/







































































































































































































































































































































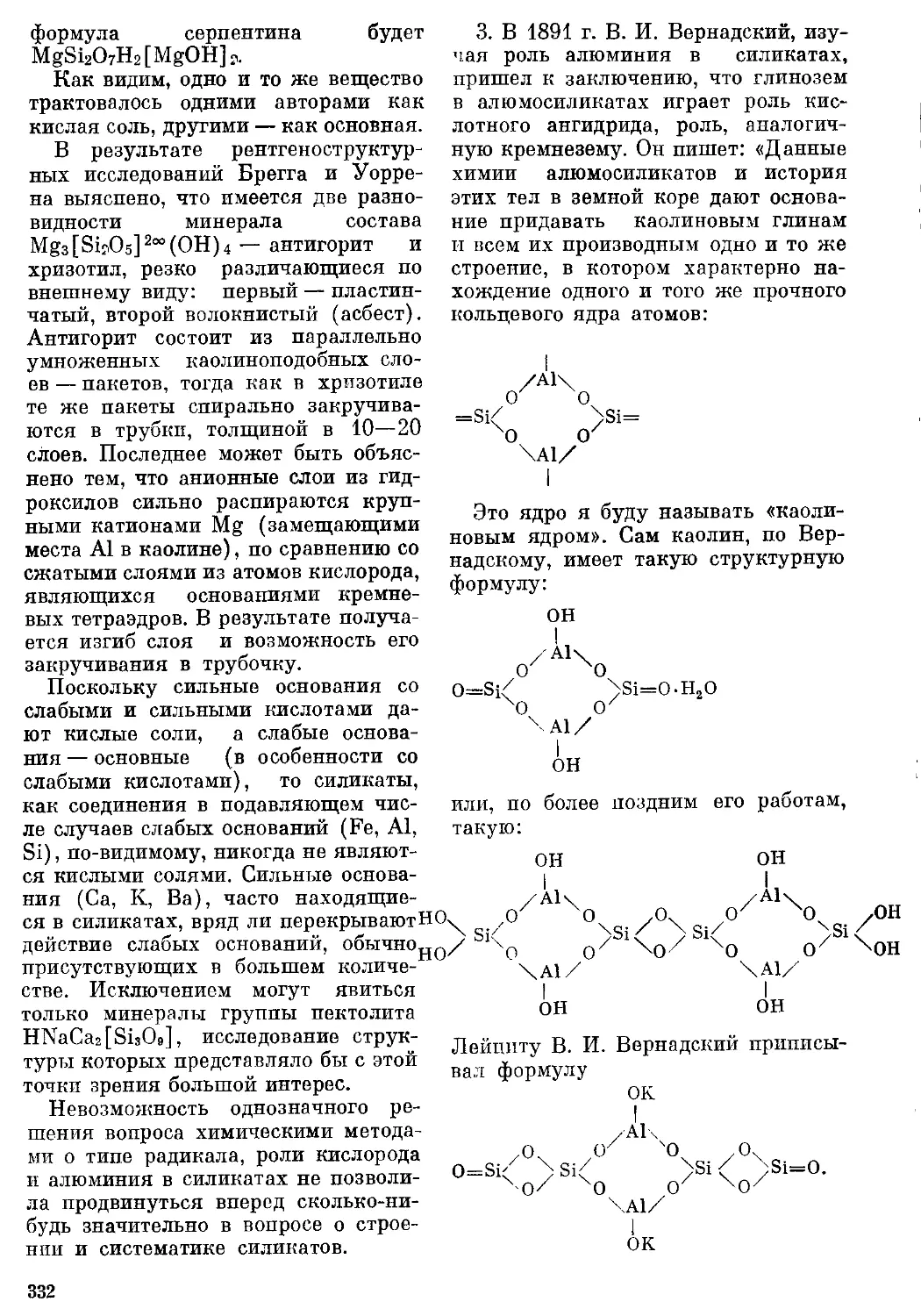

















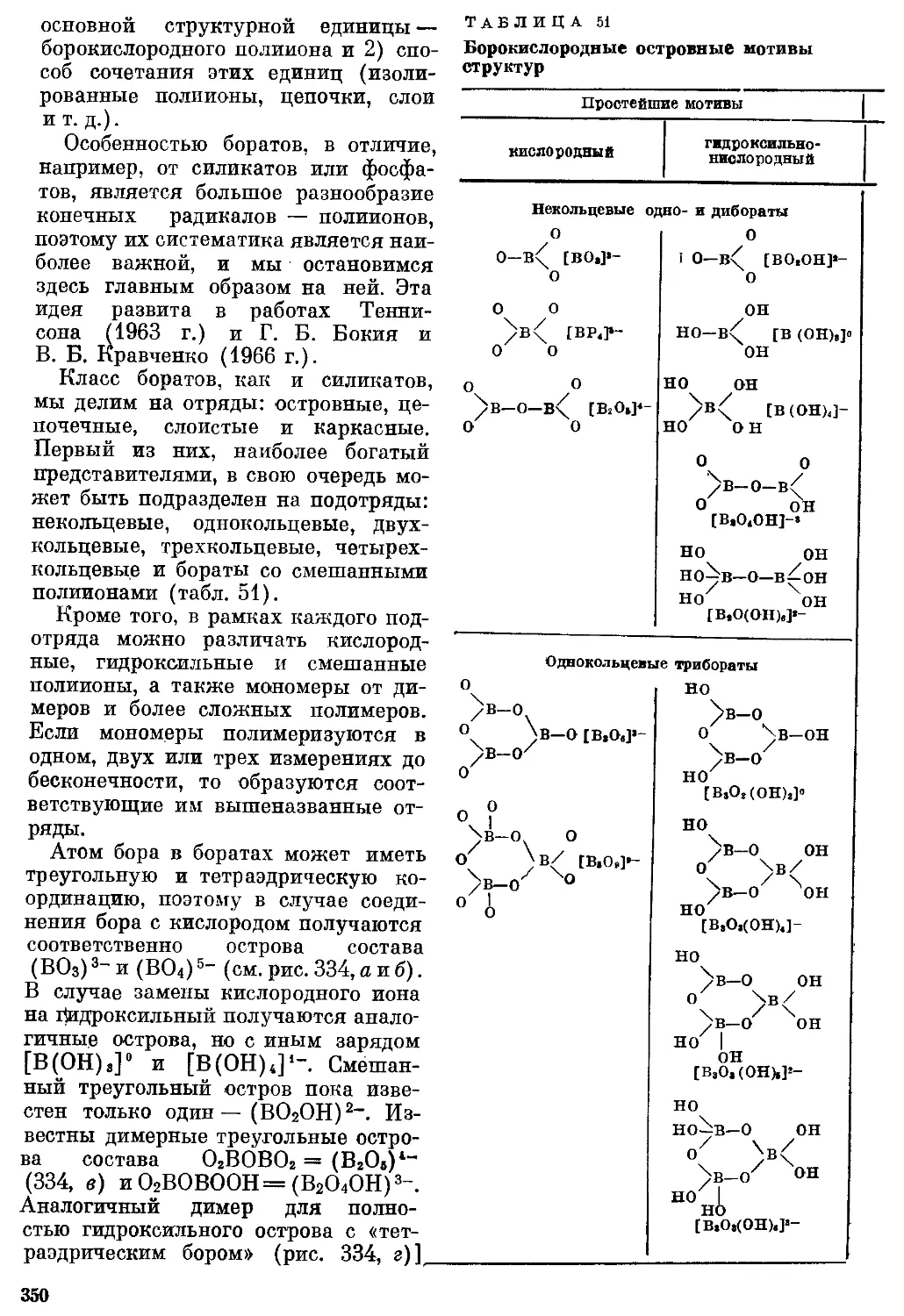











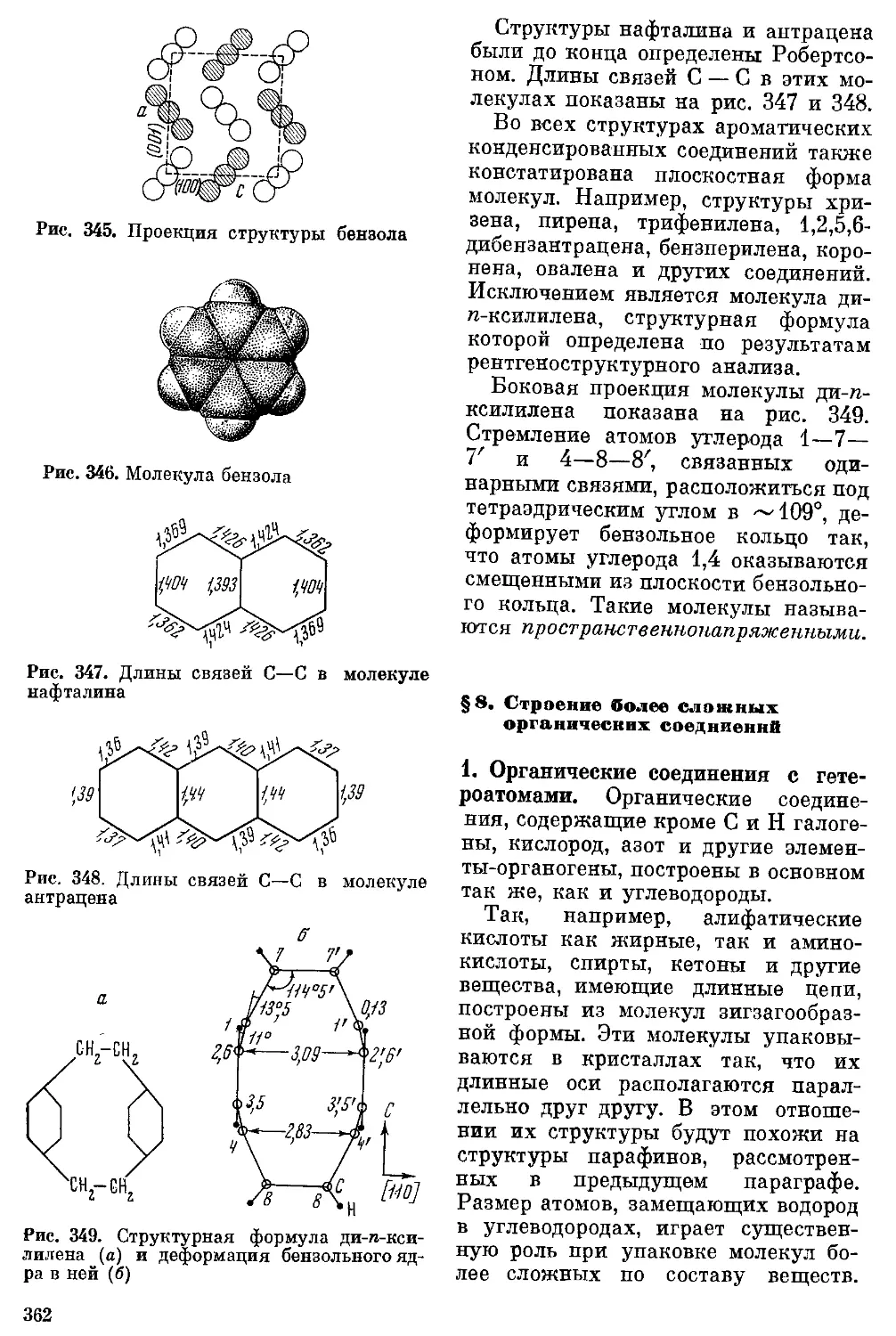























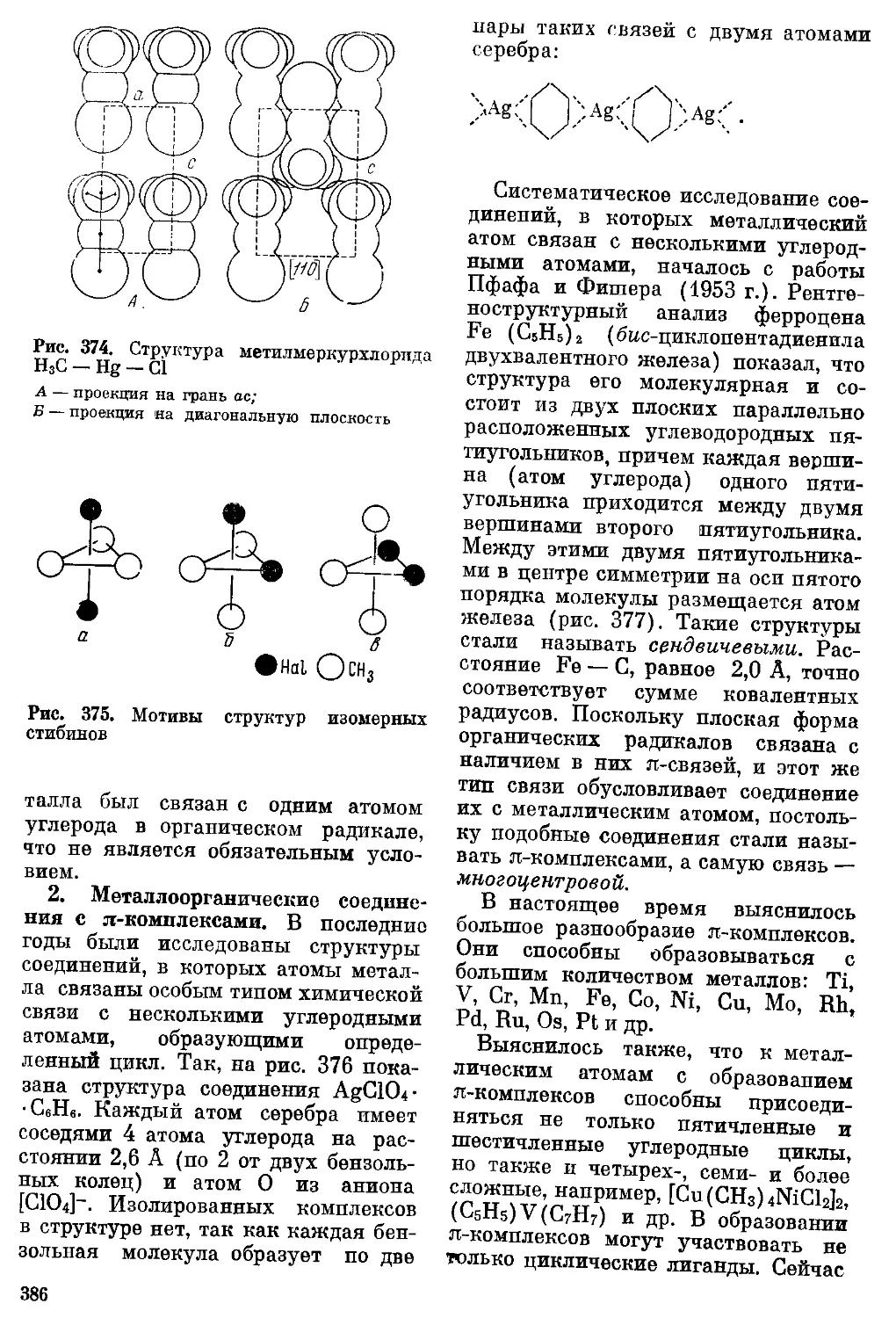



















Текст
АКАДЕМИЯ НАУК СССР
Институт
раднотехввкв в электронвкв
Г. В. БОКИЙ
КРИСТАЛЛОХИМИЯ
ИЗДАНИВ ТРВТЬВ,
ПЕРЕРАБОТАННОЕ И ДОПОЛНЕННОЕ
ИЗДАТЕЛЬСТВО «НАУКА»
МОСКВА
1971
УДК 548.3
Кристаллохимия. Изд. третье, перерабо-
переработанное и дополненное. Б о к и й Г. Б. Изд-
во «Наука», 1971 г., стр. 400.
Книга посвящена современной крис-
кристаллохимии. В ней рассмотрены основные
понятия кристаллохимии, изложены зако-
законы геометрической кристаллографии, опи-
описаны геометрические структуры кристал-
кристаллов. Приведены сведения о кристаллохими-
ческлх особенностях основных типов хи-
химических соединений.
Книга рассчитана на специалистов в
области физики и химии твердого тела,
преподавателей, аспирантов и студентов
вузов.
Таблиц 57. Иллюстраций 382. Библ.
235 назв.
2-5-3
284-1971 A)
ДРЕДИСЛОВИЕ К ТРЕТЬЕМУ ИЗДАНИЮ
Третье издание книги, как и преды-
предыдущие, посвящено систематическо-
систематическому изложению основ кристаллохимии.
Со времени второго издания прошло
более десяти лет. За это время в ми-
мировой литературе опубликовано мно-
много новых сведений об атомном строе-
строении кристаллов, которые в одних раз-
разделах существенно дополнили извест-
известные ранее факты, в других — застави-
заставили пересмотреть прежние представ-
представления. Эти изменения автор поста-
постарался отразить в новой книге. Значе-
Значение кристаллохимии за прошедшие
годы значительно возросло. Такие
области использования кристаллов,
как полупроводники и сверхпровод-
сверхпроводники, пьезо- и сегнетоэлектрики,
квантовая электроника и многие дру-
другие области науки и техники, потре-
потребовали более глубокого понимания
зависимости физических свойств кри-
кристаллов от их химического состава и
строения, а эти вопросы и являются
основным содержанием кристаллохи-
кристаллохимии.
Подавляющее большинство прибо-
приборов новой техники в качестве обяза-
обязательной детали содержит какой-либо
кристалл, обладающий особыми свой-
свойствами. Без этих кристаллов невоз-
невозможно существование самого прибо-
прибора, как невозможно существование
организмов, лишенных такого важ-
важнейшего органа, как сердце. Без пре-
преувеличения можно сказать, что все
приборы новой техники обязательно
содержат монокристальные «сердца».
Изучению атомного строения кри-
кристаллов, изучению зависимости физи-
физических и химических свойств от их
состава и строения посвящена на-
настоящая книга.
По сравнению с предыдущим изда-
изданием, вышедшим в 1960 г., сущест-
существенно дополнены третья и четвертая
части; в первых двух частях лишь
устранены опечатки и исправлены
другие незначительные дефекты. Вто-
Вторая половина книги значительно из-
изменена и расширена, в нее помещен
не только общепринятый, хорошо
проверенный материал, но и в не-
небольшом количестве включен мате-
материал, несколько дискуссионный, но
более близкий автору по его научный
интересам; в частности, в некоторые
разделы внесены данные, содержа-
содержащиеся в собственных оригинальных
работах. Сюда относятся вопросы
кристаллохимической систематики
веществ, изучение закономерности
трансвлияния, геометрической теории
дальтонидов и бертоллидов, строения
комплексных соединений с кратными
связями и некоторые другие.
При подготовке нового издания бы-
были использованы советы и замечания,
которые сделали автору коллеги в
устном и письменном виде. В связи
с этим автор хотел бы поблагодарить
Н. В. Белова, Е. И. Гладышевского,
Г. Г. Дворянкину, В. Б. Кравченко,
Ю. Б. Кузьму, В. И. Соболевского,
В. А. Франк-Каменецкого, С. Д. Чет-
Четверикова и И. И. Шафрановского. Ав-
Автор особенно благодарен К. П. Гуро-
Гурову, В. Ф. Дворянкину и П. И. Кри-
пякевичу, сделавшим критические за-
замечания и дополнения соответственно
в XII и XX главы. Автор также бла-
благодарен Э. Е. Буровой, оказавшей су-
существенную помощь в технической
подготовке рукописи, и И. Г. Соколо-
Соколовой, взявшей на себя труд составле-
составления библиографии и оказавшей су-
существенную помощь по устранению
неточностей в тексте.
Библиография приведена в кон-
конце книги в алфавитном порядке.
Г. Б. Бокий
ВАЖНЕЙШИЕ ТАБЛИЦЫ
Стр.
ТАБЛИЦА 4
32 видов симметрии 32
ТАБЛИЦА 5
Проекции 32 видов симметрии 33
ТАБЛИЦА 7
Пространственные группы симметрии 70
ТАБЛИЦА 11
Ионные радиусы 138
ТАБЛИЦА 17
Периодическая система элементов
Д. И. Менделеева 162
ТАБЛИЦА 19
Первые потенциалы ионизации 167
ТАБЛИЦА 20
Энергии сродства некоторых атомов к
первому электрону 168
Тетраэдрические радиусы по Полингу
и Хаггинсу 181
ТАБЛИЦА 25
Важнейшие пространственные распо-
расположения валентных связей, получаю-
получающиеся в результате различных слу-
случаев гибридизации 196
ТАБЛИЦА 28
Значения электроотрицательности 213
ТАБЛИЦА 35
Атомные радиусы 285
ТАБЛИЦА 52
Величины ковалентных и Ван-дер-
Ваальсовых радиусов неметалличе-
неметаллических элементов 355
ТАБЛИЦА 57
Ковалентные радиусы при связях раз-
разной кратности 384
ЧАСТЬ ПЕРВАЯ
ЗАКОНЫ
ГЕОМЕТРИЧЕСКОЙ
КРИСТАЛЛОГРАФИИ
ГЛАВА I
ПОНЯТИЕ О КРИСТАЛЛЕ, КРИСТАЛЛИЧЕСКОМ ВЕЩЕСТВЕ
И КРИСТАЛЛОГРАФИИ
1. Кристаллическое вещество
Находящиеся в природе вещества
обычно встречаются в одном из трех
основных агрегатных состояний: газо-
газообразном, жидком или твердом. По-
Последнее является почти идентичным
кристаллическому состоянию. Каждое
состояние отличается от другого ха-
характером движения материальных ча-
частиц относительно друг друга.
В газах наблюдается беспорядочное
движение. Притяжением материаль-
материальных частиц — молекул или атомов —
друг к другу в газе можно пренебречь
и в первом приближении считать, что
при взаимном столкновении такие
частицы отталкиваются по закону уп-
упругих шаров.
В жидкости движение частиц зна-
значительно замедлено, имеется времен-
временная упорядоченность благодаря силам
взаимодействия — силам притяже-
притяжения. Расстояния между частицами
значительно сокращены по сравнению
с расстояниями в газе, о чем легко
¦судить по совершенно различной
сжимаемости газов и жидкостей.
В кристаллах материальные части-
частицы, например молекулы, ориентиро-
ориентированы относительно друг друга. В ре-
результате этого кристалл принимает
определенную форму в виде какого-
либо многогранника. Материальные
частицы совершают тепловые колеба-
колебания около положений равновесия.
Если температура далека от темпера-
температуры плавления кристалла, то в нем,
как правило, частицы почти никогда
не движутся поступательно. В этом
заключается резкое отличие кристал-
кристалла от жидкости.
Природа сил притяжения мате-
материальных частиц во всех агрегатных
состояниях всегда электрическая. Так,
в молекулярном кристалле, т. е. в кри-
кристалле, построенном из нейтральных
частиц — молекул, относительная
ориентировка этих частиц может
быть связана, например, с наличием
у них электрических диполей (рис. 1).
-х-
^c
^Zb*~. ^*ц^ . **<, /v
Рис. 1. Проекция кристаллического много»
гранника (показана ориентировка моле*
кул)
Чтобы из беспорядочного (жидко-
(жидкого) состояния вещество перешло в
упорядоченное (кристаллическое),
всегда необходимо некоторое время.
Оно носит название времени кристал-
кристаллизации. Бели охлаждение и застыва-
застывание происходит быстрее, чем время,
необходимое для кристаллизации,
то образуется аморфное, или стекло-
стеклообразное тело, в котором частицы
остаются неупорядоченными, как в
жидкости.
2-/50
Н5(Г
Рис. 2. Прочность кристалла (в Т/мм*} то»
варенной соли в зависимости от направ-
направления
§2. Основные свойства
кристалла
Одним из основных свойств кри-
кристаллов является анизотропия, т. е.
неравносвойственность. Под этим тер-
термином понимается изменение свойств
в зависимости от направления.
Так, например, если вырезать из
кристалла поваренной соли в различ-
различных направлениях стержни попереч-
поперечным сечением в 1 мм2 и испытывать
их на разрыв, то окажется, что они
имеют различную прочность. Так,
стержень, вырезанный перпендику-
перпендикулярно к одной паре граней куба и
параллельно другим, разорвется при
приложении к нему силы в 570 Г/мм2
(рис. 2). Такой же стержень, выре-
вырезанный параллельно диагонали грани
куба, разорвется при усилии в
1150 Г/мм2. Стержень же, вырезан-
вырезанный по пространственной диагонали
куба, окажется самым прочным.
Разрыв наступит при усилии, превы-
превышающем 2150 Г/мм2. Если бы мы вы-
вырезали такие стержни из стекла или
какого-либо другого изотропного те-
тела, то независимо от направления
они разрывались бы при одинако-
одинаковой нагрузке. Этим и отличаются
анизотропные вещества от изотроп-
изотропных.
Вместе с тем кристаллы являются
телами однородными. Однородность
кристаллического вещества состоит в
том, что два его участка одинаковой
формы и одинаковой ориентировки
Рис. 3. Иллюстрация однородности кри-
кристаллического вещества
одинаковы по своим свойствам
(рис. 3).
Третьим важным свойством кри-
кристаллического вещества является его
способность образовывать плоскост-
плоскостные многогранники, так называемая
способность самоогранения. Это свой-
свойство также является следствием внут-
внутренней (атомной) упорядоченности.
Если кристалл во время роста не
встречает механических препятствий,
то он вырастает в виде выпуклого
многогранника.
И, наконец, важнейшим свойством
кристалла является его симметрия.
Подробно о ней будет сказано ниже.
§ 8. Кристалл
¦ кристаллическое вещество
В отличие от газообразного и жид-
жидкого кристаллическое состояние зна-
значительно многообразнее. Одни и те
же по составу и форме молекулы мо-
могут быть упакованы в кристаллах раз-
разными способами (рис. 4, а — г). От
способа же упаковки зависят физико-
химические свойства вещества. Та-
Таким образом, одни и те же по хими-
химическому составу вещества могут и на
самом деле часто обладают различны-
различными физическими свойствами. Для
жидкого состояния такое многообра-
многообразие совсем не характерно, а для га-
газов — невозможно.
Если взять, например, обычную по-
поваренную соль, то легко увидеть даже
без микроскопа отдельные кристалли-
кристаллики. Каждый кристаллик есть вещест-
вещество NaCl, но одновременно он имеет
и черты индивидуума. Он может быть
большим или малым, кубическим
или прямоугольно - параллелепипе-
дальным, по-разному ограненным и
т. д.
В жидкости нельзя увидеть отдель-
отдельных индивидуумов — капель, в кри-
кристаллическом же веществе они ви-
видимы.
Если посмотреть под микроскопом
металлический шлиф (рис. 5), то и в
нем обнаруживаются отдельные кри-
кристаллики с присущими им индиви-
индивидуальными чертами. Если мы хотим
подчеркнуть, что имеем дело с оди-
одиночным, отдельным кристаллом — с
кристаллическим индивидуумом, то
мы называем его монокристаллом.
Под термином «кристалл» во многих
книгах подразумевается как одиноч-
одиночный кристалл, так и кристаллическое
вещество. Чтобы подчеркнуть, что
иногда мы имеем дело со скоплением
многих кристаллов, пользуются тер-
термином кристаллический агрегат.
Показанный на рис. 5 метал-
металлический шлиф является хорошим
примером кристаллического агрегата.
В нем отдельные кристаллы почти не
огранены. Это часто имеет место при
быстрой кристаллизации, начавшейся
одновременно во многих точках рас-
расплава. Растущие кристаллы являются
препятствием друг другу и мешают
правильному огранению каждого из
них.
\r x X 1С
в
Рис. 4. Различные способы упаковки одно-
однородных частиц
Рис. 5. Фотография металлического шли*-
фа с большим увеличением
4. Кристаллография
Кристаллография занимается изу-
изучением многообразия кристаллов, кан
ботаника — многообразием растений,,
химия — химических соединений и
т. п.
Она выявляет признаки единства
(законы) в этом многообразии; ис-
исследует свойства и строение (струк-
(структуру) одиночных кристаллов и кри-
кристаллических агрегатов. Кристалло-
Кристаллография изучает протекающие в кри-
кристаллах явления, взаимодейстие кри-
кристалла со средой, изменения, происхо-
происходящие в кристаллах под влиянием тех
или иных воздействий. Одним словом,
кристаллография является наукой,
всесторонне изучающей кристалли-
кристаллическое вещество.
Обычно кристаллографию делят на
три раздела: геометрическая кристал-
кристаллография, химическая кристаллогра-
кристаллография (кристаллохимия) и физичес-
кая кристаллография (кристаллофи-
(кристаллофизика). Последние два раздела могут
изучаться независимо друг от друга,
но оба они базируются на первом, без
знания которого невозможно их ра-
рациональное изложение.
В силу того что кристаллическое
вещество, в отличие от других, некри-
некристаллических веществ, имеет упоря-
упорядоченную атомную структуру и ани-
анизотропно, методы кристаллографии
резко отличаются от методов других
наук. Симметрия проявляется во
внешней форме кристаллов, в их
структуре, в физических явлениях,
протекающих в кристаллах, во
взаимодействии кристалла с окру-
окружающей средой, в изменениях,
претерпеваемых кристаллом под
влиянием внешних воздействий.
Поэтому особенностью метода кри-
кристаллографии является последова-
последовательное применение принципа сим-
симметрии во всех случаях. Благодаря
этому весьма специфическому методу
кристаллография является самостоя-
самостоятельной наукой, связанной с другими
частичным совпадением задач и пред-
предмета исследования в конкретных слу-
случаях.
Нельзя изучать кристалличес-
кристаллическое вещество вне процесса его образо-
образования, вне связи с жидкой и газооб-
газообразной фазой. Эти процессы изучает
физическая химия, так как любой
процесс или положение равновесия
зависят от физико-химических усло-
условий среды. Относительное располо-
расположение атомов и молекул в кристалли-
кристаллическом веществе зависит от качества
•самих атомов, от их химической при-
природы. Отсюда тесная связь с химией,
особенно со стереохимией. Атомы и
молекулы в кристаллах образуют гео-
геометрически правильные комплексы.
Совокупность их определяет форму
кристаллов в виде многогранников.
Многогранники же изучаются мате-
математикой и, в первую очередь, геомет-
геометрией. Очевидна, конечно, связь кри-
кристаллографии с физикой, особенно
с теми ее разделами, которые зани-
занимаются изучением различных свойств
твердых тел. В последние годы интен-
интенсивно развивается промышленность,
использующая монокристаллы с раз-
различными свойствами: оптическими,
электрическими, механическими и
т. п. Связь кристаллографии с химией,
физической химией и физикой на-
настолько тесная, что не позволяет про-
провести даже условных границ между
этими науками.
Очевидна связь кристаллографии
и с геологическими дисциплинами,
прежде всего с минералогией, геохи-
геохимией и петрографией. Подавляющее
количество минералов кристаллично,
и так как многие из них известны в
виде хорошо образованных кристал-
кристаллов, то на заре своей истории кри-
кристаллография рассматривалась как
часть минералогии. Внешняя форма
кристаллов остается до сих пор важ-
важнейшим диагностическим признаком
минералов. А кристаллохимическое
исследование атомных структур ми-
минералов является основой современ-
современной их систематики. Некоторые кри-
кристаллографические методики иссле-
исследования играют важную роль в гео-
геологических дисциплинах; так, на-
например, кристаллооптический анализ
является сейчас основным методом
петрографии.
§ 5. Распространенность
кристаллического вещества
В давние времена считалось, что
кристаллы представляют собой
большую редкость. Действительно,
нахождение в природе крупных од-
однородных кристаллов — явление не-
нечастое. Однако мелкокристалличе-
мелкокристаллические вещества встречаются весьма
часто. Выше было сказано, что твер-
твердое состояние материи обычно экви-
эквивалентно кристаллическому состоя-
состоянию. Так, например, почти все гор-
горные породы: граниты, песчаники, из-
известняки и т. п. кристалличны. Кри-
сталличны почти все руды, являю-
являющиеся сырьем металлургической про-
промышленности. Кристалличны также
и те продукты металлургической
10
промышленности, которые получают-
получаются в результате переработки руд, —
все металлы и их сплавы. Из мелких
кристалликов состоят также все стро-
строительные материалы. Большинство
твердых продуктов химической про-
промышленности также кристаллично
(квасцы, селитра, купорос, сода, наф-
нафталин и т. д.), а жидкие химические
продукты, например ряд продуктов
нефтяных производств или неоргани-
неорганические кислоты, легко могут быть по-
получены в кристаллическом состоянии
при низких температурах. По мере
совершенствования методов исследо-
исследования (сначала визуальные методы,
затем микроскопия, рентгеновский
анализ, электронография и т. п.)
кристалличными оказывались веще-
вещества, считавшиеся до того аморф-
аморфными.
Сейчас мы знаем, что даже некото-
некоторые части организма кристалличны,
например роговица глаза. Рентгено-
структурное и кристаллохимическое
исследование белков и вирусов вы-
выросло в особое научное направление.
Несомненно дальнейшее внедрение
кристаллографических методов ис-
исследования в химию и биологию.
§6. Кристаллизация.
Производство монокристаллов
Особое место в кристаллографии за-
занимает кристаллизация. Процессы
получения кристаллов из газообраз-
газообразных, жидких и твердых фаз изуча-
изучаются сейчас весьма интенсивно. Они
нужны не только для развития тео-
теории фазовых переходов. Сейчас мы
являемся свидетелями интенсивного
роста промышленности монокристал-
монокристаллов во всем мире.
Многообразие кристаллов и их
специфические свойства в настоящее
время интенсивно используются в
науке и технике. Известно, например,
что для изготовления оптических
призм и линз, пропускающих ультра-
ультрафиолетовые и инфракрасные лучи,
используются монокристаллы NaCl,
LiF, CaF2, SiO2 и др. Получить эти
вещества в виде крупных совершен-
совершенных монокристаллов—задача весь-
весьма трудная. Между тем техника тре-
требует от нас с каждым днем все боль-
больших и больших количеств новых мо-
монокристаллов. Широкое распростра-
распространение получили в промышленности
пьезокристаллы кварца БЮг, сегнето-
вой соли dH^KNa • 4Н2О и др. Для
точных приборов, в частности для
часов, подшипники изготовляются из
рубинов — АЬОз с небольшой при-
примесью СггОз.
Новый скачок в развитии промыш-
промышленности монокристаллов рубинов
произошел после того, как они стали
использоваться в качестве лазеров.
Применение различных кристаллов
для квантовых генераторов, люмине-
люминесцентных кристаллов и монокристал-
монокристаллов полупроводников уже создало це-
целую зпоху в науке и промышлен-
промышленности.
Получение кристаллов обычно свя-
связано с применением сверхчистых ве-
веществ, поэтому задачи кристаллогра-
кристаллографов, химиков и физиков слились в
этой проблеме воедино. Следователь-
Следовательно, специалистам, работающим в об-
области производства монокристаллов,
а также студентам химических вузов
и факультетов, готовящимся к по-
подобной работе, необходимо знание
основ кристаллографии и особен-
особенно — кристаллохимии.
ГЛАВА II
ЗАКОН ПОСТОЯНСТВА ДВУГРАННЫХ УГЛОВ
В КРИСТАЛЛАХ
1. Первые работы,
носвнщенные научению
внешней форны кристаллов
Хорошо ограненные кристаллы очень
давно привлекали внимание челове-
человечества. Нахождение их в природе в
древности связывалось с фантастиче-
фантастическими представлениями. Хорошо об-
образованные кристаллы наделялись
особыми мистическими свойствами.
Им приписывалась способность исце-
исцелять от болезней или, наоборот, вы-
вызывать некоторые заболевания, вли-
влиять на судьбу человека и т. п. Такое
особое отношение к кристаллам спо-
способствовало их коллекционированию,
а последнее вызывало более деталь-
детальные наблюдения их формы, что и
привело, в конце концов, к настоя-
настоящим научным обобщениям.
Уже в 79 г. нашего летоисчисления
Плиний Старший упоминает о пло-
скогранности и прямореберности
кристаллов. Этот вывод и может счи-
считаться первым обобщением геометри-
геометрической кристаллографии.
С тех пор на протяжении многих
столетий весьма медленно и посте-
постепенно накапливался материал, поз-
позволивший в конце XVIII в. открыть
важнейший закон геометрической
кристаллографии — закон постоян-
постоянства двугранных углов в кристал-
кристаллах.
Этот закон связывается обычно с
именем французского ученого Роме
де Л'Иля, который в 1783 г. опубли-
опубликовал монографию, содержащую
обильный материал по измерению
углов природных кристаллов.
Для кристаллов каждого вещества
(минерала), изученного им, оказа-
оказалось справедливым положение, что
углы между соответственными гра-
гранями во всех кристаллах одного и то-
того же вещества являются постоян-
постоянными. Заслуга Роме де Л'Иля как
раз и заключается в том, что он рас-
распространил закон постоянства углов
на все вещества, кристаллы которых
он мог достать и измерить. Эта ра-
работа явилась плодом всей его долгой
жизни. Ее результаты он системати-
систематически докладывал ученым в Париже.
Эти сообщения явились первыми лек-
лекциями по кристаллографии.
Не следует думать, что у Роме
де Л'Иля не было предшественни-
предшественников. История открытия закона по-
постоянства углов прошла огромный,
почти двухвековой путь, прежде чем
этот закон был отчетливо сформу-
сформулирован и обобщен для всех кри-
кристаллических веществ. Так, напри-
например, И. Кеплер уже в 1615 г. указы-
указывал на сохранение углов в 60° меж-
между отдельными лучиками у снежи-
снежинок. В 1669 г. Н. Стенсен открыл
закон постоянства углов в кристал-
кристаллах кварца и гематита. Годом позже
Э. Бартолин сделал тот же вывод
применительно к кристаллам каль-
кальцита, а в 1695 г. Левенгук — к кри-
кристаллам гипса. Он показал, что и
у микроскопически малых и у боль-
больших кристаллов гипса углы между
соответственными гранями одина-
одинаковы.
В России закон постоянства углов
был открыт М. В. Ломоносовым для
12
кристаллов селитры A749 г.), пи-
пирита, алмаза и некоторых других
минералов. Одновременно Ломоно-
Ломоносов предложил стройную гипотезу о
молекулярном строении селитры,
ю чем подробнее будет сказано
ниже.
Весьма многочисленные иссле-
исследования кристаллов химических
соединений, полученных в лабора-
лаборатории, были проведены преемником
Ломоносова по кафедре химии в
Российской академии наук Т. Е.
Ловицем.
В качестве примера рассмотрим
рисунок различных по форме кри-
кристаллов кварца SiO2 из работы Роме
де Л'Иля (рис. 6). Все кристаллы
обладают тем свойством, что углы
между соответственными гранями
постоянны. Грани у отдельных кри-
кристаллов могут быть развиты по-
разному. Грани, наблюдающиеся на
одних кристаллах, могут отсутство-
отсутствовать на других. Но если мы будем
измерять углы между соответствен-
соответственными гранями (например, а, Ъ и с
на рис. 6), то значения этих углов
будут оставаться постоянными не-
независимо от формы кристалла.
b
\
с
/
U-J
с
'-
\J
1
и
и
-у
с
Рис. 6. Рисунок из книги Роме де Л'Иля
A783 г.), иллюстрирующий закон постоян-
постоянства углов на примере различных по фор-
форме кристаллов кварца
Одинаковые простые формы обозначены соот-
соответственно буквами а, б и с
§2. Методы намерения
кристаллов
Всю работу по измерению кристал-
кристаллов Роме де Л'Иль провел с по-
помощью прикладного гониометра, изо-
изобретенного его учеником Каранжо.
Этот прибор (рис. 7) представляет
собой транспортир с линейкой, вра-
вращающийся вокруг оси, проходящей
через его центр. Прикладывая кри-
кристалл так, чтобы одна из его граней
касалась края транспортира, а вто-
вторая — подвижной линейки, можно
прочесть по шкале транспортира от-
отсчет, соответствующий двухгран-
двухгранному углу между гранями кристал-
кристалла. На рисунке показано измерение
угла между гранями а и Ъ в кри-
кристаллах кварца, изображенных на
рис. 6. Ясно, что точность измере-
измерения кристаллов с помощью при-
прикладного гониометра невелика. Она
достигает величины примерно в пол-
полградуса.
Достаточно высокую точность дал
отражательный гониометр, изобре-
изобретенный в 1809 г. Волластоном.
Принцип его сводится к тому, что
на вращающемся столике, снабжен-
снабженном лимбом, определенным образом
ориентируется измеряемый кри-
кристалл (рис. 8). В плоскости, парал-
параллельной лимбу, располагаются две
оптические трубы. Из одной на кри-
кристалл падает параллельный пучок
света, который после отражения от
грани кристалла попадает в другую
трубу. Разность отсчетов между от-
отражениями от двух граней дает угол
между ними. Точность такого изме-
измерения достигает секунды. Однако
преимущество отражательного го-
гониометра перед прикладным не
только в точности измерения. Для
работы на отражательном гониомет-
гониометре требуются кристаллы значитель-
значительно меньшей величины. Это обстоя-
обстоятельство позволило значительно рас-
расширить круг исследуемых ве-
веществ.
Недостатком отражательного го-
гониометра Волластона является то,
13
что он снабжен только одним лим-
лимбом и позволяет производить изме-
измерения не всего кристалла, а лишь
одной его зоны (под зоной подразу-
подразумевается совокупность граней, пе-
пересекающихся в параллельных реб-
ребрах). Для измерения граней, распо-
расположенных в разных зонах, приходи-
приходилось каждый раз переклеивать и за-
заново юстировать (т. е. совмещать
нужное направление в кристалле с
осью прибора) кристалл, что, ко-
конечно, снижает точность и удлиня-
удлиняет время полного измерения кри-
кристалла.
Последним этапом в усовершен-
усовершенствовании методики измерения кри-
кристаллов было изобретение Е. С.
Федоровым A889 г.) двукружного
(теодолитного) гониометра. Этот
прибор не имеет указанного выше
недостатка. Все кристаллографиче-
кристаллографические лаборатории мира перешли
постепенно на измерение кристал-
кристаллов с помощью двукружного гонио-
гониометра.
Федоров одновременно с разра-
разработкой методики измерения на дву-
кружном гониометре разработал си-
систему математической обработки
результатов этих измерений.
Кристалл К (рис. 9) при помо-
помощи взаимно-перпендикулярных пря-
прямых и цилиндрических салазок
(юстирный — Ю и центрирный — Ц
аппараты) приводится в определен-
определенное положение по отношению к оси <р
гониометра. Вращая кристалл вме-
вместе с юстирным и центрирным аппа-
аппаратами вокруг осей ф и р, мы можем
поставить любую его грань в такое
положение, при котором отсвет от
нее попадает в зрительную трубу.
Это положение будет фиксироваться
двумя отсчетами — по лимбам <р
и р. Поставив последовательно в та-
такое положение все грани кристалла
и взяв для каждой из них отсчеты по
Ф и р, мы будем иметь численные
величины, характеризующие все
углы между гранями измеряемого
кристалла, иначе — их сферические
координаты.
Рис. 7. Измерение угла между гранями
кристалла кварца с помощью прикладно-
прикладного гониометра
Рис. 8. Принцип устройства отражательно-
отражательного гониометра
К — кристалл;
S — источник света;
С — коллиматор;
/ — окуляр;
Н — градуированный лимб;
п — нониус;
о,, о2 — грани кристалла;
JVi, N2 — нормали к граням
Рис. 9. Схематическое изображение дву-
двукружного гониометра
К — кристалл;
Ц — центрировочные,
Ю — котировочные салазки на гониометриче-
гониометрической головке
14
§3. Методы вычисления крае галлов
Под вычислением кристаллов подра-
подразумевается система математической
обработки результатов измерения
кристаллов на гониометре.
Е. С. Федоров одновременно с
разработкой методики измерения
кристаллов на двукружном гонио-
гониометре разработал и систему соответ-
соответствующих вычислений. Графические
методы расчетов кристаллов нашли
свое завершение в работах другого
замечательного русского кристалло-
кристаллографа — Ю. В. Вульфа.
При обработке результатов изме-
измерения кристалла пользуются обыч-
обычно следующим приемом. Центр
кристалла помещается в центр шара
(рис. 10). Из центра кристалла опус-
опускаются перпендикуляры на все его
грани и продолжаются до пересече-
пересечения с шаром. После этого кристалл
можно отбросить. Мы его заменили
пучками прямых, или полупрямых.
При этом исчезают отличия в форме
граней различных кристаллов. Это
ясно видно из рис. 11, где в плоскос-
плоскости, перпендикулярной главной оси,
показаны разрезы трех кристаллов
кварца. Если заменить каждый из
этих кристаллов кристаллическим
пучком, то эти пучки будут тождест-
тождественны между собой, несмотря на
различие в формах кристаллов. Кри-
Кристаллической пучок характеризует
набор углов между гранями кристал-
кристалла, т. е. сохраняет наиболее важную
его характеристику, соответствую-
соответствующую закону постоянства углов.
Угол между прямыми в этом пучке
является дополнительным до 180° к
углу между гранями.
После отметки точек на шаре
(рис. 10) можно отбросить и кристал-
кристаллический пучок, так как сферический
угол между точками на шаре отвеча-
отвечает углу между соответственными пря-
прямыми кристаллического пучка.
В последней стадии эти точки со
сферы проектируются на ее эква-
экваториальную плоскость (рис. 12). По-
Получаем так называемую стереогра-
Рис 10. Принцип сферической проекцию
Ш
\mm
Рис. 11. Иллюстрация параллельности со-
соответственных граней кристаллов кварца'
с различной внешней огранкой
1—111 — вид сверху,
IV — все три проекции наложены друг
на друга
Рис. 12. Принцип стереографической про-
проекции
Черные кружки — выходы нормалей на шаре,
белые — их проекции на экваториальную плос-
плоскость (стереографическая проекция)
15
у =270° fl=aO°
V
у =90°
Рис. 13. Сетка Вульфа
-фическую проекцию кристалла. Та-
Таким образом мы заменяем трехмер-
трехмерный образ двумерным. Чтобы опре-
определить по проекции углы между гра-
гранями, надо воспользоваться специаль-
специальной сеткой — сеткой Вульфа. Сама
проекция точек делается обычно на
восковке, под которую подкладывает-
ся транспарант — сетка Вульфа
(рис. 13).
Для измерения угла между двумя
точками на стереографической про-
проекции совмещаем центр восковки с
центром сетки Вульфа и вращаем
первую относительно второй, пока
точки не попадут на один из мери-
меридианов сетки Вульфа. По меридиа-
меридиану отсчитываем угол. В сетке Вульфа
деления проведены через 2°, так что
работа на ней позволяет вести вы-
вычисления до 'АЛ Промежуток между
линиями делится на глаз на 4 части.
Диаметр сетки 20 см.
§4. Отклонения от закона
постоянства углов
По мере совершенствования методи-
методики и повышения точности измере-
измерения кристаллов выяснилось, что за-
закон постоянства углов оправдывает-
оправдывается лишь приблизительно. В одном
и том же кристалле углы между оди-
одинаковыми по типу гранями слегка от-
отличаются друг от друга.
Ниже в качестве примера приве-
приведены результаты измерения хорошо
образованного кристалла шпинели
MgA^CU- Они имеют форму октаэдра
(рис. 14). В этом многограннике
имеется 12 равных друг другу дву-
двугранных углов G0°31'44"), соответ-
соответствующих 12 его ребрам. Разброс
измеренных величин углов равен
Рис. 14. Октаэдр
4'35". Отклонения от идеального зна-
значения -М'56" и —2'39":
70°31'25" 70°33'40* 70°31'50" 70°31'15"
70'29'05" 70*31'20" 702'35" 70°31'45*
700'30" 70'32'00" 70°32'00" 70°32'50*
Как сказано выше, для измерения
был взят очень хорошо образованный
кристалл. У многих веществ отлоне-
аия двугранных углов между соответ-
соответственными гранями достигают 10—
20', а в некоторых случаях и градуса.
Грани реального кристалла никог-
никогда не представляют собой идеальных
плоских поверхностей. Нередко они
бывают покрыты ямками или бугор-
бугорками роста, в некоторых случаях
грани представляют собой кривые
поверхности, например у кристал-
кристаллов алмаза. Иногда замечаются на
гранях плоские участки, положение
которых слегка отклонено от пло-
плоскости самой грани, на которой они
развиваются. Эти участки называют
в кристаллографии вицинальными
гранями, или просто вициналями.
Вицинали могут занимать большую
часть площади нормальной грани, а
иногда даже полностью заменить
последнюю. Часто на гранях наблю-
наблюдаются ступеньки, иногда имеющие
спиральную форму (рис. 15). Таким
образом, можно говорить о скульп-
скульптуре граней, являющейся предме-
предметом изучения особого раздела кри-
вталлографии — морфологии внешней
формы кристаллов.
Наблюдаются, конечно, и более
закономерные изменения двугран-
двугранных углов кристаллов, например в
зависимости от температуры.
В табл. 1 приведены значения уг-
углов между гранями а и Ъ (рис. 6)
в кварце при разной температуре.
ТАБЛИЦА i
Изменение угла между гранями
в кристалле кварца в зависимости
от температуры
t, °С
-166
0
21
100
200
Угол
1281'54"
128в12'51"
128в13'12"
128в13'36"
128°14'54»
t, "С
300
400
500
550
575
Угоя
128в16'12"
128в17'54'
128в20'12"
128в22'00"
128в23'18'
Рис. 15. Спираль на грани @001) кристал-
кристалла кварца (фото Л. И. Цинобера)
В заключение этого параграфа
необходимо сказать о случаях резко-
резкого изменения углов кристаллов, ко-
которое наступает при полиморфном
превращении веществ (см. гла-
главу XIII), явлении, открытом позже
2 Кристаллохимия
формулировки закона постоянства
углов. Одно и то же вещество при
полиморфном превращении скачком
меняет свои свойства: например, пе-
переход ромбической серы в моно-
моноклинную сопровождается увеличени-
увеличением удельного объема Av = 0,014 см3/г
и термическим эффектом в 3,12 кал/г.
Еще резче меняются свойства кри-
кристаллического углерода при перехо-
переходе алмаза в графит. Плотность алма-
алмаза 3,5, графита 2,2; твердость алма-
алмаза 10, графита 1 и т. д.
При полиморфном превращении
наряду со скачкообразным измене-
изменением физических свойств скачком
меняется и внешняя форма кристал-
кристаллов, при этом совокупность двугран-
двугранных углов одной модификации мо-
может совсем не соответствовать сово-
совокупности двугранных углов другой.
Учитывая все сказанное выше,
можно так сформулировать закон по-
постоянства углов:
во всех кристаллах, принадлежа-
принадлежащих к одной полиморфной модифика-
модификации данного вещества, при одинако-
одинаковых условиях углы между соответ-
соответственными гранями (и ребрами) по-
постоянны.
Г Л А В А III
СИММЕТРИЯ КРИСТАЛЛОВ
§ 1. Понятие о симметрии
Вскоре после опубликования «Кри-
«Кристаллографии» Роме де Л'Иля его
младший соотечественник Гаюи кри-
критически переработал весь материал
этой книги. На этой основе ему уда-
удалось открыть второй важнейший за-
закон геометрической кристаллогра-
кристаллографии — закон рациональности отноше-
отношений параметров (см. главу V).
Одновременно он обратил внима-
внимание на то, что кристаллы являются
симметричными телами. Результатом
развития этой идеи явилось матема-
математически строгое учение о симметрии
кристаллов.
Симметричной фигурой называет-
называется такая фигура, в которой отдель-
отдельные части мысленно могут быть сов-
совмещены друг с другом посредством
симметрического преобразования.
На рис. 16, а изображена сим-
симметричная фигура, а на рис. 16, б —
несимметричная.
Симметрическим преобразованием
называется такое преобразование,
при котором равные части фигуры
совмещаются друг с другом. Так,
отразив левую половину фигуры
(рис. 16, а) в плоскости, перпендику-
перпендикулярной чертежу (пунктирная ли-
линия), мы совместим ее с правой
частью фигуры. Правая часть фигу-
фигуры, отразившись при этом в той же
плоскости, совпадет с левой частью
фигуры. В результате такого симме-
симметрического преобразования фигура
совместится сама с собой.
Каждому симметрическому преоб-
преобразованию соответствует некоторый
геометрический образ. Эти геометри-
геометрические образы называют элементами
симметрии. В разобранном примере
таким геометрическим образом будет
плоскость симметрии. На рис. 16, а
она располагается перпендикулярно
чертежу. Ее след показан пунктирной
линией.
§ 2. Элементы симметрии
На рис. 17 показаны различные сим-
симметричные фигуры. Равнобедренный
треугольник (а) имеет плоскость
симметрии такую же, как домик на
рис. 16, а. Фигура, изображенная на
рис. 17, б, плоскости симметрии не
имеет. Однако она тоже симметрич-
симметрична. Если повернуть фигуру на 180°
вокруг линии, перпендикулярной чер-
чертежу и проходящей через центр фи-
фигуры, то нижняя ее часть совместится
с верхней и наоборот. Эта линия бу-
будет называться осью симметрии. Сим-
Симметрическому преобразованию —
повороту — будет отвечать геометри-
геометрический образ — ось симметрии. По-
Порядком оси называется число совме-
совмещений фигуры при повороте на 360°.
При повороте на 180° фигура 17, б
совместится один раз сама с собой.
Рис. 16. Симметричная (о) и несимметрич-
несимметричная {б) фигуры
2* 19
Точка А при этом совместится с В и
В с А. При повороте на следующие
180° фигура вновь совместится сама
с собой. Минимальный угол поворота,
при котором происходит совмещение
фигуры, называется элементарным
углом поворота оси. Для разобранно-
разобранного случая элементарный угол а ра-
равен 180°, а порядок оси и = 3607а = 2.
Такая ось симметрии называется
двойной осью симметрии, или осью
симметрии второго порядка.
Фигура 17, в обладает осью сим-
симметрии третьего порядка, или трой-
тройной осью симметрии. Элементарный
угол поворота для нее равен
120°=36073.
На рис. 17, г — е показаны фигу-
фигуры, в центрах которых проводят оси
четвертого, пятого и шестого поряд-
порядков. Как было установлено на опыте, в
кристаллах не может быть осей сим-
симметрии 5-го, 7-го и более высоких
порядков. Это эмпирическое правило
затем было строго доказано на осно-
основании теории решетчатого строения
кристаллов (см. главу VI).
Кроме осей и плоскостей симмет-
симметрии, в кристаллах могут быть и дру-
другие элементы симметрии. Одним из
таких элементов симметрии являет-
является центр симметрии, или центр ин-
инверсии.
Симметрическое преобразование,
отвечающее центру симметрии, есть
отражение в точке. На рис. 18 изоб-
изображен косой параллелепипед. Эта
фигура обладает центром симмет-
симметрии — точка С.
Для получения отражения в плос-
плоскости мы из каждой точки отража-
отражаемой фигуры опускаем на плоскость
отражения (плоскость симметрии)
перпендикуляр и продолжаем его на
равные расстояния (рис. 19). Для
получения отражения в точке мы
соединяем все точки фигуры ABDE
(рис. 18) с точкой С и продолжаем
их на соответственно равное рас-
расстояние. В результате получаем
обратнопараллельное изображение
фигуры A'B'D'E'.
Кроме перечисленных выше эле-
Рис. 17. Различные симметричные фигуры
ментов симметрии, в кристаллогра-
кристаллографии встречаются также сложные
оси симметрии: инверсионные и зер-
зеркально-поворотные. Им соответству-
соответствует операция поворота с одновремен-
одновременной инверсией или отражением в
плоскости.
На рис. 20, а изображен шар, на
котором нанесены точки, получаю-
получающиеся из точки 1 в результате пово-
поворота ее вокруг оси третьего поряд-
порядка Ьъ — точки 2 ж 3. Эта фигура об-
обладает тройной поворотной осью,
имеющей, следовательно, элементар-
элементарный угол поворота 120°.
На рис. 20, б — аналогичная фигу-
фигура, но имеющая ось шестого поряд-
D' . В*
Рис. 18. Фигура, обладающая центром сим-
симметрии
Рис. 19. Действие плоскости симметрии
20
ка Z/б и, следовательно, элементар-
элементарный угол поворота 60°.
Зеркально-поворотная ось шестого
порядка Лб показана на рис. 20, в.
Точка 1 после поворота на 60° еще
не совпадает с точкой 2. Для их
совпадения ее необходимо затем
отразить в плоскости чертежа,
тогда она из верхней части сфе-
сферы переместится в нижнюю и совпа-
совпадет там с точкой 2. (Точки, находя-
находящиеся на верхней полусфере, обозна-
обозначены кружками, на нижней — крес-
крестиками.) При этой же операции точ-
точка 2 после поворота фигуры на 60°
окажется под точкой 3, с которой
она совпадает только после отраже-
отражения в плоскости чертежа. При по-
последующем симметрическом преобра-
преобразовании точка 3 совпадает с точкой
4, 4 с 5, 5 с 6 я 6 с 1. В результате
фигура совместится сама с собой.
При полном повороте (на 360°) сов-
совмещение фигуры самой с собой
произойдет 6 раз. Надо обратить
внимание, что фигура в не имеет
отдельно ни оси 6-го порядка, ни
плоскости симметрии; она имеет од-
одну зеркально-поворотную ось шестого
порядка. Одновременно этот элемент
симметрии содержит в себе ось треть-
третьего порядка и центр симметрии. Так,
при элементарном повороте вокруг
оси L3 и последующей инверсии точ-
точка 1 совместится с точкой 6, 6 с 5
и т. д. Следовательно, зеркально-по-
зеркально-поворотная ось шестого порядка явля-
является одновременно инверсионной
осью третьего порядка, т. е. Л6 = Л\.
Инверсионная ось шестого поряд-
порядка показана на рис. 20, г. Точка 1
после поворота на 60° попадет в по-
положение а, а при инверсии — под
плоскость чертежа в положение 2. За-
Затем из положения 2 в положение 3
через вспомогательную точку с
и т. д. Следует обратить внимание, что
фигура г не имеет ни поворотной
оси шестого порядка, ни центра сим-
симметрии; она имеет одну инверсион-
инверсионную ось Lg, но этот элемент симмет-
симметрии содержит в себе ось L3 и пло-
плоскость симметрии. Следовательно,
Рис. 20. Действие осей симметрии:
а — тройной поворотной L3;
б — шестерной поворотной Le;
в — шестерной зеркально-поворотной Л,;
г — шестерной инверсионной Ьв
инверсионная ось шестого порядка
является одновременно зеркально-
поворотной третьего порядка, т. е.
На рис. 21 изображены фигуры,
имеющие оси симметрии Лб и L^.
Для всех операций, связанных с
математической обработкой экспери-
экспериментальных наблюдений и изучения
симметрии кристаллов, достаточно
знания какого-либо одного типа
сложных осей симметрии — зеркаль-
зеркально-поворотных или инверсионных.
Разные авторы предпочитают тот
или иной тип осей в различных
случаях, поэтому знание их необхо-
необходимо.
Рис. 21. Фигуры, обладающие осями сим-
симметрии
а — шестерной зеркально-поворотной;
б — шестерной инверсионной
21
ТАБЛИЦА 2
Элементы симметрии н их обозначения
Элемент симметрии
Обозна-
Обозначение
Изображение по отношению
к плоскости чертежа
перпендикулярное
параллельное
Плоскость симметрии
Центр симметрии
Двойная поворотная ось симметрии
Тройная поворотная ось симметрии
Четверная поворотная ось симметрии
Шестерная поворотная ось симметрии
Четверная инверсионная или зеркально-
поворотная оси симметрии
Шестерная инверсионная ось симметрии
Тройная инверсионная или шестерная зер-
зеркально-поворотная оси симметрии
Р(=т)
С(=1)
U (= 2)
и (= з)
U (= 4)
U (= 6)
// =
С • о
ф
о
*
А
О
С • о
• 1
I—I
Зеркально-поворотные оси воспри-
воспринимаются несколько легче инвер-
инверсионных, их легче определить на мо-
моделях, поэтому при изучении внеш-
внешней формы кристалла ими часто
пользуются. Инверсионными осями
удобно пользоваться при изучении
атомной теории структуры кристал-
кристаллов и базирующейся на ней кристал-
кристаллохимии. Поэтому мы в дальнейшем
будем пользоваться только инвер-
инверсионными осями.
Четверные зеркально-поворотные
оси полностью соответствуют четвер-
четверным инверсионным: Л 4 = Li.
Двойная инверсионная ось экви-
эквивалентна плоскости симметрии, т. е.
Ьг =Р, двойная эеркально-поворот-
ная — центру симметрии: Л2=С.
Для формальной полноты картины
следует еще указать на ось первого
порядка Li. Очевидно, что этой осью
обладают все фигуры, ибо любая из
них после поворота на 360° совпадает
сама с собой. Других элементов сим-
симметрии в кристаллах быть не может.
В табл. 2 собраны все встречаю-
встречающиеся в кристаллографии элементы
симметрии и их обозначения.
Оси симметрии были введены в
кристаллографию Вейссом в 1804 —
1809 гг. Позднее A815 г.) он пред-
предложил деление кристаллов на 6 си-
систем — сингоний (см. ниже).
§3. Сложение элементов симметрии.
Виды Сиииетрии
Симметрические преобразования или
элементы симметрии, им соответ-
соответствующие, являются математиче-
математическими образами, допускающими
22
проведение с ними определенных
математических операций или пре-
преобразований. Так, в частности, эле-
элементы симметрии можно складывать.
Сложение элементов симметрии при-
приводит к появлению равнодействую-
равнодействующего элемента. Равнодействующее
же преобразование (или элемент
симметрии) сразу дает тот резуль-
результат, к которому приводит последова-
последовательное применение исходных эле-
элементов симметрии. Например, после-
последовательное отражение в двух пло-
плоскостях, пересекающихся под уг-
углом а, эквивалентно повороту вокруг
линии пересечения этих плоскостей
на угол 2а в направлении от плоско-
плоскости первого отражения к плоскости
второго отражения.
На рис. 22 / и // — следы двух пло-
плоскостей, перпендикулярных к пло-
плоскости чертежа и пересекающихся
под углом а. Точка А после отраже-
отражения в плоскости / совместится с точ-
точкой В, которая, отразившись в пло-
плоскости //, попадает в положение С.
Из равенства прямоугольных тре-
треугольников А 01 и IOB, а также ВОН
и НОС следует, что угол
АОС=2а. Следовательно, последова-
последовательное отражение точки А в пло-
плоскостях I ж II действительно эквива-
эквивалентно повороту вокруг О на угол 2а.
Справедлива и обратная теорема:
каждый поворот вокруг оси на угол
2а эквивалентен последовательному
отражению в двух плоскостях, пере-
пересекающихся по этой оси под углом а.
Из прямой теоремы можно сделать
несколько важных выводов. Так, ес-
если плоскости располагаются под пря-
прямым углом, то последовательное от-
отражение в них эквивалентно поворо-
повороту на 180° (рис. 23), или иначе — ли-
линия пересечения таких плоскостей
является осью второго порядка Ьг-
Если плоскости симметрии пересека-
пересекаются под углом 45°, то равнодей-
равнодействующим элементом симметрии бу-
будет ось четвертого порядка.
Из сказанного следует, что элемен-
элементы симметрии находятся не в произ-
произвольных сочетаниях друг с другом,
Рис. 22. Отражение в двух плоскостях 1
и //, пересекающихся под углом а, экви-
эквивалентно повороту вокруг линии их пере-
пересечения на угол 2а
,r у
-л
с с,
1
Рис. 23. Линия пересечения двух плоско-
плоскостей, располагающихся под прямым уг-
углом, является осью второго порядка
Рис. 24. Фигура обладает не только двумя
взаимно перпендикулярными плоскостями
симметрии, но и двойной поворотной осью,
возникающей в результате пересечения
этих плоскостей. Формула симметрии
Ьг2Р
а только в определенных. Так, на-
например, нет такой фигуры, которая
обладала бы только двумя взаимно
перпендикулярными плоскостями
симметрии и не обладала бы одно-
одновременно двойной поворотной осью.
На рис. 24 изображена такая фигура,
обладающая двумя плоскостями сим-
симметрии, расположенными перпенди-
23
\
Рис. 25. Фигура обладает четырьмя плос-
плоскостями симметрии, расположенными под
углом 45°, и осью симметрии четвертого
порядка, являющейся осью их пересече-
пересечения. Формула симметрии L44P
Л
Рис. 26. Возникновение центра симметрии
С в точке пересечения ?2 с перпендику-
перпендикулярной ей плоскостью симметрии Р
Рис. 27. Фигура, обладающая формулой
симметрии L2PC
кулярно к плоскости чертежа, и
осью второго порядка, являющейся
линией их пересечения. Далее, не
может быть фигуры, имеющей Ьц и
одну из плоскостей — / или //. В ре-
результате сложения оси Li с проходя-
проходящей через нее плоскостью симметрии
возникает вторая плоскость, перпен-
перпендикулярная к первой и проходящая
также через ось. Таким образом, фи-
фигура (рис. 24) всегда имеет три эле-
элемента симметрии. Полная совокуп-
совокупность элементов симметрии фигуры
называется видом симметрии или
классом.
В математике вместо термина вид
симметрии часто пользуются его си-
синонимом точечная группа симмет-
симметрии. Происхождение этого термина
связано с тем, что при любых сим-
симметрических преобразованиях у
многогранника по крайней мере од-
одна точка остается на месте (не пере-
перемещается).
Если фигура в проекции будет
иметь форму не ромба, а квадрата
(рис. 25), то ее вид симметрии будет
характеризоваться осью симметрии
L4 и четырьмя плоскостями симмет-
симметрии, проходящими через нее. Кратко
это записывается формулой симмет-
симметрии L4AP. Формула симметрии фигу-
фигуры, изображенной на рис. 24, будет
L22P.
Если плоскость симметрии рас-
располагается перпендикулярно к Lj
(рис. 26), то в результате их сложе-
сложения L%-\-P возникает центр сим-
симметрии. Точка А после поворота
около оси L\ попадает в положение
В, а после отражения в плоскости Р
совмещается с точкой D. Соединим
точку С, точку пересечения оси с
плоскостью, с А, В и D и проведем
в плоскости Р прямую СЕ. Следует
доказать, что точка С является цент-
центром симметрии, т. е. что отражение
точки А в точке С совместит первую
с точкой D. Или, иными словами,
требуется доказать, что точки А, С
и D лежат на одной прямой и отре-
отрезок AC —CD. Из построения легко
видеть, что треугольники DEC и
24
Рис. 28. Сложение поворотов около осей
L и М эквивалентно повороту около оси N
(плоскость / заштрихована)
--Л
I
I
i
8
Рис. 29. Два последовательных отражения
в одной и той же плоскости равносильны
отсутствию отражения
т
М
Рис. 30. Пересечение двух осей Ь2 под пря-
прямым углом даст третью ось Lz, пересекаю-
пересекающуюся в той же точке под прямыми уг-
углами к первым двум
ЕСВ, а также BCF и FCA попарно
равны и равны между собой. Отсюда
доказывается равенство отрезков АС
и CD. Далее, угол ECF — прямой. Он
является суммой двух углов ЕСВ и
BCF. Если к ним прибавить 2 таких
же угла, то получим, что ACD дей-
действительно равен 180°.
Была доказана теорема L2-\-P=C
(рис. 26). На том же чертеже также
легко доказать, что La-\-C=P или
C-\-P=L2. На рис. 27 показана фигу-
фигура, обладающая видом симметрии
L2PC.
Если складывать две пересекаю-
пересекающиеся оси, то равнодействующим
элементом симметрии будет также
ось, проходящая через ту же точку
пересечения.
Каждую иэ осей L и М (рис. 28)
с элементарными углами поворота 2а
и 2C соответственно можно заменить,
по первой теореме, двумя плоскостя-
плоскостями отражения с углами аир. По-
Поскольку взаимная ориентировка обе-
обеих пар плоскостей отражения произ-
произвольна, то мы выберем их так, чтобы
две из этих плоскостей совпали. Тог-
Тогда поворот около L на 2а мы заме-
заменим последовательным отражением
в двух плоскостях / и //, пересека-
пересекающихся под углом а, а поворот око-
около М на 2C — отражением в двух
плоскостях // и ///, пересекающихся
под углом р. Записать это можно
так: поворот L + поворот М = отра-
отражение / + отражение // + отраже-
отражение // + отражение ///. Два после-
последовательных отражения в одной и
той же плоскости (//) равносильны
отсутствию отражения: точка А пос-
после первого отражения совмещается
с точкой В (рис. 29), а второе отра-
отражение в той же плоскости возвра-
возвращает ее в исходное положение А.
Поэтому два поворота — L и М —
равносильны двум отражениям /+
-{-III, а эти отражения можно заме-
заменить поворотом около линии их пе-
пересечения N (рис. 28) на угол 2у.
В этой теореме, если ее доказывать
строго, необходимо учитывать направ-
направления отражений, о которых мы ни-
ничего не говорили. Эта теорема впер-
впервые строго была доказана Эйлером.
Частными случаями ее будет пересе-
пересечение двух осей Ь2 под прямым
углом. Равнодействующей осью бу-
будет являться третья ?г, пересекаю-
пересекающаяся в той же точке под прямыми
углами к первым двум (рис. 30).
25
Если оси ?г пересекаются под уг-
углом 60, 45 или 30°, то равнодейству-
равнодействующими осями симметрии будут Ьг,
L4 или Le соответственно.
§4. Схема вывода
82 видов симметрии
Как было сказано выше, Гаюи пер-
первый высказал идею о том, что кри-
кристаллы являются симметричными
многогранниками. Дальнейшим раз-
развитием учения о симметрии кристал-
кристаллов явились работы Вейсса. В период
1804—1809 гг. он эмпирически уста-
установил наличие различных осей сим-
симметрии в кристаллах, а в 1815 г.
предложил деление кристаллов на 6
систем (сингоний).
В 1867 г. русский академик
А. В. Гадолин строго математическим
j путем вывел 32 вида симметрии кри-
\ сталлов. Интересно отметить, что эм-
эмпирически был установлен к тому
времени только 20—21 вид симмет-
симметрии. В настоящее время имеются
примеры кристаллов всех 32 видов
симметрии. После того как результа-
результаты исследования Гадолина прочно
вошли в науку, была найдена работа
Гесселя, сделанная на 30 лет ранее
Гадолина и содержавшая аналогич-
аналогичный вывод.
ТАБЛИЦА 3
Схема вывода видов симметрии
Зная три теоремы о сложении эле-
элементов симметрии, доказанные в
предыдущем параграфе, можно по-
получить представление о математиче-
математическом выводе видов симметрии, по-
понять принцип этого вывода.
Этот вывод мы прежде всего раз-
разделим на две части: в первой будут
выведены виды симметрии с одной
главной осью симметрии (с осью
третьего и более высоких порядков)
и без главных осей, во второй час-
части — с несколькими главными ося-
осями.
В первой колонке табл. 3 выписа-
выписаны все 8 возможных осей симметрии.
Инверсионная ось Li содержит в
себе центр инверсии, а ось L\— пло-
плоскость симметрии, перпендикулярную
к ней. Это обстоятельство иногда
подчеркивается тем, что после наи-
наименования оси ставится знак С или
Р соответственно, как это и сделано
в первом столбце табл. 3. Такая сим-
символика является нестрогой, т. е. в
других случаях аналогичных элемен-
элементов симметрии мы не указываем:
например, ось L2, содержащуюся в
каждой четной поворотной оси (Li
или Ls). Избежать такой двойствен-
двойственности легко, если в каждом виде сим-
симметрии указывать только порож-
порождающие элементы симметрии, т. е.
тот минимум, который в результате
1
Li
и
U
Li
U
L-C
h
L-P
6
2
+ c
UPC121
_[3]
LiPC
LtPC
(L-C)
_[4]
_M
3
+ \\P
P
U2P
L33P
L«4P
LSP
з 2
4
6
4
{L-3U3PC)
3
(L-2L22P)
4
(L-3L24P)
6
5
+L2PC
(ЬгРС)
З^ЗРС
_[3]
Z.44L25PC
LSLJPC
(L-SU3PC)
_[4]
26
Рис. 31. Прибавление центра симметрии
к оси L3 превращает ее в инверсионную
ось Li
Рис. 32. Прибавление плоскости симметрии
параллельно инверсионной оси Li вызы-
вызывает появление двойных осей L2, перпен-
перпендикулярных к Li
сложения дает всю формулу сим-
симметрии. Указание на С и Р при
осях Ll и Li имеет скорее педагоги-
педагогическое значение, так как именно эти
элементы симметрии на моделях кри-
кристаллов учащиеся будут находить
скорее и легче, чем сами инверсион-
инверсионные оси.
Все указанные в первом столбце
табл. 3 оси могут присутствовать
в фигурах в единственном числе.
В этом случае они будут являться
восемью простейшими видами сим-
симметрии. К этим осям симметрии мы
последовательно будем прибавлять
другие элементы симметрии (верхняя
строка табл. 3). Действуя таким об-
образом, мы заведомо не пропустим ни
одного вида симметрии, но некото-
некоторые из них, очевидно, получим в таб-
таблице несколько раз. Вывод будем
проводить по колонкам сверху вниз
и слева направо. Те виды симметрии,
которые будут появляться второй раз
(и последующие разы), будем сразу
ставить в скобки и при окончатель-
окончательном подсчете не учитывать. Если
прибавление нового элемента сим-
симметрии изменяет характер оси, то
соответствующую клетку прочерки-
прочеркиваем, так как все возможные оси
выписаны в первой колонке и нового
вида симметрии, очевидно, в таком
случае не получим. Все примечания
в таблице последовательно с выво-
выводом будут разбираться.
Если к каждой из осей прибавить
центр симметрии (вторая колонка),
то мы получим несколько новых ви-
видов симметрии.
[1]. Прибавление С к оси первого
порядка, т. е. к фигуре, не имеющей
элементов симметрии, дает вид сим-
симметрии, имеющий только центр сим-
симметрии.
[2]. Прибавление к L2 центра сим-
симметрии дает по второй теореме (§3)
вид симметрии с формулой L2PC.
[3].Если к оси Ь^ прибавить центр
симметрии, то это превратит ее в
инверсионную ось Li (рис. 31), а
таковая ось уже имеется в первой ко-
колонке, так что нового вида симмет-
симметрии в этой клетке таблицы мы заве-
заведомо не получим.
[4] .Прибавление С к Li или L& пре-
превращает эту ось в La или в L& соот-
соответственно.
Прибавление к осям симметрии
плоскости симметрии может быть
осуществлено параллельно оси, пер-
перпендикулярно к оси и наклонно.
Прибавление плоскости симметрии
параллельно главной оси (столбец 3)
по первой теореме приводит к появ-
появлению новых плоскостей, причем об-
общее число плоскостей будет равно
порядку оси.
[5]. Можно показать, что прибавле-
прибавление плоскости симметрии параллель-
параллельно к инверсионным осям Zf и Li вы-
вызовет также появление двойных осей
симметрии, перпендикулярных к
главной оси и расположенных
по биссектрисам углов между плоско-
плоскостями симметрии. El и Pi (рис. 32)
заданы. Так как L\ есть одновремен-
одновременно и L%, то перпендикулярно к Pi
появится равнодействующая пло-
плоскость Рп. Задав произвольно точку 1,
с помощью плоскостей симметрии
и Li мы получим точки 2, 3 и. 4.
Этим бы дело и ограничилось, если
бы главная ось была простой Li. Она
переводила бы точку 1 в положение
4, а точку 2 — в положение 3. Но так
как ось является инверсионной, то
точка 1 после поворота по часовой
стрелке на 90° и инверсии попадает
27
Рис. 33. Прибавление плоскости симмет-
симметрии наклонно к главной оси вызывает
появление второй главной оси
Рис. 34. Lh, Lm, Ln — места выходов на
шаре осей разного наименования. Проведе-
Проведение через них дуг разбивает шар на сеть
сферических треугольников
а-
Н
4
А
ГУ
Рис 35. Три взаимно перпендикулярные
плоскости, разбивающие шар на 8 октан-
октантов
в положение 1' (под плоскость черте-
чертежа) . Соответственно появляются точ-
точки 2', 3' и 4'. Число точек при этом
удваивается (8 вместо четырех).
Легко видеть, что фигура обладает
дополнительными элементами сим-
симметрии — двумя осями симметрии L2,
перпендикулярными к главной оси.
Ьъ переводит точку 1 сразу в поло-
положение 3', точку 2 — в положение 4'
и т. д. Аналогично действует и L*1.
Центра симметрии фигура не имеет.
Но он имеется у фигуры, содержа-
содержащей вместо Ll ось Lj.
[6]. Прибавление Р параллельно 1л
дает три плоскости симметрии, про-
проходящие через главную ось. В пере-
пересечениях этих плоскостей с четвертой,
перпендикулярной главной оси, по-
появляются равнодействующие еси Li.
Прибавление плоскости симметрии
перпендикулярно главной оси не дает
новых видов симметрии. При этом
у простых четных осей появляются
С в качестве равнодействующего
элемента симметрии и виды симмет-
симметрии те же, что и в столбце 2.
Прибавление же Р к осям L3 и L%
приводит к превращению их в оси
другого наименования, уже имею-
имеющиеся в первом столбце. Следователь-
Следовательно, такое прибавление также не при-
приведет к новым видам симметрии. Для
Li и Li справедливы оба сделанные
замечания.
Прибавление плоскости симметрии
наклонно к главной оси производить
нельзя, так как это вызовет появле-
появление второй главной оси того же наи-
наименования (рис. 33), а мы услови-
условились выводить пока, виды симметрии
с одной главной осью.
В четвертом столбце выписаны ви-
виды симметрии, получающиеся от
прибавления к главным осям перпен-
перпендикулярной оси L2. Прибавлять L2
параллельно главным осям бесполез-
бесполезно, так как при этом нечетные оси
становятся осями вдвое большего
наименования, а четные оси и без
того содержат ось 1,2, а прибавление
наклонной L2 дало бы вторую глав-
28
ную ось. Прибавление к Li перпен-
перпендикулярной L% приводит, по теореме
Эйлера, к появлению трех других Ьг,
также перпендикуляных к 1ц.
Для полноты вывода мы должны
еще прибавить одновременно по два
элемента симметрии, но так как сло-
сложение двух элементов симметрии
обязательно приводит к появлению
третьего — равнодействующего, то в
последнем столбце (пятом) мы долж-
должны просуммировать все элементы
симметрии из столбцов 2, 3 и 4. При-
Прибавление этих элементов симметрии
в иной ориентировке или же других
элементов симметрии не приводит к
новым видам симметрии.
.Подсчет полученных видов симмет-
симметрии показывает, что в столбце A)
их 8, во B) — 4, в C) — 8, в D) — 4
ив E) — 3, а всего 27.
Вторая часть вывода будет посвя-
посвящена видам симметрии с нескольки-
несколькими главными осями.
Если имеется несколько главных
осей, пересекающихся в одной точке,
то, вращая весь этот пучок вокруг
всех его осей, можно получить двоя-
двоякий результат. Во-первых, может
случиться, что через некоторое число
поворотов оси одного наименования
начнут совпадать, и система, следова-
следовательно, будет иметь конечное число
осей; во втором случае число осей
может оказаться бесконечно боль-
большим. Очевидно, что нас может инте-
интересовать только первый случай, так
как кристаллы являются всегда ко-
конечными многогранниками и имеют,
следовательно, конечное число эле-
элементов симметрии.
Если такой пучок поместить в
центр шара, то на его поверхности
окажутся выходы осей (рис. 34).
Предположим, что сначала мы имели
только две оси — Lm и Ln. При пово-
повороте каждой из них вокруг другой
мы получим тЬпта.пЬт. Эту операцию
будем повторять до тех пор, пока оси
одного наименования не станут сов-
совпадать друг с другом. Тогда поверх-
поверхность шара окажется заполненной
конечным числом точек — выходов
осей. Складывая эти оси друг с дру-
другом, мы можем получить новую сп-
стему осей Lk, которые в частном слу-
случае могут оказаться и осями Lz.
В результате вся поверхность шара
окажется испещренной точками —
выходами осей. Эти точки мы можем
соединить дугами, которые будут яв-
являться следами плоскостей, проходя-
проходящих через оси и пересекающихся в
центре шара. Вследствие этого вся
поверхность шара окажется разбитой
на сеть сферических треугольников.
Процесс разбиения можно провести
таким образом, чтобы в вершинах
треугольников располагались оси
Lm, Ln и Lh (рис. 34). Исследуем воз-
можные варианты.
Прежде всего вспомним, что сум-
сумма углов сферического треугольника
больше 2d. На рис. 35 шар разбит
тремя взаимно-перпендикулярными
плоскостями на 8 сферических тре-
треугольников — октантов с суммой
углов, равной 270°. Если мы начнем
уменьшать угол в точке А, т. е. при-
приближать дугу АС к АВ, как это пока-
показано стрелкой, то сумма углов сфери-
сферического треугольника ABC начнет
уменьшаться, при этом углы в точ-
точках С ж В будут оставаться прямы-
прямыми. В пределе, когда сферический
треугольник выродится в дугу, сум-
сумма его углов станет равной 2d (по
90° при С и В и 0° при А). Таким
образом мы приходим к выводу, что
сферические треугольники LmLnLh
(рис. 34) имеют сумму углов, боль-
большую 2d.
Мы знаем, что в кристаллах могут
существовать оси L6, Li, L% и L2 (для
упрощения рассуждения инверсион-
инверсионные оси могут рассматриваться как
простые соответствующего наимено-
наименования), а плоскости, их соединяю-
соединяющие, составляют, следовательно, углы
в 30, 45, 60 и 90° соответственно (так
как угол между плоскостями отра-
отражения, заменяющими поворот около
оси, вдвое меньше элементарного
угла поворота оси). Этих сведений
достаточно, чтобы исследовать те сфе-
сферические треугольники LmLnLh, ко-
29
Рис. 36. Вид симметрии с формулой З^г^з
Рис. 37. Вид симметрии с формулой
iLJL^iL
*-
Рис. 38. Расположение осей симметрии
в тетраэдре
Рис. 39. Расположение осей симметрии
в кубе
Рис. 40. Вид симметрии с формулой
3L2iL33PC
Рис. 41. Вид симметрии с формулой
3LiL6L9PC
Рис. 42. Вид симметрии с формулой
3L24L36P
торые могут встретиться в кристалло-
кристаллографии. Прежде всего одна из трех
вершин обязательно должна быть Ь%,
так как любой набор осей Lz, L* и L&
не дает сумму углов треугольника
больше Id. Но она должна быть толь-
только одна, так как мы выводим виды
симметрии с несколькими главными
осями. Легко видеть, что в вершине
треугольника не может быть оси L&,
так как даже при максимальном зна-
значении третьего угла F0°) мы не по-
получим сферического треугольника:
90°+30°+60° = 2 d, а надо, чтобы
сумма его углов была более 2d. Из
сказанного следует, что в кристал-
кристаллах, имеющих несколько осей выс-
высшего порядка (т. е. порядка 3 и вы-
выше) , не встречаются оси шестого
порядка. Если исходный треуголь-
треугольник будет иметь две оси второго
порядка и только одну ось шестого по-
порядка, то мы и придем к виду сим-
симметрии с одной главной осью, а не
с несколькими. Очевидно, он уже был
получен в первой части вывода.
Испытывая такпм же образом дру-
другие случаи, мы придем только к
двум возможным вариантам:
1) 90°+ 60°+60°, соответствующий
осям L2+L3+L3;
2) 90° + 60°+45°, соответствующий
осям L2+Lz-\-Li.
В вершинах исходного треугольни-
треугольника не могут располагаться даже три
Lz — оси высшего порядка, дающие
максимальные углы в 60°.
На рис. 36 и 37 показаны эти два
возможных случая. Исходные сфери-
сферические треугольники обозначены
жирными линиями. Поворачивая тре-
треугольник вокруг всех осей, мы полу-
получаем, в конце концов, пространствен-
пространственное расположение всех осей симмет-
симметрии ЗЬг4&3 в первом случае и
3L44L36L2 — во втором. Это будут
два новых вида симметрии. Такие
наборы осей симметрии имеются у
правильного тетраэдра (рис. 38) и
куба (рис. 39).
Дальнейший вывод новых видов
симметрии сводится к прибавлению
30
к этим двум дополнительных эле-
элементов симметрии С и Р; осей Z-2
прибавлять уже не надо, так как все
возможные случаи осевых видов
симметрии уже получены.
Прибавление С к первому осевому
виду симметрии (рис. 36) дает, со-
согласно 2-й теореме (§3), три плоско-
плоскости симметрии, перпендикулярные
каждой оси Z-2 и проходящие, сле-
следовательно, через две другие Ьъ
(рис. 40). Формула симметрии его
ЗЬгАЬзЗРС. Соответственно у второго
осевого вида симметрии получится
9 плоскостей симметрии: 3 — перпен-
перпендикулярно осям Li и 6 — перпенди-
перпендикулярно L2. Формула симметрии
будет 3UAL36L29PC (рис. 41). При-
Прибавление плоскости симметрии к осе-
осевым видам симметрии нужно произ-
произвести так, чтобы при этом не возни-
возникало новых осей симметрии. Это
можно осуществлять или задавая Р
через тройные оси, или между ними.
Если задать в обоих осевых видах
симметрии (рис. 36 и 37) Р между
осями третьего порядка, то они ока-
окажутся перпендикулярными к четным
осям L2 и L^ вызовут появление цен-
центров симметрии, и мы придем к уже
разобранным видам симметрии (рис.
40 и 41). Если же прибавляемая Р
пройдет через оси L3, то получится
новый вид симметрии 3L2ALZ6P (рис.
42). Этим и будут исчерпаны все воз-
возможные виды симметрии. Общее чис-
число их — 32.
§ 5. Систематика видов
Симметрии
Вывод видов симметрии, схема кото-
которого была только что изложена, до
некоторой степени подсказывает и
название их.
Виды симметрии, имеющие только
главные оси, называются примитив-
примитивными (табл. 4). Прибавление к ним
центра симметрии приводит к «цен-
«центральным» видам симметрии, приба-
прибавление плоскости и оси симметрии —
соответственно к планалъным и акси-
аксиальным. Виды с максимальным ко-
количеством дополнительных элемен-
элементов симметрии называются «план-
аксиальными». Если главной осью
является инверсионная ось симмет-
симметрии, то это также подчеркивается в
названии. Так получаются «инверси-
«инверсионно-примитивные» и «инверсионно-
планалъные» виды симметрии (табл.
4).
Изображение всех видов симмет-
симметрии дано в табл. 5. Номера, данные
в ней, соответствуют номерам табл. 4.
Кроме того, в табл. 5 указаны федо-
федоровские названия видов симметрии,
весьма распространенные в русской
и иностранной литературе. Ниже
полной формулы симметрии приведе-
приведено международное обозначение видов
симметрии. Для него используются
цифровые обозначения не всех эле-
элементов симметрии, а только важней-
важнейших или даже порождающих.
Последние являются теми элемента-
элементами симметрии, которыми мы пользо-
пользовались при выводе видов сим-
симметрии (см. табл. 3). Буква т в ме-
международных обозначениях является
начальной буквой слова mirroi— зер-
зеркало.
Все виды симметрии делятся на
три категории: низшую, среднюю и
высшую. В низшую попадают виды
симметрии, не имеющие осей высше-
высшего порядка (выше чем 3) (см. табл. 4
и 5); в среднюю — виды симметрии,
имеющие одну ось высшего порядка;
в высшую — с несколькими осями
высшего порядка. В частности, по-
последние всегда имеют 4?з. Низших
видов симметрии 8, средних — 19 и
высших — 5.
Каждая категория подразделяется
на сингонии. В низшей категории их
3: триклинная, моноклинная и ромби-
ромбическая. В кристаллах триклинной
сингонии нет ни осей, ни плоскостей
симметрии; у моноклинных кристал-
кристаллов может быть как ось, так и пло-
плоскость симметрии, но не может быть
нескольких одинаковых элементов
симметрии: нескольких осей или пло-
плоскостей. Однако последнее условие
обязательно для ромбических кристал-
31
ТАБЛИЦА 4
Таблица 32 видов симметрии
Категория
Низшая
Средняя
Высшая
Сингония
Триклинная
Моноклинная
Ромбическая
Тригональная
Тетрагональная
Гексагональная
Кубическая
Виды симметрии
прими-
примитивный
1
и
9
и
14
и
21
и
28
4Z.83Lg
центральный
2
С
10
ис
15
LtPC
22
UPC
29
ЦлЪЬ&РС
{планальный
3
Р
6
Lj2P
11
WZP
16
L»4P
23
Lo6P
30
ЬиЗЬъбР
аксиальный
4
La
7
31*
12
ЬЗЬа
17
L44La
24
ЬббЬа
31
3L44L.6L2
планаксиальны й
5
LjPC
8
3L»3PC
13
X.3L23PC
18
L«4La5PC
25
L66L27PC
32
3^41.6^9^0
инвер-
сионно-
прими-
тивный
19
h
26
L-=UP
в
инверсионно'
планальный
20
L-2Lj2P
4
27
L-3L23P = ЫЪиЬР
в
Примечание. Инверсионные оси третьего порядка в таблице не указаны. Они присутствуют в 10- и 13-м видах симметрии. В куОичесиой син-
гонии инверсионные оси вообще не указаны. В тексте и в остальных таблицах наименование видов симметрии дано по Е. С. Федорову.
ТАБЛИЦА 5
Проекции 32 видов симметрии
Проекция
Символ
Название
Проекция
Символ
Название
Триклшшая сингоийя
С
I
Моноэдриче-
ский
Пинакои-
дальный
Моноклинная сингония
м
1 !
Р
2 или т
2
UPG
2/т
Диэдриче-
ский безос-
безосный
Диэдриче-
ский осевой
Призмати-
Призматический
Ромбическая сингоння
L&P
тт.2 или
тт.
3L2
222
Ромбо-пира-
мидальпый
Ромбо-тетра-
эдрическвй
Ромбо-дшш-
рамидаль-
ный
10
11
12
13
14
15
16
17
18
19
20
Тригональиая сиигония
и
3
3
Зт
32
Зт
Тригональ-
но-пирами-
дальный
Ромбоэдри-
Ромбоэдрический
Дитриго-
нально-пи-
рамидаль-
ный
Тригональ-
но-трапецо-
эдрический
Тригональ-
но-скалено-
эдрический
Тетрагональная сингония
4
UPC
Цт
t
4 mm или
4m
L.4L,
422 или
Цхптт
4
L-2L&P
Шт
3 Кристаллохимия
Тетраго-
нально-пи-
рамидаль-
ный
Тетраго-
нально-ди-
цирамидяль-
Дитетраго-
нально-пи-
рамидаль-
ный
Тетраго-
42 нально-тра-
пецоэдриче-
ский
Дитетраго-
нально-ди-
пирамидаль-
ный
Тетраго-
нально-тет-
раэдрический
Тетраго-
нально-сиа-
лепоэдриче-
ский
33
ТАБЛИЦА 5 (окончание)
Проекция
Символ
Название
Проекция
Символ
Название
Гексагональная сингония
21
22
24
25
26
27
6
LGi>C
6/1Я
LSP
Qmm
Z*f,6Z<2
622 или 62
L&LJPC
Щттт
L =ЦР
б6
LJiL&P =
g
= L33L24P
8m2
Гексаго-
нально-пи-
рамидаль-
ный
Гексаго-
Гексагонально- ди-
пирамидаль-
ный
Дигексагот
нально-пи-
рамидаль-
ный
Гексаго-
нально-тра-
пецоэдричес-
кий
Дигексаго-
нально-ди-
пирамидаль-
ный
Тригональ-
но-дипира-
мидальный
Дитригона-
льно-дипи-
рамидаль-
ный
Кубическая сингоиия
4L33L2
23
3?
2/т 3 или
З
43m
432 или 43
Пентагон-
тритетра-
эдрический
Дидодека'
эдрическя i
Гексатетра-
эдрический
Пентагон-
триокта-
эдрический
Гексокта-
эдрический
лов: каждый кристалл ромбической
сингонии имеет несколько одинако-
одинаковых элементов симметрии.
Средняя категория имеет 3 синго-
сингонии, называемые по типу главной
оси: тригоналъная Ь$ или Zj , тетраго-
тетрагональная Li или Zj и гексагональная
L6 или Li.
Высшая категория включает одну
сингонию — кубическую, характери-
характеризующуюся несколькими осями выс-
высшего порядка, в частности, каждый
ее вид симметрии имеет 4?з.
Наиболее симметричный вид сим-
симметрии каждой сингонии называется
голоэдрическим видом симметрии.
ГЛАВА IV
ФОРМА КРИСТАЛЛИЧЕСКИХ МНОГОГРАННИКОВ
§1. Понятие простой формы
Изучение внешней формы кристал-
кристаллов началось прежде изучения сим-
симметрии, однако только после из-
известного завершения этого учения
(вывод 32 видов симметрии) появи-
появилась надежная основа для создания
геометрического учения о внешней
форме кристаллов. Основным поня-
понятием его является понятие простой
формы.
Простой формой называется мно-
многогранник, который может быть по-
получен из одной грани с помощью
элементов симметрии. В качестве
примера возьмем какой-либо вид
симметрии, например L&P (рис. 43,
а ж б), и проведем произвольно —
косо по отношению к элементам сим-
симметрии — какую-либо плоскость —
грань кристалла 1 на рис. 43, в.
В стереографической проекции это
будет точка 1 (рис. 43, а). Рис. 43, д
изображает вид сверху фигуры в.
Отразив грань 1 в плоскости сим-
симметрии 1, получим грань 2. После
отражения граней 1 ж 2 в плоскости
// получим грани 3 ж 4. Далее все
будет повторяться; так, если грань 1
(в) или ее проекцию в виде точки 1
(а) повернуть около оси Li на 180°,
то получим точку 3, которая уже
была ранее получена. Новых граней
(точек) мы не получим. В результа-
результате применения всех элементов сим-
симметрии мы получили из грани 1 еще
три грани, а всего, следовательно, че-
четыре грани. Этот многогранник, со-
состоящий из четырех одинаковых
граней, и является простой формой.
В данном конкретном случае он на-
называется ромбической пирамидой.
Простые формы могут быть общи-
общими и частными в зависимости от
того, как расположена исходная
грань по отношению к элементам
симметрии. Если она расположена
косо, как в нашем примере, т. е. в
общем положении, то и простая фор-
форма, полученная из нее, будет общей.
Если же исходная форма располо-
расположена параллельно или перпендику-
перпендикулярно к элементам симметрии, то
получается частная простая форма.
Так, например, основание пирами-
Рис. 43. Получение простой формы (ром-
(ромбической пирамиды) из одной грани
о помощью элементов симметрии
а — элементы симметрии ромбо-пирамидально-
го вида симметрии в стереографической
проекции;
б — те же элементы симметрии в пространстве;
в — получение ромбической пирамиды;
г — комбинация ромбической пирамиды с мо-
ноэдром;
9 — ромбическая пирамида (вид сверху)
35
ды 5 (рис. 43, г) является частной
простой формой, ибо эта грань пер-
перпендикулярна L2 и обеим плоскос-
плоскостям симметрии. Отражение ее в пло-
плоскостях симметрии и вращение во-
вокруг оси Z/2 дают совмещение ее
самой с собой. Эта частная про-
простая форма состоит из одной грани
и называется моноэдром. Моно —
по-гречески один, эдр — грань, т. е.
это одногранник. Для понимания
названий простых форм полезно
знать греческие названия простых
чисел: 1 — моно, 2 — ди, 3 — три,
4 — тетра, 5 — пента, 6 — гекса,
7 — гепта, 8 — окта, 10 — дека, 12 —
додека.
Федоровские названия видов сим-
симметрии (табл. 5, стр. 33) определя-
определяются названием общей простой фор-
формы в данном виде симметрии.
Простые формы бывают открыты-
открытыми и закрытыми. Закрытая форма
может одна образовать кристалли-
кристаллический многогранник (см., напри-
например, куб, рис. 39). Одна открытая
простая форма замкнутого много-
многогранника образовать не может (см.,
например, ромбическую пирамиду,
рис. 43, в). Кристалл в этих случаях
огранен гранями нескольких про-
простых форм, составляющих комбина-
комбинацию простых форм. Так, например,
кристалл на рис. 43, г представляет
собой комбинацию двух простых
форм: ромбической пирамиды и мо-
ноэдра, первая состоит из четырех
граней, вторая — из одной.
Закрытые простые формы могут,
конечно, тоже входить в комбина-
комбинацию. Ниже мы познакомим с много-
многочисленными примерами такого ро-
рода. Число простых форм, встречаю-
встречающихся во всех сипгониях, равно 47.
Рис. 44. Простые формы низших сингоний
а — моноэдр;
б — пинакоид;
в — диэдр;
г—ромбическая пирамида;
9 — ромбическая призма;
е — ромбический тетраэдр;
ж — ромбическая дипирамида;
з — стереографическая проекция ромбической
дипирамиды
г-5). На рис. 44 изображены все
простые формы кристаллов низших
сингоний. Моноэдр показан на рис.
44, а.
По две грани имеют пинакоид
(б) и диэдр (в). В первом случае
грани параллельны друг другу, во
втором — пересекаются. Четырех-
Четырехгранных простых форм три: знако-
знакомая нам ромбическая пирамида (г),
ромбическая призма (д) и ромби-
ромбический тетраэдр (е). Во всех этих
названиях имеется слово «ромби-
«ромбический», так как сечением всех трех
§2. Простые формы
низших сингоний
Простые формы удобно рассматри-
рассматривать в порядке увеличения числа их
граней.
С простейшим случаем мы уже по-
познакомились — это моноэдр (рис. 43,
Рис. 45. Комбинация ромбической пирами-
пирамиды с двумя моноэдрами
36
фигур является ромб (см. г, д и е).
Последняя простая форма (ж) име-
имеет 8 граней и называется ромби-
ромбической дипирамидой. Этот много-
многогранник не является комбинацией
двух ромбических пирамид. Он по-
получается из одной грани с помощью
элементов симметрии ромбо-дипира-
мидального вида симметрии ЪЬч
ЗРС(э). Из всех семи простых форм
кристаллов низшей категории только
две закрытые — ромбические тет-
тетраэдр и дипирамида, остальные —
открытые.
Форма граней одной и той же
простой формы в зависимости от
комбинации может сильно варьиро-
варьировать, поэтому она не является ха-
характерным признаком простой фор-
формы. Так, например, ромбическая пи-
пирамида, комбинирующаяся с одним
моноэдром, имеет треугольные грани
(рис. 43, г), а в комбинации с двумя
мотюэдрами (рис. 45) они четырех-
четырехугольные.
Для простой формы характерны-
характерными являются число граней и ориен-
ориентировка их друг к другу и к элемен-
элементам симметрии.
§3. Простые формы
средних синround
Как было сказано выше, кристаллы
средних сингоний характеризуются
наличием одной оси высшего поряд-
порядка, поэтому сеченпе простых форм
средних сингоний будет иметь эти
же оси.
Типы таких сечений показаны в
верхней строке рис. 46. В зависи-
зависимости от сечения получаем 6 призм:
тригоналъную, дитригоналъную, тет-
тетрагональную, дитетрагональную, гек-
гексагональную и дигексагоналъную.
Если исходной геометрической фор-
формой будет пирамида или дипирамида,
то соответственно получаем простые
формы, называющиеся тригоналънои
пирамидой, дитригоналъной пирами-
Рис. 46. Призмы, пирамиды и дипирами-
ды средних сингоний
. Сечение
Ис-
Исходная^
простая'
Призма
Триган
Дитригон
Тетрагон
Дитетрагон
Гегсагон
Дигексаган
-J.
Пирамида,
Дипирамида.
37
дой и т. д., или тригоналъной дипира-
мидой, дитригоналъной дипирамидой
и т. д. (нижняя строка).
Кроме этих 18 простых форм, в
средних сингониях встречаются еще
и другие. На рис. 47 изображены
Рис. 47. Трапецоэдры
а — тригональный;
б — тетрагональный;
е — гексагональный
Рис. 48. Стереографические проекции три-
гональной дипирамиды (о), трапецоэдров
(б —левая форма, в —правая форма),
ромбоэдра (г)
три трапецоэдра: тригональный, тет-
тетрагональный и гексагональный. Эти
фигуры отличаются от соответ-
соответствующих дипирамид тем, что ниж-
нижняя половина их находится не точно
под верхней, а смещена относитель-
относительно нее на некоторый угол. На рис. 48
изображены тригональная дипира-
мида (а), тригональный трапецоэдр
(б) и их стереографические проек-
проекции. Угол смещения ф произволь-
произвольный. Он может быть осуществлен по
часовой стрелке или против (б и в).
Соответствующие трапецоэдры при
одинаковых по величине, но разных
по направлению углах ф отличаются
друг от друга как левая рука от лра-
вой и называются соответственно
левыми или правыми. Такие фигуры
в кристаллографии называются энан-
тиоморфными.
Следующая простая форма, кото-
которую мы рассмотрим, называется
ромбоэдром. Он похож на трапецо-
трапецоэдр, но отличается от него тем, что
нижние его грани располагаются как
раз посередине между верхними
и наоборот (рис. 48, г). Эта фигура
может быть получена из куба (рис.
49, б) путем деформации его вдоль
одной из осей L3. В случае деформа-
деформации сжатия получается тупой ром-
ромбоэдр (а), в случае растяжения —
острый (в).
Встречается в средних сингониях
и тетраэдр, но, в отличие от ромби-
ромбического (рис. 44, г), он имеет квад-
квадратное сечение (рис. 50). В результа-
результате удвоения каждой грани тетраэдра
и ромбоэдра получаются тетрагональ-
Рас. 49. Тупой (а) и острый (в) ромбоэд-
ромбоэдры, получающиеся сжатием и растяжени-
растяжением куба (б) по оси третьего порядка
38
Рис. 50. Тетрагональный тетраэдр
Рис. 51. Скаленоэдры тетрагональный (а)
и гексагональный (б)
ный и соответственно гексагональ-
гексагональный скаленоэдры (рис. 51).
Кроме перечисленных в этом па-
параграфе 25 простых форм, в сред-
средних сингониях могут встречаться
моноэдр и пинакоид, описанные в
предыдущем параграфе. В средних
сингониях они всегда являются част-
частными формами, грани которых пер-
перпендикулярны к главным осям.
§4. Простые формы
кубической сингонии
В кубической сингонии могут быть
только свои специфические простые
формы. Ни одна из простых форм
низшей или средней категории не
встречается в кубических кристал-
кристаллах. Точно так же ни одна простая
форма кубической сингонии не
встречается в кристаллах других син-
сингонии.
Некоторые простые формы куби-
кубической сингонии мы уже знаем, на-
например куб (рис. 39), октаэдр
(рис. 14) и тетраэдр (рис. 38).
Тетраэдр кубической сингонии от-
отличается от тетрагонального и ром-
ромбического тетраэдра тем, что его
грани являются равносторонними
треугольниками, тогда как у тетра-
тетрагонального тетраэдра они являются
равнобедренными, а у ромбическо-
ромбического — произвольными треугольниками
с тремя неравными ребрами.
Если взять за исходные простые
формы тетраэдр и октаэдр, то можно
получить ряд производных простых
форм (рис. 52). В верхней строке
показаны формы граней. Первой
изображена грань правильного (ку-
(кубического) тетраэдра — равносторон-
равносторонний треугольник. Если вместо одной
грани появляются три, то фигура на-
называется тритетраэдр, если шесть —
гексатетраэдр. Так как тритетраэдров
мюжет быть несколько, то перед на-
названием указывается форма каждой
из получающихся граней. Грани
тритетраэдров могут быть треуголь-
треугольные, четырехугольные и пятиуголь-
пятиугольные, соответственно фигуры, имею-
Рис. 52. Простые формы кубической син-
сингонии, производные от тетраэдра и окта-
октаэдра
Тетраэдр
Тетраэдр
Тригонгритеграэдр
Тетрагонтритетраздр
Пентагрнтритеграэвр
Гексатетраэдр
Октаэдр
Октаэдр Тригантриактаздр Тетрагантриокгаэдр Пвнтагонт'риоктаэдр Ге'ксоктаэдр
39
Рис. 53. Куб (гексаэдр) (а) и тетрагекса-
эдр (б)
Рис. 54. Ромбо-додекаэдр (а), пентагон-
додекаэдр (б), дидодекаэдр (в)
щие такие грани, получают название
тригон-тритетраэдр, тетрагон-тритет-
раэдр и пентагон-тритетраэдр.
Те же самые по форме грани мо-
могут быть и у октаэдров (нижняя
строка). Их названия получаются
таким же образом, как и для тетра-
тетраэдров. Соответственно получим сле-
следующие 5 простых форм кубической
сингонии: октаэдр, тригон-триокта-
эдр, тетрагон-триоктаэдр, пентагон-
триоктаэдр и гексоктаэдр.
Общее число граней у всех про-
простых форм легко может быть высчи-
высчитано, если учитывать их название.
Тетраэдр и октаэдр имеют соответ-
соответственно 4 и 8 граней, так как тетра
по-гречески — 4, окта — 8, а эдр —
Рис. 55. Комбинация октаэдра с кубом (а)
и с ромбододекаэдром (б) (белые кружки
на проекции — грани куба)
грань. Все тритетраэдры будут иметь
по 12 граней CX4), а триоктаэд-
ры — по 24 Cx8). Гексатетраэдр
также имеет 24 грани FX4), а гек-
гексоктаэдр — 48. Это — максимальное
число граней, которое может иметь
простая форма.
Кроме этих десяти простых форм,
в кубической сингонии может быть
еще 5: куб (или гексаэдр) и тетра-
гексаэдр (или «пирамидальный куб»)
(рис. 53, а и б) и три додекаэдра
(рис. 54).
Додека — по-гречески 12, додека-
додекаэдр — двенадцатигранник. Если фор-
форма грани у додекаэдра ромб (рис.
54, а), фигура называется ромби-
ромбическим додекаэдром (или ромбо-
ромбододекаэдром), если пятиугольник
(б) — пентагональным додекаэдром
(или пентагон-додекаэдром). В ре-
результате удвоения каждой грани
пентагонального додекаэдра полу-
получается 24-гранник (рис. 54, в), назы-
называющийся дидодекаэдром.
Тетраэдр, куб, октаэдр, ромбо-доде-
ромбо-додекаэдр и пентагон-додекаэдр являют-
являются важнейшими простыми формами
кристаллов кубической сингонии.
Остальные формы встречаются зна-
значительно реже.
На рис. 55 показано несколько
комбинаций простых форм кубиче-
кубической сингонии и их стереографиче-
стереографические проекции.
§ 5. Возможные грани
Каждая грань кристалла представля-
представляет собой плоскость, па которой рас-
располагаются атомы. На рис. 56 изо-
изображен кристалл NaCl и его атомная
структура. Когда кристалл растет от
а к б, то все грани передвигаются
параллельно самим себе, так как на
них откладываются все новые и но-
новые слои атомов. По этой причине
параллельно каждой грани в струк-
структуре кристалла располагается огром-
огромное количество атомных плоскостей,
которые когда-то в начальных ста-
стадиях роста тоже располагались на
40
В Q-p-O-Q-O-O-Q-O B
*—•—•—©—±—i—e —ф
о-р-о-ф-р-Р-о-о
р-р-р-р-р--о-о-о
S—S—в—S—S—в—•—в
р-р-р-р-р-Р-с-р
Рис. 56. Кристалл NaCl и его атомная
структура
гранях кристалла, но в процессе рос-
роста оказались внутри него.
Ребра кристалла представляют со-
собой прямые, на которых атомы рас-
располагаются в ряд. Таких рядов в кри-
кристалле тоже огромное количество и
они параллельны действительным
ребрам кристалла (рис. 56, а, б ж в).
Кристаллический многогранник
обычно представляет собой комбина-
комбинацию нескольких простых форм, грани
(или ребра) которых являются дейст-
действительными гранями (или ребрами).
Грань, которой на данном кристал-
кристалле нет, но которая может оказаться
на других кристаллах того же веще-
вещества, называется возможной гранью.
Возможной гранью может быть пло-
плоскость, проходящая через два дейст-
действительных или возможных ребра
кристалла. Так, возможной гранью
кубического кристалла (рис. 56, в) бу-
будет грань ABDC, проходящая через
действительные ребра АВ и CD. Эта
грань также будет атомной плоско-
плоскостью (рис. 56, г) и поэтому на дру-
других кристаллах может проявиться в
виде реальной грани. Точно так же,
если возьмем две реальные грани,
которые на данном кристаллическом
многограннике не пересекаются, то
линия, параллельная линии их пере-
пересечения, будет возможпым ребром
кристалла.
Совокупность граней, пересекаю-
пересекающихся в параллельных ребрах кри-
кристалла, называется поясом, или зо-
зоной. Так, на рис. 56, а, б А грани ку-
куба, пересекающиеся в вертикальных
ребрах, будут представлять одну из
зон кристалла. Параллельная этим
ребрам линия называется осью зоны.
Каждая грань кристалла принадле-
принадлежит по крайней мере двум поясам.
Так, передняя грань куба принадле-
принадлежит зоне, ось которой параллельна
ребру АВ и одновременно — второй
зоне с осью, параллельной ребру АЕ.
Все грани одной зоны проектируют-
проектируются на стереографической проекции на
одной дуге большого круга (рис. 55).
Сама дуга есть проекция оси зоны.
Каждая ось зоны будет возможным
ребром, поэтому каждая ось симмет-
симметрии будет также возможным ребром,
а плоскость симметрии — возмож-
возможной гранью.
Нормаль к плоскости симметрии
также всегда будет возможным реб-
ребром, а плоскость, нормальная к оси
симметрии, — возможной гранью.
Если в кристалле имеются 4 не-
непараллельных грани, то из них мож-
можно вывести бесконечное количество
возможных граней.
§6. Двойники
и закономерные сроетки
Очень часто кристаллы одного и того
же вещества срастаются друг с дру-
другом закономерным образом, образуя
так называемый двойник. При этом
обычно возникают дополнительные
элементы симметрии, называющие-
называющиеся в этом случае двойниковым эле-
ментол симметрии.
41
Рис. 57. Двойниковое срастание
а — индивидуальный кристалл;
б — двойник;
в — полисинтетический двойник
Рис. 58. Двойник по шпинелевому закону
Рис. 59. Срастание двух разных веществ
а — закономерное расположение кристаллов KJ
на слюде (слева показан одиночный кри-
кристаллик);
б — атомные сетки калия в структуре KJ;
« — атомные сетки калия в структуре слюды
Так, на рис. 57, а показан индиви-
индивидуальный кристалл, имеющий ось L2
перпендикулярно плоскости чертежа.
Этот кристалл может срастись со вто-
вторым таким же кристаллом так (рис.
57, б), что получающийся сросток —
двойник — будет иметь плоскость
симметрии т., параллельную оси ?г
и перпендикулярную плоскости чер-
чертежа. Такой плоскости симметрии у
индивидуальных кристаллов нет,—
это двойниковая плоскость симмет-
симметрии. Если сросток состоит из многих
кристаллов (в), закономерно череду-
чередующихся друг с другом, то он называ-
называется полисинтетическим двойником.
Двойникование кристаллов явля-
является очень распространенным явле-
явлением в природе. Многие вещества,
получающиеся в лаборатории, также
часто имеют двойники как простые,
так и полисинтетические.
На рис. 58 показано двойниковое
срастание октаэдрических кристал-
кристаллов. Такие двойники часто встреча-
встречаются у шпинели MgAl2O4 и называ-
называются поэтому шпинелевыми.
Иногда наблюдаются закономер-
закономерные сростки кристаллов разнородных
веществ (эпитаксия). Так, например,
если взять каплю водного раствора
KJ и испарить ее на свежем изломе
слюды KAl2[Si3A10io] (ОН)г, то по-
полученные кристаллы йодистого ка-
калия будут ориентированы парал-
параллельно друг к другу и вполне зако-
закономерно по отношению к определен-
определенным кристаллографическим на-
направлениям в кристаллах слюды
(рис. 59, а). Это объясняется тем,
что плоскости срастания будут иметь
сходное расположение атомов. Так,
в разобранном примере атомы
(ионы) калия в структуре KJ в
плоскости, перпендикулярной Ls
(грань октаэдра), располагаются
по гексагональному закону (рис. 59,
б). Атомы (ионы) калия в структу-
структуре слюды в плоскости, параллельной
грани пинакоида, имеют сходное
расположение (рис. 59, в).
ГЛАВА V
ЗАКОН ЦЕЛЫХ ЧИСЕЛ
И АНАЛИТИЧЕСКИЕ МЕТОДЫ ОПИСАНИЯ
КРИСТАЛЛИЧЕСКИХ МНОГОГРАННИКОВ
§ 1. Открытие закона целых чисел
в кристаллографии
После опубликования работ Роме де
Л'Иля по измерению кристаллов
младший его соотечественник Р.-Ж.
Гаюи в кратчайший срок A784 —
1801 гг.) переработал этот материал
и открыл второй эмпирический закон
геометрической кристаллографии —
закон рациональности отношений
параметров (закон целых чисел).
Трудно переоценить значение этого
закона в кристаллографии.
Его открытие было первым прямым
доказательством прерывного строения
материи, оно предшествовало откры-
открытию закона целых чисел в химии
(Дальтон, 1808 г.). Установлено пря-
прямое влияние Гаюи на Дальтона.
Гаюи не остановился только на
опытной стороне своего открытия. Он
сделал существенную попытку про-
проникнуть в тайну строения вещества,
создав для объяснения закона рацио-
рациональности отношений параметров
стройную для того времени теорию
строения кристаллов из многогран-
многогранных молекул, имеющих различные
размеры по разным направлениям.
Эти материалистические выводы не-
несравненно глубже чисто эмпириче-
эмпирических обобщений Роме де Л'Иля, цели-
целиком стоявшего на идеалистических
позициях и боявшегося изучать то,
что «скрыто от нас самой приро-
природой» — внутреннее строение кристал-
кристаллов.
Сущность закона сводится к сле-
следующему. Пусть кристалл (рис. 60, в)
представляет собой комбинацию двух
простых форм — двух ромбических
дипирамид. Отдельно эти простые
формы показаны на рис. 60, а ж б.
Относительная величина обеих про-
простых форм может быть различной,
тогда и многогранники, получающие-
получающиеся от их комбинации, тоже будут
отличными друг от друга.
На рис. 60, в, г, д и е показаны
комбинации двух дипирамид а ж б,
причем величина дипирамиды а
оставлена постоянной, а дипирамиды
б в направлении от е к д ж далее
к г и в постепенно увеличивающейся.
В результате такого роста кристалла
расстояние п по нормали от центра
до грани дипирамиды б все время
возрастает — rai<! «2< гсз< щ.
Вследствие закона постоянства углов
угол б между гранями дипирамид
а и б и угол A80°—б) между норма-
нормалями п ж т остаются во всех случаях
строго постоянными. Из этого следует,
что размер той или иной грани
в комбинации и связанное с ним
расстояние от центра не имеют для
кристалла существенного значения.
Гораздо важнее расположение граней
относительно друг друга или, что то
же самое, относительно координатных
осей кристалла. За координатные оси
можно выбрать в данном конкретном
примере три взаимно-перпендику-
взаимно-перпендикулярные двойные оси Ьч (рис. 61).
Как бы далеко ни отстояла грань
от центра кристалла, отношение длин
отрезков О А : ОВ : ОС, отсекаемых
гранью на координатных осях, оста-
останется постоянным, в соответствии с
законом постоянства углов. Сами
отрезки (параметры) О А \, ОВ\ и ОС\
43
Рис. 60. Две ромбические диппрамиды и их
комбинации (внизу даны контуры про-
проекция вдоль оси X. Жирными линиями
выделена проекция полученного много-
многогранника)
(рис. 60, г) пе будут соответственно
равны отрезкам О А 2, ОВ2 и OCi (рис.
60, д) ж отрезкам ОА3, ОВг и ОСЪ
(рис. 60, е), но отношения этих отрез-
отрезков всегда будут равны, т. е.
О Ах ф ОАг ф ОА3,
OBi = OBi ф ОВг,
О& ф ОСг ф ОС3,
НО
О Ах : ОВг : ОСг = ОА2 : ОВг : Od =
= ОАг : ОВг : ОС3. A)
Из всего вышесказанного следует,
что перемещение граней параллель-
параллельно самим себе не влияет на соотно-
соотношение A). При росте кристаллов
грани именно перемещаются парал-
параллельно самим себе, при этом отно-
относительное перемещение отдельных
граней может сильно меняться в
зависимости от условий роста, но
углы наклона по отношению друг к
другу и координатным осям остают-
остаются постоянными.
Закон рациональности отношений
параметров (закон целых чисел)
может быть сформулирован так:
Отношение отрезков (параметров),
отсекаемых гранью кристалла на трех
координатных осях, равно отноше-
отношению целых и взаимно простых чи-
чисел, при условии, что эти параметры
измерены особыми единицами для
каждой из осей. За единицы измере-
измерения должны быть взяты параметры
некоторой другой грани кристалла.
Грань, параметры которой приня-
приняты за единицы измерения парамет-
параметров остальных граней, называется
единичной гранью.
На рис. 62 изображены две грани
дипирамид а ж б (рис. 60). Для удоб-
удобства рассуждения грань ABC сдви-
сдвинута параллельно самой себе так,
чтобы один из параметров совпал
для обеих граней ОЬ=ОВ.
Из сформулированного закона
следует, что если измерить отрезок
(параметр) ОА параметром Оа,
ОВ — параметром Ob и ОС — пара-
параметром Ос и взять их отношения
ОА1Оа:ОВ,ОЪ: ОС/Ос, то оно будет
равно отношению простых целых
Рис. 61. Выбор координатных осей в мно-
многограннике
чисел. В нашем примере:
ОА ов ОС
Оа
2 * 3 я
Отрезки Оа, Ob и ОС не равны друг
другу. И мы их используем как еди-
единицы (масштабы) измерения — каж-
каждый для одной определенной оси.
Гаюи показал, что закон целых
чисел — закон рациональности отно-
отношений параметров — является таким
же общим законом для всех кристал-
кристаллов, как и закон постоянства дву-
двугранных углов. Для всех измерен-
измеренных к тому времени кристаллов и
для всех граней каждого кристалла
им была показана справедливость
этого закона.
Гаюи не остановился на простом
констатировании закона, он сделал
Рпс. 62. Положение граней двух дипира-
дипирамид относительно координатных осей
попытку объяснить его, исходя из
молекулярных представлений. В его
представлении молекулы вещества
имели форму многогранников, ана-
45
Рис. 63. Грани кубического кристалла, от-
отсекающие на осях OY и OZ равные отрез-
отрезки (а) и отрезки в отношении 2:1 (б)
логичных кристаллическим много-
многогранникам. Ему было известно свой-
свойство многих кристаллов при ударах
раскалываться по плоскости (явле-
(явление спайности). Таким свойством,
например, обладают кристаллы по-
поваренной соли. Если ударить молот-
молотком по кристаллу NaCl, то он рас-
рассыплется на осколки, имеющие фор-
форму прямоугольных параллелепипе-
параллелепипедов и, в частности, кубиков. Гаюи
представлял себе, что если продол-
продолжать дробление дальше и дальше и
получать все более мелкие и мелкие
осколки в форме кубиков, то, в кон-
конце концов, придем к мельчайшим
далее неделимым частицам — моле-
молекулам, которые будут иметь ту же
форму.
На рис. 63, а показан разрез ку-
кубического кристалла, у которого
проекция грани СВ отсекает на
осях OY и OZ равные отрезки (пара-
Рис. 64. Грань октаэдра, построенная из
молекул, имеющих форму куба (по Гаюи)
метры). Вторая грань того же кри-
кристалла С В' (б) отсекает отрез-
отрезки ОВ' и ОС, отношение отрез-
отрезков ОВ'/ОВ : ОС/ОС -2:1. На
рис. 64 показан рисунок Гаюи, ил-
иллюстрирующий грань октаэдра, по-
построенную из «молекулярных» ку-
кубов. В общем случае, по представле-
представлениям Гаюи, размеры таких нарал-
лелепипедальных молекул могут
быть различными в разных направ-
направлениях и несоизмеримыми друг с
другом. Они будут иметь форму не
кубов, как в разобранном выше при-
примере, а параллелепипедов,— прямо-
прямоугольных в случае прямоугольной
системы координат, т. е. будут иметь
форму спичечной коробки или кир-
кирпича. На рис. 65 показан разрез
кристалла, построенного из таких
кирпичиков. Отрезок ОС не равен
отрезку ОВ (рис. 65, а), потому что
ребро с кирпичика не равно ребру Ъ,
но если измерять отрезок ОС пара-
параметром с, а отрезок ОВ — парамет-
параметром Ь, то грань ВС и в этом случае
будет единичной гранью (как и в
случае рис. 63, а), ибо отношение
ОС/с : ОВ/Ъ= 6/1 : 6/1 = 1 : 1. Соответ-
Соответственно грань СВ' (рис. 65, б) дает
то же отношение, что и соответст-
соответствующая грань на рис. 63, б. Разни-
Разница будет только в том, что масштабы
измерения (единичные параметры)
по осям Z и Y в последнем случае
будут разными, а числовые величи-
величины полученных отношений будут
оставаться одинаковыми и постоян-
постоянными.
Исходные параллелепипеды могут
быть и косоугольными. Тогда и си-
система координат должна быть косо-
косоугольной (рис. 66, а и б). Но и в
этом случае также сохранятся все
числовые соотношения, характери-
характеризующие закон рациональности отно-
отношений параметров.
Как ни кажутся нам сейчас наив-
наивными представления Гаюи о много-
многогранных молекулах, мы не должны
преуменьшать значение этой работы,
значение открытия закона рацио-
рациональности отношений параметров.
46
§ 2. Кристаллографические символы
Рис. 65. Грани ромбического кристалла,
отсекающие на осях OY и OZ неравные
отрезки в отношении 1:1 (а) и 2: 1 (б)
Это нервый закон целых чисел, от-
открытый в естествознании. Его откры-
открытие является первым прямым дока-
доказательством прерывного, «молекуляр-
«молекулярного» строения материи. В самом
деле, если бы материя (кристаллы)
не была построена из отдельных тож-
тождественных друг другу частиц, то
было бы необъяснимо существование
такого закона. Влияние этого откры-
открытия на все области знания, и в пер-
первую очередь на химию, весьма вели-
велико. Дальтон, открывший позже (в
1S08 г.) закон целых чисел в химии,
бывал в предшествовавшие годы в
Париже, где слушал лекции Гаюи,
поэтому влияние открытия закона
целых чисел в кристаллографии на
открытие закона целых чисел в хи-
химии не подлежит сомнению. Оба эти
закона вытекают и являются след-
следствием одних и тех же причин — пре-
прерывного строения материи.
Положение грани в пространстве
(т. е. по отношению к системе ко-
координат) однозначно определяется
величинами отрезков (параметров),
отсекаемых ею на координатных
осях. Так, положение грани ABC
(рис. 62) однозначно определяется
отрезками ОА, ОВ и ОС, если систе-
система координат (XYZ) задана. Если
выберем соответствующие масштабы,
т. е. единичные параметры, которыми
будем измерять по каждой из осей, то
положение грани ABC будет подчи-
подчиняться закону целых чисел. В ка-
качестве единичных параметров могут
быть взяты отрезки, отсекаемые на
тех же осях другой гранью того же
кристалла, т. е, отрезки Оа, Ob и Ос
(рис. 62).
При этом абсолютные размеры от-
отрезков ОА, ОВ и ОС теряют всякое
значение, и можно представить грань
ABC перемещающейся параллельно
самой себе в любое положение. Обыч-
Обычно принимается положение, когда
грань проходит через начало коорди-
координат. В этом случае весь кристалл
представляется системой плоскостей
(граней), проходящих через начало
координат.
В кристаллографии для определе-
определения положения грани в пространстве
берется не прямое отношение целых
чисел, например (в случае рис. 62)
ОА ОВ ОС о / о *
dT:W:W = 8:4:3' а обратные
им величины, т. е.
1 1 1 ..,
Рис. 66. Грани триклинного кристалла,
отсекающие на осях OY n OZ неравные
отрезки в отношении 1:1 (а) и 2:1 (б)
ОА/Оа " ОВ\ОЪ • ОС/Ос.
Каждая из этих величин называ-
называется индексом символа грани по
данной координатной оси, а сово-
совокупность трех индексов называется
символом грани. Символ грани ста-
ставится в круглые скобки без каких-ли-
каких-либо знаков между индексами. Так,
символ грани ABC будет C68). Если
индекс является двузначным числом,
47
то он отделяется точками. Так, на-
например, символ A0.2.3) показывает,
что по оси X индекс равен 10, по оси
Y — 2 и по оси Z — 3. Для подавля-
подавляющего большинства реальных гра-
граней кристалла индексы символа
обычно бывают меньше 10.
Обратные величины приняты в
кристаллографической символике
вследствие того, что для граней, па-
параллельных одной или двум коорди-
координатным осям, индексами символа бу-
будут нули, а не бесконечности, что
гораздо удобнее при всех математи-
математических операциях с этими величи-
величинами.
Для нахождения символа той или
иной грани кристалла надо прежде
всего выбрать координатные оси и
единичную грань, затем определить
величины отрезков, отсекаемых на
осях X, Y и Z исковой гранью; из-
измерить эти отрезки соответствую-
соответствующими отрезками единичной грани;
взять отношения, обратные найден-
найденным, и привести их к целым числам.
Полученные величины будут яв-
являться индексами символа данной
грани.
Каждая грань простой формы по-
получает свой символ, отличающийся
от символа другой грани той же
простой формы переменой индексов
символа но осям и знаками. Число-
Числовые значения индексов символа для
всей простой формы остаются одни-
одними и теми же. Так, на рис. 67 пока-
показаны куб и октаэдр с обозначением
символов каждой грани, в нижней
части рисунка даны их стереографи-
стереографические проекции. Шесть граней куба
имеют следующие символы: A00),
@10), @01), A00), @10), @01).
Если желательно символом показать
всю простую форму, то символ ста-
ставится в фигурные скобки. Так, сим-
символ куба будет {100}. Под этим сим-
символом подразумеваются все 6 выше-
написанных символов. Символом ок-
октаэдра будет {111}. Под этим симво-
символом подразумеваются все его 8 гра-
граней: A11), A11), AЦ), (ГЦ),
A11), A11), A11), AЙ).
48
Рис. 67. Кристаллографические символы
куба (а) и октаэдра (б)
§ 3. Математическое определение
символов грани
Для точного определения символов
грани обычно используется теорема
косинусов Вульфа. Согласно этой
теореме, индексы символа грани
прямо пропорциональны косинусам
углов, которые составляют нормаль
к данной грани с соответствующими
осями координат. За единицу изме-
измерения косинусов для каждой оси на-
надо принимать косинус угла, который
образует с данной осью нормаль к
единичной грани.
На рис. 68 изображены две грани
кристалла аЬс и ABC. Первая грань
принята за единичную. Требуется
найти символ грани ABC.
На каждую грань из начала ко-
координат опущены нормали On и ON
соответственно. Нормаль On дает с
осями X, Y и Z углы fa, ц\ и vi, где
Рис. 68. К определению символа грани
ABC (abc — единичная грань)
=/LnOZ. Нормаль ON с осями X, Y
и Z образует соответственно углы Л,
ц и v, где X—/LNOX и т. д.
Пусть символ грани ABC будет
(pqr). Требуется доказать, что
COS X COS [A COS V
Р'- Я'-r = :
B)
¦ COS [il ' COS Vl
По определению символа имеем
_ _J 1 1
Р'-Ч'-г~ ОА/Оа '• ОВ/ОЬ : ОС/Ос ~
Оа ОЬ Ос
= ~ОА '¦ О В '¦ ОС ¦
Из шести прямоугольных треуголь-
треугольников следует, что
Оа = Оп: cos Хг OA—ON: cos X
ОЬ = Оп: cos Hi и OB •= ON : cos p
Oc = On: cos vi ОС = ON : cos v.
Откуда
Oa ~: On : cos
On
ON
cos Я,
cos Xi
ОА~ ON: cosX
соответственно
Ob On cos fi Ос On cos v
OB ~ ~ON ' cos Hi и ОС = [ON ' cosvi *
Подставляя эти отношения в урав-
уравнение B) и сокращая на On/ON, по-
получаем
cos X cos Ц cos v
Р-Я r = costa. ' cosjii ' cos vi '
что и требовалось доказать.
Таким образом, если в избранной
системе координат задано положение
единичной и любой другой грани кри-
кристалла, то, найдя величины косинусов
углов между нормалями к этим гра-
граням и координатными осями, можно
определить символ любой грани.
§4. Установка кристаллов
Под термином «установка кристал-
кристаллов» подразумеваются правила выбо-
выбора координатных осей и единичной
грани в кристаллах разных сингоний.
Установка кристалла проводится в
строгом соответствии с его симметри-
симметрией.
За координатные оси всегда выби-
выбираются действительные или возмож-
4 Кристаллохимия
ные ребра кристалла и, в частности,
оси симметрии.
Проще всего установка кубиче-
кубических кристаллов, где за координат-
координатные оси принимаются три двойные
или четверные взаимно-перпендику-
взаимно-перпендикулярные поворотные оси, за единич-
единичную грань — грань октаэдра или тет-
тетраэдра. Они отсекают на всех трех
координатных осях равные отрезки.
Координатные оси обозначаются X,
Y и Z. Координатные углы — а, |5 и у,
?ри этом а лежит против оси X, т. е.
это угол YOZ, р — против Y (угол
XOZ) и Y — против Z (угол XOY).
Кратко установка кристаллов куби-
кубической сингоний может быть записа-
записана так: а = р=-у=9О°, а = Ь=с, где
а, Ъ ж с — отрезки, отсекаемые еди-
единичной гранью на осях X, Y и Z.
В кристаллах триклинной синго-
сингоний нет никаких элементов симмет-
симметрии, кроме центра, поэтому любые
три ребра кристалла (действитель-
(действительные или возможные), не лежащие в
одной плоскости, могут быть взяты за
координатные оси, любая грань, пе-
пересекающая все три координатные
оси,— за единичную. Соответственно
сказанному установка триклинных
кристаллов будет записана так: аф
Ф$Фу = 90°, афЪфс. Описы-
Описывать такой кристалл надо в косоуголь-
косоугольной системе координат и измерять
параметры по каждой из координат-
координатных осей своим масштабом.
В табл. 6 собраны правила уста-
установки кристаллов всех сингоний.
В тригональной и гексагональной
сингониях удобно применять систе-
систему координат с 4 осями. Дополни-
Дополнительная ось U берется под углом
120° к осям X и Y (см. рис. 69).
С точки зрения аналитической гео-
геометрии она не нужна. Индекс симво-
символа по этой оси не независим. Легко
доказать, что он равен сумме индек-
индексов по первым двум осям с обратным
знаком U= — (X-\-Y). Однако, чтобы
символы граней всей простой формы
имели одинаковые индексы, прихо-
приходится вводить дополнительную
ось.
49
ТАЛЛИЦА 6
Установка кристаллов
Сингония
Выбор координатных осей
Координатные
углы и единичные
параметры
Единичная грань
Кубическая
Тетрагональная
Ромбическая
Моноклинная
Триклинная
Тригональная и
гексагональная
Три взаимно-перпендикуляр-
взаимно-перпендикулярные оси Li или Ьц
L4 или L- выбирается за ось Z.
За X и У — перпендикулярные
L, перпендикулярные норма-
нормали к плоскостям симметрии или
перпендикулярные ребра
Три оси Ьг, или — одна Lt за Z
и две нормали к плоскостям
симметрии за X и У
L2 или нормаль к плоскости ва
ось Y. За X и Z — два дейст-
действительных или возможных реб-
ребра, лежащих в плоскости сим-
симметрии или в плоскости, нор-
нормальной к оси симметрии.
В последние годы наметилась
тенденция Ьг располагать верти-
вертикально, т. е. принимать за ось Z
Три действительных или воз-
возможных ребра кристалла
За ось Z —1.6, L g , Lt или L-,
за оси X, Y и U — три оси ?2,
или нормали к плоскостям сим-
симметрии, или три ребра, распо-
расположенные под 60° друг к другу
и лежащие в плоскости _|_ к Z
(см. описание в тексте)
а = р = у
а = Ъ = с
афЬф с
афЪфс
90
Грань октаэдра или
тетраэдра
Грани пирамиды, ди-
пирамиды или тетра-
тетраэдра
Грань ромбических
пирамиды, дипирами-
ды или тетраэдра
Грани ромбической
призмы или диэдра
фРфГ
афЬфс
а = Р = Э
Г = 120°
о = Ь фс
Грани пинакоида или
мояоэдра
Грани соответствую-
соответствующих пирамиды, дипи-
рамиды или ромбоэдра
Все вычислительные операции про-
проще всего производить в системе ко-
координат XyZ, а затем вписать четвер-
четвертый индекс символа, руководствуясь
написанным выше соотношением.
Ца рис. 69 показана проекция гек-
гексагональной призмы {1120} с указа-
указанием символов всех ее шести граней:
A120) ,B110), A210), A120),B110),
BН0)
{1210)
г „_ (HZO)
/(МО)
Рис. 69. Проекция и символы гексагональ-
гексагональной призмы
B110). Если отбросить третий ин-
индекс, то символы граней будут^(НО),
'B10), A20), A10), B10), A20), и
по ним нельзя будет сказать, что они
принадлежат шести граням одной
простой формы. По виду общего
символа {110} нельзя будет непосред-
непосредственно заметить, что другие грани
той же простой формы будут иметь
индексы не единицы, а двойки.
Для кристаллов тригональной
сингонии существует и другая уста-
установка. За оси координат выбираются
три ребра ромбоэдра или пирамиды.
В этом случае а = р = у =90° и
а = Ь = с. Единичной гранью явля-
является грань пинакоида или моноэдра.
Она расположена перпендикулярно
к главной оси и отсекает поэтому
на ребрах одинаковые отрезки.
ЧАСТЬ ВТОРАЯ
ГЕОМЕТРИЧЕСКАЯ
ТЕОРИЯ СТРУКТУРЫ
КРИСТАЛЛА
глава vi
КРИСТАЛЛИЧЕСКАЯ РЕШЕТКА
§1. Понятие
кристаллической решетки
Наблюдающаяся геометрическая пра-
правильность форм кристаллов застави-
заставила исследователей искать причины ее
в закономерном внутреннем (атом-
(атомном) строении. Уже И. Ньютон в
своей «Оптике» в 1675 г. писал:
«Нельзя ли предположить, что при
образовании кристалла частицы не
только установились в строй и ряды,
застывая в правильных фигурах, но
также посредством некоторой поляр-
полярной способности повернули свои оди-
одинаковые стороны в одинаковом на-
направлении».
В последующее время в работах
Гюйгенса, Ломоносова, Гаюи и Вол-
ластона мы находим идеи, которые в
еще более ясной форме предвосхи-
предвосхищают понятие кристаллической ре-
решетки. В 1813 г. Волластон предло-
предложил заменить многогранные молеку-
молекулы Гаюи шарами или просто матема-
математическими точками. В результате бы-
было создано представление о кристалле
как о пространственной решетке.
Для построения пространственной
решетки достаточно задать в прост-
пространстве четыре точки О, А, В, С так,
чтобы на одной прямой было не боль-
больше двух точек, а в одной плоско-
плоскости — не больше трех. Другие точки,
или узлы решетки, получим из дан-
данных параллельными переносами их
по направлениям OX, OY, OZ на рас-
расстояния О А, ОВ, ОС (рис. 70). Сле-
Следовательно, решетка есть бесконеч-
бесконечное, трехмерное периодическое обра-
образование.
Совокупность узлов, расположен-
расположенных на прямой, определяемой двумя
произвольными узлами решетки, на-
г
Рис. 70. Построение пространственной ре-
решетки
51
зывается рядом, расстояние между
ближайшими точками ряда — пара-
параметром ряда. Плоскости, определяе-
определяемые тремя произвольными узлами,
не лежащими на одной прямой, назы-
называются сетками; а параллелограммы,
построенные по узлам сетки,— пет-
петлями; параллелепипеды, вершины
которых заняты узлами решетки,—
ячейками решетки. Ячейка называет-
называется примитивной, или простой, если
• узлы решетки располагаются только
в вершинах ячейки, и сложной, если
узлы решетки содержатся также где-
либо внутри или на поверхности
ячейки.
В одной и той же решетке можно
выбрать различными способами бес-
бесконечное множество примитивных
ячеек, отличающихся друг от друга
по величине ребер и углам между ни-
ними. Объем примитивной ячейки,
однако, не зависит от ее формы и яв-
является величиной постоянной для
данной решетки, так как он пред-
представляет собой тот объем, кото-
который приходится на один узел ре-
решетки.
Пространственную решетку можно
представлять себе либо как бесконеч-
бесконечную систему узлов, либо как беско-
бесконечную систему параллелепипедов,
целиком заполняющих пространство.
Оба представления не вполне эквива-
эквивалентны друг другу; в частности, сим-
симметрия системы параллелепипедов
неправильно отражает истинную сим-
симметрию гексагональных кристаллов,
чего нельзя сказать о системе узлов.
На этом основании мы будем в даль-
дальнейшем рассматривать пространст-
пространственную решетку предпочтительно
как систему узлов и считать линии и
плоскости, проводимые внутри ре-
решетки, как вспомогательные элемен-
элементы, не входящие в решетку.
Идея замены многогранных моле-
молекул Гаюи математическими (безраз-
(безразмерными) точками безусловно была
прогрессивной, так как никаких ме-
методов, позволявших изучить форму
частиц (атомов и молекул), в то вре-
время не было. Вместе с тем эта идея
позволяла заниматься математиче-
математической стороной вопроса о пространст-
пространственном расположении узлов решетки,
о симметрии решеток. Полный вывод
всех возможных случаев кристал-
кристаллических решеток был сделан в
1855 г. О. Бравэ (см. ниже 14 реше-
решеток Бравэ).
§2. Кристаллический многогранник
и решетка кристалла
Существует соответствие между тер-
терминами, употребляемыми при описа-
описании кристаллических многогранников
и кристаллических решеток. Это мож-
можно легко уяснить из рис. 71.
Кристаллический многогранник'
ограничен конечным числом граней.
Каждой грани кристалла в кристал-
кристаллической решетке отвечает серия па-
параллельных плоских сеток. Число па-
параллельных плоских сеток в этой
серии бесконечно, так же как беско-
бесконечно и число таких серий, ибо через
любые три узла решетки можно про-
провести плоскую сетку и параллельно
ей бесконечное число таких же пло-
плоских сеток. Каждая сетка будет воз-
возможной гранью кристалла.
Грани кристалла пересекаются в
ребрах. Плоские сетки — в рядах.
Число ребер в кристаллическом мно-
многограннике всегда конечно, число ря-
рядов в решетке бесконечно велико.
Каждому ребру кристалла соответст-
соответствует в решетке бесконечная серия
параллельных рядов. Кроме того, бу-
будет бесконечно много других серий
рядов, параллельных возможным реб-
ребрам кристалла. Ребра в кристалличе-
кристаллическом многограннике, пересекаясь, об-
образуют вершины. Ряды же в решет-
решетке пересекаются в узлах. Можно,
конечно, в решетке провести ряды,
пересекающиеся в точке, не являю-
являющейся узлом (см. ряды а и б на
рис. 72). Но всегда можно выбрать
ряды б\, бг и т. д., параллельные б и
идущие от узлов ряда а. Сказанное в
равной мере, конечно, относится и к
пересечению плоскостей.
52
S
Рис. 71. Кристаллический многогранник
(а) и кристаллическая решетка (б)
Рис. 72. Различные серии рядов в кри-
кристаллической решетке
Число узлов в кристаллической ре-
решетке мы также мыслим себе беско-
бесконечным, иначе узлы, расположенные
на гранях, будут отличаться от узлов,
расположенных внутри решетки, а
мы всегда говорим о тождествен-
тождественности узлов. Следовательно, всякую
кристаллическую решетку мы мыс-
мыслим неограниченной, т. е. простираю-
простирающейся до бесконечности по всем трем
направлениям. В этом смысле мы го-
говорим уже не о кристалле, а о кри-
кристаллическом пространстве.
§3. Трансляция
Вследствие строгой периодичности
кристалла во всех трех измерениях
одинаковые материальные частицы —
Рис. 73. Различные по величине и на-
направлению трансляции в одной и той же
решетке
структурные элементы — закономер-
закономерно повторяются.
Эта повторяемость схематически
может быть описана при помощи
трансляций — симметрических пре-
преобразований, характеризующих па-
параллельный перенос всей структуры.
Элементом симметрии, отвечающим
новому симметрическому преобразо-
преобразованию, будет ось трансляции. Для
точной характеристики периодично-
периодичности кристалла необходимо указать
направление трансляций и их вели-
величину. Надо всегда иметь в виду, что
в литературе термин «трансляция»
используется как для обозначения
симметрического преобразования, так
и элемента симметрии.
На рис. 73 изображена структура
кристалла. Для простоты и наглядно-
наглядности взят пример двумерной структу-
структуры. Направления и величины различ-
различных трансляций указаны векторами
t\, г2, h,... Любой параллельный пере-
перенос структуры (простирающейся до
бесконечности) в направлении век-
векторов t\, h,... характеризуется опреде-
определенной величиной переноса.
Естественно, что в любой структу-
структуре число таких направлений — осей
трансляций — будет бесконечно.
Совокупность всех трансляций в
кристаллической структуре состав-
составляет трансляционную группу, назы-
называемую иначе группой переносов, или
кристаллической решеткой.
Само собой разумеется, что для ха-
характеристики периодичности кри-
кристалла нет необходимости брать все
возможные трансляции. Для этой це-
цели достаточно выбрать в пространст-
пространстве три не лежащие в одной плоско-
плоскости трансляции, которые для просто-
простоты можно мыслить пересекающимися
в одной точке — в начале координат.
На этих трансляциях можно постро-
построить параллелепипед, которым можно
характеризовать решетку кристалла.
Весь кристалл в этом случае окажет-
окажется разбитым мысленно на равные па-
параллелепипеды, находящиеся в па-
параллельном положении, касающиеся
друг друга целыми гранями и без
53
промежутков заполняющие все про-
пространство. Отсюда ясно, что термин
«параллелепипедальная система» мо-
может употребляться в известном смыс-
смысле как синоним решетки.
Естественно, что поскольку решет-
решетку можно построить на любых трех
трансляциях, не лежащих в одной
плоскости, то и параллелепипедаль-
ные системы могут быть выбраны для
данной структуры бесконечно разно-
разнообразными способами. В нашем при-
примере (рис. 73, а) вспомогательные
линии — ребра параллелограмма (в
пространстве — параллелепипеда) —
построены на трансляциях U и t5.
Конечно, систему вспомогательных
линий можно было бы провести в на-
направлениях любых двух векторов t,
не лежащих на одной прямой, напри-
например t\ и U (рис. 73, б).
Необходимо помнить, что исход-
исходный параллелепипед повторяемости,
а с ним и всю параллелепипедальную
систему можно переносить в кристал-
кристаллическом пространстве параллельно
самой себе.
Начало координат, т. е. вершину
параллелепипеда повторяемости, или,
что то же самое, узел решетки, мы
можем помещать в любую точку кри-
кристаллической структуры. Помещение
его в ту или иную точку определяет-
определяется только удобством вычисления.
На рис. 74 изображена структура
соединения, состоящего из двух раз-
различных типов атомов химических
элементов — «белого» и «черного».
Начало координат выбрано в центре
тяжести «белого» атома а. Размеры
ячейки определяются величинами
трансляций t\ и ti. Начало координат
можно поместить в центре тяжести
«черного» атома Ь. Форма и размеры
элементарного параллелепипеда, как
это легко видеть на рисунке, конечно,
останутся прежними. Можно поме-
поместить начало координат в середину
отрезка, соединяющего центры «бе-
«белых» и «черных» атомов (точка с),
и везде в направлениях, параллель-
параллельных ребрам предыдущих параллеле-
параллелепипедов (пунктирных), на расстоя-
54
Рис. 74. Различные способы выбора начала
координат в структуре
ниях t\ и ti мы будем встречать точ-
точки с\, а и т.д., тождественные точки
с, делящие линии, соединяющие
центры атомов а и Ъ, пополам. Мож-
Можно поместить начало координат в точ-
точку, делящую этот отрезок в отноше-
отношении 2:3 или вообще m:n, и всегда мы
будем получать в параллельном на-
направлении тождественные точки на
расстояниях t\ и t2. Таким образом,
можно бесконечно различным спосо-
способом выбирать начало координат, но
всегда будем получать одинаковые по
размерам и одинаково ориентирован-
ориентированные параллелепипеды повторяемости.
Неправильно трактовать структу-
структуру кристалла как систему нескольких
решеток, вставленных одна в другую
(первая — «белая» и вторая — «чер-
«черная» решетки). Таких систем можно
выбрать для данного примера не толь-
только эти две, но, как было показано вы-
выше, бесконечно много. Но раз таких
систем может быть бесконечно много,
то надо из этого многообразия суметь
Рис. 75. Различные параллелепипеды (па-
(параллелограммы) повторяемости в одной
и той же решетке
выбрать одну. Из бесконечно разно-
разнообразных по форме и размерам парал-
параллелепипедов повторяемости (рис.
75), характеризующих одну и ту же
решетку, мы для удобства работы ос-
останавливаемся на одном. Точно так
же каждую структуру будем характе-
характеризовать одной единственной решет-
решеткой определенной симметрии и опре-
определенного размера. Под размерами
решеток подразумеваются размеры
их элементарных ячеек. Начало же
координат будем помещать в такую
точку структуры, которая окажется
удобной для проведения вычислений.
Часто бывает удобно поместить его
в центр тяжести атома, тогда, будучи
совмещенным с узлом решетки, он
получит координаты @00), что зна-
значительно упрощает все вычисления.
Но в двухатомной молекулярной
структуре (рис. 76) удобнее для рас-
расчета поместить начало координат в
точку, делящую линию, соединяю-
соединяющую два атома, пополам. В этом слу-
случае координаты атомов будут отли-
отличаться только знаками, и расчет бу-
будет производиться проще, чем в слу-
случае помещения начала координат в
центре тяжести одного из атомов.
Из сказанного должно быть ясно,
что с узлами решетки связаны мате-
материальные частицы структуры, но со-
совершенно не обязательно считать, что
непосредственно в узлах располага-
располагаются материальные частицы.
Решетку кристалла следует вос-
воспринимать как математическую аб-
абстракцию, аналогичную понятию эле-
элемента симметрии, употребляющегося
при описании кристаллических мно-
многогранников; при помощи такого по-
понятия решетки можно удобно (мате-
(математически) описывать периодичность
кристаллической структуры. Понятие
«решетки кристалла» недопустимо
путать с понятием «структуры кри-
кристалла». Под структурой мы понима-
понимаем конкретное расположение матери-
материальных частиц в кристалле. Число
различных типов решеток равно 14.
Число различных структурных ти-
типов бесконечно велико.
Р
Р
Р
Рис. 76. Молекулярная структура (начало
координат выбрано в центре тяжести
молекулы)
Основное свойство решетки — ее
периодичность — проявляется в том,
что любые ее два узла можно совме-
совместить друг с другом при помощи
трансляции. При таком совмещении
все остальные узлы решетки совме-
совместятся с другими узлами той же ре-
решетки. Таким образом, вся решетка
совместится сама с собой, и мы не
сможем отличить начальное положе-
положение решетки от ее конечного положе-
положения.
Иэ этого свойства решетки выте-
вытекает важное следствие о невозможно-
невозможности присутствия в кристаллах осей
пятого, седьмого и более высоких по-
порядков. Доказать это положение
можно следующим образом.
Предположим, что в кристалле
имеется Ь^. Тогда, взяв в решетке
такого кристалла один из узлов, бли-
ближайших к этой оси, мы обязаны полу-
получить вокруг нее 5 узлов на том же
расстоянии (см. 1—5 на рис. 77).
Между любыми двумя узлами решет-
2о-
о',
/о-
Рис. 77. К доказательству невозможности
в кристаллах осей 5-го порядка
55
ки имеется трансляция (например,
1—5). При трансляционном переносе
все узлы решетки должны совме-
совместиться друг с другом. Если сущест-
существует трансляция 1—5, то узел 2 при
таком переносе должен переместиться
параллельно 1—5 на то же расстоя-
расстояние. Стрелка, проведенная от него в
направлении, параллельном 1—5, не
попадает в узел. Ее конец оказыва-
оказывается внутри пятиугольника, т. е. на
расстоянии, более коротком, чем рас-
расстояние ?>ъ—2, Ls—2 и т.д., а эти
расстояния приняты по условию за
кратчайшие. Следовательно, в решет-
решетке не может быть осей пятого поряд-
порядка. Так же доказывается невозмож-
невозможность в решетке осей L7 и более вы-
высоких порядков.
§4. Плоское сетки решетки
В общем случае петля плоской сетки
будет параллелограммом (рис. 78, а).
Однако ввиду того, что кристалличе-
кристаллические решетки могут обладать разной
симметрией, то и плоские сетки могут
иметь разные оси (в проекции точки)
и плоскости (в проекции линии) сим-
симметрии.
Рис. 78. Возможные случаи плоских сеток
Каждый параллелограмм харак-
характеризуется тремя величинами: двумя
ребрами — а и Ь и углом у. Частные
случаи получаются в зависимости от
возможных случаев симметрии.
Симметрия случая афЪ, у ф90°
(рис. 78, а) характеризуется наличи-
наличием только осей L2 в центрах паралле-
параллелепипедов.
Если афЪ, y=90o (рис. 78, б),
то через зти оси будут проходить по
две взаимно-перпендикулярные пло-
плоскости.
Возможна, конечно, и вторая сет-
сетка с симметрией предыдущего глу-
чая, если а = Ь и у ф 90° (рис. 78, в).
Сетка получит ось L4, если а — Ъ
и у = 90° (рис. 78, г). При этом каж-
каждый параллелограмм будет иметь 4
плоскости симметрии, проходящие
через L^ Такая же симметрия будет
характеризовать каждый узел.
Если а = Ь и у = 60° или, что то
же самое, y = 120°, то получим пя-
пятую сетку (рис. 78, д). Как мы гово-
говорили выше, параллелограммы в сетке
есть результат вспомогательного по-
построения. Сетка есть совокупность уз-
узлов, а линии (ребра параллелепипеда)
можно проводить через узлы беско-
бесконечным числом способов. Для нагляд-
наглядности проведем на рис. 78, д верти-
вертикальную систему линий, соединяю-
соединяющих узлы (пунктирные прямые).
В этом случае выявится симметрия
Le6P для узлов и LS3P для центров
треугольников.
Этими пятью вариантами исчер-
исчерпываются все случаи симметрии пло-
плоских сеток решеток. Обычно бывает
удобнее вести вычисления в прямо-
прямоугольной системе координат, поэтому
случаям бив (рис. 78), имеющим ;
одинаковую симметрию, придается и
одинаковая установка: аФЬ,у = 90°.
Отличие их заключается в том, что
для случая в в центре петли оказы-
оказывается дополнительный узел, и па-
параллелограмм из примитивного дела-
делается центрированным (рис. 79). На
каждый примитивный параллело-
параллелограмм приходится 1 узел, на центри-
центрированный — 2.
56
Форму петли плоской сетки всег-
всегда можно определить, если известна
форма грани кристалла, параллель-
параллельная этой сетке.
Покажем это на примере. На рис. 80
изображена пунктиром одна из ре-
реальный граней ромбоэдра кварца.
Рис. 79. Два способа выбора элементарно-
элементарного параллелограмма в ромбической сетке
Рис. 80. Определение формы петли плос-
плоской сетки на грани ромбоэдра кварца
Рис. 81. Определение формы ячейки
Рис. 82. Построение ячейки решетки
Возьмем произвольную точку О и
проведем через нее пучок прямых,
параллельных ребрам грани. Две из
этих прямых ОХ и OY примем за оси
сетки, а на третьей — О А — отметим
произвольно узел сетки а. Этими дан-
данными определяется вся сетка, а сле-
следовательно и ее петля.
Указанное построение может слу-
служить опытным подтверждением сет-
сетчатой структуры грани кристалла,
так как при всей произвольности
сделанного построения все линии
пучка обязательно будут проходить
через узлы сетки. Из изложенного
легко видеть, что определение формы
петли сетки по форме грани возможно
лишь в том случае, если грань имеет
по меньшей мере три ребра, среди ко-
которых нет параллельных друг другу.
Аналогично может быть определе-
определена и форма ячейки решетки. Для это-
этого достаточно провести из произволь-
произвольно выбранной точки О (рис. 81) четы-
четыре ребра OX, OY, OZ, ОА параллель-
параллельно каким-либо четырем ребрам
кристалла, отметить на ребре ОА про-
произвольную точку а и, приняв ее за
конец диагонали параллелепипеда,
построить самую ячейку и отвечаю-
отвечающую ей решетку. (В общем случае
это будет форма сложной ячейки.)
Проделав это построение, можно убе-
убедиться, что все другие ребра кристал-
кристалла будут параллельны рядам решетки,
а все грани кристалла — параллельны
ее сеткам.
На практике приходится иметь
дело не с ребрами, а с гранями кри-
кристалла. Измерив на гониометре углы
между какими-либо четырьмя граня-
гранями кристалла, нетрудно по ним по-
построить сначала тетраэдр, а затем и
параллелепипед (рис. 82). Выбор че-
четырех граней в этом случае ограничи-
ограничивается только одним условием, чтобы
в одной зоне было не более двух гра-
граней.
Что касается размеров петли пло-
плоской сетки и ячейки решетки, то они,
как увидим далее (гл. VIII), опре-
определяются с помощью рентгеновских
лучей.
57
§5. 14 решеток Бравэ
Способом, указанным в предыду-
предыдущем параграфе, можно найти фор-,
мы ячеек кристаллических реше-
решеток для кристаллов всех 7 сингоний|
(рис. 83). Характеристика этих ячеек
(соотношение величин ребер и уг-
углов, обычно называемых параметра-
параметрами решетки) целиком совпадает с
данными табл. 6 по установке кри-
кристаллов (см.стр. 50).
При этом число ячеек будет боль-
больше числа сингоний (семи). Это сле-
следует хотя бы из того, что даже пло-
плоских ромбических сеток имеется две
(см. § 4). Аналогично этим двум пло-
плоским случаям надо испытывать каж-
каждую из 7 решеток на возможность
наличия в них дополнительных узлов
в центрах граней или в центрах самих
параллелепипедов.
Таким образом, наличие дополни-
дополнительных узлов в других местах ячей-
ячейки невозможно, так как их появле-
появление вызвало бы резкое изменение
симметрии решетки.
Элементарные ячейки в решетках
Бравэ выбираются так, чтобы сим-
симметрия их оставалась такой же, как и
всей решетки, число прямых углов
было бы максимальным, а объем
ячейки минимальным.
Исследовав последовательно все
сочетания 7 решеток с пятью случая-
случаями симметрии [плоских сеток, можно
получить математически однознач-
однозначный ответ о числе возможных прост-
пространственных решеток. Эту задачу,
как было сказано выше, решил
О. Бравэ в 1855 г.
Рассмотрим подробнее ромбиче-
ромбические решетки.
Комбинируя ячейку рис. 83, в с
плоскими сетками (рис. 78, б и в) и
принимая во внимание, что в трех-
трехмерной ячейке может быть еще узел
в центре ячейки, легко получим 4
ячейки Бравэ (рис. 84). Они называ-
называются: примитивная — Р, базоцентри-
рованная — С, гранецентрирован-
ная — Р и объемноцентрирован-
ная — /.
Рис. 83. Формы ячеек пространственных
решеток
о — кубической сингоний;
б — тетрагональной; д — триклннной;
в — ромбической; е — гексагональной;
г — моноклинной; ж — тригональной
•К7
7
Рис. 84. Четыре ромбические решетки
Бравэ
а — примитивная;
б — базоцентрированная;
в — гранецентрированная;
г — объемноцентрировавная
Рис. 85. Ромбическая ячейка не может
иметь центрированными две пары граней
Рис. 86. Базоцентрированная тетрагональ-
тетрагональная ячейка сводится к вдвое меньшей при-
примитивной
58
Легко показать, что не могут быть
одновременно центрированными две
пары граней (рис. 85). В этом слу-
случае мы имели бы трансляцию АВ,
при которой в силу основного свой-
свойства решетки все узлы решетки дол-
должны были бы совместиться друг с
другом. Однако в дважды центриро-
центрированной ячейке этого свойства решетки
нет, ибо при трансляции АВ узел С
ни с каким узлом не совпадает.
Следовательно, подобная ячейка
не является ячейкой какой-либо ре-
решетки.
Таким образом, четырьмя случая-
случаями (рис. 84) исчерпываются все воз-
возможные ромбические решетки.
Тетрагональных решеток только
две — Р и /. Базоцентрированная те-
тетрагональная решетка сводится к
примитивной (рис. 86). Для этого
новые оси X и Y следует выбрать под
45° к старым осям X' и У. При этом
новая ячейка Р будет вдвое меньше
старой ячейки С. Точно таким же
способом доказывается тождество
гранецентрированной тетрагональной
ячейки с объемноцентрированной
(рис. 87). Моноклинных решеток
также две, кубических — три, а в
триклшгаой, тригональной и гексаго-
гексагональной сингонии — по одной.
Все 14 решеток Бравэ изображены
на рис. 88.
Во всех сингониях, кроме гексаго-
гексагональной, ячейки Бравэ являются па-
параллелепипедами, поэтому часто тер-
термин «элементарная ячейка» употреб-
употребляется как синоним элементарного
параллелепипеда. В гексагональной
решетке также часто выбирается
прямоугольный параллелепипед
(рис. 89,6), который обозначается С
и называется о_ртогексагональной
ячейкой (с Ъ = яУЗ). В других случа-
случаях выбирается примитивный парал-
параллелепипед (рис. 89,<?) с а = Ъ и углом
Y = 120°.
Каждую ромбоэдрическую ячейку
можно заменить гексагональной, и
наоборот. Ромбоэдрическая ячейка
заменяется втрое большим паралле-
параллелепипедом гексагональной оингонии
Рис. 87. Гранецентрированная тетраго-
тетрагональная ячейка сводится к вдвое меньшей
объемноцентрированной
Рис. 88.14 решеток Бравэ
а — триклинная; моноклинные:
б — примитивная,
в — базоцентрированная; ромбические:
г — примитивная,
д — базоцентрированная,
е — гранецентрированная,
ж — объемноцентрированная; тетрагональные:
г — примитивная,
и — объемноцентрированная,
к — гексагональная,
р л — тригональная; кубические:
м — примитивная,
н — гранецентрированная,
о — объемноцентрированная (отмечены те уг-
углы, которые отличаются от 90°)
59
§6. Понятие
о кристаллохимнческош анализе
Рис. 89. Выбор ячейки в гексагональной
сингонии
а — гексагональная ячейка Бравэ;
б — ортогексагональная ячейка;
в — примитивный параллелепипед
(равным 7з объема полной гексаго-
гексагональной ячейки с а = Ь^=с и у^^О0),
имеющим два дополнительных уз-
узла на пространственной диагонали
(рис. 90, а), а гексагональная — ром-
ромбоэдрической того же объема, имею-
имеющей два дополнительных узла внутри
ячейки, расположенных на главной
оси (рис. 90, б).
На рисунке изображены три гекса-
гексагональные ячейки, расположенные
друг над другом (боковые ребра не
изображены).
Рис. 90. Связь между гексагональной
и ромбоэдрической ячейками
а — переход от ромбоэдрической ячейки к гек-
гексагональной;
б — переход от гексагональной ячейки к ром-
ромбоэдрической
В кристаллической решетке можно
выделить бесконечно большое число
плоских сеток. Через любые три уз-
узла решетки, не лежащие на одной
прямой, можно провести плоскость, и
эта плоскость (плоская сетка) будет
возможной гранью кристалла. Число
различных плоских сеток в кристал-
кристалле бесконечно велико, а число реаль-
реально существующих граней всегда
весьма ограниченно. Разные серии
сеток будут отличаться друг от друга
ретикулярной плотностью, т. е. чис-
числом узлов, приходящихся на едини-
единицу площади. Бравэ предположил, что
грани кристалла являются сетками
с наибольшей ретикулярной плотно-
плотностью. Эта гипотеза обычно известна
под названием правила, или закона
Бравэ. Однако Бравэ не предложил
способа определения типа решетки в
реальных случаях. Гипотеза продол-
продолжала оставаться лишь догадкой. Она
была в известной мере решена Е. С.
Федоровым при создании кристалло-
химического анализа. Е. С. Федоров
разработал стройную систему, по ко-
которой можно было, опираясь на гипо-
гипотезу Бравэ, определить структуру
кристалла, т. е. найти тип решетки
Бравэ у кристаллов того или ииого
вещества. Для этого прежде всего
изучалась внешняя форма кристал-
кристаллов исследуемого вещества. На осно-
основании этого изучения составлялся
список граней: вначале выписыва-
выписывались грани, встречающиеся на каж-
каждом кристалле, затем — грани, обыч-
обычно наблюдающиеся, затем — грани,
встречающиеся все реже и реже. Для
каждого типа решетки были составле-
составлены таблицы сеток, начиная от сеток
с максимальной ретикулярной плот-
плотностью и далее со все уменьшающей-
уменьшающейся плотностью. Сопоставляя список
символов граней, найденных на кри-
кристаллах определяемых веществ, со
списком теоретических плотностей,
можно сделать вывод о типе решетки
Бравэ у кристаллов конкретных ве-
веществ.
60
Это рассуждение может претендо-
претендовать на известную строгость только
для кристаллов кубической синго-
нии, так как максимальную плот-
плотность у простой кубической решетки
имеет сетка A00), у центрирован-
центрированной—A10) и гравецентрирован-
ной—A11). В кристаллах средней
и низшей категорий вопрос сильно
осложняется тем, что теоретическая
плотность сеток сильно зависит от
отношений осей и величин коорди-
координатных углов.
Для придания методу универсаль-
универсальности Федорову пришлось доказать
на огромном экспериментальном ма-
материале, что все кристаллы по своим
углам приближаются к кубическому
или гексагональному типам, что у
них можно выделить зоны, аналогич-
аналогичные призмам тригональной, тетраго-
тетрагональной или гексагональной синго-
ний, что отклонение от этих идеаль-
идеальных значений у реальных кристал-
кристаллов низших сингоний встречается
тем реже, чем сильнее само отклоне-
отклонение. Это обобщение известно под наз-
названием закона кристаллографических
пределов, который может быть сфор-
сформулирован так: все кристаллы иде-
идеальны или приближаются к идеаль-
идеальным.
Расположив все известные к тому
времени кристаллы по степени откло-
отклонения их от идеальных, Федоров по-
получил возможность определять по
этим таблицам вещество по внешней
форме его кристаллов. Метод такого
определения и получил название
кристаллохимического анализа.
Как сказано выше, в кристаллохи-
мическом анализе попутно решался
вопрос об определении структуры
кристалла (т. е. типа решетки Бра-
вэ), что, вообще говоря, является не
обязательным, если ставить себе
целью только идентификацию веще-
вещества по внешней форме его кристал-
кристаллов, поэтому во всех последующих
методах такого рода, являющихся
развитием кристаллохимического
анализа Федорова (например, метод
А. К. Болдырева или Т. В. Баркера),
эта промежуточная стадия (опреде-
(определение типа решетки) опущена. По-
Последнее обстоятельство действитель-
действительно целесообразно, так как, помимо
некоторой излишней промежуточной
работы, определение типа решетки
Бравэ в кристаллохимическом анали-
анализе лишь вероятно, а в рентгеновском
методе оно достоверно. Но так или
иначе метод Федорова, который был
разработан в 1910 г., явился первым
методом определения типа решетки
у конкретных кристаллических ве-
веществ. Определяя тип структуры
кристаллов, Федоров всегда пользо-
пользовался понятием параллелоэдра, ко-
которое лишь до известной степени
аналогично понятию решетки.
Если начать мысленно раздувать
узлы решетки, то, все время увеличи-
увеличивающиеся в объеме, они в какой-то
момент времени столкнутся, — меж-
между ними появится плоская грань, гра-
грани же в дальнейшем пересекутся в
вершинах. В результате, вместо без-
безразмерного узла решетки мы полу-
получим многогранник, тесно прилегаю-
прилегающий к аналогичным соседним,— это
и будет параллелоэдр.
Параллелоэдрами называются оди-
одинаковые выпуклые многогранники,
целиком заполняющие пространство
в параллельном положении. Теория
параллелоэдров была создана Е. С.
Федоровым в конце XIX в. Паралле-
лоэдры имеют всегда попарно равные
и параллельные грани. Грани у па-
параллелоэдров могут быть либо четы-
четырехугольными, либо шестиугольны-
шестиугольными. Существенно различаются парал-
лелоэдры числом пар граней. По это-
этому признаку можно выделить четы-
четыре основных типа параллелоэдров с
тремя, четырьмя, шестью и семью
парами параллельных граней. Назы-
Называются они соответственно трипарал-
лелоэдрами, тетрапараллелоэдрами,
гексапараллелоэдрами и гептапарал-
лелоэдрами. Наиболее симметричные
основные параллелоэдры показаны
на рис. 91. Все другие параллелоэдры
могут быть получены из наиболее
симметричных основных путем од-
61
Рис. 91. Наиболее симметричные паралле-
лоэдры
о — трипараллелоэдр;
б — тетрапараллелоэдр;
в — гексапараллелоэдр;
г — гептапараллелоэдр
Рис. 92. Заполнение пространства гекса-
гексагональными призмами (а) и соответствую-
соответствующая решетка (б)
•
•
a
•
•
Рис. 93. Заполнение пространства ром-
ромбододекаэдрами (а) и соответствующая
решетка (б)
V
Рис. 94. Заполнение пространства кубоок-
таэдрами (а) и соответствующая решет-
решетка (б)
нородной деформации, т. е. растяже-
растяжениями и сдвигами.
Параллелоэдр выражает собой:
форму и величину области простран-
пространства, приходящейся на каждый узел
решетки. Представить себе, как за-
заполняется пространство кубами,—
просто. Взяв центры тяжести таких
кубов и отбросив затем самые кубы,
мы получим простую кубическую ре-
решетку. Заполнение пространства
гексагональными призмами (тетра-
параллелоэдрами) показано на рис.
92, а. Взяв центры тяжести этих
призм, мы придем к гексагональной
решетке (рис. 92, б). Заполнение
пространства ромбическими додека-
додекаэдрами (гексапараллелоэдрами) по-
показано на рис. 93, а, ему соответ-
соответствует гранецентрированная кубиче-
кубическая решетка (рис. 93, б). Запол-
Заполнение пространства кубооктаэдрами
(гептапараллелоэдрами) показано на
рис. 94, а, ему соответствует объемно-
центрированная решетка (рис. 94,6).
Другим решеткам Бравэ будут от-
отвечать параллелоэдры того же типа,
т. е. с тремя, четырьмя, шестью и
семью парами параллельных граней,
но эти параллелоэдры будут менее
симметричны, чем только что опи-
описанные. Так, тетрагональной решет-
решетке будет отвечать параллелоэдр в
форме тетрагональной призмы. Он
может быть получен из куба путем
деформации последнего (растяжения
или сжатия) вдоль оси четвертого
порядка. Ромбоэдр же получится в
результате деформации куба по трой-
тройной оси и т. д.
Тетрагональный параллелоэдр мож-
можно превратить в ромбический путем
растяжения (сжатия) в направле-
направлении, перпендикулярном к грани
призмы. К ромбическому параллело-
эдру мы придем также и от гексаго-
гексагональной призмы, растягивая или
сжимая ее по одной из осей второго
порядка, перпендикулярной к оси
шестого порядка. От ромбического
параллелоэдра к моноклинному мож-
можно прийти в результате сдвига, что
схематически показано на рис. 95.
62
Растяжение
t
а
1
1
/
Сдвиг
71
1
Рис. 95. Превращение симметричного па-
раллелоэдра в менее симметричный по-
посредством растяжения и сдвига (а). Наи-
Наименее симметричный трипараллелоэдр (б)
Рис. 96. Схема преобразования кубическо-
кубического и гексагонального типов кристаллов в
низшие типы в результате деформаций
параллелоэдров
Для превращения моноклинного па-
раллелоэдра в триклинный необхо-
необходим второй сдвиг под некоторым уг-
углом к первому. В результате всех
этих сдвигов и растяжений мы смо-
сможем перейти от куба к триклинному
косоугольному параллелепипеду
(рис. 95, б).
Из сказанного ясно, что все решет-
решетки тетрагональной, тригональной,
ромбической, моноклинной и три-
клинной сингоний являются подчи-
подчиненными четырем наиболее симмет-
симметричным параллелоэдрам. Из этих
четырех параллелоэдров один (гек-
(гексагональная призма) относится к
гексагональной сингоний, а осталь-
остальные три — к кубической.
Соответственно этому все кристал-
кристаллы, по Федорову, делятся на два ти-
типа: кубический и гексагональный.
Кубический тип имеет три структу-
структуры, каждая из которых может встре-
встретиться у кристаллов, деформирован-
деформированных по оси третьего или четвертого
порядка (тригональных или тетра-
тетрагональных). Таким образом, кубиче-
кубический тип кристаллов делится на
шесть групп. Из этих групп, а также
из гексагонального типа, в результа-
результате дальнейших деформаций выво-
выводятся решетки низших сингоний (см.
схему рис. 96).
ГЛАВА VII
ТЕОРИЯ СТРУКТУРЫ КРИСТАЛЛОВ В. С. ФЕДОРОВА
§1. Краткие сведения о теории
Законы симметрии, о которых гово-
говорилось выше, распространяются не
только на внешнюю форму кристал-
кристаллов. Им подчинено и их внутреннее,
атомное, строение. Внешняя форма
является только следствием внутрен-
внутреннего строения кристалла.
В 1890 г., задолго до первых опре-
определений структур кристаллов, Е. С.
Федоровым были выведены строго
математическим путем все возможные
сочетания элементов симметрии в
пространстве. Е. С. Федоров и А.
Шенфлис доказали, что таких про-
пространственных групп симметрии мо-
:жет быть только 230. Этот вывод стал
впоследствии незыблемой основой
современной кристаллохимии — тео-
теорией атомной структуры кристаллов.
Изучение внутренней симметрии
кристалла сложнее изучения внешней
симметрии, так как значительно уве-
увеличивается разнообразие элементов
симметрии и, кроме того, в атомных
структурах приходится считаться с
бесконечным числом тождественных
элементов симметрии: параллельно
каждой плоскости или оси симметрии
имеется бесконечное количество пло-
плоскостей и осей, а соответственно, и
центров симметрии.
Последнее обстоятельство можно
понять из рассмотрения простейшей
атомной модели тетрагонального кри-
кристалла, спроектированной вдоль оси
четвертого порядка (рис. 97, а). Назо-
Назовем прямую, проходящую через се-
серию атомов, атомным рядом. В каж-
каждом атомном слое, образованном се-
серией атомных рядов, располагаются
атомы, связанные по четыре осью
симметрии четвертого порядка. Все
атомы одинаковы, и, следовательно,
мы можем считать, что через каж-
каждый из них перпендикулярно плос-
плоскости чертежа проходит ось четвер-
четвертого порядка. А это будет верно в
том случае, если мы не будем счи-
считаться с внешним ограничением кри-
кристалла и предположим, что кристалл
простирается до бесконечности. От-
Отсюда следует, что построение теории
обязательно для бесконечно большо-
большого кристалла или, как говорят, для
кристаллического пространства.
Многообразие элементов симмет-
симметрии, с которым необходимо считаться
при изучении внутренней структуры
кристаллов, возрастает. Кроме тех
элементов симметрии, которые харак-
характеризуют внешнюю форму кристал-
кристаллов, здесь появляются новые. Важней-
Важнейшим из них является трансляция
(т. е. параллельный перенос), о кото-
которой мы подробно говорили выше
(стр. 53).
Перенос как составная часть может
входить в другие элементы симмет-
симметрии. В структуре, кроме обычных
плоскостей симметрии, имеются так
называемые плоскости скользящего
отражения. На рис. 97, б первые обо-
обозначены сплошными линиями, а вто-
вторые — пунктирными. Чтобы совмес-
совместить атом 1 с атомом 2 (рис. 98), до-
достаточно атом 1 отразить в плоскости
(плоскость тп); а для того, чтобы сов-
совместить его с атомом 3 при помощи
плоскости скользящего отражения
а, необходимо не только отразить его
64
в этой плоскости, но и перенести па-
параллельно ей на половину трансля-
трансляции. Обозначение плоскостей сколь-
скользящего отображения а, Ъ или с соот-
соответствует переносу вдоль оси X, Y
или Z. Поворотные оси симметрии в
структуре могут остаться поворотны-
поворотными или же превратиться в винтовые,
включающие в себя перенос. На
рис. 99 изображены двойная поворот-
поворотная ось и двойная винтовая. В верх-
верхней части рисунка оси лежат в пло-
плоскости чертежа, в нижней — перпен-
перпендикулярно к ней*. Все элементы
симметрии, включающие в себя пере-
перенос, обычно называются элементами
симметричности.
Поскольку число элементов сим-
симметрии, характеризующих внутрен-
внутреннюю симметрию кристаллов, больше,
чем число элементов симметрии внеш-
внешней формы, то количество сочетаний
из них также значительно больше 32,
а именно 230.
Чтобы понять, каким образом они
получаются, можно обратиться к од-
одному из простейших видов симмет-
симметрии, скажем, ромбо-пирамидальному
Ь%2Р. Можно думать, что в этом ви-
виде симметрии могут существовать
только три производные федоровские
группы:
1) с двумя зеркальными плоскостя-
плоскостями симметрии — mm;
2) с одной зеркальной и второй
скользящей — та;
3) с двумя скользящими — ab.
Однако это не совсем так. Разбе-
Разберем только второй случай, когда одна
из плоскостей будет плоскостью
скользящего отражения. Оказывает-
Оказывается, что группа та лишь одна из воз-
возможных групп. Картина усложняет-
усложняется, если принять во внимание на-
направление скольжения. На рис. 100
изображены три ячейки, каждая из
которых ограничена плоскостью зер-
зеркального отражения и плоскостью
* Индекс '/г около второй точки внизу
указывает расстояние ее по высоте от
плоскости чертежа; Т — величина транс-
трансляции, параллельной оси симметрии.
5 Кристаллохимия
о
О
а
о о
о о
о о
о о о
о о
о о
о о
Рис. 97. Структура тетрагонального р^
сталла, спроектированная вдоль главной
оси
/ I
о—Г
I
i *
i
Рис. 98. Зеркальная плоскость симметрии
и плоскость скользящего отражения
0
0
С
1
I
I
aI
'r
2
2
1 2
О о
о of/2
/
о
0
4
1
2
0
0
0 4» 0
Рис. 99. Двойная поворотная и двойная
винтовая оси
1
тп
Рис. 100. Возникновение трех различных
пространственных групп в зависимости
от направления скольжения плоскостей
симметрии
65
4
в
в
1/2 »
1/2»
1/2»
1/2
; »
»
в
1/2»
1/2»
1/2»
»
в
»
1/2»
1/2»
! в
! »
»
1/2 »
1/2»
I «
|
!»
•
б
0
в
I в
1 <§ь
в
в
в
в
в
в
в
скользящего отражения. Для просто-
простоты не будем изображать производных
элементов симметрии, в частности,
осей симметрии. На первом рисунке
направление скольжения параллель-
параллельно плоскости чертежа, на втором —
перпендикулярно, в последнем — по
диагонали прямоугольника, постро-
построенного на предыдущих направлениях
скольжения. Как видно, в одном
разобранном нами случае будет не
одна, а три федоровские группы.
Если мы зададим какую-либо точку
внутри ячейки (рис. 101, а), то, «раз-
«размножая» ее (т. е. получая производ-
производные точки) при помощи элементов
симметрии, получим правильную си-
систему точек. Обращает на себя вни-
внимание то обстоятельство, что во всех
трех случаях правильные системы
точек будут различными, т. е. в про-
пространстве они располагаются по-раз-
по-разному, хотя исходная точка во всех
случаях выбрана одинаково — в ле-
левом нижнем углу ячейки.
Эти правильные системы точек и
соответствуют различным случаям
закона расположения атомов в кри-
кристаллах.
В одной и той же федоровской
группе симметрии может быть не-
несколько вариантов расположения то-
точек в зависимости от положения ис-
исходной точки по отношению к эле-
элементам симметрии. Так же различно
Рис. 101. Различные правильные системы
точек
может быть и число точек, приходя-
приходящихся на одну ячейку. Это число на-
называется кратностью правильной си-
системы точек. На рис. 101, б, соответ-
соответствующем группе та, двойным круж-
кружком изображена новая исходная точ-
точка — 2. Расположение точек этой си-
системы иное, чем в системе 1, и число
их в два раза меньше. Это — новая
правильная система точек, характер-
характерная для той же федоровской группы.
По этой системе также могут распо-
располагаться атомы в кристаллическом
пространстве. Точки могут быть рас-
расположены на элементах симметрии
(частное положение) и вне их (об-
(общее положение). Положение точек
на элементах симметричности со
скольжением (на винтовых осях и
плоскостях скользящего отражения)
является общим*.
Представим себе химическое со-
соединение типа АХг, которое кри-
кристаллизуется в федоровской группе,
изображенной на рис. 101, б. В
большинстве случаев атомы элемен-
элемента А будут располагаться в систе-
системе 2, а атомы элемента X — в систе-
системе 1. Подробный элементарный вы-
вывод всех федоровских групп симмет-
симметрии для одного вида симметрии дай
в книге Г. Б. Бокия и М. Н. Порай-
Кошица «Практический курс рент-
* См. сноску на стр. 65.
66
геноструктурного анализа» A951г.).
Краткий вывод всех групп сделан
акад. Н. В. Беловым в работе «Клас-
«Классный метод вывода пространствен-
пространственных групп симметрии» (Труды Ин-
Института кристаллографии, № 6,
1951г.).
На рис. 102—104 собраны услов-
условные обозначения всех элементов
симметричности, необходимые для
изображения 230 федоровских групп.
Сами эти группы показаны иа рис.
105—135. Ради экономии места на
каждом чертеже группы изображена
не вся элементарная ячейка, а только
ее минимальная независимая область
(обычно 'Д ячейки). Для работы с
группами по этим таблицам читатель
должен нарисовать для себя на от-
отдельном листе бумаги интересующую
его группу целиком.
Современные хорошо разработан-
разработанные на основе этой теории методы
эксперимента позволяют надежно
определять внутреннее строение
твердых тел — с большой степенью
точности определять координаты
атомов и измерять, следовательно,
расстояния между ними.
В настоящее время надежно опре-
определено около 20 000 структур кри-
кристаллов. Среди этого множества
структур нет ни одной, которая про-
противоречила бы теории Федорова.
Величие работы Федорова в том
и заключается, что он на многие годы
вперед опередил современную ему
науку и предсказал все возможные
случаи расположения атомов в про-
пространстве, в то время как A890 г.)
многие вообще считали недоказанным
реальное существование самих ато-
атомов. Заканчивая краткое изложение
этой замечательной работы, хочется
подчеркнуть, что она имеет не толь-
только познавательное, теоретическое
значение. Достаточно вспомнить,
что почти все свойства твердых тел
зависят от двух причин: от их хими-
химического состава и от кристаллическо-
кристаллического строения. Мы часто не знаем еще
форм этой зависимости, но у нас нет
сомнений, что она существует.
t 1
f-
о ¦
ф h
Рис. 102. Условные обозначения осей сим-
симметричности, расположенных перпендику-
перпендикулярно и параллельно плоскости чертежа
. m \m
а,Ь
п
—¦-. d
Рис. 103. Условные обозначения плоско-
плоскостей симметричности, расположенных пер-
перпендикулярно и параллельно плоскости
чертежа
W-
n.d
ЧЗт
Рис. 104. Условные обозначения элемен-
элементов симметричности, расположенных косо
по отношению к плоскости чертежа
5*
67
Если бы не было теории Федорова,
то не было бы и надежды когда-либо
научиться создавать твердые тела с
заранее заданными, нужными для
промышленности свойствами. Твер-
Твердой основой для открытия новых за-
законов, связывающих свойства веще-
вещества с их составом и строением, явля-
являются периодический закон химиче-
химических элементов и геометрическая
теория структуры кристаллов.
§2. Федоровские
группы симметрии
Существует несколько способов обо-
обозначения пространственных групп
симметрии: по Федорову, по Шёнф-
лису, современный международный
символ и др.
Международный символ включает
тип решетки Бравэ и те элементы
симметрии, которых достаточно,
чтобы представить по символу всю
пространственную группу симмет-
симметрии.
Во всех сингониях на первом месте
обязательно стоит большая буква, по-
показывающая тип решетки Бравэ:
Р — примитивная, F — гранецентри-
рованная, / — объемноцентрирован-
ная, А, В, С — базоцентрированная
в соответствующей ориентировке.
При обозначении элементов симмет-
симметрии предпочтение отдается плоско-
плоскостям, и только в случае их отсутст-
отсутствия вводится символ оси.
Символы низшей, средней и выс-
высшей категорий следует рассмотреть
отдельно.
Низшая категория. В триклинной
и моноклинной сингониях с одним
элементом симметрии стоит обозначе-
обозначение его после символа решетки: Pi,
С2, Ртп. В моноклинной сингонии в
виде симметрии с центром инверсии,
образованным при пересечении оси
с перпендикулярной к ней плоско-
плоскостью, после символа решетки стоит
наименование оси, а затем — перпен-
перпендикулярной к ней плоскости. Пер-
Перпендикулярность выражена наклон-
наклонной чертой, например, P2\lm. В ром-
ромбической сингонии отражены имею-
имеющиеся элементы симметрии вдоль
координатных направлений. При
этом на первом месте стоит обозна-
обозначение оси вдоль направления X или
перпендикулярной к X плоскости, на
втором — оси вдоль Y и т. д. На-
Например, Pmml, Abm2, Cmcm, P222i.
Обозначения пространственных
групп приведены в соответствии со
вторым изданием Международных
таблиц A952 г.). В (старых изданиях
не вводилось наименование оси
вдоль третьего координатного на-
направления в символе ромбо-пирами-
дального вида симметрии, так что
первые два примера обозначались
как Pmm и АЪтп.
Средняя категория. Для различ-
различных сингонии средней категории
характерны оси высшего порядка.
Поэтому сразу же после символа
решетки стоит обозначение главной
оси. Если перпендикулярно главной
оси проходит плоскость симметрич-
симметричности, то наименование ее вводится
в символ непосредственно за осью
и отделяется наклонной чертой.
В тетрагональной сингонии сле-
следующая за ними буква показывает
тип плоскости, параллельной сторо-
стороне элементарного квадрата (или
оси), а последняя буква — диагонали
квадрата: например, Mi/acd, PA2m,
Micd (в последних двух примерах
нет перпендикулярной плоскости).
Так, символ /4j/acd показывает, что
в обьемноцентрированной тетраго-
тетрагональной ячейке перпендикулярно
главной оси располагается плоскость
симметричности типа а, параллельно
главной оси и стороне элементарного
квадрата — плоскость типа с, а по
диагонали квадрата — типа d.
В старых изданиях Международных
таблиц, кроме обычных установок Р
и /, пользуются соответственно уста-
установками С и F. Оси ячейки в послед-
последних повернуты на 45° по отношению
к осям обычной установки. В связи
с этим порядок букв в символах уста-
установок С и F обратный, например:
68
14/тгас/ге — FA/mmc и P42tc = C4c2i.
Установки С и F обычно употребля-
употребляются в трех группах тетрагонально-
скаленоэдрического вида симметрии,
в которых параллельно кратчайшей
трансляции проходят не оси 2, а пло-
плоскости, например P4m2 = C42m.
В гексагональной сингонии поря-
порядок букв остается таким же, как в
тетрагональной. На первом месте сто-
стоит буква, символизирующая пло-
плоскость симметричности, перпендику-
перпендикулярную главной оси (если таковая
плоскость в группе есть). Затем сто-
стоит буква, характеризующая пло-
плоскость, проходящую по длинной диа-
диагонали ромба, и на последнем ме-
месте — по короткой диагонали ромба.
Старое и новое написания символа
пространственных групп тригональ-
ной и гексагональной сингонии отли-
отличаются друг от друга. В старых изда-
изданиях Международных таблиц, на-
например, не всегда обозначены винто-
винтовые оси. Учтен только порядок глав-
главной оси F). Кроме того, в символе
тригональных и гексагональных
групп в новом написании обязатель-
обязательно ставятся три знака элементов сим-
симметричности вдоль трех главных на-
направлений. В старом написании ог-
ограничивались в некоторых группах
двумя. Поэтому, например, группа
Р6422 в старых изданиях обознача-
обозначалась как С6^2. Сказанное выше отно-
относится и к тетрагональной сингонии.
Если вдоль одного из направлений
элемент симметричности отсутствует,
то в группах тригональной сингонии
ставится 1, например Р3с1, Р31т.
В старых изданиях, кроме установ-
установки С, в тригональной и гексагональ-
гексагональной сингониях употреблялась уста-
установка Н, оси в которой (ребра ячей-
ячейки) расположены под углом в 30° к
осям обычной установки. В установке
Н на первом месте оказывается, та-
таким образом, буква, символизирую-
символизирующая тип плоскости симметричности,
проходящей по короткой диагонали
ромба, например CQ/mmc=HQ/mcm.
Установка Н обычно употреблялась в
случае отсутствия плоскости симмет-
симметричности, проходящей по длинной
диагонали ромба, например С31т —
=НЪт.
В последнем издании установка Н
исключена. Отсутствующий элемент
симметричности заменяется единицей
(как написано выше). Тип решетки
в этом случае только один — прими-
примитивный, поэтому во всех группах
тригональной и гексагональной син-
сингонии символ решетки Р.
Необходимо отметить, что для осей
порядок написания букв обратный.
Так, в группе С32 ось 2 проходит по
короткой диагонали ромба.
Буквой R в группах тригональной
сингонии обозначается ромбоэдриче-
ромбоэдрическая решетка.
Высшая категория. В кубической
сингонии цифра 3 символизирует
наклонные оси третьего порядка.
Цифры или буквы, стоящие перед
цифрой 3, определяют оси или пло-
плоскости, параллельные грани куба
элементарного параллелепипеда.
Знак, стоящий после цифры 3, указы-
указывает на плоскость (ось) симметрич-
симметричности, параллельную диагональной
трансляции (т. е. параллельную гра-
грани ромбододекаэдра). Если послед-
последняя отсутствует, то место за циф-
цифрой 3 остается пустым. Примеры:
Р32, Um, P432.
В табл. 7 собраны все 230 федо-
федоровских (пространственных) групп
симметрии. После порядкового номе-
номера во втором столбце дается символ
группы по Федорову, затем по Шён-
флису и, наконец, международный
символ. Если старое и новое написа-
написания символа отличаются, в скобках
дан символ в старом обозначении.
В третьем столбце цифрами ука-
указаны кратности правильных систем
точек и координаты одной (исход-
(исходной) точки.
На рисунках 105—135 изображе-
изображены все 230 пространственных групп
симметрии.
69
ТАБЛИЦА 7
Пространственные группы симметрии
1
2
3
4
5
6
7
8
9
ТРИКЛИННАЯ СИНГОНИЯ
Моноэдрический вид симметрии
Is
с\
Pi
1: (a) 000
ГГииакоидальвый вид симметрии
2s
с\
рТ
1: (a) 000 F) 00 2(c)°2°
wJooWJ|o(/)|oJ
wo44(*)ttt
МОНОКЛИННАЯ СИНГОНИЯ
Диэдрическии осевой вид симметрии
3s .11
рг
la
Cl
P2i
4s
сг
л л
1 1
2: (e) xyz
2: (a) xyz
1
2 : (a) 0t/0 F) Oy у
4:(e)*»*}+(oOO;||o)
Диэдрическии безосный вид симметрии
5s ...... 1
Pm
\h
PC
6s
c\
Cm
th
c\
Co
l:(a) i0iF)iyz
2: (с) жуг
2: (a) xyz
2: (a) xOz \ r 1 1 \
4: F)жг/г J + @00;"T°y
/ И 1
i:(a)xyz+ @00; yyOl
]
10
11
12
13
14
Триаматический вид симметрии
7s
c\h
P2/m
Zh
C\h
рг\с
la
P2itm
3a
8s
c\h.
C2/m
1 1
1: (aH00FH^ 0(c) 00 ^
Id)- 00 (e) -~0(/H --
2 22-22
«4°4<*> tii
2:(iHt/0(/-)Tt/0(A;Ht/-
11 1
i:@)xyz
11 1
2 : (a) 000 F) у у 0 (c) 0 у 0
1 111
(d) у 00 (e) 0 у -g (/) 2 2/ 4
4 : (g) жуг
1 1
2 : (a) 000 F) у 00 (с) 00 у
11 1
4: (/) *y*
1 1
2 : (a) 000 (Ь) у 00 (с) 00 у
1 1
4: (е) zyz
2 : (a) 000 F) 0 у 0
1 111
(сH0у(<*H 2 2
1 1 111
4: (е) 4Т° ^^44 2
(g) Ot/0 (ft) 0t/ у
@ хОг
+
8 : (/) «2/г J
+ @00; 44"°
Tl 2 г
70
ТАБЛИЦА 7 (продолжение)
15
ih
С2/с
4: (a) 000FH у 0
(e) °3/ -4
8: (/) xyz
4-
000; 4 4 '
16
РОМБИЧЕСКАЯ СИНГОНИЯ
Ромбо-пирамидальный вид симметрии
1 1
17
18
19
20
21
13s
(Ртт)
Ртт2
1 : (aH0z(&Hyz(c) у02
(d) -5--5-;
5А
(Рсс)
Рес2
6А
С%о
(Рта)
Рта2
lh
(Aw)
PneZ
8А
(Am)
(A&)
jPSa2
4:
1:
4:
2
4-
2
4
2
4
2
4
(t) xyz
(a) OOz (&) 0 у z (с) у
1
1
(j) ZJ/Z
(a) OOz F) 0 у z (с) -у
(d) ZJ/Z
(a) OOz (Ь) у Oz
(c) xyz
(aH0z(&Hy z
: (c) zi/z
: (a) OOz (b) 0 у z
: (c) zi/z
22
23
24
25
26
27
28
29
9a
(Pmc)
Pmc2i
10a
(Pmn)
PmnZi
Ha
(Pea)
PeaZi
12a
C9
(Pna)
PnaZi
14s
(Cmm)
Cmm'l
10A
(CcJ)
Ccc2
13a
(Cmc)
CmeZx
15s
(Amm)
AmtnZ
2:(aHj/z(b)yy*
4 : (c) xj/z
2: (a) Oj/z
4: (b) xyz
4: (a) xyz
4 : (a) zj/z
2 : (a) OOz (b) 0 у z
1 1
(e) Oj/z I
8 : (/) sj/z j
+ (ooo;44<
4 : (a) OOz (b) 0 у z
J..L
8 : (d) гуг
4-
+ 000;yy(
1 ^
2 : (a) OOz F) у Oz
4 : (c) zOz (d) Oyz ^ +
(e)Y»«
8 : (/) xyz J
000; О55
71
ТАБЛИЦА 7 (продолжение)
30
31
32
33
34
35
36
37
HA
{АЪт)
Ahml
12ft
2c
AtnaZ
13ft
(Aba)
17s
(Fmm)
Fmm2
16ft
c19
(Fdd)
Fdd2
16s
СЦ
(Imm)
Imm2
14ft
cfv
(Ima)
Ima2
15ft
си
(Iba)
IbaZ
4 : (a) OOz (b) у Oz
1
8 : (d) xyz
1 1 1 \
+ @00;0yy)
4 : (a) OOz (&) ¦?- yz I _|_
8 : (c) sj/z ^
+ (OOO; 0 2 2)
4:<«H0«l / J_JA
8 : (&) xyz J + ^°00' ° 2 2 J
4: (a) OOz
1 1
(d) xOz
lQ:(e)xyz
+ (OOO; 0 --; - 0s; -5O]
J
8 : (a) OOz \
16 : (b) zt/z J +
/ 111111 A
{ 22
2:(aH0z(bHyz |
4 : (c) zOz (d) Oj/z 1 "t"
8 : (e) xyz J
( 111^
+ (о°о;ут2;
4:(aH0z(&)jj/z 1
8 : (c) xyz j
/ 111^
+{ • 2 2YJ
4 : (a) OOz (b) 0 у z 1 +
8 : (c) zj/z J
38
39
40
41
42
43
Ромбо-тетраэдрический вид симметрии
9s
P222
4a
P222!
7a
D\
J?2i2i2
8a
D\
P212l2l
10s
C222
5a
^2
C222i
1 : (a) 000 (&) g 00 @H^0
, . 1 11 11
\Q>J \J\J ty \6\ IT сТ" \J) О ^
(гH±±(Д)±±±
1 1
1 1 1
1 11
@) у j/0 (p) у У у (?) 00 z
1 111
(г) у Oz (s) 0 2 z (t) у yz
4 : (и) xyz
2:(a)z00(&)a:y 0 (c) 0t/-J
1 1
(fl) 2 у &
4: (e) xyz
2 : (aH0z(&Hys
4: (c) zyz
4 : (a) xyz
1 1
2 : (a) 000 (b) 0 у 0
(с) у 0 g (d) 00 j
1
4 : (e) zOO (/) zO ^
1
(g)OyO(h)Oy у
@00z(/Hyz
1 1
( ) 4 4 z
¦ 4-
1
8 : (/) zt/z J
4 : (a) zOO (&) Oj/ T 1 4.
+ foOO;
TT°J
72
ТАБЛИЦА 7 (продолжение)
44
45
46
F
47
12s
J?222
11s
Dl
J222
6a
D\
омбо-дипв
18s
Pmmm
4 : (a) 000 (b) 00 у
,111,, 113
(*) 444^444
8 : (e) zOO (/) OyO , ,
А Л '
± i
(&j \J\JZ (>^l ~~7—7~ 2
li.ii
16 : (k) xyz )
+ [оОО;Оуу;|оУ|о)
2 : (a) 000 (b) -к- 00 1
\ / ' dt
1 1
(cH0y(dHy0
1
4 : (e) zOO (/) xO у
1
(g) OyO (ft) у yO
(tH0z(/Hyz
¦ +
8 : (k) xyz J
/ 1 1 1 \
+ [ °' 2 2 г)
1 1 ^
{cH\z
8 : (d) xyz
+
in
рамидальный вид симметрии
1 1
l:(aH00 (Ь) —00 (с) 00 у
11 1 11
11 111
(?) 0 J у (л) " " Т
1 1
2 : (/) г00 (/) z0 — (к) х у 0
11 " 1
1 11
(o)yy0(p)yyy(?H0z
А 1 11
(О 2ZW 2 ZW2 2Z
4 : (и) Oyz (v) y yz (w) xOz
Л Л
1 1
(z) z у z (y) zyO (z) xy у
8 : (a) xyz
48
49
50
51
52
17ft
Реет
18ft
D\h
Phan
19ft
Dlh
Pnnn
14a
D5
Pmma
15a
Pmna
11 1
2 : (a) 000 (Ь)у у 0 (с) 0 у 0"
(d) у 00 (e) 00 -g (/) у 0 ^
11111
1.11 1
4: (t)zO-^-(/) x-g-^ (k)Oy^
11 11
1 1
(oHy z(p) yOz(g) zyO'
8 : (r) xyz
1 1 1
2 : (a) 000 (b) у 00 (с) у 0у
(d) 00 у
1 1 „ 111
4:(e) TT0(/L42^)a:0(>
1 Л Л 1
(ft) zO у (i) 0y0 (/) Oy у
1
(/cH0z(ZHyz
8 : (m) zyz
2 : (a) 000 (Ь) у 00 (с) 00 y
1
111 333
1
(/)yy0(ftH0z(ZHy z
8: (m) xyz
1 1
2:(aH00(&Hy0(cH0y
n 1 1 in ii
№°2 2 W 4 Oz (/) 4 2 г
4 : (g) OyO (ft) Oy у (t) zOz
1 1
8: (I) xyz
1 11
2 : (a) 000 (&) у 00 (с) у у О
(dHy0
1 1 j_
(ft) Oyz
8 : (i) zyz
73
ТАБЛИЦА 7 (продолжение)
53
54
55
56
57
58
59
60
16a
n8
Pcea
17a
n8
Pnna
22a
РЪат
23a
7I1
Pbem
24a
Pmmn
25a
Л12
Pnnm
26a
*K
Pben
27a
rI0
Peon
4:(aH00FHy О
1 11
(<*)-4-°2(е)ттг
8 : (/) xyz
4 : (a) 000 (&) 00 у (с) -j Oz
8: (e) xyz
2 : (a) 000 (&) 00 у (с) 0 у 0
1 1
4:(eH0z(/Hyz(g)a:y0
(А) *У у
8 : (.) zyz
4: (a) 000 (b) yOO(c)a:-|-O
8 : (e) xyz
2 : (a) OOz (b) 0 у z
4:
11. HI
8 : (g) zi/z
1 1
2 : (a) 00 (&) 00 y(c) 0 у 0
4 : (в) 00г (/) 0 -j ж (g) zyO
8: (A) xyz
4:(aH00(&Hy0(CHt/-|-
8 : (d) xyz
4: (a) 000 FH0 y
8: (e) xyz
1 1
61
62
63
64
28a
*й
Pnma
29a
n15
Pbca
19s
Cmmm
20h
Cccm
4: (a) 000FH0 у (e)Jj!
8 : (d) xyz
4 : (a) 000 F) 00 y
8: (c) zj/z
2 : (a) 000 (Ь) у 00
11 1
(C)y0y(dH0y
11
111
(g) аЮО (/г) ж0 у
(iHj/0(/Hyy
(ft) OOz (Z) 0 у z
8:(™)xTz(re)°
(o) a;Oz (p) xyO
+
16 : (r) ij/z
:(aH0^FHy^ }
(c) 000 (d) 0 у 0
.. 1 1 ... 1 3
(i) OOz (/) 0 у z
16 : (m) zj/z
74
ТАБЛИЦА 7 (продолжение)
65
66
67
68
21A
Cmma
22h
22
Сева
18a
Cmcm
19a
Cmca
4: (a) -J-OO(b) X0"!
(c) 000 (d) 00 у
11 111
(gH|z
1 \ +
8 : (A) xOO (i) xO у
1 11
(/) 4 У° («) 4 3/ 2
1
(Z)T0z(mHi/z
(re)xTz
16: (o) xyz
ЛЛЛ ,!.ч ЛЛ 1 1
4 : (a) 000 (&) 00 у 1
11 11
16) XUU ( / J Ut/U
1 'i
(g) 00z (/г) 4 4 z
16 : (t) хуг
¦ +
1 ^
+ { ' 2 2 "У
4 : (a) 000 (&) 0 у 0 1
1
1 1
(/) Oj/z (g) xj/ -j
16 : (/г) xyz )
+ ^ ' 2 2 J
4:(aH00F)y00 1
1 1
&:(c)TT0(d)x0O
1 1
¦ +
16 : (g) xyz J
/ i i ^
+ 000; у
69
70
71
21s
Fmmm
24/г
?J4
Fddd
20s
Ал
4 : (a) 000 F) 00 у
л1 1 ! a 1
8: (cH4~ W4 °~
11 111
(g) xOO (A) 0y0
(i) OOz
.11 11
16-0) 4 4 г (A) 4 У1
1 1
(Z) x -г- " (m) Otfz
(ra) xOz @) xj/O
32 : (j,) xyz
( 11 1 1
+ 1000; О22; 2°2;
8 : (a) 000 (b) 00 у
111 5 5 5
(е)г00(/H!/0
(g) OOz
32 : (A) xyz
1 11_ 1 1_ 1
1 1 1
2 : (a) 000 (&) 0 у у
11 11
(c)yy0(d)y0^
1
4:(e)x00(/)o;-0
(g)OyO(A)Oj/y
(iH0z(/)y0z
111
(m) xOz (n) xj/O
16 : @) xyz
/ 1
+
Г2°)
i
1 Л
г J
.+
)
1 1'
+(ooo;yyyy
75
ТАБЛИЦА 7 (продолжение)
72
73
74
23ft
ГJ6
Ibam
20а
Imma
21а
4:(«H0f (bJ-gO-j-
(c) OOO (d) у 00
„..111 .1
(g)O»-j(A)OOx
@0yz(/)zj/0
16 :
@004
тт
4 : (а) 000 (b) 00 у
111 ИЗ
<с> 444^444
8:(/)zOO (g)TyT
(hHyz(i)xTz
16 : (/) xyz
111
8:(аH00(&)ТТТ
(с) хО -j(d)Ty0
16 : (/) xyz
Т z
[_
j
1 1 1
ТЕТРАГОНАЛЬНАЯ СИНГОНИЯ
Тетрагонально-пирамидальный вид
симметрии
1 1
1 : (а) ООг (&) у у z
1
2 : (с) 0 у г
4: (d) xyz
4: (а) xyz
75
76
22s
о\
Pk
30а
о\
P4i
77
78
79
80
31а
о\
JflZ
33а
о\
Р4г
23s
01
14
32а
01
Х4х
4: (а) xyz
11
Дитетрагонально-пирамидальный вид
симметрии
1: (a) 00z (&) у у z
81
82
83
84
85
24s
/>i
Civ
Pimm
25ft
/->5
Civ
Pice
26ft
Л2
С4Г
Pibm
27ft
а
J»4wc
36а
(P4mc)
jP4ii»lC
2 : (с) у 0г
4:
8 :
(e) a;0z (/)zys
2: (а) ООг (&)ууг
4: (cHy z
8: (d) xyz
2 : (a) OOz (Ь) у 0г
4: (с) г, у + х, г
8 : (d) xyz
2 : (a) OOz
4:(&Hyz
8 : (с) xyz
2 : (a) 00z (&) у у z (с) 0 у г
4 :
8 : (/) zyz
е) г у
76
ТАБЛИЦА 7 (продолжение)
86
87
88
89
¦90
¦91
-92
-93
37a
(P4cm)
38a
/i4
CiV
39a
4»
(P4bc)
РЬгЪс
25.9
Clv
I4mm
jfeL
34a
C«
(/4/nd)
J4imd
35a
C4r
(J4ed)
Hied
1 1
2 : (a) OOz (Ь) у у
1
8: (e) xyz
2: (a) OOz
1
4 : (&) 0 у z (с) ш
8: (d) xyz
4 : (a) OOz (&) 0 у z
8: (c) xyz
2 : (a) OOz
1
4 : (&) 0 у z
8 : (c) zzz (d) zOz
16 : (e) xyz
r
+ 000;
4 : (a) OOz (&) у 0z
8:(c) г, у+ г,
16 : (rf) xyz
/AAA.
v
8 : (b) Oj/z i _f. j 000
16 : (c) zi/z j ^
8 : (a) OOz 1 /АЛА
16:l&;^} + @00
г
+
1 1 i\
TTYJ
z
+
1.1. L)
ill)
t) t) П У
Zi ^r " /
1 1 1"\
; 2 2 2j
Тетрагонально-тетраэдрическии вид
симметрии
26s
Pi
1: (a) 000 (&) 00 у
1 1 JL
2:(eH0z(/)yy,
4: (/г) xyz
(o)yy0
-(gHyz
94
Г
(
95
96
97
98
27s
II
?етрагона.т
.имметрии
28s
Cift
РЦт
гт
c\h
РЦп
41a
c2ih
Ptym
42a
ptn
2 : (a) 000 (b) 00 y
w4iw4r
4:(eH0z(/H yz
8: (g) xyz
• +
( . l 'l l\
1ьно-дипирамидальный вид
1 11
1 : (aH00(&H0y (c)yy0
111
2:(eHy0(/Hyy(gH0z
(AL-y*
1 . 1
8: (I) xyz
2 : (a) 000 (&) 00 у (с) 0 у z
11 111
4= (d)-^0 («L 4 2(/H0z
8: (g) zi/z
11 1
2: (a) 000 (&)y — 0(cHy0
11 1 HI
(dH^2 (eH0 4(/) 224
11 1
4 : (g) OOz (/г)у у z (j) 0 у z
(/) ^3/0
8 : (k) xyz
2 : (a) 000 (&) 00 у
111 113
8 : (g) zj/z
77
ТАБЛИЦА 7 (продолжение)
99
100
29s
C\h
ХЦт
40а
С%-
X4i/a
2: (а) 000 F) 00 у
1 11
4: (cHy 0(dHy-?-
(e) OOz
111 1
¦ (/) 4 4 4 W 2Z
(/г) zj/0
16 : (t) zj/z
+
/ 1 1 1A
+ @00;yyyJ
4: (a) 000(&H0 у
11 15
8 : (c) ° T 8 W T 8
(e) OOz
16: (/) xyz
л \
Тетрагонально-трапецоэдрический вид
симметрии
101
102
103
30s
(Р42)
Р422
43a
(P42X)
Р42Х2
44а
D\
(¦P4i2)
1: (a) 000 (b) 00 у (с) у у 0
111
2: (e)y00(/)—0y (gH0z
1 1
1 1
1 1 „1
(I) xOO (m)x у у (га) zO ^
(о) г тт 0
* ' 2
8: (р) xyz
1 1
2 : (а) 000 F) 00 у (с) 0 у г
4 : (d) OOz (e) zzO (/) хх у
8 : (g) xyz
1 3
4 : (а) 0z0 F) у хО (с) zz -g-
8: (d) xyz
10
10
106
107
108
109
45а
Dl
(Р4з2)
jP4322
47а
(РЫ)
48а
(P4i2i)
49а
jD8
(P4»2i)
50а
31s
(J424)
J422
4 : (а) 0z0 (Ь) у z0 (с) xi
8: (d) zj/г
1 1
2: (аH00(Ь)уу0(с)
11 1
1 1
1 1
1 1
(те) z ^ 0 (га) хх -^ (о)
8: (р) xyz
4: (а) zzO
8 : F) xyz
4: (а) ххО
8: F) zj/z
2 : (а) 000 F) 00 у
5
с"8"
1
ОуО
111
1
\
з
4 : (с) OOz (d) 0 у z (e) izO
8: (g) xyz
1 А
2 : (а) 000 F) 00 у
1 11
4:(cHy0(dHyT
(е) OOz
(ft) zOO @ zO у
1 1
16: (к) xyz
1 1 '
+ 000; у -j
) +
1 \
-_\
78
ТАБЛИЦА 7 (продолжение)
110
111
112
ИЗ
114
46a
Df
(J4i2)
T4i22
1
4 : (a) 000 (&) 00 —
8 : (c) OOz (d) zzO
11 > "+"
(е)м:0(/J: 4 g
16:(g)zt/z
i 1 A
+ @00; -7TT
~^ Z 2 2/
Тетрагонально-скаленоэдрический вид
симметрии
32.9
D2d
РШт
30/г
Dld
P42c
52a
D3
PlZim
53a
PlL
1
2
4
8
2
4
8
2
4
8
2
4
8
j j j J
:(aH00(b)yTT(CH0Y
1 1
(d) 2 2 °
1 11
:(e) у 00(/)y0y(gH0«
1 1
Wit»
:@*00(/)Syy(*)*0y
1 1
: (o) a;j/z
1 1 1 111
11 11
(iHTT(eH00(/J20
1 11. 11
:(g) x0^{h)rlXi(i) x — -?
(/Hг-|-(/сH0г@4тг
1
: (n) xyz
: (a) 000 FH0 у (сH у г
: (d) OOz (e) x, -j + x, z
¦ (/) xyz
: (a) 000 F) 00 -j
: (c) OOz (d) 0 у z
: (e) яг/z
115
116
117
118
33s
D%i
(Cl2m)
P4m2
31A
(Ci2c)
-P4c2
32h
(Ci2b)
р\ьг
33A
(C42ra)
P4n2
1:
2:
4:
8:
2:
4 ¦
8:
2:
4:
8:
2:
4:
8:
(a) 000 F
(d) 00 -g-
(e) OOz (/)
(A) xxO (j)
1
(k)x-z
(I) xyz
(a) 00 — (
11
i_
1 1
(A) 2 2 z
(/) zj/z
1 1
l l
Ill
1
«yO")*0*
6)-r-
Z Z f
)
3
1
(t) 0 2 z
(aH00(&H0 —(c)C
о 1 1
(eH0z(/)
1
W *. 2
(i) syz
(a) 000 F)
1 3
W°T1
(e) OOz (/)
(*)*• T
(i) xyz
4»
1
-(с) 000
1
1
1
1 1
00T(cHTT
1
г, у —-
+ *.-(
1
h)t)\z
79
ТАБЛИЦА 7 (продолжение)
119
120
1
121
1122
35s
(F42m)
llmt
34ft
(Fl2c)
34s
Du
51o
Djd
1Ш
2 : (a) 000 (b) 00 у
11 13
4 : («) OOz (/) 0 у s
1 1
@ xOz
16: (/) zj/z
+
[ft ¦ 1 1 M
4 : (a) 00 -^ F) 000
1
1 1
16: (j) xyz
/ 1 1
_|_ 1 000; -Q- -p
,+
M
2:(aH00(&H0y \
1 1T1
(e) OOz
1
1
(ft) 0 у z (/) xxz
16: (/) xyz
+ @00;y4
1
4 : (a) 000 (&) 00 у
1 1
16 : (e) zi/z
+
4)
+
+ @00; ТГ vt
1 \ 2 2 2/
Дитетрагонально-дипирамидальный вид
симметрии
123
124
125
JPifmmm
35ft
JPijmee
36ft
Pifnbm
1: (a) 000 (ЬH0 у (с)уу0
1 1 1
2:(eHyy(/Hy0Ur)OOz
1 1
4 : (j) 0 yz (,!)ххП(к)хх Y
1 1
1 1
8 : (p) xyO (?) гуу (r) xxz
(s)xOz(t)x yz;
16: (и) zi/z
1 111
1 1
(d)yyO
1 1
1.1 1
16: (л) xyz
2 : (a) 000 (b) 00 у (с) 0 у 0
1 1
(i>iV "ll 1,
4: (eL|0(/)|Ty:(gH02
(ftH yz
i
8:(l)xxO(j)xx-j (k)x00
1 1
(Z) xO у (m) ж, у + x, z
16 : (n) zi/z
ТАБЛИЦА 7 (продолжение)
12
127
128
129
130
ъп
Pi/nnc
54а
Pi/mbm
55а
Pi/nmm
56а
D\h
Pi/mne
57а
Pi/nec
2 : (а) 000 F) 00 у
4: (с) y00(d)y0-|-(eH0Z
111 1
8:(/)ттт (г)тОг
(/г) стО (*) жОО (/) хО у
16 : (fc) xyz
111
2:(аH00FH0у(сHуу
(d)OyO
4:(еH0*(/H у*
(g)x,y + s, 0
1 1
(ft) я, у +х, у
1
1 .
/ЬЛ 7* ¦¦' — —Г" aCi 2
16 : (I) xyz
2: (a) 000F) 00у (c)Oyz
11 111
4: (d) X T ° W T T T
8: (g) гяО (fe) xx у @ 0*г
1
16:(ft)*yz
2:(aH00(bH0y
4-(eH —0(«ft0—-(e)OOx
1 11
8 : (/) 0 у z (g) x, у + я, х
(Л) *у0
16: (i) яуг
4:(а)ООх(Ь)°00(с)Оуг
1
(
16: (g) xyz
131
132
133
60а
(Pi/mmc)
Palmitic
61a
(Pi/mcm)
Pii/mcm
62a
(Pi/nnm)
Pk^nwm
2:(aH00F)yy0(oHy0
(tfHyy(eH0|
«ITT
1 1 1
11
(/)*00(fc)zyy
8: (n) nj(e) Oxz
1
(p) у xz (?) гуО
16: (г) гуг
2: (a) 000 F) 00 x
(C)yy0(d)yy|
4- 11,1
1 1
(l)xxT
8 : (k) 0 у z (Z) xO x
1 1
(o) xxz
16: (p) xyx
1
2: (a) 000 F) 00 у
1 11
4:(cHyO(d)OyT
1113 3 3
(e) T 4 4 "' 4 4 T
(«) 00л
1 1
8 : (h) 0 у г (i) x00 (/) гО у
1 1
(ft)*,y +e, T
1 3 ^
16: (n) xyz
Кристаллохимия
81
ТАБЛИЦА 7 (продолжение)
134
135
136
137
138
63а
ТI1
Dih
(Р4/пЪс)
Ptynbc
64а
т\14
(РА/тпт)
Р4г/тпт
65а
(Pi/ncm)
Pii/ncm
66а
(Pi/mbc)
Р&г/тЬс
67а
nl5
Vih
РШтс)
P42/nmc
4:<аH-тг-г
(о) 0 -j 0 (d) 000
8:(«)TTT(/)°TZ
(*H0.(A)eOj@*o4-
16: (ft) xyz
2: (a) 000 FH0 y
4:(oH-TrO(dH-rr-r
(e)OOz(f)xxO(g)x?O
8:(fcHy*(f
16: (fc) xwz
4
4:(aH0TFH00(o)TT
8:
-j +x, z
16: (/) xyz
4:(aH00FH0-|-(cHy0
1 1
8:(eH0«(/H y(z)
.1 1
i6:(i)xyx
2 : (a) 000 FH0^-
4 : (c) OOz (d) 0 -j z
S:le)-r-r-r(f)xai
6: (h) xyz
139
140
141
142
37»
ТЦттт
38Л
58a
(Ii/acd)
Xii/acd
59a
(/4/amd)
liifamd
2 : (a) 000 F) 00 ^
4 : (с) О ^r
(e)OOz
8;(/)ттт
(«H{»(A)«0
1 .
16 : (*) ж, 4" + *» "Г
(n)Osz
32: (о) zyz
(m) ггг
+
(«ю-,4-1
ТУТ
4:(«H0T6@)T^
(с) 000 (d) 0 |-О
111
8:(e)-r-r-r
16:A)мт(|-
(к)хуО
32 : (m) xyz
+
3;ттт)
8 : (а) 000 F) 00 —
16:(CH-J4(dH0;
1 1 ... 1
32:
+
ЭОО;у 2 2/
1 \
4 : (а) 000 F) 00 у
8: -1 * --1 5
е @0)г
16 •¦(/)* т4
(Л) Огг
32 : (*) агг/*
,1И\
3>TTTJ
82
ТАБЛИЦА 7 (продолжение)
ТРИГОНАЛЬНАЯ СИИГОИИЯ
Тригоиально-пирамидальный вид
симметрии
12 2 1
1 : (aH0zF)y у z(c)yy
3:
3: (а
3: (a) xyz
1: (a) xxx
3: F) xyz
Дитригонально-пирамидальиый вид
симметрии
1 2
143
144
145
146
38s
(сз3)
рз
68а
С2
3
69а
(СЗ,)
РЗа
39»
т
147
148
149
150
151
152
40s
(C3m)
39Л
(СЗс)
41»
**.
(ЯЗт)
40Л
(ЯЗс)
Р31с
42»
ЕЗт
41А
ВЗс
1:
21
3: (d) xxz
6 : (е) xyz
2:(aH0zF)yyi
6: (d) xyz
1: (a) OOz
3: (c) zOz
1 2
2 : (a) OOz (Ь) у у ;
6 : (с) xyz
1: (a) xxx
3: F) xxi
6: (с) xyz
2: (a) xxx
6: F) zi/z
2 1
Тригонально-трапецоэдрический вид
симметрии
1 : (a) 000 (Ь) 00 -j
1 2
2 : (с) 00z (d) у у z
3 : (е) х00 (/) хО у
6 : (g) xj/z
1 5
3:(а)хО-тг(Ь)хО-рг
&:(c)xyz
153
154
155
156
157
158
159
44s
Dl
(С32)
Р321
70a
(C3i2)
P3i2i
71a
(C3,2)
i»3221
45s
»l
(Я32)
P312
72a
(Я312)
73a
(Я322)
46s
B32
3 : (a) xO у (Ь) zO -g-
6: (o) xyz
1 2
1: (a) 000. (bH0-7jr (c) y-g-O
12 1
2 1
Шит
III
2:
3 : (/) x,
6 : (I) xyz
-1 -¦i
6:(o)^z i
_2 - 1
3 : (a) xx у (о) хх -g-
6 : (c) syz
111
l^aJOOO^yjy
2:(с)ггг
6: (/) xyz
83
ТАБЛИЦА 7 (продолжение)
160
161
162
163
164
165
Ромбоэдр
51s
cL
(СЗ)
РЗ
52s
с*
ИЗ
ический вид симметрии
1
1 : (а) 000 F) 00 у
1 2
2 : (с) 00z (d) 3 3 z
3:(е)у00(/) уОу
6: (g) xyz
1: (а) 000 F) у уу
2: (с) ххх
1 11
3:(d)y00(e) Оуу
6: (/) хух
Дитригонально-скаленоэдрический вид
симметрии
55s
let)
P3ml
45ft
(СЪс)
PSel
56s
(ЯЗго)
46Л
^Id
(ЯЗс)
jP31c
1: (а) 000 F) 00 у
2:(oH0z(d)i-T*
3:(е)|оо(/)|оу
1
12: (/) xyz
1
2:(а)"ОО-^-F)ООО
1 2
4: (oH0i(d) y-j-z
1 1
6:(e)y00(/)x0T
1: (а) 000 (Ь) 00 у
12 12 1
2: (с)-д "з 0 (d)-g-g-у
(eH0z
1 11
3:(/)y00(g)y0y
1 2
•( ) 3 з z
6 : (t) ггО (/) xs у (ft) *0z
1
2: (а) 00-^- (Ь) 000
12 1 2 11
1 2
4:(eH0z(/)-3--3-j
6:(г)уОО(Л)гг4-
12: (i) да/z
166
167
57s
да
47Л
¦^3d
l:(aH00(b)yyy
2: (с) хгх
1 12
3:(d)y00(eHyy
6 : (/) xxQ (g) xx у (h) xxz
12: (i) xyz
111
4 4 4
4: @) ггя
1 11
61 (d) ТГ 00 (в) «г iT1 — э; -т-
12 : (/) zyz
ГЕКСАГОНАЛЬНАЯ СИНГОНИЯ ?f
(
168
169
170
171
172
173
Гексагонально-пирамидальный вид
симметрии
49*
C\
(C6)
i»6
74a
75a
(C68)
76a
(C6j)
JPbt
77a
78a
CJ
(C68)
i>68
1: (a) OOi
1 2
: F) y-g-i
3: (с) у 0*
6: (d) si/i
6: (a) xyx
6: (a) xyx
3:(aH0i(b)yyz
6: @) xyx
1 1
3:(aH0iF)yyi
6 : @) zyz
1 2
6: (c) xyx
ТАБЛИЦА 7 (продолжение)
Дигексагонально-пирамидальный вид
симметрии
174
175
176
177
50*
(Сбтто)
Рбтт
44А
С2
°в»
(Сбсс)
.Рбсс
79а
Г4
(СЪтс)
.Рбздае
80а
С
(Сбст)
1: (а) 00z
2: (b)i-| z
3:(e)y0z
6 : (d) xOz (e) xxz
12: (/) xyz
2: (aH0z
1 2
4:(ЬI-Т2
6 : (с) у Oz
12:(d)xj/Z
2 : (a) OOz F) -s- "о" г
О О
6: (о) яг*
12: (d) xyz
2: (a) OOz
1 2
4:(Ь)у-д-г
6: (с) xOz
12: (d) xyz
Гексагонально-дипирамидальный вид
симметрии
178
179
53s
<4
(Сб/т)
Рб/т
81а
<&
(С6з/т)
-Рбз/J»
1: (a) 000 FH0 у
12 12 1
2:(c)TY0(d)TTT
(e) OOz
3:(/)-oOO(gL-o4
...12
4:(fc)TTz
e^OyOzO^a^t^xi/y
12: @ xyz
2: (a) 00^- F) 000
12 1 2 11
(°) T T T(d) T T 4
1 2
4:(eH0z(/)TTz
6:(г)^-00(Л)гуТ
12:(i)xyz
Гексагонально-трапецоэдрический вид
симметрии
180
181
182
183
184
54s
D\
(Ш)
.P622
82a
D\
(C6i2)
i»6i22
83a
(C652)
i»6522
84a
D\
(C6,2)
i»6222
85a
JD»
(C642)
.Р6«22
1: (a) 000 (b) 00 у
12 12 1
2;WlI°Wll2
(eH0z
1 11
3:(/)-200te)T0T
1 2
4:(fc)TT*
1 1
6 : (?) у 0z (/) z00 (A;) xO у
(l)xx-0(m)x2-j
12: («) xyz
1
6 : (a) г00 (Ь) х, 2x, -?
12: (C) si/z
6:(а)*ОО(Ь)г,2г,-^
12: (с) xyz
3 : (a) 000 F) 00 у (с) у 00
(d)y0y
6:(eH0z(/)y0z(g)sOO
(Л)жОу (t)x, 2x, 0
у
12: Gc) syz
3 -. (a) 000 F) 00 у (с) у 00
1 1
(i)y0y
R • 1„\Пп, 14\ —Oz (в) xOO
о ; (e) vvz [j j у и \& / *^
/fc\-»^ /#\ * 9т П
(Л) Л) у у) г, ?с, и
(i)x, 2г. тг
12: (ft) xyz
85
ТАБЛИЦА 7 (продолжение)
185
с
186
187
188
86а
Dl
(С682)
.Р6.22
2 : (a) 000 F) 00 -j
12 1 12 3
1 2
4:(eH0z(/)T?Z
1
12: (i) xyz
^игексагональио-динирамидальный вид
имметрии
58»
D\h
(CfJ/mmm
Рб/ттп,
48fc
&lh
(C6/mcc)
.Рб/mce
87a
D\h
(C6n?em)
P&ifmcm
1: (a) 000 F) 00 у
12 12 1
» ' 3 3 ''332
(eH0z
1 11
3:(/)yOO(g)yOT
12
4:(Л) 3 3 z
1 1
6 : (i) у Oz (/) s00 (ft) xO у
i
(l)x, 2x, 0(m) xt 2x, —
2
12 : (n) xOz @) x, 2x, z
1
(p) xyO (?) xy у
24 :(r)*yz
2: (a) 00-^-F) 000
121 12
4: (c) 3 з 4 W з з °
(e) OOz
111
6 : (/) у 0 -|- (g) у 00
1 2
.1 .1
(fc) x, 2x, T (I) xyO
2A:(m)xyz
1
2: (a) 00 у F) 000
12 112
4: («) 3 з 4 (») з з °
(e)OOz
6:(/)y00(g)x0-|-
1 2
.1
(A;) xOz
2A:(l)xyz
189
88а
7}4
(C6/mmc)
2 : (а) 000 FH0 \
12 112 3
(с) з з 4 1 ' 3 3 4
4:(.H0.(/,4|-.
6 : (g) у 00 (Л) г, 2х, т
1
12 ' (i) ж00 f/Л 2iw ——
(ft) г, 2х, z
24: A) xyz
Тригонально-дипирамидальныи вид
симметрии
190
43«
(С6)
JF*6
1: (а) 000 F) 00 у
12 12 1
(") 3 3 ^3 3 2
2 1 2 11
1 2
2:(gHOz(A)TTz
*v* ' 0 о
2 1
(J) уз"*
1
3 : (/) хуО (к) ху у
6: (I) хуг
Дитригонально-пирамидальныи вид
симметрии
191
48s
(C6m2)
-P6w2
1:(аH00FHРу(с)-з-у0
12 1 2 1
W_3 3 2 (е) 3 3 °
2 11
Ш 3 3 2
1 2
t2 = (?) 00z (Л) з з z
¦2 l)-i
(i) 3 з г
1
2
12: (o) zyz
ТАБЛИЦА 7 (продолжение)
192
193
194
43Л
(С6с2)
i»6c2
47s
D3h
{#6то2)
42Л
(Я6о2)
РЬ2с
1 12
2 : (а) 000 F) 00 -|- (о) -j -g- 0
12 1 2 1
2 11
"'334
12 2 1
6 :(/•)*«) (Л) в» J
12: @ xyz
А
1: (а) 000 F) 00 у
12 12 1
1
3:(/)xOO(g)x у
1
12: (/) xyz
1 12 1
2 : (а) 000 (Ь) ОО-^(с) 3 3 4
2 11
00 , А_2_г
1
12: (i)x'/z
КУБИЧЕСКАЯ СИНГОНИЯ
195
196
Пентагон-тритетраэдрическии вид
симметрии
59s
7-1
.Р23
89а
Л,
1 1 1
11 1
4 : (е) хм
1 1
1 1
(г) г 2 2
12 : (/) xyz
4: (а) ххт
12: F) xyz
197
198
199
200
201
61s
7>j
^23
60*
7-s
J23
90а
J2i3
111
4:(аH00F)ууу
111 3 3 3
(с) т т т^) tit
16: (е) xxx
1 1
24 : (/) х00 (?) х т Т
48:(h)xy. I
+(ооо;ууо4от;о1т)
2: (а) 000
1 1
6:(ЬHуу
8: (с) ххх
12: (d)x00(e)x у 0
24: (/) xyz
¦ +
1 1 1 1\
+ (,000; у у у)
8:(а) xzz j
12: F)xO-j|+(oOO;yy yj
24 : (с) xyz )
^идодекаэдрический вид симметрии
62s
п
РтЗ
49fe
п
РпЗ
1:(аH00(Ь)ууу
3:(cHyy(d)y 00
1 1
1 1
(А)хуу
8: (г) хгх
\2:(j)Q*yz(k)-jyz
24 : (I) xyz
2 : (а) 000
111 333
1 1
6:(dHyy
8: (е) xxx
1
12:(/)x00(g)zy0
24: (h) xyz
87
ТАБЛИЦА 7 (продолжение)
202
203
204
205
206
91а
п
РаЗ
64s
п
РтЗ
50h
п
Fd3
63s
п
1тЗ
92а
п
1аЗ
4:(аH00(Ь)ууу
8: (с) ххх
24: (d) xyz
4:(аH00(Ь)ууу
111
"(СL 4 4
2A:(d)O~(e)x00
32: (/) ххх
1 1
4 4
96: (i) xyz
/11 1 1
+^000, у у 0; 2 2 '
8: (а) 000 F) у у у
111 5 5
32: (е) ххх
48: (/) аЮО
96: (g) xyz
^-(ооо^-о-О;^0^1 с
2 : (а) 000 v
6:(ЬHуу
111
8: И Т Т Т
12 : (d) xOO (е) хО у
16: (/) ххх
24: (g) Qyz
48: (h) xyz
> +
1 i)
5
8
,
-
+
+ («»•¦ ттт)
8:(аH00(Ь)ттт
16: (о) хаот
1
48 : (е) яуг
+ @00; -
1
2"
+
1
2 '
]
207
208
209
210
"¦ексатетр!
65s
п
Plbm
51Л
Ti
а
Р43п
67s
т\
ШЗт
52fe
Т\
Fl3c
1эдрический вид симметрии
111
1 1 1
4: (е) жхт
1 1
6 : (/) жОО (g) х у у
12 : (Л) а; у 0 (i) ггг
24: (/) xyz
2: (а) 000
11 11
6 : (Ь) ° 2 2 Vе) 4 2°
1 1
(d) 4 ° 2
8:(е)*х*
1 1
12 : (/) xOO (g) х у 0 (Л) гО -^
24: (i) xyz
111
4 : (а) 000 F) у у у
111 3 3 j$_
(C)ITT^'4 4 4
16 : (е) zzx
1 1
48: (h) xxz
96 : (i) тЗ/z <
/ 1111 .-jii.1
+y ° '2 2 • 2 2 ' 2 2,/
1 1 1
8: (o)OOO(b) jjj
1 1 1
24 : (c) 0 T -j (d) jOO
32 : (e) жгт
г— i
96 : (fe) хуг
+
/ 1111 1 1\
88
ТАБЛИЦА 7 (продолжение)
211
212
213
,
214
215
66s
T%
llSm
93a
1*
J43d
2: (a) 000
1 1
8: (с) ххх
12 ± i-0(e)iOO
24 : (/) x у 0 (g) ххх
48: (Л) zyz
( i
+ @00; у
3 1 7 1
12:(a)T0T(b)-g-0T
16: (с) ххх
24: («0*0 4"
48: (e) xyz
+ foOO;y-
Пентагои-триоктаэдрический вид
симметрии
68s
О1
.P432
94a
(i>483)
.Р4з32
95a
О7
-P4i32
l:(aH00(b)yyy
11 1
3:(cHyy(d)y 00
6 : (e) xOO (/) x у у
12:(Л)х y0(i)O«0}
24: (к) xyz
111 55
8: (с) ххх
A A
12 : (d) -g- , x, ^- — x
24 : (e) хуг
3 3 3 7 7
4:(a)~8~ 8"?F)"8""8 "
8 : (o) xzx
12 : (d) 1 , x, 1 + x
24: (e) xyz
ч
1
2
'
1
2
1
2
5
8
7
8~
M
+
t)
216
217
218
98a
O*
JP4232
70s
O3
(/•43)
F432
97a
Oi
Fh32
2: (a) 000
111 333
11 11
6 : (d) 0 yy(e) -?0~2
1 1
(/) 4 2 °
8 : (g) xxx
A A
11
1 1
(") 4 ' ж' 2 — *
1 1
(I) -? , x, у + x
24 : (rn) xyz
4:(aH00F)yyy >,
8:(«>TTT
24 • (d\ 0 — — П 00
• I ) 4 4 Iе/г
32: (/) xxx
48 : (g) Ozx (h) у гг
1 1
96:0')*»*
Л А \
L8:(aH00(b)yyy '
5 5 5
32 : (е) ххх
48 : (/) хОО
1 1
9&:{h)xyz
! 1111 1 1\
+ 000; у-р 0;у0у,0уу)
I
89
ТАБЛИЦА 7 (продолжение)
219
220
221
222
69s
О5
(/43)
Г432
96a
0»
(/4i3)
J4i32
2 : (a) 000 >
6:(ЬHуу
111
12:(d)Xy 0(е)х00
16: (/) ххх
1
24 : (g) x — 0 (h) Oxx
1 1
(l)T,x,Y-x
48 : (/') xyz *
+ @00;yy-
¦ ' ' 8 8 8 * ' 8 8 8
11 5 1
12:(c)-g-0T(d)-g-0T
16: (e) xxx
24 . /л xq A
4
1 1
1 1
48 : (I) xyz j
I)
Гексоктавдрический вид симметрии
71*
°h
РтЗт
53Л
o\
РпЗп
l:(aH00(b)yyy
1 1 1-х
3 : (o) 0 у у (d) у 00
1 1
6 : (e) xOO (/) x у у
8: (г) «г
12:(ft)*y0(iHzz(/)y гг
24 : (к) Oyz [1) у 1/z (m) xxz
48 : (n) хуг
2: (a) 000
6:(ЬHуу
111
8:(C)TTT
1 1
16 : (/) xxx
1
48: (i) xyz
223
224
225
102a
o\
РтЗп
103a
.
jPnSm
73s
o\
Fm3m
2 : (a) 000
6:FHyy(C)|0y
1 1
1 1 1
1 1
12:(/)гОО (g)xO 2 («)* 2 °
16: (i) xxx
1 1
24 :(/)¦?,*, y+;r(A;Hi/z
48: (I) xyz
2: (a) 000
111 333
4: (ь)тт~4~(с)ттт
1 1
6:(dHyy
8:(e)xxx
1 1
12:(/)T°"^
111
24 : (h) xO у @ x» я> ~ ~ x
1 i, ft)
w) 4 ' *• 2 "• * '
48: (i) гз/г
1111
4: (a) 000F)-у у
1 1 j_
• («) 4 4 4
1 1
24:(<iHx (e)s0°
32: (/) xxx
1 1
@ у xx
96 : (/) Oyz (k) xxz
192: (I) xyz
¦ +
* A
f 000' 110' — 0 — • 0 — —
\ '2 2 >2 2> 2 2
ТАБЛИЦА 7 (окончание)
226
227
228
54Л
ol
Fm3c
100a
ol
FdZm
101a
o\
Fd3c
8:(a)-?-jT(bH00 '
24 : (с)-f00(((Hji
48:(e)z00 (/)z -j-?
64: (g) ажв
1
96 : (h) -r xx IX) Oyz
+
1
m-.a) xyz
. /nm и iAi л i i \
+ \ООО;2-2О;2°2;ОТТ)
1 1 1 >
8:(aH00F)TTi
<U L, Ct
16- M1 4 *
5 5 5
(^"8"?"8"
oL : (e)xxx
48: (/) гОО
96: (g) xxz
(A)-g-, «,-4 -ж
192:(«)xi/z
/ 1111
> +
1 4 ,
+ ^22^2°2=0tij
16 : (a) 000 \
32 : F) 4- 4 4-
W8 8 8
3 3 3
W 8"T8"
48:(d)^00
64: (e) xxx
96: (/) xOO
1 1
(?) У . X, -? — I
192:(A)xj/2
+ (о00;^0;|о|;0
> +
2 2/
229
230
72e
ImZm
99A
J«3d
2: (a) 000
6 • (Ъ\ о -г тг
yj. ^i/^ 2 2
8:«>Ш
Tt *t "i
1 1
12: (d)^0^*00
16: (/) Axx
\
24:(g)z0y
w/ it
(h) Oxx
1 1
48:(t)-?-, ж, у —г
(/Hi/z(A)^z
96:(ZJ2/z . J
+ @0°: TYTJ
16 :(я) 000 )
111
1 ' 8 8 8
24:(c)-g-0-|-
/л 3 n *
32 : (e) xxx
48:(/)*0-J-
1 1
(g) "g- > ж, j — <r
96: (A) i?/z
¦ +
,f000 1 1 1\
-I- ^uuu, 2 2 2/
91
/ Pi
PI
3
PZ
4 PZ
: i 1
i 1 ;
: 1 :
5 C2
\ \
6 Pm
7 Pc
11 P2/c
iZ P2/ /m
9 Cc
-114
10 P2/m
i4 CZ/m 15 С 2/с ~
Рис. 105. Федоровские группы тржклинной
И МОНОКЛИННОЙ СИНГОНИЙ
IB Pmm Z
21 РЬа 2
¦+-¦
26 Стт2
1!1
¦ if t
31 Am a 2
36 Imq 2
¦¦¦?
¦i
/7
Pmc 21
-t—г—
t 1 f
j—f ;
37 Jba ?.
-*-
j
1
*
18
i
23
6
9
—I—
-f-
Pma
Pmn
-4--
—t
1
i
2
1
9
—i
-f
Cmc 21
-i
Л
t #
24 Pea 2.
4mm •
-Э-
fdd2
• i ¦
20 Pnn 2
i
25 ч -°/7a 2i
-f
-f ^ -4—*-
—f-—
SS [mm 2 с
Рис. 106. Федоровские группы ромбо-пи-
рамидального вида симметрии
92
t f f
-/A
—/A -•-
I/a
I
f
I I'M
42 С222
чз с 2гг.
44 F222
-w
45 1222
ЧВ I 2^/,
Рис. 107. Федоровские группы ромбо-тет-
раэдрнческого вида симметрии
47 Рттт
г
Si Pmma
it РЬзт
Реет
—f—I
414
-1/4 о
у 14
\ т\
^
i
49 РЬап
\П4 ф\
53 Рсса
i
/С/7/7/7
-1/4 —о 1
i/A
-/A
_ _|..
57 Рттт 58 Рппт
\114
-Ф о
_* i.
52 Pbca
Рис. 108. Примитивные федоровские груп-
группы ромбо-дипирамидального вида симмет-
симметрии
93
—<—
63 tmmm
t \ t
-§—i—$~w
-4—-?—|—/д -if-—4—dji-
4—!—i+w -\—j^-f
f f f7\\ f |
-p- i -9—/A
4 1= Ф—//*
-0
-114
\ \ \
-p—I—?-
?7 Лтеот
"m
—1/Ч
7/ Immm
68 Cmca
— -4 f I —
Рис. 109. Непримитивные федоровские
группы римбо-дипирамвдального вида сим-
симметрии
¦
7^
¦
75
< >
i
РЧ
$
О
14
77
80 I4f Рис. ПО. Федоровские группы тетраго-
тетрагонально-пирамидального вида симметрии
94
81 PiJ mm
8Б Р4гст
91 I4tmm
92 I4cd '
83 РЧ Ьт
88 P4zbc 89 14 mm
Рис. 111. Федоровские группы дитетраго-
нально-пирамидального вида симметрии
Ф
93 P4
ф
94
<> 1/4
Рис. 112. Федоровские группы тетраго-
нально-тетраэдрического вида симметрии
75 Р4,
78 Р4,
¦
79 14
80
Рис. 113. Федоровские группы тетраго-
нально-дипирамидального вида симметрия
•5
у
/
у
/lu
t
1/4
101 r> 22
Ч
/
ч
V
m
/14
A/8
4-
3/8 1/8 3/8
/ 4 /
1/8
104 P132Z 'OS рчг22
3/8
/1/4
109 142:
9
НО Г 1,22
3/8
/111
*
У
3/8
+
./А
f \
10В Pli 2,2
107 P4322
108
Ч
Рис.114. Федоровские группы тетрагональ-
но-трапецоэдрического вида симметрии
/¦
X
¦111 P12tc
Ч У \J? \J>
1/4 1/1//4 //41/4 /I4//4
1/4 \ i ТЧ
//1
//7 /'V^
1/4
\
/
<*
I
X
\
•1/1-1 к. 1
¦/II : ^Г :
/Hi 7
у
<•> j <
/18 Pin 2
J/* //? да
\ \
\
\
-^ 3/8
ли' Л/т?^ Ч
/А
//* /*, —'/* X
. / ¦•-..
Ф
122 I42d
Рис. 115. Федоровские группы тетраго-
нально-скаленоэдрического вида симметрии
'9G
123 РЧ/ттт
127 РЧ/mbm
У
№
131 P4z/mmc
1/4
i Ж
135 Р4г/тпт
\JI4
li
P4/mcc
132
139 14I mm m
I ..¦¦¦
140 /4 I mem
*У Ч»/ |
—m'jb—
/ j ¦••¦
Ж i w
Ф
У
—71
¦¦¦¦¦;¦ '
X
P4/nnc
*.•¦
Ш Р4г/тт
^0-
/Л
У 1 X
\ ! /
14,/атй
i'lic. 116. Федоровские группы дитетраго
нально-дипирамидального вида симметрии
7 Кристаллохимия
97
РЗ
1ft P3f
¦If 5 P3,
1ЧБ R3
Рис. 117. Федоровские группы тригональ-
но-пирамидального вида симметрии
Р3т1
If 8 P3d
149 Р31т
ж
¦ISO P3ic
151 R3m
'¦¦¦v
¦i-
Ш R3c
Рис. 118. Федоровские группы дитриго-
нально-пирамидального вида симметрии
Рис. 119. Федоровские группы тригональ-
но-трапецоэдрического вида симметрии
Рис. 120. Федоровские группы ромбоэдри-
ромбоэдрического вида симметрии
Рис. 121. Федоровские группы дитриго-
нально-скаленоэдрического вида симмет-
симметрии
168 PB 169 PS,
171 РВЬ 172
Рис. 122. Федоровские группы гексагональ-
иЗ P6j но-пирамидального вида симметрии
9!)
471 P6 mm
: \
v"
/^
f
176 ¦ PBj/nc
177 PS3 cm
Рис. 123. Федоровские группы дигексаго-
нально-ппрамидального вида симметрии
178 РВ/т 17S РБ3/т
Рис. 124. Федоровские группы гексаго-
нально-дипирамидального вида симметрии
\f/
* Федоровские группы геьсагоналъ
но-трапецоэдрического вида симметрии
100
w.
i89' Pff,/mmc
~l
PBjmcm
Рис. 126. Федоровские группы дигексаго-
нально-дипирамидального вида симметрии
190 Р 6
Рис. 127. Федоровская группа тригональ-
но-;пгаирамидальиого вида симметрии
Ш PBZm
^¦1*
:<. ^,J ...'¦«
PBZc
П<
Рис. 128. Федоровские группы дитриго-
нально-дипирамидального вида симметрии
101
^L
195 P23
t Г1
,r*
^
К
196 P2f3
114 ПЧ 1
:S.\
» «^
f/A \1/ч
\ , -
W
w
к
5ЧК
Рис. 129. Федоровские группы пентагон-
тритетраэдрического вида симметрии
¦//*
198 123
199 I2J
7Ш РтЗ
114
//77 J"
^ /ff/5
С 1 О —О О
202 "' РаЗ*г 203 РтЗ
Рис. 130. Федоровские группы дидодекаэд-
рического вида симметрии
Кроме обозначенных на рисунке элементов
симметрии, см. также дополнительно для со-
соответствующей федоровской группы: *Р23;
*2р2,з; *з^23; *4Г23; *572,3
102
Рис. 131. Федоровские группы гексатетраэд-
рпческого вида симметрии
Кроме обозначенных на рисунке элементов
симметрии, см. также дополнительно для со-
соответствующей федоровской группы: *Р23;
«F23; «723; *472,3
*-&*
-fi-
гт
./
w
Рис. 132. Примитивные федоровские груп-
группы пентагон-триоктаэдрического вида сим-
симметрии
Кроме обозначенных на рисунке элементов
симметрии, см. также дополнительно для со-
соответствующей федоровской группы тройные
винтовые оси: *Р23; *2Р2,3
РЧг32*
103
//г
Z20
/</ 32
'3
Рис. 133. Непримитивные федоровские
группы пентагон-триоктаэдрического вида
симметрии
Кроме обозначенных на рисунке элементов сим-
симметрии, см. также дополнительно для соответ-
соответствующей федоровской группы тройные винто-
винтовые оси: *F23; «123; *=12,3
ffl
Рис. 134. Примитивные федоровские труп
пы гексоктаэдрического вида симметрии
Кроме обозначенных иа рисунке элементов сим-
симметрии, см. также дополнительно для соответ-
соответствующей федоровской группы: *Р432; *2Р4232
224 РпЗт*2
104
m м
lilil ffl
114
//W J/77
230 la 3d
U
Рнс. 135. Непримитивные федоровские
группы ! ексоктаэдрического вида симмет-
симметрии
Кроме обозначенных па рисунке элементов сим-
симметрии, см. также дополнительно для соответ-
соответствующей федоровской группы: *F43; для фе-
федоровской группы Fm3c начало координат в
группе F43 следует поместить в СД'Д'Д); *2F4i32:
«1432; *44i32
ГЛАВА VIII
ЭКСПЕРИМЕНТАЛЬНАЯ ПРОВЕРКА
ГЕОМЕТРИЧЕСКОЙ ТЕОРИИ КРИСТАЛЛОВ.
РЕНТГЕНОСТРУКТУРНЫЙ АНАЛИЗ
§1. Первые определения
атомных структур кристаллов
при помощи рентгеновских лучей
В 1912 г. Лауэ доказал, что рентге-
рентгеновские лучи аналогичны по своей
природе лучам света, но отличаются
от последних значительно меньшей
(примерно в 10000 раз) длиной вол-
волны. Длины волн рентгеновских лучей
оказались одного порядка с меж-
межатомными расстояниями в кристал-
кристаллах. В том же году В. Л. Брегг и не-
несколько позже Г. В. Вульф вывели
формулу, связывающую межплоско-
межплоскостные расстояния в кристаллах d с
длиной волны рентгеновских лучей
X и углами скольжения 8. Одновре-
Одновременно В. Г. Брегг и В. Л. Брегг оп-
определили экспериментально величи-
величины d для разных кристаллов. Схема
опыта Бреггов показана на рис. 136,
где S — источник рентгеновских лу-
лучей, К — испытуемый кристалл,
0 — угол скольжения (дополнитель-
(дополнительный до 90° к углу падения), / —
ионизационная камера. Кристалл
монтирован на оси, перпендикуляр-
перпендикулярной к плоскости чертежа. Поворота-
Поворотами около этой оси можно изменять
углы падения рентгеновских лучей
на кристалл. Вокруг оси может вра-
вращаться и камера /, с помощью кото-
которой улавливается отраженный луч.
Опыты, проделанные на этой уста-
установке, показали, что рентгеновские
лучи отражаются от граней кристал-
кристалла не под всеми углами, а лишь под
некоторыми. Для различных веществ
и для разных граней кристалла одно-
одного вещества углы, при которых про-
происходит отражение рентгеновских
лучей, вообще различны. Анализ по-
полученных измерений привел к сле-
следующему объяснению (рис. 137).
Кристалл можно себе представить
как серию плоских сеток, отстоящих
друг от друга на одинаковом рассто-
расстоянии. Обозначим это расстояние че-
через d. Плоскость чертежа перпенди-
перпендикулярна к плоскостям сеток. Su S2,
S3... — параллельный пучок рентге-
рентгеновских лучей; АС — ВС= А есть раз-
разность хода двух лучей, идущих пос-
после отражения в соседних плоскостях
в одном направлении AT. Из прямо-
прямоугольных треугольников ABC и ACD
получаем:
Д = АС A — cos 26) = 2HCsin2 0,
AC =d/sinQ, A=2dsin6. A)
Если А равны целому числу волн
п, то отраженные лучи в результате
интерференции будут максимально
усиливать друг друга, и их можно бу-
будет обнаружить с помощью иониза-
ионизационной камеры. Для этого случая
уравнение A) примет вид:
Д = пк = 2 d sin 0. B)
Рис. 136. Схема опыта Бреггов
106
Если угол падения не будет удов-
удовлетворять уравнению B), то суммар-
суммарного отражения не произойдет, так
как лучи, отраженные отдельными
плоскостями, взаимно погасят друг
друга.
Уравнение B) указывает на воз-
возможность определения межплоскост-
межплоскостных расстояний в кристаллах, если
известны длины волн рентгеновских
лучей X, порядок отражения п и уг-
углы скольжения 8. Для первого отра-
отражения d=V2sin.8i. После открытия
Лауэ и вывода основной формулы
рентгеновского анализа Бреггами и
Вульфом последовало чрезвычайно
быстрое развитие структурного ана-
анализа. С помощью рентгеновских лу-
лучей В. Г. и В. Л. Бреггам удалось
определить межатомные расстояния
в кристаллах и взаимное расположе-
расположение атомов для целого ряда ве-
веществ, т. е. определить их кри-
кристаллическую структуру. Одной из
первых была определена структура
меди.
Кристаллы меди принадлежат к
кубической сингонжи. Для опреде-
определения структуры кубического кри-
кристалла необходимо найти расстоя-
расстояния d между плоскими сетками ку-
куба {100}, ромбического додекаэдра
{110} и октаэдра {111}. Этих трех
величин вполне достаточно, чтобы
однозначно определить тип решетки
Бравэ.
Отношение межплоскостных рас-
расстояний для простой кубической ре-
решетки (рис. 138) следующее:
a У 2 а VI
rfiou ¦ duo '¦ dux — а, : g • 3 —
= 1 : 0,71 : 0,58;
для гранецентрированной кубиче-
кубической решетки:
а_ a V2 а УЪ
Лоо : dno : dm == 2 ' 4 ' 3
= 1 : 0,71 : 1,15;
для объемноцентрированной куби-
кубической решетки:
а а У 2 а /3
dvx, : duo '¦ dm = ~<Г: 2 ' 6
= 1 : 1,41 ; 0,58.
Рис. 137. Отражение рентгеновских лучей
от серии плоских сеток кристалла
л
1
1
[id
Z—
\
1
Л t
—— Д •-
-в
Рис. 138. Межплоскостные расстояния
для простой кубической решетки
Рис. 139. Структура меди
Из рентгенограммы получены та-
такие величины:
dm: dm : dm = 1.80А : 1.28А : 2.07А =
= 1 : 0,71:1,15.
Эти отношения удовлетворяют гра-
гранецентрированной решетке с длиной
ребра куба а=3,60А (рис. 139).
После определения типа и разме-
размеров ячейки Бравэ необходимо под-
подсчитать число атомов (или моле-
молекул — для сложных веществ), входя-
входящих в эту ячейку. Для этого надо
воспользоваться плотностью веще-
вещества. Число атомов п в ячейке опре-
определится по формуле
n =
где р — плотность вещества, М —
ого молекулярный (атомный) вес,
у _ объем ячейки Бравэ. Число
1,66-10~24 есть вес атома водорода
в граммах.
Подставив в эту формулу значе-
значения для меди, получим:
C,60K-10-24-8,96 _
1,66. КГ24- 63,6 ~ ' "
107
Число атомов в ячейке должно
быть обязательно целым. Из приве-
приведенного расчета видно, что для
структуры меди оно равно 4. На од-
одну гранецентрированную ячейку,
как легко убедиться, приходится 4
узла. Поэтому единственно возмож-
возможным распределением атомов меди в
гранецентрированной ячейке явля-
является распределение их по узлам ре-
решетки.
Каждый узел решетки занят од-
одним атомом меди. В сложных струк-
структурах с одним узлом решетки часто
бывает связана целая группа ато-
атомов.
Следует заметить, что вышепри-
вышеприведенная формула может быть ис-
П0льзована для определения моле-
молекулярного веса вещества.
§2. Кристалл
как дифракционная решетка
Из элементарного рассмотрения
дифракционной картины известно,
что если падающие лучи П (рис.
140) расположены под углом i к ли-
линии отверстий А\, А2... Ап, располо-
расположенных на растоянии а друг от дру-
друга, то дифрагированные лучи будут
распространяться в направлении
ДД в том случае, если разность хода
СА% — А]В будет равна целому числу
длин волн света п%, т. е.
a (cos ф — cos i) = rik,
C)
где п может принимать последова-
последовательно значения 0,1,2,3, и т. д. в зави-
зависимости от изменения угла i. Таким
образом, если известна длина волны
К, порядок отражения п и углы па-
падающего (i) и дифрагированного ср-
луча, то по формуле C) легко опре-
определить расстояние между отверс-
отверстиями — период идентичности.
Аналогичным образом можно
представить себе дифракцию рент-
рентгеновских лучей от одномерного
кристалла, т. е. от ряда атомов, рас-
расположенных на прямой через рав-
равные промежутки (рис. 141). Одна-
Однако, поскольку направление в пло-
плоскости чертежа ничем не отличается
от других направлений, расположен-
расположенных под углом ф к линии атомов
А\Ач...Ап, то дифрагированные лучи
будут образовывать конус вокруг
этой линии. Угол конуса будет за-
зависеть от величины п (числа длин
волн, укладывающихся в величине
разности хода, на рис. 141 п= 1).
Так как условие дифракции будет
выполняться для разных п, то мы
будем иметь не один дифракцион-
дифракционный конус, а несколько (рис. 142).
Рис. 140. Условие отражения лучей от ди-
дифракционной решетки
Рис. 141. Дифракция рентгеновских лучей
атомным рядом
п=2
Рис. 142. Дифракционные конусы, созда-
создаваемые атомным рядом
108
Дифракция от плоской сетки по-
показана на рис. 143. В общем случае
сетка определяется двумя периода-
периодами идентичности а и b и углом у.
Показаны один дифракционный ко-
конус от ряда А и один от В.
Первому условию отвечает урав-
уравнение
¦I (COS фа — COS h) = пк,
второму
Ь (cos фг — cos h) — тк.
Для дифракции от двухмерной
сетки оба условия должны удовлет-
удовлетворяться одновременно. В этом слу-
случае сетка рассеивает в одной и той
ж© фазе. Дифракционными направ-
направлениями будут линии пересечения
обоих конусов (рис. 143).
В случае дифракции от трехмер-
трехмерного (т. е. реального) кристалла
должны одновременно выполняться
три условия:
a (cos ф1 ¦— cos i\) = пк,
Ь (cos ф2 — cos h) = тк,
«(cos фз — cos г'з) = IX.
D)
Однако выполнения их в общем
случае нет, так как пересечение
третьей системы конусов совсем не-
необязательно будет проходить по тем
же линиям, что и первых двух.
Для того чтобы вое три условия
удовлетворялись одновременно, т. е.
чтобы пространственная решетка
рассеивала рентгеновские лучи в
одной и той же фазе, надо в каком-
то интервале непрерывно менять
либо длину волны рентгеновских
лучей, либо углы падения. В первом
cnyqae для получения рентгено-
рентгенограммы используется не характери-
характеристическое (монохроматическое) из-
излучение, а «белое» (не монохрома-
монохроматическое) (метод Лауэ). Недостат-
Недостатком его является то, что лучи, соот-
соответствующие дифракционным мак-
максимумам и дающие на рентенограм-
ме темные пятна, имеют разные
длины волн, что очень осложняет
Рис. 143. Дифракционная картина, созда-
создаваемая атомной сеткой
учет всех факторов, влияющих на
интенсивность пятен.
Во втором случае используются
монохроматические рентгеновские
лучи, но кристалл медленно и рав-
равномерно вращается вокруг оси, сов-
совпадающей с каким-либо кристалло-
кристаллографическим направлением (метод
вращающегося кристалла). Тогда
при каких-то особых положениях,
при особых углах, удовлетворяющих
сразу трем уравнениям D), возни-
возникает кратковременная «вспышка» —
дифрагированный луч, оставля-
оставляющий на рентгенограмме след в ви-
виде темного пятна. Этот метод не
имеет того недостатка, которым об-
обладает метод Лауэ. Поэтому он ис-
используется в рентгеноструктурном
анализе гораздо шире. Метод Лауэ
обычно используется только для оп-
определения симметрии кристалла или
для ориентировки неограненного
кристаллического осколка.
§3. Методика определения
параметров н типа решетки
Наиболее удобным методом опреде-
определения параметров решетки являет-
является метод вращения. В этом методе
кристалл помещается в цилиндри-
цилиндрическую камеру. Какое-либо его кри-
кристаллографическое направление
(пусть для конкретности это будет
ось с) совмещается с осью цилинд-
цилиндра. Во время съемки фотографиче-
фотографическая пленка прилегает к внутрен-
109
ней стороне камеры. Монохромати-
Монохроматический рентгеновский луч направ-
направлен перпендикулярно к оси враще-
вращения.
Дифрагированные лучи должны
лежать на семействе конусов (рис.
144), удовлетворяющих условию
С COS ф = пк,
где с — период идентичности кри-
кристалла в направлении, совпадающем
с осью вращения. Вершина конуса
совпадает с кристаллом, а ось —
с осью вращения (рис. 145). Каж-
Каждый конус будет пересекать цилинд-
рическую пленку по прямой, назы-
называемой слоевой линией. Углы <р оп-
определяются по расстоянию эквато-
экваториальной (нулевой) слоевой линии
до первой п= 1, до второй п = 2 и т. д.
Диаметр камеры, конечно, должен
быть известен. Слоевая линия будет
не сплошной, как было бы при ди-
дифракции от одномерного кристалла,
а прерывистой, т. е. состоящей из
отдельных пятен. Каждое пятно
удовлетворяет не одному C), а сра-
сразу трем условиям D), как это было
сказано в предыдущем параграфе.
Рентгенограмма вращения пока-
показана на рис. 146. По ней, пользуясь
формулой B), легко определить пе-
периоды идентичности кристалла, т. е.
параметры его решетки.
Для определения типа решетки не-
необходимо знание параметров а, Ъ и с.
Затем получают рентгенограммы вра-
вращения вдоль плоских диагоналей гра-
граней и по пространственной диагонали.
В случае центрировки решетки по
некоторым из зтих направлений па-
параметр окажется вдвое меньшим, чем
вычисленный из значений а, Ъ и с в
предположении примитивной ячейки.
Для определения параметров ре-
решетки нет необходимости осуществ-
осуществлять полное вращение кристалла на
360°. Для получения слоевых линий
достаточно кристалл колебать на 15—
20°. Это значительно сократит экспо-
экспозицию и ускорит работу. Такой ме-
метод съемки называется методом ко-
колебаний, или качаний.
Рис. 144. Возникновение рентгенограммы
вращения
Рис. 145. Определение периода идентич-
идентичности кристалла по рентгенограмме вра-
вращения
Рис. 146. Рентгенограмма вращения
110
Тип решетки можно определить и
по одной или двум рентгенограммам
вращения, если последние проинди-
цировать. Под индицированием пони-
понимается определение символов отра-
отражающих граней (точнее, систем пло-
плоских сеток, параллельных действи-
действительным или возможным граням
кристалла) и порядка отражения
[т.е. п в формуле B)].
На системе индицирования оста-
останавливаться не будем. Скажем толь-
только, что при вращении кристалла во-
вокруг осей а, Ъ или с соответственно
первый, второй или третий индексы
символа на нулевой слоевой линии
равны 0, на первой — 1 и т. д. При
индицировании обязательно будут
рефлексы с символом, имеющим об-
общий делитель, например 220. Такой
символ обозначает второй порядок
отражения от системы плоских сеток
A10).
В отличие от символов граней, ко-
которые ставятся в круглых скобках,
символы дифракционных максимумов
пишутся без скобок.
Можно показать, что в примитив-
примитивной решетке встречаются любые сим-
символы hkl. В объемноцентрирован-
ной — только такие, в которых
h + к + I = In (четному числу).
В гранецентрированнои присутствуют
отражения с h + к = 2п, к + I = 2п
и h-\-l=2n или, иначе говоря, отра-
отражения со всеми четными или всеми
нечетными индексами (нуль считает-
считается четным числом).
Когда эти суммы равны нечетному
числу, то элементарные дифрагиро-
дифрагированные лучи гасят друг друга, и сум-
суммарный дифракционный эффект ста-
становится в этом случае равным нулю.
Понять эту закономерность мож-
можно на простом примере (рис. 147).
Системой «черных» шаров показана
примитивная ячейка be. В верхней
части рисунка дана схема отражения.
Разность хода АВ + ВС = пХ. Если
решетку центрировать «белыми» ша-
шарами, то разность хода А'В' + В'С
будет в два раза меньше, чем
АВ + ВС, поэтому первое отражение
Рис. 147. Центрирование ячейки «белыми»
шарами вызывает погасание нечетных
порядков отражения
при угле ¦& (и любое нечетное) будет
погашено, так как разность хода бу-
будет при этом равна половине длины
волны. Четные отражения сохранят-
сохранятся. Центрирование ячейки «белыми»
узлами приводит к появлению посере-
посередине между отражающими плоско-
плоскостями из «черных» шаров новой си-
системы отражающих плоскостей из
«белых» шаров.
Другим очень распространенным
методом является метод порошка (ме-
(метод Дебая — Шеррера). Образец в
этом случае приготовляется в виде то-
тоненького цилиндрика из спрессованно-
спрессованного порошка и облучается монохро-
монохроматическим рентгеновским пучком
лучей. Рентгенограмма получается в
этом случае потому, что среди массы
кристаллических зерен, беспорядочно
расположенных в цилиндрическом
образце, всегда найдется какое-то ко-
количество зерен под таким углом, ко-
который удовлетворяет одновременно
трем условиям D). Ясно, что слое-
слоевых линий при этом уже не получит-
получится, а получатся сплошные конуса
с углом в 4г% осью которых будет
рентгеновский пучок (рис. 148). В пе-
пересечении с цилиндрической пленкой
такая система коаксиальных конусов
дает систему кривых, изображенных
на рис. 149. Это так называемая де-
баеграмма, или рентгенограмма по-
порошка. Проиндицировав такую рент-
рентгенограмму, также можно определить
размер и тип решетки Бравэ. Этот
метод удобен тем, что не требует об-
111
Рис. 148. Возникновение дебаеграммы
Рис. 149. Схема дебаеграммы А1
разца в виде монокристалла, но он
имеет и существенные недостатки,
так как такую рентгенограмму гораз-
гораздо труднее проиндицировать. Это
удается довольно легко для высоко-
высокосимметричных кристаллов (кубиче-
(кубических) , труднее — для кристаллов
средних сингоний и не всегда воз-
возможно для низкосимметричных
кристаллов.
§4. Методика оиределеиия
пространственных групп
симметрии
Если рассматривать пространствен-
пространственные группы одного вида симметрии
(см. табл. 7 и рисунки 105—135), то
легко обнаружить, что они отличают-
отличаются друг от друга наличием тех или
иных плоскостей скользящего отра-
жепия и винтовых осей.
Эти элементы симметрии дают на
рентгенограммах характерные погаса-
погасания. Под этим термином подразумева-
подразумеваются отсутствующие на рентгено-
рентгенограммах пятна определенного симво-
символа. С системой «погашенных» отра-
отражений мы встречались уже при
определении типа решетки. Как было
сказано в предыдущем параграфе, в
примитивной решетке возможны лю-
любые отражения hkl, а в объемноцен-
трированной только такие, которые
удовлетворяют условию h-\-k + l=2n.
В этой решетке пятно с символом 100
будет отсутствовать, но может при-
присутствовать пятно с символом 200. От
сеток, параллельных граням (НО), на
рентгенограммах могут присутство-
присутствовать пятна как первого порядка 110,
так и второго и более высоких: 220,
330 и т. д.
Винтовые оси создают закономер-
закономерные погасания среди отражений от
систем плоских сеток, перпендику-
перпендикулярных к ним. Так, двойная винтовая
ось, параллельная оси Z кристалла,
даст погасание тех отражений 00/,
при которых I будет нечетным чис-
числом; четверная винтовая ось даст по-
погасания в направлении 00Z всех от-
отражений, за исключением тех, у ко-
которых I кратно четырем. Это объясня-
объясняется тем, что винтовые оси создают
дополнительные, «вставленные» в
ячейку плоскости, отражающие рент-
рентгеновские лучи. На рис. 150 эти до-
дополнительные плоскости, перпенди-
перпендикулярные к винтовым осям, показа-
показаны пунктиром. По этой же причине
плоскости скользящего отражения
также создают закономерные погаса-
погасания в системе плоских сеток с симво-
символами (МО), (МЛ) или @Ы). В насто-
настоящее время имеются хорошо разрабо-
разработанные схемы, позволяющие по
наличию на рентгенограммах харак-
характерных погасаний определить про-
пространственную группу симметрии.
Из последних примеров видим, что
для определения пространственных
групп симметрии наибольшее значе-
значение имеют отражения от серий пло-
плоских сеток, параллельных координат-
координатным плоскостям. На рентгенограмме
вращения (рис. 146) такой плоскости
отвечает нулевая (экваториальная)
слоевая линия. По этой причине в
рентгеноструктурном анализе часто
производится съемка только таких
112
Рис. 150. Действие винтовых осей вызыва-
вызывает закономерное погасание отражений
нулевых (для каждой из координат-
координатных осей) слоевых линий. Есть спо-
способ съемки, при котором все слоевые
линии, за исключением одной, могут
быть отгорожены ширмой, и пятна от
них на фотопленку не попадают. Ка-
Камера устроена таким образом, что
нулевая слоевая линия рентгенограм-
рентгенограммы вращения разворачивается на всю
плоскость пленки. Для этой цели в
камере не только вращается кристалл,
но движется или вращается пленка.
Это так называемые методы разверт-
развертки слоевых линий. Камеры называ-
называются рентгенгониометрами. При такой
съемке весьма облегчается процесс
индицирования рентгенограмм. Этот
метод съемки является поэтому наи-
наиболее распространенным при исследо-
исследовании атомных структур сложных ве-
веществ, обычно дающих низкосим-
низкосимметричные кристаллы.
§5. Определение положения атомов
в кристаллической решетке
После определения параметров ре-
решетки, ее типа и пространственной
группы кристалла можно приступить
к определению положения атомов.
Рис. 151. Замена атомной решетки на мо-
молекулярную влияет лишь на интенсив-
интенсивность отражений
Для этой цели необходимо прежде
всего рассмотреть вопрос, как будут
отражаться рентгеновские лучи от
плоскостей кристаллической решетки,
если в последней имеется несколько
типов атомов и, следовательно, они
не располагаются в узлах решетки.
На рис. 151, а изображена решетка
D ячейки), на рис. 151, б те же ячей-
ячейки решетки заполнены молекулами
вещества состава АВ («черные» и «бе-
«белые» шары). Рассмотрим какое-либо
отражение от диагональной серии
плоскостей с межплоскостным рас-
расстоянием d, стеды которых перпен-
перпендикулярны плоскости чертежа.
Допустим, что на решетку (а) пада-
падают лучи под таким углом 0, что про-
происходит отражение от серии плоско-
плоскостей с межплоскостным расстояни-
расстоянием d. В сложной структуре (б) под
таким же углом 0 также будет про-
происходить отражение, так как этот
угол зависит только от величины
межплоскостного расстояния d (оди-
(одинакового для случаев а и б). Разница
будет заключаться в следующем. Для
всех плоскостей одной системы (на-
(например, А) отраженные лучи будут
находиться в одинаковой фазе друг
с другом. То же, конечно, будет и в
отношении системы В плоскостей.
Однако отражения от системы А и
системы В, в общем случае не будут
находиться в одинаковой фазе, так
что общая амплитуда волн, отражен-
отраженных от обеих систем плоскостей, бу-
будет меньше, чем в том случае, если бы
атомы А и В располагались в одних
и тех же плоскостях в испытуемом
направлении, т. е. если бы величина
х была равна 0. Если величина х бу-
будет равна 1/zd, то отражения четного
порядка от плоскостей А и В будут
иметь одинаковую фазу, а отражения
нечетного порядка будут иметь про-
противоположные фазы и поэтому будут
погашены, т. е. пятна на рентгено-
рентгенограмме будут отсутствовать, если А
и В — атомы одного и того же эле-
элемента. Если атомы А и В принадле-
принадлежат разным элементам, то такие от-
отражения будут максимально ослаб-
8 Кристаллохимия
113
ленными, но не равными нулю. Это
обстоятельство связано с тем, что ин-
интенсивность отраженных от атомов
лучей зависит от числа электронов
атома, равного атомному номеру эле-
элемента.
Это так называемый атомный фак-
фактор /, который всегда необходимо
учитывать при проведении рентгено-
структурного анализа, fxФfъ, если
А и В — разные химические элемен-
элементы. Величина / характеризует рас-
рассеивающую способность атома и за-
зависит от числа л распределения в нем
электронов.
В общем случае х Ф 1Ш. Разность
фаз между лучами, отраженными от
плоскостей А и В, для отражения
первого порядка будет 2nx/d, для
п-го отражения 2nnx/d. Из рассмот-
рассмотренного примера видно, что число и
расположение пятен на рентгенограм-
рентгенограмме зависит от размеров ячейки, типа
решетки и пространственной группы.
Интенсивность пятен зависит от ко-
количества, типа и взаимного располо-
расположения атомов в решетке.
В разобранном примере предпола-
предполагалось, что для атомных плоскостей
всех типов отраженные волны имеют
одинаковые начальные фазы. В дей-
действительности же, в общем случае,
имеет место сдвиг начальных фаз для
разных атомных плоскостей, завися-
зависящий от координат этих атомов или
соответственно от места выбора нача-
начала координат в элементарной ячейке.
Поэтому и начальная фаза резуль-
результирующего луча остается нам неиз-
неизвестной.
В центросимметричном кристалле,
если начало координат расположить
в центрах симметрии, происходит та-
такая компенсация сдвигов фаз, при ко-
которой начальные фазы принимают
значения 0 или я. Поэтому проблема
неопределенности начальных фаз пе-
переходит в проблему определения зна-
знаков результирующих (суммарных для
всех сортов атомов) структурных
амплитуд, что значительно упрощает
весь ход рентгеноструктурного ана-
анализа.
Существует хорошо разработанная
система подсчета интенсивностей от-
отраженных рентгеновских лучей, если
известно положение атомов, в решет-
решетке. Поэтому, если мы на основании
каких-либо соображений можем сде-
сделать предположение о размещении
атомов в ячейке, то правильность та-
такого предположения можно прове-
проверить. Для этого рассчитывают интен-
интенсивность всех рефлексов, исходя из
сделанного предположения о разме-
размещении атомов, и сравнивают с экспе-
экспериментально измеренными интенсив-
ностями пятен на рентгенограмме.
Если совпадение есть, то выдвинутый
нами в качестве предположения ва-
вариант расположения атомов в ячейке
правилен, если совпадения нет — вы-
выдвинутый вариант неправилен. Сле-
Следует сделать другое предположение
о расположении атомов в ячейке и
опять подвергнуть его проверке рас-
расчетом. При выдвижении варианта
надо учитывать федоровскую группу
симметрии и возможные для нее пра-
правильные системы точек. Такой метод
расшифровки структур кристаллов
получил название метода проб и оши-
ошибок. Он был долгое время единствен-
единственным методом рентгеноструктурного
анализа.
§6. Гармонический метод
рентгеиоструктурного анализа
Если амплитуды волн в каком-либо
направлении hkl и относительные фа-
фазы для каждого рода атомов извест-
известны, как, например, в случае, когда
известны координаты атомов в ячей-
ячейке, то можно вычислить и результи-
результирующую структурную амплитуду Fhk[
по формуле сложения простых гар-
гармонических колебаний, имеющих
одинаковую частоту:
F*=(fA cosq>A +/B c0S(Pb + • • •)" +
+(/А sin фА + /в sin фв +.. .)*, E)
где фд — фаза для атомов, А, фв — фа-
фаза для атомов В.
114
Если кристалл имеет центр инвер-
инверсии и начало координат выбрано в
нем, то формула E) значительно
упрощается, так как в этом случае
синусная часть ее пропадает:
/•;= 2/А cosq>A +2/B coscpB + • • •
Как было сказано в предыдущем
параграфе, сопоставляя вычисленные
значения структурных амплитуд с
экспериментальными (с интенсивно-
стями_ / пятен рентгенограммы,
F ~ "|/7), можно проверить правиль-
правильность выдвинутого варианта струк-
структуры. Возможны и другие расчеты,
в частности появляется возможность
вычислить распределение электрон-
электронной плотности в кристалле.
Как впервые доказал В. Л. Брегг,
поскольку электронная плотность в
кристалле является периодической и
непрерывной функцией, то она может
быть разложена в ряд Фурье. Теоре-
Теоретический расчет показал, что коэффи-
коэффициентами в этом ряду являются
структурные амплитуды.
Следовательно, если структурные
амплитуды известны, то можно под-
подсчитать электронную плотность в
каждой точке элементарной ячейки.
Максимумы (сгустки) плотности ука-
укажут на местонахождение атомов.
При подсчете электронной плотно-
плотности нет необходимости считать ряд
для очень большого числа точек в
ячейке. Практика показывает, что для
подсчета распределения электронной
плотности в кристалле обычно доста-
достаточно каждую координатную ось эле-
элементарной ячейки разделить на 48
(или 60) частей. Тогда весь объем
ячейки окажется разбитым на 483 па-
параллелепипедов. Вычислив электрон-
электронную плотность для каждого из них,
находим места сгустков электронов,
определяющих положения атомов.
Обычно для сокращения работы
вычисляют электронную плотность
не по всей ячейке, а только в проек-
проекциях на две координатные плоскости.
Из соответствующих двух проекций
можно получить 3 координаты для
каждого атома, т. е. однозначно опре-
определить его положение в пространстве.
Выражение для проекции электрон-
электронной плотности на плоскость X у эле-
элементарной ячейки центросимметрич-
ного кристалла имеет вид:
h к
—со
F)
где S — площадь проекции ячейки;
хну — доли ячейки.
Положительный знак структурной
амплитуды означает, что начальная
фаза отраженного луча равна нулю,
т. е. совпадает с фазой луча, исходя-
исходящего из начала координат элементар-
элементарной ячейки (узла решетки). Отрица-
Отрицательный знак структурной амплитуды
означает противоположность фазы
луча, отраженного кристаллом, и лу-
луча, исходящего из начала координат.
На полученной проекции центры
клеток, имеющих равные значения
р, соединяются линиями так, как это
делается на топографических картах,
где линии соединяют разные высоты.
Высокие максимумы электронной
плоскости, аналогичные горным вер-
вершинам на топографической карте,
Рис. 152. Проекция электронной плотно-
плотности [PtfNHshCUj-ifuc на плоскость YZ
8*
115
соответствуют центрам атомов (рис.
152).
К сожалению, метод анализа Фу-
Фурье пока не является прямым, так
как из эксперимента (из рентгено-
рентгенограмм) можно получить лишь абсо-
абсолютные значения структурных амп-
амплитуд \Fhko\, а начальные фазы
(знаки) заранее неизвестны. По этой
причине нельзя найти координаты
атомов, подставляя в ряды Фурье
значения |^щ)|, полученные из из-
измеренных интенсивностей. А знаки
можно найти только тогда, когда за-
заранее известно положение атомов.
Таким образом мы можем решить
только обратную задачу и, следова-
следовательно, использовать F-ряды лишь
для проверки и дальнейшего уточ-
уточнения выдвинутого варианта (см.
ниже). В этом случае метод Фурье
почти не имел бы преимуществ по
сравнению с методом проб и ошибок.
В настоящее время имеется ряд ра-
работ, посвященных проблеме опреде-
определения знаков структурных амплитуд.
Окончательное решение этой проб-
проблемы сделает метод построения ря-
рядов электронной плотности прямым
методом рентгеноструктурного ана-
анализа.
В настоящее время получил боль-
большое распространение метод построе-
построения .Р2-рядов, предложенный Паттер-
соном. Формула ^2-ряда для проек-
проекции на плоскость ху имеет вид:
- 2 2
h к
cos
h -
Рис. 153. Происхождение максимумов
на проекции Паттерсона
о — проекция структуры;
б — проекция Паттерсона
Направление и расстояние максиму-
максимумов от начала координат соответст-
соответствуют всем межатомным векторам
структуры.
На рис. 153, а извбражена проек-
проекция элементарной ячейки некоторой
гипотетической структуры, содержа-
содержащей 4 атома. На рис. 153, б показано
расположение максимумов в проек-
проекции Паттерсона, соответствующей
этой структуре. Как видно, количест-
количество максимумов значительно возросло
[вместо N стало N(N—1)]. Обрат-
Обратный переход от б к а является до-
довольно сложной задачей. Поэтому рас-
расшифровка проекций рядов Паттер-
В этом случае нет необходимости
знать что бы то ни было, кроме того,
что дает эксперимент. Недостаток
метода Паттерсона заключается в
том, что максимумы на проекции
^2-ряда уже не отвечают положени-
положениям атомов. Число таких максимумов
гораздо больше числа атомов в ячей-
ячейке. Эти максимумы определяются
атомными факторами пар атомов А „ ,к, _
и В, А и С, В и С и т. д. и вектор- *?j*^ ГтТрсонаГ'М"~ ^
ными расстояниями между ними, на плоскость YZ
А ш
ии
116
сона представляет часто весьма боль-
большие трудности.
В ряде случаев, однако, примене-
применение этого метода дает блестящие ре-
результаты. Так, если в химической
формуле исследуемого вещества один
атом — тяжелый, а остальные — лег-
легкие, как это имеет место в большин-
большинстве комплесных соединений, напри-
например Pt(NH3JCl4, то задача опреде-
определения структур решается однознач-
однозначно, так как высота максимумов на
паттерсоновской проекции пропор-
пропорциональна произведению атомных
номеров. Среди многих максимумов,
отвечающих межатомным векторам,
соединяющим тяжелый атом с лег-
легкими, можно без труда найти боль-
больший максимум, отвечающий меж-
межатомному расстоянию между тяжелы-
тяжелыми атомами (во взятом примере это
будет вектор Pt—Pt, рис. 154).
Зная положение в ячейке тяжело-
тяжелого атома, определяем знаки струк-
структурных амплитуд ряда Фурье, сде-
сделав предположение, что они обуслов-
обусловлены именно тяжелым атомом. По-
Построением проекции ряда Фурье мо-
могут быть найдены максимумы для сле-
следующего по атомному номеру эле-
элемента. В нашем примере это будет
С1. После этого по приблизительным
координатам атома С1 вносятся по-
поправки в знаках для некоторого ко-
количества структурных амплитуд, не-
неправильно рассчитанных при учете
положения только одного (тяжелого)
атома. Затем можно построить вто-
вторую, более точную проекцию элек-
электронной плотности. В зависимости
от сложности структуры этот прием
может повторяться большее или
меньшее число раз (обычно не более
3—4 раз). В этом заключается су-
существо метода последовательных при-
приближений, в настоящее время явля-
являющегося основным методом рентге-
ноструктурного анализа.
Все эти расчеты чрезвычайно тру-
трудоемки. Для их ускорения в послед-
последние годы стали широко применяться
быстродействующие электронные вы-
вычислительные машины. В результате
значительного усовершенствования
методики рентгеноструктурного ана-
анализа и вычислительной техники в
последние годы расшифрованы струк-
структуры многих сложных химических
соединений. Результаты этих иссле-
исследований являются фактическим ма-
материалом современной кристаллохи-
кристаллохимии.
ЧАСТЬ ТРЕТЬЯ
ОСНОВНЫЕ ПОНЯТИЯ
КРИСТАЛЛОХИМИИ
Г Л А В А IX
РЕЗУЛЬТАТЫ ПЕРВЫХ
РЕНТГЕНОСТРУКТУРНЫХ ИССЛЕДОВАНИЙ КРИСТАЛЛОВ
$1. Три простейшие
кристаллические структуры
чистых металлов
Одной из первых структур, опреде-
определенных методом рентгеновского ана-
анализа, была структура меди.
Проведенное исследование показало,
что в структуре меди решетка Бравэ
является гранецентрированной куби-
кубической. Длина ребра кубаа=3,61А*.
На одну элементарную ячейку при-
приходится четыре атома. Поскольку
число узлов в кубической гранецен-
трированный ячейке тоже равно че-
четырем, то единственным возможным
расположением атомов меди в кри-
кристаллической структуре будет распо-
расположение их по узлам решетки (рис.
155). Аналогичную структуру имеют
• Данные по размерам решеток и меж-
межплоскостных расстояний выражаются в
ангстремах AА = 1~8 см). В 1946 г. было
введено обозначение кХ для прежней
единицы неточного ангстрема, встречаю-
встречающегося в работах, вышедших из печати
до 1947 г. 1А = кХ • 1,00202. В дальней-
дальнейшем в тексте значок А будет во многих
случаях опускаться.
многие другие металлы, например,
Au(a=4,07A), Ag(a=4,08), Al(a=
=4,04), Pt(a=3,92) и т. д.
Структура a-Fe показана на
рис. 156. Элементарная ячейка
этой структуры — объемноцентриро-
ванный куб, сторона которого равна
2,86 А. На одну ячейку приходятся
два атома (кратко это записывается
п=2). Поскольку каждая ячейка со-
содержит два узла, то, как и в струк-
структуре меди, единственным возможным
расположением атомов в структуре
является расположение их по узлам
решетки. Аналогичную структуру
имеют и другие металлы, например
Na(a=4,28), К(а=5,33), Ва(а =
=5,01), |5-Ti(a=3,32A) и т. д.
Третьей важнейшей структурой
будет структура Mg (рис. 157).
Ячейка магния гексагональная; а —
=3,20 А, с=5,20А. На каждую ячей-
ячейку приходится шесть атомов: три
располагаются в вершинах и в цент-
центре базисных граней и три — в цент-
центрах тех трех (из шести) тригональ-
ных призм, на которые можно мыс-
мысленно разбить гексагональную ячей-
ячейку (рис. 157, а). При этом «заселен-
118
ные» и «пустые» призмы чередуются
между собой.
Нужно заметить, что выбор начала
координат в структуре в известной
мере произволен, поэтому структуру
магния можно себе представить ина-
иначе, если придать исходному атому
магния координаты не @00), как
показано на рисунке, а Gз 2/з lU),
как это обычно принято.
Аналогичную структуру имеют
а-Со(а = 2,51; с = 4,07 А) а-Ве(а =
=2,28; с=3,58 А) и т. д. Часто стру-
структуры гексагональных кристаллов
изображают не полной гексагональ-
гексагональной ячейкой, а примитивным парал-
параллелепипедом, составляющим одну
треть ее (рис. 157, б).
Федоровские пространственные
группы симметрии трех вышеопи-
вышеописанных структур следующие: для
Си — Fm3m, для a-Fe — 1тЗт и для
Mg —
Рис. 155. Структура меди
Рнс. 156. Структура a-Fe
а °... I I I ~к
Рис. 157. Структура магния
о—полная гексагональная ячейка;
б — примитивная ячейка
§2. Число атомов, нряходящихея
на одну ячейку структуры
Чтобы правильно понимать структу-
структуру кристаллов, необходимо научить-
научиться подсчитывать на пространствен-
пространственной модели или по рисунку число
атомов каждого химического элемен-
элемента, приходящихся на одну элемен-
элементарную ячейку.
Так, например, на одну гранецент-
рированную кубическую ячейку меди
(рис. 155) приходятся четыре атома.
Каждый атом, расположенный в вер-
вершине элементарного куба, принадле-
принадлежит восьми ячейкам (рис. 158). Вер-
Вершин у куба 8, следовательно, от ато-
атомов, располагающихся в них, на долю
той ячейки, которую мы избрали
в качестве исходной, приходится
8 • 7в = 1 атом. Каждый атом, распо-
располагающийся в центре грани, принад-
принадлежит двум ячейкам. Следовательно,
6 атомов, расположенных в центрах
граней, дадут на избранную ячейку
6 • 7г=3 атома. Итак, всего на каж-
каждую ячейку структуры приходится
4 атома.
В структуре a-Fe одна ячейка со-
содержит только два атома. Один полу-
получаем из восьми атомов, расположен-
расположенных по вершинам ячейки, а второй,
расположенный в центре ячейки,
принадлежит ей целиком.
В структуре Mg на полную гекса-
гексагональную ячейку приходится шесть
атомов. Каждый из двенадцати ато-
атомов, расположенных в вершинах
гексагональной призмы, принадле-
принадлежит шести ячейкам (рис. 159). Они
дают 2 атома на ячейку A2-7б)-
Два атома, расположенных в цент-
центрах базисных граней, принадлежат
одновременно двум ячейкам и дают
в сумме еще один атом B-72). Три
внутренних атома целиком принад-
принадлежат одной ячейке. Всего, следо-
следовательно, на ячейку приходится
шесть атомов B+1 + 3).
Если в качестве ячейки выбран
примитивный параллелепипед, рав-
равный по объему 7з гексагональной
ячейки, то он содержит 2 атома. Это
119
/
/
/
/
/1
л
ш
/
7
/
7
/
Рис. 158. Принадлежность каждого узла,
находящегося в вершине кубической ячей-
ячейки, восьми соседним ячейкам
Рис. 159. Принадлежность каждого узла,
находящегося в вершине гексагональной
ячейки, шести соседним ячейкам
можно было бы подсчитать и непо-
непосредственно по рис. 157, б. Каждый
из восьми атомов, располагающихся
по вершинам примитивного парал-
параллелепипеда, принадлежит восьми та-
таким ячейкам, все они дают в сумме i
один атом на ячейку. Второй атом
целиком находится внутри паралле-
параллелепипеда.
§ 3. Число правильных систем
точек в структуре
Подсчитывая число атомов, прихо-
приходящихся на одну ячейку, мы разби-
разбивали их на группы, расчеты для ко-
которых вели раздельно. Так, напри-
например, в структуре меди мы разбили
4 атома на две группы — 1 и 3; два
атома железа тоже разбили на две
группы — по одному в каждой;
шесть атомов магния разбили на три
120
группы — 2, 1 и 3. Не следует ду-
думать, что эти атомы в каком-то
смысле отличаются друг от друга.
Тождественность всех атомов в
структурах Си и a-Fe легко доказы-
доказывается тем, что они располагаются
по узлам соответствующих реше-
решеток и могут быть совмещены
друг с другом в результате простых
трансляций. В структуре магния
они также тождественны, хотя и
располагаются не только по верши-
вершинам примитивных параллелепипе-
параллелепипедов повторяемости. Все эти атомы
связаны друг с другом другими эле-
элементами симметрии — осями сим-
симметрии, центрами симметрии или
плоскостями скользящего отраже-
отражения,—с помощью которых они могут
быть совмещены друг с другом. Все
они располагаются по точкам одной
правильной системы и потому должны
считаться кристаллохимически оди-
одинаковыми. Этот результат для начи-
начинающего изучать кристаллохимию
может показаться само собой разу-
разумеющимся и потому излишним. Од-
Однако такой поспешный вывод делать
не следует, так как в последующих
главах книги будут приведены при-
примеры структур простых веществ,
в которых атомы действительно кри-
кристаллохимически различны. Они,
как говорят в кристаллохимии, «не-
«нескольких сортов». Так, например,
в структурах графита, (З-Мп и мно-
многих других веществ имеются атомы
двух сортов. Любой атом одного сор-
сорта не может быть совмещен с ато-
атомом другого сорта никакими сим-
симметрическими преобразованиями.
Они кристаллохимически различны
и принадлежат к разным правиль-
правильным системам точек. В некоторых
случаях у таких атомов м»жет быть
различно и электронное строение.
§4. Структура кристалла
и структурный тип
Под структурой кристалла мы пони-
понимаем конкретное пространственное
расположение материальных частиц
(атомов, ионов, молекул). На рис.
155, 156 и 157 показаны структуры
Си, a-Fe и Mg. На каждом рисунке
указан масштаб, с помощью которо-
которого на пространственных моделях,
изображенных на этих рисунках,
можно определить расстояние между
двумя любыми интересующими нас
атомами. Так, для структуры меди
кратчайшее расстояние между дву-
двумя атомами будет равно 2,55А. Для
структуры золота это расстояние
будет равно 2,87А. Изменив соответ-
соответственно масштаб рис. 155, мы полу-
получили бы структуру золота. Если нас
интересуют не абсолютные межатом-
межатомные расстояния, а лишь относитель-
относительное расположение атомов или атом-
атомных групп в кристаллах, то мы го-
говорим о структурном типе. Рис. 155
мог бы изображать структурный тип
меди, для этого пришлось бы толь-
только отбросить масштаб. В структур-
структурном типе меди кристаллизуются,
кроме золота, серебро, У"желез° и
многие другие простые вещества.
Все эти структуры одинаковы с
точностью до подобия. Однако это
имеет место только для простейших
структур кристаллов кубической
сингонии. В структурных типах дру-
других сингоний сохранение подобия
параллелепипедов повторяемости не
обязательно, обязательным являет-
является сохранение симметрии. Так, на-
например, к структурному типу маг-
магния принадлежит как (З-Са (а=3,98;
с = 6,52 A; cja = 1,65), так и а-Ве
(а=2,28; с=3,58 А; ф= 1,57).
Структурный тип именуют обычно
по названию одного из веществ, кри-
кристаллизующихся в нем. В литера-
литературе термин «структура» часто ис-
используется как синоним термина
«структурный тип». В следующих па-
параграфах дается описание важней-
важнейших структурных типов.
§ S. Структуры алмаза в графита
Структура алмаза изображена
на рис, 160. Ее можно описать сле-
следующим образом: элементарная ку-
Рис. 160. Структурный хил алмаза
бическая ячейка мысленно разбива-
разбивается на 8 малых кубов (октантов)
тремя взаимно перпендикулярными
плоскостями, проходящими через
центр ячейки параллельно ее граням.
Во всей элементарной кубической
ячейке алмаза располагаются восемь
атомов. Половина из них D атома)
занимает узлы гранецентрирован-
ной кубической решетки, а вторая
половина D атома) располагается
в центрах четырех октантов (из
восьми). По каждому координатному
направлению «заселенные» октанты
равномерно чередуются в структуре
с «незаселенными». Федоровская
(пространственная) группа струк-
структуры алмаза — FdSm. Все атомы
углерода располагаются по точкам
одной правильной системы, следо-
следовательно, они кристаллохимически
тождественны друг другу.
Необходимо отметить, что расстоя-
расстояние С — С равно 1,54 А, и их тет-
раэдрическое окружение является
аналогичным для всех предель-
предельных алифатических и алицикличе-
ских соединений.
Структура графита показа-
показана на рис. 161. Она состоит из от-
отдельных слоев. Атомы углерода в
слое расположены по вершинам пра-
правильных шестиугольников. Центры
шестиугольников остаются пустыми.
Взаимная ориентировка слоев тако-
такова, что три вершины шестиугольни-
шестиугольника одного слоя располагаются над
центром шестиугольника следую-
следующего слоя. Таким образом, полная
вертикальная трансляция равна
удвоенному расстоянию между слоя-
121
Рис. 161. Структурный тип графита
ми. Федоровская группа симметрии
графита — PQzlmmc.
Структуры, подобные графиту, в
которых расстояния между атомами
в одном слое значительно меньше
расстояния между слоями, называ-
называются слоистыми структурами.
У графита эти расстояния 1,42 и
3,39А. По аналогии со структурой ал-
алмаза структура графита является про-
прототипом ароматических соединений.
В структуре графита атомы двух
«сортов»: один сорт атомов прихо-
приходится над и под пустым гексагональ-
гексагональным кольцом соседних слоев, вто-
второй — имеет в соседних слоях по од-
одному ближайшему атому.
§6. Простейшие структуры
соединений тииа АХ
Большинство структурных типов
простейших бинарных неорганиче-
неорганических соединений с общей формулой
АХ было определено в первые годы
применения рентгеноструктурного
анализа, так как кристаллы этих сое-
соединений обычно имеют высокую сим-
симметрию — кубическую или гексаго-
гексагональную, что значительно облегчает
полное определение их структуры.
На рис. 162 изображена структу-
структура NaCl, строение которой стано-
становится понятным, если мысленно раз-
разбить элементарную кубическую ячей-
ячейку на восемь малых кубов (октантов)
и распределить атомы (ионы) натрия
и хлора по вершинам всех малых
кубов, строго чередуя их друг с дру-
другом. Нетрудно также видеть, что
отдельно взятые атомы натрия (пра-
(правильная система точек, которую зани-
занимают атомы натрия) располагаются
по узлам гранецентрированной куби-
кубической решетки. Атомы хлора распо-
располагаются по точно такому же закону.
Эти две правильные системы только
сдвинуты друг относительно друга
вдоль координатной оси на величи-
величину а. С равным основанием можно
считать «черные» шарики за атомы
натрия, а «белые» •— за атомы хлора,
или же наоборот.
В одной элементарной ячейке со-
содержится 4 атома Na и 4 атома С1.
Кратко это записывается так: п = 4.
Величина п определяет число фор-
формульных единиц (в данном случае
единиц NaCl) в ячейке. В литера-
литературе, однако, для выражения этой
величины общепринят термин «моле-
«молекула». В такой терминологии содер-
Нсится принципиальная ошибка, кото-
которая вносит. большую путаницу и ме-
мешает правильному представлению
о строении кристаллических ве-
веществ. В кристаллах поваренной со-
соли нет обособленных двухатомных
групп NaCl, которые можно было
бы назвать молекулами. Исследова-
Исследование показало отсутствие таких моле-
молекул у подавляющего большинства
неорганических соединений.
В молекулярных структурах тер-
термины «формульная единица» и «мо-
«молекула» совпадают. Так, например,
в случае кристаллической структуры
бензола и=4 означает, что в одной
ячейке содержатся 4 молекулы СбНб.
Федоровская группа структурного
типа NaCl — FmZm.
Структурный тип CsCl изо-
изображен на рис. 163. Он весьма похож
на структурный тип a-Fe. В структу-
структуре a-Fe два одинаковых атома распо-
располагаются по вершинам кубической
ячейки и в центре ее. В структуре
CsCl эти места заняты разными
атомами (ионами). Оба эти положе-
положения эквивалентны. С одинаковым
основанием «белые» и «черные» ша-
122
• Na ®Cl
«Cs
Рис. 162. Структурный тип NaCl
Рис. 163. Структурный тип CsCl
Cl
рики можно считать как атомами
цезия, так и атомами хлора. Ес-
Если совместить начало координат с
центром тяжести атома цезия, то его
координаты будут @00), а коорди-
координаты атома хлора ('/г 7г 7г).
Ничего не изменится, если мы по-
поместим начало координат в центре
тяжести атома хлора. Тогда коорди-
координаты атома цезия будут Gг, 7г 7а).
Несмотря на большое внешнее
сходство структурных типов a-Fe и
CsCl, они существенно отличаются
друг от друга. В структуре a-Fe име-
имеется трансляция от вершины эле-
элементарного куба в центр его. В струк-
структуре же CsCl такой трансляции нет.
По этой причине элементарная ячей-
ячейка в первой структуре будет объем-
ноцентрированной, а у второй —
примитивной. Различны, конечно, и
федоровские группы симметрии:
1тЗт у a-Fe и РтЗт у CsCl.
Химическое соединение состава
ZnS в природе обычно встречается в
двух модификациях: кубической
(цинковая обманка, или сфалерит)
и в гексагональной (вюртцит).
Структура цинковой об-
обманки (рис. 164) очень сходна со
структурой алмаза. Атомы одного
элемента (безразлично, цинка или
серы) занимают узлы гранецентри-
рованной кубической ячейки,
а атомы второго элемента — центры
четырех (из восьми) малых кубов.
Пустые октанты чередуются с засе-
заселенными во всех трех координатных
направлениях. Оба положения так
же эквивалентны друг другу, как
эквивалентны положения, занимае-
занимаемые атомами натрия и хлора в
структуре NaCl или атомами цезия
и хлора в структуре CsCl. Структура
сфалерита и структура алмаза харак-
характеризуются одинаковой решеткой
Бравэ — гранецентрированной куби-
кубической. Однако их пространственные
группы симметрии различны: Fd3m
у алмаза и FA3m у сфалерита.
На рис. 165 изображена струк-
структура вюртцит а. Атомы одного
элемента располагаются так же, как
атомы магния в структуре металли-
металлического магния, т. е. по вершинам
гексагональной призмы, в центрах
базисных граней и в центрах трех
(из шести) тригоналъных призм, на
которые мысленно можно разбить
элементарную гексагональную ячей-
ячейку. Атомы второго элемента распо-
располагаются в тех же трех, уже занятых
атомами первого элемента, триго-
тригоналъных призмах и на всех верти-
вертикальных ребрах примитивных парал-
параллелепипедов. Они занимают такие
положения в структуре, что оказы-
оказываются на равных расстояниях от
четырех ближайших атомов первого
элемента. Все положения, занятые
атомами каждого элемента, состав-
составляют одну правильную систему то-
точек. Обе системы, занятые атомами
цинка и серы, эквивалентны между
собой так же, как и в случае поварен-
поваренной соли, CsCl и др. Федоровская
группа симметрии Рбзтс. Этот струк-
структурный тип иногда называется струк-
структурным типом цинкита ZnO.
На рис. 166 изображен струк-
структурный тип никелина NiAs.
Строение NiAs можно описать сле-
следующим образом: элементарная ячей-
ячейка состоит из двух коротких гекса-
гексагональных призм, составленных осно-
основаниями (такие ячейки часто назы-
называют «двухэтажными»); атомы нике-
никеля занимают все вершины и центры
базисных граней каждой из этих ко-
коротких гексагональных призм. Всю
ячейку, как обычно, мы мысленно
123
Рис. 164. Структурный тип сфалерита ZnS
«Zn
Рис. 165. Структурный тип вюртцита ZnS
(или цинкита ZnO)
•Ni О Аз
Рис. 166. Структурный тип NiAs
Рис. 167. Структурный тип BN
124
разбиваем на шесть тригональных
призм. Атомы мышьяка располага-
располагаются во всех шести призмах, по три
в каждом этаже. Таким образом,
в каждом этаже оказывается три
призмы заселеннных и три пустых.
Под (и над) каждой заселенной
призмой располагается пустая приз-
призма другого этажа и наоборот — под
(и над) каждой пустой призмой пер-
первого этажа располагается заселенная
призма другого этажа. Правильные
системы точек, занимаемые атомами
никеля и мышьяка, не эквивалентны
друг другу, поэтому белые шары на
рисунке символизируют положения,
занимаемые атомами мышьяка, а чер-
черные — атомами никеля. В этом суще-
существенное отличие структурного типа
NiAs от предыдущих. Федоровская
группа симметрии Рбз/пг/гас.
Последней структурой, рассматри-
рассматриваемой в этом параграфе, будет
структура нитрида бора BN
(рис. 167). Она весьма сходна со
структурой графита. В ней имеются
бесконечные плоские слои гексаго-
гексагональных колец. В каждом кольце три
вершины заняты атомами одного эле-
элемента, а три другие вершины — ато-
атомами другого элемента.
В отличие от структуры графита
кольца из разных слоев расположены
точно друг над другом, при этом по
вертикали (вдоль оси третьего поряд-
порядка) атомы бора и азота чередуются.
Следовательно, период повторяемости
вдоль оси с равен толщине двух сло-
слоев. Федоровская группа РВто2.
§7. Координационное число
и координационный
многогранник
Координационным числом данного
атома называется число ближайших
соседних атомов. Если речь идет о
координационном числе иона, то под-
подразумевается число ближайших окру-
окружающих его ионов противоположного
знака. Если эти ближайшие атомы
или ионы соединить друг с другом
прямыми линиями, то в общем случае
Рис. 168. Координационное число 12
в структуре типа меди
а — окружение атома меди в структуре;
б — координационный многогранник кубоокта-
эдр
Рис. 169. Координационное число 12
в структуре типа магния
о — окружение атома магния в структуре;
б — гексагональный аналог кубооктаэдра;
s — кубооктаэдр, поставленный на ось третьего
порядка
получится многогранник, носящий
название координационного много-
многогранника. Атом или ион, для которо-
которого производится подсчет координа-
координационного числа, располагается в слу-
случае высокой симметрии структуры в
центре координационного многогран-
многогранника, по вершинам которого располо-
расположены координированные атомы или
ионы.
Легко подсчитать на модели цин-
цинковой обманки (см. рис. 164), что
число ближайших соседних атомов
будет 4. Каждый атом цинка окру-
окружен четырьмя атомами серы и каж-
каждый атом серы — четырьмя атомами
цинка. Структура алмаза также бу-
будет характеризоваться координа-
координационным числом 4. В обеих структу-
структурах ближайшие соседние атомы будут
располагаться по вершинам тетраэб-
ра, т. е. координационный многогран-
многогранник для обоих типов будет тетраэд-
тетраэдром.
В структуре NaCl (рис. 162) мы
встречаемся с координационным чис-
числом 6. Каждый ион Na окружен
шестью ионами хлора, расположен-
расположенными по вершинам октаэдра. То же
окружение характерно для ионов
хлора относительно ионов натрия.
Структура NiAs также характеризу-
характеризуется координационным числом 6. На
кратчайшем расстоянии от каждого
атома никеля находятся шесть ато-
атомов мышьяка, и наоборот. Координа-
Координационные же многогранники у них
различны: у атома никеля — октаэдр,
у атома мышьяка — тригоналъная
призма.
Структура CsCl характеризуется
координационным числом 8. Ионы
противоположного знака, окружаю-
окружающие данный ион, располагаются по
вершинам куба. Тем же координаци-
координационным числом и многогранником ха-
характеризуется структурный тип a-Fe.
В структурном типе меди каждый
атом имеет координационное число 12
(рис. 168, а), форма координаци-
координационного многогранника — кубоокта-
кубооктаэдр (рис. 168, б). Координационным
числом 12 характеризуются также
структуры типа магния (рис. 157).
Координационный многогранник
этой структуры изображен на
рис. 169. Кристаллографически он
представляет собой комбинацию
двух тригональных дипирамид и
125
пинакоида с такими же углами меж-
между гранями, как в кубооктаэдре. Ря-
Рядом с этим многогранником для срав-
сравнения изображен кубооктаэдр, по-
поставленный па ось третьего порядка
(в). Совершенно очевидно, что оба
многогранника получаются один из
другого поворотом одной половины
фигуры (например, нижней относи-
относительно верхней) на угол в 60°. Мно-
Многогранник (б) специального назва-
названия не имеет. Часто его по аналогии
с кубооктаэдром называют гексаго-
гексагональным кубооктаэдром.
Структуры графита и борнитрида
могут служить примером структур с
координационным числом 3. Каждый
атом располагается в центре равно-
равностороннего треугольника.
Совершенно ясное в применении
к кубическим структурам понятие
координационного числа теряет свою
простоту и наглядность, как только
мы переходим к более сложным и
менее симметричным структурам.
Так, например, структурный тип Mg
в идеальном случае, т. е. в предполо-
предположении, что атомы имеют шаровую
форму, имел бы отношение осей
с/а = 1,633. Однако даже металли-
металлический магний имеет константы ре-
решетки а = 3,202А и с = 5,199, от-
откуда с/а = 1,624. Следовательно,
шесть атомов из 12 находятся на
кратчайшем расстоянии — 3,190, а
шесть следующих — на расстоянии
3,202. Сходную с магнием структуру
имеет кадмий. Однако отклонение от
идеального отношения с/а в структу-
структуре Cd достигает уже значительной
величины: а = 2.973А, с = 5,607 и
с/а = 1,886. В такой структуре, стро-
строго говоря, считать координационное
число равным 12 нельзя, так как рас-
расстояния разбиваются на две груп-
группы — 2,97 и 3,29. Обычно в этих слу-
случаях пишут, что координационное
число равно F+6). Такой способ
записи указывает, что шесть атомов
находятся на кратчайшем расстоя-
расстоянии от атома, избранного за начала
координат, а следующие шесть —
на несколько большем.
Е
Рис. 170. К определению координацион-
координационных чисел в структуре графита (а); два
многогранника, соответствующие двум
различным атомам углерода / и // в
структуре графита
Еще сложнее становится вопрос
при определении координационных
чисел в структурах графита и бор-
нитрида. В первом приближении ко-
координационное число каждого атома
в этих структурах равно 3. Это крат-
кратчайшее расстояние в структуре гра-
графита равно 1,42, в структуре BN —
1,45А. Но координационное число 3
не может образовать замкнутого ко-
координационного многогранника, по-
поэтому ясно, что при «втором прибли-
приближении» необходимо будет учесть
также и атомы, находящиеся в
смежных слоях. Оказывается, что в
структуре графита атомы разобьют-
разобьются на два сорта. Один сорт атома (/)
будет иметь координационное чис-
число 5 C+2) с расстоянием 1,42 и 3,39,
а второй (//) — координационное
число 15 C+12). Однако и это не
будет окончательным решением, так
как атом, избранный за начало, име-
имеет в своем же слое 9 атомов на рас-
расстояниях более коротких, чем рас-
расстояния до ближайших атомов из со-
соседних слоев C,39); шесть на рас-
расстоянии 2,46 и три на расстоянии
2,84А (рис. 170).
126
В структуре BN мы встречаемся
со всеми теми же затруднениями и,
кроме того, к ним присоединяются
еще и другие. Ближайшими тремя
атомами у бора будут атомы азота, и
наоборот. Но на следующем по вели-
величине расстоянии находятся уже
одноименные атомы. Возникает но-
новый вопрос, учитывать эти отноше-
отношения атомов или нет. Различные авто-
авторы решают этот вопрос по-разному.
А. В. Шубников предложил находить
координационное число следующим
образом. Атом, избранный за начало
координат, мысленно соединяют ли-
линиями со всеми остальными атома-
атомами в структуре и затем проводят
плоскостиг перпендикулярные к этим
линиям. Если определяется коорди-
координационное число в структуре просто-
простого вещества, то плоскости проводятся
на середине расстояния между дву-
двумя атомами; в случае ионного соеди-
соединения это расстояние делится про-
пропорционально радиусам ионов, меж-
между которыми проведена линия. В
результате пересечения плоскостей
вокруг избранного атома образуется
выпуклый многогранник, число гра-
граней которого и будет характеризо-
характеризовать его координационное число.
Чтобы довести высказанную идею
до конца, следует указать, что, кроме
числа, можно учитывать величины от-
отдельных граней, выражая их, напри-
например, в процентах по отношению ко
всей площади поверхности фигуры.
Все эти вопросы подняты для то-
того, чтобы продемонстрировать труд-
трудности, возникающие при попытке
уточнить понятие координационного
числа. В большинстве случаев для
наших целей достаточно первого при-
приближения, которого и будем придер-
придерживаться в дальнейшем.
§8. Простейшие структуры
типа AXj и АХ
• Со Of
Рис. 171. Структурный тип флюорита CaF2
На рис. 171 изображен структур-
структурный тип флюрита CaF2. Ато-
Атомы (ионы) кальция располагаются
Рис. 172. Структурный тип рутила TiO2
по узлам гранецентрированной куби-
кубической решетки, атомы (ионы) фто-
фтора — в центрах каждого октанта.
Координационное число атомов каль-
кальция —8, координационный много-
многогранник—куб; координационное
число атомов фтора—4, координа-
координационный многогранник — тетраэдр.
Координационные числа в структур-
структурном типе CaF2 сокращенно записыва-
записываются (8,4). Федоровская группа сим-
симметрии Fm3m. В этом структурном
типе кристаллизуется много различ-
различных по составу веществ, в частности
ТЬОг, по которому иногда и называ-
называют этот структурный тип. Окислы и
сульфиды щелочных металлов —
Li2O, NazO, Na2S и др.— имеют ана-
аналогичную структуру, но места, зани-
занимаемые в структуре CaF2 катионами,
в этих структурах занимают анионы,
и наоборот. Такие пары структурных
типов часто называют антиизоморф-
антиизоморфными.
На рис. 172 изображен струк-
структурный тип рутила — одной из
127
«Si О О
Рис. 173. Структурный тип кристобалита
«С ©О
Рис. 174. Структурный тип двуокиси уг-
углерода СОг.
Рис. 175. Кубическая ячейка с четырьмя
тройными непересекающимися Друг с
другой осями симметрии третьего по-
порядка
• Fe as
Рис. 176. Структурный тип пирита FeS2
модификаций ТЮг. Атомы титана
расположены по вершинам и в цент-
центре ячейки, атомы кислорода — по од-
одной из диагоналей базисных граней
ячейки и по перпендикулярной к ней
диагонали в плоскости, параллель-
параллельной базису и проходящей через
центр ячейки. Координационное чис-
число F,3). Координационные много-
многогранники — октаэдр и треугольник.
Федоровская группа симметрии
Примером структурных типов с
координационными числамиD,2) мо-
могут служить многочисленные кри-
кристаллические модификации ЭЮг.
Опишем наиболее симметричную из
них — тип кристобалита
(рис. 173). Атомы кремния распола-
располагаются в кубической ячейке так же,
как атомы углерода в структуре ал-
алмаза. В промежутке между каждой
парой ближайших атомов кремния
находится атом кислорода. Коорди-
Координационный многогранник у крем-
кремния — тетраэдр, у кислорода — ган-
гантель.
Далее рассмотрим структурный
тип СО 2 (рис. 174). Кристалличес-
Кристаллическая двуокись углерода имеет куби-
кубическую решетку, атомы углерода в
которой занимают узлы гранецентри-
рованной ячейки. Атомы кислорода
образуют гантель, в середине которой
расположен атом углерода. Коорди-
Координационные числа B,1). Если разбить
ячейку на 8 малых кубов и в каждом
малом кубе выбрать по одной прост-
пространственной диагонали (по одной
тройной оси) так, чтобы эти диагона-
диагонали при продолжении до бесконечнос-
бесконечности не пересекали бы друг друга (см.
рис. 175), то мы получим представле-
представление о направлении молекул О = С =
= 0 в кристалле. Этот тип (мотив)
расположения материальных частиц
удлиненной формы встречается во
многих структурных типах. Кратко
мы будем его называть «расположе-
«расположением по четырем тройным непересе-
непересекающимся осям».
Структуры, подобные СОг, в ко-
которых можно выделить отдельные
128
Рис. 177. Три аспекта гранецентрирован-
ной кубической ячейки
молекулы, носят название молеку-
молекулярных структур.
Геометрически весьма сходна со
структурой СОг структура пи-
пирита РеЭг (рис. 176). Как было
сказано выше, ячейку гранецентри-
рованной кубической решетки (рав-
(равно как и соответствующую ей пра-
правильную систему точек) можно рас-
рассматривать в различных аспектах.
На рис. 177 показана гранецентриро-
ванная ячейка в трех аспектах. В
случае (а) исходная точка правиль-
правильной системы помещена в начало ко-
координат и имеет координаты @00);
в случае (б) такого совпадения нет, и
координаты исходной точки ('/г 00);
в случае (в) координаты исходной
ТОЧКИ G4 V* 74).
Структура пирита FeS 2 отлича-
отличается от структуры СО 2 тем, что цен-
центры тяжести атомов железа (ионов
Fe2+) и центры тяжести групп S2
(молекулярных ионов S22~) занима-
занимают в структуре положения а и б, а
не одно из них, как это имело место
в структуре СОг. Симметрия обеих
структур остается, конечно, одинако-
одинаковой (федоровская группа РаЗ), ибо
прямая, соединяющая оба атома се-
серы в каждой группе S22-, совпадает
с одной из тройных осей малых ку-
кубов. Все группы Эг2" в структуре рас-
расположены по четырем тройным не-
непересекающимся осям. Если груп-
группу S22- считать за одну структурную
единицу, то структура FeS2 будет
аналогична структуре NaCl. Коорди-
Координационное число атома железа по
отношению к отдельным атомам се-
серы также равно шести. Однако каж-
каждый атом серы окружен только тремя
атомами железа.
9 Кристаллохимия
§ 9. Классификация структур
по координационным числам
Структуры бинарных соединений
удобно классифицировать по коорди-
координационным числам (сокращенно
к.ч.). Для соединений типа АХ в
качестве дополнительного признака
при классификации можно исполь-
использовать еще симметрию.
Так, в кубическом ряду соедине-
соединений состава АХ будут находиться
три основных структурных типа с
координационными числами 8, б и 4
(табл. 8). Гексагональный ряд начи-
начинает структурный тип NiAs с 6,
а заканчивает структурный тип
BNc3.
ТАБЛИЦА 8
Классификация структурных типов АХ
по координационным числам
Координационное
число
Структурные
типы кубичес-
кубического ряда
Структурные
типы гексаго-
гексагонального ряда
8
CsCl
6
NaCl
NiAs
4
ZnS
сфале-
сфалерит
ZnO
3
BN
Структуры соединения АХ2 и
более сложных бинарных соедине-
соединений также удобно классифицируются
по координационным числам (см.
табл. 9).
ТАБЛИЦА 9
Классификация структурных типов
по координационным числам
Координаци-
Координационное число
Структур-
Структурный тип
(8,4)
CaF2
F.3)
ТЮ2
рутил
D.2)
SiO2
кристоба-
лит
B.D
со2
129
§10. Структуры с параметрами
и ©ез параметров
Структурный тип рутила
(рис. 172) имеет федоровскую груп-
группу симметрии Pbzlmnm и п=2. Два
атома титана в ячейке занимают ме-
места одной правильной системы точек
с координатами @00; УгУг'/г). Четы-
Четыре атома кислорода также расположе-
расположены по точкам одной правильной си-
системы. Точки этой системы имеют ко-
координаты (ххО; ххО; Уг + х, х\ч — х,
Уг; 72 —я, lh + x, 2/2). Величина х
может изменяться, но симметрия
структуры при этом сохраняется.
Разные вещества, кристаллизующие-
кристаллизующиеся в этом структурном типе, могут
иметь различные значения парамет-
параметра х, выраженного в долях ячейки.
Для точного знания расположения
атомов требуется определение не
только структурного типа и констант
решетки, но еще и параметра (в на-
нашем примере параметра х). Структу-
Структуры такого типа называются структу-
структурами с параметрами.
Структура рутила является
структурой с одним параметром. Бо-
Более сложные структуры, характеризу-
характеризующиеся несколькими параметрами,
называются многопараметрическими.
В отличие от параметрических струк-
структур, структуры типа NaCl, NiAs,
CsCl, ZnS и др. называются структу-
структурами без параметров. В этих струк-
структурах знания структурного типа и
констант решетки достаточно для
определения положения всех атомов
в пространстве. Изменение хотя бы
таблица ю
одной координаты одного из атомов,
например координат атомов Na
в структурном типе NaCl с сохране-
сохранением координат другого атома, при-
привело бы к резкому изменению сим-
симметрии структуры и к появлению-
другого структурного типа.
В структурах без параметров
расстояние между атомами, выра-
выраженное в долях ячейки, всегда оста-
остается постоянным. В структуре с пара-
параметрами оно меняется в зависимости
от значений параметров.
Изменение параметра в структур-
структурном типе с параметрами влечет за
собой изменение формы координа-
координационных многогранников. В табл. 10
приведены константы решетки и
величины параметров х, найден-
найденные Н. В. Беловым и В. И. Мокеевой
для трех соединений, кристаллизу-
кристаллизующихся в структурном типе рутила.
Координационный многогранник у
атома металла только в структуре
MgF2 является достаточно близким
к идеальному октаэдру, в чем можно
убедиться, подставляя параметр
х = 0,31 в формулы, определяющие
расстояния между атомами Me—О.
Для двух атомов О, лежащих
в плоскости XY, эта зависимость
выражается формулой dMe-2O = ха ' 2?
для четырех остальных
- 2х + т) + Т
В структуре ЭпОг два атома кисло-
кислорода из шести, окружающих атом
олова, оказываются ближе, чем четы-
Кристаллохимические параметры веществ, кристаллизующихся в структурном
типе рутила
Вещество
Рутил ТЮг
Седлаит MgF2
Касситерит БпОг
4
4
4
а
,58
,87
,72
2
3
3
с
,95
,30
,17
0
0
0
с/а
,64
,68
,67
0
0
0
X
,33
,31
,26
Растоянж
к. ч. 4
1,86
1,98
2,25
А-Х, А
к. ч. 2
2,14
2,04
1,74
130
ре остальных. Вследствие этого в
структуре намечается обособление
молекул ЭпОг. В структуре ТЮг че-
четыре атома кислорода из шести ока-
оказываются ближе к атому Ti, чем два
остальных. Такое значение парамет-
параметра х обусловливает в этой струк-
структуре обособление цепочек [ТЮг] «>.
Величина параметра у параметри-
параметрических структур может меняться с
изменением термодинамических усло-
условий, например с температурой.
§11. Вычисление
межатомных расстояний
и валентных углов
в структурах
Всякую структуру кристалла удобно
описывать математически, если принять
за направление координатных осей систе-
системы X, Y и Z ребра элементарной ячейки.
Тогда положение каждого атома в прост-
пространстве можно описать координатами xyz,
атом принимается за математическую то'1-
ку, и расстояние между атомами является
иа самом деле расстоянием между их цен-
центрами тяжести или между точками федо-
федоровских правильных систем.
Вычисление межатомных расстояний в
ортогональной системе координат весьма
просто. Вычисление производится по фор-
формуле, выражающей расстояние между
двумя математическими точками с изве-
известными координатами:
где Х\у&\ и x2y2Z2 — координаты первого
и второго атомов. Обычно координаты
атомов даются в долях ячейки. Перед рас-
расчетом межатомных расстояний их необхо-
необходимо перевести в ангстремы.
Эта формула применима для расчета
расстояний между атомами в кристаллах
кубической, тетрагональной и ромбиче-
ромбической сингоний.
В случае гексагональной, моноклинной
и триклинной структур определение меж-
межатомных расстояний усложняется, по-
поскольку их система координат косоуголь-
косоугольная, и в расчет входит угол между осями.
В общем случае (триклинная синго-
ния) расчет производится по формуле
<Р= (ха — ziJ 4- (У2 — J/iJ + (z2 — ziJ +
+ 2 (Х2 — X!) (т/2 — l/l) COS f +
+ 2 (Х2 — Z)) (Z2 — Zi) COS P +
+ 2 (т/г — l/i) (za — zi) cos a,
где а, р и v — углы между соответствую-
соответствующими координатными осями.
Для гексагональных и моноклин-
моноклинных кристаллов формула несколько упро-
упрощается, так как два косинуса становятся
равными нулю. Так, для моноклинных ре-
решеток формула будет иметь вид:
= (х2 - x
+ 2 (ха —
+ (i/2 - i/aJ + (га
(z2 — zi) cos P;
для гексагональных —
d* = (X2 — X-if + (j/2 — yif + (z2 - Zif -
— (xt — Xi) (j/2 — J/l).
Знание атомных координат в структу-
структуре дает возможность вычислять валент-
валентные углы атомов. Связь между координа-
координатами атомов и валентными углами ср в об-
общем случае выражается формулой
cosq>=
+
ab cos T
+ X2Y2)
AB
+
ac cos p (XiZi + X2Z2)
+ AB +
be cos ot (YiZi +YtZz)
-\ AB '
где
A =
2a2 + У2Ь2 + Z\c* + 2ab cos
+ 2ac cos pZiZi + 26c cos aYiZi)'/2,
В = (ХУ + Yy + Zlc2 + 2ab cos TX2V2 +
+ lac cos №Z2 + 26c cos a YaZaO',
Xi=(x2—xi), Yi==(y2—yi), Zi=(z2—*i),
Z2=(a;2—x3), F2= A/2— Уз), Z2=(z2—*»),
ai P. ^ — углы между соответствующими
координатными осями, а Ъ, с — параметры
ячейки.
Расчет валентных углов в структурах
моноклинных и гексагональных кристал-
кристаллов значительно упрощается, так как ис-
исчезает часть слагаемых. В случае же ор-
ортогональных систем формула принимает
совершенно простой вид. Так, для кубиче-
кубических решеток
cosq> =
где
VCD
Практически удобнее вести расчет от-
отдельными этапами, вычисляя прежде
нужные расстояния между атомами. Оп-
9*
131
ределение валентного угла в этом случае
производится по тригонометрической фор-
формуле, дающей нужный угол, если извест-
известны три стороны треугольника (три меж-
межатомных расстояния):
COS ф!,2 =
где п, гг, гз — межатомные расстояния. Эта
формула имеет ряд преимуществ перед
предыдущей, так как расчет по ней более
прост, особенно если известны некоторые
межатомные расстояния в структуре. Кро-
Кроме того, наглядность формулы позволяет
без труда сделать однозначный выбор
между тупым и острым углом (cos ф мо-
может быть со знаком плюс или минус), в то
время как предыдущая формула требует
дополнительного анализа.
§12. Структура кристалла,
кристаллическая решетка
и иравильиаи система точек
Из теории кристаллических реше-
решеток (параллелепипедальных систем
точек) известно, что исходный (т. е.
любой) параллелепипед повторяемо-
повторяемости, а с ним и всю параллелепипе-
дальную систему можно мысленно
переносить в кристаллическом про-
пространстве параллельно самому себе.
При таком переносе конечная систе-
система ничем не будет отличаться от
исходной. Начало координат, т. е.
вершину параллелепипеда, или, что
то же самое, узел решетки, можно
представлять себе помещенным в лю-
любой точке кристаллической структу-
структуры. Часто бывает удобно поместить
его в центре тяжести атома, тогда
этот атом получает координаты @00),
крайне упрощающие все вычисления.
В структуре соединения более или
менее сложного химического состава,
допустим АХ, атомы одного элемента
(А) иногда можно совместить с уз-
узлами решетки, но тогда атомы друго-
другого элемента (X) обязательно окажут-
окажутся в промежутках между узлами, и
в общем случае их координаты будут
иметь отличные от нуля значения
(xyz). Можно поместить узел на сере-
середине расстояния между атомами А и
X, тогда координаты A— (xyz), a
координаты X— {хуЦ. Такое рас-
расположение осей координат может
оказаться более удобным при вычис-
вычислении. Так, например, в структуре
пирита (рис. 176) узел решетки
удобно бывает расположить в сере-
середине промежутка между атомами
серы. Хотя в этом случае в узле нет
никакого атома, но в нем располага-
располагается центр тяжести группы S2.
Из сказанного ясно, что с узлами
решетки могут быть связаны мате-
материальные частицы структуры, но со-
совершенно не обязательно считать,
что они располагаются непосредст-
непосредственно в узлах. Решетка кристалла
есть математическое абстрактное по-
понятие, аналогичное понятию элемента
симметрии, употребляющемуся при
описании конкретных кристалличе-
кристаллических многогранников. С помощью
понятия решетки математически
удобно описывать периодичность кри-
кристаллической структуры. Число раз-
различных типов решеток — 14, число
различных структур или структурных
типов — бесконечно велико.
Структуры меди, алмаза, NaCl,
CaF2 (рис. 155, 160, 162, 171) имеют
одинаковый тип решетки—гранецен-
трированный куб, хотя структуры их
существенно отличаются друг от дру-
друга. С другой стороны, близкие струк-
структуры a-Fe и CsCl (рис. 156 и 163) име-
имеют различные решетки — центриро-
центрированную кубическую и примитивную
кубическую.
Нельзя путать понятия решетки
кристалла и структуры кристалла.
В литературе часто приходится встре-
встречать такие термины, как, например,
«алмазная решетка». Это неправиль-
неправильно, ибо решетка в структуре алма-
алмаза — гранецентрированная кубиче-
кубическая, такая же, как у многих других
упомянутых выше кристаллических
веществ. Термин «алмазная решетка»
не имеет никакого смысла.
Следует также отличать понятие
правильной системы точек от поня-
понятия решетки. Путаница здесь часто
происходит вследствие того, что одна
и та же модель служит нам для де-
демонстрации структуры кристаллов,
132
решетки Бравэ и правильной системы
точек. Рис. 155 может изображать
структуру меди, тогда каждый шарик
в нем символизирует один атом меди.
Но тот же рисунок может изображать
и решетку Бравэ. — гранецентриро-
ванный куб, тогда каждый шарик бу-
будет символизировать узел решетки.
В этом случае можно сказать, что
такую же решетку имеет структура
NaCl (рис. 162), цинковой обманки
(рис. 164), CaF2 (рис. 171), алмаза
(рис. 160). Вместе с тем этот же ри-
рисунок может демонстрировать одну
из федоровских правильных систем
точек. Можно сказать, что в этой
правильной системе располагаются
атомы в структуре меди, или атомы
кальция в структуре CaF2, углеро-
углерода в структуре СОг, железа в струк-
структуре FeS2, платины в структуре
K2PtCl6 и т. д. Но во всех перечис-
перечисленных структурах атомы второго
элемента — фтора в структуре CaF2,
кислорода в структуре ССЬ и т. д.—
располагаются по точкам другой фе-
федоровской правильной системы.
Следовательно, могут быть разными
и решетки у этих структур, напри-
например гранецентрированная кубичес-
кубическая у CaF2 и примитивная кубичес-
кубическая у СОг и FeS2, хотя правильная
система точек, по которой распола-
располагаются атомы кальция, атомы угле-
углерода и атомы железа в этих трех
различных структурных типах, одна
и та же.
§ 13. Основные выводы^
сделанные на оенонанни
первых определений
структур кристаллов
Важный вывод, который оказалось
возможным сделать после первых
прямых определений кристалличе-
кристаллических структур, касается экспери-
экспериментального подтверждения теории
Федорова. То, что до 1912 г. казалось
лишь догадкой, математической аб-
абстракцией, полностью подтвержда-
подтверждалось и наполнилось материалистиче-
материалистическим содержанием. Это был период
величайшего триумфа теории. Не все
предусмотренные Федоровым возмож-
возможные расположения атомов в твердых
телах были в те годы найдены. Мно-
Многие из них не найдены и до сих пор.
Важно то, что не было найдено ни од-
одного случая, который противоречил
бы закону Федорова, хотя сейчас
определено уже около 20 000 струк-
структур. Нет сомнений в том, что через
несколько лет будут найдены пред-
представители всех предусмотренных Фе-
Федоровым групп симметрии. Уместно
вспомнить, что когда А. В. Гадолин в
1867 г. вывел все случаи симметрии
кристаллов C2), то известно было
только 20, а в настоящее время мы
знаем примеры веществ для всех 32
видов симметрии.
Важнейшим выводом для химии
явилось установление факта отсутст-
отсутствия молекул в кристаллах подавляю-
подавляющего большинства неорганических
веществ. Из всех разобранных нами
в этой главе структур лишь в струк-
структуре СОг мы смогли констатировать
наличие молекул. Во всех остальных
случаях выделить молекулы в струк-
структуре оказалось невозможным. Каж-
Каждый атом натрия в структуре NaCl
оказался связанным равноценными
связями с шестью атомами хлора, а
каждый атом хлора — с шестью ато-
атомами натрия, т. е. ни о каком обособ-
обособлении молекул NaCl не может быть и
речи. Столь привычные для нас пред-
представления химии оказались невер-
неверными.
Дальнейшие работы показали,
что у большинства органических ве-
веществ молекулы сохраняются и в
кристаллической структуре. Но вы-
вывод об отсутствии молекул в кристал-
кристаллах подавляющего большинства неор-
неорганических соединений остается
справедливым и надежно подтвер-
подтвержденным результатами исследования
нескольких тысяч структур. С другой
стороны, кристаллохимия полностью
подтвердила существование комп-
комплексных радикалов в неорганических
соединениях, например СОз2", SC42",
РО43-, PtCl62-, Pt(NH3L2+ и т. п.
ГЛАВА X
ФАКТОРЫ, ОПРЕДЕЛЯЮЩИЕ СТРУКТУРУ КРИСТАЛЛОВ
§1. Установление различных типов
химической связи
После того как было определено не-
некоторое количество различных струк-
структур бинарных соединений, возник
вопрос об определении размеров ато-
атомов. Естественно было считать форму
атомов в первом приближении шаро-
шаровой и характеризовать ее радиусом
определенной величины. Метод рент-
геноструктурного анализа позволяет
достаточно точно определять меж-
межатомные расстояния, но он не может
дать сведений о размерах отдельных
атомов. Так, например, было твердо
установлено, что расстояние между
атомами натрия и хлора в структуре
поваренной соли равно 2,81А. Однако
знания только типа структуры и меж-
межатомных расстояний недостаточно
для установления размеров отдель-
отдельных атомов, в данном случае натрия
и хлора, так как сумма rNa+rci==
= 2,81 может удовлетворять бес-
бесконечному числу значений вели-
величин слагаемых. Прямое же реше-
решение п уравнений с п неизвестными
типа гк + гС1=3,14; rNa + />=2,31;
гк + ?'г=2,66 и четвертое, выписан-
выписанное выше, невозможно, так как полу-
получается неопределенная система урав-
уравнений.
Вместе с тем совершенно ясно, что
достаточно знать размер хотя бы
одного атома, чтобы узнать величины
всех остальных.
Попытка определить размеры ато-
атомов, исходя из межатомных расстоя-
расстояний в чистых металлах, потерпела
неудачу.
Полученную таким способом систе-
систему атомных радиусов очень скоро
пришлось оставить, так как обнару-
обнаружилось большое количество противо-
противоречий, приведших к мысли о полной
несостоятельности идеи, положенной
в основу системы. В самом деле, ра-
радиус атома меди, определенный из
структуры металлической меди,
оказывается равным 1,27. Вычитая
эту величину из найденного зна-
значения межатомных расстояний в
структуре CuCl B,35), получают для
атома хлора радиус 1,08. Если проде-
проделать аналогичные определения раз-
размера атома хлора в структуре NaCl,
воспользовавшись размером атома
натрия A,86), определенного из
структуры металлического натрия, то
легко получить значение 0,95. От-
Отклонение от предыдущего значения
для атома хлора превышает 0,15, что,
конечно, недопустимо, так как точ-
точность определения межатомных рас-
расстояний даже в те годы была порядка
0,01. С другой стороны, непонятным
казался и тот факт, что размеры
анионов получались меньшими, чем
размеры катионов.
Теория электростатической валент-
валентности была создана за несколько лет
(Коссель, 1916 г.) до описываемых
работ по кристаллохимии A920 г.)
и к этому времени достаточно прочно
вошла в химию. С позиции электро-
электростатической теории строения неорга-
неорганических веществ более естествен-
естественным было ожидать, что размер анио-
анионов (например, хлора) будет больше,
чем катионов (например, натрия),
так как порядковый номер натрия —
134
11, а хлора — 17. Кроме того, атом
натрия потерял один электрон и, сле-
следовательно, удерживает остальные
прочнее, чем атом хлора, получивший
липший электрон.
Все высказанные выше соображе-
соображения привели, с одной стороны, к вы-
выводу о несостоятельности указанной
идеи определения размеров атомов, с
другой стороны, они послужили толч-
толчком к развитию новых идей, так как
показали, что представление об ато-
атомах как о шарах определенного раз-
размера может быть использовано толь-
только для определенных групп соедине-
соединений. Атом одного и того же химиче-
химического элемента может находиться в
различных электронных состояниях,
в зависимости от типа химического
соединения, и иметь, следовательно,
различные размеры. Размеры атомов
патрия или меди в металлических
кристаллах могут существенно отли-
отличаться от размеров их ионов в струк-
структурах соединений типа NaCl и CuCl.
Характер связи атомов в металличе-
металлических кристаллах, очевидно, может
существенно отличаться от характера
связи атомов в солях.
Надо сказать, что до рентгено-
структурных исследований химиками
почти не изучались вещества с метал-
металлической связью (исключением яв-
являлись работы Н. С. Курнакова и его
школы). Одной из причин было то,
что интерметаллические соединения
совсем не подчиняются правилам
валентности. Кроме того, в химии в
это время господствовала гипотеза
о молекулярном строении всех слож-
сложных веществ. Считалось, что в моле-
молекулах действуют химические силы, а
между молекулами — физические.
Под химическими связями под-
подразумевались силы, связывающие
атомы в молекулы, а под физически-
физическими — силы, обусловливающие кри-
кристаллизацию веществ. Интерметал-
Интерметаллические соединения не считались
поэтому химическими.
Изучение кристаллических струк-
структур различных веществ привело, с
одной стороны, к более дробному под-
подразделению типов межатомных свя-
связей, с другой стороны, стерло грани-
границы между физическими и химически-
химическими силами. Кроме металлической и
ионной связей, были установлены ко-
валентная (гомеополярная) и оста-
остаточная связи. Гомеополярная связь,
первая теория которой была предло-
предложена в 1916 г. Льюисом, проявляется
между атомами большинства молекул
органических соединений и между
атомами в таких кристаллах, как
алмаз. Остаточная, или Ван-дер-
Ваальсова, связь обусловливает сцеп-
сцепление между атомами в кристаллах
инертных газов и между молекулами
в кристаллах органических соедине-
соединений. Из приведенных выше приме-
примеров видно, что силы связи одного и
того же типа могут связывать ато-
атомы как в молекулы (например,
СО2), так и в кристаллы (например,
алмаз).
Ионные связи между атомами
могут существовать как в молекулах,
так и в кристаллах (например, в
случае паров NaCl и кристалла).
Подробнее типы химических свя-
связей в кристаллах будут рассмотрены
в XII главе.
§2. Гетер одссиичсскис
и гомодеемичеекие структуры
В некоторых структурах между ато-
атомами могут существовать различные
по типу связи. Такие структуры
называются гетеродесмическими. В
отличие от них структуры, в которых
между всеми атомами действуют силы
одного типа, называются гомодесми-
ческими. Примером структур первого
типа может служить структура СОг,
второго типа —' структура NaCl. Гете-
родесмические структуры характери-
характеризуются низкими координационными
числами и резкой разницей в. меж-
межатомных расстояниях — кратчайшее
расстояние до одного-трех ближай-
ближайших атомов резко отличается от сле-
следующего по величине расстояния
между аналогичными атомами. Так,
например, кратчайшее расстояние
135
между атомом углерода и каждым из
атомов кислорода в кристаллической
структуре СОг равно 1,О6А, ближай-
ближайшее расстояние от этого же атома
углерода до атомов кислорода из дру-
других молекул равно 3,14. Резкая раз-
разница в этих расстояниях объясняет-
объясняется различным типом связи и различ-
различной силой взаимодействия между
указанными атомами. Между атома-
атомами углерода и кислорода в молекуле
действуют ковалентные силы, между
молекулами — остаточные.
Вторым примером гетеродесмиче-
ских структур может служить ранее
описанная структура графита. Рас-
Расстояние между двумя ближайшими
атомами углерода в слое равно 1,42,
кратчайшее расстояние между атома-
атомами из разных слоев равно 3,39.
§3. Эффективные радиусы ионов
Еще Ломоносов в 1749 г. предложил
считать молекулы («корпускулы»)
шарами. Кристаллы он представлял
себе как совокупность шарообразных
соприкасающихся друг с другом мо-
молекул. Но первые модели структуры
кристаллов NaCl и других веществ из
шарообразных «атомов» различных
размеров были построены только
в 1906—1907 гг. Барлоу и Попом.
Эти представления получили даль-
дальнейшее развитие после опубликова-
опубликования ряда работ по изучению электрон-
электронного строения атома.
Многочисленные опыты показали,
что в кристаллах типа NaCl струк-
структурные частицы, слагающие кри-
кристалл, являются ионами. Атом нат-
натрия, после отщепления от него ва-
валентного электрона, становится по-
положительно заряженным ионом с ус-
устойчивой внешней электронной обо-
оболочкой, аналогичной электронной
оболочке благородного газа (неона).
Атом хлора, присоединяя один элек-
электрон, заряжается отрицательно и по-
получает аналогичную устойчивую кон-
конфигурацию внешних электронов. Со-
Состояние равновесия между ионами
натрия и хлора наступает в результа-
результате уравновешивания сил притяжения
между разноименными ионами и сил
отталкивания, возникающих между
отрицательно заряженными электрон-
электронными оболочками обоих ионов. При
симметричном окружении каждого
иона в кристалле ионами противо-
противоположного знака можно с большой
степенью точности считать эти ионы
несжимаемыми шарами и размер их
характеризовать величиной радиуса.
Не следует, однако, смешивать этот
эффективный радиус с расстоянием
наружной оболочки атома (иона) от
ядра.
§ 4. Определение ионных
и атомных радиусов
Как было сказано выше, с помощью
рентгеновского анализа нельзя опре-
определить размеры ионов в кристалли-
кристаллических структурах. Измеряя межпло-
межплоскостные расстояния, можно полу-
получить сумму радиусов катиона га и
аниона гх *. Так, например, расстоя-
расстояние Na|—С1=2,81. Какая часть из
этого расстояния приходится на до-
долю т-Na, какая на долю га—сказать
нельзя. Заранее можно предполагать,
что размер анионов будет, в общем,
больше размера катионов, так как
анионы имеют, по сравнению с ней-
нейтральными атомами, лишние электро-
электроны, в то время как катионы содержат
меньшее число электронов, чем ней-
нейтральные атомы. Кроме того, очевид-
очевидно, что при переходе от одного эле-
элемента к другому внутри одной под-
подгруппы периодической системы эле-
элементов будет иметь место увеличе-
увеличение размеров ионов с возрастанием
атомного номера. Об этом можно су-
судить по кривым атомных объемов.
Определить размеры радиусов
анионов можно следующим способом.
Если взять вещества с малыми катио-
катионами и большими анионами, то мож-
можно ожидать, что межплоскостные рас-
* Начальными буквами алфавита обычно
обозначаются катионы, последними —
анионы.
136
Рис. 178. Схема определения радиуса иона
стояния кристаллов будут обусловле-
обусловлены только размерами анионов. Ма-
Маленькие же катионы будут распола-
располагаться в пустотах между анионами.
Этот случай будет иметь место тогда,
когда анионы касаются друг друга
(рис. 178). Сравнивая межатомные
расстояния двух соединений, кри-
кристаллизующихся в структуре хлори-
хлористого натрия (Mg—О 2,10 и Мп—О
2,24), можно сделать вывод, что
ион марганца больше, чем ион магния.
Возьмем анион большего размера,
чем кислород, например селен. Тогда
соответствующие межатомные рас-
расстояния будут: Mg — Se 2,73 и
Мп — Se 2,73.
Очевидно, что в этих структурах
межатомные расстояния обусловле-
обусловлены только размерами иона селена.
Отсюда можно вычислить радиус
иона двухвалентного селена. Он
равен 2,73-/2/2 = 1,92 (см. рис. 178).
'Зная же размеры хотя бы одного
аниона, можно определить радиусы
других ионов. Для этого надо выбрать
структуры, в которые входит извест-
известный анион и у которых межатомные
расстояния обусловлены суммой ра-
радиусов Га + гх. Таким путем можно
определить радиусы всех ионов. Так,
например, Са—Se = 2,96, откуда
Са2+=1,О4; Са—0 = 2,38, откуда
О2" = 1,34.
Эта работа была проделана для
большинства химических элементов
В. М. Гольдшмидтом в 1926 г. Он
воспользовался не радиусом селена
Se2~ = l,92, определенным этим спо-
способом, а радиусами ионов фтора и
кислорода: F~= 1,33 и О2~ = 1,32, по-
полученными в 1928 г. Вазашерна из
рефрактометрических данных. Оба
метода дают хорошо совпадающие
между собой результаты. Так, напри-
например, Гольдшмидтом получены значе-
значения радиусов ионов для Se2~ = l,91,
для Са2+=1,06.
Атомные же радиусы металлов мо-
могут быть определены непосредствен-
непосредственно из данных рентгеноструктурного
анализа. Для этого достаточно поде-
поделить пополам найденное эксперимен-
экспериментально межатомное расстояние. Так,
например, кратчайшее расстояние
между двумя атомами в структуре
меди равно 2,55, откуда радиус атома
меди равен 1,27. Кратчайшее расстоя-
расстояние между атомами в структуре маг-
магния равно 3,20. Радиус атома магния
равен 1,60.
§ 5. Ионные радиусы
химических элементов
Ниже приведена таблица ионных ра-
радиусов большинства химических эле-
элементов периодической системы Мен-
Менделеева.
В настоящее время в литературе
имеется несколько таблиц ионных
радиусов. Объясняется это обстоятель-
обстоятельство главным образом тем, что в ос-
основу таблицы авторами кладутся раз-
разные величины исходных ионных ра-
радиусов. Как было сказано выше,
Гольдшмидт положил в основу табли-
таблицы величины ионных радиусов фто-
фтора A,33) и кислорода A,32). Расчет-
Расчетный же радиус иона кислорода, по
Полингу, равен 1,40. Отсюда резкое
расхождение в значениях ионных ра-
радиусов двухвалентных и трехвалент-
трехвалентных металлов, определяемых главным
образом из структур их окислов, в
таблицах Гольдшмидта и Полинга.
Нет сомнения в том, что В. М. Гольд-
Гольдшмидт преуменьшил значение ра-
радиуса иона кислорода, но и величина
1,40, несомненно, слишком велика.
Н. В. Белов и Г. Б. Бокий приняли
среднюю величину для иона кислоро-
кислорода 1,36 и, 1учтя новые данные по
i37
ТАБЛИЦА 11
Ионные радиусы
Периоды
Под
1а
На
Ша
IVa
Va
Via
Vila
Villa
Li
1+ 0,68
Na
1+ 0,98
Be
2+ 0,34
Mg
2+ 0,74
К
1,33
Ca
2+ 1,04
Sc
3+ 0,8?
Ti
2+ 0,78
3+ 0,69
4+ 0,64
V
2+ 0,72
3+ 0,67
4+ 0,61
5+ 0,4
Cr
2+ 0,83
3+ 0,64
6+ 0,35
Mn
2+ 0,91
3T 0,70
4+ 0,52
7+@,46)
Fe
2+ 0,80
3+ 0,67
Co
2+ 0,78
3+ 0,64
Rb
1+ 1,49
Sr
2+ 1,20
Y
3+ 0,97
Zr
4+ 0,82
Nb
4+ 0,67
5+ 0,66
Mo
4+ 0,68
6+ 0.65
Tc
Ru
4+ 0,62
Rh
3+ 0,75
4+ 0,65
Cs
+ 1,65
Ba
2+ 1,38
La
3+ 1,04
¦+ 0,90
Hf
4+ 0,82
Та
¦ @,66)
w
4+ 0,68
6+ 0,65
Re
6+ 0,52
Os
4+ 0,65
Ir
4+ 0,65
Fr
Ra
2+ 1,44
Ac
3+1,11
T
Ku
Лантаноиды
Актиноиды
3+
4+
Се
1,
0,
02
88
3+
Рг
1,
00
Nd
3+ 0,
99
3+
Рт
@,98)
Sm
3+ 0
97
Зн
Ей
v 0,
97
rr
Th
3+ 1,
4+ 0,
08
95
3+
4+
Ра
1,
0,
Об
91
3+
4+
U
1,
0,
04
89
Np
3+ 1
4+ 0,
02
88
Ри
3+ 1,
4+ 0,
01
86
3
4
Am
*" 1
*о,
00
85
В скобках приведены значения вычисленных радиусов.
Для благородных газов даны значения атомных радиусов.
:138
группы
Villa
Ni
2+ 0,74
Pd
4+ 0,64
Pt
4+ 0,64
Gd
3+ 0,94
Cm
Ib
Cu
1+ 0,98
2T 0,80
Ag
1+ 1,13
Au
1+ A,37)
Tb
3+ 0,89
Bk
9+
9+
2+
3+
lib
Zn
0,83
Cd
0,99
Hg
1,12
Dy
0,88
Cf
3+
3+
3+
1+
3+
1+
3+
3+
Illb
в
@,20)
Al
0,57
Ga
0,62
In
1,30
0,92
Tl
1,36
1,05
Ho
0,86
Es
IVb
С
4- 0,2
4+ @,15)
4- B,60)
Si
4+ 0,39
Ge
2+ 0,65
4+ 0,44
Sn
2+ 1,02
4- 0,67
Pb
2+ 1,26
4+ 0,76
Er
3+ 0,85
Fm
3+
5+
3"
3+
5+
3-
3T
5+
3-
3+
5+
3-
3+
5+
3+
Vb
N
0,15
1,48
P
0,35
1,86
As
0,69
@,47)
1,91
Sb
0,90
0,62
2,08
Bi
1,20
@,74)
2,13
Tu
0,85
\ld
2~
2-
6+
2"
4+
6+
2-
4+
6+
Vlb
0
1,36
s
1,82
@,29)
Se
1,93
0,69
0,35
Те
2,11
0,89
@,56)
Po
Yb
0,81
No
1-
1+
1-
1-
7+
1-
7+
1-
7+
3^
Vllb
H
1,36
0,00
F
1,33
Cl
1,81
@,26)
Br
1,96
@,39)
J
2,20
@,50)
At
Lu
0,80
Lr
0
(
0
0
0
VHIb
He
1,22
Ne
) 1,60
Ar
1,92
Kr
1,98
Xe
2,18
Rn
13»
/
3
и
I
a h
©
а
Be
о
®
И Л IF I U Ш
Ь с b a b a t> a t а а
?+ 5+ 5+ Л + Л+ I + Г +
t/ I/ lJ V 7 J J
Се Pr Nd Pm Sm
Рис. 179. Относительные величины ионных
радиусов химических элементов
структурам простейших бинарных
соединений, составили сводную таб-
таблицу ионных радиусов (табл. 11).
В основу ее положена эксперимен-
экспериментальная система Гольдшмидта. В тех
случаях, когда расхождение с гольд-
шмидтовскими значениями меньше
0,02, оставлена прежняя величина.
Теоретически вычисленные значения
ионных радиусов помещены в табли-
таблице только в том случае, если неизвест-
неизвестны экспериментальные. В таблице
они поставлены в скобки. Радиусы
ионов приведены для координацион-
координационного числа 6. Объяснение этому бу-
будет дано в последующих параграфах.
При составлении этой таблицы уч-
учтены все те валентные состояния эле-
элементов, которые приведены в табли-
таблицах предшествующих авторов, т. е., в
частности, и многовалентные (напри-
(например, шестивалентная сера и др.).
Здесь значок S6+ следует рассматри-
рассматривать как указание на величину ва-
валентности, а не зарядности. Харак-
Характер связи S—О в ионе SO42- явля-
является ковалентным, а не ионным. Ска-
Сказанное относится в известной мере
также и к переходным металлам.
Так, например, расстояние Fe—S в
пирите можно вычислить как сумму
соответствующих ковалентных радиу-
радиусов.
На рис. 179 воспроизведена перио-
периодическая таблица Менделеева с гра-
графическим изображением относитель-
относительных размеров ионов для многих хи-
химических элементов. Из рассмотре-
рассмотрения этого рисунка и из данных
табл. 11 легко сделать вывод о том,
что размеры катионов в общем слу-
случае меньше размеров анионов.
Радиусы ионов редкоземельных
элементов равномерно уменьшаются
от La3+=l,04 до Lu3+=0,8O, несмот-
несмотря на возрастание порядкового номе-
номера. Это явление было открыто Гольд-
шмидтом и носит название ланта-
140
ноидного сжатия. Объяснение его
следует искать в электронном строе-
строении атомов редкоземельных эле-
элементов.
Во всех группах периодической си-
системы радиусы одинаково построен-
построенных ионов возрастают с увеличением
атомного номера элемента, но за
счет лантаноидного сжатия радиусы
элементов 3-го большого периода ока-
оказываются приблизительно равными
радиусам элементов 2-го большого
периода, например:
гТа5+' гМо«+
= Гх
Внутри одного ряда периодической
системы при переходе к следующему
по номеру элементу, ион которого
имеет больший положительный заряд,
размеры катионов уменьшаются:
Na+ = 0,98; Mga+ = 0,74;
Al3+ — 0,57; Si1+ =0,39.
В последние годы эффект, анало-
аналогичный лантаноидному сжатию, был
найден для последних тяжелых эле-
элементов, начиная от № 89 — актиния.
Этот эффект по аналогии можно наз-
назвать актиноидным сжатием.
§6. Метод изображения
кристаллических структур
шарами разных размеров
Как только были определены разме-
размеры ионов большинства химических
элементов, сразу же возник новый ме-
метод изображения структур кристал-
кристаллов. Структура, изображенная по это-
этому методу, представляет собой сово-
совокупность шаров разных радиусов, у
которых соблюдены относительные
размеры. При этом разноименные
шары соприкасаются друг с другом.
На рис. 180 показана структура CaF2.
В ней сохранены относительные раз-
размеры радиусов Са2+ A,04) и F~
A,33).
Очевидно, что этот способ изобра-
изображения структур точнее отражает
внутреннее строение кристалла, чем
обычный способ изображения струк-
структур одинаковыми шарами значитель-
Рис. 180. Структура CaF2 (размеры ячей-
ячейки и ионов даны в одном масштабе)
но меньшего размера, чем полусумма
межатомных расстояний. Однако при
новом способе изображения большие,
соприкасающиеся друг с другом ша-
шары заслоняют внутренние участки
ячейки и делают всю структуру мало
наглядной. По этой причине такой
метод изображения структур редко
используется в кристаллохимии.
§ 7. Геометрические пределы
устойчивости структур
с различными
координационными числами
Координационное число зависит от
относительных размеров централь-
центрального иона и соседних с ним. Устойчи-
Устойчивой структура кристалла будет тогда,
когда каждый ион соприкасается
только с ионами противоположного
знака. Такой случай в проекции на
плоскость показан на рис. 181, а. Если
размер центрального иона (допустим,
катиона) будет уменьшаться, то в мо-
момент, когда окружающие анионы со-
соприкоснутся друг с другом, структура
станет менее устойчивой (рис. 181, б).
Если заменить катион на другой,
меньшего размера, то последний при-
приобретает возможность свободно пере-
перемещаться в промежутке между ани-
анионами. Такое положение создает не-
неустойчивость структуры и может по-
повлечь за собой перемену координа-
координационного числа, т. е. полную пере-
перегруппировку ионов. Произойдет это
вследствие того, что ион, размер кото-
которого меньше, чем размер межанион-
иой пустоты, в какой-то момент вре-
141
Рнс. 181. Схема, иллюстрирующая степень
устойчивости структур
а — устойчивая структура — каждый ион со-
соприкасается только с ионами противопо-
противоположного знака;
б — менее устойчивая структура — анионы ка-
касаются друг друга;
в — д — неустойчивая структура — свободное
перемещение катиона приводит к уменьше-
уменьшению координационного числа
Рнс. 182. Определение предела устойчиво-
устойчивости структур с координационным числом 6
Рис. 183. Определение предела устойчиво-
устойчивости структур с координационным числом 8
мени приблизится к двум анионам
(если рассматривать картину в одной
плоскости, рис. 181, в) и удалится
от двух других. В следующий момент
один из двух более удаленных анио-
анионов приблизится к катиону, оттолк-
оттолкнув второй анион (г). А это приведет
уже к перемене координационного
числа C вместо 4) и полной пере-
перегруппировке ионов в структуре (д).
Пределы устойчивости различных
координационных чисел легко вычис-
вычислить. Рассмотрим предел устойчиво-
устойчивости для координационного числа 6.
Шесть анионов, окружающих катион,,
располагаются по вершинам октаэдра.
Сечение октаэдра через центры четы-
четырех анионов показано на рис. 182^
Диагональ квадрата 2га+2гх, а сторо-
сторона 2г*]/2, откуда ra :rx=f2—1 = 0,41.
Это отношение будет нижним пре-
пределом устойчивости структур с ко-
координационным числом 6. Если ра-
радиус аниона будет меньше размера
катиона, то предел га '¦ гх будет обрат-
обратной величиной только что найден-
найденного значения, т. е. будет равен
1/0,41=2,41. Одновременно это от-
отношение будет верхним пределом
устойчивости структур с координа-
координационным числом 6. Однако в ин-
интервале ОТНОШеНИЙ Га '. Гх ОТ 0,41 ДО
2,41 будут находиться переделы ус-
устойчивости структур с координацион-
координационным числом 8. Нижний предел
определяется __ из уравнения
2га+2гх = 2гжу3 (см. рис. 183).
Он равен 0,73. Верхний предел устой-
устойчивости структур с координационным
числом 8 определяется обратной ве-
величиной 1/0,73 = 1,37.
В табл. 12 указаны пределы отно-
отношений радиусов ионов для различных
координационных чисел. Приводятся
два предела отношений га:гх — пер-
первый, когда катион меньше аниона, и
второй — для обратного отношения.
Пределы га : гх, большие единицы,
могут реализоваться только для ко-
координационного числа 8, так как мак-
максимальный размер катиона 1,65 (Cs+),
а минимальный размер аниона—1,33
(F~). В этом случае га : гх = 1,25.
142
ТАБЛИЦА 12
Предельные значения отношений радиусов
ионов для различных координационных
чисел
К. ч.
2
3
4
6
8
Форма
окружения
Гантель
Треуголь-
Треугольник
Тетраэдр
Октаэдр
Куб
от 0
Отношение
ДО
ДО оо
от 0,
4,45
от 0
2,41
от 0
1,37
от 0,
15
ДО
,22
До
,41
До
73
0,15
ДО
6,45
ДО
4,45
ДО
2,41
до 1
и от
0,22
0,41
0,73
37
6
и
и
и
,45
от
от
от
Следовательно, вторые пределы отно-
отношений радиусов ионов, указанные в
последнем столбце таблицы, для одно-
одноатомных ионов получены быть не мо-
могут. Однако они могут иметь некото-
некоторый физический смысл в случае
комплексных катионов (например,
[Со(ГШз)б]3+ и т. д.). При некото-
некоторых, конечно, сугубо ориентировоч-
ориентировочных, расчетах форма таких сложных
ионов мюжет быть принята за шар, и
размер их тогда может быть охарак-
охарактеризован радиусом. Посмотрим на
примере галоидных солей щелочных
металлов, насколько оправдываются
эти геометрические пределы:
нений га : гх выше предела 0,73,
является подтверждением правилу
геометрических пределов. Из этой таб-.
лицы можно сделать вывод, что ве-
вещества не всегда имеют структуру с
максимальным из возможных окру-
окружений (см., например, RbBr, RbCl,
KF и др.).
Надо иметь в виду, что нижний и-
верхний пределы для каждого ко-
координационного числа существенно,
отличаются по своему характеру.
Так, например, структура типа
NaCl (к. ч. 6) геометрически устой-
устойчива в пределах от 0,41 до 0,73. Если
перейден нижний предел, то.
структура действительно делается
неустойчивой вследствие касания
анионов друг с другом. Если же пе-
перейден верхний предел, то такого
касания нет вплоть до отношения,
равного 2,41, но внутри этого интер^
вала @,41—2,41) будут находиться
пределы для следующего (больше-
(большего) координационного числа (рис,
184). Если отношение радиусов до-
достигнет значения 0,73, то чисто гео-
геометрических представлений будет
недостаточно, чтобы обосновать не-
необходимость смены координационно-
координационного числа. Для этого потребуется
привлечение энергетических сообра-
соображений, о которых речь будет ниже.
Поэтому в том факте, что в нашей
таблице выше значения 0,73 имеет-
Соединение
Га ¦ ГХ
Соединение
Га : Гх
LiJ
0,31
LiBr
0,35
<0,41
NaF
0,74
LiCl
0,
(CsJ)
0,
75
38
NaJ
0,45
RbBr
0,76
>0,73
NaBr
0,50
RbCl
0,82
LiF
0,51
>o,
(CsBr)
0,84
NaCl KJ
0,54 0,60
41<0,73
(CsCl)
0,91
KBr
0,68
KF
1,00
RbJ
0,68
RbF
1,12
KC1
0,73
CsF
1,24
Большинство соединений, приве-
приведенных в таблице, кристаллизуется в
структуре NaCl (координационное
число 6). Исключение составляют три
соединения, имеющие структуру с ко-
координационным числом 8. Они заклю-
заключены в скобки. Для всех трех соеди-
ся шесть соединении со структурой
типа NaCl, не следует видеть несо-
несостоятельности геометрического под-
подхода к решению вопроса о зависи-
зависимости координационного числа от
отношения радиусов ионов. Эти слу-
случаи не могут считаться такими же
143..
0.12
0/5/0,9/ L,73
Рис. 184. Пределы устойчивости для раз-
различных координационных чисел
исключениями, какими являются
три случая, у которых пределы от-
отношений радиусов лежат ниже зна-
значения 0,41. Эти последние исклю-
исключения легко могут быть поняты
после рассмотрения параграфов, по-
посвященных изучению явления поля-
поляризации ионов в кристаллах.
§ 8. Поляризация ионов
До сих пор мы представляли себе
ионы несжимаемыми шарами, при-
причем считали, что центр тяжести
отрицательного заряда совпадает с
центром тяжести положительного за-
заряда атомного ядра. В действитель-
действительности такое представление справед-
справедливо лишь в первом приближении.
Если ион будет находиться в элек-
электрическом поле, то центры тяжести
противоположных электрических за-
зарядов разойдутся, образуя диполь.
Форма иона, следовательно, откло-
отклоняется от шаровой. Дипольный мо-
момент |д, пропорционален напряжен-
напряженности поля Е и измеряется произве-
произведением сдвигаемого заряда Ze на
дипольное расстояние d между цен-
центрами зарядов: \х, = аЕ = Zed. Коэф-
Коэффициент пропорциональности а, на-
называется коэффициентом деформи-
деформируемости иона, или поляризуе-
поляризуемостью. Его величина приблизитель-
приблизительно постоянна для данного иона во
всех структурах. Ниже приведены
значения а • 1024:
Из приведенных данных видно,
как изменяется коэффициент а в
подгруппах периодической системы
и по периоду. В кристаллах каждый
ион всегда находится в электрическом
поле других ионов. Взаимодействие
двух ионов разных знаков схематиче-
схематически показано на рис. 185.
Положительно заряженный ион
будет отталкивать ядро аниона и
притягивать больше отрицательных
зарядов на ближайшей к нему сто-
стороне отрицательного иона. Вслед-
Вследствие этого плотность собственного
электронного облака с этой стороны
у него будет меньше. В результате
одностороннего действия поляриза-
поляризации шаровая форма иона будет на-
нарушена. Расстояние между ионами
будет уменьшенным по сравнению
с суммой радиусов ионов га + гх.
Чем больше радиус иона, тем легче он
будет поляризоваться.
Из таблицы ионных радиусов (табл.
11) видно, что этому условию хо-
хорошо удовлетворяют анионы. Кати-
Катионы же, напротив, характеризуются
меньшими размерами и часто боль-
большими зарядами. В силу этого они
поляризуются значительно слабее,
но способность поляризовать со-
соседние ионы тем сильнее, чем они
меньше и чем больше их заряд. Ка-
Катионы с конфигурацией наружной
электронной оболочки, отвечающей
благородному газу, например Na+,
Са2+ и т. п., поляризуют соседние
ионы и сами поляризуются слабее,
чем катионы с 18-электронной
внешней оболочкой, например Си+,
Ag+ и др.
В изолированной ионной молекуле
(рис. 185) поляризационное дей-
действие имеет однорторонний харак-
характер и вызывает образование диполя
и укорочение расстояния между
центрами ионов. В кристаллической
]+
075
02-
3,12
Na+
0,21
g2_
7,25
K+
0,87
Se2"
8,4
Rb+
1,81
Te"-
9,6
Cs+
2,79
F~
0,99
Na+
0,21
0,12
СГ Br~
3,05 4,17
Al3+
0,065
J-
6,28
0,043
444
Рис. 185. Взаимодействие двух ионов
а — без учета эффекта поляризации;
E — о учетом эффекта поляризации
Рис. 186. Поляризация иона в кристалле
решетке необходимо считаться с од-
одновременным действием на один
ион нескольких симметрично распо-
расположенных ионов (рис. 186). Поэто-
Поэтому образование диполя в кристалле
не обязательно, но обязательно сок-
сокращение расстояния между ионами
и часто также уменьшение коорди-
координационного числа.
Сокращение расстояний между
ионами вследствие поляризации
можно проследить на галоидных со-
соединениях Ag (табл. 13). Радиус
иона Ag+=1,13. Ион серебра яв-
является сильно поляризующим ио-
ионом, так как он имеет 18-электрон-
ную оболочку. Ионы же галоидов
легко поляризуются.
ТАБЛИЦА 13
Поляризация ионов в галоидных
соединениях серебра
Соеди-
Соединение
AgF
AgCl
AgBr
AgJ
Наблю-
Наблюденное
расстоя-
расстояние о
Ag-X, A
2,46
2,77
2,88
2,99
Сумма
радиусов
ионов, А
2,46
2,94
3,09
3,33
Сокраще-
Сокращение рас-
расстояния,
0
5,8
6,8
10,3
Тип
структуры
NaCl
NaCl
NaCl
ZnS
(сфалерит)
§0. Зависимость размеров атомов
н ионов
от координационных чисел.
Структурный тип иеровскнта
Выше мы рассматривали зависи-
зависимость координационного числа от
размеров радиусов ионов. Сейчас
рассмотрим обратную зависимость.
При изложении вопроса о поляриза-
поляризации мы указали, что представление
об ионах как о несжимаемых ша-
шарах справедливо лишь в первом
приближении. Поляризация ионов
в той или иной мере имеет место
во всех кристаллах. Сильнее всего
она проявляется в отдельных моле-
молекулах. В этом случае мы формально
можем считать для обоих ионов
А и X координационное число рав-
равным 1. С увеличением координа-
координационного числа одностороннее
уменьшение расстояния А — X за-
затрудняется, и мы априори должны
ожидать увеличения этого расстоя-
расстояния с увеличением числа ближай-
ближайших ионов противоположного зна-
знака.
Изменение расстояния А — X с из-
изменением координационного числа
удобнее всего исследовать на двух
модификациях вещества, отличаю-
отличающихся координационными числами.
В качестве примера приведем рас-
расстояние между NH4+ и С1- в двух
модификациях хлористого аммония
(табл. 14). Возможны также анало-
ТАБЛИЦА 14
Зависимость межатомных расстояний
от координационного числа
Соединение
NH4C1
NH4C1
Структур-
Структурный тип
NaCl
CsCl
Координа-
Координационное
число
6
8
Расстоя-
Расстояние „
А—X, А
3,27
3,35
гичные изменения расстояний меж-
между соответственными ионами в двух
веществах, различающихся по хи-
химическому составу. Такой пример
рассмотрен в табл. 15. Из приведен-
10 Кристаллохимия
145
ТАБЛИЦА 15
Зависимость межатомных расстояний
от координационного числа
Соединение
SrO
SrZrO3
Структур-
Структурный тип
NaCl
СаТЮз
К. ч. по
отноше-
отношению к кис-
кислороду
6
12
Расстояние
А-Х, А
2,57
2,89
ных в таблице двух веществ первое
кристаллизуется в структурном ти-
типе NaCl, второе — в структурном ти-
типе перовскита — СаТЮз (рис. 187).
Элементарная ячейка СаТЮз при-
примитивная, кубическая. Федоровская
группа РтЗт. Начало координат
обычно выбирают в центре тяжести
атомов титана, которые в этом слу-
случае занимают все вершины элемен-
элементарного куба, в центре которого
расположен атом кальция. Атомы
кислорода распределяются по сере-
серединам всех ребер, создавая вокруг
атома кальция координационный
многогранник в форме кубооктаэд-
ра (к. ч. 12).
В. М. Гольдшмидт в результате
анализа ряда структур, аналогич-
аналогичных приведенным в табл. 14 и 15,
определил зависимость межатомных
расстояний от координационного
числа.
Как это следует из табл. 16, вели-
величины изменений межатомных рас-
расстояний в зависимости от координа-
координационного числа для металлов не-
несколько отличаются от соответству-
соответствующих величин для ионных кристал-
кристаллов. Для ионных соединений самым
распространенным координацион-
пых числом является 6. Поэтому в
табл. 16 расстояние А — X для к. ч. 6
принято за 100%. Большинство же
металлов кристаллизуется в струк-
структурах с к. ч. 12, поэтому межатомное
расстояние А — А для к. ч. 12 при-
принято за 100%- По этой же причине
в основной таблице ионных радиу-
радиусов (табл. 11) все приведенные ве-
величины относятся к координацион-
координационному числу 6. Для нахождения рас-
расстояний между атомами с иными
координационными числами необхо-
необходимо в полученную (в результате
суммирования атомных или ионных
радиусов) величину внести поправ-
поправку на координационное число, поль-
пользуясь данными табл. 16.
ТАБЛИЦА 16
Зависимость межатомных расстояний
от координационного числа
Для ионных кристаллов
Координа-
Координационное
число
12
8
6
4
Расстоя-
Расстояние А—X,
112
103
100
94
Для металлов
Координа-
Координационное
число
12
8
6
4
Расстоя-
Расстояние А—А,
100
98
96
88
Рис. 187. Структурный тип перовскита
CaTiOs
§10. Слоистые структуры
Если анион легко поляризуется, а
катион обладает сильными поляри-
поляризующими свойствами, то среди сое-
соединений АХг и более сложных появ-
появляются структуры совершенно спе-
специфического характера. Это так на-
называемые слоистые структуры.
Представителями таких структур
являются CdCl2, CdJ2, M0S2 и др.
Прежде всего остановимся н а
структуре С<Пг* (рис. 188).
* CdJ2 кристаллизуется в трех полиморф-
полиморфных модификациях. Здесь описана одна
из них.
146
Рис. 188. Структурный
тип CdJ2
Рис. 189. Структурный
тип CdCl2
Рпс. 190. Структурный
тип M0S2
Она характеризуется координацион-
координационными числами 6 (октаэдр) и 3 (три-
гональная пирамида). Слой атомов
кадмия располагается между двумя
слоями из атомов иода, в результате
чего образуются тройные спои. Силы
связи внутри тройных слоев зна-
значительно больше, чем между двумя
тройными слоями, вследствие чего
у С(Пг наблюдается совершенная
спайность, параллельная базису (ге-
(гексагональная ячейка Бравз в струк-
структуре CdJ2 показана пунктиром).
Каждый третий слой анионов повто-
повторяет первый, каждый четвертый —
второй и т. д. Федоровская группа
симметрии РЗт. Подобную структу-
структуру имеют также TiS2, S11S2, TiSe2,
PbJ2, Mg(OHJ, Mn(OHJ и др.
Структурный тип CdCl2
очень близок к описанному только
что структурному типу CdJ2. В обо-
обоих случаях координационные чис-
числа — 6 и 3, координационный много-
многогранник у кадмия — октаэдр. Трой-
Тройные слои из анионов и катионов так-
также одинаковы. Отличаются оба ти-
типа различной взаимной ориентацией
тройных слоев. В структуре CdCl2
тройные слои располагаются друг
над другом так, что только четвер-
четвертый слой анионов повторяет первый,
пятый — второй и т. д. (рис. 189).
Вследствие этого элементарная
ячейка структуры CdCl2 пе гексаго-
гексагональная, а ромбоэдрическая. Федо-
Федоровская группа R3m.
Структурный тип молиб-
молибденита MoS2 (рис. 190) также име-
имеет координационные числа 6 и 3
и состоит из тройных слоев. Одна-
Однако, в отличие от CdCl2 и CdJ2, ме-
металлический атом имеет координа-
координационный многогранник не октаэдр,
а тригональную призму. Федоров-
Федоровская группа PQ/mmc. Подробнее этот
структурный тип будет описан в
следующей главе.
ю*
14?
§ 11. Влияние ноляриаации
на структуру кристаллов
Разбирая типы структур, различных
соединений, мы касались вопроса о
влиянии поляризации на структуру
кристаллов лишь попутно. Более
детально это влияние можно просле-
проследить на схеме В. М. Гольдшмидта
для соединений АХг.
Из схемы (рис. 191) можно сде-
сделать вывод, что возрастание поляри-
поляризации и параллельное уменьшение
координационного числа влечет за
собой переход от типичных ионных
(координационных) структур к мо-
молекулярным. Внутри группы соеди-
соединений с отношением га : гх, лежа-
лежащим в одних и тех же пределах
критических значений, увеличение
поляризации ведет к появлению
слоистых структур CdCl ->- CdJ2 ->
MoS2 и структур типа пирита
S
Структурные типы ДХ2
-П 74 1 Структура ^
"• | рутила—wCdCI2-»-
I Структура
\ флюорита
1 Струур
| рутила—wCdCI2-»-CdJ2—"ст-руктуоа
MOS2
(слоистые
структуры)
,41 \ Структура SiOj
С02
22 I СтРУктУРа С02
\ (молекулярная структура)
Увеличение поляризации
Рис. 191. Влияние поляризации на струк-
структуру кристаллов
Влияние поляризации выражает-
выражается также в уменьшении координа-
координационных чисел. Это особенно на-
наглядно можно проследить на приме-
примере Са(ОН)г. Радиус Са!+= 1,04, ра-
радиус (ОН)~ = 1,4. Отношению
га '¦ гх — 0,74 должна бы соответст-
соответствовать структура типа флюори-
флюорита (8,4), но из-за наличия постоян-
постоянного диполя образуется слоистая
структура типа CdJ2 с координаци-
координационными числами 6 и 3. Поскольку
поляризация приводит к образова-
образованию диполей, то наличие постоянно-
постоянного диполя, например, у радика-
радикала ОН", равнозначно сильной поля-
поляризации.
§12. Факторы, определяющие
структуру кристаллов
(правило Гольдшмидта)
Выше мы разобрали факторы, вли-
влияющие на структуру кристаллов.
По Гольдшмидту, структура кри-
кристалла определяется числом его
структурных единиц, соотношени-
соотношением их размеров и их поляризацион-
поляризационными свойствами.
Под структурной единицей сле-
следует понимать атом или ион, а иног-
иногда также группу атомов — молекулу
или же комплексный ион. В ионных
соединениях атомный номер элемен-
элемента и его валентность не являются
факторами, сколько-нибудь сущест-
существенно влияющими на структуру кри-
кристалла (иллюстрацию к правилу
см. § 7—11 настоящей главы).
ГЛАВА XI
ТЕОРИЯ ПЛОТНЕЙШИХ ШАРОВЫХ УПАКОВОК
§1. Гексагональная ¦ кубическая
илотиейшие шаровые упаковки
Геометрическая задача о макси-
максимальном заполнении пространства
шарами имеет бесконечное множест-
множество решений. Из них два решения, о
которых сейчас только и будет идти
речь, имеют для кристаллографии
наибольшее значение.
Плюский слой шаров, плотней-
шим образом прилегающих друг к
другу, представлен на рис. 192. Что-
Чтобы наложить плотнейшим образом
на первый слой второй, каждый шар
второго слоя следует поместить в
углубление между тремя шарами
первого слоя. Это показано на
рис. 193,6. Для большей ясности ша-
шары несколько раздвинуты. При нало-
наложении третьего слоя шаров возмож-
возможны два варианта. В варианте а каж-
каждый шар третьего слоя лежит на
трех шарах второго слоя таким обра-
образом, что под шаром третьего слоя
нет шара в первом слое. В вариан-
варианте б каждый шар третьего слоя так-
также лежит на трех шарах второго
слоя, но под каждым шаром третье-
третьего слоя оказывается шар в первом
слое. Плотность заполнения прост-
пространства шарами в обоих вариантах,
конечно, одинакова, одинаково и
координационное число шаров A2),
но симметрия в расположении ша-
шаров различна.
Вариант а отвечает кубической
сингонии (гранецентрированной ре-
решетке Бравэ), б — гексагональной
сингонии.
Процент занятого шарами прост-
пространства, при условии их касания,
для обоих вариантов равен 74,05.
Промежутки составляют немногим
более одной четверти общего объема.
На рис. 194 изображены плотнейшие
упаковки (кладки).
Рис. 192. Слой шаров, плотнейшим обра-
образом прилегающих друг к другу
Рис. 193. Проекции двух основных плот-
нейших упаковок шаров
а — кубическая;
б — гексагональная
Рис. 194. Плотнейшие упаковки шаров
по кубическому (а) и гексагональному
(б) законам
149
Между описанными упаковками
существует и практически важное
различие. В гексагональной структу-
структуре имеется лишь одно направление,
нормально к которому расположены
плотнейшие плоские слои, тогда
как в кубической таких направле-
направлений 4, соответственно четырем объ-
объемным диагоналям куба. Это обсто-
обстоятельство приводит к существен-
существенным физическим различиям, напри-
например, у металлов, кристаллизующихся
в том или другом типе структур.
§ 2. Тины нустот
в шаровых унаковках
Свободное пространство между ша-
шарами в плотнейших упаковках соот-
соответствует пустотам двух родов. Одни
окружены четырьмя шарами и име-
имеют, следовательно, координационное
число 4, другие располагаются меж-
между шестью шарами, т. е. имеют коор-
координационное число 6 (рис. 195).
Центры четырех шаров, между ко-
которыми образуется пустота первого
рода, располагаются по вершинам
тетраэдра, поэтому пустоты с коор-
координационным числом 4 носят назва-
название гетраэдрических пустот. Центры
шести шаров, замыкающих пустоту
второго рода, расположены по вер-
вершинам октаэдра и называются окта-
эдрическими.
Размеры этих пустот обусловлены
нижними критическими значениями
отношений радиусов для координа-
координационных чисел 4 и 6 (см. табл. 12,
стр. 143). Если радиус шаров упа-
упаковки принять за единицу, то радиу-
радиусы шаров, которые могут быть поме-
помещены в тетраэдрические и октаэдри-
ческие промежутки, будут выра-
выражаться числами 0,22 и соответствен-
соответственно 0,41. На п шаров, уложенных
плотнейшим образом, приходятся
п октаэдрических пустот и 2п
тетраэдрических, т. е. на 1 шар
плотнейшей упаковки приходятся
Рис. 195. Тетраэдрические и октаэдриче-
ские пустоты между шарами в плотней-
плотнейших упаковках
1 октаэдрическая и 2 тетраэдри-
тетраэдрические пустоты. Это относится как
к кубической упаковке, так и к гек-
гексагональной. Обе структуры отлича-
отличаются друг от друга не числом и раз-
размерами пустот, а их взаимным рас-
расположением. На рис. 196, а показан
один шар из структуры плотнейшей
кубической упаковки и окружаю-
окружающие его 6 октаэдрических и 8 тетра-
тетраэдрических пустот в виде шариков,
которые могут быть помещены в эти
пустоты. Рис. 196,6 изображает шар
из структуры плотнейшей гексаго-
гексагональной упаковки в аналогичном
окружении.
Рис. 196. Шар из плотнейших кубической
(а) и гексагональной (б) упаковок, окру-
окруженный малыми шарами из тетраэдриче-
тетраэдрических и средни*"* — из октаэдрических
пустот
!50
§ 3. Многослойные упаковки.
Способы обозначения
плотнейшпх шаровых упаковок
Зная строение двух простейших упа-
упаковок шаров, легко понять, что чис-
число различных упаковок бесконечно
велико. В самом деле, в гексагональ-
гексагональной плотнейшей упаковке третий
слой повторяет первый, следо-
следовательно, упаковка двухслойная.
В кубической упаковке четвер-
четвертый слой повторяет первый, и
упаковка, следовательно, трехслой-
трехслойная. Четырехслойную упаковку мож-
можно получить укладкой первых трех
слоев шаров по «кубическому» зако-
закону, а четвертый уложить таким обра-
образом, чтобы он повторял второй (ина-
(иначе говоря, второй, третий и четвер-
четвертый слои будут уложены по гексаго-
гексагональному закону). Четырехслойную
упаковку иногда называют «топазо-
«топазовой», так как впервые она была отк-
открыта у минерала топаза. Пятислой-
ную упаковку можно получить, на-
наложив первые три слоя по кубиче-
кубическому закону, а последующие два —
по гексагональному. Очевидно, что
плотность всех этих упаковок одина-
одинакова, а число разнообразных случаев,
отличающихся друг от друга, в пер-
первую очередь числом слоев, повторя-
повторяющихся в направлении главной оси
упаковки (направление, перпенди-
перпендикулярное плотнейшим слоям), будет
бесконечно велико. Так, легко себе
представить не только двух-, трех-,
четырех- и пятислойные упаковки,
но и шести-, семислойные и т. д.
Обозначив каждый слой упаковки
буквами А, В или С и условившись
слой, повторяющий какой-либо из
предыдущих, обозначать одинаковой
с ним буквой, мы придем к весьма
простому и удобному обозначению
упаковок. Двухслойная будет обо-
обозначаться рядом ...АВАВАВ.., трех-
трехслойная ...АВСАВС, четырехслой-
ная ...АВСВАВСВ.., пятислойная
...АВСАВАВСАВ... и т. д. Трех букв
достаточно, чтобы изобразить любую
многослойную упаковку. Начальный,
т. е. любой, слой мы можем обозна-
обозначать какой угодно из этих букв. Важ-
Важно соблюдать последовательность
букв. Так, например, четырехслойную
упаковку мы можем обозначить не
только так, как было указано выше,
но и иначе, а именно ...АВ AC ABAC...
и этот способ записи тождествен с
предыдущим. Слой А при первом ва-
варианте записи четырехслойной упа-
упаковки обозначен во втором варианте
через С, соответственно слой В обо-
обозначен теперь буквой А и слой С —
буквой В. Последовательность слоев
осталась, конечно, одинаковой, в чем
можно убедиться, подписав оба ряда
букв друг под другом:
ВСВА
ABAC
ВСВ .
ABAC.
Расстояние между двумя верти-
вертикальными чертами указывает величи-
величину трансляции вдоль главной оси,
выраженную числом слоев упаковки.
Первая черта может быть поставлена
в любом месте, в частности на букве
или между двумя буквами, вторая
должна отстоять от нее в данном слу-
случае на четыре буквы, т. е. на четыре
слоя. Среди многослойных упаковок
могут быть разные упаковки с одина-
одинаковым числом слоев. Так, например,
имеются две шестислойные упаковкь
...АВСАСВ... и ...АВАВАС... Перейти
от одной упаковки ко второй, анало-
аналогично тому, как это мы сделали для
двух вариантов написания четырех-
четырехслойной упаковки, нельзя. Эти упа-
упаковки не могут быть совмещены друг
с другом. Относительное расположе-
расположение слоев в них различно. Это — две
различные упаковки, в то время как
четырехсложная упаковка одна.
Внимательно рассматривая буквен-
буквенные выражения (формулы) плотней-
ших шаровых упаковок, нетрудно ви-
видеть, что любой шар (буква) может
находиться или между повторяющи-
повторяющими друг друга слоями шаров, как в
гексагональной плотнейшей упаков-
упаковке, т. е. между одинаковыми буквами,
или между двумя слоями шаров, не
151
повторяющими друг друга, как в ку-
кубической плотнейшей упаковке, т. е.
между разными буквами. Располо-
Расположение пустот вокруг избранного ша-
шара и любого шара рассматриваемого
слоя будет в первом случае такое же,
как вокруг шара в плотнейшей гек-
гексагональной упаковке, а во втором
случае — как вокруг шара в куби-
кубической упаковке. Поэтому эти шары
(или, точнее, слои) удобно обозна-
обозначать соответственно буквами г ж к.
Таким образом, мы приходим к ново-
новому обозначению шаровых упаковок.
Ниже даются сопоставления обо-
обозначений для первых шести упаковок:
п = 2 ... АВАВАВ...
г г г г г г
п = Ъ ... ABC ABC ...
к ккккк
л = 4 ... АВАСАВ . . .
к гкг кг
п = Ъ ... ABC AB ABC . . .
гкккггкк
п = 6 A). .. АВСАСВАВС . . .
г к кг к к гкк
B)... ABABACABA .. .
кгггкгкгг
Семислойных упаковок — три, вось-
мислойных — шесть и т. д.
Недостатком обозначения упако-
упаковок буквами к ж г, в отличие от обо-
обозначения буквами А, В ж С, является
то, что непосредственно из формулы
не виден порядок упаковки («слой-
ность»). Преимущество нового спо-
способа заключается в более легком об-
обнаружении элементов симметрии упа-
упаковки.
§4. Предварительные замечания
о симметрии шаровых
упаковок.
Кубическая п.ютпейшая
шаровая упаковка
В слое плотно упакованных шаров
(рис. 197) через центр каждого ша-
шара перпендикулярно к слою прохо-
проходит ось шестого порядка и шесть
152
Рис. 197. Элементы симметрии плотнейше-
го слоя шаров
плоскостей симметрии. Чероз каж-
каждую пустоту проходят оси третьего
порядка и по три плоскости симмет-
симметрии. Если перейти ко второму, треть-
третьему и т. д. слоям и помещать над
пустотами шары новых слоев, то лег-
легко видеть, что ось шестого порядка,
присутствующая в изолированном
(первом) слое, превратится в ось
третьего порядка в любой трехмер-
трехмерной плотнейшей упаковке. При этом
исчезнут три плоскости симметрии
из шести. Оси третьего порядка
и плоскости симметрии, проходившие
через пустоты в первом слое, ника-
никаких изменений не претерпят. Таким
образом, в любой многослойной упа-
упаковке мы будем иметь три систе-
системы осей третьего порядка (проходя-
(проходящие через центры шаров и центры
пустот обоих типов) с проходящими
через них плоскостями симметрии.
Каждая из плоскостей симметрии яв-
является общей для всех трех осей.
Эти оси симметрии в частных случа-
случаях могут быть шестерными зеркаль-
зеркально-поворотными, инверсионными или
шестерными винтовыми осями, но
при всех обстоятельствах они будут
включать в себя поворотную ось
третьего порядка и три плоскости
симметрии, проходящие через нее.
До сих пор рассматривались эле-
элементы симметрии, перпендикуляр-
перпендикулярные к плоскости исходного слоя ша-
шаров. В действительности же, кроме
этих элементов симметрии, разные
плотнейшие упаковки будут иметь
и другие элементы симметрии. Вы-
Выше, говоря о типах пустот в плотней-
гпих упаковках, мы указывали, что
шары плотнейших упаковок могут
быть двух типов — гик. При этом
симметрия их различна. В частности,
шар типа г будет иметь только одну
ось третьего порядка, а шар типа
к — четыре оси третьего порядка.
Единственная поворотная ось выс-
высшего порядка в шаре типа г будет
обязательно совпадать с главной
осью упаковки, а вся упаковка бу-
будет иметь одну главную ось треть-
третьего порядка (точнее, три системы
параллельных осей третьего поряд-
порядка), если в формуле, составленной
буквами гиге, есть хотя бы одна
буква г. Из сказанного следует, что
только одна упаковка ...кккк... будет
принадлежать к кубической синго-
нии. Федоровская пространственная
группа ее — Fm3m. Остальные плот-
нейшие упаковки будут принадле-
принадлежать к гексагональной сингонип. Тер-
Термин «гексагональная сингония» ис-
используется в широком смысле слова,
т. е. под ним подразумеваются обе
подсингонии — собственно гексаго-
гексагональная и тригональная.
Вывод всех случаев симметрии
плотнейших шаровых упаковок
впервые был сделан Н. В. Беловым.
§5. Фёдоровские группы симметрии
г енсаг опальных
шаровых упаковок
Помня о том, что любая плотнешпая
шаровая упаковка обязательно име-
имеет в качестве главной оси ось треть-
третьего порядка, через которую прохо-
проходят три плоскости симметрии, не-
нетрудно выбрать из 12 видов сим-
симметрии гексагональной сингонии
пять, удовлетворяющих этому свой-
свойству. Такими видами симметрии бу-
будут следующие:
L66P=6mm,
Дальнейший вывод может быть
сведен к выбору пространственных
групп у этих пяти видов симметрии,
что легко сделать, пользуясь табл. 7
(стр. 83—87).
Очевидно, что симметрия групп
должна удовлетворять тому же тре-
требованию. Ниже собраны все про-
пространственные группы указанных ви-
видов симметрии. Подчеркнуты те
группы, которые имеют зеркальные
плоскости симметрии, проходящие
через оси третьего порядка всех
трех систем (см. рис. 118, 121, 123,
126, 128). Это и будут 7 простран-
пространственных групп плотнейших упако-
упаковок гексагональной сингонии:
1) 3m: P3ml, P3cl, Р31т,
Р31с, ДЗт, ЛЗс
2) Зт: P3ml, P3cl, РЫт,
Р31с, ДЗт, ДЗс
3) 62т: Р6т2, РЪс2, Р62т, Рб2с
4) бтт: Рбттга, Рбсс, Р&зтс,
5) 6/ттт:Р6/ттт, Рб/тсс, Рбз/тст,
3m,
Остальные группы отпадают, так
как некоторые из них имеют не
зеркальные плоскости симметрии,
а плоскости скользящего отраже-
отражения; затем, зеркальные плоскости
проходят не через все оси третьего
порядка, и, наконец, вследствие на-
наличия поворотных осей шестого по-
порядка, которых не может быть
в плотнейших упаковках.
До 8-слойных упаковок включи-
включительно встречаются только 4 груп-
группы: D-^PSmi, D\h=PQzlmmc,
D *h =PEm2 и Qf=Fm3m. Группы
Dla—RBm и C^=P3ml встречаются
впервые в 9-сло иных упаковках. Груп-
Группа C\v — PQzmc встречается впер-
впервые только в 12-слойных упаковках.
Группа Ch—R3m встречается пер-
первый раз только среди 21-слойных
упаковок.
153
I в. Элементы симметрии
плотпейшпх шаровых упаковок
Выше мы указали 8 федоровских
(пространственных) групп симмет-
симметрии, возможных в плотнейших упа-
упаковках.
Поскольку к кубической сингонии
принадлежит только одна упаков-
упаковка — трехслойная ...АВСАВС... или
...кккк..., имеющая пространствен-
пространственную группу Fm3m, то не представля-
представляет труда разобраться в том, где и ка-
какие элементы симметрии будут про-
проходить в пространстве, заполненном
шарами по этому закону. Переходя
же к гексагональным упаковкам, мы
встречаемся с тем обстоятельством,
что в каждую группу попадает бес-
бесконечное множество упаковок с раз-
различными периодами идентичности.
Вопрос, следовательно,сводится к то-
тому, чтобы найти, в каких слоях или
между какими слоями располагаются
дополнительные (к основному комп-
комплексу РЗ) элементы симметрии: пло-
плоскости, перпендикулярные к главной
оси, и центры симметрии. Про-
Производные двойные оси, конечно, лег-
легко могут быть найдены в результате
сложения плоскостей симметрии.
Обозначение плотнейших упаковок
при помощи букв гик позволяет
без чертежа и модели находить эти
дополнительные элементы симмет-
симметрии.
Так, через центры шаров каж-
каждого слоя, обозначенного буквой г и
разбивающего всю упаковку на две
симметричные части, проходит плос-
плоскость симметрии, перпендикулярная
к главной оси. Если буква гс разбива-
разбивает всю формулу упаковки на две сим-
симметричные части, то в центрах шаров
этого слоя будут располагаться цент-
центры симметрии. Центры симметрии
будут находиться также и между
слоями в тех случаях, когда пара
одинаковых букв кк или гг делит фор-
формулу упаковки на две зеркально рав-
равные части. В качестве примера при-
приведем одну из 9-слойных и одну из
12-слойных упаковок:
В
X
АВАВСАВАВ
гггкккггг
X X
ВАВСАВСАСВАСВА
i i
кгкккккгкккккг
II X II X ||
Вертикальной чертой обозначена
граница периода, двойной чертой —
плоскость симметрии, крестиком —
центр симметрии.
§ 7. Правильные системы точек
в плотнейших
шаровых упаковках
В упаковках двух- и трехслойных
все шары располагаются по точкам
одной федоровской правильной си-
системы, т. е. они кристаллохимически
тождественны. Однако для упаковок
высоких порядков слоиности эта осо-
особенность может не соблюдаться. Этот
факт легко показать на примере пя-
тислойной упаковки, имеющей федо-
федоровскую группу Р3т\. В примитив-
примитивном параллелепипеде решетки этой
упаковки содержатся 5 атомов, а
кратность 5 невозможна ни в одной
федоровской группе. В группе Р3т1
имеются кратности: 1, 2, 3, 6 и 12.
Следовательно, шары плотнейшей пя-
тислойной упаковки кристаллохими-
кристаллохимически не (могут быть тождественными,
они различаются физически, в част-
частности своей симметрией. Такие упа-
упаковки следует считать упаковками
из двух (или более) типов шаров
одного размера. Условно станем
считать такие шары окрашенными
в разные цвета, а всю упаковку —
упаковкой разноцветных шаров.
Разноцветные шары не могут быть
совмещены друг с другом никакими
симметрическими преобразованиями,
мыслимыми в данной пространствен-
пространственной группе. Так как шары в гс-слой-
ных упаковках обязательно несколь-
нескольких типов «цветов», то их, очевидно,
можно распределить по местам упа-
упаковки разными способами и, в част-
частности, так, что симметрия ее станет
154
иной, например повысится до куби-
кубической.
Высота слоя в шаровой упаковке
равна yQ-d/З, где d— диаметр шара.
Отношения длины трансляции вдоль
пространственной диагонали куба к
длине трансляции вдоль диагонали
грани (т. е. в -слое плотнейшей упа-
упаковки) для трех типов кубических
решеток Р, I и F равны соответст-
соответственно: У6/2, У674 и УбГ
Если в слое имеется трансляция
между двумя ближайшими шарами,
т. е. ее длина равна d, то может осу-
осуществиться только одно из трех вы-
выписанных выше отношений, именно,
в трехслойной упаковке — ЗУ6/3. От-
Отсюда можно сделать однозначный
вывод, что из тождественных («од-
(«одноцветных») шаров можно сложить
одну плотнейшую упаковку — трех-
трехслойную (т. е. кубическую) с прост-
пространственной группой Fm3m.
Однако если взять шары двух цве-
цветов и распределить их таким образом,
чтобы они, чередуясь друг с другом,
обеспечивали трансляцию в слое, рав-
равную 2d, то при трехслойной упаков-
упаковке мы получим соотношение Уб/2, удо-
удовлетворяющее примитивной решетке,
I и придем, следовательно, к новой
I пространственной группе для этой
упаковки. Для этого случая можно
привести и реальный пример —
структуру Cu3Au.
Если взять в слое трансляцию,
равную Ad, то отношение в трехслой-
трехслойной упаковке приведет нас к объем-
ноцентрированной кубической ре-
решетке и к новой пространственной
группе для плотнейших упаковок.
Полная диагональ куба будет равна
шести слоям. Для этого случая мы
будем иметь четыре упаковки: двой-
двойную кубическую, тройную гексаго-
гексагональную и две шестислойных. Сим-
Симметрия последних трех упаковок,
конечно, останется гексагональной,
хотя элементарный ромбоэдр у них
будет иметь форму куба. Однако
двойная кубическая упаковка >. ша-
шарами двух цветов может сохранить
кубическую симметрию при объем-
ноцентрированной ячейке, т. е. будет
принадлежать к еще одной новой
пространственной группе. Процесс
усложнения можно, очевидно, про-
продолжить до бесконечности.
Из сказанного легко прийти к вы-
выводу, что кубических плотнейших
упаковок из разноцветных шаров од-
одного размера может быть бесконечно
много. Разноцветные шары можно за-
закономерным образом распределить
по местам плотнейших кубических
гс-слойных упаковок (где п кратно
трем) таким образом, что сохранится
кубическая симметрия, но не обяза-
обязательно гранецентрированная ре-
решетка.
Упаковки с высокими значениями
га, как было показано выше, обяза-
обязательно построены из разноцветных
шаров. С этой точки зрения новые
кубические упаковки, имеющие две
федоровские группы, не являющиеся
ни одной из восьми указанных выше
групп симметрии, равноценны дру-
другим упаковкам с я>4.
Естественна, конечно, возможность
получения с помощью разноцветных
шаров и других (не кубических) упа-
упаковок, в частности иных гексагональ-
гексагональных (кроме 7, описанных выше), а
также тетрагональных, ромбических,
моноклинных и триклинных.
§ 8. Значение теорпп
шаровых упаковок
для кристаллохимии
При изучении простейших структур
мы уже встретились с тем явлением,
что кристаллы многих химических
элементов построены по принципу
плотнейшей упаковки. Плотнешпая
кубическая упаковка характерна
для кристаллов: Си, Ag, Аи, Са, Sr,
Al, Th, Pb, Nb, v-Fe, cc-Co, Ni, Rh,
Pd, Ir, Pt и др. Плотнейшую гекса-
гексагональную кладку имеют: Be, Mg,
fi-Ce, Tl, Ti, Zr, Hf, fi-Cr, B-Co, Ru, Os
и др. Кристаллический Sm имеет
9-слойную упаковку ...АВАВСВСАС...
155
Принцип плотнейшей упаковки
остается справедливым и для ионных
соединений. Размеры анионов, как
правило, значительно больше разме-
размеров катионов. В ионных структурах
анионы располагаются по одному из
законов плотнейшей кладки, ка-
катионы же располагаются в проме-
промежутках между анионами, в пустотах.
Этим объясняется тот факт, что са-
самыми распространенными координа-
координационными числами для катионов яв-
являются 4 и 6. Но катионы обычно не
заполняют всех пустот между анио-
анионами.
Идея плотнейшей анионной клад-
кладки оказалась очень плодотворной
при описании известных структур и
при определении новых. Так, напри-
например, структура NaCl образована
плотнейшей кубической упаковкой
ионов хлора с заполнением всех ок-
таэдрических пустот ионами натрия.
Тетраэдрические пустоты остаются
свободными. Структура NiAs харак-
характеризуется плотнейшей гексагональ-
гексагональной кладкой ионов мышьяка с за-
заполнением всех октаэдрических пус-
пустот ионами никеля. В структуре цин-
цинковой обманки мы имеем плотней-
шую кубическую кладку из ионов
серы; половина тетраэдрических пу-
пустот занята атомами цинка. Вторая
модификация ZnS — вюртцит — ха-
характеризуется плотнейшей гексаго-
гексагональной укладкой ионов серы с за-
заполнением половины тетраэдриче-
тетраэдрических пустот ионами цинка.
Структуры АХг могут быть полу-
получены из плотнейшей анионной упа-
упаковки с заполнением половины окта-
октаэдрических пустот. Причем это запол-
заполнение может происходить различны-
различными способами. Например, пустоты
могут быть заполнены рядами — че-
через один слой, через два и т. д.; слоя-
слоями — через один слой или зигзагооб-
зигзагообразно и т. п., что приводит к их боль-
большому многообразию. Структуры А2Х
могут быть получены плотнейшей
укладкой анионов и заполнением
всех тетраэдрических пустот катио-
катионами.
156
Это имеет место, например, в
структурах Li2O, Na2O и т. д.
Структуры соединений АгХз могут
быть получены плотнейшей укладкой
анионов и заполнением, предполо-
предположим, Уз октаэдрических пустот ка-
катионами, см., например, AI2O3.
Самые сложные структуры силика-
силикатов часто могут быть интерпретиро-
интерпретированы с помощью плотнейшей кисло-
кислородной упаковки с заполнением ка-
катионами промежуточных пустот (пи-
роксены, амфиболы).
Принцип плотнейшей укладки по-
получил подтверждение для веществ
с ненаправленными связями между
структурными единицами, особенно
для металлов и ионных соединений.
Он остается справедливым и для
структур молекулярных, в частности
органических соединений, хотя в этом
случае применение его осложняется
тем, что форма сложных молекул
обычно сильно отличается от шара
(см. гл. XIX, § 6). Если же форма
структурных единиц шаровая или
близкая к шару, то структуры ве-
веществ с Ван-дер-Ваальсовыми связя-
связями геометрически ничем не отличают-
отличаются от металлических структур. Так,,
например, гелий кристаллизуется в
плотнейшей гексагональной упаков-
упаковке, а остальные инертные газы — в
кубической.
Принцип плотнейшей упаковки
явился еще одним подтверждением
предложенного Е. С. Федоровым раз-
разделения кристаллов на два типа: ку-
кубический и гексагональный.
§ О. Метод изображения
структурных типов
С помощью многограпппков.
Структуры па тетраэдров
и октаэдров
Поскольку расположение анионов
обычно определяется одним из двух
вариантов плотнейшей упаковки ша-
шаров, то нет необходимости показы-
показывать это на модели. Достаточно ука-
указать только на тип кладки. Положе-
Положение же катионов, занимающих пус-
Рис. 198. Типы катион-
ных многогранников —
тетраэдров и октаэдров
(по Л. Полингу)
Рис. 199. Структурный
тип CdCb, изображен-
изображенный с помощью коорди-
координационных многогранни-
многогранников
Рис. 200. Структурный
тип NaCl
Рис. 201. Структурный
тип NiAs
Рис. 202. Структурный
тип сфалерита ZnS, по-
построенный из шаров (а)
и из многогранников
Рис. 203. Структурный
тип вюртцита ZnS
Рис. 204. Структурный
тип рутила ТЮг в двух
аспектах
тоты между шарами, а также коор-
координационные числа катионов необ-
необходимо особенно подчеркивать, что-
чтобы показать разницу между теми
или иными структурами. Л. Полинг
достигает этого тем, что центры ани-
анионов, окружающих катион, соединяет
линиями. В результате получается
¦многогранник, число вершин которо-
которого дает координационное число кати-
катиона, а пространственное распределе-
распределение многогранников наглядно пока-
показывает взаимное расположение ка-
катионов.
На рис. 198 показаны катионы с
координационными числами 4 и 6 —
тетраэдр и октаэдр. Шары являются
анионами плотнейшей кладки. На
рис. 199 изображена структура CdCl2,
выполненная по этому методу.
Полиэдрический метод применяет-
применяется главным образом при описании
структурных типов, а не отдельных
структур. Ребра тетраэдров, октаэд-
октаэдров и других фигур берутся одина-
одинаковыми, а сами фигуры неискажен-
неискаженными, хотя в действительности они
часто отличаются от правильных
форм. Масштаб модели лишь при-
приблизительно отвечает относительным
размерам межатомных расстояний в
структуре.
Этот метод особенно удобен при
описании структурных типов слож-
сложных соединений, например сили-
силикатов.
Рис. 205. Структурный тип CdJ2 (двух-
(двухслойной модификации)
Рис. 206. Структурный тип коррунда А12Оз
На рис. 200—206 показаны извест-
известные уже нам структуры: NaCl, NiAs,
сфалерит ZnS, вюртцит ZnS, рутил
ТЮ2, СсМг, корунд AI2O3, изображен-
изображенные при помощи координационных
тетраэдров и октаэдров.
§10. Структуры со сложными
координационными
многогранниками
Н. В. Белову удалось распространить
принцип плотнейшей укладки на
весьма сложные соединения. Для это-
этого ему пришлось определить формы
катионных многогранников с редко
встречающимися координационными
числами (рис. 207). Рассмотрим не-
несколько примеров структур таких со-
соединений.
1. Атомы никеля в структуре при-
природного миллерита NiS располагают-
располагаются в центре многогранника, имеюще-
имеющего форму, похожую на половину ок-
октаэдра (координационное число 5)
(а).
2. В структуре M0S2 в силу особен-
особенности электронной оболочки молибде-
молибдена координационному числу 6 отве-
отвечает тригональная призма (б). Струк-
Структура представляет собой чередова-
чередование слоев тригональных призм, за-
заполненных ионами молибдена, и пус-
пустых слоев из октаэдров. Это отража-
отражает слоистый характер структуры
(рис. 208).
3. Многогранник, отвечающий ко-
координационному числу 7, представ-
представляет собой комбинацию тригономет-
тригонометрической призмы с половиной окта-
октаэдра (рис. 207, в). Структуры этого
типа тесно связаны со структурами,
имеющими координационное число 5.
В зависимости от того, где находится
центральный атом — ближе к центру
призмы или к центру пирамиды
(половины октаэдра),— получаются
структуры с координационным чис-
числом 7 или 5.
Примером структуры с координа-
координационным числом 7 является структу-
структура антимонита Sb2S3.
158
Рпс. 207. Многогранники Н. В. Белова
Рис. 208. Структурный тип MoS2
Рис. 209. Структурный тип СаТЮз
Рис. 210. Структурный тип КзГСо(ГТО2)в]
4. В структуре CsCl все ионы цезяя
занимают центры всех кубов (г).
В структуре CaF2 ионы кальция за-
занимают центры половинного числа
всех кубов. G подобными же кубами
из кислородных ионов вокруг Са мы
встречаемся в скаполитах.
5. Атомы меди в структуре СиА1г
и калия в структуре KHF2 имеют
координационное число 8 и помеща-
помещаются в многограннике, называемом
томсоновским кубом. Он может быть
получен из куба путем поворота верх-
верхней квадратной грани относительно
нижней на некоторый угол (д).
6. Ионы циркония в структуре цир-
циркона ZrSiC>4, ионы кальция в струк-
структуре шеелита CaWC>4 и ангидрита
GaSO4 помещаются в многогранниках
с координационным числом 8 (е).
Вторые «катионы» располагаются в
тетраэдрах.
Следующие четыре многогранника
характеризуют катионы, имеющие
координационное число 12. Эти слу-
случаи встречаются у катионов большого
размера. Такие катионы сами зани-
занимают положение шаров плотнейшей
кладки.
7. Кубооктаэдр (ж) характерен для
кальция в структуре СаТЮз (рис.
209), для цезия — CS3TI2CI9, для ка-
калия — КгРМЗб. В последнем струк-
структурном типе кристаллизуется боль-
большое количество веществ, в частности
одна из модификаций (NH^SiFe.
8. Примером гексагонального ана-
аналога кубооктаэдра (с плоскостью
симметрии, перпендикулярной трой-
тройной оси) (з) может служить коорди-
координационный многогранник иона
аммония в другой модификации
(NPL^SiFe или иона натрия в мине-
минерале сведенборгите NaBe4Sb07.
9. Многогранник в форме притуп-
притуплённого тетраэдра (лавесовский по-
полиэдр) (и) встречается в металличе-
металлических структурах таких соединений,
в которых радиус атома одного эле-
элемента в У2 раза больше радиуса атома
другого элемента (объем в два ра-
раза больше), например в структуре
MgCu2. Если формулу написать в ви-
159
де СигСиг, то ей отвечала бы обычная
плотнейшая укладка атомов меди.
Вместо этого мы имеем MgCu2, т. е.
в исходной плотнейшей укладке каж-
каждая пара отсутствующих атомов ме-
меди замещается одним крупным ато-
атомом, в данном случае атомом магния.
Другие примеры: КЫг, РЬАиг, BiAu2,
СиВег и минерал мальдонит Au2Bi.
Из тех же многогранников, но насла-
наслаиваемых по несколько другим зако-
законам, построены структуры MgZn2 и
MgNi2.
10. Ион калия в структуре
Кз[Со(]>Ю2)б] помещается в центре
двадцатигранника (в). Этот двадца-
двадцатигранник не представляет правиль-
правильного икосаэдра, а является комбина-
комбинацией пентагон-додекаэдра {201} с ок-
октаэдром {111}. Он имеет 8 равносто-
равносторонних и 12 равнобедренных тре-
треугольников. Атом кобальта, окружен-
окруженный шестью атомами азота, имеет тот
же многогранник из атомов кисло-
кислорода.
Вся структура представлена на
рис. 210. Верхняя часть структуры
отодвинута вверх, чтобы яснее были
видны составляющие ее многогран-
многогранники.
ГЛАВА XII
ТИПЫ ХИМИЧЕСКИХ СВЯЗЕЙ В КРИСТАЛЛАХ
В первом параграфе гл. X (стр. 134)
уже кратко упоминалось о различных
типах химической связи в кристал-
кристаллах. В настоящей главе этот вопрос
будет рассмотрен подробно.
Если для понимания ионной связи
достаточно знания периодического
закона и старых (классических)
принципов теории валентности, то
для объяснения ковалентной связи
этих сведений уже совершенно не-
недостаточно. Даже для элементарного
объяснения этого типа связи необхо-
необходимо привлекать квантовую теорию
вещества (волновую механику). По-
Поэтому в настоящей главе специаль-
специальный параграф (§4) посвящен изло-
изложению основных принципов волно-
волновой (квантовой) механики. Читатели,
знакомые с основами волновой меха-
механики, могут пропустить этот параг-
параграф без ущерба для понимания даль-
дальнейшего текста.
§ 1. Периодическая система
химических элементов
и строение атома
1. Электронная структура атомов.
После открытия Менделеевым
A869 г.) периодического закона хи-
химических элементов и наглядного
изображения его в виде таблицы
(табл. 17) был выполнен ряд работ,
благодаря которым постепенно выяс-
выяснялась физическая сущность, лежа-
лежащая в основе открытой закономерно-
закономерности.
В 1911 г. Резерфорд эксперимен-
экспериментально доказал, что каждый атом со-
11 Кристаллохимия
стоит из положительно заряженного
ядра, окруженного электронами.
Через несколько лет в результате ра-
работы Мозли A913 г.) выяснилась
природа менделеевского атомного но-
номера; атомный номер химического
элемента оказался равным значению
положительного заряда ядра атома
этого элемента, выраженного в еди-
единицах абсолютного значения заряда
электрона. Другими словами, атом-
атомный номер равен числу электронов,
окружающих ядро.
Еще раньше появились первые ра-
работы по квантованию энергии — сна-
сначала применительно к излучению
«абсолютно черного тела» (Планк,
1901 г.), а после объяснения законов
фотоэлектрического эффекта (Эйн-
(Эйнштейн, 1905 г.)—применительно ко
всем системам атомных размеров.
Важнейшим шагом в этом направле-
направлении явились работы Бора A913 г.),
применившего принцип квантования
к проблеме строения атома. В качест-
качестве наглядной модели атома в этой
теории используют обычно солнеч-
солнечную систему, где в центре находится
ядро (Солнце), а вокруг, по орбитам
движутся электроны (планеты).
Однако в дальнейшем от такой на-
наглядной модели пришлось отказаться.
Если в солнечной системе орбиты
всех планет можно точно рассчитать,
то в атоме определить точное поло-
положение движущегося электрона невоз-
невозможно, т. е. нельзя говорить о впол-
вполне определенной орбите его движе-
движения вокруг ядра. Это является след-
следствием существующего в микромире
принципа неопределенности (Гейзен-
161
берг, 1927 г.), согласно которому для
всех элементарных частиц, в том чис-
числе и электронов, нельзя одновремен-
одновременно определить положение и импульс
(а следовательно, и скорость).
Из этого принципа, однако, не сле-
следует, что движение электронов в ато-
атоме совсем произвольно. Напротив,
электроны должны находиться во
вполне определенных областях про-
пространства, расположенных вокруг
атомного ядра. Эти области, заменив-
заменившие первоначальные боровские орби-
орбиты, обычно называют орбиталями.
Такие области образуют некоторые
замкнутые пространственные слои
вокруг ядра, которые принято назы-
называть оболочками. Электроны вокруг
ядра образуют оболочечную структу-
структуру. На каждой оболочке может нахо-
находиться только вполне определен-
определенное число электронов. Если атом не
возбужден, то электроны, вообще го-
говоря, занимают оболочки по порядку,
начиная с самой внутренней. Оболоч-
Оболочкам, начиная с самой внутренней,
присвоены порядковые номера 1, 2,
3 и т. д.* Номер наружной оболочки,
в которой еще имеется злектрон, со-
соответствует номеру периода таблицы
Менделеева, в котором расположен
элемент данного атома.
Номер оболочки обычно обознача-
обозначают буквой п и называют главным
квантовым числом. Это число есть
одна из характеристик квантового со-
состояния атомного электрона, оно ха-
характеризует принадлежность элект-
электрона к определенной электронной
оболочке.
Квантовое состояние электрона ха-
характеризуется еще рядом других ве-
величин, о которых будет сказано ниже.
Важно отметить, что число таких воз-
возможных квантовых состояний для
каждой оболочки вполне определен-
определенное и равно 2п2, причем в каждом
электронном состоянии может нахо-
находиться не более одного электрона
(принцип запрета Паули, 1925 г.).
* В спектроскопии оболочки принято так-
также обозначать буквами К, L, M, N, О...
162
Отсюда становится понятным прин-
принцип электронного строения атомов
элементов периодической системы
Менделеева. У самого легкого элемен-
элемента — водорода — первая оболочка за-
занята одним электроном. У следую-
следующего элемента — гелия — имеется
два электрона, и, следовательно, пер-
первая оболочка занята полностью.
С лития, имеющего три электрона, на-
начинается заполнение второй оболоч-
оболочки, в которой может находиться
2 -22=8 электронов. Дальнейшее за-
заполнение этой оболочки осуществля-
осуществляется в ряду элементов Be, В, С, N, О,
F и заканчивается в элементе Ne. Да-
Далее начинается заполнение третьей
оболочки, в которой может находить-
находиться 2-32 = 18 электронов, затем чет-
четвертой оболочки B-42=32 электро-
электрона) и т. д.
Электронные оболочки можно раз-
разбить на подоболочки (группы
электронов). Каждая подоболочка ха-
характеризуется момептом количества
движения электрона вокруг ядра (ор-
(орбитальным моментом). Эта величина
квантована, причем так, что квадрат
момента количества движения может
принимать только следующие зна-
значения:
Р2 = 1A + i)h2,
где h — постоянная Планка, деленная
на 2я, а I называется азимутальным
(или орбитальным) квантовым чис-
числом. Число I может принимать толь-
только следующие значения:
1 = 0,1,2....,в-1.
Соответствующие подоболочки
(и группы электронов) часто обозна-
обозначаются символами s, p, d, f и т. д.
Электроны с одинаковыми кван-
квантовыми числами п и I имеют одина-
одинаковую энергию. Энергия атомных
электронов квантована, т. е. значе-
значения энергии электронов на разных
подоболочках отличаются на конеч-
конечную величину. Эти значения образу-
образуют дискретную последовательность
(дискретный спектр энергии).
Число I характеризует также сим-
симметрию орбиталей, т. е. симметрию
областей, где наиболее вероятно рас-
расположены электроны данной подобо-
лочки. s-Подоболочка имеет сфериче-
сферическую симметрию, в ней могут нахо-
находиться максимально два электрона.
р-Подоболочка образует октаэдричо-
скую розетку, в ней могут находить-
находиться шесть электронов. Общее число
квантовых состояний в подоболочке
равно Zi=2Bl-\-l), т. е. для d и /
соответственно 10 и 14.
Заметим, что индивидуальные хи-
химические свойства элементов суще-
существенным образом связаны с характе-
характером симметрии внешних подоболочек
атома, с характером пространственно-
пространственного (углового) распределения электро-
электронов внешней подоболочки.
Орбитальный момент количества
движения есть векторная величина.
Однако квантовое число I определяет
только абсолютное значение этого
вектора:
но не его ориентацию в пространстве.
Ориентация в пространстве этого век-
вектора в общем случае остается неопре-
неопределенной. Только во внешнем магнит-
магнитном поле можно фиксировать величи-
величину составляющей орбитального
момента количества движения элект-
электрона вдоль этого магнитного поля.
Установлено, что энергия взаимодей-
взаимодействия орбитального движения элект-
электрона с магнитным полем квантована,
и соответственно квантована про-
пространственная ориентация вектора
момента количества движения. Эта
ориентация характеризуется кванто-
квантовым числом т (или тг), которое мо-
может принимать только следующие
значения: т = 0, ±1, ±2, ..., ±1.
Число т принято называть магнит-
магнитным квантовым числом. Проекция
орбитального момента на направле-
направление магнитного поля (скажем, вдоль
координатной оси z) может прини-
принимать только следующие значения:
Р. = mi.
Состояние электрона характеризу-
характеризуется еще одпим квантовым числом s,
называемым спином. Для грубой на-
наглядности спиновое состояние срав-
сравнивают с собственным моментом ко-
количества движения электрона (вра-
(вращения). Составляющая этого момен-
момента вдоль определенной оси может при-
принимать только два значения: ±й/2.
Соответствующее квантовое число
принимает значения s = ± '/г. Сог-
Согласно принципу Паули, в одном и
том же состоянии не могут быть два
электрона. Поэтому при одинаковых
h, l и т значения s у двух электро-
электронов противоположны по знаку
(s = -\-llz и s = — 7г). Говорят, что
спины двух электронов в этом случае
антилараллельны. Это обозначают
символом (| \) или знаком (-\ ).
Иногда говорят не об электронном
квантовом состоянии, а о квантовой
ячейке. Ячейка характеризуется
квантовыми числами п, I, т. В ней,
следовательно, может находиться
максимально два электрона с антипа-
антипараллельными спинами. Общая харак-
характеристика электронных состояний
атома приведена в табл. 18.
С помощью такой классификации
электронных состояний атома и с
учетом принципа запрета Паули лег-
легко описать электронное строение ато-
атомов в периодической системе элемен-
элементов. В каждом последующем элемен-
элементе периодической системы число
атомных электронов на единицу
больше. Новый электрон занимает
следующее по порядку электронное
состояние, но при том, однако, усло-
условии, чтобы получаемая электронная
конфигурация приводила бы к мини-
минимальной эпергпи атомной системы.
Если это условно не выполняется, то
электрон занимает квантовое элект-
электронное состояние не в указанном в
табл. 18 порядке, а такое, которое
соответствует минимальной энергии
атомной системы.
На рис. 211 показаны электронные
строения атомов элементов начала
периодической системы. Как видно
из рисунка, нарушение порядка рас-
11*
163
ТАБЛИЦА 18
Характеристика электронных состояний атома (во всех случаях *= + 7г)-
n
1
2
3
i
0
0
1
0
1
2
Символ
Is
2s
2p
3s
Ър
Ы
m
0
0
—1
0
+1
0
—1
о
+1
—2
—1
0
+1
+2
n
4
i
0
1
2
3
Символ
is
Ap
id
4/
m
0
—1
0
+1
—2
0
+1
+2
—3
2
—1
0
+1
+2
+3
положения электронов впервые об-
обнаруживается у калия, у которого
последний (валентный) электрон
расположен на 4«-подоболочке, а не
на З^-подоболочке. Такое нарушение
(отсутствие электронов на Зй-под-
оболочке) имеется и у последующих
химических элементов, и только на-
начиная со Sc «с опозданием» начина-
начинается заполнение Зй-подоболочки.
Второй особеннностью электронно-
электронного строения в периодической системе
элементов является то, что в послед-
последней (валентной) подоболочке элек-
электроны сначала последовательно за-
заполняют квантовые состояния с раз-
разными т (ячейки), но с одинаковыми
s (т. е. спины у валентных электро-
электронов при таком заполнении парал-
параллельны) ; это, например, имеет место
Рис. 211. Последовательность заполнения
электронами электронных оболочек ато-
атомов элементов начала периодической си-
системы
164
у С и N. Только после того, как во
всех квантовых ячейках окажется по
одному электрону, начинают запол-
заполняться квантовые состояния с анти-
антипараллельными спинами («спарива-
(«спаривание спинов»). Это, например, прояв-
проявляется у О ж F. Описанная особен-
особенность называется правилом Гунда
A931 г.). Наличие в атомах неспа-
ренных электронов позволяет объ-
объяснить магнитные свойства простых
веществ и химических соединений.
2. Основные сведения об ядре и
электроне. В настоящем разделе для
полноты картины мы кратко сообщим
основные данные о структуре ядер
и об электроне.
Ядра всех атомов химических эле-
элементов состоят из протонов и нейтро-
нейтронов. Каждый протон имеет положи-
положительный заряд, равный + 1,6-ДО-19 в,
нейтрон заряда не имеет. Масса про-
протона равна 1,679 -10~24 г, масса нейт-
нейтрона равна 1,675 • 10~24 г, т. е. немно-
немного меньше массы протона. Каждый
атом химического элемента имеет
строго постоянное и вполне опреде-
определенное число протонов в ядре (это чи-
число равно атомному номеру элемен-
элемента). Число же нейтронов в ядрах
атомов одного и того же химического
элемента может несколько разли-
различаться. Этому соответствует наличие
в природе нескольких изотопов хими-
химических элементов. Так, например, хо-
хорошо известны три изотопа водоро-
водорода: протий, ядро которого состоит
только из одного протона; дейтерий
с ядром из одного протона и одного
нейтрона и тритий, в ядре которого
содержится один протон и два нейт-
нейтрона. Различные изотопы элементов
отличаются, таким образом, друг от
друга своими атомными весами.
Физическая природа взаимодейст-
взаимодействия нейтронов и протонов в ядрах
атомов до сих пор полностью не вы-
выяснена. Однако известно, что это
очень сильное взаимодействие. По
этой причине можно с достаточной
строгостью считать ядро весьма ком-
компактным образованием и характери-
характеризовать его определенным линейным
размером,- называемым радиусом яд-
ядра. Радиусы атомных ядер оказались
порядка 10~12—10~13 см. Для сравне-
сравнения напомним, что линейные разме-
размеры атома имеют порядок величи-
величины 10~8 см.
О структурных особенностях элек-
электрона говорить почти невозможно.
Описание поведения электрона мож-
можно дать только в рамках волновой ме-
механики, которая указывает лишь об-
область его наиболее вероятного на-
нахождения. При этом можно опреде-
определить среднюю скорость его движения
в этой области. Она оказывается ог-
огромной. Так, например, электрон
атома водорода имеет среднюю ско-
скорость 2 • 10~ъсм/сек *. При такой сред-
средней скорости «облет» пространства,
где он может находиться, занимает
время порядка \.Qrxl сек. Поэтому
удобно ввести модельное представле-
представление об облаке размазанного заряда
электрона, окружающего ядро. Фор-
Форма этого облака определяется орби-
талями, которые волновая механика
определяет вполне точно.
Следует все же заметить, что по-
поскольку электрон заряжен, он может
взаимодействовать с другими заря-
заряженными частицами на далеких рас-
расстояниях. Это взаимодействие элект-
электрической природы(кулоновское взаи-
взаимодействие) и сравнительно слабое.
Однако установлено, что на очень ма-
малых расстояниях (порядка 10~13 см)
проявляется сильное взаимодействие
с другими частицами. Это можно
рассматривать как проявление струк-
структурной особенности электрона (нали-
(наличия у него конечных размеров).
Масса электрона равна 9,1-10~28 г,
т. е. почти в две тысячи раз меньше
массы протона. Отсюда можно сде-
сделать заключение, что практически вся
масса атома сосредоточена в ядре.
3. Нормальное и возбужденное со-
состояние атомов. Ионы. Как уже отме-
отмечалось, электроны в атомах в нор-
нормальном состоянии заполняют внут-
Скорость света ~ 3 • 1010 см/сек.
165
реинис оболочки («нижние» элек-
электронные состояпия) так, что атом в
нормальном состоянии имеет мини-
минимальную энергию. Все остальные
электронные состояния остаются пус-
пустыми. Переход на них электрона из
занятого состояния соответствует по-
поглощению атомной системой вполне
определенных порций энергии. Состо-
Состояние атома, в котором его энергия не
минимальная, а повышена из-за пере-
перехода электрона в ранее незанятое
состояние, называется возбужденным
состоянием. Атом в возбужденном
состоянии обычно обозначается сим-
символом химического элемента со знач-
значком звездочки. Например, Н* означа-
означает возбужденный атом водорода. При
возбуждении атома водорода в зависи-
зависимости от величины поглощаемой энер-
энергии возможен переход электрона с
орбитали Is, соответствующей нор-
нормальному состоянию атома, на орби-
орбитали 2s, 2p и т. д. Возможно также
постепенное возбуждение атома: сна-
сначала переход с орбиталп Is на орби-
таль 2s, затем с 2s на 2р и т. д. Для
примера на рис. 212 показано элект-
электронное строение атома углерода в
нормальном и возбужденном состоя-
состояниях.
Для каждого электрона в атоме
существует вполне определенная мак-
максимальная энергия, при возбуждении
которой электрон уже не будет связан
с атомной системой. При этой энер-
1s 2s
Рис. 212. Электронное строение атома уг-
углерода в нормальном и возбужденном
состояниях
1s 2s 2p
шшшп
Рис. 213. Электронное строение атома
и иона натрия
гии возбуждения электрон покидает
атом. Соответствующую эпергию воз-
возбуждения называют энергией иониза-
ионизации атома, а сам атом называют по-
положительным ионом (или катионом)
соответственно тому, что он приобре-
приобретает положительный заряд. Остав-
Оставшиеся электроны не компенсируют
положительный заряд ядра. Оче-
Очевидно, заряд иона равен по величине
и обратен по знаку заряду электрона
(или суммарному заряду всех по-
потерянных электронов, если потерян
не один, а несколько электронов).
Следует обратить внимание, что
атом может поглощать энергию толь-
только вполне определенными порциями.
Поэтому, соответственно условиям
возбуждения атома", возможны случаи
перехода электронов как с внешних,
так и с внутренних оболочек. Анало-
Аналогично и выбивание электрона из
атома («ионизация») может проис-
происходить как с внешних, так и с внут-
внутренних оболочек. Однако в последнем
случае за очень короткий последую-
последующий промежуток времени (порядка
10~8 сек) происходит перестройка
электронной структуры так, чтобы
энергия иона стала минимальной,
чему соответствует переход электро-
электрона с внешних оболочек на незанятое
состояние во внутренней оболочке.
Процесс приводит в конце концов
к тому, что незанятое состояние ока-
оказывается на самой внешней подобо-
лочке. Для примера на рис. 213 по-
показаны электронные строения атома
и иона натрия.
Энергия ионизации различна для
отрыва разных электронов. Она наи-
наименьшая для самого внешнего элек-
электрона и увеличивается для последу-
последующих. Обычно энергию ионизации
измеряют в электронвольтах, а саму
энергию в этом случае называют по-
потенциалом ионизации, причем энер-
энергию ионизации самого внешнего элек-
электрона называют первым потенциалом
ионизации, следующего электрона —
вторым потенциалом ионизации и т. д.
В качестве примера приведем первые
четыре потенциала ионизации атома
166
алюминия: 5,98, 18,8; 28,5 и 120,0 эв.
Обращает на себя внимание резкое
возрастание потенциала ионизации
для отрыва электрона от устойчивой
заполненной восьмиэлектронной обо-
оболочки.
В табл. 19 приведены первые потен-
потенциалы ионизации элементов. На рис.
214 они изображены графически. Из
графиков отчетливо видна связь ве-
величины первого потенциала иониза-
ионизации с положением элемента в перио-
периодической системе.
ТАБЛИЦА 19
Первые потенциалы ионизация атомов*
Атомы способны принять на внеш-
внешнюю оболочку один или несколько
дополнительных электронов. При
этом атом превращается в отри-
отрицательный ион, называемый также
анионом. У разных атомов такой про-
процесс происходит с поглощением или
выделением энергии. Величина этой
энергии называется энергией сродст-
сродства атома к электрону. Энергия срод-
сродства к первому присоединяющемуся
электрону у многих атомов положи-
положительна (т. е. присоединение про-
Атом
н
Не
Т *
ы
Be
В
С
N
О
F
Ne
Na
Mg
Al
Si
P
S ,
Cl
Ar
К
Ca
Sc
Ti
V
Cr
Mn
Потенциал
иониза-
ионизации, эв
13,595
24,581
5„390
Q ЭОГ)
8,296
11,256
14,53
13,614
17,418
21,559
5,138
7,644
5,984
8,149
10,484
10,357
1.3,01
15,755
4,339
6,111
6,54
6,82
6,74
6,764
7,432
Атом
Fe
Со
Ni
Си
Zn
Ga
Ge
As
Se
Br
Kr
Rb
Sr
Y
Zr
Nb
Mo
Tc
Ru
Rh
Pd
Ag
Cd
In
Sn
Потенциал
иониза-
ионизации, зв
7,87
7,86
7,633
7,724
9,391
6,00
7,88
9,81
9,75
11,84
13,996
4,176
5,692
6,38
6,84
6,88
7,10
7,28
7,364
7,46
8,33
7,574
8,991
5,785
7,342
Атом
Sb
Те
J
Хе
Cs
Ва
La
Се
Рг
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tu
Yb
Lu
Hi
Та
W
Re
Потенциал
иониза-
ионизации, эв
8,639
9,01
10,454
12,127
3,893
5,210
5,61
6,9
5,76
6,3
5,6
5,67
6,16
6,7
6,8
—
6,08
6,14
6,2
6,15
7
7,88
7,98
7,87
II
I Атом
1
Os
Ir
Pt
Au
Hg
Tl
Pb
Bi
Po
At
Rn
Fr
Ra
At
Th
Pa
U
Потенциал
иониза-
ионизации, эв
8,7
9
9,0
9,22
10,43
6,106
7,415
7,287
8,43
9,2
10,746
3,98
5,277
6,86
6,95
6,08
* По справочнику «Энергии разрыва химических связей. Потенциалы ионизации и сродство к
улектрону», М., Изд-во АН СССР, 1962.
167
Е,з6
Рис. 214. Величины первых потенциалов
ионизации для атомов химических элемен-
элементов
10
15
Z0 25
Атомные номера
3D
исходит с выделением энергии).
Но присоединение второго электрона,
как правило, требует затраты энер-
энергии, т. е. энергия сродства в этом
случае отрицательна. В табл. 20 при-
приведены энергии сродства некоторых
атомов к первому электрону. Следует
обратить внимание, что у некоторых
атомов металлов 6-подгрупп периоди-
периодической системы энергия сродства до-
достигает значительной величины.
4. Основные целочисленные соотно-
соотношения в химии. Открытие периодиче-
периодического закона химических элементов
явилось в известном смысле обобще-
обобщением многих целочисленных соотно-
соотношений в химии. Эти соотношения под-
подготовили почву для открытия закона,
легли в его основу и благодаря ему
же получили новое освещение и
обоснование.
Среди ряда понятий химии, в кото-
которых проявляются целочисленные за-
закономерности, важнейшим является
понятие валентности. Под валент-
валентностью данного химического элемен-
ТАБ ЛИЦА 20
Энергии сродства некоторых атомов к первому электрону*
Атом
Н
Не
Li
Be
В
С
N
О
Сродство
к электрону,
эв
0,75
0,19
0,82
-0,19
0,33
1,2
0,05
1,465
Атом
F
Ne
Na
iWg
Al
Si
Сродство
к электрону,
эв
3,57
-0,57
0,47
-0,32
0,52
1,46
Атом
Р
S
С1
к
Си
Se
Вг
Ag
Сродство
к электрону,
эв
0,77
2,2
3,76
0,82
2,4
1,7-3,7
3,54
2,5
Атом
Sb
Те
I
Аи
Hg
Т1
Bi
Сродство
к электрону,
а»
2
2-3,6
3,29
2,01
1,54
2,1
0,7
По справочнииу «Энергии разрыва химических связей. Потенциалы ионизации и сродство к
электрону». М., Изд-во АН СССР, 1962 г.
168
та подразумевается число атомов
Н, которое необходимо для образова-
образования с одним атомом рассматриваемо-
рассматриваемого элемента химического соединения
или для замещения атома этого эле-
элемента в химическом соединении.
Введению в химию понятия валент-
валентности (Франкланд, 1853 г.) пред-
предшествовало открытие других цело-
целочисленных соотношений. В полемике
с Бертоле A801 — 1807 гг.) Пруст
сформулировал закон постоянства со-
состава химических соединений. В пе-
период 1803 —1808 гг. Дальтон ввел
понятие эквивалентов, т. е. весовых
количеств элементов, соединяющих-
соединяющихся с одной весовой частью водорода,
и сформулировал закон кратных отно-
отношений. В 1808 г. Гей-Люсак, изучая
химические реакции между газами,
открыл, что объемы полученных газов
относятся к объемам исходных газов
как простые целые числа (закон объ-
объемных отношений).
В последующие годы гипотеза Аво-
гадро A811 г.), электрохимическая
теория Берцелиуса A845 г.) и рабо-
работы Канниццаро A840—1860 гг.) при-
привели к понятиям молекулярных и
атомных весов и позволили сформу-
сформулировать понятие валентности.
Определение валентности большин-
большинства химических элементов позволило
обоснованно интерпретировать фор-
формулы существующих химических сое-
соединений, число которых Bi настоящее
время составляет уже несколько мил-
миллионов. В этом и заключается огром-
огромное значение понятия валентности.
Однако уже из работ Берцелиуса и
Канниццаро следовало, что одной и
той же валентности какого-либо эле-
элемента следует приписать различный
электрохимический характер в раз-
разных химических соединениях. Так,
например, роль водорода в соедине-
соединениях типа NaH, Нг, НгО и НС1 су-
существенно различна. В первом сое-
соединении он проявляет себя как отри-
отрицательный ион (Na+ Н~), в послед-
последнем соединении — как положитель-
положительный ион (Н+С1~, по аналогии с
Na+Cl~), во втором и третьем же сое-
соединениях приписать атому водорода
какой-либо заряд невозможно.
Это обстоятельство показывает, что
число, характеризующее валентность
элемента, еще недостаточно, чтобы
полностью охарактеризовать способ-
способность атома к образованию химиче-
химических связей с атомами других хими-
химических элементов. В последние годы
эту способность стали характеризо-
характеризовать несколько по-иному, хотя наз-
название для характеристики оставили
прежнее — валентность. Чтобы новое
содержание понятия валентности мо-
можно было бы отличить от старого, в
котором главным образом подчерки-
подчеркивалась целочисленность связей, для
старого понятия стали пользоваться
термином формальная валентность.
В главе X § 1 мы уже указывали,
что неувязки в теории валентности
привели первоначально к разграни-
разграничению типов химической связи, а в
дальнейшем — к развитию теории ва-
валентности именно по линии теории
химической связи.
В заключение этого раздела еще
отметим, что высшая формальная
валентность большинства химических
элементов равна номеру группы пе-
периодической системы, в которой этот
элемент находится. Д. И. Менделеев
называл эту валентность элементов
их характеристической валентностью.
§ 2. Ионная связь
1. Понятие ионной связи. Тот факт,
что инертные (благородные) газы
в обычных условиях почти не способ-
способны вступать в химические соедине-
соединения, говорит о большой устойчивости
наружных электронных оболочек
атомов этих элементов. Наружная
оболочка атома гелия содержит два
электрона, у атомов остальных
инертных газов наружная оболочка
содержит по восьми электронов.
Исследование электронной струк-
структуры атомов в химических соедине-
соединениях показывает, что в ряде случаев
электронная структура у них анало-
169
Положительный заряд ядра,
Л? 18 36
о Нейтральные атомы
• Ионы
Н ILilВIN | ГИл| Р]СМ К |ScmMnfCo|Cu|GnMs|B71blYlNb|fclRhI/lgfln[sij| J
Не Be С 0 NeMgSiS ArCnTt Cr Fe Ni Zn Бе Se KpSfZp Md RuPdCri SnTe Xe I
Рис. 215. Электронные оболочки элементов
в наиболее обычных валентных состоя-
состояниях
гична электронной структуре атомов
инертных газов. Такие атомы в соеди-
соединениях находятся в виде ионов, при-
приобретая или теряя электроны, в ре-
результате чего их наружная оболочка
содержит число электронов, соответ-
соответствующее наиболее устойчивому элек-
электронному состоянию. Наиболее часто
при этом встречаются анионы и ка-
катионы с наружной оболочкой из вось-
восьми электронов, реже — из 18 элек-
электронов. Число потерянных или при-
приобретенных электронов характери-
характеризует валентность атома в данном сое-
соединении. Такая связь иллюстрирова-
иллюстрирована на рис. 215.
Процесс образования из атомов
различных химических элементов сое-
соединения с таким типом связи по су-
существу может быть разбит на два эта-
этапа: 1) образование из нейтральных
170
атомов положительных и отрицатель-
отрицательных ионов и 2) притяжение разнои-
разноименных ионов с образованием проч-
прочного химического соединения.
Основы изложенных выше пред-
представлений были развиты Косселем
A916 г.), а соответствующая хими-
химическая связь была названа ионной
(или полярной) связью.
Межатомные расстояния в ионных,
кристаллах определяются уравнове-
уравновешиванием сил притяжения между
анионами и катионами и сил оттал-
отталкивания между их электронными
оболочками. Изучение структур ион-
ионных кристаллов указывает на нена-
ненаправленность ионных связей и на
ненасыщаемость их. Каждый ион
стремится окружить себя максималь-
максимальным количеством ионов противолож-
ного знака. Это число определяется
соотношением размеров ионов, а не их
химическими свойствами (см. струк-
структуры NaCl и CsCl на рис. 162 и 163).
То обстоятельство, что у приведенных
в качестве примера веществ соеди-
соединяющиеся друг с другом элементы
одновалентны, не означает, что струк-
структурные химические формулы их бу-
будут Na—С1 и Cs—С1. Такая формула
будет неверной, так как в структуре
NaCl каждый ион окружен шестью
ионами противоположного знака,
в структуре CsCl — восемью. Подроб-
Подробнее этот вопрос будет рассмотрен в
гл. XIV.
2. Энергия решетки ионных кри-
кристаллов. В ионных кристаллах ионы,
как имеющие заряд, все взаимодейст-
взаимодействуют между собой по закону Кулона
с силой
где Z\e и Z& — заряды взаимодейст-
взаимодействующих ионов, образующих кри-
кристалл; е—абсолютная величина заря-
заряда электрона; Zi и Z2 по абсолютной
величине равны формальной валент-
валентности соответствующих атомов.
Ионные кристаллы образованы из
ионов двух сортов — положительно и
отрицательно заряженных. Величина
г есть расстояние между двумя рас-
рассматриваемыми ионами. Если это
расстояние будет бесконечно велико,
то сила F равна нулю. На конечном
расстоянии сила взаимодействия двух
противоположно заряженных ионов
отрицательна, что соответствует при-
притяжению; ионы стремятся сблизить-
сблизиться, на наикратчайшее допустимое
расстояние; оно для них будет соот-
соответствовать устойчивому связанному
состоянию. Сила взаимодействия оди-
одинаково заряженных ионов положи-
положительна, что соответствует отталкива-
отталкиванию. Ионы стремятся разлететься и
ни на каком конечном расстоянии не
образуют устойчивого соединения.
Таким образом, устойчивому образо-
образованию соответствует отрицательная
полная энергия системы. Такое усло-
условие реализуется при образовании ион-
ионного кристалла. Энергию решетки,
т. е. энергию взаимодействия меж-
между всеми ее ионами, можно пред-
представить себе следующим образом.
Очевидно, эта энергия равна по вели-
величине и обратна по знаку энергии, ко-
которую необходимо затратить, чтобы
полностью разрушить кристалличес-
кристаллическую структуру, т. е. чтобы разве-
развести все ионы на бесконечные рас-
расстояния.
Отсюда сразу следует, что энергия
решетки равна работе, которая совер-
совершается при сведении ионов из бес-
бесконечности в кристаллическую струк-
структуру. Работа при постоянной силе F
равна произведению силы на путь.
При изменении силы с расстоянием
формула для работы имеет более
сложный интегральный характер.
Так, работа, совершаемая при сбли-
сближении двух ионов до расстояния R,
равна
F(r)dr.
Подставляя сюда выражение для F
по закону Кулона, получим:
fi
Таким образом, работа просто равна
энергии системы из двух ионов на
расстоянии R. Точно так же опреде-
определяется энергия и всей кристалли-
кристаллической структуры. Задача заклю-
заключается только в правильном опре-
определении расстояний R между иона-
ионами при устойчивом кристалличе-
кристаллическом образовании. Если предпо-
предположить, что между ионами действуют
только кулоновские силы, то ионам
следует приписать конечные «не-
«непроницаемые» для других ионов
размеры. Ионы представляются как
несжимаемые шары. Когда расстоя-
расстояние между ионами становится равным
сумме их радиусов /?=ri+r2, между
ними сразу возникают бесконечно
большие силы отталкивания некуло-
новской природы, препятствующие их
171
дальнейшему сближению. Расчет
энергии в этом приближении можно
лучше всего понять на конкретном
примере, скажем, структуры ионного
кристалла NaCl.
Рассмотрим какой-нибудь положи-
положительный ион натрия (заряд его равен
+е). Он окружен шестью отрицатель-
отрицательными ионами хлора (заряд каждого
из них равен — е), находящимися на
расстоянии R. Энергия взаимодей-
взаимодействия рассматриваемого иона с этими
6 ионами хлора равна
Далее, на расстоянии R}/2 от рассмат-
рассматриваемого иона натрия расположены
12 ионов натрия. Энергия взаимодей-
взаимодействия рассматриваемого иона с этими
12 ионами равна
Далее, на расстоянии Л]/3 располо-
расположены 8 ионов хлора. Соответствую-
Соответствующая энергия взаимодействия равна
На расстоянии ЛУ4 расположены
шесть ионов натрия. Соответствую-
Соответствующая энергия равна
На расстоянии i?]/5 расположены 24
иона хлора. Энергия взаимодействия
равна
И так можно продолжать и далее.
По отношению к выбранному иону
остальные ионы расположены по так
называемым координационным сфе-
сферам с вполне определенными радиуса-
радиусами и числами ионов на сфере (коор-
(координационными числами).
Для каждой кристаллической
структуры эти характеристики хоро-
хорошо известны.
Окружение иона хлора повторяет
окружение иона натрия (см. рис. 216),
с тем только различием, что бли-
ближайшими соседями будут ионы
натрия, затем ионы хлора и т. д.
Рис. 216. Схема структуры NaCl (к вы-
вычислению структурного коэффициента)
Энергия взаимодействия иона хлора
со всеми остальными ионами, следо-
следовательно, будет равна соответствую-
соответствующей энергии для иона натрия.
Отсюда вытекает, что полная энер-
энергия решетки просто равна энергии
взаимодействия какого-либо одного
иона со всеми остальными, умножен-
умноженная на число ионов в решетке и де-
деленная на 2 для учета того факта, что
в этой схеме одно и то же взаимодей-
взаимодействие между двумя ионами подсчиты-
вается дважды: один раз, когда один
из ионов выбран как центральный,
а другой — как периферийный, и вто-
второй раз — при противоположном вы-
выборе.
Энергия одного иона в случае ре-
решетки NaCl в приближении учета
только кулоновского взаимодействия
равна, таким образом,
A)
Знакопеременный ряд в скобках
(обозначим его через а) связан со
структурой кристалла. Для каждой
конкретной кристаллической струк-
структуры члены этого ряда разнятся по
вполне определенному закону, и ал-
алгебраическая сумма ряда имеет впол-
вполне определенную величину. Для
структур типа NaCl сумма ряда равна
1,748. Подсчет полной энергии решет-
решетки затруднителен из-за того, что не-
172
известно полное число ионов в решет-
решетке. Поэтому принято энергию решет-
решетки определять или для единицы
объема или для одной граммолекулы
вещества. Число ионов в граммолеку-
ле равно числу Авогадро Л^, умно-
умноженному на число ионов в соответ-
соответствующей молекуле (число ионов в
формульной единице 2то); это число
для различных типов структур при-
приведено в табл. 21.
ТАБЛИЦА 21
Коэффициенты а, 2т и а для различных
типов структур
Структурный
тип
NaCl
CsCl
ZnS
(сфалерит)
CaF2
TiO2
(рутил)
CdJ2
A1,Q»
(корунд)
а
1,75
1,76
1,64
2,52
2,40
2,36
4,17
2m
2
2
2
3
3
3
5
а
1,75
1,76
1,64
1,68
1,60
1,57
1,67
Таким образом, энергия решетки
в общем случае неоднократно заря-
заряженных ионов
Параметр а обычно называют
структурной постоянной или посто-
постоянной Маделунга. Для каждого типа
кристаллических структур эта по-
постоянная имеет вполне определен-
определенное значение (для ряда структур зна-
значения а приведены в табл. 21).
В частности, для структуры NaCl
а= 1,748 и полная энергия решетки
NaCl в рассматриваемом приближе-
приближении равна
—1,748 -^-N. C)
Возможно уточнение изложенной мо-
модели на учет сил отталкивания неку-
лоновской природы. В этой модели
ионы уже не считаются несжимаемы-
несжимаемыми шарами. О действительном харак-
характере и величине этих сил отталкива-
отталкивания можно судить по эксперимен-
экспериментальным данным о сжимаемости
кристаллов. Их величина очень резко
падает с увеличением расстояния
между ионами. Первоначально Борн
предложил для энергии отталкивания
простейшую формулу В/г71, где п го-
гораздо больше 2 в соответствии с ука-
указанным выше опытным фактом. Зна-
Значение п находится эмпирически. Для
разных кристаллов в соответствии с
опытом значения п оказались в пре-
пределах от 5 до 12. Величина п нахо-
находится в определенной корреляции с
положением элемента по периодам
таблицы Менделеева.
Выражение для энергии решетки
с учетом борновской формулы для
энергии отталкивания некулоновской
природы имеет следующий вид:
ZV
Л
D)
Коэффициент р в этой формуле, как
и а, учитывает структурные особен-
особенности расположения ионов. Вычисле-
Вычисление этого коэффициента аналогично
вычислению постоянной Маделунга а.
Только теперь все члены сходяще-
сходящегося ряда, образующего коэффициент
C, имеют одинаковые знаки, так как
каждый член этого ряда соответству-
соответствует отталкиванию пары ионов незави-
независимо от того, имеют ли они одинако-
одинаковые или разные знаки. Однако вычис-
вычисление этого коэффициента не явля-
является обязательным.
Следует учесть, что кристалл есть
равновесная система, для которой
расстояние между ионами таково, что
потенциальная энергия системы (т. е.
энергия решетки) имеет минимум.
Этот минимум определяется из усло-
условия обращения в нуль первой произ-
производной от энергии по расстоянию при
равновесном расстоянии R между
ионами:
du
dfi =0'
173
или в развернутой форме:
N
R2 a--tfH
=0.
E)
Отсюда получаем
Подставляя это выражение для Бр1
в формулу D), получим
F)
Эта формула не является чисто тео-
теоретической, так как при расчете по
этой формуле значение параметра п
определяется эмпирически из опыт-
опытных данных по сжимаемости.
В развитие этой теории Борн, Май-
ер и Гельмгольц предложили описы-
описывать зависимость энергии отталкива-
отталкивания от расстояния выражением вида
Бе~л/р. Входящие сюда параметры
В и р так же, как и прежде, опреде-
определяются опытным путем. Однако
с теоретической точки зрения послед-
последняя формула предпочтительнее, так
как р варьируется гораздо меньше,
чем параметр п при переходе от од-
одного кристалла к другим. В то время
как п меняется в пределах от 5 до 12,
относительное изменение р не превы-
превышает 6%, т. е. величина р остается
почти постоянной.
3. Приближенные формулы энергип
решетки кристаллов в форме, удобной
чля расчета. Систематизированный
анализ формул Борна, выполненный
А. Ф. Капустинским, дал ему возмож-
возможность предложить весьма упрощен-
упрощенную формулу, удобную для конкрет-
конкретных расчетов. Прежде всего он обра-
обратил внимание на то обстоятельство,
что у значительного числа элементов
п равно или близко к 9. В то же вре-
время изменение п от этого среднего
значения на ±3 меняет вычисленное
значение энергии решетки только на
3—5 %• Поэтому в пределах такой
точности в качестве параметра п
можно выбрать постоянную величи-
величину, равную 9.
Далее, Капустинский заметил, что
коэффициент а зависит главным об-
образом от числа ионов в формульной
единице 2пг. Поэтому его можно за-
заменить на приблизительно постоян-
постоянный коэффициент
а=2а/2тп.
Величина а изменяется с измене-
изменением координационного числа при-
приблизительно так же, как изменяются
при перемене координационного чис-
числа радиусы ионов. Так, например,
при изменении координационного
числа с 6 на 8 радиус увеличивается
на 3%, а коэффициент а — на 1,2%;
при переходе от координационного
числа 6 к 4 радиус уменьшается на
6 % t а а — на 7 % и т. д. Поэтому ес-
если в формуле Борна расстояние меж-
между ионами R заменить суммой ион-
ионных радиусов га+гх, полученных для
координационного числа 6, то коэф-
коэффициент а станет действительно
практически постоянной величиной,
равной 1,748. Подставляя в форму-
формулу F) численные значения для N, е,
а ж п, получим простую формулу для
расчета энергии решетки (в ккал/
/моль):
G)
Капустинский предложил также при-
приближенную форму записи второй
формулы Борна — Майера, учитыва-
учитывающей экспоненту в члене отталкива-
отталкивания. По Капустинскому параметр р
этой формулы лучше всего аппрокси-
аппроксимируется значением р=0,345. Тогда
формулу можно приближенно пред-
представить в виде
U = 287 •
0,345 ,
1- - - I- (8)
Для расчета энергии решетки по
этой формуле необходимо знать со-
состав вещества, т. е. число ионов в
«молекуле» 2пг и их валентности
Zi и Ъг, а также иметь таблицу ион-
ионных радиусов для координационного
числа шесть. Таким образом, эта фор-
174
мула имеет более общее значение, чем
формула F). Она может быть назва-
названа бесструктурной, или универсаль-
универсальной.
Согласно П. В. Грушевицкому, для
сложных по составу кристаллов энер-
энергию решетки по порядку величины
можно оценить, разбивая формулу
вещества на нейтральные группы
и оценивая энергию каждой группы
отдельно. Например, энергия решет-
решетки анортита CaAl2Si2Os рассматрива-
рассматривается как сумма энергии следующего
вида:
ТАБЛИЦА 22
Оценка энергетических характеристик
(в ккал/моль)
А. Е. Ферсман обратил внимание,
что по порядку величины оценка
энергии решетки может быть выпол-
выполнена по совсем упрощенной форме
(9)
Величина В, приближенно равная
Z2/2, может рассматриваться как по-
потенциал полной ионизации. Эту вели-
величину Ферсман назвал эк (сокращение
от слов энергетическая константа).
В формуле Ферсмана величину
256 эк можно рассматривать как
вклад в энергию решетки от соответ-
соответствующих ионов. Аналогично трак-
трактуется второе слагаемое в формуле
Ферсмана.
Заслугой Ферсмана является по-
постановка проблемы о вычислении
энергии кристалла по двум инкри-
ментам, свойственным катиону и ани-
аниону. Простое суммирование эк'ов мо-
может рассматриваться, конечно, только
как грубое приближение. Сама схема
расчета является, очевидно, не обо-
обоснованной, так как не существует
математических правил, могущих
привести к такой формуле из исход-
исходной.
Можно еще ввести дополнитель-
дополнительную энергетическую константу, рав-
равную 1 эк, деленной на валентность,
и названной вэк (впервые такого ти-
типа константу ввел В. К. Семенченко
в 1927 г.). В табл. 22 приводятся
Катионы
эк
вэк
Одновалентные
Cs
К
Na
Li
Ag
Си
0,30
0,36
0,45
0,55
0,60
0,70
0,30
0,36
0,45
0,55
0,60
0,70
Двухвалентные
Ва
Са
РЬ
Cd
Си
Fe
Mg
Zn
1,35
1,75
1,65
2,00
2,10
2,12
2,15
2,20
0,67
0,87
0,82
1,00
1,05
1,06
1,07
1,10
Трехвалентные
Сг
А1
Fe
В
4,75
4,95
5,15
6,00
1,58
1,65
1,71
2,00
Четырехвалентные
Sn
РЬ
Ti
Mo
Si
С
7,90
7,95
8,40
8,50
8,60
12,20
1,97
1,99
2,10
2,12
2,15
3,05
Пятивалентные
Р
V
14,40
16,45
3,39
3,29
Шеоти валентные
s
21,90
3,65
Семивалентные
Re
28,10
4,01
Анионы
эк
«эк
Одновалентные
J
NOs
Вг
С1
CN
Н
F
ОН
0,18
0,19
0,22
0,25
0,25
0,32
0,37
0,37
0,18
0,19
0,22
0,25
0,25
0,32
0,37
0,37
Двухвалентные
SO4
СгО4
СОз
Те
Se
S
О
0,70
0,75
0,78
0,95
1,10
1,15
1,55
0,35
0,38
0,39
0,47
0,55
0,57
0,75
Трехвалентные
РО4
AsO4
BOS
As
Р
N
1,50
1,53
1,68
2,65
2,70
3,60
0,50
0,51
0,56
0,88
0,90
1,20
Четырехвалентные
ZrO4
TiO4
SiO4
2,30
2,45
2,75
0,58
0,61
0,69
значения эк ов и вэк ов некоторых
ионов.
4. Экспериментальные методы
проверки формул для энергии ре-
решетки. Проверка формулы для энер-
энергии решетки может быть выполнена
с помощью кругового процесса Бор-
на — Габера (рис. 217).
Энергия, которая затрачивается
извне на то, чтобы процесс произо-
произошел, отмечена знаком минус. Если
процесс идет самопроизвольно и при
этом энергия выделяется, то она обо-
обозначается знаком плюс. При образо-
образовании N!aCl из NaTB +V2 С12 выде-
выделяется энергия (-\-Q). Следователь-
Следовательно, при противоположном процессе
энергия будет затрачиваться (—Q).
Если мы «обойдем» круговой про-
процесс в одном направлении, то сумма
энергии всегда будет равна нулю:
откуда:
где а — теплота сублимации металла,
I — потенциал ионизации металла,
D — теплота диссоциации молекулы
неметалла, Е — сродство к электро-
электрону неметалла, Q — теплота реакции.
Все эти величины определяются
экспериментально; точность их опре-
определения составляет 3—5 %• Круго-
Круговой процесс дает возможность про-
проконтролировать правильность теоре-
теоретических расчетов.
Сопоставление результатов вы-
вычисления энергии решетки по раз-
различным формулам дано в табл. 23.
NaCl,,,,
ТАБЛИЦА 23
Энергия (в ккал(моль) решеток, полу-
полученная экспериментально и вычисленная
по Борну, Капустинскоыу и Ферсману
Вещество
LiF
CaF2
AIF3
CdF2
NaCl
KC1
РЬС1г
NaBr
NaJ
CdJa
ZnO
AUOs
SiOs
Na2S
ZnS
^эксп
240,1
617,2
1440,0
661,9
180,4
164,4
521,3
171,7
160,8
495,5
970
3618
3097
524
852
617,7
—
628,7
179
163
534
171
158
977
3708
516
818
240,6
624,7
1616,0
650,2
183,6
162,0
490,6
174,1
161,0
471,1
952,5
4063
3593
564,7
796,8
236
638
1408
701
179
156
550
171
161
604
960
3724
2995
524
857
(Na,,,
Рис. 217. Круговой процесс Борна — Га-
Габера
Часто круговой процесс исполь-
используется для определения тех или
иных величин, входящих в него.
Ясно, что, вычислив энергию решет-
решетки по одной из формул, можно опре-
определить величину любого члена урав-
уравнения, если известны величины всех
остальных. Действительно, с по-
помощью кругового процесса впервые
была определена энергия сродства
к электрону. Часть членов — энер-
энергия ионизации и энергия сродства
к электрону — являются атомными
константами, другие — теплота дис-
диссоциации или энергия сублимации —
характеризуют простые вещества,
и только энергии решеток и теплоты
образования относятся к сложным
веществам. Таким образом, первые
четыре величины могут быть зара-
заранее найдены экспериментально или
вычислены для всех химических
элементов. Экспериментальное опре-
определение двух последних величин для
всех веществ затруднительно ввиду
того, что число сложных химиче-
химических соединений очень велико и с
176
каждым днем увеличивается в ре-
результате новых синтезов. Поэтому
теоретическая возможность расчета
теплот образования химических со-
соединений весьма важна. Формула
кругового процесса и используется
главным образом для определения
теплот образования соединений. Од-
Однако такой расчет стал возможным
только благодаря знанию величин
энергии решеток.
§3. Элементарные представлении
о ««валентной связи
1. Понятие ковалентной связи. Вось-
миэлектронная устойчивая оболочка
атома может быть получена у обоих
атомов соединения не только за счет
отдачи избыточных (сверх восьми)
наружных валентных электронов
или соответствующего принятия на
наружную оболочку недостающих
(до восьми) электронов, но также
и за счет «совместного использова-
использования» этими атомами пары (или не-
нескольких пар) электронов (Льюис,
1910 г.). Этот способ достижения
Рис. 218. Схематическое изображение мо-
молекулы СЬ (направления спинов электро-
электронов наружных оболочек обозначены зна-
знаками + и —)
устоычивых электронных оболочек
характерен, например, для галогенов,
СС14 и др. (рис. 218, 219). Химиче-
Химическая связь, образующаяся при этом,
носит название гомеополярной или
ковалентной связи.
Если при ионном (полярном)
тине связи характерно, что соединя-
соединяющиеся атомы проявляют противо-
противоположную электрическую природу —
одни легко теряют один или несколь-
несколько своих валентных электронов,
а другие легко приобретают избы-
избыточные электроны, — то при кова-
ковалентной связи тенденция к потере
или присоединению электронов у
обоих соединяющихся атомов при-
приблизительно одинакова.
В идеальном случае ковалентная
связь образуется между одинаковы-
одинаковыми атомами (например, в молекуле
водорода). В каждом случае такая
связь характеризуется наличием
общей электронной пары с антипа-
антипараллельными спинами. В структур-
структурных химических формулах каждая
такая общая электронная пара
Н Н
Н:Н, rCi-.Ci: :N:H, H:C:H,
Н Н
: С1:
:Ci:C: Cl
Н:
Н:
С::С
:Н
:Н
обозначается штрихом:
Н Н
Н—Н, Cl—Cl, N—Н , Н—С—Н,
\ I
н н
С1 Н Н
\
с=с
С1
н
\
н
Рис. 219. Схематическое изображение мо-
молекулы СС14
Устойчивая электронная струк-
структура из 8 электронов по предложе-
предложению Льюиса называется октетом.
2. Координационная связь. С по-
помощью ковалентных связей из ато-
атомов могут образовываться не только
нейтральные молекулы, но также ра-
радикалы и комплексные ионы. При
12 Кристаллохимия
Г77
этом электроны, образующие кова-
лентную пару, до образования связи
могли принадлежать как разным ато-
атомам, так и одному атому, участвую-
участвующему в связи или же даже могут
быть заимствованы у постороннего
атома (обычно металлического), не
участвующего в образовании связи
в комплексном ионе. чТак, в комп-
комплексном ионе SC>42~
октет у атома серы достроен за счет
двух дополнительных электронов,
например, атома Mg в соединении
MgSO4. Это соединение гетеродесми-
ческое, и его структурную формулу
скорее следует писать в виде
Mg2
0
0
0
\
0
/ \ /
, а не в виде Mg S
V \>
хотя бы из-за тождественности всех
атомов кислорода в структуре этого
соединения. Во всех случаях, допу-
допускающих двойственную трактовку
типа химической связи (ионную или
ковалентную), в соединениях осуще-
осуществляется промежуточный тип свя-
связи, подробно о котором будет сказа-
сказано ниже.
В некоторых случаях централь-
центральный атом в комплексном ионе не по-
получает извне дополнительных элек-
электронов (как это имело место с ато-
атомом серы в сульфатном ионе), а, на-
напротив, он сам отдает избыточный
(против октета) электрон. Это имеет
место, например, в аммонийном ионе
(NH4L", в соединениях типа NH4C1
и др.
В том случае, когда ковалентная
связь осуществляется за счет элек-
электронной пары, принадлежащей одно-
одному атому, участвующему в связи,
она обычно называется координа-
координационной и обозначается не черточ-
черточкой, а стрелкой. По предложению
Сиджвика атом, отдавший оба элек-
электрона на образование ковалентной
связи, называется донором, а атом,
присоединившийся к донору и не
давший для образования связи своих
электронов, акцептором. Надо сразу
же оговориться, что эта разновид-
разновидность ковалентной связи отличается
от обычной только генетически. Пос-
После образования связи никакой разни-
разницы между ковалентной и координа-
координационной связями нет, поэтому нельзя
никакими физическими методами
отличить их друг от друга.
Координационная связь часто
встречается в комплексных (коорди-
(координационных) соединениях. Прочную
координационную связь с комплек-
сообразующим атомом металла часто
Н
дают молекулы аымиака :N:H.
Н
В качестве примера укажем соеди-
соединения двухвалентной платины типа
Pt(NH3JCl2. Структурная формула
последнего соединения имеет вид:
Cl NH3
\
Pt
Cl
\
NH3
Поскольку, как сказано выше,
нет физической разницы между
обычными ковалентными связями и
координационными, стрелки в струк-
структурных формулах обычно опускают-
опускаются и заменяются штрихами.
3. Структура простых веществ с
ковалентными связями. Существен-
Существенное отличие ковалентной связи от
ионной заключается в том, что число
электронов, осуществляющих кова-
лентные связи, у каждого атома
ограниченно, и, следовательно, эта
связь является насыщаемой. В слу-
случае ионной связи один катион может
притягивать сколько угодно окружа-
окружающих его анионов, и наоборот.
В случае же ковалентной связи, на-
например в случае водорода, один из
его атомов, соединившись со вторым
атомом, образует молекулу Н2, кото-
которая уже не будет реагировать с
третьим свободным атомом водорода.
178
По этой причине атомы элемен-
элементов последних 6-подгрупп периоди-
периодической системы могут образовывать
8 — Л^ ковалентных связей, где ./V —
номер группы, равный числу валент-
валентных электронов у этих атомов. При
этом число связей будет одновремен-
одновременно равно координационному числу
К атомов этих элементов в молеку-
молекулах и кристаллах.
Руководствуясь правилом К=8—N
(Юм-Розери, 1928 г.), легко понять
особенность кристаллических струк-
структур многих простых веществ. Так,
кристаллические структуры водорода
и галогенов построены из двухатом-
двухатомных молекул. Кристаллические струк-
структуры элементов VI-6 подгруппы по-
построены из многоатомных молекул
или бесконечных цепей (рис. 220),
оба эти случая удовлетворяют коорди-
координационному числу 2 (.?=8—6=2).
Простые вещества элементов
V-Ь подгруппы имеют кристалличе-
кристаллические структуры, построенные из сло-
слоев, а у элементов IV-6 подгруппы (ал-
(алмаз, Si, Ge, a-Sn) кристаллические
структуры представляют собой трех-
трехмерную тетраэдрическую вязь (рис.
160, стр. 121). В этом случае каждый
атом образует 4 ковалентные связи,
отдавая в общее пользование для
каждой связи один из своих валент-
валентных электронов и получая второй
электрон от соседнего атома. Для
алмаза, например, это схематически
можно изобразить так:
\ /
с
/ \
\ /
с
/ \
Рис. 220. Схематическое изображение мо-
молекул хлора и серы и цепочки атомов се-
селена
В приведенных примерах прояв-
проявляется также вторая (кроме насы-
насыщаемости) особенность ковалентной
связи. Отдельные связи ориентиро-
ориентированы относительно друг друга под
определенными углами (валентные
углы), которые и определяют на-
направленность ковалентной связи.
В частности, этим обстоятельством
объясняется то, что кристаллические
структуры с ковалентными связями
часто характеризуются низкими ко-
координационными числами и не под-
подчиняются законам плотнейших упа-
упаковок. Направленность ковалентной
связи, хорошо известную из экспери-
экспериментальных определений кристал-
кристаллических структур, удалось удовле-
удовлетворительно объяснить только на ос-
основе строгой квантовой теории (кван-
(квантовой химии).
4. Кристаллические структуры
бинарных и более сложных химиче-
химических соединений с ковалентной
связью. В главе IX, § 5 была описа-
описана структура алмаза, а в § 6 структу-
структура двух модификаций ZnS — сфале-
сфалерита и вюртцита. Для всех этих
структур характерно, что каждый
атом окружен по тетраэдру четырьмя
другими атомами таким образом, что
структура в целом представляет со-
собою непрерывную трехмерную тетра-
тетраэдрическую вязь. В случае алма-
алмаза тетраэдрическая координация
объясняется тем, что каждый
атом образует четыре равноценные
ковалентные связи, поскольку угле-
углерод находится в IV группе периоди-
периодической системы (8—4 = 4). Однако,
12*
179
как сказано в пункте 2 настоящей
главы, для образования ковалентной
связи совершенно не обязательно,
чтобы оба ее электрона принадлежа-
принадлежали разным атомам. Ковалентная
связь может быть и донорно-акцеп-
торного типа. Важно только, чтобы
в случае бинарного соединения с
тетраодрической координацией сум-
сумма валентных электронов была бы
равна восьми (правило Гримма —
Зоммерфельда, 1926 г.), т. е. по два
на каждую связь.
В структуре ZnS атом серы отдает
для образования четырех ковалент-
ных связей шесть электронов, а атом
цинка — только два. Этот случай
отмечен стрелками.
*Zn
' \
Схемы координационной связи в
соединениях А1Р и AgJ, кристалли-
кристаллизующихся в структурном типе ZnS,
изобразятся следующим образом:
\ /
Л1
/ \
\ /
р
/ \
и
ч /
Ag
/ \
\ /
J
/ \
Правило образования кристалли-
кристаллических структур сложных соедине-
соединений с ковалентной связью было
сформулировано в 1964 г. Пирсоном:
¦ = 8.
В формуле Пирсона 2 у — сумма
валентностей всех атомов в химиче-
химической формуле соединения; Ъа — чис-
число электронов, участвующих в связи
янион—анион; Ьс — число электро-
электронов катиона, не участвующих в свя-
связи (или образующих связь катион —
катион); па — число анионов в хими-
химической формуле соединения.
Здесь следует обратить внимание
на то, что атомы, вступающие в ко-
валентную связь с соседними атома-
атомами, совсем не обязательно исполь-
используют все свои валентные электроны.
Так, из рис. 220 (см. стр. 179) видно,
что в молекуле С1г на каждом атоме
остается шесть C пары) электронов,
не участвующих в связи. Из того же
рисунка видно, что в молекуле серы
Ss остается две пары электронов на
атом.
Аналогичные случаи наблюда-
наблюдаются и у металлических атомов. Так,
в соединении PbS свинец имеет фор-
формальную валентность 2, и, следова-
следовательно, в связи участвует только два
из четырех валентных электронов. То
же имеет место и в структуре соеди-
соединения TISe (рис. 221). Как видно из
рисунка, атомы Т1 в соединении
TISe оказываются двух типов: одни
имеют координационное число 8,
а другие — 4. Эти факты позволяют
сделать заключение о том, что Т1Г
одновалентен, а Т1ц — трехвалентен.
Следовательно, атомы Т1Г не исполь-
используют двух своих валентных электро-
электронов.
Проверка правила Пирсона для
PbS и Т^Эег дает соответственно:
4 + 6-2 2.3 + 2-6-2
¦ j = 8 и g = 8-
Для всех элементов 6-подгрупп
величина I>v в формуле Пирсона яв-
является просто суммой характеристи-
характеристических валентностей.
Следует иметь в виду, что пра-
правильно пользоваться этой формулой
можно, если известна кристалличе-
кристаллическая структура соединения. Структу-
Структура позволит оценить, какую роль
(более электроположительную или
более электроотрицательную) в хи-
химическом соединении играют эле-
элементы IV-Ь и даже V-Ь подгрупп.
on OSe
Рис. 221. Структура TISe
180
И только после выяснения этого об-
обстоятельства можно, не боясь оши-
ошибок, использовать правило Пирсона.
Для соединений металлов l-а и
На подгрупп, а также металлов 1-Ъ
IV-6 подгрупп с элементами IV-Ь —
VII-& подгрупп применение этого
правила не вызывает сомнений. Не-
Некоторые затруднения (иногда непрео-
непреодолимые) возникают лишь при при-
применении этого правила к соединени-
соединениям с переходными металлами.
Г. Б. Бокий и Р. Ф. Клевцова
A965 г.) специально исследовали
структуру соединения Zn4Sb3 (|3-фа-
за в системе Zn—Sb), для которого
нельзя подобрать валентную форму-
формулу, удовлетворяющую правилу Пир-
Пирсона. Если предположить, что все ато-
атомы сурьмы трехвалентны, то (8+
+ 15)/3 = 7,7. При втором возможном
предположении, что два из трех ато-
атомов сурьмы образуют молекулярный
поп Sb2''~, получается (8 + 15 + 2)/3 =
= 8,3. Определение атомной структу-
структуры этого соединения покапало, что
кратности у атома цинка и сурьмы
следующие: Zni—8, Znn—8, Zrim—
8; Sbi—8, Shu—8, Sbm—4, т. е. фор-
формула должна иметь вид Zii24Sb2o =
= Zn6Sb5 = Znli2oSb. Исследуемая фа-
фаза на диаграмме состояния имеет
область гомогенности от Zni,2eSb до
Zn^sSb, т. е. полученный состав вы-
выходит за границы этой фазы. Анализ
структуры показал, что часть атомов
цинка замещает изоморфно (см. ниже)
Sbi и Sbiu, а атомы Sbn образуют
молекулярные ионы Sb24~. Формулу
этого соединения следовательно надо
писать так:
т. е. этот состав (соответствующий
химическому анализу) лежит в обла-
области |3-фазы. Проверка правила дает:
2у + Ьа — Ъс A2,4 + 4,8-5L-2
4,8
38,4
4,8
Приведенный пример является
первым случаем применения правила
Пирсона для соединений с дробными
коэффициентами в химических фор-
формулах, как видим, правило и для
этих случаев хорошо выполняется.
Поэтому кажется целесообразным
называть это правило обобщенны:-!,
правилом валентности для соедине-
соединений, с ковалентной связью.
Все развитые в настоящем пара-
параграфе представления носят сугубо
качественный характер. Пользуясь
ими, нельзя производить каких-либо
количественных вычислений. Однако
благодаря простоте и наглядности
они получили большое распростране-
распространение и широко используются для эле-
элементарного рассмотрения вопросов
строения кристаллов с ковалентным
типом связи.
5. Система тетраэдрических кова-
лентных «радиусов». Для подсчета
межатомных расстояний в структу-
структурах с ковалентной связью система
ионных радиусов становится уже не-
непригодной. Лучше в этом случае
пользоваться системой ковалентных
«радиусов». Так как большинство
металлов fr-подгрупп имеет в струк-
структурах сульфидов и их аналогов ко-
координационное число 4 и координа-
координационный многогранник — тетраэдр,
то соответствующая система «ради-
«радиусов» называется системой «тетра-
«тетраэдрических радиусов»:
Тетраэдрические радиусы
по Полингу и Хаггинсу
В С N О F
0,88 0,77 0,70 0,66 0,64
Al Si P S C1
0,26 1,17 1,10 1,04 0,99
Си Zn Ga Ge As So Br
1,35 1,31 1,26 1,22 1,18 1,14 1,11
Ag Cd In Sn Sb Те J
1,53 1,48 1,44 1,40 1,36 1,32 1,28
Аи Hg Tl Pb Bi
1,50 1,48 1,47 1,46 1,46
Здесь же приведены данные и
для неметаллических элементов. Для
181
Рис. 222. Расположение соседних молекул
Х2 в кристаллах
легких атомов «радиусы» практи-
практически совпадают с нормальными ко-
валентными радиусами для ординар-
ординарных связей, для тяжелых — имеются
небольшие отклонения.
Слово «радиусы» в настоящем раз-
разделе поставлено в кавычки, так как
атомы, связанные, например в моле-
молекуле СЬ, ковалентной связью, распо-
расположены друг от друга на минималь-
минимальном расстоянии 2г (рис. 222). Из-за
строгой направленности ковалентной
связи размер атома по всем другим
направлениям превышает это мини-
минимальное расстояние. В этих направ-
направлениях у него уже нет ковалентной
связи, и его взаимодействие характе-
характеризуется ван-дер-ваальсовской сфе-
сферой с определенным радиусом R (по-
(поскольку ван-дер-ваальсовская связь
является связью ненаправленной).
Понятие о ван-дер-ваальсовских свя-
связях будет дано в последующем.
§ 4. Физические основы
ковалеитной связи
1. Основные принципы квантовой ме-
механики. Теория Бора, которая кратко бы-
была охарактеризована в предыдущем пара-
параграфе, позволила вычислить положение
(частоты) спектральных линий атома во-
водорода. Однако эта теория не могла объ-
объяснить спектры других атомов. Даже для
гелия удавалось с помощью этой теории
получить только качественные соотноше-
соотношения. Совсем не удалось согласовать тео-
теорию со спектром молекулярного водорода.
Даже для атомарного водорода можно
было рассчитать только частоты, но не
удавалось определить интенсивность ли-
линий и их тонкую структуру, наблюдаемую
с помощью спектральных приборов боль-
большой разрешающей способности.
Поэтому на смену теории Бора пришла
современная квантовая механика, нача-
начало развития которой было положено опу-
опубликованной в 1926 г. работой Шрединге-
ра по теории электрона.
Истоки представлений современной
квантовой механики следует искать в по-
полемике начала двадцатых годов о связи
волновых и корпускулярных свойств ма-
материи. Эта полемика была завершена ра-
работой Де-Бройля A924 г.), который свобод-
свободному электрону приписал определенную
длину волны Л, величина которой опреде*
ляется формулой:
Я, = —, A0)
где р — импульс электрона, h — постоян-
постоянная Планка. Для классической частицы
с массой т импульс можно выразить че-
через скорость частицы v: p = mv. Тогда со-
соотношение Де-Бройля примет вид:
h
mv
(И)
С другой стороны, «волне» с длиной вол-
волны к принадлежит также и частота v, ко-
которая, согласно Де-Бройлю, определяется
по формуле:
Е
' = "*"
A2)
где Е — энергия частицы.
Таким образом, имеем соотношение, свя-
связывающее энергию частицы с волновой
характеристикой (частотой)
Это соотношение подобно по своей форме
постулированной Бором формуле для
энергии атомной системы (связанного в
атоме электрона):
Е = nhv,
где п — целое число.
Представления Де-Бройля о волновой
трактовке частицы (электрона) в 1927 г.
получили подтверждение в опытах по
дифракции электронов.
Для ориентировки укажем, например,
что для электрона с энергией 20 вв длина
волны по Де-Бройлю будет равна 2,7X
X К)-8 ем, т. е. имеет порядок величины
длины орбиты электрона в атоме водоро-
водорода в нормальном состоянии (первая бо-
ровская орбита). Действительно, радиус
первой боровской орбиты равен оо =
= 0,529 • 10~8 см, а ее длина равна 2яао =
— 3,39 • 10~8 см. Вообще волны Де-Бройля
имеют ту любопытную особенность, что
можно дать усложненное соотношение
182
между этой волной и длиной любой орби-
орбиты теории Бора.
Следует обратить внимание, что поня-
понятие совершенно свободная частица есть
фикция. Однако это понятие применимо
при вполне определенных условиях.
В классической физике, очевидно, частицу
можно считать свободной, если потенци-
потенциальная энергия ее взаимодействия мала
по сравнению с ее кинетической энергией,
изменение кинетической энергии проис-
происходит только в результате «столкновений»
с другими частицами (влияние внешнего
силового поля не учитывается), т. е. об-
область взаимодействия много меньше дли-
длины свободного движения частицы между
такими взаимодействиями. Эти рассужде-
рассуждения можно с определенной модификацией
распространить и на представления
Де-Бройля.
Строго говоря, говорить о волновом
процессе с определенной длиной волны
можно только в том случае, если волна
распространяется на бесконечном протя-
протяжении. Однако если область взаимодейст-
взаимодействия мала по сравнению со средним расстоя-
расстоянием между частицами, то, когда на этих
расстояниях укладывается много длин волн,
можно говорить о свободной частице и ее
длине волны Де-Бройля. Другими слова-
словами, необходимо, чтобы заметное измене-
изменение силового поля, действующего на ча-
частицу, проявлялось бы только на рассто-
расстояниях, много больших де-бройлевской
длины волны. В атомной системе это ус-
условие не выполняется и приписать свя-
связанному электрону определенную длину
волны нельзя. Но если рассматривается
стационарная система (энергия системы
не изменяется), то можно ввести вторую
волновую характеристику — частоту. Дру-
Другими словами, в связанных стационарных
системах для частицы можно указать
только энергию, но не импульс, для сво-
свободной же частицы можно указать обе
эти величины.
Следует, однако, иметь в виду, что в
предельном случае очень малых длин
волн, когда де-бройлевская длина волны
сравнима с размерами рассматриваемой
частицы, квантовая механика переходит
в свой классический прототип — реляти-
релятивистскую механику или механику Ньюто-
Ньютона (в нерелятивистском случае).
Для классической механики характер-
характерно, что если заданы начальные условия
для частицы (начальное положение в про-
пространстве и начальная скорость), а также
внешние условия, в которых происходит
движение частицы (силовое поле во всех
точках пространства и во все моменты
времени), то можно строго рассчитать по-
положение и скорость частицы в любой по-
последующий момент времени.
В квантовой механике все определяет-
определяется по-иному. В этой теории нельзя задать
начальные условия в классическом смыс-
смысле. Поэтому теряет смысл и задача стро-
строгого расчета положения и скорости частицы
в последующие моменты времени. Одна-
Однако волновые свойства частиц дают оп-
определенную информацию об этих величи-
величинах.
Эти свойства дают возможность опреде-
определить нахождение частицы в момент вре-
времени t в какой-либо точке пространства
или вероятность обнаружить у частицы
определенное значение ее скорости. Кван-
Квантовая механика характеризует статисти-
статистические результаты опытов с очень боль-
большим числом частиц, находящихся в оди-
одинаковых условиях. Именно эта статистич-
ность проявляется в волновом аспекте
квантовой механики. Корпускулярные же
свойства частиц проявляются в законах
взаимодействия между ними и в зако-
законах действия на них внешних силовых
полей.
Невозможность задания в квантовой
механике начальных условий связана со
специфической особенностью квантовых
систем — так называемым принципом не-
неопределенности (Гейзенберг, 1927 г.). Со-
Согласно этому принципу, точность опреде-
определения координаты частицы х и соответ-
соответствующей ей составляющей импульса
рх ограничена условием
&х&р > k/2n.
A3)
Это условие характеризует и ограничен-
ограниченную точность информации, которую можно
получить о частице. Например, если с по-
помощью прибора удалось зафиксировать
точно положение частицы в пространст-
пространстве, то эта фиксация связана с неконтро-
неконтролируемым воздействием прибора на состо-
состояние движения частицы, и скорость ее или
импульс становятся полностью неопреде-
неопределенными. Максимальная информация в
принципе достижима лишь с точностью
Да; Ар = й/2я.
A4)
2. Уравнение Шредингера. Классическая
механика основана на постулатах Ньюто-
Ньютона. Основное уравнение, определяющее
движение материальной точки, не выводит-
выводится, а именно постулируется. Аналогичное
имеет место и для квантовомеханического
уравнения движения Шредингера. Чтобы
лучше понять интерпретацию уравнения
Шредингера, полезно напомнить основные
идеи классической механики Ньютона.
В классической механике под уравне-
уравнением движения частицы понимается урав-
уравнение, решая которое при заданных на-
начальных условиях, можно найти простран-
пространственную координату и импульс (или ско-
скорость) частицы в любой последующий мо-
момент времени.
183
Сравнение движения Ньютона есть ма-
математическое выражение постулата Нью-
Ньютона, согласно которому в каждой прост-
пространственной точке и в каждый момент
времени частица испытывает ускорение
а3 пропорциональное внешней силе Л\
действующей на частицу в этой прост-
пространственной точке в рассматриваемый
момент времени:
формулой
dU
F = — —- = — grad U\
dr
в одномерном случае (ска.кем, сила ,T,ei'icT-
вует вдоль оси х)
у =
dU
где т — масса частицы. Так как
- dv cPr
а =а=~шг'
то уравнение можно также написать в
виде:
Oh _>
Здесь v — вектор скорости частицы, г —
радиус-вектор пространственной точки на-
нахождения частицы. Для составляющих по
осям координат получим:
т dt2 =
dh,
m~W = FV
dH
dt2
Решение уравнения такого типа имеет
вид:
и
x(t) = x (t0) -\-(t- to) vx (to)
( t'
и
Как видим, для определения x(t) требует-
требуется знание начальных условий, т.е. x(to)
и vx (t0).
Работа, производимая над частицей си-
силой F на бесконечно малом пути ds, равна
скалярному произведению силы на путь:
F ds. Работа, производимая на конечном
пути, равна интегралу по этому пути от
элементарной работы:
При действии силового поля на свободную
частицу меняется энергия последней, при-
причем у свободной частицы вся энергия есть
кинетическая энергия ее движения:
mv2/2. Если частица (точечное тело) имеет
потенциальную энергию U, за счет измене-
изменения которой производится работа, то сила
действия при этой работе определяется
В некоторых случаях за счет уменьшения
потеициальной энергии частицы увеличи-
увеличивается ее кинетическая энергия. При этом,
если система замкнута (т. е. частица изо-
изолирована от внешних воздействий), полная
энергия частицы не должна изменяться
(закон сохранения энергии). Математиче-
Математически в одномерном случае это получает-
получается следующим образом.
Из условия F — —dU / dx следует dU ---
= —Fdx и
U — Uo = —\ Fdx.
Но по закону Ньютона F ¦¦
Поэтому можно написать
\ Fdx = га » —гг dx = т \.
г г,г
= ОТ \ 27^27 ^= -^Т-
ma — mdvldtj
Таким образом,
плп
mv2
Этот результат показывает, что для замк-
замкнутой (изолированной) системы сумма ки-
кинетической и потенциальной энергии, т. е.
полная энергия системы есть величина по-
постоянная.
Законы сохранения (в том числе и за-
закон сохранения энергии) являются самы-
самыми общими законами природы. Они оста-
остаются в силе и для систем, описываемых
квантовой, а не классической механикой.
Поэтому величина полной энергии изуча-
изучаемой квантовой системы существенным об-
образом входит в аппарат квантовой теории
стационарных систем, каковыми, в част-
частности, являются изолированные системы.
Если заданы условия, в которых осуще-
осуществляется движение изучаемой частицы
(скажем, электрона), т. е. указано сило-
184
вое поле, действующее па него, то квапго-
вомехапическая задача заключается в оп-
определении возможных значений энергии
(спектра) изучаемой частицы и характера
состояпия движения частицы. Эта задача
как раз решается анализом и решением
уравнения Шредингера. Под изучением со-
состояния движения следует понимать изу-
изучение вероятностного распределения воз-
возможных значений скоростей или прост-
пространственного положения частицы. Такой
тип информации диктуется волновыми
свойствами частицы и статистическим ха-
характером квантовой механики, о чем гово-
говорилось в предыдущем разделе.
Стационарное уравнение Шредингора
имеет вид:
ЯГ (х, у, z) = ЕГ (х, у, z),
A5)
где Н — оператор Гамильтона частицы, пп-
ляющийся аналогом фунцпи Гамильтона
классической мехапики, \|з — волновая
функция частицы (подробно смысл этой
функции будет объяснен ниже).
Прежде всего можно отметить анало-
аналогию с классическим соотношением для
функции Гамильтона.
Н (х, у, z, px, py, pz) = Е,
которое при известной форме функции
Гамильтона определяет совокупность зна-
значений х, у, z, рх, ру, pz, которые могут
принадлежать рассматриваемой частице
при заданной энергии Е.
Однако в уравнении Шредингера функ-
функция Н заменена оператором, форма кото-
которого такова, что уравнение имеет анало-
аналогию с волновым уравнением. Именно, в
самом общем случае, когда с классической
точки зрения частица должна была бы об-
обладать кинетической и потенциальной
энергией (в квантовой механике опреде-
определенной величиной является только полная
энергия, а кинетическая и потенциальная
энергии порознь не имеют определенных
значений), оператор Гамильтона частицы
имеет вид:
# = —¦
z),
где V2 — оператор Лапласа, V2 = дг1дх2 +
+ а2 / ду2 + З3 / dz2; h = h / 2я, h — посто-
постоянная Планка. Уравнение Шредингера, та-
таким образом, имеет форму
или
2т
V2T + — (E-U) V = 0.
Шредингера аналогично уравнениям вод-
новых процессов. Поэтому целесообразно
использовать «наглядные аналогии», чтобы
лучше уяснить себе различные особенно-
особенности уравнения Шредингера. Такие нагляд-
наглядные аналогии будут рассмотрены в следую-
следующих разделах, где подробно разбираются
одномерные, двухмерные и трехмерные
механические колебания (колебания стру-
струны, мембраны и «пылевого шара»),
3. Колебания струны *. Рассмотрим стру-
струпу, натянутую вдоль оси х. Пусть как-ьч-
нибудь точка этой струны получила не-
небольшое вертикальное смещение вдоль <>си
у. Струна начнет совершить колебания.
Найдем уравнение движения такой колеб-
колеблющейся струны. Найдем силу, которая
действует на элемент струны ds в направ-
направлении оси. Именно эта сила оугроделмет
Рис. 223. К выводу уравнения колебания
струны
колебательное движение струны. Очевидно,
величина этой силы равна (см. рис. 223):
f = Т sin q>2 — Т sin q>i = Т (sin фа — sin cpi),
где Т — сила натяжения струны, которая
по абсолютной величине постоянна во всех
точках струны и направлена по касатель-
касательной к линии струны в данной точке. Если
смещение струны невелико, то углы qii и
Ф2 малы, и синусы от этих углов можно за-
заменить тангенсами; последние же опреде-
определяют наклон кривой у (х) в соответствую-
соответствующих точках, т. е. равны ду/дх в точках х
и х + дх. Таким образом получим:
Производная ду/дх есть функция от х.
Разложим эту функцию в точке х + дх в
ряд Тейлора и ограничимся линейным
приближением:
у\ /ду\
A8)
Таким образом, вертикальная составляю
щая результирующей силы натяжений.
* Подробнее см., например, Ф. Морз «Ко-
Именно в последней форме уравнение лебание и звук». М.— Л., ОНТИ, 1949.
185
действующая на элемент поверхности стру-
струны, равна
¦ Tdx
Idi ~д^\х"=Т['дх^\/х-
Обозначим массу единицы длины стру-
струны через т. Тогда масса элемента струны
длиной dt равна mds. Если углы <р малы,
то можно принять ds ~ dx и mds « mdx.
Уравнение движения элемента струны
есть просто уравнение движения Ньютона,
которое с учетом вышеизложенного запи-
запишется следующим образом:
A9)
B0)
Для решения этого уравнения предполо-
предположим, что у можно представить в виде про-
произведения функции, зависящей только от
координат, на функцию, зависящую толь-
только от времени, т, е. у = W (х) 0 (t). Тогда
уравнение B0) сводится к виду
дх2
m d'v
Тв ~W
B1)
Поскольку х и t — независимые перемен-
переменные, то левая часть уравнения, зависящая
только от х, может быть равна правой ча-
части уравнения, зависящей только от t, при
любых х и t только в том случае, если обе
части уравнения равны одной и той же по-
постоянной. Приравнивая левую и правую
части уравнения B1) такой постоянной,
получим два отдельных уравнения для
V (х) и для 0 (t):
T~d^~ = coast- To"d7
= const.
Удобнее ввести новую постоянную со, по-
положив const = — mu>2/T. Тогда уравнения
примут вид:
d-Q
=0. B2)
Решениями этих уравнений служат про-
простые синусоиды и косинусоиды. Комбини-
Комбинируя их, получим выражение для у.
Выбранная форма записи для постоян-
постоянной (const) удобна тем, что тогда в реше-
решениях вида
1 sin со
B3)
(здесь введено обозначение Т/т = vz, ве-
величину со можно отождествить с угловой
частотой).
Легко проверить, что величина v имеет
размерность скорости и связана с длиной
волны соотношением
v = &> %/2п.
Решение можно также записать в виде
или
/ 2яж -
у = cos I at — —s— I,
причем сразу отсюда получается, что
функция у при заданном t имеет одно и
то же значение при х = пХ для всех целых
значений п.
Таким образом, если струна отклоняет-
отклоняется от положения равновесия, то ее коле-
колебание образует волну, распространяющую-
распространяющуюся вдоль струны с определенной скоростью
v. Уравнение B2), описывающее движение
струны, называется волновым уравнени-
уравнением. Ему удовлетворяет бесчисленное мно-
множество решений вида B3). Отбор решений
возможен, если заданы так называемые
граничные условия. Пусть, например,
струна закреплена в конечных точках
х = 0 и х = L (L — длина струны). Тог-
Тогда на функцию у накладываются следую-
следующие граничные условия:
у @, t) = ЧГ @) в (t) = 0, B4)
B5)
о = т AN@ = 0.
Так как приж=0 W @) =0, то решение не
может содержать косинусов. Так как при
a:=i4r(L) = 0, то при t = 0 sin (m/v) =0,
откуда
coL
= —я
А
= ЛЯ.
Отсюда получаем
А = 2L/n,
B6)
т. е. возможны колебания струны только
по вполне определенным (дискретным)
наборам значений длины волны.
В случае закрепленной струны волна,
возникшая на одном ее конце, дойдя до
другого конца, отразится. Возникнут две
бегущие волны (прямая и отраженная),
движущиеся в противоположных направ-
направлениях. В результате интерференции (на-
(наложения) этих волп возникает стоячая
волна. В точках струны, где эти волны
полностью компенсируют друг друга, стру-
струна остается неподвижной. Такие точки на-
называются узлами (или узловыми точками).
Из условия B6) вытекает, что при п = 1
будут только два узла (на концах) и рас-
расстояние между ними равно половине дли-
длины волны, т. е. X = 2L; при п = 2 будут
186
три узла и % = L и т. д. Набор дискретных
возможных длин волн называется спект-
спектром. Дискретность спектра показывает,
что длины волн «квантованы».
4. Колебания мембраны и «пылевого ша-
шара». Двумерным аналогом колеблющейся
струны может служить колеблющаяся мем-
мембрана. Волновое уравнение для мембраны
имеет вид:
0*1/ о о U
нение имеет вид:
тде Т — натяжение мембраны, а — массо-
массовая плотность мембраны (имеющая ту же
размерность, что и плотность m одномер-
одномерного случая).
Это уравнение можно записать также
в виде
о
т
B7)
где V2 — двумерный оператор Лапласа:
V2 = д2/д2 д*/д2
Как и в случае волнового уравнения
для струны, уравнение B7) тоже допуска-
допускает разделение переменных (времени и
пространственных координат). После та-
такого разделения уравнение, в которое вхо-
входят только пространственные переменные,
запишется в виде:
3@-
(*. У) + ~Tf~ ЧГ (х, у) = 0.
B8)
В колеблющейся мембране, закреплен-
закрепленной по краям, в отличие от колеблющейся
струны, закрепленной у концов, будут не
узловые точки, а узловые линии. Каждое
состояние колебания мембраны можно ха-
характеризовать числом и формой таких уз-
узловых линий (они могут быть концентри-
концентрическими, расположенными в шахматном
порядке и т. п.). Полная характеристика
узловых линий дается двумя наборами
целочисленных значений (которые обо-
обозначаются через п ж I). Простейшие слу-
случаи изображены на рис. 224.
Трехмерным аналогом колеблющейся
струны может служить колебание упруго-
упругого шара, состоящего из частиц, находяще-
находящегося во взвешенном состоянии («пылевой
шар»). Соответствующее волновое урав-
п=1, 1=0 т)=2. г=0 т)=2, 1=1 г=?,, 1=2
Рис. 224. Узловые линии, возникающие
.при колебании круглой мембраны
V"F (х, у, z) + -у- ЧГ (х, у, ж) = 0, B9)
где У2=а2/9ж2 + дУду2 + d*ldz\
В колеблющемся шаре узловые элементы
образуют двухмерные поверхности, и для
их характеристики требуются три набора
целочисленных значений. В механических
колебаниях функция ^(х, у, z) соответст-
соответствует амплитуде изучаемой системы в точ-
точке х, у, z. В случае стоячих волн сам про-
процесс колебания во времени описывается
другим уравнением, которое мы здесь под-
подробно не рассматривали, так как для изу-
изучения связанных атомных систем оно не
представляет особого интереса.
5. Волновая функция электрона. Урав-
Уравнение Шредингера по форме сходно с
трехмерным уравнением для «пылевого
шара». Однако функция W в уравнении
Шредингера имеет совершенно другой фи-
физический смысл, чем амплитуды классиче-
классических волновых уравнений. Эту функцию
называют волновой функцией по формаль-
формальной аналогии с классической функцией W.
Однако такой простой интерпретации эта
функция не имеет. Физический смысл
имеет величина x?(x,y,zJdV, означаю-
означающая вероятность того, что электрон (или
другая изучаемая частица) будет обнару-
обнаружен в физически элементарном объеме
dV, содержащим точку (х, у, z).
Легче всего смысл волновой функции
иллюстрировать на примере свободного
(не связанного) электрона.
Функция U в уравнении Шредиигера
A8) есть оператор потенциальной энер-
энергии. Для свободного электрона этот опера-
оператор должен быть тождественно равен ну-
нулю. Тогда уравнение Щредингера прини-
принимает вид:
или
= 0.
C1)
Для свободного электрона его полная
энергия совпадает с понятием кинетиче-
кинетической энергии. Поэтому в предельном слу-
случае классической механики Е — p2/2mt где
р — импульс электрона. Сообразуясь с этим
классическим соответствием, оператор
V
в квантовой механике называют опе-
оператором шшуульса (в одномерном слу-
случае этот оператор имеет вид — i/h • д/дх).
Свободной частице, как мы зпаем,
можно приписать определенный им-
импульс, если не фиксировать положение ча-
частицы в пространстве. Пусть частица име-
187
ет импульс р; выберем координатную ось
х вдоль этого направления импульса (т. о.
сведем задату к одномерному случаю).
Тогда, согласно соотношению Де-Бройля,
этот импульс выражается через длину вол-
волны: р = hj'k. Таким образом, формально
вместо Е можно написать
'lm
J.m
и уравнение принимает вид
Й2 62Т 1 / /г 2
2т
tm
или
дх-
-1-
\ к
C2)
(напомним, что h-2nh). Как видим, мы
пришли к полному аналогу волнового
уравнения для струны. Аналогично в двух-
двухмерном и трехмерном случаях можно от
уравнения Шредипгера для свободной ча-
частицы формально прийти к Волковым
уравнениям для мембраны и «пылевого
тара». Отсюда видна причина, по которой
уравнение Шредингера называют волно-
волновым. Такая аналогия используется также
при решении уравнения Шредингера.
Рассмотрим решепие уравнения Шре-
дингера для свободного электрона в одно-
одномерном случае. Уравнение имеет вид:
C3)
Решить это уравнение —> значит найти до-
дозволенные значения Е (спектр энергии),
при которых решение существует, и вид
фупкции хУ(х), соответствующий каждому
дозволенному значению Е. Дозволенные
значения Е называются собственными
значениями энергии уравнения Шрединге-
Шредингера, а соответствующие решения: для XY —
собственными функциями.
Для свободного электрона имеется
только одно очевидное ограничение на
значения Е: значения энергии Е не могут
быть отрицательными, т. е. они могут быть
любыми положительными значениями (не-
(непрерывный спектр энергии). Общее реше-
решение при Е >. 0 имеет вид:
Ч? (х) = A cos kx -\- В sin kx, (З^)
где к = i2mE / h, а А и В — постоянные,
которые находятся из условия норми-
нормировки.
Мы уже отмечали, что величины |4rj2rfV
есть вероятность нахождения электрона в
объеме dV около определенной простран-
пространственной точки. Интеграл от этой величи-
величины по всему объему V есть вероятность
нахождения электрона где-то внутри всего
объема. Но это есть достоверное событие-,
т. е. такая вероятность равпа единице.
Соотношение, выражающее этот факт, т. о.
называется условием нормировки, а регао-
iii'G ураш.о-'ня Шрелппгера 4f, удовлетво-
удовлетворяющее этому условию, называется норми-
нормированной собственной ;шга волпозой)
функцией.
Из условия нормировки пля свободного
электрона получаете Л = I / f V, В = i}V
(i — мнимая единица), так что
1
(х) = —-т~ (cos kx + i yin kx).
C6)
Отсюда пс.'.у.аем:
| T (x) B = -у- (cos2 kx -I- sin2 kx) = -y, C7)
т. е. вероятность нахождения свободного
Эчсктрон.: в любой точке пространства од-
одна и та же. Такагг неопределенность есть
следствие принципа неопределенности
Гейзонберга. Свободному электрону дан-
Hoii энергии Е можно приписать яттолпе
оп'!оделоиный импульс и поэтому его по-
положение в пространстве полностью ш1-
орпедглеино. Урав;1с:;ие Ш;)олш!г.м1я лля
свободного электрона для проблем хими-
химической связи но представляет особого ин-
интереса. Электрон связан в атоме взаимо-
взаимодействием с ядром (а также взаимодейст-
взаимодействием с другими электронами, однако в пер-
первом приближении последнее взаимодейст-
взаимодействие не учитывают). Поэтому для таких
проблем следует рассматривать уравнение
Шредингера в виде A7), содержащем опе-
оператор потенциальпой энергии U. Это урав-
уравнение наиболее удобно записывать в «вол-
«волновой» форме:
2то (Е — U)
К"
C8)
Вид решения этого уравнения существен-
существенным образом зависит от формы оператора
потенциальной энергии U. Только в про-
простейших случаях решение уравнения не
представляет затруднений. Для иллюстра-
иллюстрации в следующем разделе мы рассмотрим
самую простую модель связанного элект-
электрона: одномерное движение электрона в
потенциальном ящике. Эта модель очень
полезна для задач физики атома.
6. Простейшая модель связанного элект-
электрона. Рассмотрим одномерное движение
электрона в прямоугольном «потенциаль-
«потенциальном ящике», т. е. потенциальная функция
которого в силовом поле имеет вид: (см.
рис. 225): U = 0 при 0 < х < a, U = Un
при х ^ 0, i^e.
Анализ одномерного стационарного
уравнения Шредингера сразу показыва-
188
°0
X
Рис. 225. Схема одномерного прямоуголь-
прямоугольного потенциального ящика
ет, что в завпсимости от значения энер-
энергии Е возможны 2 типа решений, имею-
имеющих разный физический смысл. При
Е ^ Uo частица «не чувствует ящика* и
ведет себя как свободная. Мы опять при-
приходим к уравнению Шредингера для сво-
свободной частицы в форме C1), если за
начало отсчета энергии Е (нуль энер-
энергии) принять не дно ящика, а значение
Uo. При E<U0 будут уже другие усло-
условия. Из физических соображений очевид-
очевидно, что тогда частица не может оказать-
оказаться вне ящика, т. е. |vF[2 = 0 при х ^ О и
х ^ а. Но такое условие возможно, толь-
только если ? = 0 при х ^0 и х^а *.
Таким образом уравнение Шредингерп
на интервале 0<ж<а сводится к уравне-
уравнению вида:
при «граничных условиях»
Y @) = У (а) = 0.
Общее решение уравнения такой формы
мы привели в предыдущем разделе:
Чг (х) = A cos kx
0
В sin kx,
где к =
Но теперь это решение должно удовлет-
удовлетворять граничным условиям. Условие
\Р@) дает /1=0. Из условия Чг(о)=0 по-
получается sin ка = 0, откуда ка = гея, где
п — целое положительное число начиная
с единицы. Таким образом, находим
спектр энергии (учитывая, что Е=
- HVI2m):
2та2
¦ ге\
C9)
Эти значения энергии определяют уров-
* Строго говоря, в квантовой механике
это верно только при U-*-co, при конеч-
конечном же Uo функция Ч' «проникает» в
область ж^Оиж^аи быстро там за-
затухает, однако такое уточнение для нас
несущественно.
ни энергии в потенциальном ящике, при-
причем отсчет энергии ведется начиная от
дна ящика.
Уравнение C9) показывает, что энер-
энергия в рассматриваемом примере оказы-
оказывается квантованной величиной и соот-
соответственно кваптованпо состояние движе-
движения электрона.
Выражение для собственной функции
соответствующей ге-му уровню энергии,
имеет вид:
яге
-sin-
D0)
Здесь множитель перед синусом есть яв-
явное выражение для постоянной В, ко-
которое находится из условия нор.мн-
ровки.
Заметим еще, что расстояние между
соседними уровнями энергии определяет-
определяется по формуле:
Я/г2
Из этой формулы видно, что чем меньше
ширина а потенциального ящика, тем
больше расстояние между уровнями.
§ 5. Ковалентная связь
в молекулах и кристаллах
1. Решение уравнения Шрединге-
Шредингера для атома водорода. Даже для
простых атомных систем (например,
для атома водорода) точное решение
уравнения Шредингера получить
весьма трудно. Поэтому метод реше-
решения мы опишем здесь только в общих
чертах.
В последнем разделе предыдуще-
предыдущего параграфа было показано, что
энергия электрона в одномерном по-
потенциальном ящике квантована, т. с.
образует дискретный ряд значений,
характеризуемый целочисленным
числом п, называемым квантовым
числом. При этом нужно обратить
внимание, что получаемое решение
во многом аналогично решению вол-
волнового уравнения для колеблющейся
струны, закрепленной по концам.
Это решение храктеризуется, таким
образом, одним параметром — квап-
189
Qfh
X/
Рис. 226. Переход от декартовой системы
координат к сферической (угол Z О г обо-
обозначим ¦&).
товым числом п. В двухмерных зада-
задачах связанного электрона решение
аналогично решению волнового
уравнения для колеблющейся мем-
мембраны, закрепленной по краям, так
что решение характеризуется двумя
квантовыми числами. Наконец, в
трехмерном случае решение харак-
характеризуется тремя квантовыми чис-
числами.
Как мы уже отмечали, характер
решения уравнения Шредингера за-
зависит от вида оператора потенциаль-
потенциальной энергии. В случае атома водоро-
водорода выражение для этого оператора
(потенциальной энергии электрона в
электрическом поле заряда ядра) яв-
является довольно простым: взаимо-
взаимодействие происходит по закону Ку-
Кулона и потенциал имеет вид: — ег/г,
где г — расстояние электрона от яд-
ядра. Заметим, что такая форма по-
потенциала верна только при действии
«голого» ядра на атомный электрон;
если кроме рассматриваемого элек-
электрона имеются еще другие атомные
электроны, то форма потенциала
будет гораздо сложнее.
Таким образом, уравнение Шре-
Шредингера для атома водорода прини-
принимает вид:
теме координат, в которой оператор
Лапласа имеет вид
а
дх2
Г ~Я„2 1~ "~ЯТ2 •
В этой системе г= Ух* + уг + z2 , так
что уравнение получается очень
сложным по форме. Однако оно вы-
выглядит гораздо проще, если перейти
от декартовой системы координат к
сферической, где положение прост-
пространственной точки характеризуется
расстоянием от центра системы ко-
координат г и двумя углами 0, ф
(см. рис. 226). Между старой и новой
системами координат имеется соотно-
соотношение ¦¦ i t
ж= г sin d cos <р,
у = г sin ¦& sin cp,
z =r cos d.
Если выбрать в новой системе ко-
координат центр в ядре, то расстоя-
расстояние от ядра до электрона и будет
координатой г новой системы. Тогда
решение можно искать в виде:
Y(r, «, ф) = Л(г)в(#)Ф(ф). D2)
Уравнение разбивается на три
отдельных уравнения относительно
функций R, в, Ф. Все три решения
оказываются квантованными, т. е.
характеризуются своими квантовыми
числами. Решение для Ф характе-
характеризуется так называемым магнит-
магнитным квантовым числом тп; решение
для в характеризуется азимуталь-
азимутальным квантовым числом I (а также
магнитным квантовым числом тп);
решение для R характеризуется
главным квантовым числом п (а так-
также азимутальным квантовым чис-
числом I). Появление второй характе-
характеристики указывает на взаимосвязь
решений.
Таким образом, общее решение за-
записывается в виде:
= Rn, С")
п
Для водорода энергия системы зави-
Уравнение Шредингера мы рань- сит только от главного квантового
гяе представляли в декартовой сие- числа п, но не зависит от квантовых
190
Рис. 227. Типы атомных орбиталей
а — s-орбиталь;
б — р-орбитала
чисел I и т. Состояния электрона,
описываемые различными собствен-
собственными волновыми функциями, но
имеющие одинаковую энергию, на-
называются вырожденными. Таким об-
образом, у водорода все состояния,
отличающиеся азимутальным, маг-
магнитным и спиновым квантовыми
числами, но имеющие одно и то же
главное квантовое число, вырожде-
вырождены. Однако такое вырождение сни-
снимается при возмущении системы.
Например, у других атомов электрон
движется в поле ядра и в поле дей-
действия других атомных электронов.
Действие других электронов и есть
то возмущение, которое приводит к
различию значений энергии с одина-
одинаковыми п, но с разными азимуталь-
азимутальными квантовыми числами I. Снятие
вырождения по т происходит при
воздействии на атомную систему
магнитного поля.
Итак, для водорода можно опреде-
определить спектр энергии как зависящий
от квантового числа п (Еп), в общем
же случае спектр определяется на-
набором значений энергии, завися-
зависящих от га и Z (Eni).
Как и в старой квантовой теории
Бора, размещение электронов по
квантовым состояниям определяется
принципом минимума свободной
энергии системы и принципом Пау-
Паули, согласно которому у атома не
может быть двух и более электронов
с одинаковыми значениями всех че-
четырех квантовых чисел (п, I, m, s),
характеризующих состояния элект-
электрона.
2. Аналитические выражения вол-
волновых функций водородоподобных
атомов и их геометрическая интер-
интерпретация.
Для водородоподобных атомов, да-
даже если не учитывать возмущения,
вносимого электронами на состояние
рассматриваемого электрона, общее
выражение решения уравнения
Шредингера довольно сложно. Одна-
Однако для собственных функций с не-
небольшими значениями п, I, и т эти
выражения имеют сравнительно
простой вид. В табл. 24 приведены
некоторые из этих выражений для
водородоподобного атома с зарядом
ядра Ze (е — абсолютная величина
заряда электрона, Z — атомный но-
номер химического элемента). В табли-
таблице приняты следующие обозначения:
а0 — радиус первой боровской орби-
орбиты для атома водорода, равный
h2/me2 = 0,529 А р = Zr/ao, т. е. р есть
расстояние электрона от атомного
ядра, выраженное в единицах ao/Z.
Примеры атомных орбиталей пока-
показаны на рис. 227.
Как видно из табл. 24 и рис. 227, а,
собственные функции s-орбиталей
не зависят от углов ft и ф и, следова-
следовательно, обладают сферической сим-
симметрией. Три />-орбитали похожи на
гантели. Они имеют ясно выражен-
выраженный направленный характер, причем
191
Рис. 228. Пять d-орбиталей
их оси взаимно перпендикулярны.
Поэтому в принципе направления
этих осей можно принять за коорди-
координатные оси х, у, z. Именпо этим
объясняется, почему соответствую-
соответствующие орбитали обозначены индекса-
индексами х, у, z.
На рис. 227, б показаны пример-
примерные граничные поверхности для
этих атомных орбиталей. Все три р-
орбитали совершенно эквивалентны,
за исключением их направлений.
Поэтому в общем случае произволь-
ТАБЛИЦА 24
Выражения для Т-функции
Выражения для
—1
is- /itч,
2p2 4 |/2
Xcosd
r _ 1 /2 Л'.
ре "х
Xsin
vir
V. ~Pk
/ Z 3
7=\—\
31г
\
X
1 81 |/"Зя
X B7 — 183 + 2р2)е-р/з
ную орбиталь р-типа с осью симмет-
симметрии, не совпадающей с заранее вы-
выбранными осями х, у, z, можно пред-
представить в виде линейной комбинации
базисных ^-орбиталей, направленных
по этим координатным осям. Если
рассматривать W и WPx, WPy, WPz как
векторы, то в линейном представле-
представлении W = aWPx+ |И'р„, + Г%ъ; коэф-
коэффициенты а, Р, у можно рассматри-
рассматривать как направляющие косинусы.
В табл. 24 не приведены орбитали
d-типа (для I = 2, т = 0, ±1,_±2).
Однако знание этих орбиталей су-
существенно для понимания свойств
комплексных соединений и так на-
называемых переходных металлов.
Имеется пять различных собствен-
собственных функций d-типа в соответствии
с пятью возможными значениями
магнитного квантового числа. Одна-
Однако все пять функций соответствуют
одному и тому же значению энергии
(о вырождении по магнитному кван-
квантовому числу мы уже говорили вы-
выше). Форма d-орбиталей приведена
на рис. 228.
3. Приближенные методы решения
уравнения Шредингера для много-
многоэлектронных атомов. Метод валент-
валентных связей.
Поскольку строгое решение урав-
уравнения Шредингера возможно только
для простейших случаев (например,
таких, как Н), а в химии приходится
решать очень сложные задачи, то
дальнейшее развитие теории кова-
лентной связи пошло по линии раз-
развития приближенных методов реше-
решения уравнения Шредингера, исполь-
192
зующих эмпирические закономерно-
закономерности и дополнительные постулаты.
Здесь мы опишем так называемый
метод валентных связей. Развитие
этого метода было положено извест-
известной работой Гайтлера и Лондона
A927 г.). В последующие годы этот
метод особенно интенсивно развивал-
развивался Полингом.
Основой метода валентных связей
является предположение, что кова-
лентная химическая связь характе-
характеризуется перекрыванием валентных
атомных орбиталей атомов, участву-
участвующих в связи. Так, например, хими-
химическая связь атомов А и В, образо-
образованная парой электронов в s-состоя-
ниях с противоположными спинами,
на рис. 229 представлена областью
перекрытия двух окружностей, кото-
которые изображают орбитали электро-
электронов, принадлежащих ядрам, располо-
расположенным в точках а и Ъ (область
перекрывания заштрихована). Оче-
Очевидно, что в случае s-состояшш обо-
обоих валентных электронов, образую-
образующих связь, величина области
перекрывания зависит только от рас-
расстояния между ядрами. Величина об-
области перекрывания характеризует
силу связи.
Простейшая схема ковалентной
связи, изображенная на рис. 229,
реализуется в молекуле водорода Щ.
В 1927 г. В. Гайтлер и Ф. Лондон вы-
выполнили квантовомеханический рас-
расчет основного стационарного состоя-
состояния молекулы Нг. Было установле-
установлено, что молекула водорода образуется
из двух атомов водорода только в
случае, если у этих атомов спины их
s-электронов антипараллельны (син-
глетное состояние). Этот результат
дал возможность объяснить две важ-
важнейшие особенности ковалентной
связи: 1) притяжение между двумя
одинаковыми нейтральными атома-
атомами; 2) насыщенность связей, т. е.
невозможность присоединения к мо-
молекуле Нг третьего атома.
Насыщаемость ковалентной связи
можно понять на основе принципа
Паули. Ковалентная связь характе-
13 Кристаллохимия
ризуется перекрыванием орбиталей,
но, следовательно, в области перекры-
перекрывания не могут быть два электрона,
имеющих одинаковыми все четыре
квантовые числа. У атома водорода в
основном состоянии га=1,1=0, т?г=О.
Поэтому различаться могут только
спины. Но различных ориентации
для спина только две, поэтому в ко-
ковалентной связи не может участво-
участвовать электрон третьего атома.
В случае образования ковалентной
связи электронами р-типа степень
перекрывания зависит не только от
расстояния между атомными ядрами,
но и от направления, по которому
происходит сближение атомов. Проч-
Прочная химическая связь осуществляет-
осуществляется при вполне определенной взаим-
взаимной ориентации орбиталей электро-
электронов атомов, вступающих в связь.
Ориентация должна быть такова,
чтобы достигалось максимальное пе-
перекрывание орбиталей электронов,
образующих связь, и минимальное
перекрывание орбиталей остальных
электронов. При такой взаимной
ориентации достигается минимум
свободной энергии системы. Таким
образом, согласно этой схеме связи
Рис. 229. Схема химической связи между
двумя атомами, образованной парой элект-
электронов в s-состояниях
Рис. 230. Схема химической связи между
двумя атомами, у одного из которых ва-
валентный электрон находится в р-состоя-
нии, а у другого — в s-состоянии
193
Рис. 231. Схема химической связи между
двумя атомами, валентные электроны ко-
которых находятся в р-состоянжи
Рис. 232. Расположение гантелей р-элект-
ронов, участвующих в образовании я-свя-
зи
Рис. 233. Схема строения1 молекулы эти-
этилена
всегда ориентированы относительно
друг друга под определенными ва-
валентными углами. Аналогичное име-
имеет место и для связи, образуемой
d-электронами.
Идея образования ковалентных
связей за счет максимально возмож-
возможного перекрывания орбиталей собст-
собственных волновых функций связываю-
связывающих электронов было впервые сфор-
сформулирована Л. Полингом.
Если атом с одним валентным элек-
электроном в s-состоянии вступает в
связь с другим одновалентным ато-
атомом с электроном в р-состоянии, то
ковалентная связь образуется в на-
направлении вытянутости облака р-
электрона (рис. 230), так как при
этом обеспечивается максимальное
перекрывание орбиталей, что и при-
приводит к наибольшей связи.
В молекуле Н2О два р-электрона
атома кислорода образуют две кова-
лентные связи с двумя s-электрона-
ми двух атомов водорода. Перекрыва-
Перекрывание орбиталей соответствует схеме,
показанной на рис. 230. Пример свя-
связи между двумя ^-электронами, ког-
когда связь направлена по оси двух ган-
гантелей, вытянутых по одной прямой,
показана на рис. 231. Такая связь,
например, наблюдается в молекуле
перекиси водорода ЩОг. Таким об-
образом, перекрывание двух сфер
(s-s-связь), сферы и гантели
(s-p-связь) и двух гантелей
(р-р-связъ) имеет то общее свойство,
что электронное облако, описываю-
описывающее вероятностное распределение
двух электронов, участвующих в об-
образовании ковалентной связи, сим-
симметрично относительно вращения
вокруг линии связи. Такого типа ко-
валентные связи обычно называются
а-связями. В частности, такого типа
а-связи возникают у молекулы
воды.
Существует еще другой тип пере-
перекрывания волновых функций р-элек-
тронов, который приводит к так
называемой я-связи. Этот тип хими-
химической связи возникает в случае
кратных связей, т. е. когда между
двумя атомами насыщаются несколь-
несколько (две или три) связи. На рис. 232
показано расположение гантелей ор-
орбиталей р-электронов, образующих
я-связь.
На рис. 233 показана схема строе-
строения молекул этилена (Н2С=СН2),
атомы углерода в которой связаны
двойной связью (а-связь и я-связь),
а атомы водорода связаны с углеро-
углеродом только а-связью (а-связь на ри-
рисунке показана линиями).
В случае я-связи атомные орбита-
пи р-типа перекрываются меньше,
194
a s+ p
б s + 2n
s + Up
3q
1q
1
Рис. 234. Схема, i показывающая возник-
возникновение электронного состояния в ре-
результате гибридизации состояний (а)
ь. ирос1Ч)йшио случаи гибридизации (б)
г s + 2р
д s + Зр +• 2й
чем в случае 6-связи, и потому в
общем случае я-связь является бо-
более слабой связью, чем а-связь меж-
между теми же атомами. Следовательно,
энергия двойной связи всегда мень-
меньше удвоенной энергии а-связи.
Метод валентных связей не явля-
является единственным полуэмпириче-
полуэмпирическим методом анализа уравнения
Шредингера для трактовки ковалент-
ной связи в сложных соединениях.
Среди других методов безусловно
заслуживает внимания метод моле-
молекулярных орбит. Идея метода заклю-
заключается в том, что каждому валент-
валентному электрону, участвующему в хи-
химической связи, приписывается не
атомная волновая функция, а волно-
волновая функция электрона в системе
двух ядер (или ионов), находящихся
на том расстоянии, на котором они
находятся в рассматриваемой моле-
молекуле. Идея обобществления электро-
электронов (электрон принадлежит сразу
обоим ядрам) при химической связи
является весьма плодотворной.
По сравнению с методом валент-
валентных связей метод молекулярных ор-
орбит имеет ряд преимуществ. Однако
обсуждение этого вопроса выходит за
рамки настоящей книги, так как в
этой книге мы задались целью дать
как можно более наглядное изложе-
изложение теорий химической связи, а метод
молекулярных орбит мало нагля-
нагляден. Именно по этой причине для
тз*
195
популяризации физических идеи,
лежащих в основе понимания кова-
лентной связи, мы ограничились из-
изложением метода валентных связей
как наиболее паглядного.
4. Гибридизация связей. В § 1
была приведена электронная струк-
структура атома углерода: Is22s22p2. В
нормальном состоянии атом углеро-
углерода имеет два неспарепяых р-элект-
рона. Отсюда следует ожидать, что
нормальной его валентностью бу-
будет 2. Действительно, иногда угле-
углерод имеет такую валентность. На-
Например в СО. Однако в подавляющем
большинстве своих соединений уг-
углерод имеет валентность 4.
Чтобы объяснить такую валент-
валентность, следует предположить, что
при образовании четырехвалентного
соединения атом углерода перехо-
переходит в возбужденное состояние, при
котором один 2.9-электрон переходит
на 2/ьуровень, причем не спарива-
спариваясь с имеющимися на этом уровне
электронами. В результате у углеро-
углерода окажется четыре неспаренных
электрона и он будет иметь следую-
следующую структуру: Is22s2ps (три р-элек-
трона находятся в состояниях
2рх, 2ру, 2pz). Для осуществления
такого перехода необходимо затра-
затратить энергию в 96 ккал/молъ. Оче-
Очевидно, эта энергия полностью черпа-
черпается из энергии, выделяющейся при
образовании двух дополнительных
связей. Иначе не существовало бы
четырехвалентного углерода (его
существование было бы энергетиче-
энергетически невыгодным).
Возникает вопрос о пространст-
пространственной ориентации четырех образу-
образующихся таким образом связей. Ка-
Казалось бы, что исходя из общих фи-
физических соображений, три ^-связи
должны быть направлены взаимно
перпендикулярно, а четвертая связь
(s-связь), как лишенная свойств на-
направленности, должна существенно
отличаться от трех /ьевязей. Весь
опыт органической химии противо-
противоречит такому предположению. Со
времени Вант-Гоффа и Лебеля
A900 г.) известна равноценность
всех четырех связей атома углерода.
Это доказывается как чисто химиче-
химическими методами (установившими
факт отсутствия изомеров в соедине-
соединениях CX3Y и CX2Y2), так и физиче-
физическими методами (из анализа враща-
тельно-колебательных спектров).
Чтобы согласовать все известные
факты с теорией ковалентной связи,
Л. Полингом A931 г.) была выдви-
выдвинута идея о гибридизации связей.
Согласно этой идее, вместо исходных
собственных функций всех валент-
валентных электронов надо' ввести новые
функции, являющиеся линейными
комбинациями исходных, причем
число этих гибридных орбиталей
равно числу исходных. При я/?3-гиб-
ридизации четыре новых одинаковых
валентных направления расположе-
расположены от центра к вершинам тетраэдра
(в центре находится ядро углерода),
все углы между связями тогда полу-
получаются одинаковыми, равными
109°32'. Такая конфигурация свя-
связей энергетически выгоднее исход-
исходной, связи прочнее, т. к. происходит
ТАБЛИЦА 25
Важнейшие пространственные расположе-
расположения валентных связей, получающиеся в
результате различных случаев гибридиза-
гибридизации
рди-
:он-
тсло
Its
2
3
3
4
4
5
5
6
6
Используемые
атомные
орбитали
sp или dp
sp2, dp2 или
d4
d2p
sp3 или d?s
dsp*
dsj? или
d?sp
d*s
d4f
disp
Результир^/ющая
(гибридная) орбиталь
Лпнейная
Плоская тригональная
Т ригона льно-пирами-
дальная
Тетраэдрическая
Плоская тетрагональная
Тригонально-дипирами-
дальная
Тетрагонально-пирами-
Тетрагонально-пирамидальная
Октаэдрическая
Тригонально-призмати-
ческая
196
большее перекрытые электронных
орбиталей, чем при связях s—s, s—р
ир—р.
Изменение формы орбиталей схе-
схематически показано на рис. 234.
Идея гибридизации нашла подтвер-
подтверждение в некоторых приближенных
методах решения уравнения Шре-
дингера. Как правило, гибридизация
захватывает только электронные
состояния с одним значением кван-
квантового числа I или двух соседних
значений. В работе Кимбола были
определены пространственные ори-
ориентации валентностей для различных
случаев гибридизации; эти результа-
результаты приведены в табл. 25.
§ в. Металлическая селяь
1. Понятие металлической связи. Ме-
Металлы, в отличие от всех других
кристаллических твердых тел, обла-
обладают характерными физическими
свойствами и особенными кристал-
кристаллическими структурами. Металли-
Металлические кристаллы обладают высокой
электропроводностью и теплопровод-
теплопроводностью, а кристаллические структу-
структуры обычно удовлетворяют требова-
требованиям плотнейших упаковок и харак-
характеризуются, следовательно, больши-
большими координационными числами. Сое-
Соединения, образующиеся из несколь-
нескольких металлических элементов, от-
отличаются по характеру связи от
всех других классов химических ве-
веществ. Обычные представления о ва-
валентности элементов не способны
объяснить химический состав боль-
большинства интерметаллических соеди-
соединений. Состав интерметаллических
фаз часто не подчиняется закону
простых кратных отношений и мо-
может варьировать в широких преде-
пределах. Этот факт говорит о том, что
связь между атомами в металличе-
металлических кристаллах (и жидких распла-
расплавах) не ограничивает соотношение
элементов ни численно, ни простран-
пространственно. Каждый атом в металле стре-
стремится окружить себя максималь-
максимально boimo/Кным числом соседних ато-
атомов. Такую связь принято называть
металлической связью.
Из высокой электропроводности
и теплопроводности металлов можно
сделать заключение, что, по край-
крайней мере, часть электронов имеет
возможность свободно перемещать-
перемещаться по кристаллу и уже под действи-
действием слабого электрического поля или
небольшого градиента (перепада)
температуры может образовываться
направленный поток электронов
(Друде, 1902 г.). Согласпо теории
Друде, строение металлов можно
представить себе как совокупность
положительно заряженных ионов
(атомных остовов кристаллической
структуры), между которыми свобод-
свободно перемещаются электроны, подчи-
подчиняющиеся газовым законам («элект-
(«электронный газ»).
Большие координационные чис-
числа у атомов в кристаллических струк-
структурах говорят о том, что металличе-
металлическая связь, как и ионная, в отличие
от ковалентной связи, не обладает
свойством направленности. В этом
отношении металлическая связь
сходна с ионной.
Широкие области гомогенности
пнтерметаллических фаз всегда явля-
являлись затруднением при научении хи-
химии металлов. По существу говоря,
этот раздел химии успешно стал раз-
развиваться лишь благодаря работам
Н. С. Курнакова по физико-химиче-
физико-химическому анализу и развитию кристал-
кристаллохимии, так как ведущим призна-
признаком интерметаллического соединения
оказался не его состав, а кристалли-
кристаллическая структура.
Рассмотрим более подробно при-
природу металлической связи на приме-
примере кристалла лития.
Свободный атом лития имеет
следующую электронную структуру:
Is22s. Грубо можно предположить,
что в кристалле лития два ls-элект-
рона каждого атома, т. е. электроны
внутренней оболочки мало подвер-
подвержены воздействию окружающих ато-
атомов, но 2х-электроны уже не при-
197
надлежат отдельному атому, а при-
принадлежат кристаллической решетке
в целом. Говорят, что электроны
обобществлены. Именно с пекоторы-
ми особенностями энергетического
спектра таких обобществленных элек-
электронов в металлах связаны специ-
специфические особенности металлической
связи. Обобществленные электроны
ответственны также за высокую
электропроводность и теплопровод-
теплопроводность лития.
Можно образно сказать, что металл
лития состоит из положительно за-
заряженных атомных (ионных) осто-
остовов, которые погружены в «море
электронов» («электронный газ»),
причем эти «бывшие» 2в-электроны
атомов лития ведут себя как вырож-
вырожденный газ, т. е. как система частиц,
подчиняющаяся не классическим, а
квантовым статистическим законам
(квантовой статистике).
Ионы Li+ (атомные остовы), имею-
имеющие электронную структуру Is2,
расположены по узлам объемноцент-
рированной кубической решетки.
Известно, что радиус иона Li+ со-
составляет 0, 68 А. Длина связи Li—Li
в молекуле Li2 в газе равна 2,674 А,
а в кристалле лития расстояние меж-
между ближайшими соседями составляет
3,03 А. Однако увеличенную длину
связи в кристалле по сравнению с
молекулой нельзя рассматривать как
признак более слабой связи. Энер-
Энергия связи в кристалле равна
39 ккал/молъ, а в молекулярном
газе эта энергия составляет
13 ккал/молъ. Металлическая связь
осуществляется через электроны, об-
образующие газ почти свободных элек-
электронов (так называемые электроны
проводимости). Атом в решетке, та-
таким образом, связан даже сильнее,
чем в молекуле.
В любом кристалле в нормальных
условиях в 1 си3 содержится при-
примерно 1022 атомных остова. Такого
же порядка концентрация электро-
электронов проводимости. Строгая теория
показывает, что все электроны кри-
кристаллической решетки являются
198
обобществленными: спектр всех
электронов получается по одной и той
же схеме одноэлектронного прибли-
приближения (один электрон в системе
всех ядер). Однако роль «внешних
и внутренних обобществленных
электронов» в химической связи
разная. Это лучше всего можно по-
понять с точки зрения аналогии с
ковалентной связью. Собственные
функции внутренних электронов,
«приписанных» к разным ядрам, не
перекрываются и, значит, не дают
вклада в связь, а собственные функ-
функции внешних электронов перекрыты,
и они ответственны за связь (следу-
(следует, однако, предупредить, что эта
аналогия очень условная, физиче-
физический смысл этих связей очень разли-
различен).
Точное квантовомеханическое опи-
описание металлической связи представ-
представляет сложную задачу, рассмотрение
которой выходит за рамки настоящей
книги. Поэтому в следующих разде-
разделах будут описаны лишь общие пред-
представления, на основе которых может
быть понята металлическая связь —
так называемая зонная модель крис-
кристалла. Следует сразу же оговориться*
что зонная модель описывает поведе-
поведение электронов в кристаллах с любым
типом связи, но нас будут интересо-
интересовать только те аспекты этой теории,
которые непосредственно связаны с
теорией металлической связи.
2. Энергетический спектр электро-
электронов в кристаллах. Энергетический
спектр электронов в кристаллах
имеет свои специфические особенно-
особенности. Легче всего эти особенности мож-
можно понять следующим образом.
Рассмотрим систему, состоящую
из двух одинаковых ядер, удаленных
на очень большое («бесконечно боль-
большое») расстояние друг от друга, и из
одного электрона. Электрон притяги-
притягивается ядрами и может оказаться или
около одного ядра или около другого.
В обоих случаях он будет вести себя
как атомный электрон. Спектр энер-
энергии такого электрона, как мы знаем,
состоит из дискретных уровней энер-
гии, но теперь в рассматриваемой
системе один и тот же спектр полу-
получается дважды: один раз, когда рас-
рассматриваемый электрон находится у
одного ядра, а другой,— когда у
другого ядра. В то же время волновые
функции в этих двух случаях будут
разными. Говорят, что уровни энер-
энергии электрона в такой системе дваж-
дважды вырождены. Если ядра сближать,
то возникнет взаимодействие чисто
квантовой природы (так называемый
обменный эффект), и в результате
дважды вырожденный уровень оказы-
оказывается расщепленным на два отдель-
отдельных уровня энергии, причем чем бли-
ближе ядра, тем сильнее возмущение и
тем значительнее расщепление. Ана-
Аналогичное имеет место в системе из
трех одинаковых ядер и одного элект-
электрона: здесь происходит расщепление
трижды вырожденного уровня на три
разных уровня. По такой же схеме
рассматривают и кристалл. Прибли-
Приближенно допускают, что в задаче о
спектре энергии наличие многих
электронов в системе является не
очень существенным, побочным фак-
фактором и при определении энергетиче-
энергетического спектра можно рассматривать
систему из N ядер, образующих крис-
кристаллическую решетку, и одного элек-
электрона. Это — так называемое одно-
электронное приближение, на основе
которого до самого последнего време-
времени была построена вся электронная
теория кристаллов. Только такие яв-
явления как ферромагнетизм и сверх-
сверхпроводимость потребовали создания
многоэлектронной теории. Для тео-
теории химической связи в кристаллах
одноэлектронное приближениие дает
вполне удовлетворительные резуль-
результаты.
Итак, рассматривается кристалл из
N ядер и одного электрона. Исход-
Исходным является дискретный атомный
спектр энергии электрона. Однако
каждый уровень энергии в кристалле
iV-кратно расщеплен. При этом учи-
учитывается, что самые нижние уровни
соответствуют внутренним электро-
электронам атома, эти электроны испытыва-
испытывают слабое воздействие со стороны
других ядер кристалла, и в соответст-
соответствии с этим эффект расщепления та-
таких уровней очень мал. Последую-
Последующие уровни расщепляются все зна-
значительнее. Для самых верхних уров-
уровней возможно даже перекрытие об-
областей расщепления.
Расщепление уровней всегда не-
необходимо, если рассматривать зада-
задачу с точки зрения принципа Паули.
Действительно, в системе из N ядер,
если бы не было расщепления
уровней, на каждый уровень энер-
энергии одноэлектронного приближения
можно было бы по принципу Паули
поместить только 2BZ+1) элект-
электрон, где 2Z+1 означает число различ-
различных значений квантового числа
т при данном значении кванто-
квантового числа I, а множитель 2 соот-
соответствует двум противоположным
ориентациям спина. Если же есть
iV-кратное расщепление, то на об-
образующиеся N уровней можно по-
поместить iV-2BZ+l) электрон [по
2BZ+1) на каждый уровень]. Если,
кроме того, учесть возмущающее
действие электронов друг на друга,
то и эти N уровней расщепляются
на 2Z+1 отдельных уровня, так что
на каждом уровне может быть толь-
только по два электрона с противопо-
противоположными ориентациями спина.
Но даже для самых высоких
уровней максимальное расщепле-
расщепление составляет конечную величину,
в то же самое время число возникаю-
возникающих уровней необычайно велико,
так как число N есть число узлов кри-
кристаллической решетки, которое прак-
практически бесконечно велико. Поэтому
уровни, возникающие при расщепле-
расщеплении, расположены настолько густо,
что их рассматривают как квазине-
квазинепрерывные. Совокупность таких уров-
уровней, получаемая из одного вырож-
вырожденного уровня, называется полосой
энергии.
Таким образом, одноэлектронный
спектр энергии кристаллов состоит
из полос, разделенных в общем слу-
случае интервалами «запрещенных зна-
199
Рис. 235. Типичный одноэлектроппый
спектр энергии металлов (полосы заштри-
заштрихованы)
чений энергии» (см. рис. 235). При
этом самые нижние полосы очень
узкие. Этому соответствует локали-
локализация электронов вблизи ядер.
Это по значит, что можно указать
точно, где находится электрон. Мож-
Можно лишь утверждать, что электрон
находится в области около одного
из N ядер системы. Для последую-
последующих полос энергии области локали-
локализации электрона около ядер расши-
расширяются. Наконец, у валентных
электронов эти области становятся
настолько широкими, что захватыва-
захватывают все пространство кристалла.
Электрон может с равной вероятно-
вероятностью оказаться в любой точке кри-
кристалла. Именно с этой точки зрения
валентный электрон может рассмат-
рассматриваться как «свободный». Следует
помнить, что в этой схеме обобщест-
обобществляются, т. е. принадлежат решетке
в целом все электроны (электроны в
этой схеме нельзя приписать опреде-
определенным атомным остовам), но толь-
только электроны самых внешних оболо-
оболочек оказываются совершенно нело-
кализованными в пространстве.
3. Зоны Бриллюэна. В предыду-
предыдущем разделе мы выяснили, что
спектр энергии электрона в кри-
кристалле имеет вид полос, разделен-
разделенных запрещенными областями энер-
энергии. Теория дает возможность рас-
рассчитать этот спектр. Если известен
аакон взаимодействия электрона с
ионами решетки (потенциал решет-
решетки), то в принципе можно опреде-
определить энергию электрона как функ-
функцию так называемого волнового век-
вектора к:Е = Е(к). Составляющие
этого вектора по оси х, у, z, т. е. кх,
ку, kz, имеют размерность if см. Уста-
Установлено, что при вполне определен-
определенных значениях к функция Е(к)
терпит разрыв, т. е. величина Е ме-
меняется скачком. Такой скачок и со-
соответствует области запрещенных
значений энергии. Значении к, при
которых функция Е{к) терпит раз-
разрыв, определяются типом решетки.
Соответствующая теория была раз-
развита Блохом A928 г.). В теории
Блоха рассматривается уравнение
Шредипгера для одного электрона
в поле кристаллической решетки.
Поскольку решетка имеет вполне
определенную периодическую струк-
структуру, то такой лее периодичностью
должен обладать и потенциал ре-
решетки, стоящий в уравнении Шре-
дингера:
D4)
— -^- V2T + СТ = ЕЧГ.
Если а, Ь, с есть основные век-
векторы решетки, то периодичность
выражается формулой
U (г г- nw-\- пгЬ + пзс) = U (г), D5)
где пх, п,г, Пз — целые числа. Нали-
Наличие такой периодичности облегчает
решение. Даже не зная точного ви-
вида U, можно показать, что решенле
будет иметь вид:
Т = и-* (г) eikr , Ds )
причем функция Uh(r) имеет та-
такую периодичность, что и потен-
потенциал U.
Решение такого вида называется
функцией Блоха. При таком реше-
решении собственные значения Е тоже
являются функциями волнового век-
вектора к.
В теории Блоха обращается вни-
внимание на то, что в задании векто-
200
pa к содержится определенный
произвол. Действительно, из усло-
условия
D7)
(г
пга
+ nsc) = и-, (г)
К
идет условие для
)— eik T Ut{r):
-, G-f- ma
К
-f- nsc) =
У). D8)
В то же время для каждой кристал-
кристаллической решетки с основными век-
векторами а, Ъ, с всегда можно ука-
указать так называемую обратную ре-
решетку с основными векторами а*,
Ъ*, с* такими, что по определению,
имеет место равенство
а а =ЪЪ = ее =1,
Т. е.
_ Л
гЪ Ь*
__ 2гегсс __ л
Поэтому в качестве решения уравне-
уравнения Шредингера мы могли бы выб-
выбрать и функцию
где ^ 1'=к + 2nmi'a* + 2nm^b* +
+ 2ятизс*, mi, пг2, mz — любые целые
числа, потому что эта функция тоже
удовлетворяла бы основному усло-
условию D8).
Такой произвол в выборе парамет-
параметра решения неудобен. Поэтому при-
принято параметр к ограничивать значе-
значениями, лежащими внутри элементар-
элементарной ячейки обратной решетки (т. е.
решетки с основными векторами
a*, b*, с*). Например, в случае прос-
простой кубической решетки с периодом
а значения к тогда ограничены ус-
условиями:
кк
Такая ограниченная область значе-
значений в пространстве вектора к назы-
называется приведенной зоной Бриллю-
эна или просто зоной Бриллюэна.
При ограничении значений векто-
вектора к потребовалось ввести второй
параметр для характеристики блохов-
ских собственных функций и собст-
собственных значений энергии. Почему
это потребовалось, поясним более на-
наглядно для случая простой кубиче-
кубической решетки.
В первоначальном решении век-
вектор к мог принимать любые значе-
значения. При этом вид функции и~(г) и
значения энергии Е(к) в области
—я/2 <; кх, ку, kz <! я/2 (первая зона
Бриллюэна), в области я/2 ' кх, ку,
/с2<2я/2 и —2л/2<кх, ку, kz<
^ —я/2 (вторая зона Бриллюэна) и
в последующих зонах ие совпадали
между собой. При ограничении обла-
области значений параметра <к, чтобы не
потерять решения, соответствующие
второй и следующим зонам, решения
записывают в виде
Параметр п указывает номер ис-
исходной зоны Бриллюэна. Для новых
функций Еп (к) можно ввести усло-
ьие периодичности, если распрост-
распространить эти функции на все значения
к, т. е.
Еп (X + 2кт7а + '1птф + 2лт3с) = Ег1 (к)
(схематически этот результат пока-
показан на рис. 235).
Значения функций Еп (к) с задан-
заданным п (рис. 236) образуют полосу
Рис. 236. Схема получения кривых Еп(к)
201
энергии, которая в общем случае
отделена областями запрещенных
значений энергии от соседних полос
энергии, соответствующих функциям
Еп(к) с другими значениями п. Эти
полосы энергии те же самые, о кото-
которых мы говорили в предыдущем раз-
разделе. Там мы подчеркивали, что пер-
первоисточником этих полос были диск-
дискретные атомные уровни энергии.
Поэтому часто индекс п отмечают не
цифрой, указывающей номер исход-
исходной зоны Бриллюэна, а буквальным
символом соответствующего дискрет-
дискретного уровня энергии атомного элек-
электрона. Например, говорят о 4 s-полосе
(Eid(k)), Зй-полосе (Е3а (к)) и т. д.
Для полной характеристики элект-
электронных свойств кристалла сущест-
существен характер зависимости энергии
Еп от волнового вектора к в послед-
последней заполненной зоне и степень за-
заполнения этой зоны электронами.
Для металлов характерно то, что
функцию Еп(к) для последней
заполненной зоны можно предста-
представить в виде
D9)
где т* имеет размерность массы и
тем ближе к истинной массе элект-
электрона, чем слабее электроны связаны
с атомными остовами решетки; эту
величину называют эффективной
массой. Абсолютная величина вол-
волнового вектора имеет размерность
1/см, ее произведение на постоянную
Планка имеет размерность импуль-
импульса. Таким образом, у металлов энер-
энергию электронов последней заполнен-
заполненной зоны можно представить форму-
формулой, по форме совершенно анало-
аналогичной формуле для энергии совер-
совершенно свободного электрона. По
этой причине соответствующие элект-
электроны называют почти свободными
электронами.
Второй не менее важной особенно-
особенностью металлов является не полное
заполнение электронами верхней зо-
зоны (называемой у металлов зоной
проводимости). Чтобы лучше по-
понять, что подразумевается под тер-
термином «полное или неполное запол-
заполнение зоны Бриллюэна электронами»,
необходимо снова привлечь предста-
представления, связанные с принципом Па-
Паули.
Мы уже отмечали, что величина
hk имеет размерность импульса р.
Если характеризовать состояние
электрона параметром к, то точность
определения этого параметра ограни-
ограничена принципом Паули, подобно со-
соответствующему ограничению для
точности определения импульса.
Действительно, нусть кристалл имеет
объем V с линейными размерами Ьх,
Lv, Lz. Электрон может находиться
в любой точке объема кристалла, т. е.
точность определения его простран-
пространственной координаты (скажем, х)
равна Lx. Тогда, согласно принципу
Паули, точность определения коорди-
координаты импульса дается условием
откуда
Таким образом, все пространство
вектора к можно разбить на ячей-
ячейки объемом Акх ¦ Аку • Akz =
— l/(LxLvLz) =1/V. Каждая такая
ячейка характеризует определенное
значение вектора к, т. е. характери-
характеризует определенное состояние элект-
электрона внутри рассматриваемой зоны.
Дополнительной характеристикой со-
состояния электрона будет его спин
(две возможные ориентации). Сле-
Следовательно, в каждой ячейке в прост-
пространстве вектора к, согласно принци-
принципу Пазили, может находиться не более
двух электронов. Полное заполнение
означает, таким образом, случай, ког-
когда число электронов в зоне равно
удвоенному числу ячеек. Неполное
заполнение получается, когда элект-
электронов меньше.
Электроны в первую очередь запол-
заполняют те ячейки, которые соответству-
соответствуют меньшей энергии (такие ячейки
202
Рис. 237. Первая и вторая зоны Бриллюэ-
на для структуры меди
находятся ближе к центру зоны).
В металлах электроны незаполненной
полностью зоны не касаются поверх-
поверхности зоны, а внутри зоны образуют
собственную поверхность равной
(и максимальной для них) энергии.
Эта поверхность в пространстве век-
вектора А называется поверхностью Фер-
Ферми. Вообще говоря, она может иметь
сложную форму. Но если между энер-
энергией и волновым вектором имеется за-
зависимость типа формулы D9), то
поверхность Ферми будет сфериче-
сферической или близка к ней. Следует сде-
сделать замечание, что вблизи границ
зоны Бриллюэна, которые всегда
имеют не сферическую (а подчас и
довольно сложную) форму (см. при-
примеры на рис. 237), соотношение D9)
выполняется плохо, и размещение
электронов по ячейкам подчиняется
весьма усложненным правилам.
Однако для металлов все же наибо-
наиболее характерно, что электроны пос-
последней зоны можно рассматривать
как почти свободные, вся совокуп-
совокупность этих электронов участвует в
образовании металлической связи, а
та часть электронов, которая распо-
расположена в квантовых ячейках вбли-
вблизи поверхности Ферми, ответственна
за специфические физические свой-
свойства металлов (электропроводность,
теплопроводность и т. п.).
В следующем разделе последнее
будет кратко пояснено.
4. Некоторые физические свойства
металлов в свете зонной теории.
Электропроводность металлов связа-
связана, как известно, с тем, что внешнее
электрическое поле, приложенное к
металлическому проводнику, вызыва-
вызывает в нем электрический ток, который
есть не что иное, как направленный
поток электронов. Электрическое поле
ускоряет электроны, т. е. увеличива-
увеличивает их энергию, которая затем в ре-
результате рассеяния электронов на
тепловых колебаниях атомных осто-
остовов теряется электронами и идет на
разогревание кристалла (джоулево
тепло). На языке зонной теории этот
процесс означает, что под действием
поля электроны стремятся перебрать-
перебраться в соседние квантовые ячейки, соот-
соответствующие большей энергии. Но
для такого перехода необходимо, что-
чтобы в соседних ячейках были бы неза-
незанятые места (как то требует прин-
принцип Паули). Такие ячейки имеются
только у поверхности Ферми. Поэто-
Поэтому только электроны, расположенные
около поверхности Ферми, участвуют
в процессе электропроводности. Оцен-
Оценка числа таких электронов показала,
что в металлах оно очень велико.
Аналогично и теплопроводность
связана с тем, что электроны вблизи
поверхности Ферми способны при-
приобретать добавочную тепловую энер-
энергию и переносить ее в определенном
направлении.
Такая трактовка физических
свойств металлов помогла понять
также физические свойства кристал-
кристаллов других типов.
У изоляторов все зоны подразде-
подразделены или на полностью занятые или
на полностью пустые, причем ин-
интервал области запрещенных значе-
значений энергии между двумя соседни-
соседними зонами настолько велик, что
электропроводность у таких кристал-
кристаллов отсутствует, а теплопроводность
осуществляется совершенно другим
механизмом.
У полупроводников ситуация иная.
При абсолютном нуле энергии в по-
полупроводнике, как и в изоляторе, все
зоны или полностью заполнены или
полностью пустые. Последняя запол-
заполненная зона называется валентной
зоной. Однако запрещенный интер-
интервал энергии между полосами энер-
203
гии этой зоны и последующей (пу-
(пустой) зонами невелик. Поэтому уже
при комнатных температурах неко-
некоторое количество электронов за счет
теплового возбуждения перебрасы-
перебрасывается в квантовые ячейки пустой
зоны, где эти электроны ведут себя
совершенно точно так же, как элект-
электроны проводимости в металлах (где
понятие валентной зоны и зоны про-
проводимости совпадают). Поэтому эта
первая пустая зона в полупроводни-
полупроводниках носит название зоны проводи-
проводимости. Механизм электропроводно-
электропроводности и электронной теплопроводности
здесь такой же, как в металлах; глав-
главное же различие заключается в том,
что число таких электронов прово-
проводимости мало и эффект от них неве-
невелик; кроме того, эффект должен по
определенной закономерности уве-
увеличиваться с температурой, связан-
связанной с увеличенным перебросом. По-
Поэтому температурная зависимость
этих свойств у металлов и полупро-
полупроводников резко различна.
Имеется некоторый критерий, свя-
связанный с зонной теорией, для клас-
классификации кристаллов. У металлов
зоны не полностью заполнены п ча-
часто наблюдается перекрытие верх-
верхних полос. У остальных крисгал.ч-.-.в
при абсолютном нуле температура
(Т = 0) зоны или заполнены, или
пустые, причем если расстояние
между полосой энергии, псо^едпеГ:
заполненной зоны и полосой зноргии
первой пустой зоны много больше
кТ (где к — постоянная Больцмана),
то кристалл есть изолятор, в про-
противном случае кристалл обладает
свойствами полупроводника. Для
ориентировки укажем, что при ком-
комнатной температуре величина кТ
примерно равна 0,025 эв.
Следует оговориться, что здесь мы
коснулись свойств полупроводников
только с точки зрения их классифи-
классификации по зонной теории. Вообще же
говоря особенности свойств полу-
полупроводников гораздо сложнее. Под-
Подробнее об этом будет изложено в
гл. XII § 8, разделе 4.
5. Энергия решетки металличе-
металлического кристалла. Энергия решетки
металлического кристалла при абсо-
абсолютном нуле температуры слагается
из кинетической энергии электронов
проводимости и потенциальных энер-
энергий взаимодействия атомных осто-
остовов с электронами проводимости
(притяжение), энергии взаимного
электростатического отталкивания
атомных остовов, других видов взаи-
взаимодействия между атомными осто-
остовами и энергии взаимного отталки-
отталкивания электронов. Теори:! энергии
решетки металлического кристалла
еще не достигла такого состояния,
как теория ионных и молекулярных
кристаллов. Мы до сих пор не имеем
формулы, позволяющей производить
аналогичные вычисления энергии для
металлов. Поэтому ограничимся рас-
рассмотрением одного частичного слу-
случая — структуры металлического нат-
натрия, рассмотренного Вигнером и
Зейтцем A934 г.).
Все пространство в структуре
натрия (объемноцентрпрованная
кубическая) можно разбить на
многогранники — параллелоэдры.
В каждом параллелоэдре будет на-
находиться один ион натрия и в сред-
среднем один электрон проводимости.
Последнее обстоятельство связано с
тем, что электроны, отталкиваясь
друг от друга, будут стремиться рав-
равномерно распределиться по зеему
объему кристалла. Потенциальная
энергия решетки будет тогда состав-
составляться из энергии взаимодействия
катиона с электроном в одном парал-
параллелоэдре и энергии взаимодействия
параллелоэдров друг с другом. Пос-
Последней составляющей приближенно
можно пренебречь, так как каждый
параллелоэдр будет электростатиче-
электростатически нейтральным, а взаимодействие
нейтральных многогранников не
может быть очень большим.
Упомянутые выше авторы приме-
применили для случая натрия волновое
уравнение Шредингера для расчета
энергии взаимодействия атомного
остова с электроном в одном парал-
204
- оч -
-0,5
iOxs/ra
¦S'10
Рис. 238. Энергия решетки металлическо-
металлического натрия (по Вигнеру и Зейтцу)
лелоэдре. Их результаты показаны
графически (кривая 1) на рис. 238.
По оси ординат отложена энергия
Е в единицах Ридберга A рид-
берг = 13,25 эв), а по оси абсцисс
расстояние между атомами в атом-
атомных единицах rjro, где г, — радиус
шара, равновеликого параллелоэдру,
а го — радиус первой боровской орби-
орбиты атома водорода.
Кинетическая энергия электронов
в тех же координатах показана кри-
кривой 2. Суммарная кривая 3 дает
результат вычисления энергии ре-
решетки. Справедливость теории про-
проверяется по расположению минимума
на кривой 3. Этот минимум дейст-
действительно отвечает кратчайшему меж-
межатомному расстоянию в кристалличе-
кристаллической структуре металлического нат-
натрия.
6. Электронная эмиссия погранич-
пограничного слоя металла. Электроны прово-
проводимости почти свободно движутся
внутри металла. Их энергию можно
трактовать как кинетическую энер-
энергию. Однако эти результаты в теории
получаются только для электронов в
объеме, когда не учитывается гранич-
граничный эффект. Как показал И. Е. Тамм
A931 г.), в пограничном слое необ-
необходимо учитывать потенциальную
энергию притяжения. В результате
в пограничном слое электрон уже не
оказывается свободным.
Грубо наглядно это можно объяс-
объяснить следующим образом. В погра-
пограничном слое притяжение атомными
остовами (положительными ионами)
изнутри металла не уравновешива-
уравновешивается притяжением поверхностных
ионов. Поэтому электрон уже огра-
ограничен в своем движении (не свобо-
свободен), он «не выпускается» из кри-
кристалла наружу. Однако потенциаль-
потенциальная энергия такой связи имеет хотя
и большую, но все же конечную ве-
величину. Поэтому, если передать
электронам проводимости добавоч-
добавочную энергию, то они смогут выйти
из кристалла.
С точки зрения зонной теории,
электронам необходимо передать
энергию, поднимающую электрон с
уровня энергии Ферми в область не-
непрерывного спектра энергии (выше
области полосового спектра энергии,
характерного для металла). Такая
необходимая энергия называется ра-
работой выхода, а сам процесс носит
название электронной эмиссии. Это
явление широко применяется в сов-
современной технике (электронные
приборы). .
Дополнительная энергия может
быть передана электронам различны-
различными способами. При бомбардировке
поверхности металлов пучком быст-
быстро летящих электронов бомбардиру-
бомбардирующие электроны передают свою
энергию электронам металла и неко-
некоторые из последних в состоянии по-
покинуть металл. Такой же результат
получается за счет поглощения све-
световой энергии. Явление вырывания
электронов при облучении называ-
называется фотоэмиссией или внешним фо-
фотоэффектом.
Наиболее распространенным в
технике типом эмиссии является
термоэмиссия, когда энергия, иду-
идущая на возбуждение электронов, по-
получается за счет повышения темпе-
температуры самого металла. Однако для
осуществления термоэмиссии необхо-
необходима температура выше 2000°С, т. е.
температура, при которой не пла-
плавятся только тугоплавкие металлы.
Поэтому в термоэлектронных прибо-
приборах использовались только такие
металлы. Лишь в самые последние
205
годы температуру термоэмиссии уда-
удалось значительно понизить за счет
специальных покрытий поверхности
металлов, при которых уменьшается
работа выхода.
§ 7. Остаточная связь
1. Понятие остаточной (Ван-дер-
Ваальсовой) связи. При выводе за-
законов для газов обычно делается до-
допущение, что молекулы взаимодейст-
взаимодействуют друг с другом только при столк-
столкновениях. Такое допущение есть
идеализация, и сами законы носят
название законов идеальных газов.
К реальным газам эти законы приме-
применимы лишь в известном приближении
и только в определенных условиях
(не очень высокие давления и плот-
плотности газов). Но даже в этих услови-
условиях можно наблюдать отклонения по-
поведения реальных газов от идеаль-
идеальных. Ван-дер-Ваальс A873 г.) иссле-
исследовал эти отклонения и объяснил их
тем, что в теории реальных газов сле-
следует учитывать взаимодействия меж-
между молекулами не только посредством
столкновений. Силы такого дополни-
дополнительного взаимодействия названы
Ван-дер-Ваальсовыми, или остаточ-
остаточными силами.
В молекулярных кристаллах так-
также существуют силы такой природы.
Их называют остаточной связью.
Среди простых (одноатомных) кри-
кристаллических веществ остаточная
связь обнаружена только у инертных
газов в твердой (кристаллической)
фазе. Число же кристаллических
структур с остаточной связью у мо-
молекулярных соединений чрезвычай-
чрезвычайно велико. В частности, кристаллп-
ми с такой связью являются Нг, Ог,
N2 и молекулярные кристаллы боль-
большинства органических соединений.
Именно остаточная связь, действую-
действующая между молекулами, обеспечива-
обеспечивает кристаллическую структуру таких
соединений. Все инертные газы и
многие молекулярные вещества с
простыми симметричными молекула-
206
Ми кристаллизуются в структуры с
плотнейшими упаковками. Этот факт
указывает, что остаточные связи не-
насыщаемы и пенаправленны. Поэто-
Поэтому к таким кристаллам применима
теория плотнейших упаковок. Эта
теория применима также (но с неко-
некоторыми усложнениями) и к молеку-
молекулярным кристаллам из более слож-
сложных и несимметричных молекул.
В молекулярных кристаллах мож-
можно выделить два типа сил: внутримо-
внутримолекулярные силы и силы между
молекулами. Последними силами как
раз являются остаточные связи. Как
правило, эти силы намного слабее,
чем внутримолекулярные, но в то же
время именно ими определяются
многие важные физические свойства
таких кристаллов (температура
плавления, твердость, тепловое рас-
расширение и др). Низкие температуры
плавления молекулярных кристал-
кристаллов, их малая твердость и значитель-
значительное тепловое расширение свидетель-
свидетельствуют о чрезвычайной слабости
Ван-дер-Ваальсовых сил по сравне-
сравнению с силами других типов связи,
О сравнительной величине остаточ-
остаточной связи по сравнению с внутримо-
внутримолекулярной можно судить по теплота
сублимации молекулярного кристал-
кристалла и энергии диссоциации соответст-
соответствующих молекул (следует, однако,
отметить, что внутримолекулярные
силы обычно исследуются у вещест-
вещества в жидкой или газообразной фазе,
но это мало влияет на оценочный
результат). Так, у молекулярного во-
водорода в твердой фазе теплота субли-
сублимации равна 0,5 ккал/моль, а энер-
энергия диссоциации молекулы водорода
составляет около 100 ккал/молъ, т. е.
намного больше.
2. Энергия решетки кристалла с
Ван-дер-Ваальсовой связью. В тео-
теории реальных газов Ван-дер-Ваальса
выражение для энергии взаимодейст-
взаимодействия содержит два члена. Один из них
соответствует притяжению между
молекулами, а другой отталкиванию.
В случае кристаллов с Ван-дер-Ва-
Ван-дер-Ваальсовой связью вклад от энергии
отталкивания в энергию решетки в
целом лучше всего представлять так
же, как и у ионных кристаллов, т. е.
в виде:
где г — расстояние между центрами
молекул. С/отт определяется по ко-
коэффициентам сжимаемости кри-
кристаллов. Истолкование члена притя-
притяжения было получено постепенно. В
настоящее время установлено, что в
выражении для энергии решетки
член, соответствующий притяжению,
состоит из трех слагаемых:
Us.
Все три слагаемых связаны с диполь-
ным взаимодействием, но происхож-
происхождение диполей различно.
Если молекулы полярны, т. е. об-
обладают собственным дипольным
электрическим моментом, то между
диполями разных молекул возникает
взаимодействие притяжения. Рас-
Расстояние между молекулами много
больше, чем размер диполя. Поэто-
Поэтому энергию взаимодействия между
двумя диполями определяют в моде-
модели точечных диполей, в которой эта
энергия обратно пропорциональна
шестой степени расстояния между
диполями. Коэффициент пропорцио-
пропорциональности зависит от взаимной
ориентации молекул. Поэтому это
взаимодействие названо ориентаци-
онным взаимодействием. Соответст-
Соответствующий член в энергии решетки
имеет вид:
Величина А положительна и зависит
от величины дипольного момента мо-
молекул, а также от температуры. Ве-
личипа А тем больше, чем больше
дипольный момент и чем меньше
температура. Последнее связано с
тем, что из-за теплового движения
(или колебания) молекул ориента-
ориентация диполей относительно друг друга
все время меняется, и в целом эф-
эффект от этого слабеет. Чем выше тем-
температура, тем больше эффект тепло-
теплового движения.
Теорию ориентационного взаимо-
взаимодействия развил ученик Ван-дер-
Ваальса Кеезом. Кроме врожденных
диполей, у молекул может возник-
возникнуть наведенный дипольный момент
под воздействием окружающих не-
неполярных молекул. Наведенные ди-
польные моменты взаимодействуют
между собой совершенно точно, так
же, как и врожденные диполи. Энер-
Энергия такого взаимодействия дает в
энергию решетки вклад, равный
где постоянная В так же, как и по-
постоянная А, должна зависеть от
дипольного момента молекул и, кро-
кроме того, от поляризуемости молекул,
т. е. от способности молекул созда-
создавать из себя наведенный диполь.
Величина В, очевидно, меньше вели-
величины А.
Взаимодействие такого типа на-
называется индукционным взаимодей-
взаимодействием. Теорию этого эффекта раз-
развил Дебай A920 г.).
Наконец, даже у нейтрального
атома, если он входит в систему,
т. е. образует с другими атомами
газ, жидкость или твердое тело, слу-
случайным образом может появиться
дипольный момент в результате не-
несовпадения «центра тяжести» обла-
облака размазанных зарядов его оболо-
чечных электронов и заряда ядра.
Такого же типа дипольный момент
может возникнуть и у молекул. Этот
момент имеет флюктуационную при-
природу и существует очень кратковре-
кратковременно. Но за это время он может
вызвать поляризацию соседней моле-
молекулы, и между новым наведенным
диполем и флюктуационно возник-
возникшим диполем возникает взаимодей-
взаимодействие. Это взаимодействие опять бу-
будет зависеть от расстояния между мо-
молекулами как 1/г6. Силы такого взаи-
взаимодействия называют дисперсион-
дисперсионными.
Теорию этого эффекта развил
Лондон. Вклад в энергию решетки
207
от дисперсионных сип можно пред-
представить в виде;
U3 = — С/г*,
где постоянная С положительна и
зависит от поляризуемости молекул
и от собственной частоты колебаний
молекул.
В полярных молекулах наиболее
сильна ориентационная связь, в не-
неполярных — дисперсионная. Для
иллюстрации в табл. 26 приведены
энергии трех типов связей (отне-
(отнесенные к единице длины связей)
для ряда веществ.
При подсчете энергии решетки
молекулярных кристаллов по фор-
формуле
выяснилось, что величина С/г при-
примерно одинакова по порядку вели-
величины с энергией С/Отт и обратна по-
последней по знаку. Поэтому С/г и
С/отт взаимно компенсируют друг
друга и для ориентировочных оце-
оценок энергии решеток можно пользо-
пользоваться формулой
Результаты такого расчета для ряда
веществ приведены в табл. 27.
В этой же таблице указаны опыт-
опытные значения, полученные из опре-
определения теплоты сублимации. Срав-
Сравнение результатов с данными для
энергии решеток ионных кристаллов
(см. табл. 23 на стр. 176) еще раз
показывает, насколько слабы силы
связи в молекулярных кристаллах.
ТАБЛИЦА 26
Три составляющих энергии Ван-дер-Ваальсовой связи (орг-10~со)
Соединение
HCl
НВг
HJ
Н2О
NHs
СО
Дипольный
момент
1,03
0,78
0,38
1,84
1,50
0,12
Поляризуемость
а-Ю4
2,63
3,58
5,4
1,48
2,21
1,99
Ориентационнтлй
эффект
18,6
6,2
0,35
190,0
84,0
0,0034
Наведенный
эффект
5,4
4,05
1,68
10,0
10,0
0,057
Дисперсион-
Дисперсионный эффект
105,0
176,0
383,0
47,0
93,0
67,5
ТАБЛИЦА 27
Энергия решеток некоторых молекулярных структур {ккал]моль)
Вещество
Ne
Аг
Кг
С12
о2
Рассчитанная
энергия
решетки
0,47
2,08
3,0
7,0
1,46
Наблюденная
теплота
сублимации
0,59
2,03
2,80
7,43
2,06
Вещество
N2
HCl
НВг
СН4
Рассчитанная
энергия
решетки
1,64
4,0
4,5
2,42
Наблюденная
теплота
сублимации
1,86
5,05
5,52
2,70
208
§ 8. Промежуточные типы связей
1. Понятие о промежуточных типах
связей. В предыдущих параграфах
были рассмотрены основные типы
химической связи. Однако в реальных
веществах они редко встречаются в
чистом виде. Как правило, у боль-
большинства химических соединений тип
связи носит промежуточный (пере-
(переходный) характер и только в боль-
большей или меньшей степени прибли-
приближается к одному из идеальных типов.
Эту мысль можно графически иллю-
иллюстрировать треугольником (рис.
239, а), вершины которого символи-
символизируют три типа химической связи:
ионную, ковалентную и металличе-
металлическую. Реальные же вещества по типу
химической связи в них будут распо-
располагаться на ребрах этого треугольни-
треугольника или даже на его плоскости. Если
бы имелись методы точного определе-
определения доли участия в реальной связи
тех или иных идеальных типов свя-
связей, то каждое вещество можно было
бы изобразить на диаграмме рис.
239, а в виде точки. Но к сожалению,
таких методов в настоящее время
еще нет. В отдельных случаях, в осо-
особенности когда речь идет о сравнении
двух веществ, качественно все же
можно сказать, у которого из них
связь будет, например, более ионной
и менее ковалентной.
Промежуточный тип связи между
ионной и ковалентной можно рас-
рассмотреть на примере галоидоводород-
ных кислот. Кислоты НХ можно схе-
схематически изобразить как ковалент-
ные соединения Н:Х: или же как
м и
Рис. 239. Схемы, иллюстрирующие пере-
переходные случаи между ионными, ковалент-
ными и металлическими связями (а) и
между этими связями и ван-дер-ваальсов-
ским типом химических связей (б)
ионные Н+:Х:~. Очевидно, что при-
приближение к одному из этих двух
крайних случаев у разных кислот бу-
будет различным. Поскольку поляриза-
поляризация ионов не бывает равна нулю, то
нельзя говорить о чисто ионном ха-
характере связи. Так, HF будет более
ионной, чем НС1, а последняя более
ионной, чем НВг; наиболее гомеопо-
лярная связь уШ.
Существуют взаимные переходы
между ковалентной и металлической
связями, с одной стороны, и между
ионной и металлической,— с другой.
Промежуточный характер между
ионной и Ван-дер-Ваальсовой связя-
связями типичен для водородной связи
(см. ниже).
Некоторые авторы (например
Р. Эванс) считают возможным поло-
положить тип химической связи в основу
кристаллохимической классификации
веществ. Несостоятельность подоб-
подобной классификации вытекает из ска-
сказанного нами выше о промежуточ-
промежуточных типах связи в реальных струк-
структурах. Объективного критерия для
выбора из двух или трех типов связи
одного, определяющего группу, в ко-
которую следует отнести соединение,
нет, а, следовательно, отнесение таких
соединений в ту или иную группу не
является объективным. Надо также
иметь в виду, что мы до сих пор не
имеем простых, хорошо разработан-
разработанных методов, позволяющих надежно
определять тип химической связи.
Это еще больше затрудняет приме-
применение классификации, основываю-
основывающейся на типах химической связи.
Однако это обстоятельство не долж-
должно умалять той огромной роли, кото-
которую играет теория химической свя-
связи в кристаллохимии.
Четвертый тип связи — ван-дер-ва-
альсовский — в графическом изобра-
изображении помещается в вершине тетра-
тетраэдра, основанием которого служит
только что описанный треугольник
(рис. 239, б). Следует отметить, что
при любом типе связи всегда сущест-
существует дополнительная остаточная
14 Кристаллохимия
209
связь. Однако ее в этих случаях мож-
можно не учитывать, так как энергия
остаточной связи значительно мень-
меньше, чем энегрия остальных типов
связи.
2. Водородная связь. Если атом во-
водорода осуществляет притяжепие
двух электроотрицательных атомов,
то такая связь носит название водо-
водородной. Водородная связь не является
пятым типом химической связи. Для
описания ее могут быть использованы
уже известные нам понятия и терми-
термины. Однако некоторые особенности
этой связи заставляют нас излагать
ее отдельно.
Водородная связь первоначально
была открыта в жидкостях и газах.
Было установлено, что некоторые ор-
органические кислоты, спирты и дру-
другие соединения, имеющие активные
группы, содержащие водород, очень
склонны давать стабильные димеры.
Так, например, димеры муравьиной
или уксусной кислот являются устой-
устойчивыми даже выше температуры ки-
кипения. Для объяснения зтого явления
была предложена следующая схема:
ОН ... О
/ V,
R-C
С—R
О НО
Оба атома водорода осуществляют
дополнительную связь с атомом кис-
кислорода второй молекулы, в результате
чего и образуется димер. Поскольку
эти димеры сохраняются в парах и в
расплаве, то тем более они должны
сохраняться в кристалле. К сожале-
сожалению, с помощью рентгеноструктурно-
го анализа мы не можем непосред-
непосредственно обнаружить атом водорода
ввиду ничтожно малой рассеивающей
способности последнего. Поэтому о
водородной связи мы судим по ненор-
ненормально малому межмолекулярному
расстоянию между двумя электроот-
электроотрицательными атомами, если при
этом число таких ненормально малых
расстояний совпадает с числом соот-
соответствующих атомов водорода в хи-
химической формуле исследуемого ве-
-100
- HF
-200- H,E ^
_1 L
i 2 3 i 5
Периоды
Рис. 240. Зависимость температур плавле-
плавления гидридов от состава
щества. Вторым признаком водород-
водородной связи является угол Н—О—Нг
равный 109°.
Водородная связь может сущест-
существенно влиять на физические свойства
соединений. На рис. 240 графически
изображена зависимость температур
плавления некоторых гидридов от
состава. Из рисунка видно, что толь-
только три соединения резко выпадают
из общей закономерности. Это соеди-
соединения водорода с наиболее электроот-
электроотрицательными элементами: кислоро-
кислородом, азотом и фтором. Объяснение
этому следует искать в образовании
водородных связей.
Энергия водородной связи варьиру-
варьирует в достаточно широких преде-
пределах — от 5 до 10 ккал/молъ, соответ-
соответственно варьируют и межатомные
расстояния. Так, расстояние между
двумя атомами кислорода, связанны-
связанными водородной связью, может ме-
меняться от 2,54 А в КН2РО4 до
2,76 А в структуре льда. Чем короче
межатомное расстояние, тем сильнее-
связь. Энергия водородной связи в 10
раз больше энергии Ван-дер-Ваальсо-
вой связи и в 10 раз меньше кова-
лентной или ионной связи. Напомним,
что теплота сублимации кристалли-
кристаллического водорода равна 0,5 клал/
/моль, а энергия диссоциации моле-
молекулы водорода —102, 6 ккал/молъ.
Связь атома водорода, присоеди-
присоединившегося к атому кислорода с обра-
образованием гидроксила, можно рассмат-
210
ривать как ковалентную :О:Н или
же как ионную :О:2~Н+- Атом водо-
водорода, как было сказано в предыдущем
параграфе, присоединяется к атому
кислорода так, что угол R — О — Н
составляет 109°. Однако этот признак
недостаточен, чтобы трактовать эту
связь как ковалентную, так как ос-
основное свойство — насыщаемость ко-
валентной связи — не позволяет при-
присоединить к атому водорода другой
атом кислорода, а при водородной
связи это всегда имеет место. Ион-
Ионный тип связи, напротив, хорошо со-
согласуется с характером водородной
связи. Атом водорода теряет свой
единственный электрон и, превратив-
превратившись в протон, в стремлении окру-
окружить себя возможно большим числом
соседних ионов противоположного
знака, полностью экранируется уже
двумя атомами (рис. 241), так как
размер его, по сравнению с разме-
размерами всех других атомов, ничтожно
мал. В этом случае расстояния от
протона до центров тяжести кисло-
кислородных атомов обычно считаются
одинаковыми, например, в структуре
КН2РО4 они равны, как было сказано
выше, 1,27 А.
Однако водородные связи не всегда
имеют одинаковую длину. Так, в
структуре льда расстояние между
двумя атомами кислорода равно
2,76 А. В таких случаях водородную
связь скорее следует считать остаточ-
остаточной связью. В самом деле, в изолиро-
изолированной молекуле воды расстояние от
ядра кислорода до протона, как это
следует из изучения инфракрасных
спектров, равно 0,9 А, т. е. оно почти
на 0,4 А короче самого короткого рас-
расстояния в соединениях с водородной
связью. В этом случае протон являет-
является положительным полюсом одного
диполя, а атом кислорода другой мо-
молекулы — отрицательным полюсом
второго диполя. Притяжение обус-
обусловливает димеризацию или же
взаимную ориентировку молекул в
кристаллах. Поскольку дипольное
взаимодействие значительно слабее
ионного, то расстояние между двумя
атомами кислорода будет больше, и
нет оснований считать, что протон
будет находиться как раз в середине
между двумя атомами кислорода. Та-
Рис. 241. Экранирование протона двумя
атомами кислорода
ким образом, эта связь будет несим-
несимметричной. В таких случаях, напри-
например, в структуре льда, протон может
и не быть связанным с одним каким-
либо кислородным атомом, а «пере-
«перескакивать» от одного к другому. Не-
Несимметричные, более слабые водород-
водородные связи некоторые авторы предла-
предлагают называть гидроксильными
связями.
В обоих крайних типах водородной
связи протон имеет координационное
число 2, поэтому всякая водородная
связь является направленной связью,
чем формально напоминает ковалент-
ные связи.
Рассмотрим структуру кристалли-
кристаллического льда в качестве примера сое-
соединения с водородными связями (рис.
242). Ее можно представить себе ана-
аналогичной структуре вюртцита, в
которой все места атомов цинка и
Рис. 242. Структура льда
14*
211
серы заняты атомами кислорода,
удерживаемыми друг относительно
друга четырьмя водородными связя-
связями, обусловливающими тетраэдриче-
ское окружение. Каждая молекула
воды имеет, следовательно, «тетраэд-
рическую форму». При этом две вер-
вершины тетраэдра будут нести положи-
положительные заряды и две — отрицатель-
отрицательные. Федоровская группа симмет-
симметрии — Рб/ттс.
Такое расположение атомов кисло-
кислорода обусловливает наличие в струк-
структуре широких каналов. Пустоты в
этих каналах таковы, что в них могут
размещаться шары, диаметр которых
равен диаметру шаров, из которых
построены «стенки» канала. Начало
плавления льда есть процесс, при ко-
котором молекулы воды «проваливают-
«проваливаются» в каналы. Этим обстоятельством
объясняется всем известная анома-
аномалия удельного веса льда и воды.
3. Ионно-ковалентные связи. Элек-
Электроотрицательность. Если атомы А и
В образуют молекулу с чисто ион-
ионными связями (А+В~), то предпола-
предполагается, что атом А потерял электрон,
а атом В приобрел его. При этом
предполагается, что оба иона пред-
представляют собой несжимаемые шары,
так что сумма их радиусов равна меж-
межатомному расстоянию в молекуле.
Однако давно стало ясным, что такие
допущения неоправданны. Недоста-
Недостаток чисто ионных представлений пы-
пытался исправить Фаянс путем введе-
введения понятия поляризации ионов (см.
гл. X, §8), в результате чего в харак-
характере связи начинает проявляться «ко-
валентность». Однако такая попытка
подойти к пониманию ионно-кова-
лентных связей, если так можно вы-
выразиться, «со стороны ионных свя-
связей» не получила в дальнейшем боль-
большого развития. Гораздо более эффек-
эффективным оказался подход «со стороны
ковалентных связей».
Ковалентная связь может быть
вполне симметричной только в слу-
случае, когда оба атома, образующие
связь, вполне одинаковы (в молеку-
молекулах типа Аг). В молекуле же из не-
212
одинаковых атомов (типа АВ) долж-
должна наблюдаться асимметрия связи.
«Центр тяжести связи» не будет со-
совпадать с серединой расстояний меж-
между атомами, т. е. в связи будет наблю-
наблюдаться некоторая полярность (напом-
(напомним, что связь обусловлена перекры-
перекрытием облаков распределения зарядов
электронов, образующих связь).
Относительную величину этого сме-
смещения, выраженную в процентах,
некоторые авторы называют степенью
ионности связи.
Приведенные выше рассуждения
убедительно свидетельствуют о том,
что реальные связи в молекулах из
неодинаковых атомов обязательно
должны быть промежуточными меж-
между ионными и ковалентными.
Для количественной характеристи-
характеристики степени ионности связи обычно
вводится понятие электроотрицатель-
электроотрицательности (Л. Полинг, 1932 г.). Под этим
термином понимается относительная
способность атома в молекуле к при-
притяжению валентных электронов. Из
относительности этого понятия опре-
определение его абсолютной величины
очень условно. Поэтому для количе-
количественных определений исходным яв-
является определение разности энергии
связи двух разных рассматриваемых
атомов и полусуммы энергий связей
между соответствующими одинако-
одинаковыми атомами, образующими чисто
ковалентную связь:
АЕ = Е (А — В) — -2 [Е (А—А) +Е (В—В)]_
Величина АЕ очевидно равна те-
теплоте реакции.
= 2АВ.
Поэтому для вычисления АЕ мож-
можно пользоваться непосредственно теп-
лотами реакций, а не энергиями свя-
связей, которые надежно определены
только для очень небольшого числа
соединений.
Из сравнения величины АЕ для
целого ряда соединений с одним и тем
же атомом судят об электроотрица-
электроотрицательности разных атомов относитель-
ТАБЛИЦА 28
Значения электроотрицательности
Перио-
Периоды
1
4
5
6
1а
и
0 95
Na
0,9
К
0,8
Rb
0,8
Cs
0,75
Fr
«у
Па
Be
V
I,2
Ca
',°
Sr
1,0
Ba
0,9
Ra
0,9
111а
Sc
1,3
Y
1,2
La ,
1,2*
Aci +
У It
1
2
3
4
Лантаноипы т
1
Актиноиды J J
lVa
Ti
* 0,9
* 1,1
* 1,3
* I,6
Zr
1,5
Hf
1,4
Ce
1,2
Th
V 1,0
4' 1,4
Vd
- V
3* 1,4
4* 1,7
5* 1,9
Nb
1,7
Ta
3* 1,3
5* 1,7
Pr
1,2
Pa
3* 1,3
5' 1,7
Via
' Cr
1* 1,0
2M,4
3*1,6
4* 2,2
6* 2,4
Mo
4* 1,6
6* 2,1
W
4* 1,6
6* 2,0
Nd
1,3
и
4* 1,4
6* 1,9
Подгруппь
Vila
Mn
2* 1,4
3* 1,5
4* 2,1
7* 2,5
Tc
5* 1,9
7* 2,3
Re
5* 1,8
6* 2,1
7* 2,2
Pm
1,3
Np
4* 1,4
6* 1,9
Villa
Fe
2* 1,7
3* 1,8
Ru
2,0
Os
2,1
Sm
1,3
Pu
1 3
Co
1,7
Rh
4
Ir
Eu
1,2
Am
1,1
Ni
i,8
Pd
Pt
2,2
Gd
1,3
Cm
1,4
Ift
Cu
1*1,8
2* 2,0
Af?
i,9
Au
2,3
Tb
1,3
Bk
1,3
lift
7,n
¦1,6
Cd
>,7
Hg
l,8
Dy
l,»
a
1,3
III*
в
2,0
Al
V
Ga
1,6
In
1,7
Tl
14,4
3* 1,9
Ho
1,3
F.s
IV ft
С
2,6
Si
b9
Ge
2,0
Sn
2* 1,7
4* 1,9
Pb
24,6
4* 1,8
Er
1,3
Fm
Vb
N
3,0
P
2,1
¦ As
2,0
Sb
3* 1,8
5* 2,1
Bi
1,8
Tu
'l3
Md
VI*
0
3,5
s
2,6
Sr
Те
2,1
Po
2,0
Yb
1,2
Nn
viift
H
2,15
F
3,9
CI
3,0
Br
2,9
J
2,6
At
2,2
Lu,
Lr
но эталонного атома, электроотрица-
электроотрицательность которого принята за еди-
единицу.
Этим методом Полинг, а затем и
другие исследователи определили
электроотрицательности большинства
атомов периодической системы. Ре-
Результаты современных оценок элект-
роотрицательностей приведены в
табл. 28 (таблица была составлена в
1956 г. Горди и Тамасом и обновле-
обновлена в 1962 г. С. С. Бацановым).
Электроотрицательности могут
быть определены не только из тер-
Рис. 243. Кривая зависимости процента
ионности химической связи от разности
электроотрицательностей
мохимических данных, но и из дан-
данных спектрального анализа, измере-
измерений потенциалов ионизации и срод-
сродства к электрону, а также рядом
других способов. Во всех случаях
получались достаточно согласованные
результаты *.
Ценность понятия электроотрица-
электроотрицательности заключается в том, что
между этой величиной и степенью
ионной связи имеется определенная
зависимость. На рис. 243 показана
такая зависимость, найденная Полин-
гом и характерная для соединений
водорода с галогенами (типа НС1).
Как видно из этого рисунка, зависи-
зависимость близка к линейной. Только в
предельных случаях близости к чисто
* Подробно о методике и результатах экс-
экспериментов можно прочесть в книге
С. С. Бацанова «Электроотрицательность
элементов и химическая связь» (Ново-
(Новосибирск, 1962 г) и в сборнике материа-
материалов по дискуссии, посвященной поня-
понятию электроотрицательности, организо-
организованной ВХО им. Менделеева (Новоси-
(Новосибирск, 1965 г. ).
213
ионной или чисто ковалентной связям
наблюдается отклонение от линейно-
линейного хода.
В заключение как пример прак-
практической ценности понятия электро-
электроотрицательности упомянем работу
А. С. Поваренных A958 г.), в которой
установлена прямая зависимость ме-
между величиной электроотрицатель-
электроотрицательности кристаллов и их показателем
преломления. Тем самым доказано
влияние характера химической связи
(степени ионности) на оптические
свойства кристаллов. Результаты при-
приведены в табл. 29.
4. Ковалентно-металлические свя-
связи. Полупроводники. В § 6 мы уже
упоминали, что наряду с металлами,
обладающими большой электропро-
электропроводностью, и изоляторами, которые
практически ток не проводят, суще-
существует широкий класс кристалличе-
кристаллических веществ, обладающих слабой
(по сравнению с металлами) электро-
электропроводностью, причем температурная
зависимость электропроводности та-
таких кристаллов резко отлична от тем-
температурной зависимости электропро-
электропроводности металлов (с повышением
температуры электропроводность это-
этого класса веществ увеличивается, а у
металлов она уменьшается). Такие
вещества названы полупроводни-
полупроводниками.
Как уже отмечалось, свойства полу-
полупроводников (в том числе и характер
электропроводности) могут быть по-
поняты на основе зонной теории.
Для напоминания на рис. 244 при-
приведена схема полосовой структуры
спектра энергии электрона в металлах
и изоляторах и заполнение этого спек-
спектра электронами. Для изоляторов ха-
характерно, что полосы или полностью
заняты, или полностью пустые.
У полупроводников спектр энергии
аналогичный изоляторам. Только ме-
между валентной полосой и полосой про-
проводимости интервал энергии невелик
(порядка 1 эв; например, у типичных
полупроводников, германия и крем-
кремния, он составляет 0,7 и 1,15 эв соот-
соответственно).
При абсолютном нуле температуры
заполнение полос (зон) электронами
в полупроводниках точно такое же,
как и в изоляторах: часть зон полно-
полностью занята, а часть — полностью
ТАБЛИЦА 29
Зависимость показателя преломления кристаллов от состояния ионно-ковалентной
связи
Соединение
КВг
RbCl
KJ
CsCl
LiBr
RbF
LiJ
CsF
MgS
CaO
CaTe
BaS
S атомных
весов
119,01
120,94
166,02
168,37
86,86
104,48
133,86
151,91
56,39
56,08
167,69
169,43
Удельный
вес
2,75
2,80
3,13
3,18
3,46
3,89
4,06
4,64
2,65
3,35
4,34
4,31
Межатомное
расстояние
3,29
3,27
3,53
3,55
2,74
2,82
3,00
3,00
2,60
2,40
3,17
3,19
% ковалентной связи
36
28
48
26
43
3
54
2
65
19
75
24
Д = 8
Д = 22
Д = 40
Д =52
Д = 46
А =24
Показатель
преломления
1,559
1,494
1,667
1,534
1,784
1,396
1,955
1,448
2,271
1,838
2,510
2,155
Д =0,065
Д =0,133
Д =0,388
Д =0,507
Д =0,433
Д =0,355
214
Уровень
Ш/////У///Ш
, V '//////////У////
Рис. 244. Энергетический спектр метал-
металла (а) и изолятора (б)
пустая (последняя заполненная
полоса называется валентной полосой,
а первая пустая — полосой проводи-
проводимости). Однако с повышением темпе-
температуры ситуация резко изменяется.
Чтобы лучше пояснить изучаемое
явление, напомним, что в газах все
молекулы совершают тепловые хаоти-
хаотические движения с некоторой средней
скоростью, определяемой температу-
температурой. При этом все молекулы имеют
разную скорость около этой средней
скорости. Разброс значений скоростей
тем больше, чем выше температура.
Довольно аналогичная картина на-
наблюдается для электронного газа в
металлах, только здесь средняя ско-
скорость определяется не температурой,
а энергией уровня Ферми, но величи-
величина разброса по-прежнему связана с
температурой. Разброс здесь возмо-
возможен потому, что имеются незаполнен-
незаполненные уровни энергии в полосе, на кото-
которые могут попасть электроны при
«разрыхлении» их положений в поло-
полосе проводимости за счет теплового
возбуждения.
В полупроводниках при очень низ-
низких температурах (но все-таки от-
отличных от нуля) соответствующий
разброс в значениях скоростей (и
энергий) электронов должен быть не-
невелик, и он приходится на область
запрещенных значений энергии в ин-
интервале между полосами. Но при бо-
более высоких температурах (скажем,
комнатных) разброс достигает уже
заметной величины, и часть электро-
электронов благодаря этому перейдет из ва-
валентной зоны в зону проводимости.
Число таких электронов, вначале
незначительное, быстро увеличивает-
увеличивается с ростом температуры. Эти элект-
электроны в зоне проводимости ведут себя
так же, как и электроны проводимо-
проводимости металлов. Они обеспечивают ме-
металлическую электропроводность (но
менее значительную из^за малого об-
общего числа таких электронов); они
же, очевидно, должны приводить и
к появлению в веществе типично ме-
металлической связи, но уже как до-
дополнительной к основной связи, ко-
которая у полупроводников ковалент-
ная. Таким образом, при очень низ-
низких температурах в полупроводниках
химическая связь почти целиком ко-
валентная, а с повышением темпера-
температуры к этой связи все более приме-
примешивается металлическая связь.
Полупроводники обладают еще од-
одной важной особенностью. Когда из
валентной зоны в зону проводимости
переходит небольшое число электро-
электронов, в валентной зоне появляется та-
такое же небольшое число вакантных
мест («дырок»). Формально можно
представить себе, что в этих вакант-
вакантных местах находятся сразу две ча-
частицы: одна с зарядом электрона, а
вторая с равным ему, но положитель-
положительным зарядом (так что суммарный
заряд по-прежнему равен нулю).
Тогда валентная зона электронов
окажется полностью заполненной, но
над этим однородным «фоном» будет
находиться положительный заряд.
Теория и опыт показали, что энер-
энергию такой положительно заряженной
частицы можно представить форму-
формулой,
Е =
2т*
E1)
где к — волновой вектор; т* мож-
можно рассматривать как эффектив-
эффективную массу частицы с таким зарядом.
Таким образом, эти частицы описы-
215
ваются совершенно так же, как и
почти свободные электроны, и, сле-
следовательно, они также должны да-
давать вклад в электрические свойст-
свойства полупроводников. Электропровод-
Электропроводность, например, определяется как
электропроводность за счет электро-
электронов проводимости («электронная
электропроводность»), так и за счет
этих положительных частиц («ды-
(«дырочная электропроводность»). Вели-
Величина вклада от каждой части зависит
от сравнительной величины их эф-
эффективных масс, а они существенным
образом связаны с характером потен-
потенциала кристаллической решетки.
У некоторых полупроводников эф-
эффективная масса дырок очень велика
по сравнению с эффективной массой
электронов проводимости, и поэтому
основной вклад в электрические свой-
свойства дают электроны проводимости.
Такие полупроводники называются
электронными полупроводниками или
полупроводниками п-типа (от слова
negativ — негативный, т. е. отрица-
отрицательный). В противоположном слу-
случае (большая эффективная масса
электронов проводимости) основной
вклад дают дырки. Такие полупро-
полупроводники называются дырочными по-
полупроводниками или полупроводни-
полупроводниками р-типа (positiv — позитивный,
т. е. положительный).
Рассмотренные выше полупровод-
полупроводники с узкой областью запрещенных
значений энергии между валентной
полосой и полосой проводимости яв-
являются так называемыми собственно
полупроводниками. К ним, напри-
например, принадлежат германий и крем-
кремний. Однако существует класс ве-
веществ с гораздо более широким за-
запрещенным интервалом, благодаря
которому нет прямого переброса элек-
электронов из валентной полосы в поло-
полосу проводимости. И все же эти ве-
вещества относят к классу полупровод-
полупроводников (такие вещества даже более
распространены, чем собственно по-
полупроводники, и они находят очень
широкое применение в современной
технике).
216
Рис. 245. Различные типы полупроводни-
полупроводников
С точки зрения зонной теории та-
такие вещества являются изолятора-
изоляторами. Для чистых веществ это дейст-
действительно так. Полупроводниковые
свойства они приобретают за счет
примесей.
Если в таком веществе имеется
ничтожное количество примесных
атомов (число примесных атомов
должно быть очень мало, чтобы эти
атомы не взаимодействовали между
собой), то одноэлектронный спектр
энергий вещества наряду с полосами
будет иметь дискретные уровни энер-
энергии («примесные уровни»). В част-
частности, эти уровни могут оказаться
в области запрещенного интервала
энергии между валентной полосой и
полосой проводимости. Эти уровни
энергии могут быть как занятыми
электронами, так и свободными. По-
Полупроводниковые свойства возникают
только в двух специальных случаях
примесных уровней.
Если над валентной полосой и
близко от нее расположен примесный
уровень, не занятый электронами
A на рис. 245, а), то из валентной
зоны на этот уровень могут перейти
электроны, и в валентной полосе по-
появятся дырки (обозначенные круж-
кружками) , в результате чего вещество
приобретает свойства полупроводни-
полупроводника р-типа. Соответствующие при-
примесные уровни называются акцеп-
акцепторными.
Если под полосой проводимости и
близко от нее расположен примесный
уровень B на рис. 245,6), занятый
электронами, то с этого уровня в по-
полосу проводимости могут перейти эле-
электроны, и вещество приобретает свой-
свойства полупроводника и-типа. Соответ-
Соответствующие примесные уровни назы-
называются донорными.
Искусственным подбором приме-
примесей можно регулировать полупровод-
полупроводниковые свойства таких веществ.
На рис. 245 показаны схемы энерге-
энергетических спектров и переходов элект-
электронов в полупроводниках различного
типа.
В заключение обратим внимание
еще на одно обстоятельство. Химиче-
Химическая связь в полупроводниках про-
проявляется почти полностью как кова-
лентная. Доля металлической связи
незначительна. Это связано с незна-
незначительным числом «носителей» за-
зарядов, ответственных за металличе-
металлическую связь. Но в то же время это не-
незначительное число носителей заря-
зарядов создает вполне измеримые элект-
электрические свойства, такие же, как у
металлов. Объяснение такого разли-
различия заключается в том, что электри-
электрические свойства (например, электро-
электропроводность) определяются не толь-
только числом «свободных» носителей за-
зарядов, но и их подвижностью, т. е.
средней добавочной скоростью, со-
создаваемой единичным внешним элек-
электрическим полем. У полупроводников
эта подвижность имеет довольно
большую величину.
5. Ионно-металлические связи.
На рис. 239, а типы возможных свя-
связей мы изобразили треугольником.
В предыдущих разделах мы обсуди-
обсудили характер промежуточной связи и
свойства соответствующих веществ
для случаев, отвечающих на диаг-
диаграмме положениям вблизи двух ребер
треугольника. Положение у третьего
ребра кратко будет рассмотрено в
этом разделе. Оно соответствует пере-
переходным ионно-металлическим свя-
связям. Этот случай пока что изучен
наименее полно.
С точки зрения зонной теории, нет
качественной разницы между кова-
лентными и ионными кристаллами.
Поэтому особенных изменений в
свойствах спектра энергии не следу-
следует ожидать. Однако количественные
характеристики в этих двух предель-
предельных случаях могут сильно разли-
различаться.
Так, ионные кристаллы, как пра-
правило, имеют гораздо более широкий
интервал запрещенных значений
энергии между полосами, чем кова-
лентные кристаллы. Например, в
ионном кристалле NaCl ширина за-
запрещенного интервала энергии со-
составляет ~ 6 зв (напомним, что для
типичных собственно полупроводни-
полупроводников ширина интервала имеет порядок
величины 1 эв). Поэтому при тех тем-
температурах, при которых ковалент-
ные кристаллы проявляют себя как
полупроводники, переброс электронов
в ионных кристаллах практически
еще не происходит, и они по отноше-
отношению к электронной проводимости ве-
ведут себя как изоляторы. В принципе
при повышении температуры можно
осуществить условия, при которых.
будет происходить переход электро-
электронов из валентной зоны. Однако ока-
оказалось, что такие температуры для
всех ионных кристаллов намного-
выше их температуры плавления, так
что металлические свойства ионные
кристаллы не проявляют.
Однако электропроводность у ион-
ионных кристаллов существует. Это не
электронная электропроводность, а
ионная (так называемый электропе-
электроперенос) . Механизм электропроводно-
электропроводности заключается в следующем.
В кристаллах (реальных) всегда су-
существуют вакантные узлы кристал-
кристаллической решетки, концентрация
этих вакансий тем больше, чем выше
температура. При наложении внеш-
внешнего электрического поля заряжен-
заряженные ионы совершают перескоки в ва-
вакансии так, что в результате полу-
получается направленный перенос элек-
электрического заряда. Подвижность
ионов неодинакова, и перенос в ос-
основном осуществляется ионами од-
одного знака (например, в кристалле'
NaCl электроперенос осуществляется
217
ионами Na+, только при очень высо-
высоких температурах в переносе начина-
начинают участвовать также и ионы С1~).
Таким образом, вклада металличе-
металлической составляющей в ионные кристал-
кристаллы ни в чем нельзя обнаружить, если
идти по ребру треугольника
диаграммы от ионной вершины к ме-
металлической.
Но, с другой стороны, если идти в
противоположном направлении, от
металлической вершины, то можно
обнаружить интерметаллические со-
соединения, в которых проявляется не-
некоторое участие в химических связях
ионной составляющей.
Такие соединения могут образовы-
образовываться между самыми электроположи-
электроположительными металлами, характеризуе-
характеризуемыми наинизшими потенциалами
ионизации, и металлами Ь-подгрупп,
у которых малы (того же порядка)
энергии сродства к электрону (см.
табл. 20). В качестве примера можно
назвать соединения NaCu, NaAu,
CsAu и др., принадлежащие к струк-
структурному типу CsCl. Еще в начале
XX в. А. А. Байков указывал, что
медь в интерметаллических соедине-
соединениях с щелочными и щелочноземель-
щелочноземельными элементами играет роль, ана-
аналогичную галогенам в неорганических
соединениях тех же металлов. Мож-
Можно предполагать, что металлы I-b
подгруппы в таких соединениях мо-
могут достраивать свою s-оболочку до
устойчивой двухэлектронной конфи-
конфигурации, приобретая таким образом
отрицательный заряд.
Некоторые соединения щелочнозе-
щелочноземельных металлов с оловом и свин-
свинцом (например, MggSn и Mg2Pb) так-
также можно отнести к этой категории
соединений. Для таких соединений,
в отличие от подавляющего боль-
шипства интерметаллических соеди-
соединений, справедливы правила валент-
валентности и характерна принадлежность
к структурным классам, типичным
для ионных соединений.
Интересно отметить, что среди та-
таких интерметаллических соединений
встречаются вещества с очень узким
запрещенным интервалом энергии
между полосами, т. е. эти вещества
проявляют полупроводниковые свой-
свойства. Это показывает большой диапа-
диапазон свойств веществ с промежуточ-
промежуточным характером химической связи.
ГЛАВА XIII
ИЗОМОРФИЗМ И ПОЛИМОРФИЗМ
§ 1. Истории открытии
Явление изоморфизма было открыто
в 1819 г. Э. Митчерлихом. Материа-
Материалом ему послужили кристаллы
КН2РО4, KH2As04 и NH4H2PO4. Им
было обнаружено, что эти близкие по
составу вещества кристаллизуются
в одинаковых формах. Митчерлих
назвал их изоморфными. В букваль-
буквальном переводе термин «изоморфизм»
означает «равноформенность». Через
год он установил, в результате более
точных измерений, что кристаллы
изоморфных веществ не строго одина-
одинаковы, а лишь близки друг к другу по
форме. Так, для трех указанных
солей углы между главной осью
и нормалью к грани A01) будут
соответственно 43°12', 43°10' и 45°12'.
Таким образом, изоморфными вещест-
веществами можно назвать твердые вещест-
вещества, сходные по химическому составу
и имеющие близкие по форме кри-
кристаллы.
Явление изоморфизма привлекло к
себе внимание Д. И. Менделеева.
Исследованию его была посвящена
первая диссертация Д. И. Менделеева
«Изоморфизм в связи с другими
¦отношениями кристаллической фор-
формы к составу» A856 г.).
Явление полиморфизма было так-
также открыто Э. Митчерлихом A822 г.).
Сущность его заключается в том, что
некоторые вещества * в различных
условиях способны образовывать раз-
* Термином «полиморфизм» мы будем
пользоваться как для сложных, так и
для простых веществ, не вводя для по-
последних специального термина аллотро-
аллотропия.
ные по симметрии и по форме кри-
кристаллы. Общеизвестным примером
являются две кристаллические фор-
формы углерода: графит и алмаз. Каж-
Каждая из этих форм называется поли-
полиморфной модификацией. Отдельные
полиморфные модификации иногда
очень резко отличаются друг от друга
по своему атомному строению и физи-
физическим свойствам. Так, например,
графит принадлежит к гексагональ-
гексагональной сингонии, алмаз — к кубической:
графит черного цвета, непрозрачен,
хорошо проводит электрический ток;
алмаз прозрачен, электрического тока
не проводит; графит является одним
из самых мягких минералов, алмаз —
самый твердый из всех известных
веществ; удельный вес графита 2,22,
алмаза — 3,51. Митчерлиху был изве-
известен полиморфизм серы и углекисло-
углекислого кальция. Сера кристаллизуется при
одних условиях в ромбических, а при
других — в моноклинных кристаллах.
СаСОз известен в гексагональной мо-
модификации (кальцит) и в ромбиче-
ромбической (арагонит).
Явление полиморфизма чрезвы-
чрезвычайно распространено. Почти все ве-
вещества при известных условиях мо-
могут быть получены в различных по-
полиморфных модификациях.
§2. Д«рентгеновские работы
но наоморфиаиу
В качестве примера явления изомор-
изоморфизма можно привести ряд углекис-
углекислых солей двухвалентных металлов.
Многие иэ них кристаллизуются в од-
одном и том же классе симметрии и
219
Рис. 246. Ромбоэдр кальцита
имеют кристаллы, характерной фор-
формой которых является ромбоэдр (рис.
246). Ромбоэдр, как известно, с гео-
геометрической точки зрения характери-
характеризуется одним углом. Для разных
веществ изоморфной группы угол а
имеет различные значения:
Вещество
ZnCOs
103*28'
MgCQs
1031'
FeCOs
103°04'
Вещество МпСОз CdCOs СаСОз
а 102°50' 102-30' 101*55'
Было обнаружено, что многие изо-
изоморфные вещества могут образовы-
образовывать друг с другом однородные кри-
кристаллические фазы переменного со-
состава. По аналогии с жидкими систе-
системами они были названы Вант-Гоф-
фом твердыми растворами A890 г.).
В то время многие считали, что
способность образовывать твердые
растворы является непременным
свойством изоморфных веществ, по-
поэтому до сих пор термины «изоморф-
«изоморфные вещества», «смешанные кристал-
кристаллы», «твердые фазы переменного сос-
состава», «твердые растворы» нередко
употребляются как синонимы.
Существенным моментом явилось
установление близости молекулярных
объемов у изоморфных веществ
(Ю. В. Вульф). Физические свойства
смешанных кристаллов зависят от
свойств чистых компонентов и от
относительного содержания их в изо-
изоморфной смеси. Так, по Реджерсу
220
A889 г.), удельный вес смешанного
кристалла
Vi
где d\ и d% — удельные веса компонен-
компонентов, a Vi и V2 — их молекулярные
объемы. Следует отметить, что ли-
линейная зависимость между удельным
весом и молекулярным объемом сме-
смешанного кристалла дает, по Ю. В.
Вульфу, возможность открывать по-
полиморфные модификации, не сущест-
существующие при обыкновенных услови-
условиях, и определять их удельные веса.
По мере дальнейших исследований
выяснилось, что не все вещества,
имеющие близкие по форме кристал-
кристаллы (или даже одинаковые как, напри-
например, все кристаллы кубической син-
гонии), обладают способностью да-
давать твердые растворы. Для обозначе-
обозначения таких веществ был предложен
термин изогонизм, т. е. равноуголь-
ность. Близок к этому понятию и тер-
термин изотипия *. Изотипными веще-
веществами называются такие, у которых
при совершенно различном химиче-
химическом составе симметрия кристаллов
одинакова, и очень близки геометри-
геометрические константы и формы кристал-
кристаллов. Примером таких веществ могут
служить металлический магний
и окись цинка. Теперь хорошо извест-
известна причина такого необычайного
сходства кристаллов этих веществ.
Магний кристаллизуется в гексаго-
гексагональной плотнейшей упаковке.
В окиси цинка атомы кислорода
располагаются по точкам той же упа-
упаковки, а атомы цинка — в тетраэд-
рических пустотах.
Не всегда два изоморфных веще-
вещества могут смешиваться друг с дру-
другом в любых пропорциях — раство-
растворимость может быть ограниченной.
* Термин «изотипия» иногда использует-
используется некоторыми авторами в ином смыс*
ле — как синоним термина «изострук-
турность>>.
Это обстоятельство подчеркивается
терминами совершенный и несовер-
несовершенный изоморфизм (А. К. Болды-
Болдырев). На рис. 247 показаны диаграм-
диаграммы состояния для пар веществ:
а) не дающих ни твердых растворов,
ни соединений; б) дающих непре-
непрерывные твердые растворы и в) с огра-
ограниченной смешиваемостью в твер-
твердом состоянии (промежуточный
случай). Последняя диаграмма инте-
интересна еще и тем, что на ней отчет-
отчетливо видно изменение (уменьшение)
растворимости одного компонента в
другом в твердом состоянии с пони-
понижением температуры. Очень инте-
интересная система NaCl — КС1 была
изучена Н. С. Курнаковым и С. Ф.
Жемчужным A901 г.). При высо-
высоких температурах эти соединения
образуют непрерывный ряд твердых
растворов. С понижением темпера-
температуры начинается распад твердых рас-
растворов, а при комнатной температуре
оба вещества совсем не смешиваются
друг с другом.
К этому же времени было найдено
немало случаев, когда вещества, дале-
далекие друг от друга по химическим
и кристаллографическим свойствам,
образовывали друг с другом фазы
переменного состава в более или
менее широком интервале составов,
например углерод в железе.
Все зти новые факты не укладыва-
укладывались в рамки старых понятий и при-
приводили нередко к большой путанице
Рис. 247. Диаграммы состояния систем
а — компоненты не образуют твердых раство-
растворов;
б — компоненты дают непрерывные твердые
растворы;
¦в — случай ограниченной смешиваемости
в терминологии. Говоря по существу,
понятие изоморфизма включает в
себя два совершенно различных явле-
явления: 1) способность некоторых хими-
химически сходных веществ кристаллизо-
кристаллизоваться в сходных формах и 2) способ-
способность образовывать твердые фазы
переменного состава. Некоторые
изоморфные вещества одновременно
обладают обоими свойствами, но, во-
вообще говоря, это не обязательно и
одно свойство закономерно не выте-
вытекает из другого. Имеется много изо-
изоморфных веществ, которые обладают
только одним из них и вовсе лише-
лишены другого.
§3. Дорентгеновскне работы
но полннорфнзму
Развитие термодинамической теории
фазовых равновесий (Дж. В. Гиббс)
и последовавшее за ним чрезвычайно
бурное развитие экспериментальных
исследований систем обусловило
более глубокое изучение явлений изо-
изоморфизма и полиморфизма.
Каждая полиморфная модификация
имеет свою область существования
на фазовой диаграмме. Переход от
одной модификации к другой, напри-
например ромбической серы в моноклин-
моноклинную (рис. 248), связан со скачко-
скачкообразным изменением свойств (на
рисунке линии равновесия мета-
стабильных фаз показаны пункти-
пунктиром). Взятый пример будет характе-
характеризоваться, в частности, изменением
(увеличением) удельного объема
AV=0,014 и термическим эффектом
в 3,12 кал[г.
Обычно различают монотропные
и энантиотропкые превращения. Для
первых характерен переход, который
не может быть повторен в обратном
направлении, для вторых взаимные
переходы легко осуществляются с
изменением условий (температуры
и давления). В качестве примера мо-
нотропного превращения обычно при-
приводят полиморфизм углерода. Алмаз
легко может быть превращен в гра-
221
Рис. 248. Диаграмма состояния серы
фит, обратный переход осуществлен
лишь в последние годы при очень вы-
высоких давлениях и температурах.
Примером энантиотропных превра-
превращений может служить переход серы
от ромбической к моноклинной и об-
обратно. Не всегда такой переход осуще-
осуществляется легко, часто наблюдается
задержка превращения, в результате
чего фаза становится термодинами-
термодинамически метастабильной. Однако на
практике метастабильные фазы мо-
могут быть чрезвычайно устойчивыми
и внешне не проявлять своей мета-
стабильности. Достаточно вспомнить
метастабильный характер стекла, ко-
которое может сотнями лет не перехо-
переходить в устойчивое кристаллическое
г/с
/SO
Ш
во
40
о
Z0
1 Раствор
- fi-pamb
, %
Рис. 249. Диаграмма состояния сии/емы
NH4NO3 — Н2О
состояние. Превращение метаста-
метастабильной формы металла в стабиль-
стабильную можно задержать при помощи
закалки.
Как бьшо сказано выше, некоторые
вещества имеют несколько поли-
полиморфных модификаций. В качестве
примера рассмотрим диаграмму со-
состояния NH4NO3 —Н2О (рис. 249).
Кривая имеет несколько точек изло-
излома, отвечающих определенным тем-
температурам перехода одной модифи-
модификации в другую. Этими точками кри-
кривая разделяется на участки, кото-
которыми определяются границы устой-
устойчивости данной модификации.
Азотнокислый аммоний имеет четыре
модификации: в пределах темпера-
температур от —18 до +32° С устойчива р-
ромбическая модификация, от 32 до
84° С — а-ромбическая, от 84 до
125° С — тригональная и выше
125°— кубическая.
Если исходное состояние раствора
изобразить фигуративной точкой Ъ, то
при охлаждении раствора кристалли-
кристаллизация начнется в точке Ьь причем
выпадает кубическая модификация
NH4NO3. При дальнейшем охлажде-
охлаждении раствора фигуративная точка бу-
будет двигаться по кривой вниз до точ-
точки 6г- В этой точке система инва-
инвариантна, так как в равновесии нахо-
находятся три фазы (две твердых ш
раствор). При снижении теплосодер-
теплосодержания системы температура остается
постоянной, пока кубическая моди-
модификация не перейдет в тригональную.
После исчезновения кубической фазы
продолжается кристаллизация триго-
нальной модификации, фигуративная
точка перемещается вниз по кривой,
растворимости. Следующие останов-
остановки происходят при 84° С (переход
тригональной модификации в а-ром-
бическую) и при 32° С, когда а-ром-
а-ромбическая модификация переходит в
Р-ромбическую. При — 18° С процесс
заканчивается кристаллизацией эв-
эвтектической смеси (З-модификации
NH4NO3 со льдом.
222
§ *. Первые ревтгеноструктурвые
исследовании изоморфных
веществ
Применение рентгеновских лучей для
определения внутренней (атомной)
структуры кристаллов У. Бреггом
A913 г.) позволило установить, что
изоморфные вещества обычно имеют
сходное пространственное расположе-
расположения между аналогичными атомами у
RbCl имеют одну и ту же структуру,
т. е. ионы металла и ионы хлора распо-
располагаются в кристаллическом прост-
пространстве одинаковым образом относи-
относительно друг друга. Однако расстоя-
расстояния между аналогичными атомами у
этих веществ несколько различны.
Так, например, кратчайшее расстоя-
расстояние между атомами натрия и хлора
а/2 (в структуре NaCl) равно 2,81 А,
между атомами калия и хлора —
3,14 А, между атомами рубидия и
хлора —3,27 А.
Выяснилось, что твердые растворы
могут образовываться только в том
случае, когда аналогичные межатом-
межатомные расстояния у нескольких веществ,
имеющих одинаковую структуру, не-
несильно отличаются друг от друга. Если
же отличие велико, то твердые раство-
растворы не образуются вовсе или образуют-
образуются только в определенных условиях.
Так, например, NaCl и КС1 дают твер-
твердые растворы только при высокой
температуре, при обычных темпера-
температурах твердые растворы распадаются.
Твердые растворы КС1 — RbCl могут
существовать при обычных условиях.
Между тем все три вещества кристал-
кристаллизуются в кубах и имеют, следова-
следовательно, одинаковые по форме кри-
кристаллы.
У веществ, образующих друг с дру-
другом твердые растворы такого типа, не-
некоторые физические свойства сходны,
в частности, всегда близки их молеку-
молекулярные объемы, так как только при
этом условии возможна устойчивость
всего кристалла. Поэтому, если неко-
некоторые составные части структуры (ио-
(ионы) будут заменены другими, сход-
сходными с ними по физическому дейст-
действию, то получающаяся при такой за-
замене форма кристаллов по своим,
геометрическим константам будет
весьма близка к форме кристаллов
исходного вещества, о чем говорил
еще Митчерлих. После первых же
рентгеноструктурных работ выясни-
выяснилось подчиненное значение сходства
внешней формы кристаллов и важ-
важность принадлежности изоморфных
веществ к одному структурному типу.
Такие вещества логичнее называть
изоструктурными веществами, а не
изоморфными.
Замещающие друг друга атомы (ио-
(ионы) А и В располагаются в структу-
структуре твердых растворов (А, В)Х стати-
статистически. Это значит, что в данном
объеме на га частиц А приходится тп
частиц В, причем частицы обоих сор-
сортов занимают одинаковые места в
структуре. Любое количество частиц
А может быть заменено частицами В.
Твердые растворы состава 50% АХ+
+50% ВХ отличаются от химическо-
химического соединения АВХг. В соединении'
АВХг атомы А и В закономерно че-
чередуются между собой во всей струк-
структуре, тогда как в твердом растворе
(А, ,В)Х каждое из положений (А, В)
может быть замещено как А, так и В
по закону вероятности, т. е. в этом
случае вероятность того, что положе-
положение (А, В) будет замещено ионом А
или В, равно 0,5; при 30% АХ ш<
70% ВХ соответствующие вероятно-
вероятности будут 0,3 и 0,7. Чтобы выразить
эту мысль в терминах теории структу-
структуры Е. С. Федорова, надо сказать, что
в твердых растворах атомы А и В рас-
располагаются по точкам одной правиль-
правильной системы, а в химическом соедине-
соединении — по двум разным.
Изменение констант решеток изо-
изоморфных веществ с составом (рис.
250) происходит в первом приближе-
приближении линейно (правило Вегарда). Это-
правило математически, конечно, про-
противоречит правилу аддитивности мо-
молекулярных объемов изоморфных ве-
веществ (см. § 2). Однако поскольку и
молекулярные объемы, и линейные
размеры кристаллических ячеек у
22*
_l I L_J_
A Cocmad, % В
"Рис. 250. Изменение констант решеток изо-
изоморфных веществ с составом
изоморфных веществ чрезвычайно
•близки, то обоими этими правилами
практически можно пользоваться для
проведения приблизительных расче-
расчетов молекулярных объемов или кон-
констант решеток смешанных кристал-
кристаллов.
§ 5. Структурная классификация
типов нолииорфиаиа
Результаты рентгеновских исследо-
исследований различных полиморфных моди-
модификаций многих веществ позволяют
предложить следующую структур-
структурную классификацию типов полимор-
полиморфизма.
1. Структуры полиморфных моди-
модификаций отличаются друг от друга по
координационным числам. Примером
может служить полиморфизм хлори-
хлористого аммония. Одна из модификаций
кристаллизуется в структурном типе
CsCl (к. ч. 8), вторая — в структурном
типе NaCl (к. ч. 6). К этому же типу
относятся структуры Р- и у-железа
(Р-модификация имеет к. ч. 8, у — 12)
ш, конечно, полиморфизм углерода —
алмаз и графит.
2. Структуры отличаются типом
плотнейшей упаковки, но координа-
координационное число остается одним и тем
же у всех модификаций. Например,
5 модификаций ZnS, 8 модификаций
карборунда, 3 модификации С<Лг и
т. п. Этот случай полиморфизма часто
называют полигипией. Три модифи-
модификации ТЮ2— рутил, брукит и ана-
таз — отличаются друг от друга типом
упаковки: рутил имеет гексагональ-
гексагональную упаковку, анатаз — кубическую,
брукит — четырехслойную (топазо-
(топазовую). При такой смене упаковки мо-
может, конечно, иметь место и неболь-
небольшой поворот некоторых атомных
групп в структуре, но в данном слу-
случае это будет второстепенным фактом.
3. Тип упаковки и координацион-
координационные числа в структурах сохраняются,
но меняется мотив расположения ка-
катионов (колумбит и моссит ГеШ)гОб).
4. Менее заметная разница в струк-
структурах полиморфных веществ наблю-
наблюдается в тех случаях, когда одна из
модификаций отличается от другой
небольшим поворотом некоторых
структурных групп в процессе поли-
полиморфного превращения. Это имеет ме-
место в различных модификациях ЭЮг:
у р-кристобалита угол Si — О — Si
180°, а у кварца 160°. Еще мень-
меньшим поворотом отличаются друг от
друга структуры а- и р-кварца.
К этому типу следует отнести и по-
полиморфизм некоторых органических
алифатических соединений, являю-
являющихся результатом изменения накло-
наклона длинноцепочечных молекул в
момент полиморфного превраще-
превращения.
5. Особый тип полиморфизма свя-
связан с вращением молекул или радика-
радикалов в кристаллической решетке. Вра-
Вращение молекул детально изучено в
кристаллических парафинах. На рис.
251, а изображен разрез, перпендику-
перпендикулярный оси с ромбической ячейки
кристаллов СгэНво. Это вещество име-
имеет также высокотемпературную гек-
гексагональную модификацию, аналогич-
аналогичную по структуре ромбической. Об
отклонении ромбической структуры
от гексагональной можно судить по
отклонению угла г|) от 60°. Удалось
осуществить переход ромбической ре-
решетки в гексагональную при медлен-
медленном повышении температуры, так
как коэффициенты расширения в
направлении осей а, Ъ и с довольно
сильно отличаются. Кристалл при
нагревании расширяется, так что
угол г|) приближается к 60°. Когда он
становится равным 60°, появляется
возможность вращения молекул око-
около их длинной оси (рис. 251, б).
ИЗА
Рис. 251. Полиморфизм парафина С2вНво
В этот момент кристаллы становятся
гексагональными, что доказано как
рентгеновскими, так и оптическими
исследованиями. Оптическая инди-
индикатриса в этот момент делается одно-
одноосной. Гексагональная структура со-
соответствует плотной укладке вра-
вращающихся цилиндрических молекул.
Дж. Бернал исследовал вращение
молекул в кристаллических спиртах,
в частности в С12Н25ОН. Это вещество
при 24° С кристаллизуется в гексаго-
гексагональной структуре. При понижении
температуры до 16° С оно переходит в
моноклинную модификацию. В моно-
моноклинной ячейке углеродные цепи на-
наклонены к базису под некоторым уг-
углом. Если нагревать моноклинную мо-
модификацию до температуры плавле-
плавления, то не происходит перехода веще-
вещества в гексагональную модификацию.
Последняя может быть получена
только из расплава. Необратимость
здесь связана с тем, что одного вра-
вращения недостаточно для получения
гексагональной структуры; необходи-
необходимо изменить и угол наклона к ба-
базису.
Вращение молекул повышает сим-
симметрию решетки. Вещества с неболь-
небольшими изометричными молекулами
часто имеют высокосимметричные мо-
модификации, связанные с вращением
молекул. Таковы, например, N2, HC1 и
др. Вращение молекул в органических
кристаллах — очень распространен-
распространенное явление. В неорганических крис-
кристаллах с ионной связью также имеет
место вращение ионов; так, например,
в структуре NH4NO3 в интервале тем-
температур от 84 до 125° С ион [NO3]~
вращается вокруг тройной оси. Рас-
Рассчитанные интенсивности отражен-
отраженных рентгеновских лучей совпадают
с наблюденными на рентгенограммах,
если предположить, что рассеиваю-
рассеивающая масса кислородных атомов рас-
расположена по тору, бесконечная ось
симметрии которого совпадает с трой-
тройной осью кристалла. В интервале от
125° С до температуры плавления
A69,6° С) ион NOi" принимает шаро-
шаровую форму вследствие беспорядочно-
беспорядочного вращения около одной точки, а не
около оси, как в предыдущем случае.
Высокотемпературная модификация
(кубическая) NH4NO3 обусловлена
беспорядочным вращением обоих ио-
ионов NH4+ и NO3".
При изменении структуры всегда
меняется в той или иной степени тип
химической связи. Последнее обстоя-
обстоятельство сказывается на изменении
физико-химических свойств, причем
тем значительнее, чем резче меняется
тип связи, и особенно сильно в том
случае, когда одна из модификаций
имеет гомодесмическую структуру, а
вторая — гетеродесмическую (напри-
(например, алмаз и графит). Изменение ва-
валентного угла кислорода в структурах
кварца и тридимита, конечно, тоже
связано с изменением характера хи-
химической связи от более ковалентной
к более ионной.
6. Наш обзор был бы неполным, если
ничего не сказать о «полиморфизме»,
не связанном с изменением структу-
структуры. Этот тип превращения характери-
характеризуется тепловым эффектом и скачко-
скачкообразным изменением других свойств
и по этой причине, следовательно,
может быть причислен к полиморф-
полиморфным превращениям. Известным при-
примером такого рода превращения явля-
является потеря магнитных свойств у же-
железа при а—»- Р-превращении при тем-
температуре 770° С. Структуры обеих мо-
модификаций — объемноцентрирован-
ный куб — совершенно одинаковы.
Многие примеры таких гомео-
морфных полиморфных превращений
у веществ, содержащих водород, по-
видимому, только случайно отнесены
к этой категории. Изменение направ-
направления или характера водородных свя-
Т5 Кристаллохимия
225
зей или, тем более, вращения прото-
протонов констатировать методом рентге-
ноструктурного анализа нельзя. По-
Поэтому превращения, связанные с эти-
этими явлениями, только кажутся гомео-
морфными. В действительности они
относятся к одной из предыдущих
категорий. Условия существования
тех или иных полиморфных модифи-
модификаций веществ связываются с тер-
термодинамическими условиями — в
первую очередь с температурой и
давлением (см., например, диаграм-
диаграмму состояния серы, рис. 248).
В последние годы выяснилась ог-
огромная роль микропримесей (или
весьма незначительных отклонений
стехиометрического состава от иде-
идеального) для стабилизации тех или
иных полиморфных модификаций
веществ. Так, например, выяснилось,
что природный алмаз всегда содер-
содержит то или иное количество азота
(до 0,15%). Кристаллизации ромби-
ромбической модификации СаСОз (арагони-
(арагонита) способствует микропримесь
стронция. Весьма незначительный
избыток одного из компонентов (по-
(порядка 10~5%) в соединениях ти-
типа ZnS стабилизирует те или иные
модификации этих веществ.
§ в. Условия,
необходимые для проявления
изоморфизма
Многочисленные исследования изо-
изоморфных веществ показывают, что
образование смешанных кристаллов
возможно тогда, когда замещающие
друг друга частицы обладают сход-
сходным характером взаимодействия, так
как только при этом условии возмож-
возможна устойчивость всего кристалла.
Близость размеров замещающих
друг друга частиц является первым
необходимым условием для проявле-
проявления изоморфизма. Однако к этому
условию нельзя подходить чисто ме-
механически и заранее предвидеть,
какие элементы с какими будут да-
давать твердые растворы. Вопрос
этот гораздо сложнее. На замеща-
емость одного химического элемента
другими большое влияние оказывают
«молекулярные объемы», или разме-
размеры элементарных ячеек, в структурах
обоих компонентов.
Ион натрия, например, не может
заменить в простейших структурах
ион лития, так как разница в раз-
размерах радиусов (Na+ = 0,98, Li+=
=0,68) этих ионов сильно сказыва-
сказывается на размерах элементарных яче-
ячеек: а для NaCl равно 5,63 А, для
LiCl — 5,13. Но те же ионы могут за-
заменить друг друга в сложных соеди-
соединениях LiMnPO4 и NaMnPCt, так
как относительная разница в разме-
размерах элементарных ячеек в этом слу-
случае будет значительно меньшей. Ве-
Вещества, образующие друг с другом
твердые фазы переменного состава,
обычно кристаллизуются в одном
классе симметрии и имеют сходные
решетки с близкими параметрами.
Но самое понятие сходства, близости
и т. п. не может быть определено
точно. Так, например, если вещест-
вещество А кристаллизуется в кубической
гранецентрированной решетке, а ве-
вещество В — в ромбоэдрической, с
острым углом ромбоэдра, близким
к 60°, то, казалось бы, между ними
не может существовать твердых ра-
растворов, так как слишком велико
различие по симметрии этих реше-
решеток. Однако известны случаи образо-
образования твердых растворов таких со-
соединений. Объяснить это можно
очень легко, если вспомнить, что
всякая гранецентрированная кубиче-
кубическая решетка будет иметь примитив-
примитивный параллелепипед повторяемости
в форме острого ромбоэдра с углом
в 60°. Поэтому если размеры такого
ромбоэдра у вещества А близки к
размерам ромбоэдра у вещества В,
то твердые растворы (АВ) могут
наблюдаться несмотря на кажущее-
кажущееся столь резким различие по симмет-
симметрии. Сходство решеток надо пони-
понимать в федоровском смысле, т. е. го-
гораздо шире, чем понимал это Гримм
и др. Очень показательны в этом от-
отношении непрерывные твердые рас-
226
творы индия, имеющего тетрагональ-
тетрагональную решетку с отношением осей
с/а=1,08, и металлов, кристалли-
кристаллизующихся в гранецентрированных
кубических ячейках, размеры кото-
которых близки к константам решетки
индия, например таллия (Е. С. Ма-
Макаров) .
Из сказанного следует, что сим-
симметрия кристаллов не играет боль-
большой роли при образовании смешан-
смешанных кристаллов. Гораздо более важ-
важным фактором являются размеры и
формы ячеек кристаллических реше-
решеток.
Большую роль играет поляризация
замещающих друг друга компонен-
компонентов. Для образования смешанных
кристаллов поляризационные свой-
свойства замещающих друг друга частиц
должны быть близкими (Гольд-
шмидт). В настоящее время необхо-
необходимо расширить трактовку этого
пункта и говорить не только о поля-
поляризации, но и вообще о сохране-
сохранении типа химической связи.
Ион одновалентной меди и ион
натрия имеют одинаковые размеры
@,98), однако, в силу резкой разни-
разницы в типах химической связи у со-
соединений меди и натрия, изоморф-
изоморфные смеси натриевых и медных со-
солей практически не встречаются.
Соли натрия являются ионными со-
соединениями, соли меди — ковалент-
ными. Атом меди, кроме того, обра-
образует, как правило, небольшое число
C—4) ковалентных связей. Так,
NaCl — структура ионная с коорди-
координационным числом 6, a CuCl — струк-
структура ковалентная с координацион-
координационным числом 4 (тип сфалерита ZnS).
Для нее справедливо все то, что ска-
сказано относительно структуры AgJ
в § 4 главы XII о типах химиче-
химической связи.
Для образования смешанных кри-
кристаллов среди ионных соединений
необходимо, конечно, чтобы знаки
зарядов замещающих друг друга
компонентов совпадали. Геометриче-
Геометрически, однако, мыслим случай, когда
у разных веществ с одним и тем же
структурным типом места располо-'
жения анионов и катионов взаимно
заменены. Так, например, мы хоро-
хорошо знаем структурный тип CaF2. Но
в этом же структурном типе кристал-
кристаллизуются и Na2O. Места, занятые
катионами кальция в структуре
CaF2, в структуре Na2O занимают
анионы кислорода, и, наоборот, ме-
места, занятые анионами фтора, в
структуре Na2O занимают катионы
натрия.
Такое явление называется анти-
антиизоморфизмом. Ясно, что антиизо-
антиизоморфные вещества никогда не обра-
образуют фаз переменного состава.
§7. Предел
изоморфной замеотиноотн.
Морфотровна ¦ иолнморфнам
Как было сказано выше, изоморф-
изоморфное замещение в химически анало-
аналогичных веществах возможно только
при том условии, что типы химиче-
химической связи замещающих друг друга
частиц близки, а размеры не выхо-
выходят за известные пределы. Если пре-
пределы перейдены, то изоморфизм не
проявляется. Гольдшмидтом уста-
установлена серия рядов, подтверждаю-
подтверждающих сказанное. Возьмем, например,
BaZi-Оз, кристаллизующийся в струк-
структурном типе перовскита СаТЮз.
В этом соединении цирконий (Zr4+=
= 0,82) можно заменить на олово
(Sn4+=0,67), или титан (Ti4+=
= 0,64), при этом структурный тип
сохраняется. Можно барий (Ва2+ =
= 1,38) заменить на стронций
(Sr2+ = 1,20) или на кальций (Са2+ =
= 1,04). Все производные вещества
будут составлять одну изоморфную
группу. Однако если заменить каль-
кальций на магний (Mg2+=0,74), то из-
изменение радиусов будет настолько
велико, что предел возможности изо-
изоморфного замещения будет перейден,
и мы получим вещество новой струк-
структуры (морфотропия).
Под морфотропией понимается
резкое, но закономерное изменение
кристаллической формы и структуры
Т5*
227
в зависимости от ^закономерного из-
изменения химического состава. Гольд-
шмидт рассматривает полиморфизм
как частный вид морфотропии.
В данном случае сравниваются меж-
между собой две структуры одного и того
же вещества в двух состояниях, от-
отвечающих разным термодинамиче-
термодинамическим условиям. До тех пор, пока из-
изменения условий невелики и не пе-
переходят известных границ, вещество
остается изоморфным самому себе в
первоначальном состоянии, но как
только граница дозволенных дефор-
деформаций перейдена, происходит поли-
полиморфное превращение (автоморфо-
тропия).
Приведенное рассуждение показы-
показывает, что явления полиморфизма,
изоморфизма и морфотропии тесно
связаны друг с другом и являются
следствием общих причин. Очень хо-
хорошим примером могут служить кар-
карбонаты двухвалентных металлов и
изоструктурные с ними нитраты ще-
щелочных металлов:
Эта граница морфотропная, так как
RbNO3 известен в одной полиморф-
полиморфной модификации. Такой границы
нет в карбонатах, поскольку размеры
бария не превышают предела суще-
существования структурного типа араго-
арагонита.
Изучение изоморфных рядов, пре-
пределов изоморфизма и морфотропии
дает нам прекрасную иллюстрацию
закона перехода количественных из-
изменений в коренные качественные.
§ 8. Влинние изотопного состава
на кристаллическую структуру
Идеальным примером изоморфизма
замещения должны явиться систе-
системы, содержащие переменное коли-
количество изотопов какого-либо хими-
химического элемента. Заранее можно
предвидеть, что эффект изменения
констант решеток в таких системах
должен быть весьма незначительным.
Однако это изменение, как показы-
показывает опыт, вполне надежно может
Соединение
т катиона
Соединение
г катиона
MgCOa
0,74
СаСО»
1,04
LiNOs
0,68
CoCOs
0,78
NaNOa
0,98
FeCOa
0,80
ZnCOs
0,83
CaCOa SrCOs
1,04
KNOs
низкотемп.
1,33
1,20
KNOa
высокотемп.
1,33
МпСОз
0,91
PbCOs
1,26
RbNOi
1,49
CdCOa
0,99
BaCOs
1,38
CsNOs
1,65
В таблицу внесены вещества в по-
порядке возрастания размеров изомор-
фнозамещающих друг друга ионов.
Граница изоморфного замещения у
карбонатов проходит как раз на
кальциевой соли. Слева от этой гра-
границы вещества имеют структуру ти-
типа кальцита, справа — арагонита.
Для нитратов эта граница проходит
по калиевой селитре. Ее высоко-
высокотемпературная модификация имеет
структуру арагонита, низкотемпера-
низкотемпературная — кальцита. Оба вещества,
СаСОз и KNOa, полиморфны. Вторая
смена структурного типа в нитратах
происходит между ШЧОз и RbNO3.
быть определено с помощью совре-
современных рентгеновских прецизионных
методов измерения межатомных рас-
расстояний в кристаллах. Лучше всего
изучены структуры, в которых водо-
водород замещен на дейтерий, поэтому
только о таких структурах мы и бу-
будем здесь говорить.
Водород в химических соедине-
соединениях, как известно, может быть в
нескольких существенно различных
состояниях и, следовательно, выпол-
выполнять весьма разные функции. Край-
Крайними случаями будут соединения
водорода типа гидридов щелочных
металлов, где водород является анио-
228
ном, и соединения, в которых водо-
водород образует водородную связь, яв-
являясь, как сказано выше, катионом.
Первый случай, насколько о нем
можно судить на примере гидрида
лития, характеризуется уменьшени-
уменьшением межатомных расстояний при за-
замене водорода на дейтерий. Обрат-
Обратное соотношение получается при
замене водорода, образующего водо-
водородную связь. Так, константа решет-
решетки LiH, кристаллизующегося в
структурном типе поваренной соли,
равна 4,085 А, а для LiD эта вели-
величина равна 4,065. В тетрагональных
структурах KH2As04, KH2PO4 и
NH4H2PO4, являющихся классиче-
классическими примерами структур с корот-
короткими водородными связями (расстоя-
(расстояние 0...Н...0 у КН2РО4 равно 2,54),
при замене водорода на дейтерий
происходит увеличение длин водо-
водородных связей на 0,0080, 0,0097 и
0,0100 А соответственно. В структу-
структурах с гидроксильными связями изо-
изотопный эффект проявляется таким
же образом, что и в структурах с ко>-
роткими водородными связями, но
по величине он значительно меньше.
Чрезвычайно интересный случай
полиформизма КН2РО4 при замене
водорода на дейтерий наблюдал Убе-
лоде в 1939 г. При некоторых усло-
условиях соединение KD2PO4 кристалли-
кристаллизуется в моноклинных кристаллах с
восемью молекулами в элементарной
ячейке: а=7,37; 6=14,73; с=7,17
А и р = 92°, в то время как структу-
структура КН2РО4 тетрагональная с а =7,43
и с = 6,97 А. Этот пример оправдыва-
оправдывает рассмотрение изотопного эффекта
в главе об изоморфизме, так как слу-
служит наглядным доказательством на-
наличия у химических веществ поли-
полиморфных (морфотропных) превраще-
превращений, связанных с заменой в структуре
одного изотопа другим. Поскольку
методика измерений межатомных
расстояний из года в год совершен-
совершенствуется, и точность измерений воз-
возрастает, то в ближайшее время при-
придется, видимо, в некоторых случаях
учитывать изотопный состав веществ,
особенно для тех из них, которые
употребляются в качестве стандар-
стандартов, например NaCl. Для этого со-
соединения учет изотопного состава
особенно важен, так как хлор имеет
очень большое, по сравнению с дру-
другими элементами, содержание не-
неглавного изотопа.
§0. Изовалеитный
и гетеровалеитиый изоморфизм
Первые исследователи изоморфизма
придавали большое значение валент-
валентности замещающих друг друга ком-
компонентов в изоморфной смеси. Равен-
Равенство их валентности считалось обя-
обязательным условием. Например, в изо-
изоморфных соединениях КН2РО4 и
KH2ASO4 как фосфор, так и мышьяк
пятивалентны, в соединениях ZnSO< •
•7Н2О и FeSO4-7H2O двувалентный
цинк замещается двувалентным желе-
железом. Позднейшие работы, однако, по-
показали, что валентность не играет оп-
определяющей роли в образовании изо-
изоморфных веществ и что часто заменя-
заменяющие друг друга ионы имеют раз-
различную валентность. Случаев такого
гетеровалентного изоморфизма очень
много, например FeCOe—ЭсВОз. Дву-
Двувалентное железо замещается трех-
трехвалентным скандием, четырехвалент-
четырехвалентный углерод — трехвалентным бором.
Замещающие друг друга ионы имеют
близкие размеры, и это оказывается
достаточным для образования сме-
смешанных кристаллов.
В. И. Вернадский задолго до нача-
начала рентгеноструктурных исследова-
исследований кристаллов высказал идею заме-
замещения трехвалентным алюминием
четырехвалентного кремния в сили-
силикатах, что впоследствии блестяще
оправдалось на опыте.
В полевых шпатах Na+ и Si4"*" за-
замещаются на Са2+ и А13+ с образова-
образованием непрерывного ряда твердых
растворов NaAlSieOs—CaAl2Si2O8
(альбит—анортит). В разобранных
примерах сумма валентностей заме-
замещающих элементов одинакова. Это
не обязательно. Изоморфные смеси
229
образуют, например, СаТЮ3 с KMgFs,
BaSO4 с KBF4 и др.
Во всех этих случаях изоморфизм
обусловлен близостью размеров заме-
замещающих друг друга компонентов.
Только одинаковой «потребностью
в объеме» (Гольдшмидт) объясняет-
объясняется также замещение некоторых эле-
элементов друг другом в природных ми-
минералах; способные замещать друг
друга элементы располагаются в пе-
периодической системе Менделеева по
диагонали (закон диагональных ря-
рядов А. Е. Ферсмана). Так, литиевые
минералы часто содержат примесь
магния, магниевые — скандия, каль-
кальциевые — иттрия и т. д.
Существенный шаг в вопросе изу-
изучения гетеровалентного изоморфизма
был сделан В. Г. Хлопиным и
Б. А. Никитиным. Им удалось обна-
обнаружить у таких систем новое явле-
явление — нижний порог смешиваемости,
сущность которого заключается в том,
что при образовании смешанных кри-
кристаллов в системах КСЮ4 и BaSO4
или KBF4 и SrSO4 наблюдается раз-
разрыв смешиваемости. Если концент-
концентрация одного из компонентов стано-
становится очень малой, то смешанные
кристаллы не образуются — явление,
которое никогда не наблюдается при
образовании истинных изовалентных
смешанных кристаллов. Объяснить
подобное явление можно тем, что
такие пары изоструктурных веществ
дают «смешанные кристаллы», в ко-
которых не происходит замещения атом
за атом, а один иэ компонентов
«вкраплен» в кристаллы другого,
образуя нечто вроде «коллоид-
«коллоидного» твердого раствора, а не истин-
истинного.
§10. Изоморфизм
о заполнением пространства
Выше мы говорили, что анионы в
ионных кристаллах обычно занимают
места шаров при плотнейшей их
укладке. Однако для таких структур,
как NaCl, формально можно считать,
230
что эти места заняты с равным пра-
правом либо ионами натрия, либо иона-
ионами хлора.
Вопрос о том, какой из компонен-
компонентов (анионы или катионы) следует
считать лежащим на местах центров
шаров плотнейшей упаковки, теряет
смысл, когда размеры катионов и
анионов близки друг к другу. Обычно
анионы больше катионов, и тогда ло-
логичнее считать упаковку анионов с
заполнением пустот в ней катиона-
катионами. Но в таких соединениях, как
KF(K+=1,33 и F-=l,33), кристал-
кристаллизующихся в структурном типе
NaCl, этот вопрос уже несуществен.
В редких случаях, когда катион пре-
превышает по размеру анион, например
CsF(Cs+=l,65), можно говорить о
катионной плотнейшей упаковке.
Аналогичный случай имеет место
в структурном типе CaF2. Некоторые
представители этого структурного
типа имеют обратное отношение
га:гх, например BaF2(Ba2+=1,38).
Поэтому есть основание считать, что
в структуре CaF2 места шаров плот-
плотнейшей упаковки занимают катионы
кальция, а анионы фтора располага-
располагаются в тетраэдрических пустотах.
Так как на п шаров приходится га
октаэдрических пустот и 2п тетраэд-
тетраэдрических, то в структуре CaF2 заня-
заняты все тетраэдрические пустоты,
а октаэдрические остаются свободны-
свободными. Этим объясняется странный, на
первый взгляд, факт образования
изоморфных смесей между CaF2 и
YF3. Ионы иттрия весьма близки по
размерам к ионам кальция (Y3+=
= 0,97; Са2+=1,04), поэтому в сме-
смешанных кристаллах ионы обоих эле-
элементов могут замещать друг друга.
При распределении ионов YF3 в
структуре CaF2 иттрий замещает по-
положение ионов кальция; две трети
ионов фтора заполняют все тетраэд-
тетраэдрические пустоты, а одна треть их
располагается в оставшихся до этого
пустыми октаэдрических пустотах.
Этот случай изоморфизма называется
изоморфизмом с заполнением прост-
пространства.
§11. Твердые растворы
второго рода.
Структуры внедрения
Твердые растворы замещения иначе
называются твердыми растворами
первого рода. В отличие от них, фазы
переменного состава,, в которых ато-
атомы одного элемента не заменяют в
структуре атомы второго, а распола-
располагаются в промежутках между ними,
называются твердыми растворами
внедрения, или твердыми растворами
второго рода. Этот тип твердых рас-
растворов встречается в тех случаях,
когда размеры атомов обоих компо-
компонентов резко отличаются друг от
друга. Он особенно характерен для
систем, в которых один компонент
является металлом, а второй — не-
неметаллом, причем размер атома не-
неметалла значительно меньше разме-
размера атома металла. Наименьшие ато-
атомы будут у следующих элементов:
Н@,46), N@,71) и С @,77). Они
часто образуют с металлами твердые
растворы второго рода, носящие со-
соответственно названия гидридов,
нитридов и карбидов. Эти вещества
играют очень важную роль в техни-
технике, являясь основой промышленно-
промышленности тугоплавких сплавов.
Типичным твердым раствором вне-
внедрения является сталь — раствор уг-
углерода в железе. Твердый раствор
углерода в У"м°ДиФикаЦии железа,
имеющей структуру плотнейшей
упаковки, называется а у с т е н и-
том, Температура превращения
¦р-модификации железа в у-шо]щ$ж-
кацию равна 920° С. Однако аустенит
претерпевает превращение при го-
гораздо более низкой температуре.
А при закалке или при добавлении
третьего компонента (легированные
стали) можно вовсе избежать пре-
превращения аустенита. При медленном
охлаждении до 700° С он превраща-
превращается в перлит — механическую
смесь феррита и цементита. Фер-
Феррит есть твердый раствор внедрения
углерода в структуру а-, р-железа.
Максимальное содержание углерода
в нем 0,06%. Избыточное количест-
количество углерода образует с железом оп-
определенное химическое соединение —
цементит^ состава FeeC
Если задержать превращение аус-
аустенита до 150° G, то при этой темпе-
температуре превращение пойдет иным пу-
путем. В данном случае образуется
твердая сталь, которая называется
мартенситом. Мартенсит есть
пересыщенный твердый раствор уг-
углерода в a-Fe. Эта фаза может со-
содержать до 1,6% углерода. Она ха-
характеризуется тетрагональной ре-
решеткой с отношением осей с/я = 1,07.
Взятый пример характеризуется
небольшой растворимостью неметал-
неметаллического компонента в металле.
Есть однако много примеров, когда
растворенные атомы занимают все
пустоты какого-либо типа в упаков-
упаковке (часто плотнейшей) металличе-
металлических атомов. Ясно, что в этом слу-
случав отношение компонентов оказыва-
оказывается простым стехиометрическим, ж
такие фазы по своей структуре уже
не отличаются от нормальных хими-
химических соединений. Они часто назы-
называются структурами внедрения. При-
Примером структур внедрения может
служить ScN и многие другие. У на-
названного вещества атомы металла
располагаются по точкам плотней-
плотнейшей кубической упаковки, а атомы
неметалла занимают все октаздриче-
ские пустоты. В результате образу-
образуется структурный тип NaCl.
Под влиянием растворения неме-
неметаллического компонента металл в
структуре внедрения может иметь
структуру, не свойственную ему в
чистом виде. Так, например, метал-
металлический тантал кристаллизуется в
центрированной кубической упаков-
упаковке, а в структуре внедрения ТаС ато-
атомы тантала располагаются по точ-
точкам кубической плотнейшей удаков-
ки. Это явление, следовательно, сход-
сходно с явлением своеобразного поли-
полиморфизма.
Иногда пустоты между металличе-
металлическими атомами занимают двухатом-
двухатомные «молекулы» неметалла Н2 или
231
С2. В этих случаях структура внедре-
внедрения, очевидно, в пределе оказывает-
оказывается отвечающей составу МеХг. При-
Примером могут служить ЬаСг, ТЬСг,
ZrKb и др. Конечно, структура внед-
внедрения того же состава может полу-
получиться и при заполнении всех тет-
раэдрических пустот в плотнейшей
упаковке, но тип структуры будет
уже иной (CaF2, а не NaCl).
Твердые растворы внедрения
встречаются не только у металлов.
Хорошим примером для неорганиче-
неорганических соединений является раствор
натрия в окиси вольфрама, так назы-
называемые натрий-вольфрамовые брон-
бронзы. Структура WO3 геометрически
сходна со структурой перовскита
СаТЮз. Атомы кислорода располага-
располагаются у обоих веществ одинаково, а
атомы вольфрама распределены по
местам, занятым в структуре перов-
перовскита атомами титана. Места же
кальция в структуре СаТЮ3 остают-
остаются в случае структуры WO3 свобод-
свободными. В эти места и внедряются ато-
атомы металлического натрия. В пре-
предельном случае, при заполнении всех
пустых мест, получается стехиомет-
рическое соотношение, и состав твер-
твердого раствора становится точно
NaWO3. В этом случав его можно
считать определенным химическим
соединением, таким же, как СаТЮз.
Очевидно, что фазы переменного со-
состава такого типа обусловлены пере-
переменной валентностью, в данном слу-
случае вольфрама. При замещении ато-
атомами натрия известного процента
пустот часть атомов вольфрама из
шестивалентного состояния перехо-
переходит в пятивалентное. Таким образом,
твердые растворы внедрения делают
возможным непрерывный переход от
одного структурного типа — WO3 — к
другому — СаТЮ3, от одного опреде-
определенного химического соединения к
другому.
Это — новое представление в хи-
химии, сущность которого стала понят-
понятной в результате развития кристал-
кристаллохимии. В данном явлении можно
видеть пример диалектической свя-
связи — непрерывного перехода между
двумя узлами (определенными хи-
химическими соединениями) на непре-
непрерывной кривой составов.
Заканчивая настоящий параграф,
можно еще раз указать на различие
между твердыми растворами первого
и второго рода. «В твердых растворах
первого рода понятие растворителя
и растворенного вещества теряет
смысл, в твердых растворах второго
рода эти понятия сохраняют свое
значение. Парадоксальным примером
могут служить структуры цеолитов,
содержащие, как известно, перемен-
переменное количество воды. Здесь раствори-
растворителем будет сам цеолит, а растворен-
растворенным веществом — вода. Этот тип
твердых растворов внедрения полу-
получается в том случае, когда пустоты в
структуре располагаются «столба-
«столбами», образуя «трубы», в которые и
могут попадать молекулы воды. Вто-
Вторым существенным отличием будет
то, что для твердых растворов заме-
замещения необходимо сходство типов
химической связи у замещающих
компонентов. В твердых растворах
внедрения тип связи обоих компонен-
компонентов может быть совершенно иным.
§12. Твердые растворы вычитания.
Дефекты струвтуры
Известны многочисленные примеры
веществ, у которых содержание од-
одного из компонентов отклоняется от
стехиометрического состава. Так,
давно уже было установлено, что
пирротин всегда содержит избыточ-
избыточное количество серы по сравнению
со стехиометрическим составом FeS.
Поэтому химиками предлагались
многочисленные варианты формул
типа Fe6S7 или FenSn+i, имеющих
целью подчеркнуть наблюдающиеся
аномалии состава. Однако химиче-
химических методов было недостаточно,
чтобы объяснить причину этого явле-
явления или хотя бы отдать себе отчет
в этом вопросе со структурной точ-
точки зрения. Предположение о том,
232
что в таких структурах имеет место
внедрение избыточного количества
атомов серы в промежутки между
другими атомами, было опровергнуто
непосредственным экспериментом.
Методом реятгеноструктурного
анализа было показано, что структу-
структура пирротина (тип NiAs) не представ-
представляет собой твердого раствора внедре-
внедрения. Атомы серы располагаются по
местам шаров гексагональной плот-
нейшей упаковки независимо от того,
имеется ли их равное количество с
числом атомов железа или избыточ-
избыточное. В случае избытка атомов серы
появляется некоторое количество
свободных октаэдрических пустот,
которые в нормальных структурах
все заняты атомами металла. Следо-
Следовательно, если в соединении увеличи-
увеличивается избыточное содержание серы,
то в структуре такого соединения
происходит увеличение числа неза-
незанятых октаэдрических пустот. Таким
образом, формулу пирротина следует
скорее писать не FenSn+i, a Fen-iSn
или Fei-sS. Последняя гораздо луч-
лучше передает сущность аномалии
состава *.
Фазы переменного состава такого
типа часто называются твердыми
растворами вычитания, а их структу-
структуры — дефектными, или дефицитны-
дефицитными. Появление их, очевидно, так же
как и в некоторых случаях твердых
растворов внедрения, связано с пе-
переменной валентностью одного из
элементов. Так, в пирротине при из-
избыточном содержании серы против
нормального стехиометрического со-
соотношения 1:1 часть атомов железа
из двухвалентного состояния перехо-
переходит в трехвалентное.
Большую работу по изучению твер-
твердых растворов вычитания и дефект-
дефектных структур проделал Е. С. Мака-
Макаров. Результаты его работы графи-
* Если характер дефектов в структуре не
установлен, то могут быть употреблены
оба способа написания формулы, так
как они в этом случае выражают толь-
только пределы области гомогенности фазы
переменного состава.
• Nt
Te,Sb,Sn,ln,aw.%
Рис. 252. Области существования твердых
фаз переменного состава в системах:
Ni — In, Ni — Sn, Ni — Sb и Ni —Те
чески представлены на рис. 252.
Рисунок представляет собой изобра-
изображение участков диаграмм состояний
четырех систем. В верхней части
рисунка изображены три структур-
структурных типа Ni2ln, NiAs(NiSb) и
CdJ2(NiTe2). Пунктирными верти-
вертикальными прямыми указаны составы,
отвечающие этим структурам. За-
Заштрихованные прямоугольники пока-
показывают области существования фаз
переменного состава в системах
Ni — In, Ni — Sn, Ni — Sb и Ni — Те.
Так, например, фаза NiSb может су-
существовать в области составов, на не-
несколько процентов превышающих
состав 1: 1 как в ту, так и в другую
сторону. Слева от пунктирной пря-
прямой фаза содержит избыток никеля
и является структурой внедрения.
Избыточное количество атомов ни-
никеля располагается в промежутках
между атомами сурьмы. Заполнение
всех пустот этого типа привело бы
к появлению нового структурного
типа Ni2ln. Однако в системе Ni — Sb
этот структурный тип не реализует-
реализуется. Вправо от пунктирной прямой в
системе Ni — Sb образуется твердый
233
раствор вычитания, который в пре-
пределе привел бы к структурному типу
CdJ2 (верхний правый рисунок). Он
действительно реализуется в системе
Ni — Те. Между двумя структурными
типами NiAs(NiTe) и CdJ2(NiTe2)
осуществляется непрерывный пе-
переход.
Твердая фаза переменного состава
в системе Ni—Sb показывает, что
между твердыми растворами внедре-
внедрения и вычитания нет принципиаль-
принципиальной разницы, — это две стороны од-
одного и того же явления.
Некоторые фазы (например, в си-
системе Ni—Sn, а также FeS и Сигв)
могут существовать только в дефект-
дефектном состоянии. Область составов фазы
в системе Ni—Sn не пересекает ни
одной пунктирной прямой, отвечаю-
отвечающей простому рациональному соста-
составу и чистому, не дефектному, струк-
структурному типу.
§ 13. Структуры
с дробным количеством атомов
в элементарной ячейке
Имеется много веществ, которые мо-
могут существовать только в дефект-
дефектных структурах, например у-А12Оз.
Изучение этого соединения показало,
что его структура весьма близка к
структуре шпинели MgAl4O3.
Элементарная ячейка шпинели
содержит 8 атомов Mg, 16 А1 и 32
атома О. В структуре y-AUOs в эле-
элементарной ячейке тоже 32 атома
кислорода, и они занимают те же
места, но в этой же ячейке только
217з атомов алюминия. Последние
статистически занимают места маг-
магния и алюминия в структуре шпине-
шпинели, причем в среднем 22/з этих мест
на одну элементарную ячейку оста-
остаются свободными. Изучение дефект-
дефектной структуры у-АЬОз объяснило
давно известный факт образования
непрерывных твердых растворов
шпинели с окисью алюминия.
Те же взаимоотношения имеют
место в структурах окислов железа:
ТАБЛИЦА 30
Структурные типы шпинелей и
Структурный
тип
Нормальный
Дефектный
Нормальный
Дефектный
Химичес-
Химический
состав
Fe,O,
Fe2Os
CesS4
Ce2S3
Состав,
приведен-
приведенный к
одинако-
одинаковому ко-
количеству
кислорода
(или серы)
Fee0i2
Fe8Oi2
CeeSi2
CegSi2
Число
атомов в
элементар-
элементарной
ячейке
Fe24O«
Fe2iyO,,
CeiaSij
Ce101/iSie
Fe3O4 имеет структуру шпинели,
Y-FeaO» — дефектную структуру
Y-A12OS. Вторым примером дефект-
дефектных структур такого же типа являет-
является структура CesS4. Сопоставление
структур Y-F2O3, Ce3S4 и шпине-
шпинели приведено в табл. 30.
В дефектном структурном типе
СегЭз кристаллизуются многие суль-
сульфиды актиноидов, например АЭ
PU2S3, А
§ 14. Внутренние
твердые растворы.
Автонаоиорфиые вещества
Совсем не обязательно, чтобы харак-
характер дефекта в дефектных структурах
сводился к наличию в структурах пу-
пустот, не занятых атомами. Дефекты
могут быть и иного характера. Луч-
Лучше всего это можно продемонстриро-
продемонстрировать на структурах различных шпи-
шпинелей.
Некоторое своеобразие структуры
шпинели заключается в том, что
больший по объему ион магния @,74)
располагается в меньшей тетраэдри-
ческой пустоте, а меньший ион алю-
алюминия @,57) — в больших октаэдри-
ческих пустотах. Как показали рабо-
работы Барса и Позняка A932 г.), в гал-
лиевой шпинели MgGaaO4 (raa=0,62)
половина атомов галлия располага-
234
«тся в тетраэдрических пустотах, за-
заменяя, следовательно, места, занятые
в обычной шпинели атомами магния.
Вторая половина атомов галлия и
все атомы магния занимают октаэдри-
ческие пустоты, располагаясь стати-
статистически, по закону случая, т. е. так
же, как располагаются замещающие
друг друга компоненты в твердых
растворах первого рода. Поэтому та-
такой случай дефектных структур удоб-
удобно называть внутренним твердым
раствором, или автоивоморфными ве-
веществами. По химическому составу
такие фазы будут соответствовать
истинному химическому соединению,
но по своему строению будут ана-
аналогичны твердым растворам заме-
замещения.
Автором совместно с М. А. Порай-
Кошицем при изучении структур ком-
комплексных соединений было обнару-
обнаружено, что структура K^tPtCUB^]-
транс представляет собой внутрен-
внутренний твердый раствор. Ось комплекса
Br — Pt — Br располагается в струк-
структуре по закону случая во всех трех
координатных направлениях, обусло-
обусловливая кубическую симметрию кри-
кристаллов.
Соединение LiFeCb имеет структу-
структуру NaCl. Все катионы вследствие бли-
близости размеров (Li+=0,68 и Fe3+=
= 0,67) занимают статистически ме-
места атомов натрия в структуре пова-
поваренной соли.
Оба типа дефектов — с незанятой
частью пустот и со статистическим
распределением нескольких элемен-
элементов по одной правильной системе то-
точек — могут встретиться в структуре
одновременно. Так, например,
Ag2HgJ4 кристаллизуется в струк-
структурном типе, близком к ZnS, с про-
пропуском 25% октаэдрических пустот
и при статистическом заполнении
атомами серебра и ртути остальных
75%.
Дефектные структуры и внутрен-
внутренние твердые растворы могут, конечно,
образовывать с другими соединения-
соединениями фазы переменного состава, в част-
частности, твердые растворы замещения,
например, непрерывные твердые ра-
растворы АЬОз со шпинелью или твер-
твердые растворы LiFeO2 и Li2Ti03 с MgO.
ГЛАВА XIV
КЛАССИФИКАЦИЯ СТРУКТУРНЫХ ТИПОВ
§1. Предварительные замечания
о классификации
структурных типов
Всякая классификация, и в частности
классификация структурных типов,
должна обязательно учитывать, что
отдельные индивидуумы являются
лишь «узлами», часто связанными
друг с другом непрерывными перехо-
переходами. Проведение между ними опре-
определенных границ вызывает большие
затруднения, ибо резкая разница в
одних признаках может оказаться со-
совсем нерезкой или даже условной и
случайной — в других. Следователь-
Следовательно, в самой природе классифициру-
классифицируемых объектов лежит отсутствие рез-
резких границ, а между тем всякая клас-
классификация обязательно включает в
себя момент резкого разграничения.
Выше мы видели, как отдельные объ-
объемы, отдельные структурные типы
непрерывно переходят один в другой.
Несколько примеров такого рода бы-
было описано нами в параграфах, посвя-
посвященных твердым растворам внедре-
внедрения, вычитания и дефектным струк-
структурам. Тем более нет резкой разницы
между разными группами структур-
структурных типов. А в нашу задачу как раз
и входит разделение структурных ти-
типов на определенные группы — кате-
категории, т. е. проведение резких границ
там, где их нет' в действительности.
Вынужденно поступая таким образом,
мы всякий раз будем указывать на те
непрерывные переходы, которые свя-
связывают выделенную нами (по тем или
иным признакам) группу с соседни-
соседними. Это, как нам кажется, должно
сгладить известный формализм, при-
присущий всякой классификации, и на-
нашей, в частности.
По какому же признаку следует
гругшировать структурные типы —
типы кристаллических соединений?
Кристаллохимия внесла в химию точ-
точные знания межатомных рас-
расстояний. Этот признак может быть
положен в основу классификации.
Пользуясь этим признаком, можно
прежде всего разделить все структур-
структурные типы на 5 категорий (отрядов):
1) координационные; 2) островные;
3) цепочечные; 4) слоистые; 5) кар-
каркасные.
Чтобы отнести ту или иную струк-
структуру к одной из пяти категорий, мож-
можно предложить следующий наглядный
способ. Представим себе, что имеется
модель структуры, сделанная с соблю-
соблюдением масштаба в межатомных рас-
расстояниях. Тогда, мысленно взяв цир-
циркулем расстояние, равное наименьше-
наименьшему межатомному расстоянию в струк-
структуре, мы станем поочередно переме-
перемещать ножки циркуля с одного «атома»
на другой, передвигаясь по всем воз-
возможным направлениям в структуре.
При этом могут оказаться 5 различ-
различных вариантов.
1. Действуя таким образом, мы
сможем «обойти» все атомы структу-
структуры (например, в структуре а-, |3- или
y-Fe или в структуре NaCl и т. п.).
Эти структуры мы будем называть
координационными, так как они будут
характеризоваться большими коорди-
координационными числами с правильными
координационными многогранниками.
2. Противоположный результат по-
236
лучится в том случае, если мы сможем
«обойти» ограниченное число атомов
в структуре, например только атомы
одной молекулы. Этот случай может
быть иллюстрирован на структуре се-
серы. Кристаллическая ромбическая се-
сера построена из восьмиатомных моле-
молекул. Кратчайшее расстояние между
атомами в молекуле серы равно 2,ЮА,
а наименьшее расстояние между ато-
атомами из разных молекул равно 3,30.
Следовательно, поместив одну ножку
циркуля в «атом» серы и передвигая
его от атома к атому, циркуль все
время будет «кружиться» в пределах
одной молекулы и не сможет обойти
всех атомов структуры. Эти восемь
атомов образуют замкнутую моле-
молекулу — «остров», поэтому структуры
такого типа мы будем называть
островными структурами.
3. Может оказаться, что, действуя
таким образом, мы сможем передви-
передвигать циркуль до бесконечности в од-
одном направлении (например, структу-
структура селена) или же в одной плоскости
(например, в структуре графита);
Структура селена построена из беско-
бесконечных цепей с кратчайшим межатом-
межатомным расстоянием в цепи — 2,34 и
кратчайшим межатомным расстояни-
расстоянием между цепями, равным 3,53 А. В
структуре графита кратчайшее меж-
межатомное расстояние в слое равно 1,42,
а между слоями — 3,39. Сказанного
достаточно, чтобы понять основную
характерную черту цепочечных и сло-
слоистых структур.
4. Более сложными будут каркас-
каркасные структуры. Каркасная структура
будет у NaWO3. Взяв циркулем рас-
расстояние W—О, мы сможем обойти все
«атомы» вольфрама и кислорода во
всей структуре, в трех измерениях, но
при этом ножка циркуля ни разу не
попадет в «атом» натрия. Мотив
структуры [WOe]"" будет трехмер-
трехмерным. Если бы мы взяли соединение
не с пятивалентным вольфрамом
NaWOe, а с шестивалентным — \?0з,
то у последнего вещества структура
оказалась бы не каркасной, а коорди-
координационной. Выше мы говорили, что
в натрий-вольфрамовых бронзах со-
содержание натрия по отношению к
содержанию вольфрама в атомных
процентах может меняться от 0 до
100. Следовательно, в этом случав
имеется постепенный переход от ко-
координационной структуры к каркас-
каркасной.
§2. Грунпы структурных тинов
с нейтральными и заряженными
структурными мотивами
У гомодесмических веществ из всех
пяти категорий структурных типов
могут быть только координационные
структуры. Все остальные будут ха-
характерны для гетеродесмических. Ха-
Характер связи для последних четыре!
категорий заведомо должен быть иным
внутри мотива структуры и между
мотивами. Напомним, что при описа-
описании типов химической связи указыва-
указывалось, что ковалентную связь по цело-
целому ряду признаков, как, например, по
ее направленности и насыщенности,
можно противопоставить трем осталь-
остальным: металлической, ионной и Ван-
дер-Ваальсовой. При классификации
структурных типов вновь встречаемся
с этим противопоставлением. В самом
структурном мотиве наиболее харак-
характерным типом химической связи бу-
будет ковалентная, часто дающая посте-
постепенные переходы к ионной. Между
структурными мотивами будут дейм-
вовать остаточная, металлическая или
ионная связи, причем из этих трех
наиболее характерной будет остаточ-
остаточная. В тех случаях, когда между мо-
мотивами осуществляется остаточная
или металлическая связь (например,
в структуре графита), между струк-
структурными мотивами нет дополнитель-
дополнительных атомов. Эти мотивы уже сами по
себе являются валентно-насыщенны-
валентно-насыщенными и электростатически нейтральны-
нейтральными. Их поэтому условно можно на-
назвать «молекулярными» (в том
смысле, что они не будут нести за-
заряда, т. е. будут нейтральными, как
молекулы). Если же связь между
мотивами будет ионной, то в проме-
237
жутках между ними могут оказать-
оказаться дополнительные атомы (ионы),
усложняющие структуры. В этом слу-
случае островные, цепочечные, слоистые
или каркасные мотивы не будут «мо-
«молекулярными». Они окажутся заря-
заряженными, т. е. будут ионами.
В соответствии с этим последние
четыре категории структурных типов
можно подразделить на две группы
каждую: одна группа объединяет
структурные типы, у которых мотивы
являются валентно-насыщенными —
нейтральными (мы условились их на-
называть «молекулярными»); вторая —
структурные типы, мотивы которых
представляют сложные ионы («ион-
(«ионные»). В островных структурах фор-
форма островов — молекул и ионов — мо-
может быть весьма различной, и это
может быть положено в основу даль-
дальнейшей классификации. В частности,
они могут быть с центральным атомом
или без него — кольчатые молекулы
или ионы. Кроме того, они могут быть
изометричными, вытянутыми в одном
или в двух направлениях, плоскими
или гофрированными и т. д.
Примером цепочечных «молекуляр-
«молекулярных» структур является селен, при-
примером цепочечных «ионных» — амфи-
амфиболы и пироксены (см. гл. XVIII, § 6).
«Молекулярную» слоистую структуру
будут иметь графит и СсИг. Характер
связи между слоями у графита — ме-
металлический, у Gd J2 — остаточный
(дипольный). «Ионной» слоистой
структурой обладает, например, слю-
слюда KAl2[Si3AlO10] (ОН) 2.
Для каркасных структур более ха-
характерным будет ионное взаимодей-
взаимодействие между самим каркасом и атома-
атомами, находящимися вне каркаса, так
как нейтральный «молекулярный»
каркас ничем не отличается от коорди-
координационных структур. Хорошим при-
примером ионных каркасных структур
может служить приведенная выше
структура NaWO3 или же структура
полевых шпатов. Структура ортокла-
ортоклаза K[AlSisO8] похожа на структуру
одной из модификаций SiO2: располо-
расположение атомов в каркасном мотиве
[AlSi3O8] — очень сходно с располо-
расположением их в структуре SiO2, так как
в полевых шпатах алюминий занима-
занимает те же места в структуре, что и
кремний. Но замена lU атомов крем-
кремния на алюминий придает каркасу
[AlSi3O8]~ заряд, равный единице.
Есть однако каркасные структуры,,
по своей природе близкие к «моле-
«молекулярным». Примером последних
могут служить цеолиты — анальцим
Na[AlSi2O6] ¦ Н2О или десмин
Ga[Al2Si7Oi8]-7H2O. Структура их та-
такова, что при трехмерном располо-
расположении атомов, аналогичном каркасу
полевых шпатов, включая и внеш-
внешние ионы Na или Са, в ней остается
еще много пустых промежутков
(труб). В них могут попадать поляр-
полярные молекулы (обычно воды), кото-
которые в переменном количестве всегда
присутствуют в структурах цеолитов.
Вещества, кристаллизующиеся в
структурах такого рода, имеют не-
небольшую плотность, так как они не
подчиняются законам плотнейшей
упаковки.
Ионные каркасные структуры, как
и ионные островные, цепочечные и
слоистые, также могут давать посто-
постоянные переходы к координационным
структурам в зависимости от заряда
и размера внешних ионов. Об этом
подробнее будет сказано ниже.
§3. Гранжцы нрнменнмостн
принятой классификации
структурных типов
Принятый нами прием — обход с
помощью циркуля мотива структу-
структуры — строго справедлив только для
простейших веществ, состоящих из од-
одного или двух типов атомов. Как толь-
только переходим к структурам более
сложных соединений, то сразу же
сталкиваемся с неравенством меж-
межатомных расстояний между разными
атомами в мотиве. Этим неравенством
приходится пренебрегать. Так, на-
например, если в кремнекислородном
мотиве часть атомов кремния заме-
замещается на алюминий (Si4+=0,39;
238
Рис. 253. Молекула бензола
Рис. 254. Молекула пиридина
А13+=0,57), как это имеет место в
структурах полевых шпатов, то, хо-
хотя расстояние А1—О будет больше
расстояния Si—О, мы этой разницей
пренебрегаем и считаем алюминий
входящим в алюмокремнекислород-
ный мотив по той причине, что он в
структуре играет одинаковую роль с
кремнием, имеет ту же тетраэдриче-
скую координацию, размеры его и
валентность несильно отличаются от
размеров и валентности кремния. Та-
Таким образом, положив в основу
классификации структурных типов
минимальные расстояния между ато-
атомами, необходимо учитывать при
этом размер атомов (ионов) и их ва-
валентность.
Структуру бензола мы, конечно,
посчитаем молекулярной. Структур-
Структурную единицу будет представлять вся
молекула СбНб (рис. 253), а не группа
СН, хотя расстояние С—Н«1,05, рас-
расстояние С—С л; 1,42. В молекуле пи-
пиридина (рис. 254) расстояние С—N
будет несколько меньше расстояния
С—С, но мы во всех этих и аналогич-
аналогичных им случаях разницей в расстояни-
расстояниях пренебрегаем. Если в молекуле на-
наряду с ординарными связями — С —С—
A,54) присутствуют двойные С = С
A,32) или тройные — С==С— A,20),
то мы считаем одним островом всю
молекулу, т. в. учитываем укороче-
укорочение связи с увеличением кратности
ее. В островных структурах с кова-
лентными связями в молекуле и ос-
остаточными межмолекулярными в
этом отношении недоразумений не
будет, так как межмолекулярные
расстояния нередко чуть ли не вдвое
больше внутримолекулярных.
В ионных соединениях вопрос
сложнее, так как отпадает один кри-
критерий — тип связи, а остальные —
межатомные расстояния и валент-
валентность — могут резко меняться при
замене одного атома на другой.
В этом случае неизвестно, что счи-
считать пределом, где будет проходить
граница. Последнее можно показать
на примере структуры силикатов.
Они изучены наиболее полно. Можно
считать, тоже в какой-то мере услов-
условно, что если разница в валентностях
электроположительных элементов
превосходит единицу, а следова-
следовательно, и расстояние между их ато-
атомами тоже резко отличается (по-
(порядка 0,5А), то эти большие рас-
расстояния уже не следует считать вхо-
входящими в основной мотив структуры.
Так, например, ортоклаз K[AlSi3Os]
можно без натяжки считать принад-
принадлежащим к каркасной ионной струк-
структуре.
Ион калия и аналогичные ему
ионы, не входящие в алюмокремне-
кислородный мотив, мы будем назы-
называть внешними ионами.
Та же картина будет и в слюде.
Ионы калия «цементируют» двух-
двухмерные листы состава [(S13AIO10) •
•А12(ОНJ]. В обоих случаях разница
между валентностями электрополо-
электроположительных атомов была равна двум
или даже трем, поэтому выделение
основного мотива структуры не
239
представляет труда. Но если вместо
калия в структуре будут находиться
двухвалентные металлы, как быть
тогда? Считать ли их ионы внешни-
внешними или нет? Ведь увеличение ва-
валентности меняет характер связи,
делая его все более ковалентным.
Где же проходит граница?
Есть основание считать, что гра-
граница чаще бывает более резкой меж-
между двух- и трехвалентными элемен-
элементами, чем между одно- и двухва-
двухвалентными или трех- и четырехва-
четырехвалентными. Однако и здесь имеется
элемент условности. Например, трех-
трехвалентные редкоземельные элементы
обычно изоморфно замещают кальций
и играют, следовательно, в структу-
структурах одинаковую кристаллохимичес-
кую роль. При разнице валентности
в единицу решающим фактором бу-
будет размер атома (иона). Поэтому
замена во внешней сфере иона
Na+ @,98) большим ионом Са2+ A,04)
еще не меняет категории структур-
структурного типа.
Однако маленький двухвалентный
ион Ве2+ @,34), соизмеримый с ио-
ионом А13+ @,57), уже не может счи-
считаться внешним ионом не только в
алюмосиликатах, но и в чистых сили-
силикатах.
«Бестелесный» протон, образую-
образующий водородную связь, обычно ме-
меняет категорию структурного типа.
Так, например, СаСО3 является ост-
островным структурным типом, ибо
комплексы (СО3J~ цементируются
в структуре ионами Са2+. Структура
ЫаНСОз является цепочечной, ана-
аналогичной MgSiOe, ибо группы СОз
«скреплены» в цепочку водородной
связью. Между этими цепочками
располагаются ионы натрия. Струк-
Структура льда из-за водородных связей
является координационной, а струк-
структура НгЭ — молекулярной, так как
водород с атомами серы водородной
связи не образует. Структура льда
вместе с тем весьма сходна с каркас-
каркасными структурами вследствие нали-
наличия в ней «труб», обусловливающих
низкую плотность льда.
§4. Метод изображения
структурных тниов формулами
Существует символика для краткой
записи структурных типов. Так,
структура NaCl будет записана сле-
следующим образом: [NaCl6/J3°°. Это
значит, что координационное число
в структуре 6, и каждый атом (ион)
С1 окружен шестью атомами (иона-
(ионами) натрия. Значок Зоо показывает,
что структура NaCl координационная,
т. е. этот мотив распространяется
в трех измерениях до бесконеч-
бесконечности.
Две модификации MnS, кристал-
кристаллизующиеся в структурных типах
сфалерита и поваренной соли, полу-
получат соответственно символы
[MnSvJ300 и [MnSvJ300. Структура
CaF2 будет записана в виде
[CaFs/4]300.
Бесконечная кремнекислородная
тетраэдрическая цепочка пироксено-
вого типа состава [SiO3]n получит
символ [SiOVs+V,]00- Первая дробь
показывает, что из четырех кисло-
кислородных атомов (сумма числителей),
окружающих атом кремния, два де-
делятся между двумя атомами крем-
кремния, и два принадлежат только одно-
одному атому кремния. Мотив в виде
сдвоенного тетраэдра, например
Si2O7, получит символ [SiOyJ+3/,]2.
§ 5. Структурные химические
формулы
Структурная химическая формула в
отличив от обычной (брутто-форму-
лы) должна отражать строение хи-
химических веществ. Впервые струк-
структурные формулы были введены в на-
науку А. М. Бутлеровым для органиче-
органических (молекулярных) соединений.
Без них невозможно было бы понять
разницу в строении различных изо-
изомеров, полимеров и вообще веществ
с одинаковым валовым составом.
Идеи Бутлерова в начале XX в. бы-
были распространены на область комп-
комплексных неорганических соединений
240
Вернером и Чугаевым. Так, хлоро-
платинат калия вместо записи в фор-
форме, отвечающей двойной соли 2KG1-
• PtCLi, стали писать K2[PtCl6]. Фи-
Физико-химические исследования под-
подтвердили такую структурную форму-
формулу. В частности, это вещество при
растворении в воде диссоциирует на
три иона: 2К+ и комплексный ион
[РЮ1бр~. Кристаллохимия также
подтверждает правильность указан-
указанной структурной формулы. Кристал-
лохимическая структура хлороплати-
ната калия построена из упакован-
упакованных плотнейшим образом ионов
fPtCl]2-, в промежутках между кото-
которыми располагаются ионы К+. Ко-
Координационное число атомов калия
12, т. е. каждый атом окружен две-
двенадцатью атомами С1.
Полное описание структурного ти-
типа, наподобие того, как это было сде-
сделано в предыдущем параграфе, не
всегда необходимо при химических
исследованиях. В некоторых случаях
это может иметь значение, в других —
является ненужным усложнением
формулы. Однако структурные фор-
формулы должны отражать действитель-
действительное строение соединений и являться
краткой записью, по которой можно
судить о некоторых важных свойст-
свойствах соединений.
Важность правильного написания
структурных формул особенно хоро-
хорошо иллюстрируется на примере сили-
силикатов. Отношение Si к О, рав-
равное 1 : 3, может получаться различ-
различным сочленением кремнекислородных
тетраэдров Саз^зОд], ВезАЫ^бОш]
или Mg2[SiO3b. Бесконечные радика-
радикалы в виде листов состава [Si2Os]
встречаются, например, в тальке
Mg3[Si205]1, @HJ и в слюдах. От-
Отношение Si к О, равное 1:4, вовсе не
указывает на существование изоли-
изолированных кремнекислородных тет-
тетраэдров. Так, например, ортосиликат-
ную формулу диоптаза H2Cu[SiO4]
в результате определения его кри-
кристаллической структуры Н. В. Бело-
Беловым пришлось записывать как мета-
силикатную: Cu6[Si6Oi8]-6Н2О.
Часто близкие по составу вещества
имеют весьма различные свойства,
что является следствием различия
их кристаллических структур. Так,
например, Na2CO3 и Na2SiO3 различа-
различаются тем, что у первой соли имеется
островной радикал СОз2", а у вто-
второй — радикал SiO32~ имеет форму
цепочки бесконечной протяженности.
Эти особенности структур подчерки-
подчеркиваются соответствующим написани-
написанием структурных формул: Na^COs] и
Na2[SiO3]1". Весьма сходные по
составу силикаты NaAlSi2O6 и
KAlSi2O6 имеют разные структуры и
совершенно различные физические
свойства. Их структурные формулы
NaAl[SiO3]2lo° и K[AlSi2O6]3°° объяс-
объясняют это различие.
Даже в области органической хи-
химии имеются случаи, когда правиль-
правильная формула соединения была опре-
определена только в результате рентгено-
структурного исследования. Так была
установлена формула адамантана
СюШб, молекула которого представ-
представляет собой тетраэдр, в вершинах ко-
которого располагаются метиловые
группы СН, а в серединах ребер —
метиленовые группы СНг (см.
стр.361).
Очень часто кристаллохимия, под-
подтверждая структурные формулы ор-
органических соединений, вносит суще-
существенные дополнения. Так, например,
было доказано, что структура поли-
тена построена из бесконечных цепо-
цепочек, составленных из СНг-звеньев.
Структурную формулу этого соедине-
соединения следует писать [СН2]1со.
Во многих учебниках органической
химии для нафталина давались две
равноценные формулы
Правда, в последние годы на основа-
основании косвенных соображений хими-
химики-органики пришли к симметричной
структурной формуле. Между тем
точные рентгеноструктурные опреде-
определения межатомных расстояний в мо-
16 Кристаллохимия
241
лекуле нафталина (см. рис. 347,
стр. 362) позволяют непосредственно
выбрать первую формулу и отбро-
отбросить вторую как неверную, как не
отвечащую характеру распределе-
распределения двойных и простых связей и как
не имеющую центра симметрии, ко-
который в действительности в молеку-
молекуле есть. Однозначность определения
структурных формул органических
соединений по кристаллохимическим
данным в ближайшее время, несом-
несомненно, получит признание. Даже
простое указание межатомных рас-
расстояний в молекулах органических
соединений позволяет учитывать
взаимное влияние атомов и, следова-
следовательно, правильнее судить о физиче-
физических свойствах этих веществ и глуб-
глубже понимать их реакционную способ-
способность.
В области комплексных соединений
имеются случаи просто неверного
определения строения веществ, сде-
сделанных на основании химических
данных. Так, например, считавшееся
yuc-изомером K[Co(NH3J(NO2L]
оказалось на основании прямого
рентгеноструктурного определения
гракс-изомером. Соединение, которо-
которому химики приписывали формулу
К2 [RuCl5OH], оказалось двухъядер-
ным комплексным соединением
K4[Cl5RuORuCl5]H2O.
ГЛАВА XV
ЗАВИСИМОСТЬ ФИЗИКО-ХИМИЧЕСКИХ СВОЙСТВ
ТВЕРДЫХ ВЕЩЕСТВ ОТ СТРОЕНИЯ КРИСТАЛЛОВ
§ 1. Зависимость
фиаино-химичвских свойств
твердых веществ
от тина химической связи
в кристаллах
Ряд физических свойств, в том числе
термические, механические и элек-
электрические (например, теплопровод-
теплопроводность и электропроводность в метал-
металлах), существенно зависит от типа
химической связи.
О прочности кристаллов проще
всего можно судить по их механиче-
механическим и термическим свойствам. Чем
прочнее кристалл, тем больше его
твердость и тем выше его температу-
температура плавления. Если изучать измене-
изменение твердости с изменением состава
в ряду однотипных веществ и сопо-
сопоставлять полученные данные с соот-
соответствующими значениями для тем-
температур плавления, то заметим «па-
«параллелизм» в изменении этих свойств.
По этой причине некоторые из меха-
механических и термических свойств
удобно рассматривать одновременно.
В гетеродесмических соединениях
некоторые свойства, например, ме-
механическая прочность органических
соединений, зависят только от одного
(слабейшего) типа связи. Вторым ти-
типом связи — гомеополярным — в
этом случае можно пренебречь. Оп-
Оптические свойства органических кри-
кристаллов, напротив, будут зависеть
от внутримолекулярных сил, а Ван-
дер-Ваальсовы силы связи при изу-
изучении оптических свойств можно не
принимать во внимание.
Физические свойства веществ мож-
можно подразделить на две группы:
структурно чувствительные и струк-
структурно нечувствительные свойства.
Первые зависят от атомной структу-
структуры кристаллов, вторые — главным
образом от электронного строения
атома и типа химической связи. При-
Примером первых могут служить меха-
механические свойства, примером вто-
вторых — электрические и оптические.
Так, хорошая электропроводность
металлов, обусловленная наличием
свободных электронов, будет наблю-
наблюдаться не только в кристаллах, но и
в расплавленных металлах.
Ионный характер связи проявляет-
проявляется, в частности, в том, что многие
соли, например галоидные соли ще-
щелочных металлов, растворяются в по-
полярных растворителях, диссоциируя
на ионы. Однако факт отсутствия
растворимости не может еще служить
доказательством наличия у соедине-
соединения неполярной связи. Так, энергия
связи, например, у окислов настолько
больше энергии связи щелочных га-
логенидов, что диэлектрическая по-
постоянная воды уже недостаточна для
отрыва ионов от кристалла.
Кроме того, некоторые соединения,
преимущественно с гомеополярным
типом связи, под влиянием большой
диэлектрической постоянной поляр-
полярного растворителя могут в растворе
диссоциировать на ионы, хотя в кри-
кристаллическом состоянии иониыии
соединениями они могут и не быть
(например НС1, НВг).
16*
243
§ 2. Электрические свойства
Наличие в металлах свободных элек-
электронов обусловливает их специфи-
специфические физические свойства: электро-
электропроводность, теплопроводность, не-
непрозрачность и блеск (отражательная
способность). Электроны, свободно
передвигаясь в металле, не могут
выйти наружу из-за потенциального
барьера. Для преодоления электроном
этого барьера необходимо затратить
работу. Если при этом затрачивается
лучистая энергия, то эффект отрыва
электрона вызывает так называемый
фотоэлектрический эффект. Анало-
Аналогичный эффект наблюдается и у го-
меополярных соединений. Вырван-
Вырванный из молекулярной орбиты элект-
электрон, оставаясь внутри кристалла, обу-
обусловливает у последнего металличе-
металлическую проводимость (внутренний фо-
фотоэлектрический эффект). В нор-
нормальных же условиях (без облуче-
облучения) такие соединения не являются
проводниками электрического тока
ни в кристаллическом, ни в расплав-
расплавленном состояниях.
В ионных кристаллах также наб-
наблюдается внутренний фотоэлектриче-
фотоэлектрический эффект, причем энергия отрыва
электрона равна ионизационному
потенциалу. Без облучения ионные
кристаллы, так же как и гомеополяр-
ные, электрического тока не прово-
проводят. Но, в отличие от гомеополярных,
ионные вещества проводят электри-
электрический ток в расплаве, где проводи-
проводимость обусловлена передвижением
самих ионов.
Совсем особые свойства имеют ве-
вещества с дефектными структурами.
Для примера рассмотрим электро-
электропроводность AgJ. Это вещество изве-
известно в трех модификациях. Две
низкотемпературные модификации
принадлежат к структурным типам
сфалерита и вюртцита. Высокотемпе-
Высокотемпературная модификация, устойчивая
от 145,6° С до температуры плавле-
плавления E52° С), имеет дефектную
структуру. Атомы (ионы) иода рас-
располагаются по узлам центрированной
кубической упаковки, а атомы (ио-
(ионы) серебра располагаются в пусто-
пустотах. Поскольку число пустот в ячей-
ячейке больше числа шаров упаковки,
катионы имеют возможность пере-
передвигаться по всей решетке, подобно
жидкости или газу. Эти особенности
структуры и создают особые свойст-
свойства веществ. Электропроводность мо-
модификации AgJ типа ZnS вблизи тем-
температуры превращения A42,4° С)
равна 0,00033 см2/молъ. Превращение
AgJ в высокотемпературную моди-
модификацию сопровождается скачкооб-
скачкообразным повышением электропровод-
электропроводности в несколько тысяч раз A,31
при 146,5° С). Далее, с повышением
температуры электропроводность
увеличивается, доходя вблизи темпе-
температуры плавления до 2,64. Интерес-
Интересно отметить, что эта величина превос-
превосходит величину электропроводности
расплава B,36 при 554° С).
§3. Оптические свойства
Оптические свойства ионных кри-
кристаллов весьма близки к свойствам
этих веществ в растворе. Электриче-
Электрическая составляющая падающего света
деформирует электронную оболочку
иона. Мерой этой деформации (поля-
(поляризации) является рефракция отдель-
отдельных ионов. Очевидно, что рефракция
ионов будет возрастать с увеличением
размеров и уменьшением стабильно-
стабильности электронной оболочки, например,
с увеличением заряда анионов (см.
табл. 31). Из таблицы следует, что
основная доля в рефракции ионного
соединения принадлежит анионам.
В симметричных (координацион-
(координационных) структурах, так же как и в ра-
растворах, ионная рефракция является
аддитивным свойством поляризуемо-
поляризуемости данных ионов. В несимметричных
структурах рефракция обусловлена
не только деформацией ионов элек-
электрической составляющей светового
луча, но также деформацией за счет
электрических полей соседних ионов,
вызывающих одностороннюю поляри-
поляризацию. Этот эффект используется в
244
ТАБЛИЦА 31
Зависимость рефракции от размера
и заряда иона
Ион
Радиус, А
Рефракция, см*
F"
1,33
2,5
С1-
1,81
9,0
Вг~
1,96
12,6
J-
2,20
19,0
Ион
Радиус, А
Рефракция, см3
02-
1,36
7,0
F"
1,33
2,5
Ne
1,60
1,00
Na+
0,98
0,5
Mg-
0,74
0,3
кристаллохимии для суждения о
структуре кристалла.
Ионные кристаллы, за исключени-
исключением солей редкоземельных и переход-
переходных металлов, обычно прозрачны и
бесцветны.
Гом е опо л ярны е вещества
существенно отличаются по оптиче-
оптическим свойствам от ионных вследствие
наличия у них электронов, принадле-
принадлежащих одновременно двум атомам.
Прочность такой связи сильно варь-
варьирует: у алмаза она весьма прочна, у
кремния или ZnS — слабее, у олова
настолько непрочна, что это вещество
обладает многими металлическими
свойствами. Уменьшение прочности
связи влечет за собой абсорбцию в
более длинноволновой части спектра.
Алмаз абсорбирует только в ультра-
ультрафиолетовой части спектра, поэтому
он прозрачен и бесцветен. Фотоэлек-
Фотоэлектрическая проводимость этих веществ
имеет место в том случае, если их
освещать лучами с длинами волн, со-
соответствующими их полосе поглоще-
поглощения. Алмаз обладает фотоэлектриче-
фотоэлектрической проводимостью в ультрафиоле-
ультрафиолетовой части спектра, кремний — в ви-
видимой, а для олова характерна уже
металлическая проводимость.
Гомеополярные соединения облада-
обладают высокими показателями преломле-
преломления, что создает (как говорят в мине-
минералогии) характерный смолистый,
жирный алмазный блеск. Они часто
бывают окрашены и непрозрачны.
В растворах характер распределения
электронов существенно отличен от
распределения их в кристаллах, по-
поэтому оптические свойства, в частно-
частности поглощение света, будут здесь со-
совершенно иными. Этим гомеополяр-
гомеополярные соединения существенно отлича-
отличаются от ионных.
Влияние степени ионности на ве-
величины показателей преломления
кристаллов с переходным (ионно-ко-
валентным) типом связи было рас-
рассмотрено в XII главе, § 8, стр. 214.
Молекулярные соедине-
соединения в твердом, жидком и газообраз-
газообразном состояниях имеют приблизитель-
приблизительно одинаковые оптические свойства.
Соединения с Ван-дер-Ваальсовой
связью обычно прозрачны и часто бес-
бесцветны. Если структура построена из
линейных (большинство парафинов)
или плоских (тг-дихлорбензол) моле-
молекул, приблизительно параллельных
ДРУГ ДРУГУ, то у кристаллов наблюда-
наблюдается резкая оптическая анизотропия
положительного знака в первом слу-
случае и отрицательного — во втором.
Оптические свойства кристаллов
широко используются для определе-
определения положения неизометрических
молекул в структуре.
§4. Ковкость металлов
Металл является ковким, если он
склонен к пластической деформации.
Под пластической деформацией пони-
понимают способность отдельных участков
кристалла скользить относительно
ДРУГ друга при внешнем воздействии.
Такое скольжение, очевидно, будет
осуществляться легче всего, если
один плотн^ейший шаровой слой будет
скользить по второму плотнейшему
слою. Случай скольжения друг отно-
относительно друга двух неплотнейших
слоев менее благоприятен для пласти-
пластической деформации. В этом случае
шары одного слоя будут провали-
245
ваться глубже в пустоты между ша-
шарами соседнего слоя, что будет спо-
способствовать их механическому тормо-
торможению. Поэтому, при прочих равных
условиях, металл со структурой цен-
центрированного куба будет менее ков-
ковким, чем металл со структурной гране-
центрированного куба.
Металлические изделия всегда
представляют собой мелкокристалли-
мелкокристаллический агрегат с беспорядочным рас-
расположением кристаллов. Очевидно,
что для характеристики способности
металлов к пластической деформации
важен второй структурный фактор —
число направлений, нормально к ко-
которым расположены плотнеишие слои
в упаковке. Чем это число выше, тем
больше вероятность, что направление
скольжения в одном кристаллическом
зерне совпадет (или будет близким)
с аналогичным направлением в сосед-
соседнем зерне, ибо для осуществления
пластической деформации в куске ме-
металла скольжение должно пройти че-
через большое число кристаллов. Выше
мы подчеркивали разницу в структу-
структурах гексагональной и кубической
плотнейших упаковок. В гексагональ-
гексагональной имеется только одно направление
плотнейших слоев шаров, перпенди-
перпендикулярное главной оси, а в кубической
таких направлений четыре — перпен-
перпендикулярно четырем тройным осям.
Таким образом, пластическая дефор-
деформация, начавшаяся в одном кристалле
металла с гексагональной плотнейшей
упаковкой, может легко задержаться
на границе с другим кристаллом, так
как мала вероятность, что и у сосед-
соседнего зерна плоскость плотнейшей ша-
шаровой упаковки будет близка к соот-
соответствующей плоскости первого кри-
кристалла. Наличие четырех плоскостей
с плотнейшей укладкой шаров в каж-
каждом кристалле металла с плотнейшей
кубической упаковкой значительно
увеличивает вероятность совпадения
(или близости) двух из них в сосед-
соседних кристаллах. Таким образом, наи-
наиболее ковкими металлами будут те,
которые имеют структуру плотней-
плотнейшей кубической упаковки.
246
§ &. Сиайиость
Первая попытка объяснить явление
спайности в кристаллах принадлежит
О. Бравэ. Исходя из развитой им тео-
теории кристаллических решеток, он вы-
высказал гипотезу, что плоскости спай-
спайности проходят параллельно сеткам
с наибольшей ретикулярной плот-
плотностью, ибо такие сетки отстоят в ре-
решетке друг от друга на максимальных
расстояниях. Эта идея была бы верна,
если бы структурной единицей в кри-
кристаллах являлись изометричные мо-
молекулы, как это думал Бравэ. В этом
случае, очевидно, максимальное рас-
расстояние между ними определяло бы
наиболее слабые связи и обусловли-
обусловливало существование по этим направ-
направлениям плоскостей спайности. Одна-
Однако такой упрощенный подход к явле-
явлению спайности может оправдаться
только в простейших частных слу-
случаях, например в графите.
Одним из основных результатов
экспериментальной проверки теории
структуры кристаллов, о котором мы
говорили выше, явилось установление
факта отсутствия молекул в кристал-
кристаллах большинства неорганических сое-
соединений. Кристаллы их построены из
атомов, не группирующихся, как пра-
правило, друг с другом в молекулы, и их,
следовательно, нельзя отождествлять
с узлами решетки. Атомы связаны с
узлами решетки, но располагаются по
гораздо более сложным законам — по
законам правильных систем точек.
Так, например, константы гексаго-
гексагональной решетки металлического маг-
магния следующие: а = 3,20; с = 5,20А;
с/а=1,62А. Если бы атомы магния
располагались по узлам решетки, то
мы вправе были бы, руководствуясь
гипотезой Бравэ, ожидать спайности
по базису. Однако металлический
магний хорошей спайностью не обла-
обладает. Атомная структура его постро-
построена по типу плотнейшей шаровой
гексагональной упаковки с очень
близким к идеальному отношением
осей с/а, равным 1,633. Это показыва-
показывает, что никаких резких аномалий в
Рис. 255. Разрез структуры сфалерита (ZnS)
нормально к граням куба, ромбододека-
ромбододекаэдра и октаэдра
межатомных расстояниях в структу-
структуре магния нет и что нет никаких ос-
оснований ожидать хорошей спайности
по базису. В структурах же цинка и
кадмия отношение с/а значительно
превосходит идеальное A,86 и 1,89
соответственно) и, следовательно, си-
силы связи между атомами в направле-
направлении главпой оси существенно слабее
сил связи в любом перпендикулярном
направлении. Для рассмотренных ме-
металлов можно ожидать спайность по
базису. И действительно, эти метал-
металлы имеют весьма совершенную спай-
спайность по указанному направлению.
Учет новых экспериментальных
данных по атомным структурам кри-
кристаллов был сделан Ю. В. Вульфом
при разработке им теории спайности.
Суть идеи Вульфа легко понять из
сравнений спайности у цинковой об-
обманки и алмаза. ZnS имеет спайность
по ромбическому додекаэдру {110},
алмаз — по октаэдру {111}.
На рис. 255 показаны разрезы
структуры ZnS нормально к граням
куба, ромбододекаэдра и октаэдра.
Максимальные расстояния между
«атомными слоями» будут по октаэд-
октаэдру @,866, если длину ребра элемен-
элементарного куба принять за единицу).
Очевидно, что для алмаза эти направ-
направления и должны явиться направле-
направлениями спайности. В структуре ZnS
па этом расстоянии располагаются
глои из разных атомов (Zn и S).
Силы связи между такими слоями,
очевидно, должны быть большими,
чем силы связи между слоями, нахо-
находящимися ближе друг к другу, но на
которых находятся одновременно оба
типа атомов и которые являются поэ-
поэтому более валентно насыщенными,
как это имеет место на плоскостях,
параллельных граням ромбододекаэд-
ромбододекаэдра. Поэтому в сфалерите плоскости
спайности параллельны {110}.
§6. Коэффициенты
механического сжатии
и термического расширения
Для веществ, характеризующихся
сильными связями и структурными
типами плотнейших упаковок, сжи-
сжимаемость будет минимальной. Для
металлов величина сжатия, по дан-
данным Рейса, колеблется от 0,3-10~'до
4,5 ¦ 10~6 см2/кГ. Тот же порядок ве-
величин наблюдается у тугоплавких
окислов [@,5—1)-Ю-'6 см2/кГ].
Неорганические соли обычно ха-
характеризуются структурами плотней-
плотнейших упаковок, но если в их состав
входят одновалентные щелочные ме-
металлы со слабыми ионными силами,
то они имеют сжимаемость, значи-
значительно большую, чем в вышеприве-
вышеприведенных случаях. Так, для солей ще-
щелочных металлов она колеблется от 1
до 6. Сжимаемость самих щелочных
металлов, кристаллизующихся в ку-
кубической центрированной упаковке,
характеризуется величинами от 9 до
61. Молекулярные органические сое-
соединения имеют сжимаемость в
B0—50) • 10~6 см21кГ. Ниже показа-
показано влияние типа структуры солей ще-
щелочных металлов на сжимаемость:
Соединение
А —X, А
Коэффициент сжатия а-108
Процент заполнения пространства ионами
• В скобки взято соединение, структур-
структурный тип которого отличается от струк-
структурного типа всех остальных соедине-
LiCl
2,57
3,50
78,66
NaCl
2,81
4,18
65,66
КС1
3,14
5,65
56,01
RbCl
3,27
7,40
50,16
(CsCl) •
3,57
5,90
68,44
ний, приведенных в таблице. Это отно-
относится и к остальным таблицам.
247
Соединение
А —X, А
3-10*
NaF
2,31
39
NaCl
2,81
40
NaBr
2,94
43
NaJ
3,18
48
Для солей, имеющих структуру вать на следующем ряду галоидных
типа NaCl, сжимаемость возрастает солей натрия:
с увеличением межатомных расстоя-
ний.
Для структуры типа CsCl коэф-
коэффициент сжатия значительно мень-
меньше, чем можно было бы ожидать,
принимая во внимание монотонное
изменение коэффициента для пред-
предшествующих CsCl веществ. Это свя-
связано с тем, что структуры типа плот-
нейших упаковок для соединений с
большими катионами являются го-
гораздо менее плотными, чем структу-
структура типа CsCl. Последнее обстоятель-
обстоятельство обусловливает переход при вы-
высоких давлениях галогенидов рубидия
от структурного типа NaCl к струк-
структурному типу CsCl
объема до 15%.
Соединения с сильно поляризую-
поляризующим катионом имеют меньший коэф-
коэффициент сжатия, так как, несмо!ря
на сравнимые межатомные расстоя-
расстояния, анионы в этих С1руктурах уже
с сокращением
ненормально сжаты за счет поляри-
поляризации. Это можно проследить на гало-
галоидных солях серебра:
§ 7. Твердость и температура
плавления
Оба свойсава, твердость и температу-
температура плавления, характеризуют проч-
прочность соединения — механическую
или термическую. У металлов и солей
твердость и температуры плавления
варьируют в широких пределах, од-
однако можно сказать, что вещества
этих классов соединений более твер-
твердые и характеризуются более
высокими температурами плавления,
чем органические вещества. Для по-
последних, как известно, характерна их
мягкость и низкие температуры плав-
плавления, что является следствием сла-
слабости Ван-дер Ваальсовых сил. Удоб-
Удобнее всего проследить влияние отдель-
Соединение AgCl NaCl AgBr NaBr (AgJ) NaJ
A —X, A 2,77 2,81 2,88 2,98 2,99 3,23
Коэффициент сжатия а-106 2,40 4,18 2,74 5,09 4,14 7,01
Коэффициенты термического рас-
расширения у тех же групп соединений
изменяются аналогично коэффициен-
коэффициентам сжатия. Металлы, кристаллизу-
кристаллизующиеся в структурных типах шгот-
нейших упаковок, тугоплавкие окис-
окислы и сульфиды имеют коэффици-
коэффициент объемного расширения порядка
0,0001—0,001. Того же порядка ко-
коэффициенты у неорганических солей
@,0004—0,001). Коэффициенты объ-
объемного расширения у щелочных ме-
металлов колеблются от 0,0018 до 0,0025.
Самых больших значений они дости-
достигают у молекулярных органических
соединений — порядка 0,003. Изме-
Изменение коэффициента объемного рас-
расширения с изменением межатомного
расстояния можно проиллюстриро-
ных факторов на твердость и темпе-
температуры плавления на примере неор-
неорганических соединений. Увеличение
межатомного расстояния при сохра-
сохранении типа структуры у вещества с
одинаковой валентностью атомов вле-
влечет за собой снижение твердости и
температуры плавления:
Соединение (BeO) MgO СаО SrO ВаО
А —X, А 1,65 2,10 2,40 2,57 2,77
Твердость 9,0 6,5 4,5 3,5 3,0
по Моосу
Т. пл., °С 2570 2800 2585 2430 1923
Выше приведены вещества, кристал-
кристаллизующиеся в структурном типе NaCl
за исключением ВеО, кристаллизую-
248
щегося в структурном типе вюртцита.
Характер химической связи в первом
приближении можно считать ионным.
Для ковалентных соединений изме-
изменение твердости и температур плав-
плавления с увеличением межатомных
расстояний имеет тот же характер,
что и для ионных:
С —С С —Si Si —Si Ge — Ge
A —A, A 1,54 1,89 2,35 2,43
Твердость 10,09 9,5 7,0 6,0
Т. пл.,°С 3500 2700 1420 958
Все вещества, приведенные в табли-
таблице, кристаллизуются в структурном
типе алмаза — сфалерита.
смена координационного типа на сло-
слоистый заметно влияет на изменение
физических свойств:
Структурный тип
Соединение
А —X, А
Т. пл., #С
CaF2
SrF2
2,58
1280
CdCU
MgCh
2,63
712
Замена 8-электронного иона на 18-
электронный (или вообще на «ион»
не типа благородного газа) влечет за
собой слабое изменение твердости,
но достаточно сильное уменьшение
температуры плавления;
Структурный тип
Соединение
А - X, А
Т. пл., "С
Структурный тип
Соединение
А —X, А
Т. пл., °С
Твердость
NaCl
NaCl
2,81
840
NaCl
NaF
2,31
992
3,0
ZnS
CuCl
2,34
422
2,5
NaCl
AgCl
2,77
455
NaCl
SrO
2,57
2430
3,5
NaCl
AgBr
2,87
422
ZnS
CdS
2,52
1750
3,0
NaCl
BaO
2,77
1923
3,5
NaCl
LiCl
2,57
614
ZnS
CdTe
2,80
1041
3,0
NaCl
AgF
2,48
435
NaCl<
PbS
2,97
1120
3,0
Влияние валентности на твердость
и температуру плавления можно про-
проследить на веществах с приблизитель-
приблизительно равными межатомными расстоя-
расстояниями:
Соединение
А —X, А
Твердость
Т. пл., °С
NaF
2,31
3,5
988 •'
MgO
2,10
6,5
2800
ScN
2,23
7,5
TiC
2,23
8,5
3180
У взятых веществ характер химиче-
химической связи с изменением валентно-
валентности, конечно, существенно меняется.
Замена одного координационного
структурного типа на другой, напри-
например NaCl на ZnS или CaF2 на ТЮ2,
при прочих близких характеристиках
слабо сказывается на твердости и на
температуре плавления. Напротив,
§ 8. Влияние водородной связи
на физико-химические
свойства веществ
Водородная связь является слабой
связью, ее энергия примерно равна
5—10 ккал/молъ, поэтому создает-
создается неправильное представление, что
влияние водородной связи на физико-
химические свойства соединений всег-
всегда незначительно. Действительно, в
тех случаях, когда мы сравниваем ее
с ионными или ковалентными связя-
связями, учет водородной связи вносит
только незначительную поправку в
конечный результат расчета, ибо ее
энергия в несколько десятков раз
меньше энергии ионной или кова-
лентной связи. Однако если мы бу-
будем сравнивать водородную связь с
остаточной, то соотношение будет об-
24»
ТАБЛИЦА 32
Температура плавления и кипения
некоторых молекулярных гидридов
Соеди-
Соединение
HF
НС1
НВг
HJ
Н2О
HSS
HaSe
Н2Те
Т. пл.
-83
-112
-88
—51
0
—86
—66
—51
Т. кип.
420
—84
—67
-35
+100
-60
—41
2
Соеди-
Соединение
NH8
РНз
AsH3
SbHa
СН«
SiH4
GeH4
SllH*
Т. пл.
—78
-134
—116
-88
—184
—185
—166
—150
Т. кип.
-33
-88
—62
—17
—162
—112
-88
—22
ратным, и в этом случае ее влияние
может быть существенным. Приме-
Примером могут служить некоторые свой-
свойства молекулярных соединений.
В табл. 32 приведены соответствую-
соответствующие данные для различных гидридов.
Аномалия температур плавления
и кипения у соединений фтора, кис-
кислорода и азота хорошо объясняется
наличием водородных связей не толь-
только в твердом, но и в жидком и газо-
газообразном состояниях. Из данных
таблицы можно было бы ожидать для
НгО температуру плавления порядка
—100°, если эту величину попы-
попытаться получить экстраполировани-
экстраполированием кривой, соединяющей температу-
температуры плавления НгТе—НгЭе—H2S (см.
рис. 240). Такое огромное несоответ-
несоответствие экстраполированной и действи-
действительной температур плавления льда
легко понять, если сопоставить кине-
кинетическую энергию молекул, которая
имеет значение порядка нескольких
десятых долей ккал/молъ, с энергией
водородной связи. Ясно, что этой
энергии будет совершенно недоста-
недостаточно, чтобы разорвать водородные
связи, но достаточно для преодоления
Ван-дер-Ваальсовых связей.
§ О. Эффект экранирования ионов
Молекула с ионной связью, будучи
в целом нейтральной, может пред-
представить собой сильный диполь. К от-
350
рицательному концу такой молекулы
может быть притянут положитель-
положительный конец второй молекулы; сбоку
к ней могут близко подходить и
прочно удерживаться другие молеку-
молекулы. Вещество с такой структурой бу-
будет иметь малую упругость пара и
довольно легко кристаллизоваться,
температура плавления его будет
высокой.
Рассмотрим теперь молекулы типа
CF4. В данном случае маленький
центральный атом углерода, окру-
окруженный со всех сторон более круп-
крупными и более электроотрицательны-
электроотрицательными атомами фтора, не может при-
притягивать отрицательные концы дру-
других молекул, поэтому никакой склон-
склонности к ассоциации у подобных мо-
молекул не будет. Соединения такого
типа легколетучи и имеют низкие
температуры плавления. Обычно их
называют закрытыми соединения-
соединениями, в противоположность открытым
соединениям типа NaCl.
Если центральный атом (ион)
мал, а наружные, экранирующие его
ионы велики, то вещество будет об-
обладать всеми свойствами закрытого
соединения. Вещество с тем же ти-
типом формулы, но с большим цент-
центральным атомом (ионом) и неболь-
небольшими экранирующими ионами может
обладать свойствами открытого сое-
соединения. Так, например, четыре
иона хлора полностью экранируют
ион олова: Sn4+=0,67, С1" = 1,81,
га:гх = 0,37. Температура кипения
SnGl4 равна 114° С. Четыре иона фто-
фтора не могут полностью экранировать
ион олова, и соединение S11F4 кипит
при 705° С.
Аналогичный вывод можно сде-
сделать, сопоставляя соединения алю-
алюминия и бора. В соедиении А1СЦ
меньший по размеру ион алюминия
экранируется тремя ионами хлора,
поэтому температура кипения А1С1»
мала (81° С). A1F3 кипит при темпе-
температуре выше 1000° С, так как экрани-
экранирование неполное. Ион бора так мал
(В3+ = 0,20), что его полностью эк-
экранируют три иона фтора. Поэтому
при обычных температурах BF3 яв-
является газом. Температура плавле-
плавления BFS равна — 127° С.
Экранирование имеет место, ко-
конечно, не только для молекул, но и
для комплексных ионов. Однако яв-
явления, которые влечет за собой эк-
экранирование, в этом случае отлича-
отличаются от вышеописанных. В частно-
частности, мы не можем здесь связывать
упругость пара, температуру кппе-
яия и другие свойства с экранирова-
экранированием иона, так как комплексный ион
(например, SO42~) может прочно
удерживать катионы в кристалле
электростатическими силами.
Размеры экранирующих ионов
обусловливают состав и заряд комп-
комплексного иона. Рассмотрим пример
комплексных анионов кислородных
кислот. Отношения радиусов S6+
@,29), Si4+ @,39) и Р5+ @,35) к ра-
радиусу иона кислорода соответственно
равны 0,21, 029 и 0,26. Отсюда следу-
следует, что окружение будет происходить
четырьмя кислородными ионами,
поскольку для координационного
числа 4 отношение га : гх лежит в
пределах от 0,22 до 0,41. Комплекс-
Комплексными ионами будут [SiO4]4~,
[PO4]3-,[SO4]2-.
Невероятен ион [ЭОб] и геомет-
геометрически невероятна конфигурация
[ЭОб]6"". И действительно, неизвест-
неизвестны ни сама ортосерная кислота, ни
ее соли, в то время как имеются со-
соли ортотеллуровой кислоты Те (ОН) б,
например Ag6Te06 (Те6+=0,56А,
fa : г*=0,41), и ортоиодной — HsJOe,
например Ag5JO6 (J7+~Te6+). Отно-
Отношение радиуса иона С4+@,2А) к ра-
радиусу кислорода равно 0,15, что
удовлетворяет координационному
числу 3. Отсюда следует возможность
существования иона (СОзJ~ и не-
невозможность иона (СО4L~.
Из приведенных примеров видно,
что рассмотрение вопроса экраниро-
экранирования даже с чисто электростатиче-
электростатических позиций (как это делали Кос-
сель, Гольдшмидт и Ван-Аркель),
позволяет получить кое-какие каче-
качественные закономерности.
§10. Растворимость
В предыдущих параграфах были
рассмотрены некоторые свойства, ха-
характеризующие прочность кристал-
кристаллических веществ — твердость, тем-
температура плавления и др. Раствори-
Растворимость также характеризует проч-
прочность и поэтому наряду с ними мо-
может быть рассмотрена в этой главе.
Соль будет растворима в воде, если
притяжение ионов молекулами воды
будет больше энергии решетки.
Растворимость галоидных солей
щелочных металлов при 0° С показа-
показана в табл. 33. Если данных для 0° С
в литературе нет, то в скобках ука-
указана другая температура. Из данных
таблицы видно, что прямой зависимо-
зависимости растворимости от величины ион-
ионных радиусов нет. Это очевидно, так
как растворимость зависит не только
от величин энергии решетки, но и от
величин теплоты гидратации. По-
Последняя же зависит от размеров
ионов иначе, чем энергия решетки.
Приблизительные данные для теплот
гидратации Н можно получить путем
измерения теплот растворения L:
H^U — L,
где U — энергия решетки.
Повышенная растворимость LiCl,
Ш, NaBr, NaJ может быть объяс-
объяснена ослаблением сил связи между
ионами в решетке, так как отноше-
отношение их радиусов га : гх близко к кри-
критическому для структуры типа
NaCl @,41). В этих структурах рез-
ТАБЛИЦА 33
Растворимость галоидных солей
щелочных металлов
3
Е
«
я
К
Li
Na
К
Rb
Cs
0,1
1,0
16
12
24
F
A8°
A5°
A8°
A8е
A8°
С)
С)
С)
С)
С)
Анионы
С1
16
6,1
3,7
6,4
9,6
1
4
5
6
Вг
11
5
8
0B5°
С)
11
10
7,6
6,5
1,7
J
G«С)
251
ко возрастают силы отталкивания
соприкасающихся или почти сопри-
соприкасающихся анионов. Повышенная
растворимость KF, RbF и CsF
объясняется малой плотностью (рых-
(рыхлостью) структур. Все они, если су-
судить по отношению га : гх, должны
были бы иметь структурный тип
CsCl, а в действительности кристал-
кристаллизуются в структурном типе NaCi.
Увеличение валентности ионов
влечет за собой резкое снижение рас-
растворимости, так как энергия решет-
решетки с увеличением валентности возра-
возрастает значительно быстрее, чем
энергия гидратации. Поэтому окислы
и сульфиды двувалентных металлов
в отличие от галогенидов щелочных
металлов почти нерастворимы в во-
воде, хотя кристаллизуются они в ана-
аналогичных структурных типах.
Начиная с трехвалентных катио-
катионов сказывается эффект экранирова-
экранирования. Влияние валентности катиона и
эффект экранирования ионов ясно
видны из следующих данных:
Соединение NaF CaF2 A1C13 ThCl4
Растворимость 1,0 0,0002 6,0 хорошо
растворим
с 2,31 2,37 2,38 2,76
Соединение NaCl BaF2
Растворимость 6,1 0,009
ra + rx 2,79 2,71
РгСЬ SnJ*
4 2 Хорошо
растворим
2,81 2,87
Влияние электронного строения
ионов на растворимость весьма ин-
интересно. К. У. Стиллвелл считает,
что здесь, по-видимому, решающую
роль играет деформация катионом
аниона в решетке или молекул воды
в растворе. Можно различать четыре
случая в зависимости от того, поля-
поляризуется анион легче или труднее,
чем молекула воды, и от того, явля-
является ли катион слабо поляризующим
(т. е. имеет большие размеры, малый
заряд и 8-электронную оболочку)
или же сильно поляризующим (ма-
(малые размеры, большой заряд и не
i
1
ZJ9
BaSO,
ч
f
Анионы полтин
е/лся по сравнейин.
смолекияБми воды
Труднее
В-1Г6
Пай
6,01
140
-NaF
to
Рис. 256. Схема, иллюстрирующая влияние
различных факторов на растворимость
8-электронную оболочку). Эти четы-
четыре случая представлены на средней
части схемы на рис. 256. Легче воды
поляризуются С1~, Вг~, J-, ОН~,
труднее F~ и комплексные ионы ти-
типа SO42-, CIO4-, PtCl62- и т. п.
Если анион поляризуется легче,
чем вода, то для веществ, содержа-
содержащих сильно поляризующие катионы,
энергия решетки будет возрастать
быстрее, чем энергия гидратации,
поэтому вещества, содержащие легко
поляризующиеся анионы и малые по
размеру катионы с большими заряда-
зарядами или с 18 электронами в наружной
оболочке, будут иметь растворимость,,
значительно меньшую, чем аналогич-
аналогичные соли, но с большими катионами,
имеющими 8-электронную оболочку
(случай А-+С, рис. 256). Стрелка
указывает направление увеличения
растворимости.
Если сравнить растворимость со-
солей А и В, т. е. двух соединений с
сильно поляризующим катионом
(например, Ag+), но у одного анионы
будут деформироваться легче, чем
молекулы воды (например, С1~), то
растворимость солей фтора будет
больше растворимости солей хлора.
В солях хлора дополнительная (за
счет деформации аниона) энергия
будет энергией решетки, а в солях
фтора — энергией гидратации.
Ион натрия слабо поляризует, по-
поэтому дополнительной энергии за
счет поляризации не будет ни в
структуре NaF, ни в воде. Ион се-
серебра, напротив, поляризует сильно.
Поскольку молекулы воды поляризу-
поляризуются сильнее, чем F~, то Ag+ будет
252
стремиться перейти в растворитель.
Поэтому растворимость AgF будет
больше растворимости NaF (D-*-B).
Направление растворимости D—>-С
будет определяться величиной энер-
энергии решетки. Расстояние Na—С1
больше, энергия решетки соответст-
соответственно меньше и растворимость боль-
больше, чем в NaF. Дополнительная энер-
энергия за счет поляризуемости ионом
натрия анионов в кристалле и моле-
молекул воды в растворе очень мала и на
процесс растворимости практически
не влияет. Ниже приведены значения
растворимости некоторых солей (в
моль!л воды), иллюстрирующие ска-
сказанное выше:
Na+
Ag+
T1+
SO42-
20H-
F"
1,
14,
3,
IV
2,
1,
0
0
6
lg2+
19
5-10-*
ci-
6,1
6-10"
9-10"
CaJ+
1,3-
2,5-
e
3
lO
10
Вг"
11,3
4,5-10"'
8,5-10-*
Sr2+
6-10-*
3,3.10-«
J"
10,6
1,3-10-8
6-10-5
Ba2+
6,3-10-e
2,5-10-i
Поскольку О2- и S2- поляризуются
легче, чем вода, их соединения с
й-металлами (например, Zn, Cd
и т. д.) менее растворимы, чем окис-
окислы и сульфиды а-металлов (Mg, Ca
и т. д.).
Растворимость, как было показано
выше, сильно зависит также от типа
структуры. Вещества, имеющие сло-
слоистые и молекулярные структуры,
растворяются лучше, чем вещества,
имеющие координационные структу-
структуры. На этом в качественном анализе
основано отделение при помощи иона
хлора (добавления НС1) 6-катионов
одновалентных (Ag+) и самых круп-
крупных двувалентных (РЬ2+) от осталь-
остальных й-двувалентных (Zn, Cd) и дру-
других й-катионов с большей валент-
валентностью. Первые образуют с хлором
нерастворимые координационные
структуры (AgCl, РЬСЬ), а вторые —
легко растворимые слоистые и моле-
молекулярные структуры (ZnCl2, CdCl2).
Последние элементы осаждаются
ионом S2- при добавлении сероводо-
сероводорода (например ZnS).
ГЛАВА XVI
СТРОЕНИЕ РЕАЛЬНОГО КРИСТАЛЛА
§1. Идеальный и реальный
кристалл
1. Кристаллическое и аморфное со-
состояние. До сих пор мы рассматрива-
рассматривали кристалл как тело, построенное из
атомов, расположенных по идеаль-
идеальным законам геометрии. В действи-
действительности такой подход является во
многих отношениях абстрактной иде-
идеализацией, результатом принятого
понятия однородности кристалличе-
кристаллической среды, полошенного в основу
учения о форме кристаллов, их сим-
симметрии. В действительности сущест-
существует непрерывный переход от иде-
идеально-правильного в геометрическом
и физическом смысле кристалла к
телам с полностью неупорядоченным
расположением атомов — аморфным,
стеклообразным твердым телам.
Здесь следует сразу же оговориться,
что у реальных веществ в таких со-
состояниях существует определенная
степень упорядоченности, в особен-
особенности касающаяся ближнего поряд-
порядка. Поэтому если допустить анало-
аналогичную абстракцию, которую мы до-
допускали до сих пор в отношении кри-
кристалла, то и аморфное состояние сле-
следует несколько идеализировать и в
первом приближении считать его
идеально неупорядоченным. Тогда
оба этих состояния займут крайние
положения на воображаемой прямой,
характеризующей постепенные пере-
переходы между двумя пределами. Вслед
за идеальным кристаллом на этой
линии будут располагаться реальные
кристаллы, которые займут на ней
значительный участок. Часть реаль-
реальных кристаллов будет примыкать к
почти идеальным. Степень их неупо-
неупорядоченности может быть весьма не-
незначительной, а по мере нарушения
упорядоченности мы будем все
дальше и дальше уходить от крайней
точки, символизирующей идеальный
кристалл. Как сказано выше, реаль-
реальные аморфные тела тоже сохраняют
известную степень упорядоченности,
поэтому они будут располагаться на
той же линии, но вблизи второго ее
конца, отклоняясь от него все даль-
дальше по мере возрастания степени упо-
упорядоченности, по мере приближения
к кристаллическому состоянию.
2. Отклонения внешней формы ре-
реальных кристаллов от идеальных
геометрических законов. В первой
части этой книги, посвященной гео-
геометрии кристалла, уже упоминалось
о реальном кристалле, о степени от-
отклонения его формы от идеальных
геометрических форм. Уже пер-
первые работы, посвященные изучению
внешней формы кристаллов, показа-
показали, насколько она может быть разно-
разнообразна и «неправильна» у кристал-
кристаллов одного и того же вещества (см.
рис. 6, стр. 13). Однако пренебреже-
пренебрежение небольшими отклонениями от не-
некоей идеальной формы привело к
открытию закона постоянства углов.
Однако и этот закон оправдывался
только с определенной точностью.
На примере хорошо образованного
кристалла шпинели (см. рис. 14, стр.
17) можно проследить степень от-
отклонения реального кристаллическо-
254
го многогранника от геометрически
правильного. Причина таких откло-
отклонений лежит в нарушениях внутрен-
внутренней (атомной) структуры кристалла,
о которой мы можем судить, изучая,
например, реальные грани кристал-
кристаллов.
При сильном увеличении грань
внешне даже весьма совершенного
кристалла представляет собою дале-
далеко не идеальную геометрическую
плоскость. На ней можно различить
отдельные участки — блоки, откло-
отклоненные от идеального положения на
тот или иной, обычно небольшой
угол. В примере шпинели отклоне-
отклонения двухгранных углов достигли 4,5'.
Это значит, что большинство блоков,
выходящих на поверхность соответ-
соответствующих граней, имели отклонения
такого порядка. Отдельные же блоки
могли отклоняться на значительно
большие углы.
Все это привело к представлениям
о том, что реальный кристалл явля-
является как бы «мозаикой» из блоков.
Только в отдельных блоках строение
кристалла представляется идеальным
(во всяком случае более идеальным,
чем во всем кристалле в целом). Са-
Сами же блоки по отношению друг к
другу несколько разориентированы
и между ними, следовательно, про-
проходят какие-то разупорядоченные
зоны.
Мозаичное строение кристаллов
нашло подтверждение в опытах по
определению прочности кристаллов
в зависимости от их величины. Эти
опыты показали, что прочность кри-
кристаллов значительно возрастает с
уменьшением их размеров. При раз-
размерах порядка 10~5 см (что соответ-
соответствует размеру отдельных блоков)
прочность кристаллов достигает зна-
значений, в сотни раз превышающих
прочность макроскопических кри-
кристаллов, и хорошо согласуется с тео-
теоретически рассчитанной прочностью.
Очевидно, что реальные большие
кристаллы разрушаются прежде все-
всего по границам блоков, связанных
друг с другом более слабыми сила-
силами, чем силы, действующие внутри
блоков, где атомные структуры яв-
являются, согласно этой теории, гораз-
гораздо более совершенными.
Однако мозаичное строение кри-
кристаллов является не единствен-
единственным способом нарушения идеальной
структуры.
Вторым нарушением идеальной
геометрии кристалла, которое здесь,
следует сразу же упомянуть и кото-
которое также можно наблюдать на гра-
гранях реальных кристаллов, является
спиральная форма их поверхности.
Впервые такие спирали на гранях
кристаллов наблюдал Г. Г. Леммлейп
A955 г.). На рис. 15 (стр. 17) была
уже приведена фотография такой
спирали на грани пинакоида кристал-
кристалла кварца.
Все эти характерные нарушения
идеальной структуры кристаллов яв-
являются, конечно, следствием наруше-
нарушений, первоначально возникающих в
атомной структуре кристаллов, к
описанию которых мы и переходим
в следующих параграфах.
§2. Точечные дефекты
в атомной структуре
кристалла
1. Точечные дефекты в металличе-
металлических кристаллах. В металлическом
кристалле, в котором атомы располо-
расположены по узлам решетки, простей-
простейшим точечным дефектом является
случай, когда один из узлов решетки
оказывается не занятым атомом (рис.
257, а). Такой точечный дефект на-
называется вакантным местом или сво-
свободной вакансией. В результате об-
образования такого точечного дефекта
произойдет некоторое перемещение
соседних с дефектом атомов (рис.
257, б), и решетка окажется напря-
напряженной. Область, в которой атомы
заметно сместились от своих идеаль-
идеальных положений, занимает несколько
(до 10) межатомных расстояний. На
эту область рассредоточен точечный
дефект. На рис. 257, б такая область
255
f ис. 257. Точечный дефект в структуре
металлического кристалла в виде свобод-
свободной вакансии (а). Смещение соседних с
вакансией атомов из своих первоначаль-
первоначальных (идеальных) положений (б).
показана пунктирной окружностью.
На свободную вакансию может пере-
перескочить соседний атом, это будет
равнозначно тому, что вакансия пе-
переместилась на место этого атома.
Движение дефектов в кристаллах
весьма характерно. Чем выше темпе-
температура, тем более вероятен этот про-
процесс.
Рис. 258. Точечный дефект в структуре
металлического кристалла в виде внедрен-
внедренного в структуру дополнительного атома
(а). Смещение соседних с ним атомов из
своих первоначальных положений (б).
При комнатных температурах
в таких металлах, как свинец или
кадмий, каждая вакансия осущест-
осуществляет приблизительно один скачок в
сутки, яо вблизи температуры плав-
плавления такие скачки осуществляются
десятки тысяч раз в секунду. В ре-
результате подобных передвижений
вакансии могут объединиться и об-
образовать более крупные макроско-
макроскопические нарушения, но могут и ис-
исчезать, если при перемещении им
удается достигнуть поверхности кри-
кристалла. Среднее число вакансий в
обычном металлическом кристал-
кристалле объемом в 1 см3 равно 1012 —
1014.
Противоположным случаем будет
точечный дефект, связанный с внед-
внедрением межузельного атома (рис.
258, а). Как и в случае образования
256
вакансии, вокруг этого дефекта об-
образуется область искажения решет-
решетки (рис. 258,6). Попавший в между-
узлие атом сможет под влиянием
тепловых колебаний (его самого и со-
соседних с ним атомов) переместиться
в соседнее положение. Если на своем
пути он встретит вакансию и попа-
попадает в нее, то два дефекта в кристал-
кристалле при этом исчезнут. Таким образом
в реальном кристалле все время по-
появляются и исчезают точечные де-
дефекты.
2. Точечные дефекты в ионных
кристаллах. Аналогичные дефекты
(см. раздел 1 настоящего §) суще-
существуют и в кристаллах с ионной свя-
связью. В отличие от дефектов металли-
металлического кристалла дефекты в ионных
кристаллах несут на себе определен-
определенный заряд. На рис. 259, а показаны
дефекты (обозначенные пунктирны-
пунктирными кружками), связанные с появле-
появлением вакансий (дефекты Шотки), а
на рис. 259, б — дефекты, возник-
возникшие от попадания отдельных ионов
в междоузлие (дефекты Френкеля).
Из-за наличия в структуре положи-
положительных и отрицательных ионов на
вакансию, обозначенную на рис.
259, а стрелкой 1, может попасть
только анион, а стрелкой 2 — катион.
Энергия образования дефектов Шот-
Шотки в щелочногалоидных кристал-
кристаллах порядка 2 эв, дефектов Френке-
Френкеля — 1,5 эв.
Дефекты структуры такого рода
были описаны нами в главе об изо-
изоморфизме (глава XIII). По существу
вся эта глава уже посвящена реаль-
реальному кристаллу. Для дефектных
структур, например пиротина Fei_xS,
характерно наличие определенного
количества дефектов в катионной
части, равное половинному числу
трехвалентных атомов железа в этом
веществе.
Дефекты Френкеля в ионных кри-
кристаллах могут, как правило, созда-
создаваться только катионами в силу того,
что они по размеру значительно
меньше анионов. Движение катиона,
обозначенного стрелкой 1 (рис.
17 Кристаллохимия
0 0/0
б
5 0 Ф
0 .'V i © © 9 © ©, 0 © 0
0 © (-) © 0 0 ©
е © 0 е е © е
^э
О 9
© © ©;
Рис. 259. Типы точечных дефектов в струк-
структуре ионного кристалла
а — появление вакансии (дефект Шотки);
б — внедрение дополнительных ионов между
ионами, занимающими нормальные поло-
положения в структуре (дефекты Френкеля)
259, б), перебрасывает дефект в со-
соседнее положение. Но катион может
вытолкнуть в междоузлие и соседний
с ним катион, а сам занять его мес-
место (стрелка 2).
Поскольку в реальном кристалле
обычно сосуществуют дефекты обоих
типов (Шотки и Френкеля), по-
постольку замещения вакансий и пе-
перескоки атомов по междоузлиям мо-
могут осуществляться параллельно.
3. Точечные дефекты в кристал-
кристаллах с ковалентным и ван-дер-вааль-
совским типом связей. В кристаллах
с ковалентным типом связи по су-
существу имеют место те же случаи.
Образование вакансий в них связано
с обрывом химических связей и появ-
появлением ненасыщенных валентностей
у атомов, находящихся на границе с
вакансией. По этой причине вакан-
вакансия несет на себе некоторый элект-
электрический заряд.
Ван-дер-ваальсовская связь, как
связь ненаправленная, сходна в этом
отношении с металлической. Поэтому
точечный дефект молекулярного кри-
кристалла (пропущенная молекула)
сходен с аналогичным дефектом ме-
металлического кристалла. Однако здесь
возможны дефекты, связанные с не-
неправильным положением крупных
молекул весьма сложной формы. На
рис. 260, а показана полностью упо-
257
Рис. 260. Идеальный (в) и дефектный (б)
молекулярный кристалл
рядоченная структура кристалла из
длжнноцепочечных молекул. Оси мо-
молекул расположены перпендикулярно
плоскости чертежа. На рис. 260, б
порядок расположения молекул на-
нарушен, хотя центры тяжести их
остались на своих местах. Этот слу-
случай нарушения возможен только в
органических и в неорганических
кристаллах с крупными неправиль-
неправильной формы группировками атомов
(молекул или радикалов). Пунктир-
Пунктирной окружностью на рис. 260, б по-
показана вакансия, образовавшаяся в
результате отсутствия одной из мо-
молекул.
Рис. 261. Искажения, появляющиеся в
структуре в результате замещения одного
из атомов основного вещества посторон-
посторонним атомом
о — атом примеси большего размера;
б — атом примеси меньшего размера;
• — искажение решетки в результате внедре-
внедрения атома меньшего размера между ато-
атомами основного вещества
4. Точечные дефекты, связанные с
примесными атомами. До сих пор
мы рассматривали дефекты, получаю-
получающиеся в идеально чистых кристаллах
простых веществ и индивидуальных
химических соединениях. Но это
лишь предельная абстракция, тако-
таковых веществ в действительности не
существует. Каждое вещество содер-
содержит то или иное количество приме-
примесей, т. е. атомов постороннего веще-
вещества.
Современная техника требует весь-
весьма чистых веществ, однако добиться
полного отсутствия примесей или их
наличия в количестве менее, чем
10~8%, удалось лишь для очень не-
немногих простых веществ. Самые чи-
чистые химические соединения содер-
содержат примесей или избытка одного из
своих компонентов значительно боль-
больше приведенной выше величины.
А это значит что в кристаллах раз-
размером в 1 см3 обычно содержится при-
примесных атомов не менее чем 1015.
Попадая в кристалл, посторонние
258
атомы создают в нем дефекты, ана-
аналогичные описанным выше.
На рис. 261, я и б показаны иска-
искажения, появившиеся в решетке в
результате замещения одного из ато-
атомов основного вещества посторонним
атомом (атомом примеси). На рис.
261, в показан случай внедрения
постороннего атома. Как правило,
внедренные атомы имеют размеры
меньше, чем атомы основного ве-
вещества.
Для ионных и ковалентных кри-
кристаллов характерны случаи, когда
атомы примеси имеют другую валент-
валентность. Если замещающий атом имеет
большую валентность (например,
Са2+, замещающий Na+ в NaCl), то
это вызывает точечный дефект (ва-
(вакансию) в катионной части структу-
структуры, так как сумма положительных и
отрицательных валентностей в кри-
кристаллах обычно соблюдается весьма
точно.
Замещающий атом высшей валент-
валентности в кристаллах с ковалентной
связью, например, атом азота в кри-
кристалле алмаза, сохраняет одну нена-
ненасыщенную валентность. В результате
этого в кристалл может внедриться
(в структурах кристаллов с ковалент-
ковалентной связью обычно имеется много
крупных пустот) атом какого-либо
другого вещества, который будет ком-
компенсировать эту валентность. Таким
образом, один тип дефектов может
повлечь и обычно влечет за собой
появление других.
§ 8. Дислокации
1. Линейные дислокации. Первона-
Первоначально понятие о дислокации в кри-
кристаллах было введено в физику твер-
твердого тела Тейлором и Орованом в
1930 г. для описания процесса пла-
пластической деформации металлов. Од-
Однако вскоре выяснилось гораздо бо-
более общее значение теории дислока-
дислокаций.
Действительно, проще всего ис-
искусственно получить дислокацию в
кристалле, если подвергнуть его пла-
пластической деформации. Однако со-
совершенно аналогичные дислокации
могут возникать в кристалле само-
самостоятельно в процессе его роста. Что
же представляет собою линейная ди-
дислокация в кристалле?
Представим себе, что мы начинаем
сдвигать половину кристалла относи-
относительно второй неподвижной его по-
половины (рис. 262,а). (Направление
сдвига показано стрелкой.) В резуль-
результат© сдвига половила кристалла мо-
Рис 262 Смещение атомных плоскостей Рис. 263. Двухмерная схема дислокацга
в кристалле в результате сдвига в результате сдвига
а — положение до сдвига;
б — положение после сдвига
3
3
3
3
3
3
3
г
2
z
z
z
г
z
z
f
1
/
/
/
6
i
3
3
3
г
г
г
2
3
3
3
3
j
/
/
Z
г
z
Z
1
1
1
5
5
и
s
з
3
2
р
з
1
Т7»
259
жет переместиться па одно межатом-
межатомное расстояние (рис. 262, б), и ре-
решетка кристалла в конечном итоге
вновь станет идеальной. Однако ряд
атомов 1, 1, 1... при этом разорвется,
связь между атомами (соединенны-
(соединенными на рис. 262, б пунктирной линией)
будет нарушена, но вместо нее обра-
образуются совершенно аналогичные свя-
связи между рядами 1, 1, 1... и 2, 2, 2,
затем 2, 2, 2... я 3, 3, 3... и т. д. за
счет перемещения соответствующих
цепочек атомов на одно межатомное
расстояние. В большинстве случаев
процесс идет не так. Деформация
сжатия в направлении сдвига приво-
приводит к тому, что нарушается связь
между атомами в каком-либо ряду
внутри кристалла (на рис. 263 в ря-
ряду 5, 5, 5...).
В результате в кристалле возникает
как Пы вставленная полуплоскость
(верхняя часть рядов 5, 5, 5...), назы-
Рис. 264. Появление дислокации в кри-
кристалле
Рис. 265. Схема появления винтовой дис-
дислокации в кристалле
260
ваемая обычно экстраплоскостью.
В пространстве это показано на
рис. 264. Сдвиг в кристалле произо-
произошел не по всей плоскости, а только
на части ее ABDC. Дислокация, обоз-
обозначенная на рис. 263 значком «-L»,
перпендикулярна к плоскости черте-
чертежа. Она имеет линейную протяжен-
протяженность (линия АВ на рис. 264), поэто-
поэтому носит название линейной дислока-
дислокации. На рисунке она выглядит как то-
точечный дефект. И, забегая вперед, сле-
следует сказать, что точечные дефекты
часто могут служить источником ли-
линейных дислокаций во время роста
кристалла, причем длина дислокации
в отличие от точечных дефектов мо-
может простираться на миллионы меж-
межатомных расстояний и пронизывать
кристаллы из конца в конец. Вокруг
дислокации образуется область
(шнур) наиболее деформированной
напряженной части кристалла. Тол-
Толщина шнура того же порядка, что и
у точечных дефектов, т. е. несколь-
нескольких межатомных расстояний.
2. Винтовая дислокация. Понятие
о винтовой или спиральной дислока-
дислокации было введено в физику твердого
тела Бюргерсом в 1939 г. Такая дис-
дислокация также может возникнуть за
счет сдвига части кристалла на один
межатомный параметр (или кратное
их число). На рис. 265 в плоскости
сдвига ABCD одна часть кристалла
опустилась на один параметр решет-
решетки СС и соответственно DDr. Ось дис-
дислокации АВ в этом случае будет па-
параллельна направлению сдвига, а не
перпендикулярна, как это было в слу-
случае линейной дислокации. Вокруг оси
винтовой дислокации также будет
располагаться область наиболее иска-
искаженного участка кристаллической ре-
решетки размером в несколько меж-
межатомных расстояний. На рис. 266 по-
показан один виток такой дислокации
на участке решетки, где имеет место
максимальная деформация ячеек. На-
Начало и конец изогнутой стрелки (сим-
(символизирующей один шаг спирали)
расположены на соседних узлах од-
одного вертикального ряда решетки.
Направление-
сдвига часта
нрис/лаллЪ-
ческои ре -
шетки
В
Направление
движения' _
д&с'лдкаций
Рис. 266. Один виток винтовой дислокации
j
1
-Li i~t—
i
-
4
h
H
Hi
П
111
±111
Г
¦
1
1 П
А1ГП
1
1
i
Рис. 267. Появление дислокации в резуль-
результате срастания двух блоков в кристалле
а — первоначальные состояния в блоках, решет-
решетки не нарушены;
б — на границе срастания блоков возникла дис-
дислокация
Горизонтальный ряд узлов оказы-
оказывается разорванным и сдвинутым
по вертикали на один параметр. Ле-
Левее этой стрелки решетка почти не
нарушена. Если мы начнем обходить
эту стрелку, то через 180° встретим
опять практически ненарушенный
участок решетки, причем ряд
будет представлять собою почти
не искаженную прямую линию. Об-
Обходя по стрелке дальше область мак-
максимальных деформаций, мы будем
двигаться по винтовой кривой все
глубже и глубже в середине кристал-
кристалла, опускаясь на один параметр при
каждом обороте на 360°.
Выше мы описали два крайних слу-
случая дислокаций: линейной и винто-
винтовой. Реальные случаи, как правило,
представляют собою комбинацию этих
двух идеальных случаев. При этом
образуется криволинейная дислока-
дислокация с произвольными кривыми по-
поверхностями скольжения. Описать та-
такую дислокацию в каждом конкрет-
конкретном случае часто бывает трудно. Од-
Однако отдельные ее части обычно уда-
удается свести к описанным выше иде-
идеальным случаям.
3. Образование и движение дисло-
дислокаций. Выше было рассмотрено обра-
образование дислокаций за счет пластиче-
пластической деформации кристалла, однако
это далеко не единственная причина
их образования. Дислокации часто
появляются в, процессе роста кристал-
кристалла. Если отдельные блоки растущего
Рис. 268. Возникновение дислокации (б)
в местах скопления дефектов (а) в кри-
кристалле
2С1
т
Рис. 269. Возникновение и перемещение
дислокации в процессе сдвига
а — первоначальное состояние;
б — появление дислокации;
в — движение дислокации;
г — исчезновение дислокации на поверхности
кристалла
1
\
1
а
1
j
В
Рис. 270. Взаимодействие двух днижущих-
ся дислокаций в кристалле
а — положения дислокаций до перемещения;
б — образование серии точечных дефектов
в результате рекомбинации дислокаций
кристалла слегка разориентированы,
то на их стыке могут образоваться
дислокации. На рис. 267, а по-
показаны два блока с ненарушенными
решетками. В результате срастания
этих блоков по общей границе возни-
возникают линейные дислокации (рис.
267,6).
Дислокации могут возникнуть в
местах скопления дефектов кристал-
кристалла. Рис. 268 демонстрирует этот слу-
случай. В результате деформации струк-
структуры около этого дефекта, на его ме-
месте возникает дислокация (случайб).
Как и точечные дефекты, дислокации
могут перемещаться в кристалле. На
рис. 269, а—г последовательно пока-
показано, как под действием сил, сдвига-
сдвигающих нижнюю половину кристалла
относительно верхней, возникшая ли-
линейная дислокация перемещается
внутрь кристалла, выходит в конце
концов на его поверхность, где и ис-
исчезает. Это исчезновение дислокаций
на поверхности кристалла вполне
аналогично исчезновению вакансий, о
которой мы говорили выше (§3 на-
настоящей главы). В процессе пере-
передвижения дислокации взаимодейст-
взаимодействуют друг с другом. На рис. 270, а
показано взаимодействие двух линей-
линейных дислокаций противоположного
знака. В результате их рекомбинации
происходит образование ряда дефек-
дефектов — вакансий (рис. 270, б) или меж-
узельных атомов. Этот процесс
энергетически выгоден, так как упру-
упругие деформации кристаллической ре-
решетки при рекомбинации дислокаций
существенно уменьшаются.
§*. Зависимость
фианко-хнннческпх свойств
кристаллов
от реальной структуры
1. Дислокации и прочность материа-
материалов. Предыдущая глава была посвя-
посвящена рассмотрению вопроса о зави-
зависимости физико-химических свойств
кристаллов от их идеальной структу-
структуры. Однако многие свойства кристал-
кристалла зависят не столько от его идеаль-
идеальной структуры, сколько от тех дефек-
дефектов, которые всегда присутствуют в
реальных кристаллах. Рассмотрим
кратко зту зависимость.
Изучение дислокаций проводилось
особенно интенсивно в связи с разра-
разработкой теории прочности металлов.
Повышение прочности обусловливает
увеличение долговечности и надежно-
надежности работы деталей машин, снижает
их вес и увеличивает, следовательно,
полезную грузоподъемность различ-
различных видов транспорта, повышает ско-
скорость его движения и т. д.
262
Наличие даже небольшого числа
дислокаций в металлах, пронизываю-
пронизывающих в нем значительные участки,
часто бывает достаточным, чтобы сни-
снизить его теоретическую прочность на
несколько (порядков. Эта идея нашла
свое подтверждение после открытия
того факта, что нитевидные кристал-
кристаллы, так называемые «усы», обладают
почти идеально правильным безде-
бездефектным атомным строением и по
своей прочности приближаются к
наивысшей теоретически возможной
прочности.
Однако это не единственный путь
повышения прочности конструкцион-
конструкционных металлов. При пластической де-
деформации, от которой как раз и тре-
требуется уберечь металлические изде-
изделия, происходит движение дислока-
дислокаций, поэтому можно увеличить проч-
прочность, если затруднить эти движения.
Последнее можно сделать, например,
за счет увеличения количества дефек-
дефектов в кристаллах путем введения по-
посторонних атомов (легирующие до-
добавки) , а также за счет соответствую-
соответствующей обработки, в первую очередь,
термической. Все эти вопросы в на-
настоящее время очень широко освеще-
освещены в литературе *.
2. Методы обнаружения дефектов
кристаллов. В ряде случаев обнару-
обнаружение дислокаций и других дефектов
основывается на определенных физи-
физико-химических свойствах кристаллов,
зависящих от самих дефектов.
На месте выхода дислокации на
поверхность грани кристалла имеется
максимальное напряжение решетки,
и, следовательно, требуется несколь-
несколько меньшая энергия для отрыва ато-
атомов от этого места, чем для отрыва
атомов участка грани с совершенной
структурой. Подбирая условия для
такого эксперимента, можно наблю-
наблюдать результаты под микроскопом.
Наибольшее распространение среди
методов обнаружения дислокаций по-
получил метод травления. Хорошие ре-
* Дислокации в кристаллах. Библиогра-
Библиографический указатель, вып. 2. М., «Нау-
«Наука», 1966 г.
зультаты для некоторых веществ да-
дает и метод испарения. В местах вы-
выхода дислокаций в результате трав-
травления или испарения получаются ям-
ямки травления, которые могут быть до-
доведены до размеров, хорошо видимых
под микроскопом. Однако наиболее
тонкие наблюдения были сделаны при
электронномикроскопических иссле-
исследованиях.
Очень подробно разработаны сей-
сейчас всевозможные методы «декориро-
«декорирования», для чего требуется подбор
подходящих примесей (например,
цветных), которые осаждаются преж-
прежде всего на дислокациях. Непрозрач-
Непрозрачные примеси, осаждающиеся на де-
дефектах прозрачного кристалла, дела-
делают эти дефекты видимыми под мик-
микроскопом. В прозрачных, главным
образом, изотропных кристаллах
удается выявить области резких на-
напряжений на границах дислокаций в
поляризованном свете. Этот метод
особенно удобен для изучения несо-
несовершенств кристаллов внутри всего
их объема.
Существует и много других мето-
методов, носящих, однако, как правило,
более частный характер.
3. Зависимость электрических
свойств полупроводниковых кристал-
кристаллов от их реальной структуры. В кри-
кристаллах с гомеополярным типом свя-
связи каждая дислокация влечет за со-
собой появление ряда разорванных свя-
связей вдоль линии дислокации. Число
разорванных связей на один пара-
параметр решетки для веществ со струк-
структурой алмаза (Si, Ge) в зависимости
от типа плоскости скольжения, на-
направления оси дислокации и некото-
некоторых других дополнительных условий,
представлено в табл. 34 (Хорнстр,
1958 г.).
Неспаренные электроны с разор-
разорванных связей могут либо перейти
в зону проводимости, либо присоеди-
присоединить к себе второй электрон в силу
стремления атома образовать вокруг
себя октет. В первом случае дислока-
дислокация будет действовать как ряд доно-
доноров, во втором как ряд акцепторов.
263
ТАБЛИЦА 34
Число разорванных связей на один
параметр ячейки
Плоскость
скольжения
A11)
A11)
A00)
A00)
A10)
A10)
Ось
дислокации
[110]
[211]
[НО]
[100]
[100]
[211]
Число разорванных
связей на один
параметр ячейки
1,41
0,82 или 1,6.3*
0 » 2,83
0 » 2,0
0 » 2,0
0 » 1,63
Два значения в зависимости от дополнитель-
дополнительных условий.
В кремнии и германии, в частности,
дислокации являются акцепторами.
Если на дислокации заняты все ак-
акцепторные уровни, то лиши дислока-
дислокации оказываются заряженными отри-
отрицательно, а это обстоятельство уже
оказывает сильное влияние на рас-
рассеяние электронов проводимости и,
следовательно, на подвижность элек-
электронов и «дырок».
Кроме того, разорванные на дисло-
дислокации связи уменьшают концентра-
концентрацию носителей электрических заря-
зарядов. Если принять во внимание, что
на дислокациях и других дефектах
структуры скапливаются атомы при-
примесей, то легко представить себе, на-
насколько сильно могут влиять дисло-
дислокации на электрические свойства по-
полупроводниковых веществ, принимая
во внимание, что в плохих кристал-
кристаллах число выходов дислокаций на
1 смг поверхности может достигать ве-
величины 105 и даже выше. Характер
точечного дефекта (см. случаи, разо-
разобранные на рис. 261) может также
по-разному влиять на электрические
свойства полупроводников. Один и
тот же примесный атом, попав в по-
положение, отмеченное на рис. 261 а, в,
или на дислокацию, может проявить
прямо противоположные свойства.
В частности, в одном положении он
может играть роль донора, в другом—
акцептора или быть нейтральным.
4. Оптические свойства ионных
кристаллов, зависящие от дефектов
кристалла. Специфическими оптиче-
оптическими свойствами обладают дефект-
дефектные ионные кристаллы. Особенно
подробно в этом отношении изучены
кристаллы щелочно-галоидных соеди-
соединений.
Давно было известно, что нагрева-
нагревание NaCl или КС1 в паре соответ-
соответствующего щелочного металла вызы-
вызывает их заметное окрашивание соот-
соответственно в желтый и синий цвет.
В их спектрах поглощения удалось
обнаружить широкую область погло-
поглощения, максимум которой для NaCl
имел значение 4650 А и для КС1 —
5630 А. Эти полосы были названы F-
полосами.
В 1937 г. Де-Бур так объяснил по-
появление F-полос: атомы натрия или
калия из паров, проникая в кристал-
кристаллы соответствующих солей, создают
там точечные дефекты в анионной ча-
части, в которые попадают электроны
для компенсации валентности. Это и
есть F-центры. Получившаяся систе-
система с точки зрения квантовой механи-
механики весьма сходна с атомом водорода.
Кроме основного состояния, такой
электрон имеет ряд дискретных воз-
возбужденных уровней. F-поглощение
соответствует переходу электрона из
основного состояния в первое возбуж-
возбужденное состояние. Эта гипотеза была
впоследствии подтверждена многими
исследователями. Кроме F-центров
были найдены V-, R\~, R%-, М- и N-
центры, связанные с различными де-
дефектами кристалла. Так, например, V-
центр связан с появлением вакансии
в катионной части структуры, которая
служит ловушкой «дырок». F-центры
приводят к появлению соответствую-
соответствующих полос поглощения.
5. Влияние дефектов на анизотро-
анизотропию свойств кристаллов. Не только
оси дислокаций располагаются по оп-
определенным кристаллографическим
направлениям. Очень часто и точеч-
точечные дефекты выстраиваются в ряды
определенного символа. Следствием
этого может служить повышенная
264
анизотропия кристалла в этом направ-
направлении по сравнению с анизотропией
более совершенного кристалла. Осо-
Особенно заметно это бывает в том слу-
случае, когда в дефекты закономерно по-
попадают примеси. Последние могут да-
даже вызвать оптическую анизотропию
в кубических кристаллах. Хорошо
известна подобная анизотропия в пи-
пирамидах роста кубических кристал-
кристаллов, если последние кристаллизова-
кристаллизовались с примесью.
Те макродефекты, о которых бы-
было упомянуто в первых главах
книги, являются следствием микро-
микродефектов их структур. Спираль на
грани @001) кварца (см. рис. 15,
стр. 17) несомненно является
следствием спиральной дислока-
дислокации, появившейся во время роста
этого кристалла.
Основным положением химии
является утверждение, что свойст-
свойства любого химического соединения
будут всегда одинаковыми, незави-
независимо от способа его получения.
Это остается справедливым и для
молекул. Однако для твердого ве-
щесдва немолекулярного строения,
а таковых среди неорганических
соединений подавляющее боль-
большинство, это утверждение прихо-
приходится признать устаревшим, если
речь идет о тонких изменениях не-
некоторых свойств (например, элект-
электрических свойств полупроводнико-
полупроводниковых соединений).
Кристаллы, полученные разны-
разными методами, существенно отлича-
отличаются друг от друга своей реальной
структурой, а следовательно, и
свойствами. В зависимости от ма-
материала тигля, состава исходных
реактивов и т. п. в полученный кри-
кристалл всегда попадает какое-то ко-
количество микропримесей. Послед-
Последние могут весьма существенно вли-
влиять на физические свойства полу-
полученного кристалла.
Даже если предположить, что со-
состав кристаллов абсолютно одина-
одинаков, то ;и тогда они могут сущест-
существенно отличаться друг от друга
своей реальной структурой, как то:
степенью упорядоченности, числом
и направлением дислокаций, напря-
напряжениями, вызванными ростом и
т. п., т. е. они могут иметь и всегда
имеют разную степень совершенст-
совершенства, зависящую, в частности, от ско-
скорости кристаллизации. Некоторые
электрические свойства, например,
подвижность носителей тока, суще-
существенно различны у монокристал-
монокристаллов и поликристаллических агрега-
агрегатов одного и того же вещества.
В отличие от жидкостей и газов ин-
индивидуальность кристаллов всегда
должна учитываться при исследо-
исследовании твердых тел.
ЧАСТЬ ЧЕТВЕРТАЯ
КРИСТАЛЛОХИМИЯ
ПРОСТЫХ ВЕЩЕСТВ
И ХИМИЧЕСКИХ
СОЕДИНЕНИЙ
ГЛАВА XVII
КРИСТАЛЛОХИВ1ИЧЕСКИЕ ЗАКОНОМЕРНОСТИ
В ПЕРИОДИЧЕСКОЙ СИСТЕМЕ ЭЛЕМЕНТОВ Д. И. МЕНДЕЛЕЕВА
§ 1. Предварительные аанечанпя
В качестве основы для систематиче-
систематического обзора структур простых ве-
веществ была принята развернутая
форма менделеевской таблицы
(рис. 271). Эта форма удобна тем, что
в левой части таблицы группируются
элементы с типичными металлически-
металлическими свойствами, а в правой — с неме-
неметаллическими. Распределение на под-
подгруппы, принятое в таблице, наиболее
обоснованно как с точки зрения общ-
общности химических свойств элементов,
так и с точки зрения кристаллохими-
ческих особенностей, что будет пока-
показано ниже.
При рассмотрении таблицы легко
убедиться, что работа по определению
кристаллических структур химиче-
химических элементов в основном: закончена.
Число «белых пятен» в таблице очень
невелико. Уже одно это обстоятельст-
обстоятельство само по себе должно побуждать ис-
исследователей к отысканию каких-то
общих закономерностей.
Все многообразие типов структур
химических элементов можно свести
всего лишь к шести основным случа-
случаям, указанным в условных обозначе-
обозначениях. Элементы левой части таблицы
имеют простые структуры плотней-
ших шаровых упаковок и близких к
ним, вследствие ненаправленности
металлической связи. Кристалличе-
Кристаллические структуры элементов правой ча-
часта таблицы значительно разнообраз-
разнообразнее и сложнее вследствие направлен-
направленности и ограниченности числа кова-
лентных связей. Выделять среди них
отдельные структурные типы нецеле-
нецелесообразно. В их многообразии трудно
будет заметить общие черты.
Обозначения, принятые в таблице,
объединяют структуры элементов в
более крупные группы с общими важ-
важными структурными признаками.
С этой точки зрения первые три
структурных типа следовало бы так-
также объединить в одну группу струк-
структур типичных металлов. В этом слу-
случае число условных знаков сократи-
сократилось бы до четырех. Однако
поскольку сами структурные типы в
в этой группе весьма резко отличают-
отличаются друг от друга, и каждый структур-
266
ный тип представлен многочисленны-
многочисленными примерами, то нам казалось
целесообразным дать в этой части
таблицы более детальное подразделе-
подразделение.
Некоторые значки элементов в таб-
таблице окружены двумя или даже тре-
тремя окружностями или многоугольни-
многоугольниками. Эти условные ообозначения
относятся к полиморфным модифика-
модификациям. Низкотемпературной модифи-
модификации соответствуют внутренний
многоугольник или крут, высокотем-
высокотемпературной модификации — наруж-
наружный.
Если для того или иного элемента
известны несколько модификаций, но
структуры окончательно установле-
установлены, скажем, только для двух, то при-
приводятся данные, относящиеся к этим
двум модификациям. Кроме того, мо-
нотропные модификации не выделя-
выделяются особо, а включаются в таблицу
наряду с энантиотропными, если
структура их надежно определена.
В этих случаях относительное распо-
расположение многоугольников в одной
клетке таблицы становится, конечно,
несколько произвольным.
Незаконченные исследования
структур приводятся только тогда,
когда нет литературных данных, по
которым можно было бы судить о за-
законченном исследовании структуры
хотя бы одной модификации этого
простого вещества (см., например,
исследование структуры кристалличе-
кристаллического кислорода).
Рис. 271. Кристаллические структуры про-
простых веществ
1 — гексагональная плотиейшая упаковка;
2 — кубическая плотнейшая упаковка;
3 — кубическая центрированная упаковка;
4 — молекулярная структура;
5 — структура с координационным числом
К - 8 — N;
в — прочие структуры
/в it Ша
7а Ша
П ЕЬ ШЬ ПЬ 7Ь М ШЬ ШЬ
0 ®
:'в' © ф Щ) f (ne)
E (si4) Ф Ф @ ®
Лантаноиды
Th
(g) ($
®ш
Pm [SmJ QjjiJ
/¦;¦¦ ¦¦JS('
в
Cm
Bk
Cf
Es
Fm f
»1d No
Lr
О
О
)B
267
§ 2. Кристаллические структуры
истинных металлов
Обратимся к левой части таблицы.
Здесь наиболее распространенными
типами структур являются: 1) тип
гексагональной плотиейшей упаков-
упаковки; 2) тип кубической плотнейшей
упаковки; 3)тип кубической объемно-
центрированной упаковки.
Процент заполнения пространства
шарами для первых двух 74,05, для
третьей — 68,01.
Как для гексагональной, так и для
кубической плотнейших упаковок
координационное число равно 12, для
центрированной кубической — 8. Вы-
Высокие координационные числа обус-
обусловлены ненаправленностью металли-
металлической связи, следствием чего являет-
является стремление металлических атомов
окружить себя максимально возмож-
возможным числом соседних атомов.
Элементы левой части таблицы, как
правило, имеют структуру одного из
указанных трех типов. Исключением
являются а- и C-U, Pa, все три моди-
модификации марганца и а- и C-Np.
Ha примере а-Mn можно убедиться
в том, что равноценность всех атомов
в кристаллической структуре просто-
простого вещества не обязательна. В самом
деле, 58 атомов Мп, приходящихся на
одну ячейку, распадаются на четыре
группы, или четыре «сорта»: по 2, 8,
24 и 24 атома. Никакими симметри-
симметрическими преобразованиями нельзя
совместить атомы одного сорта с ато-
атомами других сортов. Это обстоятель-
обстоятельство позволяет предполагать, что
электронное состояние у этих атомов
тоже различное. Как ни своеобразен
структурный тип а-Mn, все же видно
большое сходство его с нормальными
структурами металлов: та же высокая
симметрия (кубическая), те же боль-
большие координационные числа. Струк-
Структура а-Mn имеет усложненный струк-
структурный тип объемноцентрированной
кубической решетки.
Условное обозначение в виде точеч-
точечного пунктира отвечает сложным
структурам. Однако если такая слож-
сложная структура близка к какому-либо
простому структурному типу, то фор-
форме фигуры, окружающей значок эле-
элемента, придается соответственно фи-
фигура этого структурного типа. В разо-
разобранном случае условным обозначе-
обозначением для структуры а-Mn будет
пунктирный восьмиугольник.
Структура a-U принадлежит к ром-
ромбической сингонии. Но она весьма
близка к нормальной структуре гек-
гексагональной плотнейшей упаковки,
что и отмечено пунктирным шести-
шестиугольником.
(З-Мп кристаллизуется в кубиче-
кубической решетке. Его структура весьма
близка к структурному типу плотней-
плотнейшей кубической упаковки. Атомы в
структуре р-Mn двух сортов, но оба
имеют координационные числа 12 с
несколько различными расстояниями
(от 2,36 до 2,67). К этому же струк-
структурному типу приближается структу-
структура Y-Mn. Ее решетка весьма близка
к гранецентрированной кубической,
но отличается от последней деформа-
деформацией по оси четвертого порядка.
Вследствие этой деформации симмет-
симметрия структуры тетрагональная. Отно-
Отношение осей в гранецентрированном
аспекте с/а' = 0,93, т. е. весьма близ-
близко к единице.
Разобрав таким образом все струк-
структуры элементов, расположенных в
левой части таблицы, можно сделать
заключение о большом сходстве всех
этих структур между собой. Имею-
Имеющиеся отклонения от простейших
трех типов не так значительны, чтобы
повлиять на этот вывод.
§3. Особенности структурных типов
Т-Мп, Hg и Zn
Структуру \"Мп, несмотря на боль-
большое сходство ее со структурой плот-
плотнейшей кубической упаковки, необ-
необходимо считать новым структурным
типом. При самой незначительной де-
деформации куба, например, в направ-
направлении оси четвертого порядка, резко
(скачком) меняется симметрия эле-
268
ментарной ячейки. В частности,
в структуре -у-Мп пропадают две оси
четвертого порядка и все оси третье-
третьего. Решетка из кубической превраща-
превращается в тетрагональную. В тетрагональ-
тетрагональной же структуре, по правилам Бравэ,
нужно выбирать не гранецентриро-
ванную ячейку, а объемноцентриро-
ванную вдвое меньшего объема. Опи-
cainie структуры у-Mn делается
обычно в гранецентрированном аспек-
аспекте только для того, чтобы подчеркнуть
большое сходство этой структуры со
структурой плотнейшей кубической
упаковки.
К тому же структурному типу
принадлежит In. Однако по сравне-
сравнению со структурой Y~Mn знак дефор-
деформации кубической решетки противо-
противоположный, вследствие чего отноше-
отношение осей в гранецентрированном
аспекте получается несколько боль-
большим единицы (с/а' = 1,08).
Очень интересна структура Hg.
Ртуть кристаллизуется в ромбоэдри-
ромбоэдрической решетке, которая, однако,
весьма близка к кубической гране-
центрировапной. Элементарная гра-
нецентрированная кубическая ячейка
в качестве примитивного параллеле-
параллелепипеда имеет острый ромбоэдр с yi-
лом а = 60°. Любая деформация та-
такого ромбоэдра (в данном случае
речь идет о деформации вдоль глав-
главной оси) влечет за собой исчезнове-
исчезновение целого ряда элементов симмет-
симметрии решетки; в частности, пропада-
пропадают 3Д осей третьего порядка и все
оси симметрии четвертого порядка.
Это обстоятельство влечет за собой
выбор в качестве элементарной ячей-
ячейки, по правилам Бравэ, уже не этого
искаженного куба, превратившегося
в ромбоэдр, а примитивного ромбоэд-
ромбоэдра, имеющего в этом случае ту же
симметрию и вчетверо меньший объ-
объем. Структура ртути, таким образом,
может быть получена из плотнейшей
кубической упаковки, если послед-
последнюю деформировать (сжимать) по
оси третьего порядка до тех пор, по-
пока примитивный ромбоэдр не изме-
изменит своего утла с 60 до 72°32'.
Гораздо сложнее со структурами
типа гексагональной плотнейшей
упаковки. Деформация вдоль главной
оси шестого порядка не влечет за со-
собой изменения симметрии (отноше-
(отношение с/а, в частности, меняется у од-
одного и того же вещества с темпера-
температурой) и поэтому нет такого жест-
жесткого критерия, каким мы пользова-
пользовались при описании структур, полу-
получающихся в результате деформации
куба.
На рис. 272 показаны шаровая гек-
гексагональная плотнейшая упаковка и
структуры элементов, у которых отно-
отношение осей с/а максимально откло-
отклоняется от этого идеала. Возникает
вопрос, какими отклонениями с/а от
идеального значения 1,633 можно пре-
пренебречь и какие отклонения следует
считать уже выходящими за пределы
данного структурного типа. Для отве-
ответа на этот вопрос обратимся к рис. 273,
представляющему собой распределе-
распределение по группам периодической систе-
системы тех элементов, которые кристалли-
кристаллизуются в подобных гексагональных
структурах. По оси ординат отложе-
отложены значения с/а. Параллельно оси
абсцисс проведена линия идеального
отношения с/а =1,633.
Легко видеть, что значения с/а
для структур 28 элементов распола-
располагаются вблизи линии идеального зна-
значения, обычно несколько ниже ее.
Соответственные значения для Zn и
Cd расположены совершенно в сто-
стороне. Особенно хорошо это видно,
если спроектировать все значения с/а
иа ось ординат и получить на ней
Идеальная решетна Cd
г/а = 1,57 с/а = /, 633 с/а = 1,89
Рис. 272. Идеальная гексагольная шаро-
шаровая упаковка и максимально отклоняю-
отклоняющиеся от нее структуры Be и Cd
269
с/а
1,90
1,80
1,70
iseV
'т
jfi33
i
г, г'
о
ее Be
i
Са «г
<xSc
i
е
т°ьс
i
Но°Ти°
I
Hf
1 I !
gRe
Тс @Ru
Os
, i
О Bi
¦°аСо
i i
oCd
°Zn
о
«Tl
1 1
la la Ша.
Wa r,-.
Рис. 273. Распределение элементов, кри-
кристаллизующихся в гексагональных струк-
структурах, по группам периодической системы
Ша Ша Ib ЛЬ
пределы отклонений. Этот график
дает возможность сделать вывод, что
все элементы, указанные на
рис. 273, за исключением Zn и Cd,
принадлежат к одному структурному
типу. Zn и Cd только приближаются
к нему аналогично тому, как у-Мп,
la и Hg приближаются к структур-
структурному тину плотнейшей кубической
упаковки.
Разобранные выше структуры
у-Мп, In и Hg, а также все рассмот-
рассмотренные гексагональные структуры
могут быть удобно интерпретирова-
интерпретированы, если шары плотнейшей упаковки
заменить соответственно эллипсои-
эллипсоидами вращения.
§4. Кристаллические структуры
элементов б-иодгрупи
Перейдем теперь к рассмотрению
кристаллических структур элементов
правой части менделеевской таблицы
(рис. 271).
Руководящим принципом здесь яв-
является правило Юм-Розери A930 г.)
о координационном числе атомов в
кристаллических структурах неметал-
неметаллических элементов (см. XII гл.,
стр. 179). Координационное число
в этих структурах К — 8 —./V, где JV—
номер группы периодической си-
системы элементов. Это правило являет-
является следствием тенденции атомов ок-
окружать себя октетом электронов за
счет образования общих пар электро-
электронов с соседними атомами. Каждая па-
пара электронов, принадлежащая од-
одновременно двум соседним атомам,
обусловливает ковалентную связь
между ними.
Из этого следует, что координаци-
координационное число атомов галогенов и водо-
водорода в кристаллической структуре
должно быть равно единице. Дейст-
Действительно, хлор, бром и иод имеют
структуры, в узлах решеток которых
располагаются двухатомные молеку-
молекулы и, следовательно, каждый атом
имеет одного ближайшего соседа.
Расстояние С1 — С1 в кристалличе-
кристаллической молекуле равно 2,02, в той же
структуре расстояние между бли-
ближайшими атомами из разных моле-
молекул равно 3,34. Водород кристаллизу-
кристаллизуется в плотнейшей гексагональной
структуре, в узлах которой также
располагаются двухатомные моле-
молекулы.
В VI-Ь подгруппе координацион-
координационное число должно быть равно 2. Оно
может быть осуществлено в замкну-
замкнутых кольцеобразных молекулах или в
бесконечных цепях. Оба варианта в
270
действительности и наблюдаются:
так, например, в ромбической сере и
в моноклинных сере и селене в узлах
решетки располагаются кольцевые
восьмиатомные молекулы. Расстоя-
Расстояние между соседними атомами в
структуре ромбической серы рав-
равно 2,10, а между атомами из разных
молекул — 3,30. В структуре селена
эти расстояния соответственно равны
2,34 и 3,53. Структуры гексагональ-
гексагональных селена и теллура построены из
бесконечных цепей с расстоянием
между соседними атомами у селена
2,32, у теллура — 2,86, а между ато-
атомами соседних цепей соответственно
3,46 и 3,74. Обе структуры полония
не удовлетворяют правилу К = 8—N.
В V-fe подгруппе координационное
число должно быть равно. 3. Это дей-
действительно имеет место в структурах
Р (черного), As, Sb и Bi. Все эти ве-
вещества имеют структуры, построен-
построенные из гофрированных слоев. Рас-
Расстояния между атомами внутри одно-
одного слоя всегда меньше, чем расстоя-
расстояния между атомами, находящимися в
соседних слоях:
Расстояние между р As Sb Bi
атомами
(черный)
в слое, А 2,17 2,51 2,87 3,10
соседних слоев, 3,87 3,15 3,37 3,47
А
В V-Ь подгруппе могут быть также
молекулярные структуры, но они
должны быть построены из четырех-
четырехатомных тетраэдрических молекул,
чтобы удовлетворять правилу Юм-
Розери. Это действительно имеет
место для низкотемпературной моди-
модификации фосфора, представляющей
собой плотнейшую кубическую упа-
упаковку таких молекул.
Типичным представителем элемен-
элементов IV-Ь подгруппы является угле-
углерод, имеющий в модификации алма-
8а структуру с координационным
числом 4. К тому же структурному
типу принадлежат и остальные эле-
элементы этой группы; Si, Ge, Sn, за
исключением свинца, который кри-
кристаллизуется в плотнейшей кубиче-
кубической структуре.
Структуры графита и белого олова
построены сложнее, чем это требу-
требуется согласно правилу К = 8 — N.
Рассматривая с этих позиций
структуры элементов подгрупп \-Ь,
VI-Ь и VII-Ь, видим, что многие из
них, удовлетворяя правилу К=8 — N,
одновременно являются молекуляр-
молекулярными. На рис. 271 этот факт отмечен
тем, что правая половина окружно-
окружности в условных обозначениях сплош-
сплошная, а левая изображена пунктирной
линией, соответствующей структу-
структурам с К. — & — N. Однако у некоторых
элементов этих подгрупп число кова-
лентных связей меньше, чем можно
было бы ожидать по правилу Юм-
Розери. В этом случае структуры ых
молекулярные, но неподчиняющиеся
этому правилу (см., например, кисло-
кислород и азот).
Правило К = 8 — N можно распро-
распространить и на элементы VII 1-й под-
подгруппы и формально считать, что
атомы благородных газов в кристал-
кристаллических структурах имеют коорди-
координационное число, равное нулю, т. е.
атомы в их молекуле не имеют бли-
ближайших соседей, связанных с ними
ковалентными связями. Эти вещест-
вещества имеют «одноатомные» молекулы,
которые связаны друг с другом Ван-
дер-Ваальсовыми силами. С этой точ-
точки зрения структуры благородных га-
газов следует отметить в таблице так
же, как и структуры VII-Ь подгруппы.
В таблице не выделяются специ-
специально молекулярные структуры типа
плотнейших упаковок. Однако по-
поскольку Ван-дер-Ваальсовы силы,
как и металлические, являются нена-
ненаправленными, то мы вправе ожидать
и действительно имеем среди молеку-
молекулярных структур химических эле-
элементов структуры, в которых моле-
молекулы упакованы по одному из типов
плотнейших упаковок. Такие струк-
структуры встречаются во всех группах,
где имеются молекулярные структу-
структуры. В Vll-ft подгруппе таким приме-
271
ром является структура водорода,
который кристаллизуется по типу
гексагональной плотнейшей упаков-
упаковки с идеальным отношением осей
с/а = 1,633. Это по-видимому, являет-
является следствием вращения молекул в
кристаллической решетке прп любой
температуре вплоть до абсолютного
нуля. Типы кубической и гексаго-
гексагональной плотнейших упаковок моле-
молекул наблюдаются в кристаллических
структурах кислорода, азота и даже
фосфора, где низкотемпературная
модификация представляет собой
плотнейшую кубическую упаковку
молекул 1*4, построенных тетраэдри-
чески. На рис. 271 все подобные
структуры показаны как молекуляр-
молекулярные. В обозначениях указывается
лишь наиболее существенный мо-
момент — подчиняется ли данная моле-
молекулярная структура правилу К =
= 8 — N. Тип упаковки, как второ-
второстепенный факт, специально не под-
подчеркивается.
Структуры благородных газов, как
указано было выше, можно рассмат-
рассматривать как молекулярные, с «одно-
«одноатомными» молекулами, формально
подчиняющиеся правилу К=8 — ./V,
и с этой точки зрения онп обознача-
обозначаются так же, как структуры галоге-
галогенов.
Элементы Ill-b подгруппы должны
были бы иметь по правилу К = 8 — N
координационное число 5, но, как
известно из теории кристаллических
решеток, в структурах не может
быть осей симметрии пятого порядка
или многогранников с пятью тож-
тождественными вершинами. Поэтому,
если бы даже тенденция атомов ок-
окружать себя пятью соседями была
весьма сильной, то и в этом случае
не могли образоваться столь правиль-
правильные структуры, как в рассмотренных
выше случаях для иных координа-
координационных чисел. Нужно также иметь
в виду, что даже сильно искаженные
структуры с координационным чис-
числом 5 из-за недостатка валентных
электронов не могут быть обусловле-
обусловлены только ковалентными связями.
Кроме того, lll-b подгруппа нахо-
находится уже достаточно далеко от эле-
элементов с ярко выраженными неметал-
неметаллическими свойствами и поэтому
необходимо, наряду с ковалентными
тенденциями, считаться и с сильно
выраженным стремлением к образо-
образованию нормальных металлических
структур. В результате «борьбы»
этих противоположных тенденций
в данной подгруппе появляются из
ряда вон выходящие, единственные в
своем роде «уродливые» структуры,
такие, например, как галлий, индий,
бор.
Структуры А1 и TI нормальные ме-
металлические. О них будет сказано
ниже.
Наконец, остаются элементы П-6
подгруппы. Их структуры кратко бы-
были рассмотрены в § 3 данной главы.
По правилу К = 8 — N следует ожи-
ожидать координационного числа 6.
Правда, у этих элементов, как и у
элементов предыдущей группы, не
хватает валентных электронов для
образования нужного количества ко-
валентных связей, да и металличе-
металлические тенденции выражены у них до-
достаточно ярко. Поэтому их структу-
структуры весьма близки к нормальным
структурам металлических элементов
с координационным числом 12. Од-
Однако, как указывалось выше, их все
же нельзя отнести к структурным ти-
типам плотнейших шаровых упако-
упаковок — они лишь приближаются к
ним. Вместо координационного числа
12 имеются две группы соседних ато-
атомов: 6 на кратчайшем расстоянии и
6 — на несколько большем. Так, для
Zn, Cd и Hg соответствующие значе-
значения межатомных расстояний будут:
2,66 и 2,91; 2,97 и 3,29; 3,00 и 3,47.
Это дает формальное право считать
структуры Zn,Cd и Hg удовлетворяю-
удовлетворяющими правилу К = 8 — N.
Таким образом, подводя итог обзо-
обзору структур химических элементов
правой части менделеевской таблицы,
можно сказать, что большинство эле-
элементов, находящихся здесь, имеет
молекулярные структуры или струк-
272
туры, подчиняющиеся правилу
К = 8 — N. Многие из этих структур
элементов, удовлетворяя правилу Юм-
Розери, одновременно являются мо-
молекулярными. Имеющиеся некоторые
сложные структуры не нарушают
общей гармонии.
§ 5. Особенность элементов
III-'; и IV-5 подгрупп,
имеющих типичные
структуры металлов
Четыре элемента, стоящие в правой
части менделеевской таблицы, име-
имеют структуры типа плотнейших упа-
упаковок: А1, а- и P-Tl, Pb, a также In,
имеющий очень близкую к кубиче-
кубической плотнейшей упаковке тетраго-
тетрагональную структуру. Объясняется этот
факт известной гипотезой, согласно
которой указанные элементы в кри-
кристаллическом состоянии не отщепля-
отщепляют всех своих валентных элементов.
Соли четырехвалентного свинца го-
гораздо менее устойчивы, чем двува-
двувалентного. Соли трехвалентного тал-
таллия менее устойчивы, чем соли
одновалентного. Атомы этих элемен-
элементов, входя в кристаллическую струк-
ТУРУ> отдают в общее пользование
только часть валентных электронов,
которых не может хватить на обра-
образование нужного количества кова-
лентных связей. Подтверждением не-
неполного отщепления электронов мо-
могут служить аномальные межатом-
межатомные расстояния в структурах простых
веществ. На рис. 274 по оси абсцисс
отложены атомные номера элементов
3, 5 и 6 периодов таблицы Менделе-
Менделеева, по оси ординат — межатомные
расстояния. Как видим, эти расстоя-
расстояния у Pb, Tl, A1 и In больше, чем
следовало бы ожидать по ходу кри-
кривой, соединяющей на диаграмме точ-
точки, отвечающие соответствующим
значениям межатомных расстояний
соседних с ними элементов. Недоста-
Недостаточное количество коллективизиро-
коллективизированных электронов в структурах Pb,
Tl, In и А1 по сравнению с соответ-
соответствующим количеством у других
J
12 °14 45 47 49
11 13 44 46 48 50
' '
М Ru Pd Cd Sn
Mg Si Rh Ag
А
77 79 81 83
76 78 80 82
i i i i i i i
Os Pt Hg Pb
1p Au Tl Bt
Атомный номер
Рис. 274. Аномалия в межатомных рас-
расстояниях у Pb, Tl, A1 и In
элементов тех же подгрупп приво-
приводит к ослаблению связи между ато-
атомами. В результате межатомные рас-
расстояния получаются несколько завы-
завышенными (приблизительно на 0,15 А)
по сравнению с соседними элемента-
элементами, полностью отщепляющими в кри-
кристаллическом состоянии свои валент-
валентные электроны.
Кроме указанных выше ненормаль-
ненормальных свойств алюминия, аномалию его
можно видеть, по Юм-Розери, из
сравнения температур плавления А1
и соседних с ним элементов:
Na
97°С
Mg
650°С
А1
658° С
Si
1420°С
Увеличение температуры плавле-
плавления AI по сравнению с магнием все-
всего на 8° противоречит обычному по-
повышению температур плавления
элементов коротких периодов с уве-
увеличением валентности элемента.
§ в. Распределение элементов
по подгруппам
периодической системы
на основании
нристаллохимических данных
В этом параграфе будет указана
возможность использования общих
закономерностей кристаллических
18 Кристаллохимия
273
структур простых веществ для обос-
обоснования распределения элементов по
подгруппам периодической системы.
Дело в том, что в химической литера-
литературе фигурируют до 10 вариантов та-
таблицы Менделеева, причем авторы
обычно не считают нужным обосно-
обосновывать тот или иной вариант разделе-
разделения на подгруппы. Одни и те же эле-
элементы у одних авторов оказываются в
главных, у других — в побочных под-
подгруппах. Кроме того, и объем каждой
из этих подгрупп не остается по-
постоянным.
Эти варианты сводятся к различ-
различному расположению элементов корот-
коротких периодов над элементами длин-
длинных. Особенно часто «перестанов-
«перестановкам» подвергаются элементы корот-
коротких периодов II, III и IV групп, а
также водород. Иногда элементы ко-
коротких периодов размещают посере-
посередине между подгруппами элементов
длинных периодов. Кроме того, обыч-
обычно не выделяют подгруппу VIH-fe,
считая ее нулевой группой. Все это
создает большую путаницу в учебной
литературе.
Первая попытка использовать кри-
сталлохимические закономерности
для обоснования варианта, изо-
изображенного на рис. 271, сделана на-
нами (Бокпй, 1942 г.). Появившиеся
в последующие годы описания новых
экспериментальных работ по опреде-
определению структур простых веществ це-
целиком подтверждают правильность
предложенного распределения эле-
элементов на подгруппы.
Почти к такому же варианту напи-
написания периодической системы эле-
элементов пришли независимо друг от
друга и другие исследователи — Абег
и Панет A933 г.) и Б. В. Некрасов
A935 г.).
Вариант Некрасова отличается от
варианта, предложенного автором
настоящей монографии, только на-
наименованием подгруппы инертных
газов: Некрасов считает необходимым
выделить их в отдельную (нулевую)
группу, автор причисляет их к Vlll-fr
подгруппе. В этом есть своя логика:
274
если на внешней оболочке атома на-
находится один электрон, подгруппу
называют первой, два — второй, ...,
семь — седьмой, если же 8— то ее
следует называть восьмой, а не нуле-
нулевой, как это обычно делают. Благород-
Благородные газы заканчивают периоды, а
не начинают новые. Некоторые авто-
авторы идут еще дальше и даже в самой
нулевой группе различают две под-
подгруппы. Это уже является принципи-
принципиальной ошибкой.
В последние годы рядом авторов
,было получено; ецного химических
соединений благородных газов. Это
обстоятельство тоже является осно-
основанием, чтобы не считать эти элемен-
элементы совершенно исключительными,
требующими выделения их в отдель-
отдельную особую группу.
Недостатком разделения элементов
по подгруппам на основании физиче-
физических методов исследования является
то, что для разных свойств получа-
получаются разные варианты таблицы. Так,
например, по своим спектральным
свойствам водород аналогичен щелоч-
щелочным металлам, а гелий — щелочнозе-
щелочноземельным. Поэтому оба эти элемента
в таблице периодической системы в
работах, посвященных спектроскопи-
спектроскопическим исследованиям химических
элементов, помещаются в первой и во
второй группах, где по этим свойст-
свойствам им и надлежит быть. Однако на-
нахождение гелия во второй группе при
классификации, учитывающей не спе-
спектральные, а какие-либо другие физи-
физические свойства, оказывается совер-
совершенно неоправданным.
Общим недостатком обычных хи-
химических методов разделения элемен-
элементов по подгруппам является то, что
свойства каждого элемента изучают-
изучаются в процессе реакции, т. е. во взаи-
взаимодействии с другими элементами, а
от характера этих последних и от
внешних условий зависит поведение
испытуемого элемента. Только крис-
таллохимический метод лишен этого
недостатка. Этим методом изучены
кристаллические структуры металлов
и неметаллов в состоянии, когда ато-
мы данного элемента находятся «во
взаимодействии» только с атомами
того же самого элемента, что пред-
представляет, конечно, определенное пре-
преимущество.
Кроме того, по сравнению с други-
другими химическими методами, которые
всегда изучают вещество в процессе
превращения и поэтому могут быть
названы «динамическими», этот ме-
метод является «статическим», так как
он изучает вещество в состоянии по-
покоя. Благодаря статичности резуль-
результаты, полученные из изучения струк-
структур простых веществ, весьма удобно
использовать для целей систематики
элементов.
Теперь можно перейти к самому
распределению элементов по под-
подгруппам.
VHI-fr подгруппа включает все
благородные газы. К VII-& подгруп-
подгруппе, кроме всех галогенов, бесспорно
относится также и водород. В VI-6
подгруппе находятся элементы, удо-
удовлетворяющие, как и в предыдущих
случаях, правилу К=8—N, а так-
также О и Ро. Элементы V-b подгруппы
характеризуются молекулярными
слоистыми структурами с расстоя-
расстояниями между атомами в слое мень-
меньшими, чем между атомами из раз-
разных слоев (N, Р). Элементы V-a под-
подгруппы — V, Nb и Та, в отличие от
вышеуказанных, кристаллизуются
в объемноцентрированных кубиче-
кубических структурах. IV-fr подгруппу со-
составляют С, Si, Ge, Sn и Pb с харак-
характерными алмазными структурами
(координационное число 4) для боль-
большинства из них. Отнесение угле-
углерода и кремния, как это делается не-
некоторыми авторами, к подгруппе Ti,
Zr, Hf и Th, имеющих типичные ме-
металлические структуры с координа-
координационным числом 12, следует приз-
признать совершенно неправильным. Как
было сказано выше, в подгруппу
lll-b по ряду кристаллохимических
соображений мы относим В, Al, Ga,
In и Т1. Эти элементы имеют в том
или ином отношении аномальные
кристаллические структуры. От
них отделяем подгруппу 111-а — Sc,
Y, La.
Принадлежность В и Al к подгруп-
подгруппе Ga, а не Sc, можно подтвердить,
также сходством наружной элект-
электронной оболочки, т. е. пользуясь ме-
методом, аналогичным методу Некра-
Некрасова. В самом деле, строение наруж-
наружной оболочки у В, А1 и Ga будет
2s22p\ 3s2Spx и As4p\ в то время
как у Sc — 3d'4s2.
Соответствующей разницы у эле-
элементов II группы нет, поэтому этот
критерий в данном случае не может
быть принят во внимание. Во II
группе Zn, Gd и Hg как вещества,
имеющие аномальные металлические
структуры, удовлетворяющие до из-
известной степени правилу ,ЙГ=8 — N,
составляют подгруппу b; Be, Mg, Ga,
Sr и Ва составляют подгруппу а и
характеризуются нормальными ме-
металлическими структурами.
1-6 подгруппа (Си, Ag, и Аи) ха-
характеризуется гранецентрирован-
ными кубическими структурами; 1-а
(Li, Na, К, Rb, Cs) в обычных усло-
условиях — объемноцентрированными.
Минимальные межатомные расстоя-
расстояния у элементов подгруппы Ь B,55;
2,88 и 2,87) также резко отлича-
отличаются от соответствующих расстояний
у элементов подгруппы а C,03; 3,71;
4,62; 4,87 и 5,26), что, впрочем, ха-
характерно и для других групп.
Таким образом, по структурным
кристаллохимическим данным 1-а,
И-а, Ш-Ь, IV-6, V-Ь, VI-6, VII-Ь и
VIII-Ь подгруппы являются главны-
главными, остальные — побочными.
§ 7. О классификации
химических соединений
Прежде чем говорить о классифика-
классификации химических соединений, необхо-
необходимо сгруппировать химические эле-
элементы в периодической системе
Д. И. Менделеева, т. е. провести в таб-
таблице какие-то «границы» между
элементами, которые объединили бы
более близкие друг другу элементы
и разделили бы более далекие.
18*
275,
Проведение любых границ в таб-
таблице, конечно, условно. В зависимо-
зависимости от внешних условий и от окру-
окружающей среды один и тот же элемент
будет проявлять себя по-разному:
в одних условиях он может быть
близок к одним элементам, в дру-
других условиях — к другим. Проведе-
Проведение резких границ — неизбежный не-
недостаток всякой классификации. Мо-
Может быть по этой причине до сих пор
в химии нет одной какой-либо обще-
общепринятой классификации химиче-
химических соединений.
Однако выделение и совместное
рассмотрение определенных групп
соединений неизбежно. Отдельно рас-
рассматриваются всегда соединения ме-
металлических элементов с неметал-
неметаллическими элементами, отдельно рас-
рассматриваются и органические соеди-
соединения.
Некоторые авторы предлагают
класть в основу классификации тип
химической связи. Недостатки этой
системы были выше обсуждены (см.
стр. 209). Автор данной монографии
положил в основу классификации
периодическую систему элементов
Д. И. Менделеева. Тот факт, что не-
некоторые из групп или областей со-
соединений по этой классификации со-
совпадают с группами, принятыми дру-
другими авторами, не может считаться
ее недостатком или слабой сто-
стороной.
Напротив, поскольку целью всякой
классификации является выделение
и группировка сходных по свойствам
химических соединений, то совпаде-
совпадение (частичное или полное) для оп-
определенных групп соединений вполне
возможно.
Таблица Менделеева позволяет для
подавляющего большинства соеди-
соединений сразу находить область, в ко-
которую входит данное соединение,
или же она допускает выбор одной
из двух областей для переходных по
свойствам соединений. Последнее
обстоятельство лежит в самой при-
природе элементов, составляющих эти
соединения. Их «пограничное» поло-
положение обусловливает то, что они лег-
легче других реагируют на внешние
воздействия и, в зависимости от ус-
условий и среды, проявляют свойства то
одной области соединений, то другой.
Ясно, что описывать такие соедине-
соединения следует в одном месте (с одной
группой соединений), но ссылка на
это описание обязательно должна
иметься и при описании другой груп-
группы соединений. Иногда удобно рас-
рассматривать группы переходных сое-
соединений отдельно, после описания
значительно больших по объему
групп типичных соединений.
При проведении границ в периоди-
периодической таблице нужно считаться с
особенностями кристаллических
структур простых веществ. Однако
слишком формальным и поэтому не-
неправильным было бы положить в ос-
основу классификации структурные
типы.
Один и тот же структурный тип
также может быть обусловлен раз-
различными причинами и поэтому может
быть характерен для совершенно
разных по своей природе простых
или сложных веществ. Так, напри-
например, металлическая медь и аргон
кристаллизуются в структурном типе
плотнейшей кубической упаковки;
NaCl и SnSb имеют весьма близкие
структуры; одним и тем же струк-
структурным типом характеризуются
CsCl, BeCu, LaCd и т. д. Правда,
сходные между собой химические сое-
соединения кристаллизуются обычно в
одинаковых или близких структур-
структурных типах, поэтому игнорировать
структурные типы при классифика-
классификации нельзя, но необходимо помнить,
что число структурных типов во мно-
много раз меньше числа химических со-
соединений и поэтому одни и те же
структурные типы могут встречаться
у разных по своей природе химиче-
химических соединений. Ясно также, что
сходные по своей природе химиче-
химические соединения, особенно те, кото-
которые кристаллизуются в одинаковых
или близких структурных типах,
будут иметь, в частности, один и
376
тот же или, по крайней мере, близ-
близкий тип химической связи. Но этот
факт будет уже вторичным, подчи-
подчиненным. Он будет являться следст-
следствием принятой классификации со-
соединений, а не ее основой. Это не зна-
значит, конечно, что природа химиче-
ческой связи будет игнорироваться.
Этому вопросу необходимо уделить
при описании определенных обла-
областей или групп химических соеди-
соединений должное внимание.
§ 8. Классификация
двойных (бинарных)
и более сложных
химических соединений
Какие же границы можно провести
в периодической таблице химических
элементов? Какие имеются в таблице
области, объединяющие наиболее
близкие между собой химические
элементы?
Важнейшая вертикальная граница
в таблице проходит через элементы
IV-ft подгруппы (рис. 271). Струк-
Структуры большинства элементов IV-Ь
подгруппы подчиняются правилу
К = 8 — N, но в отличие от элемен-
элементов, стоящих от грапицы справа,
структуры их гомодесмические, и в
этом отношении они сходны с метал-
металлическими элементами, находящими-
находящимися слева от границы.
Итак, справа от границы распола-
располагаются элементы с гетеродесмиче-
скими структурами, подчиняющиеся
правилу К = 8 — N, слева — элемен-
элементы с гомодесмическими или почти
гомодесмическими структурами, не
подчиняющиеся строго правилу К =
= 8 — N. Сами же элементы IV-6
подгруппы имеют черты тех и дру-
других: с одной стороны, их структуры
являются гомодесмическими; с дру-
другой стороны, большинство из них под-
подчиняется правилу К = 8 — N. Двой-
Двойственность природы этих элементов
сказывается, конечно, не только в
структурах их простых веществ, но
и в ряде других химических свойств.
Так, например, в таких соединениях,
как ЭпОг и РЬОг, кристаллизующих-
кристаллизующихся в структурном типе рутила или
PbF2, кристаллизующихся в струк-
структурном типе флюорита (CaF2), эле-
элементы IV-fe подгруппы играют элек-
электроположительную роль. В таких
же соединениях, как, например,
MgaSn или MgaPb, кристаллизующих-
кристаллизующихся в структурном типе антифлюори-
антифлюорита (Na2O), те же элементы IV-Ь
подгруппы играют электроотрица-
электроотрицательную роль.
Граница, проходящая по углероду,
обозначена пунктиром, так как для
углерода гораздо более характерной
будет структура графита, а не алма-
алмаза. Все же сказанное выше относит-
относится к структуре алмаза.
Справа от этой границы уже не
встречается «металлических» струк-
структурных типов.
Следовательно, граница, проходя-
проходящая через IV-fe подгруппу, отделяет
металлические элементы (металлы)
от неметаллических (неметаллов).
Установления одной вертикальной
границы уже достаточно для про-
проведения первого деления бинарных
химических соединений на три
области.
1) Область интерметаллических со-
соединений, т. е. соединений элементов,
находящихся слева от границы, меж-
между собой.
2) Область соединений металличе-
металлических элементов с неметаллическими.
Эти соединения обычно подчиняются
правилам валентности и составляют
главный предмет изучения неоргани-
неорганической химии и минералогии. Поэто-
Поэтому мы будем называть эти соединения
неорганическими, пользуясь этим
термином в более узком смысле сло-
слова, чем это обычно принято.
Объединение этих двух групп в од-
одну,, как это делается многими автора-
авторами, на том только основании, что все
эти соединения не являются орга-
органическими соединениями, непра-
неправильно.
3) Третью область соединений по
аналогии с предыдущими можно бы-
было бы составить из неметаллических
277
элементов. Однако по свойствам эти
элементы значительно резче отлича-
отличаются друг от друга, чем металлы. По-
Поэтому для выделения более однород-
однородных частей приходится провести еще
«диагональную границу», идущую от
бора к астату (№ 85). Справа от этой
границы расположены элементы, у
которых кристаллические структуры
хотя бы для одной модификации мо-
молекулярные (или сложные, где
К < 8 — N, например графит). Неко-
Некоторые авторы называют их элемента-
элементами-органогенами. Соединения этих
элементов составляют область моле-
молекулярных, или органических соеди-
соединений в более широком смысле сло-
слова, чем это обычно принято, т. е. в
эту область попадают соединения
элементов-органогенов не только с
углеродом, но и друг с другом. Об-
Область этих соединений мы будем на-
называть областью органических соеди-
соединений и их аналогов.
Для элементов, стоящих ниже ди-
диагональной границы (Sb, Bi, Те, Ро),
сохраняем старый термин — «полу-
«полуметаллы». Если их структуры не под-
подчиняются правилу Юм-Розери, то
К > 8 — N. Соединения этих элемен-
элементов друг с другом не составляют осо-
особой области соединений, так как по
своим свойствам они приближаются
то к первой, то ко второй, то к третьей
группам более типичных соединений.
Рассматривать соединения полуме-
полуметаллических элементов, так же как и
элементов, через которые проходят
«границы», удобнее отдельно, после
того как рассмотрены области типич-
типичных соединений. Для структуры бо-
бора, находящегося на самой диагональ-
диагональной границе, К 3g 8 — Л^.
Диагональная граница имеет зна-
значение не столько для классификации,
сколько для объединения веществ со
сходными электрическими свойства-
свойствами, поскольку металлический харак-
характер элементов нарастает в периодиче-
периодической таблице справа налево и сверху
вниз.
Для первой области соединений бу-
будет характерна металлическая связь.
Вторая область будет характеризо-
характеризоваться постепенным переходом от ти-
типичных ионных связей (типа NaCI)
к типичным ковалентным (типа PtS).
Соединения третьей области обычно
имеют гетеродесмические структуры
с ковалентной связью внутри молекул
и остаточной — между молекулами.
Соединения полуметаллических эле-
элементов по своему характеру будут
приближаться то к одной, то к дру-
другой, то к третьей из этих областей в
зависимости от свойств соединяющих-
соединяющихся элементов и внешних условий.
В свою очередь в рамках областей
могут быть выделены и более мелкие
«семейства» элементов. Это особенно
важно сделать для металлов, состав-
составляющих около трех четвертей всех
элементов. Металлы удобно делить на
а-металлы (Ма) и fr-металлы (М&) и
отделять среди первых щелочные и
щелочноземельные (Ms) от переход-
переходных (М<г) с выделением из последних
лантаноидов и актиноидов (М/), ко-
которые удобно рассматривать вместе с
металлами II 1-я подгруппы (металлы
вообще будем обозначать буквой А
или же другими начальными буквами
алфавита).
Типичные неметаллические эле-
элементы будем обозначать буквой X
или вообще последними буквами ал-
алфавита, полуметаллы — буквой Т.
Самую простую систематику бинар-
бинарных соединений, которая позволила
бы обозреть все многообразие их,
можно развить, если выписать через
равные промежутки на двух коорди-
координатных осях символы всех химиче-
химических элементов (например, в порядке
атомных номеров) и разделить затем
всю площадь координатного полу-
полуквадрата на клетки. Тогда каждая
клетка будет отвечать определенной
паре химических элементов. Эта пара
элементов может вовсе не давать друг
с другом химических соединений или
же может иметь одно или несколько
соединений. Если в каждой клетке
выписать все бинарные соединения
между соответствующими элемента-
элементами, то получим полную и весьма про-
278
стую систематику двойных соедине-
соединений. Недостаток такой системы не
только в ее громоздкости. Она нехо-
нехороша и тем, что близкие по характе-
характеру строения и свойствам соединения
окажутся разобщенными между со-
собой. На площади координатного полу-
полуквадрата нельзя будет выделить об-
областей близких между собой химиче-
химических соединений.
Кроме того, такая систематика бу-
будет слишком формальной, так как в
ее основе нет химического принципа.
Она не базируется на периодическом
законе.
Однако если объединить заранее
химические элементы и выписывать
их не в порядке атомных номеров, а
по подгруппам (сверху вниз) перио-
периодической системы, то такая система-
систематика будет в значительной мере ли-
лишена обоих указанных недостатков
(см. схему). В такой последователь-
последовательности и целесообразно давать обзор
соответствующих групп бинарных со-
Схема классификации веществ
Более сложные соединения ти-
типа FeCu2SnS4 или K2VF5O будут
примыкать соответственно к ука-
указанным группам тройных и могут
быть описаны сразу после них.
Отдельную группу составят, ко-
конечно, сложные соединения с нейт-
нейтральными, частицами, например
кристаллогидраты.
Прежде чем переходить к дета-
деталям классификации, необходимо
сделать существенную оговорку.
Бесспорно, что в развернутой форме
периодической системы слева груп-
группируются металлы, а справа — не-
неметаллы. Но где проходит граница?
Выше было сказано, что наиболее
четкая граница проходит по элемен-
элементам IV-Ь подгруппы. Однако, конечно,
это тоже в какой-то мере условно,
как и всякое проведение границ в
любой системе с непрерывным изме-
изменением свойств. Для большинства
соединений проведенная нами грани-
граница оправдывается.
Простые вещества
Двойные соединения
Тройные
Четверные
А
АВ
ABC
ABCD
ах:
АВХ'
АВСХ
AXY
ABXY
AXYZ
X
XY
XYZ
WXYZ
единений с описанием их физико-хи-
физико-химических свойств. Причем тройные
и более сложные соединения элемен-
элементов, принадлежащих одной области,
будут иметь много общего с соответ-
соответственными бинарными соединениями
и поэтому обзор их можно давать
вслед за соответствующими бинарны-
бинарными соединениями, т. е. описание про-
проводить не по строкам,, а по столбцам.
Тройные соединения металличе-
металлических элементов с неметаллически-
неметаллическими следует разделить на две части:
в первую войдут соединения с дву-
двумя металлическими и одним неме-
неметаллическим элементом, например
FeCuS2, во вторую — с одним ме-
металлическим и двумя неметалличе-
неметаллическими, например BaSO-t.
Проведение границы по IV-fr под-
подгруппе оправдывается для подавля-
подавляющего большинства соединений, но
не для всех. В некоторых случаях
элементы V-6 и даже Vl-fr подгрупп,
в особенности наиболее тяжелые, в
некоторых соединениях проявляют
металлический характер. И соедине-
соединение с ними металлов приводит к по-
появлению фаз металлического харак-
характера, которые, с точки зрения автора,
и следовало бы называть м е т а л л и-
дами.
Даже самые электроотрицательные
элементы — кислород и фтор — в не-
некоторых соединениях с металлами мо-
могут образовывать фазы, состав кото-
которых не подчиняется правилам валент-
валентности, а свойства близки к металли-
279
ческим. Таковы, например, субокис-
субокислы типа МозО, W3O, соединения ти-
типа Ag2F, карбиды, нитриды, гидриды
и некоторые другие аналогичные со-
соединения. Для всех этих веществ
термин «металлиды» показывает, что
несмотря на то, что в их состав вхо-
входят неметаллические элементы, фи-
физические свойства у таких соедине-
соединений близки к свойствам металлов или
инт ер металлических соединений, и
они существенно отличаются по сво-
своим электрическим свойствам от дру-
других соединений металлов с неметал-
неметаллами, большинство из которых явля-
являются либо изоляторами, либо полу-
полупроводниками.
§ О. Кристаллохимическая
систематика
химических соединений
1. Принципы кристаллохтшческой
систематики. В основе кристаллохи-
мической систематики лежит хими-
химический состав и атомная кристалли-
кристаллическая структура веществ.
Эти два фактора — состав и струк-
структура — определяют многие физиче-
физические свойства веществ, а ведь приме-
применение твердых тел связано именно с
наличием у них определенной ком-
комбинации физических свойств, следо-
следовательно, такая систематика облег-
облегчает выбор веществ для практиче-
практического использования. Примером мо-
могут служить алмаз и графит, облас-
области применения которых, несмотря на
одинаковый состав, совершенно раз-
различны из-за резкого отличия их фи-
физических свойств, связанных с их
атомным строением. Химический со-
состав следует положить в основу на
первоначальных стадиях создания
систематики, о чем будет сказано
ниже.
Всякая систематика должна преж-
прежде всего определить объекты, подле-
подлежащие рассмотрению. Автор пред-
предполагает систематизировать химиче-
химические соединения, находящиеся в
кристаллическом состоянии. Но до
280
сих пор мы не имеем общепринятого
определения химического соедине-
соединения. Пока речь идет о соединениях
молекулярного строения (например,
СО2, H2S, CH4 или даже координа-
координационных соединений постоянного
состава типа NaCl или CaSOi) вопрос
как будто ясен. Но большинство не-
неорганических соединений, в особен-
особенности интерметаллических, имеет
переменный состав, поэтому опреде-
определение понятия химического индиви-
индивидуума в этом случае сделать гораздо
труднее.
С точки зрения автора, понятие
химического индивидуума близко к
понятию фазы (в этом отношении
автор согласен с соответствующими
высказываниями Н. С. Курнакова),
но не тождественно этому понятию.
Если два вещества А и В в физико-
химической системе дают непрерыв-
непрерывные твердые растворы (А, В), то фа-
фаза будет одна, а веществ, по крайней
мере, два. Многие авторы предлага-
предлагают считать здесь три вещества: от О
до 337з% А, от 33 7з до 662/з% А п
от 662/з до 100% А. Разрешить этот
вопрос можно только формально, за-
заранее договорившись, где мы усло-
условимся поставить границы.
Таким образом, по существу мож-
можно считать, что в такой-то системе
отдельными веществами (простыми
веществами или химическими соеди-
соединениями постоянного или перемен-
переменного состава) будут считаться все
фазы, кроме фаз, простирающихся
непрерывно от одного компонента до
другого. В трехкомпонентных систе-
системах отдельные фазы могут прости-
простираться не только от одного компо-
компонента до другого, но и от одного би-
бинарного соединения, расположен-
расположенного на границе фазовой диаграм-
диаграммы, до другого. Еще более сложные
случаи могут быть в многокомпонент-
многокомпонентных системах. Как было сказано
выше, для всех этих фаз надо фор-
формально условиться о границах и счи-
считать, что в таких системах сущест-
существует 2, 3 или более индивидуальных
веществ. Если в твердых растворах
происходит упорядочение, то сверх-
сверхструктуру приходится считать от-
отдельным индивидуумом.
Для неживой природы весьма ха-
характерен непрерывный переход од-
одного вида в другой, и не только ви-
видов, но и более высоких, таксономиче-
таксономических (систематических) категорий. С
этим фактом приходится считаться. В
этом и заключается существенная
граница между кристаллохимиче-
ской систематикой веществ (или дру-
других объектов неживой природы) и
систематиками биологических объек-
объектов. Остановимся прежде всего на
систематике соединений постоян-
постоянного состава.
2. Определение класса и подклас-
подкласса. Выделение классов химических
соединений обычно делается по хи-
химическому составу, причем в каче-
качестве элементов, определяющих клас-
классы, берутся неметаллы. По ним да-
дается и название классов. Так, обыч-
обычно различают классы окислов, суль-
сульфидов, фторидов и т. д. Соли кисло-
кислородных кислот объединяются в
классы нитратов, карбонатов и суль-
сульфатов. .Автор не предполагает менять
эту систему коренным образом, но
ее принципы надо выдержать более
строго, руководствуясь при этом пе-
периодическим законом Менделеева.
Классы объединяются в разделы, на-
например раздел солей кислородных
кислот.
3. Предлагаемая систематика клас-
классов. Из схемы (см. стр. 279) вид-
видно, что все простые вещества раз-
разделяются на две группы: А и X;
двойные соединения — на три груп-
группы: АВ, АХ, XY; тройные соедине-
соединения — на Четыре группы: ABC, ABX,
AXY, XYZ; четвертые соединения —
на пять групп: ABCD, ABCX, ABXY,
AXYZ, XYZW и т. д. Эту таблицу
можно было бы продолжить до бес-
бесконечности, однако в этом, как по-
показывает практика, нет никакой
нужды.
Все столбцы быстро «выклинива-
«выклиниваются» по той причине, что отдель-
отдельные химические элементы, входящие
в конкретное химическое соедине-
соединение, изоморфно замещают друг дру-
друга и таким образом сводят сложные
соединения к более простым.
Если в схеме провести верти-
вертикальные линии за первым столбцом
и последним, то выделяются три
группы соединений: первый стол-
столбец — интерметаллические соедине-
соединения, последний столбец — соедине-
соединения, составленные из неметалличе-
неметаллических элементов и, в частности, из эле-
элементов-органогенов, а в центре таб-
таблицы будут располагаться неоргани-
неорганические соединения в узком смысле
этого слова.
Если рассмотреть столбцы только
центральной части схемы, то легко
заметить, что первый столбец ха-
характеризуется тем, что у него один
неметаллический атом и один, два,
три и т. д. металлических; во втором
столбце группируются вещества, у
которых два неметаллических атома
и один, два, три и большее количест-
количество металлических; в третьем столб-
столбце — три неметаллических атома и
несколько металлических.
Систематика неорганических сое-
соединений основывается, главным об-
образом, на их анионной части. Анио-
Анионов гораздо меньше, чем катионов, и
они резче отличаются друг от друга
по свойствам. Это выражается, в ча-
частности, в том, что при переходе от
одной подгруппы к другой (в правой
части рис. 271) мы всегда встреча-
встречаемся с изменением на единицу ва-
валентности, в то время как металлы
тех же периодов обладают перемен-
переменной валентностью.
В один класс объединяют соедине-
соединения с однотипными анионами. Автор
тоже придерживается этой системы,
но с разделением на подклассы. Сле-
Следовательно, если рассматривать ве-
вещество, попадающее в первый стол-
столбец схемы, то подкласс, к которому
относится данное вещество, опреде-
определяется тем элементом, который под-
подразумевается под значком X. Если
X — кислород, то получим подкласс
окислов; если X — сера, то подкласс
281
будет называться соответственно под-
подклассом сульфидов; если X — хлор,
то получим подкласс хлоридов
и т. д.
В один класс будем относить все
вещества, у которых анионы принад-
принадлежат к одной подгруппе периоди-
периодической системы. Так, соединения ме-
металлов с кислородом, серой, селеном
и теллуром будем относить к одному
классу MXVI, где XVI любой элемент
VI-Ъ подгруппы.
Для конкретности обратимся к си-
систематике бинарных соединений.
Раздел галоидных соединений часто
бывает разбит на два подкласса: фто-
фториды и соли прочих галоидоводород-
ных кислот С1*, гдеС1*—Cl, BrmraJ.
Действительно, соединения наиболее
легких неметаллических элементов
(в нашем примере фторидов) резко
отличаются по своим свойствам от
остальных, и поэтому целесообраз-
целесообразность сведения соединений наиболее
тяжелых элементов в один подкласс
оспаривать трудно.
Раздел, в котором анионы являются
элементами VI-6 подгруппы, делится
на окислы и на соединения других
элементов VI-Ь подгруппы. Окислы
должны быть таким же подклассом
в этом классе, как фториды в преды-
предыдущем.
Сульфиды, селениды и теллури-
ды (подкласс S*) можно рассмат-
рассматривать совместно, хотя теллуриды
иногда имеют специфику, отсутствую-
отсутствующую у сульфидов и селенидов.
Совершенно аналогично тому, как
мы отнесли в разные классы соеди-
соединения с одновалентными и двухва-
двухвалентными анионами, мы обязаны вы-
выделить и соединения с трехвалент-
трехвалентными анионами.
4. Количество таксонов между
классом и видом. Если с понятиями
вида и класса был вопрос более или
менее ясен, то вопрос о промежуточ-
промежуточных звеньях в систематике исследо-
исследован значительно меньше. До сих пор
оставался неясным вопрос, какое ко-
количество таксономических (система-
(систематических) категорий — таксонов —
должно быть между классом и ви-
видом. В биологии, как известно, их
три: отряды, семейства и роды.
Конечно, для систематики объек-
объектов неживой природы число их мо-
может быть и другим. Ниже будет по-
показано, что и здесь целесообразно вы-
выделение трех таксонов, что, без-
безусловно является случайным совпа-
совпадением.
5. Понятие рода. В ряде работ ав-
автор уже указывал, что «химические
соединения или виды» целесообразно
группировать по структурным типам.
Можно условиться, что к одному «ро-
«роду» относятся вещества, принадле-
принадлежащие одному структурному типу.
Это вполне логично, так как физико-
химические свойства веществ опре-
определяются не только химическим со-
составом, но и структурой.
Род будет определяться структур-
структурным типом, вид — химической фор-
формулой. Влияние структурного типа
на физические свойства химических
соединений можно видеть на примере
солей щелочных металлов (см. табл.
на стр. 247).
Вещества, близкие по химическо-
химическому составу и принадлежащие к од-
одному структурному типу, имеют
близкие физические свойства. Веще-
Вещества же, близкие по химическому со-
составу, но принадлежащие к разным
структурным типам, по физическим
свойствам обычно различаются друг
от друга более резко.
6. Понятие семейства. Объедине-
Объединение родов в семейства — наиболее
трудная часть систематики. Можно
предложить объединение по сходным
координационным числам и много-
многогранникам, и для неорганических сое-
соединений в качестве основного эле-
элемента опять-таки выбрать анион.
Для интерметаллических соединений,
по-видимому, следует выбрать атом
с наименьшим координационным
числом (см. ниже). Такое объедине-
объединение, вероятно, позволит сблизить
вещества по физическим свойствам,
так как многие из них определяются
ближним порядком. Этот вопрос
282
требует еще дополнительной разра-
разработки.
7. Понятие отряда. Каждый класс
химических веществ или минералов
делится на пять групп, играющих
роль, аналогичную биологическим
«отрядам». Этими пятью «отрядами»
будут являться наиболее характерные'
группы структурных типов, опреде-
определяющие основной мотив структуры:
координационные, островные, цепо-
цепочечные, слоистые и каркасные (см.
главу XIV, § 1).
Выделение отрядов по одному
признаку целесообразно потому, что
их структурные особенности опреде-
определяют основные физические свойства.
Так, если мы возьмем в качестве ха-
характеристики оптические свойства,
то окажется, что вещества со слои-
слоистыми структурами будут одноосны-
одноосными или псевдоодноосными, отрица-
отрицательными. Цепочечные структуры ха-
характерны для веществ, имеющих по-
положительную индикатрису. Остров-
Островные, координационные и каркасные
структуры будут характеризоваться
малым двупреломлением, в то время
как цепочечные и слоистые — боль-
большим.
ГЛАВА XVIII
КРИСТАЛЛОХИМИЯ ИНТЕРМЕТАЛЛИЧЕСКИХ СОЕДИНЕНИЙ
§1. Система «металлических»
радиусов атомов
Величины «металлических», или
атомных, радиусов получаются деле-
делением пополам кратчайших межатом-
межатомных расстояний в структурах чистых
металлов, характеризующихся коор-
координационным числом 12. Если струк-
структура металла характеризуется иными
координационными числами, то для
получения величины атомного радиу-
радиуса недостаточно знания половины
кратчайшего межатомного расстоя-
расстояния. В это значение должна быть
внесена поправка на координацион-
координационное число. Величины поправок были
указаны в табл. 16 на стр. 146.
На основе значений межатомных
расстояний была составлена таблица
атомных радиусов (Г. Б. Бокий,
1953 г.) для всех металлов подгруппы
а и первых трех подгрупп Ъ периоди-
периодической системы элементов и для РЬ
(см. табл. 35).
Для металлов Al, T1 и РЬ радиусы,
вычисленные из межатомных расстоя-
расстояний их кристаллических структур,
по-видимому, завышены приблизи-
приблизительно на 0,15 А вследствие того, что
атомы этих элементов в чистом
металле не полностью отщепляют
свои валентные электроны. Однако
в твердых растворах и интерметал-
интерметаллических соединениях они могут быть
полностью ионизированы.
Данные для элементов IV—VI-Ь
подгрупп и для Ga взяты из справоч-
справочников (см., например, справочник
Данса и Лакса). Определение атом-
атомных радиусов полуметаллических
элементов производится обычно на
основании величин межатомных рас-
расстояний в соединениях этих элемен-
элементов с металлами, если соответству-
соответствующие структуры относятся к одному
из структурных типов, характерных
для нормальных ингерметаллическнх
соединений.
§2. Тины взаимодействия
металлических атомов
в двойных системах
Атомы разных металлов могут взаи-
взаимодействовать между собой различ-
различным образом. В одном случае они
могут химически не реагировать
друг с другом ни в твердом, ни в жид-
жидком состояниях. Такая бинарная сис-
система будет представлять собой два
не смешивающихся друг с другом
жидких слоя. Другим крайним случа-
случаем будет образование определенного
химического соединения между двумя
взятыми металлами. Между Э1ими
предельными случаями будут распо-
располагаться системы, в которых два
металла, смешиваясь в жидком сос-
состоянии, образуют эвтектику в твер-
твердом, и системы, в которых металлы
образуют непрерывный ряд твердых
растворов. Таким образом, если рас-
распределить системы в порядке увели-
увеличения химического взаимодействия
компонентов, получится следующий
ряд: а) металлы не взаимодейству-
взаимодействуют друг с другом ни в твердом, ни
в жидком состояниях; б) металлы
смешиваются в жидком состоянии,
а в твердом образуют эвтектику;
284
и
a
a,
k
о
В
-о
>
"О
>
>
-о
>
S
-о
1—(
>
в
>
>
>
в
III
в
в
Пе-
иоды
о.
СО
м-*
о
о
*«-
о
о
о
cq оэ
о
<^-
о
«3
"^
3!
со -
со
со
CQ
от
0 го-
го
« со
о
*z-
о -
со
р^
О CD
СД "*
та1 ОЭ
СО
^:-
со
CD
dco
тЗсо
CD
^""
О02
*:-
. о
1Л
со "^.
1Л
d
Он
О
а5
. со
Йи™.
ш со
о
а:-
ее оо
—J ^^
те tT<]
и -
(М
СО
^о^
* со
о
«5
=:-
Зсо
г!-
в) металлы образуют друг с другом
твердые растворы во всем интервале
составов; г) металлы образуют меж-
между собой одно или несколько интер-
интерметаллических соединений.
Между этими случаями, конечно, не
существует резких границ. Металлы
могут не реагировать при одних усло-
условиях, но реагировать при других или
же реагировать друг с другом только
в более или менее узкой области со-
составов. Твердые растворы могут быть
не только непрерывными, но и охва-
охватывать более широкую или узкую
область составов. Интерметалличе-
Интерметаллические соединения могут буть устойчи-
устойчивыми или же образовываться в ре-
результате распада твердых растворов
(сверхструктуры). Интерметаллпче-
ские фазы могут иметь узкую об-
область гомогенности и в этом отноше-
отношении быть весьма сходными с обычны-
обычными неорганическими соединениями
или же иметь очень широкую область
гомогенности, т. е. давать широкую
область твердых растворов с обои-
обоими компонентами или с одним из
них.
Все эти случаи стирают резкие
границы между указанными выше
типами взаимодействия. Тем не менее
методически удобнее рассмотреть их
раздельно, иллюстрируя сказанное
типичными примерами и указывая на
возможные отклонения, на постепен-
постепенные переходы к другим типам и их
перекрывание между собой.
§ 8. Системы металлов,
образующих два шидких слоя
или эвтектику
Металлы, не смешивающиеся друг
с другом ни в твердом, ни в жидком
состояниях или же смешивающиеся
в жидком состоянии, но в твердом не
дающие ни твердых растворов, ни
соединений, а только эвтектику, обыч-
обычно имеют резко отличающиеся по раз-
размерам атомы. Если радиусы атомов
металлов близки, то это способствует
образованию твердых растворов. Так,
например, К и Li образуют два жид-
жидких слоя, а К с Rb и Cs дает непре-
непрерывный ряд твердых растворов
(Гы = 1,55; rNa = 1,89; гк = 2,36;
гвь = 2,48; rcs = 2,68). Магний
(г = 1,60) дает в широких пределах
твердые растворы с Li, а в системах
с Na и К образуют два жидких слоя.
Резкая разница в электронном
строении двух атомов также пре-
препятствует образованию твердых рас-
растворов и способствует появлению
двух жидких слоев или эвтектик. Так,
например, в системах Си — V, Сг, Мо,
W образуются два жидких слоя, в си-
системе Си — Ti — эвтектика. Анало-
Аналогично меди ведет себя серебро с выше-
вышеперечисленными металлами. Та же
картина наблюдается в системах Си и
Ag с далеко отстоящими от них ме-
металлами VIII-а подгруппы.
Существенную роль играет также
разность температур плавления ме-
металлов.
Элементы, не полностью отщепляю-
отщепляющие свои валентные электроны (Т1,
РЬ) вследствие особенности электрон-
электронного строения, чаще образуют с дру-
другими металлами эвтектики или два
жидких слоя. Так, например, в систе-
системах, содержащих свинец, металл
VI-a — VIII-a и первых Ь-подгрупп,
часто наблюдаются два жидких слоя
(РЬ с Сг, Mn, Fe, Co, Ni, Cu, Zn и др.)
или эвтектика (РЬ с W, Ag, Cd и др.).
С металлами первых а-подгрупп
вследствие их сильной электрополо-
электроположительности свинец образует много
прочных интерметаллических соеди-
соединений, структуры которых нередко
принадлежат к обычным структур-
структурным типам бинарных неорганических
соединений — типа NiAs, CaF2 и др.
§4. Структурная характеристика
твердых растворов
и интерметаллических
соединений
На рис. 275 изображена схема, пока-
показывающая различные случаи сочета-
сочетаний двух типов атомов в кристалле.
Относительное количество «белых» и
«черных» атомов на всрх рисунках
286
00*000000*0
oooooooooo
оооо* о о • о о о
0*00000000
ооооооооооо
00000000*0
оо*оо*оо *ло о
ООоО*ООООо
ооооооооо оо
о о о оооо о • о"
000*0000 ООО
0*000 00000
оооооо**оо*
0*00000000
00*0 0-000 00 0
оооооо*ооо
ооо о • о о о о о о
оооооооо**о
о*о*оооооо
ООО ОООООООО
в
• оо*оо*оо*о
ООООООО 00 О
ооооооооооо
о*оо*ооо*о
ооооооооооо
00*0000000
• 00000*00* О
oooooooooo
оооо ооооооо
О • О О 9 О О • О О
ООО ОООООООО
о оооооооо о
• о о • о о • о о в о
oooooooooo
ооооооооооо
о ооо*оо*оо
О О • ОООООООО
оооо о • о о о о
» о о • о о о о о*о
о о оооо оооо
ооооооооооо
ооооо оооооо
оо*ооо*оОо
О* ОООООООО*
0000*00000
0000000*000
• ооооооооо
ооо*о ооооо о
oooooooooo
о о • о о о • о о • о
ооо*оооооо
• оо ооооо ооо
ооооо о о • о о
ооооо*ооо*о
0*00000000
000о*°0 0'000
о о • о о о • о * о
00000*000 ОО
oooooooooo
оо*оо°ооооо
ооооо* оо»о
OOOOQOO ОООО
г
• оо*оо*оо • о
oooooooooo
ооооо°оооо о
о • о о • о о • о о
о о ооо о о о о о а
oooooooooo
• о о • о о • о о • о
0-0 ОООоО ООО
ооооооооооо
о*оо*оо*оо
о о ооооооооо
ооооооооо о
• оо*оо*оо*о
oooooooooo
ооооооооооо
о* оовоо#оо
ооооо о о оооо
ооооо°оооо
• о о • о о • о о • о
oooooooooo
ооооооооооо
Рис. 275. Схема различных случаев рас-
распределения двух типов атомов в кри-
кристалле
А — полностью неупорядоченная структура;
В — структура с ближним порядком;
В — не полностью упорядоченная структура;
Г — полностью упорядоченная структура
одинаково, но расположение их раз-
различно. В случае А распределение ато-
атомов совершенно произвольно, вероят-
вероятность встретить «белый» и «черный»
атомы в любой точке кристалличе-
кристаллической структуры пропорциональна от-
относительному количеству тех и дру-
других атомов. Этот рисунок соответст-
соответствует полной неупорядоченности. От
него резко отличается случай полно-
полностью упорядоченного взаимного рас-
расположения атомов в простран-
пространстве (Г). Таковы структуры многих
неорганических соединений. Однако
между этими двумя случаями можно
расположить еще два промежуточных.
В случае Б имеется упорядочен-
упорядоченность в ближайших координационных
сферах — упорядоченность ближнего
порядка. На рисунке не найдется ни
одной пары «черных» атомов, распо-
располагающихся на кратчайшем друг к
другу расстоянии а или же на рас-
расстоянии ail. Все имеющиеся сведе-
сведения о тонком строении твердых ра-
растворов указывают на то, что именно
такое расположение характеризует
подавляющее большинство твердых
растворов.
Случай В характеризуется не
только ближним порядком, в нем на-
наблюдается и дальний порядок. Однако
он не достигает 100%. Большинство
интерметаллических соединений ха-
характеризуется именно такой степенью
упорядоченности, причем ее часто
выражают определенным процентом
от идеальной упорядоченности.
Из сказанного ясно, что не сущест-
существует резких границ между твердым
раствором и соединением. Упорядо-
Упорядоченные твердые растворы и не пол-
полностью упорядоченные соединения
являются теми самыми случаями,
которые обычно реализуются в при-
природе и в лаборатории.
§ 5. Двойные металлические
системы
с неограниченной взаимной
растворимостью компонентов
в твердом состоянии
Металлы с металлами дают непрерыв-
непрерывные твердые растворы почти исклю-
исключительно типа замещения. Твер-
Твердые растворы внедрения наблю-
наблюдаются у металлов с неметалличе-
неметаллическими элементами, характеризующи-
характеризующимися весьма малыми размерами ато-
атомов (Н, В, С, N). Твердые растворы
вычитания обычно наблюдаются
у металлов с некоторыми неметалли-
неметаллическими элементами, например, с се-
серой, селеном и, гораздо реже, между
двумя металлическими элементами,
например в системе Ni:— А1. Об этом
типе твердых растворов будет сказа-
сказано позже.
В настоящем параграфе будет ра-
разобран самый типичный для металлов
случай — твердые растворы замеще-
замещения. Необходимым условием образо-
образования непрерывных твердых раство-
растворов является принадлежность кри-
кристаллических структур обоих метал-
металлов к одному структурному типу или,
в исключительных случаях, к двум
очень близким, например In и Т1.
287
i Соотад
Рис. 276. Типы Д1 грамм состояния с не-
непрерывными твер;* ми растворами
Кроме этого условия, необходима бли-
близость размеров атомов. Отклонение
значений атомных радиусов не долж-
должно превышать 10—12% *• Типы диа-
диаграмм состояния непрерывных твер-
твердых растворов представлены на
рис. 276.
Примером для случая а являются
системы раздела I табл. 36. Тип диа-
диаграммы б наблюдается в системах
раздела III. Среднее отклонение зна-
значений атомных радиусов в обоих ти-
типах непрерывных твердых растворов
порядка 5%.
Промежуточные п смешанные слу-
случаи между а и б собраны в разделе
IV табл. 36. Диаграмма с минимумом
(рис. 276, в) встречается в разделе II
той же таблицы. Они допускают не-
несколько большее среднее отклонение
в атомных размерах, порядка 6%.
По-видимому, системы с таким типом
диаграмм приближаются к системам
с ограниченными твердыми раство-
растворами или к системам с эвтектикой.
В эту же группу попадают две изве-
известные системы с непрерывными твер-
твердыми растворами, в которых оба эле-
элемента не принадлежат соседним под-
подгруппам, но находятся в одном перио-
периоде: V—Fe и Сг—Fe.
Для всех примеров, рассмотренных
выше, помимо уже упомянутых
двух условий, характерно и третье —
условие химической близости элемен-
элементов, образующих твердый раствор, на
что впервые указал Н. С. Курнаков
еще до того времени, как появилась
* Во всех последующих расчетах боль-
больший радиус атома принимается за
100%.
возможность сформулировать первые
два требования. Действительно, оба
элемента, образующие непрерывные
твердые растворы, обычно находятся
в одной подгруппе периодической си-
системы химических элементов или в
одном периоде, причем, как правило,
в соседних подгруппах (Г. Б. Бокий,
1954 г.).
На примере непрерывных твердых
растворов, величины отклонений
атомных радиусов которых составля-
составляют 12% (К —Cs) и 11% (Си-Аи),
мы видим, что если фактор химиче-
химической близости благоприятен, то он
может до известной степени компен-
ТАБЛИЦА 36
Условия образования непрерывных
твердых растворов
Система "j
[ Со — Ni
Со —Pd
Со—Pt
Rh-Pt
Ni - Си
Pd-Ag
Pd —Au
у Ag _ Аи
Отклонение
[чина
онения
усов, %
gsgg
Sssa
1,0
9,0
9,5
3,0
3,0
5,0
5,0
0
от 0
До 9,5о/о
K-Rb
K—Cs
Rb —Cs
V —Fe
III Cr — Fe
Mn —Co
Mn —Ni
Fe —Pd
1 Си — Аи
Отклонение
до 12%
5,
12,0
7,5
6,0
1,0
4,0
4,5
8,0
11,0
от 1
Система
f Mo_W
Mn_ Fe
Fe —Co
Fe—Ni
Fe—Pt
III { Ni —Pt
Mg-Cd
Pd — Cu
Pt Au
In_Tl
( In - РЬ
Отклонение
ДО 10%
С Са — Sr
TV
I V
Ti—Zr
Zr-Hf
Fe-Rh
Fe— Ir
Rh— Pd
Ir—Pt
i Pd pt
Отклонение
:чина
онения
ных
усов, %
1,0
3,0
1.0
1.5
8.5
10.0
2,5
6,5
4,0
3,0
5,0
от 1
2,0
8,5
0,5
6,0
6,5
2,0
2,0
0,5
от 0,5
до 8,5о/о
288
сировать неблагоприятный размер-
размерный фактор. Иногда он может даже
компенсировать такой неблагоприят-
неблагоприятный фактор, как необходимость изо-
структурности.
Только большой химической бли-
близостью индия и таллия можно объяс-
объяснить образование между ними непре-
непрерывных твердых растворов с посте-
постепенным переходом от тетрагональной
структуры индия к кубической струк-
структуре таллия. Твердые растворы
In — Т1 были изучены методом рент-
геноструктурного анализа Е. С. Ма-
Макаровым.
Интересно отметить, что Т-элемен-
ты (полуметаллы) никогда не обра-
образуют непрерывных твердых растворов
с Мь- и тем более с Ма-металлами *.
Необходимость химической близо-
близости элементов для образования непре-
непрерывных твердых растворов подтверж-
подтверждается также фактом наличия твер-
твердых растворов в системах Pt — Аи,
Pd — Аи, Ni — Си и отсутствием их в
системах Со — Си и Fe — Си, несмот-
несмотря на близость размеров атомов и
принадлежность Fe, Co и Си к одно-
одному структурному типу. Только с со-
соседним по периоду элементом — ни-
никелем — медь дает непрерывные
твердые растворы.
Резюмируя все сказанное, можно
сформулировать следующие три пра-
правила, необходимые для образования
непрерывного ряда твердых раство-
растворов.
1. Оба металла должны принадле-
принадлежать к одному структурному типу
(или, как исключение, к двум весьма
близким).
2. Величины отклонений атомных
радиусов обоих компонентов не долж-
должны превышать 10—12%.
3. Оба элемента должны быть хи-
химически близкими друг к другу.
* Здесь и в дальнейшем будем придер-
придерживаться следующих обозначений: Ма
и Мь — металлы а- и 6-подгрупп соответ-
соответственно; М3 — щелочные и щелочнозе-
щелочноземельные; Md — металлы переходных
групп; М/ — лантаноиды, актиноиды и
металлы 111-а подгрупп.
19 Кристаллохимия
§ в. Влияние полиморфизма металлов
на тип диаграмм
с твердыми растворами.
Твердые растворы железа
с другими металлами
Первым условием возможности обра-
образования непрерывных твердых ра-
растворов между двумя металлами яв-
является принадлежность их к одному
структурному типу. Однако если один
из компонентов (или оба) в зависи-
зависимости от температуры (или других
термодинамических факторов) может
существовать в двух или более струк-
структурных типах, то это обстоятельство
может существенно изменить харак-
характер диаграмм, простейшие случаи ко-
которых были рассмотрены в предыду-
предыдущем параграфе.
Учитывая возможные случаи поли-
полиморфизма (аллотропии) металлов,
все металлические системы можно
подразделить на три следующие ка-
категории:
1. Высокотемпературные модифи-
модификации обоих металлов принадлежат к
одному структурному типу, например
Со —Ni, Ag —Au, Ti — Zr, Cr — Fe
и др.
2. Одна из модификаций одного
компонента принадлежит к тому же
структурному типу, который имеет
одна из модификаций другого компо-
компонента. Эти модификации обоих метал-
металлов не должны быть высокотемпера-
высокотемпературными, т. е. система не должна при-
принадлежать к первому тину, например
Fe — Мп.
3. Оба элемента не имеют ни одной
модификации с одинаковым типом,
например Ru — Pt, V — Со и др. Оче-
Очевидно, что в таких системах могут
быть широкие области твердых рас-
растворов, но непрерывными они быть
не могут.
Сказанное лучше всего может быть
проиллюстрировано на примере си-
систем, содержащих в качестве одного
из компонентов железо. Эти системы
представляют большой промышлен-
промышленный интерес и поэтому изучены наи-
наиболее полно. Железо, как известно,
имеет 3 полиморфные модификации,
289
Атомн. % С,"
40 ВО
100
Рис. 277. Система с непрерывным твердым
раствором на основе а-, |3- и 6-Fe
1 Рис. 278. Система с непрерывным твердым
' раствором на основе Y~Fe
I Рис. 279. Система с ограниченным твер-
—| дым раствором, в которой а-, {5- и 6-Fe
| объединены в одну фазу
Рис. 280. Система с ограниченным твер-
твердым раствором, в которой «- и {5-Fe раз-
разобщены с б-модификацией
40 60
Вес. % О
100
О
1534
иоо
юоо
гоо
40 60
Атомн. % Ni
из которых а, |3 и б принадлежат к
одному структурному типу, а у — к
другому.
Все бинарные системы железа с
металлами можно разделить на 4 ка-
категории:
\. Системы с непрерывными твер-
твердыми растворами на базе а-, C- и
б-модификаций: V—Fe, Cr—Fe
(рис. 277).
2. Системы с непрерывными твер-
твердыми растворами на базе Y-модифи-
кации: Fe—Pd, Fe—Pt и др.
(рис. 278).
3. Системы с ограниченными твер-
j дыми растворами, в которых а- C- и
I б-модификации объединены в одну
фазу: Ti—Fe, Mo—Fe и др.
(рис. 279).
4. Системы с ограниченными твер-
твердыми растворами, в которых а-моди-
фикация разобщена с б-модифика-
б-модификацией: Zr — Fe и др. (рис. 280).
Системы железа с металлами под-
подробно рассмотрены в монографии
И. И. Корнилова A951 г.).
290
Атомн. % Fe
JO SO
20
40
Атпмн. % Fe
BO 80
100
1900
{500
1100
700
I
1855е
\r
\ \
-\—ч
W-z
I
\2,5
e-Zr
I Г
I
Жидность
A
t
a-Z/>+Fe2
i
t-a
C^
f
Ш0"
I
—
i
лЛ
1S39°
$A
Жидн+r-fs
960°
гнитное
1 1
r-Fe,
1
—l-
i
I
20
40
Bec.L
BO
, Fe
80
/00
Fe
§ 7. Изменение констант решеток
твердых растворов
Поскольку структура непрерывного
ряда твердых растворов замещения
двух металлов такая же, как у чи-
чистых компонентов, то естественно
ожидать равномерного изменения
констант решетки твердого раствора
в зависимости от состава. Простей-
Простейшая зависимость величины констан-
константы решетки от состава выражается
прямой линией. Эта зависимость ча-
часто называется правилом Вегарда.
Константа твердого раствора ка-
какого-либо промежуточного состава
ff/77 %
e== 100 '
где п\ и п2 константы решеток чистых
компонентов, а с\ и сг молярные кон-
концентрации в процентах обоих ком-
компонентов в сплаве.
К такому идеальному случаю при-
приближаются системы Pt—Аи, Pd—Аи
и др. Обычно же кривая реальных
значений констант решеток отклоня-
отклоняется от прямой и проходит выше нее
(Си—Pd, Си—Аи) или ниже (Ag—
Аи). Первый случай соответствует
положительному отклонению, вто-
второй — отрицательному.
На рис. 281 изображено несколько
кривых, показывающих зависимость
констант решеток твердых растворов
от состава.
Как правило, сплавы с выпуклой
линией ликвидуса дают отрицатель-
отрицательные отклонения, а сплавы с вогну-
вогнутой — положительные.
Если известна кривая зависимости
константы решетки от состава, то
можно легко определить состав спла-
сплава, используя результаты рентгено-
рентгенографического измерения. Этим мето-
методом часто пользуются в металлогра-
металлографии для определения границ раство-
растворимости твердых растворов.
Рис. 281. Зависимость константы решетки
Ni — А1 от состава
19* 291
§8. Ограниченные
твердые растворы
Как было сказано выше, для образо-
образования непрерывных твердых раство-
растворов между металлами необходимы
три условия: кристаллы обоих ком-
компонентов должны принадлежать к
одному структурному типу (или к
двум очень близким); величины от-
отклонений атомных радиусов компо-
компонентов не должны превышать 10—
12%; оба элемента должны быть
химически близкими.
Несоблюдение одного из этих трех
условий приводит к тому, что между
компонентами образуются только
ограниченные твердые растворы.
Ширина области существования твер-
твердых растворов может варьировать в
очень широких пределах. Причины
большей или меньшей растворимости
компонентов друг в друге будут
разобраны ниже.
Примерами систем, в которых оба
компонента принадлежат к разным
структурным типам, но имеют более
или менее благоприятный объемный
фактор и химическое сходство, могут
служить Li—Mg, Os—Pd, Cd—Jig,
Ag—Cd, Ag—Hg, Cu—Zn, Zn—Al.
Все они характеризуются широкой
областью существования твердых
растворов.
Неблагоприятный объемный фак-
фактор отрицательно влияет на образова-
образование твердых растворов. Если откло-
отклонение размеров атомов лежит в
пределах 10—15%, то области твер-
твердых растворов еще достаточно широ-
широки. В качестве примера см. данные
для систем, содержащих железо
(табл. 37). Дальнейшее увеличение
разницы в атомных размерах приво-
приводит к резкому снижению взаимной
растворимости компонентов.
Влияние химической близости
элементов на образование твердых
растворов можно проследить по дан-
данным табл. 38. В ней собраны раство-
растворимости металлов l-а, 11-а, и \-Ъ под-
подгрупп в металлах YV-b подгруппы.
Ясно, что химическая близость у ме-
металлов подгрупп b будет большей, и
при прочих приблизительно равных
условиях область существования
твердых растворов будет большей,
чем в системах Ms—Мь.
Влияние относительной валентно-
валентности можно проследить по данным
табл. 39. Здесь выбраны системы
Ms—Мь и Мь—Мь, так как валент-
валентность элементов Md в металлическом
состоянии далеко не всегда ясна.
Данные этой таблицы, за редким
исключением, подтверждают ту
ТАБЛИЦА 37
Влияние объемного фактора
на растворимость при одинаковых
структурных типах
Система
Fe_Sc
Fe_Ti
Fe_Zr
Fe - Th
Fe_Nb
Fe_Ta
Fe_Mo
Fe_W
Fe —Au
Fe —Al
Величина
отклонения
атомных
радиусов, %
23
13,5
21
30
13
13,5
9,5
10
12,5
12
Максимальная
растворимость
в железе, %
Нет
6,9
<1
Нет
8,2
2,0
23
13
4,76
52,8
ТАБЛИЦА 38
Влияние химической близости
компонентов на растворимость
Тип
металлов
М8-Мь
мь-мь
Система
Li — Pb
Mg — Sn
Mg_Pb
Си —Si
Си —Ge
Си —Sn
Ag —Ge
Ag-Sn
Величина
отклоне-
отклонения атом-
атомных ради-
радиусов, %
11,5
1,0
8,5
4,5
8,0
19,0
3,5
9,0
Максимальная
растворимость
элементов IV-b
подгруппы,
ат. %
<1 Pb
3,5 Sn
3,9 Pb
14,0 Si
12,0 Ge
9,26 Sn
6,5 Ge
12,2 Sn
292
ТАБЛИЦА 39
Влияние относительной валентности на растворимость
Тип
металлов
М8-Мь
м„-мь
/"( Tw л m Д -а ш f\
система
Be —Си
Be-Ag
Mg_Cu
Mg —Ag
Си — Zn
Cu_Cd
Си —Al
Си— Si
Си —Sn
Ag_Zn
Ag-Cd
Ag-Al
Ag-Sn
Величина
отклонения
атомных
радиусов, %
11,5
21,5
20,0
10,0
8,0
18,0
10,5
4,5
19,0
3,5
8,0
0,8
9,0
Граница раствори-
растворимости в металле
с низшей валентно-
валентностью, ат. %
16,6 Be
3,5 Be
6,5 Mg
30,0 Mg
38,4 Zn
1,7 Cd
20,4 Al
14,0 Si
9,26 Sn
37,8 Zn
42,5 Cd
20,4 Al
12,2 Sn
Граница раствори-
растворимости в металле
с высшей валентно-
валентностью, ат. %
<2,0 Си
10 Ag
0,01 Си
3,9 Ag
2,3 Си
0,12 Си
2,5 Си
<2,0 Си
0,4 Си
6,3 Ag
6,2 Ag
18,7 Ag
<0,1 Ag
мысль, что атому с высшей валент-
валентностью легче «имитировать» атом с
низшей валентностью, чем наоборот.
Более вероятно, что атом с высшей
валентностью не будет использовать
всех своих валентных электронов для
образования металлической связи,
чем то, что атом с низшей валент-
валентностью будет образовывать связь за
счет невалентных электронов.
Предел образования ограниченного
твердого раствора определяется элек-
электронной концентрацией; под элект-
электронной концентрацией подразумева-
подразумевается отношение числа свободных
валентных электронов к числу ато-
атомов. С нею связана величина свобод-
свободной энергии кристалла, которая ми-
минимальна, если сплав находится в
состоянии равновесия. Каждой кри-
кристаллической решетке сплава отвеча-
отвечает свое специфическое зонное строе-
строение, а зона Бриллюэна может
вместить только определенное коли-
количество электронов. При заполнении
зоны наступает момент, когда даль-
дальнейшее прибавление электронов при-
приводит к очень резкому увеличению
энергии. В этот момент может насту-
наступить изменение кристаллической
структуры сплава. Новая структура
по своему зонному строению будет
более благоприятна для большей
электронной концентрации. Так, для
гранецентрированной решетки пер-
первая энергетическая зона имеет форму
кубооктаэдра. Эта структура остает-
остается устойчивой до значения электрон-
электронной концентрации, равной приблизи-
приблизительно 1,4.
Справедливость сказанного выше
можно проверить сопоставлением
предельных значений растворимости
металлов с разной валентностью в
одном и том же металле. Очевидно,
чем выше валентность растворяе-
растворяемого элемента, тем меньшее количе-
количество его может быть растворено.
Для примера возьмем сплавы меди с
Zn, Ga, Ge, As и серебра с Cd, In, Sn,
Sb. Зная предельную величину элек-
электронной концентрации для гранецен-
гранецентрированной кубической решетки
A,4), можно легко рассчитать пре-
пределы растворимости вышеуказанных
многовалентных элементов (табл.
40).
Вполне удовлетворительное со-
совпадение рассчитанных и определен-
определенных из опыта величин предельной
293
ТАБЛИЦА 40
Влияние электронной концентрации
на предельную растворимость
Раствори-
Растворитель
Си
Растворяе-
Растворяемый элемент
Zn
Ga
Ge
As
Cd
In
Sn
Sb
Предельная раство-
растворимость
рассчит.
40
20
13,3
10
40
20
13,3
10
, ат. %
экспер.
38,4
20
12
7
40
20
12
7
концентрации твердых растворов
подтверждает справедливость приве-
приведенных выше соображений.
f О. Явление «старения» сплавов
Некоторые сплавы обладают спо-
способностью заметно изменять физиче-
физические свойства во времени. Это явле-
явление носит название «старения».
Хорошим примером могут служить
сплавы алюминия с медью (основа
так называемого дюралюминия).
При высоких температурах алюми-
алюминий растворяет медь. Максимальное
содержание меди при 548° С равно
5,65%. При комнатной температуре
эта величина падает приблизительно
до 0,2%. Однако с помощью закалки
можно сохранить большое содержа-
содержание меди и при низких температурах.
При этом выяснилось, что если за-
закалка проведена при температуре
ниже 100° С, то такой сплав начина-
начинает со временем изменять свои свой-
свойства: прочность его возрастает.
Детальные исследования этого про-
процесса показали, что в этих условиях
ио плоскости куба кристаллов т в ер до-
го раствора начинают собираться ато-
атомы меди, образующие участки толщи-
толщиной в один-два слоя и протяженностью
в несколько десятков ангстрем. Эти
«двухмерные» участки создают в
сплаве каркас и обусловливают повы-
повышенную прочность. В обычных усло-
условиях процесс на этом заканчивается,
хотя получающаяся структура сплава
в термодинамическом отношении не
является стабильной.
Можно искусственно создать усло-
условия, позволяющие проследить даль»
нейшие изменения в сплаве на пути к
достижению термодинамического рав-
равновесия. Так, закалка при 200° С дает
начало существованию новой мета-
стабильной в'-фазы, имеющей состав
СиА12 и структуру типа CaF2. Разме-
Размеры ячейки этой фазы таковы, что пло-
плоскости, в которых располагаются ато-
атомы А1, по форме и размерам соответ-
соответствуют плоскостям из атомов А1 в
твердом растворе. Таким образом,
твердый раствор может непрерывно
переходить через общие плоскости из
атомов А1 в в'-фазу. Эта фаза само-
самостоятельно существовать не может, и
если связь с твердым раствором нару-
нарушается, то она превращается в ста-
стабильную форму СиА12, имеющую спе-
специфическую структуру (см. стр.313).
§ 10. Тнердые растворы вычитания
Твердые растворы вычитания в
интерметаллических фазах подробно
изучены в системах Со—А1 и Ni—А1.
Фаза Ni—А1, имеющая область гомо-
гомогенности от 40 до 55 ат. % А1, кристал-
кристаллизуется в структурном типе GsCl.
В области с содержанием А1 менее
50 ат. % эта фаза представляет собой
нормальные твердые растворы заме-
замещения. Атомы Ni, находящиеся в фа-
фазе сверх 50%, статистически замеща-
замещают атомы А1.
Если исследовать в этой области
константы решетки твердых раство-
растворов и их плотности в зависимости от
состава (рис. 282), то, как и следует
ожидать, по мере увеличения содер-
содержания А1 константа решетки будет
возрастать (гм — 1,43 и rN1 = 1,24),
а плотность падать (ат. вес. А1—26,97,
294
о
а, А
2,886
2,878
2,870
2,862
2,854
,/
/
[
/
f
/1
/
J
2фазы
45
5
П
\
5
\
Н
0
/
\
5
N
ч
•г.
6
6,6
6,4
5,8%
5,6^
Г
13Ы
Рис. 282. Зависимость константы решет-
решетки A) и плотности B) твердого раствора
Ш—А1 от состава
Ni—58,69). Плавный ход кривых на-
нарушается вблизи состава 50%. На
кривой параметров решетки обнару-
обнаруживается максимум, а на кривой плот-
плотностей — резкий излом. Кривая в этом
месте вдет гораздо круче вниз, чем
экстраполяционная кривая (пунктир-
(пунктирная линия), вычисленная в предполо-
предположении, что в области от 50 ат. % А1
и выше существуют твердые растворы
замещения. Такой ход кривых одно-
однозначно указывает, что в области высо-
высоких концентраций алюминия имеют
место твердые растворы вычитания.
При избыточном (сверх 50%) коли-
количестве А1 структура делается дефект-
дефектной — часть мест, которые должны
были бы быть заняты в соединении
NiAl атомами Ni, остаются пустыми.
Эти пустоты статистически распреде-
распределяются по всему объему кристалла.
Как показал С. Т. Конобеевский,
для твердых растворов вычитания
остается справедливым правило пре-
пределов электронной концентрации, если
последнюю относить не к числу ато-
атомов, а подсчитывать число электронов,
приходящихся на каждую ячейку. В
самом деле, если по вершинам ячейки
расположены атомы алюминия, а в
центре — атом никеля, то электрон-
яая концентрация будет 3, так как
валентность Л1 равна трем, а нике-
никеля — нулю. Если же ячейка пустая,
то на одну такую ячейку все равно
приходятся три электрона.
Пустоты в дефектных структурах
не всегда распределяются статисти-
статистически — они могут быть упорядочены.
В этом случае элементарная ячейка
и симметрия кристаллов могут изме-
измениться. Как впервые указал Хегг,
структуры у~Фаз являются, по су-
существу, дефектными упорядоченны-
упорядоченными сверхструктурами типа CsCl.
В системе Ni—А1 в области, приле
гающей к точке состава 60 ат. % А1,
имеется фаза №2А13. В структуре ее
7з «никелевых» мест остается неза-
незанятой. Все эти незанятые места опре-
определенным образом располагаются в
плоскостях A11). Структура из куби-
кубической (NiAl) становится тригональ-
ной (Ш2А13).
§11. Двойные металлические
системы о тремя и о большим
количеством твердых фаз
Если в двойной металлической сис-
системе из расплава кроме компонентов
может кристаллизоваться третья твер-
твердая фаза — химическое соединение *,
то соответствующие диаграммы отно-
относятся к одному из основных двух ти-
типов (рис. 283, а, б). Случай б отлича-
отличается от случая а тем, что соединение
состава AmBn на диаграмме плавко-
плавкости имеет скрытый максимум (пунк-
(пунктирная кривая), в отличие от явного
максимума в случае а. Промежуточ-
Промежуточное соединение АтВ„ может образо-
образовать твердые растворы с обоими ком-
компонентами или с одним из них. Диаг-
Диаграммы, соответствующие случаям а и
б, по с твердыми растворами, показа-
показаны соответственно в случаях виз.
Случай в отличается от г еще и тем,
что в последнем вся область сущест-
существования третьей твердой фазы лежит
за пределами ординаты химического
соединения АтВ„. Н. С. Курнаков
* Такие соединения называются интер-
интерметаллическими, или металлическими.
295
Yi
- 1
1
1
a.
V"
i
1
1
6
—?—
i
N
Рис. 283. Типы двойных металлических си-
систем с тремя твердыми фазами
jor С u, am. %
то 10—° -50 -70 - 90
900
700
500
300
ыОо
!° Рриертс-Аистен и Po3j
Hup на нов1/Шемчи>нныи .,
№д
Л
\\
>—•
=3
I
^\
=3
{
4
ОС
\
~^
ОС'
тидкз-к
\
о 20
Аи
но во во
См, бес. %
Си
Рис. 284. Диаграмма состояния системы
Си —Аи
Аи
рассматривает две возможные трак-
трактовки этого случая — д и е. Экспери-
Экспериментальные кривые в обоих случаях
почти одинаковы. Разница между ни-
ними заключается в пунктирных (тео-
(теоретических) кривых. В первом слу-
случае третью фазу он трактует как твер-
твердый раствор компонента А в соеди-
соединении AmBn- Во втором случае — как
твердый раствор компонента А в не-
несуществующей в чистом виде на
опыте В'-модификации компонента В.
Вовсе не обязательно, чтобы третья
фаза имела бы упорядоченную струк-
структуру. Она может быть твердым рас-
раствором с кристаллической структурой,
отличной от структур чистых компо-
компонентов. Этот случай имеем в системах
Си—Zn, Си—Ga, Си—Ge, когда вслед-
вследствие достижения предельного значе-
значения электронной концентрации наря-
наряду с твердым раствором меди с другим
компонентом (а-фаза) появляется
новый твердый раствор — C-фаза с
иной кристаллической структурой (в
приведенном примере — объемноцен-
трированной кубической). Между дву-
двумя твердыми растворами а и C распо-
располагается двухфазная область (а + C).
Естественно, конечно, что в двухком-
понентньтх системах таких промежу-
промежуточных фаз постоянного и переменно-
переменного состава может быть несколько.
До сих пор мы рассматривали слу-
случаи выпадения третьих твердых фаз
из жидкого расплава. ;В действитель-
действительности они могут выпадать также из
твердых растворов [см. диаграммы со-
состояния Си—Аи (рис. 284), Ni—Аи
(рис. 285) и Pt—Ag (рис. 286)]. Вы-
Выпадающие новые твердые фазы могут
или сохранить в основном структур-
структурный тип «материнского» твердого
раствора (Cu3Au, рис. 287), или дать
слегка искаженную, связанную с по-
понижением симметрии, структуру
(CuAu, см. рис. 296), или могут при-
принадлежать к другому структурному
типу.
Рис. 285. Диаграмма состояния системы
Ni —Аи
296
{°C
1900
1500
1100
700
Pi 25 50 75 100
Дд, am. %
Рис. 286. Диаграмма состояния системы
Pt —Ag
Рис. 287. Структура Cu3Au
§ 12. Особенности строения
интерметаллических
соединений.
Отношения между
интерыеталлическнми
соединениями
и твердыми растворами
Для неорганических соединений
характерна весьма высокая степень
упорядоченности. Так, в структуре
NaCl трудно себе представить, что
в каком-то участке атом хлора ока-
окажется окруженным атомами хлора.
Вследствие ионного характера свя-
связи в кристалле поваренной соли по-
попавший «не на свое место» ион бу-
будет в процессе кристаллизации вы-
вытолкнут одноименными ионами.
Поэтому структуру NaCl можно
представить построенной из строго
чередующихся между собой ионов
противоположного знака, и упоря-
упорядоченность ее должна быть близка к
100%.
В интерметаллических соедине-
соединениях картина иная. Очень часто бли-
ближайшими соседними атомами в струк-
структуре могут оказаться одноименные
атомы. Так, например, в структурном
типе СизАи (рис. 287) даже в идеаль-
идеальном случае у каждого атома меди из
двенадцати ближайших соседних ато-
атомов восемь будут атомами меди и
только четыре — атомами золота. По-
Поскольку в системе Си — Аи имеют ме-
место непрерывные твердые растворы,
то при составе 75 ат. % Си и 25 ат. %
Аи каждый атом меди будет окружен
в среднем девятью атомами меди и
тремя атомами золота. Проявление
химизма между медью и золотом при-
приводит, очевидно, к тому, что в струк-
структуре соединения понижается число
однородных атомов в первой коорди-
координационной сфере.
Меньший антагонизм между одно-
одноименными атомами в соединениях с
металлической связью по сравнению
с соединениями с ионной связью при-
приводит к тому, что даже в химическом
соединении не все места одной пра-
правильной системы точек в структурном
типе оказываются занятыми атомами
одного химического элемента. Так,
например, места в центрах граней в
структуре Cu3Au (рис. 287), заняты
не на 100% атомами меди, а приблизи-
приблизительно на 80—90%)- Аналогично не
все места в вершинах элементарных
параллелепипедов заняты атомами зо-
золота. В реальной структуре часть ато-
атомов золота располагается в центрах
граней ячейки и, соответственно,
часть атомов меди располагается в ее
вершинах. Степень упорядоченности
не достигает 100%, а составляет лишь
большую или меньшую часть. Сте-
Степень упорядоченности зависит от не-
нескольких причин: от химической бли-
близости компонентов, от скорости кри-
кристаллизации соединения и т. п. Если
интерметаллическая твердая фаза об-
образовалась из расплава, то при про-
прочих равных условиях упорядочен-
упорядоченность в ней будет более высокой, чем
у фаз, образующихся из твердых рас-
растворов.
Можно ли при описании подобных
случаев говорить о наличии интерме-
интерметаллических соединений? Многие ав-
авторы предпочитают пользоваться тер-
297
что —^rr*^-—
Hz 20^40 ВО 1
"" am.%
Рис. 288. Диаграмма диссоциации воды
мыном «упорядоченные твердые рас-
растворы», «сверхструктуры» или нейт-
нейтральным термином «интерметалличе-
«интерметаллическая фаза», избегая слова «соедине-
«соединение».
Автору кажется вполне возможным
использование термина «соединение»,
который в этом отношении согласен
с взглядами Д. А. Петрова, приводя-
приводящего в качестве аналогии диаграмму
диссоциации воды (рис. 288). Выше
4700° К в газовой фазе состава 662/з ат.
% Н2 и 33 7з ат.% О2 присутствуют
только молекулы водорода и кислоро-
кислорода. При понижении температуры по-
появляются молекулы НгО. Никто не
станет отрицать появления здесь хи-
химического соединения. Три вещества
образуют газовую фазу. При дальней-
дальнейшем понижении температуры мы мо-
то
1400
то
1000
800
дОО
ж
ее
I
-
/V+ ?
7
ct+fi
I I
Ni
20 40
AVam.%
Рис. 289. Диаграмма состояния системы
Ni — A1
жем при известных условиях прийти
к тому, что вся фаза (на 100%) ока-
окажется состоящей из газообразных мо-
молекул НгО. Точно так же фаза состава
75 ат.% Си и 25 ат.% Аи (рис. 284)
может состоять из соединения СизАи
и твердых растворов — (Си, Аи),
(СизАи, Си), (Си3Аи, Аи). В зависи-
зависимости от условий процентное отно-
отношение соединения и твердых раство-
растворов может и будет, конечно, меняться.
Но ниже какой-то температуры
C95° С) вряд ли можно отрицать на-
наличие в фазе интерметаллического
соединения Cu3Au.
Из разобранных примеров видно,
что нет и не может быть проведено
резкой границы между понятиями
«твердый раствор» и «интерметалли-
«интерметаллическое соединение». Это — разные
случаи проявления химизма между
двумя элементами. И нет поэтому ни-
ничего удивительного в том, что в ряде
случаев одна фаза охватывает область
от интерметаллического соединения
до одного из его компонентов. При-
Примером может служить диаграмма со-
состояния системы Ni — А1 (рис. 289).
Ниже температуры 1100° С однород-
однородная фаза распадается на два твердых
раствора аг и а", осуществляющихся
на основе алюминия и Ni3Al (струк-
(структурный тип СизАи). Выше этой тем-
температуры существуют непрерывные
твердые растворы между соединени-
соединением и одним из компонентов.
§13. Процесс упорядочения
в иитернетадлических фазах
Детальные исследования твердых фаз
в системе Си — Zn показали, что в об-
области высоких температур C-фаза
CuZn, относящаяся к структурному
типу a-Fe, полностью не упорядочена.
В области же низких температур
упорядоченность достигает весьма
высоких значений, и вполне целесо-
целесообразно относить ее к структурному
типу CsCl. Упорядоченная фаза CuZn
обычно обозначается как CsCl. Про-
Процесс перехода неупорядоченного со-
298
Рис. 290. Схема полностью неупорядочен-
неупорядоченной (а) и полностью упорядоченной (б)
структур
стояния в упорядоченное и обратно
осуществляется в определенной об-
области температур, а не при одной
температуре, как обычно осу-
осуществляются фазовые переходы, свя-
связанные, например, с полиморфизмом
вещества. На рис. 290 схематически
показны полностью неупорядочен-
неупорядоченная структура а и полностью упоря-
упорядоченная Ь. Вероятность найти, на-
например, «белый» атом в вершинах
элементарных параллелепипедов или
в центрах их одинакова и равна 50%
для случая а. Для случая Ъ она рав-
равна 100% для вершин параллелепипе-
параллелепипедов и 0% —для их центров.
На каждом рисунке одно из мест в
структуре оставлено пустым (кре-
(крестик). Такие «пустоты» в некотором
количестве всегда имеются в реаль-
реальных кристаллах. Если вычислить
энергию перескока соседнего с пу-
пустотой «белого» атома в случаях а и
б, то окажется, что в случае неупо-
неупорядоченного состояния энергия мень-
меньше, чем в случае упорядоченного со-
состояния. Чем меньше упорядочен-
упорядоченность, тем легче она нарушается. Вы-
Высокий порядок препятствует возник-
возникновению беспорядка. Если постепен-
постепенно повышать температуру, то снача-
сначала можно наблюдать медленное и
незначительное увеличение беспоряд-
беспорядка. По мере его увеличения дальней-
дальнейшее разупорядочение происходит все
быстрее, лавинно нарастая, и, нако-
наконец, почти внезапно, упорядоченность
исчезает в сравнительно узкой обла-
области температур. Так, например, ин-
интенсивное разупорядочение фазы
CuZn начинается при температуре
около 390° С и заканчивается в ин-
интервале 20° С. (Детальные рентгено-
рентгенографические исследования процессов
упорядочения в системе Си—Аи были
проведены Н. В. Агеевым. Количе-
Количественная теория упорядочения
твердых растворов была развита
Л. Д. Ландау и Е. М. Лифшицем.)
Структурную сторону процесса
упорядочения очень детально иссле-
исследовали Бредлей и Джей A932 г.) на
примере системы Fe — А1.
На рис. 291 изображена ячейка с
четырьмя эквивалентными положе-
положениями — а, Ь, с, d. Если все эти
положения занимают одинаковые ато-
атомы (например, атомы железа), то ри-
рисунок будет изображать 8 элементар-
элементарных ячеек структуры cc-Fe. При со-
составе 50 ат. % Fe и 50 ат.% А1 ато-
атомы Fe занимают положения а и ft,
а атомы А\ — с и d. В этом случае
рисунок изображает 8 элементарных
ячеек интерметаллического соедине-
соединения FeAl, кристаллизующегося в
структурном типе CsCl. Если к чисто-
чистому железу постепенно прибавлять все
большее и большее количество алю-
алюминия, то в закаленных сплавах (со-
(соответствующих состоянию сплава при
высоких температурах) в интервале
от 0 до 25 ат. % А1 наблюдается чисто
статистическое распределение атомов
алюминия в структуре железа, т. е.,
иначе говоря, имеет место типичный
твердый раствор. Вероятность встре-
встретить атом А1 в положениях а, Ъ, с или
d пропорциональна количеству рас-
растворенного алюминия. На рис. 292
эта зависимость изображается пря-
прямой линией A, 2). По оси абсцисс от-
отложен состав сплава в атомных про-
процентах А1; по оси ординат — вероят-
вероятность нахождения атомов А1 в каком-
либо положении — а, Ъ, с или d, выра-
выраженная в процентах.
Сплавы, содержащие 25 ат.% А1,
представляют собой твердые растворы
А1 в cc-Fe. Дальнейшее прибавление
алюминия приводит к упорядочению
твердого раствора. Атомы алюминия
начинают занимать положения с и d
(кривая 3), вытесняя из них атомы
299
О а • b ®c ©d
Рис. 291. Четыре эквивалентных положе-
положения в ячейке
10 Ц9 ЦЗ Z0,7Z%4Z8,3 32,6
kifiec. %
Рис. 292. Процесс упорядочения в систе-
системе Fe — А1
железа в положения а и Ь. Этот про-
процесс заканчивается при составе
50 ат.% А1, при котором получается
структура FeAl с упорядочением,
близким к 100%. Таково поведение
атомов в структуре a-Fe при высоких
температурах.
Если аналогичные исследования
производить с отожженными сплава-
сплавами, т. е. создавать условия, благопри-
благоприятные для упорядочения, то процесс
упорядочения начнется гораздо рань-
раньше. В отожженных сплавах, начиная
уже с состава 18 ат. % А1, происходит
резкое упорядочение. Атомы А1 начи-
начинают занимать одно из положений —
с, вытесняя атомы железа (кривая 4).
При составе 75 ат.% Fe и 25 ат.%
А1 получается новый структурный
тип Fe3Al (см. рис. 301). Однако сте-
степень упорядоченности не достигает
100%. Часть положений с еще зани-
занимают атомы железа. Дальнейшее при-
прибавление алюминия приводит к пол-
полному вытеснению атомов железа из
300
этого положения. Однако 100%-ное
заполнение положений с атомами
алюминия осуществляется только при
составах, значительно более богатых
А1, чем состав FeeAl. Начиная с
25 ат.% А1 атомы алюминия начи-
начинают занимать также положение d,
покидая при этом позицию с (кри-
(кривая 5). При 38 ат.% А1 вероятность
найти атомы А1 в положениях end
одинакова. В области > 38 ат. % А1
отожженные сплавы не отличаются
от закаленных.
§14. Дальтониды и бертолляды
С 1900 г. Н. С. Курнаков с сотрудни-
сотрудниками начал экспериментально изу-
изучать химические взаимоотношения
металлов друг с другом. Основная
трудность установления в таких си-
системах химических соединений за-
заключается в том, что при взаимодей-
взаимодействии металлов очень часто образовы-
образовывались твердые фазы переменного
состава, которые затруднительно
было характеризовать какими-либо
определенными формулами. Эти фазы
или примыкали к компонентам, т. е.
образовывали ограниченные твердые
растворы на основе компонентов, или
же были отделены от компонентов,
как это показано в средней части всех
диаграмм, изображенных на рис. 283.
Условимся их называть третьими, или
промежуточными, фазами. В даль-
дальнейшем будем говорить именно о них.
Н. С. Курнаков обратил внимание
на то, что учение о фазах не делает
принципиального различия между хи-
химическими индивидуумами постоян-
постоянного и переменного состава, и предло-
предложил эти индивидуумы называть
соединениями постоянного и пе-
переменного состава. Примеры соедине-
соединений третьих фаз постоянного состава
изображены на рис. 283, а и б, а при-
примеры третьих фаз соединений пере-
переменного состава в — е.
Все учение Курнакова о бертолли-
дах и далътонидах относится в основ-
основном к третьим фазам переменного
состава, о которых речь ниже.
Состав
к i
Рис. 293. Различные случаи кривых «сос-
«состав — свойство» для основных типов хи-
химических соединений
В дальнейшем Н. С. Курнаков и
его сотрудники выяснили, что для
установления рационального состава
таких фаз целесообразно использо-
использовать не только плавкость, но также и
другие физические свойства сплавов,
такие, как твердость, электропровод-
электропроводность и др.
В случае определенных соединений
и соединений постоянного состава на
изотермах диаграмм «состав — свой-
свойство» появляются особые точки, от-
отвечающие рациональному составу
(семейства кривых а, Ъ, рис. 293).
Курнаков предложил называть их
«сингулярными» точками. При изме-
изменении факторов равновесия наклоны
кривых могут меняться, семейства
кривых а и Ъ для одних свойств мо-
могут образовывать минимум, для дру-
других — максимум, но положение син-
сингулярной точки, отвечающей составу
соединения, остается постоянным.
В случае неопределенных соедине-
соединений диаграмма «состав — свойство*
будет «несингулярного» типа. Она мо-
может быть совсем без максимума (г),
но может и иметь максимум (в). Од-
Однако кривая «состав — свойство» в
этом случае будет более пологой и, са-
самое главное, максимум на кривой бу-
будет смещаться, в зависимости от
состава, при изменении факторов рав-
равновесия. Фазы такого типа Н, С. Кур-
Курнаков предложил называть бертолли-
дами, в отличие от далътонидов, или
определенных соединений. Таким об-
образом, к 1913 г. понятия дальтонидов
и бертоллидов были уже введены в хи-
химию. Было показано, что бертоллиды
являются такими же химическими
индивидами, как и дальтониды.
Из сказанного выше следует, что
понятия дальтонид и бертоллид отно-
относятся к характеристике фаз, причем
фаз переменного состава. Поэтому ис-
использование этих терминов примени-
применительно не к фазам, а к компонентам
будет неправильным. Молекулярные
соединения не могут иметь перемен-
переменного состава, поэтому учение
Н. С. Курнакова к ним не относится.
Однако если изучаются фазы, образо-
образованные двумя молекулярными соеди-
соединениями, например, органическими,
т. е. системы, в которых молекуляр-
молекулярные соединения являются компонен-
компонентами, то учение Н. С. Курнакова к та-
таким системам полностью применимо.
То обстоятельство, что дальтониды
характеризуются обычно составом,
отвечающим сингулярной точке, при-
приводит часто к большой путанице.
Говоря о рациональном составе даль-
дальтонидов и противопоставляя их ирра-
иррациональным по составу бертоллидам,
авторы часто забывают, что рацио-
рациональный состав дальтонидов относит-
относится лишь к одной ординате — к сингу-
сингулярной точке и что вне этой точки
состав их тоже уже не подчиняется
закону постоянства состава, т. е. он
в такой же мере, как и для бертолли-
бертоллидов, является иррациональным. Раз-
Разница здесь заключается лишь в том,
что у дальтонида есть сингулярная
точка, отвечающая рациональному со-
составу и максимуму на кривой
301
свойств, а у бертоллидов такой рацио-
рациональной точки нет; и если их состав
проходит через одну или несколько
точек рациональных отношений, то
этим отношениям не отвечают особые
точки на кривой свойств и этими точ-
точками (составами) нельзя характери-
характеризовать бертоллид.
Я. С. Курнаков, следуя определе-
определению Бертолле, писал, что ввиду не-
невозможности приписать бертоллидам
какой-либо постоянный и рациональ-
рациональный состав, их следует рассматривать
в целом как химическое соединение
переменного состава. Это первое по
времени определение бертоллидов,
в котором сделана попытка проник-
проникнуть в сущность понятия. Однако это
определение весьма несовершенно.
Ширина области гомогенности фаз
не является признаком, характеризу-
характеризующим принадлежность ее к дальтони-
дам или бертоллидам. Эта область мо-
может быть очень узка, и тем не менее
фаза не обязательно будет при этом
дальтонидом.
С 1913 по 1928 г. II. С. Курнаков с
учениками продолжал интенсивно
разрабатывать высказанные ранее
идеи. Для этого он сделал несколько
попыток проникнуть в сущность по-
понятия бертоллид как понятие менее
ясное, чем дальтонид, и в известном
смысле ему противопоставляемое.
В статье «Растворы и сплавы»
Курнаков предлагает рассматривать
самостоятельно существующие твер-
твердые фазы переменного состава (бер-
Am. %
Am. %
Рис. 294. Трактовка бертоллидов как твер-
твердых растворов на базе «мнимого» соеди-
соединения (а) или «мнимой» модификации
компонента (б)
толлпды) как определенные соедине-
соединения в состоянии диссоциации или же
как твердые растворы определенных
соединений или полиморфных моди-
модификаций компонентов, которые явля-
являются неустойчивыми в свободном со-
состоянии,—так называемые «мнимые
соединения» (рис. 294).
Это определение противопоставляет
бертоллиды дальтонидам. Кроме того,
в нем содержится важный момент о
самостоятельном существовании бер-
толлида, т. е. подчеркивается разрыв
между бертоллидом и другими фазами
системы, в частности, фазами, образо-
образованными на базе компонентов.
В нескольких работах Н. С. Кур-
Курнаков A936, 1940 гг.) пишет о воз-
возможности непрерывных переходов
дальтонидов в бертоллиды при измене-
изменении условий, например, при более
высоких температурах. Впоследст-
Впоследствии эти переходы реально были об-
обнаружены Н. В. Агеевым и Е. С. Ма-
Макаровым A943 г.) в нескольких сис-
системах, см., например, Fe—Sb—Ni.
На рис. 293 собраны различные
типы кривых «состав — свойство»: се-
семейства кривых а, б, и в имеют
максимумы, г — без максимумов. Се-
Семейство кривых а характеризует
наличие определенных соединений,
семейство кривых г типично для твер-
твердых растворов. Индексами 1 отмече-
отмечены кривые, оба конца которых рас-
располагаются на составах, отвечающих
простым кратным отношениям или
компонентам системы. Если у кри-
кривых только один конец удовлетворя-
удовлетворяет этим требованиям, то такие кри-
кривые обозначены идексами 2 ж 3, вто-
второй конец у них иррационален. Ин-
Индексы 4 проставлены у тех кривых,
у которых оба конца иррациональны
(наиболее распространенный слу-
случай).
Многие фазы имеют кривые типа
34, и тогда возникает вопрос: как их
трактовать. Является ли этот отре-
отрезок аналогичным отрезком кривой
типа а\, б\, в\ или Z\.
Трактовка фаз с кривой этого ти-
типа вызывает наибольшие трудности.
302
В самом деле, если бы можно было
экстраполировать ее в обе строны
или хотя бы в одну, то вопрос был
бы разрешен.
Н. С. Курнаков, исходя из теоре-
теоретических соображений, предвидел
два возможных решения и в обоих
случаях такие фазы назвал бертол-
лидами. Однако он не предложил
метода, позволяющего однозначно
отнести фазы к тому или иному типу:
к типу твердого раствора на основе
«мнимого соединения» или к типу
твердого раствора в неустойчивой мо-
модификации компонента. Н. С. Курна-
Курнаков эту двойственность решения,
вероятно, считал принципиально не-
неразрешимой. Действительно, разли-
различить эти случаи, пользуясь метода-
методами только физико-химического ана-
анализа, по-видимому, невозможно. Во
всяком случае, все попытки учеников
Курнакова, сделанные в этом на-
направлении, оказались тщетными. Не-
Некоторые из них даже стали отрицать
рациональность курнаковского тер-
термина «мнимые соединения».
В действительности вопрос, по-ви-
по-видимому, не обстоит столь безнадеж-
безнадежно. Если использовать сведения, ко-
которые дает нам современная крис-
кристаллохимия, базирующаяся на гео-
геометрической теории структуры крис-
кристаллов, то по закономерному измене-
изменению структуры с составом мы можем
находить эти предельные структуры
«мнимых соединений» Курнакова
и тем самым во многих конкрет-
конкретных случаях решать вопрос одно-
однозначно.
Химические индивидуумы в твер-
твердом состоянии существуют в виде
простых веществ, химических со-
соединений постоянного и переменного
состава с узкой или широкой обла-
областью гомогенности. Нередко возни-
возникают трудности в определении при-
природы фаз переменного состава. Од-
Однако сочетание метода физико-хими-
физико-химического анализа с данными об атом-
атомной структуре веществ позволяет
эти трудности преодолеть (Г. Б. Бо-
кий, 1956).
Рассмотрим, например, атомное
строение дальтонидов и бертоллидов
на примере двойных соединений,
изображенных на рис. 252. Итак,
дальтониды — фазы переменного
состава, характеризующиеся сингу-
сингулярной точкой, являющейся пересе-
пересечением двух кривых на диаграмме
«состав — свойство». Эта точка отве-
отвечает рациональному составу. При-
Примером дальтонида на рис. 252 (стр.
233) будет фаза в системе Ni—Sb,
дальтоновская точка которого отве-
отвечает составу NiSb. Область гомоген-
гомогенности этой фазы простирается по обе
стороны от точки, отвечающей соста-
составу 50 : 50 %. Несмотря на неболь-
небольшую протяженность фазы (порядка
10%), кристаллохимические сведе-
сведения о ее строении позволяют легко
определить пределы, до которых мог
бы дойти состав этой фазы, если бы
замещения в одной и второй пра-
правильных системах точек продолжа-
продолжались бы до конца. Это будут ШгЭЬ
и №ЭЬг со структурами N12I11 и
ШТег соответственно. Оба написан-
написанные выше состава являются состава-
составами курнаковских «мнимых соедине-
соединений». Три других фазы переменного
состава, изображенные на рис. 252,
являются бертоллидами без макси-
максимума на кривой свойств. Причем в
системе Ni — Те фаза имеет протя-
протяженность от одного реального преде-
предела — NiTe до другого — №Тег.
В системе Ni — In реализуется толь-
только один предел Nijn, второй предел
является мнимым соединением Niln,
и на диаграмме он не реализуется.
В системе Ni — Sn оба предела не ре-
реализуются — Ni2Sn и NiSn являются
«мнимыми соединениями».
Наибольшие трудности вызвала
структурная теория бертоллидов с
максимумом, не отвечающим опреде-
определенному химическому составу и сме-
смещающемуся в зависимости от изме-
изменения термодинамических условий
(Г. Б. Бокий, 1963). Этот случай
можно иллюстрировать на примере,
аналогичном системе Fe — А1 (рис.
292).
303
a
с
-
a
A
I
/
/
/
g
—•
20 40 Б0 SO 100%
/IB
11
о
о
г
О
О
h 1
Рис. 295. Смещение максимума на кривых
«состав — свойство» в зависимости от из-
изменения условий кристаллизации
На рис. 295 схематически пока-
показана диаграмма состояния «состав —
свойство» такого бертоллида (а) и
возможная атомная структура его
(б). Точки, отвечающие различным
составам на диаграмме, и распреде-
распределение атомов компонентов А и В по
различным положениям в кристал-
кристаллической структуре, показаны на
рис. 295, б. Из этой схемы видно, что
чем позже в твердых растворах на-
начинается упорядочение (точки a, d,
g...), тем больше отклоняется макси-
максимум от состава 1 : 1 (точки с, е, ?...).
На схеме кривые ас, de, gi, а также
сЪ, ef показаны приблизительно
параллельными друг другу; вероят-
вероятно, в действительности это не всегда
бывает так. Возможно, что вторая се-
серия кривых при заполнении положе-
положения 1 не достигает кривой аЪ, а идет
каким-то образом непосредственно к
точке Ь (см. пунктирные линии от е к
Ъ, от i к Ь...). При таком механизме
заполнения пространства атомами
особенно наглядно подчеркивается
тот факт, что чем позже в твердых
растворах начинается упорядочение,
тем правее располагается максимум,
и кривая становится более пологой.
Таким образом, с выяснением при-
природы бертоллидов, имеющих макси-
максимум на кривой «состав — свойство»,
по всей видимости, завершается атом-
ноструктурная теория дальтонидов и
бертоллидов, а завершение ее геомет-
геометрической части открывает новые
возможности для продолжения иссле-
исследований в области физической химии
и, в частности, в термодинамике.
Мы недостаточно знаем причины,
побуждающие конкретные твердые
растворы к началу упорядочения.
В самом деле, почему в системе же-
железо — алюминий упорядочение начи-
начинается с 18 ат. % А1, а в ферритах
типа МпхРез_жО4, имеющих структу-
структуру шпинели, при малых значениях х
степень обращенности структуры
близка к нулю и неожиданно (почти
скачком) возрастает до 75% при
х ~ 0,8. Это убедительно было пока-
показано в 1962 г. Ю. 3. Нозиком нейтро-
нографическим методом.
Дальнейшее развитие идей Курна-
кова, применение их для разработ-
разработки новых методов весьма перспек-
перспективны.
§ 15. О систематике соединений
переменного состава
При разработке систематики объек-
объектов неживой природы, кроме непре-
непрерывности изменения состава, мы
встречаемся еще и с непрерывностью
изменения геометрических форм, ха-
характеризующих атомную структуру
кристаллов: параметров их решеток,
форм координационных многогран-
многогранников и т. п., что, в частности, может
привести к непрерывному переходу
одного структурного типа в другой.
Как известно, кубических решеток
Бравэ три: примитивная, центриро-
центрированная и гранецентрированная. Ес-
Если по узлам этих решеток распола-
располагаются атомы, то мы излучим три
структурных типа: Ро, a-Fe и Си.
Нетрудно показать, что деформаци-
деформацией вдоль оси третьего порядка мож-
можно получить из любого названного
304
структурного типа два других. У ме-
меди примитивной ячейкой будет ром-
ромбоэдр с утлом в 60°. Сжимая его,
можно получить ромбоэдр с углом в
90°, т. е. простой куб. А при даль-
дальнейшем сжатии до угла в 109° —
структурный тип a-Fe. По пути мы
получим и структурный тип Hg с уг-
углом ромбоэдра, равным 72°32'.
Из гранецентрированной решетки
можно получить центрированную,
если сжимать первоначальный куб
вдоль оси четвертого порядка. В объ-
емноцентрированном тетрагональном
аспекте гранецентрированная ячей-
ячейка имеет отношение осей с/а = 1,41.
Сжатием мы приводим это отноше-
отношение к единице. Промежуточный
структурный тип Y"Mn с отноше-
отношением осей с/а = 1,31 (см. табл. 42).
Многие интерметаллические со-
соединения имеют промежуточные зна-
значения отношений параметров реше-
решеток и при изменении термодинами-
термодинамических условий могут непрерывно
заполнять промежутки между этими
крайними значениями.
Непрерывные переходы можно
осуществить не только от одного
структурного типа к другому, но и
от одного семейства к другому или
от одного отряда к другому.
В качестве примера рассмотрим
уже фигурировавшие ранее три ве-
вещества, принадлежащих к структур-
структурному типу рутила (см. главу IX,
§ 10). Изменение параметра в струк-
структурном типе с параметрами, как
было там сказано, влечет за собой из-
изменение формы координационных
многогранников. Только координа-
координационный октаэдр у атома магния в
структуре MgF2 является достаточ-
достаточно близким к идеальному. В струк-
структуре SnCb два атома кислорода из
шести, окружающих атом олова, ока-
оказываются ближе, чем четыре осталь-
остальных. Вследствие этого в структуре
намечается обособление молекул
впОг. В структуре ТЮг четыре атома
кислорода из шести оказываются бли-
ближе к атому Ti, чем два остальных.
Такое значение параметра х обуслов-
20 Кристаллохимия
ливает в этой структуре обособление
цепочек [ТЮг]».
Величина параметра х может ме-
меняться не только от вещества к ве-
веществу, но она будет разной у одно-
одного и того же вещества в различных
термодинамических условиях.
По этой причине в данном вопро-
вопросе тоже следует формально условить-
условиться, какие изменения считать доста-
достаточными, чтобы отнести вещество
в ту или иную таксономическую
группу. Когда разница очень вели-
велика, тогда вопрос ясен. Например, в
структуре СОг ближайшее расстоя-
расстояние С—О равно 1,14, а следующее
уже 3,12, т. е. превышает кратчай-
кратчайшее почти втрое, поэтому ясно, что
мы имеем дело с молекулярной сис-
системой. А как считать структуру SnOa,
когда разница в расстояниях не дос-
достигает и 30%?
В § 9 главы XVII была изложена
система, в которой предложены ос-
основы классификации соединений
постоянного состава. Как уже неод-
неоднократно говорилось, многие фазы
на диаграмме состояния имеют оп-
определенную область гомогенности,
более узкую или более широкую. По
Н. С. Курнакову, эти фазы могут
быть дальтонидами или бертоллида-
ми. Если первые еще можно харак-
характеризовать постоянным составом,
хотя бы относящимся лишь к одной
«сингулярной» точке, то вторые да-
даже в том случае, если область гомо-
гомогенности их достаточна узка, ка-
каким-либо определенным составом,
лежащим в этой области гомоген-
гомогенности, характеризовать нельзя. Это
уже было со всей очевидностью яс-
ясно Н. С. Курнакову. Существо та-
таких фаз он предложил трактовать
как твердые растворы либо в несу-
несуществующем «мнимом» соединении,
либо в несуществующей «мнимой»
модификации компонента. Все дело
затруднялось тем, что в арсенале
физико-химического анализа было
очень мало средств для такого отне-
отнесения конкретных фаз к одному или
другому типу бертоллида и для точ-
305
ного определения состава «мнимого»
соединения в первом случае. Как
уже указывалось ранее (Г. Б. Бо-
кий, 1963 г.), такая возможность
появилась в результате развития
кристаллохимии. Целочисленность
коэффициента в формуле химиче-
химического соединения (закон Дальтона)
определялась в основном правилами
целочисленной валентности. Но ког-
когда выяснилось, что в одном соеди-
соединении атомы одного элемента могут
иметь разную валентность, причем
соотношение их может меняться в
зависимости от условий синтеза и
равновесных термодинамических ус-
условий, например, в сульфиде желе-
железа, то стало ясно, что выразить фор-
формулу такого соединения целочис-
целочисленными коэффициентами невозмож-
невозможно. В этом примере формулу такого
соединения пишут в виде Fej-^S, где
х колеблется от 0 до 0,17.
Для всех соединений, химиче-
химические формулы которых нельзя выра-
выразить целочисленными коэффициен-
коэффициентами, мы предлагаем использовать
кристаллохимическую целочислен-
целочисленность — кратность правильных сис-
систем точек. Целочисленность валент-
валентности и коэффициентов в формулах
используется в химии очень давно,
а кристаллохимическая (структур-
(структурная) целочисленность практически
до сих пор в химии не использо-
использовалась.
С точки зрения кристаллохимии
фазы такого типа характеризуются
тем, что в одной правильной системе
точек, занятой атомами железа, на-
начали освобождаться места в резуль-
результате замещения некоторых атомов
двухвалентного железа атомами
трехвалентного. В предельном случае
трехвалентных атомов железа в этой
правильной системе 34%, а 49% за-
занято атомами двухвалентного железа,
и 17% мест пустует.
Фазы переменного состава со
структурной точки зрения могут
быть трех типов: твердые растворы
замещения, внедрения и вычитания.
Во всех случаях кратности правиль-
правильных систем точек целочисленны, по-
поэтому даже фазы переменного соста-
состава можно представить в виде форму-
формулы с целочисленными коэффициен-
коэффициентами.
Замещаться или освобождаться та
или иная правильная система точек
может либо до конца (что отразится
на химической формуле соединения),
либо ограничение будет наложено
правилами валентности и так, что
сохранится целочисленность коэффи-
коэффициентов в химической формуле. Этот
второй случай, очевидно, имеет мес-
место в взятом примере сульфида же-
железа. Предельным (нереализуемым
экспериментально) случаем будет
формула Fe?S3.
В той же работе A963 г.) мы ука-
указывали, что в случае установления
типа твердого раствора кристаллохи-
мический метод дает в руки исследо-
исследователя простой путь определения
состава курнаковских «мнимых» со-
соединений (или «мнимых» модифика-
модификаций компонентов) в конкретных слу-
случаях. Всегда в силу целочисленности
кратностей правильных систем то-
точек для «мнимых соединений» коэф-
коэффициенты в формулах будут цело-
целочисленными.
Таким образом, каждую фазу пе-
переменного состава можно характери-
характеризовать пределами (реальными или
мнпмыми) с целочисленными коэф-
коэффициентами в формулах. Для бертол-
лидов таких пределов два (в бинар-
бинарных системах), для дальтонидов —
тоже два. Кроме того, дальтотгиды
характеризуются еще составом, соот-
соответствующим целочисленной дальто-
новской точке.
Итак, любую фазу мы можем ха-
характеризовать одним, двумя или тре-
тремя целочисленными составами. Пер-
Первый случай относится к фазам по-
постоянного состава, второй — к бер-
толлидам и третий — к дальтонидам.
В последних двух случаях осущест-
осуществляется непрерывный переход меж-
между крайними пределами или только
на ограниченном участке. В случае
дальтонидов, очевидно, в формуле
306
соединения должны быть указаны
границы этого участка.
В качестве примера вернемся к
ряду бинарных систем, атомные
структуры соединений в которых бы-
были исследованы Е. С. Макаровым
(см. главу XIII, § 12). Их целочис-
целочисленные мнимые пределы выписаны
ниже в круглых скобках; в квад-
квадратных скобках указаны те струк-
структурные типы (роды), к которым
принадлежит, стоящее рядом соеди-
соединив:
[NiAs]NiTe — NiTe2 [CdJ2],
[NiJnjNiJn — Ni1,,In(NiIn)[NiAs],
[Ni2In](Ni2Sn)Ni1,7Sii-
Ni1|2Sn(NiSn)[NiAs],
[Nijn] (Ni2Sb)Ni.,2Sb-NiSb
[NiAs] -Nio,7Sb(NiSb2) [CdJ2].
Резюмируя все изложенное выше,
можно сказать, что любую фазу би-
бинарной системы переменного состава
можно интерпретировать двумя или
тремя постоянными составами. Для
фаз тройных и многокомпонентных
систем соответственно будет и боль-
большее число предельных составов. Вся
кристаллохимическая систематика
фаз переменного состава сведется к
систематике соединений постоянного
состава.
Чем ближе с химической и струк-
структурной точки зрения будут друг к
ДРУГУ предельные целочисленные
соединения, тем незначительнее бу-
будут отклонения физико-химических
свойств на всей протяженности фазы
переменного состава, например, в
случае, если оба предельных соеди-
соединения относятся к одному структур-
структурному типу (роду).
Если же предельные соединения
относятся к разным структурным
типам (родам), семействам и отря-
отрядам, то изменение физико-химиче-
физико-химических свойств в таких фазах перемен-
переменного состава будет происходить с
составом более резко.
§1в. Важнейшие структурные тины
бинарных интерметаллических
соединений
1. О систематике структур интерме-
интерметаллических соединений. Одни и те
же по составу сплавы могут в одних
условиях иметь упорядоченные
структуры, в других — неупорядо-
неупорядоченные. Но описывать элементарные
ячейки структурных типов прихо-
приходится в идеализированном виде, т. е.
так, как будто бы упорядоченность
в них достигает 100%. В свете всего
вышесказанного о процессе и степе-
степени упорядочения интерметалличе-
интерметаллических фаз нам кажется, что такое опи-
описание не вызовет путаницы пред-
представлений о природе этих соедине-
соединений.
Структуры интерметаллических
соединений относятся ко всем кате-
категориям структур, перечисленным в
главе XIV (§1). Учитывая, что доля
ковалентной связи в интерметалли-
интерметаллических соединениях сравнительно не-
невелика, группы, в которые соединя-
соединяются атомы в структурах интерме-
интерметаллических соединений (каркасы,
слои, цепочки, острова) не обособле-
обособлены четко друг от друга; часто выде-
выделение таких групп затруднительно,
так как расстояния внутри группы не-
ненамного меньше расстояния между
атомами различных групп. В резуль-
результате этого интерметаллические кар-
каркасные, слоистые, цепочечные и
островные структуры имеют много
общего с координационными. Поэто-
Поэтому при классификации структур ин-
интерметаллических соединений целе-
целесообразно пользоваться признаком
координации атома. Все сказанное
до сих пор о структурах интерметал-
интерметаллических соединений относится так-
также ко многим структурам чистых
металлов, поэтому прежде чем перей-
перейти к систематическому описанию
структур интерметаллических соеди-
соединений, рассмотрим отдельно те
структуры соединений, которые вы-
выводятся из известных уже нам (гла-
(глава XVII) структур металлов.
20*
307
Атомы в структуре определённогб
интерметаллического соединения
(или металла) могут быть как близ-
близкой, так и резко различной величи-
величины. Другими словами, координаци-
координационные числа в структуре одного ин-
интерметаллического соединения мо-
могут быть одинаковыми, а в структуре
другого — различными. В последнем
случае возникает вопрос: координа-
координация какого атома — большего или
меньшего — должна лежать в основе
классификации структурных типов?
Анализ координационных чисел в
структурах; интерметаллических со-
соединений приводит к выводу, что раз-
разнообразие координационных харак-
характеристик более крупных атомов
слишком велико для того, чтобы эти
характеристики можно было поло-
положить в основу классификации. В то
же время атомам меньшего разме-
размера свойственно небольшое число ко-
координационных многогранников. По
этой причине структуры интерме-
интерметаллических соединений, составлен-
составленные из атомов различного размера,
лучше классифицировать по коорди-
координационным характеристикам атомов
меньшего размера, располагая со-
соединения в порядке увеличения раз-
различия в размерах атомов. Системати-
Систематика всех структурных типов интер-
интерметаллических соединений была
предложена П. И. Крипякевичем
A963 г.).
2. Структурные типы интерметал-
интерметаллических соединении, сходные со
структурами чистых металлов. Для
структурных типов интерметалличе-
интерметаллических соединений иногда характерна
чрезвычайная близость их друг к дру-
другу и к некоторым структурным ти-
типам чистых металлов. Целесообраз-
Целесообразно поэтому при описании отдельных
типов объединить их в группы —
«семейства». Часто детали структур-
структурного типа для отдельных соединений
определены не до конца. В этом слу-
случае можно говорить о принадлежно-
принадлежности определяемой структуры только
к данному семейству, не уточняя во-
вопроса о принадлежности к опреде-
определенному структурному типу. Описа-
Описание семейства начнем со структур-
структурных типов, у которых в качестве ро-
доначального структурного типа бу-
будет тип меди.
Среди структур чистых металлов
имеются представители, очень незна-
незначительно отличающиеся от структур-
структурного типа меди (кубической плотней-
шей упаковки). Они получаются в
результате незначительной, но за-
заметной деформации. Так, например,
структурный тип у-Мп может быть
получен из структурного типа меди
в результате небольшого сжатия по
оси четвертого порядка, а структур-
структурный тип Hg — в результате неболь-
небольшого сжатия по тройной оси. В этих
структурных типах могут кристалли-
кристаллизоваться неупорядоченные бинарные
фазы, причем составы их могут за-
заходить за пределы составов 1:1.
В случае упорядочения фазы может
быть получено несколько новых
структурных типов из одного исход-
исходного, в зависимости от состава и
симметрии ее кристаллов. Причем од-
одна элементарная ячейка новой
структуры может соответствовать
или одной ячейке старой структуры
(см. структуры Си и СизАи, тип I в
табл. 41, или нескольким (тип II).
Так, из структурного типа Си по-
получается структурный тип СизАи
(рис. 287). Тетрагональный аналог
этого структурного типа — ЭгРЬз —
воспроизводится таким же образом
ТАБЛИЦА 41
Структурные типы семейства меди
Характер
' деформации
решетки исход-
исходного структур-
структурного типа
Тип I
Тип II
Ко,
Си
СизАи
Pt7Cu
«я
¦у-Мп
CuAu
SrPbs
TiAU
ZrAlj
к
8
ЯП
о«
Он О,
PtCua
PtCu
308
из структурного типа у-Mn. Тетра-
Тетрагональной решеткой характеризует-
характеризуется и структурный тип CuAu (рис.
296). Удвоенную ячейку по сравне-
сравнению со БгРЬз с иным мотивом упоря-
упорядочения имеет структурный тип
TiAb (рис. 297). Учетверенная ячей-
ячейка будет у структурного типа ZrAb
(рис. 298), у которого, кроме того,
атомы Zr слегка сдвинуты из идеаль-
идеального положения. Структура PtCu3
аналогична структуре SrPbs, но при-
принадлежит к ромбической сингонии;
такого структурного типа для просто-
простого вещества нет. Структурный тип
PtCu получается из структурного ти-
типа Hg за счет удвоения ячейки вдоль
каждого ребра и упорядочения, как
показано на рис. 299.
Итак, структурные типы семейст-
семейства меди могут быть представлены в
виде следующей схемы (табл. 41).
Все тетрагональные структурные
типы (и, в частности, тип GuAu), по-
получающиеся вследствие деформации
кубических типов, можно рассматри-
рассматривать как в гранецентрированном, так
и в объемноцентрированном аспекте
(табл. 42).
ТАБЛИЦА 42
Различные аспекты тетрагонально-
искаженных структурных типов семейств
Си и a-Fe
Структур-
Структурный тип
Си
Y-Mn
a-Fe
Отношение с/а в тетраго нальной
установке
гранецентриро-
ванный аспект
1,00
0,93
0,71
центрированный
аспект
1,41
1,31
1,00
Таким образом, семейство струк-
структурного типа Си непрерывно перехо-
переходит в семейство типа a-Fe, которое
может быть представлено в виде сле-
следующей схемы (табл. 43).
Аналогичные семейства получают-
получаются у структурных типов Mg и a-Mn.
К последнему относятся структура
Рис. 296. Структурный тип CuAu
Рис. 297. Структурный тип TiAl3
Рис. 298. Структурный тип ZrAlj
«Я
Рис. 299. Структурный тип PtCu
309
•Tl ONa
Рис 300. Структурный тип NaTl
•41 OFe
Рис. 301. Структурный тип FejAl
о
77
Рис. 302. Структурный тип Сг2А1
ТАБЛИЦА 43
Структурные типы семейства
Тип I
Тип II
О
9
в
н
К р,
a-Fe
CsCl
NaTl
рис. 300)
FeaAl
рис. 301)
g 5
% n
S я
Ен и
Pa
MnAu
Сг2А1
(рис. 302)
AuCd
Р.З
Ni2Al»
у-латуни и родственные ей вещест-
вещества с формулой A;cBi3-*. Все эти се-
семейства в зависимости от присуще-
присущего им координационного многогран-
многогранника будут рассмотрены ниже.
3. Структурные типы с кубоокта-
эдрической координацией. В основе
структурных типов с кубооктаэдри-
ческой координацией лежат плотней-
шие упаковки шаров одинакового
размера (гл. XI). Для чистых метал-
металлов известны в настоящее время
структуры с 2-, 3-,. 4- и 9-слойными
упаковками (структурные типы Мп,
Си, La и Sm соответственно; см. гла-
главу XVII); для интерметаллических
соединений — с теми же упаковками
и, кроме того, с 6-, 8-, 12- и 15-слой-
ными. Координационный многогран-
многогранник во всех этих структурах — кубок-
таэдр и (или) его гексагональный
аналог. Структуры металлов и ин-
интерметаллических соединений, от-
относящиеся к данному классу (см.
раздел 2 настоящего параграфа), мо-
могут представлять собой как недефор-
мированные, так и деформирован-
деформированные плотнейшие упаковки. Распреде-
Распределение атомов различных компонен-
компонентов в структурах интерметаллических
соединений может быть неупорядо ¦
ценным (в этом случае они принад-
принадлежат к соответствующим структур-
структурным типам чистых металлов), но
чаще всего эти структуры упорядочен-
ны. Из одного исходного типа чисто-
310
го металла, как сказано выше, мо-
может быть получено несколько струк-
структурных типов с упорядоченным рас-
распределением атомов (и различным
составом, т. е. соотношением коли-
количеств атомов двух или больше сор-
сортов), так называемых сверхструктур-
сверхструктурных типов. Сверхструктуры одного
состава, производные определенной
плотнейшей упаковки, могут отли-
отличаться друг от друга (кроме дефор-
деформации) структурой плотноупакован-
плотноупакованного слоя, т. е. способом распределе-
распределения атомов различных компонентов
в слое (Н. Л. Смирнова, 1952—
1959); в большинстве случаев вся
структура состоит из слоев одинако-
одинакового типа, реже — из одинаковых па-
пакетов слоев. Таким типам (например,
АиСиз, TiAU n ZrAU, производным
типа Си) свойственна неодинаковая
симметрия и неодинаковый размер
элементарной ячейки.
Структурные типы с кубооктаэд-
рической координацией можно сис-
систематизировать по симметрии и раз-
размеру элементарной ячейки по типу
плотнейшей упаковки и (в случае
сверхструктур) по составу и по ти-
типу структуры плотноупакованного
слоя.
К данному классу структурных
типов могут принадлежать не толь-
только те соединения, компоненты кото-
которых имеют очень близкие атомные
радиусы (таких комбинаций метал-
металлов известно очень мало), но и со-
соединения с отношением атомных ра-
радиусов компонентов, заметно отлича-
отличающимися от 1 (например, с r?r/rAi =
= 1,21); в структурах последних раз-
размеры атомов выравниваются (эффек-
(эффективные атомные радиусы становятся
равными).
4. Структурные типы с кубической
координацией. Структурные типы с
кубической координацией возника-
возникают из кубической структуры типа
a-Fe. Из нее выводится ряд типов,
характеризующихся различными со-
составами, симметрией или размером
элементарной ячейки, собранных в
табл. 44. Сюда же относятся куби-
кубические типы Ti7Sb2 и ЫггРЬз, с а'—-
= За и 6а соответственно. Всем ти-
типам свойственно координационное
число всех атомов, равное 8 + 6
(8 атомов в вершине куба и 6 атомов
на более далеких расстояниях в вер-
вершинах октаэдра).
5. Структурные типы с икосаэдри-
ческой координацией атомов мень-
меньшего размера. При взаимодействии
металлов, атомы которых имеют раз-
различные размеры, могут образовывать-
образовываться, кроме структур с кубооктаэдри-
ческой координацией (см. стр. 310),
также структуры, в которых атомы
различных компонентов сохраняют
свои размеры. В таких случаях атом
меньшего размера (X) приобретает
к.ч. 12 и икосаэдрическую коорди-
координацию (отношение радиуса атома,
находящегося в вершине икосаэдра,
к радиусу атома в центре икосаэд-
икосаэдра — при условии соприкосновения
всех атомов — составляет приблизи-
приблизительно 1,08), а более крупный атом
(R) приобретает к. ч. от 14 до 24
(тем более высокое, чем выше отно-
отношение атомов радиусов) (П. И. Кри-
пякевич, 1960 г.). Координационные
многогранники с 14, 15 и 16 верши-
вершинами, так же как и икосаэдр, имеют
лишь треугольные грани; это так на-
называемые нормальные многогранни-
многогранники (Белов, 1947 г., Крипякевич,
1957, 1960 гг.; Франк и Каспер,
1959 г.).
Структуры с икосаэдрической ко-
координацией свойственны не только
интерметаллическим соединениям,
но и некоторым модификациям ме-
металлов — a-Mn, C-Mn, C-W (точнее
W3O), p-U. Структуры этих модифи-
модификаций состоят из атомов различно-
различного размера (к.ч. 12—16; см. гл.
XVII, § 2); каждой из них отвечает
сверхструктура, имеющая представи-
представителей среди интерметаллических со-
соединений (%-фаза TisRe24, зт-фаза
системы Сг — Ni — Si, Cr3Si, cr-фаза
V2Ni).
Из структурных типов, состоящих
из крупных атомов, с к.ч. 16 и
больше, наиболее распространены
311
ТАБЛИЦА 44
Систематика структур интерметаллических соединений
Семейства (структурные типы
имеющие одинаковые
К. ч.
12
12
12
12
10
8 (+6)
8 (+2)
6
к. ч. и к. м.)
К. м.
Гексагональный
аналог кубоокта-
эдра (г. п. у.)
Кубооктаэдр
(к. п. у.)
Оба типа кубоок-
таэдров
Икосаэдр
Пентагональная
призма
Куб
Томсоновский куб
Тригональная
призма
Род (структурный
Mg, Mg3, Cd, MnCu2Sn
Си, т-Мп, CusAu, CuAu, ЭгРЬз,
TiAb, ZrAl3, AlCug, PtCu
La, NisTi D ел. у.); F ел.
(8 ел. у.); Sm (9 ел. у.); A2 ел
o-Mn, P-Mn, WSO (P-W), 3-L
a-V2Ni, MgCu2, MgZn2, MgNis
ТЬМшг, NaZni3
MnAle, Mn2Hg5
ТИП)
PlCu3, Hg, Pt7Cu,
y.); Au75Cdi(iI.ni6
y.); A5 ел. у.)
I, Y-Ti6Re24,Cr3Si,
, CaCu6, ThNii?,
a-Fe, Pa, CsCl, MnAu, NaTl, Fe»Al, Cr2Al, AuCd,;
TbSbai Li22Pba, f-латуни : CusZ
CuAb
CrB (TIJ), A1B2, a-ThSi2
П8, CU9AI4
Отношение
радиусов
атомов
1-1
1—1
1,05-1
—
1-1
г—1,
x-i,
,23
,21
,50
37
56
89
Примечание: к. ч. — координационное число; к. м. — координационный многогранник; г. п. у.—
гексагональная плотнейшая упаковка; х — нижняя граница неизвестна.
типы MgZnn и MgNi2 — типы так
называемых; фаз Лавеса (к.ч. 16);
СаСи5, Th2Nii7, TI1M1112 (к.ч. 20);
NaZni3 (к.ч. 24). Отношение дейст-
действительных атомных радиусов компо-
компонентов, образующих фазы Лавеса, на-
находится в довольно широком интер-
интервале (~ 1,05—1,50).
Наиболее важным является струк-
структурный тип MgCu2 (рис. 303). Он
имеет кубическую ячейку. Атомы
магния располагаются по алмазному
закону. В каждом пустом октанте
структуры алмаза расположен пра-
правильный тетраэдр из атомов Си, при-
причем центры тяжести Сщ-тетраэдров
находятся в центрах октантов. Для
структуры характерна пространст-
пространственная тетраэдрическая вязь из ато-
атомов Си. Атом Mg окружен 12 атома-
атомами Си, образующими лавесовский
полиэдр (рис. 207, и), и 4 атомами
Mg; атом Си окружен 6 атомами Си
и 6 атомами Mg. В этом структурном
типе кристаллизуется большое число
соединений, для большинства кото-
которых характерна узкая область гомо-
гомогенности.
К структурному типу MgCu2 близ-
близки типы MgZn2 и MgNi2, которые
отличаются от него типом упаковки,
когда в основе структуры MgCu2 ле-
лежит 3-слойная плотнейшая упаковка;
Структуры MgZll2 И MgNi2 ВЫВОДЯТСЯ
соответственно из 2- и 4-слойной упа-
упаковок.
6. Структурные типы с дефектной
кубической координацией. Структу-
Структуры с дефектной кубической коорди-
координацией этого класса образуются при
вычитании части атомов из структур
с кубической координацией, т. е.
структуры типа a-Fe или ее сверх-
сверхструктур. В первом случае образуют-
образуются, между прочим, кубические струк-
структуры 7"латУнеи (Cu5Zn8, CueAU),
содержащие 52 атома в элементарной
ячейке (из ячейки типа a-Fe с утро-
312
•Cu OMg
Рис. 303. Структурный тип MgCu2
• А1. О Си
Рис. 304. Структурный тип СиАЦ
енным ребром, содержащей 54 атома,
вычитаются 2 атома), во втором слу-
случае — тригональная структура типа
№гА13 (вычитание :/з атомов Ni из
определенных положений структуры
NiAl типа CsCl). Структуры вычита-
вычитания, аналогичные №2А1з, целесо-
целесообразно рассматривать как основан-
основанные на простой кубической кладке
соприкасающихся атомов большего
размера (А1); атомы меньшего
размера (Ni) находятся в части ку-
кубических пустот кладки.
7. Структурные типы с тетраго-
тетрагонально-антипризматической коорди-
координацией атомов меньшего размера.
При увеличении разности в размерах
атомных радиусов двух компонентов
соединения кубическая координация
(которой свойственно rRlrx = 1,37)
уступает место тетрагонально-анти-
призматической (rR/rx^ 1,56). Струк-
Структуры с тетрагонально-антипризмати-
тетрагонально-антипризматическим окружением атома меньшего
размера атомами большего размера
довольно широко распространены
как среди собственно интерметалли-
интерметаллических соединений, так и среди ме-
таллоподобных боридов, силицидов и
германидов. Самое большое число
структур относится к структурному
типу СиА12 (рис. 304). В тетраго-
тетрагональной структуре СиАЬ атомы Си,
имеющие тетрагонально-антипризма-
тетрагонально-антипризматическую координацию, соединены
в цепи, параллельные оси Z; анти-
антипризмы сочленяются друг с другом
квадратными гранями в колонны.
Так как антипризмы несколько сплю-"
щены, в координационную сферу ато-
атома меди входят, кроме 8 атомов А1,
еще 2 атома Си из центров соседних
многогранников; расстояние Си — Си
даже меньше расстояния Си — А1.
Атомы А1 имеют координационный
многогранник с 15 вершинами
A1 А1 + 4 Си), идентичный с много-
многогранником, свойственным части ато-
атомов V в cr-фазе V2NL
8. Структурные типы с тригональ-
но-призматической координацией
атомов меньшего размера. Дальней-
Дальнейшее увеличение разности между ве-
величинами атомных радиусов приво-
приводит к тригонально-призматическому
окружению меньших атомов атомами
большего размера (при одновремен-
одновременном наличии контактов R — R и
R — X отношение атомных радиусов
равно 1,89). Призмы могут соединять-
соединяться друг с другом вершинами, ребра-
ребрами, треугольными или прямоуголь-
прямоугольными (квадратными) гранями. Приз-
Призмы, соединенные гранями, образуют
колонны, стены, штабеля или слои.
При соединении призм прямоуголь-
прямоугольными гранями возникают связи меж-
между атомами меньшего размера (чаще
всего Со, Ni, Cu, Hg, В, Al, Ga, Si, Ge).
Эти связи соединяют атомы указан-
указанных элементов в пары, прямоуголь-
прямоугольные группы Х4, зигзагообразные це-
цепи (простые, двойные или разветв-
разветвленные), сетки, каркасы. Наиболее
широко распространены структуры
следующих типов (указаны способы
соединения атомов меньшего размера
313
и к.ч. атомов большего размера) :
U3Si2 (пары; к.ч. 15 и 12) СгВ = Ш
(цепи; к.ч. 17), А1В2 (сетки; к.ч. 20),
a-ThSi2 (каркас; к.ч. 20). В структу-
структурах А1Вг и a-ThSi2 призмы заполня-
заполняют все пространство без пропусков,
в структуре СгВ промежутки между
призмами, соединенными в стены,
заняты пустыми тетрагональными
пирамидами н тетраэдрами.
В структурах интерметаллических
соединений встречаются и другие
(кроме указанных в разделах) ко-
координационные многогранники ато-
атомов меньшего размера, например
10-верпшнник (в структуре МпА1б),
пентагональная призма (в структуре
Mn2Hg5), но они сравнительно мало
распространены.
При отношении атомных радиусов,
большем, чем то, которое свойственно
тригональной призме, возникают
структуры с октаэдрической (гя/гх =
= 2,42) и тетраэдрической координа-
координацией (ги/гх = 4,45). Они свойствен-
свойственны, кроме интерметаллических со-
соединений (типа NiAs, CaF2 и др.),
также полуметаллическим и соле-
образным соединениям. Эти структу-
структуры рассматриваются в других разде-
разделах. Для них характерен направлен-
направленный тип связи, т. е. переход от ме-
металлической связи к ковалентной.
§ 17. Природа интериеталлпчесвпх
соединений
1. Электронные соединения. Выше
говорилось о большой роли электрон-
электронной концентрации при образовании
твердых фаз переменного состава.
Указывалось, что превышение из-
известных пределов электронной кон-
концентрации приводит к изменению
структурного типа. Так, например,
увеличение электронной концентра-
концентрации в твердом растворе (Си, Zn) за
счет прибавления цинка к меди мо-
может происходить только до кон-
концентрации 1,4. Следовательно, в ин-
интервале электронных концентраций
от 1 до 1,4 устойчивой будет а-фаза
твердого раствора со структурным
типом плотнейшей кубической упа-
упаковки. Увеличение концентрации
сверх значения 1,4 приводит к смене
структурного типа. В системе обра-
образуется Р-фаза, имеющая структуру
кубической объемноцентрированной
упаковки со статистическим распре-
распределением атомов. Этот структурный
тип обладает другим зонным строени-
строением, позволяющим принять в первую
зону большее число электронов.
В некоторых системах вместо струк-
структуры кубической центрированной
упаковки появляются структуры ти-
типа (З-Мп со статистическим распреде-
распределением атомов. Подсчет, проведен-
проведенный Джонсом, показал, что при кон-
концентрации 1,5 структура кубической
центрированной упаковки делается
неустойчивой, что в свою очередь
приводит к новой смене структурно-
структурного типа: р-фаза сменяется /у~Фазои-
Если электронная концентрация в си-
системе продолжает увеличиваться, то
при значениях, близких к 1,62, про-
происходит новая смена структурного
типа: ^"Фаза заменяется е-фазой,
имеющей структуру гексагональной
плотнейшей упаковки со статисти-
статистическим распределением атомов. Эту
закономерную смену структурных
типов в зависимости от электронной
концентрации Г. В. Курдюмов пред-
предлагает называть «концентрационной
аллотропией» (полиморфизмом).
Все перечисленные выше проме-
промежуточные фазы имеют специфиче
ское для них зонное строение и ха-
характеризуются следующими элект-
электронными концентрациями: р-фаза
3:2 = 1,5; ^-фаза 21 :13 = 1,62 и
е-фаза 7:4= 1,75. При подсчете
электронной концентрации надо
иметь в виду, что в разных соедине-
соединениях один и тот же элемент может
иметь различную валентность. Так,
например, элементы VIH-a подгруп-
подгруппы в интерметаллических соединени-
соединениях обычно имеют валентность, рав-
равную нулю, но в некоторых соедине-
соединениях они одновалентны (в ВеСо,
ШзЭп и др.) и даже двухвалентны.
314
2. Соединения, определяющиеся
объемным фактором. Ненаправлен-
Ненаправленность металлической связи обуслов-
обусловливает стремление каждого атома
окружить себя максимальным чи-
числом соседних, что приводит к струк-
структурам с большими координационны-
координационными числами. Было давно установлено,
что соединения состава АВг со струк-
структурами типа фаз Лавеса имеют
структурные типы с тетраэдрической
вязью одного из компонентов в тех
случаях, когда объем атома А при-
приблизительно в два раза больше объ-
объема атома В; отношение радиусов
з
атомов обоих компонентов У2 :1 =
= 1,260 или 1,225, если это отноше-
отношение рассчитывать из межатомных
расстояний (Шульце, 1939 г.).
Н. В. Белов A947 г.) установил, что
каждый атом компонента А занима-
занимает в структуре этих соединений сра-
сразу два места плотнейшей упаковки
и обладает специфическим координа-
координационным многогранником типа при-
притуплённого тетраэдра (лавесовского
полиэдра; см. структуры MgCu2 и др.,
рис. 207, и). Многие из них характе-
характеризуются большим процентом за-
заполнения пространства, чем плотней-
шие упаковки пз шаров одного
размера.
К этой категории соединений от-
относятся и некоторые интерметалли-
интерметаллические фазы, кристаллизующиеся
в структурном типе CsCl. Для этого
структурного типа характерно отно-
отношение га : гх = 0,73 -т- 1.37 или, счи-
считая всегда размер меньшего атома
за 1, возможные отношения размеров
будут колебаться от 1 до 1,37. При-
Принимая, однако, во внимание, что
в двойных металлических системах
при отношении радиусов атомов,
близком к 1 (до 1,10), образуются
твердые растворы, то соединений со
структурой типа CsCl следует ожи-
ожидать в интервале 1,10—1,37. Этот
предел почти совпадает с пределом
для соединений АВ2 A,09—1,34), как
показали Г. Б. Бокий и Э. Е. Вай-
штейн A943 г.), предположив воз-
возможность деформации атомов в
структурах фаз Лавеса. Таким обра-
образом, определенное соотношение объ-
объемов атомов может приводить к обра-
образованию в системе соединений АВ и
АВ2 с указанными структурными
типами.
Структуре типа CaCu5(RX5) свой-
свойственно отношение эффективных
атомных радиусов rRlrx, равное 1,44
(отношение объемов равно 3).
Структуры типов Th2Ni17 и ТЪМп12
образуются из нее при замещении
соответственно Уз и У2 более круп-
крупных атомов парами менее крупных
CRX5 -vRrX2Xi5; 2RX5 -»- RX2X10).
Величина отношения атомных ра-
радиусов влияет не только на величину
максимального к.ч. в структурах
данного класса, но и на состав соеди-
соединений: чем больше отношение эф-
эффективных атомных радиусов гк/гх,
свойственное данному типу RXn, тем
больше величина п, т. е. содержание
менее крупных атомов (Крипякевич,
1952, 1957, 1960 гг.).
3. Соединения, обусловленные ко-
валентными силами. Структуры со-
соединений с ковалентными связями
характеризуются низкими координа-
координационными числами. Эти соединения
нередко имеют структуры, типич-
типичные для неорганических соедине-
соединений — CaF2 и NiAs. В структурном
типе NiAs оба сорта атомов имеют
координационное число 6. Однако
атомы более электроположительно-
электроположительного элемента занимают места
с координационным многогранником
в форме октаэдра, т. е. многогран-
многогранника, часто встречающегося как в
соединениях с направленными (ко-
(ковалентными) связями, так и с не-
ненаправленными (ионными). Более
электроотрицательный элемент за-
занимает место, у которого координа-
координационный многогранник имеет форму
тригональной призмы, характерной
только для атомов, образующих ко-
валентные связи. Интересно, что
именно эту позицию занимают наи-
наименее электроположительные метал-
металлы, расположенные в IV-Ь и Ш-Ь
315
подгруппах — Ge, Sn, In. Этот факт
является лишним подтверждением
наличия у этих соединений ковалеит-
ных связей между атомами. Для ин-
интерметаллических соединений, крис-
кристаллизующихся в структурном типе
CaF2, характерным является то, что
элемент, более способный образовы-
образовывать ковалентные связи (т. е. стоя-
стоящий правее в периодической систе-
системе), всегда имеет координационное
число 4 (PtAb, AuAl2) AuGa2, Аи1пг).
4. Соединения, обусловленные
ионными силами. Тот же структур-
структурный тип CaF2 может быть обуслов-
обусловлен ионными силами. Для интерме-
интерметаллических соединений это может
иметь место у соединений самых
электроположительных металлов М5
с наиболее электроотрицательными
Miv-ь (Mg2Si, Mg2Ge, Mg2Sn, Mq2Pb).
В этих случаях сохраняются даже
нормальные правила валентности.
Однако это необязательно. Поскольку
интерметаллические соединения, в
отличие от неорганических, почти
всегда являются проводниками, а не
изоляторами, и имеют, следовательно,
свободные электроны, то очевидно,
нельзя ожидать образования в атомах
устойчивых групп электронов, удов-
удовлетворения правил валентности и со-
сочетаний атомов друг с другом в опре-
определенных кратных отношениях. Так,
например, в системе Mg — Аи в об-
области составов, близких к 50%, об-
образуется фаза, имеющая структуру
CsCl и обусловленная электроста-
электростатическим притяжением между поло-
положительно заряженными атомами
Mg и отрицательными атомами золо-
золота, хотя предположение о существо-
существовании в ней ионов типа Mg2+ и
особенно Аи2" совершенно не выдер-
выдерживало бы критики. Как уже гово-
говорилось в главе XII, элементы 1-Ь
подгруппы (Си, Ag и Аи) в интерме-
таллических соединениях могут иг-
играть роль, аналогичную галогенам в
неорганических соединениях, что
связано с возможностью дополнить
свою s-оболочку до устойчивой двух-
электронной конфигурации.
В настоящем параграфе были рае-
зобраны четыре основных типа ин-
интерметаллических соединений:
а) электронные соединения; б) соеди-
соединения, обусловленные объемными
соотношениями составляющих их
атомов; в) соединения с ковалентны-
ми связями; г) соединения с ионны-
ионными связями. Неправильно было бы
думать, что этими четырьмя груп-
группами исчерпываются все типы ин-
интерметаллических соединений и что
существуют резкие границы между
ними. Все эти факторы могут играть
более важную или более второсте-
второстепенную роль при образовании соеди-
соединения и, как правило, присутствуют
одновременно. Учет одного из них и
пренебрежение остальными может
привести к односторонности и несо-
несовершенству представлений о приро-
природе интерметаллических; соединений.
Один и тот же структурный тип
может быть обусловлен разными
факторами. Так, структурный тип
CsCl встречается среди электронных
соединений (CuZn); среди соедине-
соединений, определяющихся в первую оче-
очередь объемными факторами (MgSr);
среди соединений, существование
которых обусловлено ионными сила-
силами (MgAu). Тип CaFj, может встре-
встречаться как у «ионных» соединений
(MgnGe), так и у «ковалентных»
(Auln2).
Сказанного достаточно, чтобы по-
понять неправильность отождествления
типов интерметаллических соедине-
соединений со структурными типами, что
часто можно видеть в книгах по ме-
металлографии.
Структурные типы и типы соедине-
соединений должны обязательно излагаться
отдельно.
Интересно, что структурный тип
CsCl более характерен для интерме-
интерметаллических фаз, чем для обычных
неорганических соединений, число
представителей которых не превыша-
превышает 15, причем некоторые из них су-
существуют только в необычных со-
состояниях, например при высоких
температурах или давлениях. Веро-
316
¦ятно, целесообразнее было бы для
интерметаллических фаз называть
этот структурный тип не CsCl, а, на-
например, GuZn.
Особая упорядоченность FesAl бы-
была описана в § 13 этой главы. Фор-
Формально в этом случае мы приходим к
структурному типу BiF3, в котором
все места плотнейшей упаковки за-
заняты атомами алюминия, а атомы
железа занимают все тетраэдриче-
ские и октаэдрические пустоты.
Из других структурных типов не-
неорганических соединений среди ин-
интерметаллических фаз осуществля-
осуществляется тип NiAs и близкие к нему.
Структурные типы ZnS, NaCl,
ТЮ2 и слоистые не имеют предста-
представителей среди интерметаллических
соединений.
Немногочисленные интерметалли-
интерметаллические соединения кристаллизуются
в структурных типах, характерных
для боридов и силицидов.
ГЛАВА XIX
КРИСТАЛЛОХИМИЯ НЕОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
§1. О классификации
бинарных соединении
Классификация бинарных соедине-
соединений постоянного состава проще все-
всего может быть сделана на основе
учета относительного количества
атомов одного и другого элементов,
входящих в данное соединение.
Так, можно рассматривать в опре-
определенной последовательности следу-
следующие ряды соединений:
АВ, АВ2, ABs, АВ4, АВ6, АВо, АВ7,
АВ8, и т. д.;
А2В8, А2Ва, A^j, A2B9 и т. д.;
АзВ4, A3B5, AsBj, АзВ8 и т. д.;
А4В6, A4B7, А4В» и т. д.
Однако не все эти сочетания воз-
возможны. Ограничения в эти ряды вно-
вносятся теорией структуры кристал-
кристаллов и теорией валентности.
По теории Федорова, в 230 груп-
группах симметрии возможны правиль-
правильные системы точек только с опреде-
определенным отношением кратностей:
AiBi, AiB2, AiBs, A1B4, AiBe, AiB8;
AiBl2, A1B10, A1B24, AiB48;
A2B3, AsB4, АзВ8.
Теория валентности исключает из
этих тринадцати отношений четыре
(средняя строка). Это обстоятельст-
обстоятельство необходимо иметь в виду, посколь-
поскольку в данном разделе мы предполага-
предполагаем рассматривать только неоргани-
неорганические соединения в узком смысле
этого слова, а к большинству из них
приложимы правила валентности.
Однако реально среди «невалентных»
соединений такие случаи встречают-
встречаются. Так, например, в структуре UB12
атомы урана занимают одну правиль-
правильную систему точек с кратностью 4,
а атомы В — вторую правильную сис-
систему с кратностью 48.
Отсутствие электроположительной
валентности, более высокой, чем 8, и
электроотрицательной — более высо-
высокой, чем 4, исключает из первона-
первоначальной таблицы еще ряд формул;
в результате приходим к следующе-
следующему ряду:
АВ, АВ2 АВз, АВ4, (АВ6), АВа, (АВ7),
АВЙ, А2Вз, (А2В6), (А2В7), А8В4,
(АзВ6), (А»В7), А3В8.
В скобках поставлены формулы с
таким соотношением, при котором
атомы по крайней мере одного пз
элементов не могут занимать точкп
одной правильной системы. Это зна-
значит, что такие бинарные химиче-
химические соединения кристаллизуются в
структурном типе одного из тройных
(или более сложных) соединений и
со структурной точки зрения, строго
говоря, и должны были бы рассмат-
рассматриваться среди структур тройных
соединений, а не двойных. Возьмем,
например, PBrs. В структуре этого
соединения имеются тетраэдрические
комплексы РВз'4+ п отдельно распо-
расположенные ионы Вг~. Формулу такого
бинарного соединения следовало бы
писать как тройного, т. е. [РВг4]Вг.
То же относится к бинарному соеди-
соединению, у которого по крайней мере
318
один из элементов имеет различную
валентность, например РезСч. Пра-
Правильно написанная формула должна
выглядеть так:
Fe2+Fe23+04.
Структуры подобных соединений бу-
будут принадлежать к одному из струк-
структурных типов тройных соединений:
во взятом примере — к типу шпинели
MgAl2O4.
Соединение типа А3В4 могло
бы кристаллизоваться в структур-
структурном типе бинарного соединения толь-
только в том случае, если бы оно состав-
составлялось из элементов с валентностью
три или четыре. Исключением из
этого правила могут служить только
соединения, имеющие дефектные
структуры. Не следует, однако, ду-
думать, что даже простейшие соедине-
соединения, скажем типа АВг, обязательно
кристаллизуются в таком структур-
структурном типе, в котором атомы элемента
А располагаются по точкам одной
правильной системы, а атомы
элемента В — по точкам другой, с
кратностью вдвое большей, чем у
первой.
В структурах простых веществ мы
встречаемся с элементами, атомы ко-
которых располагаются в кристаллах
по нескольким правильным систе-
системам (например, а и Р-Mn, и др.). Тем
более, такие случаи возможны в би-
бинарных и более сложных соедине-
соединениях.
Таким образом, это будет третьим
случаем, когда двойное соединение
будет иметь структуру тройного.
Если первые два случая можно зара-
заранее определить из отношения коэф-
коэффициентов в формуле и из валентно-
валентности входящих в нее элементов, то тре-
третий случай устанавливается только
после определения кристаллической
структуры.
В третьей части книги описыва-
описывалось достаточно большое число би-
бинарных неорганических соединений.
Поэтому нет необходимости в этой
части книги повторяться.
Перейдем теперь к более сложным
соединениям.
§ 2. Тройные и более сложные
неорганические соединения
Из схемы (см. стр. 279) известно,
что тройные неорганические соеди-
соединения делятся на две группы: АВХ и
AXY. В первой группе два металли-
металлических элемента образуют соедине-
соединение с одним неметаллическим, во
второй — один металлический эле-
элемент с двумя неметаллическими.
В структурном отношении эти груп-
группы также существенно отличаются.
Пример первой из них мы уже зна-
знаем — это перовскит СаТ1Оз. Для
структур этой группы характерно то,
что координационные числа катио-
катионов обычно больше числа анионов
в химической формуле и поэто-
поэтому никаких особых групп атомов
(комплексов) и структуре нет. Так,
в структуре перовскита Са имеет
координационное число 12, a Ti — 6,
в то время как число атомов кисло-
кислорода, приходящееся на каждый из
металлических атомов, равно трем.
Для второй группы соединений,
напротив, весьма характерно об-
образование комплексов — радика-
радикалов. Примером могут служить струк-
структура кальцита СаСОз (рис. 305) или
тенардита Na2SO4 (рис. 306). В обе-
обеих структурах атомы неметалличе-
неметаллических элементов образуют радикалы
СОз2~ в первом случае и
SG2~ — во втором. Карбонатный
ион имеет форму плоского тре-
треугольника, сульфатный ион — тетра-
тетраэдра (рис. 307).
Иногда такого типа структуры
удобно описывать как бинарные. Так,
например, структура кальцита весь-
весьма сходна со структурой типа NaCl.
Места, занимаемые в СаСОз ионами
кальция, аналогичны местам Na+
в NaCl; вместо же СГ~ в
структуре кальцита расположен
пон СО32~. Так как последний не
имеет шаровой формы и скорее мо-
может быть аппроксимирован к сплюс-
сплюснутому эллипсоиду вращения или
овалоиду, то вся структура СаСОз
отличается от структуры NaCl тем,
319
С°0
Рис. 305. Структура кальцита СаС03
Рис. 306. Структура тенардита Na2SC>4
U3| |0U4|
Рис. 307. Конфигурация ионов СО32-
Рис. 308. Ячейка структуры NaCl и СаСОз
<D
Du Sn S
Рис. 309. Структуры сфалерита ZnS, халь-
халькопирита Cub'eS2 и станнина Cu2FeSnS4
что элементарный куб NaCl дефор-
деформирован (сжат) по оси третьего по-
порядка и превращен в тупой ромбо-
ромбоэдр (рис. 308). Вместо шарового
иона С1~, через который проходили
4 оси Ь%, в структуре СаСОз сохра-
сохраняется одна ось Ьз, проходящая че-
через центр иона (СОзJ~.
В такой же мере и структура
NaC104 аналогична структуре NaCl.
Более сложные структуры из не-
нескольких металлических элементов и
одного неметаллического также со-
сохраняют черты бинарного неоргани-
неорганического соединения. Так, на рис.
309 показаны структуры сфалерита
ОСа
»Мд
Рис. 311. Структура Th(OHJSO4
ZnS, халькопирита CuFeS2 и станни- Рис- 31°- Структура доломита CaMg(COsJ
на Cu2FeSnS4. Металлические ато-
атомы упорядочены друг относительно
друга. Чем они ближе друг другу
по химическим свойствам, тем более
вероятна неупорядоченность.
Аналогично СаСОз и Na2SC>4 пост-
построены и более сложные химические
соединения типа ABXY. Например,
структуру доломита CaMg(COaJ
(рис. 310) можно получить из струк-
структуры кальцита СаСОз, если взять
две ячейки по оси Z, и атомы Са
в четных горизонтальных слоях за-
заменить на атомы Mg (ср. с рис. 305).
Структура Th(OHJSO4 (рис. 311)
построена из тетраэдрических SCv
и сферических ОН~-анионов. Сфери-
Сферическая симметрия приписывается
гидроксилу из-за трудности опреде-
определения положения протона. Коорди-
Координационное число Th4+ равно 8 DОН~
и 40 из сульфатных ионов).
До сих пор рассматривались слу-
случаи, когда неметаллические атомы
образовывали комплексные анионы.
Реже они могут образовывать и ком-
комплексные катионы, наиболее распро-
распространенным из которых является
аммонийный ион (NH4)+. В этом
ионе протоны часто (в особенности
при повышенных температурах) мо-
могут вращаться, в результате чего та-
такой ион аппроксимируется шаром.
Тогда аммоний становится весьма
похожим на катион щелочного ме-
металла К+ или Rb4", которые он часто кими шариками)
Рис. 312. Структура КНгРО» (положение
атомов водорода показано белыми малень-
21 Кристаллохимия
321
изоморфно и замещает. В таком слу-
случае вещество, построенное из трех
неметаллических элементов, стано-
становится похожим на обычную бинар-
бинарную соль. Так, например, NH4CI вы-
выше 184° С имеет структуру типа NaCl,
а ниже этой температуры — типа
CsCl. В тех случаях, когда образу-
образуется водородная связь, как, напри-
например, в случае фтористого аммония,
иону аммония уже нельзя приписать
шаровую форму. Он имеет тогда тет-
раэдрическую форму. Структура
NH4F изоструктурна вюртциту. Во-
Водородные связи могут скрепить в
трехмерный каркас комплексные
анионы. Так, например, скреплены
тетраэдрические ионы РО43" в струк-
структуре КН2РО4 (рис. 312). Если рас-
рассматривать эти комплексы изолиро-
изолированно, то они располагаются в струк-
структуре так же, как атомы Sn в его бе-
белой модификации. Каждый атом О
из одной группы РО4 связан с другим
О из другой группы РО4 водородной
связью. Внутри этого трехмерного
каркаса расположены ионы К+.
Значительно реже центром ком-
комплексного аниона являются металли-
металлические атомы, хотя для некоторых
из них это характерно. Например,
Be весьма охотно образует анион
BeF42-, а Сг—СгО42-. Оба аниона тет-
раэдрической формы и изоструктур-
ны с сульфатным ионом.
Соединения, содержащие такие
ионы, по своим свойствам весьма
близки к комплексным соединениям
(см. гл. XXI).
§3. Структурная систематика
класса сульфатов
В качестве примера структурной
систематики сложных соедине-
соединений рассмотрим класс сульфатов
(Г. Б. Бокий и Л. И. Горогоцкая,
1966 г.). Этот класс избран нами вви-
ввиду важности его для минералогии и
неорганической химии.
Поскольку сульфатный ион (в от-
отличие от ортосиликатного иона
Рис. 313. Структура пальмиерита К2РЬ-
•(SOib (белыми кружками обозначены
ионы калия)
[S1O4]4"", см. ниже), является всег-
всегда изолированным комплексом, по-
постольку все сульфаты формально
должны быть отнесены к островным.
Однако благодаря различной проч-
прочности других полиэдрических эле-
элементов структур в структурах со-
соединений этого класса могут, быть вы-
выделены и более сложные мотивы.
Рис. 314. Структура гипса CaSCU ¦
а — проекция на плоскость XZ;
б — проекция яа плоскость YZ
322
В связи с этим в основу выделения
того или иного структурного мотива
в сульфатах нами положен принцип
объединения SO4 —тетраэдров с наи-
наиболее прочными полиэдрами, обыч-
обычно октаэдрами, двух- и более высо-
высоковалентных катионов.
Например, в пальмиерите —
K2Pb(SO4b (рис. 313) выделен сло-
слоистый мотив структуры, проявляю-
проявляющийся в облике и спайности минера-
минерала и обусловленный чередованием
более прочно связанных слоев из
БО^тетраэдров и РЬ-октаэдров * с
более «рыхлыми» слоями из К-де-
сятиверпшнников* *.
Принцип объединения наиболее
прочных полиэдров двух сортов при
выделении мотива структуры приме-
применим, конечно, не только к сульфатам.
В водных сульфатах, например,
определяющим структурный мотив
признаком является характер кон-
консолидации в пространстве различных
катионно-кислородных полиэдров в
группировки, не связанные между
собой при помощи общих анионов.
Например, в гипсе (рис. 314)
БО-гтетраэдры объединены с Са-вось-
мивершинниками в слои, которые
соединяются друг с другом при по-
помощи водородных связей. Напомним,
что на слоистый характер структуры
гипса, несмотря на изолированное
положение БО^тетраэдров, давно бы-
было обращено внимание (А. С. Пова-
Поваренных, 1956 г.; Е. С. Лазаренко,
1963 г.). В то же время упомянутый
выше принцип не был последова-
последовательно применен к классификации
других сульфатных структур.
Все рассмотренные структуры
сульфатов отнесены нами к остров-
островным, сложно-островным, цепочеч-
цепочечным, слоистым и каркасным.
Распределение структур по моти-
мотивам приведено в табл. 45.
* Условимя далее М-полиэдрами назы-
называть полиэдры вокруг двух- и более вы-
соковалетных катионов
** Полиэдры вокруг одновалентных катио-
катионов обозначаются как R-полиэдры.
ТАБЛИЦА 45
Распределение структур сульфатов
по группам структурных типов
Группа
структурных типов
Островной
Сложно-островной
Цепочечный
Слоистый
Каркасный
И
в*»
III
18
3
5
8
4
о а
?5
36
6
7
17
4
SB.
21
7
1
7
1
Среди островных структур даль-
дальнейшее различие проводится по
принципу взаимного расположения
катионных М-или R-полиэдров. Та-
Такой принцип оправдан, по нашему
мнению, тем, что взаимное располо-
расположение катионов играет важную роль
в формировании структурного мо-
мотива соединений и в его внешних
проявлениях, сказывающихся в ани-
анизотропии физических свойств, ориен-
ориентировке спайности и проч. На этом
основании выделены структуры с
изолированным положением SO4-
тетраэдров и М-полиэдров (рис.
315), структуры с цепочками (рис.
316), слоями (рис. 317), каркасами
(рис. 318) из катионных М- или
В-полиэдров.
В сложно-островных структурах
острова представляют собой либо
замкнутые кольцевые группировки
(структура леонгардтита MgSCU •
• 4НгО, рассмотренная Н. В. Беловым
как пример неорганических молеку-
молекулярных структур), либо открытые
группировки (в изученных структу-
структурах М-полиэдр, заключенный меж-
между двумя ЭОгтетраэдрами, рис.319).
Цепочечный мотив структур фор-
формируется либо в результате чередова-
чередования ЭО4"тетраэдров и М-полиэдров
в цепочке (рис. 320), либо в резуль-
результате дополнительного «скрепления»
цепочек из М-полиэдров SCU-TeTpa-
эдрами, присоединяющимися к ним
сбоку (рис. 321).
21»
823
X
Й
0
о
Рис. 315. Тип структуры с изолированным
положением сульфатных тетраэдров и Mg-
полиэдров [структура шенита K2Mg(SO.,J-
•6Н2О]
Рис. 316. Пример цепочечной структуры
кизерита (структура MgSO4-H2O)
Рис. 317. Пример слоистой структуры
(структура CdSOO
В слоистых структурах степень
совершенства поверхности плотных
слоев различна. Так, структуры гип-
гипса (рис. 314) и пальмиерита (см.
рис. 313) характеризуются гладки-
гладкими слоями. Параллельно им прохо-
проходит весьма совершенная спайность.
В структуре сингенита K2Ca(SO4J-
• Н2О (рис. 322) более плотные
слои, состоящие из вСч-тетраэдров
и Са-полиэдров, имеют «шипы» в ви-
виде наружных БС^-тетраэдров, глу-
глубоко проникающих в «рыхлую» про-
прокладку, состоящую из К-полиэдров.
Параллельно этим слоям проходит
плоскость совершенной спайности.
Подобные слои наблюдаются в
структуре натрохальцита —
NaCu2OH(SO4J • Н2О. Еще менее со-
совершенна поверхность слоев в алу-
алуните, вантгоффите и глауберите.
Пример каркасной структуры по-
показан на рис. 323.
Современная кристаллохимиче-
ская классификация должна опреде-
определяться главнейшими характеристи-
характеристиками веществ — составом и структу-
структурой. Применительно к классу
сульфатов мы попытались этого до-
достигнуть, расположив все рассмот-
рассмотренные соединения, главным обра-
образом, минералы в виде таблицы, где
в горизонтальных рядах соединения
объединены по химическому призна-
признаку, а в вертикальных — по структур-
структурному (табл. 46). Место химического
соединения в классификации, таким
образом, в равной мере определяет-
определяется как составом, так и структурой.
По химическому составу нами вы-
выделены сульфаты одновалентных ка-
катионов, кислые сульфаты однова-
одновалентных катионов, сульфаты двух-
и четырехвалентных катионов, а
также двойные сульфаты без доба-
добавочных и с добавочными анионами.
Внутри этих наиболее крупных под-
подразделений, имеющих характер под-
подклассов, выделяются водные и без-
безводные сульфаты. Сходное деление
сульфатов по химическому принци-
принципу принято, например, А. Н. Винчел-
лом и Г. Винчеллом A953 г.).
324
V\-mi
тетраэдр
Рис. 318. Пример каркасной структу-
структуры [ деталь структуры чухровита
(Ca3TR) A12SO4F,2 • 11Н2О (ОН) 1
Рис. 320. Пример цепочки из чередующих-
чередующихся сульфатных тетраэдров и М-полиэдров
(структура CuSO4-5H2O)
Сульфаты меди — брошантит
Cu4(OHNSO4 и долерофанит
CU2OSO4 — отнесены нами к двой-
двойным на основании присутствия в
этих минералах двух кристаллохими-
чески различных сортов атомов меди.
Для простых безводных сульфа-
сульфатов характерны разновидности ост-
островных структур. Последние харак-
характерны также для моногидратов и для
многоводных простых сульфатов (с
количеством молекул воды, равным
или больше 6). Водные простые суль-
сульфаты с промежуточным количеством
молекул воды (от двух до пяти) ха-
характеризуются более сложными мо-
мотивами: сложноостровным, цепочеч-
цепочечным, слоистым, каркасным. Отмечен-
Отмеченная закономерность вызвана тем, что
мотив водных сульфатов во многом
Рис. 319. Пример сложно-островной струк-
структуры [структура лоонита K2Mg(SO4J • 4Н2О]
Рис. 321. Пример цепочек из Mg-полиэд-
ров и SOi-тетраэдров [структура уклон-
сковита NaMg(SO4) (ОН)]
325
ТАБЛИЦА 46
Схема классификации'сульфатов
Химический тип
Простые
сульфаты
R-катионов
Кислые
сульфаты
R-катионов
Простыв
сульфаты-
М ^-катио-
^-катионов
Простые
сульфаты
М*+-катио-
нов
Безводные
R2SO4
Водные
R2S0«.nH20
Безводные
RHSO4
Безводные
MSO4
Водные
MS04.nH20
Водные
М (SO«)S.
• bHjO
Мотив структуры
Островной
С изолирован-
;¦ ным
""положением
М- или R-no-
лиэдров
Гексагидрит
MgSOt-6H2O
Ретгерсит
a-NiSO«.
•6Н2О
Мелантерит
FeSO«-7H2O
Эпсомит
MgSO4- 7H20
5С?цепочками
из М- или
R-полиэдров
Мирабилит
Na2SO4.
• 10Н2О
NiSOt
Халькоциа-
нит
CuSOt
Кизерит
MgSO«-H2O
HgS0«-H20
Со слоями
из М-по-
лиэдров
CdSO«
С каркасами
из М- или
R-полиэдров]
Тенардит
Na2S04
Аркантит
a-K2SO4
Мер кал лит
RHSOt
Ангидрит
CaSO4
Барит BaSO«
Сложно-
островной
i
1
I
Леонгардит
MgSO4-4H2O
5 Цепочечный
Халькантит
CuSO4-5H2O
Слоистый
Гипс
CaSOt.2H2O
Циркосуль-
фат
Zr(SO«).
• 4Н,0
Каркасный
CuSO4-3HaO
ТАБЛИЦА
45 (оковчание)
X имичсскийтип
Двойные
сульфаты
5ез добавоч-
добавочных анионов
Двойные
сульфаты
с добавоч-
добавочными анио-
анионами
Безводные
RmMn (SO4)P
Водные
RmMn(SO1)p-
•гаНгО
Безводные
RmMn(SO4)p.
•(OH,F)e
Водные
RmMn(SO4)p-
.(ОН, F)a.
• nH2O
Мотив структуры
Островной
С изолировав-
ным
положением
М- или R-no-
.. лиэдров
Квасцы
RAl(SO4)a.
• 12Н2О
Шенит
K2Mg (SO4J.
• 6Н2О
С цепочками
из М- или
R-полиэдров
Со слоями
из М-по-
лиэдров
С каркасами
из М- или
R-полиэдров
Кридит
СазА12 (ОН,
F)i0SO4-
• 2Н2О
Чухровит
(CasTR).
•Al2S0«-Fi2-
.1Ша0@Н)
Сложно-
островвой
Леонит
K2Mg (SO4J-
• 4Н2О
Астраханит
Na2Mg(SOj)»-
• 4Н,0
Цепочечный
Кренкит
Na2Cu(SO4J-
• 2Н2О
Краузит
KFe (SO4J-
• Н2О
Линарит
PbCu(SOi).
•(ОНJ
Уклонско-
вит
NaMg (SO4).
.(ОН)-2Н2О
Слоистый
Пальмиерит
К2РЬ (SO^b
Вантгоффит
NaeMg (SO4L
Глауберит
Na2Ca (SOt)t
Сингенит
К2Са (SO4J-
• H2OY
ш
Алунит
КАЬ (ОН)в-
•(SO4J
Натрохаль-
цит
NaCu2 (ОН).
• (SO4J-H2O
Каркасный
Лангбейнит
K2Mg3 (SO4K
Долерофа-
нит
Cu2OSO4
Брошантит
Cu4(OHNSO4
Примечание: в таблице изоструктурвые соединения ве указаны.
Рис. 322. Структура сингенита K2Ca(SC>4)i-
•Н20
в — проекция яа плоскость YZ;
б — проекция на плоскость XZ
Рис. 323. Каркасная структура [структура
брошантита Cu4SO4(OH)e]
определяется взаимным расположени-
расположением молекул НгО и.атомов кислорода,
не связанных общими катионами.
Так как атомы кислорода образуют
окружение серы, а молекулы воды —
окружение катионов М, то при коли-
количестве молекул НгО, равном или
большем координационного числа
катиона, БО^тетраэдры становятся
«полностью изолированными» от
других полиэдров.
Для двойных сульфатов наиболее
характерен слоистый мотив струк-
структур, обусловленный чередованием
слоев из SO4 и М-полиэдров со слоя-
слоями из крупных R-полиэдров. Слои-
Слоистые структуры наблюдаются и у со-
соответствующих моногидратов (син-
генит). Возрастание количества воды
приводит к образованию сложноост-
ровного типа структуры (леонит,
астраханит) и далее при геНгО^
^к. ч. М-атома — к образованию
островного типа структуры.
§4. Правила Полинга
для структур
ионных кристаллов
Большинство неорганических сое-
соединений подчиняется правилам ва-
валентности. Это дает формальное ос-
основание считать их построенными из
ионов. Для структур таких соедине-
соединений Л. Полингом было сформулиро-
сформулировано несколько правил.
Первое из них является правилом
Гольдшмидта, изложенным на
стр. 148.
Второе правило, носящее название
правила электростатической валент-
валентности, гласит, что в устойчивой ион-
ионной структуре валентность каждого
аниона точно или приблизительно
равна сумме валентных усилий этого
аниона с соседними с ним катиона-
катионами. Под валентным усилием подра-
подразумевается дробь Z/K, где Z — ва-
валентность катиона, а К — его коор-
координационное число.
Для проверки этого правила не-
необходимо определить координацион-
координационные числа всех катионов по отноше-
328
нию к анионам и анионов по отноше-
отношению к катионам и затем составить
соответствующее уравнение. Напри-
Например, в структуре перовскита Са2+
имеет координационное число 12, от-
откуда каждое усилие связи на 1 02~
получается равным 2/i2 = '/•• Ато-
Атомы титана имеют валентность 4 и ко-
координационное число 6, откуда уси-
усилие связи будет 4/«. Каждый атом
кислорода окружен четырьмя атома-
атомами Са и двумя атомами TL Отсюда
сумма валентных усилий катионов,
сходящихся на каждом атоме кисло-
кислорода, будет 4 • 7« + 2 • 7в = 2, т. е.
равна валентности аниона. Следова-
Следовательно, для структуры перовскита
правило электростатической валент-
валентности точно выполняется.
В структуре шпинели каждый
атом кислорода окружен 3Alvt и
lMg^-. Римскими цифрами пока-
показаны координационные числа этих
катионов. Сумма валентных усилий,
сходящихся на каждом атоме кис-
кислорода, для структуры шпинели бу-
будет 3 • 7в + 1 • U = 2, т. е. правило
выполняется точно. В более слож-
сложных структурах оно выполняется
лишь приблизительно, но обычно с
точностью до V».
Третье правило говорит о том,
что наличие в координационной
структуре общих ребер и особенно
граней для двух соседних координа-
координационных полиэдров катионов умень-
уменьшает устойчивость структуры.
Этот эффект особенно существен
для многовалентных катионов с низ-
низкими координационными числами.
Рис. 324. Возможные сочетания двух тет-
тетраэдров и двух октаэдров через общие
вершины, ребра и грани (по Полингу)
На рис. 324 в верхнем ряду нарисо-
нарисованы тетраэдры с общей вершиной,
ребром и гранью, в нижнем ряду
приведен аналогичный рисунок для
октаэдров. Если расстояния между
центрами фигур для крайних левых
рисунков положить равными едини-
единице, то для тетраэдров, соединенных
ребром, эта величина будет 0,58, а
для соединенных гранью — 0,33. Для
ряда октаэдров аналогичные величи-
величины будут иметь следующие значения:
0,71 и 0,58. Ясно, что при таком сбли-
сближении двух многовалентных катио-
катионов, которое особенно сильно у тет-
тетраэдров, развиваются такие большие
силы отталкивания, что структура
кристалла делается неустойчивой.
По этой причине ион состава (БЮз)
никогда не имеет форму сдвоенных
по ребру тетраэдров.
Полингом сформулированы и дру-
другие правила, не имеющие, однако,
такой универсальности, как два из-
изложенных.
§ S. Особенности структур
с преимущественно
ковалентиым типом свяаи
В предыдущем параграфе были рас-
рассмотрены правила, пригодные для
строения ионных кристаллов. Не
следует думать, что Полинг пред-
представлял себе кристаллы построенны-
построенными из чистых ионов Са2+, А13+, О2~
и в особенности Si4+. Во всех разо-
разобранных случаях под терминами
«ион», «ионная связь» и т. п. подра-
подразумевается, что связь в значительной
степени ионная, но совсем не обяза-
обязательно предельно ионного типа.
В тех случаях, когда вместо окис-
окислов рассматриваются сульфиды, се-
лениды или теллуриды, да еще при
этом в качестве электроположитель-
электроположительных частиц структуры фигурируют
атомы металлов VIII-а и всех Ь-под-
групп, то связь в таких структурах
будет уже весьма далекой от ионной,
и правила Полинга в применении к
ним не всегда будут оправдываться.
Так, структуры ZnS, CuFeS2 и
329
eptos
Рис. 325. Структура PtS
подчиняются всем трем
перечисленным выше правилам. Но,
например, структура PtS не подчиня-
подчиняется первому правилу, так как квад-
квадратная конфигурация у Pt, характер-
характерная для PtS, не может осуществлять-
осуществляться ни при каком соотношении
размеров ионов (рис. 325). Из этого
не следует, что перечисленные выше
структуры ионного типа. Дело в том,
что есть координационные много-
многогранники, которые не могут встре-
встречаться в ионных кристаллах, напри-
например, квадрат, но другие координаци-
координационные многогранники могут в рав-
равной степени встречаться как в ион-
ионных соединениях, так и в предельно
ковалентных, например, тетраэдр.
В таких структурах форма коорди-
координационного многогранника не может
служить критерием для суждения о
типе связи.
-с *Si OS
Рис. 326. Структура SiS2
Не может быть структуры типа
NiAs у чисто ионных соединений,
так как и в этом случае один из ко-
координационных полиэдров не окта-
октаэдр, а тригональная призма, прису-
присущая соединениям с ковалентным ха-
характером связи.
Все природные соединения соста-
состава S1O2 имеют координационные
структуры, в которых каждый атом
кремния расположен в центре тет-
тетраэдра из четырех кислородов, со-
соединенных друг с другом через общие
вершины. Соединение же SiS2 имеет
цепочечную структуру, в которой
каждый тетраэдр S1S4 имеет по два
общих ребра с соседними тетраэдра-
тетраэдрами и, следовательно, для нее третье
правило Полинга уже не оправдыва-
оправдывается (рис. 326).
§6. Кристаллохимия силикатов
1. Введение. Силикаты являются од-
одним из наиболее сложных по хими-
химическому составу классов неорганиче-
неорганических веществ. Они включают веще-
вещества, содержащие Si и О вместе со
многими элементами. Тот факт, что
большинство силикатов нераствори-
нерастворимо в воде, весьма затруднял струк-
структурно-химическое изучение их обыч-
обычными химическими методами. Ни для
одного из классов веществ не было
предложено такого количества раз-
различных вариантов гипотетических
структурных формул, как для клас-
класса силикатов. И можно сказать беэ
преувеличения, что все эти струк-
структурные формулы оказались неверны-
неверными. По этой причине химику весьма
важно знать кристаллохимические
методы и результаты исследований
силикатов, позволившие объективно
определить структуры кристалличе-
кристаллических веществ и тем самым понять их
химическую сущность.
Только появление рентгенострук-
турного анализа позволило выяснить
правильные структурные формулы
этих соединений. Расшифровки
структур силикатов явились триум-
330
фом кристаллохимии, коренным об-
образом изменившими прежние хими-
химические представления о природе си-
силикатов.
Силикаты составляют более трети
всех минералов. Почти все породо-
породообразующие минералы, составляю-
составляющие земную кору, являются силика-
силикатами. Не меньшее значение силика-
силикаты имеют и в промышленности.
Керамическая промышленность,
промышленность строительных ма-
материалов, использующая кирпич, це-
цемент и др., целиком базируются на
силикатах.
Трудность химического изучения
силикатов связана прежде всего со
сложностью их химического состава.
В силикатах отношение кислотного
ангидрида к основаниям колеблется
в широких пределах. В силикатах
нередко встречаются одновременно
3, 4 и большее число различных ка-
катионов. Для силикатов чрезвычайно
характерны изоморфные замещения
как катионов, так и самого кремния
и вхождение в силикаты других ани-
анионов (О2- ОН-, F-, SO42-, СО32-
и др.) и нейтральных частиц — ЩО
и др.
2. История изучения силикатов.
История химии силикатов состоит как
бы из пяти этапов.
1. Эмпирическое написание фор-
формул силикатов в виде окислов позво-
позволило накопить сведения об их со-
составе:
Na2O-AhO3-6SiO2 — альбит,
CaO-Al2O3-2SiO2 — анортит,
2HsCK3Mg0.2SiC>2— серпентин,
2BeO-SiC>2 — фенакит и т. д.
В результате установления соста-
состава появилась возможность создать
первую классификацию силикатов,
основывающуюся на следующем от-
отношении:
Число атомов О, связанных с Si _
Число атомов О, связанных ~
с прочими элементами
Так, если
л = 1—моносиликаты: фенакит, анортит;
л = 2 — дисиликаты: диопсид CaO-MgO-
• 2SiO2, лейцит KsO-AUOs^SiO*;
п = 3 — трисиликаты: альбит и т. д.
п < 1 — субсиликаты: серпентин.
Недостатком этой классификации
явилось то, что весьма близкие ве-
вещества (например, альбит и анортит)
оказывались в разных разделах, а в
один раздел попадали вещества весь-
весьма различные.
2. Второй этап связан с представ-
представлением о силикатах как о солях по-
поликремневых кислот.
Исходной кислотой считалась ор-
токремневая I^SiCj. Поликремневые
кислоты выводились из нее по урав-
уравнению
п = 1, т = 1: НгЭЮз — метакремневая
кислота;
п = 2, т = 1: HeSi2C>7 — ортодикремневая
кислота;
п = 2, т = 3 : H2Si2C>5 — метадикремневая
кислота я т. д.
Силикаты считались солями этих
кислот. На этом этапе оказалось
возможным еще больше упорядо-
упорядочить экспериментальный материал,
но вопрос о природе силикатов оста-
оставался нерешенным в той же мере,
как и раньше, так как определение
типа кислотного радикала остава-
оставалось совершенно произвольным, так
же как полный произвол существо-
существовал и в вопросе о роли водорода и
кислорода и химической формул? си-
силиката.
В качестве примера В. С. Соболев
приводит трактовку разными авто-
авторами химической формулы серпен-
серпентина, имеющую состав 2H2O-3MgO-
• 2SiG. Раммельсберг писал ее
H2Mg3Si2O8-H2O, трактуя, очевидно,
как моногидрат кислой соли орто-
кремневой кислоты.
Кларк и Шнейдер этот же мине-
минерал трактуют как основную соль,
придавая формуле следующий вид:
[MgSiO4]2H3MgOH. По Чермаку,
331
формула серпентина будет
MgSi2O7H2[MgOH]?.
Как видим, одно и то же вещество
трактовалось одними авторами как
кислая соль, другими — как основная.
В результате рентгеноструктур-
ных исследований Брегга и Уорре-
Уоррена выяснено, что имеется две разно-
разновидности минерала состава
Mg3[Sir,05]2oo(OHL — антигорит и
хризотил, резко различающиеся по
внешнему виду: первый — пластин-
пластинчатый, второй волокнистый (асбест).
Антигорит состоит из параллельно
умноженных каолиноподобных сло-
слоев — пакетов, тогда как в хризотиле
те же пакеты спирально закручива-
закручиваются в трубки, толщиной в 10—20
слоев. Последнее может быть объяс-
объяснено тем, что анионные слои из гид-
роксилов сильно распираются круп-
крупными катионами Mg (замещающими
места А1 в каолине), по сравнению со
сжатыми слоями из атомов кислорода,
являющихся основаниями кремне-
кремневых тетраэдров. В результате получа-
получается изгиб слоя и возможность его
закручивания в трубочку.
Поскольку сильные основания со
слабыми и сильными кислотами да-
дают кислые соли, а слабые основа-
основания — основные (в особенности со
слабыми кислотами), то силикаты,
как соединения в подавляющем чис-
числе случаев слабых оснований (Fe, A1,
Si), по-видимому, никогда не являют-
являются кислыми солями. Сильные основа-
основания (Са, К, Ва), часто находящие-
находящиеся в силикатах, вряд ли перекрывают НО.
действие слабых оснований, обычно„п/
присутствующих в большем количе-
количестве. Исключением могут явиться
только минералы группы пектолита
HNaCa2[Si3O9], исследование струк-
структуры которых представляло бы с этой
точки зрения большой интерес.
Невозможность однозначного ре-
решения вопроса химическими метода-
методами о типе радикала, роли кислорода
и алюминия в силикатах не позволи-
позволила продвинуться вперед сколько-ни-
сколько-нибудь значительно в вопросе о строе-
строении и систематике силикатов.
3. В 1891 г. В. И. Вернадский, изу-
изучая роль алюминия в силикатах,
пришел к заключению, что глинозем
в алюмосиликатах играет роль кис-
кислотного ангидрида, роль, аналогич-
аналогичную кремнезему. Он пишет: «Данные
химии алюмосиликатов и история
этих тел в земной коре дают основа-
основание придавать каолиновым глинам
и всем их производным одно и то же
строение, в котором характерно на-
нахождение одного и того же прочного
кольцевого ядра атомов:
=Si<"
/А1\
О О
O (Г
\А1/
Это ядро я буду называть «каоли-
«каолиновым ядром». Сам каолин, по Вер-
Вернадскому, имеет такую структурную
формулу:
ОН
/А1\
О
О
ХО 0х
\А1/
ОН
или, по более поздним его работам,
такую:
Si<
,0
ОН
/A1v
\А1/
I
ОН
,оч о
1< >Si<
\(К ХО
он
\А1/
ОН
ОН
Лейциту В. И. Вернадский приписы-
приписывал формулу
ОК
,-АЬ
O=Si
О.
о
NO
о
\А1/
ОК
Si=O.
332
Как видим, эти формулы были на-
навеяны успехами органической химии.
В зтот период всем неорганическим
соединениям приписывалось мо-
молекулярное строение, что в общем
оказалось неверным. Неверными по-
поэтому оказались и молекулярные
формулы силикатов. Однако работы
Вернадского имели большое положи-
положительное значение, так как выяснение
роли алюминия в алюмосиликатах
существенно облегчило расшифровку
кристаллохимических структур та-
таких сложных веществ, каковыми яв-
являются, например, алюмосиликаты.
Четырехчленные кольца в каркасах
полевых шпатов оказались постро-
построенными аналогично каолиновому яд-
ядру. Существенная разница заключа-
заключается в том, что эти кольца не явля-
являются изолированными. Во многих
позднейших работах В. И. Вернад-
Вернадский говорит о четвертой «побочной»
валентности алюминия, подчеркивая
этим еще большую его аналогию с
кремнием. Эта идея также получила
известное подтверждение тому, что
А1, изоморфно замещая в алюмоси-
алюмосиликатах Si, аналогично последнему
имеет координационное число 4.
В последних работах В. И. Вернад-
Вернадский структурную формулу каолина
писал так:
Н2О
/А1\
O=Si< О >Si=O.
ХО | О/
\А1/
Н2О
Все точно установленные факты о
строении силикатов получены с по-
помощью рентгеноструктурного анали-
анализа, о чем речь пойдет ниже. Сейчас
следует отметить, что В. И. Вернад-
Вернадский весьма высоко оценил этот но-
новый метод и во многом способство-
способствовал развитию его в СССР. В 1923 г.
он писал: «Уже сейчас эти исследо-
исследования резко меняют наши представ-
представления о химическом строении мине-
минералов и должны лечь в основу всех
представлений в этой области».
4. Если идеи о каолиновом ядре
в алюмосиликатах были навеяны
главным образом успехами органи-
органической химии, то введение в химию
силикатов таких понятий, как «по-
«побочные» валентности, координацион-
координационное число и т. п., связаны с успеха-
успехами стереохимии комплексных сое-
соединений, которыми мы обязаны в
первую очередь А. Вернеру и
Л. А. Чугаеву. Работами этих ученых
и их многочисленных последователей
было выяснено, что четырехвалент-
четырехвалентная платина в своих соединениях
имеет координационное число 6. По
аналогии многие стали приписывать
кремнию в силикатах координацион-
координационное число 6. Так, например, Каль в
1926 г. каолин представлял себе по-
построенным так:
Ah
0 0 On
Н20—Si—0—Si—H2 0
\
о _.
Считалось, что структурная формула
топаза вполне отвечает этим пред-
представлениям. Ее изображали следую-
следующим образом:
ОН
О J О
Ч/
j
он
Все эти формулы оказались невер-
неверными. Причина неудач заключалась
в том, что в химию кремния были
механически перенесены стереохи-
мические схемы, заимствованные из
области комплексных соединений
(главным образом, платины и кобаль-
кобальта) и молекулярных органических
веществ. Как в химии комплексных
соединений, так и в органической
химии заключения о строении дела-
делаются на основании изучения поведе-
поведения веществ в жидком или раство-
растворенном состояниях. Такой подход к
333
силикатам экспериментально не мог
быть осуществлен, так как большин-
большинство из них не растворимы в обычных
растворителях (вода, спирт и т. д.),
а плавятся при очень высоких тем-
температурах. Основным агрегатным со-
состоянием для силикатов является
кристаллическое состояние.
Авторы стереохимии силикатов не
имели в своем распоряжении таких
опытных фактов, какие имелись
в распоряжении исследователей, изу-
изучавших химию углерода, кобальта
или платины. Это обстоятельство
приводило к тому, что структурные
формулы силикатов являлись плодом
обычно довольно необоснованной
фантазии, что со временем оконча-
окончательно дискредитировало химичес-
химическое решение проблемы строения и
классификации силикатов.
5. Современный этап развития
этой проблемы связан с успехами
кристаллохимии. В настоящее время
рентгеноструктурный и электронно-
графический анализы являются поч-
почти единственными средствами экспе-
экспериментального изучения строения си-
силикатов.
Особые химические свойства крем-
кремния определяются его положением в
периодической системе элементов (на
пересечении двух границ — верти-
вертикальной и диагональной, рис. 271,
стр.267).
Силикаты занимают промежуточ-
промежуточное положение между солями более
сильных кислот и двойными окисла-
окислами, о структурах которых говорилось
в § 2 настоящей главы.
3. Основные черты строения сили-
силикатов. В настоящем разделе мы крат-
кратко сообщим основные характерные
черты строения силикатов, получен-
полученные при помощи рентгеноструктур-
ного анализа.
Экспериментальные данные по
строению силикатов связаны, глав-
главным образом, с именем В. Л. Брэгга,
который в период 1926—1931 гг. ис-
исследовал многие структуры силика-
силикатов и, опираясь на работу Ф. Махач-
ки, предложил первую основатель-
основательную систематику силикатов. Эти ра-
работы показали, что в силикатах ни-
никогда нет молекул. Атом кремния
всегда имеет координационное число-
4, т. е. ближайшими атомами, окру-
окружающими его, являются 4 атома кис-
кислорода, образующие тетраэдр.
Если сравнить ортосиликатный ион
SiO*4" с другими тетраэдрическими
ионами, о которых была речь в пре-
предыдущем параграфе, например, с
РО43~ или с SO42~ и др., то выясняет-
выясняется, что он имеет наибольший размер
и характеризуется наименьшей внут-
внутренней силой связи, т. е. является
менее устойчивым. По этой причине
силикаты по своим свойствам, по
сравнению с другими солями кисло-
кислородных кислот, приближаются к
окислам. Радикалы [SiO4]4~ охотно
объединяются друг с другом через
общие атомы кислорода, нейтрали-
нейтрализуя валентность последних. В резуль-
результате образуются более сложные ра-
радикалы поликремневых солей. Такое
объединение радикалов [SiO^4" про-
происходит только путем обобщения вер-
вершин тетраэдра, а не ребер или гра-
граней. Однако в каждом тетраэдре SiO«
могут быть обобщены 1, 2, 3 или все
четыре вершины. В результате полу-
получается большое разнообразие отноше-
отношений Si: О в силикатах. Различные по
форме сочетания взаимно связанных
ЗЮ4-тетраэдров носят название крем-
некислородного мотива структуры.
Не все атомы кислорода в силика-
силикате обязательно входят в кремнекисло-
родный мотив. Часть из них может
оставаться вне этого мотива — это
так называемые свободные ионы кис-
кислорода. Силикаты такого строения
особенно близки к окислам, в кото-
которых все ионы кислорода в этом смыс-
смысле «свободны». Одновалентные ионы
ОН~, F~ и нейтральные частицы, на-
например НгО, почти никогда не вхо-
входят в кремнекислородные мотивы.
Алюминий и некоторые другие эле-
элементы могут замещать в кремнекис-
лородном мотиве часть атомов Si.
При этом образуется алюмокремне-
кислородный мотив. Сложность
334
строения силикатов определяется
тем, что А1 отнюдь не всегда имити-
имитирует в силикатах кремний. В каждом
конкретном случае приходится выяс-
выяснять его роль.
Кремнекислородные и алюмокрем-
некислородные мотивы — радика-
радикалы — не всегда бывают конечных раз-
размеров, как это характерно для дру-
других солей кислородных кислот. Они
часто образуют бесконечные цепи,
листы или трехмерные каркасы.
Изучение структур таких силикатов
невозможно обычными химическими
методами, так как при переводе в
другое агрегатное состояние неизбеж-
неизбежно разрушение радикалов бесконеч-
бесконечной протяженности. Современная си-
систематика силикатов базируется на
характере кремнекислородных ради-
радикалов. Все силикаты делятся на 2
большие группы:
а) с кремнекислородными радика-
радикалами конечных размеров и
б) бесконечных размеров.
4. Строение силикатов с Si — О-
радикалами конечных размеров.
Строение кремнекислородных моти-
мотивов конечных размеров показано на
рис. 327 и 328.
Каждому типу кремнекислородно-
го мотива соответствуют группы
структурных типов, собранные в
табл. 47.
Изоморфное замещение кремния
алюминием в силикатах, имеющих
радикалы конечных размеров, не
характерно. Оно обычно для силика-
силикатов с кремнекислородными радика-
радикалами более сложного строения.
В рассмотренной же группе сили-
силикатов замещение алюминием части
атомов Si наблюдается обычно толь-
только в силикатах с самыми крупными
кольцами — шестичленными и
сдвоенными шестичленными. При-
Примером первого может служить во-
воробьевит CsBesAbtSisAlOis], второ-
второго — осумелит (высокотемператур-
(высокотемпературный кордиерит) CaMg2Als [Al8Si9O80].
5. Строение силикатов с Si — О-ра-
дикалами бесконечных размеров.
Строение кремнекислородных цепо-
цепочек и листов показано на рис. 329 и
330. Алюмосиликаты чаще относятся
к группе цепочечных соединений,
чем к кольчатым.
В роговых обманках замещение
Si на А1 достигает 25%.
Рис. 327. Строение кремнекислородных мо-
мотивов конечных размеров
Рис. 328. Изображение кремнекислород-
кремнекислородных мотивов конечных размеров тетраэд-
тетраэдрами
4
м<
LSiD3j
Рис. 329. Типы кремнекислородных цепо-
цепочек
335
Рис. 330. Типы кремнекислородных слоев
ТАБЛИЦА 47
Классификация силикатов с Si — О-мотивами конечных размеров
Форма радикала
Тетраэдр
Сдвоенный тетра-
тетраэдр
5 сочлененных
тетраэдров
Трехчленное коль-
кольцо
Четырехчленное
кольцо
Шестичленное
кольцо
Сдвоенное шести-
шестичленное кольцо
Химиче-
Химический
состав
радикала
[SiOi]
[Si2O,]
[Si5Oi,]
[Si3O9]
[Sie0ie]
[Si1203o]
Заряд
ради-
радикала
4—
6—
12-
6—
8—
12—
12—
Заряд
на 1 атом
Si
4—
3—
2»/.-
2—
2—
2
1—
Силикаты
без дополнительных
анионов
Оливин
MgFe [SiO4]
Гранат
А12Саз [SiO4]3
Тортвейтит
Sc2[SUO7]
Бенитоид
BaTi[Si3Oe]
Берилл
Be3Al2 [Si6Oi8]
Миларит
КСагВегА! [Si^Oao)
Силикаты
с дополнительными
анионами или нейтраль-
нейтральными частицами
Топаз
Al2 [SiO4] (ОН, FJ
Ильваит
CaFe(in)Fe(n)[SUO7].
• ООН
Зуниит*
Ali3Si502o(OH, FI8C1
Кайнозит
Ca2Ce2[Si4012] Соз-Н2О
Диоптаз
CuelSieOwl-BHuO
• В структуре характерны островные группы [SUOu], составленные из центрального тетраэдра,
сочлененного с 4 другими тетраэдрами. Все атомы А1 находятся в октаэдрах. Структура кубиче-
кубическая.
Сдвоенная цепочка (пояс) из че-
четырехчленных колец [Si205]lo°
встречена пока только у алюмосили-
алюмосиликатов — силлиманита и муллита. Мо-
Мотив [S16O17] 1о° впервые обнаружен
Н. В. Беловым и X. С. Мамедовым
в 1955 г. Примеры соединений с бес-
бесконечными цепочечными радикала-
радикалами собраны в табл. 48, в которой
химические формулы написаны в
идеализированном виде, т. е. в них
изоморфные замещения либо совсем
не указаны, либо указаны только са-
самые распространенные.
Рассматривая ряд поясных силика-
силикатов по мере усложнения кольца —
4-членное, 6-членное и т. д.
(Si2O5, SuOh, Si6Oi7 и т. д.), легко
видеть, что в пределе такой пояс ра-
разобьется на 2 цепочки состава SiCb.
Таким образом, пироксеновый мотив
является последним членом этого
ряда.
В табл. 48 приведены также струк-
структуры с листовыми кремнекисло-
родными мотивами. Имеется очень
большое число листовых силикатов
с шестичленными кольцами. Это
многочисленные слюды, пиро-
пирофиллит Al2[Si4Oio] (ОН)г, тальк
Mg3[Si4Oi0] (ОН) 2, хлорит AlMgs-
[AlSi3Oio] (ОНK и др. Пример листо-
листовых силикатов с четырех- и восьми-
членными кольцами известен только
один — апофиллит.
Для многих листовых силикатов
весьма характерно замещение Si на
А1, достигающее иногда 50% и более
(см., например, группу слюд). В му-
мусковите это замещение достигает
25%, в Маргарите СаА^А^гОю]200'
(ОН)г — 50%, а в ксантофиллите
Ca2Mg5Al [Al5Si302o]2°° • (ОН) 4 коли-
количество А1 в алюмокремнекислородном
листе даже превышает 50%.
Из сказанного в разделах 4 и 5 сле-
следует, что такая замена облегчается
по мере усложнения кремнекислород-
ного радикала.
Симметрия мотива тетрагональная.
Все минералы, имеющие этот мотив,
обладают спайностью по @01), что
делает их сходными со слюдами.
Особое место среди силикатов за-
занимают силикаты группы мелилита.
Их алюмокремнекислородный лист,
имеет пятичленные кольца (рис. 330),
Чисто кремниевый мотив имел бы со-
состав [Si3O7] 2°°, однако в этой группе
два из трех атомов Si заменены на А1.
Такой мотив встречается, в частности,
в структуре геленита Ca2[Al2Si07] 2o°.
Силикаты с таким мотивом явля-
являются в некотором смысле уже пере-
переходными от слоистых к каркасным.
В типичных слоистых мотивах в каж-
каждом вЮ^тетраэдре три вершины из
четырех являются поделенными меж-
между двумя атомами кремния. В каркас-
каркасных силикатах каждая из четырех
вершин делится между двумя атома-
атомами кремния. В мотиве [Si3O7] одна
треть тетраэдров имеет поделенными
все 4 вершины, а 2/а — три вершины
из четырех.
В тетраэдрах со всеми поделен-
поделенными вершинами А1 может изо-
изоморфно замещаться на Mg, тогда
второй атом А1 для компенсации ва-
валентности замещается на Si. До кон-
конца такой процесс может быть доведен
в искусственном минерале акерма-
ните Ca2MgSi2O7, являющемся уже
диортосиликатом. Природный мине-
минерал мелилит представляет собой фазу
переменного состава на основе геле-
геленита и акерманита.
6. Строение каркасных силикатов.
В каркасных силикатах присутствие
А1 является обязательным, так как
в чисто кремниевом каркасе валент-
валентности Si полностью компенсируются
валентностями О и такой каркас ста-
становится валентно нейтральным.
В этом случае мы приходим к струк-
структурам различных модификаций SiO2.
Эти структуры, как известно, не под-
подчиняются правилам плотнейшей упа-
упаковки. Следовательно, каркасные
структуры являются «рыхлыми»,
«ажурными» структурами. Они со-
содержат крупные пустоты, в которых
размещаются катионы большого раз-
размера и небольшого заряда, способные
заполнить объем этих пустот и ком-
компенсировать заряд каркаса, получив-
22 Кристаллохимия
337
ТАБЛИЦА 48
Клавсвфикацня силикатов с Si — О-мот ивам и бесконечных размеров
Форма радикала
Цепочка
Сдвоенная цепоч-
цепочка с 4-членными
кольцами
Сдвоенная цепоч-
цепочка с 6-членяыми
кольцами
Сдвоенная цепоч-
цепочка с 8-членными
кольцами
Лист с 6-членны-
ми кольцами
Лист с 4- и 8-член-
8-членными кольцами
Лист с 5-членны-
ми кольцами
Химический
состав
радикала
[SiO3]100
[SijOs]100
[SiAi]100
[Si6O17]lo°
[Si2O5]2o°
[Si O5]200
[Si3O,]200
Заряд
на
1 атом
Si
2—
1-
1Y»-
173-
1—
1—
7»-
Силикаты без
добавочных
анионов
Диопсид
MgCa [SiOB]i°°
Силикаты с добавочными
анионами или
нейтральными частицами
Рамзаит
Na2Ti2[SiO3]i°°O3
Тремолит
CaaMg5[SuOu]Jco(OH)s
Базальтическая роговая
обманка
NaCajMgs [Si4Ou]JooOOH
Ксонотлит
Са6 fSie017]1C0 (ОН)г
Каолинит
Ala[Si,O5]2co(OHL
Апофиллит
KCailSijOsjfF.SHjO
А1 (В, Ве)-силикаты
без добавочных
анионов
Авгит
(Mg,Fe)-
•Ca[(Si,Al)O3]J°°
Силлиманит
Al [AlSiOs]100
Андалузит
Al [AlSiOs]200
Геленит
Саа [AUSiO7]2oc
Al (В, Ве)-силикаты
с добавочными анионами
Роговая обманка
^5Oo/oCaa(Mg, FeM-
[SUOu]Jf30(OH,F),+^50%
NaCa3 (Mg, FeL (Fe, Al).
• [AlSisOnlffO, OH, F)a
Мусковит
КАЬ [AlSisOio]200 (OHJ
шийся в результате замены части
атомов Si на А1 или другие элементы,
способные имитировать Si (напри-
(например, В или Be).
Классифицируются каркасные си-
силикаты по характеру петель. Обычно
различают 3 группы:
1) Петли каркаса имеют только
шестиугольную форму, как у моди-
модификаций SiC>2 — тридимита и кристо-
балита. По-видимому, сходной со
структурой тридимита является
структура нефелина NafAlSiCU] 3°°.
Структура чкаловита Na2[BeSi2O6]3°°
является производной от высокотем-
высокотемпературного кристобалита. Его ячей-
ячейка получается в результате утроения
ребер а и Ъ в C-кристобалите. Ионы
Na+ располагаются в крупных пусто-
пустотах, имеющихся в каркасе.
2) Во второй группе петли в карка-
каркасе четырех- и шестиугольные. Важ-
Важнейшим представителем является со-
содалит Na8[AlSi04]63ooCl2. Сюда,
по-видимому, относится и ультрама-
ультрамарин (NaCaL-8[AlSiO4]6(SO4, Cl, S).
3) В последнюю группу попадают
силикаты, в каркасе которых петли
имеют четырехугольную и восьми-
восьмиугольную формы. Эта группа явля-
является важнейшей, так как к ней отно-
относятся все полевые шпаты, а также
некоторые боросиликаты, например
данбурит Ca[B2Si2O8]300.
7. Структурные формулы силика-
силикатов. В предыдущих пунктах были рас-
рассмотрены основные кремнекисло-
родные мотивы, встречающиеся
в структурах силикатов. В некоторых
веществах, конечно, возможно и одно-
одновременное нахождение нескольких
мотивов в структуре. Такой случай,
например, наблюдали Н. В. Белов
и И. М. Руманова в структуре эпидо-
та Ca2Al2FeSi3Oi2OH. Оказалось, что
в этой структуре присутствуют одно-
одновременно как ортосиликатные ради-
радикалы, так и диортосиликатные. На ос-
основании рентгеноструктурного иссле-
исследования формула эпидота должна
быть написана так:
Ca2A]2Fe [Si2O7] [SiO4]OOH .
Полное рентгеновское исследование
силиката, и вообще любого другого
сложного вещества, позволяет одно-
однозначно написать его структурную
формулу и, кроме того, часто и уточ-
уточнить его состав. Так, например, уче-
ученые в течение десятков лет спорили
о химической природе каламина,
имеющего состав H2Zn2Si0s. Часть
из них считала его ортосиликатом
и писала его формулу как Zii2Si@4-
¦ НгО. Другая часть считала этот ми-
минерал основной солью метакремневой
кислоты и соответственно писала его
формулу ZnSiO8'Zn(OH)![. Для под-
подтверждения своей точки зрения каж-
каждая группа приводила ряд доводов.
Рентгеноструктурное исследование
показало, что кремнекислородный
мотив в этой структур© [S12O7] и фор-
формулу вещества следует писать как
Zn4[Si2O7](OHJ-H2O. Далеко не
всегда при отношении Si: О = 1: 4 ве-
вещество обязательно будет ортосили-
ортосиликатом. Например, рамзаит, имеющий
состав Na2Ti2Si2O9, т. е. отношение
О : Si>4, оказался не ортосиликатом,
а щелочным метасиликатом, имею-
имеющим структурную формулу
Na2Ti2- [SiO,],O« (H. В. Белов и
Л. М. Беляев).
До работы Н. В. Белова и Р. И.
Смирновой формула куспидина при-
принималась как CaaSis0e • 3Ca(F, ОНJ,
и он относился к группе кольчатых
силикатов. В результате этой работы
выяснилась его структурная форму-
формула —Ca4[Si2O7]F2. В этом веществе,
по-видимому, нет изоморфного заме-
замещения F на ОН, которое в других си-
силикатах часто наблюдается. Обе фор-
формулы близки, что можно усмотреть,
если первую из них написать как
Ca2Si2O6Ca2(F, ОН) 4, но отнюдь не
тождественны.
В качестве последнего примера
можно указать на формулу ксонот-
лита, которую разные авторы писа-
писали как 3CaSiO3-H2O или 5CaSiO3-
¦ Н2О. Оказалось же она бСаЭЮз • НгО
или, точнее, Cas^isOn] (ОН) 2.
8. Особенности структур силика-
силикатов, содержащих крупные катионы.
22*
339
В предыдущем разделе были приве-
приведены примеры силикатов с брут-
то-формулой ортосиликатного соста-
состава, но содержащих в структурах не
БЮггруппы, а диортогруппы-812О7.
Многие из таких силикатов были
исследованы Н. В. Беловым с со-
сотрудниками. При этом выяснилось,
что почти все они являются силика-
силикатами кальция или же содержат дру-
другие крупные катионы — Na, Zr, Ti,
Nb, Та, Mn, TR.
H. В. Белов обратил внимание на
то, что ребра БЮ^тетраэдра близки
по размеру B,55 — 2,70) к ребрам
октаэдра, если последний построен
атомами кислорода вокруг неболь-
небольшого катиона — Mg, Fe, Al B,7 —
2,8). Если же в структуре силика-
силиката присутствует крупный катион
(например, кальциевый или натри-
натриевый) с размером ребра порядка 3,8,
то с ним хорошо может сочетаться
ребро диортогруппы, имеющей фор-
форму тригональной призмы, высотою
порядка 4А (рис. 331).
В свете этих чисто структурных
результатов Н. В. Белов A953 г.)
сделал, казавшееся ранее пара-
парадоксальным, заключение, что строи-
строительной основой силикатов служат
не кремнезм и не анионные крем-
кремнекислородные радикалы, а наобо-
наоборот, катионы, обычно укладываю-
укладывающиеся в стержни из кислородных
октаэдров (вокруг каждого катио-
катиона), и к этим основным архитектур-
архитектурным конструкциям лишь приспоса-
приспосабливаются кремнекислородные ради-
радикалы. Цепочки, ленты, сетки и даже
кольца достаточно прочны, но не
жестки и легко деформируются,
приспосабливаясь к различным кон-
конструктивным условиям, создавае-
создаваемым расположением ведущих катио-
катионов.
Классическая (с 30-летним ста-
стажем) брэгговская кристаллохимия
силикатов, основывавшаяся целиком
на структурах с мелкими катионами
(Mg, Ре, А1), является сейчас для
нас лишь первой главой этой кри-
кристаллохимии. Идеи, изложенные вы-
Рис. 331. Сочетание диортогруппы Si2O7
с крупным октаэдром
ше, Н. В. Белов назвал второй гла-
главой этого раздела кристаллохимии.
Группа Si2O7 не обязательно долж-
должна быть изолированной. Сдвоенное
шестерное кольцо миларита S112O30
с этой точки зрения следует рас-
рассматривать как конденсацию шести
групп Si2O7 FSi2O7—6-2 атомов кис-
кислорода).
Усложнение формы цепочек со-
состава (SiOs)le>, представленное на
рис. 332, также связано с выделе-
выделением в них групп Si2O7 по одной (б),
по две (в) и по три (г) на одинте-
траэдр, не входящий в группу Si2O7
и «смотрящий» в противоположную
сторону по сравнению с тетраэдра-
тетраэдрами, входящими в группу Si2C>7. Та-
Такие формы цепочек связаны с нали-
наличием в структурах различно распо-
Рис. 332. Различные формы цепочек со-
состава [SiO3]1°°
340
ложенных октаэдров из крупных ка-
катионов и с разным способом компен-
компенсации избытка расстояния между
кислородными атомами в , группе
сдвоенного тетраэдра Si2O7 и окта-
октаэдра.
С этих же позиций могут быть
рассмотрены мотивы из лент, слоев
и каркасов.
9. Элементы, имитирующие крем-
кремний в силикатах. До сих пор рас-
рассматривался главным образом состав
кремнекислородных мотивов, но
почти не затрагивался вопрос о дру-
других элементах, которые характерны
для силикатов. В силикатах весьма
часто встречаются следующие эле-
элементы Li, Na, К, Be, Mg, Ca, Ti, Zr,
Mn, Fe, Zn, B, Al, Si, 0, H, F.
Более всего похожи на структуры
силикатов структуры германатов
и фосфатов. Однако Р и Ge или сов-
совсем не встречаются в силикатах,
или находятся там в виде очень
небольших примесей и поэтому не
играют какой-либо существенной
роли.
Из перечисленного выше списка
важнейших для силикатов химиче-
химических элементов некоторые обладают
способностью имитировать кремний.
Это прежде всего А1, В и Be, кото-
которые так же, как и кремний, имеют
координационное число по отноше-
отношению к атомам кислорода 4 и коорди-
координационный многогранник — тетра-
тетраэдр. Размеры таких тетраэдров тоже
близки к размерам кремнекислород-
кремнекислородных тетраэдров. Из этих трех элемен-
элементов-имитаторов кремния наиболее
близки по размерам В и Be, поэтому
аналогия между ними и Si более пол-
полная. Существует мнение, что В в си-
силикатах часто имеет координацион-
координационное число 3. Оно основано на том, что
в структуре борной кислоты и других
боратов бор действительно имеет та-
такое координационное число. Однако
для силикатов число 3 у бора неха-
нехарактерно.
Атом (ион) А1 крупнее атома Si,
поэтому А1 в силикатах встречается
как в тетраэдрах, так и в октаэдрах.
В первом случае вещества называ-
называются алюмосиликатами, во втором —
силикатами алюминия. «Тетраэдриче-
ский» А1 изоморфно замещает Si и,
вероятно, бор; «октаэдрический» А1
является аналогом Fe, Mg и других
металлов. В некоторых силикатах,
например в мусковите, А1 играет од-
одновременно обе эти роли, и такие ве-
вещества называют алюмосиликатами
алюминия (и других металлов, встре-
встречающихся в них).
Как ни важен геометрический при-
признак для разделения силикатов на
алюмосиликаты и силикаты алюми-
алюминия, он не может претендовать на
большую строгость, и существующая
двойственность в трактовке структу-
структуры алюмо-, боро-, бериллосиликатов
лежит в самой природе этих веществ.
В некоторых случаях описанные
выше координационные критерии не
могут играть решающей роли в оп-
определении типа структуры. С таким
случаем мы встречаемся, например,
в низкотемпературном кордиерите,
являющемся полным структурным
аналогом берилла:
Be3Ala[Si6Oi8] — берилл,
AkMg2 [SisAlOis]— кордиерит.
Берилл никогда не может быть на-
назван алюмосиликатом, поскольку А1
в этой структуре занимает позицию
с координационным числом 6. В кор-
кордиерите же А1 занимает позицию,
аналогичную позиции Be в берилле,
т. е. имеет координационное число 4
и координационный многогранник —
тетраэдр. В этом случае координация
трех атомов А1, находящихся вне
кремнекислородного мотива, ничем
не отличается от координации того
атома А1, который входит в шести-
членное кольцо. Если же рассматри-
рассматривать все атомы А1 совместно с Si, то
вместо кольчатого структурного ти-
типа получаем для кордиерита каркас-
каркасный тип. При первом подходе мы
должны будем назвать кордиерит
кольчатым алюмосиликатом магния
и алюминия, при втором — каркас-
каркасным алюмосиликатом магния.
341
Бериллий как элемент, имеющий
в силикатах координационное число 4
и координационный многогранник
тетраэдр, также является имитатором
кремния. По этой причине сам берилл
можно называть кольчатым силика-
силикатом бериллия и алюминия или же бе-
риллосиликатом алюминия с каркас-
каркасной структурой.
Все сказанное в отношении А1
может быть распространено и на В.
Кристаллохимическое отличие В от
А1 заключается в том, что А1 имеет в
силикатах координационные числа
6 и 4, а бор — 4 или 3. Бор, так же
как и А1, имитирует кремний. Иллю-
Иллюстрацией этого могут служить, напри-
например, анортит Са [ Al2Si2Os] 3°° и данбу-
рит Ca[B2Si208]3o°, которые имеют
весьма сходные структуры. Правда,
для данбурита возможна и другая
трактовка структуры — СаВг^гО^О.
Рассмотрим еще одну группу ми-
минералов:
ВвзАЬ [SieOi8] — берилл,
СвАЬВез [AlSijOig] — воробьевит,
КСа2ВеаА1 [впгОзо] — миларит,
(Na,Ca) (Mg,FeKB3Al6 [Si6Oi8]O9 OHL —
турмалин.
В турмалине такие же шестичлен-
ные кольца, как в берилле, но не по-
полярные. Они объединяются В-тре-
угольниками и А1-октаэдрами.
В этом случае, когда в радикале
часть атомов занята трехвалентными
(В, А1) или двувалентными (Be) эле-
элементами, часть атомов кислорода мо-
может замещаться гидроксилами или
фтором, чего почти никогда не наб-
наблюдается в чисто кремниевых ради-
радикалах.
Среди А1 (В, Ве)-силикатов наи-
наибольшую группу составляют каркас-
каркасные. Но в их каркас никогда не входят
ионы 0Н~ и F".
Аналоги орто- и диорто-радикалов
среди А1, В, Ве-силикатов, содержа-
содержащие изолированные ионы типа
[AlOt]5- и [А12О7]3~, не известны.
Какой же критерий положить в
основу классификации, чтобы одноз-
однозначно относить А1(В,Be)-силикаты в
определенную группу? В. С. Соболев
в качестве такого критерия предлага-
предлагает считать сходство структур алюмо-
алюмосиликата с соответствующим силика-
силикатом. Например, данбурит считать бо-
росиликатом потому, что его боро-
кремнекислородный мотив аналоги-
аналогичен мотиву анортита.
Согласиться с этим нельзя. Этот
критерий заставил бы нас считать
данбурит силикатом бора до тех пор,
пока структура анортита не была бы
определена, и боросиликатом—после
определения структуры анортита.
Мы в качестве критерия предлагаем
следующее: если все кислородные
ионы в структуре силиката могут
быть объединены в мотив, включаю-
включающий только кремний, то элементы-
имитаторы кремния остаются вне
этого мотива, и такие минералы сле-
следует отнести в группу силикатов
алюминия, бора, бериллия. Если же
при использовании всех атомов крем-
кремния остаются свободные (неохвачен-
(неохваченные) ионы кислорода, а иногда и ОН
или F, но они могут быть включены
в А1 (В, Be)-кремниевый мотив, то
это — А1(В, Be)-силикаты.
Так, например, берилл — силикат
Be и А1, а кордиерит — алюмосиликат
А1 и Mg, данбурит — боросиликат Са,
миларит — силикат К, Са, Be, Al, a
турмалин алюмоборосиликат с двой-
двойным шестичленным кольцом, а не
силикат с простым шестичленным
кольцом, и т. д.
10. Важнейшие катионы, встречаю-
встречающиеся в силикатах. Тетраэдрическая
координация в силикатах встречает-
встречается также у Ti, Fe и Zn. Однако разме-
размеры таких тетраэдров значительно пре-
превышают размеры кремнекислород-
ных.
Некоторые авторы, однако, счи-
считают, что Ti может в небольших коли-
количествах изоморфно замещать Si.
Обычным же координационным
числом для большинства металлов в
силикатах является 6 и координа-
координационным многогранником — октаэдр.
Такую координацию имеют: Li, Ms?,
342
Ca, Ti, Zr, Mn, Fe2+, Fe3^ Al. Все эти
элементы в той или иной степени изо-
изоморфно замещают друг друга.
Кальций крупнее магния, поэтому
он иногда имеет координационные
многогранники, являющиеся переход-
переходными от октаэдра (к. ч. 6) к томсо-
новскому кубу или другим фигурам,
отвечающим координационному чис-
числу 8. Координационное число 8
встречается также у Zr4+ и редко —
у Fe2+.
Крупные одновалентные катионы
Na+ и К+ имеют координационные
числа 8 и более высокие. Они встре-
встречаются в каркасных силикатах, на-
например в полевых пшатах или в цео-
цеолитах, где имеются крупные полости,
или же располагаются между слоями
в слоистых силикатах. Например, в
слюдах маленький ион Li+ имеет ко-
координационное число 6. Он замещает
в слюдах Mg2+, Fe2+, Fe3+, A13+ и
никогда — К+.
Выше мы разобрали роль катионоп
в структурах силикатов, остановимся
теперь на роли главнейших анионов.
11. Важнейшие дополнительные
анионы в силикатах. Как было ска-
сказано раньше, главнейшей анионной
частью являются кремнекислородные
мотивы. Вне этих мотивов могут оста-
оставаться избыточные анионы О2~, ОН~
и F~, имеющие близкие размеры и
изоморфно замещающие друг друга.
Число свободных (находящихся вне
кремнекислородного мотива) О2~ не-
непосредственно не определяется хими-
химической формулой. Как указывалось
выше, рамзаит Na2Ti2Si2O9 не явля-
является ортосиликатом с одним свобод-
свободным ионом кислорода, а является ме-
тасиликатом с тремя свободными
ионами 02~. Вещества такого типа
будем называть оксисиликатами. Ио-
Ионы гидроксила и фтора всегда нахо-
находятся вне кремнекислородного моти-
мотива, но могут входить в алюмо-(В.Ве)-
силикатный мотив.
В силикатах встречаются некото-
некоторые анионы кислородных кислот:
СОз2~, SO42"", РО43~ и значительно ре-
реже — МоО42~ и AsQ33~. Еще реже на-
наблюдаются бескислородные анионы
Эг2", С1~ и др. Из нейтральных ча-
частиц в силикатах чаще всего встре-
встречаются молекулы воды, например, в
натролите Na2[Al2Si30io]3oo-2H20.
Ранее формулы содалита и гаюина
писали соответственно 3NaAlSiOt •
•NaCl и 3NaAlSiO4-CaSO4, но на са-
самом деле отдельных молекул NaCl,
CaSO4 в структуре этих соединений
нет, поэтому правильно эмпириче-
эмпирические формулы этих минералов следу-
следует писать Na4[AlSi04l33ooCl 0
Na3Ca[AlSi04]33ooS04.
В сложных силикатах, в состав ко-
которых входит несколько типов катио-
катионов с различной валентностью, не-
несколько типов анионов с различной
валентностью и нетральные частицы,
замечается следующая закономер-
закономерность (Г. Б. Бокий, 1960 г.): наибо-
наиболее многовалентные катионы окруже-
окружены в структуре анионами с макси-
максимальной валентностью. Одновалент-
Одновалентные же катионы стремятся быть окру-
окруженными нейтральными частицами,
если таковые имеются. Так, напри-
например, в ; структуре апофиллита
KCa4Sis02oF • 8Н2О атомы кремния
окружены четырьмя атомами кисло-
кислорода, а К — восемью молекулами во ¦ ,
ды, Са имеет к.ч. 7 D О + 1 F +
+ 2Н2О).
Частным случаем этого правила
является то хорошо известное обстоя-
обстоятельство, что одновалентные анионы
ОН~ или F~ почти никогда не входят
в кремнекислородный мотив.
Если в структуре нейтральных час-
частиц нет, то одновалентные катионы
обычно окружаются теми атомами
кислорода из кремнекислородного мо-
мотива, которые делятся между двумя
атомами кремния (или Si, Al), т. е.
валентности которых уже почти пол-
полностью насыщены. Примером может
служить К+ в структуре мусковита.
Повышение координационного числа
катиона эквивалентно понижению ва-
валентности. Так, если в структуре име-
имеются два катиона равной валентности,
но с разными координационными чис-
числами, то катион с меньшим координа-
343
ционным числом будет связан с атома-
атомами кислорода, а катион с большим
координационным числом может
иметь в своей координационной сфе-
сфере и одновалентные анионы (напри-"
мер, два сорта А1 в структуре слюд).
Эта закономерность, конечно, спра-
справедлива не только для силикатов, но
и для других сложных неорганиче-
неорганических веществ.
12. Плотнейшие упаковки в сили-
силикатах. Очень распространено мнение,
что к структурам силикатов неогра-
неограниченно применимы принципы плот-
нейших шаровых упаковок. В дейст-
действительности это далеко не так.
В плотнейших упаковках, как из-
известно, есть 2 типа пустот: октаэдри-
ческие и тетраэдрические, для кото-
которых координационные числа соответ-
соответственно 6 и 4. Отсюда следует, что
структуры, в которых катионы имеют
иные к. ч., заведомо не будут подчи-
подчиняться закону шготнейшей упаковки.
Катионы, имеющие к. ч. 4 и 6, обычно
больше, чем пустоты, поэтому они раз-
раздвигают шары упаковки, не нарушая
существенно взаимного расположения
шаров упаковки. Сами шары плотней-
шей упаковки имеют к. ч. 12, поэтому
особенно крупные катионы могут
иногда занимать место шара упаков-
упаковки. В этом случае может образовать-
образоваться единая анионно-катионная упа-
упаковка.
В силикатах плотнейшую упаковку
составляют ионы кислорода, гидрок-
еильные ионы или изоморфно замеща-
замещающие их ионы фтора.
Все структуры силикатов, с точки
зрения теории плотнейших упаковок,
можно разделить на 4 группы.
1) Структуры подчиняются плот-
нейшим упаковкам.
2) Структуры, в которых плотней-
шая упаковка охватывает не все анио-
анионы (О2-, 0Н~ и F~) силиката. Часть
из них располагается в пустотах меж-
между шарами плотнейшей упаковки.
Это — структуры, являющиеся плот-
нейпшми упаковками с лишними ани-
анионными шарами.
3) Структуры, в которых анионы
распределяются по местам плотней-
плотнейших шаровых упаковок, но не занима-
занимают всех этих мест, т. е. оз таких струк-
структурах имеются крупные пустоты,
соизмеримые по размеру с размерами
шаров упаковки. В эти пустоты могут
попадать крупные катионы, крупные
анионы (одноатомные или комплекс-
комплексные) и нейтральные частицы, напри-
например молекулы воды.
4) Структуры в целом не подчиня-
подчиняются законам плотнейших упаковок.
Среди этих структур иногда можно
выделить участки, например слои, для
которых плотнейшая анионная упа-
упаковка сохраняется.
Впервой группе встречаются
структуры, в основе которых лежат
разные типы упаковок. Так, гексаго-
гексагональной плотнейшей упаковкой ха-
характеризуются минералы группы фор-
форстерита — клиноюмита:
форстерит — MgaSiO*,
норбергит — Mg2 [SiO4] .Mg (OH)a ,
хондродит — 2Mg2 [SiO4].Mg(OHJ ,
юмит — ЗМ ga[SiO4] • Mg(OHJ,
клиноюмит — 4Mg2 [SiO4]-Mg(OHJ .
В основе структуры дистена
Al2[SiO4]O лежит кубическая плот-
плотнейшая упаковка. Такой же упаков-
упаковкой характеризуйся амфиболы:
Ca2Mg5[Si40ii]2l00 — тремолит,
NaCa2Mg5[Si4Oi,]2I°°OOH — базаль-
тическая роговая обманка. В струк-
структурах пироксенов тоже кубическая
плотнейшая упаковка, однако коорди-
координационный многогранник ионов Са
представляет уже весьма искаженный
октаэдр, приближающийся по форме
к томсоновскому кубу, т. е. много-
многограннику, характерному для коорди-
координационного числа 8 (например, диоп-
сид CaMg[SiO3]2100). Все ионы кис-
кислорода и гидроксила в структуре
ставролита FeAl4[SiO4]2O2(OHJ так-
также расположены по местам кубиче-
кубической плотнейшей упаковки.
Четырехслойной упаковкой ха-
характеризуется структура топаза
Al2[Si04] (ОН) 2, шестислойной
(АВСАСВ) — структура рамзаита
Ma2Ti2[SiO,]2l0O0,.
344
Ко второй группе относят-
относятся, например, структуры силлима-
силлиманита AlJAlSiOs] 1о° и тортвейтита
Sc2[Si2O7j. Структура силлиманита
сходна со структурой рутила ТЮг, в
основе которой лежит гексагональ-
гексагональная плотнейшая упаковка. В отличие
от рутила, в структуре силлиманита
«лишний» атом кислорода, соединяю-
соединяющий 2 тетраэдра, располагается в
октаэдрических пустотах, остающих-
остающихся в рутиле пустыми. Тот же харак-
характер структуры и у тортвейтита. Свя-
Связующий атом кислорода группы
[Si2C>7] занимает центр октаэдра. Осо-
Особенно характерен этот случай для
силикатов (и их аналогов) с крупны-
крупными катионами. Как было сказано в
разделе 8, с октаэдром, в центре ко-
которого расположен крупный катион,
лучше согласуется по размерам не
[SiO,j] -группа, a [Si2Oy] или анало-
аналогичная ей [Р2О7]. Кубической плот-
нейшей упаковкой, например, харак-
характеризуется структура ZrP2O7, где
крупные октаэдры циркония сочета-
сочетаются с вписанными в октаэдры груп-
группами [Р2О7].
Силикатов, составляющих третью
группу, в структурах которых ани-
анионы не занимают всех мест плотней-
шей шаровой упаковки, довольно
много. Примером гексагональной
плотнейшей упаковки, у которой про-
пропущена '/4 часть шаров, может слу-
служить гемиморфит ZiLi[Si2O7] (ОН)г'
ШО. В эту же категорию попадает
большая группа каркасных силика-
силикатов, например, производных от струк-
структуры кристобалита. В структурах со-
содалита Nat[Al3Si3Oi2]3coCl и гаюина
Na5[Al3Si30i2]3ooS04 имеются боль-
большие пустоты, соответствующие четы-
четырем шарам плотнейшей упаковки. В
структуре содалита эта пустота заня-
занята четырьмя ионами Na, в центре
которых расположен ион G1; таким
образом, 4 места упаковки заняты
пятью атомами.
Структуру турмалина можно пред-
представить себе в виде очень сильно
искаженной кубической плотнейшей
упаковки с пропусками.
Рис. 333. Три возможные формы шести-
членных колец
Большинство структур силикатов
не подчиняется закону плотнейших
упаковок, даже с теми оговорками,
которые сделаны для предыдущих
двух групп.
К этой категории можно было бы
отнести и турмалин, так как кольча-
кольчатые силикаты, вообще говоря, не под-
подчиняются этой закономерности (см.,
например, берилл ВезАЬ^бО^] и
бенитоид BaTi[Si3O9]).
Слоистые силикаты также могут
быть представлены как плотнейшие
упаковки только с большой на-
натяжкой. Например, в мусковите
KAl2[AlSi30io]2oo(OHJ и других слю-
слюдах пакеты могут быть представлены
в виде очень сильно искаженной упа-
упаковки типа АВСА. Ионы калия име-
имеют координационное число 12. Они
соединяют два пакета и в упаковку
поэтому входить не могут. Припи-
Приписать им координационное число 6
можно лишь в. том случае, если пу-
пустые кольца в слое будут иметь не
гексагональную форму (рис. 333, а),
а тригональную (рис. 333, б). Вероят-
Вероятно, в действительности реализуется
промежуточный случай, и такое коль-
кольцо принимает дитригональную форму
(рис. 333, в), но дитригон гораздо
ближе к гексагону, чем к тригону.
В других слоистых силикатах нет
промежуточных ионов, аналогичных
ионам К в слюдах, а сами пакеты, по-
видимому, имеют кольца почти стро-
строго гексагональной формы. Таковы:
тальк Mg3[Si40io]2oo(OHJ, пирофил-
пирофиллит Al2[Si40io]2oo(OHJ) каоли-
каолинит Al2[Si2O5]2o<>(OHL, хлорит
AlMg5[AlSi3O10]2oo(OH)8 и др.
345
Часто нарушают плотнейшую упа-
упаковку крупные катионы Са2+, Zr4+, K+
и др. Они не помещаются в октаэдри-
ческую пустоту упаковки из 02~ ио-
ионов, раздвигают их, образуя коорди-
координационные многогранники с 8 и боль-
большим числом вершин. В эту группу
следует отнести циркон Zr[SiO4],
гранат Ca3Al2[Si04]3, куспидин
Cat[Si207]F2 и каркасные силикаты, в
частности, полевые шпаты, например,
ортоклаз KjAlSiaOe]300 и близ-
близкий к ним по структуре данбурит
Ca[B2Si2O8]300 и др.
Строение самих каркасов в кар-
каркасных силикатах, как было сказано
выше, сходно со строением различ-
различных модификаций SiO2. Эти же
последние, как известно, не подчи-
подчиняются закону плотнейпшх упако-
упаковок. В некоторых из них (например,
в кристобалите) только 7г мест в
плотнейшей упаковке занята атома-
атомами кислорода. Конечно, и в плотней-
плотнейшей упаковке можно так занять 'Д
тетраэдрических пустот, что в запол-
заполненных тетраэдрах каждая вершина
будет общей для двух соседних за-
заполненных тетраэдров. При этом об-
образуется каркас с плотнейшей
упаковкой. Такое расположение,
например, характерно для CuSi-тет-
раэдров в структуре халькопирита.
Однако такое расположение в S1O4-
тетраэдрах будет сопряжено с огром-
огромным отталкиванием двух атомов Si
соседних тетраэдров. Валентный угол
кислорода увеличится от 109° до пре-
предела — 180°. При этом атомы, зани-
занимавшие первоначально места плот-
плотнейшей упаковки, переместятся,
объем структуры резко возрастет, и
плотнейшая упаковка расстроится.
Шары в ней теперь будут занимать
только половину мест плотнейшей
упаковки.
В халькопирите такого процесса
«передвижки» анионов не наступает,
во-первых, из-за значительно более
слабого отталкивания между двумя
атомами меди, чем между двумя ато-
атомами Si, вследствие меньшего заряда
и большего расстояния Си — S, чем
Si — О, и, во-вторых, вследствие то-
того, что в халькопирите каждый атом
S имеет тетраэдрическую координа-
координацию — 2Cu+2Fe, а атомы кислорода
в ЭЮг — гантельную — 2Si.
13. Изоморфизм в классе силика-
силикатов. В заключение необходимо еще
раз вернуться к вопросу, затронуто-
затронутому в начале настоящего параграфа,
а именно — к освещению причин
неудач изучения силикатов чисто
химическими методами. Как уже го-
говорилось выше, одной из причин
является чрезвычайная сложность
состава этого класса веществ, слож-
сложность, связанная с наличием в сили-
силикатах изоморфных замещений как
изовалентных, так и гетеровалент-
ных.
Для последних А. Е. Ферсма-
Ферсманом было предложено правило «диа-
«диагональных рядов» в периодической
системе химических элементов. Оно
связано с тем, что размер ионов у
соседних элементов сильнее отлича-
отличается по горизонтали и по вертикали,
чем по диагонали. Ниже представле-
представлена часть менделеевской таблицы с
наиболее важными для силикатов
элементами, в которой указаны диа-
диагональные ряды, характерные для ге-
теровалентного изоморфизма в сили-
силикатах (по В. С. Соболеву):
Ряды изоморфного замещения элементов
Li
Na
0,98\
К
1,33\
Rb ^
1,49
Cs
165
Be.
0,34
Mg
074
Ca
\ Sr"
\
Ba
1)8
В
0,20
''¦•Al
XSc ^^
Y ^~~~^~-
La P.3.
1,04 102-080
С
Si
039
Ti
Zr
0,82
Hf
0.82
N
P
V
0 4
Nb
0,66
Та
0.66
Для того чтобы произошла замена
одного химического элемента на дру-
другой с иной валентностью, необходима
компенсация заряда. Ниже собраны
важнейшие случаи замещающих друг
друга пар химических элементов
346
ТАБЛИЦА 49
Данные анализа роговой обманки
Окисел
SiO2
тю2
А1203
Fe2O3
FeO
MnO
MgO
CaO
Na2O
K2O
F
H2O+
Поправка
на F2 = О
Вес. %
оквела
40,26
3,77
13,23
7,19
6,48
0,18
13,18
12,20
1,73
0,56
0,09
1,25
100,12
~~ 0,04
100,08
Относитель-
Относительные
молярные
количества
окислов *
670
47
129
45
90
3
327
218
28
6
4
70
Относитель-
Относительное
количество
кислорода
в окисле
1340
94
387
135
90
3
327
218
28
6
4
70
2702
~ 2
2700
Число атомов
кислорода,
рассчитанное
на 24 **
11,9
0,8
3,44
1,2
0,77
0,03
2,9
1,95
0,25
0,05
0,05
0,65
24,025
~ 0,025
24,000
Относитель-
Относительное
количество
катионов
670
47
258
90
90
3
327
218
56
12
140
Число атомов
катионов
5,96
0,4
2,30
0,8
0,77
0,03
2,9
1,95
0,5
0,1
1,3
• Множитель 1000/М.
•• Общий делитель 2702 : 24 = 112,6.
в силикатах:
Na+ + Si4+ =
Са2+ + А13+ = Се3+
2Са2+ = Na+ + Се»+,
Li+ + А13+ = Са1* +
Li+ + Al8+ =
Знание изоморфных замещений
позволяет правильно составлять
структурные формулы силикатов по
результатам химического анализа.
Приведем пример расчета анализа
силиката и нахождение его правиль-
правильной структурной химической форму-
формулы (табл. 49 заимствована у В. G. Со-
Соболева с небольшими изменениями).
Написание правильной структур-
структурной формулы прежде всего возможно
потому, что по кристаллографиче-
кристаллографическим данным известно, что анализи-
анализировалась роговая обманка. Атомная
структура ее показывает, что анион-
анионная часть состоит из кремнекисло-
родного мотива [Si4Oii]lo° и изоли-
изолированных гидроксильных ионов и
ионов кислорода. Катионы делятся
на две группы в отношении 2:5 или
на три (в случае базальтической ро-
роговой обманки) в отношении 1:2:5.
Идеальные формулы для этих слу-
случаев соответственно будут
Ca2Mg5 [Si4On]g (OH)g
и NaCaaMgs [Si«Ou]g О (ОН)
К Mn Fe2+ Al F (А)
Fe3+
Al
Tl
В третьей графе таблицы вычис-
вычислены относительные молярные коли-
количества окислов. В качестве множите-
множителя взято отношение 1000/М. В чет-
четвертой — найдено относительное
атомное количество кислорода. При
суммировании первого и третьего
столбцов необходимо взять поправку
на фтор, который, как известно, в
347
силикатах замещает гидроксил. Для
внесения поправки 2 атома F «заме-
«заменяем» в анализе на атом О.
Дальнейший расчет был бы невоз-
невозможен, если бы не было известно,
что анализируется роговая обманка и
что в ее структуре имеется 24 ато-
атома О. Поскольку структура роговой
обманки подчиняется правилам плот-
нейшей упаковки, то мы вправе
считать, что налицо все 24 атома
кислорода и что в анионной части
этого силиката нет дефектов.
Для нахождения числа атомов кис-
кислорода (графа 5) находим общий де-
делитель 2702 : 24 = 112,6.
Из данных для атомного количества
катионов находим соответственное
число атомов катионов (графа 7). По
этим числам можно составить струк-
структурную формулу анализированного
силиката с учетом возможных изо-
изоморфных замещений. При этом преж-
прежде всего должны учитываться воз-
возможности небольшого изоморфного
замещения магнием кальция и каль-
кальцием — натрия, что показано в фор-
формуле (А) изогнутыми стрелками.
Ниже этой формулы указаны наибо-
наиболее вероятные изоморфные замеще-
замещения тех элементов, которые обнару-
обнаружены в результате анализа.
По этим данным и данным таблицы
можно составить правильную химиче-
химическую формулу проанализированного
силиката:
(Nao,5K0,i)o,6
(O0,65OHl,3Fo,05) .
Весь Si включается в кремнекис-
лородный мотив. Недостающее коли-
количество дополняется А1, избыток кото-
которого, очевидно, изоморфно замещает
Mg.
Из приведенной формулы видно,
что восьмое катионное место в струк-
структуре базальтической роговой обманки
заполняется не все (на 0,6). Соответ-
Соответственно «свободного» кислорода тоже
меньше единицы, а гидроксила —
больше единицы.
Небольшой избыток катионов, заме-
замещающих Mg @,12), может быть обу-
обусловлен тем, что часть атомов Mg, как
известно, может замещать Са, а по-
последний может занимать и восьмое
катионное место в структуре, т. е.
изоморфно замещать Na. Этот неболь-
небольшой избыток может и просто объяс-
объясняться ошибками анализа, так как в
качестве примера взят конкретный
реальный анализ минерала. Как ви-
видим из разобранного примера, по дан-
данным только химического анализа, без
серьезного кристаллохимического ана-
анализа, невозможно нахождение пра-
правильной структурной формулы слож-
сложных веществ.
14. Зависимость физических
свойств силикатов от их структуры.
Сходство химических формул не всег-
всегда обусловливает сходство свойств.
Только знание атомных структур и,
следовательно, правильных структур-
структурных формул позволяет понимать
сходство и различие физических
свойств. В качестве примера рассмот-
рассмотрим две пары веществ, отличающихся
друг от друга тем, что в одном из них
натрий заменен на калий (табл. 50).
В первой паре физические свойства
почти одинаковы, во второй резко от-
отличаются. Это объясняется тем, что
первая пара имеет сходные структу-
структуры каркасного типа, вторая же пара
характеризуется резко различными
структурными типами. Правильно на-
написанные их структурные формулы
будут соответственно NaAl[Si2O6]1IX>
HK[AlSi2O6]300.
Хорошим примером, иллюстрирую-
иллюстрирующим ту же мысль, могут служить две
модификации LiAlSi2O6, имеющие
следующие структурные формулы:
LiAl[Si2O6] и Li[AlSi2O6].
Кристаллохимия силикатов может
служить примером современного эта-
этапа развития химии одного из важней-
важнейших классов химических соединений.
Сейчас мы являемся свидетелями
того, как в результате новых экспе-
экспериментальных определений структур
бороводородов в корне ломаются те
химические схемы их строения, кото-
348
ТАБЛИЦА 50
Зависимость свойств силикатов от структуры
Силикат
Альбит
NaAlSisO8
Микроклин
KAlSiaOg
Жадеит
NaAlSi2Oe
а-Лейдит
KAlSi2O6
Оптические
константы
ng = 1,536
пт = 1,529
Пр = 1,525
rig — Ир = 0,011
и* = 1,526
пт = 1,524
пр = 1,519
ng — np = 0,007
ng = 1,667
nm = 1,659
np = 1,654
пг —гер = 0,013
пг = 1,509
Пр =1,508*
ng — np = 0,001
Физические константы
Твердость 6—6,5
Спайность @01) и @10)
совершенная
р = 2,61
t пл. 1118° С
Твердость 6—6,5
Спайность @01) и @10)
совершенная
р = 2,57
t пл. 1170° С
Твердость 6,5—7
Спайности A10) средняя
р = 3,3-3,4
Твердость 5—6
Спайности нет
р = 2,45—2,50
Сингония
Триклинная
Триклинная
Моноклинная
Кубическая
• В кристаллах лейцита наблюдается аномальное двупреломление.
рые предлагались для них на осно-
основании результатов чисто химических
исследований. Вместе с определением
структур кристаллохимия бороводо-
родов заставляет по-новому рассмат-
рассматривать такие старые проблемы химии,
как, например, проблему валентности.
Нет сомнения в том, что кристалло-
химические методы и для других
классов соединений окажутся столь
же плодотворными, как и для классов
силикатов, бороводородов и т. п. Сов-
Современный этап развития химии во
многом обязан кристаллохимии, и нет
сомнения в том, что кристаллохими-
ческие представления в ближайшие
годы еще глубже будут внедряться в
химию.
§ 7. Кристаллохимия боратов
История изучения строения боратов
во многом напоминает соответствую-
соответствующую историю для силикатов с той,
однако, разницей, что она сдвинута
по времени приблизительно на 25—
30 лет. Так, в 1947 г. А. В. Николаев
в специальной монографии, посвя-
посвященной природным боратам, рассмат-
рассматривал их как соли гипотетических
полиборных кислот. (Для силикатов
аналогичная концепция развивалась
до 20-х годов нашего столетия.)
Структурные формулы этих кислот
выводятся с использованием пред-
представления о цепочках из атомов бора
к треугольной координации. Идея
Вант-Гоффа о четырехкоординиро-
ванном атоме бора, к сожалению, бы-
была почти забыта. Следует отметить,
что ни одна из предсказанных
А. В. Николаевым структурных фор-
формул не подтвердилась при структур-
структурном анализе боратов.
В основу кристаллохимической
систематики боратов целесообразно
положить два признака: 1) строение
349
основной структурной единицы —
борокислородного полииона и 2) спо-
способ сочетания этих единиц (изоли-
(изолированные полиионы, цепочки, слои
и т. д.).
Особенностью боратов, в отличие,
например, от силикатов или фосфа-
фосфатов, является большое разнообразие
конечных радикалов — полиионов,
поэтому их систематика является наи-
наиболее важной, и мы остановимся
здесь главным образом на ней. Эта
идея развита в работах Тенни-
сона A963 г.) и Г. Б. Бокия и
В. Б. Кравченко A966 г.).
Класс боратов, как и силикатов,
мы делим на отряды: островные, це-
цепочечные, слоистые и каркасные.
Первый из них, наиболее богатый
представителями, в свою очередь мо-
может быть подразделен на подотряды:
некольцевые, однокольцевые, двух-
кольцевые, трехкольцевые, четырех-
кольцевые и бораты со смешанными
полиионами (табл. 51).
Кроме того, в рамках каждого под-
подотряда можно различать кислород-
кислородные, гидроксильные и смешанные
полиионы, а также мономеры от ди-
меров и более сложных полимеров.
Если мономеры полимеризуются в
одном, двух или трех измерениях до
бесконечности, то образуются соот-
соответствующие им вышеназванные от-
отряды.
Атом бора в боратах может иметь
треугольную и тетраэдрическую ко-
координацию, поэтому в случае соеди-
соединения бора с кислородом получаются
соответственно острова состава
(ВОз) 3~ и (В04M" (см. рис. 334, а ж б).
В случае замены кислородного иона
на гйдроксильный получаются анало-
аналогичные острова, но с иным зарядом
[В(ОН),]0 и [В(ОНL]'- Смешан-
Смешанный треугольный остров пока изве-
известен только один—(ВОгОНJ". Из-
Известны димерные треугольные остро-
острова состава О2ВОВО2 = (В2О5L"
C34, в) иО2ВОВООН=(В2О4ОНK-.
Аналогичный димер для полно-
полностью гидроксильного острова с «тет-
раэдрическим бором» (рис. 334, г)]
ТАБЛИЦА 51
Борокислородвые островные мотивы
структур
Простейшие мотивы
кислородный
гидроксильно-
нисяородный
Некольцевые одно- и дибораты
О
0-ъ( [во,р-
0
О О
/В\ [ВР4]»-
0 О
о о
>В—О—В\ [ВгО,]*-
ох о
0
I 0—В\ [BOtOH]»-
0
он
НО—В<( [В (ОН),]»
он
но он
/В<( [В (ОН),]-
но он
0 0
/В-О-В<(
0 ОН
[В.О.ОН]-»
но он
но^в-о-в^он
НО ОН
[В,О(ОН)в]«-
Одвокольцевые трибораты
0 НО
>в_о
0 \в—0 [В»0«]»~
/В—(Г
0
0 °
^В—0. 0
>в_о/ Х°
0 о
)>В—0
0 "VB—ОН
/В—0
но
[В,Ог(ОН)г]»
но
Ъъ-о он
0 >в/
)>В-0 ОН
но
но
)>В—0 ОН
0 ^ В'
Ъв—о он
но 1
он
[В,О,(ОН),р-
но
но^в—о он
0 ^в\
Nb-o7 он
но 1
но
[В,0,(ОН).Р-
350
ТАБЛИЦА 51 (окончание)
Сложные мотивы
гидроксильно-квслородный
Двухкольцевоб тетраборат
ОН
О-В-0
НО—В в В—OH[B.Ot(OHLp-
/
Двужкольцевые пентабораты
НО ОН
В—О в—В:
О \в/ О В»О,(ОНL]-
ч. / \ /
/В—О О—В\
но он
ч
но
ч
о'
но
в—о о—в
он
/
Vb<
I
О [В,О.(ОН,)]'-
I
он
он
но
в—о о—в
он
О \В<^ О [В.06(ОН),]>
Ъв-о о-в<(
он I I он
он он
Четырехкольцевой тетраборат
ОН
В
в-
о/чо
но о
ч
в
он
он
Бораты со снетаииьши полиионаии
НО ОН
>
в-о о-в
>
о \в/ о
ЧВ—О О—В<^ [В6О7(ОН),]«-
1 1
\
в
но
НО ОН
\в-о о-в/
7 >< х
><
\в-о о-в/
°
но он
>в-о о-вгон
О \в/ О [В,Ов@Н),]<-
но
>в_о/ \>-в<
он он
он
Трехкольцевоб гексаборат
ОН
В
он о о он
V
/ ч /
О О и
в в в
[В,О7(ОНN]>-
Чо
Чон
он
[В,Ои(ОН),р-
но он
\в—о о—в/
< X >°
\в-о о-в
НО О О А„ ОН
\ / \ / он
но—в в
!, о
/I
но
[В,О1
S51
Рис. 334. Треугольный (а), тетраэдриче-
ский (б) и другие некольцевые борокис-
борокислородные острова (в—в)
Ok,
Рис. 337. Борокислородные цепочки (а)
и слои (б) из колец
Рис. 335. Однокольцевые борокислородные
острова
Рис. 336. Двухкольцевые борокислородные
острова
имеет состав (ОНKВОВ(ОНKВОВ-
•(ОН)з = [В2О(ОНN].
Образование цепочек из «треуголь-
«треугольных» и «тетраэдрических» атомов бо-
бора, получающихся при дальнейшей
конденсации мономеров, иллюстриру-
иллюстрирует рис. 334,9 и е. Формулы их со-
соответственно будут
B ^2
() (И
Весьма разнообразны кольцевые
радикалы. В табл. 51 они системати-
систематизируются нами по числу колец в
острове: однокольцевые, двухкольце-
вые, трехкольцевые и т. д.
Однокольцевые острова собраны
на рис. 335, а—д.
Двухкольцевые острова показаны
на рис. 336, а—з. Отдельные кольца
уже среди двухкольцевых радикалов
могут соединяться друг с другом че-
через общую вершину (рис. 336, в — з)
и через общее ребро (а и б). В более
сложных радикалах наблюдается со-
соединение колец не только через об-
общий атом бора, но и через общий
атом кислорода (см. табл. 51). Даль-
Дальнейшая конденсация кольцевых ра-
радикалов приводит к образованию
цепочек, слоев (рис. 337) и каркасов.
А. Ф. Горбов A960 г.) и В. Б. Крав-
Кравченко A963 г.) показали смену по-
полиионов в структурах боратов, обра-
образующихся из растворов с разным от-
отношением компонентов и с разным
рН. В результате такого подхода
удалось проследить некоторые гене-
генетические взаимоотношения между
боратами.
23 Кристаллохимия
ГЛАВА XX
КРИСТАЛЛОХИМИЯ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
И ИХ АНАЛОГОВ
§ х. Ковалентные
и Ван-дер-Ваальсовы «радиусы»
неметаллических элементов
Структуры простых веществ неме-
неметаллических элементов и их соедине-
соединений обычно являются гетеродесми-
ческими. Характеризовать межатом-
межатомные расстояния в таких структурах
приходится по крайней мере двумя
величинами — ковалентными и Ван-
дер-Ваальсовыми «радиусами». Тер-
Термин «радиус» в геометрическом смыс-
смысле не может быть оправдан для ко-
валентной связи и употребляется
лишь по аналогии с металлическими
или ионными радиусами. Под этим
термином подразумевается та доля
в межатомном расстоянии, которая
приходится на тот или иной элемент,
атомы которого связаны ковалентны-
ковалентными связями с другими атомами. Сам
же атом в этом случае теряет форму
шара.
В табл. 52 собраны величины Ван-
дер-Ваальсовых и ковалентных «ра-
«радиусов» для всех неметаллических
элементов. С увеличением кратности
связи уменьшается величина «ра-
«радиуса». Точность значений порядка
0,02-0,05 А.
Простое сопоставление Ван-дер-
Ваальсовых радиусов (табл. 44) с
ионными радиусами анионов (табл.
11, стр. 138) указывают на их боль-
большую близость. Разница обычно не
превышает величину 0,02—0,03 А
(исключением являются расчетные
радиусы реально не существующих
анионов, например С4~). Л. Полинг
обратил внимание также на то, что
Ван-дер-Ваальсов радиус обычно
превышает ковалентный радиус то-
того же элемента при ординарной связи
на величину порядка 0,8 А. Обоими
этими правилами можно пользовать-
пользоваться для получения приблизительных
значений радиусов для тех элемен-
элементов, для которых отсутствуют соот-
соответствующие экспериментальные ве-
величины.
Говоря же о всей системе Ван-дер-
Ваальсовых радиусов в целом, надо
иметь в виду, что она выводилась,
исходя из наблюдающихся мини-
минимальных межатомных расстояний.
Однако если молекулы достаточно
сложной формы, то они не могут
упаковаться в кристалле так, чтобы
все расстояния между соседними мо-
молекулами оказались бы равновесны-*
ми — часть из них будет завышена,
а часть соответственно занижена,
так как атомы не представляют со-
собой несжимаемые сферы. По этой
причине «кристаллические» Ван-дер-
Ваальсовы радиусы имеют не-
несколько заниженные значения по
сравнению с истинными значениями,
о которых мы, к сожалению, знаем
пока еще очень мало, за исключени-
исключением Ван-дер-Ваальсовых радиусов
благородных газов.
По мере накопления эксперимен-
экспериментальных значений межмолекулярных
расстояний и их анализа удалось
установить довольно резкую анизо-
анизотропию Ван-дер-Ваальсовых разме-
размеров атомов. Межмолекулярный «ра-
«радиус» в направлении продолжения
ковалентной связи значительно мень-
меньше, чем в перпендикулярном направ-
354
лении. Угловая зависимость для ато-
атома углерода по Достровскому, Юзу
и Ингольду A946 г.) выражается
формулой
где ¦& — угол между определяемым
радиусом До и направлением кова-
лентной связи. Это значит, что при
максимальном значении Rmax = 1,9
(в направлении, перпендикулярном
к ковалентной связи) на протяжении
связи, т. е. при ¦& = 180°, его величи-
величина едва достигнет значения 1,1, а под
Z45 или 135° —1,5 А.
Чем больше величина электроотри-
электроотрицательности атома (т. е. чем боль-
большая доля ионности в связи), тем ани-
анизотропия делается меньше. Коэффи-
Коэффициент 0,4 в вышеприведенной форму-
формуле для углерода заменяется на 0,2
для атомов галогенов. Хорошей ил-
ТАБЛИЦА 52
Величины ковалентных и Ван-дер-Ваальсовых
элементов (в А)
В
0,89
0,80
С
0,77
0,72
0,69
0,61
1,7—1,9
Si
1,17
Ge
1,22
Sn
1,40
Pb
1,46
N
0,70
0,66
0,63
0,55
1,57*
P
1,10
1,9
As
1,21
2,0
Sb
1,41
2,2
Bi
1,51
1,69
О
0,66
0,61
0,59
1,38
S
1,04
0,94
1,8
Se
1,17
2,0
Те
1,37
2,2
H
0,28
1,17*
F
0,64
0,35
Cl
0,99
1,80
Br
1,14
1,95
J
1,33
2,15
«радиусов» неметаллических
He
1,22
Ne
1,60
Ar
1,92
Kr
1,98
Xe
2,18
Связь
Ординарная
Межмолекулярная
Ординарная
Полуторная
Двойная
Тройная
Межмолекуляриая
Ординарная
Двойная
Межмолекулярная
Ординарная
Межмолекулярная
Ординарная
Межмолекулярная
Ординарная
Межмолекулярная
По данным А. И. Китайгородского.
23*
355
люстрацией анизотропии межмолеку-
межмолекулярных размеров атома иода в иод-
содержащих молекулярных кристал-
кристаллах может служить работа Т. Л. Хо-
цяновой, А. И. Китайгородского и
Ю. Г. Стручкова A953 г.). Размер
атома иода связи оказался на 0,5 А
меньше, чем в перпендикулярном
направлении.
§ 2. Предварительные замечания
о молекулярных структурах
Структуры простых веществ элемен-
элементов-органогенов, т. е. элементов,
стоящих в таблице Д. И. Менделеева
(стр. 267) справа сверху от диаго-
диагональной границы, являются, главным
образом, молекулярными. Таковы же
структуры и у большинства соедине-
соединений этих элементов друг с другом.
Молекулярные соединения характе-
характеризуются тем, что между атомами в
молекулах действуют ковалентные
связи, а между молекулами — оста-
остаточные. Поэтому каждую молеку-
молекулярную структуру необходимо ха-
характеризовать двумя системами ве-
величин — внутримолекулярными (ко-
валентными) расстояниями и меж-
межмолекулярными (Ван-дер-Ваальсовы-
ми). Остаточные силы являются си-
силами ненаправленными, и поэтому
молекулы стремятся упаковаться в
структурах плотнейшим образом.
Если молекулы «одноатомны», как у
благородных газов, или вращаются,
как в кристаллической структуре во-
водорода или у высокотемпературной
модификации азота, то структуры
получаются в виде идеальных плот-
нейших шаровых упаковок. Если же
молекулы малосимметричны, то и
структуры обычно имеют низкую
симметрию.
Знание формы молекул и меж-
межатомных расстояний в них для хи-
химии более важно, чем знание упако-
упаковок молекул в кристалле. Поэтому
при работе с молекулярными соеди-
соединениями приходится пользоваться
данными о строении молекул, полу-
полученными не только рентгенострук-
турным методом, но и другими ме-
методами (спектральным, электроно-
графическим и др.), тем более, что
определение структур молекулярных
соединений часто затруднено тем,
что при обычных условиях эти сое-
соединения газообразны или жидки. Ра-
Работа же при низких температурах
представляет известные эксперимен-
экспериментальные трудности. При этом надо
помнить, что если молекулы сущест-
существуют в парах, расплаве или раство-
растворах, то это еще не значит, что они
обязательно присутствуют и в кри-
кристаллах. Так, например, в кристал-
кристаллах NaCl и NaBr никаких молекул
нет. Межатомные расстояния в струк-
структурах этих веществ B,81 и 2,98 со-
соответственно) сильно отличаются от
соответствующих межатомных рас-
расстояний в молекулах паров B,51 и
2,64).
Конфигурация молекул в газооб-
газообразном и кристаллическом состояни-
состояниях также может быть весьма различ-
различной. Так, например, для серы в парах
характерны линейные молекулы S2
с расстояниями 1,92, тогда как в кри-
кристаллах имеются кольчатые молеку-
молекулы Se с расстояниями 2,10. Триго-
нально-пирамидальная молекула РС15
в парах имеет расстояния Р — С1 2,10
и 2,25. В кристаллах же существуют
тетраэдрические катионы [РС14]+ и
октаэдрические анионы [РС1в]~.
Даже в тех случаях, когда форма
молекулы не меняется с изменением
агрегатного состояния вещества, меж-
межатомные расстояния, определенные
разными методами, получаются не-
несколько отличными. Объясняется это,
в частности, также и тем, что опре-
определение ведется при разных темпера-
температурах. Для брома и иода в парах най-
найдены межатомные расстояния 2,28 и
2,66 соответственно. Те же расстоя-
расстояния, определенные в кристаллах, ока-
оказались равными 2,27 и 2,70.
356
§8. Формы простейших молекул
и комплексных ионов
Двухатомные молекулы разных ве-
веществ могут отличаться друг от дру-
друга только величиной атомов, т. е.
межатомными расстояниями.
Трехатомные же молекулы могут
быть или линейными, например СОг,
или изогнутыми (уголковыми), на-
например F2O, СЬО, Н2О, H2S и др.
Четырехатомные молекулы XY3
могут быть плоскими, например BF3,
ВС13 или пирамидальными — NH3,
РН3, РС13 и др.
Молекулы состава XY4 обычно тет-
раэдрические — СН4, СС14, SiCU и
т. д., а молекулы состава XYe — окта-
эдрические, например SF6, SeFe.
Молекулы XY5, как было указано
выше, в газообразном состоянии ча-
часто имеют форму тригональных дипи-
рамид состава PGI5, PFs и др. Однако
при кристаллизации молекулы распа-
распадаются на ионы: PCls — на PCL(+ и
РС16, РВг5 — на РВг4+ и Вг-.
Элементы-органогены очень часто
дают устойчивые многоатомные
ионы такой же формы, как вышеука-
вышеуказанные формы молекул. Так, напри-
например, широко распространены линей-
линейные ионы — ОН-, CN-, N3-, SCN- и
т. д.; изогнутые ионы — NH2~, N02~
и др.; плоские треугольные N03~,
СО32~ и др.; пирамидальные — ЭОз2"
1Ог~ и др.; тетраэдрические — NH4+,
SO42-, РО43-, СЮ4- и др. Эти и по-
подобные им комплексные ионы обыч-
обычно входят в качестве самостоятель-
самостоятельных структурных единиц в структу-
структуры тройных и более сложных неорга-
неорганических соединений. Таковы, в ча-
частности, соли кислородных кислот.
§ 4. Валентные углы
Атомы элементов Vl-b подгруппы
при образовании двух ковалентных
связей и атомы элементов V-b
подгруппы — трех ковалентных
связей с атомами элементов VII-6
подгруппы дают валентные углы,
приближающиеся к 90°. Однако от-
отталкивание валентно не связанных
между собой атомов может сущест-
существенно изменить это значение. Очевид-
Очевидно, отталкивание будет заметным, ес-
если размер атома Xv или Xvi будет
недостаточно большим для того, что-
чтобы при соблюдении валентного угла
90° воспрепятствовать атомам Yvh
касаться друг друга своими Ван-дер-
сание атомов Y происходит, то ва-
валентный угол нарушается. Изменение
валентного утла в зависимости от ве-
величин атомов, входящих в молекулу,
Ваальсовыми сферами. Если такое ка-
показано в табл. 53.
Т АБ Л ИЦА 53
Изменение валентного угла в зависимости
от величины
Соедине-
Соединение
НаО
H2S
HaSe
С12О
атомов
Расстоя-
Расстояние
Y-
0
1
1
-X
97
35
68
Расстоя-
Расстояние
Y-
1
1
2
-Y
,54
,99
,83
Величи-
Величина
2rY
2
2
2
3
34
34
34
62
Валент-
Валентный
угол в*
105
92
90
115
В тетраэдрических молекулах и
ионах, например в СЕЦ и NH4+, ва-
валентные углы равны 109,5°.
Интересно отметить, что валентные
углы у атомов разных элементов де-
деформируются приблизительно одина-
одинаково. Это впервые замечено автором
на структуре PtS (Г. Б. Бокий,
1952 г.). Несмотря на резкую разни-
разницу в электронном строении обоих эле-
элементов и на разницу пространствен-
пространственной конфигурации (тетраэдрической
у серы и квадратной у платины) и
величин валентных углов, деформа-
деформация их приблизительно одинакова.
Тем более вероятна одинаковая спо-
способность к деформации валентных уг-
углов у элементов-органогенов. Этим
объясняется приблизительное сохра-
сохранение угла в 109° между одинарны-
одинарными связями у углерода в алифатиче-
алифатических соединениях.
357
Между простой и двойной связями
валентный угол равен 120° как в али-
алифатических, так и в ароматических
соединениях. Между двумя двойны-
двойными связями угол равен 180°, т. е. мо-
молекула СОг линейна. Такой же угол
между простой и тройной связями,
например, в производных ацетилена.
С помощью таблиц ковалентных и
Ван-дер-Ваальсовых «радиусов» и ва-
валентных углов можно достаточно
точно строить модели молекул. При
этом построении надо только учиты-
учитывать влияние валентно не связанных
между собой атомов.
§ 5. Классификация
молекулярных структур
Многие не очень сложные молекуляр-
молекулярные структуры отличаются той осо-
особенностью, что молекулы в них рас-
располагаются по точкам одной правиль-
правильной системы. Этот эксперименталь-
экспериментальный факт можно положить в основу
классификации. В первом приближе-
приближении молекулу (для конкретности —
центр тяжести молекулы) можно при-
принять за точку. Так как в некоторых
простейших правильных системах
точки располагаются по узлам реше-
решеток, то мы получаем в этом случае
структурные типы, сходные со
структурными типами простых ве-
веществ. На этой стадии классифици-
классифицирования мы, следовательно, еще
отказываемся от разграничения типов
структур по форме и по симметрии
молекул или по числу атомов, входя-
входящих в молекулу.
Для структур, у которых доказано
вращение молекул в кристаллах или
в силу специфических особенностей
метода рентгеноструктурного анализа
не определены положения легких ато-
атомов (главным образом, водорода),
первая стадия классификации будет
окончательной. Число групп, на кото-
которые разобьются все молекулярные
структуры, на этой стадии будет срав-
сравнительно невелико.
Перечислим главнейшие из них,
1. Центры тяжести молекул распо-
располагаются по точкам кубической плот-
нейшей упаковки (Р-НС1, H2S, СЩ,
СО2 и др.)
2. Молекулы располагаются по уз-
узлам кубической центрированной упа-
упаковки (SiF4).
3. Молекулы располагаются по ме-
местам атомов углерода в структуре ал-
алмаза (AS2O3, а-НгО при температуре
жидкого воздуха, и др.).
4. Молекулы располагаются по ме-
местам атомов Zn и S в структуре вюрт-
цита, гексагонального аналога струк-
структуры алмаза (р-НгО — обыкновен-
обыкновенный лед и др.).
5. Молекулы располагаются по ме-
местам гексагональной плотнейшей упа-
упаковки (Н2, N2, p-CO и др.).
Дальнейшее подразделение можно
сделать по симметрии кристаллов.
Так, например, тетрагональную
структуру HJ (с/а — 1,08) можно
рассматривать после кубической
структуры C-НС1; после тетрагональ-
тетрагональных структур можно рассматривать
ромбические; например а-НС1
(а:Ь:с = 0,94: 1:1,07), и т. д.
Вторая стадия классификации дол-
должна учесть действительный тип ре-
решетки Бравэ и федоровскую группу.
Так, например, в структуре СОг цент-
центры тяжести молекул совпадают с уз-
узлами кубической гранецентрирован-
ной решетки, но действительная ре-
решетка Бравэ этой структуры — при-
примитивная, федоровская группа Раб.
В структурах а-СО и NH3 центры тя-
тяжести молекул только приблизитель-
приблизительно совпадают с узлами гранецентри-
рованной решетки. Федоровская груп-
группа их P2i3. Только после разделения
по федоровским группам целесообраз-
целесообразно делить структуры по форме и по
симметрии молекул и по числу ато-
атомов в них. Эти факторы находят свое
отражение в структуре, в ее симмет-
симметрии, в принадлежности структуры к
той или иной федоровской простран-
пространственной группе.
358
§в. Применение принципов
плотней шей упаковки
к молекулярным кристаллам
Поскольку Ван-дер-Ваальсовы силы
являются ненаправленными силами,
то априори можно ожидать, что мо-
молекулы в кристалле будут стремить-
стремиться окружить себя максимальным чис-
числом соседних молекул, в результате
чего упаковка будет, вероятно, стре-
стремиться к плотнейшей. Тот факт, что
кристаллические структуры благород-
благородных газов, построенных из «одноатом-
«одноатомных молекул», являются плотнейши-
ми упаковками (у гелия гексагональ-
гексагональная, у остальных — кубическая), под-
подтверждает сказанное выше предложе-
предложение.
Первые попытки применения идей
плотной упаковки молекул в кристал-
кристаллах органических соединений принад-
принадлежат ученику Е. С. Федорова про-
профессору Б. П. Орелкину. В своей ра-
работе, посвященной кристаллической
структуре трифенилбензола A929—
1930 гг.), он показал, что молекулы в
кристалле упакованы весьма плотно
и что нельзя правильно передать
строение органического кристалла,
если считать каждый атом за сферу.
При упаковке молекул выпуклость
одной из них обязательно попадает
во впадину другой.
Эти идеи в последние годы были
широко развиты А. И. Китайгород-
Китайгородским. Он показал, что в подавляющем
числе структур органических веществ
молекулы имеют координационное
число 12, а в слое — 6 (рис. 338).
Рис. 338. Способ плотнейшей упаковки
молекул в органическом кристалле. Ко-
Координационное число в слое 6
Молекула считается входящей в
координационную сферу, если она
соприкасается с центральной молеку-
молекулой. Число точек соприкосновений с
каждой молекулой не имеет значения.
На рис. 338 центральная молекула
касается каждой из пяти молекул в
одной точке, а в одной, отмеченной
звездочкой,— в двух точках.
Определив форму и размер моле-
молекул, а также объем элементарной
ячейки, без труда можно вычислить
процент1 занимаемого ими простран-
ТАБЛИЦА 54
Таблица вероятных федоровских групп симметрии для кристаллов органических
соединений
Собственная
симметрия мо-
молекулы
Симметрия мо-
молекулы в кри-
кристалле
Федоровская
группа и крат-
кратность п-поло-
жения, зани-
занимаемого моле-
молекулой
1,2 т, mm,
222
1
ф. гр.
pi
P2i
Р2ф
Рса
Рпа
Р 2i2121
п
2,4
2,4
4
4
4
4
1,2/m, mmm
1
Ф. гр.
71
P2i/c
С 2/с
РЪса
п
1,2
2,4
4
4
mm
2
ф. гр.
С 2/с
Р 2i2i2
РЪпс
п
4
2,4
4
т
ф. гр.
Рте
Стс
Рпта
п
4
4
4
222
2
ф. гр.
С 2/с
Р 2i2i2
Pbcn
п
4
2,4
4
359
ства в ячейке кристалла. Эта величи-
величина колеблется от 60 до 80%. Если
вспомнить, что в плотнейшей шаро-
шаровой упаковке шарами заполнено
74,05%, то станет ясным, что прин-
принцип плотнейшей упаковки для моле-
молекулярных кристаллов, в общем, оп-
оправдывается.
Исходя из принципов плотнейшей
упаковки для фигур произвольной
формы и учтя собственную симмет-
симметрию этих фигур, А. И. Китайгород-
Китайгородский нашел вероятные федоровские
группы для кристаллов органических
соединений (табл. 54). Подсчет по-
показывает, что таких групп 12.
§7. Строение углеводородов
В качестве примера простейшего
углеводорода рассмотрим структуру
метана. На рис. 339 показана молеку-
молекула СШ. Высокая симметрия молеку-
молекулы обусловливает кубическую сим-
симметрию структуры метана. Федоров-
Федоровская группа РтпЪт, п = 4, а = 5,89
(при — 180° С). Центры молекул
расположены по узлам гранецентри-
рованной решетки. Расстояние С—Н
не определялось. По-видимому, моле-
молекулы в кристаллах вращаются.
Во всех алифатических соедине-
соединениях, например в парафинах, атомы
углерода располагаются по зигзаго-
зигзагообразной цепочке с расстоянием С—С,
приблизительно равным 1,54, и с
углом С—С—С, близким к тетраэдри-
ческому углу — ЮЭ'Д0. Форма цепоч-
цепочки показана на рис. 340, а проекция
ее вдоль оси — на рис. 341. Упаковы-
Упаковываются алифатические цепи в проек-
проекции вдоль оси цепи двумя способами
(рис. 342).
Детально был исследован парафин
СгэНбо. Ромбическая ячейка его име-
имеет размеры: а = 7,45; Ъ = 4,95 и с =
= 77,2. Федоровская группа Рптщ.
В 1951 г. Б. К. Вайнштейн и
3. Г. Пинскер электронографическим
методом определили структуру пара-
парафина: а = 7,41, Ъ = 4,96 и подпериод
с'=2,54. Расстояние С—С в молекуле
Рис. 339. Молекула метана СН4
Рис. 340. Форма цепочки из атомов угле-
углерода в алифатических соединениях
Рис. 341. Проекции цепочки алифатиче-
алифатического соединения вдоль ее оси
Рис. 342. Упаковка алифатических цепо-
цепочек в проекции вдоль оси цепочки (нуля-
(нулями и единицами обозначены различные
уровни)
а — винтовая ось направлена вдоль короткого
периода ячейки;
б — винтовая ось направлена вдоль длинного
периода ячейки
Рис. 343. Молекула адамантана
360
оказалось равным 1,52, С — Н —
— 1,17, углы С — С — С - 110° и Н —
— С — Н — 105°. Расстояние С — С
между молекулами 4,18 и 4,20. Крат-
Кратчайшее расстояние Н—Н равно 2,49.
Молекулы алифатических соедине-
соединений совершают тепловые колебания
вокруг оси с. Вблизи температур пла-
плавления осуществляется вращение мо-
молекул. Тогда симметрия кристаллов
вместо ромбической становится гекса-
гексагональной.
Из алищгклических соединений
остановимся на структуре адаманта-
на СюШб, структурная формула кото-
которого не могла быть установлена хи-
химическими методами. Высокая сим-
симметрия кристаллов, как выяснилось,
являющаяся следствием высокой
симметрии молекул, позволила опре-
определить структуру на сравнительно
ранней стадии ретгеноструктурного
анализа. Кубическая ячейка имеет
а = 9,426, п = 4и федоровскую груп-
группу F43/ra. Ниже показана формула
молекулы адамантана:
сн.
Ч
На рис. 343 представлена молекула
адамантана в пространстве; на рис.
344 его кристаллическая структура
без атомов водорода. Расстояния С—С
равны 1,54. Определение структуры
позволило дать адамантану рацио-
рациональное название — трициклодекан.
Структуры ароматических соедине-
соединений изучены лучше, чем других клас-
классов органических соединений, а
структура самого бензола определена
только в 1954 г. Коксом и Смитом.
Правда, из прежних работ были изве-
известны параметры ячейки, федоровская
группа и та особенность этой струк-
структуры, что молекулы находятся в цен-
центрах симметрии. Последнее обстоя-
обстоятельство являлось доказательством
равноценности связей между всеми
атомами углерода. На рис. 345 пока-
показана проекция структуры бензола, на
рис. 346 — его молекула. Геометриче-
Геометрические константы этой структуры
при —3°С следующие: а=7,460, Ъ =
—9,666, с=7,034 А, федоровская
группа РЪса. Расстояния Ci — Сг =
=4,377; Са—Сз-1,382; С3-С4 =
= 1,374; среднее 1,378. Углы
C6CiC2 = 119°28/; С,С2Сз = 120°49';
С2СзС4=г119°42/. Все атомы углерода
лежат в одной плоскости. Неполное
совпадение расстояний и углов мо-
может объясняться эффектом, завися-
зависящим от взаимодействия молекул в
кристалле, а также ошибками экспе-
эксперимента. Расстояния С — Н не опре-
определялись.
Ранее были определены структуры
дифенила, дифенилбензола, п,п'-бис-
(фенил) дифенила и симметричного
трифенилбензола. Во всех случаях
наблюдалась плоская форма молекул
с расстояниями С — С в кольце
1,39—1,42 и между кольцами 1,47—
1,48.
Если сопоставить расстояния меж-
между двумя углеродными атомами, сое-
соединяющими бензольные кольца в мо-
молекулах дибензила, стильбена и тола-
на, то получим значения: С—С 1,58;
С = С 1,33 и С — С 1,19.
Рис. 344. Структура адамантана (без ато-
атомов водорода)
361
Рис. 345. Проекция структуры бензола
Рис. 346. Молекула бензола
Рис. 347. Длины связей С—С в молекуле
нафталина
Рис. 348. Длины связей С—С в молекуле
антрацена
с-н.-сн.
/ Z Ъ
Рис. 349. Структурная формула ди-га-кси-
лилена (а) и деформация бензольного яд-
ядра в ней (б)
Структуры нафталина и антрацена
были до конца определены Робертсо-
ном. Длины связей С — С в этих мо-
молекулах показаны на рис. 347 и 348.
Во всех структурах ароматических
конденсированных соединений также
констатирована плоскостная форма
молекул. Например, структуры хри-
зена, пирена, трифенилева, 1,2,5,6-
дибензантрацена, бензперилена, коро-
нена, овалена и других соединений.
Исключением является молекула ди-
/г-ксилилена, структурная формула
которой определена по результатам
рентгеноструктурного анализа.
Боковая проекция молекулы ди-п-
ксилилена показана на рис. 349.
Стремление атомов углерода 1—7—
7' и 4—8—8', связанных оди-
одинарными связями, расположиться под
тетраэдрическим утлом в ~109°, де-
деформирует бензольное кольцо так,
что атомы углерода 1,4 оказываются
смещенными из плоскости бензольно-
бензольного кольца. Такие молекулы называ-
называются пространственнонапряженными.
§8. Строение более сложных
органических соединенна
1. Органические соединения с гете-
роатомами. Органические соедине-
соединения, содержащие кроме С и Н галоге-
галогены, кислород, азот и другие элемен-
элементы-органогены, построены в основном
так же, как и углеводороды.
Так, например, алифатические
кислоты как жирные, так и амино-
аминокислоты, спирты, кетоны и другие
вещества, имеющие длинные цепи,
построены из молекул зигзагообраз-
зигзагообразной формы. Эти молекулы упаковы-
упаковываются в кристаллах так, что их
длинные оси располагаются парал-
параллельно друг другу. В этом отноше-
отношении их структуры будут похожи на
структуры парафинов, рассмотрен-
рассмотренных в предыдущем параграфе.
Размер атомов, замещающих водород
в углеводородах, играет существен-
существенную роль при упаковке молекул бо-
более сложных по составу веществ.
362
Рис. 350. Деформация атомом хлора од-
одного из атомов водорода в структуре 1,5-
дихлорантрацена
oTTttll
°c
Рис. 351. Структура дигидрата щавелевой
кислоты (СООНJ-2Н2О
Рис. 352. Распределение водородных свя-
связей в структуре щавелевой кислоты
Это особенно существенно для С1-,
Вг-, J-производных, так как размеры
атомов этих элементов значительно
больше, чем у других элементов-ор-
элементов-органогенов. Сравним, например, фор-
мы молекул нафталина и антрацена
(рис. 347 и 348) и 1,5-дихлорнафта-
лина и 1,5-дихлорантрацена (рис. 357
и 358), структуры которых были оп-
определены А. И. Китайгородским и
С. С. Кабалкиной. Если построить
проекцию молекулы 1,5-дихлорантра-
1,5-дихлорантрацена (рис. 350), то видно, что атом
С1, не помещаясь между атомами во-
водорода, сильно деформирует один из
них. В результате формы молекул
дихлорнафталина и дихлорантрацена
отличаются от форм молекул наф-
нафталина и антрацена (рис. 358, ж).
Вторым фактором, накладываю-
накладывающим особый отпечаток на кристалли-
кристаллические структуры органических ве-
веществ, является электроотрицатель-
электроотрицательность. Если в составе вещества име-
имеются ионы гудроксила или молекулы
воды, а также наиболее электроотри-
цательпые элементы — F, О и N, то,
как правило, в структуре образуется
система водородных связей. Водород-
Водородные же связи сильно изменяют физи-
физико-химические свойства веществ
(см. стр. 249). В качестве при-
примера рассмотрим структуру ди-
дигидрата щавелевой кислоты (рис.
351) и распределение в ней
водородных связей (рис.
352). Аналогично распределение во-
водородных связей и в структуре ди-
дигидрата диацетилендикарбоновой кис
лоты НООС-С = С-С = С-СООН-
•2Н2О (рис.353).
2. Структуры, в которых определе-
определено положение атомов водорода. К со-
сожалению, фиксация положения ато-
атомов водорода методом рентгенострук-
турного анализа даже в молеку-
молекулах органических соединений почти
невозможна. Теоретические подсче-
подсчеты, подтвержденные эксперимен-
экспериментально, показывают, что отношение
значений электронной плотности в
центре атома водорода и углерода
при построении трехмерных рядов
Фурье рн/ро ~ 'Аз, т. е. пики от
атомов водорода приблизительно в
13 раз меньше, чем пики от атомов
углерода. Этим и объясняются труд-
трудности обнаружения водорода на кар-
363
Рис. 353. Распределение водородных свя-
связей в структуре дигидрата диацетиленди-
карбоновой кислоты (С2СООНJ-2Н2О
тах (или в объемах) электронной
плотности. Точность определения
положения водорода в кристалличе-
кристаллических структурах органических соеди-
соединений методом рентгеноструктурно-
го анализа поэтому не превышает
0,1 А. При использовании методов
структурной электронографии
Фн/фс~ 1/4,5, что позволяет точнее
определять положение легких атомов
в присутствии тяжелых.
Это обстоятельство удачно ис-
использовано Б. К. Вайнштейном
A954 г.) при определении положе-
положения атомов водорода в структуре ди-
кетошшеразпна. Структура этого ве-
вещества представляет интерес в свя-
связи с важной ролью дикетопиперази-
новых группировок в строении бел-
белка. С кристаллохимической точки
зрения она интересна тем, что в ней
имеются атомы водорода как обра-
образующие, так и не образующие водо-
водородные связи.
Структура дикетоштеразина была
определена рентгеновским методом
еще в 1938 г. Р. Б. Кори, который
но характеру расположения молекул
в цепях и сокращению межатомного
расстояния N...0 из соседних моле-
молекул констатировал наличие водо-
водородных связей:
СН2 2'.s5 СН2
CO/4NH OC/\NH
CO HN4/CO
СН2 СН2
Опуская все методические тонко-
ти, которые пришлось преодолеть
Б. К. Вайнщтейну для выявления
положения атомов водорода в струк-
структуре, приведем сейчас итоговый
трехмерный синтез Фурье потенциа-
потенциала молекулы дикетопиперазина
(рис. 354). На рисунке сплошные
линии проведены через 15в, пунк-
пунктирные — через 7,5 в, первая сплош-
сплошная линия — через 15 в.
Обращает на себя внимание тот
факт, что при правильной форме пи-
пиков от тяжелых атомов, пики от ато-
атомов водорода вытянуты, что, видимо,
следует связать с тепловым движе-
движением. Те атомы водорода, которые
не участвуют в водородных связях,
совершают тепловые колебания во-
вокруг атома углерода как центра по
какому-то участку сферы. Атомы же
водорода, участвующие в водородной
связи, совершают тепловые колебания
в основном по линии связи и имеют
меньшую амплитуду колебания в
перпендикулярном направлении.
В молекуле дикетопиперазина рас-
расстояния С—Н, равные 1,09 и 1,11, и
N—Н, равное 0,98, хорошо совпадают
со стандартными значениями 1,09 и
1,00 А соответственно.
3. Строение органических соедине-
соединений с немолекулярными структурами.
До сих пор говорилось о молекуляр-
молекулярных структурах, которыми действи-
действительно характеризуется большинство
органических соединений. Однако,
как выяснилось в последнее время,
универсальность молекулярных пред-
представлений не оправдывается даже
364
1 A
Рис. 354. Трехмерный синтез Фурье по-
потенциала молекулы дикетошшеразина
для этой области химии. Существуют
целые группы органических веществ,
у которых атомы, соединяясь друг с
другом, образуют бесконечные цепи.
По-видимому, такое строение явля-
является характерным для всех волокни-
волокнистых веществ, как искусственных,
так и естественных, в частности,
шелка, каротина и др. Простейшим
прообразом всех структур такого ро-
рода является полиэтилен (политен),
структура которого
СНа СН2 СН2 СН2
СН2 СН2 СН2 СН2 СН2....
аналогична парафинам, в которой бес-
бесконечные СН2-цепи вытянуты вдоль
осп с. Политен кристаллизуется в
ромбической сингонии (а = 7,40, в =
= 4,93 и с = 2,534). Федоровская
группа Рпат, п=4 (СН2).
Существуют структуры органиче-
органических соединений с бесконечными
слоями и с бесконечными трехмер-
трехмерными каркасами, в которых атомы
связаны ковалентными силами. Та-
Такие структуры в этом смысле анало-
аналогичны структурам силикатов, о кото-
которых говорилось в предыдущей главе.
Не подлежит сомнению, что органи-
органических соединений немолекулярного
строения очень много. Широкое ис-
исследование их, по-видимому, задер-
задерживалось из-за сложности объектов
и необходимости применения иных
методов, кроме тех, которые обычно
использует препаративная органиче-
органическая химия.
§0. Ионные структуры соединений
элементов-органогенив
Выше говорилось о том, что в случае
образования элементами-органогена-
элементами-органогенами комплексных ионов их тройные
соединения становятся весьма сход-
сходными с бинарными солями. Так, на-
например, NH4CI по своим свойствам
весьма сходен с КС1 и выше 184 °С
принадлежит к тому же структурно-
структурному типу.
Такие комплексные ионы харак-
характерны и для других элементов ор-
органогенов. Выше мы говорили о
структурах РС15, РВг5, которые
также являются ионными:
[РС14]+[РС16]- ж [РВг4]+Вг-. Кри-
Кристаллическая структура N2O5 по-
построена из линейного катиона NG+ и
треугольного аниона NGy.
N(CH3LC1O4 имеет структуру типа
NaCl- [N(CH3L] + [C1O4]-.
Многие органические соединения,
которым обычно приписывается мо-
молекулярное строение, в действитель-
действительности построены из ионов. Так, на-
например, структурная формула гидро-
гидрохлорида анилина обычно изобража-
изображается так: / ^NH2-HC1- Такое изоб-
изображение заставляет предполагать,
365
Рис. 355. Структура «-ка-
прината калия построе-
построена из анионов [СН3 (СНг) е-
•СОг]~ и катионов К+
СН1
сн
СН
Vc
СН LH2 D D
СН |
СН
I
csinfi
Рис. 356. Проекции
структуры пенициллина
что в структуре этого вещества су-
существуют молекулы / \nh2 и
НС1. Однако это не так. Структура
построена из катионов [¦'^NHsl*
и анионов С1~. Аналогичное строе-
строение имеют и все Na- или К-соли
органических кислот. Так, например,
к-капринат калия СНз
О—К
в действительности построен из анио-
анионов [СНз(СН2)8СО2]- и катионов К+
(рис. 355). Ион К+ окружен шестью
атомами кислорода. Расстояния С—О
в карбоксильной группе одинаковы,
и, следовательно, связи полуторные.
Тот же случай встречается в
структуре бензилпенициллида калия,
из которой было определено строе-
строение пенициллина. Структурная фор-
формула пенициллина, как известно, не
могла быть однозначно установлена
чисто химическими методами, не-
несмотря на то, что над ее определе-
определением работали очень крупные хими-
химики, затратившие много усилий и
времени. На рис. 356 показаны
две проекции аниона пенициллина
(Д. Хочкин с сотрудниками, 1952 г.).
§ 10. Зависимость характера связи
от межатомных расстояний
Помимо решения вопроса о струк-
структурных формулах органических сое-
соединений, рентгеноструктурный ана-
анализ может дать сведения о природе
химической связи и взаимном влия-
влиянии атомов.
Известно, что каждому типу связи
приблизительно соответствует опре-
определенное межатомное расстояние.
Так, если в большом числе структур
алифатических соединений измерять
расстояние С—С, то всегда будем
получать величину, близкую к 1,54.
Если же простая связь будет нахо-
находиться между кратными, то величи-
величина ее может значительно умень-
уменьшиться за счет уменьшения кова-
лентного радиуса в состоянии
s/^-гибридизации @,74 А) по сравне-
сравнению с s/ьгибридизацией @,77А) в
алифатических соединениях. Допол-
Дополнительное укорочение ординарной
связи происходит в таких системах
за счет сопряжения — перераспреде-
перераспределения электронной плотности, приво-
приводящего к частичному выравниванию
двойных и ординарных связей. В аро-
ароматических соединениях в результате
сочетания обоих этих эффектов
длина простой связи обычно не
превышает величины 1,45—1,46.
Чрезвычайно резкое укорочение про-
простой связи наблюдалось в структуре
дигидрата диацетилендикарбоновой
кислоты, где расстояние между угле-
углеродными атомами типа =С—С=
равно 1,33. Оно даже меньше обыч-
обычного расстояния С=С«1,35. Этот
пример показывает, что длина связи
не всегда является однозначной ха-
характеристикой ее формальной крат-
кратности. Конечно, и свойства связи су-
существенно изменяются, если сохраня-
сохраняется ее кратность, но резко изменится
длина. В некоторых случаях рав-
равным по длине связям следует припи-
приписывать разную кратность, например,
межатомному расстоянию ~ 1,4 А мо-
может отвечать как двойная, так и про-
простая связи; в отдельных случаях
можно предположить, что меньшей
по длине связи будет соответствовать
меньшая кратность. Это, конечно,
исключительные случаи. В общем же
случае, при прочих равных условиях,
более коротким межатомным расстоя-
расстояниям отвечает большая кратность
связи.
Для согласования всего имеющего-
имеющегося экспериментального материала по
межатомным расстояниям молекул
органических соединений с теорией
строения Бутлерова необходимо сде-
сделать одно неоднократно высказывав-
высказывавшееся ранее допущение о возможно-
возможности существования в ароматических
соединениях не только одно- и двух-
двухкратных связей, но и выравненных
промежуточных, в частности полутор-
полуторных, обозначенных в нижеследую-
367
щих схемах знаком-. Выравненные
промежуточные связи обычно обоз-
обозначаются пунктиром. Постараемся
показать взаимное влияние атомов и
возможность изображения молекулы
одной структурной формулой. По-
Последнее, конечно, возможно только
в том случае, если имеются экспери-
экспериментальные данные по межатомным
расстояниям, определенные с прием-
приемлемой точностью. Для изображения
структурных формул ароматических
соединений достаточно использовать
три типа связи: простые, двойные и
полуторные. При прочих равных ус-
условиях атомы, связанные этой пос-
последней связью, очевидно, располага-
располагаются на расстояниях, промежуточ-
промежуточных между расстояниями, получаю-
получающимися при простой и двойной
связях.
Гипотеза Кекуле предопределяла
существование в ароматических сое-
соединениях углеродных атомов только
одного типа атома углерода \С = .
Назовем его типом А. Высказанная
выше гипотеза предусматривает на-
наряду с существованием атомов типа
А существование второго типа ато-
атомов 5>С—. Назовем его типом Б.
При проверке опытного материала
будем базироваться на идее сохра-
сохранения определенной симметрии
молекул.
Изучение симметрии сложных
ионов и молекул комплексных соеди-
соединений позволило сделать вывод, что
при вхождении в кристалл много-
многоатомные структурные единицы те-
теряют свою симметрию в определен-
определенной последовательности: легче всего
теряются оси высоких порядков, за-
затем плоскости и в последнюю оче-
очередь — центр симметрии. Если он
есть в комплексном ионе или моле-
молекуле, то он обычно сохраняется и
при вхождении иона в кристалл. Тем
более это правило должно сохранить
свое значение для органических сое-
соединений, так как межмолекулярные
силы (из-за которых, главным обра-
образом, и происходит потеря симметрии
.368
молекулами при вхождении их в
кристаллы) в органических соедине-
соединениях значительно слабее, чем в не-
неорганических.
Рассмотрим с позиций всего вы-
вышесказанного структуру молекулы
нафталина. Долгое время для нее
предлагались две «равноценные»
структурные формулы а и б с разной
локализацией двойных связей (рис.
357). В последние годы, правда, ста-
стали приводить для нафталина симмет-
симметричную структурную формулу на ос-
основании косвенных соображений.
Однако межатомные расстояния и
наличие центра симметрии в молеку-
молекуле нафталина (в) указывают на пре-
преимущество структурной формулы б
перед а. Следует, однако, отметить,
что разница в расстояниях ...С = С... =
= 1,37 ± 0,02 А и ...С-С... = 1,41 ±
±0,02 А очень мала, что согласуется
с химическим поведением нафталина
как ароматической системы. Цент-
Центральная двойная связь несколько
длиннее среднего значения для
двойной связи, краевые ординар-
ординарные — несколько короче обычного.
Этим определяется близость их вели-
величин.
Структура молекулы 1,5-дихлор-
нафталина (рис. 357, г) подтвержда-
подтверждает, что локализация двойных связей
в ней выражена ярче, чем в незаме-
незамещенной молекуле.
Непреодолимые трудности встре-
встречаются при попытках распределить
U39 У 39 1,37
Рис. 357. Структурные формулы и меж-
межатомные расстояния в нафталине (а — в)
и 1,5-дихлорнафталине (г)
Рис. 358. Структурные формулы и меж-
межатомные расстояния в антрацене и его
производных
двойные связи в молекуле антраце-
антрацена, если пользоваться формулой Ке-
куле для строения бензольного ядра.
Ни один из вариантов а и б
(рис. 358) не может быть согласован
с существованием у молекулы антра-
антрацена центра симметрии (г), на кото-
который определенно указывают рентге-
ноструктурные исследования Синкле-
Синклера, Робертсона и Матьисона. Однако
те же данные хорошо согласуются со
структурной формулой д, построен-
построенной из атомов углерода типа Б. Рас-
Расстояния между атомами, связанными
полуторной связью, равны 1,39 ±
± 0,03 А. Замещение двух атомов во-
водорода на атомы хлора в положени-
положениях 1,5 сохраняет центр симметрии в
молекуле ж, но приводит к перегруп-
перегруппировке связей (з). Увеличение
двух расстояний С—С (ж) до 1,45 А
в центральном шестиугольнике свя-
связано с отталкиванием атомов водо-
водорода от атомов хлора.
Иная перегруппировка связей в
молекуле антрацена (рис. 358) на-
наблюдается при замещении двух поло-
положений — 9 и 10 — кислородом. Меж-
Межатомные расстояния в молекуле ан-
трахинона, по данным Сена, показа-
показаны на рис. в. Распределение связей
показано на рис. е. Здесь атомы кис-
кислорода связаны двойными связями.
Расстояние ...С = О... равно 1,15 А.
В карбоксильных же группах связи
кислорода с углеродом одинаковы, и,
очевидно, они полуторные, что и
следовало бы показать на структур-
структурной формуле соответствующим зна-
0
ком R—С<< . Расстояние С -г- О
О
равно 1,25 А. При простой связи рас-
расстояние С—О возрастает до 1,4 А и
выше.
Возвращаясь к структурам антра-
антрацена и его производным, можно от-
отметить, что в зависимости от типа и
положения заместителя число угле-
углеродных атомов типа А и Б может су-
существенно меняться. Так, в антраце-
антрацене все атомы углерода типа В (см. д).
В 1,5-дихлорантрацене 8 атомов уг-
углерода типа А и 6 атомов типа Б.
В антрахиноне 12 атомов углерода
типа Б и только 2 — типа А. Для
этой молекулы можно было бы пред-
предложить распределение связей по Ке-
куле (см. и), согласовав его с псев-
псевдосимметрией молекулы. Однако та-
24 Кристаллохимия
369
кое распределение связей будет на-
находиться в резком противоречии с
межатомными расстояниями.
Из всего сказанного следует, что
частичная локализация двойных свя-
связей в бензольных кольцах ароматиче-
ароматических соединений не является строго
постоянной. Под влиянием различ-
различных заместителей валентные элект-
электроны могут перегруппировываться,
что приводит к иной локализации
л-связей или к полной их делокали-
зации по отдельным бензольным
кольцам (з, е) или по всей молекуле
(д).
Из всего сказанного в этом пара-
параграфе и в § 7 следует, что формула
самого бензола должна быть написа-
написана так, как это показано на
рис. 359, а. Весьма многочисленные
химические данные, а также и кри-
сталлохимические, подтверждают
Рис. 359. Структурная формула беязопа
равноценность всех связей в моле-
молекуле бензола. Из этого, однако, не
следует, что обязательно у всех без
исключения производных бензола
также будут равноценны все связи.
На примере антрацена мы видели,
что от характера заместителей зави-
зависит то или иное расположение свя-
связей в антраценовом ядре, поэтому
можно ожидать, что в каких-то слу-
случаях бензольное ядро примет форму
б. В одном случае такой тип бензоль-
бензольного ядра, по-видимому, обнаружен.
Имеется в виду структура 2,2'-ди-
хлорбензидина
(Д. Л. Смейр, 1948 г.). Межатомные
расстояния показаны на рис. 360.
Разница в расстояниях С = С A,29;
/,53
ч
\
Cl
US'
иэ°
f
118°
<f
Cl
щ
\/гз°
щ
ЦЧ).
г is)
iZ9
V
ПО
°/
k
Рис. 360. Межатомные расстояния в струк-
структуре 2,2'-дихлорбензидина
1,30 и 1,32) и С-С A,43 1,45 и
1,46) значительно превосходит ошиб-
ошибку измерения @,3). Было бы важно
подтвердить (или опровергнуть) дан-
данные для этой принципиальной струк-
структуры, используя современную мето-
методику.
§11. Пространственные
затруднения в молекулах.
Конформации
Форма молекулы не всегда бывает
идеальной. Даже длины связей меж-
между одноименными атомами при той
же кратности не остаются одинако-
одинаковыми, например, в сопряженных си-
системах (см. § 10).
Но и при сохранении длин связей
молекула может иметь различные
формы — конформации — за счет по-
поворотов отдельных ее частей вокруг
ординарных связей, отклонений свя-
связи от плоскости бензольного ядра и
изменений величин валентных углов.
В § 7 мы рассмотрели деформацию
молекулы в результате уравновеши-
уравновешивания валентных усилий, приводя-
приводящих, в частности, к изменению иде-
идеальных валентных углов. В этом па-
параграфе мы рассмотрим главным об-
образом конформации, вызванные от-
отталкиванием валентно не связанных
между собой атомов.
Всякая «перегрузка» молекулы пз-
за сильного отталкивания валентно
не связанных атомов приводит к по-
повышению потенциальной энергии мо-
молекулы. Однако перегрузка не всегда
приводит к тем конформационным
370
искажениям (по сравнению с идеаль-
идеальными моделями), о которых мы го-
говорили выше. Так, например, в тет-
раэдрических молекулах четырех-
хлористого углерода CCU хлороформа
СНС13 и хлористого метилена СЩСЪ
расстояния С1 —С1 порядка 2,9, тог-
тогда как равновесное расстояние по-
порядка 3,6. Форма же молекул при
этом сохраняется почти идеально
тетраэдрической. Самые точные из-
измерения показывают увеличения
тетраэдрического угла С1—С—С1 в
последних двух случаях только на
1—2°, что лишний раз показывает,
что электронные оболочки разных
атомов (в нашем примере Н и С1) де-
деформируются почти одинаково.
Если атомы деформируются только с
одной стороны, то изменение валент-
валентных углов гораздо более значительно
(см. табл. 53,стр. 357).
Конформационные искажения поч-
почти не могут повлиять на длины свя-
связей. Уменьшение потенциальной
энергии напряженной молекулы, осу-
осуществляющееся за счет расталкива-
расталкивания мешающих друг другу атомов
(или групп атомов), приводит к из-
изменению валентных углов, а если
речь идет о плоских (копланарных)
молекулах, например молекулах
ароматических соединений, то и к на-
нарушению копланарности за счет ис-
искривлений самих бензольных ядер
или же вывода из плоскости ядер бо-
боковых атомов или их групп.
Искажение копланарности моле-
молекул удобнее всего иллюстрировать на
примерах многоядерных конденсиро-
конденсированных систем. На рис. 361 изобра-
изображена молекула дибензофенантрена.
Если бы молекула была идеально
плоской, то расстояние между двумя
валентно не связанными атомами уг-
углерода (отмеченными стрелками)
должно было бы быть 1,4 вместо
3,6 А — равновесного. Реально это
расстояние за счет расталкивания ва-
валентно не связанных атомов увели-
увеличивается от 1,4 до 3,0 А, но при этом
сильно нарушается копланарность
молекулы (см. рис. 361), а от этого
резко уменьшается энергия я-связи
за счет уменьшения перекрывания
облаков р-электронов, что приводит
к общему увеличению потенциальной
энергии молекулы.
На примере молекулы хлоранила
(рис. 362) видны отклонения в раз-
разные стороны от плоскости бензоль-
бензольного ядра валентно не связанных ато-
атомов хлора и кислорода.
Поворот части молекулы вокруг
ординарной связи удобно иллюстри-
иллюстрировать на примере дифенила
и его галоидопроизводных типа
В случае дифенила расталкивание
атомов водорода приводит к поворо-
повороту одного бензольного ядра относи-
относительно второго на угол 42°.
Рис. 361. Молекула дибензофенантрена
(+0,053)
О
(-0,0/)
1,342
121°22'Z i22°23'
(-0,01)
Cl-
(+0.057)
О
(+0,059)
Рис. 362. Молекула хлоранила
24*
(-005^ >
37*
В случае же галоидозамещенных
дифенила аналогичные углы поворо-
поворота для F-, С1-, Вг- и J-производ-
ного соответственно будут 60, 74, 75
и 79°.
Аналогичные повороты около двой-
двойной связи наблюдаются очень редко
и сравнительно на небольшие углы.
От степени перегруженности молекул
зависят некоторые их свойства. Так,
например, в монографии М. С. Нью-
мана A960 г.) указывается, что ско-
скорость рацемизации оптически актив-
активных о-производных дифенила изме-
изменяется обратно пропорционально из-
изменению перегруженности. С точки
зрения конформации проанализиро-
проанализировано, в каких положениях дополни-
дополнительные заместители сильнее замед-
замедляют рацемизацию, в каких — слабее.
В обзоре Р. Л. Авояна и др.
A960 г.) подробно описаны исследо-
исследования пространственных затрудне-
затруднений и конформации в ароматических
молекулах.
Конформационный анализ позво-
позволил удовлетворительно предвидеть
реальную геометрическую форму до-
достаточно сложных органических мо-
молекул.
ГЛАВА XXI
КРИСТАЛЛОХИМИЯ СЛОЖНЫХ ХИМИЧЕСКИХ СОЕДИНЕНИЙ.
КРИСТАЛЛОГИДРАТЫ, КОМПЛЕКСНЫЕ, МЕТАЛЛООРГАНИЧЕСКИЕ
И КЛАТРАТНЫЕ СОЕДИНЕНИЯ
§ 1. Строение кристаллогидратов
Выше, в гл. XII, уже говорилось, что
в молекуле воды заряды расположе-
расположены по тетраэдру. Две вершины несут
положительный заряд, две — отрица-
отрицательный. На рис. 363 показано строе"
ние молекулы воды по Берналу и
Фаулеру, выведенное ими из данных
спектроскопии, наблюдений диполь-
ного момента и изучения других фи-
физических свойств. Протоны глубоко
внедрились в кислородный ион, ядро
которого вследствие этого сместилось
из центра О в О. Все расстояния ука-
указаны на рисунке.
Тетраэдрическое распределение
зарядов молекулы воды обычно со-
сохраняют и в кристаллогидратах.
В этом случае две вершины «тетраэд-
«тетраэдра» каждой молекулы воды обраще-
обращены к положительно заряженным
структурным единицам, две дру-
другие — к отрицательно заряженным.
При этом часто наблюдаются контак-
контакты двух молекул воды друг с другом.
В этом случае, как и в структуре
льда, положительный участок одной
молекулы обязательно контактирует
с отрицательным участком другой.
Если катион, вокруг которого распо-
расположены молекулы воды, является
многовалентным, то в таких случаях
часто оба отрицательных заряда мо-
молекулы воды (две вершины «тетраэд-
«тетраэдра») объединяются в одну и распре-
распределение зарядов вместо тетраэдриче-
ского становится треугольным. Две
вершины «треугольника» несут поло-
положительные заряды (протоны),
третья — компенсирующий их отри-
отрицательный заряд.
Каждый кристаллогидрат можно
выразить схемой, показывающей рас-
распределение зарядов на структурных
единицах. Так, на рис. 364 показана
такая схема для NiSO-i-7H2O по Би-
версу и Шварцу A935 г.). Каждый
атом Ni окружают 6 молекул воды,
причем 4 имеют треугольную коорди-
координацию (тип А), а 2 — тетраэдриче-
скую (тип В). Если заряд атома Ni,
равный двум, поделить между шестью
молекулами воды так, что «треуголь-
«треугольные» получают в два раза больше,
чем «тетраэдрические», то 4 молеку-
молекулы воды получают по 2 части и 2 —
по одной. Иначе, для определения ча-
части заряда, приходящегося на 1 до-
долю связи, надо поделить заряд атома
Ni на 10 частей.
Соответственно положительные
участки молекул воды получают уси-
усилия связи, равные Vs и 2/s заряда. На
Рис. 363. Строение молекулы воды (по
Берналу и Фаулеру)
373
-схеме направления стрелок показа-
показаны от положительного заряда к отри-
отрицательному. У 1-й, 2-й, 3-й и 6-й мо-
молекул воды направления стрелок
определяются к двум другим молеку-
молекулам однозначно из-за ее «треуголь-
яой» формы. Соответственно опреде-
Тип А
Тип В
¦Рис. 364. Схема распределения зарядов
для NiSCv7H2O (по Биверсу и Шварцу)
Рис. 365. Слой молекул Н2О в структуре
1пРз-ЗНгО (разрез параллельно АВ на
рис. 366,о)
Маленький белый кружок соответствует эаря-
ДУ (+)> заштрихованный — эаряду (—)
ляются направления у 5-й, 7-й и, на-
наконец, у 4-й молекул. Положитель-
Положительные концы молекул воды направлены
к атомам кислорода из сульфатного
иона. Два из них получают от моле-
молекул воды 2/б заряда и 2 — по 3/s.
Остальная часть компенсируется ва-
валентностью серы: 13/б для первых
двух и 12Д для двух других.
Систематизировать кристаллогид-
кристаллогидраты лучше всего по признаку взаим-
взаимного расположения молекул в струк-
структурах: а) молекулы воды изолирова-
изолированы друг от друга; б) молекулы воды
составляют обособленные группы;
в) молекулы воды образуют цепи;
г) — слои; д) — трехмерные каркасы.
Чтобы правильно находить типы
взаимного расположения молекул во-
воды в кристаллогидрате, нужно обяза-
обязательно учитывать расстояния. При
самых прочных водородных связях
расстояние 0...Н...0 не бывает короче
2,52; в структуре льда оно равно 2,76.
Когда расстояние достигает или пре-
превышает ЗА, говорить о наличии кон-
контактов нельзя.
Обычно случаи «а» и «б» наблюда-
наблюдаются у кристаллогидратов с малым
количеством кристаллизационной во-
воды, случаи «г» и «д» — с большим.
Примеры кристаллогидратов с изо-
изолированными молекулами воды бу-
будут рассмотрены в следующем пара-
параграфе, посвященном строению комп-
комплексных соединений. Изолированную
группу в виде шестичленного кольца
наблюдали Н. В. Белов, В. П. Буту-
Бутузов и Н. И. Головастиков в структу-
структуре диоптаза Cue [Sie0i8] • 6Н2О.
В качестве примера структуры
кристаллогидрата, в котором молеку-
молекулы воды образуют гофрированный
лист, может служить InF8-3H2O
(рис. 365. Г. Б. Бокий и Т. С. Хода-
шева, 1956 г.). Структура построена
из цепи октаэдров — 1пЁз • 2Н2О. Один
ион фтора является общим для двух
октаэдров. Третья молекула НгО
не контактирует с In (рис. 366).
Строение кристаллогидратов орга-
органических соединений почти ничем не
отличается от строения неорганиче-
374
Ниже приведено несколько фор-
формул комплексных соединений, удов-
удовлетворяющих перечисленным выше
требованиям:
К2 [PtCh], [РЦШзНСЬ], [Co(NH3)eNO2]Cl,.
Ог
Рис. 366. Структура InF3-3H2O
о. — проекция вдоль оси ХУ\
б — проекция вдоль оси ZY
ских. В них, может быть, чаще на-
наблюдается система водородных свя-
связей, на которую мы выше уже ука-
указывали, когда шла речь о дигидрате
щавелевой и дигидрате ацетиленди-
карбоновой кислот.
§ 2. Строение
комплексных соединенна
1. Определение комплексных соеди-
соединений. До сих пор нет единой точки
зрения на вопрос, какие соединения
следует называть комплексными. Ав-
Автор данной работы предлагает назы-
называть комплексными такие тройные
или более сложные соединения,
которые имеют следующие признаки:
а) всегда имеется группа атомов,
связь между которыми более тесная,
чем между другими атомами; эта
группа в структурных формулах
обычно ставится в квадратные скобки
и называется комплексом;
б) в комплексе атомы группиру-
группируются вокруг одного атома, называе-
называемого центральным атомом, или компг
лексобразующим;
в) центральный атом является
атомом металла. Координационное
число центрального атома обычно
больше его формальной валентности.
Комплекс может иметь как поло-
положительный, так и отрицательный за-
заряд. Он может быть и нейтральным.
В этом случае структура будет мо-
молекулярной.
Если соединение не удовлетворяет
одному или нескольким из выше-
вышеуказанных условий, то мы не будем
считать его комплексным.
- Комплексные соединения можно
прежде всего различать по числу
комплексов, приходящихся на одну
формульную единицу. Выше было
написано три комплексных соедине-
соединения. Каждое из них характеризует-
характеризуется наличием одного комплекса в фор-
формульной единице. Хорошо известны
комплексные соединения с двумя
комплексами в формульной едини-
единице, например [Fe(CN)e] [Co(H2O)e].
Известны также аналогичные соеди-
соединения с тремя комплексами и т. д.
От комплексных соединений тако-
такого типа следует отличать двухъядер-
ные, трехъядерные и т. п. комплек-
комплексные соединения, характеризующие-
характеризующиеся тем, что оба комплекса соединены
друг с другом общим аддендом, назы-
называемым в этом случае мостиком, на-
например, [ (NH3) 5CoNH2Co (NH3) 5] С15.
В кратком обзоре комплексных
соединений остановимся только на
простейших случаях. Классифициро-
Классифицировать соединения будем прежде
всего по типу комплекса, т. е. по
его координационному числу и фор-
форме.
Наиболее распространенным яв-
является координационное число 6 и
координационный многогранник —
октаэдр. В качестве примера струк-
структур такого рода рассмотрим структу-
структуру K2PtCle (рис. 367). В комплекс-
комплексном ионе по трем взаимно перпенди-
перпендикулярным направлениям на равных
расстояниях от Pt расположены
6 атомов хлора. Это показывает, что
никакой разницы между главной и
«побочной» валентностями нет. В вод-
водном растворе ото вещество, как
известно, диссоциирует на три иона:
2К+ и [1ЧС1б]2~. Это может служить
375
Рис. 367. Структура K2PtCle
подтверждением, что связь между
Pt и С1 в комплексе ковалентная,
а между [PtCle]2" и К+ ионная. Об
этом же говорят и межатомные рас-
расстояния. Расстояние Pt — С1 = 2,33 А
почти совпадает с суммой ковалент-
ных радиусов 1,31+0,99 = 2,30 и
сильно отличается от суммы
ионных 0,64 + 1,81 = 2,45.
Таким образом, кристаллохими-
ческие данные подтвердили основ-
основные идеи стереохимии о строении
комплексных соединений, но одно-
одновременно и позволили их существен-
существенно дополнить, внеся в качественные
стереохимические схемы количест-
количественный элемент —величины меж-
межатомных расстояний.
Большую роль в стереохимии сыг-
сыграли исследования характера ионно-
ионного распада комплексных соединений
в зависимости от состава комплекс-
комплексного иона. Вернеру и Миолатти уда-
удалось доказать справедливость напи-
написания структурных формул для хлор-
амиачных переходных рядов четы-
четырехвалентной и двухвалентной пла-
платины и трехвалентного кобальта в
виде комплексных соединений, а не
двойных солей, как их писали рань-
раньше. До этих работ формулу хлорпла-
тината калия представляли как
PtCLi-2KCl. Из такого написания
можно было сделать неправильный
вывод о неравноценности атомов хло-
хлора в этом соединении или сделать бо-
более неправильный вывод о том, что
такое соединение состоит из молекул
PtCl4 и КС1. Г. Б. Бокий, М. А. По-
рай-Кошиц, Г. А. Кукина и Л. А. По-
Попова исследовали структуры многих
соединений хлораммиачного пере-
переходного ряда четырехвалентной пла-
платины и полностью подтвердили спра-
справедливость заключений Вернера и
Миолатти об их строении. Кроме то-
того, если посмотреть на ряд
[K2[PtCle], K[PtCl5NH3]-H2O,
[PtCUfNHsfe] (цис и транс),
[PtCb (NHs)b] СЬН2О (реберный),
[PtC]2(NH3L]Cl2(ff"c и транс),
[PtCl (ШзM]С1з-Н2О, [Pt(NHs)e]CI4-H2O,
то легко видеть, что в том случае,
когда в наружной сфере находится
один ион, то обязательно присутству-
присутствует еще молекула кристаллизацион-
кристаллизационной воды (см. моноаммин и триам-
мин). Это обстоятельство ускользну-
ускользнуло от внимания химиков. Роль этой
воды станет ясной, если обратимся к
структурам этих соединений. Кри-
Кристаллы обоих членов ряда принадле-
принадлежат к триклинной сингонии, но они
псевдокубические и по структуре
весьма близки к хлорплатинату ка-
калия. Структура КгРКШб представляет
собой единую плотнейшую кубиче-
кубическую упаковку К и С1. Ионы К могут
быть заменены на Rb, Cs, NH4 Tl;
вместо С1 может быть Вг или J, а для
некоторых центральных атомов ме-
металлов и F. Все эти вещества образу-
образуют очень большую изоструктурную
группу. Размер катионов очень ве-
велик, это самые крупные катионы. По
своему размеру они соизмеримы с
размерами анионов. Именно по этой
причине они и образуют единую
плотнейшую упаковку с анионами.
Если оставить незанятой какую-то-
часть упаковки, то вся структура
станет неустойчивой. И лишь вхож-
вхождение одной молекулы воды делает
структуру устойчивой. Роль этой во-
воды — заполнить место в плотнейшей
упаковке.
376
В этом случае молекула воды оста-
остается во внешней сфере. В некоторых
комплексах она может быть и адден-
дом, например, в [Cc^NHsb-HaOJCls.
Многие структуры комплексных
соединений можно рассматривать
как структуры бинарных соедине-
соединений. Для этого нужно весь комплекс
считать одной структурной едини-
единицей. В этом случае структуры хлор-
платината калия и его аналогов бу-
будут принадлежать к структурному
типу антифлюорита. В нем комплек-
комплексные ионы составляют плотнейшую
кубическую упаковку, а ионы внеш-
внешней сферы занимают тетраэдриче-
ские пустоты этой упаковки. Комп-
Комплексные ионы не имеют шаровой
формы, поэтому в такой упаковке
тетраэдрические пустоты больше ок-
таэдрических. Так, например, гексам-
мин кобальта [Co(NH3)e]Cl3 имеет
аналогичную структуру. Ионы
С1 занимают в кубической упа-
упаковке [Co(NH3)e]3+ все тетраэдриче-
ские и октаэдрические пустоты. Ана-
Аналогична структура и K3[Co(NO2)e].
В ней тоже октаэдрическая пустота
немного меньше тетраэдрических.
Г. Б. Бокий и Л. А. Попова дока-
доказали изоструктурность соединения
(NH4JNa[Rh(NO2N] с написанным
выше соединением кобальта и объяс-
объяснили, почему не удалось синтезиро-
синтезировать соединение Na2(NH4)>
• [Ш1(Ж>2)б]. Если бы такое соедине-
соединение существовало, то меньшие, ионы
должны были бы располагаться в
больших пустотах, а большие — в
меньших. Структура такого соедине-
нения должна была бы быть весьма
неустойчивой.
Правилам шштнейшей упаковки
подчиняются и молекулярные струк-
структуры диамминов [PtCl4(NH3J]. Цис-
изомер кристаллизуется в гексаго-
гексагональной плотнейшей упаковке
(рис. 368), транс-язоиетр — в кубиче-
кубической (рис. 369).
Структура пентаммина Чугаева
[Pt(NH3MCl]Cl3-H2O (рис. 370) пра-
правилам плотнейшей упаковки не под-
подчиняется.
Рнс. 368. Структура [PtCl4(NHsJl-if«c
ONH3 #[
Рис. 369. Структура [PtClt(NH3)г]-транс
PI
Рнс. 370. Структура пентаммина Чугаева
[Pt(NH3NCl]Cl3-H2O
377
Рис. 371. Структура соли Гро [Pt(NH3LCl2]
СЬ
Интересные явления наблюдали
Г. Б. Бокий и М. А. Порай-Кошиц
A948 г.). При исследовании струк-
структуры г^анс-изомера тетраммина
[Pt(NH3LCl2]Cl2 (рис. 371) было об-
обнаружено, а затем уточнено Е. М. Ро-
Романовой резкое уменьшение меж-
межатомных расстояний С1—С1 из раз-
разных комплексов: 3,48 А вместо нор-
нормального 3,62. Полученная величина
еще раз подтверждает ковалентный
характер связи Pt—Cl, вызывающий
уменьшение размеров С1 в направле-
направлении линии связи.
При изучении структуры
K.2[PtGl4Br2]-Tране, синтезированного
А. В. Бабаевой, было обнаружено, что
кристаллы принадлежат к кубиче-
кубической сингонии, и размер их элемен-
элементарной ячейки имеет промежуточное
значение между Кг[Р1;С1б] и
K2[PtBr6]. Объяснить эти факты мож-
можно только тем, что ось Вг—Pt—Вг
в кристалле статистически распола-
располагается по всем трем координатным
направлениям. Это своеобразный
случай автоизоморфизма.
До сих пор обсуждались главным
образом комплексные соединения с
одпоатомными аддендами (в случае
аммиака методом рентгеноструктур-
ного анализа удается определить
только координаты N), содержащи-
содержащими в наружной сфере 2 иона (тип
CaF2). Есть аналоги и других про-
простейших структур. Так, например,
Fe[Fe(CN)e] имеет структурный тип
NaCl, структура Ba[SiF6] представ-
представляет собой тригонально искаженный
структурный тип CsCl. Двукомплекс-
ное соединение [№(Н2О)б] [SnCV]
кристаллизуется в том же структур-
структурном типе.
Г. С. Жданов и 3. В. Звонкова ис-
исследовали структуры ряда родани-
дов. Они показали, в частности, что
не существует изороданид-ион типа
S—C=N, а роданогруппа всегда име-
имеет строение S = С = N с расстояни-
расстояниями S—С = 1,58 A, C-N = 1,24A.
Эта группа к платине присоединяет-
присоединяется серным концом, к Со — азотным.
Очень интересный результат полу-
получили Матьисон, Меллор и Стефенсон
A952 г.) при исследовании структу-
структуры K2RuClsOH, которому химики
приписывали форму, аналогичную
хлорплатинату калия. Оказалось, что
гидроксальных групп в нем нет,
а имеются двухъядерный комплекс с
кислородным мостиком и молеку-
молекула кристаллизационной воды. Струк-
Структурная формула его будет
K4[Cl5RuORuCl5]-H2O.
Часто комплексные соединения
имеют координационное число 4.
При этом возможна тетраэдрическая
или квадратная конфигурация комп-
комплекса.
А. Вернер и Л. А. Чугаев много-
многочисленными работами, главным обра-
образом связанными с изомерией, дока-
доказали, что двухвалентная платина име-
имеет квадратную координацию. Впо-
Впоследствии это было подтверждено и
рентгеноструктурным анализом на
структуре K2PtCl4 (рис. 372). Тетра-
эдрическую координацию имеют не-
некоторые комплексные соединения
двухвалентного кобальта.
Для соединений двухвалентного Со
типа СоС12А2, где А — амин, извест-
известно по две формы (а — синие, Р —
фиолетовые). Многие авторы припи-
приписывали кобальту в этих соединениях
квадратную координацию и считали
эти формы за цис- и граис-изомеры.
Аналогичные а- и C-формы имеет и
соединение с двумя молекулами па-
ратолуидина.
378
Рис. 372. Структура K2PtCl4
Г. Б. Бокий, Т. И. Малиновский и
А. В. Аблов исследовали структуру
СоСЬ • 2C7H9N, которая оказалась мо-
молекулярной, с тетраэдрической коор-
координацией у кобальта. Так как тетра-
эдрическое строение исключает воз-
возможность цис- и траис-изомерии,
C-формы будут иметь иное строение.
Ранее Б. К. Вайнштейном была ис-
исследована структура СоС12-2НгО,
оказавшаяся цепочкой:
Н2О С1ШО С1Н2О С1ШО
. ..Лс°\' /Сс/ ^Со^ "/Со^....
Н2О 'HjO .uH2O Н2О
Другие р-формы тоже имеют цепо-
цепочечное строение, аналогичное дигид-
рату. В последнем случае атом Со
имеет координационное число 6, и со-
соединение, строго говоря, не является
комплексным, так как изолирован-
изолированные комплексы отсутствуют. Этим и
отличаются а- и C-формы диаминов
двухвалентного кобальта.
С точки зрения теории химической
связи нет существенной разницы
между цепочечными соединениями и
соединениями с изолированными
комплексами, поэтому многие авторы
рассматривают их совместно, хотя
здесь уже несомненно имеет место
переход от чисто комплексных соеди-
соединений к обычным неорганическим
соединениям.
Другие координационные числа
встречаются в комплексных соеди-
соединениях реже. Из них стоит упомя-
упомянуть координационное число 2 для се-
серебра (структура K[Ag(CNJ]).
Аналогичные структуры имеют
некоторые соединения ртути.
Так, соединение Hg(NH3JCl2 содер-
содержит линейные комплексные катионы
[H3N-Hg-NH3]2+ и анионы С1~ во
внешней сфере.
Во всех приведенных выше приме-
примерах центральным атомом являлся
<2~металл.
Трем наиболее часто встречаю-
встречающимся координационным многогран-
многогранникам соответствует в этом случае
гибридизация типа cPsp3 для октаэд-
октаэдра, sp3 или d3s — для тетраэдра и
dsp2 — для квадрата.
В последние годы в нашей стране
и за рубежом было определено очень
много структур комплексных соеди-
соединений, в том числе и мало устойчи-
устойчивых, ранее почти не привлекавших
внимание исследователей. В резуль-
результате этих работ было найдено боль-
большое количество необычных коорди-
координационных многогранников. Так, ис-
искаженные октаэдры были найдены у
некоторых сложных соединений пла-
платины, кобальта, рения, ванадия, ме-
меди и др.
Координационное число 7 наблю-
наблюдалось у ниобия для двух координа-
координационных многогранников по одной
грани тригональной призмы —
K3[NbOF6] и K2[NbF7] соответствен-
соответственно. Координационное число 8 и мно-
многогранник в виде томсоновского куба
наблюдались у циркония,' например в
структуре тетраацетилацетоната цир-
циркония.
В табл. 55 перечислены главные
комплексообразующие атомы пере-
переходных металлов. Из таблицы видно,
что в интервале do—ds у централь-
центральных атомов преобладают большие ко-
координационные числа —8, 7 и 6.
Многогранники в виде искаженных
тетраэдров у Vv и Revn характерны
только для комплексных соединений
с кратными связями. Октаэдр (пра-
(правильный и искаженный) является
наиболее характерным координаци-
379
ТАБЛИЦА 55
Координационные числа и координационные многогранники комплексообразующпх
атомов переходных металлов и металлов 6-подгрупп
Эле-
Элемент
do
Scrir
Tiiv
ZrIV
yV
Nbv
Mo
di
ylV
ReVI
di,
Rev
d3
Crni
ReIV
Cr"
Mn111
Re111
RuIV
OsIV
d&
Mn11
Fe111
Координационное число
8
Д
т. к.
т. к.
7
СЛ
сл, с л
сл
сл
6
о, о. и.
0
0. И.
0
0
0
о, о. и.
0
0
о
0, 0. И.
0, 0. И.
5
т. п.
4
т. и.
т. и.
т
т
3
2
Эле-
Элемент
de
FeIr
Со111
Rhin
1гШ
PtIV
d
Co11
Rhn
d8
Cu111
Ni"
Pdn
Pt11
d»
Cu"
dio
Cu1
Ag1
Zn11
Cdn
Hg11
Координационное число
8
7
6
0
0
0
0
0
0
0
0, 0. И.
0. И.
0. И.
о, о.и.
0, 0. И.
0
0, 0. И.
5
т. п.
т. п.
т. п,
тр. д.
тр. д.
тр. д.
тр. д.
т. п.
т, т. и.
4
т
т. к.
т. к.
к.
к.
т, т.и.
т, т.и.
т, т. и.
т
3
2
тр
г
г
Примечание. Обозначения координац ионных многогранников: д — додекаэдр; т. к. — томсонов-
екий куб; о —октаэдр; т. п. — тетраг ональная пирамида; тр. д. — тригональная дипирамида;
т—тетраэдр; к — квадрат; тр — треугольник; г —гантель; и — искаженный; ол —сложный.
онным многогранником для всех
d-металлов. При увеличении числа
электронов, в интервале ^6—^ю на-
начинают преобладать меньшие коор-
координационные числа: 5, особенно ха-
характерно 4, а при di0 фигурируют
уже числа 3 и 2. Особенно разнооб-
разнообразны координационные многогран-
многогранники у двухвалентной меди.
2. Трансвлияние. Открытие зако-
закономерности трансвлияния (И. И. Чер-
Черняев, 1926 г.) позволило осущест-
осуществить направленный синтез и с его по-
помощью получить сотни новых ком-
комплексных соединений. Ряд трансвлия-
трансвлияния И. И. Черняева с небольшими
дополнениями, сделанными в после-
последующие годы, имеет следующий вид:
С2Н4 > GO > NO" > J" > Вг- > СГ >
> Еп/2 > NH3 > Н20,
где En — этилендиаммин. В комплек-
комплексных соединениях он занимает два
координационных места.
380
Сущность закономерности транс-
трансвлияния сводится к тому, что один и
тот же лиганд, стоящий напротив
сильно трансвлияющего лиганда
(в гране-положении) лабилизуется
сильнее (и замещается, следователь-
следовательно, легче), чем лиганд того же соста-
состава, но стоящий рядом (в ^мс-положе-
нии) к сильно траневлияющему ли-
ганду. Каждый лиганд из вышеупо-
вышеупомянутого ряда вытесняет другие,
стоящие от него справа, а сам он вы-
вытесняется лигандами, стоящими от
него слева.
Если пользоваться закономерно-
закономерностью трансвлияния, то легко понять,
как синтезируются цис- и гранс-изо-
меры соединения состава
Pt(NH3bCl2:
KPtNHsCb —-* Pt (NH2JC12
цис
|NH3
Pt (NH3JCb<-— Pt (NH3KC1
mpanc
Если на PtNHsCb действовать аммиа-
аммиаком, то он заместит один атом хлора,
соседним с уже имеющимся в ком-
комплексе аммиаком, так как они ла-
билизуют друг друга сильнее, чем ам-
аммиак лабилизует атом хлора, стоя-
стоящий от него в гране-положении.
В результате получится ^мс-изомер.
Если на триаммин Pt(NH3KCl дейст-
действовать хлором, то он заместит тот ам-
аммиак, который стоит напротив уже
имеющегося в комплексе атома хло-
хлора, так как этот атом хлора сильно ла-
лабилизует аммиак, расположенный к
нему в гране-положении. В результа-
результате получится гранс-изомер.
Эту идею можно иллюстрировать
на примере структуры K[PtNHsCb].
Как уже говорилось выше, с точки
зрения правила трансвлияния, три
атома хлора в этом соединении не-
неравноценны, поэтому различие меж-
между ними должно сказаться на меж-
межатомных расстояниях. Следовало
ожидать, что более сильно лабилизо-
ванные лиганды должны дальше от-
отстоять от центрального атома. Иссле-
Исследование структуры этой соли
(Г. Б. Бокий, Б. К. Вайнштейн и
А. А. Бабарэко, 1951 г.) дало обнаде-
обнадеживающие результаты: расстояния
Pt — Cl по координатам С1—Pt—Cl
и NH3 — Pt — Cl оказались разными:
2,35 и 2,32 А соответственно. Для
нахождения веществ, у которых была
бы большая разница в расстояниях,
в дальнейшем мы брали более транс-
влияющие лиганды, чем хлор, напри-
например, С2Н4 или NO2~ (Г. Б. Бокий и
Г. А. Кукина, 1954—1964 гг.). Эти
кристаллохимические работы позво-
позволили внести количественный элемент
в чисто качественные химические
представления (табл. 56).
ТАБЛИЦА 56
Расстояния между атомами платины
и галогена
Соединение
К [PtNH3Cl3]
K[PtC2H4Cl3].H2O
K[PtC2H4Br3]-H2O
PtNH3C2H4Br2-4"c
Координата
NH3 — Pt
Cl-Pt
Cl —Pt
C2H4 — Pt
Br —Pt
C2H4 — Pt
NHb —Pt
C2H4 — Pt
-Cl
-Cl
-Cl
-Cl
— Br
-Br
— Br
— Br
Pt-r,
2,32
2,35
2,28
2,39
2,42s
2,52
2,41
2,51
Исследование кристаллохимиче-
ских особенностей соединений, содер-
содержащих нитрогрупггу, привело к уста-
установлению того факта, что NGvrpynna
в соединениях четырехвалентной
платины обладает пониженным
трансвлиянием (более низким, чем
хлор). Вначале это было установлено
рефрактометрическим (Г. Б. Бокий и
С. С. Бацанов, 1954 г.), а затем и
рентгеноструктурным методом
(Г. Б. Бокий и Г. А. Кукина, 1957 г.).
Установление этого факта привело к
выводу, что ряд силы трансвлияния,
приведенный выше, не является та-
таким универсальным, каким он счи-
считался на протяжении более 25 лет. Си-
Сила трансвлияния некоторых лиган-
дов зависит от валентности централь-
381
ного атома. Этот неожиданный ре-
результат через несколько лет был под-
подтвержден химическими методами.
Объяснение этого факта следует ис-
искать в стерических препятствиях,
возникающих в комплексах, содержа-
содержащих в качестве лигандов многоатом-
многоатомные группы и невозможности обра-
образования я-связей (Г. Б. Бокий,
И. Б. Берсукер, 1963 г.).
3. Комплексные соединения с крат-
кратными связями. В начале этого па-
параграфа, давая определение комплек-
комплексного соединения, автор указывал,
что обычно координационное число
центрального атома больше его фор-
формальной валентности. Это действи-
действительно всегда так, если в качестве
лигандов в комплексные соединения
входят одновалентные ионы и ней-
нейтральные группы, а таковых подав-
подавляющее большинство. Однако в не-
некоторые комплексные соединения
входят двух- и трехвалентные лиган-
ды. Если они при этом занимают од-
одно координационное место, как, на-
например, в случае одноатомных ли-
лигандов (кислорода или азота), то ко-
координационное число центрального
атома может быть равно и даже
меньше его формальной валентности,
например K2[OsVINCl5] или
К[ОзушОз№|. Этот тип комплексных
соединений характерен для многова-
многовалентных (VI—VIII) металлов, вы-
выступающих в качестве центральных
атомов. В этих случаях между цент-
центральным атомом металла и лиганда
образуются кратные связи.
Формально такие соединения даже
противоречат теории А. Вернера, в
основе которой лежит предположе-
предположение об идентичности всех связей в
комплексе (как главных, так и по-
побочных). Фактически же они явля-
являются такими же объектами координа-
координационной химии, как и обычные ком-
комплексные соединения.
Некоторые лиганды, например ни-
трозогруппа N0, присоединяясь к
центральному атому, в разных соеди-
соединениях могут образовывать связи
разных кратностей. Я. К. Сыркину и
В. И. Беловой A958 г.) принадле-
принадлежит теоретическое рассмотрение во-
вопроса и установление двух крайних
типов электронного строения нитро-
зогруппы (о, б);
а в t
Промежуточный случай в (вероятно,
наиболее распространенный в соеди-
соединениях рутения) в структурном отно-
отношении трудно отличим от случая б.
Работами советских авторов (Г. Б. Бо-
Бокий, Н. А. Парпиев, М. А. Порай-Ко-
шиц, В. С. Сергиенко, Т. С. Ходашо-
ва) и зарубежных (Симонсен, 1965 г.)
установлено, что в структурах нитро-
зосоединений рутения расстояние
Ru —N лежит в пределах 1,67—
1,80 А, а угол Ru —N —О близок к
180° A67—180°). Аналогично строе-
строение комплексных соединений дру-
других переходных металлов. Угол
Me—N—О, равный 121°, был найден в
структуре [CoCl (NO) (C2H8N2) 2] СЮ4
Снийдером и Уивером A970 г.).
Кратные связи хорошо изучены
для неметаллических элементов в их
соединениях друг с другом и очень
мало изучены в соединениях ме-
металл — неметалл. Мы (Г. Б. Бокий,
Л. О. Атовмян и Т. С. Ходашова,
1952 г.) высказали предположение,
что образование устойчивых группи-
группировок Me — О, О — Me — О, Me — N,
Me — О — Me, Me — N — Me и др.
должно явиться характерной особен-
особенностью химии комплексных соедине-
соединений тяжелых металлов, содержащих
О и N в качестве лигандов. Эти груп-
группировки, аналогичные О — Me — О,
давно известны для урана.
В 1952 г., исходя из предположе-
предположения об осмиловой группировке в сое-
соединении КгОэО^НгО, мы предло-
предложили структурную формулу для не-
него в виде K2[OsO2(OHL], что и было
подтверждено в 1962 г. М. А. Порай-
Кошицем, Л. О. Атовмяном прямым
рентгеноструктурным анализом. В
структуре K2OsNCls нами обнаруже-
обнаружена линейная группировка N=Os—С1,
аналогичная О = Os = О.
382
В структуре NaNH4[MoO3C2O4]-
• 2Н2О (Л. О. Атовмян и Г. Б. Бокий,
1962 г.) атом Мо сдвинут из центра
октаэдра к одной из граней, поэтому
три расстояния Мо — О короче трех
остальных (три коротких расстояния
лежат в пределах 1,82—1,88, три
длинных в пределах 2,23—2,24). Это
обстоятельство дает возможность
формального выделения тригонально-
пирамидальной группировки [МоО3],
аналогичной [БОз]. Если соединяют-
соединяются неметаллические элементы с не-
неметаллическими, то образуются изо-
изолированные группировки; если ж©
неметаллические элементы соединя-
соединяются с металлическими, то такой ло-
локальной группировки не образуется,
так как тяжелые металлы способны
образовать какое-то количество до-
дополнительных связей из-за наличия
у них незаполненных d-орбит.
В заключение этого раздела приво-
приводим таблицу ковалентных «радиусов»
металлов для разных кратностей
(Г. Б. Бокий и Л. О. Атовмян, 1961 г.).
Интересно, что ковалентные «ра-
«радиусы» металлов с увеличением
кратности уменьшаются более разко,
чем «радиусы» неметаллов (см.
табл. 57).
4. О связи металл — металл. В те-
течение всего длительного периода вре-
времени, когда в химии господствовали
препаративные химические методы,
связи металл — металл почти не ис-
исследовались. Одним из немногих при-
примеров, который приводился в курсах
неорганической химии, были галоид-
галоидные соединения ртути, образующие
молекулы типа Г—Hg—Hg—Г.
Определение структур простейших
соединений fr-металлов (например,
сульфидов Ga и In) методами рент-
геноструктурного анализа показало,
что подобные образования встречают-
встречаются довольно часто.
Во всех подобных случаях химиче-
химическая связь Me — Me осуществляется
за счет s- и р-электронов, так как у
^-металлов II—IV групп d-оболочка
заполнена. В последние годы в ре-
результате совершенствования методи-
методики рентгеноструктурного анализа и
применения его к изучению атом-
атомных структур сложных комплексных
соединений, круг веществ, в которых
наблюдаются связи Me — Me, значи-
значительно расширился.
Выяснилось, что очень многие пе-
переходные металлы, во всяком случае
металлы IV-a — VIII-a подгрупп, а
также металлы 1-Ь подгруппы спо-
способны давать комплексные соедине-
соединения, имеющие связи Me — Me. При
этом в связи могут вовлекаться s-,
р- и d-электроны с образованием а-
п- и б-связей. Последнее обстоятель-
обстоятельство указывает на то, что связи Me —
— Me могут быть весьма разнообраз-
разнообразными, и расстояния Me — Me для од-
одного и того же металла варьируются
в широких пределах. Сопоставлять
расстояния Me — Me в комплексных
соединениях надо с кратчайшим меж-
межатомным расстоянием в структуре со-
соответствующего простого вещества
или с суммой двух атомных радиусов.
Простейшим случаем таких соеди-
соединений будет Си(СНзСООJ'НгО, су-
существующее в виде димеров. Струк-
Структура его определена в 1953 г. Дж.
Ван-Никерком и Ф. Шёнингом и
уточнена И. Броуном и Дж. Дуни-
цем в 1961 г. Схематически она изо-
изображена на рис. 373. Расстояние
Си — Си в этой структуре 2,45 А. Ана-
Аналогичные димеры дают соединения
Rh, Сг и Мо. Расстояния Me — Me
Рис. 373. Строение димера [Си(СН3СООJ-
•Н2О]2
383
ТАБЛИЦА 57
Ковалентные радиусы (в А) прн связях разной кратности
V
Н,13D)
50,96D)
Nb
Та
Сг
,17D)
,93 D)
Мо
,29D)
,22D)
W
•1,25D)
,21D)
Мп
'1,12D)
70,96 D)
Тс
Re
4,24D)
'1,08D)
1,12E)
Fe
el,19E)
в1,05E)
Ru
41,36F)
*1,17F)
Os
'1,42F)
81,25F)
,05F)
Со
Rh
1г
Ni
Pd
Pt
Си
Ag
Аи
Zn
Cd
Hg
В
0,83
0,66i
Al
1,26
Ga
In
Tl
С
0,77
0,65,
0,604
Si
1,17
1,04
0,985
Ge
1,22
1,17
1,12,
Sn
1,40
1,355
1,314
Pb
1,46
1,43
1,398
N
0,7
0,62
0,54b
P
1,10
0,96
As
1,21
1,15
Sb
1,40
Bi
1,46
О
0,66
0,604
0,524
S
1,04
0,836
Se
1,17
1,01
Те
1,37
Po
F
0,64
Cl
0,99
0,76
Br
1,14
J
1,33
1,19
At
Связь
Ординарная
Двойная
Тройная
Ордвн арна я
Двойная
Тройная
Ординарная
Двойная
Тройная
Ординарная
Двойная
Тройная
Ординарная
Двойная
Тройная
и
61,42G)
0,976B)
Примечания. Цифра слева вверху у значения радиуса металла указывает формальную валентность, цифра оправа в скобках — координацион-
координационное число металла в соединении, из которого рассчитан радиус. Значение радиусов для Si, Ge, Sn и Pb при двойных связях рассчитаны теорети-
теоретически. Таблица составлена по данным следующих авторов: для V — Пальмер A938 г.); для Сг — Пальмер A938 г.) и Ганиц A956 г.); для V и Сг —
Пальмер A938 г.).
для родиевого соединения 2,38 А
(М. А. Порай-Копгац и А. С. Анциш-
кина, 1962 г.). Расстояние Me—Me в
этих структурах меньше, чем в чис-
чистых металлах (для Си — 2,56, для
Rh — 268 А). В соединениях этого
типа лиганды образуют мостики, стя-
стягивающие металлические атомы и
упрочняющие связь Me — Me.
Есть, однако, соединения, димери-
зация в которых осуществляется непо-
непосредственно за счет связи Me — Me.
Такова, например, структура
Mea^C^Re — ReCl4] • 2Н2О, опреде-
определенная Ф. А. Коттоном и С. В. Гар-
рисом A965 г.). Расстояние Re—Re,
равное 2,22 А, весьма мало. В метал-
металле это расстояние равно 2,74 А. Это
дает основание считать, что связь
Re — Re четырехкратная. Всего в
комплексе 24 валентных электрона.
Из них 16 расходуются на 8 связей
Me — С1, 8 электронов идут на обра-
образование связи Me Ш Me (одна а-,
две — я- и одна б-связи). Наличие
б-связи предопределяет призматиче-
призматическое расположение атомов О. Стери-
ческие препятствия — отталкивание
противоположных атомов хлора друг
от друга — не позволяют атомам ме-
металла сблизиться еще больше. Трой-
Тройная связь Re=Re найдена Козьми-
ным, Кузнецовым и Поповой
A965 г.) для двух модификаций
(CsH5NH)HReBr4 со средним рассто-
расстоянием Me — Me » 2,24. Обе структу-
структуры построены из катионов пиридония
(C5H5NH)+ и димерных анионов
[HBr4Re=ReBr4H]-.
Аналогичные структуры наблюда-
наблюдаются у комплексных соединений, где
в качестве лигандов фигурируют кар-
карбонильные группы, например,
[Fe2(CO)8]2-, Rh2(COLCl2, Fe2(CO)9
и др.
В некоторых случаях в кристалли-
кристаллических структурах образуются цепоч-
цепочки — Me — Me — Me —, например, в
структуре соли Магнуса
[Pt(NH3L][PtCl4]. Известны и сое-
соединения, в которых связь Me — Me
образована разными элементами, на-
например, [Си (NH8) 4] [PtCl4].
25 Кристаллохимия
§ 3. Строение металлоорганнческих
соединений
1. Строение простейших металлоор-
ганическнх соединений. В предыду-
предыдущем параграфе были рассмотрены
структуры комплексных соединений.
Их внутрисферными лигандами мо-
могут быть радикалы или молекулы
как неорганические, так и органи-
органические. Если в последнем случае
присоединение лиганда к централь-
центральному атому происходит через атом
углерода, то такое соединение будет
металлоорганическим. Таким обра-
образом, нет существенной разницы меж-
между комплексными и металлооргани-
ческими соединениями. Как в тех,
так и в других связь между металли-
металлическим атомом и лигандами кова-
лентная.
В § 2 (стр. 379) приводилась в ка-
качестве примера комплексного соеди-
соединения с координационным числом 2
структура Hg(NH3JCl2. Линейная
форма сохраняется и у молекулы ме-
таллоорганических соединений ме-
тилмеркурхлорида Н3С—Hg—С1
(рис. 374), этилмеркурхлорида, этил-
меркурбромида, бутил- и пропил-
меркурхлорида, дифенилмеркурида
и др. аналогичных соединений, изу-
изученных А. И. Китайгородским и
Грденичем A948 и 1949 гг.).
Очень интересны структуры сти-
бинов. Триметилдихлорстибин, а
также триметилдибром- и дииодсти-
бины имеют молекулярную структу-
структуру, в. которой для атома Sb осущест-
осуществляется координационное число 5 в
форме тригональной дипирамиды
(рис. 375, а).
Атомы галогена расположены на
полюсах, а метальные группы — в
экваториальной плоскости. Мысли-
Мыслимы, конечно, и два других изомера
(рис. 375, б, в).
Известны аналогичные соединения
с фенильными группами в качестве
лигандов, у которых можно предпо-
предполагать те же изомеры.
В разобранных примерах металло-
органических соединений атом ме-
385
Рис. 374. Структура метилмеркурхлорида
Н3С — Hg — С1
А — проекция на грань ас;
Б — проекция на диагональную плоскость
%
S
Рис. 375. Мотивы структур изомерных
стибинов
талла был связан с одним атомом
углерода в органическом радикале,
что не является обязательным усло-
условием.
2. Металлоорганические соедине-
соединения с jt-комплексами. В последние
годы были исследованы структуры
соединений, в которых атомы метал-
металла связаны особым типом химической
связи с несколькими углеродными
атомами, образующими опреде-
определенный цикл. Так, на рис. 376 пока-
показана структура соединения AgClGv
¦С6Нб. Каждый атом серебра имеет
соседями 4 атома углерода на рас-
расстоянии 2,6 А (по 2 от двух бензоль-
бензольных колец) и атом О из аниона
[ClOJ". Изолированных комплексов
в структуре нет, так как каждая бен-
бензольная молекула образует по две
пары таких связей с двумя атомами
серебра:
>Ag;'f ;Ag:'
Систематическое исследование сое-
соединений, в которых металлический
атом связан с несколькими углерод-
углеродными атомами, началось с работы
Пфафа и Фишера A953 г.). Рентге-
ноструктурный анализ ферроцена
Fe (С5Н5J (бме-циклопентадиенила
двухвалентного железа) показал, что
структура его молекулярная и со-
состоит из двух плоских параллельно
расположенных углеводородных пя-
пятиугольников, причем каждая верши-
вершина (атом углерода) одного пяти-
пятиугольника приходится между двумя
вершинами второго пятиугольника.
Между этими двумя пятиугольника-
пятиугольниками в центре симметрии на оси пятого
порядка молекулы размещается атом
железа (рис. 377). Такие структуры
стали называть сендвичевыми. Рас-
Расстояние Fe — С, равное 2,0 А, точно
соответствует сумме ковалентных
радиусов. Поскольку плоская форма
органических радикалов связана с
наличием в них я-связей, и этот же
тип связи обусловливает соединение
их с металлическим атомом, постоль-
постольку подобные соединения стали назы-
называть зт-комплексами, а самую связь —
многоцентрово й.
В настоящее время выяснилось
большое разнообразие зт-комплексов.
Они способны образовываться с
большим количеством металлов: Ti,
V, Сг, Mn, Fe, Co, Ni, Cu, Mo, Rh,
Pd, Ru, Os, Pt и др.
Выяснилось также, что к метал-
металлическим атомам с образованием
зт-комплексов способны присоеди-
присоединяться не только пятичленные и
шестичленные углеродные циклы,
но также и четырех-, семи- и более
сложные, например, [Си(СНзL№СЬ]2,
(С5Нб)У(С7Н7) и др. В образовании
зт-комплексов могут участвовать не
только циклические лиганды. Сейчас
386
Рис. 376. Структура AgC104-C6H6
Рис. 377. Молекула ферроцена
всеми, а только частью своих угле-
углеродных атомов; в этом случае такие
лиганды по типу связи не отличают-
отличаются от ациклических.
Простейшим случаем л-комплекса
будет соединение металла с этиле-
Н Н
ном типа
гДе л-связь
Иг
Рис. 378. Карта электронной плотности
в структуре соли Цейзе
известно большое количество соеди-
соединений упомянутых выше металлов с
ациклическими органическими со-
соединениями или радикалами.
Циклические лиганды в ряде слу-
случаев присоединяются к металлу не
обозначена стрелкой. Этот случай
был предсказан Чаттом и Данкан-
соном на основании данных инфракра-
инфракрасной спектроскопии и впервые на-
надежно экспериментально доказан
Г. Б. Бокием и Г. А. Кукиной в
структуре соли Цейзе K[PtXj2HiCls] •
¦НгО, о которой говорилось в преды-
предыдущем параграфе (стр. 381). Ниже
приводится карта электронной плот-
плотности детали этой структуры, из ко-
которой ясно видно боковое присоеди-
присоединение молекулы этилена к атому
платины (рис. 378). Этот простейший
тип связи в я-комплексах называют
теперь грехценгровым.
Весьма интересны аллильные
л-комплексы, в которых лиганд типа
СН2 = СН2—СН2 может присоеди-
присоединяться к металлу двумя атомами
углерода с образованием л-связи, а
третьим атомом углерода — ординар-
ординарной связью (а-связь). Возможен и
другой вариант: из-за полной дело-
кализации электронов атом металла
образует с тремя атомами углерода
лиганда две симметричные л-связи:
Чаще наблюдается случай, когда
образуется одна л- и одна а-связи:
С
Весьма близки
по
своей природе к л-комплексам ком-
комплексные соединения, содержащие в
25* 387
качестве лигандов карбонильные
группы. Часто карбонилы при-
присутствуют одновременно с я-лиганда-
ми, например, (СН2=СН—CN) •
•Fe(CO)., C4H*Fe(COK и др.
Как считать координационное чис-
число центрального атома, когда в ка-
качестве лиганда присутствует я-ли-
ганд? По-видимому, следует усло-
условиться, что такие лиганды занимают
одно координационное место. Так,
в структуре соли Цейзе (см. рис.
378) мы считали, что координация у
платины продолжает оставаться чет-
четверной по квадрату, а не пятерной.
Естественно, что чем крупнее я-ли-
ганд, тем больше он создает препят-
препятствий для образования достаточно
большого координационного числа.
В структуре ферроцена Fe(C5H5J
координационное число Fe следует
считать равным двум.
§4. Клатратные
и другие сложные
химические соединения
Под термином «клатратное соедине-
соединение» понимается соединение двух
или большего числа молекулярных
соединений, в которых одна молеку-
молекула большая, а вторая — небольшая,
например, ЗС6Н4(ОНJ • М, где
M-SO2, H2S, HCN, HC1, НВг,
HCOOH, CH3CN и др.
В подобных соединениях оба типа
молекул сохраняются (рис. 379).
Одни из них в результате системы
водородных связей образуют кар-
каркас. Так, на рис. 380 ясно видны
О
о
о
О
Рис. 379. Два типа молекул в клатратных
соединениях
«шестиугольники», образованные во-
водородными связями гидроксильных
молекул гидрохинона. На рис. 381
показана одна из пустот в каркасе
гидрохинона, образованная системой
водородных связей. В эти пустоты
как раз и попадают целиком неболь-
небольшие молекулы другого вещества.
В этом отношении клатратные соеди-
соединения похожи на кристаллогидраты,
в особенности на те из них, в кото-
которых молекула воды заполняет пусто-
пустоту в плотнейшей упаковке.
В разобранных выше примерах
оба исходные вещества (гидрохинон
и сероводород) молекулярны, поэто-
поэтому конечный продукт можно было бы
называть димолекулярным. В этом
случае подобные клатратные соеди-
соединения составили бы одну группу с
димолекулярными соединениями мо-
мочевины с парафинами. Однако совер-
совершенно необязательно, чтобы каждое
из исходных веществ было бы моле-
молекулярного строения. Трехмерный
каркас или слой могут быть состав-
составлены не только дипольными или во-
водородными связями, но и ковалент-
ными и ионными.
Примером такого соединения мо-
может служить структура Ni(CNJNH3-
•С«Нв (рис. 382).
Но даже и в том случае, когда ис-
исходные вещества состоят из молекул,
последние могут претерпеть сильные
изменения при образовании ими
сложного соединения. Так, например,
соединению кумарина с сулемой
приписывали формулу
Н
\
\т
• А н
о—с=о
В результате рентгеноструктурно-
го анализа, проведенного Ю. Т. Струч-
Стручковым A951 и 1953 гг.), оказалось,
что молекулы сулемы в нем сохраня-
сохраняют свою индивидуальность, но связь
Hg — С1 в ней значительно ослабе-
ослабевает, становится более ионной. Об
этом можно судить по увеличенному
388
О 1 Zk
Рис. 380. Образование «шестиугольников»
водородных связей гидроксиламинных мо-
молекул гидрохинона
Рис. 381. Образование водородными свя-
связями пустот в каркасе гидрохинона (бе-
(белыми кружками показаны атомы кисло-
кислорода)
Рис. 382. Структура Ni(CNJNHs-C,H.
расстоянию Hg — С1, равному 2,33 А
(в самой структуре сулемы это рас-
расстояние равно 2,21), и по нарушению
прямолинейности молекулы С1 —
— Hg — С1 (угол становится равным
171°). Более ионный, чем в сулеме,
характер атома ртути позволяет осу-
осуществить почти ионную связь с кето-
кислородом молекулы кумарина —
расстояние Hg — О равно 2,38 А.
Таким образом, можно считать, что
вещество имеет промежуточный ха-
характер между димолекулярным и
комплексным, в котором в ртути
осуществляется координационное
число 3.
В некоторых клатратных соедине-
соединениях пустоты могут располагаться
друг над другом с образованием
широких труб, в которых могут раз-
размещаться мелкие молекулы, в част-
частности молекулы воды. В этом отно-
отношении клатратные соединения по
своему строению становятся неотли-
неотличимыми от цеолитов, о которых шла
речь в разделе, посвященном крис-
кристаллохимии силикатов.
Клатратные соединения обладают
рядом специфических свойств, благо-
благодаря которым в последние годы к
ним привлекается большое внима-
внимание. Особый интерес представляют
также клатратные соединения, в
пустотах которых находятся метал-
металлические атомы. В некоторых клат-
ратах серебра, в частности, наблюда-
наблюдалась сверхпроводимость, которая до
сих пор наблюдалась лишь у метал-
металлов или у интерметаллических сое-
соединений с особыми структурами.
Заканчивая книгу, можно повто-
повторить ту же мысль, которая высказы-
высказывалась уже при описании структур
силикатов: без кристаллохимических
исследований невозможно было бы
сделать сколько-нибудь разумных
предположений о структуре таких
сложных соединений, как силикаты,
некоторые органические соединения,
я-комплексы, клатраты и пр. Крис-
таллохимический подход к проблеме
позволил выяснить не только фор-
формально-геометрические закономер-
закономерности их строения, но и обнаружить
и детально исследовать новые типы
химического взаимодействия, кото-
которые ускользали от внимания иссле-
исследователей, пока они пользовались в
своей работе только препаративны-
препаративными химическими методами.
ЛИТЕРАТУРА
Аеоян Р. Л., Стручков Ю. Т., Дашев-
ский В. Г. ЖСХ, 7, № 2, 289 A966).
Агеев Н. В. Химия металлических спла-
сплавов. М.—Л., Изд-во АН СССР, 1941.
Агеев Н. В., Макаров Е. С. Изв. АН СССР,
№ 2, 87 A943).
Агеев Н. В., Шойхет Д. Н. Ann. Phys.,
23, 90 A935).
Атовмян Л. О., Андрианов В. Г. ЖСХ, 3,
№ 6, 635 A962).
Атовмян Л. О., Бокий Г. В. ДАН СССР,
143, № 2, 342 A962).
Пацанов С. С. Электроотрицательности
элементов и химическая связь. Новоси-
Новосибирск. Изд-во СО АН СССР, 1962.
Белов Н. В. Структура ионных кристал-
кристаллов и металлических фаз. М., Изд-во
АН СССР, 1948.
Белов Н. В. Труды Ин-та кристаллогра-
кристаллографии АН СССР, 6, 25 A951).
Белое Н. В. ДАН СССР, 88, № 6, 953
A952).
Белов Н. В. ЖСХ, 1, № 1, 39 A960).
Белов Н. В., Бокий Г. Б. Материалы I
совещания по кристаллохимии. М.,
Изд-во АН СССР, 1954.
Белов И. В., Мокеева В. М. Труды Ин-та
кристаллографии АН СССР, 5, 13
A949).
Бокий Г. Б. Природа, № 1, 28 A942).
Бокий Г. Б. Состояние теории химическо-
химического строения в органической химии. М.,
Изд-во АН СССР, 1952.
Бокий Г. Б. ДАН СССР, 89, № 3, 459
A953).
Бокий Г. Б. Успехи химии, 23, вып. 5,
605 A954).
Бокий Г. Б. ЖНХ, 1, вып. 6, 1150 A956).
Бокий Г. Б. ЖНХ, 8, вып. 5, 1033 A963).
Бокий Г. Б. ЖСХ, 4, № 1, 73 A963).
Бокий Г. Б. Принципы кристаллохимиче-
ской систематики арсенидов, сульфи-
сульфидов, арсеносульфидов и их аналогов.
Новосибирск. Изд-во СО АН СССР, 1964.
Бокий Г. Б., Атовмян Л. О. ЖСХ, 2, № 3,
308 A961).
Бокий Г .Б., Атовмян Л. О., Ходашова
Т. С. ДАН СССР, 128, № 1, 78 A959).
Бокий Г. В., Бацанов С. С. ДАН СССР, 95,
№6, 1205 A954).
Бокий Г. Б., Берсукер И. В. ЖСХ, 4, № б,
934 A963).
Бокий Г. В., Вайнштейн Э. Е. Изв.
АН СССР, ОХН, № 4, 241 A943).
Бокий Г. Б., Вайнштейн Б. К., Бабарвко
А. А. Изв. АН СССР, № 6, 667 A951).
Бокий Г. Б., Горогоцкая Л. И. ЖСХ, 8,
№4,662 A967).
Бокий Г. В., Клевцова Р. Ф. ЖСХ, 6,
№6,866 A965).
Бокий Г. Б., Кравченко В. Б. ЖСХ, 7,
№6, 920 A966).
Бокий Г. Б., Кукина Г. А. Кристаллогр.,
2, вып. 5, 400 A957).
Бокий Г. Б., Кукина Г. А. ДАН СССР,
135, №4, 840 A960).
Бокий Г. Б., Малиновский Т. И., Аблов
А. В. Кристаллогр., 1, 248 A956).
Бокий Г. В., Парпиев Я. Л. Кристаллогр.,
2, вып. 8,691 A957).
Бокий Г. Б., Попова Л. А. Изв. АН СССР,
ОХН, №2, 89 A945).
Бокий Г. Б., Порай-Кошиц М. А. ДАН
СССР, 64, 133 A949).
Бокий Г. Б., Порай-Кошиц М. А. Прак-
Практический курс рентгеноструктурного
анализа. М., Изд-во МГУ, 1951.
Бокий Г. Б., Порай-Кошиц М. А. Куки'
на Г. А., Попова Л. А. Изв. АН СССР,
серия физ., 15, № 2, 170 A951).
Болдырев А. К. Кристаллография. М.— Л.,
ОНТИ, 1934.
Бутлеров А. М. О химическом строении
вещества, Соч., т. 1. М., Изд-во АН СССР
1953.
Вайнштейн Б. К. Труды Ин-та кристал-
кристаллографии АН СССР, вып. 10, 117
A954).
Вайнштейн Б. К., Пинскер 3. Г. Труды
Ин-та кристаллографии АН СССР, № 6,
163 A951).
Вернадский В. И. Очерки геохимии, 4-е
изд. М.— Л., Горгеонефтеиздат, 1934.
Вернадский В. И. Земные силикаты, алю-
алюмосиликаты и их аналоги, 2-е изд.
М.— Л, Изд-во АН СССР, 1937.
Вернер А. Новые воззрения в области
неорганической химии. Л., ОНТИ, Хим-
теорет., 1936.
390
Винчелл А., Винчелл Г. Оптическая ми-
минералогия. Под ред. Д. С. Белянкина.
М, ИЛ, 1953.
Вулъф Ю. В. Изоморфизм.— Приложение
к книге Д. И. Менделеева «Основы хи-
химии», т. 2, 9-е изд. М., Госиздат, 1928. '
Вулъф Ю. В. Избранные работы по кри-
кристаллофизике и кристаллографии. М.—
Л., Гостехиздат, 1952.
Гадолин А. В. Зап. С.-Петерб. минералог,
о-ва, 2-я серия, ч. 4, 112 A867).
Гольдшмидт В. М. Основные идеи геохи-
геохимии, вып. 1. Под ред. А. В. Ферсмана.
М., Госхимтехиздат, 1933.
Горбов А. В. Труды ВНИИГ, 40, 392 A960).
Грденич Д. Р., Китайгородский А. И.
ЖФХ, 23, 1161 A949).
Грушевицкий П. В. Уч. зап. ЛГУ, № 178,
серия геолог., вып. 4 A954).
Гюйгенс X. Трактат о свете. Пер. под
ред. В. Фредерикса. М.— Л., ОНТИ,
1935.
Дальтон Д. Сборник избранных работ по
атомистике. М., Госхимиздат, 1940.
Дислокации в кристаллах. Библиографи-
Библиографический указатель, вып. 2. М., «Наука»,
1966.
Жданов Г. С, Звонкова 3. В. Успехи хи-
химии, 22, 3 A953).
Инголъд X. Принцип электронной теории
органических реакций. Сб. «Электрон-
«Электронная теория в органической химии». М.,
ОНТИ, 1936.
Канниццаро С. Обзор развития понятия
об атоме, частице и эквиваленте и раз-
различных систем формул. Киев, 1873.
Капустинский А. Ф. Z. Kristallogr., 86,
359 A933).
Китайгородский А. И. Органическая кри-
кристаллохимия. М., Изд-во АН СССР,
1955.
Китайгородский А. И., Грденич Д. Р.
Изв. АН СССР, ОХН, № 2, 262 A848).
Китайгородский А. И., Кабалкина С. С.
ДАН СССР, 72, 899 A950).
Конобеевский С. Т. Уч. зап. МГУ, 24, 13
A943).
Корнилов И. И. Железные сплавы. М.,
Изд-во АН СССР, 1956.
Кравченко В. Б. ЖСХ, 4. 280. 761 A963).
Крипякевич П. И. ДАН СССР, 85, 321
A952).
Крипякевич П. И. Исследование по кри-
кристаллохимии металлических соедине-
соединений с высокими координационными
числами. Автореферат канд. дисс.
Львов, Изд-во Львовского Гос. ун-та,
1957.
Крипякевич П. И. Крпсталлогр., 5, 79,
273 A960).
Крипякевич П. И. ЖСХ, 4, № 1, 117
A963).
Курнаков Н. С. Растворы и сплавы. При-
Приложение к книге Д. И. Менделеева
«Основы химии», т. 2, 9-е изд. М., Гос-
Госиздат, 1928.
^Курнаков Н. С. Введение в физико-хими-
* ческий анализ. 3-е изд. ОНТИ, 1936.
Курнаков Н. С. Успехи химии, 5, 3
A936).
Курнаков Н. С. Собрание избранных ра-
работ, т. 2. Л., Хнмтеорет., 1939.
Курнаков Н. С. Жемчужный С. Ф. Изо-
Изоморфизм соединений калия и натрия.
СПБ., 1905.
Курнаков Н. С, Жемчужный С. Ф. ЖРФО,
39, 211 A907).
Лазаренко Е. К. Основы генетической ми-
минералогии. Львов. Изд. Львовского
ун-та, 1963.
Ландау Л. Д., Ливщиц Е. М. Статисти-
Статистическая физика. М., Гостехиздат, 1951.
Леммлейн Г. Г., Дукова Е. Д. ДАН
СССР, 102, 77 A955).
Ловиц Г. Е. Избранные труды по хи-
химии и химической технологии. М.— Л.,
Изд-во АН СССР, 1955.
Ломоносов М. В. Полное собрание сочи-
сочинений, т. 2. М.—Л., Изд-во АН СССР,
1951.
Макаров Е. С. Строение твердых фаз с
переменным числом атомов в элемен-
элементарной ячейке. М.— Л., Изд-во АН СССР,
1947.
Макаров Е. С. Изв. АН СССР, ОХН, 1950,
485.
Менделеев Д. И. Изоморфизм в связи с
другими отношениями кристаллической
формы к составу. СПБ., 1856.
Менделеев Д. И. Основы химии, изд. 1-е.
СПБ., 1869-1871.
Менделеев Д. И. Основы химии, изд.
9-е. М.— Л., 1928.
Море Ф. Колебание и звук. М.—Л., Гос-
Гослитиздат, 1949.
Николаев А. В. Физико-химическое изу-
изучение природных боратов. М., Изд-во
АН СССР, 1947.
Новик Ю. 3. Нейтронографическое иссле-
исследование системы марганцевых ферри-
ферритов. Автореферат канд. дисс. М., 1962.
Ньюман М. С. Пространственные эффекты
в органической химии. Пер. с англ. под
ред. А. Н. Несмеянова. М., ИЛ, 1960.
Ньютон И. Оптика. Пер. с англ. под ред.
С. И. Вавилова. М., ГИЗ, 1927.
Орелкин Б. П. Ргос. Roy. Soc, 144A, 630
A934).
Поваренных А. С. Зап. Всесоюзн. мине-
минералог, об-ва, ч. 87, № 3, 348 A958).
Поваренных А. С. Изв. АН СССР, серия
геолог., № 12, 91 A956).
Порай-Кошиц М. А., Анцышкина А. С.
ДАН СССР, 146, № 5, 1102 A962).
Порай-Кошиц М. А., Романова Е. М. Изв.
АН СССР, сектор платины, 28, 282
A954).
Потенциалы ионизации и сродство к элект-
электрону. Справочник. М., Изд-во АН СССР,
1962.
Смирнова Н. Л. Кристаллогр., 1, вып. 5,
502 A956).
391
Смирнова Н. Л. Кристаллогр., 4, вып. 1,
20 A959).
Смирнова Н. Л. Кристаллогр., 1, вып. 2,
165 A965).
Соболев В. С. Введение в минералогию
силикатов. Львов, Изд. Львовского гос.
ун-та, 1949.
Стручков Ю. Т., Китайгородский А. И.
Изв. АН СССР, ОФМН, 15, 176 A951);
ДАН СССР, 93, 675 A953).
Сыркин Я. К., Белова В. И. Изв. АН СССР,
ОХН, 12,1492 A958).
Тамм И. Е. ЖЭТФ, 3, 34 A933).
Федоров Е. С. Зап. С.-Петерб. минералог.
о-ва, 2-я серия, ч. 21, 1 A885).
Федоров Е. С. Зап. С.-Петерб. минералог.
о-ва, 2-я серия, ч. 26, 458 A890).
Федоров Е. С. Зап. С.-Петерб. минералог.
о-ва, 2-я серия, ч. 28, 1 A891).
Федоров Е. С. Симметрия и структура
кристаллов. Серия «Классики науки».
Л.—М., Изд-во АН СССР, 1949.
Ферсман А. Е. Геохимия. М.—Л., ОНТИ,
Химтеорет, 1936.
Ферсман А. Е. Периодический закон Мен-
Менделеева в геохимии. Труды Юбилейного
Менделеевского съезда, т. 1. М., Изд-во
АН СССР, 1936.
Ферсман А. Е. ДАН СССР, 2, № 8—9, 556
A935).
Ферсман А. Е. Изв. АН СССР, вып. 35,
№ 10, 1425 A935).
Ферсман А. Е. ДАН СССР, 3, № 4, 173
A935).
Хоцянова Т. Я., Китайгородский А. И.,
Стручков Ю. Г. ЖФХ, 27, 780 A953).
Черняев И. И. Изв. Ин-та платины, 4, 213
A926).
Чугаев Л. А. Исследования в области ком-
комплексных соединений. М., 1906.
Шубников А. В. Изв. АН СССР, 515 A922).
Юм-Розери В. Структура металлов и спла-
сплавов. М.—Л., ОНТИ, 1938.
Avogadro A. J. Phys. Chim. d'Histoire Na-
turelle et des Arts, 7 A812).
Barlow ]., Pope W. J. J. Chem. Soc, 89,
1675, 1906; 91, 1150, 1907.
Barth T. F., Posnjak E. Z. Kristallogr., 82,
325 A932).
Bartholin E. Experimenta Crystalli Islan-
dici disdiaclastici gnibus mira et insoli-
ta refractio detegitur. Kopenhagen, 1669.
Berzelius 1. 1. Lehrbuch der chemie. Dre-
sen und Leipzig, 1845.
Bloch F. Z. Phys., 52, 555 A928).
Block F. Z. Phys., 59, 208 A930).
Bloch F. Die Elektronentheorie der Metal-
le. Leipzig, 1933.
Boer J. H. de. Rec. Trav. Chim. Pays. Bas.,
36, 301, 1937.
Bohr N. Phil. Mag., 26, 1 A913).
Born M. Verhandle Dtsch. phys. Ges., 21,
13 A919).
Born M. Z. phys. Chem., 37, 863; 38, 803
A926).
392
Born M., Lande A. Ber., 45, 1048 A948).
Bradley A. J., Jay A. H. Proc Roy. Soc,
A136, 210 A932).
Bragg W. L. Proc. Cambr. Phil. Soc, 17, 43
A913).
Bragg W. L. Atomic Structure of Mine-
Minerals. Oxford, Univ. Press, 1937.
Bragg W. H., Bragg W. L. The crystalline
state, vol. 1. London, 1933.
Bravais A. Etudes cristallographiques. Pa-
Paris, 1866.
Brillouin L. Die Quantenstatistik. Berlin,
1931.
Brown I. D., Dunitz J. D. Acta Cryst., 14,
N5, 480 A961).
Burgers 1. M. Proc. Kong. Nederl. Akad.
Wetensch., 42, 293, 378 A939).
Burgers 1. M. Proc. Phys. Soc, 52, 23
A940).
Burgers J. M. Proc. Kong. Nederl. Acad.
Wetensch., 50, 452, 595, 719, 858 A947).
Chatt J., Duncanson L. A. Nature, 178, 997
A956).
Chlopin V., Nikitin B. Z. phys. Chem.,
A145, 137 A929).
Clarke F. W. The Data of Geochemistry.
US. Geol. Sur., Bull., 770 A924).
Corey R. B. J. Amer. Chem. Soc, 60, 1598
A938).
Cotton F. A., Harris C. B. Inorg. Chem. U,
N3, 330 A965).
Cox E. G., Proc. Roy. Soc, A135, 491
A932).
Cox E. G., Cruickshank D. W. /., Smith
J. A. S. Nature, 175, 766 A955).
D'Ans J., Lax E. Taschenbuch fur che-
miker und Physiker. Berlin, 1943.
Davisson С J., Germer L. H. Nature, 119,
538 A927).
De Broglie L. Ann. phys., 3, 22 A925).
Debye P. Z. Phys., 21, 178 A920).
Drude P. Ann. Phys., 1, 566 A900).
Einstein A. Ann. Phys., 17, 132 A905).
Fafans K., loos I. Z. Phys., 23, 1, A924).
Fischer W., Pffaf Z. Naturforsch., 7b, 377
A952).
Frank F. C, Rasper 1. S. Acta Crystallogr.,
12, 483 A959).
Frankland E. Ann., 77 A851).
Friedrich W., Knipping P., Lane M. Ann.
Phys., 41, 971 A912).
Gibbs W. Amer. J. of Science and Arts, 31,
246 A861).
Goldschmidt V. M. Skr. Norske Vidensk.
Acad. (Oslo). I kl., 2, 112 A926).
Gordy W., Thomas W. J. Chem. Phys., 24,
439 A956).
Haber F. Verhandl. Dtsch. phys. Ges., 21,
750 («19).
Hagg G. Nova Acta Reg. Soc. Sci., 7, 3
A929).
Hagg G. Z. phys. Chem. (BJ9, 192 A935).
Найу R. Y. Essai d'une theorie sur la
structure des cristaux. Paris, 1784. ,
Heisenberg W. Z. phys., 43, 172 A927).
Heitler W., London F. Z. phys., Chem., 44,
455 A927).
Hessel J. F. С Krystallometrie oder Kry-
stallonomie und Krystallographie. Leipzig,
1897.
Hodgkin D. С Acta Kryst., 5, 770 A952).
Hornstra J. 1. Phys. Chem. Solids, 5,' 129
A958).
Hugging M. J. Amer. Chem. Soc, 75, 4123
A953).
Hume-Rothery W. The Metallic State. Ox-
Oxford. Univ. Press, 1931.
Jones H. Proc. Roy. Soc, (A) 144, 225
A934).
Jones. H. Proc. Roy. Soc, 49, 250 A937).
Kekule A. Lehrbuch der organischen Che-
mie, Bd I. Erlangen, 1861.
Keppler I. Strenasen de Nive Sexangula.
1619.
Kossel W. Ann. Phis., 49, 229 A916).
Kossel W. Valenzkrafte und Routgenspekt-
ren. Berlin, 1924.
Leeuwenhoek V. A. Arcana nature detecta.
1695.
Lewis G. J. Amer. Chem. Soc, 38, 762
A916).
London F. Z. Phys., 63, 245 A930).
London F. Z. phys. Chem., 11, 222 A931).
Machatschki F. Zbl. Min. Ceol. Palaont,
A97 A928).
Mathieson A., Mellor D. P., Stephen-
son N. 5. Acta Cryst., 5, 185 A952).
Mayer J., Helmholz L. Z. phys. Chem.,
75, 1 A932).
Mitscherlich E. Ann. Chim. Phys., 14, 172
A820).
Mitscherlich E. Ann. Chim, Phys., 19, 350
A822).
Mitscherlich E. Ann. Chim. Phys., 24, 264
A823).
Moseley H. Phil. Mag., 26, 1024 A913).
Moseley H. Phil. Mag., 27, 703 A914).
Orowan E. Z. Phys., 89, 614, 635 A934).
Pauli W. Z. Z. Phys., 31, 765 A925).
Pauling L. J. Amer. Chem. Soc, 49, 765
A927).
Pauling L. J. Amer. Chem. Soc, 51, 1010
A929).
Pauling L. J. Amer. Chem. Soc, 53, 1367
A931).
Pauling L. J. Amer. Chem. Soc, 54, 3570
A932).
Pauling L. Phys. Rev., 54, 899 A938).
Pauling L. Proc. Roy. Soc, A196, 343
A949).
Pauling L. J. Amer. Chem. Soc, 69, 542
A947).
Pauling L. General Chemistry. California,
1948.
Patterson A. L. Z. Krist., 90, 543 A935).
Pearson W. B. Acta Cryst., 17, 1 A964).
Planck M. Verhandle Dtsch. phys. Ges.,
2, 237 A900).
Prammelsberg K. F. Handbuch der Rristal-
lographisch — physikalischen Chemie.
Leipzig, 1881—1882.
Robertson J. M. Acta Cryst., 2, 238 A949).
Robertson J. M. Nature, 164, 915 A949).
Robertson J. M. Acta Cryst., 3, 251 A950).
Rome de Lisle. Cristallographie. Paris,
1783.
Rutherford E. A. Phil. Mag.,21, 669A911).
Schoenflies A. Theorie der Krystallstruc-
tur. Berlin, 1923.
Schroedinger E. Ann. Phys., 79, 36, 489;
80, 437; 81, 109 A926).
Sen S. N. Indian J. Phys., 22, 347 A948).
Shulze G. E. Z. Electrochem. 45, 849
A939).
Sidgwick N. V. The Electronic Theory of
Valency. Oxford, 1929.
Simonsen S. H. J. Inorg. Nucl. Chem.,
27, 309 A965).
Smare D. L. Acta Cryst, 1, 150 A948).
Snyder D. A., Weaver D. L. Inorg. Cnem.,
9, 2760 A970).
Stensen N. De solido intra solidum na-
tura liter contento. Firenze, 1669.
Stillwell С W. Crystal Chemistry N. Y.—
London, 1938.
Taylor G. I. Proc. Roy. Soc, A145, 362
A934).
Tennison C. Fortschr. Mineral., 41, 64
A963).
Tscherman G. Lehrbuch der Mineralogie.
Wien — Leipzig, 1921.
Ubbelohde A. R. Proc. Roy. Soc, A173,
417 A939).
Van Arkel A. E., de Boer J. H. Chemische
Bindung als Electrostatisch Verschinsel.
Amsterdam, 1930.
Vegard L. Z. Phys., 5, 17 A921).
Van't Hoff J. H. Vorlesungen iiber theore-
tische und physinalische Chemie. Bra-
Braunschweig, 1900.
Van Nickerk I. N., Schoening F. R. L. Acta
Cryst., 6, 609 A953).
Wasastjerna J. A. Soc. Sci. Fenn. Comm.
Phys. Math. I, 38, 22 A923).
Weiss С S. Dynamische Anschnitt der
Rrystallisation. Leipzig, 1804.
Weiss С S. Ubersichtliche Darstellung der
verschieden naturalistsch Abteilungen
der Krystallisations systeme.— Berlin,
1814-1815.
Werner A., Miolati A. Z. phys. Chem., 12,
48 A893).
Werner A., Miolati A. Z. phys. Chem., 14,
510 A894).
Wigner E., Seitz F. Phys. Rev., 43, 804
A933).
Wollaston W. H. Description of a reflecti-
reflective goniometer.— Philosophical Trans.
of the Roy. Soc of London, 1809.
Wulff G. Z. Kryst., 36, 1 A902).
Wulff G. Z. Phys., 14, 217 A913).
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Автоизоморфные вещества 234
Автоморфотропия 228
Агрегат кристаллический 9
Аллотропия 219
— концентрационная 314
Анализ кристаллохимический 61
Ангстрем 118
Анизотропия 8
Антиизоморфизм 227
Атомный фактор 114
Бравэ закон 60
Бертоллиды 300
Бриллюэна зоны 200
Вакансия 255
Валентность 168, 169
— побочная 375
Валентные углы 357
Валентных связей метод 192
Вид симметрии 22
Вицинали 17
Время кристаллизации 7
Вульфа сетка 16
Вульфа теорема косинусов 48
Гибридизация состояний 195
Голоэдрический вид симметрии 34
Гониометр 13
Грань возможная 40
— действительная 41
— единичная 45
Группа переносов 53
— трансляционная 53
Дальтониды 300
Двойник 41
— полисинтетический 42
Двойниковый элемент симметрии 41
Дислокация 259
— винтовая 260
— криволинейная 261
— линейная 259
Закон Бравэ 60
— кристаллографических пределов 61
— постоянства углов 12—18
— рациональности отношений параметров
(закон целых чисел) 43
Зона 41
Изоморфизм 219
— совершенный и несовершенный 221
—изовалентный и гетеровалентный 229,
346
— с заполнением пространства 230
Изоструктурные вещества 223
Индекс символа грани 47
Ионы 165
Каолиновое ядро 332
Категория 31
— высшая 31
394
— низшая 31
— средняя 31
Класс 281
Ковкость 245
Комплекс 375
Комплексобразующий атом 375
Конформации 370
Координационное число 124
Координационный многогранник 124
Коэффициент механического сжатия 247
— термического расширения 247
Кратность правильной системы точек 66
Кристалл идеальный 254
— мозаичный 255
— реальный 254
Кристаллизация 11
Кристаллогидраты 373
Кристаллохимический анализ 60
Кристаллография 9
Метод в ращения 109
— гармонический 114
— качания (колебания) НО
— Лауэ 109
— Паттерсона 116
— порошка 111
— последовательных приближений 117
— Фурье 115
— проб и ошибок 114
— развертки слоевых линий 113
Монокристалл 9, 11
Морфотропия 227
Однородные тела 8
Ось зеркально-поворотная 20
— зоны 41
— инверсионная 20
— симметрии 20
— трансляции 53
Отряд 282
Параллелоэдры 61
Параметр решетки 58
— ряда 51
Период идентичности 108
Плоскость скользящего отражения 64
— симметрии 19
Погасания 112
Полиморфизм 219
Политипия 224
Полуметалл 278
Полупроводник 214
Поляризация ионов 145
Потенциал ионизации атомов 167
Пояс см. зона
Правило Вегарда 223
— Гольдшмидта 148
— Полинга для структур ионных кристал-
кристаллов 328
— электростатической валентности 328
— Юм-Розери 270
Правильная система точек 66
Пределы устойчивости структур 143
Проекция Паттерсона 116
¦— стереографическая 15
Простая форма 35
— — общая 35
— — частная 35
Пространственнонапряженные молекулы
362
Пространственные группы симметрии 64
Пустоты октаэдрические 150
— тетраэдрические 150
Радиусы ионные 137
— ковалентные 181
— металлические 284
— эффективные 136
Растворимость 251
Ретикулярная плотность 60
Решетка кристаллическая 51, 53
— пространственная 51
— Бравэ 58
Род 280
Ряд 52
Связь химическая 134
— ван-дер-ваальсовская (остаточная) 206
— водородная 210
— донорно-акцепторная 178
— ионная 169 -•••ij.fi
— ковалентная или гомеополярная 177
— координационная 177
— металлическая 197
— многоцентровая 386
— л-связь 387
— промежуточная 209
— трехцентровая 387
Семейство 282
Сетка плоская 56
Сжатие актиноидное 141
— лантаноидное 140, 141
Символ грани 47
Симметрическое преобразование 19
Симметрическая фигура 19
Симметрия 8, 19
— шаровых упаковок 153
Синглетное состояние 193
Сингония 31
Скульптура граней 17
Слоевая линия 110
Слюды 42
Соединения переменного состава 304
— мнимые 305
Спайность 246
Способность самоогранения 8
Сростки закономерные 42
Стереографическая проекция 15
Структура кристалла 120, 148
— гетеродесмическая 135
— гомодесмическая 135
— внедрения 231
— дефектная 232
— каркасная 237, 337
— координационная 236
— молекулярная 128
— неупорядоченная 299
— островная 237, 353
— с дробным количеством атомов в эле-
элементарной ячейке 234
— с параметрами и без параметров 130
— сендвичевая 386
— слоистая 146, 237, 337, 353
— упорядоченная 295
— цепочечная 237, 335, 353
Структурная амплитуда 114
Структурный коэффициент'(Маделунга)
173
Структурный тип 120
— антиизоморфный 127
Твердость 248
Твердые растворы 231, 286
— — второго рода 231
— — вычитания 232
— — внутренние 235
— — ограниченные 292
— — первого рода 231
Температура плавления 248
Точечная группа симметрии 24
Трансвлияние 380
Трансляционная группа 53
Трансляция 53
Узел решетки 54
Упаковка шаров плотнейшая 149, 344,
359
— — — гексагональная^149
— — — кубическая 149
— — — многослойная 151
Упорядочение 298
Установка кристаллов 49
Федоровские группы симметрии 68
Формульная единица 122
Центр симметрии 20
Центральный атом 375
Шредингера уравнение 183
Электронная концентрация 293
— эмиссия 205
Электронные оболочки атома 162
Электроотрицательность 212
Элементы симметрии 19
Элементы симметричности 65
Энергия* решетки 171
— сродства к электрону 168
Эпитаксия 42
Эффект дисперсионный 207
— индукционный или наведенный 207
— ориентационный 207
— экранирования ионов 250
Эффективная масса электрона 202
Юстировка кристалла 14
Ячейка решетки 52
— базоцентрированная 58
— гранецентрированная 58
— примитивная 58
— объемноцентрированная 58
— центрированная 58
— элементарная 58
ОГЛАВЛЕНИЕ
Предисловие к третьему изданию
Важнейшие таблицы в книге
Часть перв ая
Законы геометрической ирп-
оталлографпп
Глава I
Понятие о кристалле, кристаллическом
веществе и кристаллографии
§ 1. Кристаллическое вещество
§ 2. Основные свойства кристалла
§ 3. Кристалл и кристаллическое ве-
вещество
§ 4. Кристаллография
§ 5. Распространенность кристалли-
кристаллического вещества
§ 6. Кристаллизация. Производство
монокристаллов
Глава IV
Формы кристаллических многогранни-
двугранных углов
Глава II
Закон постоянства
в кристаллах
§ 1. Первые работы, посвященные
изучению внешней формы кри-
кристаллов
§ 2. Методы измерения кристаллов
§ 3. Методы вычисления кристаллов
§ 4. Отклонения от закона постоян-
постоянства углов
Глава III
Симметрия кристаллов
§ 1. Понятие о симметрии
§ 2. Элементы симметрии
§ 3. Сложение элементов симметрии.
Виды симметрии
§ 4. Схема вывода 32 видов симмет-
симметрии
§ 5. Систематика видов симметрии
7
7
7
8
8
9
ков
§ 1. Понятие
§ 2. Простые
НИИ
§ 3. Простые
НИИ
§ 4. Простые
гонии
простой формы
формы низших синго-
формы средних синго-
формы кубической син-
§ 5. Возможные грани
§ в. Двойники
ки
Глава V
[ и закономерные срост-
35
35
36
37
39
40
41
10
11
12
12
13
15
Закон целых чисел и аналитические ме-
методы описания кристаллических много-
многогранников 43
§ 1. Открытие закона целых чисел в
кристаллографии 43
§ 2. Кристаллографические символы 47
§ 3. Математическое определение
символов грани 48
§ 4. Установка кристаллов 49
Часть -вторая
Геометрическая теория струк-
структуры кристалла 51
16
19
19
19
22
26
31
Глава VI
Кристаллическая решетка
§ 1. Понятие кристаллической ре-
решетки
§ 2. Кристаллический многогранник
и решетка кристалла
§ 3. Трансляция
§ 4. Плоские сетки решетки
§ 5. 14 решеток Бравэ
§ 6. Понятие о кристаллохимиче-
ском анализе
51
51
52
53
56
5S
60
396
Глава VII
Теория структуры кристаллов Е. С. Фе-
Федорова 64
§ 1. Краткие сведения о теории 64
{ 2. Федоровские группы симметрии 68
Глава VIII
Экспериментальная проверка геомет-
геометрической теории кристаллов. Рентгено-
структурный анализ 106
| 1. Первые определения атомных
структур кристаллов при помо-
помощи рентгеновских лучей 106
§ 2. Кристалл как дифракционная
решетка . 108
§ 3. Методика определения парамет-
параметров и типа решетки 109
§ 4. Методика определения про-
пространственных групп симмет-
симметрии 112
§ 5. Определение положения ато-
атомов в кристаллической решетке 113
§ 6. Гармонический метод рентгено-
структурного анализа 114
Часть третья
Основные иоиятпя кристалло-
кристаллохимии 118
Глава IX
Результаты первых рентгеноструктур-
пых исследований кристаллов 118
§ 1. Три простейшие кристалличе-
кристаллические структуры чистых метал-
металлов 118
§ 2. Число атомов, приходящихся на
одну ячейку структуры 119
§ 3. Число правильных систем точек
в структуре 120
§ 4. Структура кристалла и струк-
структурный тип 120
§ 5. Структура алмаза и графита 121
§ 6. Простейшие структуры соеди-
соединений типа АХ 122
§ 7. Координационное число и коор-
координационный многогранник 124
§ 8. Простейшие структуры типа
АХ2 и А2Х 127
§ 9. Классификация структур по ко-
координационным числам 129
§ 10. Структуры с параметрами и без
параметров 130
§11. Вычисления межатомных рас-
расстояний и валентных углов в
структурах 131
§12. Структура кристалла, кристал-
кристаллическая решетка и правильная
система точек 132
§13. Основные выводы, сделанные
на основании первых определе-
определений структур кристаллов 133
Глава X
Факторы, определяющие структуру
кристаллов 134
§ 1. Установление различных типов
химической связи 134
§ 2. Гетеродесмические и гомодес-
мические структуры 135
§ 3. Эффективные радиусы ионов 136
§ 4. Определение ионных и атомных
радиусов 136
§ 5. Ионные радиусы химических
элементов 137
§ 6. Метод изображения кристалли-
кристаллических структур шарами раз-
разных размеров 141
§ 7. Геометрические пределы устой-
устойчивости структур с различными
координационными числами 141
§ 8. Поляризация ионов 144
§ 9. Зависимость размеров атомов и
ионов от координационных чи-
чисел. Структурный тип перов-
скита 145
§10. Слоистые структуры 146
§11. Влияние поляризации на струк-
структуру кристаллов 148
§12. Факторы, определяющие струк-
структуру кристаллов (правило
Гольдшмидта) 148
Глава XI
Теория плотнейших шаровых упа-
упаковок 149
§ 1. Гексагональная и кубическая
плотнейпше шаровые упаковки 149
§ 2. Типы пустот в шаровых упаков-
упаковках 150
§ 3. Многослойные упаковки. Спосо-
Способы обозначения плотнейших
шаровых упаковок 151
§ 4. Предварительные замечания о
симметрии шаровых упаковок.
Кубическая плотнейшая шаро-
шаровая упаковка 152
§ 5. Федоровские группы симметрии
гексагональных шаровых упа-
упаковок 153
§ 6. Элементы симметрии плотней-
ших шаровых упаковок 154
§ 7. Правильные системы точек в
плотнейших шаровых упаков-
упаковках 154
§ 8. Значение теории шаровых упа-
упаковок для кристаллохимии 155
§ 9. Метод изображения структур-
структурных типов с помощью много-
многогранников. Структуры из тетра-
тетраэдров и октаэдров 156
§10. Структуры со сложными коор-
координационными многогранниками 158
Г л а в а XII
Типы химических связей в кристаллах 161
§ 1. Периодическая система химиче-
химических элементов и строение ато-
атомов 161
§ 2. Ионная связь 169
§ 3. Элементарные представления о
ковалентной связи 177
§ 4. Физические основы ковалент-
ковалентной связи 182
§ 5. Ковалентная связь в молекулах
и кристаллах 189
§ 6. Металлическая связь 197
§ 7. Остаточная связь 206
§ 8. Промежуточные типы связи 209
Глава XIII
Изоморфизм и полиморфизм 219
§ 1. История открытия 219
§ 2. Дорентгеновские работы по изо-
изоморфизму 219
§ 3. Дорентгеновские работы по по-
полиморфизму 221
§ 4. Первые рентгеноструктурные
исследования изоморфных ве-
веществ 223
§ 5. Структурная классификация ти-
типов полиморфизма 224
§ 6. Условия, необходимые для про-
проявления изоморфизма 226
§ 7. Предел изоморфной заместимо-
сти. Морфотропия и полимор-
полиморфизм 227
§ 8. Влияние изотопного состава на
кристаллическую структуру 228
§ 9. Изовалентный и гетеровалент-
ный изоморфизм 229
§10. Изоморфизм с заполнением про-
пространства 230
§11. Твердые растворы второго рода.
Структура внедрения 231
§12. Твердые растворы вычитания.
Дефектные структуры 232
§13. Структуры с дробным количест-
количеством атомов в элементарной
ячейке 234
§14. Внутренние твердые растворы.
Автоизоморфные вещества 234
Глава XIV
Классификация структурных типов 236
§ 1. Предварительные замечания о
классификации структурных
типов 236
§ 2. Группы структурных типов с
нейтральными и заряженными
структурными мотивами 237
§ 3. Границы применимости приня-
принятой классификации структур-
структурных типов 238
§ 4. Метод изображения структур-
структурных типов формулами 240
§ 5. Структурные химические фор-
формулы 240
Глава XV
Зависимость физико-химических
свойств твердых веществ от строе-
строения кристаллов 243
§ 1. Зависимость физико-химиче-
физико-химических свойств твердых веществ
от типа химической связи в
кристаллах 243
§ 2. Электрические свойства 244
§ 3. Оптические свойства 244
§ 4. Ковкость металлов 245
§ 5. Спайность 246
§ 6. Коэффициенты механического
сжатия и термического расши-
расширения 247
§ 7. Твердость и температура плав-
плавления 248
§ 8. Влияние водородной связи на
физико-химические свойства ве-
веществ 249
§ 9. Эффект экранирования иоиов 250
§10. Растворимость 251
398
Глава XVI
Строение реального кристалла
§ 1. Идеальный и реальный кри-
кристалл
§ 2. Точечные дефекты в атомной
структуре кристалла
§ 3. Дислокации
§ 4. Зависимость физико-химиче-
физико-химических свойств кристаллов от ре-
реальной структуры
Часть четвертая
Кристаллохимия иростых ве-
веществ п химических соедине-
соединений
Глава XVII
Кристаллохвмические закономерности
в периодической системе элементов
Д. И. Менделеева
§ 1. Предварительные замечания
§ 2. Кристаллические структуры ис-
истинных металлов
§ 3. Особенности структурных типов
y-Mn, Hg и Zn
§ 4. Кристаллические структуры
элементов 6-подгрупп
§ 5. Особенность элементов Ill-b и
lY-b подгрупп, имеющих типич-
типичные структуры металлов
§ 6. Распределение элементов по
подгруппам периодической си-
системы ¦ на основании кристалло-
химических данных
§ 7. О классификации химических
соединений 275
§ 8. Классификация двойных (би-
(бинарных) и более сложных хи-
химических соединений 277
§ 9. Кристаллохимическая система-
систематика химических соединений 280
Глава XVIII
Кристаллохимия интермсталлических
соединений 284
§ 1. Система «металлических» ра-
радиусов атомов 284
§ 2. Типы взаимодействия металли-
металлических атомов в двойных систе-
системах 284
§ 3. Системы металлов, образующих
два жидких слоя или эвтек-
эвтектику 286
§ 4. Структурная характеристика
254 твердых растворов и интерме-
интерметаллических соединений
254 § *>• Двоииые металлические систе-
системы с неограниченной взаимной
255 растворимостью компонентов в
259 твердом состоянии
§ 6. Влияние полиморфизма метал-
металлов на тип диаграммы с твер-
262 дыми растворами. Твердые ра-
растворы железа с другими метал-
металлами
§ 7. Изменение констант решеток
твердых растворов
Ограниченные твердые растворы
Явление «старения» сплавов
Твердые растворы вычитания
Двойные металлические систе-
системы с тремя и большим количе-
количеством твердых фаз
Особенности строения интерме-
интерметаллических соединений. Отно-
Отношения между интерметалличе-
интерметаллическими соединениями и тверды-
твердыми растворами
Процесс упорядочения в интер-
интерметаллических фазах
Дальтониды и бертоллиды
О систематике соединений пе-
переменного состава
Важнейшие структурные типы
бинарных интерметаллических
соединений
Природа интерметаллических
соединений
266
266
266
268
268
270
273
273
§8.
§ 9.
§10.
§11.
§12.
§13.
§44.
§15
§16.
§17.
Глава XIX
Кристаллохимия неорганических со-
§ 1. О классификации бинарных со-
соединений
§ 2. Тройные и более сложные неор-
неорганические соединения
§ 3. Структурная систематика клас-
класса сульфатов
§ 4. Правила Полинга для структур
ионных кристаллов
§ 5. Особенности структур с преиму-
преимущественно ковалентным типом
связи
§ 6. Кристаллохимия силикатов
§ 7. Кристаллохимия боратов
287
289
291
292
294
294
295
297
298
300
304
307
314
318
318
319
322
328
329
330
349
399
Глава XX
Кристаллохимия органических соеди-
соединений и их аналогов 354
§ 1. Ковалентные и Ван-дер-Вааль-
совы «радиусы» неметалличе-
неметаллических элементов 354
§ 2. Предварительные замечания о
молекулярных структурах 356
§ 3. Формы простейших молекул и
комплексных ионов 357
§ 4. Валентные углы 357
§ 5. Классификация молекулярных
структур 358
§ 6. Применение принципов плот-
нейшей упаковки к молекуляр-
молекулярным кристаллам 359
§ 7. Строение углеводородов 360
§ 8. Строение более сложных орга-
органических соединений 362
§ 9. Ионные структуры соединений
элементов-органогенов 365
§10. Зависимость характера связи от
межатомных расстояний 367
§11. Пространственные затруднения
в молекулах. Конформации 370
Глава XXI
Кристаллохимия сложных химиче-
химических соединений. Кристаллогидраты,
комплексные, металлоорганические
и клатратные соединения 373
§ 1. Строение кристаллогидратов 373
§ 2. Строение комплексных соеди-
соединений 375
§ 3. Строение металлоорганических
соединений 385
§ 4. Клатратные и другие сложные
химические соединения
Литература
Предметный указатель
388
390
39л
Георгий Борисович Бокий
КРИСТАЛЛОХИМИЯ
Издание 3-е, переработанное и дополненное
Утверждено к печати
Институтом радиотехники и электроники
Академии наук СССР
Редактор Р. А. Баранова
Художник Э. Л. Эрман
Художественный редактор Н. Н. Власик
Макет и техническое редактирование
Э. Л. Куниной
Сдано в набор 24/XI 1970 г.
Подписано к печати 19/VTI 1971 г.
Формат 70Xl00'/u. Бумага Ni 1.
Усл. печ. л. 32,65. Уч.-изд. л. 27,7.
Тираж 11 000. Т-10640. Тип. зак. 1544.
Цена 2 р. 34 к.
Издательство «Наука».
Москва К-62, Подсосенский пер., 21
2-я типография издательства «Наука».
Москва Г-99, Шубинский пер., 10
Исправления и опечатка
Стр.
69
250
291
337
342
355
365
Колонка
Слева
Справа
Справа
Слева
Слева
Таблица
Справа
Строка
1 СВ.
8 сн.
1 и 2 сн.
4 сн.
21 сн.
14 сн.
9 сн.
Напечатано
14
меньший
Ni—А1 от состава
мотива
не полярныя
0,35
NaCl-[
Должно быть
/4
наибольший
от состава
мотива мелилита—
полярные
1,35
NaCl: [
Кристаллохимия