirodoved. fak. Masarykovy univ., 1958, 9, 397.
350. Sommer L.— Z. anorg. Chem., 1958, 164, 299.
351. Sommer L., Hnilickova M.— Bull. Soc. Chim. France, 1959,
1, 36.
352. Sommer L.— Chem. Listy, 1956, 50, 1729.
353. Sommer L.— Chem. Listy, 1956, 50, 1702.
354. Sommer L.— Coll. Czech. Chem. Communs, 1963, 28, 2393.
355. Sommer L.— Z. anorg. Chem., 1963, 321, 191.
356. Sommer L.— Coll. Czech. Chem. Communs, 1963, 28, 2716.
357. Sommer L.— Coll. Czech. Chem. Communs, 1963, 28, 2102.
358. Traver A.— Chim. Ind., 1932, 3, 345.
359. T г a u b e R.— Z. Chem., 1963, 3, 194.
360. Tarig M. S., Ahmad N., Rahman S. M. F.— Z. anorg. Chem.,
1965, 336, 110.
361. Thiele К.-H., Muller J.— Z. Chem., 1964, 4, 273.
362. Tomas V., В о d e a C.— Rev. roumaine chim., 1969, 14, 959.
363. Tomas V., Bodea C.— Rev. roumaine chim., 1969, 14, 141.
364. Tomas V., Bodea C.— Rev. roumaine chim., 1969, 14, 405.
365. Umapathy P.— J. Indian Chem. Soc., 1968, 45, 1006.
366. V i a 1 1 e t P.— Compt. rend. 88° Congr. nat. Soc. savant. Clermont-Fer-
rand, 1963, Sec. Sci, 1. Paris, 1964, 145.
367. V i a 1 1 e t P.—Compt. Rend., 1961, 252, 1789.
368. Ventkateswarlu V., Rao С. B.— Current Sci., 1960, 29, 136.
369. W e e 1 e r A.— Z. anal. Chem., 1884, 23, 410.
370. W e e 1 e r A.— Ber., 1882, 15, 25, 92.
371. W e i s s 1 e r A.— Ind. Eng. Chem. Analyt. Ed., 1945, 17, 695.
148
372. Wendling E., La v i 11 a n dr e J.— Bull. Soc. chim. France, 1967,
2142.
373. Wendling E.— Rev. chim. miner., 1968, 4, 909.
374. Weiss A., Ruthardt R.— Z. Naturforsch., 1969, 24b, 355.
375. Z i к m u n d M., Stephickova L. et al.— Proc. 2-nd Conf. Co-
ordinat. Chem. Smolenice-Bratislava, 1969, Bratislava, 1969, 283; РЖХим.,
1970, 12B101.
376. Zikmund M., Stepnickova L., Hrhciarova K-— Chem.
zvesti, 1968, 22, 917.
377. Ziegler M., Glemser O., Baeckmann A.— Angew. Chem.,
1958, 70, 500.
У
ГЛАВА III
ЭЛЕКТРОХИМИЧЕСКОЕ ПОВЕДЕНИЕ ТИТАНА
В электрохимическом отношении титан отличается небольшим
энергетическим барьером при переходе катионов с поверхности като-
да в водные растворы и очень высоким барьером разряда гидрати-
рованных ионов. Обмен катионами титана между расплавами без-
водных солей и катодом не встречает значительных энергетических
затруднений. Поэтому титан легко переходит с поверхности катода
в раствор и расплав, а выделяется на катоде только при электролизе
солевых расплавов. Из водных растворов электрохимическое осаж-
дение металлического титана на катоде, как правило, не происходит.
СТАНДАРТНЫЕ ЭЛЕКТРОДНЫЕ ПОТЕНЦИАЛЫ
В ВОДНЫХ РАСТВОРАХ
Стандартные электродные окислительно-восстановительные по-
тенциалы титана, вычисленные на основании термодинамических
данных, приведены в табл. 5. ФорбесиХолл [247] вычислили нор-
мальный окислительно-восстановительный потенциал Ti2+/Ti3+ при
температуре 0° С, который оказался равным —0,16 в.
Таблица 5
Стандартные электродные окислительно-
восстановительные потенциалы титана в водных
растворах при 25°С [88,272]
Электрод Электродная реакция Стандартный потенциал, в
Ti/Ti+3 Ti — Зе = Ti 3+ —1,75
Ti/TiF2-" Ti + 6F~ — 4е = TiF2-" — 1,24
Т17Н+6 Ti + Н2О—24е = ТЮ2 + 4Н+ —0,95
Ti2+/Ti3+ Ti2+ — 1е = Ti3+ —0,376
Ti3+/Н+ Ti3+ + Н2О— 1е= Ti2++ 2Н+ -0,1
Экспериментально в водных растворах определены только стан-
дартные окислительно-восстановительные потенциалы перезарядки
ионов титана. В результате измерения электродвижущей силы в
сернокислых растворах определен нормальный потенциал Ti3+/Ti4+
при температуре 18° С (—0,05 в) [233]. В. С. Сырокомский с соав-
150
торами [149] определил величины формального потенциала Ti3+/Ti4+
в разбавленных солянокислых и сернокислых растворах при ком-
натной температуре (—0,1 в). А. Г. Стромберг и А. И. Картушин-
ская [148] вычислили стандартный окислительно-восстановительный
потенциал по реакции
ТЮС1+ + 2Н+ + е Ti3++H2O + Cl
из данных полярографических измерений. Он оказался равным
0,455 в.
Прибавление к водным растворам титановых солей комплексо-
образующих агентов повышает формальный окислительно-восста-
новительный потенциал перезарядки ионов титана и смещает его
Таблица 6
Влияние комплексообразования на формальный потенциал ТН3/Т1’4+ в разных
средах
Центральный ион Комплексообра- зующий агент Концентрация комплексообразу- ющего агента, моль/ л Потенциал по от- ношению к нор- мальному водо- родному элект- роду, в Величина скач- ка по сравнению с формальным потенциалом, в
1-н. H2SO4
Ti3-b K2so4 0,083 —0,246 0,146
у j3+ НСООН 6,520 —0,247 0,147
Ti3+ (NH4)2SO4 0,440 —0,286 0,486
Ti3+ СНзСООН 5,360 —0,356 0,256
Ti4+ HF 4,876 +0,124 0,224
Ti4+ NH4F 1,860 +0,362 0,462
- 1-н. НС1
Tis+ nh4ci 0,570 —0,162 0,062
Ti4+ nh4f 3,620 +0,504 0,604
2-н. H2SO4
'pjS-f- KCl 0,342 —0,156 0,046
Ti4+ nh4f 2,420 +0,524 0,634
в отрицательную сторону (табл. 6) [149]. Формальный окислительно-
восстановительный потенциал Ti+3/Ti4+ составляет в 6-н. H2SO4
— 0,246, а в 10-н. НС1 — 0,279 в. По величине скачка формального
потенциала перезарядки ионов титана относительно формального по-
тенциала электрода Ti3+/Ti4+ в разбавленных соляной и серной кис-
лотах можно судить о комплексообразовании ионов титана с различ-
ными веществами. Как видно из табл. 6, специфическими комплек-
сообразователями для Ti3+ в водных растворах служат добавки
KCl, NH4C1, K2so4) (NH4)2SO4i НСООН, СНзСООН, а для'Т14+ —
151
I
HF и NH4F. Вследствие смещения окислительно-восстановитель-
ного потенциала перезарядки ионов титана под действием комплексо-
образователей добавка сульфата аммония к раствору Ti2(SO4)3
замедляет окисление титана на воздухе. Восстановление Ti4+ в
присутствии HF и NH4F вследствие комплексообразования затруд-
няется.
ЭЛЕКТРОХИМИЧЕСКОЕ ВОССТАНОВЛЕНИЕ ТИТАНА
ИЗ ВОДНЫХ РАСТВОРОВ
Вследствие большой отрицательной величины нормального по-
тенциала и высокого термодинамического барьера разряда ионов
на катоде электрохимическое восстановление титана из водных
растворов до металла оказывается практически невозможным. При
электролитическом восстановлении титана в солянокислых [266] и
в сернокислых [233] растворах наблюдалось образование только
ионов Ti3+. Изучено влияние на восстановление Ti4+ материала
катода [241] и оказалось, что на катодах из различных материалов
(золота, амальгамированного серебра, свинца и цинка) также
имело место только образование ионов Ti+ . Осаждение металли-
ческого титана при электролизе кислых и щелочных растворов
титана не обнаружили и другие авторы [81].
Восстановление титана до металла возможно при условии, ког-
да он образует сплавы с материалом катода [77—79] или одновремен-
но с ионами титана разряжаются ионы других металлов, например
кобальта [77]. Совместное осаждение титана и кобальта происходит
из борфтористоводородных солей обоих металлов. Н. Г. Кудрявцев
с соавторами [80] из борфтористоводородных электролитов получил
сплав титана и кобальта с содержанием от 1 до 30% Ti.
На катодах из свинца, меди и платины, по данным [81], обра-
зуется пленка из металлического титана толщиной 3—4 мкм, после
чего выход по току падает до нуля. Авторы считают, что появление
титановой пленки на катодах перечисленных металлов происходит
вследствие деполяризации разряда ионов при образовании сплавов
титана с материалом катода. После насыщения поверхности катода
титаном деполяризация исчезает и разряд ионов титана прекра-
щается; на катоде начинает выделяться водород.
По данным потенциостатических исследований [41, 42], титан в
разбавленных сернокислых и солянокислых растворах с концентра-
цией менее 1,0 и 0,1 моль/л соответственно, а также в растворах
хлорной кислоты (0,1—10 моль/л) подвергается самопассивирова-
нию. На анодных потенциостатических кривых область активного
растворения титана отсутствует. В растворах с концентрацией 5—
13 моль!л H2SO4 и 10 моль!л НО наблюдается активное растворение
титана и его пассивация с последующим выделением на аноде кис-
лорода. При потенциалах выделения кислорода более 2 в пленка
152
из низших окислов титана на аноде превращается в двуокись ти-
тана. Легирование титана ванадием и алюминием сопровождается
возрастанием критического тока пассивации [297]. Ионы фтора
подавляют перепассивацию титана в щавелевокислых растворах и
способствуют увеличению анодного тока в пассивной области.
При низких частотах переменного тока (менее 5000 гц) электрод-
ный потенциал титана смещается в положительную, а при высо-
ких — в отрицательную сторону [180, 181]. В отличие от постоянного
тока, для которого смещение потенциала электрода в положитель-
ную сторону приводит к пассивации анода, при переменном токе
анодное смещение потенциала сопровождается увеличением кор-
розии.
Анодное растворение титана в плавиковой и кремнефтористово-
дородной кислотах сопровождается переходом его в раствор в трех-
валентном состоянии [1].
Катод из титана по величине перенапряжения на нем водорода
близок к меди и серебру. Коэффициенты в уравнении Тафеля для
титана при температуре 25° С таковы: а = 0,96, b = 0,110 [68].
Выделение водорода на титане идет в соответствии с теорией замед-
ленной рекомбинации [141]. При плотности тока 10-5—10--1 а!’см2
на нитриде титана перенапряжение водорода также подчиняется
уравнению Тафеля [147].
В связи с разработкой методов химического анализа выполнено
значительное количество работ по изучению полярографического
восстановления титана в водных растворах. В 0,065-н. серной кис-
лоте имеет место необратимое полярографическое восстановление
Ti4+ до Ti3+ [308, 309]. Потенциал полуволны Ei/2 = —0,78 в от-
носительно насыщенного каломельного электрода. О необратимос-
ти волн восстановления титана в сернокислых растворах свидетель-
ствует независимость потенциала полуволны от периода капания,
отсутствие перегиба на анодно-катодной волне смеси Т^3+ и Ti4+
и совпадение потенциалов катодного и анодного зубцов на осцилло-
графических кривых [256, 267]. На фоне 10-мол. NH4HSO4 ионы
Ti4+ восстанавливаются необратимо, что объясняется сильным
комплексообразованием сульфата аммония с ионом Ti3+. В раство-
рах серной кислоты и сульфата аммония восстановление титана на
ртутном капельном электроде сопровождается образованием трех
волн [255]. Появление этих волн связывается с восстановлением до
Ti3+ различных форм титана, а именно: TiO2+, Ti (OH)SO^ и
Ti (OH)2SO4+-
E. И. Крылов с соавторами [75, 76] при катодном восстановлении
титана в сернокислых растворах получил отчетливо выраженные
волны, отвечающие необратимым процессам:
Ti4+ + e -> Ti3+
И Ti3+ + e -> Ti2+.
153
Лингей и Кеннеди [271] в разбавленных сернокислых растворах
титана также получили необратимые полярографические волны.
Восстановление титана в 4-мол. H2SO4 протекает обратимо. Потен-
циал полуволны равен —0,05 в относительно насыщенного кало-
мельного электрода.
Струбл 1301] в0,01-н. солянокислом растворе наблюдал необрати-
мую катодную волну восстановления Ti4+ до Ti3+ с £1/2 = —0,81 в
относительно насыщенного каломельного электрода. Эту же волну
получил Колоузек [268] в 0,1-н. НС1. В солянокислых растворах
авторы наблюдали появление необратимой анодной волны окисле-
ния Ti3+ до Ti4+ с £1/2 = 0,14 в. В насыщенном растворе хлористого
кальция получена анодная волна при том же потенциале полуволны
[268].
В 0,2-н. солянокислых растворах Лингей и Кеннеди [271] на-
блюдали раздельное появление катодной (Ti4+) и анодной (Ti3+)
волн, которые с увеличением концентрации НС1 сближаются. В 1-н.
солянокислых растворах с высокой концентрацией хлористого
калия обе волны необратимы. По мнению авторов, это служит до-
казательством существования в водных растворах ионов оксихло-
рида титана, для восстановления которых, например по реакции
Т[ОСГ — е 4- 2Н+ + ЗСГ = TiClr + Н2О,
требуются ионы Н+ и С1~.
При полярографическом восстановлении Ti4+ на фоне 0,1-н.
NH4C1 и НО (pH 0,6—1,5) имеются две волны [273]. Первая из них
исчезает при добавлении многовалентных катионов. Добавка
5 об. % этиленгликоля улучшает форму полярограммы и позволяет
определить содержание титана в растворе полярографическим ме-
тодом при pH 9—12.
В растворах хлорной кислоты на ртутном капельном электроде
получена полярографическая волна восстановления ТЮ2+ [255].
В 7-н. НС1О4 появлялись катодная и анодная волны; обе волны не-
обратимы [271].
Исследованы каталитические волны Ti4+ в присутствии С1ОГ-
иона [183]. Определены константы скорости окисления Ti3+ ионами
СЮГ в присутствии органических кислот, KCNS и H2SO4.
В разбавленной фосфорной кислоте титан дает необратимые ка-
тодные и анодные волны [271]. В растворах с концентрацией 1—
10 моль!л Н3РО4 получаются обратимые волны с £1/2 = —0,25 в
относительно насыщенного каломельного электрода (1 моль/л Н3РО4).
Полярографическое восстановление титана в пирофосфорной кисло-
те изучали Д. И. Курбатов [701 и Атма с соавторами [221].
Гото и Исич [47] также отметили, что Ti4+ в концентрированной
фосфорной кислоте на ртутном капельном катоде дает четкие диф-
ференциальные волны восстановления. Величина предельного тока
154
пропорциональна концентрации титана в растворе. В концентри-
рованной фосфорной кислоте при температуре 60—120° С полу-
чаются четкие волны окисления Ti3+ с £ 1/2 = —0,39 в (100° С, ртут-
ный каломельный электрод) [46]. Получена обратимая катодная и
анодная волны в оксалатных растворах титана с pH < 3 [292, 311],
а также в растворах титана в винной и лимонной кислотах. Потен-
циал полуволны в растворе с концентрацией 0,1-мол. КгСгС^ и
1-мол. H2SO4 равен — 0,17 в относительно насыщенного каломель-
ного электрода. Вообще же потенциал полуволны относительно
насыщенного каломельного электрода зависит от pH и концентрации
щавелевой кислоты и выражается уравнением [292]
£1/2 = — 0,25 + 0,080 pH + 0,020 log [Н2С2О4] (*)•
В растворах этилендиаминтетрауксусной кислоты Ti3+ и Ti44"
образуют волны, отвечающие одноэлектронным процессам. По-
тенциалы полуволн их зависят от pH среды и концентрации комп-
лексона. Восстановительно-окислительный процесс обратимый. При
полярографическом восстановлении титана в уксуснокислых рас-
творах получена необратимая катодная волна [292, 294].
Хабаши [254] изучил полярографическое восстановление титана
на фоне 0,5-мол. уксусной кислоты в присутствии салициловой
кислоты, пирокатехина, пирогаллола, ацетилсалициловой, метил-
салициловой, щавелевой и малеиновой кислот и получил поляро-
граммы, состоящие из двух волн. Менее отрицательная кинетиче-
ская волна, по мнению автора, соответствует восстановлению комп-
лекса Ti4+ с органическим лигандом. Вторая (диффузная) волна по-
является в результате восстановления ионов титанил a TiO2+. В при-
сутствии бензойной кислоты, фенола, о-фенилдиамина, и-амино-
бензойной и янтарной кислот на полярограммах восстановления
титана в уксуснокислых растворах появляется только вторая волна.
В 0,1-мол. растворе роданида калия появляются обратимые ка-
тодные и анодные волны с Е i/2 = —0,45 в относительно насыщен-
ного каломельного электрода. Согласно [307], в роданидных раство-
рах происходит обратимое восстановление титана по -реакции
[TiOHCNS]2+ 4- Н+ + е [TiCNS]2+ + Н2О.
Вилкинсон с соавторами [315] в 0,1-мол. хлорной кислоте полу-
чил обратимые катодную и анодную волны для [Ti (С5Н5)]2~ с £ц2 =
— —0,44 в относительно насыщенного каломельного электрода.
Полярографическое восстановление бпс-(циклопентадиенил)дихлор-
титана в солянокислом растворе наблюдали И. А. Коршунов и
И. И. Малюгина [82].
Полярографические волны при катодном восстановлении титана
на ртутном капельном электроде получены в растворах молочной,
салициловой, фталевой, муравьиной кислот [292], в растворах
глицерина и нитрилтриуксусной кислоты [223], в смеси винной и
155
пирофосфорной кислот [227], в нитрилтриуксусной кислоте [293],
в смеси 1,2-диаминоциклогексан-М,М,Ы',Ы'-тетрауксусной и ук-
сусной кислот [294], в 1,2-пропандиолкарбонате [2491, диметил-
сульфоксиде и N,N-диметилформамиде 1250]. С помощью пере-
меннотоковой полярографии определены константы скорости вос-
становления Ti4+ в 1-мол. винной кислоте (/<$ = 2,26 • см/сек),
в 8-мол. серной кислоте (Ks = 3,39 • 10“3 см/сек) и в 0,1-мол. ща-
велевой кислоте с примесью 0,2-мол. KBr (Ks = 6,66 • 10~3 см/сек)
[182].
В сухом ацетонитриле на фоне (C2H5)4NBr Олвер и Росс [283]
наблюдали более глубокое полярографическое восстановление ти-
тана. На полярограмме Ti3+ они отметили две волны, для одной из
которых Еу2 = —1,3 в относительно насыщенного каломельного
электрода, а вторая с более отрицательным значением потенциала.
По мнению авторов, первая волна вызвана восстановлением Ti34"
до Ti2+, а вторая — Ti2+ до металла. Из полученных данных они
рассчитали величину потенциала восстановления Ti3+ до Ti2+ в
водных растворах (около— 2 в относительно насыщенного каломель-
ного электрода).
Имеется сводка потенциалов полуволн полярографического вос-
становления титана в водных растворах [69, 312].
В растворах комплексного соединения Ti (IV) с перекисью во-
дорода при pH 3—5 в области потенциалов 0,3—0,1 в наблюдается
каталитическая волна [179]. Величина предельного тока каталити-
ческой волны зависит от pH раствора и концентрации титана. Ка-
талитически активным является комплекс [TiO (НО2)]+, восстанав-
ливающийся на ртутном капельном катоде. В присутствии ЭДТА
каталитическая волна появляется при более низких значениях pH
и обусловлена восстановлением тройного комплекса [TiA (НО2)1,
где Н4А—ЭДТА.
Полярографическим методом изучен состав ионов титана в при-
сутствии фтористого натрия [296]. При pH < 2 имеет место равно-
весие Ti4+ Ti (ОН)з~, а при 2 < pH > 6 — равновесие Ti (ОН)з"«^
TiO2+. Волна восстановления Ti (ОН)з“ носит кинетический ха-
рактер. Полярографическое восстановление TiO2+ наблюдается
только при pH > 6, так как волна разряда этих ионов с Еу2 —
= —1,35 в относительно насыщенного каломельного электрода
при pH < 6 закрыта током разряда ионов водорода.
АНОДНОЕ ОКИСЛЕНИЕ ТИТАНА
Как и многие другие металлы, титан в водных растворах может
подвергаться анодному окислению. Исследовано анодное окисление
металлического титана в водных растворах муравьиной кислоты
и показано, что при низких анодных потенциалах на поверхности
156
анода образуется окисная пленка [232, 295]. Методом рентгенострук-
турного анализа установлено, что она состоит из двуокиси титана
со структурой анатаза. В концентрированных растворах муравьиной
кислоты на аноде образуется слой желтого осадка, состоящего из
аморфной двуокиси титана.
И. Л. Розенфельд с соавторами [1461 изучил состав окислов при
анодном окислении титана методом фотоэлектрической поляризации.
Оказалось, что в растворах 0,1-н. КОН и 1,0-н. Na2SO4 на кривых
(г, ср) имеются три характеристические области, отвечающие пасси-
вации, задержке ср и дальнейшей пассивации. Отрицательный знак
величины фотоэлектропроводности свидетельствует об электронном
типе проводимости пленки на титане, что свойственно низшим его
окислам.
Адсорбированные на аноде кислород и ОН-группы окисляют
присутствующие в сернокислых растворах ионы Fe2+ и Ti3+ [144].
Это приводит к увеличению тока при достижении <р = 1,25 в, ре-
гистрируемого на потенциостатических кривых.
Фокс и соавторы [248] исследовали анодные покрытия титана ме-
тодами электроно- и рентгенографии. По их данным, внутренний слой
покрытий состоит из рутила, а внешний — из анатаза. Покрытие тита-
на двуокисью титана методом анодирования изучал Хикмотт [253].
ЭЛЕКТРОДНЫЕ ПОТЕНЦИАЛЫ В СОЛЕВЫХ РАСПЛАВАХ
Электродные потенциалы титана изучались преимущественно в
расплавах галогенидов щелочных и щелочноземельных металлов в
связи с разработкой методик электролитического получения и ра-
финирования металла.
Величины стандартных электродных потенциалов зависят не
только от температуры, но и от природы расплава, в котором рабо-
тает электрод. Причиной изменения величин электродных потен-
циалов под влиянием природы расплавов служит комплексообра-
зование титана с компонентами солевых сплавов. Н. А. Логинов
и соавторы [90] показали, что в расплавах LiCl, КО и CsCl с уве-
личением радиуса катиона щелочного металла стандартный элек-
тродный потенциал смещается в более отрицательную сторону.
Равновесный потенциал металлического титанового электрода
в хлоридных расплавах (эвтектическая смесь LiCl — КО и экви-
мольная смесь КО — Nad) до содержания в сплаве 6 вес. % Ti
изменяется в соответствии с уравнением
Е = Ет1«/т,2+ + 2,3 log [Ti2+] (в),
где [Ti2+] — мольно-долевая концентрация титана [164]. В этих
расплавах превалируют ионы двухвалентного титана, а расплавы
ведут себя как идеальные растворы. В смешанных хлоридно-фторид-
ных расплавах потенциал титанового электрода сильно смещен в
157
сторону потенциала выделения щелочных металлов из-за сильного
комплексообразования [162]. Равновесный потенциал титанового
электрода в смешанных хлоридно-фторидных расплавах не зависит
от концентрации титана в расплаве [176]. Титановый электрод в
этих расплавах вследствие образования труднорастворимых фто-
ридов M2TiF4 (М — щелочной металл) ведет себя как электрод вто-
рого рода, обратимый относительно ионов фтора [175].
Фленгас [246] определил следующие стандартные окислительно-
восстановительные потенциалы титана в расплаве эквимольной сме-
си КС1—NaCl при температуре 670° С относительно хлорного элек-
трода сравнения: Е°тр/т12+ = —1,967, £°Ti«/Ti3+ = —1,898,
E°Tio/Ti44- = —1,549, £°Ti2+/Ti3+ = —1,762 и £°Ti3+/Ti4+ =
-—1,202 в.
Мензис с соавторами [274] изучил стандартный окислительно-
восстановительный потенциал Ti3+/Ti4+ в эвтектической смеси
LiCl — КО методом потенциометрического титрования TiCl3 во-
дородом. При бесконечном разбавлении и температуре 500° С окис-
лительно-восстановительный потенциал, по его данным, равен
—1,550 в *. М. В. Смирнов с соавторами [174] методом потенцио-
метрического титрования TiCl3 водородом в эквимольном расплаве
КО — NaCl определил формальное значение окислительно-вос-
становительного потенциала при 700° С (B°Ti2+/Ti3+= —1,726 в).
Это подтверждено данными [91], согласно которым £т12+/Т13+ =
— —1,807 в. Авторы [174] нашли также выражение для темпера-
турной зависимости стандартных потенциалов (в):
Ет^щ2+ = — 2,371 + 5,73 • КГ4?,
£п«/п2+ = — 2,156 + 3,82 • КГ4?,
где Ti — температура в ° К.
М. В. Смирнов и Н. А. Логинов [87, 177] определили окислитель-
но-восстановительный потенциал Ti347Ti4+ в эквимольной смеси
КО — NaCl при 750—890° С и вычислили температурную зависи-
мость стандартных электродных потенциалов в этом интервале тем-
ператур. Она выражается следующими уравнениями (в);
£0io/Ti+2 = — 2,382 + 4,83 • КГ4Т,
£п»/п3+ = — 2,158 + 3,16 • КГ4?,
ЕТР/ТН+ = — 1,987 + 3,84 • 1(Г4?,
£°Ti2+/Ti3+ = - 1,592 + 2,82 • КГ4?,
£°Ti3+/Ti4+ = - 1,473 + 5,86 • 1(Г4?.
___________ 1
* Потенциалы в расплавах измерены относительно хлорного электрода срав-
нения.
158
Температурная зависимость формального окислительно-вос-
становительного потенциала Ti3+/Ti4+ в расплаве CsCl при концен-
трации 0,13—0,32% Ti3+ и 0,06—1,24% Ti4+ в интервале 730—
930° С выражается уравнением [178]
£°Ti3+/Ti4+ = - 1,210 + 2,26 • 10-4Т (в).
На основании измерений электродвижущих сил установлена кон-
станта реакции между металлическим титаном и его двух- и трех-
валентными ионами в расплаве хлоридов лития, калия и цезия.
На величину константы равновесия оказывает влияние величина
радиуса катиона щелочного металла соли-растворителя. Найдено
общее выражение для величины константы равновесия:
log Л = —у------YT~r----Н---------~г— ’
где Т — температура, ° К; г — радиус иона щелочного металла.
При одинаковой температуре константа равновесия уменьшает-
ся с увеличением радиуса иона щелочного металла соли-раствори-
теля, что связано со смещением равновесия в сторону диспропорцио-
нирования двухвалентного титана на металлический и трехвалент-
ный.
Формальный окислительно-восстановительный потенциал
Ti2+/Ti3+ в эквимольной смеси КВг—NaBr оказался значительно
меньшим, чем в расплаве КО — NaQ [92]. В бромидном расплаве
при температуре 700° С он равен —1,56 в вместо —1,807в в хлорид-
ном расплаве [91].
ЭЛЕКТРОХИМИЧЕСКОЕ ВОССТАНОВЛЕНИЕ ТИТАНА
В БЕЗВОДНЫХ СОЛЕВЫХ РАСПЛАВАХ
Электрохимическое восстановление титана в солевых расплавах
в послевоенные годы интенсивно изучалось как в нашей стране, так
и за рубежом. В периодической печати опубликовано большое число
научных статей. Еще больше предложений по получению титана и
соединений титана низшей валентности имеется в патентной ли-
тературе. Подробную сводку опубликованных работ по электрохи-
мии титана в расплавленных средах составила Гитман [45].
СОСТАВ СОЛЕВЫХ РАСПЛАВОВ ЭЛЕКТРОЛИТНЫХ ВАНН
Для электролитического получения титана предложено восста-
новление его в солевых расплавах из галогенидов (TiF3, TiF4, K2TiF6,
TiCl2,TiCl3, TiCl4, TiI4) и окислов (TiO, TiO2). Столь ограниченный вы-
бор объясняется тем, что многие соединения титана неустойчивы при
высокой температуре (например, сульфаты, хлораты, нитраты), а
159
Таблица 7
Состав солевых ванн, предложенных для электролитического получения
титана
Солевой состав ванн Соединения, в виде которых титан вво- дится в ванну Литература
LiCl + КС1
LiCl + NaCl + КС1
LiCl + KC1
LiCl + NaCl
LiCl + NaCl + KC1
LiCl + KF + NaCl+ KC1+
+ MgCl2+ CaCh
LiCl + KC1
Lil + Nal + KI
Na2TiFe
NaF + KaTiFe
NaF + NaCl
NaF.+ NaCl + KC1
NaCl
NaCl
NaCl
NaCl
NaCl + Na4P2O?
NaCl + KaTiFe
NaCl -L- K2TiF6 + KC1
NaCl + KC1
NaCl + KC1
NaCl + KC1
NaCl + KC1
NaCl + KC1 + MgCl2
NaCl 4- KC1 + MgCl2 + CaCla
NaCl + MgCl2
NaCl + CaCl2
NaCl + CaCl2
NaCl + CaCl2 +
NaCl + SrCla
Na3AlFe
NaCl + A1C13
KC1
KC1 + MgCla
BaCI2
BaCla
MgFa
CaF2
KaTiF6
TiCla + TiCl3
TiCl3
TiCl4
TiCl4
TiCl4
TiCl4
Til4
TiCl4 + (Ha)
TiCl4 + (Ha)
TiCla
TiCl2
KaTiFe
TiCla
TiCl4
TiO
TiO2
TiO2
TiCl4
TiF3, TiCl3
KaTiFe
TiCla
TiCl4
TiCla, TiCl3, TiCL
TiCl4
TiCl4
TiCla, TiCl3, TiCL
TiCl4
TiCl2, TiCl3, TiCl4
TiCl4
TiO2
TiCl3, TiCl4, TiOa
TiCl4
TiOa
TiCl3 + TiCl4
TiO
TiCI3, TiCl4
TiCl4
TiCl4
[237, 240]
[11, 12, 126, 290]
[11, 230, 252, 259, 302,
314, 289]
[119]
[113, 132, 134, 205, 260]
[206]
[202,231 ]
[101]
[206]
[206]
243]
243]
21, 83, 109, 112, 229, 305,
306]
[110]
[16, 24, 25, 97, 105, 121,
131, 205, 243]
[108]
155
74, 84, 86]
[111]
[12, 63, 226, 243]
23, 226, 237, 239, 240]
310]
31, 63, 131, 134, 238]
50, 63, 213, 219]
[13]
[134]
[9, 102, 217]
134]
122, 225]
208]
214, 261]
4, 63, 64, 65, 151]
6, 280, 285]
57, 58, 216
50, 62, 304
108]
63]
198]
26]
[131]
160
Продолжение табл. 7
Солевой состав ванн Соединения, в виде которых титан вво- дится в ванну Литература
СаС12 Галогениды тита- [18, 96, 107, 155, 189,
на, титанаты ще- лочных металлов, TiO, TiO2 298]
СаС12 + СаО TiOa [135]
А1С13 TiCl4 [22]
Галогениды щелочных металлов Низшие галогени- [8, 16, 25, 34, 117, 190,
ды, TiO2 194, 199, 207]
Низшие галогени- [16, 25, 34, 117, 194,
ды, TiO 199] X
TiO2 [99, 190, 195, 207]
Галогениды щелочных метал- лов + А1С13 или MgCl2 TiCl4 [98]
Галогениды щелочных металлов и кальция TiO2 [114]
Фториды щелочных металлов TiO2, TiCl4, TiO [10, 15, 17, 26, 184, 200, 216, 299]
Фториды щелочноземельных ме- таллов To же [15, 26, 204, 216]
Фтортитанаты щелочных метал- лов TiCl4 [8, 20, 26, 187]
Фтортитанаты щелочноземельных металлов MTiFe [26, 37]
Смесь фтортитанатов и галогени- дов щелочных металлов M2TiFe [10, 24, 29, 188, 316]
Хлориды щелочных металлов Низшие хлориды, [15, 19, 30, 31, 106, 127,
TiO2, TiCl4 128, 129, 133, 138, 140, 166, 189, 201, 210, 215, 288, 305]
Хлориды щелочноземельных ме- Низшие хлориды, [14, 15, 30, 31, 106, 127,
таллов TiO2, TiCl4 128, 129, 133, 166, 201, 212, 215]
Бромиды щелочных металлов Низшие бромиды и хлориды тита- на, TiCl4 [30, 127]
Бромиды щелочноземельных ме- Низшие бромиды [30, 127]
таллов и хлориды титана, Т1С14
Йодиды щелочных металлов TiCl4 [30]
Йодиды щелочноземельных ме- таллов TiCl4 [30]
11 1-1669
161
Таблица 8
Состав солевых ванн для электролитического рафинирования титана
Солевой состав ванны
Растворимый анод, подвер-
гающийся рафинированию
Хлориды щелочных и редкозе-
мельных элементов (церий)
Галогениды щелочных металлов
Галогениды щелочных и щелочнозе-
мельных металлов
Фториды щелочных и щелочноземель-
ных металлов
Хлориды щелочных и щелочноземель-
ных металлов
Бромиды щелочных и щелочноземель-
ных металлов
NaF + NaCl + А1С13 + TiO2
NaCl 4- KCl
NaCl + KCl + MgCl2 + TiCl2
NaCl + CaCl2
NaCl + K2TiF6
NaCl + KCl + TiCl2
NaCl + TiCl2
NaCl + TiO2 + TiCl3
K2TiF6 + TiF4
KCl + CaCl2
KCl + TiCl3
MgCl2
MgCl2 + CaCl2
CaF2
CeF3 + CeTiF6
Фторидные соли
Фосфаты, фториды и бораты щелочных
металлов
Титанаты, щелочных и щелочноземель-
ных металлов
Хлориды и бромиды
Литература
[218]
[211, 299]
[100,137,186,
269]
[28, 32]
[28, 32, 33,
115,136,185]
[33, 136,244]
[52]
[51, 67, 95,
152, 157]
[199, 265]
209]
[27, 120,
130]
[279]
[2,51,61,65,
104, 123, 258,
264]
[124]
116
118
[38]
139]
209
125
125
262
TO, 17, 184,
299]
[196]
[19]
Низшие хлори-
ды + KaTiFe
Сплавы титана, загряз-
ненный титан
Загрязненный титан
То же
Сплавы титана, загряз-
ненный титан
Загрязненный титан
Сплавы А1 — Ti и
Zn — Ti
Загрязненный титан
То же
» »
» в
» »
» »
Загрязненный титан
То же
в »
» в
» в
» в
» в
в в
TiO2
TiO2
TiO2
TiCl4
электрохимическое восстановление некоторых из них (нитратов,
боратов) приводит к образованию металлических соединений и
сплавов.
Электролитом для электрохимического получения и рафиниро-
вания металлического титана могут служить галогениды щелочных
и щелочноземельных металлов и их смеси, фтортитанаты щелочных
и щелочноземельных металлов в смеси с галогенидами щелочных
162
металлов, галогениды алюминия (А1С13, Na3AlF6) в чистом виде или в
смеси с фторидами и хлоридами натрия и калия, фосфаты и бориды
щелочных металлов в смеси с фторидами и хлоридами натрия и ка-
лия, титанаты щелочных и щелочноземельных металлов в смеси
с хлористым кальцием и др. (табл. 7 и 8).
Наибольший интерес для электрометаллургии титана представ-
ляют электролиты на основе хлористого натрия и эквимольной смеси
хлористых натрия и калия. Они отличаются относительно невысоки-
ми температурами плавления, вместе с тем термостойки, имеют вы-
сокую электропроводность, не вызывают коррозии аппаратуры, хо-
рошо растворяют низшие хлориды титана и легко отделяются от
электролитических осадков промыванием водой. В расплавах из
хлористого натрия и эквимольной смеси хлористого натрия и калия
удобно вести электролиз хлоридов титана и рафинирование ме-
талла. Для электролиза окисных соединений титана из-за малой их
растворимости в расплавленных хлоридах щелочных металлов при-
меняются хлоридно-фторидные ванны из смеси хлористого натрия
и фтортитаната калия.
НАПРЯЖЕНИЕ РАЗЛОЖЕНИЯ
Величина напряжения разложения определяет энергетические
затраты электролитического процесса. Напряжение разложения со-
единений титана рассчитано по термодинамическим данным и измере-
но экспериментально. Термодинамический расчет дает величину
напряжения разложения в идеальных условиях. Экспериментально
найденные величины напряжения разложения зависят от природы
электролита, материала электродов, а также в известной мере и от
методики проведения опытов. Термодинамический расчет напряже-
ния разложения проводится по уравнению
AF®
Е =_______—
С zF ’
где Е — напряжение разложения, в\ АТ? — свободная энергия
образования соединения при температуре Т\ z — изменение заряда
ионов при электролизе; F — число Фарадея (96 500 к). Различные
авторы для расчета напряжения разложения соединений титана
пользовались различными исходными данными. Полученные ими
величины поэтому имеют несколько различные значения (табл. 9).
По величине напряжения разложения соединения титана в порядке
ее уменьшения располагаются в следующий ряд: TiF4 > TiCl4 >
> TiO2,TiF3 > TiCl3 > Ti2O3, TiF2 > TiCl2 > TiO, из которого сле-
дует, что теоретически наиболее энергоемким процессом получе-
ния металлического титана является электролиз фторидов, а с наи-
меньшей затратой электроэнергии протекает электролиз окислов
титана.
Результаты экспериментального определения напряжения раз-
ложения соединений титана в солевых расплавах приведены в
11*
163
Таблица 9
Величины напряжения разложения титановых соединений
Реакция Напряже- ние раз- ложения, в Темпера- тура, °C Литера- тура
TiF4 TiF3 + -^F3 0,35 727 [240]
TiF4 Ti + 2F2 1,84 727 [240]
TiFs & TiFa + 4F« & 2,59 727 [240]
TiF3 Ti + 1-^-Fa - 2,33 727 [240]
TiF2 Ti + F2 2,02 727 [240]
TiCl4 Ti + 2C12 1,44 727 [240]
TiCl4 Ti + 2Cla 1,58 700 [36]
TiCl4 Ti + 2Cla 1,54 800 [36]
TiCl4 Ti + 2Cla 1,50 900 [36, 43]
TiCl4 Ti + 2C12 1,46 1000 [36]
TiClg Ti + lyCla 1,71 700 [36,43]
I TiCl3 Ti + l-E-Cla 1,66 800 [36]
TiClg Ti + l-|ci2 1,59 900 [36]
1,54 1000 [36]
TiCl3 Ti + l^-Cla
TiCl2 Ti + CI2 1,89 700 [36,43]
TiCl2 Ti + Cl2 1,82 800 36]
TiCl2 Ti + Cl2 1,77 900 36]
TiCla Ti + Cla 1,72 1000 36
TiCla Ti + Cla 1,67 727 [240]
TiCl4 Ti + 2Cla 1,67 860 [192]
TiClg TiCla + jCla 1,34 800 [38]
1 , TiCl3 Ti + 1-^C12 1,69 800 [38]
TiClg Ti + 1-^-Cla 1,68 860 [192]
TiCla Ti + Cl2 1,80 800 [38]
TiCla Ti + Cla 1,91 860 192]
TiO2 Ti + O2 1,29 1000 [40]
J 2TiO2 Ti2O3+ туОг 1,10 900 [281]
TiO2 (Рутил) Ti ((3) + O2 1,90 900 281]
TiO2 Ti -- O2 1,81 900 [94]
TiOa Ti + Oa 1,77 1000 94]
TiOa Ti + Oa 1,72 1100 94]
TiOa Ti — Og 1,68 1200 [94]
TiO2 Ti + O2 1,792 800 193]
164
Продолжение табл. 9
Реакция Напряже- ние раз- ложения, в Темпера- тура, °C r Литера- тура
TiO2 + * Ti — O2 1,772 850 [193]
TiO2 > Ti + O2 1,731 900 [193]
1
TiO • Ti + -^Ог 2,56 900 [94]
TiO Ti -j—gO2 2,52 1000 [94]
TiO^> 1 Ti + ~2 O2 2,48 1100 [94]
TiO<± Ti + 4-Os 2,44 1200 [94]
2TiOs 1 Ti2O3 + ~2^2 0,36 727 [240]
Ti2O3 ; 1 a 2TiO + g-Os 0,67 727 [240]
TiO<± 1 Ti + ~2 O2 1,28 727 [240]
табл. 10. Они не соответствуют приведенному ряду, так как разряд
многовалентных ионов титана на катоде может протекать ступенча-
то. Величина напряжения разложения зависит также от деполяри-
зации анода.
КАТОДНЫЕ ПРОЦЕССЫ ПРИ ЭЛЕКТРОЛИТИЧЕСКОМ
ВОССТАНОВЛЕНИИ ТИТАНА
Электролиз титана изучен при температуре, которая ниже его
температуры плавления, так как он тугоплавкий металл и при вы-
соких температурах очень активно взаимодействует с различными
веществами. При электролизе расплавов ниже 1000° С титан вы-
деляется на катоде в виде порошкообразного продукта или друз
сросшихся кристаллов. В случае применения катодов из расплав-
ленных металлов (цинка, свинца, олова и др.) образуются жидкие
сплавы титана.
Металлический титан при электролизе может получаться в ре-
зультате непосредственного разряда ионов и как продукт вторичной
реакции химического восстановления первоначально выделившими-
ся на катоде металлами. Механизм электролитического осаждения
титана очень сложный, так как титан обладает переменной валент-
ностью и восстанавливается до различных степеней окисления.
С расплавами солей титан образует комплексные соединения, за счет
чего потенциал выделения его на катоде может изменяться. Кроме
разряда ионов на катоде и металлотермического восстановления
165
о
*“4
сз
а
S
X
к
ч
о
о
со
о
со
О
св
СО
X оз S
К Р. х
СХ о «
со
X
О)
ч
СП
сч СЧ сч сч
СП ООО
•••"* *—ч «—м > -ч
О СЧ LQ LO LQ
Оазо^^
СОСЧСОСЧСЧ^СЧОО^^СЧ
ГЮ СО СО Ю —.СО СО СО СО СО СО СО 00 ОО 00 00 ОО 00 ОО
' TF О Г- О О О О О О О О О О О О О
СЧ СЧ —' —. —<счсчсчсчсчсчсч
Tf xf Tf Tt>
OO oo 00 oo
OOOiO Ю c
О О О СЧ СЧ СО
00 00 00 Ь- 00
о о о о LO о
Ю со со СО СЧ СЧ
ю 00 oo oo t- ь-
о
м*
о
I оооооооооооооооо
ОСЧОСЧООООООЮОООЮОО
ot^ О О OOOOQOO о о оооо—* оооо
00 «—• •—« »—« ♦
^^ьЬпииииииии^ООООООООООООООО
^^^РнРнньннРннннРРннннньнньн
* Продуктом восстановления были низшие окислы титана.
166
первично выделившимися металлами образование металличе-
ского титана в электролизной ванне возможно в результате дис-
пропорционирования соединений низшей валентности, например
по реакции
Механизму электролиза титана посвящено значительное коли-
чество работ. В основном механизм электролиза хлоридов, фтори-
дов и окислов титана в расплаве солей выяснен, хотя в деталях
между мнениями отдельных авторов нет еще полного согласия. Как
показало изучение формы кривых ток — напряжение, механизм
электролиза соединений титана зависит от состава расплава, под-
вергающегося электролизу, и условий проведения процесса (темпе-
ратуры, плотности тока, материала электродов, конструкции электро-
лизера и т. п.). В интервале температур 700—900° С титан выделя-
ется на катоде в виде кристаллов различной крупности (форма их
бывает игловидной, пластинчатой, призматической и октаэдри-
ческой) и в виде мелкодисперсных осадков [48].
Электролиз фторидов титана
Наиболее детально изучен электролиз фтортитаната калия K2TiF6,
который отличается благоприятными физико-химическими свой-
ствами и доступен для промышленного производства в значитель-
ных масштабах.
Колк с соавторами [270] на основе анализа вольт-амперных кри-
вых, снятых в расплаве K2TiFe + NaCl, заключил, что электрохи-
мическое восстановление фтортитаната калия протекает по схеме
что подтвердилось в результате изучения кривых напряжения раз-
ложения и напряжение поляризации — время [237, 240]. В распла-
ве K2TiF6—КС1—LiCl наблюдается ступенчатое изменение потен-
циала титанового электрода, что можно объяснить протеканием
процесса Ti4+ -> Ti3+.
Дроссбах и Росбергер [239], изучив напряжение разложения и
потенциалы отдельных электродов, пришли к заключению, что в
расплаве K2TiF6—КС1—NaCl на катоде происходит последовательное
восстановление титана до металла:
[TiFe]2- + е = [TiFj- + 2F~,
[TiF4]— + Зе = Ti + 4F~
При полярографическом восстановлении K2TiFe в расплаве
LiCl—КО не получена катодная волна вплоть до потенциала раз-
ложения фона [223]. На этом основании сделано предположение,
что восстановление фтортитаната калия до металлического титана
167
Рис. 2. Поляризация молибденово-
го катода в расплавленной смеси
NaCl+ KCl + K2TiF6 при темпера-
турах 700 (/, 2) и 800° С (5, 4):
1, 3 - 5; 2, 4 — 20 вес.% K2TiFe.
является вторичным процессом и протекает под действием щелоч-
ных металлов, получающихся при электролизе.
Вурм и соавторы [313], подвергая электролизу фтортитанаты
калия и натрия в расплаве КС1—NaCl, наблюдали образование
двойных фторидов трехвалентного титана K2NaTiF6, K3TiF6,
Na3TiFe и выделение на катоде совместно с титаном металлического
натрия. Исходя из этих наблюдений, они предположили, что элек-
тролиз фтортитанов протекает в две стадии: вначале на катоде об-
разуются соединения трехвалентного титана, которые затем вос-
станавливаются до металла за счет
вторичной реакции со щелочным
металлом.
К иной точке зрения пришли
М. В. Смирнов и Л. А. Циовкина
[170] при изучении поляризации мо-
либденового катода в хлоридном
расплаве K2TiF6. Они показали,
что механизм электролиза K2TiFe
зависит от концентрации титана в
электролите и температуры элек-
тролиза. По их данным, в расплаве
эквимольной смеси KCl + NaCl,
содержащей 5 и 20% K2TiF6, ста-
ционарный потенциал молибденово-
го электрода устанавливается при
—1,0-Н—1,2 в (т-т° С)
(рис. 2). Когда катодная плотность
тока превышает предельный ток
саморазрядки молибдена, наблю-
даются наклоны поляризационных кривых, связанные с перезаряд-
кой ионов TiFl~ до TiFg-.
Выделению металлического титана на катоде в результате пря-
мого разряда ионов мешает образование осадков труднораствори-
мых в расплаве двойных фторидов двухвалентного титана состава
M2TiF4, где М — щелочной металл, которые образуются при взаимо-
действии ионов TiF|" с ионами щелочных металлов:
TiF|~ + 2M+ + e -> M2TiF4 + 2F~
При небольших концентрациях титана эта реакция «замазывает»
выделение металлического титана на катоде, особенно при низких
температурах (700° С), когда растворимость M2TiF4 в расплаве'
мала. Волна первичного разряда ионов титана наблюдается только
при температуре 800° С и высокой концентрации в расплаве титана
(20% K2TiF6) при потенциале около —2,4 в (рис. 2, кривые 2 и 4).
При низкой температуре и малой концентрации титана потенциал
катода с увеличением плотности тока смещается в отрицательную
168
сторону до потенциала разряда щелочного металла около — 3,2 в
(кривые /, 3).
Таким образом, при высокой температуре и высокой концентра-
ции K2TiFe в расплаве возможен прямой разряд ионов титана на
катоде до металла (после предварительного восстановления до двух-
валентного состояния). При малой концентрации K2TiF6 в расплаве
и низкой температуре на катоде происходит разряд ионов щелочных
металлов и восстановление титана до металла как вторичный про-
цесс.
Электролиз хлоридов титана
Колк с соавторами [270] на основании исследования формы вольт-
амперных кривых считает, что восстановление титана в хлоридных
расплавах также протекает ступенчато, что согласуется с данны-
ми [299].
Б. Ф. Марков с соавторами [93] на кривых ток — напряжение
в системе TiCl4—KCl—NaCl при температуре 720° С обнаружил три
скачка потенциала. По мнению авторов, один из этих скачков
(<^Л в) соответствует восстановлению TiCl4 до TiCl2, второй (^2 в) —
разряду ионов Ti2+ до металла, а третий — восстановлению фона.
Восстановление титана в хлоридных расплавах до металла проте-
кает, таким образом, в две стадии. Осадок металлического титана
на катоде начинает выделяться при напряжении на ванне 1,8 в.
Это подтверждается и тем, что в хлоридных расплавах., нахо-
дящихся в контакте с металлическим титаном, последний существует
преимущественно в форме двухвалентных ионов [128, 165, 177, 222,
275]. При анодном растворении большая часть титана также пере-
ходит в электролит в виде ионов Ti2+ [165]. Потенциал титанового
электрода в расплаве K2TiF6—KCl—LiCl после выдержки расплава
над титановым металлическим порошком изменяется и металли-
ческий титан по отношению к такому расплаву -ведет себя как не-
растворимый электрод [291].
Дин [235] считает, что осаждение титана на катоде при рафини-
ровании его в расплаве NaCl является химическим процессом, про-
текающим в две стадии:
TiCl3 + Na = TiCl2 + NaCl,
TiCl2 + 2Na = Ti + 2NaCl.
Бокрис с соавторами [233] на поляризационных кривых раство-
ров TiCl4, TiCl3 и TiCl2 в эвтектических смесях LiCl — КО при тем-
пературе 380—500°C получили четкие обратимые волны восста-
новления TiCl3 до TiCl2. Четыреххлористый титан дает одну поля-
рографическую волну при температуре 400° С. Двуххлористый
титан полярографических волн до потенциала разложения фона не
дает. Отсюда авторы сделали вывод, что восстановление хлоридов
169
титана на катоде протекает с образованием только TiCl2, а металли-
ческий титан выделяется в результате вторичного процесса.
Окада и соавторы [287] при полярографическом восстановлении
TiCl3 в эвтектической смеси LiCl — КС1 получили две волны, от-
вечающие процессам Ti3+ -> Ti2+ и Ti -> Ti°. На полярограмме
восстановления Ti4+ имеется только одна волна, соответствующая
процессу Ti4+ —> Ti°. Авторы, однако, полагают, что восстановле-
ние титана в хлоридных расплавах является вторичным процессом,
протекающим по реакции
4Li + TiCl4 = Ti + 4Li+ + 4СГ.
Согласно [243], осаждение титана на катоде происходит как вто-
ричный процесс, так как трех- и двухвалентный титан с хлор-ионом
образуют прочные комплексы, разряд которых на катоде малове-
роятен.
А. И. Иванов и В. Г. Гопиенко [62] считают, что осаждение ти-
тана на катоде при электролизе четыреххлористого титана в хло-
ридных расплавах может протекать как электротермический, так
и электролитический процесс. Преобладание того или иного про-
цесса зависит от условий проведения электролиза.
Аналогичной точки зрения на механизм выделения титана на
катоде придерживаются Г. И. Звиададзе и Д. М. Чижиков [59].
На кривых ток — напряжение они не обнаружили ступенчатого раз-
ряда ионов Ti3+. По их данным, потенциал разряда титана на ка-
тоде зависит от концентрации Ti3+ в расплаве. При температуре
730° С и малой концентрации Ti3+ потенциал разряда ионов титана
на катоде выше, чем потенциал разряда ионов щелочных металлов.
Осаждение металлического титана из расплава в этих условиях про-
исходит за счет восстановления титана щелочными металлами. При
более высокой температуре разряд ионов щелочных металлов на
катоде происходит при более отрицательном потенциале, чем раз-
ряд ионов титана по реакции Ti34" —Ti°, и на катоде становится
возможным непосредственное осаждение титана.
М. В. Смирнов с соавторами [150] для выяснения механизма осаж-
дения титана из хлоридных расплавов изучили зависимость катод-
ной поляризации от плотности тока, температуры и концентрации
титана в электролите. Поляризация исследовалась в эквимольном
расплаве NaCl — КО, содержащем растворенные хлориды титана
TiCl2, TiCl3 и TiCl4, на молибденовом катоде относительно хлорного
электрода сравнения.
В расплавах TiCl2—NaCl—КО, содержащих более 2,47% TiCl2)
при плотностях тока менее Ю”1—10~2 а!см2 электролиз протекает
при потенциалах, соответствующих выделению металлического ти-
тана на катоде, т. е. менее —1,8 в (рис. 3). При более высоких плот-
ностях тока прикатодный слой обедняется ионами Ti2+ и электрод'
170
ный потенциал за счет концентрационной поляризации сдвигается
до потенциала выделения щелочного металла (—3,43 в при 700° С).
В расплавах с малой концентрацией титана потенциал молибде-
нового катода при малых плотностях тока сдвинут примерно на
0,5 в в положительную сторону относительно потенциала выделе-
ния металлического титана (рис. 3, кривые /, 4}. Однако этот сдвиг,
по мнению авторов, объясняется
деполяризацией катода в результа-
те окисления металла примесями
кислорода и влаги, которые нельзя
полностью удалить из расплава в
условиях проведения опыта с малой
концентрацией титана.
Авторы [164, 177] вычислили потен-
циал равновесного титанового элек-
трода в условиях электролиза рас-
плава TiCl2—NaCl—КС1 по приведен-
ной выше формуле
-// -ZZ -2У -5,0 -5,4
Потенциалу
Рис. 3. Поляризация молибдено-
вого катода в расплавленной сме-
си NaCl + КС1 + TiCl2 при тем-
пературах 700 (/, 2, 3) и 800° С
(4, 5, 6):
1,4 — 0,4; 2, 5 — 2,47; 3, 6 —
9,85 вес. % TiClt.
и сравнили полученные величины с
опытными данными (табл. 11). При
плотности тока менее КГ"1—102~ а!см2
численные значения равновесных по-
тенциалов титанового электрода сов-
пали с опытными значениями молиб-
денового катода. При этом потенциал
выделения титана на катоде примерно
на 1 в более положителен, чем потен-
циал разряда ионов щелочных металлов. Отсюда делается вывод,
что из хлоридных расплавов TiCl2 возможен прямой разряд ио-
нов Ti2+ на катоде.
На поляризационных кривых молибденового катода при элек-
тролизе расплава TiCl2—NaCl—КО имеется по три участка, отве-
чающих различным электродным процессам (рис. 4). При плот-
ности тока менее 10-1—10“2 а/см2 потенциал молибденового катода
положительнее потенциала титанового электрода. Соответствующие
участки на поляризационных кривых отвечают перезарядке ионов
Ti3+ до Ti2+. Величина потенциала молибденового электрода оп-
ределяется при этом величиной окислительно-восстановительного
потенциала системы Ti3+/Ti2+. Согласно [170], стандартный элек-
тродный потенциал Е°т3+/т12+ при температуре 700—800 ° С равен
около 1,7 в относительно хлорного электрода сравнения.
С повышением потенциала молибденового электрода до 1,8—
2,4 в начинается выделение металлического титана, чему на поляри-
зационных кривых отвечают наклонные участки. Из расплавов
171
с трехвалентным титаном осаждение металла на катоде сопровожда-
ется значительной концентрационной поляризацией. Причиной ее
является высокая прочность комплексных ионов трехвалентного
Таблица 11
Потенциалы (в) равновесного титанового и
молибденового электродов при электролизе смеси
TiCl2—NaCl—КС1
Концентрация TiCl2 в рас- плаве, вес.% Титановый электрод Молибденовый электрод Потенциал молибденово- го электро- да, в
Температура 700° С
0,40
2,47
9,85
—2,09
—2,02
—1,96
—2,10
—2,02
—1,91
—2,07
— 1,97
— 1,86
Температура 800° С
0,40
2,47
9,85
—2,07
— 1,98
—1,91
—2,07
—1,97
— 1,86
титана [TiCl6]3 • Металлический титан из хлоридных расплавов,
содержащих ионы Ti+3, осаждается при плотности тока 0,1 а!см2.
В интервале потенциалов 1,8—2,4 в на молибденовом катоде наблю-
дается, таким образом, непосредственный разряд ионов Ti3+ до
металла.
Третий участок на поляризационных кривых отвечает быстрому
возрастанию отрицательного потенциала до начала выделения ще-
Рис. 4. Поляризация молибденового
катода в расплавленной смеси
NaCl + КС1 + TiCl2 при температу-
рах 700 (/) и 800° С (2, 3):
1, 5—4,52; 2 — 0,516 вес.% TiCl3.
Рис. 5. Поляризация молибденового ка-
тода в расплавленной смеси NaCl +
4- КС1+ TiCl4:
1 — 720° С, 0,635 вес.% Т1С14; 2 — 750° С,
без добавки TiCl4; 3— 770° С, 0,635 вес.%
TiCl4.
172
лочных металлов (—3,3 в). На этом участке происходит совместное
осаждение титана и щелочных металлов и, возможно, образование
титана за счет восстановления его щелочными металлами.
В расплавах TiCl4—NaCl—КС1 при малых плотностях тока на
молибденовом катоде устанавливается стационарный потенциал
вследствие растворения материала электрода по реакции
Мп I nTi4+ — Мпп+ -4-
iuUtb I 'И ‘расплав — /¥1ирасплав I т ‘расплав*
Потенциал катода начинает возрастать лишь тогда, когда ток за счет
восстановления ионов титана Ti4+ до Ti3+ превысит диффузионный
ток саморастворения молибденового электрода (рис. 5). По данным
авторов, стандартный окислительно-восстановительный потенциал
£’°Ti4+/Ti3+ при температуре 700—800° С составляет около —0,8 в.
Поэтому в солевых расплавах четырехвалентный титан легко вос-
станавливается до трехвалентного. Потенциал перезарядки ионов
Ti3+ до Ti2+ и разряда их до металла заканчивается в области пре-
дельного диффузионного тока перезарядки ионов Ti4+ до Ti3+.
Вследствие малой растворимости четыреххлористого титана в хло-
ридном расплаве при температуре 700—800° С (0,2—0,3% Ti) осаж-
дение металла за счет непосредственного разряда ионов Ti4+ на
катоде возможно только при плотности тока 0,5 а!см2 (при условии
непрерывной подачи TiCl4 в электролит). Из расплавов при больших
плотностях тока наряду с металлическим титаном осаждаются ще-
лочные металлы.
Выделение титана из хлоридных расплавов четырехвалентного
титана из-за малой подвижности ионов [TiClc~], в виде которых ти-
тан находится в расплаве, сопряжено с большой концентрационной
поляризацией. Из сопоставления поляризационных кривых молиб-
денового электрода в расплаве TiCl4—NaCl—КС1 и не содержащих
титана хлоридов видно, что доля тока перезарядки ионов щелочных
металлов до субионов (2 М+-> М^“) мала. Поэтому количество обра-
зующегося титана за счет восстановления субионами щелочных
металлов составляет всего несколько процентов от общего количе-
ства выделившегося металла.
Б. Г. Рассохин с соавторами [142] нашли, что в интервале тем-
ператур 810—983° С величина формального окислительно-восста-
новительного потенциала в расплавленном КО выражается урав-
нением
£°i3+/Ti4+ = 1,371 + 4,3 • IO-4?(в).
Числа переноса в расплаве TiCl3 с NaCl, КО, RbCl и CsCl и вы-
ход по току при электролизе титана закономерно возрастают при
переходе от NaCl к RbCl [54].
Металлический титан при температуре 720—900° С, судя по из-
менению величины электродвижущей силы элемента с хлорным
электродом, взаимодействует с хлоридным расплавом [145]. Анализ
173
расплавов указывает на присутствие в них титана в двух- и трехва-
лентном состоянии. Кинетика и некоторые другие параметры элек-
тролиза титана в хлоридных расплавах рассмотрены в работе [56].
Электролиз иодидов титана
Механизм разряда ионов титана из иодных расплавов еще не
исследован.
Фортин с соавторами [245] измерил напряжение разложения со-
единений титана в эквимольной смеси расплавленных Nal + KI
при температуре 725° С с железным катодом и алюминиевым ано-
дом. Напряжение разложения найдено равным 1,7 в. Авторы пола-
гают, что оно обусловлено разрядом ионов Ti2+ на катоде. В ванне,
состоящей из смеси KI + KF, наблюдается два скачка напряжения
разложения при 0,5 и 1,7 в, которые связываются с образованием
соединений TiF2 и Til2.
Электролиз расплавов, содержащих
окислы титана
Электролиз окислов титана изучался в расплавах галогени-
дов щелочных и щелочноземельных металлов, боридов, фосфа-
тов и т. п.
При электролизе двуокиси титана в расплавленном криолите
на катоде осадки металлического титана не выделяются [276, 277].
По данным рентгеноструктурных исследований, образовавшиеся
при электролизе осадки состояли из низших окислов титана.
При электролизе фторидно-хлоридных расплавов, содержащих
ионы Ti4+, TiO2-1- и Т[2Оз+, наряду с восстановлением титана до
низших степеней валентности происходит разряд на катоде оксика-
тионов и образование низших окислов титана [161 ]. Согласно [281 ],
электролиз сплава ТЮ2—K2TiF6—KF протекает с образованием на
катоде полуторной окиси и металлического титана [281].
И. П. Бардин и А. А. Казайн [39, 85] отмечают, что катодный
осадок, полученный при электролизе сплава ТЮ2—K2TiF6—NaCl,
имеет форму груши и состоит из титана и электролита, находяще-
гося между кристаллами металла. На основании изучения кривых
катодной поляризации, они полагают, что при электролизе сплавов
ТЮ2—K2TiFe—NaCl на катоде происходит разряд ионов натрия
и комплексных ионов [TiFe]2— до металла.
Природа катодного процесса зависит от состава электролита и
времени электролиза [85]. Вначале электролиза потенциал катода
бывает равен около 2 в. При большой концентрации двуокиси ти-
тана в расплаве происходит восстановление ее на катоде до низших
окислов, выпадающих в осадок. Из электролита с малой концентра-
цией двуокиси титана в начале электролиза на катоде выделяется
174
металлический натрий. Натрий восстанавливает K2TiFe и образует-
ся осадок труднорастворимого двойного фторида K2NaTiFe. Выход
по току в начальный период электролиза вследствие протекания
этого процесса мал. По мере увеличения объема катода за счет на-
копления металла потенциал его уменьшается до 1,5—1,7 в и начи-
нается разряд ионов [TiF6]2“ до металла.
Это, однако, противоречит наблюдениям М. В. Смирнова с соав-
торами, который в нескольких работах отмечает независимость по-
ляризации катода от образования на нем порошкообразных метал-
лических осадков.
П. И. Ануфриева и А. И. Иванов [4] полагают, что образование
металлического титана при электролизе расплава TiO2—СаС12 про-
исходит вследствие вторичного восстановления двуокиси титана
металлическим кальцием, осаждающимся на катоде.
Согласно [1931, при электролизе расплава двуокиси титана в
буре на катоде происходит непосредственный разряд ионов титана
до металла. Г. А. Меерсон и М. В. Смирнов [94] в ванне из сплава
2В2О3 + СаО + CaF2, содержащего 15 вес. % ТЮ2, на кривой нап-
ряжения разложения обнаружили один скачок потенциала. Напря-
жение разложения ТЮ2 в ванне, по их измерениям, при температуре
1050° С равно 1,62 в, что ниже теоретически вычисленной величины
(см. табл. 9). Поэтому авторы считают, что при электролизе на ка-
тоде происходит одновременный разряд ионов Ti4+ и В3+ и обра-
зование диборида титана TiB2, за счет чего происходит деполяриза-
ция катода и снижение напряжения разложения TiO2 на ванне ниже
теоретической величины.
Ю. К. Делимарский и Т. Н. Капцова [53] изучили восстановле-
ние двуокиси титана в расплаве метафосфата натрия методом снятия
полярограмм. Они обнаружили ступенчатое восстановление титана
по реакциям
Ti4+ + е -> Ti3+,
Ti3+ + Зе -> Ti°.
Т. Н. Капцова [71 ] получила полярограммы восстановления TiO2
в расплаве силикатного стекла, соответствующие протеканию двух-
и четырехэлектронных процессов.
Хронопотенциометрическим методом определена средняя ве-
личина коэффициента диффузии ионов титана в расплаве NaCl—
—K2TiF6—TiO2 [55]. При 85° С она равна 3,4 • 10"~6 смЧсек.
АНОДНЫЕ ПРОЦЕССЫ ПРИ ЭЛЕКТРОЛИЗЕ
СОЕДИНЕНИЙ ТИТАНА В РАСПЛАВЕ СОЛЕЙ
Химизм анодных процессов зависит от состава электролита, ма-
териала анода и условий электролиза.
175
I
Нерастворимые аноды
В качестве нерастворимых анодов при электролизе титансодер-
жащих расплавов применяются графит, платина, вольфрам и неко-
торые другие материалы. Анодными продуктами электролиза фто-
ридов и хлоридов титана обычно являются фтор или хлор. Из сме-
шанных фтор идно-хлор идно-окисных расплавов на аноде могут
выделяться различные газы в зависимости от составов ванны, тем-
пературы электролиза и анодной плотности тока.
Анодный выход по току при электролизе хлоридных расплавов
с графитовым анодом, если в аналите не содержится низших хлори-
дов титана, практически составляет 100% [67]. Следовательно,
единственным анодным процессом в этих условиях является раз-
ряд ионов хлора.
При электролизе фторидно-хлоридно-окисных расплавов в обыч-
ных условиях на аноде выделяется кислород. Согласно [39, 85], вы-
деление кислорода на аноде происходит по схеме
2TiOFl- + 4F~ — 4е = 2TiF|" + 20.
Если анод графитовый, то он в таких расплавах не будет «нераство-
римым», так как реагирует с кислородом с образованием СО и СО2.
При этом, однако, в ванну не переходят ионы, принимающие уча-
стие в электрохимическом процессе. За счет окисления графита
кислородом наблюдается анодная деполяризация и снижение по-
тенциала разряда кислорода до небольшой величины (0,3—0,5 в) [85].
Электролиз фторидно-оксидных расплавов также происходит с вы-
делением на аноде кислорода [281].
Растворимый титановый анод
В отношении механизма анодного растворения титана имеются
две точки зрения. Дин [234, 235] анодное растворение титана в хло-
ридных расплавах представляет как химический процесс:
Ti + 2NaCl -> 2Na + TiCl2.
Ионы трехвалентного титана в расплаве, по мнению автора, по-
являются в результате окисления двуххлористого титана хлори-
стым натрием:
TiCl2 + NaCl -> TiCl3 + Na.
Другие авторы рассматривают анодное растворение титана как
электрохимический процесс. Так, М. В. Каменецкий [73] показал,
что степень окисления титана при анодном растворении в расплаве
NaCl—КО зависит от плотности тока. Если анодная плотность то-
ка менее 0,35 а/см2, титан переходит в расплав преимущественно
в виде двухвалентных ионов; при плотности тока более 1,3 а!см2
образуются трехвалентные ионы титана. В расплавленном хлори-
стом натрии при анодной плотности тока менее 0,7 а/см2 в результате
анодного растворения преимущественно образуются ионы Ti2+ [44].
176
М. В. Смирнов и Л. А. Циовкина [165] изучили поляризацию
титановых анодов в расплаве эквимольной смеси NaCl — КО с
добавками фтористого натрия и показали, что электролиз расплав-
ленных хлоридов сопровождается значительной концентрационной
поляризацией титановых анодов, достигающей 0,5 в (рис. 6). При
малой плотности тока (менее 0,2 а!см2) наблюдается линейная за-
висимость между логарифмом плотности тока и анодной поляриза-
цией:
Ф = а + b log д’»
где ф — анодный потенциал; а и b — постоянные величины; i —
плотность тока, а!см?. Постоянная Ь по своей величине близка к
»
Рис. 6. Поляризация титанового
анода в расплавленной эквимоль-
ной смеси NaCl + КС1 при 700 (7),
750 (2) и 800° С (5).
-2,6 -2,4 -2,2 -2,0 -1,8
Потенциал, 6
Рис. 7. Поляризация титанового анода
в расплавленной эквимольной смеси
NaCl + КС1 с добавкой NaF при темпе-
ратуре 700° С:
/ — без добавки NaF; 2 — 5; 3 — 10; 4 —
20% NaF.
RT
значению 2,3—, следовательно, концентрационная поляризация
при малых плотностях тока определяется в основном диффузией из
прианодного слоя в расплав ионов двухвалентного титана. С уве-
личением плотности тока выше 0,2 а!см2, анодный потенциал тита-
нового электрода возрастает и по достижении плотности тока 2—
3 а/см2 становится постоянным, так как расплавы чистых солей рас-
творяют металлы без заметной поляризации [160].
Примесь фтористого натрия при малых плотностях тока смещает
потенциал титанного анода в отрицательную сторону (рис. 7). Сме-
щение потенциала титанового анода, по мнению авторов [165],
12 1-1669
177
объясняется образованием в хлоридно-фторидных расплавах не-
растворимых осадков двойных фторидов двухвалентного титана
с фторидами калия и натрия.
С повышением плотности тока анодный потенциал возрастает за
счет концентрационной поляризации, связанной с диффузией ионов
фтора из объема электролита в прианодный слой. В области предель-
ного тока диффузии ионов фтора потенциал анода резко возрастает
и приближается к поляризации в чисто хлоридных расплавах. Из-за
недостатка фтора титан может связываться с ионами хлора. Потен-
циал анода при этом быстро растет до величины, характерной для
хлоридных расплавов.
Таким образом, при электролизе хлоридно-фторидных расплавов
с малой плотностью тока на аноде образуются фториды, а при боль-
шой — хлориды титана. Концентрационная поляризация титановых
анодов в смешанных фторидно-хлоридных расплавах достигает
значения 0,9 в.
Валентность металла, с которой он переходит в расплав при анод-
ном растворении, отвечает средней валентности его ионов в расплаве,
находящемся в равновесии с металлом при данной температуре
[72, 150, 168, 220]. Средняя валентность титана в эквимольной смеси
NaCl — КО при температуре 701—975° С и содержании титана
0,23—5,64% изменяется от 2,03 до 2,13 [175,177].
Для выяснения, с какой валентностью титан переходит в рас-
плав при анодном растворении, М. В. Смирнов и Л. А. ЦиОвкина
[165] изучили анодный выход по току. Экспериментальные данные
выхода по току при анодном растворении титана в расплаве NaCl—
КС1 с анодной плотностью тока около 0,1 а!см2 при температуре
707—932° С практически совпали с данными расчета по уравнению
температурной зависимости константы диспропорционирования по
реакции [177]
3Ti
2Ti3+ _j_ Ti
Авторы, однако, не объяснили, чем вызвано увеличение выхода
Ti3+ при повышении анодной плотности тока, отмеченное М. В. Ка-
менецким [73].
Средняя валентность титана при анодном растворении в хлорид-
но-фторидных расплавах равна 2,1 [165].
Катодный процесс при рафинировании титана аналогичен про-
цессу электролиза двуххлористого титана [3]. Рафинирование ти-
тана из титановых отходов и из расплавов бромидов щелочных ме-
таллов рассмотрено в работах [49, 244].
Растворимые аноды из окислов титана
Токопроводящие растворимые аноды могут быть приготовлены
из низших окислов титана Ti2O3 и TiO, обладающих хорошей элек-
тропроводностью, и из смеси двуокиси титана с углем. Электрохи-
178
I
мическое поведение этих анодов, представляющее большой интерес
для электрометаллургии титана, специально исследовано М. В. Смир-
новым с соавторами.
Потенциалы анодов из TiO и Ti2O3 в расплаве эвтектической
смеси LiCl — КО при температуре 500° С и малой плотности тока
равны соответственно — 1,94 и —1,5 в относительно хлорного элек-
трода сравнения [60, 163]. С повышением плотности тока потенциал
анода из Ti2O3 сдвигается в положительную сторону на 0,2—0,3 в
(рис. 8). Потенциал анода из TiO также сильно сдвигается в
положительную сторону и при плотности тока около 6 0,1см2 совпа-
дает с потенциалом анода из Ti2O3.
Электролиз анодов из TiO2 и Ti2O3 вплоть до плотности тока
3—5 oJcm2 не сопровождается выделением газообразных продуктов
[163, 169]. Наблюдения за изменением анодов и содержанием титана
в электролите позволили установить следующий механизм электро-
литического растворения окислов титана. При анодной плотности
тока 0,05 а!см2 в результате электролиза TiO в раствор переходят
ионы Ti2+ с 100%-ным выходом по току. С увеличением плотности
тока наряду с двухвалентными ионами в расплав начинают пере-
ходить ионы трехвалентного титана. Соотношение между ионами
Ti2+ и Ti3+, переходящими в раствор, не было постоянным. Одно-
временно на поверхности анода образуется корочка из окислов ти-
тана высшей валентности. Она состоит из осадков фиолетового и
белого цветов, т. е. из окислов Ti2O3 и TiO2.
При электролизе анодов Ti2O3 в аналит переходит титан в виде
трех- и четырехвалентных ионов. На поверхности анода образует-
ся белая корочка из двуокиси титана.
Химизм анодного растворения низших окислов титана выра-
жается, таким образом, следующими уравнениями:
ЗТЮ (анод) — tie -> Ti2O3 (анод) 4- Tin+ (расплав),
2Ti2O3 (анод) — пе -> ЗТЮ2 (анод) + Tin+ (расплав).
Из расплава, полученного анодным растворением низших окислов
титана, на катоде осаждается металлический титан. На этом осно-
вании авторы [163, 169] отрицают переход титана при анодном рас-
творении в расплав в виде оксикатионов типа TiO2+ и П20з+.
Аноды из смеси окислов TiO и Ti2O3 и углерода при малой плот-
ности тока ведут себя так же, как и аноды из чистых окислов, не
содержащих углерода [173]. Поляризационные кривые их (рис. 9)
мало отличаются от поляризационных кривых анодов из чистых
окислов титана (см. рис. 8). Углекислый газ при электролизе окис-
но-угольных анодов начинает выделяться не в начале, а спустя
некоторое время после включения ванны, и тем больше, чем меньше
плотность тока.
Аноды из смеси TiO2 + С при плотности тока 0,006—0,06 а! см2
12*
179
растворяются с переходом в расплав ионов трехвалентного титана.
Растворение анодов из Ti2O3 + С при тех же плотностях тока со-
провождается образованием также ионов четырехвалентного тита-
на. Путем термодинамических расчетов авторы [173] показали, что
анодная реакция
ЗТЮ — Зе = Ti2O3 + Ti3+
предпочтительнее другим возможным реакциям:
TiO + !/2С - Зе = Ti3+ + % СО2>
TiO + С — Зе = Ti3+ + СО.
-2,0 -1,6 -1,2.
Потенциал^
Рис. 8. Поляризация
анодов в расплаве
LiCl + KCl при тем-
пературе 500° С:
/ — анод из TiO; 2 —
анод из Ti2O3.
-1,6 -1,3-1,0 -0,7-0,4
Потенциал,б
Рис. 9. Поляризация окисно-
угольных анодов в эквимольной
смеси NaCl + KCl:
1 — анод из TiO 4~ С при 830° С;
2 — анод из Ti2O3 + С при 805° С;
3 — анод из TiO2 4- С при 700° С.
Растворение анода из Ti2O3 + С может протекать по трем реак-
циям, равновероятным в термодинамическом отношении:
2Ti2O3 — Зе = 3TiO2 J- Ti3+,
72 Ti2O3 + % С — Зе = Ti3+ + % СО2,
1/, Ti2O3 + ®/2 С — Зе = Ti3+ + 3/, СО.
Фактическая доля участия каждой из этих реакций в процессе анод-
ного растворения определяется кинетикой процесса. При малых
плотностях тока протекает первая реакция. Когда содержание кис-
лорода в поверхностном слое частиц Ti2O3 приблизится к предель-
ному, потенциал анода достигает величины около —1,3 в и начина-
ет протекать вторая реакция, о чем можно судить по содержанию
180
в анодных газах двуокиси углерода. Третья реакция практически
не реализуется вследствие разряда ионов хлора.
Анодные процессы, протекающие при растворении анода из
TiO2 + С, изучили М. В. Смирнов и С. Ф. Пальгуев [103, 159, 171 ].
Вследствие малой величины окислительно-восстановительного
потенциала Tj3+/Tj4+
при малых плотностях тока анодное раство-
рение двуокиси титана сопровождается восстановлением титана до
трехвалентного состояния:
TiO, + С — Зе = Ti3+ + СО,.
При больших плотностях тока
из-за малой скорости диффузии
кислорода в частицах двуокиси
титана наблюдается значительная
поляризация анода (см. рис. 9), в
результате чего на аноде начина-
ют разряжаться ионы хлора,
вступающие в реакцию хлори-
рования окисно-угольной смеси.
Вследствие адсорбации ионов
хлора на частицах углерода по-
-7J -1,5 -1,1 -0,9 -0,7—0,5
Потенциал,0
Рис. 10. Поляризация анода из
смеси TiO2 + С в расплаве
NaCl+KCl (/) и в смешанном хло-
рид но-фтор ид ном расплаве (2).
ктрода сравнения. Доля титана,
тенциал анода достигает величи-
ны хлорного электрода, и в даль-
нейшем электролиз протекает с
выделением на аноде смеси га-
зов СО2 + С12 при потенциале
0,1—0,2 в относительно хлорного
переходящего в расплав в четырехвалентном состоянии, по мере
возрастания анодного потенциала увеличивается. Соотношение меж-
ду трех- и четырехвалентным титаном в расплаве устанавливается
в соответствии с величиной окислительно-восстановительного потен-
циала, определяемого уравнением
Примесь фтористого натрия к хлоридному электролиту сдвига-
ет потенциал анода TiO2 + С в отрицательную сторону (рис. 10).
Сдвиг потенциала объясняется образованием в расплаве фторидных
комплексов трехвалентного титана, более прочных по сравнению
с хлоридными комплексами.
Растворимый анод из нитрида титана
Анодное растворение нитрида титана в эвтектическом расплаве
LiCl + КО идет по двум реакциям [163, 167]:
TiN (анод) — 4е -> Ti4+ (расплав) + % N2,
TiN (анод) — Зе -> Ti3+ (расплав) -ф l/2 N2.
Преимущественно протекает реакция с образованием четырехва-
лентного титана. Механизм анодного растворения нитрида титана
181
I
был выяснен М. В. Смирновым с соавторами в результате изуче-
ния анодных поляризационных кривых.
При плотности тока менее 1,5 • 10-3 а/см2 потенциал нитридно-
го анода изменяется мало (рис. 11). В интервале плотности тока
0,002—0,2 а!см2 наблюдается значительная поляризация анода,
потенциал его возрастает на 0,6—0,7 в, но при дальнейшем
Рис. 11. Поляризация анода из
нитрида титана в эвтектичес-
кой смеси LiCl + КС1 при тем-
пературе 753 (/), 652 (2) и
550° С (5).
-// -1,0 -0,0
Потенциал, б
Рис. 12. Поляризация анода из
карбида титана в эвтектичес-
ком расплаве LiCl + КС1 при
температуре 800 (/), 650 (2)
и 530° С (<?).
повышении плотности тока заметно не изменяется. Причину поля-
ризации анода из нитрида титана авторы усматривают в обеднении
его поверхности титаном.
Испытуемый образец нитрида титана имел состав Ti 1,22-1,27 N,
т. е. представлял твердый раствор титана в нитриде TiN. При малых
плотностях тока он вел себя как металлический титан, для которого
электродный потенциал по отношению к расплаву, содержащему
ионы Ti4-1, по данным термодинамических расчетов, составляет
около — 1,2 в (температура 550—750° С). По мере обеднения по-
верхности анода титаном состав его изменяется до предельного
TiN, после чего начинается выделение азота и потенциал анода при-
нимает постоянную величину.
Нитрид титана может найти применение для получения метал-
лического титана. Так, при электролизе нитрвдсодержащнх титано-
вых шлаков получены осадки с содержанием 98—99% металличе-
ского титана [153].
182
Растворимый анод из карбида титана
Карбид титана предложен для получения металлического ти-
тана электролизом растворимых анодов [224, 242, 257]. При анод-
ном растворении карбида титана в электролите из смеси NaCl 4- КО
с добавкой LiCl титан переходит в расплав в виде двух- и трехва-
лентных ионов. М. В. Смирнов и Ю. Н. Краснов [172] изучили ва-
лентность титана, переходящего в эквимольный расплав NaCl 4~
4- КС1. Оказалось, что анодное растворение карбида титана сопро-
вождается переходом в хлоридный расплав в основном ионов Ti4+.
Небольшое количество титана переходит в расплав в виде ионов
Ti3+. Как видно из поляризационных кривых (рис. 12), при анодной
плотности тока менее 0,003 и более 0,1 а!см2 потенциал карбида
титана в расплаве LiCl 4~ КО изменяется незначительно. В про-
межуточном интервале плотности тока наблюдается значительная
поляризация анода, достигающая 0,5—0,6 в.
Отсутствие изменения поляризации анода из карбида титана
при низкой плотности тока авторы объясняют тем, что в этих усло-
виях переход ионов титана с поверхности электрода в расплав
компенсируется диффузией титана из внутренних слоев анода и
встречной диффузией углерода. Возрастание потенциала при плот-
ности тока более 0,003 а!см2 является результатом недостаточной
диффузии титана и углерода в поверхностном слое анода. Рост по-
ляризации прекращается после накопления значительной концен-
трации ионов Ti4+ в прианодном слое и начала выделения TiCl4 из
расплава.
/
Анодное растворение сплавов титана
Кроме описанных анодов изучено анодное растворение спла-
вов титана с примесью железа, марганца, кремния, ниобия, алюми-
ния и других металлов [35, 89, 95, 104, 152, 154, 156, 191, 228, 236,
265, 278], а также рафинирование чернового металла или отходов
металлообработки, загрязненных азотом, кислородом и углеродом
[2, 5, 38, 61, 151, 153, 157, 235, 258, 263, 264, 279, 300]. Однако ра-
боты по рафинированию сплавов титана носят преимущественно тех-
нологический характер и в них обычно излагаются только результа-
ты исследований по получению металлического титана.
А. Б. Сучков с соавторами [152] показал, что при рафинировании
анодов из бинарных сплавов Ti —Fe, Ti —Si и Ti — Nb достигает-
ся их полное разделение на компоненты. Кальций при рафинирова-
нии титановых сплавов накапливается в электролите (рафинирова-
ние ведется при 760 ° С, электролит—эквимольная смесь NaCl 4~ КО).
Дин [235] сообщил, что при рафинировании титана карбиды и
нитриды титана, марганца, хрома, алюминия и ванадия остаются
в анодном шламе и не переходят в электролит. А. П. Пичуков [1041
183
показал возможность очистки титана методом электролитического
рафинирования от примесей железа, меди, кремния, молибдена и
олова. В. А. Суходский и Н. И. Ануфриева [156] сообщили, что хром
при содержании его в титане 4,3% при анодном рафинировании не
переходит на катод.
Во всех опубликованных работах отмечается, что анодное ра-
финирование является надежным методом для очистки титана от
примеси кислорода, азота и углерода. Очистка титана от марганца
и алюминия методом анодного рафинирования малоэффективна [35,
89, 95, 104, 153, 156, 238, 291].
В. С. Балихин и В. А. Резниченко [35] изучили зависимость
анодного потенциала титанового и алюминиевого электродов от
плотности тока и показали, что с повышением плотности тока кон-
центрационная поляризация титанового электрода вследствие по-
падания в прианодный слой ионов Ti3+ и Ti2+ растет быстрее, чем
алюминиевого анода, так как алюминий образует прочный хлор и д-
ный комплекс (А1С14)~. TiAl имеет более положительный потенциал
анодного растворения, чем титан и алюминий.
По данным В. Т. Мусиенко с соавторами [95], при электролизе
анодов из алюмотитановых сплавов оба металла переходят в хло-
ридный расплав. Степень загрязнения катодных осадков титана
алюминием зависит от плотности тока и концентрации низших хло-
ридов титана в электролите. Авторы показали возможность получе-
ния титановых осадков из алюмотитановых анодов, содержащих
менее 3% AI.
ПОБОЧНЫЕ ПРОЦЕССЫ, СОПРОВОЖДАЮЩИЕ
ЭЛЕКТРОЛИЗ ТИТАНА В РАСПЛАВЛЕННЫХ СРЕДАХ
Металлический титан при высоких температурах легко вступает
во взаимодействие с кислородом, азотом, углеродом и другими не-
металлами. Электрохимические процессы в расплаве солей в при-
сутствии этих примесей могут маскироваться или даже изменять
свое направление. Поэтому все электрохимические исследования
титана в расплавленных солях ведутся обычно в отсутствие воздуха,
паров влаги и других взаимодействующих с ним газов. Для защиты
продуктов электролиза от действия агрессивных газов опыты про-
водят в атмосфере аргона или гелия.
На стенках электролизеров при рафинировании титана нередко
образуется налет металла. В. А. Суходский [158] показал, что оса-
док титана на стенках электролизера образуется в результате вос-
становления ионов титана низшей валентности металлическим нат-
рием:
Ti"+ + nNa -> Ti + nNa+.
Натрий может появляться в электролите вследствие разряда его
ионов на катоде. На аноде, кроме растворения металлического
184
титана при его электролитическом рафинировании, происходит
окисление натрия, растворенного в электролите.
Образование металлического титана в объеме электролита и
на поверхности электролизера наблюдали М. В. Смирнов, и
Л. А. Циовкина [170]. Внекатодное восстановление титана до ме-
талла, по их мнению, происходит за счет субионов щелочных ме-
таллов, образующихся на катоде и диффундирующих в расплав.
ЭЛЕКТРОЛИЗ ТИТАНА ИЗ НЕВОДНЫХ РАСТВОРОВ
Электрохимическое поведение титана в неводных растворах
изучено еще недостаточно. Гутман и Новотный [251] провели опы-
ты по электролизу растворов двуххлористого и четырех хлор исто го
титана, а также бис-(циклопентадиенил)титандихлорида в раз-
личных неводных растворителях, надеясь получить в качестве осад-
ка на катоде металлический титан. Однако металл в этих опытах
не был получен.
Ошиба [284] изучил электролиз хлоридов титана в жидком
аммиаке. При комнатной температуре напряжение разложения
двухлористого титана в жидком аммиаке (анод из графита, катод
из металлического титана) равно 2,1—2,2 в. На катоде выделяет-
ся металлический титан, но в виде мелкого пирофорного порошка.
Литература
1. А г а п о в А. М., Мельников А. М., Кузьмин Л. Л.— Изв.
высш. уч. зав. Хим. и хим. технол., 1968, И, 75.
2. А н у о р и е в а Н. И.— Труды ВАМИ, 1963, 50, 177.
3. Ануфриева Н. И.— В кн.: Титан и его сплавы, 9. Изд-во АН СССР,
М., 1963, 242.
4. Ануфриева Н. И., Иванов А. И.— Изв. АН СССР. Отд. техн,
наук, металлургия и топливо, 1960, 4, 9.
5. Ануфриева Н. И., Гопиенко В. Г. и др.— Труды ВАМИ, 1963,
5, 167.
6. Австралийский пат. № 154266, от 3. 12. 53.
7. Австралийский пат. № 157574 от 22.07.54; РЖХим., 1955, 43569.
8. Австралийский пат. № 160638 от 3.02.55; РЖХим., 1955, 55731.
9. Австралийский пат. № 160980 от 17.02.55; РЖМ, 1957, 500.
10. Австралийский пат. № 167070 от 8. 03.56.
11. Австралийский пат. № 213225 от 20.09.56; РЖХим., 1959, 79227.
12. Австралийский пат. № 2132226 от 20.09.56; РЖХим., 1959, 82868.
13. Англ. пат. № 678807 от 11.05.51; Франц, пат. 1036927 от 29.04.53.
14. Англ. пат. № 682919 от 19.11.52.
15. Англ. пат. № 698151 от 7.10.53; РЖХим., 1955, 7895.
16. Англ. пат. № 712742 от 28.07.54; С. А., 49, 29110.
17. Англ. пат. № 713446 от 11.08.54; С. А., 49, 2910.
18. Англ. пат. № 724198 от 16.02.55; С. А., 49, 11473.
19. Англ. пат. № 734094 от 27.07.55.
20. Англ. пат. № 736567 от 15.06.53; С. А., 50, 3987.
21. Англ. пат. № 741958 от 14.12.55; С. А., 53, 11863.
22. Англ. пат. № 756943 от 19.06.53; С. А., 51, 6403.
23. Англ. пат. № 775870 от 13.06.53; С. А., 51, 13624.
24. Англ. пат. № 771679 от 18.01.54; С. А., 51, 8556.
185
25. Англ. пат. № 777892 от 14.06.55; С. А., 55, 546.
26. Англ. пат. № 812817 от 19.05.55; С. А., 53, 21293.
27. Англ. пат. № 823125 от 1.07.57; С. А., 54, 5298.
28. Англ. пат. № 825951 от.2.08.56; РЖХим., 1961, 20К161.
29. Англ. пат. № 826667 от 20.01.60.
30. Англ. пат. № 832416 от 9.09.57.
31. Англ. пат. № 832190 от 6.04.60; РЖХим., 1961, ЗК245.
32. Англ. пат. № 847878 < от 9.05.58; РЖХим., 1961, 17К179.
33. Англ. пат. № 856588 от 26.03.57.
34. Англ пат. № 855665 от 25.03.57.
35. Балихин В. С., Резниченко В. А.— В кн.: Титан и его сплавы,
9. Изд-во АН СССР, М., 1963, 220.
36. Белов С. Ф., Скляренко С. И.— В кн.: Сборник научных трудов
Гиредмет, 1. Металлургиздат, М., 1959, 329.
37. Бельгийский пат. 538989 от 30.07.59.
38. Б а й м а к о в Ю. В., Каменецкий М. В., Смирнов В. В.—
Изв. высш. уч. зав. Цветная металлургия, 1960, 3, 81.
39. Б а р д и н И. П., К а з а й н А. А.— Изв. АН СССР. Отд. техн, наук,
металлургия и топливо, 1960, 4, 3.
40. Б е л я е в А. И. Физико-химические процессы при электролизе алюминия.
Металлургиздат, М., 1947, 8.
41. Б а й т а л о в Д. А., Файзуллин Ф. Ф.— В кн.: Сборник аспирант-
ских работ. Казанский ун-т. Естественные науки и химия. Изд. Казанск.
ун-та, Казань, 1966, 100.
42. Б а й т а л о в Д. А., Файзулин Ф. Ф.— Учен. зап. Казанск. ун-та,
1965, 124, 31.
43. Бару В. Е., Белов С. Ф., Рузинов Л. П., Самсон Ю. И.
Физическая химия расплавленных солей. Металлургиздат, М., 1965, 297.
44. Г и т м а н Е. Б.— Укр. хим. ж., 1962, 28, 1116.
45. Г и т м а н Е. Б. Электрохимия титана в расплавленных солях. «Наукова
думка», К-, 1965.
46. Гото М., И с и и Д.— J. Chem. Soc. Japan. Pure Chem. Sec., 1969, 90, 376.
47. Г о т о М., И с и и Д.— J. Chem. Soc. Japan. Pure Chem. Sec., 1968, 89, 864.
48. Гоп иен ко В. Г., Ануфриева Н. И., Ключникова Е. Ф.—
Ж. прикл. хим., 1966, 39, 577.
49. Г о п и е н к о В. Г., Ануфриева Н. И., Христюк Г. П.—
Труды ВАМИ, 1965, 54, 55, 369.
50. Г о п и е н к о В. Г., Иванов А. И.— Изв. АН СССР. Отд. техн наук,
металлургия и топливо, 1960, 4,15.
51. Гопиенко В. Г.— В кн.: Титан и его сплавы, 6. Изд-во АН СССР, М.,
1961, 194.
52. Гр аци а н сь Ki й Н. Н., Вовкогон А. П.— Зап. 1н-ту xiM.
АН УРСР, 1940, 7, 173.
53. Делимарский Ю. К-, Капцова Т. Н.— Ж. неорг. хим., 1958,
3, 2751.
54. Д е л и м а р с ь к i й Ю. К«, Чернов Р. В.— ДАН УРСР, 1961, 1508.
55. Делимарский Ю. К-, Погребной П. А.— Укр. хим. ж.,
1967, 33, 1308.
56. Елютин В. П., Лысов Б. С., Андреев Ю. Я.— Электрохимия,
1967, 3, 432.
57. Железнов А. И., Максименко Б. Н.— Легкие металлы, 1935,
4, 24.
58. Делезнов А. И.— Легкие металлы, 1935, 1, 17.
59. 3 в и а д а д з е Г. Н., Чижиков Д. М.— В кн.: Титан и его сплавы,
4. Изд-во АН СССР, М., 1960, 153.
60. И в а н о в с к и й Л. Е., Смирнов М. В.— Изв. Восточн. филиалов
АН СССР, 1957, 10, 68.
61. И в а н о в А. И., Пичуков А. П.— В кн.: Титан и его сплавы, 8.
Изд-во АН СССР, М., 1962, 227.
186
62. И в ан ов А. И., Гопиен ко В. Г.— Труды ВАМИ, 1957, 40, 388.
63. И в а н о в А. И., Мауритс И. И., Гопиенко В. Г.— Труды
ВАМИ, 1957, 40, 380.
64. И в а н о в А. И., Ануфриева Н. И.— В кн.: Титан и его сплавы,
6. Изд-во АН СССР, М., 1961, 131.
65. И в а н о в А. И.— В кн.: Титан и его сплавы, 6. Изд-во АН СССР, М.,
1961, 124.
66. Иванов М. В., Максимов В. С.— Электрохимия, 1965, 1, 727.
67. Иванов А. И., Кухарева И. Г.— В кн.: Титан и его сплавы,
8. Изд-во АН СССР, М., 1962, 220.
68. Ким Л. П., Муртазаев А. М.— ДАН УзССР, 1964, 10, 30.
69. Крюкова Т. А., Синякова С. И., Арефьева Т. В. Поля-
рографический анализ. Госхимиздат, М., 1959.
70. Курбатов Д. И.— Изв. СО АН СССР, 1959, 5, 81.
71. Капцова Т. Н. Физическая химия расплавленных солей и шлаков.
Изд. ГНТИ, М., 1962, 469.
72. Комаров В. Е., Смирнов М. В., Барабошкин А. Н.—
Труды Ин-та электрохимии УФАН СССР, 1962, 3, 25.
73. Каменецкий М. В.—Научн. докл. Высшей школы. Металлургия,
1958, 2, 104.
74. К о л о м и ц к и й Ф. М., Пономарев В. Д.— Изв. высш. уч. зав.
Цветная металлургия, 1959, 5, 106.
75. Крылов Е. И., Колеватова В. С., Самарина В. А.— ДАН
СССР, 1954, 158, 593.
76. Крылов Е. И., Колеватова В. С.— Ж- аналит. хим., 1956, 11,144.
77. Кочергин С. М., Победимский Г. Р.— Ж- прикл. хим., 1960,
33, 238.
78. Кудрявцев Н. Т., Головчанская Р. Г., Барабашки-
н а Н. К-—Труды МХТИ, 1961, 32, 272.
79. Кудрявцев Н. Г., Головчанская Р. Г., Барабашки-
н а Н. К.—ДАН СССР, 1960, 132, 636.
80. Кудрявцев Н. Т., Головчанская Р. Г., Савостьяно-
ва В. М.— Изв. высш. уч. зав. Хим. и хим. технол., 1966, 791.
81. Кудрявцев Н. Т., Головчанская Р. Г.— ДАН СССР, 1963,
148, 1339.
82. Коршунов И. А., Малюгина Н. И.— Ж- общ. хим., 1964, 34, 734.
83. Канадский пат. № 511842 от 12.04.55.
84. К а з а й н А. А., Хромов А. Д.— В кн.: Титан и его сплавы, 2.
Изд-во АН СССР, М., 1959, 103.
85. К а з а й н А. А.— В кн.: Титан и его сплавы, 8. Изд-во АН СССР, М.,
1962, 207.
86. К а з а й н А. А., Хромов А. Д. Металлургия СССР (1917—1957),
1. Металлургиздат, М., 1958, 633.
87. Л оги н ов Н. А., Смирнов М. В.— Труды ин-та электрохимии
УФАН СССР. 1962, 3, 17.
88. Л а т и м е р В. Окислительные состояния элементов и их потенциалы в
водных растворах. ИЛ, М., 1954.
89. Л у к а ш и н В. И., Р е з н и ч е н к о В. А., ХромовА. Д.— Изв.
АН СССР. Отд. техн, наук, металлургия и топливо, 1960, 4, 29.
90. Л о г и н о в Н. А., Смирнов М. В., Россохин Б. Г.— Труды
ин-та электрохимии УФАН СССР, 1965, 7, 9.
91. М а р к о в Б. Ф., П о д а ф а Б. П.— Укр. хим. ж., 1963, 29, 600.
92. М а р к о в Б. Ф.. Под а фа Б. П.— Укр. хим. ж., 1965, 31, 873.
93. Марков Б. Ф., Гитман Е. Б., Белякова Е. П.— Укр. хим.
ж., 1961, 27, 39.
94. М е е р с о н Г. А., С м и р н о в М. В Химия редких элементов, 2.
Изд-во АН СССР, М., 1955, 130.
95. М у с и е н к о В. Т., О р л о в В. М., Сорокин Т. И.— Изв. АН СССР.
Отд. техн, наук, металлургия и топливо, 1961, 6, 30.
I 187
96. Немецкий пат. № 150557 от 1903; Англ. пат. № 13759 от 17.06.1904.
97. Немецкий пат. № 615951 от 16.07.35.
98. Норвежский пат. № 97209 от 12.12.60; РЖМ, 1961, 11Г126.
99. Норвежский пат. № 84401 от 11.10.54; РЖХим., 1961, 55732.
100. Норвежский пат. № 85139 от 7.03.55.
101. Норвежский пат. № 94293 от 20.07.59; РЖХим., 1960, 53339.
102. Норвежский пат. № 84928 от 31.01.55.
103. Пальгуев С. Ф., Смирнов М. В.— Труды Ин-та химии и метал-
лургии УФАН СССР, 1954, 8, 47.
104. Пичуков А. П.— В кн.: Титан и его сплавы, 6. Изд-во АН СССР,
М., 1961, 185.
105. Пат. ФРГ № 1103600 от 17.10.52; С. А., 56, 11366.
106. Пат. ФРГ № 1016452 от 20.03.58; РЖХим., 1959, 35607.
107. Пат. США № 2707169 от 26.04.55.
108. Пат. США № 2707170 от 26.04.55; С. А., 53, 12163.
109. Пат. США № 2731402 от 17.01.56; С. А., 50, 6228.
ПО. Пат. США № 2749295 от 5.06.56; РЖХим., 1958, 18624.
111. Пат. США № 2731404 от 17.01.56; С. А., 50, 6229.
112. Пат. США № 2813068 от 12.11.57; С. А., 52, 6986.
113. Пат. США № 2789943 от 23.04.57; С. А., 54, 9380.
114. Пат. США № 2777763 от 15.01.57; С. А., 51, 6493.
115. Пат. США № 2785066 РЖХим., 1959, 14921.
116. Пат. США № 2798844 С. А., 51, 17979.
117. Пат. США № 2780593 С. А., 51, 6403.
118. Пат. США № 2783196 РЖХим., 1958, 61641.
119. Пат. США № 2864749 РЖМ, 1960, 5255.
120. Пат. США № 2821506 РЖМ, 1959, 12479,
121. Пат. США № 2848397 С. А., 52, 19623.
122. Пат. США № 2848395 С. А., 52, 19623.
123. Пат. США № 2901410 С. А., 53, 21293.
124. Пат. США № 2909472 РЖМ, 1961, 1г180.
125. Пат. США № 2917440 РЖМ, 1961, 1г178.
126. Пат. США № 2898275 РЖХим., 1960, 85495.
127. Пат. США № 2875038 С. А., 53, 12145.
128. Пат. США № 2943032 РЖМ, 1961, 8г 139.
129. Пат. США № 2951021 РЖХим., 1961, 14к215.
130. Пат. США № 2955078 РЖМ, 1961, 11Г128.
131. Пат. США № 2921890 РЖХим., 1961, 4к220.
132. Пат. США № 3082159 С. А., 58, 10981.
133. Пат. США № 2951795 РЖХим., 1961, 17к23.
134. Пат. США № 2975111 ИЖХим., 1962, 5к211.
135. Пат. США № 3003934 РЖМ, 1962, 10г102.
136. Пат. США № 3029193 РЖХим., 1963, 13Л239.
137. Пат. США № 3036961 РЖХим., 1963, 17Л195.
138. Пат. США № 3002905; Offic. Gaz. U. S. Patent Office, 1961, 771. 1, 214.
139. Пат. США № 2986502; Offic. Gaz. U. S. Patent Office, 1961. 766, 5, 1304.
140. Пат. США № 3019174; Offic. Gaz. U. S. Patent Office, 1962, 774, 5, 1166.
141. Петренко А. Т., Харитонов Е. Г.— Всесоюзная конференция
по электрохимии, 1969. «Мицниереба», Тбилиси, 1969, 517.
142. Россохин Б. Г., Смирнов М. В., Логинов Н. А.— Труды
Ин-та электрохимии УФАН СССР, 1966, 8,29.
143. Рассохин Б. Г., Смирнов М. В., Логинов Н. А.— Труды
Ин-та электрохимии УФАН СССР, 1965, 7, 17.
144. Р у с к а л Ю. С., Клинов И. Я.— Изв. высш. уч. зав. Хим. и хим.
технол., 1969, 12, 1244.
145. Родякин В. В., Волынская М. П.— В кн.: Сборник трудов
Всесоюзного н.-и. и проектного ин-та титана, 2. «Металлургия», М., 1968, 77.
146. Розенфельд И. Л., Акимов А. Г., Оше Е. К-— Защита ме-
таллов, 1969, 5, 217.
188
147. Семен о в-К обзарь А. А., Обол он чи к В. А.— Всесоюзная
конференция по электрохимии, 1969. «Мицниереба», Тбилиси, 1969, 580.
148. Стромберг А. Г., К ар ту ш ин ск а я А. И.— В кн.: Теория
и практика полярографического анализа. Изд-во «Штинца», Кишинев, 1962,
341.
149. С ы р о к о м с к и й В. С., Силаева Е. В., Авилов В. Б.—
Зав. лабор., 1949, 15, 896.
150. Скиба О. В., Смирнов М. В., Рыжик О. А.— Труды Ин-та
электрохимии УФАН СССР, 1962, 3, 41.
151. С т р е л е ц X. Л., Войницкий А. И. и др.— Легкие металлы.
Сборник материалов техн, информ, под ред. В. В. Болдина и
В. М. Гуськова, 1957, 4, 114.
152. С у ч к о в А. Б., Б о р о к Б. А. и др.— Цветные металлы, 1959, 6, 57.
153. С у ч к о в А. В., Борок Б. А. и др.— Цветные металлы, 1959,
8, 50.
154. Сучков А. Б., Борок Б. А., Ермакова Т. Н.— В кн.: Титан
и его сплавы, 6. Изд-во АН СССР, М., 1961, 180.
155. Скляренко С. И., Л и п к е с Я. М.— Ж- прикл. хим., 1940,
13, 51.
156. Суходский В. А., Ануфриева Н. И.— Труды ВАМИ, 1962,
48, 188.
157. Суходский В. А., ГопиенкоВ. Г.— Труды ВАМИ, 1960, 46,
146.
158. С у х о д с к и й В. А.— Изв. АН СССР. Отд. техн, наук, металлургия
и топливо, 1960, 4, 63.
159. Смирнов М. В., Пальгуев С. Ф.— Труды Ин-та химии и ме-
таллургии УФАН СССР, 1954, 8, 57.
160. Смирнов М. В., Ивановский Л. Е.— Ж- физ. хим., 1957, 31,802.
161. Смирнов М. В., Пальгуев С. Ф., Волченкова 3. С.—
Изв. Восточных филиалов АН СССР, 1957, 3, 94.
162. Смирнов М. В., Ивановский Л. Е., Ошина Л. Д.— Тру-
ды Ин-та химии УФАН СССР, 1958, 2, 53.
163. Смирнов М. В., Ивановский Л. Е., Краснов Ю. Н.—
Труды Ин-та химии УФАН СССР, 1958, 2, 177.
164. Смирнов М. В., Ивановский Л. Б., Логинов Н. А.—
ЛАН СССР 1958 121 685
165. С м и р н о’в М.’ В..’ Ц и о в к и н а Л А.— Изв. СО АН СССР, 1958, 9,
17.
166. Смирнов М. В., Ю ш и н а Л. Д., Ивановский Л. Е.— Тру-
ды Ин-та химии УФАН СССР, 1958, 2, 161.
167. Смирнов М. В., Краснов Ю. Н.— Ж- неорг. хим., 1958, 3, 1876.
168. Смирнов М. В., Ч у креев Н. Я-—Ж- физ. хим., 1958, 32, 2165.
169. Смирнов М. В., Ивановский Л. Е.— В кн.: Титан и его спла-
вы, 2. Изд-во АН СССР, М., 1959, 100.
170. Смирнов М. В., Ц и о в к и н а Л. А.— Изв. СО АН СССР, 1959,
10, 74.
171. С м и р н о в М. В., Пальгуев С. Ф.— Труды Ин-та электрохимии
УФАН СССР, 1960, 1, 29.
172. Смирнов М. В., Краснов Ю. Н.— Ж- неорг. хим., 1960, 5, 1241.
173. Смирнов М. В., Краснов Ю. Н.— Труды Ин-та электрохимии
УФАН СССР, 1960, 1, 35.
174. Смирнов М. В., Ц и о в к и н а Л А., Логинов Н. А.—
ДАН СССР, 1961, 136, 1388.
175. Смирнов М. В., Логинов И. А., Циовкина Л. А.— Тру-
ды Ин-та электрохимии УФАН СССР, 1961, 2, 29.
176. С м и р н о в М. В., Л о г и н о в Н. А., Циовкина Л. А.— Тру-
ды Всесоюзного совещания по физической химии расплавленных солей
и шлаков. Изд. ГНТИ, М., 1962, 337.
177. Смирнов М. В., Логинов Н. А.— Изв. СО АН СССР, 1962, 4, 64.
189
178. Смирнов М. В., Рассохин Б. Г., Логинов Н. А.— Тру-
ды Ин-та электрохимии УФАН СССР, 1966, 9, 49.
179. Торопова В. Ф., Заббарова Р. С.— Изв. высш. уч. зав. Хим.
и хим. технол., 1969, 12, 1487.
180. Томашов И. Д., Струков Н. М.— ДАН СССР, 1963, 152, 1177.
181. Томашов Н. Д., Модестова В. Н., П л а в и ч Л. А., Авер-
бух А. Б.— Коррозия металлов и сплавов, 2. «Металлургия», М., 1965,
80.
182. Танака К-, Моринага К-, Накано К-—J. Chem. Soc. Japan.
Pure Chem. Sec., 1968, 89, 1060.
183. Танака K-, Моринага К., Накано К-— J- Chem. Soc. Japan.
Pure Chem. Sec., 1969, 90, 478.
184. Французский пат. № 1093160 от 2.05.65.
185. Французский пат. № 1115435 от 24.04.56.
186. Французский пат. № 1064893 от 18.05.54.
187. Французский пат. № 1087968 от 2.03.55.
188. Французский пат. № 1093161 от 2.05.55.
189. Французский пат. № 1066602 от 8.06.54.
190. Французский пат. № 1051539 от 18.01.54.
191. Хромов А. Д., Лукашин В. И., Резниченко В. А.—
В кн.: Титан и его сплавы, 6. Изд-во АН СССР, М., 1961, 169.
192. Хромов А. Д., К а з а й н А. А.— В кн.: Титан и его сплавы, 4.
Изд-во АН СССР, М., 1960, 140.
193. Ш е н ь Ц и н ь-н ан, Олейник В. А., Д е л и м а р с к и й IO. К- —
Укр. хим. ж., 1962, 28, 599.
194. Швейцарский пат. № 335202 от 14.02.59.
195. Шведский пат. № 145872 от 22.06.54.
196. Шведский пат. № 148777 от 8.02.55; РЖМ, 1956, 6298.
197. Шведский пат. № 142488 от 13.10.53.
198. Японский пат. № 3859 от 29.09.52; С. А. 47, 8561.
199. Японский пат. № 5954; РЖМ, 1960, 20060.
200. Японский пат. № 854 от 11.02.54; РЖМ, 1957,2140.
201. Японский пат. № 7553 от 19.10.55; С. А., 52, 8803.
202. Японский пат. № 6851 от 27.09.55; РЖХим., 1958, 65091.
203. Японский пат. № 8005 от 15.09.56; РЖХим., 1959, 54079.
201. Японский пат. № 7809 от 8.09.56; РЖМ, 1959, 26540.
205. Японский пат. № 5854 от 18.07.56; РЖХим., 1959, 5386.
206. Японский пат. № 5702 от 14.07.56; РЖХим., 1959, 5385.
207. Японский пат. № 8257 от 24.09.56; РЖХим., 1959, 54083.
208. Японский пат. № 1302 от 25.02.56; С. А., 54, 6403.
209. Японский пат. № 2655 от 11.04.56; С. А., 54, 8556.
210. Японский пат. № 8903 от 19.10.57; РЖХим., 1960, 53338.
211. Японский пат. № 606 от 30.01.57; РЖХим., 1960, 23090.
212. Японский пат. № 5053 от 17.07.57; РЖМ,1960, 25950.
213. Японский пат. № 5405 от 24.07.57; РЖМ, 1960, 25951.
214. Японский пат. № 1406 от 27.02.57; РЖМ, 1960, 17368.
215. Японский пат. № 2357 от 18.04.57; РЖХим., 1960, 23089.
216. Японский пат. № 1504 от 7.03.57; РЖМ, 1960, 20057.
217. Японский пат. № 503 от 28.01.57; РЖМ, 1959, 26539.
218. Японский пат.№ 39554 от 20.06.57; РЖМ, 1960, 20060.
219. Японский пат. № 53 от 17.01.59; РЖМ, 1960, 20056.
220. Юшина Л. Д., Смирнов М. В.— ДАН СССР, 1957, 115, 949.
221. A t m a R., Kumar S., Sinha В.— Indian J. Chem., 1964, 2, 314.
222. Alpert M. B., Schultz F. J., Sullivan W. F.— J. Elect-
rochem. Soc., 1957, 104, 555.
223. В о c k r i s J. O’ M., H i 1 1 s G. I., N e n z i e s I. A., G о u n g L.—
Nature, 1956, 178, 654.
224. В e t t F. L., W о r n e r H. K.— Proc. Austral. Inst. Mining Metallurgy,
1956, 178,1; Экспресс-информация ВИНИТИ. Цветная металлургия, 1957, 16.
190
225. В e 11 F. L, Hickman B. S., Willis G. M., Worner H. K.—
В кн.: Извлечение и очистка редких металлов. Сборник докладов на симпози-
уме по металлургии редких металлов в Лондонском институте горного дела и
металлургии в 1956 г. Атомиздат, М., 1960, 466.
226. Brenner A., Senderoff S.— J. Electrochem. Soc., 1952, 99, 224.
227. С i h а 1 i k J., D о 1 e z a 1 J., Simon V., Z у k a J.— Chem. Listy,
1954, 48, 28.
228. Cordner G., Worner H. W.— Austr. J. Appl. Sci., 1951, 2, 358;
Титан. Сборник переводных статей. ИЛ, М., 1954, 101.
229. Calusaru A., Craiu V., Anghel Р. Din. comun. ICECHIM,
1957—1962, vol. 2, s. 1, Inst, cercetari chimice, s. a., 72—84; РЖХим., 1965,
17Б722.
230. Chem. Eng., 1952, 59, 103; 1953, 60, 103.
231. Chem. Eng. News, 1960, 38, 28.
232. Cheseldine D. M.— J. Electrochem. Soc., 1964, 111, 1005.
233. Diethejm B., Foerster F.— Z. phys. Chem., 1908, 62, 129.
234. Dean R. S.— Light Metal Age, 1957, 15, 20.
235. Dean R. S.— Metal. Ind., 1957, 90, 143, 193; Проблемы современной ме-
таллургии. Сборник переводов и обзоров иностранной периодической литера-
туры, 4. ИЛ, М., 1957, 41.
236. Dean R. S.— Metal Ind., 1957, 90, 165.
237. Dean R. S., Collet V/., Macawley F., Resnek L.,
Bronn R., Horstein J., Goodlok С. Электролитический титан.
ИЛ M. 1957.
238. Drossbach Р.— Z. Elektrochem., 1955, 59, 512.
239. Drossbach P., R os b er ger E.— Z. Elektrochem., 1956, 60, 470.
240. Drossbach P.— Z. Elektrochem., 1954, 58, 686.
241. Emmanuel-Zavizziano H., H a i s i n s k у M.— Compt. Rend.,
1936, 203,161.
242. Ervin G. J., Ueltz H. F., Washburn M. E.— J. Electrochem.
Soc., 1959, 106, 144.
243. Ehrlich P., Kuhn 1 H.— Z. anorg. Chem., 1959, 298, 176.
244. Ehrlich P., Gutsche V/., К u h n 1 H.— Z. anorg. Chem., 1961,
312, 70.
245. Fortin B. J., Wurm J. Y., Gravel L., Potvin R. J.— J.
Electrochem. Soc., 1959, 106, 428.
246. F 1 e n g a s S. N.— Ann. N. Y. Acad. Sci., 1960, 79, 853.
247. Forbes G. S., Hall L. P.— J. Am. Chem. Soc., 1924, 46, 385.
248. Fox M., Hanson R. H., Munro B. S.—Brit. J. Appl. Phys.,
1965, 16, 1213.
249. Gutmann V., К о g e 1 n i g AL, M i c h 1 m а у r M.— Monatsh.
Chem., 1968, 99, 707.
250. Gutmann V., Michlmayr M.— РЖХим., 1969, 6Б, 1485.
251. Gutmann V., N о w о t n у H.— Monatsh., 1958, 89, 331.
252. Gardner G., Worner H. V/.— Austral. J. Appl. Sci., 1951, 2, 358.
253. H i c k m о t t T. W.— J. Electrochem. Soc., 1966, 113, 1223.
254. H a b a s h у G. M.— J. Chem. U. A. R., 1961, 4, 169.
255. H a b a s h у G. M.— Coll. Czech. Chem. Communs, 1960, 25, 3166.
256. Habashy G. M.— Z. anorg. Chem., 1960, 306, 312.
257. Hickman B. S., W i 1 1 i s G. M.— Proc. Austral. Inst. Mining Meta-
lurgy, 1956, 178, 29.
258. Hunter M. A.—J. Wash. Acad. Sci., 1958, 48.
259. Head R. B.— Proc. Austral. Inst. Mining Metallurgy, 1956, 178, 21.
260. Head R.— J. Electrochem. Soc., 1961, 108, 806.
261. Huppertz W.— Electrochem. Metal. Ind., 1955, 3, 35.
262. Hatschek R. L.— Iron. Age, 1953, 172, 73.
263. Jaffe R. L.— Mod. Metals, 1958, 14, 46.
264. Kiihnl H., Ehrlich P., U i h 1 e i n R.— Z. anorg. Chem., 1960,
306, 246.
191
4
265. Kroll W. J.— Chem. Ind., 1960, 43, 1314.
266. Knecht E.— Ber., 1903, 36, 166.
267. Kumar S., Sinha В. C.— Centr. Glass and Ceram. Res. Ind. Bull.,
1958, 5, 18.
268. К о 1 о u s e к M.— Coll. Czech. Communs, 1939, 11, 592.
269. К а к e r D., Nettle J.— U. S. Bur. Mines, AIME, Annual Meeting, 1956,
270. К о I к A. 1., Sibert M. E., Steinberg M. A.— Electrochem.
Soc., spring Meeting, 1955.
271. Lingane J. J., Kennedy J. H.— Analyt. Chim. Acta, 1956, 15,
294.
272. Latimer W. M., Hildebrand J. H Referativ book of inorganic
Chemistry. N. Y., 1947, 315.
273. L i e q e о i s C.— J. Chim. phys. et phys.-chim. biol., 1969, 66, 1416.
274. Menzies I. A., Hills G. J., Young L., В о с к r i s J.— Trans.
Farad. Soc., 1959, 55, 1580.
275. Mellgren S., Opie W.— J. Metals, 1957, 9, 266.
276. Mergault P.— Compt. Rend., 1955, 241, 1755.
277. Mergault M. P.— Bull. Soc. franc, electiciens, 1956, 6, 689.
278. Nettle J., Hill T., Baker D.— U. S. Bur. Mines Rept. Invest.,
1958, 5410; C. A., 1958, 18018.
279. N e t t 1 e J. R., Baker D. H., Wartman F. S.— Metal. Ind.,
1957, 91, 47; C. A., 1957, 6398.
280. Nicolaus H. O.— Techn. Rundschau, 1960, 52, 13.
281. Ono K., Matsushima T., Hata K-, К u r i у a m a T.—Sci.
Rept. Res. Inst. Tohoku Univ., 1957, A9, 227; C. A., 1957, 17522.
282. Okada S., К a wane M., H ash ino T.— Bull. Eng. Res. Inst.
Kyoto Univ., 1956, 9, 27; РЖМ, 1957, 21200.
283. О 1 v e r J. W., Ross J. W.— J. Am. Chem. Soc., 1963, 85, 2565.
284. О s h i b a T.— J. Chem. Soc. Japan. Ind. Chem. Soc., 1959, 62, 985; РЖХим.,
1960, 46405.
285. Opie W. R., Svanstrom K. A.— Trans. Metallurg. Soc. AIME,
1959/1960, 215, 253.
286. Ogawa G.— J. Metal. Finish. Soc., 1958, 9, 323.
287. Okada S., Kawane M., H a s h i n о T.— Z. Elektrochem., 1958,
62, 437.
288. Okada S., Kawane M., H a s h i n о T.— J. Chem. Soc. Japan.
Ind. Chem. Sec., 1960, 63, 48.
289. Okada C., Kawane M., T akahashi M.— Bull. Eng. Res.
Inst. Kyoto Univ., 1954, 6, 57.
290. Okada S., Kawane M., T akahashi M., H a s h i n о T.— J.
Chem. Soc. Japan. Ind. Chem. Sec., 1935, 56, 410; РЖМ, 1956, 7379.
291. Okada S., Kawane M., H a s h i n о T.— РЖМ, 1957, 21202.
292. P e c s о к R. L.— J. Am. Chem. Soc., 1951, 73, 1304.
293. Pecsok R. L., Maverick E. F.— J. Am. Chem. Soc., 1954, 76, 358.
294. P r i b i 1 R., R о u b a 1 Z., S v a t e к E.— Chem. Listy, 1952, 46, 396.
295. Piggott A. R., Leckie H., Shreir L. L.— Corros. Sci., 1965,
5, 165.
296. Rale a R., Lucie B.— Ann. stint, univ. Jasi. Soc. I, 1965, Cll, 23.
297. Radovici О., C i о 1 a c S.— Rev. roumaine chim., 1969, 14, 297.
298. Sibert M. E., McKenna Q. H., Steinberg M. A., Wainer E.—
J. Electrochem. Soc., 1955, 102, 252.
299. Sibert M. E., Steinberg M. A.— J. Metals, 1956, 8, 1162.
300. Sibert M. E., Steinberg M. A.— В кн.: Проблемы современной
металлургии, 3. ИЛ, М., 1957, 64.
301. Strubl R. —Coll. Czech. Chem. Communs, 1938, 10, 475.
302. Sinha H. N., W о г n e г H. K-— Trans. Ind. Inst. Metals, 1955/56, 9, 123.
303. Sinha H. N.— Trans. Ind. Inst. Metals, 1958, 11, 81.
304. Sinha H. N.— Trans. Ind. Inst. Metals, [959, 12, 165.
192
305. Schwabe К.— Freiberger Forschungsh., 1956, В., 17, 5; РЖМ, 1957, 11916.
306. Steinberg M. A., Carlton S. S., S i b e r t M. E., WainerE.—
J. Electrochem. Soc., 1955, 102, 332.
307. Tribalot S., Delafosse D.— Anal. Chim. Acta, 1958, 19, 74.
308. Tanaka R., Tamamushi R. Sbornik I mezinarodniho polarogra-
fickeho sjezdu w Praze, I. Praha, 1951, 486.
309. T a m a mush i R.— Bull. Chem. Soc. Japan, 1950, 23, 250.
310. T a d e n u m a H., Ikeda H., Fujita K.— J. Chem. Soc. Japan.
Ind. Chem. Sec., 1956, 59, 356.
311. Vandenbosh V.— Bull. Soc. Chim. Belg., 1949, 58, 532.
312. V 1 с e к A. A.— Chem. Listy, 1956, 50, 400.
313. Wurm J. G., Gravel L., Potvin R.— J. Electrochem. Soc.,
1957, 104, 301.
314. Worn er H. K-, Willis G. M., Worn er H. W.— J. Electroctem.
Soc., 1953, 100, 8.
315. W i 1 к i n s о n G., Pauson P. L., Birmingham J. M., С о t-
t о n F. A.— J. Am. Chem. Soc., 1953, 75, 1011.
316. Yoshizawa S., Hashino T.— Techn. Rept. Res. Inst. Kyoto Univ.,
1959, 9, 45.
13 1-1669
ГЛАВА IV
\ • ' ... А ' •
КРАТКИЕ СВЕДЕНИЯ О МЕТОДАХ
ПРОИЗВОДСТВА ТИТАНА И ЕГО СОЕДИНЕНИЙ
В настоящей главе описаны методы обогащения титановых руд,
производства титановых шлаков, четыреххлористого титана, дву-
окиси титана, металлического титана и ферротитана. Остальные
виды титановой продукции производятся еще в небольших количе-
ствах. Для их получения используются препаративные методы, опи-
санные в предыдущих главах монографии. Рассматриваемые в этой
главе вопросы освещены в книгах [24, 38, 47, 48, 237] и в несколь-
ких тематических сборниках статей.
ОБОГАЩЕНИЕ ТИТАНОВЫХ РУД
Титановые руды обычно характеризуются низким содержанием
полезных компонентов. С целью получения высококачественных
минеральных концентратов их обогащают. В крупных промышлен-
ных масштабах в ряде стран налажено обогащение только титано-
вых руд, содержащих ильменит и рутил, а обогащение сфеновых
и перовскитовых руд проводилось пока в опытных масштабах.
Промышленная технология производства сфеновых и перов-
скитовых концентратов еще не разработана. Титановые концентра-
ты: ильменитовый (аризонитовый) и рутиловый производятся из
песков морского происхождения и коренных пород. Черные мор-
ские пески, содержащие ильменит, образуются в результате раз-
рушения коренных пород и отлагаются в прибрежной полосе
современных или древних морей. Кроме ильменита эти пески со-
держат нередко рутил, циркон, монацит, касситерит, силикатные
минералы и кварц.
Обогащение морских песков и получение из них ильменита, ру-
тила и концентратов сопутствующих минералов производится с
применением гравитационного, электромагнитного и электроста-
тического методов. Обычно эти методы комбинируются. На совре-
менной обогатительной фабрике [123, 145] обогащаются морские
пески древнего происхождения, содержащие аризонит, рутил и
циркон. По принятой на фабрике схеме темноцветные минералы
отделяются от кварцевого песка на спиральных сепараторах, аризо-
нит и рутил от циркона — на электростатических сепараторах,
циркон от сопутствующих минералов — на спиральных сепарато-
194
pax. Отделение аризонита, имеющего более высокую магнитную
восприимчивость, от немагнитного рутила производят на магнитных
сепараторах с сильным магнитным полем.
Пески, состоящие из ильменита, вначале также подвергаются
обработке на спиральных сепараторах для отделения от кварца.
Темноцветные минералы поступают после сушки на магнитную
Таблица 12
Химический состав ильменитовых концентратов
Компонен- ты Иршинское место- рождение Кусинское месторож- дение Самотканское место- рождение Централь- ное ме- сторожде- ние Волчан- ское ме- сторожде- ние
1 2 1 2
ТЮ2 51,00 54,50 41,60 61,20 63,10 58,00 62,00
FeO 34,30 17,71 34,50 — 4,75 Не опре- деляли
Fe2O3 9,40 23,30 12,10 27,50 26,50 28,18 28,88
SiO2 1,75 1,12 3,31 1,94 2,70 3,05 0,63
А12О3 0,50 1,02 3,56 2,60 3,49 1,15 1,29
Сг2О3 — — — 3,20 1,46 0,28 0,72
МпО 0,81 0,61 0,80 1,20 0,96 — —
СаО 0,47 Следы Следы Следы 0,33 0,52 0,35
MgO 1,20 0,73 3,20 0,80 1,55 0,19 0,44
SO2 0,22 — —• — — —
V2Os 0,17 0,10 0,20 — 0,12 0,19 0,36
Р2О5 0,02 1 1 — 0,32 0,064
s Потери при прокал- — — — — 0,70 0,007 0,016 — -
ке + Н2О — — — — — — ——•
Сумма 99,82 99,11 99,97 98,44 100,22 96,646 94,737
Литерату- ра [17] [182] [184] [182] [118] [35] [35]
сепарацию, в результате которой получается ильменитовый концен-
трат. Остальные минералы после отделения ильменита разделяются
методами гравитации и электростатической сепарации.
Разработана также и комбинированная схема обогащения мор-
ских песков, по которой темноцветные минералы получают в виде
коллективного концентрата методом флотации [781. Коллективный
концентрат разделяется на компоненты электросепарацией.
Породы коренных титановых месторождений кроме ильменита
содержат обычно титаномагнетит, магнетит и силикатные минералы
(полевые шпаты, роговую обманку, пироксены, гранат, биотит).
Руда измельчается на шаровых или стержневых мельницах до
20 меш. На магнитных сепараторах со слабым магнитным полем
отделяются сильномагнитные минералы (титаномагнетит и магне-
тит). Немагнитный остаток подвергается обогащению на гравита-
ционных столах, в результате которого получается ильменит, а
остальные минералы уходят в хвосты. С целью повышения качества
13*
195
ильменитового концентрата продукт после гравитационного обога-
щения переочищается на магнитных сепараторах с сильным магнит-
ным полем.
Содержание двуокиси тита-
на в ильменитовом концент-
рате зависит от минералоги-
ческого состава руд, из кото-
рого он получен. Пески Само-
тканского ильменитового мес-
торождения содержат хром-
шпинелиды [146]. Описанные
схемы обогащения песков не
обеспечивают эффективную
очистку ильменита от хром-
шпинелидов и получение кон-
центратов с малой примесью
хрома.
Ильменит из россыпных
месторождений Флориды, Вир-
Траванкора (Индия) и Житомирской области (УССР)
Таблица 13
Состав рутилового концентрата из песков
Самотканского месторождения [145]
Содержание, вес.%
Компонент
Образец 1 Образец 2
96,55
92,17
0,34
0,96
0,64
0,10
0,02
0,01
0,43
2,68
0,31
0,11
0,11
TiO2
ZrO2
SiO2
AI2O3
Сг2О3
P2O5
Nb2O5
гинии (США),
содержит около 50% TiO2, уральский ильменит — около 42% TiO2,
ильменит из прибрежных песков Австралии, Индии (близ Квилона)
и Днепропетровской области — около 60% TiO2 (табл. 12). Состав
рутилового концентрата представлен в табл. 13.
ПРОИЗВОДСТВО ТИТАНОВЫХ ШЛАКОВ
Кроме ильменита и рутила для производства двуокиси титана
и четыреххлористого титана в больших масштабах используются
титановые шлаки, получаемые из ильменитовых концентратов или
необогащенных железо-титановых руд. Промышленное производ-
ство титановых шлаков впервые было создано в Канаде (г. Сорель)
фирмой Quebec Iron and Titanium Corporation из руды Квебекского
месторождения [287, 307]. Запасы руды в этом месторождении со-
ставляют 136 млн. т с содержанием 35% TiO2 и 40% Fe [220].
В 1963 г. [221] эта фирма обогащенную ильменитовую руду до содер-
жания 37% TiO2 и 42% Fe плавила на 8 электропечах мощностью
по 16 Мгв. В результате получается низкофосфористый чугун и ти-
тановый шлак с содержанием 70,5% TiO2 и 14% FeO, который ис-
пользовали для производства двуокиси титана. Производство ти-
танового шлака.этой фирмой в 1963 г. составило 464 тыс. т [166].
В 1965 г. было проплавлено 1,19 млн. т руды и получено 494 тыс. т
титанового шлака и 337 тыс. т чугуна [220].
Титановые шлаки в зависимости от исходного сырья могут со-
держать 20—40, около 70 и 85—87% TiO2. Низкопроцентные тита-
новые шлаки получаются при восстановительной плавке титаномаг-
196
нетитовых концентратов [14]. Практи-
ческого применения они еще не нашли. —
Шлаки с содержанием 70% ТЮ2
типа канадских разлагаются серной =*
кислотой, подобно ильмениту, и при- *
меняются для производства двуокиси о
титана сернокислотным методом. Вы- а
сокотитановые шлаки, содержащие |
более 80% ТЮ2, применяются для про- g.
изводства четыреххлористого титана g
вместо природного рутила [182].
Титановые шлаки получаются при g
восстановлении ильменита твердым |
углеродом в электрических печах. 1
И. П. Бардин и В. А. Резниченко
[15] провели термодинамический ана- Л
лиз восстановления ильменита твер- |
дым углеродом по реакциям: £
FeO • TiO2 + С = Fe + TiO2 + СО, g
3/4FeO • TiO2 + C = 3/4Fe + |
ч-Чад + со, |
% FeO • TiO2 + C = 2/3 Fe + |
+ % Ti2O3 + CO, |
V2 FeO • TiO2 + C = v2 Fe + g
+ % TiO + CO. I
Изменения свободной энергии, вы-
численное для этих реакций, приведе- *
ны в табл. 14. Восстановление FeO • £
. ТЮ2 (ильменита) твердым углеро- &
дом до температуры 603° С протека- m
ет с образованием металлического же- о
леза и двуокиси титана. Выше этой §
температуры наряду с железом обра- о
зуется окисел Ti3O5, а выше 1242° С— и
Ti2O3. Реакция восстановления иль- |
менита в электропечи идет за счет g
прямого взаимодействия с твердым |
углеродом и при действии окиси yr- s
лерода. Косвенное восстановление
окисью углерода протекает только
до момента расплавления шихты. За
его счет отнимается 20—30% кисло-
рода, связанногос окислами железа в
шихте [1711. После расплавления
197
происходит только прямое восстановление окислов железа твердым
углеродом.
Окислы титана, получающиеся при восстановлении ильменита,
способны образовывать с окислами FeO, MgO, МпО и СаО химические
соединения и твердые растворы. Вследствие этого восстановление
ильменитовых и других титансодержащих материалов углеродом —
сложный процесс, приводящий к образованию соединений, которые
определяют минералогический (фазовый) состав титановых шлаков.
Основными минералами титановых шлаков являются аносовит
(твердый раствор на основе Ti3O5), тагировит (твердые растворы на
основе Ti2O3), твердые растворы на основе ортотитаната магния,
перовскит, сфен, ильменит, титанавгит и форстерит.
Аносовит был открыт в титановых шлаках К. X. Тагировым
[76, 178, 204]. Д. С. Белянкин и В. В. Лапин [16] считают, что ано-
совит твердый раствор (Mg, Fe, Mn)O • 2TiO2, так как межплоскост-
ные расстояния изученного ими образца были близки к таковым
оротитаната магния MgO • 2TiO2. Г. С. Жданов и А. А. Русаков
[76, 177] на основании рентгеноструктурного исследования при-
шли к заключению, что аносовит является низшим окислом тита-
на состава Ti3O5. К. X. Тагиров и А. В. Руднева с соавторами
[178, 180, 204] уточнили состав аносовита и выразили его формулой
п [(Al, Fe, Ti)2O3 • TiO2] • т [(Mg, Fe, Ti)O • 2TiO2].
Аносовит образует удлиненные призматические кристаллы, имею-
щие в поперечном сечении форму правильных ромбов [13]. В про-
ходящем свете непрозрачен и окрашен в черный цвет. В отражен-
ном свете оптические свойства кристаллов зависят от степени вос-
становления шлака.
По данным Г. С. Жданова и А. А. Русакова [76, 177], исследо-
вавших монокристалл аносовита с содержанием 88,3% ТЮ2, кото-
рый получил К. X. Тагиров из шлака при температуре 1400—
1500° С, он имеет ромбическую решетку с параметрами а = 3,754,
b — 9,474, с = 9,734 A, z = 4, ррентг = 4,29 г/см3. Авторы считают,
что исследованный ими образец является высокотемпературной мо-
дификацией аносовита. Другие авторы [85, 209] для исследованных
ими образцов аносовита получили рентгенограммы, не совпадаю-
щие с данными Г. С. Жданова и А. А. Русакова. Е. Ф. Белецкий
[182] полагает, что это несовпадение может быть объяснено суще-
ствованием двух полиморфных модификаций Ti3O5.
Аносовит — самый распространенный минерал титановых шла-
ков. Состав его изменяется в широких пределах. По данным
А. В. Рудневой [182], в титановых шлаках встречается три разно-
видности аносовита: магний-алюминий-аносовит, титан-(111)-ано-
совит и железистый аносовит (встречается в шлаках с высоким со-
держанием окиси железа). Присутствие аносовита в титановых шла-
ках определяет температуру их плавления. Температура плавления
аносовита зависит от отношения в нем Ti2O3 : TiO2 и изменяется,
по данным К. X. Тагирова, от 1670 до 1720° С.
198
Аносовит также определяет вскрываемость титановых шлаков
серной кислотой. Влияние химического состава на вскрываемость
аносовита серной кислотой детально еще не изучено. Хорошо раз-
лагаются серной кислотой титановые шлаки, содержащие около
70% TiO2 (общая) и 10—12% FeO.
А. В. Руднева [178] разработала метод выделения аносовита
из титанового шлака в чистом виде. Шлак, кроме аносовита, содер-
жит корольки железа и стекло. Для удаления металла измельчен-
ный шлак кипятят 1,5—2,0 ч с 10%-ной соляной кислотой. Сили-
катное стекло растворяется при кипячении с 30%-ным КОН.
Рис. 13. Зависимость минералогического состава шлаков от содержания
в них TiO2 и условий плавки [182]:
/ — аносовитовые шлаки (плавка брикетов с недостатком восстановителя — неф-
тяного кокса); II — шлаки, содержащие Ti2Os (плавка брикетов с избытком неф-
тяного кокса); III— шлаки, содержащие твердые растворы на основе Ti2O3 (плав-
ка агломерата с нефтяным коксом); IV — магнезиально-титановые шлаки (плавка
брикетов с антрацитом). 7\ — Ti2O3; Tz — твердый раствор на основе Ti2O3,
OP — ортотитанат магния.
Существование твердых растворов на основе Ti2O3 в титановых
шлаках впервые установила А. В. Руднева с соавторами [178, 179,
204]. По данным кристаллических и рентгеноструктурных исследо-
ваний, полуторная окись Ti2O3 образует неограниченный ряд твер-
дых растворов с гейкелитом MgTiO3, а возможно, и с другими
минералами типа ильменита.
Твердый раствор на основе Ti2O3 — тагировит [176] встречается
в заводских шлаках при содержании выше 68% TiO2. Количествен-
ное содержание его зависит от условий плавки. Чистая полуторная
окись встречается только в высокопроцентных шлаках, содержащих
79—84% TiO2. Твердые растворы на основе Ti2O3 при охлаждении
окисляются кислородом воздуха, в результате чего образуется ру-
тил, а шлак рассыпается на мелкий порошок [15].
199
А. В. Руднева с соавторами [178, 179] в силикатных титановых
шлаках обнаружила изотропные кристаллы кубической формы,
являющиеся твердым раствором на основе ортотитаната магния и
ортотитаната железа. Состав их выражается формулой 2(Mg, Fe)O •
• TiO2. Минералы перовскит CaTiO3 и титанит (сфен) CaTiSiO5
встречаются в шлаках, содержащих кальций и кремний. В шлаках
с повышенным содержанием алюминия присутствует титанавгит
т (СаО • MgO • 2SiO2) • п [СаО (А1, “
Табл ица 15
Фазовый состав литого и гранулированного
титанового шлака [201]
Наименование фазы Литой шлак, % Гранули- рованный шлак, %
Аносовит 40—45 25—30
Перовскит 20—25 5—10
Уфанат 5—10
Коричневый минерал 1—5 3—5
Титанавгит 2—5 2—3
Сфен 1—2 0—1
Стекло 5—10 45—30
Ti)2O3 • SiO2]. Шлаки с повы-
шенным содержанием FeO
(более 8—10%) содержат иль-
менит [178].
Кроме перечисленных ми-
нералов в отдельных работах
указывалось на образование
в титановых шлаках минера-
лов байковита (предполагае-
мый состав MgO • Ti2O3) [178],
имманита [14], уфанита [201 ],
минерала «х» [182] и др. Одна-
ко существование их нуждает-
ся еще в подтверждении.
Минералогический состав
титановых шлаков зависит от
химического состава, степени
восстановления двуокиси тита-
на, условий плавкий режима охлаждения (рис. 13). Постоянной со-
ставной частью титановых шлаков является только аносовит. Осталь-
ные минералы присутствуют в шлаке при определенном химиче-
ском составе и определенных условиях их плавки.
При резком охлаждении титановых шлаков (гранулированием
при выливании расплавленного шлака в холодную воду) получается
значительное количество стекловидной фазы (табл. 15) и уменьшает-
ся образование перовскита, плохо разлагаемого серной кислотой.
Грануляция улучшает качество титановых шлаков, предназна-
ченных для производства двуокиси титана сернокислотным ме-
тодом.
Химический состав титановых шлаков зависит в основном от
состава сырья и метода плавки (табл. 16). В типичных промышленных
шлаках, получаемых из ильменита Самотканского месторождения,
содержится 86—88% TiO2 и 5,9—6,0% FeO [117].
Плавки титановых шлаков можно проводить с флюсами (сода,
мел, поваренная соль, известь) для понижения температуры плав-
ления и вязкости и без них. В качестве восстановителя используют-
ся малозольный антрацит и нефтяной кокс. Шихта может плавиться
без специальной подготовки и после брикетирования или окускова-
ния методом агломерации. Связующими для изготовления брикетов
служат каменноугольный пек и сульфитно-целлюлозная барда.
200
Плавка окускованной шихты позволяет снизить расход электро-
энергии, так как проводится с закрытым колошником.
Температура плавления шлака, полученного из окускованной
шихты, изменяется в пределах от 1425 до 1630° С [182]. При пере-
восстановлении шлака температура плавления его повышается.
Таблица 16
Химический состав (вес.%) титановых шлаков
Компонент Опытные шлаки из ильменита Шлак из кусинско- го ильме- нита Шлак из 1 самотканского ильменита Шлак из титаномаг- нетита
2 3
TiO2 67,0 45,96 87,5 87,50 90,10 (валовый) 37 *
Ti2O3 — 24,73 — — (28,40)
SiO2 4,5 5,94 2,8 2,79 2,35 12
А12О3 4,8 7,13 5,1 5,11 3,00 19
Fe2O3 — 6,58 — — 2,70 (валовый) —
FeO 6,2 — 3,2 3,18 — 16
MnO —- 1,65 0,3 1,15 1,10 —
CaO 10,0 0,32 — 0,29 0,67 3
MgO 7,2 7,41 1,0 2,16 2,60 11
S 1,2 — — 0,008 —
Fe 1,48 — —-- 0,81 —
Cr2O3 — — 1,7 1,68 1,45 —
V2O5 — — — 0,16 — —
Сумма 100,9 101,20 100,9 104,03 — 98
Литература [337] [178] [Н8] [34] [178]
Таблица 17
Расход электроэнергии при плавке титановых шлаков [182]
Шихта Ильменит с содержанием 40—42% ТЮ2 Ильменит с содержанием 60—63% TiO2
кеч на 1 т шлака % тю, кеч на 1 т шлака % TiO2
Порошковая 5275 84,0 2450 84,5
Брикеты 3040 78,1 1930 —
Агломерат 4520 77,3 i —- —
Шлаки из неокускованной шихты имеют более высокую температу-
ру плавления — около 1730—1750° С [182]. Расход электроэнер-
гии на плавку шлака зависит от содержания двуокиси титана в
исходном концентрате и в шлаке, а также от способа его плавки
(табл. 17).
При плавке титановых шлаков одновременно получается чу-
гун, состав которого зависит от исходного сырья. Так, в чугуне,
201
полученном при плавке хромосодержащего ильменита, повышено
содержание хрома (табл. 18).
Распределение хрома между шлаком и чугуном зависит от со-
отношения объемов чугуна и шлака (кратности шлака), температу-
ры плавки и расхода углерода. Переход хрома в чугун обычно со-
ставляет 30—50%. При плавке хромсодержащего ильменита Само-
тканского месторождения получается чугун с содержанием 1,0—
1,2% Сг и шлак с содержанием 1,2—2,5% Сг2О3 [182]. Увеличение
Таблица 18
Содержание примесей в чугуне, полученном при электроплавке титановых
шлаков [118, 182]
Исходное сырье С Ti Si Cr Мп S Р V
Хромсодержащий иль- менит (порошковая 1,22 0,070 0,32 1,15 0,050 0,16 0,11
шихта) 1,10 0,111 0,30 1,06 0,087 0,20 0,20 0,0012
Кусинский ильменит (порошковая шихта) 3,30 0,710 0,41 " 11 0,290 0,17 0,1200
расхода восстановителя позволяет снизить примесь хрома в титано-
вом шлаке, но получить безхромистые титановые шлаки из хром-
содержащего ильменита, по-видимому, будет трудно [183].
С. И. Денисов с соавторами [73] снял баланс выплавки титаново-
го шлака из ильменита состава 45,6% TiO2, 34,8% FeO, 19,4% Fe2O3,
Таблица 19
Баланс двуокиси титана и железа при электроплавке
титанового шлака
Продукт плавки TiO2, % Fe, %
Титановый шлак 91,7 4,86
Чугун Пылевидные продукты в отходя- 4,1 91,11
щих газах 2,2 1,71
Неучтенные потери 2,0 1 2,32
9,2% А12О3 на электропечи 10,5Мгв (табл. 19). По их данным, извле-
чение титана в шлак при электроплавке составляет 91,7%. Общее
извлечение титана в молотый шлак, подготовленный для приготов-
ления брикетов, равно 83,8%.
Хранение титанового шлака на воздухе сопровождается окисле-
нием низших окислов титана до двуокиси. Аносовит в результате
окисления при температуре ниже 750° С превращается в анатаз,
202
а при более высокой температуре — в рутил [32, 33]. Хлорирование
титанового шлака газообразным хлором при 800° С приводит к
удалению из него в виде хлоридов около 50% примесей железа, маг-
ния, алюминия и кремния.
Производство титанового шлака электровосстановлением руд и
концентратов связано с большой затратой электроэнергии. С целью
уменьшения расхода электроэнергии выплавку титанового шлака
в электропечах рекомендуется сочетать с предварительным восста-
новлением железа в руде при нагревании с углем или с природным
газом в трубчатых печах или печах кипящего слоя. Метод двухсту-
пенчатого восстановления не нашел пока промышленного использо-
вания, очевидно, из-за сложности аппаратурного оформления, вы-
званной сочетанием двух существенно различных процессов. Расход
электроэнергии при выплавке титанового шлака может быть сни-
жен примерно на 8%, если заменить электропечи с открытым колош-
ником на агрегаты закрытого типа [71]. Однако снижение энерго-
затрат за счет совершенствования электроплавки титановых руд и
концентратов, по-видимому, неосуществимо.
Титановые шлаки нашли широкое применение в производстве
четыреххлористого титана и двуокиси титана сернокислотным ме-
тодом. Как сырье для производства четыреххлористого титана по
ряду существенных показателей титановые шлаки уступают при-
родному рутилу, получаемому при обогащении руд в виде высоко-
процентных концентратов. Более эффективным сырьем для произ-
водства четыреххлористого титана является техническая двуокись
титана типа искусственного рутила. Производство ее может быть
налажено в крупных масштабах из различных видов титанового
сырья сернокислотным, солянокислотным и другими методами [47].
ВОССТАНОВИТЕЛЬНЫЙ ОБЖИГ ИЛЬМЕНИТОВЫХ
КОНЦЕНТРАТОВ
Ильменитовые концентраты, получаемые из руд многих место-
рождений, содержат минерал, подвергшийся изменению в резуль-
тате окислительных процессов, происходящих при разрушении ко-
ренных пород. В измененном ильмените значительная часть железа
находится в трехвалентном состоянии. Иногда при минералообра-
зовании все связанное с ильменитом железо полностью окисляется
до трехвалентного и ильменит превращается в новую минеральную
разновидность типа аризонита. Концентраты с минералом, подверг-
шимся изменению, содержат некоторую часть титана в виде минера-
лов лейкоксена и рутила. Они реагируют с серной кислотой менее
энергично, чем неизмененный ильменит, и малопригодны для про-
изводства двуокиси титана по сернокислотному методу. С целью
повышения реакционной способности измененных ильменитовых
концентратов при разложении серной кислотой предлагается под-
203
вер гать их восстановительному обжигу для переведения окисного
железа в закисное, реагирующее с образованием титаната двухвалент-
ного железа FeTiO3 [93, 94, ПО, 111, 2231. Измененный ильменит
житомирских месторождений при восстановительном обжиге в те-
чение 1 ч полностью превращается в продукт, реагирующий с сер-
ной кислотой так же энергично, как и неизмененный минерал
[НО]. Восстановительный обжиг его рекомендуется проводить в
атмосфере водорода и окиси углерода при 450—550° С, природного
газа при 650—750е С, а также при действии твердого углерода при
900—1000° С.
Энергичное восстановление ильменита протекает под действием
газообразных восстановителей в расплавах хлоридов щелочных ме-
таллов. Скорость восстановления в расплаве солей бывает в не-
сколько раз больше, чем при взаимодействии газов с порошкообраз-
ным продуктом [72].
ПРОИЗВОДСТВО ЧЕТЫРЕХХЛОРИСТОГО ТИТАНА
Четыреххлористый титан — важный полупродукт титановой
промышленности. Он используется для производства металличе-
ского титана, двуокиси титана и титанорганических соединений.
Особенно крупных масштабов использование четыреххлористого
титана приобрело в металлургии титана. Оба промышленных мето-
да производства металлического титана (магний- и натрийтермиче-
ский) базируются на восстановлении четыреххлористого титана.
В последние годы усиленно разрабатывается и внедряется в про-
мышленное производство получение двуокиси титана окислением
четырех хлор исто го титана кислородом. Большое количество четы-
реххлористого титана, по-видимому, в скором времени станет по-
треблять промышленность органического синтеза. Четыреххлори-
стый титан является исходным продуктом для получения различ-
ных титанорганических соединений и полимеров, обладающих
ценными техническими свойствами.
Все известные в настоящее время методы получения четырех-
хлористого титана сводятся к хлорированию титансодержащих ма-
териалов каким-либо хлорирующим агентом с последующей очист-
кой технического продукта от содержащихся в нем примесей. Ис-
ходными материалами для получения четыреххлористого титана
могут служить двуокись титана [138, 232—234, 320, 3561, рутил
[300, 301 ], титановый шлак [14, 202, 269], карбид титана [202, 270],
ильменит [23, 289], ферротитан [3471, сфен [40, 139, 140] и др. Со-
временные методы промышленного производства четыреххлористого
титана основаны на хлорировании рутила и высокотитанового шла-
ка с содержанием 85—87% ТЮ2. Наиболее высокие технико-эко-
номические показатели получаются при хлорировании рутила. Ти-
тановые шлаки используются только при недостатке рутила.
204
В качестве хлорирующих агентов используются газообразный
хлор [23, 40, 138—140 , 202 , 232, 270, 289, 301, 320 , 347], четырех-
хлористый углерод [356] и хлороформ [300]. В современном про-
мышленном производстве хлорирующим агентом служит отходя-
щий хлор-газ от электролиза металлического магния или сжижен-
ный хлор.
Восстановителем при хлорировании титансодержащих материа-
лов в препаративных опытах используется древесный и сахарный
уголь. В промышленном производстве восстановителем служит неф-
тяной кокс.
Технический четырех хлор истый титан бывает загрязнен хлори-
дами железа, алюминия, фосфора, серы, ванадия, кремния, а также
фосгеном, хлоропроизводными углеводородов, газообразным хло-
ром и оксихлоридами титана. Для очистки четереххлористого тита-
на от примесей применяется многократная перегонка над металли-
ческой ртутью или калием [260, 326], над амальгамой натрия [300],
а также обработка металлическим натрием [316, 349] или амальга-
мой натрия [232] с последующей перегонкой. Высокая степень
очистки четыреххлористого титана от оксихлорида титана и приме-
сей хлоридов фосфора, серы и кремния возможна только в резуль-
тате многократной дистилляции или ректификационной перегонки.
Технология промышленного производства четыреххлористого ти-
тана в литературе освещена недостаточно. Имеется термодинами-
ческий расчет процесса хлорирования двуокиси титана с газообраз-
ным хлором в присутствии углерода [62] и окиси углерода [139].
Произведен термодинамический анализ процесса хлорирования
ильменита [315] и описано хлорирование титанового шлака в шахт-
ных электропечах [185].
И. А. Глухов с соавторами [52] провел термодинамический ана-
лиз реакций хлорирования соединений титана в атмосфере хлора
и хлоридов серы и установил, что взаимодействие двуокиси титана
и рутила с хлоридами серы протекает при более низкой температуре
и требует меньших затрат энергии, чем хлорирование газообразным
хлором в присутствии нефтяного кокса [51]. Изменение степени
хлорирования двуокиси титана (х) от времени (т) при температуре
240—300° С описывается уравнением
х = Кт.
Зависимость константы скорости реакции К от температуры вы-
ражается уравнениями
2,3 1g К = ----20,47 (хлорирование ТЮ2),
2,31 1g К = 1(^20--22,19 (хлорирование рутила).
Кажущаяся энергия активации равна 16,6 ккал!моль при хлориро-
вании двуокиси титана и 20,31 ккал!моль при хлорировании рутила.
205
Вопросу кинетики хлорирования двуокиси титана смесью газообраз-
ного хлора и хлоридов серы посвящена работа [217].
Хлорирование титансодержащих материалов протекает очень
интенсивно при пропускании газообразного хлора через расплав
хлоридов калия, натрия, их смеси и карналлита, в котором эти мате-
риалы суспензированы совместно с измельченным нефтяным кок-
сом. Добавка хлорного железа к расплаву хлоридов калия и натрия
увеличивает скорость хлорирования двуокиси титана в несколько
раз [30, 218]. Механизм хлорирования в расплаве солей изучен еще
недостаточно. Согласно [31], при хлорировании в расплаве солей
происходит образование промежуточного растворимого оксихло-
ридного соединения. Взаимодействуя с окисью углерода и раство-
ренным в расплаве хлором, оно образует TiCl4 и СО2. В присут-
ствии хлорида железа двуокись титана в расплаве солей хлорирует-
ся не элементарным хлором, а хлорным железом.
С. П. Зезянов и В. И. Ильичев [79] изучили кинетику хлориро-
вания двуокиси титана в расплаве хлоридов калия и натрия газо-
образным хлором в присутствии окиси углерода. Оказалось, что
скорость реакции пропорциональна концентрации окиси углерода
в газовой фазе. Скорость хлорирования максимальная при отноше-
нии С12 : СОобъемн = 1:3. Кинетика хлорирования двуокиси тита-
на смесью С12 + СО и фосгеном проходит через максимум при 560
и 460° С соответственно [90].
Основными продуктами реакции хлорирования двуокиси титана
четыреххлористым углеродом являются TiCl4, СОС12 и СО2 [120].
Образования оксихлоридов при хлорировании двуокиси титана че-
тыреххлористым углеродом в заметных количествах не наблюдается.
Окислы хрома при хлорировании титановых шлаков превраща-
ются в трихлорид СгС13 и неустойчивый тетрахлорид хрома СгС14
[54]. Тетрахлорид хрома в растворе четыреххлористого титана в
течение 4—5 суток распадается на элементарный хлор и трихлори-
стый хром, выделяющийся в виде мелкой суспензии. При хлориро-
вании хромсодержащих высокотитановых шлаков в пылевых ка-
мерах и в оросительных конденсаторах из газовой фазы осаждается
до 90% хрома.
В. А. Безворитный и А. Б. Безукладников [11 ] изучили хлориро-
вание ярегского флотационного концентрата, состоящего из 49,5%
ТЮ2, 36,12% SiO2, 5,7% Fe2O3, 3,6% А12О3, 1,1% СаО и0,71% MgO.
Авторы пытались полностью прохлорировать двуокись титана и
двуокись кремния в расплаве карналлита при добавке в качестве
катализатора буры. Однако хлорирование титана и кремния в
расплаве солей протекает селективно. При температуре 800° С в ре-
зультате хлорирования получается смесь тетрахлоридов, в которой
содержится только 10% SiCl4. Двуокись кремния в расплаве кар-
наллита взаимодействует с газообразным хлором значительно медА
леннее, чем двуокись титана. Она остается в расплаве преимуще-
ственно в виде непрохлорированного остатка.
206
Хлорирование титанового шлака в расплавленном карналлите,
а также схема укрупненной установки для хлорирования титансо-
держащих материалов описаны в работах [29, 107, 108].
Ванадий при хлорировании титансодержащих материалов пе-
реходит в газовую фазу в виде VOC13, VC14, VC13 и VC12 [55].
О форме нахождения ванадия в жидком четыреххлористом титане
достоверных данных еще не имеется. По-видимому, в тетрахлориде-
сырце он содержится в виде оксихлорида VOC13, образующего с
TiCl4 смеси с неограниченной растворимостью в жидком состоянии
[122].
Тетрахлорид титана от ванадия очищают с помощью медного
порошка [147]. Каталитически активную медь готовят обработкой
ее летучими спиртами, альдегидами и кетонами. В английском патен-
те [4] предлагается очищать четыреххлористый титан от примеси
ванадия, хрома и железа с помощью сероводорода. Пары TiCl4 в
смеси с H2S пропускаются через псевдоожиженный слой твердых
частиц при температуре 140—300° С и атмосферном давлении.
' Псевдоожиженный слой получается из кварцевого песка. Недавно
предложен новый метод очистки четерыххлористого титана от ва-
надия восстановлением его в газовой фазе водородом и природным
газом [103].
Г. В. Серяков с соавторами [189, 190] разработал метод очистки
четыреххлористого титана от примесей FeCl3 и А1С13 пропусканием
газообразных продуктов через колонный аппарат, заполненный кус-
ками поваренной соли. И. С. Морозов [112] предложил для очистки
четыреххлористого титана от железа, алюминия и ванадия пропу-
скать пары его через расплав хлористого калия. Ванадий в этом
расплаве, судя по ИК-спектрам поглощения, связывается хлори-
стым калием в виде двойной соли K2VOC14. Применение солевого
расплава позволяет получать весьма чистый продукт с содержанием
0,001—0,003% V и менее 0,001% А1 и Fe.
Примеси органических веществ в тетрахлориде титана разру-
шаются при действии брома и хлора [252].
Описание производства четыреххлористого титана хлорирова-
нием рутила на заводе французской фирмы в г. Танне (близ города
Лиолуза в Эльзасе) приведено в статье [77], и описание техноло-
гии производства и очистки четыреххлористого титана — в статье
[126].
Титансодержащие материалы хлорируются в виде брикетов
в шахтных электропечах (рис. 14) или в виде измельченной шихты
в расплавленных солях продуванием через расплав газообразного
хлора.
Исходные титановые материалы рутил или титановый шлак
измельчают на шаровых мельницах до крупности помола 0,09 мм
(—80%), а нефтяной кокс — до крупности 0,15 мм (—80%). Измель-
ченные материалы смешивают в необходимой пропорции. Расход
восстановителя определяется из расчета связывания кислорода
207
всех окислов, содержащихся в шихте, в окись и двуокись углерода
при соотношении СО : СО2 = 5 : 1—8 : 1. Расход кокса, обычно,
составляет 17—20% к весу прокаленных брикетов.
Шихту смешивают со связующим, нагревают до 90—110° С и
брикетируют. В качестве связующего для производства брикетов
используют каменноугольный пек (в количестве 7—8%) или кон-
центрат сульфитно-спиртовой барды с удельным весом 1,26 (в коли-
честве 10—14%).
Т1С14
Рис. 14. Схема производства четырех хлор истого титана.
Брикетирование производится на кольцевых прессах под дав-
лением до 300 кПсм2. Брикеты для удаления влаги и летучих
веществ прокаливают в железных кюбелях, нагреваемых в печах
со съемным сводом топочными газами при температуре 800—850° С
в течение 12 ч.
В. П. Печенкин [134] предложил проводить совместный размол
титанового шлака и восстановителя. Это обеспечивает наиболее тес-
ное смешение инградиентов и получение брикетов более однород-
ного состава. Рекомендовано также непрерывное смешение шихты
208
со связующими материалами и непрерывный обжиг брикетов в шахт-
ной печи при температуре 1000—1200° С, обогреваемой продуктами
неполного сжигания природного газа.
Шахтные электропечи, применяющиеся при хлорировании бри-
кетов из титанового шлака, работают полунепрерывно. Необходимая
температура для хлорирования поддерживается за счет теплоты
реакции и электрического подогрева. Электрический подогрев осу- г
ществляется с помощью двух рядов угольных электродов, распо-
ложенных друг над другом в нижней части печи. Верхний пояс
электродов работает при пуске печи, а нижний — все время, что-
бы предотвратить охлаждение и затвердевание расплава на по-
дине.
Брикеты загружаются в верхнюю часть печи периодически.
Хлор подается непрерывно через фурмы, расположенные между
нижним и верхним поясом электродов. Температура в хлораторе
поддерживается в пределах 900—1200° С. После накопления зна-
чительного количества непрохлорированного остатка печь останав-
ливается и очищается. Хлор-газ в шахтной электропечи фиксирует-
ся практически полностью.
Паро-газовая фаза, состоящая из четыреххлористого титана и
хлоридов железа, алюминия, кремния, ванадия, фосфора, серы и
др., из шахтной электропечи поступает в конденсационную систему.
Вначале она проходит через пылевую камеру, в которой осаждают-
ся уносимые из печи частицы пыли и значительное количество хлор-
ного железа. Эти частицы, судя по электронным фотографиям, со-
стоят из шарообразных зерен первичной кристаллизации размером
2—6 мкм [58].
Затем хлориды конденсируются в оросительных конденсаторах
при температуре не выше 70° С. Конденсаторы орошаются охлаж-
денным четыреххлористым титаном. В оросительных конденсаторах
конденсируются твердые хлориды (FeCl3 + А1С13) и значительная
часть TiCl4. Окончательная конденсация четыреххлористого титана
и значительной части четыреххлористого кремния производится
в поверхностных конденсаторах, охлаждаемых водой или охлажден-
ным до —15° С рассолом хлористого кальция.
Отходящие из конденсаторов газы состоят в основном из СО,
СО2 и небольших количеств TiCl4, НО и SiCl4. В них, обычно, со-
держится также небольшое количество фосгена (до 15 мг/л), обра-
зующегося при температуре ниже 500—600° С в результате взаимо-
действия окиси углерода с хлором [170]. После конденсации TiCl4
газы подвергаются газоочистке и выбрасываются в атмосферу.
Пульпа из оросительных холодильников поступает в аппараты
для отделения твердых хлоридов в сгустители типа Дорра. Твердые
хлориды выводятся из сгустителей и подвергаются утилизации.
Сначала они нагреваются для отгонки TiCl4, который возвращается
в процесс, а остаток, состоящий в основном из FeCl3 и А1С13, нейтра-
лизуется и выбрасывается на свалку.
14 1-1669
209
Б. А. Галицкий и соавторы [56] для отделения пульпы от че-
тыреххлористого титана предложили использовать фильтрацию че-
рез металлокерамические фильтры под давлением 2 ати. Ско-
рость фильтрации составляет 0,5—2,0 м3 фильтрата на 1 м3 филь-
трующей поверхности в 1 ч.
Сырой четерех хлор истый титан, полученный по описанной схе-
ме, содержит следующие примеси (%): Si 0,02—0,3, TiOCl2 0,04—
0,15, Al 0,01—0,1, СОС12 0,005—0,15, Fe 0—0,2, Cl2 0,03—0,08,
V 0,01—0,1, S 0—0,03. Так как содержание примесей в сыром
Ванадиевый кек Вода
Рис. 15. Комплексное использование меднованадиевых кеков тита-
нового производства.
четыреххлористом титане превышает допустимые пределы, его под-
вергают специальной очистке.
Очистка четыреххлористого титана от ванадия производится об-
работкой медным порошком или сероводородом. Одновременно че-
рез жидкость продуваются пары воды, чтобы вызвать частичный
гидролиз четыреххлористого титана и этим облегчить коагуляцию
взвеси хлористого алюминия. После отстаивания четыреххлори-
стый титан очищается от кремния и оксихлорида титана ректифика-
ционной перегонкой и дистилляцией, в результате чего содержание
примесей может быть уменьшено до 1 • 10—4%. Вместо отстаива-
ния рекомендуется также фильтрация через стеклянную вату [1].
Химизм процесса очистки четыреххлористого титана от ванадия
изучен еще недостаточно. И. Н. Рубан с соавторами [172] считает,
что при существующей технологии оксихлорид ванадия гидроли-
зуется с образованием поливанадиевых кислот переменного состава,
которые вместе с другими продуктами гидролиза дают коллоидно-
дисперсную смесь.
210
Ванадиевые кеки, получаемые при очистке четыреххлористого
титана, имеют примерно следующий состав (%): 12, 8 TiO2, 7,0 V2O5,
26,2 Си, 0,38 SiO2, 1,06 Д12О3, 0,36 Fe, 45,2 Cl [91 ]. Они являются
ценным продуктом, из которого можно извлекать ванадий и медь.
X. А. Курумчин и А. П. Яценко [91 ] разработали метод регенера-
ции меди из ванадиевых кеков с попутным извлечением ванадия
(рис. 15).
Л. В. Денисов и соавторы [70] предлагают извлекать ванадий
из растворов после выщелачивания ванадиевых кеков экстракцией
его ди-2-этилгексилфосфорной кислотой. Л. А. Нисельсон с соав-
торами [121] разработал схему ректификационной очистки окси-
хлорида ванадия от четыреххлористого титана и других примесей.
Производство четыреххлористого титана является сравнительно
молодой отраслью промышленности. Технология получения четырех-
хлористого титана еще несовершенна. Большим тормозом является
низкое качество титанового сырья, особенно титановых шлаков.
Несмотря на высокое содержание двуокиси титана в шлаках (80—
87%), при хлорировании их все же получается значительное коли-
чество (^ 200 кг на 1 т TiCl4) побочных продуктов: хлоридов же-
леза, алюминия, кремния, кальция, магния и др. На образование
этих хлоридов затрачивается значительное количество хлора. Ути-
лизация их при крупных масштабах производства представляет
большие трудности и до настоящего времени технически не разре-
шена. Вследствие этого первым и основным условием совершенство-
вания технологии производства четыреххлористого титана являет-
ся повышение качества титановых шлаков или замена их на техни-
ческую двуокись титана с содержанием 97—98% TiO2
Большой интерес представляют и поиски принципиально новых
методов получения четыреххлористого титана, например из серно-
кислых растворов путем конверсии сульфатов в хлориды [156—160,
341].
ПРОИЗВОДСТВО ДВУОКИСИ ТИТАНА
I Промышленное производство двуокиси титана началось после того,
кай выяснилось, что она обладает хорошими молярными свойствами
и может применяться взамен свинцовых и цинковых белил.
Впервые в качестве титанового пигмента было предложено ис-
пользовать обожженный при температуре 500° С и затем размоло-
тый ильменит [266]. Методы получения двуокиси титана в качестве
белого титанового пигмента были разработаны Ибсеном [284] в
Норвегии, а также Росси и Бартоном [330] в США.
Несколько ранее Увейнтрауб [354] предложил метод получения
двуокиси титана из ильменита (которая предназначена для исполь-
зования в электротехнической промышленности), основанный на
разложении его серной кислотой с последующим гидролизом раство-
ра сульфата титана. Данный метод стал протипом современного
14*
211
метода производства пигментной двуокиси титана, по которому
измельченный ильменит смешивается с двукратным количеством
93%-ной серной кислоты и нагревается в железном котле до 100° С.
При этой температуре начинается бурное взаимодействие ильмени-
та с серной кислотой, заканчивающееся образованием спека твер-
дых сульфатов титана и железа. Спек выщелачивается водой для
переведения сульфата титана в раствор. Одновременно с сульфатом
титана растворяются и сульфаты железа. В полученном растворе
трехвалентное железо восстанавливается до двухвалентного действи-
ем металлического цинка, сернистым газом или гипосульфитом нат-
рия. Затем раствор разбавляется водой до концентрации 0,5—3,0%
TiO2 и нагревается до кипения.
В результате гидролиза сульфата титана образуется гидроокись,
которую отфильтровывают, промывают водой и прокаливают до
превращения ее в двуокись титана. Получают весьма чистую дву-
окись титана с малым содержанием примесей.
Этот метод был усовершенствован Вейзманом и Блюменфельдом
[353], предложившими восстанавливать трехвалентное железо в рас-
творе железными стружками, а также Мекленбургом [299] и Блюмен-
фельдом [231], разработавшими методы гидролиза сульфата титана
с применением специально приготовленных или получаемых в про-
цессе гидролиза зародышей.
В настоящее время сернокислотный метод производства двуоки-
си титана широко используется в промышленности. Первый завод
титановых пигментов мощностью 1000 т в год был построен в Нор-
вегии в 1916 г. фирмой Titan Со A/S [312]. В 1919 г. производ-
ство титановых пигментов было начато в США фирмой Titan Pig-
ment Со.
В настоящее время двуокись титана производится главным об-
разом для получения белых пигментов различного назначения.
Технология производства двуокиси титана является специфической,
рассчитанной на получение технического продукта с определен-
ными свойствами. Вообще двуокись титана можно получить многи-
ми способами, однако продукт с высокими молярно-техническими
свойствами удается получить только при гидролизе сульфатов или
хлоридов титана в водных растворах с применением специально
приготовленных зародышей или в результате сжигания четырех-
хлористого титана высокой чистоты.
На большинстве заводов двуокись титана получается по серно-
кислотному методу, в основе которого лежит гидролиз сульфата
титана. Хлорный метод (сжигание четыреххлористого титана) до
настоящего времени применяется в небольших масштабах, так как
в экономическом отношении он уступает сернокислотному методу.
Другие методы производства титановых пигментов не нашли при-
менения либо вследствие того, что не обеспечивают получение про-
дукции высокого качества, либо по техническим и экономическим
соображениям.
212
Двуокись титана, предназначенная для производства твердых
сплавов и различных соединений титана, получается по схемам,
применяющимся в производстве титановых пигментов, но частично
упрощенным за счет исключения некоторых специфических опе-
раций.
СЕРНОКИСЛОТНЫЙ МЕТОД ПРОИЗВОДСТВА ТИТАНОВЫХ ПИГМЕНТОВ
Разложение титансодержащих концентратов серной кислотой
Сырьем для производства титановых пигментов служат ильмени-
товый концентрат с содержанием 40—60% TiO2, титановые шлаки
с содержанием около 70% TiO2 и рутил. Ильменит и титановые шла-
ки применяются для производства двуокиси титана сернокислотным
методом, рутил — для производства титановых пигментов хлорным
методом (через четырех хлор истый титан).
К качеству сырья предъявляются определенные требования.
Ильменит и титановые шлаки должны содержать не более 0,1%
Сг2О3, так как хром является вредной примесью, придающей белым
пигментам серое окрашивание. Нежелательна также примесь фос-
фора, ухудшающая дисперсность продукта, а также ванадия и мар-
ганца.
Лучшими сортами ильменита считаются концентраты, содержа-
щие высокий процент железа в закисной форме. Они более легко раз-
лагаются серной кислотой и не требуют больших затрат железных
стружек на восстановление окисного железа. Ильменит, подверг-
шийся частичному изменению в гипергенных условиях, содержа-
щий примесь лейкоксена и рутила, серной кислотой разлагается
плохо, и при переработке таких концентратов выход двуокиси тита-
на бывает заниженным.
Хорошо разлагаются серной кислотой титановые шлаки, содер-
жащие около 70% TiO2 и 10—12% FeO. Высокотитановые шлаки
содержат примесь рутила и плохо разлагаются серной кислотой.
Нежелательной примесью в титановых шлаках является окись
алюминия, присутствие которой в растворах сульфата титана по-
нижает их стабильность. |
Химизм сульфатизации титансодержащих материалов частично
рассмотрен нами в первой части этой книги (стр. 396). Взаимодейст-
вие ильменита с серной кислотой протекает с выделением тепла. Ве-
личина теплового эффекта реакции зависит от степени измененности
ильменита и концентрации серной кислоты. При комнатной темпе-
ратуре ильменит реагирует с серной кислотой очень медленно, но
реакция ускоряется при нагревании. По достижении определенной
температуры наступает самопроизвольное разложение с разогре-
ванием массы до температуры кипения. Температуру начала само-
разогревания (температуру начала разложения [47]) можно исполь-
зовать для характеристики реакционной активности ильменита.
213
Рис. 16. Изменение температуры
пульпы иршинского ильменитового
концентрата с 85%-ной серной кис-
лотой в период саморазогревания.
Сопоставимые величины температуры начала разложения по-
лучаются при определении ее по стандартной методике. С помощью
термографического анализа, проведенного в одинаковых условиях
[47], найдено, что разложение малоизмененного ильменита, состоя-
щего преимущественно из кричтонита (FeTiO3), 75—93%-ной сер-
ной кислотой начинается с саморазогревания при 108—123° С.
Измененный ильменит, состоящий из аризонита (Fe2O3 • 3TiO2),
начинает разлагаться серной кислотой с саморазогреванием при
162—169° С.
> Реакционную активность ильменита, зависящую от степени его
измененности, можно характеризовать и соотношением в концент-^
рате окисного и закисного железа
или содержанием в нем кричтони-
та. Однако из-за сложности мине-
ралогического состава концентра-
тов, содержащих измененный ми-
нерал, истинное содержание в них
кричтонита, по данным химических
анализов и даже фазового анализа
[211], определить трудно. Более
надежные результаты при опреде-
лении содержания кричтонита мо-
жет дать, по-видимому, измерение
мессбауэровских спектров [276], но
методика минералогического анали-
за с помощью у-рассеяния оконча-
тельно еще не разработана.
Исследование разложения ильменитового концентрата с помощью
записи термограмм на пирометре Курнакова показало, что входя-
щие в состав минерала окислы железа и титана реагируют с сер-
ной кислотой селективно [36]. Судя по термограммам нагревания,
при разложении кричтонита вначале реагирует с серной кислотой
закись железа (температура 130—160° С), а затем в реакцию всту-
пает двуокись титана (195—200 ° С). Окись железа в измененном мине-
рале ведет себя при сульфатизации как самостоятельный компонент.
При большой массе реагирующих материалов взаимодействие
ильменитового концентрата с серной кислотой как самопроизволь-
ный процесс начинается при более низкой температуре. На опытной
установке Крымского завода пигментной двуокиси титана в реакто-
ре с загрузкой 0,6 т зарегистрирована температура начала разло-
жения иршинского ильменитового концентрата в интервале 50—
60° С (рис. 16). Измерения в лабораторных условиях при навеске
ильменита 200 г дали для этого же образца температуру -начала
реакции 89° С. Однако скорость химической реакции при большой
массе реагирующих материалов вначале нарастает очень медленно.
Закипает реагирующая масса только спустя 3—4 ч после того мо-
мента, когда зарегистрировано ее саморазогревание.
214
Реакционная способность титанового шлака при взаимодействии
с серной кислотой, как показали В. А. Резниченко с соавторами
[186], зависит от состава твердого раствора аносовитовой фазы и
наличия других фазовых составляющих. Существенное влияние на
взаимодействие титанового шлака с серной кислотой оказывает со-
держание низших окислов титана в составе твердого раствора и
примесь окиси магния. Титанаты магния по уменьшению скорости
растворения в серной кислоте располагаются в ряд 2MgO • TiO2,
MgO • TiO2, MgO • 2TiO2. Малой реакционной способностью с сер-
ной кислотой обладают шлаки, содержащие тагировит, свободную
Ti2O3, перовскит и рутил.
Самопроизвольное разогревание реагирующей массы (ильмени-
тового концентрата или титанового шлака) с серной кислотой ши-
роко используется на большинстве современных заводов как ме-
тод сульфатизации титансодержащего сырья в аппаратах периоди-
ческого действия без внешнего обогрева. Этот метод, несмотря на
простоту аппаратурно-технологического оформления, отличается
существенными недостатками. Он применим только для переработ-
ки легковскрываемых титансодержащих материалов, к числу ко-
торых относится неизмененный ильменит и некоторые виды титано-
вых шлаков.
Измененные ильменитовые концентраты из-за малой величины
теплового эффекта при взаимодействии их с серной кислотой и ма-
лой скорости реакции при температуре ниже 160—170° С в аппара-
тах периодического действия вскрываются в недостаточной степени,
а растворы сульфата титана при выщелачивании продуктов их раз-
ложения получаются с высоким кислотным фактором.
В аппаратах периодического действия возникают большие за-
труднения при регулировании температуры процесса, что нередко
приводит к чрезмерному перегреванию реагирующей массы и выбро-
су ее из аппарата выделяющимися в большом количестве газообраз-
ными продуктами реакции. Известны случаи образования плотного,
малопористого спека (козлов), трудновыщелачиваемого водой.
Эти недостатки периодического процесса могут быть устранены
в результате использования в производстве сырья соответствующего
состава и поддержания постоянства его качества. Более радикаль-
ной мерой при совершенствовании метода вскрытия титансодержа-
щего сырья серной кислотой является переход к использованию не-
прерывной технологии с внешним обогревом.
В патентной литературе описано несколько методов непрерыв-
ного разложения ильменитового концентрата серной кислотой,
основанных на разогревании реагирующей массы за счет теплоты
растворения олеума водой или обработкой гидролизной кислотой.
В качестве аппаратов для непрерывного разложения рекомендуется
использовать вращающиеся трубчатые печи, противни [244], шаро-
вые мельницы, аппараты шнекового типа [305], вращающиеся сме-
сители [336]. Однако эти методы, за редкими исключениями [333],
215
не нашли промышленного применения, что объясняется трудностя-
ми, связанными с необходимостью точного дозирования реагирую-
щих компонентов и с транспортированием в аппаратах реагирующей
массы.
В последние годы изучена сульфатизации титансодержащих ма-
териалов в аппарате шнекового типа [41], в печи с кипящим слоем
[141] и в аппарате кольцевого типа [44]. Наиболее перспективным
для сульфатизации титансодержащих материалов оказались аппара-
ты кольцевого типа. Принцип действия аппарата кольцевого типа
заключается в непрерывной подаче заранее приготовленной смеси
ильменитового концентрата с серной кислотой в обогреваемый до
необходимой температуры вращающийся стальной реактор в виде
желобообразного кольца. Благодаря непрерывной загрузке пульпы
в зону с высокой температурой аппарат позволяет вести разложение
ильменита без непрерывного перемешивания реагирующей массы,
чем устраняется основной недостаток, присущий сульфатизаторам
[41, 141, 244, 305, 331].
На Крымском заводе пигментной двуокиси титана разработан
промышленный метод непрерывного разложения ильменитового кон-
центрата серной кислотой в аппаратах кольцевого типа [42].
Он пригоден для сульфатизации серной кислотой ильменитовых
концентратов различного качества, так как позволяет поддержи-
вать в зоне реакции необходимую температуру за счет внешнего
обогрева топочными газами. Измененные ильмениты при непрерыв-
ном методе сульфатизации разлагаются с получением спеков, даю-
щих при выщелачивании растворы сульфата титана с низким кис-
лотным фактором.
Восстановление титана и железа
в сернокислых растворах
На существующих заводах восстанавливают трехвалентное же-
лезо до двухвалентного и часть четырехвалентного титана до трех-
валентного в сернокислых растворах перед гидролизом сульфата
титана металлическими железными стружками, чугунным порошком
и другими отходами металлообработки. При использовании для
производства двуокиси титана в качестве сырья неизмененных иль-
менитов или титанового шлака расход железа на восстановление
бывает небольшим и метод химического восстановления экономи-
чески оправданным.
Если же сырьем для производства двуокиси титана служит
измененный ильменитовый концентрат, содержащий значительный
процент железа в трехвалентном состоянии, то расход железного
скрапа в качестве восстановителя увеличивается до 200 кг и более
на 1 т готовой продукции. Увеличивается и выход железного ку-
пороса в качестве побочного продукта. В связи с этим приобретает
216
актуальность замена химического метода восстановления железа
и титана в сернокислых растворах на электролитический.
Опыты по электролитическому восстановлению железа и титана
в сернокислых растворах были поставлены еще в тридцатых годах
[501. Электролиз проводили в электролизере со свинцовыми элек-
тродами и диафрагмой из пористого фарфора, отделяющей анодное
пространство от катодного. При температуре 50° С и плотностях
тока 1—6 а/дм2 на катоде и 0,83—2,52 а/дм2, на аноде выход по току
при электролитическом восстановлении железа в производственных
растворах составлял 86,7—100%. В анодном пространстве электро-
лизера получалась серная кислота с концентрацией 48—50%, со-
державшая только небольшую примесь титана (0,4—0,5 г/л TiO2),
которую авторы предлагали использовать, выводя ее из процесса.
В другой работе [219] было показано, что выход по току увели-
чивается с повышением температуры, а электролиз без анодной
диафрагмы из-за низкого выхода по току практически невозможен.
Е. П. Белякова и соавторы [19] провели повторные опыты по
электролитическому восстановлению железа и титана в сернокислых
растворах?-Авторы показали, что идея использования анодной сер-
ной кислоты из-за сложности при ее отводе из электролизера являет-
ся технически нереальной. Они разработали конструкцию анодной
ячейки, в которой серная кислота, выделяющаяся при электролизе,
не задерживается, а самопроизвольно инфильтруется в катодное
пространство. На основе данной конструкции анодной ячейки раз-
работан промышленный метод восстановления железа и титана в
сернокислых растворах. Электролиз ведется при следующих усло-
виях: температура электролита 50—60° С, электроды свинцовые,
катодная и анодная плотности тока 4,5 а!дм\ напряжение на элек-
тролизере 4,0 в.
Электролитическое восстановление трехвалентного железа до
двухвалентного в указанных условиях протекает с выходом по току
70%, расход электроэнергии на восстановление 1 кг железа 3 кеч.
Выход по току при восстановлении титана составляет 50%. По тех-
нико-экономическим показателям электролитический метод вос-
становления железа и титана в сернокислых растворах при перера-
ботке измененных ильменитовых концентратов обладает существен-
ными преимуществами перед химическим методом восстановления
железными стружками и чугунным порошком.
При изучении окисления трехвалентного титана в сернокислых
растворах кислородом воздуха установлено, что реакция окисления
титана—нулевого порядка [130, 131]. Скорость окисления увеличи-
вается с повышением температуры и не зависит от содержания при-
месей сульфата калия и ниобия.
217
Гидролиз сульфата титана
Физико-химические основы гидролитического разложения суль-
фата титана уже рассматривались нами в первой части книги. Со
времени опубликования ее получены новые данные, характеризую-
щие этот процесс.
П. И. Медведев и соавторы [119] изучили кинетику гидролиза
сульфата титана в зависимости от условий приготовления титановых
зародышей. Они показали, что скорость гидролиза сульфата титана
увеличивается с ростом pH титанового зародышевого раствора и
повышением температуры. Кинетика процесса описывается уравне-
нием реакции первого порядка с константами скорости 2,2 • 10-2 X
X мин~х при 110° С и 4,6 • 10~2 мин~х при 120° С. Энергия акти-
вации процесса составляет 21 ккал!моль.
В технологических растворах, полученных из измененного иль-
менита после разложения его серной кислотой с примесью сульфата
аммония, гидролиз сульфата титана также описывается уравнением
реакции первого порядка. Величина константы скорости реакции
зависит от количества добавленного титанового зародышевого раст-
вора [87]. Под действием титановых зародышей скорость гидролиза
увеличивается в десятки раз.
Разработан новый метод приготовления титановых зародышей,
основанный на нейтрализации растворов сульфата титана аммиа-
ком [47]. По этому методу технологический раствор сульфата титана
разбавляют водой до концентрации TiO2 25 г!л. К нему добавляют
сульфат аммония или эквивалентное количество серной кислоты
из расчета, чтобы в готовом зародышевом растворе концентрация
сульфата аммония была равной 140—160 г!л. Раствор нагревают
до температуры 40—50° Сив него медленно добавляют концен-
трированный раствор аммиачной воды с содержанием 200—
260 г!л NH3 при непрерывном перемешивании до pH 2,85—3,0.
Полученную пульпу гидроокиси титана нагревают до 65—70° С и
выдерживают при этой температуре 1,5 ч. Затем раствор охлаждают
до комнатной температуры и используют по назначению.
Продолжая развивать точку зрения о том, что в концентрирован-
ных растворах титан находится в форме гидроксокомплексов,
Ю. Д. Долматов [671 изучил состояние титана в растворе методами
потенциометрического и кислотно-основного титрования, измере-
ния вязкости и электропроводности. По данным его исследований,
в растворах сульфата титана доминируют ионы Ti (ОН)2+ и
Ti (ОН)з*. С повышением концентрации сульфата титана равно-
весие сдвигается в сторону образования ионов Ti (ОН)з\ Гидролиз
сульфата титана при нагревании растворов приводит к уменьшению
заряда иона Ti (ОН)14~Х)+ до 0,25 и образованию поликатионов,
а нагревание растворов — к деполимеризации ионов титана.
218
Кинетика гидролиза
Ю. Д. Долматов и соавторы [22, 68, 69], рассматривая гидролиз
сульфата титана как топохимическую реакцию, охарактеризовали
его математическим уравнением в виде двойной логарифмической
зависимости выхода от времени нагревания раствора. Это уравне-
ние не содержит величин, учитывающих влияние титановых заро-
дышей на кинетику процесса. Кроме того, двойная логарифмиче-
ская зависимость относится к малочувствительным функциям. Ис-
пользование для анализа экспериментальных данных дает мало
сведений о механизме гидролиза. Ввиду сложности процесса гидро-
лиза сульфата титана кинетику его удобнее изучать по отдельным
стадиям, не пытаясь описать одним математическим уравнением.
Следуя этому принципу, разделим процесс гидролиза на три стадии
и рассмотрим с точки зрения кинетики каждую из них в отдель-
ности-—------ -Л-------------------------- ----------------„
Гидролитическое разложение сульфата титана — первая ста-
дия этого процесса — характеризуется изменением в растворе кон-
центрации молекул яр но-дисперсного титана. Изучать кинетику гид-
ролитического разложения титана стало возможным после разработ-
ки метода определения концентрации молекулярно-дисперсного
титана в растворе. Однако гидролиз сульфата титана в присутствии
сульфата аммония и при использовании титановых зародышевых
растворов протекает без образования заметных количеств коллоид-
ной гидроокиси титана.
Поэтому еще до разработки пероксидного метода определения
молекулярно-дисперсного титана удалось описать кинетику гидро-
литического разложения сульфата титана по изменению общей
концентрации титана в растворе.
Автором [43] было показано, что гидролиз сульфата титана опи-
сывается уравнением реакции первого порядка. Аналогичной за-
висимости подчиняется гидролиз сульфата титана, проводимый в
присутствии титановых зародышей [119]. В отсутствие титановых
зародышей изменение концентрации молекул яр но-дисперсного ти-
тана описывается уравнением реакции первого порядка [106].
Второй стадией гидролиза сульфата титана, согласно предложен-
ного* нами механизма, является мицеллообразование. Полагая, что
скорость мицеллообразования прямо пропорциональна концентра-
ции немицеллярной гидроокиси титана и объему мицеллы, можно
написать
----= kCV,
л
где С — концентрация немицеллярной гидроокиси титана; V —
объем мицеллы; t — время; k — коэффициент пропорциональности.
Исходя из этого соотношения после подстановки значения V из
219
упрощенного уравнения Релея, получаем уравнение кинетики
мицеллообразования [46, 105]:
В этом уравнении а — величина постоянная; Сд — начальная кон-
центрация немицеллярной гидроокиси титана; Сп — концентрация
центров мицеллообразования — число титановых ‘зародышей в еди-
нице объема; Vo — объем титанового зародыша; р — плотность дис-
персной фазы; R90 — коэффициент рассеяния. Оно связывает эк-
спериментально определяемые величины (начальную концентрацию
Таблица 20
Влияние добавленного искусственно приготовленного титанового раствора на
начальные размеры мицелл и параметры кинетики мицеллообразования
Добавлено титанового зародышево- го раствора, % Начальные размеры мицелл с сп • 10 Масса мицеллооб- разования, % Доля самопроиз- вольно возника- ющих зародышей, % 1 - а 1 со з 1 S k сп
О О СО ° Средний радиус R • 107, см
0 8,75 2,75 0,850 1,29 100,0 0,700 0,82
0,25 14,70 5,27 0,162 1,73 86,3 0,157 0,97
1,00 98,80 6,17 0,119 2,03 50,3 0,167 1,40
3,00 120,00 6,60 0,345 7,20 58,3 0,690 2,00
немицеллярной гидроокиси титана и коэффициент рассеяния) с
числом титановых зародышей в единице объема, объемом титанового
зародыша (начальным объемом мицеллы) и массой мицеллообразо-
вания. Последняя представляет собой массу титановых зародышей
в единице объема и численно равна произведению рСл70.
Пользуясь этим уравнением, можно анализировать эксперимен-
тальные данные при изучении кинетики гидролиза, характеризовать
эффективность титановых зародышевых растворов, определять оп-
тимальный расход их для проведения гидролиза. Пример анализа
экспериментальных данных с помощью уравнения кинетики мицел-
лообразования приведен в табл. 20. Исследовался гидролиз сульфа-
та титана в растворе с концентрацией 100 г! л ТЮ2 и отношением
SO3 : TiO2 = 1,5 при температуре 70° С. В этом растворе при гид-
ролизе возникает 0,85 • 1016 центров мицеллообразования в 1 см3
со средним радиусом 2,75 нм. Масса мицеллообразования составляет
1,29% от общего содержания титана в растворе:
При добавлении искусственно приготовленных титановых за-
родышей доля самопроизвольно возникающих центров мицелло-
образования, на основе которых образуются мицеллы гидроокиси
220
титана, уменьшается, а средний радиус титановых зародышей ра-
k
стет. Увеличивается также отношение , являющееся в уравне-
нии кинетики мицеллообразования мерой скорости реакции. Одна-
ко снижение доли самопроизвольно возникающих титановых за-
родышей в растворе (центров мицеллообразования) происходит толь-
ко до некоторого предела, составляющего около 50% и отвечающего
1% добавленных искусственно приготовленных зародышей. До-
бавка большего количества искусственно приготовленных титано-
вых зародышей с точки зрения снижения доли самопроизвольно
возникающих при гидролизе центров мицеллообразования беспо-
лезна, хотя и приводит к увеличению массы мицеллообразования
и ускорению реакции мицеллообразования.
По уравнению кинетики мицеллообразования экспериментально
определен средний радиус частиц немицеллярной гидроокиси ти-
тана, равный 0,75 нм. Немицеллярная гидроокись титана является,
таким образом, дисперсной фазой с малым размером частиц, отли-
чающимся менее чем на один порядок от размеров атомов титана
и кислорода.
Третья стадия гидролиза сульфата титана — коагуляция гид-
роокиси титана. Экспериментально коагуляцию гидроокиси титана
можно изучать методом светорассеяния и по изменению концентра-
ции в растворе коллоидно-дисперсной фазы. Первый метод позво-
ляет изучать кинетику коагуляции в скрытый период ее протекания.
В период образования осадка кинетику процесса можно характери-
зовать изменением концентрации коллоидно-дисперсной гидрооки-
си титана в растворе.
Исследование кинетики коагуляции гидроокиси титана в скры-
тый период ее протекания методом светорассеяния показало, что
процесс описывается уравнением реакции второго порядка [105].
Причем константа скорости коагуляции не зависит от количества
добавленных в раствор титановых зародышей. Вызвано это, вероят-
но, тем, что в коагуляции принимают участие преимущественно
мицеллы, выросшие до определенного размера на основе центров
мицеллообразования еще до того времени, когда начинается их
укрупнение за счет взаимного слипания.
Изменение концентрации коллоидно-дисперсной гидроокиси ти-
тана в период образования осадка гидроокиси существенно зависит
от отношения SO3 • TiO3 в растворе, добавки титановых зароды-
шей и примесей солей [106]. При гидролизе сульфата титана без
добавки титановых зародышей и отношении SO3 : ТЮ2 = 1,25 (кон-
центрация TiO2 в растворе 100 г! л) первоначально выпавший оса-
док гидроокиси титана подвергается пептизации. При дальнейшем
нагревании раствора до кипения пептизировавшаяся гидроокись
титана коагулируется повторно уже необратимо в условиях экспе-
римента.
221
Механизм действия титановых зародышей
Пользуясь методом светорассеяния, автор и Е. П. Лыков изу-
чили поведение титановых зародышевых растворов при добавлении
к раствору серной кислоты с целью выяснения механизма действия
их при гидролизе сульфата титана. Оказалось, что в растворе 10%-
ной серной кислоты, концентрация которой близка к составу интер-
мицеллярной жидкости, агрегаты гидроокиси титана зародышевого
золя разукрупняются до частиц меньшего размера. Кинетика раз-
Рис. 17. Изменение радиуса кол-
лоидных частиц гидроокиси титана
в 10%-ной серной кислоте:
1 — гидроокись титана, осажденная из
сернокислого раствора едким натром
при 20° С и pH 4,0; 2, 3 — зародыше-
вый титановый раствор, выдержанный
при 60° С 1 и 6 ч соответственно.
укрупнения зависит от температу-
ры и способа приготовления тита-
нового зародышевого раствора, в
частности от продолжительности
термической обработки. При ком-
натной температуре разукрупнение
агрегатов идет до среднего радиуса
частиц 2—6 нм (рис. 17). Зароды-,
шевые растворы, подвергавшиеся
при их приготовлении термической
обработке меньшее время, разукруп-
няются до частиц с меньшим ра-
диусом.
Пр и тем пер ату р е выше 50—60 ° С
через 10—15 мин после смешения
зародышевого раствора с серной
кислотой разукрупнение агрегатов
гидроокиси титана приостанавли-
вается. Образовавшиеся частицы с
радиусом менее 10 нм начинают коа-
гулировать в результате взаимного
слипани#.
Механизм действия титановых зародышевых растворов при гид-
ролизе сульфата титана состоит, таким образом, в разукрупнении
агрегатов мицелл до размера 'зародышей мицеллообразования, на
основе которых в последующем вырастают мицеллы метатитановой
кислоты. В процессе гидролиза специально приготовленные золи и
осажденная щелочами гелеобразная гидроокись титана в качестве
титановых зародышей принципиально ведут себя одинаково. Гидро-
титановых зародышей принципиально ведут себя одинаково. Гидро-
окись титана, однако, при смешивании с 10%-ной серной кислотой
претерпевает не только разукрупнение агрегатов, но и в значитель-
ной степени растворяется, переходя в молекулярно-дисперсное
состояние. Золи гидроокиси титана, подвергшиеся термической обра-
ботке в процессе приготовления зародышевых растворов, при сме-
шении с 10%-ной серной кислотой сохраняют свою природу. Агре-
гаты гидроокиси титана, содержащиеся в них, не растворяются до
молекул яр но-дисперсного состояния.
222
Прокаливание гидроокиси титана
Пигментная двуокись титана из гидроокиси получается прока-
ливанием последней при температуре 800—1000° С, в результате
чего от гидроокиси титана отщепляются гидроксильные группы, а
также химически связанные вода и сульфогруппы. При прокалива-
нии формируются частицы кристаллической двуокиси титана со
структурой анатаза или рутила с определенными физико-техни-
ческими свойствами.
При производстве пигментов со структурой анатаза прокалива-
нию предшествует солевая обработка гидроокиси титана-раствором
сульфата калия и фосфорной кислоты. Солевая обработка замед-
ляет превращение анатаза в рутил, а также предотвращает спекание (
частиц и придание им жесткости на ощупь.
Формирование структуры рутила при прокаливании гидроокиси
титана ускоряется при добавке к ней рутилизирующих зародышей.
Роль последних выполняет специально полученная гидроокись ти-
тана. Рутилизация ускоряется при добавке к гидроокиси титана
окиси цинка и некоторых других минерализаторов.
Физико-химические процессы, протекающие при прокаливании
гидроокиси титана, изучены еще недостаточно. Исследования в
основном направлены на выяснение закономерностей изменения
размеров частиц гидроокиси титана при прокаливании и кинетики
перехода анатаза в рутил.
При нагревании гидроокиси титана до 750° С размеры частиц
увеличиваются от 0,006 до 0,1—0,5 мкм [275]. Прокаливание ана-
таза выше 900° С приводит к превращению его в рутил. Размер
кристаллов при этом увеличивается до 1 мкм при температуре
1100° С и 1,8 мкм при 1150° С.
Изучено поведение гидроокиси титана при прокаливании мето-
дами рентгенографии, электронографии и рассеяния рентгеновских
лучей [20]. Установлено, что образующаяся при гидролизе сульфа-
та титана гидроокись состоит из блоков (частиц) размером 6—10 нм
и имеет структуру анатаза. При нагревании до 650—700° С она
превращается в двуокись титана также со структурой анатаза. Раз-
меры блоков при этом увеличиваются незначительно. Выше 800° С
анатаз превращается в рутил в виде удлиненных призматических
кристаллов [21].
Гидроокись титана, осажденная из растворов щелочами и ста-
ревшая при комнатной температуре длительное время, при про-
каливании ведет себя так же, как полученная гидролизом из кипя-
щих растворов. Свежеосажденная гидроокись титана при прокали-
вании дает частицы размером более 100 нм.
Кинетика превращения анатаза в рутил описывается уравне-
нием [335]
а = 1 — e~btn,
223
где а — выход рутила за время /; b и п — константы. Энергия
активации фазового перехода для образца анатаза, полученного
сернокислотным методом, равна 105 ккал/моль и хлорным мето-
дом — 107 ккал/моль. Большая величина энергии активации — ре-
зультат существенного влияния температуры на скорость фазового
перехода.
Поверхностная обработка
Некоторые свойства титановых пигментов, например светостой-
кость, улучшаются при обработке поверхности частиц (модифициро-
вании) некоторыми неорганическими и органическими соединения-
ми. В качестве веществ для поверхностной обработки двуокиси ти-
тана используются соединения алюминия и кремния.
Частицы двуокиси титана при модифицировании покрываются
пленкой гидроокисей, осаждаемых из растворов щелочных солей
действием разбавленных кислот и двуокиси углерода. Диспергируе-
мость и гидрофобность двуокиси титана в синтетических связующих
повышаются при обработке ее кремнеорганическими жидкостями.
Диспергируемость двуокиси титана в водоэмульсионных красках
улучшается при обработке уксусной кислотой. При получении не-
мелящей двуокиси титана рекомендуется покрывать ее частицы
пленкой гидроокиси алюминия, вес которой в пересчете на А12О3
составляет 0,5—5% к весу ТЮ2 [96]. Обработанный продукт про-
каливают при 500—800° С 10 мин для частичного обезвоживания
гидроокиси алюминия.
Н. Н. Стремилова и соавторы [193] сопоставили эффективность
поверхностной обработки двуокиси титана в зависимости от метода
нанесения пленки на поверхность частиц. Они показали, что осаж-
дение алюмокремниевых пленок нейтрализацией щелочных рас-
творов кислотами придает двуокиси титана большую фотоактив-
ность, чем осаждение методом карбонизации.
Схема технологического процесса
Схема производства титановых пигментов по сернокислотному
методу включает следующие основные операции: разложение сырья
серной кислотой и получение раствора сульфата титана, гидролиз
сульфата титана, фильтрация и промывание метатитановой кислоты,
прокаливание метатитановой кислоты, размол и поверхностная
обработка двуокиси титана, регенерация гидролизной серной кис-
лоты (рис. 18). Эти операции лежат в основе производства двуокиси
титана различного назначения на всех заводах, хотя детали техно-
логического процесса на заводах различных фирм имеют отличия.
Двуокись титана имеет кристаллическую структуру анатаза и ру-
тила.
При гидролизе сульфата титана в сернокислых растворах полу-
чается гидроокись титана, которая после прокаливания превра-
щается в двуокись титана со структурой анатаза [1361. Анатаз яв-
ляется метастабильной модификацией двуокиси титан!а. При дли-
тельном нагревании он превращается в рутил. Гидролиз хлоридов
титана позволяет получить двуокись титана со структурой рутила.
Рутил образуется также сжиганием четырех хлор исто го титана
кислородом.
Подвергая метатитановую кислоту и прокаленную двуокись
титана различным видам обработки, можно получить титановые
пигменты с различными техническими свойствами (табл. 21).
Ильменит
Серная кислота
.. ...................................................
I Разложение ильменита и получение раствора сульфата титана |
, ___________Зародышевый раствор
I Гидролиз сульфата титана~
Фильтрация и промывание метатитановой, кислоты
Нейтрализация
и утилизация
серной кислоты
Прокаливание
метатитановой
кислоты
-....... ,
Размол и поверхностная
обработка двуокиси титана
Титановые пигменты
В канализацию или
на производство
суперфосфата
Рис. 18. Схема производства титановых пигментов сернокислотным
методом.
Технология и аппаратура производства титановых пигментов
сернокислотным методом описаны в монографиях [24, 237], поэтому
вкратце остановимся на основных операциях производства двуокиси
титана.
Ильменитовый концентрат или титановый шлак размалывают на
шаровых мельницах сухого помола. Размолотый материал должен
содержать не менее 97% частиц с размером до 44 мкм.
Разложение ильменита и титанового шлака серной кислотой
на большинстве заводов производят в аппаратах периодического
действия (типа котлов) при температуре 180—200° С без внешнего
обогрева. Реакционная масса разогревается до нужной температуры
за счет теплоты реакции. Первоначальное нагревание реагирующей
массы до начала протекания самопроизвольного процесса произво-
дят в результате разбавления 93%-ной серной кислоты водой до
концентрации 85—88%.
На некоторых заводах ильменит разлагают серной кислотой
в непрерывно действующих аппаратах шнекового типа [283].
1 5 1-1669
225
Таблица 21
Технические условия на двуокись титана (ГОСТ 9808-65)
Показатель Норма для марок
А-1 АВ А-01 р-1 Р-01 Р-02
Белизна, условные единицы (не ме- нее) 96,0 96—97 96,0 94,0 94,0 94,0
Цветность (отте- нок) По эталону — — — —
Содержание дву- окиси титана, % (не менее) 98 98 94,5 98,5 95 93
Содержание ру- тильной формы, % (не менее) Не опреде- ляется — 95 95 95
Содержание влаги, % (не более) 0,5 0,5 0,5 0,3 0,3 0,3
Содержание водо- растворимых со- лей, % (не более) 0,5 0,5 0,5 0,3 0,3 0,3 *
pH водной вытяж- ки 6,5—8,0 6,5—7,5 6,5—8,0 6,5—8,0 6,5—8,0 6,5—8,0
Маслоемкость, г масла на 100 г пигмента (не бо- лее) 30 Не опреде- ляется 30 25 25 25
Разбеливающая способность, ус- ловные единицы (не менее) 1150 То же 1200 1500 1650 1650
Укрывистость в пе- ресчете на сухой пигмент, г/л/2 (не менее) 45 -* » » 40 40 40 40
Остаток после мок- рого просева на сите с сеткой № 0045К, % (не более) 0,2 0,1 0,05 0,2 0,05 0,05
Перетираемость ме- тодом «клина», условные едини- цы (не более) 15 Не опреде- ляется 10 20 10 10
Содержание частиц Не 94 Не Не Не Не
размером до опреде- опреде- опреде- опреде- опреде-
1 мкм, % (не ме- нее) ляется ляется ляется ляется ляется
Стабильность 0,5% -ной водной суспензии дву- окиси титана, % (не менее) То же 94 То же То же То же То же
226
Продолжение табл. 21
Показатель Норма для марок
А-! АВ А-01 Р-1 Р-0) Р-02
Водоемкость, % (не Не опре- 94 Не опре- Не опре- Не опре- Не опре-
более) деляется деляется деляется •деляется деляется
Содержание РегО3, % (не более) То же 0,02 То же То же То же То же
Содержание Р2О5, % (не более) » » 0,5 » » »- » » » » »
Обозначения: А-1 — двуокись титана анатазной формы, необработан-
ная; АВ — двуокись титана анатазной формы, необработанная (вискозная);
А-01 — двуокись титана анатазной формы, обработанная соединениями алюминия
и кремния; Р-1 — двуокись титана рутильной формы, необработанная; Р-01 —
двуокись титана рутильной формы, обработанная соединениями алюминия и
кремния; Р-02 — двуокись титана рутильной формы, обработанная соединениями
алюминия, кремния и цинка.
Разогревание реагирующей массы в этих аппаратах происходит
за счет разбавления водой 20 %-го олеума.
При разложении ильменитового концентрата и титанового шла-
ка серной кислотой протекают следующие основные реакции:
ТЮ2 4- H2SO4 - TiOSO4 • Н2О,
FeO + H2SO4 = FeSO4 • H2O,
Fe2O3 + 4H2SO4 = Fe2 (SO4)3 • H2SO4 • 3H2O.
Обычно на разложение 1 m ильменитового концентрата расхо-
дуется 1,6—1,8 т серной кислоты в пересчете на 100%-ную, на раз-
ложение титанового шлака — 2,3—3,0 т [59]. Серная кислота для
разложения берется с некоторым избытком, который необходим для
обеспечения высокой степени разложения концентрата (94—96%)
и получения стабильного раствора с кислотным фактором около
0,75 (под кислотным фактором понимается мольное отношение сво-
бодной серной кислоты, не связанной титаном в виде титанилсуль-
фата, к двуокиси титана, находящейся в растворе).
Исследованию процесса разложения ильменита серной кислотой
посвящены работы [25, 137, 215, 229]. Способ разложения титановых
шлаков серной кислотой описан в [322].
Образовавшиеся сульфаты титана, железа и др. растворяются
в воде при температуре 60—70° С. Полученный раствор с концент-
рацией 120—130 г!л ТЮ2 фильтруется для отделения от неразло-
жившихся минералов. Предварительно в нем восстанавливается
трехвалентное железо до двухвалентного железным скрапом. Чтобы
предохранить двухвалентное железо от окисления кислородом воз-
духа до трехвалентного состояния восстанавливается также часть ти-
тана (3—4 г!л ТЮ2). Основная масса железа из раствора выделяется
15*
227
при охлаждении в виде семиводного железного купороса. Кон-
центрация железа снижается при этом до 0,3—0,35 г FeO на 1 г TiO2
в растворе. Очищенный от железа раствор сульфата титана упа-
ривается до концентрации 220—250 г!л TiO2 и подвергается гид-
ролизу.
Раствор сульфата титана, получаемый в производстве двуокиси
титана, является метастабильным. Как видно из диаграммы состоя-
ния системы TiO2—SO3—Н2О (глава VII, ч. I), при установлении рав-
новесия сульфат титана в этом растворе должен гидролизоваться
с образованием гидроокиси титана и серной кислоты:
TiOSO4 + /гН2О = TiO2 • nH2O + H2SO4.
При температуре ниже 70° С гидролиз сульфата титана однако
не идет из-за высокой энергии активации. При нагревании раство-
ров до кипения (107—110° С) образуется осадок гидроокиси титана,
получившей название метатитановой кислоты.
Так как гидролиз сульфата титана протекает в сильно кислых
растворах (область устойчивости гидроокиси титана при 100° С
простирается до концентрации 40% H2SO4), в которых сульфаты
металлов, содержащиеся в технических растворах, не разлагаются,
он был использован для очистки титана от железа, марганца, ва-
надия и других примесей.
В производстве пигментной двуокиси титана гидролиз является
одной из важнейших операций. От того, как проведен гидролиз,
зависит выход и качество полученной продукции.
Впервые гидролиз сульфата титана для производства двуокиси
титана был предложен Уейтраубом [354], а затем использован для
получения титановых пигментов Росси и Бартоном [330].
В патентной литературе описано большое число различных ме-
тодов гидролиза сульфата титана, которые однако можно свести к
двум основным разновидностям.
По одним из них, к числу которых относится метод Блюменфель-
да [231], гидролиз раствора сульфата титана, предварительно упа-
ренного до концентрации 200 г/л TiO2, производится кипячением
после разбавления водой. По другим методам, например по методу
Мекленбурга [299], гидролизуются растворы сульфата титана, со-
держащие 120—150 г/л TiO2. Для ускорения процесса и получения
продуктов определенного качества применяются зародыши, пред-
ставляющие из себя специально приготовленный золь гидроокиси
титана. Наибольшее распространение в производстве титановых
пигментов получили комбинированные методы, основанные на ис-
пользовании гидролиза предварительно упаренных растворов с
использованием зародышей и разбавления [28].
По методу итальянской фирмы Мантекатини, нашедшему при-
менение в СССР, на гидролиз поступают растворы сульфата титана,
содержащие 220—240 г/л ТЮ2 (в том числе 3—4 г/л в трехвалентном
состоянии) с отношением FeO : TiO2 = 0,3—0,35 и кислотным фак-
228
тором 0,7—0,75. Раствор подогревается глухим паром до температу-
ры 60° С, после чего добавляется окись сурьмы в количестве 0,2%
от содержания в растворе TiO2 и вливается зародышевый раствор
(0,2—0,5% по весу ТЮ2). Затем раствор нагревают до температуры
кипения (107—110° С) и кипятят 1 ч. За это время гидролизуется
около 40% сульфата титана. После этого раствор разбавляют го-
рячей водой в течение 1 ч до концентрации ТЮ2 160—170 г!л и ки-
пятят еще 1 ч. К пульпе метатитановой кислоты добавляют 0,1%
(к весу TiO2) цинкового порошка, пульпу охлаждают и направля-
ют на фильтрацию и промывку.
Зародышевый раствор готовят, обычно, осаждением гидроокиси
титана из сернокислого раствора едким натром. При осаждении
соблюдается определенная скорость добавления едкого натра и тем-
пературный режим. В результате получается коллоидный раствор
гидроокиси титана с концентрацией около 20 г/л TiO2, который
может храниться, не изменяя своих свойств, в течение суток.
Анатазный зародышевый раствор готовят следующим образом.
Упаренный раствор сульфата титана с температурой 20—30° С за-
ливают в бак и разбавляют водой, температура которой 15—20° С,
до удельного веса 1,1 (1 : 5). Время разбавления 30—40 мин.
В течение 40—60 мин раствор нагревают глухим паром до 40° С
и в течение 2 ч при температуре 40—45° С нейтрализуют едким нат-
ром с концентрацией около 50 г!л до pH 3. Затем раствор подогре-
вают за 30—40 мин до 60° С. Зародыши созревают при 55—60° С
в течение 4 ч. За 1,5—2 ч раствор охлаждают до 25—30° С, после
чего он готов к употреблению. Концентрация ТЮ2 в зародышевом
растворе около 20 г/л.
Паста метатитановой кислоты перед прокаливанием подвергает-
ся солевой обработке. По рецепту фирмы Мантекатини, солевая об-
работка анатазной метатитановой кислоты производится добавле-
нием к пасте с концентрацией 250—300 г! л TiO2 окиси сурьмы
в количестве 0,1%, фосфорной кислоты 0,26—0,86% и сульфата
калия 1,0% (к весу TiO2). К рутиловой метатитановой кислоте
добавляется раствор семиводного сульфата цинка 0,5—0,8%
и рутилизующие зародыши 2% (к весу TiO2). Паста нейтрализует-
ся аммиаком до pH 3—3,5.
Для приготовления рутилизирующих зародышей в реактор за-
гружается промытая метатитановая кислота и концентрированный
раствор едкого натра. Расход едкого натра вычисляется из уравне-
ния
10 ТЮ2 • SO3 ЮН2О + 7NaOH = 2,5Na2Ti4O9 +
+ Na2SO4+ 13,5Н2О
и берется с полуторакратным избытком. Суспензию нагревают за
50—60 мин до 104° С и выдерживают при этой температуре 3—4 ч.
Массу разбавляют холодной водой (1 : 1), осадок титаната натрия
отфильтровывают и промывают водой. После этого титанат натрия
229
смешивают с водой (1 : 1) и нейтрализуют концентрированной со-
ляной кислотой при 15—20° С до pH 2,5—2,8. Суспензию за 1,0—
1,2 ч нагревают паром до 45° С и для предотвращения окисления
железа добавляют раствор треххлористого титана. Затем произво-
дится пептизация гидроокиси титана. К пасте добавляют цинковый
порошок в количестве 0,2% к весу ТЮ2 и концентрированную со-
ляную кислоту до отношения (молярного) НО : TiO2= 1,125. Пасту,
нагретую острым паром до 90° С (за 30 мин), разбавляют горячей
водой (90—95° С) 1 : 2 и нагревают при температуре 90—100° С
1 ч. Полученный зародышевый раствор имеет концентрацию TiO2
около 50 г/л.
Для кристаллизации двуокиси титана с анатазной или рутиль-
ной структурой метатитановую кислоту прокаливают при 850—
950° С в трубчатых печах, обогреваемых топочными газами. Полу-
ченный продукт размалывают и выпускают в качестве товарного
продукта. Некоторые сорта анатаза и рутила, отличающиеся повы-
шенной атмосферо- и светостойкостью, подвергаются после размола
поверхностной обработке, которая заключается в смешении двуоки-
си титана с растворами сульфатов цинка и алюминия, жидкого
стекла, соды и едкого натра.
Недостатком сернокислотного метода производства двуокиси ти-
тана в обычном его виде является образование в качестве отхода
производства большого количества гидролизной серной кислоты
(после фильтрации метатитановой кислоты) примерно следующего
состава (%):
Нг5О4 TiOSO4 FeSO4 AI2O3 MgSO4 CaSO4 Прочие сульфаты
17,5 1,2 7,5 2,1 3,0 0,2 0,9
Она бывает загрязнена также примесями хрома и ванадия, вслед-
ствие чего ее нельзя после упаривания повторно использовать для
разложения ильменита.
Для утилизации гидролизной серной кислоты предложено мно-
го различных способов: термическое разложение в смеси с огарком
и углистым колчеданом [114], нейтрализация аммиаком с целью
получения железо-окисных пигментов и сульфата аммония [249],
упаривание до концентрации 55% H2SO4 и использование после
отделения осадка сульфата железа для производства суперфосфата
и др. Однако на большинстве заводов гидролизная серная кислота
не утилизируется, а нейтрализуется и сливается в водоемы или спу-
скается в моря.
Другим недостатком производства двуокиси титана по серно-
кислотному методу является большой выход сульфата железа в
виде побочного продукта, который до настоящего времени не нахо-
дит эффективного использования.
Однако несмотря на эти недостатки, которые, по-видимому,
можно устранить в результате усовершенствования процесса (пе-
реход на использование вместо ильменита титановых шлаков, при-
230
менение для очистки титана от примесей высаливания двойного
сульфата титанила и аммония), сернокислотный метод позволяет
производить продукцию по невысокой себестоимости.
Примерный расход сырья, материалов, электроэнергии и пр.
при производстве двуокиси титана следующий (в виде побочных
продуктов получаются 8—10 т гидролизной серной кислоты и 3 tn
железного купороса на 1 т двуокиси титана):
Ильменит с содержанием 49% ТЮ2, т 2,4
Серная кислота 92,5%-ная техническая, т 4,2
Серная кислота 92,5%-ная аккумуляторная, т 0,26
Сернокислый алюминий, кг 50
Сернокислый цинк, кг 37
Жидкое стекло, кг 13
Сернокислый калий, кг 9
Сода, кг 31
Едкий натр, кг 65
Трехокись сурьмы, кг 5
Соляная кислота 30%-ная, кг 100
Аммиачная вода 25%-ная, кг 70
Цинк порошкообразный, кг 5
Чугунные опилки, т 0,16
Древесная мука, т 0,11
Электроэнергия, кеч 1360
Пар 5—10 атм, мгк 4,0
Природный газ, л/3 950
Вода неочищенная, м3 310
Вода очищенная, м3 37
ХЛОРНЫЙ МЕТОД ПРОИЗВОДСТВА ТИТАНОВЫХ ПИГМЕНТОВ
Исходным полуфабрикатом для производства пигментной дву-
окиси титана хлорным методом служит четыреххлористый титан, из
которого двуокись титана можно получать методом гидролиза или
сжиганием кислородом при высокой температуре. Четыреххлори-
стый титан гидролизуется при нагревании водных растворов и в га-
зовой фазе под действием паров воды.
Промышленные методы производства титановых пигментов ме-
тодом гидролиза еще не разработаны. Различные схемы проведения
процесса описаны только в патентных изданиях. Общим недостат-
ком их является неудовлетворительное решение проблемы исполь-
зования образующихся при гидролизе хлористого водорода или
соляной кислоты, которые не удается вернуть для повторного ис-
пользования в производстве двуокиси титана.
На воздухе четыреххлористый титан, по данным рентгеногра-
фических, ИК-спектроскопических и термографических исследо-
ваний [243], с парами воды образует нестойкие соли состава
Ti (ОН)ХС14_Х, разлагающиеся в последующем до гидроокиси тита-
на с выделением соляной кислоты.
Л. Н. Шегров [218] изучил гидролиз тетрахлорида титана при на-
гревании смеси его с небольшим количеством воды при температуре
231
20—150° С.. Если гидролиз ведется при молярном отношении
Н2О : TiCl4 = 1 : 1, то получаются осадки ждл то pg „цвета перемен-
ного состава.^Нагревание их Приводит к выделению в газовую
фазу паров НО и TiCl4. При большем расходе воды (отношение
Н2О : TiCl4 =1:2 или 1 : 4) образуются оксихлориды титана
и выделяется соляная кислота.
Гидролиз четыреххлористого титана парами воды при температу-
ре 25—75° С на воздухе приводит к образованию вязкой жидкости,
затвердевающей при длительном стоянии в белую массу. Парофаз-
ный гидролиз при 1000—1200° С позволяет получать двуокись ти-
тана с пигментными свойствами, поэтому его можно использовать
при условии нахождения эффективного метода утилизации хлори-.
сто го водорода.
Сжигание четыреххлористого титана кислородом протекает по
реакции
TiCl4 + О2 - TiO2 + 2CL.
Выделяющийся хлор может быть возвращен в производство для по-
лучения четыреххлористого титана, и поэтому метод сжигания уже
нашел применение в производстве. Использование хлорного метода
получения титановых пигментов в промышленности сдерживается
ограниченностью природных запасов рутила, служащего основным
эффективным сырьем для получения четыреххлористого титана.
Важнейшей операцией в производстве двуокиси титана хлор-
ным методом является сжигание тетрахлорида титана. Оно произ-
водится при температуре 900—1200° С [5—7, 99, 115, 116, 250] на
горелках специальной конструкции, обеспечивающих поддержание
температуры реакции в заданных пределах и выдерживание про-
дуктов сгорания в течение определенного времени [38].
Для получения продукта с высокой дисперсностью пребывание
двуокиси титана в зоне высоких температур не должно превышать
0,01—5 сек [99]. В зависимости от условия сжигания тетрахлорида
титана получаемая двуокись титана имеет структуру рутила или
анатаза. Опыты японских исследователей показали [113], что сме-
шение исходных реагентов при температуре 400°C приводит к обра-
зованию анатаза с размером частиц 0,5—1,0 мкм. Предварительное
нагревание их до 1000° С при сжигании дает продукт с содержанием
рутила до 60%. Добавка к четыреххлористому титану хлористого
алюминия в количестве 1—5% [99] уменьшает фотохимическую ак-
тивность двуокиси титана.
В английском патенте [9] для снижения фотоактивности двуокиси
титана предлагается добавлять к четыреххлористому титану перед
его сжиганием четыреххлористый кремний. Двуокись титана с раз-
мером частиц 0,2—0,4 мкм получается сжиганием смеси TiCl4 и
SiCl4 при 900—1050° С. Добавка четыреххлористого кремния к
четыреххлористому титану составляет 0,5—4,0%. Полученный пос-
ле сжигания продукт прокаливается дополнительно 0,2—2,0 ч при
1100—1300° с.
232
Примесь четыреххлористого кремния к четыреххлористому ти-
тану используется для регулирования размеров частиц двуокиси
титана, получаемой сжиганием [213]. Примесь хлористого алюми-
ния ускоряет переход анатаза в рутил. Содержание рутила в го-
товом продукте зависит от концентрации зародышевых кристаллов,
возникающих на первой стадии реакции. Энергия активации горе-
ния четыреххлористого титана колеблется в пределах 22—
26,5 ккал!моль.
Титановые пигменты, получаемые при сжигании четыреххло-
ристого титана, содержат до 0,6% О [57]. Водная суспензия такого
продукта имеет pH < 7, и он не пригоден для приготовления кра-
сок. Десорбцию хлора из пигмента рекомендуется проводить про-
каливанием его при температуре 300—900° С [57, 99]. Содержание
хлора при этом понижается до 0,1%. Такой продукт имеет pH вод-
ной вытяжки 5—6,8 и пригоден для изготовления красок и эмалей.
Пигментная двуокись титана, полученная хлорным методом, для
повышения физико-технических свойств нуждается, как и при сер-
нокислотном методе производства, в поверхностной обработке со-
единениями кремния и алюминия [74].
На действующем заводе в г. Танне (Эльзас, Франция) принята
следующая схема получения пигментной двуокиси титана хлорным
методом [63]. Четыреххлористый титан, получаемый из рутила, очи-
щается от примесей ректификационной перегонкой. Ванадий по-
глощается активированным углем и удаляется с кубовым остатком.
Тетрахлорид титана сжигается при температуре 900° С. Пылевид-
ная двуокись титана осаждается в электрофильтре.
Хлор из отходящих газов улавливается с помощью однохлори-
стой серы по реакции
S2C12 + С12 = 2SC12
при температуре 220° С под давлением 10 атм в присутствии при-
меси иода (0,001%) как катализатора. В последующем двуххло-
ристая сера разлагается:
2 SC12 = S2C12 + Cl2
с выделением хлора, который возвращается на хлорирование ру-
тила.
Совершенствование методов производства
двуокиси титана
Научные исследования в области производства двуокиси титана
направлены на усовершенствование сернокислотного метода и раз-
работку других методов производства, которые могли бы базиро-
ваться на использовании рутила, лейкоксена, перовскита, аризони-
та и сфена. ч.
Канадская фирма Континенталь Тайтениум [251 ] разрабатывает
метод производства двуокиси титана, основанный на двухстадий-
ном разложении ильменита нагреванием с серной кислотой под
233
давлением и с применением гидролиза сульфата титана также при
нагревании растворов под давлением.
В работе И. С. Рассудовой с соавторами [168] и в канадском
патенте [100] описаны методы получения двуокиси титана с рутиль-
ной структурой из растворов сульфата титана путем введения перед
гидролизом некоторых добавок. Разработан метод получения дву-
окиси титана гидролизом двойного сульфата титанила и аммония
(NH4)2TiO (SO4)2 • Н2О [330], который позволяет перерабатывать
титановые концентраты, содержащие примеси ниобия, хрома и др.
с одновременной утилизацией гидролизной серной кислоты.
Предложены щелочные методы для разложения рутилизирован-
ных руд: спекание концентратов с содой при 700—1000° С [210],
сплавление с едким натром при 350—550° С [148], нагревание с
раствором едкого натра под давлением (концентрация NaOH
40—50%, Т : Ж = 1 : 2, температура 650° С) [27]. Образующиеся
при щелочной обработке титанаты натрия легко разлагаются сер-
ной кислотой.
В канадском патенте [101 ] описан циклический метод получения
двуокиси титана, по которому ильменит выщелачивают соляной кис-
лотой при температуре около 70° С. Остаток, обогащенный дву-
окисью титана, хлорируют с углем и полученный четыреххлористый
титан гидролизуют в водном растворе. Хлорное железо, накопленное
в первом растворе и полученное при хлорировании, объединяют,
восстанавливают металлическим железом и подвергают электроли-
зу с целью получения металла. Соляная кислота и хлор после гид-
ролиза возвращаются в цикл.
Предпринимаются попытки разработать новые методы производ-
ства технической двуокиси титана, предназначенной для произ-
водства тетрахлорида титана. Разработан сернокислотный метод
получения технической двуокиси титана типа искусственного ру-
тила из ильменитового концентрата Самотканского месторождения
[47]. Метод позволяет получать продукт с содержанием 97—98%
TiO2 и малым количеством примесей ванадия, хрома и алюминия.
Продукт можно эффективно использовать взамен природного ру-
тила и титанового шлака при производстве четыреххлористого ти-
тана. В работах [17, 18] описаны опыты по получению техни-
ческой двуокиси титана обработкой ильменита соляной кислотой
при атмосферном давлении и при нагревании до температуры 250—
270° С под давлением 60—70 атм.
Б. Н. Мелентьев с соавторами [92] предложил метод получения
технической двуокиси титана из перовскитового концентрата,
основанный на автоклавном разложении его азотной кислотой. Ме-
тод позволяет комплексно использовать полезные составляющие
перовскита с выпуском в качестве побочного продукта азотного
минерального удобрения.
Разработан метод получения технической двуокиси титана из
лейкоксеновых концентратов с содержанием 80—85% TiO2 обра-
234
боткой их едким натром под давлением [212] и спеканием с содой
при температуре 1000—1100° С с последующим выщелачиванием
спека водным раствором соды и разбавленной соляной кислотой
[63, 64]. Предложен метод получения технической двуокиси
титана сплавлением ильменита с сульфатом натрия и углем [214].
Проделаны опыты по переработке сфена сплавлением с кремне-
фтористым натрием [124], хлорированием [39] и разложением сер-
ной кислотой [45].
Иозеф и Метью [286] разработали способ получения четыреххло-
ристого титана хлорированием карбидизированного ильменита.
Обожженный на воздухе ильменит спекают при температуре 1000° С
с углем, измельчают, смешивают с небольшим количеством NaCl и
иода и хлорируют. Образовавшийся четыреххлористый титан отго-
няют при температуре 200—250° С, а в остатке получают карбид
железа, используемый как технический продукт.
В патентах [98, 150] описаны новые методы приготовления ти-
тановых зародышевых растворов. Зародышевый материал для про-
изводства рутила предлагается получать прокаливанием двойного
сульфата титанила и аммония, образующегося из четыреххлористого
титана и сульфата аммония, при температуре 900° С 1—2 «/[152].
IO. Д. Долматов [66] изучил сорбцию ионов Сг3+ из технических
растворов сульфата титана на различных материалах с целью раз-
работки метода очистки титана от примеси хрома. Сорбенты добав-
лялись в количестве 0,25 г на 100 мл раствора сульфата титана с
концентрацией 120 г/л ТЮ2 и перемешивались в течение 10 мин.
Затем определяли остаточную концентрацию хрома в растворе.
Заметные количества хрома адсорбировались на окиси алюминия,
свежеосажденных СаСО3 и SrCO3, катионитах КУ-1 и КУ-2. Сили-
кагель и двуокись титана ионы Сг3+ в заметных количествах из
сернокислых растворов не адсорбируют.
В патенте [95] отмечается повышение степени гидролиза .суль-
фата титана в разбавленных растворах при добавке примесей нио-
бия и тантала в количествах 0,2—2,0%.
Таки и Кунитоми [346] получили двуокись титана в виде пуши-
стого порошка, предназначенного для синтеза драгоценных камней.
Синтез двуокиси титана проводили нагреванием гидроокиси титана,
полученной обработкой четыреххлористого титана серной кислотой,
при 600° С и прокаливанием смеси двуххлористого титана и суль-
фата аммония при 850° С.
В канадском патенте [97] описан способ синтеза двуокиси титана
с размером частиц менее 0,5 мкм, имеющей структуру рутила. Про-
дукт получается прокаливанием двойного сульфата титанила и
аммония при температуре 850—1100° С в течение 1—2 ч до полного
удаления аммонийных и сульфатных групп.
Н. Ш. Сафиуллин и Э. К. Беляев [195—197] разработали метод
очистки измененных хромсодержащих ильменитов от примеси хро-
ма спеканием с карбонатом натрия. В результате окислительного
235
обжига самотканских ильменитовых концентратов с добавкой 30—
35% Na2CO3 при температуре 900—950° С в течение 2—2,5 ч зна-
чительная часть трехвалентного хрома, входящего в состав ильме-
нита, окисляется до шестивалентного состояния. Одновременно
получается титанат состава Na2O • Fe2O3 • 3TiO2, аналогичный ми-
нералу фрейденбергиту. При выщелачивании сплава водой примесь
хрома переходит в раствор в виде хромата натрия. Остаток от вы-
щелачивания реагирует с кислотами более энергично, чем необра-
ботанный концентрат, и может быть использован для получения
технической и пигментной двуокиси титана.
А. М. Боброва с соавторами [12] изучила коагуляцию щламов в
растворах сульфата титана после выщелачивания продуктов раз-
ложения ильменита серной кислотой. В них содержится 20—30 г!л
твердых продуктов во взвешенном состоянии. Наилучшие резуль-
таты получены при использовании в качестве коагулянтов сульфа-
нола НП-1 —смеси натриевых солей оксисульфокислот с алкиль-
ными остатками, и препарата СКО — продукта сульфатизации ку-
бовых остатков, образующихся при дистилляции фталевого альде-
гида.
Для осветления растворов сульфата титана, полученных из изме-
ненного ильменита, активными коагулянтами оказались поверх-
ностно-активные вещества и столярный клей [89].
Несмотря на обширные исследования по разработке новых ме-
тодов получения титановых пигментов, ни один из них не доведен
до промышленной стадии использования.
I
ПРОИЗВОДСТВО МЕТАЛЛИЧЕСКОГО ТИТАНА
Промышленное производство металлического титана началось
после второй мировой войны сначала в США, а затем в Англии,
Японии и ФРГ. Металлический титан производится также и в СССР.
Титан может быть получен восстановлением его из различных
соединений, однако данная задача оказалась значительно труднее,
чем это казалось вначале. При высокой температуре титан — очень
активный металл. Он легко соединяется с кислородом, азотом и
углеродом, примеси которых ухудшают его механические свойства.
Сравнительно чистый металл можно получить только при восста-
новлении его в вакууме или в атмосфере инертных газов (гелия
или аргона).
Титан обладает большим сродством к кислороду (рис. 19). Толь-
ко очень немногие металлы, например Са, Ва и А1, обладают боль-
шим сродством к кислороду, чем титан, и пригодны для восстанов-
ления его из окислов. Титан склонен также к образованию с кисло-
родом твердых растворов, в которых сродство к кислороду повы-
шается с уменьшением его содержания. Восстановление титана из
окислов металлами затрудняется еще тем, что титан с многими ме-
таллами, например с алюминием, образует сплавы.
236
Сродство титана к галоидам значительно меньше, чем к кислоро-
ду (рис. 20). Из галогенидов титан может быть восстановлен натрием,
1_______।____1____1____1----1-----1----1---1----1—‘
0 200 400 600 800 1000 1200 1400 1600 1800 2000 t,QC
Рис. 19. Изменение свободной энергии металлов к кислороду в рас-
чете на 1 моль кислорода.
калием, магнием, кальцием и другими металлами, не образующими
с титаном твердых растворов.
В литературе описаны многочисленные попытки получения ти-
тана из его соединений методами восстановления. Сделан термоди-
намический анализ восстано-
вительных реакций в системах
MgCl2 — TiCl4—Mg и NaCl—
TiCl4—Na [3081. Имеется
обзор зарубежных патентов
по получению металлического
титана и его сплавов за 1962—
1966 гг. [37 J.
Давиль и Шрётер [261 ] при
восстановлении двуокиси ти-
тана водородом смогли полу-
чит^ только закись TiO, а
Вейс [348] при температуре
900° С — восстановить дву-
окись титана до Ti3O5. При
действии металлического нат-
рия [230, 323] и магния [295,
325] двуокись титана также
восстанавливается только до
Рис. 20. Изменение свободной энергии ме-
таллов к галогенидам в расчете на 1 моль
галогена.
низших окислов.
При восстановлении рутила углеродом в электропечи полу-
чен титан со значительной примесью карбида [302—304], а при
237
восстановлении рутила кремнием — сплав титана, содержащий
3% Fe и 20,4% Si [279, 3091.
Многие исследователи пытались получить металлический титан
из его окислов восстановлением металлическим алюминием [271,
272, 328, 338, 352, 355], но при этом образовывался не чистый ме-
талл, а сплавы титана с алюминием.
Более успешными были опыты по восстановлению двуокиси ти-
тана металлическим кальцием и гидридом кальция. Так, при вос-
становлении двуокиси титана металлическим кальцием при 1000° С
в атмосфере аргона получен металл 98%-ной чистоты [2931, а при
восстановлении двуокиси титана гидридом кальция СаН2 при 1000—
1200° С — металл 95%-ной чистоты [109].
Фрейндлих и Вихра [268], восстанавливая двуокись титана гид-
ридом кальция, получили металл высокой чистоты. Реакция вос-
становления начинается при 500° С и быстро заканчивается при
800° С. Восстановление при 900° С длится всего 5 мин.
При восстановлении двуокиси титана атомарным водородом
[274] получается гидроокись титана.
Многие авторы получали титан восстановлением фтортитана-
та калия K2TiF6 металлическим калием или натрием [236, 273, 340,
350, 351]. Историческое значение этого метода состоит в том,
что, пользуясь им, Берцелиус [236] получил металлический титан
впервые.
Водород начинает реагировать с четыреххлористым титаном при
температуре 750° С [104]. Однако продуктами восстановления
являются низшие хлориды титана. В. В. Карпухин и Э. А. Королев
[88] в органоводородной плазме получили из TiCl4 треххлористый
титан с выходом 42%.
Штахлер и Бахран [339] восстанавливали TiCl4 водородом до
TiCl3 и TiCl2 при температуре 1100° С, а двуххлористый титан под-
вергали диспропорцианированию с образованием металла по реак-
ции
2TiCl2 = Ti + TiCl4.
При температуре 100—170° С TiCl4 восстанавливается до метал-
лического титана амальгамой натрия [153]. В эфирном растворе
TiCl4 восстанавливается металлическим магнием до низших хлори-
дов [334]. Металлический титан получен диспропорционированием
двубромистого титана при 500—600° С [359], а также при восста-
новлении Til2 водородом [263].
Из числа галогенидов для производства металлического титана
наибольший интерес представляет четыреххлористый титан. Впер-
вые опыты по получению металлического титана из четыреххлори-
стого титана восстановлением металлическим натрием проведены Де-
виллем [262]. Получение титана из TiCl4 восстановлением металли-
ческим натрием описано в работах [235, 280, 291, 298, 317—319,
324]. Хунжер [2801 применил натрийтермию для получения тита-
на в крупнолабораторных масштабах.
238
I
Кроль [125] предложил получать титан из TiCl4 восстановлением
металлическим кальцием. Вскоре он запатентовал метод восстанов-
ления TiCl4 металлическим магнием в атмосфере аргона [149]. Этот
метод, описанный в статьях [292, 294, 295], стал первым промышлен-
ным способом производства металлического титана.
Известны методы получения титана, основанные на металло-
термическом восстановлении его из различных соединений алюми-
нием с последующей очисткой сплавов от примеси алюминия кипя-
чением с едким натром [253] или селективным хлорированием под
действием А1С13, TiCl4 и НО [327].
Кроме металлотермии выполнено большое число исследований
с целью разработки электролитических методов получения титана
электролизом галогенидов и окислов из расплава солей [60, 82].
Однако до настоящего времени электролитические методы производ-
ства титана не нашли практического применения. Электролизом
TiCl4 не удается получить металл с малым содержанием примесей
[81 ]. В промышленности металлический титан производится только
магнийтермическим [75, 126] и натрийтермическим [61, 49, 310] ме-
тодами. Для рафинирования металла применяются иодидный и
электролитический методы.
Восстановление титана из четыреххлористого титана металли-
ческим магнием производится в герметически закрытых стальных
реакторах в атмосфере аргона или гелия [203, 294, 357, 358]. Из
реактора эвакуируют воздух и в нем создается атмосфера гелия или
аргона. Затем в реактор заливают металлический магний, подни-
мают температуру до 700—800° С и подают четыреххлористый ти-
тан. Четыреххлористый титан реагирует с магнием по реакции
Tiaras ~i~ 2М§ЖИДК = 2М§С12жидк TiTB.
Механизм этой реакции носит автокаталитический характер
[127, 181]. Образующиеся кристаллы металлического титана при
температуре реакции свариваются в губчатую массу.
В. В. Родякин и В. И. Скрыпнюк [173] различают три периода
в протекании реакции восстановления четыреххлористого титана
металлическим магнием, характеризующиеся различной величиной
энергии активации. В первый период скорость процесса лимитиру-
ется скоростью химической реакции между TiCl4 и магнием. Во вто-
рой период восстановление TiCl4 зависит от скорости подвода маг-
ния через слой образовавшегося продукта. В третий период на
восстановительный процесс существенное влияние оказывает обво-
лакивание титана вязким расплавом хлористого магния. Б. С. Тим-'
ченко и Н. Е. Мальцева [207] нашли, что оптимальная температура
магнийтермического восстановления TiCl4 находится в интервале
930—940° С. Выше 1150° С металлический титан образует сплавы
с железом.
Когда магний прореагирует и восстановление титана закончится,
образовавшийся хлористый магний из реактора сливается. Реактор
239
подключается к вакуумному насосу и из него при температуре
1020° С [175] отгоняют остаток хлористого магния и непрореаги-
ровавший металлический магний. После этого реактор охлаждают
и полученную титановую губку выгружают.
Как видно из этого описания, технология производства титана
магнийтермическим методом сравнительно простая. Однако металл
высокого качества получается только тогда, когда для его произ-
водства используются исходные материалы с малым содержанием
примесей, а в процессе восстановления и вакуумной дистилляции
не допускается соприкосновение нагретой губки с воздухом.
Таблица 22
Содержание примесей (%) в четыреххлористом титане, магнии и титановой губке
Компонент Четыреххло- ристый титан Магний Титановая губка
сырец рафинирован- ный из рафиниро- ванного маг- ния из магния- сырца
О 0,00243 0,0043 0,0044 0,048 0,045
N - — 0,0012 0,0016 0,0186 0,0176
Fe 0,00304 0,0350 0,0250 0,050 0,083
Si 0,00314 0,0080 0,0070 0,029 0,037
Al 0,0010 — — —
V 0,00394 —- — * * --
Ni ' 1 0,0010 0,0010 11
С ' — — 0,0150 0,0174
COC12 менее 0,021 1 11 — —
Cl — — 0,091 0,091
В условиях магнийтермического восстановления металлический
титан ассимилирует только часть вносимых с сырьем кислородсо-
держащих примесей [191]. Кислород и азот, наиболее существенно
влияющие на качество металла, накапливаются в титановой губке
не из исходных материалов, а в процессе ее получения. Это хоро-
шо видно из табл. 22, в которой сопоставлены химические анализы
исходного сырья и полученной из него титановой губки. Содержание
кислорода и азота в последней на порядок выше, чем в исходных
материалах.
Для снижения примеси кислорода в титане делались попытки
раскисления металла. Термодинамические расчеты показывают, что
раскислителями титана могут быть цирконий, иттрий, лантан,
гадолиний, кальций и бор [133]. Опыты по раскислению титана
гадолинием, лантаном и иттрием дали противоречивые результаты.
Введение 0,1% бора привело к снижению содержания кислорода
в металле, но увеличило его хрупкость. Присадка циркония снижает
примесь кислорода в титане. Раскисление титана углеродом дает
положительный эффект при температуре 1200° С и остаточном дав-
лении в вакууме 10~3 мм рт. ст. [194].
240
Недостатком магнийтермического метода получения титана яв-
ляется приваривание губки к стенкам реактора в результате высо-
кой температуры процесса, затрудняющей ее извлечение из ре-
актора.
Натрийтермический метод получения титана основан на восста-
новлении четыреххлористого титана по реакции
TiCl* + 4№жидк = TiTB + 4NaCl + 209,8 ккал.
В энергетическом отношении натрийтермический процесс уступает
магнийтермическому, так как расход электроэнергии на производ-
ство натрия с учетом расходных коэффициентов больше на 25%,
чем на производство необходимого для восстановления магния.
Однако восстановление четыреххлористого титана натрием начина-
ется с заметной скоростью при значительно более низкой температу-
ре (около 200° С), чем восстановление магнием, поэтому процесс
можно проводить без спекания губки с привариванием ее к стенкам
реактора.
Генри и Бекер [281] показали, что при восстановлении металли-
ческим натрием образуются две фазы: a-фаза, состоящая из раство-
ра натрия в расплавленном NaCl, и P-фаза, представляющая собой
раствор низших хлоридов в NaCl. Из термодинамических соображе-
ний можно ожидать, что TiCl4 вначале восстанавливается натрием
до Т1С13 и TiCl2. Эти продукты конденсируются в холодной части
ванны вместе с газообразным NaCl. Затем низшие хлориды титана
восстанавливаются растворенным в NaCl металлическим натрием.
Изменение свободной энергии реакции
TiCl4 + 4Na = Ti + 4NaCl
в интервале температур 200—700° С по расчетам В. А. Шубина и
В. А. Пазухина [216] выражается уравнением
AF = —221 200 + 65Т (кал/моль).
Восстановление низших хлоридов титана промежуточного со-
става натрием подчиняется закономерностям натрийтермического
восстановления TiCl3. После восстановления тетрахлорида титана
,на 50% процесс протекает, как при восстановлении TiCl2 [198].
Восстановление четыреххлористого титана металлическим нат-
рием протекает нормально при одновременной и строго регулируе-
мой подаче в реактор обоих компонентов [49]. Одинаково недопу-
стимо как накопление в реакторе избытка металлического натрия,
приводящее к развитию реакции восстановления в газовой фазе с
образованием мелкодисперсного металла, так и образование избыт-
ка четыреххлористого титана, способствующего образованию низших
хлоридов, загрязняющих губку.
Восстановление четыреххлористого титана натрием произво-
дится в стальном реакторе (рис. 21), который помещен в нагрева-
тельную печь [61]. В него заливают для начала реакции 100—200 кг
металлического натрия, после чего реактор начинают нагревать.
16 1-1669
241
Когда температура наружных стенок реактора достигает 500—
550° С, в него через патрубок в крышке начинают подавать четырех-
хлористый титан, а затем — металлический натрий. Дозировка
реагирующих веществ регулируется так, чтобы в реакторе всегда был
небольшой избыток металлического натрия. Температура в реакто-
ре по мере протекания процесса за счет теплоты реакции возрастает
от 500 до 1000° С, поэтому обогрев реактора необходим только при
пуске его в работу. Затем необходимая температура в реакторе под-
Рис. 21. Реактор для восстанов-
ления четырех хлор истого титана
металлическим натрием:
1 — реторта; 2 — водяное охлажде-
ние; 3 — крышка, 4 — ловушка на
линии стравливания инертного га-
за; 5 — зарыпка из хлористого нат-
рия; 6 — экран.
держивается только за счет теплоты
реакции и регулируется изменением
скорости подачи реагирующих веществ.
Образующаяся при восстановлении
губка не спекается в королек (в от-
личие от магнийтермического процес-
са), а имеет зернистое строение. Наи-
более высококачественная губка со-
стоит из зерен размером около 3 мм.
После окончания восстановления
расплав и губку из реактора сливают
и охлаждают и массу обрабатывают
0,5—1,0%-нойсоляной кислотой. Хло-
ристый натрий растворяется. Титано-
вую губку промывают водой и сушат
при 100—110° С [128].
Расход металлического натрия для
восстановления четыреххлористого ти-
тана определяется так называемым
коэффициентом расхода, зависящим
от растворимости натрия в расплаве
NaCl. Перешедший в раствор натрий
в количестве нескольких процентов
от общего его расхода теряется, так
как не принимает участия в процессе
восстановления.
Изучена возможность получения металлического титана вос-
становлением TiCl4 раствором металлического натрия в жидком
аммиаке при температуре —40° С [252, 296]. По этому методу ме-
таллический титан образуется наряду с азотистыми соединениями,
вероятно, аминами или аминосоединениями.
Титан высокой чистоты получается методом иодидного рафини-
рования технической губки [26, 169, 228], которое основано на тер-
мическом разложении иодидов титана при температуре выше
1200° С на металлический титан и иод. В свою очередь металличе-
ский иод при 175—550° С обладает способностью реагировать с
титаном с образованием летучих иодидов (Til4, TiI3, TiI2). В аппара-
тах иодидного рафинирования титана осуществляется перенос его
в виде летучих иодидов из зоны с температурой 300—500° С и тер-
242
мическое разложение последних на нагретой титановой проволоке
[126].
На современных промышленных установках методом иодидного
рафинирования получаются титановые прутки диаметром до 40 мм
и весом до 15 кг.
Электролитическому рафинированию подвергается бракованная
титановая губка, сплавы титана, отходы металлообработки титана
и его сплавов, низкосортная титановая губка, полученная электро-
лизом двуокиси титана. Электролитом служит хлористый натрий
Таблица 23
Допустимое содержание примесей (вес. %) в титановой губке
различных марок по техническим условиям СГУ 72-02-61
Компонент ТГ-2 ТГ-1 тг-о ТГ-00
N 6-10-2 5-10—2 3-10“2 3-Ю-2
Fe З.ЦГ"1 3-Ю-1 1,5- Ю-1 1,5.10—2
О 2-10“1 2-Ю-1 1.10~1 1 • 10“х
Si 1 • 10-1 1 • 10“х 5-10—2 5-10_2
С 6-10—2 6-10“2 5-10“2 5-10“2
С1 1,2 - IO-1 1 ю-1 8-10“2 6-Ю-2
Предел прочности, кг/мм2 60 53 46 38
Твердость по Бринелю 185 165 145 115
или смесь хлористых натрия и калия. В процессе рафинирования
титан очищается от кислорода, азота, углерода, железа и других
примесей. От примеси алюминия титан очищается плохо.
Электролитическое рафинирование титана производится в гер-
метически закрытых электролизерах в атмосфере аргона или гелия.
В патентной литературе описано значительное количество конструк-
ций электролизеров для рафинирования титана [60]. Приведем крат-
кое описание одного из них [2, 10].
Электролизер представляет собой стальную ванну, футерован-
ную огнеупорным кирпичом и обогреваемую переменным током с
помощью вспомогательных электродов. Катод — пластинчатый,
анод — вертикальный плоскопараллельный. Подлежащий рафини-
рованию анодный титан загружается в виде кусков в полый анод ив
бункера анодного материала, смонтированного по периферии ван-
ны и снабженного вакуумными шиберами и дозаторами.
Электролитическое рафинирование титана проводится с соблю-
дением следующего примерного режима [200]: электролит — экви-
мольная смесь NaCl + КО, температура электролита 800° С, анод-
ная плотность тока 0,2—0,5 а!см\ катодная плотность тока 3,0 а1см2.
Проблема использования титановых отходов и бракованной ти-
тановой губки не может быть полностью разрешена с помощью
электролитического рафинирования. Необходим поиск и других
16*
24В
методов очистки титана. Перспективным методом для рафинирова-
ния титана могут оказаться реакции переноса на основе галогени-
дов низшей валентности. Эффективное отделение титана от алюми-
ния имеет место при хлорировании титаналюминиевых сплавов че-
тыреххлористым титаном в среде твердых хлоридов щелочных и
щелочноземельных металлов [53]. Термодинамические основы раз-
деления сплавов титана и алюминия на основе хлоридов рассмотре-
ны в работе [192]. Эффективность электролитического рафинирова-
ния титаналюминиевых сплавов повышается после предваритель-
ного гидрирования их [129].
В СССР металлический титан производится по магнийтермиче-
скому методу. Технические условия на металл приведены в табл. 23.
ч •
ПРОИЗВОДСТВО ФЕРРОТИТАНА
Ферротитаном называется сплав титана с железом, содержащий
20—30% Ti. Он применяется для конечного раскисления и дега-
зации ответственных конструктивных сталей. Применение его осно-
вано на способности титана связывать кислород и азот в нераство-
ри1^ые в стали соединения (окислы и нитриды титана); титан также
дает прочный карбид с углеродом. Добавка ферротитана в хромо-
никелевые нержавеющие стали предотвращает образование карби-
дов хрома, вызывающих появление интерметаллитной коррозии.
Небольшие количества титана (0,1%) измельчают структуру стали.
Впервые ферротитан был получен Росси [329, 331, 332] нагре-
ванием ильменита с углем в тигельной электрической печи и алю-
мотермическим восстановлением ильменита. По первому методу по-
лучается сплав железа, титана и'углерода, содержащий 15—20% Ti
и 5—8% С. Алюмотермическим методом получается безуглероди-
стый ферротитан.
Ферротитан в настоящее время производится в значительных
масштабах алюмотермическим методом [132]. Калориметрические
аспекты выплавки ферротитана рассмотрены в статье [188]. Опре-
делены технические условия на ферротитан. '
ОБЛАСТИ ПРИМЕНЕНИЯ ТИТАНА И ЕГО СОЕДИНЕНИИ
Металлический титан и его сплавы используются в настоящее
время главным образом в военном и гражданском самолетостроении,
ракетостроении, для строительства морских судов и в химическом
машиностроении [48, 80, 143, 265]. Из титана был изготовлен вым-
пел, доставленный советской ракетой на Луну. Из него также воз-
двигнут величественный монумент в Москве в честь советских кос-
монавтов — покорителей космоса.
Вследствие своих исключительных свойств — легкости, корро-
зионной устойчивости и большой механической прочности —титан
244
может найти широкое применение для изготовления лопаток ком-
прессоров и последних ступеней паровых турбин, насосов для пе-
рекачки органических кислот, хлоридных растворов, влажного хло-
ра, разбавленных растворов соляной и серной кислот. Он может
служить заменителем нержавеющей стали в производстве оборудо-
вания для пищевой, винодельческой, фармацевтической и других
отраслей промышленности.
Из титана изготовляются токоведущие части электролизеров в
производстве хлора, так как они при анодной поляризации покры-
ваются окисной защитной пленкой, не проводящей ток [222]. Ти-
тан применяется для раскисления стали [267].
Таблица 24
Свойства белых пигментов
Характеристика Цинковые белила Свинцовые белила Титановые белила
Анатаз Рутил
Показатель преломления Удельный вес Укрывистость, г/см2 Маслоемкость 1 рода 1,95—2,05 5,5 100 12—14 1,94—2,09 6,4—6,8 130—200 9—12 2,49—2,56 3,84 38—40 20—25 2,62—2,90 4,20 35 17
Большая часть производимой двуокиси титана используется для
производства масляных красок и эмалей в качестве белого пигмента.
По молярно-техническим свойствам двуокись титана превосходит
ранее широко распространенные цинковые и свинцовые белила
(табл. 24).
Отличительной особенностью двуокиси титана является высокая
укрывистость, равная 30—50 г!м2. По укрывистости титановые
пигменты превосходят цинковые и свинцовые. Вследствие высокой
укрывистости расход двуокиси титана для окраски одной и той же
поверхности в 3—5 раз меньше, чем цинковых и свинцовых белил.
В лакокрасочной промышленности двуокись титана использу-
ется в форме анатаза и рутила с наполнителями (сульфат бария,
тальк, сульфат кальция и др.).
Анатаз отличается повышенной чувствительностью к действию
света. Окрашенные титановыми белилами в форме анатаза поверх-
ности подвергаются так называемому мелению — разрушению кра-
сочной пленки под действием света. Рутил фотохимически менее
активен, чем анатаз. Изготовленные на его основе покрытия не
подвергаются мелению. Вследствие меления анатазовых покрытий
они применяются преимущественно для покраски внутри помеще-
ний.
Рутил совместим со всеми видами масляных, смоляных и эфир-
целлюлозных лакокрасочных материалов и применяется для изгото-
вления белых и цветных пигментов, предназначенных для наружных
245
покрытий [226]. В полуглянцевых, матовых и грунтовочных
красках, где требуются наполнители, например масляные краски
для стен, наиболее эффективен рутил-кальциевый пигмент. Он по-
лучается совместным осаждением TiO2 и CaSO4.
Двуокись титана с размером частиц 10—60 ммкм используется для
матирования волокна искусственного шелка (целлюлозы) в массе
и на поверхности [258]. Добавка двуокиси титана составляет 2—3%.
При меньшей норме расхода двуокись титана вызывает деструкцию
целлюлозы [227, 314].
Согласно [343], в основе фотохимического действия TiO2 ле-
жит реакция
TiO2 -J- hv = Ti2O3 + О *.
Образующийся по этой реакции атомарный кислород вызывает окис-
ление целлюлозы. При содержании 2—3% TiO2 проявляется защит-
ное действие двуокиси титана.
Двуокись титана, наряду с мелом, каолином, гипсом и др. ми-
неральными пигментами, применяется при производстве белой бу-
маги [164, 230, 238, 241, 242, 297]. Добавка двуокиси титана к бу-
маге в количестве 1,5% от веса волокна повышает отражение света
на 20% [3131. Губер [278] рекомендует для более прочного удержа-
ния двуокиси титана осаждать ее на волокне методом гидролиза.
Двуокись титана является лучшим веществом для производства
матовых эмалей [247]. Она применяется также как усилитель для
силиконовых каучуков [102].
При нагревании железа с двуокисью титана до температуры
1200° С происходит титанирование железа [142]. На поверхности
металла образуются фазы Fe3Ti и TiC, что повышает твердость ти-
танированных образцов. В результате спекания двуокиси титана
с окисью хрома в молярном отношении 1 : 3 при температуре 1300° С
получен термистор [224].
Кроме белых пигментов двуокись титана используется для при-
готовления цветных красок. Получен стойкий к свету и кислотам
пигмент «Солнечный желтый С» при прокаливании смеси 4—5% NiO,
15% Sb2O5 и TiOo [259], а также окрашенные пигменты в системе
NiO—ZnO—TiO2," СоО—А12О3—TiO2, ZnO—FeO—TiO2 [288 ]
и др.
Многие соединения титана предложено использовать в качестве
катализаторов. Четыреххлористый титан является катализатором
при окислительной ароматизации карбоциклических соединений.
Например, при окислении тетралина твердым КСЮ4 температура
реакции понижается в присутствии TiCl4 с 370—380 до 200° С [3211.
Под влиянием TiCl4 происходит циклизация каучука через л-ком-
плексы [83]. Четыреххлористый титан служит катализатором при
перегруппировке фенил- и о-толлилацетенов в 4-оксиацетофенон
и 2-окси-З-метилацетофенон [255] и при реакции Фриделя — Крафт-
са [254].
246
Ванадат титана служит катализатором при получении бензо-
нитрила из бензальдегида [861 и 3-пиколина [187]. Перекисный ком-
плекс [TiO (ООН) ]+ катализирует окисление тиосульфата пере-
кисью водорода до сульфата [13]. В присутствии иона Ti4+
фосфорно- и мышьяковомолибденовые кислоты восстанавливаются
при комнатной температуре [285]. Материалы, содержащие низ-
ший галогенид титана, являются катализаторами полимеризации
пропилена [8]. Ванадиево-титановый катализатор ускоряет окисле-
ние пирена при температуре 370—450° С [65].
Двуокись титана в форме анатаза и рутила проявляет каталити-
ческие свойства в реакциях дегидратации этилового и пропилово-
го спиртов [162, 205]. Аналогичными свойствами обладают катали-
заторы на основе TiO2 и Nb2O3 [206]. В присутствии двуокиси тита-
на ускоряется окисление метиленового голубого до лейкоформы
[135]. Двуокись титана является катализатором при конденсации
уксусного альдегида с образованием СН3СН=СНСНО [277].
Четыреххлористый титан используется для синтеза пуромицина
[239, 240]. В английском патенте [3] описан метод придания огне-
стойкости тканям после пропитки их ацетохлоридом титана. Суль-
фат титана применяется для дубления кож. При обработке жела-
тиновых студней растворами титанилсульфата с основностью 75—
85% происходит сшивание белковых частиц [1].
Алкилтитанаты, в частности бутилтитанат, применяются для
термостойких покрытий [248, 344]. Циклогексил и олеилтитанаты
с мочевинными и меламиновыми смолами образуют хорошее покры-
тие с прекрасным блеском.
Соли титана, наряду с другими микроэлементами, влияют на
качество вина [208].
Частично восстановленная двуокись титана TiO2_х обладает
полупроводниковыми свойствами [114]. Полупроводники на основе
двуокиси титана отличаются большой шириной запрещенной зоны
ДЕ = 3,06 эв и потому могут работать в широком температурном
интервале окружающей среды (от —60 до +300° С).
Выпрямители на основе двуокиси титана изготовляются окисле-
нием металлического титана. Ленты очищенного и обезжиренного
металла нагреваются в атмосфере паров воды при температуре
600—850° С около 2 ч, отжигаются при 200—500° С и охлаждаются
в парах воды до 100° С, а затем на воздухе. Образующаяся при
первичной окислительной обработке пленка нестехиометрической
двуокиси титана обладает малым электрическим сопротивлением и
потому имеет слабые выпрямительные свойства. Для увеличения
электросопротивления полупроводниковой двуокиси титана про-
изводится повторное окисление ее. Оно осуществляется нагрева-
нием в атмосфере кислорода или воздуха, но лучшие результаты
дает анодное окисление [163]. В качестве электролита при анодиро-
вании используют 5%-ный раствор NaOH, растворы винной кисло-
ты и некоторых солей. Через электролит пропускается переменный
247
ток с напряжением 150 в и плотностью 0,2—1 alcM*. Пленку ди-
электрической двуокиси титана можно наносить на полупроводнико-
вую поверхность, смачивая ее титановыми ортоэфирами с последую-
щим гидролизом под действием паров воды и нагревая для тер-
мической обработки.
Полупроводниковые элементы на основе двуокиси титана вы-
держивают рабочее напряжение 11—25 в, плотность прямого тока
400—200 ма/см2 при падении напряжения 1,5 в и применяются в
радиотехнической промышленности.
Анодированный металлический титан с пленкой двуокиси тита-
на на поверхности используется для изготовления электролитиче-
ских конденсаторов.
НЕКОТОРЫЕ ДАННЫЕ ИЗ ЭКОНОМИКИ ПРОИЗВОДСТВА
И ПОТРЕБЛЕНИЯ ТИТАНА И ЕГО СОЕДИНЕНИЙ
В настоящее время промышленность производит в больших
масштабах металлический титан и его сплавы, четыреххлористый
титан, двуокись титана, ферротитан и в небольших количествах не-
которые другие соединения титана. Сырьевые ресурсы позволяют
развить производство титановой продукции в практически неогра-
Таблица 25
Производство ильменита и рутила в США
после второй мировой войны [245]
ниченных масштабах. Совет-
ский Союз располагает круп-
ными запасами ильменитовых,
аризочитовых, рутиловых, пе-
Год
Ильменит,
тыс. т
Рутил, тыс.
т
1947 304,9 4,68
1951 463,4 9,91
1955 520,0 8,33
1956 667,1 10,90
1957 710,3 9,66
1961 *709,8 8,20
1962 725,8 7,60
ровскиювых исфеновых руд.
Основными производителя-
ми титанового сырья в капи-
талистическом мире являются
США, Канада, Норвегия, Япо-
ния, Индия и Австралия. Про-
изводство ильменита и рутила
в США после Второй мировой
войны значительно возросло
(табл. 25).
Другой крупный произво-
дитель титанового сырья —
Канада выплавляет титановые шлаки, используемые внутри страны
и идущие на экспорт. В 1961 г. в Канаде добыто 784 тыс. т ти-
тановой руды, которая переработана на титановые шлаки.
Мировое производство ильменита в 1962 г. составило 2086, ру-
тила — 136 тыс. т [84], а в 1965 г. уже достигло 2505 и 256 тыс. т
соответственно (табл. 26) [306].
Основным производителем рутила в капиталистических странах
является Австралия. Добыча рутила в 1961 г. составила 101 [165],
а в 1962 г.— 114 тыс. т [166].
248
Производство металлического титана в капиталистических стра-
нах, начатое в 1947 г., за десятилетие достигло 20 тыс. тв год,
однако затем наступил спад, вызванный ограничением в потреб-
лении, и в настоящее время находится на уровне 8—10 тыс. т в год
Таблица 26
Производство титановых концентратов (тыс. т)
Страна • 1963 г. 1964 г. 1965 г.
Ильменит
США 806 908 907
Канада (титановые шлаки) 344 494 495
Испания 51 40 32
Норвегия 242 271 282
Португалия 0,1 0,1
Финляндия 94 116 107
Индия 26 11 30
Малайя (экспорт) 149 131 124
Япония (титановые шлаки) 0,9 1,9 2,9
Цейлон 19,1 46 49
Мальгашская республика 3,7 3 6,3
Сенегал 12,2 1,9
ЮАР 28 —
ОАР 0,5 —
Австралия 207 312 470
Всего 1 1984 2336 2505
Рутил
США 10,8 7,3 5,4
Бразилия 0,3 0,3 0,3 1 о
Индия 1,9 1,9 1,3
Сенегал 0,3 ' - -
ЮАР 1,3 — —
Австралия 187 183 219
Всего 202 192 226
(табл. 27). Фирмы, производящие металлический титан, перечне
лены в табл. 28.
Таблица 27
Производство титана (тыс. т) в капиталистических странах [84, 264, 342]
Страны 1950 г. 1955 г. 1956 г. 1957 г. 1958 г. 1959 г. I960 г. 1961 г. 1962 г.
США 0,05 6,7 13,2 15,5 4,2 3,5 4,8 6,1 6,1
Англия 0,5 1,0 1,0 1,1 0,5 1,1 0,5 1,1 0,5 0,3
ФРГ —.1 ——-. 0,5 0,5 0,5 0,5 0,5
Япония 1 — 1,2 2,5 3,1 1,6 2,5 2,4 2,3 1,5
249
Таблица 28
Фирмы, производящие титан
Фирма
Местоположение завода
Производи-
тельность»
тыс. tn в год
Du Pont de Nemours г. Ньюпорт, шт. Делавер, ——
and Co, Inc. США (магнийтермиче- ский метод)
Reactiv Metals г. Аштабьюла, шт. Огайо, США (натрийтермиче- ский метод) 2,4
Titanium Metals Corp, of America г. Гендерсон, шт. Невада, США (магнийтермиче- ский метод) 6,4
Осака Тайтениум Япония —-
Того Тайтениум Япония "
Imperial Chemicl Indastril г. Уилтон, Сев. Йоркшир, Англия (натрийтерми- ческий метод) 1,54
Основным потребителем металлического титана является США.
Основные отрасли промышленности в США, потребляющие титан,
следующие (% от общего потребления):
Военное самолетостроение 60
Ракетостроение 25
Гражданское самолето- 10
строение
Химическое машино-
строение 5
В табл. 29 приведено количество потребленной титановой губки.
В СССР губчатый титан выпускается по техническим условиям
СГУ 72-02-61 марок ТГ-2, ТГ-1, ТГ-0 и ТГ-00 (см. табл. 23).
।
I
I
I
I
I
।
Таблица 29
Потребление титана в США
Год Потребление титановой губ- ки, тыс. tn Цена титано- вой губки, доллар{кг Литература
1956 1957 1958 1959 1960 1961 1962 4,7 5,1 2,3 2,9 4,6 6,3 6,5 7,6 6,05 4,95 4,0 3,5 [264] 264 264 264 264 225 [225
250
Таблица 30
Фирмы, производящие двуокись титана [250]
Фирма Местоположение завода Производительность, тыс. т в год
Titangeselschaft GmbH
Farbenfabriken Bayer AG
Pigment-Chemie GmbH
85
54
18
ФРГ
Леверкузен
Юрдинген
Гомберг на Рейне
БЕЛЬГИЯ
| Ландербругге
Derives du Titano S. A.
15
ФРАНЦИЯ
Fabrikues de Produits Chimique de Thann et de Mulhouse S. A. Страссбург-Танн 13,5
Le Produitsdu Titane S. A. Гавр 36
British Titan Products Ltd. Кале 25 (запланировано)
N. V. Titandoixydfabrick
Mantecatini
Societta Metalchimica
Meridionale Finanziaria
Breda
British Titan Products Ltd.
Laporte Titanium
ГОЛЛАНДИЯ
| Ботлен (Роттердам)
ИТАЛИЯ
Спинетто-Маренго
Апулия
АНГЛИЯ
Гринсби
Биллингам
Стел л и нгбор роу
12
27
(8-12)
86,4
20,3
30,5
Titan Со S. А.
НОРВЕГИЯ
| Фридрикштат
Pigmentos de Titanium Ltd.
ПОРТУГАЛИЯ
| Синиш
ФИНЛЯНДИЯ
Vnorikemia Оу
«Dow-Unquinesa» Union
Quimica del Norte de Espana
Cromogenia у Quinica Curtiente
Nevin S. A.
| Район Пори
ИСПАНИЯ
Бильбао
Барселона
Барселона
15
15
16
6
251
Продолжение табл. 30
Фирма Местоположение завода П роизводительность тыс. т в год
ЧЕХОСЛОВАКИЯ
Spolek pro chemiskow a hutni | Хрустов | 3
Vyrobu
Всего Европа I 447
National Lead Со (Titanium Pig-
ment Corp.)
Du Pont de Nemours and Co, Inc.
American Cyanamid Co.
Glidden Co.
New Jersey Zinc Co.
American Potach Chemical Corp.
Gadot Titania Corp.
Canadian Titanium Pigments
British Titan Products (Canada)
Ltd.
Continental Titanium Corp.
США
Сайревил, Нью-Джерси
Сант Луис, Миссури
Балтимора, Мариленд
Еджи Мур, Делавер
Нью-Джорсонвилл,
Теннеси
Калифорния
Пиней Риве, Виргини/
Саванаг, Джорджия
Ховкинс Роинт, Мэри-
ленд
Глоцисте Сити, Нью-
Джерси
Калифорния
Огайо
КАНАДА
Квебек
»
Квебек
Всего Северная Америка
АРГЕНТИНА
«Titanit» Cia. Industrial de Pig-
mentos у Afines, A. S.
American Gianamid
(название фирмы еще не известно)
Пилар (близ Буэнос-Ай-
реса)
Cia. Petroquimica Brasilleira
Cia. Quimica CiL
Du Pont do Brasil S. A;
БРАЗИЛИЯ
Сан-Паоло
Эспир ито-Санто
МЕКСИКА
Pigmentos у Productos Quimicos
S. A.
* Строительство не закончено.
(167) 274
(Ю7)
(85) 192
(55)
(45)
(7)
(18) 81
(63)
51
43
23
36
22,5
18
6,5*
747
(Ь2)
(Ю)
I
Проект
4,5
Проект
5,5
252
Продолжение табл. 30
Фирма Местоположение завода Производительность, тыс. т в год
Travancoro Titanium Products
Ltd.
Botanium Ltb.
Fuji Titan Co Ltd.
Furukawa Mining Co Ltd.
Ishihara Sangyo Kaisha Ltd.
Mitsui Mining Co Ltd.
Sakoi Chemikal Industry Co Ltd.
Teikoku Kako Co Ltd.
Titan Kagyo Co Ltd.
индия
Карела
Бомбей
(запланировано)
Япония
Хиратеука-Кобэ
Осака
Йокаси
Хиби
Осака
Окайяма
Ямачиси
АВСТРАЛИЯ
Australian Titan Products
Laporte Titanium (Australia) Ltd.
Тасмания
южно-американская республика
South African Titan Products
(Pty.) Ltd.
Всего прочие страны
Мировая производительность
8
4,5
7,8
9,6
36
0,6
3
9,6
7,2
0,012
0,010
106,3
1300
Умбогинтвини
Производство четыреххлористого титана в США составило
26,6 тыс. т в 1961 г. и 28,8 тыс. т в 1962 г. [225].
Двуокись титана является более массовым продуктом титановой
промышленности, чем металлический титан. Мировое производство
двуокиси титана в 1962 г. (без СССР) превысило 1 млн. т. Произ-
водственные мощности по двуокиси титана в 1965 г. составляли
1,7млн. т в год [257], в том числе в США 725 тыс. т в год [311 ].
Перечень зарубежных фирм, производящих двуокись титана, при-
веден в табл. 30.
Основным производителем двуокиси титана ком мире являются США: в капиталистичес-
1955 г. 1956 г. 1957 г. 1958 г. 1959 г.
Производство ТЮ2, 409 тыс. т 478 457 404 506
Стоимость титановых пигментов в США в (в долларах за 1 т) [246]: Рутил-кальциевый пигмент 30%-ный Рутил-кальциевый пигмент 50% -ный , Двуокись титана (анатаз) Двуокись титана (рутил) 1958 г. 210 320 596 618 составляла ч
253
Структура потребления двуокиси титана США следующая (% от
общего потребления):
Пигменты, эмали, лаки
Бумажное производство
Производство каучука
Производство линолеума
Кожевенное производство
Текстильное производство
Замазки для сварочных электродов
Прочее
75
10
2
2
2
1,5
1,5
6
Двуокись титана производится преимущественно по сернокис-
лотному методу. Получение двуокиси титана сжиганием четырех-
хлористого титана может базироваться только на использовании
природного рутила, запасы которого невелики, поэтому оно не по-
лучило развития, хотя некоторые иностранные фирмы к созданию
хлорного метода производства двуокиси титана проявляют некото-
рый интерес. По хлорному методу двуокись титана производит фир-
ма de Thann et du Mulhouse во Франции (завод производительностью
18 тыс. т [250]) и английская фирма British Titanium Products
Ltd. (завод производительностью 25 тыс. т в г. Кале, Франция
[250]). Лицензию на производство двуокиси титана хлорным мето-
дом приобрела фирма Cabot Titania Corp. (США) [250]. По хлорному
методу предполагает создать производство двуокиси титана фирма
American Potash Chemical Corp, в Лос-Анжелосе [250].
Производство двуокиси титана в крупных масштабах создается
в СССР. В основу его положен сернокислотный метод.
* *
♦
Химия титана за последние 20—25 лет развилась в обширную
область знаний. В мировой научной литературе накоплен огромный
фактический материал, который не мог быть освещен полностью в
настоящей монографии, и выход ее в свет не исключает необходи-
мости с ознакомлением с первоисточниками и другими обзорными
работами.
Из приведенных в этой книге данных следует, что еще не все
проблемы химии титана разработаны в достаточной мере. Особен-
но слабо освещены в научных работах состояние титана в водных
и неводных растворах и характер равновесий в гомогенных фазах,
особенно в концентрированных растворах и в газообразных смесях
при температуре выше 1000° С. Не приобрели еще соответствующего
размаха работы по синтезу неорганических соединений титана, изу-
чению закономерностей их образования, химического и кристалли-
ческого строения. Недостаточно изучены методы синтеза и свойства
полимерных неорганических й элементоорганических соединений
титана.
254
Все эти проблемы представляют в настоящее время наибольший
интерес для исследований в области химии титана. При их прора-
ботке следует ожидать получение новых интересных результатов,
имеющих не только частное, но и общехимическое значение.
Литература
1. Арбузов Г. А., Старосельский П. И., Кузнецова 3. И.—
Научн. труды Моск, технолог, ин-та легкой пром., 1955, 5, 41.
2. Ануфриева Н. И., Гопиенко В. Г., Христюк Г. П., Ива-
нов А. И., Светличный Б. Н.— Труды ВАМИ, 1963, 50, 167.
3. Англ. пат. № 698742 от 21.10.1953.
4. Англ. пат. № 866771 от 3.05.61; РЖХим., 1962, ЗК74.
5. Англ. пат. № 686570 от 28.01.1956; РЖХим., 1955, 7849П.
6. Англ. пат. № 707560 от 21.04.1954; РЖХим., 1956, 2391П.
7. Англ. пат. № 698159 от 7.10.1953; РЖХим., 1955, 2733П.
8. Англ. пат. № 1014322; РЖХим., 1966, 23Л197.
9. Англ. пат. № 734448; РЖХим., 1957, 17368П.
10. Авторское свидетельство СССР № 138746; Бюлл. изобр., 1961, 11, 51.
11. Безворитный В. А., Безукладников А. Б.— Цветные ме-
таллы, 1967, 9, 72.
12. Б о б р о в а А. М., Соколова Л. А., Карпенко Р. Н.— Лако-
красочные материалы и их применение, 1962, 3, 45.
13. Б а б к о А. К., Литвиненко В. А.— Укр. хим. ж., 1963, 29, 618.
14. Бардин И. П., Резниченко В. А.— Металлургия СССР, 1. Изд-
во АН СССР, М., 1958, 583.
15. Бардин И. П., Резниченко В. А.— В кн.: Титан и его сплавы,
2. Изд-во АН СССР, М., 1959, 23.
16. Белянкин Д. С., Лапин В. В.— ДАН СССР, 1951, 80, 421.
17. Б е л я к о в а Е. П., Двернякова А. А.— Укр. хим. ж., 1963, 29,
220.
18. Б е л я к о в а Е. П., Двернякова А. А.— Укр. хим. ж., 1964, 30,
880.
19. Белякова Е. П., Горощенко Я. Г., Зосимович Д. Н.,
Антонов С. П., Двернякова А. А., Кладницкая К. Б.,
Широкова Г. А. Научно-техническая конференция по обсуждению
исследовательских работ в области производства пигментной двуокиси титана
сернокислотным методом. Сумское обл. изд-во, Сумы, 1968, 6.
20. Б и р ю к Л. И., Горощенко Я. Г., Калиниченко Н. В.,
Хандрос Э. Л.— Укр. хим. ж., 1971, 37, 1063.
21. Бирюк Л. И., Горощенко Я. Г., Калиниченко Н. В.—
Укр. хим. ж., 1971, 37.
22. Б о б ы р е н к о Ю. Я., Ш е й н к м а н А. И., Долматов Ю. Д.—
Ж- прикл. хим., 1967, 41, 716.
23. Бурксер Е. С., Александров Г. П.— Редкие металлы, 1933,
2, 14.
24. Беленький Е. Ф., Р и с к и н И. В. Химия и технология пигментов.
Госхимиздат, Л., 1960.
25. Богуславский И. Н., Богуславская Б. И.— Лакокрасоч-.
ная индустрия, 1934, 3, 20.
26. Баташев В. И., Резниченко В. А.— В кн.: Титан и его сплавы,
8. Изд-во АН СССР, М., 1962, 167.
27. Б е к к е р м а н Л. И.— Отраслевое совещание по проблеме производства
двуокиси титана. Тезисы докладов. Изд-во «Транспорт», Челябинск, 1964, 39.
28. Бородина М. Л., Гомозова В. Г., Михайлова Ю. В., 3 о-
лотухина Л. Н.— Лакокрасочные материалы и их применение, 1961,
4, 16.
255
29. Безукладников А. Б.— Ж- прикл. хим., 1962, 35, 2380.
30. Безукладников А. Б.— Ж- прикл. хим., 1967, 40, 291.
31. Безукладников А. Б., Ж а г л о в В. С.— Тезисы докладов кон-
ференции по химии и технологии титана. «Наукова думка», К., 1969, 34.
32. Васютинский Н. А., Бережко А. В.— Ж- прикл. хим., 1967,
10, 2446.
33. Васютинский Н. А., Сидоренко А. П., РысьеваЮ. Н.~
Изв. АН СССР. Металлы, 1967, 6, 63.
34. Васютинский Н. А., Мовсесов Э. Е.— Изв. АН СССР. Металлы,
1965, 1, 82.
35. В а с ю т и н с к и й Н. А., Блинов Б. С., Сидоренко А. П.,
Денисов С. И., Распопин В. Г., Лекалова Л. И. Метал-
лургия и химия топлива. «Металлургия», М., 1968, 5.
36. Веселкова Е. А., Старикова Г. А.— Ж- прикл. хим., 1969,
42, 38.
37. Гастаулин 3. Г. Новое в производстве металлического титана. Изд-во
Центрального н.-и. ин-та информации и технико-эконом. исследований цвет-
ной металлургии, М., 1970.
38. Г а р м а т а В. А., Г у л я н и ц к и й Б. С., Крамник В. Ю., Л и п -
кес Я. М., Сер яков Г. В., Сучков А. Б., Хомяков П. П.
Металлургия титана. «Металлургия», М., 1968.
39. Горощенко Я. Г., Удэ Э. О., Карпенко О. А.— В кн.: Ти-
тан и его сплавы, 9. Изд-во АН СССР, М., 1963, 123.
40. Горощенко Я. Г., Удэ Э. О., Карпенко О. А.— В кн.: Титан
и его сплавы, 9. Изд-во АН СССР, М., 1963, 127.
41. Горощенко Я- Г., Мотов Д. Л., Трофимов Г. В., Бело-
кос ко в В. И.— Изв. Карельского и Кольского филиалов АН СССР,
1959, 4, 135.
42. Горощенко Я- Г., Степанов В. Н., Пашу та Ю. С.— Укр.
хим. ж., 1972, 38.
43. Горощенко Я* Г. Физико-химические исследования переработки
редкоземельных титанониобатов сернокислотным методом. Изд-во АН СССР,
М.—Л., 1960, 126.
44. Г о р о щ е н к о Я- Г., Майоров В. Г., Воробейчик А. И.,
Челпанов Л. Г.— В кн.: Титан и его сплавы, 9. Изд-во АН СССР, М.,
1963, 162.
45. Горощенко Я. Г., Кондратович Н. М.— В кн.: Титан и его
сплавы, 9. Изд-во АН СССР, М., 1963, 149.
46. Горощенко Я- Г.— Тезисы докладов конференции по химии и технол.
титана. «Наукова думка», К-, 1969, 5.
47. Горощенко Я. Г., Белякова Е. П. и др. Техническая двуокись
титана и ее получение сернокислотным методом. «Наукова думкд», К-, 1968,
53.
48. Г а л и ц к и й Б. А., Абелев М. М., Шварц Г. Л., Шыл-
к и н Б. Н. Титан и его сплавы в химическом машиностроении. «Машино-
строение», М., 1968.
49. Гуляницкий Б. С. Получение титана натрийтермическим способом
(обзор зарубежной литературы). Изд. ЦИИНЦветмет, М., 1959.
50. Г р а ч е в К. Я., Ремпе ль С. И.— Ж- прикл. хим., 1937, 10, 1355.
51. Глухов И. А., Шалухина Л. М., Давыдовская Р. М.—
ДАН Тадж. ССР, 1969, 12, 26.
52. Г л у х о в И. А., Шалухина Л. М., Давыдовская Р. М.,
Пулатов М. С.— Изв. АН Тадж. ССР. Отд. физ.-мат. и геол.-хим. наук,
1968, 4, 54.
53. Г о п и е н к о В. Г.— Труды ВАМИ, 1963, 51, 117.
54. Г а л и ц к и й Н. В., Гуськов В. М.— Труды ВАМИ, 1965, 54—55,
376.
55. Г а л и ц к и й Н. В., Лебедева Г. Н.— Тезисы докладов конфе-
ренции по химии и технологии титана. «Наукова думка», К-« 1969, 25.
256
56. Г а л и цк и й Н. В., Дмитриев Ю. М., Улизько Е. С.— Те-
зисы докладов конференции по химии и технологии титана. «Наукова думка»,
К., 1969, 36.
57. Галицкий Н. В., Лебедев Г. Н., Печеник Т. С.— Тезисы
докладов конференции по химии и технологии титана. «Наукова думка»,
К., 1969, 62.
58. Г а л и ц к и й Н. В., Прохорове. Т.— Коллоид, ж., 1964, 26, 163.
59. Голландский пат. № 73894 от 15.01.1954; РЖХим., 1956.
60. Г и т м а н Е. Б. Электрохимия титана в расплавленных солях. «Наукова
думка», К-, 1965.
61. Гуськов В. М., Войницкий В. И. и др.— Цветная металлургия.
Научно-техн, бюллетень, 1964, 4 (249), 32.
62. Г о д н е в И. Н., Памфилов А. В.— Ж- общ. хим., 1937, 7, 1264.
63. Дмитровский Е. Б., Резниченко В. А., Соломаха В. П.—
В кн.: Титан и его сплавы, 5. Изд-во АН СССР, М., 1961.
64. ДмитровскийЕ. Б.,Б урмистроваТ. М.,Рез ни чен коВ. А.—
В кн.: Титан и его сплавы, 8. Изд-во АН СССР, М., 1962, 14.
65. Д у п л я к и н В. К-, С е м б а е в Д. X., Суворов Б. С.— Изв.
АН Каз.ССР. Сер. хим., 1969, 65.
66. Д о л м а т о в Ю. Д.— Ж- прикл. хим., 1967, 40, 254.
67. Д о л м а т о в Ю. Д.— Ж- прикл. хим., 1969, 42, 1725.
68. Д о л м а т о в Ю. Д., Бобыренко Ю. Я., Шейкман А. И.—
Ж. прикл. хим., 1967, 40, 11.
69. Д о л м а т о в Ю. Д., Бобыренко А. И., Шейкман А. И.—
ЖВХО, 1966, 11, 351.
70. Д е н и с о в а Л. В., Седова Л. П., Сорокин И. П. — Тезисы
докладов конференции по химии и технологии титана. «Наукова думка», К.»
1969, 26.
71. Д е н и с о в С. И., О д е г о в Е. В., Шахрай И. М., Мовсе-
сов Э. Г., Печенкин В. П. Металлургия и химия титана. «Металлур-
гия». М., 1969, 51.
72. Д е н и с о в С. И., Дегтярев В. С.— Ж-' прикл. хим., 1967, 40,
767.
73. Денисов С. И., Дегтярев В. С., Сидоренко А. В.— Цвет-
ная металлургия. Бюлл. МЦМ СССР, М., 1967, 11, 36.
74. Ермолаева Т. А., Полотнюк И. А., Федотова И. М.,
Филатова В. В.— Тезисы докладов конференции по химии и технологии
титана. «Наукова думка», К-, 1969, 7.
75. Ж е м р е к У. Д. Процессы и аппараты химико-металлургической техно-
логии редких металлов. Атомиздат, М., 1965.
76. Ж Д а н о в Г. С., Русаков А. А.— Труды Ин-та кристаллографии
АН СССР, 1954, 9, 165.
77. 3 е ф и р о в А. П., Невский Б. 3.— Цветные металлы, 1957, 7, 91.
78. 3 а д о р о ж н ы й В. Г., К а ш к а р о в И. Ф., К о п а е в А. А., Т и -
ще н к о А. Г.— Цветные металлы, 1965, 8, 7.
79. Зезянов С. П., Ильичев В. И.— Изв. высш. уч. зав. Цветная
металлургия, 1966, 5, 65.
80. И т е л ь с о н Г. М., Жилкин В. Б. Титановое оборудование в произ-
водстве никеля. Мурманское книжное изд-во, Мурманск, 1963.
81. Иванов А. И., Тимофеев В. В.— Труды ВАМИ, 1965, 54—55, 361.
82. Иванов А. И., Суходский В. А. Электролитическое получение
титана. Изд. ЦИИНЦветмет, М., 1961.
83. И л ь и ч е в 3. Ф., Харламов Е. Н., Словохотова Н. А.—
ДАН СССР, 1965, 164, 581.
84. Каганович С. Я.— Цветная металлургия. Научно-техн, бюлл., 1964,
3, 36.
85. Куцев В. С., О р м о н т Б. Ф.— Ж- физ. хим., 1955, 29, 597.
86. Кагорлицкий А. Д., Суворов Б. В., Рафиксов С. Р.—
Ж. прикл. хим.. 1959, 32, 288.
17 1-1669
257
87. Коз а чек Н. Н., Горощенко Я. Г., Лыков Е. ЩПарах-
н е в и ч Л. А.— Укр. хим. ж., 1968, 34, 140.
88. Карпухин В. В., Королев Э. А.— Изв. высш. уч. зав. Цветная
металлургия, 1968, 6, 66.
89. Каменева Д. Б.— Научно-техническая конференция по обсуждению
исследовательских работ в области производства пигментной двуокиси титана
сернокислотным методом. Сумское обл. изд-во, Сумы, 1968, 5.
90. К е т о в А. Н., Печковский В. В., Колесов И. М.— Изв.
высш. уч. зав. Хим. и хим. технол., 1966, 9, 570.
91. К у р у м ч и н X. А., Я ц е н к о А. П.— Цветные металлы, 1967, 4,
72.
92. Костры к ин В. М., Мелентьев Б. Н., Резничен-
ко В. А.— В кн.: Минеральное сырье, 16. «Недра», М., 1966, 63.
93. К о м а р о в О. К-, Богатов А. Д., Баскаева О. Г.— Научные
труды. Гиредмет, 1966, 16, 117.
94. Комаров О. К-— Научные труды. Гиредмет, 1966, 16, 114.
95. Канадский пат. № 506184; РЖХим., 1956, 11307П.
96. Канадский пат. № 515417; РЖХим., 1957, 17369П.
97. Канадский пат. № 514603; РЖХим., 1957, 12366П.
98. Канадский пат. № 606111; РЖХим., 1956, 5368П.
99. Канадский пат. № 517816, 1955.
100. Канадский пат. № 506112; РЖХим., 1956, 5369П.
101. Канадский пат. № 506094; РЖХим., 76466П.
102. К алуженина К- Ф., Ермолаева Т. А.— Труды Ин-та резин,
промышл., 1957, 4, 42.
103. Лебедев Г. Н., Серебрякова А. В., Старшенко В. И.,
Г у з ь С. Ю. Авторское свидетельство №9177619, 14.02.1966; Изображения,
промышленные образцы, товарные знаки, 1966, 1, 86.
104. Лебедев Г. Н., Галицкий Н. В., Сергач Э. В.— В кн.:
Сборник трудов Всесоюзного н.-и. и проектного ин-та титана, 3. «Метал-
лургия», М., 1969, 62.
105. Лыков Е. П., Горощенко Я. Г.— Тезисы докладов конференции
по химии и технологии титана. «Наукова думка», К-, 1969 , 9.
106. Лыков Е. П., Горощенко Я- Г.— Научно-техническая конферен-
ция по обсуждению исследовательских работ в области производства
пигментной двуокиси титана сернокислотным методом. Сумское обл. изд-во,
Сумы, 1968, 12.
107. Мачкасов Е. И., Сулейманов Э. Н., Пономарев В. Д.—
Труды Ин-та металлургии и обогащения, 1966, 18, 62.
108. Мачкасов Е. И., Пономарев В. Д., Спивак Ю. М., С у -
лейманов Э. Н.— Цветная металлургия. Изд-во АН Каз.ССР, Алма-Ата,
1962, 51.
109. М е е р с о н Г. А., Кац Г. А., Хохлова А. В.— Ж- прикл. хим.,
1940, 13, 1770. >
ПО. Мороз Ю. А., Денисове. И., Ткаченко С. А., Дубниц-
к а я И. В.— Тезисы докладов конференции по химии и технологии
титана. «Наукова думка», К., 1969, 21.
111. Мороз Ю. А., Сидоренко А. П.— Цветные металлы, 1970, 2, 47.
112. Морозов И. С.— Ж. прикл. хим., 1969, 42, 757.
113. Мацумото А., Сакамото С. и др.— J* Chem. Soc. Japan. Ind.
Chem. Sec., 1967, 70, 2115.
114. M а к о в с к и й Ф. А., Усачев Е. П. Выпрямители на основе
полупроводниковой двуокиси титана. «Наука», М., 1966.
115. Мойнов С. Г., Резниченко В. А.— В кн.: Титан и его сплавы,
9. Изд-во АН СССР, М., 1963, 166.
116. Мелентьев Б. Н., Мойнов С. Г., Резниченко В. А.—
В кн.: Титан и его сплавы, 8. Изд-во СССР, М., 1962, 114.
117. М и ш е н е в В. А., К р а м н ы к В. Ю. и др.— В кн.: Титан и его
сплавы, 9. Изд-во АН СССР, М., 1963, 105.
258
I
118. Мо в се со в Э. Е., Быстрей ин М. Н., Брындин В. Г.
Денисове. И.— Металлургическая и горная промышленность, 1965,2, 48.
119. Медведев П. И., ГалинкерИ. С., Чиненко В. П.— Лако-
красочные материалы и их применение, 1968, 5, 10.
120. Макушкина Н. И., Петров Е. С., Найкина Р. М.— Изв.
СО АН СССР. Сер. хим., 1969, 4, 59.
121. Нисельсон Л. А., Третьякова К- А., Амирова С. А.,
МикляевА. Д.— Изв. АН СССР. Металлы, 1966, 1, 61.
122. Нисельсон Л. А., Третьякова К- В., Голубков Ю. В.,
Попов В. Д.— В кн.: Исследования в области хлорной металлургии
титана. «Металлургия», М., 1969, 68.
123. Никити н\ А. А., ДокшинВ. С., Корнеев Ф. И., Го-
да с с В. О.— Цветные металлы, 1963, 2, 8.
124. Новоселова А. В., Бацанова Л. В.— Изв. СО АН СССР,
1960, 8, 142.
125. Немецкий пат. № 674625, 1939.
126. Основы металлургии, 3. Металлургиздат, М., 1963, 242.
127. Огурцов С. В., Резниченко В. А.— В кн.: Титан и его сплавы,
4. Изд-во АН СССР, М., 1960, 132.
128. О л е с о в Ю. Г., Плахотников Н. А., Дрозденко В. А.
Гидрометаллургия титана. Изд-во Центр, н.-и. ин-та информации и технико-
эконом. исследований цветной металлургии, М., 1970.
129. О л е с о в Ю. Г., Устинов В. С., Рубцов А. Н., Демчен-
ко О. Я-—Ж- прикл. хим., 1969, 42, 222.
130. Полякова В. М., Чернявская Е. И.— Труды Ин-та химии,
17. Уральский филиал АН СССР, Свердловск, 1970, 155.
131. Полякова В. М., Чернявская Е. И.— Труды Ин-та химии,
17. Уральский филиал АН СССР, Свердловск, 1970, 148.
132. Плинер Л., Су ч альни ко в С. И., Рубинштейн Е. И.
Алюмотермическое производство ферросплавов и лигатур. Металлургиздат,
М., 1963.
133. Полин И. В., Максимов В. М., Дармограй В. В.— Цвет-
ные металлы, 1966, 9, 78.
134. Печенкин В. П.— Тезисы докладов конференции по химии и техно-
логии титана. «Наукова думка», К-, 1969, 33.
135. Памфилов А. В., Мазуркевич Я. С., Пахомова Э. П.—
Кин. и кат., 1969, 10, 915.
136. Памфилов А. В., Иванчева Е. Г., Трилетова К- Ф.—
Ж- прикл. хим., 1940, 13.
137. Памфилов А. В., Иванчева Е. Г., Аляева В. В., Собо-
лева И. М.— Ж- прикл. хим., 1938, 10, 631.
138. Памфилов А. В., Худякова А. С., Штандель Е. Г.—
Ж- общ. хим., 1935, 5, 605.
139. Памфилов А. В., Шихер М. Г.— Ж- общ. хим., 1937, 7, 2760.
140. Памфилов А. В., Штандель Е. Г.— Ж. прикл. хим., 1936,
9, 1781.
141. Пусько А. Г., Пономарев В. Д., Титова Г. И.—
Ж- прикл. хим., 1963, 36, 1665.
142. Парфенов В. А.— Сталь, 1954, 5, 463.
143. Пульцин Н. М. Титановые сплавы и их применение в машиностроении.'
Машгиз, М.— Л., 1962.
144. Печковский В. В., Амирова С. А., К а л и к о Б. С.—
Ж- прикл. хим., 1960, 33, 1976.
145. Подкосов Л. Г.— В кн.: Титан и его сплавы, 4. Изд-во АН СССР
М., 1960, 3.
146. П я т н о в В. И.— Научные труды. Гиредмет, 1963, 10, 58.
147. Пат. США № 2725350; С. А., 50, 4470.
148. Пат. США № 3069235; Offic. Gaz. U. S. Patent Office, 785, 3, 993.
149. Пат. США № 2205854.
17*
259
150. Пат. США № 2886415; РЖХим., 1961, 7П280.
151. Пат. США № 2842428; РЖХим., 7П281.
152. Пат. США № 2520454; С. А., 1950, 11046.
153. Пат. США № 2618550; С. А., 1953, 2116.
154. Пат. ФРГ № 1717035, 1970.
155. Пат. США № 3490927, 1970.
156. Пат. США № 2857242, 1958.
157. Пат. США № 2857243, 1958.
158. Пат. США № 2857264, 1958.
159. Пат. США № 2857265, 1958.
160. Пат. США № 3032393, 1962.
161. Пат. США № 3502503, 1970.
162. Пат. США № 3502595, 1970.
163. Пат. США № 3502552, 1970.
164. Пат. ГДР № 7386; РЖХим., 1956, 11419П.
165. Редкие металлы. Сб. рефератов № 12 за 1962 г. Гиредмет, М., 1963.
166. Редкие элементы, 22. Гиредмет, М., 1963.
167. Русаков А. А., Жданов Г. С.— ДАН СССР, 1951, 77, 411.
168. Рассудова Н. С., Ермолаева Т. А., Истомина В. Н.—
Лакокрасочные материалы и их применение, 1961, 1, 30.
169. Рейф м ан М. Б., Дмитриев В. Н., Лошкова М. А.— В кн.:
Научные труды. Гиредмет. Металлургиздат, М., 1959, 354.
170. Резников И. Л., Безукладников А. Б. и др.— В кн.: Титан
и его сплавы, 9. Изд-во АН СССР, М., 1963, 140.
171. Распопин В. Г., Денисове. И.— Тезисы докладов конференции
по химии и технологии титана. «Наукова думка», К-, 1969, 22.
172. Рубан И. Н., Копылова Е. А., Лукьянова Н. Д., Исла-
мов Р. С.— Тезисы докладов конференции по химии и технологии титана.
«Наукова думка», К-, 1969, 25.
173. Родякин В. В., Скрыпнюк В. М.— Изв. высш. уч. зав. Цвет-
ная металлургия, 1969, 5, 60.
174. Родякин В. В., Гармата В. А. и др.— Цветные металлы, 1965,
8, 64.
175. Родякин В. В., Кушкин Б. Н., Арутюнов Э. А., Пет-
ру н ь к о А. И.— Цветные металлы, 1965, 10, 67.
176. Руднева А. В.— ДАН СССР, 1959, 125, 149.
177. Руднева А. В.— В кн.: Титан и его сплавы, 2. Изд-во АН СССР, М.,
1959, 41. ,
178. Руднева А. В., Модель М. С., Малышева Т. Я.— В кн.:
Титан и его сплавы, 2. Изд-во АН СССР, М., 1959, 50.
179. Руднева А. В., Модель М. С., Малышева Т. Я.— ДАН
СССР, 1957, 115, 141.
180. Руднева А. В.— Труды V Совещания по экспериментальной и технщ
ческой минералогии и петрографии. Изд-во АН СССР, М., 1956.
181. Резниченко В. А., Огурцов С. В., Л о п а т и н Г. С., М е -
ликбекова С. А.— В кн.: Титан и его сплавы, 4. Изд-во АН £ССР,
М., 1960, 122.
182. Резниченко В. А., Рапопорт М. Б., Ткаченко В. А. Ме-
таллургия титана. Исследования электроплавки титановых шлаков. Изд-во
АН СССР, М., 1963.
183. Резниченко В. А., Соловьев В. И.— В кн.: Титан и.его спла-
вы, 8. Изд-во АН СССР, М., 1962, 22.
184. Резниченко В. А., Соловьев В. И.— В кн.: Титан и его спла-
вы, 2. Изд-во АН СССР, М., 1959, 29.
185. Резниченко В. А., Соло маха В. П.— В кн.: Титан и его спла-
вы, 5. Изд-во АН СССР, М., 1961.
186. Резниченко В. А., Воробейчик А. И., Соловьев В. И.—
Тезисы докладов конференции по химии и технологии титана. «Наукова
думка», К., 1969, 23.
260
187. Суворов Б. В., Кагарлицкий А. Д., Афанасье-
ва Т. А., К а н И. И.—Химия гетероциклических соединений, 1969,
1129.
188. С у ч а л ь н и к о в С. И., ПлинерЮ. Л.— Ж. прикл. хим.,
1969, 42, 2347.
189. Серяков Г. В., Мастерова А. П., Рязанова Н. М.—
В кн.: Сборник научных трудов, 1. Гиредмет. Металлургиздат, М., 1959,
314.
190. Серяков Г. В., Мастерова А. П., Рязанова Н. М., С е -
дых Т. С., К а ш п е р о в В. Ф.— В кн.: Научные труды, 6. Гиредмет.
— Металлургиздат, М., 1962, 16.
191. С а н д л е р Р. А., Холмовская Н. А.— Изв. АН СССР. Металлы,
1967, 6, 58—62.
192. Сандлер П. А., Подзоров Б. Н.— Изв. АН СССР. Металлы,
1967, 4, 202.
193. Стремилова Н. Н., Милько В. И. и др.— Тезисы докладов
конференции по химии и технологии титана. «Наукова думка», К-, 1969,
62.
194. Соловьев В. И., Резниченко В. А.— В кн.: Проблемы метал-
лургии титана. «Наука», М., 1967, 191.
195. Сафиуллин Н. Ш., Беляев Э. К.— Укр. хим. ж., 1968, 34,
217.
196. Сафиуллин Н. Ш., Беляев Э. К.— Хим. пром. Украины, 1967,
4, 6.
197. Сафиуллин Н. Ш., Беляев Э. К.— Хим. пром. Украины, 1966,
3, 5.
198. Савин В. Д.—Ж- физ. хим., 1968, 42, 94.
199. Сборник трудов по химической технологии минерального сырья Кольского
полуострова, 1. Изд-во АН СССР, М.— Л., 1959.
200. Суходский В. А., Гопиенко В. Г.— Труды ВАМИ, 1960, 46,
146.
201. Серебренникова А. В., Ефремкин В. В.— В кн.: Титан
и его сплавы, 2. Изд-во АН СССР, М., 1959, 78.
202. Сырокомский В. С. Титан и его соединения. Изд-во АН СССР,
Л., 1926.
203. Стрелец X. Л., Сергеев В. В., Форсблом Г. В.— Тру-
ды ВАМИ, 1958, 41.
204. Тагиров К. X., Руднева А. В., Модель М. С., Дмитров-
ский Е. Б.— В кн.: Сборник трудов Ин-та металлургии АН СССР, 1.
Изд-во АН СССР, М., 1957, 21.
205. Толстопятова А. А., Филатова Т. Н., Корытный Е. Ф.,
Баландин А. А.— Изв. АН СССР. Сер. хим., 1969, 1439.
206. Толстопятова А. А., Филатова Т. Н., Баландин А. А.—
Изв. АН СССР. Сер. хим., 1969, 1443.
207. Тимченко Б. С., Мальцева Н. Е.— Цветные металлы, 1967,
8, 72.
208. Ф р о л о в-Б а г р е е в А. М., Андреевская Е. Г.— Виноделие
и виноградарство СССР, 1955, 5, 12.
209. ФилоненкоН. Е., Кудрявцев М. В., Лавров И. В.—
ДАН СССР, 1952, 86, 561.
210. Французский пат. № 1095478; РЖХим., 1957, 23750П.
211. Ф е д о р о в а М. Н., Кривобудская К. С.— Зав. лабор., 1964,
30, 515.
212. Федорова М. Н.— В кн.: Титан и его сплавы, 9. Изд-во АН СССР,
М., 1963, 36.
213. Хара К о дз у.— Trans. Mining and Metallurg. Assoc. Kyoto, 1968, 16, 371.
214. Шарова A. K-, Деменев H. В., Фотиев А. А., Ива-
ки н A. A.—В кн.: Титан и его сплавы, 4. Изд-во АН СССР, М., 1960, 95.
215. Штандель И. Г.— Ж. общ. хим., 1936, 5, 1629.
261
216. Шубин В. А., Пазухин В. А.— Сборник научн. трудов Моск,
ин-та цветных металлов и золота, 1958, 31, 162.
217. Шалухина Л. М., Давыдовская Р. М., Емельяно-
ва Э. А.— Изв. АН Тадж.ССР. Отд. физ.-мат. и геол.-хим. наук, 1969, 2, 44.
218. Щ е г р о в Л. Н.— Труды Всесоюзн. н.-и. ин-та хим. реактивов и особо
чистых хим. веществ, 1964, 26, 160.
219. Щербаков И. Г., Грачев К. Я.— Ж- прикл. хим., 1937, 10, 607.
220. Экспресс-информация. Цветная металлургия. Изд. Всесоюзн. ин-та науч-
ной и техн, информации, М., 1967, 21, 21, 27.
221. Экспресс-информация ВИНИТИ. Цветная металлургия, 1963, VI, 24,
чреф. 86.
222. Якименко Л. М., Коханов Г. Н., Веселовская И. Е.,
Дшагацпанян Р. В.— В кн.: Титан и его сплавы, 10. Изд-во
АН СССР, М.. 1963, 167.
223. Яковлева С. В., Комаров О. К-, П я т н о в В. И.— В кн.:
Научные труды, 16. Гиредмет, М., 1966, 87.
224. A s о S., Onuki S., Nishiza wa H.— J. Electrochem. Soc. Japan,
1QK1 IQ OQO
225. Am. Metal. Market, 1963, 70.
226. Anderson W. B.— Paint and Varnish Prod., 1953, 43, 33, 39.
227. A g s t e r A.— Meili and Textilber., 1954, 35, 1209.
228. Van Arkel A. E. Reine Metalle. Berlin, 1939.
229. Askenasy P., Heise K-— Z. anorg. Chem., 1931, 196, 257.
230. Barclay E. A.— Tappi Monograph Series, 1954, 11, 36.
231. Blumenfeld J. Пат. США № 1504669, 1924; С. A., 1924, 3257.
232. Billy M.— Ann. Chim., 1921, 16, 5.
233. В о u r i о n F.— Compt. Rend., 1907, 145, 62.
234. В о u r i о n F.— Ann. Chim. Phys., 1910, 20, 547.
235. Botts E. D., Krauskopf F. C.— J. Phys. Chem., 1927, 31, 1404.
236. Berzelius J. J.— Pogg. Ann., 1825, 4, 1.
237. Barksdale J. Titanium, Second Edit. Ronald Press Comp., N. Y., 1949.
238. Brill H. C.— Tappi, 1956, 39, A15.
239. Baker B. R., Sh aub R. E.— J. Am. Chem. Soc., 1955, 77, 2396.
240. Baker B. R., Schaub R. E., Joseph J. P., Williams J. H.—
J. Am. Chem. Sos., 1955, 77, 12.
241. Bennett G. H.— Pulp and Paper Mag. Canada, 1953, 54, 107.
242. Berndt K-— Das Papier, 1953, 7, 230.
243. В e d e n B., Guillaume I., Martin M.-J.— Compt. Rend., 1970,
C270, 34.
244. Booge J., Krchma I. J., McKinney R.M. Пат. США№ 2098025
и 2098026; С. А., 1938,100; Канадские пат. № 390014 и 390015; С. А., 1940,6419.
245. Chem. Eng. News, September, 1958, 1.
246. Chem. Eng. News, oktober, 1958, 6.
247. Cecil P. S.— Ceramic Age, 1953, 62, 38, 40.
248. C h a f f i 1 d H. W.— Paint Manufact., 1957, 27, 25.
249. C i z i n s k у Z.— Chem. prumysl, 1960, 10, 231.
250. Chem. Ind., 1963, 15, 167.
251. Canad. Chem. Processing, august, 1961, 72; Am. Metal Market, 1964, 69, 14.
252. Сато, Авата и др.— Rept. Sci. Res. Inst., 1955, 31, 410.
253. Cue il 1 eron J., Pascaud C.— Compt. Rend., 1952, 235, 1220.
254. Cullinane N. M., Leyshon D. M.— J. Chem. Soc., 1954, 2942.
255. C u 1 1 i n a m e N. M., Lloyd E. T., T u d b a 1 1 J. K-— J. Chem. Soc.,
1954 3894.
256. Clobaugh W. S., Leslie R. T., Gilchrist R.— J. Res. Nat.
Bur. Stand., 1955, 55, 261.
257. Chem. Age, 1965, 94, 445.
258. D i t h m a г K.— Textil-Praxis, 1953, 8, 383.
259. Dickenson J.— Paint Int. Mag., 1956, 71, 42.
260. D e m о 1 у A.— Compt. Rend. Trav. Chim., 1849, 5, 325.
262
261. Dawihe W., Schroter K-— Z. anorg. Chem., 1937, 233, 178.
262. Deville H.- S. C.— Compt. Rend., 1855, 40, 1034.
263. Def acqz E., С о p a u x H.— Compt. Rend., 1908, 147, 65.
264. Eng. Min. J., 1962, 163, 2.
265. Eng. Min. J., 1963, 164, 120.
266. F a r u p P. Пат. США № 966815, 1910; С. A., 1910, 3015.
267. Foundry Trade J., 1953, 95, 336.
268. Freundlich W., В i c h a r a M.— Compt. Rend., 1954, 238, 1324.
269. Gmelin’s Handbuch. Titan, 1951, 41.
270. Georgers H., S c h a 1 t e r A.— Ber., 1909, 42, 3200.
271. Goldschmidt H.— Z. Elektrochem., 1898, 4, 494.
272. Goldschmidt H.— Lieb. Ann., 1898, 301, 19.
273. G 1 a t z e i E.— Ber., 1876, 9, 1829.
274. Glemser O., Hauschild U., Lutz G.— Z. anorg. Chem., 1952,
269, 93.
275. Gias son D. R., Johnson J. S., Sheppard M. A.— J. Appl.
Chem., 1969, 19, 46.
276. G i e b T. C., G r e e n v о о d N. N., Twist W.— Inorg. Nucl. Chem.,
1969, 31, 947.
277. Нодзу P. С., Кунимика С., Ока С., К у су н о сэ К.— J. Chem.
Soc. Japan. Ind. Chem. Sec., 1954, 57, 914.
278. Huber O.— Wochenbl. Papierfabr., 1960, 88, 299.
279. H e u m a n n B.— Stahl und Eisen, 1908, 28, 356.
280. Hunter M. A.— J. Am. Chem. Soc., 1910, 32, 330.
281. H e n r i с T. A., Baker D. H.— Metallurg. Soc. Conf., 1961, 8, 721.
282. Ind. Eng. Chem., 1961, 53, 23A.
283. Industr. Chem., 1954, 30, 602.
284. J e b s e n G. Норвежский пат. № 21693, 1916; С. A., 1916, 3141.
285. .Jean M.—Compt. Rend., 1955, 240, 2237.
286. Joseph P. T., Mathew P. M.— Chem. Communs, 1969, 8, 374.
287. Knoerr A. W.— Eng. Mining J., 1952, 153, 72.
288. Korirth E.— Angew. Chem., 1952, 64, 265.
289. Knickerbocker R. G., Gorski С. H., Kenworhy H., Star-
1 i p e r A. G.— Trans. AIME, 1951, 185, 785.
290. Konig T., Pfordten O.— Ber., 1889, 22, 2070.
291. Kern S.— Chem. News., 1876, 33, 57.
292. Kroll W., Schlechten A. W. et al.— Trans. Am. Electrochem.
Soc., 1947, 92, 187.
293. Kroll W.— Z. anorg. Chem., 1937, 234, 42.
294. Kroll W., Schlechten A. W.— Met. Ind. London, 1946, 69, 319.
295. Kroll W.— Trans. Am. Electrochem. Soc., 1940, 78, 35.
296. Kameyama N., Satoh S.-L, Awata M., Yamaguchi S.—
J. Sci. Res. Inst., 1955, 49, 35.
297. Lusseyran P.— Papeterie, 1950, 72, 305, 307, 309, 311.
298. Levy D., Hamburger L.— Z. anorg. Chem., 1914, 87, 209.
299. Mecklenburg W. Пат. США № 1758528, 1930; С. A., 1930, 3330.
300. Merz V.— J. prakt. Chem., 1866, 99, 159.
301. McTaggart F. К., С о u n s i J.— Sci. Ind. Res. India, 1945, 18, 5.
302. M о i s s a n H.— Ann. Chim. Phys., 1896, 9, 229.
303. M о i s s a n H.— Compt. Rend., 1895, 120, 290.
304. M о i s s a n H.— Bull. Soc. Chim., 1895, 13, 959.
305. M с В e r t у F. H. Пат. США № 2098054 и 1098055; С. А., 1938, 1011; Ка-
надские пат. № 390016 и 390017; С. А., 1940, 6414.
306. Mineral Trade Notes, 1966, 2, 54.
307. М о г i s s у W. В.— Canad. Paint and Varnish Mag., 1951, 25, 22.
308. N i e d e r k о r n I.— Din comun. ICECHIM, 1957—1962; 2; РЖХим., 1965,
126479.
309. Neumann B.— Z. Elektrochem., 1908, 14, 169.
310. N i e d e r k о r n I.—Din comun. ICECHIM, 1957—1962,2; РЖХ им., 1965,18B1.
263
311. Oil Paint and Drung. Rep., 1965, 189, 5.
312. Oppergaar Assur. Tekn. ukell., 1966, 113, 784; РЖХим., 1967, 7A10.
313. Osterr.-papir-Ztg., 1953, 59, 1517.
314. Pinte J., Rochas P.— Bull. Inst textill France, 1952, 31, 9.
315. Patel С. C., J e r e G. V.— Trans. Mettallurg. Soc. AIME, 1960, 218, 219.
316. P f о r d t e n O. F.— Lieb. Ann., 1887, 237, 202.
317. Petterson R. A.— Phys. Rev., 1925, 10, 56.
318. P о d s z u s E.— Z. anorg. Chem., 1917, 99, 123.
319. Potvin R., Farnham G. S.— Trans. Canad. Inst. Min. Metallurgy,
1946, 49, 516.
320. Pierre J.— Ann. Chim. Phys., 1847, 20, 5.
321. Patai S., Freitag N.— Bull. Res. Consil Israel, 1954, 4, 33.
322. P i e г c e W. M., Waring R. K., Fetterolf L. D., Quebec
Iron and Titanium., Corp C. A., 1949, 9482.
323. Ruff O., Brintzinger H.— Z. anorg. Chem., 1923, 129, 267.
324. Robinson F. C., Hutchins С. C.— Am. Chem. J., 1884, 6, 74.
325. Rutkowski W.— Prace inst. Min-wa huth., 1956, 8, 85.
326. Rose H.— Pogg. Ann., 1829, 15, 145.
327. Ramamurthy S.— J. Sci. Ing. Research, 1952, lie, 545.
328. Rossi A. J.— Elektrochem. Ind., 1903, 1, 523.
329. Rossi A. J.— Iron Age, 1896, 57, 354, 464.
330. Rossi A. J., Barton L. E. Пат. США № 1196030 1196031, 1916;
С. A., 1916, 2624.
331. Rossi A. J.— Stahl und Eisen, 1901, 21, 590.
332. , Rossi A. J.— Mineral Ind., 1901, 9, 715.
333. Roden C.— Chim. Ind., 1956, 75, 287.
334. Rheinboldt H., Schwenzer K-— Z. Phys. Chem., 1939, 140, 273.
335. Suzuki A., T u к u d a R.— Bull. Chem. Soc. Japan, 1969, 42, 1853.
336. S а к 1 a t w a 1 1 a B. D., Dann H. E., Marshall A. E. Пат. США
№ 2394470; С. A., 1946, 2595.
337. S t a d d a r d C., Cole S. S., E с к L. T., Davis C. W.— U. S. Bur.
Mines, Rept. Invest., 1950, 4750.
338. Stavenhagen A., Schuchard E.— Ber., 1902, 35, 909.
339. Stachler A., Bachran F.— Ber., 1911, 44, 2906.
340. Schneider E. A.— Z. anorg. Chem., 1895, 8, 81.
341. Schossberger F. V.— Ind. Eng. Chem., 1959, 51, 669.
342. Taylor I.— Metal Ind., 1956, 89, 429.
343. T r i e b e r E.— Svensk. papperstidn, 1955, 38, 185.
344. Thomas A.— Peintures, pigments., vernis, 1952, 28, 457.
345. Ту r ol его va P., Hanykyr V.— Sb. Vysoke skoly chem.-technol.
Praze, 1967, B10, 87.
346. Taki S., Kunitomi M.— J. Chem. Soc. Japan. Ind. Chem. Sec., 1954,
57, 26A.
347. V igouroux E., Arrivant G.— Bull. Soc. Chim., 1907, 1, 19.
348. Wyss R.— Ann. Chim., 1948, 3, 215.
349. Wagner R. F.— Ber., 1888, 21, 960.
350. Wohler F., Deville H.-S. C.— Lieb. Ann., 1850, 73, 34.
351. Wohler F., Deville H. S.— Ann. Chim. Phys., 1850, 29, 166.
352. Wohler F.— Lieb. Ann., 1860, 113, 24, 8.
353. Weizmann C., BlumenfeldJ. Англ. пат. № 209441, 1924; С. A.,
1924, 1884.
354. Weintraub G. Пат. США № 1014793, 1912; С. A., 1912, 671.
355. Watts О. P.— Trans. Am. Electrochem. Soc., 1905, 8, 101.
356. Watts C. W., Bell C. A.— J. Chem. Soc., 1878, 442.
357. Cook F. I., W a r t m a n.— Light Metal., 1952, 15, 267, 308, 309.
358. W a r t m a n F. L, Baker О. H., M e t t e 1 e I. K-, Horne V. E.—
J. Electrochem. Soc., 1954, 101, 507.
359. Young R. C., S c h u m b W. C.— J. Am. Chem. Soc., 1930, 52, 4233.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ К I ЧАСТИ
Анатаз 22
— параметры решетки 127
— превращение в рутил 132
Аризонит 20
— химический состав 25
Бромиды титана 293
----- диспропорционирование 294
Брукит 22
— параметры решетки 127
— превращение в рутил 136
Галогениды титана 235
Гексааквотитантрибромид 297
Гексааквотитантрииодид 304
Гексааминокобальтальгексабромтита-
нат 301
Гексааминотитантетраиодид 305
Гексахлортитанаты 289
Гидроксибромид титана 302
Гидроксихлориды титана 288
Гидролиз сульфата титана, схема 365
-------кинетика 362
Гидроперекись титана 120
Гидроокись титана 106
-----адсорбционные свойства 116
-----аморфная 113
-----коллоидные свойства 117
-----методы получения 108
-----мицеллы 365
----- немицеллярная 363
-----низшей валентности 106
-----состав ПО
-----состояние в растворах 115
----- старение 111
----- строение 112
Двойные сульфаты титана 370
-------и аммония 372
----------бария 389
----------кальция 371, 387
---------- магния 387
---------- натрия 370
----------переходных металлов 391
----------церия 390
Двойные фториды титана 241
Двубромистый титан 295
-----методы получения 295
-----реакция присоединения 296
-----физические свойства 295
Двуиодистый титан 303
Двуокись титана 126
----- диэлектрические свойства 140
----- кристаллическое строение 126
-----линейное расширение 143
-----люминесцентное свечение 138
-----магнитные свойства 140
-----методы получения 131
-----полупроводниковые свойства 141
-----растворимость в соленых распла-
вах 147
-----светопреломление 139
----- теплоемкость 143
-----теплопроводность 143
-----термическая устойчивость 146
-----точка плавления 142
-----упругость пара 143
----- физические свойства 136
-----фотохимическая активность 154
----- хемосорбция 156
-----химические свойства 145
----- электропроводность 141
-----энтальпия образования 144
----- энтропия образования 144
Двухлористый титан 249
-----взаимодействие с водой 254
-----двойные соли 252
-----реакции присоединения 249
----- способы получения 249
-----физические свойства 251
Диакводигидроксититаниодид 305
Даиммонийтитангексабромид 301
Ильменит 20
— химический состав 24
Йодиды титана 302
----- диспропорционирование 303
История открытия титана 5
Кинетический метод 357
Кнопит, см. перовскит
Коэффициент рассеяния 358
Метатитановая кислота 107
Оксибромиды титана 302
Оксибромиды титана, присоединение
фенилбромамина 302
Оксихлориды титана 288
Оксихлориды титана (III) 266
Ортотитановая кислота 107
265
Перовскит 25
— химический состав 26
Пер оксосульфаты титана 394
Пер оксотитанаты 177, 197
Пероксофтортитанаты 248
Пероксохлортитанаты 293
Радиус коллоидных частиц 363
Растворимость металлов в титане 82
Релея уравнение 360
Рутил 24
— параметры решетки 127
— свободная энергия образования 145
Светорассеяния метод 357
Система CsCl—TiCl3—НС1—Н2О 265
— НС1—K2TiCl6—H2O 290
— HC1—Cs2TiCl6—H2O 291
— HC1—(NH4)2TiCl6—H2O 291
— HC1—Rb2TiCl6—H2O 290
— KC1—TiCl3—HC1—H2O 264
— NH4C1—TiCl3HCl—H2O 264
— RbCl— TiCl3—HC1—H2O 265
— Ti—Ag 75
— Ti—Al 59
— Ti—As 73
— Ti—Au 79
— Ti—В 41
— Ti—Be 40
— Ti—Bi 81
— Ti—C 43
— TiCl4—HC1—H2O 289
— Ti—Co 69
— Ti—Cr 66
— Ti—Cu 71
— Ti—Fe 68
— TiF4—HF—H2O 239
— TiF4—KF—H2O 245
— Ti—Ga 72
— Tip-Ge 73
— Ti—H 36
— Ti—Hg 80
— Ti—In 70
— Ti—Ir 78
— Ti—Mn 67
— Ti—Ni 70
— Ti—Nb 74
— Ti—N 46
— Ti—О 48
— Ti—Os 78
— TiO2—SO3—(NH4)2SO4—H2O 384
— TiOSO4—(NH4)2SO4—H2O 387
— Ti—P 62
— Ti—Pb 80
_ Ti—Pd 75
— Ti—Pt 78
— Ti—Re 78
— Ti—Ru 75
— Ti—S 63
— Ti—Sb 71
— Ti—Se 73
Система Ti—Si 61
— Ti—Sn 70
— Ti—Те 78
— Ti—U 81
— Ti—Zn 72
Состояние титана в солянокислых
растворах 292
Сульфатизация титансодержащих ма-
териалов 396
Сульфаты титана 325
----- средний 334
----- гидролиз 353
-----схема гидролиза 364
-----(III) 325
-----двойные соли 331
-----комплекс с сульфатом титана
(IV) 328
-----методы получения 328
-----спектры поглощения 326
Сульфаты титана (IV), классификация
333
-----методы получения 333
-----описанные в литературе 332
----- полиионы 356
Сульфотитановая кислота 360
Сфен, химический состав 26
Титана атомный радиус 7
— гидроксокомплексы 358
— загорание в кислороде 15
— изотопы 7
— ингибиторы коррозии 16
— кинетика окисления 14
— классификация соединений 28
— коррозионная устойчивость 12
— коэффициенты линейного расшире-
ния 9
— магнитная восприимчивость 11
— магнитные моменты изотопов 7
— металлические соединения 35
— многокомпонентные металлические
соединения 84
— параметры решетки 8
— пассивация 16
— плотность 8
— поверхностное натяжение 11
— полиморфизм 8
— постоянная Холла 11
— распространенность в природе 11
— растворимость 84
— рудные месторождения 18
— сверхпроводящее состояние 10
— содержание в метеоритах 12
— комплексные соединения с переход-
ными металлами 366
— строение электронной оболочки 27
— температура кипения 9
----- плавления 9
— теплоемкость 9
— термоионная эмиссия 11
266
Титана упругость пара 9
— физиологическое действие 17
— золи 118
— тиохлорид 292
Титанаты 188
— актинидов 199
— алюминия 197
— бария 189
— бериллия 178
— висмута 204
— гафния 201
— - железа 206
— иттрия 198
— кадмия 188
— калия 173
— кальция 182
— кобальта 210
— лития 169
— магния 179
— марганца 205
— методы получения 168
— натрия 170
— никеля 210
— ниобия 203
— редкоземельных элементов 198
— рубидия 175
— свинца 202
— смешанные 211
— стронция 186
— хрома 205
— цезия 175
— цинка 184
— циркония 200
Титанилсульфат 335
Титанилсульфата дигидрат 338
— моногидрат 336
-----кристаллическое строение 337
Титанилсульфаты кислые 340
Титанит 26
Сфен, см. титанит
Титан коллоидно-дисперсный 357
— молекулярно-дисперсный 357
-----метод определения 358
Титаннитроиодид 305
Титановые минералы 21
Титаноксииодид 305
Титаноксииодид-т-трипиридин 305
Титанорубидиевые квасцы 332
Титаноцезиевые квасцы 332
Трехбромистый титан, аддукты с тет-
рагидрофураном 298
----- физические свойства 297
Тетраиодид титана, присоединение гид-
розина 305
-------диметилформамида 305
-------гексаметилентетрамина 305
------- пиридина 305
Трехбромистый дитан 296
-----аддукты с этилендиамином 298
Трехбромистый титан, двойные соли
298
-----методы получения 296
Трехиодистый титан 304
Трибромид титана, комплексы с нитри-
лами 297
-------- со спиртами 297
Треххлористый титан 254
----- аддукты с пиридином 258
-----взаимодействие с водой 261
-----двойные соли 259
-----кристаллогидраты 258
-----комплексы с мочевиной 258
-----присоединение органопроизвод-
ных 258
--------сернистого азота 258
-----реакции разложения 261
-----способы получения 254
-----физические свойства 258
Фототропия 137
Фториды титана 235
Фтортитанаты аммония 242
— калия 243
— лития 242
— меди 246
— натрия 242
— рубидия 247
— цезия 247
Хлорбромтитанаты 281
Хлориды титана 248
Центры мицеллообразования 120
Четырехбромистый титан 298
----- двойные соли 301 -
-----комплексы с диметилформами-
дом 301
----------диоксаном 301
---------- нитрилами 301
----------пиразином 301
---------- пиридином 301
----------серопроизводными 301
----------фенилен- бис- (диметилар-
сином) 301
---------- эфирами 301
-----методы получения 298
-----реакция с мочевиной 301
-----присоединение аммиака 300
--------гексаметилентетрамина 301
--------гидразина 300
-------- трибромида фосфора 301
-----соединения с аминами 301
-----сольват с SO2 301
----- физические свойства 293
-----присоединение фосфина 301
Четырех и од истый титан 304
-----методы получения 304
-----термическая диссоциация 305
-----физические свойства 305
Четырехфтористый титан 237
Четыреххлористый титан 266
г
267
Четыреххлористый титан, аддукты с
азотной кислотой 275
----------альдегидами 280
----------амидами кислот 282
----------аминами 280
----------анизолом 277
----------ароматическими углево-
дородами 277
----------гидразином 272
----------дифениловым эфиром 277
----------карбоновыми кислота-
ми 280
----------кетонами 280
---------- нитрилами 282
---------- пиридином 281
----------окислами серы 273
---------- органоарсинопроизвод-
ными 284
----------органопроизводными оло-
ва 283
----------органоселенопроизводны-
ми 282
----------органохлоропроизводны-
ми 284
----------сероводородом 275
----------силанами 283
----------сложными эфирами 278
----------тетрагидропираном 278
-----------тетрагидрофураном 278
----------фосфином 283
---------- хинолином 281
----------хлорзамещенными эфира-
ми 279
----------хлоридами фосфора 272
----------хлорокисью азота 274
----------хлорокисью селена 274
----------хлорокисью хрома 274
----------цианистым водородом 273
----------шиффовыми основаниями
282
----------этиловым эфиром 277
----------эфирами бензойной ки-
слоты 278
---------- эфирами и хлоридом циа-
на 279
------------- уксусной кислоты 278
-----комплексы с 4,4' -ди-(трет-
бутил)- дифенил-6,6'- сульфидом
281
Четыреххлористый титан, комплексы
с 2-диметиламинометил-4-метилфено-
лом 281
-----------1, Г -диокси-2-диметил ами-
нометилом 281
----------- дипиридилом 281
----------- индолом 281
----------- органическими основани-
ями 281
-----------пиперидином 281
----------- пиразином 281
---------2,4,6-(тридиметиламиноме-
тил)-фенолом 281
----------- тринизолом 281
----------- фенантралином 281
-----------формамидом 281
-----------2-диэтиламинометил-4~
трет-аминофенолом 281
-----------уротропином 281
------ действие этил нитрата 279
----- двойные соли 275
-----взаимодействие с аммиаком 271
-----------нитросоединениями 282
----- вязкость 268
-----ИК-спектры поглощения 267
-----магнитная восприимчивость 271
-----показатели преломления 270
-----растворимость хлоридов метал-
лов 285
-----реакции восстановления 285
-----реакции обменного разложе-
ния 286
-----природа химической связи 271
-----присоединение галогенов 275
-----очистка от примесей 267
-----спектры поглощения 271
-----способы получения 266
-----строение молекулы 267
-----теплоемкость 269
----- теплота испарения 269
-------- плавления 269
----- температура кипения 268
--------плавления 268
-----удельный вес 268
-----упругость пара 261
-----энергия диссоциации 270
----- энтропия плавления 269
Экстракция титана 366
268
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ КО II ЧАСТИ
Алкилортотитанаты, реакции с бис-
(ацето)ацетатами 38
Ал кил си л оксититанаты 61
— методы синтеза 61
Алкилтитанаты 31
Алкоксититанацилаты смешанные 57
бш?-(Алкокси)титаноксиды 52
— реакции переэтерификации 53
Алкоксититанхлориды, диспропорцио-
нирование 50
тетракис-(Аллокси)титан 36
Амиды ортотитановой кислоты 58
Амиды титана, каталитические свой-
ства 60
тегпракис-(Амилоксн)титан 35
трнс-(Амилокси)титанхлорид 48
Амилоксититантрихлорид 44
Изоамилоксититантрихлорид 44
Амилоксититантрихлорид 44
/иетро/шс-(Анилидо)титан 59
Анодное окисление титана 156
--------анодные покрытия 157
—-------состав анодного осадка 157
— растворение титана 153
Аносовит 198
— железистый 198
— магний-алюминиевый 198
— метод выделения из шлаков 198
Аносовит-титан-три 198
— химический состав 198
Аризонитовый концентрат 194
Арилтитанаты 37
Ацетилацетоксититантр ихлор ид 44
/лрнс-(Ацетилацетонато)титан 50
/и/7ш>(3-Ацетиленацетонато)титан 126
/прш>(Ацетилацетонато)титан (I II)
127
Ацетилацетоноксититантрихлорид 44
Ацетилацетоноксититантрихлорид-т-ди-
этилоксид 45
б/я>(Ацетилацетонокси)титандихлорид
46
/прнс-(Ацетилацетонокси)титанхлорид
48
бпс-(Ацетилацетонокси) титанди хлор ид,
комплексное строение 46
— Продукты присоединения 49
бис-( Ацети лацетонокси )титандихлорид,
реакция с трихлоридом железа
46
Ацетил ацетонокси -этоксититанди -
хлор-т-этанол 47
бис-(Ацетокси)титандихлорид 56
Ацетоксититантр и хлор ид 56
Ацилтитанаты, полимерные 57
Байковит 200
/п/7аш>(Бензил)титан 7
Бензилалюминийтитанпентахлорид 8
Бензилдиалюминийтитаноктахлорид 8
/прпс-(Бензоилацетонато)титан 127
/п/шс-(Бензоилметанацетонато)титан
127
Апетра/шс-(Бутиламидо)титан 59
бис- (Б у токси) диоксих и но л и н атотита н
134
/прш>(Бутокси)титан 50
тешракис-(втор-Бутокси)титан 35
теАпря/шс-(Бутокси)титан 33
тетракис-(пгрет-Бутокси)титав 35
тггпря/шс-(«-Бутокси)титан-т-аммин 34
дпрпс-(Бутокси)титанацетат 57
бис-(н -Бутокси)титан -бпс-(бензоил аце-
тон) 34
/прш>(н-Бутокси)титанбензоилацетон
34
Бутоксититан-6нс-(бортетр а гидр ид)-т-
тетрагидрофуран 34
тр//с-(Бутокси)титан-т-бутанол 50
бпс-(Бутокси)титандиацетат 57
бпс-(Бутокси)титандибромид 49
бг/с-(Бутокси)титандибромид-т-бутанол
49
бш>(Бутокси)титандихлорид 46
бш>(<?/пор-Бутокси)титандихлорид 46
бцс-(Бутокси)титандихлорид-т-бутанол
46
б//с-(Бутокси)титандихлорид-т-пипери-
дин 46
теАпра/шс-(н-Бутокси)титан, методы
синтеза 33
бпс-(н-Бутокси)титан-б//с-(2-оксиэтил-
амин) 34
трцс-(н-Бутокси)титан -2 -оксиэтиламин
34
269
н-Бутоксититана, /л/шс-(2-оксиэтил а-
мин) 34
тетракис-(трет-Бутокси)титан, реак-
ции с ацетобромидом 35
/ие/тгракцс-(Бутокси)титан, реакции с
триметилалюминием 34
-------- трихлоридом бора 34
--------циклооктатетр аеном 34
щетракис-(трет-Бутокси)титан, реак-
ции с этиленгликолем 35
/?1е/пракцс-(Бутокси)титан, согидрол из
69
Бутоксититантрихлорид 44
щрели-Бутоксититантрихлорид 44
етпор-Бутоксититантрихлорид 44
Бутоксититантрихлорид, продукты при-
соединения 44
Бутоксититантрихлорид, сольваты 44
Бутокси-трис-(тр имети леи локси)титан
64
/прпс-(Бутокси)титанхлорид 47
— аммиакаты 48
трис-(Бутокси)титандифтор ид 43
Восстановление титана и железа 216
— ильменита углеродом 197
--------термодинамический анализ
197
Восстановительный обжиг ильменита
203
Галогенамиды титана смешанные 59
Галогенэфиры, методы синтеза 41
Гексаацетоксидититаноксан 56
Гексаизобутоксидититаноксид 67
Гексаизопропоксидититаноксид 67
Г ексамети лбензол гексах лоротр ититан-
хлорид 7
Гексагексокситрититандиоксиполимер
67
Гексаметилдисилазан-гексаметилами-
дотитан 60
Гексаметилдисилазантитантрихлорид
60
/иелпракш>(н-Гексаокси)титан 35
лпрпс-(Гексаокси)титанхлорид 48
Гексаоксититантрихлорид 44
Гексаэтокситр ититандиоксидих лор ид
полимер 67
mpuc-(V ексафтор ацетил ацетонато)титан
(III) 127
Гексаэтоксидититаноксид 67
Гептабутокситрититандиоксих л ор ид ’по-
лимер 67
/пепгрп/шс-(Гептаокси)титан 36
Гептаэтокситр ититандиоксих л ор ид ’по -
лимер 67
Гейкилит 199
тетракис-(3-Уидроксибутокси)титан 36
/прш>(2-Гидроксибутокси)титанхлорид
48
бпс-(2,4-Г идроксипентокси)титанди-
хлорид 57
/ие/прокцс-(2-Гидроксипропокси)титан
36
Гидротитаналтетрахлоридбутонол 125
Гидролиз сульфата титана, доля само-
возникающих зародышей 220
--------кинетика 219
-------- коллоидная гидроокись ти-
тана 219
-------- константа гидролиза 220
--------коэффициент рассеяния 220
--------масса мицеллообразования
220
--------механизм действия зароды-
шей 222
-------- немицеллярная гидроокись
титана 219
--------объем титанового зародыша
220
--------параметры кинетики мицел-
лообразования 220
-------- пептизация гидроокиси ти-
тана 221
--------по методу Блюменфельда 228
--------------Мантекатини 228
-------- Мекленбурга 228
--------центры мицеллообразования
220
Двуокись титана, зарубежные фирмы
251
-----масштабы производства 253
-----хлорным методом 254
-----превращение анатаза в рутил 223
----- поверхностная обработка 224
-----производство из ильменита 212
----- полупроводников 247
-----сернокислотным методом 213
-----хлорным методом 231
----- прокаливание гидроокиси 223
-----снижение фотоактивности 232
-----совершенствование производства
233
-----солевая обработка 223
-----стоимость пигментов в США 253
-----структура потребления 254
-----------в США 250
-----технические условия 226
-----сорта титановых пигментов 226
-----хлорирование в расплаве солей
206
-----термодинамика хлорирования
205
Дейтерийметилтитантрихлорид 7
бис- (Ди а нти п ир и л мета н)тита нгекс ар о-
данид 132
бш>(Диацетилацетоксититанхлорид)
оксид-т-хлороформ 48
щетра/шс-(Дибензиламидо)титан 59
270
/иетря/шс-(Дибензилтиокарбаминат)
титан 60
Диантипирилметантитантетрароданид
132
/7ге/пракг/с-(Дибутиламидотитан) 59
Дибутокситрититандиоксид’полимер
67
Дигексаокситр ититанд иокси д ’пол имер
67
Дигидропиридинтитаноксотетрахлорид-
т-акво 128
Ди гидр оп и р и д и ндитита нтр иоксотетр а -
хлорид-т-триакво 128
Дигидропиридинтетратитангептаоксо-
тетрахлорид-т-пентаакво 128
Дигидропиридинтитанпентахлорид-ти-
та н а л этокси -ацетонитр ил -тетр ах лор ид
129
Дигидропиридинтитанэтоксипентахло-
рид 129
Дигидротитанал-бцс-(Ы-фураилдифе-
нил гидроксил амидо)-дисульфат 128
т/шс-(Диметиламидо)титан 60
/пе/тгра/шс-(Диметиламидо)титан 58
/пе/иро/сис-(Диметиламидо)титан-т-
бис-(никельтетракарбонил) 58
/пегпракпс-(Диметиламидо)титан, спектр
комбинационного рассеяния 58
бис-(Диметиламидо)титандиметилмало-
нат 58
Диметил амидотитантр их лорид-т-бш>
(диметиламин) 59
бш>(Диметиламин)борхлорид-т-бш>(ти-
тантрихлорид) 138
бис-(Диметиламин)бор хлорид’димер-т-
трис-(титантетрахлорид) 138
бш>(Диметиламингидро)бордихлорид-
т-дититангексахлорид 138
Диметиламиноди гидр ид -т-дититангек-
сахлорид 138
тетракис-(Л имети лдитиокар бамми н ат)
титан 61
Д имети лтитандих лор ид 7
Диметиламинтитандихлорид-т-икс-(ме-
тиламин) 59
/пе/тгря/шс-(Диметилтитанокси)титан 37
бш?-(Диметилфениламиногидро)титан-
гексахлорид 129
тетракис-Щ и нитронафтокси)титан 37
Ди п ир идингидротитанокситр их лор ид-т-
акво 128
Дипиридинтитанпентахлорид-т-мета-
нол 125
тетршшс-(Дипропилдитиокарбами-
нат)титан 61
Дититаноксигексахлорид-т-тетраацето-
нитрид 129
Дихлортитан-бцс-(бутанат) 56
Дихлортитан-бпс-(валериат) 56
Дихлортитан-бис-(дих лор ацетат) 56
Дихлортитан-бис-(изобутилат) 56
Дихлортитанметиламин-т-бис-(метила-
мин) 60
Дих лортитан -бпс-(три хлор ацетат) 56
Дихлортитанэтиламин-т-этиламин 60
Дихлорэфиры смешанные 47
Дихлортитанэфиры с производными фе-
нола 46
те/тг/ш/шс-(Дифениламидо)титан 59
Диметиламидо-бш>(диметиламидо)ти-
тан 60
пгрнс-(Диэтиламидо)титан 60
/пе/пря/шс-(Диэтиламидо)титан 59
— энтальпия образования 59
Диэтиламидотитантрихлорид-т-бцс-(ди-
этиламин) 59
трш>(Диэтиламиндигидро)титангекса-
хлорид 130
Апрш?-(Диэтиламиндигидро)дититанно-
нахлорид 130
те/пра/шс-(Диэтилдитиокарбаминат)ти-
тан 61
бцс-(Диэтилдитиокарбаминат)титан-
оксид 60
Диэтилметилсилоксититанантрихло-
рид 64
бис-(Диэтилметилтитаноксилокси)ти-
тандихлорид 64
Диэтилоксититаноксид 66
Диэтилоксидититантриоксид 66
Диэтилсурьматитанпентахлорид 66
Дициклопентадиенилтитаналюминий-
диэтил’димер 20
бпс-(Дициклопентадиенилтитанброми-
од)оксид-т-триакво 14
бис-(Дициклопентадиенилтитандирода-
нид)оксид 18
бш>(Дициклопентадиенилтитан)кар-
бонат 34
бш>(Дициклопентадиенилтитаннит-
ро)оксид-т-тетраакво 14
бис-(Дициклопентадиенилтитан-
хлор)нитрофенил 11
бпс-(Дициклопентадиенилтитан)оксид
18
бш>(Дициклопентадиенилтитан)суль-
фат 17
бш>(Дициклопентадиенилтитантри-
акво)-бнс-(тетрафенилборат) 18
Дициклопентадиенилтитанхлорид 11
Дициклопентадиенилтитанхлорид’ди-
мер 20
/пе/пря/шс-(Изоамилокси)титан, методы
получения 35
тегтгракцс-(Изобутоксититанат) 35
бш>(Изобутокси)титандихлорид 46
— аддукты 46
/п/шс-(Изобутокси)титанхлорид 15
271
/прш>(Изобутокси)триизобутилсилок-
сититан 64
Изопропиламидо-бцс-(диметиламидо)ти-
тан 60
бш>(Изопропокси)диоксихинолинато-
титан 134
те/лрякис-(Изопропокси)титан 6
трцс-(Изопропокси)титанацетат 57
/п/шс-(Изопропокси)гексаизопропил-
дисилазан 60
б«с-(Изопропокси)титан-бш>(гексаме-
тилдисилазан) 60
/прмс-(Изопропокси)титангексаметил-
дисилазан 60
Изопропоксититанборгидрид-т-тетра-
гидрофуран 33
/пе/пра/шс-(Изопропокси)титан-т-диа-
мин 33
б«с-(Изопропокси)титандиацетат 57
бш>(Изопропокси)титандибензоилаце-
тон 33
трцс-(Изопропокси)титанбензоилаце-
тон 33
бис-(Изопропокси) титандихлорид
46
б«с-(Изопропокси)титандихлорид, ад-
дукты 46
бпс-(Изопропокси)титандихлорид-т-изо-
пропинол 46
Изопропоксититан-трш>(2-оксиэтил-
амин) 33
бис-(Изопропокси)титан-бнс-(2-окси-
этиламин) 33
/п/шс-(Изопропокси)титан-2-оксиэтил -
амин 33
гпетракис-(\Лзъи р опокси)титан -т-п ир и -
дин 33
Изопропоксититантрибромид-т-изопро-
пилацетат 49
Изопропокси-бш?-(триметилсилокси)ти-
тан 63
/п/шс-(Изопропокси)триметилсилокси-
титан 63
Изопро покситита нтр и хлор ид, аддукты
44
Изопропоксититантрихлорид, синтез и
свойства 44
/тгрш>(Изопропокси)титанхлорид 47
— аддукты 47
цфш>(Изопропокси)титанфторид 43
тетршшс-(Изопропокси)титан-т-этил-
амин 33
тетракис-(у1зъыр опокси)титан -т-эти л -
диамин-1,2 33
ИК-спектры поглощения связи Ti—N
60
Ильменит измененный 213, 216
— степень измененности 213
— производство концентратов 249
Ильменитовый концентрат 194
-----разложение серной кислотой 213
-----химический состав 195
бнс-(Инденил)титангалогениды 7
Инденилтитандихлороксид 7
Инденилтитантрихлороксид 7
Кальциевотитановый комплекс с ти-
роном 124
Катализаторы наттовские 21
Комплексные соединения титана 90
Комплексы титана с ал кил сульфидами
и алкиламинами 61
Комплекс триметаллический
Ga—Ti—Ga 34
Комплексы циклопентадиенилтитана с
р-дикетонами 17
Кричтонит 214
Лейкоксен 213
Литийтитан-трпс-(дипиридил), аддукт
132
Литийциклопентадиенилтитан-mpwc-
(этоксид) 13
о-Метанол фенокси-яг/шс-(бутокси)ти-
тан 57
о-Мета нол фенокси -т/шс-(пропокси)ти -
тан 57
о-Метанолфенокси-пг/шс-(этокси)титан
57
бш>(Метилацетоацетокси)титандихло-
рид 47
Метиламидометилимидтитанхлорид 59
бцс-(Метил ар си нтр имети л енарси ндиме-
тил)-т-титантетраиодид 139
бщ>(Метиларсинтриметиленарсиндиме-
тил)-т-бис-(титантетрахлорид) 62
бш>(Метиларсинтриметиленарсиндиме-
тил)-т-/п/шс-(титанфторид) 139
/пелпракцс-(Метилдифенилсилокси)ти-
тан 62
/пе/прякмс-(Метилдиэтилсилокси)титан
61
Метилтитандихлорид-т-дипиридин 65
Метилтитантрихлорид 7
Метилтрихлортитан 6
бш>(Метилциклопентадиенил)титан-
дихлорид 14
Метилциклопентадиенилтитантрихло-
рид 13
Метил циклопентадиенил-циклопента-
диенилтитандихлорид 17
Метилпентациклодиенилтитан-этокси-
дихлорид 13
Метилокси-дп/шс-(этокси)титан 38
Метилциклопентадиенилтитан-бцс-(это-
кси)хлорид 13
Методы утилизации гидролизной сер-
ной кислоты 230
/пе/пра/шс-(Метокси)титан 31
6/7С-(Метокси) титан дихлор ид 45
272
4
Метоксититантрихлорид 43
Механизм электролитического осажде-
ния титана 167
Молекулярно-дисперсный титан 219
Монохлорэфиры смешанные 48
Напряжение разложения соединений
титана 163
----------- в расплавах солей 164
а-Нафтил трибутоксититан 6
тегпратшс-(Нафтокси)титан 37
бпс-(Нафтокси)титандихлорид 46
бпс-(о-Нитрофенокси)титандихлорид 120
л«-Нитрофеноксититантрихлорид 120
Нонадекаэтоксигептатитанпентоксид 53
те/пря/шс-(Нонаокси)титан 36
Окислительно-восстановительные по-
тенциалы, нормальный Ti2^/Ti3-^ 150
— ------Ti3+/Ti4+ 151
----титана, стандартные 150
— формальный 158
быс-(8-Оксибензокси)титандибромид 49
те/пра/шс-(8-Оксихинолинато)титан 134
т/шс-(Оксихинолинато)титанбромид
135
бш>(8-Оксихинолинато)титандибромид
134
трцс-(8-Оксихинолинато)титаниодид
135
/п/шс-(8-Окси х и н о л и н ато)титанфтор и д
135
бис-(8-Оксихинолинато)титандифторид
134
бис-(8-Оксихинолинато)титандихлорид
134
тетра/шс-(2-Оксиэтиламин)титан 32
/пе/и/7а/шс-(2-Оксиэтиламин)титан-т-
2-гидроксиэтиламин 32
Оксоацетилацетонатотитан (111)’ди-
мер 127
Оксодиацетилацетонатотитан’полимер
127
бис-{ 1 -Оксо, 1,3-диметилпропенокси)-
бис-(триметилсилокси)титан 64
бис-( 1 -Оксо, 1,3-дифенилпропенокси)-
бис-(триметилсилокси)титан 64
бмс-(Оксодихинолинатотитан)-т-акво
134
ще/прак«с-(Оксодихинолинатотитан)-т-
акво 134
Оксо-бцс-(фенилселендиоксо)титан 139
те/пракцс-(Октамидо)титан 59
гиетрокнс-(Октадекаамидо)титан 59
бпс-(Октафенилтетрасиликопентаок-
со)титан 62
Октаэтокситрититандиоксид 67
Олигомеры со связью 73
— титановые линейные 70
Оксо-бцс-(нафтилселендиоксо)титан 139
Пентагидротитанал -N -фур аи лдифенил-
гидрокси л амин-тр исул ьфат-тр игид-
росульфат 128
Пентадиенилгексаметиламидотитан’ди-
мер 60
бш?-(Пентадиени л) гексаметил амидоти-
тан’димер 60
Пентаметилбензолтитандихлоргидро-
ксид 65
Пе нтамети л ци к л опента д иен и л тита н -
оксогидроксид’полимер 13
Пентаметилциклопентадиенилтитан-
оксохлор ид’полимер 13
Пентаметилциклопентадиенилтитан-
трихлорид 13
бш?-(Пентафторпропионат)титан-
оксид’полимер 57
Перовскит 198
Пески морские черные 194
Пиридинтитантетрахлор ид-т-димета-
нол 124
Полибутил титанаты, методы получе-
ния 67
Полимеры титана с салицилацетатом 68
— титанкремнийорганические, приме-
нение 70
— титановые линейные 69
-----с хелатными комплексами 69
Полиорганосилоксититаноксаны, мето-
ды получения 68
Полиорганотитаноксаны 66
Полиорганотитаноксилоксаны, кон-
денсация 70
— методы получения 69
Полиорганофосфонилоксититанокса-
ны 71
Полититанаты фосфинатные 73
Перенапряжение водорода на титане
153
-----------коэффициенты управле-
ния Тафеля 153
Полярография титана в растворах вин-
нокислых 155
-----------карбоновых кислот 155
-----------лимоннокислых 155
----------- роданидных 155
-----------сернокислых 153
-----------солянокислых 154
-----------уксуснокислых 155
-----------фосфорнокислых 154
-----------хлорнокислых 154
-----------ЭДТА 155
-----------щавелевокислых 155
Применение титана и его соединений
244
Производные со связью титан — угле-
род 5
--------титан — элемент — углерод
29
18 1—1669
273
Пропилацетоксититантрихлорид 56
Пропиленоксититантрихлорид 44
Пропиленоксититантрихлор ид-т-пипе-
ридин 44
6пс-(Пропионат)титаноксид’полимер 57
/т?/шс-(Пропокси)диметилпропоксисило-
ксититан 64
/прш>(Пропокси)трипропилсилоксити-
тан 64
/прш>(Пропокси)титан 50
/пелпра/шс-(н-Пропокси)титан 33
/пе/тг/7шшс-(Пропокси)титан 32
/п/шс-(Пропокси)титанбромид 49
б«с-(Пропокси)титандибромид 49
/пр«с-(Пропокси)титандипропилсило-
ксиоксотитан-дпрпс-(пропокси) 64
/п/шс-(Пропокси)титан-т-дипропинол 50
бпс-(Пропокси)титандихлорорид-т-пи-
перидин 45
бш>(Пропокси)титандихлорид-т-про-
пинол 45
Пропоксититантрибромид 48
дп/шс-(Пропокси)титантриметилсилок-
си-оксо-/п/шс(пропокси)-титан 64
Пропоксититантрихлорид 44
бш?-(Пропокси)титандихлорид 45
Пропоксититантрихлорид-т-пипери-
дин 44
Изопропоксихлортитаноксид 47
Реакции телп/яг/шс-(диалкиламидо)ти-
тана 59
— титантетрахлорида с триалкилфос-
фатами 66
— триэтаноламинотитаната с хлора-
мином 65
Рутил, производство концентратов 249
Рутиловый концентрат 194
Рутиловый концентрат, химический
состав 196
Свойства белых пигментов 245
Связь многоцентровая 9
Сернокислотный метод производства
титановых пигментов, расход сырья и
материалов 231
Серопроизводные титана 60
Смешанные алкокси- и силоксититаны
63
Соединения титана с силанами 61
Структура сэндвичевая 9
Сульфатизации непрерывная 216
Сульфатизации периодическая 215
Сульфат титана, гидролиз 212
----- кинетика 218
-----состояние в растворах 218
Сфен 198
Тагировит 198
Тетраалкоксититан 30
Тетрааминотитан 58
Тетрагидропиридинтетратитантриоксо-
тетрадекахлорид-т-дипиридин-т-три-
акво 128
Тетракупферронатотитан 135
Тетраметилтитан 5
Тетра-(М-нитрозофенилгидроксилами-
нат)титан 135
тетршшс-(Тетрапропиламин)дититан-
нонахлорид 130
Тетрафенилтитанат 6
/прш>(1,2-Тетрафторпропокси)титан-
аминохлорид 46
т/шс-(1,2-Тетрафторпропокси)титан-
хлорид 47
бис-(1,2-Тетрафторпропокси)титан-
аминхлорид 46
1,2-Тетрафторпропоксититанандиамин-
хлорид 44
бш>(1,2-Тетрафторпропокси)титанди-
хлорид 46
трис-( 1,2-Т етрафтор пропокси)титан-
нитрогидротитан-/л/шс-(1,2-Тетра-
фторпропокси)титанхлорид 47
1,2-Т етрафторпропоксититантр ихлорид
44
Тетраэтиламинтитанпентахлорид 129
Т етр аэти л ами нтита нтетр абром ид 130
Т етр аэтил аминтитантетр абр омид-т-ди -
ацетонитрил 130
Тетраэтиламинтитантетрахлорид 130
Т етр аэти л ами нтитантетр ах лор ид-т-ди-
ацетонитрил 130
Тетраэти лами нтетрах лор и дметоксити-
тан-т-ацетонитрил 129
Тетраэтиламинтитанпентахлорид-т-аце-
тонитрил 129
б«с-(Тетр аэти л амин) титантетрахлор-
оксид 129
Тетраэтиламинтитантетрахлоридбро-
мид-т-диацетонитрил 130
Тетраэтиламинтитанэтилокситетра-
хлор ид 129
Тетр аэти л амин-циклопентадиен и лти-
тан-бис-(дисульфодикарбодициан) 18
Тетр аэтилсурьмадититан нонахлор ид 66
бнс-(Т етр аэтилсур ьма)титанпентах л о-
рид 66
6uc-(Y етр аэтилсур ьма)титангексахло-
рид 66
Т етр аэтилсур ьматитантетр ах лор ид 66
Титана азидокомплексы 94
— аквокомплексы 91
Титана (III) аминокомплексы, магнит-
ные свойства 130
Титана барьер анодного растворения
150
-----разряда ионов на катоде 150
Титанавгит 198
Титаназотдимагнийдихлорид-т-тетра-
гидрофуран 65
274
Титана каталитическая полярографи-
ческая волна 156
Титана (III) комплексы с ализарином
125
-----ацетил ацетоном 126
-----диантипир ил-о-оксифенил мета-
ном 132
-----дипиколиновой кислотой 130
-----дитизоном 125
-----диэтилтетрамином 130
-----кокателином 125
-----никотинамидом 130
-----со спиртами, фенолами, нафтола-
ми 122
-----с протокатехиновой кислотой 125
-----эриохромчерным 137
Титана комплексы борнокислые 93
----- бутанольные 119
-----виннокислые 107
----- глиоксиматные 138
-----карбамидные 131
-----молибдатные 94
-----молочнокислые 109
-----муравьинокислые 102
-----перекисно-молоновые 99
--------оксалатные 100
--------фторидные 100
-----перекисные 95
--------бесцветные 95
--------малеиновые 99
--------каталитическое действие 98
--------окрашенные 95
— — — смешанные 98
-------- строение 95
--------с ЭДТА 100
-----роданид-диантипирилметановые
92
-----роданидные, константа образо-
вания 91
--------упрочнение внешнесферными
ионами 92
--------экстракция 92
-----с азокрасителями 136
-------- ализарином 135
-----салицилатно-глицериновые 113
--------маннитовые 113
----- — пиридиновые 111
--------трибутилфосфатные 111
----- салицилатные 109
-----с алифатическими эфирами 133
--------амидами кислот послойные
130
-------- аминами 130
--------Р- и у-аминоспиртами 119
-------- антипирином 130
--------арсинопроизводными 139
-------- аскорбиновой кислотой 114
— 3-бензил-4,5-диоксикумарином 136
—-------боропроизводными 138
Титана комплексы с 1-бутеном 133
-----— транс-2-бутаном 133
-------- бутиллитием 138
--------ванилином 120
--------виолуровыми кислотами 118
--------галловой кислотой 120
--------гидроксамовой кислотой 117
--------глицерином 124
-------- гликолевой кислотой 118
--------глицериновой кислотой 118
--------селеномолибдатные 94
--------с двухатомными спиртами 125
--------диантипирилметаном 128
--------1,2-дигидроксибензен-3,5-ди-
сульфонатом 124
--------дигидроксикумарином 136
--------Р-дикетонами 126
--------М,М-диметилформамидом,
спектры ЯМР 129
--------3,4-диоксибензойной кисло-
той 118
--------1,8-диоксинафталином 124
----- диокситартроновой кислотой 118
--------2,5-диокси-З-ундецил-п-бен-
зохиноном 128
--------3,4-диокси-4-х лор азобензо-
лом 136
--------ди-/г-толилтиовиолуровой ки-
слотой 119
--------1,1-дифенилэтиленом 133
--------цис-транс-транс-л,отк?а^^-
ном 133
-------- дифенилвиолуровой кисло-
той 118
--------о-дифенилом 125
--------изобутеном 133
-------- карбоксилкаротиноидами 134
--------карбонильными соединения-
ми 127
--------карбоксилгалланилидом 131
--------г кверцетином 137
-------- коричной кислотой 117
--------криптосантином 137
-------- кумарином 136
-------- купферроном, неокупферро-
ном и n-фенилкупфероном 135
-------- маннитом 124
--------миндальной кислотой 118
-----„ — моносахаридами 133
--------Р -нафтолом 119
--------нейтральные 90
--------непредельными углеводоро-
дами 133
--------4-нитро-2-аминофенолом и 1-
фенол-3-мет ил-5-пирозалоном 136
--------1 -нитрозо-2-нафтол -3,6-дису-
льфокислотой 1Л7
--------одноатомными нафтолами 119
-----------спиртами 119
18*
275
Титана комплексы с фенолами 119
--------2-окси-1 -нафтол -о-амино-
нафтолом 119
—-------олигосахаридами 133
--------папаверином и пирокатехи-
ном 129
-----2 (a-N-пипер идино)-изопро-
п ил -4 -окситетр аги дрофу р а ном-3 128
-----пиридилазорезорцином 137
-----> — пирогалкарбоновой кислотой
121
—-------------и антипирином 121
--------и пирамидоном 121
--------пирогаллоном и его производ-
ными 120
--------пирокатехином-3-дисульфо-
кислотой 123
-----— пирокатехином и диантипи-
рилметаном 123
--------------его производными 121
-------------- пирамидоном 123
--------------метилсалицилатом 122
-----------— салициловым альдеги-
дом 122
--------------бензоил метанол ом 122
-------------- пиколиновой кислотой
123
--------------пиридином 123
--------------константы нестойкос-
ти 122
-------------- пиколиновой кисло-
той 118
--------пирокатехиновым фиолето-
вым 123
-------- пропиленом 133
--------протокатехиновой кислотой,
см. 3,4-диоксибензойной кислотой
-------- с салицилальдоксином 114
--------с селенопроизводными 139
--------транс-2 -стильбеном 133
-------- со спиртом 133
--------сульфатные 93
--------константы нестойкости 93
-----с тетраметил эти леном 133
-----сульфатные со смешанными ли-
гандами 93
--------тетрафенилэтиленом 133
-------- тимолом 120
--------тиогидроксамой кислотой
117
--------тироном, см. 1,2-дигидрокси-
‘бензиден-3,5-дисульфонатом
-------- тироном 123
-------- триметилэтиленом 133
-----— триоксиглутаровой кислотой
109
—-------2,2,7-триокси-9-фенилфлуоро-
>ном 124
Титана комплексы с трифен и л эти ле-
ном 133
-------- трополоном
--------фенантралином 133
--------о-фенилен-бис-(диэтил) арси-
ном 139
--------1-фенил-З-метил-4-бензолпи-
р азол и ном 129
--------фенилфлуороном, см. 2,2,7-
триокси-9-фенилфлуороном •
-----— p-фосфонкарбонильными со-
единениями 139
--------фосфорпроиз водными 139
--------фталоцианином 136
—— — хромотроповой кислотой 115
--------циклогександиолами 133
--------цис-транс-транс-циклогокси-
ном 133
--------транс-транс-транс-циклоге-
ксеном 133
--------транс- транс- транс-цикло &о-
декатр иеном 133
--------шиффовыми основаниями
111
--------эмберлином, см. 2,5-диокси-
3-ундецилбензохинон
--------ЭДТА 131
--------эриохромчерным 137
--------эскулатином 133
-------- эфирами 133
-------- фенолантипирином 119
-----уксуснокислые 102
-----фосформолибдатные 94
----- фенола 119
-----фторидные, константа нестойкос-
ти 98
-----фторэтанольные 119
-----хлоридные 92
----- цианидные 92
-----щавелевокислые с ЭДТА 103
----- яблочнокислые 109
-----янтарнокислые 109
Титаналдибром-бш>(дипиридилпири-
дин)бромид 132
Титаналдибром-б«с-(дипиридилпири-
дин)бромид-/п-хлороформ 132
Титаналгексаакво’трианион 91
Т итаналгекса-(2,3-метил-1 -фенил-5-пи-
разолон) трибромид-т-диэтанол
130
Т итаналгекса-(2,3-метил-1 -фенил-5-пи-
разолон) трииодид 130
Титаналгексаэтиленмочевина-триио-
дид 130
Титаналгексаэтиленмочевина-трихло-
рид-т-диэтанол 130
Титаналдибромид-бис-(дипиридилпи-
ридин)-титаналтетрабромиддипири-
дилпиридин 132
.276
Т итаналдипиридилди хлорид- хлорид
132
Титаналкилы 5
Титаналтрибромид-бш>(дипиридилпи-
ридин)-бромид-т-ацетонитрил 132
Т итаналтрибромиддипиридилпири-
дин’димер 132
Титаналтрихлорид-додекапрено-Р-ка-
ротинхлорид 134
Титаналтри хлор ид-т-трипропанол 125
Титанал-Г4-фураилдифенилгидроксил-
амин-дихлор дихлорид 128
Титана оксалаты 104
Титана оксалаты двойные, термическая
устойчивость 106
Титана пероксосоли 101
— равновесные электродные потенци-
алы 158
Титанарилы 5
Титана стандартные электродные по-
тенциалы 157
— тартраты 108
— формиат 102
Титанацилаты 55
— основные 57
— полимерные 67
— применение 68
Титанацетанилидатотрихлорид 126
Титан-2,4-бутандиамин-тетрахлорид
129
Титангалогеноэфиры 49
Титангексаметилендиаминтетрабромид
129
Титангексаметилендиаминтетрахлорид
129
Титандиацетонатодибромид 126
Титандиацетонатодииодид 126
Т итандиацетил ацетонатодифторид 126
Титандиацетонатодихлорид 126
Т итандибензоил ацетанилидатоди-
хлорид 129
Т итандикверцетинанодигидроксид 137
Титанди-(о-нафтилдиамин) тетрабромид
129
Титанди-(о-толидин) тетрабромид 129
Титанди-(о-фенилендиамин) тетрабро-
мид 129
Титандихлор-бцс-(бензоилацетат) 31
бпс-(Титанди хлор циклопентадиенил)
оксид 12
бис-(Титандициклопентадиенилхлор)-т-
цинкдихлорид 14
Титан-трис-(дипиридил) 132
Титанил-бис-(метилсульфат) 93
Титан-М-индолил 59
3 итан-М-карбозил 59
Титан металлический, иодидное рафи-
нирование 242
•----масштабы производства 249
Титан металлический, методы получе-
ния 237
-----области потребления 249
-----натрийтермический метод полу-
чения 241
-----магнийтермический метод полу-
чения 239
— — производство по методу Кроля
239
-----сродство к галогенидам 237
----------- кислороду 237
-----технология производства нат-
рийтермическим методом 241
-----фирмы-производители 250
----- электролитический метод полу-
чения 239
-----электролитическое рафинирова-
ние 243
Титаннитромагнийхлорид-т-пиридин 65
Титановая губка 239
-----допустимые примеси 243
-----марки в СССР 249
----- раскисление 240
Титановые зародыши, методы приго-
товления 229
-----получение осаждением аммиа-
ком 218
-----рутилизирующие 229
Титановые пигменты 211
-----требование к качеству сырья 213
— руды обогащение 194
— концентраты минеральные 194
— соли карбоновых кислот 55
— соли нафтеновых кислот 55
— Титановый концентрат самотканс-
кий 196
-----ярегский 206
Титаноксихинолинатотрихлорид-т-пи-
-ридин 135
Титанокси^инолинатотрихлорид-т-
(8-оксихинолин) 135
Титаноксиэфиры 54
Титаноксо-Г4-оксипиридин-ди хлорат
128
Титанорганические соединения 5
— сополимеры 73
Титансодержащие материалы, поведе-
ние примесей 206
----- хлорирование 205
— — разложение серной кислотой*
214
Титансульфатхлормети л сульфат 93
Титантетра-(о-анизидин)тетрабромид
129
Титантетра- (n-анизидин) тетра-бро-
мид 129
Титантетраанилинтетрабромид 129
Т итантетрабензоламинтетрабромид 129
Титантетрабензидинтетрабромид 129
277
Титантетр абр оми д-т-(^ш>)транс-
1,2-б«с-(диметил)арсиноэтилен 139
Т итантетрабромид-т-дифталимид 130
Т итантетрабромид-т-су кцинами д 130
Титантетрабромид-метиларсин-бис-
(триметилен-диметиларсин) 65
Т итантетрабромид-т-триацетанилид 126
Титантетр аброми д-т-фтал амид 130
Титантетрабромид-т-1,2-циклогекса-
фталимид 130
Титантетрадианизидинтетрабромид 214
Титантетрагалогениды, аддукты с 8-ок-
сихинолином 135
Титантетрагалогенидные комплексы с
антроном 127
--------бензатроном 127
-------- нитроантроном 127
Т итантетр а- (а- нафти л ами н) тетр абр о-
мид 130
Титантетра-(Р-нафтиламин)тетрабро-
мид 130
Титантетра-(ж-толуидин)тетрабромид
130
Титантетра-(п-толуидин)тетрабромид
129
Титантетра-(о-толуидин)тетрабромид
129
Титантетра-(о-фенэтидин)тетрабромид
129
Титантетра-(п-фенэтидин)тетрабромид
129
Т итантетр афтор ид-т-ацетальдегид 129
Т итантетрафторид-т-бензал ьдегид 127
Т итантетр афтор ид-т-ди мети л кетон 127
трш>(Титантетрафторид)-т-метилар-
син 139
Титантетрахлорид-т-ацетанилид’димер
128
Т итантетрахлорид-т-ацетонитрил 129
Титантетрахлор ид-т-п-ацетофенилме-
ти л кетон 137
Титантетрахлорид-т-бензамид’димер
128
Титантетрахлорид, восстановление маг-
нием 239
-----натрием 241
-----в жидком аммиаке 242
Титантетрахлорид-т-5-М-диалкиламо-
фурфуролы 137
Титантетрахлорид-т-дибензофенон,
см. дифен ил кетон
Титантетрахлорид-т-М,Ы-диметилаце-
тамид 128
Титантетрахлор ид-т-диметил глиоксим
138
Т итантетр а хл ор и л-т-бис-(ди метил -
глиоксим) 138
Титантетрахлориддипиридилпиридин
132
Т итантетрахлорид-т-1,3-дипиридино-2-
(4-хинолин)пропан 129
Титантетрахлор ид-т-бщ>(дифенилке-
тон), гидролиз 137
Титантетрахлорид-т-дифурфурол 137
Титантетрахлорид-т-ди-2-(Р-диэтил-
аминоэтил)-5-винилпиридин 129
Титантетр ахл орид-т-2-ф-диэтиламино-
этил) хинолин 129
Титантетрахлорид-т-4-(Р-диэтиламино-
этил)хинолин 129
Титантетрахлор ид-т-алюминийдиэтил-
хлорид 8
бг/о(Титантетрахлорид)-т-Р-апа-8-ка-
ротиналь 134
Титантетрахлорид-т-Р-апа-8-кароти-
наль 134
Т итантетрахлорид-т-кантаксантин 133
Титантетр а хлорид, масштабы производ-
ства в США 253
бш>(Титантетрахлорид)-т-метиларсин,
б«с-(триметилен-диметиларсин) 65
Титантетрахлорид, методы очистки от
ванадия 207
бис-(Т итантетрахлорид) -т-оксамид 128
Титантетрахлорид-т-(8-оксихинолин)-т-
пиридин 135
Титантетрахлорид-т-4-(Р-пиперидино-
этил) хинолин 129
Титантетрахлорид производство, ва-
надиевые кеки 211
-----в г. Тане 207
-----восстановители 205
— — связующее для брикетов 208
-----совершенствование технологии
211
-----титановое сырье 204
— — хлорирующие агенты 205
Титантетрахлорид, разложение водой
232
Т итантетр ахл ор ид-т- р одоксанти н 134
бис- (Т итантетр а хлор ид) - т- р одоксанти н
134
— сжигание кислородом 232
— содержание примесей 210
— сольволиз метанолом 119
Т итантетрахлорид-т-сукцинамид 130
Титантетрахлорид-т-п-толилметилкетон
127
Титантетрахлор ид-т-толилпропилке-
тон 127
Титантетрахлорид-т-триацетанилид
126
Титантетрахлорид-т-триэтил алюминий
8
бис-(Т итантетрахлорид)-т-фталимид 130
Титантетр ахл ор и д-т-фтал имид 130
Т итантетр ахл ор ид-т-1,2-ци кл огексан-
фталимид 130
278
I
i
Титантетрахлорид-т-этилацетат 32 /
Титантетрацетанилидатобромид 126
Титантриацетанилидатоиодид 126
Т итантриацетани л и датохл ор и д 126
Титантриацетанилидатохлорат 126
Титантрииодид-т-гексакарбамид 131
Т и тантр иио ди д-т-мети л арси н-бис- (тр и -
метилен-диметиларсин) 65
Титантрихлорид-т-триэтилалюминий 8
Титантрихлор ид-т-диметанол 125
Титантрихлорид-т-диметилглиоксим
138
Титантрихлорид-т-дитетрагидрофу-
ран-т-бензол 133
Титантрихлор ид-т-изобутанол 125
Титантрихлор ид-т-пентаметанол 125
Титантрихлорид-т-(сурьматетраэтил-
хлорид)’полимер 66
Титантрихлорид-т-тетрагидрофуран
133
Титантрихлорид-т-тетраизопропил 125
Титантрихлорид-т-тетраметанол 125
Титантри хлор и д-т-тетраэтанол 125
Титантрихлорид-т-трибутилфосфат 139
Титантрихлорид-т-тризопропил 125
Титантрихлороксо’дианион 128
Титанфтал оцианинатооксид 136
Титанфтал оцианинатодихлорид 136
Титанфтал оцианинатохлорид 136
Титан-бнс-(Ы-фураилдифенилгидро-
ксиламидо)ди хлорид 128
Титан-бш>(М-фураилдифенилгидро'-
ксиламидо) сульфат 128
Титанхлорацилаты, каталитическая ак-
тивность 57
Титанэтилендиаминтетрабромид 129
Титанэтилендиаминтетрахлорид 129
Титанэфиры 30
— амиловых спиртов 35
— гексиловых спиртов 35
— гидролиз 50
— двойные 50
— с изомерами гетиловых спиртов 36
— с изомерными октиловыми спирта-
ми 36
— механизм гидролиза 50
— применение, каталитические си-
стемы 54
— производных бензола 37
— полимеризация 53
— скорость гидролиза 51
— смешанные 39
— со смешанными оксигруппами 38
— трехвалентного титана 50
— устойчивость к гидролизу 51
— цепочечное строение 52
— энтальпия образования 37
Триалкилсилоксигалогенотитаны, ме-
тоды получения 64
Три ацетил ацетонатотитан (III) 127
бш>(Трибутоксититанокси)борфенил 58
Тригидротитанал-М-фураилдифенил-
гидроксиламин-трисульфат-гидро-
сульфат 128
бис-ft риизопропиламингидро) титан-
гексахлорид 129
Трикалийтитаналгексацианид 92
Трикалийтитаналгексадиамид-т-дика-
лийцианид 92
Трикалийтитаналгексацианид-т-калий-
цианид 92
Триметиламинтитанхлорид 58
Триметилсилазантитанхлорид 60
бис-(Триметилсилокси) дититантриок-
сид’полимер 69
тетракис-фриметилсиланокси)титан 61
бис- (Т р и метил си л ан окси) тита н ди хл о-
рид 64
Триметилсилоксититантрибромид 65
Триметилсилоксититантрихлорид 64
/прш?-(Триметилсилокси)титанхлорид
64
Трипиридинтитангексабромид 130
Трипиридинтитангексахлорид 130
Трипиридинтитантетрахлориддибро-
мид 130
/тге/тгрс/шс-(Трипропилсилокси) титан 62
бш>(Трифторацетат)титаноксид’поли-
мер 57
/прцс-(Трифтордиацетат)титаноксид-т-
бис-(трифторацетат) 57
/прш>(Трифтортеноилацетонато)титан
127
Три хлорацетоксититантри хлор ид 56
/прис-(/пре/п-Трихлорметан-диметилме-
токси)титанхлорид 48
Трихлораттитанацетат-т-нитрозил хло-
рид 57
Трихлортитанбутират 57
Трихлортитанвалериат 56
Трихлортитан-дихлорацетат 57
Трихлортитан-изобутират 57
Трихлортитанпропионат-т-нитрозил-
хлорид 57
Трихлортитановые эфиры производных
фенола 45
Трихлортитан-трихлорацетат 57
Т ри хлорфеноксититан 120
бцс-(Триэтиламиногидро)титангекса-
хлорид 129
/пе/пря/шс-(Триэтилсилокси)титан 61
Триэтилсурьмадихлорид-т-(титантет-
рахлорид)’полимер 66
Уфанит 200
/пг/пракгц>(Фенилацетат)титан 58
Фенилацетоксититантрихлорид 56
бцс-(Фенилдиметиламингидро)титан-
гексахлорид 129
279
те/прбмшс-(Фенилдиметилсилокси)ти-
тан 62
Фенилтитандихлорид-т-дипиридин 65
те/ирдкис-(Фенилдиэтилокси)титан 62
бис-(Фенилсалицилокси)титанди-
хлорид 47
Фенилтризопропоксититан 61
X тетракис-(Фенокси)титан 37
/?2£/пря>шс-(Фенокси)титан-т-гидрохло-
рид 120
бш>(Фенокси)титандихлорид 46
Феноксититантрихлорид 45
Феноксититантрихлорид, комплексы с
диэтиловым эфиром 45
/прнс-(Фенокси)титанхлорид 48
/ирш>(Фенокси)титанхлорид, производ-
ные фенола 48
тетршшс-(Фенокси)титан- т-фенол 120
Ферротитан 244
Фиксация азота 24
Фиксация НС1 при реакции этерифи-
кации 30
Форстерит 198
Фосфор нотитанмолибденовая кислота
94
Фталоцианин титана 65
Хлоралкоксититаны, комплексные соли
119
лирпс-(1-Хлорпропокси)титанхлорид 47
б«с-(п-Х лорфенокси)титанди хлорид
120
Циглера — Натта катализаторы 25
пгг/пракш>(Циклогексанокси)титан 9
Циклогексоксититантрихлорид 45
/лрш>(Циклооктатетранен)дититан 9
б«с-(Циклопентадиенил)аллилтитан 11
бмс-(Циклопентадиенил)германийтри-
фенил 24
бмс-(Циклопентадиенил)дикарбонил-
титан 16
бш>(Циклопентадиенил)-бш?-(н-диме-
тиламинофенил) титан 16
те/пракнс-(Циклопентадиенил)дити-
тан-бутан ди ат 57
/тге/ирак^с-(Циклопентадиенил)дити-
тан-бутэндиат-2 57
бас-(Циклопента диен и л-бис-(дифен и л)
титан 16
Циклопентадиенил-дихлорциклопен-
таенилтитандихлорид 14
Циклопентадиенил-тетрахлорцикло-
пентатитандихлорид 14
бпс-(Циклопентадиенил)-бнс-(метил)
титан, теплота сгорания 17
/лрш>(Циклопентадиенил)титан 10
/72в/72ракнс-(Циклопентадиенил)титан 10
бпс-(Циклопентадиенил)титаналлил 16
бш>(Циклопентадиенил)титаналюми-
нийдиэтил 20
бис- (Ци кл опента диен и л)титанами -т-
диимин-15
бис-(Циклопентадиенил)титанацетат 57
бш>(Циклопентадиенилтитанацетилаце-
тонатжелезо) (олово, ртуть, цинк,
кадмий)хлориды 20
Циклопентадиенилтитан-трш>(аце-
токсид) 13
бш>(Циклопентадиенил)титан-бцс-(аце-
токсид) 16
бмс-(Циклопентадиенил)титан-б«с-
(бензилсульфид) 18
бис-(Циклопентадиенил)титанборгид-
рид 18
бш?-(Циклопентадиенил)титанбор дей-
терид 18
бш>(Циклопентадиенил)титанборте-
трафторид 18
бш>(Циклопентадиенил) титанборте-
трахлорид 18
бис-(Ци кл опентадиен ил) титанбром-
бор дибромид 138
Циклопентадиенилтитан-тр«с-(бу-
токсид) 15
бис-(Циклопентадиенил)титанхлорбор-
дихлорид 138
бмс-(Циклопентадиенил)титан, валент-
ное состояние 10
бмс-(Циклопентадиенил)титангалоге-
ниды 22
бш>(Циклопентадиенил)титангидро-
ксобромид-т-акво 17
бш>(Циклопентадиенил)титандека-
нат 57
бис-(Циклопентадиенил)титандибен-
зоат 23
Циклопентадиенилтитандибромид 14
бш>(Циклопентадиенил)титандибро-
мид 17
бис-(Циклопентадиенил)титандигид-
рид, анион 11
бш>(Циклопентадиенил)титандидейте-
рид, анион 11
бно(Циклопентадиенил)титандиизо-
роданид 18
б«с-(Циклопентадиенил)титандиизосе-
леноцианид 18
бш>(Циклопентадиенил)титандиизоци-
анат 18
Циклопентадиенилтитандииодат 14
бнс-(Циклопентадиенил)титан’ димер
23
бнс-(Циклопентадиенил)титандиметил-
аминтетрафенилборат 18
бш>(Циклопентадиенил)титандипири-
дин-/пе/тгра/шс-(фенил)борат 18
Циклопентадиенилтитандироданид 11
бис-(Циклопентадиенил)титандисуль-
фогидрид 19
280
бш>(Циклопентадиенил)титандисуль-
фодикарбодициан 18
бш>(Циклопентадиенил)титан-т-бш>
(дифенилацетилен) 17
Циклопентадиенилтитан дифторид 13
бпс-(Циклопента диенил)титандифторид 17
бш?-(Циклопентадиенил)титандирода-
нид 14
бис-(Циклопентадиенил)титанди хлор-
алюминийдихлорид 20
бцс-(Циклопентадиенил)титандихлор-
галлийдихлорид 24
Циклопентадиенилтитанди хлорид 11
бш>(Циклопентадиенил)титанди хло-
рид 10
бпс-(Циклопентадиенил)титандихло-
рид-т-алюминийтрихлорид 22
бщ>(Циклопентадиенил)титандихлор-
алюминийхлорэтил 20
бпс-(Циклопентадиенилтитандихло-
рид)-бпс-(алюминийдиэтил) 20
бш>(Циклопентадиенил)титандихло-
рид-т-бш>(алюминийтрихлорид) 22
бш>(Циклопентадиенил)титандихло-
ридалюминийэтилхлорид 21
бш>(Циклопентадиенил)титандихло-
рид, замещение метиллитием 60
бнс-(Циклопентадиенил)титандихло-
рид, комплексы с алюминийтрихло-
ридом 21
— реакции присоединения 22
бщ>(Циклопентадиенил)титандихло-
рид-бнс-(диэтилалюминий) 21
бщ>(Циклопентадиенил)титанди хло-
рид, теплота сгорания 17
бш>(Циклопентадиенил)титанкарбо-
ксилат 57
бщ>(Циклопентадиенил)титан-бш>
(метил)’анион 11
бш>(Циклопентадиенил)титан-бщ>(ме-
тил сульфид) 18
бцс-(Циклопентадиенил)титанметил-
хлорид 16
бпс-(Циклопентадиенил)титаноксид 11
бш>(Циклопентадиенил)титаноксипи-
крат 17
Циклопентадиенилтитаноксохлорид 12
бнс-(Циклопентадиенил)титанолово-
трифенил 23
бцс-(Циклопентадиенил)титанпента-
сульфид 14
бцс-(Циклопентадиенил)титанпентафе-
нилгидроксид 16
бцс-(Циклопентадиенил)титан-бпс-
(пентафторфенил) 16
б^с-(Циклопентадиенил)титанпента-
фторфенилфторид 16
бш?-(Циклопентадиенил)титанпента-
фторфенилхлорид 16
бцс-(Циклопентадиенил)титанпента-
фторфенил этокси д 16
бцс-(Циклопентадиенил)титанполи-
сульфид (и = 3—5) 20
Циклопентадиенилтитанполисульфид’
полимер 20
бш>(Циклопентадиенил)титан-бпс-(про-
пил)’анион И
бнс-(Циклопентадиенил) титан-бнс-
(пропилсульфид) 18
Циклопентадиенилтитан-/п/шс-(про-
поксид) 15
Циклопентадиенилтитанроданидхло-
рид 11
бцс-(Циклопентадиенил)титанстеарат57
бис-(Циклопентадиенил)титанскварат 20
бпс-(Циклопентадиенил)титан-т-тера-
гидрофуран 10
бпс-(Циклопентадиенил)титан-бпс-(п-
толилсульфид) 18
бш>(Циклопентадиенил)титантрибор-
октагидрид 18
Циклопентадиенилтитантрибромид 13
Ци клопентадиен и лтитан-хп/жо (триме-
тил силоксид) 15
бцс-(Циклопентадиенил)титан-бщ>(три-
фтор ацетоксид) 17
Циклопентадиенилтитантрихлоридалю-
минийдихлорид-т-алюминийтри-
хлорид 22
Циклопентадиенилтитантрихлорид 13
Циклопентадиенилтитантрихлоридалю-
минийдихлорид 22
Циклопентадиенилтитантрихлорид, па-
раметры решетки 12
— реакция с метиллитием 19
— теплота сгорания 17
бцс-(Циклопентадиенил)титанфенил-
бромид 16
Циклопентадиенилтитан-/лр«с-(фенил-
сульфид) 19
бш>(Циклопентадиенил)титан-бш>(фе-
нилсульфид) 18
Циклопентадиенилтитанфенилсульфид-
хлорид 19
бис-(Циклопентадиенил) титанфенил-
формиат 57
бнс-(Циклопентадиенил) титанформиат
57
/пе/пра/шс-(Циклопентадиенилтитан-
хлорат) 15
бш>(Циклопентадиенил)титан-бцс-
(хлор ацетоксид) 17
бно(Циклопентадиенил) титан-б«с-
(п-хлор бенз и л сульфид) 18
бпс-(Циклопентадиенил) титанхлорди-
фенилборид 138
бнс-(Циклопентадиенил)титанхлор-т-
диэтилалюминийхлорид 21
281
бис-(Циклопентадиенил)титанхлорид,
реакции с карбонатами 19
-------- солями дитиокарбонатов 20
бис-(Циклопентадиенил)титан хлорид,
комплексы с дипиридилом 23
бш>(Циклопентадиенил)титанхлорме-
тилимин-т-икс-метиламин 16
бис (Циклопентадиенил)титанциклопен-
тадиенилхлорид-т-этил алюминий-
дихлорид 21
бис-(Циклопентадиенил)титан-бш>
(этил)’анион 11
б«с-(Циклопентадиенил)титан-бг/с-
(этилацетоксид) 17
Ци кл опентадиен и л титан-бис- (это-
ксид) 15
Циклопентадиенилтитанэтоксиди хло-
рид 19
Циклопентадиенилтитан-бнс-(этокси)
хлорид 12
бнс-(Циклопентадиенил)титанэтокси-
хлорид 15
бнс-(Циклопентадиенил)-циклопента-
сульфотитан 19
бис-(Циклопентадиенил)хлортитансви-
нецтрифенил 24
бис-(Циклопентадиенил)хлортитан-
оловотрифенил 24
бис-(Циклопентадиенил)хлортитангер-
манийтрифенил 24
Циклопентадиенилтриметоксититан 12
бис-(Циклопентадиенил)-бис-(п-тол-
титан 16
бнс-(Циклопентадиенил)-бнс-(ж-толлил)
титан 16
бнс-(Циклопентадиенил)циклотетра-
сульфотитан 20
бнс-(Циклопентадиенил)циклотрисуль-
фотитан 19
Циклопентадиенильные соединения ти-
тана 9
-----с металлической связью
24
бнс-(Циклопентадиенил)-бнс-(этил)ти-
тан, теплота сгорания 17
Шлаки высокотитановые 197
Шлаки низкотитановые 196
— титановые 196
-----баланс двуокиси титана 202
-----кратность 202
-----минералогический состав 198
-----окисление на воздухе 202
-----применение 203
-----производство в Канаде 196
-----разложение серной кислотой
213
-----распределение хрома 202
----- расход электроэнергии 201
-----реакционная способность 215
Шлаки титановые, содержание двуоки-
си титана 196, 197
-----температура плавления 201
-----фазовый состав 200
-----флюсовая плавка 200
-----химический состав 201
— — хлорирование в расплаве кар-
нолита 207
Электродные процессы при восстанов-
лении титана 165
----- из расплава солей 167
Электролиз титана в расплавленных
средах 184
-----из неводных растворов 185
-----с нерастворимыми анодами 176
-----с растворимыми анодами 176
-----из металлических сплавов 183
----- нитрида 181
» — — окислов 178
Электролитическое восстановление ти-
тана в солевых расплавах 159
--------состав электролита 159
--------в водных растворах 152
--------катодные пленки 152
— — — самопассивирование 152
--------в сернокислых растворах 152
--------солянокислых растворах 152
бис-(Этил ацетоацетокси) титанди хлорид
56
Этилендиаминатные комплексы титана
(III) 131
бис-(Этилендиамин)титандихлорид-т-
бмс-(диэтиламин) 59
/пе/праяис-(Этилдиметилсилокси)титан
61
бш>(Этилдиметилаллокси)титандихло-
рид 64
Этилдиметилсилоксититантрихлорид 64
Этилтитаналюминийпентахлор-бис-
(дипиридил) 9
Этилтитантрихлорид 7
бис-(Этокси)-бис-(метилбензокси) ти-
тан 57
/пр«с-(Этокси)метилбензоксититан 57
/ие/пршшс-(Этокси)титан 31
тр«с-(Этокси)титан 50
/?грис-(Этокси)титанбромид 49
трис-(Этокси) титанбромид-т-пипери-
дин 49
тетршшс-(Этокси)титан, ассоциация 32
б«о(Этокси)-б«с-(бензил)титан 7
т/шс-(Этокси)титанбензои л ацетат 31
тетршшс-(Этокси)титан-т-диамин 32
/прис-(Этокси)титанхлорид-т-диамин 47
бис- (Этокси) -титан дибензоил ацетат 31
бно(Этокси)титандибромид 49
бш>(Этокси)диоксихинолинатотитан
134
б«с-(Этокси)титандифторид 43
282
(Этокси) титанди хлор ид 45
б«с-(Этокси)титандихлорид, продукты
присоединения 45
Этоксихлортитаноксид 54
Этоксититан-/прш?-(2-оксиэтиламин) 32
бш?-(Этокси)титан-б«с-(2-оксиэтил-
амин) 32
т/шо(Этокси)титан-2-оксиэтиламин 32
тетрп/шс-(Этокси)титан-т-пиридин 32
mempa/o/c-(Этокси) титан, полимериза-
ция 32
трис- (Этокси) титан хл ор и д-т- ами н-47
/тгрмс-(Этокси)титанхлорид-т-пипери-
дин 47
Этоксититантрибромид 48
бис-(Этокси)титандибромид, аддукты 49
Этоксититантрифторид 42
Этоксититантри хлорид 43
— аддукты 43
— продукты присоединения 43
Этоксититантри хлор ид-т-этил ацетат 32
Этоксититантрихлорид-т-этоксиаце-
тат 43
zzzpwc-(Этокси) титанфтор ид 43
/прцс-(Этокси)титанхлорид 47
/прцс-(Этокси)титанхлорид, аддукты 47
трис- (Этокси) титан хл ор и д-т-тр и ами н
47
/ирис-(Этокси)титанхлорид-т-этанол 47
/пе/иршшс-(Этокси)титан-т-этиламин 32
/пе/пршшс-(Этокси)титан-т-этилдиа-
мин-1,2 32.
*
ОГЛАВЛЕНИЕ
Предисловие.......................................................... 3
Глава I. Титанорганические соединения ............................... 5
Производные со связью титан — углерод............................ 5
Титаналкилы и титанарилы...................................... 5
Циклопентадиенильные производные титана....................... 9
Реакции замещения ....................................... 15
Реакции присоединения.................................... 20
Соединения со связью металл — металл..................... 24
Фиксация азота .......................................... 24
Катализаторы Циглера — Натта и другие каталитические реакции 25
Производные со связью Ti—Э—С.................................... 29
Эфиры ....................................................... 30
Алкилтитанаты ........................................... 31
Арилтитанаты ............................................ 37
Титанэфиры со смешанными оксигруппами.................... 38
Галогенэфиры ............................................ 41
Состояние титангалогенэфиров в растворах................. 49
Спектральные и магнитные свойства........................ 49
Диспропорционирование алкоксититанхлоридов .............. 50
Двойные титанэфиры ...................................... 50
Эфиры трехвалентного титана.............................. 50
Гидролиз титанэфиров .................................... 50
Каталитические системы и применение титанэфиров.......... 54
Ацильные соединения ..................................... 55
Амиды ортотитановой кислоты ................................. 58
Соединения с серопроизводными................................ 60
Соединения с силанами ....................................... 61
/пг/пршшс,-Триалкил(арил)силоксититаны................... 61
Алкокситриалкилсилоксититаны ............................ 63
Триалкилсилоксигалогентитаны ............................ 64
Другие титанорганические соединения ..................... 65
Полимерные органические соединения титана.................... 66
Полиорганотитаноксаны ................................... 66
Полимерные титанацилаты ................................. 67
Полиорганосилоксититаноксаны ............................ 68
Полиорганотитансилоксаны ................................ 69
Полиорганофосфонилоксититаноксаны........................ 71
Сополимеры ненасыщенных титанорганических соединений ... 73
Литература ..................................................... 74
Глава II. Комплексные соединения титана............................. 90
Комплексные соединения с неорганическими лигандами.............. 91
Аквокомплексы ............................................... 91
Роданидные комплексы......................................... 91
Цианидные комплексы ......................................... 92
Хлоридные комплексы.......................................... 92
Сульфатные комплексы......................................... 93
Борнокислые комплексы ....................................... 93
Азидокомплексы .............................................. 94
Молибдатные и фосфор молибдатные комплексы................... 94
Перекисные комплексы......................................... 94
Смешанные перекисные комплексы .............................. 98
285
Комплексы с органическими и смешанными лигандами................101
Муравьинокислые комплексы...................................102
Уксуснокислые комплексы ....................................102
Щавелевокислые комплексы....................................102
Виннокислые комплексы ......................................107
Лимоннокислые комплексы.....................................108
Комплексы с яблочной и янтарной кислотами...................109
Молочнокислые комплексы ....................................109
Комплексы с триоксиглутаровой кислотой......................109
Салицилатные комплексы.................................... 109
Комплексы с аскорбиновой кислотой...........................114
Комплексы с хромотроповой кислотой..........................115
Комплексы с тиогидроксамовой и гидроксамовой кислотами ... 117
Комплексы с коричной кислотой...............................117
Комплексы с сульфокислотами.................................117
Комплексы с пиколиновой кислотой............................118
Комплексы с миндальной кислотой.............................118
Комплексы с глицериновой, гликолевой и диокситартронрвой ки-
слотами ....................................................118
Комплексы с диоксибензойной кислотой........................118
Комплексы с виолуровыми кислотами...........................118
Комплексы со спиртами, фенолами и нафтолами.................119
Комплексообразование с одноатомными спиртами............119
Комплексы с одноатомными фенолами и нафтолами...........119
Комплексы с пирогаллолом и его производными.............120
Комплексы с пирокатехином и его производными............121
Комплексы с резорцином..................................124
Комплексы с диоксинафталином............................124
Комплексы с глицерином и маннитом.......................124
Комплексы с тироном.....................................124
Комплекс с фенилфлуороном...............................124
Комплексные соединения титана (III) со спиртами, фенола-
ми, нафтолами и их производными.........................125
Комплексы с Р-дикетонами....................................126
Комплексы с карбонильными соединениями......................127
Комплексы с аминами, амидами и их производными..............128
Комплексы с этилендиаминтетрауксусной кислотой..........131
Карбамидный комплекс ...................................131
Комплекс с карбоксилгалланилидом........................131
Комплексы с диантипирилметаном..........................131
Комплексы с дипиридилом.................................132
Комплексы с фенантралином...............................133
Комплексы с эфирами......................................г . 133
Комплексы с непредельными углеводородами....................133
Комплексы с оксихинолином...................................134
Комплексы с купферроном, неокупферроном и п-фенилкупферро-
ном ........................................................135
Ализариновые комплексы .....................................135
Комплексы с азокрасителями..................................136
Комплексы с фталоцианином ..................................136
Комплексы с кумарином.......................................136
Комплексы с кверцетином.................'...................137
Комплексы с эриохромчерным..................................137
Комплексы с пиридилазорезорцином............................137
Комплекс с криптоксантином .................................137
Глиоксиматные комплексы.....................................138
Комплексы с элементоорганическими соединениями..................138
Комплексы с бутиллитием....................................-138
Комплексы с борпроизводными.................................138
286
Комплексы с селенпроизводными...............................139
Комплексы с арсинпроизводными...............................139
Комплексы с фосфорпроизводными..............................139
Литература .....................................................140
Глава III. Электрохимическое поведение титана.......................150
Стандартные электродные потенциалы в водных растворах...........150
Электрохимическое восстановление титана из водных растворов .... 152
Анодное окисление титана....................................156
Электродные потенциалы в солевых расплавах......................157
Электрохимическое восстановление титана в безводных солевых
расплавах ......................................................159
Состав солевых расплавов электролитных ванн.................159
Напряжение разложения.......................................163
Катодные процессы при электролитическом восстановлении тита-
на .....................................................165
Электролиз фторидов титана ............................. 167
Электролиз хлоридов титана ..............................169
Электролиз иодидов титана .............................. 174
Электролиз расплавов, содержащих окислы титана...........174
Анодные процессы при электролизе соединений титана в расплаве
солей ..................................................175
Нерастворимые аноды .....................................176
Растворимый титановый анод...............................176
Растворимые аноды из окислов титана......................178
Растворимый анод из нитрида титана.......................181
Растворимый анод из карбида титана.......................183
Анодное растворение сплавов титана.......................183
Побочные процессы, сопровождающие электролиз титана в расплав-
ленных средах ..............................................184
Электролиз титана из неводных растворов.....................185
Литература .....................................................185
Глава IV. Краткие сведения о методах производства титана и его соеди-
нений ..........................................................194
Обогащение титановых руд.........................................194
Производство титановых шлаков ...................................196
Восстановительный обжиг ильменитовых концентратов . 203
Производство четыреххлористого титана............................204
Производство двуокиси титана.....................................211
Сернокислотный метод производства титановых пигментов .... 213
Разложение титансодержащих концентратов серной кислотой 213
Восстановление титана и железа в сернокислых растворах . . 216
Гидролиз сульфата титана.................................218
Кинетика гидролиза ......................................219
Механизм действия титановых зародышей....................222
Прокаливание гидроокиси титана......................... 223
Поверхностная обработка..................................224
Схема технологического процесса..........................224
Хлорный метод производства титановых пигментов ....... 231
Совершенствование методов производства двуокиси титана .... 233
Производство металлического титана ......................... 236
Производство ферротитана ....................................244
Области применения титана и его соединений.......................244
Некоторые данные из экономики производства и потребления титана
и его соединений у...............................................248
Литература ......................................................255
Предметный указатель к I части.......................................265
Предметный указатель ко II части.....................................269
Яков Гаврилович Горощенко
ХИМИЯ ТИТАНА
Печатается по постановлению ученого совета
Института общей и неорганической химии АН УССР
Редактор Л. П. Кругля к.
Художественный редактор И. В. Козий.
Оформление художника В. И. М о х н а т о в а.
Технический редактор Н. П. Рахлина.
Корректор Л. М. Тищенко.
Сдано в набор 19.VII. 1971 г. Подписано к печати 14.1.1972 г.
БФ 01111. Зак. № 1-1669. Изд. № 9. Тираж 1300.
Бумага № 1, 6ОХ9О*/1в. Печ. физ. листов 18,0.
Услов. печ. листов 18,0. Учетно-изд. листов 19,77.
Цена 2 р. 22 к.
Издательство «Наукова думка», Киев, Репина, 3.
Киевский полиграфический комбинат,
Комитета по печати при Совете Министров УССР, ул. Довженко, 3.