


























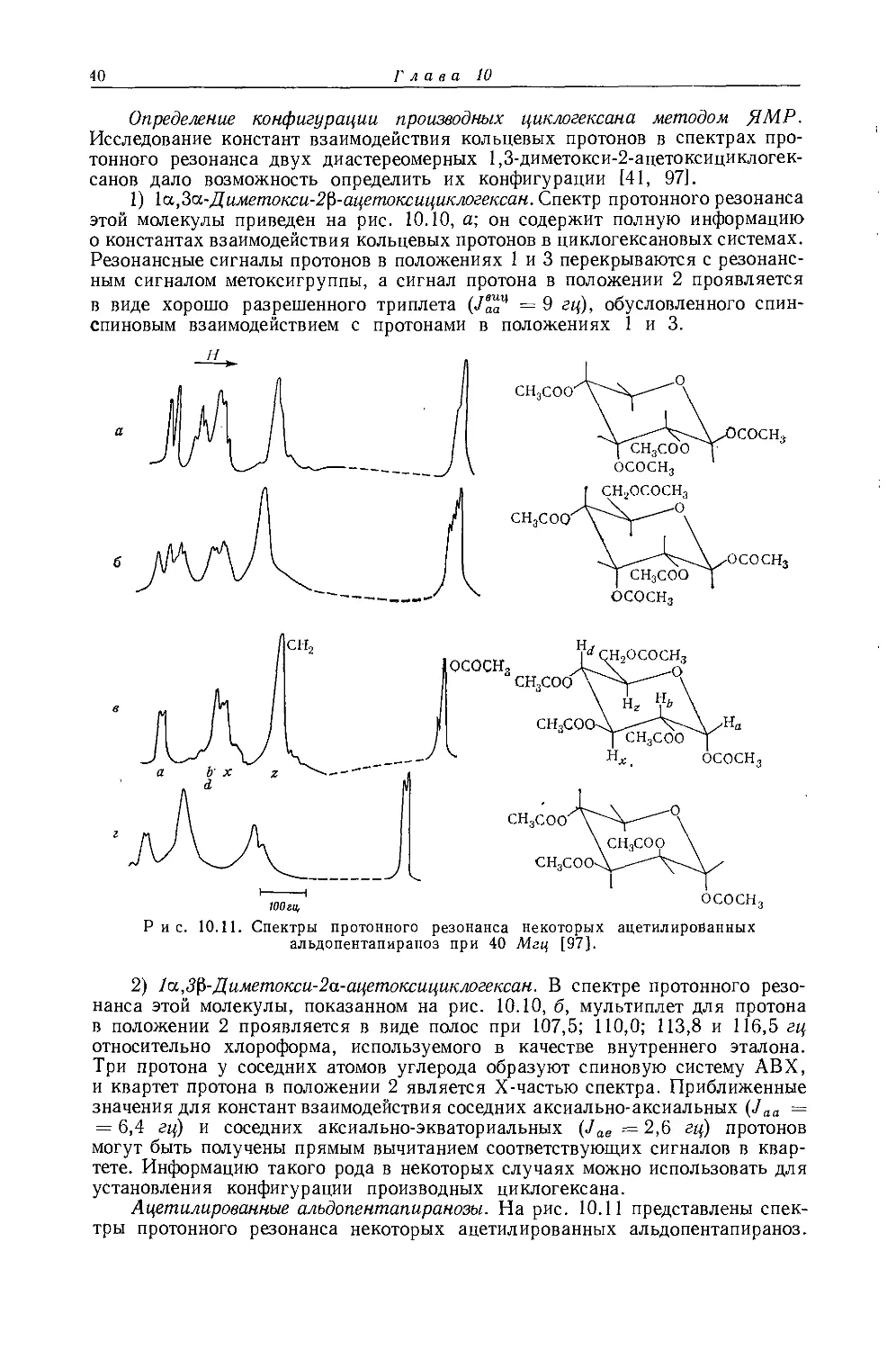








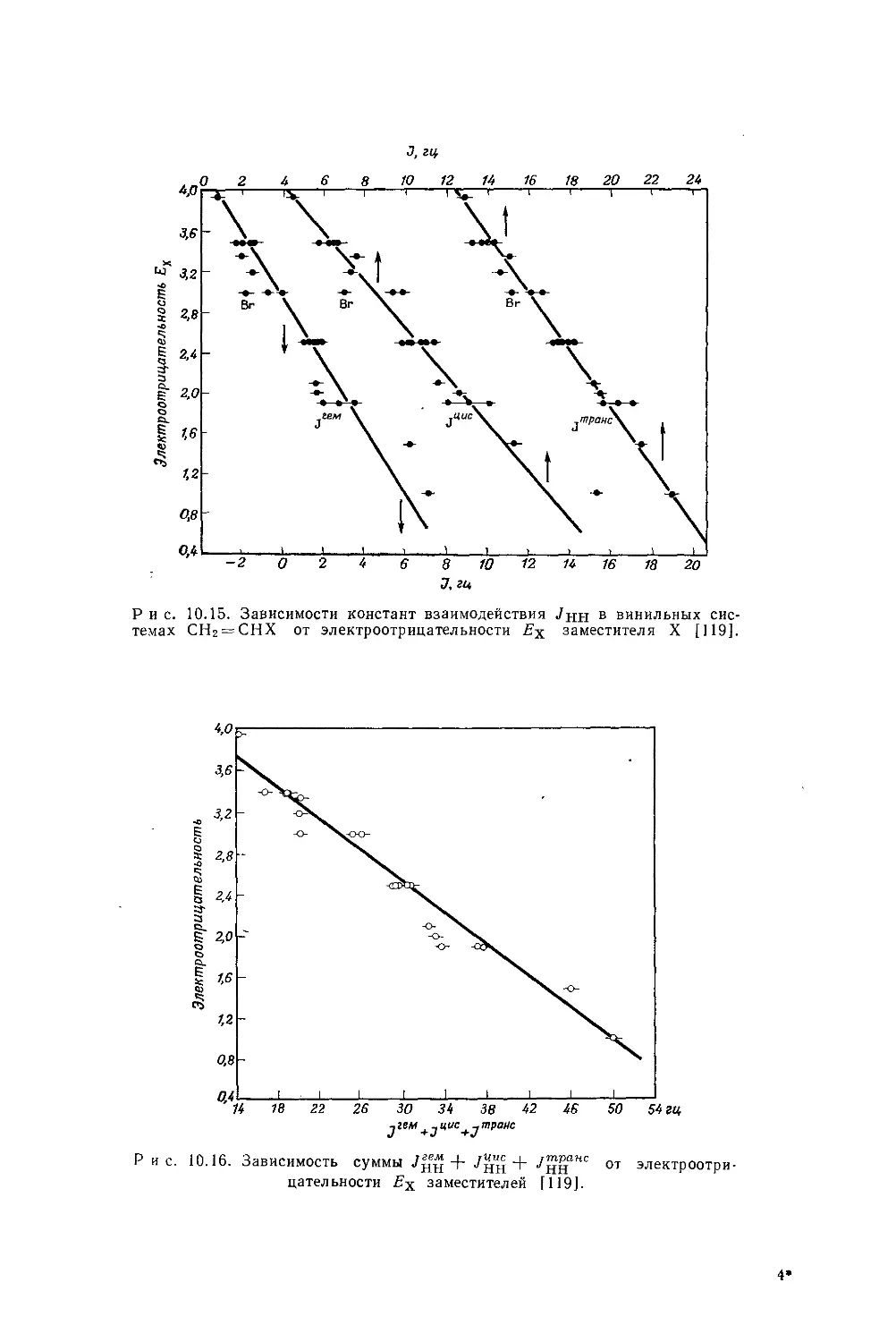












































































































































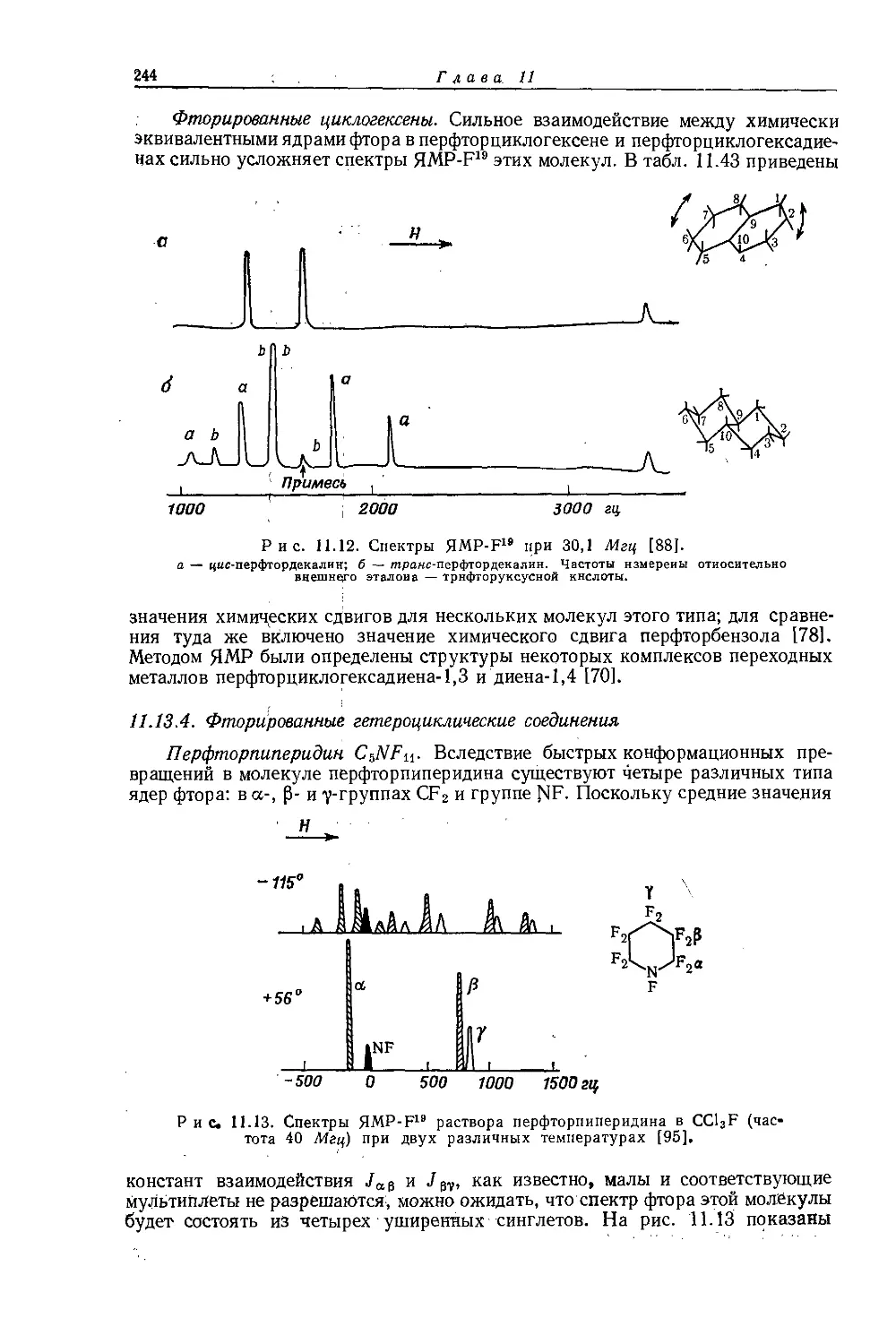









































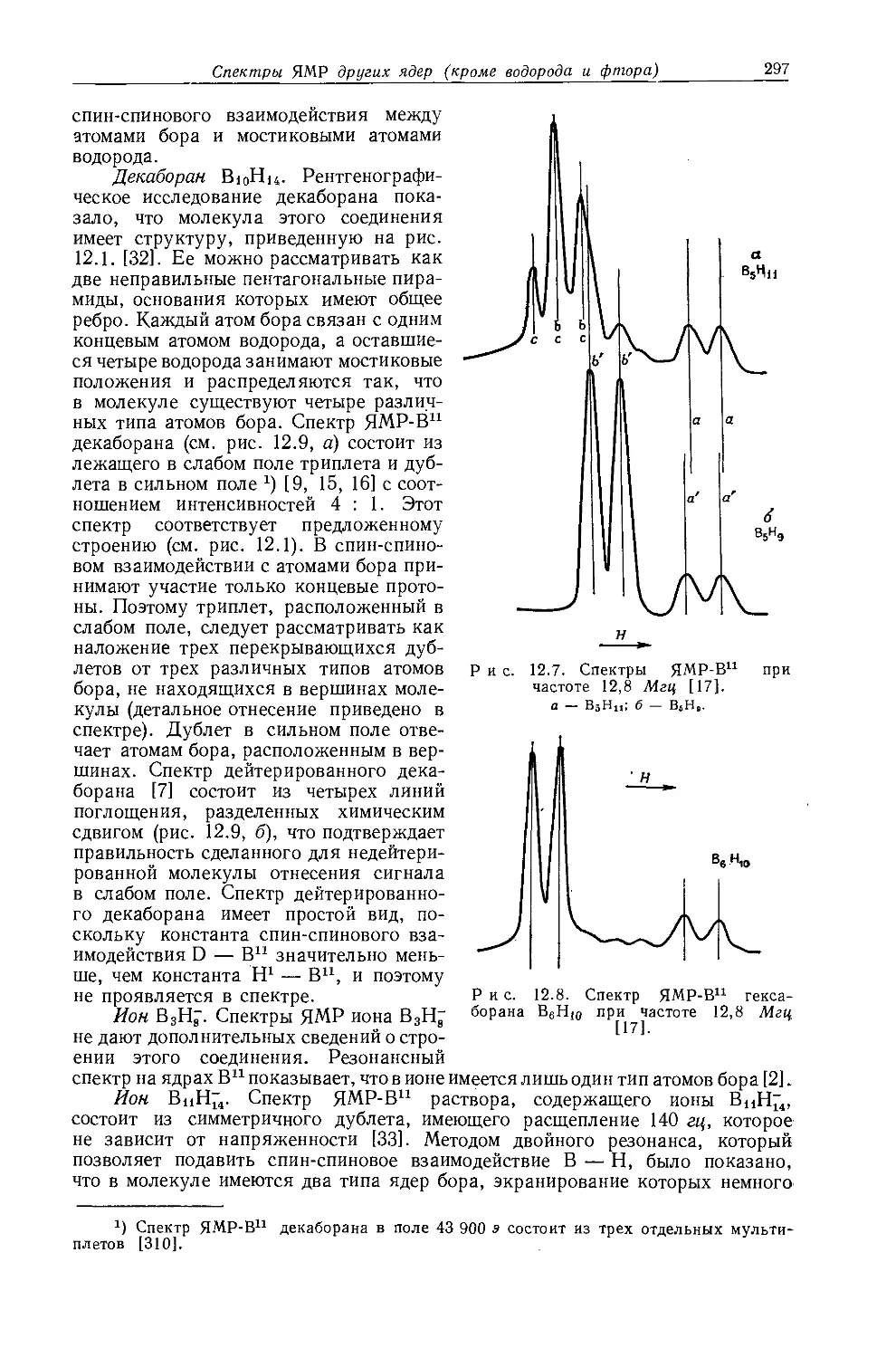
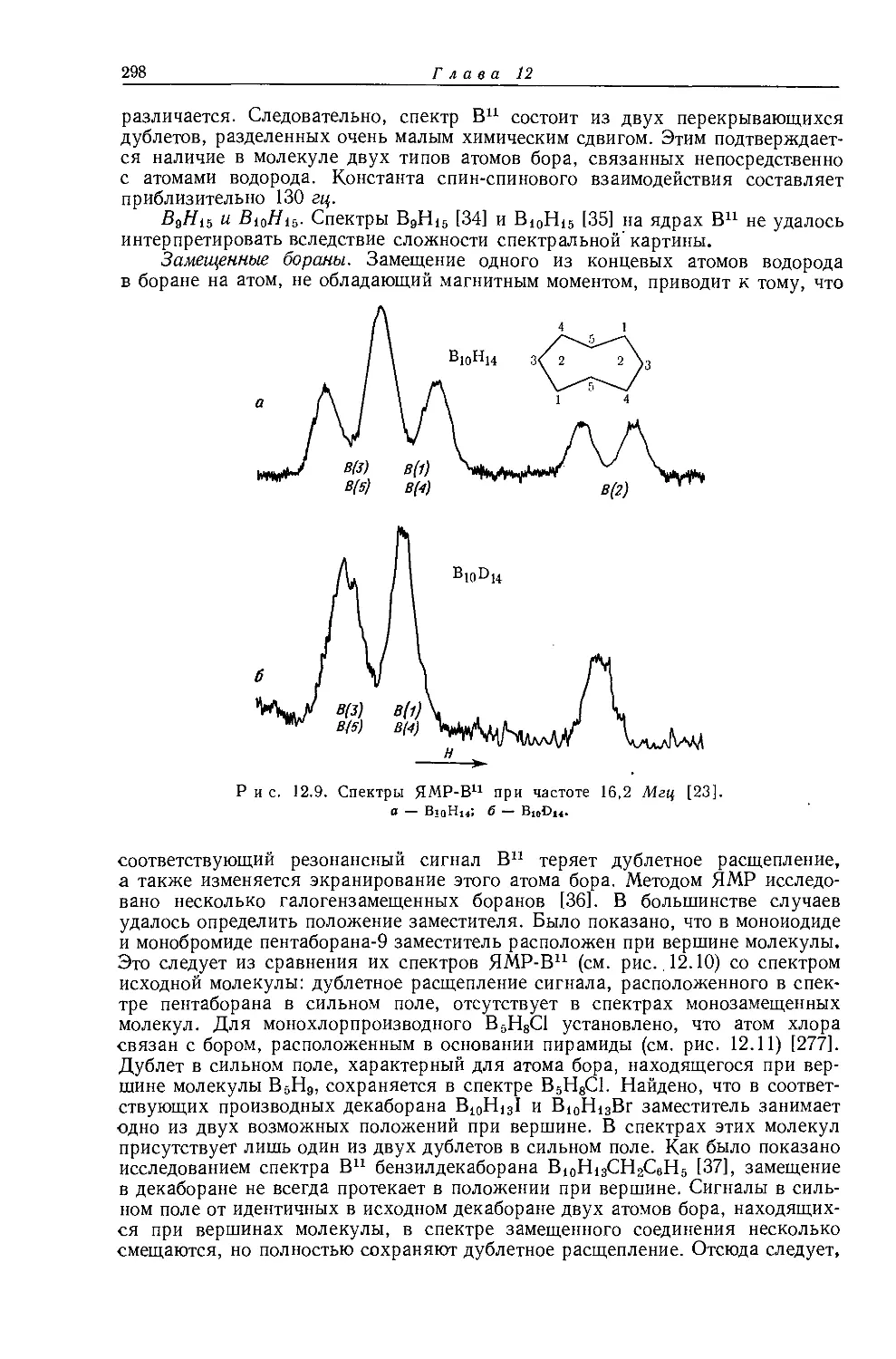
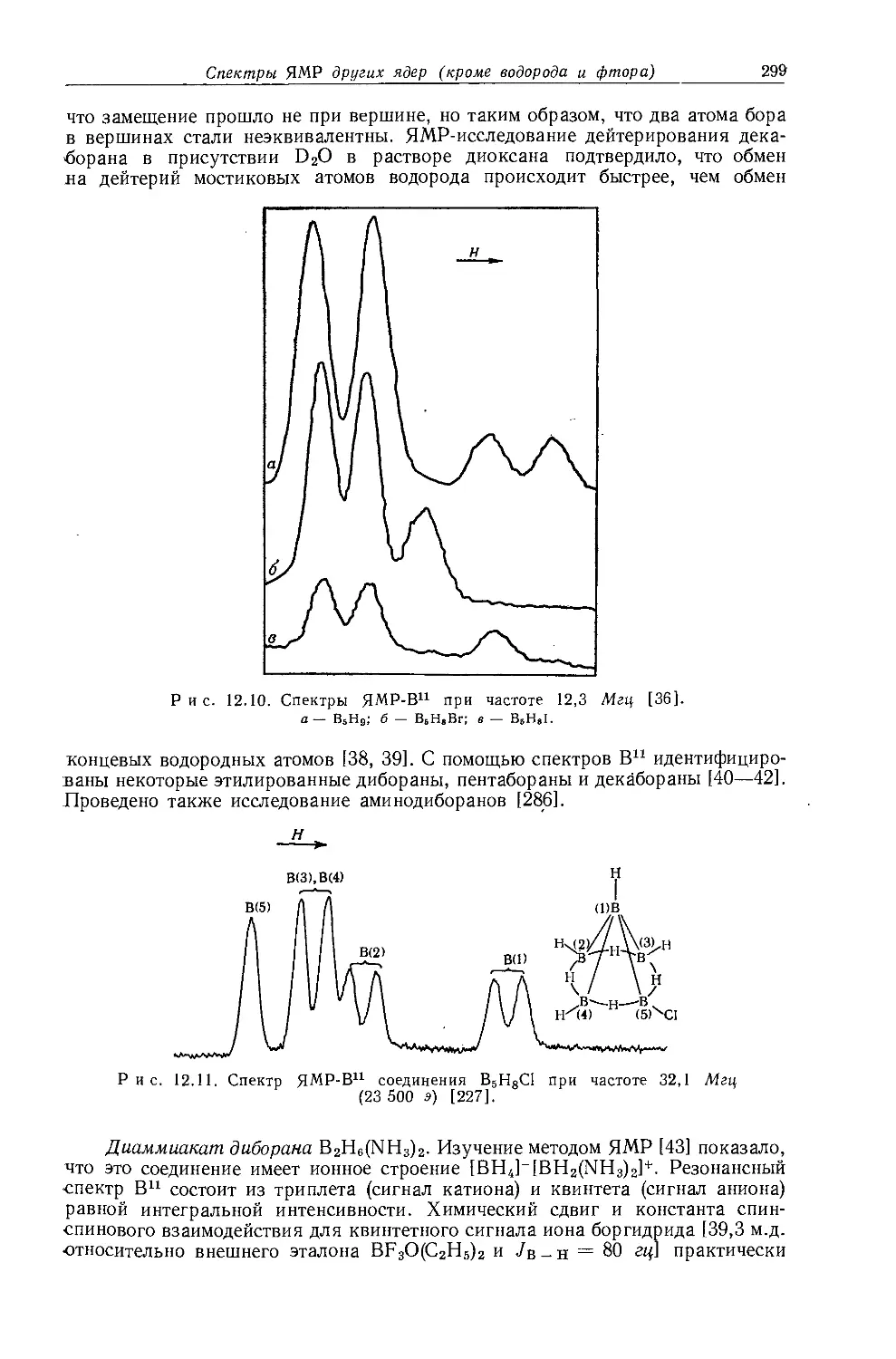





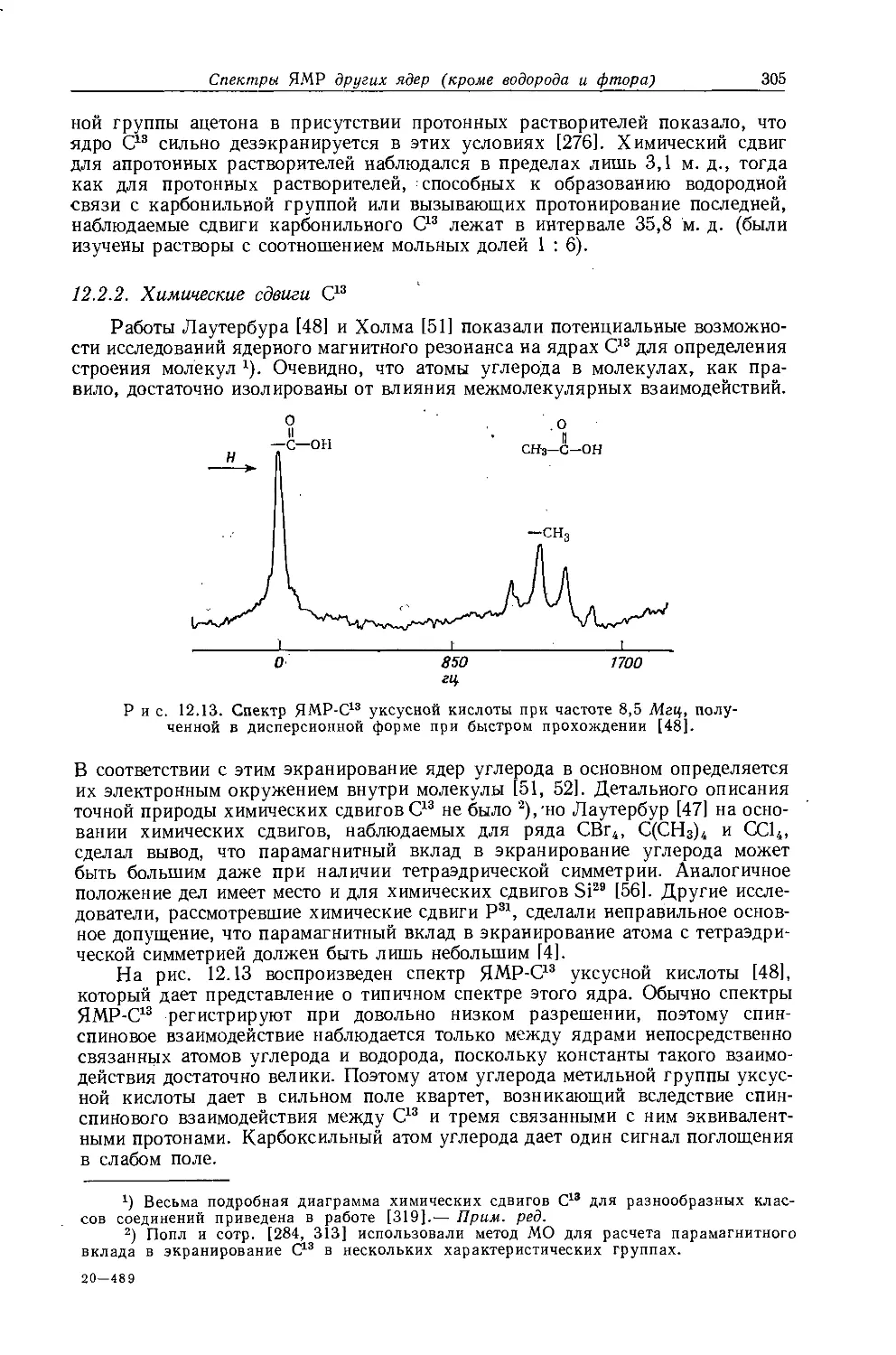












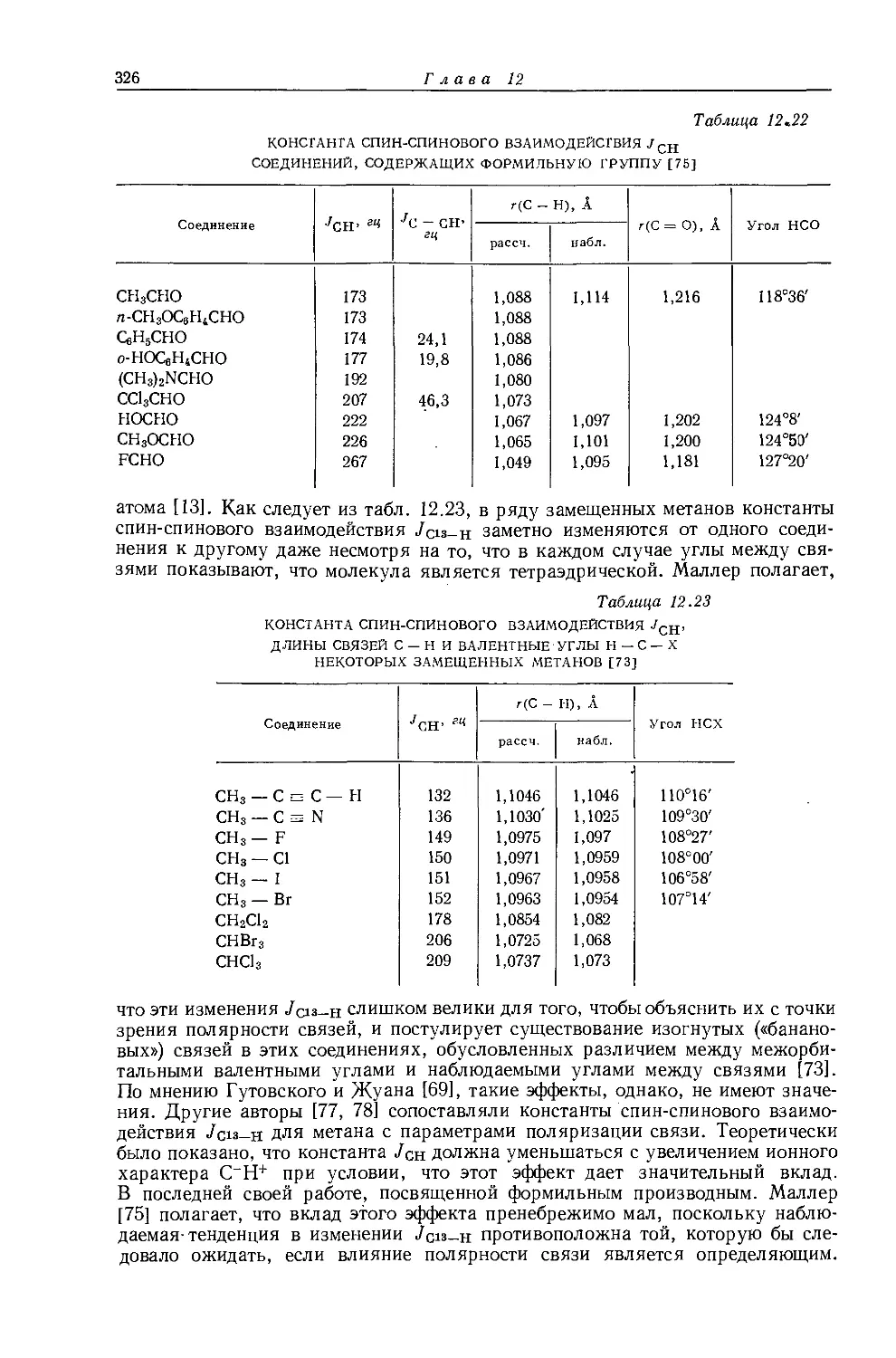


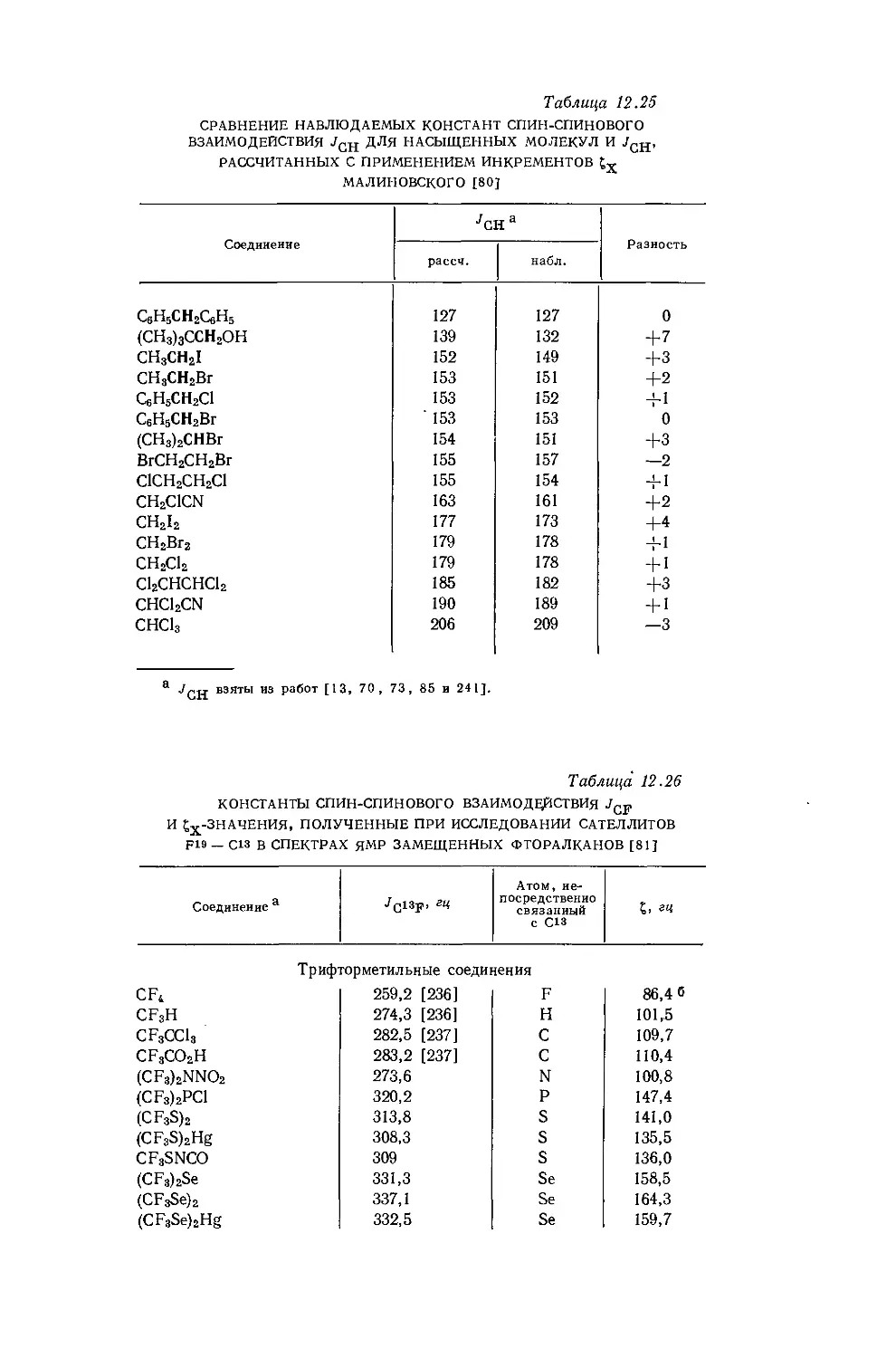
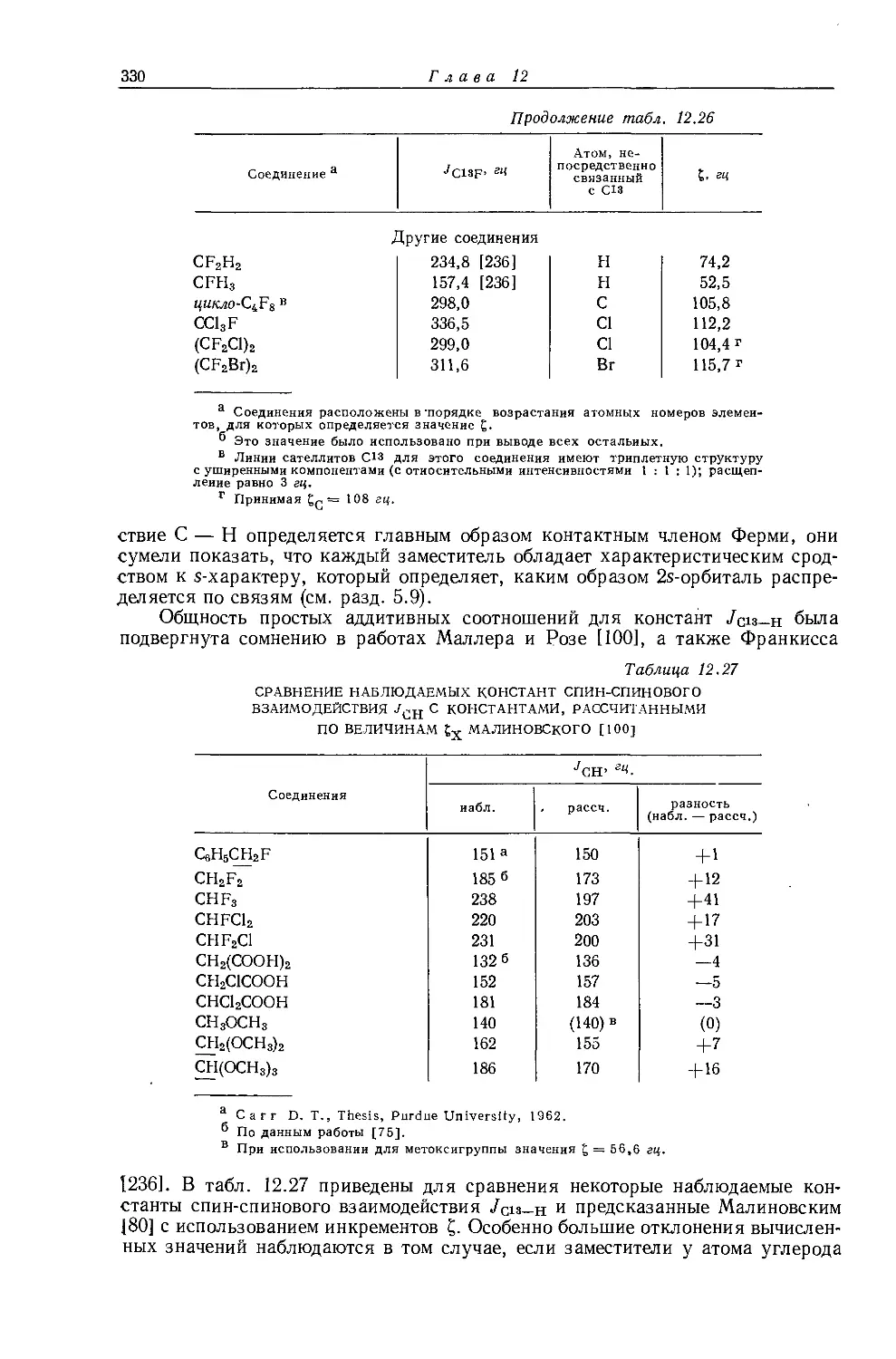


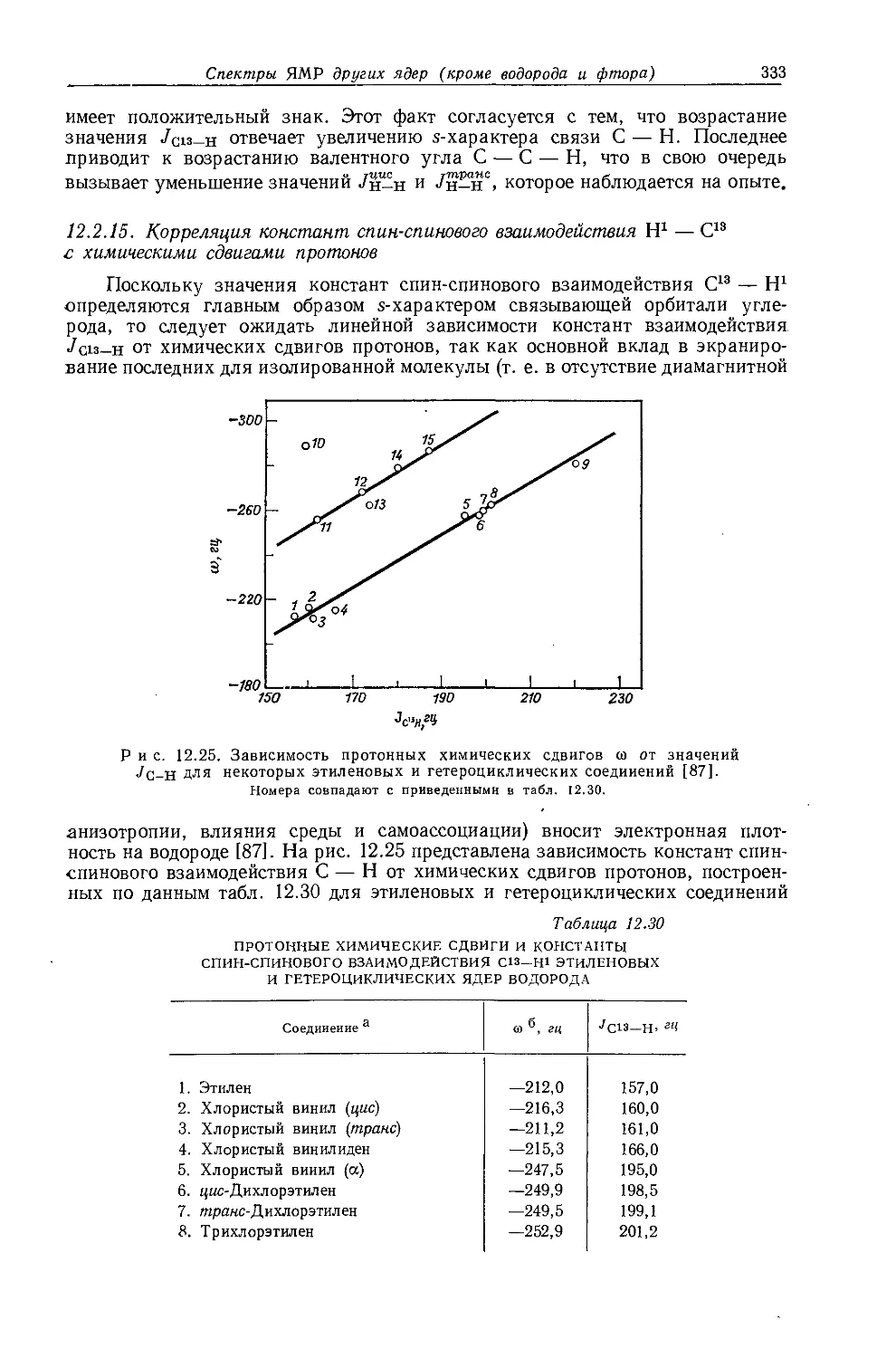






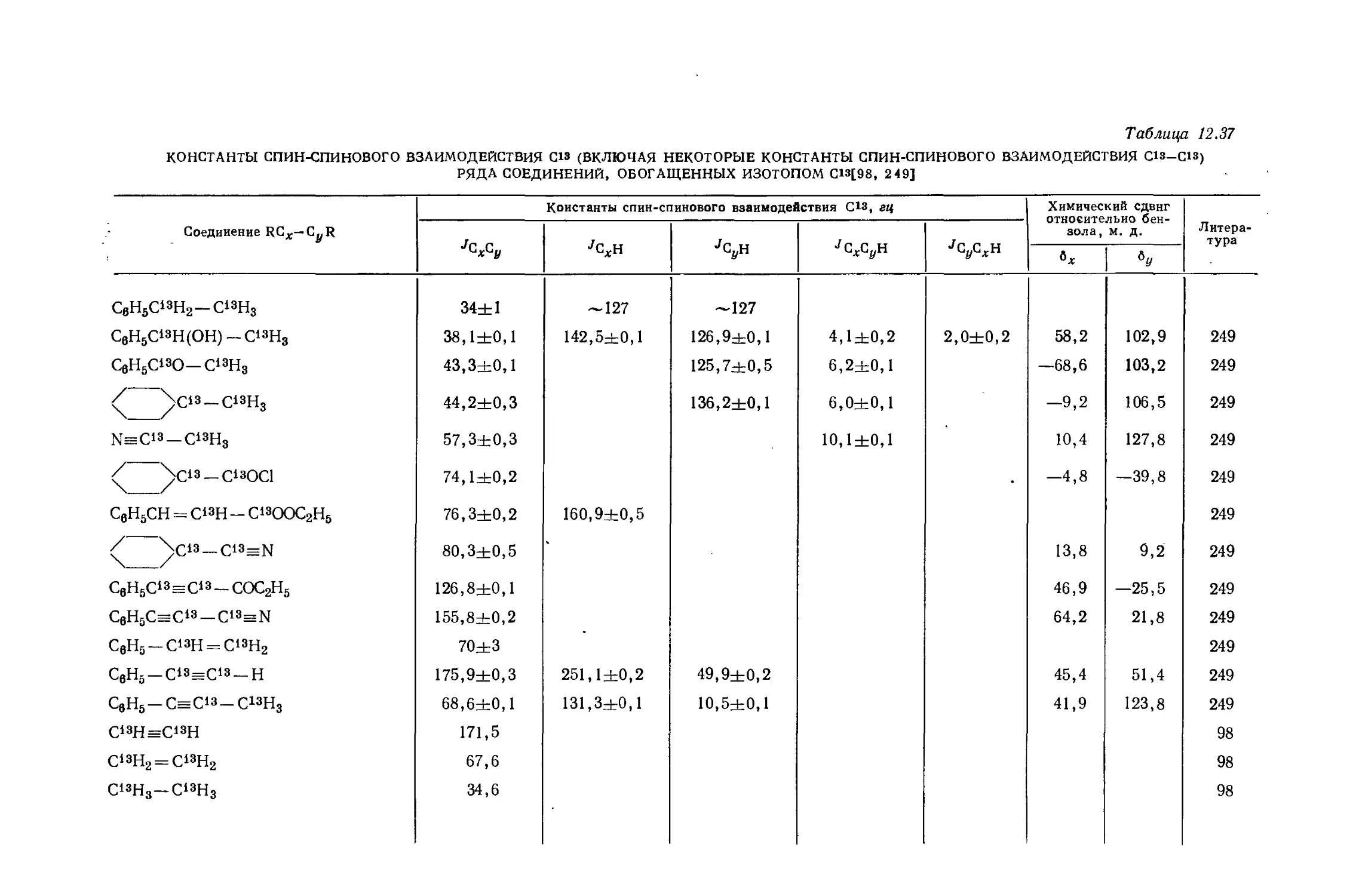



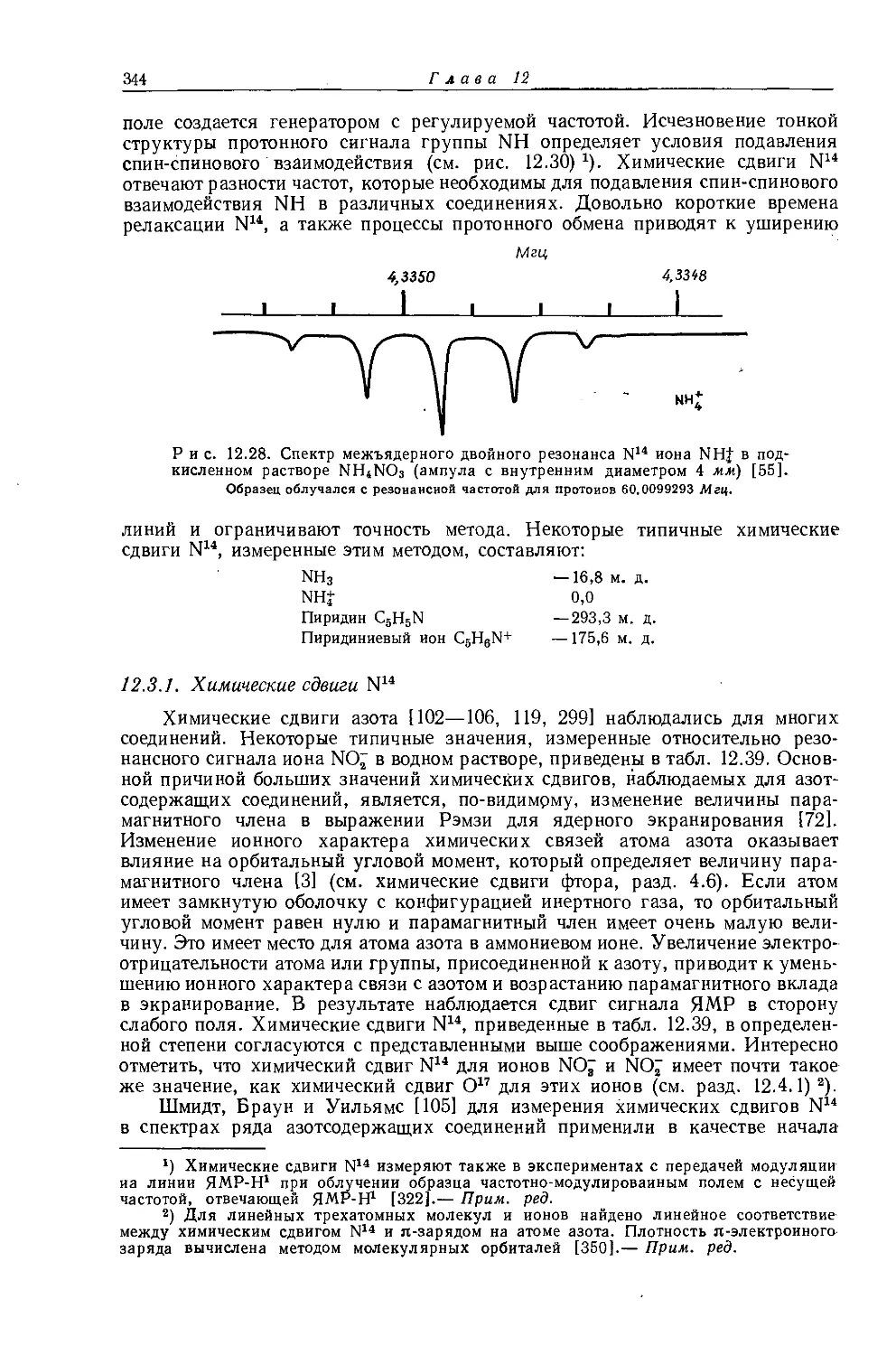
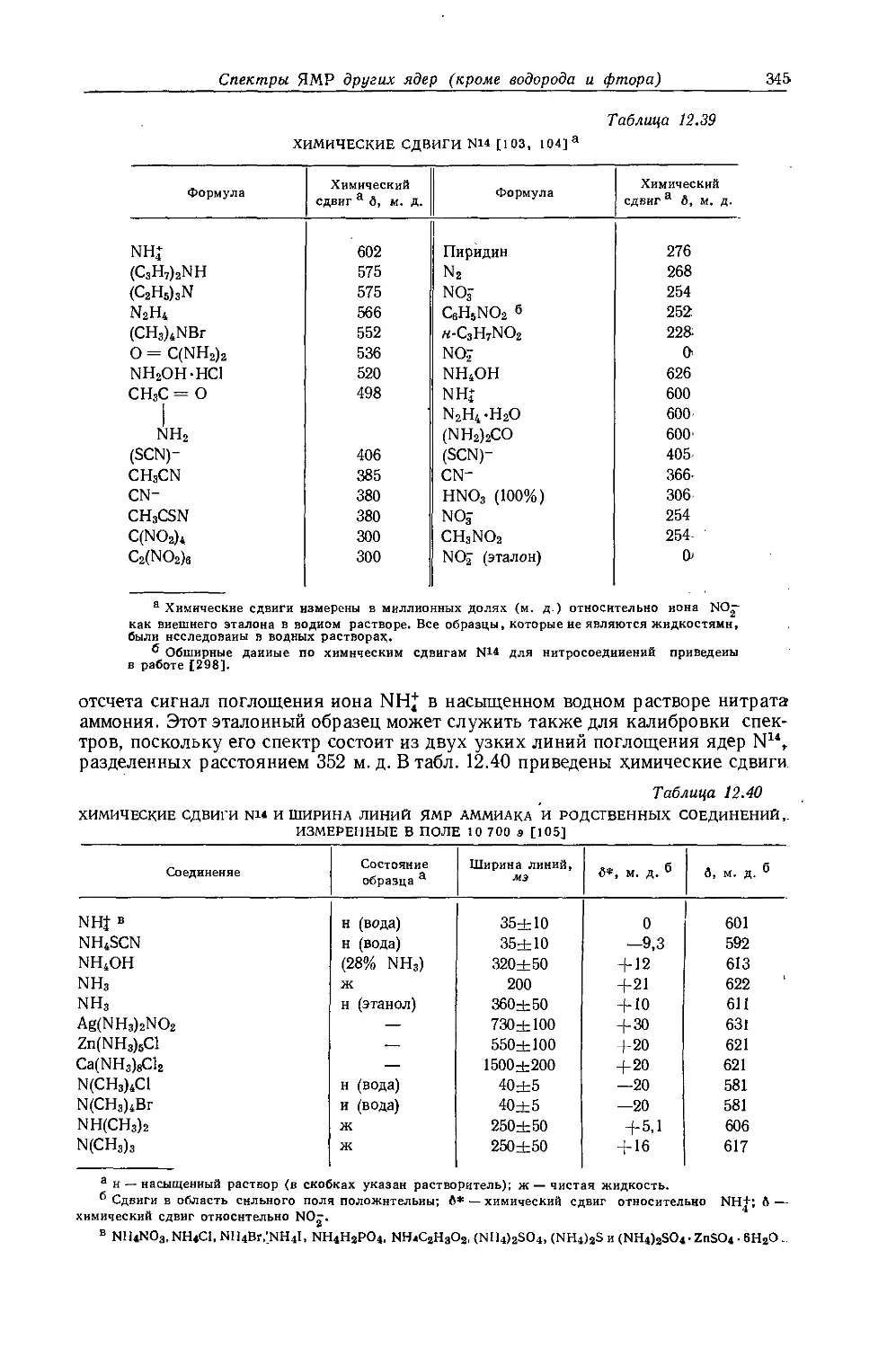


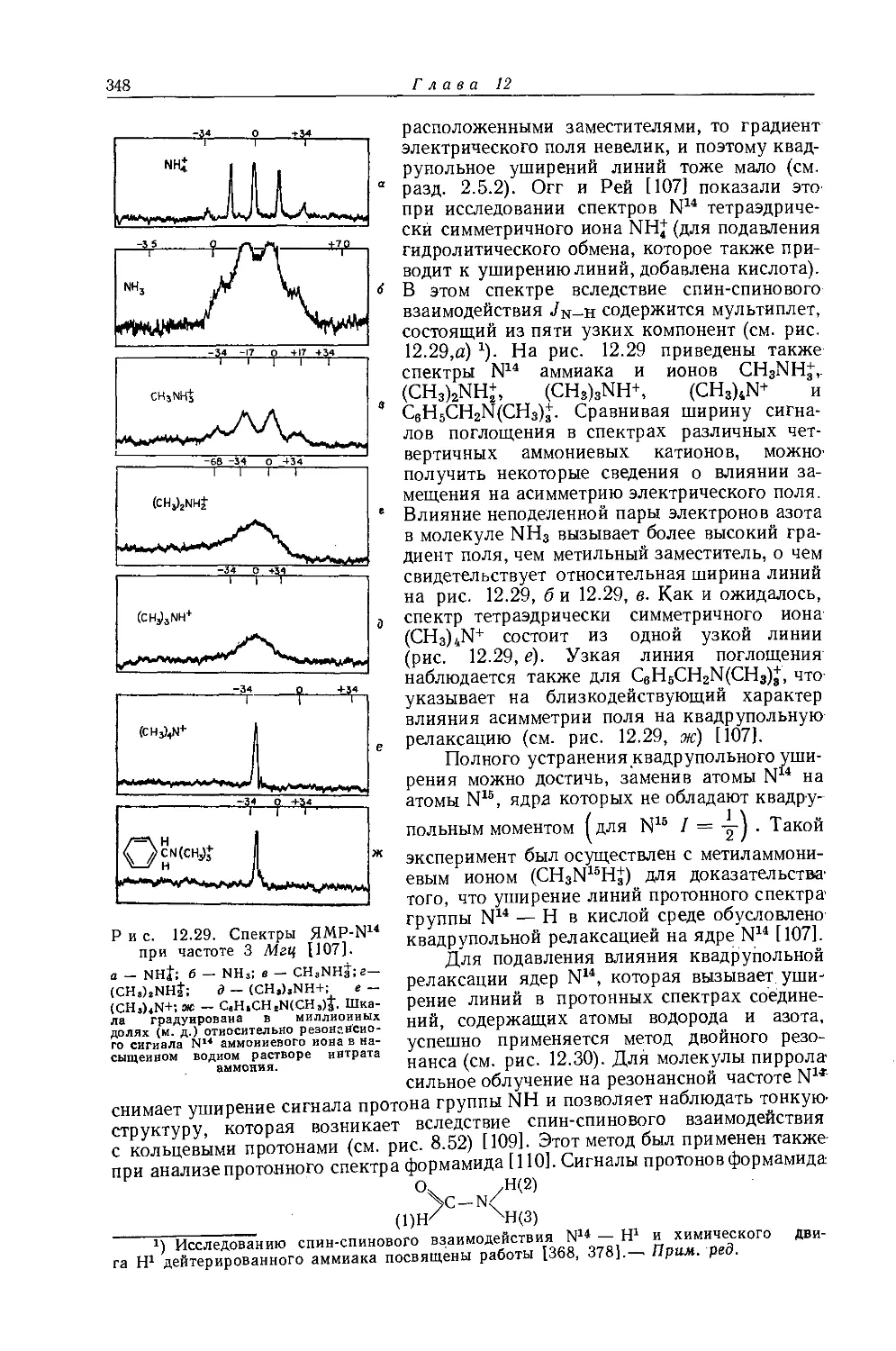




































































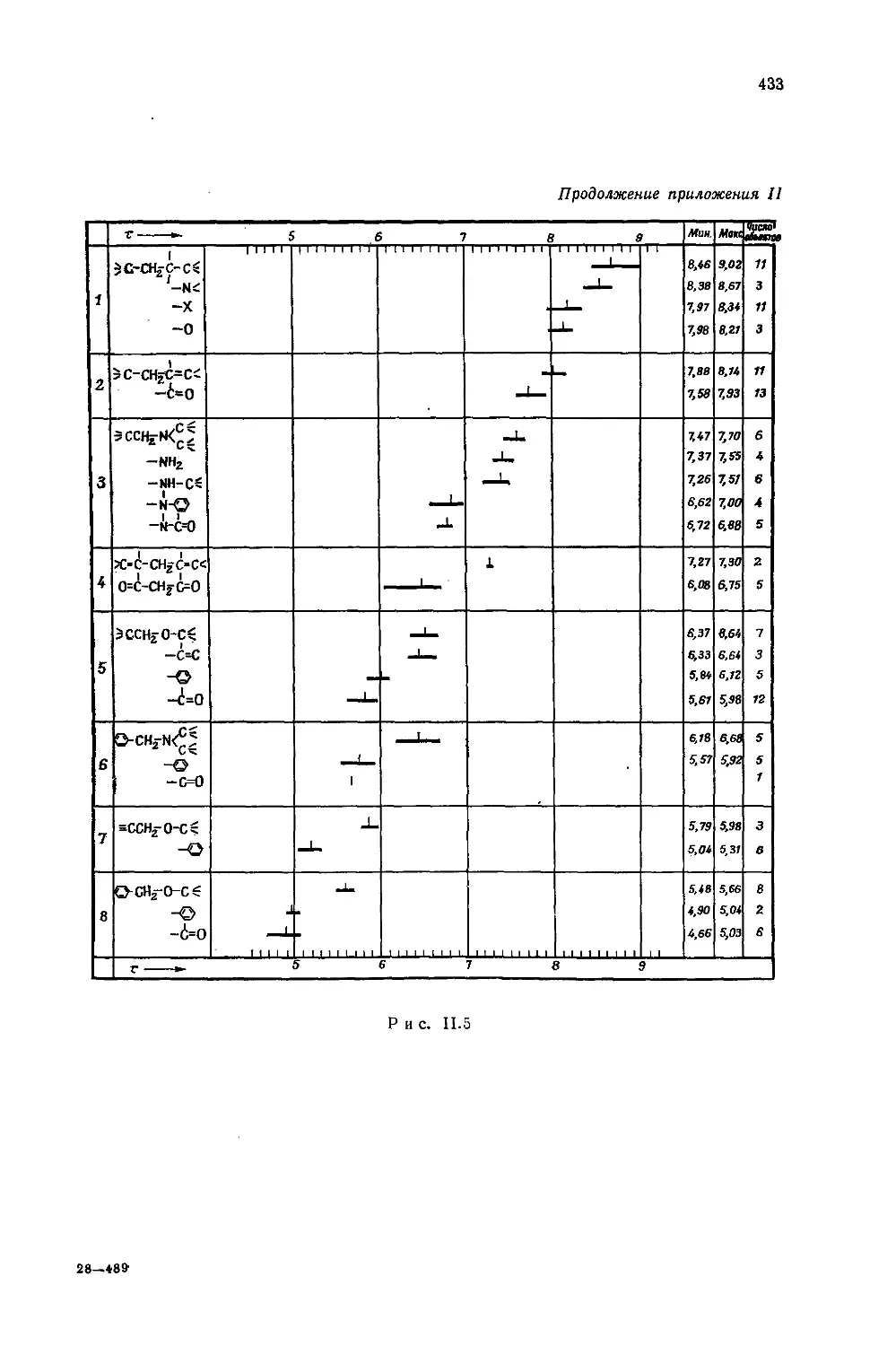

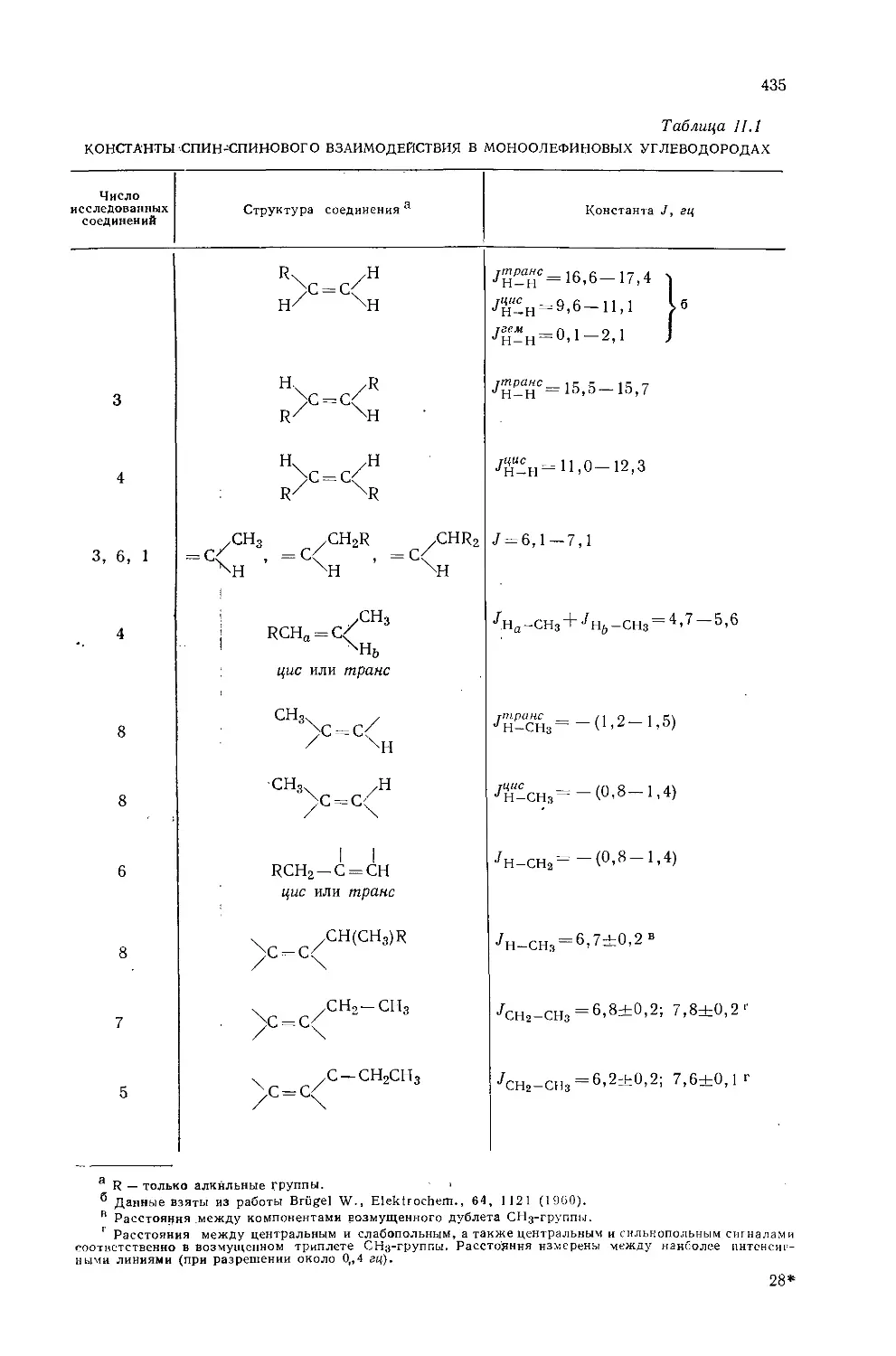

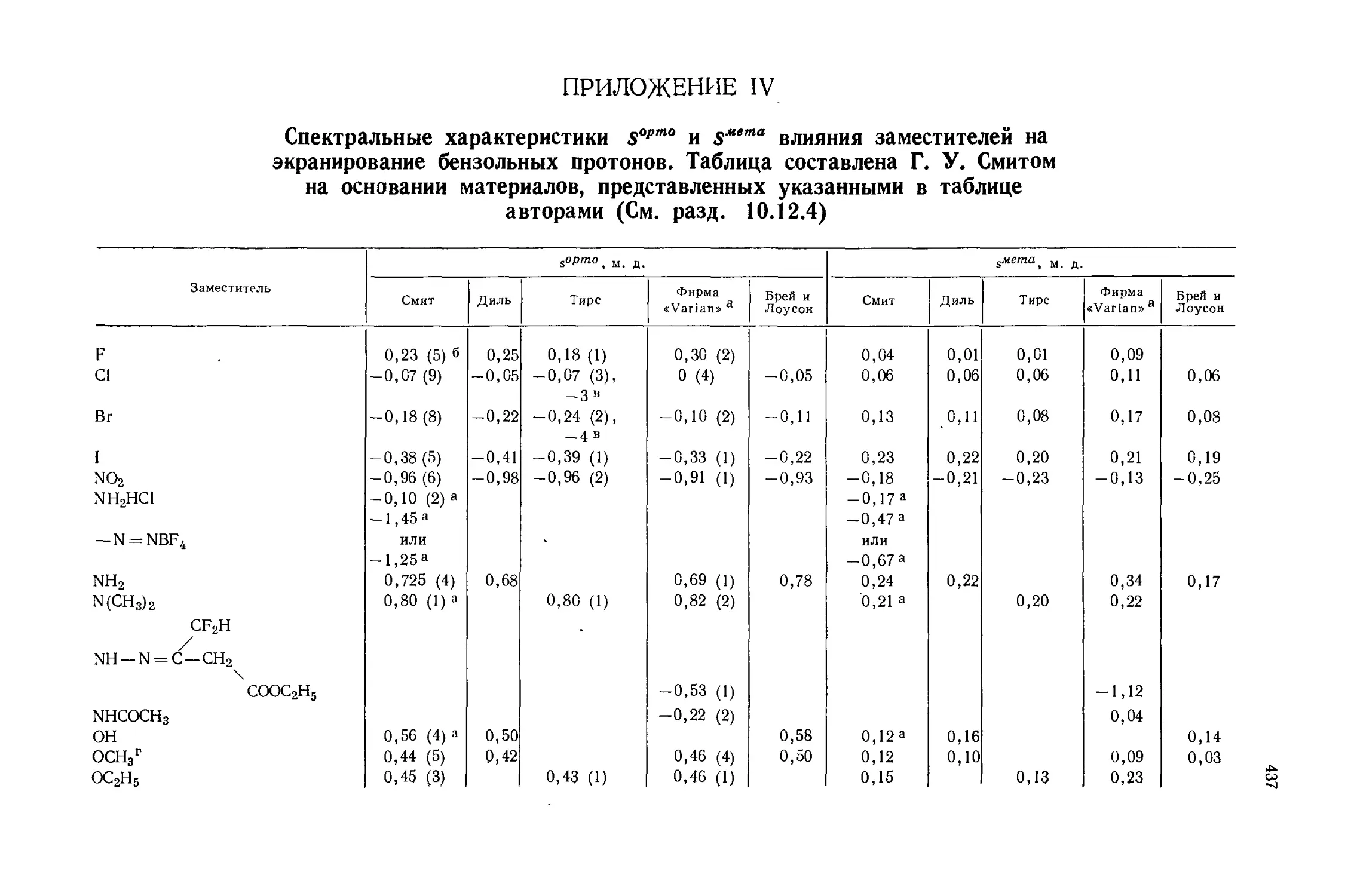

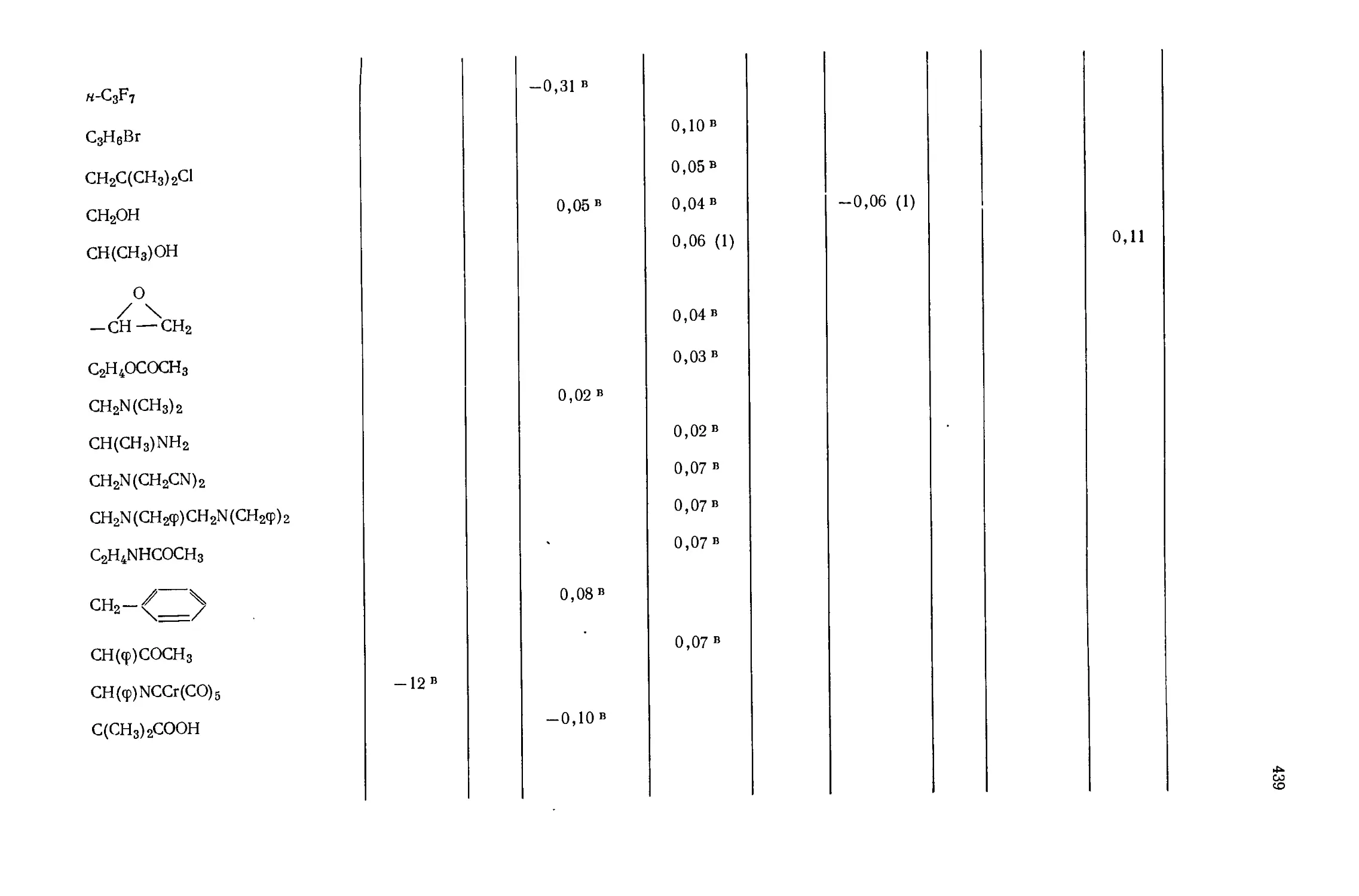
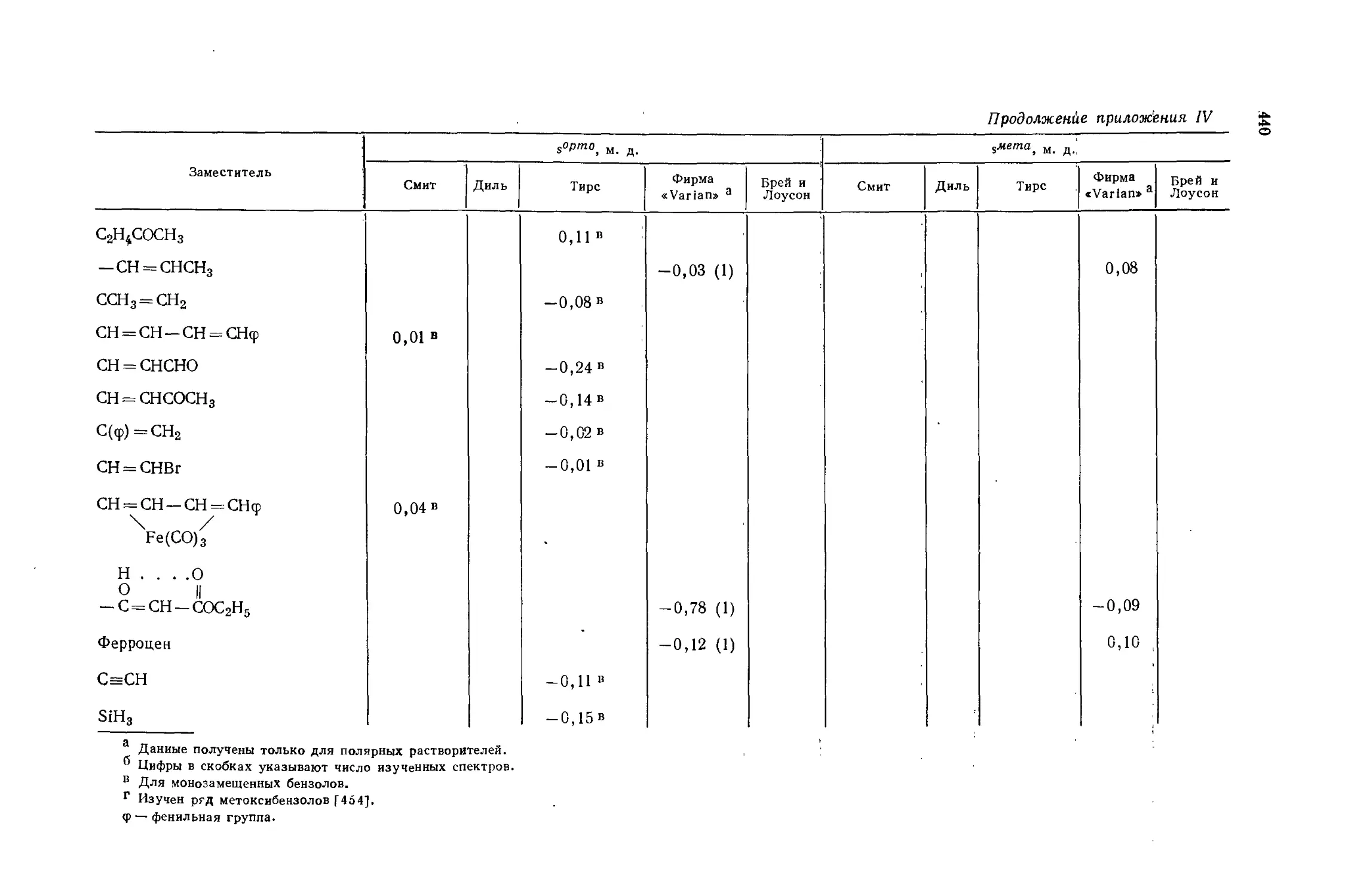
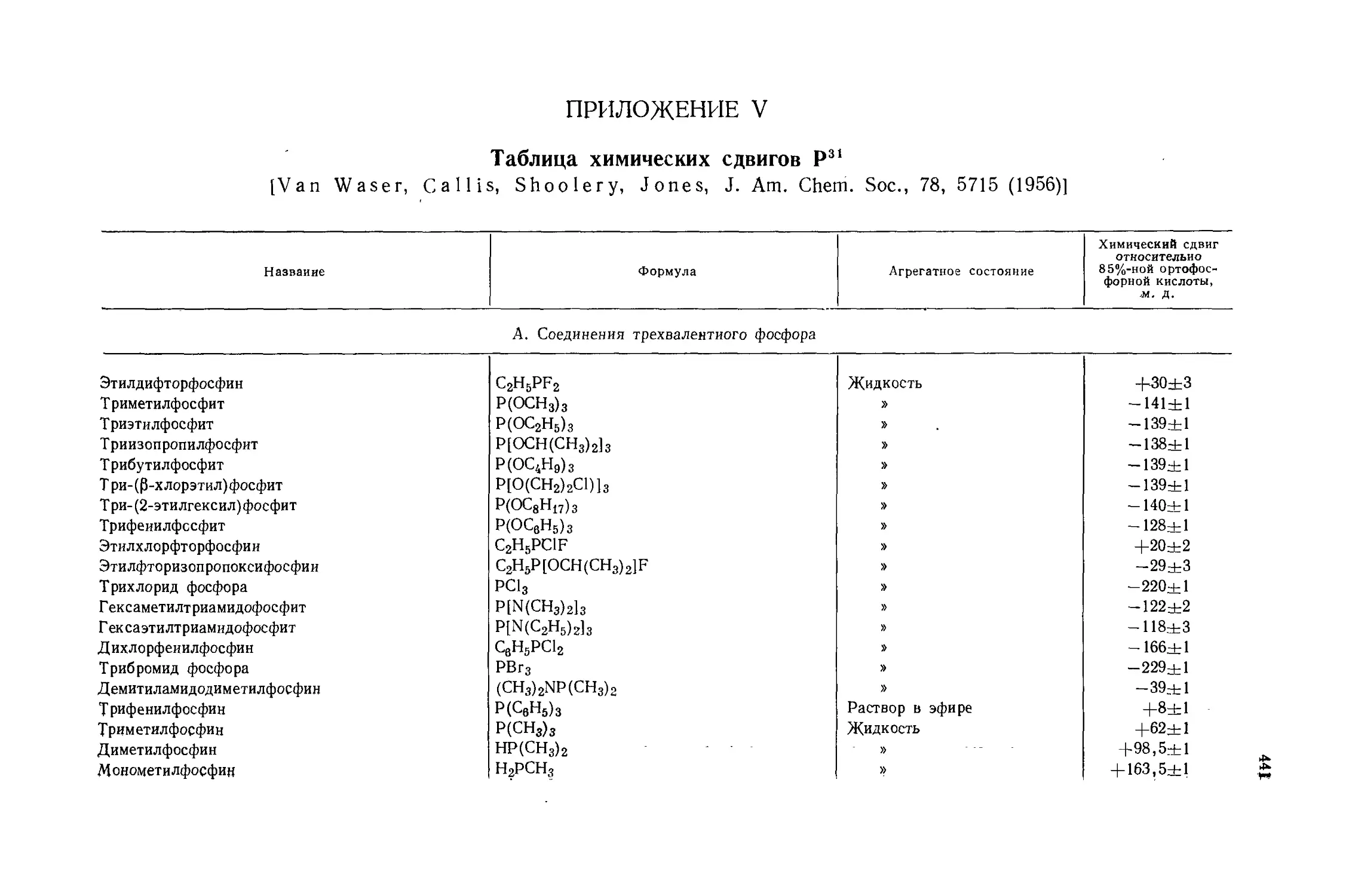
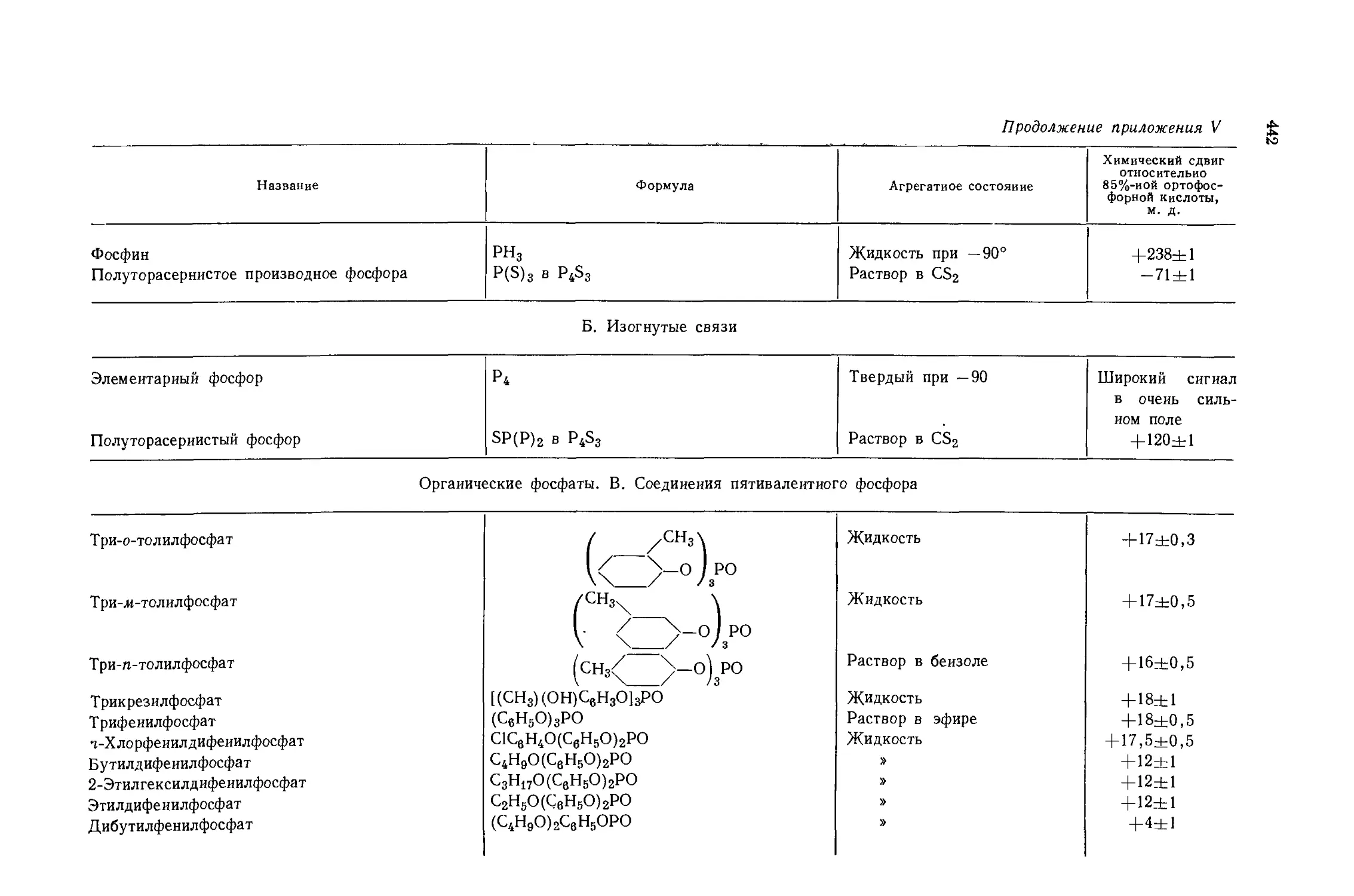

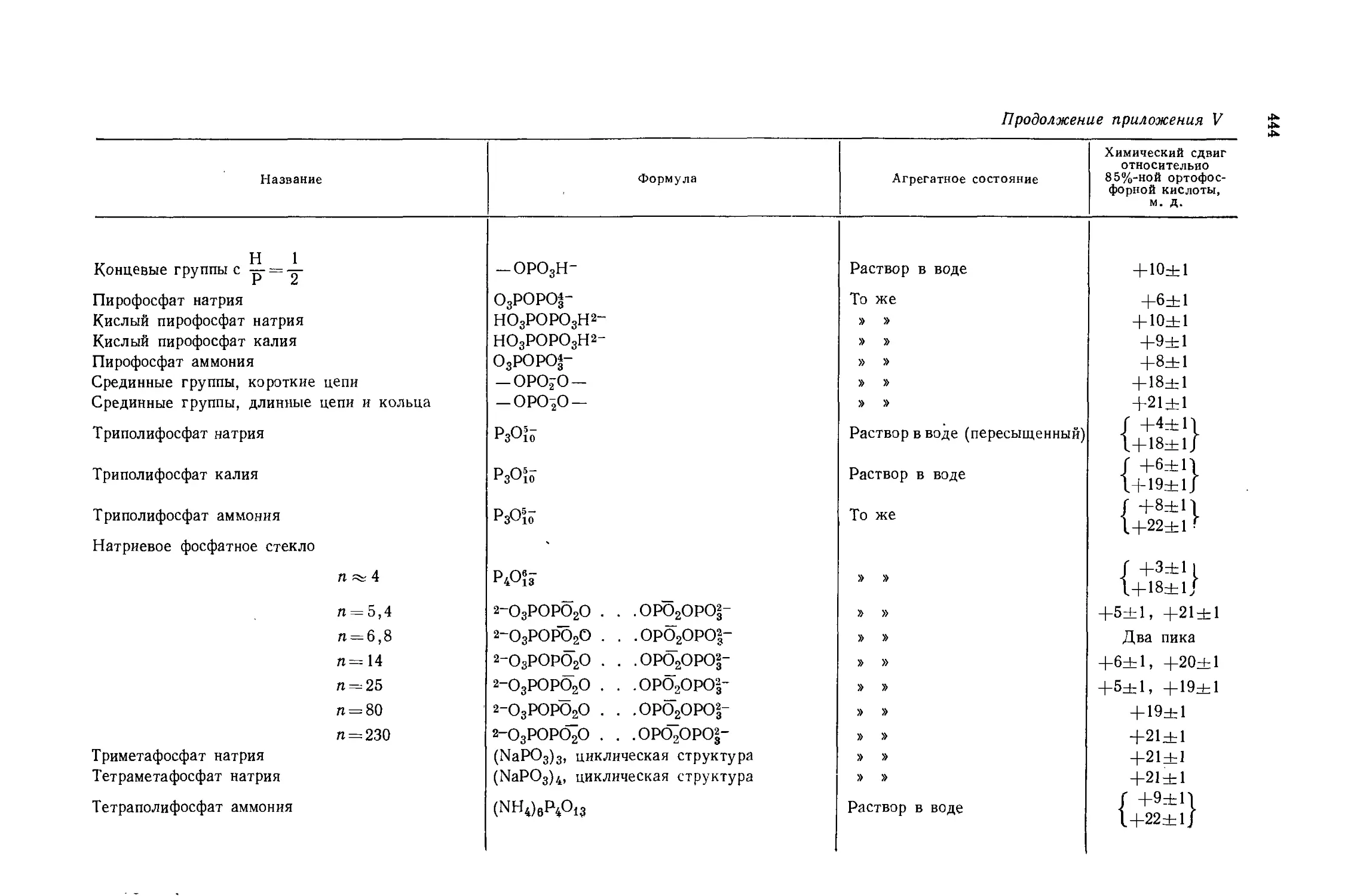
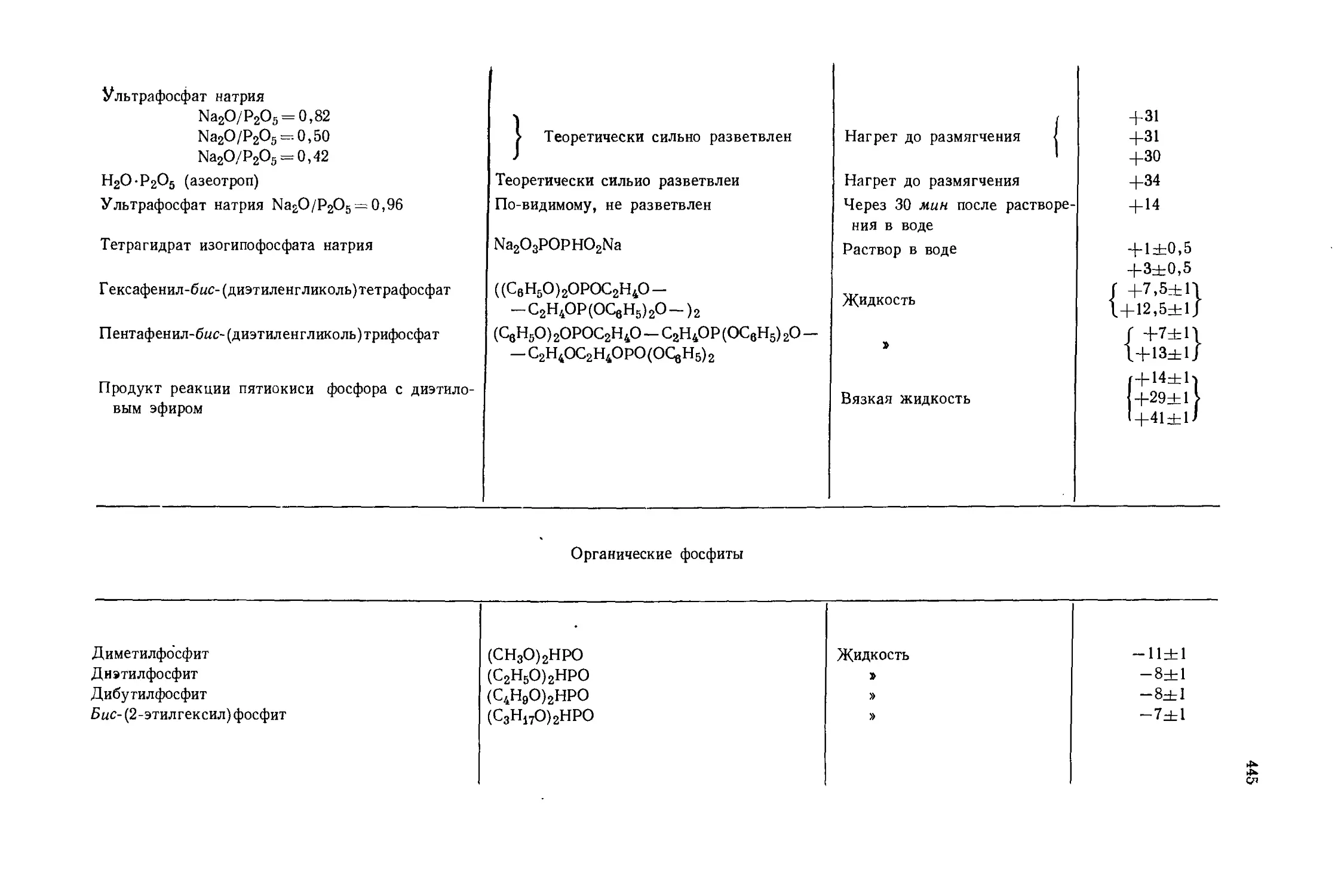
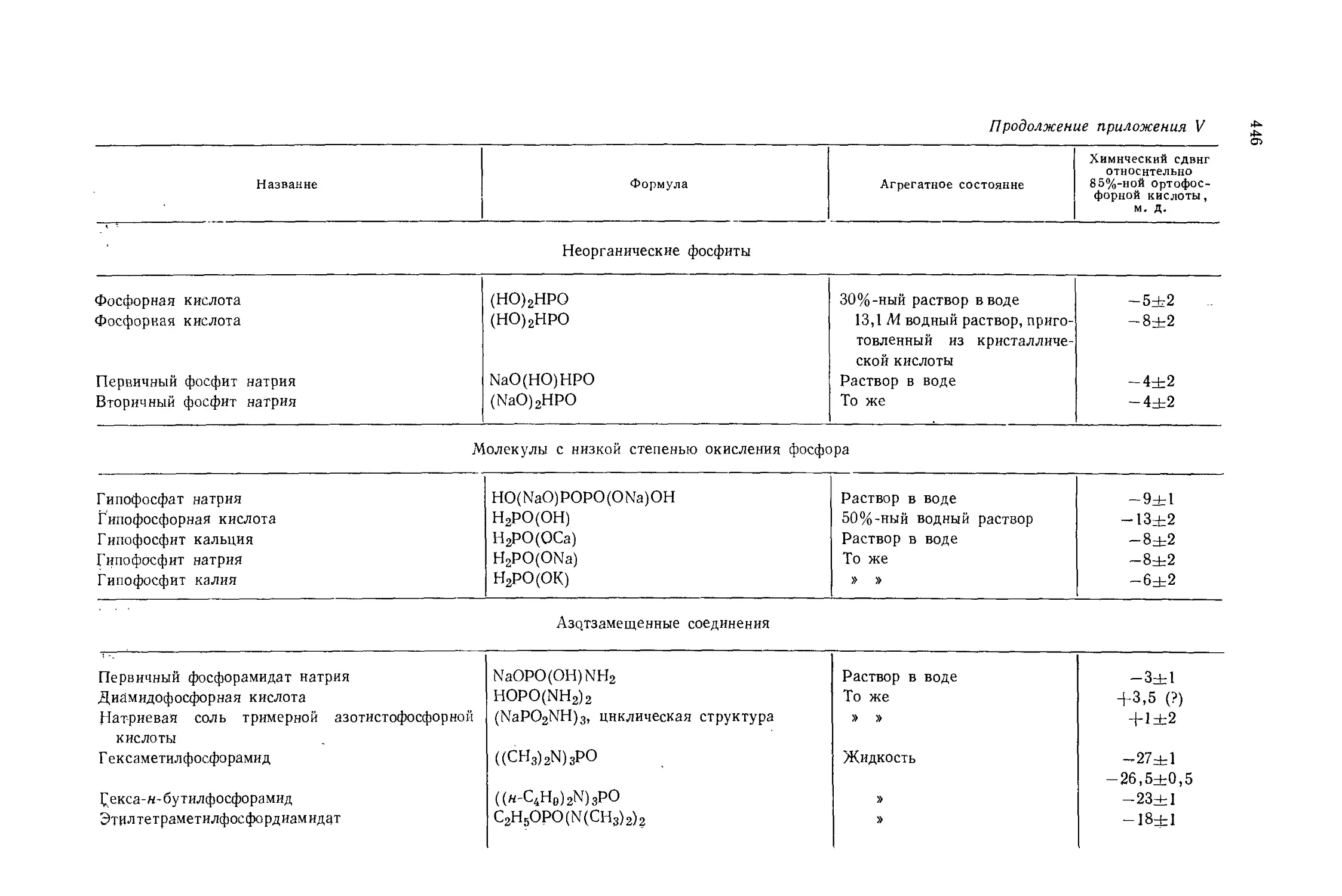

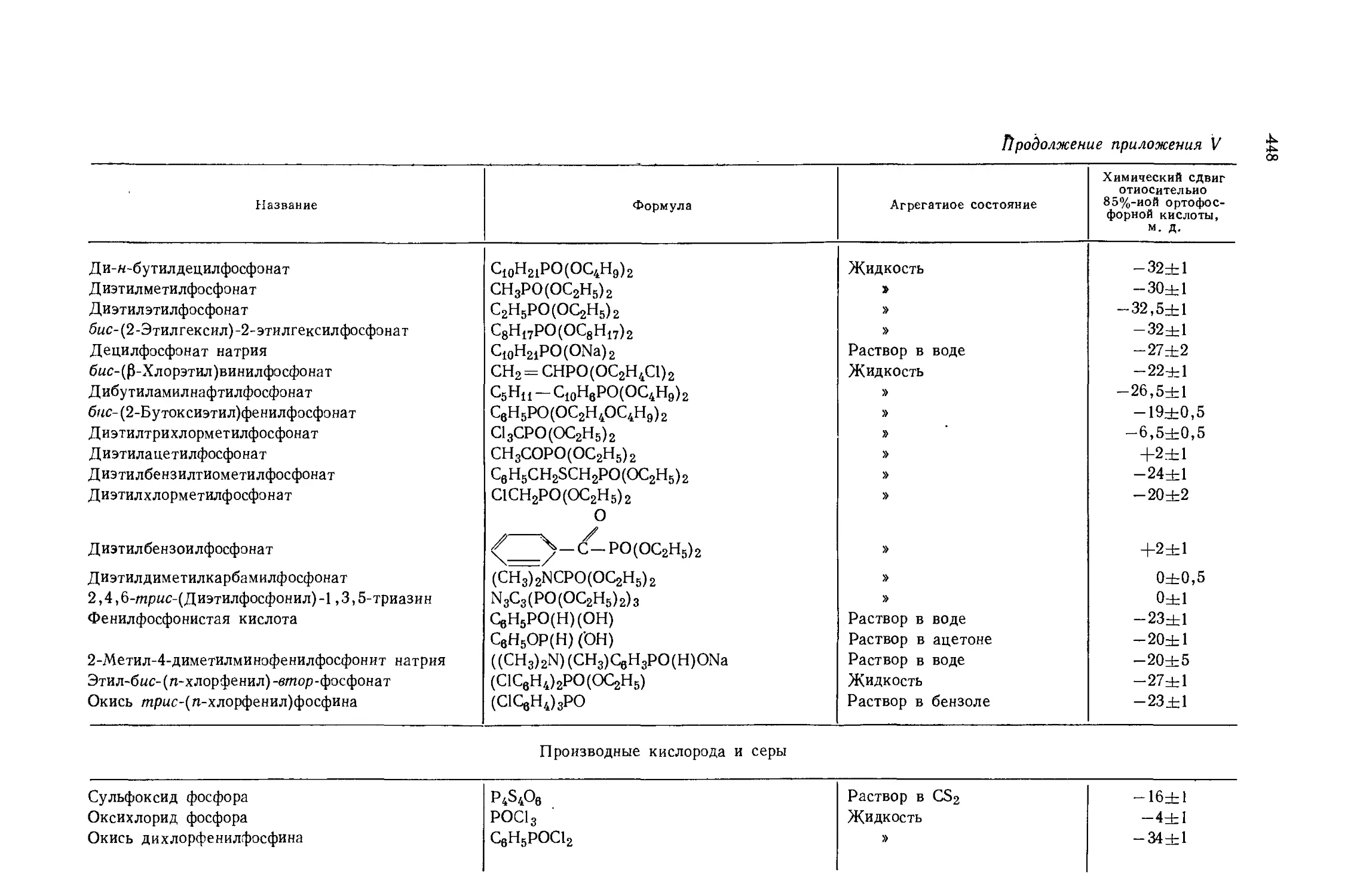
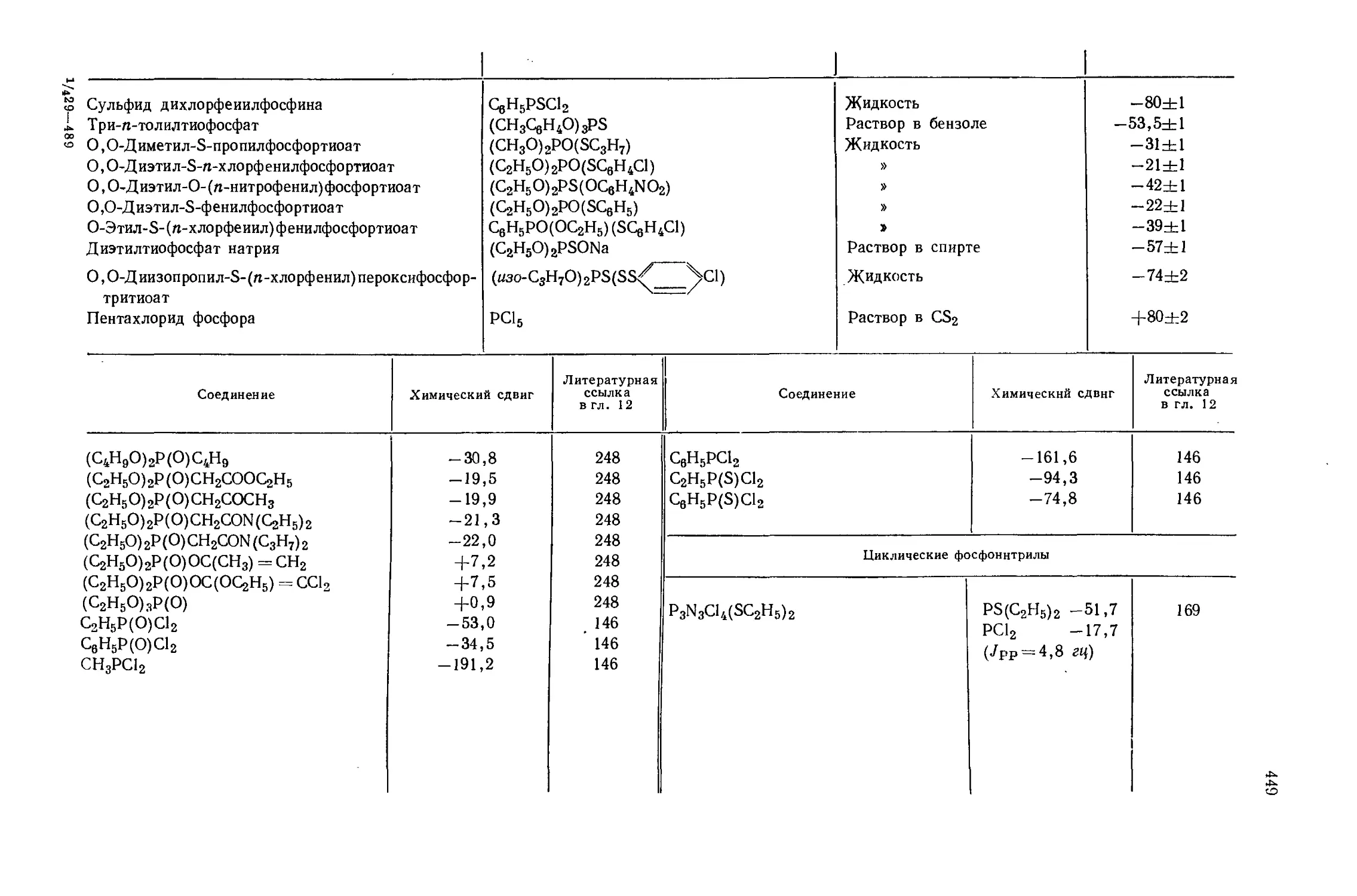


















Автор: Эмсли Дж. Финей Дж. Сатклиф Л.
Теги: распространение световых лучей отражение преломление поглощение излучение политика химия
Год: 1969




ИЗДАТЕЛЬСТВО
«М И Р»
HIGH RESOLUTION NUCLEAR MAGNETIC RESONANCE SPECTROSCOPY
In Two Volumes
J. W. Emsley, J. Feeney, L. H. Sutcliffe
VOLUME 2
PERGAMON PRESS
OXFORD • LONDON • EDINBURGH • NEW YORK PARIS • FRANKFURT
1966
Дж. Эмсли, Дж. Финей, Л. Сатклиф
СПЕКТРОСКОПИЯ
ЯДЕРНОГО
МАГНИТНОГО
РЕЗОНАНСА
ВЫСОКОГО
РАЗРЕШЕНИЯ
Том 2
Перевод с английского канд. физ.-мат. наук Ю. С. КОНСТАНТИНОВА, канд. хим. наук В. С. ПЕТРОСЯНА, канд. хим. наук Ю. А. УСТЫНЮКА, канд. физ.-мат. наук Э. И. ФЕДИНА
Под редакцией канд. физ.-мат. наук В. Ф. БЫСТРОВА и доктора хим. наук проф. Ю. Н. ШЕЙНКЕРА
ИЗДАТЕЛЬСТВО «МИР» • МОСКВА • 1969
УДК 535.338:32
Двухтомная монография посвящена стремительно развивающемуся физическому методу исследования строения молекул и химических процессов. Благодаря непрерывному-росту числа ЯМР-спектрометров в лабораториях быстро увеличивается круг научных работников, активно использующих спектры магнитного резоиаиса. Книга может служить исчерпывающим пособием по ЯМР-спектроскопии. В данном томе обобщен и систематизирован громадный фактический материал (цитируется более 1000 работ). Приведено большое число таблиц и графиков, облегчающих работу читателю; очень ценна не публиковавшаяся ранее таблица т-значений Тнрса для различных органических соединений.
Книга предназначена для химиков-органиков и физикохимнков — научных работников, преподавателей, аспирантов и студентов старших курсов химических вузов.
Редакция литературы по химии
Инд. 2-5-4
ГЛАВА 10
Корреляции спектров ЯМР-Н1 со строением молекул
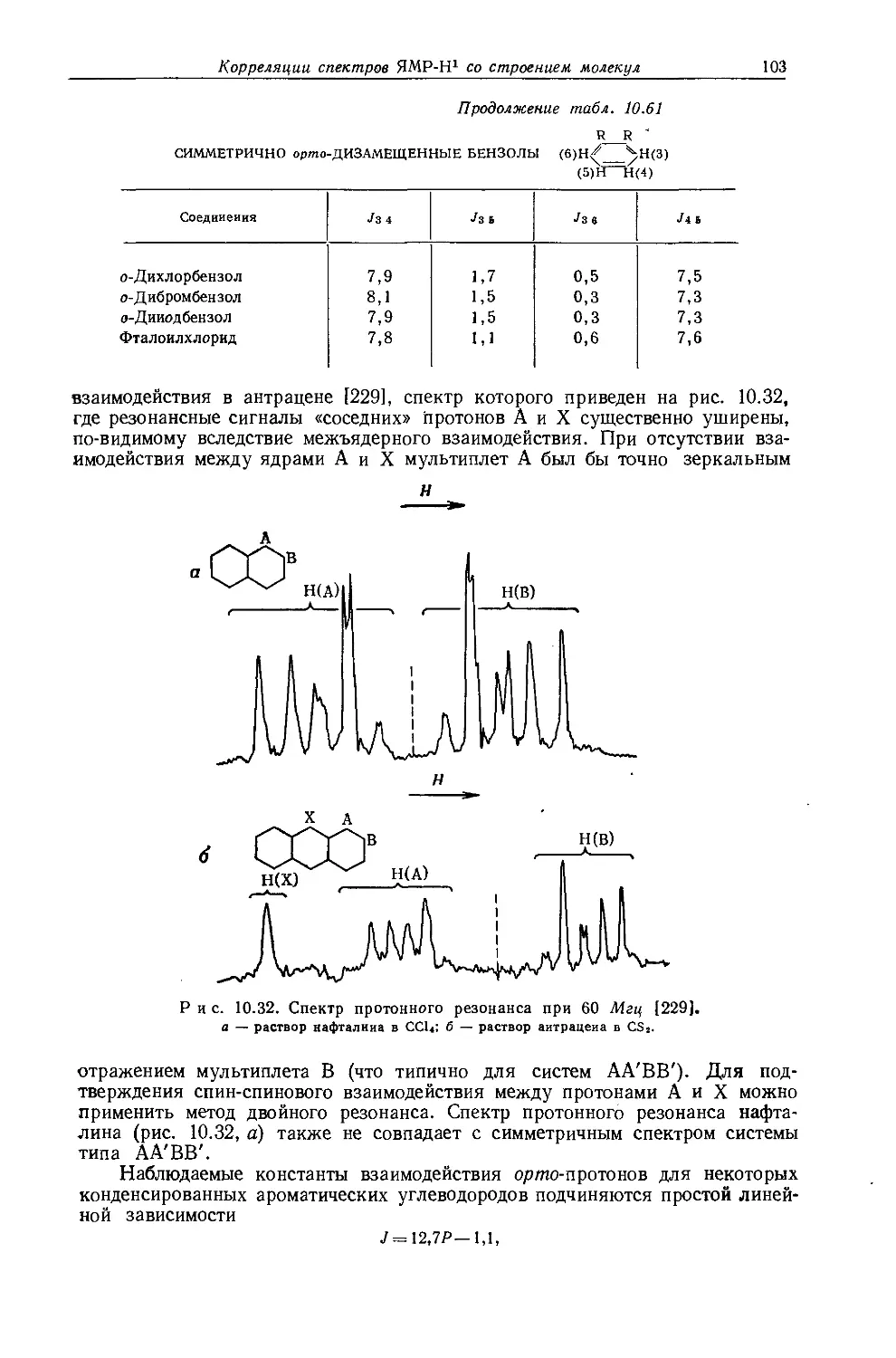
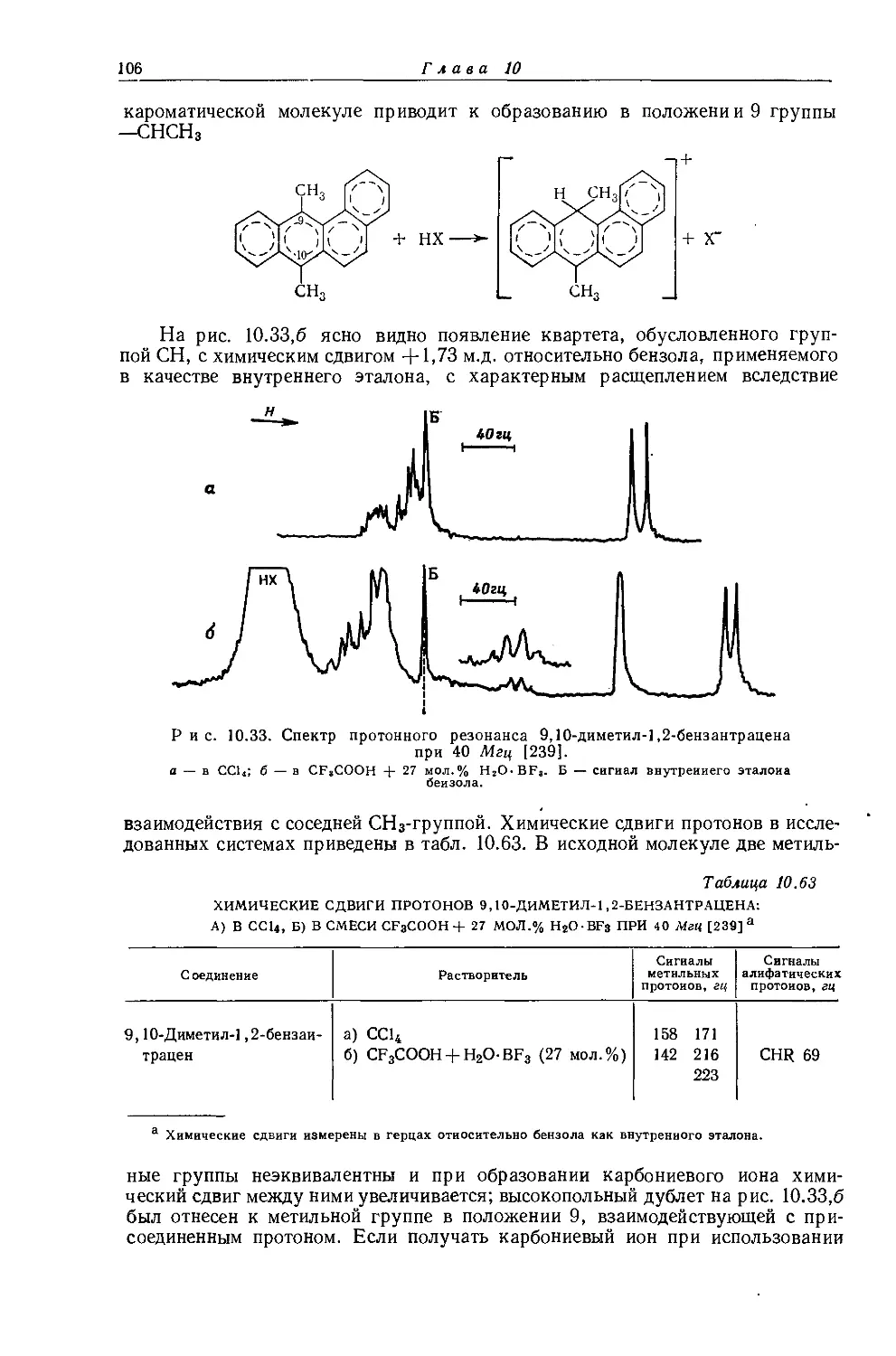
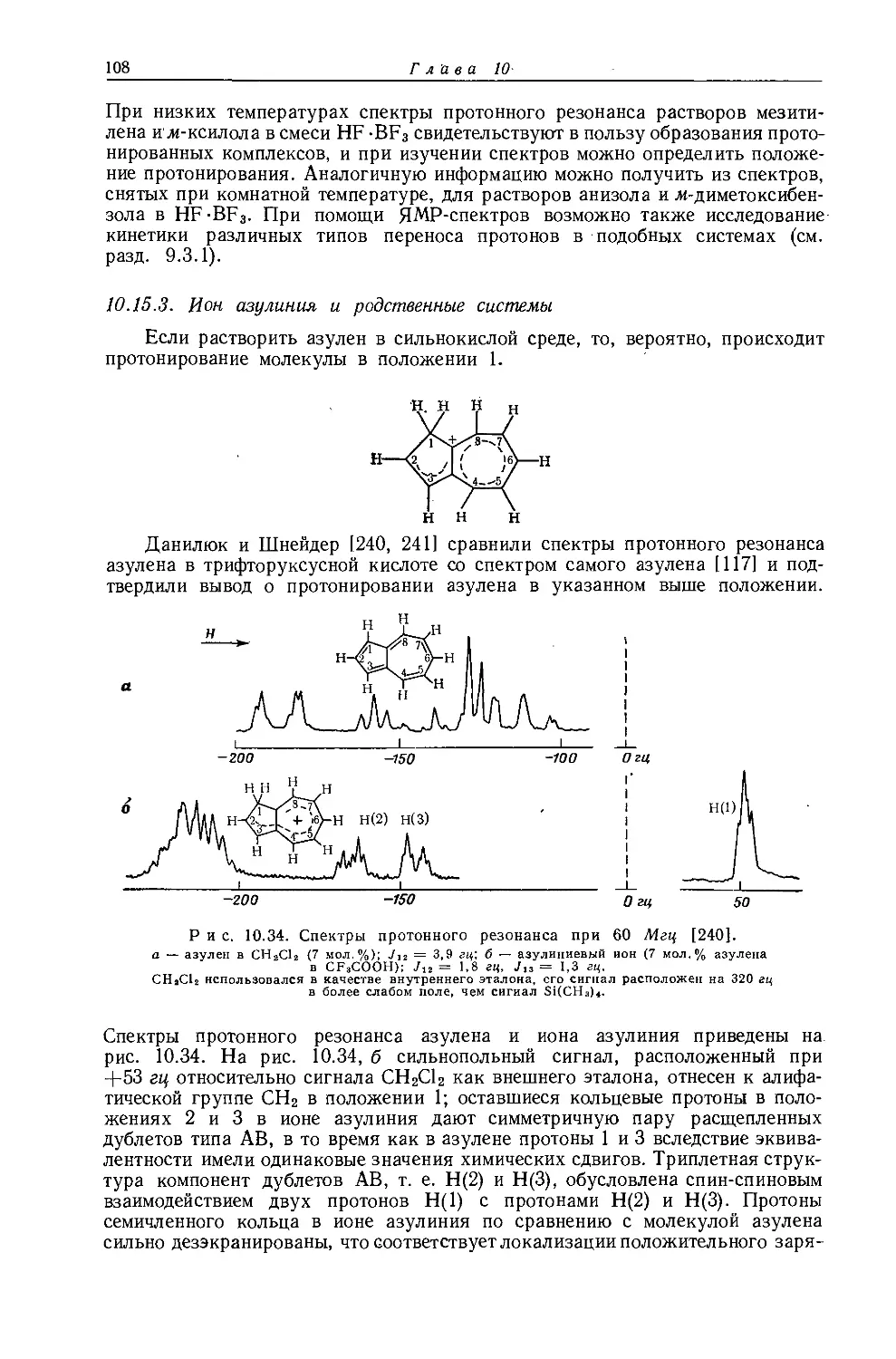
Ядра, обладающие магнитными моментами, могут служить источником подробной информации о своем электронном окружении. Эта информация может быть получена из спектров их резонансного поглощения. Несмотря на то что имеется много случаев теоретической интерпретации наблюдаемого экранирования ядер и спин-спинового взаимодействия этих ядер, часто приходится ограничиваться лишь качественным пониманием этих явлений. Поэтому значительная часть ценной информации, потенциально содержащейся в спектрах ЯМР, остается в настоящее время недоступной. До тех пор пока квантовомеханическое рассмотрение не сможет достаточно точно предсказывать спектральные параметры молекул, содержащих большое число атомов, мы должны довольствоваться корреляциями между спектральными и структурными параметрами. В литературе уже имеются сообщения о многих корреляциях такого типа, поэтому целью настоящей главы является собрание их воедино вместе с характеристическими значениями протонных химических сдвигов и констант спин-спинового взаимодействия Н — Н для различных классов молекул. Корреляции между спектральными и структурными параметрами чрезвычайно полезны для теоретического понимания спектральных констант. Более того, из эмпирических корреляций часто можно предсказать химические сдвиги и константы спин-спинового взаимодействия для молекул с неизвестным строением. Знание многих корреляций позволяет извлечь из данных ЯМР наибольшую информацию. Большинство значений т, приведенных в этой главе, взято из неопубликованной работы Тирса (см. приложение I). Где это было возможно, значения химических сдвигов указаны в т-шкале. Химические сдвиги, полученные экстраполяцией от одного эталона к другому, ненадежны из-за отсутствия поправки на влияние растворителя и объемную диамагнитную восприимчивость. Для случаев, когда возможны значительные расхождения такого рода и соответствующие пересчеты не были проведены, указан приближенный коэффициент такого пересчета.
На экранирование ядер водорода в молекуле могут влиять различные факторы (детальное рассмотрение этих вопросов приведено в гл. 3 и 4). При отсутствии влияния объемной магнитной восприимчивости и межмолекулярных взаимодействий главными эффектами экранирования являются:
1. Диамагнитное экранирование ядра электронной оболочкой, в которой находится это ядро. Соседняя электроотрицательная группа будет влиять на электронную оболочку посредством индуктивного эффекта через связи (см. разд. 3.4) и, таким образом, изменять величину этого экранирования.
2. Экранирование соседними анизотропными группами. Величина такого экранирования существенна, если протоны находятся вблизи от центра магнитной анизотропии молекулы. Такой центр может быть обусловлен атомом, группой или связью, обладающими различными значениями поперечной %п и продольной Хпр компонент магнитной восприимчивости [например, аромати
6
Глава 10
ческие кольца, связи С = О, С С, С — X (где X — галоген)]. В соответствии с этим ядра водорода будут экранированы или дезэкранированы в зависимости от относительных значений компонент восприимчивости соседней системы и от геометрии молекулы (см. разд. 4.4).
3. Эффекты внутримолекулярного электрического поля. Если молекула имеет постоянный дипольный момент, то обусловленное им электрическое поле может влиять на экранирование ядер водорода, взаимодействуя с их электронными оболочками (см. разд. 3.9).
4. Сдвиги, обусловленные связями С — С. Для ядер водорода в алканах Дейли [7] предположил, что следует принимать во внимание дополнительный фактор дезэкранирования, названный им «сдвигом, обусловленным связями С — С». Природа этого эффекта не ясна, однако его нельзя рассматривать как эффект анизотропии связи С — С. Было сделано предположение, что если связь С — Н заменить связью С — С, то энергия электронного возбуждения существенно изменится таким образом, что парамагнитный вклад в экранирование увеличится.
АЛКАНЫ
10.1. Химические сдвиги производных алканов
10.1.1. Корреляция протонных химических сдвигов метильных производных СН3Х с электроотрицательностью заместителей
Многие исследователи пытались связать протонные химические сдвиги в алкильных группах с электроотрицательностью заместителей. В замещенных алканах корреляции такого типа следует ожидать, если диамагнитный вклад в экранирование протонов является преобладающим эффектом экранирования, поскольку в этом случае электронная плотность вокруг ядер водорода зависит главным образом от близости и природы электроноакцепторного заместителя. Увеличение электроотрицательности заместителя будет уменьшать электронную плотность вокруг соседних ядер водорода и, следовательно, дезэкраниро-вать их. Впервые зависимость протонных химических сдвигов от электроотрицательности заместителей в алкильных производных наблюдал Шулери [1]. Доказательства такой зависимости были найдены также Мейером, Сайка и Бутовским [2]. Позднее Мейер и Бутовский [3] исследовали ряд метильных производных и обнаружили обратную тенденцию в поведении химических сдвигов с увеличением электроотрицательности заместителей, после чего стало очевидно, что необходимо рассматривать и другие эффекты экранирования. Так, существенное влияние на протонные химические сдвиги оказывают объемная магнитная восприимчивость вещества и магнитная анизотропия соседних групп, обусловленная движением электронов в этих группах, связях и атомах (см. разд. 4.4). Влияние внутримолекулярных электрических полей на экранирование ядер водорода в алканах не рассматривалось [24].
Метилгалогениды. Оллред и Рохов [4] подробно исследовали спектры протонного резонанса ряда метилгалогенидов. Для того чтобы избежать влияния объемной диамагнитной восприимчивости, они применяли растворы очень низкой концентрации в четыреххлористом углероде (наиболее часто употребляемый в ЯМР-спектроскопии растворитель, что обусловлено его химической инертностью и магнитной изотропией). Если для всех исследуемых метилгалогенидов использовать один и тот же растворитель, то в сильно разбавленных растворах влияние объемной диамагнитной восприимчивости для них будет одинаково. Разбавление образцов исключает, кроме того, эффекты экранирования, обусловленные слабыми межмолекулярными взаимодействиями (типа водородной связи). На практике для получения химических сдвигов при
Корреляции спектров ЯМР-Н1 со строением молекул
7
•бесконечном разбавлении необходимо экстраполировать протонные химические сдвиги, измеренные для ряда разбавленных растворов.
Химические сдвиги для метилгалогенидов приведены в табл. 10.1. Как видно из таблицы, химические сдвиги чистых метилгалогенидов очень близки друг к другу, но при разбавлении образцов четыреххлористым углеродом
Таблица 10.1
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ НЕКОТОРЫХ МЕТИЛЬНЫХ
ПРОИЗВОДНЫХ [4]
Соединение Химический сдвиг 6 протонов СН3-группы, м. д. (Н2О — внешний эталон) Т ДЛЯ 0%-НОГО раствора в ССЦ а
чистое вещество 0%-ный раствор в ССЦ
ch3no2 1,19 0,48 5,69
CH3F 2,10 0,53 5,74
(CH3)2SO4 1,03 0,85 6,06
(СН3)2СО3 1,36 1,02 6,23
CH3OCeH5 1,83 1,10 6,31
СН3СООСН3 1,65 1,14 6,35
(CH3)2SO3 1,48 1,21 6,42
(CH3O)4Si 1,50 1,23 6,44
(СН3О)3В 1,64 1,26 6,47
сн3он 1,85 1,39 6,60
СН3ОСН3 2,63 1,55 6,76
СН3С1 2,10 1,74 6,95
€Н3СОВг 2,15 1,98 7,19
СН3Вг 2,02 2,10 7,31
СН3СОСвН5 3,00 2,25 7,46
СН3С6Н5 3,25 2,47 7,68
€Н31 2,00 2,60 • 7,81
(СН3СО)2О 3,04 2,60 7,81
СНзСООН 3,19 2,69 7,90
(СН3)2СО 3,34 2,70 7,91
СН3СООСН3 3,28 2,78 7,99
CH3CN 3,15 2,89 8,10
CH3C(NOH)CH3 2,98 8,19
(CH3)3N 3,18
(СН3)4С 4,35 3,85 9,06
а т = 5,21 + бн2о-
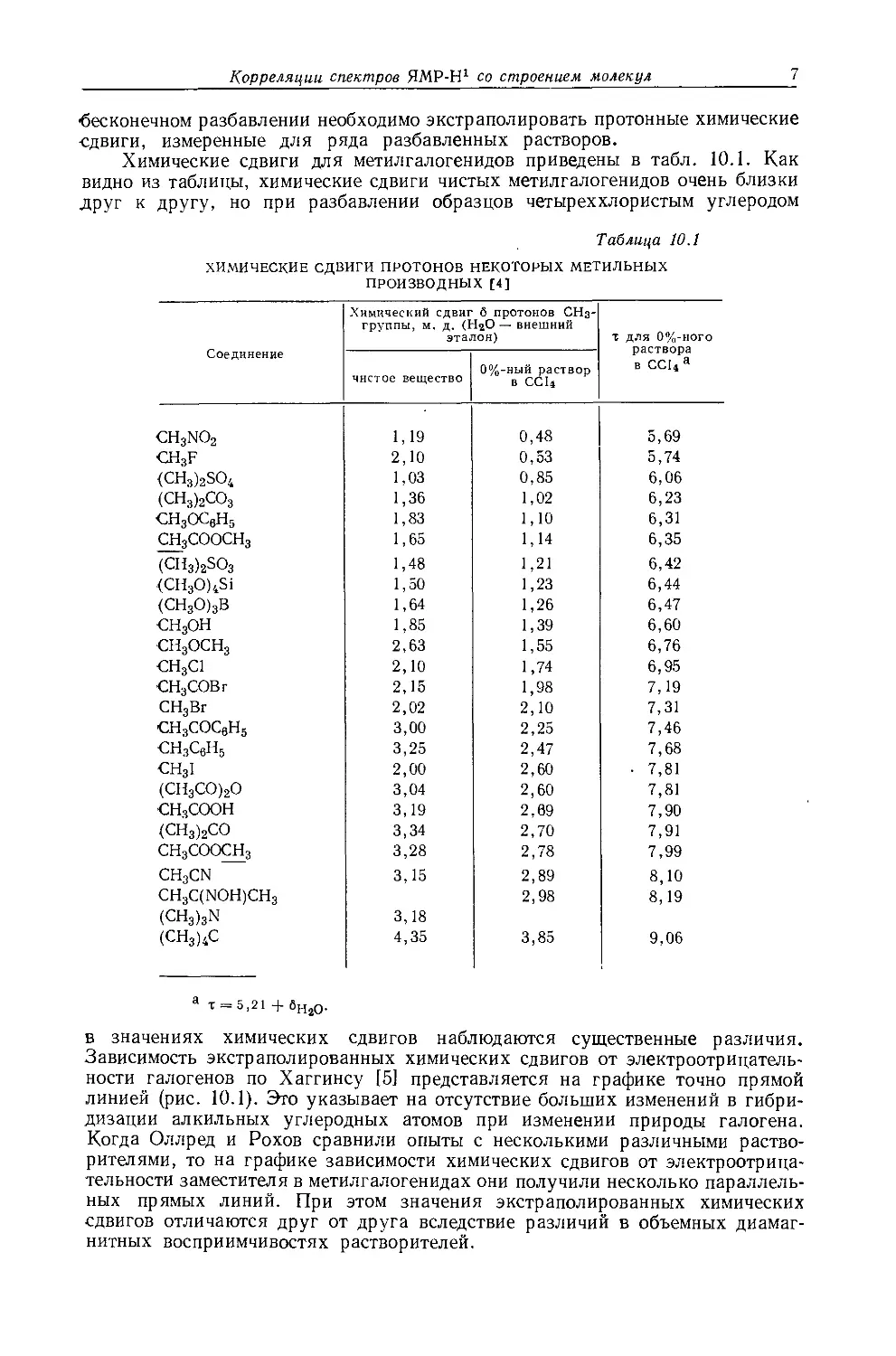
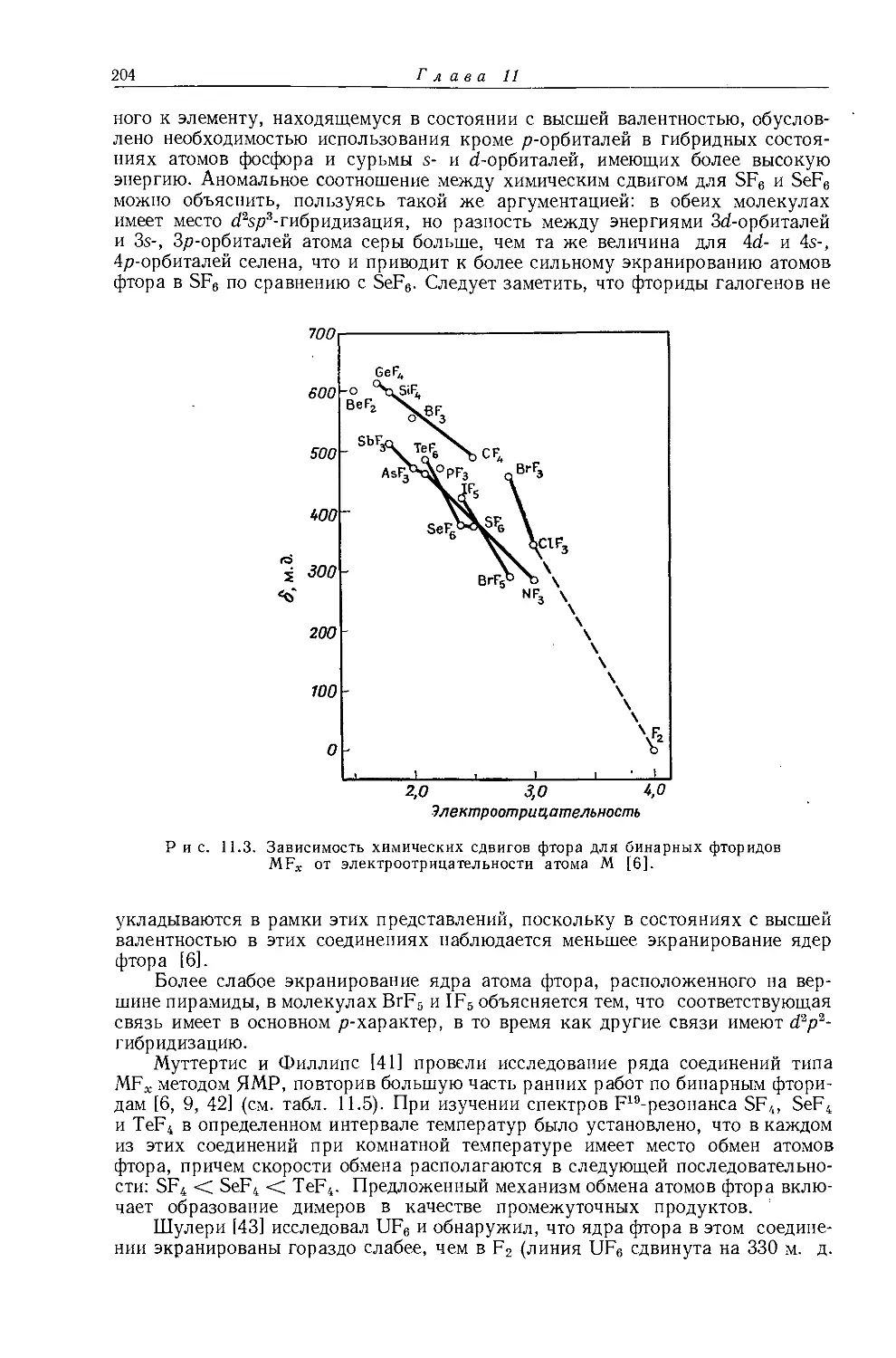
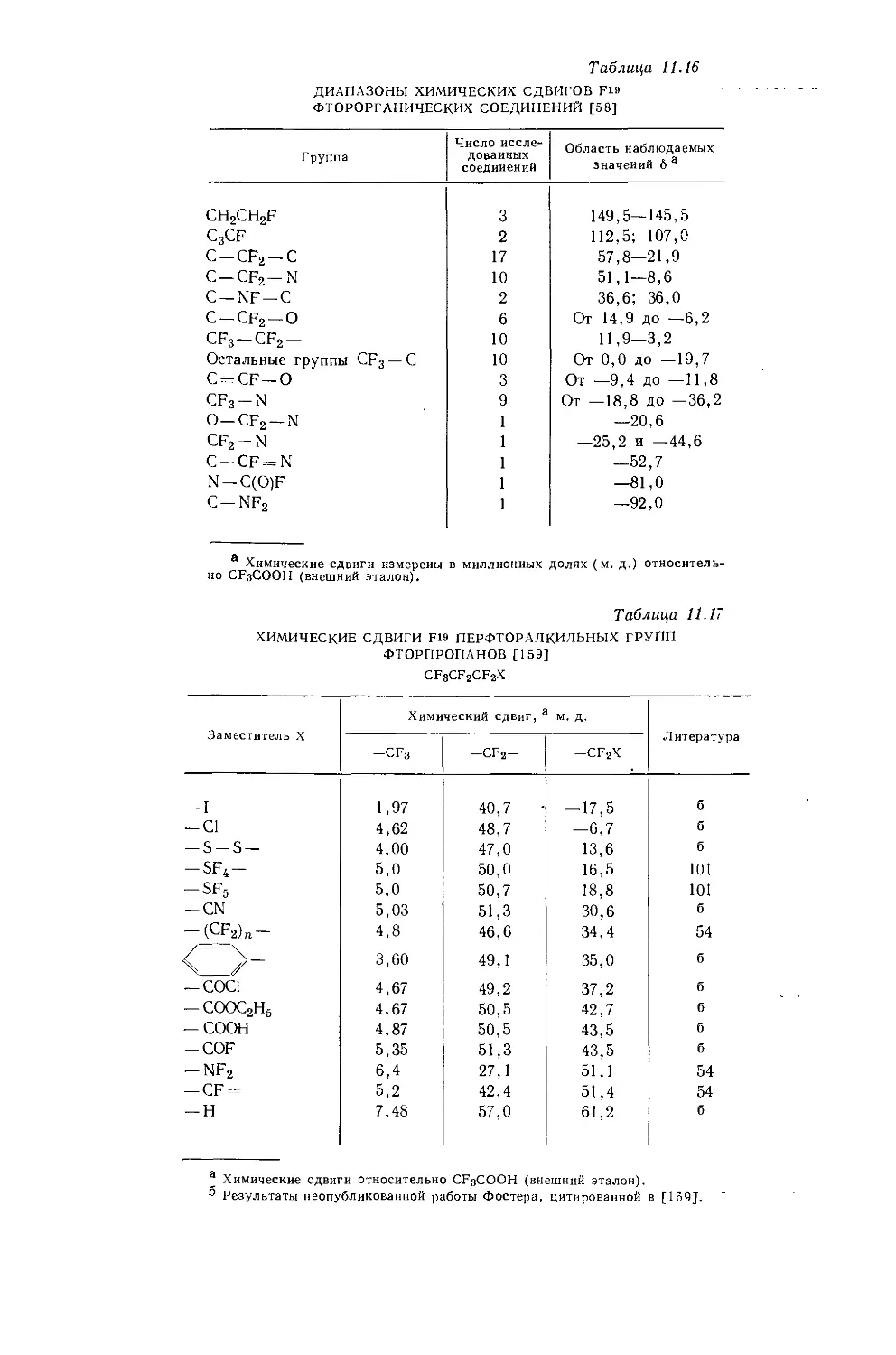
в значениях химических сдвигов наблюдаются существенные различия. Зависимость экстраполированных химических сдвигов от электроотрицательности галогенов по Хаггинсу [5] представляется на графике точно прямой линией (рис. 10.1). Это указывает на отсутствие больших изменений в гибридизации алкильных углеродных атомов при изменении природы галогена. Когда Оллред и Рохов сравнили опыты с несколькими различными растворителями, то на графике зависимости химических сдвигов от электроотрицательности заместителя в метилгалогенидах они получили несколько параллельных прямых линий. При этом значения экстраполированных химических сдвигов отличаются друг от друга вследствие различий в объемных диамагнитных восприимчивостях растворителей.
8
Глава 10
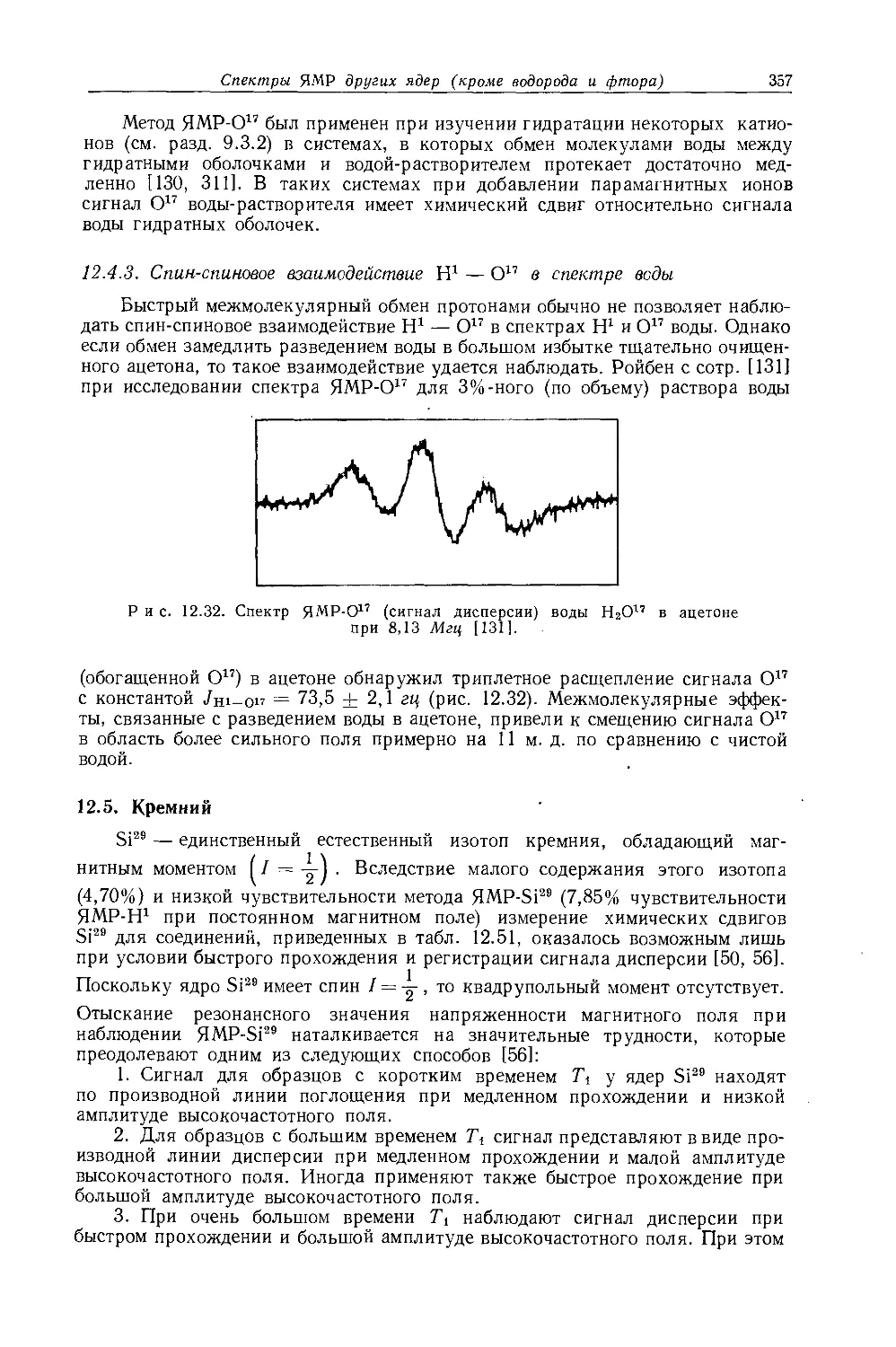
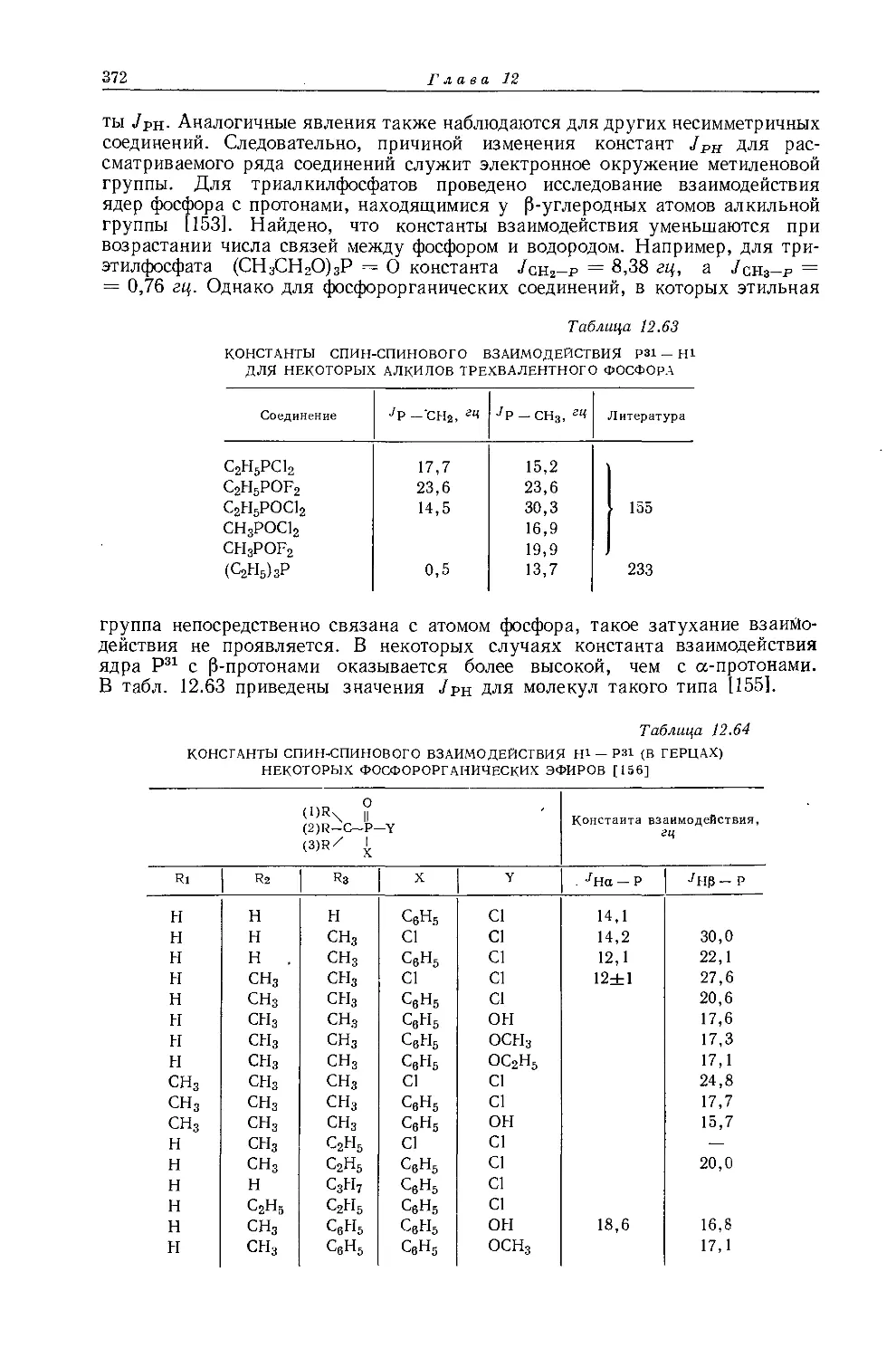
Другие метильные производные. В табл. 10.1 приведены протонные химические сдвиги ряда метильных производных, измеренные относительно сигнала воды, применявшейся в качестве внешнего эталона. Значения химических сдвигов даны как для чистых веществ, так и для разбавленных растворов в четыреххлористом углероде. Оллред и Рохов [4] качественно объяснили различия химических сдвигов, наблюдавшиеся для разных заместителей, изменением гибридизации связи при изменении электроотрицательности заместителя. Для метильных производных с приблизительно одинаковой гибридизацией химические сдвиги коррелируются с электроотрицательностью. Следует, однако, отметить, что приведенная на рис. 10.1 линейная корреляция химических сдвигов метильных протонов с электроотрицательностью заместителя
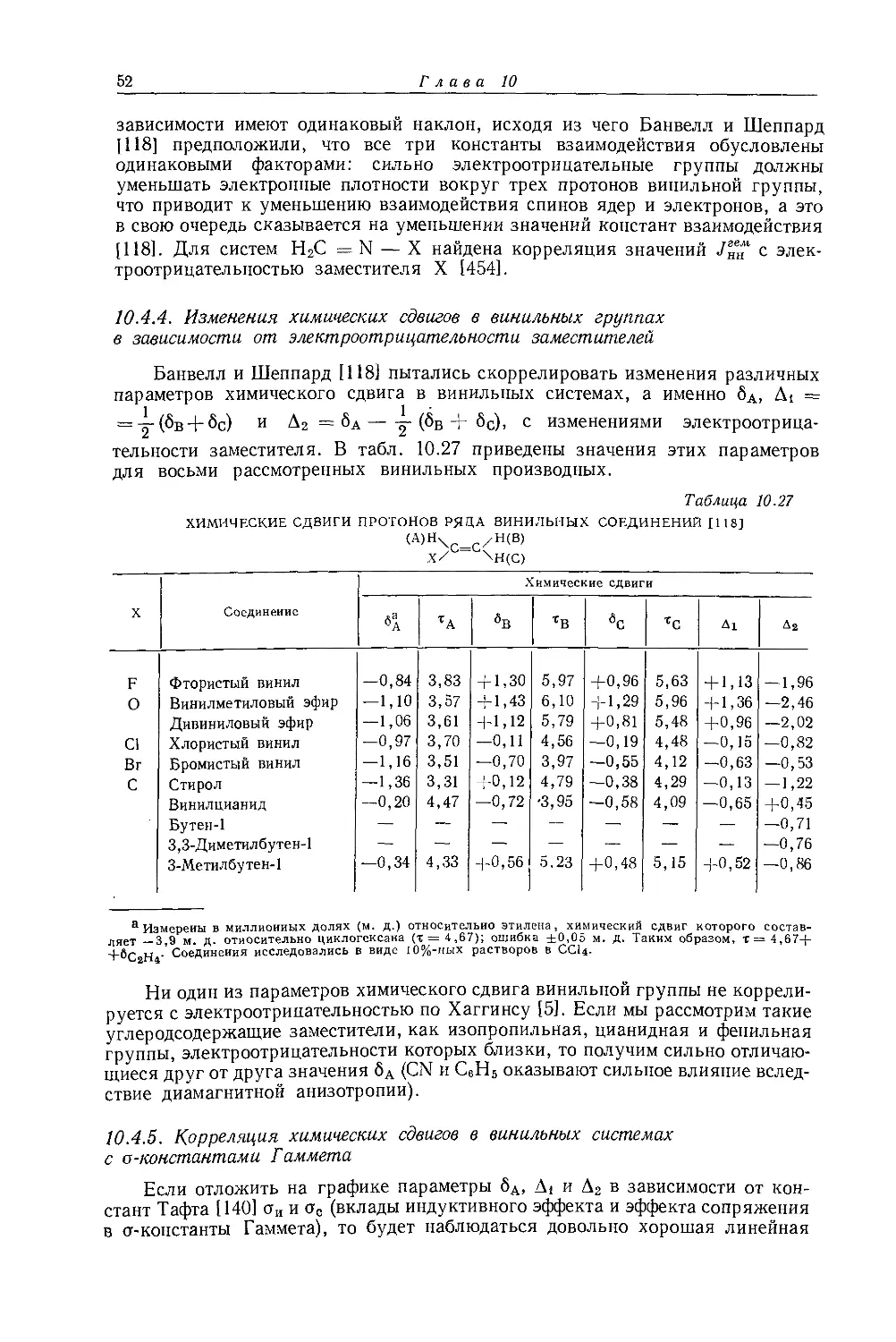
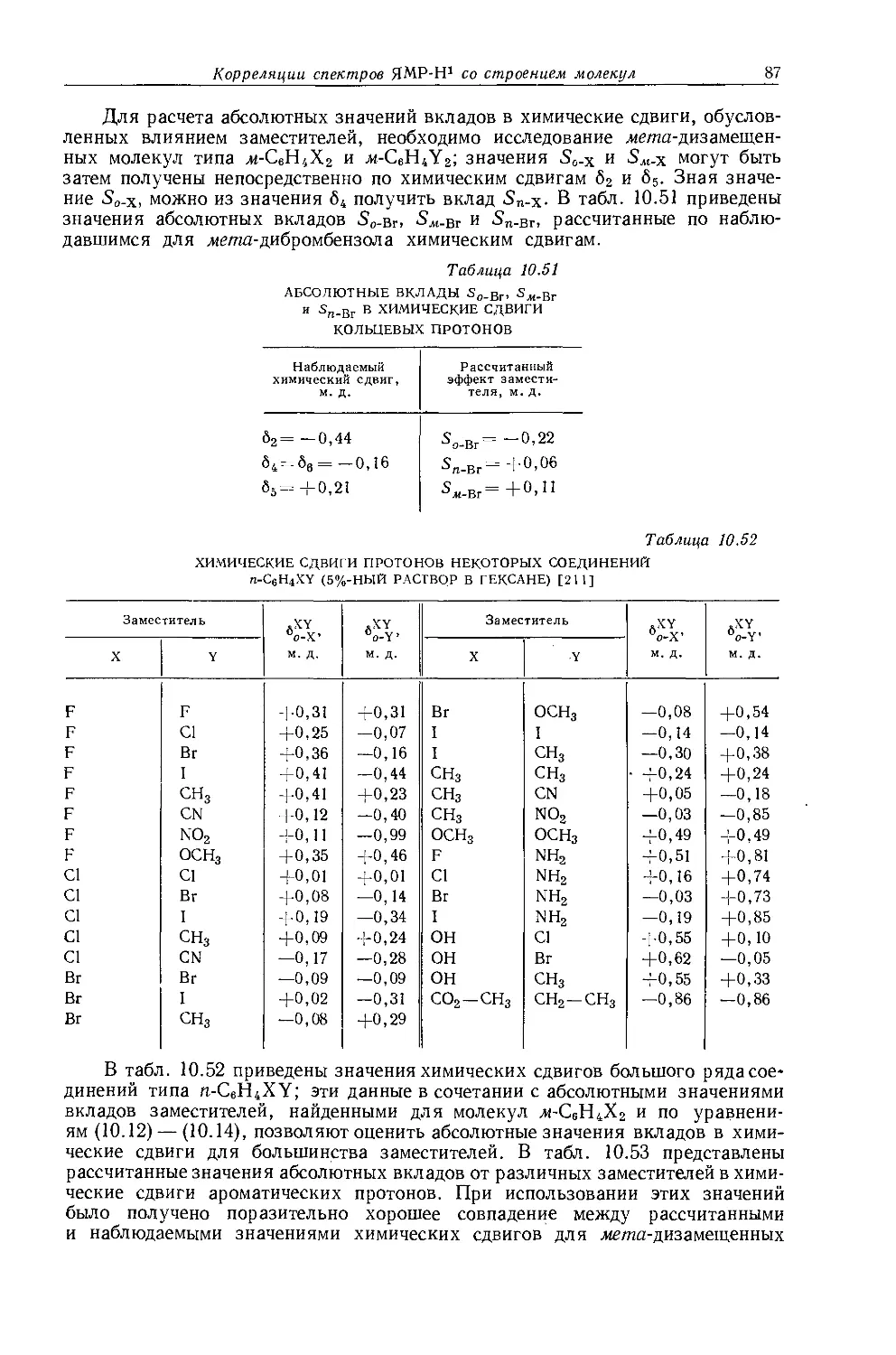
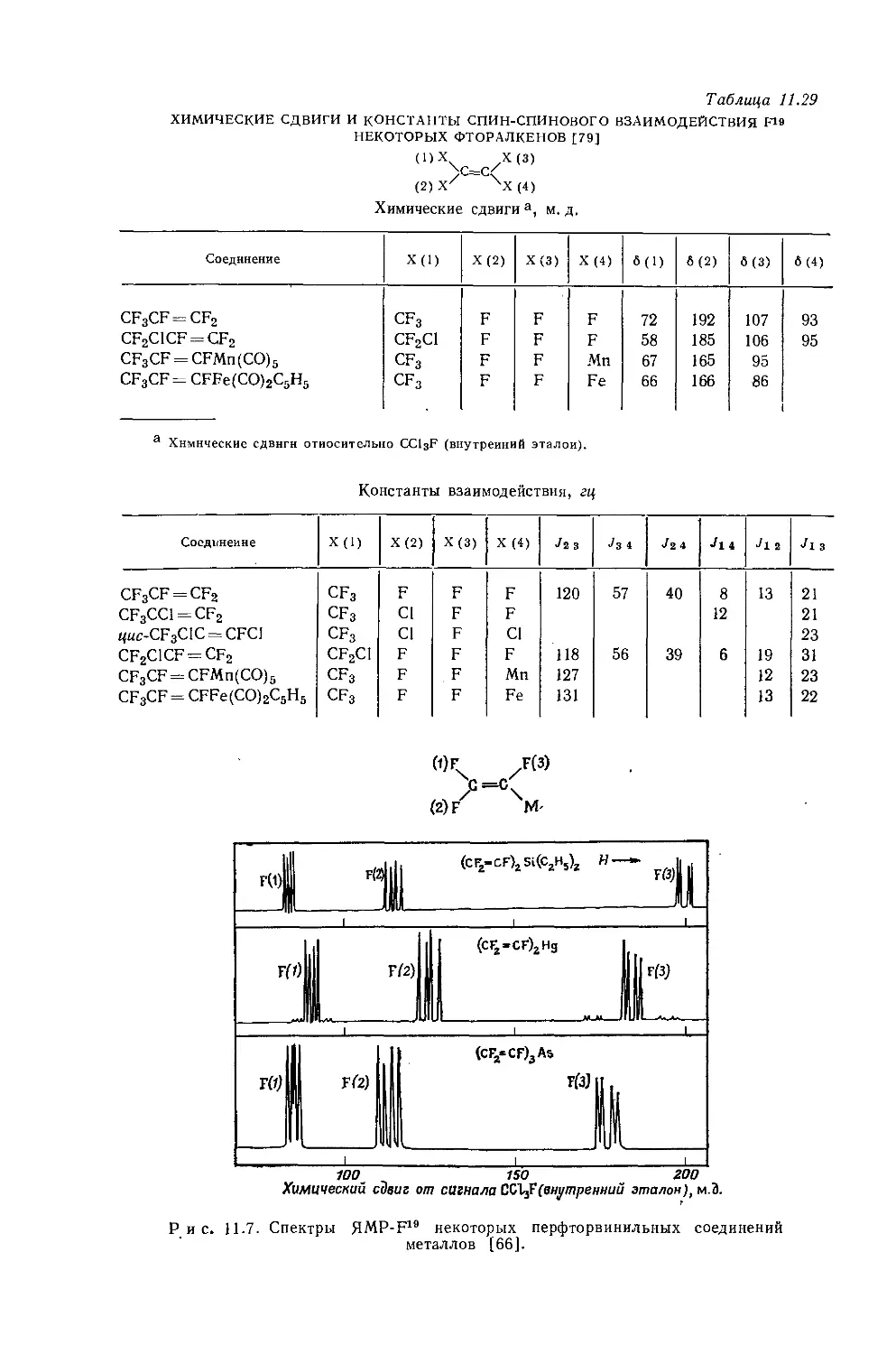
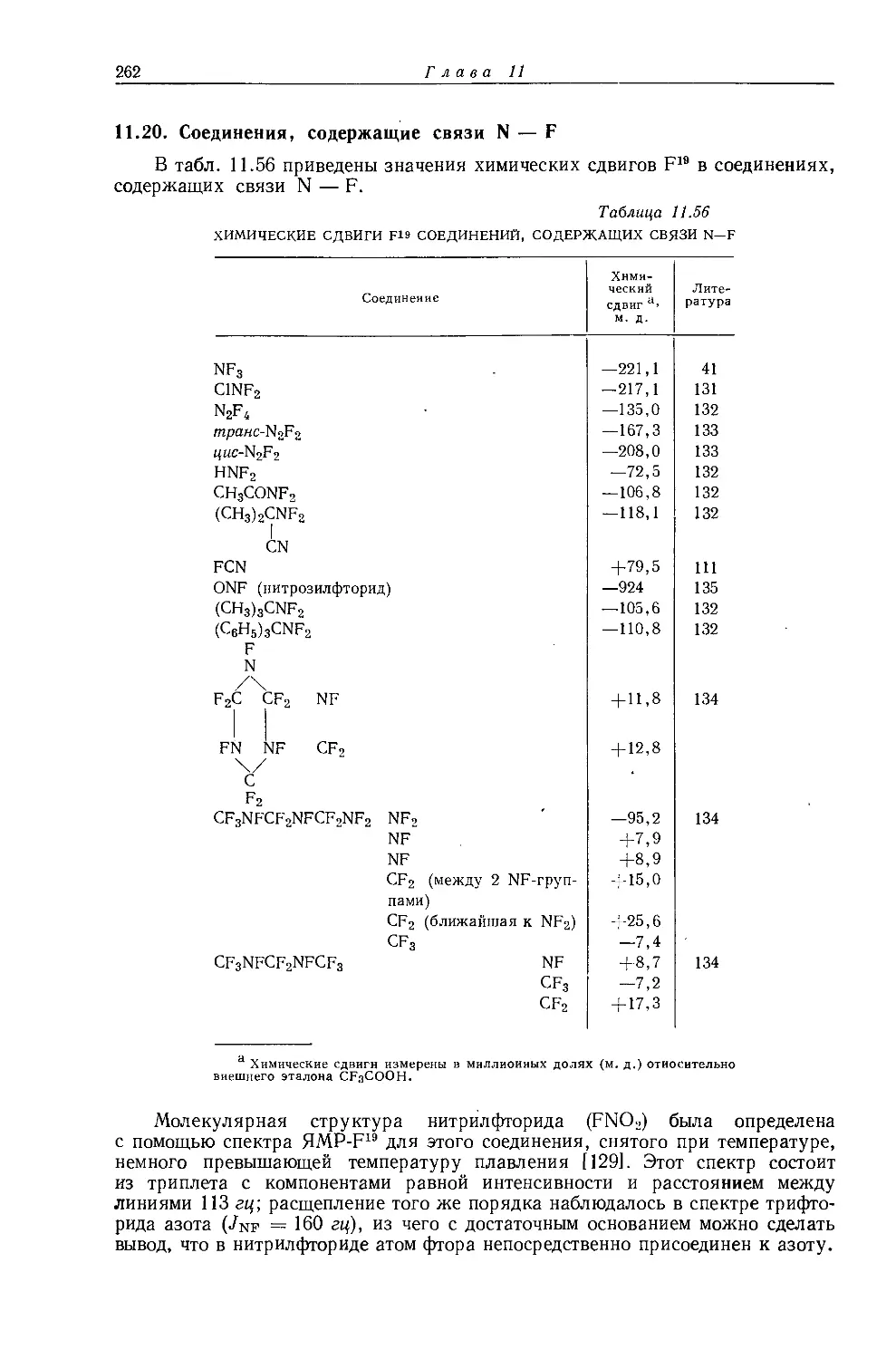
Рис. 10.1. Зависимость химических сдвигов протонов в метилгалогенидах от электроотрицательности галогенов по Хаггинсу [4].
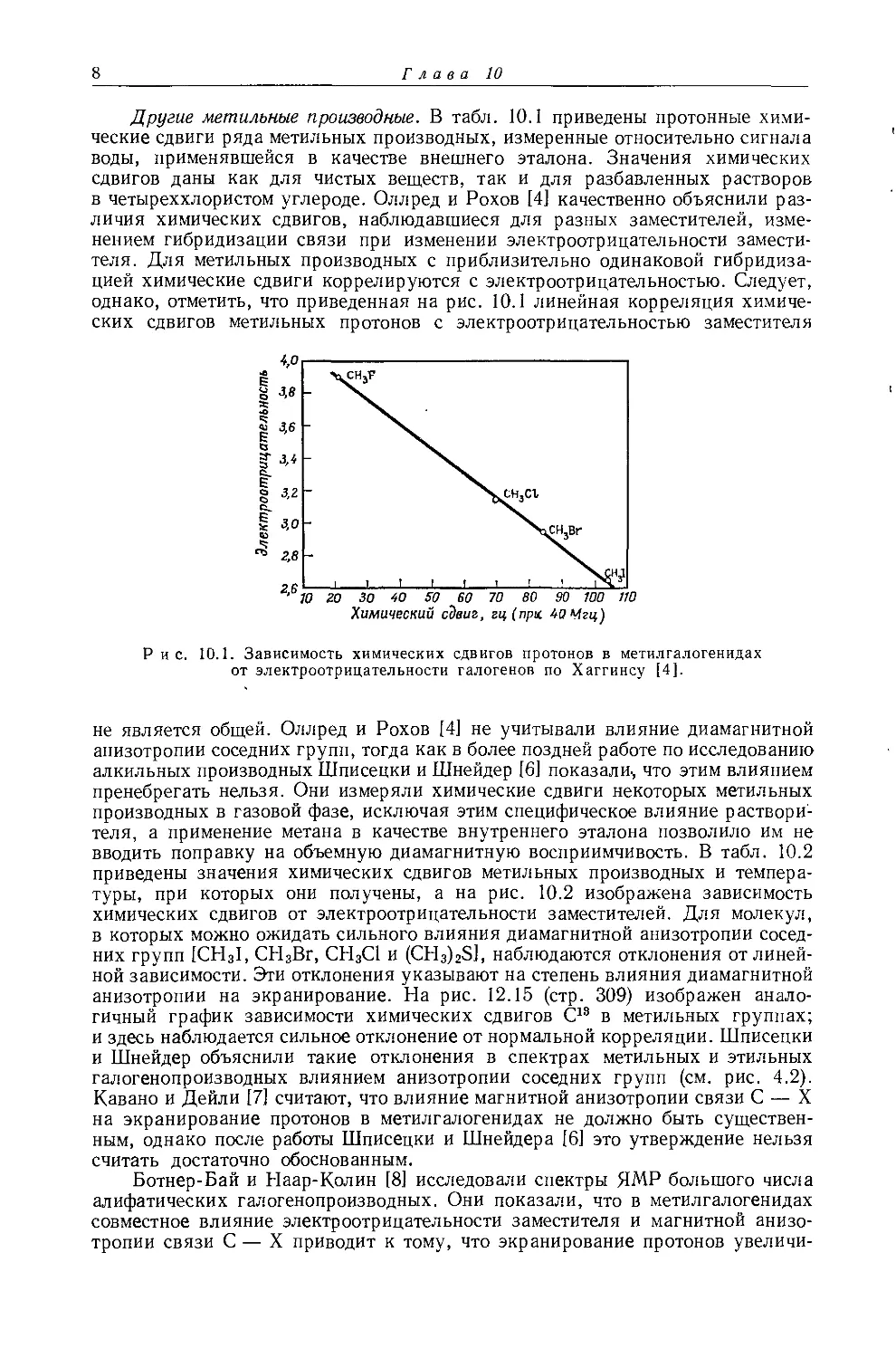
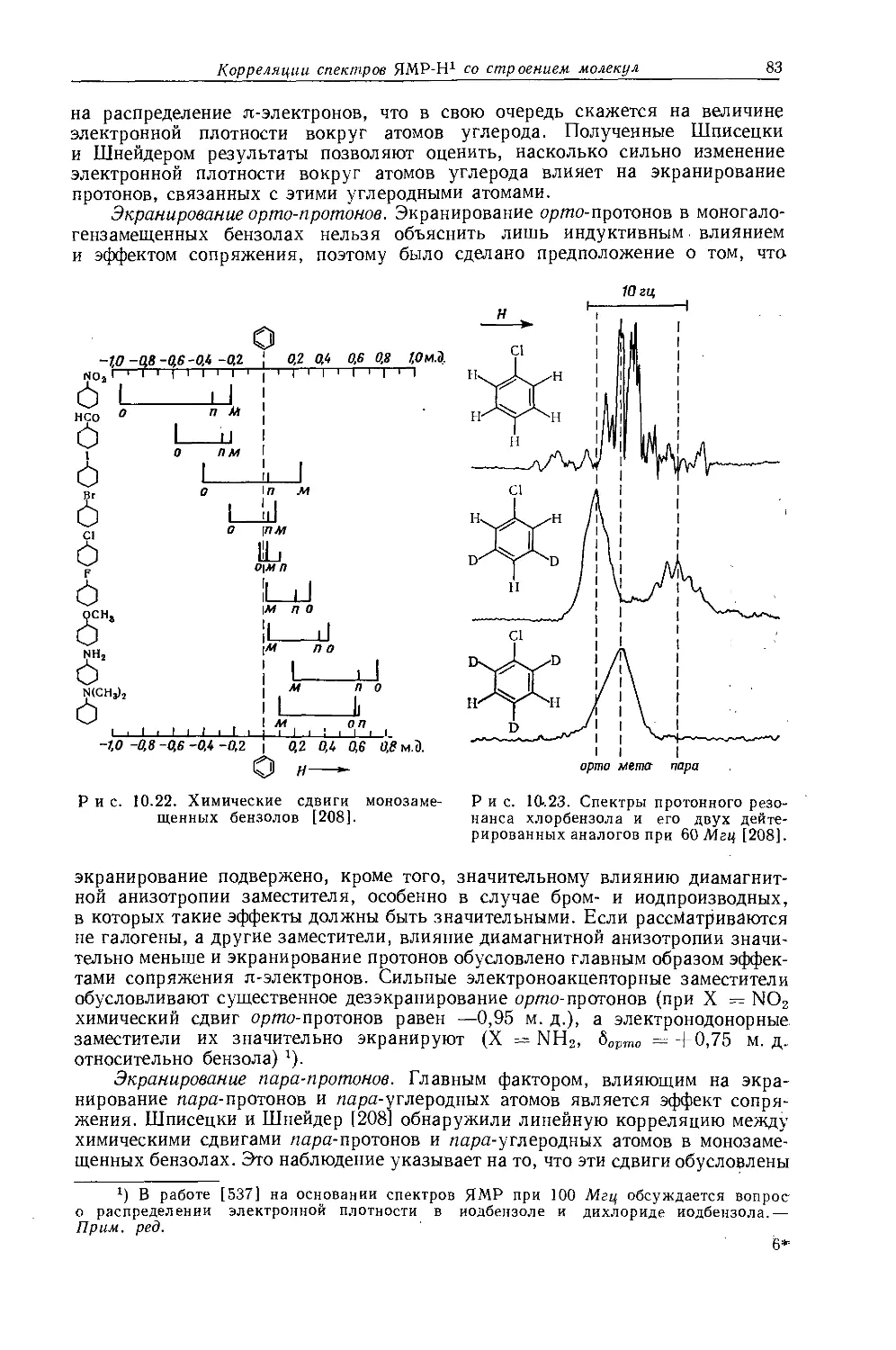
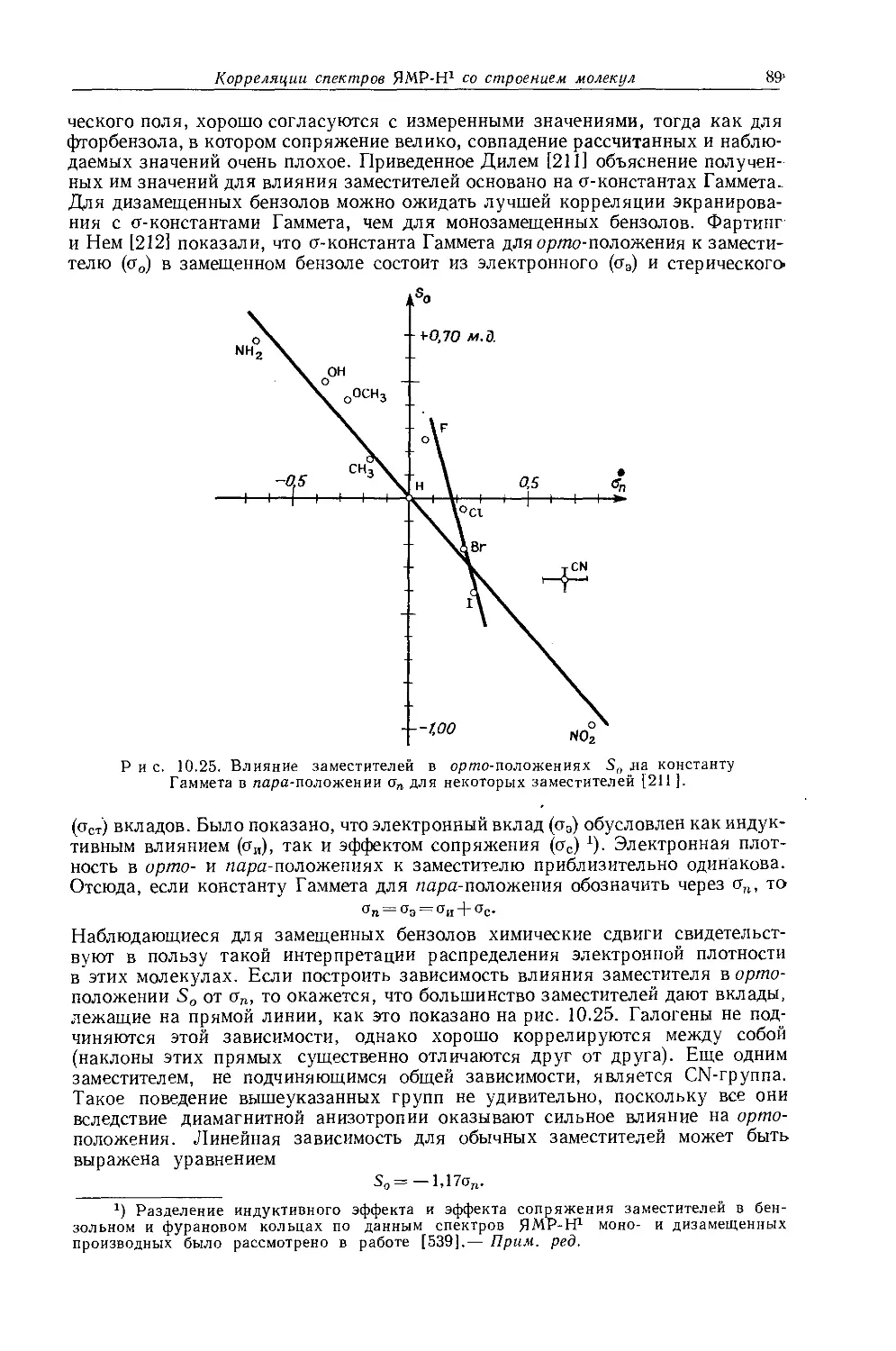
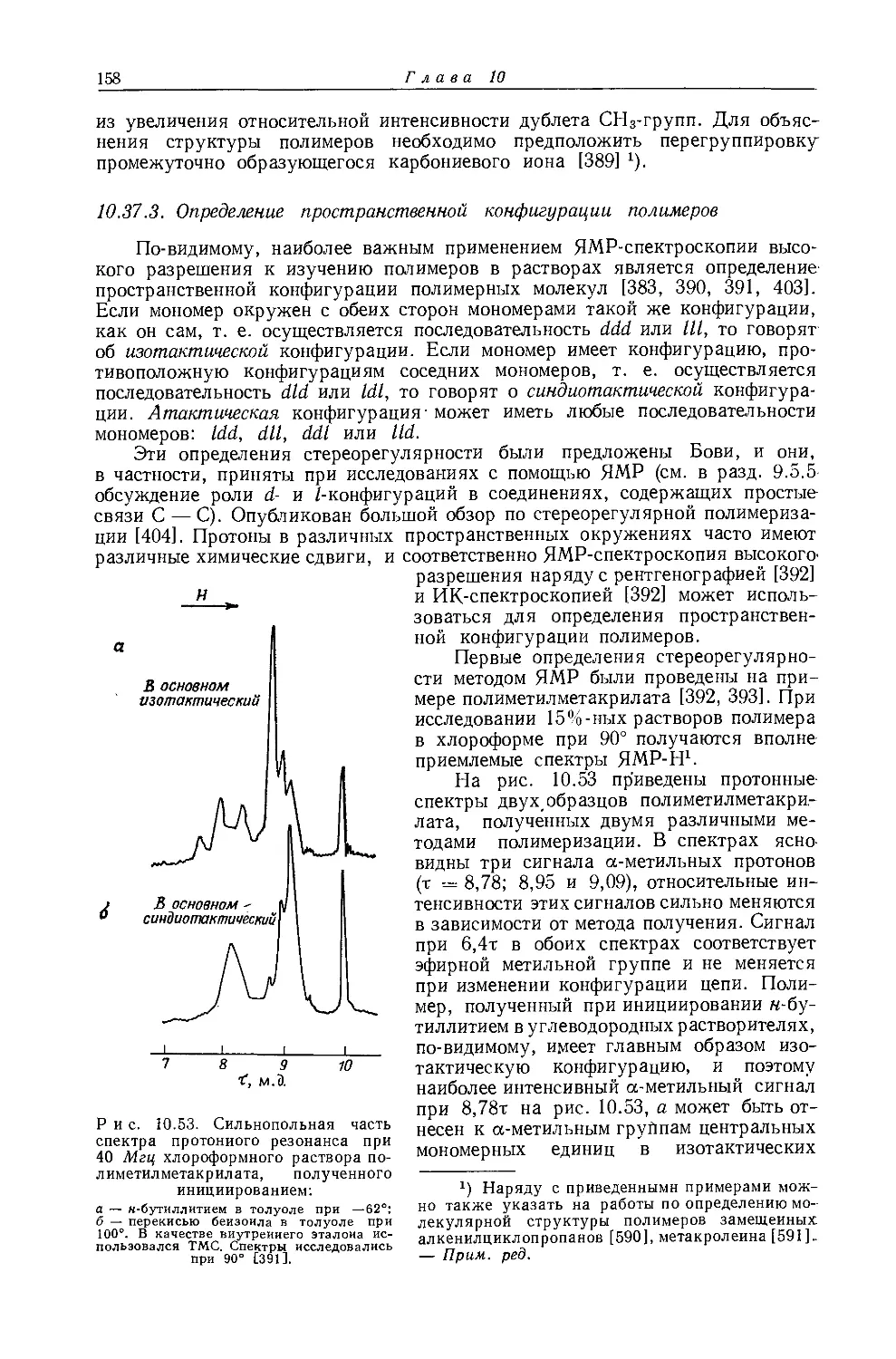
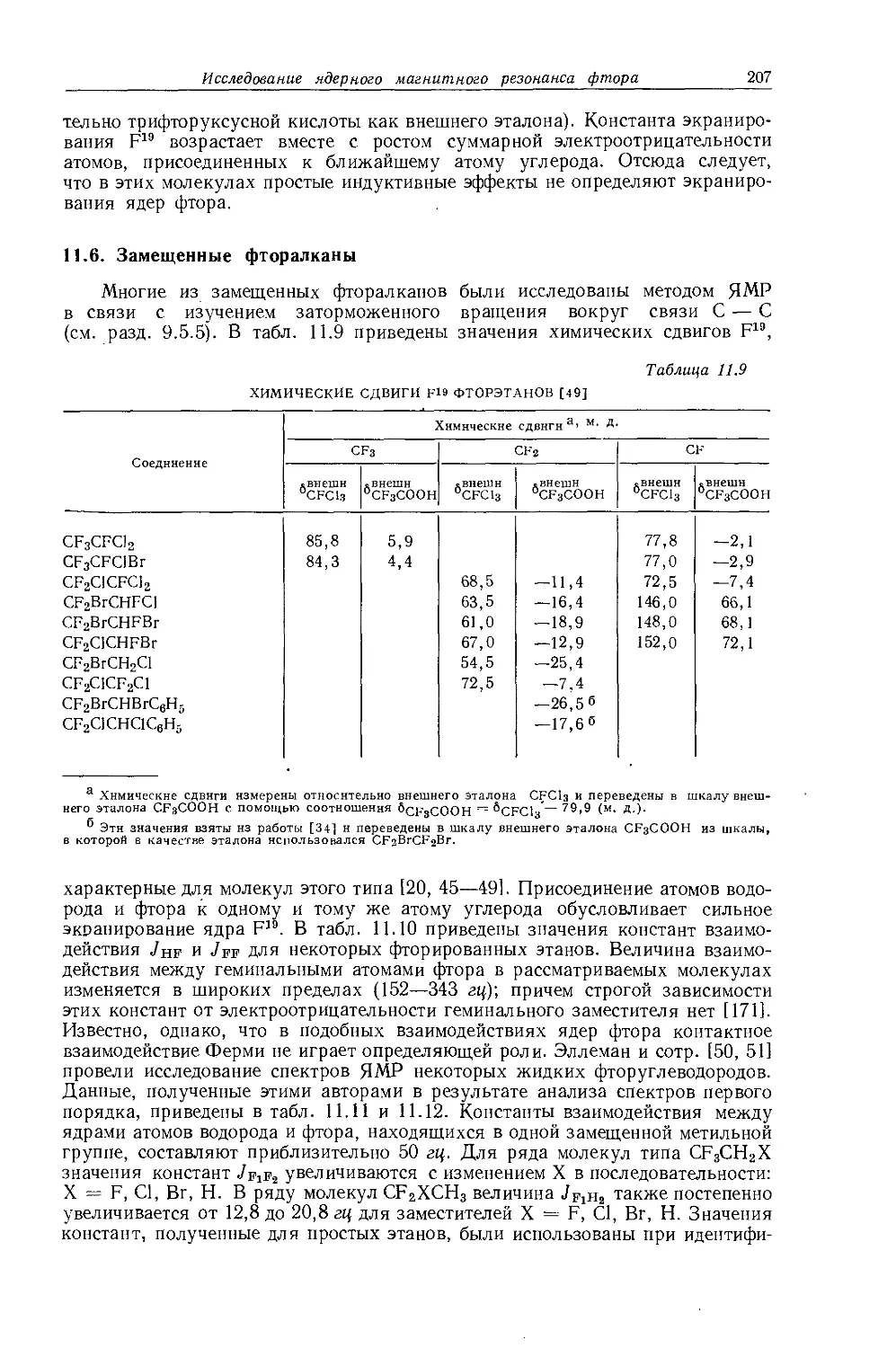
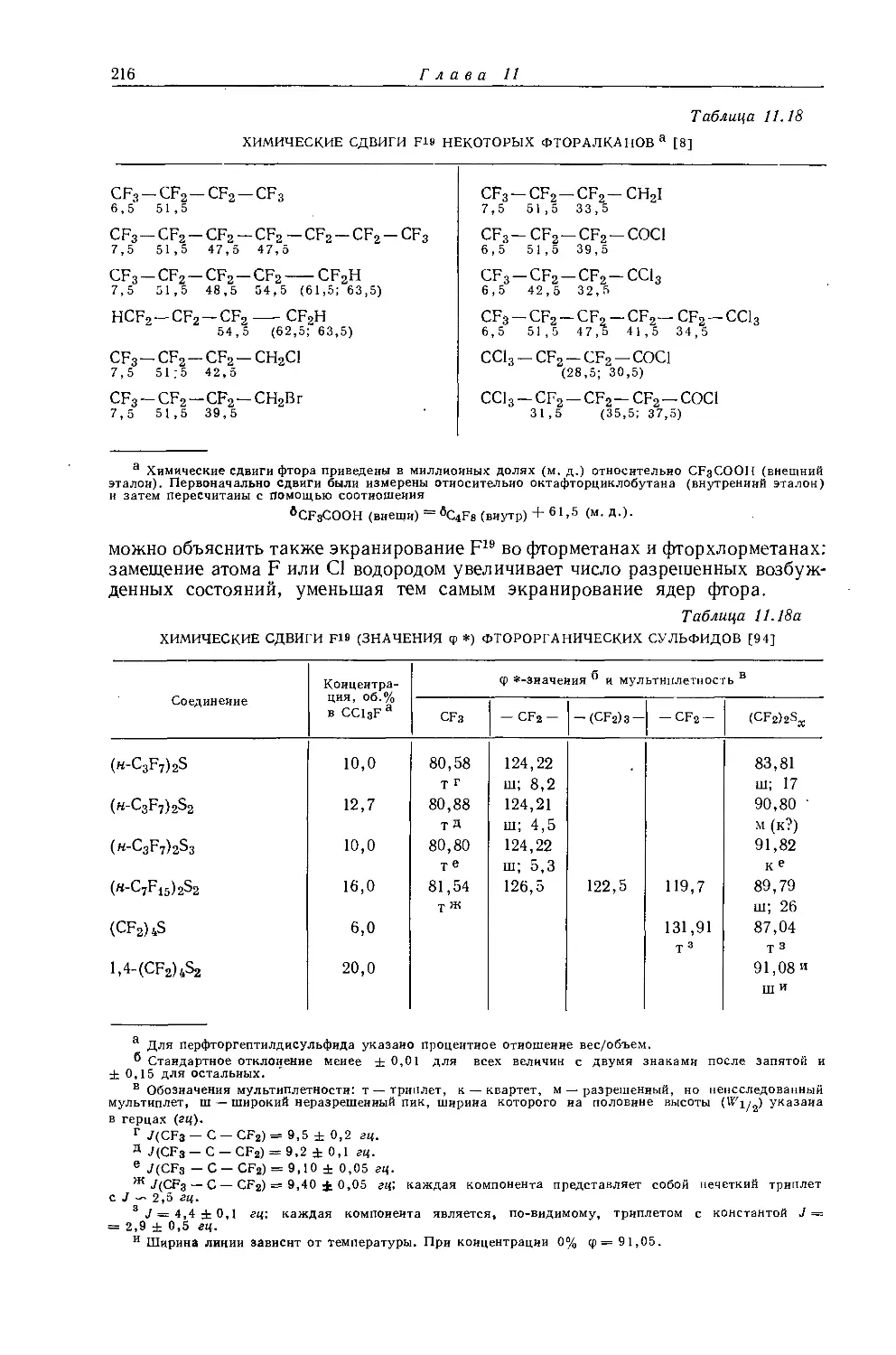
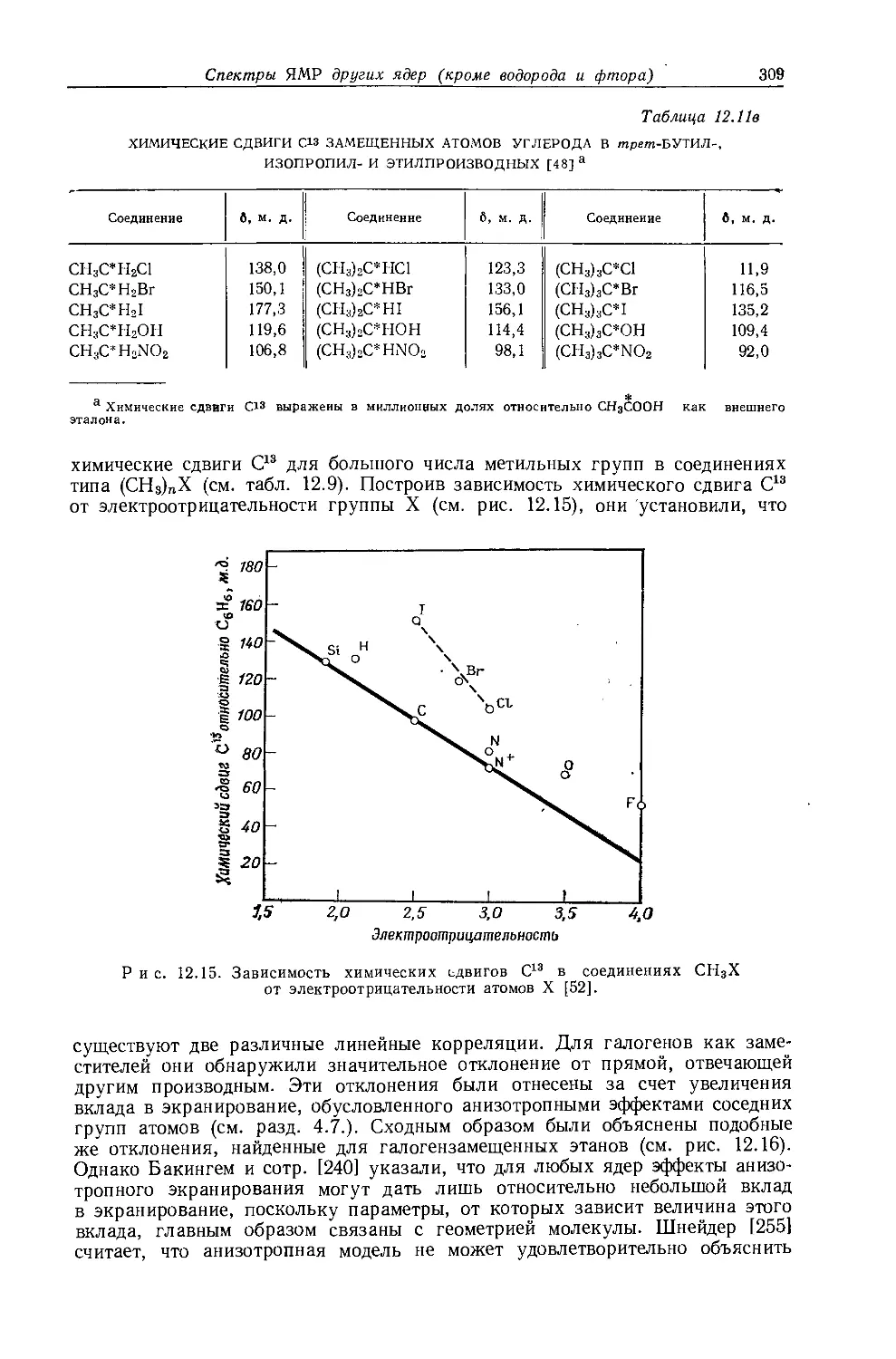
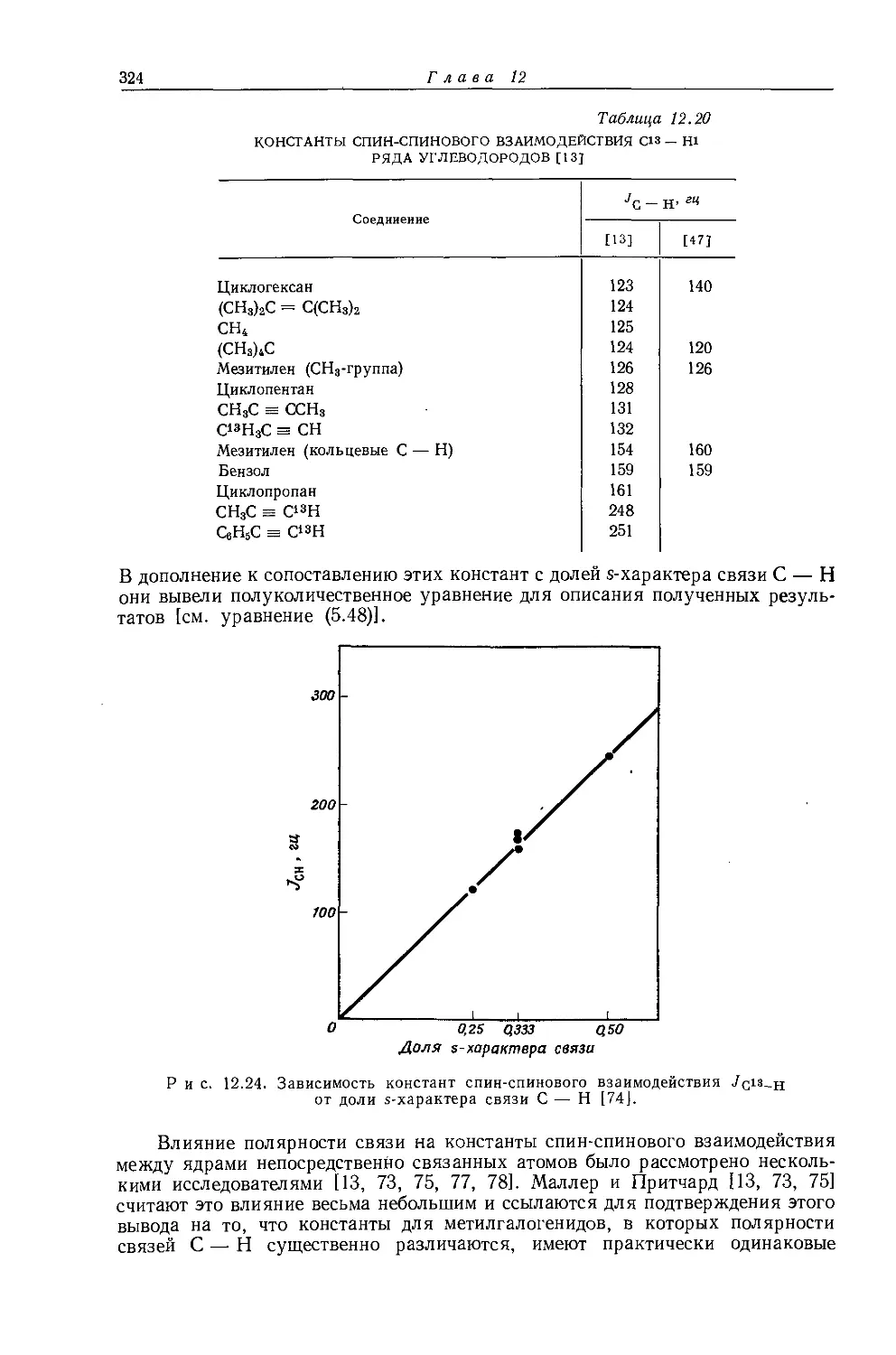
не является общей. Оллред и Рохов [4] не учитывали влияние диамагнитной анизотропии соседних групп, тогда как в более поздней работе по исследованию алкильных производных Шписецки и Шнейдер [6] показали., что этим влиянием пренебрегать нельзя. Они измеряли химические сдвиги некоторых метильных производных в газовой фазе, исключая этим специфическое влияние растворителя, а применение метана в качестве внутреннего эталона позволило им не вводить поправку на объемную диамагнитную восприимчивость. В табл. 10.2 приведены значения химических сдвигов метильных производных и температуры, при которых они получены, а на рис. 10.2 изображена зависимость химических сдвигов от электроотрицательности заместителей. Для молекул, в которых можно ожидать сильного влияния диамагнитной анизотропии соседних групп [СН31, СН3Вг, СН3С1 и (CH3)2S], наблюдаются отклонения от линейной зависимости. Эти отклонения указывают на степень влияния диамагнитной анизотропии на экранирование. На рис. 12.15 (стр. 309) изображен аналогичный график зависимости химических сдвигов С13 в метильных группах; и здесь наблюдается сильное отклонение от нормальной корреляции. Шписецки и Шнейдер объяснили такие отклонения в спектрах метильных и этильных галогенопроизводных влиянием анизотропии соседних групп (см. рис. 4.2). Кавано и Дейли [7] считают, что влияние магнитной анизотропии связи С — X на экранирование протонов в метилгалогенидах не должно быть существенным, однако после работы Шписецки и Шнейдера [6] это утверждение нельзя считать достаточно обоснованным.
Ботнер-Бай и Наар-Колин [8] исследовали спектры ЯМР большого числа алифатических галогенопроизводных. Они показали, что в метилгалогенидах совместное влияние электроотрицательности заместителя и магнитной анизотропии связи С — X приводит к тому, что экранирование протонов увеличи-
Корреляции спектров ЯМР-Н1 со строением молекул
9
Таблица 10.2
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ НЕКОТОРЫХ МЕТИЛЬНЫХ ПРОИЗВОДНЫХ
В ГАЗОВОЙ ФАЗЕ [6]
Соединение Температура, °C Химический сдвиг та, м. д.
сн4 22 0
CH3F 22 —4,00
СН3С1 22 —2,71
СН3Вг 92 —2,32
СН31 109 — 1,85
(СН3)2О 22 —3,10
(CH3)3N 120 —2,03
(СН3)4С НО —0,83
СН3СН3 22 —0,75
(CH3)2s 108 — 1,90
CH3CN 150 — 1,53
(СН3)2СО 140 — 1,82
СН3СНО 122 — 1,79
ch3no2 166 —3,91
(СН3) 4S i 100 +0,13
(CH3)4Ge 120 0,00
(CH3)4Sn 100 + 0,10
(CH3)4Pb 170 —0,57
а Химические сдвиги измерены относительно резонанс-иого сигнала СН<, взятого в качестве внутреннего эталона; значения т получаются по уравнению т=9,7 Н-дсщ*
вается в ряду от хлорида к иодиду. Таким образом, индуктивный эффект заместителя является главным фактором, влияющим на экранирование
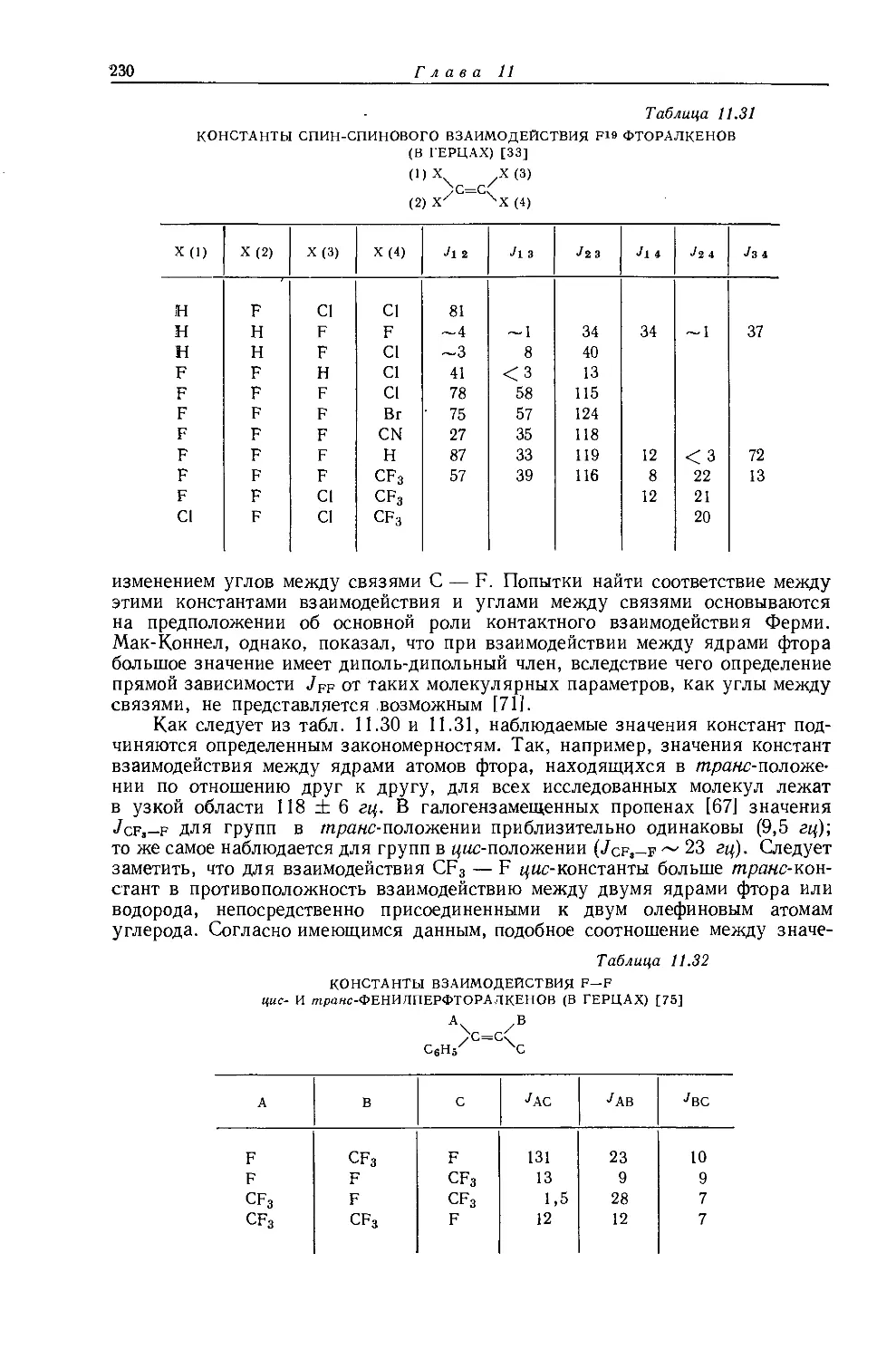
Рис. 10.2. Зависимость химических сдвигов протонов в метилгалогенидах от электроотрицательности галогенов по Хаггинсу [6].
протонов в метильных производных, лишь при отсутствии влияния диамагнитной анизотропии соседних групп 1). Оллред и Мак-Кой [435] рассмотрели протон
J) Значения протонных химических сдвигов замещенных метанов типа CHXYZr исправленные с учетом анизотропии магнитной восприимчивости заместителя, а также константы Jq13—н в этих соединениях хорошо коррелируются с индуктивными о* константами Тафта [493, 494].— Прим. ред.
10
Глава 10
ные химические сдвиги метильных производных Zn, С, N, О и F и объяснили полученные результаты главным образом полярностью связей. Опубликованы :и другие работы по исследованию алканов [466, 471] *).
10.1.2. Корреляция химических сдвигов с электроотрицательностью .заместителей в этильных производных
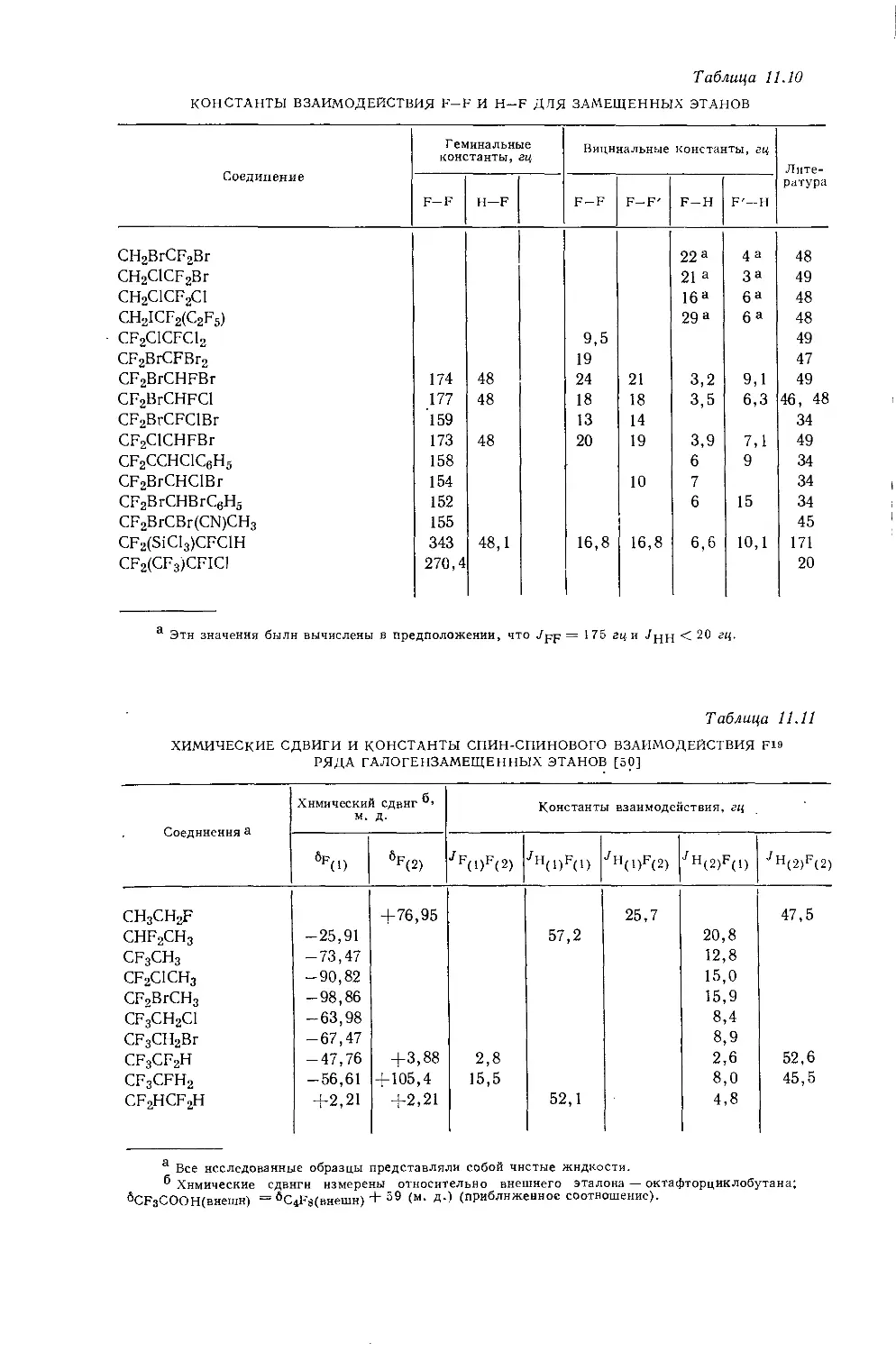
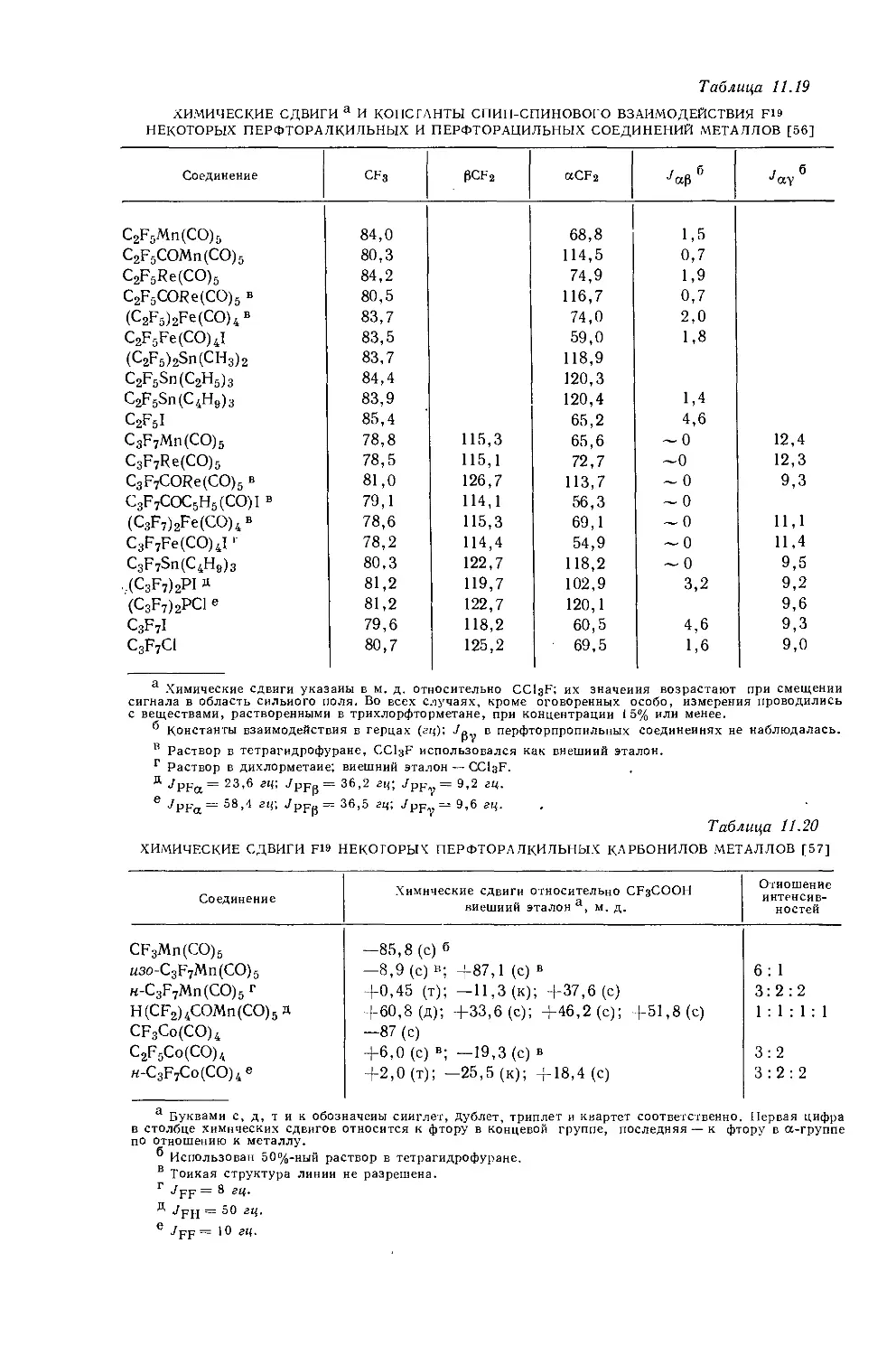
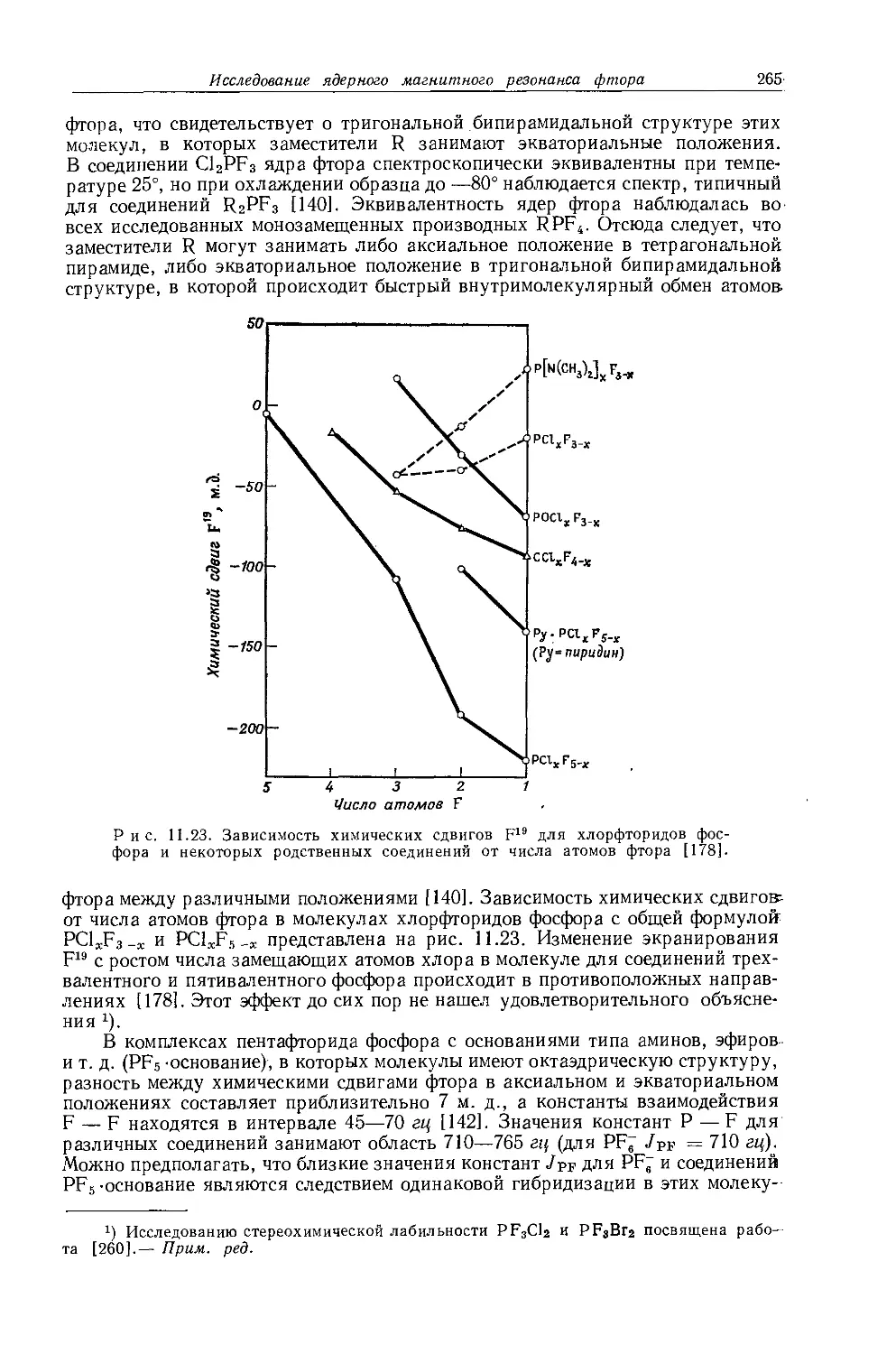
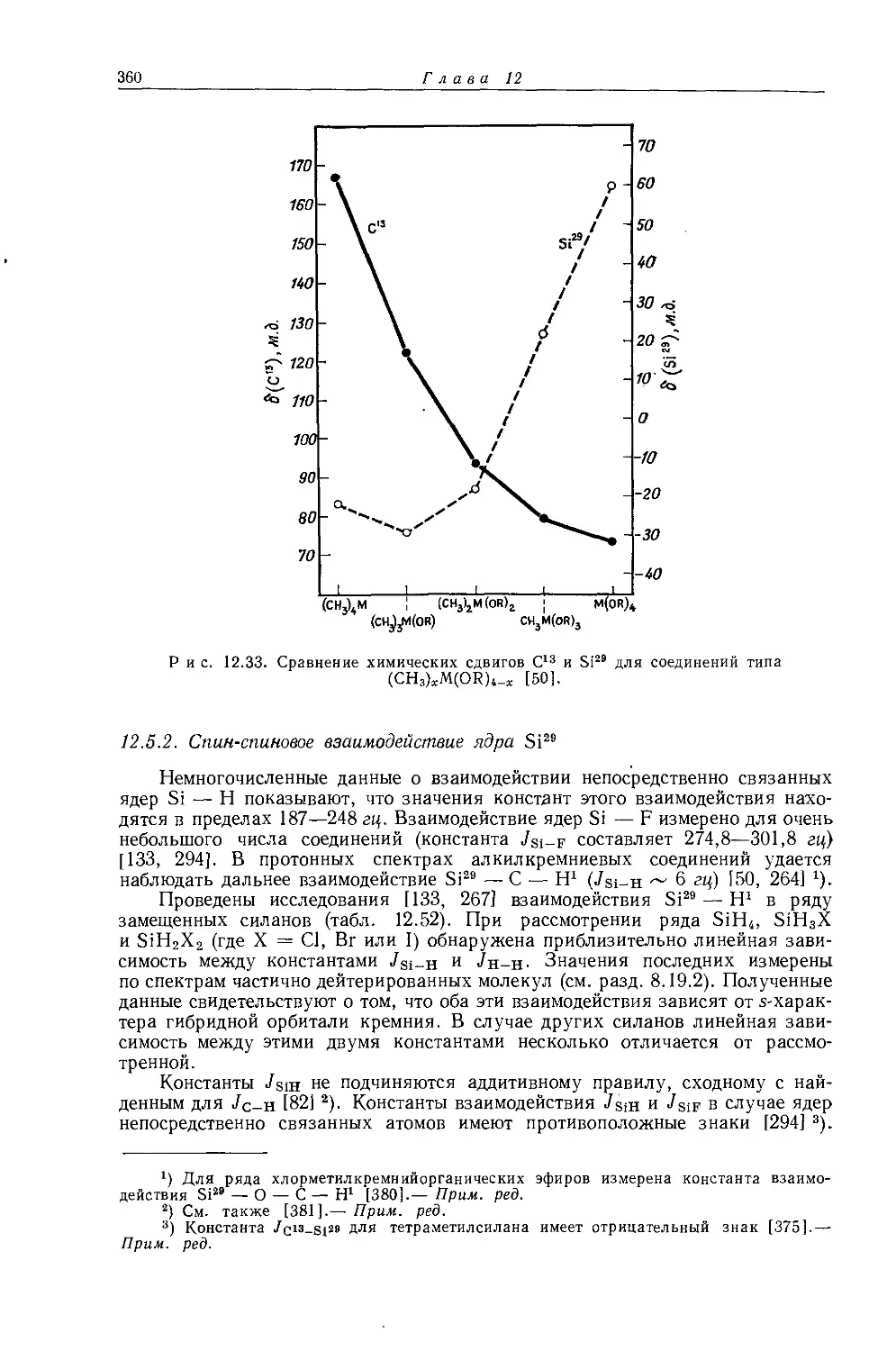
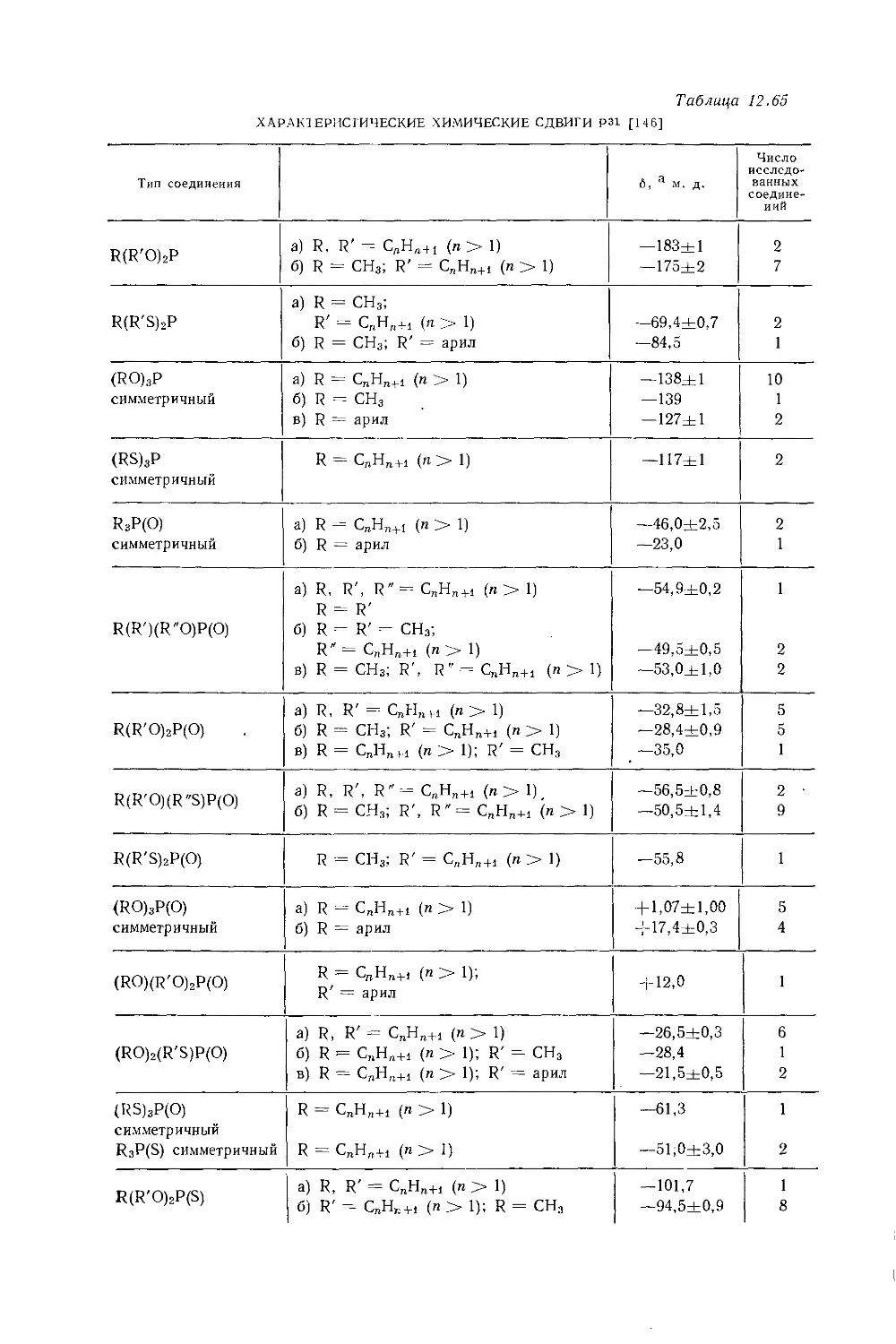
Дейли и Шулери [9] исследовали спектры ЯМР большого числа этильных производных с целью построения зависимости экранирования а-метиленовых протонов от электроотрицательности заместителей по Хаггинсу. Для того чтобы исключить влияние объемной диамагнитной восприимчивости, они измерили химические сдвиги а-СН2-группы относительно Р-СН3-группы, как
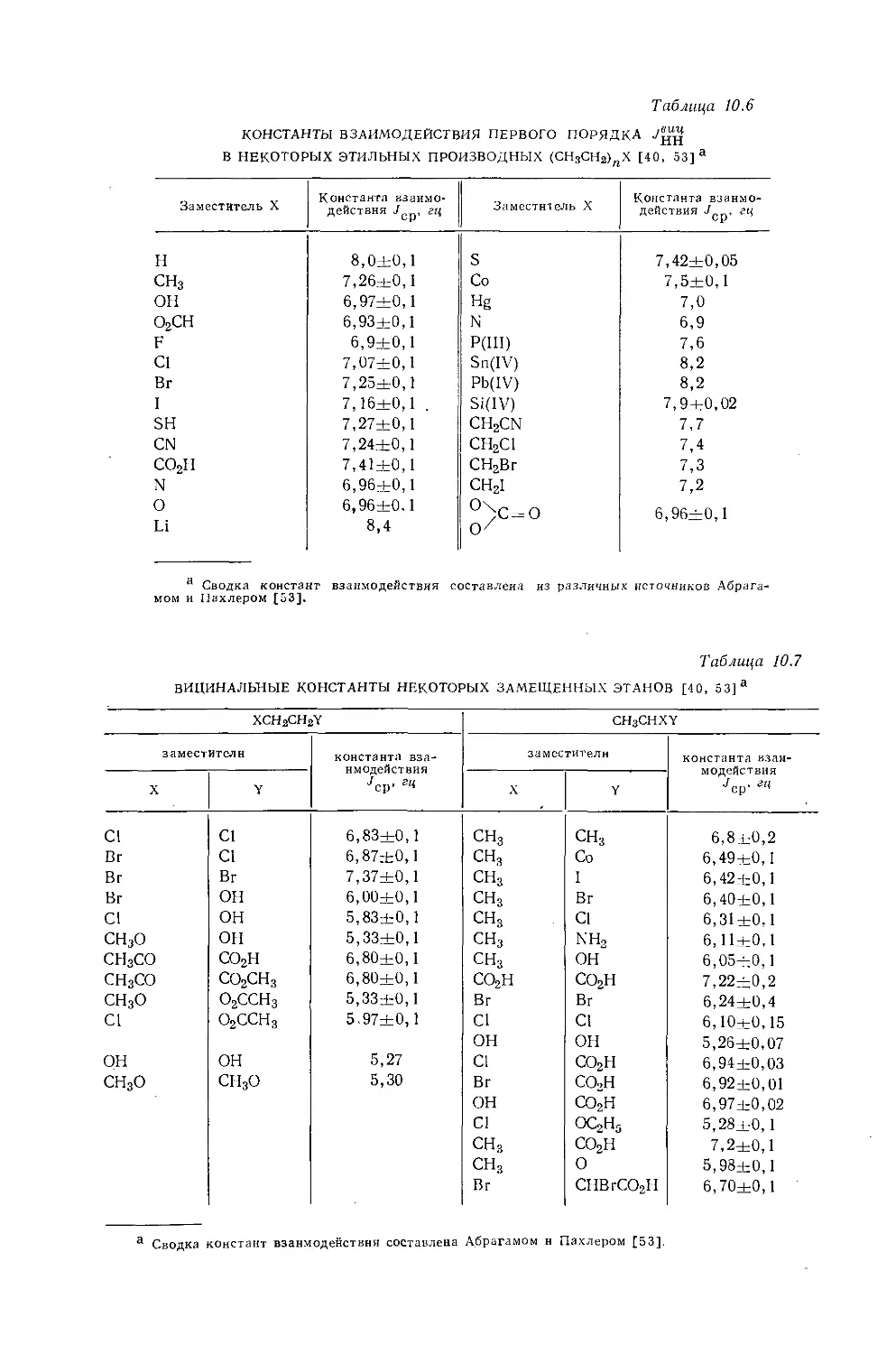
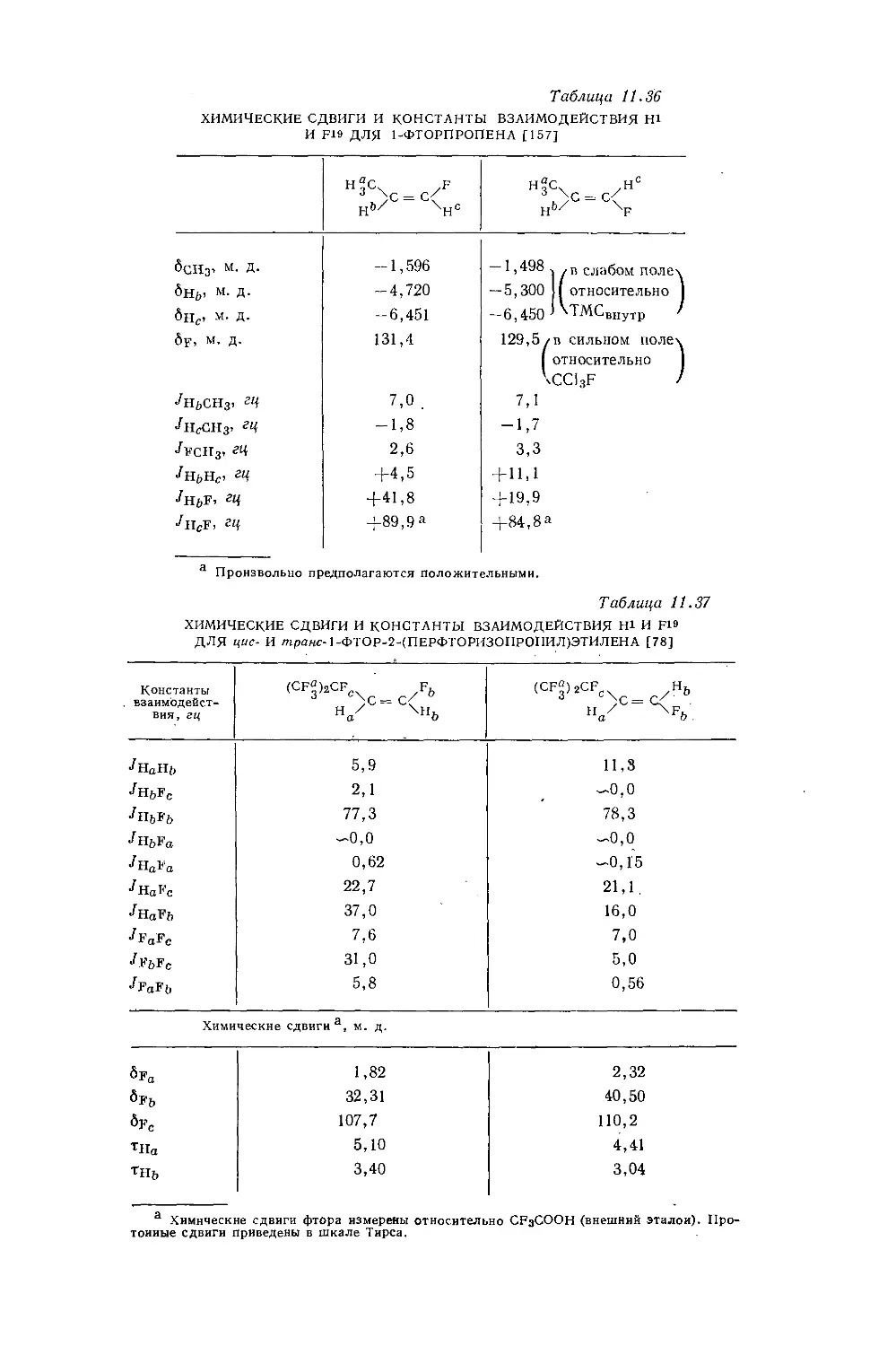
Рис. 10.3. Типичный спектр первого порядка протонного резонанса этильной группы.
это показано на рис. 10.3. Позднее эта работа была повторена многими другими исследователями, и в табл. 10.3 приведены значения химических сдвигов для четырнадцати этильных производных, экстраполированные к бесконечному разбавлению в четыреххлористом углероде. Результаты для растворов в четыреххлористом углероде соответствуют уравнению Дейли — Шулери [9], которое отражает наблюдаемое линейное соотношение:
Электроотрицательность = 0,684Д+ 1,78, (10.1)
где Л = 6СНз-бСн2 (м. Д-).
Это уравнение может быть использовано для предсказания электроотрицательности заместителя, если известен внутренний химический сдвиг его этильного производного (см. табл. 10.3). При использовании корреляций такого рода следует с осторожностью относиться к группам, которые могут обладать магнитной анизотропией и влиять на экранирование. Если бы внутренний химический сдвиг А был обусловлен только индуктивным эффектом, то следовало бы допустить, что влиянию заместителя подвержена лишь а-СН2-группа и вклад диамагнитной анизотропии соседних групп в экранирование несуществен. Однако эти допущения неправомерны: Оллред и Рохов исследовали разбавленные растворы этилгалогенидов в четыреххлористом углероде и показали, что химические сдвиги метильных групп не одинаковы.
т) Спектры ЯМР-Н1 метильных производных кремния, германия и олова исследованы также в работах [495—499]. Показано, что химический сдвиг метильных групп определяется индуктивным эффектом, магнитной анизотропией заместителя, присоединенного к атому металла, и Лт-взаимодействием последнего с заместителем.— Прим. ред.
Корреляции спектров ЯМР-Н1 со строением молекул
11
Таблица 10.3
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ3 НЕКОТОРЫХ АЛКИЛЬНЫХ СОЕДИНЕНИЙ RX [7]
Заместитель X T, M. Д. Рассчитанные электро-отрица-тельно-сти Т, м. Д.
R = CH3 R = C2H6 R — ИЗО-С3Н7 R -- Н-С3Н7
a-CH2 р-снз а-СН р-снз а-СН2 ДСН2 ?-СНз
С1 —2,83 6 —3,25 6 — 1,106 3,25 —3,92 6 — 1,32 6 —3,25 —1,58 -0,83
Вг —2,47 6 -3,15 6 —1,436 2,96 —3,98 б — 1,50 6 —3,12 -1,67 —0,83
I — 1,93 6 —2,93 6 — 1,65 6 2,66 —4,02 6 — 1,67 6 —2,93 — 1,65 —0,80
ОН —3,17 6 —3,37 6 —0,95 6 3,43 —3,72 —0,93 —3,27 — 1,30 —0,70
CN — 1,756 —2,12 — 1,08 2,49 —2,45 — 1,12 —2,07 — 1,48 —0,88
соон — 1,856 —2,13 —0,93 2,60 —2,33 —0,98 —2,08 — 1,45 —0,77
С6Н5 —2,12 6 —2,40 6 —0,98 6 2,75 —2,67 6 — 1,02 6 -2,37 — 1,42 —0,73
-О- —3,02 6 —3,15 6 —0,92 6 3,31 —3,33 —0,85 —3,05 — 1,32 —0,70
—S—- — 1,876 —2,27 — 1,02 2,64 —2,70 — 1,02 —2,20 — 1,37 —0,75
сно — 1,956 —2,23 -0,90 2,69 —2,17 6 —0,90 б —2,12 — 1,42 —0,75
nh2 —2,23 —2,52 —0,87 2,91 —2,85 6 —0,80 б —2,38 — 1,20 —0,70
-s2- —2,45 —1,12 2,69 —2,40 — 1,48 -0,80
NO3 —4,27 — 1,17 3,90 —4,93 — 1,15 —4,15 — 1,55 —0,82
no2 —4,07 6 —4,15 — 1,35 —2,50 6 — 1,30 6 —4,05 — 1,78 —0,80
F —4,05 6
H 0,00 —0,63 — 1,12 —0,68
а Химические сдвиги измерены в миллионных долях (м. д.) относительно бензола, экстраполированы к бесконечно разбавленным растворам в ССЦ и затем пересчитаны относительно метана. Значения Т получаются по уравнению т = 9,78 + бсщ-
® См. приложение I.
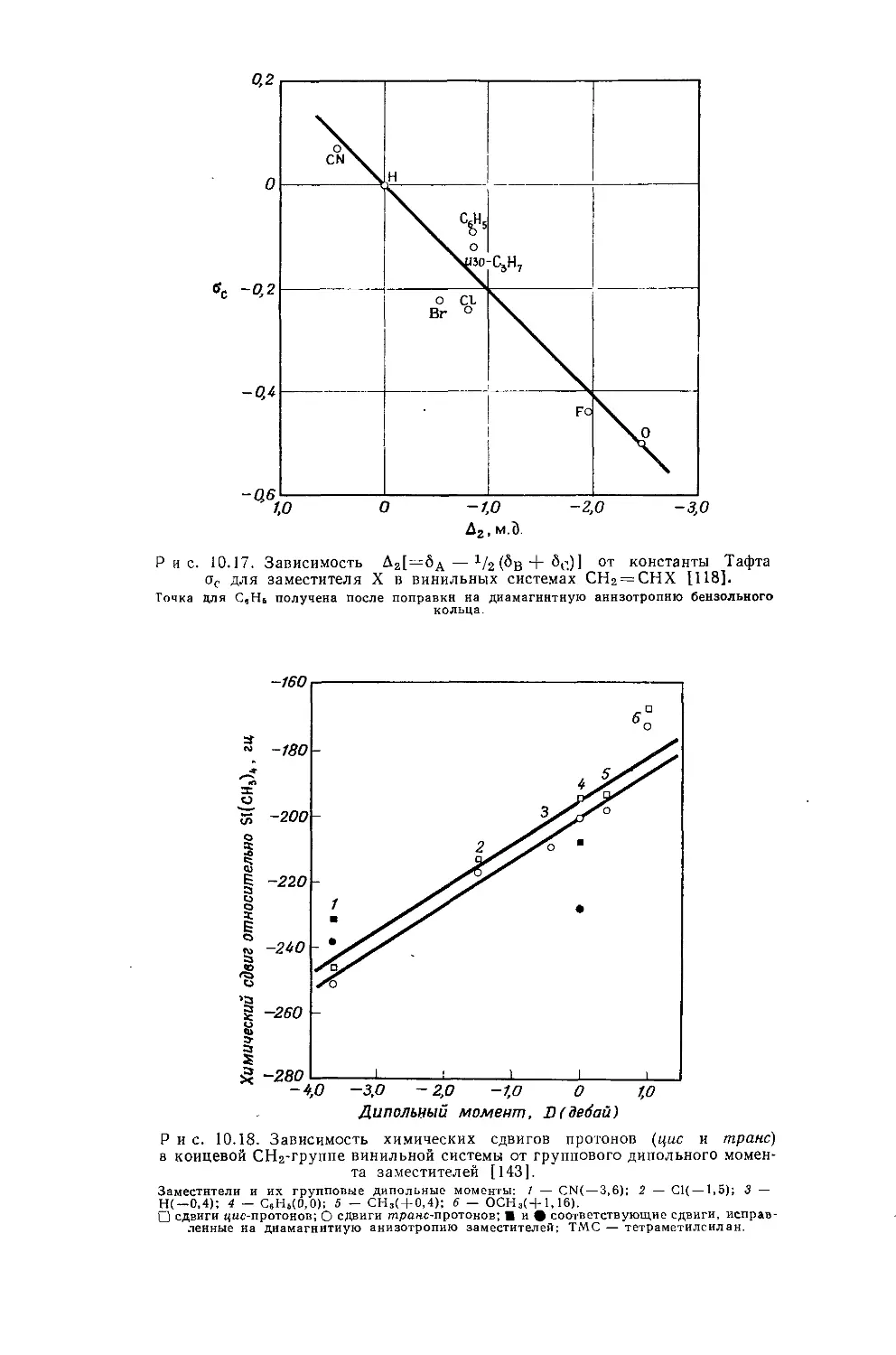
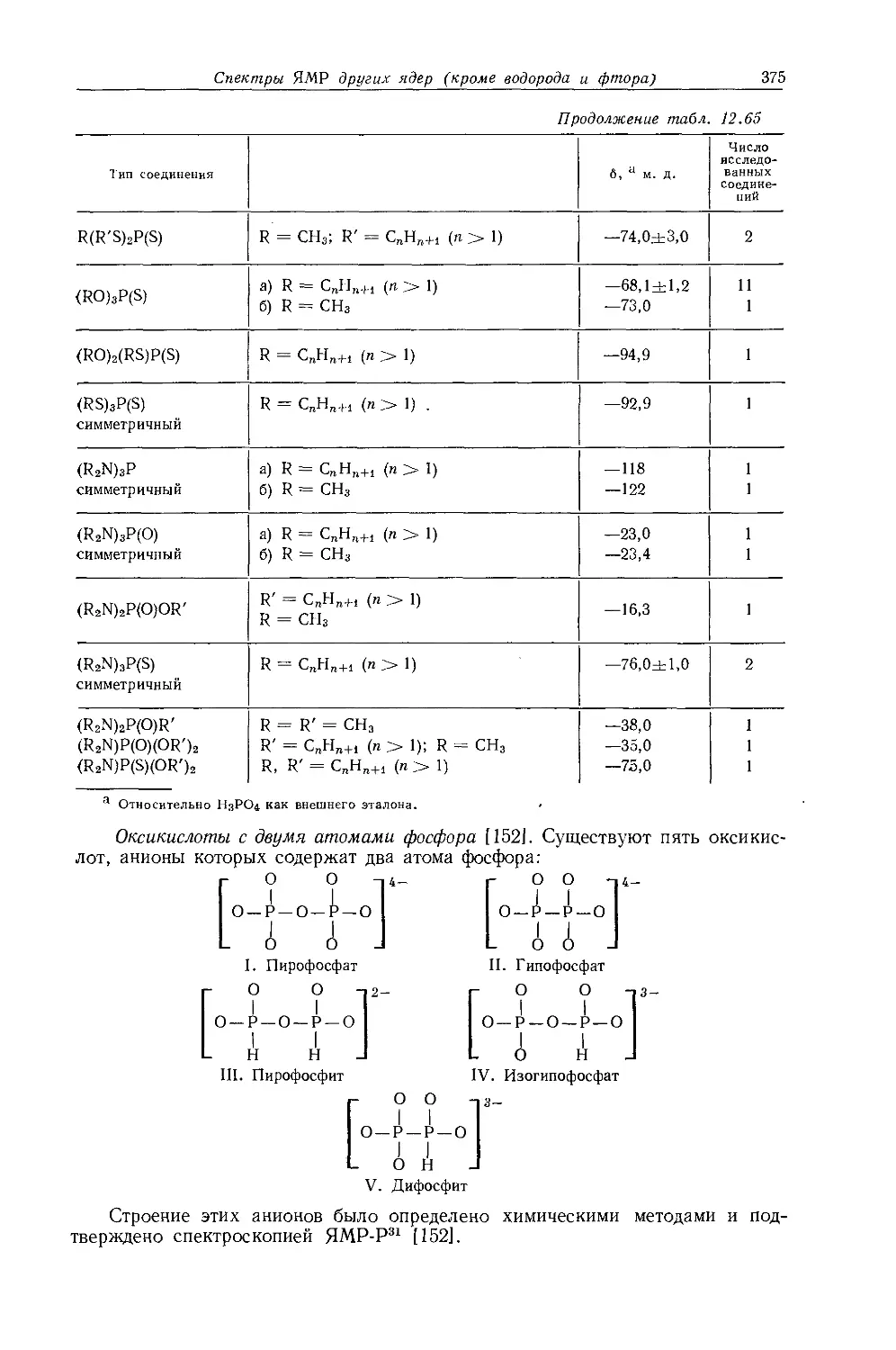
В действительности химические сдвиги метильных групп в этилгалогенидах говорят о том, что экранирование протонов в р-положенйи увеличивается с увеличением электроотрицательности заместителя. Изучение табл. 10.3 и 10.4 показывает, что химические сдвиги метильных групп в этильных производных изменяются в пределах 1 м. д. Шписецки и Шнейдер объяснили линейную корреляцию Д с электроотрицательностью тем, что вклады диамагнитной анизотропии в экранирование СН2- и СН3-групп в этильных производных приблизительно одинаковы по величине, но противоположны по знаку и, таким образом, случайно взаимно сокращаются.
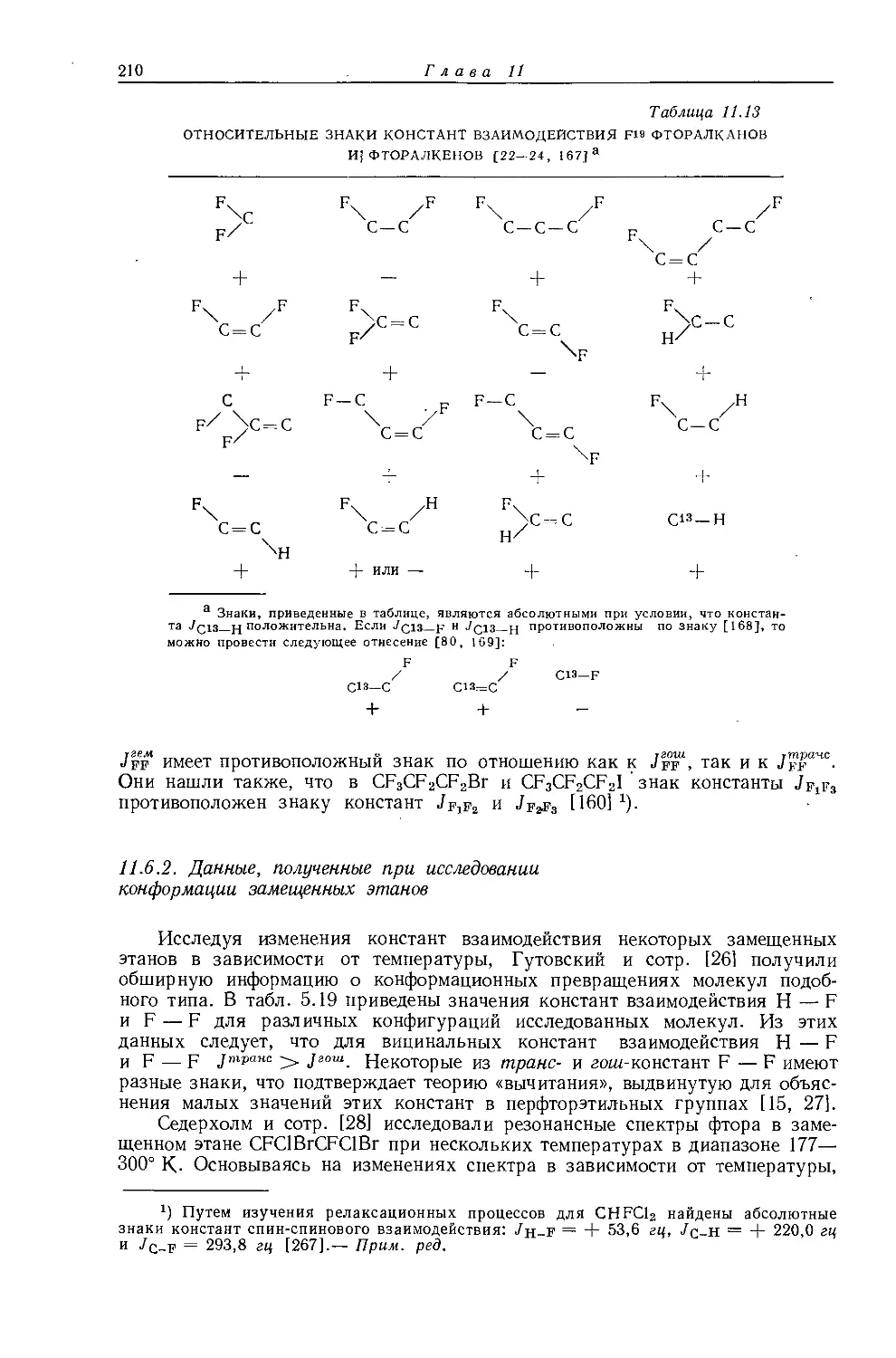
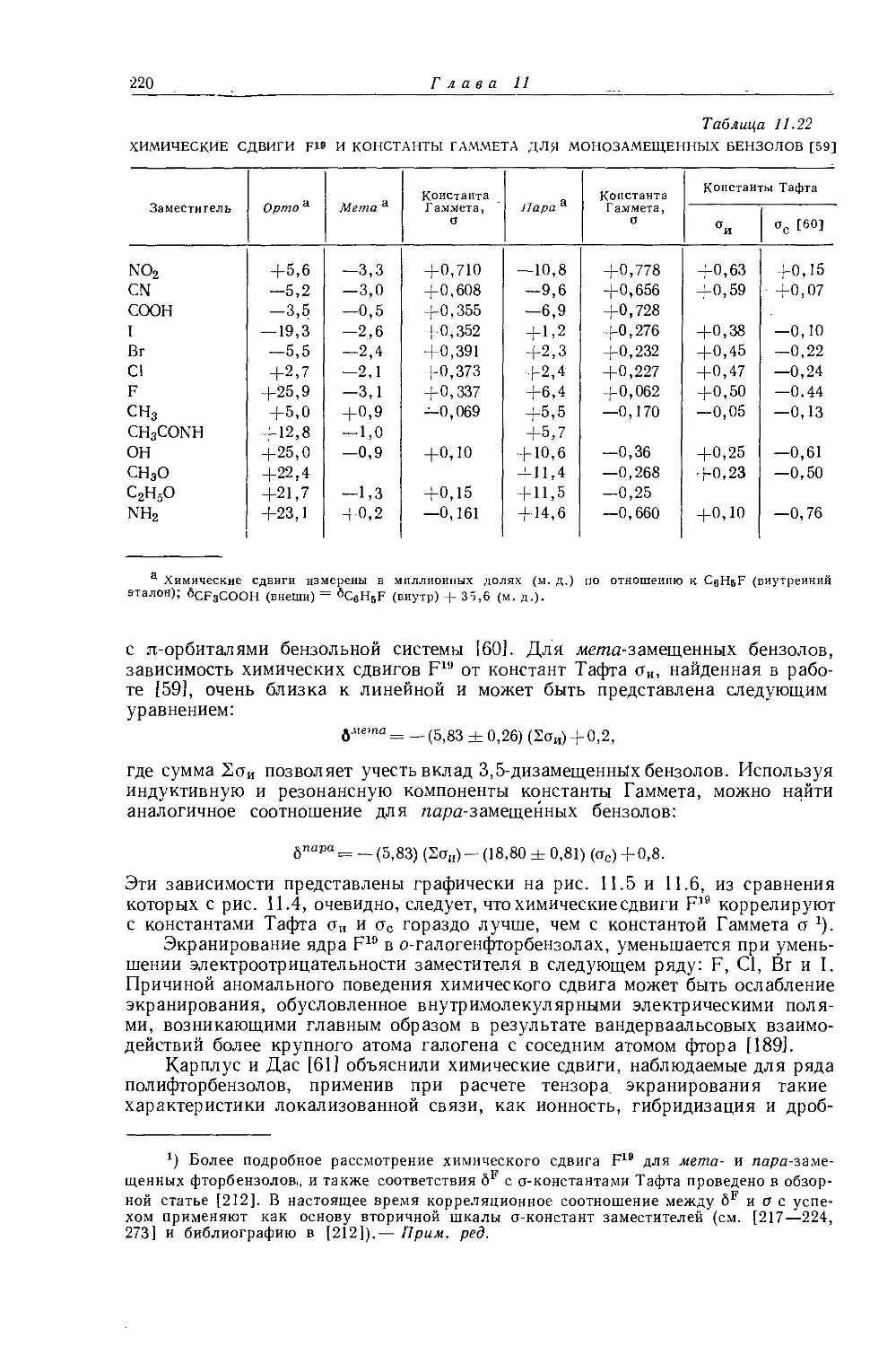
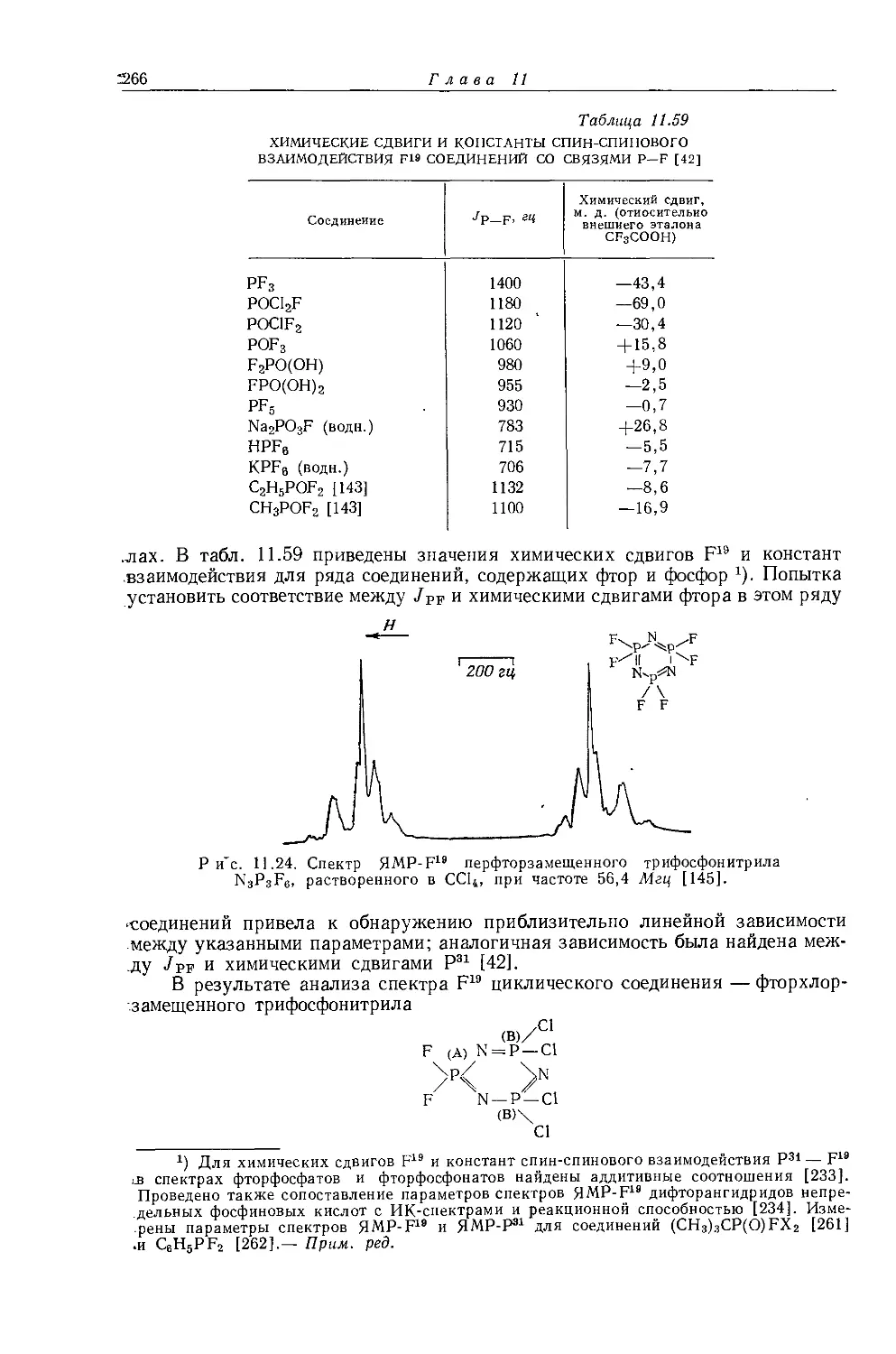
Если построить зависимость химических сдвигов метиленовых и метильных групп газообразных этильных производных от электроотрицательности заместителя, то получатся ожидаемые линейные соотношения (см. рис. 10.4 и табл. 10.4). Прямая зависимости химического сдвига <х-СН2-протонов от электроотрицательности заместителей имеет наклон, аналогичный наблюдавшемуся для метильных производных, однако фактические химические сдвиги меньше соответствующих сдвигов для метильных производных. Этого и следовало ожидать ввиду большей электроотрицательности метильной группы по сравнению с атомом водорода. И здесь галогениды обнаруживают аномальные химические сдвиги вследствие влияния диамагнитной анизотропии соседних групп. Как можно видеть на рис. 10.4, зависимость химических сдвигов метильной группы от электроотрицательности заместителя в этильных производных линейна. Такая зависимость, хотя и гораздо менее ярко выраженная, чем для <х-СН2-групп, характерна для всех этильных производных, за исключением обладающих магнитноанизотропным центром (S, I, Br, С1 и т. д.). Имеется значительное сходство между химическими сдвигами, наблю-
12
Глава 10
Таблица 10.4
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ ГАЗООБРАЗНЫХ СОЕДИНЕНИЙ СН3СН2Х (ОТНОСИТЕЛЬНО МЕТАНА) [6]
Соединение Температура, °C Химический сдвиг та, м. д.
СН2 СН3
ch3ch2f 40 —4,23 -1,14
СН3СН2С1 ИЗ -3,22 -1,29
СН3СН2Вг 130 -3,12 -1,47
СН3СН21 168 -2,97 -1,66
(СН3СН2)2О 95 -3,30 -1,02
(CH3CH2)3N 150 -2,39 -0,90
(CII,CII2)4C . 220 -1,27 -0,72
(СН3СН2)Н 22 -0,75 -0,75
СН3СН2СН2СН3 95 -1,21 -0,81
(CH3CH2)2S 170 -2,38 -1,10
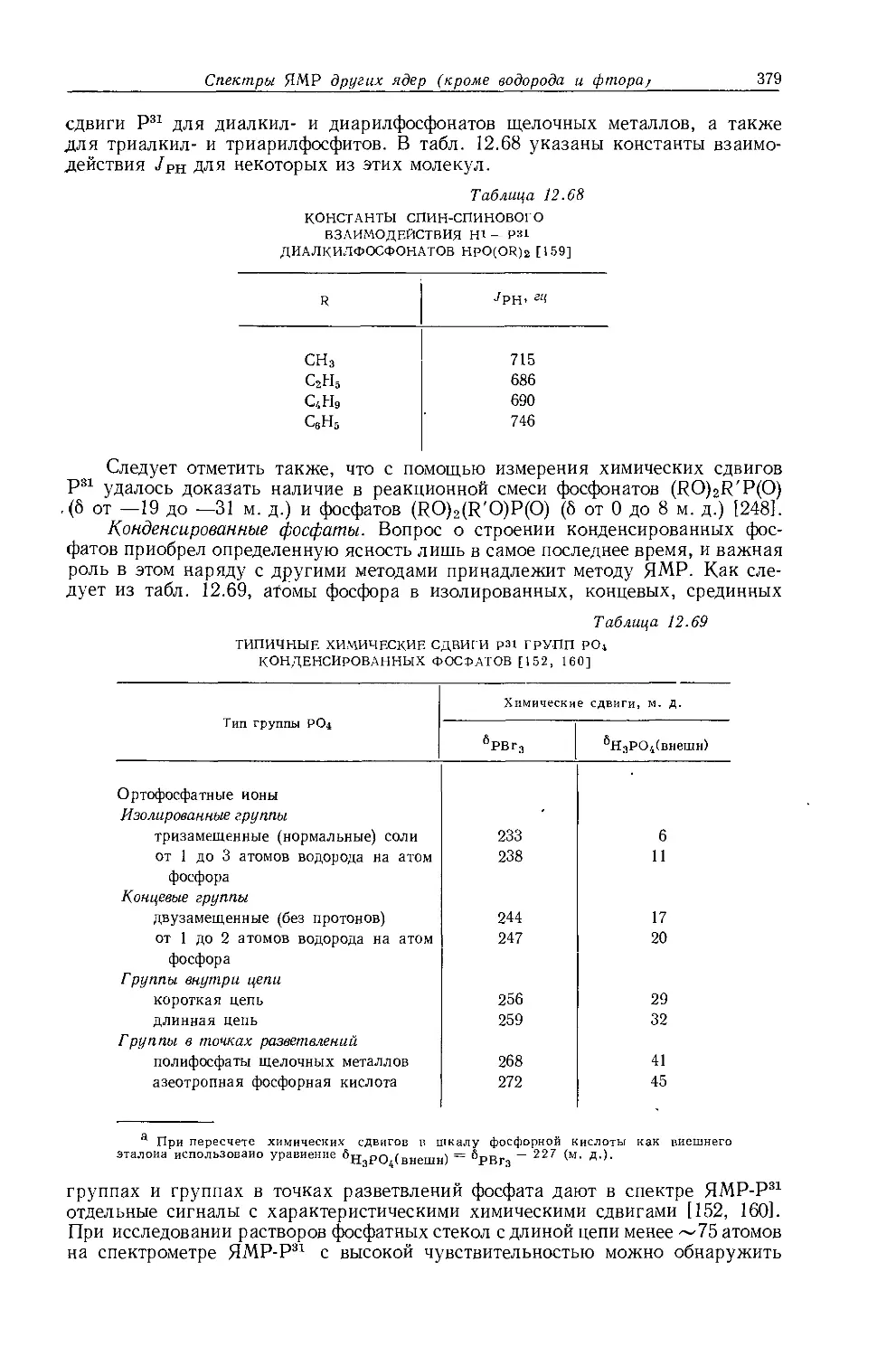
CH3CH2CN 155 -1,96 -1,08
(CH3CH2)4Si 220 -0,57 -0,90
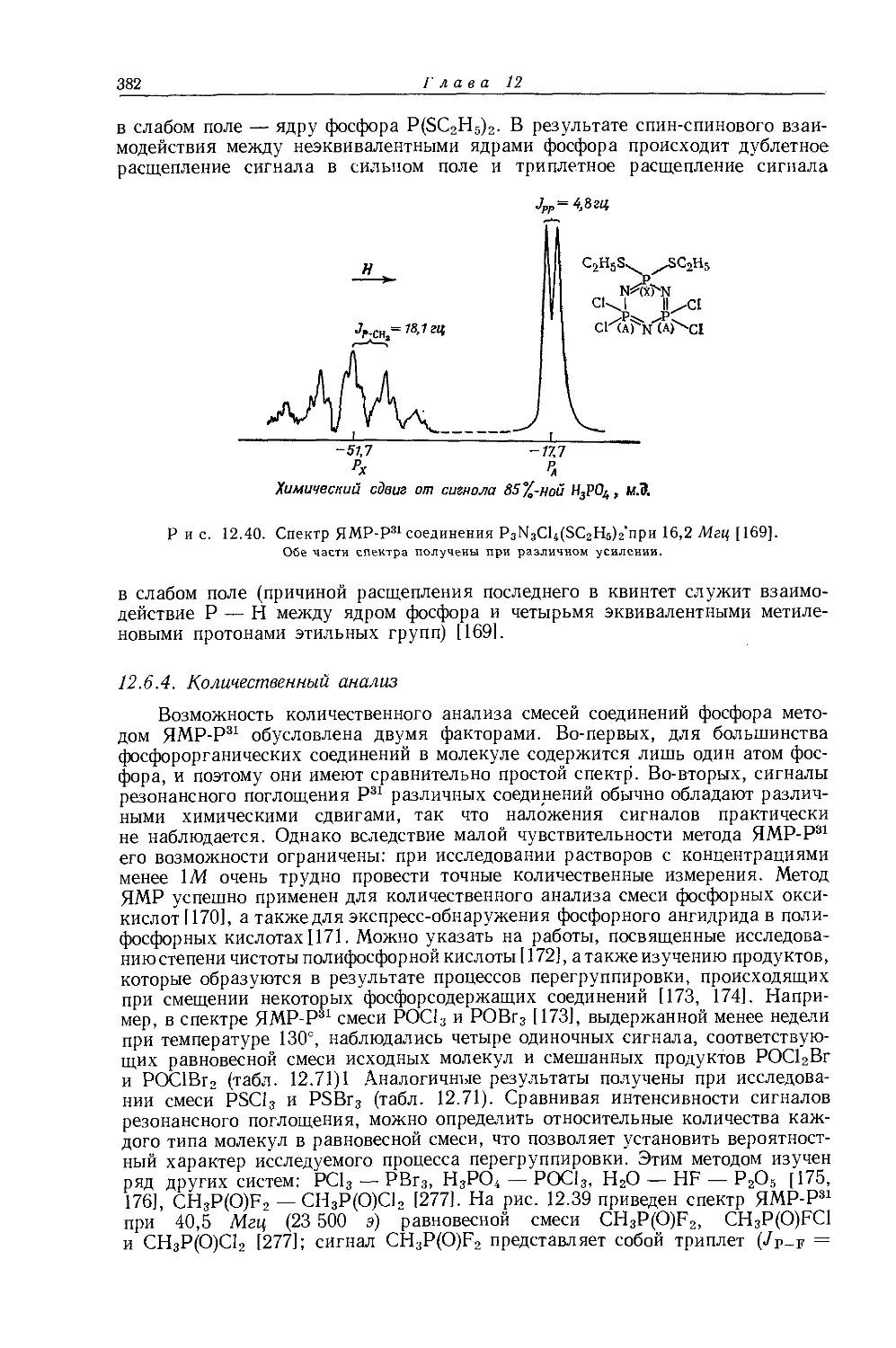
(CH3CH2)4Pb 145 -0,74 -0,74
а Значения получаются по уравнению т = 9,78 4~ (ВНутр)'
даемыми в спектрах ЯМР-Н1 и ЯМР-С13 (ср. рис. 10.4 и рис. 12.16). Абсолютные химические сдвиги <х-СН2- и 0-СН3-протонов в этильных соединениях, экстраполированные к бесконечно разбавленным растворам в четыреххлористом углероде, не могут быть объяснены лишь влиянием электроотрицатель-
Р и с. 10.4. Зависимость химических сдвигов протонов в соединениях типа СН3СН2Х от электроотрицательности заместителя X [6].
ности и магнитной анизотропии, поэтому Кавано и Дейли [7, 83] предположили, что следует учитывать еще один фактор, влияющий на экранирование и обусловленный связью С — С. Некоторые данные в пользу этого предположения получены при измерении химических сдвигов ал кил галогенидов, представленных в табл. 10.3. При переходе от метильных к этильным и изопропильным
Корреляции спектров ЯМР-Н1 со строением молекул
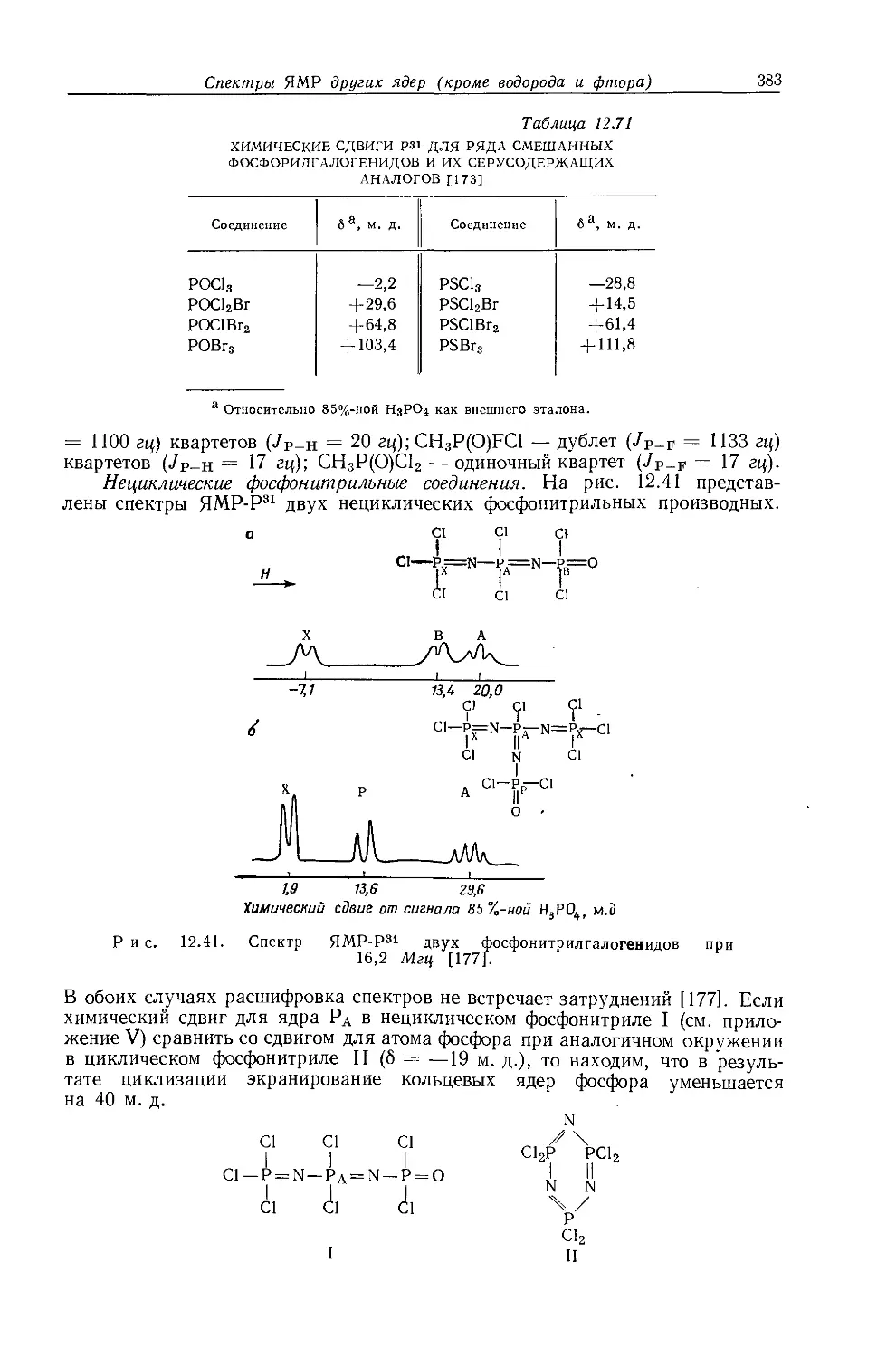
13
производным происходит уменьшение экранирования сс-протонов, причем оно прямо пропорционально числу связей С — С. Так, химические сдвиги изопропильных производных определяются по уравнению
ба(«зо-С3Н7Х) = б(СН3Х)— 2б(С- С)х ’ (1 ° 2)
где 6(с-С)х = 6(СН3Х) — 6а(сн3сн2х)- Такой большой сдвиг, обусловленный С—С-связями, нельзя объяснить влиянием магнитной анизотропии. Аналогичные сдвиги, обусловленные С — С-связями, наблюдались в экранировании а-протонов монозамещенных циклогексильных производных [83, 379] (см. разд. 10.3.6). Сдвиг, обусловленный С — С-связями, рассматривается не как результат влияния магнитной анизотропии, обусловленной простой связью С — С: предполагается, что при замене связи С — Н связью С — С происходит изменение энергии электронного возбуждения Е, которое влияет на парамагнитный член в уравнении экранирования Рэмзи [10] (см. разд. 3.5) и обусловливает значительное изменение в полном экранировании. Вследствие линейной зависимости внутреннего химического сдвига от электроотрицательности заместителя в этильных производных, это влияние равным образом должно сказываться на а- и Р-протонах; оценки вклада сдвига, обусловленного С — С-связями, в экранирование протонов проведены Кавано и Дейли {7, 11]. Шписецки и Шнейдер [61 приводят убедительное доказательство того, что любое отклонение от прямой зависимости между химическими сдвигами и электроотрицательностями заместителей обусловлено влиянием анизотропии соседних групп. Разногласия, которые имеются у различных авторов по этому вопросу, показывают, что более глубокое понимание данного явления станет доступным только после того, как удастся преодолеть ограничения существующей модели, в которой химические сдвиги рассматриваются как результат нескольких независимых локальных вкладов.
Следует отметить, что лишь в немногих случаях рассматривалось влияние внутримолекулярных электрических полей в алканах. Машер [24] показал, что в этаноле слабое экранирование <х-СН2-протонов может быть объяснено полностью на этой основе. В галогензамещенных соединениях большое влияние на экранирование протонов оказывают, по-видимому, вандерваальсовы силы внутри молекулы, в частности увеличение таких дисперсионных эффектов будет обусловливать уменьшение экранирования [448].
Этильные производные металлов. При исследовании спектров ЯМР-Н1 этильных производных некоторых металлов было показано, что использование
Таблица 10.5
ВНУТРЕННИЕ ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ В НЕКОТОРЫХ этильных ПРОИЗВОДНЫХ МЕТАЛЛОВ
Соединение бсн3~бсн2> м. Д. Литература
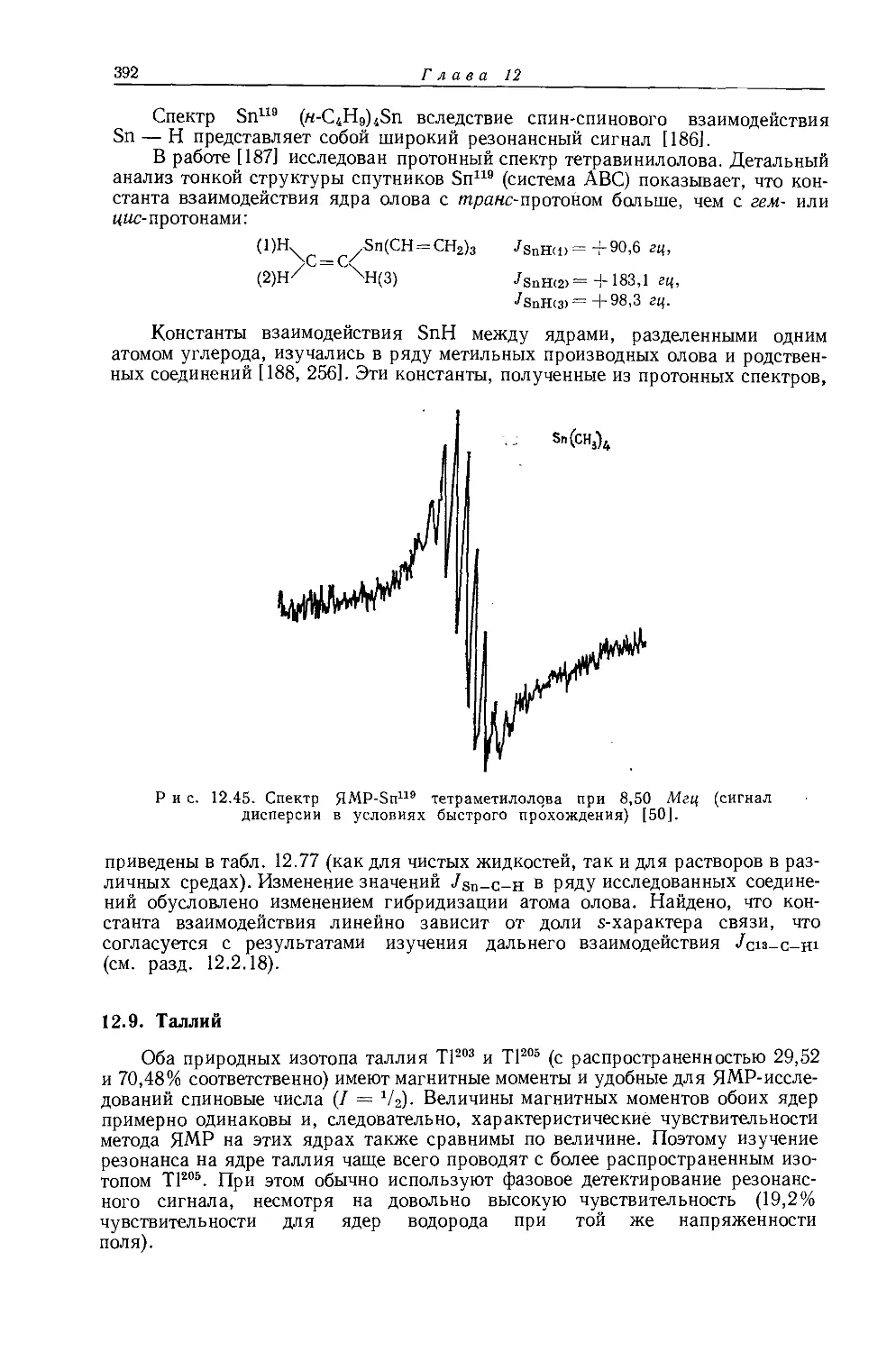
Zn(C2H5)2 -0,848 12
Ge(C2H5)4 -0,307 12
LiC2H5 — 2,190 349
SiCl3(C2H5) +0,195 12
SiCl2(C2H5)2 +0,015 12
Ga(C2H5)3 -0,560 12, 14
Si(C2H5)4 -0,420 15
значений внутренних химических сдвигов в уравнении (10.1) не дает ожидаемых для металлов значений электроотрицательностей. В табл. 10.5 приведены внутренние химические сдвиги для некоторых жидких этильных производных. Разделение резонансных сигналов групп СН3 и СН2 в этильных производных
14
Глава 10
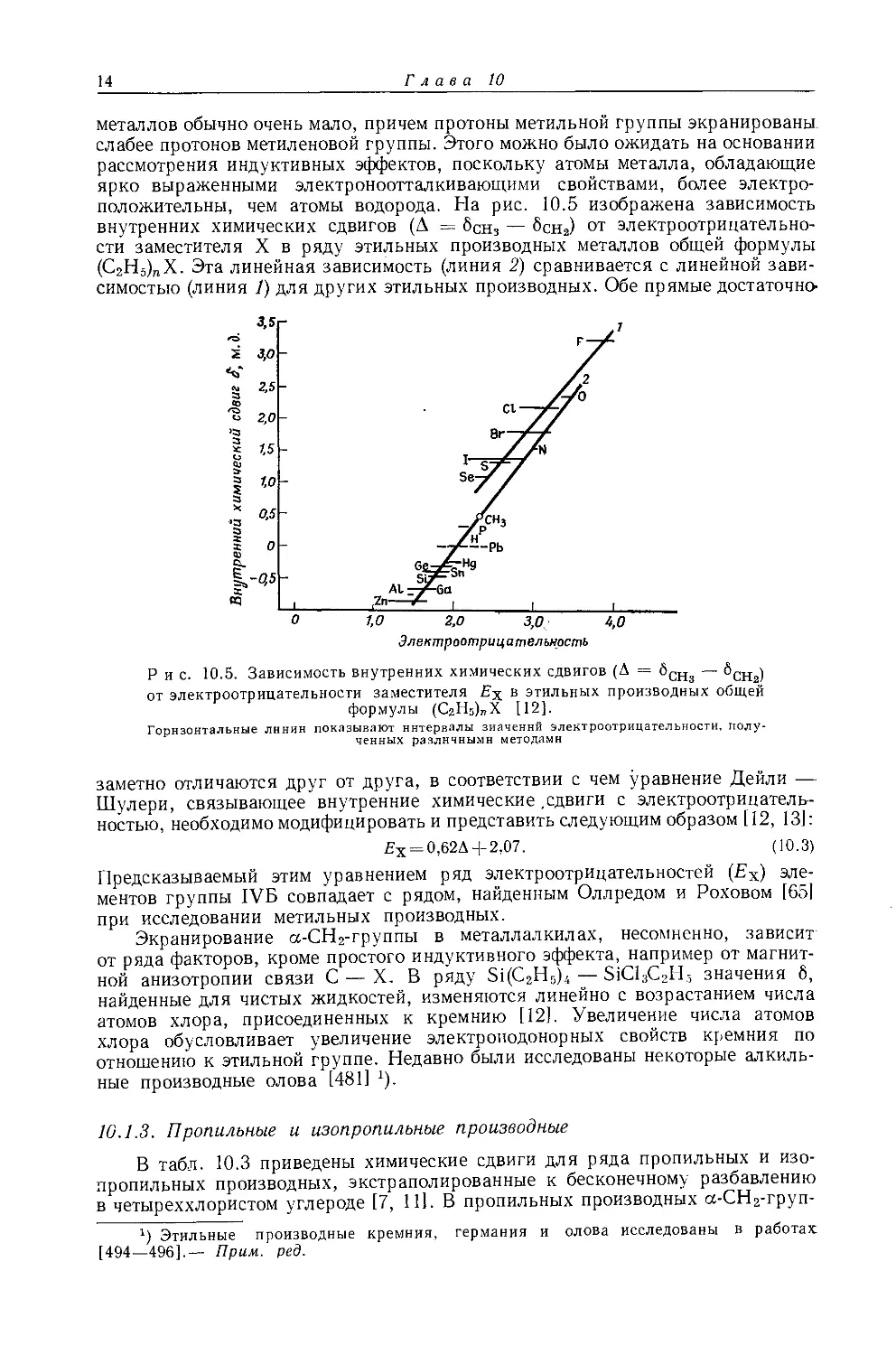
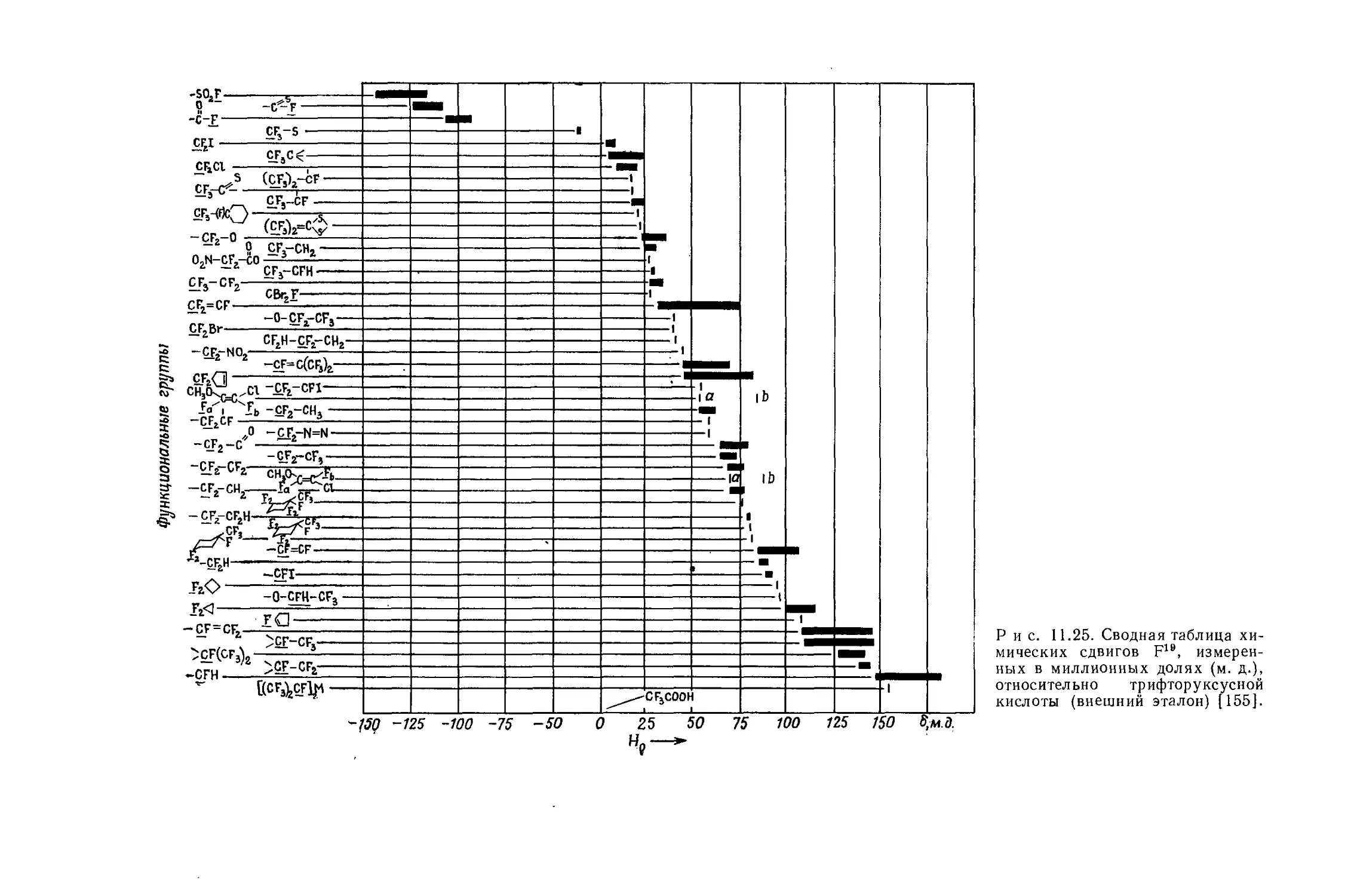
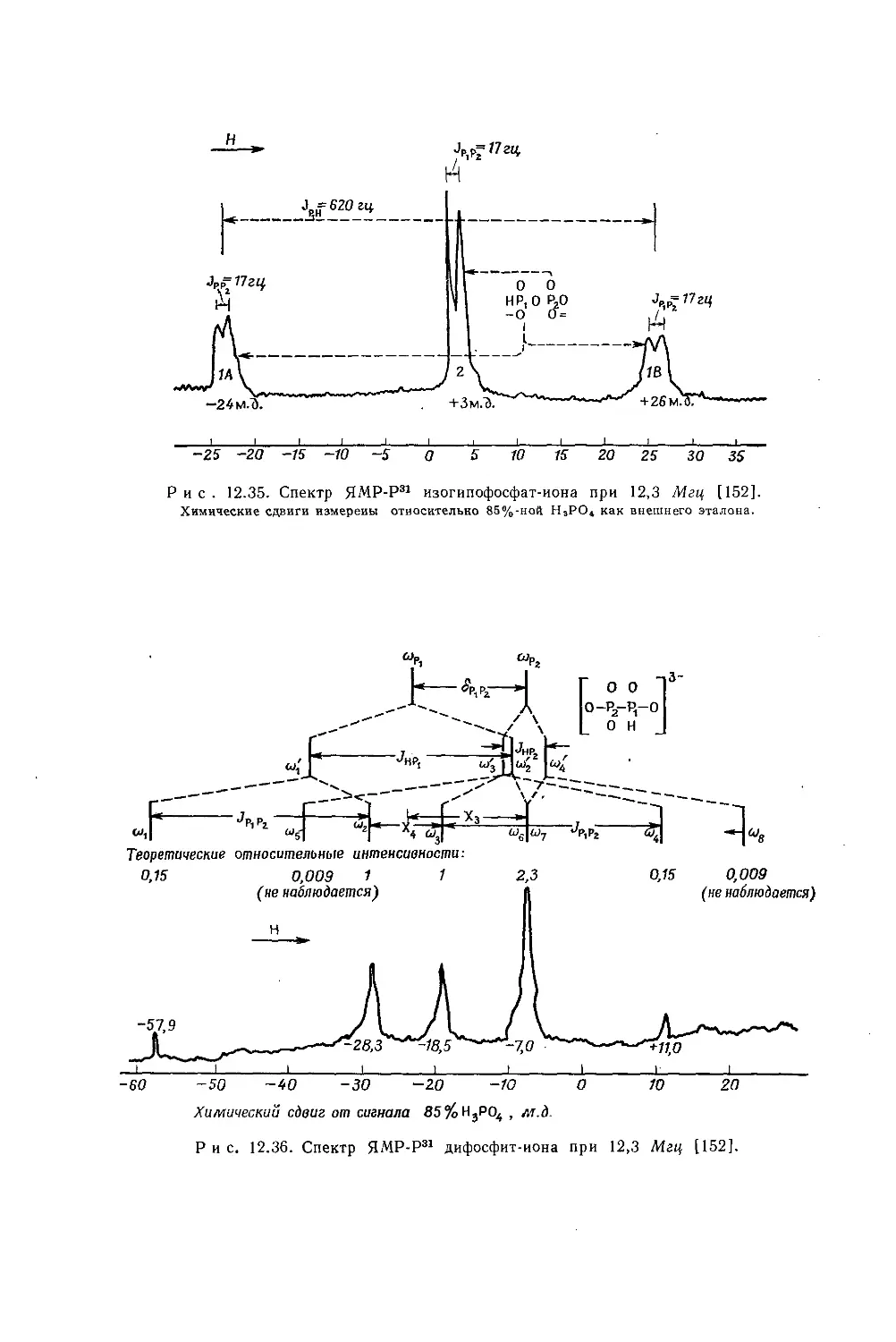
металлов обычно очень мало, причем протоны метильной группы экранированы слабее протонов метиленовой группы. Этого можно было ожидать на основании рассмотрения индуктивных эффектов, поскольку атомы металла, обладающие ярко выраженными электроноотталкивающими свойствами, более электроположительны, чем атомы водорода. На рис. 10.5 изображена зависимость внутренних химических сдвигов (Д = бСн3 — 6сн2) от электроотрицательности заместителя X в ряду этильных производных металлов общей формулы (С2Н5)ПХ. Эта линейная зависимость (линия 2) сравнивается с линейной зависимостью (линия 1) для других этильных производных. Обе прямые достаточно-
Рис. 10.5. Зависимость внутренних химических сдвигов (А = §СНз — $сн2) от электроотрицательности заместителя £х в этильных производных общей формулы (С2Н5)„Х [12].
Горизонтальные линии показывают интервалы значений электроотрицательности, полученных различными методами
заметно отличаются друг от друга, в соответствии с чем уравнение Дейли — Шулери, связывающее внутренние химические .сдвиги с электроотрицательностью, необходимо модифицировать и представить следующим образом [12, 13]:
£х = 0,62Д-]-2,07. (10.3)
Предсказываемый этим уравнением ряд электроотрицательностей (£х) элементов группы IVB совпадает с рядом, найденным Оллредом и Роховом [651 при исследовании метильных производных.
Экранирование <х-СН2-группы в метал л ал кил ах, несомненно, зависит от ряда факторов, кроме простого индуктивного эффекта, например от магнитной анизотропии связи С — X. В ряду Si(C2H5)4 — SiCl3C2H3 значения 6, найденные для чистых жидкостей, изменяются линейно с возрастанием числа атомов хлора, присоединенных к кремнию [12]. Увеличение числа атомов хлора обусловливает увеличение электронодонорных свойств кремния по отношению к этильной группе. Недавно были исследованы некоторые алкильные производные олова [4811 J).
10.1.3. Пропильные и изопропильные производные
В табл. 10.3 приведены химические сдвиги для ряда пропильных и изопропильных производных, экстраполированные к бесконечному разбавлению в четыреххлористом углероде [7, 11]. В пропильных производных <х-СН2-груп-
-1) Этильные производные кремния, германия и олова исследованы в работах [494—496].— Прим. ред.
Кор реляции спектров ЯМР-Н1 со строением молекул
15.
пы имеют химические сдвиги, аналогичные химическим сдвигам метиленовых протонов в соответствующих этильных производных, тогда как концевые метильные группы почти нечувствительны к изменению электроотрицательности заместителей. Поскольку наблюдаемые химические сдвиги нельзя объяснить лишь влиянием заместителей, по-видимому, имеются существенные вклады в экранирование других эффектов (необходимо рассматривать как влияние-магнитной анизотропии, так и сдвиг, обусловленный связями С — С). Для рассматриваемых молекул необходимо учитывать еще один важный фактор — наличие вращательной изомерии.
10.1.4. Вклады диамагнитной анизотропии связей С — Н и С — С в экранирование ядер водорода
Возможность вкладов диамагнитной анизотропии связей С — Н и С — С в экранирование ядер водорода рассматривалась несколькими авторами [16—19] (см. также разд. 4.4.2). Ги и Тийё [19] рассчитали величины продольной и поперечной восприимчивостей связей С — НиС — Си, используя эти величины, оценили вклады диамагнитной анизотропии в экранирование. Вариационный расчет Тийё дал для Дус~с значение +1,21 -10"30 слёмолекула, тогда как экспериментально полученные значения [16, 21, 22, 159] изменяются от +2-10"30 до +1O-1O'30 см3!молекула. Экспериментальная оценка этого-параметра осложняется трудностью разделения вкладов различных эффектов экранирования (таких, как диамагнитная анизотропия связей С — Н). Во всех случаях значение Д%с-с положительно, однако Попл [26] отметил, что такое явление несовместимо с тем, что для алифатических углеводородов с длинной цепью ось наибольшего диамагнетизма направлена вдоль цепи. Недавно-Цюрхер [25] провел расчеты Д%с-с методом наименьших квадратов и получил значения, близкие к полученным Тийё. Он показал также, что Дус н может быть значительной. Согласно Тийё, Дус-Н может изменяться от 0,24-1О"30' до 1,50-1О"30 смЧмолекула в зависимости от выбора волновой функции. Теория Попла об анизотропных вкладах в экранирование ядер не предсказывает вклада простых связей С — С, что обусловлено априорным-характером расчета [26]. В настоящее время невозможно дать более определенную оценку эффекта экранирования, обусловленного анизотропией простой связи С — С. Однако он намного меньше, чем для двойных и тройных связей, из-за сильной делокализации заряда ст-связи, обусловленной перекрыванием 5р3-гибридизо-ванных орбиталей.
10.2. Константы спин-спинового взаимодействия для производных алканов
Константы взаимодействия часто указывают на стереохимию взаимодействующих ядер и поэтому представляют особый интерес для химиков, интересующихся строением молекул. В настоящее время абсолютные и относительные знаки констант взаимодействия могут быть определены достаточно надежно, и эта информация существенно увеличивает как практическое, так и теоретическое значение корреляций между константами взаимодействия и другими молекулярными параметрами.
Константы взаимодействия в более простых незамещенных углеводородах (этан, этилен, ацетилен) имеют большее теоретическое значение, чем соответствующие константы для замещенных молекул. Эти константы могут быть получены лишь путем изотопного замещения, которое обусловливает магнитную неэквивалентность ядер без существенного изменения электронной конфигурации исходной молекулы [28, 42, 430] (например, CH3D или Н2С13 =С12Н2).
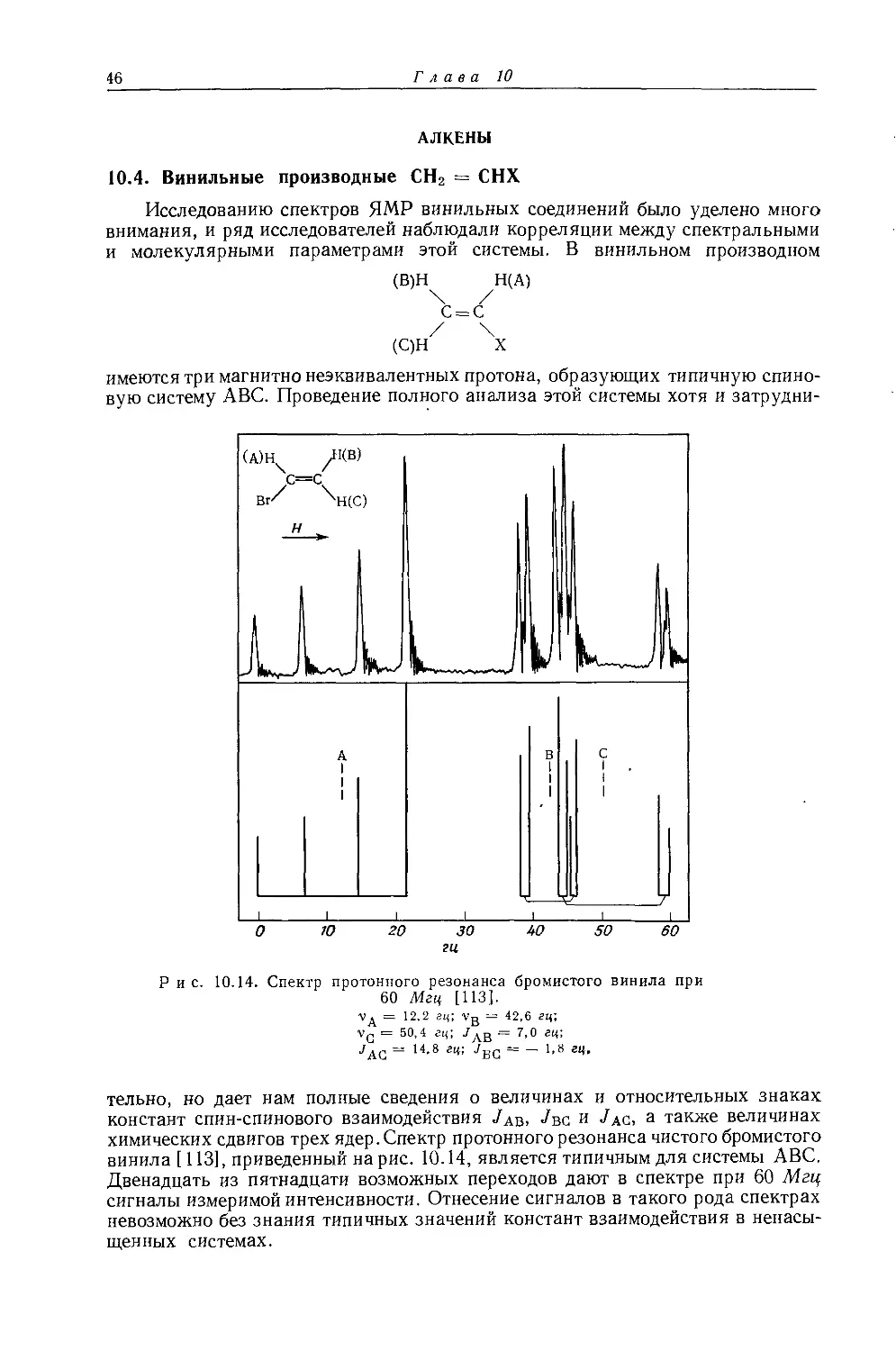
Банвелл и Шеппард [34] опубликовали обзор по константам взаимодействия Н — Н, наблюдаемым в спектрах производных углеводородов.
16
Глава 10
10.2.1. Константы геминального взаимодействия протонов у насыщенного атома углерода
Используя метод валентных связей, Карплус и сотр. [28—30] вывели соотношение между константами взаимодействия геминальных протонов и валентным углом Н — С — Н. Это соотношение было усовершенствовано Гутовским и Сомерсом [31], которые ввели поправку на колебательные эффекты в СН2-группе. Согласно теории, константы геминального взаимодействия Н — Н в производных алканов положительны и того же знака, что и константы вицинального взаимодействия. Однако это утверждение неправильно, так как экспериментальные результаты [32] четко указывают на то, что /нн в алканах отрицательна. При расчете констант взаимодействия геминальных протонов производится сложение довольно больших членов с разными знаками, что не позволяет получить точный результат. Несоответствие между рассчитанными и экспериментальными значениями /нн можно объяснить использованием либо неточных молекулярных волновых функций, либо грубых приближений, сделанных при расчете (см. разд. 5.3.2).
Мак-Коннел [33] использовал для решения этой проблемы метод молекулярных орбиталей и также получил положительные значения /нн- Но, применив векторную модель Дирака, Мак-Коннел показал, что константы взаимодействия геминальных протонов могут быть отрицательными [35].
В табл. 5.5 и 5.6 приведены константы геминального взаимодействия протонов для некоторых замещенных метанов и этанов [34]. Для симметричных молекул константы геминального взаимодействия определяются при помощи дейтерирования [28, 36, 37]. Для четырех из исследованных молекул удалось показать, что константа геминального взаимодействия отрицательна по сравнению с константой вицинального взаимодействия (которая, как полагают, имеет абсолютный положительный знак) [32]. Поэтому разумно предположить, что константы геминального взаимодействия, указанные в табл. 5.5, имеют отрицательный знак [34] в противоположность теоретически предсказанным значениям (см. разд. 5.3.3).
10.2.2. Константы вицинального взаимодействия протонов
Константы спин-спинового взаимодействия, наблюдаемые в спектрах алканов, необходимо рассматривать как усредненные значения констант взаимодействия в различных вращательных изомерах. Карплус [39] использовал метод валентных связей для расчета вклада контактного взаимодействия в спин-спиновое взаимодействие двух протонов у соседних атомов углерода (т. е. вицинальных протонов) с двугранным углом ср (см. разд. 5.3.1) и получил соотношения
Jнн (контакт) =8,5 cos2 ф — 0,28 гц (0° ф 90°), (10.4)
Унн (контакт) = 9,5 cos2 ф—0,28 гц (90° ф 180°) (10 5)
Так как контактный член дает, несомненно, наибольший вклад в спин-спиновое взаимодействие протонов, то соотношения (10.4) и (10.5) предсказывают значения констант взаимодействия, находящиеся в хорошем согласии с экспериментальными значениями. В табл. 5.1 приведены типичные значения предсказанных констант вицинального взаимодействия для различных двугранных углов.
Чтобы получить приближенное теоретическое значение •/сн3-сн3 Для этана, можно просто усреднить выражение константы взаимодействия, учитывая внутреннее вращение:
(7= [2^нн' (Ф = 60°)4" (*₽= 180°)]/3 = 4,2 гц
Шеппард и Линден-Белл [42] измерили константу взаимодействия /сн3-сн3 в этане по спутникам С13Н, и полученное ими значение (J = 8,0 гц) оказалось
Корреляции спектров ЯМР-Н1 со строением, молекул
17
намного больше теоретического значения. Ботнер-Бай и Глик [40] аналогично измерили значения констант взаимодействия /сн3-сн2 в этильных производных (7сн3—сн2 = 6,0—7,4 гц).
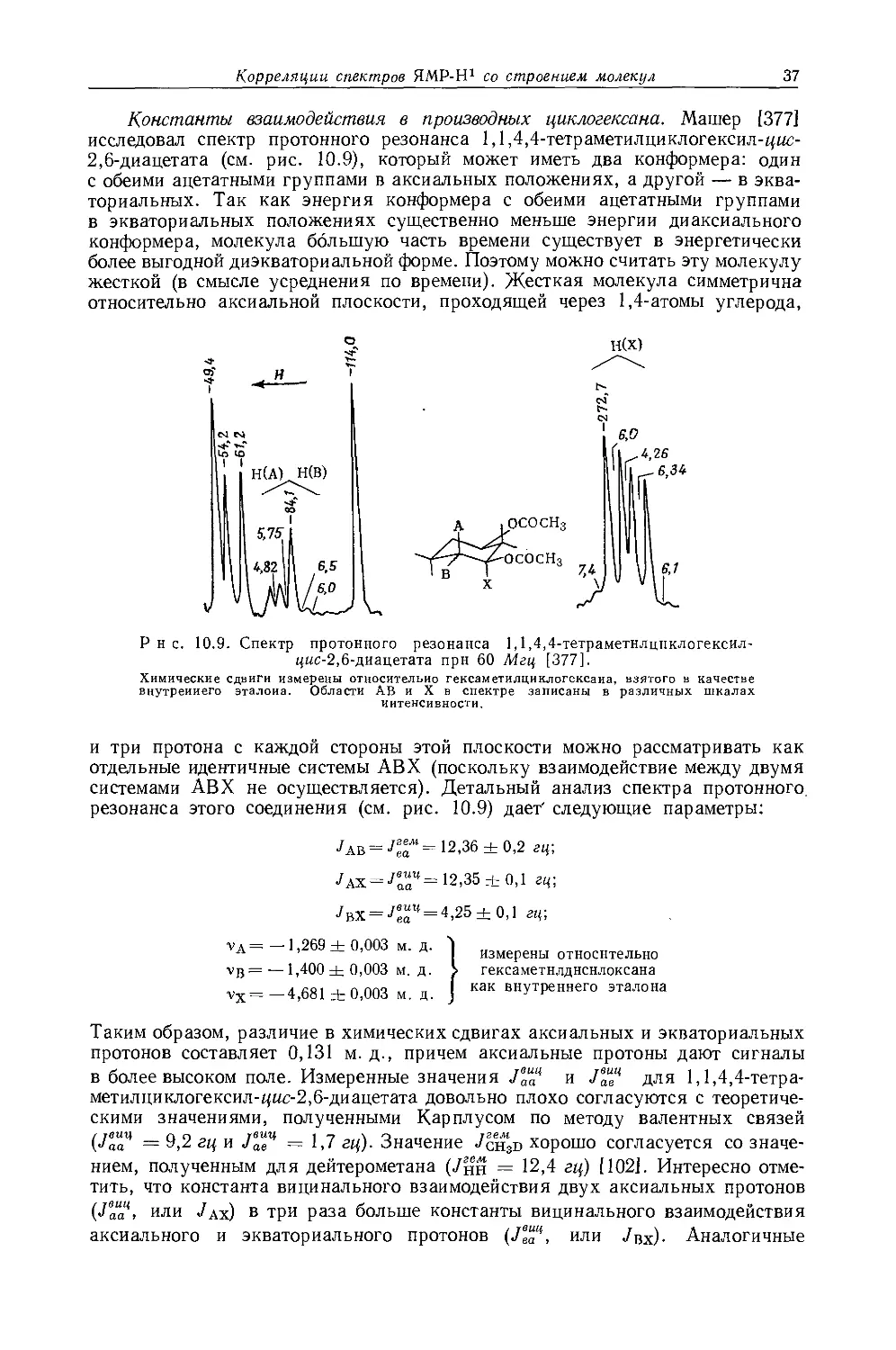
Уравнения (10.4) и (10.5) могут быть применены более успешно для определения констант спин-спинового взаимодействия в молекулах, где конформация (а следовательно, и двугранный угол) известна, как, например, для некоторых циклогексильных производных. При исследовании ряда ацетилированных сахаров Лемье и сотр. [41] удалось измерить константы взаимодействия между двумя аксиальными протонами у соседних атомов углерода (Jaa = = 5—8 гц, <р = 180°), двумя экваториальными протонами у соседних атомов углерода (Jee = 3—4 гц, <р = 60°) и аксиальным и экваториальным протонами у соседних атомов углерода (Jae = 2—3 гц, ф = 60°). Эти значения хорошо согласуются со значениями, предсказанными теоретически (см. табл. 5.1). Однако Машер [377] показал, что значения и Jeda4 в 1,1,4,4-тетраметил-циклогексил-2,6-диацетате плохо согласуются с теоретическими значениями Карплуса (о константах взаимодействия в производных циклогексана см. в разд. 10.3.6). Очевидно, уравнения Карплуса (10.4) и (10.5) для предсказания значений Тнн в системе Н — С — С — Н' обычно дают значения меньшие, чем экспериментальные. Шуг, Мак-Магон и Бутовский [43] использовали уравнения Карплуса, чтобы оценить влияние крутильных колебаний и реориентаций вокруг связи С — С на усредненные константы взаимодействия в замещенных этанах. Они показали, что крутильные колебания обусловливают температурную зависимость констант транс- и гош-взаимодей-ствия, причем зависимости этих двух констант имеют противоположные знаки. Однако для молекул, обладающих осью симметрии третьего порядка, усредненная константа взаимодействия не зависит от температуры вследствие погашения колебательных.эффектов за счет внутреннего вращения (типичными молекулами такого рода являются этан и 1-замещенные этаны). Поэтому в этил-нитрате константа 7сн3-сн2 равна 6,92 гц в интервале температур до 100° [43]. Некоторые другие исследователи использовали уравнение Карплуса для изучения конформации пяти- [44—46] и шестичленных [47—49] колец.
Ботнер-Бай и Глик [40] опубликовали значения констант взаимодействия первого порядка для ряда алканов, приведенные в табл.’ 10.6 и 10.7. Для этильных и изопропильных соединений константы взаимодействия /Сн3-сн линейно связаны с электроотрицательностью заместителя. Следует отметить, что константы взаимодействия Н — Н для этих молекул были получены при допущении, что наблюдаемые протонные спектры являются спектрами первого порядка. Конрой [51] показал, что значения /ц-с-с-н' будут зависеть от электронной плотности на протонах, что находится в согласии с наблюдениями Ботнер-Бая и Глика.
В настоящее время для широкого круга молекул получены более точные значения констант вицинального взаимодействия [15, 52, 486]; для этанов предложено [34, 42] улучшенное выражение для связи между константой взаимодействия и электроотрицательностью:
7сн3-сн2 = 7,9-°,7Л£, (10.6)
где Д£ (= £х — -^н) — разность электроотрицательностей заместителя X и атома водорода, а 7,9 гц соответствует наблюдаемому вицинальному взаимодействию в этане. Уравнение (10.6) может предсказать значения 7сн3-сн2 с точностью ±0,3 гц.
Абрагам и Пахлер [53] предложили более общую корреляцию значений /нн в фрагментах CH — СН с суммой электроотрицательностей атомов, связанных непосредственно с атомами углерода. Они показали, что соотношение
7 = 8,0—1,02£
(Ю.7)
2=489
Таблица 10.6
КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ ПЕРВОГО ПОРЯДКА
В НЕКОТОРЫХ ЭТИЛЬНЫХ ПРОИЗВОДНЫХ (СН3СН2)ПХ [40, 53] а
Заместитель X Константа взаимодействия Jnr., гц ср Заместитель X Константа взаимодействия Jnr., гц ср
н 8,0+0,1 S 7,42+0,05
СН3 7,26+0,1 Со 7,5+0,1
ОН 6,97+0,1 Hg 7,0
о2сн 6,93+0,1 N 6,9
F 6,9+0,1 Р(Ш) 7,6
С1 7,07+0,1 Sn(IV) 8,2
Вг 7,25+0,1 Pb(IV) 8,2
I 7,16+0,1 Si(IV) 7,9+0,02
SH 7,27+0,1 ch2cn 7,7
CN 7,24+0,1 CH2C1 7,4
СО2Н 7,41+0,1 CH2Br 7,3
N 6,96+0,1 CH2I 7,2
О 6,96+0.1 °')c=o 6,96+0,1
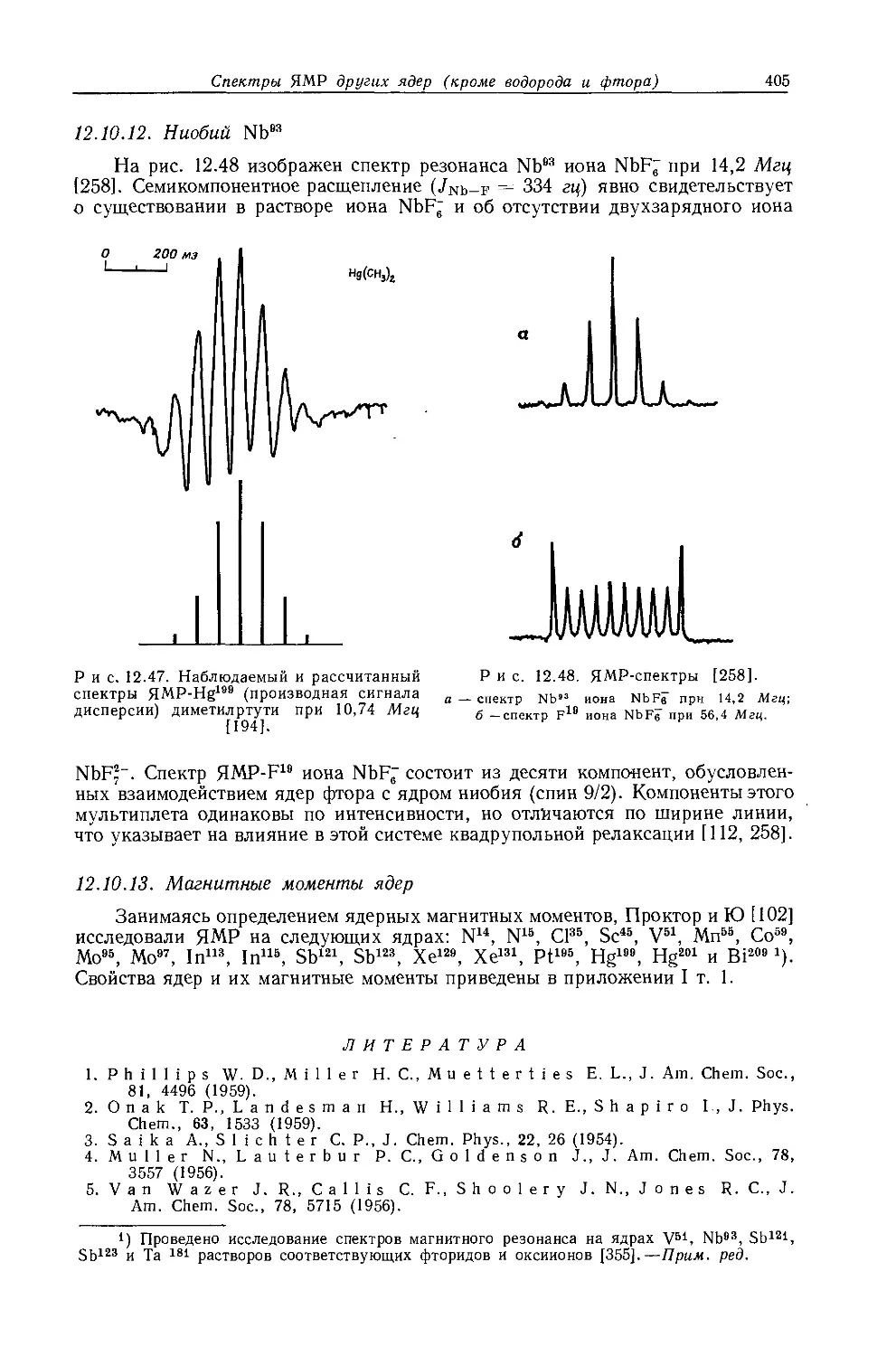
Li 8,4 о/
а Сводка констант взаимодействия составлена из различных источников Абрага-мом и Пахлером [53].
Таблица 10.7
ВИЦИНАЛЬНЫЕ КОНСТАНТЫ НЕКОТОРЫХ ЗАМЕЩЕННЫХ ЭТАНОВ [40, 53] а
xch2ch2y ch3chxy
заместители константа взаимодействия Jcp’ гч заместители константа взаимодействия 4ср. гц
X Y X Y
Cl Cl 6,83+0,1 сн3 сн3 6,8±0,2
Br Cl 6,87+0,1 сн3 Со 6,49+0,1
Br Br 7,37+0,1 сн3 I 6,42+0,1
Br OH 6,00+0,1 сн3 Вг 6,40+0,1
Cl OH 5,83+0,1 сн3 С1 6,31+0.1
CH3O OH 5,33+0,1 СН3 nh2 6,11 + 0,1
CH3CO CO2H 6,80+0,1 сн3 он 6,05+0,1
CH3CO co2CH3 6,80+0,1 СО2Н СО2Н 7,22±0,2
CH3O O2CCH3 5,33+0,1 Вг Вг 6,24±0,4
Cl O2CCH3 5.97+0,1 С1 С1 6,10+0,15
он он 5,26+0,07
OH OH 5,27 С1 СО2Н 6,94+0,03
CH3O CH3O 5,30 Вг СО2Н 6,92+0,01
он СО2Н 6,97+0,02
С1 ОС2Н5 5,28±0,1
сн3 СО2Н 7,2+0,1
сн3 о 5,98±0,1
Вг СНВгСО2Н 6,70+0,1
а Сводка констант взаимодействия составлена Абрагамом и Пахлером [53].
Корреляции спектров ЯМР-Н1 со строением молекул
19
адекватно описывает константы вицинального взаимодействия Н — Н в этильных производных и этанах общей формулы XCH2CH2Y и CH3CHXY (см. табл. 10.6 и 10.7), несмотря на то что оно и не является лучшим линейным соотношением для каждого из этих классов молекул. Для молекул общей формулы ХСН2СН2Х, чтобы получить значения /ни, необходимо исследовать в спектрах спутники С13Н [50].
Конрой [51] использовал приближение МО для получения соотношения между константами взаимодействия 7Нн- и двугранным углом в системах Н — С — С — Н'. Полученная им теоретическая кривая позволяет предсказывать порядок величины константы взаимодействия для разных двугранных углов. В большинстве случаев имеется хорошее согласие между наблюдаемыми и рассчитанными значениями /н-с-с-н'-
10.2.3. Знаки констант взаимодействия протонов
Следует отметить, что для некоторых классов соединений найденную зависимость константы взаимодействия от валентного угла нужно применять с осторожностью, поскольку имеются случаи, когда это соотношение не выполняется (диоксоланы [54], эпихлоргидрин [55], 2,2-метациклофан [561, диэтил-сульфит [571). Во всех случаях константы взаимодействия геминальных и вицинальных протонов отличаются по знаку, тогда как, согласно теории Карплуса, они должны иметь одинаковые знаки. Исходя из этого, можно предположить, что некоторые теоретические расчеты неверны. Теорией предсказываются абсолютные знаки констант взаимодействия, однако из спектров
Таблица 10.8 АБСОЛЮТНЫЕ ЗНАКИ КОНСТАНТ ВЗАИМОДЕЙСТВИЯ ПРОТОНОВ
а положительна [463].
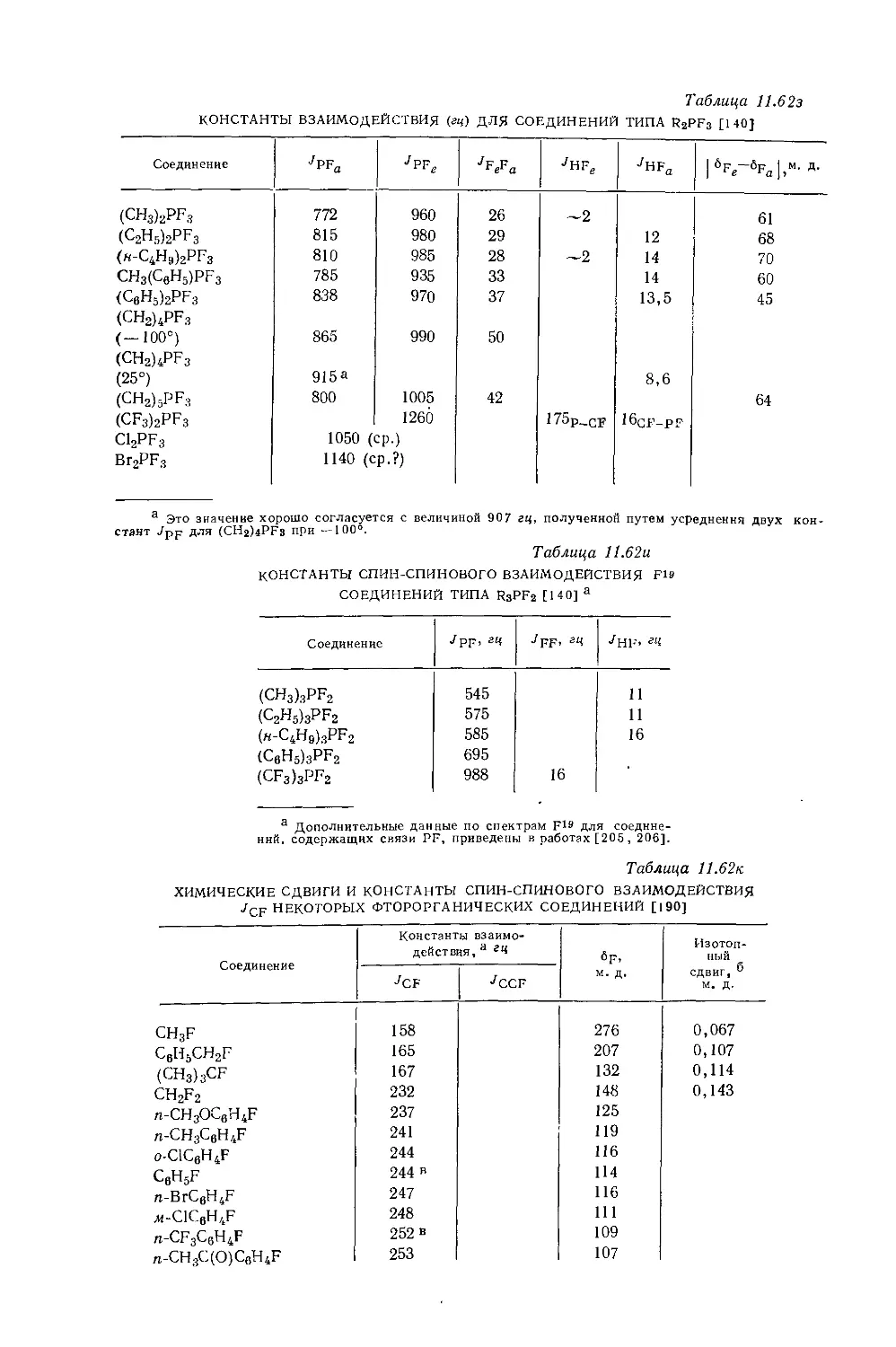
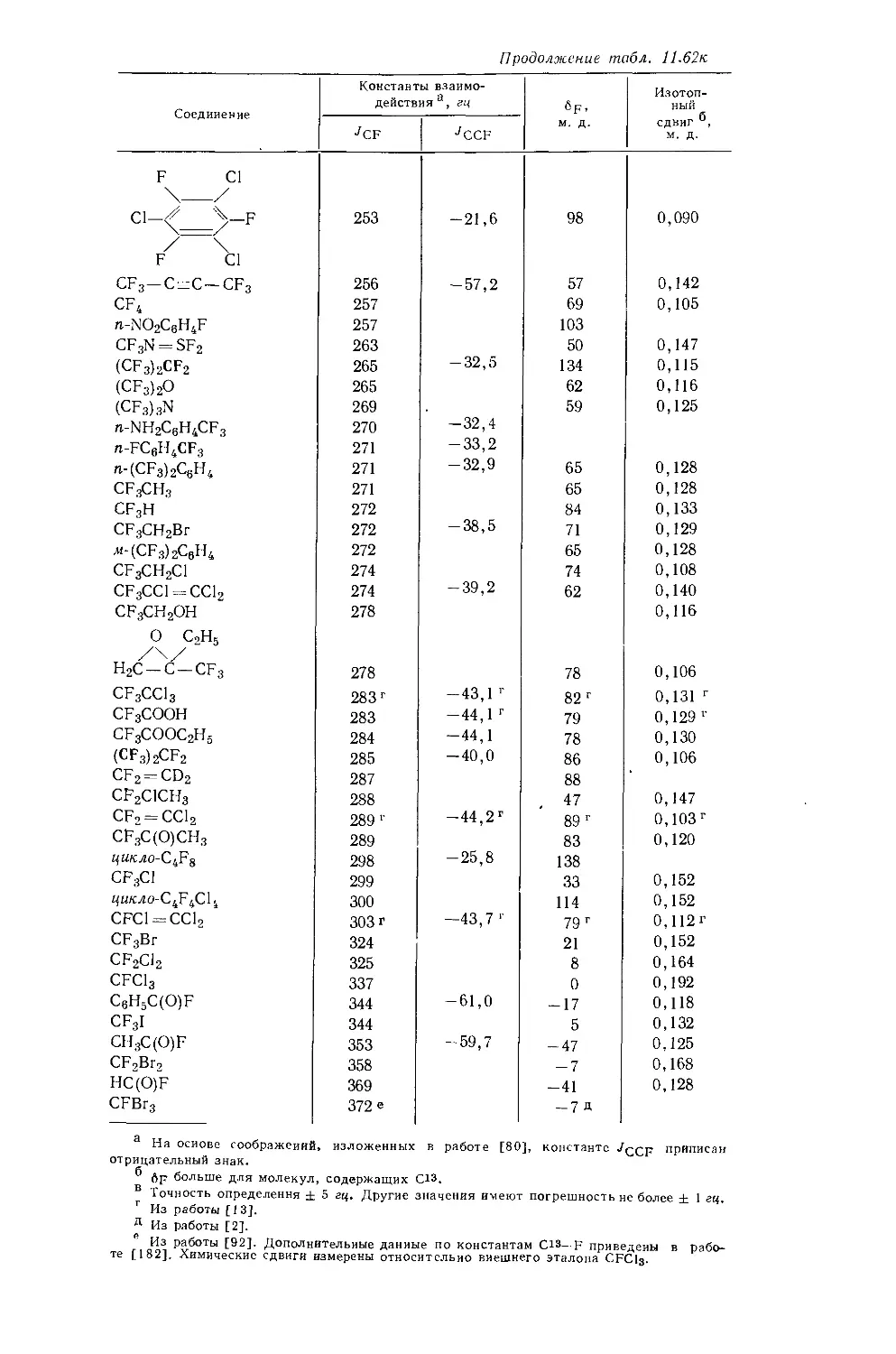
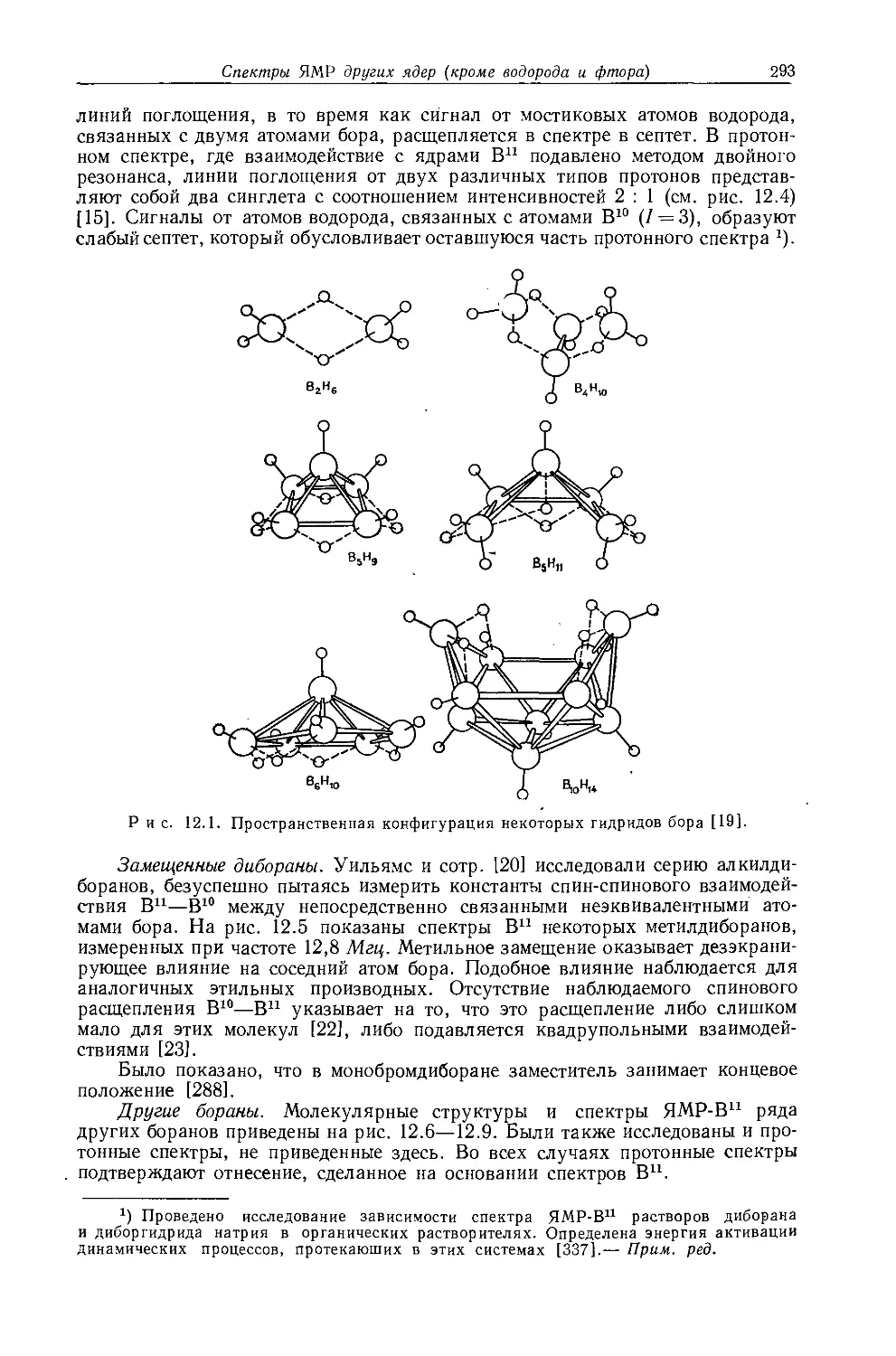
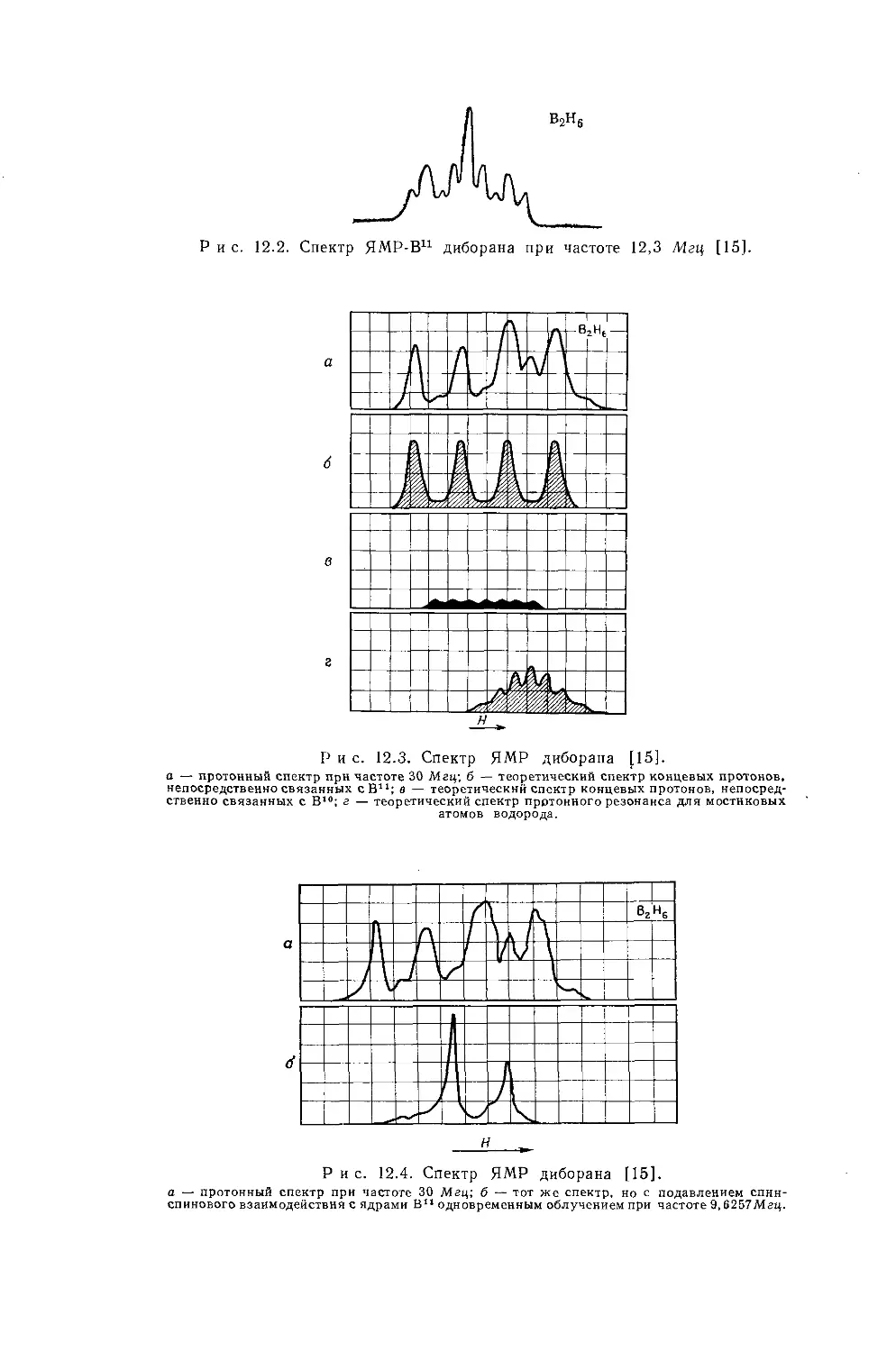
б Измеренная константа имеет очень малое отрицательное значение (0,3 гц)} поэтому возможно что знаки констант в молекулах этого типа не всегда будут одинаковыми, *
в См. работу [38].
2*
20
Глава 10
обычно можно получить лишь относительные знаки этих констант. Для экспериментального определения абсолютных знаков констант взаимодействия Карплус [58] предложил косвенный метод, успешно примененный для большого числа молекул Лаутербуром и Курландом [32]. Теория предсказывает для констант взаимодействия между непосредственно связанными углеродным и водородным атомами (Тен) положительный знак, поэтому часто для одной и той же молекулы возможно определение относительных знаков Унн и Усн, а абсолютные знаки /н-н могут быть выведены из относительных. Определив абсолютные знаки констант взаимодействия протонов в некоторых группах, их можно использовать, чтобы установить абсолютные знаки констант взаимодействия для молекул, в которых содержатся эти группы и относительные знаки констант взаимодействия уже известны. В табл. 10.8 приведены абсолютные знаки констант взаимодействия протонов, полученные подобным образом для некоторых систем. Более прямой метод определения абсолютных знаков констант взаимодействия сводится к тому, что наблюдают, как влияет на спин-спиновые мультиплеты помещение образца в сильное электрическое поле. К алканам этот метод до сих пор не удалось применить для определения абсолютных знаков /н-н и /н-н-
Ввиду того что в алканах /нн имеет знак, противоположный тому, который предполагают Карплус и сотр. [28—30], необходимы дальнейшие расчеты. В настоящий момент имеются данные, подтверждающие обоснованность теоретических расчетов для /нн. Понимание абсолютных знаков различных констант позволяет с большей уверенностью оперировать с эмпирическими корреляциями между константами /н-н и молекулярными параметрами.
10.2.4. Константы дальнего взаимодействия протонов в насыщенных системах
Для протонов, разделенных более чем тремя насыщенными связями, значительное спин-спиновое взаимодействие не характерно. Однако в некоторых молекулах такое взаимодействие встречается. Например, резонансный сигнал альдегидных протонов в димере метакролеина (I)
z(\ ZCH3 н__/ \Z J
11 II IXCHO н с /н НзС“\/<Н I
н н
представляет собой дублет с независимым от поля расщеплением в 1,3 гц [59]. Расщепление обусловлено, по-видимому, дальним взаимодействием с одним из кольцевых протонов в [5-положении к альдегидной группе. Исследуя дейтерированные молекулы, можно исключить возможность взаимодействия олефинового и альдегидного протонов.
Другим примером дальнего взаимодействия протонов служит 1,2-дибром-2-фенилпропан [59] с конфигурацией, в которой, по-видимому, объемистые атомы брома расположены в транс-положении по отношению друг к другу, как это показано в структуре II. Из спектра протонного резонанса этой моле-
Вг
н3с<Схс6н5.
(1)Н^/>Н(2)
Вг
И
Корреляции спектров ЯМР-Н1 со строением молекул
21
кулы очевидно, что один из двух неэквивалентных атомов водорода, Н(1) или Н(2), взаимодействует с С-метильной группой (/сн3-н = 0,65 гц). Исследуя специально дейтерированные 1,2-дибром-2-фенилпропаны (III и IV), можно показать, что дальнее взаимодействие /сн3-н имеет место, если атомы водорода
Вг
НзС-^^СеНз HXC>D
Вг
III
Вг н3с^/с^с6н3 D^C>H
Вг
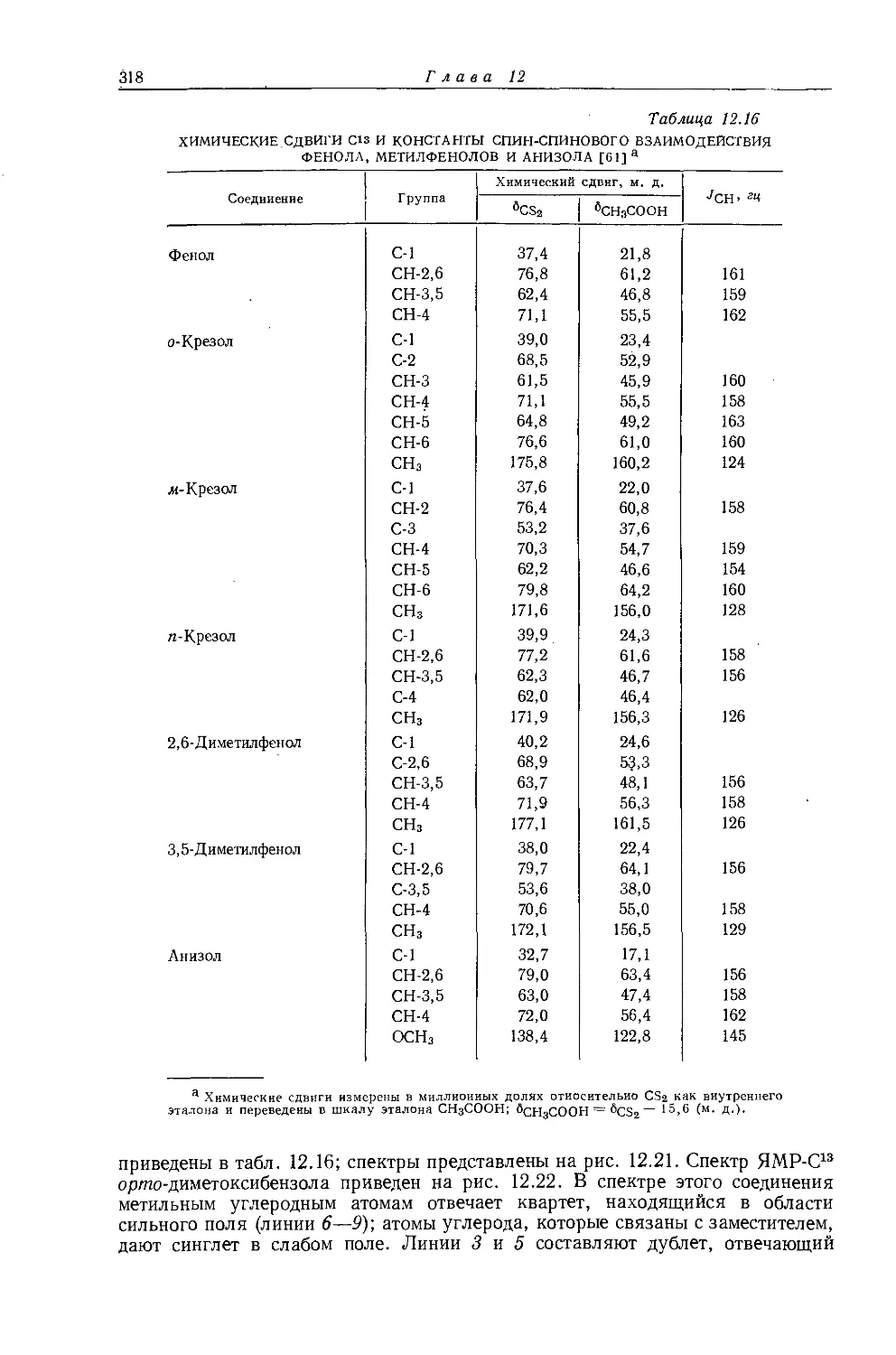
IV
находятся в транс-положении к метильной группе. Для обоих приведенных примеров (I и II) характерно наличие лишь одной конформации.
Интересным было наблюдение константы дальнего взаимодействия в ацетоне по спутникам С13Н (JCh3-ch3 = 0,54 гц) [60]. До тех пор дальнее спин-спиновое взаимодействие протонов наблюдали лишь для молекул, существующих в предпочтительных конформациях, тогда как в ацетоне при комнатной температуре происходит свободное вращение вокруг связи С — С. Карплус [61, 62[ показал, что большие константы дальнего взаимодействия протонов между ядрами, разделенными л-связями, могут быть объяснены участием низко-лежащих Зл-состояний ист — л-взаимодействиями в связях С — Н [63]. Было сделано предположение о том, что низколежащие состояния могут вносить значительный вклад во взаимодействие протонов также в ст-связанных системах, геминальных по отношению к ненасыщенной связи, как, например, в ацетоне.
Появление дальнего спин-спинового взаимодействия в димере метакролеина [59] и в метилформиате [64] может быть обусловлено влиянием соседних карбонильных групп.
Константа дальнего взаимодействия через напряженное тетрациклобутановое кольцо была измерена для производных экзо-бицикло-[2,1,1]-гексано-ла-5 (V) [368]. Взаимодействие протонов в положениях а и а' производных V
характеризуется константой 6,8—8,1 гц. Однако, хотя напряжение кольца фиксирует ядра в выгодных для взаимодействия положениях, оно не является необходимым фактором, обусловливающим дальнее взаимодействие. Этот вывод был подтвержден при сравнении стереохимии соединения V и метил-а,р-дибромизобутирата (VI), в котором константа дальнего взаимодействия 7н-сн3 равна 0,8 гц [59]; это гораздо меньше, чем константа дальнего взаимодействия для соединения V, вследствие того, что метильные протоны проводят лишь одну треть времени в выгодном для дальнего взаимодействия положении (т. е. транс-положении к взаимодействующим с ними протонам) и, кроме
22
Глава 10
того, два углеродных атома, к которым присоединены взаимодействующие протоны, находятся в соединении VI на гораздо большем расстоянии друг от друга, чем в циклобутановом кольце V. Интересно, что для эндо-бицикло-12,1,1] -гексанола-5 (VII) константы дальнего взаимодействия не могли быть измерены в связи с их очень малой величиной.
Анет [369] определил константы дальнего взаимодействия между протонами, разделенными четырьмя ст-связями в 2-эндо-3-эндо-камфандиоле-2,3 (VIII) (Л в = 1,4 г<| и J3 5 = 1,0 гц) х).
Константы дальнего взаимодействия были измерены также для сопряженных диенов [444], аценафтенов [432] и некоторых насыщенных соединений [423].
10.3. Другие алкильные соединения
10.3.1. Метильные производные элементов группы IVБ
Оллред и Рохов [65] с целью исследования электроотрицательностей элементов группы IVB измерили химические сдвиги метильных производных углерода, свинца, германия, олова и кремния путем экстраполяции к бесконечному разбавлению в четыреххлористом углероде. Приняв значения электроотрицательностей углерода и кремния за 2,60 и 1,90 соответственно [5] и предположив линейную зависимость между метильными химическими сдвигами и электроотрицательностью заместителя, они получили значения электроотрицательностей для остальных элементов группы IVB. Значения химических сдвигов соединений, приведенных в табл. 10.9, указывают на то, что электроотрицательности уменьшаются в следующем порядке: С > Pb > Ge > > Sn > Si; но такой ряд противоречит ряду, предсказанному другими мето-
1) На основании изучения знаков и величин констант дальнего спин-спинового взаимодействия для производных полихлорбицикло-[2,2,1 [-гептенов найдена стереохимическая зависимость констант J для фрагмента Н — С — С — С — Н [500, 501].— Прим. ред.
Корреляции спектров ЯМР-Н1 со строением молекул
23
дами [66]. Шписецки и Шнейдер [6] исследовали химические сдвиги Н1 и С13 газообразных метильных производных элементов группы IVB (исключив, таким образом, влияние анизотропии элементов группы IVB на экранирование ядер Н1 и С13 в этих соединениях) и по химическим сдвигам определили значения электроотрицательностей, аналогичные найденным Оллредом и Роховом и также плохо согласующиеся со значениями, полученными другими методами, особенно для первых членов ряда. Для более ясного понимания эффектов экранирования в такого рода молекулах Браун и Уэбстер [66] исследовали спектры протонного резонанса ряда метилхлорпроизводных углерода, кремния, олова, а также ди-трет-бутилового эфира, гексаметилэтана и соответствующих производных кремния, германия и олова. Результаты, полученные ими, представлены в табл. 10.9 и 10.10. Все измерения проведены в разбавленных
Таблица 10.9
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ МЕТИЛЬНЫХ ПРОИЗВОДНЫХ ЭЛЕМЕНТОВ ГРУППЫ IVB [06]
Соединение а T, M. Д. Соединение T, M. Д.
(СНз)4С 9,073+0,005 ! (CH3)eGe2 9,788+0,005
(CH3)4Si 10,000 (CH3)eSn2 9,790+0,005
(CH3)4Sn 9,930+0,005 (CH3)eSi2O 9,950+0,005
(CH3)4Ge 9,873 6 (CH3)eC2O 8,752+0,005
(СН3)6С2 9,130+0,007 (CH3)6Ge2O 9,700+0,010
(CH3)6Si2 9,963+0,002 (CH3)6Sn2O 9,730+0,002
а Соединения были исследованы в виде 2 — 10%-ных растворов в четыреххлористом
углероде.
б По данным работы [65].
Таблица 10,10 химические сдвиги протонов НЕКОТОРЫХ алкильных производных
УГЛЕРОДА, КРЕМНИЯ И ОЛОВА (СН3)а;МС14_х
Соединение a T, M. Д. Соединение a , T, M. Д.
(CH3)4c 9,073+0,002 (CH3)2SiCl2 9,200+0,002
(CH3)3CC1 8,404 + 0,005 CH3SiCl3 8,858+0,005
(CH3)2CC12 7,828±0,014 (CH3)4Sn 9,930+0,005
CH3CC13 7,257+0,007 (CH3)3SnCl 9,368+0,012
(CH3)4Si 10,000 (CH3)2SnCl2 8,835+0,007
(CH3)3SiCl 9,578+0,002 CH3SnCl3 8,353+0,005
а Соединения были исследованы в виде 2—10%-ных растворов в четыреххлорнстом углероде.
растворах в четыреххлористом углероде с тетраметилсиланом в качестве внутреннего эталона. Было показано, что замещение метильных групп в тетра-метилпроизводных углерода, кремния и олова на более электроотрицательные заместители приводит к уменьшению экранирования оставшихся метильных протонов, как и ожидалось на основании представлений об индуктивных эффектах. При последовательном замещении метильных групп на атомы хлора внутри каждого ряда экранирование остальных метильных протонов уменьшается приблизительно одинаково. Уменьшение экранирования в ряду углевода сильнее, чем в ряду кремния. Такая же закономерность наблюдалась
24
Глава 10
при сравнении экранирования протонов в связях Si — Н и С — Н соответственно в метилхлорсиланах и их углеродных аналогах, что было объяснено двоесвязностью кремния с атомами хлора [66]. Для соединений олова такая двоесвязность незначительна, поэтому наблюдается изменение химических сдвигов, такое же, как в производных углерода х).
В гексаметильных производных элементов группы IVB метильные протоны во всех случаях, за исключением гексаметил этан а, экранированы слабее, чем в тетраметильных производных. Введение атома кислорода между двумя центральными атомами приводит к уменьшению экранирования для всех четырех рядов молекул.
10.3.2. Алкильные производные элементов, имеющих ядерные магнитные моменты
При исследовании алкильных производных элементов, ядра которых обладают магнитными моментами, методом ЯМР можно получить весьма интересную информацию. Алкильная группа, связанная с изотопом X, дает резонансный спектр, каждая компонента которого расщепится в дублет в том случае, если немагнитный изотоп X заменить на магнитный (со спиновым
Усиление поля
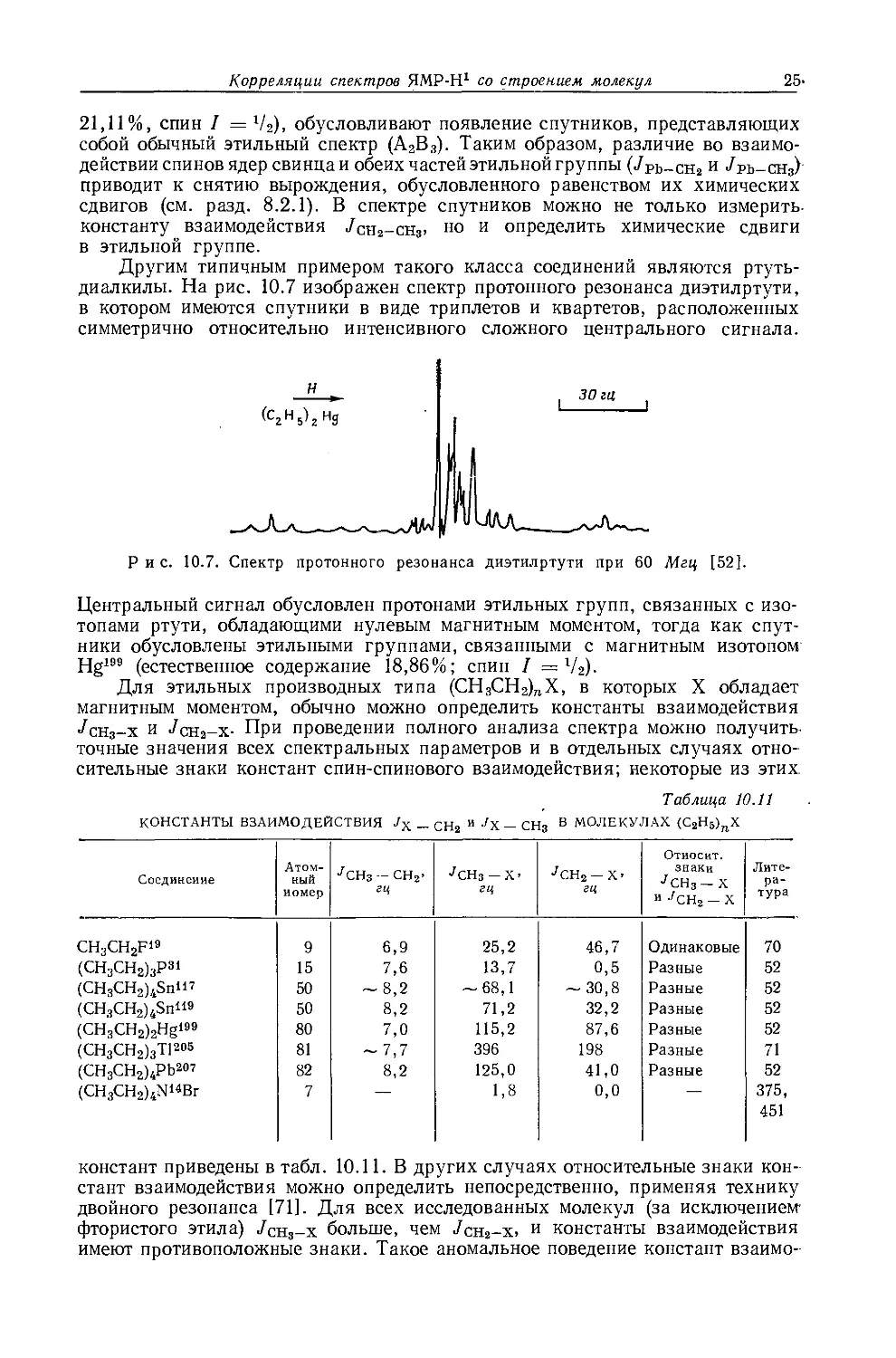
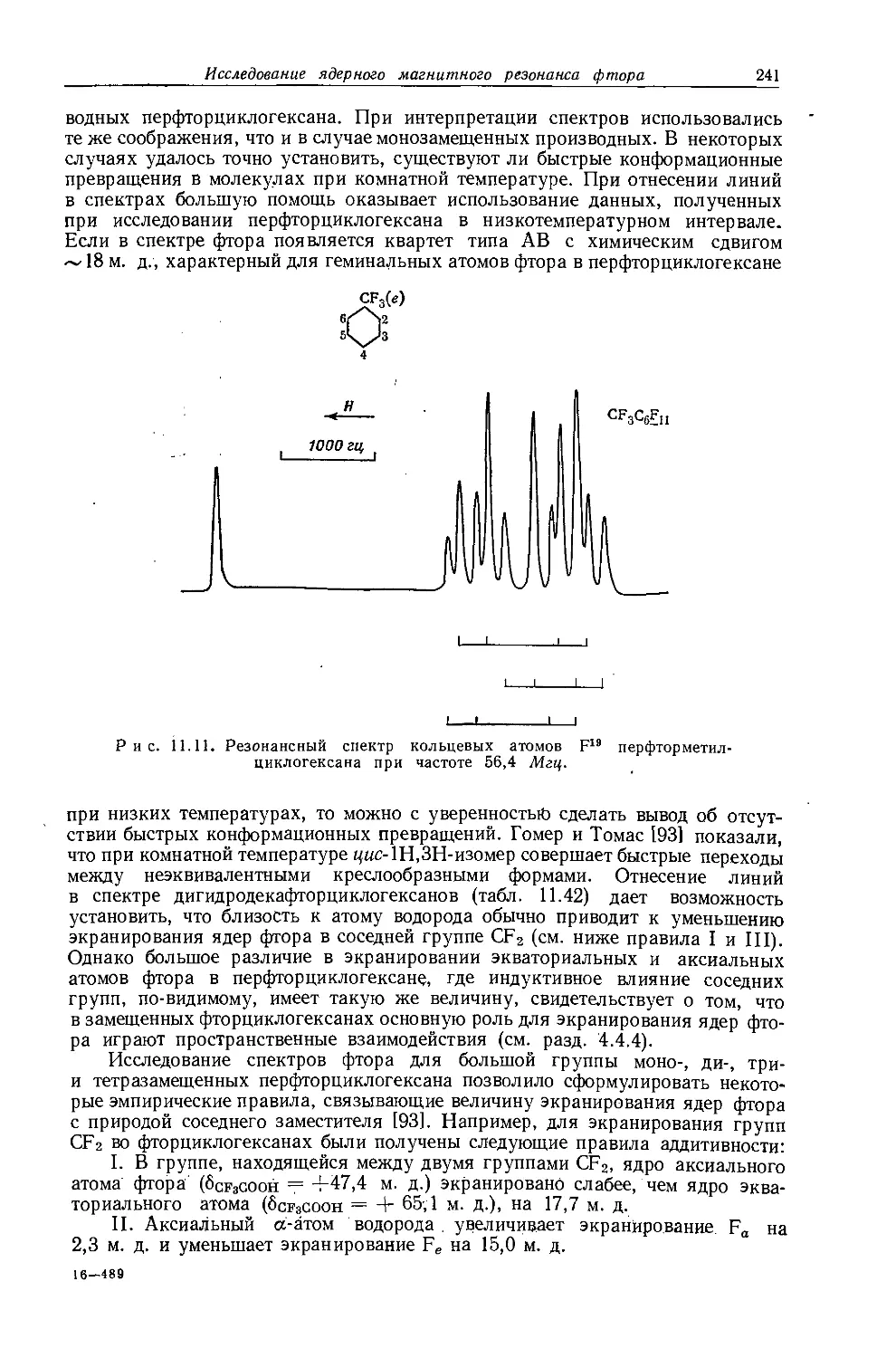
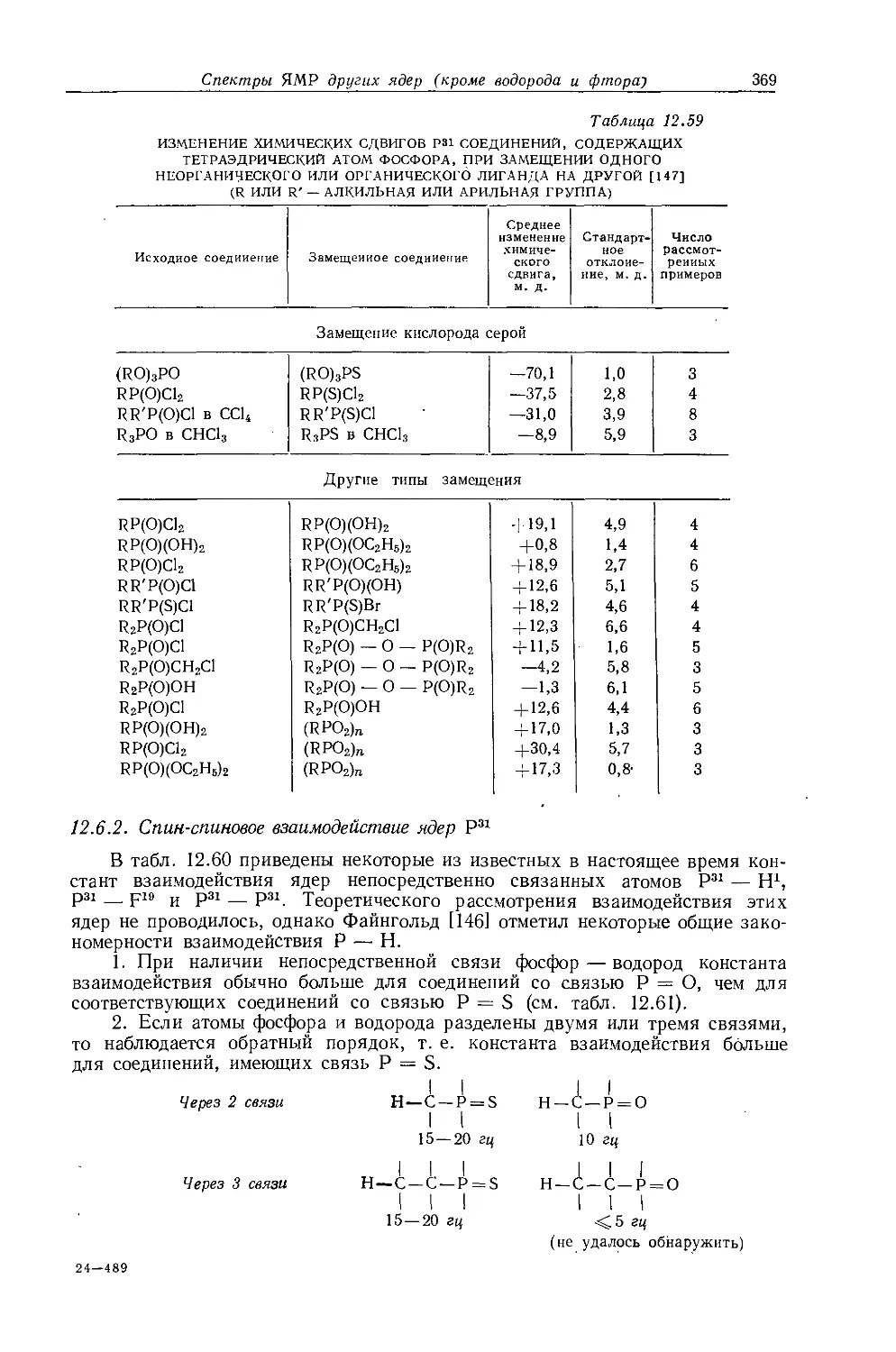
Рис. 10.6. Спектр протонного резонанса тетраэтилсвинца при 40 Мгц [52].
числом I =1/2). Уже в 1956 г. Бейкер [67] показал, что исследование спутников в спектрах ЯМР алкилов металлов с магнитными моментами дает информацию об алкильных группах, которая не может быть получена другими методами. На рис. 10.6 приведен спектр протонного резонанса тетраэтилсвинца при фиксированной частоте 40 Мгц. Легко видеть, что спектр состоит из интенсивной центральной полосы, относительно которой симметрично расположены две пары слабых спутников. Этильные группы, связанные с немагнитными изотопами свинца, дают интенсивный неразрешенный сигнал в центре спектра. Эти этильные группы не дают обычного «этильного» спектра ввиду того, что химический сдвиг между группами СНз и СН2 очень мал. Однако этильные группы, связанные с магнитным изотопом РЬ207 (естественное содержание
х) См. примечание на стр. 10. — Прим. ред.
Корреляции спектров ЯМР-Н1 со строением молекул
25-
21,11%, спин I = 1/2), обусловливают появление спутников, представляющих собой обычный этильный спектр (А2В3). Таким образом, различие во взаимодействии спинов ядер свинца и обеих частей этильной группы (/₽ь-сн2 и ^рь-сн3) приводит к снятию вырождения, обусловленного равенством их химических сдвигов (см. разд. 8.2.1). В спектре спутников можно не только измерить, константу взаимодействия /сн2-сн3, но и определить химические сдвиги в этильной группе.
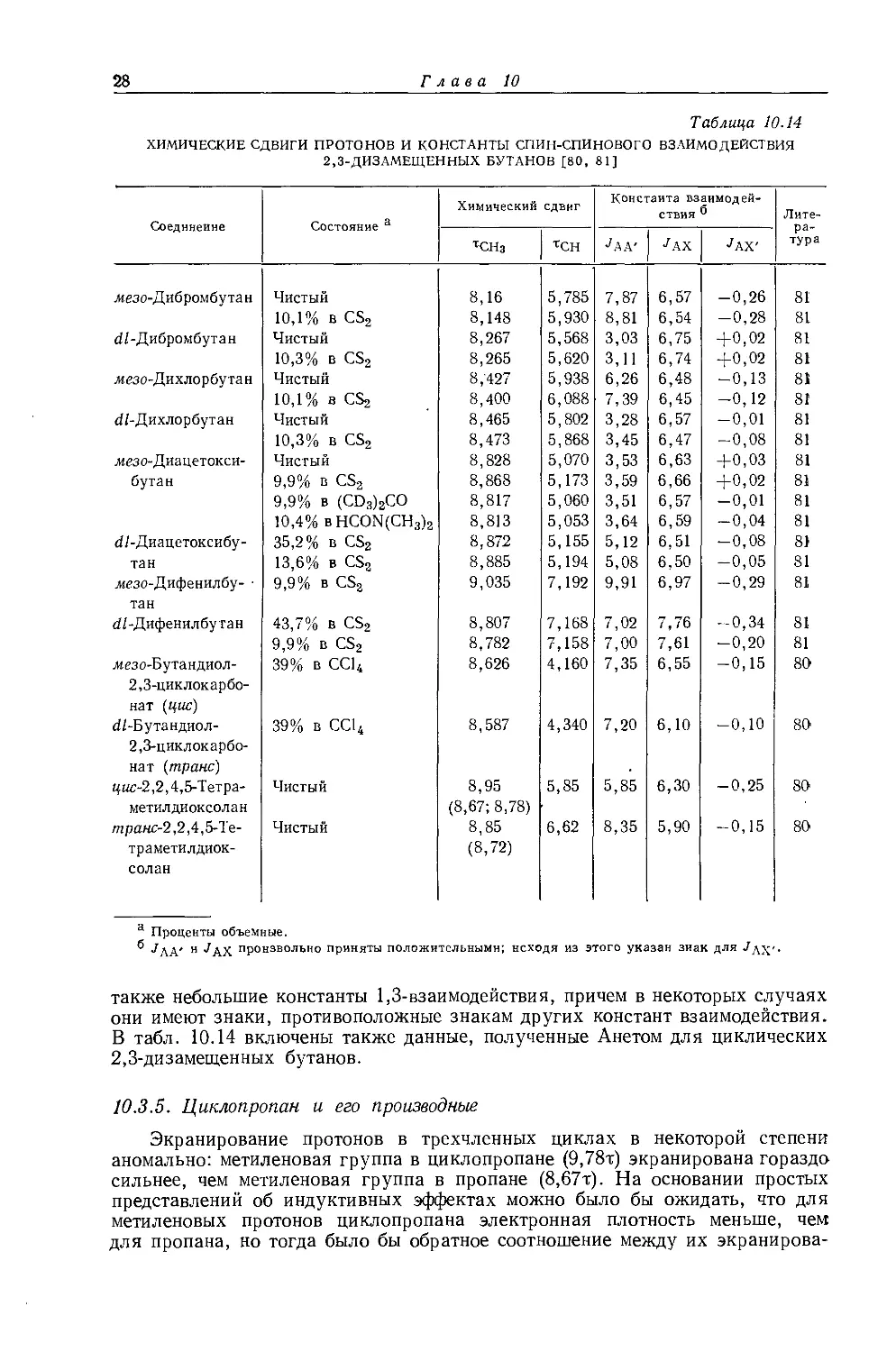
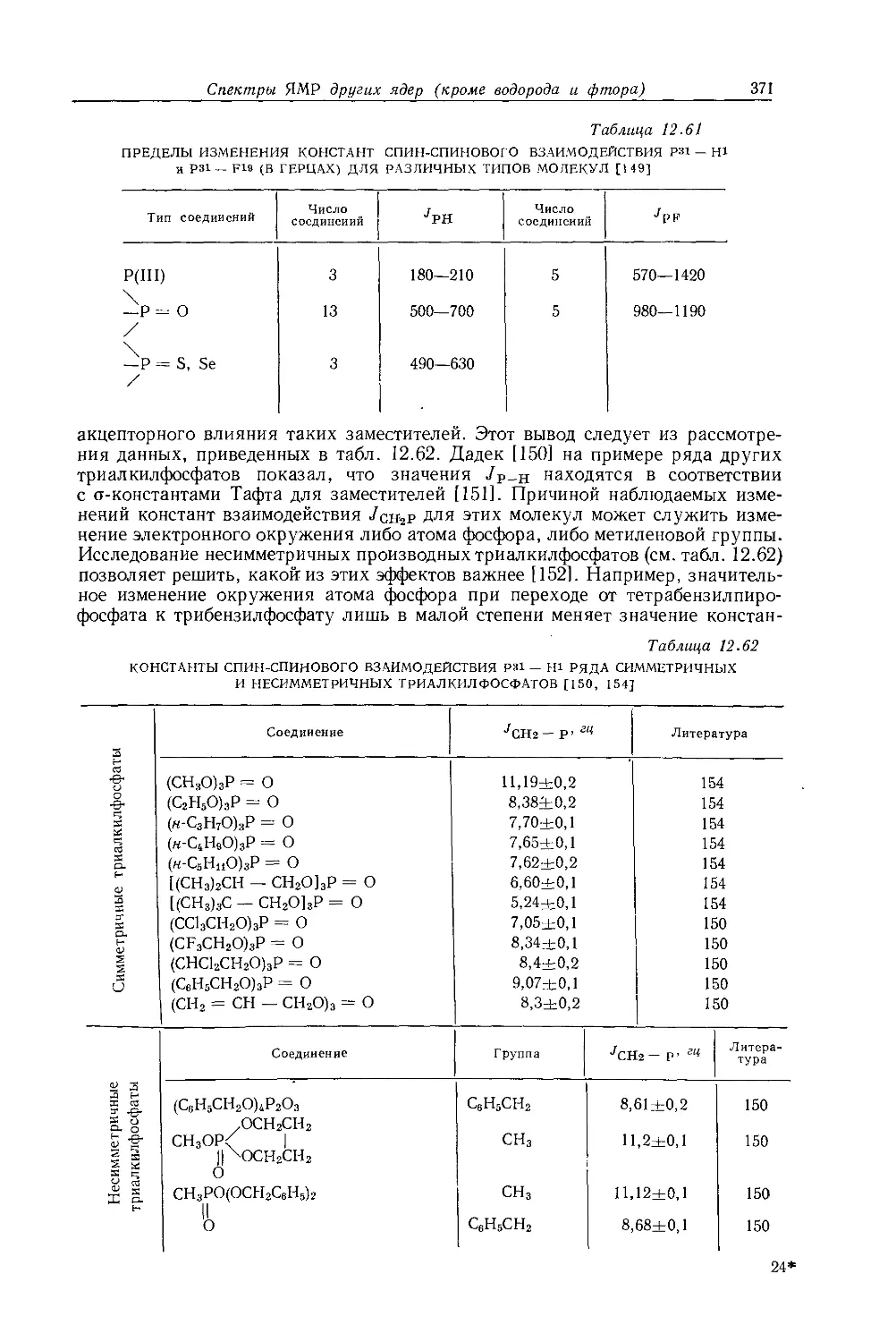
Другим типичным примером такого класса соединений являются ртутьдиалкилы. На рис. 10.7 изображен спектр протонного резонанса диэтилртути, в котором имеются спутники в виде триплетов и квартетов, расположенных симметрично относительно интенсивного сложного центрального сигнала.
Рис. 10.7. Спектр протонного резонанса диэтилртути при 60 Мгц [52].
Центральный сигнал обусловлен протонами этильных групп, связанных с изотопами ртути, обладающими нулевым магнитным моментом, тогда как спутники обусловлены этильными группами, связанными с магнитным изотопом Hg199 (естественное содержание 18,86%; спин I = V2).
Для этильных производных типа (СН3СН2)геХ, в которых X обладает магнитным моментом, обычно можно определить константы взаимодействия /сн3-х и /сн2-х- При проведении полного анализа спектра можно получить-точные значения всех спектральных параметров и в отдельных случаях относительные знаки констант спин-спинового взаимодействия; некоторые из этих
Таблица 10.11
КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ Jx — СН2 и 7Х — СН3 в МОЛЕКУЛАХ (С2Н5)ПХ
Соединение Атомный иомер JCH3 - сн2. гц JCH3 - х. гц JCH2- х. гц Относит, знаки JCH3- X и JCH2 — X Литература
CH3CH2F‘9 9 6,9 25,2 46,7 Одинаковые 70
(СН3СН2)3Р31 15 7,6 13,7 0,5 Разные 52
(CH3CH2)4Sn117 50 ~ 8,2 — 68,1 ~30,8 Разные 52
(CH3CH2)4Sn119 50 8,2 71,2 32,2 Разные 52
(CH3CH2)2Hg199 80 7,0 115,2 87,6 Разные 52
(СН3СН2)3Т1205 81 ~ 7,7 396 198 Разные 71
(CH3CH2)4Pb207 82 8,2 125,0 41,0 Разные 52
(CH3CH2)4N14Br 7 — 1,8 0,0 — 375,
451
констант приведены в табл. 10.11. В других случаях относительные знаки констант взаимодействия можно определить непосредственно, применяя технику двойного резонанса [71]. Для всех исследованных молекул (за исключением-фтористого этила) 7сн3-х больше, чем 7сн2-х, и константы взаимодействия имеют противоположные знаки. Такое аномальное поведение констант взаимо
26
Глава 10
действия было объяснено влиянием занятых d-орбиталей. Если d-электроны участвуют в образовании химической связи между атомом X и алкильной группой, они могут вызывать большие изменения как в величине, так и в знаке контактного вклада Ферми в константу спин-спинового взаимодействия. Предполагается, что в образовании связи С — X участвуют Sd-электроны при X = Pb, Т1 и Hg, 4d^TCKTpoHbi при X = Sn и неподеленная пара электронов при X = Р [70]. Нарасимхан и Роджерс [52] предложили качественное объяснение констант взаимодействия X — Н, основанное на приближении дальнего молекулярно-орбитального взаимодействия, постулированного Мак-Коннелом 133]. Они предположили, что различные одноэлектронные члены дают значительные вклады в контактное взаимодействие Ферми между метильными протонами и ядрами X. Однако Эванс [422], исходя из того, что относительные знаки двух констант взаимодействия X — Н аналогичны наблюдаемым в алканах геминальным и вицинальным константам, предположил, что константы взаимодействия X — Н не аномальны, как это казалось ранее. Он считает, например, что константы взаимодействия Т1205 — Н1 обусловлены контактным взаимодействием Ферми через бх-электроны атома таллия.
В работе [72] были исследованы спектры протонного резонанса диметиль-ных и диэтильных производных Cd и Se. Константы взаимодействия металл — протон в диэтилкадмии имеют свойства, аналогичные свойствам констант в других алкильных производных металлов, т. е. /м-сн3 больше /м-сн2-Исследование некоторых пропильных производных металлов [73] также показывает, что /м-а-сн2 меньше, чем /м-р-сн2 (см. табл. 10.12); следовательно, это явление справедливо не только для метильных групп.
Таблица 10.12
КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ ДЛЯ НЕКОТОРЫХ н-ПРОПИЛЬНЫХ ПРОИЗВОДНЫХ МЕТАЛЛОВ (н-С3Н7)пХ [73]
Cd Sn Hg Pb
7Х _ с — н’ гЧ 51,6 49,1 95,0. 40,5
7Х_с —с — Н’ 60,2 67,2 110,3 102,4
J0.3.3. Другие алкильные производные металлов
При исследовании спектров ЯМР диалкильных производных ртути было показано, что во всех исследованных соединениях ядра ртути взаимодействуют с Р-СН-протонами сильнее, чем с а-СН-протонами [69, 74]. Сводка измеренных констант взаимодействия дана в табл. 10.13. Хаттон и сотр. [424] измерили константы взаимодействия Hg199 — Н1 в широком ряду соединений CH3HgX и CH3CH2HgX: при этом было обнаружено сильное увеличение констант взаимодействия с увеличением электроотрицательности Х-заместителя [).
Исследование спектра протонного резонанса бромистого бутенилмагния в растворе диэтилового эфира показало, что структура этого реактива Гриньяра почти точно описывается формулой СН3СН = СН —CH2MgBr [75]. Алифатическая группа СН2, связанная непосредственно с магнием, дает дублет в сильном поле (т = 9,3 м. д.), что указывает на значительную электронную плотность вокруг протонов.
х) В работах [528—530] исследовано влияние растворителей на химические сдвиги протонов и константы спин-спинового взаимодействия 7нг-Н в молекулах диметил-, дибензил- и дифенилртути. В работе [502] обнаружена линейная зависимость между рКа ряда углеводородов и значениями JHg-H в соответствующих ртутьорганических •соединениях.— Прим. ред.
Корреляции спектров ЯМР-Н1 со строением молекул
27
Таблица 10.13
КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ Ш — Hgl99, ИЗМЕРЕННЫЕ ДЛЯ РЯДА АЛКИЛЬНЫХ СОЕДИНЕНИЙ РТУТИ [69, 74]
Соединение а Анз - Hg ^а-СНг - Hg Ah2X - Hg
C2H5HgCH3 129,4 (₽-CH3) 96 93,7 (a-CH3)
C2H5HgCH2Cl 152,2 119,1 46,1
C2H5HgCH2Br 134,8 95,8 39,3
C2H5HgCH2I 159,2 125,8 40,2
•ClCH2Hg(W-C4H9) 115,3 45
ClCH2HgCH3 115,6 51,9
•ClCH2HgCH2Cl 60
CH3HgCH3 6 102
<2H5HgC2H5 6 120 91
(«-C3H7)2Hg 6 90 (a)
108 (P)
(u3O-C3H7)2Hg 6 78(a)
126 (₽)
€H3HgCN в 178,0
CH3HgI в 200,0
CH3HgC104 в 233,2
а Значение J для диалкильных производных ртути было получено в лаборатории авторов, если это не оговорено особо.
б По данным работы [69].
в По данным работы [424]; все соединения были исследованы в виде 5 (мол.)%-ного раствора впириднне.
Спектр ЯМР бромистого аллилмагния (спиновая система АХ4) показывает, что это соединение существует в виде смеси быстро взаимопревращаю-щихся аллильных изомеров:
СН2 = СН—CH2MgBr BrMgCH2 — СН = СН2.
Применение спектроскопии ЯМР к бромистому у,у-диметилаллилмагнию показало, что он также претерпевает подобные превращения. При охлаждении образца синглет, соответствующий метильным протонам, превращается в симметричный дублет [77, 78]. На основании спектра ЯМР было также показано, что бутиллитий в эфирном растворе существует в виде сольватированного димера [440].
10.3.4. 2,3-Дизамещенные бутаны
Анет [79, 80] установил, что два метиновых протона в мезо- и г//-2,3-дизаме-щенных н-бутанах общей формулы CH3CHRCHRCH3 магнитно неэквивалентны вследствие того, что они по-разному взаимодействуют с разными концевыми метильными группами. Поэтому для полного анализа спектров протонного резонанса этих молекул их следует представлять как системы Х3АА'Х;. Такой анализ был проведен для ряда 2,3-дизамещенных н-бутанов [80, 81], и по величинам констант спин-спинового взаимодействия JAA' между неэквивалентными метановыми протонами были сделаны качественные оценки содержания вращательных изомеров. В табл. 10.14 приведены константы спин-спинового взаимодействия, полученные по спектрам этих молекул. Измерены
28
Глава 10
Таблица 10.14
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ И КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ 2,3-ДИЗАМЕЩЕННЫХ БУТАНОВ [80, 81]
Соединение Состояние а Химический сдвиг Константа взаимодействия б Литература
тсн3 ТСН + л' 4 АХ 4 АХ’
.мезо-Дибромбута н Чистый 8,16 5,785 7,87 6,57 -0,26 81
10,1% в CS2 8,148 5,930 8,81 6,54 -0,28 81
dl-Дибромбутан Чистый 8,267 5,568 3,03 6,75 +0,02 81
10,3% в CS2 8,265 5,620 3,11 6,74 +0,02 81
жезо-Дихлорбутан Чистый 8,427 5,938 6,26 6,48 -0,13 81
10,1% в CS2 8,400 6,088 7,39 6,45 -0,12 81
dZ-Дихлорбутан Чистый 8,465 5,802 3,28 6,57 -0,01 81
10,3% в CS2 8,473 5,868 3,45 6,47 -0,08 81
мезо- Диацетокси- Чистый 8,828 5,070 3,53 6,63 +0,03 81
бутан 9,9% в CS2 8,868 5,173 3,59 6,66 +0,02 81
9,9% в (CD3)2CO 8,817 5,060 3,51 6,57 -0,01 81
10,4% bHCON(CH3)2 8,813 5,053 3,64 6,59 -0,04 81
dZ-Диацетоксибу- 35,2% в CS2 8,872 5,155 5,12 6,51 -0,08 81
тан 13,6% в CS2 8,885 5,194 5,08 6,50 -0,05 81
.иезо-Дифенилбу- 9,9% в CS3 9,035 7,192 9,91 6,97 -0,29 81
тан
dZ-Дифенилбутан 43,7% в CS2 8,807 7,168 7,02 7,76 -0,34 81
9,9% в CS2 8,782 7,158 7,00 7,61 -0,20 81
.иезо-Бутандиол- 39% в СС14 8,626 4,160 7,35 6,55 -0,15 80
2,3-циклокарбо-
нат (цис)
dZ-Бутандиол- 39% в СС14 8,587 4,340 7,20 6,10 -0,10 80
2,3-циклокарбо-
нат (транс)
цис-2,2,4,5-Тетра- Чистый 8,95 5,85 5,85 6,30 -0,25 80
метилдиоксолан (8,67; 8,78)
транс-2,2,4,5-Те- Чистый 8,85 6,62 8,35 5,90 -0,15 80
траметилдиок- (8,72)
солан
а Проценты объемные.
6 4АА' н 4АХ произвольно приняты положительными; исходя из этого указан знак для /дх'-
также небольшие константы 1,3-взаимодействия, причем в некоторых случаях они имеют знаки, противоположные знакам других констант взаимодействия. В табл. 10.14 включены также данные, полученные Анетом для циклических 2,3-дизамещенных бутанов.
10.3.5. Циклопропан и его производные
Экранирование протонов в трехчленных циклах в некоторой степени аномально: метиленовая группа в циклопропане (9,78т) экранирована гораздо сильнее, чем метиленовая группа в пропане (8,67т). На основании простых представлений об индуктивных эффектах можно было бы ожидать, что для метиленовых протонов циклопропана электронная плотность меньше, чем для пропана, но тогда было бы обратное соотношение между их экранирова
Корреляции спектров ЯМР-Н1 со строением молекул
29
нием. Несмотря на то что Уолш [82] предположил для циклопропанового кольца небольшой л-электронный характер, сильное увеличение экранирования метиленовых групп циклопропана вряд ли обусловлено влиянием наведенных кольцевых токов. Можно показать, что такой эффект очень мал и должен вызывать дезэкранирование в положениях, в которых находятся кольцевые протоны [83]. До сих пор не предложено какого-либо удовлетворительного объяснения этих аномальных химических сдвигов. Имеются данные, свидетельствующие об аналогичном аномальном экранировании в эпоксидах [83, 84] г).
Влияние размеров кольца на экранирование кольцевых протонов в циклических молекулах. Уиберг и Нист [348] исследовали спектры ЯМР-Н1 некоторых циклоалканов, циклоалкенов и циклоалканонов. Полученные ими результаты приведены в табл. 10.15. Для того чтобы исключить возможность каких-либо
Таблица 10.15 ХИМИЧЕСКИЕ Сдвиги ПРОТОНОВ НЕКОТОРЫХ ЦИКЛОАЛКАНОВ, ЦИКЛОАЛКЕНОВ И ЦИКЛОАЛКАНОНОВа [348]
Число атомов углерода в кольце Цикло-алкаиы Циклоалкеиы
= сн сн2с = Дт 6 ссн2с Дг 6
3 9,78 (с) 2,99 (т) 9,08 (т) 0,70 — —
4 8,04 (с) 4.03(c) 7.46(c) 0,56 — —
5 8,49 (с) 4,40 (т) 7,72 (м) 0,77 8,10(м) 0,39
6 8,56 (с) 4,41(т) 8,04(м) 0,53 8,35(м) 0,20
7 8,46 (с) 4,29(т) 7,89 (ш) 0,57 8,38 (ш ) 0,08
8,51 (ш ) -0,05
8 8,46 (с) 4,44 (т) 7,89 (ш) 0,54 8,50 (ш ) -0,04
Циклоалканоны
сн2- с = О дг б С - сн2 - С Дг 6
4 6,97 (т) 1,07 8,04 (к) 0,00
5 7.94(c) 0,55 7.98(c) 0,51
6 7,78 (ш) 0,78 8,21 (ш) 0,35
7 7,62 (ш) 0,84 8,34 (ш) 0,12
8 7,70 (ш) 0,76 8,19(ш) 0,27
8,50 (ш) -0,04
а Буквы указывают на число компонент резонансного сигнала: с — синглет, т — три-плет, к — квинтет, м — не полностью разрешенный -мультиплет, ш — широкий неразрешенный мультиплет. Возможная ошибка равна ±0,02.
б Сдвиг измерен относительно соответствующего циклоалкана.
эффектов межмолекулярного экранирования, исследовались разбавленные растворы этих соединений в четыреххлористом углероде. В ряду циклоалканов наблюдается значительное уменьшение экранирования кольцевых протонов при уменьшении размеров кольца. Исключение составляет циклопропан, протоны которого экранированы гораздо сильнее, чем в больших циклических соединениях, причем это экранирование настолько велико, что его нельзя объяснить влиянием диамагнитной анизотропии соседних связей С — С,
х) В работе [503] проведены расчеты анизотропии магнитной восприимчивости циклопропанового и окисного колец и показано, что учет межатомных эффектов позволяет объяснить сдвиг протонов в спектрах этих молекул.— Прим. ред.
30
Глава 10
как это делали в случае различий химических сдвигов циклопентана, циклогексана и метиленовых протонов в открытой цепи [159]. В циклоалкенах олефиновые протоны экранированы почти одинаково для 5- — 8-членных циклов, тогда как в циклобутене проявляется меньшее экранирование (возможно, вследствие большей деформации валентных углов), а в циклопропене это экранирование еще меньше. Химические сдвиги олефиновых протонов циклопропена можно объяснить при помощи модели кольцевых токов. Следует иметь в виду, что ядра С13 в трехчленной циклической молекуле окиси этилена экранированы на 30 м. д. сильнее, чем ядра С13 в шестичленных циклах и молекулах с открытыми цепями [300].
Замещенные циклопропаны. Транс-дибромциклопропан (I) дает обманчиво
I
простой спектр ЯМР-Н1, на основании которого в более ранней работе [85] был сделан вывод о том, что константы взаимодействия цис- и транс-протонов в этой молекуле одинаковы. Гутовский и Грант [86] показали, что можно провести детальный анализ спектра системы АА'ХХ', если использовать для цис- и транс-констант неравные значения. В последующих исследованиях
----,! ।
217,0 100,4
Химический сдвиг относительно , гц
Рис. 10.8. Спектр протонного резонанса 1,1-дихлор-2-метоксициклопро-пана при 60 Мгц [90].
циклопропановых производных [87—90] было показано, что константы взаимодействия ^ис-протонов больше, чем соответствующие константы транс-протонов (см. табл. 10.16). Оказалось, что эти данные являются общими для производных циклопропана и могут быть использованы в конформационном анализе молекул. Грэхэм и Роджерс [90] исследовали спектры ЯМР ряда замещенных циклопропа'нов; полученные ими результаты приведены в табл. 10.16. Спектр ЯМР-Н1 типичного циклопропанового производного приведен, на рис. 10.8. При изучении данных, приведенных в табл. 10.16, можно видеть, что на цис- и транс-константы взаимодействия природа заме-
Соединение
С1
Таблица 10.16
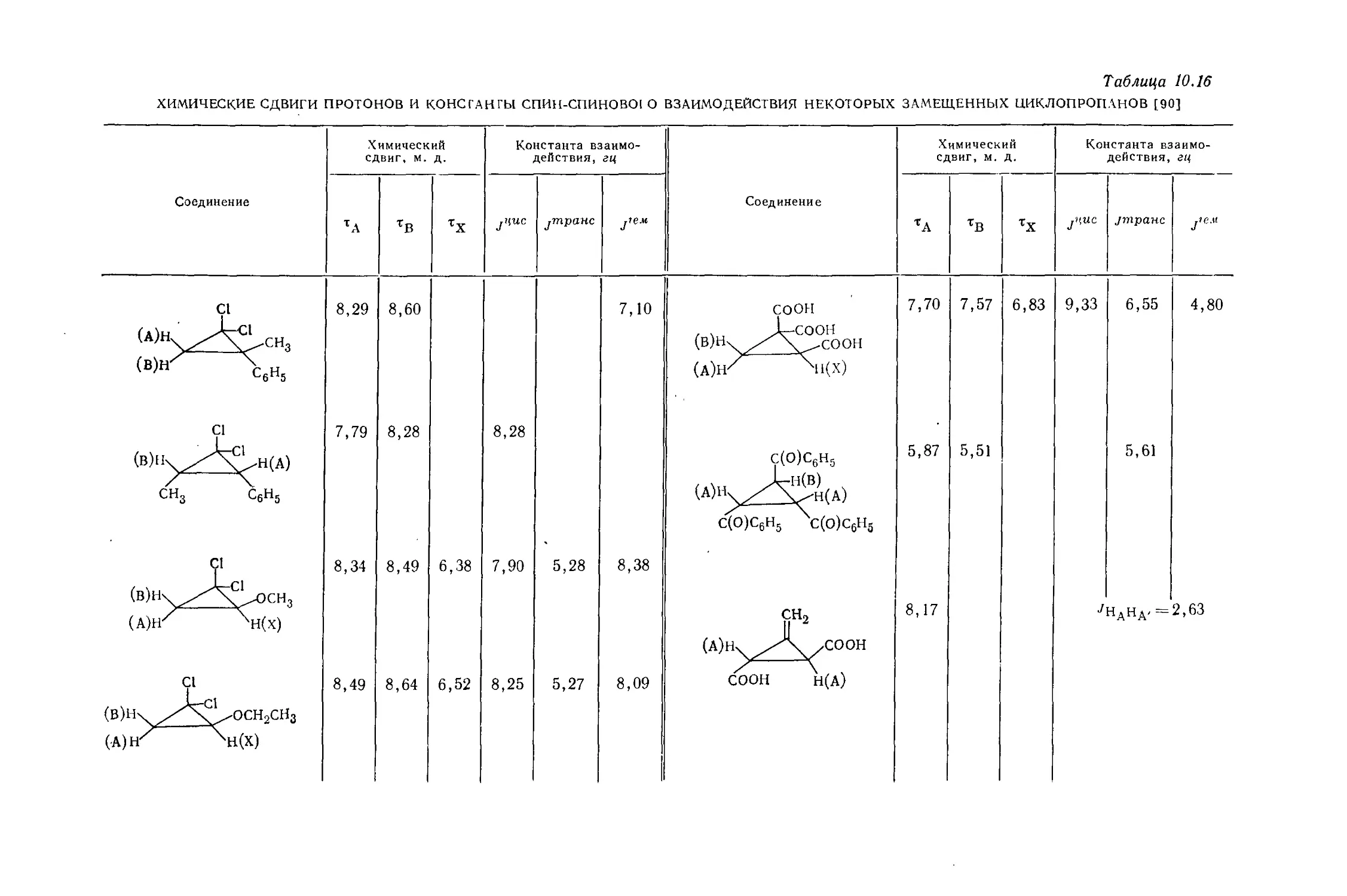
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ И КОНСГАНГЫ СПИН-СПИНОВО1 О ВЗАИМОДЕЙСТВИЯ НЕКОТОРЫХ ЗАМЕЩЕННЫХ ЦИКЛОПРОПАНОВ [90]
Химический сдвиг, м. д. Константа взаимодействия, гц
ТА ТВ тх jUUC jm.pa.Hc j’eM Соединение
8,29 8,60 7,10 (в)нх СООН JL-COOH /^\_^-СООН
(a)hz \1(Х)
7,79 8,28 8,28 С(О)С6Н5 с(о)с6н5 \(о)с6н5
8,34 8,49 6,38 7,90 5,28 8,38 сн2
(А)Н, ./А /СООН
8,49 8,64 6,52 8,25 5,27 8,09 СООН Н(А)
Химический сдвиг, м. д. Константа взаимодействия, гц
ТА ТВ тх J,tuc jmpanc J,e"
7,70 7,57 6,83 9,33 6,55 4,80
5,87 5,51 5,61
8,17 J НАНА' = 2,63
.32
Глава 10
стителя влияет очень мало, тогда как химические сдвиги существенно от нее зависят. Химический сдвиг протонов в незамещенном циклопропане составляет 9,78т, следовательно, заместители, приведенные в табл. 10.16, вызывают .дезэкранирование протонов [85]. Магнитно анизотропные группы типа фенила или карбонильной группы оказывают значительное дезэкранирующее влияние -на протоны кольца [90].
Замещенные циклопропаны существуют в жестко закрепленных конформациях, благодаря чему на них можно изучать зависимость констант спин-спинового взаимодействия от углов. Измеренные значения цис- и транс-констант в циклопропанах находятся в хорошем согласии со значениями, рассчитанными по методу валентных связей [39, 92, 433, 441, 467]. Из табл. 10.16 можно видеть, что константы геминального взаимодействия более чувствительны к природе заместителя, чем соответствующие константы вицинального взаимодействия.
Было показано, что константу геминального и вицинального взаимодействия Н — Н в циклопропане имеют противоположные знаки [91] (как и в случае ациклических алканов).
В работе [88] применили спектроскопию ЯМР для определения конфигурации некоторых хлорциклопропанов. Это было сделано при допущении, что константа тра«с-взаимодействия больше, чем константа ^ис-взаимодействия, что противоречит выводам Грэхэма и Роджерса [90]. Теоретические предсказания указывают на большую величину tfizc-взаимодействия, а недавно Хаттон и Шефер [350] достаточно однозначно показали, что так оно и есть на •самом деле.
Таблица 10.17
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ НЕКОТОРЫХ МОНОЗАМЕЩЕННЫХ ЭПОКСИДОВ [S3, 84] (В) Н^______________ZY
(A) h/\oz\h (X)
Y тв ТА ТХ
Н 7,42 7,42, 7,42
осн3 7,00 7,06 6,63
С6Н5 7,19 7,40 6,39
соон 7,01 7,07 6,53
С1а 7,17 7,25 5,10
CN 6,98 6,89 6,50
а По данным работы [83].
Эпоксиды. В табл. 10.17 приведены химические сдвиги протонов для некоторых монозамещенных эпоксидов. Показано, что кольцевые протоны экранированы сильнее соответствующих протонов в ациклических эфирах [83 , 84, 93, 484] 1).
Константы геминального и вицинального взаимодействия в эпоксидах имеют противоположные знаки.
Я Пространственное строение некоторых производных циклогексана и декалина, содержащих в качестве одного из заместителей эпоксидную группу, было установлено с помощью спектров ЯМР в работе [504]. Спектры эпоксидных производных бициклических терпенов рассмотрены в работах [505—507]. На основании полученных данных сделаны выводы о конформации исследованных соединений и диамагнитном влиянии эпоксидного кольца. В работе [508] химические сдвиги N-производных этиленимина рассмотрены с точки зрения инверсии цикла и электронного строения молекул— Прим. ред.
Корреляции спектров ЯМР-Н1 со строением молекул
33
10.3.6. Производные циклогексана
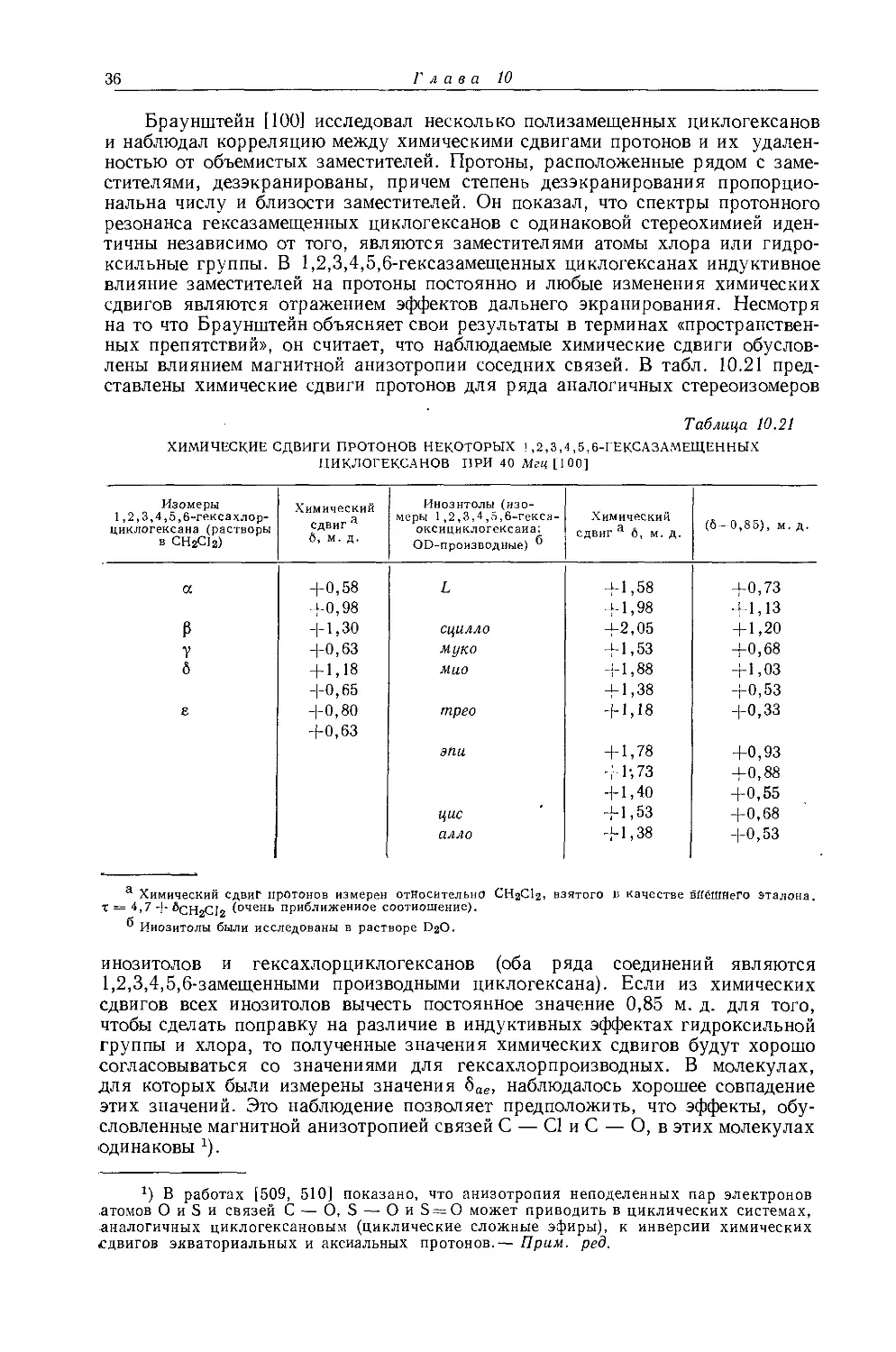
Циклогексан представляет собой неплоскую молекулу, в которой напряжение в цикле минимально, когда молекула имеет кресловидную конформацию, как это показано в разд. 4.4.2. В такой форме молекулы у каждого атома углерода имеется два положения заместителей — экваториальное и аксиальное. Если при комнатной температуре в циклической молекуле происходят быстрые превращения одной конформации в другую (см. разд. 9.7.1), то каждый заместитель будет проводить в аксиальном и в экваториальном положениях равные промежутки времени. Когда такой эффект преобладает над остальными, спектр ЯМР может фиксировать лишь усредненное окружение магнитных ядер, участвующих во взаимопревращении. Если же скорость взаимопревращения становится достаточно малой для того, чтобы при помощи метода ЯМР можно было «видеть» магнитные ядра отдельно в каждом из конформационных окружений, становится возможным проведение оценок экранирования в аксиальном и экваториальном положениях: молекулы этого типа можно рассматривать как фиксированные конформеры и наблюдать отдельные сигналы для протонов в аксиальном и экваториальном положениях. В разд. 9.7 приведены данные по использованию ЯМР для исследования скоростей конформационных взаимопревращений в циклогексановых системах. Исследование циклогексильных производных [94—96, 434, 452] в широком интервале температур часто может указать на конформационную подвижность молекулы (т. е. фиксированная она или взаимопревращающаяся). В этом разделе будут рассмотрены другие наблюдения конфигурационных и конформационных эффектов в производных циклогексана.
Химические сдвиги протонов в производных циклогексана. Экранирование метиленовых протонов в циклогексане (т = 8,564) существенно меньше экранирования СН2-групп в ациклических алканах с неразветвленной цепью (т = 8,75 как для «-гексана, так и для «-гептана). Джекман и Уайли [85] считают, что это различие обусловлено вкладом в экранирование эффектов, обусловленных диамагнитной анизотропией соседних связей С — С. На этой же основе можно объяснить различие в экранировании протонов в аксиальном и экваториальном положениях. Протоны в аксиальном положении обычно экранированы сильнее протонов в экваториальном положении приблизительно на 0,1—0,6 м. д. (см. табл. 10.18). Джекман [85] рассчитал вклад диамагнитной анизотропии в экранирование протонов, использовав значение (%п — Хпр) =
Таблица 10.18
РАЗЛИЧИЕ В ЭКРАНИРОВАНИИ МЕЖДУ АКСИАЛЬНЫМИ И ЭКВАТОРИАЛЬНЫМИ ПРОТОНАМИ (6ае) ПРОИЗВОДНЫХ ЦИКЛО1 ЕКСАНА [85, 96—98]
Соединения 6ае, м. д.
Циклогексан [96] 6-1,2,3,4,5,6-Гексахлорциклогексан [97] е-1,2,3,4,5,6-Гексахлорциклогексан [97] 0,46 0,51 0,20
.иио-Инозитолгексаацетат [97] цис- и транс-4-тре/п-Бутилциклогек-санол [97] цис- и /пранс-4-тре/п-Бутилциклогек-силацетат [97] Андростерон и эпиандростерон [98] Па- и ПР-Оксипрогестерон [98] 0,20 0,13 0,40 0,45 0,43
3-489
34
Глава 10
— + 5,5-10-30 см3!молекула для простых связей С — С. Вследствие того что поперечная компонента магнитной восприимчивости (/п) больше продольной (/пр), дальнодействующие эффекты экранирования, обусловленные простой связью С — С, противоположны наблюдаемым для тройной связи С = С, у которой поперечная компонента магнитной восприимчивости меньше продольной. Джекман [85] рассчитал значение 6ое = 0,40 м. д. для различия в экранировании аксиальных и экваториальных положений, что отлично согласуется с экспериментальными значениями, приведенными в табл. 10.18. Машер [171 также объяснил различие в химических сдвигах в циклогексанах влиянием магнитной анизотропии.
Мориц и Шеппард [22] предположили, что внутренние химические сдвиги в пропане, изобутане и циклогексане могут быть объяснены влиянием магнитной анизотропии связей С — Си, исходя из этого предположения, рассчитали значение Д/с-с = 7,0-10-30 см3/молекула. Для циклогексана они сделали те же предположения, что и Джекман [85] (см. разд. 4.4.2): лишь связи С(2) — С(3) и С(5) — С(6) дают вклады в различное экранирование аксиальных и экваториальных протонов у углеродного атома С(1) и вклад связей С — Н равен нулю. Различие в экранировании аксиальных и экваториальных протонов, как это было показано при исследованйи спектра циклогексана при низкой температуре, составляет 0,46 м. д. и может быть объяснено влиянием магнитной анизотропии связей С — С, если предположить, что Д/с-с = = 7-Ю"30 см3/молекула. Это значение несколько выше значения, предсказанного Ботнер-Баем и Наар-Колином [8] (5,5-10-30 см3!молекула) на основании различия химических сдвигов в циклогексане и циклопентане, и много больше значения, рассчитанного теоретически (1,21-10-30 см3/молекула). Значение Д/с-с = уд . ю~30 см3/молекула полностью объясняет различие в экранировании протонов в пропане (0,4375 м. д.) даже без учета различия индуктивных эффектов групп СН2 и СН3, на котором строили свои выводы Нарасимхан и Роджерс [16]. Различие в химических сдвигах в других алканах можно объяснить аналогичным образом. В мезо- и Л-формах 2,3-дигалогенбутанов имеется различие в химических сдвигах метиновых, а также и метильных протонов, что не может быть обусловлено индуктивными эффектами, так как они одинаковы в обоих изомерах. Кроме того, влияние на экранирование магнитной анизотропии связей углерод — галоген будет также одинаково в обоих изомерах, поэтому разумно предположить, что наблюдаемые различия в химических сдвигах обусловлены магнитной анизотропйей связей С — С. Наблюдаемые различия в химических сдвигах легко объяснить, если для Д/с-с использовать значение 7,0 •10~30 см3/молекула, учитывая при этом конформацию молекул. Совпадение значений Д/с-с для циклогексана и 2,3-дигалогенбутанов свидетельствует о том, что кольцевые токи в циклогексане незначительны. Хотя вышесказанное еще не доказывает того, что магнитная анизотропия связи С — С является единственным источником дальнего экранирования, все же можно предположить, что она может вызывать значительное экранирование. Несмотря на успех данной модели в предсказании различий химических сдвигов кольцевых протонов в циклогексане [85] и метилциклогексане [95], для диметилциклогексанов [95] эта модель не смогла объяснить химических сдвигов кольцевых протонов. Маллер и Тош [95] исследовали спектры протонного резонанса для семи диметилциклогексанов в широком интервале температур и показали, что:
1) «нормальное» различие в химических сдвигах аксиальных и экваториальных протонов в циклогексане (~0,46 м. д.) становится гораздо меньшей величиной в диметилциклогексанах, имеющих один аксиальный метильный заместитель;
2) для таких пар соединений, как метилциклогексан и транс- 1,4-диметил-циклогексан, химические сдвиги аксиальных протонов в соответствующих положениях молекул сильно отличаются друг от друга;
Корреляции спектров ЯМР-Н1 со строением молекул
35
3) в цис- 1,3-диметилциклогексане некоторые кольцевые протоны дают сигналы, сдвинутые на 0,6 м. д. в более сильное поле по сравнению с метильными протонами.
Ни одно из этих наблюдений нельзя объяснить вкладами индуктивного эффекта и магнитной анизотропии связей.
Машер [23, 24] предположил, что наблюдаемые различия в экранировании аксиальных и экваториальных протонов в циклогексане могут существенна зависеть от влияния внутримолекулярных электрических полей.
Кавано и Дейли [7] высказали идею, что вклад в экранирование протонов в алканах дает также сдвиг, обусловленный связями С — С. Природа этого сдвига неясна, однако ясно, что это не влияние магнитной анизотропии.
Таблица 10.19
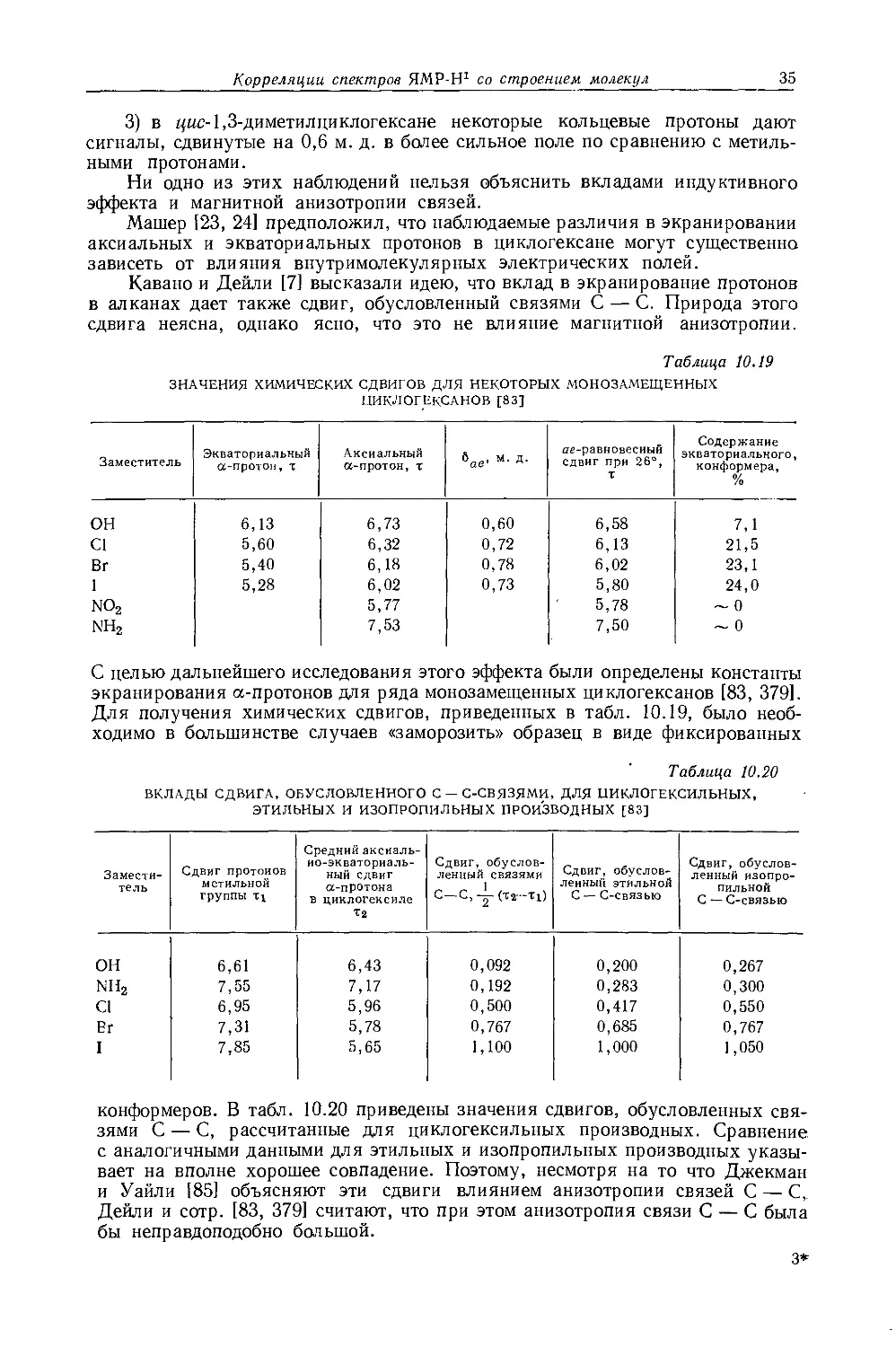
ЗНАЧЕНИЯ ХИМИЧЕСКИХ СДВИГОВ ДЛЯ НЕКОТОРЫХ МОНОЗАМЕЩЕННЫХ ЦИКЛОГЕКСАНОВ [83]
Заместитель Экваториальный а-протон, т Аксиальный а-протон, т 6ое, м. Д. ае-равновесиый сдвиг при 26°, Т Содержание экваториального, конформера, %
он 6,13 6,73 0,60 6,58 7,1
С1 5,60 6,32 0,72 6,13 21,5
Вг 5,40 6,18 0,78 6,02 23,1
1 5,28 6,02 0,73 5,80 24,0
no2 5,77 5,78 ~ 0
nh2 7,53 7,50 ~ 0
С целью дальнейшего исследования этого эффекта были определены константы экранирования а-протонов для ряда монозамещенных циклогексанов [83, 379]. Для получения химических сдвигов, приведенных в табл. 10.19, было необходимо в большинстве случаев «заморозить» образец в виде фиксированных
Таблица 10.20
ВКЛАДЫ СДВИГА, ОБУСЛОВЛЕННОГО С — С-СВЯЗЯМИ, ДЛЯ ПИКЛОГЕКСИЛЬНЫХ, ЭТИЛЬНЫХ И ИЗОПРОПИЛЬНЫХ ПРОИЗВОДНЫХ [83]
Заместитель Сдвиг протонов метильной группы Т1 Средний аксиаль-ио-экваториаль-ный сдвиг а-протона в циклогексиле т2 Сдвиг, обусловленный связями С —С, Д- (Т2--Т1) Сдвиг, обусловленный ЭТИЛЬНОЙ С — С-связью Сдвиг, обусловленный изопропильной С — С-связью
он 6,61 6,43 0,092 0,200 0,267
nh2 7,55 7,17 0,192 0,283 0,300
Cl 6,95 5,96 0,500 0,417 0,550
Er 7,31 5,78 0,767 0,685 0,767
I 7,85 5,65 1,100 1,000 1,050
конформеров. В табл. 10.20 приведены значения сдвигов, обусловленных связями С — С, рассчитанные для циклогексильных производных. Сравнение с аналогичными данными для этильных и изопропильных производных указывает на вполне хорошее совпадение. Поэтому, несмотря на то что Джекман и Уайли [85] объясняют эти сдвиги влиянием анизотропии связей С — С, Дейли и сотр. [83, 379] считают, что при этом анизотропия связи С — С была бы неправдоподобно большой.
3*
36
Глава 10
Браунштейн [100] исследовал несколько полизамещенных циклогексанов и наблюдал корреляцию между химическими сдвигами протонов и их удаленностью от объемистых заместителей. Протоны, расположенные рядом с заместителями, дезэкранированы, причем степень дезэкранирования пропорциональна числу и близости заместителей. Он показал, что спектры протонного резонанса гексазамещенных циклогексанов с одинаковой стереохимией идентичны независимо от того, являются заместителями атомы хлора или гидроксильные группы. В 1,2,3,4,5,6-гексазамещенных циклогексанах индуктивное влияние заместителей на протоны постоянно и любые изменения химических сдвигов являются отражением эффектов дальнего экранирования. Несмотря на то что Браунштейн объясняет свои результаты в терминах «пространственных препятствий», он считает, что наблюдаемые химические сдвиги обусловлены влиянием магнитной анизотропии соседних связей. В табл. 10.21 представлены химические сдвиги протонов для ряда аналогичных стереоизомеров
Таблица 10.21
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ НЕКОТОРЫХ 1,2,3,4,5,6-ГЕКСАЗАМЕЩЕННЫХ ЦИКЛОГЕКСАНОВ ПРИ 40 Мгц [100]
Изомеры 1,2,3,4,5,6-гексахлор-циклогексана (растворы в CH2CI2) Химический сдвиг а 6, м. д. Инозитолы (изомеры 1,2,3,4,5,6-гексаоксицикл огексаи а; OD-производные) ® Химический сдвиг а б, м. д. (6—0,85), м. д.
а 4-0,58 L + 1,58 +0,73
4-0,98 + 1,98 + 1,13
₽ 4-1,30 сцилло +2,05 + 1,20
У 4-0,63 муко + 1,53 +0,68
6 4-1,18 Мио + 1,88 + 1,03
4-0,65 + 1,38 +0,53
£ 4-0,80 трео + 1,18 4-0,33
4-0,63 эпи + 1,78 +0,93
+ 1-.73 +0,88
+ 1,40 +0,55
цис + 1,53 4-0,68
алло + 1,38 4-0,53
а Химический сдвиг протонов измерен относительно CH2CI2, взятого В качестве внешнего эталона. Т = 4,7 " (°чень приближенное соотношение).
& Инозитолы были исследованы в растворе D2O.
инозитолов и гексахлорциклогексанов (оба ряда соединений являются 1,2,3,4,5,6-замещенными производными циклогексана). Если из химических сдвигов всех инозитолов вычесть постоянное значение 0,85 м. д. для того, чтобы сделать поправку на различие в индуктивных эффектах гидроксильной группы и хлора, то полученные значения химических сдвигов будут хорошо согласовываться со значениями для гексахлорпроизводных. В молекулах, для которых были измерены значения 6ае, наблюдалось хорошее совпадение этих значений. Это наблюдение позволяет предположить, что эффекты, обусловленные магнитной анизотропией связей С — С1 и С — О, в этих молекулах одинаковы 1).
*) В работах [509, 510] показано, что анизотропия неподеленных пар электронов атомов О и S и связей С — О, S — Ои5 = О может приводить в циклических системах, аналогичных циклогексановым (циклические сложные эфиры), к инверсии химических сдвигов экваториальных и аксиальных протонов.— Прим. ред.
Корреляции спектров ЯМР-Н1 со строением молекул
37
Константы взаимодействия в производных циклогексана. Машер [377] исследовал спектр протонного резонанса 1,1,4,4-тетраметилциклогексил-г(нс-2,6-диацетата (см. рис. 10.9), который может иметь два конформера: один с обеими ацетатными группами в аксиальных положениях, а другой — в экваториальных. Так как энергия конформера с обеими ацетатными группами в экваториальных положениях существенно меньше энергии диаксиального конформера, молекула большую часть времени существует в энергетически более выгодной диэкваториальной форме. Поэтому можно считать эту молекулу жесткой (в смысле усреднения по времени). Жесткая молекула симметрична относительно аксиальной плоскости, проходящей через 1,4-атомы углерода,
Р н с. 10.9. Спектр протонного резонанса 1,1,4,4-тетраметнлцнклогексил-цис-2,6-диацетата прн 60 Мгц [377].
Химические сдвиги измерены относительно гексаметилциклогексаиа, взятого в качестве внутреннего эталона. Области АВ и X в спектре записаны в различных шкалах интенсивности.
и три протона с каждой стороны этой плоскости можно рассматривать как отдельные идентичные системы АВХ (поскольку взаимодействие между двумя системами АВХ не осуществляется). Детальный анализ спектра протонного, резонанса этого соединения (см. рис. 10.9) дает' следующие параметры:
7ав = С-“= 12,36 ±0,2 гц-
4ах = С1»= 12,35 ±0,1 гц-
•/ВХ = С1, = 4>25±°,1 гц-
va=—1,269 ± 0,003 м. д. vb = — 1,400 ± 0,003 м. д.
Ух=—4,681 ±0,003 м. д.
измерены относительно
> гексаметнлднснлоксана как внутреннего эталона
Таким образом, различие в химических сдвигах аксиальных и экваториальных протонов составляет 0,131 м. д., причем аксиальные протоны дают сигналы в более высоком поле. Измеренные значения и J™4 для 1,1,4,4-тетра-метилциклогексил-г(ис-2,6-диацетата довольно плохо согласуются с теоретическими значениями, полученными Карплусом по методу валентных связей (^ааЦ = 9,2 гц и JaeU = 1,7 гц). Значение хорошо согласуется со значением, полученным для дейтерометана (7нн = 12,4 гц) [102]. Интересно отметить, что константа вицинального взаимодействия двух аксиальных протонов (Jaa1, или Jах) в три раза больше константы вицинального взаимодействия аксиального и экваториального протонов (Jea4, или 7вх)- Аналогичные
38
Глава 10
наблюдения [478] были сделаны при исследовании диоксана [101] х) и ацетилированных сахаров [97], в которых конформации «кресла» аналогичны существующим в циклогексанах; в случае сахаров значения и J™'1 равны 2—4 гц, а Уда4 составляют 5—8 гц [97].
Конфигурационные эффекты в ацетилированных углеводах. Лемье и сотр. [41, 97, 103] наблюдали большое число интересных конфигурационных эффектов в спектрах протонного резонанса ацетилированных углеводов, которые в конформационном отношении подобны циклогексанам. Ацетилированные
гц
Рис. 10.10. Спектры протонного резонанса при 40 Мгц [97].
а — 1а,3а-диметокси-2р-ацетоксициклогексаи; б — 1а, ЗР-диметокси-2а-ацетоксицикло-гексан. Оба соединения исследованы в растворе в хлороформе; растворитель использовался в качестве внутреннего эталона. А и В — спектры, снятые при разной степени усиления.
производные удобны для исследования, ибо они не усложняют спектров кольцевых протонов нежелательной тонкой структурой спин-спинового взаимодействия. Наиболее интересные моменты проведенного исследования заключаются в следующем:
1) Для метильных групп в экваториальных и аксиальных ацетатных заместителях наблюдалось различие в химических сдвигах приблизительно на 0,13—0,25 м. д. Например, в протонном спектре фиксированной молекулы пентаацетата a-D-глюкопиранозы, приведенном на рис. 10.11, в, в области сильного поля видны два резонансных сигнала метильных групп, отстоящих
Изучение конформации молекул 1,3-диоксанов, 1,3-диоксоланов и некоторых циклических сложных эфиров с помощью спектров протонного резонанса было проведено в работах [511—514].— Прим. ред.
Корреляции спектров ЯМР-Н1 со строением молекул
39
друг от друга на 0,15 м. д. Менее интенсивный сигнал в более слабом поле обусловлен (судя по его интенсивности) одиночной аксиальной ацетатной группой, тогда как интенсивный сигнал в более сильном поле относится к метильным протонам экваториальных ацетатных групп. Такая интерпретация подтверждается спектрами большого числа ацетилированных углеводов, а также 1,2,3,4,5,6-гексаацетоксициклогексанов известной конфигурации.
2) Химические сдвиги экваториальных протонов на 0,20 м. д. больше химических сдвигов аксиальных протонов (см. рис. 10.10).
3) Пара геминальных протонов (АВ) у углеродного атома в а-положении (по отношению к кислороду кольца) в тетраацетате P-D-ксилопиранозы различается по химическим сдвигам гораздо сильнее (0,61 м. д.), чем это обычно бывает для экваториально-аксиальной пары протонов. По-видимому, такое большое различие в химических сдвигах обусловлено влиянием магнитной анизотропии связи С — О.
Различие в химических сдвигах, от 0,25 до 0,65 м. д. для экваториальных и аксиальных протонов у а-углеродного (по отношению к кислороду кольца) атома наблюдалось также в спектрах некоторых аномерных пар ацетилированных альдопираноз (аномеры отличаются друг от друга тем, что ацетатная группа в a-положении по отношению к кислороду кольца аксиальна в одном аномере и экваториальна в другом).
4) В молекулах типа циклогексана, имеющих жесткую структуру, константы спин-спинового взаимодействия двух аксиальных протонов (аа) у соседних атомов углерода в два-три раза превышают константы взаимодействия вицинальных протонов, имеющих какую-либо другую ориентацию (ае или ее).
На основе приведенных эмпирических принципов можно получать значительную полезную информацию как о конфигурации, так и о конформации производных циклогексана. Кроме того, большую пользу может принести наблюдение относительной ширины линий в спектрах протонного резонанса производных циклогексана [23, 104—106]. В молекулах, претерпевающих быстрые взаимопревращения, константы взаимодействия кольцевых протонов имеют значения, средние между константами взаимодействия в фиксированных конформациях, что и обусловливает меньшую ширину линий для молекулы по сравнению с конформерами. В некоторых спектрах широкие резонансные сигналы жесткого конформера обусловлены перекрыванием сигналов с близкими химическими сдвигами. Быстрые взаимопревращения молекул усредняют химические сдвиги экваториального и аксиального протонов, и это также приводит к сужению линий.
цис- и транс-^-трет-Бутилциклогексанолы и ацетаты. Молекулы этого типа существуют в известных конформациях, в которых объемистая трет-бутильная группа занимает предпочтительно экваториальное положение. Лемье и сотр. [97] исследовали протонные спектры этого класса молекул и опубликовали данные по ширине линий резонансных сигналов протона в положении 1. Было показано, что полуширина этого сигнала в rfizc-спирте и rfizc-ацетате (~7 гц) составляет лишь треть от полуширины сигнала для /пранс-изомеров (~22 гц при 40 Мгц). Причина уширения линий заключается, по-видимому, в неразрешенной мультиплетной структуре, обусловленной спин-спиновым взаимодействием, и так как константы спин-спинового взаимодействия Н — Н значительны для ядер, разделенных вплоть до трех связей, то аксиальный 1-протон в транс-изомерах более сильно взаимодействует с соседними метиленовыми протонами, чем в rfuc-изомерах. Это является следствием того, что аксиальные протоны у соседних атомов углерода имеют константы взаимодействия гораздо большие, чем протоны в других ориентациях х).
-1) Измерение ширины линий резонансных сигналов протонов для установления конфигурации производных rnpern-бутилциклогексапа и циклогексанола было проведено также в работе [515].— Прим. ред.
40
Глава 10
Определение конфигурации производных циклогексана методом ЯМР. Исследование констант взаимодействия кольцевых протонов в спектрах протонного резонанса двух диастереомерных 1,3-диметокси-2-ацетоксициклогек-санов дало возможность определить их конфигурации [41, 97].
1) 1а.,За-Диметокси-2^>-ацетоксициклогексан. Спектр протонного резонанса этой молекулы приведен на рис. 10.10, а; он содержит полную информацию о константах взаимодействия кольцевых протонов в циклогексановых системах. Резонансные сигналы протонов в положениях 1 и 3 перекрываются с резонансным сигналом метоксигруппы, а сигнал протона в положении 2 проявляется в виде хорошо разрешенного триплета (</ааЧ =9 гц), обусловленного спин-спиновым взаимодействием с протонами в положениях 1 и 3.
Рис. 10.11. Спектры протонного резонанса некоторых ацетилированных альдопентапираноз при 40 Мгц [97].
2) 1а,3^>-Диметокси-2а-ацетоксициклогексан. В спектре протонного резонанса этой молекулы, показанном на рис. 10.10, б, мультиплет для протона в положении 2 проявляется в виде полос при 107,5; 110,0; 113,8 и 116,5 гц относительно хлороформа, используемого в качестве внутреннего эталона. Три протона у соседних атомов углерода образуют спиновую систему АВХ, и квартет протона в положении 2 является Х-частью спектра. Приближенные значения для констант взаимодействия соседних аксиально-аксиальных (Jaa = = 6,4 гц) и соседних аксиально-экваториальных (Jae = 2,6 гц) протонов могут быть получены прямым вычитанием соответствующих сигналов в квартете. Информацию такого рода в некоторых случаях можно использовать для установления конфигурации производных циклогексана.
Ацетилированные альдопентапиранозы. На рис. 10.11 представлены спектры протонного резонанса некоторых ацетилированных альдопентапираноз.
Корреляции спектров ЯМР-Н1 со строением молекул
41
Все они для удобства могут быть разбиты на четыре отдельные области. Аномерные протоны (у замещенного ацетатной группой атома углерода, соседнего с кислородом кольца) дают сигнал с относительной интенсивностью 1 в области ~3,9—4,7т. Их слабое экранирование обусловлено тем, что атом углерода, у которого они находятся, связан с двумя атомами кислорода. Существенным подтверждением правильности отнесения сигнала является расщепление его в дублет вследствие сильного взаимодействия аномерных протонов с одиночным протоном в положении 2. Во всех случаях величина этой константы взаимодействия хорошо согласуется с предсказанными для известных ориентаций двух ядер величинами. Три протона у вторичных атомов углерода дают сигналы соответствующей интенсивности в области от ~4,6 до 5,4т, тогда как
Рис. 10.12. Спектр протонного резонанса тетра-О-ацетил-1,4-дидезокси 1,4-динитронеоинозитола при 60 Мгц [107].
Растворитель — CDCh» эталон — Si(CH3)<-
сигналы метиленовых протонов ацетилированных альдопентапираноз нахо-дятся в области ~5,8—6,5т. Наконец, очень узкие сигналы в области ~8,0— 8,5т обусловлены метильными протонами ацетатных заместителей. Таким образом, изучение ЯМР подтверждает циклические структуры и аномерные конфигурации, предложенные для этих молекул.
1,2,3,4,5,6-Гексахлорциклогексаны. Лемье и сотр. [97] исследовали спектры протонного резонанса для пяти изомеров 1,2,3,4,5,6-гексахлорциклогексанов. Все протоны в p-изомере эквивалентны, и поэтому наблюдается одиночный резонансный сигнал. В симметрично замещенном у-изомере быстрое взаимопревращение конформеров усредняет экранирование протонов, что также обусловливает появление одиночного сигнала. Как 6-, так и е-изомер дают простые спектры, в которых экваториальные протоны экранированы слабее аксиальных. Очень сложный спектр наблюдается для «-изомера (система АА' ВВ' СС') с наиболее тонкой структурой для резонансных сигналов «-протонов. Аналогичные спектры были получены для сходных гексаацетатов.
Тетра-О-ацетил-1,4-дидезокси-1,4-динитронеоинозитолы. Лихтентхалер и Фишер [107] применили метод ЯМР для определения конфигурации нескольких новых производных циклогексана. Исследование протонного спектра тетра-О-ацетил-1,4-дидезокси-1,4-динитронеоинозитола показывает, что молекула имеет нео-1,4-конфигурацию, как показано на рис. 10.12. Сильнопольный дублет обусловлен двумя различными типами метильных групп в ацетатных заместителях, причем две ацетатные группы находятся в экваториальном и две — в аксиальном положениях. Шесть кольцевых атомов водорода образуют три
42
Глава 10
пары эквивалентных протонов (А, В и Е). Н(А) и Н(В) дают пару дублетов равной интенсивности; большее расщепление, наблюдаемое в каждой паре дублетов, является характеристическим для аксиально-аксиального спин-спинового взаимодействия, тогда как меньшее взаимодействие обусловлено взаимодействием соседних экваториальных протонов. Это позволяет предположить, что протоны Н(А) и Н(В) занимают четыре аксиальных положения, тогда как протоны Н(Е) находятся в двух экваториальных положениях. Резонансный сигнал двух эквивалентных экваториальных протонов Н(Е) представляет собой триплет вследствие спин-спинового взаимодействия с двумя соседними аксиальными протонами Н(А) и Н(В).
На основании исследования спектров протонного резонанса была определена конфигурация ряда дезоксиаденозинов [108].
Производные ацетоксихолестанона. Уильямсон и Джонсон [109] исследовали ряд изомерных а-ацетоксикетонов (производных ацетоксихолестанона-3 и ацетоксихолестанона-2) и обнаружили существенно большие константы цис- и транс-взаимодействия Jeu4, чем наблюдавшиеся ранее для циклогексановых производных. Они нашли, что Jae имеют значения до 7,4 гц, a Jaa — до 13,1 гц. Авторы сочли необходимым изменить параметры в уравнениях Карплуса (10.4) и (10.5), описывающих зависимости констант взаимодействия J0'14 от двугранных углов, с тем чтобы получить разумные значения валентных углов. Для некоторых молекул оказалось, что протоны в экваториальных положениях дают сигналы в более высоком поле, чем соответствующие аксиальные протоны, что находится в противоречии с обычным поведением таких протонов. Наблюдаемое явление было объяснено влиянием жесткости кольца, а также вкладом в химический сдвиг магнитной анизотропии карбонильной группы (см. структуру I)
Производные монозамещенных циклогексанов. При комнатной температуре циклогексилгалогениды существуют в виде смеси быстро взаимопревращаю-щихся конформеров с объемистыми заместителями, проводящими большую часть времени в экваториальном положении; подробности такого процесса уже обсуждались (см. разд. 9.7) [ПО, 111]. Аналогичное явление имеет место для метилциклогексанов. При исследовании протонных спектров метилциклогексана и его 2,2,6,6-тетрадейтерированного аналога (см. рис. 10.13) было показано, что константа взаимодействия метильных протонов с протоном, связанным с тем же атомом углерода, 7сн3-н равна 6.8 гц.
Расщепление резонансного сигнала метильных протонов в дублет в спектре метилциклогексана составляет лишь 4,7 гц, что кажется несовместимым с остальной тонкой структурой спектра. Анет [112] показал, что в насыщенных соединениях, содержащих группы СНСН3, очень часто расщепление резонансных сигналов метильных протонов в дублет не равно константе взаимодействия 7сн-сн3, даже если химический сдвиг между двумя типами ядер велик по сравнению с соответствующей константой спин-спинового взаимодействия. Это происходит потому, что необходимо рассматривать и подвергать детальному анализу влияние других магнитных ядер, сильно взаимодействующих с протонами СН или СН3. При соответствующем дейтерировании молекулы можно отделить фрагмент СНСН3 от других протонов молекулы и тогда наблюдается более резкое дублетное расщепление метильного сигнала (см. спектр
Корреляции спектров ЯМР-Н1 со строением молекул
43
протонного резонанса 2,2,6,6-тетрадейтерометилциклогексана, показанный на рис. 10.13). Сообщение Машера [105] о неожиданно малых значениях ^сн-сн3 в диметилциклогексанах теперь можно понять в свете работы Анета. Аналогичный эффект наблюдается также в спектре 3-метилциклогексанона, в котором широкий сильнопольный сигнал при 9,0т, обусловленный СН3-протонами, при соответствующем дейтерировании молекулы превращается в узкий дублет [65, 112].
Дизамещенные циклогексаны. Браунштейн и Миллер [106] показали, что методом ЯМР можно определить стереохимию дизамещенных циклогексанов при условии, что положения заместителей известны. Быстрое взаимопревращение производных циклогексана может привести к усреднению магнитного
8,0 9,0
Р и с. 10.13. Спектры протонного резонанса раствора в СС14 при 60 Мгц [112]. а — метнлцнклогексан; б — 2,2,6,6-тетрадейтерометилинклогексаи. Сигналы метильных протонов исследованы прн медленном прохождении и показаны справа.
окружения кольцевых протонов таким образом, что для ядер, которые в фиксированных конформерах были бы экранированы в различной степени, наблюдается одиночный резонансный сигнал. Другим, более тонким следствием быстрого взаимопревращения является его влияние на константы спин-спинового взаимодействия протонов у соседних атомов углерода. В фиксированной конформации такое взаимодействие гораздо больше, чем в быстро взаимопре-вращающейся молекуле. Влияние спин-спинового взаимодействия сказывается главным образом на уширении сигналов кольцевых протонов, а результатом быстрого взаимопревращения будет сужение резонансных сигналов. Поэтому при исследовании протонных спектров цис-транс-пары дизамещенных циклогексанов известной стереохимии всегда более узкие сигналы наблюдаются для молекул, способных к взаимопревращению. Из дизамещенных молекул наиболее часто претерпевают взаимопревращения такие конформеры, в которых
44
Глава 10
заместители расположены в аксиально-экваториальных положениях по отношению друг к другу. Для метильных заместителей такой случай осуществляется в цис-1,2-, транс-1,3- и цис-1,4-диметилциклогексанах, для которых в результате быстрого взаимопревращения молекул наблюдаются узкие резонансные сигналы [105]. Наоборот, транс-1,2-, цис-1,3- и транс- 1,4-соединения должны существовать главным образом в виде молекул с обеими метильными группами в экваториальных положениях и в их спектрах вследствие отсутствия быстрого взаимопревращения будут наблюдаться широкие резонансные сигналы. В спектрах протонного резонанса диметилциклогексанов кольцевые протоны молекул типа ее дают широкие слабопольные резонансные сигналы (ширина линий до 10—15 гц при 40 Мгц), в то время как для быстро взаимопре-вращающихся молекул типа ае в этой области наблюдаются относительно-узкие резонансные сигналы (ширина линий до 5—9 гц при 40 Мгц). Таким образом, если положения заместителей известны, то измерения ширины линий могут дать информацию о конфигурации молекулы. В табл. 10.22 приведена
Таблица 10.22
ШИРИНА РЕЗОНАНСНЫХ СИГНАЛОВ КОЛЬЦЕВЫХ ПРОТОНОВ В СПЕКТРАХ ДИЗАМЕЩЕННЫХ ЦИКЛОГЕКСАНОВ ПРИ 40 Мгц [105]
Соединение Конформация Ширина линии, гц
Метилциклогексанол
цис-1,2 а, е 8,4
транс-]. ,2 е, е 22,0
цис-1,3 е, е 37,8
транс-1,3 а, е 12,5
цис-1,4 а, е 14,3
транс-1,4 е, е 38,4
Диметилциклогексан
цис-1,2 а, е 4,8
транс-1,2 е, е 14,1
цис-1,3 е, е , 12,8
транс-1,3 а, е 8,6
цис-1,4 а, е 4,8
транс-1,4 е, е 19,2
Циклогександиол
цис-1,2 а, е 7,1
транс-1,2 е, е 20,2
Циклогександиацетат
цис-1,2 а, е 10,3
транс-1,2 е, е 35,0
Циклогександибензоат
транс-1,2 е, е 16,0
ширина резонансных сигналов кольцевых протонов для некоторых дизамещен-ных циклогексанов известной конфигурации, причем во всех случаях наблюдается совпадение ширины линий с вышеприведенными рассмотрениями. Маллер и Тош [95] с успехом исследовали конформации быстро взаимопревра-щающихся дизамещенных циклогексанов типа ае, охлаждая образцы до температуры ниже —100°х).
х) Стереохимия монохлорпроизводных метил-, этил- и диметилциклогексанов исследовалась с помощью спектров ЯМР-Н1 в работе [516].— Прим. ред.
Корреляции спектров ЯМР-Н1 со строением молекул
45
Декалины. Конформации цис- и транс-декалинов находят свое отражение в спектрах ЯМР. Дыс-декалин существует в виде быстро взаимопревращающей-ся молекулы и дает одиночный узкий резонансный сигнал; транс-декалин имеет жесткую структуру и дает широкий, плохо разрешенный резонансный сигнал, приведенный на рис. 9.25. При охлаждении раствора цыс-декалина в сероуглероде до —117° одиночный резонансный сигнал, наблюдаемый при комнатной температуре, превращается в дублет вследствие «замораживания» конформаций [95].
10-Метилдекалолы. Машер [23] исследовал спектры протонного резонанса четырех изомеров 10-метилдекалолов-2, и полученные результаты (см.
Таблица 10.23
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ а АНГУЛЯРНЫХ МЕТИЛЬНЫХ ГРУПП НЕКОТОРЫХ
10-МЕТИЛДЕКАЛОЛОВ [23]
Соединение б, м. д. т-Значе-ния
1О-Метил-цис-декалол-2-цис -0,927 9,02
10-Метил-чис-дека лол-2 -транс -0,900 9,05
10-Метил-транс-декалол-2-цис -0,784 9,17
10-Метил-тра«с-декалол-2-транс -0,758 9,19
а Химические сдвиги измерены в миллионных долях (м. д.) относительно гексаметилдисилоксана (ГМДС), взятого в качестве внутреннего эталона; т = 9,95 4- брмДС'
табл. 10.23) отчетливо указывают на существенную зависимость экранирования ангулярных метильных групп от конфигурации кольца. Резонансный сигнал ангулярной метильной группы в спектре транс-декалола сдвинут на 0,142 м. д. в область более сильного поля от соответствующего сигнала для /{ыс-декалола. Это наблюдение оказывает существенную помощь при отнесении С-метильных групп в стероидах. Небольшое дополнительное влияние на
10-Метил-транс-декалол-2-цис 10-Метил-цис-декалол-2 цис
экранирование метильных протонов оказывают гидроксильные группы в цис-и транс-положениях по отношению к метильной группе (если гидроксильная группа при С2 расположена в цыс-положении по отношению к метильной группе, то резонансный сигнал СН3-протонов сдвинут на 0,026 м. д. в область более слабого поля по сравнению с тем случаем, когда оба заместителя находятся в транс-положении). Машер [105] наблюдал аналогичную картину и для экранирования метильных групп в метил циклогексанолах.
46
Глава 10
АЛКЕНЫ
10.4. Винильные производные СН2 = СНХ
Исследованию спектров ЯМР винильных соединений было уделено много внимания, и ряд исследователей наблюдали корреляции между спектральными и молекулярными параметрами этой системы. В винильном производном
(В)Н Н(А)
С = С
(Сщ/ \
имеются три магнитно неэквивалентных протона, образующих типичную спиновую систему АВС. Проведение полного анализа этой системы хотя и затрудни-
Р и с. 10.14. Спектр протонного резонанса бромистого винила при 60 Мгц [ИЗ].
~ 12,2 гц; Vg — 42,6 гц;
vG = 50,4 гц; Jдв — 7,0 гц;
JAG = 14,8 гц; == — 1,8 гц.
тельно, но дает нам полные сведения о величинах и относительных знаках констант спин-спинового взаимодействия </дв, /вс и Jac, а также величинах химических сдвигов трех ядер. Спектр протонного резонанса чистого бромистого винила [113], приведенный на рис. 10.14, является типичным для системы АВС. Двенадцать из пятнадцати возможных переходов дают в спектре при 60 Мгц сигналы измеримой интенсивности. Отнесение сигналов в такого рода спектрах невозможно без знания типичных значений констант взаимодействия в ненасыщенных системах.
Корреляции спектров ЯМР-Н1 со строением молекул
47
При исследовании спектров различных дизамещенных олефинов можно найти относительные величины трех констант взаимодействия; было показано, что для винильных производных, в которых заместитель не является металлом, константа геминального взаимодействия имеет наименьшее значение, а константа транс-взаимодействия — наибольшее значение: (от —3,5 до
— 2,5 гц) < Jab (от+4,0 до + 12,0 гц) < JacT"0 (от +12 до +19 гц). Значения констант вицинального взаимодействия находятся в весьма хорошем качественном согласии с теоретически предсказанными значениями [39, 621 (J4«c __ 6,1 гц и jm-ранс _ 11,9 гцу Некоторые типичные константы
Таблица 10.24
НЕКОТОРЫЕ ТИПИЧНЫЕ КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ В ОЛЕФИНОВЫХ СОЕДИНЕНИЯХ [30, 114]
Соединение Константы взаимодействия протонов, гц
,гем JHH .транс JHH JHH
mpa«c-CeH5CH = СНСНО 15,6
тра«с-СН3СН = СНСНО 16,2
Н2С = СВгСН2Вг 2,1
Н2С = С(ОСН3)С(ОСН3) = сн2 2,1
цис-СвН5СН = СНСОСНз 12,0
цис-СН3СО2СН = CHCN [114] 11,5
mpa«c-CH3CO2CH = CHCN [114] 15,0
в дизамещенных олефинах [301 приведены в табл. 10.24. В некоторых системах константы взаимодействия обнаруживают существенную зависимость от природы растворителя [474—477].
10.4.1. Корреляция между константами геминального взаимодействия Jhh и валентными углами Н — С — Н
Гутовский и сотр. [30] показали, что между константой взаимодействия Jhh и валентным углом Н — С — Н в СН2-группе существует корреляция. Они применили метод валентных связей и вывели кривую зависимости рассчитанных значений Jhh от валентных углов Н — С — Н. На основании имеющихся экспериментальных данных для алканов и алкенов можно показать, что теоретическая кривая плохо согласуется с наблюдаемыми константами. Это расхождение особенно наглядно в случае производных алканов, для которых теория предсказывает большие положительные значения Jhh в противоположность экспериментально наблюдаемым отрицательным значениям [32, 58]. Для алкенов расчеты оказались более успешными, поскольку они предсказывают уменьшение значений Jhh с увеличением валентных углов Н — С —-Ни объясняют появление как отрицательных, так и положительных небольших констант взаимодействия. Однако очевидные ошибки, допущенные при теоретическом рассмотрении в случае алканов, делают расчет некорректным. Более точные предсказания валентных углов в группе СН2 на основании констант взаимодействия Jhh в дальнейшем, вероятно, удастся получить при совершенствовании общего теоретического рассмотрения констант взаимодействия геминальных протонов.
48
Глава 10
10.4.2. Константы вицинального взаимодействия в алкенах
Расчеты констант вицинального взаимодействия протонов проведены с большим успехом, чем аналогичные расчеты констант геминального взаимодействия [39].
Карплус [39] предсказал, что константы взаимодействия 7нп"1С и +/+ должны содержать вклады о-связей величиной + Н,9и+6,1гц соответственно, а также вклады л-связей величиной + 1,5 и 4-1,5 гц соответственно (последний вклад обусловлен о — ^-конфигурационным взаимодействием) [62]. Несмотря на то что рассчитанные значения (13,4 и 7,6 гц) для транс- и цис-констант взаимодействия несколько ниже экспериментальных значений (19,0 и 11,7 гц)
[42],
предсказанное
отношение
juuc jmpanc
является правильным.
Аналогичная
ситуация наблюдается для транс- -и гош-констант вицинального взаимодей-
ствия в случае этана.
Гутовский и Порт [116] рассчитали влияние изменения валентного угла Н — С — С на константы вицинального взаимодействия в замещенных этиленах. Изменение угла от 120 до 125° должно привести к уменьшению вклада о-связи в значение </цн от +6,1 до +4,0 гц. На основании этого можно объяснить константы взаимодействия орто-протонов в семи-, шести- и пяти-член-ных ароматических кольцах (12,7 и 3,7 гц соответственно [117]).
Значения и ++ в винильных соединениях СН2 = СНХ линейно зависят от электроотрицательности заместителя X согласно соотношениям
JgPa«c= 19,0—3,ЗДе (гц), 7НН=11’7—4,0Д£ (гц),.
(10.8)
(10.9)
где 19,0 и 11,7 гц — наблюдаемые транс- и цис-константы взаимодействия в этилене; АЕ — разность электроотрицательностей заместителя X и атома водорода [34, 108, 119, 120].
Транс- и цис-константы взаимодействия в винильных системах более чувствительны к изменениям электроотрицательности заместителя, чем константы вицинального взаимодействия в насыщенных системах [34].
Знаки констант взаимодействия в ненасыщенных системах. С помощью определения относительных знаков констант взаимодействия Н — Ни С13 — Н в алкенах Лаутербур и Курланд [32] смогли вывести абсолютные знаки констант взаимодействия Н — Н (см. табл. 10.25).
Их выводы основаны на предположении, что константы взаимодействия <7с1з-н между непосредственно связанными ядрами положительны.
Интересным примером детального изучения, в котором для определения относительных знаков констант взаимодействия были применены и анализ спектра, и двойной резонанс, является исследование 1-хлорбутадиена-1,2 (I)
(Х)Н3С С1
С = С = С
(B)HZ \(А)
Детальный анализ [176] спектров ЯМР-Н1 при 40 и 60 Мгц позволяет определить только относительные знаки Jах и 4х, тогда как двойной резонанс [177] дает лишь относительные знаки JАВ и jBX; таким образом определяются относительные знаки всех констант (+/дх, +/вх и —J'ab)-
Корреляции спектров ЯМР-Н1 со строением молекул
49
Таблица 10.25
АБСОЛЮТНЫЕ ЗНАКИ И ЗНАЧЕНИЯ НЕКОТОРЫХ КОНСТАНТ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ В НЕНАСЫЩЕННЫХ СОЕДИНЕНИЯХ
Знаки констант взаимодействия протонов в X—СН=СНз
X J, гц Литература
цис транс гем
н + П,7 + 19,1 +2,5 42
СН3 + 10,02 + 16,81 +2,08 124
СН3СН2 + 10,4 + 17,4 + 1,9 125
(СН3)2СН + 10,4 + 17,3 + 1,6 118
(СНз)зС + 10,8 + 17,5 + 1,4 125
СС13 + 10,07 + 16,05 —0,42 126
CeHs +и,о + 17,8 + 1,1 127
2,4,6-(СН3)3СвН2 + 11,6 + 18,0 +2,3 127
CN + 11,8 + 18,0 + 1,0 118
СН3О +6,8 + 14,4 -2,2 118
СН2 = СНО +6,4 + 14,0 -1,8 118
СН3СОО +6,5 + 14,1 -1,5 127
Константы взаимодействия протонов в системах
—сн=сн—сн—
Соединение J а Jb J с Литература
цис транс цис транс
сн3—сн = сн2 + 10,02 + 16,81 +6,40 -1,75 —1,33 124
траис-СНзСН = СН — СНО + 15,3 Р6,80 — 1,60 128
триис-СНз — СН = СН—СООН + — 128
цис-СН2С1 —СН = СНС1 +7,20 + 7,2 , —0,9 129, 130
транс-СН2С1 — СН = CHCI + 13,1 +7,1 —0,5 129, 130
транс-СНз—СН = СН — С6Н6 +6,6 — 1,8 131
10.4.3. Корреляция между константами взаимодействия и электроотрицательностями заместителей в ванильных производных
Брюгель и сотр. [132] провели анализ спектров протонного резонанса 131 винильного производного. Полученные ими результаты широко использовались для демонстрации существования корреляций между параметрами спектров ЯМР и структурой молекул. В табл. 10.26 собраны значения констант взаимодействия в винильных производных для большого ряда заместителей, различных по своей электроотрицательности [119]. Если построить зависимость одной из трех констант взаимодействия /ни, Jhh или J^aHC от электроотрицательности заместителя Ех, то будет наблюдаться обратно пропорциональная зависимость этих двух параметров [118, 119]. На рис. 10.15 показаны три приблизительно линейные зависимости J от Ех- Однако неясно, будет ли линейной такая зависимость в общем случае. Значения J для бромистого винила намного меньше значений, предсказанных линейным соотношением. Разброс на различных графиках может быть обусловлен неточностями в определении констант взаимодействия (поскольку для нахождения констант взаимодей-
4 — 489
50
Глава 10
Таблица 10.26
КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ ПРОТОНОВ ВИНИЛЬНЫХ ПРОИЗВОДНЫХ [119]
X £x Число изученных образцов гц гц jmpaHc гц Литература
F 3,95 a 3 —3,2 +4,65 + 12,75 132, 133
30, 120
С1 3,2a 2 — 1,4 +7,3 + 14,6 132, 133
Вг 3,0a 4 — 1,8 +7,1 + 15,2 132, 133
113, 30
OR (алкил) 3,5a 17 — 1,9 +6,7 + 14,2 132, 133
OR (арил) 3,5 a 13 —1,5 +6,5 + 13,7 132
OOCR 3,5 6 12 — 1,4 +6,3 + 13,9 132
Фосфатная группа 3,56 5 —2,3 4~5,8 + 13,2 132
no2 3,35 B 1 —2,0 +7,6 +15,0 132
nr 3,06 5 0 +9,4 + 16,1 132, 30
COOR 2,56 14 + 1,7 + Ю,2 + 17,2 132
CN 2,56 1 +1,3 + 11,3 + 18,2 134
COR 2,56 2 + 1,8 +11,0 + 18,0 132, 30
R (алкил) 2,56 18 + 1,6 +ю,з + 17,3 132, 124,
R (арил) 2,56 9 + 1,3 +н,о + 18,0 132
Пиридил 2,56 3 + 1,1 + 10,8 + 17,5 132
Сульфониевая группа 3,0b 9 —0,6 +9,9 + 16,6 132
Sn 1,9 a 4 +2,8 + 14,1 +20,3 132, 135.
As 2,1 a 4 + 1,7 + H,6 + 19,1 132
Sb 2,0a 1 +2,0 + 12,6 + 19,5 132
Pb 1,96 1 +2,0 + 12,1 + 19,6 132
Hg 1,96 1 +3,5 + 13,1 +21,0 135
Al : O(C2H5)2 1,56 1 +6,3 + 15,3 +21,4 135
Li 1,06 1 +7,1 + 19,3 +23,9 136
Ge 1,0 1 +ю,о + 16,0 +ю,о 427
а По данным работы [5].
б По данным работы [155].
в По данным работы [137].
ствия при анализе системы АВС необходимо проведение большого объема; вычислительных работ); это подтверждается большим разбросом на графике /нн — Ех, где неточности определения должны быть больше вследствие малых значений Jhh- Наилучшая корреляция наблюдается для J^h"10 и где ошибки влияют наименьшим образом. Однако при построении зависимости суммы трех констант взаимодействия SJ = (Jhh + Жн + Jhh ис) (гц) от электроотрицательности заместителей Ех (см. рис. 10.16) было получено убедительное доказательство того, что разброс на рис. 10.15 обусловлен неточностями в измерении констант взаимодействия [30, 119, 132]. Несмотря на то что сумма 2J определяется точно при непосредственном изучении спектра, на линейном графике существует значительный разброс. Все же приближенное линейное соотношение оказывает существенную помощь для определения правильного значения SJ, когда имеются какие-то сомнения (как, например, в случае тетравинилсилана [132, 138, 442]). Следствием линейной зависимости, которая существует между отдельными константами взаимодействия и электроотрицательностью заместителя £х, является линейная зависимость между самими константами взаимодействия. На рис. 10.15 все три линейные
Рис. 10.15. Зависимости констант взаимодействия Унн в винильных системах СН2 = СНХ от электроотрицательности Ех заместителя X [119].
Рис. 10.16. Зависимость суммы Удд + ^дн + /нн‘НС от электроотрицательности Ех заместителей [119].
4*
52
Глава 10
зависимости имеют одинаковый наклон, исходя из чего Банвелл и Шеппард [118] предположили, что все три константы взаимодействия обусловлены одинаковыми факторами: сильно электроотрицательные группы должны уменьшать электронные плотности вокруг трех протонов винильной группы, что приводит к уменьшению взаимодействия спинов ядер и электронов, а это в свою очередь сказывается на уменьшении значений констант взаимодействия [118]. Для систем Н2С = N — X найдена корреляция значений с электроотрицательностью заместителя X [454].
10.4.4. Изменения химических сдвигов в винильных группах в зависимости от электроотрицательности заместителей
Банвелл и Шеппард [118] пытались скоррелировать изменения различных параметров химического сдвига в винильных системах, а именно бА, А! = = (^в + бс) и Д2 = бА-------(бв + бс), с изменениями электроотрица-
тельности заместителя. В табл. 10.27 приведены значения этих параметров для восьми рассмотренных винильных производных.
Таблица 10.27
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ РЯДА ВИНИЛЬНЫХ СОЕДИНЕНИЙ [118] (А)Н\Г_Г/Н(В) х/ \Н(С)
X Соединение Химические сдвиги
«1 ТА дВ тв дС тс А1 &2
F Фтористый винил —0,84 3,83 + 1,30 5,97 +0,96 5,63 + 1,13 — 1,96
О Винилметиловый эфир —1,10 3,57 + 1,43 6,10 + 1,29 5,96 + 1,36 —2,46
Дивиниловый эфир —1,06 3,61 + 1,12 5,79 +0,81 5,48 +0,96 —2,02
С1 Хлористый винил —0,97 3,70 —0,11 4,56 —0,19 4,48 —0,15 —0,82
Вг Бромистый винил —1,16 3,51 —0,70 3,97 —0,55 4,12 —0,63 —0,53
С Стирол —1,36 3,31 +0,12 4,79 —0,38 4,29 —0,13 — 1,22
Винилцианид —0,20 4,47 —0,72 •3,95 —0,58 4,09 —0,65 +0,45
Бутен-1 — — — — — — — —0,71
3,3-Диметилбутен-1 —- — — — — — — —0,76
З-Мети лбу тен-1 —0,34 4,33 +0,56 5.23 +0,48 5,15 +0,52 —0,86
а Измерены в миллионных долях (м. д.) относительно этилена, химический сдвиг которого составляет — 3,9 м. д. относительно циклогексана (т —4,67); ошибка ±0,05 м. д. Таким образом, т=4,67-|-+6С2Н4* Соединения исследовались в виде 10%-ных растворов в СС1<.
Ни один из параметров химического сдвига винильной группы не коррели-руется с электроотрицательностью по Хаггинсу [5]. Если мы рассмотрим такие углеродсодержащие заместители, как изопропильная, цианидная и фенильная группы, электроотрицательности которых близки, то получим сильно отличающиеся друг от друга значения бА (CN и СеН5 оказывают сильное влияние вследствие диамагнитной анизотропии).
10.4.5. Корреляция химических сдвигов в винильных системах с а-константами Гаммета
Если отложить на графике параметры бА, Ai и А2 в зависимости от констант Тафта [140] ои и ос (вклады индуктивного эффекта и эффекта сопряжения в о-константы Гаммета), то будет наблюдаться довольно хорошая линейная
Корреляции спектров ЯМР-Н1 со строением молекул
53
зависимость между значениями ос и Д2, как это показано на рис. 10.17. Корреляция других параметров химического сдвига с ои и ос менее удовлетворительна. В табл. 10.28 приведены значения электроотрицательностей по Хаггинсу и значения констант Тафта ои и ос для рассмотренных заместителей.
Таблица 10.28
ЗНАЧЕНИЕ ЭЛЕКТРООТРИЦАТЕЛЬНОСТЕЙ Е ПО ХАГГИНСУ И КОНСТАНТ ТАФТА аи И ас ДЛЯ РЯДА ЗАМЕСТИТЕЛЕЙ [118]
Е F>O>Cl>Br>C 3,9 3,5 3,15 2,95 2,6
CN > F > Cl > Br > ОСН3 > С6Н5 > Н > трет-С^9 0,59 0,5 0,47 0,45 0,23 0,10 0,0 —0,17
CN > Н > С6Н5 > mpem-CJJg > Br > Cl > F > ОСН3 0,07 0,0 —0,09 —0,12 —0,22 —0,24 —0,44 —0,50
10.4.6. Корреляция химических сдвигов с групповыми дипольными моментами в винильных системах
Редди, Мендел и Голдстейн [143] нашли линейную зависимость между химическими сдвигами концевых СН2-групп в винильных системах и групповыми дипольными моментами [142] заместителей. На рис. 10.18 изображены графики, иллюстрирующие эту линейную зависимость.
Бакингем [27] объяснил влияние растворителя на изменение химических сдвигов в бромистом виниле и цис- и транс-1,2-дихлорэтилене изменением полярных эффектов, сопровождающих изменения диэлектрической проницаемости растворителя.
10.4.1. Аномальные химические сдвиги протонов в винильных системах
Было замечено, что некоторые заместители, в винильных производных вызывают аномальные химические сдвиги винильных протонов, несовместимые с ранее выведенными линейными соотношениями. Тремя такими заместителями являются группы—feN, —СеН5 и —OR. Каковы же причины наблюдаемого несоответствия?
Винилцианид СН2 =CHCN. Если исходить из простых представлений об электроотрицательностях, группа CN, будучи сильно электроотрицательной, должна оттягивать электронную плотность от протона при а-углеродном атоме сильнее, чем от протонов при |3-углеродном атоме. Следовательно, Р-протоны должны быть более экранированными, как это наблюдается в случае хлористого винила [138, 144]. Однако Голдстейн и сотр. [143, 145] наблюдали резонансное поглощение а-протонов винилцианида в более сильном поле, чем для p-протонов (это означает, что а-протоны экранированы сильнее). Попытки объяснить это аномальное явление наличием резонансных структур
—С = N и —C = N
показывают, что а-протоны при таком рассмотрении должны быть еще менее экранированными. По-видимому, сильное экранирование а-протонов обусловлено значительным влиянием диамагнитной анизотропии тройной связи в группе — С = N [143, 146]. Это влияние должно вызывать увеличение экранирования всех протонов молекулы, но степень экранирования каждого из них зависит от расстояния до электрического центра тяжести связи — С = N
0,2
Рис. 10.17. Зависимость Д2[=6А — х/2 (6В + бс.) ] от константы Тафта ос для заместителя X в винильных системах СН2 = СНХ [118].
Точка для СвН6 получена после поправки на диамагнитную анизотропию бензольного кольца.
Рис. 10.18. Зависимость химических сдвигов протонов (цис и транс) в концевой СН2~группе винильной системы от группового дипольного момента заместителей [143].
Заместители и их групповые дипольные моменты: / — CN( —3,6); 2 — С1( —1,5); 3 — Н(—0,4); 4 — С6Н6(0,0); 5 - СН3( + 0,4); 6 — ОСН3(+1,16).
□ сдвиги цис-протонов; О сдвиги транс-протонов; I и ф соответствующие сдвиги, исправленные на диамагнитную анизотропию заместителей; ТМС — тетраметилсилан.
Корреляции спектров ЯМР-Н1 со строением молекул
55
и от величины угла, образованного линией, соединяющей этот центр с протонами, и осью связи — С = N. Уайпл и сотр. [144] рассчитали вклады диамагнитной анизотропии связи — С = N в экранирование трех протонов в винилцианиде и показали, что при учете этих поправок наблюдаемые химические сдвиги протонов уже не кажутся аномальными. В табл. 10.29 приведены химические сдвиги протонов винилцианида и его метильных производных.
Таблица 10.29
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ ВИНИЛЦИАНИДОВ И ИХ МЕТИЛЬНЫХ ПРОИЗВОДНЫХ [168]
Хими- -ческий сдвиг а (1)Н\С=С/Н(3) (2)Н/ \Н(4) Н\ /Н >с=с( н/ \сн3 Нх /Н >С=С( н/ \CN н\с_с/снз Н/ \CN СНз\с=С/Н Hz C\CN н\ „/Н CH3/C-C\CN
—211,5 —198,5 6 —231,6 —221,6 —251,0
ш2 —211,5 — 195,0 6 —238,7 —225,8 —261,3
<о3 —211,5 —219,0 —208,7 —206,6
со 4 —211,5 —229,3 6
аВ герцах (гц) при 40 Мгц-, внутренний эталон — Si(CHg)4. 6 По данным работы [124].
Стирол СН2 = СНСвН5. Для винильных протонов в стироле также были -обнаружены аномальные химические сдвиги и было предположено, что это явление обусловлено влиянием диамагнитной циркуляции электронов в ароматическом кольце (т. е. диамагнитными кольцевыми токами) [147, 227]. Считается, что сопряжение в молекуле стирола обусловливает расположение винильной группы и бензольного кольца в одной плоскости; тогда расчет дает для расстояний протонов А, В и С от центра кольца
Н(А)
^С-Н(В)
У i(C)
значения 3,5; 4,9 и 3,8 А соответственно. Банвелл и Шеппард [118], используя выражение Попла [227] (см. разд. 3.7), внесли необходимые поправки в значения химических сдвигов винильных протонов; величины этих поправок следующие: бд = +0,7 м. д.; бв = +0,2 м. д. и бс = + 0,55 м. д. Для «-метилстирола копланарность винильной и ароматической систем несколько нарушена (по данным УФ-спектра а-метилстирола) [148]. Такое искажение молекулы может вызвать сближение ядра В с ароматическим кольцом, в результате чего ядро В становится менее экранированным, чем ядро С (в противоположность отнесениям в стироле) (см. табл. 10.27). В пользу такого отнесения свидетельствовала более тонкая структура сигнала протона С по сравнению с сигналом лротона В (/н-снз обычно больше, чем /н-сн3, но это справедливо не для всех случаев). Из спектра протонного резонанса О-метилиндулина [149, 150] также следует, что в а-замещенных стиролах экранирование протонов В и С противоположно экранированию этих протонов в стироле. Однако Робертс и Девис [151] на примере селективно дейтерированных а-метилстиролов
CgHg Н С6Н5 D
\ = С и \ = С ЩС7 Х' D НзС"7 ХН
56
Глава 10
показали, что олефиновые протоны, расположенные в т/га«с-положении к метильной группе, экранированы слабее соответствующих ^«с-протонов. Значения 7н-сн3 оказались больше, чем значения 7й-сн3-
Виниловые эфиры СН2 = CHOR. При исследовании спектров ЯМР-Н1 многих виниловых эфиров
Н\ /Н
С = С
н/
было показано, что протоны концевых метиленовых групп дают сигналы в аномально высоких полях [115, 118, 153]. Химический сдвиг по сравнению с химическими сдвигами концевых метиленовых групп в других винильных производных на 0,5 м. д. больше [115].
Таблица 10.30
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ И КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ НЕКОТОРЫХ ЭФИРОВ [115]
<в>н\ с-с/Н(А)
(С)Н/ \OR
Ra JBC JAB JAC ТВ тс ТА тв тс
Декалил ±1,2 — 5,65 5,12 0,525
mpem-Бутил —0,1 ±6,2 ±13,2 6,15 5,80 3,72 0,355
Изопропил — 1,2 ±6,9 ±13,7 6,12 5,85 3,73 0,264
Циклогексил — 1,6 ±5,7 ±14,7 6,15 5,90 3,87 0,266
Фурфурил — 1,9 ±6,6 ±14,2 6,15 5,90 3,60 0,252
2-Этилгексил — 1,8 ±6,1 ±12,9 6,20 5,95 3,60 0,249
изо-Бутил — 1,7 ±6,3 ±14,2 6,12 5,93 3,56 0,190
и-Бутил — 1,8 ±6,9 ±14,4 6,05 5,85 3,47 0,190
Этил — 1,7 ±6,9 ±14,9 6,12 5,93 3,55 0,187
Метил —2,2 ±6,6 ±14,4 6,15 6,00 3,62 0,154
2-Хлорэтил —2,7 ±6,6 ±16,2 5,95 5,83 3,50 0,120
а Константа транс-взаимодействия Jдр произвольно выбрана положительной, и знаки двух других констант в каждой молекуле определены относительно Jдр.
В табл. 10.30 собраны спектральные параметры, наблюдаемые в спектрах ЯМР некоторых винильных производных. Причина сильного экранирования концевых метиленовых протонов заключается, по-видимому, в делокализации электронов, выражаемой резонансными структурами —
сн3—О—СН = СН2 и сн3—о=сн—сн2.
Такая делокализация приводит к тому, что электронная плотность на концевом атоме углерода становится близкой к электронной плотности на атоме углерода в алканах [115, 118]. Однако существенных изменений в стереохимии винильной группы не происходит, так как наблюдаемые константы взаимодействия являются характерными для винильной системы (см. табл. 10.26). Заместители в табл. 10.30 расположены в порядке возрастания их электроотрицательностей; при этом константы взаимодействия уменьшаются с увеличением электроотрицательности заместителя, как и следовало ожидать из эмпирического рассмотрения констант взаимодействия в винильных системах (разд. 10.4.3). Поэтому алкильные группы в алкоксизаместителях
Корреляции спектров ЯМР-Н1 со строением молекул
57
рассматриваются как электроноотталкивающие группы, не вступающие в сверхсопряжение.
Существует также корреляция между электроотрицательностью заместителей в алкоксигруппах и разностью химических сдвигов протонов концевой группы СН2 (6ВС); однако между самими химическими сдвигами тв и тс и электроотрицательностью заместителей корреляции нет. Возможно, что внутренний химический сдвиг 6ВС обусловлен главным образом влиянием диамагнитной анизотропии связи С — О на экранирование р-^ис-протона.
10.4.8. Хлористый винил и хлорэтилены
При сравнении химических сдвигов протонов хлористого винила с химическим сдвигом протонов этилена (т = 4,71) можно видеть, что введение атома хлора изменяет химические сдвиги а-, fi-цис- и р-тра«с-прото нов на —0,90; —0,12 и 0 м. д. соответственно (для указанных положений значения т составляют 3,81; 4,59 и 4,71 м. д.) [123]. Эти результаты позволяют предположить,, что электроотрицательный атом хлора сильно оттягивает (индуктивный эффект)' электроны из a-положения, делая его менее экранированным, тогда как в p-положениях индуктивное влияние погашается сопряжением с неподелен-ной парой электронов [123].
Таблица 10.31
ХИ1МИЧЕСКИЕ сдвиги ПРОТОНОВ И КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ РЯДА ХЛОРЭТИЛЕНОВ [123]
Соединение т /нн’ гч JC13H> Sli
ЦиС-С2Н2С12 3,72 5,3 198,5
/яраис-С2Н2С12 3,76 12,1 199,1
1,1-С2Н2С12 4,62 166
С2НС13 3,68 201,2
С2Н3С1 а 3,81 гем —1,3 а 195
$-цис 4,59 цис 7,5 fi-цис 160
ft-транс 4,71 транс 14,7 Р-транс 161
В табл. 10.31 приведены значения химических сдвигов и констант спин-спинового взаимодействия для ряда хлорэтиленов. Введение нескольких атомов хлора в молекулу этилена слабо влияет на экранирование «-протона.
Положение хлора в хлорэтиленах легко установить по расщеплениям от взаимодействия СВ * * * * 13Н: протоны, находящиеся по отношению к хлору в «-положении, имеют константы взаимодействия 7С-н = 195—201 гц, тогда, как для p-протонов 7С-н = 160—166 гц.
10.4.9. Пропен-1, бутен-1 и гексен-1
При исследовании большого числа олефиновых соединений Ботнер-Бай и Наар-Колин [124] изучили спектры ЯМР-Н1 винильных соединений с алкильными группами в качестве заместителей (соединения типа алкен-1). Исследование этих соединений представляло интерес с точки зрения, во-первых, влияния заместителей на химические сдвиги протонов в цис-, транс- и гем-
положениях по отношению к заместителю и, во-вторых, влияния изомерии вращения вокруг связи между тригональным и тетраэдрическим атомами
углерода на параметры спектров ЯМР. Все изученные соединения дают сложные спектры (спектр протонного резонанса пропена-1 при 60 Мгц приведен на рис. 10.19), и для получения точных значений химических сдвигов и констант взаимодействия необходимо проведение полного анализа спектров-
-58
Глава 10
Пропен-1 представляет собой систему ABCD3, и для получения согласия между рассчитанным и наблюдаемым спектром необходимо применение вычислительной техники. Обозначив протоны следующим образом:
(3)Н СН3(4, 4', 4")
\ = C/Z
(2)HZ H(l)
можно сделать отнесения сигналов в спектре (см. рис. 10.19). Шестнадцать -слабопольных сигналов обусловлены протонами (1), и, используя приближение первого порядка, можно найти приближенные значения констант: эти шестнадцать сигналов можно рассматривать как дублет (Л 3 = ~ 17 гц) дублетов (Ji 2 = ~ 10 гц), каждый сигнал которых представляет собой квартет
Р и с. 10.19. Спектр протонного резонанса (при 60 Мгц) олефиновых протонов в пропене-1 [124].
Растворитель — CCi4; внутренний эталон — Si(CH3)4.
<(Jt 4 = ~ 6 гц). Изучение мультиплетов в сильном поле показывает, что они также представляют собой пару дублетов, сигналы которых расщеплены в квартеты; исходя из этого можно получить приближенные значения J2 3, J2 4 и J3 4. При непосредственном изучении спектра можно получить, кроме того, приближенные значения химических сдвигов. Используя приближенные значения параметров, можно построить пробную матрицу (см. гл. 8) и затем использовать вычислительную машину для модификации этой матрицы до тех пор, пока не будут получены решения, совпадающие с измеренными значениями. Элементы полученной таким образом матрицы и дадут точные значения химических сдвигов и констант взаимодействия. Было обнаружено, что некоторым константам взаимодействия следует приписать обратные (по отношению к обычно принятым) знаки для того, чтобы получить теоретический спектр, хорошо согласующийся с наблюдаемым. В табл. 10.32 указаны химические -сдвиги и константы взаимодействия для чистых образцов пропена-1, бутена-1 и гексена-1. Были исследованы также разбавленные растворы этих соединений в четыреххлористом углероде и показано, что для протонов 2 наблюдаются небольшие сдвиги в более сильное поле по сравнению со сдвигами протонов 1 и 3. Данные для растворов приведены в табл. 10.32 в столбцах «стандарт». Изучение табл. 10.32 показывает, что изменение алкильной группы оказывает очень малое влияние на химические сдвиги протонов или на их константы взаимодействия. При введении в молекулу этилена одной алкильной группы происходят следующие изменения химических сдвигов:
г (1)—г (этилена) = —0,402 м. д., х (2)—т (этилена) = 4-0,476 м. д., х(3)—х (этилена) = 4-0,399 м. д., где х (этилена) = 4,650 м. д.
Корреляции спектров ЯМР-Н1 со строением молекул
59
Таблица 10.32
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ ПРОТОНОВ ПРОПЕНА-1, БУТЕНА-1 И ГЕКСЕНА-1 [124]
(4)
(3)Н\ /ОВД
(2)Н/ \Н (1)
Химический сдвиг Пропен- 1, R — Н Бутен-I, R = СНз Гексен-1, R = С3Н7
чистый стандарт а чистый стандарт чистый стандарт
т(1) 4,27 4,27 4,21 4,22 4,26 4,27
г(2) 5,12 5,12 5,13 5,13 5,13 5,12
т(3) 5,04 5,04 5,05 5,06 5,06 5,06
т(4) 8,34 8,28 8,02 8,00 7,97 7,98
^СНз (концевая) 9,02 9,01 9,14 9,10
Константы взаимодействия
J12, гц 10,02 10,32 10,23
J 1з, гЧ 16,81 17,23 17,03
J14, гц 6,40 6,22 6,55
J23’ гЧ 2,08 1,96 2,23
^24> гц -1,33 -1,26 -1,18
J34, гц -1,75 -1,66 -1,51
а «Стандарт»— разбавленные растворы алкенов в ССЦ.
Ботнер-Бай и Наар-Колин [124] суммировали главные факторы, определяющие экранирование в органических молекулах:
1) близость атомов или групп, отталкивающих или притягивающих электроны по индуктивному механизму или вследствие сопряжения [9, 154, 156];
2) близость групп, обладающих диамагнитной анизотропией [146, 157—160];
3) существование в молекуле постоянных электрических полей, способных возмущать электронные орбитали вблизи протонов [8, 161];
4) близость поляризуемых групп, что приводит к заметному среднему значению квадрата электрического поля на протонах и уменьшает их экранирование [162]. Существует также парамагнитный вклад связей С — Н, который обычно не рассматривают, поскольку его доля в экранировании постоянна [141].
Ботнер-Бай и Наар-Колин провели детальное рассмотрение относительной роли различных факторов, обусловливающих экранирование в случае алкенов-1. Полезно рассмотреть их подход к решению проблемы.
1) Алкильные группы в алкенах могут подавать электроны к двойной связи либо по индуктивному механизму, либо по резонансному (т. е. путем гиперконъюгации). Если бы индуктивное влияние было единственным, то. поскольку оно должно сильно ослабевать по мере увеличения расстояния от алкильной группы, наибольшая электронная плотность была бы на ближайшем атоме углерода и в результате протоны 1 у этого углеродного атома были бы сильно экранированы. Однако опыт показал, что протоны 2 и 3 экранированы сильнее протона 1, что свидетельствует о влиянии других факторов, -обусловливающих экранирование протонов в этих молекулах. То, что |3-про-тоны экранированы сильнее а-протонов, может быть следствием эффекта гиперконъюгации, предусматривающего существование резонансных форм:
60
Глава 10
Н\ /СНгН
С = С н/ \н
+
Н\ ,СН2Н
и С —С
н/ \н
2) Другая попытка объяснить наблюдаемые для пропена-1 химические сдвиги основана на рассмотрении диамагнитной анизотропии одиночной связи С — С. Однако этот эффект, по-видимому, не имеет значения, так как он предусматривает дезэкранирование р-протонов 2 по сравнению с двумя другими винильными протонами, что находится в противоречии с экспериментальными наблюдениями.
3) Дипольный момент в алкенах-1, по-видимому, направлен вдоль линии, изображенной [163] стрелкой
Для того чтобы рассчитать вклады в экранирование винильных протонов, Ботнер-Бай и Наар-Колин предположили, что этот дипольный момент можно заменить постоянным электрическим полем, действующим в этом же направлении. Было предположено, что электрическое поле сдвигает электронную плотность вдоль связи в сторону протонов, увеличивая тем самым их экранирование. Такое рассмотрение предусматривает величины и знаки химических сдвигов, совпадающие с экспериментальными значениями, и, так как дипольные моменты различных алкенов-1 очень близки друг к другу, можно было-предсказать наблюдаемую на опыте независимость химических сдвигов от природы алкена.
4) Для определения вклада в экранирование, обусловленного близостью поляризуемых групп, используется следующее выражение:
Дп= —1,5- 10-18ае/г6, где Да — изменение экранирования (безразмерная величина); а — поляризуемость группы (см3); е — потенциал ионизации (эрг); г — расстояние до-группы (см), оно должно быть < 2 А для того, чтобы этот эффект был значительным. Несмотря на то что данный эффект приводит к качественному согласию химических сдвигов винильных протонов с наблюдаемыми величинами, любой разумный выбор значений а и е приводит к заниженным величинам химических сдвигов.
На основании изложенных рассуждений был сделан вывод, что наличие в молекуле постоянного диполя (эффект 3) позволяет дать наилучшее объяс нение наблюдаемых для пропена-1 химических сдвигов.
10.4.10. Константы спин-спинового взаимодействия в алкенах-1
Исследование констант взаимодействия, приведенных в табл. 10.32 и 10.33, показывает, что значения, полученные для алкенов-1, существенно-не отличаются от типичных значений, наблюдаемых для винильных систем. В бутене-1 имеется возможность вращательной изомерии вокруг связи между sp3- и з/?2-гибридизованными атомами углерода. Константы взаимодействия Ji ь, J2 4 и Jз 4 зависят от изомерии молекулы и показывают, является ли данный вращательный изомер преобладающим при данной температуре. Информацию такого рода можно получить при использовании соотношений Карплуса [39] [см. уравнения (10.4) и (10.5)], коррелирующих значения J и двугранные углы. Так, константы взаимодействия, наблюдаемые для жидкого бутена-1, указывают на то, что при комнатной температуре существуют равные концентрации трех вращательных изомеров [164].
Корреляции спектров ЯМР-Н1 со строением молекул
61
Таблица 10.33
КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ3 ПРОТОНОВ НЕКОТОРЫХ ОЛЕФИНОВЫХ СОЕДИНЕНИЙ [124]
(3>H\r_r/CH2R<4)
(2)Н/ ХН(1)
Соединение 712 713 714 723 724 7з4
Нх /СН2С1 >с=с< СИ гт 13,1 13,1 7,1. 7,3 0,5 — 1,2
Ск /СН2С1 >с=с< Н/ н 7,2 7,0 7,2 7,4 0,9 —1,2
II /СН2С1 >с=с< Н/ С1 1,9 1,9 (0,3; 0,9) (0,3; 1,0)
Н. /СН2Вг >с=с< Н/ хн 2,2 (0,3; 1,0)
Н\ /СвН5 >с=с< Н/ ч-i 10,6 11,3 17,2 17,8 1,6 1,2
н. /CN V=c< Hz ri 11,28 18,19 1,27
а Значения констант взаимодействия приведены в герцах (гч). Таблица составлена по данным раз-личных авторов.
Ботнер-Бай и сотр. [165] исследовали спектры протонного резонанса ряда алкилэтиленов; результаты их исследования приведены в табл. 10.34. Для транс-ориентированных протонов, находящихся у соседних тригонального и тетраэдрического атомов углерода, константа взаимодействия имеет значение jmva-нс _~ и 5 гц, тогда как гош-ориентированные протоны имеют значение = 3,7 гц. Используя эти значения и экспериментальные константы спин-спинового взаимодействия протонов у соседних тригонального и тетраэдрического атомов углерода, можно предсказать, какой из вращательных изомеров является предпочтительным. Величина констант дальнего взаимодействия аллильного протона и концевых метиленовых протонов также зависит от вращательной изомерии молекулы, причем взаимодействие будет наименьшим, когда аллильный атом водорода и группа СН2 образуют скошенную конформацию.
10.5. Дизамещенные олефины
Характеристические значения для гем-, цис- и'транс-констант взаимодействия, наблюдаемые в винильных системах, в дизамещенных олефинах изменяются незначительно, и соответственно этому проблема определения конфигурации геометрических изомеров часто может быть однозначно решена методом ЯМР высокого разрешения. По мере возрастания числа точно измеренных спектров олефиновых соединений применение ЯМР для определения геометрической изомерии этиленовых соединений становится чрезвычайно полезным 1). Штелинг [378] собрал значения химических сдвигов и констант
Я Систематическое исследование спектров протонного резонанса цис- и транс-алкенов-1, -2, -3, -4 и 1- и 3-замещенных циклогексенов было проведено Липпмаа и сотр. [517].— Прим. ред.
Таблица 10.34
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ ПРОТОНОВ НЕКОТОРЫХ А ЛКИЛЗАМЕЩЕННЫХ ОЛЕФИНОВ [165]
HJ4) R (4')
(3) Rx ZCX
С=С R (4)
(2) RZ \н (1)
R (2) R (3) R (4) R (4') Состояние а Химические сдвиги Константы взаимодействия
т (1) г (2) т (3) т (4) /12 /13 /14 /23 ^24 /34
Н н СН3 СН3 ч р 4,271 4,276 5,186 5,186 5,088 5,102 (8,170)в (8,170)в 10,37 17,22 6,41 1,74 — 1,17 — 1,43
Н н Н ч р 4,280 4,295 5,075 5,074 5,058 5,064 (8,000)в (8,000) в 10,13 17,02' 7,00 2,05 —1,15 —1,43
Н н Н трет-С^д ч р 4,223 4,231 5,032 5,036 5,055 5,061 8,082 8,075 10,02 17,10 7,46 2,37 —0,94 —1,32
н н mpem-C4H9 трет-С^д ч р 4,260 4,262 5,013 5,020 5,214 5,221 8,434 8,435 9,97 17,01 10,65 2,63 —0,10 —0,63
сн3 СНз mpem-C4H9 /?грет-С4Н9 ч р 4,775 4,792 (8,443) г (8,438)г (8,270)г (8,276)г 8,107 8,125 —1,25Д —1,25 Д 11,37 е е е
а ч — чистое вещество; р — 10%-ный раствор в ССЦ. б Фиксированная частота спектрометра 60 Мгц.
в Предполагаемые значения (без анализа).
г Отнесение неоднозначно.
Д /Н—СН3*
е Не разрешено; вероятно, меньше 0,2 гц.
Корреляции спектров ЯМР-Н1 со строением молекул
63:
взаимодействия (см. приложение II) для большого ряда моноолефинов. Геометрические изомеры ди- и тризамещенных олефинов могут характеризоваться химическими сдвигами групп Р-СН3 и сс-СН [378].
10.5.1. цис- и транс-Дизамещенные олефины
В молекулах типа
С1\ /С1 Н\ /С1
С=С и С=С н/ \н С1/ \н
химические сдвиги протонов различны, и это различие называют «дифференциальным экранированием». Для вышеприведенных молекул «дифференциальное экранирование» тчис — ттраис = — 0,08 м. д. Джекман и Уайли объяснили эти различные сдвиги дальним экранированием, обусловленным влиянием диамагнитной анизотропии [166]. В табл. 10.35 приведены дифференциальные-сдвиги для четырех цис-транс-пар а,р-дизамещенных олефинов.
Таблица 10.35
ДИФФЕРЕНЦИАЛЬНЫЙ ХИМИЧЕСКИЙ СДВИГ (,хцис — tmpaHC) ДЛЯ ЧЕТЫРЕХ цис-транс-ПАР а,0-ДИЗАМЕЩЕННЫХ ОЛЕФИНОВ СНХ = СНХ [166]
X СНз С1 Вг со2сн3
^тиране* Д* —0,03 —0,08 —0,38 +0,525
Вклад эффекта диамагнитной анизотропии в экранирование обратно пропорционален кубу расстояния (г) от экранированных ядер до электрического центра^ тяжести группы электронов (см. разд. 4.4). Если это расстояние больше 3 А, дальнее экранирование будет пренебрежимо мало (за исключением экранирования в ароматических кольцах). Для всех цис-дизамещенных олефинов Р-протоны находятся далеко от центра связи углерод — заместитель и не-испытывают дальнего экранирования. В транс-дизамещенных олефинах расстояние между, p-протоном и центром связи углерод — заместитель гораздо короче (~2,4 А), и поэтому наблюдается вклад связи углерод — заместитель в экранирование. В связи с этим дифференциальные химические сдвиги, приведенные в табл. 10.35, можно объяснить вкладом, имеющим место в транс-изомере. В диметильных производных дифференциальное экранирование очень-мало, так как в транс-изомере нет вклада в дальнее экранирование, что является следствием очень небольшой величины угла 0 в этой молекуле. В дигалогенопроизводных связь углерод — галоген по своей природе биполярна, и расстояние г нужно измерять не от центра связи углерод — галоген, а от ядер галогена. Это приводит к малому и отрицательному значению члена (3 cos2 0 — 1). Как в транс-дихлор-, так и в транс-дибромэтиленах осуществляется парамагнитный вклад в экранирование р-протонов.
Из других дизамещенных олефинов изучены траас-стиролы [428], транс-пропены [428] и а- и Р-замещенные коричные кислоты [429].
10.5.2. 2-Замешрнные пропены
Джекман и Уайли [166] измерили химические сдвиги и дифференциальные химические сдвиги олефиновых протонов в ряду 2-замещенных пропенов» общей формулы
(А)Н ZCH3(X) с=с
(В)н/ XR
<64
Глава 10
Таблица 10.36
ХИМИЧЕСКИЕ СДВИГИ ПРОГОНОВ НЕКОТОРЫХ 2-ЗАМЕЩЕННЫХ ПРОПЕНОВ [166] (Л)Н\ /ОД, (Х) (В) н/ c\r
Химические сдвиги Химические сдвиги
R Т(СНз) гв ТА ТВ — ХА R т(СНз) ТВ ТА ХВ ~ ХА
СН2С(СН3)3 8,22 5,37а 5,20а 0,17 СОС1 7,97 3,52 3,98 -0,46
С1 7,85 4,92 4,92 0,00 COCH3 8,20 3,96 4,16 -0,20
Вг 7,70 4,67 4,47 0,20 сно 7,90 3,62 3,92 -0,30
CN 7,97 4,28а 4,24 а 0,04 с6н5 7,86 4,72 4,98 -0,20
со2сн3 8,10 3,96 4,51 -0,55 00ССН3 8,09 5,38 5,38 0,00
conh2 8,06 4,24 4,63 -0,39
а Отнесение неоднозначно.
В табл. 10.36 приведены химические сдвиги для некоторых молекул такого типа. Дифференциальные химические сдвиги, наблюдаемые для этого ряда соединений, проявляют некоторую аналогию с химическими сдвигами соответствующих 1,2-дизамещенных производных (например, при R = С1, Вг, СООСН3): все рассмотренные производные метакриловой кислоты (R = CONH2, СОС1 и СООСН3) имеют близкие дифференциальные сдвиги; для соответствующих альдегидов и метилкетонов наблюдаемые значения тв — гд также достаточно велики. Во всех случаях дифференциальные сдвиги обусловлены дезэкранированием p-протона, находящегося в цмс-положении к карбонильной группе, в результате влияния диамагнитной анизотропии последней.
Дифференциальные сдвиги наблюдаются для Р-протонов в 2-замещенных алкенах и при других заместителях:
1) неопентильной группе (следовательно, эта группа большую часть времени пребывает, по-видимому, в конформациях, в которых связи С — С ориентированы так, что цис-^-протоны испытывают существенное влияние диамагнитной анизотропии этих связей);
2) фенильной группе, в которой аромати-ческие кольцевые токи по-разному влияют на экранирование p-протонов. Нитрильная и ацетатная группы показывают очень малую разницу во влиянии на экранирование Р-протонов; в обоих случаях эффективные центры электронных облаков удалены от р-про-тонов настолько далеко, что влияние их диамагнитной анизотропии становится несущественным. Спектры ЯМР 2-замещенных пропенов были исследованы также и другими авторами, которые наблюдали хорошее совпадение полученных ими данных с данными Джекмана и Уайли [166]. В табл. 10.37 приведены значения химических сдвигов и констант взаимодействия, полученные при детальном анализе систем АВХ3, образуемых этими молекулами [145]. Было показано, что в спектре протонного резонанса 2-хлорпропена, в котором отнесения сигналов могут быть сделаны достаточно однозначно на основании отнесений в спектре хлористого винила (см. табл. 10.31), ^н—снз больше, чем •/н-снз- Эти данные указывают на аналогию в величинах констант взаимодействия с данными, полученными для бутена-1 [125]. В 2-хлорпропене обе константы дальнего взаимодействия Ун-снз одинаковы по знаку [145], и имеются данные, свидетельствующие о том, что они отрицательны (см. табл. 10.8). Во всех исследованных соединениях величины обеих констант дальнего взаимодействия Jh-снз отличаются примерно на 0,6 гц.
Что касается экранирования протонов в 2-замещенных пропенах, приведенных в табл. 10.37, то было показано, что заместитель R влияет на экранирование протонов при двойной связи сильнее, чем на протоны метильной
Корреляции спектров ЯМР-Н1 со строением молекул
65
Таблица 10.37
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ ПРОТОНОВ НЕКОТОРЫХ 2-ЗАМЕЩЕННЫХ ПРОПЕНОВ [145]
R Химический сдвиг, м. д. Константа взаимодействия, гц
л°са 6А ТА х СО 6В тв я СО бх ТХ J АХ 7вх 7ав
Вг —0,42 4,54 —0,24 4,72 2,82 7,78 1,4 0,8 2,0
С1 0,03 4,99 0,00 4,96 2,99 7,95 1,38 0,65 1,25
ОСОСНз 0,53 5,49 0,45 5,41 3,19 8,15 1,2 0,6 1,0
ОСН3 1,3 5,26 1,3 6,26 3,31 8,27
сно, — 1,08 3,88 —0,78 4,18 3,27 8,23 1,6 1,0 1,1
CN —0,58 4,38 —0,47 4,49 3,15 8,11 1,7 1,15 1,0
СН3 0,42 5,38 0,42 5,38 3,36 8,32 1,25 1,25
СН2С1 0,20 5,16 0,05 5,01 3,24 8,20 1,5 0,85 1,6
СН2Вг 0,20 5,16 0,01 4,97 3,21 8,17 1,5 0,8 1,5
СН21 0,24 5,20 —0,09 4,87 3,17 8,13 1,5 0,8 1,5
а 6 03 — химические сдвиги, измеренные относительно НгО (внешний эталон) для разбавленных раст-воров в циклогексане. Обозначение параметров дано в соответствии со спиновой системой АВХз, в которой А выбрано таким образом, чтобы было | J АХ | > | J gX т = 4 ,96 ^Н^О*
группы. Несмотря на то что значения химических сдвигов качественно согласуются со значениями, предсказанными при рассмотрении индуктивного и мезомерного эффектов, между химическими сдвигами олефиновых протонов и константами Гаммета для заместителей не наблюдалось корреляции. Экранирование p-протонов, находящихся в ^«с-положении по отношению к группе СН2Х, гораздо сильнее зависит от различных факторов, чем экранирование соответствующих /праяс-протонов. Это явление согласуется с идеей Джекмана и Уайли [166] о вкладах в экранирование, обусловленных дальним влиянием диамагнитной анизотропии.
Для некоторых 2-замещенных пропенов при изменении растворителя наблюдалось специфическое влияние среды, что было использовано при отнесении сигналов в спектрах.
10.5.3. 2,3-Дизамещенные пропены
Уайпл, Голдстейн и Мак-Клюр [167] исследовали ряд 2,3-дизамещенных пропенов для определения влияния аллильного замещения на параметры спектров ЯМР. Эти молекулы имеют общую формулу
уСН2Х с = с н/ \у
и их спектры ЯМР-Н1 обычно можно анализировать как систему АВХ2.
В р-метилаллилгалогенидах (Y = СН3, X = галоген) олефиновые протоны, проявляющие сильное взаимодействие с метильными протонами, слабо взаимодействуют с протонами галогенметильной группы и наоборот. Здесь константы взаимодействия ./ц-сн3 также больше констант ^н-сн3-
В табл. 10.38 приведены химические сдвиги и константы взаимодействия для исследованных 2,3-дизамещенных пропенов, и можно видеть, что заместители в положении 2 сильнее влияют на экранирование всех протонов, чем заместители в положении 3. Это можно было предсказать для олефиновых 5-489
66
Глава 10
Таблица 10.38
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ ПРОТОНОВ РЯДА
2,3-ДИЗАМЕЩЕННЫХ ПРОПЕНОВ [167]
Н\ /Y )С=С'
Н/ \СН2Х
Заместитель X Химический сдвиг а, м. д. Константа взаимодействия, гц
Аоо 6А ХА А СО бв тв А со 6Х ТХ JAX JBX JAB
Y = = С1
I —0,44 4,52 —0,17 4,79 1,05 6,01 0,7 1,4
Вг —0,45 4,51 —0,25 4,71 1,08 6,04 0,9 1,5
С1 —0,48 4,48 —0,27 4,69 1,01 5,97 1,2 0,6 1,5
ОН —0,38 4,58 —0,19 4,77 0,96 5,92 1,5 1,0 1,4
Y = Вг
Вг —0,91 4,05 —0,51 4,45 0,96 5,92 1,1 2,0
С1 —0,94 4,02 —0,51 4,45 0,92 5,88 1,5 0,9 1,9
Y = СН3
I —0,09 4,87 0,24 5,20 1,25 6,21 0,8 1,5
Вг 0,01 4,97 0,20 5,16 1,25 6,21 0,9 1,5
С1 0,05 5,01 0,20 5,16 1,16 6,12 1,0 1,6
а Химические сдвиги измерены в миллионных долях (м. д.) относительно Н2О (внешний эталон) для разбавленных растворов в циклогексане. Обозначение параметров дано в соответствии со спиновой системой ABXj, в которой А выбрано таким образом, чтобы было | J| < | |.
протонов, однако для протонов метиленовой группы на основании рассмотрения индуктивного влияния аллильных заместителей следовало ожидать большего экранирования. Влияние заместителя в положении 2 на химические сдвиги и константы взаимодействия олефиновых протонов совершенно не зависит от степени аллильного замещения и очень близко к наблюдаемому в 2-заме-щенных пропенах. Константы дальнего взаимодействия </н-сн2х в аллильных производных меньше и уменьшаются в ряду X = ОН > Cl > Вт > I. Эти константы дальнего взаимодействия зависят от природы растворителя, причем их значения увеличиваются по мере уменьшения полярности растворителей. В полярных средах возможно существование вращательных изомеров с различными дипольными моментами, поэтому изменение констант взаимодействия с изменением природы растворителя можно объяснить такими конформационными эффектами 1).
10.5.4. Влияние метильных групп на спектры протонного резонанса производных этилена
Редди и Голдстейн [168] исследовали спектры протонного резонанса десяти метильных производных этилена, чтобы выяснить влияние метильной группы на параметры спектров (см. табл. 10.29, 10.39 и 10.40). Все исследованные
9 Спектры ЯМР-Н1 аллилсиланов общей формулы (СН2 = CHCH2)nSi(CH2X)4_n. были изучены в работе [518].— Прим. ред.
Корреляции спектров ЯМР-Н1 со строением молекул
67
Таблица 10.39
РАССЧИТАННЫЕ И НАБЛЮДАЕМЫЕ ХИМИЧЕСКИЕ СДВИГИ а ЦИАНПРОПЕНОВ [168]
Соединение Положение протона по отношению к иитрилъ-ной группе Химический сдвиг относительно Si(CHjj)4 (внутренний эталон), гц
рассчит. б рассчит. в иабл.
а-Метакрилонитрил цис —222,2 —225,2 —225,8
транс —218,6 —221,6 —221,6
транс-Кротонитрил а —206,0 —209,0 —208,7
₽ —256,5 —259,5 —261,3
цис-Кротонитрил а —202,5 —205,5 —206,6
₽ —249,4 —252,4 —251,0
а Измерены в герцах (гц) при фиксированной частоте 40 Мгц.
б Без дополнительной поправки иа строение —3 гц.
в С дополнительной поправкой иа строение —3 гц.
Таблица 10.40
РАССЧИТАННЫЕ И НАБЛЮДАЕМЫЕ
ХИМИЧЕСКИЕ СДВИГИ а
ХЛОРПРОПЕНОВ
Соединение Положение протона по отношению к атому хлора Химический сдвиг
рассчит. б набл.
2-Хлорпропен цис -200,0 -201,0
транс -198,5 -199,8
транс-1 -Хлорпропен-1 а -234,8 -235,3
₽ -234,3 -232,8
цис-1 -X лорпропен-1 а -231,3
₽ -229,3
а Указаны в герцах (гц) при фиксированной частоте -40 Мгц-, внутренний вта-лои — тетраметилсилан.
б Для этих соединений ие было необходимости в поправке на строение.
молекулы являются производными пропена-1 общих формул:
Н3С\ ,Н н3сх Н С=С С=С
х/ н/ \х
и имеют одну из следующих
н/
В тех случаях, когда можно было ожидать влияния концентрации на сдвиг, была проведена экстраполяция химических сдвигов к бесконечному разбавлению в инертном растворителе. Было показано, что введение метильной группы обусловливает одинаковое и характерное изменение экранирования в каждом из положений этиленовой системы. Рассмотрим конкретные примеры.
Винилцианиды и цианпропены. В табл. 10.29 приведены значения химических сдвигов для этилена, пропилена, винилцианида и цианпропенов (относительно тетраметилсилана и при 40 Мгц). В пропилене транс-протоны экранированы на 3,5 гц, а в винилцианиде на 7,1 гц сильнее цыс-протонов. Если эти два эффекта аддитивны, то для 2-цианпропена сигнал протонов в цис-положении к нитрильной группе должен находиться на 3,6 гц в более слабом поле, чем сигнал /npawc-протонов. Экспериментально наблюдается различие в 4,2 гц в том же направлении, что хорошо согласуется с теоретическим значе
5*
68
Глава 10
нием (ошибка эксперимента равна ±0,5 гц). При аналогичном рассмотрении химических сдвигов протонов в пропилене, винилцианиде, цис- и тра«с-крото-нитрилах можно предсказать значения химических сдвигов в производных кротонитрила. Для того чтобы предсказать не внутренние, а истинные значения химических сдвигов, нужно использовать уравнение
Д, = Д( (пр.) —211,5 гц, (10.10)
где А/ — влияние метильной группы на химический сдвиг i-ro протона, Аг (пр.) — химический сдвиг протона в том же положении относительно СН3-группы в пропилене и 211,5 гц — химический сдвиг этилена относительно тетраметилсилана при 40 Мгц. Итак, для влияния метильной группы мы получаем следующие значения:
a-положение —17,8 гц
0-положение (транс к СН3-группе) ±16,5 гц
0-положение (цис к СН3-группе) ± 13,0 гц
Теперь мы можем предсказать химические сдвиги протонов в цианпро-пенах, прибавляя вышеприведенные значения к химическим сдвигам соответствующих протонов в винилцианиде; при этом получатся значения, приведенные в табл. 10.39. Все шесть предсказанных значений больше, чем предполагалось, на 3 гц, и для того, чтобы скомпенсировать это несоответствие (предполагая, что оно постоянно), можно модифицировать уравнение (10.10) следующим образом:
Д: = Дг (пр.)—214,5 гц. (10.11)
Очень хорошо согласуются предсказанные и наблюдаемые химические сдвиги в системах бромистый винил —бромпропены и хлористый винил—хлорпропены (см. табл. 10.40).
10.6. Тризамещенные олефины
10.6.1. 1-Замешанные изобутены
Для того чтобы выяснить возможность определения геометрической конфигурации родственных молекул по химическим сдвигам аллильных протонов, в работе [166] был исследован ряд соединений общей формулы
R ,СН3 '
С = С н/ ХСГ13
В табл. 10.41 приведены химические сдвиги олефиновых протонов и двух Р-метильных групп в исследованных соединениях. Эффекты дальнего экрани-
Таблица 10.41
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ НЕКОТОРЫХ 1-ЗАМЕЩЕННЫХ ИЗОБУТЕНОВ [ 1 66]
R г(Р-СНз) j R г(Р-СНз)
олефиновый Н цисл транс а т — цис хтранс олефиновый Н цис а транс а хцис х транс
С(СН3)3 4,87 8,38 6 8,32 6 0,06 СОСН3 4,03 7,94 8,14 —0,20
Вг 4,22 8,25 8,25 0,00 сно 4,37 7,89 8,09 —0,20
СО2СН3 4,38 7,88 8,16 —0,28 ООССНз 3,21 8,35 8,35 0,00
СОС1 3,99 7,88 8,03 —0,15 с=сн 4,83 8,12 6 8,20 6 —0,08
а Изобутены, безусловно, не существуют отдельно в цис- и транс-формах, но метильные группы в (CHeJjC^sCHR дают два различных сдвига; это обозначается цис и транс в соответствии с тем, находится метильйая группа в цис- или /прдяг-положении к R.
® Цис- и тяпнс-отиесения не однозначны.
Корреляции спектров ЯМР-Н1 со строением молекул
69
рования, обусловленные свободно вращающимися метильными группами, малы, и ими можно пренебречь. Если рассматриваются молекулы, содержащие группы с большой магнитной анизотропией (например, если R = СООСН3, СНО, СОСНз), то сильные эффекты дальнего экранирования не дают возможности использовать ЯМР для определения геометрической конфигурации молекул типа —(CH3)C=CR— [169].
Таблица 10.42
КОНСТАНТЫ цис- И mpaw-ВЗАИМОДЕЙСТВИЯ Н—СН3 В ЗАМЕЩЕННЫХ
ПРОПЕНАХ И ИЗОБУТЕНАХ [166]
RC(CH3)=CHg ,транс ‘'Н-СНз* гц Тцис ’'Н-СНз’ гц /нн, гц RC(CH3)=CH2 гтранс •'Н-СНз’ гц гцис J Н-СНз’ гц
R = Вг 0,8 1,4 1,4 R = С(СН3)3 1,4 1,4
СО3СН3 0,9 1,45 1,8 Вг 1,5 1,5
conh2 0,95 1,5 1,8 СОгСНз 1,3 1,3
СОС1 0,90 1,5 0,0 СОС1 1,4 1,2
СОСН3 0,7 1,3 0,7 ООССН3 1,55 1,55
СНО 1,5 1,0 1,0
Измеренные значения констант цис- и транс-взаимодействия Н — СН3 в замещенных пропенах и изобутенах (см. табл. 10.42) показывают, что цис-значения обычно бывают больше; однако имеющиеся исключения не позволяют использовать такого рода данные для определения, конфигураций.
10.6.2. а,$-Ненасыщенные эфиры
Исследование методом ЯМР большого ряда ^ас-транс-изомерных пар а,р-ненасыщенных эфиров позволило установить важность вкладов дальней анизотропии в экранирование протонов в олефиновых системах [169].
В табл. 10.43 приведены химические сдвиги для 24 молекул этого типа
СН3^ /Н с=с СН3СООСН2^ ХСООСН3
Как уже было показано, карбонилсодержащие группы в олефиновых системах обладают ярко выраженными эффектами дальнего экранирования по отношению к ^нс-Р-водородным атомам и цос-р-метильным группам, что позволяет использовать эти эффекты для определения конфигурации замещенного олефина. Изучение табл. 10.43 показывает, что значения т Р-протонов в разных молекулах очень чувствительны к стереохимии заместителей, причем Р-водород дезэкранирован на величину 0,5—0,9 м. д. в изомере, в котором он находится в цас-положении к карбоалкоксигруппе, по сравнению с соответствующим транс-изомером. Для цас-Р-метильной группы измеренные химические сдвиги указывают на постоянное дезэкранирование при переходе от цис- к транс-изомеру на величину ~0,25 м. д. для всех исследованных соединений. Поэтому, если имеются оба изомера а,р-ненасыщенных эфиров, обычно по спектрам протонного резонанса этих соединений легко определить их конфигурации.
Авторы работы [170] также исследовали спектры ЯМР некоторых а,|3-нена-сыщенных эфиров; полученные результаты включены в табл. 10.44, А. Они использовали более точный метод анализа наблюдаемых спектров, что позволи-
Таблица 10.43
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ цис-транс-ИЗОМЕРНЫХ ПАР а,В-НЕ11АСЫЩЕННЫХ ЭФИРОВ Г169]
СНзч .И
СНзСООСНз/ хсоосн3
Эфир Р-Н, Да fJ-СНз, Да Эфир р-сн3, д a
Метиловый эфир метакриловой кислоты 4,51 I 3,96 J 0,55 Диметнловый эфир цис-|3-метилглутаконовой кислоты 8,04 ч > 0,24 6
Метиловый эфир Р,Р-диметилакриловой кис- 8,16 Н 0,28 Диметиловый эфир транс-Р-метилглутаконовой 7,80
лоты 7,88 кислоты
Диметнловый эфир малеиновой кислоты 3,86 1 0,53 Этиловый эфир цис-р-ионилиденуксусной кислоты 7,98 . 0,27
Диметиловый эфир фумаровой кислоты 3,33 1 Этиловый эфир транс-р-ионилиденуксусной кислоты 7,71
Диметиловый эфир цитраконовой кислоты 4,25 | 0,94 7,96 j 0,24 Метиловый эфир цис-гераниевой кислоты 8,27 • 0,25
Дйметиловый эфир мезаконовой кислоты 3,31 J 7,72 Метиловый эфир транс-гераниевой кислоты 8,02
Диметиловый эфир диметилмалеиновой кис- 8,11 ч Метиловый эфир цис-фитеновой кислоты 8,14 0,25
лоты ► 0,10 Метиловый эфир транс-фитеновой кислоты 7,89
Диметиловый эфир диметилфумаровой кис- 8,01 Метиловый эфир цис-а, p-диметилкоричной кислоты 8,05
ЛОТЫ 3,57 | Метиловый эфир транс-а, Р-диметилкоричной кис- 7,75 0,30
Метиловый эфир цис-кротоновой кислоты 0,62 7,86 ! 0,25 ЛОТЫ
Метиловый эфир транс-кротоновой кислоты 2,95 J 8,11 Диметиловый эфир цис(а,Р)-транс(у,6)-р-метил- 8,00 ч
Метиловый эфир ангеликовой кислоты 4,02 1 0,85 8,03 . 0,25 муконовой кислоты > 0,25
Метиловый эфир тиглиновой кислоты 3,27 / 8,27 Диметиловый эфир транс(а,$)-транс(у,6)-Р-метил- 7,75
муконовой КИСЛОТЫ
аТ — т = Д, где индексы цис и транс характеризуют положение протона (или протонов) относительно карбоксильной группы и необязательно относятся транс цис к конфигурации эфира.
6 Для группы 3-СН2СО2СН3 Хтранс н тиис составляют 6,28'и 6,935 соответственно; Д = 0,665.
Таблица 10.44
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ И КОНСТАНТЫ ДАЛЬНЕГО СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ
А. Ненасыщенные кислоты н эфиры [148, 170]
Метнлмезаконат
Метнлцнтраконат
J™PaHC=\,56
J«"c=l,64
Б. Другие олефиновые соединения [148]
Соединение Константа 1«,3-взаимо-действия, гц Литература
.транс J\,3 1ЦиС J 1,3
Бутен-1 1,3 1,9 125
«-Метилстирол 0,7 1,4 171
•Чис-Пропенилбензол 1,4 171
транс-Пропеннлбензол 1,9 131
цис-цис-Днметнлмуконат 1,3 172
транс-транс- Днметнлмуконат 0,7 172
транс-1,2,З-Трнхлорпропен-1 а 0,8 129
цис-1,2,3-Т рнхлорпропен-1 0,4 129
цис-1,3-Днхлорпропен-1 6 0,9 129
1,2 173
транс-1,3-Дихлорпропен-1 0,5 129
1,2 173
а транс характеризует ориентацию двух атомов хлора при двойной связи. ® цис характеризует ориентацию двух хлорсодержащнх групп.
72
Глава 10
ло получить более надежные значения констант взаимодействия. На рис. 10.20 приведен спектр протонного резонанса олефиновых и метильных протонов в цис-метилкротонате. Анализ системы АВХ3 дает значения, почти точно совпадающие со значениями химических сдвигов и констант взаимодействия, наблюдаемыми в спектре. Обращает на себя внимание значительная ошибка в значениях констант взаимодействия, полученных в предположении, что спектр полностью отвечает первому порядку. Ее важно учитывать, особенно если придается значение малым различиям в константах взаимодействия. В парах исследованных авторами изомерных эфиров было показано, что константы взаимодействия Jh-chs могут быть меньше, равны или больше констант <7н-сн3
(2) Н\ /Н (з)
(1) ИдУ V00CH3 (4)
Олефиновые протоны р- Метильные протоны
Н----->-
Рис. 10.20. Спектр протонного резонанса олефиновых и .метильных протонов в цнс-метилкротонате прн 60 Мгц [170].
и, по-видимому, значения таких констант дальнего взаимодействия не следует применять для определения геометрической конфигурации. В табл. 10.44,Б приведены некоторые константы дальнего взаимодействия Н — Н, полученные по спектрам олефиновых молекул [148].
10.7. Дальнее спин-спиновое взаимодействие через олефиновые системы
Спин-спиновое взаимодействие протонов, разделенных четырьмя связями, часто наблюдается между метильными и 0-протонами в пропенах и их производных (см. табл. 10.44).
Хоффман [174] объяснил существование таких дальних взаимодействий наличием сверхсопряжения между орбиталями метильной группы и л-орби-талями (о а—л-взаимодействиях см. в разд. 5.5). Если такой механизм действительно имеет место, можно ожидать, что:
1) взаимодействие л-электрона одного из ненасыщенных атомов углерода с протонами метильной группы, находящейся у того же атома углерода, будет приблизительно равно его взаимодействию с протоном, непосредственно связанным с этим ненасыщенным углеродным атомом;
2) константы взаимодействия Jн-с==с-сн3 через тройную связь будут в два раза больше соответствующих констант через двойную связь [174, 175]. Эти ожидания подтвердились в экспериментах: во-первых, константа взаимо-
Корреляции спектров ЯМР-Н1 со строением молекул
73.
действия /снз-снз (=1 гц) в тиглиновом альдегиде имеет величину того же порядка, что и константа Тн-снз в /лракс-кротоновом альдегиде (1,6 гц),.
Тиглиновый альдегид
транс-Кротоновый альдегид
во-вторых, в пропаргилхлориде НС = С — СН2С1 константа взаимодействия /н-сн2 равна 2,6 гц.
Снайдер и Робертс [176] наблюдали дальнее 1,4-взаимодействие в трех замещенных алленах:
(СН3)2С = С = СН2
(СН3)2С = С = СНС1 СН3СН = С = СНС1
X В А
4= 3,03 ± 0,06 гц
Ц 4 = 2,14 ± 0,08 гц
Л з = ’— 5,8 ± 0,1 гц (Jдв) 71 4= +2,4 ±0,1 гц (/ах)
Противоположные знаки констант 1,3- и 1,4-взаимодействия в спектре СН3СН = С = СНО совпадают с теоретическими предсказаниями Карплуса [62] для констант дальнего взаимодействия в л-электронных системах. Величины этих констант взаимодействия объясняются сверхсопряжением, причем, считается, что важные вклады дают структуры II и III
Н----------Н
И----------Н | I
СН3 —С = С = СН <--> СН2=С—С = С <----> СН2—С = С—С:
I I I I I I
X Y X Y X Y
I II III-
Если рассматриваются эти структуры, то значение Ju = 3,9 гц, предсказанное Карплусом на основании теоретического расчета, уменьшится и может совпасть, с экспериментально наблюдаемым значением = 3,0 гц [176, 62].
. В винилформиате (IV) можно обнаружить несколько констант дальнего-взаимодействия Н — Н [370]: Ji4 = 0,6 гц; = 1,7 гц; J3i = 0,8 гц.
(1)
IV
л-Электронная система, по-видимому, является проводником таких взаимодействий, и, как было отмечено, траис-ориентированные протоны взаимодействуют сильнее цис-ориентированных протонов. Банвелл и Шеппард. [34] отметили, что дальнее взаимодействие в олефиновых системах наиболее эффективно для протонов, разделенных «зигзагообразными» связями (как в случае винилформиата) 1).
Эксперименты по двойному резонансу показали, что олефиновые протоны в циклогексене взаимодействуют как с а-, так и с р-метиленовыми протонами
х) На основании величины дальних констант взаимодействия была установлена стереохимия 3-замещенных хинуклидннов с семициклической двойной связью [519] — Прим. ред.
74
Глава 10
кольца [371]. Однако в циклопентене такого взаимодействия нет. Существует несколько примеров констант дальнего взаимодействия, которые нельзя объяснить взаимодействием а — л [34, 262] (см. разд. 5.5). Константы дальнего взаимодействия наблюдались в замещенных фуранах (разд. 10.16), хинолинах (разд. 10.20) и пиридинах (разд. 10.18).
10.8. Другие олефины
10.8.1. Сопряженные диены [459]
Как мы уже видели, применение метода молекулярных орбиталей [33] для расчета констант спин-спинового взаимодействия протонов (см. гл. 5) для всех дальних констант взаимодействия Н — Н дает положительные знаки. Этот вывод находится в противоречии с данными, полученными при использовании векторной модели Дирака, .которая предсказывает чередование знаков констант взаимодействия вдоль цепи углеродных атомов [178, 35]. Метод валентных связей [179] предсказывает для спин-спиновых взаимодействий через л-электронную систему в четной чередующейся углеводородной цепи положительные константы взаимодействия протонов, разделенных нечетным числом углерод-углеродных связей, и отрицательные константы взаимодействия для протонов, разделенных четным числом углерод-углеродных связей. Александер [125] и другие исследователи [124] провели полный анализ спектра протонного резонанса бутена-1 и показали, что константа взаимодействия протонов при углеродных атомах 1 и 3 имеет отрицательный знак.
Элвидж и Джекман [172] исследовали цис,цис-диметилмуконат и показали, что знаки констант взаимодействия чередуются вдоль цепи. Этот сопряженный диен имеет структуру
12 3 4
СН3ООС — СН = СН—СН = СН— СООСН3.
Константы взаимодействия для этой молекулы следующие: Ji2 = + H,8 гц, ,/13 = —1,3 гц и J23 = +11,3 гц. Для других изомеров этой молекулы наблюдаются аналогичные знаки констант взаимодействия.
10.8.2. Олефиновые производные металлов
Винильные производные. Исследование спектров ЯМР винильных производных металлов представляло значительный интерес, особенно с точки зрения получения информации о природе связи металл — углерод в этих соединениях. В табл. 10.26 и 10.45 приведены параметры спектров ЯМР некоторых из иссле-
Таблица 10.45
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ НЕКОТОРЫХ винильных ПРОИЗВОДНЫХ МЕТАЛЛОВ [135] <В)Н\Г_Г/Н(А> (С)Н/ \х
Соединение Химический сдвиг а, м. д.
н (А) Н(В) Н (С)
Дивинил ртуть —5,87 —5,07 —4,53
Тетравинилолово —4,79 —4,60 —4,15
Эфират тривинилалюминия —4,79 —4,54 —4,18
а Химические сдвиги измерены относительно циклогексана, взятого в качестве внешнего эталона.
Корреляции спектров ЯМР-Н1 со строением молекул 75
дованных винильных производных металлов [135, 136, 152]. Как можно видеть, химические сдвиги протонов не обнаруживают простой корреляции с электроотрицательностями металлов. Сильное экранирование винильных протонов в эфирате тривинилалюминия можно объяснить малой электроотрицательностью алюминия, однако большое различие в химических сдвигах винильных протонов в производных ртути и олова, в которых электроотрицательности Sn(IV) и Hg(II) одинаковы [135], не поддается аналогичному объяснению. Возможная причина аномальных сдвигов заключается в различных диамагнитных восприимчивостях металлов.
Методом ЯМР были исследованы винильные производные ртути [135], олова [135] и таллия [422], причем получены следующие константы взаимодействия протонов с металлами:
(1)НХ /X >с=с< (2)HZ ХН(3) •^XH(l)’ гтранс ... JXH(2)’ ги> рем ?11
(CH2 = CH)2Hg 159,6 295,5 128,5
(CH2 = CH)4Sn 90,6 183,1 98,3
(СН2 = СН)2Т1+ 805 1618 842
(СН2 = СН)Т12+ 1806 3750 2004
Константы транс-взаимодействия X — Н имеют наибольшие значения, так же как и константы mpawc-взаимодействия Н — Н в винильных системах, однако константы гем- и цис-взаимодействия X — Н в отличие от соответствующих констант взаимодействия протонов имеют приблизительно одинаковые значения х).
Виниллитий [136] дает обычный протонный спектр ЯМР типа АВС, отнесения в котором были сделаны при помощи эмпирических корреляций электроотрицательности заместителя с константами взаимодействия Н — Н. При анализе спектра были получены следующие спектральные параметры:
(В)Н^ ,Н(А) 6а = 0 м. д.; 6в== +0,511 м. д.; 6с= + 1,19 м. д.
С=С
(С)н/ ^Li /ав=19,3 гц; /ас = 23,9 гц; = гц.
Из такого отнесения сигналов следует, что винильный протон Н (А), расположенный рядом с литием, экранирован слабее остальных, чего нельзя было ожидать на основании простых индуктивных представлений. Большую роль в экранировании протонов Н(А) могут играть наведенные электронные токи. При исследовании спектра ЯМР аллиллития было показано, что это соединение при комнатной температуре существует в виде смеси быстро взаимо-превращающихся аллильных изомеров. Аналогичным образом ведет себя бромистый аллилмагний (см. разд. 10.3.3).
При исследовании винильных производных кремния * 2) были получены следующие значения констант взаимодействия для винильных протонов 1180, 442]:
(В)Ну /Н(А)
С = С /ав=14,6 гц; /ас = 20,4 гц; /вс = 3,8 гц.
(С)н/ XSi(CH3)3
х) В работе [520] исследованы спектры протонного резонанса Р-хлорвииильиых производных ртути.— Прим. ред.
2) Рассмотрение химических сдвигов протонов в спектрах вииилсилаиов проведено в работе [541].— Прим. ред.
76
Глава 10
Циклопентадиенильные производные. Известно, что ферроцен имеет сэнд-вичевую структуру (I) с двумя свободно вращающимися вокруг оси пятого-порядка циклопентадиенильными кольцами, что хорошо согласуется с его протонным спектром ЯМР, в котором наблюдается один резонансный сигнал 1181] х). В спектрах протонного резонанса Sn(C5H5)2 и РЬ(С5Н5)2 также имеется лишь один резонансный сигнал с химическим сдвигом соответственно 4,38-и 4,83 м. д. относительно циклогексана [182]. Если молекулы имеют а-свя-занные структуры (II), то циклопентадиенильные кольца должны давать, сложные спектры из-за имеющихся в кольце магнитно неэквивалентных ядер. Однако молекулы типа л-С6Н5(а-С6Н5)Ее(СО)2 дают в спектрах лишь два резонансных сигнала — по одному для каждого типа циклопентадиенильных. колец [181].
II
резонанса дициклопентадие-
Аналогичным образом в спектре протонного нилртути Hg(C6H5)2, которая должна иметь линейную a-связанную структуру, также наблюдается одиночный резонансный сигнал [183]. Следовательно, наличие одиночных резонансных сигналов в спектрах ЯМР-Н1 циклопентадиенильных производных не может служить однозначным доказательством л-связанной структуры. Отсутствие сложных мультиплетов в спектрах ЯМР-Н1 некоторых ст-связанных циклопентадиенильных производных указывает на 1,2-миграцйю металла по циклопентадиенильному кольцу со скоростью, достаточной для того, чтобы кольцевые протоны стали эквивалентными [181] 2). Соединения с л-связанными циклопентадиенильными кольцами у атома переходного металла дают одиночный резонансный сигнал в области 2,5 — 3,1 м. д. относительно бензола; протоны в a-связанных циклопентадиенильных группах экранированы слабее. Спектр протонного резонанса л-циклопентадиенилцик-лопентадиенренийдикарбонила Ci0Hi0Re(CO)2 очень сложен; в нем имеется одиночный сигнал протонов л-связанного циклопентадиенильного кольца и пять мультиплетов, обусловленных протонами второго С6Н5-кольца, которое-не является л- или a-связанным с атомом рения, а присоединено к нему за счет донорных свойств одной из двойных связей [184].
Несколькими исследователями [185, 460, 482, 483] было изучено большое-количество циклопентадиенильных производных других переходных металлов,, и во всех случаях спектры протонного резонанса подтверждали структуры,, полученные с применением других методов 3).
10.8.3. Метильные группы в сопряженных полиенах
Исчерпывающее исследование химических сдвигов метильных протонов для 64 замещенных сопряженных полиенов проведено Барбером и сотр. [186]. Резонансные сигналы метильных групп чувствительны к их молекулярному окружению, и при анализе спектров можно получать полезную информацию-
!) Спектры ЯМР-Н1 ферроцена н ряда его производных были исследованы также-в работах [522—526]. Корреляционный анализ химических сдвигов в монозамещенных ферроценах имеется в работе [527].— Прим. ред.
2) Иная точка зрения на строение дициклопентадиеннлртутн и данные о спектрах ЯМР-Н1 некоторых циклопентадиенильных соединений ртути приведены в работе [531].-^ Прим. ред.
з) О спектрах ЯМР-Н1 титаноценов см. в работах [525, 526]; данные о спектрах цйклопеитадиенилтрикарбонилов Мп и Re приведены в работе [532].— Прим. ред,.
Корреляции спектров ЯМР-Н1 со строением молекул
77
о положении метильных групп в молекуле. Метильные группы, связанные с олефиновым атомом углерода, обычно имеют значения т = 8,30 — 8,40. Однако метильные группы у неконцевых двоесвязных атомов углерода полиеновой цепи дают сигналы при т =7,95 — 8,15 (концевые метильные группы несколько более экранированы, для них т = ~8,2). Хорошим примером полиена, содержащего как концевые, так и «внутрицепные» метильные группы, может служить ликонен. Строение ликонена и химические сдвиги различных метильных групп этой молекулы следующие:
10.8.4. Кольцевые токи в аннуленах
Аннулены представляют собой циклические молекулы, у которых в кольцах чередуются одиночные и двойные связи. В таких молекулах, как [14]-анну-лен (I), можно было бы ожидать возникновения кольцевых токов, если бы циклический скелет молекулы обладал ароматическими свойствами (см. разд. 4.5). Такие кольцевые токи привели бы к сильному экранированию внутренних протонов и к необычно слабому экранированию внешних протонов. Однако спектр протонного резонанса [14]-аннулена содержит одиночный сигнал с химическим сдвигом, аналогичным химическому сдвигу олефиновых протонов в циклооктатриене, что свидетельствует о неароматичности системы [3651. Вместе с тем в спектре протонного резонанса [18]-аннулена (II) наблюдаются два широких резонансных сигнала — один для внутренних и один для внешних
протонов (х = 1,1 и 11,8 соответственно), что указывает на ароматический характер молекулы [3651. При исследовании спектров ЯМР-Н1 некоторых дегидроаннуленов было также показано, что они имеют ароматический характер [365].
АЦЕТИЛЕНЫ
10.9. Ацетилен и родственные молекулы
Аномально высокое экранирование ацетиленовых протонов, промежуточное между экранированием протонов в алканах и алкенах, было предметом нескольких теоретических исследований. В настоящее время твердо установлено, что такое высокое экранирование обусловлено влиянием диамагнитной
Таблица 10.46
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ НЕКОТОРЫХ АЦЕТИЛЕНОВЫХ ПРОИЗВОДНЫХ, ИЗМЕРЕННЫЕ ДЛЯ РАЗБАВЛЕННЫХ РАСТВОРОВ В СС14 И ЦИКЛОГЕКСАНЕ. [189]
Соединение Химический сдвиг, т-значения Константа взаимодействия
= СН - СН2 JHH’ г1(
НС = ССН2С1 а 7,76 6,06 2,6
НС ССН2Вг а 7,76 6,32 2,7
НС = ССН21 а 7,81 6,48 2,8
НС = ССН2ОН 6 7,67
НС = СС (СН3)=СН2б 7,29
НС = СС6Н6 б 7,07
а Измерено в виде разбавленных растворов в циклогексане [189].
б Измерено в виде разбавленных растворов в СС14 (см. приложение I).
Таблица 10.47
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ И КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ В СПЕКТРАХ НЕКОТОРЫХ МОНОЗАМЕЩЕННЫХ АЦЕТИЛЕНОВ, ИССЛЕДОВАННЫХ В ВИДЕ ЧИСТЫХ ЖИДКОСТЕЙ [188]
Константа
Соединение взаимодействия, гц = СН, м. д. м. д. Состояние
НС = ССН2С1 2,65 — 1,19 7,37 Жидкий
НС = ССН2Вг 2,70 — 1,31 7,25 »
НС = ССН2ОН 2,50 — 1,32 7,24 »
/СО—
hc = cch2-n< 2,55 —0,375 8,18 Разбавленный рас-
Xco~U твор в бензоле
НС = ССН2—О—J 2,51 — 1,19 7,37 Жидкий
О
НС = CCH2-N(CH3)2 2,51 —1,02 7,54 »
HCe=C-CH2-N(C2H5)2 2,55 »
+ /СН3
НС = CCH2N $-СН3 I-Хс2н5 2,60 Раствор в воде
НС = ССН(ОН)СН3 2,15 —1,23 7,33 Жидкий
НС = ССН(ОН)С6Н5 2,20 — 1,02 7,54 Раствор в СС14
НС =5 CCH(CH3)N(C2H5)2 2,20 Жидкий
НС = С —CH(CH3)N 7 О 2,10 »
НС = CCH(CH3)NHCH(CH3)2 2,20 —1,21 7,35
НС = CCH(CH3)NH(h-C4H9) 2,10 Раствор в СС14
НС = CCH(CH3)N(CH3)2 2,00 Раствор в СС14
а Химические сдвиги были измерены и миллионных долях (м. д.) относительно циклогексана, взятого в качестве внутреннего эталона; т = 8,56 +^СвН12'
Корреляции спектров ЯМР-Н1 со строением молекул
79
анизотропии тройной связи (см. разд. 4.4). Исследование т-значений ацетиленовых протонов показывает, что они близки к химическим сдвигам метиленовых и метиновых протонов (см. табл. 10.46), и поэтому определение ацетиленовых протонов методом ЯМР довольно затруднительно. Однако химические сдвиги ацетиленовых протонов сильно зависят от природы растворителя, что может быть использовано для их характеристики. Например, добавление пиридина к разбавленному раствору монозамещенного ацетилена в четыреххлористом углероде приводит в большинстве случаев к дезэкранированию ацетиленовых протонов примерно на 1 м. д. [187].
В табл. 10.47 приведены химические сдвиги ацетиленовых протонов в ряду монозамещенных ацетиленов, исследованных в виде чистых жидкостей [188]: изменение химических сдвигов составляет лишь 0,3 м. д., что указывает на слабую зависимость экранирования ацетиленовых протонов от заместителя, если предположить, что внутримолекулярные взаимодействия в различных соединениях одинаковы. В литературе имеется лишь незначительное количество данных по исследованию ацетиленов' в разбавленных растворах в инертных растворителях (см. табл. 10.46); наименьшие химические сдвиги наблюдаются в молекулах, в которых тройная связь сопряжена с другими электронными системами *).
10.9.1. Гамгенопроизводные метилацетилена ХСН2С = СН
Галогенметилацетилены дают в спектрах протонного резонанса два мультиплета: дублет и триплет — с отношением интенсивностей 2:1. Типичным
HCsCCH2Bt
Рис. 10.21. Спектр протонного резонанса пропаргнлбромнда при 40 Мгц [189].
примером является спектр пропаргнлбромнда, показанный на рис. 10.21. В табл. 10.46 приведены химические сдвиги и константы спин-спинового взаимодействия для трех пропаргилгалогенидов [189]. Как указано в табл. 10.46, химические сдвиги получены для разбавленных растворов в циклогексане с использованием тетраметилсилана в качестве внутреннего эталона и, по-види-
J) Спектры ЯМР-Н1 1-, 2-, 3- н 4-алкннов исследовали сьв работе Лнппмаа и сотр. [517].— Прим. ред.
so
Глава 10
мому, очень слабо, зависят от межмолекулярных взаимодействий. Как и следовало ожидать из рассмотрения индуктивных эффектов, экранирование протонов —СН2Х уменьшается с увеличением/^электроотрицательности галогена (аналогично изменению экранирования а-СН2-протонов в алкилгалогени-дах [8]). При изменении природы галогена наблюдаются лишь небольшие изменения химических сдвигов ацетиленовых протонов (~0,05 м. д.). Этот эффект по величине аналогичен влиянию галогенов на экранирование у-про-тонов в ряду «-пропилгалогенидов [8]. Все три константы взаимодействия в пределах ошибки эксперимента равны, и малому различию между ними нельзя придавать никакого значения.
10.10. Константы спин-спинового взаимодействия в ацетиленах
В табл. 10.46 и 10.47 приведены константы дальнего спин-спинового взаимодействия, измеренные для. ряда ацетиленовых соединений (во всех случаях взаимодействующие ядра разделены четырьмя связями). Было показано, что в общем случае константы взаимодействия почти не зависят от природы заместителей. Например, молекулы общей формулы RCH2C = СН имеют значения 7сн2-сн, равные 2,5—2,7 гц, тогда как для производных типа RR'CHC = СН 7сн-сн = 2,0—2,2 гц. Уменьшение констант взаимодействия при введении нового заместителя может быть обусловлено изменением валентных углов [188]. Согласно Карплусу [62], в этиленовых, алленовых и ацетиленовых соединениях существенный вклад во взаимодействие протонов вносят л-электроны. Карплус показал также, что если л-электроны существенно влияют на механизм взаимодействия, то константы имеют положительные знаки в том случае, когда взаимодействующие протоны разделены четным числом атомов углерода, и отрицательные — в противном случае. Для ацетиленовых систем имеется вполне хорошее согласие между наблюдаемыми константами и теоретическими оценками Карплуса, например:
J (теория) J (эксперимент)
Н—С=С—Н +4,6 9,1
Н — CsC — СН/ —3,7 , от—2,1 до—2,9
/-НС — С=С—СН</ +2,9 2,9. (знак неизвестен)
Для этилацетилена НС = СС2Н5 при полном анализе спектра А2В3С было показано, что две наблюдаемые константы имеют противоположные знаки: 7сн3—сн2 = +7,2 гц; 7сн2-сн =—2,4 гц и 7снз—сн =0 гЦ [190]. Из спектра метилацетилена было найдено, что константа взаимодействия 7сн3-сн = = 2,8 ± 0,3 гц [192] близка по величине к соответствующей константе в этил-ацетилене (/сн2-сн = —2,4 гц) и других замещенных ацетиленах. Из этого следует, что электронная структура системы — С — С = СН при -замещении изменяется незначительно [190]. Снайдер и Робертс [193] показали, что протоны в полиацетиленах и алленах обнаруживают необычное дальнее спин-спиновое взаимодействие: для этих молекул были измерены константы спин-спинового взаимодействия протонов, разделенных несколькими (от пяти до девяти) связями (см. табл. 5.9). По-видимому, можно считать, что механизм дальнего взаимодействия протонов в полиацетиленовых системах связан со сверхсопряжением. Это дополняет карплусовскую схему спин-спинового взаимодействия, основанную на методе валентных связей. Для объяснения
Корреляции спектров ЯМР-Н1 со строением молекул
81
больших констант дальнего спин-спинового взаимодействия постулируется, что в молекулах типа
^СН— (С=С)П — сн/
важную роль играет резонансная структура
Н----------Н
>С=(С = С)П = С/
Гофман и Гроновиц [174, 175] также считают, что о — л-взаимодействия вносят значительные вклады в константы спин-спинового взаимодействия. Согласно утверждению этих авторов, взаимодействие метильных протонов с другими протонами, разделенными тройной связью, должно быть приблизительно в два раза больше взаимодействия таких протонов, разделенных двойной связью, при условии, что гиперконъюгационный механизм взаимодействия является преобладающим. Действительно, наблюдаемые константы взаимодействия полностью подтверждают это. Константы дальнего взаимодействия в винилацетиленах [194, 195] служат также подтверждением теории, согласно которой основным фактором, влияющим на взаимодействие протонов в ацетиленовых и олефиновых системах, является сверхсопряжение.
(1)Н /14 = —2,4 гц
Х'С
С Н(2)
С —С Ji2 = 0,8 гц
(4)Н ХН(3) J13 = 1,0 гц
Возможность обнаружения сверхсопряжения методом ЯМР, несомненно, будет стимулировать новые исследования констант дальнего взаимодействия этого типа.
10.11. Влияние растворителей на химические сдвиги протонов в ацетиленах
Ацетилен может самоассоциировать [196], а также образовывать слабые водородные связи с молекулами, содержащими электроотрицательные центры (например, с ацетоном, ацетонитрилом, пиридином) [187, 189, 197] х). Поэтому не удивительно, что химические сдвиги протонов в ацетилене и его производных существенно зависят от природы растворителя. Эти эффекты детально рассмотрены в разд. 10.39.
АРОМАТИЧЕСКИЕ МОЛЕКУЛЫ
10.12. Производные бензола
10.12.1. Влияние растворителей на спектры протонного резонанса ароматических соединений
Несмотря на значительное число работ, посвященных исследованию экранирования протонов в ароматических молекулах, большинство полученных результатов не имеют большого теоретического значения из-за того, что в экспериментах не было исключено влияние межмолекулярных взаимодействий на экранирование. Такие взаимодействия можно свести к минимуму,
*) Водородные связи, образуемые ацетиленовыми соединениями, были исследованы с помощью спектроскопии ЯМР-Н1 также в работах [533—535].— Прим. ред.
6-489
82 Глава 10-
исследуя бесконечно разбавленные растворы образцов в инертных растворителях. Прежде чем приступать к изучению спектров ЯМР ароматических соединений, необходимо выяснить, в какой мере влияют на них межмолекулярные взаимодействия (см. разд. 10.39.2) [198—201, 207].
10.12.2. Монозамещенные бензолы
Шписецки и Шнейдер [208] детально исследовали спектры ЯМР-Н1 и ЯМР-С13 ряда монозамещенных бензолов, чтобы выяснить влияние заместителей на химические сдвиги в этих молекулах. Исследуя все образцы в виде 5%-ных растворов в циклогексане, авторы надеялись исключить какие-либо межмолекулярные взаимодействия. В более ранних исследованиях этого класса соединений [154, 156, 198, 203, 204, 209, 210] было высказано предположение о том, что экранирование ароматических протонов зависит от индуктивного влияния и эффекта сопряжения заместителей. Шписецки и Шнейдер [208] нашли, необходимым для полного объяснения наблюдаемых химических сдвигов рассмотреть влияние магнитной анизотропии заместителя. В табл. 10.48
Таблица 10.48
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ а НЕКОТОРЫХ МОНОЗАМЕЩЕННЫХ БЕНЗОЛОВ, ИЗМЕРЕННЫЕ ДЛЯ 5%-НЫХ РАСТВОРОВ
В ЦИКЛОГЕКСАНЕ [208]
Заместитель Сдвиг протона относительно бензола
орто мета пара
F + 18,5 + 1.4 + 13,0
С1 — 1,2 + 1,5 +7,0
Вг — 13,4 +5,1 + 1,8
I —24,0 + 15,0 + 2,0
ОСН3 + 26,0 +2,4 + 22,0
nh2 +45,3 + 12,2 +37,5
N(CH3)2 +36,0 +6,0 +36,9
сно —34,8 —12,8 — 16,5
no2 —56,9 — 12,5 —20,0
а Даиы в герцах (гц) относительно бензола при 60 Мгц. Положнтель-иые сдвиги указывают на большее по сравнению с бензолом экранирование протонов, отрицательные сдвиги указывают иа меньшее экранирование.
приведены химические сдвиги для орто,- мета- и /шря-протонов в исследован-ных монозамещенных бензолах. Отнесения химических сдвигов проводились при помощи селективно дейтерированных молекул х). (В табл. 12.14 даны химические сдвиги С13 в этих молекулах.) На рис. 10.22 химические сдвиги протонов представлены в виде диаграммы. Спектры протонного резонанса хлорбензола и его двух дейтерированных аналогов приведены на рис. 10.23. Дейтерирование приводит к существенному уширению линий, обусловленному неразрешенной мультиплетной структурой и квадрупольными эффектами.
По спектрам ЯМР-С13 можно определить относительные электронные плотности на каждом углеродном атоме. Изменение природы заместителя будет сопровождаться эффектом сопряжения, влияющим главным образом
*) При помощи селективного дейтерирования удалось определить химические сдвиги протонов в фенилмеркурхлориде [536].— Прим. ред.
Корреляции спектров ЯМР-Н1 со строением молекул
83
на распределение л-электронов, что в свою очередь скажется на величине электронной плотности вокруг атомов углерода. Полученные Шписецки и Шнейдером результаты позволяют оценить, насколько сильно изменение электронной плотности вокруг атомов углерода влияет на экранирование протонов, связанных с этими углеродными атомами.
Экранирование орто-протонов. Экранирование орто-протонов в моногало-гензамещенных бензолах нельзя объяснить лишь индуктивным влиянием и эффектом сопряжения, поэтому было сделано предположение о том, что
Рис. 10.22. Химические сдвиги монозамещенных бензолов [208].
п м
11
м по
м по
I_____J
м по
О
! 0,2 0,i О,Б 0,8 to МЛ
। | "г | । I । | । ~1
1 1 । । । । ।
0,2 0,1, О,Б 0,8 м.Э.
Рис. 10-. 23. Спектры протонного резонанса хлорбензола и его двух дейтерированных аналогов при 60 Мгц [208]
экранирование подвержено, кроме того, значительному влиянию диамагнитной анизотропии заместителя, особенно в случае бром- и иодпроизводных, в которых такие эффекты должны быть значительными. Если рассматриваются не галогены, а другие заместители, влияние диамагнитной анизотропии значительно меньше и экранирование протонов обусловлено главным образом эффектами сопряжения л-электронов. Сильные электроноакцепторные заместители обусловливают существенное дезэкранирование орто-протонов (при X = NO2 химический сдвиг орто-протонов равен —0,95 м. д.), а электронодонорные заместители их значительно экранируют (X = NH2, 6орто = +0,75 м. д. относительно бензола) г).
Экранирование пара-протонов. Главным фактором, влияющим на экранирование пара-протонов и пара-углеродных атомов является эффект сопряжения. Шписецки и Шнейдер [208] обнаружили линейную корреляцию между химическими сдвигами пара-протонов и пара-углеродных атомов в монозамещенных бензолах. Это наблюдение указывает на то, что эти сдвиги обусловлены
х) В работе [537] на основании спектров ЯМР при 100 Мгц обсуждается вопрос о распределении электронной плотности в иодбензоле и дихлориде иодбензола.— Прим. ред.
6*‘
84
Глава 10
одним и тем же фактором, а именно изменениями л-электронной плотности на napa-углеродном атоме.
Экранирование мета-протонов. Отсутствие корреляции между химическими сдвигами мета-протонов и мета-углеродных атомов указывает на то, что экранирование мета-протонов зависит в существенной степени от всех трех факторов — индуктивного влияния, эффекта сопряжения и магнитной анизотропии. Оценки вклада в экранирование мета-протонов, обусловленного «электрическим полем», постулированного Бакингемом [27], хорошо согласуются с данными для одних молекул (нитробензол) и хуже для других (иод-и бромбензолы). Шписецки и Шнейдер считают, что экранирование орто-и пара-протонов за счет эффекта «электрического поля» подавляется другими эффектами. Хотя мета-протоны в некоторой степени подвержены влиянию «электрического поля», на экранировании мета-углеродных ядер этот эффект сказывается крайне слабо.
10.12.3. Корреляция химических сдвигов протонов с о-константами Гаммета в монозамещенных бензолах т)
Химическая реакционная способность в мета- и пара-положениях в различных замещенных бензолах может быть выражена о-константами Гаммета [202]. Если эти константы каким-либо образом связаны с электронными плот-
76
12
«о' 4 ‘О
О
-8
0,6
0,4 л 5 0,2
3 -
“о о
-0,2
N(CH,)2
Химический „ сдвиг пара-С
\oF о \ rfl сн, V _
н °
1 Ч COCH,
д*. °
Ж
нсоо
-NHQ^NfcHj^ Химический ~
сдвиг пара-Н1 Чосм3
НСОо
J___L.
=1
-0,8 -0,4 О 0,4 0,8
е пара
Рис. 10.24. Зависимость химических сдвигов napa-Н1 и пара-С13 моиозаме-щеииых бензолов от а-констант Гаммета [208].
ностями на мета- и пара-углеродных атомах, то можно ожидать наличия корреляций о-констант Гаммета (или модифицированных констант Тафта [140], отражающих индуктивное влияние и эффект сопряжения) с химическими сдвигами соответствующих ядер Н1 и С13.
Многие исследователи пытались показать существование таких корреляций [140, 154, 156, 203—206]. Шписецки и Шнейдер критически рассмотрели возможность корреляций такого типа для монозамещенных бензолов. Вследствие того что константы Гаммета получены при исследовании дизамещенных бензолов, они не могут быть корректно применены к монозамещенным производным. Кроме того, константы Гаммета получены на основании кинетических данных и, по-видимому характеризуют распределение электронной плотности в переходном состоянии, а не в невозбужденной молекуле. Наконец, экранирование протонов и углеродных атомов обусловлено не только электронной плотностью вокруг
ядер (возможны также вклады диамагнитной анизотропии и „парамагнетизма, не обнаруживающие корреляций с химической реакционной способностью). Несмотря на эти возражения, было показано, что химические сдвиги пара-С13 и пара-Н1 в монозамещенных бензолах обнаруживают приблизительно линейную корреляцию с о-константами Гаммета (см. рис. 10.24). Химические сдвиги мета-протонов очень плохо коррелируются с о-константами Гаммета и Тафта.
До работы Шписецки и Шнейдера была опубликована работа Корио
Д По вопросам корреляции химических сдвигов в спектрах ЯМР с о-коистаитами см. обзор В. Ф. Быстрова в книге [538]; корреляция химических сдвигов с л-плотиостями рассмотрена в работе [593].— Прим. ред.
Корреляции спектров ЯМР-Н1 со строением молекул
85
и Дейли [156], в которой была предпринята попытка оценить относительные электронные плотности в орто-, мета- и пара-положениях к различным заместителям для большого ряда монозамещенных бензолов. К сожалению, в данной работе не было предпринято мер для исключения межмолекулярных взаимодействий, поэтому трудно говорить о теоретическом значении этих измерений.
10.12.4. Дизамещенные производные бензола
Диль [211] показал, что химические сдвиги протонов в мета- и пара-диза-мещенных бензолах, измеренные для разбавленных (5 мол.%) растворов в гексане, могут быть рассчитаны эмпирически в предположении, что влияние заместителя на химические сдвиги аддитивно. Влияние заместителя X (в молекуле С6Н5Х) на экранирование орто-, мета- и пара-протонов обозначим соответственно через S0-x, $м-Х и Sn.x. Аналогично для заместителя Y в молекуле (C6H5Y) будем иметь S0-y, Sm-y и Sn.y. Если влияние заместителей аддитивно, то для трех пара-дизамещенных бензолов, имеющих заместители X, Y и Z, влияние заместителей на химические сдвиги будет определяться следующими выражениями:
n-CeH4XY: dxx=S0.x+S^_Y; 60x? = S0.Y + Slt.x; (10.12)
n-C6H4YZ:6Y2=S0.Y + Sjtt.z; d™ = 5^+5^; (10.13)
n-C6H4XZ : 6x/x=S0.x-i-S.„.z; 6XZZ = S^ + S^, (10.14)
а химические сдвиги между неэквивалентными ядрами в молекуле запишутся в виде
AXY = 6 SX-Y = 5о-Х + Sm-Y — So-Y~~ SM-X’
Д YZ = S?-Y— = So-Y + 5ai-Z — 5o-Z ~ Sm-Y'>
a — °o-X °o-Z_— °o-X ’ dAt-Z °o-Z °ai-X-
Если вышеприведенные соотношения верны, то при условии, что известны значения Axz и AYZ, можно рассчитать различия в химических сдвигах AXY, так как
axy=axz_ayz
В табл. 10.49 приведены наблюдаемые и рассчитанные различия между химическими сдвигами неэквивалентных протонов в ряду дизамещенных
Таблица 10.49 предсказанные и наблюдаемые различия между ХИМИЧЕСКИМИ СДВИГАМИ НЕЭКВИВАЛЕНТНЫХ ПРОТОНОВ В РЯДУ napa-ДИЗАМЕЩЕННЫХ БЕНЗОЛОВ n-C6H4XY [211]
Заместитель дХУ Заместитель ДХУ
X Y набл. рассчит. X Y иабл. рассчит.
С1 F —0,32 —0,31 CN СНз —0,23 —0,26
Вг F —0,52 —0,53 no2 F — 1,10 — 1,00
Вг С1 —0,22 —0,21 no2 СН3 —0,82 —0,83
I F -0,85 —0,86 ОСН3 F +0,11 +0,10
I С1 —0,53 —0,53 ОСН3 Вг +0,62 +0,63
I Вг —0,33 —0,33 nh2 F +0,30 +0,22
СН3 F —0,18 —0,17 nh2 С1 +0,58 +0,54
СН3 Вг +0,37 +0,35 nh2 Вг +0,76 +0,68
СНз С1 +0,15 +0,14 nh2 I + 1,04 + 1,10
СН3 I +0,68 +0,69 он С1 +0,45 +0,48
CN F —0,52 —0,42 он Вг +0,67 +0,66
CN С1 —0,11 —0,09 он СНз +0,22 +0,29
86
Глава 10
бензолов: наблюдается хорошее совпадение для большинства исследованных молекул [211]. Все наблюдаемые химические сдвиги приведены относительно сигнала, обусловленного 5 (мол.)%-ным раствором бензола в гексане. На самом деле химические сдвиги были измерены относительно хлороформа, применяемого в качестве внешнего этанола, и пересчитаны в шкалу, соответствующую бензолу в качестве внутреннего этанола. Несмотря на то что во всех таких случаях делаются поправки на диамагнитную восприимчивость, использование хлороформа позволяет избежать внутримолекулярных взаимодействий, которые имели бы место, если бы в качестве внутреннего этанола использовали непосредственно бензол.
Рассмотрение одних лишь лара-дизамещенных бензолов не дает возможности рассчитать абсолютную величину влияния заместителей. Чтобы произвести такой расчет, необходимо рассматривать химические сдвиги в мета-диза-мещенных бензолах с точки зрения влияния заместителей S0.x, Su.x и т. д. Если для мгта-дизамещенной молекулы м-С6Н4Х2 принять обозначения
Н X нОн Н X то для вкладов в химические сдвиги, обусловленных влиянием заместителей, можно написать выражения:
S2 = 2SO.X,
64 = se = S0_x + Sn_x,
65 = 25^.
Если рассматривается napa-дизамещенная молекула
Н Н
то вклады будут следующие:
= 63 = = б6 = •S0.x + •$„_х=дн.
Следовательно, эти соотношения позволяют легко проверить концепцию аддитивности, поскольку, если она верна, химический сдвиг протонов в п-СбН4Х2 должен быть равен среднему значению химических сдвигов б2 и б5 в молекуле м-С6Н4Х2. Изучение табл. 10.50 ясно показывает, что для большинства исследованных соединений концепция аддитивности применима.
Таблица 10.50
НАБЛЮДАЕМЫЕ И РАССЧИТАННЫЕ ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ В МОЛЕКУЛАХ п-С6Н4Х2 (5%-НЫЙ РАСТВОР В ГЕКСАНЕ) [211]
Заместитель .И-С0Н4Х2 -Е-(бг + бб). м. д. п-СвНдХг м. д.
Вг (—0,44+0,21)/2= —0,115 —0,09
С1 (—0,10+0,08)/2 = —0,01 +0,01
ОСН3 (+0,76+0,17)/2= +0,465 +0,49
СО2СН3 (—1,49—0,19)/2= — 0,84 —0,86
Корреляции спектров ЯМР-Н1 со строением молекул
87
Для расчета абсолютных значений вкладов в химические сдвиги, обусловленных влиянием заместителей, необходимо исследование жта-дизамещен-ных молекул типа л-С6Н4Х2 и At-C6H4Y2; значения S0.x и S«_x могут быть затем получены непосредственно по химическим сдвигам б2 и б5. Зная значение S0.x, можно из значения б4 получить вклад Sn-x- В табл. 10.51 приведены значения абсолютных вкладов S0_Br, Д«-вг и Sn.Br, рассчитанные по наблюдавшимся для ж/иа-дибромбензола химическим сдвигам.
Таблица 10.51
АБСОЛЮТНЫЕ ВКЛАДЫ S0.Br, S^.Br и S„.Br В ХИМИЧЕСКИЕ СДВИГИ КОЛЬЦЕВЫХ ПРОТОНОВ
Наблюдаемый химический сдвиг, м. д.
Рассчитанный эффект заместителя, м. д.
62= —0,44
64 = б6 = —0,16
65= +0,21
50-вг= —0,22
5„.вг= +0,06
5ж.вг=+031
Таблица 10.52
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ НЕКОТОРЫХ СОЕДИНЕНИЙ п-С6Н4ХУ (5%-НЫЙ РАСТВОР В ГЕКСАНЕ) [211]
Заместитель • XY 6o-X’ M. Д. • XY 6o-Y’ M. Д. Заместитель axy Ort V» o-X M. Д. • XY 6o-Y' м. Д.
X Y X Y
F F +0,31 +0,31 Br OCH3 —0,08 +0,54
F С1 +0,25 —0,07 I I —0,14 —0,14
F Вг +0,36 —0,16 I CH3 —0,30 +0,38
F I +0,41 —0,44 CH3 CH3 +0,24 +0,24
F СН3 +0,41 +0,23 CH3 CN +0,05 —0,18
F CN +0,12 —0,40 CH3 no2 —0,03 —0,85
F no2 +0,11 —0,99 OCH3 OCH3 +0,49 +0,49
F осн3 + 0,35 +0,46 F nh2 +0,51 +0,81
С1 Cl +0,01 +0,01 Cl nh2 +0,16 +0,74
С1 Br +0,08 —0,14 Br nh2 —0,03 +0,73
С1 I +0,19 —0,34 I nh2 —0,19 +0,85
С1 CH3 +0,09 +0,24 OH Cl +0,55 +0,10
С1 CN —0,17 —0,28 OH Br +0,62 —0,05
Вг Br —0,09 —0,09 OH CH3 +0,55 +0,33
Вг I +0,02 —0,31 CO2 —CH3 CH2—CH3 —0,86 —0,86
Вг CH3 —0,08 +0,29
В табл. 10.52 приведены значения химических сдвигов большого ряда соединений типа n-C6H4XY; эти данные в сочетании с абсолютными значениями вкладов заместителей, найденными для молекул м-С6Н4Х2 и по уравнениям (10.12) — (10.14), позволяют оценить абсолютные значения вкладов в химические сдвиги для большинства заместителей. В табл. 10.53 представлены рассчитанные значения абсолютных вкладов от различных заместителей в химические сдвиги ароматических протонов. При использовании этих значений было получено поразительно хорошее совпадение между рассчитанными и наблюдаемыми значениями химических сдвигов для .мета-дизамещенных
88
Глава 10
Заместитель So з.н Sn
nh2 +0,68 +0,22
он +0,50 +0,16
ОСН3 +0,42 +0,10 +0,33
F +0,25 +0,01
СН3 +0,17 +0,13 +0,17
С1 —0,05 +0,06 +0,13
Таблица 10.53
АБСОЛЮТНЫЕ ВКЛАДЫ НЕКОТОРЫХ ЗАМЕСТИТЕЛЕЙ В ХИМИЧЕСКИЕ СДВИГИ КОЛЬЦЕВЫХ ПРОТОНОВ [211]
Заместитель So Sn
Вг —0,22 +0,11 +0,06
CN —0,35 —0,13
I —0,41 +0,22
СО2СН3 —0,74 —0,10 —0,20
no2 —0,98 —0,21
молекул. Наилучшим образом это. можно показать на примере л-хлорнитро-бензола:
NO2 I —0,98/4—0,98 — 0,21^—0,21
+ 0,06
+ 0,13,4\ —0,05
-г 0,06!^JL Cl
— 0,05
no2
— 0,85/4—0,103
—ojsl^J—Cl
(Набл.)
(Рассчит.)
Для opmo-дизамещенных бензолов совпадение намного хуже; причем это наблюдается для всех замещенных бензолов, обладающих значительными дипольными моментами. Например, в случае 1,2,3-трихлорбензола (р, = 0,8D) рассчитанные химические сдвиги сильно отличаются от измеренных значений
С1
С,\А/С1 4-0,14^+0,14 + 0,25
(Рассчит.)
С1 С1\А/С| - O.Ut' J,— 6,07 - +0,21
(Набл.)
Напротив, в случае симметричного 1,2,4,5-тетрахлорбензола, не имеющего дипольного момента, наблюдается отличное совпадение между наблюдаемыми и рассчитанными значениями химических сдвигов.
р, -—0,32 р, р, —0,28 р,
\у\/
I SI IJI
Cl/'^'O+l С1/_>/>С1
(Рассчит.) (Набл.)
Аналогичная ситуация имеет место и для мезитилена.
Диль [211] не пытался объяснить влияние заместителей в бензоле наличием электрического поля, обусловленного постоянным дипольным моментом ароматической молекулы, поскольку такое приближение дает плохие результаты для некоторых замещенных бензолов (например, галогенбензолов) и, кроме того, не отражает эффектов сопряжения в бензольных кольцах. Приближение Бакингема [480], по-видимому, справедливо лишь в тех случаях, когда эффекты сопряжения невелики. Так, для нитробензола, в котором сопряжение мало, значения химических сдвигов, предсказанные в приближении электри-
Корреляции спектров ЯМР-Н1 со строением молекул
89>
ческого поля, хорошо согласуются с измеренными значениями, тогда как для фторбензола, в котором сопряжение велико, совпадение рассчитанных и наблюдаемых значений очень плохое. Приведенное Дилем [211] объяснение полученных им значений для влияния заместителей основано на о-константах Гаммета. Для дизамещенных бензолов можно ожидать лучшей корреляции экранирования с а-константами Гаммета, чем для монозамещенных бензолов. Фартинг и Нем [212] показали, что о-константа Гаммета для орто-положения к заместителю (оо) в замещенном бензоле состоит из электронного (оэ) и стерического-
Рис. 10.25. Влияние заместителей в орто-положениях So ла константу Гаммета в пара-положении <т„ для некоторых заместителей [211 ].
(ост) вкладов. Было показано, что электронный вклад (оэ) обусловлен как индуктивным влиянием (еги), так и эффектом сопряжения (<тс) Д Электронная плотность в орто- и лара-положениях к заместителю приблизительно одинакова. Отсюда, если константу Гаммета для лара-положения обозначить через оп, то
<ТП = 0э = Ои -р ос-
Наблюдающиеся для замещенных бензолов химические сдвиги свидетельствуют в пользу такой интерпретации распределения электронной плотности в этих молекулах. Если построить зависимость влияния заместителя ъорто-положении So от оп, то окажется, что большинство заместителей дают вклады, лежащие на прямой линии, как это показано на рис. 10.25. Галогены не подчиняются этой зависимости, однако хорошо коррелируются между собой (наклоны этих прямых существенно отличаются друг от друга). Еще одним заместителем, не подчиняющимся общей зависимости, является CN-rpynna. Такое поведение вышеуказанных групп не удивительно, поскольку все они вследствие диамагнитной анизотропии оказывают сильное влияние на орто-положения. Линейная зависимость для обычных заместителей может быть выражена уравнением
So = 1,17оп.
Разделение индуктивного эффекта и эффекта сопряжения заместителей в бензольном и фурановом кольцах по данным спектров ЯМР-Н1 моно- и дизамещенных производных было рассмотрено в работе [539].— Прим. ред.
90
Глава 10
Аналогичные линейные зависимости найдены для влияния заместителей в мета- и пара-положениях SM и Sn:
SM = — 0,30on + 0,06, Sn= — 0,49on + 0,10.
Брей и Лоусон [213] вслед за Дилем предприняли попытку разработать схему для предварительной оценки химических сдвигов протонов в полизаме-щенных бензолах. Полученные ими результаты во многом отличаются от данных, приведенных Дилем [211]. Было показано, что галогены в общем случае дают неудовлетворительное согласие между рассчитанными и измеренными химическими сдвигами и что лщ/па-дизамещенные соединения также
Таблица 10.54
НАБЛЮДАЕМЫЕ И РАССЧИТАННЫЕ ХИМИЧЕСКИЕ СДВИГИ а НЕКОТОРЫХ ЗАМЕЩЕННЫХ ФЕНОЛОВ [213]
Соединения Положение в кольце Химический иабл. СДВИГ, м. д. рассчит.
ОН 1
H3C-|Z\-CH3 3,5 0,35 0,50
5k/3 4 ОН | 4 0,53 0,54
Н3С-/\-СН3 5 0,53 0,59
4 он 4 0,75 0,77
н3с-У\-сн3 5U3 СН3 он 3,5 0,69 0,73
б/\-СН3 6 1,00 1,03
НзС-^-СНз 4 4 0,85 0,91
а Химические сдвиги измерены в миллионных долях (м. д.) относитель-но бензола в качестве внешнего эталона. Образец и эталон исследовались в виде разбавленных растворов в СС14.
не согласуются с предполагаемой схемой аддитивности. Во многих других случаях (см. табл. 10.54) схема аддитивности дает блестящее согласие рассчитанных и измеренных химических сдвигов (приведенные в таблице химические сдвиги получены для сильно разбавленных растворов с бензолом в качестве внешнего эталона). Показано, что схема аддитивности не позволяет
Корреляции спектров ЯМР-Н1 со строением молекул
91
предсказать химические сдвиги в пентазамещенных бензолах и 2,6-дизаме-щенных фенолах. Удивительно, что дальнейшее замещение в последних приводит к уменьшению различий между наблюдаемыми и рассчитанными значениями (см. табл. 10.54). Смит [214] также получил значения So и SM, используя приближение, аналогичное использованному Дилем; в приложении IV приведены такие значения, полученные различными исследователями.
Мартин [215] провел точное измерение химических сдвигов протонов для большого числа mz/ш-дизамещенных бензолов и показал, что полученные значения подчиняются соотношению
6 = 4(Ri) + yi(Ri) dM (R4),
где 6 — химический сдвиг протонов в орто-положении к заместителю R4 и в люта-положении к заместителю R4, do и dM — характеристические параметры экранирования орто- и .мета-протонов, а у — эмпирическая константа. Преимуществом этой работы по сравнению с работами Диля и других исследователей является учет у, так как не всегда у = 1. В табл. 10.55 приведены
Таблица 10.55
ПАРАМЕТРЫ ЗАМЕСТИТЕЛЕЙ В пара-ДИЗАМЕЩЕННЫХ БЕНЗОЛАХ [215]
Заместитель <1о, М. д. “м' м- д’ Y
nh2 0,768 0,271 0,70
ОСН3 0,477 0,108 0,67
СН3 0,183 0,107 0,91
С1 0,000 0,065 1,00
Вг —0,159 0,134 1,03
I —0,363 0,265 1,10 •
no2 —0,955 —0,155 1,20
CN —0,27 —0,100
СНО —0,54 —0,195
СОСНз —0,64 —0,091
СОС1 —0,83 —0,156
некоторые из полученных Мартином параметров экранирования для пара-дизамещенных бензолов.
Дейли и сотр. [485, 487] объяснили химические сдвиги в замещенных бензолах влиянием кольцевых токов и л-электронных плотностей.
10.12.5. Галогензамещенные бензолы
Штелинг [216] исследовал спектры протонного резонанса 55 галоген-замещенных бензолов. К сожалению, были исследованы неразбавленные растворы этих соединений в инертном растворителе (использовались 50%-ные растворы в СС14), что существенно уменьшает теоретическое значение полученных данных.
Несмотря на это, на основании полученных значений химических сдвигов, приведенных в виде диаграммы на рис. 10.26, можно сделать некоторые
92
Глава 10
обобщения. Что касается кольцевых протонов, то их химические сдвиги обусловлены главным образом влиянием заместителей в орто-положениях по отношению к рассматриваемым ядрам, причем это влияние приблизительно аддитивно.
Заместители в кольце
F, СН3
T-Шкала {_____________i________1
I I I u I III I 11 н I i; 11 и | 11 r r
Сдвиг относительно CgH6_______о
Заместитель, от но- F
сительно которого ука- IM заны положения протонов:СН^-^- о,мр
м,п
1(Р
С1, сн3
Cl —
сн3 -
-СН3 I
J м,п
OZOM,n
Вг, СН3
Вг-*~
СН, -
I, СН3
7 —
снг -
м,по,м,п
о,м,п
- СН3 II
СН2С1, сн3
ch2cl —
СН2Вг, СН3
СН2Вг — сн3 —
СН2С1, С1
м,п o/ipMp o ____________
2 0 W }
? м ? [ Протоны
I " ,[ кольца
мп О,М,П 0J4 oZ)
Протоны I кольца
I о,mJ?2
„ (п20,М,Л
Протоны кольца Щ
_________
СН,С1 — п (°,м,л '-"Z'-'- Протоны J в
Г] — кольца 1
“ I В
м,п
-сн2-] |*J СТ о м,п
о м,п
-СН2- I I
_________м~п о2м,п
~сн2— !
о м,п ~СН3 1| м,п
о м,п
м.п
ом,п
-сн3 II
М.П
2
3
5
6
<НС12
CHCLZ -* Протоны .
кольца I СН
СНС1СН3
СНСШ3+П ОМ" кольца
-сн3|
СН2СН2С1
CL — Кольцо
t-Шкала
Свейг относительно СМ
а
—СН2— | 0
.......
в
1-сн2-
Л
та
Химический сдвиг, м.д.
Рис. 10.26. Протонные химические сдвиги некоторых замещенных бензолов [216].
Пример', в 1-хлор-З-метилбензоле протон в положении 2 расположен орто по отношению к С1 и орто к СНз» протон в положении 4 — пара к С1 и орто к СНз, а СНз — мета к С1.
Было, показано, что химический сдвиг метиленовых протонов в соединениях типа СвН5СН2Х почти не зависит от природы галогена X. Орто-заместители типа атомов галогенов X, групп СН2Х и СН3 обусловливают дезэкранирование протонов —СН2Х. Экранирование метильных протонрв. под влиянием opmo-заместителей X и СН2Х изменяется аналогичным образом-однако присутствие второй метильной группы в орто-положении вызывает увеличение их экранирования [217]. Это явление обусловлено влиянием диамагнитной анизотропии галогенсодержащих заместителей х). Ч
Ч В работе [541] проведено изучение влияния заместителей на экранирование СН2-протонов в замещенных бензилмеркургалогенидах.— Прим. ред.
Корреляции спектров ЯМР-Н1 со строением молекул
93
10.12.6 Замещенные ксилолы
Химические сдвиги протонов ряда 2,6-диметил-1-замещенных бензолов, исследованных в виде разбавленных растворов в четыреххлористом углероде, были измерены в работе [210]; полученные результаты приведены в табл. 10.56. Из спектра 2,6-диметилдейтеробензола было найдено, что резонансные сигналы мета-протонов при введении метильных групп сдвинуты в сторону сильного поля на 0,17 м. д. по сравнению с пара-протонами. Поэтому в значения AvM, приведенные в табл. 10.56, введена поправка на эту величину, и исправленные велйчины Av'M также приведены в табл. 10.56. Попытки рассчитать
Таблица 10.56
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ В РЯДУ 2.6-ДИМЕТИЛ-1-ЗАМЕЩЕННЫХ БЕНЗОЛОВ [210]
Заместитель а а 71 °с 6 ТСН3
CN 12,9 0,3 2,6 0,10 7,52
Вг 16,8 16,8 6,5 —0,22 7,61
1 18,4 14,4 8,1 —0,11 7,54
F 22,5 26,5 12,2 —0,44 7,76
D 24,8 14,5 14,5 0,00 7,73
ОН 25,8 39,0 15,5 —0,60 7,88
nh2 29,0 47,2 18,7 —0,76 7,89
no2 12,5 5,2 2,2 +0,05 7,68
соон 16,0 7,0 5,7 + 0,06 7,55
ОСН3 23,5 29,0 13,2 —0,20 7,75
NHCH3 25,5 35,8 15,2 —0.39 7,76
N(CH3)2 26 26 15,7 —0,21 7,76
а и Дуд — химические сдвиги (в герцах при 60 Мгц) относительно бензола, взятого в качестве внутреннего эталона, для мета- и пяра-прото-нов соответственно.
б По данным работы Taft R. W., D е п о N. С., S k е 1 1 A. S., Ann. Rev. of Phys. Chem., 9, 292 (1958). Значения для последних пяти заместителей неправлены с учетом стернческнх препятствии сопряжению. Соединения исследованы в виде разбавленных растворов в СС!<.
экранирование ядер в мета- и пара-положениях в 2,6-диметиланилине и Л\ЛГдиметил-2,6-диметиланилине при помощи уравнений Бакингема [480] привели к результатам, плохо согласующимся с наблюдаемыми значениями. Неспособность теории электрического поля объяснить экранирование ароматических протонов проявилась также в случае 2,6-диметилфторбензола, где •теория некорректно предсказала меньшее экранирование пара-протонов по •сравнению с .мета-протонами [210].
В работе [210] было показано, что существует лишь грубая корреляция между значениями Av(t, Avn и о-константами Гаммета. Однако при рассмотрении только тех молекул, которые содержат группы, не оказывающие стери-ческих препятствий сопряжению, можно получить очень хорошие линейные зависимости Av’M и Avn от вкладов констант Тафта ои (индуктивный эффект) и ос (эффект сопряжения) в о-константы Гаммета. Наблюдаемые химические сдвиги удовлетворяют простым уравнениям
Av^t=14,5—17jb—10<тс (гц при 60 Мг{),
Avn=14,5—17аи—46ас.
94
Глава 10
Из этих уравнений можно ожидать линейной зависимости между разницей в экранировании пара- и жша-протонов (Av„ — Av^) и константой Тафта ос; как видно из рис. 10.27, такая зависимость действительно имеет место. Следовательно, наблюдаемые химические сдвиги мета- и пара- протонов в замещенных ксилолах, по-видимому, зависят главным образом от индуктивного влияния и эффекта сопряжения заместителей.
Рис. 10.27. Линейная зависимость разности химических сдвигов (в герцах при 60 Мгц) мета- и пара-протоноъ от констант Гаммета [210].
Опубликованы также характеристические химические сдвиги для метил-и этилзамещенных бензолов и некоторых конденсированных ароматических молекул [363]. Такие данные очень полезны для установления структуры молекул, в которых неясно расположение алкильных заместителей
10.12.7. Оксибензолы
Для изучения распределения л-электронной плотности в оксибензолах были исследованы спектры протонного резонанса фенола, гидрохинона, катехина, резорцина и пирогаллола и измерены химические сдвиги кольцевых протонов относительно бензола как внешнего эталона [373]. Межмолекулярные влияния электрических полей исключались путем исследования молекул в двух различных растворителях и экстраполяцией к среде с единичной диэлектрической проницаемостью. Были рассчитаны влияния внутримолекулярных электрических полей, обусловленных дипольными моментами связей С — О и О — Н, и вклады этих влияний [27] в химические сдвиги протонов были вычтены из наблюдаемых значений. Изменения исправленных значений химических сдвигов были целиком приписаны влиянию перераспределений л-электронной плотности (см. разд. 4.5.5).
10.12.8. Ароматические алкоксисоединения
Хискок [218] опубликовал значения протонных химических сдвигов для обширного ряда производных анизола и фенетола (см. табл. 10.57 и 10.58). Химические сдвиги метильных протонов в мета- ш/ш/ш-замещенных анизолах обнаруживают приблизительно линейную, корреляцию с соответствующими
Корреляции спектров ЯМР-Н1 со строением молекул
95
о-константами Гаммета. Для этого класса соединений существенным оказалось влияние растворителей, и поэтому химические сдвиги были измерены для разбавленных растворов образцов в инертных растворителях. Спектры ЯМР ароматических алкоксисоединений были исследованы также Куном и Кассиди [219].
Таблица 10.57
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ, ИЗМЕРЕННЫЕ ДЛЯ АНИЗОЛА И РОДСТВЕННЫХ СОЕДИНЕНИЙ [218]
Соединение Химический сдвиг протонов3
кольцо метоксигруппы метильная группа
Анизол 2,88—3,3 6,34
о-Метиланизол 3,08—3,36 6,35 7,86
.и-Метиланизол 3,03—3,55 6,38 7,77
л-Метиланизол 3,03; 3,35 6,38 7,80
о-Аминоанизол 3,48 6,34
о-Нитроанизол 2,30—3,00 6,16
.я-Нитроанизол 2,25—2,85 6,16
л-Нитроанизол 1,92; 2,82 6,16
л-Хлоранизол 2,88; 3,35 6,36
о-Броманизол 2,50—3,25 6,18
л-Броманизол 2,74; 3,35 6,36
о-Иоданизол 2,28—3,36 6,22
о-Метоксибензойная кислота 6 2,30—3,20 6,22
л-Метоксибензойная кислота 6 2,58; 3,17 6,24
Метил-о-метоксибензоат 2,30—3,15 6,20 6,20
Метил-л-метоксибензоат 2,13; 3,20 6,22 6,24
о-Ацетамидоанизол в 3,2 6,24 7,95
л-Ацетамидоанизол в 2,65; 3,20 6,30 • 7,95
л-Метоксибензальдегид 2,32; 3,12 6,21
о-Фениланизол 2,55—3,23 6,37
.я-Диметоксибензол 3,05—3,75 6,37
л-Диметоксибензол 3,32 6,43
л-Циананизол 2,53; 3,12 6,18
л-Метоксибензиловый спирт 2,95; 3,36 6,39
2,4-Динитроанизол г 5,90
2,4-Дих лоранизол 2,75; 2,90; 3,30 6,23
2,5-Диметокситолуол 3,48 6,33; 6,37 7,87
2,3-Диметоксибензальдегид 2,70—3,30 6,22
2-Амино-5-нитроанизол я 2,25; 2,35; 3,40 6,13
2-Иод-5-нитроанизол я 2,12; 2,45; 2,50 6,11
2-Окси-5-формиланизол 2,65; 3,00 6,13
2-Окси-5-пропениланизол 3,35 6,26
Все спектры сняты для 10%-ных растворов в ССЦ, если не оговорено особо. Химические сдвиги кольцевых протонов приведены в т-шкале в виде интервалов, в пределах которых простая интерпретация наблюдающихся мультиплетов невозможна.
6 В качестве сольватирующего агента добавлено 10% гексаметнлтриамидофосфата.
в 5%-ный раствор в CDCI3.
г 0,5%-ный раствор в ССЦ. Малая величина отношения сигнала к шуму не позволяет выделить сигналы кольцевых протонов.
Д 2%-ный раствор в CDCI3.
96
Глава 10
Таблица 10.58
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ, ИЗМЕРЕННЫЕ ДЛЯ ФЕНЕТОЛА И РОДСТВЕННЫХ СОЕДИНЕНИЙ [2 18]
Соединение Химический сдвиг протонов а
кольцо метиленовая группа метильная группа метильные заместители у кольца
Фенетол 2,88—3,3 6,13 8,68
о-Метилфенетол 3,18—3,42 6,11 8,66 7,86
л!-Метил фенетол 3,03-3,50 6,13 8,68 7,77
n-Метил фенетол 3,10; 3,36 6,15 8,69 7,80
о-Аминофенетол 3,48 6,10 8,68
м-Аминофенетол 3,15—4,02 6,20 8,73
л-Аминофенетол 3,50; 3,65 6,20 8,73
о-Диэтоксибензол 3,30 6,07 8,65
м-Диэтоксибензол 3,05—3,77 6,13 8,67
п-Диэтоксибензол 3,35 6,15 8,70
п-Нитрофенетол 1,85; 3,10 2,93 8,57
а Все соединения исследованы в виде 10%-ных растворов в ССЦ. Химические сдвиги кольцевых протонов приведены в т-шкале в виде интервалов, в пределах которых простая интерпретация наблюдающихся мультиплетов невозможна.
J0.12.9. Ароматические альдегиды
В табл. 10.59 и 10.60 приведены химические сдвиги формильного протона для широкого ряда ароматических альдегидов [220]. Экранирование формильного протона определяется в существенной степени влиянием магнитной
Таблица 10.59
ХИМИЧЕСКИЕ СДВИГИ ФОРМИЛЬНЫХ ПРОТОНОВ
НЕКОТОРЫХ мета- И пара-ЗАМЕЩЕННЫХ
БЕНЗАЛЬДЕГИДОВ [220]
Альдегид та
Бензальдегид 0,04 (+0,02)
ж-Толуиловый альдегид 0,10 (+0,03)
n-Толуиловый альдегид 0,11 (+0,03)
n-Анисовый альдегид 0,19 (+0,02)
jn-Хлорбензальдегид 0,06 (+0,02)
п-Хлорбензальдегид 0,06 (0,00)
3,4-Дихлорбензальдегид 0,09 (—)
jn-Бромбензальдегид 0,09 (+0,01)
л-Диметиламинобензальдегид 0,35 (+0,05)
jn-Фторбензальдегид 0,05 6 (+0,02)
л-Фторбензальдегид 0,08 (+0,01)
л- Бензилоксибензальдегид 0,12 (+0,05)
3,5-Диметоксибензальдегид 0,18 (+0,04)
3,4,5-Триметоксибензальдегид 0,27 (+0,02)
Пиперональ 0,26 (+0,02)
а Сдвиги измерены в м. д. для 5%-ных растворов в ССЦ с ТМС в качестве внутреннего эталона. Цифры в скобках представляют собой сдвиг (м. д.), наблюдаемый при разбавлении 20%-ных' растворов в ССЦ до 5%-ных растворов.
б Это центр наблюдаемого дублета; J — 1,8 гц.
Корреляции спектров ЯМР-Н1 со строением молекул
97
Таблица 10.60
ХИМИЧЕСКИЕ СДВИГИ ФОРМИЛЬНЫХ ПРОТОНОВ НЕКОТОРЫХ ор/ло-ЗАМЕЩЕННЫХ БЕНЗОЛОВ
И ПОЛИЦИКЛИЧЕСКИХ АЛЬДЕГИДОВ [220]
Альдегид та
Салициловый альдегид 0,14 (+0,05)
Бензальдегид 0,04 (+0,02)
о-Толуиловый альдегид —0,18 (+0,03)
о-Нитробензальдегид —0,37 (+0,03)
о-Анисовый альдегид —0,39 (+0,01) б
о-Хлорбензальдегид —0,45 (+0,04)
Мезитиловый альдегид —0,49 (+0,04)
1-Нафтойный альдегид —0,31 (+0,07)
а Сдвнгн измерены для 5%-ных растворов в ССЦ с Si(CH3)4 в качестве внутреннего эталона. Цифры в скобках представляют собой сдвиг (м. д.), наблюдаемый прн разбавлении 20%-ных растворов в ССЦ до 5%-ных растворов.
6 Это центр наблюдаемого дублета J ~ 0,8 гц, обусловленного взаимодействием с одним нз кольцевых протонов.
анизотропии как карбонильной группы, так и ароматического кольца, а также поляризацией связи С — Н, приводящей к уменьшению электронной плотности вокруг формильных протонов. Для объяснения наблюдаемых констант экранирования ароматических альдегидных протонов было сделано предположение, что влияние диамагнитной анизотропии карбонильной группы во всех молекулах одинаково и что изменения в экранировании обусловлены относительными величинами двух противоположных эффектов: дезэкранирования, вызванного кольцевыми токами, и экранирования, определяемого изменениями поляризации связи С — Н. Альдегидный протон — СНО в ацетальдегиде имеет значение т = 4-0,27, а в бензальдегиде его т-значение равно 4-0,04; следовательно, можно предположить, что влияние кольцевого тока на экранирование сильнее влияния поляризации связи' С — Н. Воздействие кольцевого тока для этого ряда молекул можно считать постоянным и, таким образом, непостоянство экранирования обусловлено изменениями поляризации связи С — Н, что является следствием электронных влияний различных по природе заместителей. Электронодонорные заместители уменьшают электроположительность атома углерода карбонильной группы и увеличивают экранирование формильных протонов.
Для мета- и пара-замещенных бензальдегидов формильные протоны имеют значения т в области от 4-0,35 до —0,20, а химические сдвиги приблизительно линейно зависят от о-констант Гаммета. Opmo-замещенные имеют соответствующие значения % в области от —0,20 до —0,50; для этих молекул получены некоторые данные, свидетельствующие о стерических препятствиях эффекту сопряжения. Для ароматических альдегидов проявляется влияние растворителя, аналогичное тому, которое наблюдали Шефер и Шнейдер [201] для других ароматических молекул. В растворах в четыреххлористом углероде химические сдвиги альдегидных протонов очень слабо зависят от концентрации, что позволяет предположить отсутствие сильных межмолекулярных водородных связей.
В спектрах opmo-замещенных бензальдегидов наблюдались константы дальнего спин-спинового взаимодействия протонов [372]; формильные протоны взаимодействуют с кольцевыми протонами в положениях 3 и 5 (см. разд. 10.14). Это ясно видно из спектра протонного резонанса 2,4-дихлорбензальдегида, показанного на рис. 10.28, в котором слабопольный дублет обусловлен формильными протонами.
7-489
Рис. 10.28. Спектр протонного резонанса 2,4-Дихлорбензальдегида при 60 Мгц [372].
2,6 -Динитротолуол
3 - Хлоризофталилхлорид
3,4-Дихлорбензонитрил
2- Нитро-4-хлорбензонитрил
2 -Иод - 5-нитробензонитрЬл
2,5-Дибромнитробензол
4 ~ Амино-2-нитротолуол.
3,4-Динитр офенол
2 - Нитро -4 - хлоранилин
2,5-Динитрофенол
2 - Амино-5-нитротолуол
2,4-Д инитроанилин
3,5-Дибромбензойная кислофа
3,5-Дихлоранилин
3 -Этил- 5-метилфенол
3,5-Дикарбоксинитробензол
—----Н
Рис. 10.29. Химические сдвиги протонов (относительно Н2О в качестве внешнего эталона) в некоторых тризамещенных бензолах, исследованных в виде жидкостей или растворов в ацетоне или диметилсульфоксиде [221]. Значения сдвигов исправлены на объемную диамагнитную восприимчивость, т = 5 2 + SH2O.
Корреляции спектров ЯМР-Н1 со строением, молекул
99
10.12.1 0. Тризамещенные бензолы
Лин и Ричардс [221] измерили химические сдвиги протонов для большого ряда тризамещенных бензолов. На рис. 10.29 приведены химические сдвиги, измеренные относительно воды, использовавшейся в качестве внешнего эталона. Поскольку не был исключен вклад межмолекулярных взаимодействий в экранирование кольцевых протонов (так как не были исследованы образцы малой концентрации в инертных растворителях), ценность проделанной работы невелика. Однако ясно, что увеличение числа электроотрицательных заместителей приводит к уменьшению экранирования ароматических протонов. Для некоторых молекул было показано, что вклады в экранирование, обусловленные тремя заместителями, аддитивны.
10.12.1 1. Дифенилы
Браунштейн [222] исследовал спектры ЯМР некоторых 2- и 4-галоген-дифенилов. Некоторые резонансные сигналы в спектрах протонного резонанса 2-галогендифенилов находятся в области более сильного поля, чем для исходного дифенила; это явление было объяснено пространственным взаимодействием атома галогена с орто-водородными атомами в незамещенном кольце,.
н
0,21 м.д.
СсН6 (эталон)
Рис. 10.30. Спектр протонного резонанса о-терфенила в СС1* при 40 Мгц [223].
Гофман и сотр. [223] исследовали спектры протонного резонанса дифенила, о- и ^-терфенилов. Дифенил и jn-терфенил дают сложные спектры тйпа АА'ВВ'С, однако о-терфенил дает очень простой спектр [223], состоящий из двух узких сигналов с отношением интенсивностей 4 : 10 (см. рис. 10.30). Единственное возможное объяснение этого спектра заключается в том, что все протоны одного из колец эквивалентны, а протоны двух других колец экранированы сильнее первых четырех протонов. Гофман считает, что вследствие больших констант взаимодействия в ароматических системах для появления одиночного резонансного сигнала не обязательна полная эквивалентность ядер. При расчете вкладов в экранирование кольцевых протонов кольцевыми токами было показано, что в о-терфениле две боковые фенильные группы перпендикулярны к центральному кольцу. Результаты, полученные для дифенила и л<-терфенила, не позволяют провести конформационный анализ такого типа.
7*
100
Глава 10
10.12.1 2. Инден
В спектре протонного резонанса индена, показанном на рис. 10.31, резонансные сигналы протонов пятичленного кольца интерпретируются по первому порядку [224]. Вследствие того что эти протоны слабо взаимодействуют друг с другом (система спинов АРХ2), при анализе спектра протонного резонанса этого соединения нельзя получить относительные знаки констант взаимодействия. Эллеман и Менетт [224], применив метод двойного резонанса, показали, что Jах имеет знак, противоположный знаку JАр и 7рх- Химические сдвиги и константы взаимодействия для протонов пятичленного кольца будут следующими:
тА = 3,334; тР = 3,865; тх = 7,008;
/Ар= 4-5,58 гц-, /ах= —1»98гч; /рх=+2,02 г^.
На рис. 10.31 можно видеть константу дальнего взаимодействия (0,52 гц), проявляющуюся в мультиплете А и отнесенную к взаимодействию протонов
Рис. 10.31. Спектр протонного резонанса индена при 60 Мгц [224].
Химические сдвиги (гц) измерены относительно Si(CH3)4. взятого в качестве внутреннего эталона. Области спектра А и Р записаны при усилении в два раза .большем, чем области Хг и ароматических протонов.
А с ароматическими протонами в положении 4 [450]. В области Х2 также можно обнаружить две дальние константы взаимодействия между метиленовыми и ароматическими протонами.
10.12.1 3. Кольцевые токи в ароматических молекулах
В разд. 4.3 и 4.5 на примере различных молекул с ароматическими свойствами проведено детальное рассмотрение вопроса о влиянии диамагнит-.ных кольцевых токов в ароматических соединениях на экранирование протонов^ Некоторые исследователи [225, 226] усовершенствовали предложенное первоначально Поплом [227] уравнение для расчета влияния кольцевого тока, рассматривая магнитное поле как обусловленное циркуляцией л-элек-тронов пр окружности радиуса а, а не как магнитный диполь. Используя такое приближение (см. разд. 3.8), Джонсон и Бови [225] рассчитали вклады в экранирование протонов, расположенных в различных положениях вблизи от ароматического кольца х). Полученные ими результаты приведены в приложении II в первом томе.
Теоретический расчет химических сдвигов, обусловленных л-электронными кольцевыми токами, позволил объяснить аномальные химические сдвиги фенильных протонов в полистироле [542].— Прим. ред.
Корреляции спектров ЯМР-Н1 со строением молекул
101
10.13. Полициклические ароматические молекулы
Бернстейн, Шнейдер и Попл [228] исследовали спектры ЯМР некоторых полициклических ароматических углеводородов и показали, что наблюдаемые химические сдвиги хорошо согласуются с величинами, полученными из рассмотрения кольцевых токов в сопряженных циклических системах (см. разд. 4.3). Большинство соединений были исследованы в виде расплавленных образцов, поскольку в то время исследователи еще не располагали данными о сильных межмолекулярных взаимодействиях в таких системах. Позднее другие исследователи [229, 437] провели полный анализ спектров протонного резонанса молекул этого типа, используя разбавленные растворы углеводородов в инертных растворителях. Рассчитанные при рассмотрении кольцевых токов сдвиги хорошо согласовались с наблюдаемыми значениями (см. табл. 4.9).
10.14. Спин-спиновое взаимодействие в ароматических молекулах
Протоны в производных бензолах взаимодействуют друг с другом с характеристическими значениями констант, зависящими от положения протонов в молекуле относительно друг друга. Интервалы значений констант спин-спинового взаимодействия
jhh ° ~ 7-°—9,2 гц; J^a ~ 1,1 — 3,1 гц; ~ 0,0—0,7 гц
почти не зависят от природы заместителей [221]. Значения констант взаимодействия играют важную роль при определении характера замещения в ароматических системах методом ЯМР. Изменение констант взаимодействия при варьировании заместителей мало: однако для разных рядов замещенных бензолов наблюдается линейная зависимость констант взаимодействия от суммы электроотрицательностей заместителей, связанных с кольцом [406].
Расчет констант взаимодействия из сложных спектров протонного резонанса замещенных бензолов возможен лишь при детальном анализе спектров. В табл. 10.61 приведены константы взаимодействия ароматических протонов, полученные при таком анализе [234]: очевидно, что они находятся в ожидаемом интервале характеристических значений для орто-, мета- и пара-констант [221, 230—233]. Относительные знаки трех констант взаимодействия одинаковы для всех исследованных молекул: Бакингем и Мак-Лахлан [38] показали, что J0^10 в n-нитротолуоле имеет абсолютный положительный знак и, по-видимому, все константы взаимодействия в производных бензола положительны.
Мак-Коннел [179, 191] использовал метод валентных связей для оценки относительной величины л-электронного вклада в константы взаимодействия протонов в ароматических системах. Он пришел к выводу, что в нафталине и замещенных бензолах взаимодействие орто- и мета-протонов осуществляется главным образом через о-связи, тогда как взаимодействие пара-протонов, а также дальнее взаимодействие (J16, 7, Угв и J2i в нафталине не наблюда-
ются) осуществляется главным образом через л-электронную систему (см. разд. 5.6). Расчет л-электронных вкладов в константы взаимодействия протонов в бензоле и нафталине дает небольшие положительные константы для протонов, разделенных нечетным числом связей С — С, и небольшие отрицательные значения в противоположном случае.
Взаимодействие протонов различных колец в конденсированных ароматических углеводородах непосредственно не наблюдалось (за исключением хинолинов; см. разд. 10.20). Однако имеются косвенные доказательства такого
Таблица 10.61
КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ КОЛЬЦЕВЫХ ПРОТОНОВ В АРОМАТИЧЕСКИХ МОЛЕКУЛАХ [234]
(2) (3)
Н Н
napa-ДИЗАМЕЩЕННЫЕ БЕНЗОЛЫ
(1)R<^>R(4) Н Н
(6) (5)
Заместители Ji 3 Ji Б Ju Ji 5
nh2, осн3 8,5 0,5 2,8 2,8
nh2, ci 8,6 0,4 2,8 2,5
NH2, Вг 8,5 0,3 2,9 2,3
nh2, I 8,5 0,3 2,9 2,2
nh2, no2 9,0 0,3 2,6 2,3
OCH3, Cl 8,8 0,3 3,1 2,5
OCH3, Br 8,7 0,3 3,1 2,5
OCH3, I 8,9 0,4 3,0 2,4
OCH3, no2 9,0 0,3 2,7 2,7
Cl, Br 8,4 0,4 2,5 2,5
Cl, I 8,4 0,4 2,4 2,4
Cl, no2 8,7 0,3 2,8 2,2
Br, I 8,6 0,3 2,5 2,2
Br, NO2 8,9 0,4 2,6 2,2
CN, OCH3 8,8 0,4 2,2 2,7
CN, Cl 8,4 0,4 2,1 2,1
COCH3, OCH3 8,7 0,4 2,6 2,3
COCH3, Cl 8,4 0,5 2,2 2,2
COCI, OCH3 8,9 0,3 2,7 2,4
COCI, Cl 8,5 0,5 2,3 2,3
CHO, OCH3 8,6 0,4 2,5 2,1
CHO, Cl 8,3 0,4- 2,1 2,1
CH3, nh2 8,1 0,5 2,4 2,4
CH3, OCH3 8,3 0,4 2,7 2,4
CH3, Cl 8,4 0,4
CH3, Br 8,1 0,6 2,3 2,3
CH3, I 7,9 0,6 2,4 2,0
CH3, no2 8,5 0,4 2,3 2,1
СИММЕТРИЧНО лета-ДИЗАМЕЩЕННЫЕ БЕНЗОЛЫ
R_Н(6) (2)Н\==/Н(5)
R Н(4)
Соединения Ji 5 Ji 5 Ji 4
л-Дибромбензол 8,1 0,4 1,8
л-Дииодбензол 7,7 0,3 1,6
M-Динитробензол 8,3 0,5 2,2
Изофталоилх чорид 7,9 0,6 1,9
Корреляции спектров ЯМР-Н1 со строением молекул
103
Продолжение табл. 10.61 R R ' СИММЕТРИЧНО opmo-ДИЗАМЕЩЕННЫЕ БЕНЗОЛЫ (6)Н<^__^Н(З)
(5)НН(4)
Соединения Ji 4 Ji Б Ji в Ji Б
о-Дихлорбензол 7,9 1,7 0,5 7,5
о-Дибромбензол 8Д 1,5 0,3 7,3
о-Дииодбензол 7,9 1,5 0,3 7,3
Фталоилхлорид 7,8 1,1 0,6 7,6
взаимодействия в антрацене [229], спектр которого приведен на рис. 10.32, где резонансные сигналы «соседних» протонов А и X существенно уширены, по-видимому вследствие межъядерного взаимодействия. При отсутствии взаимодействия между ядрами А и X мультиплет А был бы точно зеркальным
н
----->»
Рис. 10.32. Спектр протонного резонанса при 60 Мгц (229]. а — раствор нафталина в CCU; б — раствор антрацена в CSa.
отражением мультиплета В (что типично для систем АА'ВВ'). Для подтверждения спин-спинового взаимодействия между протонами А и X можно применить метод двойного резонанса. Спектр протонного резонанса нафталина (рис. 10.32, а) также не совпадает с симметричным спектром системы типа АА'ВВ'.
Наблюдаемые константы взаимодействия орто-протонов для некоторых конденсированных ароматических углеводородов подчиняются простой линейной зависимости
7 = 12,7Р—1,1,
104
Глава 10
где Р — л-порядок соответствующей связи С — С, который может быть рассчитан при использовании метода молекулярных орбиталей. Это наблюдение не согласуется с выводом Мак-Коннела о том, что взаимодействие орто-протонов в ароматических молекулах осуществляется главным образом через сг-связи.
Константы дальнего взаимодействия наблюдались в спектрах орто-замещенных бензальдегидов
<4 /Н с
в которых резонансные сигналы альдегидных протонов появляются [372] в виде дублетов вследствие их взаимодействия с кольцевыми протонами, расположенными через пять связей (J = 0,31 — 0,83 гц). При исследовании различных замещенных ароматических альдегидов было показано, что в одних молекулах альдегидный протон взаимодействует с протоном в положении 5, а в других — с протоном в положении 3. Для наблюдения такого дальнего взаимодействия необходимо, чтобы заместитель находился в орто-положении к альдегидной группе.
10.15. Ароматические ионы
Исследования спектров ЯМР ароматических ионов позволили впервые получить некоторые прямые доказательства о строении этих соединений, о чем до сих пор удавалось судить лишь косвенным образом [83, 208, 229, 235— 246, 248, 250, 251]. Для того чтобы дать представление о подобных исследованиях, обсудим некоторые типичные примеры.
10.15.1. Триарилкарбониевые ионы
Триарилкарбониевые ионы могут образовыЬаться при действии сильных кислот на триарилкарбинолы
АгзСОН-|-2Н+ Аг3С+-|-Н3О+.
Замещение в арильной группе сильно влияет на стабильность иона; так три-n-онизилкарбинол имеет константу ионизации, которая в 3 • 107 раз превышает соответствующую константу для незамещенного трифенилкарбинола. Существуют различные точки зрения на строение триарилкарбониевого иона; некоторые данные позволяют предположить, что лишь одно или два бензольных кольца сопряжены с центральным атомом углерода, тогда как другие данные свидетельствуют в пользу симметричной «пропеллерной» структуры, в которой л-орбитали бензольных колец одинаково перекрываются с р-орбиталями центрального атома углерода [235]. В табл. 10.62 приведены химические сдвиги протонов в ряду замещенных арилкарбониевых ионов [236]. Для молекул с водородсодержащими заместителями ни в одном случае не наблюдалось мультиплетных резонансных сигналов, обусловленных протонами заместителей. Несмотря на то что такое явление свидетельствует в пользу симметричной молекулы «пропеллерного» вида, оно все же не исключает структур с одним сопряженным кольцом, существующих в виде смесей быстро взаимопревращающихся изомеров. Охлаждение растворов три-п-анизилметил-и три-п-толилметилкатионов до —80° не изменяет вида резонансных сигналов заместителей. Во всех случаях резонансные сигналы заместителей представ-
Корреляции спектров ЯМР-Н1 со строением молекул
105
Таблица 10.62
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ НЕКОТОРЫХ ЗАМЕЩЕННЫХ ТРИАРИЛКАРБОНИЕВЫХ ИОНОВ [236] а
Соединение, из которого получен катион Сдвиг ароматических протонов Сдвиг протонов заместителя
Трифенилкарбинол — 101,0 —
я-Толилдифенилкарбинол —98,0 4-Ю2,5
Ди-я-толилфенилкарбинол —94,75 4-104,5
Три-я-толилкарбинол —93,5 4-105,5
jh-Тол илдифенил карбинол —95,5 4-И2,5
Три-л-толилкарбинол —89,0 4-112,5
я-Анизилдифенилкарбинол —96,0 +42,0
Три-я-анизилкарбинол —87,5 +47,0
я-треяг-Бутилфенилдифенилхлорметан —101,0 + 151,0
трет- Бутил бензол —76,0 + 161,5
Анизол —68,0 4-59,5
Толуол —69,0 + 124,5
а Химические сдвиги измерены в герцах относительно воды как внешнего эта-лона при рабочей частоте 40 Мгц; поправка на объемную диамагнитную восприимчивость не делалась. Соединения исследованы в виде 10%-ных растворов в смеси, содержащей 1,5 М трифторуксусную кислоту в ангидриде этой кислоты.
С „ , VH2O (внешн)
т — °’2 “I In •
ляют собой одиночные сигналы, тогда как протоны бензольных колец дают хорошо разрешенные мультиплеты. В карбониевых ионах протоны как бензольных колец, так и заместителей дезэкранированы по сравнению с аналогичными протонами в исходных молекулах.
О’Рейлли и Лефтин [237] также исследовали спектры протонного резонанса трифенилкарбониевого иона, и полученные ими результаты свидетельствуют в пользу эквивалентности всех колец, имеющих время, жизни больше 10~2 сек. Наблюдаемые химические сдвиги для протонов колец как в трифенил-, так и в дифенилметилкарбониевых ионах были объяснены электронным строением частиц и наличием в них кольцевых токов.
Дел, Воган и Берри [238] исследовали на примере нескольких дейтерированных трифенилкарбониевых ионов эквивалентность трех бензольных колец. Изучая 3,5-дидейтеро- и 3,4,5-тридейтеросоединения, в которых все кольца дейтерированы одинаково, эти авторы сделали вывод, что три кольца действительно эквивалентны за время снятия спектра ЯМР (время жизни больше 10'2 сек). Спектр протонного резонанса 3,5-дейтеропроизводного состоит из двух резонансных сигналов с отношением интенсивностей 1 : 2 (причем менее интенсивный сигнал находится в более слабом поле), тогда как 3,4,5-тридейтеропроизводное дает лишь одиночный сигнал. /шря-Протоны экранированы наиболее слабо, а орто-протоны — наиболее сильно, мета-протоны экранированы немного слабее орто-протонов. Были измерены и и константы взаимодействия между кольцевыми протонами в ароматических ионах: J0.M = 1,8 гц и JM.n = 3,0 гц-, измерить значение J0_n не удалось.
10.15.2. Карбониевый ион 9,10-диметил-1,2-бензанпграцена и другие ароматические карбониевые ионы
Этот ароматический карбониевый ион был получен при растворении 9,10-диметил-1,2-бензантрацена в смеси либо CF3COOH + Н2О BF3, либо HF + BF3 [239]. Методом ЯМР было доказано, что присоединение протона
106
Глава 10
кароматической молекуле приводит к образованию в положении 9 группы —СНСНз
На рис. 10.33,6 ясно видно появление квартета, обусловленного группой СН, с химическим сдвигом +1,73 м.д. относительно бензола, применяемого в качестве внутреннего эталона, с характерным расщеплением вследствие
Рис. 10.33. Спектр протонного резонанса 9,10-диметил-1,2-бензантрацена при 40 Мгц [239].
а — в СС14; б — в CF3COOH + 27 мол.% H2O-BF3. Б — сигнал внутреннего эталона бензола.
взаимодействия с соседней СН3-группой. Химические сдвиги протонов в исследованных системах приведены в табл. 10.63. В исходной молекуле две метиль-
Таблица 10.63
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ 9,10-ДИМЕТИЛ-1,2-БЕНЗАНТРАЦЕНА: А) В ССЦ, Б) В СМЕСИ CF3COOH + 27 МОЛ.% H2OBF3 ПРИ 40 Мгц [239]а
Соединение Растворитель Сигналы метильных протонов, гц Сигналы алифатических протонов, гц
9,10-Диметил-1,2-бензаи-трацен а) СС14 б) CF3COOH + H2O-BF3 (27 мол.%) 158 171 142 216 223 CHR 69
а Химические сдвиги измерены в герцах относительно бензола как внутрениого эталона.
ные группы неэквивалентны и при образовании карбониевого иона химический сдвиг между ними увеличивается; высокопольный дублет на рис. 10.33,6 был отнесен к метильной группе в положении 9, взаимодействующей с присоединенным протоном. Если получать карбониевый ион при использовании
Корреляции спектров ЯМР-Н1 со строением молекул
107
в качестве растворителя полностью дейтерированной кислоты, то к атому углерода в положении 9 присоединяется дейтерий и квартета группы СН в спектре не наблюдается.
Аналогичным образом были получены другие ароматические ионы, и методом ЯМР было доказано существование в них алифатических протонов. Полученные результаты собраны в табл. 10.64. В некоторых случаях имеют место обменные процессы типа
АН+ + А А + АН+, (10.15а)
АН+ + Х- А4-НХ, (10.156)
Таблица 10.64
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ (В ГЕРЦАХ ПРИ 40 Мгц)
НЕКОТОРЫХ КАРБОНИЕВЫХ ИОНОВ (ОТНОСИТЕЛЬНО БЕНЗОЛА
КАК ВНУТРЕННЕГО ЭТАЛОНА) [239]
Соединение Растворитель Сигналы метильных протонов,гц Сигналы алифатических протонов, гц
Пентаметилбензол СС14 202
cf3cooh+h2obf3 196, 199, 204 Не наблюда-
(30 мол.%) лись
HF+BF3 180, 187, 198 СН2 102
Г ексаметилбензол HF+BF3 193 CHR 131
Г ексаэтилбензол СС14 СН2 193
СН3 246
HF + BF3 СН2 185 CHR 120
СН3 254
9,10-Диметилаитрацен CCI4 166
cf3cooh+h2obf3 148, 223 CHR 102
(17 мол.%) 231
Пиреи HF + BF3 СН2 122
3,4-Бензпирен cf3cooh+h2obf3 СН2 160
(19 мол. %)
влияющие на ширину резонансных сигналов СН2-группы. Так , в спектр протонного резонанса пентаметилбензолониевого иона
СН3
в HF + BF3 наблюдается широкий резонансный сигнал алифатической группы СН2.
Равновесие (10.15а) осуществляется главным образом в растворах сильно основных ароматических молекул в сильных кислотах, причем большое влияние на равновесие оказывает кислотность растворителя.
При помощи смеси HF -BF3 удалось осуществить протонирование таких молекул, как мезитилен, анизол, м-диметоксибензол и м-ксилол [364] х).
J) Ионы рассматриваемого типа удается получить также при взаимодействии мези-тилена и других ароматических углеводородов с бромистым алюминием и бромистым водородом, что было показано с помощью спектров ЯМР [543, 544, 594, 595].—Прим. ред.
108
Глава 10
При низких температурах спектры протонного резонанса растворов мезити-лена и л-ксилола в смеси HF -BF3 свидетельствуют в пользу образования протонированных комплексов, и при изучении спектров можно определить положение протонирования. Аналогичную информацию можно получить из спектров, снятых при комнатной температуре, для растворов анизола и л-диметоксибен-зола в HF-BF3. При помощи ЯМР-спектров возможно также исследование кинетики различных типов переноса протонов в подобных системах (см. разд. 9.3.1).
10.15.3. Ион азулиния и родственные системы
Если растворить азулен в сильнокислой среде, то, вероятно, происходит протонирование молекулы в положении 1.
Данилюк и Шнейдер [240, 241] сравнили спектры протонного резонанса азулена в трифторуксусной кислоте со спектром самого азулена [117] и подтвердили вывод о протонировании азулена в указанном выше положении.
Рис. 10.34. Спектры протонного резонанса при 60 Мгц [240].
а — азулен в СН2С12 (7 мол. %); J12 = 3,9 гц', б — азулиниевый ион (7 мол. % азулена в CF3COOH); J12 = 1,8 гц, J13 = 1,3 гц.
CH2CI2 использовался в качестве внутреннего эталона, его сигнал расположен на 320 гц в более слабом поле, чем сигнал Si(CH3)<-
Спектры протонного резонанса азулена и иона азулиния приведены на рис. 10.34. На рис. 10.34, б сильнопольный сигнал, расположенный при +53 гц относительно сигнала СН2С12 как внешнего эталона, отнесен к алифатической группе СН2 в положении 1; оставшиеся кольцевые протоны в положениях 2 и 3 в ионе азулиния дают симметричную пару расщепленных дублетов типа АВ, в то время как в азулене протоны 1 и 3 вследствие эквивалентности имели одинаковые значения химических сдвигов. Триплетная структура компонент дублетов АВ, т. е. Н(2) и Н(3), обусловлена спин-спиновым взаимодействием двух протонов Н(1) с протонами Н(2) и Н(3). Протоны семичленного кольца в ионе азулиния по сравнению с молекулой азулена сильно дезэкранированы, что соответствует локализации положительного заря
Корреляции спектров ЯМР-Н1 со строением молекул 109
да главным образом на атомах углерода семичленного кольца. Исследование дейтерированных азулиниевых ионов методом ЯМР [242] подтвердило отнесения, приведенные на рис. 10.34, б.
Спектры протонного резонанса 4,6,8-триметилазулена, гвайазулена и их сопряженных кислот также свидетельствуют в пользу монопротонирования пятичленного кольца [241].
В спектре протонного резонанса иона гвайазулиния
Н Н сн3 н
Н—<2 / + б)—Н
СН3 Н СН(СН3)2 наблюдается чрезвычайно большая константа дальнего взаимодействия метиленовых протонов в положении 1 с метильными протонами в положении 3 (Jн2с-с-с-сн3 = 2,0 гц) [241]. Большая величина константы, по-видимому, •обусловлена делокализацией электронов связи С — Н вследствие сверхсопряжения. Если метильную группу в положении 3 заменить на атом водорода, как в ионе азулиния, то константа взаимодействия +з = 1,3 гц. Это явление служит убедительным доказательством того, что спин-спиновое взаимодействие в таких ионах осуществляется по ол-механизму [241] (см. разд. 10.7).
10.15.4. Циклопентадиенил-анион С5Н” и тропилий-катион С7Н^
Спектры протонного резонанса ионов С5Н5~ и С7П+ были исследованы независимо несколькими исследователями [99, 239, 248]. Было показано, что узкий одиночный сигнал в спектре C5H5_Na+ имеет химический сдвиг, равный +1,85 м.д. относительно бензола как внутреннего эталона, тогда как резонансный сигнал протонов тропилий-катиона при тех же условиях имеет сдвиг —1,9 м.д. в более слабом поле х). Сам циклопентадиен дает очень сложный спектр ЯМР, состоящий из двух мультиплетных сигналов при +0,84 и +4,4 м. д. относительно бензола как внутреннего эталона. Если исследовать спектр ЯМР смеси циклопентадиена и циклопентадиенил-аниона, то можно видеть, что имеет место простое сложение компонент спектров и нет никаких данных в пользу быстрого обмена между анионом и исходной молекулой.
В табл. 10.65 приведены химические сдвиги для ряда солей С5Н" и С7Щ; во всех случаях ионы дают одиночные резонансные сигналы [248]. В табл. 10.65 приведены также химические сдвиги для некоторых сэндвичевых соединений — ферроцена, рутеноцена, осмоцена и магноцена.
Другие ЯМР-исследования [248] показали, что положительные заряды на атомах, с которыми связаны протоны, обычно приводят к уменьшению экранирования последних (см. табл. 10.66). Следовательно, наблюдаемые химические сдвиги для ионов С5Н‘ и C7Ht могут быть объяснены простым электрическим взаимодействием между зарядом, локализованным на л-орби-талях атома углерода, и электронами связи С — Н. Френкель и сотр. [248] использовали электростатическую модель для объяснения химических сдвигов этих ионов и предсказания распределения заряда в ароматических молекулах.
Дейли, Гавер и Нейкам [83] исследовали возможности корреляции химических сдвигов протонов с электронными плотностями в ароматических ионах С5Н5~, С7Н7 и С8Н|". Эти системы удобны для такого исследования тем, что
х) Недавно были исследованы спектры протонного резонанса катионов дитропи-лия [545] и винилтропилия [546].— Прим. ред.
110
Глава 10
Таблица 10.65
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ СОЕДИНЕНИЙ
С ЦИКЛОПЕНТАДИЕНИЛ-АНИОНОМ (С5Н~) И ТРОПИЛИЙ-КАТИОНОМ (С7н+) [248]
Соединения Растворитель Концентрация, % Хим. сдвиг а, 6±0 ,004 м. д.
C7H+C1OJ Диметилсульфоксид 2 -1,938
С7Н+С1Ог Ацетонитрил 2 -1,873
C7H|BF4 Диметилсульфоксид 2 -1,934
СуЩВРг Ацетонитрил 2 -1,873
C5HjNa+ Диметилсульфоксид 2 1,970
C5H-Na+ Ацетонитрил 2 1,785
С5НД<а+ Тетрагидрофуран 2 1,724
С5НДл+ • » 2 1,733
(C5H5)2Mg » 2 1,494
(C5H5)2Mg Этиловый эфир 1 1,089
(C5H5)2Mg Бензол 1 0,819
(C5H5)2Mg Циклогексан 1 1,185
(C5H5)2OS Тетрагидрофуран 2 2,696
(C5H5)2Ru » 2 2,805
(C5H5)2Fe » 2 3,200
а Химические сдвиги измерены в м. д. относительно бензола как внутреннего эталона»
Таблица 10.66
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ НЕКОТОРЫХ ЧАСТИЦ С ПОЛОЖИТЕЛЬНЫМ ЗАРЯДОМ [248]
Конечное состояние (А) Начальное состояние (В) Протон Сдвиг 6ДВ, M. Д.
CH3NH+ ch3nh2 CH3 -0,30 a
(CH3)2NHJ (CH3)2NH CH3 -0,44 a
(CH3)3NH+ (CH3)3N CH3 — 0,70a
NH3CH2CO7 NH2CH2COj CH2 -0,29 6
nh3ch2co2h NH3CH2COj CH2 -0,50 6
CH3COHNHCH3 CH3CONHCH3 HCH3 -0,37»
a Grunwald Е., Loewenstein Н., Meiboom S., J. Chem. Phys., 27, 64 1 (1957).
6 Takeda M„ Jardetzky О., J. Chem. Phys., 26, 1346 (1957).
B Fraenkel G., Niemann C., Proc. Natl. Acad. Sci. U.S., 44, 688 (1958.)
для них известны заряды, симметрично распределенные по кольцу. Однако, прежде чем коррелировать химические сдвиги с электронными плотностями, необходимо рассмотреть два дополнительных эффекта, а именно вклады в экранирование, обусловленные кольцевым током, и влияние растворителя. Для ароматических ионов вклад кольцевого тока отличается от соответствующего вклада в бензоле. Величину этого вклада можно оценить, используя соотношение
Корреляции спектров ЯМР-Н1 со строением молекул
111
где — значение константы экранирования, приведенное в таблице Джонсона и Бови [225] (см. т. 1, приложение II) для соответствующего расстояния между центром кольца и протонами; ал — радиус исследуемого кольца; аь — радиус бензольного кольца; It — отношение величин кольцевых токов иона и бензола; 0,63 — эмпирическая константа, найденная Джонатаном и сотр. [229]. Это соотношение было использовано для определения ошибок, обусловленных кольцевым током, для пяти-, шести-, семи- и восьмичленных ароматических ионов [229]. Влияние растворителя было исключено путем вычитания химического сдвига в газовой фазе, исправленного на сдвиг Ван-дер-Ваальса (который определяется как средний химический сдвиг метана и этана при бесконечном разбавлении в растворителе), из химического сдвига, полученного при бесконечном разбавлении в индивидуальном растворителе. В табл. 10.67 приведены химические сдвиги для трех ароматических ионов,
Таблица 10.67
КОРРЕЛЯЦИЯ ХИМИЧЕСКОГО СДВИГА С ЭЛЕКТРОННОЙ ПЛОТНОСТЬЮ В АРОМАТИЧЕСКИХ ИОНАХ [83]
Ион Химический сдвиг
наблюдаемый относительно внутреннего эталона — бензола исправленный на влияние растворителя исправленный на влияние кольцевого тока отличие в л-заря-дах по сравнению с бензолом отношение хим, сдвиг электронн. плотность
С5НГ + 1,731 + 1,60 + 1,43 +0,200 7,15
с7Ш -1,87 -1,70 -1,59 -0,143 11,13
с8нг + 1,575 + 1,48 +2,42 +0,250 9,70
Среднее 9,3
поправки на кольцевые токи и влияние растворителя и показано, что среднее отношение химического сдвига к электронной плотности равно 9,3 м.д. на заряд электрона. Аналогичные исследования были проведены другими авторами, причем полученные ими результаты хорошо согласуются с вышеприведенными [99, 243, 244]. При делении наблюдаемых химических сдвигов [208] (исправленных на влияние кольцевого тока) неальтернантных углеводородов и азулена на 9,3 были определены изменения электронных плотностей в различных положениях молекул относительно бензола [83]; при этом получено качественное совпадение значений со значениями, рассчитанными другими методами [245, 246]. Аналогичные расчеты были с успехом проведены для пиридина, хинолина, изохинолина и пиримидина [83]. Шефер и Шнейдер [991 также применили метод ЯМР для оценки электронных плотностей в ароматических ионах (см. разд. 4.5.5.).
Однако несмотря на то, что имеется разумное согласие между наблюдаемыми и рассчитанными электронными плотностями, точно определить электронные плотности по измерению химических сдвигов невозможно ввиду многих трудностей, встречающихся в таких сложных системах.
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
10.16. Фуран и его производные
Спектр протонного резонанса чистого фурана состоит из двух триплетов, на основании чего в ранних работах был сделан вывод, что обе константы взаимодействия Jab и Ja-b- равны. Редди и Голдстейн [249] показали, что спектр фурана в ацетоне состоит из двух квартетов, а на основании исследо-
112
Глава 10
вания расщеплений С13 — Н удалось показать, что константы Jab и Ja-b' различны. Константы взаимодействия, найденные таким образом, совпадают с данными, полученными при анализе системы АА'ВВ' ранее рассмотренного спектра (см. разд. 8.16.2). В табл. 10.68 приведены константы взаимодей-
Таблица 10.68
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ ПРОТОНОВ ФУРАНА И МЕТИЛЗАМЕЩЕННЫХ ФУРАНОВ
ствия и химические сдвиги для фурана и его двух метильных производных [252]. Как можно было ожидать из рассмотрения, основанного на представлениях об электроотрицательности, протоны при а-углеродных атомах (по отношению к кислороду) экранированы слабее соответствующих ^-протонов. Метильные группы увеличивают экранирование протонов, как и следовало ожидать для электронодонорного заместителя. В 2-метилфуране метильные протоны почти так же сильно взаимодействуют с соседним кольцевым протоном (72з = = 1,1 гц), как протоны 4 и 5 между собой (J46 = 1,9 гц).
Лин и Ричардс [253] при исследовании ряда замещенных фуранов показали, что константы взаимодействия кольцевых протонов имеют характеристические значения, а именно:
J34 — 3,1—3,8 гц,
1,7—2,0 гц, J35 = 0,7—1,0 гц.
Таким образом, константы взаимодействия в фуранах значительно меньше соответствующих констант в производных бензола.
Эксперименты по двойному резонансу показали, что все три константы взаимодействия кольцевых протонов в 2-фуранкарбоновой кислоте имеют один и тот же знак [367].
соон
о
2-Фуранкарбоновая кислота
В табл. 10.69 приведены химические сдвиги для ряда производных фурана, измеренные относительно сигнала воды (внешний эталон) [254]. Протоны, связанные с кольцевыми атомами углерода, дают сигналы в более слабых полях, чем протоны, связанные с этиленовыми атомами углерода, что объяс-
Корреляции спектров ЯМР-Н1 со строением молекул
113
Таблица 10.69
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ В НЕКОТОРЫХ ЗАМЕЩЕННЫХ ФУРАНАХ [254]
Соединение Растворитель 6а а, м. д. бр а, м. д. Отношение сигналов а: ₽
Фуран СС14 -2,45 (т) -1,38 (т) 1 : 1
С12СНСНС12 -2,80 (т) -1,75 (т) 1 =;1
СН2С12 -2,83 (т) -1,78 (т) 1 : 1
2-Метилфуран СН2С12 -2,60 (м) -1,63; -1,35 (м) 1 :2
2,5-Диметилфуран СС14 -0,875 (с)
СН2С12 -1,21 (с)
Ментофуран СС14 -2,13
СН2С12 -2,40
Фурфурол СС14 -2,53 -1,41 1 :2
СН2С12 -2,78 -1,63 1 : 2
СС14 -2,90 (с) -2,40; -1,78 (д, к) 1 :2
СН2С12 -3,05 (с) -2,60; -1,93 (д, к) 1 :2
2-Фуранкарбоновая СН2С12 -2,90 (к) — 2,65; —1,88 (к, к) 1 :2
кислота
3-Фуранкарбоновая CDC13 -3,40; -2,73 -2,08 2 : 1
кислота
2-Метил-З-фуранкар- CDC13 -2,53 -1,98 1 : 1
боновая кислота
4-Метил-З-фуранкар- CDC13 -3,40; -2,58
боновая кислота
Кафестол СН2С12 -2,60 (д) -1,58 (д) 1 : 1
Колумбии СН2С12 -2,83 -1,73 2 : 1
Лимонин CDCI3 или СН2С12 -2,83 -1,75 2 : 1
а Буквы с, д, т, к, м, обозначают синглет, дублет, триплет, квартет, мультиплет соответственно. Отсутствие такого индекса отвечает одиночной линии, которая, возможно, является неразрешенным мультиплетом. 6 измерены в миллионных долях относительно воды как внешнего эталона, т = 5,2 + + 6Н2О (внешн) (очень приближенное соотношение).
няется влиянием кольцевого тока. Это положение хорошо иллюстрируется сравнением т-значений для фурана и дигидропирана
ZH 3,75 /\/н 5,46
J/~H 2,70 | ||
О ХО/ХчН 3,78
Абрагам и Бернстейн [255, 256] исследовали большое число замещенных фуранов, полученные ими данные приведены в табл. 10.70 и 10.71. Изучение табл. 10.70 показывает, что константа взаимодействия J3 t значительно возрастает при увеличении электроотрицательности заместителя в положении 2 (соединения расположены в порядке возрастания электроотрицательности заместителей). Аналогичная, хотя и менее четкая корреляция наблюдалась для замещенных пирролов [256]. Абрагам и Бернстейн нашли интересную корреляцию между константой J3 4 и суммой углов (0! + 02), а также между константой J2 з и суммой углов (02 + 03)
(4)Н Н(3)
\ 8181/02 (5)Н-<^ ^>^Н(2) oz
8—489
114
Глава 10
Таблица 10.70
КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ ПРОТОНОВ (гц) НЕКОТОРЫХ ЗАМЕЩЕННЫХ ФУРАНОВ [256J
Соединение J2 4 Ji 5 ^3 4 J;i 5 J4 5
2-Метилфуран 3,12(0,10) 1,03(0,07) 1,90(0,08) ^СН3-Н(з)—0,91
2-Фуранметан-тиол 3,25 0,73 1,91 7СН2-Н(з)=0’79>
2-Фурфурил-амин 3,18(0,10) 0,76(0,10) 1,94(0,10) •/сн2-н(з)=0.84-
2-Фурфурол 2-Фуранакрило-вая кислота 2-Фуранакро-леин 3,17(0,05) 3,44 (0,07) 3,46(0,08) 0,84(0,07) 1,90 (0,05) 1,84 (0,08) 1,83(0,04)
2-Аминофуран 3,57(0,07) 0,72(0,07) 1,76(0,04)
Этиловый эфир 2-фуранкарбо-новой кислоты 3,47(0,09) 0,91 (0,06) 1,76 (0,05)
2-Фуранкарбо-новая кислота 3,51(0,07) 0,86(0,09) 1,73(0,08)
Этил-2-фуроил-ацетат 3,60(0,09) 0,82(0,10) 1,72(0,05)
2-Фурфурол 3,62 (0,05) 0,82(0,06) 1,65(0,08) ^сн-н(5)=0’Во 81
2-Нитрофуран 3,66 (0,10) 0,98(0,07) 1,92(0,07)
2-Фуроилтри-фторметилке-тон 3,70(0,06) 0,76(0,06) 1,70(0,04)
2-Фуроилхлорид 5-Хлорметил-фУРфуРол З-Фураикарбо-новая кислота 2-Метил-З-фу-ранкарбоно-вая кислота 4-Метил-З-фуранкарбо-новая кислота 2,3-Фурандикарбоновая кислота 0,8(0,1) 1,5(0,1) 1,6(0,1) 3,79(0,07) 3,67(0,07) 0,79(0,07) 1,70(0,05) 1,8(0,1) 1,94(0,1) 1,83(0,1) 7снз-Н(5)= !>*
Во многих случаях протоны заместителей взаимодействуют с соседними кольцевыми протонами (аналогичное дальнее взаимодействие наблюдается в про-
стых олефиновых системах). Было показано, что химические сдвиги для замещенных фуранов зависят от природы растворителя (см. табл. 10.71, где химические сдвиги приведены в миллионных долях относительно соответствующего резонансного сигнала исходной молекулы), и это сильно ограничивает возможности построения зависимостей химических сдвигов от природы заместителей [256]. Тем не менее для 2-замещенных фуранов в ацетоновом растворе получены линейные соотношения между химическими сдвигами, которые
Таблица 10.71
ВЛИЯНИЕ ЗАМЕСТИТЕЛЕЙ НА ХИМИЧЕСКИЕ СДВИГИ КОЛЬЦЕВЫХ ПРОТОНОВ ЗАМЕЩЕННЫХ ФУРАНОВа [256] 4 3
Соединение Чистые жидкости ® 10%-иый раствор в ацетоне в 10%-иый раствор в CH2CI2 г
*3 Й4 б5 Й2 6з 64 б5 6з 64 65
2-Метилфуран 4-0,38 4-0,08 +0,10 +0,435 + 0,153 +0,205 +0,43 +0,15 +0,23
2-Фуранметантиол 4-0,15 4-0,03 +0,03
2-Фурфуриламин 4-0,13 -0,05 -0,08 4-0,242 + 0,100 +0,137
2-Фурфурол -0,04 -0,01 -0,05 4-0,151 +0,076 +0,089 +0,15 +0,15 +0,05
Фуран 0,00 0,01 0,00 0,000 0,000 0,000 0,000 0,00 0,00 0,00
2-Фуранакриловая кислота -0,408 -0,144 -0,136
2-Фуранакролеин -0,551 -0,209 . -0,226
Этиловый эфир 2-фуранкарбоновон кислоты -0,85 -0,18 -0,33 -0,774 -0,183 -0,226
2-Фуранкарбоновая кислота -0,795 -0,181 -0,220 -0,88 -0,10 -0,08
Этил-2-фуроилацетат -1,13 -0,35 -0,33 -0,959 -0,255 -0,298
Фурфурол -1,10 -0,35 -0,48 -0,987 -0,294 -0,366 -0,83 -0,15 -0,23
2-Нитрофуран -1,08 -0,43 -0,33 -1,065 -0,402 -0,318
2-Фу роилтрифторметилк етон -1,198 -0,373 -0,454
2-Фуроилхлорид -1,20 -0,38 -0,48
5-Хлорметилфурфурол -1,10 -0,45
2,5-Диметилфуран 4-0,50 4-0,50
3-Фуранкарбоновая кислота Д -0,70 -0,39 -0,17
2-Метил-З-фуранкарбоновая кислота д -0,29 +0,08
4-Метил-З-фуранкарбоновая кислота Д -0,52 +0,20
2,3-Фурандикарбоновая кислота Д 1 -0,64 —0,42
а Химические сдвиги измерены в миллионных долях относительно соответствующего сигнала фурана.
б Истинные значения для фурана относительно диоксана (внутренний эталон): Н(2) = — 3,83 м. д. и Н (3) = — 2,75 м. д., в Истинные значения для фурана относительно ацетона (внутренний эталон); Н (2) = — 5,48 м. д. и Н (3) =—4,36 м. д.
® Неизвестная концентрация, очень разбавленный раствор.
116
Глава 10
могут быть представлены следующим образом:
6з = 3,0864—0,42,
65= 1,2364+о,ю.
Влияние заместителей в 2-замещенных фуранах и в производных бензола различно. По-видимому, экранирование протонов в фуране зависит от двух факторов: 1) электроотрицательности заместителей; 2) сопряжения заместителей с электронной системой кольца1). Увеличение электроотрицательности заместителя приводит к уменьшению экранирования всех кольцевых протонов, причем дезэкранирование а-протонов наибольшее. Эффекты сопряжения возмущают л-электронную систему кольца и таким образом влияют на экранирование кольцевых протонов. Несмотря на то что влияние диамагнитной анизотропии в этом исследовании не учитывалось, наблюдавшиеся химические сдвиги качественно объяснялись на основе изложенных соображений. Было также показано, что для полизамещенных фуранов химические сдвиги кольцевых протонов можно приблизйтельно оценить, предположив, что вклады различных заместителей в экранирование аддитивны.
10.17. Пиррол и его производные
Спектр протонного резонанса кольцевых протонов пиррола, изображенный на рис. 10.35 и 8.52, состоит из двух мультиплетов, причем слабополь-
Пиррол (жидк.) Н-Дейтеропиррол(жидк}
25%-ный раствор в ацетоне
8 гц
20%-ный раствор в ацетоне
Рис. 10.35. Спектры протонного резонанса пиррола и N-дейтеропиррола при 40 Мгц [255].
ный мультиплет обусловлен а-протонами [257] (см. также разд. 8.19.5). Влияние квадрупольного момента ядер азота приводит к сильному уширению сигнала NH, но не устраняет спин-спинового взаимодействия протона при
х) Как следует из данных работ [547, 548], в соединениях типа 2-фурилсиланов сопряжение заместителя с электронной системой фуранового кольца может иметь характер d—рл-взаимодействия, вносящего свой вклад в экранирование кольцевых протонов.— Прим. ред. i
Корреляции спектров ЯМР-Н1 со строением молекул
117
азоте с кольцевыми протонами. Это взаимодействие сравнимо с взаимодействием кольцевых протонов и сильно усложняет спектры. Абрагам и Бернстейн [255] сумели избежать такого усложнения спектра, исследуя протонный резонанс N-дейтеропиррола (см. рис. 10.35). Тонкую структуру можно получить также при исследовании пиррола в растворителях типа Ы.Ы-диметилформамида или диметилсульфоксида, которые вызывают быстрый обмен протонов NH по механизму основного катализа [258].
Спектр протонного резонанса пиррола первоначально был объяснен в предположении, что константы взаимодействия J2 з и J2 4 равны; однако в более поздних исследованиях спектров замещенных пирролов [256, 258]
Таблица 10.72
КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ ПРОТОНОВ (гч) ЗАМЕЩЕНЫХ ПИРРОЛОВ [258] 4______________________________3
5\ /2 н
Заместители
Другие константы
з-сн3, 4-СО2С2Н5
2-СН3, З-СО2С2Н5
2-СНО, 5-СН3
2-СНО
2-СОСН3 2-СН3 3-СО2СН3
3,75
3,80
3,75
3,40
3,10
2,40
2,40
2,45
2,80
1,40
1,35
1,50
1,40
2,20 Ji 5 = 3,05;
Л 4 ~ /45!
J1 з = 2,50 j
•/сн3-П(2)— *’00
•/СН3-Н(4) = 0’65; ^СНз-Н(3) = 0’45
/сно-н<6)=1>15
J14-~/45; /15-3,00
было показано, что это не так. В табл. 10.72 приведены константы взаимодействия, найденные из спектров замещенных пирролов. Гроновиц с сотр. [258] приводит следующие средние значения для констант взаимодействия кольцевых протонов: <73 4 = 3,40 — 3,80; J4 6 = 2,40 —'3,10; J2 5 = 1,95 — 2,20; J24 = 1,35 — 1,50 гц. Можно показать, что спектр протонного резонанса пиррола (рис. 10.35) находится в согласии с этими средними значениями [256]. При расшифровке спектров замещенных пирролов большую помощь оказывает знание характеристических значений констант взаимодействия кольцевых протонов. При рассмотрении химических сдвигов и констант взаимодействия в спектре протонного резонанса монозамещенного продукта тиоцианирования пиррола было показано, что образуется 2-замещенное производное [259]. Подобные структурные определения станут более надежными, после того как накопится большое количество данных по характеристическим значениям констант взаимодействия в замещенных пирролах. Следует отметить, что, зная значения констант в простых пирролах, нельзя предсказать константы взаимодействия кольцевых протонов в конденсированных пирролах, поскольку было показано [260], что в некоторых конденсированных пирролах J46 принимает значения ~3,5 гц. Коэн и Мак-Лахлан [261] определили относительные знаки констант взаимодействия кольцевых протонов в 2-карбоксипирроле и показали, что все они одинаковы. В 2-метилпирроле метильные протоны взаимодействуют с протонами в положениях 3 (<72 3 = 0,65 гц) и 4 (<72 4 = 0,45 гц); однако в 2,3- и 2,5-диметилпроизводных взаимодействие такого типа обнаружить не удалось.
Хоффман и Гроновиц [262] обратили внимание на близкие значения констант взаимодействия кольцевых протонов J3 4 в фуранах, пирролах и тиофенах.
118
Глава 10
В табл. 10.73 собраны типичные значения констант взаимодействия протонов в ароматических молекулах. Была предпринята попытка объяснить величины констант взаимодействия J2 4 и J2 5 в пирролах изменением гибридизации атома азота [262].
Таблица 10.73
ТИПИЧНЫЕ КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ ПРОТОНОВ (гц) В МОНОЦИкЛИЧЕСКИХ АРОМАТИЧЕСКИХ СОЕДИНЕНИЯХ а
4,94-5,8 +
а Для фуранов, тиофенов и пирролов были определены лишь относительные знаки констант взаи-модействия, ио, по-видимому, абсолютные знаки положительны. Пределы коистаит взаимодействия обусловлены значениями, приведенными в этой главе.
10.17.1. Химические сдвиги в пирролах
В настоящее время еще не выяснено окончательно влияние растворителей на экранирование протонов в пирролах, и поэтому мы не можем детально обсуждать влияние различных факторов на химические сдвиги протонов в этих молекулах. В табл. 10.74 приведены т-значения для ряда пирролов, исследованных в растворах в диоксане [258]. При сравнении химических сдвигов протонов в замещенных пирролах и в самом пирроле можно видеть, что электроноакцепторные заместители дезэкранируют кольцевые протоны, тогда как электронодонорные заместители оказывают противоположное влияние. Абрагам и сотр. [263], исследуя влияние хлористоводородной кислоты
Корреляции спектров ЯМР-Н1 со строением молекул
119
Таблица 10.74
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ В НЕКОТОРЫХ ЗАМЕЩЕННЫХ ПИРРОЛАХ [258]
4 3
5 /2
N
Н
Заместители Концентрация в диоксане, % Химические сдвиги в, м. д. Боковые цепи
Т2 Тз т4 Т5
Нет 34 3,27 3,83 3,83 3,27
3-СН3, 4-СО2С2Н5 22 3,53 2,70 Т3-СН3^7 В’76
2-СНз, 3-СО2С2Н5 27 3,56а 3,50 а
2-СНО, 5-СН3 29 •3,07 4,01 тсно = 9>79 +н3 = 7+2
2-СНО 34 3,07 3,74 2,87 ^сно — О.бЗ
2-СОСНз 29 3,10 3,81 2,98
2-СНз 32 4,25 4,06 3,48 ТСНз = 7’76
3-CO2CH3 26 2,56 3,43 3,24
:2,5-(СН3)2; 3,4-(SCN)2 10 тсн3 = 7’70
I-CH3, 2,5-(СН3)2 39 б 4,41 4,41 Т2,5-СН3 = 7’99 Т1-СН3==6’95
а Отнесение неоднозначно.
б Эти сдвиги получены в растворе в циклогексане.
в Химические сдвиги измерены относительно сигнала диоксана; последний использован в качестве растворителя: т = б°"^?ан + 6’43'
на различные соединения, измерили химические сдвиги в некоторых метильных производных пиррола. С помощью метода ЯМР было показано, что под действием НС1 происходит протонирование молекулы по а-углеродному атому
R(4) R(3)
\ Z R(2)
н
В табл. 10.75 проведено сравнение химических сдвигов кольцевых протонов в монозамещенных пирролах и тиофенах [258]. Несмотря на то что для двух рядов соединений использовались разные растворители, совпадение между экранированием соответствующих протонов удивительно хорошее.
При введении метильной группы в молекулы тиофена, фурана и пиррола химические сдвиги кольцевых протонов изменяются на +0,75 м.д. Этот эффект аналогичен влиянию метильной группы на p-протоны в этиленовых системах общей формулы СН2 = СХСН3 и, по-видимому, является типичным для систем с «р2-гибридизованными атомами углерода. В пропилене, где влияние метильной группы определяется путем сравнения с химическими сдвигами в этилене .[265], вклад СНз-группы в экранирование mpawc-протона составляет +0,425 м.д. и цис-протона +0,325 м.д. (полный вклад равен +0,75 м.д.). В табл. 10.76 приведены химические сдвиги для метильных производных
120
Глава 10
X
Таблица 10.75 ХИМИЧЕСКИЕ СДВИГИ КОЛЬЦЕВЫХ ПРОТОНОВ ЗАМЕЩЕННЫХ ПИРРОЛОВ И ТИОФЕНОВ [258]
4 3
Соединение Химические сдвиги а, м. д.
Й2 бз 64 65
2-Метилпиррол +0,42 +0,23 + 0,21
2-Метилтиофен + 0,37 +0,24 + 0,28
2-Пирролальдегнд -0,76 -0,09 -0,40
2-Тиофенальдегид -0,65 -0,10 -0,45
2-Ацетилпиррол -0,73 -0,02 -0,29
2-Ацетилтиофен -0,57 0,00 — 0,2(5
З-Карбометоксипиррол -0,71 -0,40 -0,03
3-Карбометокситиофен -0,78 -0,47 +0,05
а Химические сдвиги измерены относительно соответствующих сигналов а- и p-протоиов пиррола и тиофена.
Таблица 10.76 ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ МЕТИЛЗАМЕЩЕННЫХ ФУРАНОВ, ПИРРОЛОВ И ТИОФЕНОВ3 [264]
4 3
5 к'
Соединение Т2 Тз Т4 Тб тСНз(2) тСНз(3)
Фуран 2,7 3,75 3,-75 2,70
2-Метилфуран 4,19 3,90 2,87 7,83
2,5-Диметилфу ра н 4,31 4,31 7,86
Тиофен 2,85 3,02 3,02 2,85
2-Метилтиофен 3,37 3,25 3,11 7,62
З-Метилтнофен 3,29 3,25 2,97 7,83
2,5-Диметилтиофен 3,61 3,61 7,70
Пиррол 3,49 3,85 3,85 3,49
2-Метилпиррол 4,24 4,02 3,66
а Тетраметилсилан использовался и в качестве растворителя и
в качестве эталона.
фурана, пиррола и тиофена [264]. Все образцы исследованы в виде разбавленных растворов в тетраметилсилане. Редди и Голдстейн [264] считают, что вклад метильной группы в экранирование протонов в этиленовой системе является мерой заряда, перенесенного заместителем в эту систему, возможно, в результате сверхсопряжения. При замене этиленовой системы на сопряженную структуру возможна делокализация заряда по этой сопряженной системе. Некоторое уточнение этой гипотезы можно сделать, зная химический, сдвиг ароматических протонов в толуоле: резонансный сигнал этих протонов, находится на 0,15 м.д. в более сильном поле, чем резонансный сигнал бен
Корреляции спектров ЯМР-Н1 со строением молекул
121
зольных протонов, а для пяти ароматических протонов полный сдвиг составит 0,75 м.д., что абсолютно совпадает с полным вкладом, наблюдаемым в этиленовой системе. Известно, что все рассмотренные в табл. 10.76 молекулы обладают ароматическими свойствами, и на их примере можно исследовать подобные-эффекты. При изучении данных, приведенных в таблице, можно видеть, что в этих соединениях указанные эффекты действительно имеют место. Влияние-замещения на различные кольцевые протоны зависит в некоторой степени от гетероатома, и наблюдаемые химические сдвиги лишь крайне приближенно,, что соответствует нашим знаниям о сопряжении в различных молекулах, отражают это влияние [264].
10.17.2. Порфирины
Порфирины, представляющие собой класс сложных органических соединений, интенсивно изучались методом ЯМР [267, 268, 271]. Скелет этих молекул имеет общую форму в виде замкнутой сопряженной системы.
Возникающий в результате сопряжения кольцевой ток сильно влияет на химические сдвиги протонов, расположенных как на самом порфириновом' кольце, так и вблизи него. На рис. 10.36 приведен спектр протонногсо
। । > । । । ।_____।__i__i—।_।__।_।—1_
О 5 10 15
•С, м.Э.
Рис. 10.36. Спектр протонного резонанса копропорфирина-1 в CDCh при 60 Мгц [269].
резонанса типичного порфирина, копропорфирина-1, где заместители Р — = СН2СН2СООСНз и М = СН3 [269]. Сигнал в очень слабом поле (т = = 0,04 м.д.) обусловлен четырьмя метиновыми протонами: таким образом, вследствие влияния кольцевого тока метиновые протоны дезэкранированы на 2,7 м.д. по сравнению с протонами незамещенного бензола. Широкий сигнал в очень сильном поле (т = 13,89 м.д.) обусловлен протонами NH, сильно-экранированными вследствие влияния кольцевого тока (смещение на 13,00 м.д. в сильное поле относительно протонов NH в пирроле). Подвержены влиянию-кольцевого тока и метильные группы в кольцах: они имеют химический сдвиг т = 6,45 м.д., указывающий на их меньшее экранирование по сравнению с обычными метильными группами у ненасыщенного атома углерода. Попытки:
122
Глава 10
дать количественное описание влияния кольцевых токов на экранирование протонов в порфиринах были не очень успешными, однако предсказание относительного экранирования протонов возможно [268]. Спектр протонного резонанса копропорфирина-1 подтверждает, что четыре пиррольных кольца эквивалентны вследствие таутомерного равновесия, включающего в себя обмен протонов NH со скоростью, большей 200 сек-1. Методом ЯМР было исследовано большое число порфиринов и измерено влияние заместителей на химические сдвиги протонов в этих молекулах [269, 270].
10.17.3. Индолы
Коэн и сотр. [272] показали, что метод ЯМР может быть успешно применен для определения структуры индолов (I)
Н(3) СН3 н
I II III
На рис. 10.37. показан спектр протонного резонанса индола. При исследовании •спектров метилзамещенных индолов известной структуры можно видеть (рис. 10.37), что два сильнопольных триплета в спектре индола обусловлены протонами Н(2) (т = 3,32) и Н(3) (т = 3,62). Триплетное расщепление обусловлено взаимодействием протонов Н(2) и Н(3) не только друг с другом, но и с протоном NH. Это подтверждается тем, что в спектре N-метилиндола (рис. 10.37, б) триплетное расщепление исчезает и протоны 2 и 3 дают пару дублетов. Из спектров, приведенных на рис. 10.37, можно сделать вывод об отсутствии индолениновой таутомерии в основном состоянии производных индола. Например, 3-метилиндол дает спектр, ожидаемый для структуры II, а не для структуры III, в которой протоны СН3-группы у насыщенного атома углерода дали бы резонансный сигнал с т = 8,60.
10.18. Пиридин и родственные молекулы
Бернстейн, Попл и Шнейдер [273] провели детальный анализ сложного спектра ЯМР пиридина (на рис. 10.38 показана часть спектра, записанного при 100 Мгц). Отнесение сигналов протонов 2 и 3 сделано при исследовании специфически дейтерированных пиридинов. Показано, что менее всего экранированы протоны в положении 2 и что протоны в положении 4 экранированы слабее протонов в положении 3: аналогичный порядок экранирования 2-, 3-и 4-углеродных атомов наблюдается в спектре ЯМР-С13 пиридина [205]. При анализе спектра протонного резонанса пиридина были рассчитаны константы взаимодействия
•^23 = 5,5 гц-, гц', J34 = 7,5 гц;
J25 = 0,9 гц; /35=1,6гц; 726 = 0,4 гц.
Хорошо известно, что в спектрах ЯМР пиридинов проявляется ярко выраженное влияние растворителя [275], и вследствие того, что ни в одном из исследований таких систем не было сделано поправок на влияние растворителя (путем использования малой концентрации в инертном растворителе), имеющиеся значения химических сдвигов не могут быть использованы для количественных оценок.
Необходимо рассмотреть также влияние кольцевого тока на экранирование протонов пиридина. Холл, Хардиссон и Джекман [247, 366] попытались оценить величину вклада кольцевого тока в экранирование протонов некоторых гетероциклических соединений при помощи теории самосогласованных
.__ХН (3,62)
ОС ,
N H (з з2) н
Индол (в СС1Л)
&52)
ЧДДн (3J8)
сн, (6,63)
N-Метилиндол (снз^^_
_ (в СВС13)_______________|
6,63 10,
а.н (3,87)
Асн, (7,80) н
2-Метилиндол (в CDCi3)
7,80
^-^снз (7,70)
О-Ан (3,20) н
З-Метилиндол (в СВС13)
2,82 3,20
t, м.Ъ.
7,70
Рис. 10.37. Спектры протонного резонанса индола и некоторых метил-замещенных индолов при 60 Мгц (т-значения приведены на спектрах) [272].
Рис. 10.38. Спектр резонанса протонов Н(3), Н(4) и Н(5) пиридина при 100 Мгц.
124
Глава 10
молекулярных орбиталей. Результаты, которые они получили, указывают на влияние заместителей на величину кольцевого тока. Если электроотрицательность группы, присоединенной к кольцу, возрастает, то увеличивается и кольцевой ток вследствие возрастающего противодействия электроотрицательного центра эффекту локализации элементов у азота кольца.
Брюгель [274] исследовал спектры протонного резонанса более чем 150 производных пиридина; в табл. 10.77 приведены химические сдвиги для некоторых из исследованных соединений. Вообще говоря, химические сдвиги
Таблица 10.77
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ В НЕКОТОРЫХ МОНОЗАМЕЩЕННЫХ
ПИРИДИНАХ [274]
Соединение а Химические сдвиги м. д.
t2 тз Т4 Тб те
Пиридин 1,71 в 3,23 2,85
4-Метилпиридин 1,45 2,96
4-Хлорпиридин 1,38 2,73
4-Цианопиридин 0,95 2,00
4-Амииопиридин 1,56 3,36
З-Метилпиридин 1,46 2,55 2,87 1,46
3-Хлорпи{]идин 1,27 2,27 2,71 1,36
З-Цианопиридин 0,78 1,53 2,19 0,91
2-Метилпиридин 3,00 2,57 3,08 1,49
2-Хлорпиридин 2,51 2,17 2,61 1,42
2-Аминопиридин 3,30 2,56 3,40 1,89
а Соединения исследовались в виде чистых жидкостей, когда это было возможно; твердые вещества исследовались в растворе в диметилсульфоксиде.
б т-Значения, экстраполированные от химических сдвигов, измеренных относительно воды как внешнего эталона.
в Сдвиг сигнала протона Н(2) в слабое поле был объяснен влиянием магнитной анизотропии атома азота и внутримолекулярного электрического поля, обусловленного локальным дипольным моментом неподеленной электронной пары азота [465].
отражают изменение электроотрицательности заместителя, однако в не-которых случаях важно учитывать влияние анизотропии. Из спектров 2-за-мещенных производных найдено, что заместители СН3 и NH2 вызывают увеличение экранирования кольцевых протонов, тогда как группа CN дезэкранирует их [276—278]. Эти наблюдения можно объяснить индуктивным влиянием и эффектом сопряжения данных заместителей [276]х).
Наблюдаемые в замещенных пиридинах константы взаимодействия аналогичны константам в самом пиридине, за исключением соединений, содержащих сильные электронодонорные заместители в положении 3 [274].
В работе [279] был проведен детальный анализ системы ABCD спектров протонного резонанса некоторых 3-замещенных пиридинов, исследованных в виде чистых жидкостей * 2). Полученные результаты приведены в табл. 10.78. Спектр протонного резонанса 3-бромпиридина при 40 Мгц приведен на рис. 10.39 и представляет собой типичный спектр 3-замещенного пиридина
1) Корреляция химических сдвигов протонов в 2-замещенных пиридинах и пиразинах с л-плотностями на углеродных атомах рассмотрена в работе [593].— Прим. ред.
2) Проведено исследование ряда замещенных 3-оксипиридинов в растворах органических соединений и в воде. Данные сопоставлены с распределением электронной плотности и реакционной способностью веществ [549—553].— Прим. ред.
Корреляции спектров ЯМР-Н1 со строением молекул
125
Таблица 10.78
КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ ПРОТОНОВ (гц) НЕКОТОРЫХ З-ЗАМЕЩЕННЫХ ПИРИДИНОВ [2 79]
(QH/\/\h(D)
Н(В)
(А)НХ /\/R
R JAB JAC JBC JAD JBD JCD
СОСН3 7,99 4,87 1,79 0,83 2,12 0,00
СОН 7,85 5,00 1,81 0,88 2,02 0,00
Вг 7,81 4,76 1,44 0,81 2,39 0,30
С1 8,22 4,69 1,52 0,71 2,49 0,30
1279]. Спектр пиридин-3-карбоксиальдегида подтверждает взаимодействие альдегидных протонов с p-протонами (J = 0,44 гц), а не с расположенными ближе к ним а- и у-протонами.
Ковалевский и сотр. [362] исследовали также сложные спектры типа ABCD, даваемые некоторыми 2-замещенными пиридинами. Величину типич-
н(в)
10гц
Спектр протонного резонанса 3-бромпирИдина при 40 Мгц [279].
Рис. 10.39.
пых констант взаимодействия в таких соединениях можно оценить при рассмотрении значений, полученных при анализе ABCD-спектра протонного резонанса 2-хлорпиридина:
Н(С) (Л)нАН<В> <djhAc1
Jab Jac Jad Jbc jbd Jcd
0,96 .7,22 4,67 7,75 0,75 1,98 гц
Методом ЯМР были исследованы также а-, |3- и у-пиколины, причем было показано, что влияние метильных протонов как на химические сдвиги, так и константы спин-спинового взаимодействия мало [280, 281]. В спектрах пиколинов не наблюдалось взаимодействия метильных и кольцевых протонов. В табл. 10.79 приведены спектральные параметры, полученные при полном анализе спектров пиколинов. В случае сложных спектров некоторую помощь при отнесении сигналов оказывают эксперименты по двойному резонансу.
На рис. 10.40 приведен спектр протонного резонанса 1.2,5,6-тетрагидропиридина и несколько спектров, показывающих изменения, происходящие в них при использовании двойного облучения некоторых протонов [282]. При изучении спектров двойного резонанса можно легко отнести резонансные
Рис. 10.40. Спектр протонного резонанса 1,2,5,6-тетрагидропиридина [282]. а — при 100 Мгц без двойного резонанса; б — сигнал олефиновых протонов при облучении протонов у атома С(2); в — сигнал олефиновых протонов при облучении протонов-у атома С(5); г — сигнал олефиновых протонов при облучении протонов у атомов С(2> и С<5); д — сигнал олефиновых протонов при облучении олефиновых протонов; е — сигнал протона С(2) при облучении протона С(5) н олефиновых протонов.
Корреляции спектров ЯМР-Н1 со строением молекул
127
сигналы, соответствующие протонам при С (3) и С (4). Мультиплет при 575 гц (т = 4,25) находится в олефиновой области спектра и представляет собой квартет типа АВ с дополнительной тонкой структурой, обусловленной спин-спиновым взаимодействием с протонами у С (2) и С (5). Квартет типа АВ обусловлен резонансными сигналами олефиновых протонов, разность химических сдвигов которых мала по сравнению с их константой взаимодействия. На рис. 10.40 сильнопольные компоненты мультиплета АВ при облучении протона у атома С (2) сужаются, и если предположить, что J2 з> 73 5, то это указывает на большее экранирование протонов у С (3) по сравнению с протонами у С (4). Этот вывод подтверждается при облучении протона у С (5), когда сужаются слабопольные компоненты мультиплета АВ. Спектр может быть еще более упрощен при использовании множественных облучений. Шуле-ри [282] обсудил и другие примеры использования двойного резонанса для подтверждения правильности отнесения сигналов в спектрах протонного резонанса.
10.19. Влияние растворителей на спектры протонного резонанса ненасыщенных гетероциклических соединений
Шефер и Шнейдер [275] показали, что экранирование протонов в ненасыщенных гетероциклических соединениях сильно зависит от влияния растворителя: эти эффекты обсуждаются в разд. 10.39.3.
10.20. Хинолины
В табл. 10.80 приведены химические сдвиги кольцевых протонов ряда алкилзамещенных хинолинов, растворенных в гексане, ацетоне и бензоле [283]. Изучение табл. 10.80 показывает, что специфическое влияние растворителей.
Таблица 10.80
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ в РЯДУ АЛКИЛЗАМЕЩЕННЫХ ХИНОЛИНОВ В ГЕКСАНЕ, АЦЕТОНЕ И БЕНЗОЛЕ [283] а
Соединение Растворитель Н(2) Н(3) Н(4) Н(5) Н(6) Н(7) Н(8)
Хинолин н-Гексан -1,32 0,33 -0,45 -0,60
Ацетон -1,42 0,02 -0,62 -0,80
Бензол -0,88 1,10 0,35 -0,42
7-Этилхинолин н-Гексан -1,30 0,38 -0,40 -0,07 0,22 -0,43
Ацетон -1,42 0,07 -0,77 -0,37 0,00 -0,43
Бензол -0,98 1,02 0,22 0,42 -0,35
6,8-Диметилхи- н-Гексан -1,28 0,33 -0,33 0,20 0,20
нолин Ацетон -1,35 0,10 -0,62 0,07 0,07
Бензол -0,95 0,98 0,25
5,8-Диметилхи- н-Гексан -1,32 0,32 -0,60 0,38 0,18
НОЛИН Ацетон -1,43 0,02 -0,85 0,22 0,03
Бензол -1,02 0,97 0,05
5,7-Диметилхи- н-Гексан -1,27 0,37 -0,57 0,43 -0,25
НОЛИН Ацетон -1,38 0,08 -0,85 0,25 -0,23
Бензол -0,98 0,95 0,05 0,93 -0,20
а Химические сдвиги (м. д.) измерены для 5 (мол.) %-ных растворов относительно CHCI3 (внешний, эталон), сделана поправка на объемную диамагнитную восприимчивость; т = 3,5 + б CHCI3 (виешн ).
128
Глава 10
Продолжение табл. 10.80
Соединен не Растворитель Н(2) Н(3) Н(4) Н(5) Н(6) Н(7) Н(8)
4,6-Диметилхи- «-Гексан -1,10 0,52 -0,13 0,12 -0,50
нолин Ацетон -1,18 0,25 -0,33 -0,05 -0,45
Бензол -0,88 1,13 0,38 0,62 -0,45
3,8-Диметилхи- н-Гексан -1,20 -0,17
НОЛИН Ацетон -1,30 -0,48
Бензол -0,97 0,47
2,8-Диметилхи- н-Гексан 0,38 -0,32
НОЛИН Ацетон 0,13 -0,60
Бензол 0,10 0,27
•очень велико, особенно в случае бензола. Несмотря на отсутствие какого-либо удовлетворительного объяснения предпочтительного влияния растворителя на отдельные части молекул, можно видеть, что влияние растворителя •становится больше с увеличением расстояния между атомами азота и водорода. Однако нет никаких простых соотношений, объясняющих, почему экранирование протонов 5, 6 и 7 в бензольных растворах меньше, чем экранирование протонов 3 и 4. Специфическое влияние растворителя может быть с успехом использовано при интерпретации сложных спектров замещенных хинолинов. В табл. 10.81 приведены константы спин-спинового взаимодействия, определенные из спектров алкилхинолинов [283]. Константа взаимодействия орто-протонов J2 3 = 4,0 ± 0,2 гц гораздо меньше аналогичной константы производных бензола (~ 8 гц).
Таблица 10.81 КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ КОЛЬЦЕВЫХ ПРОТОНОВ (гц) НЕКОТОРЫХ АЛКИЛЗАМЕЩЕННЫХ ХИНОЛИНОВ [283]
5 *
Js 8 N
Соединение Дгза J24a J3 4a Ji 8 в Ji 7 7 ./7 8 Jq 8
Хинолин 4,1 + 1,8± 8,5± 1,о± 8,2± 2,1 +
±0,2 ±0,2 ±0,3 ±0,2 ±0,3 ±0,2
2,8-Диметилхи- 4,2+ 2,2±
нолин ±0,2 +0,2
4,6-Диметилхи- 1,8± 8,5+
нолин ±0,2 ±0,2
5,7-Диметилхи- 4,0± 1,6± 8,4± 0,5
нолин ±0,1 ±0,1 +0,1
5,8-Диметилхи- 4,1 + 1,7± 8,4± 7,2±
НОЛИН ±0,1 +0,1 ±0,2 ±0,3
6,8-Диметилхи- 4,1± 1,8± 8,2±
НОЛИН ±0,1 ±0,1 ±0,2
7-Этилхинолин 4,1± 1,8± 8,3+ 0,8+ 8,4-J-
±0,1 +0,1 ±0,2 +0,2 ±0,1
А
Jg з, *^2 4 и J34 в хииолйие имеют одинаковые относительные знаки [426].
Корреляции спектров ЯМР-Н1 со строением молекул
129
В спектрах ЯМР 5,7-дихлор- и 5,7-диметилхинолинов наблюдалось спин-спиновое взаимодействие протонов в различных кольцах [284]. Спектры ЯМР 8-метилхинолина, 5,7-диметилхинолина и 5,7-дихлорхинолина приведены на рис. 10.41; химические сдвиги и константы взаимодействия, определенные из спектров, даны в табл. 10.82. Во всех спектрах квартеты А и В
8-Метитинолин
2,00 згоо f
Рис. 10.41. Спектры протонного резонанса при 60 Мгц [284].
а — 8-метилхинолнн; б — 5,7-дихлорхинолии; в — 5.7-диметнлхннолин. В спектрах а и в не показаны резонансные сигналы СН3-групп.
были отнесены соответственно к протонам 2 и 3, а сигнал С, отнесенный к протону 4, расщеплен подобно сигналам А и В, что указывает на взаимодействие протонов 2, 3 и 4 друг с другом. Однако сигнал С в спектре дизамещенных
Таблица 10.82
ХИМИЧЕСКИЕ СДВИГИ (т-ЗНАЧЕНИЯ) И КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ (гц) ПРОТОНОВ НЕКОТОРЫХ ЗАМЕЩЕННЫХ ХИНОЛИНОВ [284]
Соединение Т2 Тз Т4 те Т8 тметил Тг 3 Тз 4 Т2 4 Т4 8 Те 8
8-Метилхино-лин 1,28 2,88 2,18 7,23 4,1 7,9 1,8
5,7-Дихлорхи-нолин 1,18 2,66 1,61 2,50 2,05 4.2 8,5 1,6 0,8 2,0
5,7-Диметилхи-нолин 1,38 2,95 2,09 3,08 2,43 7,65 7,57 3,9 8,6 1,7 0,8
9^489
130
Глава 10
хинолинов представляет собой октет, что указывает на взаимодействие протона 4с каким-то другим протоном, отличным от протонов 2 и 3. Так как никаких других протонов в пиридиновом кольце нет, дополнительное расщепление может быть лишь следствием взаимодействия с протонами другого кольца. Другие отнесения, указанные на спектрах, основаны на эмпирических рассмотрениях влияния заместителей СН3 и С1 (сигнал D обусловлен протоном 8 и сигнал Е — протоном 6). Поэтому можно предположить, что в дихлорпроиз-водном протон 8 взаимодействует с протоном 4, тогда как для диметильного производного из-за того, что резонансные сигналы этих протонов неразре-шены и уширены, нельзя установить, взаимодействует протон 4 с протоном 6 или с протоном 8. Шефер [283] также наблюдал небольшую константу J48 («1 гц) в метилхинолинах.
Сейферт [355] измерил химические сдвиги кольцевых протонов в замещенных хинолинах и нашел хорошую линейную зависимость химических сдвигов отдельных протонов от их внеплоскостных колебательных частот С — Н. Были изучены также мононитропроизводные хинолинов [489].
Методом ЯМР изучены также другие азотсодержащие гетероциклические соединения: 4-замещенные пиридины [285], пиридин-1-оксиды [285], пирро-лины [286], пиридоны-2 и родственные соединения [287, 425] х). Из рассмотрения вкладов в экранирование кольцевых токов стала возможной количественная оценка ароматичности колец 2-пиридонов [287].
10.21. Тиофен и его замещенные
Тиофен и его замещенные были предметом исследования в работах многих авторов [174, 233, 253, 262, 288—298, 468, 470]. При исследовании образцов тиофена, дейтерированного в определенных положениях, Гроновиц и Хоффман [289] сделали однозначные отнесения сигналов в спектре ЯМР тиофена.
Юга
Л р
Рис. 10.42. Спектр протонного резонанса тиофена при 40 Мгц [289].
Константы взаимодействия в тиофене имеют одинаковые знаки [261] и равны J2 з =5,15 гц; /2 4 = 1,05 гц; J2 5 =2,75 гц и J3i =3,45 гц (точность всех полученных значений составляет ±0,2 гц). Чистый тиофен дает сложный спектр типа АА'ВВ', приведенный на рис. 10.42, причем мультиплет «-протонов находится на 0,125 м.д. в более слабом поле, чем мультиплет р-протонов [289]. Моно- и дидейтерированные тиофены дают более простые спектры протонного резонанса, позволяющие сделать однозначное отнесение сигналов (рис. 10.43). В табл. 10.83 и 10.84 приведены значения химических сдвигов
0 Среди азотсодержащих гетероциклических соединений, изучавшихся методом ЯМР-Н1, можно упомянуть также производные оксипиримидина [554, 555], аминотиа-золин, его алкильные и ацильные производные и их соли [556, 557], органилсилатра-ны [558].— Прим. ред.
Корреляции спектров ЯМР-Н1 со строением молекул
131
и констант спин-спинового взаимодействия для монозамещенных тиофенов [293]. Значения химических сдвигов получены для сильно разбавленных растворов в циклогексане, где межмолекулярными взаимодействиями можно пренебречь. Химические сдвиги, указанные в табл. 10.83, измерены относительно резонансных сигналов а- и Р-протонов тиофена; приведены также
Рис.
10гц
10.43. Спектры
10 гц
протонного резонанса двух дидейтерированных фенов при 40 Мгц [289].
тио-
экстраполированные т-значения. Детальное исследование влияния диамагнитной анизотропии заместителей на экранирование кольцевых протонов показало, что этот вклад в общее экранирование протонов невелик; следова-
Таблица 10.83 ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ ЗАМЕЩЕННЫХ ТИОФЕНОВ ОТНОСИТЕЛЬНО СИГНАЛОВ а- И р-ПРОТОНОВ ТИОФЕНА [293] а
4 3 5\~/2
Заместитель 2-Замещенные тиофены З-Замещенные тиофены
6з 6* Й2 в4 бе
no2 -0,82 +0,03 -0,30 -0,95 -О’, 60 -0,03
SO2C1 -0,73 -0,06 -0,45
CN -0,47 0,00 -0,28 —0,63 -0,20 -0,15
СНО -0,65 -0,10 -0,45 -0,79 -0,45 -0,03
СОСНз -0,57 0,00 -0,28 -0,68 -0,47 +0,02
СОСгН5 -0,55 0,00 -0,28 -0,70 -0,48 +0,04
СОС1 -0,88 -0,06 -0,44 -1,05 -0,50 -0,03
СО2СН3 -0,70 +0,05 -0,20 -0,78 -0,47 +0,05
SCN -0,30 +0,05 -0,28 -0,25 -0,05 -0,05
С- СН -0,15 +0,16 +0,12
I -0,13 +0,33 -0,01 -0,06 0,00 +0,19
Вг 4-0,05 +0,27 +0,11 +0,12 +0,08 +0,10
2-Тиенил -0,08 +о,п +0,15
SH 0,00 +0,20 +0,07 +0,22 +0,20 +о,ю
SCH3 4-0,03 +0,18 +0,05 +0,33 +о,ю +0,03
СН3 +0,37 +0,24 +0,28 +0,45 +0,22 +0,14
ОСН3 +0,94 +0,43 +0,82 + 1,10 +0,38 +0,20
ОС2Н5 +0,92 +0,43 +0,81
ОС(СН3)3 +0,77 +0,38 +0,65 +0,78 +0,36 +0,22
nh2 +0,95 +0,45 +0,85 + 1,25 +0,53 +0,25
а- и p-протонов тиофена: измерения были эк-ШЛООК _ Г» <1 1 I В Г Л л л . «
Химические сдвиги измерены относительно сигналов с; ;; p.„H„lu„uo 1иифена. измерения оыли экстраполированы к бесконечному разбавлению в циклогексановом растворе- t,=2 844-U пчта.-истинные значения т для а- и (3-протоиов равны 2,81 и 2,96 соответственно (приложение I) ’ ' 3'
9*
132
Глава 10
Таблица 10.84
КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ ПРОТОНОВ (ги) МОНОЗАМЕЩЕННЫХ ТИОФЕНОВ [292]
4_3
2-ЗАМЕЩЕННЫЕ ТИОФЕНЫ 5Sy /X
S
Заместители Ji 4 Ji 5 6s Взаимодействие в боковой цепи
no2 4,0 1,7 5,1
SO2C1 3,8 1,4 5,1
CN 4,5 4,5
СНО 3,8 5,0 *^н<5) - х =0,9
СОСН3 3,6 1,1 5,0
сос2н5 3,6 1,2 5,0
СОС1 3,4 1,4 5,0
со2сн3 3,6 1,3 5,0
SCN 3,7 1,4 5,2
с = сн 3,5 1,4 5,1 •^н(З) — х = 0’4
I 3,8 1,3 5,0
Вг 4,0 1,7 5,0
4 X
З-ЗАМЕЩЕННЫЕ ТИОФЕНЫ о/' /2
s
Взаимодействие
Заместители Ji 4 Ji 5 As в боковой цепи
no2 1,5 3,2 5,4
CN 1,4 2,8 5,1
СНО 1,4 2,7 5,2 •^н(5) —Х = 0,7
СОСН3 1,3 2,8 5,1
СОС1 1,4 2,9 5,1
СО2СН3 1,3 2,9 5,1
SCH 1,5 3,0 5,0
I 1,2 2,9 4,9
тельно, химические сдвиги различных кольцевых протонов обусловлены главным образом электронным окружением этих протонов. Наблюдаемые химические сдвиги хорошо согласуются с индуктивным влиянием и эффектом сопряжения данных заместителей. В 3-замещенных тиофенах химические сдвиги протонов 4 меньше химических сдвигов протонов 2, тогда как последние больше химических сдвигов протонов 3 в 2-замещенных тиофенах.
В 2-метилтиофене [262] константа взаимодействия протона 3 с метильными протонами равна 1,00—1,15 гц, тогда как протон 4 взаимодействует с метильными протонами с константой 7н-сн3=0,2—0,5 гц, причем эти константы имеют противоположные знаки. Аналогичным образом в 3-метилтиофене метильные протоны взаимодействуют с протоном 2 (0,9—1,25 гц) и с протонами 4 и 5 (0,4—0,5 гц). Этим константам дальнего взаимодействия было уделено много внимания из-за возможности взаимодействия по механизму сверхсопряжения [174, 297]. Расчет показал, что если эти константы дальнего взаимодействия обусловлены сверхсопряжением, то метильные протоны в 2,3-диметилтиофене должны взаимодействовать друг с другом с константой
Корреляции спектров ЯМР-Н1 со строением молекул
133
•Л:н3-сн3 = 1 гц [297]; /сщi-снз такого порядка наблюдалась для 2,3-диме-тил-4,5-дибромтиофена [405]
В г СН3
Br xsz СН3
спектр ЯМР-Н1 которого представляет собой два квартета с Jchs—сн3 = 0,75 гц.
10.22. Тиазол и его метильные производные
Тиазол представляет собой пятичленное кольцо с двумя гетероатомами — азотом и серой:
(4)Н
\_N / К (5)H/\S/XH(2)
При исследовании спектров ЯМР-Н1 ряда метилзамещенных тиазолов было сделано однозначное отнесение сигналов в спектре самого тиазола [266]. В табл. 10.85 приведены значения химических сдвигов и констант спин-спинового взаимодействия для ряда тиазолов. Кольцевые протоны 2 и 4 дают более широкие сигналы, чем протон 5, вследствие влияния квадрупольного момента
Таблица 10.85
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ ПРОТОНОВ НЕКОТОРЫХ ЗАМЕШЕННЫХ ТИАЗОЛОВ [266]
Соединение Химические сдвиги а, м. д. Константы взаимодействия, гц
62 Т2 64 т4 65 Тб Ji 4 Ji 5 Ji 5
Тиазол 2-Метилтиазол 4-Метилтиазол 2,4-Диметилтиазол -3,85 -3,67 1,4 1,5 -2,83 -2,53 2,4 2,7 1111 — ч- NO NO СИ ОО О 60 --J О 00 СИ 3,0 3,1 3,4 3,6 . 0 3,1 3,6 1,8 2,0
а б обозначает химический сдвиг, измеренный в миллионных долях относительно воды как внешнего эталона, т = 5,2 -j- 6pj2o (внешн).
соседнего атома азота. Влияние атома азота сказывается также и на значениях констант взаимодействия J2 4 и J2 5: несмотря на то что протон 2 отделен от протонов 4 и 5 одинаковым числом связей, взаимодействие через атом серы (J2 5 = 2,0 гц) значительно превышает взаимодействие через атом азота (724 слишком мала для обнаружения).
СЛОЖНЫЕ ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Метод ЯМР может дать много важных сведений о строении сложных органических молекул с неизвестной структурой, однако для проведения полного структурного анализа обычно необходимо применение и других физических методов. Наибольшую пользу метод ЯМР приносит на начальной стадии исследования, когда с его помощью можно охарактеризовать различные ядра в молекуле и оценить их относительные количества, и на конечной стадии исследования при выборе одной из нескольких возможных структур. Из
134
Глава 10
спектров ЯМР можно получить лишь небольшую информацию о скелете молекулы в целом, но иногда оказывается возможным указать на присутствие в ней отдельных циклических компонентов (ароматическое, циклогексановое, гетероциклическое кольца). В некоторых случаях из спектров ЯМР можно получить сведения о конформации молекул. Ниже обсуждаются некоторые примеры применения метода ЯМР для установления строения сложных органических соединений.
10.23. Алкалоиды
Спектры ЯМР-Н1 алкалоидов лунакрина и Лунина [299] указывают на следующие структуры:
Лунакрнн
Лунин
На рис. 10.44 показаны спектры протонного резонанса этих алкалоидов, и полезно более детально ознакомиться с отнесением резонансных сигналов в этих спектрах.
Рис. 10.44. Спектры протонного резонанса лунакрина и Лунина в CDC13 при 60 Мгц [299].
Сдвиги измерены относительно бензола (внешний эталон).
Лунакрин. Спектр ЯМР-Н1 лунакрина состоит из семи хорошо разрешенных мультиплетов, обозначенных на рис. 10.44 буквами от а до g. Исходя из значений химических сдвигов резонансные сигналы а и b отнесены к прото
Корреляции спектров ЯМР-Н1 со строением молекул
135
нам ароматических колец, мультиплет с — к а-протону в дигидрофурановом кольце, два интенсивных сигнала d — к N-метильным и О-метильным протонам, сложный мультиплет е — к p-протонам дигидрофуранового кольца и мультиплеты fug— к протонам боковой цепи. Группы линий е и с представляют собой спектры типа АВХ, обусловленные двумя (J-протонами (А, В) и а-протоном (X) дигидрофуранового кольца. Полный анализ этой области спектра дает следующие значения параметров: бдв = 0,278 м.д.; Jав = = 15,3 гц; Jax = 9,9 гц; 7вх = 8,7 гц. Компоненты в Х-части спектра (область с) претерпевают дальнейшее дублетное расщепление вследствие взаимодействия с другим ядром Y со спином I = (Jxy — J гц). Это наблюдение позволяет предположить, что алкильный остаток боковой цепи представляет собой изопропильную группу, что подтверждается наличием сигналов f и g. Интересной особенностью спектра является наличие двух неэквивалентных сигналов g метильных протонов в изопропильной группе; эта неэквивалентность метильных групп обусловлена асимметрией всей молекулы. Сигналы а и b обусловлены тремя неэквивалентными ароматическими протонами (система АВХ). Анализ этой части спектра дает значения Jax = 7 гц, Jab = = 7 гц, /вх = 2 гц и 6ав =0,15 м.д. Зная характеристические значения констант взаимодействия ароматических протонов, можно сделать вывод, что три ароматических протона должны находиться рядом друг с другом. Отнесение сигнала метоксильных протонов, расположенных рядом с азотом хинолинового кольца, сделано на основании химических сдвигов, полученных для замещенных хинолинов известного строения.
Лунин. Изучение спектра ЯМР-Н1 лунакрина помогает получить информацию о строении родственных алкалоидов. Строение алкалоида Лунина, содержащегося в листьях растений, определено именно таким образом. На рис. 10.44 приведен спектр Лунина, который, как можно видеть, очень похож на спектр лунакрина. Новый сигнал при +23 гц (относительно бензола как внешнего эталона) обусловлен метилендиоксигруппой (—СН2О2), что следует из значения химического сдвига и интенсивности сигнала и согласуется с молекулярной формулой соединения. Вследствие почти полной идентичности сигналов с, е, f и g в обоих спектрах был сделан вывод, что в а-изопропил-дигидрофуран-4-хинолиновой системе никаких изменений по сравнению с лунакрином не происходит. Этот вывод подтверждается изучением сигналов ароматических протонов а и Ь, свидетельствующих о наличии лишь двух протонов, находящихся в орто-положении друг к другу = 8,5 гц). Как и в спектре лунакрина, один из ароматических протонов дает сигналы в очень слабом поле; этот факт свидетельствует о том, что он находится в в положении 5 4-хинолиновой системы.
10.24. Стероиды
Исследованию стероидов методом ЯМР посвящено большое число работ, направленных на определение числа, положения и ориентации метильных групп в стероидной молекуле. Такое определение становится возможным благодаря тому, что установлены некоторые эмпирические соотношения между химическими сдвигами и положением метильных групп в различных стероидных молекулах известного строения [98, 491]. При помощи метода ЯМР можно определить характеристические химические сдвиги для экваториальных и аксиальных протонов, используя их для установления конфигурации заместителей. Для получения подобной информации на основе спектра ЯМР-Н1 при частоте 60 Мгц необходимо иметь 5—25 мг вещества, а для получения информации о метильных группах достаточно и 1 мг.
Изучение спектров протонного резонанса двух типичных 3-кетоаллопрег-нанов, показанных на рис. 10.45, может принести большую пользу при дальнейшей интерпретации протонных спектров стероидов [301, 303]. В сильно
136
Глава 10
польной области спектра (т = 7,3—9,3) можно видеть сложный широкий мультиплет, над которым возвышается несколько узких сигналов. Этот широкий сигнал обусловлен протонами, связанными непосредственно со скелетом колец: уширение обусловлено, во-первых, малыми временами релаксации таких протонов при отсутствии быстрого молекулярного вращения и, во-вторых, спин-спиновым взаимодействием протонов с близким экранированием. В противоположность этому ангулярные метильные группы, имеющие большие
Химический сдвиг относительно Si (СН3)+, гц
Рис. 10.45. Спектры протонного резонанса двух 3-кетоаллопрегнанов в CDC13 при 60 Мгц [301].
времена релаксации вследствие их быстрого вращения, дают интенсивные узкие сигналы. Из других сигналов, отделяющихся от широкого мультиплета, можно отметить сигналы метиленовых протонов, находящихся рядом с двойными связями, олефиновых протонов и протонов гидроксильных групп, а также протонов у атомов углерода, связанных с атомами кислорода, фтора, хлора и азота [301]. Вследствие сложности широкого «метиленового» мультиплета из него можно получить лишь ограниченную информацию. Однако эта область («фингерпринтовая», или, как ее иначе называют, «область отпечатка пальцев») имеет характерный вид для молекулы каждого стероида, и поэтому незначительные изменения в структуре молекулы существенно изменяют форму этого мультиплета [98]. Так, введение метильной группы в молекулу 3-кетоаллопрегнана приводит к существенным изменениям неразрешенной области спектра (см. рис. 10.45).
Наиболее значительный вклад в определение строения стероидных молекул дают характеристические химические сдвиги ангулярных метильных групп С (18) и С (19) [98, 302]. Как видно из табл. 10.86, в которой приведены хими-
Таблица 10.86
ХИМИЧЕСКИЕ СДВИГИ МЕТИЛЬНЫХ ГРУПП С(18) И С( 1 9) НЕКОТОРЫХ СТЕРОИДОВ [98]
Стероид а ^С(18), м. д. бС(1 9), м. д.
дб(в)-Холестен-3(3-ацетат 5,70 5,38
Д4-Андростен-17Р-ол-3-он 5,60 5,20
Д4-Андростен-17|3-о л-17а-метил-3-он 5,50 5,20
19-Нортестостерон 5,58 —
Д4-Прегнен-9а-фтор-11 р ,21 -диол-3,20-дион-21 -ацетат 5,45 4,83
Д4-Прегнен-21-ол-З,11,20-трион 5,73 5,00
Д4-П регнен-3,20-дион-21 -ацетат 5,70 5,20
д5<в)-Прегнен-Зр-ол-20-он 5,75 5,38
Д4-Прегнен-3,20-дион 5,80 5,25
Аллопрегнан-3,11,20-трион 5,83 5,20
П регнан-3,11,20-трион 5,78 5,18
Прегнан-3,20-дион 5,75 5,38
Аллопрегнан-3,20-дион 5,78 5,38
Д4-Прегнен-11 (З-ол-3,20-дион 5,50 4,93
Д4-Прегнен-11а-ол-3,20-дион 5,70 5,10
Д4-Прегнен-11 а-ацетокси-3,20-дион 5,70 5,15
Д5<6), 1в-Прегнадиен-ЗР-ол-20-он 5,50 5,35
Д6<в,-Прегнен-Зр-метокси-20-он 5,78 5,40
д5<в)-п регнен-ЗР,21 -диацетокси-20-он 5,73 5,38
д5(в)-Прегнен-Зр,21-диол-20-он-21-ацетат 5,73 5,38
д5(в), 1в-п регнадиен-Зр-ацетат-20-он 5,48 5,35
Аллопрегнан-3,20-дион-21 -ацетат 5,73 5,38
Д4-Прегнен-3,11,20-трион 5,78 5,00
Д5<6 >-А ндростен-ЗР-ол-17-он 5,50 5,38
Д5(в,-Андростен-Зр-ацетат-17-он 5,50 5,35
Аллопрегнан-11а-ол-3,20-дион 5,73 5,25
Стигмастерилацетат 5,70 5,40
Д5<в)-П регнен-Зр-ацетат-20-он 5,78 5,38
Д4-Андростен-3,11,17-трион 5,55 4,98
Эстрон 5,55 —
Андростан-ЗР-ол- 17-он 5,55 5,53
5,6-Дигидроэргостерилацетат 5,85 5,58
Д4-Прегнен-17а-ол-3,20-дион 5,68 5,18
Прегнан-4-хлор-17а-ол-3,11,20-трион 5,85 5,20
Д4-Прегнен-11Р, 21 -диол-3,20-дион 5,45 4,93
Прегнаи-11а-ол-3,20-дион 5,78 5,28
Андростан-За-ол-17-он 5,58 5,53
Этиохолан-За-ол- 17-он 5,58 5,48
Д4,17(20>-Прегнадиен-11р,21-диол-3-он 5,23 4,93
Д4,9 (И >.]-[ регнадиен-17а ,21 -диол-3,20-дион 5,75 5,08
Д ^Аллопрегнен-] 7а,21 -диол-3,11,20-трион-21 -ацетат 5,75 5,13
Прегнан-За-ацетокси-11,20-дион 5,83 5,25
Прегиан-За-ол-11,20-дион 5,83 5,30
д4,9(И)_11регнадиен-17а,21-диол-3,20-дион-21-ацетат 5,80 5,05
Эргостерин 5,78 5,45
Д5-Холестен-3-он 5,70 5,23
д5<в>,7-Холестадиен-Зр-ацетат 5,78 5,48
а Растворы в CDCls; в качестве внешнего эталона использовался бензол; т = 3,5 + (виешн)
138
Глава 10
ческие сдвиги групп 18-СН3 и 19-СН3, эти сдвиги очень чувствительны к природе и положению других заместителей [98]. Различия в экранировании метильных протонов, по-видимому, сильно зависят от влияния диамагнитной анизотропии соседних групп [23]. Как установили некоторые исследователи [301, 304], химические сдвиги метильных групп С (18) и С (19) могут быть рассчитаны, если предположить, что влияние соседних заместителей на экранирование метильных протонов аддитивно х). Сломп и Мак-Келлар [302] исследовали ряд стероидов с боковыми углеводородными цепями и, несмотря на сложность метильной области спектров, смогли определить положение резонансных сигналов ангулярных метильных групп.
Конформационное положение скелетных протонов, как правило, определить невозможно, поскольку их резонансные сигналы находятся в пределах широкой неразрешенной части спектра. Однако если к атому углерода, с которым связан водород, присоединен также атом кислорода, то резонансный сигнал пре тона смещается в слабое поле и становится легко различимым. Шулери и Роджерс [98] исследовали спектры протонного резонанса ряда оксистероидов (см. табл. 10.87) и показали, что химические сдвиги протонов
Таблица 10.87
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ У ЗАМЕЩЕННЫХ АТОМОВ УГЛЕРОДА В РЯДУ ГИДРОКСИЛЬНЫХ ПРОИЗВОДНЫХ СТЕРОИДОВ
Соединение Положение протона Ориентация группы ОН Конформация протона а Химический сдвиг м. д.
Андростерон С(3) За е 2,28
Эпиандростерон С(3) зр а 2,83
5-Изоандростерон С(3) За а 2,83
За-Оксипрегнан-11,20-дион С(3) За а 2,90
11Р-Оксипрогестерон С(11) пр е 1,95
Оа-Фторкортикостеронацетат С(11) ПР е 1,93
Кортикостерон С(11) пр е 2,05
Д4,17<20)-Прегнадиен-11р,21-диол-3-он С(11) пр е 1,95
11а-Оксипрогестерон С(11) 11а а 2,38
11а-Оксипрегнан-3,20-дион С(11) Па а 2,38
11а-Оксиаллопрегнан-3,20-дион С(11) Па а 2,50
19-Нортестосте рои С(17) 17₽ а 2,75
d а ~ аксиальный; е — экваториальный.
& Химические сдвиги измерены относительно внешнего стероидов в CDCI3; X = 3,5 + бс6Н6(внешн)-
эталона — бензола. Исследовались растворы
гидроксильных групп сильно зависят от того, находятся ли эти протоны в аксиальном или экваториальном положении. Используя эмпирические данные такого типа, часто можно получить полезные сведения о конформации стероидных молекул неизвестного строения.
Ричардс и сотр. [351] исследовали спектры ЯМР некоторых 11-кето-стероидов.
Я Подробные таблицы с указанием аддитивных вкладов различных заместителей в химические сдвиги метильных групп С (18) и С (19) приведены в работах [559, 560]. Вклады, обусловленные шестичленным кольцом D в £)-гомостероидах, были определены в работе [561].— Прим. ред.
Корреляции спектров ЯМР-Н1 со строением молекул
139
10.25. Жирные кислоты
Гопкинс и Бернстейн [305] исследовали методом ЯМР большой ряд жирных кислот, в которых функциональные группы, такие, как окси-, ацетокси-, эпокси- и другие, дают резонансные сигналы с характеристическими химическими сдвигами. При использовании значений химических сдвигов, приведенных в табл. 10.88, была определена структура некоторых жирных кислот в природных глицеридных маслах.
Таблица 10.88
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ ЖИРНЫХ КИСЛОГ [305]
Тип протонов Химический сдвиг 6 а’ М. д.
Концевые СН3 +0,43
СН2 в цепи 0,00
СН2 в a-положении к СООН -1,00
— ОСН2 — -3,00
1 — ОСН I -4,00
— сн=сн — -4,1
сн2-сн=сн — -0,75
Сопряженные СН = СН -4,6
СН3 в сложиоэфирной группе -2,3
а Измерен относительно сигнала СНз-групп в цепи длннноцепочечных жирных кислот; т=8,75-]-6 (весьма приближенное соотношение).
10.26. Аминокислоты и пептиды
В работах [306 — 308] были исследованы спектры ЯМР ряда аминокислот в воде и трифторуксусной кислоте с целью получения характеристических значений химических сдвигов, необходимых для установления структуры белков. Некоторые из этих значений указаны в табл. 10.89. При изучении спектральных параметров может быть получена информация о распределении зарядов в молекуле, индуктивном влиянии полярных групп и положительных зарядов, скорости протонного обмена с растворителем, силе оснований и в некоторых случаях о конформации молекул ]).
10.27. Другие исследования* 2)
На рис. 10.46 приведен спектр протонного резонанса (при 100 Мгц) птерокарпина вместе с двумя предполагаемыми структурами этой молекулы [282]. Анализ спектра свидетельствует в пользу структуры II [283, 352, 353].
Проводится исследование конформационных состояний аланиновых дипептидов [576] и циклических депсипептндов [577].— Прим. ред.
2) Наряду с отмеченными в этом и предыдущих разделах примерами приложения спектроскопии ЯМР к изучению природных соединений можно упомянуть также работы, посвященные исследованию схизаидрина [562], природных кумаринов [563, 565], гликозидов женьшеня и акантопанакса [566—572], сесквитерпенового лактона — гроссгеми-на [573], продуктов изомеризации аскаридола [574], антибиотика оливииа [575].— Прим. оед.
Таблица 10.89
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ АМИНОКИСЛОТ И ПЕПТИДОВ В РАСТВОРЕ В CF3COOH [308] а
Соединение TCONH TNH + ТСН2, СНз
Глицин Диглицин Тетраглицин 2,05 (д; J = 6 гц) 2,05 (м) 2,25 (м) 2,46 (м, ш) 2,46 (м, ш) 2,49 (м, ш) 5,80 (к; 7 = 6 гц) 5,71 (м, ш) 5,68 (м, ш)
TCONH XNH + ха-СН ТСН, СН2 тсн3
DL-Аланин DL-Валилглицин L-Валил-Ь-валин (2) (1) 1,97 2,15 (д; 7-4 гц) 2,52 (м, ш) 2,58 (м, ш) 2,52 (м, ш) 5,54 (м; 7 = 5 гц) 5,64 (м, ш) (1) 5,64 (м, ш) (2) 5,26 (м, ш) 7,55 (м) 7,61 (м) 8,15 (д; 7 = 7 гц) 8,75 (д; 7 = 6 гц) (1) 8,91 (д; 7 = 7 гц) (2) 8,52 (д; 7 = 7 гц)
TCONH тарил TNH+ та-СН ГСН, СН2, СНз
DL-Фенилаланин Этиловый эфир N-ацетил- L-тирозина 1,63 (д; 7 = 8,5 гц) 2,60 3,00 2,60 5,40 (м, ш) 5,03 (к; 7=7 гц) 6,56 (д; 7= 7 гц) СН3СОО 8,01 СН2 6,80 (д, 7 = 6 гц) СН2О 5,63 (к, 7 = 7,5 гц) СН3 8,66 (т, 7 = 7,5 гц)
TSH TNH+ та-СН ТСН2, СНз
L-Цистеин 8,06 (т; 7 = 9 гц) 2,34 (м, ш) 5,22 (к; 7 = 5 гц) 6,54 (д, д; 7 = 5 гц, 7 = 9 гц)
а Мультиплетность резонансных сигналов обозначена следующим образом: д — дублет; т — триплет; к — квартет; м — неразрешенный мультиплет. Если ширина сигнала превышает 10 гц, ои обозначен как широкий (ш^.
Корреляции спектров ЯМР-Н1 со строением молекул
141
Три ароматических протона 5, 6 и 8 образуют систему АВХ, для которой можно сделать однозначное отнесение сигналов, как это показано на рис. 10.46. Сигналы остальных ароматических протонов (3' и 6') не проявляют и следов расщепления, что свидетельствует о их пара-положении по отношению друг к другу в структуре II (взаимодействие орто-протонов равно 8 гц).
Семикарбазоны и 2,4-динитрофенилгидразоны альдегидов и кетонов могут быть охарактеризованы химическими сдвигами протонов N = С — Н [309,
I Птерокарпин Л
Химический сдвиг от сигнала Si(CH3)4, гц Н-------------------------->-
Рис. 10.46. Спектр протонного резонанса птерокарпина при 100 Мгц [282].
310] . В некоторых случаях из спектров ЯМР можно получить сведения о стереоизомерии, обусловленной связью—C=N—, и соотношении син-анти-изо-меров [310]. Методом ЯМР с успехом были изучены также пурины [311, 312], ротеноны [313], глицериды [407], замещенные нафталины [408], углеводы [490], норборнены [479] и [1—nl-парациклофаны [409].
10.28. Определение €-метильных групп методом ЯМР
Анет [112] детально изучил вопрос об определении С-метильных групп методом ЯМР высокого разрешения. Метильные группы, связанные непосредственно с насыщенным атомом углерода, дают интенсивные сигналы с характеристическим значением т ~ 9,0 м.д.
Если насыщенный углеродный атом связан, кроме метильной группы, с одним или несколькими атомами водорода, резонансный сигнал метильных протонов расщепляется вследствие спин-спинового взаимодействия протонов. C-Метильные группы у атомов углерода, не связанных с атомами водорода, обычно дают одиночные сигналы, но для некоторых молекул наблюдается слабое взаимодействие метильных протонов с протонами, отделенными четырьмя связями [59]. Например, в структуре I
Н3С О
. ус/ \ СН3
Н-' I 4 /
НзС\| ^СНз-СН3 = 0’5 гц
/ \ / СЕ3
Н ХО
I
142
Глава 10
протоны двух неэквивалентных геминальных метильных групп взаимодействуют слабо. Молекулы, содержащие группу ^СНСН3, дают в спектрах сильно расщепленные дублеты (J ~ 6—8 гц). Однако, если две группы протонов (СН и СН3) взаимодействуют сильно (см. разд. 8.13.3), то молекула дает спектр типа АВ3 и простое дублетное расщепление для метильных протонов больше не наблюдается. Для получения точных значений спектральных параметров в этих случаях необходимо проведение полного анализа спектров.
Довольно часто молекулы, содержащие группы ^)СНСН3, дают сложные
спектры, даже если протоны СН- и СН3-групп лишь слабо взаимодействуют друг с другом (т. е. разность химических сдвигов сигналов, соответствующих
Рис. 10.47. Спектры протонного резонанса при 60 Мгц [112].
а — метилянтарная кислота; б — (3-метнлглутаровая кислота в CF3COOH. Сигналы групп СООН не показаны. Сигналы метильных протонов при медленном прохождении показаны справа.
СН- и СНз-группам, гораздо больше константы взаимодействия между ними). Это явление обусловлено тем, что протоны —СН сильно взаимодействуют с другой группой протонов в молекуле. Для проведения полного анализа системы необходимо рассматривать все протоны, взаимодействующие с протонами, с которыми взаимодействуют метильные протоны. Поэтому может наблюдаться кажущееся взаимодействие протонов, даже если константа взаимодействия между ними равна нулю. (Машер и Кори [314] провели детальное исследование такого кажущегося взаимодействия в трехспиновых системах.) Некоторые примеры таких явлений приведены в спектрах протонного резонанса метилянтарной кислоты и Р-метилглутаровой кислоты, показанных на рис. 10.47. Несмотря на аналогию в строении этих молекул, форма сильнопольного резонансного сигнала в двух спектрах совершенно разная. В метилянтарной кислоте этот сигнал представляет собой четкий дублет: разность химических сдвигов а- и двух а'-протонов достаточно велика, чтобы на резонансный сигнал метильных протонов не могли влиять какие-либо другие
Корреляции спектров ЯМР-Н1 со строением молекул
143-
ядра, кроме третичного протона. В p-метилглутаровой кислоте а-, а'- и р-про-тоны лишь слабо отличаются по экранированию друг от друга и дают один широкий резонансный сигнал. В этом случае для метильных протонов наблюдается сложный сигнал [112].
В результате такого поведения групп ^>СНСН3 расщепление сигнала метильных протонов часто бывает меньше истинной константы спин-спинового взаимодействия 7н-сн3- Анет [112] объяснил этим аномальные константы взаимодействия в метилциклогексане, а также сильно изменяющиеся значения констант взаимодействия в 6а- и бр-метилстероидах [303].
СОЕДИНЕНИЯ, в КОТОРЫХ ВОДОРОД СВЯЗАН С АТОМАМИ, ОТЛИЧНЫМИ ОТ УГЛЕРОДА
10.29. Водород, связанный с атомами кислорода
В разд. 9.4 было показано, что такие атомы водорода часто образуют водородную связь, в результате чего большой вклад в их экранирование вносят межмолекулярные взаимодействия. Единственно приемлемыми значениями химических сдвигов такого типа будут значения, полученные при бесконечном разбавлении в инертных растворителях, таких, как четыреххлористый углерод или циклогексан, в которых молекулы обычно существуют в мономерной форме. Вторым фактором, усложняющим изучение гидроксильных систем, является возможность обмена водорода, связанного с атомом кислорода. Кроме смещения резонансного сигнала, это приводит также к исчезновению его тонкой структуры. Вследствие этих трудностей лишь в небольшом числе работ сделаны попытки скоррелировать химические сдвиги протонов этого типа со строением молекул. Главное место в исследованиях гидроксильных систем методом ЯМР отведено водородной связи и обменным процессам [315—317] (см. разд. 9.4.4). В табл. 10.90 приведены экстраполированные к бесконечному разбавлению в инертном растворителе химические сдвиги для ряда молекул, содержащих оксигруппы. Интересной особенностью химиче-
Таблица 10.90
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ В ОН-СЙСТЕМАХ
ПРИ БЕСКОНЕЧНОМ РАЗБАВЛЕНИИ В ССЦ а
Соединение т, м. д. Литература
СН3ОН 9,5
С2Н5ОН 9,3
/прет-С4Н9ОН 9,3
Фенол 5,8 315
о-Крезол 5,5 315
п-Хлорфенол 5,3 315
ж-Хлорфенол 5,5 315
о-Хлорфенол 4,4 315
Метилсалицилат —0,58
Салициловый альдегид —0,95
Карбоновые кислоты (димеры) —2,2 315, 316, 319
а Химические сдвиги измерены относительно циклогексана (внутренний эталон) в растворах в ССЦ и затем переведены в т-шкалу [85].
144
Глава 10
ских сдвигов замещенных фенолов является их постоянство (т ~ 5,5); однако в молекулах, для которых можно было бы предсказать сильную внутримолекулярную водородную связь (например, в орто-хлорфеноле), наблюдается существенное уменьшение экранирования. Разбавление в инертном растворителе не разрушает внутримолекулярных водородных связей.
Патерсон и Типман [360] измерили химические сдвиги гидроксильных протонов в ряду пара-замещенных фенолов, применяя экстраполяцию их к бесконечному разбавлению в четыреххлористом углероде. Эти данные приведены в табл. 10.91, причем экстраполяция справедлива вследствие того,
Таблица 10.91
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ
В РЯДУ пара-ЗАМЕЩЕННЫХ
ФЕНОЛОВ [360]
Соединение
Химический сдвиг ОН а, м. д.
С8Н5ОН п-С1С6Н4ОН п-ВгС8Н4ОН п-1С8Н4ОН
—2,84
—2,90
—2,91
—2,92
а Химические сдвиги ОН измерены относительно циклогексана (внутренний эталон) н экстраполированы к бесконечному разбавле-НИЮ в ССЦ; т = 8,564 + 6Сен12 (внутр)-
что исследования проводились при малых концентрациях образцов. Зависимость экранирования ОН-протонов от природы заместителей в кольце не наблюдалась.
Гутовский и сотр. [318] измерили химические сдвиги ОН-протонов для фенола, р-нафтола, 9-фенантрола и некоторых хелатных орто-замещенных производных каждого из этих соединений, применяя экстраполяцию к бесконечному разбавлению в четыреххлористом углероде. Различие в экранировании ОН-протонов в хелатных и исходных молекулах (А6) авторы объяснили наличием внутримолекулярных водородных связей. Показано, что значения А6 пропорциональны порядку связей между атомами углерода ароматического кольца, у которых находятся заместители [318] ').
В карбоновых кислотах положение резонансного сигнала протона ОН-группы зависит от разбавления лишь при концентрации, меньшей 25 мол.%: это наблюдение соответствует известному свойству таких молекул образовывать стабильные димеры и существовать в мономерной форме лишь при сильном разбавлении. Ривс [319] измерил химические сдвиги протонов —СООН для некоторых простых карбоновых кислот, существующих в виде димеров (чистые жидкости), и показал, что химические сдвиги одинаковы для всех молекул (т = — 2,2+ 0,1); следовательно, окружение протонов в различных молекулах с водородной связью приблизительно одинаково. Известно, что уксусная кислота образует в жидком состоянии не димеры, а полимеры, что находит свое отражение в значении химического сдвига протона СООН-группы (т =—1,55); при разбавлении в четыреххлористом углероде химический сдвиг увеличивается дот-— 2,1, т. е. до значения, ожидаемого для димера.
’) Проведено исследование водородной связи пространственно затрудненных фенолов [578, 579]. Химические сдвиги ОН мономерных молекул подвержены влиянию пространственного электронного отталкивания, которое приводит к уменьшению экранирования протона группы ОН [580].— Прим. ред.
Корреляции спектров ЯМР-Н1 Со строением молекул
145
10.30. Водород, связанный с атомами азота
N14 имеет спин 7= 1 и поэтому обладает квадрупольным моментом. В связи с этим под влиянием квадрупольной релаксации происходит уширение резонансных сигналов протонов, связанных непосредственно с атомом азота. Такого уширения сигнала можно избежать либо методом двойного резонанса [257], либо исследованием симметричных молекул типа иона аммония, в котором отсутствуют градиенты электрического поля [320]. Если протон NH-rpyn-пы быстро обменивается с другими протонами системы (например, в аминах в присутствии следов воды), наблюдается острый резонансный сигнал. Водород, связанный с азотом, может образовывать водородную связь, и химические сдвиги для мономера могут быть получены лишь при исследовании разбавленных растворов в инертном растворителе [161, 321, 322] *) (разд. 9.4.4).
Для протонов NH мономерного моноэтиламина в четыреххлористом углероде т =9,17 (СС14 реагирует с аминами; измерения проводились до начала заметного протекания реакции) [321, 322]. При охлаждении аминов химический сдвиг протонов NH остается постоянным вплоть до температуры замерзания: эти значения брались в качестве химического сдвига ассоциата «-мера; сводка таких значений дана в табл. 10.92. Ассоциативный сдвиг
Таблица 10.92
АССОЦИАТИВНЫЙ ХИМИЧЕСКИЙ СДВИГ НЕКОТОРЫХ АМИНОПРОИЗВОДНЫХ
Соединение Ассоциативный сдвиг, м. д. Литература
Моноэтиламин 0,995 322
Диэтиламин 0,990 322
Изобутил амин >0,87 322
Аммиак 1,05 161
(разница в химических сдвигах мономера и n-мерд) равен во всех случаях 1,0 м. д. Это значение хорошо согласуется с ассоциативным сдвигом, наблюдавшимся для жидкого и газообразного аммиака [161]. Ввиду наличия многих факторов, затрудняющих исследование протонов NH методом ЯМР, не удивительно, что до сих пор не получено никаких корреляций химических сдвигов NH со строением молекул. Керн и Липскомб [323] рассчитали экранирование протонов NH, однако полученные значения плохо согласуются с экспериментальными данными.
Как мы уже видели, протоны, связанные с атомами азота, могут давать широкие сигналы вследствие влияния квадрупольной релаксации (ширина сигнала в формамидах и пирролах равна 10—75 гц при 40 Мгц) и узкие сигналы вследствие обменных процессов; имеется еще возможность расщепления сигнала NH в триплет вследствие спин-спинового взаимодействия с ядрами N14 (7 -- 1). Такие триплетные расщепления наблюдались в спектрах протонного резонанса безводного аммиака [324] и ионов аммония в кислых растворах [325].
При наблюдении уменьшения ширины линий при повышении температуры Робертс [326] показал, что часто наблюдаемое уширение резонансных сш налов протонов NH действительно обусловлено квадрупольной релаксацией, а не промежуточными скоростями обмена протонов. Для многих азотсодержа-
Я Исследованы химические сдвиги NH при разбавлении для циклических иминов [580] и пиразола [581].— Прим. ред.
10-489
146
Глава 10
щих молекул широкий сигнал NH при достаточно высоких температурах расщепляется в триплет. В табл. 10.93 приведены константы взаимодействия
Таблица 10.93
ВЛИЯНИЕ ТЕМПЕРАТУРЫ НА ПАРАМЕТРЫ СПЕКТРОВ ПРОТОННОГО РЕЗОНАНСА НЕКОТОРЫХ NH-СОДЕРЖАЩИХ МОЛЕКУЛ [326]
Соединение Сигнал NH при 35° Приблизительная температура перехода синглета в триплет, °C ^триплета » гц
Формамид Широкий синглет 50+10 60±4
Ацетамид То же 175+10 56+5
N -Мети лформамид » » Не наблюдается 60+5
до 250°
N -Метилацетамид » » 225+20 60 + 5
Сукцинимид >250
Пиррол Очень широкий синглет 50±25 55+5
Метиламмонийхлорид а Широкий триплет < 0 49+2
Этиламмоиийхлорид а То же < о 50+2
Диметиламмонийхлорид а Очень широкий триплет <0 53+3
Солянокислый пирролидин а То же 25+5 52+4
а Приблизительно 50%-ные растворы в воде, содержащие две капли концентрированной HCI на 0,5 мл раствора.
протонов NH-групп и температурные изменения для некоторых молекул. Использование двойного резонанса позволило однозначно определить природу уширения сигналов NH.
Для идентификации водорода в группах ОН и NH методом ЯМР необходимо исследовать растворы низкой концентрации в инертных растворителях. Если в спектре имеются сигналы, чрезвычайно чувствительные к изменению концентрации и температуры, то эти сигналы, несомненно, обусловлены протонами этого типа. Добавление к образцу небольшого количества кислоты приведет к значительному изменению химического сдвига этих сигналов, и это свойство можно использовать при отнесении сигналов. Другой быстрый метод определения сигналов —ОН и —NH заключается во встряхивании образца с небольшим количеством тяжелой воды (D3O): быстрый обмен водородов, связанных с атомами кислорода и азота, на дейтерий приведет к уменьшению интенсивностей интересующих нас сигналов.
10.31. Водород, связанный с атомами бора
Опубликовано лишь небольшое число примеров химических сдвигов для групп типа ВН. В диборане [436] твн2 =6,05 м.д. и твнв = 10,53 м.д.; константа взаимодействия мостиковых и концевых протонов в диборане равна ~7 гц.
Химический сдвиг т протонов ВН-группы в боразоле и родственных соединениях равен 5,5 м. д. По-видимому, в экранирование кольцевых протонов в боразоле существенный вклад вносят ароматические кольцевые токи [327].
Керн и Липскомб [323, 328] рассчитали экранирование протонов в диборане: полученные ими результаты приблизительно совпадают с экспериментально наблюдаемыми значениями экранирования мостиковых и концевых протонов ВН (см. разд. 12.1.3).
Корреляции спектров ЯМР-Н1 со строением молекул
147
10.32. Водород, связанный с атомами серы
Показано, что резонансный сигнал протона SH-группы в этилмеркаптане при переходе от чистой жидкости к раствору в СС14 смещается в более сильное поле. Это служит доказательством образования в чистой жидкости водородных связей. Различие в химических сдвигах протонов SH-групп в чистой жидкости и в разбавленном растворе в четыреххлористом углероде равно 0,38 ± ± 0,015 м. д. [329]. Работ по исследованию атомов водорода, связанных непосредственно с атомами серы, очень мало [217, 329—331], и лишь в случае этилмеркаптана сделана оценка влияния водородной связи на химические сдвиги протонов SH. Однако ввиду небольшого различия в химических сдвигах в чистой жидкости и в разбавленном растворе - этилмеркаптана можно лишь незначительно расширить область значений химических сдвигов в алифатических тиолах (т =8,4—8,8), указанную Чемберленом [217] (в разбавленных растворах в СС14 наблюдаются значения т = 8,4—9,2). В тиофеноле химический сдвиг протонов SH равен т = 6,4, однако в данном случае не делалось никакой поправки на вклад межмолекулярных взаимодействий в экранирование протонов SH в этой молекуле [217].
Недавно [462] опубликованы данные по химическим сдвигам протонов в некоторых алифатических (tsh = 7,8—9,0) и ароматических (т = 6,3—7,0) тиолах, исследованных в виде разбавленных растворов в четыреххлористом углероде х).
10.33. Водород, связанный с атомами кремния
' Исследование влияния органических групп и атомов хлора в некоторых замещенных силанах показывает, что для объяснения экранирования протонов в этих молекулах необходимо, кроме электроотрицательности заместителей, учитывать и другие факторы [332]. В табл. 10.94 приведены химические
Таблица 10.94
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ Si —Н В ТРИЗАМЕШЕННЫХ СИЛАНАХ RgSiH [332] а
R Мультиплет -иость пика т, м. д. R Муль-ти~ плет-ность пика т, м, д. R Мультиплет -ность пика т, м. д.
СН3 10 6,149+0,014 я-С4Н9 м 6,336 +0,002 п-С8Н4С1 1 4,625+0,014
с2н5 7 6,388+0,012 нзо-С4Н9 7 6,151 +0,007 СН2С6Н5 7 5,969+0,014
н-С3Н7 7 6,316±0,014 СН2=СН- м 5,672±0,012 п-СН2С6Н4СН3 7 6,023+0,014
нзо-С3Н7 м 6 6,701+0,014 CgHs 1 4,579+0,005 п-СН2СяН4С1 7 6,013+0,012
п-С6Н4СНз 1 4,649+0,012 С1 1 3,848+0,002
а Дополнительные данные по химическим сдвигам протонов Si—Н приведены в работах [461 , 488]. ® м означает мультиплет, в котором нельзя было подсчитать точное число линий.
сдвиги протонов Si — Н в ряду тризамещенных силанов [332]. В общем случае увеличение электроотрицательности заместителей вызывает уменьшение экранирования протонов Si — Н. Однако некоторые сдвиги аномальны; например, в соответствии с выводами этой работы для винильных и фенильных производных должен соблюдаться следующий порядок электроотрицательностей: С2Н5 < СН=СН2 < С6Н5, тогда как в других исследованиях [333] обнару-
1) Найдено, что константы вицинального HS — СН и дальнего HS — С — СН взаимодействий имеют положительный знак [582].— Прим. ред.
10*
148
Глава 10
живается обратный порядок. Аномальное экранирование в винильных производных было объяснено образованием л-связи р — d между 8р2-гибридизован-ным атомом углерода и кремнием [332, 180], а результаты для фенильных производных были объяснены в терминах теории валентных связей вкладом резонансных структур типа
= Si = <f Cl
Однако многие аномалии, найденные для химических сдвигов протонов в замещенных силанах, наблюдаются также в спектрах соответствующих производных углерода: следовательно, предположение о наличии в кремниевых соединениях л-связи р — d, сделанное на основании данных ЯМР, далеко не однозначно [439].
Если в замещенных силанах изменять природу алкильной группы, то экранирование протонов Si — Н будет изменяться в следующем порядке: СНз = изо-С4Н9 < rt-C3H7 = «-С4Н9 < С2Н5 < изо-С3Н7, который хорошо согласуется с ожидаемыми индуктивными эффектами *)• Было сделано предположение, что отклонения от ожидаемых индуктивных эффектов могли быть обусловлены сверхсопряжением. Химические сдвиги протонов в замещенных гидридах кремния менее чувствительны к изменениям природы заместителей по сравнению с химическими сдвигами соответствующих производных углерода [410].
Зависимость т-значений протонов связи Si — Н от колебательных частот Si — Н линейна для всех тризамещенных силанов, за исключением винильных и фенильных производных. Следовательно, оба эти параметра обусловлены, по-видимому, одинаковыми эффектами, отличающимися от простых индуктивных влияний [332].
Уэбстер [332] исследовал соединения с общими формулами (CH3)xCl3_xSiH, (СН3)ЖС13_ЖСН, (СН3)ж(С6Н5)з_ж51Н и (СНз)ж(С8Н5)3_жСН и показал, что последовательное замещение метильных групп на электроотрицательные атомы хлора обусловливает дезэкранирование протонов Si — Н и С — Н. Каждое последующее замещение оказывает меньшее влияние на экранирование, чем предыдущее, причем экранирование протонов С — Н по сравнению с протонами Si — Н изменяется гораздо сильнее. Последний эффект, по-видимому, обусловлен стремлением кремния к образованию двойной связи, что приводит к локализации электронной плотности на атоме кремния и поэтому уменьшает влияние замещения метильных групп на атомы хлора. Аналогичные эффекты наблюдаются в фенильных производных силана.
При исследовании констант взаимодействия Н — D в дейтерированных силанах, SiH3D и SiHD3, было показано, что в SiH4 Jhh =2,75 ± 0,15 гц [361]. Константы геминального взаимодействия Н — Н были измерены аналогичным образом и для замещенных силанов; эти значения вместе с другими константами взаимодействия приведены в табл. 12.52. Попытки объяснить изменения геминальных констант взаимодействия на основе простого метода валентных связей исходя из изменения валентных углов Н — Si — Н оказались безуспешными. Эта неудача была объяснена влиянием d-орбиталей кремния на спин-спиновое взаимодействие [361]; однако более вероятно, что такой подход к решению этой проблемы вообще несостоятелен [58, 56].
10.34. Водород, связанный с атомами германия, олова и свинца
Дрейк и Джолли [359] исследовали методом ЯМР некоторые германы. Протоны в группах —GeH3 имеют химические сдвиги, равные —3,3 м. д.
Я Корреляция химических сдвигов протонов Si — Н и Ge — Н с индуктивными константами заместителей в замещенных гидридах кремния и германия была проведена в работах [583, 584].— Прим. ред.
Корреляции спектров ЯМР-Н1 со строением молекул
149
относительно тетраметилсилана (внешний эталон), тогда как соответствующие сдвиги для групп —SiH3 равны от —3,3 до —3,6 м. д., а для групп —СН3 они составляют около —1,1 м. д. Протоны в группах ^)GeH2 экранированы сильнее протонов —GeH3 на 0,193 м. д. Такое поведение аналогично свойствам силанов, но противоположно свойствам алканов, что позволяет предположить следующий ряд изменения электроотрицательностей: С > Н > > Si Ge *).
Каэж и Флиткрофт [374] исследовали спектры протонного резонанса некоторых станнанов и родственных соединений, включая (СН3)3РЬН; полученные
Таблица 10.95
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ ПРОТОНОВ В НЕКОТОРЫХ СТАННАНАХ И РОДСТВЕННЫХ СОЕДИНЕНИЯХ [374]
Производные олова Химические сдвиги (т-значення) и константы взаимодействия Н-Н (гц) Константы взаимодействия, гц
TSn-H (мульт.) а TSn—СНз (мульт.) а JHCSnH Snii9—н SnH7— н С13.Ц Snli9-СНз Snll7—СНз
SnH46 CH3SnH3B (CH3)2SnH2B (CH3)3SnH в (CH3)4Sn (CH3)3Sn-,Li+r 6,15(1) 5,86 (4) 5,24 (7) 5,27(10) 9,73 (4) 9,83 (3) 9,82 (2) 9,86(1) Ю,4 (1) 2,7 2,53 2,37 1931 1852 1758 1744 1846 1770 1682 1664 130 126,5 128,5 128 t 58,0 56,5 54 2 55,5 54,5 51,5
14
Производные свинца гРЬН (мульт.) ТрЬСНз (мульт.) 7НСРЬН РЬ207—н Cis—н РЬ2О7-СНз
(СН3)3РЬН б (CH3)4Pb 2,32(10) 9,15(2) 9,13 1,4? 2379 136 132,5 66,7 60,5
а В скобках указана мультиплетность сигналов,
б Химические сдвиги измерены для 9%-ного раствора в циклопентане, содержащем 9% толуола (внутренний эталон), CHg-группа которого в этих условиях имеет Т= 7,68.
в Чистая жидкость прн -|-40о.
гПриблизительно 0,1 М раствор в CH3NH2, для СНз-протонов которого Т= 7,77.
ими результаты приведены в табл. 10.95. Протоны, связанные с оловом и свинцом (т = ~ 6 и ~2 соответственно), экранированы слабее протонов, связанных с углеродом в аналогичных производных. На рис. 10.48 показан спектр протонного резонанса триметилплюмбана (СН3)3РЬН. Метильные и гидридный протоны дают два раздельных мультиплета с правильным отношением интенсивностей, и для обоих наблюдаются спутники РЬ207Н. Метильные протоны взаимодействуют с гидридным протоном, в результате чего резонансный сигнал метильных протонов расщеплен в дублет (7н-сн3 = 1,47 гц).
Исследованы также спектры ЯМР смешанных гидридов кремния, германия, мышьяка и фосфора (например, SiH3PH2, SiH3AsH2, GeH3PH2 и GeH3AsH2) [411].
1) В работе [585] с помощью спектров ЯМР-Н1 исследовалось строение эфиратов тригалогенгерманов.— Прим. ред.
150
Глава 10
Ривс и Уэллс [449] вывели следующее соотношение для констант взаимодействия Jx-н и атомного номера Zx для элементов IV группы (X = С, Si, Sn и Pb):
(/x-H/Px)l/2 = °-676Zx + 8,0.
Аналогичное соотношение получено для констант взаимодействия 7х-сн[449].
(сн3)3рЬн
(сн3)3рьн
2,379 гц
.с6н5сн3
WH3,
Рис. 10.48. Спектр протонного резонанса (СН3)3РЬН-при 40 Мгц и температуре —50° [374].
а — общий вид спектра, включая сигналы внутреннего эталона — толуола; б — растяну- ' тый спектр протона группы РЬН; в — растянутый спектр протонов группы СН3 — Pb со спутниками РЬ207Н и С13Н.
10.35. Водород, связанный с атомами переходных металлов
Протоны, связанные непосредственно с атомами диамагнитных переходных металлов, экранированы очень сильно, а их сигналы смещены в сильное поле от сигнала тетраметилсилана на 5,9—17,3 м. д. и, следовательно, могут быть легко отнесены в спектрах ЯМР-Н1 [323, 334—3411. Например, гидрид бисциклопентадиенилрения (C5H5)2ReH дает два сигнала с отношением интенсивностей 10 : 1 [334]. В табл. 10.96 приведены химические сдвиги гидридов для некоторых молекул этого типа. Сильное экранирование гидридных протонов, по-видимому, обусловлено внедрением протона в электронную плотность металла [338]. Керн и Липскомб [323, 336, 455] дали теоретическое объяснение химического сдвига гидридного протона в НСо(СО)4; ими показано, что экранирование зависит от электронной плотности вокруг протона и от распределения электронов по 4s- и 4р-орбиталям атома кобальта.
Исследованы спектры ЯМР-Н1 протонированных комплексов некоторых карбонилов переходных металлов (таких, как [л-С5Н5Мо(СО)3]2Н+ и ]n-C5H5Ru(CO)2]2H+) [354]; протонирование приводит к образованию связи металл — водород, о чем свидетельствует появление резонансных сигналов в очень сильном поле (т = 28,58—38,07).
Корреляции спектров ЯМР-Н1 со строением, молекул
151
Таблица 10.96
ХИМИЧЕСКИЕ СДВИГИ ГИДРИДНЫХ ПРОТОНОВ В ГИДРИДАХ ПЕРЕХОДНЫХ МЕТАЛЛОВ
Соединение Эталон Химический сдвиг, м. д. т Литература
(C5H5)2ReH С6Н5 в толуоле 20,5 23,6 334
НСо(СО)4 То же 17,3+2,0 20,4 335
H2Fe(CO)4 » » 17,4 20,5 337
НСг(СО)3С5Н5 » » 13,1 16,2 338
НМо(СО)3С5Н5 » » 12,8 15,9 338
HW(CO)3C5H5 » » 14,7 17,8 338
HPtCl [Р(С2Н5)3]2 н20 22,0 27,2 339 , 342
[HCo(CN)5]3- н2о . 17,4 22,6 340
[HRh(CN)5]3- н20 15,6 20,8 340
НМп(СО)5 н2о 13,0 18,2 341
Т— 5,2±6pi2Q (ВИешн).
ДРУГИЕ ИССЛЕДОВАНИЯ
10.36. Контактные химические сдвиги в парамагнитных частицах
Спектры ЯМР-Н1 парамагнитных молекул указывают на очень большие различия в экранировании разных протонов в этих молекулах. Химические •сдвиги в этих соединениях охватывают область ~400 м. д., что значительно •больше области изменения химических сдвигов для диамагнитных молекул (~20 м. д.). Иногда в спектрах парамагнитных молекул наблюдаются широкие резонансные сигналы, обусловленные малыми временами релаксации протонов, что является следствием возникновения больших- флуктуирующих магнитных полей, вызывающих быструю релаксацию (см. разд. 2.5.1). Большие различия в экранировании ядер, наблюдающиеся в 9тих системах, можно количественно объяснить исходя из изотропного контактного взаимодействия между протонами и неспаренной спиновой плотностью, переместившейся в молекулу лиганда с атома парамагнитного металла, с которым связан этот .лиганд (см. разд. 3.16) [343, 344, 457]. Наличие небольшой неспаренной спиновой плотности в различных частях лиганда было качественно объяснено Милнером и Праттом [343]. При рассмотрении комплекса этилендиамина •с никелем
/NH2—СН212+
(H2O)4n/ |
xnh2-ch2
можно сделать вывод о том, что Sd-орбитали Ni(II) в присутствии октаэдрических групп лигандов расщепляются на две группы: три орбитали типа Z2g и две орбитали типа eg с более высокой энергией. Восемь Sd-электронов распределяются так, чтобы полностью занять ^-орбитали, а на двух её-орбита-лях размещаются по одному электрону. Эти два неспаренных электрона поляризуются в магнитном поле таким образом, что их спины ориентируются по полю. Связи N1 — N лежат вдоль той же оси, что и орбиталь eg, поэтому имеется возможность переноса электронной плотности от азота к никелю. Вследствие поляризации одиночного неспаренного электрона eg в магнитном поле перенос электрона азота в связи N1 — N будет происходить преимуще
152
Глава 10
ственно со спином, антипараллельным спину электрона никеля. Это приводит к ориентации по полю небольшого избытка спиновой плотности, оставшейся на атоме азота. Этот избыток называется положительной спиновой плотностью и приводит к уменьшению диамагнитного экранирования ядер азота. Если на связывающих орбиталях имеется небольшое количество неспаренных электронов, то электроны, связанные с атомами водорода, присоединенными к азоту, будут иметь антипараллельную ориентацию и спиновая плотность может переноситься в другие части молекулы по спин-поляризационному механизму. Отрицательная спиновая плотность на атомах водорода вызывает
-С1Ь^ >nh2 .
।________А_________I_______I——।—
-200 -100 0 100 200 м.Э.
Рис. 10.49. Спектр протонного резонанса водного раствора комплекса 1 : 1 никеля(П) с этилендиамином при 56,4 Мгц [343].
сильное экранирование протонов этих атомов, что объясняет появление сигналов этих протонов в спектрах ЯМР-Н1 комплексов никеля с этилендиамином в очень сильных полях (см. рис. 10.49).
Аналогичное рассмотрение логически приводит нас к отрицательным спиновым плотностям на а-углеродных атомах, и следовательно, к положительным спиновым плотностям на атомах водорода, связанных с «-углеродными атомами, что объясняет большие сдвиги сигналов метиленовых групп, связанных с атомами азота, в слабое поле (см. рис. 10.49). Аналогичным образом были исследованы и многие другие комплексы Ni(II) и Со(Н), причем в некоторых случаях была получена информация о строении комплексов и относительных количествах различных частиц.
Итон и сотр. [344—346] исследовали изотропное контактное взаимодействие в спектрах ЯМР растворов некоторых аминотропониминатов Ni(II) в дейтерохлороформе и наблюдали гораздо более узкие, сигналы, чем Милнер и Пратт. Ион Ni(II) в этих комплексах быстро изменяет конфигурацию от плоской квадратной (диамагнитный) до тетраэдрической (парамагнитный), что благоприятно влияет на времена релаксации протонов. Малая ширина резонансных сигналов позволяет наблюдать их тонкую структуру, что делает отнесение линий в спектре более однозначным [464]. Значения констант спин-спинового взаимодействия очень близки к значениям соответствующих констант в исходной молекуле хинолина.
На рис. 10.50 приведен спектр протонного резонанса Ы,Ы-ди(6-хинолил)-аминотропонимината Ni(II). В спектре имеется девять сдвигов контактного взаимодействия, из значений которых можно рассчитать спиновые плотности в каждом положении молекулы (см. разд. 3.16). Этот метод дает значения, хорошо согласующиеся со значениями, полученными расчетом по методу валентных связей. При исследовании большого ряда молекул можно сделать оценку относительной легкости передачи электронной плотности через группы, образующие сопряженную систему. Показано, что электронная плотность может передаваться через центральный атом металла.
При измерении контактных сдвигов была проведена оценка распределения спиновой плотности для никелевых хелатов N-ж-толилсалицилальдиминов, но резонансные сигналы в их спектрах очень широки и тонкая структура, обусловленная спин-спиновым взаимодействием, не наблюдается [357]. Контактные химические сдвиги для N-метилсалицилальдимина Ni(II) зависят от концентрации, что объясняется изменением магнитной восприимчивости при разбавлении [358].
Корреляции спектров ЯМР-Н1 со строением, молекул
153
С помощью измерения контактных сдвигов протонов [438] в некоторых тетраэдрических комплексах Со(И) были оценены спиновые плотности *). При исследовании спектров ЯМР-F19 соответствующих фторпроизводных N-замещенных аминотропониминатов Ni(II) были получены [356, 431] константы контактного взаимодействия F19 для орто-, мета- и пара-замещенных фторбензолов. Простая зависимость, которая существует между константами
а
<;
Рис. 10.50. Спектры протонного резонанса при 60 Мгц в CDC13 с ТМС в качестве внутреннего эталона [344]. ,,
а — N,М'-ди-(6-хииолил)-амииотропоиимииат Ni(II); б — 6-хинолильная группа в комплексе амииотропоиимииата Ni(II).
контактного взаимодействия протонов и спиновыми плотностями на соседних атомах углерода в ароматической молекуле, неприменима для констант кон-тактного взаимодействия фтора из-за сильного л-связывания фтора с ароматической системой.
10.37. Исследования растворов полимеров* 2)
Несколько сотен работ, посвященных исследованию твердых полимеров-методом ЯМР низкого разрешения, показали, что такие исследования полезны при определении относительных положений протонов в молекулах, величины сегмента и степени кристалличности полимерных образцов. Исчерпывающие обзоры по этим вопросам опубликованы Пауэлсом [380], Слихтером [381], Зауэром и Вудвордом [382]. Относительно небольшое число работ было посвя
х) Парамагнитные сдвиги в спектрах были изучены также для пиридинатов Со(П) [586] и некоторых М,М-диалкилдитиокарбаматов [587]. В работе [588] рассмотрено соответствие закономерностей делокализации спиновой плотности, полученных из данных спектров ЭПР радикалов и ЯМР молекул и комплексов парамагнитных ионов.— Прим. ред.
2) Обширный материал по вопросам исследования полимеров методом ЯМР как на низком, таки на высоком разрешении приведен в книге Слонима и Любимова [589].— Прим. ред.
154
Глава 10
щено исследованиям полимеров методом ЯМР высокого разрешения главным образом из-за малой растворимости полимеров и уширения резонансных сигналов, которое не позволяло проводить полный анализ спектра. Уширение резонансных сигналов происходит в результате того, что молекулярное движение полимеров слишком мало для полного усреднения дипольного уширения. Поэтому часто полимеры в растворах исследуют при высоких температурах, что позволяет использовать растворы большей концентрации и уменьшает их вязкость. Однако в большинстве случаев ширина резонансного сигнала определяется величиной сегментов в полимерах. Если для полимера удастся найти подходящий растворитель, то спектроскопию ЯМР можно применять для определения структуры полимера, как это делается при определении строения больших органических молекул. Поскольку экранирование магнитных ядер чувствительно к слабым изменениям в их пространственном окружении, для одинаковых ядер полимерной цепи с различной стереохимической конфигурацией часто наблюдаются разные резонансные сигналы. Так, 15%-ный раствор полиметилметакрилата в хлороформе (см. разд. 10.37.3) при 90° дает три резонансных сигнала, соответствующих а-СН3-группам в изотактическом, синдиотактическом и атактическом фрагментах полимерной цепи. Изменение интенсивностей этих сигналов позволяет определить стереорегулярность различных образцов с точностью ±5%.
Методом ЯМР исследованы также полимеры и сополимеры этилена [443]. 10%-ные растворы сополимеров (например, этилена с этил акр ил атом) в дифениловом эфире нагревали до 240°, затем помещали в обычный спектрометр ЯМР без термостатированного датчика и, прежде чем раствор успевал остыть, быстро записывали спектры очень хорошего качества. Этот метод имеет очевидные ограничения, однако он пригоден для качественных и количественных определений состава.
При исследовании ширины линий в спектрах протонного резонанса иногда можно получить полезную информацию о подвижности сегментов в полимерной молекуле.
Для того чтобы показать возможности метода при решении этой проблемы, разберем несколько типичных примеров исследований растворов полимеров методом ЯМР. В разд. 11.24 приведены некоторые примеры исследования фторированных полимеров.
10.37.1. Сегментное движение в полимерах
На рис. 10.51 показан спектр ЯМР-Н1 высокого разрешения полистирола (мол. вес ~30 ООО), растворенного в четыреххлористом углероде [383]. На этом же рисунке приведен спектр ЯМР-Н1 относительно небольшой молекулы кумола С6Н5СН(СН3)2. Очевидно, что ширина сигналов в спектре полимера больше ширины сигналов в спектре кумола, что вполне согласуется с более слабым меж- и внутримолекулярным движением больших полимерных молекул в растворе по сравнению с молекулами кумола. Однако ширина сигнала бензольных протонов в полимере значительно уже (0,005 э) остальных сигналов и, кроме того, ширина этих сигналов не зависит от молекулярного веса образца 1383, 384] в области 2000—1 000 000. Ширина рассматриваемых линий не зависит также от концентрации раствора (ниже 50%), что указывает на ее независимость от макроскопической вязкости раствора. Если бы ширина сигнала бензольных протонов была обусловлена движением всей полимерной молекулы, то сигнал был бы значительно шире наблюдаемого и его ширина увеличивалась с увеличением молекулярного веса полимера. Следовательно, узкие резонансные сигналы можно объяснить движением сегментов в пределах полимерной молекулы. Ширина сигнала определяется локальной вязкостью в непосредственной близости от каждого сегмента. Если соседние сегменты не препятствуют движению друг друга, то локальная вязкость не зависит ни
Корреляции спектров ЯМР-Н1 со строением молекул 155
от молекулярного веса полимера, ни от концентрации раствора. В противоположность этому ширина сигналов небольших молекул зависит от вязкости в пределах обычно встречающихся вязкостей (см. разд. 2.7). Однако если растворитель дает резонансный сигнал, то его ширина линии сильно зависит от вязкости раствора полимера, что указывает на трудность реориентации молекул растворителя в этом растворе. Одияма [384] показал, что ширина сигналов фенильных и метиленовых протонов полистирола в растворе в четыреххлористом углероде не зависит от длины полимерной цепи. Измерения ширины сигналов указывают на движение в цепи сегментов, содержащих около десяти мономерных единиц [384].
СЛ
j__।__।__।_।__।__।__। । ।
1 23 4 567 8Э 10
С, м.с).
Рис. 10.51. Спектры протонного резонанса при 40 Мгц [383].
а — 10%-ный раствор кумола в СС14: б — 15%-иый раствор полистирола в СС14. Сигнал при 10 м. д. соответствует протонам внутреннего эталона — ТМС.
Скорости, энтальпии и энтропии активации для различных видов движения, совершаемых полимерными цепями, можно определять непосредственно при измерении времен спин-решеточной релаксации 7\ в широком интервале температур (см. разд. 2.4). Например, было показано, что бензольные кольца, связанные со случайными клубками полимерных цепей, вращаются в 40— 50 раз медленнее, чем молекулы свободного бензола [383].
Полимерные молекулы, в которых подвижность сегментов мала, дают в спектрах широкие сигналы. Так, спектры протонного резонанса природных белков с большим молекулярным весом (~65 000) разрешены очень плохо вследствие заторможенного движения сегментов. Частично закрученные поли-пептидные цепи очень сильно переплетаются, образуя компактные сферы или эллипсоиды. В таких случаях ширина сигналов определяется вращением всей молекулы и для молекул с большим молекулярным весом реориентация их затруднена. Молекулы белков типа альбумина бычьей сыворотки можно развернуть, растворяя их в растворе дейтеромочевины в D2O или в трифторуксусной кислоте; тогда они дают хорошо разрешенные спектры ЯМР-Н1 вследствие достаточной подвижности сегментов [385, 386].
Бови и Тирс [387] исследовали протонные спектры водных растворов полиакриламида и полиметакриламида и обнаружили, что резонансные сигналы протонов при азоте уже соответствующих сигналов в спектрах низкомолекулярных амидов. Сужение линий объяснено частичным исчезновением квадрупольной релаксации N14, являющейся причиной уширения сигналов протонов NH. По-видимому, беспорядочное движение полимерных сегментов достаточно медленно, и электрический квадрупольный момент ядер N14 может эффективно взаимодействовать с молекулярной решеткой. Поэтому в протонном спектре водного раствора полиакриламида в области амидных протонов
156
Глава 10
наблюдается дублет в противоположность широкому одиночному сигналу для низкомолекулярных амидов. Если исследовать низкомолекулярный амид в вязком растворителе, то в его спектре также наблюдается два сигнала. Два отдельных сигнала, наблюдаемых для полимера, соответствуют двум амидным протонам, неэквивалентным вследствие заторможенного вращения вокруг связи С — N. Спин-спиновое взаимодействие этих двух протонов, по-видимому, очень мало. При исследовании слияния амидного дублета с изменением температуры были определены скорости вращения амидных групп и энергии активации при различных значениях pH.
10.37.2. Определение молекулярной структуры полимеров
Некоторые исследования высокомолекулярных соединений в растворах имеют много общего с обычными исследованиями строения сложных органических соединений методом ЯМР. Для иллюстрации этого положения разберем два типичных примера.
Сополимеры бутадиен — изопрен. Спектры протонного резонанса сополимеров бутадиен — изопрен (мол. вес 100 000—200 000), исследованных
Рис. 10.52. Спектр протонного резонанса 5%-ного и изопрена (1 : 1) в СС14 при 60 Мгц
сополимера бутадиена [388]:
в виде 5%-ных растворов в СС14 [388], могут быть использованы для характеристики протонов в различных частях молекулы. Измерением интенсивностей резонансных сигналов с точностью +3% можно определить состав сополимера. Для такого сополимера найдены следующие формы:
СН, СН3 СН,
-сНг4-сн.снг сн2Д сн-сн,- -снД^сн-с^-
цис и транс
—СН2 —СН-СН = СН2 —сн2 —сн = сн —сн2 — ' цис и транс
На рис. 10.52 показан хорошо разрешенный спектр протонного резонанса сополимера бутадиена и изопрена 1:1; для различных типов протонов наблюдалось семь отдельных сигналов, как это показано в табл. 10.97. Отнесения сигналов сделаны на основании химических сдвигов, полученных на небольших модельных соединениях.
Аналогичным образом были изучены полиизопрен, полибутадиен и сополимеры винилиден — хлористый винил [458].
Поли-(п — \)-метилалкены-1. Методом ЯМР было исследовано строение полимерных (п—1)-метилалкенов-1 (типа полиизобутилена и поли-3-метил-
Корреляции спектров ЯМР-Н1 со строением, молекул
157
Таблица 10.97
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ ХАРАКТЕРИСТИЧЕСКИХ ГРУПП В СОПОЛИМЕРАХ БУТАДИЕНА И ИЗОПРЕНА 1 : 1 [388]
Номер сигнала а Химический сдвиг 6, т-значение Тип протонов
1 4,70 —сн=сн2
—сн=сн—
2 4,97 — сн = сн2
сн3
—сн=с—
сн3
3 5,35 сн2=с—
4 8,02 сн—с=с
5 8,42 СН3—С —с
6 8,74 сн—с—с
7 9,08 СН3 —С—с
а Номера сигналов соответствуют спектру на рис. 10.52.
6 Химические сдвиги измерены относительно сигнала ТМС — внутреннего эталона (10 м. д.).
бутена-1), полученных катионной полимеризацией в гомогенной среде с хло-
ристым алюминием при различных температурах. Можно легко определить
геминальные метильные группы
С(СН3)2 и изолированные метиленовые
группы, так как они дают резонансные сигналы, не имеющие тонкой структуры. Появление дублета в метильной области указывает на присутствие группы СН — СН3 (включая изопропильную). В молекулах, в которых подвижности
геминальных метильных групп мешают значительные пространственные препятствия, наблюдается существенное дезэкранирование метильных протонов по сравнению с молекулами, в которых возможно свободное вращение этих метильных групп (аналогичное дезэкранирование наблюдается и для пространственно затрудненных метиленовых групп). Введение двух или нескольких метиленовых групп между геминальными метильными группами существенно ослабляет пространственное напряжение, и тогда метильные протоны имеют нормальные значения химических сдвигов (это было установлено при исследовании протонных спектров небольших модельных соединений). Так, в спектре протонного резонанса полиизобутилена пространственно затрудненные метильные группы имеют низкие значения химических сдвигов (т = 8,9) по сравнению с геминальными метильными группами в поли-3-метилбутене-1 (т = 9,17); это наблюдение указывает на разделение геминальных метильных групп в последнем полимере более чем одной метиленовой группой. При катионной полимеризации более длинных цепей мономеров (например, 5-метил-гексен-1) при —73° наблюдается увеличение числа концевых изопропильных групп в полимере, сопровождающееся удлинением цепи мономера. Это обусловливает появление в спектре дублета в метильной области, помимо одиночного сигнала геминальных метильных групп.
Если проводить полимеризацию при более высоких температурах (0°), то содержание концевых изопропильных групп увеличивается, что следует
158
Глава 10
из увеличения относительной интенсивности дублета СН3-групп. Для объяснения структуры полимеров необходимо предположить перегруппировку промежуточно образующегося карбониевого иона [389] L).
ции [404]. Протоны в различных различные химические сдвиги, и
Рис. 10.53. Сильнопольная часть спектра протонного резонанса при 40 Мгц хлороформного раствора полиметилметакрилата, полученного инициированием:
а — н-бутиллитием в толуоле при —62°: б — перекисью бензоила в толуоле при 100°. В качестве виутреииего эталона использовался ТМС. Спектры исследовались при 90° [391].
10.37.3. Определение пространственной конфигурации полимеров
По-видимому, наиболее важным применением ЯМР-спектроскопии высокого разрешения к изучению полимеров в растворах является определение пространственной конфигурации полимерных молекул [383, 390, 391, 403]. Если мономер окружен с обеих сторон мономерами такой же конфигурации, как он сам, т. е. осуществляется последовательность ddd или III, то говорят об изотактической конфигурации. Если мономер имеет конфигурацию, противоположную конфигурациям соседних мономеров, т. е. осуществляется последовательность did или Idl, то говорят о синдиотактической конфигурации. Атактическая конфигурация - может иметь любые последовательности мономеров: Idd, dll, ddl или lid.
Эти определения стереорегулярности были предложены Бови, и они, в частности, приняты при исследованиях с помощью ЯМР (см. в разд. 9.5.5 обсуждение роли d- и /-конфигураций в соединениях, содержащих простые-связи С — С). Опубликован большой обзор по стереорегулярной полимериза-— пространственных окружениях часто имеют
соответственно ЯМР-спектроскопия высокого' разрешения наряду с рентгенографией [392] и ИК-спектроскопией [392] может использоваться для определения пространственной конфигурации полимеров.
Первые определения стереорегулярности методом ЯМР были проведены на примере полиметилметакрилата [392, 393]. При исследовании 15%-ных растворов полимера в хлороформе при 90° получаются вполне приемлемые спектры ЯМР-Н1.
На рис. 10.53 приведены протонные спектры двух,образцов полиметилметакри-лата, полученных двумя различными методами полимеризации. В спектрах ясно видны три сигнала а-метильных протонов (т == 8,78; 8,95 и 9,09), относительные интенсивности этих сигналов сильно меняются в зависимости от метода получения. Сигнал при 6,4т в обоих спектрах соответствует эфирной метильной группе и не меняется при изменении конфигурации цепи. Полимер, полученный при инициировании н-бу-тиллитием в углеводородных растворителях, по-видимому, имеет главным образом изотактическую конфигурацию, и поэтому наиболее интенсивный а-метильный сигнал при 8,78т на рис. 10.53, а может быть отнесен к а-метильным группам центральных мономерных единиц в изотактических
х) Наряду с приведенными примерами можно также указать на работы по определению молекулярной структуры полимеров замещенных алкенилциклопропанов [590], метакролеина [591]-— Прим., ред.
Корреляции спектров ЯМР-Н1 со строением, молекул
159
последовательностях или триадах (ddd или III). Резонансный сигнал при 9,09т обусловлен метильными группами центральных мономерных единиц в синдиотактических последовательностях, а сигнал при 8,95т — а-метильными группами в атактических конфигурациях. Измерение относительных интенсивностей этих резонансных сигналов дает соотношение различных звеньев в полимерной цепи. Метиленовые группы в синдиотактических конфигурациях дают одиночные сигналы, так как при усреднении во времени оба протона находятся в одинаковом химическом окружении (см. рис. 10.53, б). В изотактической последовательности два метиленовых протона обычно экранированы различным образом и дают пару дублетов типа АВ (см. разд. 8.13.1). Для изотактического полиметилметакрилата три компоненты квартета АВ, обусловленного изотактическими метиленовыми протонами, расположены в сильном поле с центром при 8,14т; четвертая компонента, вероятно, наложилась на сигнал а-метильных протонов. Различие химических сдвигов неэквивалентных метиленовых протонов составляет 0,60 м. д., а константы взаимодействия J = 15,5 гц (см. рис. 10.53, a) J).
Метод ЯМР был применен также для определения геометрической изомерии в поли(этилен-1,4-циклогександикарбоксилате) [376] и стереорегулярности полипропилена [472] и поливиниловых эфиров [473].
10.37.4. Применение двойного резонанса для определения строения полимеров
В винильных полимерах, имеющих лишь один a-заместитель, осуществляется сильное спин-спиновое взаимодействие между протонами у а-и Р-углеродных атомов. Это взаимодействие часто усложняет спектры до
------1---------1----------1---------1_________I__________1_
5 6 7 8 9 10
•С, М.Э.
Рис. 10.54. Спектры протонного резонанса при 60 Мгц [394].
а — поливинилхлорид в хлорбензоле (15%-ный раствор; при 170°); б — поли-сс-дейтеро-винилхлорид (при 156°); в —сс-протоиы в поливинилхлориде при облучении р-протонов (при 150°); г — fl-протоны в поливинилхлориде при облучении сс-протонов (при 150°).
В качестве внутреннего эталона использовался гексаметнлдисилоксан.
такой степени, что становится невозможно наблюдать различные мультиплеты, обусловленные протонами в различных стереохимических последовательностях. Бови и сотр. [394] использовали метод двойного резонанса для подавления спин-спинового взаимодействия а- и p-протонов в поливинилхлориде, поливинилфториде и поливинилметиловом эфире, что дало возможность с успехом определить стереорегулярность этих соединений. На рис. 10.54, а
х) Исследование стереорегулярности полимеров метил- и бутилметакрилата с помощью ЯМР-Н1 проводилось в работе [592].— Прим. ред.
160
Глава 10
приведен спектр протонного резонанса поливинилхлорида в растворе в хлорбензоле при 170° и 60 Мгц. Джонсен [395] в более ранней работе отнес два перекрывающихся сильнопольных триплета (с центрами при т = 7,78 и 7,96)
к мезо- и рацемическим метиленовым группам соответственно и предположил, что протоны в каждой СН2-группе магнитно эквивалентны. Сильнопольный триплет был произвольно отнесен к рацемическим метиленовым протонам как находящимся в синдиотактических последовательностях. а-Протоны дают квинтет с центром при 5,53т. Бови и сотр. [394] сделали отнесения сигналов метиленовых протонов, подавив взаимодействия а- и Р-протонов. При этом наблюдались два сильнопольных синглета, разделенных на 0,20 м. д. (см. рис. 10.54, г). Спектр двойного резонанса метиленовых протонов аналогичен спектру протонного резонанса поли-а-дейтеровинилхлорида [396], приведенному на рис. 10.54, б. Если облучать метиленовые протоны, то в области а-протонов наблюдаются три отдельных сигнала при т 5,48; 5,59 и 5,71 (рис. 10.54, в). При рассмотрении влияния селективного облучения мезо-и рацемических протонов на область а-протонов три слабопольных сигнала можно отнести к синдиотактической, атактической и изотактической триадам
соответственно (в предположении, что верно отнесены сигналы p-протонов). Поэтому квинтет |3-протонов в действительности представляет собой три перекрывающихся квинтета. Для выяснения степени стереорегулярности данного полимера можно проводить измерение интенсивностей в спектре двойного резонанса.
Тинчер [397] подверг сомнению правильность предположения Джонсена о том, что изотактические метиленовые протоны в поли-
fl
Химический сдвиг, м.Э.
Рис. 10.55. Спектры протонного резонанса при 60 Мгц [398].
а — изотактический полипропилен в п-дихлорбеизоле (при 175°); б — изотактический поли-2,3,3,3-тетрадейтеропропилеи в 2-хлортиофёие (при 110°); в — образец иеразделеииого иа фракции поли-2,3,3,3-тетрадей теропропиле-иа в 2-хлортиофеие (при 110°). Внутренний эталон — гексаметилдисилок-саи, т = 9,95.
винилхлориде магнитно эквивалентны, и пока-зал, что наблюдаемый спектр можно объяснить, допустив неэквивалентность метиленовых протонов. Однако эксперименты по двойному резонансу показали, что в пределах разрешения данного эксперимента • два протона имеют одинаковые химические сдвиги.
10.37.5. Использование дейтерирования при определении стереорегулярности полимеров
Полипропилен. Спин-спиновое взаимодействие протонов у соседних атомов углерода в полимерных цепях, как, например, в полипропилене, сильно усложняет спектры протонного резонанса. Замещение некоторых атомов водорода на дейтерий может значительно упростить спектр, так как величины констант взаимодействия Н — D малы и не вызывают заметных расщеплений сигналов [398]. На рис. 10.55, а показан сложный спектр протонного резонанса изотактического полипропилена [398]. Сато и сотр. [399] провели полный анализ спектров такого типа, что дало им возможность определить стереорегулярность различных образцов полипропилена. В противоположность рис. 10.55, а спектр изотактического поли-2,3,3,3-тетрадейтеропропилена, приведенный на рис. 10.55, б, представля
Корреляции спектров ЯМР-Н1 со строением молекул
161
ет собой типичный спектр двух неэквивалентных взаимодействующих протонов, образующих систему АВ (см. разд. 8.13.1). Каждый протон в метиленовой группе дает дублет и вследствие сильного взаимодействия ядер интенсивности компонент дублета не равны. В спектре протонного резонанса не разделенного на фракции поли-2,3,3,3-тетрадейтеропропилена, показанном на рис. 10.55, в, в дополнение к квартету АВ изотактической метиленовой группы наблюдается интенсивный сигнал синдиотактической метиленовой группы. Измерением интенсивностей соответствующих сигналов в спектре дейтерированного полипропилена можно определить соотношение изотактических и синдиотактических триад. Полипропилен в работе [398] был получен из мономера с использованием типичных катализаторов Циглера, а фракции разного стереоизомерного состава разделялись путем экстракции растворителем (диэтиловым эфиром и «-гептаном).
Полистирол. Спектр протонного резонанса изотактического полистирола (мол. вес 200 000), растворенного в различных растворителях (CS2, CDC13, CSH6 и т. д.), содержит два сигнала, соответствующих алифатическим протонам, причем слабопольный сигнал соответствует а-протонам, а сильнопольный сигнал — р-протонам [401, 402]. Такое отнесение было подтверждено исчезновением слабопольного сигнала при замещении а-протона на дейтерий [401]. Атактический полистирол дает лишь один сигнал в алифатической области спектра во всех растворителях, за исключением ароматических, в случае которых снова видны два сигнала [401].
Вследствие сильного взаимодействия а- и p-протонов в полистироле, уширяющего их резонансные сигналы, методом ЯМР невозможно определить различные стереохимические конфигурации молекулы [184, 400, 401]. Браун-штейн и сотр. [400] исследовали стереорегулярность полистиролов, прибегнув к замещению водорода на дейтерий. Если на дейтерий заместить р-протон стирола, исчезает тонкая структура сигналов а-протона. Поэтому в протонном спектре полидейтеростирола, полученного из мономера и исследованного в растворе в бензоле при 100°, наблюдаются отдельные сигналы для а-протонов в изотактической, синдиотактической и атактической конфигурациях [400].
10.38. Эмпирическая оценка химических сдвигов
Как мы уже видели (гл. 4), квантовая механика' не может быть использована для расчета экранирования протонов в молекулах (кроме простейших) вследствие нашего недостаточного знания необходимых волновых функций. Поэтому, очевидно, желательно было бы иметь эмпирический метод оценки химических сдвигов протонов в молекулах. Примас, Арндт и Эрнст [347] предложили простую схему для химических сдвигов протонов, связанных с углеродом. Эта схема основана на предположении, что химический сдвиг протона можно рассматривать как сумму нескольких характеристических вкладов различных групп и свойств молекулы. Несмотря на отсутствие теоретического обоснования такого аддитивного поведения, нельзя не признать успеха схемы в предсказании химических сдвигов. Значение т для протонов в молекуле определяется простым выражением
т = То + 2 cjTj’
где Та = 9,067; Tj — вклад группы или свойства в химический сдвиг; — число раз, которое данная группа встречается в молекуле. Примас определил вклады Tj, рассмотрев обширные данные, собранные Тирсом (см. приложение I), и введя их в вычислительную машину, запрограммированную для решения большого числа линейных уравнений.
Для применения схемы углеродный скелет молекулы нумеруется, начиная от атома углерода, с которым связан протон (углерод 1); далее нумерация углеродных атомов идет в соответствии с числом связей, отделяющих данный 11 — 489
162
Глава 10
Таблица 10.98
ВКЛАДЫ В ХИМИЧЕСКИЕ СДВИГИ ОТ РАЗЛИЧНЫХ ЗАМЕСТИТЕЛЕЙ И ГРУПП [347]
Заместитель Характеристический вклад Число образцов
То = 9,067 284
Нет Нет 21
1 Т1 = —0,248 96
2 Т2 = —0,244 137
3 Т3= —0,147 56
4 Т4 = —0,006 10
Заместитель а Положение Характеристический вклад Число образцов
-CR3 3 Т5 =4-0,038 по
Двойная связь 1 Тв = —3,802 21
2 Т7 = —0,583 31
3 Т8= —0,203 7
Тройная связь 1 Т9= —1,032 3
2 Т10= —0,694 4
= О 1 Ти = —8,536 6 7
2 Т12=—1,021 21
3 Т13= —0,004 11
—ОСН3 2 Т14= +0,373 6
-OCH2-CR3 2 Т15=+0,237 7
3 Т16= —0,210 6
—ОН 1 Т17=—2,467 8
2 Т18= — 0,048 8
3 Т19= —0,235 5
— О—СО—CR3 1 Т20= -2,931 28
2 Т21= —0,041 11
3 7*22 ~ 4-0,086 5
— F 2 Т23= -0,089 9
3 Т24= -0,131 9
-Cl 1 Т25=-2,170 29
2 Т26=-0,254 19
3 Т27= —0,177 12
— Вг 1 Т28=-1,995 24
2 Т29= —0,363 16
3 Т30= -0,023 7
— I 1 Т31=-1,846 10
2 Т32= —0,388 5
—nh2 2 Т33= —0,094 3
— О—CR3 1 Т34= +1,434 в
а R может быть любой группой, включая R = Н.
® Это значение можно использовать только тогда, когда вторым заместителем в положении 1 является —О—CRs с Т34.
в Следует рассматривать только в сочетании с 7ц.
атом углерода от протона, как показано ниже; считается, что каждый углеродный атом вплоть до углерода 4 вносит вклад в химический сдвиг. Атом углерода в положении 4 рассматривается как заместитель у атома углерода 3. Учитываются только вклады от заместителей у первых трех углеродных ато-
Корреляции спектров ЯМР-Н1 со строением молекул
163
№ Заместитель Характеристический
вклад
1 — С2 Т{
2 — С2—С3 т2
3 -С3/0 \Сз Тз
4 /С3 — С2^-С3 \С3 т^
мов; они приведены в табл. 10.98. Способ применения схемы легче понять при изучении типичных примеров в табл. 10.99.
Таблица 10.99
НАБЛЮДАЕМЫЕ И РАССЧИТАННЫЕ ХИМИЧЕСКИЕ СДВИГИ ДЛЯ РЯДА МОЛЕКУЛ [347]
1) СН3СО-СОСН3
’с' = 7'о + 7’2 + 7'5+7'12+7'13 = 7,83
Значение, измеренное Тирсом: т = 7,77
2) СН3СН2\ уСООС2Н5 С
CHgCH./ ХСООС2Н5
г = То + 7\ + т4+ Т5 + 2Т13 + 2Т1в = 8,42
Значение, измеренное Тирсом: т = 8,16
3) СН3СН2ч /СООСН2СН3 С
СНзСН;/ ХСООСН2СН3
т=7\)-|-7Т~р7'2о = 5,89
Значение, измеренное Тирсом: т = 5,85
4) НС=ССН2Вг
т= То + T2 + Т ю + Т’гя = 6,13
Значение, измеренное Тирсом: т = 6,18
5) (СН3)2С==СНСН2СН2СНСН2СНО
СН3
т = То + Tt + Т3 + Т& + Т12 = 7,69
Значение, измеренное Тирсом: т = 7,81
6) CF2C1CHC12
т = То -| 7’1 272з 2 7 25 + Т'гв = 4,05
Значение измеренное Тирсом: т = 4,08
Для большинства из 284 рассмотренных соединений получено отличное совпадение предсказанных и наблюдаемых химических сдвигов, причем наименее надежные предсказания сделаны для галогензамещенных молекул. В ряд исследованных соединений не были включены циклические молекулы, кислоты и молекулы, содержащие гетероатомы в скелете (за исключением простых и сложных эфиров, ангидридов кислот). Для альдегидов, кетонов, простых и сложных эфиров, ангидридов кислот и ацил галогенидов было предположено, что в т-значения вносят свой вклад заместитель R = О, OCOR, ОСН3, ОС2Н5 и т. д.
Помимо этого общего метода оценки химических сдвигов, для отдельных классов соединений был предложен ряд других методов; некоторые из них 11*
164
Глава 10
обсуждались в соответствующих разделах этой главы. При отсутствии подходящих модельных соединений такие эмпирические расчеты химических сдвигов могут быть очень полезными при определении структуры молекул методом ЯМР.
10.39. Влияние среды в жидкостях
Константа экранирования ядер в данной молекуле зависит от распределения электронов в молекуле, а также от природы окружающей среды (см. разд. 3.9 и 3.10). Наблюдаемая константа экранирования о представляет собой сумму значений для изолированной молекулы и вклада окружающей среды, так что
° — Череда + ^молекула>
где Омолекула — значение, полученное для газообразного соединения при низких давлениях, оСреда — разность значений, полученных для газообразного соединения при низких давлениях и для раствора данного соединения в каком-либо растворителе, причем в обоих случаях отсчет ведется от одного и того же внешнего эталона.
Бакингем, Шефер и Шнейдер [200] предположили, что оСреда представляет собой сумму
Череда = + Оа + Що + ЧЕ + °Н> (10.16)
где оь пропорциональна объемной магнитной восприимчивости среды (см. разд. 3.3). Величина оь зависит от формы ампул [см. уравнение (3.6)] и равна нулю, если они сферические. Большинство исследований по ЯМР проводится в цилиндрических ампулах и в этом случае
2
щ> = у лхв, (10.17)
где Хв — объемная восприимчивость среды.
оа является следствием ненулевого усреднения анизотропии диамагнитной восприимчивости молекул растворителя. Эта составляющая особенно важна для растворителей с дискообразными молекулами, таких, как ароматические соединения, и растворителей с линейными молекулами, таких, как сероуглерод. Величина оа зависит от природы анизотропии и обсуждалась в разд. 3.10 (где оа обозначалась как Д269). Для дискообразных молекул оа может быть приближенно описана уравнением
оа=—2л (хн- Х±)/(3/?з),
а для линейных молекул
Ча = П (Хц — Х_ц)/(ЗЯ3),
где R — расстояние между ядрами растворенной молекулы и центром молекулы растворителя, п — число растворенных молекул в пределах расстояния R. Бензол является примером растворителя с дискообразными молекулами, и для метана, растворенного в бензоле, показано, что [(хц — Х1) = = — 9-Ю-29; п = 2; R ~ 4,5А] оа = 1,3 м. д. Для метана в сероуглероде [(Хц — Х1) = — 5-Ю-29; п = 2; R ~ 4,0А] оа = — 0,5 м. д. Абрагам [419] рассчитал вклад магнитной анизотропии растворителей с цилиндрическими молекулами в химические сдвиги: для бензола он получил значение оа, хорошо согласующееся со значением, которое получил Бакингем с сотрудниками, тогда как значение оа для сероуглерода значительно меньше указанного Бакингемом. Хотя формула предсказывает правильные знаки вкладов, наблюдаемые значения для неполярных растворенных веществ всегда больше предсказанных.
Корреляции спектров ЯМР-Н1 со строением молекул
165
ow — вклад слабых вандерваальсовых взаимодействий между растворителем и растворенным веществом [200, 421]. В <sw вносят вклады два эффекта: а) взаимодействие между растворенными веществами и растворителем (в их равновесной конфигурации), приводящее к возмущению, по-видимому, к разрыхлению электронного окружения ядра растворенного вещества; это приводит к уменьшению диамагнитного члена в уравнении Рэмзи для о (см. разд. 3.5) и поэтому к отрицательному значению ow; б) отклонение от равновесной конфигурации молекул растворителя приводит к изменению во времени электронного окружения молекул растворенного вещества. Если растворенное вещество представляет собой атом в S-состоянии, взаимодействие будет искажать электронную симметрию, что приведет к возникновению парамагнитного члена в константе экранирования (см. разд. 3.5). Это зависящее от времени взаимодействие будет наиболее эффективным для ядер в атомах, но может быть также важным для ядер в молекулах с симметричным окружением, как, например, в аксиально симметричных .связях X — Н. Эффект «а» не зависит от температуры, но эффект «б» возрастает при увеличении температуры, делая значение ow более отрицательным. Исследование метана в СС14 при —27 и +95° показывает, что эффект «б» для него невелик; однако для атомов типа Хе129 этот эффект сильно зависит от температуры.
оЕ определяется влиянием электрического поля на константу экранирования [492]. Бакингем [480] показал (см. разд. 3.9), что приложенное электрическое поле влияет на константу экранирования протона в связи X — Н. Поэтому при растворении полярной молекулы в среде с диэлектрической проницаемостью 8 возникает поле реакции, которое стремится уменьшить значение константы экранирования и сдвинуть резонансный сигнал в слабое поле. Даже молекулы, не обладающие постоянным дипольным моментом, могут обусловливать появление поля реакции при условии, что молекулы обладают полярными группами (см. разд. 3.9). В обоих случаях при использовании модели Онзагера для расчета поля реакции оЕ пропорциональна (8-1)/(8+1).
Последний член он в уравнении (10.16) является следствием специфического (типа водородной связи) взаимодействия растворителя с растворенным веществом и обычно бывает наибольшим членом. Специфическое влияние растворителя такого типа уже рассматривалось в разд. 9.4 и здесь обсуждаться не будет.
Бакингем и др. [200] проверили уравнение (10.16) для различных случаев растворения как полярных, так и неполярных веществ в разных растворителях. В качестве неполярного соединения они выбрали метан и измерили его химические сдвиги в нескольких растворителях относительно внешнего эталона хлороформа. Затем эти сдвиги были пересчитаны относительно газообразного метана по уравнению (10.17) для значения оь. В табл. 10.100 приведены полученные значения химических сдвигов, а в последнем столбце — сдвиги относительно газообразного метана, по которым можно найти значение (^среда—оь). Для симметричных неполярных молекул типа метана члены Он и оЕ равны нулю и на величину Av (СН4раСтв — СН4газ) влияют только (ра + ow). Вместо того чтобы рассчитывать значение ow и сравнивать его с Av(CH4PacTB— СН4Газ), Бакингем и сотрудники [200] использовали значение теплоты испарения растворителя при точке кипения как меру вандервааль-сова взаимодействия. Это главным образом мера взаимодействия между молекулами растворителя, пропорциональная тем не менее взаимодействию между молекулами растворителя и молекулами растворенного вещества при условии, что осуществляется простое вандерваальсово взаимодействие. Теплота испарения Нъ при температуре кипения Ть рассчитывалась по эмпирическому соотношению Гильдебранда и Скотта [412]
Я&= 17,ОТЬН-0,ООЭТь.
Таблица 10.100
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ РАЗБАВЛЕННЫХ РАСТВОРОВ СН4 В РАЗЛИЧНЫХ РАСТВОРИТЕЛЯХ (ПРИ 60 Мгц) [200]
Растворитель —хв-106 Химические сдвиги, гц
измеренные (относительно СНС13) исправленные (относительно СНС1з) исправленные (относительно GH4 газ)
1. СН4 (газ) 0 538,2 446,5 0
2. Неопентан 0,554 а 460,9 438,7 —7,8
3. Циклопентан 0,661 а 445,9 437,1 —9,4
4. н-Гексан 0,586 452,9 434,7 —11,8
5. Циклогексан 0,631 445,2 432,7 —13,8
6. транс-Бутен-2 0,45 а 469,4 437,3 —9,2
7. ч«с-Бутен-2 0,475а 468,2 436,1 —10,4
8. Ацетон 0,460 469,9 435,9 —10,6
9. Диэтиловый эфир 0,547 458,1 435,0 —11,5
10. Этилацетат 0,547 455,9 432,9 —13,6
11. Этилнитрат 0,485а 463,4 432,7 —13,8
12. Триэтиламин 0,610 а 449,0 433,8 — 12,7
13. Пропин 0,470а 460,6 427,9 —15,6
14. Ацетонитрил 0,486а 449,6 418,9 —27,6
15. Бутин-2 0,519 а 448,5 421,9 —24,6
16. Сероуглерод 0,681 418,8 412,5 —34,0
17. Диацетилен 0,401 а 450,0 408,7 —37,8
18. Дицианоацетилен 0,417а 440,5 401,2 —45,2
19. Бензол 0,626 470,5 457,3 + Ю,8
20. Толуол 0,631 466,8 454,3 +7,8
21. Хлорбензол 0,707 456,3 453,3 +6,8
22. Нитробензол 0,598 479,8 462,6 + 16,1
23. Нитрометан 0,473 а 476,8 444,4 —2,1
24. Нитроэтан 0,497 469,2 440,0 —6,5
25. Бром 1,17 а 369,4 424,6 —21,9
26. СН3Вг 0,73б 424,5 424,5 —22,0
27. СН31 0,918 395,2 418,6 —27,9
28. СН2С12 0,733 425,5 425,7 —20,8
29. СН2Вг2 0,946 392,0 419,0 —27,5
30. CH2I2 1,16 350,2 404,1 —42,4
31. СНС13 0,731 423,1 423,1 -23,4
32. СНВг3 0,913 383,9 406,7 —39,8
33. СС14 0,684 427,2 421,3 —25,2
34. СВг4 (100°) 0,863в 396,0 412,5 —34,0
35. СН2С1Вг 0,839а 408,4 421,9 —24,6
36. СНС1Вг2 0,792а 396,8 404,4 —42,1
37. СС13Вг 0,752а 415,5 418,1 —26,4
38. SiCl4 0,796 426,1 434,2 — 12,3
39. SnCl4 0,986 395,8 427,8 — 18,7
а Оценены по константам Паскаля и путем интерполяции данных для родственных, соединений.
® Из работы Bothner-By A. A., G 1 i с k R. Е., J. Chem. Phys., 26, 1647 (1957).
в Плотность измерена при 100° и равна 2,943 г/мл.
Корреляции спектров ЯМР-Н1 со строением молекул
167
На рис. 10.56 показана зависимость Ду(СН4раств— СН4газ) от Нь для растворителей, приведенных в табл. 10.100. На рисунке видна линейная зависимость для растворителей со сферическими молекулами, а растворители с дискообразными и линейными молекулами дают точки, расположенные •сверху и снизу от прямой, что указывает на дополнительное влияние значения аа для этих растворителей. Галогенсодержащие растворители из рис. 10.56 исключены; для них зависимость Ду(СН4раств — СН4газ) также линейна, но их точки лежат ниже зависимости для других растворителей (см. рис. 10.57). Не было предложено никакого объяснения этого явления; оно может указывать просто на то, что значения магнитных восприимчивостей неправильны.
Для проверки справедливости уравнения (10.16) в случае полярных молекул Бакингем и сотр. [200] измерили химические сдвиги ацетонитрила CH3CN в различных растворителях. Они попытались выделить полярный эффект аЕ из всех остальных, не используя растворители, для которых можно было ожидать больших значений оа и ow, т.. е. ароматические, с линейными молекулами и галогенсодержащие растворители, и приняв за эталон раствор -CH3CN (5 мол.%) в «-гексане. Значения сдвигов, измеренные относительно внешнего эталона хлороформа, приведены в табл. 10.101; причем для них учтена поправка на разность объемных диамагнитных восприимчивостей Был построен график зависимости величины Av(CH3CNpaCTB—СН3СМн_Гексан)
Таблица 10.101
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ (ПРИ 60 Мгц) 5%-НЫХ РАСТВОРОВ CH3CN В РАЗЛИЧНЫХ РАСТВОРИТЕЛЯХ (ОТНОСИТЕЛЬНО ВНЕШНЕГО ЭТАЛОНА СНС13) [200]
Растворитель -Х„-106 . Химический сдвиг, гц
измеренное значение исправленное значение
1. н-Гексан 0,586 358,7 340,5
2. Циклогексан 0,576 353,7 334,3
3. гщс-Бутен-2 0,475а 367,3 335,3
4. транс-Бутен-2 0,475а , 371,3 339,3
5. Циклогексан 0,576 353,7 334,3
6. Трнметиламин 0,610а 344,8 329,6
7. Диэтиловый эфир 0,547 353,2 330,1
8. Этилформиат 0,537 350,3 326,0
9. Циклогексанои 0,614 338,3 323,7
10. Ацетон 0,460 357,0 323,0
11. Ацетальдегид 0,393 369,1 326,8
12. Четыреххлористый углерод 0,684 323,8 317,9
13. Хлороформ 0,731 319,2 319,2
14. Хлористый метилен 0,733 320,9 321,1
15. Этилхлорид 0,664а 341,3 332,9
16. Ацетонитрил 0,486а 344,8 314,1
17. Нитрометан 0,473а 366,6 334,2
18. Бензол 0,626 434,2 421,0
19. Мезнтилен 0,652а 417,8 407,8
20. Нитробензол 0,598 364,8 348,1
21. Бромбензол 0,771 376,2 381,2
22. Бензонитрил 0,658 361,4 352,3
а Оценены по константам Паскаля и путем интерполяции данных для родственных •соединений.
20-
10 -
О
\ -ю
* -20
-----о 3Я
- 6&^-^о8
о 7 9 .
-СН3СЕСН
с6н6
° СвН6СНз°Сбн5С1
снэыо2
° С2Н5Ы02
38 5 и
C6H5N02 о
-30 §
сн3с°ссн3 о
о CS2
НС-ХС5СН
39
сн3с;м
Л -50
о
N1CCSCCSN
5 6 7 8 9 ю
Теплота растворения, ккал /моль
Рис. 10.56. Зависимость химического сдвига метана (в герцах при 60 Мгц относительно газообразного метана в разбавленных растворах в различных растворителях) от теплоты испарения растворителя [200].
Точки, соответствующие различным растворителям, пронумерованы согласно табл..
Ю.ЮО.
Рис. 10.57. Зависимость химического сдвига метана (в герцах при 60 Мгц относительно газообразного метана, в разбавленных растворах в галогенированных растворителях) от теплоты испарения растворителя [200].
Растворители пронумерованы согласно табл. 10.100.
Корреляции спектров ЯМР-Н1 со строением молекул
169*
от (в — 1)/(2е + 2,5) для нескольких растворителей; при этом зависимость получалась линейной (см. рис. 10.58). Абрагам [413] показал, что химические сдвиги CH3CN и СН4 в 5%-ных растворах в одинаковых растворителях содержат одинаковые вклады оа, ow и оь, следовательно, различие в сдвигах, обусловленных растворителем, вызвано разными значениями оЕ или он. В табл. 10.102 приведены химические сдвиги относительно внешнего эталона
о
о
0,1 0,2 0.3
2e + Z.5
ОЛ 0,5
Рис. 10.58. Зависимость химического сдвига CH3CN в растворах (5 мол.%) в различных растворителях (в герцах при 60 Мгц относительно CH3CN в 5%-ном растворе в н-гексане) от (е — 1)/(2е + 2,5).
Растворители пронумерованы согласно табл. 10.101 [200].
СНС13 для растворов CH3CN и СН4 (5 мол. %) в различных растворителях,, в том числе в некоторых растворителях, обладающих большими значениями оа. Разность химических сдвигов Av(CH3CN — СН4)раСтв определялась для каждого растворителя и затем все сдвиги откладывались на шкале, за нулевое положение которой было принято Av в «-гексане. Для ацетонитрила
Таблица 10.102
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ (ПРИ 60 Мгц) CH3CN И С1Ц В РАЗЛИЧНЫХ РАСТВОРИТЕЛЯХ (ОТНОСИТЕЛЬНО ВНЕШНЕГО ЭТАЛОНА СНС13) [413]
Растворитель Химический сдвиг, гц Av(CH3CN — СН4)
ацетонитрил метан (абсолютн.) (относит.)
н-Гексан 358,7 452,9 —94,2 0
транс-Бутен-2 371,3 469,4 —98,1 —3,9
ЧИс-Бутен-2 367,3 468,2 —100,9 —6,7
Ацетон 357,0 469,9 —112,9 —18,7
Диэтиловый эфир 353,2 458,1 —104,9 —10,7
Триэтиламин 344,8 449,0 —104,2 — 10,0
Ацетонитрил 344,8 449,6 —104,8 — 10,6
Бензол 434,2 470,5 —36,3 +57,9
Нитробензол 364,8 479,3 —114,5 —20,3
Нитрометан 366,6 476,8 —110,2 — 16,9
СН2С12 320,9 425,5 — 104,6 —10,4
СНС13 319,2 423,1 —103,9 —9,7
СС14 323,8 427,2 —103,4 —9,2
170
Глава 10
(p/а ~ 1,0-10е; по = 1,34)
ая=-11 [(е-1)/(е + 0,9)]-4,3[(е-1)/(е + 0,9)]2,
и если отложить Av(CH3CN — СН4) против (в — 1)/(в + 0,9), то получается зависимость, изображенная на рис. 10.59. Кривая, приведенная на рисунке, рассчитана из вышеприведенного уравнения. Совпадение между теорией и экспериментом очень хорошее, исключение составляет бензол, для которого наблюдаются аномально большие сдвиги в сильные поля, по-видимому обусловленные большими значениями он.
Диль и Фримен [420] усовершенствовали теорию Бакингема для вклада <тЕ, введя учет формы растворенных молекул (см. разд. 3.9).
Рис. 10.59. Различие значений химических сдвигов CH3CN и СН4 в разных растворителях (относительно н-гексана) от (е — 1)/(е + 0,9) [413].
Абрагам [419] показал, что обусловленные алифатическими растворителями сдвиги йодистого метила можно количествейно объяснить влиянием членов оь, оа и оЕ [см. уравнение (10.16)].
Хруска и сотр. [447] исследовали растворы цис- и транс-дихлор- и дибром-этиленов в смеси диоксан/вода. При изменении соотношения диоксана и воды диэлектрическая проницаемость системы меняется в области от 2 до 10, и считая, что разность Д = 6чис — обусловлена лишь влиянием электрического поля реакции, можно было проверить уравнение Бакингема: было показано, что уравнение правильно описывает влияние растворителей.
Ламброзо и сотр. [445] измерили химические сдвиги в некоторых полярных молекулах, обусловленные влиянием поля реакции, и сдвиги в некоторых неполярных газах, обусловленные вандерваальсовыми взаимодействиями, причем последние оказались больше предсказанных на основании теоретической модели Ховарда и сотр. [446]. Сдвиги в полярных молекулах также оказались больше значений, рассчитанных по теории поля реакции Бакингема [27, 200].
10.39.1. Влияние растворителей на спектры протонного резонанса ацетиленовых соединений
Исследование ИК-спектров ацетилена показало [196], что он способен а самоассоциации с образованием слабых водородных связей между слабокислыми атомами водорода и л-электронами тройной связи. Монозамещенные
Корреляции спектров ЯМР-Н1 со строением молекул
171
ацетилены ведут себя таким же образом, и в жидких ацетиленах, по-видимому, осуществляются ассоциации типа
R
С
R-C=C—Н
Вследствие сильного влияния диамагнитной анизотропии тройной связи ацетиленовых систем наблюдается существенное дезэкранирование ацетиленовых протонов, обусловленное их близостью к тройной связи. Добавление инертного растворителя типа циклогексана уменьшает самоассоциацию, что приводит к возрастанию экранирования ацетиленовых протонов. Ричардс и Хаттон [197] показали, что пропаргилхлорид С1СН2С =. СН и фенилацетилен именно таким образом ведут себя при растворении в циклогексане (сдвиг, обусловленный растворением, равен 0,4 м. д.). Другие растворители оказывают гораздо более сильное влияние на экранирование ацетиленовых протонов
Таблица 10.103
ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ (Т-ЗНАЧЕНИЯ) ПРОПАРГИЛХЛОРИДА, ФЕНИЛАЦЕТИЛЕНА И БЕНЗОИЛАЦЕТИЛЕНА В РАЗЛИЧНЫХ РАСТВОРИТЕЛЯХ [197j
Растворитель Пропаргилхлорид Феиилаце-тилеи = СН Беизоил-ацетилеи = СН
=сн сн2
Чистое соединение 7,41 5,87 6,91 6,42 а
Циклогексан 7,81 6,13 7,26 6,90
N ,N-Диметилацетамид — — 5,78 4,63
М,Г\'-Диметилформамид 6,38 5,61 5,65 4,89
Пиридин — — ' 5,90 4,80
Ацетон 6,94 5,77 6,39 5,49
Диоксан 7,07 5,87 6,50 5,87
Ацетонитрил 7,20 5,83 6,65 5,75
Нитробензол 7,10 5,79 6,56 6,05
Нитрометан 7,27 5,82 6,74 6,18
Фторбензол 7,75 6,26 7,08 6,99
Тиофен 7,92 6,38 7,22 7,28
Бензол 8,12 6,58 7,29 7,42
Толуол 8,16 6,62 7,36 7,43
а Насыщенный раствор в ССЦ.
вследствие специфических взаимодействий между растворителем и растворенным веществом. В табл. 10.103 приведены химические сдвиги для пропаргил-хлорида, фенилацетилена и бензоилацетилена в различных растворителях. Для сравнения химических сдвигов в различных растворителях в качестве эталона был выбран сигнал ацетиленовых протонов каждого соединения в разбавленном циклогексановом растворе. Некоторые растворители способствуют дезэкранированию ацетиленовых протонов; показано, что все эти
172
Глава 10
растворители содержат сильные электроотрицательные центры, которые могут действовать как акцепторы протонов и способствуют образованию слабых водородных связей типа
;с=о . . .Н —С = С —R.
Диамагнитная циркуляция вокруг ацетиленовых протонов подвержена влиянию сильного электрического поля карбонильной группы, вызывающему дезэкранирование протонов. Ацетиленовый протон в пропаргилхлориде при использовании ацетонитрила в качестве растворителя также дезэкранируется, но в гораздо меньшей степени; это явление объясняется тем, что дезэкранирование, обусловленное электроотрицательной группой С = N, почти полностью-компенсируется влиянием диамагнитной анизотропии этой группы, которая увеличивает экранирование, как это показано на схеме
Н
С—СН2С1
Увеличение экранирования ацетиленовых протонов вызывают также ароматические растворители, содержащие сильно полярные заместители (например, пиридин и нитробензол). Если ароматическая молекула содержит сильную-электронооттягивающую группу, то образование слабой водородной связи сопровождается уменьшением экранирования ацетиленовых протонов. Бензол, толуол и тиофен увеличивают экранирование ацетиленовых протонов. Следует отметить, что СН2-группа пропаргилхлорида в бензольном растворе подвержена влиянию в той же степени, что и ацетиленовые протоны. Это позволяет исключить возможность специфической ассоциации типа
В работе [187] было исследовано изменение экранирования ацетиленовых протонов при разбавлении монозамещенных ацетиленов пиридином: во всех случаях при переходе от сильно разбавленного раствора в СС14 к аналогичному раствору в пиридине наблюдалось уменьшение химических сдвигов ацетиленовых протонов на 1 м. д. (см. табл. 10.104). По-видимому, это обусловлено-влиянием кольцевого тока пиридинового кольца на ацетиленовые протоны вследствие образования слабой водородной связи между азотом и ацетиленовыми водородами.
Исследовано также влияние среды на резонансный сигнал ацетиленовых протонов в пропаргилбромиде [189]. Наблюдаемые сдвиги объяснены образованием специфических водородных связей.
Корреляции спектров ЯМР-Н1 со строением молекул
173
Таблица 10.104
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ ПРОТОНОВ МОНОЗАМЕЩЕННЫХ АЦЕТИЛЕНОВ В ССЦ И В ПИРИДИНЕ [187]
R т (в ССЦ), м. д. т (в пиридине), м. д. Дт, м. д. J а, гц
н 8,20 >0,5
н-С3Н7 8,21 2,7
W-C4H9 8,27 2,4
Н-СдНц 8,25 7,30 0,95 2,1
С1СН2 7,60 2,7
ВгСН2 7,67 2,7
1СН2 7,81 2,8
носн2 7,67 . 6,82 0,85 2,7
НОС(СН3)2 7,72 6,80 0,92 >0,5
СН3ОСН2 7,63 6,71 0,92 2,5
СН3ОС(СН3)2 7,67 6,75 0,92 >0,5
С6Н5ОСН2 7,99 6,60 1,39 2,35
о=сн 8,Н >0,5
С6Н5 7,07 6,03 1,04 >0,5
сн2=сн 7,08 >0,5
НОСН2СН2 8,08 2,4
а Некоторые значения J, полученные и растворителя.
в ССЦ, и в пиридине, не зависят от природы
10.39.2. Влияние растворителей на спектры протонного резонанса ароматических соединений
Если чистый бензол разбавлять инертным растворителем типа СС14, то одиночный сигнал ароматических протонов постепенно смещается в область более слабых полей (смещение при бесконечном разбавлении составляет 0,75 м. д.) [198, 199]. Ботнер-Бай и Глик [198] предположили, что это уменьшение экранирования обусловлено разрывом слабых межмолекулярных связей между бензольными кольцами. В чистой жидкости эти связи, по-видимому, удерживают плоскости колец приблизительно параллельно друг другу в положениях, в которых протоны экранируются под влиянием кольцевых токов соседней молекулы; такой эффект будет действовать равным образом на все ароматические ядра [200, 201]. Аналогичный, хотя и менее ярко выраженный сдвиг наблюдается в спектре протонного резонанса толуола при разбавлении. Шефер и Шнейдер [201] исследовали влияние растворителей на спектры-протонного резонанса ряда замещенных бензолов; полученные ими данные приведены в табл. 10.105. Значительное изменение химических сдвигов ароматических протонов, наблюдавшееся при изменении природы растворителей, подчеркивает необходимость указания растворителя, в котором проводилось каждое конкретное измерение. Без этого данные о химических сдвигах часто оказываются бесполезными. На рис. 10.60 приведены спектры протонного резонанса н-метилнитробензола в различных растворителях [201]; эти спектры характеризуют общую тенденцию изменения химических сдвигов протонов в таких молекулах под влиянием растворителей. Спектр кольцевых протонов в н-метилнитробензоле представляет собой пару дублетов типа АВ. Раствор (5 мол.%) в инертном растворителе, гексане, рассматривается как раствор «изолированных» молекул, не вступающих ни в какие межмолекулярные взаимодействия, кроме вандерваальсовых; спектр протонного резонанса «изо-
174
Глава 10
Таблица 10.105
ХИМИЧЕСКИЕ СДВИГИ КОЛЬЦЕВЫХ ПРОТОНОВ3 пара-ДИЗАМЕЩЕННЫХ БЕНЗОЛОВ В РАЗЛИЧНЫХ РАСТВОРИТЕЛЯХ [201]
Соединение ® Чистая жидкость Растворы (5 мол.%)
в гексане в ацетоне в бензоле
орто мета орто мета орто мета орто мета
NO2\ZZ/ch3 —34,4 14,6 —38,2 —2,6 —0,3 71,9
SO2CI<^ )?СН3 —23,8 7,2 -33,0 —8,8 8,7 69,3
сн3со^ )сн3 —4,9 35,3 — 19,5 19,5 —25,4 9,2 3,1 53,1
СК<^ )СН3 0 • 15,0 3,0 16,4 —10,9 4,7 42,2 58,4
f/ Sch, в 27,6 26,6 38,9 28,3 27,8 16,4 62,2 62,2
С1^ ^>сн3 20,2 33,2 19,6 29,0 10,9 16,1 43,0 66,0
Вг^ >СН3 20,4 44,8 9,8 31,6 1,8 18,0 34,9 70,3
^'СН3 -3,6 37,0 —10,7 25,1 21,8 76,8
NQ>^ -12,2 18,5 -26,2 4,0 42,5 70,9
SQjCl^ ^>F -35,3 16,5 —49,3 -9,1 20,7 88,7
BrX 18,7 47,3 5,1 36,3 —8,6 18,8 48,3 80,9
а _ _ , г , VCHC13 (внешн)
3,5+ 60
б Водород обозначен орто и мета по отношению к заместителям, указанным с левой стороны формулы.
в Сигнал лета-протонов расположен в более слабом поле,, чем сигнал орто-протонов.
лированной» молекулы показывает, что в ней ароматические протоны экранированы слабее, чем в чистой жидкости. Если в качестве растворителя используется ацетон, наблюдается сдвиг сигналов всех кольцевых протонов в слабое поле; бензол же как растворитель обусловливает противоположное смещение сигналов. Следует также отметить, что влияние растворителя на экранирование орто- и мета-протонов различно и внутренний сдвиг этих протонов (один относительно другого) при варьировании растворителя изменяется. В ацетоне и в бензоле влияние растворителя сильнее сказывается на экранировании л/ста-протонов. Неподеленная пара электронов кислородного атома ацетона обусловливает его сильные донорные свойства н-типа, тогда как молекула бензола представляет собой слабый донор электронов л-типа. Шефер и Шнейдер попытались найти фактор, определяющий влияние растворителя на экранирование ароматических протонов, рассмотрев все возможные способы взаимодействия растворителя с исследуемым веществом. Среди рассмотренных факторов были:
а) чисто магнитный вклад, обусловленный взаимодействием растворенного вещества с растворителем в магнитном поле (<та);
б) влияние пространственных эффектов объемистых заместителей в ароматическом кольце на взаимодействие растворителя с растворенным веществом (<тш);
Корреляции спектров ЯМР-Н1 со строением молекул
175
в) полярное взаимодействие растворенного вещества с растворителем как следствие изменения диэлектрической проницаемости среды (<тЕ);
г) специфическое молекулярное взаимодействие, включающее образование комплекса, в котором растворитель и растворенное вещество имеют определенные ориентации относительно друг друга (ор).
Поведение пара-дизамещенных ароматических соединений в растворах гексана и бензола можно качественно объяснить, если допустить, что в гексановом растворе слабые межмолекулярные силы между ароматическими молекулами не могут удерживать молекулы вместе и что экранирование соответствует отсутствию межмолекулярного влияния кольцевых токов. Разбавление замещенных ароматических соединений бензолом (имеющим по сравнению с ними меньший молекулярный объем) увеличивает возможность действия межмолекулярных сил, обусловливающих фиксированные относительно друг
---L_^_l ................
-40 -20 0 20 40 60 80 гц
СНС13 эталон
Р и с. 10.60. Спектр протонного резонанса л-метилнитробензола при 60 Мгц в различных растворителях [201].
а — чистая жидкость при 80°: б — раствор в ацетоне (5 мол.%); в — раствор в гексаие(5 мол.%); г — раствор в бензоле (5 мол.%).
друга положения ароматических молекул, в которых кольцевые протоны подвержены воздействию межмолекулярных влияний кольцевых токов. Однако, если бы такая картина исчерпывающе описывала все эффекты, то
орто- и лото-протоны одинаковым образом испытывали бы на себе влияние кольцевого тока. Кроме того, наблюдаемый в разбавленном бензольном растворе сдвиг значительно больше, чем это можно было ожидать из различия молекулярных объемов растворенного вещества и растворителя. Влияние пространственных затруднений на обусловленные растворителем химические сдвиги не может быть очень большим, так как влияние растворителя на экранирование орто-
и жта-протонов в дизамещенных ароматических соединениях с разными по объему заместителями (как в napa-mpem-бутилтолуоле) одинаково. Что касает-
ся вклада электрического поля в экранирование полярных молекул, то он может быть велик лишь в случае растворителей с большими диэлектрическими проницаемостями; у гексана и бензола диэлектрические проницаемости малы, и поэтому такие эффекты также будут малы. Ацетон имеет большую диэлектрическую проницаемость, поэтому полярные эффекты могут быть значительными. Таким образом, оставалось предположить, что наблюдаемое влияние растворителя обусловлено образованием слабых водородно-связанных комплексов типа
X
176
Глава 10
Химические сдвиги ароматических протонов молекул вещества, растворенного как в бензоле, так и в ацетоне, зависят от температуры, чего и следовало ожидать, если имеет место ассоциация указанного типа. Мы уже видели, что бензол может образовывать с молекулами типа хлороформа слабые комплексы [207], в которых связь С — Н хлороформа направлена перпендикулярно плоскости бензольного кольца. В таком положении сигнал протонов хлороформа под влиянием кольцевого тока смещается в более сильное поле х).
10.39.3. Влияние растворителей на спектры протонного резонанса ненасыщенных гетероциклических соединений
Шефер и Шнейдер [275] показали, что экранирование кольцевых протонов в ненасыщенных гетероциклических соединениях очень сильно зависит от растворителя. Если исследуются растворы в гексане, то наблюдаемые химические сдвиги можно считать свободными от вкладов межмолекулярных взаимодействий. Если химические сдвиги, полученные для бензольного и ацетонового растворов, в которых имеют место специфические взаимодействия растворителя с растворенным веществом, сравнить с химическими сдвигами в гексановом растворе, то можно получить некоторую информацию о природе межмолекулярных взаимодействий. В табл. 10.106 приведены химические сдвиги кольцевых протонов в некоторых гетероциклических соединениях относительно
Таблица 10.106
ХИМИЧЕСКИЕ СДВИГИ КОЛЬЦЕВЫХ ПРОТОНОВ В НЕКОТОРЫХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЯХ [275] а
Соединения Раствор (5 мол.%)
в гексане в ацетоне в бензоле
а 6 а 6 а Р
Пиррол 0,935 1,307 0,673 1,358 1,403 1,455
N-н-бутилпиррол 0,997 1,452 0,757 1,433 1,275 1,418
Тиофен 0,263 0,450 —0,018 0,297 0,837 0,982
Фуран 0,155 1,197 —0,102 1,027 0,730+0,05 1,727
4-Метилпиридин —0,093 0,533 —0,983 0,283 —0,683 1,183
4 N 2: — 1 ,270 — 1 ,612 -0,5 50
5^ %2 4: 0,232 —0 ,223 1,112
5: -0,438 —0,537 0,052
5 4 2: — 1,320 — 1,422 —0,882
'б/Х/\з 3: 0,325 0,022 1,102
7I II 12 4. —0,443 —0,617 0,343
8 N 8: —0,600 —0,807 —0,415
а Химические сдвиги измерены в миллионных долях (м. д.) относительно внешнего эталона СНС1з :и исправлены на разницу в объемных диамагнитных восприимчивостях; т= 3,5 -|- &сНС1з (внеши)-
хлороформа как внешнего эталона, исправленные на разность объемных диамагнитных восприимчивостей [275]. На рис. 10.61 проиллюстрированы сильные изменения в протонном спектре пиррола при использовании различных растворителей; рисунок показывает, как можно использовать растворите
х) В работе [540] на основании спектров протонного резонанса обсуждаются вопросы сольватации дифенилиодониевых соединений.— Прим. ред.
Корреляции спектров ЯМР-Н1 со строением молекул
177
ли для разрешения перекрывающихся мультиплетов. Слабопольный квартет на рис. 10.61 обусловлен а-протонами, подверженными влиянию растворителя сильнее остальных протонов. Добавление ацетона приводит к смещению сигнала а-протонов в слабое поле по сравнению с раствором в гексане. Бензол влияет на а-протоны противоположным образом, увеличивая их экранирование. Тиофен и фуран в этом отношении очень похожи на пиррол, т. е. их а-протоны также подвержены влиянию ацетона и бензола сильнее, чем 0-протоны. Однако в случае 4-пи-колина наибольшее влияние растворителя испытывает протон 3 (орто-ао-ложение по отношению к СН3-группе). Это явление аналогично по знаку и величине влиянию растворителей на пара-замещенные толуолы (см. разд. 10.39.2) и может быть объяснено образованием водородной связи между растворителем и 0-протоном пиколина. Для хинолина влияние растворителя (бензол) сильнее всего сказывается на протонах 3 и 4, тогда как протон 8 меньше всего подвержен этому влиянию. В тиазоле наибольшему влиянию растворителя подвержен протон, больше всего удаленный от атома азота (положение 5).
Таким образом, бензол и ацетон по-разному влияют на тиофен, фуран и пиррол, с одной стороны, и на пара-дизамещенные бензолы и 4-пиколины, с другой стороны. В случае пятичленных гетероциклов сильнее всего подвержены влиянию растворителя прото-
зталон ----->.
Рис. 10.61. Спектры протонного резонанса кольцевых протонов пиррола при 60 Мгц [275].
а — раствор в ацетоне (5 мол. %); б — раствор в гексане (5 мол. %); в — раствор в бензоле (5 мол.%), г — раствор в бензоле (20 мол.%). Спектры приведены в обычной шкале (с поправкой на объемные диамагнитные восприимчивости) относительно сигнала хлороформа.
ны, расположенные рядом сэлектроот-
рицательным атомом. Разумно предположить, что водородная связь между группой NH в пирроле и молекулой растворителя сильнее будет влиять на а-протоны, чем на 0-протоны. Это, однако, не является главным фактором, объясняющим экранирование, так как в молекуле N-n-бутилпиррола, где водородной связи нет, наблюдается аналогичное влияние растворителя. На основании
этого можно сделать вывод о том, что водородная связь осуществляется главным образом между молекулами растворителя и а-протонами пиррола.
10.40. Влияние среды в газах
Обычно принимается, что константы экранирования в газообразных соединениях соответствуют константам экранирования изолированных молекул. Это справедливо для газов при низких давлениях, но при высоких давлениях химические сдвиги в газообразных соединениях сильно зависят от влияния среды [414, 415]. В случае неидеального газа константа экранирования может быть разложена в ряд обратной зависимости от молярного объема V,n 12—489
178
Глава 10
[415] [см. уравнение (3.142), где о обозначена как £<’>]
а=а°+ (’ЙгМй) +’"’ (10-18)
где о0 — константа экранирования для изолированной молекулы, а о2 мала и ею можно пренебречь; оу обусловлена столкновением двух молекул и дается уравнением
01 Л’о опара ехр ( —j dx2, (10.19)
в котором Ао — число Авогадро > &пара — константа экранирования частиц 1, обусловленная присутствием частиц 2, р — межмолекулярная потенциальная энергия в конфигурации т2, dx2 — интеграл по всем конфигурациям молекулы 2 в ампуле с образцом. В смеси газов, состоящей из х молекул 1, содержащих протоны, для которых исследуется константа экранирования, и (1 —х) молекул 2,
a = a0-l-l/m1 {-wp-Hl —х) с[2-••}+•••,
где индексы 11 и 12 указывают на двойные столкновения между присутствующими частицами. Для небольших значений х о}2 можно приравнять оу, а обусловлена пятью вкладами, аналогичными вкладам в уравнении (10.16)
Щ = (°1)ь + (°1)а + (Щ)и> + (О1)е + (щ)н-
Для цилиндрических образцов
2
(°1)Ь ~ "д’
где %т = XvIM. — молярная диамагнитная восприимчивость.
(<Jnapa)a Для аксиально симметричных молекул 2 дается выражением (3.53) [---з (хц — %д_) (3 cos2 02 — 1) г-3]; применив это выражение и потенциал
Штокмайера для р в уравнении (10.19), Бакингем и сотр. [200] получили для (стД, выражение
(а1)а= —( 1080 ) (^|| —{ т2^9 (у) + У* ("Тб-) ^15 (</) + •••} >
где
Hi и р2 —дипольные моменты молекул 1 и 2 соответственно, Нп(у) — функции, табулированные Бакингемом и Поплом [416].
(<Гпара)Е дается уравнением (<тПара)Е = — АЕ — BE2 (см. разд. 3.9). Бакингем и сотр. [200] показали, что для полярных веществ, растворенных в неполярном газе,
- (4 «) ".Л (в.) [(*) Н, и + (-Д-) (10.20)
а для неполярных веществ, растворенных в полярном газе,
<»>>= - [4 «)". w+(f)". w] • о»-’»
В уравнениях (10.20) и (10.21) 02 — молекулярный квадрупольный момент; а2 — поляризуемость молекул газа, используемого в качестве растворителя.
Эмпирически показано, что (<Т1)Ш отрицательна, и, допустив уменьшение экранирования вследствие флуктуирующего электрического поля в молекулах газа-растворителя, среднее квадратичное значение (F2) которых не усредняется до нуля, можно получить приближенное значение (<Ji)u,. Бакингем
Корреляции спектров ЯМР-Н1 со строением молекул
179
и сотр. [200] показали, что
<F2>= W2 ,
где /2— потенциал ионизации молекул растворителя, а г — расстояние между двумя молекулами. Поэтому
(Чпара)ш = В (У72),
и подстановка этого выражения в уравнение (10.19) дает
= а2/2 {Н^ + (^) + • • } • (10.22)
Qi для бинарной смеси газов может быть получена при измерении химических сдвигов протонов как функции давления; тогда
’="(-£) (,0-23>
где М — молекулярный вес молекул 1, р — плотность (г/ои3), о выражена в миллионных долях (м. д.). Значение оу, полученное из уравнения (10.23), будет зависеть от мольной доли растворенного вещества в смеси газов, поэтому необходимо определить оу для ряда различных мольных долей и затем экстраполировать к нулевой концентрации растворенного вещества. В работе [414] были измерены при бесконечном разбавлении значения (оОнабл для неполярных углеводородов СН4 и С2Н6 и полярного НС1; полученные результаты приведены в табл. 10.107. В последнем столбце табл. 10.107 даны величины
Таблица 10.107
ЗНАЧЕНИЯ (Ч1)набл ДЛЯ НЕКОТОРЫХ ДВОЙНЫХ СМЕСЕЙ ГАЗОВ [414]
Растворенное вещество Растворитель (°1)иабл’ Ю6» смЗ/моль (О1)ь-106, см?/моль [(°1)набл - «Т1)Ь]Х 106, 'СМЪ/М.ОЛЪ
сн4 сн4 —42 —36- —6
сн4 НС1 —58 —47 —11
С2Н4 С2Н4 —50 —39 —11
С2Н6 С2Н6 —74 —56 —18
НС1 сн4 —62 —36 —26
НС1 С2н6 —92 —56 —36
НС1 Кг —94 —61 —33
НС1 со2 —98 —44 —54
НС1 SF6 — 114 —92 —22
НС1 Хе —139 —95 —44
НС1 НС1 —150 —46 —104
НС1 H2S — 176 —53 — 123
НС1 OCS — 176 —68 —108
[(аОнабл — (<71)ь1, которые для растворенных углеводородов [где (ot)a и (oi)B пренебрежимо малы] могут быть сведены к (сц)и,. Значение В может быть получено подгонкой уравнения (10.22) к данным для СН4, С2Н4 и С2Н6; найденное таким образом В равно (1,0 ± 0,3) • 10-18 эл.-ст. ед.
Для полярного НО как (О1)ш, так и (п^е дают вклады в [(оНабл — (оДьК и подгонка уравнений (10.20) и (10.22) к наблюдаемым данным дает значения А и В для этой молекулы. Значения, полученные Бернстейном и сотр. [414],
12*
180
Глава 10
следующие:
А = (40,4 + 2)-10-12 эл.-ст, ед.,
В= (0,38 ± 0,1)-10-18 эл.-ст. ед.
Бернстейн и сотр. [417, 418] определили (<Т1)Набл и, следовательно, А и В для протонов и ядер фтора в растворенных соединениях; полученные ими значения приведены в табл. 10.108. Петракис и Бернстейн [418] обсудили физический смысл значений А и В. Они предположили, что значение А характеризует легкость смещения заряда в направлении связи X — Н или X — F; следовательно, значение А положительно в связях X — Ни отрицательно для X — F.
Таблица 10.108
ЗНАЧЕНИЯ ПАРАМЕТРОВ СВЯЗИ А И В ДЛЯ РАЗЛИЧНЫХ ПОЛОЖЕНИЙ СВЯЗИ [418]
Связь A- 1012, эл.-ст. ед. B- 1018, эл.-ст. ед.
CH (в неполярных углеводородах) l,0±0,3
CH (в CHF3) 2,92+0,8 0,84+0,3
HC1 40,4+2,0 0,38±0,1
Атом H 0,74
CF (в CF4) 16,4+2,1
CF (в CHF3) —9,9+3,6 15,1+3,0
SF (в SF6) 29,5+2,4
SiF (в SiF4) 43,5+5,1
Значение В зависит от поляризуемости атома Н или F, следовательно, оно характеризует легкость смещения заряда в направлении, перпендикулярном направлению связи, и будет увеличиваться с увеличением степени двоесвяз-ности связи X — F.
ЛИТЕРА ТУР. А
1. Shoolery J. N., J. Chem. Phys., 21, 1899 (1953).
2. М е у е г L. Н., S a i k a A., G u t о w s к у Н. S., J. Am. Chem. Soc., 75, 4567 (1953).
3. Meyer L. H., Gu tows ky H. S., J. Phys. Chem., 57, 481 (1953).
4. A 1 1 r e d A. L., Rochow E. G., J. Am. Chem. Soc., 79, 5361 (1957).
5. Huggins M. L., J. Am. Chem. Soc., 75, 4123 (1953).
6. Spiesecke H., Schneider W. G., J. Chem. Phys., 35, 722 (1961).
7. Cavanaugh J. R., Dailey В. P., J. Chem. Phys., 34, 1099 (1961).
8. В о t h n e r - В у A. A., N a a г - С о 1 i n C., J. Am. Chem. Soc., 80, 1728 (1958).
9. Dailey В. P., Shoolery J. N., J. Am. Chem. Soc., 77, 3977 (1955).
10. Ramsey N. F., Phys. Rev., 78, 699 (1950).
11. Cavanaugh J. R., Dailey В. P., J. Chem. Phys., 34, 1094 (1961).
12. Narasimhan P. T., R ogers M. T., J. Am. Chem. Soc., 82, 5983 (1960).
13. Shoolery J. N., Lecture Notes, Varian Associates Conference (1957).
14. В г о w ns t e i n S., S m i t h В. С., E h r 1 i c h G., Laubengayer A. W., J. Am. Chem. Soc., 81, 3826 (1959).
15. McGarvey B. R., S 1 о m p G., J. Chem. Phys., 30, 1586 (1959).
16. N a r as i m h a n P. T., R ogers M. T., J. Chem. Phys., 31, 1302 (1959).
17. M и s h e r J. I., J. Chem. Phys., 35, 1159 (1961).
18. T i 1 1 i e и J., Ann. Phys., 2, 471, 631 (1957).
19. G и у J., T i 1 1 i e и J., J. Chem. Phys., 24, 1117 (1956).
20. W h i t m a n D. R., Saunders M., О n s a g e r L., D и b b H. E., J. Chem. Phys., 32, 67 (1960).
21. Reddy G. S., Goldstein J. H., J. Chem. Phys., 38, 2736 (1963).
22. M о r i t z A. G., Sheppard N., Mol. Phys., 5, 361 (1962).
23. Musher J. 1., J. Am. Chem. Soc., 83, 1146 (1961).
Корреляции спектров ЯМР-Н1 со строением молекул
181
24. Musher J. I., J. Chem. Phys., 37, 34, 192 (1962).
25. Ziircher R. F., Discuss. Faraday Soc., 34, 66 (1962).
26. P о p 1 e J. A., Discuss. Faraday Soc., 34, 7 (1962).
27. В u с к i n g h a m A. D., Can. J. Chem., 38, 300 (1960).
28. К a r p 1 u s M., Anderson D. H., Farrar T. C., G u t о w s к у H. S., J. Chem. Phys., 27, 597 (1957).
29. К a r p 1 u s M., Anderson D. H., J. Chem. Phys., 30, 6 (1959).
30. Gutowsky H. S., Karplus M., Grant D. M., J. Chem. Phys., 31, 1278 (1959).
31. Gutowsky H. S., Mo c h e 1 V. D., Somers B. G., J. Chem. Phys., 36, 1153 (1962).
32. Lauterbur P. C., Kurland R. J., J. Am. Chem. Soc., 84, 3405 (1962).
33. M с С о n n e 1 1 H. M., J. Chem. Phys., 24, 460 (1956).
34. В a n w e 1 1 C. N., Sheppard N., Discuss. Faraday Soc., 34, 115 (1962).
35. M с С о n n e 1 1 H. M., J. Chem. Phys., 23, 2454 (1955).
36. Barfield M., Grant D. M., J. Am. Chem. Soc., 83, 4726 (1961).
37. Bernstein H. J., Sheppard N., неопубликованные данные, цитируемые в [34].
38. Buckingham A. D., McLauchlan К. A., Proc. Chem. Soc., 144 (1963).
39. Karplus M., J. Chem. Phys., 30, 11 (1959).
40. Glick R. E., Bothner-By A. A., J. Chem. Phys., 25, 362 (1956).
41. L e m i e u x R. U., К u 1 1 n i g R. K-, Moir R. Y., J. Am. Chem. Soc., 80, 2237 (1958).
42. Sheppard N., Lynden-Bell R. M., Proc. Roy. Soc., A 269, 385 (1962).
43. S c h u g J. C., McMahon P. E., Gutowsky H. S., J. Chem. Phys., 33, 843 (1960).
44. Abraham R. J., McLauchlan K- A., Mol. Phys., 5, 195 (1962).
45. A b r a h a m R. J., Hall L. D., Hough L., McLauchlan K- A., Chem. and Ind., 213 (1962).
46. Abraham R. J., McLauchlan K- A., Hall L. D., Hough L., J. Chem. Soc., 1962 , 3699.
47. Musher J. L, J. Chem. Phys., 34, 594 (1961).
48. Williamson K- L., J ohnson W. S., J. Am; Chem. Soc., 83, 4623 (1961).
49. Abraham R.J., Holker J.S. E., J. Chem. Soc., 1963 , 806.
50. Sheppard N., Turner J. J., Proc. Roy. Soc., A 252, 506 (1959).
51. Conroy H., Advances in Organic Chemistry, Vol. II, Interscience, N.Y., 1960.
52. N ar as i m h a n P. T., R о g e r s M. T., J. Chem. Phys., 34, 1049 (1961).
53. A b r a h a m R. J., Pachler K- G. R., Mol. Phys., 7, 165 (1964).
54. Fraser R. R., Lemieux R. U., Stevens J.D., J. Am. Chem. Soc., 83, 3901 (1961).
55. R e i 1 1 у C. A., S w a 1 e n J. D., J. Chem. Phys., 35, 1522 (1961).
56. G u t о w s к у H. S., J u an C., Discuss. Faraday Soc., 34, 52 (1962).
57. Kaplan F., Roberts J. D., J. Am. Chem. Soc., 83, 4666 (1961).
58. К a r p 1 u s M., J. Am. Chem. Soc., 84, 2458 (1962).
59. Roberts J. D., Lutz R. P., D a v i s D. R., J. Am. Chem. Soc., 83, 246 (1961);
Davis D. R., Roberts J. D., J. Am. Chem. Soc., 84, 2252 (1962).
60. Holmes J. R., К ivelson D., J. Am. Chem. Soc., 83, 2959 (1961).
61. Karplus M-, J. Am. Chem. Soc., 82, 4431 (1960).
62. К a r p 1 u s M., J. Chem. Phys., 33, 1842 (1960).
63. McConnell H. M., J. Chem. Phys., 24, 764 (1956).
64. Fraenkel G., J. Chem. Phys., 34, 1466 (1961).
65. A 1 1 r e d A. L., R о c h о w E. G., J. Inorg. Nucl. Chem., 5, 264 (1958).
66. Brown M. P., Webster D. E., J. Phys. Chem., 64 , 698 (1960).
67. В а к e r E. B., J. Chem. Phys., 26, 960 (1957).
68. N a r a s i m h a n P. T., R о g e r s M. T., J. Chem. Phys., 31, 1430 (1959).
69. Jaffe H. H., Reynolds G. F., Flautt T. J., Dessy R. E., J. Chem. Phys., 30, 1422 (1959).
70. Stafford S. L., Baldeschwieler J. D., J. Am. Chem. Soc., 83, 4473
(1961).
71. Maher J. P., E v a n s D. F., Proc. Chem. Soc., 208 (1961).
72. Klose G., Ann. Phys., 8, 220 (1961).
73. Klose G., Ann. Phys., 10, 391 (1963).
74. Feeney J., Sutcliffe L. H., неопубликованные данные.
75. Nordlander J. E., Young W. G., Roberts J.D., J. Am. Chem. Soc., 83, 494 (1961)
76. Nordlander J. E., Roberts J. D., J. Am. Chem. Soc., 81, 1769 (1959).
77. Whitesides G. M., Nordlander J. E., Roberts J . D., J. Am. Chem. Soc., 84, 2010 (1962).
78. Whitesides G. M., Nordlander J. E., Roberts J.D., Discuss. Faraday Soc., 34, 185 (1962).
182
Глава 10
79. A n е t F. A. L., Proc. Chem. Soc., 327 (1959).
80. A n e t F. A. L., J. Am. Chem. Soc., 84, 747 (1962).
81. Bo th ner-B у A. A., N a a r - Co 1 i n C., J. Am. Chem. Soc., 84, 743 (1962).
82. W a 1 s h A. D., Trans. Faraday Soc., 45, 179 (1949).
83. D a i 1 e у В. P., Gawer A., N eikam W. C., Discuss. Faraday Soc., 34, 18
(1962).
84. R e i 1 1 у C. A., S w a 1 e n J. D., J. Chem. Phys., 32, 1378 (1960).
85. J а с к m a n L. M., Nuclear Magnetic Resonance Spectroscopy, Pergamon Press,
London, 1959.
86. G u t о w s к у H. S., G r a n t D. M., J. Chem. Phys., 34, 699 (1961).
87. Робертс Дж., Введение в анализ спектров ЯМР высокого разрешения (спин-спиновое взаимодействие), ИЛ, М., 1963.
88. С 1 о s s G. L., С 1 о s s L. Е., J. Am. Chem. Soc., 82, 5723 (1960).
89. S т i d t J., d e Boer Th. J., Rec. Trav. Chim., 82, 1235 (1960).
90. G r a h a m J. D., R о g er s M. T., J. Am. Chem. Soc., 84, 2249 (1962).
91. H u t t о n H. M., S c h a e f e r T., Can. J. Chem., 41, 684 (1963).
92. К a r p 1 u s M., J. Phys. Chem., 64, 1793 (1960).
93. Fraser R. R., Can. J. Chem., 40, 1483 (1962).
94. H a r r i s R. K-, Sheppard N., Proc. Chem. Soc., 418 (1961).
95. Muller N., Tos ch W. C., J. Chem. Phys., 37, 1167 (1962).
96. J e n s e n F. R., Noyce D.S., Sederholm С. H., Berlin A. J., J. Am. Chem. Soc., 82, 1255 (1960); 84, 386 (1962).
97. Lemieux R. U., К u llnig R. K-, Bernstein H.J..Schneider W. G.,
J. Am. Chem. Soc., 80, 6098 (1958).
98. Shoolery J. N., Rogers M. T., J. Am. Chem. Soc., 80, 5121 (1958).
99. Sc h a f e r T., S c h n e i d e r W. G., Can. J. Ciem., 41, 966 (1963).
100. Brownstein S., J. Am. Chem. Soc., 81, 1606 (1959).
101. Cohen A. D., Sheppard N., Turner J. J., Proc. Chem. Soc., 118 (1958).
102. G u t о w s k у H. S., M e у e r L. H., M с С 1 u r e R. E., Rev. Sci. Instrumets, 24, 644 (1953).
103. Lemieux R. U., К u 1 1 n i g R.K,Bern stein H.J..Schneider W. G-, J. Am. Chem. Soc., 79, 1005 (1957).
104. Musher J. L, Richards R. E., Proc. Chem. Soc., 230 (1958).
105. Musher J. L, Spec t. Acta, 16, 835 (1960).
106. Brownstein S., Miller R., J. Org. Chem., 24, 1886 (1959).
107. Lichtenthaler F. W-, Fischer H. O. L., J. Am. Chem. Soc., 83, 2005 (1961).
108. L e e W. W., Benitez A..Anderson C. D., Goodman L., Baker В. B., J. Am. Chem. Soc., 83, 1906 (1961).
109. Williamson K- L-, J ohnson W. S., J. Am. Chem. Soc., 83, 4623 (1961).
110. R e e v e s L. W., Stromme К- O., Can. J. Chem., 38/ 1241 (1960).
111. Reeves L. W., Stromme K- O., Trans. Faraday Soc., 57, 390 (1961).
112. A n e t F. A. L., Can. J. Chem., 39, 2262 (196b).
113. S c h a e f e r T., S c h n e i d e r W. G., Can. J. Chem., 38, 2066 (1960).
114. Corey E. J., частное сообщение.
115. Feeney J., Ledwith A., Sutcliffe L. H., J. Chem. Soc., 1962, 2021.
116. Gutowsky H. S., Porte A. L.,J. Chem. Phys., 35, 839 (1961).
117. S c h n e i d e r W. G., Bernstein H. J., Pople J. A., J. Am. Chem. Soc., 80, 3497 (1958).
118. В a n w e 1 1 C. N., S h e p p a r d N., Mol. Phys., 3, 351 (1960).
119. S c h a e f e r T., Can. J. Chem., 40, 1 (1962).
120. Sheppard N., В a n w e 1 1 C. N., Proc. Roy. Soc., A 263, 136 (1961).
121. Dowl ing J. M.,Stoicheff В. P., Can. J. Phys., 37, 703 (1959).
122. Bak B., Christensen D., Hansen-Nygaard L., Rostrup-Anderson J., Spect. Acta, 13, 120 (1958).
123. Whipple E. B., Stewart W. E., Reddy G. S., Goldstein J. H., J. Chem. Phys., 34, 2136 (1961).
1 24. В о t h n e r - В у A. A., N а а г - С о 1 i n C., J. Am. Chem. Soc., 83, 231 (1961).
125. Alexander S., J. Chem. Phys., 28, 358 (1958).
126. Castellano S., Caporiccio C.,J. Chem. Phys., 36, 566 (1962).
127. Bishop E. O., Richards R. E., Mol. Phys., 3, 114 (I960).
128. Freeman R., Mol. Phys., 4, 385 (1961).
129. Mortimer F. S., J. Mol. Spect., 3, 335, 340 (1959).
130. Cohen A. D., Sheppard N., Proc. Roy. Soc., A 252, 488 (1959).
131. Fessenden R. W., Waugh J. S., J. Chem. Phys., 30, 944 (1959).
132. В r ti g e 1 W., A n k e 1 Th., Krtickeberg F., Z. Electrochem., 64, 1121 (1960).
133. В a n w e 1 1 C. N., Sheppard N., Turner J. J., Spect. Acta, 16, 794 (1960).
134. Castellano S., W a u g h J. S., J. Chem. Phys., 34, 295 (1961).
135. Moore D. W., H a p p e J. A., J. Phys. Chem., 65, 224 (1961).
Корреляции спектров ЯМР-Н1 со строением молекул 183
136. Johnson С. S., Weiner М. A., Waugh J.S., Seyferth D., J., Am. Chem. Soc., 83, 1306 (1961).
137. Clifford A. F., J. Phys. Chem., 63, 1227 (1959).
138. Castellano S., Waugh J.S.,J. Chem. Phys., 35, 1900 (1961).
139. Reddy G. S., Goldstein J. H., J. Chem. Phys., 35, 380 (1961).
140. Taft R. W., J. Am. Chem. Soc., 79, 1045 (1957).
141. П о п л Дж., Шнейдер В., Бернстейн Г., Спектры ядерного магнитного резонанса высокого разрешения, ИЛ, М., 1962.
142. Smyth С. Р., Dielectric Behavious and Structure, McGraw-Hill, N.Y., 1955.
143. Reddy G. S., Mandell L., Goldstein J. H., J. Am. Chem. Soc., 83, 1300 (1961).
144. Whipple E. B., Stewart W. E., Reddy G. S., Goldstein J. H., J. Chem. Phys., 34, 2136 (1961).
145. Whipple E. B., Goldstein J; H., Mandell L., J. Am. Chem. Soc., 82, 3010 (1960).
146. P о p 1 e J. A., J. Chem. Phys., 24, 1111 (1956).
147. Johnson С. E., В о v e у F. A., J. Chem. Phys., 29, 1012 (1958).
448. Fraser R. R., Can. J. Chem., 38, 549 (1960).
149. Feeney J., Sutcliffe L. H., неопубликованные данные.
150. Beach W. F., R i c h a r d s J. H., J. Org. Chem., 26, 3011 (1961).
151. Davis D. R., Roberts J. D., J. Am. Chem. Soc., 84, 2252 (1962).
152. Hobgood R. T., Goldstein J. H., Reddy G. S., J. Chem. Phys., 35, 2038 (1961).
153. Hobgood R. T., Reddy G. S., Goldstein J. H., J. Phys. Chem., 67, 110 (1963).
154. Bothner-By A. A., Glick R. E., J. Am. Chem. Soc., 78, 1071 (1956).
155. Gordy W., Thomas W.J.O., J. Chem. Phys., 24, 439 (1956).
156. Corio P. L., D a i 1 e у В. P., J. Am. Chem. Soc., 78, 3043 (1956).
157. P о p 1 e J. A., Proc. Roy. Soc., A 239, 550 (1957).
158. McConnell H. M., J. Chem. Phys., 27, 226 (1957).
159. Bothner-By A. A., N a a r - С о 1 i n C., Ann. N. Y. Acad. Sci., 70, 883 (1958).
160. Narasimhan P. T., R о g e r s M. T., J. Phys. Chem., 63, 1388 (1959).
161. Schneider W. G., Bernstein H. J., Pop 1 e J. A., J. Chem. Phys., 28, 601 (1958).
162. Bothner-By A. A., J. Mol. Spect., 5, 52 (1960).
163. Smith J. W., Electric Dipole Moments, Butterworth, London, 1955, p. 198.
164. Whipple E. B., J. Chem. Phys., 35, 1039 (1961).
165. Bothner-By A. A., N a a r - Co 1 i n C.,Giinther H.,J. Am. Chem. Soc..
84 , 2748 (1962).
166. Jackman L. M., Wiley R. H., J. Chem. Soc., 1960, 2881.
167. Whipple E. B., Goldstein J. H., McClure G. R., J. Am. Chem. Soc., 82, 3811 (1960).
168. Reddy G. S., Goldstein J. H., J. Am. Cherp. Soc., 83, 2045 (1961).
169. Jackman L. M., Wiley R. H., J. Chem. Soc., 1960, 2886.
170. Fraser R.R., McGreer D. E., Can. J. Chem., 39, 505 (1961).
171. См. ссылку[141], гл. И.
172. E 1 v i d g e J. A., Jackman L. M., Proc. Chem. Soc., 89 (1959).
173. В a n w e 1 1 C. N., Cohen A. D., Sheppard N., Turner J. J., Proc. Chem. Soc., 266 (1959).
174. Hoffman R. A., Mol. Phys., 1, 326 (1958).
175. Hoffman R. A., G г о п о w i t z S., Acta Chem. Scand., 13, 1477 (1959).
176. Snyder Е.1., Roberts J.D.,J. Am. Chem. Soc., 84, 1582 (1962).
177. M a n a t t S. L., E 1 1 e m a n D. D., J. Am. Chem. Soc., 84, 1579 (1962).
178. Bak B., Shoolery J. N., Williams G. A., J. Mol. Spect., 2, 525 (1958).
179. McConnell H. M., J. Chem. Phys., 30, 126 (1959).
180. Hobgood R. T., Goldstein J. H., Reddy G. S., J. Chem. Phys., 35, 2038 (1961).
181. Piper T. S., W i 1 k i n s о n G., J. Inorg. Nucl. Chem., 3, 104 (1956).
182. Dave L. D., Evans D. F., Wilkinson G., J. Chem. Soc., 1959, 3684.
183. Strohmeier W., Lemon R. M., Z. Natur., 14a, 109 (1959).
184. G г e e п M. L. H., Wilkinson G., J. Chem. Soc., 1958, 4314.
185. Green M. L. H., Pratt L., Wilkinson G., J. Chem. Soc., 1959, 3753.
186. Barber M. S., Davis J. B., Jackman L. M., Weedon В. C. L., J. Chem. Soc., 1960, 2870.
187. Kreevoy M. M., Charman H. B., Vinard D. R., J. Am. Chem. Soc., 83, 1978 (1961).
188. Hatton J. V., Richards R. E., Trans. Faraday Soc., 56, 315 (1960).
189. Whipple E. B., Goldstein J. H., Mandell L., Reddy G. S., McClure G. R., J. Am. Chem. Soc., 81, 1321 (1959).
190. Narasimhan P. T., R о g e r s M. T., J. Chem. Phys., 33, 727 (1960).
184
Глава 10
191. McConnell Н. М., J. Mol. Spect., 1, 11 (1957).
192. S h о о 1 e г у J. N., J. Mol. Spect., 63, 110 (1960).
193. Snyder E. I. .Roberts J. D., J. Am. Chem. Soc., 84, 1582 (1962).
194. Snyder E. I..Altman L.J., Roberts J.D.,J. Am. Chem. Soc., 84, 2004 (1962).
195. Hirst R. C., G r a n t D. M., J. Am. Chem. Soc., 84, 2009 (1962).
196. Brand J. C. D., E g 1 i n t о n G., Morman J. F., J. Chem. Soc., 1960, 2526.
197. Richards R. E., Hatton J. V., Trans. Faraday Soc., 57, 28 (1961).
198. Bothner-By A. A., Glick R. E., J. Chem. Phys., 26, 1651 (1957).
199. Zimmerman J. R., Foster M. R., J. Phys. Chem., 61, 282 (1957).
200. Buckingham A. D., Schaefer T., Schneider W. G., J. Chem. Phys., 32, 1227 (1960).
201. Schaefer T., Schneider W. G., J. Chem. Phys., 32, 1218 (1960).
202. Hammett L. P., Trans. Faraday Soc., 34, 156 (1938).
203. Taft R. W-, E h r e n s о n S.,Lewis I. C., G 1 i с к R. E., J. Am. Chem. Soc., 81, 5352 (1959).
204. Chart o n - К о e c h 1 i n M., Leroy M. A., J. chim. Phys., 56, 850 (1959).
205. L a u t e r b u r P. C., Ann. N.Y. Acad. Sci., 70, 841 (1958).
206. Gutowsky H. S., McCall -D. W., McGarvey B. R., Meyer L. H., J1. Amer. Chem. Soc., 74 , 4809 (1952).
207. Korinek G. J. .Schneider W.G., Can. J. Chem., 35, 1157 (1957).
208. Spiesecke H., Schneider W. G., J. Chem. Phys., 35, 731 (1961).
209. Yamaguchi I., Hayakawa N., Bull. Chem. Soc. Japan, 33, 1128 (1960).
210. Fraser R.R., Can. J. Chem., 38, 2226 (1960).
211. Diehl P., Helv. Chim. Acta, 44, 829 (1961).
212. Farthing A. C., N a m B., Steric Effects in Conjugated Systems, Butterworth, London, 1958, p. 131.
213. В г e у W. S., L a ws о п K. D., частное сообщение.
214. S m i t h G. W., частное сообщение.
215. Martin J. S., D a i 1 e у В. P., J. Chem. Phys., 39, 1722 (1963).
216. S t e h 1 i n g F. C., Anal. Chem., 35, 773 (1963).
217. Chamberlain N. F., Anal. Chem., 31, 56 (1959).
218. Heatchcock C., Can. J. Chem., 40, 1865 (1962).
219. Kun K- A., Cassidy H. G., J. Org. Chem., 26, 3223 (1961).
220. К 1 i n с к R. E., S t о th er s J. B., Can. J. Chem., 40, 1071 (1962).
221. L e a n e J. B., Richards R. E., Trans. Faraday Soc., 55, 707 (1959).
222. Brownstein S., J. Am. Chem. Soc., 80, 2300 (1958).
223. Hoffman R. A., К i n e 1 1 P. О., В e r g s t г о m G., Arkiv. Kemi, 15, 333 (1960).
224. E 1 1 e m a n D. D.,, M a n a t t S. L., J. Chem. Phys., 36, 2346 (1962).
225. Johnson С. E., Bovey F. A., J. Chem. Phys., 29, 1012 (1958).
226. Waugh J. S., Fessenden R. W., J. Am. Chem. Soc., 79, 846 (1957).
227. P о p 1 e J. A., J. Chem. Phys., 24, 1111 (1956).,
228. Bernstein H. J., Schneider W. G., P о p 1 e J. A., Proc. Roy. Soc., A 236, 515 (1956).
229. J onathan N., Gordon S., D a i 1 e у В. P., J. Chem. Phys., 36, 2443 (1962).
230. Schaefer T. P., Richards R. E., Trans. Faraday Soc., 54, 1280 (1958).
231. Nageswara Rao B. D., Venkateswarlu P., Proc. Indian, Acad. Sci., 54, 1 (1961).
232. Richards R. E., Schaefer T., Mol. Phys., 1, 331 (1958).
233. D i s c h 1 e г В., E n g 1 e r t G., Z. Natur., 16a, 1180 (1961).
234. Martin J., Dailey В. P., J. Chem. Phys., 37, 2594 (1962).
235. Sharp D. W. A., Sheppard N., J. Chem. Soc., 1957, 674.
236. M о о d i e R. B., Connor T. M., Stewart R., Can. J. Chem., 37, 1402 (1959).
237. O’R e i 1 1 у D. E., L e f t i n H. P., J. Phys. Chem., 64, 1555 (1960).
238. Dehl R., Vaughan W. R., Berry R. S., J. Org. Chem., 24, 1616 (1959).
239. MacLean C., van der W a a 1 s J. H., M а с к о r E. L., Mol. Phys., 1,
247 (1958).
240. Danyluk S. S., Schneider W. G., J. Am. Chem. Soc., 82, 997 (1960).
241. Danyluk S. S„ S c h n e i d e r W. G., Can. J. Chem., 40, 1777 (1962).
242. Lauterbur P. С., частное сообщение.
243. Spiesecke H., Schneider W. G., Tetrahedron Letters, 468 (1961).
244. MacLean С., M а с к о r E. L., J. Chem. Phys., 34, 2208 (1961).
245. J u 1 g A., J. chim. Phys., 52, 377 (1955).
246. Pariser R., J. Chem. Phys., 25, 1112 (1956).
247. Hall G. G., Hardisson A., Jackman L. M., Discuss. Faraday Soc., 34, 15 (1962).
248. Fraenkel G., Carter R. E., McLachlan A. M., R i c h a r d s J. H., J. Am. Chem. Soc., 82, 5846 (1960).
249. Reddy G. S., Goldstein J. H., J. Am. Chem. Soc., 84, 583 (1962).
Корреляции спектров ЯМР-Н1 со строением молекул
185
250. М а с L е а п С., Mackor Е. L., Mol. Phys., 4, 241 (1961).
251. Leto J. R., Cotton F. A., Waugh J. S., Nature, 180, 978 (1957).
252. Reddy G. S., G о 1 d s t e i n J. H., J. Phys. Chem., 65, 1539 (1961).
253. L e a n e J. B., Richards R.E., Trans. Faraday Soc., 55, 518 (1959).
254. Corey E. J., Slom p G., Dev S., Tobinaga S., Glazier E. R., J.
Am. Chem. Soc., 80, 1204 (1958).
255. Abraham R. J., Bernstein H. J., Can. J. Chem., 37, 1056 (1959).
256. Abraham R. J., Bernstein H. J., Can. J. Chem., 39, 905 (1961).
257. Varian Technical Information Bulletin № 50.
258. Gronowi tz S., Hornfeldt A., G e s t b 1 о m B., Hoffman R. A., Arkiv. Kemi., 18, 133 (1961).
259. Gronowi tz S., Hornfeldt A., G e s t b 1 о m B., Hoffman R. A., Arkiv. Kemi., 18, 151 (1961).
260. T u i t e R. J., J о s e у A. D., Snyder H. R., J. Am. Chem. Soc., 82, 4360 (1960).
261. Cohen A. D., McL auchl an K. A., Discuss. Faraday Soc.., 34, 132 (1962).
262. Hoffman R. A., Gronowi tz S., Arkiv. Kemi., 16, 563 (1960).
263. Abraham R. J., Bullock E., M i t r a S. S., Can. J. Chem., 37, 1859 (1959).
264. Reddy G. S., G о 1 d s t e i n J. H., J. Am. Chem. Soc., 83, 5020 (1961).
265. Reddy G. S., Goldstein J. H., J. Am. Chem. Soc., 83, 2045 (1961).
266. T a u r i n s A., Schneider W. G., Can. J. Chem., 38, 1237 (1960).
267. Ellis J., Jackson A. H., Kenner G. W., Lee J., Tetrahedron Letters, № 2, 23 (1960).
268. Abraham R.J., Mol. Phys., 4, 145 (1961).
269. Becker E. D., Bradley R. B., Watson C. J., J. Am. Chem. Soc., 83, 3743 (1961).
270. Abraham R. J., Jackson A. H., Kenner G. W-, J- Chem. Soc., 1961,
3468.
271. Becker E. D., Bradley R. В., J. Chem. Phys., 31, 1413 (1959).
272. Cohen L. A., Daly J. W., К n у H., W i t к о p В., J. Am. Chem. Soc., 82, 2184 (1960).
273. Bernstein H. J., Pople J. A., Schneider W. G., Can. J. Chem., 35,
1487 (1957).
274. Briigel W-, Z. Elektrochem., 66, 159 (1962).
275. Schaefer T., Schneider W. G., J. Chem. Phys., 32, 1224 (1960).
276. Freymann M., Freymann R., Libermann D., Compt. Rend., 250, 2185 (1960).
277. Freymann M., Freymann R., Compt. Rend., 248, 677 (1959).
278. Freymann M., Freymann R., Arch, des Sci., 12, 207 (1959).
279. Kowalewski V. J., deKowalewski D. G., J. Chem. Phys., 36, 266 (1962).
280. Baker E. B., J. Chem. Phys., 23, 1981 (1955).
281. Nageswara Rao B. D., Venkateswarlu P., Proc. Indian Acad. Sci., 54, 305 (1961).
282. Shoolery J. N., Discuss. Faraday Soc., 34, 104 (1962).
283. Schaefer T., Can. J. Chem., 39, 1864 (1961).
284. A n e t F. A. L., J. Chem. Phys., 32, 1274 (1960).
285. Katritzky A. R., Lagowski J. M., J. Chem. Soc., 1961, 43.
286. Bonnett R., McGreer D. E., Can. J. Chem., 40, 177 (1962).
287. E 1 v i d g e J. A., Jackman L. M., J. Chem. Soc., 1961, 859.
288. G г о n о w i t z S., Hof f ш an R. A., Arkiv. Kemi., 13, 279 (1958).
289. Gronowi tz S., Hoffman R. A., Arkiv. Kemi., 15 , 45 (1959).
290. Gronow i tz S., Hoffman R. A., Acta Chem. Scand., 13, 1687 (1959).
291. Gronowitz S., Hoffman R. A., Arkiv. Kemi., 16 , 501 (1960).
292. Gronowitz S., Hoffman R. A., Arkiv. Kemi., 16, 515 (1960).
293. Gronowitz S., Hoffman R. A., Arkiv. Kemi., 16, 539 (I960).
294. Takahashi K-, M a t s u к i Y., Mashiko T., Hazato G., Lecture
to the Annual Meeting of the Chemical Society of Japan, Tokyo, April, 1958.
295. Fujiwara S., Katayama M., Hayashi S., Shimizu H., N i s h i -m u r a S., Bull. Chem. Soc. Japan, 32, 201 (1959).
296. Corio P. L., W e i n b e r g I., J. Chem. Phys., 31, 569 (1959).
297. Hoffman R. A., Gronowitz S., Arkiv. Kemi., 16, 471 (1960).
298. Hoffman R. A., Arkiv. Kemi., 17, 1 (1961).
299. Goodwin S., Shoolery J. N., Johnson L. F.,J. Am. Chem. Soc., 81. 3065 (1959).
300. L a u t er b u r P. C., Determination of Organic Structures by Physical Methods, Vol. 2, Editors F. C. Nachod and W. D. Phillips, Academic Press, New York, 1962, Chapt. 7.
301. S 1 о m p G., частное сообщение.
302. S 1 о m p G., Mac К el 1 ar F. A., J. Am. Chem. Soc., 84, 204 (1962).
186
Глава 10
303. S 1 о m р G., McGarvey В. R., J. Am. Chem. Soc., 81, 2200 (1959).
304. Ziircher R.F., Helv. Chim. Acta, 44, 1380 (1961).
305. Hopkins C. Y., Bernstein H. J., Can. J. Chem., 37, 775 (1959).
306. Takeda M., J ardetzky O., J. Chem. Phys., 26, 1346 (1957).
307. J ardetzky O., J ardetzky C. D., J. Biol. Chem., 233, 383 (1958).
308. Bovey F. A., Tiers G. V. D., J. Am. Chem. Soc., 81, 2870 (1959).
309. Curtin D. Y., G о u r s e J. A., Richardson W. H., Rinehart K. L., J. Org. Chem., 24, 93 (1959).
310. Karabatsos G. J., Graham J. D., Vane F. M., J. Am. Chem. Soc., 84, 753 (1962) '
311. J ardetzky C. D., J. Am. Chem. Soc., 82, 229 (1960).
312. Jardetzky C. D., J ardetzky O., J. Am. Chem. Soc., 82, 222 (1960).
313. Crombie L., Lown J. W., J. Chem. Soc., 1962, 775.
314. Musher J. I., С о г e у E. J., Tetrahedron, 18, 791 (1962).
315. Huggins С. M., Pimentel G. C., Shoolery J. N., J. Phys. Chem., 60, 1311 (1956).
316. Cohen A. D., Reid C. J. Chem. Phys., 25, 790 (1956).
317. Reeves L. W., Trans. Faraday Soc., 55, 1684 (1959).
318. Porte A. L.,Gu towsky H. S., Hunsberger I. M., J. Am. Chem. Soc., 82 , 5057 (1960).
319. Reeves L. W., Trans. Faraday Soc., 55, 1684 (1959).
320. Ogg R. A., R a у J. D., J. Chem. Phys., 26, 1339 (1957).
321. Feeney J., Sutcliffe L. H., Proc. Chem. Soc., 118 (1961).
322. Feeney J., Sutcliffe L. H., J. Chem. Soc., 1962, 1123.
323. К e r n C. W., L i p s с о m b W. N., J. Chem. Phys., 37, 260 (1962).
324. Ogg R. A., J. Chem. Phys., 22 , 560 (1954).
325. Gut о ws к у H. S., S a i к a A., J. Chem. Phys., 21, 1688 (1953).
326. Roberts J. D., J. Am. Chem. Soc., 78, 4495 (1956).
327. Watanabe H., I t о K-, К u b о M., J. Chem. Phys., 34, 1043 (1961).
328. Kern C. W., L i p s с о m b W. N., J. Chem. Phys., 37, 275 (1962).
329. For sen S., Acta Chem. Scand., 13, 1472 (1959).
330. Meyer L. H., S a i к a A., G u t о w s к у H. S., J, Am. Chem. Soc., 75, 4567 (1953).
331. Abraham R. J., P о p 1 e J. A., Bernstein H. J., Can. J. Chem., 36, 1302 (1958).
332. Webster D. E., J. Chem. Soc., 1960, 5132.
333. Dippy J. F. J., Chem. Rev., 25, 151 (1939).
334. Green M. L. H., P r a t t L., W i 1 к i n s о n G., J. Chem. Soc., 1958, 3916.
335. Friedel R. A., Wender I., S h u f 1 er S. L., Sternberg H. W-, J. Am. Chem. Soc., 77, 3951 (1955).
336. Stevens R. M., К e r n C. W., Lipscomb W. N.; J. Chem. Phys., 37, 279 (1962).
337. Cotton F. A., Wilkinson G., Chem. and Ind., 1305 (1956).
338. Piper T. S., Wilkinson G., J. Inorg. Nucl. Chem., 3, 104 (1956).
339. Chatt J., Duncanson L. A., Shaw B. L., Proc. Chem. Soc., 343 (1957).
340. Griffith W. P., W i 1 к i n s о n G., J. Chem. Soc., 1959 , 2757.
341. Cotton F, A., Down J. L., Wilkinson G., J. Chem. Soc., 1959, 833.
342. Feeney J., Lee J., Sutcliffe L. H., неопубликованные данные.
343. Milner R. S., P r a t t L., Discuss. Faraday Soc., 34, 88 (1962).
344. Eaton D. R.,Josey A. D., Phillips W. D., Benson R. E., Discuss. Faraday Soc., 34, 77 (1962).
345. Eaton D. R., Josey A. D., Phillips W. D., Benson R. E., J. Chem. Phys., 37, 347 (1962).
346. Eaton D. R., Phillips W. D., Caldwell D. J., J. Am. Chem. Soc., 85, 397 (1963).
347. Primas H., Ernst R., Arndt R., Paper presented at the International Meeting of Molecular Spectroscopy, Bologna, Sept., 1959.
348. Wiberg К. B., N i s t B. J., J. Am. Chem. Soc., 83, 1226 (1961).
349. Brown T. L., Dickerhoof D. W., В a f u s D. A., J. Am. Chem. Soc., 84,
1371 (1962).
350. Hutton H. M., Schaefer T., Can. J. Chem., 40, 875 (1962).
351. Cox J. S. G., Bishop E. O., Richards R. E., J. Chem. Soc., 1960, 5148.
352. Bredenberg J. B., Shoolery J.N., Tetrahedron Letters, 285 (1961).
353. Perrin D. D., P e r r i n D. R., J. Am. Chem. Soc., 84, 1922 (1962).
354. Davison A., McFarlane W., Pratt L., Wilkinson G., J. Chem. Soc., 1962, 3653.
355. S e i f f e r t W., Ang. Chem. Int. Ed., 1, 215 (1962).
356. Eaton D. R., J о s e у A. D., P h i 1 1 i p s W. D., Benson R. E., Mol. Phys., 5, 407 (1962).
Корреляции спектров ЯМР-Н1 со строением молекул
187
357. La Lancette Е. A., Eaton D. R., Benson R. Е., Phillips W. D., J. Am. Chem. Soc., 84, 3968 (1962).
358. Holm R. H., J. Am. Chem. Soc., 83, 4683 (1961).
359. Drake J. E., Jolly W. L., J. Chem. Soc., 1962, 2807.
360. Paterson. W. G., Tip man N. R., Can. J. Chem., 40, 2122 (1962).
361. Ebsworth E. A. V., Turner J. J., J. Chem. Phys., 36, 2628 (1962)
362. Kowalewski V. J., de Kowalewski D. G.,J. Chem. Phys., 37, 2603 (1962).
363. Freymann R., Dvolaitzky M., Jacques J., Compt. Red., 253, 1436 (1961).
364 MacLean С., M а с к о r E. L., Discuss. Faraday Soc., 34, 165 (1962).
365. Jackman L. M., Sondheimer F., Amiel Y., Ben-Efraim D. A., G a о n i Y., Wolovsky R., Bothner-By A. A., J. Am. Chem. Soc., 84, 4307 (1962).
366. Hall G. G., Hardisson A., Proc. Roy. Soc., A268, 328 (1962).
367. Freeman R„ W h i f f e n D. H., Mol. Phys., 4, 321 (1961).
368. Wiberg К- B., L о w r v B. R., N i s t B. J., J. Am, Chem. Soc., 84, 1594 (1962).
369. A n e t F. A. L., Can. J. Chem., 39, 789 (1961).
370. Schaefer T., J. Chem. Phys., 36, 2235 (1962).
371. E 1 1 e m a n D. D., Manati S. L., J. Am. Chem. Soc., 83, 4095 (1961).
372. Kowalewski V. J., de Kowalewski D. G., J. Chem. Phys., 37, 1009 (1962).
373. S h u g J. C., D e с к J. C., J. Chem. Phys., 37, 2618 (1962).
374. Kaesz H. D., Flitcroft N., J. Am. Chem. Soc., 85, 1377 (1963).
375. Anderson J. M., Baldeschwieler J. D.. Dittmer D. C., Phillips W. D., J. Chem. Phys., 38, 1260 (1963).
376. Chien J. C. W., W a 1 к e r J. F., J. Polym. Sci., 45, 239 (1960).
377. Musher J. I., J. Chem. Phys., 34, 594 (1961).
378. S t eh 1 i n g F. С., частное сообщение.
379. N e i к a m W. C., D a i 1 e у В. P., J. Chem Phys., 38, 445 (1963).
380. P о w 1 e s J. G., Polymer, 1, 219 (1960).
381. SI i ch ter W. P., Advances in Polymer Science, 1, 35 (1958).
382. Sauer J. A., Woodward A. E., Rev. Mod. Phys., 32, 88 (1960).
383. Bovey F. A., Tiers G. V. D., F i1i povi ch G., J. Polym. Sci., 38, 73 (1959).
384. О d i у a m a A., J. Phys. Soc. Japan, 14, 777 (1959).
385. Saunders M., Wishnia A., Kirkwood J. G., J. Am. Chem. Soc., 79, 3289 (1957).
386. J ardetzky O., Jardetzky C. D., J. Am. Chem. Soc., 79, 5322 (1957).
387. Bovey F. A., Tiers G. V. D., J. Polym. Sci., Al, 849 (1963).
388. Hung Yu Chen, Anal. Chem. 34, 1134 (1962).
389. Edwards W. R., Chamberlain N. F., J. Polym. Sci., Al,-2299, (1963).
390. Bovey F. A., Tiers G. V. D., Advances in Polymer Science, 3, 139 (1963).
391. Bovey F. A., Tiers G. V. D., J. Polym. Sci., 44, 173 (1960).
392. F о x T. G., Goode W. E., Gratch S., Hu ggett С. M., Kincaid J. F.. Spell A., Stroupe J. D., J. Polym. Sci., 31, 173 (1958).
393. J о h n s e n LL, T e s s m a r K., Kolloid Zeitschrift, 168, 160 (1960).
394. Bovey F. A., Anderson E. W., Douglass D. C., Manson J. A., J. Chem. Phys., 39, 1199 (1963).
395. J о h n s e n U., J. Polym. Sci., 54, S6 (1961).
396. Bovey F. A., Tiers G. V. D., Chem. and Ind., 1826 (1962).
397. Tincher W. C„ J. Polym. Sci., 62, S148 (1962).
398. S t e h 1 i n g F. C., Paper № 45 , Division of Polymer Chemistry, American Chem. Soc. National Meeting, Washington, March, 1962; J. Polym. Sci., A2, 1815 (1964).
399. Satoh S., Chujo R., Ozeki T., Nagai E., J . Polym. Sci., 62, S101 (1962).
400. Brownstein S., Bywater S., Worsfold D. J., J. Phys. Chem., 66, 2067 (1962).
401. Yoshino T., Kyogoku H., Komiyama J., Manabe Y., J. Chem. Phys., 38, 1026 (1963).
402. Kern R.J., Pustinger J. V., Nature, 185, 236 (1960).
403. N i s h i о к a A., Watanabe H., Yamaguchi L, Shimizu H., J. Polym. Sci., 45, 232 (1960).
404. В a w n С. E. H., L e d w i t h A., Quart. Rev., 16, 361 (1962).
405. Gronowitz S.,Gestblom B., Hoffman R. A., Acta Chem. Scand., 15, 1201 (1961).
406. Cox P. F., J. Am. Chem. Soc., 85, 380 (1963).
407. Chapman D., J. Chem. Soc., 1963, 131.
408. Wells P. R., J. Chem. Soc., 1963, 1967.
409. Cram D. J., Singer L. A., J. Am. Chem. Soc., 85, 1084 (1963).
410. Ebsworth E. A. V., Turner J.J., J. Phys. Chem., 67, 805 (1963).
188
Глава 10
411. D г а к е J. Е., J о 1 1 у W. L., J. Chem. Phys., 38, 1033 (1963).
412. Hildebrand J. H., Scott R. L., Solubility of Non-electrolytes, Reinhold, New York, 1950, p. 427.
413. Abraham R. J., J. Chem. Phys., 34, 1062 (1961).
414. Raynes W. T., В u с к i n g h a m A. D., В e r n s t e i n H. J., J. Chem. Phys.,
36, 3481 (1962).
415. Gordon S., Dailey В. P., J. Chem. Phys., 34, 1084 (1961).
416. Buckingham A. D., P о p 1 e J. A., Trans. Faraday Soc., 51, 1173 (1955).
417. P e t г a к i s L., Bernstein H. J., J. Chem. Phys., 37, 2731 (1962).
418. Petrakis L., Bernstein H. J., J. Chem. Phys., 38, 1562 (1963).
419. Abraham R.J., Mol. Phys., 4, 369 (1961).
420. Diehl P., Freeman R., Mol. Phys., 4, 39 (1961).
421. Bothner-By A. A., J. Mol. Spect., 5, 52 (1960).
422. Maher J. P., Evans D. F., Proc. Chem. Soc., 176 (1963).
423. van Meurs N., Spect. Acta, 19, 1695 (1963).
424. Hatton J. V., S c h n e i d e r W. G., S i e b r a n d W., J. Chem. Phys., 39,
1330 (1963).
425. J atdetzky O., J. Am. Chem. Soc., 85, 1823 (1963).
426. Paterson W. G., В i g a m G., Can. J. Chem., 41, 1841 (1963).
427. Cawley S., Danyluk S. S., Can. J. Chem., 41, 1850 (1963).
428. К a s i w a g a H., Niva J., Bull. Chem. Soc. Japan, 36, 405 (1963).
429. К a s i w a g a H., Nakagawa N., Niva J., Bull. Chem. Soc. Japan, 36, 410 (1963).
430. Graham D. M., Holl o.w а у С. E., Can. J. Chem., 41, 2114 (1963).
431. Eaton D. R., Josey A. D., Sheppard W. A., J. Am. Chem. Soc., 85, 2689 (1963).
432. Dewar M. J. S., F a h e у R. C., J. Am. Chem. Soc., 85, 2704 (1963).
433. Wiberg К- B., N i s t B. J., J. Am. Chem. Soc., 85, 2788 (1963).
434. A n e t F. A. L., В a n n a r d R. A. B., Hall L. D., Can. J. Chem., 41, 2331 (1963).
435. McCoy C. R., Allred A. L., J. Inorg. Nucl. Chem., 25, 1219 (1963).
436. Gaines D. F., Schaeffer R., T e b b e F., J. Phys. Chem., 67, 1937 (1963).
437. Memory J. D., С о b b T. B., J. Chem. Phys., 39, 2386 (1963).
438. Horrocks W. D., La Mar G. N., J. Am. Chem. Soc., 85, 3512 (1963).
439. Ebsworth E. A. V., Frankiss S. G., J. Am. Chem. Soc., 85, 3516 (1963).
440. Cheema Z. K., Gibson G. W., Eastham J,F.,J. Am. Chem. Soc., 85, 3517 (1963).
441. Patel D. J., Howden M. E. H., Roberts J. D., J. Am. Chem. Soc., 85, 3218 (1963).
442. Summit! R., Eisih J. J., Trainor J. T., Rogers M. T., J. Phys. Chem., 67, 2362 (1963).
443. Porter R.S., Nicksic S. W., Johnson J. F., Anal. Chem., 35, 1948 (1963).
444. Bishop E. O., Musher J. I., Mol. Phys., 6, 621 (1963).
445. Lumbroso N., W u T. K-, Dailey В. P., J. Phys. Chem., 67, 2469 (1963).
446. Howard В. B., L i n d e г В., E m e r s о n M. J., J. Chem. Phys., 36, 485 (1962).
447. H r u s к a F., В о с к Е., S с h а е f е г Т., Can. J. Chem., 41, 3034 (1963).
448. Schaefer Т., Reynolds W. F., Y onemoto T., Can. J. Chem., 41, 2969 (1963).
449. Reeves L. W., W e 1 1 s E. J., Can. J. Chem., 41, 2698 (1963).
450. E 1 v i d g e J. A., Foster R.G..J. Chem. Soc., 1963 , 590.
451. Franck-Neumann M., Lehn J. M., Mol. Phys., 7, 197 (1964).
452. Brownstein S., Can. J. Chem., 40, 870 (1962).
453. Zweig A., Lehnsen J. E., Lancaster J. E., N e g 1 i a M. J., J. Am. Chem. Soc., 85, 3940 (1963).
454. Shapiro B. L.. Ebersole S. J., Karabatsos G. J., Vane F. M., M a n a t t S. L., J. Am. Chem. Soc., 85, 4041 (1963).
455. Lohr L. L., L i p s с о m b W. N., Inorg. Chem., 3, 22 (1964).
456. Dietrich M. W. Keller R. E., Anal. Chem., 36, 258 (1964).
457. Holm R. H., Chakravorty A., Dudek G. O., J. Am. Chem. Soc., 86,
379 (1964).
458. Chujo R., S a toh S., N a g a i E., J. Polym. Sci., A2, 895 (1964).
459. Hobgood R. T., Goldstein J. H., J. Mol. Spect., 12, 76 (1964).
460. Rausch M. D., M a г к V., J. Org. Chem., 28, 3225 (1963).
461. Ebsworth E. A. V., Turner J. J., Trans. Faraday Soc., 60, 256 (1964).
462. Marcus S. H.. M i 1 1 e r S. L, J, Phys. Chem., 68, 331 (1964).
463. H r u s к a F„ S c h a e f e r T., R e i 1 1 у C. A., Can. J. Chem., 42, 697 (1964).
464. Eaton D., R., Josey A. D., Phillips W. D., Benson R. E., J. Chem. Phys., 39, 3513 (1963).
465. Gil V. M. S., M u r r e 1 1 J. N., Trans. Faraday Soc., 60, 248 (1964).
Корреляции спектров ЯМР-Н1 со строением молекул
189
466. Н о f m a n W., Stefaniak L., Urbanski Т., Witanowski М., J. Am. Chem. Soc., 86, 554 (1964).
467. Williamson К- L,, Lanford C. A., Nicholson C. R., J. Am. Chem. Soc., 86, 762 (1964).
468. Mathis С. T., Goldstein J. H., J. Phys. Chem., 68, 571 (1964).
469. Smith G. W., J. Mol. Spect., 12, 146 (1964).
470. E 1 v i d g e J. A., Foster R. G., J. Chem. Soc., 1964 , 981.
471. Massey A. G., Randall E. W., Shaw D., Spectrochim. Acta, 20, 379 (1964).
472. S t e h 1 i n g F. C., J. Polym. Sci., A2, 1815 (1964).
473. Brownstein S., Wiles D. M.,J. Polym. Sci., A2, 1901 (1964).
474. Bates P., C a w 1 e у S., D a n у 1 u к S. S., J. Chem. Phys., 40, 2415 (1964).
475. Watts V. S., R e d d у G. S., G о 1 d s t e i n J. H., J. Mol. Spect., 11, 325 (1963).
476. Shapiro N. L., Kopchick R.M., Ebersole S. J.,J. Chem. Phys., 39, 3154 (1963).
477. Shapiro B. L., Ebersole S. J., Kopchick R. M., J. Mol. Spect., 11, 326 (1963).
478. H u i t r i с А. С., С a r r J. B., Trager W. F., N i s t B. J., Tetrahedron, 19, 2145 (1963).
479. Laszlo P., von R agne Schle ye r R., J. Am. Chem. Soc., 86, 1171 (1964).
480. Buckingham A. D., Can. J. Chem., 38, 300 (1960).
481. Clark H. C., Kwon J. T., Reeves L. W., Wells E.J., Inorg., Chem., 3, 907 (1964).
482. Mark V., Rausch M. D., Inorg. Chem., 3, 1067 (1964).
483. Levenberg M. I., Richards J . H., J . Am. Chem. Soc., 86, 2634 (1964).
484. Lehn J., Riehl J., Mol. Phys., 8, 33 (1964).
485. Dailey B. P„ J. Chem. Phys., 41, 2304 (1964).
486. Ebersole S., Castellano S., Bothner-By A. A., J. Phys. Chem., 68, 3430 (1964).
487. W u T. K-, Dailey В. P., J. Chem. Phys., 41, 2796 (1964).
488. van Dyke С. H., MacDiarmid A. G., Inorg., Chem., 3, 1071 (1964).
489. Black P. J., Heffernan M. L., Austral. J. Chem., 17, 558 (1964).
490. McCasland G. E., Furuta S., Johnson L. F., Shoolery J. N., J. Org. Chem., 29 , 2354 (1964).
491. Б x а к к a H., Уильямс Д., Применение ЯМР в органической химии, изд. «Мир», М., 1966.
492. Laszlo Р., Musher J.I., J. Chem. Phys., 41, 3906 (1964).
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
493. Молин Ю. Н., Лёшина Т. В., Мамаев В. П., ДАН СССР, 163, № 2, 402 (1965).
494. Лёшина Т. В., Молин Ю. Н., Мамаев В. П., сб. «Реакционная способность органических соединений», 3, вып. 1, 52 (1966).
495. Егорочкин А. Н., Миронов В. Ф., Воронков М. Г., Журн. структ. хим., 7, 450 (1966).
496. Егорочкин А. Н., Хидекель М. Л., Пономаренко В. А., Задорожный Н. А., Изв. АН СССР, ОХН, 1868 (1963).
497. Егорочкин А. Н., Хидекель М. Л., Пономаренко В. А., Зуева Г. Я., Р а з у в а е в Г. А., Изв. АН СССР, ОХН, 373 (1964).
498. Егорочкин А. Н., Хидекель М. Л., Разуваев Г. А., Изв. АН
СССР, Сер. хим., 437 (1966).
499. Егорочкин А, Н., Хидекель М. Л., Разуваев Г. А., Миро-
нов В. Ф., Кравченко А. Л., Изв. АН СССР, ОХН, 1312 (1964).
500. Быстров В. Ф., Степаняиц А. У., J. Mol. Spectr., 21, 241 (1966).
501. Быстров В. Ф., Степанянц А. У., Яблонский О. П., сб. «Кванто-
вая химия и радиоспектроскопия молекул», изд. «Наука», М., 1967 .
502. Белецкая И. П., Федин Э. И., Федоров Л. А., К в а с о в Б. А., Реутов О. А., Изв. АН СССР, сер. хим., 1, 221 (1967).
503. Самитов Ю. Ю., Зав. лабор., № 2, 167 (1962).
504. Машер Дж., А х р е м А. А., ДАН СССР, 134, Ns 2, 354 (1960).
505. Арбузов Б. А., И с а е в а 3. Г., С а м и т о в Ю. Ю., ДАН СССР, 137, Ns 3, 589 (1961).
506. Арбузов Б. А. .Самитов Ю. Ю., Исаева 3. Г., ДАН СССР, 150, № 5, 1036 (1963).
507. Арбузов Б. А., Вильчинская А. Р., Самитов Ю. Ю., Юлдашева Л. К., ДАН СССР, 164, Ns 5, 1041 (1965).
508. Быстров В. Ф., Костяновский Р. Г., Паньшин О. А., Степанянц А. У., Южакова О. А., Опт. и спектроск., 19, 217 (1965).
190
Глава 10
509. Арбуз
510. С а м и т 511. Арбуз в а Л. С а м и т С а м и т Богатский А. В. .Самитов (1966).
Портнова С. Л., Ш е й н к е р
Камер ни цки й А. В., ЖОрХ, 3, 44 (1967). С а м и т № 2, 103 (1964). Л и n п м а а Э., № 4 (1966).
Егорочкин хов Г. Г., М и Яхонтов Л. кер Ю. Н., Рубцов М. В., ДАН СССР, 168, Н есмея в о в А, Н., Бор и со в А. Е., Ф еди Р е у * ‘ ---- -
Разу нов Сами Двор бин Двор. .
в а Л. П., Н е с м е я и о в А. Н., ДАН СССР, 169, № 5, 1083 (1966). Несмеянов А. Н., Кочеткова Н. С., Пет д и н Э. Н., ДАН СССР, 152, 875 (1963).
Несмеянов А. Н.,Кочеткова Н. С., Федин Петровский П. В., ДАН СССР (1968).
Дворянцева Г. Г., Губин С. П., Шейнкер собиость органических соединений, Петрося " ~ " сер. хим., Б е л е ц к а Реутов Петрося Дворянцева Г. Г., кер Ю. Н., Н е с м е я но в А. Н., ДАН СССР, i 66, 868 (1966).
Нес ... - " "
А н
512.
513.
514.
515.
516.
517.
518.
519.
520.
521.
522.
523.
524.
525.
526.
527.
528.
529.
530.
531.
532.
533.
534.
535.
536.
537.
538.
539.
540.
541.
542.
543.
544.
545.
546.
547.
548.
о в Б. А., Самитов Ю. Ю., Tetrahedron Letters, № о в Ю. Ю., ДАН СССР, 164, № 2, 347 (1965).
Ю. Ю., С а н ч у г о в а Н. С., 95 (1965).
Р. М., Журн. структ. хим., 5, № Р. М-, Журн. структ. хим., 5, № Ю. Ю., Г а р к о в ---
о в Б. А., С а м и т о в К-, ДАН СССР, 165, № ов Ю. Ю., Аминова о в Ю. Ю., Аминова
3,
Ю. Н., А х р е м
о
в Ю. Ю., М а
Алла
8, 473 (1963).
Ю л д а ш е -
2, 207 (1964).
4, 538 (1964).
и к Н. Л., ЖОрХ, 2, 1335
А. А., Прохода А. М.,
медов Ф. А., Муси
М., Изв. АН Эст. ССР,
н а
сер.
А. Н., р о н о Н., Мастафанова Л. И.,
А. А., Азерб. хим. журнал,
физ.-мат. и техн.
Разуваев Г. А.,
Хидекель М. Л., в В. Ф., Изв. АН СССР, ОХН, 1521 (1964). ~ ~ к. <
Турчин 1085 (1966). н Э. И., ~
П е
то в О. А., ДАН СССР, 178, 1094 (1968). ваев Г. А., Егорочкин А. Н., X В. Ф., Изв. АН СССР, ОХН, 928 (1964).
т о в Ю. Ю., А м и н_о в а Р. М., ДАН СССР, 156, я н ц е в а
С. П., Ш е й_н к~е р^Ю.Н., ДАН СССР, 160, 1075 (1965).* । я н ц е в а " " ”
и д е к е
л ь
м
Ф.,
т р
М.
наук, 15,
П
Ш
о с я н
Л., М
е т у -
е и н -
В. С.,
и
Р
о -
. № 1, 142 (1964).
Г. Г., П о р т н о в а С. Л., Грандберг К- И.,
Г. Г., Портнова С. Л., Шейн к.ер Ю. Н., Юр
н В. С., Беспалов 2127 (1967).
я И. П., Ф О. А., ДАН н В. С., Р е
е я н о в и с и м о в 174 , 368 (1967). а к о в Н.
А.
К.
Еле
Петров А. А., Е
В.,
Г
ь
ровский П. В., Ф
3, 86 (1966).
Б. П., Реутов
у-
е -
е -
Э. И., Леонова Е. Л.,
Ю. Н., Реакционная спо-
О. А.,
Изв. АН СССР,
Л. А., Федин Э. Н., К 351 (1967).
е д о р о в СССР 174 у то в О. А., ЖОрХ, 3, 2112 (1967).
Турчин К- Ф., Матери кова
в а с о в Б. А.,
Р. Б., Шейн-
Н., Д в о р я Н., К о л о б о
н ц е в а в а
Г. Г., Пустыльник Ж- П., Н. Е., Ш е й н к е р Ю. Н., ДАН СССР,
Опт. и спектроск., 16, 148, 797 (1964).
Лебедев В. Б., Опт. и спектроск., 16,
А.,
В.,
Петров А. л с а к о в 1013* (1964); 17, 679 (1964).
Петров А. А., Елсаков Н. В., 3
Теор. и экспер. хим., 1, вып. 5, С"”
Петросян В. С., Реутов
Петросян В. С., Реутов
Жданов Ю. А., Минкин химии, Изв. Ростовского ун-та,
Г а в а р Р. А., Страд ынь Я- П., Хим. гетероциклич. соед., № 1, 15 (1965).
Петросян В. С., Р е у т о в О. А., ДАН СССР, 175, 613 (1967).
Петросян В. С., Розенберг В. И., Бундель Ю. Г., Реутов О. А., Изв. АН СССР, сер. хим., 1958 (1968).
Сергеев Н. М., Г р и м б е р г А. Н., Журн. структ. хим., 7, № 3, 356 (1966).
К о п т ю г В. А., Р е з в у х и н А. И., 3 а е в Е. Е., АН СССР, ОХН, № 9, 1700 (1963).
Коптюг В. А., Резвухин А. И., Заев Е. Е., М о л и н 34, 3999 (1964).
А х р е м И. С., Ф е д и н Э. И., Квасов Б. А., В о л ь п гоп Letters, 5265 (1967).
Скороходов С. С., Гладковский туров А. С., Изв. АН СССР, ОХН, № 7, Егорочкин А. Н., Лукевиц Э. Я., циклич. соед., 499 (1965).
Егорочкин А. Н., Лукевиц Э. Я., В симпозиум по кремнийорганической химии (1965).
Н.
1авгородный В. С.,Лебедев В.Б., 697 (1965).
О. А., Изв. АН СССР, сер. хим., 1962 (1968).
О. А., Изв. АН СССР, сер. хим., 1361 (1967).
В. И., Корреляционный анализ в органической 1966.
Молин
и н М.
Г. А., Слыв 1273 (1963). Воронков
Ю. Н., Изв.
Ю. Н., ЖОХ,
Е., Tetrahed-
ина С. Г., X а ч а -
М. Г., Хим. гетеро-
оронков М. Г., Международный (Прага), Научные сообщения, 212
Корреляции спектров ЯМР-Н1 со строением молекул
191
549. Д юм а.е в К- М., Смирнов Л. Д., Быстров В. Ф., Изв. АН СССР, ОХН, 883 (1962).
550. Смирнов Л. Д., Лезина В. П., Быстров В. Ф., Д ю м а е в К- М., Изв. АН СССР, ОХН, 752 (1963).
551. Л е з н н а В. П., Быстров В. Ф., Смирнов Л. Д., Дюмаев К- М., Теор. и экспер. хим., 1, 281 (1965).
552. Дюмаев К- М., Смирнов Л. Д., Лезина В. П., Быстров В. Ф., Теор. н экспер. хим., 1, 290 (1965).
553. Смирнов Л. Д., Л е з и и а В. П., Быстров В. Ф., Дюмаев К. М., Изв. АН СССР ОХН, 198, 1836 (1965).
554. Хейфец Г. М., Хромов - Борисов Н. В., Кольцов А. И., ДАН СССР, 166, 3, 635 (1966).
555. Хейфец Г. М., X р ом о в - Б о р и со в Н. В., Кольцов А. И., ЖОрХ, 2, 8, 1516 (1966).
556. Шейнкер Ю. Н., Пересленн Е. М., Кольцов А. И., Баженов Н. М., Волькенштейн М. В., ДАН СССР, 148, 878 (1963).
557. Пересленн Е. М., Шейченко В. И., Шейнкер Ю. Н., ЖФХ, 40, 38 (1966).
558. Егорочкнн А. Н., П е с т у н о в'н ч В. А., Воронков М. Г., Зел-чан Г. И., Хим. гетероциклических соед., 300 (1965).
559. Ziircher R.F., Helv. Chim. Acta, 46, 2054 (1963).
J., Rock S., Steroids, 3, 243 (1964).
560. С о h е n А.
Портнова С. Л., Шейнкер Ю. Н., Ржезннков В. М., Ананченко С. Н., Т о р г о в И. В., ДАН СССР, 166, 125 (1966).
К о ч е т к о в Н. К-, X о р л н н А. Я., Ч и ж Изв. АН СССР, ОХН, вып. 850 (1962).
Ш е й и к е р Ю. Н., П ------
1382 (1964).
Перельсон М. Е., ДАН СССР, 159, № 1, Кузнецова Г. А., •Шейнкер Ю. Н., 566. Еляков Г. Б., Стр № 48, 3591 (1964).
561.
562.
563.
564.
565.
о
в
О. С., Шейченко В. И.,
е к Г. Ю., П е р е л ь с
о
н
е к
М. Е., ДАН СССР, 158, № 6,
Г. Ю., Шейнкер Ю. Н.,
Никонов Г. К., П 154 (1964).
А б ы ш е в А. 3., П е р е л ь с о н М. Е., П е к Г. Ю., Хнмня прнродн. соед., № 5, 310 (1966).
н г и н а Л. И., Уварова Н. И., Tetrahedron Letters,
567. ДзнзенкоА. К-, Заев Е. Е., Е
В., ДАН СССР, 156, № 1, 92 (1964).
А. К-, Н е ф е д о
ляков Г. Б., Молин Ю. Н., Воевод
в а М. Ю., Еляков Г. Б., ДАН СССР, 162, № 3,
Л. А., Д з и з е
н к о
А. К-, Е л я к о в Г. Б., ДАН СССР, 165,
о А.
с к и й В.
568. Дзнзенко 569 (1965).
569. Е л я к о в а 562 (1965).
570. Еляков Г. Б., Д з и з е н к 141 (1966).
571. Елякова Л. А., Дзнзенко прнродн. соед., № 1, 32 (1966).
572. Елякова Л. А., Дзнзенко
К.,
А.
А.
Елкин Ю. Н., Tetrahedron Letters, № 1,
К., Сова
К., Сова
В.-В., Еляков
В.
В., Еляков
Г.
Г.
Б., Хим.
Б., Хим.
прнродн. соед., № 3, 149 (1966).
573. Рыбалко К- С., Шейченко В. И., ЖОХ, 35, 580 (1965).
574. Данилова А. С., Кольцов А. И., ЖОрХ, 1, № 5, 965 (1965).
575. Берлин Ю. А., Васина И. В., Клящнцкнй Б. А., Колосов М. Н., Пек Г. Ю., Пнотровнч Л. А., Чу пру нова О. А., ДАН СССР, 167, 1054 (1966); Tetrahedron Letters, № 14, 1425 (1966).
576. Быстров В. Ф., Иванов В. Т., Овчинников Ю. А., Пек Г. Ю.,
577.
Портнова С. Л., Симпозиум ЮПАК по химии природных соединений, Стокгольм, 1966.
Овчннннко
Acta Chim. Hung., 44, 211 (1965).
578. Б ы с т _ . .
ДАН с т с т с т
579. Б ы
580. Б ы
581. Б ы
63
582. Б ы
в Ю. А., Иванов В. Т., П е к Г. Ю., Шемякин М. М.,
В. Ф., Дюмаев К- М., Лезина В. П., Никифоров Г. А., 148, 1077 (1963).
В. Ф., Лезина
В.
В.
Ф., Ф.,
ров СССР, ров ров ров
(1964). с т р о в н я и ц А.
583. X и д е к е л р о ж н ы й Н. А., Р а ИЗО (1963).
584. Егорочкнн А. Н.,
Л е з н н а Г р а и д б е
В. У., ь М. Л., Е
Ф-, Ю ж а
В. П., В. П., р г И.
Опт. и спектроск., 16, 1004 (1964).
Опт. и спектроск., 16, 790 (1964).
И., Шарова Г. И., Опт. и спектр., 17,
вский Р. Г., Паньшин О. А., С т
е п а -
К о с т я и о
к о в а О. А., Опт. и спектроск., 19, 217 (1965).
горочкнн А. Н., Пономаренко В. А., 3 а дозу в а е в Г. А., Петров А. Д., Изв. АН СССР, ОХН,
Хндекель М. Л., Пономаренко В. А., 3 у е -
в а Г. Я., Свнрежева С. С., Разуваев Г. А., Изв. АН СССР, ОХН, 1865 (1963).
192
Глава 10
585. Нефедов О. М., Колесников С. П., Шейченко В. И., ДАН СССР, 162, 589 (1965).
586. Заев Е. Е., Скубневская Г. И., Молин Ю. Н., Ж. структ. хим., 6, № 4, 639 (1965).
587. Заев Е. Е., Ларионов С. В., Молин Ю. Н. , ДАН СССР, 168, № 2, 341 (1966).
588. Заев Е. Е., М о л и н Ю. Н., Жидомиров Г. М., Воеводский В. В., ДАН СССР, 1967.
589. Слоним И. Я., Любимов А. Н., Ядерный магнитный резонанс в полимерах, изд. «Химия», М., 1966.
590. Лишаиский И. С., Зак А. Г., Федорова Е. ф., Хачатуров А. С., Высокомолек. соед., 7, 966 (1966).
591. К о т о н М. М., Андреева И. В., Гетманчук Ю. П., М а д о р -с к а я Л. Я., Покровский Е. И., Кольцов А. И., Филатова В. А., Высокомолек. соед., 7, 12, 2039 (1965).
592. Баженов Н. М., В ол ькеиштейн М. В., Хачатуров А. С., Высокомолек. соед., 5, № 7, 1025 (1963).
593. ДворянцеваГ. Г., Лезина В. П., Быстров В. Ф., Ульянова Т. Н., С ы р о в а Г. П., Шейнке р'Ю. Н., Изв. АН СССР, сер. хим., 994 (1968).
594. К о п т ю г В. А., РезвухинА. И., Шубин В. Г., Корчагина Д. В., ЖОХ, 35, 864 (1965).
595. К о п т ю г В. А., РезвухинА. И., Крисин А. П., Исаев И. С., Жури, структ. хим., 8, 624 (1967).
ГЛАВА 11
Исследование ядерного магнитного резонанса фтора
11.1. Введение
Быстрый рост числа научных публикаций, посвященных различным аспектам спектроскопии магнитного резонанса на ядрах F19, особенно в последние годы, свидетельствует о повышении интереса к этой области исследований. Химические сдвиги и константы спин-спинового взаимодействия для фтора намного больше, чем для ядер водорода. Поэтому исследование фторсодержащих молекул часто позволяет получить о структуре и динамических процессах, происходящих в молекулах, более детальные сведения, чем могут дать аналогичные исследования протонсодержащих молекул (см. разд. 9.5.5 и 11.13.3). Теоретические представления о природе химических сдвигов и констант спин-спинового взаимодействия для фтора менее глубоки, чем для протонов, главным образом из-за сложности электронной конфигурации фтора.
Фтор с атомным весом 19 — единственный встречающийся в природе изотоп этого элемента — является идеальным объектом для спектроскопии ЯМР. Несмотря на то что линии поглощения F19 несколько менее интенсивны и обычно более склонны к насыщению по сравнению с линиями протонного резонанса, спектры ЯМР-F19 могут быть легко получены с помощью стандартных спектрометров ЯМР, выпускаемых промышленностью. Спин I ядра F19 равен у, и потому квадрупольный момент отсутствует. Благодаря одновалентности и высокой реакционной способности фтор образует большое число соединений, которые удобны для исследования; к тому же многие из этих соединений при температуре 25° являются жидкостями. Диапазоны наблюдаемых значений констант спин-спинового взаимодействия и химических сдвигов для фтора намного шире, чем для протонов, поскольку для Н1 и F19 они определяются различными факторами. Интенсивность сигналов в чистых фторсодержащих жидких соединениях достаточно высока, что позволяет получить хорошее разрешение (в спектрах ЯМР-F19 при частоте 56,4 Мгц можно довольно легко разрешить линии с расщеплением 0,3 гц). Соединения, содержащие несколько неэквивалентных групп ядер фтора, между которыми существует косвенное спин-спиновое взаимодействие, в большинстве случаев дают простые спектры первого порядка (см. рис. 11.1), поскольку химические сдвиги между этими группами велики по сравнению с соответствующими константами спин-спинового взаимодействия. Большое различие между значениями химических сдвигов и констант спин-спинового взаимодействия, наблюдаемое в большинстве спектров ЯМР-F19, вызывает необходимость использования двух скоростей развертки спектра: большой (~30 гц-сек-1) для измерения химических сдвигов и малой (~0,2 гц-сект1, как для протонных спектров) для определения параметров тонкой структуры линий.
При изучении спектров ЯМР-F19 используются как внутренние, так и внешние эталоны. В качестве внешнего эталона чаще всего применяется трифторуксусная кислота. Большинство химических сдвигов, определенных 13—489
194
Глава И
по отношению к этому стандарту, приводится в литературе без учета диамагнитных поправок (несмотря на то что эти поправки малы, ими не всегда можно пренебрегать) [1]. Филипович и Тирс [2] показали, что химические сдвиги F19 следует определять при бесконечном разведении в трихлорфторметане CC13F, используя это вещество в качестве внутреннего эталона (см. разд. 7.2.2). Растворы в CCI3F с концентрацией менее 10% можно рассматривать как
н
(CF3)ZCF Вг
%г-сг^ 8,9гц
+1,15 м.Э.
+ 66,9 мл).
Рис. 11.1. Спектр ЯМР-F19 (CF3)2CFBr при частоте 56,4 Мгц.
Химические сдвиги указаны по отношению к внешнему эталону CF3COOH.
растворы с бесконечным разведением. В тех случаях, когда исследование разбавленных растворов проводить не удается, можно воспользоваться экстраполяцией к бесконечному разведению. Значительное несоответствие между величинами химических сдвигов фтора, измеренными относительно внешнего и внутреннего эталонов (табл. 11.1), указывает на необходимость введения единой системы эталонирования резонансных спектров F19 с использованием внутреннего эталона.
Таблица 11.1
ХИМИЧЕСКИЕ СДВИГИ F19, ИЗМЕРЕННЫЕ ОТНОСИТЕЛЬНО CCI3F, ИСПОЛЬЗОВАННОГО в КАЧЕСТВЕ ВНУТРЕННЕГО И ВНЕШНЕГО ЭТАЛОНА [2]
Соединение ^внешн a Фвнутр 6 Разность Д(р
C6H5SO2F -65,38 -65,497 +0,12
CFBr3 -8,85 -7,397 — 1,45
BrCF2CF2Br +63,32 +63,403 +0,08
C6H5CF3 +63,95 +63,747 +0,20
CFC12CFC12 +67,27 +67,750 -0,48
CF3CO2H +78,45 +76,539 + 1,91
C6H6F + 113,68 + 113,123 +0,56
(CF2)4 + 138,03
(C2H3)3SiF + 176,78 + 176,24 +0,54
h-C6Hi3F +219,42 +219,022 +0,40
CF2Br2 —6,768
CF3CCI3 +82,204
а Фвнешн — химические сдвиги (м. д.) для чистых жидкостей, измеренные относительно внешнего эталона CCI3F.
б Фвнутр—химические сдвиги (м. д.) относительно внутреннего эталона CClgF, экстраполированные к бесконечному разведению в этом соединении.
Исследование ядерного магнитного резонанса фтора 195
Эванс [1] исследовал влияние растворителей на экранирование ядер F19. В некоторых случаях это влияние было настолько велико, что его нельзя было объяснить изменениями объемной диамагнитной восприимчивости (которые учитывались). Эванс предположил, что причиной дополнительного экранирования может служить увеличение парамагнитного члена, обусловленное присутствием полярных молекул растворителя, и рекомендовал осторожно относиться к применению CC13F в качестве внутреннего эталона. В тех случаях, когда необходимо получить очень точные значения химических сдвигов F19, другие авторы [3] предлагают проводить экстраполяцию к бесконечному
Таблица 11.2
ХИМИЧЕСКИЕ СДВИГИ В СПЕКТРАХ F19 НЕКОТОРЫХ ФТОРОРГАНИЧЕСКИХ СОЕДИНЕНИЙ, ОБУСЛОВЛЕННЫЕ БЕСКОНЕЧНЫМ РАЗВЕДЕНИЕМ В БРОМОФОРМЕ И ЧЕТЫРЕХХЛОРИСТОМ
УГЛЕРОДЕ (ИЗМЕРЕННЫЕ ПО ОТНОШЕНИЮ К РАСТВОРАМ В ГЕПТАНЕ) [1]
Соединение CHBr3 ссц • Соединение СНВгз ССЦ
6a ^коррб 6a ^KOpp б 6a ^KOpp 6 6a ^коррб
,'CH3)3CF 5,95 5,18 2,55 2,32 c6h5cf3 4,03 3,26 1,70 1,47
cf2 = cci2 4,87 4,10 2,01 1,78 CC13F 2,83 2,06 1,30 1,07
cf4 4,55 3,82 1,95 1,72 (CC12F)2 2,71 1,94 1,05 0,82
C6HhF (C6H6)3CF 4,53 4,03 3,76 3,26 1,88 1,70 1,64 1,47 CBr3F 1,57 0,80 0,81 0,58
а б (м. д.) измерены относительно соответствующих линий для разбавленного раствора в гептане.
б 6КОрр — значения химических сдвигов с учетом поправок на объемную восприимчивость.
разведению для нескольких различных растворителей. Увеличение парамагнитного члена при растворении обусловлено, по-видимому, изменением средней энергии электронного возбуждения ДЕ, возникающим из-за возрастания дисперсионных сил взаимодействия между молекулами вещества и растворителя [1]. В некоторых других системах была обнаружена приблизительно такая же зависимость химических сдвигов F19 от растворителя: с уменьшением концентрации раствора наблюдалось постепенное уменьшение экранирования ядер фтора [1]. В табл. 11.2 приведены значения химических сдвигов для различных фторорганических соединений, растворенных в СНВг3 и СС14. Особенно большое изменение химических сдвигов F19 было отмечено при исследовании бензотрифторида в интервале температур от —29 до 159° (при повышении температуры наблюдалось линейное возрастание экранирования с коэффициентом 1,06-10 2 м. д.1град) [1]. Увеличение давления газообразного пер-фторметана также сопровождалось изменением химического сдвига [при этом экранирование уменьшалось с коэффициентом (1,15 + 0,15)-10-2 м. д./атм]. Таким образом, увеличение расстояний между молекулами, наблюдаемое при нагревании образца или понижении давления, приводит к увеличению экранирования ядер фтора, что согласуется с ослаблением дисперсионных сил при увеличении межмолекулярных расстояний в системе [1]. В молекулах, которые содержат атомы ([пора, находящиеся в непосредственной близости от других групп, наблюдаются внутримолекулярные взаимодействия, подобные упомянутым выше межмолекулярным взаимодействиям. Если внутренние вандерваальсовы силы между атомами велики, экранирование ядер фтора уменьшается. Ховард и сотр. [4] оценили влияние межмолекулярных дисперсионных сил на химические сдвиги F19. Вычисленные ими поправки находятся в пол у количественном согласии с экспериментальными значениями.
13*
196
Глава И
Глик и Эренсон [3] нашли соответствие между «исправленными» значениями сдвигов, обусловленных растворителем, и молярными поляризуемостя-ми молекул растворителя для некоторых галогензамещенных метанов. Эванс [1], однако, показал, что при определении химических сдвигов, обусловленных растворителем, важную роль играют также размеры молекул последнего, и корреляция, наблюдавшаяся Гликом и Эренсоном, обусловлена поляризуе-мостями галогензамещенных метанов, отнесенными к их молярным объемам. Об этом свидетельствует тот факт, что многие сдвиги, вызванные растворителем, зависят от температуры, в то время как молярные поляризуемости от температуры не зависят. К такому же заключению пришел Ховард [4] в результате теоретического исследования влияния межмолекулярных дисперсионных сил на химические сдвиги F18.
Химические сдвиги фтора, приведенные в этой главе, считаются положительными, если соответствующая линия поглощения смещена в сторону более сильного поля относительно сигнала эталона.
11.2. Химические сдвиги ядер фтора
Как было показано выше, основной вклад в экранирование ядер фтора вносит парамагнитный член второго порядка в выражении, полученном Рэмзи для экранирования ядер (см. разд. 4.6). В большинстве фторсодержащих соединений вклад диамагнитного члена составляет только 1% наблюдаемого химического сдвига, в то время как парамагнитный член, согласно Сайка и Слихтеру, имеет значительную величину во всех соединениях, кроме полностью ионизованных фторид-ионов [5]. Величина парамагнитного сдвига зависит от степени ионности связи, и, как было показано на примере большого ряда бинарных фторидов [6] (для которых экранирование атомов фтора уменьшалось с ростом электроотрицательности присоединенных атомов), теория согласуется с экспериментом. Рост электроотрицательности присоединенного атома приводит к уменьшению ионного характера связи между ним и атомом фтора, увеличивая тем самым парамагнитный вклад в экранирование. Существуют, однако, ионные соединения фтора (см. нитрозилфторид, разд. 11.20), в которых наблюдается сильное парамагнитное дезэкранирование, обусловленное наличием низколежащих электронных энергетических уровней с соответствующей симметрией [7]. Таким образом, ' кроме электроотрицательности заместителя, на экранирование ядер фтора могут влиять другие факторы. Эванс предположил, что внутримолекулярные дисперсионные силы, действующие между присоединенными группами и ближайшими атомами фтора, могут изменить экранирование за счет увеличения парамагнитного вклада. Дисперсионные силы особенно велики, когда рассматриваемые заместители находятся в непосредственной близости от атома фтора, и, следовательно, эффект будет наиболее заметным для групп большого размера. Было отмечено, что объемистые группы действительно оказывают влияние на экранирование ядра фтора, причем противоположное тому, которого можно было бы ожидать исходя из соображений электроотрицательности заместителей [8]. Дополнительный вклад в экранирование ядер фтора вносит появление частичной двоесвязности и ионных свойств в насыщенных галогензамещенных фторуглеродных производных 19, 10]. Эти эффекты также приводят к химическим сдвигам ядер фтора в направлении, противоположном определенному электроотрицательностью заместителей. Можно рассчитать, однако, вклады в химический сдвиг F18, обусловленные как дисперсионными силами при наличии групп большого размера, так и частичной двоесвязностью, учитывая низколежащие электронные энергетические уровни. Если эти уровни обладают симметрией, допускающей смешивание с основным состоянием, то это может привести к значительному парамагнитному экранированию. Бутовский и Хоффман показали также, что использование орбиталей с более высокой энергией в гибридных
Исследование ядерного магнитного резонанса фтора 197
орбиталях атомов, присоединенных к фтору, приводит к возрастанию экранирования ядра F19 [6].
Во фторсодержащих молекулах с сопряженными связями разница в экранировании ядер F19 определяется главным образом плотностями зарядов л-электронов и порядками связей С — F [172, 173]. Дело обстоит иначе в случае ядер фтора, находящихся в орто-положении по отношению к заместителям в ароматических кольцах, где заметный вклад в экранирование вносят, по-видимому, вандерваальсовы дисперсионные эффекты [189].
Тирс сообщил о влиянии изотопного замещения водорода дейтерием на химические сдвиги фтора: сигнал от группы —CF2D в «-C3F7D оказался смещенным на 0,60 м. д. в сторону более сильного поля по отношению к резонансной линии «-C3F7H. Гутовский [12] отнес это увеличение сдвига за счет различия в амплитудах колебаний атомов водорода и дейтерия в этой молекуле. Изотопный эффект наблюдался также в системах F — С — D(H) [179], F —С13(С12) [13, 80, 81], F —С —CF(C12) [13, 80, 81], F — Si29(Si28) [14], F — S33(S32) [180] и F — S34(S32) [180]. В ряде случаев ядро фтора, присоединенного к более тяжелому атому, экранируется сильнее. Это согласуется с предсказанными различиями в амплитудах нулевых колебаний: более тяжелый изотоп обладает меньшей амплитудой колебаний, что приводит к ослаблению поляризации электронов атома фтора и увеличению экранирования его ядра [180].
11.3. Константы спин-спинового взаимодействия F19
В гл. 5 показано, что косвенное спин-спиновое взаимодействие F — F определяется не только контактным взаимодействием Ферми; значительный вклад может вносить также дипольный эффект. Поэтому теоретический расчет констант спин-спинового взаимодействия с ядрами фтора сопряжен с известными трудностями. Однако для нескольких простых систем в этом направлении был достигнут определенный успех (см. разд. 5.8). Попл [164] рассчитал константы взаимодействия F — F и Н — F для ряда фторсодержащих соединений и показал, что электрон-орбитальное взаимодействие хотя и вносит заметный вклад, но не играет определяющей роли для константы спин-спинового взаимодействия. Это видно из табл. 5.13, в которой экспериментально наблюдаемые значения Jff и JHf [9, 19, 32—34] сопоставлены с вычисленными значениями вкладов, обусловленных электрон-орбитальным взаимодействием (см. разд. 5.8.2).
Константы взаимодействия с ядрами фтора не всегда монотонно уменьшаются с ростом числа связей между взаимодействующими ядрами. В качестве примера можно рассмотреть спектр ЯМР-F19 соединения (CF3)2NCF2CF3^ в котором ядра фтора группы CF2 очень слабо взаимодействуют с ядрами соседней группы CF3 (Jcf2-cf3 <1 гЧ), но сильно взаимодействуют с более удаленной группой (CF3)2N (J = 16 гц) [15]. Подобный эффект наблюдался также в спектре перфторметилциклогексана СР3СвРи: ядра фтора группы CF3 сильнее взаимодействуют с двумя соседними кольцевыми группами CF2, чем с атомом фтора, присоединенным к тому же атому углерода; это можно видеть из спектра на рис. 11.2, представляющего собой квинтет дублетов (JCf3-cf2 — 14,2 гц и Jcf3-cf = 6,1 гц) [16]. Такая же явная аномалия наблюдается, как правило, для фторалканов [17], дифторбензолов [19] и циклобутанов [18]. Для фторалканов подобное поведение констант взаимодействия является следствием слабого взаимодействия между ядрами атомов фтора, присоединенных к соседним атомам углерода [17, 201. Некоторые примеры очень малых констант взаимодействия F — F приведены в табл. 11.3. Полагают, что значение константы взаимодействия F — F в значительной степени зависит от пространственного удаления взаимодействующих атомов фтора [17] и что малость вицинальных констант для перфторэтильных групп является
198
Глава И
следствием большого удаления метильных и метиленовых ядер фтора друг от друга (2,73 А). Константы спин-спинового взаимодействия между ядрами фтора, находящимися в положениях 1,3 свободной углеродной цепи, когда при некоторых конформациях расстояния между ядрами фтора сокращаются, имеют значение 7—10 гц. Замещение промежуточного атома углерода в цепи атомом азота увеличивает константу взаимодействия до 10—17 гц. В циклических соединениях ядра атомов фтора в тех же положениях имеют, как правило меньшие константы взаимодействия. Для замещенных этанов константы взаимодействия между фторами при соседних атомах углерода гораздо больше, чем в перфторэтильных производных (например, в CF2BrCBrFCl Jcf2-cf = = 18 гц). Стерические взаимодействия в таких молекулах, по-видимому,
и
Рис. 11.2. Спектр ЯМР-F19 группы CF3 чистого перфторметилциклогексана при частоте 56,4 Мгц.
приводят к сближению атомов фтора, увеличивая тем самым спин-спиновое взаимодействие F — F [17]. Эванс [22] определил относительные знаки констант Jf-f для замещенных фторэтанов и показал, что константа Jffu , с одной стороны, и обе константы Jpp и Jpp , с другой стороны, имеют противоположные знаки. Доказательство того, что взаимодействие F — F осуществляется по механизму пространственного взаимодействия, основывается главным образом на экспериментально наблюдаемой зависимости численных значений констант Jpp от расстояния между взаимодействующими ядрами. Однако если принять во внимание разные относительные знаки констант взаимодействия F — F, то установить простую зависимость между величиной константы и расстоянием между ядрами не удается. Это доказывает, что взаимодействие между ядрами фтора осуществляется в основном через химические связи, а не через пространство. В работе [198] было показано, что для геминальных констант, по-видимому, в одинаковой мере имеют значение оба механизма; в случае вицинальных констант в качестве основного механизма рассматривается взаимодействие через химическую связь, и наконец, если ядра разделены более чем тремя связями, то для объяснения взаимодействия привлекается пространственный механизм х). Можно ожидать, что вклад механизма взаимодействия через химическую связь будет уменьшаться с ростом электроотрицательности заместителей. Такая тенденция действительно была обнаружена для констант Jepp в широком ряду фторэтанов.
Влияние температуры на константы взаимодействия. Гутовский и сотр. [192] показали, что колебательные движения атомов должны влиять на константы взаимодействия, и поэтому следует ожидать заметной зависимости констант от температуры. Такие изменения наблюдались для констант взаимодействия F — F и Н — F [191]. Для молекулы CF3CFC12, конформация кото-
Возможность передачи спин-спинового взаимодействия через пространство рассмотрена для F19 — F19 в работах [263, 264], для Н1 — F19— [265, 266].— Прим. ред.
Таблица 11.3
КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ F19 ВО ФТОРАЛКАНАХ [17]
№ п/п и литература Соединение Константы взаимодействия, гц
1а cf3-cf2 >N-CF3 CF3— CF/ a b c Jab~ 10,2 Jac —6,8 Дс= 15,8
2а CF3 — CF24 \n —CF2 —CF3 cf3-cf2/ a b b a Jdb + Jabf ~ 13,6
За CF3—CF2—0—CF2—CF3 a b ba Jab + Jab'
4 [162] CF3—CF2—COOH a b Jab = 1,38
5 [15] ZCFs CF3— CF2N< a b c i o*'< p p"4 O Q Cy " Va О __ CP
6 CF3-CF2-CF2-NF2 a b c d LQ C4 " да' — 2 — V II II II II V Л Q 'в o 'tJ 43
7 CF3—CF2—CF2—COOH a b c Jab 1. J ac = 9,9 Jbc 1
8 CF3 — CF2 — CF2—COOC H3 a b c p" A 11 A H- <© H-o
9 CF3—CF2 —CFCH3 a b c ab A 1 Ac = 10,8 Jbb' =270,4 Ac=14,6 Ада=14,6
10 (CF3)3CF a b Jab = 4,0
200
Глава 11
Продолжение табл. 11.3
№ п/п и литература Соединение Константы взаимодействия, гц
11 CF3-CF2-CF2H(D) а Ь с J ab 'С 1 J(LC 1 ^&с = 4,0 (4,5)
12 zCF2 — cf2. -CF3-N< V xCF2-CF2/ a be Jab- 13,6
13 ,cf2—cf2. CF3—Ny >CF2 \cf2-cf2/ a bed /аЬ=16,4
14 /CF2-CF2 cf3-cf2-n< | \cf2-o a b c e d LQ LO оэ 02 1О V II II V II II V V V II hO V'tJ Ь О "У D-a у
15 [163] C6H6-CH-CH2 1 1 cf2-cf2 a b /А А
16 [34] CF2Br — CFBrCl a c b Jab= 159 J ас — 13 ^Ъс = 14
17 [46, 48]
CF2Br- CFHC1
а с b
Jab=177
J ас = 18
Jbc = 18
а Вследствие сильного взаимодействия между группами СР2 ядра фтора трифторме-тильиых групп CFg «видят» только комбинированные спиновые состояния групп CFg, и потому из мультиплетного расщепления линий в спектре можно определить лишь сумму двух констант: Jab + Jab. [21].
рой не оказывает влияния на константы взаимодействия, величина JFF уменьшается от 6,1 гц при —65° до 5,6 гц при 90°. Сходным образом для CF3CF2COOH значение JFF составляет 1,7 гц при —29° и 1,3 гц при 85°. Отсюда следует, что изучение конформационных состояний молекул, опирающееся на измерения зависимости констант взаимодействия от температуры, может привести к ошибочным результатам, если предполагать, что константы для различных изо-
Исследование ядерного магнитного резонанса фтора
201
меров от температуры не зависят. Подобные эффекты наблюдались для фтор-алкенов [191] типа CF2 = CFCOF, для которого при —105° могут быть выморожены два различимых методом ЯМР изомера:
П
1------<2,0гч-
Значения Jab и Jax, наблюдаемые при —105° для каждого из изомеров, оказываются меньшими, чем их средние значения при комнатной температуре (Jab = 6,0 гц и Jax = 36,8 гц). Это можно объяснить, только предположив, что константы взаимодействия в отдельных изомерах зависят от температуры [191]. На константы взаимодействия F — F могут влиять также изменения растворителя [201].
Константы дальнего взаимодействия F19. Измерено [190] большое число констант дальнего взаимодействия для ядер фтора; некоторые из них приведены в табл. 11.4.
Таблица 11.4.
КОНСТАНТЫ ДАЛЬНЕГО ВЗАИМОДЕЙСТВИЯ F —F [190]
Соединение Константы взаимодействия, гц
Н—C—C—F Н—С—С—С—F F--F
CH3C(O)F 6,9
CF3CH2C1 8,5
CF3CH2Br 9,0
CF3CH2OH 8,9
CF3CH3 12,8
CF2C1CH3 14,8
(CH3)3CF 20,4
n-CH3C(O)CeH4F 8,7 5,5
n-NO2CeH4F 8,2 4,8
n-CH3OCeH4F 7,8 4,8
n-CF3C6H4F 8,5 5,1
n-BrCeH4F 8,1 4,9
CF3 —C=C —CF3 2,2
(CF3)2O 8,0
(CF3)3N 10,8
cf3 = sf2 10,7
Константа дальнего взаимодействия Н — F между ядрами, разделенными пятью насыщенными связями, наблюдалась в спектре 1,1-дифтор-2,2-дихлор-З-фенил-З-метилциклобутана [37]
С12 f2
Сн/
Н2
I
202
Глава 11
В этой молекуле два атома фтора неэквивалентны, и один из них взаимодействует с протонами группы СН3, находящейся в положении 3 (JHf = 2 гц). При исследовании спектра этой молекулы, стереоспецифически меченной дейтерием в положении 4, удалось установить, что с метильной группой, по-видимому, взаимодействует фтор, находящийся в цис-положении относительно метильной группы. Другие авторы [38] измерили константы дальнего взаимодействия Н — F через 5 и 6 насыщенных связей в стероидах типа
II
В спектре 6-0-фторстероида (II) наблюдается взаимодействие между ядром фтора и ангулярной метильной группой при С(19) с константой Jhf = 4,3 гц, причем взаимодействующие группы также находятся в цис-положении. Таким образом, вполне возможно, что механизм дальнего взаимодействия Н — F в насыщенных системах связан со стереоспецифическими взаимодействиями.
В большом числе случаев константы дальнего взаимодействия F — F были обнаружены между ядрами, разделенными 5 насыщенными связями. Константа взаимодействия между ядрами фтора через 6 связей была найдена в спектре ЯМР для (СРз)2МСР2СРВгСРз (Jf-c-n-c-c-cf = 1,7 гц) [174] х).
11.4. Бинарные соединения фтора
Как уже было указано выше, увеличение электроотрицательности атома, непосредственно связанного с атомом фтора в бинарных фторидах, приводит к уменьшению экранирования F19 в ряду соединений с элементами, имеющими одинаковую валентность и принадлежащими 'к одной группе периодической таблицы. Этот вывод подтверждается приведенными в табл. 11.5 данными по химическим сдвигам F19 для некоторых бинарных фторидов. Графики зависимости химических сдвигов фтора от электроотрицательностей присоединенных элементов для нескольких рядов бинарных фторидов, приведенные на рис. 11.3, почти во всех случаях представляют собой приблизительно прямые линии [6]. Внутри каждой группы при увеличении заряда ядра элемента М магнитное экранирование присоединенного атома фтора уменьшается. Существует несколько исключений из этого общего правила: например, в молекуле SFe ядра фтора экранированы сильнее, чем в SeFe. В некоторых случаях с помощью спектра ЯМР-F19 можно подтвердить молекулярную структуру бинарного соединения. Спектр пентафторида иода состоит из двух линий с отношением интенсивностей 4:1. Это позволяет сделать вывод, что молекула IF5 имеет форму тетрагональной пирамиды, которая была установлена ранее другими методами [40]. Аналогичный спектр ЯМР-F19 и, следовательно, аналогичную структуру имеет молекула BrF5. В обоих случаях наблюдается мультиплетность линий поглощения, обусловленная спин-спиновым взаимодей-
х) В спектре Fq —— CFg = CF4CI наблюдалось спин-спиновое взаимодействие между ядрами фтора Fc и Рд, разделенными семью химическими связями (Jдс = = 2,8 гц для гщс-изомера и 2,0 гц для транс-изомер а) [229].— Прим. ред.
Исследование ядерного магнитного резонанса фтора
203
Таблица 11.5
ХИМИЧЕСКИЕ СДВИГИ F19 ДЛЯ БИНАРНЫХ ФТОРИДОВ а [6,41]
Соединение ^СРзСООН (внешн)’ м. д. [41] ^F2 (внешн)’ м. Д. [6] Взаимодействие M—F
M JjVlF’ гч
NF3 -219 4-285,0 N14 160
C1F3 -193; -81 4-343,4
BrF3 -54,3 4-461,1
PF3 -42,3 4-463,7 p31 1441
AsF3 -35,0 4-469,1
SbF3 -23,9 4-507,9
BF3 +54,2 +555,5 ВИ 10
sf4 -195; -148
SeF4 -141
TeF4 -51,4
cf4 -11,9 +491,0
bf4- 4-71,0
SiF4 4-833 +598,9 Si" 178
GeF4 4-99,0 +608,8
BrF5 -349; -219 + 290,5; +152,9
if5 -138; -95,8 + 418,2; +368,9
pf5 -0,7 +502,1 p31 916
AsF5 -11,3
SbF5 4-6,8; 4-26,2 +537,3
4-52
MoFg -355 Mo95, Mo97 44
WFg -242 W183 48
SeFg -128 + 372,7 Se77 1400
SFg -127 + 375,6
TeFg -20,6 + 485,9 Те125 3688
Tei23 3052
PFr -11,6 рз1 710
SiF*- 4-49,8 Si" 110
AsF^ -18,1 As73 930
SbF6- 4-32,3 Sb‘2i 1843
BeF2 + 599,1
F“ +548,2
f2 0,0
HF +625,0
IF7 +261,5
а Данные, приведенные в таблице, позволяют найти приближенное соотношение между сдвигами, определенными относительно двух эталонов: ^сРзСООН=^р2 (внешн)~ 504 м. д.
ствием: менее интенсивная из двух линий сдвинута в область слабого поля. Перекрывание мультиплетных линий в спектре F19 гептафторида иода (Jjf = = 2100 гц- см. [200]) не дало возможности однозначно подтвердить пента-гональную бипирамидальную структуру этой молекулы [40].
При изменении валентности элемента, присоединенного к фтору, для бинарных фторидов наблюдаются изменения в экранировании ядер фтора (см. химические сдвиги для тригалогенидов и пентагалогенидов фосфора и сурьмы в табл. 11.5). Увеличение экранирования ядра фтора, присоединен-
204
Глава 11
лого к элементу, находящемуся в состоянии с высшей валентностью, обусловлено необходимостью использования кроме р-орбиталей в гибридных состояниях атомов фосфора и сурьмы s- и d-орбиталей, имеющих более высокую энергию. Аномальное соотношение между химическим сдвигом для SFe и SeFe можно объяснить, пользуясь такой же аргументацией: в обеих молекулах имеет место б?2хр3-гибридизация, но разность между энергиями Sd-орбиталей и 3s-, Зр-орбиталей атома серы больше, чем та же величина для 4d- и 4s-, 4р-орбиталей селена, что и приводит к более сильному экранированию атомов фтора в SFe по сравнению с SeFe. Следует заметить, что фториды галогенов не
Рис. 11.3. Зависимость химических сдвигов фтора для бинарных фторидов MFX от электроотрицательности атома М [6].
укладываются в рамки этих представлений, поскольку в состояниях с высшей валентностью в этих соединениях наблюдается меньшее экранирование ядер фтора [6].
Более слабое экранирование ядра атома фтора, расположенного на вершине пирамиды, в молекулах BrF5 и IF5 объясняется тем, что соответствующая связь имеет в основном р-характер, в то время как другие связи имеют сРр1-гибридизацию.
Муттертис и Филлипс [41] провели исследование ряда соединений типа MFX методом ЯМР, повторив большую часть ранних работ по бинарным фторидам [6, 9, 42] (см. табл. 11.5). При изучении спектров Р19-резонанса SF4, SeF4 и TeF4 в определенном интервале температур было установлено, что в каждом из этих соединений при комнатной температуре имеет место обмен атомов фтора, причем скорости обмена располагаются в следующей последовательности: SF4 < SeF4 < TeF4. Предложенный механизм обмена атомов фтора включает образование димеров в качестве промежуточных продуктов.
Шулери [43] исследовал UFe и обнаружил, что ядра фтора в этом соединении экранированы гораздо слабее, чем в F2 (линия UFe сдвинута на 330 м. д.
Исследование ядер ново магнитного резонанса фтора
205
в область более слабого поля от линии F2). Это отвечает большому парамагнитному вкладу в константу экранирования, обусловленному малой энергией электронного возбуждения ДЕ гексафторида урана [44] 1).
11.5. Галогенопроизводные фторуглеродов
При исследовании соединений этого типа ряд авторов [9, 10] получили аномальные значения сдвигов F19. Можно было бы ожидать, что последовательное фторирование метана приведет к уменьшению ионного характера связей С — F и к ослаблению экранирования ядер фтора. Изучение химических сдвигов для фторметанов, приведенных в табл. 11.6, показывает, что зависимость
Таблица 11.6
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ
F19 ФТОРМЕТАНОВ [9, 158]
Соединение Химический сдвиг F19, м. д. а Константы взаимодействия, гц [158]
6CFi (внешн) а 19] «PCClsF (внутр) [158] JHF ^C13F JCi3H
CH3F +210,0 +271,9 46,4 157,5 149,1
ch2f2 +80,9 + 143,4 50,2 235 184,5
CHF3 + 18,2 +78,6 79,7 274,3 239,1
cf4 0,0 +63,3 — 259,2 —
а eCF3COOH (внешн) = eCFi (внешн) “ 11 ’9 (м- «•)•
такого типа действительно преобладает. Протонные спектры этих соединений свидетельствуют о том, что ионность связи С — Н уменьшается по мере того, как ионность связи С — F растет [9].
Таблица 11.7
ХИМИЧЕСКИЕ СДВИГИ F19
ФТОРХЛОРМЕТАНОВ [9]
Соединение Химический сдвиг ®CF4 (внешн) a, м. д.
CFC13 -76,7
CF2C12 -60,4
CF3C1 -36,8
cf4 0,0
®СР3СООН(внешн) ^CF4 (внешн) 11.9 (м. д.) (соотношение между шкалами определено весьма приближенно).
Следовало ожидать также, что постепенное фторирование тетрахлорметана будет сопровождаться аналогичным изменением химических сдвигов F19, поскольку хлор, подобно водороду, менее электроотрицателен, чем фтор. Из табл. 11.7 следует, однако, что для ряда фторхлорметанов наблюдается зависимость, обратная той, которую можно было предсказать из соображений
г) Проведено систематическое изучение спектров ЯМР-F19 растворов фторидов элементов IV [250], а также V и VI [271] групп периодической таблицы. Измерены химические сдвиги F19 для жидких и газообразных F2 (—422 и —419 м. д. соответственно), OF2 (—249 и —248 м. д.) и NF3 (—142 и —138 м. д.) относительно CFC13 [251]. Рассмотрению диамагнитного вклада в экранирование двухатомных молекул F2 и FC1 посвящена работа Девиса [253].— Прим. ред.
206
Глава 11
электроотрицательности. Кроме того, исходя из простых индуктивных представлений, следовало полагать, что химический сдвиг фтора в CFC13 будет занимать промежуточное положение между химическими сдвигами для CFH3 и CF4 (т. е. CFF3). Для объяснения аномальных химических сдвигов оказалось необходимым постулировать участие в молекулярной структуре галогензаме-щенных метанов ионных структур с двойными связями типа
С1- С1+
II
Cl — C = F+ и Cl —С F-
I I
С1 С1
I II
Предполагалось, что атомы фтора обладают большей способностью к образованию, двойных связей, чем атомы хлора, и потому структура I должна играть основную роль в общей структуре молекулы. Для обычных фторметанов экранирование F19 определяется в основном индуктивным эффектом. Во фторгало-генпроизводных метана индуктивные эффекты проявляются гораздо меньше, и, следовательно, вклад двоесвязных структур в общую структуру молекулы может оказаться определяющим для экранирования ядер фтора. Это должно привести к ослаблению ионности связей С — F при уменьшении числа атомов фтора в молекуле. Однако более вероятно, что уменьшение экранирования ядер фтора обусловлено увеличением числа низколежащих возбужденных уровней в молекулах СН3Х после замещения атома фтора или хлора на атом водорода (см. разд. 11.8) [56].
Аналогичные данные для фторхлорметанов были получены в работе [10], авторы которой исследовали более широкий круг фторуглеводородных молекул (см. табл. 11.8, где представлены химические сдвиги, измеренные относи-
Таблица 11.8
ХИМИЧЕСКИЕ СДВИГИ F19 ФТОРХЛОРАЛКАНОВ
[10]
Соединение
^CF3COOH (внешн)» М. Д.
CC13F
CF3COOH FsC^
CC12F ccif2—ccif2 C12FC\ ccif2 CF3
CC12
CC1F
\f3
CF3 I CF
—78,6
0,0 +7,33 —0,33 —6,00 —6,00
— 10,1 -5,33
I I
F2c CF
V'cFg
F2
+42,4
—3,73
—6,00
+ 109,6
+47,9
Исследование ядерного магнитного резонанса фтора
207
тельно трифтор уксусной кислоты как внешнего эталона). Константа экранирования F19 возрастает вместе с ростом суммарной электроотрицательности атомов, присоединенных к ближайшему атому углерода. Отсюда следует, что в этих молекулах простые индуктивные эффекты не определяют экранирования ядер фтора.
11.6. Замещенные фторалканы
Многие из замещенных фторалканов были исследованы методом ЯМР в связи с изучением заторможенного вращения вокруг связи С — С (см. разд. 9.5.5). В табл. 11.9 приведены значения химических сдвигов F19,
Таблица 11.9
ХИМИЧЕСКИЕ СДВИГИ F19 ФТОРЭТАНОВ [49]
Соединение Химические сдвиги a’
CF3 cf3 CF
A внеш и °CFC13 двнешн °CF3COOH двнешн °CFC13 двнешн °CF3COOH свнешн °CFC13 двнешн °CF3COOH
cf3cfci2 85,8 5,9 77,8 —2,1
CF3CFClBr 84,3 4,4 77,0 —2,9
CF2C1CFC12 68,5 —11,4 72,5 —7,4
CF2BrCHFCl 63,5 — 16,4 146,0 66,1
CF2BrCHFBr 61,0 — 18,9 148,0 68,1
CF2ClCHFBr 67,0 —12,9 152,0 72,1
CF2BrCH2Cl 54,5 —25,4
CF2C1CF2C1 72,5 —7,4
CF2BrCHBrC6H5 —26,5 6
CF2C1CHC1C6H5 —17,6 6
а Химические сдвиги измерены относительно внешнего Эталона CFCI3 и переведены в шкалу внеш-него эталона CF3COOH с помощью соотношения = ^CFC13*— 79,9 (м. д.).
6 Эти значения взяты нз работы [34] н переведены в шкалу внешнего эталона CF3COOH из шкалы, в которой в качестве эталона использовался СРзВгСРгВг.
характерные для молекул этого типа [20, 45—49]. Присоединение атомов водорода и фтора к одному и тому же атому углерода обусловливает сильное экранирование ядра F19. В табл. 11.10 приведены значения констант взаимодействия JHf и Jff для некоторых фторированных этанов. Величина взаимодействия между геминальными атомами фтора в рассматриваемых молекулах изменяется в широких пределах (152—343 гц); причем строгой зависимости этих констант от электроотрицательности геминального заместителя нет [171]. Известно, однако, что в подобных взаимодействиях ядер фтора контактное взаимодействие Ферми не играет определяющей роли. Эллеман и сотр. [50, 51] провели исследование спектров ЯМР некоторых жидких фторуглеводородов. Данные, полученные этими авторами в результате анализа спектров первого порядка, приведены в табл. 11.11 и 11.12. Константы взаимодействия между ядрами атомов водорода и фтора, находящихся в одной замещенной метильной группе, составляют приблизительно 50 гц. Для ряда молекул типа CF3CH2X значения констант Jfif2 увеличиваются с изменением X в последовательности: X = F, Cl, Br, Н. В ряду молекул CF2XCH3 величина JfiH2 также постепенно увеличивается от 12,8 до 20,8 гц для заместителей X = F, Cl, Br, Н. Значения констант, полученные для простых этанов, были использованы при идентифи-
Таблица 11.10
КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ F-F И Н-F ДЛЯ ЗАМЕЩЕННЫХ ЭТАНОВ
Соединение Г еминальные константы, гц Вицинальные константы, гц Литература
F-F H-F F-F F—Fz F-H F'-H
CH2BrCF2Br 22 a 4 a 48
CH2ClCF2Br 21 a 3a 49
CH2C1CF2C1 16a 6a 48
CH2ICF2(C2F5) 29 a 6a 48
cf2cicfci2 9,5 49
CF2BrCFBr2 19 47
CF2BrCHFBr 174 48 24 21 3,2 9,1 49
CF2BrCHFCl 177 48 18 18 3,5 6,3 46, 48
CF2BrCFClBr 159 13 14 34
CF2ClCHFBr 173 48 20 19 3,9 7,1 49
CF2CCHC1C6H5 158 6 9 34
CF2BrCHClBr 154 10 7 34
CF2BrCHBrCeH5 152 6 15 34
CF2BrCBr(CN)CH3 155 45
CF2(S1C13)CFC1H 343 48,1 16,8 16,8 6,6 10,1 171
CF2(CF3)CFICI 270,4 20
а Эти значения были вычислены в предположении, что Jpp = 175 гц и /pjH < 20 гц.
Таблица 11.11
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ F19 РЯДА ГАЛОГЕНЗАМЕЩЕННЫХ ЭТАНОВ [50]
r Соединения a Химический сдвиг б, M. Д. Константы взаимодействия, гц
6f(1) 6F(2) Jf(1)f(2) Jh(1)F(1) JH(1)f(2) Jh(2)f(1) Jh(2)f(2)
ch3ch2f +76,95 25,7 47,5
chf2ch3 -25,91 57,2 20,8
CF3CH3 -73,47 12,8
CF2C1CH3 -90,82 15,0
CF2BrCH3 -98,86 15,9
CF3CH2C1 -63,98 8,4
CF3CH2Br -67,47 8,9
cf3cf2h -47,76 +3,88 2,8 2,6 52,6
cf3cfh2 -56,61 + 105,4 15,5 8,0 45,5
cf2hcf2h +2,21 +2,21 52,1 4,8
а Все исследованные образцы представляли собой чистые жидкости.
б Химические сдвиги измерены относительно внешнего эталона — октафторциклобутана; 6СР3СООН(внешн) = 6С4Р8(внешн) + 59 (“• Д-) (приближенное соотношение).
Исследование ядерного магнитного резонанса фтора
209
Таблица 11.12
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ F19 ГАЛОГЕНЗАМЕЩЕННЫХ ПРОПАНОВ [5V]
Химические сдвиги а, м. д.
Константы взаимодействия, гц
Соединение
chf2cf2chf2 cf3ch2cf3 CF3CHC1CF3 CF3CH2CH3 chf2cf2ch2f cf3cf2ch2f cf3cf2chf2 cf2cicf2ch2f CF2BrCF2CH2F
CHF2CF2CHFBr | CHF2CF2CHFC1 {
+3,27
—71,69
—65,22
—66,21
+3,24
—51,00
—52,22
—65,64
—71,25 +0,66
+ 1,17 + 1,66
+ 1,97
—0,20
—7,54
—7,97 — 1,98
— 13,76 — 16,60
—9,23
—5,89
+3,27
—71,69
—65,22
+ 109,01
+ 108,17
+2,86 + 103,57 + 105,15
+24,22
+20,77
13,9
15,2
4,5
15,1
15,5
16,7
45,9
46,0
52,1
45,9
46,0
48,7
12,0
11,7
4,5
11,8
11,7
47,5
a Химические .сдвиги измерены относительно внешнего эталона — октафторциклобутана; 6CF3COOH (внешн) = 6C4F8 (внешн) + 59 (м. д.) (приближенное соотношение).
кации более сложных спектров фторированных пропанов (см. табл. 11.12), бутанов, пентанов, гексанов и октанов [51].
Исследование спектров ЯМР дифторметильных групп CHF2X большого числа молекул показало, что константы взаимодействия Jhf значительно возрастают с увеличением электроотрицательности заместителя X (наблюдалось изменение Jhf в пределах 53,4—75,6 гц) [25]. По-видимому, изменения величины этих геминальных констант определяются в основном электроотрицательностью первого атома заместителя X х).
11.6.1. Относительные знаки констант спин-спинового взаимодействия F19 во фторалканах
Для того чтобы расширить теоретические представления о константах взаимодействия Н — F и F — F, необходимо знать относительные знаки этих констант в различных молекулах. Развитие метода двойного резонанса дало возможность определить относительные знаки констант взаимодействия для большого числа различных молекул. Данные об относительных знаках констант взаимодействия фтора, полученные разными авторами, сведены в табл. 11.13 [22, 23, 24, 167]. Эксперименты по двойному резонансу показали, что обе константы взаимодействия Н — F в 1,1,1,2-тетрафторэтане CF3CFH2 имеют одинаковый знак. Аналогичные результаты были получены для CHFBrCF2Br и CF3CF2CHF2 [22]. Определение относительных знаков констант Н — Н и F — F привело к другому результату — геминальные и вицинальные константы имеют разные знаки. Эллеман и Менетт [23] подтвердили этот результат, показав, что в CFBr2CF2Br при температуре —110°
х) Проведен расчет экранирования F19 для ряда фторированных алифатических соединений путем сопоставления с вычисленными электронными зарядами атомов фтора [254].— Прим. ред.
14-489
210
Глава 11
Таблица 11.13
ОТНОСИТЕЛЬНЫЕ ЗНАКИ КОНСТАНТ ВЗАИМОДЕЙСТВИЯ F19 ФТОРАЛКАНОВ И| ФТОРАЛКЕНОВ [22—24, 167]а
F\ /F
С — С
F\ /F
С-С-С
F-
>с = с
Fz
+
F — С
Х'с = с/
+
F\ /Н
С = С
+ или —
F\ Н
С-С
С13 —н
а Знаки, приведенные в таблице, являются абсолютными при условии, что констан-та ^С13—н положительна* Если F н </(31з____н противоположны по знаку [168], то
можно провести следующее отнесение [80, 169]:
F / С13—С
+
F С13=С/
CIS—F
Jff* имеет противоположный знак по отношению как к JfT, так и к J™pa4C. Они нашли также, что в CF3CF2CF2Br и CF3CF2CF2I знак константы JF1f3 противоположен знаку констант и [160] 1).
11.6.2. Данные, полученные при исследовании конформации замешанных этанов
Исследуя изменения констант взаимодействия некоторых замещенных этанов в зависимости от температуры, Гутовский и сотр. [26] получили обширную информацию о конформационных превращениях молекул подобного типа. В табл. 5.19 приведены значения констант взаимодействия Н —F и F — F для различных конфигураций исследованных молекул. Из этих данных следует, что для вицинальных констант взаимодействия Н — F и F — F > 1гош. Некоторые из транс- и гош-констант F — F имеют
разные знаки, что подтверждает теорию «вычитания», выдвинутую для объяснения малых значений этих констант в перфторэтильных группах [15, 27].
Седерхолм и сотр. [28] исследовали резонансные спектры фтора в замещенном этане CFCIBrCFCIBr при нескольких температурах в диапазоне 177— 300° К- Основываясь на изменениях спектра в зависимости от температуры,
!) Путем изучения релаксационных процессов для CHFC12 найдены абсолютные знаки констант спин-спинового взаимодействия: Jh_f = + 53,6 гц, /с-Н = + 220,0 гц и Jц-F = 293,8 гц [267].— Прим. ред.
Исследование ядерного магнитного резонанса фтора 211
эти авторы определили, что при комнатной температуре спектр представляет собой наложение спектров быстро вращающихся dl- и мезо-изомеров, а при низких температурах — суперпозицию спектров от различных ротамеров двух изомеров. Самое слабое экранирование наблюдалось для ядра фтора, находящегося в гош-положении по отношению к атомам Вг и F (0,00 м. д.), в то время как экранирование фтора в гош-положении к атомам F и С1 (6,5 м. д.) было больше, чем для фтора в гош-положении к атомам Вг и С1 (3,8 м. д.). На основании этих результатов можно предположить, что изменение экранирования происходит за счет пространственных эффектов, поскольку индуктивный эффект для фтора во всех этих молекулах приблизительно одинаков ]).
11.7. Фторуглеводородные соединения, содержащие азот
В табл. 11.14 и 11.15 обобщены данные по спектрам ЯМР-F19 ряда фтор-углеводородов, содержащих азот [52, 53, 58] 2). В табл. 11.14 включены значения химических сдвигов F18 для двух фторпиримидинов [53]; очень часто спектры ЯМР-F18 позволяют дать характеристику замещенных фторпиримидинов.
Я Исследованы химические сдвиги и константа взаимодействия F — F и Н — F для хлорированных 2,2-дифторпропанов [268] и монофторированных спиртов [269].— Прим. ред.
2) Спектры ЯМР-F18 ряда М-(перфторалкил)перфторпиперидинов и М-(перфтор-алкил)перфторморфолинов, М'-М-ди(перфторалкил)перфторпиперазинов и других азотсодержащих соединений см. [208—210].— Прим. ред.
Таблица 11.14
ХИМИЧЕСКИЕ СДВИГИ F19 ФТОРУГЛЕРОДНЫХ СОЕДИНЕНИЙ АЗОТА [52, 53] а
Соединение Интенсивность Химический сдвиг Тонкая структура
(CF3CO)2NCH3 CH3CON(CF3)2 CgH5CON(CF3)2 CO(CF2)3CONC6H5 f a 1 5,57 -21,3 -21,5 42,1 2 неэквив. 'квартета Синглет » Триплет
^7“ t b a b k 2 57,1 Квинтет
CO(CF2)3CONCO(CF2)3CONCO(CF2)3CO cd a b (a 2 41,7 Синглет
Ь 1 46,7 »
c 2 49,0 Триплет
d 4 55,6 Квинтет
(CF3CO)4N2 1 -2,1 Синглет
1 2,0 »
1 1 4,1 10,6 » »
BrCF2CF2CF2CONCO a 1 -14,2 Триплет трип-
a b c ‘ b 1 40,1 летов Триплет
c 1 38,7 »
BrCF2CF2CF2CONH2 a 1 -15,1 Триплет трип-
a b c b 1 42,1 летов Триплет
c 1 42,2 Триплет
а Химические сдвиги (м. д.) измерены относительно CFgCOOH (внешний эталон).
14*
Продолжение табл. 11.14
Соединение
Интенсивность
Химический сдвиг
Тонкая структура
2,4,6-Трифторпиримидин [53] Fa
NN
Fb\./Fc
N Тетрафторпиримидин [53] Fa
NZ>,Fd Fbi^Jpc N
-6,3
-31,3
95,2
Таблица 11.15
ХИМИЧЕСКИЕ СДВИГИ Fit* ЦИКЛИЧЕСКИХ И НЕЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ АЗОТА [58]
Соединения, содержащие азот Г руппа Химический сдвиг а Тонкая структура б
Д. Циклические
Ь с CF2 (a) 56,7 М
a /CF2CF2 cf2< >nf xcf2cf/ CF2(b) CF2 (c) 54,5 32,7 М м
NF 36,6 м
b c CF2 (a) 56,2 м
a ,cf2cf2v cf2cf2 cfZ >n-n< >cf2 ^cf2cf2' xcf2cf/ CF2 (b) CF2 (c) 55,3 19,1 м м
b c CF2(a) 57,8 м
a /CF2CF2, cf2< >ncf3 X^CF/ CF2 (b) CF2 (c) 55,2 16,8 м Квартет
CF3 -25,3 Квинтет
a b CF2 (a) 9,6 м
cf2cf2 O\ /NCF3 ХСР2СР/ CF2 (b) CF3 17,0 —24,0 Квартет Квинтет
a b CF2 (a) 4,8 м
/CF2CF2 O< >NF X^CF/ CF2 (b) NF 33,7 36,0 м м
aCF2-O CF2 (a) 14,9 м
CF2 (b) 22,2 Квинтет
bCF2 CF2C CF2(c) —20,6 м
\cF2CF2 CF2 (d) 8,6 Квинтет
d CF3 10,6 Триплет
Исследование ядерного магнитного резонанса фтора
213
Продолжение табл. 11.15
Соединения, содержащие азот Г руппа Химический сдвиг a Тонкая структура 6
Б. Нециклические
abed е (CF3CF2CF2CF2)2NCF3 CF3 (д) CF3 (e) 5,0 —26,5 Триплет м
CF2(ft) 25,0 м
CF2 (c) 21,9 м
CF2 (d) 10,0 м
а Ь (CF3CF2)2NCF3 CF3 (a) CF3(ft) 7,7 —25,3 м м
CF2 16,8 м
а b CF3CF2CF2NF2 CF3 CF2 (a) 6,4 27,1 Триплет Квартет
CF2(b) 51,1 Триплет
nf2 —92,0 м
(CF3)2NC(O)F CF3 —20,0 Дублет
CF —81,0 Септет
(CF3)2NC(O)N(CF3)2 CF3 —20,4 Нет
(CF3)2CHC = N CF3 — 10,6 Дублет
a b c d CF3CF2CF2CF = NCF3 CF3 (a) CF3(d) 5,2 —36,2 Триплет Дублет
CF2(b) 42,4 Квинтет
CF2 (c) 51,4 Дублет
CF —52,7 м
cf2=ncf3 CF3 —18,8. м
CF —44,6 Дублет
CF —25,2 Дублет
HN(CH3CH2F)2 CF 145,5 м
а Химические сдвиги измерены в миллионных долях (м. д.) относительно CFgCOOH (внешний эталон).
б м — широкий неразрешенный мультиплет.
11.8. Другие производные фторуглеводородов
Маллер, Лаутербур и Сватос [54] провели исследование спектров F19 для большого числа фторированных органических соединений. Полученные ими данные приведены вместе с результатами других авторов [159] в табл. 11.16 и 11.171).
Резонансные линии групп —CF3, CF2 и CF сильно смещены относи-
тельно друг друга, и их химические сдвиги имеют следующие значения (отно-
х) Многочисленные данные по химическим сдвигам F19 фторорганических соединений собраны Ю. С. Константиновым [252].— Прим. ред.
Таблица ltJ6
ДИАПАЗОНЫ ХИМИЧЕСКИХ СДВИГОВ F19 ФТОРОРГАНИЧЕСКИХ СОЕДИНЕНИЙ [58]
Группа Число исследованных соединений Область наблюдаемых значений д а
ch2ch2f 3 149,5—145,5
C3CF 2 112,5; 107,0
С —CF2 —с 17 57,8—21,9
c-cf2-n 10 51,1—8,6
C-NF-C 2 36,6; 36,0
С —CF2 —О 6 От 14,9 до —6,2
cf3—cf2— 10 11,9—3,2
Остальные группы CF3 — С 10 От 0,0 до —19,7
C = CF—О 3 От —9,4 до —11,8
CF3 —N 9 От —18,8 до —36,2
0—CF2 —N 1 —20,6
cf2=n 1 —25,2 и —44,6
C-CF=N 1 —52,7
N —C(O)F 1 —81,0
C —nf2 1 —92,0
а Химические сдвиги измерены в миллионных долях (м. д.) относительно CF3COOH (внешний эталон).
Таблица 11.17
ХИМИЧЕСКИЕ СДВИГИ F19 ПЕРФТОРАЛКИЛЬНЫХ ГРУПП
ФТОРПРОПАНОВ [159]
CF3CF2CF2X
Заместитель X Химический сдвиг, a м. д. Литература
-CF3 -CF2- -CF2X
-I 1,97 40,7 — 17,5 6
-CI 4,62 48,7 —6,7 6
-S-S- 4,00 47,0 13,6 6
-sf4- 5,0 50,0 16,5 101
-sf5 5,0 50,7 18,8 101
-CN 5,03 51,3 30,6 6
-(CF2)n- 4,8 46,6 34,4 54
1 /X Я 1 \z 3,60 49,1 35,0 6
— coci 4,67 49,2 37,2 6
— COOC2H5 4,67 50,5 42,7 6
— COOH 4,87 50,5 43,5 6
— COF 5,35 51,3 43,5 6
-nf2 6,4 27,1 51,1 54
— CF 5,2 42,4 51,4 54
— H 7,48 57,0 61,2 6
Химические сдвиги относительно CF3COOH (внешний эталон).
б Результаты неопубликованной работы Фостера, цитированной в [159J.
Исследование ядерного магнитного резонанса фтора
215
сительно CF3COOH):
CF3 ОТ — 14 до + 8 м. д.;
С —CF2—С от + 27,1 до + 57 м. д.
С >CF —С CZ от + 107 до 4' 113 м. д
Химические сдвиги протонов для аналогичных водородсодержащих алкильных групп располагаются в обратном порядке. Количество заряда, которое может притянуть к себе атом фтора от связанного с ним атома углерода, уменьшается по мере возрастания числа атомов фтора, присоединенных к этому атому углерода. С увеличением замешанности углерода фтором ионный характер связи С — F ослабляется и, следовательно, экранирование ядра фтора постепенно уменьшается. При изменении заместителей, присоединенных к этим алкильным группам, наблюдаются изменения химических сдвигов F18, которые не всегда легко объяснить, так как группы больших размеров, находящиеся в непосредственной близости от атома фтора, могут вызвать так называемое «стерическое дезэкранирование» (repulsive deshielding) его ядра. Стерическое дезэкранирование, по-видимому, является следствием дисперсионных сил, действующих между группой большого размера и данным атомом фтора. Этот эффект приводит к тому, что экранирование фтора отличается от ожидаемого на основании электроотрицательности заместителей. Примером может служить более сильное экранирование группы CF3 в (CF3)3CF по сравнению с экранированием в (CF3)4C. Следовало бы ожидать, что атом фтора, будучи более электроотрицательным, чем группа CF3 [55], сильнее уменьшит электронную плотность на трифторметильных группах в (CF3)3CF, а это в свою очередь должно привести к меньшему экранированию ядер фтора групп CF3 в этом соединении по сравнению с (CF3)4C. Термин «стерическое дезэкранирование» был введен Тирсом [8], который исследовал ряд фторуглеродных соединений, содержащих заместители больших размеров. Как показал Тирс, отталкивательные стерические взаимодействия приводят к уменьшению экранирования, что эквивалентно удалению электронов от рассматриваемого атома фтора. Эванс [1] объяснил этот эффект изменениями парамагнитного’вклада в экранирование, обусловленными внутримолекулярными дисперсионными силами, действующими между группами большого размера и атомами фтора. В табл. 11.18 приведены полученные Тирсом значения химических сдвигов F18 ряда фторуглеводородов, измеренные относительно трифторуксусной кислоты. Рассмотрение химических сдвигов для перфторалкилхлоридов, -бромидов и -иодидов дает следующий ряд «эффективной способности заместителей притягивать электроны»: I > Br > Cl > F > CF2I > CF2Br > CF2C1 > CF3. Большие размеры некоторых из этих групп объясняют, по-видимому, их меньшую способность к притяжению электронов от соседней фторсодержащей алкильной группы (т. е. более низкую электроотрицательность). Подобные «стерические» эффекты проявляются также в тех случаях, когда между группой большого размера и исследуемым атомом фтора находится группа — СН2 или —CF2.
В настоящее время, однако, распространено мнение, что при непосредственном присоединении группы CF2 к атому хлора, брома или иода экранирование фтора определяется главным образом наличием в молекуле низколежа-щих возбужденных уровней [56]. Использование орбиталей со все большей энергией при изменении заместителя в последовательности X = F, Cl, Br, I приводит к соответствующему увеличению доступности низколежащих возбужденных состояний в группах —CF2X. Если низколежащие состояния обладают необходимой симметрией для смешивания с основным состоянием в присутствии магнитного поля, то можно ожидать появления большого парамагнитного сдвига в сторону слабого поля. С помощью этих представлений
216
Глава 11
Таблица 11.18
ХИМИЧЕСКИЕ СДВИГИ F19 НЕКОТОРЫХ ФТОРАЛКАНОВ3 [8]
cf3-cf2-cf2-cf3 6,5 51,5 CF3-CF2-CF2-CH2I 7,5 51,5 33,5
CF3 - CF2 - CF2 - CF2 - CF2 - CF2 - CF3 7,5 51,5 47,5 47,5 CF3—CF2—CF2 —COCI 6,5 51,5 39,5
CF3 - cf2 - cf2 - cf2 — cf2h 7,5 51,5 48,5 54,5 (61,5; 63,5) CF3-CF2-CF2-CC13 6,5 42,5 32,5
hcf2—cf2—cf2 — cf2h 54,5 (62,5; 63,5) CF3 - cf2 - cf2 - cf2- cf2 - CC13 6,5 51,5 47,5 41,5 34,5
cf3-cf2-cf2-ch2ci 7,5 51; 5 42,5 CC13 — CF2 — cf2 — COCI (28,5; 30,5)
CF3 — CF2—CF2 — CH2B r 7,5 51,5 39,5 CC13 — cf2 — cf2 — cf2—COCI 31,5 (35,5; 37,5)
а Химические сдвиги фтора приведены в миллионных долях (м. д.) относительно CFgCOOH (внешний эталон). Первоначально сдвиги были измерены относительно октафторциклобутана (внутренний эталон) и затем пересчитаны с помощью соотношения
^CFgCOOH (виещи) ^C4Fs (внутр) + 61,5 (м.д.).
можно объяснить также экранирование F18 во фторметанах и фторхлорметанах: замещение атома F или С1 водородом увеличивает число разрешенных возбужденных состояний, уменьшая тем самым экранирование ядер фтора.
Таблица 11.18а
ХИМИЧЕСКИЕ СДВИГИ F19 (ЗНАЧЕНИЯ ср*) ФТОРОРГАНИЧЕСКИХ СУЛЬФИДОВ [94]
Соединение Концентрация, об. % В CClgF a Ф *-зиачеиия и мультнплетность
CF3 — CF2 — — (СЕг)з — — cf2- (СР2)23Ж
(h-C3F7)2S 10,0 80,58 124,22 83,81
т г ш; 8,2 ш; 17
(h-C3F7)2S2 12,7 80,88 124,21 90,80 ’
Т Д ш; 4,5 м (к?)
(h-C3F7)2S3 10,0 80,80 124,22 91,82
т е ш; 5,3 к е
(h-C7F15)2S2 16,0 81,54 126,5 122,5 119,7 89,79
т ж ш; 26
(CF2)4S 6,0 131,91 87,04
<р 3 т 3
1,4-(CF2)4S2 20,0 91,08й
ITT и
а Для перфторгептилдисульфнда указано процентное отношение вес/объем.
б Стандартное отклонение меиее ± 0,01 для всех величин с двумя знаками после запятой и ±0,15 для остальных.
в Обозначения мультиплетностн: т — триплет, к — квартет, м — разрешенный, но неисследованный мультиплет, ш — широкий неразрешенный пик, ширина которого иа половине высоты (W\/^ указана в герцах (гц).
г J(CF3 — С — CF2) = 9,5 ± 0,2 гц.
д J(CF3 —С — CF2) = 9,2 ± 0,1 гц.
е J(CFg — С — CF2) = 9,10 ± 0,05 гц.
ж J(CFg — С — CF2) = 9,40 ± 0,05 гц: каждая компонента представляет собой нечеткий триплет с J - 2,5 гц.
3J = 4,4±O,1 гц: каждая компонента является, по-видимому, триплетом с константой J = == 2,9 ± 0,5 гц.
и Ширина линии зависит от температуры. При концентрации 0% ср= 91,05.
Исследование ядерного магнитного резонанса фтора
217
Близость атомов фтора к атомам азота и кислорода в молекуле уменьшает экранирование ядер фтора, чего следовало ожидать, учитывая обычный индуктивный эффект (например, сигнал фтора в группах С — NF — С смещен
в сторону слабого поля приблизительно на 70 м. д. по отношению к резонансу
с\
>CF —С). cZ
группе
В табл. 11.18а представлены значения химических сдвигов для ряда фтор-органических сульфидов. Резонансные линии F18 групп — CF2 — S — в основном расположены в диапазоне 80—95ф* и занимают среднее положение между линиями, наблюдаемыми для аналогичных групп CF2 в изоэлектрон-ных соединениях фосфора и хлора [94]. Хотя представление о смешивании низколежащих возбужденных состояний с основным состоянием объясняет сильное экранирование, наблюдаемое для фтора в группе — CF2S —, положение максимумов УФ-поглощения для этих молекул указывает на то, что значения ф* должны располагаться в обратном порядке. Поэтому, если экранирование определяется эффектом «смешивания» состояний, то «низколежащие электронные состояния'», соответствующие максимумам УФ-поглощения, не обладают, по-видимому, необходимой симметрией, чтобы оказать влияние на экранирование *).
11.9. Перфторалкильные и перфторацильные соединения металлов
Химические сдвиги и константы спин-спинового взаимодействия F19 для нескольких молекул этого типа приведены в табл. 11.19 [56]. Если группа — CF2— связана непосредственно с металлом переходной группы, экранирование ядер фтора уменьшается, что обусловлено, вероятно, наличием низколежащих возбужденных электронных состояний для связей углерод — металл. Для этих молекул наблюдаются большие сдвиги в область слабого поля, которые имеют тот же порядок величины, что и для перфторалкилгалогенидов (см. табл. 11.19). Величину наблюдаемых парамагнитных сдвигов нельзя объяснить ни диамагнитной составляющей экранирования, ни влиянием магнитной анизотропии соседнего атома, которые слишком малы. Парамагнитный вклад может иметь достаточную величину в том случае, если молекула обладает несколькими низколежащими возбужденными состояниями, симметрия которых позволяет им смешиваться с основным состоянием при наложении магнитного поля. При этом в смешивании принимают участие частично заполненные d-орбитали, что для a-группы CF2 обусловливает парамагнитный вклад около 50 м. д. Введение карбонильной группы между атомом переходного металла и группой CF2 приводит к исчезновению парамагнитного сдвига. Тот же эффект наблюдается, если группа CF2 присоединена к атому олова или фосфора. Хотя электроотрицательность атома, непосредственно связанного с группой CF2, несомненно оказывает некоторое влияние на экранирование ядер фтора, должны быть приняты во внимание также другие факторы. Например, химические сдвиги группы a-CF2 в соединениях олова и фосЗюра имеют одинаковый порядок величины, несмотря на то что электроотрицательности этих атомов сильно отличаются. В то же время, поскольку в этих молекулах нет частично заполненных d-орбиталей, парамагнитная составляющая экранирования невелика [56].
Мак-Клеллан [57] провел исследование спектров ЯМР-F18 некоторых перфторалкильных и перфторацильных производных карбонилов марганца и кобальта. Полученные им значения химических сдвигов приведены в табл. 11.20.
г) Проведено также исследование спектров F19 полигалогенированных эфиров, синтезированных на основе перфторизобутилена [211].— Прим. ред.
Таблица 11.19
ХИМИЧЕСКИЕ СДВИГИ а И КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ F19 НЕКОТОРЫХ ПЕРФТОРАЛКИЛЬНЫХ И ПЕРФТОРАЦИЛЬНЫХ СОЕДИНЕНИЙ МЕТАЛЛОВ [56]
Соединение CFg ₽CF2 aCF2 7 «6 6 J 6 Jay
C2F5Mn(CO)5 84,0 68,8 1,5
C2F5COMn(CO)5 80,3 114,5 0,7
C2F5Re(CO)5 84,2 74,9 1,9
C2F5CORe(CO)5 в 80,5 116,7 0,7
(C2F5)2Fe(CO)4B 83,7 74,0 2,0
C2F5Fe(CO)4I 83,5 59,0 1,8
(C2F5)2Sn(CH3)2 83,7 118,9
C2F5Sn(C2H5)3 84,4 120,3
C2F5Sn(C4H8)3 83,9 120,4 1,4
c2f5i 85,4 65,2 4,6
C3F7Mn(CO)5 78,8 115,3 65,6 — 0 12,4
C3F7Re(CO)5 78,5 115,1 72,7 -0 12,3
C3F7CORe(CO)5 в 81,0 126,7 113,7 ~ 0 9,3
C3F7COC5H5(CO)I в 79,1 114,1 56,3 ~ 0
(C3F7)2Fe(CO)4B 78,6 115,3 69,1 ~ 0 11,1
C3F7Fe(CO)4I ' 78,2 114,4 54,9 — 0 11,4
C3F7Sn(C4H8)3 80,3 122,7 118,2 — 0 9,5
,.(C3F7)2PI Д 81,2 119,7 102,9 3,2 9,2
(C3F7)2PCI e 81,2 122,7 120,1 9,6
C3F7I 79,6 118,2 60,5 4,6 9,3
c3f7ci 80,7 125,2 69,5 1,6 9,0
а Химические сдвиги указаны в м. д. относительно CClgF; их значения возрастают при смещении сигнала в область сильного поля. Во всех случаях, кроме оговоренных особо, измерения проводились с веществами, растворенными в трихлорфторметане, при концентрации 15% или менее.
б Константы взаимодействия в герцах (гц); в перфторпропильных соединениях не наблюдалась.
в Раствор в тетрагидрофуране, CCI3F использовался как внешний эталон.
г Раствор в дихлорметаие; внешний эталон — CCI3F.
Д Jрр^ = 23,6 гц; Jрр^ = 36,2 гц; Jрр^ = 9,2 гц.
® Jрра — 58,4 гц; JPFp ~ 36,5 гц; Jрр^, = 9,6 гц.
Таблица 11.20
ХИМИЧЕСКИЕ СДВИГИ F19 НЕКОТОРЫХ ПЕРФТОРАЛКИЛЬНЫХ КАРБОНИЛОВ МЕТАЛЛОВ [57]
Соединение Химические сдвиги относительно CF3COOH внешний эталон а, м. д. Отношение интенсивностей
CF3Mn(CO)5 —85,8 (с) б
u30-C3F7Mn(CO)5 —8,9 (с) в; 4-87,1 (с) в 6 : 1
H-C3F7Mn(CO)5 r 4-0,45 (т); —11,3 (к); 4-37,6 (с) 3:2:2
H(CF2)4COMn(CO)5 Д 4-60,8 (д); 4-33,6 (с); 4-46,2 (с); 4-51,8 (с) 1 : 1 : 1 : 1
CF3Co(CO)4 -87 (с)
C2F5Co(CO)4 4-6,0 (С) В; —19,3 (с) в 3:2
«-C3F7Co(CO)4 e 4-2,0 (т); —25,5 (к); 4-18,4 (с) 3:2:2
Буквами с, д, т и к обозначены синглет, дублет, триплет и квартет соответственно. Первая цифра в столбце химических сдвигов относится к фтору в концевой группе, последняя — к фтору в а-группе по отношению к металлу.
6 Использован 50%-ный раствор в тетрагидрофуране.
в Тонкая структура линии не разрешена.
г Jрр = 8 гц.
д ^FH = 50 гц.
® Jрр = Ю гц.
Исследование ядерного магнитного резонанса фтора
219
Таблица 11.21
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ F1» ФТОРАЛКИЛОВ РТУТИ [152]
Соединение Константы взаимодействия, гц Химические сдвиги a, м. Д.
JHg-CF3 JHg- cf2 JHg-CH2 7Hg-CF 6CF3 6cf2 eCF
(C2F5)2Hg (CF3CH2)2Hg (CF3CHF)2Hg 71 224 161 770 130 480 +6,0 +28,9 —0,6 +31,9 + 146,3
а Химические сдвиги измерены относительно CF3COOH (внешний эталон).
Химические сдвиги и константы спин-спинового взаимодействия для фтора трех фторалкильных соединений ртути представлены в табл. 11.21. Интересно отметить, что ядра фтора группы CF3 в перфторэтиле взаимодействуют с ядром ртути слабее, чем фтор группы CF2, в то время как для взаимодействия ядер Hg — Н нефторированных алкилпроизводных ртути наблюдается противоположная картина (см. разд. 10.3.2).
11.10. Фторированные ароматические соединения
11.10.1 Химические сдвиги F18 в замещенных фторбензолах
Заместитель в молекуле бензола оказывает различное влияние на орто-, мета- и пара-положения молекулы. В орто-положении влияние заместителя осуществляется через индуктивный, резонансный (эффект сопряжения) и стерический эффекты, для мето-положения в основном играет роль индуктивный эффект, для пара-положения — эффект сопряжения. Некоторые авторы предприняли попытки получить сведения об электронном распределении в ароматических системах путем исследования спектров ЯМР-F18 производных фторбензола. Ряд мета- и пара-замещенных-фторбензолов был изучен Гутовским и сотр. [59], которые провели корреляцию химических сдвигов F18 с константами Гаммета [58] для этих молекул (см. табл. 11.22). В обоих случаях наблюдалось приблизительно линейное соответствие, определяемое уравнениями
^Mercia__q J метa
anapa= __0,05606”opa + 0,271,
где омета и (jnapa — константы Гаммета, а 8мета и б”ара — химические сдвиги F18 для мета- и пара-замещенных фторбензолов соответственно. На рис. 11.4 представлена зависимость химических сдвигов фтора от констант Гаммета ст для мета- и пара-замещенных фторбензолов [60].
Значения 8''1ара, 8мета и 8°г‘то оказываются приблизительно пропорциональными друг другу, если любая пара из этих параметров сравнивается в пределах ряда соединений, для которого предполагается пропорциональность индуктивного и мезомерного эффектов.
Тафт [60] провел разделение константы Гаммета ст на индуктивную (сти) и резонансную (стс) части, определив сти как меру влияния свободной энергии заместителя (по отношению к атому водорода), проявляющуюся в его способности притягивать или отдавать электроны либо непосредственно, либо через ст-связи. Сходным образом резонансная часть <тс характеризует способность заместителя к притяжению или отталкиванию электронов при сопряжении
220
Глава 11
Таблица 11.22
ХИМИЧЕСКИЕ СДВИГИ F18 И КОНСТАНТЫ ГАММЕТА ДЛЯ МОНОЗАМЕЩЕННЫХ БЕНЗОЛОВ [59]
Заместитель Opmo a Merna a Константа Гаммета, о Пара а Константа Гаммета, о Константы Тафта
°и + [60]
no2 +5,6 —3,3 +0,710 —10,8 +0,778 +0,63 +0,15
CN —5,2 —3,0 +0,608 —9,6 +0,656 +0,59 +0,07
COOH —3,5 —0,5 +0,355 —6,9 +0,728
I — 19,3 -2,6 +0,352 + 1,2 +0,276 +0,38 —0,10
Br —5,5 —2,4 +0,391 +2,3 +0,232 +0,45 —0,22
Cl +2,7 —2,1 +0,373 + 2,4 +0,227 +0,47 —0,24
F +25,9 —3,1 +0,337 +6,4 +0,062 +0,50 —0.44
CH3 +5,0 +0,9 —0,069 +5,5 —0,170 —0,05 —0,13
CH3CONH + 12,8 —1,0 +5,7
OH +25,0 —0,9 +о,ю + Ю,6 —0,36 +0,25 —0,61
CH3o +22,4 + 11,4 —0,268 +0,23 —0,50
C2H5O +21,7 —1,3 +0,15 + 11,5 —0,25
nh2 +23,1 +0,2 —0,161 + 14,6 —0,660 +0,10 —0,76
а Химические сдвиги измерены в миллионных долях (м. д.) элон); 6CF3COOH (внеши) ~ ^CgHjF (внутр) + 35,6 (м. д.).
по отношению к CgHgF (внутренний
с л-орбиталями бензольной системы [60]. Для .яета-замещенных бензолов, зависимость химических сдвигов F10 от констант Тафта сти, найденная в работе [59], очень близка к линейной и может быть представлена следующим уравнением:
6Л1еша = __(5)83 ± 0 2б) (2аи) + 0,2,
где сумма 2сти позволяет учесть вклад 3,5-дизамещенных бензолов. Используя индуктивную и резонансную компоненты константы Гаммета, можно найти аналогичное соотношение для пара-замещенных бензолов:
6пара= —(5,83) (2оп)-(18,80 ± 0,81) (ас) 4-0,8.
Эти зависимости представлены графически на рис. 11.5 и 11.6, из сравнения которых с рис. 11.4, очевидно, следует, что химические сдвиги F19 коррелируют с константами Тафта сти и стс гораздо лучше, чем с константой Гаммета ст 2).
Экранирование ядра F19 в о-галогенфторбензолах, уменьшается при уменьшении электроотрицательности заместителя в следующем ряду: F, С1, Вг и I. Причиной аномального поведения химического сдвига может быть ослабление экранирования, обусловленное внутримолекулярными электрическими полями, возникающими главным образом в результате вандерваальсовых взаимодействий более крупного атома галогена с соседним атомом фтора [189].
Карплус и Дас [61] объяснили химические сдвиги, наблюдаемые для ряда полифторбензолов, применив при расчете тензора, экранирования такие характеристики локализованной связи, как ионность, гибридизация и дроб-
х) Более подробное рассмотрение химического сдвига F19 для мета- и парц-заме-щенных фторбензолов,, и также соответствия с о-константами Тафта проведено в обзорной статье [212]. В настоящее время корреляционное соотношение между и о с успехом применяют как основу вторичной шкалы о-констант заместителей (см. [217—224, 273] и библиографию в [212]).— Прим. ред.
j-l;4-NO2 oN02
оСНО ocN .3-F.5-F
3-F, 5-1*
F*£VqHN°z
left \CN
H3CV'* I 3-г-4'1 ми Н0*х* > 3-F,4-CI
- CHjCICHO
*сн3 boi
CH.o °Cl,Br о о
о °СН3
3-F,4-NH2 oF
ООН ООСН3
°NH2 ________I_________I______I_______I L__ 0,8 -0,4 О 0,4 0,8 1,2
-16
—12
-8
—4 О 4 8 12 16
Sff
Рис. 11.4. Зависимость химических сдвигов фтора для м- ип-фторбензолов от константы Гаммета о [60].
• лета-заместители; О лара-заместители.
Рис. И.5. Соответствие между химическими сдвигами F18 и константами Тафта аи и ас для п-фторбензолов [60].
222
Глава И
ный порядок связи. Проссер и Гудмен [172] расширили эти представления и получили выражение (4.32), описывающее химический сдвиг для сопряженных соединений как функцию плотности п-электронов на атоме фтора и связанном с ним атоме углерода, а также порядка связи С — F. Полученное
CNo°n0z. °сн3со
НЖ^СНзС0 СН3 СН°С1 °! °с6н5
ОСН3
O3-F.4- N02
^•'3,5-Е, ^*<3-65-1
О
3-F/-CI Л4~Гг5-с1з
°сг,вг
.'<f&3-CF.,4-F
3-NH,,4-F
F 2
8Н о оЗ- F.4-NH, 0СН3
оын2
—16
-12
-8
-4 О 4 8
12
16
-0,4 0 0,4 0,8 1,2 1,6
Гбц
Рис. 11.6. Зависимость химических сдвигов F19 для п- и л«-фторбензолов от индуктивного параметра Тафта аи [60].
ф л/ета-заместители; о пара-заместители.
выражение хорошо согласуется с экспериментальными значениями сдвигов для соединений типа /г-Х—C6H4F [173, 193] J).
11.10. 2. Спин-спиновое взаимодействие во фторированных ароматических соединениях
Бак, Шулери и Уильямс [29] определили константы взаимодействия JHf в различных дейтерофторбензолах между ядрами в орто-, мета- и /гара-поло-жениях. Получены следующие значения: J0/™ — 9,4 гц, Jj^na = 5,8 гц и 7рнРа = 0,0 ± 0,5 гц. Эти данные позволили провести полный анализ протонного спектра монофторбензола, который рассматривался как система типа АВВ'СС'Х [30].
Значения констант Jhf и для большого числа замещенных фтораро-матических соединений, полученные Бутовским и сотр. [19], приведены
') Измерены химические сдвиги F19 для фторзамещенных трифенилкарбониевых ионов [255].— Прим. ред.
224
Глава И
в табл. 11.23. В некоторых случаях константы /нг водной молекуле имели различные знаки. Основными факторами, определяющими величину орто-констант, являются положения взаимодействующих ядер по отношению к другим ядрам в кольце и природа взаимодействующих ядер [19, 183]. Меньшее влияние оказывает изменение заместителей в ароматическом кольце. Диапазоны изменения констант взаимодействия указаны в табл. 11.24. При
Таблица 11.24
КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ F19 ДЛЯ ЗАМЕЩЕННЫХ БЕНЗОЛОВ (В ГЕРЦАХ) [19, 189]
взаимодействии ядер фтора в ароматических молекулах играют роль как сг-, так и л-электронные системы, причем ни одной из них нельзя отдать предпочтения. Уильямс и Гутовский исследовали влияние различных факторов, определяющих взаимодействия F — F и Н — Нв ароматических молекулах, и пришли к выводу, что основной вклад в них вносит контактное взаимодействие Ферми для пятиэлектронной системы (величины наблюдаемых констант показывают, что связь С — F приблизительно на 5% определяется 25-электронами фтора) [31]. Попл [164] показал, что для фторбензолов заметное влияние на величину констант F — F и Н — Н оказывает спин-орбитальное взаимодействие (см. табл. 5.13).
Для п-фтортолуола константа /нг гораздо больше, чем химические сдвиги между неэквивалентными протонами, что приводит к усреднению наблюдаемых значений ГНТ° и J^ma: ± (j<£™ + = ± 7,4 гц [35]. Однако
в протонном спектре вполне возможны значения констант взаимодействия Н — F, которые согласуются с обычными для ароматических соединений: /н™ = g g и j^>™a = уд гц Разбавление /г-фтортолуола различными растворителями позволяет увеличить химический сдвиг между неэквивалентными протонами; получающиеся при этом сложные спектры в некоторой степени свидетельствуют о том, что J^10 J'^na 135].
Относительные знаки констант спин-спинового взаимодействия в ароматических соединениях. Эванс [165], применив метод двойного резонанса, определил относительные знаки констант спин-спинового взаимодействия F — F и Н — F в 2,3-дихлор-1,4,5-трифторбензоле и 2-хлор-1,4,5-трифтор-3-нитробензоле:
1 t jopvao. —. тмета. — /пара.
1. ztJpp , “T'*'FF ’ JFF ’
n t jopmo, , тмета . , тпара
2- ±JHF » ±JHF ’ •
Данные для констант взаимодействия F — F противоречат результатам расчета методом молекулярных орбиталей (Уильямс и Гутовский [31]), согласно
Глава И
225
которым знаки всех трех констант Jfp должны быть положительными. Экспериментально наблюдаемые относительные знаки констант взаимодействия F — F согласуются с полученными ранее путем детального анализа спектров ЯМР фторароматических соединений [19, 29].
11.10. 3. Замещенные бензотрифториды
Ряд авторов [59, 63] измерили химические сдвиги фтора для некоторых монозамещенных бензотрифторидов. Влияние заместителей на величину химических сдвигов в этих соединениях было приблизительно на порядок меньше наблюдаемого во фторбензолах и, кроме того, противоположно • по знаку. Последний факт пока не нашел объяснения. Константы взаимодействия F — F, измеренные для ряда 6-замещенных 2-фторбензотрифторидов, оказались сильно зависящими от природы заместителя (JF-cf3 = 13—34 гц) [184]. Величины констант приблизительно пропорциональны химическим сдвигам группы CF3 и размерам заместителя.
11.10. 4. Полизамещенные производные фторбензолов
Значения химических сдвигов F19 для ряда полизамещенных фторбензолов приведены в табл. 11.25. В некоторых случаях химические сдвиги обладают
Таблица 11.25
ХИМИЧЕСКИЕ СДВИГИ F19 ПОЛИЗАМЕЩЕННЫХ ФТОРБЕНЗОЛОВ [59]
Заместители Химический сдвиг a, M. Д. Заместители Химический сдвиг a, M. Д. Заместители Химический сдвиг a, м. Д.
3-NO2, 4-NH2 + 14,3 2,4-(F)2 +30,4 2-CF3, 4-F +7,4
2-NO2, 4-NH2 +21,6 3,4-(F)2 +2,4 3-CF3, 4-F +4,0
3,5-(F)2 —5,5 2,5-(F)2 +20,9 2,4,6-(CH3)3 + 15,0
3-F, 5-1 —4,5 2-C1, 4-F +8,6 2,5-(Cl)2, 4-F +5,5
2,4-(NO2)2 —5,4. 3-C1, 4-F +3,9 ' 2,5-(Br)2, 4-F —2,2
2-NH2, 4-F +28,7 2-C1, 5-F —2,0 2,4,5-(F)3 +26,7
3-NH2, 4-F +5,8 3-F, 4-C1 —2,0 2-Br, 4,5-(F)2 —4,3
2-NH2, 5-F + 18,3 2-Br, 4-F +0,8 2,4-(F)2, 5-Br +27,5
3-F, 4-NH2 + 11,5 3-Br, 4-F +4,0 2,5-(F)2, 4-Br +20,6
2-NO2, 4-F + 10,3 2-1, 4-F — 12,3 2,5-(F)2, 3-CF3 + 17,7
3-NO2, 4-F +2,0 3-1, 4-F +4,5 2,4-(F)2, 6-CF3 +32,0
2-NO2, 5-F —1,1 2-1, 5-F —23,6 3,4-(F)z, 5-CF3 —0,7
3-F, 4-NO2 — 14,9 3-F, 4-1 —3,0 2,4,6-(CH3)3, 3-F +9,6
а Химические сдвиги измерены относительно C3HjF (внутренний эталон). Наблюдаемое ядро фтора находится в положении 1.
свойством аддитивности, что позволяет предсказывать их величину путем суммирования известных вкладов от отдельных заместителей [59]. Химические сдвиги для ряда ароматических полифтораминопроизводных были измерены Татлоу и сотр. [65] для подтверждения структуры этих молекул.
Методом ЯМР были исследованы также некоторые перфторароматические соединения типа С6Н5Х; типичные значения сдвигов приведены в табл. 11.26 [64, 189, 196, 197]. Сдвиги F19 для ядер в орто-положениях не согласуются с предсказанными теорией, учитывающей только плотности л-электронов;
15-489
226
Глава И
Таблица 11.26
ХИМИЧЕСКИЕ СДВИГИ F1» МОНОЗАМЕЩЕННЫХ ПЕРФТОРБЕНЗОЛОВ CeF6X [189]а
Заместитель X d°Pm0, м. д. 6naPat М.Д. м. д.
F 162,28 162,28 162,28
С1 140,61 156,11 161,48
Вг 132,54 154,65 160,60
I 119,18 152,53 159,65
Sn(CH3)3 122,30 152,75 160,67
HgCH3 121,91 153,52 160,05
CeF5 138,25 150,27 160,76
NHCH3 161,89 173,07 165,21
H 138,89 153,50 162,06
а Точность ± 0,08 м. д. Химические сдвиги измерены относительно CFCI3 (внутренний эталон), концентрация которого составляла 5 мол.% в растворе исследуемого вещества в ССЦ той же концентрации.
подобные аномалии наблюдаются также для сдвигов в хлорфторбензолах и обусловлены вандерваальсовыми взаимодействиями [189] х).
11.10. 5. Фторзамещенные нафталины
Измерение химических сдвигов фтора в а- и 0-фторнафталинах [62] показало, что ядра фтора в этих молекулах оказываются более экранированными, чем в монофггорбензоле. Поскольку для химических сдвигов Н1 в ароматических молекулах наблюдается обратная зависимость, этот факт был объяснен отсутствием заметного влияния кольцевых токов на экранирование ядер фтора в ароматическом кольце. Хотя экранирования ядер Н1 и F19 за счет кольцевых токов имеют одинаковый порядок величинй, изменения химических сдвигов фтора, обусловленные другими факторами, настолько велики, что влиянием кольцевых токов можно пренебречь.
11.11. Фторалкены
11.11.1. Перфторвинильные производные
Значения химических сдвигов и констант спин-спинового взаимодействия для ряда перфторвинильных производных металлов и металлоидов приведены в табл. 11.27, 11.28 и 11.29 [66]. Во всех исследованных молекулах константы экранирования ядер фтора располагаются в следующий ряд: crF1 < crF2 < < сгр3, что, по-видимому, может служить доказательством слабой корреляции между наблюдаемыми сдвигами F19 и природой заместителя а). Замещение бором вызывает существенное уменьшение экранирования концевой группы = CF2, что, вероятно, вызвано резонансным эффектом притяжения электронов атомом бора [66]. На рис. 11.7 изображены спектры ЯМР-F19 трех типичных * 2
х) Спектры ЯМР-F19 ряда полифторхлорбензолов см. в работах [213, 214], некоторых эфиров и фенолов на основе полифторбензолов см. в работах [215, 216].— Прим. ред.
2) Рассмотрению химических сдвигов трифторвинильных соединений и выведению йа их основе аддитивной системы для вычисления сдвигов в спектрах ди- и монофтор-олефинов посвящены работы [225—226, 256].— Прим. ред.
Таблица 11.27
ХИМИЧЕСКИЕ СДВИГИ F19 НЕКОТОРЫХ ПЕРФТОРВИНИЛЬНЫХ СОЕДИНЕНИЙ [66]
(1)F\ ZF(3)
с = с
(2) FZ
Соединение Химический сдвиг a, м. д.
F(l) F(2) F(3)
cf2=cfbci2 71,6 87,9 184,5
cf2=cfbf26 72,8 99,8 206,61
(CF2=CF)3B 72,7 91,1 185,9»
(CF2 = CF)2Hg 89,9 124,5 185,0
(CF2 = CF)3As 84,8 112,7 177,0
(CF2 = CF)2Si (C2H5)2 83,5 114,3 199,7
(CF2 = CF)2Ge(CH3)2 86,6 118,6 195,5
(CF2 = CF)4Ge 80,1 112,7 196,5
(CF2 = CF)2Sn(CH3)2 85,9 121,2 194,6
(CF2 = CF)Sn(C4H9)3 88,1 123,3 192,7
(CF2=CF)zSn(CeH5)2 84,3 118,6 193,2
CF2 = CFC1 [68]» 105 121 145
cf2=cfh b 103 127 185
CF2 = CFC8H5b 102 133 193
а Измерен относительно CCI3F (внутренний эталон). Все вещества исследовались в виде растворов в CCI3F с концентрацией 10 мол.%.
б Химический сдвиг для группы BFg равен 86,7 м. д. при концентрации 25%; химические сдвиги при концентрации 10% отличаются менее чем на 1 м. д.
в Приближенные значения.
Таблица 11.28
ПРИБЛИЖЕННЫЕ ЗНАЧЕНИЯ КОНСТАНТ ВЗАИМОДЕЙСТВИЯ F— F ДЛЯ ПЕРФТОРВИНИЛЬНЫХ СОЕДИНЕНИЙ МЕТАЛЛОВ [66]
(1)F F(3)
Хе—
(2)FZ ХМ
Соединение гц гц jmpaHC^ гц
CF2 = CFBC12 7 19 114
CFo = CFBF2 18 — if 117
(CF2 = CF)3B <5 24 1 110
(CF2 = CF)2Hga 75 37 109
(CF2 = CF)2Si(C2H6)2 62 26 117
(CF2 = CF)2Ge(CH3)2 72 32 118
(CF2 = CF)4Ge 71 32 118
(CFz = CF)2Sn(CH3)26 75 34 116
(CF2=CF)Sn(C4H9)3 79 34 115
(CF2 = CF)2Sn(C6H5)2 68 34 118
а ^Hgl99— F (1)=223 г'<1 7Hgl99—F (2)=1 7 гЧ’ 7Hgl99—F (3)=82° гч-
6 JSn—F(l)=29 г(<; 7Sn—F (2)=25 гч: 7Sn -F (3)=208 ги-
15»
Таблица 11.29
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ FM НЕКОТОРЫХ ФТОРАЛКЕНОВ [79]
(1)Х X (3) \с—с'
(2)Х7 ХХ(4)
Химические сдвиги а, м. д.
Соединение X(l) X(2) X(3) X (4) 6(1) 6(2) 6(3) 6(4)
cf3cf=cf2 CF3 F F F 72 192 107 93
cf2cicf=cf2 CF2C1 F F F 58 185 106 95
CF3CF = CFMn(CO)5 CF3 F F Mn 67 165 95
CF3CF= CFFe(CO)2C5H5 CF3 F F Fe 66 166 86
а Химические сдвиги относительно CCI3F (внутренний эталон).
Константы взаимодействия, гц
Соединение X(l) X(2) X(3) X (4) Jzs J3 4 Jz 4 Ji 4 Ji z 71 3
cf3cf=cf2 CF3 F F F 120 57 40 8 13 21
cf3cci = cf2 CF3 Cl F F 12 21
4uc-CF3C1C = CFC1 CF3 Cl F Cl 23
cf2cicf = cf2 CF2C1 F F F 118 56 39 6 19 31
CF3CF = CFMn(CO)5 CF3 F F Mn 127 12 23
CF3CF = CFFe(CO)2C5H5 CF3 F F Fe 131 13 22
Рис. 11.7. Спектры ЯМР-F19 некоторых перфторвинильных соединений металлов [66].
Исследование ядерного магнитного резонанса фтора
229
перфторвинильных соединений металлов; каждый спектр состоит из трех квартетов линий, что характерно для трехспиновых систем со слабым взаимодействием типа АРХ. Квартет в спектре (CF2 =CF)3As, расположенный в сильном поле, имеет тонкую структуру, обусловленную дальним взаимодействием между магнитно неэквивалентными ядрами фтора в различных перфторвинильных группах (эта молекула представляет собой спиновую систему типа АА'А"РР'Р"ХХ'Х"). Подобные взаимодействия были обнаружены также при исследовании спектров фтора других перфторвинильных соединений металлов [66].
11.11.2. Другие фторалкены
В табл. 11.30 приведены химические сдвиги фтора для четырех галогензамещенных пропенов, измеренные относительно резонансной линии группы
Таблица 11.30
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ F19 ГАЛОГЕНЗАМЕЩЕННЫХ ПРОПЕНОВ [67] а (1)ХЧ X (3) (2) ХХ ХХ (4)
Э ®СЕзСООН=®СЕзСЕ2СООН ~ (м‘ Д-)> химические сдвиги измерены в миллионных долях
(м. д.) относительно сигнала СЕз-группы CF3CF2COOH (внешний эталон). Константы взаимодействия в герцах.
CF3 в CF3CF2COOH [67]. Атомы фтора, находящиеся в /пранс-положении по отношению к группе CF3, экранированы слабее, чем атомы фтора в ^ис-положе-нии. В CF3CF=CF2 атом фтора, находящийся в a-положении по отношению к CF3, экранирован очень сильно. Путмен, Андерсон и Шарки [69] показали, что спектр фтора 1,1,4,4-тетрафторбутадиена CF2=CHCH=CF2 состоит из одной широкой линии поглощения (химический сдвиг по отношению к CF3COOH составляет от +7,9 до 14,4 м. д.) х).
11.11.3. Константы взаимодействия для фторалкенов
Для фторированных олефинов спин-спиновое взаимодействие исследовано более подробно, чем химические сдвиги фтора. В ряде случаев была дана теоретическая интерпретация наблюдаемых значений констант спин-спинового взаимодействия (см. разд. 5.8.1). В табл. 11.31 приведены значения различных констант спин-спинового взаимодействия, найденные в результате анализа спектров ЯМР-F19 некоторых фторированных этиленов и пропенов [33, 67]. Большое различие в значениях констант взаимодействия между ядрами атомов фтора, присоединенных к одному атому углерода (эти константы изменяются в пределах от 16 до 87 гц), в известной степени обусловлено, по-видимому,
х) Исследованы также спектры ЯМР-F19 изомеров фторстнльбена [227], аф-днфтор-Р-хлорстирола [228, 229] н Р-фторстирола [230].— Прим, ред-
230
Глава И
Таблица 11.31
КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ F19 ФТОРАЛКЕНОВ (В ГЕРЦАХ) [33] (1)Х X (3)
_С'
(2) ХУ Ух (4)
X (1) X (2) Х(3) X (4) Jl 2 Jl 3 Jz3 Jl 4 Jz 4 Js 4
Н F С1 С1 81
Н Н F F ~4 ~1 34 34 ~1 37
Н Н F С1 ~3 8 40
F F Н С1 41 < 3 13
F F F С1 78 58 115
F F F Вг ' 75 57 124
F F F CN 27 35 118
F F F Н 87 33 119 12 < 3 72
F F F CF3 57 39 116 8 22 13
F F С1 CF3 12 21
С1 F С1 CF3 20
изменением углов между связями С — F. Попытки найти соответствие между этими константами взаимодействия и углами между связями основываются на предположении об основной роли контактного взаимодействия Ферми. Мак-Коннел, однако, показал, что при взаимодействии между ядрами фтора большое значение имеет диполь-дипольный член, вследствие чего определение прямой зависимости JFF от таких молекулярных параметров, как углы между связями, не представляется возможным [71].
Как следует из табл. 11.30 и 11.31, наблюдаемые значения констант подчиняются определенным закономерностям. Так, например, значения констант взаимодействия между ядрами атомов фтора, находящихся в транс-положении по отношению друг к другу, для всех исследованных молекул лежат в узкой области 118 ± 6 гц. В галогензамещенных пропенах [67] значения для групп в транс-положении приблизительно одинаковы (9,5 гц)', то же самое наблюдается для групп в ^uc-положении (JCf,-f ~ 23 гц). Следует заметить, что для взаимодействия CF3 — F ^мс-константы больше транс-кон-стант в противоположность взаимодействию между двумя ядрами фтора или водорода, непосредственно присоединенными к двум олефиновым атомам углерода. Согласно имеющимся данным, подобное соотношение между значе-
Таблица 11.32
КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ F—F цис- И транс-ФЕНИЛПЕРФТОРАЛКЕНОВ (В ГЕРЦАХ) [75]
А\ /В
>С=Сч
С6Н5/ ю
A в c JAC 7 AB JBC
F CF3 F 131 23 10
F F CF3 13 9 9
CF3 F CF3 1,5 28 7
CF3 CF3 F 12 12 7
Исследование ядерного магнитного резонанса фтора
231
ниями цис- и транс-констант проявляется также в протонных спектрах пропенов [72, 73].
Значения констант F — F, измеренные для некоторых цис- и транс-фенил-перфторолефинов, приведены в табл. 11.32 [75]. Поскольку предварительно была проведена тщательная идентификация спектров изомеров, полученные относительные значения цис- и транс-констант взаимодействия F — F и F — CF3 подтверждают найденные другими авторами соотношения между их величинами во фторалкенах, а именно:
I тЦис । I ттранс ।
I J F—F I I VF—F I’
I juuc । \ I ттранс । I JF-CF3 I । JF-CF3 I*
В работах [76—78] были исследованы относительные величины констант взаимодействия Н — F для фторалкенов. Из табл. 11.33 следует, что значения Jhf лежат в диапазоне 72—85 гц, изменяется от 0 до 20 гц, a Jhf™c — от 12 до 53 гц.
Таблица 11.33
КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ Н—F НЕКОТОРЫХ ФТОРАЛКЕНОВ
Соединение J8eM ?T1 ’’HF* ,гц J^aHC, гц Литература
fhc=ch2 +84,7 +20,1 +52,4 77
f2c=ch2 ~1 34 33
fcic=ch2 8 40 33
ci2c=chf 81 33
f2c=chci < 3 13 33
f2c=chf 72 < 3 12 33
цис-CHF = CHF a +72,7 6 +20,4 188
транс-CHF = CHF B +74,3 6 +4,4 188
JHH == Т 2’° гц ’ JFFC= ± 18,7 гч.
б Предполагаются положительными.
в j™Pa"c = ±9,5 гц, J™PaHC = Т 124,8 гц.
Таблица 11.34
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ F19 И Н1 цис- И транс-2-ФТОР-3-ХЛОРПРОПЕНА-2 [79]
Н3с. .Cl с = с F/ ЧН Т. кип. 30° Н3С ,Н с = с fz \л Т. кип. 60°
S ( = CH), м. д. S ( —CH3), м. д. S ( —F), м. д. 7pH’ гЧ ^FCHs’ ^НСНз> гЧ 1,99 5,86 12,66 10,8 16,85 1,14 1 92 ] б'31 / ($• (^4)4—внутренний эталон) 10,18 (CF3COOH—внешний эталон) 24,2 16,3 1,26
В табл. 11.34 приведены данные, полученные Рейлли при исследовании спектров Н1 и F19 цис- и транс-2-фтор-3-хлорпропена-2 [76], из которых можно сделать вывод, что значение J'hf меньше, чем Фнранс.
232
Глава 11
Койл, Стаффорд и Стоун [74] провели анализ спектра ЯМР-F19 цис-1,2-дифторэтилена CFH = CFH и получили следующие результаты:
др (относительно CFC13) = 168 м. д.,
J*“c=18,6 гц; 1™»тс = 20,1 гц; 3^ = 71,9 гц.
Бальдешвилер и сотр. [188] исследовали спектры ЯМР цис- и транс-1,2-дифторэтилена в газовой фазе и в результате их подробного анализа, включающего измерение ширины линий, определили относительные знаки констант взаимодействия (табл. 11.33).
Таблица 11.35 химические сдвиги И КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ для цис- И тран<?-2-ХЛОРГЕПТАФТОРБУТЕНА-2 [82]
CF3CC1 = CFCF3
1-CF3
ф * а 71з 714
транс цис +64,804 +64,180 24,72 8,67 1,27 11,32
4-CF3
ф * а ^41 7 43
транс цис +68,350 +66,274 1,33 11,43 5,49 7,74
3-F
ф * а 7з1 7з4
транс цис + 113,669 + 106,761 24,76 8,09 5,38 8,09
а Химические сдвиги измерены в миллионных долях (м. д.) относительно СС!зР (внутренний эталон) в растворе, содержащем 20 об.% исследуемого вещества.
Изучение спектра ЯМР-F19 цис- и транс-2-хлоргеп‘гафторбутена-2 позволяет различить цис- и транс-константы взаимодействия фтора с трифторметильной группой (см. табл. 11.35) [82]. Значения констант согласуются с найденными ранее для фторалкенов. Кроме того, было обнаружено взаимодействие между ядрами групп CF3, разделенных двойной связью, и установлено следующее соотношение между константами:
I rm-ранс t „ | гцис । I •'CF3-CF3I I •'CF3-CF3 I-
Подробный анализ спектров ЯМР-Н1 и ЯМР-F19 цис- и транс-1-фтор-пропенов [157] дал возможность определить спектральные параметры, приведенные в табл. 11.36, из которой следует, в частности, что
. гтпранс । \ । гцис I
I JF-СНз I t/F—СН3 *
Таблица 11.36
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ Н1 И F19 для 1-ФТОРПРОПЕНА [157]
н?сч .F 3 )с = с< Н6/ \нс н?с. ,нс 3 >с = с< НЬ/ XF
бенз’ м- д- — 1,596 — 1,498 . . в СЛабом полех
6нй, м. д. -4,720 — 5,300 II относительно j
бнс, м. д. -6,451 — 6,450 J 'ТМСвнутр
м. д. 131,4 129,5/в сильном полех
[ относительно ]
\CCI3F /
7нйсн3, гц 7,0 . 7,1
7нсснз> гц -1,8 -1,7
7fch3, гц 2,6 3,3
•1ньнс, гц +4,5 + Н,1
+т+, гц +41,8 + 19,9
^нст, гц +89,9 а +84,8а
а Произвольно предполагаются положительными.
Таблица 11.37
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ Н1 И F19 для цис- И транс- 1-ФТОР-2-(ПЕРФТОРИЗОПРОПИЛ)ЭТИЛЕНА [78]
Константы (CF«)2CF Fb (CF«)2CF Hb
взаимодействия, гц ° ;C = C< н / \нь a 0 ° ,c= c< +/ .
5,9 11,3
JKb?c 2,1 -0,0
JHbFb 77,3 78,3
JHbFa -0,0 -0,0
JHaVa 0,62 -0,15
JHaFc 22,7 21,1
JHaFb 37,0 16,0
Jvavc 7,6 7,0
Ji'bFc 31,0 5,0
JFaVb 5,8 0,56
Химические сдвиги а, м. д.
SFa 1,82 2,32
SFb 32,31 40,50
6fc 107,7 110,2
THa 5,10 4,41
THb 3,40 3,04
а Химические сдвиги фтора измерены относительно CF3COOH (внешний эталон). Протонные сдвиги приведены в шкале Тирса.
234
Г л а в а 11
Интересные сведения о константах взаимодействия F — F и Н — F были получены из резонансных спектров цис- и транс- 1-фтор-2-(перфторизопро-пил)этиленов (см. табл. 11.37) [78]. Хотя спектры Н1 и F19 для этих соединений являются спектрами первого порядка, они сильно усложнены спин-спиновым взаимодействием. На рис. 11.8 показан спектр двух ядер фтора в грс-изомере:
(cf3)2cf^c=c^f, на^ ''и»
И
, гогц t
ч II II 1.1. 1.1. Illi Illi II .1.1 II II
г,
Рис. 11.8. Теоретический и экспериментальный спектры ЯМР-F19 цис-1-фтор-2-(перфторнзопропил)этилена при частоте 56,4 Мгц [78].
а — спектр ядра Fc; б — спектр ядра Fb.
мультиплет ядра Fc (дублет дублетов септетов дублетов; см. рис. 11.8, а) состоит из 56 частично перекрывающихся линий, положение которых определяется следующими константами: 1рьрс =31,0 гц; Jhofc =22,7 гц; Jfc(Cf3)2= 7,6 г/; /ньрс =2,1 гц. Вид спектра ядра Fb (дублет дублетов дублетов септетов; см. рис. 11.8, б) определяется такими значениями констант: JnbFb = 77,3 гц; JHoFb = 37,0 гц; 7рьрс = 31,0 гц; ^Fb(CF3)2 = 5,8 гц [195] 1).
11.11.4. Относительные знаки констант взаимодействия
F19 во фторалкенах
Эванс [161] определил относительные знаки некоторых констант взаимодействия для фторалкенов с помощью метода двойного резонанса; полученные им данные приведены в табл. 11.38. Такое же соотношение между знаками констант, по-видимому, имеет место в большинстве фторалкенов. Результаты определения относительных знаков для широкого ряда соединений [24] приведены в табл. 11.13.
Эллеман и Менетт [153] показали, что для 1,1,2-трифтор-2-бромэтилена знак константы J™PaHa противоположен знаку и Рис. 11.9 иллю-
') Н. М. Сергеев и Н. С. Савченко исследовали зависимость вицинальных констант Jhf и от электроотрицательности заместителей во фторолефинах [231].— Прим. ред.
Исследование ядерного магнитного резонанса фтора
235
Таблица 11.38
ОТНОСИТЕЛЬНЫЕ ЗНАКИ КОНСТАНТ ВЗАИМОДЕЙСТВИЯ F19 НЕКОТОРЫХ ФТОРАЛКЕНОВ [161, 24]
CF2 = CFC1
CF2 = CFH
cf2=cfcf3
11 2 = тгем , JFF ±
73 4 = тгем , JHF
11 2 = тгем , JFF
7з 4 = тгем — JFF3 +
± Лз = 4р ± •^2 4 = ^HF + j13=4f ± /2 4 = 4^3 ±
J2 3=J^aHC +
12 3 = тпгранс -j- JFF +"
Л 4 = J™?aHC ±
123 — ттранс —- JFF ч"
1ц = тпгранс , JFF3
формы мультиплетной линии при
стрирует изменения щении определенных участков спектра
Для определения относительных знаков констант взаимодействия Н — F и F — F применяется метод двойного гетероядерного резонанса: спектр ЯМР для одного из ядер Н1 (или F10) снимается при одновременном облучении образца сильным полем с частотой, которая является резонансной для соответствующей группы в спектре другого ядра F19 (или Н1).
11.11.5. Сведения, получаемые при исследовании сателлитов, обусловленных взаимодействием С13 — F
Исследование сателлитов C13F в спектре ЯМР-F19 цис- и транс-С£С\ = = CFC1 дало возможность измерить относительные значения цис- и транс-констант взаимодействия F — F (см. табл. 11.39) [80]. По этим данным трансконстанта оказалась больше, чем цис-константа, что согласуется с результатами Рейлли [76]. Кроме того, было доказано, что константы С13 — F и С13—CF имеют противоположные знаки.
Тирс [81] провел анализ сложных мультиплетов C13F в спектрах F19 цис-и транс-2,3-дихлоргексафторбутена-2 (C1CF3C = CCF3CI); полученные им результаты приведены в табл. 11.40. Взаимодействие между ^ис-трифторме-тильными группами приблизительно на порядок сильнее взаимодействия между этими группами, находящимися в транс-положении [81, 82].
1) Аналогичный результат получен путем анализа спектра ЯМР-F19 при частоте 21,35 и 94,077 Мгц для CF2 = CF — ОС2Н5 [232].— Прим. ред.
избирательном насы-
Рис. 11.9. Спектр ЯМР-F19 1,1,2-трифтор-2-бромэтилена при частоте 56,4 Мгц [153].
а — исходный спектр после фазового детектора; б — спектр ядра А при облучении линий 5 и 6 и спектр ядра Р при облучении линий 2 и 4; й = 1008 гц; VHi/2n = 150 гц; в — спектр ядра А при облучении линий 9 и 10 в области л; Й = 2565 гц; тН1/2л = 150 гц; г — внд спектров двойного резонанса, предсказываемый теорией для случаев б н в, если ^АХ и ^РХ* а также АХ н ^РХ имеют раз-личные знаки; д — вид спектров двойного резонанса в тех же случаях, когда знаки констант одинаковы
235
Глава 11
Таблица 11.39 Таблица 11.40
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ спин-спинового ВЗАИМОДЕЙСТВИЯ цис- И mpaw-CFCl = CFC1 [80] ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ F19 цис- И транс- 2,3-ДИХЛОРГЕКСАФТОРБУТАНА~2*[8 1 ]
цис транс цис транс
<р, м. д. + 105,079+0,004 + 119,602+0,003 <р, м. д. +60,446 +63,730
JC13-F19’ +299,26+0,27 +289,57+0,42 Д<р (C^Fg-C^Fg), +0,137 +0,132
J cis—pig, гЦ - 38,10+0,27 — 53,85 + 0,42 м. д.
JPF' ’ 37,50+0,10 129,57+0,14 ^cisF’ гЧ 275,3 275,7
^CFa-CFs’ г1‘ 13,4 1,44
а Произвольно принята положительной.
11.12. Фторацетилены
Ядро фтора, присоединенного к ацетиленовому атому углерода, сильно экранировано электронами, и его резонансная линия оказывается смещенной в сторону сильного магнитного поля. Химический сдвиг резонанса фтора для фторацетилена F — С =С—Н составляет +181,3 м. д. по отношению к резонансу CF3COOH (внешний эталон); константа взаимодействия 7hf для фторацетилена составляет 21 гц [83].
11.13. Фторированные циклические соединения
Метод ядерного магнитного резонанса применялся для исследования молекулярной структуры большого числа циклических’молекул [16, 84—89, 93]. В тех случаях, когда циклические молекулы не являются плоскими (как, например, производные циклогексана), возникают проблемы, связанные с существованием конформационных изомеров, которые также могут быть исследованы методом ЯМР [16, 86—89, 93].
11.13.1. Производные циклопропана
Филлипс [85] исследовал ЯМР-F19 1-метил-2,2-дифторциклопропана при частоте 30 Мгц и показал, что спектр согласуется с циклической структурой молекулы. Этот спектр приведен на рис. 11.10, в. Два геминальных атома фтора в этой молекуле неэквивалентны из-за их различного положения по отношению к метильной группе, вследствие чего спектр состоит из двух дублетов типа АВ (см. разд. 8.13.1) с химическим сдвигом 16,8 м. д. и геминальной константой взаимодействия между ядрами фтора, равной 157 гц. Тонкая структура линий обусловлена спин-спиновым взаимодействием ядер фтора с протонами в соседних группах СН и СН2. Линии, расположенные в более сильном поле, имеют заметное дублетное расщепление в результате взаимодействия с группой СН, в то время как компоненты спектра в слабом поле уширены нз-за взаимодействия с обеими соседними группами. Этот спектр не дает возможности отнести пары линий в сильном и слабом поле к определенным ядрам фтора.
Исследование ядерного магнитного резонанса фтора
237
Н --
____л
8fT
Рис. 11.10. Спектры ЯМР-F19 при 30 Мгц [85].
а — 1,1-диметил-2,2,3,3-тетрафторциклобутан; б — 1-фенил-2,2;3,3-тетрафторциклобутан;
$ — 1-метил-2,2-дифторциклопропан. Химические сдвиги измерены в герцах относительно внешнего эталона — тетрафторциклобутаиа.
11.13.2. Моно- и дизамещенные производные тетрафторциклобутана
Спектр F19 1,1,2,2-тетрафторциклобутана состоит из одной линии, что свидетельствует об одинаковом экранировании ядер фтора в этой молекуле [85]. Этого можно было ожидать, если молекула имеет плоскую структуру или совершает быстрые переходы между двумя неплоскими структурами.
При соответствующем замещении эквивалентность ядер фтора в молекуле нарушается, что следует из спектров моно- и дизамещенных производных циклобутана. Иллюстрацией может служить спектр Р19-резонанса 1,1-диметил-2,2,3,3-тетрафторциклобутана, изображенный на рис. 11.10, а. Замещение двумя метильными группами не нарушает эквивалентности геминальных атомов фтора в каждой из групп CF2, но приводит к появлению химического сдвига между линиями этих групп. Линия, находящаяся в более слабом поле, расщеплена в триплет из-за взаимодействия с двумя эквивалентными протонами группы СН2. Менее симметричное замещение молекулы приводит к появлению неэквивалентности двух атомов фтора в группе CF2. Каждая пара неэквивалентных геминальных атомов фтора дает два дублета типа АВ, как показано на рис. 8.7. Примером может служить также спектр фтора 1-фенил-2,2,3,3-тетрафторциклобутана (рис. 11.10,6). В этом случае неэквивалентны все четыре атома фтора, и спектр состоит из двух перекрывающихся квартетов типа АВ.
238
Глава 11
Исследуя ряд замещенных тетрафторциклобутанов, можно получить сведения, позволяющие провести в спектрах идентификацию линий групп CF2. Так, например, линии /, 3, 7 и 8 на рис. 11.10, б отвечают группе CF2 в положении 2, а линии 2, 4, 5 и 6 — группе CF2 в положении 3. В табл. 11.41 содержатся
Таблица 11.41
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ НЕЭКВИВАЛЕНТНЫХ ГЕМИНАЛЬНЫХ ЯДЕР ФТОРА В ТЕТРАФТОРЦИКЛОБУТАНАХ [85]
Соедииеиие J3 а 63 а J’i 6 62 6
f2 f ^2
224 7,43 230 23,8 CH2C1
f2 f F2 4 w2
206 10,0 206 23,5 C«HS
F F2 2 H. H f1 211 8,50 202 22,5 C —CH2
f2 H F2 f 2 210 2,90 213 22,4 OCH,
I F ?2 2 H2 4 219 5,53 ' 218 21,0 CH2OH
f2 f2 h 1 H 2
220 7,73 220 20,5 CH = CH2
f2 4 4
а J2 и J3 — константы взаимодействия (гц) между двумя неэквивалентными геминальными ядрами фтора в положениях 2 и 3 соответствеиио.
® 62 и 6з — химические сдвиги (м. д.) между сигналами двух неэквивалентных геминальных ядер фтора.
данные, полученные из спектров ЯМР-F19 некоторых моно- и дизамегценных тетрафторциклобутанов [85]. В нескольких случаях резонансные линии фтора имеют тонкую структуру,' обусловленную спин-спиновым взаимодействием ядер фтора с соседними протонами.
Среднее значение констант взаимодействия между ядрами геминальных атомов фтора в тетрафторциклобутанах составляет 211 гц. Было установлено также, что разница в экранировании геминальных ядер фтора в группе CF2 обычно больше для монозамещенных производных тетрафторциклобутана, чем
Исследование ядерного магнитного резонанса фтора
239
для дизамещенных. Это согласуется с представлением о том, что экранирование в таких молекулах может определяться либо асимметричным индуктивным эффектом, либо непосредственным взаимодействием фтора с заместителем, поскольку в каждом из этих случаев значения химических сдвигов фтора в дизамещенных производных будут приблизительно равны разности между химическими сдвигами для двух соответствующих монозамещенных производных циклобутанов.
В более ранней работе Шулери [84] исследовал несколько замещенных фторциклобутанов со структурами типа
CF2 —СН2 CF2—CHR
II II
СС12—CHR СС12—СН2
где R обозначает группы: — Si(OC2H5)3, — Si(CH3)3 и — Si(CH3)2R'. Он показал, что во всех исследованных молекулах геминальные атомы фтора неэквивалентны вследствие несимметричного замещения атомов, присоединенных к циклическим атомам углерода плоского кольца. Молекула
CF2—СН2
F2—С—S i (СН3)3
I н
обладает двумя различными парами неэквивалентных геминальных атомов фтора; спектр ЯМР-F18 для этой молекулы очень похож на изображенный на рис. 11.10, б.
Интересным примером молекул, в которых не наблюдается монотонного уменьшения констант взаимодействия Н — F с ростом числа связей между соответствующими атомами, являются замещенные циклобутены, исследованные Шартсом и Робертсом [18]. В молекулах типа
С6Н5С—CFC1 С6Н5С—СН2
II I “ II I
НС—CF2 FC—CF2
взаимодействие Н — F между ядрами атомов, расположенных по диагонали и разделенных четырьмя связями (Jhf = 8 — 12 гц), значительно больше взаимодействия между спинами соседних атомов водорода и фтора через три связи (Jhf — 1—2 гц).
Методом ЯМР были исследованы также некоторые фторсодержащие производные циклобутанов [90] с общей формулой
H2C-CRR'
F2C—CFX
Молекулы циклобутана и замещенных циклобутанов имеют слегка изогнутое кольцо, что должно проявляться в экспериментах по ядерному резонансу. Филлипс [92] наблюдал температурную зависимость спектра ЯМР-F18 1-хлор-2,2,3,3-тетрафторциклобутана
F2C—СН2
I I
F2C—СНС1
которую он объяснил изменением заселенностей более высоких колебательных уровней изогнутого кольца при изменении температуры х).
В спектре F1® перфторциклобутана наблюдается узкая синглетная линия вплоть до температуры —102°, что указывает на быструю инверсию молекулы [257].— Прим. ред.
240
Глава И
11.13.3. Фторированные циклогексаны
Молекулы циклогексана и его производных имеют форму неплоских шестичленных циклов, обусловленную стремлением кольцевых атомов углерода занять положения, в которых связи С — С образуют друг с другом тетраэдрические углы. Наиболее устойчивой в этом смысле является структура, имеющая форму «кресла» (см. разд. 9.7.1). В этой структуре связи атомов углерода, не принимающие участия в образовании кольца, могут занимать экваториальное (е) и аксиальное (а) положения по отношению к кольцу. Экваториальные связи направлены к периферии молекулы, аксиальные — вверх и вниз относительно кольца. Одновременное вращение вокруг отдельных связей С — С приводит к тому, что заместитель, первоначально находившийся в аксиальном положении, переходит в экваториальное положение, т. е. для монозамещенных производных циклогексана существуют две формы молекулы, каждая из которых характеризуется определенным пространственным расположением атомов и называется конформационным изомером.
Тирс [94] показал, что образование кольца приводит к сдвигу резонанса группы —CF2— в область сильного поля приблизительно на 10 м. д. Так, например, в перфторциклогексане (133,25<р) и перфторциклобутане (134,94q>) группы CF2 экранированы сильнее, чем те же группы, расположенные в середине длинной цепи н-перфторалкилов (ср ~ 122,5). Аналогичные эффекты наблюдались для циклических фторорганических сульфидов (см. табл. 11.18а).
Перфтор циклогексан СвН12. Спектр ЯМР-F19 перфторциклогексана состоит из одной широкой линии поглощения. При охлаждении раствора этого вещества в трихлорфторметане и трифторметилбензоле до —66° эта линия превращается в квартет типа АВ [86]. При комнатной температуре происходят быстрые переходы между конформациями циклогексана (см. разд. 9.7), вследствие чего времена жизни атомов фтора в аксиальном и экваториальном положениях становятся одинаковыми и настолько малыми, что наблюдается одна линия поглощения. При понижении температуры времена жизни атома фтора в каждом из положений увеличиваются до тех пор, пока при —66° скорость взаимных превращений становится достаточно малой для наблюдения отдельных резонансных линий от атомов в аксиальных и экваториальных положениях. Химический сдвиг между линиями поглощения для атомов фтора, находящихся в этих положениях, составляет 18,2 м. д.; константа взаимодействия между геминальными атомами фтора равна 284 + 1 гц- Последняя значительно превосходит по величине аналогичные константы во фторциклобутанах (Jff = = 220 гц) [85] и фторциклопропанах (JFf = 160 гц) [85].
Монозамещенные перфторциклогексаны. Монозамещенные молекулы циклогексана при комнатной температуре имеют обычно преимущественную структуру, в которой заместитель занимает экваториальное положение. В этой «фиксированной» структуре существуют три различные пары неэквивалентных геминальных ядер фтора, что можно видеть на примере структуры молекулы перфторметилциклогексана, изображенной на рис. 11.11. Очевидно, что в данном случае атомы фтора в положениях (2, 6), (3, 5) и 4 неэквивалентны; кроме того, два атома фтора, присоединенные к каждому из углеродных атомов, также неэквивалентны, поскольку они находятся в экваториальном и аксиальном положениях. Таким образом, в спектре ЯМР этой молекулы каждая из трех групп неэквивалентных геминальных атомов фтора дает квартет типа АВ [16] (см. рис. 11.11). Аналогичные спектры фтора наблюдались и в других монозамещенных перфторциклогексанах, в том числе в моногидро- и монохлорпро-изводных (существует убедительное доказательство существования быстрых конформационных превращений молекулы моногидроперфторциклогексана при комнатной температуре) [16, 87].
Дигид'роперфторциклогексаны CBFц>Н2. В работах [16, 87, 93] было проведено исследование спектров ЯМР -F19 полного набора изомеров дигидропроиз
Исследование ядерного магнитного резонанса фтора
241
водных перфторциклогексана. При интерпретации спектров использовались те же соображения, что и в случае монозамещенных производных. В некоторых случаях удалось точно установить, существуют ли быстрые конформационные превращения в молекулах при комнатной температуре. При отнесении линий в спектрах большую помощь оказывает использование данных, полученных при исследовании перфторциклогексана в низкотемпературном интервале. Если в спектре фтора появляется квартет типа АВ с химическим сдвигом ~ 18 м. д., характерный для геминальных атомов фтора в перфторциклогексане
।—।----1 I
!__I-------1_I
Рис. 11.11. Резонансный спектр кольцевых атомов F19 перфтор метилциклогексана при частоте 56,4 Мгц.
при низких температурах, то можно с уверенностыЪ сделать вывод об отсутствии быстрых конформационных превращений. Гомер и Томас [93] показали, что при комнатной температуре ^ис-1Н,ЗН-изомер совершает быстрые переходы между неэквивалентными креслообразными формами. Отнесение линий в спектре дигидродекафторциклогексанов (табл. 11.42) дает возможность установить, что близость к атому водорода обычно приводит к уменьшению экранирования ядер фтора в соседней группе CF2 (см. ниже правила I и III). Однако большое различие в экранировании экваториальных и аксиальных атомов фтора в перфторциклогексане, где индуктивное влияние соседних групп, по-видимому, имеет такую же величину, свидетельствует о том, что в замещенных фторциклогексанах основную роль для экранирования ядер фтора играют пространственные взаимодействия (см. разд. 4.4.4).
Исследование спектров фтора для большой группы моно-, ди-, три-и тетразамещенных перфторциклогексана позволило сформулировать некоторые эмпирические правила, связывающие величину экранирования ядер фтора с природой соседнего заместителя [93]. Например, для экранирования групп CF2 во фторциклогексанах были получены следующие правила аддитивности:
I. В группе, находящейся между двумя группами CF2, ядро аксиального атома фтора (6Cf3cooh = +47,4 м. д.) экранировано слабее, чем ядро экваториального атома (6cf3cooh = + 65,1 м. д.), на 17,7 м. д.
II. Аксиальный а-атом водорода, увеличивает экранирование Fa на 2,3 м. д. и уменьшает экранирование Fe на 15,0 м. д.
16—489
Таблица 11.42
ХИМИЧЕСКИЕ СДВИГИ F19 И ИХ ОТНЕСЕНИЕ ДЛЯ НЕКОТОРЫХ ДИГИДРО ДЕКАФТОРЦИКЛОГЕКСАНОВ [87]
Соединение Отнесение а Порядковый номер линии Химический сдвиг, гц
1 1265
Цис-1Н,ЗН, т. кип. 90,9° 2 1570
2, 2' 4 1790
Н 6 2050
б/\,2 4, 6 3 1650
51 1н 5 1930
6 2050
4 4', 6' 8 2330
5, 5' 7 2250
10 5730
I'’3' { 11 5770
Ч«с-1Н,4Н, т. кип. 85,4° 1 1670
2, 3, 5, 6 j 2 1950
Н 2', 3', 5', 6' - 3 2080
6м2 4 2370
\/3 1' 4' / 5 5940
Н 1 ’ 4 1 6 5980
транс-1Н,2Н, т. кип. 70° 4а, 5а 1 1790
3 2070
6|/Ч]На 2 1860
Зе, За, бе, ба ! 4 2155
\/3 L 5 2420
4 4е, 5е и 6 2550
7 . 284&
г 8 5850
le, 2е j 9 5900
Смесь транс- 1Н,ЗН и
транс- 1Н,4Н, т. кип. 79°
Н 2, 2' 1 1815
/ X 2 2010
6, ,2 4, 4 , 6,б' j 3 2085
5\/Н 5, 5' 5 2255
4 1 3 / 6 5860
1,3 j 7 5905
На
б/\2 2е, 2а, Зе, За 4 2100
1 Г 5е, 5а, бе, ба 4 2100
V 1е, 4е 8 9 6120 6165
а Штрихом обозначены атомы, занимающие транс-положение по отношению к ближайшему атому водорода в молекуле, совершающей конформационные переходы. Скобками' объединены компоненты мультиплетной структуры линий. Химический сдвиг CgFia равен* + 2200 гц (55 м. д.) относительно CF3COOH (внешний эталон). Некоторые из приведенных отнесений были сделаны в работе [93].
Исследование ядерного магнитного резонанса фтора
243
III. Экваториальный а-атом водорода уменьшает экранирование Fa на 4,3 м. д. и Fe — на 10,5 м. д.
IV. Аксиальный 0-атом водорода увеличивает экранирование Fe приблизительно на 4 м. д. 1).
Таблица 11.43
ХИМИЧЕСКИЕ СДВИГИ F19 НЕКОТОРЫХ
ФТОРЦИКЛОГЕКСЕНОВ [78] а
Соединение Отнесение Химические сдвиги F19, м. д.
Yi
40,9
55,6
74,6
Перфторциклогексен
Y| ]а
76,9
86,0
48,6
Перфторциклогексадиен-1,3
;а
80,9
36,6
Перфторциклогексадиен-1,4
88,0
Перфторбензол
а Химические сдвиги измерены в м. д, ний эталон).
& Этн отнесения предположительны.
относительно CFgCOOH (внеш-
цис- и транс-Перфтордекалин. Исследование этих молекул методом ядерного резонанса подтвердило, что при комнатной температуре t^uc-изомер совершает быстрые конформационные превращения, в то время как транс-изомер образует жесткую конформацию (см. рис. 11.12) [88]. В транс-изомере существуют две группы неэквивалентных геминальных атомов фтора, каждая из которых в спектре F19 этой молекулы дает квартет типа АВ (рис. 11.12, б). Один из квартетов АВ (отмеченный на рисунке буквами а) характеризуется химическим сдвигом 18,2 м. д., который наблюдался также в спектре фиксированного конформера CeF12 при низких температурах, что является подтверждением жесткости этой молекулы; оставшаяся пара дублетов может быть поэтому отнесена к атомам фтора, соседним по отношению к ангулярному положению. В цис-изомере из-за быстрых превращений существуют две различные группы эквивалентных геминальных атомов, которые в спектре фтора дают две синглетные линии поглощения, как показано на рис. 11.12, а.
х) Исследованию влияния электростатического поля заместителей на химический сдвиг F18 для моно- и дизамещеииых перфторциклогексанов посвящена работа Эмсли [258].— Прим. ред.
16*
244
Глава И
Фторированные циклогексены. Сильное взаимодействие между химически эквивалентными ядрами фтора в перфторциклогексене и перфторциклогексадие' нах сильно усложняет спектры ЯМР-F19 этих молекул. В табл. 11.43 приведены
Рис. 11.12. Спектры ЯМР-F18 при 30,1 Мгц [88].
а — цис-перфтордекалин; б — /пранс-перфтордекалин. Частоты измерены относительно внешнего эталона — трнфторуксусной кислоты.
значения химических сдвигов для нескольких молекул этого типа; для сравне-ния туда же включено значение химического сдвига перфторбензо л а [78]. Методом ЯМР были определены структуры некоторых комплексов переходных металлов перфторциклогексадиена-1,3 и диена-1,4 [70].
11.13.4. Фторированные гетероциклические соединения
Перфторпиперидин CsNFu- Вследствие быстрых конформационных превращений в молекуле перфторпиперидина существуют четыре различных типа ядер фтора: в а-, р- и у-группах CF2 и группе ]4F. Поскольку средние значения
Н
Рис. 11.13. Спектры ЯМР-F19 раствора перфторпиперидина в CC13F (частота 40 Мгц) при двух различных температурах [95].
констант взаимодействия Ja6 и Jвт, как известно, малы и соответствующие мультиплеты не разрешаются, можно ожидать, что спектр фтора этой молёкулы будет состоять из четырех уширенных синглетов. На рис. 11.13 показаны
Исследование ядерного магнитного резонанса фтора
246
спектры фтора для раствора перфторпиперидина в CC13F, снятые на частоте 40Л4гц при разных температурах [95]. При температурах выше —40° наблюдаются четыре отдельные линии поглощения, указывающие на то, что в молекуле происходят быстрые конформационные превращения. При понижении температуры линия группы NF остается неизменной, а линии а-, р- и у-групп CF2 превращаются в квартеты типа АВ, поскольку ядра геминальных атомов фтора становятся неэквивалентными. Неизменность линии поглощения от группы NF даже при температуре —115° показывает, что атом фтора в этой
Таблица 11.44
ХИМИЧЕСКИЕ СДВИГИ Й ГЕМИНАЛЬНЫЕ КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ F1S ПЕРФТОРПИПЕРИДИНА [95]
Ядро фтора Константа взаимодействия, гц Химический сдвиг а, м. д.
i J ae Fa Fe NF
F2y a *. . 185 + 113,7 + 105,2
F2|/X|F2₽ p 278 + 140,9 + 122,1
F2\ /F2a N F Y NF 284 + 142,1 + 125,1- + 112,9
’Химические сдвиги измерены относительно CCI3F (внутренний эталон).
группе совершает быстрое движение. В табл. 11.44 приведены значения химических сдвигов и констант спин-спинового взаимодействия фтора для перфторпиперидина; эти величины подобны измеренным для фторциклогексанов [16].
(CF2)tPe(CO)t [96]. В этом циклическом соединении к атому металла сим-
,CF2-CF2
(CO)4Fe< |
+cf2-cf2
метрично присоединены группы СО, и потому спектр фтора, как и следовало ожидать, состоит из двух мультиплетов (химические сдвиги относительно внутреннего эталона — трихлорфторметана — составляют: 6а = + 70,6 м. д. и 6В = + 136,9 м. д.; Ja$ = 2,4 гц).
При несимметричном замещении гетероциклических соединений, например, для молекулы
R CF2-CF2
/Со\ I ос cf2—cf2
6<x = +67,5 м. д.,
6В == + 135 м. д.,
где R — циклопентадиенил, геминальные атомы фтора в a-положении неэквивалентны и в спектре F19 дают квартет типа АВ с бдв = 6.25 м. д. и J = 218 гц: При этом только одно из ядер фтора в a-положении взаимодействует с ядрами фтора в p-положении (JaB = 4,3 гц).
Фторированные гетероциклические производные серы, селена и фосфора. Креспан и Лангкамерер [97] провели исследование спектров ЯМР некоторых фторированных гетероциклических производных (см. табл. 11.45). Резонансный
246
Глава И
спектр фтора для молекулы
I I Р
FaC7 XCF2 \ I
f2c cf2
р7
I
указывает на существование фиксированной конформации, в которой ядра геминальных атомов фтора в каждой из групп CF2 неэквивалентны (Jf(1>f(2) = = 280 гц, JpF(i) = 128 гц и JPF(2) = 54 гц). При нагревании образца до 100°
Таблица 11.45
ХИМИЧЕСКИЕ СДВИГИ F19 НЕКОТОРЫХ ФТОРИРОВАННЫХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ [97] а
Соединение
б, м. д.' CF2-M
б, м. д. **
—С—CF2—С
+ 19,7 +6,44
+23,4
+ 18,2
+24,4
+46,3
+64,9
а Химические сдвиги измерены относительно CFCI2CFCI2 (внешний эталон); бСРзСОон (виешн) = eC12FCCFCl2(внешн) — 12>° (“• Д-) (это соотношение является весьма приближенным).
никаких изменений в спектре не наблюдалось, что свидетельствует о высоком потенциальном барьере, препятствующем взаимным превращениям из аксиальной конформации в экваториальную.
Другие циклические соединения. Ито и сотр. [156] измерили химические сдвиги фтора для 2- и 3-фторпиридинов, 1-фторпирена и циклогексилфторида и показали, что величина химических сдвигов хорошо соответствует л-электрон-ной плотности кольцевого атома углерода, к которому присоединен фтор. Это дает возможность предположить, что изменения экранирования обусловлены в основном полярностью о-связи CF 1).
х) Исследованы константы спин-спинового взаимодействия F1’ — Н1 ряда фторированных гликопираноз [259].— Прим. ред.
Исследование ядерного магнитного резонанса фтора
247
11.14. Метил- и этилфторсиланы RxSiF4_x
Шнелль и Рохов [98] исследовали зависимость магнитного экранирования атомов фтора во фторсиланах от природы и числа алкильных заместителей х). Данные, полученные ими, приведены на рис. 11.14 в виде графика. Эти авторы считают, что двумя наиболее важными факторами, определяющими экранирование ядер фтора во фторсиланах, являются: индуктивный эффект, который должен приводить к увеличению экранирования при введении в молекулу электронодонорной алкильной группы, и противоположный ему эффект,
Рис. 11.14. Зависимость химических сдвигов F19 метил- и этилфторсилаиов RxSiFi-x от числа алкильных заместителей [98].
6CF3COOH = 6<C2H5>2S1F2 + 66,6 (м' д,)
обусловленный d„— ря-взаимодействием в связи Si — F [99]. Результаты измерений, о которых упоминалось выше, не дают возможности оценить относительный вклад каждого из этих эффектов; однако наличие минимумов на кривых рис. 11.14 указывает на то, что эти эффекты противоположны по знаку. Ядра фтора в этильных производных экранированы сильнее, чем в метильных, чего и следовало ожидать на основании простых представлений об индуктивном эффекте.
11.15. Пентафторид сурьмы
При комнатной температуре пентафторид сурьмы представляет собой вязкую полимерную жидкость; возможная структура этой молекулы была установлена с помощью метода ЯМР [100]. При —10° спектр F19 этой молекулы состоит из трех линий поглощения с отношением интенсивностей 1:2:2, что указывает на присутствие трех неэквивалентных групп фтора (см. рис. 11.15).
х) См. также [272].— Прим. ред.
248
Глава И
Подобный спектр может отвечать молекуле с длинной цепью, составленной из групп SbF?, что показано на рис. 11.16: каждые две из этих групп имеют
Химический сдвиг от сигнала CF3C00H (внешний эталон)
Рис. 11.15. Спектры ЯМР-F1® пентафтор нда сурьмыпрн частоте 40 Мгц[100]. а — при температуре около —10°; б.— при температуре около 80®.
один общий атом фтора (типа А). Из четырех остальных атомов фтора два (типа В) находятся в mpawc-положении по отношению к А и два (типа С) —
Рис. 11.16. Предлагаемая
структура полимерного пеи-тафторида сурьмы [100].
в транс-положении по отношению друг к другу. Тонкая структура линий поглощения возникает из-за взаимодействий между неэквивалентными ядрами фтора, которые характеризуются следующими значениями констант: 7дв ~ 70 гц, /вс « 130 гц и /дс < 30 гц. При комнатной температуре спектр F19 этой молекулы состоит из одной линии, что указывает на быстрый обмен атомов фтора. На рис. 11.15,6 представлен спектр F19 этого соединения при температуре около 80°.
11.16. Соединения, содержащие связи F — S
11.16.1 Гексафторид серы и его монозамещенные производные
В гексафториде серы атомы фтора расположены в углах правильного октаэдра (рис. 11.17} и молекула имеет бипирамидальную тетрагональную структуру. При замещении одного из ато
Исследование ядерного магнитного резонанса фтора
249'
мов фтора оставшиеся атомы образуют пирамиду с квадратным основанием, что приводит к различному экранированию ядер фтора, находящихся в. основании и вершине этой пирамиды (см. рис. 11.17, б). Исследование спектров ЯМР-F19 монозамещенных молекул гексафторида серы (SF5X) подтверждает указанную структуру, поскольку спектры состоят из, двух групп линий с отношением интенсивностей 1 : 4. Константы взаимодействия между
а б в
г
Рис. 11.17. Молекулярная структура SFe и некоторых его производных.
ядрами атомов фтора, находящимися в основании и вершине пирамиды, сравнимы по величине с химическим сдвигом между соответствующими линиями поглощения (даже при частоте 56,4 Мгц). Поэтому спектры фтора, наблюдаемые для молекул SF5X, относятся к спектрам типа АВ4 (см. разд. 8.13.4) [101]. Для этих молекул спектр ядра А состоит из 9 линий, а спектр ядер В — из 12 линий [103]. Пример спектра системы АВ4, типичный для молекул этого класса, изображен на рис. 11.18.
Меррилл и сотр. [102] исследовали Спектры F19 ряда пентафторидов серы; полученные ими данные приведены в табл. 11.46. Линии поглощения ядер фтора, присоединенного непосредственно к атомам серы, смещены в область слабого поля. Во фторалкильных производных, как, например, C3F7SF5 и и C4F9SF5, ядра фтора группы a-CF2 взаимодействуют с базисными (в основании пирамиды) и аксиальными (при вершине) ядрами фтора группы —SF5. Для CF3SF5 константа взаимодействия между CF3 и базисными ядрами фтора составляет 22 гц, в то время как соответствующая константа ’взаимодействия
Рис. 11.18. Спектр ЯМР-F18 SF5OCeH5 при' частоте 40 Мгц [105].
с аксиальным ядром фтора равна 6,4 гц. В некоторых алкильных производных —SF5 (например, CICH2CH2SF5) протоны группы а-СН2 взаимодействуют только с базисными ядрами фтора в группе SF6 (Jch2-fb = 6,8 гц) [103]. Во всех исследованных молекулах константа взаимодействия между базисными и аксиальными ядрами фтора близка к 150 гц. Ядро атома фтора, расположенного при вершине пирамиды, обычно экранировано сильнее базисных ядер, однако для алкильных производных наблюдается обратная картина (см. табл. 11.47). [103, 104]. При изменении заместителя возникают значительные изменения величины экранирования базисных ядер фтора. Приблизительно сходные
250
Глава И
Таблица 11.46
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ МЕЖДУ ЯДРАМИ ФТОРА В ОСНОВАНИИ И ВЕРШИНЕ ПИРАМИДАЛЬНЫХ МОЛЕКУЛ ДЛЯ МОНОЗАМЕЩЕННЫХ ПРОИЗВОД-
НЫХ ГЕКСАФТОРИДА СЕРЫ SFBX [102]
Соединение 6 a, M. Д. /дв> гч
F в основании F при вершине
SF5Br -88,2 —5,00 143,1
SF5C1 —68,4 —4,88 148,5
cf3sf5 18,4 —5,43 145,4
sf5oso2f — 14,8 1,63 153,5
(Sf5)2so4 —15,1 0,43 153,4
fc2h4osf5 —2,48 —17,98 153,8
fcic2h3osf5 —2,69 —15,93 154,7
c2f5osf5 —15,2 —3,35 152,8
C5F9OSFb — 14,7 —4,00 154,9
fc2ci4osf5 —14,8 —6,37 154,9
cf3osf5 —11,4 —4,23 153,0
sf5osf5 — 14,0 —4,53 150
sf6oosf5 0,75 0,16
а Химические сдвиги, приведенные здесь, получены для 50%-ных растворов в SFB и потому могут содержать небольшие ошибки. Однако изменения сдвига в родственных соединениях SO3F2 и SFgOF составляли менее 0,3 м, д. при изменении концентрации SFe от 10 до 90 мол.%. Положительные сдвиги соответствуют линиям, смещенным в область сильного поля относительно сигнала SFe. 6cF3COOH (внешн) = eSFe “ 127 (“• Д-) (весьма приближен-ное соотношение).
Таблица 11.47
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ МЕЖДУ ЯДРАМИ ФТОРА ПРИ ВЕРШИНЕ И В ОСНОВАНИИ ПИРАМИДЫ ДЛЯ РЯДА АЛКИЛЬНЫХ ПРОИЗВОДНЫХ С ОБЩЕЙ ФОРМУЛОЙ SFBR [103]
Соединение <p* a, M. Д. JABi ги-
F прн вершине F в основании
ch2=chsf5 —80,9 —59,3 147,8
CH3CHC1CH2SFb —82,2 —64,9 144,2
cich2ch2sf6 —81,1 —64,3 144,4
а Значения <р* измерены относительно внутреннего эталона ®CF3COOH = fCFCls — 78.6 (м. д.).
CFCI3;
изменения экранирования были отмечены для фторалканов, когда к группе CF2 присоединены такие же заместители. Направление изменения, однако, противоположно ожидаемому на основании электроотрицательности [8, 103]. Так, например, уменьшение экранирования наблюдается в следующем ряду заместителей: F > CF3 > С1 > Вг. Нерегулярные изменения экранирования ядер фтора, расположенных при вершине пирамиды группы —SF5, сравнительно невелики. Если полагать, что индуктивный эффект одинаков для всех атомов фтора в группе —SF5, то, поскольку базисные атомы фтора расположены ближе к заместителю, чем атом фтора при вершине пирамиды, возникает мысль
Исследование ядерного магнитного резонанса фтора
251
о возможности объяснения больших вариаций в экранировании базисных ядер пространственным взаимодействием с заместителем. Однако влияние магнитной анизотропии соседнего атома слишком мало, чтобы объяснить наблюдаемое экранирование, поэтому более вероятно, что значительное дезэкранирование базисных атомов фтора обусловлено низколежащими возбужденными электронными состояниями, обладающими подходящей симметрий для смешивания с основным состоянием [102, 56].
Таблица 11.48
ХИМИЧЕСКИЕ СДВИГИ F1» ПЕРФТОРАЛКИЛЬНЫХ ПРОИЗВОДНЫХ ГЕКСАФТОРИДА СЕРЫ [101]
Группа C2F5SF5 C3F7SF5 C4F9SF5 (C2F6)2SF4 (C3F7)2SF4 o/cf2cf2x '4cf2cf2/ >sf4
- CF3 С—CF2 —S 5,3 15,5 5,0 18,8 5,2 18,0 4,5 21,2 5,0 16,5 22,2
й й о л 71 ’’111 СП 1 I о о Ч] 4] ьэ м 1 1 о о —118,7 а-?- —137,5 50,7 б 46,5 49,5 б —104,0 50,0 —106,1 2,4 —123,0 в —94,8
а Значения в для ядер фтора при вершине и в основании пирамиды SFj получены в результате ана-лиза мультиплетной структуры спектра.
® Резонансный сигнал SFg имеет точно такой же вид, как для CaFgSFg.
в Эти значения соответствуют центрам триплетов, возникающих вследствие неэквивалентности ядер фтора в группе SF4 этой молекулы.
Таблица 11.49
ХИМИЧЕСКИЕ СДВИГИ F19 ПЕРФТОРАЛКИЛЬНЫХ ПРОИЗВОДНЫХ
ГЕКСАФТОРИДА СЕРЫ [104]а
Соединение 6sf 6 6sf4 6CF3S eCF2-s 6cf2-c ®CF2C—s eCF2CC—S
(CF3CF2)2SF4 -105,0 21,0 4,5
•cf3sf4cf2cf3 -99,7 -11,4 21,4 4,5
cf3sf4cf2cooch3 -99,7 -12,7 12,1
CFgCFzSFs —138,0 -118,5 24,0 5,6
T 3 a
CF3CF2CF2CF2SF5 —136,9 -119,5 17,8 (a) 5,0 46,2 (P) 48,7 (y)
cf3sf4cf2sf5 —141,6 -103,9 -125,8 B -11,9 —10,3
а Химические сдвиги измерены относительно внешнего эталона CF3COOH.
6 Химический сдвиг фтора при вершине пирамиды SFg.
в Химический сдвиг фтора в основании пирамиды SF5.
В табл. 11.48—11.50 приведены значения химических сдвигов и констант спин-спинового взаимодействия фтора для некоторых перфторалкильных производных гексафторида серы [101, 104]. В этих молекулах взаимодействие между ядрами фтора, разделенными тремя или четырьмя связями, осуществляется, по-видимому, как через химические связи, так и через пространственные взаимодействия. Для взаимодействия через пять связей преобладает последний механизм.
252
Глава 11
Таблица 11.50 КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ F—F ПЕРФТОРАЛКИЛЬНЫХ ПРОИЗВОДНЫХ
ГЕКСАФТОРИДА СЕРЫ [104] а
Соединение V) 1 CO b О сю 1 64 [I-о сю 1 о СО Ьч О 64 Ьч О 1 о со Ь-О 64 Ьч О 1 и о СО Ь-О ю в сю 1 СО 0-О ю Ьч О д л ь. 1 Ь,в сю 64 Ь О 1 00 О
cf3cf2sf4 15,70 9,33 < 1
cf3sf4cf2cf3 24,00 15,10 9,40
cf3sf4cf2cooch3 23,50 13,60
cf3cf2sf5 14,36 8,56 4,82 152,19 < 1
CF3CF2CF2CF2SF5 г 17,0 10,80 2,42 4,93 2,47 145,96 <1
cf3sf4cf2sf5 P 22,92 21,42 5,28 151,87
а Значения констант указаны в герцах.
б SFa обозначает вершинный атом фтора в группе SF&.
В ^SF — F — константа взаимодействия между вершинным атомом фтора и атомами фтора в осно-а Ъ
вании группы SFg.
г Для объяснения наблюдаемого мультиплета константа ^CFg—С—SF4 в этом соединении должна* быть равна 8 — 9 гц. При установлении соответствия между отдельными парами групп и константами взаимодействия были сделаны определенные предположения об ожидаемых относительных величинах некоторых из констант.
Д Для объяснения мультиплетности сигнала группы SF4 с химическим сдвигом —103,9 м. д. необходимо, чтобы константа взаимодействия Jgp4_с—SF4 в ЭтоМ соединении составляла .около 10—12 гц.
Ряд авторов [ЮЗ, 105, 106] для интерпретации спектров F19 производных SFsX провели подробный анализ системы АВ4, применив метод сложных частиц [107]. В молекуле SF5OSO2F [105] ядра фтора группы —SO2F взаимодействуют как с аксиальными (J = 0,9 гц), так и с базисными ядрами F19 группы SF5 (J = 7,2 гц)', большое различие между величинами этих констант может быть объяснено различным влиянием пространственного механизма на взаимодействие F — F [17, 20]. , '
11.16.2. Дизамещенные производные гексафторида серы
В дизамещенных производных гексафторида серы заместители обычно-находятся в транс-положении, а оставшиеся атомы фтора образуют плоскую квадратную структуру, в которой все четыре атома эквивалентны (рис. 11.17,в). При отсутствии спин-спинового взаимодействия с ядрами заместителей спектр F19 для этих молекул состоит из одиночной линии поглощения. Структурой указанного типа обладают перфтор алкильные производные гексафторида серы.
В некоторых молекулах заместители располагаются в соседних углах октаэдра, как показано на рис. 11.17,а для циклического соединения CF2CF2CX3F2CF2SF4. При этом угол между связями С — S — С составляет приблизительно го0 и эквивалентность четырех атомов фтора нарушается, поскольку два из них лежат в плоскости С — S — С, а остальные два — выше и ниже этой плоскости. В спектре F19 этой молекулы наблюдаются два триплета равной интенсивности, причем расщепление в триплетах возникает в результате взаимодействия двух различных пар эквивалентных ядер фтора между собой. Данные по химическим сдвигам в этой молекуле приведены в табл. 11.48; отнесение триплетов к определенным ядрам фтора оказалось невозможным [101].
Спектр F19 цис-SF4(SO3F)2 (структура I) является типичным для системы А2В2Х2 и служит подтверждением того, что эта молекула имеет необычную
Исследование ядерного магнитного резонанса фтора
253
^ис-конфигурацию [108]
О
/ав= 156 гц,
6ав=501 гц при частоте 40 Мгц.
Спектр F19 молекулы CF3SF4SF4CF3 состоит из триплета от групп CF3, смещенного на —5,5 м. д. относительно резонанса трифторуксусной кислоты, использованной в качестве внешнего эталона, и двух мультиплетов (квинтетов или септетов), расположенных в области слабого поля, в которой обычно наблюдаются сигналы группы S — F (6cf3cooh = — 95,5 и —116 м. д.) [109]. Причина возникновения в этом соединении неэквивалентности атомов фтора, связанных с серой, осталась невыясненной.
11.16. 3. Тетрафторид серы
Спектр ЯМР-F19 тетрафторида серы [ПО, 111], полученный при температуре —110°, состоит из двух триплетов с внутренним химическим сдвигом -48 м. д. и расщеплением 78 гц между компонентами тонкой структуры. Это
50 гц
Рис. 11.19. Половина спектра ЯМР-F19 тетрафторида серы, расположенная в слабом поле, при частоте 56,4 Мгц и при температуре —101° [175].
свидетельствует о наличии двух различных типов атомов фтора и согласуется с данными ИК-спектроскопии [112], согласно которым молекула имеет неплоскую структуру (Сг»). Эта молекула имеет тригональную бипирамидаль-ную структуру, в которой одно из экваториальных положений в ней занимает неподеленная пара электронов:
254
Глава 11
Низкотемпературный спектр F19 тетрафторида серы был исследован также другими авторами [175], и пара триплетов наблюдалась вплоть до —60°. При охлаждении образца до —101° все пики триплетов расщеплялись в дублеты (см. рис. 11.19), что следовало ожидать для системы типа А2В2 (см. разд. 8.13.8) с константой взаимодействия Jab = 76,3 гц и химическим сдвигом v06ab = = 2935 гц (при частоте 56,4 Мгц). Поскольку отношение J/v06 мало, наблюдаемое расщепление второго порядка является несколько неожиданным и возникает, по-видимому, за счет больших абсолютных значений J и v06. При комнатной температуре в SF4 происходит быстрый обмен атомов фтора; исследование спектров ЯМР этой молекулы в диапазоне температур дало возможность найти теплоту активации обменного процесса: 4,5 ± 0,8 ккал!моль. Эксперименты по ЯМР показали, что молекула SeF4 имеет такую же структуру, как SF4 [41, 110].
11.16. 4. Перфторалкильные производные SF4
Значения химических сдвигов и констант спин-спинового взаимодействия для соединений этого типа приведены в табл. 11.51 и 11.52 [113]. Симметрия
Таблица 11.51
ХИМИЧЕСКИЕ СДВИГИ F19 ПЕРФТОРАЛКИЛЬНЫХ
ПРОИЗВОДНЫХ SF2 и SF1 [113]а
Соединение 6sf2 6SF 6CF3 6CF
[(CF3)2CF]2SF2 —56,9 4-4,97 +76,4
(CF3)2CFSF2CF3 —53,8 4-8,25 +100
4-5,38 (изо-С3Р7)
(CF3)2CFSF3 —129 — 13,5 4-6,47 +97,4
(CF3)2CFSF 4-293 +6,10 +91,1
[(CF3)2CF]2S +11,0 +99,0
(CF3)2CFSOCF(CF3)2 +7,87- +94,8
+ 14,2 +71,8
(CF3)2CFS(O)OH +5,33 + 115
(CF3)2CFS(O)OC2H5 +4,76 + 113
а Химические сдвиги измерены в миллионных долях (м. д.) относительно CFCI2CFCI2; eCF3COOH (внешн) = eC12FCCFC12 (внешн) — 12’° (м- Д-) весьма приближенное соотио-шение).
монозамещенных производных (I) не дает возможности определить по спектрам F19, в каком из двух положений — аксиальном или экваториальном — находится заместитель. Спектр F19 для SF в дизамещенной молекуле [(CF3) 2CF]2SF2
Исследование ядерного магнитного резонанса фтора
255’
Таблица 11.52
КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ F-F (В ГЕРЦАХ) ПЕРФТОРАЛКИЛЬНЫХ ПРОИЗВОДНЫХ SF2 И SF4 [ИЗ]
Соединение
[(CF3)2CF]2SF2 (CF3)2CFSF3 (CF3)2CFSF2CF3
(CF3)2CFSF (CF3)2CFSOCF(CF3)2
(CF3)2CFS(O)OH (CF3)2CFS(O)OC2H5
10
1
19
12 (U3o-C3H7)
4,0
2,8
28
0,7
10
0,4
22
0,4
7,2
4,3 (U3o-C3H7)
9,3
11 (CF'—CFg)
2 (CF'-CF3)
8,0
8,0
20
состоит из одной мультиплетной линии. Это дает серьезное основание предполагать, что диаксиальное (Па) или диэкваториальное (Пб) расположение-заместителей в этих молекулах более вероятно, чем аксиально-экваториальное (II в).
Следует заметить, однако, что при наличии усредняющих процессов, например межмолекулярного обмена заместителями, спектр подобного типа может наблюдаться независимо от того, какая из форм замещения является доминирующей. В сульфенилфториде (CF3)2CFSF—моно замещен ном производном дифторида серы — обнаружено очень сильное экранирование ядер (+281 м. д. относительно внешнего эталона — CF3COOH; см. табл. II.51 и 11.52). Этот большой сдвиг был объяснен смешиванием основного состояния с низколежащими возбужденными состояниями, обусловленным активацией несвязывающих электронов серы.
11.17. Другие фторсодержащие соединения серы
Спектры F19 фторсульфоната фтора SO3F2, тионилтетрафторида SOF4 и гипофторита пентафторида серы SOF6 были исследованы для конденсированной фазы газообразных образцов при низких температурах, причем химические сдвиги фтора измерялись относительно перфторциклобутана [114].
Фторсулъфонат фтора. Спектр F19 для этого соединения состоит из двух дублетов равной интенсивности, что позволяет приписать молекуле следующую структуру: SO2F -OF [114].
Тионилтетрафторид. Спектр F19 этого соединения, содержащий синглетную линию поглощения, указывает на то, что молекула имеет структуру F4S = О, а не SF3OF. Первая из этих структур имеет форму пирамиды с квадратным основанием. Однако эксперименты по ЯМР не позволяют с полной определенностью исключить возможность существования второй структуры, поскольку эквивалентность ядер фтора может быть следствием быстрых обменных процессов [114]. Более однозначные сведения о структуре этого соединения могут быть получены, по-видимому, из ЯМР-экспериментов, проводимых при разных температурах.
Гипофторит пентафпюрида серы. Спектр F19 этого соединения типичен для спиновой системы АВ4Х [103]; это дает основание предполагать, что молекула имеет структуру SF5OF, в которой пять атомов фтора, непосредственно-соединенных с серой, образуют тетрагональную пирамиду (см. рис. 11.17, б) [114, 115].
*256
Г л а в. a 11
SsOgFz. Это соединение, получаемое при реакции двуокиси фтора и серы < пероксидисульфурилдифторидом, судя по спектру F19, состоящему из одной линии с химическим сдвигом —31,7 м. д. относительно перфторциклопентана, имеет следующую возможную структуру [116]:
ООО FSOSOSF ООО
Тиокарбоновые кислоты. Химические сдвиги F19, измеренные в двух тиокарбоновых кислотах [117], приведены в табл. 11.53, в которой для сравнения даны значения химических сдвигов некоторых водородсодержащих аналогов.
Таблица 11.53
ХИМИЧЕСКИЕ СДВИГИ F19 НЕКОТОРЫХ ФТОРАЛКИЛЬНЫХ КАРБОКСИЛЬНЫХ КИСЛОТ [117]а
Fl f2 F3 f4
1 2 3 CF3CF2CF2COOH 5,0 42,8 49,9
cf3cf2cf2cosh 4,4 38,7 48,8 —
H(CF2)4COOH 41,8 47,0 51,8 60,8
H(CF2)4COSH 37,6 45,8 50,8 60,3
а Химические сдвиги измерены в миллионных долях (м. д.) относительно трнфторуксусной кислоты (внешний эталон).
Тиоспирты. В тиоспиртах C3F7CH2SH и H(CF2)4CH2SH константы взаимодействия протонов группы SH и ядер фтора в ближайшей группе CF2 составляют <1 гц [118].
Методом ЯМР были исследованы также многие другие соединения серы [181].
11.18. Соединения фтора с другими галогенами
Некоторые из этих веществ были в числе первых фторсодержащих соединений, исследованных методом ЯМР высокого разрешения [6]. Спектры F19-резонанса BrF5, IF5 и IF7 уже обсуждались выше (см. разд. 11.4). Для пентафторидов было установлено, что молекулы имеют структуру тетрагональной пирамиды. Гептафторид иода имеет сложный спектр F19, указывающий на неэквивалентность всех ядер фтора, однако более определенные сведения о строении этой молекулы получены не были.
11.18.1. Трифторид хлора
Исследование ЯМР-F19 для C1F3 при различных температурах показывает, что при комнатной температуре в этой молекуле происходит быстрый обмен атомов F [119]. Согласно данным микроволновой спектроскопии [120] и исследований дифракции электронов [121], молекула C1F3 имеет симметрию С2о. В этой плоской молекуле две связи С1—F длиннее третьей, и, следовательно, магнитное окружение одного из ядер фтора отличается от окружения двух .других. Можно поэтому ожидать, что спектр ЯМР-F19 будет состоять из двух линий, соответствующих неэквивалентным типам ядер фтора. При комнатной
Исследование ядерного магнитного резонанса фтора
257
температуре спектр F18 состоит из двух очень широких линий, которые при охлаждении образца до —60° расщепляются и образуют спектр, типичный для
систем АВ2 (см. разд. 8.13.2). Подробный анализ низкотемпературных спектров показывает, что химический сдвиг между линиями от неэквивалентных ядер фтора в этой молекуле равен ~110 м. д., а константа спин-спинового взаимодействия составляет 403 гц * 1). Спектр фтора, снятый при частоте 40 Мгц, приближается к спектру, характерному для системы АХ2: дублет в слабом поле соответствует двум эквивалентным ядрам фтора, взаимодействующим с третьим ядром, в то время как это последнее дает триплет, обусловленный взаимодействием с пер-- выми двумя. При более низких частотах,
2220 1794 -963-1396-1783
Рис. 11.20. Спектры ЯМР-F19 трифторида хлора, снятые при температуре —60° и при различных рабочих частотах [119].
а — при 10,0 Мгц; б — при 30,0 Мгц; в — при 40,0 Мгц. Сдвиги измерены относительно S Fe.
Рис. 11.21. Изменения спектра ЯМР-F19 C1F3 в температурном интервале от—40 до 4-60° при частоте 30 Мгц [119].
30 и 10 Мгц, химический сдвиг между неэквивалентными группами ядер фтора становится сравнимым с константой взаимодействия между ними и наблюдается сложный спектр типа АВ2.
4 Недавние исследования C1F3 в газообразном состоянии дают значения 125,9 м.д. для химического сдвига и 441 гц для константы взаимодействия [194].
1 7—489
258
Глава It
На рис. 11.21 показаны изменения в спектре C1F3, происходящие при повышении температуры образца от —40 до +60°. Прежде всего исчезают мультиплетные расщепления отдельных линий, и в конце концов эти линии сливаются в одну. Причиной наблюдаемых изменений спектра в зависимости от температуры является быстрый обмен атомов фтора между химически неэквивалентными положениями. Можно оценить величины средних времен жизни атома фтора в каждом из этих положений при температурах, когда исчезает мультиплетность сигналов (—15°) и происходит слияние линий (-р60°), и, пользуясь этими данными, определить энергию активации обменного процесса в этом температурном интервале (4,8 ккал/моль). По данным Хэймера [122], температура, при которой исчезает мультиплетность линий в C1F3, изменяется от образца к образцу, что указывает на наличие в системе каких-то примесей (вероятно, HF). В одном из образцов мультиплетность сохранялась до 0°. Будет ли происходить обмен в трифториде хлора в отсутствие фтористого водорода, неизвестно, однако если он все же имеет место, то энергия активации должна быть значительно выше, чем 4,8 ккал/моль [122]. Изучение резонанса F19 в системе HF/C1F3 свидетельствует о существовании быстрого обмена атомов фтора. Аналогичные эксперименты с бинарными смесями каких-либо фторсодержащих молекул HF, C1F3, C1F и BrF3 показывают, что во всех случаях происходит быстрый взаимный обмен атомов фтора [122].
11.18.2. Трифторид брома
Спектр Р19-резонанса BrF3 при комнатной температуре состоит из одной линии поглощения, и если структура этой молекулы подобна структуре ClF3r то можно предположить, что в трифториде брома также происходит обмен атомов фтора [119]. Высокая температура плавления этого вещества (8,8°)-препятствует проведению экспериментов с чистым BrF3 при низких температурах. Исследование обмена в этой системе можно было бы выполнить с применением инертного растворителя, который бы позволил проводить измерения при охлаждении образца.
11.18.3. Пентафториды иода и брома
Пентафториды иода и брома IF5 и BrF5 имеют, как известно, структуру тетрагональной пирамиды
F I /Р FZ \F Окружение четырех атомов фтора, образующих основание пирамиды, отличается от окружения вершинного атома фтора, что находит отражение в спектре ЯМР-F19- В обоих случаях наблюдаются два мультиплета с отношением интенсивностей 1 : 4. Квинтет в слабом поле отвечает одиночному атому фтора при вершине пирамиды, который в одинаковой мере взаимодействует с четырьмя эквивалентными ядрами базисных атомов фтора. Последние в свою очередь дают дублет, смещенный в область сильного поля. Если в пентафториде иода при комнатной температуре и происходит обмен, то его скорость недостаточно велика для того, чтобы вызвать исчезновение мультиплетной структуры линий, обусловленной спин-спиновым взаимодействием (JF-f = 81 гц). Нагревание образца до 115° приводит к слиянию мультиплетной структуры, обусловленному быстрым обменом атомов фтора, и, следовательно, их время жизни в каждом из положений при этой температуре можно определить из выражения т= l/(4nJF_F) = 1/(4л-81) = 9,8-10-4 (сек).
Попытки определить температуру слияния двух линий в спектре IF5 оказались неудачными из-за экспериментальных трудностей. Согласно оценкам, получен
Исследование ядерного магнитного резонанса фтора
259
ным из сравнения температурных зависимостей ширины линий в пентафториде иода и C1F5 (эксперименты с IF5 проводились при температурах до 175°), температура слияния линий приблизительно равна 195°, что приводит к значению энергии активации процесса 13 ккал! моль. Пентафторид брома имеет такой же спектр F19, как IF5, однако при нагревании образца до 180° уширения линий не наблюдается, из чего можно заключить, что энергия активации процесса обмена атомов фтора в соединениях брома имеет гораздо большую величину [119]. Хэймер [122] показал, что при смешивании BrF5 с другими фторидами, например HF, C1F3, C1F и BrF3, его спектр не изменяется, что может служить свидетельством отсутствия быстрых обменных процессов в указанных смесях.
11.18.4. Перхлорилфторид FC1O3 и родственные соединения
Спектр F19 этого соединения интересен тем, что в нем проявляется спин-спиновое взаимодействие между ядрами фтора и хлора (ядра обоих изотопов хлора С135 и С137 имеют спиновое число I = у) [123]. ЯдраС!35 и С137 обладают квадрупольными моментами, взаимодействие которых с несимметричными градиентами электрического поля внутри молекулы приводит к уменьшению времен релаксации. Ядро хлора в перхлорилфториде находится в тетраэдрическом окружении трех атомов кислорода и одного атома фтора, вследствие чего молекула не обладает дипольным моментом, и градиент электрического поля, действующего на это ядро, равен нулю. Это исключает возможность быстрой квадрупольной релаксации ядра хлора и позволяет наблюдать взаимодействие F — Cl (Jf_ci35 =310 гц). Спектр фтора состоит из двух перекрывающихся квартетов, обусловленных взаимодействиями F — С135 и F — С137. Ядро фтора в этом соединении дезэкранировано почти так же сильно (бсгзсоон(внешн) = —320 м. д.), как в молекуле F2. Отсутствие дипольного момента свидетельствует о том, что электроотрицательность группы СЮ3 и фтора имеет величину одного порядка, и это согласуется с наблюдаемым сдвигом резонанса F19 в область слабого поля [6]. Другие авторы [166] измерили химические сдвиги F19 для некоторых родственных соединений, например для FC1O4 (—225,9 м. д.), для FOF (—250,0 м. д.), для FC1O3 (—287,0 м. д.) и для FOC1O3 (—225,9 м. д.), используя в качестве внутреннего эталона CFC13. Между значениями химических сдвигов для FC1O3, которые указываются различными авторами, существует очень большое несоответствие (~45 м. д.) [123, 166].
11.19. Соединения, содержащие связи В —F
11.19.1. Трифторид бора и родственные соединения
Ранние исследования спектра фтора в BF3 не выявили какой-либо тонкой структуры наблюдаемой синглетной линии поглощения, обусловленной взаимодействием В — F. Отсутствие мультиплетного расщепления было отнесено за счет взаимодействия квадрупольного момента бора с градиентами электрического поля внутри молекулы [42, 124]. Однако в последних экспериментах с трифторидом бора [125] в присутствии других тригалогенидов бора было показано, что линия поглощения фтора представляет собой квартет (7ВР = = 15 ± 2 гц), и потому прежние неудачи в обнаружении мультиплетной структуры можно объяснить недостаточной разрешающей способностью спектрометров. Изучая смесь ВС13 и BF3 при комнатной температуре, Койл и Стоун [125] обнаружили в спектре F19 (рис. 11.22, а) наряду с линией от исходного вещества дополнительные линии поглощения от смешанных галогенидов бора BC12F и BC1F2 (см. также табл. 11.54). В этой бинарной системе существует неустойчивое равновесие между четырьмя соединениями, и всякая попытка выделить смешанные галогениды бора приводила к неудаче вслед-17*
260
Глава И
ствие того, что они имеют тенденцию к быстрому диспропорционированию до BF3 и ВС13. Аналогичные данные были получены при исследовании спектров F19 бинарной системы ВВг3—BF3 (см. рис. 11.22, б). В тройной смеси BF3—
Рис. 11.22. Спектры ЯМР-F19 при частоте 40 Мгц для смешанных галогенидов бора, образующихся при комнатной температуре в смесях.
а — BF3 — ВС13; б — BF3 — BBr3; b -- BF3 — ВС13 — ВВг;!. Сигнал от BF3 смещен в сторону сильного поля иа 50,4 м.д. относительно сигнала от CF3COOH (внешний эталон).
ВС13—ВВг3, кроме смешанных галогенидов, найденных в бинарных смесях, было обнаружено неизвестное ранее соединение BBrCIF (см. рис. 11.22, в). В табл. 11.54 приведены значения констант взаимодействия В — F, полученные из спектров смешанных галогенидов бора. Кроме того, были исследованы
Таблица 11.54
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ FH> СОЕДИНЕНИЙ, СОДЕРЖАЩИХ СВЯЗИ В- F [125, 126]
Соединение Химический сдвиг Константа взаимодействия ^В11—F19, гЧ Литература
6BF3 (внутр) m. д. (относительно CCI3F при бесконечном разведеини) х а OCF3COOH (внешн)
ch3bf2 +68,8 —9,8 77 1
c2h5bf2 +74,6 — 4,0 81 126
Я-СдН7Вр2 + 72,8 —5,8 81
ch2=chbf2 +88,6 +ю,о 67
BF3 0 + 127 +48,4 15+2 У
BF2C1 —51,5 —1,1 34
BF2Br —68,4 —18,0 56 125
BFC12 —99,0 —48,6 74
BFCIBr —114,8 —64,4 92
BFBr2 — 130,4 —80,0 108 J
а Химические сдвиги были переведены в шкалу внешнего эталона CF3COOH.
спектры F19 некоторых органических дифторборанов [64, 126]. Как следует из табл. 11.54, ядра фтора в этих молекулах дезэкранированы по сравнению с ядрами в трифториде бора. Это означает, что однократное замещение трифторида бора органической группой сопровождается оттягиванием электронов
Исследование ядерного магнитного резонанса фтора
261
от двух оставшихся атомов фтора. Подобный эффект невозможно объяснить, если принимать во внимание только электроотрицательность заместителя. Поэтому для объяснения химических сдвигов в данном случае необходимо учитывать образование л-связей. При замещении атома фтора в трифториде бора органической группой происходит смещение л-электронов донорных атомов фтора к бору, и вполне вероятно, что это может привести к дезэкранированию атомов фтора. В органических дифторборанах связь В — С также в небольшой степени обладает л-характером, причем в исследованных молекулах наименьший вклад л-связи имеет место в винильном производном, что и объясняет более сильное экранирование фтора в этом соединении.
Койл и сотр. [127, 207] провели измерения химических сдвигов F18 в соединениях, содержащих связь В — F; полученные ими данные сведены в табл. 11.55. Поскольку органические группы обычно являются худшими
Таблица 11,55
ХИМИЧЕСКИЕ СДВИГИ F19 СОЕДИНЕНИЙ СО СВЯЗЯМИ В—F [127]
Соединение Химический сдвиг а, М. д. Соединение Химический сдвиг а, М. д. Соединение Химический сдвиг а» м. д.
BF3 0 СН2 = СН —CH2-BF2 —52 (CH3)3N-BF3 +37
с6н6—bf2 —35 h-C3H7BF2 —54 (CH3)3N-BC2H5F2 +38
ch2=ch-bf2 —38 с4н9—bf2 —54 (CH3)3N-BF2CH=CH2 +39
cf2=cf—bf2 —40 c2h6bcif —99
(CH3)2BF —107
а Химические сдвиги измерены в миллионных долях (м. д.) относительно внутреннего эталона CCI3F и затем переведены в шкалу BF3;
SBF3=6CFC13 (внутр) + 127 <м- Д-).
Были исследованы разбавленные растворы.
донорами л-электронов, чем атомы фтора, замещение последних этими группами в производных трифторида бора приводит к оттягиванию электронов от оставшихся атомов фтора и уменьшению экранирования последних, которое наблюдается экспериментально.
11.19.2. Ион BF;
Благодаря высокой симметрии иона BF" квадрупольное уширение линий в спектре F19 этого соединения отсутствует. Спектр F19 этого иона наблюдался в водном растворе BF4BF3CF3 ([BFJ[BF3CF3]) и представлял собой квартет (Jbf = 4,8 гц), компоненты которого имели гораздо меньшую ширину, чем линии поглощения в других соединениях со связью В — F [128]. Химические сдвиги F19, измеренные относительно внешнего эталона CF3COOH, и константы спин-спинового взаимодействия В —• F для этой молекулы [BF4]_[BFgCF"]+ имеют следующие значения:
6BFI = 72,3 М’Д'; 6bf3 = 77>3 м. д.; SCFs= — 2,06 м. д.;
7g р = 4,8 гц, Jg р, = 39,0 гц‘, 7gF„ = 34,0 гц.
Величина 7b-f для иона BF^ зависит от присутствия и концентрации катионов [204] 1).
х) При изменении концентрации компонент бинарного растворителя вода — ацетонитрил наблюдается изменение знака /в-f Для тетрафторбората серебра [270].— Прим. ред.
262
Глава 11
11.20. Соединения, содержащие связи N — F
В табл. 11.56 приведены значения химических сдвигов F18 в соединениях, содержащих связи N — F.
Таблица 11.56
ХИМИЧЕСКИЕ СДВИГИ FW СОЕДИНЕНИЙ, СОДЕРЖАЩИХ СВЯЗИ N—F
Соединение Химический сдвиг а» м. д. Литература
NF3 —221,1 41
C1NF2 —217,1 131
n2f4 —135,0 132
mpawc-N2F2 — 167,3 133
4«c-N2F2 —208,0 133
hnf2 —72,5 132
ch3conf2 — 106,8 132
(CH3)2CNF2 — 118,1 132
CN
FCN +79,5 111
ONF (нитрозилфторид) —924 135
(CH3)3CNF2 — 105,6 132
(C6H5)3CNF2 — 110,8 132
F
N
F2C CF2 NF + 11,8 134
| | FN NF CF2 + 12,8
f2
cf3nfcf2nfcf2nf2 nf2 —95,2 134
NF +7,9
NF +8,9
CF2 (между 2 NF-rpyn- + 15,0
пами)
CF2 (ближайшая к NF2) +25,6
CF3 —7,4
CF3NFCF2NFCF3 NF +8,7 134
CF3 —7,2
cf2 + 17,3
а Химические сдвиги измерены в миллионных долях (м. д.) относительно внешнего эталона CF3COOH.
Молекулярная структура нитрилфторида (FNO2) была определена с помощью спектра ЯМР-F19 для этого соединения, снятого при температуре, немного превышающей температуру плавления [129]. Этот спектр состоит из триплета с компонентами равной интенсивности и расстоянием между линиями 113 гц', расщепление того же порядка наблюдалось в спектре трифторида азота (Jnf = 160 гц), из чего с достаточным основанием можно сделать вывод, что в нитрилфториде атом фтора непосредственно присоединен к азоту.
Исследование ядерного магнитного резонанса фтора
263
В молекуле фторциана FCN, где такая связь отсутствует, константа взаимодействия Jnf равна 33 гц (6рр3соон = + 79,5 м. д.) [111].
Вид спектров F19 для молекул NF3 и FCN зависит от температуры [41]. Охлаждение образца трифторида азота вызывает слияние компонент триплета при температурах ниже —180°, что объясняется зависимостью времени квадру-польной релаксации ядра азота от температуры [130].
Химический сдвиг F19 в нитрозилфториде O=N—F — один из самых •больших сдвигов в область слабого поля [—420 м. д. относительно F2 или —924 м. д. по отношению к CF3COOH (внешний эталон)], что является неожиданным, если принять во внимание преимущественно ионный характер связи N — F [135]. Хемилюминесцентный анализ этого соединения выявил наличие низколежащего электронного состояния, которое должно обладать соответствующей симметрией для «смешивания» с основным состоянием в присутствии магнитного поля. Этим объясняется, почему парамагнитный вклад в экранирование в данном случае имеет большую величину. Таким образом, установление качественного соответствия между ионным характером связи и химическими сдвигами F19 (см. разд. 11.4) имеет смысл только для таких молекул, у которых разность энергий между основным и низколежащим возбужденным электронными уровнями имеет один порядок величины.
Изучение спектров F19 транс- и ц«с-М2Р2 позволило подтвердить, что эти молекулы имеют структуру I и II [133, 136]:
F
= f/n = n'\f,
I II
Наблюдаемые спектры характерны для спиновых систем типа АА'ХХ', что подтверждается также экспериментами по двойному резонансу с облучением образца на резонансной частоте N14. В табл. 11.57 указаны химические сдвиги и константы спин-спинового взаимодействия для изомеров N2F2 [136]. Хими-
Таблица 11.57
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ F1» ИЗОМЕРОВ N2F2 и РОДСТВЕННЫХ СОЕДИНЕНИЙ [136]
Соединение Химический сдвиг а» м. д. Коистаиты взаимодействия, гц
F19 N14 JNF +'Г JFF
n2f4 б —59,8 +39,8
транс-N 2F2 —94,9 —75,2 ±136 ±73 322
4uc-N2F2 —133,7 —13,3 ±145 ±37 99
NF3 б — 146,9 0,0 155
а Химические сдвиги фтора измерены относительно внутреннего эталона CCI3F. Химические сдвиги N14 измерены по отношению к NF3.
б Результаты неопубликованной работы Рэиделла и Бальдешвилера, цитируемые в [136].
ческие сдвиги N14 измерены с помощью метода двойного резонанса [137], причем было установлено, что в каждой из молекул ядра азота эквивалентны. Первоначально предполагалось, что молекула, которой приписана структура R
ifwc-изомера (II), может быть 1,1-дифтордиазином, = N. Однако данные, Fz
полученные методом двойного резонанса, не подтверждают этого предположе
264
Глава 11
ния, и, кроме того, в молекуле фтордиазина вряд ли имеет место эквивалентность ядер атомов азота.
Из сравнения значений констант взаимодействия N — F, приведенных в табл. 11.57, можно заключить, что валентные углы F — N — N в цис-и /пракс-изомерах N2F2, по-видимому, сильно отличаются по величине.
11.21. Соединения, содержащие связи О — F
В табл. 11.58 приведены химические сдвиги и константы спин-спинового взаимодействия для ряда гипофторитов [115]. При частоте 40 Мгц этим соеди-
Таблица 11.58
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ, ПОЛУЧЕННЫЕ
ИЗ СПЕКТРОВ F19 НЕКОТОРЫХ ГИПОФТОРИТОВ ПРИ ЧАСТОТЕ 40 Мгц [115]
Соединение Химический сдвиг (относительно SFe) а, м. д. Константа взаимодействия J, гц
для F2 или OF для SF или CF
F2 —365,5
of2 —217,0
fso2of —187,5 SF 21,5 ^SO2F-OF 6,1
sf5of —131,5 SF 1,75 7sf—OF 6,0
SF4 3,64 ^SF4—OF 17,4 ^SFi—SF6 155,0
CF3OF —92,5 CF3 128,5 ^CF3—OF 33’2
а Сдвиг считается положительным при смещении соответствующей линии в область сильного поля. При изменении концентрации раствора SO3F2 в SFg до бесконечного разведения возникает сдвиг в сторону сильного поля на 0,4 м. д. Сдвиги SFgOF при тех же условиях изменяются еще меньше. Другие соединения исследовались в виде разведенных растворов; экстраполяция к бесконечному разведению не проводилась.
6 Атом F19 в SF при вершине группы —SFs. 6CF3COOH (внешн) = eSFg~ — 127 (м. д.) (приближенное соотношение).
нениям отвечают спектры F19 первого порядка, исключение составляет SF5OF (система АВ4Х). Химические сдвиги для группы —OF позволяют сделать вывод, что заряд атома фтора отрицателен [115].
11.22. Соединения, содержащие связи Р — F
Пентафторид фосфора имеет, как известно, бипирамидальную тригональную структуру [138, 139]. Поэтому следовало бы ожидать, что спектр фтора этого соединения будет состоять из двух линий с отношением интенсивностей 2 : 3, отвечающих неэквивалентным группам атомов фтора. Однако в действительности спектр содержит одиночный дублет, расщепление между компонентами которого обусловлено взаимодействием с ядром фосфора (Jpf = 930 гц\ 6gf3gooh = — 0,68 м. д.) [42, 140]. Большие химические сдвиги между аксиальными и экваториальными ядрами F19 наблюдались в спектрах производных R2PF3 [140]. Это позволяет предположить, что эквивалентность ядер фтора в молекуле PF5 обусловлена внутримолекулярными переходами атомов фтора между аксиальным и экваториальным положениями в тригональной бипирами-дальной структуре [140, 141]. Муттертис и сотр. [140] исследовали спектры F19 большого числа моно-, ди- и тризамещенных фторидов фосфора (V) (см. табл. 11.62е—11.62и). По спектрам дизамещенных алкильных и арильных производных R2PF3 обнаружено существование двух различных групп атомов
Исследование ядерного магнитного резонанса фтора
265-
фтора, что свидетельствует о тригональной бипирамидальной структуре этих молекул, в которых заместители R занимают экваториальные положения. В соединении С12РРз ядра фтора спектроскопически эквивалентны при температуре 25°, но при охлаждении образца до —80° наблюдается спектр, типичный для соединений R2PF3 [140]. Эквивалентность ядер фтора наблюдалась во всех исследованных монозамещенных производных RPF4. Отсюда следует, что заместители R могут занимать либо аксиальное положение в тетрагональной пирамиде, либо экваториальное положение в тригональной бипирамидальной структуре, в которой происходит быстрый внутримолекулярный обмен атомов
Рис. 11.23. Зависимость химических сдвигов F19 для хлорфторидов фосфора и некоторых родственных соединений от числа атомов фтора [178].
фтора между различными положениями [140]. Зависимость химических сдвигов^ от числа атомов фтора в молекулах хлорфторидов фосфора с общей формулой PC1XF3_X и PC1xF5_x представлена на рис. 11.23. Изменение экранирования F19 с ростом числа замещающих атомов хлора в молекуле для соединений трехвалентного и пятивалентного фосфора происходит в противоположных направлениях [178]. Этот эффект до сих пор не нашел удовлетворительного объяснения г).
В комплексах пентафторида фосфора с основаниями типа аминов, эфиров и т. д. (PF5 -основание), в которых молекулы имеют октаэдрическую структуру, разность между химическими сдвигами фтора в аксиальном и экваториальном положениях составляет приблизительно 7 м. д., а константы взаимодействия F — F находятся в интервале 45—70 гц [142]. Значения констант Р — F для различных соединений занимают область 710—765 гц (для PF, JPF = 710 гц). Можно предполагать, что близкие значения констант JpF для PF~ и соединений PF5 -основание являются следствием одинаковой гибридизации в этих молеку-
0 Исследованию стереохимической лабильности PF3CI2 и РР3ВГг посвящена работа [260].— Прим. ред.
266
Глава 11
Таблица 11.59
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ F19 СОЕДИНЕНИЙ СО СВЯЗЯМИ P-F [42]
Соединение Jp p, гц Химический сдвиг, м. д. (относительно внешнего эталона CF3COOH)
PF3 1400 —43,4
POC12F 1180 —69,0
pocif2 1120 —30,4
POF3 1060 + 15,8
F2PO(OH) 980 +9,0
FPO(OH)2 955 —2,5
pf5 930 —0,7
Na2PO3F (водн.) 783 +26,8
HPFe 715 -5,5
KPFe (водн.) 706 —7,7
C2H5POF2 [143] 1132 —8,6
CH3POF2 [143] 1100 —16,9
.лах. В табл. 11.59 приведены значения химических сдвигов F19 и констант .взаимодействия для ряда соединений, содержащих фтор и фосфор '). Попытка установить соответствие между JPP и химическими сдвигами фтора в этом ряду
Ри'с. 11.24. Спектр ЯМР-F19 перфторзамещенного трифосфонитрила N3P3F6, растворенного в СС14, при частоте 56,4 Мгц [145].
‘соединений привела к обнаружению приблизительно линейной зависимости между указанными параметрами; аналогичная зависимость была найдена между ^pf и химическими сдвигами Р31 [42].
В результате анализа спектра F19 циклического соединения — фторхлор-.замещенного трифосфонитрила
(В)/С1
F (A) N = P-C1
X >
F N —Р —Cl
(В)\
С1
х) Для химических сдвигов F19 и констант спин-спинового взаимодействия Р31— F19 :В спектрах фторфосфатов и фторфосфонатов найдены аддитивные соотношения [233]. Проведено также сопоставление параметров спектров ЯМР-F19 дифторангидридов непредельных фосфиновых кислот с ИК-спектрами и реакционной способностью [234]. Измерены параметры спектров ЯМР-F19 и ЯМР-Р31 для соединений (СН3)3СР(О)РХ2 [261] .и C6H5PF2 [262].— Прим. ред.
Исследование ядерного магнитного резонанса фтора
267
были вычислены следующие константы взаимодействия между ядрами фтора и фосфора [144]:
^fp(A) = 934 гц-,
^FP(B) ~ 14 г(6
^Р(А)Р(В) = —'100 ги!-
Перфторзамещенный трифосфонитрил N3P3Fe представляет собой систему АА'А"Х2Х'Х" и дает сложные спектры фтора и фосфора [145]. На рис. 11.24 показан спектр F19 этой молекулы при частоте 56,4 Мгц-, хотя он и дает возможность найти приближенное значение большой константы взаимодействия Р — F (Jpp = 935 гц), для определения остальных констант необходимо провести полный анализ спектра. В нециклических молекулах, например (CH3)2NPF2, JPF ж 1200 гц [202].
11.23. Комплексные соединения
11.23.1. Комплексы трифторида бора
Огг и Диль [146] провели исследование спектров F19 нескольких комплексов трифторида бора с электронодонорными молекулами типа воды, спиртов, эфиров и кетонов. Они изучали смеси двух донорных жидкостей, к которым добавляли некоторое количество трифторида бора, недостаточное для насыщения доноров. При низких температурах, когда обмен происходит медленно, в этой системе наблюдались две достаточно узкие линии поглощения. Например, для комплекса BF3 со смесью метанола и этанола спектр F19 состоял из двух линий, отстоящих друг от друга на 2,0 м. д. Измерения интенсивностей линий поглощения в смесях разных составов показали, что между двумя донорными жидкостями и их соответствующими комплексами существует обратимое равновесие. Это дало возможность получить значения констант равновесия при разных температурах и вычислить соответствующие изменения энтальпии и стандартной энтропии. При более высоких температурах сигналы уширяются и затем сливаются, что указывает на быстрый обмен группами.BF3 между двумя комплексами. Исследование формы линий при температуре слияния сигналов позволяет определить скорость обмена и энергию активации (см. разд. 9.1.3).
При введении трифторида бора в систему наблюдались большие сдвиги в протонных спектрах донорных молекул. Несомненно, что в тех случаях, когда протоны являются единственными магнитными ядрами в акцепторных молекулах, для исследования комплексообразования нельзя использовать спектроскопию ЯМР, поскольку изменения протонных сдвигов для акцепторных молекул слишком малы (в противоположность сдвигам F19 в BF3).
11.23.2. Комплексы тетрафторидов металлов
Спектры F19 ряда комплексов, образованных органическими донорными молекулами и тетрафторидами титана, олова, кремния, германия и молибдена, были исследованы для определения природы комплексообразования и структур комплексов [147]. Спектры комплексов TiF4 и SnF4 с моно- и дифункциональ-ными донорными молекулами [например, (CH3)2SO, аминами и амидами] состоят из двух триплетов равной интенсивности. При изменении температуры образцов от +15° до точки плавления растворителей (в качестве последних использовались эфиры, нитрилы или избыток основания, с которым образуется комплекс) изменений в спектрах F19 не возникало. Вид спектров свидетельствует о наличии в этих молекулах двух неэквивалентных пар одинаковых атомов фтора и является серьезным доказательством октаэдрической симметрии молекул, в которых заместители занимают ^ис-положения. Комплексы титана
268
Глава 11
1 : 2 и 1 : 1 образуют октаэдрические структуры типа I и II. Экранирование ядер экваториальных атомов фтора, расположенных в одной плоскости с заместителями, отличается от экранирования остальных двух ядер фтора, занимающих положения выше и ниже этой плоскости, причем каждой из пар ядер отвечает триплетная линия поглощения (JFF =36 гц для TiF4 ^СгЩОН).
F
F | основание
/Т‘\
F | основание
F
1
Комплекс 1 :2 основание = (CH3)2SO
основание
11
Комплекс 1 :1
основание = этилендиамин
Нагревание некоторых комплексов SnF4 и TiF4, растворенных в эфире, выше комнатной температуры приводит к исчезновению триплетной структуры отдельных линий поглощения, уширению этих линий и, наконец, их слиянию, что указывает на быстрый обмен лигандов.
Спектры растворов различных комплексов SiF4 и GeF4 в эфире или сильном основании состоят из одной линии поглощения в широком температурном интервале от точки замерзания растворителя до температур, значительно превышающих комнатную. Можно предполагать, что синглетный спектр возникает вследствие быстрого лигандного обмена, однако причиной его возникновения может быть также симметричность молекулы, возникающая при транс-замещении октаэдрической структуры.
Спектры F19 комплексов SF4 дают мало сведений, поскольку основность третичных аминов недостаточна для того, чтобы вызвать заметные изменения в спектре этого слабого акцептора (см. рис. 11.19). Комплексы третичных: аминов с SF4 обладают синглетным спектром F19, характеристики которого остаются неизменными в интервале температур от —100 до - 50°.
Спектры F19 двухосновных комплексов (MoF4^основание) состоят из одной широкой линии поглощения.
11.23.3. Другие комплексы фторидов металлов
Методом ЯМР были исследованы водные растворы комплексов фторидов металлов (Th, Al и Zr), и сделана попытка установить корреляцию между наблюдаемыми химическими сдвигами F19 и различными параметрами этих соединений [148, 149]. Однако простой корреляции с электроотрицательностью иона металла или стабильностью комплекса установить не удалось. Несмотря на то что ThF3+ и A1F2+ гораздо более стабильны, чем ZrF+, резонансная линия F19 последнего соединения занимает промежуточное положение между линиями двух первых. Однако атомы фтора в катионах металлов с большим атомным номером, как правило, дезэкранированы, и существует приблизительное соответствие между химическими сдвигами и значениями Aid (где А — атомный номер металла катиона и d — межатомное расстояние). Спектры некоторых комплексов состоят из нескольких линий поглощения, что свидетельствует о наличии в системе более одного вида комплексов. В эквимолярной смеси водных растворов A1(NO3)3 и NaF были обнаружены две линии поглощения фтора, относящиеся к двум различным комплексам: A1F2+ и A1F+. Атомы фтора в этой системе не принимают участия в быстром обмене, что дает возможность с помощью химического сдвига между соответствующими линиями оценить нижний предел среднего времени жизни атома фтора в каждом из комплексов (8,0-10-3 сек). Измерение относительной интенсивности линий поглощения позволяет определить относительную концентрацию двух комплексных ионов.
Исследование ядерного магнитного резонанса фтора
269
Комплексы гексафторида теллура с третичными аминами изучались методом ЯМР при различных температурах [154]. Наблюдаемый спектр F19 соединения TeFe -2N(CH3)3 свидетельствует о том, что молекула имеет октаэдрическую структуру и при температуре —180° существует в трех различных геометрически изомерных формах (были получены три пика поглощения). При комнатной температуре наблюдается одиночная линия, что является серьезным доказательством существования быстрого обмена молекул амина х).
11.24. Фторсодержащие полимеры
Как было показано выше, возможности ЯМР при исследовании водородсодержащих полимеров существенно ограничены большой шириной линий в протонных спектрах этих соединений, а также трудностью выбора подходящих растворителей (см. разд. 10.37). В случае фторированных полимеров эти трудности могут быть частично преодолены, поскольку, во-первых, химические сдвиги F19 обычно значительно превышают ширину линий поглощения и, во-вторых, многие из водородсодержащих растворителей, которые нельзя использовать при исследовании протонных спектров из-за того, что их собственные спектры перекрываются со спектрами полимеров, оказываются подходящими для фторированных полимеров [150].
Таблица 11.60
ХИМИЧЕСКИЕ СДВИГИ F19 ФТОРУГЛЕРОДНЫХ ГРУПП В ПОЛИМЕРАХ
Г руппа Образец Химический сдвиг относительно CF3COOH (внешн.) а, м. д.
1 F-C-H Политрифторэтилен 131,7
1 F —С —С1 Кель-F 50,0 .
' 1 F — С— F 1 Перфтордекановая кислота 41,7
*CF3 Перфтордекановая кислота 3,3
F—С —Н Поливинилфторид 103,3
1 F-C —F 1 Поливинилиденфторид 13,3
а Химические сдвиги измерены с погрешностью 0,8 м. д.
Нейлор и Ласоски [150] провели исследование большого числа фторированных полимеров в виде 10%-ных растворов в диметилформамиде или бута-ноне-2. В табл. 11.60 и 11.61 приведены значения химических сдвигов фтора
1) Химические сдвиги F19 измерены для водных растворов HF [235—241], для растворов фторида цезия с целью сопоставить с каталитической активностью [242], для ионных кристаллов [243, 244], для систем KHF2 — KF — Н2О [245], для водных рас--творов, содержащих ионы F“, HF4, NHJ [246], для кремнийфторидов [247].— Прим. ред.
270
Глава 11
Таблица 11.61
ХИМИЧЕСКИЕ СДВИГИ F19 ФТОРУГЛЕРОДНЫХ ГРУПП В ПОЛИМЕРАХ
Г руппа Образец Химический сдвиг относительно CF3COOH (внеши.) а» М. д.
Поливинилфторид
103,3
Н F Н -LLL
н н н
F F F
I I I -С-С-С-I I I F Н F
Н F Н
I I I С-С-С
I I I
Н F Н
F F F
—С—С—С— I I I Н F Н
F F F
Политрифторэтилен
Поливииилидеифторид
Политрифторэтилен
Перфтордекановая кислота
131,7
13,3
38,3
41,7
I I I
F F F
F F СН3 Н
F F
Поли (2-метил-1,1,4,4-тетрафторбутадиен)
а Химические сдвиги измерены с погрешностью 0,8 м. д.
30,0
35,0
для полимеров. Спектр F19 поливинилиденфторида указывает на то, что мономе.-ры в этой молекуле образуют структуру типа «голова к голове».
В результате исследования спектра F19 раствора поливинилиденфторидов в N.N-диметилацетамиде при частоте 56,4 Мгц Вильсон [176] определил химические сдвиги групп CF2, находящихся в различном окружении:
- CF2 - СН2 - CF* - СН2 - CF2, — СН2 — СН2—CF* — СН2 - CF2, — CF2 - СН2—CF* - CF2 — СН2, — СН2—СН2 — CF* — CF 2 — СН2,
<р*= +91,6 м. д.,
Ф*= +94,8 м. д.,
<р*= М 13,6 м. д., гр* = у 115,9 м. д.
Эти данные показывают, что химические сдвиги F19 определяются не только ближайшими, но и более удаленными группами полимерной цепи. Измерение интенсивностей отдельных линий дает возможность установить относительное содержание структур «голова к голове», «хвост к хвосту» и «голова к хвосту» в полимерной цепи. Спектр F19 политрифторхлорэтилена, растворенного в 3,3-быс-трифторметилбифениле, при температуре 150° состоит из двух дублетов с компонентами неравной интенсивности [177]. Сравнение этого спектра с аналогичными спектрами соединений: мезо- и dZ-CF2ClCFClCF2CFQCF2Cl — дает возможность отнести компоненты дублетов к ядрам фтора в изотактическом и синдиотактическом фрагментах полимерной цепи.
Функциональные группы
-SO.F 0 -C-F -C-F -1 1 -1 L? lb lb 1 -1 _ 1 1
VI- и Г.Г Т “ 3
Vr.l ~е Г.Г Г £
।
CF-C^S——— i
~ъ “ CF, CF 1 и
лг /cV»z \ — 3
а . a
_ — 1
0 CF,- СН, п К1 ГГ —ГП — * А —
4. — 4 рг ррц
via Ы ||
L 1» и 1 п '' —° * v - - - 1
vDT, Г Г Г гл z~ ,- - -
——0 ГГ ГГ 1
СГ R|- — Z 3 1
~'*• Г.Г U гг ru 1
Г Г МЛ - ~~ 4 — z ' 1
И* NO*'
Г t ' Of и(СОД
SO __ г_,
CH,tKc ,,-Cl £Fz CFI
Fnx\ '4Fl —rr ru
nr rr ~Л 9 I
— П -Г Г KI M
au vK |1 N ГГ- Г Л 1
“ CF—гГ
rv rr -
cr^ сн,Оу~^Г|, 1Л1
—г.г. ru Frt<f^r.i _l
_2 UHZ r,
C.F Г.Г H
LTz ьг2п - >6^
jZ—f F rr *
-CRII ~"a ^CVT
*“VJ- 1 1
-Z.'v' П-Гги ГГ 1
Г «И ' - - ? 1
-г<1 ГЛ
— CF=CF,
~ 4 xr.r—r.r
XCF(CF > Г’
-СГ(.сгз)г x.„
-CFH——cr^
Пгг\ ггТм -
^^-CFjCOOH
-150 -125 -100 -75 -50 0 25 50 75 100 125 150 0,м.д.
Рис. 11.25. Сводная таблица химических сдвигов F18, измеренных в миллионных долях (м. д.), относительно трифторуксусной кислоты (внешний эталон) [155].
:272
Глава 11
Спектры ЯМР-F19 были использованы для определения структуры линейного сополимера, образованного из винилиденфторида и гексафторпропилена, а также для установления молярного соотношения мономеров в сополимере 1151]. В этих исследованиях в качестве растворителей применялись ацетон, трифторуксусная кислота или трифторуксусный ангидрид; было установлено, что увеличение диаметра ампулы с образцом до 8 мм не приводит к серьезному ухудшению разрешения.
11.25. Другие исследования
Обобщенные значения химических сдвигов F19 для различных соединений представлены в табл. 11.62 и на диаграмме рис. 11.25 [155] J).
Таблица 11.62а ХИМИЧЕСКИЕ СДВИГИ F16 НЕКОТОРЫХ ФТОРИРОВАННЫХ УГЛЕВОДОРОДОВ [50]
Формула Группа 6 a M. Д.
Фторуглероды
(CF3)4C CF3 -13,8
(CF3)3CF CF3 -1,2
CF 112,5
cf3cf2cf2cf3 CF3 6,0
cf2 51,4
•а b с de CF3(a) 4,8
CF3CF2CF2CF(CF2CF3)2 CF3(e) 3,2
CF2(&) 46,6
CF2(c) 34,4
CF2(rf) 37,4 -
CF 107,0
Перфторциклогексан cf2 • 55,0
Алкоксипроизводные перфторизобутилена
(CF3)2CHCF2OCH3 CF3 -12,8
cf2 -3,0
<cf3)2c=cfoch3 CF3 -18,9
CF -9,4
<CF3)2C = CFOCH2CH2CH3 CF3 -19,7
CF -11,8
<CF3)3 С = C(OCH2CH = CH2)2 CF3 -11,1
(CF3)2CHCF2OCH2CH2F CF3 -13,0
cf2 -6,2
CF 149,5
CF3 -19,3
a b
(CF3)2C = CFOCH2CH2F CF(a) -11,1
CF(&) 149,5
a Химические сдвиги измерены относительно внешнего эталона
CF3COOH.
1) Химические сдвиги F18 для различных фторорганических соединений см. также [248].— Прим. ред.
Продолжение табл. 11. 62а
Формула Группа 6 м. д.
Другие соединения
CF3CF2COOH CF3 7,0
cf2 46,0
CF3 11,9
(CF3CF2)2O cf2 12,9
Таблица 11.626
ХИМИЧЕСКИЕ СДВИГИ F19 НЕКОТОРЫХ ФТОРЭТАНОВ [171]
Соединение <p a, M. Д.
CF2BrCF2Br CF2C1CF2C1 CF2ICF2C1 63,4 71,2 59,6 (CF2I) 67,O(CF2C1)
CF2C1CFC12 72,2(CFC12) 68,05 (CF2C1)
CF3CFC12 84,6 (CF3) 76,8(CFC12)
а Химические сдвиги измерены относительно внутреннего эталона CFCI3 ((р-значе-ния).
Таблица 11.62в
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ F19 НЕКОТОРЫХ ПЕРФТОРАЛКАНОВЫХ ПРОИЗВОДНЫХ [78] а-
(CF3)2CFBr (CF3)2CFI cf3cf2cf2i cf3cficf2ci CF3x /CF3 >CFCF< CF2Ck xCFC12 (CF3)2CFCF(CF3)2 (CF3)2CFBr (CF3)2CFI cf3cf2cf2i 1 CF3CFICF2C1 < (CF3)2CFCF(CF3)2 6cf3 = 1,15, Sqf = 66,9 6Cf3 = —2,00, 6Cp = 70,4 6Cf3 = 2,25, 6Cf2 = 40,6, dCF2i=-17,2 6<?,f3=— 5,65, 6Cfa= —15,8, 6cfb=—12,5, 6cfi = 38.7 6cf3=-8,87, 6Cf2= —20,6, 5CF = 84,5 6cf3 =—5,54, 6cf— 103,3 •7cf3 - cf = 8,9±0,5 •7cf3 — cf = 12,5+0,1 •7cf3 - cf2 = 0,8+0,1; 7cf3 - cf2i = 9,4+0,2 •7cf3 - cf2i = 4,7+0,2 •7c,f3 - CF = H,8+0,1; JCF3 _ cfa = 9,4±0,1 7cf3 — cfb = 11 >8+0,1; Jcp _ cpA = 18,8+0,5 7cf - cFb= 18,1+0,5; Jcfa - gfb:= H6±2 •7cf3 - CF = 9,0+0,1
а Химические сдвиги измерены в миллионных долях (м. д.) относительно CF3COOH. Положительные значения соответствуют сдвигам в область сильного поля. Значения констант взаимодействия даны в герцах (гц).
18-489
Таблица 11.62г
ХИМИЧЕСКИЕ СДВИГИ Fl!> НЕКОТОРЫХ СОЕДИНЕНИЙ, СОДЕРЖАЩИХ ПЕРФТОРМЕТИЛЬНЫЕ ГРУППЫ [171]а
Соединение <p, 6 м. д. ^FF В»
(CF3)2NNO2 59,20 10,8
(CF3)2PC1 61,4 8,93
(CF3)2S 38,64 9,68
(CF3S)2 46,88 4,47
(CF3S)2Hg 21,11 < 0,2
(CF3)2AsC1 56,06 7,85
(CF3)2Se — 8,48
(CF3Se)2 38,1 3,08
(CF3Se)2Hg 15,2 < 0,2
CF3I 4,78 —
(CF3)2Hg 36,4 5,3
цикло-C^Fg 135,5 Г
а Химические сдвиги F19 трифторметильиой группы для ряда других соединений см. в работах [2 19, 221, 222, 249]. — Прим. ред.
6 Химические сдвиги измерены относительно внутреннего эталона CFCI3.
в Для определения констант взаимодействия F—F был проведен анализ спектров сателлитов C13F19.
г Спектры сателлитов C13F19 состоят нз триплетов с отношением интенсивностей 1 *. 1 : 1 и расщеплением 3,0 гц.
Таблица 11.62д
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ F19 И Р31 РЯДА ПЕРФТОРАЛКИЛЬНЫХ ПРОИЗВОДНЫХ ФОСФОРА [170]
Соединение 6pa-6 q>F6, B , J (P-cf3) r
(CF3)2PF — 123,9+0,6 66,5+0,05 89,6+0,20
(CF3)2PC1 -50,0+0,4 61,4±0,10 85,1+0,10
(CF3)2PBr -33,7+0,1 59,5+0,02 80,6+0,05
(CF3)2PI — 0,8+0,04 55,4+0,05 73,2+0,03
(CF3)2P-CN 40,7+0,2 51,3+0,20 85,6+0,10
(CF3)2P-NCO — 34,5+0,2 63,2+0,20 88,0±0,10
(CF3)2P-NCS — 61,9+0,05 87,3+0,10
(CF3)2P—OC2H5 — 92,3+0,3 65,3+0,20 86,6+0,04
(CF3)2P-N(CH3)2 —46,3+0,2 60,0+0,10 85,6+0,10
(CF3)2P —S —CF3 -12,8+0,1 55,6+0,20 • 83,8+0,40
(CF3)2P-Se-CF3 -14,1+0,1 53,7+0,10 77,2+0,10
(CF3)2PH — 47,5+0,04 68,6±0,10
(CF3)3P 2,6±0,0 50,8+0,05 85,5-0,10
(CF3)2PC13 — 78,8+0,10 193,3+0,20
(CF3)3P->O —2,3+0,15 66,2+0,05 113,4+0,10
[(CF3)3P]2Ni(CO)2 — 52,6+0,5 56,6+0,12 91,4+0,10
а В миллионных долях (м. д.) относительно 85%-ной Н3РО4.
б Положительным считается сдвиг в сильное поле.
в В миллионных долях (м. д.) относительно CCI3F.
г В герцах (гц).
Таблица JJ.62e
КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ ДЛЯ ФТОРИДОВ ФОСФОРА(У) [140]
Соединение •/HP
pf5 916
ch3pf4 967 6-7
cich2pf4 997 6—7
c2h5pf4 987 6-7
h-C4H9PF4 990 6
C8HldPF4 изомер A 955 6,5
изомер В 955 6,5
C6H5CH2PF4 1002 6-7
4uc-(C6H5)HC = CH(PF4) 940 6—7
c6h5pf4 963 < 0,5
jw-CH3C6H4PF4 960 < 0,5
n-CH3C6H4PF4 960 < 0,5
4i-C3H7C6H4PF4 963 < 0,5
n-C3H7C6H4PF4 958 < 0,5
л<-С1С6Н4РР4 960 < 0,5
n-C!C6H4PF4 960 < 0,5
2,5-(CH3)2CeH3PF4 981 < 0,5
cf3pf4 1103 12 (FC —PF)
Таблица 11.62ж
ХИМИЧЕСКИЕ СДВИГИ FH> ФТОРИДОВ ФОСФОРА(У) [1 40] а
Соединение 6CF3’ " A- ^F(moho), м. Д. ^F(e)» м- Д- ^F(a)» м* Д*
CF3PF4 -3,4 -10
(CF3)2PF3 -3,8 -7,5
(Cf3)3pf2 -12 / -16,8
ch3pf4 -31
(CH3)2PF3 +9,8 -74
(CH3)3PF2 -73
(CH2)5pf3 +6 -58
c2h5pf4 -25
(C2h5)2pf3 + 16 -52
(C2h5)3pf2 -44
h-C4H9PF4 -27
(h-C4H9)2PF3 + 13 -56
(h-C4H9)3PF2 -44
c6h5pf4 -23
(C6h5)2pf3 + 1,2 -44
(CeH5)3PF2 -39
C6H5(CH3)PF3 +5,4 -60
cich2pf4 -24
c8h15pf4 -31
c6h5ch = chpf4 -32
2,5-(CH3)2C6H3PF4 -25
Химические сдвиги измерены относительно внешнего эталона CF3COOH.
18*
Таблица 11.62з
КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ (гц) ДЛЯ СОЕДИНЕНИЙ ТИПА R2PF3 [140]
Соединение JPFa •'PF, 4Fa JHFe JHFa | 6Fe~6Fa ),“• A’
(CH3)2PF3 772 960 26 ~2 61
(C2h5)2pf3 815 980 29 12 68
(h-C4H9)2PF3 810 985 28 ~2 14 70
CH3(CeH5)PF3 785 935 33 14 60
(CeH5)2PF3 838 970 37 13,5 45
(CH2)4PF3
(—100°) 865 990 50
(CH2)4PF3
(25°) 915a 8,6
(CH2)5PF3 800 1005 42 64
(CF3)2PF3 1260 r/5p_cF 16GF_pF
ci2pf3 1050 (cp.)
Br2PF3 1140 (cp.?)
а Это значение хорошо согласуется с величиной 907 гц, полученной путем усреднения двух констант Jpp для (CH2)4PF3 при —100°.
Таблица 11.62и
КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ F19
СОЕДИНЕНИЙ ТИПА R3PF2 [140] а
Соединение Jpp, гц J pp, гц JHF’ ги-
(CH3)3PF2 545 11
(C2h5)3pf2 575 11
(w-C4H9)3PF2 585 16
(CeH5)3PF2 695
(CF3)3PF2 988 16
а Дополнительные данные по спектрам F19 для соединений. содержащих связи PF, приведены в работах [205, 206].
Таблица 11.62к.
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ JCF НЕКОТОРЫХ ФТОРОРГАНИЧЕСКИХ СОЕДИНЕНИЙ [190]
Соединение Константы взаимодействия, a ги> 6F, M. Д. Изотопный сдвиг, M. Д.
JCF JCCF
CH3F 158 276 0,067
CeH5CH2F 165 207 0,107
(CH3)3CF 167 132 0,114
ch2f2 232 148 0,143
n-CH3OCeH4F 237 125
n-CH3C3H4F 241 119
o-C!CeH4F 244 116
CeH5F 244 a 114
n-BrCeH4F 247 116
л-С1С3Н4Р 248 111
n-CF3CeH4F 252 в 109
n-CH3C(O)CeH4F 253 107
Продолжение табл. 11.62к
Соединение Константы взаимодействия8, гц 6р, м. д. Изотопный СДВИГ 6, м. д.
JCF JCCF
F С1
С1—F 253 -21,6 98 0,090
F \1
CF3-C=C-CF3 256 -57,2 57 0,142
cf4 257 69 0,105
n-NO2CgH4F 257 103
cf3n = sf2 263 50 0,147
(CF3)2CF2 265 -32,5 134 0,115
(CF3)2O 265 62 0,116
(CF3)3N 269 59 0,125
n-NH2C6H4CF3 270 -32,4
n-FCeH4CF3 271 -33,2
n-(CF 3)2C6H4 271 -32,9 65 0,128
СРзСН3 271 65 0,128
CF3H 272 84 0,133
CF3CH2Br 272 -38,5 71 0,129
л«-(СР3)2С6Н4 272 65 0,128
CF3CH2C1 274 74 0,108
cf3cci = cci2 274 -39,2 62 0,140
cf3ch2oh 278 0,116
0 c2H5
H2c-C — CF3 278 78 0,106
CF3CC13 283г -43,1 r 82 г 0,131 г
CF3COOH 283 -44,1 г 79 0,129 г
CF3COOC2H5 284 -44,1 78 0,130
(CF3)2CF2 285 -40,0 86 0,106
cf2=cd2 287 88
cf2cich3 288 . 47 0,147
cf2 = cci2 289 r -44,2Г 89 г 0,103 г
CF3C(O)CH3 289 83 0,120
цикло-C^Fg 298 -25,8 138
CF3C1 299 33 0,152
г{шсло-С4Р4С14 300 114 0,152
CFC1 = CC12 303 г —43,7 ' 79 г 0,112 г
CF3Br 324 21 0,152
CF2C12 325 8 0,164
CFC13 337 0 0,192
C6H5C(O)F 344 -61,0 -17 0,118
CF3I 344 5 0,132
CH3C(O)F 353 -59,7 -47 0,125
CF2Br2 358 — 7 0,168
HC(O)F 369 -41 0,128
CFBr3 372 е -7 Д
а На основе соображений, изложенных в работе [80], константе приписан
отрицательный знак.
б Др больше для молекул, содержащих С13.
в Точность определения ± 5 гц, Другие значения имеют погрешность не более ± 1 гц.
г Из работы [ 1 3].
Д Из работы [2].
Из работы [92]. Дополнительные данные по константам С13—F приведены в работе [182]. Химические сдвиги измерены относительно внешнего эталона CFCI3.
278
Глава 11
Таблица 11.62л
ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ F19 НЕКОТОРЫХ АЛКИЛ- И СИЛИЛСЕЛЕНИДОВ [39]
Соединение Химический сдвиг (p *, a M. Д. JHF> £4
CF3SeSiH3 Cr3SeC2H5 +25,5 +35,2 1,6
C3F7SeSiH3 C3F7SeC2H5 +77,9 (a-CF2) + 121,3 (₽-CF2) +79,5 (CF3) +88,6 (a-CF2) + 123,7 (P-CF2) +82,5 (CF3) 3,0; 1,5
а <р* измерены относительно внутреннего эталона CFCI3.
Ито и сотр. [156] провели исследование спектров F19 некоторых монофтори-рованных молекул. Были измерены химические сдвиги F19 во фторидах ксенона и родственных соединениях (6xeF2 = 629 м.д.; 6xeF4 = 450 м.д. и 6xeFe = = 310 м. д.) относительно F2[185—187]. Кроме того, в работе [203] сообщается об исследовании фторидов криптона.
ЛИТЕРА ТУРА
1. Е v a n s D. F., J. Chem. Soc., I960, 877.
2. F i 1 i р о v i с h G., Tiers G. V. D., J. Phys. Chem., 63, 761 (1959).
3. Glick R.E., Ehrenson S. J.,J. Phys. Chem., 62, 1599 (1958).
4. H о w a r d В. B., Linder B., Emerson M. T., J. Chem. Phys., 36, 485 (1962).
5. S a i k a A., S 1 i c h t e г С. P., J. Chem. Phys., 22, 26 (1954).
6. Gutowsky H. S., H о f f m a n C. J., J. Chem. Phys., 19, 1259 (1951).
7. H о 1 m e s J. R., Stewart В. B., MacKenzie J.S., J. Chem. Phys., 37, 2728 (1962).
8. T i e r s G. V. D., J. Am. Chem. Soc., 78, 2914 (1956).
9. M e у e r L. H., Gutowsky H. S., J. Phys. Chem., 57, 481 (1953).
10. S m i t h T. S., S m i t h E. A., J. Phys. Chem., 63, 1701 (1959).
11. T i e r s G. V. D., J. Am. Chem. Soc., 79, 5585 (1957).
12. G u t о w s k у H. S., J. Chem. Phys., 31, 1683 (1959).
13. Tiers G. V. D., J. Phys. Soc. Japan, 15, 354 (1960).
14. T i e r s G. V. D., J. Inorg. Nucl. Chem., 16, 363 (1961).
15, Saika A., Gutowsky H.S..J. Am. Chem. Soc., 78, 4818 (1956).
16. Feeney J., Sutcliffe L. H., Trans. Faraday Soc., 56, 1559 (1960).
17. Petrakis L., Sederholm С. H., J. Chem. Phys., 35, 1243 (1961).
18. S h a r t s С. M., Roberts J. D., J. Am. Chem. Soc., 79, 1008 (1957).
19. G u t о w s k у H. S., Holm С. H., S a i k a A., Williams G. A., J. Am. Chem. Soc., 79, 4596 (1957).
20. С г a p о L. M., Sederholm С. H., J. Chem. Phys., 33, 1583 (1960).
21. M u s h e r J. I., J. Chem. Phys., 36, 1086 (1962).
22. E v a n s D. F., Discuss. Faraday Soc., 34, 139 (1962).
23. M a n a t t S. L., E 1 1 e m a n D. D., J. Am. Chem. Soc., 84, 1305 (1962).
24. E v a n s D. F., M a n a t t S. L., E 1 1 e m a n D. D., J. Am. Chem. Soc., 85, 238
(1963).
25. Arison В. H., Shen T. Y., Trenner N. R., J. Chem. Soc., 1962, 3828.
26. G u t о w s k у H. S., В e 1 f о r d G. G., McMahon P. E., J. Chem. Phys., 36, 3353 (1962).
27. G u t о w s k у H. S., Techniques of Organic Chemistry, Editor A. Weissberger, Interscience, N. Y., Chapt. XLI, Vol. 1, Part IV, 3rd Edn, 1960.
Исследование ядерного магнитного резонанса фтора
279
28. Т h о m р s о п D. S., N е w m а г к R. A., Sederholm С. Н., J. Chem. Phys., 37, 411 (1962).
29. В а к В., S h о о 1 е г у J. N., Williams G. A., J. Mol. Spect., 2, 525 (1958).
30. Fujiwara S., Shimizu H., J. Chem. Phys , 32, 1636 (1960).
31. Williams G. A., G u t о ws к у H.S.,J. Chem. Phys., 30, 717 (1959).
32. Solomon I., Bloembergen N., J. Chem. Phys., 25, 261 (1956).
33. M с С о n n e 1 1 H. M., Reilly С. A., M c L e a n A. D., J. Chem. Phys., 24, 479 (1956).
34. Drysdale J. J., Phillips W.D.,J. Am. Chem. Soc., 79, 319 (1957).
35. S c h a ef er T., Can. J. Chem., 37, 882 (1959).
36. Abraham R.J., Bernstein H.J., Can. J. Chem., 39, 216 (1961).
37. Takahashi M., Davies D. R., Roberts J.D.,J. Am. Chem. Soc., 84, 2935 (1962).
38. С г о s s A. D., L a n d i s P. W., J. Am. Chem. Soc., 84, 1736 (1962).
39. E b s w о r t h E. A. V., Emeleus H.J., Welcman N., J. Chem. Soc., 1962, 2290.
40. Lord R. C., L у n c h M. A., S c h u m b W. C., S 1 о w i n s к i E. J., J. Am. Chem. Soc., 72, 522 (1950).
41. Muetterties E. L., Phillips.W. D.,J. Am. Chem. Soc., 81, 1084 (1959).
42. G u t о w s к у H. S., M с С a 1 1 D. W., S 1 i c h t e г С. P., J. Chem. Phys., 21, 279 (1953).
43. Shoolery J. N., Varian Technical Bulletin, № 1 (3), (1955).
44. Silbiger G., Bauer S. H., J. Am. Chem. Soc., 68, 312 (1946).
45. P h i 1 1 i p s W. D., Ann. N.Y. Acad. Sci., 70, 817 (1958).
46. L e e J., S u t с 1 i f f e L. H., Trans. Faraday Soc., 54, 308 (1958).
47. N a i r P. M., Roberts J. D., J. Am. Chem. Soc., 79, 4565 (1957).
48. Shoolery J.N.,Crawford B.,J. Mol. Spect., 1, 270 (1957).
49. Lee J., Sutcliffe L. FL, Trans. Faraday Soc., 55, 880 (1959).
50. E 1 1 e m a n D. D., Brown L. C., W i 1 1 i a m s D., J. Mol. Spect., 7, 307 (1961).
51. Elleman D. D., Brown L. C., W i 1 1 i a m s D., J. Mol. Spect., 7, 322 (1961).
52. Y о u n g J. A., Durrell W. S., Dresdner R. D., J. Am. Chem. Soc., 84,
2105 (1962).
53. Schroeder FL, Kober E., Ulrich FL, R a t z R., Agahigian FL, Grundmann C., J. Org. Chem., 27, 2580 (1962).
54. Muller N., Lauterbur P. C., Svatos G. F.,J. Am. Chem. Soc., 79, 1807 (1957).
55. Bigeleisen J., Wolfsberg M., J. Chem. Phys., 23, 1535 (1955).
56. Pitcher E., Buckingham A. D., Stone F.G. A.,J. Chem. Phys., 36, 124 (1962).
57. M с С 1 e 1 1 a n W. R., J. Am. Chem. Soc., 83, 1598 (1961).
58. H a m m e t t L. P., Chem. Rev., 17, 125 (1935).
59. Gutowsky H. S., McCall D. W., McGarvey B. R., Meyer L. FL, J. Am. Chem. Soc., 74, 4809 (1952).
60. T a f t R. W., J. Am. Chem. Soc., 79, 1045 (1957).
61. К a r p 1 u s M., D a s T. P., J. Chem. Phys., 34, 1683 (1961).
62. I s о b e T., Inukai К., 1 t о К., J, Chem. Phys., 27, 1215 (1957).
63. R i c h a r d s R. E., S c h a e f e r T., Trans. Faraday Soc., 54, 1447 (1958).
64. В о u r n A. J. R., Gillies D. G., Randall E. W., Proc. Chem. Soc., 200 (1963).
65. Brooke G. M., Burdon J., Tatlow J.C., J. Chem. Soc., 1961, 802.
66. Coyle T. D., Stafford S. L., S t о n e F. G. A., Spect. Acta, 17, 968 (1961).
67. Reilly C. A., S w a 1 e n J. D., J. Chem. Phys., 34, 2122 (1961).
68. Робертс Дж., Введение в анализ спектров ЯМР высокого разрешения (спин-спиновое взаимодействие), ИЛ, М., 1963.
69. Anderson J.L., Putnam R. Е., Sharkey W. FL, J. Am. Chem. Soc., 83, 382 (1961).
70. Hoehn FL FL, Pratt L., Watterson K- F., Wilkinson G., J. Chem. Soc., 1961, 2738.
71. McConnell FL M., J. Chem. Phys., 24, 460 (1956).
72. A 1 e x a n d e r S., J. Chem. Phys., 28, 358 (1958).
73. W h i p p 1 e E. B., Goldstein J. FL, Mandell L., J. Am. Chem. Soc., 82, 3010 (1960).
74. С о у 1 e T. D., Stafford S. L., S t о n e F. G. A., J. Chem. Soc., 1961, 743.
75. A n d r e a d e s S., J. Am. Chem. Soc., 84, 864 (1962).
76. R e i 1 1 у C. A., J. Chem. Phys., 37, 456 (1962).
77. В a n w e 1 1 C. N., Sheppard N., Proc. Roy. Soc., A 263, 136 (1961).
78. Boden N., E m s 1 e у J. W., Feeney J., Sutcliffe L. H., неопублико-
ванные данные.
79. С о у 1 e Т. D., Stafford S. L., S t о n е F. G. A., Spect. Acta, 17, 1244 (1961).
80. T i e r s G. V. D., Lauterbur P. C., J. Chem. Phys., 36, 1110 (1962).
280
Глава 11
81. Т i е г s G. V. D., J. Chem. Phys., 35, 2263 (1961).
82. Т i е г s G. V. D., J. Phys. Chem., 66, 1192 (1962).
83. M i d d 1 e t о n W. J., Sharkey W. H., J. Am. Chem. Soc., 81, 803 (1959).
84. S h о о 1 e г у J. N., Discuss. Faraday Soc., 19, 215 (1956).
85. P h i 1 1 i p s W. D., J. Chem. Phys., 25, 949 (1956).
86. T i e r s G. V. D., Proc. Chem. Soc., 389 (1960).
87. F e e п e у J., S u t с 1 i f f e L. H., J. Phys. Chem., 65, 1894 (1961).
88. T h о m a s L. F., Homer J., Proc. Chem. Soc., 139 (1961).
89. Sheppard N., Harris R. K., Proc. Chem. Soc., 418 (1961).
90. Tarrant P., Johnson R. W., В г e у W. S., J. Org. Chem., 27, 602 (1962).
91. D u n i t z J. D., Schomaker V. J., J. Chem. Phys., 20, 1703 (1952).
92. P h i 1 1 i p s W. D., Determination of Organic Structures by Physical Methods, Editors F. C. Nachod and Phillips, Academic Press, New York, 1962, Vol. 2, Chapt. 6.
93. H о m e r J., T h о m a s L. F., Trans. Faraday Soc., 59, 2431 (1963).
94. T i e r s G. V. D., J. Phys. Chem., 66, 764 (1962).
95. Reeves L. W., Wells E. J., Discuss. Faraday Soc., 34, 177 (1962).
96. Coyle T. D., King R. В., P i t c h e r E., S t a f f о r d S. L., T r e i c h e 1 P., Stone F. G. A., J. Inorg. Nucl. Chem., 20, 172 (1961).
97. К r e s p a n C. G., Langkammerer С. M., J. Org. Chem., 27, 3584 (1962).
98. Schnell E., R о c h о w E. G., J. Am. Chem. Soc., 78, 4178 (1956).
99. S t о n e F. G. A., S e у f e r t h D., J. Inorg. Nucl. Chem., 1, 112 (1955).
100. Hoffman C. J., H о 1 d e г В. E., J о 1 1 у W. L., J. Phys. Chem., 62, 364 (1958).
101. Muller N., L a u t e r b u r P. C., S v a t о s G. F., J. Am. Chem. Soc., 79, 1043 (1957).
102. Merrill С. I., W i 1 1 i a m s о n S. M., Cady G. H., Eggers D. F., Inorg. Chem., 1, 215 (1962).
103. Boden N., Feeney J., Emsley J. W., Sutcliffe L. H., Trans. Faraday Soc., 59, 620 (1963).
104. Rogers M. T., Graham J. D., J. Am. Chem. Soc., 84, 3666 (1962).
105. Packer R. J., H arris R. K-, J. Chem. Soc., 1961, 4736; 1962, 3077.
106. Chapman D., J. Chem. Soc., 1963, 131.
107. Whitman D. R., Onsager L., Saunders M., D u b b H. E., J. Chem. Phys., 32, 67 (1960).
108. Shreeve J. M., Cady G.H.,J. Am. Chem. Soc., 83, 4521 (1961).
109. Young J. A., Dresdner R. D., J. Org. Chem., 24, 1021 (1959).
НО. С о t t о n F. A., George J. W., Waugh J. S., J. Chem. Phys., 28, 994 (1958).
111. Fawcett F. S., L i p s с о m b R. D., J. Am. Chem. Soc., 82, 1509 (1960).
112. D о d d R. E., Woodward L. A., Roberts H. L., Trans. Faraday Soc., 52, 1052 (1956).
113. Rosenberg R. M., Mu et t er t i es E.L., Inorg. Chem., 1, 756 (1962).
114. D u d 1 e у F. В., S h о о 1 e г у J. N., Cady G. H., J. -Am. Chem. Soc., 78, 568 (1956).
115. C a d у G. H., M e r r i 1 1 С. I., J. Am. Chem..Soc., 84, 2260 (1962).
116. Roberts J. E., Cady G. H., J. Am. Chem. Soc., 81, 4166 (1959).
117. Sheppard W. A., Muetterties E. L., J. Org. Chem., 25, 180 (1960).
118. H a r r i s J. F., S h e p p a r d W. A., J. Org. Chem., 26, 354 (1961).
119. M u e t t e r t i e s E. L., P h i 1 1 i p s W. D., J. Am. Chem. Soc., 79, 322 (1957).
120. Smith D. F„ J. Chem. Phys., 21, 609 ((1953).
121. Burbank R. D., В e n s e у F. N., J. Chem. Phys., 21, 602 (1953).
122. Hamer A. N., J. Inorg. Nucl. Chem., 9, 98 (1959).
123. Brownstein S., Can. J. Chem., 38, 1597 (1960).
124. Ogg R. A., Discuss. Faraday Soc., 17, 215 (1954).
125. Coyle T. D., S t о n e F. G. A., J. Chem. Phys., 32, 1892 (1960).
126. Coyle T. D., Stone F. G. A., J. Am. Chem. Soc., 82, 6223 (1960).
127. Coyle T. D., Stafford S. L., S t о n e F. G. A., J. Chem. Soc., 1961, 3103.
128. Chambers R. D., Clark H. C., R e e v e s L. W., Willis C. J., Can. J. Chem., 39, 258 (1961).
129. Ogg R. A., R a у J. D., J. Chem. Phys., 25, 797 (1956).
130. P о p 1 e J. A., Mol. Phys., 1, 168 (1958).
131. Petry R. C., J. Am. Chem. Soc., 82, 2400 (1960).
132. Petry R. C., F r e e m a n J. P., J. Am. Chem. Soc., 83, 3912 (1961).
133. Colburn С. B., Johnson F. A., Kennedy A., McCallum K., Metzger L. С., P a г к e r C. O., J. Am. Chem. Soc., 81, 6397 (1959).
134. Hynes J. B., Bigelow L. A., J. Am. Chem. Soc., 84, 2751 (1962).
135. Holmes J. R., Stewart В. B., Mac Kenz i e J.S., J. Chem. Phys., 37, 2728 (1962).
136. Noggle J. H., Baldeschwieler J. D., Colburn С. B., J. Chem. Phys., 37, 182 (1962).
137. Baldeschwieler J. D., Randall E. W., Proc. Chem. Soc., 303 (1961).
138. В r a u n e H., P i n n о w P., Z. Phys. Chem., В 35, 239 (1937).
Исследование ядерного магнитного резонанса фтора
281
139. Brockway L. О., Beach J. Y., J. Am. Chem. Soc., 60, 1836 (1938).
140. Mahler W., Schmutzler R., MuettertiesE.L., Inorg. Chem., 2, 613 (1963).
141. Berry R. S., J. Chem. Phys., 32, 933 (1960).
142. Muetterties E.L.,Bither T.A..Farlow M.W.,Coffman D.D., J. Inorg. Nucl. Chem., 16, 52 (1960).
143. Feeney J., Sutcliffe L. H., неопубликованные данные.
144. Heffernan M. L., White R.F. M., J. Chem. Soc., 1961, 1382.
145. Boden N., Feeney J., Sutcliffe L. H., неопубликованные данные.
146. Ogg R. A., Diehl P., Nature, 180,1114 (1957).
147. Muetterties E. L., J. Am. Chem. Soc., 82, 1082 (1960).
148. Paulson R. E., С о n n i c k R. E., J. Phys. Chem., 63, 568 (1959).
149. Paulson R. E., С о n n i c k R. E., J. Am. Chem. Soc., 79, 5153 (1957).
150. Naylor R. E., L a s о s k i S. W., J. Polymer Sci.., 44, 1 (1960).
151. F e r g u s о n R. C., J. Am. Chem. Soc., 82, 2416 (1960).
152. Krespan C. G., J. Org. Chem., 25, 105 (1960).
153. E 1 1 e m a n D. D., M a n a t t S. L., J. Chem. Phys., 36, 1945 (1962).
154. Muetterties E. L., Phillips W. D., J. Am. Chem. Soc., 79, 2975 (1957).
155. В r a m e E. G., Anal. Chem., 34, 591 0962).
156. I t о К., Inukai К., I s о b e T., Bull. Chem. Soc. Japan, 33, 315 (1960).
157. BeaudetR. A., Baldeschwieler J. D.,J. Mol. Spect., 9, 30 (1962).
158. Frankiss S. G., J. Phys. Chem., 67, 752 (1963).
159. Brownstein S., Chem. Rev., 59, 463 (1959).
160. E 1 1 e m a n D. D., Mana tt S. L., частное сообщение, цитируемое в работе [22,
161. Е v a n s D. F., Mol. Phys., 5, 183 (1962).
162. Reilly С. A., J. Chem. Phys., 25, 604 (1956).
163. П о n л Дж., Шнейдер В., Бернстейн Г., Спектры ядерного магнитного резонанса высокого разрешения, ИЛ, М., 1962.
164. Р о р 1 е J. A., Mol. Phys., 1, 216 (1958).
165. Evans D. F., Mol. Phys., 6, 179 (1963).
166. A g a h i g i a n H., Gray A. P., V i c k e r s G. D., Can. J. Chem., 40, 157 (1962).
167. E 1 1 e m a n D. D., Manatt S. L., Pearce C. D., частное сообщение.
168. Tiers G. V. D., J. Am. Chem. Soc., 84, 3972 (1962).
169. Tiers G. V. D., J. Phys. Chem., 67, 928 (1963).
170. Packer K. J., J. Chem. Soc., 1963, 960.
171. Harris R. К., частное сообщение (1963).
172. P г о s s e r F., Goodman L., J. Chem. Phys., 38, 374 (1963).
173. Taft R. W., Prosser F., G о о d m a n L., D a v i s G. T., J. Chem. Phys., 38, 380 (1963).
174. R о g e r s M. T., S u m m i t t R. G., частное сообщение.
175. Bacon J., Gillespie R.J., Quail J. W., Can. J. Chem., 41, 1016 (1963).
176. Wilson C. W., J. Polymer Sci., Al, 1305 (1963).
177. Tiers G. V. D., В о v e у F. A., J. Polymer Sci., -Al, 833 (1963).
178. Holmes R. R., Gallagher W. P., Inorg. Chem., 2, 443 (1963).
179. Tiers G. V. D., J. Am. Chem. Soc., 79, 5585 (1957).
180. Gillespie R. J., Quail J. W., J. Chem. Phys., 39, 2555 (1963).
181. Cady G. H., Chem. Can., 15, 22 (1963).
182. Van der К e 1 e n G. P., Bull. Soc. Chim. Belg., 72, 644 (1963).
183. A r u 1 d h a s G., Venkateswarlu P., Mol. Phys., 7, 65, 77 (1964).
184. G u t о w s k у H. S., M о c h e 1 V. D., J. Chem. Phys., 39, 1195 (1963).
185. Brown T. H., Whipple E. В., V e r d i e r P. H., Science, 140, 178 (1963).
186. L a z d i n s D., Kern C. W., Karplus M., J. Chem. Phys., 39, 1611 (1963).
187. Brown T. H., Whipple E. В., V e r d i e r P. H., J. Chem. Phys., 38, 3029 (1963).
188. Flynn G. W., Matsushima M.,C r a i g N.C., В a 1 d es ch w i el erj. D., J. Chem. Phys., 38, 2295 (1963).
189. Boden N., E m s 1 e у J. W., Feeney J., Sutcliffe L. H., Mol. Phys., 8, 133 (1964).
190. Muller N., Carr D. T., J. Phys. Chem., 67, 112 (1963).
191. В г e у W. S., Ramey К. C., J. Chem. Phys., 39, 844 (1963).
192. Schug J. C., McMahon P. E., Gutowsky H.S.,J. Chem. Phys., 33, 843 (1960).
193. Taft R. W., Price E., F о x I. R., L e w i s I. C., A n d e r s о n К. K., Davis G. T., J. Am. Chem. Soc., 85, 3146 (1963).
194. Alexakos L. G., Cornwell C. D., J. Chem. Phys., 41, 2098 (1964).
195. Boden N., E m s 1 e у J. W., Feeney J., Sutcliffe L. H., Proc. Roy. Soc., A 282, 559 (1964).
196. Chambers R. D., Chivers T., J. Chem. Soc., 47, 82 (1964).
197. Fenton D. E., Massey A. G., J. Inorg. Nucl. Chem., 27, 329 (1965).
198. Ng S., Sederholm С. H., J. Chem. Phys., 40, 2090 (1964).
282
199.
200.
201.
202.
203.
204.
205.
206.
207.
208.
209.
210.
211.
212.
213.
214.
215.
216.
217.
218.
219.
220.
221.
222.
223.
224.
225.
226.
227.
228.
229.
230.
231.
232.
233.
234.
235.
Глава 11
Jameson C. J., Gutowsky H. S., J. Chem. Phys., 40, 2285 (1964).
Gillespie R. J., Q u a i 1 J. W., Can. J. Chem., 42 , 2671 (1964).
Ng S., Tang J., Sederholm С. H., J. Chem. Phys., 42, 79 (1965).
C a v e 1 1 R. G., J. Chem. Soc., 1964, 1992.
R. G., J. Chem.
F., Malm J. G., Hindman J. C., J. Am. Chem. Soc., 87, 25
С a v е 1
S с h г е (1965).
К u h 1 m Holme (1964).
Muetterti Inorg. Chem.,
" T.C., Coyle T. D., J. Chem. Phys., 41, 2612 (1964).
n
а s
Farrar
е г
п п R.
t D.
К., Gran
R., Carter R.
е s Е. L., М a h 1 3, 1298 (1964).
М., J. Phys. Chem., 68 , 3208 (1964).
Р., Р е t е г s о n G. Ё., Inorg. Chem., 3, 1748
er W., Packer K. J., Schmutzle
r R.,
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
в С. В., Степанов А. П., Пушкина Л. Н., Мазал ина О. К., ЖОХ, 36, 1613 (1966).
о в
С. A.,
С о к о л о Шабал ......
Пушкина Л. Н., К о л л е го в В. Ф.,Ст е п а Соколов С. В., ЖОХ, 36, 1619 (1966).
Плашкин В. С..Пушкина Л. Н., Колле Жури, всесоюзн. хим. общ-ва им. Менделеева, 12, Пушкина Л. Н., Юминов В. С., Степанов А. П., Соколов Применение молекулярной спектроскопии в химии, М., изд-во «Наука», стр. 218.
Быстров В. Ф. в кн. Ю. А. Жданова и В. И. Минкина «Корреляционный анализ в органической химии», Изд-во Ростовского
Молин Ю. Н., Кулакова Г. И., П л Журн. структ. хим., 5, 781 (1964).
Молин Ю. Н., Петров А. К., Кул Журн. аналит. хим., 20, 396 (1965).
Соколов С. В., Пушкина Л. Н., Г у Коллегов В. Ф., ЖОХ, 37, 181 (1967).
Татауров Г. П., Пушкина Л. Н., Губкина
Соколов С. В., ЖОХ, 37, Ягупольский Л. М., Б
СССР, 135, 377 (1960).
Быстров В. Ф., Я г у п о л к о в Ю. А., ДАН СССР, 153, Ягупольский Л. М., Быстров В. к о в Ю. А., ЖОХ, 34, 3682 (1964).
Быстров В. Ф., Южакова О. А., К
147, 843 (1962).
Орда В. В., Ягупольский н я н ц А. У., ЖОХ, 35, 1628 (1965).
Волощук В. Г., Ягупольск ров В. Ф., ЖОХ, 35, 118 (1965).
Ягупольский Л. М., Сырова р о в В. Ф., ЖОХ, 38, 2591 (1968).
Юж - - -
а к о в
а
а
б
нов
г о в
А. П., Мазал
В. Ф., Сокол № 2, 233 (1967).
о в
о в
С. А.,
С. В.,
С. В.,
1967,
университета, 1966, гл. 1, разд то ~ ~ ~ ~
н
о
в В. Ё., Якобсон
Г.
Г.
к о
к и
в
а
Г. И., Якобс о н
Г.
Г.,
н
а
Н. И., Татауров
Г.
П„
674 (1967).
ыстров В. Ф.,
Н. И., Колл
У т я н с к а
я
е г о в
В.
Ф.
Э.
3.,
ДАН
ь с к и й Л. 1321 (1963).
М„ Ст
е п а н я н ц
Ф., С т
е п а н я н ц
о
с т
ЯКОБС
к
и
и
Р.
А.
А.
У.,
У.,
Фиал-
Фиал-
Г., ДАН СССР,
л.
М„
Б ы с т
Р
о
в
В. Ф., Степа-
и
и
Л.
М., С ы
Г. П.,
р
о
в
а
Г.
П., Б ы с т -
В.
В о л о щ
У
к
В.
Г.,
Б ы с т -
с
с
к
и и
а О. А., Быстров АН СССР, сер. хим., 240 (1966).
Сергеев Н. М., Шапетько
Сергеев Н. М., Шапетько хим., 6, 300 (1965).
Шапетько Н. Н., Сергеев Н. М., П е т р и й О. П., Талалаев Махина А. А., Журн. структ. хим., 6, 159 (1965).
Сергеев Н. М., Шапетько Н. Н., П а н ~ Теор. и эксперим. хим., 1, 695 (1965).
Шапетько Н. Н., Сергеев Н. М., Панов Е. М., Журн. структ. хим., 6, 642 (1965).
Сергеев ....... ”
хим., 8, № Сергеев Сергеев Быстров н я н ц И.
Мышл я ковский Л. Н., канд. дисс., Ленинградский совета, 1966.
Утянская Э. 3., С т е п а н я н ц А. У., Винник М. ДАН СССР, 124, 1095 (1959).
ф., К О
т я н о в
р.
Г., Изв.
Н. Н., Теор.
Н. Н., Тим
эксперим.
и
о ф е ю к Г. В., Журн. структ.
хим., 2, 813 (1966).
а
Т. В.,
о в Ё. М.,
Сорокин
Сорокин
а
а
Р. С.,
Р. С.,
H. M., Шапетько H. H., Тимофеюк 1—2 (1967).
H. M., Савченко H. С., Теор. и эксперим.
H. M., Жури, структ. хим., 8, 567 (1967).
В. Ф., Неймышева А. А., Л., ДАН СССР, 156, 637 (1964).
Г. В., Журн. структ.
хим., 3, 812 (1967).
Степан
я н ц А. У., К н у -
технол. ин-т им. Лен-
H., Чирков Н. М.,
Исследование ядерного магнитного резонанса фтора
283
236. Бородин П. М., Скрипов Ф. И., Изв. вузов, сер. радиофиз., 1, 37 (1958).
237. Бородин П. М., Л е г и н Е. К., СвентицкийЕ. Н., ХусидманМ. Б., Журн. структ. хим., 4, 266 (1953).
238. Бородин П. М., Никитин М. К., Свентицкий Е. Н., Журн. структ. хим., 6, 188 (1965); сб. «Ядерный магнитный резонанс», вып. 1, 83, изд. ЛГУ, 1965.
239. Бородин П. М., Свентицкий Е. Н., сб. «Ядерный магнитный резонанс», вып. 1, 76, изд. ЛГУ, 1965.
240. Свентицкий Е. Н., Савоскина Г. П., Гладкий Ю. Г., сб. «Ядерный магнитный резонанс», вып. 2, 1967.
241. Бородин П. М., Свентицкий Е. Н.. сб. «Ядерный магнитный резонанс», вып. 2, изд. ЛГУ, 1967.
242. Соколенко В. А., Афанасьев М. Л., Молин Ю. Н., Якобсон Г. Г., сб. «Реакционная способность органических соединений», II, 216 (1965).
243. В а н - И - Ц ю, ДАН СССР, 136, 317 (1961).
244. Ван-И-Цю, Володичева М. И., Физ. тв. тела, 4, 642 (1962).
245. Ван-И-Цю, Скрипов Ф. И., ДАН СССР, 136, 58 (1961).
246. Ван-И-Цю, Журн. структ. хим., 2, 367 (1961).
247. Бородин П. М., Сингх Р., Щербаков В. А., сб. «Ядерный магнитный резонанс», вып. 2, изд. ЛГУ, 1967.
248. Бородин П. М., Скрипов Ф. И., сб. «Молекулярная спектроскопия», 1, 78, (1957); Изв. вузов, сер. радиофиз., 1, 69 (1958); сб. «Парамагнитный резонанс», изд. КГУ, 1960.
249. Быстров В. Ф., УтянскаяЭ. 3., Я г у п о л ь с к и й Л. М., Опт. и спектр., 10, 138 (1961).
250. Буслаев Ю. А., Щербаков В. А., Дяткина М. Е., Журн. структ. хим., 6, 16 (1965).
251. N е b g е n J. W., Rose W. В., Metz F. I., J. Molec. Spectrosc., 20, 72 (1966).
252. Константинов Ю. С., ДАН СССР, 134, 868 (1960).
253. Davies D. W., Nature, 207, № 4992, 75 (1965).
254. Jullien J., Stahl-Lariviere H., Bull. Soc. chim. France, 420 (1966).
255. Colter A. K-, Schuster I. I., Kurland R. J., J. Am. Chem. Soc. 87, 2278 (1965).
256. Дубов С. С., ТетельбаумБ. И., Стерлин P. H., Журн. всесоюзн. хим. об-ва им. Д. И. Менделеева, 7, 691 (1962).
257. Bauman R. Р., Bulkin В. J., J. Chem. Phys., 43, 496 (1966).
258. Е m s 1 е у J. W., Molec. Phys., 9, 381 (1965).
259. Hall L. D., Manville J. F., Chem. and Ind., 991 (1965).
260. Mahler R., Muetterties E. L., Inorgan. Chem., 4, 1520 (1965).
261. Sch mu tz 1 er R., Reddy G. S,, Z. Naturforsch., 20b, 832 (1965).
262. Schmutzler R., Chem. Ber., 98, 552 (1965).
263. Boden N., Feeney J., Sutcliffe L. H., J. Chem. Soc., 1965, 3482.
264. S e r v i s K. L., R о b e r t s J. D., J. Am. Chem. Soc., 87, 1339 (1965).
265. Cross A. D., J. Am. Chem. Soc., 85, 4011 (1964).
266. Myhre P. C., Edmonds J. W., Kruger J, D., J. Am. Chem. Soc., 88, 2459 (1966).
267. M a с k о r E. L., M a c L e a n C., J. Chem. Phys., 44, 64 (1966).
268. White H. F., Analyt. Chem., 37, 403 (1965).
269. Martin J. A., C.r. Acad. Sci., 261, 4385 (1965).
270. Gillespie R. J., Hartmann J.S.,J. Chem. Phys., 45, 2712 (1966).
271. Буслаев Ю. А., Щербаков В. А., Журн. структ. хим., 7, 345 (1966).
272, Пестун ов ич В. А., Егорочкин А. Н., Воронков М. Г., Миронов В. Ф., Скорик Ю. И., Журн. структ. хим., 6, 915 (1965).
273. Петросян В. С., Реутов О. А., ДАН СССР, 180, 876 (1968).
ГЛАВА 12
Спектры ЯМР других ядер (кроме водорода и фтора)
В принципе ядерный магнитный резонанс можно применять для исследования любого изотопа, у которого спин ядра не равен нулю. Однако по ряду причин большинство известных изотопов, удовлетворяющих этому требованию, не было изучено методом ЯМР в условиях высокого разрешения. Для наблюдения спектра высокого разрешения необходимо, во-первых, чтобы вещество, содержащее магнитные ядра, находилось в таком состоянии, где прямое диполь-дипольное взаимодействие усредняется до нуля (обычно это реализуется в газообразном или жидком состоянии). Кроме того, для успешного исследования методом ЯМР желательно, чтобы магнитное ядро обладало следующими свойствами:
1. Ядро должно иметь большой магнитный момент, так как характеристическая чувствительность метода ЯМР при постоянной напряженности магнитного поля пропорциональна величине магнитного момента в третьей степени.
2. Ядро должно иметь спиновое число I = у, поскольку ядра со спиновым числом, превышающим -i, обычно имеют квадрупольный момент, который ведет к квадрупольному уширению сигналов ЯМР. Кроме того, спектры 1
ядер со спиновым числом у проще интерпретируются.
3. Естественное содержание изотопа должно быть достаточным для обнаружения сигнала ЯМР данного ядра.
4. Время спин-решеточной релаксации для ядра в его различных окружениях должно быть достаточно коротким, чтобы избежать эффектов насыщения.
5. В исследуемом веществе не должно быть сильного взаимодействия между магнитными ядрами и неспаренными электронами (однако это не мешает исследованию некоторых парамагнитных частиц).
Необходимо исследовать вещество в жидком или газообразном состоянии. Идеальным случаем является жидкое вещество, поскольку для газа следует создавать высокие давления (и даже в этом случае невозможно обнаружить ядра с малой интенсивностью сигнала). Твердые вещества необходимо растворить в соответствующем растворителе или расплавить перед проведением эксперимента.
При отсутствии любого из этих свойств проведение измерений при высоком разрешении не обязательно становится невозможным. Очень часто тщательный подбор образцов и экспериментальных условий устраняет трудности. Повысить чувствительность можно, используя возможно большую напряженность внешнего магнитного поля при соответствующей частоте радиочастотного генератора, а также увеличивая размер образца. Оба эти приема приводят
Спектры ЯМР других ядер (кроме водорода и фтора)
285
к понижению разрешающей способности прибора, и в общем случае следует искать некоторый оптимальный вариант в отношении размеров образца.
Влияние квадрупольного уширения одного ядра на другое можно устранить методом двойного резонанса. При этом происходит подавление спин-спинового взаимодействия ядер, обладающих квадрупольным моментом, с другими ядрами (см. разд. 8.19.5). Для решения этой задачи также успешно пользуются замещением изотопа, ядро которого обладает квадрупольным моментом, на изотоп того же элемента с ядром, имеющим спин, равный нулю. Узкие линии сигналов поглощения от ядер, которые имеют квадрупольный момент, можно получить, исследуя молекулы с симметричным электрическим полем. Квадрупольное уширение возникает при взаимодействии квадрупольного момента и градиентов электрического поля в молекуле (см. разд. 2.5.2), и если градиент электрйческого поля равен нулю (как в тетраэдрическом ионе NHJ), то уширение снимается.
Если данный изотоп имеет низкое естественное содержание, то повысить интенсивность сигнала можно путем обогащения вещества этим изотопом. Дополнительное преимущество этого способа состоит в том, что имеется возможность провести избирательное обогащение некоторых групп в молекуле, что позволяет выполнить однозначное отнесение химических сдвигов.
Ядра с длинными временами релаксации обычно изучают, наблюдая сигнал дисперсии в условиях быстрого прохождения, так как при этом можно использовать более высокие значения радиочастотного поля Hi, не вызывая насыщения. Другой метод устранения насыщения состоит в уменьшении времени релаксации исследуемых ядер путем введения парамагнитных ионов или свободных радикалов в раствор. Влияние насыщения также можно свести к минимуму, если непрерывно подавать в область катушки приемника ненасыщенные ядра, используя проточный метод.
Другие методические приемы, которые позволяют проводить измерения спектров высокого разрешения при неблагоприятных условиях, рассмотрены в разделах, посвященных ЯМР на конкретных ядрах.
Большая часть из опубликованных до настоящего времени работ по ЯМР посвящена либо спектрам Н1, либо спектрам F19, так как свойства этих ядер удовлетворяют всем условиям, необходимым для успешного изучения методом ЯМР. Кроме того, большое число соединений, содержащих протоны и ядра фтора, являются легко доступными. Из числа остальных ядер, обладающих магнитным моментом, достаточно подробно изучены лишь В11, С13 и Р31. Опубликовано также несколько исследований ЯМР на других ядрах.
Все магнитные ядра, кроме водорода и фтора, обладают некоторыми свойствами, которые в различной степени препятствуют применению метода ЯМР. С этим приходиться считаться и принимать соответствующие меры для улучшения условия наблюдения сигналов ЯМР данных ядер. Сводная таблица магнитных свойств ядер приведена в приложении I т. 1.
В этой главе все химические сдвиги приняты положительными, если они отвечают сигналам, находящимся в области более высокого поля, чем сигнал эталонного соединения.
12.1. Бор
Оба встречающихся в природе изотопа бора имеют ядра, которые обладают магнитным моментом: В10, имеющий естественное содержание 18,8% и зна-з чение спина I = 3, и В11 с естественным содержанием 81,2% и спином I =~2-Исследования ЯМР высокого разрешения на ядрах бора проводятся, как правило, для более распространенного изотопа В11, поскольку его сигнал легче наблюдать как вследствие более высокого естественного содержания, так и из-за лучшей характеристической чувствительности метода ЯМР-В11. Послед
286
Глава 12
нее обусловлено тем, что ядро В11 обладает более высоким значением магнитного момента (ц = 2,6880 ядерного магнетона), чем В10 (ц = 1,8006 ядерного магнетона). При напряженности внешнего магнитного поля 14 000 э резонансные частоты ядер В10 и В11 составляют 6,405 и 19,124 Мгц соответственно. Несмотря на то что ядро В11 обладает квадрупольным моментом, уширение линий, вызываемое взаимодействием квадрупольного момента с градиентами электрического поля, обычно не слишком велико. Поскольку характеристическая чувствительность для ЯМР-В11 достаточно велика (16,5% по отношению к чувствительности для протонов при постоянной напряженности поля), то при исследовании жидких образцов в ампулах с внешним диаметром 5 мм можно получить достаточно интенсивные сигналы поглощения В11. Однако эти линии поглощения обычно значительно шире, чем линии спектров Н1 или F18, в результате чего малые константы спин-спинового взаимодействия не наблюдаются. Видимо, по этой причине определено лишь несколько констант спин-спинового взаимодействия ядра бора с протоном, разделенных более чем одной химической связью [7]. Еще одна причина уширения линий, с которой часто сталкиваются при исследовании жидких соединений бора, связана с их высокой вязкостью. В этих случаях добавление инертного растворителя позволяет получить более узкие линии поглощения. Установлено, что влиянием растворителей на экранирование ядер бора можно пренебречь, если только не протекает химическая реакция [2]. При исследованиях ЯМР-В11 в качестве эталонных соединений чаще всего применяются метилборат и эфират трехфтористого бора. Оба эти соединения легко доступны и дают острые линии поглощения. Обычно применяется метод внешнего эталона, а поправками на объемную диамагнитную восприимчивость пренебрегают.
/
12.1.1. Химические сдвиги В11
Ряд исследователей изучали спектры ЯМР соединений бора для того, чтобы найти соответствие химических сдвигов ядер бора с их электронным окружением. В табл. 12.1 приведены химические сдвиги В11 некоторых соединений относительно метилбората как внешнего эталона. Филлипс, Миллер и Муттертис [1] указали, что общие закономерности в изменениях химических сдвигов В11 можно качественно объяснить, так же как и экранирование ядер F18 [3] и Р31 [4, 5], с точки зрения изменений парамагнитного вклада в ядерное экранирование. Почти все химические сдвиги В11 лежат между расположенным в слабом поле резонансным сигналом триметилбора В (СН3)3, в котором атом бора имеет хр2-гибридизацию и незанятую атомную орбиталь pz, и расположенным в сильном поле сигналом иона ВН^, в котором атом бора имеет $р3-гиб-ридизацию. Такого соответствия следует ожидать, если вклад парамагнитного члена в общее экранирование является определяющим и степень дезэкранирования зависит от числа несбалансированных р-электронов. При образовании аддуктов ВН3 атом бора становится более экранированным, что является отражением уменьшения парамагнитного вклада в экранирование вследствие подачи электронов на незаполненную р2-орбиталь бора и изменения гибридизации от sp2 до sp3.
Как видно из табл. 12.2, экранирование В11 в ряду ВС13, ВВг3 и В13 возрастает по мере уменьшения электроотрицательности галогена. Трехфтористый бор имеет аномальный химический сдвиг В11, что можно объяснить значительным обратным донорным взаимодействием между заполненными р-орби-талями фтора и незанятой р2-орбиталью бора (частично двойной характер связи В — F) [6] х).
х) Расчет химических сдвигов В11 методом МО-ЛКАО, предложенным Карплусом и Поплом [313], проведен для смешанных галогенидов бора в приближении средней энергии возбуждения электронных состояний [336 ].— Прим. ред.
Таблица 12.1
ХИМИЧЕСКИЕ СДВИГИ ВИ И КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ ВИ—Н1 СОЕДИНЕНИЙ БОРА [1]
Соединение 6 а, м. д. +-- Н’
В(СН3)3 —68,2
В(С2Н5)з —66,6
СбН5ВС12 —35,9
С6Н5В(ОН)2 (в пиридине) —15,2
С4Н9В(ОН)2 (в ацетоне) —14,3
С6Н5В(ОС2Н5)2 — 10,4
Н СНз
/В — N4
Н3С—N< )ВН — 14,3 134
\В-н/
н СНз
н н
/В—Nx
NH< )ВН xb-n/ — 12,3 136
н н
В[М(С2Н5)2]з — 12,9
В(ОСН3)з 0
В(ОС2Н5)3 +0,6
В(ОС3Н7-н)з +0,5
В(ОС4Нд-Я)з —0,1
I ш I СП I +0,5 137 (концевой)
Hz Hr Н 48 (мостиковый)
Нх ,НЧ /Н
)в< >в( +36,7 130 (концевой)
Н/ '-Nz ''Н 29 (мостиковый)
НзС7 ХСН3
Н3С СН3
нх ,n; /Н
>В< >В< + 14,5 116
Hz 'sNz ХН
Н3с Хсн3
| ^О.ВНз + 19,0 103
^=)>N-BH3 + 31,4 90
(CH3)2NH-BH3 + 32,8 94
(СНз)зМ-ВНз + 24,9 97
(СНз)2РН-ВН3 + 55,6 96
НаВ(СбН5)4 (в воде) + 16,1
NaB(OCH3)4 (в СН3ОН) + 15,2
А1(ВН4)з + 55,1 86
NaBH4 (в 0,1 н. NaOH) +61,0 81
LiB(C ее ССоН5)4 +49,4
\O-BF3 + 19,0
NH3-BF3 (в воде) +20,2
(CH3)3N-BF3 (в смеси С6Н6—СН3ОН) + 18,6
288
Глава 12
Продолжение табл. 12.1
Соединение 6а, м. д. •+— гч
TIBF4 (в воде) + 18,8
AgBF4 (в воде) +20,3
(C2H5)2O-BF3 + 18,2
CH3OH-BF3 + 19,1
H3BO2F2 + 18,7
В1з-РС1зб + 82,4
BI3-C5H5N6 +77,4+5
(h-C4H9)3NH -В(С6Н5)4 6 + 23,9
BBr3-C5H5N6 + 23,6
ВВгз'(СгНь^О б +23,5
BF3-PC136 17,4
BF3-H2O6 — 17,2
В(С2Н5)з -(CH3)2NH б — 13,4
ВС13 -C5H5N б —8,4
ВС13-(С2Н5)20б —6,9
(С2Н5О)2ВВг б — 1,1
(СбН5)зВ б —42,6
а 6 — химический сдвиг, измеренный в миллионных долях (м. д.) относительно В(ОСНз)з как внешнего эталона.
6 Эти Химические сдвиги были измерены в миллионных долях относительно BF3 • О(С2Н5)2 как внешнего эталона. Перевод в шкалу В(ОСНз)з осуществлен по уравнению: 6В(ОСН3)з 6BF3.O(C2H5)2 + 17’4 <м- Д-) (приближенное соотношение).
Табл. 12.2—12.4 включают химические сдвиги ряда соединений бора с плоской геометрией, различающихся степенью частично двойного характера связей В — X. Из табл. 12.2 и 12.3 следует, что экранирование ядра бора возрастает с повышением частично двойного характера связи В — X [2]. Сигналы В11 производных тиоборана, имеющих тригональную геометрию, находятся в более слабом поле, чем аналогичные соединения со связями В — О,
Таблица 12.2
ХИМИЧЕСКИЕ СДВИГИ ВИ ТРИГАЛОГЕНИДОВ
БОРА [6]
Соединение Химический сдвиг б3’ м. д.
BF3 BCI3 ВВг3 В13 +6,6 —29,2 —22,7 + 23,3
а Химические сдвиги измерены в миллионных долях относительно эфирата BF3 как внешнего эталона; бв(ОСН3)3 (виеши) = 6bF3O(C2H5)2 (виешн) + + 17,4 (м. д.).
несмотря на более высокую электроотрицательность кислорода по сравнению с серой. Это можно объяснить относительно малым вкладом обратного донорного взаимодействия в соединениях, содержащих серу [8]. Если изменение гибридизации отсутствует, то увеличение электроотрицательности атомов,
Таблица 12.3
ХИМИЧЕСКИЕ СДВИГИ ВИ И КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ НЕКОТОРЫХ СОЕДИНЕНИЙ БОРА [2]
Соединение 6 а, м.д. J, гц
В5Н81 (бор при вершине) (в CS2) 55,0
В5Н11 (бор при вершине) 53,5 170 (ВН) + 5
В5Н9 (бор при вершине) 51,8 173 (ВН) ± 5
В6Н10 (бор при вершине) 51,2 182 (ВН) ± 5
В4Н10 (ВН) 40,0 154 (ВН) ± 5
NaBH4 (в воде) 38,7 82 (ВН4) + 3
LiBH4 (в эфире) 38,2 75 (ВН4) ± 3
В5Н8Вг (бор при вершине) (в CS2) 36,4
В10Н14 (положения 2 и 4) (в CS2) 34,9 158 (ВН) + 5
(CH3)2HNBH3 (в бензоле) 15,1 91 (ВН3) ± 3
В5Н9 (бор в основании пирамиды) 12,5 160 (ВН) ± 1
В5Н8Вг (бор в основании пирамиды) (в CS2) 12,5 161 (ВН) ± 5
В5Н8! (бор в основании пирамиды) (в CS2) 11,8 160 (ВН) ± 5
C5H5NBH3 (чистая жидкость) 11,5 96 (ВН3) ± 3
(CH3)3NBH3 (в бензоле) 9,1 101 (ВН3) ± 3
NaB(C6H5)4 (в воде) 8,2
В4Н10 (ВН2) 6,5 123 (ВН2) + 3
В13 (жидкий расплав) 5,5
ВдНц (ВН в основании пирамиды) 2,3 133 (ВН)
NaBF4 (в воде) 2,3
BF3-пиперидин (в CS2) 2,3
BF3 -гексаметилентетрамин 1,4
NH4BF4 (в воде) 1,8
BF3 ’O(W-C4Hg)2 0,0
BF3 -О(С2Н5)2 0,0
HBF4 (50%-ный раствор в воде) —0,1
ВР3-Р(СбН5)3 (в СНС13) —0,4
BF3 •S(C2H4C6H5)2 (в CHCI3) —0,5
В10Н14 (положения 5, 7, 8, 10) (в CS2) —0,5 141 (ВН)
Тетраацетилдиборат (в СНС13) —1,1 + 1,0
NaBO2[B(OH)Tt] (в воде) —1,3
NaBjOs (в воде) —1,3+1,0
В5Нц (ВН2 в основании пирамиды) —2,9 130 (ВН2)
LiB(OCH3)4 (в метаноле) —2,9
NaBO3 (в воде) —5,5
К2В4О7 (в воде) —7,5+1,0
НВС12-О(С2Н5)2 —7,9 152 (ВН) + 5
DBC12 *О(С2Н5)2 —8,0 ?(BD)
Na2B4O7 (в воде) —8,9
BF3 (газ) —9,4+1,0
(NH4)2B4O7 (в воде) —10,3
ВС13-О(С2Н5)2 (в эфире) —10,5
N(CH2CH2O)3B (в воде) —10,7
В10Н14 (положения 1, 3, 6, 9,) (в CS2) —12,4 138 (ВН)
КВ5О8 (в воде) —13,0
Три(о-хлорфеиил)борат (в эфире) —13,7±2,0
NaB5O8 (в воде) —14,4±1,0
В6Н10 (бор в основании пирамиды) —15,0 160 (ВН) ± 5
1 9—489
290
Глава 12
Продолжение табл. 12.3
Соединение 6 а, м. д. J, гц
Три(о-крезил)борат (в эфире) —15,0+1,0
В2Н6 (газ) — 16,6 128(ВН2) + 4
Метилметаборат (в бензоле) — 17,3
н -Бутилметаборат (в бензоле) — 17,5
В(ОСН2СН = СН2)з — 17,5
В(ОСНз)з — 18,1
В(ОС2Н5)з — 18,1
В(ОН)з (в воде) — 18,8+1,0
В(ОС2Н5)2С1 —23,3 + 1,0
НВ(ОСН3)2 —26,1 141 (ВН) ± 5
DB(OCH3)2 —26,7 24 (BD) + 4
В(ОН)2(н-С9Н19) (в эфире) —29,3+1,0
В(ОС2Н5)С12 —32,5+1,0
B2H2O3 —33,6+3,0 169 (ВН) ± 5
ВВг3 —40,1
ВС13 —47,7
В(С2Н5)3 —85±1,0
3 6В(ОСН3)з = 6BF3O(C2H5)2 + 17>4 (м- Д-) (приближенное соотношение).
Таблица 12.4
ХИМИЧЕСКИЕ СДВИГИ ВИ НЕКОТОРЫХ ТРИГОНАЛЬНЫХ СОЕДИНЕНИЙ БОРА [8]
Соединение Химический сдвиг3, м. Д.
В(СН3)з - —68,2
/S-CH2
ИЗО-С4НдВ^ | —51,9
\s — сн2
Л/-С4 Нд В (SC4 Нд-Л/) 2 —51,3
B(SC4H9-h)3 —47,9
Я-С4НдВ(ОН)2 — 14,3
В(ОСН3)з 0
а В(ОСН3)з применялся в качестве внешнего эталона.
непосредственно связанных с бором, приводит, как правило, к дезэкранированию ядра бора, т. е. химический сдвиг становится более отрицательным. (Это, однако, не следует рассматривать как доказательство того, что изменение диамагнитного вклада является определяющим для химических сдвигов В11 [2, 9].) Для четырехвалентных соединений ВН~, В(С6Н5Д, В(ОСН3Д экранирующая способность групп, непосредственно связанных с атомом бора, уменьшается в следующем ряду: Н > Саромат > О. Эту последовательность можно было бы предсказать из обычных соображений электроотрицательности, если бы действовали индуктивные эффекты. Такое же явление обнаружено в протонных спектрах углеродных аналогов [10], а также в спектрах Р31 фосфорных аналогов [5] этих соединений.
Спектры ЯМР других ядер (кроме водорода и фтора)291
Из наблюдаемых значений химических сдвигов В11 видно, что экранирование ядра бора зависит от степени заполнения свободной р2-орбитали тригонального атома бора: экранирование максимально для тетраэдрических замещенных соединений бора [11]. В табл. 12.5 приведены химические сдвиги В11
Таблица 12.5
химические сдвиги ви некоторых производных
ТРИГАЛОГЕНИДОВ БОРА [И]
Соединение Химический сдвиг а, м. д.
С2Н5ВС12 —16,0
C6H5BC12 —6,7
СН2 = СНВС12 —6,2
ВС13 0
C2H5BC1F 1,7
c2h5bf2 18,9
сн2 = chbf2 24,2
а Химические сдвиги измерены в миллионных долях относительно ВС1з как внешнего эталона; $В(ОСНз)з = ^BClg — — 29,6 (м. д.).
некоторых винильных и этильных замещенных галогенидов бора. Ядро В11 винильных производных трихлорида и трифторида бора экранировано в большей степени, чем в этильных соединениях. Это дает основание предполагать, что в винильных соединениях важным фактором экранирования является л-характер связи бор — углерод.
12.1.2. Спин-спиновое взаимодействие
Высказано предположение [12], что спин-спиновое взаимодействие между протоном и другими магнитными ядрами, связанными с. ним химической связью, должно зависеть от s-характера связи между атомами. Это подтверждается константами спин-спинового взаимодействия Н1 — В11, приведенными в табл. 12.6, из которой следует, что для атома бора в $р3-гибридизации константы меньше, чем для бора с s/r-i ибридизапией. Например, для (CH3)3NBH3 константа /вп-ш равна 97 гц, в то время как для боразола [222] ее значение составляет 136 гц. Маллер и Притчард [13] измерили константы спин-спинового взаимодействия Jcis-hi между ядрами непосредственно связанных атомов для нескольких соединений и установили, что отношение Jc (Spz) - н/^с ispsy - н составляет 1,33. Это значение совпадает с теоретическим (см. разд. 12.2.11). Для констант спин-спинового взаимодействия В — Н аналогичное отношение равно 1,37, что находится в хорошем согласии с данными для констант С13 — Н.
12.1.3. Гидриды бора
Спектры ядерного резонанса на протонах и ядрах В11 были измерены для ряда гидридов бора, и в большинстве случаев было установлено, что они согласуются с молекулярными структурами, определенными с помощью других физических методов [2, 6, 9, 14—17]. Для того чтобы подавить нежелательное мультиплетное расщепление как в протонных спектрах гидридов бора, так и в спектрах В11, широко применяется метод двойного резонанса.
В табл. 12.6 приведены химические сдвиги В11 и константы спин-спинового взаимодействия В — Н ряда боранов. Спин-спиновое взаимодействие между ядрами бора и протоном мостикового атома водорода наблюдалось 19*
292
Глава 12
Таблица 12.6
ХИМИЧЕСКИЕ СДВИГИ ВИ И КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ ВИ—Ш ГИДРИДОВ БОРА
Соединение Химический сдвиг а, м. д. Константы гц
Нч /Н, .н
>в< >в< +0,5 137,48
НХ ''Н-' хн
нч ZH4 ,н
>В< ЧВ< ' + 36,7 130,29
н/ хн
НзС^Нз
В3Н7 -О(С2Н5)2 +25,8 31+5
NaB3H8 (в воде) +46,5 32
В4Н10 ВН2 ВН ВН2 ВН
+25,0 +59,9 132 156
в5н9 Основание Вершина Основание Вершина
+30,8 +69,6 168 176
ВюН14 +6,9; +19,5; +53,9 124, 128, 159
B10H12I2 +4,0; +17,6; +60,7 136, 137
B6HU —16,2; +0,7; +49,8 134, 162, 142
а Химические сдвиги измерены в миллионных долях относительно В(ОСНз)з как внешнего эталона.
только в диборанах. Константы спин-спинового взаимодействия между атомами бора и связанными с ними концевыми атомами водорода коррелируют со свойствами связи. Измеренные значения J показывают, что в исследованных молекулах характер гибридизации связи может изменяться в пределах от sp до sp2[2] х).
Диборан В2Н6. Методами дифракции электронов [18] и ИК-спектроско-пии [21] было установлено, что электронодефицитная молекула диборана обладает мостиковой структурой (см. рис. 12. L):
Нч ,Н, ,Н >В< ')В< н/ Хн" хн
В этой структуре четыре атома водорода непосредственно связаны с атомами бора обычными ковалентными двухэлектронными связями, в то время как другие два атома водорода занимают мостиковые положения, насыщая оставшиеся валентности атомов бора. Спектры ЯМР-Н1 и ЯМР-В11 жидкого диборана (см. рис. 12.2—12.4) подтверждают мостиковую модель для этой молекулы. Спектр на ядрах В11 содержит один мультиплет, представляющий собой триплет триплетов. Большое триплетное расщепление возникает вследствие спин-спинового взаимодействия ядер каждого атома бора с двумя эквивалентными концевыми атомами водорода, связанными с ним, а малое триплетное расщепление — вследствие взаимодействия ядер атома бора с парой мостиковых водородных атомов. Дополнительное подтверждение мостикового строения диборана дает протонный спектр этой молекулы, из которого непосредственно следует существование двух различных типов атома водорода. Рис. 12.3 показывает природу компонент спектра: концевым атомам водорода, связанным с одним атомом бора (со спиновым числом у), отвечает квартет
!) Опубликована обзорная статья, посвященная спектрам ЯМР электронодефицитных молекул, в том числе и бороводородов [339].— Прим. ред.
Спектры ЯМР других ядер (кроме водорода и фтора)
293
линий поглощения, в то время как сигнал от мостиковых атомов водорода, связанных с двумя атомами бора, расщепляется в спектре в септет. В протонном спектре, где взаимодействие с ядрами В11 подавлено методом двойного резонанса, линии поглощения от двух различных типов протонов представляют собой два синглета с соотношением интенсивностей 2 : 1 (см. рис. 12.4) (15]. Сигналы от атомов водорода, связанных с атомами В10 (/ = 3), образуют слабый септет, который обусловливает оставшуюся часть протонного спектра 1).
Замещенные дибораны. Уильямс и сотр. [20] исследовали серию алкилдиборанов, безуспешно пытаясь измерить константы спин-спинового взаимодействия В11—В10 между непосредственно связанными неэквивалентными атомами бора. На рис. 12.5 показаны спектры В11 некоторых метилдиборанов, измеренных при частоте 12,8 Мгц. Метильное замещение оказывает дезэкранирующее влияние на соседний атом бора. Подобное влияние наблюдается для аналогичных этильных производных. Отсутствие наблюдаемого спинового расщепления В10—В11 указывает на то, что это расщепление либо слишком мало для этих молекул [22], либо подавляется квадрупольными взаимодействиями [23].
Было показано, что в монобромдиборане заместитель занимает концевое положение [288].
Другие бораны. Молекулярные структуры и спектры ЯМР-В11 ряда других боранов приведены на рис. 12.6—12.9. Были также исследованы и протонные спектры, не приведенные здесь. Во всех случаях протонные спектры подтверждают отнесение, сделанное на основании спектров В11.
х) Проведено исследование зависимости спектра ЯМР-В11 растворов диборана и диборгидрида натрия в органических растворителях. Определена энергия активации динамических процессов, протекающих в этих системах [337].— Прим. ред.
Рис. 12.2. Спектр ЯМР-В11 диборана при частоте 12,3 Мгц [15].
Рис. 12.3. Спектр ЯМР диборана [15].
а — протонный спектр прн частоте 30 Мгц: б — теоретический спектр концевых протонов, непосредственно связанных с В11; в — теоретический спектр концевых протонов, непосредственно связанных с В10; г — теоретический спектр протонного резонанса для мостиковых атомов водорода.
Рис. 12.4. Спектр ЯМР диборана [15].
а — протонный спектр при частоте 30 Мгц; б — тот же спектр, но с подавлением спнн-спинового взаимодействия с ядрами В11 одновременным облучением при частоте 9,6257Мгц.
Спектры ЯМР других ядер (кроме водорода и фтора)
295
Тетраборан В4Ню. Электронографическое [24] и рентгенографическое [25] исследования показывают, что тетраборан имеет молекулярную структуру, приведенную на рис. 12.1. Два атома бора связаны с двумя концевыми атомами водорода каждый, а два других — только с одним концевым атомом водорода. Каждая пара атомов бора связана двумя водородными мостиками. Исследование спектров ЯМР подтвердило такую структуру молекулы [22, 26]. В спектре В11 (см. рис. 12.6) триплет в слабом поле отвечает атомам бора, связанным с двумя концевыми атомами водорода (<7в-н = 128 гц), а дублет в сильном поле — двум другим атомам бора (Jb-н = 151 гц). Тонкая структура триплета, находящегося в слабом поле, видимо, обусловлена дополнительным спин-спиновым взаимодействием атомов бора с мостиковыми водородами (Тв-н = 29 гц). Этот тип спин-спинового взаимодействия проявляется также и в дублете, расположенном в сильном поле, но вследствие магнитной неэквивалентности мостиковых ядер водорода картина имеет более сложный вид. Первоначально полагали, что спин-спиновое взаимодействие В11—В10, которое может реализоваться в 20% молекул тетраборана, также дает существенный вклад в тонкую структуру. Однако впоследствии [23] было установлено, что на спектры ЯМР-В11 тетраборана не вли-
яет одновременное облучение образца на резонансной частоте ядра В10. Это показывает, что константы спин-спинового взаимодействия В10—В11 не проявляются в спектре В11 тетраборана [23]. Подтверждение этой точки зрения было получено при исследовании спектра дейтерированного тетраборана B4D10, который состоит из двух широких резонансных линий с соотношением интенсивностей 2 : 1, не обнаруживающих тонкой структуры за счет взаимодействия В10—В11.
Протонный спектр В4Н10 свидетельствует о том, что атомы водорода, связанные с наиболее экранированными атомами бора, также более экранированы, чем другие водородные атомы [22].
Шефферу и Теббе [27] удалось избирательно обогатить молекулу тетраборана изотопом В10, осуществив реакцию меченого триборгидрида натрия со смесью хлористого водорода и диборана, в котором соотношение изотопов бора отвечает природному. Спектр В11 тетраборана, полученного этим методом, показал, что ядра В10 занимают почти исключительно положения В — Н, так как почти полностью исчез дублет, расположенный в высоком поле.
Пентаборан-9 В5Н9. Хидберг и сотр. [28] показали, что эта молекула имеет строение тетрагональной пирамиды. Один атом бора расположен в вершине и четыре — в основании пирамиды, с каждым из атомов бора связан
сн3 сн
х
сн
CH, CH
Оконц 727гц Омост*47гц
КВН»
($‘-26,7
И с>ъ<СНз и сн"^ Д
----—' RBH 4-----
f?2B
I U Омоет 38,5гц
СН3 СЦ3 К2В • W I ^—29’2 CH3 I М ($-29,3
/I
w г7”
И2В ВН2
КВН Оиост=47,5 гц 6—20,5 ($—20,5
Омост-39,7гц 6—24,8 ($—23,5
Окони. 127гц
,BH2 Оиост—18гц ($—8,8
Оконц 1^3,7гц ™ Омост=45,2гц.
<Г=-13,6 ($—13,2
вн2
Оцонц~725,5гц Змост=4Я,7гЦ 6—3,6 ($—3,6
ЯМР-В11 некоторых
Рис. 12.5. Спектры метилборанов при частоте 12,8 Мгц [20]. Химические сдвиги 6 (в миллионных долях относительно эфирата BF3) представлены точками. Линии X и Z' обусловлены примесями, Y — сигнал 1,2-днметилдиборана, продукта диспропорционирования метилбораиа.
296
Глава 12
один концевой атом водорода, а четыре атома бора в основании пирамиды попарно связаны друг с другом мостиковыми водородами.
Шулери [15], а также другие авторы [17] исследовали спектр ЯМР-В11 пентаборана-9 (см. рис. 12.7, б) и показали, что он согласуется с тетрагонально-пирамидальным строением. Два дублета с соотношением интенсивностей 4 : 1 отвечают атомам бора в основании и в вершине соответственно. Дублетное расщепление появляется вследствие спин-спинового взаимодействия между атомами бора и единственным концевым протоном, связанным с каждым из них.
Рис. 12.6. Спектр ЯМР-В11 тетраборана (В4Н10) при частоте 32,1 Мгц (23 500 5) [227].
Пентаборан-11 В5Н41. Рентгенографическое [29] и электронографическое [30] исследования показали, что этот нестабильный гидрид имеет структуру, приведенную на рис. 12.1. Молекула содержит три типа атомов бора. Первый тип — атом бора в вершине пирамиды, связанный с единственным концевым: атомом водорода. Второй тип —два атома бора, лежащих в основании пирамиды, каждый из которых связан с одним концевым атомом водорода. Третий тип — два оставшихся атома бора в основании пирамиды, к каждому из которых присоединены по два концевых атома водорода. Дублет, расположенный в сильном поле спектра В5НИ (рис. 12.7, а), отвечает атому бора в вершине пирамиды, так как только концевые атомы водорода взаимодействуют с атомами бора [9]. Уильямс и сотр. [17] установили, что линии в спектре ЯМР-В11, помеченные а' и У, обусловлены наличием примеси В5Н9 в образце. Три оставшихся сигнала в спектре В11 были отнесены за счет перекрывания триплета, расположенного в слабом поле (сигнал с отвечает атомам бора третьего типа в группах ВН2), с дублетом (сигнал b отвечает атомам бора второго типа групп ВН).
Гексаборан В6Н10. Считают, что гексаборан имеет структуру неправильной пентагональной пирамиды с одним атомом бора в вершине и пятью атомами в ее основании [31] (см. рис. 12.1). Каждый атом бора связан с единственным концевым атомом водорода, а четыре оставшихся атома водорода занимают мостиковые положения между атомами бора, лежащими в основании. Таким образом, два атома в основании пирамиды не связаны водородным мостиком. Такая структура должна содержать четыре различных типа атомов бора. Спектр ЯМР-В11 (см. рис. 12.8) указывает на присутствие только двух типов атомов бора в соотношении 5:1. Дублет в слабом поле отвечает атомам бора в основании пирамиды, а дублет в сильном поле — атому бора в вершине пирамиды [29]. Дублетное расщепление возникает вследствие спин-спинового взаимодействия В — Н между атомами бора и концевыми атомами водорода. Отсутствие в спектре В11 триплета подтверждает, что в молекуле нет групп ВН2. Таким образом, хотя спектр ЯМР подтверждает в общих чертах предположенную модель структуры гексаборана, в нем не различаются разные типы атомов бора, лежащих в основании пирамиды. Также не обнаружено
Спектры ЯМР других ядер (кроме водорода и фтора)
297
спин-спинового взаимодействия между атомами бора и мостиковыми атомами водорода.
Декаборан Bi0Hi4. Рентгенографическое исследование декаборана показало, что молекула этого соединения имеет структуру, приведенную на рис. 12.1. [32]. Ее можно рассматривать как две неправильные пентагональные пирамиды, основания которых имеют общее ребро. Каждый атом бора связан с одним концевым атомом водорода, а оставшиеся четыре водорода занимают мостиковые положения и распределяются так, что в молекуле существуют четыре различных типа атомов бора. Спектр ЯМР-В11 декаборана (см. рис. 12.9, а) состоит из лежащего в слабом поле триплета и дублета в сильном поле х) [9, 15, 16] с соотношением интенсивностей 4:1. Этот спектр соответствует предложенному строению (см. рис. 12.1). В спин-спино-вом взаимодействии с атомами бора принимают участие только концевые протоны. Поэтому триплет, расположенный в слабом поле, следует рассматривать как наложение трех перекрывающихся дуб
Рис. 12.7. Спектры ЯМР-В11 при частоте 12,8 Мгц [17].
а — BjHu; б — BSH„.
Рис. 12.8. Спектр ЯМР-В11 гексаборана В6Н10 при частоте 12,8 Мгц [17].
летов от трех различных типов атомов бора, не находящихся в вершинах молекулы (детальное отнесение приведено в спектре). Дублет в сильном поле отвечает атомам бора, расположенным в вершинах. Спектр дейтерированного декаборана [7] состоит из четырех линий поглощения, разделенных химическим сдвигом (рис. 12.9, б), что подтверждает правильность сделанного для недейтери-рованной молекулы отнесения сигнала в слабом поле. Спектр дейтерированного декаборана имеет простой вид, поскольку константа спин-спинового взаимодействия D — В11 значительно меньше, чем константа Н1 — В11, и поэтому не проявляется в спектре.
Ион В3Н^. Спектры ЯМР иона В3Н“ не дают дополнительных сведений о строении этого соединения. Резонансный
спектр на ядрах В11 показывает, что в ионе имеется лишь один тип атомов бора [2].
Ион ВцН74. Спектр ЯМР-В11 раствора, содержащего ионы В41Н74, состоит из симметричного дублета, имеющего расщепление 140 гц, которое не зависит от напряженности [33]. Методом двойного резонанса, который позволяет подавить спин-спиновое взаимодействие В — Н, было показано, что в молекуле имеются два типа ядер бора, экранирование которых немного
В Спектр ЯМР-В11 декаборана в поле 43 900 э состоит из трех отдельных мультиплетов [310].
298
Глава 12
различается. Следовательно, спектр В11 состоит из двух перекрывающихся дублетов, разделенных очень малым химическим сдвигом. Этим подтверждается наличие в молекуле двух типов атомов бора, связанных непосредственно с атомами водорода. Константа спин-спинового взаимодействия составляет приблизительно 130 гц.
и Bi0Hi&. Спектры B9Hi5 [34] и B10Hi5 [35] на ядрах В11 не удалось интерпретировать вследствие сложности спектральной' картины.
Замещенные бораны. Замещение одного из концевых атомов водорода в боране на атом, не обладающий магнитным моментом, приводит к тому, что
Рис. 12.9. Спектры ЯМР-В11 при частоте 16,2 Мгц [23]. а — BiqHi4; б — BiqD14.
соответствующий резонансный сигнал В11 теряет дублетное расщепление, а также изменяется экранирование этого атома бора. Методом ЯМР исследовано несколько галогензамещенных боранов [36]. В большинстве случаев удалось определить положение заместителя. Было показано, что в моноиодиде и монобромиде пентаборана-9 заместитель расположен при вершине молекулы. Это следует из сравнения их спектров ЯМР-В11 (см. рис. 12.10) со спектром исходной молекулы: дублетное расщепление сигнала, расположенного в спектре пентаборана в сильном поле, отсутствует в спектрах монозамещенных молекул. Для монохлорпроизводного В5Н8С1 установлено, что атом хлора связан с бором, расположенным в основании пирамиды (см. рис. 12.11) [277]. Дублет в сильном поле, характерный для атома бора, находящегося при вершине молекулы В5Н9, сохраняется в спектре В5Н8С1. Найдено, что в соответствующих производных декаборана B10Hi3I и B10Hi3Br заместитель занимает одно из двух возможных положений при вершине. В спектрах этих молекул присутствует лишь один из двух дублетов в сильном поле. Как было показано исследованием спектра В11 бензилдекаборана B10Hi3CH2C6H5 [37], замещение в декаборане не всегда протекает в положении при вершине. Сигналы в сильном поле от идентичных в исходном декаборане двух атомов бора, находящихся при вершинах молекулы, в спектре замещенного соединения несколько смещаются, но полностью сохраняют дублетное расщепление. Отсюда следует,
Спектры ЯМР других ядер (кроме водорода и фтора)
299
что замещение прошло не при вершине, но таким образом, что два атома бора в вершинах стали неэквивалентны. ЯМР-исследование дейтерирования дека-борана в присутствии D2O в растворе диоксана подтвердило, что обмен на дейтерий мостиковых атомов водорода происходит быстрее, чем обмен
Рис. 12.10. Спектры ЯМР-В11 при частоте 12,3 Мгц [36]. a — BSH9; б — ВБН8Вг; в — В8Н,1.
концевых водородных атомов [38, 39]. С помощью спектров В11 идентифицированы некоторые этилированные дибораны, пентабораны и декабораны [40—42]. Проведено также исследование аминодиборанов [286].
н
Рис. 12.11. Спектр ЯМР-В11 соединения В5Н8С1 при частоте 32,1 Мгц (23 500 э) [227].
Диаммиакат диборана B2H6(NH3)2. Изучение методом ЯМР [43] показало, что это соединение имеет ионное строение [BH4]_[BH2(NH3)2]+. Резонансный спектр В11 состоит из триплета (сигнал катиона) и квинтета (сигнал аниона) равной интегральной интенсивности. Химический сдвиг и константа спин-спинового взаимодействия для квинтетного сигнала иона боргидрида [39,3 м.д. •относительно внешнего эталона BF3O(C2H5)2 и /ц-н = 80 гц] практически
300
Глава 12
совпадают со значениями для других боргидридов (например, для NaBH4 7В -н =82 гц; для LiBH4 JB _н = 75 гц) [1]. Аналогичные соображения можно привести в пользу того, что катион имеет строение [BH2(NH3)2]+ (6 = +14,6 м.д. и 7в-н= 120 гц). Заслуживает внимания экспериментальная особенность этого исследования — вещество было растворено в жидком аммиаке и изучалось при комнатной температуре в запаянной стеклянной ампуле х).
12.1.4. Боргидриды
Боргидрид натрия NaBH4. Протонный спектр этой молекулы содержит один мультиплет с четырьмя компонентами равной интенсивности, которые возникают вследствие спин-спинового взаимодействия четырех эквивалентных протонов с одним ядром бора (7ц-и = 82 гц) [14, 15]. Для подтверждения происхождения тонкой структуры применен метод двойного резонанса [15].
Боргидрид алюминия А1В3Н^. Согласно структуре боргидрида алюминия, предложенной на основании электронографического исследования [44] и ИК-спектров [45], атом алюминия расположен в центре равностороннего треугольника, образованного атомами бора. Четыре атома водорода соединены с каждым атомом бора, образуя его тетраэдрическое окружение. Два из этих атомов водорода составляют мостик между атомами бора и алюминия. Измерения спектров ЯМР-Н1 и В11 этого соединения не привели к однозначному подтверждению такой структуры, так как все четыре атома водорода проявляют себя как эквивалентные. В спектре на ядрах В11 наблюдается только квинтет, возникающий вследствие спин-спинового взаимодействия четырех эквивалентных атомов водорода с атомом бора [46]. Линии протонного спектра этого соединения сильно уширены вследствие квадрупольного момента ядра А127. Если снять это уширение методом двойного резонанса, то протонный спектр ясно указывает на эквивалентность всех ядер водорода. Межмолекулярный обмен концевых и мостиковых атомов водорода, протекающий со скоростью, достаточно высокой для усреднения их химических сдвигов, очевидно, также приведет к исчезнованию спин-спинового расщепления сигналов (см. разд. 9.1.3). Огг [46] предположил, что такой процесс обмена мог бы протекать по туннельному механизму, который не затрагивает электронную систему связей, и соответственно сохраняется спин-спиновое взаимодействие В — Н. Однако нет необходимости рассматривать такой механизм, поскольку наблюдаемый спектр согласуется с существованием быстрого внутримолекулярного обмена мостиковых и концевых атомов водорода внутри отдельных групп ВН4 (см. разд. 9.4.5). Подобные процессы внутримолекулярного обмена характерны для гидридов бора и их аддуктов с кислотами Льюиса [19].
12.1.5. Другие резонансные исследования В11
Ион BF7. Сигнал поглощения иона BF*" смещен на 41 м. д. в сторону более слабого поля относительно сигнала иона ВН^. Поскольку оба иона имеют одну и ту же симметрию, то этот сдвиг можно объяснить изменением диамагнитного экранирования, которое должно быть слабее для BF^. Смещение может также возникнуть из-за присутствия в этих молекулах низколежащих электронных состояний различной энергии (см. разд. 11.2). Малое отличие химического сдвига В11 для иона BF7 от сдвигов для аддуктов с эфирами и аминами указы
х) Исследование спектров В11 растворов В2оНр82[(С2Н5)3МН]2
и B2oDp82[(C2H5)2ND]2 в ацетонитриле в поле 43,9 кэ показало, что ион B20Hi‘82 состоит из двух ионов B10H_J, соединенных двумя трехцентровыми связями В — Н — В [338].— Прим. ред.
Спектры ЯМР других ядер (кроме водорода и фтора)
301
вает на то, что гибридизация орбиталей бора в комплексных соединениях примерно такая же, как в ионе BF; [2] *).
(СН^гРНВНз. Шулери [15] исследовал резонансные спектры на всех трех магнитных ядрах в соединении (СН3)2РН'ВН". Полученные им результаты представлены в табл. 12.7.
Таблица 12.7
КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ В (СНз)2РН'ВНз [15]
Спектр Константы спин-спинового взаимодействия, гц
JHP JHH' J PH' JBH" JBP J ph-
н 12 6 350 90 12
ВИ 93 50
рз! 350
12.2. Углерод
Углерод С13 является единственным встречающимся в природе изотопом углерода, который обладает магнитным моментом (спиновое число I = -у). Это ядро оказалось пригодным для успешного изучения методом ЯМР [47]. Несмотря на малое естественное содержание С13 (1,1%), большие значения времени релаксации и низкую характеристическую чувствительность метода ЯМР-С13 (1,59% характеристической чувствительности метода ЯМР-Н1 для одинакового значения напряженности поля), при соответствуюшей конструкции аппаратуры и выборе экспериментальных условий удается получить достаточно большое отношение сигнал/шум. Опубликованные исследования по резонансу на этом ядре не столь обширны, как для F19 и Р31, однако они уже позволяют представить себе широкие потенциальные возможности этой области спектроскопии ЯМР. Константы экранирования для ядер С13 изменяются в довольно широком интервале (350-10-6) и очень чувствительны к малым изменениям электронного окружения. Кроме того, константы спин-спинового взаимодействия между атомами водорода и углерода, непосредственно связанными друг с другом, также велики (~120 —269 гц). Обе эти особенности дают возможность проводить исследования резонанса С13 при таких условиях, которые позволяют повысить чувствительность за счет разрешения. Важное положение, которое занимает углерод в химии, в сочетании с доступностью углеродсодержащих соединений, без сомнения, должно стимулировать быстрый рост исследований на ядрах С13 и привести к широкому совершенствованию экспериментальных методов. Успехи в этой области, по-видимому, будут достигнуты в двух направлениях: а) повышение разрешения при условии обеспечения высокой чувствительности; б) более широкое использование -соединений, обогащенных изотопом С13 [266].
Некоторые закономерности в изменениях химических сдвигов С13 были отмечены рядом авторов [48, 49]. Например, Лаутербур [50] установил, что в замещенных метанах и этанах экранирование атома углерода уменьшается -с увеличением числа электроотрицательных заместителей у Этого атома. По способности уменьшать экранирование заместители обычно располагаются в ряд I > Вг > Н > С > С1 > О.
Я Проведено исследование зависимости константы спин-спинового взаимодействия В11 — F19 для тетрафторбората серебра от концентрации раствора [363].— Прим. ред.
302
Глава 12
12.2.1. Экспериментальные' методы измерения резонанса на ядрах С13
Для того чтобы преодолеть трудности, связанные с малым природным содержанием изотопа, применяют большие ампулы (с внешним диаметром 15 мм) и высокие значения радиочастотной мощности; Hi составляет от 10 мэ-(при наблюдении сигнала поглощения) и до 200 мэ (при наблюдении сигнала дисперсии) [47]. Оптимальные значения Н\ выбирают так, чтобы получить, максимальную чувствительность в сочетании с достаточным разрешением. Для того чтобы избежать насыщения при использовании высокой напряженности радиочастотного поля, необходимо очень быстрое прохождение сигнала (используются скорости 50—100 мэ!сек) [47]. Искажение сигнала при высоких скоростях развертки и высоких уровнях радиочастотной мощности приводит к ошибкам в определении химических сдвигов, которые легко исправить, взяв среднее значение химических сдвигов, определенное из двух спектров, один из которых записан при повышении, а другой при понижении напряженности развертывающего поля [48]. Для того чтобы произошла ядерная релаксация и вновь установилось больцмановское распределение ядер, между отдельными развертками спектра необходимо делать интервалы порядка нескольких минут. Обычно наблюдают дисперсионную форму сигнала, так как при этом легче избежать насыщения [47, 48, 51]. Однако в условиях быстрого прохождения высокий уровень радиочастотной мощности вызывает такое насыщение, которое часто искажает сигнал настолько, что исчезает половина дисперсионного сигнала. В результате спектр имеет такой же вид, как при записи сигнала поглощения. Большинство исследователей использовали внешние магнитные поля с напряженностью 10 000 э или меньше (при 10 000 э резонансная частота ядра С13 составляет 10,705 Мгц). В настоящее время применяются поля с напряженностью до 24 500 э, обеспечивающие соответствующее увеличение чувствительности [57]. Лаутербур установил, что при частоте 8,5 Мгц в условиях быстрого прохождения применение ампул с диаметром 12 мм и наблюдение дисперсионной формы сигнала позволяют обнаружить резонанс ядер С13 при концентрации 1 г-атом углерода в 1 л *). Для того чтобы преодолеть трудности, связанные с насыщением резонанса С13, были с успехом использованы проточные методы [53]. Если непрерывно подавать в зону поглощения энергии ненасыщенные ядра, то можно достичь пятикратного увеличения интенсивности сигнала. Были предприняты успешные попытки увеличить интенсивность сигналов С13, исследуя молекулы в присутствии свободных радикалов (эффект-Оверхаузера, см. разд. 3.17) [54]. Повышения чувствительности можно достигнуть также с помощью метода двойного межъядерного резонанса (метод INDOR) [55]. Если в молекуле имеются два ядра разного сорта А и X, связанные между собой спин-спиновым взаимодействием, то при развертке частоты второго сильного радиочастотного поля в области резонансных частот ядра X при одновременном наблюдении спектра ядер А при помощи слабого радиочастотного поля с соответствующей частотой должны произойти изменения в интенсивности спектра А. Так получается спектр двойного межъядерного резонанса, который точно воспроизводит спектр X при условии, что уровень мощности на частотах спектра ядра X не слишком высок.| На рис. 12.12 показан спектр INDOR-С13 для необогащенного образца CF3COOH. Ясно видно значительное повышение отношения сигнал/шум по сравнению с обычным спектром С13. Спектр F19 этого вещества состоит из одного интенсивного сигнала поглощения от молекул C12F3C12OOH и из двух пар сателлитов С13—F19 от молекул C13F3C12OOH (Jcu-fi» = 283,1 гц) и молекул C12F3C13OOH (JCi3-Fi9 = 43,53 гц) соответственно. Каждый из двух мультиплетов, приведенных на рис. 12.12,
Я Применение «метода разделения во времени» дает хорошие результаты для повышения чувствительности и для упрощения методики термостатирования образца [318].— Прим. ред.
Спектры ЯМР других ядер (кроме водорода и фтора) 303
был получен при облучении одного из сателлитов C13F в спектре F19 при развертке резонансной частоты С13.
Сигналы атомов С13, связанных с атомами хлора или азота (например, в СС14), очень слабы. Их можно наблюдать только при большой скорости развертки и при высокой радиочастотной мощности. Уширение линий в спектрах таких молекул возникает вследствие квадрупольного взаимодействия с ядрами хлора или азота (изотоп С13 имеет спиновое число J/2 и соответственно не имеет собственного квадрупольного момента) [48].
у, МЩ
Рис. 12.12. Спектр межъядерного двойного резонанса С13 (INDOR-спектр) чистой CF3COOH (в ампуле с внутренним диаметром 4 мм) [55].
Квартет при более высокой частоте отвечает молекулам CF3C13OOH (при облучении на второй частоте 56,460944 Мгц), квартет при низкой частоте — молекулам C13F3COOH (вторая частота облучения 56,460816 Мгц).
Пол и Грант [303, 304] повышали соотношение сигнал/шум в спектрах С13г подавляя спин-спиновое взаимодействие с протоном методом двойного резонанса. Помимо увеличения пиковой интенсивности из-за слияния мультиплета в синглет также наблюдалось возрастание интенсивности за счет эффекта Оверхаузера.
Для калибровки спектров С13, сигналы в которых слабы, применяют предварительную калибровку скорости развертки с использованием образца, обогащенного изотопом С13 (например, иодистый метил), а затем используют ту же скорость развертки для исследования необогащенного образца. Для калибровки используют сигнал поглощения от обогащенного образца, так как он позволяет получить более острые линии, чем сигнал дисперсии, что ведет к большей точности измерений.
Можно использовать как внутренний, так и внешний эталон. В качестве внутренних эталонов применялись диоксан и сероуглерод; было показано, что необходимо брать 20—30 об. % эталонного соединения, если только они не обогащены изотопом С13 [49]. Более распространено применение внешнего эталона, причем без учета поправок на влияние объемной диамагнитной восприимчивости. Обычно для этих целей используют сероуглерод, водный раствор углекислого калия, диметилкарбонат и уксусную кислоту.
Приведенные здесь химические сдвиги С13 были измерены относительно разных эталонных соединений. В тех случаях, когда это было возможно, сдвиги были пересчитаны к уксусной кислоте как к внешнему эталону (см. табл. 12.8—12.18). Следует отметить, что, хотя уксусная кислота является превосходным внешним эталоном, ее нельзя применять как внутренний эталон, поскольку известно, что химический сдвиг карбоксильного атома углерода зависит от растворителя. Вероятно, сероуглерод служит лучшим внешним эталоном, так как его острый резонансный сигнал лежит в более слабом поле, чем большинство других резонансных сигналов С13; его можно использовать и как внутренний эталон. Химические сдвиги, измеренные относительно уксусной кислоты, можно пересчитать в химические сдвиги относительно сероуглерода по формуле [49]
+15,6(м. д.). (12.1)
СНзСООН
304
Глава 12
Таблица 12.8
ХИМИЧЕСКИЕ СДВИГИ С13 НЕКОТОРЫХ ПРОСТЫХ СОЕДИНЕНИЙ [51]
Соединение Химический сдвиг а, м. д. Соединение Химический сдвиг а, м. д.
6СвНв 6СН3СООН бСвНв есн3соон
СН212 197 247 н-Алканы 107—127 157—177
СН31 153 203 Циклоалканы 95—105 145—155
СНзВг 126 176 Окись этилена 92 142
ch3nh2 116 166 Диоксан 55 105
СН2Вг2 111 161 R—S—C=N 15 65
(CH3S)2 109 159 Нитрилы От —2 48—64
СН3С1 107 157 ДО 14
СНзОН 81 131 Ароматические и От —10 40—60
СН2С12 76 126 олефиновые до 10
CH3NO2 53 103 (COOR)2 —31 19
СНС13 52 102 ROCOOR —36 14
СС14 35 85 rconr2 —45 5
с6н6 0 50 RCOOH —52 —2
нсоон —37 13 RCOR —79 —29
KCN (насыщ. водн. —49 1 а-Оксикислоты —90 —40
раствор) (карбонильный
углерод)
я
Химические сдвиги измерены относительно бензола н переведены в шкалу СН3СООН (внешний эталон); «сНзСООН = бСвН6 + '‘° <м- Д-1-
Водный раствор карбоната калия иногда применяется как эталонный образец. Одно из его достоинств состоит в том, что препараты этого соединения, обогащенные изотопом С13, относительно недороги. Формула пересчета имеет вид йС82 = йК2СоГ(водн) + 23,2 (м. Д.). ' (12.2)
Шписецки и Шнейдер [52] использовали в качестве внешнего эталона обогащенный С13 диметил карбонат, помещенный во внутреннюю из двух концентрических сферических ампул. К эталонному соединению добавлялось небольшое количество ацетилацетоната трехвалентного железа для уменьшения величины Л, что позволяло применять мощную звуковую модуляцию. Внешняя ампула содержала исследуемый образец. Вся система была сконструирована так, что могла вращаться внутри радиочастотной катушки. При использовании сферических образцов разность объемных диамагнитных восприимчи-востей, которую следует обычно учитывать при использовании внешнего эталона, не оказывает влияния на измеряемые химические сдвиги (см. разд. 7.3.2).
Установлено, что влияние растворителей на спектры ЯМР-С13 относительно мало для молекул, которые не содержат сильно поляризующихся групп (меньше 1 м. д.) [52]. Для таких соединений, как иодистый и бромистый метил, можно было бы ожидать значительно большего влияния растворителей. Эксперименты с разбавлением обогащенных образцов циклогексаном показали, что для этих двух замещенных метанов химический сдвиг при переходе от чистой жидкости к 5 (мол.) %-ному раствору в циклогексане составляет при отсутствии влияния объемной магнитной восприимчивости соответственно 7,3 м. д. и 3,6 м. д. в сторону более сильного поля. Считали, что таким влиянием растворителя на спектры С13 других замещенных метанов (см. табл. 12.9) можно пренебречь [52]. Однако изучение химических сдвигов для карбониль
Спектры ЯМР других ядер (кроме водорода и фтора)305
ной группы ацетона в присутствии протонных растворителей показало, что ядро С13 сильно дезэкранируется в этих условиях [276]. Химический сдвиг для апротонных растворителей наблюдался в пределах лишь 3,1 м. д., тогда как для протонных растворителей, 'способных к образованию водородной связи с карбонильной группой или вызывающих протонирование последней, наблюдаемые сдвиги карбонильного С13 лежат в интервале 35,8 м. д. (были изучены растворы с соотношением мольных долей 1 : 6).
12.2.2. Химические сдвиги С13
Работы Лаутербура [48] и Холма [51] показали потенциальные возможности исследований ядерного магнитного резонанса на ядрах С13 для определения строения молекул *). Очевидно, что атомы углерода в молекулах, как правило, достаточно изолированы от влияния межмолекулярных взаимодействий.
Рис. 12.13. Спектр ЯМР-С13 уксусной кислоты при частоте 8,5 Мгц, полученной в дисперсионной форме при быстром прохождении [48].
В соответствии с этим экранирование ядер углерода в основном определяется их электронным окружением внутри молекулы 151, 52]. Детального описания точной природы химических сдвигов С13 не было 1 2),'но Лаутербур [47] на основании химических сдвигов, наблюдаемых для ряда СВг4, С(СН3)4 и СС14, сделал вывод, что парамагнитный вклад в экранирование углерода может быть большим даже при наличии тетраэдрической симметрии. Аналогичное положение дел имеет место и для химических сдвигов Si29 [56]. Другие исследователи, рассмотревшие химические сдвиги Р31, сделали неправильное основное допущение, что парамагнитный вклад в экранирование атома с тетраэдрической симметрией должен быть лишь небольшим [4].
На рис. 12.13 воспроизведен спектр ЯМР-С13 уксусной кислоты [48], который дает представление о типичном спектре этого ядра. Обычно спектры ЯМР-С13 регистрируют при довольно низком разрешении, поэтому спин-спиновое взаимодействие наблюдается только между ядрами непосредственно связанных атомов углерода и водорода, поскольку константы такого взаимодействия достаточно велики. Поэтому атом углерода метильной группы уксусной кислоты дает в сильном поле квартет, возникающий вследствие спин-спинового взаимодействия между С13 и тремя связанными с ним эквивалентными протонами. Карбоксильный атом углерода дает один сигнал поглощения в слабом поле.
1) Весьма подробная диаграмма химических сдвигов С13 для разнообразных классов соединений приведена в работе [319].— Прим. ред.
2) Попл и сотр. [284, 313] использовали метод МО для расчета парамагнитного вклада в экранирование С13 в нескольких характеристических группах.
20 — 489
Рис. 12.14. Спектры ЯМР-С13 при частоте 15,085 Мгц соединений с естественным содержанием изотопа [57].
д — уксусная кислота (слева сигнал СНзС13ООН); б — этиловый спирт; в — пиридин (естественное содержание изотопа); г — С13Н3С12= С12Н, / — обычный спектр; 2— спектр при облучении Н1 на частоте 60 Мгц.
Спектры ЯМР других ядер (кроме водорода и фтора)307
Шулери (571 удалось записать спектры С13 в форме сигналов поглощения, используя модуляцию поля с частотой 2000 гц и наблюдая сигналы С13 на боковой полосе после соответствующей настройки фазы, приводящей к исчезновению центральной полосы. Этот метод позволяет использовать значительно более низкий уровень радиочастотной мощности, чем это требуется в методе быстрого адиабатического прохождения, и таким образом значительно уменьшить уширение линий, наблюдаемое в этом методе. При скорости развертки около 1 гц -сек~х Шулери удалось в спектрах С13 соединений с природным содержанием изотопа С13 разрешить сверхтонкое расщепление, составляющее приблизительно 3 гц. Дополнительное улучшение разрешающей способности было достигнуто за счет вращения больших ампул (см. разд. 7.3.3.). С помощью этого метода можно наблюдать в спектрах ЯМР-С13 константы дальнего спин-спинового взаимодействия между непосредственно не связанными атомами. Некоторые из великолепных спектров, полученных с помощью этого метода, приведены на рис. 12.14 [57]. На рис. 12.14, а показан спектр ЯМР-С13 уксусной кислоты: сигнал поглощения карбоксильной группы расщеплен в квартет (J = 7,5± 0,5 гц) вследствие дальнего спин-спинового взаимодействия с протонами метильной группы. Константа прямого спин-спинового взаимодействия С — Н, измеренная по квартетному расщеплению сигнала метильной группы, равна 132 гц. В спектре ЯМР-С13 этанола (рис. 12.14, б) обнаруживается не только квартетное расщепление сигнала метильной группы и триплетное расщепление метиленового сигнала, возникающие вследствие спин-спинового взаимодействия между непосредственно связанными атомами углерода и водорода, но также и более тонкая структура, обусловленная дальним спин-спино-вым взаимодействием С — Н. Тонкая структура наблюдается также в резонансных спектрах С13 пиридина и метилацетилена (см. рис. 12.14,в и 12,14, г ) 157].
12.2.3. Корреляция химических сдвигов С13 с электроотрицательностью групп, связанных с атомами углерода
В табл. 12.9 и 12.10 приведены химические сдвиги С13 метильных групп для нескольких монозамещенных метанов [48, 521. Лаутербур показал, что для ряда сходных соединений типа СН3Х, например для метил галоген идо в, существует линейное соотношение между химическими сдвигами С13 и электро-
Таблица 12.9
ХИМИЧЕСКИЕ СДВИГИ С13 СОЕДИНЕНИЙ СН3Х [52] а
Соединение бс6н6 есн3соон Соединение 6C6H0 бсн3<5оон
сн4 130,8 180,8 (CH3)4Ge 130,4 180,4
CH3F 53,3 103,3 (CH3)4Sn 137,6 187,6
СН3С1 103,8 153,8 (CH3)4Pb 131,6 181,6
СН3Вг 119,3 169,3 (CH3)2s 109,2 159,2
сн31 151,0 201,0 (CH3)2SO 85,2 135,2
(СН3)2О 69,3 119,3 CH3NO2 71,4 121,4
(CH3)3N 81,2 131,2 (CH3)2CO 104 154
(CH3)4N+Br- 72,0 122,9 CH3CN 124 174
(СН3)4С 97,1 147,1 CH3CHO 99,1 149,1
(CH3)4Si 129,0 174,0
а д выражено в миллионных долях (м. д.) по отношению к бензолу как внешнему эталону ^CHgCOOH (внешн) “ ^С0Н0 (внешн) + $0 (м- Д->-
20*
308
Глава 12
Таблица 12.10а
ХИМИЧЕСКИЕ СДВИГИ С13 ХЛОРБРОММЕТАНОВ [48]
Соединение б, м. д. Соединение б, м. д. Соединение б, м. д.
СС14 81 СНС13 98 СН2С12 124
СС13Вг 109 СНС12Вг 120 СН2С1Вг 139
СС12Вг2 142 СНС1ВГ2 143 СН2Вг2 156
СС1Вг3 170 СНВг3 168
СВг4 205
Таблица 12.106
ХИМИЧЕСКИЕ СДВИГИ-С13 ЗАМЕЩЕННЫХ МЕТАНОВ [48] а
Соединение б, м. д. Соединение 6, м. д.
СН13 316 С*Н(ОС2Н5)3 50
СН212 240 С*Н2(ОСН3)2 83
С*(ОС2Н5)4 38 (С*Н3О)2СН2 130
Я ”
а Химические сдвиги измерены относительно СН3СООН как внешнего эталона.
отрицательностью группы X. Как и для ядер F19 в бинарных фторидах [58], экранирование уменьшается с повышением электроотрицательности присоединенных групп. Было предположено, что это линейное соответствие связано с изменениями в экранировании, которые обусловлены уменьшением ионности связи С — X [48] (см. также табл. 12.11). Шписецки и Шнейдер [52] изучили
Таблица 12.11а
ХИМИЧЕСКИЕ СДВИГИ С13 МЕТИЛЬНЫХ ГРУПП В mpem-БУТЙЛ-, ИЗОПРОПИЛ-
И ЭТИЛПРОИЗВОДНЫХ [48]
Соединение б, м. д. Соединение б, м. д. Соединение б, м. д.
С*Н3СН2С1 159,9' (С*Н3)2СНС1 150,2 (С*Н3)3СС1 143,6
С*Н3СН2Вг 158,1 (С*Н3)2СНВг 149,0 (С*Н3)3СВг 140,7
С*Н3СН21 155,2 (С*Н3)2СН1 145,2 (С*Н3)3С1 136,6
С*Н3СН2ОН 159,8 (С*Н3)2СНОН 152,6 (С*Н3)3СОН 146,4
C*H3CH2NO2 166,6 (C*H3)2CHNO2 158,1 (C*H3)3CNO2 150,3
Таблица 12.116
ХИМИЧЕСКИЕ СДВИГИ С13 МЕТИЛЬНЫХ ГРУПП В ХЛОР- И БРОМЭТАНАХ [48]
Соединение д, м. д.
С*Н3СН2С1 159,9
С*Н3СНС12 145,7
С*Н3СС13 131,7
С*Н3СН2Вг 158,1
С*Н3СНВг2 142,3
Спектры ЯМР других ядер (кроме водорода и фтора)
309
Таблица 12.11в
ХИМИЧЕСКИЕ СДВИГИ СДЗ ЗАМЕЩЕННЫХ АТОМОВ УГЛЕРОДА В тргт-БУТИЛ-, ИЗОПРОПИЛ- И ЭТИЛПРОИЗВОДНЫХ [48] а
Соединение в, М. д. Соединение б, м. д. Соединение в, м. д.
СН3С*Н2С1 138,0 (СН3)2С*НС1 123,3 (СН3)3С*С1 11,9
СН3С*Н2Вг 150,1 (СН3)2С*НВг 133,0 (СН3)3С*Вг 116,5
СН3С*Н21 177,3 (СН3)2С*Ш 156,1 (СН3)3с*1 135,2
СН3С*Н2ОН 119,6 (СН3)2С*НОН 114,4 (СН3)3С*ОН 109,4
CH3C’H2NO2 106,8 (CH3)2C*HNO2 98,1 (CH3)3C*NO2 92,0
о *
Химические сдвиги С13 выражены в миллионных долях относительно СН3СООН как внешнего эталона.
химические сдвиги С13 для большого числа метильных групп в соединениях типа (СН3)ПХ (см. табл. 12.9). Построив зависимость химического сдвига С13 от электроотрицательности группы X (см. рис. 12.15), они установили, что
Рис. 12.15. Зависимость химических сдвигов С13 в соединениях СН3Х от электроотрицательности атомов X [52].
существуют две различные линейные корреляции. Для галогенов как заместителей они обнаружили значительное отклонение от прямой, отвечающей другим производным. Эти отклонения были отнесены за счет увеличения вклада в экранирование, обусловленного анизотропными эффектами соседних групп атомов (см. разд. 4.7.). Сходным образом были объяснены подобные же отклонения, найденные для галогензамещенных этанов (см. рис. 12.16). Однако Бакингем и сотр. [240] указали, что для любых ядер эффекты анизотропного экранирования могут дать лишь относительно небольшой вклад в экранирование, поскольку параметры, от которых зависит величина этого вклада, главным образом связаны с геометрией молекулы. Шнейдер [255] считает, что анизотропная модель не может удовлетворительно объяснить
310
Глава 12
наблюдаемые отклонения в химических сдвигах С13, и предполагает, что важное значение может, по-видимому, иметь влияние дисперсионных сил и сильно поляризуемых групп. В общем случае увеличение дисперсионного эффекта приводит к уменьшению экранирования 1275]. Для атомов водорода и углерода, непосредственно связанных друг с другом, обычно наблюдается удовлетворительное соответствие между химическими сдвигами протонов и С13. Это подтверждается рис. 10.2, который показывает, что для метильных групп химические сдвиги протонов изменяются в зависимости от электроотрицательности заместителя X таким же образом, как и химические сдвиги С13.
Рис. 12.16. Зависимость химических сдвигов С13 в соединениях СН3СН2Х от электроотрицательности атомов X [52].
Химический сдвиг С13 для а-углеродных атомов соединений типа (СН3)ПСН3_ПХ линейно зависит от длины связи С—X [269].
Постепенное замещение атомов водорода в метане на атомы галогена приводит к монотонному изменению химического сдвига С13, как показывают данные, приведенные в табл. 12.10а и на рис. 12.17. Все графики сходятся к значению химического сдвига метана. Ни одна из зависимостей не является линейной. Во всех случаях кривизна зависимости имеет отрицательное значение, что наблюдалось также при исследовании на ядрах Р31 [143] и Si29[ 50].
Химические сдвиги С13 хлор- и метилзамещенных метанов (а именно, СН4_хХх, где X = С1 или СН3) при замещении уменьшаются в такой последовательности, какую следовало ожидать на основании соображений электроотрицательности (т. е. экранирование уменьшается при возрастании электроотрицательности). Однако в случае бром- и иодпроизводных эта закономерность не наблюдается.
Были изучены три ряда хлорбромметанов [48] (СС1хВг4_х, СНС1хВг3_х иСН2С1хВг2_х). Химические сдвиги этих соединений приведены в табл. 12.10а, и результаты представлены в графическом виде на рис. 12.18. Для всех трех рядов найдено строго линейное соответствие между химическими сдвигами С13 и числом заместителей. Такая же линейная зависимость существует для химических сдвигов С13 метильных групп в галогенированных этанах.
Рис. 12.17. Зависимость химических сдвигов С13 для некоторых замещенных метанов СНжХ4_х от числа заместителей X [52].
Рис. 12.18. Зависимость химических сдвигов С13 хлорбромметанов СС1хВг4_х, СНС1хВг3_х и СН2С1хВг2_х от числа атомов хлора [48].
Внешний эталон — СНзС*ООН.
312
Глава 12
Из приведенных выше результатов исследований следует, что пока не будут лучше изучены эффекты, обусловливающие экранирование, необходимо относиться с осторожностью к попыткам сопоставлять молекулярные параметры с константами экранирования с С13 в галогенированных соединениях *).
12.2.4. Метил- и этилпроизводные элементов IV группы
Поскольку в тетраэдрических метильных и этильных производных С, Si, Ger Sn и Pb не должно быть вклада магнитной анизотропии центрального атома; в экранирование ядер углерода, то следовало бы ожидать, что изменение констант экранирования С13 будет происходить в соответствии с электроотрицательностью элементов IV группы [52]. Были измерены протонные спектры и спектры С13 соединений этого типа; химические сдвиги С13 приведены в табл. 12.9. Тем не менее при использовании метода, примененного ранее для определения электроотрицательностей на основании данных ЯМР [59, 60], невозможно определить надежные значения для электроотрицательности элементов IV группы.
12.2.5. Алкены
Химические сдвиги С13 для алкенов в отличие от соответствующих сдвигов; протонного резонанса весьма близки к химическим сдвигам в ароматических соединениях [50, 57]. Таким образом, анизотропное экранирование, вызванное
индуцированным кольцевым током,
которое имеет существенное значение для
СН3
ядер водорода, связанного с ароматическими кольцами, очевидно, должно быть малым для ядер С13. Были исследованы спектры ЯМР-С13 нескольких алкенов, и оказалось, что влияние заместителей
на экранирование для алкенов и ароматических молекул одинаково [50]. На
основании детального анализа спектров. Н1 и С13 этилена, обогащенного изотопом С13, определены константы спин-спино-вого взаимодействия Н — Н [279].
На рис. 12.19 представлены спектры ЯМР-С13 нескольких олефиновых соединений [232]. Установлено, что группы
СССС'-СС
= сн2,
^)СН и = С/
легко опреде-
t________I________I_______I________I
О 50 IOO 150 200
Химический сдвиг от сигнала CSZ м.З.
лить по наличию в спектре триплета,, дублета или синглета соответственно. В частности, эти данные применены для идентификации полностью замещенных олефинов, которые трудно изучать с помощью методов ИК-спектроскопии или протонного резонанса. Фридель и сотр.
®си3соон‘8с8г~'5>б(м'М
Рис. 12.19. Спектры ЯМР-С13 некоторых олефиновых углеводородов при частоте 15,085 Мгц [232].
Я Проведено исследование химических сдвигов С13 для метиленовых групп в напряженных циклических углеводородах [320]. Для химического сдвига С13 линейных алканов разработана аддитивная схема инкрементов [340].— Прим. ред.
Спектры ЯМР других ядер (кроме водорода и фтора)
313.
[232] изучили спектры некоторых диолефинов как с сопряженными, так и с кумулированными двойными связями. Они показали, что для олефинов с концевым положением двойной связи сопряжение оказывает малое влияние на химические сдвиги С13. Для кумуленов, таких, как метил аллен, первый и третий атомы углерода испытывают увеличение экранирования по сравнению с экранированием обычных олефиновых ядер углерода, в то время как экранирование второго углерода сильно уменьшается. Савитский и сотр. [273] измерили разность в экранировании (6Ч“С — fimpawc) этиленовых атомов углерода цис- и транс-изомеров, общей формулы СНХ = СНХ, где X есть Cl, Br, I или СООС2Н5. Эта разность имеет тот же знак и изменяется в том же порядке, что и разность экранирования протонов в этих соединениях. Однако разность в экранировании ядер С13 по порядку величины больше и ее нельзя объяснить влиянием анизотропии.
Савитский и сотр. [301] провели эмпирические расчеты химических сдвигов С13 ненасыщенных соединений.
12.2.6. Ацетилены
Лаутербур [50] измерил химические сдвиги С13 и константы спин-спинового взаимодействия для ряда замещенных ацетиленов. Полученные результаты приведены в табл. 12.12. Сигналы поглощения ацетиленовых атомов-
Таблица 12.12
ХИМИЧЕСКИЕ СДВИГИ С13 И КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ АТОМОВ УГЛЕРОДА, СВЯЗАННЫХ ТРОЙНОЙ СВЯЗЬЮ [50]
Соединение бс> м- А- JC13 = C12-H> гц ** 6С, м. д. (относительно CS2) г** Jci3H> гч
СН3СН2С* = С*СН2СН3 111,8
о о
II - II
СНзОСС* = с*сосн3 118,2
НОСН2С* = с**н 110,5 49 118',6 248
С1СН2С* = с**н 114,1 45 , 118,1 256
ВгСН2С* с**н 112,6 47 116,6 252
СвН6С* = с**н 108,9 44 115,3 252
(СН3)2С(ОН)С* = с*н 104,0 1а 122,5 253
(С2Н5)(СН3)С(ОН)С* S с**н 104,5 J 121,3 247
а Нельзя измерить точно из-за перекрывания пиков. Приблизительное значение 50±10 гц.
углерода расположены внутри узкой характеристической области спектра С13 (от +104 до +123 м. д. относительно CS2). Заместители по своему влиянию на экранирование С13 в алкинах располагаются в такой же последовательности, как и для замещенных алканов, за исключением того, что для алкинов их влияние несколько слабее.
12.2.7. Ароматические соединения
В спектрах ЯМР-С13 ряда замещенных ароматических соединений точно определены химические сдвиги для углеродных атомов кольца [48, 49, 61]. Спектр ЯМР-С13 бензола состоит из единственного дублета (см. рис. 12.20), дублетное расщепление обусловлено спин-спиновым взаимодействием между непосредственно связанными атомом углерода и атомом водорода (/с—н = = 160 гц). Введение одного заместителя в бензольное кольцо приводит к появ-
314
Глава 12
лению четырех различных типов атомов углерода в кольце: орто, мета, пара и атома углерода, с которым связан заместитель. Если с ароматическим кольцом связан только один метильный заместитель (например, в толуоле), то атомы углерода в орто-, мета- и пара-положениях не сильно различаются по своим химическим сдвигам, поэтому при обычном разрешении они дают один дублет с тем же расщеплением, что и в спектре бензола. Сигнал поглощения атома углерода, связанного с заместителем, представляет собой синглет, смещенный в слабое поле по отношению к другим сигналам (см. рис. 12.20)
Сене 1 бензол „А. \ С*СН3 21 1 Толуол sCH
V? п-Ксилол СН] Мезитилен
Дифенил ^Cf/ 3 VL Нафталин 10 1£
Аценафтилен 13 XV Флуорантен 14
yaf'^3 о-Ксилол R А ^сн>с‘сн3 4 4-^, м-Ксилол R А , W
7 Дурол cs, Гн j r i гс — 8 . ।' 2 / ексаметил- "Г "Ъ
Фенантрен CS2 -Цс. Пирен 12 “И у он JJL
Азулен 15 ju 2,6,8-Триметилазулен 16 Ci
н--------->•
Рис. 12.20. Спектры ЯМР-С13 ароматических молекул при частоте 8,50 Мгц [49].
и перекрывающийся с компонентой дублета остальных атомов углерода кольца, расположенной в слабом поле. Спектр ЯМР-С13 только одного из трех ксилолов — орто-производного — свидетельствует о наличии двух различных типов атомов углерода кольца, не связанных с заместителями. Но даже в этой молекуле разность химических сдвигов для двух дублетов очень мала. Установлено, что вообще для метилзамещенных бензолов химические сдвиги С13 имеют почти такое же значение, как для бензола [49], и мало меняются от одной молекулы к другой. Это следует из сравнения спектров С13 толуола, ксилолов, дурола, мезитилена и гексаметилбензола (см. рис. 12.20) и химических сдвигов, приведенных в табл. 12.13. Поскольку для этих соединений различия в химических сдвигах малы, невозможно сопоставить их с числом метильных заместителей [49].
Монозамещенные бензолы. Шписецки и Шнейдер [62] провели исследование спектров ЯМР-Н1 и ЯМР-С13 монозамещенных бензолов. В табл. 12.14 приведены химические сдвиги С13 для различных атомов углерода этих соединений, измеренные относительно бензола. Точное отнесение линий для орто-, мета- и пара-положений в спектрах монозамещенных бензолов можно провести путем исследования соответствующих дейтерированных соединений. Замещение водорода на дейтерий вызывает увеличение времени релаксации соседнего углерода. В результате при обычных значениях напряженности поля Hi, необходимых для наблюдения сигнала ядер С13 с низким естествен-
Спектры ЯМР других ядер (кроме водорода и фтора)
315
Таблица 12.13
ХИМИЧЕСКИЕ СДВИГИ С13 И КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ БЕНЗОЛА И НЕКОТОРЫХ МЕТИЛБЕНЗОЛОВ [49] а
Соедииеиия с-н С*—СНз СНз
4 S СО о «о 6СН3СООН> м. д. JCH> ги~ 4 2 СО о «о бСН3СООН, м. д. V 'И 6СН3СООН> м. д. X
Бензол Толуол о-Ксилол (3,6-СН) (4,5-СН) м-Ксилол я-Ксилол Мезитилен Дурол Г ексаметилбензол 65,0 65,2 63,6 67,2 65,8 64,5 66,1 62,0 49,4 49,6 48,0 51,6 50,2 48,9 50,5 46,4 159 156 154 .154 160 150 154 142 56,0 56,6 56,4 58,0 56,4 59,6 60,4 40,4 41,0 40,8 42,4 40,8 44,0 44,8 171,8 173,6 172,6 172,1 172,2 173,6 174,9 156,2 158,0 157 156,5 156,6 158,0 159,3 122 128 127 124 128 127 124
а Химические сдвиги измерены в миллионных долях (м. д.) относительно CS2 как виутреииего эталона и переведены в шкалу внешнего эталонаСН3СООН; 6СН3СООН (виеши) = +зг (внутр) — 15,6 (м. д.).
ным содержанием, происходит насыщение сигнала ядер углеродов, связанных с дейтерием. Для этих ядер не наблюдается сигналов поглощения в спектрах ЯМР-С13. Изменение времени релаксации обусловлено дипольным механизмом. Предполагается, что для ядер углерода уменьшение эффективности
Таблица 12.14
ХИМИЧЕСКИЕ СДВИГИ С13 РЯДА МОНОЗАМЕЩЕННЫХ
БЕНЗОЛОВ [52] а
Заместитель с-х орто мета пара
F —35,1 4-14,3 —0,9 + 4,4
С1 —6,4 —0,16 —1,0 +2,0
Вг +5,4 —3,3 —2,2 + 1,0
I + 32,3 —9,9 —2,6 + 0,4
СН3 —9,1 —0,3 —0,3 +2,8
ОСН3 —30,2 + 14,7 —0,9 + 8,1
nh2 —19,2 + 12,4 —1,3 + 9,5
N(CH3)2 —22,4 + 15,7 —0,8 + 11,8
сно —9,0 — 1,2 —1,2 —6,0
СОСНз —9,3 —0,2 —0,2 —4,2
no2 —19,6 + 5,3 —0,8 —6,0
а Химический сдвиг указан в миллионных долях относительно бензола. Положительные сдвиги отвечают более сильному, а отрицательные сдвиги— более слабому экранированию атомов углерода по сравнению с экранированием в бензоле.
дипольной релаксации превышает противоположное влияние квадрупольного механизма релаксации, обусловленного дейтроном, имеющим спин I = 1. Как и следовало ожидать, в монозамещенных бензолах заместитель оказывает наиболее сильное влияние на химические сдвиги того атома углерода,
316
Глава 12
с которым он связан. Если ввести поправку, учитывающую аномальное экранирование при замещении галогенами (что наблюдается в спектрах ЯМР-С13 замещенных метанов), то наблюдается удовлетворительное линейное соотношение между исправленными химическими сдвигами атомов углерода, связанных с заместителем, и электроотрицательностью заместителей. Химический сдвиг С13 уменьшается с увеличением электроотрицательности.
Аномальное экранирование, наблюдающееся для ядер С13 в орто-положениях относительно некоторых заместителей (таких, как галоген), слишком велико для того, чтобы его можно было объяснить влиянием анизотропии заместителя. Для иодбензола Лаутербур [253] предположил, что большой вклад в экранирование обусловлен высокой поляризуемостью атома иода (см. разд. 4.4.3). Малые сдвиги одного знака, которые наблюдаются для атомов углерода в .мета-положении замещенных бензолов, свидетельствуют о том, что в этом случае можно пренебречь влияниями индуктивного эффекта и магнитной анизотропии. Можно предположить, что в пара-положении вклады индуктивного эффекта и магнитной анизотропии в экранирование атомов углерода будут малы. Поэтому изменения экранирования пара-углеродов были отнесены за счет изменения л-электронной плотности в пара-положении,, вызванного эффектом сопряжения заместителей х). Электронодонорные заместители увеличивают экранирование атома углерода в пара-положении, а электроноакцепторные уменьшают его по сравнению с экранированием в бензоле. В ряду замещенных бензолов химические сдвиги для атомов углерода в пара-положении находятся в линейной зависимости от химических сдвигов пара-протонов. Следовательно, экранирование этих двух различных атомов одинаковым образом определяется эффектом сопряжения. Хотя подобная корреляция обнаружена и для ядер в орто-положении, она не настолько ярко выражена, как для пара-положения. Между химическими сдвигами протонов и С13 в .мета-положении не обнаружено никакого соответствия. Химические сдвиги пара-углеродных атомов показывают хорошую линейную корреляцию с соответствующими ст-константами Гаммета; для .иета-углерод-ных атомов не наблюдается такой корреляции.
Другие ароматические соединения. Лаутербур [49] исследовал резонансные спектры С13 ряда других ароматических производных, включая дифенил, нафталин, фенантрен, пирен, аценафтилен и флуорантен. Ему удалось провести частичный анализ этих спектров. Другие соединения, такие, как азулен и 4,6,8-триметилазулен, имеют спектры С13, расшифровать которые можно лишь предположительно. Для этих неальтернантных углеводородов интервал химических сдвигов С13 значительно больше, чем для альтернантных. Это дает основание предполагать, что природа экранирования ядер углерода в альтернантных углеводородах определяется главным образом изменениями л-элек-тронной плотности на атомах углерода ароматических колец: в альтернантных углеводородах л-электронные плотности на всех углеродных атомах кольца очень близки, в то время как в неальтернантных углеводородах, таких, как азулен, положение иное. Лаутербур [49] выделил л-электронные плотности на разных атомах углерода в азулене и нашел, что они хорошо коррелируются с наблюдаемыми химическими сдвигами С13. Используя то же приближение, которое применили Сайка и Слихтер [3] для объяснения химических сдвигов F19, Лаутербур приблизительно оценил влияние л-электронной плотности на экранирование рассматриваемых атомов углерода. Найденный коэффициент составил 240 м. д. на один электрон * 2).
х) Обсуждение зависимости химического сдвига С13 монозамещенных бензолов от о-констант Тафта, в частности индуктивного эффекта (ои) и эффекта сопряжения (<тс), см. в работе [316].— Прим. ред.
2) Химические сдвиги С13 для ароматических соединений были рассмотрены в работе [317] по методу Попла [284, 313] с использованием теории молекулярных орбиталей.— Прим. ред.
Спектры ЯМР других ядер (кроме водорода и фтора)
317
Спектры ЯМР-С13 дифенила и нафталина состоят из одного дублета с таким расщеплением, как для бензола. Узловым атомам углерода отвечает синглет, который перекрывается с компонентами дублета, находящимися в слабом поле. Спектры ЯМР-С13 пирена и фенантрена похожи на спектры ЯМР-С13 названных выше соединений, за исключением того, что сигналы узловых атомов проявляются более четко. В общем резонансные сигналы С13 конденсированных ароматических соединений лежат в несколько более сильном поле, чем сигнал поглощения бензола (см. рис. 12.20 и табл. 12.15).
Таблица 12.15
ХИМИЧЕСКИЕ СДВИГИ С13 И КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ ДИФЕНИЛА И НЕКОТОРЫХ АЛЬТЕРНАНТНЫХ УГЛЕВОДОРОДОВ [49] а
Соединение сн СС
6CS2- м- д- бСН3СООН> м. д. •/СН- гц 6cs2- “• Д- бСН3СООН’ м. д.
Дифенил 64,6 49,0 162 55,0 39,4
Нафталин 65,9 50,3 158 56,6 41,0
Фенантрен 65,6 50,0 158 60,2 44,6
Пирен 66,0 50,4 154 56,9 41,30
60,6 45,0
а Химические сдвиги измерены в миллионных долях относительно CS2 и переведены в шкалу внеш-* *
«его эталона СН3СООН; всн3СООН (внешн) = 6CS2 (внутр) ~ 15>6 (м-
Измерение химических сдвигов С13 для метильных групп замещенных толуолов показало, что для сходных соединений наблюдается соответствие между химическими сдвигами С13 оршо-метильной группы и сдвигами F19
, 1 Фенол С(31 о-Крезол 2 Ж® а R С(5) М-Крезол 3 Д г» ™ С(3,5) ч 4 ЛЕХ п-Крезол" 1
Я С(4) 5 Мг ж 2,0-Диметилфенол 1 6 /?1 СД51 3,5-Диметилфекол С(ЗД) 7 ж Анизол о СП2)С(4>5) 8 1,2-Диметоксибензол
Л'-----«-
Рис. 12.21. Спектры ЯМР-С13 фенолов, анизола и диметоксибензолов при частоте 8,5 Мгц [61].
в том же положении. Соответствие между химическими сдвигами С13 и значениями ст-констант Гаммета не столь явно выражено, как для химических сдвигов F19 [48].
Лаутербур [61] изучил спектры ЯМР-С13 ряда замещенных фенолов, диметоксибензолов, а также анизола. Отнесение линий и их химические сдвиги
318
Глава 12
Таблица 12.16
ХИМИЧЕСКИЕ СДВИГИ С13 И КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ ФЕНОЛА, МЕТИЛФЕНОЛОВ И АНИЗОЛА [61] а
Соединение Группа Химический СДВИГ, м. д. JCH- гч
6CS2 бсн3соон
Фенол С-1 37,4 21,8
СН-2,6 76,8 61,2 161
СН-3,5 62,4 46,8 159
СН-4 71,1 55,5 162
о-Крезол С-1 39,0 23,4
С-2 68,5 52,9
СН-3 61,5 45,9 160
СН-4 71,1 55,5 158
СН-5 64,8 49,2 163
СН-6 76,6 61,0 160
СНз 175,8 160,2 124
ж-Крезол С-1 37,6 22,0
СН-2 76,4 60,8 158
С-3 53,2 37,6
СН-4 70,3 54,7 159
СН-5 62,2 46,6 154
СН-6 79,8 64,2 160
СНз 171,6 156,0 128
я-Крезол С-1 39,9 24,3
СН-2,6 77,2 61,6 158
СН-3,5 62,3 46,7 156
С-4 62,0 46,4
СНз 171,9 156,3 126
2,6-Диметилфенол С-1 40,2 24,6
С-2,6 68,9 53,3
СН-3,5 63,7 48,1 156
СН-4 71,9 56,3 158
СНз 177,1 161,5 126
3,5-Диметилфенол С-1 38,0 22,4
СН-2,6 79,7 64,1 156
С-3,5 53,6 38,0
СН-4 70,6 55,0 158
СНз 172,1 156,5 129
Анизол С-1 32,7 17,1
СН-2,6 79,0 63,4 156
СН-3,5 63,0 47,4 158
СН-4 72,0 56,4 162
ОСНз 138,4 122,8 145
а Химические сдвиги измерены в миллионных долях относительно CS2 как внутреннего эталона и переведены в шкалу эталона СН3СООН; 6СН3СООН — ®CS2 — 15,6 (м- Д-)-
приведены в табл. 12.16; спектры представлены на рис. 12.21. Спектр ЯМР-С13 орто-диметоксибензола приведен на рис. 12.22. В спектре этого соединения метильным углеродным атомам отвечает квартет, находящийся в области сильного поля (линии 6—9)', атомы углерода, которые связаны с заместителем, дают синглет в слабом поле. Линии 3 и 5 составляют дублет, отвечающий
Спектры ЯМР других ядер (кроме водорода и фтора)3191
атомам углерода в орто-положении к метоксигруппам; линии 2 и 4 принадлежат оставшимся углеродным атомам кольца.
В спектрах ЯМР-С13 фенола сигнал кольцевого атома, к которому присоединена гидроксильная группа, сильно уширен вследствие неразрешенного дальнего спин-спинового взаимодействия С13 — Н1. В крезоле так же, как и в метилбензолах, атом углерода, соединенный с метильной группой, менее экранирован по сравнению с атомом углерода, не имеющим заместителей, и его сигнал смещен на 8 м. д. в сторону более слабого поля. Лаутербуру удалось оценить влияние метильной группы на экранирование других атомов кольца.
Рис. 12.22. Спектр ЯМР-С13 о-диметоксибензола при частоте 8,5 Мгц [63].
Он установил, что экранирование за счет метильной группы у атома углерода в пара-положении кольца составляет в среднем +2,7 м. д., а дополнительные вклады в сдвиг от мета- и opmo-метильных групп имеют среднее значение —0,4 м. д. и +0,2 м. д. соответственно. Пользуясь значениями, указанными выше для влияния метильной группы на экранирование, можно вычислить ожидаемые значения сдвигов для атомов углерода в метилбензолах. Во всех рассмотренных случаях было получено хорошее согласие с экспериментальными значениями.
Установлено [272], что химические сдвиги С13 в napa-дизамещенных бензолах подчиняются соотношению аддитивности.
Лаутербур исследовал спектры ЯМР-С13 некоторых анилинов, П,Ы-диме-тиланилинов [252], метилнитробензолов [254] и метилиодбензолов [253]. Изучены также спектры замещенных ацетофенонов [282] х).
12.2.8. Пиридин и замещенные пиридины
Спектры С13 и отнесение линий для пиридина, 3,5-лутидина и 2,4,6-кол-лидина приведены на рис. 12.14 и 12.23. На основании отнесения линий, выполненного для спектров двух последних соединений, можно расшифровать спектр самого пиридина [48]. Показано, что наименее экранированными являются атомы углерода в a-положении к азоту, в то время как атомы углерода в ^-положении экранированы сильнее. Такая же последовательность имеет место в протонном спектре пиридина.
Проведено исследование спектров С13 пиколинов, нескольких пиразинов [63], ароматических аминов и иминов [287] 2).
Ч Спектры С13 орто-, мета-, пара-анизола и его ди-орпго-производных см. в работе [321], стиролов — [341], алкилфенилкетонов — [342].— Прим. ред.
2) Расшифрованы спектры С13 пурина и его дейтеропроизводных. Химические сдвиги С13 сопоставлены с вычисленной плотностью л-электронов [343].— Прим. ред.
320
Глава 12
Рис. 12.23. Спектры ЯМР-С13 3,5-лутидина и 2,4,6-коллидина при частоте 8,5 Мгц [48].
Химические сдвиги измерены относительно СРзС*ООН.
12.2.9. Химические сдвиги С13 карбонильных соединений
В табл. 12.17 приведены химические сдвиги С18 для ряда соединений, содержащих карбонильные группы; ядро карбонильного атома углерода в кетонах экранировано значительно слабее, чем в сложных эфирах. Этот
, Таблица 12.17
ХИМИЧЕСКИЕ СДВИГИ С13 КАРБОНИЛЬНЫХ ГРУПП
Соединение б, м. д. а Литература
Муравьиная кислота (88%-ный водный 27 47
раствор)
Акриловая кислота 20 50
Уксусная кислота 16 47
RCOOH (13) 51
а-Оксикислоты —25 51
Уксусный ангидрид 27 47
Ацетат калия (насыщенный водный ра- 11 47
створ)
Диметиловый эфир ацетилендикарбоно- 40 48
вой кислоты
Диэтил карбонат 39 47
Диметил карбонат 36 50
RO2CCO2R (34) 51
<RO)2CO (29) 51
Метилбёнзоат 27 48
Спектры ЯМР других ядер (кроме водорода и фтора)
321
Продолжение табл, 12,17
Соединение б, м. д. а Литература
Винилацетат 25 50
Изопропенилацетат 25 50
Метилацетат 23 50
Метилсалицилат 22 48
Метил олеат 18 50
Этиллактат 17 50
Бутиролактон 15 50
Ацетофенон —4 47
Ацетилацетон (кетонная форма) —9 50
(енольная форма) 2 50
Ацетон —12 47
Нонанон-2 —13 50
2,4,6-Т риметнлацетофенон —13 50
r2co (-14) 51
Циклогексанон —16 50
Циклопентанон —26 50
Ацетальдегид —6 47
Диметилформамид 30 47
RCONR2 (20) 51
а Химические сдвиги измерены в миллионнах долях относительно CS2. В скобках приведены значения для классов соединений, обобщенные Холмом [51]. Межмолекуляриые водородные связи оказывают большое влияние на экранирование С13 в карбонильных группах [289].
факт может быть полезным, если необходимо идентифицировать эти две группы в молекуле неизвестной структуры [50]. Для большого ряда соединений, содержащих карбонильные группы, экранирование С13 определяется главным образом локальной электронной плотностью на атоме углерода [290] х).
12.2.10. Другие исследования ЯМР-С13
Спектры окиси и двуокиси углерода, обогащенных С13, были изучены для газообразного состояния [64]. Найдено, что химические сдвиги С13 относительно сероуглерода как внешнего эталона составляют соответственно + 11,5 и +68,6 м.д. Химические сдвиги этих простых линейных молекул могут иметь большое значение для понимания природы экранирования С13 [64]. Высокая концентрация газов была достигнута путем охлаждения образцов жидким азотом перед запаиванием ампул. В газовой фазе время релаксации С13 существенно уменьшается и соответственно проблема насыщения играет малую роль. Химический сдвиг для газообразной двуокиси углерода практически совпадает с найденными для жидкой двуокиси СО2, а также для растворов СО2, обогащенной С13, в инертных растворителях и воде [65].
В спектре С13 водного раствора двуокиси углерода, обогащенной С13, наблюдались две отдельные линии, отвечающие растворенной и гидратиро
х) Химические сдвиги С13 относительно CS2 для карбонильной группы «-бензохинона, n-нафтохинона и 2-нзобутилантрахинона см. в работе [318]. Для соединений СН3С(О) — X найдено соответствие между химическим сдвигом С13 и полярностью л-связи С = О [344]. Спектры С13 для уксусной, бензойной и 2,4,6-трнметилбензойной кислот в растворе серной кислоты и олеуме свидетельствуют об образовании ионов типа RC(OH)£ [345].— Прим. ред.
21—489
322
Глава 12
ванной формам СО2. Добавление карбангидразы в качестве катализатора приводит к слиянию этих сигналов в один в результате уширения линии, вызванного процессом быстрого обмена между двумя формами. Попытка исследовать спектры С13 карбоновых кислот оказалась неудачной из-за недостаточной чувствительности использованного прибора [65].
Исследование спектра ЯМР-С13 пентакарбонила железа Fe(CO)5 было предпринято для определения строения молекулы этого соединения. До сих пор точно неизвестно, имеет ли молекула строение тетрагональной пирамиды [67] или тригональной бипирамиды [68]. Для тетрагональной пирамиды можно было ожидать, что спектр С13 будет состоять из двух линий с соотношением интенсивностей 4 : 1, а для тригональной бипирамиды в спектре должны были бы наблюдаться две линии с соотношением интенсивностей 3 : 2. Наблюдаемый спектр состоит только из одной резонансной линии, что не удовлетворяет ни одной из двух структур. Это может быть следствием межмолекулярного обмена окиси углерода или того, что различные атомы углерода имеют совершенно различные времена релаксации. Но наиболее вероятное объяснение наличия в спектре только одного сигнала состоит в том, что химические сдвиги атомов углерода в двух окружениях равны [66]. При охлаждении вещества до —60° не наблюдается изменений в спектре С13. Это дает основание полагать, что протекание процессов обмена маловероятно [125].
Таблица 12.18
ХИМИЧЕСКИЕ СДВИГИ С13 НЕКОТОРЫХ КАРБОНИЛОВ МЕТАЛЛОВ [125]
Соединение Эталон Химический сдвиг, м. д.
Fe(CO)5 С6Н6 (внешний) —84,6
Ni(CO)4 С6Н6 (внутренний) —64,0
Fe(CO)2(NO)2 СвН6 (внешний) —79,0
В табл. 12.18 приведены химические сдвиги С13 для некоторых карбонилов металлов. Поскольку экранирование слабо зависит от атома металла, оказалось невозможным получить данные, которые указывали бы, присоединены ли молекулы СО в рассматриваемых соединениях к металлу атомом кислорода или атомом углерода [125].
12.2.11. Спин-спиновое взаимодействие между ядрами Н1 и С13
Результаты измерения константы спин-спинового взаимодействия между ядрами непосредственно связанных атомов углерода и водорода были приведены в работах ряда авторов [47, 49, 51, 73—78]. В большинстве случаев значения 7с1з—н найдены при изучении протонных спектров, так как такие измерения можно провести с более высокой точностью, чем в спектрах С13 [13, 73— 78]. Константы дальнего спин-спинового взаимодействия для атомов водорода и углерода, не связанных непосредственно друг с другом, наблюдались в протонных спектрах некоторых образцов, обогащенных изотопом С13 [57, 70, 99]. В благоприятных случаях такие константы можно измерить также в протонных спектрах необогащенных образцов [71].
Тирс определил относительные знаки констант С13 — Н1 и С13 — F19 в дихлорфторметане CHC12F, применив метод двойного резонанса. Было установлено, что эти константы вопреки ожиданию имеют разные знаки (7С1з-н = = +220,0 ± 0,1 гц ив спектре F19 Jcis-f = —293,8 ± 0,2 гц). Дублет спутника C13F, находящийся в области более сильного поля, сливался в одиночный сигнал при облучении в спектре протонного резонанса дублета спутника С13Н, расположенного в слабом поле. Для этого эксперимента по двойному резонансу
Спектры ЯМР других ядер (кроме водорода и фтора)
323
был использован прибор 1), позволяющий производить интенсивное облучение протонов с частотой 42,50 Мгц и наблюдать спектр резонанса F19 при частоте 40,00 Мгц.
Спин-спиновое взаимодействие непосредственно связанных атомов. Из теории спин-спинового взаимодействия, развитой Рэмзи [72], следует, что если спин-спиновое взаимодействие в основном определяется контактным взаимодействием Ферми, то величина константы спин-спинового взаимодействия между ядрами непосредственно связанных атомов должна находиться в прямой зависимости от «- характер а 2) связи между ними (см. разд. 5.9). Такое соответствие наблюдалось для спин-спинового взаимодействия между ядрами непосредственно связанных атомов углерода и водорода. Следовательно, контактное взаимодействие Ферми является определяющим фактором для этой системы [13, 69, 73—75, 300] 3).
Шулери [74] составил таблицу типичных констант спин-спинового взаимодействия Jcts-H для соединений, в которых гибридизация углерода изменяется от sp до sp3 (см. табл. 12.19). Он принял, что вклад 2«-орбитали в связь С — Н
Таблица 12.19
КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ С13 - hi РЯДА МОЛЕКУЛ, В КОТОРЫХ ГИБРИДИЗАЦИЯ УГЛЕРОДА ИЗМЕНЯЕТСЯ ОТ sp ДО sp3 [74]
Соединение Гибридизация JGH, ЗЦ Литература
С(СН3)4 sp3 120 47
Si(CH3)4 sp3 120 47
Циклогексен sp2 170 47
СеН6 sp2 159 47
СН3С = О sp2 174 47
1 н Метил ацетилен sp 248 .74
определяется квадратами коэффициентов перед 2«-волновой функцией в представлении метода МО-ЛКАО гибридных орбиталей sp3, sp2 и sp. Тогда, если спин-спиновое взаимодействие определяется контактным членом Ферми, то константы взаимодействия С13—Н1 должны быть равны у, у и ~ значения константы, ожидаемой для связи с чистым s-характером. На рис. 12.24 показана зависимость констант /щз-н от доли «-характера рассматриваемой связи С — Н. В соответствии с предсказанием зависимость представляет собой прямую линию, которая проходит через начало координат. Аналогичные результаты были опубликованы Маллером и Притчардом [13, 73], которые исследовали этот вопрос одновременно с Шулери. Маллер и Притчард изучили протонные спектры ряда углеводородов и определили значения констант спин-спинового взаимодействия /ци-н 113], которые приведены в табл. 12.20.
1) Серийный прибор «спин-декаплер» SD-60 производства американской компании NMR Specialities Со. Inc.
2) Теоретический расчет Jc-Н для метана и метилгалогенидов в приближении средней энергии триплетного возбуждения Д£ показал, что при сопоставлении /с-Н со степенью гибридизации, ионным характером и валентным углом необходимо принимать во внимание изменение Д£ [364].— Прим. ред.
3) Теоретический расчет константы взаимодействия С13 — Н1 проведен методом самосогласованного поля [346]. Найдено соответствие 7С1з_д и квадрата порядка связи между орбиталью атома водорода и 25-орбиталью углерода [347].— Прим. ред.
21*
324
Глава 12
Таблица 12.20
КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ С13 - Н1 РЯДА УГЛЕВОДОРОДОВ [13]
Соединение JC — Н’ гц
[13] [47]
Циклогексан 123 140
(СН3)2С = С(СН3)2 124
СН4 125
(СН3)4С 124 120
Мезитилен (СН3-группа) 126 126
Циклопентан 128
СН3С = ССН3 131
С13Н3С = сн 132
Мезитилен (кольцевые С — Н) 154 160
Бензол 159 159
Циклопропан 161
СН3С = С‘3Н 248
С6Н5С = С‘3Н 251
В дополнение к сопоставлению этих констант с долей s-характера связи С — Н они вывели полуколичественное уравнение для описания полученных результатов [см. уравнение (5.48)1.
Рис. 12.24. Зависимость констант спин-спинового взаимодействия 7с13~н от доли s-характера связи С — Н [74].
Влияние полярности связи на константы спин-спинового взаимодействия между ядрами непосредственно связанных атомов было рассмотрено несколькими исследователями [13, 73, 75, 77, 78]. Маллер и Притчард [13, 73, 75] считают это влияние весьма небольшим и ссылаются для подтверждения этого вывода на то, что константы для метилгалогенидов, в которых полярности связей С — Н существенно различаются, имеют практически одинаковые
Спектры ЯМР других ядер (кроме водорода и фтора)
325
значения (см. табл. 12.21). В табл. 12.21 приведены константы спин-спинового взаимодействия Jcw-h для большого ряда замещенных метанов.
Таблица 12.21
КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ JGH В ЗАМЕЩЕННЫХ МЕТАНАХ [73]
Соединение JC — H’ гч Соединение JC — H' гц
(CH3)3SiCN 122 (CH3)2NH(BH3) 139
(СН3)2С = СН2 126 a CH3OH 141
с2н6 126 6 CH3OCeH5 143
(СН3)2С = О 126 H2C(CN)2 145
СН3 — СвН5 126 в n-CH3O — C6H4 — CHO 145
сн3 — сно 127 r • CH3NO2 147
п-СН3 — С6Н4 — no2 127 (CH3 - 0 - C(O)CH2 -)2 147 e
(CH3)4Sn 128 Д CH3F 149 ж
(СН3)3С — CN 129 CH3C1 150
СН3 — COOH 130 CH3I 151
(CH3)3N 131 CH3Br 152
(CH3)2NH 132 CH2I2 173
ch3nh2 133 CH2C12 178
CH3CC13 134 СНВгз 206
CH3CN 136 CHC13 209
(CH3)2SO 138
а J . = 155 гц; 1,2 ± 0,1 гц.
СН(олефии) нн
6 JHH = 7’7 ± °’2 гц-
® J/-itt. . == 155 гц.
'-•«(кольцо), среди
Г •7СН(аЛьдегид) = 173 гЧ‘
Д Jjj _ gn== 53 гц (сателлиты от Snil7 и Snli9 перекрываются).
6 JCH(CH2-rpynna) = 155 гч; JHH(-CH2-CH2-) = 7,1 ± 0,4 ®4’
Ж ^HF = 45>8 ± °>5 гЧ-
Маллер [75] изучил спектры формильных соединений общей формулы ,О
X — С<^ и опубликовал значения констант /схз-н для формильной группы ХН
(см. табл. 12.22) 4). Он установил, что увеличение электроотрицательности заместителя X приводит к возрастанию /схз-н и уменьшению длины связи С — Н в формильной группе. Маллер объяснил этот эффект изменением гибридизации атома углерода при изменении заместителя X. Если в качестве меры гибридизации принять угол между связями углерода, то измеренные значения показывают, что в ряду формильных соединений происходит лишь очень небольшое изменение гибридизации. Маллер утверждает, однако, что такие предположения могут ввести в заблуждение, поскольку наблюдаемые углы между связями могут значительно отличаться от углов между орбиталями, именно которые отражают действительное состояние гибридизации
х) Найдено соответствие между Jс-н и электроотрицательностью заместителя /О
X для замещенных альдегидов X — С^^ • Предполагается, что при увеличении элек-
троотрицательности X происходит возрастание s-характера орбитали С — Н [365, 366].— Прим. ред.
326
Глава 12
Таблица 12,.2 2
КОНСТАНТА СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ J сн СОЕДИНЕНИЙ, СОДЕРЖАЩИХ ФОРМИЛЬНУЮ ГРУППУ [75]
Соединение JCH, гц JC - СН’ гц г(С - H), А г(С = О), А Угол НСО
рассч. набл.
СНзСНО 173 1,088 1,114 1,216 118°36'
п-СН3ОС6Н4СНО 173 1,088
СвН5СНО 174 24,1 1,088
0-НОСвН4СНО 177 19,8 1,086
(CH3)2NCHO 192 1,080
СС13СНО 207 46,3 1,073
носно 222 1,067 1,097 1,202 124°8'
СН3ОСНО 226 1,065 1,101 1,200 124°50'
FCHO 267 1,049 1,095 1,181 127°20'
атома [13]. Как следует из табл. 12.23, в ряду замещенных метанов константы спин-спинового взаимодействия Усхз-н заметно изменяются от одного соединения к другому даже несмотря на то, что в каждом случае углы между связями показывают, что молекула является тетраэдрической. Маллер полагает,
Таблица 12.23
КОНСТАНТА СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ JCH, ДЛИНЫ СВЯЗЕЙ С — Н И ВАЛЕНТНЫЕ УГЛЫ Н — С — X НЕКОТОРЫХ ЗАМЕЩЕННЫХ МЕТАНОВ [7 3]
Соединение JCH’ гц г(С — Н), А Угол НСХ
рассч. набл.
СН3 — С ЕЕ с — н 132 1,1046 1,1046 110°16'
СН3 — С ЕЕ N 136 1,1030' 1,1025 109°30'
СН3- F 149 1,0975 1,097 108°27'
СН3 — С1 150 1,0971 1,0959 108° 00'
СН3- I 151 1,0967 1,0958 106°58'
СН3 — Вг 152 1,0963 1,0954 107°14'
СН2С12 178 1,0854 1,082
СНВг3 206 1,0725 1,068
СНС13 209 1,0737 1,073
что эти изменения /схз-н слишком велики для того, чтобы объяснить их с точки зрения полярности связей, и постулирует существование изогнутых («банановых») связей в этих соединениях, обусловленных различием между межорбитальными валентными углами и наблюдаемыми углами между связями [73]. По мнению Гутовского и Жуана [69], такие эффекты, однако, не имеют значения. Другие авторы [77, 78] сопоставляли константы спин-спинового взаимодействия /щз-н Для метана с параметрами поляризации связи. Теоретически было показано, что константа 7сн должна уменьшаться с увеличением ионного характера С“Н+ при условии, что этот эффект дает значительный вклад. В последней своей работе, посвященной формильным производным. Маллер [75] полагает, что вклад этого эффекта пренебрежимо мал, поскольку наблюдаемая-тенденция в изменении Тщз-н противоположна той, которую бы следовало ожидать, если влияние полярности связи является определяющим.
Спектры ЯМР других ядер (кроме водорода и фтора")327
Очевидно, однако, что причина изменения константы /щз-н в настоящее время не полностью ясна х).
Если константа спин-спинового взаимодействия Лиз-н является функцией только гибридизации атома углерода, то должно выдерживаться простое соотношение между /щз-н и длиной связи С — Н [13, 73, 75]. Для простых углеводородов Маллер и Притчард [13] предложили полуколичественное уравнение, связывающее эти две величины. Для замещенных метанов уравнение, связывающее длину г связи С — Ни константу /(дз-н, имеет вид
г (С—Н)= 1,1597 — 4,17-10~4713 (12.3)
Уравнение не следует применять к другим классам соединений, кроме углеводородов. Например, все вычисленные по этому уравнению длины связей С — Н для формильных производных имеют заниженные значения [75]. Показано, что значения длин связей С — Н, вычисленные по этому уравнению, находятся в хорошем согласии с измеренными величинами (см. табл. 12.23). Кроме того, названные авторы вывели эмпирическое уравнение, связывающее электроотрицательность заместителя в монозамещенных метанах с константой 7с1з-н и длиной г связи С — X. Для соединения ряда СН3Х это уравнение имеет вид
J„13 = 22,6£х+40,1г (С-Х) + 5,5, (12.4)
и —п
где £х — электроотрицательность заместителя X по шкале Хаггинса [79]-Следует соблюдать осторожность при применении подобных соотношений для растворов, так как было установлено, что величина Jcis-h в хлороформе изменяется в широких пределах в зависимости от растворителя (от 208,1 гц в растворе циклогексана до 217,7 гц в диметилсульфоксиде) [262] 2). Увеличение константы Jcis-h происходит симбатно возрастанию способности молекул растворителя образовывать водородные связи с хлороформом. Меньшее влияние растворителя наблюдалось для константы спин-спинового взаимодействия Н — F в бромхлорфторметане. В этом случае водородная связь вызывает уменьшение константы спин-спинового взаимодействия [262].
Ривс и сотр. [291—293] вывели полезное линейное соотношение между | Лху/ухТУ | 1/2 и Zx (атомный номер) для элементов IV группы. Оно имеет силу для констант 7хн, /х—сн. /хр и Тх—с—f-
12.2.12. Аддитивные соотношения для констант спин-спинового взаимодействия Н1 2 — С13 и F19 — С13
Малиновский [80] установил, что для молекул типа CHXYZ константы спин-спинового взаимодействия Тен можно предсказать, используя простое соотношение аддитивности
4ch = ?x+Sy + ?z, (12.5)
где Сх, Cy и Cz — вклады в спин-спиновое взаимодействие, обусловленное заместителями X, Y и Z соответственно. Эти инкременты для различных заместителей можно вычислить исходя из значений констант Тен для соответствующих метильных производных. Например,
£х = ^с1з_н (СН3Х)-2гн, (12.6)
где £н равно 41,7 гц [вычислено из значения Tcis-н, наблюдаемого для метана: Тс1з-н (СН4) = 3£н = 125 гц]. В табл. 12.24 приведена сводка вычисленных
1) Маллер показал, что Jc-Н Для метильной группы галогенопроизводных толуола мало зависит от введения в орто-положен не атомов хлора [367]. С точностью ошибки измерения показано, что Jc-Н не изменяется при дейтерировании метильной группы толуола [368].— Прим. ред.
2) См. также [384].— Прим. ред.
328
Глава 12
Таблица 12.24
ЗНАЧЕНИЕ £х МАЛИНОВСКОГО ДЛЯ РЯДА ЗАМЕСТИТЕЛЕЙ, ПОЛУЧЕННЫЕ ПО КОНСТАНТАМ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ JCH СООТВЕТСТВУЮЩИХ
МЕТИЛПРОИЗВОДНЫХ [80]
Заместитель JCHa’ гЧ £х(среднее), гц
н 125 41,7
F 149 65,6
С1 150 68,6
Вг 152 68,6
I 151 67,6
ОС6Н5 143 59,6
он 141, 144 59,6
S(O)CH3 138 54,6
nh2 133 49,6
NHCH3 132 48,6
N(CH3)2 131 47,6
CN 136 52,6
CC13 134 50,6
CH2I 132 48,6
C s CH 132 48,6
CHC12 131 47,6
COOH 103, 131 47,1
CH2Br 128 44,6
CH2C1 128 44,6
CHO 127 43,6
CH3 126 42,6
c6H6 126 42,6
CH(CH3)Br 126 42,6
C(O)CH3 126, 122 40,6
C(CH3)3 124, 120 38,6
а Значения взяты из работ [13, 73, 85 и 241].
инкрементов, а в табл. 12.25 проведено сравнение для насыщенных молекул величин Jqis—н, наблюдаемых и предсказанных по уравнению (12.5). Почти во всех случаях получено превосходное согласие. По-видимому, значение зависит главным образом от того атома заместителя, к которому непосредственно присоединен атом углерода. Аналогичные попытки предсказать значения констант спин-спинового взаимодействия Тщз—f также были весьма успешны [81, 307]. В табл. 12.26 приведен список значений JCis_f для ряда соединений наряду с соответствующими инкрементами: значения инкрементов сохраняются постоянными для большинства заместителей, исключая водород. Константы 7С1з-г намного превышают соответствующие константы JCi3_H. Значение Jcis-f возрастает при увеличении атомного номера (для данной группы периодической системы) того атома заместителя, который непосредственно связан с углеродом. Иногда характерные константы спин-спинового взаимодействия Jc—f можно использовать для подтверждения предлагаемого строения молекул [81].
Гутовский и Жуан [69, 82] рассмотрели теоретические предпосылки для наблюдаемого аддитивного влияния заместителей на константы Jcis-н- Использовав приближение валентных связей и приняв, что спин-спиновое взаимодей-
Таблица 12.25
СРАВНЕНИЕ НАБЛЮДАЕМЫХ КОНСТАНТ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ JCH ДЛЯ НАСЫЩЕННЫХ МОЛЕКУЛ И JCH, РАССЧИТАННЫХ С ПРИМЕНЕНИЕМ ИНКРЕМЕНТОВ £х
МАЛИНОВСКОГО [80]
Соединение JCHa Разность
рассч. набл.
С6Н5СН2СвН5 127 127 0
(СН3)3ССН2ОН 139 132 +7
СН3СН21 152 149 +3
СН3СН2Вг 153 151 +2
СвН5СН2С1 153 152 + 1
С6Н5СН2Вг ' 153 153 0
(СН3)2СНВг 154 151 +3
ВгСН2СН2Вг 155 157 —2
С1СН2СН2С1 155 154 + 1
CH2C1CN 163 161 +2
СН212 177 173 +4
СН2Вг2 179 178 + 1
СН2С12 179 178 + 1
С12СНСНС12 185 182 +3
CHC12CN 190 189 + 1
СНС13 206 209 —3
8 ^СН взяты из Работ [13, 70 , 73, 85 и 241].
Таблица 12.26
КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ JCF И ^-ЗНАЧЕНИЯ, ПОЛУЧЕННЫЕ ПРИ ИССЛЕДОВАНИИ САТЕЛЛИТОВ F19 — С13 В СПЕКТРАХ ЯМР ЗАМЕЩЕННЫХ ФТОРАЛКАНОВ [81]
Соединение а JQisp, гч Атом, непосредственно связанный с C13 J, гч
Трифторметильные соединения
cf4 259,2 [236] F 86,4 6
CF3H 274,3 [236] H 101,5
CF3CC13 282,5 [237] С 109,7
CF3CO2H 283,2 [237] с 110,4
(CF3)2NNO2 273,6 N 100,8
(CF3)2PC1 320,2 Р 147,4
(CF3S)2 313,8 S 141,0
(CF3S)2Hg 308,3 S 135,5
CF3SNCO 309 S 136,0
(CF3)2Se 331,3 Se 158,5
(CF3Se)2 337,1 Se 164,3
(CF3Se)2Hg 332,5 Se 159,7
330
Глава 12
Продолжение табл. 12.26
Соединение а JC13F’ г:( Атом, непосредственно связанный с С13 L гц
Другие соединения
cf2h2 234,8 [236] н 74,2
CFH3 157,4 [236] н 52,5
цикло-C^Fg в 298,0 с 105,8
CC13F 336,5 С1 112,2
(CF2C1)2 299,0 С1 104,4 г
(CF2Br)2 311,6 Вг 115,7 г
а Соединения расположены в'порядке возрастания атомных номеров элемеи-тов, для которых определяется значение £.
б Это значение было использовано при выводе всех остальных.
в Линии сателлитов С!3 для этого соединения имеют триплетную структуру с уширенными компонентами (с относительными интенсивностями 1:1: 1); расщепление равно 3 гц.
г Принимая Sq = гц.
ствие С — Н определяется главным образом контактным членом Ферми, они сумели показать, что каждый заместитель обладает характеристическим сродством к s-характеру, который определяет, каким образом 2«-орбиталь распределяется по связям (см. разд. 5.9).
Общность простых аддитивных соотношений для констант Jcis-h была подвергнута сомнению в работах Маллера и Розе [100], а также Франкисса
Таблица 12.27
СРАВНЕНИЕ НАБЛЮДАЕМЫХ КОНСТАНТ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ JCH С КОНСТАНТАМИ, РАССЧИТАННЫМИ ПО ВЕЛИЧИНАМ £х МАЛИНОВСКОГО [100]
Соединения JCH> гЧ.
иабл. . рассч. разность (набл. — рассч.)
c6h5ch2f 151 a 150 +1
ch2f2 185 6 173 + 12
CHF3 238 197 +41
CHFC12 220 203 + 17
CHF2C1 231 200 +31
CH2(COOH)2 132 6 136 —4
CH2C1COOH 152 157 —5
CHC12COOH 181 184 —3
CH3OCH3 140 (140) в (0)
CH2(OCH3)2 162 155 +7
CH(OCH3)3 186 170 + 16
а Carr D. Т., Thesis, Purdue University, 1962.
б По данным работы [75].
в При использовании для метоксигруппы значения £ = 56,6 гц.
[236] . В табл. 12.27 приведены для сравнения некоторые наблюдаемые константы спин-спинового взаимодействия Jcis-h и предсказанные Малиновским [80] с использованием инкрементов С- Особенно большие отклонения вычисленных значений наблюдаются в том случае, если заместители у атома углерода
Спектры ЯМР других ядер (кроме водорода и фтора)
331
имеют высокую электроотрицательность [100]. Это заставляет предполагать, что s-характер атомной орбитали углерода, используемой для образования связи С — Н, не изменяется линейно при введении новых заместителей.
Франкисс [236] указал на то, что константы спин-спинового взаимодействия С — Н во фтор- и в некоторой степени в алкокси- и хлорпроизводных метана не подчиняются правилу аддитивности Малиновского (см. табл. 12.28).
Таблица 12.28
НАБЛЮДАЕМЫЕ И РАССЧИТАННЫЕ КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ ^СН НЕКОТОРЫХ ЗАМЕЩЕННЫХ МЕТАНОВ [236]
Соединение 7сн»
иабл. рассч.
CH2[N(CH3)2]2 136,6 137
СН2(ОС2Н5)2 161,1 155
CHa(NO2)2 169,4 169
СН2С12 178,0 175
ch2f2 184,5 173
CH(NO2)3 195,8 191
СНС13 209,0 200
CHF3 239,1 197
СН(ОС2Н5)3 185,2 170
Отклонения от этого правила особенно существенны для молекул с высокоэлектроотрицательными заместителями, имеющими атом с неподеленными парами электронов, который присоединен непосредственно к данному углероду. Отклонения слишком велики, чтобы их можно было объяснить изменениями s-характера связи С — Н, даже если рассматриваемые группы имеют высокую электроотрицательность. По-видимому, важное значение имеет взаимодействие между неподеленными парами электронов непосредственно не связанных атомов кислорода и фтора, а также других заместителей.
12.2.13. Аддитивные соотношения для констант ^cis-н атомов углерода с sp'1-гибридизацией
Малиновский [83] распространил аддитивное соотношение, выведенное им для замещенных метанов, на молекулы, содержащие атомы углерода с s/Агибридизацией. Было найдено, что инкременты £х, полученные для замещенных метанов, находятся в линейном соответствии с константами спин-спинового взаимодействия Лпз-н замещенных альдегидов ХСНО. При этом значения /щз-н для молекул ХСНО можно предсказать, пользуясь соотношением
Л18„(ХСНО) = 5,3+4,01£х. (12.7)
Vi XI
В табл. 12.29 приведен перечень рассчитанных и наблюдаемых констант
спин-спинового взаимодействия СВ * * * * 13 — Н1 для некоторых замещенных альде-
гидов. Карбонильный атом кислорода альдегидной группы вносит постоянный
вклад во все наблюдаемые константы спин-спинового взаимодействия, и поэто-
му соблюдение аддитивного соотношения, выражаемого уравнением (12.7),
не является неожиданным. Расчеты констант спин-спинового взаимодействия, основанные на преобладающем влиянии контактного взаимодействия Ферми,
332
Глава 12
Таблица 12.29
РАССЧИТАННЫЕ И НАБЛЮДАЕМЫЕ КОНСТАНТЫ 7СН НЕКОТОРЫХ ЗАМЕЩЕННЫХ АЛЬДЕГИДОВ ХСНО [83]
Заместитель ?, г'(а 7Сн- г'< 6
рассч. набл. разность
н 41,7 172,5 172 —0,5
С(СН3)з 40,6 168,1 168,6 —0,5
СН(СНз)2 39,0 в 161,7 168,9 +7,2
СН2СН3 41,0 г 169,7 170,6 +0,9
СНз 42,6 176,1 172,4 —3,7
СвН5 42,6 176,1 173,7 —2,4
СвН4С1-п 42,9 ' 177,3 175,2 —2,1
CgHiCl-Jw 43,4 179,3 177,5 — 1,8
СвН4С1-о 44,1 182,1 182,8 +0,7
СС13 50,6 208,2 207,2 —1,0
N(CH3)2 47,6 196,2 191,2 —5,0
ОСНз 54,6 224,2 226,2 +2,0
ОСН2СНз 53,4 Д 219,4 225,6 +6,2
F 65,6 268,4 267,0 —1,4
а Рассчитаны по данным для метильных производных, исключая помеченные другими буквенными индексами (см. работу [80])’
б Из протонных спектров.
в Рассчитано по 7^-д([СНз)2СНСН2С1] = 147,3 гц.
г Рассчитано по J ^jjfCHgCHaCHaCl] = 149,3 гц.
Д Рассчитано по 7{.ц[(СНзСН2)2О] = 137,7 гц.
показывают, что если интегралы перекрывания не зависят от состояния гибридизации атома углерода, то инкременты, найденные .для атомов углерода с зр3-гибридизацией, сохраняют свою величину и для углерода с зр2-гибриди-зацией. Аддитивные соотношения также наблюдались для констант спин-спинового взаимодействия С — Н sp2-атомов углерода в бензоле, замещенных пиридинах и пиримидинах [83], а также и в ароматических гетероциклах [309] *)
12.2.14. Взаимосвязь /щз-н с Jvm и Jhh для этиленов
Для алкилов и алкенов было показано, что константы спин-спинового взаимодействия JH-h и ^сы-н зависят в некоторой степени от электронной плотности на ядре водорода, а также от гибридизации углерода [82, 84—86]. Таким образом, не удивительно, что существует линейное соотношение между константами спин-спинового взаимодействия Jcis-н и Лн-н- Если на одной оси координат отложить значения /щз-н для нескольких соединений СН3Х, а на другой — значения трех констант Лн-н в соответствующих соединениях СН2 = СНХ, то наблюдается приблизительно линейная зависимость [82]. Значения JH-h возрастают при уменьшении /С-н (в предположении, что /н-нс i) * * * *
i) Проведен расчет констант 7с-Н для винилгалогенидов путем минимизации функции
перекрывания о-орбиталей при варьировании параметров гибридизации валентных орбиталей
углерода [369]. Для Zc-Н в ряду замещенных тиазолов найдено соответствие с межъядер-
ным расстоянием С—Н, эндоциклическими валентными углами и индексом свободной валент-
ности [370].—Прим. ред.
Спектры ЯМР других ядер (кроме водорода и фтора)
333
имеет положительный знак. Этот факт согласуется с тем, что возрастание значения /cis-н отвечает увеличению s-характера связи С — Н. Последнее приводит к возрастанию валентного угла С — С — Н, что в свою очередь вызывает уменьшение значений /н-н и 7н—н1С, которое наблюдается на опыте.
12.2.15. Корреляция констант спин-спинового взаимодействия Н1 — С13 с химическими сдвигами протонов
Поскольку значения констант спин-спинового взаимодействия С13 — Н1 определяются главным образом s-характером связывающей орбитали углерода, то следует ожидать линейной зависимости констант взаимодействия ^с1з-н от химических сдвигов протонов, так как основной вклад в экранирование последних для изолированной молекулы (т. е. в отсутствие диамагнитной
Рис. 12.25. Зависимость протонных химических сдвигов со от значений Jc-H Для некоторых этиленовых и гетероциклических соединений [87].
Номера совпадают с приведенными в табл. 12.30.
анизотропии, влияния среды и самоассоциации) вносит электронная плотность на водороде [87]. На рис. 12.25 представлена зависимость констант спин-спинового взаимодействия С — Н от химических сдвигов протонов, построенных по данным табл. 12.30 для этиленовых и гетероциклических соединений
Таблица 12.30
ПРОТОННЫЕ ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ C13-H1 ЭТИЛЕНОВЫХ И ГЕТЕРОЦИКЛИЧЕСКИХ ЯДЕР ВОДОРОДА
Соединение а <0 б, гц JC13—Н’
1. Этилен —212,0 157,0
2. Хлористый винил (цис) —216,3 160,0
3. Хлористый винил (транс) —211,2 161,0
4. Хлористый винилиден —215,3 166,0
5. Хлористый вииил (а) —247,5 195,0
6. цис-Дихлорэтилен —249,9 198,5
7. транс-Дихлорэтилен —249,5 199,1
8. Трихлорэтилен —252,9 201,2
334
Глава 12
Продолжение табл. 12.30
Соединение а m б, гц JC13—Н’ +
9. Винилиденкарбонат —281,0 220,1
10. Бензол —278,5 159,0
11. 2,5-Диметилтиофен —255,3 162,0
12. 2,5-Дибромтиофен —268,4 172,0
13. 2,5-Дихлортиофен —262,5 174,3
14. Тиофен (Р) —279,3 180,0
15. Тиофен (а) —286,4 187,0
16. Аллен — 181,9 168,2
а В тех случаях, когда в молекуле больше одного эквивалентного положения водорода, значения относятся к положению, обозначенному в скобках.
б При частоте 40 Мгц относительно тетраметилсилана как внутреннего эталона.
[87] . Аналогичная зависимость найдена для насыщенных соединений. Константы спин-спинового взаимодействия С — Н не зависят от магнитной анизотропии и слабо подвержены влиянию среды. Поэтому присутствие этих эффектов, имеющих большое значение для химических сдвигов Н1, вызывает отклонение от линейной зависимости, приведенной на рис. 12.25. Если для какого-либо соединения наблюдается такое отклонение, то можно провести эмпирическую оценку влияния магнитной анизотропии. Этот способ был применен для оценки эффекта анизотропии в бензоле, тиофене, ацетилене, аллене и хлорид-ной группе. В тех случаях, когда можно сравнить значения вкладов магнитной анизотропии, найденные рассмотренным выше способом, с результатами других методов, наблюдается хорошее согласие.
12.2.16. Константы спин-спинового взаимодействия С13 — F19
Большое число значений Jcis-f приведено в табл. 11.62к [270]. Для ядер атомов С и F, непосредственно связанных друг с другом, константа спин-
Таблица 12.31
ЗНАЧЕНИЯ НЕКОТОРЫХ КОНСТАНТ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ С13—F19 И ИЗОТОПНЫЕ ХИМИЧЕСКИЕ СДВИГИ Aq>F19(C13 —С12) [236]
Соединения JCF> гц A(pF19(C13— C12), м. Д.
CF3CC13 [237] 282,5 0,131
CF3COOH [237] 283,2 0,129
(CF3)2S 309,4 [81] 0,148 [238]
C13F3SCFS [239] 312,4 0,141
(CF3)2Se 331,3 [81] 0,145 [238]
CF3I [238] 344,8 0,149
CF2Br2 357,0 [81] 0,168 [238]
CF2 = CC12 [237] 288,9 0,103
CFC1 = CC12 [237] 303,1 0,112
COF2 [239] 308,35 0,121
CSF2 [239] 366,0 0,143
CF3SCFS [237] 395+4 0,16+0,05
Спектры ЯМР других ядер (кроме водорода и фтора)
335-
спинового взаимодействия, как правило, уменьшается при увеличении химического сдвига F19.
В табл. 12.31 приведены константы спин-спинового взаимодействия Jcis-f и изотопные (С13 — С12) химические сдвиги F19 (см. разд. 3.11) для ряда фторсодержащих молекул [236]. Франкисс установил, что для насыщенных соединений соблюдается линейная корреляция между этими двумя параметрами, которую можно представить следующим уравнением:
Дф (С‘3-С12)= +0,007+4,36- 10-V 13 .
G —F
где изотопный сдвиг Л<р выражен в миллионных долях (м. д.), a Jci3-f19 в герцах (гц). Для атомов фтора, связанных с sp2-атомом углерода, соответствующее уравнение имеет вид
Дф(С13 — С12) = — 0,039+5,04-10-Vc_P (м. д.).
Однако физический смысл этих соотношений остается непонятным.
12.2.17. Сателлиты С13 — Не протонных спектрах симметрично замещенных бензолов
При изучении сателлитов С13 — Н в протонных спектрах симметрично-замещенных бензолов были определены константы спин-спинового взаимодействия между протонами кольца [88]. В табл. 12.32 приведены спектральные
Таблица 12.32
КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ а НЕКОТОРЫХ СИММЕТРИЧНО ЗАМЕЩЕННЫХ БЕНЗОЛОВ
Соединение 7С13Н> ги- Другие коистаиты, гц
2,3,5,6-Тетрахлорнитробензол (в СС14) 175,0+0,4 JQ13CH ‘~ 1 j или
(в ацетоне) 176,9+0,6 ^с1зссн = 4,9±0,3
2,4,6-Трибромфенол (в ацетоне) 172,5+0,8 /^“ = 2,3+0,2
2,4,6-Трихлорфенол (в ацетоне) 171,3±0,5 J^a = 2,5+0,2
2,4,6-Тринитрофенол (в ацетоне) 175,8+0,5 7Йена=2,8±0,2
2,6-Динитро-4-хлорфенол (в эфире) 174,1+0,4 7^а = 2,8+0,2
2,6-Дибром-4-нитрофенол (в ацетоне) 172,8+1,3 /нна = 3,3±0,3
1,3,5-Трихлорбензол (в СС13Вг) 172,2+0,4 а = 2,1±0,2
1,3,5-Трибромбензол (в СС13Вг) 174,3+0,5 1,7+0,3
1,3,5-Тринитробензол (в ацетоне) 179,5+1,5 /Йен°=1,9±0,2
л-Дихлорбензол (в ацетоне) 169,0+1,0
л-Диметоксибензол (в ацетоне) 161,1 + 1,0
Бензол (чистая жидкость) 158,7+0,8
2,4,6-Триметилпиридин (чистая жидкость) 158,5+0,7 у£Нз> = 126,4+0,5
а Ошибки представляют собой стандартные отклоиеиия.
параметры, измеренные этим способом, а на рис. 12.26 показаны сателлиты С13 — Н в спектрах трех замещенных бензолов. Все производные бензола, приведенные в табл. 12.32, имеют только одну линию поглощения для ароматических протонов в спектре изотопно однородных молекул, т. е. молекул, содержащих только изотоп С12. Изучение данных табл. 12.32 показывает, что
336
Глава 12
значения констант спин-спинового взаимодействия С13 — Н для замещенных бензолов превышают константу для бензола. Тем не менее эти изменения не указывают на какое-либо существенное отклонение гибридизации атомов
Н
172,2---------------►,«-
Рис. 12.26. Спектры ЯМР-Н1 при частоте 60 Мгц при большом значении радиочастотной мощности, показывающие присутствие боковых полос С13 — Н в насыщенных растворах [88].
а — 2,3,5,6-тетрахлорнитробензола в ацетоне; б — 2,4,6-трибромфенола в ацетоне; в — 1,3,5-трнхлорбензола в трихлорбромметане. Расстояния между линиями указаны в герцах. AF — сигнал калибровки.
углерода от sp2. Большинство заместителей в этих соединениях являются электроноакцепторными группами, которые повышают s-характер связи С — Н и тем самым вызывают увеличение константы /стз-н 188].
12.2.18. Константы дальнего спин-спинового взаимодействия С13 — С — Н1
Константы спин-спинового взаимодействия между ядрами углерода и водорода, которые разделены более чем одной связью, имеют существенно меньшие значения, чем константы взаимодействия для непосредственно связанных атомов, и поэтому их весьма трудно обнаружить в спектре. Константы дальнего спин-спинового взаимодействия Jcis-c-hi были измерены при исследовании протонных спектров образцов, обогащенных изотопом С13 [70, 89, 96, 99]. Значения этих констант приведены в табл. 12.33—12.37.
Для насыщенных соединений значения констант дальнего взаимодействия 7СН лежат в интервале 0—6,5 гц, и для них не наблюдается регулярного уменьшения с увеличением числа связей, разделяющих эти два ядра. Действительно, в тех случаях, когда можно наблюдать обе константы в спектре одной и той же молекулы, оказывается, что некоторые из констант Jcis-c-hi меньше, чем Jci3-c-c-Hi- Аналогичное положение имеет место для значений 7м-н в протон-
Спектры ЯМР других ядер (кроме водорода и фтора)
337
Таблица 12.33
КОНСТАНТЫ ДАЛЬНЕГО СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ JC-H (В ГЕРЦАХ) РЯДА СОЕДИНЕНИЙ [70]
О
ю о
ю X 1 В X X X
Соединение сб К | и 1 и 1 1 и и 1 1 и
X и и о 1 и 1
о! 1 и 1 и
5 ,5 .о о
о
(СН3)2СНС13СН(СН3)2 4,1 5,1 7,2 7 1
(СН3)2СНС‘ЗН(ОН)СН(СН3)2 136 4,5 6,7 67
(СН3)2СНС13Н2СН(СН3)2 ~4,8 6,1 79
(СН3)3СС13О2Н 4,2
(СН3)3СС13О2СН3 4,4 3,5
(СН3)3СС13О2- 4,4
(СН3)3СС13Н2ОН 132 4,8
(CH3)3CC‘3D2OH 4,8
(СН3)2СНС13О2Н 5,2 5,4 7,2 75
(СН3)2СНС‘ЗО — О2СН3 5,6 5,8 3,5
(СН3)2СНС‘ЗН2ОН 140 3,8 4,8 79
(CH3)2CHC‘3D2OH 3,8
а Ошибка ие превышает ±2 гц.
6 Ошибка не превышает ±0,1 гц.
ных спектрах алкильных соединений свинца [90] и ртути [91, 92], также для Jfh в спектрах F19 фторалкилов и фторциклобутанов [93].
Был измерен ряд значений констант спин-спинового взаимодействия для атомов водорода и углерода, разделенных двумя связями. Эти данные приведены в табл. 12.34 [94—96]. Средние значения констант спин-спинового взаимодействия /си-с-н для sp3-, sp2- и sp-гибридизованного углерода равны 4,0; 5,9 и 10,6 гц, и если построить график значений УС1з-с-н от доли s-характера связи С13 — С, то получится линейная зависимость. В отличие от аналогичной линейной зависимости для непосредственно связанных атомов С и Н в этом случае прямая не проходит через начало координат. При проведении теоретических расчетов было установлено, во-первых, что наиболее важным фактором в таком спин-спиновом взаимодействии является контактное взаимодействие Ферми и, во-вторых, что замена атома водорода на атом С13 не изменяет коэффициентов волновых функций основного состояния х).
Если два атома углерода в группировке С13 — С12 — Н связаны одной простой связью, то константа дальнего спин-спинового взаимодействия имеет величину 0 — 6,5 гц [70, 75, 242], в то время как для замещенных ацетиленов X — С13 == С12 — Н значения JCi3-c-h лежат в интервале 44—51 гц [50, 89]. Маллер [242] установил, что значения JCi3_c_H в этиленовых системах С13 = = СН — колеблются от 0,0 до 15,7 гц (см. табл. 12.35). Эти результаты показывают, что порядок связи С — С не является доминирующим фактором, определяющим константы спин-спинового взаимодействия Уаз-с-н- В геометрических изомерах дихлорэтилена порядок связи и гибридизация атомов
*)
Последние исследования показали, что значения J
с s-характером связи С—С [306].
с13-с-н
не всегда коррелируются
22—489
338
Глава 12
Таблица 12.34
КОНСТАНТЫ ДАЛЬНЕГО СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ CIS—Hl РЯДА СОЕДИНЕНИЙ [96J
X
X 1 X 1 1 и 1 X
Соединение X и и 1 и 1 1
X 1 со и 1 и 1 О 1 1 и
,5 и .и и X
CH3CH2C‘3D2OH ~4 6,4+0,2 7,3+0,3
(CH3CH2)2C‘3DOH 4,0+0,1 5,3+0,2 7,3+0,3
(СН3СН2)3С13ОН 3,8±0,2 4,5+0,2 7,5+0,3
СН3СН2С13(ОН)((?Нз)2 { (а) 4,0±0,1 (Р) 4,1±0,1 4,0±0,2 7,3+0,3
а Р f CHSCH2C13(C1)(CH3)2 ( (а) 3,7±0,2 (3) 3,9±0,1 5,7+0,1 7,5+0,3
аСН3 1 3 С13Н3-С(ОН)СН2СН3 129J-3 | (а) 3,2+0,2 (Р) 3,0+0,2
аСН3 1 3 С‘3Н3-С(С1)СН2СН3 СН3С‘3ООСН2СН3 132±3 | 6,0+0,1 (а) 4,2+0,2 (Р) 3,2+0,2 3,1+0,1 0
(СН3СН2)2С‘3 = о 5,7±0,1 4,7+0,1 7,5+0,1
СН3СН2С‘3О2Н 6,4+0,2 5,5+0,2 7,4±0,1
СН3СН2С‘3О2СН3 6,5±0,3 5,3+0,1 4,0+0,05 7,4±0,1
(СН3СН2)2С13= NNH<p(NO2)£ 6,5+0,3 4,8+0,4 7,2+0,2
(CH3CH2)2C‘3=NNHCONH2 131,4 4,0+0,4 4,0+0,3 7,5+0,2
С13Н3С = СН а 3,6
СН3С S С13Н а а 3 СН3С13 = СН а 247,6 (а) 10,6 (3) 50,8 4,8
С13Н3СОН б 26,6 -
Значения взяты из работы [99]. ®Lauterbur Р. С., частное сообщение. в <р—фенильная группа.
Таблица 12.35
КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ Cis-Hl НЕКОТОРЫХ ОЛЕФИНОВЫХ СОЕДИНЕНИЙ [242]
Соединение jC1sh> JC13-C—н» гц
цис-Дихлорэтилен 198,3 15,7
транс-Дихлорэтилен 199,1 0,0
Диэтилмалеат 167,6 1,5
Диэтилфумарат 168,6 4,4
углерода должны быть достаточно постоянными, однако значения JCi3_G_H сильно различаются [242]. Константы спин-спинового взаимодействия Jqis-h и Jcis-cH обнаруживают линейную корреляцию со степенью sp-гибридизации атомных орбиталей углерода. В то же время для константы спин-спинового взаимодействия Jcw-с-сн такой корреляции не наблюдается. В табл. 12.34 приведены значения дальних констант спин-спинового взаимодействия, измеренных для ряда соединений, обогащенных С13. Отсутствует также зависимость
Спектры ЯМР других ядер (кроме водорода и фтора)
339
Таблица 12.36
КОНСТАНТЫ ДАЛЬНЕГО СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ С13-Н1, ИЗМЕРЕННЫЕ НА НЕОБОГАЩЕННЫХ
ОБРАЗЦАХ [71]
Соединение Тип протона ^С13—Н1>
Ацетонитрил Ацетат натрия Метилацетат Ацетатный 9,8—10,0 5,8—6,0 6,8—6,9
Ацетофенон Метильный 5,7
Ацетальдегид 6,0
Толуол » 5 (уширенный сигнал)
константы спин-спинового взаимодействия Jcis-c-c-h от электроотрицательности заместителя,' связанного с атомом С13. Отсутствие зависимости не связано с конформационными факторами, поскольку значения констант Jh-c—с-н, отражающие эти факторы, одинаковы для всех изученных молекул (см. табл. 12.34). Следовательно, контактное взаимодействие Ферми не является единственным механизмом, определяющим спин-спиновое взаимодействие в системе С13 — С — СН, хотя и показано, что спин-спиновое взаимодействие в значительной степени обусловлено этим фактором. Для хр3-гибридизирован-ного атома углерода в одной и той же молекуле обычно Jcis-c—сн > •/си-сн-Причина этого, по-видимому, заключается в том, что константы Jcis-c-н аномально велики. Аналогичное положение для металлалкилов [90—92], которое, как первоначально предполагали, является следствием аномально малых констант спин-спинового взаимодействия Jm-c-h, также можно объяснить указанным выше образом. Сходные результаты были получены для констант спин-спинового взаимодействия Jcis—с-сн в ряду соединений, содержащих группу СН3СН2С13 (см. табл. 12.34) [96]. Рассмотрение табл. 12.33 показывает, что существует приблизительно линейная зависимость между константами JCi3_с-сн и соответствующими значениями Jh-c-c-h в молекулах, которые содержат изопропильную группу, связанную с атомом С13 [94]. Вначале полагали, что либо контактный член Ферми определяет обе названные константы спин-спинового взаимодействия, либо для спин-спинового взаимодействия С13 — С — С — Н важными являются конформационные факторы. Однако из рассмотренных выше данных (табл. 12.34) следует, что наблюдаемая зависимость для изопропильных производных является случайной [96] *).
Измерение дальних констант спин-спинового взаимодействия С13 — Н1. Если в протонном спектре основной центральный сигнал, который отвечает водородам, связанным с немагнитным изотопом С12, имеет малую ширину (< 1,5 гц,), то константы дальнего спин-спинового взаимодействия С13Н можно наблюдать для необогащенных образцов, используя обычный промышленный спектрометр [71]. В табл. 12.36 приведены измеренные таким образом значения некоторых констант дальнего спин-спинового взаимодействия. Если бы удалось устранить из спектра интенсивный центральный сигнал ядер водорода, связанного с атомом С12, то наблюдение спутников С13Н стало бы намного проще. Андерсон [97] достиг цели путем облучения образца мощным полем с частотой резонанса С13. Это поле модулируется по частоте с частотой 2 кгц. Влияние
*) Анализ факторов, влияющих на J 13 , для систем (CH3)3CCis проведен
С1 -—С—*с—н
в работе [371]. Изучение спектров Н1 сателлитов С13 ряда замещенных альдегидов показало, что константа взаимодействия 7с13^ с н1 имеет отрицательный знак, a 7ci3 hj —
положительный. Абсолютное значение 7с13 4 возрастает от отрицательных значений
( — 4,5 гц для этана) до положительных (1,2 гц для НСС12СС12Н) [372].—Прим. ред.
22*
Таблица 12.37
КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ С13 (ВКЛЮЧАЯ НЕКОТОРЫЕ КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ С13-С13) РЯДА СОЕДИНЕНИЙ, ОБОГАЩЕННЫХ ИЗОТОПОМ С13[98, 2 49]
Соединение RCX— C^R Константы спин-спинового взаимодействия СДЗ, гц Химический сдвиг относительно бензола, м. д. Литература
JCxCy Чн Чн JCxCyH JC„C..H У Л
CeH6Ci3H2-Ci3H3 34±1 — 127 -127
CeH5Ci3H(OH) — С13Н3 38,1±0,1 142,5=1=0,1 126,9+0,1 4,1+0,2 2,0+0,2 58,2 102,9 249
С8Н5С13О-С‘3Н3 43,3+0,1 125,7+0,5 6,2±0,1 —68,6 103,2 249
/ \с13-СОД3 44,2+0,3 136,2+0,1 6,0±0,1 —9,2 106,5 249
N=C13 —СОД3 57,3±0,3 10,1+0,1 10,4 127,8 249
/ \ci3- С1зОС1 74,1+0,2 —4,8 —39,8 249
СвН5СН = СОД - С‘3ООС2Н5 76,3+0,2 160,9+0,5 249
/ \ci3 — C13=N 80,3±0,5 13,8 9,2 249
CeH5Ci3=Ci3 —СОС2Н5 126,8±0,1 46,9 —25,5 249
CeH5C=C13 —C13=N 155,8+0,2 64,2 21,8 249
свн5-сод=сод2 70+3 249
СвН5-СОДС‘3-Н 175,9+0,3 251,1+0,2 49,9±0,2 45,4 51,4 249
С8Н5-С=С‘3-С13Н3 68,6+0,1 131,3+0,1 10,5±0,1 41,9 123,8 249
сод=сод 171,5 98
сод2=сод2 67,6 98
сод3—сод3 34,6 98
Таблица 12.38
КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ С13—Н1 РАЗЛИЧНЫХ СОЕДИНЕНИЙ [75, 87]
Соединение /с-н> г« Литература
А12(СН3)в 113,0 *
Si(CH3)4 Ga(CH3)3 In(CH3)3 (в бензоле) Паральдегид (группа СН3) Hg(CH3)2 CF3CH3 Н2С(СООН)2 CeH5N(CH3)2 (группа СН3) (CH3)2S CH3SH (CH3)2NCHO (группа СН3) 118,0 122,0 126,0 127,0 130,0 130,0 132,0 135,0 138,0 138,0 138,0 75
(CH3)2SO2 (в воде) Na[B(OCH3)4] (в воде) 1,4-Диоксан В(ОСН3)3 (CH3)4NOH (в воде) H2C(CN)2 (СН3О)2СО Этиленкарбонат Паральдегид (кольцевые С — Н) Триоксан (в бензоле) Ферроцен (в CS2) CH2F2 140,0 142,0 142,0 143,0 144,0 145,0 147,0 157,0 159,0 166,0 175,0 185,0
Циклогексан 123,0
Изобутилен а-Метилвинилметиловый эфир (а) цис-Кротонитрил Кротоновый альдегид а-Метакр илон итрил а-Метилвинилметиловый эфир (метокси) Метилацетилен 1,4-Дихлорбутин-2 Пропаргил хлорид 126,0 127,0 128,0 129,0 132,0 143,0 132,0 158,1 159,0
1,4-Д ибромбутин-2 2,4-Диметилпиррол (4-мерил) 160,0 127,0 87
2,4-Диметилпиррол (2-метил) 2-Метилфуран 1,2,5-Триметилпиррол (N-метил) N-Метилпиррол 4-Метил имидазол З-Метилпиразол 2-Метилимидазол 128,0 129,0 137,0 139,5 126,5 129,0 128,5
N-Метилимидазол 3-Метилтиофен 2-Метилтиофен Тетраметилаллен Толуол 140,0 126,5 128,5 127,0 126,0
342
Глава 12
этой модуляции на протонные сигналы наблюдается с помощью фазового детектора, настроенного на частоту 2 кгц. Этим способом можно достичь более высокой чувствительности, чем другими методами, и успешно измерять химические сдвиги С13 и константы спин-спинового взаимодействия С13Н.
12.2.19. Константы спин-спинового взаимодействия С13 — С13
При исследовании протонных спектров этана, этилена и ацетилена, обогащенных изотопом С13, были измерены константы спин-спинового взаимодействия между ядрами С13 [98]. Введение ядер С13 приводит к тому, что протоны становятся магнитно неэквивалентными, например, молекула НС13 = С13Н представляет собой типичную спиновую систему АА'ХХ'. Константы спин-спинового взаимодействия С — С, приведенные в табл. 12.37, измерены главным образом в спектрах ЯМР-С13 несимметрично замещенных соединений, обогащенных изотопом С13 [98, 249]. Константа Jcu-cis этана и этилена имеет значение, которого следовало ожидать при условии, что спин-спиновое взаимодействие определяется в основном контактным членом. Полагают, однако, что значительно более высокое значение константы взаимодействия С13 — С13 для ацетилена обусловлено положительным вкладом какого-то дополнительного механизма [98]. Для сопряженных ациклических соединений значения констант спин-спинового взаимодействия С13 — С13 меньше, чем для других соединений с л-электронными связями [249].
Бернстейн и Фрей [249], пользуясь приближением, принятым Маллером и Притчардом [13] для интерпретации констант взаимодействия С13 — Н (см. разд. 5.9), показали, что для углеводородов величина Jcia-cw прямо пропорциональна произведению s-характеров атомов углерода данной связи. Для замещенных углеводородов на основании эмпирического соотношения [74], включающего величины Ущз-н Для соответствующих соединений [249], были найдены «модифицированные s-характеры» орбиталей углерода.
Для углеводородов найдено простое соответствие между константой спин-спинового взаимодействия С13 — С13 и длиной простой связи С — С [249]. Пока это соотношение, однако, имеет ограниченную область применения х).
12.2.20. Различные константы спин-спинового взаимодействия С13 — Н1
Для полноты данных в табл. 12.38 приведены значения /щз-н Для различных соединений [75, 87].
Шулери и сотр. [89] измерили шесть констант спин-спинового взаимодействия С13Н в протонном спектре метилацетилена, обогащенного ядрами С13. Наблюдаемые константы спин-спинового взаимодействия (в герцах) приведены ниже:
Видимо, не существует простого соответствия между различными константами спин-спинового взаимодействия * 2).
1) Методом двойного резонанса {С13} — Н1 показано, что 7с13 с13 = 57,3 гц имеет отрицательный знак [373].—Прим. ред.
2) Для ацетонитрила, обогащенного N15, определены величины и знаки следующих коистаит взаимодействия J 13 = 136,0 гц; 7 13_ 15= — 17,5 гц; J 13 15 = 3,0 гц;
С —-Н С =:N С — C^N
Спектры ЯМР других ядер (кроме водорода и фтора)
343
12.3. Азот
Оба изотопа азота, встречающиеся в природе, N14 (содержание 99,635%) и N15 (0,365%), имеют спин, отличный от нуля, и, следовательно, могут изучаться методом ядерного магнитного резонанса. Однако низкое природное содержание изотопа N15 препятствует наблюдению его сигнала при высоком разрешении, и поэтому данный раздел в основном будет посвящен изотопу N14.
Рис. 12.27. Спектр ЯМР-N14 жидкого аммиака при частоте 3,076 Мгц [101]. Частота модуляции равна 392 гц» Два квартета являются сигналами иа боковых полосах, центральный сигнал был подавлен регулировкой фазы.
N14 имеет спиновое число I = 1 и в соответствии с этим обладает квадру-польным моментом. Его низкий магнитный момент (ц = 0,40357 ядерного магнетона) является причиной того, что характеристическая чувствительность для ЯМР-N14 очень низка (1,01-Ю-3 от чувствительности для протонов при постоянной напряженности поля). Малая чувствительность и влияние квадру-польного уширения линий объясняют относительно малый интерес к исследованию ядра N14 методом ядерного резонанса.
Акривос [101] исследовал спектр ЯМР-N14 для безводного жидкого аммиака, представленный на рис. 12.27. Он пользовался flMP-спектрометром широких линий с рабочей частотой 3,076 Мгц. Вместо того чтобы исследовать форму производной, он применил частоту модуляции 392 гц (> | у | Hi) и настройкой фазы опорного сигнала исключил центральный сигнал. На рис. 12.27 показаны две оставшиеся боковые линии в форме сигналов поглощения. Этим способом удалось существенно повысить разрешение, поскольку отсутствовало уширение линий, которое имеет место при исследовании производной из-за конечной величины амплитуды модуляции. В спектре наблюдается квартетное расщепление, вызванное спин-спиновым взаимодействием N — Н (Jn-h = 40+1 гц).
Бейкер [551 повысил соотношение сигнал/шум в спектрах N14, применив метод межъядерного двойного резонанса (INDOR) (см. разд. 12.2.1 и 6.8). Этим методом он получил спектр иона аммония NH4 в кислых растворах NH4NO3 (см. рис. 12.28).
Бальдешвилер и Рэнделл [108] предложили для измерения химических сдвигов N14 остроумный метод с применением двойного резонанса. Этот метод особенно полезен при исследовании молекул, в которых атом азота непосредственно связан с водородом. Он заключается в наблюдении протонного спектра соединения, содержащего азот, при одновременном облучении образца полем с частотой, отвечающей резонансной частоте N14. Облучающее радиочастотное
Jri3_c_Hi= —10-° гЧ и jn15hi = 1 *8° гц [374]. Константа /gi3_s129 = 4°,5 гц для тетра-метилсилана имеет отрицательный знак [375]. Константа JH^199 с13 = 689,0 гц для диметил-
ртути имеет положительный знак [376].—Прим. ред.
344
Глава 12
поле создается генератором с регулируемой частотой. Исчезновение тонкой структуры протонного сигнала группы NH определяет условия подавления спин-спинового взаимодействия (см. рис. 12.30) х). Химические сдвиги N14 отвечают разности частот, которые необходимы для подавления спин-спинового взаимодействия NH в различных соединениях. Довольно короткие времена релаксации N14, а также процессы протонного обмена приводят к уширению
Мгц
4,3350 4.3348
Рис. 12.28. Спектр межъядерного двойного резонанса N14 иона NHJ в подкисленном растворе NH4NO3 (ампула с внутренним диаметром 4 мм) [55].
Образец облучался с резоиаисиой частотой для протонов 60,0099293 Мгц,
линий и ограничивают точность метода. Некоторые типичные химические сдвиги N14, измеренные этим методом, составляют:
NH3 —16,8 м. д.
NHJ 0,0
Пиридин C6H5N —293,3 м. д.
Пиридиниевый ион C6HeN+ —175,6 м. д.
12.3.1. Химические сдвиги N14
Химические сдвиги азота [102—106, 119, 299] наблюдались для многих соединений. Некоторые типичные значения, измеренные относительно резонансного сигнала иона NO~ в водном растворе, приведены в табл. 12.39. Основной причиной больших значений химических сдвигов, йаблюдаемых для азотсодержащих соединений, является, по-видимрму, изменение величины парамагнитного члена в выражении Рэмзи для ядерного экранирования [72]. Изменение ионного характера химических связей атома азота оказывает влияние на орбитальный угловой момент, который определяет величину парамагнитного члена [3] (см. химические сдвиги фтора, разд. 4.6). Если атом имеет замкнутую оболочку с конфигурацией инертного газа, то орбитальный угловой момент равен нулю и парамагнитный член имеет очень малую величину. Это имеет место для атома азота в аммониевом ионе. Увеличение электроотрицательности атома или группы, присоединенной к азоту, приводит к уменьшению ионного характера связи с азотом и возрастанию парамагнитного вклада в экранирование. В результате наблюдается сдвиг сигнала ЯМР в сторону слабого поля. Химические сдвиги N14, приведенные в табл. 12.39, в определенной степени согласуются с представленными выше соображениями. Интересно отметить, что химический сдвиг N14 для ионов NOg и NOg имеет почти такое же значение, как химический сдвиг О17 для этих ионов (см. разд. 12.4.1) * 2).
Шмидт, Браун и Уильямс [105] для измерения химических сдвигов N14 в спектрах ряда азотсодержащих соединений применили в качестве начала
*) Химические сдвиги N14 измеряют также в экспериментах с передачей модуляции иа линии ЯМР-Н1 при облучении образца частотно-модулироваиным полем с несущей частотой, отвечающей ЯМР-Н1 [322].— Прим. ред.
2) Для линейных трехатомных молекул и ионов найдено линейное соответствие между химическим сдвигом N14 и л-зарядом на атоме азота. Плотность л-электроиного заряда вычислена методом молекулярных орбиталей [350].— Прим. ред.
Спектры ЯМР других ядер (кроме водорода и фтора)
345
Таблица 12.39
ХИМИЧЕСКИЕ СДВИГИ N14 [103, 104] а
Формула Химический сдвиг a 6, M. Д. Формула Химический сдвигa d, M. Д.
nhj 602 Пиридин 276
(C3H7)2NH 575 N2 268
(C2h5)3n 575 NO3- 254
n2h4 566 C6H5NO2 6 252
(CH3)4NBr 552 «-c3h7no2 22&
О = C(NH2)2 536 NOT 0
nh2oh-hci 520 NH4OH 626
CH3C = 0 498 NH4 600
1 N2H4-H2O 600
nh2 (NH2)2CO 600'
(SCN)- 406 (SCN)- 405-
CH3CN 385 CN- 366-
CN- 380 HNO3 (100%) 306
CH3CSN 380 NO? 254
C(NO2)4 300 CH3NO2 254
C2(NO2)e 300 NO2 (эталон) &
а Химические сдвиги измерены в миллионных долях (м. д.) относительно иона NO— как внешнего эталона в водном растворе. Все образцы, которые не являются жидкостями, были нсследоваиы в водных растворах.
б Обширные данные по химическим сдвигам №•* для нитросоедииений приведены в работе [298].
отсчета сигнал поглощения иона NH4 в насыщенном водном растворе нитрата аммония. Этот эталонный образец может служить также для калибровки спектров, поскольку его спектр состоит из двух узких линий поглощения ядер N14r разделенных расстоянием 352 м.д. В табл. 12.40 приведены химические сдвиги
Таблица 12.40
ХИМИЧЕСКИЕ СДВИГИ NH И ШИРИНА ЛИНИЙ ЯМР АММИАКА И РОДСТВЕННЫХ СОЕДИНЕНИЙ,. ИЗМЕРЕННЫЕ В ПОЛЕ 10 700 э [105]
Соединение Состояние образца a Ширина линий, мэ 6*, м. д. ® в, м. д. б
NHJ в Н (вода) 35±10 0 601
NH4SCN н (вода) 35±10 —9,3 592
NH4OH (28% NH3) 320±50 + 12 613
NH3 Ж 200 +21 622
NH3 н (этанол) 360±50 + ю 611
Ag(NH3)2NO2 — 730±100 +30 631
Zn(NH3)5Cl — 550±100 +20 621
Ca(NH3)sCl2 — 1500±200 +20 621
N(CH3)4C1 H (вода) 40±5 —20 581
N(CH3)4Br и (вода) 40+5 —20 581
NH(CH3)2 Ж 250±50 + 5,1 606
N(CH3)3 ж 250±50 + 16 617
а и — насыщенный раствор (в скобках указан растворитель); ж — чистая жидкость.
6 Сдвиги в область сильного поля положительны; б* — химический сдвиг относительно NH+; 6 — химический сдвиг относительно NO-.
® NH4NO3, NH«C1, NH4Br,'NH4I, NH4H2PO4, ЫНдС2Н3О2, (NH4)2SO4, (NH4)2S и (NH4)2SO4-ZnSO4 • 6H2O..
346
Глава 12
N14 для исследованных названными авторами соединений наряду с относительной шириной линии, измеренной на половине амплитуды сигнала. Насыщенные растворы следующих аммониевых солей дают сигналы поглощения ядер N14 примерно в одной и той же области поля с разницей +10 мэ: NH4NO3, NH4C1, NH4Br, NH4I, NH4H2PO4i МН4СгНзО2, (NH4)2SO4, (NH4)2S и (NH4)2SO4 -ZnSO4 -4H2O. При разбавлении этих растворов химические сдвиги не изменялись. Сигнал группы NH4 в спектре насыщенного раствора NH4SCN смещен примерно на 100 мэ в более слабое поле по сравнению с сигналами других аммониевых солей. Однако при разбавлении водного раствора NH4SCN эта разница постепенно уменьшается.
Из табл. 12.40 следует, что химические сдвиги для аммиака и подобных соединений лежат в очень узком интервале (около 50 м. д.). Это дает основание полагать, что электронное окружение ядра азота во всех этих соединениях практически одинаково. Небольшие различия в химических сдвигах, которые все же имеют место для аммиака и аналогичных соединений, вероятно, вызваны небольшими изменениями величины парамагнитного вклада в экранирование и находятся в приблизительном соответствии с предполагаемой ионностью связи. Сильное экранирование ядра указывает на высокую ионность связей азота в этих молекулах по сравнению с большинством других азотсодержащих соединений. Хотя химические сдвиги для аммиака и родственных соединений изменяются не очень сильно, ширина линий составляет от 35 до 1500 мэ. Это означает, что градиент электрического поля в месте нахождения ядра азота, величиной которого определяется степень квадрупольного уширения линии, может существенно изменяться, даже если электронное окружение остается практически постоянным. Рассмотренные выше факты согласуются с тем, что атом азота в аммиаке и родственных молекулах, а также в ионе NH4 имеет .хрЗ-гибридизацию и, следовательно, ядро N14 имеет весьма близкое электронное окружение. С другой стороны, градиент электрического поля должен существенно меняться в силу различного геометрического строения названных молекул.
Упомянутые выше авторы наблюдали небольшую разницу в химических сдвигах N14 между сигналами водного раствора аммиака и жидкого аммиака, что дает возможность изучать взаимодействие между аммиаком и водой.
Сигнал ядер N14, наблюдаемый для 28%-ного водного раствора гидроокиси аммония, примерно в 10 раз шире, чем для других аммониевых солей. Его химический сдвиг (+12 м. д. относительно эталонного сигнала NH+) и ширина линии не зависят от концентрации. Это служит доказательством того, что между группой NH4 в NH4OH и в других аммониевых соединениях могут существовать структурные различия. Если прибавлять соль аммония к раствору гидроокиси аммония, то в спектре смеси наблюдается только одна широкая линия поглощения N14 для NH*. Химический сдвиг этого сигнала приближается к значению химического сдвига для добавленной соли аммония, по мере того как содержание ее в системе увеличивается. Если добавлять гидроокись аммония в водный раствор аммониевой соли, то сигнал постепенно уширяется, но не меняет положения. При смешивании N(CH3)4Br и гидроокиси аммония был получен противоположный результат: наблюдались два раздельных сигнала. Эти эксперименты доказывают присутствие и отсутствие химического обмена соответственно (см. гл. 9).
Изучение резонанса N14 привело к выяснению кинетики обмена молекулами NH3 между амминными комплексами никеля и аммиаком в водном растворе или в жидком аммиаке [257].
Шмидт, Браун и Уильямс [106] исследовали резонансные спектры N14 для ряда нитратов, нитритов и нитросоединений. Результаты их измерений приведены в табл. 12.41. Они установили, что резонансные сигналы N14 для нитратной группы в LiNO3, NaNO3, KNO3, AgNO3, Ag(NH3)2NO3, Zn(NO3)2, Pb(NO3)2. Cd(NO3)2, UO2(NO3)2 и A1(NO3)3 имеют химический сдвиг и ширину
Спектры ЯМР других ядер (кроме водорода и фтора)
347
Таблица 12.41
ХИМИЧЕСКИЙ СДВИГ N14 РЯДА НИТРАТОВ, НИТРИТОВ И НИТРОСОЕДИНЕНИЙ [106]
'Соединение Химический сдвиг a d, м. Д. Ширина линии мэ Образец B
NO; г 0 60+10 н (вода)
HNO3 28,3 60±10 70%
Cu(NO3)2-3H2O 81 480±50 5,8 M раствор в воде
Cu(NO3,)2.3H2O 145 600+200 3,5 44 раствор в этаноле
C6H5NO2 21 300+50 Ж
no2c6h4cooh 20 260+80 н (этанол)
<NO2)2C6H3COC1 15 200+50 н (эфир)
NaNO2 —240 1200+200 н (вода)
а Химические сдвиги измерены в миллионных долях (м. д.) относительно иоиа NO- как внешнего эталона.
6 Измерения проведены в поле напряженностью 10 729 э.
в и — насыщенный раствор; ж — чистая жидкость.
г NH4NO3, LINO3, NaNO3, KNO3, AgNO3, Ag(NH3)2NO3, Zn(NO3)2, Pb(NO3)2, Cd(NO3)2, UO2(NO3)2 и A1(NO3)3.
линий те же, что в насыщенном растворе NH4NO3. В противоположность более ранним результатам Холдера и Клейна [104] не было обнаружено зависимости положения сигнала NO7 в этих соединений от концентрации. Для парамагнитной соли Си(МОз)г наблюдалось большое диамагнитное смещение в сторону более сильного поля; величина смещения зависела от концентрации раствора. Сдвиг в том же направлении наблюдался также для Fe(NO3)2. Для солей марганца характерны отрицательные химические сдвиги. Химические сдвиги N44 для различных исследованных нитратов и нитрогрупп, за исключением парамагнитных солей, лежат в очень узком интервале по сравнению со всем интервалом химических сдвигов для ЯМР-N14. Это дает основание предполагать, что структура атома азота в группах NOg и NO~ одинакова. Обе группы имеют плоское строение и гибридизация четырех орбиталей атома азота должна быть примерно одинаковой. Кислород более электроотрицателен, чем азот. Это приводит к уменьшению электронной плотности вокруг ядра азота и вызывает сдвиг резонансного сигнала N14 для этих групп в более слабое поле. Повышенный ковалентный характер связи в молекуле HNO3 увеличивает электронное экранирование атома азота, резонансный сигнал которого смещен в сторону более сильного поля.
Ширина линий резонансных сигналов N14 в нитритах примерно в четыре раза больше, чем в нитратах.
Изучение резонанса N14 азид-иона Ng в растворе азида натрия показало присутствие двух концевых атомов азота и одного центрального атома. Их химические сдвиги N14 равны соответственно +278 и +129 м. д. от сигнала иона NO“ как внешнего эталона [261].
Имеются сообщения о нескольких исследованиях N15 [295—297] х).
12.3.2. Эффекты в спектрах ЯМР, обусловленные квадрупольным моментом ядра азота
Причиной квадрупольного уширения линий является процесс релаксации, вызванный взаимодействием квадрупольного момента ядра с градиентами электрического поля внутри молекулы. Если ядро окружено симметрично
4) См. также [348, 349, 374].'— Прим. ред.
348
Глава 12
Рис. 12.29. Спектры ЯМР-N14 при частоте 3 Мгц 1107].
а — NH|; б — NH3; в — CH3NHj;c— (CH3)3NHt; д - (CH,)3NH+; е -(CH3)4N+; ж — C3H3CH3N(CH3)t. Шка-
рнеположенными заместителями, то градиент электрического поля невелик, и поэтому квад-руиольное уширений линий тоже мало (см. разд. 2.5.2). Огг и Рей [107] показали это при исследовании спектров N14 тетраэдрически симметричного иона NH+ (для подавления гидролитического обмена, которое также приводит к уширению линий, добавлена кислота). В этом спектре вследствие спин-спинового взаимодействия JN-h содержится мультиплет, состоящий из пяти узких компонент (см. рис. 12.29,й) 4). На рис. 12.29 приведены также спектры N14 аммиака и ионов CH3NHJ,. (CH3)2NH4, (CH3)3NH+, (CH3)4N+ и
СвН5СН2М(СН3)з. Сравнивая ширину сигналов поглощения в спектрах различных четвертичных аммониевых катионов, можно-получить некоторые сведения о влиянии замещения на асимметрию электрического поля. Влияние неподеленной пары электронов азота в молекуле NH3 вызывает более высокий градиент поля, чем метильный заместитель, о чем свидетельствует относительная ширина линий на рис. 12.29, б и 12.29, в- Как и ожидалось, спектр тетраэдрически симметричного иона (CH3)4N+ состоит из одной узкой линии (рис. 12.29, е). Узкая линия поглощения наблюдается также для CeH5CH2N(CH3)g , что указывает на близкодействующий характер влияния асимметрии поля на квадрупольную релаксацию (см. рис. 12.29, ж) [107].
Полного устранения квадрупольного уширения можно достичь, заменив атомы N14 на атомы N15, ядра которых не обладают квадр-у-польным моментом ^для N15 I — -у) . Такой эксперимент был осуществлен с метиламмони-евым ионом (CH3N15H+) для доказательства-того, что уширение линий протонного спектра-группы N14 — Н в кислой среде обусловлено квадрупольной релаксацией на ядре N14 [107].
Для подавления влияния квадрупольной релаксации ядер N14, которая вызывает уширение линий в протонных спектрах соёдине-
ла градуирована в миллионных НИЙ содержащих ЭТОМЫ ВОДОрОДЭ И ЭЗОТЭ, долях (м. д.) относительно резонансно- г „
го сигнала N14 аммониевого иона в на- уСПСШНО Применяется МСТОД ДВОЙНОГО реЗО-сыщеином ь°Ди™0Риаяс™°Ре интрата нанса (см. рис. 12.30). Для МОЛекуЛЫ ПИРРОЛа сильное облучение на резонансной частоте N14' снимает уширение сигнала протона группы NH и позволяет наблюдать тонкую-структуру которая возникает вследствие спин-спинового взаимодействия с кольцевыми протонами (см. рис. 8.52) [109]. Этот метод был применен также при анализе протонного спектра формамида [110]. Сигналы протонов формамида
О. /Н(2)
>С — N< (1)HZ ХН(3)
!) Исследованию спин-спинового взаимодействия N14 — Н1 и химического дви-га Н1 дейтерированного аммиака посвящены работы 1368, 378]. . Прим. рео.
Спектры ЯМР других ядер (кроме водорода и фтора)
349
существенно уширены вследствие спин-спинового взаимодействия Н1 — N14 [ 110, 244] и квадрупольных эффектов. Пьетт и сотр. [ 110] устранили это уширение, применив метод двойного резонанса Н1 — {N14}. Протонный спектр, полученный при облучении образца полем с частотой N14, позволил определить две константы спин-спинового взаимодействия между протонами молекулы (Ji 2 = 13 гц и Ji з = 2,1 гц). Константа J2 з (2,4 гц) была измерена в протонном спектре формамида, обогащенного изотопом N15 [245].
Тирс и Бови [111] исследовали протонные спектры ряда N-замещенных амидов. Они обнаружили, что в некоторых случаях узкие сигналы NH наблюдаются даже при отсутствии процессов протонного обмена. Например, N-аце-тил-ОЬ-валин CH3CONHCH — COOH имеет очень узкую резонансную линию
СН(СН3)2
------->-н
Рис. 12.30. Спектр ЯМР-Н1 при частоте 40 Мгц а-протонов чистого пиридина без облучения и с облучением ядер N14 [108].
для NH-группы. В несимметричных молекулах, для которых наблюдается этот эффект, следовало бы ожидать значительного градиента электрического поля в месте расположения ядра азота. Имеются теоретические предпосылки для предположения о том, что столь сильные градиенты поля могут устранить квадрупольное уширение линии [112].
Быстрый протонный обмен, протекающий с участием водорода группы NH, вызывает, как известно, исчезновение тонкой структуры, а также сужение соответствующих линий в спектрах протонного резонанса. Даже малые следы воды (1 часть на 107 частей) приводят к инициированию протонного обмена, достаточного для исчезновения тонкой триплетной структуры в протонном спектре безводного аммиака. Триплетное расщепление, обусловленное спин-спиновым взаимодействием ядер N14 — Н1, наблюдалось в протонных спектрах тщательно высушенного жидкого аммиака [113], растворов алкиламинов в кислотах [114, 115], раствора м-бутиламина в трифторуксусной кислоте [111], а также для неразбавленных жидких формамида, ацетамида и N-метилацет-амида при высоких температурах [115].
12.3.3. Константы дальнего спин-спинового взаимодействия Н1 — N14
Константы дальнего спин-спинового взаимодействия наблюдались в протонных спектрах ряда изонитрилов (см. табл. 12.42) [116]. Это дает основание предполагать, что величина градиента электрического поля вблизи ядра азота в этих соединениях невелика и что время спин-решеточной релаксации ядра N14 должно быть сравнимо с величиной, обратной константе спин-спинового взаимодействия NH [112]. Такое расщепление не наблюдалось в спектрах аминов, амидов, нитроалканов, азотсодержащих гетероциклических соединений, нитрилов, N-окисей и алкилазидов 4 *). Только в изонитрилах симметрия
4) Дальнее спин-спиновое взаимодействие N14 — Н1 наблюдается для солей тетра-
алкиламмония [356—360] и енаммония [361].— Прим. ред.
350
Глава 12
Таблица 12.42
ПРОТОННЫЕ ХИМИЧЕСКИЕ СДВИГИ И КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ H1-N14 НЕКОТОРЫХ ИЗОНИТРИЛОВ [116]
Соединение Т ± 0,02а Мультиплетное ть JNH ± °’2’ гц JHH ± °»2> гц
(CH3)3CNC б 8,56 Триплет 3,5
CH3NC б 7,15 » 2,7
CeH5CH2NC 5,72 » 1,3
(CH3)2CHNC
сн3 8,55 Дублет триплетов 2,6 7,0
сн 6,13 Септет триплетов 1,8 7,0
CH3(CH2)3NC
а-СН2 6,70 Триплет триплетов 2,0 6,7
р-сн2 Показывает расщепление
СвНцЫС» (СН) 6,42 Широкий пик
а При 40 Мгц; Si(CHg)4 — внутренний эталон.
б Растворы в ССЦ; другие соединения исследовались в чистом виде.
в Циклогексилнзонитрнл.
электронного окружения ядра азота такова, что градиент электрического поля имеет достаточно малую величину и не вызывает размывания тонкой структуры Н—N14 из-за квадрупольной релаксации. Значения констант взаимодействия особенно интересны для изопропилизонитрила (СНз)аСНМС. В этом случае спин-спиновое взаимодействие N14 с протоном в р-положении больше, чем с протоном в a-положении. Сходная ситуация наблюдается также для некоторых металлалкилов [90, 117].
12.3.4. Определение строения молекул с помощью ЯМР-N14
Цианамиды и карбодиимиды. Изучение спектра ‘ядерного магнитного резонанса на ядрах N14 для диэтилцианамида [ 118] показало, что эта молекула имеет строение нитрила
С2Н, 2 s\n_c=n, С2Н/
а не изонитрила
Сгн5\
>N — N е= С.
СгЩ/
Спектр N14 состоит из двух линий поглощения, смещенных на +149 и 285 м. д. в сторону сильного поля относительно сигнала NO~ в растворе нитрата аммония, примененного в качестве эталона. На основании величины химического сдвига следует полагать, что сигнал, находящийся в области наиболее сильного поля, принадлежит атому азота аминогруппы [104]. Химический сдвиг сигнала N14 метилизонитрила составляет в выбранной шкале 240 м. д. Это подтверждает точку зрения, что линия в диэтилцианамиде при +149 м. д. не отвечает изонитрильному атому азота.
Спектр ЯМР-N14 для N.N'-дициклогексилкарбодиимида состоит только из одной линии поглощения при + 240 м. д. относительно эталонного сигнала NO^. Это согласуется с симметричной структурой молекулы (СвНн — N = = С = N — СвНи) с двумя эквивалентными атомами азота [118] 4).
*) Химические сдвиги для азота, присоединенного к атому углерода, см. в [351].— Прим. ред.
Спектры ЯМР других ядер (кроме водорода и фтора)
351
12.4. Кислород
О17 (естественное содержание 0,037%) — единственный изотоп кислорода с ненулевым спином = у) . Исследовать магнитный резонанс этого ядра довольно трудно, потому что, во-первых, природное содержание изотопа О17 невелико, во-вторых, это ядро обладает значительным квадрупольным моментом, в-третьих, из-за низкой чувствительности метода ЯМР-О17. Поэтому не удивительно, что имеется сравнительно немного исследований, посвященных ядру О17.
Уивер, Толберт и Лафоре [120] изучили спектры ЯМР-О17 (сигналы дисперсии) ряда кислородсодержащих соединений. Авторы нашли, что время релаксации Т\ для кислорода обычно достаточно мало, что благоприятствует проведению экспериментов в условиях медленного прохождения. В табл. 12.43-
Таблица 12.43
ХИМИЧЕСКИЕ СДВИГИ 017 НЕКОТОРЫХ РАСПРОСТРАНЕННЫХ
СОЕДИНЕНИЙ [120]
Соединение д а, м. д. Соединение в а, м. д.
Тетраметилортосиликат —20 Формамид —320
Октаметилциклотетрасилок- —80 Этилнитрат —340
сан —470
н-Бутилнитрит —170 Нитрат натрия —430
—380 SO2 (жидкость) —540
Перекись водорода (30%) —190 Ацетон —600
Уксусная кислота —220 Нитроэтан —640
Муравьиная кислота —270 Нитрит натрия —690
Формиат натрия —300 Н2О 0
a Относительно воды как внешнего эталона.
приведены химические сдвиги О17 для ряда соединений, измеренные относительно сигнала воды как эталона. В спектре этилнитрата наблюдались два-резонансных сигнала с соотношением интенсивности 1 : 2, что согласуется
\ / /О'
с присутствием в молекуле двух типов кислорода: —С — О — и —
Z ^О'
Следует также заметить, что разность химических сдвигов N14 между ионами NO” и NO” (см. табл. 12.39) практически совпадает с разностью химических сдвигов О17 для этих же соединений, однако объяснения этому наблюдению пока не найдено.
12.4.1. Химические сдвиги О17
Сигнал воды наблюдается при наибольшем для ЯМР-О17 значении напряженности магнитного поля. Это объясняется частично ионным характером связи О — Ни образованием высокосимметричной системы валентных электронов вокруг атома кислорода. Вклад парамагнитной составляющей в экранирование ядра кислорода для такой симметричной системы не может быть-большим, что и приводит к наблюдаемому диамагнитному сдвигу.
В более поздней работе, посвященной исследованию резонанса О17 большого ряда органических соединений, было найдено, что химические сдвиги кислорода для спиртов и простых эфиров близки к химическому сдвигу для воды (см. табл. 12.44). Таким образом, в тех соединениях, где кислород связан)
352
Глава 12
Таблица 12.44
ХИМИЧЕСКИЕ СДВИГИ 017 РЯДА ОРГАНИЧЕСКИХ МОЛЕКУЛ [121]
Соединение ба, м. Д. Примечание
Метанол 6 +40,0
.Этанол 0 98%
Вода 0,0 Эталон
.Эфир 0
Пропионовая кислота —210
Уксусная кислота -220
Муравьиная кислота —270
Пропионовый ангидрид —240 Более сильный сигнал
—420 Менее сильный сигнал
Ацетилацетон —235
Этилацетат —310
Концентрированная азотная кис- —460 Очень интенсивный сигнал
лота
Ацетон —600
Метилэтил кетон —600
Ацетоуксусный эфир —620 Широкий сигнал
Малоновый эфир —625
а Относительно воды как внешнего эталона.
6 Сигнал 017 метанола смещается на 3,7 м. д. в слабое поле прн разбавлении в ССЦ [271].
-с углеродом или водородом простой связью, его экранирование является, по-видимому, диамагнитным, причем величина константы экранирования кислорода не зависит от того, связан он с водородом или углеродом 1121].
Фиггис и сотр. [122] измерили химические сдвиги О17 нескольких неорганических соединений (см. табл. 12.45 и 12.46). Они обнаружили, что химиче-
Таблица 12.45
ХИМИЧЕСКИЕ СДВИГИ 017 АТОМОВ КИСЛОРОДА, СВЯЗАННЫХ С ПЕРЕХОДНЫМИ МЕТАЛЛАМИ [122]
Соединение 6 a, м. д. Наименьшая энергия электронного перехода, сл<_1
Na3VO4 —571 ±4 36 900
Na2CrO4 —835+5 26 800
К2МоО4 —540+2 44 000
Na2WO4 —420+2 50 300
NaMnO4 —1219+8 18 300
NaTcO4 —749+7 34 000
NaReO4 —569+4 43 500
RuO4 —1119+10 26 400
OsO4 —796+3 33 500
МагСггО? —1125±6 22 700
СгОгС!^ —1460+8 18 000
а Отиоснтельио воды как внутреннего эталона.
Спектры ЯМР других ядер (кроме водорода и фтора)
353
Таблица 12.46
ХИМИЧЕСКИЕ СДВИГИ 017 ДЛЯ АТОМОВ КИСЛОРОДА, СВЯЗАННЫХ С НЕПЕРЕХОДНЫМИ ЭЛЕМЕНТАМИ
Соединение Состав раствора б а, м. д.
КгСО3 7 M раствор в 0,1 н. КОН —192±4
Н2О2 30%-ный водный раствор —187+5
Р0С1з Чистая жидкость —216±2
Na2SeO4 3 М раствор в 0,1 н. NaOH —204+12
S03 «Стабилизированная» жидкость —188±1
S02 Чистая жидкость —513+5
SOC12 То же —292+2
SO2C12 » » —304+3
NaBrO3 2,4 М раствор в 0,1 н. NaOH —297+5
NaC103 То же —287+3
NaC104 » » —288±5
NH4NO3 9 М раствор в 0,1 н. NaOH —420+6
HNO3 100% —414+3
NaNO2 6 М раствор в 0,1 н. NaOH —670+6
а Относительно воды как внутреннего эталона-
ские сдвиги О17 оксианионов переходных металлов с замкнутой электронной оболочкой, а также сдвиги карбонильных атомов кислорода органических молекул находятся в линейном соответствии с наименьшей энергией электронного перехода в ультрафиолетовых и видимых спектрах (см. рис. 12.31) х). Существование подобной зависимости вполне согласуется с предположением, что основной вклад в экранирование ядра О17 в подобных соединениях вносит парамагнитная составляющая в уравнении Рэмзи (см. разд. 3.5).
Ядра атомов кислорода в кетонах (ацетоне или метилэтилкетоне) экранированы значительно слабее, чем в спиртах, вследствие возрастания величины парамагнитной составляющей, обусловленной участием р-орбитали кислорода в образовании двойной карбонильной связи [121].
В спектре ЯМР-О17 карбоновых кислот (муравьиной, уксусной и пропионовой) был обнаружен лишь один сигнал, что свидетельствует о структурной эквивалентности обоих кислородных атомов и согласуется с возможностью резонанса канонических структур I и II.
I II
Характерно, что величина химического сдвига для карбоновых кислот занимает промежуточное положение между сдвигами соединений со связью С — О и сдвигами соединений с кетонной связью С = О. Спектры О17 сложных эфиров имеют сходные параметры со спектрами карбоновых кислот [121].
На основании химических сдвигов О17 можно судить о степени двоесвязно-сти связи С — О. Например, в спектре О17 пропионового ангидрида наблюдались два резонансных сигнала с отношением интенсивностей 2 : 1 [121], что согласуется с наличием в молекуле двух типов кислородных атомов. Вместе с тем значения химических сдвигов этих сигналов свидетельствуют о том,
') О спектрах ЯМР-О17 оксианионов см. также в [352].— Прим. ред.
23—489
354
Глава 12
что связь С — О носит частично двойной характер. Другими словами, электронное строение рассматриваемой молекулы можно представить как резонанс следующих структур:
У0 сн3—сн2—с<
\о сн3-сн2-с<
СН3 — сн2 — с/ сн3—сн2—с/
0+
СН3-СН2 —с<
>0+
сн3—СН2—cZ
о
Известны успешные попытки применения ЯМР-О17 при рассмотрении медленно устанавливающихся кето-енольных равновесий [121]. Например, величина химического сдвига О17 для ацетил ацетона, практически не отличающаяся от химического сдвига карбоновых кислот, приводит к выводу
Рис. 12.31. Зависимость между химическими сдвигами ЯМР-О17 и наименьшей энергией электронного перехода для соединений, в которых кислород связан с переходным металлом [122].
ф ионы с тетраэдрической симметрией; А частицы с другой симметрией.
о том, что эта молекула существует преимущественно в виде енола. Напротив, химические сдвиги О17 ацетоуксусного и малонового эфиров имеют значения, близкие к химическим сдвигам кетонов, что свидетельствует о преобладании в равновесии кето-формы. Однако ЯМР-О17 не позволяет обнаруживать малые количества молекул в равновесной смеси таутомерных форм.
В работе Криста [124] изучены спектры ЯМР-О17 большого числа соединений (табл. 12.47 и 12.48). Составленная на основе этих данных сводная табл. 12.49 характеризует диапазон изменений химического сдвига О17 для различных химических групп.
Фиггис и сотр. [125] измерили химические сдвиги О17 и С13 карбонильных соединений некоторых металлов (табл. 12.50). Следует заметить, что полученные названными авторами результаты не позволяют с абсолютной уверенностью решить, существует ли в этом классе соединений прямая связь кислород — металл.
Таблица 12.47
ХИМИЧЕСКИЕ СДВИГИ 017 НЕКОТОРЫХ ОРГАНИЧЕСКИХ МОЛЕКУЛ [124]
Соединение 6, м. д. а Соединение 6, м. д. а
-0- 0=
Метанол +37 Метилформиат —142 —358
Этанол —5 Метил ацетат — 137 —355
w-Пропанол —1 Метилпропионат — 133 —350
изо-Пропанол —37 Диметиловый эфир малоновой — 143 —363
Этиленгликоль + 6 кислоты
Диэтиловый эфир —15 Диметиловый эфир янтарной — 151 —368
Диметиловый эфир этилен- + 25 кислоты
гликоля Этилформиат —169 —359
Диоксан + 3 Этилацетат —166 —256
Тетрагидрофуран —19 Этилпропионат —164 —350
Муравьиная кислота —254 Диэтиловый эфир щавелевой —162 —355
Уксусная кислота —254 КИСЛОТЫ
Пропионовая кислота —250 Диэтиловый эфир малоновой —166 —356
Ацетон —568 кислоты
Метилэтилкетон —563 Диэтиловый эфир янтарной — 178 —363
Циклопентанон —548 кислоты
Циклогексанон —559 «-Пропилформиат — 163 —354
Окись мезитила —555 Диэтилкарбонат — 120 —240
Диацетил —571 Диацетат этиленгликоля — 148 —351
Ацетальдегид —595 Этилхлорформиат — 170 —348
Пропионовый альдегид —581 Уксусный ангидрид —259 —393
Масляный альдегид —589 Пропионовый ангидрид —248 —390
Акролеин —583 1
а Относительно воды как внешнего эталона.
ХИМИЧЕСКИЕ СДВИГИ 017 [124]
Таблица 12.48
Соединение 6а, М. д. Соединение а, м. д.
Метилформиат —28 Этилен хлор гидрин + 4
Этилформиат —45 Эпихлоргидрин + 12
Диметоксиметан —8 Диметилсульфоксид — 13
Диэтоксиметан —43 Нитробензол —561
Ацеталь —52 Тетраметоксиснлан -30
Паральдегид — 102 Формамид —306
Ацетоуксусный эфир — 164 Мочевина (водный раствор) —205
—353 Серная кислота (98%) — 138
—565 Азотная кислота (69%) —409
Ацетилацетон —269 Хлорная кислота (60%) —288
Фуран —243
Фурфурол —237 D2O (60%) + 3
—530
а Относительно воды как внешнего эталона.
23*
356
Глава 12
Таблица 12.49
НЕКОТОРЫЕ ХАРАКТЕРИСТИЧЕСКИЕ ХИМИЧЕСКИЕ СДВИГИ 017 [124]
Диапазон химических Тип кисло-
я Соединение родного
м. д. атома
От +40 до —40 Спирты и простые эфиры — О —
От —130 до —150 Сложные метиловые эфиры — О —
От —160 до —180 Сложные этиловые и пропиловые эфиры — О —
От —240 до —260 Кислоты и ангидриды кислот — О —
От —350 до —370 Сложные эфиры О =
От —390 до —393 Ангидриды кислот О =
От —550 до —570 Кетоны О =
От —580 до —600 Альдегиды О =
а Относительно воды
как внешнего эталона.
Таблица 12.50
ХИМИЧЕСКИЕ СДВИГИ 0*7 НЕКОТОРЫХ КАРБОНИЛОВ МЕТАЛЛОВ [125]
Соединение Состав раствора 6 а, м. д.
Fe(CO)5 Чистая жидкость —388
Ni(CO)4 То же —362
Fe(CO)2(NO)2 » » —418
Мп2(СО)ю 50 об. % В CHClg —355
Co(CO)3(NO) Чистая жидкость —377
а Относительно воды как внешнего эталона.
12.4.2. Влияние парамагнитных ионов на химические сдвиги О17
Растворение парамагнитного иона Gd3+ в образце Н2О17 вызвало увеличение диамагнитного экранирования ядра О17. Подобное явление, согласно предположению Шульмана и Вилуда [126], может быть вызвано переносом электрона от лиганда Н2О17 к иону металла с образованием ионов (Н2О17)+ и Gd2+. Оргел [ 127, 128] отметил, что в результате переноса электрона от лиганда на орбиталь переходного металла, занятую одним электроном, должен наблюдаться большой парамагнитный сдвиг сигнала О17 лиганда, а диамагнитный сдвиг происходит при переносе электрона лиганда на незанятую орбиталь. Если это явление действительно существует, то в первую очередь оно должно наблюдаться для ионов редкоземельных элементов, например для иона гадолиния.
В спектрах водных растворов перхлоратов редкоземельных элементов наблюдались парамагнитные и диамагнитные химические сдвиги О17 [129]. Эти сдвиги вызваны, по-видимому, изменением изотропной части непрямого контактного сверхтонкого взаимодействия неспаренных электронов атома редкоземельного элемента с ядрами кислорода. Это взаимодействие передается через электроны связи (так называемое конфигурационное взаимодействие) и приводит к появлению неспаренной спиновой плотности на атоме кислорода (см. разд. 3.16).
Спектры ЯМР других ядер (кроме водорода и фтора) 357
Метод ЯМР-О17 был применен при изучении гидратации некоторых катионов (см. разд. 9.3.2) в системах, в которых обмен молекулами воды между гидратными оболочками и водой-растворителем протекает достаточно медленно ИЗО, 311]. В таких системах при добавлении парамагнитных ионов сигнал О17 воды-растворителя имеет химический сдвиг относительно сигнала воды гидратных оболочек.
12.4.3. Спин-спиновое взаимодействие Н1 — О17 в спектре воды
Быстрый межмолекулярный обмен протонами обычно не позволяет наблюдать спин-спиновое взаимодействие Н1 — О17 в спектрах Н1 и О17 воды. Однако если обмен замедлить разведением воды в большом избытке тщательно очищенного ацетона, то такое взаимодействие удается наблюдать. Ройбен с сотр. [131] при исследовании спектра ЯМР-О17 для 3%-ного (по объему) раствора воды
Рис. 12.32. Спектр ЯМР-О17 (сигнал дисперсии) воды Н2О17 в ацетоне при 8,13 Мгц [131].
(обогащенной О17) в ацетоне обнаружил триплетное расщепление сигнала О17 с константой Jhi-oi’ = 73,5 ± 2,1 гц (рис. 12.32). Межмолекулярные эффекты, связанные с разведением воды в ацетоне, привели к смещению сигнала О17 в область более сильного поля примерно на 11 м. д. по сравнению с чистой водой.
12.5. Кремний
Si29 — единственный естественный изотоп кремния, обладающий магнитным моментом = -у) . Вследствие малого содержания этого изотопа (4,70%) и низкой чувствительности метода ЯМР-Si20 (7,85% чувствительности ЯМР-Н1 при постоянном магнитном поле) измерение химических сдвигов Si20 для соединений, приведенных в табл. 12.51, оказалось возможным лишь при условии быстрого прохождения и регистрации сигнала дисперсии [50, 56]. Поскольку ядро Si20 имеет спин / = у , то квадрупольный момент отсутствует. Отыскание резонансного значения напряженности магнитного поля при наблюдении ЯМР-Si20 наталкивается на значительные трудности, которые преодолевают одним из следующих способов [56]:
1. Сигнал для образцов с коротким временем Л у ядер Si20 находят по производной линии поглощения при медленном прохождении и низкой амплитуде высокочастотного поля.
2. Для образцов с большим временем Л сигнал представляют в виде производной линии дисперсии при медленном прохождении и малой амплитуде высокочастотного поля. Иногда применяют также быстрое прохождение при большой амплитуде высокочастотного поля.
3. При очень большом времени Т\ наблюдают сигнал дисперсии при быстром прохождении и большой амплитуде высокочастотного поля. При этом
Таблица 12.51
ХИМИЧЕСКИЕ СДВИГИ S129 И КОНСТАНТЫ JSi_н РЯДА СОЕДИНЕНИЙ КРЕМНИЯ [50, 56]
Соединение а 6, M. Д. JSi-H> гц
[(CH3)3SiO]4Si* 81,5
SiBr4 68+5
(C2H5O)4Si 59,5
[(CH3)3SiO]3Si*CH3 42,5
(C2H5O)3SiCH3 21,5
C6H5(CH3)SiH2 14,8 200
(CH3SiHO)5 13,1 248
(CeH5)2SiH2 11,8 200
[(CH3)2SiO]x 6 0
[(CH3)2SiO]4 —2,2
[C2H5(CH3)SiO]4 —2,3
CeH5(CH3)2SiH —4,5 187
SiCl4 —6+5
[(CH3)2SiO]3 (в растворе C6H6) — 12,1
(CH3)3SiCH = CH2 —15,1
[(CH3)2SiH]2O — 17,2 210
(CH3)3SiCeH5 — 17,8
(C6H5)2CH3SiOC2H5 — 18,2
[(CH3)2SiNH]3 — 18,7
[(CH3)3Si]2NH — 19,8
(CH3)3SiCH2OH —21,8
(CH3)4Si —22,0
[(CH3)3Si]2CH2 —22,5
(CH3)3SiCH2NH2 —23,2
(CH3)3SiCH2CeH5 —23,4
(CH3)3Si(CH2)3CH3 —23,8
(CH3)3SiCH2Cl —25,5
(CH3)3SiCH2NCS —28,2
[(CH3)3Si]2O —28,7
(CH3)3SiOC2H5 —29,4
[(CH3)3Si*O]3SiCH3 —29,4
CH3SiCl3 —30+5
[(CH3)3Si*O]4Si —30,4
(CH3)3SiI —30,6
(CH3)2Si(CH2Cl)CHCl2 —31,2
(CH3)3SiBr —48,4
(CH3)3SiCl —51±5
(CH3)3SiF в —53,1
(CH3)2SiCl2 —54+5
(CH3)3SiOSO3H г —57,6
(SiH3)3N 212 u
(SiH3)2NCH3 212 Д
SiH3N(CH3)2 208 Д
а Звездочкой обозначен атом кремния, для которого измерен химиче-ский сдвиг.
б Полидиметилсилоксан (вязкость 10 сстокс) — внешний эталон.
в */si—F” 275
г Раствор [(CH3)3Si]2O в избытке H2SO4.
Д Данные получены из спектра ЯМР-Н1 [134].
Спектры ЯМР других ядер (кроме водорода и фтора)
359
Таблица 12.52 КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ НЕКОТОРЫХ СИЛАНОВ (В ГЕРЦАХ) [133]
Соединение 'НН JSi29H JHF19 ^Si29F19
SiH4 2,75+0,15 202,5+0,2
SiH3F 15,6+0,2 229,0+0,6 45,8+0,1 281,0+3,0
SiH3Cl 12,7+0,2 238,1+0,2
SiH3Br 12,7+0,5 240,5+0,3
SiH3I 11,7+0,3 240,1+0,2
SiH2F2 36,2+0,7 282+3 60,48+0,07 297,8+2,5
SiH2Cl2 27,8+0,4 288,0+0,4
SiH2Br2 26,9+0,7 289,0+0,6
SiH2I2 22,7+0,3 280,5+0,2
SiHF3 381,7+1,5 96,28+0,03 274,8+0,3
SiHCl3 362,9+0,2
(SiH3)2O 13,5+0,2 221,5+0,2
(SiH3)2S 9,43+0,13 224,0+0,3
(SiH3)3N 10,4+0,2 212+2
SiH3N(CH3)2 [134] 208
(SiH3)2NCH3 [134] 212
важно, чтобы условия были подобраны исходя из применимости к исследуемой системе уравнений Блоха для быстрого прохождения (см. разд. 2.9).
Молекулы, в которых кремний непосредственно связан с атомами азота, хлора, брома или иода, дают широкие и слабые сигналы ЯМР-Si29 вследствие эффекта квадрупольного уширения, вызванного этими ядрами (особенно ядром хлора). Для некоторых соединений (табл. 12.52) удалось наблюдать мультиплетное расщепление сигнала Si29, вызванное спин-спиновым взаимодействием с ядрами Н1 и F19. Однако точное измерение величины расщепления возможно лишь при исследовании спектров Н1 и F19 этих молекул (см. разд. 10.33). Значения констант взаимодействия JSi_P и Jsi-н для ядер непосредственно связанных атомов имеют примерно одинаковый порядок величины.
Бейкер [55] при исследовании спектра Si29 тетраметилсилана добился улучшения отношения сигнал/шум с помощью техники гетероядерного (INDOR) резонанса (см. разд. 12.2.1 и 6.8).
12 .5.1. Химические сдвиги Si29
Как следует из табл. 12.51, диапазон изменения химических сдвигов для ядер Si29 значительно меньше, чем для ядер С13. По-видимому, причиной является отсутствие кратных связей в соединениях кремния, так как для соединений углерода именно с такими связями наблюдаются наиболее значительные химические сдвиги С13 в сторону слабого поля [50]. Кроме того, еще не измерены химические сдвиги Si29 для полииодсиланов — аналогов поли-иодметанов. Именно для последних наблюдались наиболее высокие константы экранирования С13. В ряде случаев заместители в сходных по строению молекулах оказывают противоположное действие на экранирование ядер С13 и Si29. Рис. 12.33 иллюстрирует это явление на примере соединений с общей формулой (CH3)XM(OR)4 _х. Подобное поведение характерно для систем, где возможно электронодонорное взаимодействие и частичная двоесвязность с участием d-орбиталей.
360
Глава 12
Рис. 12.33. Сравнение химических сдвигов С13 и Si29 для соединений типа (CH3)XM(OR)4_X [50].
12 .5.2. Спин-спиновое взаимодействие ядра Si29
Немногочисленные данные о взаимодействии непосредственно связанных ядер Si — Н показывают, что значения констант этого взаимодействия находятся в пределах 187—248 гц. Взаимодействие ядер Si — F измерено для очень небольшого числа соединений (константа JS1_P составляет 274,8—301,8 гц) [133, 294]. В протонных спектрах алкилкремниевых соединений удается наблюдать дальнее взаимодействие Si29 — С — Н1 (Jsi-h ~ 6 гц) [50, 264] х).
Проведены исследования [133, 267] взаимодействия Si29 — Н1 в ряду замещенных силанов (табл. 12.52). При рассмотрении ряда SiH4, SiH3X и SiH2X2 (где X = Cl, Вг или I) обнаружена приблизительно линейная зависимость между константами JSi_H и Jh-h- Значения последних измерены по спектрам частично дейтерированных молекул (см. разд. 8.19.2). Полученные данные свидетельствуют о том, что оба эти взаимодействия зависят от s-характера гибридной орбитали кремния. В случае других силанов линейная зависимость между этими двумя константами несколько отличается от рассмотренной.
Константы Jsih не подчиняются аддитивному правилу, сходному с найденным для Jc-h 182] 2). Константы взаимодействия Jsih и Jsif в случае ядер непосредственно связанных атомов имеют противоположные знаки [294] 3).
х) Для ряда хлорметилкремнийорганических эфиров измерена константа взаимодействия Si29 — О — С — Н1 [380].— Прим. ред.
2) См. также [381].— Прим. ред.
3) Константа 7c13-Si2s для тетраметилсилана имеет отрицательный знак [375].— Прим. ред.
Спектры ЯМР других ядер (кроме водорода и фтора)
361
12.6. Фосфор
Открытие большой роли фосфатов в жизненно важных биохимических процессах, а также высокий интерес к неорганическим полимерам способствовали возрождению химии фосфора. Достигнутые за последние 20 лет успехи в этой отрасли химии столь велики, что химию фосфора по широте и глубине имеющихся сведений можно сравнивать только с химией углерода. Большую роль в этом сыграло изучение колебательных спектров [136] и спектров комбинационного рассеяния [137]. Применение спектроскопии ЯМР также принесло значительную пользу при исследовании соединений фосфора.
Единственный в природе изотоп фосфора Р31 имеет ненулевой спин и магнитный момент р = 1,1305 ядерного магнетона. Во внешнем магнитном поле с напряженностью 10 000 э резонансная частота ядер Рз1 равна 17,235 Мгц. Широкий диапазон изменения химических сдвигов Р31 (~500 м. д.) позволяет с успехом применять их для определения молекулярной структуры соединений фосфора. В табл. 12.53—12.59 и приложении V представлены многие из известных в настоящее время химических сдвигов Р31. Для сигналов ЯМР-Р31 состояние насыщения достигается легче, чем в случае ядер Н1 и F19, но тем не менее время релаксации фосфора обычно достаточно мало, чтобы можно было пользоваться довольно большой величиной высокочастотного поля Hi в совокупности с быстрой скоростью прохождения.
Низкую чувствительность метода ЯМР-Р31 (6,64% чувствительности метода ЯМР-Н1 при том же магнитном поле) до некоторой степени компенсируется тем преимуществом этого метода, что область изменения химических сдвигов Р31 достаточно велика. Последнее обстоятельство позволяет работать при условиях, которые дают выигрыш чувствительности ценой разрешающей способности. Например, при использовании больших размеров образца и приемной катушки можно получать спектр по обычной методике (запись линий поглощения), особенно подходящей для измерения интенсивности. Тонкую структуру спектров ЯМР-Р31, возникающую в результате спин-спинового взаимодействия Р31 с другими ядрами (например, Н1 и F19), удобнее изучать по спектрам Н1 и F19. Однако при использовании ампул из тонкостенного стекла с наружным диаметром 5 мм можно легко измерить в спектре ЯМР-Р31 константу взаимодействия величиной 5 гц. При исследовании соединений фосфора важно, чтобы концентрация ядер Р31 была не ниже 0,05 М (при напряженности поля 14 000 э), однако для уверенного измерения интенсивностей сигналов необходимы концентрации в двадцать раз более высокие [138].
На рис. 12.34 приведен пример высококачественного спектра ЯМР-Р31, полученного на спектрометре с напряженностью магнитного поля 23 500 э (40,5 Мгц). В спектре отчетливо разрешены все 10 линий мультиплета триме-тилфосфита [277], обусловленные спин-сциновым взаимодействием Р31 — Н1; помимо этого наблюдаются менее интенсивные сигналы, отвечающие взаимодействию ядер Р31 — С13. При более низких значениях напряженности поля для достижения оптимальной чувствительности (при отсутствии вращения образца) используются ампулы с наружным диаметром 15 мм.
Предложена конструкция невращающейся ячейки, которая состоит из приемной катушки диаметром 6 мм, помещенной внутри ампулы с образцом [235]; при этом раствор полностью заполняет объем катушки. Достоинство ячейки такой конструкции состоит в том, что она существенно превосходит по разрешающей способности обычные ячейки с ампулами диаметром 15 мм, не уступая им по величине отношения сигнал/шум.
Опорной линией в спектрах Р31 обычно служит узкий одиночный сигнал от 85%-ного водного раствора ортофосфорной кислоты. Этот раствор помещают в тонкий запаянный капилляр и используют в качестве внешнего эталона.
362
Глава 12
Поправка на объемную диамагнитную восприимчивость составляет менее 0,5 м. д., и ее обычно не учитывают [4].
Рис. 12.34. Спектр ЯМР-Р31 триметилфосфита при 40,5 Мгц (23 500 э) [277]. а — высокое усиление; б — усиление понижено.
В качестве руководства при рассмотрении проблем, связанных с исследованием соединений фосфора методом ЯМР, можно рекомендовать блестящие обзоры Файнгольда [146], а также Катрицкого и Джонса [149] 1).
12.6.1. Химические сдвиги Р31
Впервые химические сдвиги ядер Р31 наблюдал Найт в 1949 г. [139]. Уже первоначальные измерения показали, что величина константы экранирования для атомов фосфора, находящихся в различном окружении, не имеет простой связи с индуктивным эффектом соседних групп, поскольку не было обнаружено явного соответствия между химическими сдвигами Р31 с электроотрицательностью присоединенных атомов или групп [12, 140, 141]. В табл. 12.53 и табл. 12.54 приведены химические сдвиги фосфора для ряда фосфорорганических соединений, измеренные относительно сигнала 85%-ной ортофосфорной кислоты как внешнего эталона. Хотя химические сдвиги, отвечающие атомам фосфора в двух валентных состояниях, находятся в перекрывающихся областях спектра, ядра трехвалентного фосфора обычно менее экранированы, чем пятивалентного, и это, по мнению Гутовского и Мак-Колла [141], объясняется тем, что в экранировании ядер пятивалентного фосфора участвует большое число валентных электронов. Следует отметить, что молекулы, в которых фосфор трехвалентен, имеют более широкий диапазон химических сдвигов (~500 м. д.), чем соединения пятивалентного фосфора (~ 100 м. д.). Причиной этого, возможно, являются более значительные изменения валентных углов в соединениях трехвалентного фосфора [50].
Сравнение химических сдвигов ЯМР-Р31 для соединений PF3, РС13, РВг3 и Р13 показывает, что экранирование фосфора уменьшается в ряду F > I > > С1 > Вг. Для объяснения этой аномалии Гутовский и Мак-Колл [141] постулировали, что экранирование фосфора определяется двумя эффектами — ионным характером и степенью двоесвязности Р — X. По мере роста электроотрицательности галогенного заместителя ионный характер связи Р — F увеличивается, тогда как степень двоесвязности предполагается наиболее высокой для связи Р — F и значительно меньшей для других. Если предположить, что экранирование ядер фосфора увеличивается не только с умень-
г) См. также обзорные работы [330, 331].— Прим. ред.
Спектры ЯМР других ядер (кроме водорода и фтора)
363
Таблица 12.53
ХИМИЧЕСКИЕ СДВИГИ Р31 НЕКОТОРЫХ СОЕДИНЕНИЙ
Соединение б, а м. д. Соединение 6, a M. Д.
(СН3О)2Р(О)Н —11,3 PF3 —97,0 [141]
(С2Н5О)2Р(О)Н —7,5 РС13 —219,4
(изо-С3Н7О)2Р(О)Н —4,2 РВг3 —227,4
(C2H5O)2P(S)H —69,0 Р13 — 178,0 [141]
СН3(С2Н5О)Р(О)Н —32,6 (СН3О)РС12 — 181,0
СН3(«-С3Н7О)Р(О)Н —31,5 (СН3)(С2Н5)Р(О)С1 —72,0
CH3(uso-C3H70)P(S)H —61,3 (СН3)(С13С)Р(О)(ОС2Н5) —40,7
Н2(ОН)Р(О) —12,0 СН3(«-С3Н7О)Р(О)С1 —40,1
(НО)3Р(О) 0,00 СН3(С2Н5О)Р(О)С1 —39,5
СН3(С2Н6О)Р(О)ОН —28,5 С2Н5(С2Н6О)Р(О)С1 —45,0
С2Н5(С2Н6О)Р(О)ОН —32,5 CH3(uso-C3H70)P(0)F —16,1
CH3(C2H5O)P(S)OH —88,8 С2Н6Р(О)С12 —53,0
C2H5(C2H6O)P(S)OH —94,2 СН3Р(О)С12 —44,5
(C2H5O)2P(O)SH —24,0 С6Н5Р(О)С12 —34,5
(C2H5O)2P(S)SH —85,7 CH3P(O)F2 —27,4
CH3P(O)(OH)2 (раствор в —31,1 (С2Н5О)2Р(О)С1 —2,8
ацетоне)
«-С4Н9Р(О)(ОН)2 (раствор —32,0 РОВгз + 102,9
в ацетоне)
[(НО)2Р(О)]2О + Ю,6 РОС13 — 1,9
[(С2Н5О)2Р(О)]2О + 12,5 CH3(C2H5O)P(S)C1 —94,2
[СН3(С2Н5О)Р(О)]2О —22,4 C2H5P(S)C12 —94,3
[СН3(н-С3Н7О)Р(О)]2О —22,2 CH3P(S)C12 —79,8
[С2Н5(С2Н6О)Р(О)]2О —25,0 C6H5P(S)C12 —74,8
[CH3(C2H5O)P(S)]2O —85,4 СН3(изо-С3Н7О)Р(8)С1 —58,6
(СН3)3Р + 61,0 (C2H5O)2P(S)C1 —68,1
(С2Н5)3Р +20,4 (h-C3H7O)P(S)C12 —56,3
(н-С4Н9)3Р +32,3 (h-C4H9O)P(S)C12 —56,4
(С6н5)3р +5,88 PSC13 —28,8
СН3РС12 —191,2 [(CH3)2N]2P(O)C1 —30,3
СвН5РС12 —162,0 [(CH3)2N)P(O)(CN)(OC2H6) + 10,7
а Относительно 85%-ной Н3РО4 как внешнего
эталона.
шением ионности связи, но и по мере роста степени двоесвязности, тогда становится понятным наблюдаемый порядок изменения химических сдвигов для тригалогенидов фосфора. Сходным образом можно рассматривать спектры некоторых соединений пятивалентного фосфора, например оксигалогенидов: РОС13, POC12F, POCIF2 и POF3. В этом ряду экранирование ядер фосфора и фтора увеличивается с возрастанием числа атомов фтора в молекуле. Наблюдаемую последовательность химических сдвигов можно объяснить, если предположить, что в электронную структуру молекулы значительный вклад вносят резонансные формы I и II.
о-
р
C1Q^F+
I
О
II
+-F+
Cl
II
364
Глава 12
Таблица 12.54
ХИМИЧЕСКИЕ СДВИГИ Р31 НЕКОТОРЫХ ПРОСТЫХ
МОЛЕКУЛ [141]
Соединение й, а м. д. j Соединение 6, а м. д.
р4 (в CS2) 488 HPF6 118
Р4 450 F2PO(OH) 20,1
РН3 241 pocif2 14,8
P4S3 114 poci2f 0,0
PF3 —97 H3PO4 0,0
CH3OPF2 —111 HPO(OH)2 —4,5
P2I4 (В CS2) —170 РОС13 —5,4
Pis — 178 С2Н5ОРОС12 —6,4
С1(СН2)3РС12 —182 Н2РО(ОН) —13,8
РС13 —215 PSCls —30,8
РВг3 —222
а Относительно 85%-ной Н3РО4 как внешнего эталона.
Полагают [142], что возрастание степени двоесвязности вызывает уменьшение экранирования ядер F10. Поскольку с ростом числа атомов фтора, связанных с атомом фосфора, происходит уменьшение степени двоесвязности каждой связи Р — F, то наблюдаемый сдвиг резонанса F19 в сильное поле по мере замещения атомов хлора на фтор показывает, что этот эффект является преобладающим. Экранирование ядер фосфора, напротив, увеличивается при возрастании степени двоесвязности, что согласуется с характером электронного распределения, ожидаемого при образовании частично двойной связи ~Р = F+. Индуктивный эффект заместителя должен был бы привести к противоположному распределению заряда +Р — F-. Ван Везер [143] отметил, что в ряду заместителей с электроотрицательностью более низкой, чем у брома, наблюдается возрастание экранирования фосфора при уменьшении электроотрицательности заместителя (в согласии с ожидаемым влиянием индуктивного эффекта). Для заместителей с более высокой, чем у брома, электроотрицательностью характерно обратное влияние на экранирование Р31, что, по-видимому, обусловлено возрастанием у этих заместителей тенденции к образованию связей с частично двойным характером.
Гутовский и сотр. [12] обнаружили, что зависимость между химическими сдвигами фосфора и константами взаимодействия JP_P для тех же молекул носит линейный характер. Это означает, что оба параметра определяются электронным распределением, однако связаны с ним по-разному.
Маллер, Лаутербур и Голденсон [4] исследовали ряд фосфорсодержащих соединений и попытались провести полуэмпирическое сопоставление химических сдвигов фосфора с некоторыми свойствами связей, такими, как гибридизация, ионность, степень двоесвязности. Они не рассматривали электронную плотность на ядрах фосфора, как это делали Гутовский и Мак-Колл [141], а взяли за основу метод Сайка и Слихтера [3], примененный при рассмотрении химических сдвигов фтора. По мнению этих последних авторов, изменение экранирования ядер фтора обусловлено главным образом вкладом парамагнитного члена, и поэтому изменением диамагнитного вклада, а также влиянием других атомов можно пренебречь. В таком случае с возрастанием парамагнитного вклада происходит дезэкранирование исследуемого ядра и, таким образом, его резонансный сигнал наблюдается при более слабом поле. Если этот механизм экранирования распространить на трехвалентный фосфор
Спектры ЯМР других ядер (кроме водорода и фтора)
365
и предположить, что величина парамагнитного вклада в экранирование зависит лишь от валентных электронов, а все остальные слагаемые в уравнении Сайка и Слихтера (см. разд. 4.6)
считать постоянными, то следует ожидать, что химические сдвиги фосфора можно представить полуэмлирическим уравнением
& = а— bD, (12.9)
где D — число несбалансированных р-электронов; а и b — эмпирические константы. Значение D можно найти путем рассмотрения гибридизации и степени ионности связывающих атомных орбиталей [4].
Если для молекул типа РХ3 число электронов, отдаваемых атомом фосфора на образование каждой связи Р — X, обозначить через (1 — е), то величину е можно выразить с помощью соотношения [144]
| е | =0,16 | ХА — ХВ |+ 0,0351ХА — ХВ |г, (12.10)
где ХА — электроотрицательность атома А по Полингу. Степень гибридизации а и валентный угол X — Р — X (0) связаны уравнением
р = 3(1-а2) = -т^Ц-. (12.11)
' ' 1—cos0 '
Тогда величина D выражается следующим образом:
D=|(l+e) (1-р2) + 2р2-(1 + е) | = |р2(1-е)|. (12.12)
Подставляя в уравнение (12.11) вычисленные значения и найденные величины химических сдвигов для молекул РН3 и РС13, получаем эмпирическое уравнение
6 = 4,2-102— 1,2.103.0, (12.13)
связывающее химические сдвиги Р31 с величинами D для молекул рассматриваемого типа. Химические сдвиги, предсказываемые этим уравнением, могут иметь только положительные значения, причем максимально возможный сдвиг 4,2 -102 м. д. хорошо согласуется с экспериментально наблюдаемым значением 4,9 ПО2 м. д. для Р4. Из уравнения (12.13) следует, что для других молекул РХ3 значения б равны примерно 200 м. д., что хорошо согласуется с экспериментальными данными. Вычислению более точных значений химических сдвигов Р31 препятствует большая ошибка измерения валентного угла X — Р — — X [146].
Паркс [145] уточнил уравнение Маллера (12.12) для расчета числа несбалансированных р-электронов и представил его следующим образом:
О' = Г4-₽2]₽2(1-8). (12.14)
В этом уравнении учитывается тот факт, что при приближении гибридизации фосфора к sp3 число D' несбалансированных р-электронов стремится к нулю. Однако и уравнение Паркса представляет собой лишь приближенное соотношение, поскольку для молекул с тетраэдрической симметрией sp3 наблюдались большие парамагнитные вклады в экранирование ядер С13 [47]. Паркс, далее, модифицировал эмпирическое уравнение для определения химических сдвигов:
6= —230 4-29,0-103.exp (-46-D'), (12.15)
366
Глава 12
где 6 — химический сдвиг, измеренный относительно сигнала 85%-ной орто-фосфорной кислоты. Вычисленные значения 6 хорошо согласуются с экспериментально наблюдаемыми сдвигами (см. табл. 12.55) г).
Таблица 12.55
СРАВНЕНИЕ НАБЛЮДАЕМЫХ И РАССЧИТАННЫХ ХИМИЧЕСКИХ СДВИГОВ рз1 РЯДА СОЕДИНЕНИЙ ТРЕХВАЛЕНТНОГО ФОСФОРА [4, 145]
Соединение Химический сдвиг д, м. д.
измеренный рассчитанный
(по Маллеру) (по Парксу)
РНз -Г238 [143] +240 +230
PF3 —97 [141] —640 —114
Pls — 178 [141] — 100 —201
РС13 —219 [4] —215 —201
РВгз —227 [4] —230 —227
Если атом фосфора имеет четыре заместителя, трудности расчета величины D возрастают. Кроме того, неизвестна степень двоесвязности Р = О и Р = S [4]. В соединениях пятивалентного фосфора число несбалансированных р-электронов меньше, чем в соединениях трехвалентного фосфора, и электронное распределение более симметрично. Совместное действие этих двух факторов обусловливает более сильное экранирование ядер пятивалентного фосфора по сравнению с трехвалентным. Например, для молекулы HPF.. причинами большого химического сдвига (б = 11,8 м. д.) по сравнению с другими молекулами пятивалентного фосфора, в которых фосфор связан четырьмя связями, могут служить ее высокая октаэдрическая симметрия и увеличенное число электронов, окружающих ядро фосфора, обусловленное увеличением числа соседних атомов фтора [141].
В работе Ван Везера и сотр. [143] изучены спектры ЯМР-Р31 более чем двухсот фосфорсодержащих соединений (см. приложение V). Тот факт, что химические сдвиги Р31 для соединений трехвалентного фосфора могут находиться в очень широких пределах, объясняется значительным изменением гибридизации фосфора под влиянием заместителей (например, в РН3 атом фосфора находится в р3-состоянии, тогда как в PF3 фосфор имеет хр3-гибриди-зацию). Для соединений трехвалентного фосфора влияние заместителей на экранирование является до некоторой степени аддитивным. Приближенные значения вкладов, которые вносят различные заместители в химический сдвиг Р31 (измеренный относительно 85°в-ной ортофосфорной кислоты), схематически представлены в табл. 12.56. Пользуясь этими данными, можно вычислить
Таблица 12.56
ВКЛАДЫ РАЗЛИЧНЫХ ГРУПП В ЭКРАНИРОВАНИЕ ЯДЕР Р31 В СОЕДИНЕНИЯХ ТРЕХВАЛЕНТНОГО ФОСФОРА [143]
S
сг рх, г nr2 OR 10С6Н, Г
С6Н5 R
И
Br I
-за -во -ю -го
о го
Т
40
1-------1—
60 80 м.Э.
г) Полуколичественное согласие с экспериментальными данными получено в результате расчета химических сдвигов Р31 методом МО-ЛКАО с учетом S-, р- и d-орбита-лей [332].— Прим. ред.
Спектры ЯМР других ядер (кроме водорода и фтора) 367
химические сдвиги; обычно они с точностью ±10 м. д. совпадают с экспериментальными значениями (хотя в некоторых случаях расхождения довольно велики; например, для производных RPC1F вычисленный химический сдвиг Р31 равен примерно —85 м. д., тогда как измеренное значение составляет ±20 м. д.). Сигналы ЯМР-Р31 молекул Р4, РН3 и P2S5 находятся в значительно более сильном поле, чем для других соединений трехвалентного фосфора. Это объясняется наличием в молекулах изогнутых («банановых») связей. Однако обоснованность концепции изогнутых связей вызывает сомнения [691.
Гронвеге и др. [147] измерили химические сдвиги Р31 для двухсот фосфорсодержащих молекул и показали, что в соединениях трехвалентного фосфора последовательное замещение одного органического лиганда другим вызывает постепенное и примерно одинаковое изменение экранирования. В табл. 12.57
Таблица 12.57
ИЗМЕНЕНИЕ ХИМИЧЕСКИХ. СДВИГОВ Р31 СОЕДИНЕНИЙ ТРЕХВАЛЕНТНОГО ФОСФОРА ПРИ ЗАМЕНЕ ГАЛОГЕНА ИЛИ ОРГАНИЧЕСКОГО РАДИКАЛА [147]
Исходное соединение Замещенное соединение Изменение химического сдвига прн замещении
одного радикала Двух радикалов трех радикалов
(СН3)3Р (С2Н5)3Р —13,5 — 14,5 —13,6
(С2н5)3р (С6н5)3р —5,3 —1,6 —6,5
(СН3)3Р (С6н5)3р — 15,0 — 19,0 —21,0
(СН3)2РН (С2Н5)2РН —22,5 —21,5
(СН3)2РН (С4Н9)2РН — 19,8 — 10,3
(СН3)2РН (С6Н5)2РН —27,2 —31,2
(СН3)2РС1 (С2Н5)2РС1 — 13,8 — 13,2
(СН3)2РС1 (С6Н5)2РС1 +8,6 + 1,9
(С2Н5)2РС1 (С6Н5)2РС1 +2,0 + 15,5
(СН3)2РВг (С2Н5)2?ВГ — 10,6 — 17,7
(СН3)2РВг (С6Н5)2РВг + 10,7 . +6,2
приведены изменения химического сдвига Р31 под влиянием замены атома галогена или алкильной группы в соединениях трехвалентного фосфора. Однако вклады различных заместителей в экранирование Р31 не строго постоянны — они в определенной степени зависят от влияния других заместителей.
Для объяснения того, что для соединений пятивалентного фосфора диапазон изменений химических сдвигов значительно меньше, чем для соединений трехвалентного фосфора, было выдвинуто предположение о том, что в случае пятивалентного фосфора гибридизация остается постоянной (sp3), а меняется лишь электронное распределение л-связей в пределах остова ст-связей [143]. Для ряда соединений пятивалентного фосфора вклад заместителей в экранирование Р31 слабо зависит от природы других заместителей, однако надежную аддитивную схему построить не удается [4]. Можно лишь указать, что при замене каждого атома азота, связанного с пятивалентным фосфором, на кислород имеет место постоянное изменение химического сдвига Р31 на —11 ± 2 м. д. Если кислород заменяется менее электроотрицательными атомами азота или серы, то экранирование ядра фосфора уменьшается вследствие того, что кислород имеет большую склонность к образованию двойной связи с фосфором, чем другие элементы. По-видимому, замена кислорода на атом серы сопровождается изменением типа связи, причем тип связи может изменяться, различать-
368
Глава 12
Таблица 12.58
ИЗМЕНЕНИЕ ХИМИЧЕСКИХ СДВИГОВ Р31 СОЕДИНЕНИЙ, СОДЕРЖАЩИХ ТЕТРАЭДРИЧЕСКИЙ АТОМ ФОСФОРА, ПРИ ЗАМЕНЕ ОДНОГО
ОРГАНИЧЕСКОГО ЛИГАНДА НА ДРУГОЙ [147] (R — АЛКИЛЬНАЯ ИЛИ АРИЛЬНАЯ ГРУППА, X = CI ИЛИ Вг)
Исходное соединение Замещенное соединение Среднее изменение химического сдвига Стандартное отклонение, м. д. Число рассмотренных примеров
Замещение С6Н;,
C6H5RPSC1 C2H5RPSC1 —16,1 2,1 4
C6H5RPOC1 c2h5rpoci — 16,7 0,9 3
C6H5RPSC1 ch2cirpsci —1,1 3,3 3
C6H5RPOC1 CH2ClRPOCr —3,7 1,7 3
Замещение СН3
CHaRPOCl C2H5RPOC1 —7,0 1,8 3
ch3r2ps c2h5r2ps + 1,5 1,3 3
CH3RPSX c2h5rpsx —12,8 4,5 6
CH3RPOC1 ch2cirpoci +7,2 0,8 3
CH3RPSX ch2cirpsx +2,9 1,0 3
CH3RPOC1 c6h5rpoci + 10,8 1,3 3
CH3RPSX c6h5rpsx +5,2 2,5 4
ся для разных конкретных примеров, так как вклады в химический сдвиг при таком замещении находятся в пределах от —25 до —71 м. д. на каждый замещенный атом.
Гронвеге и сотр. [147, 148] нашли, что вклад для непосредственно связанных с пятивалентным фосфором групп в экранирование ядра Р31 уменьшается в ряду СбН5>СН2С1>СН3>С2Н5. Оказалось, что поскольку вклады этих групп не зависят от природы других заместителей, то их можно использовать для успешного предсказания химических сдвигов Р31. Соответствующие данные для соединений пятивалентного фосфора приведены в табл. 12.58 и 12.59. Катрицкий и Джонс [149] указали на ограниченность аддитивной схемы для химических сдвигов в применении к соединениям трех- и пятивалентного фосфора: при статистическом расчете вкладов в химический сдвиг, проведенном для большого числа заместителей, встретились большие стандартные отклонения, особенно в случае соединений трехвалентного фосфора.
В ряду ароматических соединений, в которых атом фосфора непосредственно присоединен к кольцу, наблюдаются аномальные значения химических сдвигов Р31 (см. приложение V), которые невозможно объяснить влиянием кольцевого тока в ароматическом кольце [146].
Атомы или группы атомов, не связанные непосредственно с атомами фосфора, оказывают весьма слабое влияние на химические сдвиги и константы взаимодействия Р31 [4, 143]. Например, химические сдвиги Р31 триалкилфосфатов (С2Н6О)3РО и (С1С2Н4О)3РО равны соответственно 0,9 и 1,3 м. д. (относительно сигнала Н3РО4) *).
1) Химические сдвиги Р31 для ряда ненасыщенных эфиров фосфиновых кислот измерены Б. И. Иониным и А. А. Петровым при изучении изомеризации этих соединений [323, 324]. Проведено исследование ЯМ Р-Р31 для растворов ряда фосфорорганических соединений [325, 326], фенилазафосфор ных кислот [333], фосфинов и дифосфинов [334].— Прим. ред.
Спектры ЯМР других ядер (кроме водорода и фтора)
369
Таблица 12.59
ИЗМЕНЕНИЕ ХИМИЧЕСКИХ СДВИГОВ Р31 СОЕДИНЕНИЙ, СОДЕРЖАЩИХ ТЕТРАЭДРИЧЕСКИЙ АТОМ ФОСФОРА, ПРИ ЗАМЕЩЕНИИ ОДНОГО
НЕОРГАНИЧЕСКОГО ИЛИ ОРГАНИЧЕСКОГО ЛИГАНДА НА ДРУГОЙ [147] (R ИЛИ R' — АЛКИЛЬНАЯ ИЛИ АРИЛЬНАЯ ГРУППА)
Исходное соединение Замещеииое соединение Среднее изменение химического сдвига, м. д. Стандартное отклонение, м. д. Число рассмотренных примеров
Замещение кислорода серой
(RO)3PO (RO)3PS —70,1 1,0 3
RP(O)C12 RP(S)C12 —37,5 2,8 4
RR'P(O)C1 в СС14 RR'P(S)C1 —31,0 3,9 8
R3PO в СНС13 R3PS в CHC13 —8,9 5,9 3
Другие типы замещения
RP(O)C12 RP(O)(OH)2 + 19,1 4,9 4
RP(O)(OH)2 RP(O)(OC2H6)2 +0,8 1,4 4
RP(O)C12 RP(O)(OC2H6)2 + 18,9 2,7 6
RR'P(O)C1 RR'P(O)(OH) + 12,6 5,1 5
RR'P(S)C1 RR'P(S)Br + 18,2 4,6 4
R2P(O)C1 R2P(O)CH2C1 + 12,3 6,6 4
R2P(O)C1 R2P(O) — 0 — P(O)R2 + 11,5 1,6 5
R2P(O)CH2C1 R2P(O) — 0 — P(O)R2 —4,2 5,8 3
R2P(O)OH R2P(O) — 0 - P(O)R2 —1,3 6,1 5
R2P(O)C1 R2P(O)OH + 12,6 4,4 6
RP(O)(OH)2 (RPO2)n + 17,0 1,3 3
RP(O)C12 (RPO2)n +30,4 5,7 3
RP(O)(OC2H6)2 (RPO2)n + 17,3 0,8- 3
12.6.2. Спин-спиновое взаимодействие ядер Р31
В табл. 12.60 приведены некоторые из известных в настоящее время констант взаимодействия ядер непосредственно связанных атомов Р31 — Н1, Р31 — F19 и Р31 — Р31. Теоретического рассмотрения взаимодействия этих ядер не проводилось, однако Файнгольд [1461 отметил некоторые общие закономерности взаимодействия Р — Н.
1. При наличии непосредственной связи фосфор — водород константа взаимодействия обычно больше для соединений со связью Р = О, чем для соответствующих соединений со связью Р = S (см. табл. 12.61).
2. Если атомы фосфора и водорода разделены двумя или тремя связями, то наблюдается обратный порядок, т. е. константа взаимодействия больше для соединений, имеющих связь Р = S.
Через 2 связи H—C-P=S | | Н—С—Р=О 1 I
15—20 гц 1 1 10 гц
Через 3 связи Н—С—С—P=S 1 1 1 Н-С-С-Р=О 1 1 1
15—20 гц 1 1 1 + 5 гц (не удалось обнаружить)
24—489
370
Глава 12
Таблица 12.60
КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ НЕПОСРЕДСТВЕННО СВЯЗАННЫХ ЯДЕР С УЧАСТИЕМ ЯДРА Р31 (В ГЕРЦАХ) а
Соединение 7PH 7pF Jpp Литература
РН3 (179) 143
PF3 (1410) 141
СН3РН2 (207) 143
(СН3)2РН (207) 143
C2H5PC1F (569) 143
(пзо-С3Н7О)2НРО 692+0,5 161
СН3(С2Н5О)НРО 518+8 146
(C2H5O)2HPS 645+8 146
СН3(изо-С3Н7О)НР8 492±8 146
HPF6 (707) 141
HPO(OH)2 707 12
H2PO(OH) 584 12
C2H5POF2 1132 155
ch3pof2 110 155
P2H4 186,5 108,2 164
г 0 0 -|3-
1 1 0—P—P—0 444 480 152
1 1 L он J
(CH3O)2HPO 690,3+0,5 161
(C2H6O)2HPO 684,1+0,5 161
CH3(C3H7O)HPO 550,1+0,5 161
H2(OH)P(O)(= H3PO2) 561+8 146
а Значения 7, заключенные в скобки, измерены с точностью ± 20 гц.
Менетт и сотр. [259], пользуясь техникой двойного резонанса и приняв константу </Р_н для непосредственно связанных атомов положительной, определили абсолютные знаки ряда констант взаимодействия Р — Н 1).
Р-Н +
Н\Р-Р/Н + (НН)
,Н Рч /Н
Р —С/ С — с/
+ или — +
Р\ /Н Рч
ХС = С/ >с = с
+ н/ +
\р р—р
нх _
-(НН)
Нч /Н Х'Р — с/
+ (НН)
н
Константа взаимодействия фосфора с протонами метиленовой группы в ряду триалкилфосфатов (RCH2O)3P = О (см. табл. 12.62) уменьшается по мере усиления электронодонорного характера алкильного заместителя [154]. Значение констант было определено по протонным спектрам этих соединений, а для триметилфосфата также по спектру ЯМР-Р31 [152].
Если, однако, заместитель R представляет собой группу с высокой электроотрицательностью, то значение константы не зависит от степени электроно-
Я Предпринята попытка определить зависимость /р_с~с-н от двугранного угла [378]. Для («-С3Н7)3Р = О найдено, что константы /рна и 4рнЗ имеют одинаковые знаки [379],— Прим. ред.
Спектры ЯМР других ядер (кроме водорода и фтора)
371
Таблица 12.61
ПРЕДЕЛЫ ИЗМЕНЕНИЯ КОНСТАНТ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ Р31 — Н1 и Р31 — Fla (В ГЕРЦАХ) ДЛЯ РАЗЛИЧНЫХ ТИПОВ МОЛЕКУЛ [149]
Тип соединений Число соединений JPH Число соединений JPF
Р(Ш) 3 180—210 5 570—1420
—р = О 13 500—700 5 980—1190
—р = S, Se 3 490—630
акцепторного влияния таких заместителей. Этот вывод следует из рассмотрения данных, приведенных в табл. 12.62. Дадек [150] на примере ряда других триалкилфосфатов показал, что значения /р-н находятся в соответствии с о-константами Тафта для заместителей [151]. Причиной наблюдаемых изменений констант взаимодействия /сн2р Для этих молекул может служить изменение электронного окружения либо атома фосфора, либо метиленовой группы. Исследование несимметричных производных триалкилфосфатов (см. табл. 12.62) позволяет решить, какой из этих эффектов важнее [152]. Например, значительное изменение окружения атома фосфора при переходе от тетрабензилпирофосфата к трибензилфосфату лишь в малой степени меняет значение констан-
Таблица 12.62
КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ Р31 — Н1 РЯДА СИММЕТРИЧНЫХ И НЕСИММЕТРИЧНЫХ ТРИАЛКИЛ ФОСФАТОВ [150, 154]
Соединение JCH2 - Р ’ гч Литература
Симметричные триалкилфосфат (СН3О)3Р = О (С2Н5О)зР = о (и-С3Н7О)3Р = О (и-С4НэО)3Р = О (н-С5НцО)3Р = О [(СНз)2СН - СН2О]3Р = о [(СН3)3С - СН2О]3Р = О (СС13СН2О)зР = о (CF3CH2O)3P = О (СНС12СН2О)зР = о (С6Н5СН2О)зР = о (СН2 = СН — СН2О)з = о 11,19±0,2 8,38+0,2 7,70+0,1 7,65+0,1 7,62+0,2 6,60+0,1 5,24±0,1 7,05+0,1 8,34+0,1 8,4±0,2 9,07+0,1 8,3+0,2 154 154 154 154 154 154 154 150 150 150 150 150
Соединение Группа JCH2 - Р ’ гц Литература
Несимметричные триалкилфосфаты (С6Н5СН2О)4Р2Оз ,ОСН2СН2 СНзОР< | |росн2сн2 О СН3РО(ОСН2С6Н5)2 'i С6Н5СН2 СНз СНз с6н5сн2 8,61+0,2 11,2+0,1 11,12+0,1 8,68+0,1 150 150 150 150
24*
372
Глава 12
ты Урн- Аналогичные явления также наблюдаются для других несимметричных соединений. Следовательно, причиной изменения констант JPH для рассматриваемого ряда соединений служит электронное окружение метиленовой группы. Для триалкилфосфатов проведено исследование взаимодействия ядер фосфора с протонами, находящимися у 0-углеродных атомов алкильной группы [153]. Найдено, что константы взаимодействия уменьшаются при возрастании числа связей между фосфором и водородом. Например, для триэтилфосфата (СН3СН2О)3Р = О константа /сн2-р = 8,38 гц, а /сн3-р = = 0,76 гц. Однако для фосфорорганических соединений, в которых этильная
Таблица 12.63
КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ Р31 — HI ДЛЯ НЕКОТОРЫХ АЛКИЛОВ ТРЕХВАЛЕНТНОГО ФОСФОРА
Соединение JP -'СН2, г,( JP — сн3, г,< Литература
с2н5РС12 17,7 15,2
c2h5pof2 23,6 23,6
С2Н5РОС12 14,5 30,3 155
СН3РОС12 16,9
CH3POF2 19,9
(С2Н5)зР 0,5 13,7 233
группа непосредственно связана с атомом фосфора, такое затухание взаимодействия не проявляется. В некоторых случаях константа взаимодействия ядра Р31 с Р-протонами оказывается более высокой, чем с а-протонами. В табл. 12.63 приведены значения /рН Для молекул такого типа [155].
Таблица 12.64
КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ Н1-Р31 (В ГЕРЦАХ) НЕКОТОРЫХ ФОСФОРОРГАНИЧЕСКИХ ЭФИРОВ [156]
(DR\ ° (2)R—С—Р—Y (3)R/ 1 А Константа взаимодействия, гц
R1 r2 Кз X Y . JHa — Р JH₽ - Р
Н н Н С6Н5 С1 14,1
Н н СН3 С1 С1 14,2 30,0
Н н . СН3 С6Н5 С1 12,1 22,1
Н СНз СН3 С1 С1 12±1 27,6
Н СНз СНз Се Ня С1 20,6
н СНз СН3 с6н5 он 17,6
н СНз СН3 с6н5 ОСНз 17,3
н СН3 СН3 с6н5 ОС2Н5 17,1
СНз СН3 СН3 С1 С1 24,8
СНз СН3 СН3 С6Н5 С1 17,7
СНз СН3 СНз С6Н5 он 15,7
н СН3 с2н5 С1 С1 —
н СН3 с2н5 С6Н5 С1 20,0
н Н с3н7 CgHg С1
н с2н5 С2Н5 С6Н5 С1
н СН3 С6Н5 С6Н5 он 18,6 16,8
н СН3 С6Н5 С6Н5 ОСНз 17,1
Спектры ЯМР других ядер (кроме водорода и фтора) 373
Обратное соотношение значений 7Рн для а- и 0-протонов найдено в спектрах протонного резонанса соединений следующего строения:
О
R1\ II
R2—С —Р —Y, R3/ 1
Л.
где R представляет собой атом водорода, алкильную или фенильную группу; Y может быть ОН, С1, ОСН3 или ОСН2СН3, а X — это С1 или фенильная группа [156, 157]. Значения /Рнр и /РН(Х приведены в табл. 12.64. Следует отметить, что в спектрах молекул, содержащих фенильную группу, наблюдается дублетное расщепление компонентов сигналов от некоторых протонов. Это свидетельствует о том, что молекулы проводят значительную часть времени в определенной преимущественной конформации [156, 157].
Необходимо заметить, что из-за участия слабокислых атомов водорода ортофосфорной кислоты в быстром обменном процессе в спектре этого соединения взаимодействие Jph не наблюдается [152] х).
12.6.3. Применение ЯМР-Р31 для структурных исследований
Изменение электронного окружения атома фосфора обычно сопровождается значительным изменением экранирования ядра Р31. Показано, что каждый класс фосфорорганических соединений обладает характерным средним химическим сдвигом Р31, значением которого можно пользоваться при определении молекулярной структуры фосфорсодержащих соединений. В результате рассмотрения обширного материала (см. табл. 12.53 и приложение V) Файнгольд [146] составил таблицу таких характеристических химических сдвигов (табл. 12.65). При приготовлении раствора исследуемого вещества следует избегать растворителей, способных образовывать водородные связи (таких, как хлороформ, спирт, вода, диоксан), поскольку последнее может существенно отразиться на значениях химических сдвигов Р31 [4]. В тех немногих случаях, когда использовались растворы в четыреххлористом углероде и сероуглероде, не наблюдалось смещения сигналов Р31 при изменении концентрации вещества [4].
Рассмотрим несколько примеров установления структуры исследуемых соединений с помощью ЯМР-Р31.
Органические фосфиты [152]. Если структуру третичных эфиров фосфорной кислоты неизменно изображают в виде (RO)3P, то для первичных и вторичных эфиров иногда полагают, что фосфор связан не стремя (RO)2POH, а с четырьмя атомами: (RO)2P(H)O. Исследование этих соединений с помощью ЯМР-Р31 показало, что более 95% молекул от общего числа находятся в форме, в которой водород непосредственно связан с атомом фосфора. Основанием для такого заключения служит отсутствие в спектре сигналов, соответствующихя ядру фосфора в группе РОН, и наличие сигналов с расщеплением J (~600 гц), характерным для атомов Р и Н, непосредственно связанных друг с ругом [152]. Дублетное расщепление свидетельствует об отсутствии быстрого обмена протонами, присоединенными к атому фосфора.
*) Некоторые данные о спин-спиновом взаимодействии ядер Р31 — Н1 см. также в [327—329]. Спин-спиновое взаимодействие Р31— Си63 (7=1190 гц) и Р31 — Си65 (7=1210 гц) наблюдалось в спектре ЯМР-Р31 раствора комплексов Cu[P(OR)3]4C1O4 (где R=CH3 и С2Н5) в метиленхлориде [335].—Ярил. ред.
Таблица 12.65
ХАРАКТЕРИСТИЧЕСКИЕ ХИМИЧЕСКИЕ СДВИГИ Р31 [146]
Тип соединения 6, a M. Д. Число исследованных соединений
R(R'O)2P a) R, R' = C„Hn+1 (n > 1) 6) R = CH3; R' = CnHn+1 (n> 1) —183±1 —175+2 2 7
R(R'S)2p a) R = CH3; R' = C„H„+1 (n > 1) 6) R = CH3; R' = арил —69,4+0,7 —84,5 2 1
(RO)3P симметричный a) R = CnHn+1 (n > 1) 6) R = CH3 в) R = арил —138+1 —139 —127+1 10 1 2
(RS)3p симметричный R = C„Hn+1 (n > 1) —117+1 2
R3P(O) симметричный a) R = CnHn+1 (n > 1) 6) R = арил —46,0+2,5 —23,0 2 1
R(R')(R"O)P(O) a) R, R', R" = C„H„+1 (n> 1) R = R' 6) R = R' = CH3; R" = CnH„+1 (n> 1) в) R = CH3; R', R" = CnHn+1 (n > 1) —54,9+0,2 —49,5+0,5 —53,0+1,0 1 2 2
R(R'O)2P(O) a) R, R' = C„Hn+1 (n> 1) 6) R = CH3; R' = CnHn+1 (n > 1) в) R — CnHn+1 (n > 1); R' = CH3 —32,8+1,5 —28,4+0,9 —35,0 5 5 1
R(R'O)(R"S)P(O) a) R, R', R" = CnHn+1 (n > 1), 6) R = CH3; R', R " = CnHn+1 (n > 1) —56,5+0,8 —50,5+1,4 2 9
R(R'S)2P(O) R = CH3; R' = C„H„+1 (n> 1) —55,8 1
(RO)3P(O) симметричный a) R = CnHn+1 (n > 1) 6) R = арил + 1,07+1,00 + 17,4+0,3 5 4
(RO)(R'O)2P(O) R = C„Hn+1 (n > 1); R' = арил + 12,0 1
(RO)2(R'S)P(O) a) R, R' = CnHn+1 (n > 1) 6) R = CnHn+1 (n > 1); R' = CH3 в) R = CnH„+1 (n > 1); R' = арил —26,5+0,3 —28,4 —21,5+0,5 6 1 2
(RS)3P(O) симметричный R3P(S) симметричный A A + + e e I I e e О О II II К К —61,3 —51,0+3,0 1 2
R(R'O)2P(S) a) R, R' = CnHn+1 (n > 1) 6) R' = C„HK+1 (n > I); R = CH3 — 101,7 —94,5+0,9 1 8
Спектры ЯМР других ядер (кроме водорода и фтора)
375
Продолжение табл. 12.65
Тип соединения 6, а м. д. Число исследованных соединений
R(R'S)2P(S) R = CH3; R' = CnH„+i (n > 1) —74,0+3,0 2
<RO)3P(S) a) R = CnHn+1 (n > 1) 6) R = CH3 —68,1 ±1,2 —73,0 11 1
<RO)2(RS)P(S) R = CnHn+1 (n > 1) —94,9 1
(RS)3P(S) симметричный R = cnHn+1 (n > i) . —92,9 1
(R2N)3P симметричный a) R = CnHn+1 (n > 1) 6) R = CH3 — 118 — 122 1 1
(R2N)3P(O) симметричный a) R = CnHn+1 (n > 1) 6) R = CH3 —23,0 —23,4 1 1
<R2N)2P(O)OR' R' = CnHn+1 (n > 1) R = CH3 —16,3 1
(R2N)3P(S) симметричный R = CnHn+1 (n > 1) —76,0=1=1,0 2
<R2N)2P(O)R' (R2N)P(O)(OR')2 (R2N)P(S)(OR')2 a Относительно H3P( Оксикислоты c лот, анионы котор1 0 0- III. R = R' = CH3 R' = CnHn+i (n > 1); R = CH3 R, R' = CnHn+1 (n > 1) как внешнего эталона. двумя, атомами фосфора [152]. Суш jx содержат два атома фосфора: 0 0 -.4- г 0 0 II II -Р —0—Р —0 0 — Р — р. II и о о J Loo . Пирофосфат II. Гипофос 0 0 -.2- Г 0 1 1 1 Р—0—Р—0 0—Р—0— 1 1 1 н н J L о Пирофосфит IV. Изогипоф Г °0 -j3~ 1 1 0—Р—Р—0 L и J —38,0 —35,0 —75,0 ествуют пять - 4- -0 фат 0 -|3~ Р —0 ч осфат 1 1 1 оксикис-
V. Дифосфит
Строение этих анионов было определено химическими методами и подтверждено спектроскопией ЯМР-Р31 [152].
__|_____I____I____I_____I____I____I____I_____I____I____I_____I____L_
—25 —20 —15 —10 -5 0 5 10 15 20 25 30 35
Рис. 12.35. Спектр ЯМР-Р31 изогипофосфат-иона при 12,3 Мгц [152].
Химические сдвиги измерены относительно 85%-ной Н3РО4 как внешнего эталона.
Теоретические относительные интенсивности:
Рис. 12.36. Спектр ЯМР-Р31 дифосфит-иона при 12,3 Мгц [152].
Спектры ЯМР других ядер (кроме водорода и фтора)
377
I и II. Спектры ЯМР-Р31 пиро- и гипофосфат-анионов состоят из одиночного сигнала, что подтверждает симметричность структуры и отсутствие водородных атомов, связанных непосредственно с фосфором (химические сдвиги относительно эталонного сигнала 85%-ной ортофосфорной кислоты составляют -j-б и —9 м. д. соответственно).
III. Спектр пирофосфит-аниона представляет собой дублет, обусловленный спин-спиновым взаимодействием ядер эквивалентных атомов фосфора с соседними непосредственно с ними связанными протонами (7РН х 660 гц). Присутствие только одного дублетного сигнала свидетельствует о симметричном строении иона, и, следовательно, об эквивалентности ядер фосфора.
IV. Спектр изогипофосфат-аниона и отнесение сигналов приведены на рис. 12.35. В результате спин-спинового взаимодействия между неэквивалентными ядрами фосфора возникает слабое дублетное расщепление каждого сигнала спектра (7РР = 17 гц). Помимо этого, резонансный сигнал ядра фосфора, связанного с протоном, расщеплен в дублет из-за взаимодействия Р — Н (/рн = 620 гц). Таким образом, спектр ЯМР-Р31 подтверждает предложенную структуру иона, поскольку он свидетельствует о неэквивалентности атомов фосфора, один из которых непосредственно связан с водородом [1521.
V. Спектр дифосфит-аниона состоит из двух мультиплетов (рис. 12.36) и является АВ-частью спектра АВХ (см. разд. 8.15.1). В результате анализа спектра найдено, что 6Р1 = —22,6 м. д.; 6Ра = —7,0 м. д. (отрицательное значение соответствует положению сигнала в области более слабого поля относительно сигнала 85%-ной ортофосфорной кислоты как внешнего эталона); /Р1р2 = 480 ± 10 гц', J = 444 гц', /Ран = 94 гц. Спектр Р31, таким образом, согласуется с предложенной структурой аниона V: два неэквивалентных атома фосфора непосредственно связаны друг с другом, причем к одному из них присоединен атом водорода [152].
Тетраполифосфат-анион Р4О*~. Этому аниону приписывают структуру с тетраэдрическим расположением кислородных атомов относительно атомов фосфора:
-О -О -О -О 0=1^!—о—ik>—о—р3—о—р4=о I II II I -о- о о о-
Ядра фосфора такой структуры должны образовать спиновую систему АА'ХХ'. Наблюдаемый спектр ЯМР-Р31 (рис. 12.37) полностью соответствует этой системе [158]. В результате детального анализа получены приведенные в табл. 12.66 константы спин-спинового взаимодействия Р — Р, значения которых, как оказалось, довольно сильно зависят от pH среды.
Таблица 12.66 константы спин-спинового ВЗАИМОДЕЙСТВИЯ Р31 — Р31 (В ГЕРЦАХ) для ТЕТРАПОЛИ ФОСФ AT-ИОН А ПРИ РАЗЛИЧНЫХ pH [158]
pH 1 Т12 = 7з4 | 1 7зз 1
8 19,9 16,7
4 19,7 16,8
2 18,7 16,5
0 16,7 15,9
378
Г лава 12
Исследовано также влияние pH среды на химические сдвиги Р31 некоторых конденсированных фосфатов [158]. Оказалось, что слабокислые протоны в соединениях НР3О*~, Н2Р3О3“, HP4OJ“ и Н2Р4О*“ находятся вблизи центральной фосфатной группы, по-видимому, более длительное время, чем предполагалось ранее.
Диалкилфосфонаты щелочных металлов. В результате исследования спектров ЯМР-Р31 было установлено, что диалкилфосфонаты имеют структуру
700 200 300 400 500
гц
11,, ,, || II
720 740 750 480 500 520
гц
Рис. 12.37. Наблюдаемый и рассчитанный спектры ЯМР-Р31 тетраполифосфита аммония при 24,3 Мгц [158].
Частоты измерены относительно сигнала 85%-ной Н3РО4 как внешнего эталона. Сигнал поглощения при 543 гц обусловлен малым количеством P40iJ.
(RO)2P(H)O, а не (RO)2POH. Однако химические сдвиги Р31 диалкилфосфона-тов щелочных металлов имеют значения, характерные для атома фосфора с тремя связями. Этот результат свидетельствует о том, что соли имеют структуру (RO)2P — О — М (где М = Li, Na или К), причем связь О — М в значительной степени ковалентна [159]. В табл. 12.67 приведены химические
Таблица 12.67
ХИМИЧЕСКИЕ СДВИГИ Р31 СОЛЕЙ ЩЕЛОЧНЫХ МЕТАЛЛОВ ДИАЛКИЛ-И ДИАРИЛФОСФОНАТОВ, А ТАКЖЕ ТРИАЛКИЛ- И ТРИАРИЛФОСФИТОВ
Соединение б , a м. д. Соединение б, а м. д.
LiOP(OC2H5)2 —145 НРО(ОСН3)2 —11
NaOP(OC2H5)2 —153 НРО(ОС2Н5)2 —8
КОР(ОС2Н5)2 —152 НРО(ОС4Н9)2 —8
LiOP(OC4H9)2 —145 НРО(ОС6Н5)2 0
NaOP(OC4H9)2 —152 Р(ОСН3)з —141
КОР(ОС4Н9)2 —153 Р(ОС2Н5)з —139
LiOP(OC6H5)2 — 142 Р(ОС4Н9)з —139
NaOP(OC6 {-[5)2 — 148 Р(ОС6Н5)з —128
КОР(ОСвН5)2 —139
а Относительно 8 5%-ной Н3РО4 как внешнего эталона.
Спектры ЯМР других ядер (кроме водорода и фтора/ 379
сдвиги Р31 для диалкил- и диарилфосфонатов щелочных металлов, а также для триалкил- и триарилфосфитов. В табл. 12.68 указаны константы взаимодействия /рН для некоторых из этих молекул.
Таблица 12.68
КОНСТАНТЫ спин-спинового
ВЗАИМОДЕЙСТВИЯ Hl — Р31
ДИАЛКИЛФОСФОНАТОВ HPO(OR)2 [159]
R Трн> г,<
СНз 715
с2н5 686
с4н9 690
С6н5 746
Следует отметить также, что с помощью измерения химических сдвигов Р31 удалось доказать наличие в реакционной смеси фосфонатов (RO)2R'P(O) .(6 от —19 до —31 м. д.) и фосфатов (RO)2(R'O)P(O) (6 от 0 до 8 м. д.) [248].
Конденсированные фосфаты. Вопрос о строении конденсированных фосфатов приобрел определенную ясность лишь в самое последнее время, и важная роль в этом наряду с другими методами принадлежит методу ЯМР. Как следует из табл. 12.69, атомы фосфора в изолированных, концевых, срединных
Таблица 12.69
ТИПИЧНЫЕ ХИМИЧЕСКИЕ СДВИГИ р31 ГРУПП РО4
КОНДЕНСИРОВАННЫХ ФОСФАТОВ [152, 160]
Тип группы РО< Химические сдвиги, м. д.
бРВг3 ®Н3РО4(внешн)
Ортофосфатные ионы
Изолированные группы
тризамещенные (нормальные) соли 233 6
от 1 до 3 атомов водорода на атом 238 11
фосфора Концевые группы двузамещенные (без протонов) 244 17
от 1 до 2 атомов водорода на атом 247 20
фосфора Группы внутри цепи короткая цепь 256 29
длинная цепь 259 32
Группы в точках разветвлений полифосфаты щелочных металлов 268 41
азеотропная фосфорная кислота 272 45
а При пересчете химических сдвигов в шкалу фосфорной кислоты как внешнего эталона использовано уравнение бн3ро4(внешн) = ®РВГз — 227 Iм- д4-
группах и группах в точках разветвлений фосфата дают в спектре ЯМР-Р31 отдельные сигналы с характеристическими химическими сдвигами [152, 160]. При исследовании растворов фосфатных стекол с длиной цепи менее ~75 атомов на спектрометре ЯМР-Р31 с высокой чувствительностью можно обнаружить
380
Глава 12
концевые фосфатные группы. Нередко вследствие косвенного спин-спинового взаимодействия Р31 — Р31 в спектрах обнаруживается тонкая структура, которая может быть весьма полезной для правильного отнесения сигналов.-Например, существование такого взаимодействия в Триполи- и тетраполифосфатах указывает на то, что связи Р — Р в этих анионах имеют ковалентный характер.
На рис. 12.38 представлены сигналы ЯМР-Р31 различных фосфатных ионов при 40,5 Мгц (23 500 э) [277]; качество спектров, полученных при таких высоких полях, очевидно, значительно лучше, чем спектров, наблюдавшихся до этого при низких полях.
Рис. 12.38. Спектр ЯМР-Р31 различных фосфат-ионов при 40,5 Мгц (23 500 э) [277].
Тиокислоты пятивалентного фосфора. Метод ЯМР позволил выяснить структуру некоторых тиокислот пятивалентного фосфора [146]. Основываясь на химическом сдвиге фосфора для кислоты (C2H5O)2POSH (б = —24 м. д.), можно заключить, что в молекуле имеется связь Р = О, а не Р = S. Напротив, кислотам CH3(C2H5O)PSOH (б = —88,8 м. д.) и C2H5(C2H5O)PSOH (б = = —94,2 м. д.) в соответствии со значениями химических сдвигов была приписана структура, включающая связь Р = S.'
Пентахлорид фосфора. Это одно из немногих соединений, исследованных методом ЯМР высокого разрешения в твердом состоянии. В спектре РС15 было обнаружено два резонансных сигнала с различными химическими сдвигами, что свидетельствует о существовании в этом соединении двух положений ядер фосфора с неодинаковым электронным окружением. Таким образом, получила подтверждение гипотеза о том, что в твердом состоянии молекула РС15 существует в виде двух ионов [РС16]~ и [РС14]+. Что касается методики эксперимента, то устранение дипольного уширения было достигнуто быстрым вращением кристаллического образца вокруг оси, составляющей угол 54°44' с направлением магнитного поля (см. разд. 7.1) [162, 163].
Дифосфин Р2Н4. В результате полного анализа спектра протонного резонанса дифосфина [164] получены следующие значения констант взаимодействия: /р_р' = —108,2 гц', /р-н = +186,5 гц', /Р'-н = ±11,9 гц. Несмотря па то что молекула имеет симметричную структуру
Нч +'
>Р-Р'< н7 хн'
оба фосфорных ядра оказались магнитно неэквивалентными, а протоны образовали две пары магнитно неэквивалентных ядер. Таким образом, спектр дифосфина отвечает спиновой системе А2А'ХХ'.
Спектры ЯМР других ядер (кроме водорода и фтора)
381
Стереоизомеры замещенных дифосфинов. В спектрах ЯМР-Р31 соединений с общей формулой
Н3С /СН3
>р—р<
Rr Ri
где Ri — алкильная или арильная группа, обнаружено два сигнала резонансного поглощения одинаковой интенсивности [165]. Для объяснения этого факта было постулировано, что молекулы существуют в двух стереоизомерных формах I и II:
Н3С СН3 Н3СХ /Rj
>Р—Р< >Р—Р<
Ri Ri Rf СН3
I II
Другие исследования. Изучение спектров ряда циклических фосфорорганических соединений (табл. 12.70) показало, что не существует простого соответствия между химическими сдвигами ядер фосфора и размером цикла [166].
Таблица 12.70
ХИМИЧЕСКИЕ сдриги Р31 РЯДА. ЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ ФОСФОРА [166] а
Соединение о №)nOpcl О о (СН2)л<^РОС2Нв о /°\ />
5-Членные циклы —167 —131 —17
6-Членные циклы —153 —128 +7
Ациклические —164 — 138 [4] + 1 [4]
а В миллионных долях относительно 85%-ной Н3РО4 как виешиего эталона.
В работе [167] была рассмотрена зависимость различных сигналов ЯМР-Р31 некоторых ди- и трифосфатов аденозина, важных для биохимических процессов, от pH среды. Авторы изучали очень разбавленные растворы фосфатов (<0,1 /И); проблема чувствительности решалась методом боковой полосы с применением синхронного детектора при очень малой скорости прохождения.
н а
Тш-1100 гц
Рис. 12.39. Спектр ЯМР-Р31 равновесной смеси CH3P(O)F2, CH3P(O)FC1 и СН3Р(О)С12 при 40,5 Мгц (23 500 э) [277].
Спектроскопия ЯМР-Р31 нашла применение также при определении характера замещения в некоторых производных фосфонитрила (см. приложение V) [168, 169, 265]. На рис. 12.40 приведен типичный для соединений этого типа спектр P3N3C14(SC2H5)2. Дублет, находящийся в области сильного поля, отвечает двум магнитно эквивалентным ядрам фосфора РС12, а мультиплет
382
Глава 12
в слабом поле — ядру фосфора P(SC2H5)2. В результате спин-спинового взаимодействия между неэквивалентными ядрами фосфора происходит дублетное расщепление сигнала в сильном поле и триплетное расщепление сигнала
Химический сдвиг от сигнала 85°/-ной Н3РО4, м.5,
Рис. 12.40. Спектр ЯМР-Р31 соединения РзЫзСЛДЗСгНвЬ'при 16,2 Мгц [169]. Обе масти спектра получены при различном усилении.
в слабом поле (причиной расщепления последнего в квинтет служит взаимодействие Р — Н между ядром фосфора и четырьмя эквивалентными метиленовыми протонами этильных групп) [169].
12.6.4 . Количественный анализ
Возможность количественного анализа смесей соединений фосфора методом ЯМР-Р31 обусловлена двумя факторами. Во-первых, для большинства фосфорорганических соединений в молекуле содержится лишь один атом фосфора, и поэтому они имеют сравнительно простой спектр. Во-вторых, сигналы резонансного поглощения Р31 различных соединений обычно обладают различными химическими сдвигами, так что наложения сигналов практически не наблюдается. Однако вследствие малой чувствительности метода ЯМР-Р31 его возможности ограничены: при исследовании растворов с концентрациями менее 1Л1 очень трудно провести точные количественные измерения. Метод ЯМР успешно применен для количественного анализа смеси фосфорных оксикислот [170], а также для экспресс-обнаружения фосфорного ангидрида в поли-фосфорных кислотах [171. Можно указать на работы, посвященные исследова-ниюстепени чистоты полифосфорной кислоты [172], а также изучению продуктов, которые образуются в результате процессов перегруппировки, происходящих при смещении некоторых фосфорсодержащих соединений [173, 174]. Например, в спектре ЯМР-Р31 смеси РОС13 и РОВг3 [173], выдержанной менее недели при температуре 130°, наблюдались четыре одиночных сигнала, соответствующих равновесной смеси исходных молекул и смешанных продуктов РОС12Вг и РОС1Вг2 (табл. 12.71)1 Аналогичные результаты получены при исследовании смеси PSC13 и PSBr3 (табл. 12.71). Сравнивая интенсивности сигналов резонансного поглощения, можно определить относительные количества каждого типа молекул в равновесной смеси, что позволяет установить вероятностный характер исследуемого процесса перегруппировки. Этим методом изучен ряд других систем: РС13 — РВг3, Н3РО4 — РОС13, Н2О — HF — Р2О5 [175, 176], CH3P(O)F2 — СН3Р(О)С12 [277]. На рис. 12.39 приведен спектр ЯМР-Р31 при 40,5 Мгц (23 500 э) равновесной смеси CH3P(O)F2, CH3P(O)FC1 и СН3Р(О)С12 [277]; сигнал CH3P(O)F2 представляет собой триплет (JP_F =
Спектры ЯМР других ядер (кроме водорода и фтора)
383
Таблица 12.71
ХИМИЧЕСКИЕ СДВИГИ Р31 ДЛЯ РЯДА СМЕШАННЫХ ФОСФОРИЛГАЛОГЕНИДОВ И ИХ СЕРУСОДЕРЖАЩИХ
АНАЛОГОВ [173]
Соединение 6 a, M. Д. Соединение 6 a, M. Д.
POC13 —2,2 PSC13 —28,8
POCl2Br + 29,6 PSCl2Br + 14,5
POClBr2 + 64,8 PSClBr2 + 61,4
POBr3 + 103,4 PSBr3 + 111,8
а Относительно 85%-ной Н3РО4
как внешнего эталона.
= 1100 гц) квартетов (/р-н = 20 гц); CH3P(O)FC1 — дублет (/р_р = 1133 гц) квартетов (7р_н = 17 гц); СН3Р(О)С12— одиночный квартет (JP_F = 17 гц).
Нециклические фосфонитрильные соединения. На рис. 12.41 представлены спектры ЯМР-Р31 двух нециклических фосфонитрильных производных.
о
CI
CI—P=N
CI
н
Cl CI
I I
-?A=N-p=°
Cl Cl
— 1 —i___________________1_________
1,9 13,6 29,6
Химический сдвиг от сигнала 85°/0-ной Н3РО4, м.З
Рис. 12.41. Спектр ЯМР-Р31 двух фосфонитрилгалогенидов при
16,2 Мгц [177].
В обоих случаях расшифровка спектров не встречает затруднений [177]. Если химический сдвиг для ядра РА в нециклическом фосфонитриле I (см. приложение V) сравнить со сдвигом для атома фосфора при аналогичном окружении в циклическом фосфонитриле II (б = —19 м. д.), то находим, что в результате циклизации экранирование кольцевых ядер фосфора уменьшается на 40 м. д.
С1 С1 С1
Cl—P = N—PA = N —Р = О
Cl Jl Cl
N z \ C12P PC12 I II N N N /
P ci2 и
384
Глава 12
Константы спин-спинового взаимодействия Р31 — Р31 в тех немногих нециклических молекулах со связью Р — N, которые к настоящему времени были исследованы [177, 250], находятся в пределах 15—45 гц. Данные по химическим сдвигам ЯМР-Р31 для большого числа соединений со связью Р — N приведены в работах [281, 283[. !
12.7. Кобальт
Единственный естественный изотоп кобальта Со59 обладает высоким магнитным моментом и мог бы быть идеальным ядром для исследования методом ЯМР, если бы он не имел большого электрического квадрупольного момента = . Несмотря на это обстоятельство, было успешно выполнено несколько
работ по резонансу Со59 и получены интересные сведения об экранировании тяжелых ядер [102, 121, 123, 178, 184]. Релаксационные процессы, обусловленные квадрупольным взаимодействием, обычно не настолько эффективны, чтобы вызвать уширение линии, превосходящее неоднородность магнитного поля [ 178]. Первые измерения химических сдвигов Со59 (см. табл. 12.72) выполнили
Таблица 12.72
ХИМИЧЕСКИЕ СДВИГИ СоМ НЕКОТОРЫХ КОМПЛЕКСОВ
КОБАЛЬТА [102]
Соединение б, м. д.
К3Со(СМ)6 (эталон) 0
Co[C2H4(NH2)2]3C13 7 300
Na3Co(NO2)6a 7 400
8 100
Co(NH3)6C13 8 300
К3Со(С2О4)3 13 000
а Для Na3Co(NO2)e обнаружено два сигнала СоМ с соот-ношенном интенсивностей 1:16; это явление было объяснено существованием молекул в двух изомерных формах. С изменением температуры соотношение интенсивностей сигналов изменялось. Гассер и Ричардс [178] исследовали равновесие, устанавливающееся между изомерами,' в широком диапазоне температур.
Проктор и Ю [102]. Очень широкий диапазон изменения экранирования ядер кобальта (13-103 м. д.) свидетельствует о преобладании парамагнитной составляющей [102, 131].
12.7.1. Химические сдвиги Со69
Гриффит и Оргел [123] применили для расчета парамагнитного вклада в экранирование ядер Со59 теорию кристаллического поля. Они показали, что этот вклад обусловлен смешиванием волновых функций низколежащих возбужденных электронных уровней и основного состояния кобальта под влиянием магнитного поля. Вычисленные изменения химического сдвига для ряда комплексов трехвалентного кобальта, вызванные парамагнитной составляющей экранирования, по порядку величины совпадают с наблюдаемыми. Если воспользоваться приближением теории сильного поля, то с основным состоянием смешивается лишь 1Т1й-состояние с конфигурацией t2geg. Энергия Tlg-состояния зависит от природы лиганда, поэтому разность энергий основного и возбужденного 1Т1й-состояний (АЕ) зависит от природы лиганда. Величина парамагнитной составляющей экранирования определяется, как извест
Спектры ЯМР других ядер (кроме водорода и фтора)
385
но [72, 181], соотношением
<тпаРа = зл1с2ДБ / <° I £2 1 °>, из которого следует, что ее величина обратно пропорциональна разности энергии Л£. Следовательно, для комплексов кобальта е лигандами, вызывающими слабые поля, парамагнитная составляющая экранирования ядра Со59 должна быть наибольшей. Действительно, в ряду комплексов трехвалентного кобальта с амином, оксалатом, этилендиамином и цианидом было найдено хорошее качественное соответствие между химическими сдвигами Со59 и энергиями уровней 1T1g [123]. Если при этом принять во внимание оценочные значения величины , то соответствие улучшается. Теория Гриффита и Оргела разрабатывалась в предположении, что диамагнитная составляющая экранирования ядер Со59 мала и ее изменениями можно пренебречь. Чтобы еще раз рассмотреть точку зрения Гриффита и Оргела об экранировании кобальта, Фриман, Меррей и Ричардс [179] провели параллельное изучение спектров магнитного резонанса Со59 и электронных спектров широкого ряда комплексов Со(Ш). Данные, относящиеся к химическим сдвигам Со59 и максимумам электронного поглощения изученных ими соединений, приведены в табл. 12.73.
Таблица 12.73
РЕЗОНАНСНЫЕ ЧАСТОТЫ Со69 и ЭЛЕКТРОННЫЕ МАКСИМУМЫ ПОГЛОЩЕНИЯ ДЛЯ НЕКОТОРЫХ СОЕДИНЕНИЙ КОБАЛЬТА (В ПОЛЕ 4370,9 а) [179]
Соединение а Резонансная частота, Мгц Положения максимума поглощения света, ммк
K3[Co(CN)e] Li[Co(NO2)4(NH3)2] [Со(еп)2С12]С1 (транс) [Со(СО3)(еп)2]Вг [Со(еп)3]С13 [Co(NO2)2(NH3)4]Cl (транс) [Со(рп)3]С13 [Co(NO2)2(NH3)4]Cl (цис) Na3[Co(NO2)6] < [Co(NH3)6]Cl3 [Co(CO3)(NH3)4]NO3 | [Со(асас)3] (в бензоле) К3[Со(С2О4)3] [Co3(CO3)3](NO3)3 4,4171 4,4478 4,4485 4,4486 4,4488 4,4489 4,4490 4,4493 4,4502 (сильный) 4,4527 (слабый) 4,4534 4,4575 4,4601 4,4731 4,4747 4,4795 311 426 505 512 470 445 470 450 480 475 | 512 597 610 645 259 347 358 340 340 358 338 425 444
а Сокращения еп, рп и асас означают этилендиамин, пропилендиамии и ацетилацетои соответственно.
В теории кристаллического поля [180] все эти комплексы Со(Ш) относятся к типу комплексов «сильного поля», где 5с(-орбитали ионов Со(Ш) расщепляются на трижды вырожденную орбиталь t2g и дважды вырожденную орбиталь eg, причем энергия последней на А£ выше, чем энергия первой орбитали. Электронная оболочка комплекса в основном состоянии имеет конфигурацию 4g, а в нижнем возбужденном состоянии — 4g^g. Энергия возбуждения при переходе из синглетного основного состояния в одно из двух возможных синглетных 25-489
386
Глава 12
возбужденных состояний tlgSg, а именно 1Tlg или 17"2Й, в обоих случаях составляет примерно Приближенное значение Л£ можно определить из электронных спектров комплексов Со(III). Если построить зависимость частоты резонанса на ядрах Со59 от длины волны наиболее низкочастотного максимума поглощения света, то наблюдается, как и предсказано теорией Гриффита
Рис. 12.42. Зависимость между химическими сдвигами ЯМР-Co59 и длинами волн низкочастотного максимума поглощения комплексов Со(Ш) [179].
О симметричные комплексы, ф несимметричные комплексы.
и Оргела, хорошая линейная корреляция (см. рис. 12.42), причем химические сдвиги симметричных комплексов лучше ложатся на прямую, чем несимметричных.
Дарматти и Канекер [182] с помощью метода Гриффита и Оргела [123] также вычислили парамагнитные вклады в экранирование Со59 для ряда комплексов Со(Ш). Оказалось, что парамагнетизм второго порядка изменяется в зависимости от лигандов в последовательности: ацетилацетонат-ион > окси-нат-ион > СО9- > Cl- > СО” > Н2О > NH3 > этилендиамин > дипиридил > > NO~ > CN-. В табл. 12.74 приведены вычисленные и измеренные химические сдвиги Со59 для ряда комплексов Со(Ш): согласие между этими значениями вполне удовлетворительное. Как и предсказывалось теорией, парамагнитная составляющая экранирования уменьшается по мере роста поля, обусловленного лигандом.
Влияние растворителей на химические сдвиги Со59. Положение сигнала ЯМР-Co69 ацетилацетоната кобальта зависит от растворителя. Однако установить корреляцию между величиной изменения химического сдвига и свойствами растворителя (например, диэлектрической проницаемостью) не удалось [179]. В сравнении с химическими сдвигами, обычно наблюдаемыми для ЯМР-Co59, сдвиги, вызываемые растворителями, пренебрежимо малы.
Температурная зависимость химических сдвигов Со59. Проктор и Ю [102] исследовали температурную зависимость химических сдвигов Со59 в соединениях КзСо(С1Ч)в, Co[C2H4(NH2)2]3C1 и Co(NH3)6C13 в диапазоне 20—80°. Аналогичные результаты получены также для других комплексов Со(Ш) [179, 121]. Например, для растворов комплекса трцс-ацетилацетоната кобальта(Ш) в хлороформе наклон кривой температурной зависимости составляет 2,97 м. т\.1град. Первоначально считали, что роль температуры сводится к изменению заселенности низколежащих возбужденных уровней энергии [102, 181]. По мнению Гриффита и Оргела [123], это объяснение является неубедительным. Они предположили, что с ростом температуры изменяется
Спектры ЯМР других ядер (кроме водорода и фтора)
387
Таблица 12.74
ХИМИЧЕСКИЕ СДВИГИ Со69 КОМПЛЕКСОВ Со(Ш) [182]
Соединение а ДЕ 6, CM~l Химический сдвиг б, м. д.
иабл. в рассчит.
[Co(NH3)6]Cl3 21 000 —8 080 —8 200
[Co(NH3)6CO3]NO3 19 670 —9 000 —9 800
[Co(NH3)6C1]C12 18 720 —9 070 — 11 100
[Co(NH3)5NO2]C12 21 840 —7 460 —7 300
[Co(NH3)4CO3]NO3 19 060 —9 070 — 10 600
i]uc-[Co(NH3)4(NO2)2]NO3 22 510 —6 880 —6 600
транс- [Co(NH3)4(N O2)2 ] NO3 22 630 —7 150 —6 500
Na3[Co(NO2)6] 20 670 —7 350 —8 500
Со(ЫНз)3(ЫО2)з 23 210 —6 940 —6 000
[Co(en)3]Cl3 21 400 —7 010 —7 700
транс- [Co(en)2(NO2)2 ] NO3 23 300 —6 350 —5 900
i]uc-[Co(en)2(NO2)2]NO3 23 000 —6 470 —6 100
[Co(dipy)3](ClO4)3 22 230 —6 620 —6 900
Co(acac)3 K3[Co(CN)e] 16 900 — 12 300 0 — 14 000
а еп — этилендиамнн, dipy — 2,2-дипиридил.
® ДЕ — разница энергий между основным и возбужденным (конфигурации dG) состояниями.
в Относительно K3Co(CN)e как внешнего эталона.
заселенность более высоких колебательных уровней. Это приводит к уменьшению степени смешивания низшего возбужденного энергетического состояния 1T1g с основным состоянием, и в результате происходит возрастание частоты резонансного поглощения Со59.
Армстронг и др. [184, 314] изучили химические сдвиги Go59 при изменении не только температуры, но и давления. По их мнению, давление влияет лишь на А£, а влияние температуры связано с термическим расширением комплекса.
12.7.2. Исследование равновесных процессов с участием кобальта
Гассер и Ричардс [178] изучили скорость обмена между комплексом трехвалентного кобальта с этилендиамином
Co(NH3)|+ + 3en Coen|+-j-6NH3.
В спектре было обнаружено два резонансных сигнала Со59, которые отвечают ионам Co(NH3)’+ и Со еп|+. Изменения интенсивностей этих сигналов во времени позволили определить константу скорости процесса. Измерение константы скорости при разных температурах дало возможность найти энергию активации процесса. В растворах комплексов Со(Ш) оба процесса — электронный и лигандный обмен — протекают довольно медленно [178].
12.8. Олово
Магнитным моментом обладают три изотопа олова: Sn115 (естественное содержание 0,35%), Sn117 (7,67%) и Sn119 (8,68%). Спин каждого из этих ядер равен 1/2, и, следовательно, квадрупольным моментом они не обладают. Оба наиболее распространенных изотопа имеют примерно одинаковые магнитные моменты, поэтому их характеристическая чувствительность для сигнала ЯМР
25*
388
Глава 12
также одинакова. Однако изотоп Sn119 имеет более высокое естественное содержание, чем Sn117, и поэтому его сигнал более интенсивен. Вследствие этого обычно используют при изучении спектров ЯМР олова изотоп Sn119.
Выбор оптимальных условий наблюдения резонанса зависит в основном от времени релаксации исследуемого ядра [185, 186]. Оказалось, что резонансный сигнал для многих оловосодержащих соединений можно наблюдать на осциллографе, если использовать высокую скорость прохождения, настройку на сигнал дисперсии и большую высокочастотную мощность (величина Hi от 2 до 50 мэ). Запись резонансного сигнала можно вести как в режиме дисперсии (при быстром прохождении, например для SnCl2 -2Н2О, или медленном прохождении, например для SnCl4), так и в режиме поглощения (SnBr4). Для обеспечения необходимой чувствительности обычно используют большие ампулы с наружным диаметром 15 мм (вращать их не пытались).
Для ряда соединений олова (например, для твердых SnBr4 и Snl4, а также для насыщенных водных растворов SnCl4 и SnF4 [186]) резонанс Sn119 наблюдать не удалось вследствие того, что сигналы оказались широкими и слабыми по пиковой интенсивности. Если обогатить SnCl2 -2Н2О изотопом Sn119 (~65% изотопа Sn119), то можно получить достаточно интенсивный сигнал даже при ампуле диаметром 3 мм и использовать его в качестве внешнего эталона [186]. Однако обычно для этой цели применяется тетраметилолово, поэтому приводимые ниже химические сдвиги даны относительно Sn(CH3)4 как внешнего эталона.
12.8.1. Химические сдвиги Sn119
В табл. 12.75 приведены химические сдвиги Sn119 для ряда неорганических и органических оловосодержащих молекул [185, 186]. Эти измерения были выполнены Лаутербуром и Бурке в плане более общего исследования элементов группы IVB. Чтобы учесть погрешности, связанные с несимметричностью сигналов, измерения химического сдвига проводились путем усреднения результатов, полученных при прохождении в сторону увеличения и уменьшения напряженности поля. Большой диапазон изменения химических сдвигов Sn119 свидетельствует о том, что экранирование ядра олова определяется главным образом парамагнитной составляющей.
Рассмотрение данных табл. 12.75 свидетельствует о том, что соединения двух- и четырехвалентного олова имеют сигналы в одинаковой области спектра ЯМР. Например, химические сдвиги SnSO4, SnCl2 и SnCl2 -2Н2О находятся в той же области, что и химические сдвиги соединений четырехвалентного олова. Для соединений ЗпС1жВг4_ж наблюдается линейная зависимость химического сдвига Sn119 от числа атомов хлора, сходная с аналогичной зависимостью для углеродных соединений такого же типа (см. разд. 12.2.3). Можно считать, что каждый галогенный атом (С1, Вг или I), присоединенный к атому олова, вносит определенный, независимый от других заместителей вклад в общий химический сдвиг Sn119. Знание этих вкладов дает возможность предсказывать химические сдвиги для смешанных галогенидов олова. Например, вычисленный химический сдвиг Sn119 для молекулы SnClBrI2 равен 1046 м. д. относительно тетраметилолова, что хорошо согласуется с экспериментальным значением 1068 м. д.
Влияние растворителя на химические сдвиги Sn119. Растворитель сильно влияет на оба параметра спектров Sn119 — химический сдвиг и константу спин-спинового взаимодействия ядра Sn119 с каким-либо другим ядром [186]. Причиной этого влияния считают способность соединений олова вступать в координационную связь с молекулами многих растворителей. Например, константа взаимодействия JgnH для соединения (CH3)2SnCl2 в водном растворе равна 98 гц, тогда как в ацетоне она уменьшается до 80 гц. Столь же ощутимо влияние растворителя отражается на химическом сдвиге Sn119 соединения
Спектры ЯМР других ядер (кроме водорода и фтора)
389
Таблица 12.75
ХИМИЧЕСКИЕ СДВИГИ Snll9 НЕКОТОРЫХ СОЕДИНЕНИЙ [185, 186]
Соединение 6 a, M. Д.
Snl4 (в сероуглероде) 1701
SnSO4 (в воде) 909
SnBr4 638
Na2[Sn(OH)6] (в воде) 592
K2[Sn(OH)e] (в воде) 590
SnCl2-2H2O (в 4,85 М растворе в воде) 521 6
SnCl2 (в тетрагидрофуране) 236
(w-C4H9)2Sn(OOCCH3)2 195
SnCl4 150
(w-C4H9)4Sn 12
H-C4H8SnCl3 3
(CH3)4Sn 0
(CH3)2SnCl2 (в ацетоне) —36
(C2H5)2SnCl2 (в ацетоне) —62
(H-C4H9)2SnCl2 (в ацетоне) —71
(H-C4H9)2SnCl2 (в сероуглероде) —114
(w-C4H9)2SnS —124
(«-C4H9)3SnCl —143
(C2H5)3SnCl —151
а Точность ±2 м. д. — химический сдвиг измерен относительно Sn(CH3)4 как внешнего эталона.
б Зависит от концентрации и pH; сигнал можно сместить в слабое поле на 200 м. д. добавлением НС1.
SnCl2-2H2O. Кроме того, найдено, что для растворов в ацетоне и в метаноле химический сдвиг зависит от концентрации соединения. 'Об образовании комплексов SnCl2 -2Н2О с галогеноводородом свидетельствует тот факт, что химический сдвиг Sn119 раствора SnCl2 -2Н2О в соляной кислоте находится в прямой зависимости от ее нормальности (диапазон изменений химического сдвига ~200 м. д.).
12.8.2. Смешанные галогениды Sn(IV)
При смешении двух галогенидов олова, например SnBr4 и Snl4, уже через несколько часов наступает полное перераспределение галогенных заместителей,
н
Рис. 12.43. Спектр ЯМР-Sn119 эквимолекулярной смеси (1 : 1) SnBr4 и Snl4 при 8,5 Мгц [186].
в результате чего образуются смешанные галогениды типа SnBr3I, SnBr2I2 и SnBrI3 [186]. В спектре ЯМР-Sn119 смеси исходных соединений в молярном соотношении 1 : 1 (см. рис. 12.43) видны резонансные сигналы каждого из
390
Глава 12
смешанных галогенидов, почти одинаково удаленные друг от друга. Измерение интенсивности сигналов позволяет оценить относительное содержание галогенидов олова в образце. Оказалось, что наблюдаемые интенсивности хорошо
Экспериментальный спектр
। 500
1000 1500 мЛ
Вычисленный спектр
SnCljBr SnCl2Br2-SnClBr,-SnCl3I SnBr4 SnCl2Br< SnClBr2I
Snl4 SnBrI3 SnCTIj SnBr2I2 SnClBrI2 SnCT2I2 SnBr5I
Рис. 12.44. Спектр ЯМР-Sn119 смеси SnCl4 — SnBr4 — Snl4 (2 : 1 : 1) при 8,5 Мгц [186].
Химические сдвиги измерены относительно (CHsl.Sn как внешнего эталона.
согласуются с количествами, которых следовало ожидать при случайном обмене заместителями.
Химические сдвиги Sn119 для многих других смешанных галогенидов олова были измерены путем подбора соответствующих исходных смесей. Полученные результаты приведены в табл. 12.76; на рис. 12.44 показан спектр Sn119 смеси SnCl4, SnBr4 и Snl4. Атомы галогенов в смешанных галогенидах
ХИМИЧЕСКИЕ СДВИГИ S11119 РЯДА
Состав исходной смеси, мольная доля Химические сдвиги компонентов
SnCl4 SnBr4 Snl4 SnCl4 SnClgBr SnCl2Br2 SnClBrg SnCl3I SnBr4
0 0 1
0 0,5 0,5 — — — — —
0 0,75 0,25 — — — — — 638
0 1 0 — — — — — 638
0,25 0,75 0 384 509 — 634
0,5 0,5 0 265 387 509 — 635
0,75 0,25 0 260 386 —
1 0 0 150 — — — — —
0,75 0 0,25 — — — 551 —
0,5 0 0,5 — — — 557 —
0,25 0,25 0,5
0,25 0,5 0,25 508 629
0,5 0,25 0,25 267 543
а Относительно тетраметилолова как внешнего эталона.
Спектры ЯМР других ядер (кроме водорода и фтора)
391
олова обмениваются довольно быстро, причем сам факт, что в спектре наблюдаются отчетливые резонансные сигналы для каждого типа молекул, позволяет оценить нижний предел времени жизни молекул между актами обмена (> 10-3 сек). Попытки проследить за ходом реакции перераспределения в смеси SnCl4 — SnBr4 оказались безуспешными, следовательно, верхняя граница времени жизни равна примерно 10 сек.
12.8.3. Константы спин-спинового взаимодействия Sn119
В табл. 10.95 приведены значения констант взаимодействия SnH, полученные из спектра ЯМР станнанов и родственных молекул [234]. По мере замещения атомов водорода на метильные группы в соединениях общей формулы (CH3)xSnH4_x взаимодействие непосредственно связанных ядер Sn и Н уменьшается (для SnH4 JsnH = 1.931 гц; для (CH3)3SnH JsnH = 1744 гц), что следует объяснять [234] уменьшением s-характера орбиталей олова в связи Sn — Н при увеличении числа метильных групп (контактное взаимодействие Ферми, определяющее Jsnii, пропорционально s-характеру гибридной орбитали олова, используемой при образовании связи Sn — Н).
Наличие в спектрах ЯМР-Sn119 тонкой структуры вследствие спин-спинового взаимодействия — явление довольно частое, хотя точно измерить константу этого взаимодействия легче из спектров Н1 или F19. В спектре ЯМР-Sn119 тетраметилолова (рис. 12.45) ясно видны 9 из 13 ожидаемых линий мультиплета с JgnH = 54 гц [50, 264].
Если взаимодействующие ядра Sn119 и Н1 разделены двумя о-связями, то константа /зпш-с-т имеет величину порядка 30—100 гц. Примечательно, что взаимодействие через три связи (Sn119 — С — С — Н1) обычно сильнее, чем через две связи. Так, например, в молекуле тетраэтилолова константы JsnH имеют следующие величины:
^8п-сн3= — 71>2 гЧ и 7sn-CH2 = +32,2 гц.
Аналогичное явление наблюдается также для других металлал-килов.
Таблица 12.76
СМЕШАННЫХ ГАЛОГЕНИДОВ ОЛОВА [186]
системы а, м. д.
SnC^Brl SnClBr2l SnBrgI SnClglz SnClBrI2 SnBr2l2 SllClIg SnBrIg Snl4
1701
— — 916 — — 1187 — 1447 1698
— — 919 — — 1192 —
— — — — —- — — — —
— — — — — — — — —
— — — — — -— — — —
— — — — — — — — —
— — — 951 — — 1347 —
— — — 955 — — 1342 — 1712
672 796 919 947 1068 1189 1330 1449 1696
666 789 913 1063 1195
663 783 937 1060
392
Глава 12
Спектр Sn119 («-C4H9)4Sn вследствие спин-спинового взаимодействия Sn — Н представляет собой широкий резонансный сигнал [186].
В работе [187] исследован протонный спектр тетравинилолова. Детальный анализ тонкой структуры спутников Sn119 (система АВС) показывает, что константа взаимодействия ядра олова с транс-протоном больше, чем с гем- или цис- протонами:
c/Sn(CH--= СН2)3 ^SnH<i> = + 90,6 гц,
(2)Н// ^H(3) ^SnH<2>= + 183,1 гц,
JSnH<3>= +98,3 гц.
Константы взаимодействия SnH между ядрами, разделенными одним атомом углерода, изучались в ряду метильных производных олова и родственных соединений [188, 256]. Эти константы, полученные из протонных спектров,
5п(сн3)4
Рис. 12.45. Спектр ЯМР-Sn119 тетраметилолова при 8,50 Мгц (сигнал дисперсии в условиях быстрого прохождения) [50].
приведены в табл. 12.77 (как для чистых жидкостей, так и для растворов в различных средах). Изменение значений Jgn-c-н в ряду исследованных соединений обусловлено изменением гибридизации атома олова. Найдено, что константа взаимодействия линейно зависит от доли s-характера связи, что согласуется с результатами изучения дальнего взаимодействия Jcis-c-hi (см. разд. 12.2.18).
12.9. Таллий
Оба природных изотопа таллия Т1203 и Т1205 (с распространенностью 29,52 и 70,48% соответственно) имеют магнитные моменты и удобные для ЯМР-иссле-дований спиновые числа (7 — 1/2)- Величины магнитных моментов обоих ядер примерно одинаковы и, следовательно, характеристические чувствительности метода ЯМР на этих ядрах также сравнимы по величине. Поэтому изучение резонанса на ядре таллия чаще всего проводят с более распространенным изотопом Т1205. При этом обычно используют фазовое детектирование резонансного сигнала, несмотря на довольно высокую чувствительность (19,2% чувствительности для ядер водорода при той же напряженности поля).
Спектры ЯМР других ядер (кроме водорода и фтора)
398
Таблица 12.77
КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ Sn — Hl НЕКОТОРЫХ МЕТИЛЬНЫХ СОЕДИНЕНИЙ ОЛОВА [188]
Соединение а t, °C Константа взаимодействия®» гц
Snll7 — СН3 Snll»- сн3
(CH3)4Sn 31 51,5 54,0
51% ССЦ 31 51,4 53,8
5% СС14 31 51,6 53,7 54 [186]
(CH3)3SnCl 40 57,4 59,7
38% ССЦ 31 56,0 58,5
62% С6Н6 31 56,5 59,2
56% Н2О 31 65,2 68,4
(CH3)2SnCl2 ПО 68,0 71,0
Насыщ. < 14% ССЦ 31 66,6 69,7
47% Н2О 31 97,4 101,9 98 [186]
CH3SnCl3 55 95,7 100,0
46% ССЦ 31 95,3 99,5
51% СвН6 31 95,4 99,9
49% Н2О 31 125,4 131,1
а Для каждого соединения в первой строке приведены результаты для чистой жидкости, затем для раствора (указаны весовые проценты).
^Точность ±0,5 %.
12.9.1. Химические сдвиги ЯМР-Т1205
Роуленд и Бромберг [189[ с помощью ЯМР-Т1205 исследовали изменения электронного окружения ядер одновалентного таллия при изменении агрегатного состояния некоторых солей (нитрата, хлорида и бромида). Им удалось обнаружить химический сдвиг таллия при переходе от твердого состояния к расплаву (при температуре несколько выше точки плавления) для трех ионных солей таллия — нитрата, хлорида и бромида. Оказалось, что в твердой фазе ядра таллия более экранированы, чем в расплаве:
для T1NO3 на 132 м. д.
для Т1С1 на 596 м. д.
для Т1Вг на 558 м. д.
Была найдена корреляция между изменением химического сдвига галогенидов таллия и электроотрицательностью ионов галогена. Полагают, что в расплаве эти соли состоят из свободных ионов таллия и галогена, и поэтому электронное окружение ядер таллия в обоих соединениях одинаково. В твердом состоянии частично ковалентный характер связи Т1Х отвечает переносу некоторого-заряда. Поэтому очевидно, что разница в экранировании коррелируется с электроотрицательностью ионов галогена.
В спектре ЯМР-Т1205 расплава комплексных солей Т12С14 и Т12Вг4 обнаружены два отдельных сигнала поглощения, которые были отнесены к ионам одно- и трехвалентного таллия (см. рис. 12.46). Этот факт подтверждает существование комплексных галогенидов таллия в виде двойных солей Т1Х-Т1Х3 (что ведет к наличию в расплавленном состоянии ионов Т1+ и Т1Х~). Можно-было бы ожидать, что при расплавлении будет происходить быстрый обмен
394
Глава 12
между обоими валентными состояниями таллия, однако наличие отдельных резонансных сигналов исключает это предположение. Изучение данной системы при различных температурах показывает, что при 500° среднее время жизни соединения в каком-либо одном валентном состоянии составляет около 10~5 сек. Интересно отметить, что в жидкой фазе ионы трехвалентного таллия
TLg CL4
Т1(Ш)
I TUD
2390М.Э.
Рис. 12.46. Спектр ЯМР-Т1206 (производная сигнала поглощения) расплава Т12С14 при 289°; частота резонанса 25,16 Мгц [189].
менее экранированы, чем одновалентного, а ионы одновалентного таллия в свою очередь в жидкой фазе менее экранированы, чем в твердой. В растворе ионы одновалентного таллия также более экранированы, чем трехвалентного, по-видимому, вследствие того, что в трехвалентном состоянии ядро таллия окружено меньшим числом электронов [190]. Сигналы резонанса Т1205, наблюдаемые в расплаве Т12С14 при 289°, приведены на рис. 12.46 [189].
Таблица 12.78
ХИМИЧЕСКИЕ СДВИГИ Т1205 РЯДА НЕОРГАНИЧЕСКИХ СОЛЕЙ ТАЛЛИЯ
Соединение б а, м. д.
Т1С1О4 +3 700 б
T1NO3 - —2 800
Т1С1 —6 100
T12SO4 —900
НСООТ1 — 11 100
Т12СОз — 11 200
T1F —7 900
Т1Вг — 10 800
тп —23 300
Кз(Т1С16) —22 300
(NH4)3(T1C16) —22 300
Zn(TlCl4)2 —29 700
TINO3 (0,3 М водный раствор; 0,0
эталон)
а Относительно 0,3 М водного раствора нитрата таллия(1).
б Положительная величина означает более сильное экранирование по сравнению с эталонным соединением.
Химические сдвиги Т1205 (табл. 12.78) измерены для ряда неорганических солей таллия в твердом состоянии [191]. В спектре твердого Т12С13 наблюдаются два резонансных сигнала с соотношением интенсивности 3:1, что согласуется с формулой этого соединения Т13(Т1С16) [191]. Спектр гидратирован
Спектры ЯМР других ядер (кроме водорода и фтора)
395
ного хлорида таллия состоит из трех сигналов, что можно объяснить присутствием ионов Т1(Н2О)х+, TICI7 и Т1С1|л При добавлении к водным растворам хлорида и бромида трехвалентного таллия растворов галогенидов наблюдаются большие изменения химических сдвигов Т1205, что было объяснено образованием комплексных ионов. Более подробно проблема комплексообразования, в том числе ионов таллия [190, 191, 195], рассмотрена в разд. 9.3.
Фиггис [190] также изучал влияние анионных добавок на химические сдвиги ЯМР-Т1205 солей трехвалентного таллия в растворе. Область изменения химических сдвигов (~1800 м. д.) не перекрывается с областью химических сдвигов солей одновалентного таллия. Столь большой диапазон изменения химических сдвигов таллия позволил заключить, что преобладающую роль в экранировании ядра таллия играет парамагнетизм второго порядка [190].
12.9.2. Спин-спиновое взаимодействие Т1205—Н1
Впервые косвенное спин-спиновое взаимодействие ядер таллия и водорода наблюдалось в спектрах протонного резонанса водных растворов диалкил-и диарилталлиевых катионов [192, 312]. При комнатной температуре в протонных спектрах триметил- и триэтилталлия тонкая структура не обнаруживается: вследствие быстрого обмена алкильными группами в спектре этих соединений наблюдается один широкий резонансный сигнал. Однако при охлаждении раствора от —60 до—100° (растворитель—-хлористый метилен) скорость обмена алкильными группами снижается настолько, что в спектре появляется тонкая структура в результате спин-спинового взаимодействия Т1 — Н. В протонном спектре охлажденной смеси триметил- и триэтилталлия наблюдались сигналы смешанных алкилов Т1(СН3)2(С2Н5) и Т1(СН3)(С2Н5)2. Это свидетельствует о том, что в исходных соединениях возможен межмолекулярный обмен алкильными группами. Интересно, что обмен метильными группами в (СН3)3Т1 подчиняется кинетическому уравнению второго порядка [263]. В табл. 12.79 приведены константы взаимодействия Т1 — Н для некоторых
Таблица ‘12.79
КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ Т1205 — hi НЕКОТОРЫХ ТРИАЛКИЛОВ ТАЛЛИЯ
Соединение сн3 С2Н6
JT1H’ г'< JT1 - сн2> Лт - сн3- гц
Т1(СН3)3 250,8
T1(CHS)2(C2H6) 223,0 242,4 472,7
Т1(СН3)(С2Н5)2 186,9 218,8 441,5
Т1(С2Н6)з 198,2 396,1
триалкильных соединений таллия. Кроме того, измерены константы спин-спинового взаимодействия Т1 — Н для некоторых галогенопроизводных (СН3)2Т1Х [285].
Следует указать, что во всех изученных этильных производных Т1 взаимодействие Т1 — С — С — Н сильнее, чем Т1 — С — Н (см. разд. 10.3.2). С помощью двойного резонанса было показано, что для катиона диэтилталлия константы этих взаимодействий имеют противоположные знаки [192]. По-видимому, это справедливо и для (С2Н5)3Т1.
Взаимодействие Т1205 — Н1 изучалось также для фенильных и винильных производных таллия [132] (см. разд. 10.8.2). В спектре (С6Н5)3Т1 наблюда
396
Глава 12
лись следующие константы взаимодействия:
J”Р™° = + 259 гч; Jtih“ = ± 80 гЧ; = ± 35 гч.
Соответствующие константы для ионов (С6Н5)2Т1+ и (С6Н5)Т12+ имеют существенно более высокие значения [132].
12.9.3. Структура этилата таллия Т1ОС2Н5
Исследование ЯМР-Т1203 и ЯМР-Т1205 жидкого, неразбавленного этилата таллия подтвердило, что это соединение имеет тетрамерную структуру [193].
Тот факт, что в резонансных спектрах Т1203 и Т1205 наблюдается взаимодействие между ядрами Т1203 и Tl205 (J = 2560 гц), также свидетельствует о полимерном строении молекулы. Теоретические спектры Т1203 и Т1205 этилата таллия, полученные наложением спектров тетрамерных молекул, содержащих различные комбинации изотопов Т1 (с учетом их естественного содержания), совпали с наблюдаемыми [194]. Спин-спиновое взаимодействие Т1 — Н отсутствует.
12.10. Другие ядра
12.10.1. Дейтерий
Большинство исследований, посвященных ядерному резонансу на дейтерии, было выполнено с помощью ЯМР на тех ядрах молекулы, с которыми дейтерий взаимодействует. Например, в случае HD константа косвенного спин-спинового взаимодействия была измерена Уимметом [196] в спектре ЯМР-Н1 (Лтп = 43 ± 0,5 гц). Стефен [197] вычислил эту константу с использованием методов валентных связей и молекулярных орбиталей. Полученное значение 49 гц хорошо согласуется с наблюдаемой величиной. Теоретический )асчет константы /но различными методами был выполнен также Ишигуро 198], который получил значение 35,2 гц (обсуждение расчетов см. в разд. 5.1). 3 работе [199] спектр ЯМР-Н1 монодейтеробензола проанализирован как система спинов АВВ'СС'Х. Экспериментально наблюдаемый спектр соответствует константам взаимодействия /дх = 0,1 гц\ /вх = 0,34 гц и Jex = = 1,21 гц. Для случая, когда, помимо одного атома дейтерия, в молекуле имеется несколько атомов водорода, показано, что в спектре протонного-резонанса обычно наблюдается лишь одна «кажущаяся» константа взаимодействия JHD, причем ее значение имеет среднюю величину от всех ожидаемых констант. Тирс [200] наблюдал в спектре протонного резонанса а-дейтеротолуола изотопный эффект дейтерирования: сигнал группы CH2D представлял собой триплет с компонентами равной интенсивности (Jch2-d = 2,38 гц) и химическим сдвигом, несколько отличающимся от химического сдвига метильной группы недейтерированного толуола (разница химических сдвигов 0,015 м. д.). Еще более значительный эффект наблюдается в спектрах ЯМР-F19 дейтерированных соединений фтора [201]. Причиной подобных изотопных сдвигов, по-видимому, служит различие нулевых колебаний, которое обусловлено
Спектры ЯМР других ядер (кроме водорода и фтора)
397
различными массами изотопов (см. разд. 3.11) [202, 203]. Маршалл [204] вычислил разницу в экранировании протона для молекул Н2 и HD: полученные результаты хорошо согласуются с экспериментальными. В литературе сообщаются химические сдвиги ЯМР-Н* 2 [305] и ЯМР-Н3 [315] некоторых соединений.
12.10.2. Щелочные металлы
Натрий Na23. Исследование ЯМР-Na23 водных растворов некоторых солей натрия (NaCl, NaF, NaBr и т. д.) не выявило каких-либо различий в экранировании натрия, выходящих за пределы ошибки измерения [205] *). Однако при изменении концентрации некоторых соединений натрия (например, NaOH, NaClOi, Nal и т. д.) в спектрах ЯМР-Na23 обнаружено изменение ширины резонансного сигнала, связанное, по-вздимому, с эффектом квадрупольной релаксации. В результате значительной квадрупольной релаксации сигналы Na23 уширяются настолько, что удается исследовать лишь почти полностью ионизированные натриевые ионы Na+.
О’Рейлли [260] исследовал ЯМР-спектры Li7, Na23, Rb87, Cs133 и Nu систем щелочной металл — аммиак 2).
Рубидий Rb87 и цезий Cs133. Гутовский и Мак-Гарви [206] наблюдали химические сдвиги Rb87 и Cs133 в кристаллических галогенидах рубидия и цезия. Полученные ими данные приведены в табл. 12.80 (в качестве эталона применялся насыщенный водный раствор соответствующего хлорида). Оказалось,
Таблица 12.80
ХИМИЧЕСКИЕ СДВИГИ Rb87 И Cs133 НЕКОТОРЫХ
ТВЕРДЫХ ГАЛОГЕНИДОВ
Соль 6 a , M. Д. Соль 6 a, M. Д.
RbF —60 CsF —90
RbCl —89 CsCl — 163 •
RbBr — 129 CsBr —208
Rbl — 149 CsI —252
а Относительно
вующего хлорида.
насыщенного водного раствора соответст-
'что ион металла в растворе более экранирован, чем в твердой фазе, и что экранирование ядер Rb87 и Cs133 в кристалле возрастает по мере роста электроотрицательности галогена. Для фторидов рубидия и цезия наблюдаются очень широкие сигналы. Диапазон химических сдвигов цезия больше, чем для рубидия, и это согласуется с предположением Гутовского и Хоффмана [207] о том, что химические сдвиги возрастают по мере роста заряда ядра. Рассмотрение данных, относящихся к галогенидам щелочных металлов, показывает, что связь металл — галоген носит частично ковалентный характер, так как в противном случае химические сдвиги Rb87 и Cs133 в солях рубидия и цезия должны были бы совпадать с химическими сдвигами соответствующих свободных ионов. Ход изменения химических сдвигов от фтора к иоду в обоих случаях соответствует ожидаемому.
х) Проведено исследование термодинамических свойств и спектров ЯМР-Н1 и ЯМР-Na23 растворов едкого натра [385].— Прим. ред.
2) Измерены химические сдвиги Na23 для кристаллов соединений галогенов [362].—
Прим. ред.
398
Глава 12
12.10.3. Алюминий Al27
В табл. 12.81 приведены химические сдвиги ЯМР-Al27 для некоторых соединений. Спектры представляли собой первую производную сигнала поглощения или сигнала дисперсии [208]. Для соединений, обладающих тетраэдрической или октаэдрической симметрией относительно атома алюминия, как,
Таблица 12.81
ХИМИЧЕСКИЕ СДВИГИ А127 ДЛЯ НЕКОТОРЫХ СОЕДИНЕНИЙ [208]
Соединение ба, м. д. Характер линии и ее ширина (э) в поле 6490 э
А1(Н2О)|+ (в водном растворе кислоты) 0 Узкая (0,05)
А1(изо-С4НвО)3 (в н-гептане) —7 Узкая (0,09)
A1F2+ (KF добавлен к водному раствору А13+) — 15 Широкая (0,3)
А1216 (в этиловом эфире) —39 Узкая (0,08)
А1(ОН)у (в водном растворе основания) —80 Узкая (0,08)
А1(изо-С4Н9)С12 (чистая жидкость) —86 Широкая (3,7)
А1(ОСН3)С12 (чистая жидкость) —90 Широкая
А1С13 (в толуоле) —91 Широкая (0,3)
А1(СН3)2С1 >А1(СНз)С12 (чистая жидкость) —93 Широкая
А12Вг6 (в этиловом эфире) —96 Узкая (0,09)
А12Вг6 (в жидком Вг2) — 101 Широкая (0,3)
AIHj (LiAlH4 в этиловом эфире) — 103 Узкая (0,06)
А12С16 (в этиловом эфире) —105 Узкая (0,10)
А1(СН3)3 (чистая жидкость) — 156 Широкая (0,4)
НД1(изо-С4Н9)2 (чистая жидкость) — 162 Широкая (8,9)
А1(С2Н6)3 (чистая жидкость) —171 Широкая (1,5)
А1(изо-С4Н9)3 (чистая жидкость) —220 Широкая (5,4)
Относительно А1(НгО)3+ как внешнего эталона.
например, в случае А1(Н2О)|+, наблюдались узкие сигналы ЯМР-Al27 с шириной линии менее 0,1 э при напряженности поля 6490 э. Для молекул, не принадлежащих к кубической точечной группе симметрии относительно атома алюминия, резонансные сигналы оказались широкими. В спектрах ЯМР-А12? свежеприготовленных эфирных растворов галогенидов алюминия наблюдались узкие симметричные сигналы, что обусловлено, по-видимому, образованием димеров А12Х6, в которых окружение атомов алюминия имеет тетраэдрическую симметрию.
Спектр ЯМР-Al27 раствора ЫА1Н4 в этиловом эфире представляет собой квинтет с константой спин-спинового взаимодействия Jaih между ядром алюминия и четырьмя эквивалентными протонами приблизительно 110 гц. О’Рейлли [208] предпринял попытку оценить экранирование алюминия в ионе А1Н“ вариационным методом с использованием волновых функций, применяющихся в методах валентных связей и молекулярных орбиталей. Было получено примерное соответствие между теоретическим и экспериментальным значениями экранирования, причем предсказанная величина экранирования оказалась несколько выше полученной на опыте. При попытке применить использованную методику расчетов к более сложным молекулам не удалось получить количественного соответствия расчетных и экспериментальных величин.
В ряду галогенидов алюминия величина парамагнитной составляющей экранирования антибатна эффективному ядерному заряду на галогене и сим-батна ковалентному характеру связи металл — галоген. Как следует из экспе
Спектры ЯМР других ядер (кроме водорода и фтора)
399
римента, величины константы экранирования А127 для молекул этого типа изменяются в ряду Cl < Br < I. Таким образом, экранирование металла в основном зависит от эффективного ядерного заряда галогена, а не от ковалентного характера связи металл — галоген.
Известно, что в растворе эквимолекулярной смеси A1(NO3)3 и NaF существуют комплексные ионы типа A1FJ и A1F2+. Однако обнаружить в спектре ЯМР-Al27 отдельные сигналы этих ионов не удалось вследствие уширения линий, наблюдающегося при взаимодействии квадрупольного момента ядра А127 с градиентом асимметричного электрического поля: был обнаружен лишь одиночный широкий сигнал [209]. z
В работе [308] исследованы спектры ЯМР-Al27 некоторых комплексов триэтилалюминия *).
12.10.4. Галогены
Ядра С135, О37, Вг81 и I127 имеют ненулевые магнитные моменты, однако эти ядра мало изучались методом ЯМР, вследствие того что обладают большим квадрупольным моментом.
Хлор С135 и С137. Первые измерения химических сдвигов С135 и С137 были выполнены Проктором и Ю [102]. Оказалось, что сигналы ЯМР-Cl35 молекул Ва(С1О4)2 и НСЮ4 смещены примерно на 900 м. д. в сторону более слабого поля по сравнению с сигналом соляной кислоты.
Масуда [210] обнаружил, что химические сдвиги ЯМР-Cl35 для ряда молекул (TiCl4, VOC13, СгО2С12 и SiCl4; см. табл. 12.82) хорошо коррелируют с константами квадрупольного взаимодействия в этих соединениях [211]. Если
Таблица 12.82
ХИМИЧЕСКИЕ СДВИГИ С135 НЕКОТОРЫХ
ЖИДКИХ ХЛОРИДОВ [2 10]
Соединение
NaCl (разбавленный водный раствор) а
SiCli
СгО2С1г
VOC13
TiCk
б, м. д.
о
— 175 —585 —760 —805
а Внешний эталон для измерения химических сдвигов.
предположить, что основную роль в экранировании ядра С135 играет парамагнитная составляющая, то химический сдвиг С135 можно рассматривать как меру делокализации р-электронов при образовании химической связи С1. В этом случае корреляция между химическим сдвигом С135 и константой квадрупольного взаимодействия оказывается вполне естественной, если учесть, что, как показали Таунс и Дейли [212], ядерное квадрупольное взаимодействие в молекуле зависит от степени участия валентных электронов в р-орбитали.
В работе [213] исследована зависимость химического сдвига ЯМР-Cl35 хлорида двухвалентного кобальта от его концентрации в водных растворах. Увеличение концентрации как иона Со(П), так и С1~ приводит к дезэкранирова-
х) См. также исследования ЯМР-Al27 систем, содержащих катализаторы циглеров-ского типа [353].— Прим. ред.
400
Глава 12
нию хлора. При введении в систему солей, не участвующих в комплексообразовании, также были обнаружены изменения химических сдвигов ЯМР-С135, свидетельствующие о важной роли гидратации в рассматриваемом равновесии. Так, при добавлении нитрат-ионов к раствору хлорида- кобальта(П) был обнаружен в спектре ЯМР-Cl35 парамагнитный сдвиг. Причиной этого явления, по-видимому, служит дегидратация ионов кобальта, которые в этом случае становятся менее способными к комплексообразованию с хлорид-ионами. Рассмотрение величин /химических сдвигов ЯМР-Cl35 позволило заключить, что в комплексе хлорида кобальта(П) имеет место изотропное контактное взаимодействие электронных и ядерных спинов.
Вертц [214] измерил амплитуды сигналов ЯМР-Cl35 для водных растворов хлористого натрия различной концентрации в присутствии ряда парамагнитных ионов. Анализ экспериментальных данных подтвердил наличие обмена гидратной воды со средой (см. разд. 9.3.2).
Демельт [211] обобщил полученные им результаты изучения спектров ядерного квадрупольного резонанса С135 и С137 хлоридов и оксихлоридов некоторых металлов (см. табл. 12.83). Было найдено мультиплетное расщепление резонансных сигналов величиной около 100 кгц. Это расщепление обусловлено взаимодействием атомов хлора, занимающих различные положения в кристаллической решетке.
Таблица 12.83
ЯДЕРНОЕ КВАДРУПОЛЬНОЕ РАСЩЕПЛЕНИЕ НА ЯДРАХ С135 И СР’ ХЛОРИДОВ И ОКСИХЛОРИДОВ НЕКОТОРЫХ МЕТАЛЛОВ [211]
Соединение а Средняя частота V С135, Мгц Число линий в мультиплете
А1С1з-О(С2Н5)2 11,30 2
SiCl4 20,35 4
SnCl4 24,09 4
SnCl4 -O2NC6H5 23,08 .6
TiCl4 6,05 4
VOC13 11,54 2
СгОгСЬ 15,68 2
M0O2CI2 15,54 1
а Все измерения (за исключением МоОгОг) были выполнены при температуре жидкого воздуха.
Масуда и Канда [215] измерили химические сдвиги С135, Вг81 и I127 в молекулах НО, НВг и HI при напряженности постоянного магнитного поля 6221 э. Оказалось, что ширина линии ЯМР галогенов растет в ряду С1 < < Br < J.
Исследование водных растворов этих галогеноводородов при различных концентрациях показало, что изменяются химические сдвиги Н1 и изотопа галогена, а также ширина резонансных сигналов. Изучение этих изменений позволило получить сведения о диссоциации соответствующих кислот.
Канда [216] рассмотрел химические сдвиги С135, Вг81 и I127 галогенидов металлов в твердой фазе с точки зрения парамагнитного вклада в экранирование, являющегося результатом частичной ковалентности связей.
Иод I127. С помощью спектроскопии ЯМР-I127 изучено равновесие, устанавливающееся при добавлении элементарного иода в водный раствор иодид-ионов с образованием трииодид-ионов [217].
I' + h 13.
Спектры ЯМР других ядер (кроме водорода и фтора) 401
При внесении в систему молекул 12 сигнал ЯМР-I127 ионов I- уширяется, и измерение его ширины позволяет вычислить константы скорости прямой и обратной реакций [217].
12.10.5. Медь Си63
Измерение ширины линии ЯМР-Cu63 растворов солей одно- и двухвалентной меди в концентрированной соляной кислоте позволило оценить константу скорости бимолекулярного процесса обмена электронами (см. разд. 9.2) [218, 219].
Cu2+ + Cu+ Cu+ + Cu2+.
12.10.6. Мышьяк As75
Исследование ЯМР-As75 жидкого AsF5 не выявило какой-либо тонкой структуры спектра: ширина оказалась ~15 э при 8,8 Мгц [220].
12.10.7. Селен Se77
Уолчли [221] измерил химические сдвиги Se77 для молекул H2SeO3 (—1504 м. д.) и H2SeO4 (—1560 м. д.) в водном растворе, применив жидкий селеноводород в качестве внешнего эталона. В этих двух соединениях ядро Se77 оказалось менее экранированным, чем в H2Se.
12.10.8. Сурьма Sb121
Спектр ЯМР-Sb121 (измерялась производная сигнала поглощения) соединения NaSbF6, растворенного в водной фтористоводородной кислоте, состоит из семи компонент, что объясняется спин-спиновым взаимодействием ядра Sb121 с шестью эквивалентными ядрами фтора в ионе SbF“ [102, 223].
12.10.9. Платина Pt195
В спектре протонного резонанса комплекса платины [(Р(С2Н5)3)2РШС11 было обнаружено спин-спиновое взаимодействие 'Pt195 — Н1 с константой •^pt-н = 1276 гц [224]. В триметильных комплексах платины, таких, как [(CH3)3PtI]4, эта константа равна примерно 70—80 гц [125] х). Если одним из лигандов платинового комплекса является этилен [226] или циклопентадиенил [227], то константа Jpt-н имеет еще более низкое значение; например, для молекулы К [C2H4PtCl3] Jpt-н = 34 гц. Константа взаимодействия Jpt-p для молекулы цыс-[((С2Н5О)3Р)2Р1С12] равна 5,70 кгц [228].
12.10.10. Ртуть Hg199
Довольно широко исследовались константы спин-спинового взаимодействия Н — Hg на примере алкилртутных соединений [268, 280] (см. разд. 10.3.3).
Химические сдвиги Hg199 ряда диалкилртутных соединений были измерены с целью оценить вклад сверхсопряжения в металл-углеродную связь [91]. Если сверхсопряжение играет основную роль, то ядро Hg199 в диметилртути должно быть наиболее экранировано. Как видно из табл. 12.84, этого не наблюдается. Была найдена, однако, грубая корреляция химического сдвига Hg199 с числом p-протонов в алкильной группе. Не исключено, что это соответствие можно связать со спин-спиновым взаимодействием в этих соединениях:
1) Для [(С2Н5)3Р1С1 ]4 найдено, что /ptCH2 и JptcH3 имеют противоположные знаки [282].— Прим. ред.
26—489
402
Глава 12
Таблица 12.84
ХИМИЧЕСКИЕ СДВИГИ HglKK СОЕДИНЕНИЙ ТИПА R2Hg [91]
R д, м. д. й/№
СНз 0
с2н5 + 330 57
С3Н7 +240 60
(СН3)2НС +640 53
а У — число g-водородных атомов.
с p-протонами ртуть взаимодействует сильнее, чем с а-протонами (см. разд. 10.3.2). Аналогичное явление наблюдал Браунстейн [243] при изучении ряда ртутьорганических соединений CH3ORHgX, где R — алкильный радикал, а X может быть Cl, Br, I или ацетат-ионом. В табл. 12.85 приведены константы взаимодействия Н — Hg, полученные в результате анализа первого
Таблица 12.85
КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ Hl-Hglso НЕКОТОРЫХ РТУТЬОРГАНИЧЕСКИХ СОЕДИНЕНИЙ ОБЩЕЙ ФОРМУЛЫ CH3ORHgX [243]
Соединение Константа взаимодействия, гц
JHg—Н(1А)> JHg-H(lB) JHg-Н(2) JHg-H(3) JHg—Н(4)
о 11 СН3ОСН2СН2— HgO— с - сн3 2 1 СН3ОСН2СН2—HgCl СН3ОСН2СН2—HgBr осн3 о 230+2 225+7 207+2 217+2 245+2 249+2
СН3 — СН—СН2—Hg—О—с -сн3 3 2 1 осн3 222+4 288± 8 0
СН3—СН—СН2 — HgC 1 осн3 223±2 338+2 0
СН3—СН —СН2—HgBr осн3 о сн3 1 II > С —СН2—Hg —О—с —сн3 сн/ 3 2 1 осн3 СН3 | > С —CH2-Hg—С1 сн/ осн3 СН3, 1 > С—СН2—Hg —Вг сн/ 205+5 215+2 203+2 204+2 295+1 0 20,8+1 21,1 + 1 21,9+1 0
Спектры ЯМР других ядер (кроме водорода и фтора) 403
порядка протонных спектров этих соединений. За исключением молекулы CH3OCH2CH2HgOCOCH3, во всех случаях наблюдалось, что ^HgiwHi < ^Hgi9»H^r причем при любых R и X константа имеет узкую область значений
203—230 гц. Хотя величина существенно зависит от X, явной законо-
мерности не найдено, за исключением того, что взаимодействие ядра ртути с метановым протоном сильнее, чем с метиленовым. Взаимодействие Hg199 — — уН1 составляет около 21 гц независимо от X во emop-бутильных соединениях и отсутствует в пропильных. Высказано предположение о том, что величина константы «^HgiooHy зависит от двугранного угла между связью, соединяющей атом ртути и а-углеродный атом, и связью, соединяющей р-углеродный атом и присоединенные к нему водород или метильную группу *). Однако это явление можно объяснить также взаимодействием спинов ядер Hg199 и Н1 через пространство. Химические сдвиги а-протонов наблюдались в области т = 7,63 — 7,95.
При исследовании спектра 3-хлормеркурфурана было найдено несколько констант взаимодействия ртути с протонами цикла [278]. Все они оказались одного знака.
4 /HgCl
5/ \2 7нг-н(2> - ± 40,4 гц,
О ^Hg-H<4> = ± 74,9 гц,
7Hg-H<5) = ± 27,9 гц.
. Шнейдер и Бакингем [194] исследовали спектры ЯМР нескольких соединений ртути, таллия и свинца (Hg199, Т1205 и РЬ207). Исходя из простой модели «атом в молекуле», авторы оценили диамагнитный и парамагнитный вклады в экранирование этих металлов и дали объяснение наблюдаемым химическим сдвигам. По их мнению, следует принимать во внимание лишь парамагнитную составляющую спара, которая должна увеличиваться по мере роста электроотрицательности атома, связанного с металлом. Теория предсказывает, что для молекул, содержащих атомы с незаполненными р- и d-орбиталями, составляющая Спара должна иметь большую отрицательную величину. Предсказанные химические сдвиги между (CH3)2Hg и Hg2+, а также между (СН3)3Т1 и Т13+ (т. е. между молекулой с ковалентными связями и ионом) находятся в удовлетворительном соответствии с экспериментальными значениями. Однако химический сдвиг между (СН3)4РЬ и РЬ2+ оказался равным 3000 м. д., тогда как по теории он должен быть пренебрежимо малым. Из этого следует, что при расчетах необходимо принимать во внимание и более высокие электронные состояния атома, например 6</-орбиталь свинца. Химические сдвиги соединений, исследованных в этой работе, приведены в табл. 12.86 * 2).
На рис. 12.47 изображен резонансный спектр Hg199 диметилртути при 10,74 Мгц: в результате взаимодействия спинов Hg199 и шести эквивалентных метильных протонов наблюдается семи компонентный мультиплет [194]. Если к чистой диметилртути добавлен пиридин, то экранирование ядра Hg199 возрастает примерно на 80 м. д. 3).
Константа дальнего взаимодействия 7Hgl99H^ для соединений типа CH3CR1(OCH3)CR2R3HgCl составляет 0—48 гц и зависит от природы заместителя [383].— Прим. ред.
2) Проведено изучение химического сдвига Н§199для водных растворов HgCl2 в зависимости от концентрации LiCl. Показана возможность присутствия ионов HgClg [354].— Прим. ред.
3) Для диметилртути измерены следующие константы спин-спинового взаимодействия •7нн=1°>44| гц; 7cl3_Hg_c_H1 = 0,0 ± 0,4 гц; JHglM_cl3 = 689,0 гц; 7с13_н1 = 129,6 гц и 7не199_с_н1= —101’4 гц [376]. —Прим. ред.
26*
404
Глава 12
Таблица 12.86
ХИМИЧЕСКИЕ СДВИГИ НЕКОТОРЫХ СОЕДИНЕНИЙ РТУТИ, ТАЛЛИЯ И СВИНЦА [194] а
РЬ207 Т1205 Hgioo
Pb(NO3)2 (в воде) •РЬ(СНз)4 0 —2980 T1(NO3)3 (в воде) Т1С13 Т1ОС2Н5 Т1(СН3)3 (в эфире) 0 —2550 —2880 —4760 Hg(NO3)2 (в воде) K2[HgCl4] (в воде) Hg(CN)2 (в пиридине) Hg(C2H5)2 Hg(CH3)2 0 —1170 —1310 —2180 —2460
а Химические сдвиги выражены в миллионных долях.
12.10.11. Свинец РЬ207
Химические сдвиги некоторых соединений свинца (см. табл. 12.87) измерили Пьетт и Уивер [229], а также Рокар и сотр. [230].
Таблица 12.87
ХИМИЧЕСКИЕ СДВИГИ РЬ207 НЕКОТОРЫХ СОЕДИНЕНИЙ СВИНЦА [229, 230]
Соединение Чистота, % б, м. д. Соединение б, м. д.
РЬ (металл) 0 РЬО (желтый) +7 400
РЬО2 (порошок) 90 + 5 800 Цирконат свинца (в твер- + 12 500
94 + 6 500 дом состоянии)
98 + 6 900 РЬ(С1О4)2 (раствор) + 14 100
РЬ(С2Н3О2)2-ЗН2О (моно- + 10 900 РЬО (в твердом состоянии, + 11 200
кристалл) красный)
РЬ(С2Н3О2)2 -ЗН2О (раствор) + 12 300 РЬ(С2О4)2 (в твердом со- + 12 300
РЬС12 (кристаллический по- + 13 800 стоянии)
рошок) РЬТе + 10 800
РЬСО3 (монокристалл) +14 400 PbS + 10 100
PbSO4 (кристаллический по- + 15 200 PbSe + 8 700
рошок)
Pb(NO3)2 (раствор) + 14 400
Pb(NO3)2-H2O + 15 200
Pb(NO3)2 (кристаллический 98 + 15 200
порошок)
Широкий диапазон изменения химических сдвигов (~ 16 000 м. д.) свидетельствует о большой парамагнитной составляющей в экранировании ядра РЬ207. Оргел [231] рассчитал величины этой составляющей для ряда солей двухвалентного свинца. Полученные им значения хорошо согласуются с экспериментальными. В тех случаях, когда окружение ядра РЬ в основном ионное, парамагнитная составляющая мала. Если же связь со свинцом до некоторой степени ковалентная, то парамагнитная составляющая возрастает (как, например, в желтой окиси свинца) [229].
Химические сдвиги соединений в твердом состоянии измерялись при регистрации производных линий, полученных с помощью фазового детектора.
Спектры ЯМР других ядер (кроме водорода и фтора)
405
12.10.12. Ниобий Nb83
На рис. 12.48 изображен спектр резонанса Nb83 иона NbF- при 14,2 Мгц [258]. Семи компонентное расщепление = 334 гц) явно свидетельствует о существовании в растворе иона NbF" и об отсутствии двухзарядного иона
О 200 мэ
'----'----' | Hg(CHj)2
Рис. 12.47. Наблюдаемый и рассчитанный спектры ЯМР-Hg188 (производная сигнала дисперсии) диметилртути при 10,74 Мгц [194].
Рис. 12.48. ЯМР-спектры [258].
а — спектр Nb93 иона NbFe при 14,2 Мгц;
б —спектр F18 иона NbFe при 56,4 Мгц.
NbF8-. Спектр ЯМР-F18 иона NbF- состоит из десяти компонент, обусловленных взаимодействием ядер фтора с ядром ниобия (спин 9/2). Компоненты этого мультиплета одинаковы по интенсивности, но отличаются по ширине линии, что указывает на влияние в этой системе квадрупольной релаксации [112, 258].
12.10.13. Магнитные моменты ядер
Занимаясь определением ядерных магнитных моментов, Проктор и Ю [102] исследовали ЯМР на следующих ядрах: N14, N15, Cl35, Sc45, V51, Мп55, Со58, Mo85, Mo87, In113, In115, Sb121, Sb123, Хе128, Xe131, Pt185, Hg188, Hg201 и Bi208 4). Свойства ядер и их магнитные моменты приведены в приложении I т. 1.
ЛИТЕРАТУРА
1. Phillips W. D., М i 1 1 е г Н. С., М u е t t е г t i е s Е. L., J. Am. Chem. Soc., 81, 4496 (1959).
2. Onak Т. Р., Landesman Н., Williams R. Е., Shapiro I, J. Phys. Chem., 63, 1533 (1959).
3. S a i k a A., Slich ter С. P., J. Chem. Phys., 22, 26 (1954).
4. Muller N., Lauterbur P. C., Goldenson J., J. Am. Chem. Soc., 78, 3557 (1956).
5. Van Wazer J. R., Callis C. F., Shoolery J. N., Jones R. C., J. Am. Chem. Soc., 78, 5715 (1956).
4) Проведено исследование спектров магнитного резонанса на ядрах V51, Nb83, Sb121, Sb123 и Та 181 растворов соответствующих фторидов и оксиионов [355].—Прим. ред.
406
Глава 12
6. Опак Т. Р., Landesman Н., Williams R. Е., S h а р i г о I., paper presented to the Division! of Inorganic Chemistry, National Meeting of A.C.S., Boston, Mass., April (1959).
7. Banister A., Greenwood N. N., J. Chem. Soc., 1965, 1534.
8. Hawthorne M. F., J. Am. Chem. Soc., 83, 1345 (1961).
9. Schaeffer R., Shoolery J. N., Jones R., J. Am. Chem. Soc., 79, 4606 (1957).
10. C h a m b e r 1 a i n N. F., Anal. Chem., 31, 56 (1959).
11. С о у 1 e T. D., S t a f f о r d S. L., S t о n e F. G. A., J. Chem. Soc., 1963, 3103.
12. G u t о w s к у H. S., M с С a 1 1 D. W., S 1 i с h t е г С. P., J. Chem. Phys., 21, 279 (1953).
13. M u 1 1 e r N., P r i t c h a r d D. E., J. Chem. Phys., 31, 768 (1959).
14. Ogg R. A., J. Chem. Phys., 22, 1933 (1954).
15. Shoolery J. N., Discuss. Faraday Soc., 19, 215 (1955).
16. W i 1 1 i a m s R. E., S h a p i г о I., J. Chem. Phys., 29, 677 (1958).
17. W i 1 1 i a m s R. E., Gibbons S. G., Shapiro I., J. Chem. Phys., 30, 320, 333 (1959).
18. Hedberg K., S ch о m a ker V., J. Am. Chem. Soc., 73, 1482 (1951).
19. W i 1 1 i a m s R. E., J. Inorg. Niicl. Chem., 20, 198 (1962).
20. Williams R. E., F i s h e r H. D., Wilson C. O., J. Phys. Chem., 64, 1583 (1960).
21. P r i c e W. C., J. Chem. Phys., 16, 894 (1948).
22. W i 1 1 i a m s R. E., G i b b о n s S. G., Shapiro I., J. Am. Chem. Soc., 81, 6164 (1959).
23. Rigden J. S., Hopkins R. C.,Baldeschwieler J. D., J. Chem. Phys., 35, 1532 (1961).
24. Jones M. E., Hedberg K.,Schomaker V.,J. Am. Chem. Soc., 75, 4116 (1953).
25. N о r d m a n С. E., L i p s с о m b W. N., J. Am. Chem. Soc., 75, 4116 (1953).
26. Ogg R. А., О. M. С. C. Tech. Report 120, Navy Contract № 52-1023c at Stanford University (1955).
27. Schaeffer R., T e b b e F. N., J. Am. Chem. Soc., 84, 3974 (1962).
28. Hedberg K-, Jones M. E., Sch om aker V., J. Am. Chem. Soc., 73, 3538 (1951).
29. L i p s с о m b W. N., J. Chem. Phys., 22, 985 (1954).
30. Hedberg K-, Jones M. E., Schomaker V., неопубликованные данные, цитируемые в работе [29].
31. Hirshfeld F. L., Eriks K-, Dickerson R. E., Lippert E. L., Lipscomb W. N., J. Chem. Phys., 28, 56 (1958).
32. Kasper J. S., L u c h t С. M., Harker D., Acta Crysfa., 3, 436 (1950).
33. A f t a n d i 1 i a n V. D., Miller H. C., Parshall G. W., Mu etter-ties E. L., Inorg. Chem., 1, 734 (1962).
34. В u r g A. B., Kratzer R., Inorg. Chem., 1,' 725 (1962).
35. D u p о n t J. A., Hawthorne M. F., Chem. and Ind., 405 (1962).
36. Schaeffer R., Shoolery J. N., Jones R., J. Am. Soc. Chem., 80, 2670 (1958).
37. Siegel B.,Mack J. L., Lowe J. U., Callaghan J.,J. Am. Chem. Soc., 80, 4523 (1958).
38. H a w t h о r n e M. F., M i 1 1 e r J. J., J. Am. Chem. Soc., 80, 754 (1958).
39. S h a p i г о I., L u s t i g M., Williams R. E., J. Am. Chem. Soc., 81, 838 (1959).
40. Figgis B. N., Williams R. L., Spectrochim. Acta, 15, 331 (1959).
41. В 1 a у N. J., W i 1 1 i a m s J., W i 1 1 i a m s R. L., J. Chem. Soc., 1960, 424.
42. Blay N. J., D u n s t a n 1., Williams R. L., J. Chem. Soc., 1960, 430.
43. О n а к T. P., S h a p i г о I., J. Chem. Phys., 32, 952 (1960).
44. Beach J. Y., Bauer S. FL, J. Am. Chem. Soc., 62, 3440 (1940).
45. Price W. C., J. Chem. Phys., 17, 1044 (1949).
46. Ogg R. A., R a у J. D., Discuss. Faraday Soc., 19, 239 (1955).
47. Lauterbur P. C.,J. Chem. Phys., 27, 217 (1957).
48. Lauterbur P. C., Annals N.Y. Acad. ScL, 70, 841 (1958).
49. Lauterbur P. C., J. Am. Chem. Soc., 83, 1838 (1961).
50. Lauterbur P. C., Determination of Organic Structures by Physical Methods, Editors F. C. Nachod and W. D. Phillips, Academic Press, New York, 1962, Vol. 2, Ch. 7.
51. H о 1 m С. H., J. Chem. Phys., 26, 707 (1957).
52. Spiesecke H., Schneider W. G., J. Chem. Phys., 35, 722 (1961).
53. Forsen S., Rupprecht A., J. Chem. Phys., 33, 1888 (1960).
54. Par ker D. J., McLaren G. A., Conradi J. J., J. Chem. Phys., 33, 629 (1960).
55. В а к e r E. B., J. Chem. Phys., 37, 911 (1962).
56. Holtzman G. R., Lauterbur P. C., Anderson J. H., Koth W., J. Chem. Phys., 25, 172 (1956).
Спектры ЯМР других ядер (кроме водорода и фтора)
407
57. S h о о 1 е г у J. N. частное сообщение.
58. L a u t е г b и г Р. С., Determination of Organic Structures by Physical Methods, Editors F. C. Nachod and W. D. Phillips, Academic Press, New York, 1962, p. 483.
59. A 1 1 r e d A. L., R ochow E. G., J. Inorg. Nucl. Chem., 5, 269 (1958).
60. Brown M. P., Webster D. E., J. Phys. Chem., 64, 698 (1960).
61. L a u t e r b u r P. C., J. Am. Chem. Soc., 83, 1846 (1961).
62. Spiesecke H., Schneider W. G., J. Chem. Phys., 35, 731 (1961).
63. Lauterbur P. С., частное сообщение.
64. Ettinger R., Blume P., Patterson A., Lauterbur P. C., J. Chem. Phys., 33, 1597 (1960).
65. Patterson A., Ettinger R., Z. Elektrochem., 64, 98 (1960).
66. Cotton F. A., DantiA., Waugh J. S., Fessenden R. W., J. Chem. Phys., 29, 1427 (1958).
67. Weiss E., Z. anorg. Chem., 287, 223 (1956).
68. Ewens R. V. G., Lister M. W., Trans. Faraday Soc., 35, 681 (1939).
69. Juan C., G u t о w s к у H. S., J. Chem. Phys., 37, 2198 (1962).
70. Karabatsos G. J., J. Am. Chem. Soc., 83, 1230 (1961).
71. M c A d a m s D. R, J. Chem. Phys., 36, 1948 (1962).
72. R a m s e у N. F., Phys. Rev., 91, 303 (1953).
73. Muller N., Pritchard D. E., J. Chem. Phys., 31, 1471 (1959).
74. S h о о 1 e г у J. N„ J. Chem. Phys., 31, 1427 (1959).
75. M u 1 1 e r N„ J. Chem. Phys., 36, 359 (1962).
76. Whipple E. B., Stewart W. E., Reddy G. S., GoldsteinJ. FL, J. Chem. Phys., 34, 2136 (1961).
77. Karplus M., Grant D. M., Proc. Nat. Acad. Sci., 45, 1269 (1959).
78. Karplus M., J. Phys. Chem., 64, 1793 (1960).
79. Huggins M. L., J. Am. Chem. Soc., 75, 4123 (1953).
80. Malinowski E. R.,J. Am. Chem. Soc., 83, 4479 (1961).
81. Harris R. K., J. Phys. Chem., 66, 768 (1962).
82. G u t о w s к у H. S., J u an C., Discuss. Faraday Soc., 34, 52 (1962).
83. M a 1 i n о w s к i E. R., P о 1 1 a r a L. Z., L a r m a n n J. P., J. Am. Chem. Soc.,
84, 2649 (1962).
84. В a n w e 1 1 C. N., Sheppard N., Mol. Phys., 3, 351 (1960).
85. Sheppard N., Turner J. J., Proc. Roy. Soc., A 252, 506 (1959).
86. С о h e n A. D., Sheppard N., Turner J. J., Proc. Chem. Soc., 118 (1958).
87. G о 1 d s t e i n J. H., Reddy G. S., J. Chem. Phys., 36, 2644 (1962).
88. H u t t о n H. M., Reynolds W. F., Schaefer T., Can. J. Chem., 40, 1758 (1962).
89. Shoolery J. N., Johnson L. F., Anderson W. A., J. Mol. Spect., 5, 110 (1960).
90. Baker E. B., J. Chem. Phys., 26, 960 (1957).
91. Dessy R.E.,Flautt T. J., Jaffe H.H., Reynolds G. F.,J. Chem. Phys., 30, 1422 (1959).
92. N a r a s i m h a n P. T., R о g e г s M. T., J. Am. Chem. Soc., 82, 34 (1960).
93. Sharts С. M., Roberts J. D., J. Am. Chem. Soc., 79, 1008 (1957).
94. Karabatsos G. J., J. Am. Chem. Soc., 83, 1230 (1961).
95. Karabatsos G. J., Graham J. D., Vane F. M., J. Am. Chem. Soc., 83, 2778 (1961).
96. Karabatsos G. J., Graham J. D., Vane F. M., J. Am. Chem. Soc., 84, 37 (1962).
97. A n d e r s о n W. A., J. Chem. Phys., 37, 1373 (1962).
98. L у n d e n - В e 1 1 R. M., Sheppard N., Proc. Roy. Soc., A 269, 385 (1962).
99. S h о о 1 er у J. N., J. Mol. Spect., 63, 110 (1960).
100. Muller N., R о s e P. L, J. Am. Chem. Soc., 84, 3973 (1962).
101. A с r i v о s J. V., J. Chem. Phys., 36, 1097 (1962).
102. Proctor W. G., Y u F. C., Phys. Rev., 81, 20 (1951).
103. M a s u d a Y., К a n d a T., J. Phys. Soc., Japan, 8, 432 (1953).
104. Holder В. E., К 1 e i n M. P., J. Chem. Phys., 23, 1956 (1955).
105. Schmidt В. M., Brown L. C., W i 1 1 i a m s D., J. Mol. Spect., 2, 539 (1958).
106. Schmidt В. M., Brown L. C., W i 1 1 i a m s D_, J. Mol. Spect., 2, 551 (1958).
107. Ogg R. A., R a у J. D., J. Chem. Phys., 26, 1340 (1957).
108. Baldeschwieler J. D., Randall E. W., Proc. Chem. Soc., 303 (1961),
109. Schoolery J. N., Varian Technical Bulletin, № 50.
НО. P i e t t e L. H., Ray J. D., Ogg R. A., J. Mol. Spect., 2, 66 (1958).
111. Tiers G. V. D., В о v e у F. A., J. Phys. Chem., 63, 302 (1959).
112. P о p 1 e J. A., Mol. Phys., 1, 168 (1958).
113. Ogg R. A., J. Chem. Phys., 22, 560 (1954).
114. Grunwald E., Loewenstein A., M e i b о о m S., J. Chem. Phys., 27, 630 (1957).
115. Roberts J. D., J. Am. Chem. Soc., 78, 4495 (1956).
408
Глава 12
116. К u n t z I. D., Schleyer P. von R., A 1 1 e г h a n d A., J. Chem. Phys., 35, 1533 (1961).
117. N а г a s i m h a n P. T., R о g e г s M. T., J. Chem. Phys., 31, 1430 (1959).
118. R a у J. D., P i e t t e L. H., H о 1 1 i s H. D., J. Chem. Phys., 29, 1022 (1958).
119. Ray J. D., О g g R. A., J. Chem. Phys., 26, 1452 (1957).
120. Weaver H. E., Tolbert В. M., La Force R. C., J. Chem. Phys., 23, 1956 (1955).
121. D harm atti S. S., Sundara Rao K. J., Vijayaraghavan R., Nuovo Cimento, 11, 656 (1959).
122. Figgis B. N., Kidd R. G., N у h о 1 m R. S., Proc. Roy. Soc., A 269, 469 (1962).
123. Griffith J. S., О r g e 1 L. E., Trans. Faraday Soc., 53, 601 (1957).
124. Christ H. A., Helv. Phys. Acta., 33, 572 (1960).
125. В r a m 1 e у R., Figgis B. N., N у h о 1 m R. S., Trans. Faraday Soc., 58, 1893 (1962).
126. Shulman R. G., W у 1 u d a B. J., J. Chem. Phys., 30, 335 (1959).
127. О r g e 1 L. E., Discuss. Faraday Soc., 26, 138 (1958).
128. Or gel L. E., J. Chem. Phys., 30, 1617 (1959).
129. Lewis W.B.,Jackson J.A..Lemons J.F.,Taube H.,J. Chem. Phys., 36, 694 (1962).
130. Jackson J. A., Lemons J. F., Taube H., J. Chem. Phys., 32, 553 (1960).
131. Reuben J., Tzalmona A., Samuel D., Proc. Chem. Soc., 353 (1962).
132. Maher J. P., Evans D. F., Proc. Chem. Soc., 176 (1963).
133. Ebsworth E. A. V., T u r n e r J. J., J. Chem. Phys., 36, 2628 (1962).
134. Ebsworth E. A. V., Sheppard N., J. Inorg. Nucl. Chem., 9, 95 (1959).
135. Downs A. J., Ebsworth E. A. V., J. Chem. Soc., 1960, 3516.
136. D a a s c h L. W., Smith D. C., Anal. Chem., 23, 853 (1951).
137. G e r d i n g H., Mearsen J. W., Rec. Trav. Clim., 76, 481 (1957).
138. Van Waze r, J. R., частное сообщение.
139. Knight W. D., Phys. Rev., 76, 1259 (1949).
140. Dickinson W. C., Phys. Rev., 81, 717 (1951).
141. Gutowsky H. S., McCall D. W., J. Chem. Phys., 22, 162 (1954).
142. Meyer L. H., Gutowsky H. S., J. Phys. Chem., 57, 481 (1953).
143. Van Wazer J. R., Callis C. F., Shoolery J. N., Jones R. C.,J. Am. Chem. Soc., 78 , 5715 (1956).
144. Коулсон Ч., Валентность, изд-во «Мир», М., 1965.
145. Parks J. R., J. Am. Chem. Soc., 79, 757 (1957).
146. F i n e g о 1 d H., Annals N. Y. Acad. Sci., 70, 875 (1958).
147. Groenweghe L. C. D., Maier L., Moedritzer K., J. Phys. Chem., 60, 901 (1962).
148. Groenweghe L. C. D., Maier L., Moedritzer K-, J. Chem. Eng. Data, 7, 307 (1962).
149. Katritzky A. R., Jones R.A. Y., Angew. Chem. (Int. Edn.), 1, 32 (1962).
150. Dudek G. O., J. Chem. Phys., 33, 624 (1960).
151. Тафт P. У., Пространственные эффекты в органической химии, ИЛ, М., 1960, гл. 13.
152. Van Wazer J. R.,Callis C. F.,Shoolery J.N., Anderson W.A., J. Am. Chem. Soc., 79, 2719 (1957).
153. Shuler W. E., Axtmann R. С., A.E.C. Research and Development Report DP 474.
154. Axtmann R. C., Shuler W. E., Eberly J. H., J. Chem. Phys., 31, 850 (1959).
155. Feeney J., Sutcliffe L. H., неопубликованные данные.
156. S i d d a I 1 T. H., Prohaska C. A., J. Am. Chem. Soc., 84, 2502 (1962).
157. S i d d a 1 1 T. H., Prohaska C. A., J. Am. Chem. Soc., 84, 3467 (1962).
158. Crutchfield M. M., Callis C. F., I r a n i R. R., Roth G. C., Inorg. Chem., 1 813 (1962).
159. Moedritzer K-. J- Inorg. Nucl. Chem., 22, 19 (1961).
160. Van Wazer J. R., Callis C. F., Shoolery J. N., J. Am. Chem. Soc., 77, 4945 (1955).
161. F i n e g о 1 d H., частное сообщение.
162. Andrew E. R., Bradbury A., E a d e s R. G., Jenks G. J., Nature, 188, 1096 (1960).
163. Andrew E. R., Eades R. G., Discuss. Faraday Soc., 34, 38, (1962).
164. Lynden-Bell R. M., Trans. Faraday Soc., 57, 888 (1961).
165. Maier L., J. Inorg. Nucl. Chem., 24, 275 (1962).
166. Jones R.A. Y., Katritzky A. R., J. Chem. Soc., 1960, 4376.
167. Cohn M., Hughes T. R., J. Biol. Chem., 235, 3250 (1960).
168. Becke-Goehring M., John K-, F 1 u c k E., Z. anorg. Chem., 302, 103 (1959).
Спектры ЯМР других ядер (кроме водорода и фтора)
409
169. Boden N., Е m s 1 е у J. W., Feeney J., Sutcliffe L. Н., Chem. and Ind., 1909 (1962) и неопубликованные данные.
170. Callis С. F., Van Wazer J. R., Shoolery J. N., Anal. Chem., 28, 269 (1956).
171. G u f f у J. С., M i 1 1 e r G. R., Anal. Chem., 31, 1895 (1959).
172. Schwarzmann E., Van Wazer J. R., J. Inorg. Nucl. Chem., 14, 296, (1960).
173. Groenweghe L. C. D., Payne J. H., J. Am. Chem. Soc., 81, 6357 (1959).
174. F 1 u c k E., Van Wazer J. R., Groenweghe L. C. D., J. Am. Chem. Soc., 81, 6363 (1959).
175. Ames D. P.,Ohashi S., C a 1 1 i s C. F., V a’n Wazer J.R.,J. Am. Chem. Soc., 81, 6350 (1959).
176. Schwarzmann E., Van Wazer J. R., J. Am. Chem. Soc., 81, 6366 (1959).
177. F 1 u c k E., Z. anorg. Chem., 315, 181 (1962).
178. Gasser R. P. H., Richards R. E., Mol. Phys., 3, 163 (1960).
179. Freeman R., Murray G. R., Richards R. E., Proc. Roy. Soc., A 242, 455 (1957).
180. О p г e л Л. E., Введение в химию переходных металлов, изд-во «Мир», М., 1964.
181. Ramsey N. F., Phys. Rev., 85, 243 (1952).
182. Dharmatti S. S., Kanekar C. R., J. Chem. Phys., 31, 1436 (1960).
183. Dharmatti S. S., Kanekar C. R., Mathur S. C., Proc, of the Peaceful Uses of Atomic Energy, Geneva (1958).
184. Armstrong J. A., Benedek G. B., Bull. Am. Phys. Soc., 6, 234, D8 (1961).
185. Burke J.J..Lauterbur P. C., Abs. Am. Chem. Soc. Meeting, San Francisco (1958).
186. BurkeJ.J..Lauterbur P. C.,J. Am. Chem. Soc., 83, 326 (1961).
187. Moore D. W., H a p p e J. A., J. Phys. Chem., 65, 224 (1961).
188. Holmes J. R., К a e s z H. D., J. Am. Chem. Soc., 83, 3903 (1961).
189. Rowland T. J..Bromberg J.P..J. Chem. Phys., 29, 626 (1958).
190. Figgis B. N., Trans. Faraday Soc., 55, 1075 (1959).
191. Freeman R., Gasser R. P. H., Richards R. E., Mol. Phys., 2, 301 (1959).
192. Maher J. P., E v a n s D. F., Proc. Chem. Soc., 208 (1961).
193. Sutton L. E., S i d g w i c k N. V., J. Chem. Soc., 1930, 1461.
194. Schneider W. G., Buckingham A. D., Discuss. Faraday Soc., 34, 147 (1962).
195. Gutowsky H. S., McGarvey B. R., Phys. Rev., 91, 81 (1953).
196. W i m e t t T. F., Phys. Rev., 91, 476A (1953).
197. Stephen M. J., Proc. Roy. Soc., A 243, 274 (1957).
198. I s h i g u г о E., Phys. Rev., Ill, 203 (1958).
199. Abraham R. J., Bernstein H. J., Can. J. Chem., 39, 216 (1961).
200. Tiers G. V. D., J. Chem. Phys., 29, 963 (1958). •
201. Tiers G. V. D., J. Am. Chem. Soc., 79, 5585 (1957).
202. Ramsey N. F., Phys. Rev., 87, 1075 (1952).
203. Gutowsky H. S., J. Chem. Phys., 31, 1683 (1959).
204. Marshall T. W., Mol. Phys., 4, 61 (1961).
205. Wertz J. E., J ardetzky O., J. Chem. Phys., 25, 357 (1956).
206. Gutowsky H. S., McGarvey B. R., J. Chem. Phys., 21, 1423 (1953).
207. Gutowsky H. S., Hoffman C. J.,J. Chem. Phys., 19, 1259 (1951).
208. O’Reilly D. E., J. Chem. Phys., 32, 1007 (1960).
209. Connick R.E., Paulson R.E.,J. Am. Chem. Soc., 79, 5153 (1957).
210. M a s u d a Y., J. Phys. Soc. Japan, 11, 670 (1956).
211. D e h m e 1 t H. G., J. Chem. Phys., 21, 380 (1953).
212. Townes С. H., D a i 1 e у В. P., J. Chem. Phys., 20, 35 (1952).
213. Chesnut D. B., J. Chem. Phys., 33, 1234 (1960).
214. Wertz J. E., J. Chem. Phys., 24, 484 (1956).
215. Masti da Y., К a n d a T., J. Phys. Soc. Japan, 9, 82 (1954).
216. К a n d a T., J. Phys. Soc. Japan, 10, 85 (1955).
217. Myers О. E., J. Chem. Phys., 28, 1027 (1958).
218. McConnell H. M., W e a v e r H. E., J. Chem. Phys., 25, 307 (1956).
219. McConnell H. M., Berger S. B., J. Chem. Phys., 27, 230 (1957).
220. Jones E. D., Uehling E. A., J. Chem. Phys., 36, 1691 (1962).
221. W a 1 c h 1 i H.-E., Phys. Rev., 90, 331 (1953).
222. Ito K-, W a t a n a b e H., К u b о M., J. Chem. Phys., 34, 1043 (1961).
223. Dharmatti S. S., Weaver H. E., Phys. Rev., 87, 675 (1952).
224. C h a t t J., частное сообщение.
225. Smith J. A. S., J. Chem. Soc., 1962, 4736.
226. Powell D. B., Sheppard N., J. Chem. Soc., 1960, 2519.
227. Shaw B. L., Sheppard N., Chem. and Ind., 517 (1961).
410
Глава 12
228. Pidcock A., Richards R. E„ Venanzi L. M., Proc. Chem. Soc., 184 (1962).
229. P i e t t e L. H„ Weaver H. E., J. Chem. Phys., 28, 735 (1958).
230. R о c a r d J. M., Bloom M., Robinson L. B., Can. J. Phys., 37, 522 (1959).
231. Orgel L. E., Mol. Phys., 1, 322 (1958).
232. Friedel R. A., Retcofsky H. L.,J. Am. Chem. Soc.., 85, 1300 (1963).
233. Narasimh an P. T., R о g e r s M. T., J. Chem. Phys., 34, 1049 (1961).
234. К a e s z H. D., F 1 i t с г о f t N,, J. Am. Chem. Soc., 85, 1377 (1963).
235. Grutchfield M. M., частное сообщение.
236. F r a n к i s s S. G., J. Phys. Chem., 67, 752 (1963).
237. Tiers G. V. D., J. Phys. Soc. Japan., 15, 354 (1960).
238. Harris R. K-, частное сообщение.
239. Downs A. J., частное сообщение, цитируемое в работе [236].
240. Pitcher Е., Buckingham A. D., Stone F. G. A., J. Chem. Phys., 36, 124 (1962).
241. Tiers G. V. D., J. Phys. Chem., 64, 373 (1960).
242. Muller N., J. Chem. Phys., 37, 2729 (1962).
243. Brownstein S., Discuss. Faraday Soc., 34, 25 (1962).
244. Baldeschwieler J.D., R'andall E.W., Chem. Rev., 63, 81 (1963).
245. Sunners B., Piette L. H., Schneider W. G., Can. J. Chem., 38, 681 (1960).
246. Groenweghe L. C. D., Payne J. H., J. Am. Chem. Soc., 83, 1811 (1961).
247. F i n e g о 1 d H., J. Am. Chem. Soc., 82, 2641 (1960).
248. Speziale A. J., Freeman R.C.,J. Org. Chem., 23, 1883 (1958).
249. Frei K-> Bernstein H. J., J. Chem. Phys., 38, 1216 (1963).
250. Becke-Goehring M., F 1 u с к E., Angew. Chem. (Int. Edn.), 1, 281 (1962).
251. Becke-Goehring M., John K., F 1 u с к E., Z. anorg. Chem., 302 , 103 (1959).
252. Lauterbur P. C., J. Chem. Phys., 38, 1415 (1963).
253. Lauterbur P. C., J. Chem. Phys., 38, 1406 (1963).
254. Lauterbur P. C., J. Chem. Phys., 38, 1432 (1963).
255. Schneider W. G., частное сообщение.
256. Brown T. L., M о r g a n G. L., Inorg. Chem., 2, 736 (1963).
257. Hunt J.P., Dodgen H. W., К 1 a n b er g F., Inorg. Chem., 2, 478 (1963).
258. Packer K. J., Mu e t t er t i es E. L.,J. Am. Chem. Soc., 85, 3035 (1963).
259. M a n a t t S. L., J u v i n а И G. L., E 1 1 e m a n D. D., J. Am. Chem. Soc., 85, 2664 (1963).
260. O’Reilly D. E., Bull. Am. Phys. Soc., 8, 531 (1963).
261. Forman R. A., J. Chem. Phys., 39, 2398 (1963).
262. Evans D. F., J. Chem. Soc., 1963, 5575.
263. Maher J. P., E v a n s D. F., J. Chem. Soc., 1963, 5534.
264. Smith G. W., J. Chem. Phys., 39, 2031 (1963).
265. Kobayashi Y., C h a s i n L. A., C 1 a p p’ L. B., Inorg. Chem., 2, 212 (1963).
266. Tiers G. V. D., J. Phys. Chem., 66, 945 (1962); 67, 1372 (1963).
267. Ebsworth E. A. V., Frankiss S. G., Trans. Faraday Soc., 59, 1518 (1963).
268. Hatton J. V., Schneider W. G., S i e b r a n d W., J. Chem. Phys.., 39, 1330 (1963).
269. Savitsky G. B., N a m i к a w a K-, J. Phys. Chem., 67, 2430 (1963).
270. Muller N., Carr D. T., J. Phys. Chem., 67, 112 (1963).
271. Reuben J., Samuel D., частное сообщение.
272. Savitsky G. В., J. Phys. Chem., 67, 2723 (1963).
273. Savitsky G. B., Namikawa K., J. Phys. Chem., 67, 2755 (1963).
274. Clark H. C.,Kwon J. T., Reeves L. W., Wells E.J., Can. J. Chem., 41, 3005 (1963).
275. Schaefer T., Reynolds W. F., Yonemoto T., Can. J. Chem., 41, 2969 (1963).
276. Maciel G. E., Ruben G. C., J. Am. Chem. Soc., 85, 3903 (1963).
277. Pier E. A., (Varian Associates), частное сообщение (1964).
278. Cohen A. D., M c L a u c h 1 a n K- A., Mol. Phys., 7, 11 (1964).
279. Graham D. M., Holloway С. E., Can. J. Chem., 41, 2114 (1963).
280. Moy D., Emerson E., Oliver J.P., Inorg. Chem., 2, 1261 (1963).
281. Nielsen M. L., Pust inger J. V., J. Phys. Chem., 68, 152 (1964).
282. Dhami K- S., S to th ers J. B., Can. J. Chem., 43, 479, 498, 510 (1965).
283. Ford С. T., D i с к s о n F. E., В e z m a n I. I., Inorg. Chem., 3, 177 (1964).
284. P о p 1 e J. A., Mol. Phys., 7, 301 (1964).
285. Hatton J. V., J. Chem. Phys., 40, 933 (1964).
286. Gaines D. F., Schaeffer R., J. Am. Chem. Soc., 86, 1505 (1964).
287. Nash С. P., M a c i e 1 G. E., J. Phys. Chem., 68, 832 (1964).
288. Gaines D. F., Schaeffer R.,J. Phys. Chem., 68, 955 (1964).
289. Maciel G. E., Savitsky G. B., J. Phys. Chem., 68, 437 (1964).
Спектры ЯМР других ядер (кроме водорода и фтора)
411
290. Stothers J. В., L a u t е г b и г Р. С., Сап. J. Chem., 42, 1563 (1964).
291. Weis E.G., Reeves L. W., J. Chem. Phys., 40, 2036 (1964).
292. Reeves L. W., J. Chem. Phys., 40, 2128 (1964).
293. Reeves L. W., J. Chem. Phys., 40, 2132, 2423 (1964).
294. D а п у 1 и к S. S., J. Am. Chem. Soc., 86, 4504 (1964).
295. Binsch G.,Lambert J. B., Roberts B.W., Roberts J.D.,J. Am. Chem. Soc., 86, 5564 (1964).
296. Lambert J. B., Binsch G., Roberts J. D., Proc. Nat. Acad. Sci., 51, 735 (1964).
297. Bourn A. J. R., R a n d a 1 1 E. W., J. Mol. Spectr., 13, 29 (1964).
298. W i tanowski M., Urbanski T., Stefanik L., J. Am. Chem. Soc., 86, 2569 (1964).
299. Herbinson-Evans D., Richards R. R., Mol. Phys., 8, 19 (1964).
300. Goldstein J. H., Hobgood R. T., J. Chem. Phys., 40, 3592 (1964).
301. Savitsky G. B., N a m i к a w a K-, J. Phys. Chem., 68, 1956 (1964).
302. Nielsen M. I., Pustinger J. V., Strobel J., J. Chem. Eng. Data, 9, 167 (1964).
303. Paul E. G., Grant D. M., J. Am..Chem. Soc., 86, 2977 (1964).
304. Paul E. G., G r a n t D. M., J. Am. Chem. Soc., 86, 2984 (1964).
305. Diehl P., L e i p e r t T., Helv. Chim. Acta, 47, 545 (1964).
306. Karabatsos G. J., Orzech С. E., J. Am. Chem. Soc., 86, 3574 (1964).
307. Malinowski E. R., Vladimiroff T., J. Am. Chem. Soc., 86, 3575 (1964).
308. Swift H. E., P о о 1 e С. P., I t z e 1 J. F., J. Phys. Chem., 68, 2509 (1964).
309. Tori K., Nikagawa T.,J. Phys. Chem., 68, 3163 (1964).
310. Pilling R. L., Tebbe F. N., Hawthorne C. F., Pier E. A., Proc. Chem. Soc., 402 (1964).
311. A 1 e i M., Jackson J. A., J. Chem. Phys., 41, 3402 (1964).
312. Maher J. P., E v a n s D. F., J. Chem. Soc., 1965, 637.
313. P о p 1 e J. А., К a r p 1 и s M., J. Chem. Phys., 38, 2803 (1963).
314. Benedeck G. B., Englman R., Armstrong J. A., J. Chem. Phys., 39, 3349 (1963).
315. Tiers G. V. D.,Brown C. A.,J ackson R. A., L a r h T. N.,J. Am. Chem. Soc., 86, 2526 (1964).
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
316. Быстров В. Ф., в кн. Ю. А. Жданова и В. И. Липкина «Корреляционный анализ в органической химии», изд. Ростовского ун-та, 1966, гл. 1, разд. 7.
317. W u Т. К-, D a i 1 е у В. Р., J. Chem. Phys., 41, 2796 (1964).
318. Липпмаа Э. Т., Паст Я., Оливсон А., Салуверс Т., Изв. АН Эст. ССР, сер. физ.-мат. и техн, наук, 15, 58 (1966).
319. Липпмаа Э. Т., Оливсон А., Паст Я., Изв. АН Эст. ССР, сер. физ-мат. и техн, наук, 14, 473 (1965).
320. Липпмаа Э. Т., Соколов В. И., Паст Я. О., Реутов О. А., ДАН СССР, 173, 358 (1967).
321. Dhami К. S., Stothers J. В., Сап. J. Chem., 44, 2855 (1966).
322. Липпмаа Э. Т., Алла М., Изв. АН Эст. ССР, сер. физ-мат. и техн, наук, 15, № 4 (1966).
323. Ионин Б. И., канд. диссертация, Ленинградский технологический институт им. Ленсовета, 1964.
324. Ионин Б. И., Петров А. А., ЖОХ, 33, 432 (1963); 34, 1174 (1964).
325. Свентицкий Е. Н., Савоскина Г. П., сб. «Ядерный магнитный резонанс», вып. 2, изд. ЛГУ, 1967.
326. Бородин П. М., Свентицкий Е. Н., Чижик В. И., сб. «Ядерный магнитный резонанс», вып. 2, изд. ЛГУ, 1967.
327. Самитов Ю. Ю., Зыкова Т. В., Труды КХТИ им. С. М. Кирова, вып. 33, 9 (1964).
328. Арбузов Б. А., Самитов Ю. Ю., Визель А. О., Зыкова Т. В., ДАН СССР, 159, 124 (1964).
329. И о н и н Б. И., Лебедев В. Б., П е т р о в А. А., ДАН СССР, 152, 1354 (1963).
330. Morin С., Bull. Soc. chim. France, 1446 (1961).
331. Smith J. A. S., Lab. Pract., 13, 947 (1964).
332. Letcher J. H., V a n Wazer J. R., J. Chem. Phys., 44, 845; 45, 2916, 2926 (1966).
333. Bock H., В a 1 t i n E., Chem. Ber., 98, 2844 (1965).
334. F 1 u c k E., I s s 1 e i b K-, Chem. Ber., 98, 2674 (1965); Z. anorg. und allgem. Chem.,
339, 274 (1965).
335. King R. W., Hu ttemann T. J., Verkade J. G., Chem. Communs., 561 (1965).
412
Глава 12
336. Armstrong D. R., Petrakis P. G., Chem. Communs., 337 (1965).
337. Gaines D. F., Inorgan. Chem., 2, 532 (1963).
338. Pilling P. L., H a w t h о r n e M. F., P i er E. A., J. Am. Chem. Soc., 86, 3558 (1964).
339. Голованов И. В., Пискунов А. К-, Усп. химии, 35, 563, (1966).
340. Paul Е., G г а n t D. М., J. Am. Chem. Soc., 85, 1701 (1963).
341. Dhami K.S.,Stothers J. В., Can. J. Chem., 43, 510 (1965).
342. Dhami K- S., S t о t h er s J. B., Can. J. Chem., 43, 498 (1965).
343. Pugmire R. J., Grant D. M., Robins R. К., P h о d e G. W., J. Am. Chem. Soc., 87, 2225 (1965).
344. Maciel G. E., J. Chem. Phys., 42, 2747 (1965).
345. Traficant D. D., Maciel G. E., J. Phys. Chem., 70, 1314 (1966).
346. Bartow D. S., Richardson J. W., J. Chem. Phys., 42, 4018 (1965).
347. Yonezawa T., Morishima I., Fujii M., Bull. chem. Soc., Japan, 39, 2110 (1966).
348. Bourn A. J. R., R a n d a 1 1 E. W., Molec. Phys., 8, 567 (1964).
349. Rogers M. T., L a - P 1 a n c h e L. A., J. Phys. Chem., 69, 3648 (1965).
350. Kent J.E., Wagner E. L., J. Chem. Phys., 44, 3530 (1966).
351. Bose M., Das N., Chatterjee N., J. Molec. Spectrosc., 18, 32 (1965).
352. Jackson J. A., Taube H., J. Phys. Chem., 69, 1844 (1965).
353. Di Carlo E. N.,Swift H. E., J. Phys. Chem., 68 , 551 (1964).
354. Godfrey P. D., Heffernan M. L., К e r r D. F., Austral. J. Chem., 17, 701 (1964).
355. Hatton J. V., Saito J., Schneider W. G., Can. J. Chem., 43, 47 (1965).
356. Anderson J. M., Baldeschwieler, J. D., Dittmer D. C., Phillips W. D., J. Chem. Phys., 38, 1260 (1963).
357. Bullock E., Tuck D. G., Woodhouse E. J., J. Chem. Phys., 38, 2318 (1963).
358. Franck-Neumann M., Lehn J. M., Mol. Phys., 7, 197 (1963).
359. Gassman P. G., Heckert D. C., J. Org. Chem., 30, 2859 (1965).
360. Lehn J. M., Franck- Neumann M., J. Chem. Phys., 43, 1421 (1965).
361. Lehn J.M.,Seher R., Chem. Comm., № 23 , 847 (1966).
362. Гречишкин В. С., Златогорский М. Л., веб. «Радиоспектроскопия тверд, тела», Атомиздат, М., 1967, стр. 166.
363. Gillespie R.J., Hartman J.S.,J. Chem. Phys., 45, 2712 (1966).
364. S a i k a A., J. Chem. Phys., 45, 2715 (1966).
365. H a m m a k e r R. M., J. Chem. Phys., 43, 1843 (1965).
366. Hammaker R. M., Canad. J. Chem., 43, 2916 (1965).
367. Muller N., J. Chem. Phys., 42, 4309 (1965).
368. Muller N., В i r k h a h n R. H., J. Chem. Phys., 43, 4540 (1965).
369. Watts V. S., Goldstein J. H., Theor. chim. acta, 4', 265 (1966).
370. Vincent E. J., Metzger J., C. r. Acad. Sci., 261, 1964 (1965).
371. Karabatsos G. J., Orzech С. E., Jr.,'J. Am. Chem. Soc., 87, 560 (1965).
372. S a c k m a n E., Dreeskamp H., Spectrochim. Acta, 21, 2005 (1965).
373. McLauchlan K. A., Chem. Communs., № 6, 105 (1965).
374. McFarlane W., Molec. Phys., 10, 603 (1966).
375. McLauchlan K. A., Molec. Phys., 11, 303 (1966).
376. McLauchlan K. A., W h i f f e n D. H., Reeves L. W., Molec. Phys., 10, 131 (1966).
377. Fraenkel G., Asahi Y., Batiz-Hernandez H., Bernheim R. A., J. Chem. Phys., 44, 4647 (1966).
378. Benezra C., Owrisson G., Bull. Soc. chim. France, № 6, 1825 (1966).
379. Duval E., RanftJ., Bene G. J., Molec. Phys., 9, 427 (1965).
380. Brune H. A., Chem. Ber., 97, 2848 (1964).
381. Yoshioka T., MacDiarmid A. G., J. Molec. Spectrosc., 21, 103 (1966).
382. Kettle S. F. A., J. Chem. Soc., 1965, 6664.
383. Kiefer E. F., W a t e r s W. L., J. Am. Chem. Soc., 87, 4401 (1965).
384. De Jeu W. H., G a u r H. A., S m i d t J., Rec. trav. chim. des pays-bas, 84, 1621 (1965).
385. Мальцев Г. 3., Малинин Г. В., Машовец В. П., Щербаков В. А., Журн. структ. хим., 6, 371 (1965).
Приложения
приложение I
т-значения для различных органических соединений [данные, приведенные в этом приложении, представлены Дж. В. Д. Тирсом и относятся к растворам соединений в СС14,причем химический сдвиг эталонного Si(CH3)4 определяется значением т= + 10,00 м. д.[
Соединение а Тб, м. д. Мульти-плетность сигнала в Концентрация В СС14 г %
^СвН4(СНО)2 —0,083 3,0
С6Н4СНО +0,035 5,0
4-С1СвН4СНО 0,067 5,0
4-СН3ОС6Н4СНО 0,199 5,0
СНзСНО 0,28 4 5,0
Цитронеллаль (СНО) 0,32 2 10,0
4-(CH3)2NC6H4CHO 0,350 5,0
СвН5СН = СНСНО 0,37 2 5,0
(СН3)2СНСНО 0,43 2 5,0
СНзСН = СНСНО 0,57 2 5,0
С6Н5СН = NN = СНС6Н6 1,45 5,0
Пиридин (2) 1,50 м 5,0
n-CeH^NC^ 1,55 2,0
4-NO2CeH4SO2Cl (3) 1,55 НК 5,0
(2) 1,75 НК 5,0
НСО2СН3 1,97 10,0
я-СвН4(СНО)2 1,98 3,0
п-СбН4(СС13)2 2,017 5,0
4-СН3ОС6Н4СО2СН3 (2) 2,10 НК 5,0
HCON(CH3)2 2,16 5,0
4-FO2SC6H4CH3 (3) 2,16 НК 5,0
4-С1СвН4СНО (2) 2,22 НК 5,0
n-CeH4(CF3)2 2,225 5,0
4-BrC6H4COCH3 (2) 2,23 НК 5,0
4-СН3ОСвН4СНО (2) 2,25 НК 5,0
Нафталин (1) 2,27 м 5,0
СвН5СН = СНСО2Н 2,28 НК 5,0
С6Н5СНО ~ 2,37 м 5,0
4-(CH3)2NC6H4CHO (2) 2,37 НК 5,0
о-С6Н4(С02СН3)2 2,38 м 5,0
Кумарин (Н, Р относительно СО2) 2,39 НК 5,0
п^СвНвСвН^Нв 2,40 4,0
Фталид (ароматические Н) 2,42 м 5,0
(С6Н5СН = СН)2СО 2,43 НК 4,0
4-ВгС6Н4СОСН3 (3) 2,43 НК 5,0
c6h5cn 2,464 5,0
н-Сз Р7СеНв 2,473 5,0
416
Продолжение приложения I
Соединение а Тб, м. д. Мульти-плетность сигнала в Концентрация в CCI4 %
C6H5CF3 2,492 5,0
4-С1С6Н4СНО (3) 2,52 НК 5,0
С6Н5СОСН3 ~ 2,52 м 3,0
С6Н5СН = СНСНО 2,54 5,0
3,5-Дибромтолуол (4) 2,57 5,0
4,4'-С1С6Н4С6Н4С1 (2, 3) 2,59 5,0
я-СеЬЩг 2,617 4,0
Фуран (2) 2,62 5,0
С6Н5СН = СНСНО 2,62 НК 5,0
С6Н5СН = СНСОСНз 2,62 НК 5,0
4-FO2SC6H4CH3 (2) 2,63 НК 5,0
Нафталин (2) 2,63 м 5,0
CeHbSiHs 2,63 м 5,0
С6Н5СНВгСН2СС13 2,64 м 5,0
С6Н5СН = СНСОСНз 2,64 5,0
n-c6H4Br2 2,664 3,0
п-СвН4(СН2С1)2 2,671 4,0
п-С6Н4(СН2Вг)2 2,677 4,0
С6Н5С = сн 2,678 3,0
С6Н5С(СН3)2ООН 2,68 6,0
Кумарин (ароматические Н) 2,69 м 5,0
С6Н5С(СН3) = СН2 2,70 м 5,0
С6Н5СН2С1 2,724 м 5,0
транс-Стильбен (ароматические Н) 2,73 5,0
Бензол 2,734 2,0
С6Н5СН2Вг 2,736 5,0
С6Н5Вг 2,74 м 5,0
Хлороформ 2,75 6,0
С6Н5С1 2,756 5,0
и-С6Н4С12 2,759 4,0
(С6Н5СН2)2О 2,76 5,0
(С6Н5)2С = сн2 * 2,76 м 5,0
С6Н5СН = СНВг 2,77 5,0
3,5-Дибромтолуол (2) 2,79 5,0
СНС12Вг 2,80 5,0
C6H5CH2N(CH3)2 2,805 5,0
Т иофен (2) 2,81 м 5,0
С6Н5С(СН3)3 2,813 4,0
1 -(С6Н5)-Циклогексанол 2,82 10,0
С6Н5СН2ОН 2,83 6,0
С6Н5СН2СН2СОСН3 2,89 5,0
(С6Н5)2СН2 2,86 5,0
С6Н5СН(СН3)2 2,879 5,0
С6Н5СН2СН3 2,888 5,0
С6Н5СН2СН2С6Н5 2,893 5,0
Толуол (ароматические Н) 2,905 5,0
(4-СН3С6Н4СН2)2О 2,93 5,0 -
СНС1Вг2 2,94 5,0
СвН5СН = СНВг 2,95 НК 5,0
417
Продолжение приложения I
Соединение а тб, м. д. Мультиплетность сигнала в Концентрация в CCI4 г, %
Тиофен (3) 2,96 М 5,0
СН3СН = СНСО2Н 2,96 нк, 4 6,0
4-Фтортолуол (2) 2,96 НК 5,0
4-Ацетокситолуол (2) 2,97 НК 5,0
о-Ксилол (ароматические Н) 2,99 5,0
СНВг = СНВг(ч«с) 3,00 5,0
n-Дициклогексилбензол (ароматические Н) 3,008 10,0
Стильбен (олефиновые транс-Н) 3,01 5,0
4-СН3СвН5СН(СН3)2 3,02 3,0
Тетралин (ароматические Н) 3,03 5,0
1,4-Декаметиленбензол (ароматические Н) 3,03 е
(СвН5СН = СН)2СО 3,05 НК 4,0
я-Ксилол (ароматические Н) 3,053 5,0
4-Фтортолуол (3) 3,08 НК, м 5,0
п-СН3ОСвН4СНО (3) 3,09 НК 5,0
л-Ксилол (ароматические Н) 3,11 4,0
4-Ацетокситолуол (3) 3,13 НК 5,0
1,4-Нафтохинон (2) 3,15 3,0
СН3СН = СНСНО 3,15 нк, 4 5,0
4-СН3ОС6Н4СО2СН3 (3) 3,16 НК 5,0
СНВгз 3,18 7,0
1,2,4-СвН3(СН3)3 3,18 5,0
1,3,5-СвН3[СН(СН3)2]3 3,21 10,0
1,4-Бензохинон 3,24 2,0
n-Крезол (ароматические Н) 3,25 3,0
Дурол (ароматические Н) 3,259 5,0
Диэтилфумарат (олефиновые Н) 3,26 5,0
С6Н5СН = СНВг 3,29 , нк 5,0
п-С6Н4(ОС2Н5)2 3,329 4,0
Норборнадиен (олефиновые Н) 3,35 10,0
Мезитилен (ароматические Н) 3,356 5,0
СвН5СН = СНСНО 3,36 нк, 2 5,0
4-(CH3)2NCeH4CHO (3) 3,37 НК 5,0
СНВг = СНВг (транс) 3,38 5,0
СвН5СНС12 3,39 5,0
Пиррол (2) 3,40 м 6,0
С6Н5СН = СНСОСНз 3,40 нк 5,0
Анилин (ароматические Н) ~3,41 м 6,0
СС12 = СНС1 3,56 10,0
СНС1 = СНС1 (цис) 3,57 5,0
Циклопентадиен (олефиновые Н) 3,58 6,0
СвН5СН = СНСО2Н 3,61 нк 5,0
2,2-Парациклофан (ароматические Н) 3,62 е
СНС1 = СНС1 (транс) 3,63 5,0
Аскаридол (олефиновые Н) 3,65 5,0
Фуран (3) 3,70 5,0
Кумарин (а-Н относительно СО2) 3,72 нк 5,0
Дигидропиран (2) 3,78 2 10,0
Диметилмалеат (олефиновые Н) 3,87 5,0
27—489
418
Продолжение приложения I
Соединение а Тб, М. д. Мульти-плетность сигнала в Концентрация в ССЦ г, %
Пиррол (3) 3,91 М 6,0
СС13СНС12 3,95 10,0
СНзСН = СНСНО 3,95 нк, 2 5,0
СНВг2СНВг2 3,97 5,0
Этилметакрилат 1-й (олефиновый Н) 3,97 5,0
(СН3)2С = СНСОСНз 4,03 м 5,0
Нонборнен (олефиновые Н) 4,06 2,0
СНС12СНС12 4,06 5,0
CF2C1CHC12 4,08 3 10,0
cf3ch2chci2 4,08 3 5,0
С1СН2СН = СНСН2С1 4,09 3 10,0
w-CjFisH 4,11 з, з 97,0 Д
СНС12СО2СН3 4,11 5,0
Дициклопентадиен (олефиновые Н, нонборненовое 4,13 ш 10,0
кольцо)
СНзСН = СНСО2Н 4,22 НК 6,0
Циклогексадиен-1,3 (олефиновое Н) 4,22 е
СН2 = CCH3CN 4,25 2 5
Циклооктатриен-1,3,5 (олефиновые Н) 4,26 е
СНС12СН2С1 4,26 3 10,0
Д2-Циклогексенол (2, 3) 4,28 щ 10,0
2,5-Диметилфуран (3) 4,30 5,0
Циклооктатетраен 4,309 3,0
а-Фелландрен (олефиновые Н) 4,36 и 10,0
Циклогексен (олефиновые Н) 4,43 3 10,0
Кротиловый спирт (олефиновые Н) 4,45 . щ 5,0
w-C7F15CCH3 — СН2 4,46 щ 10,0
Этилметакрилат (2-й олефиновый Н) 4,53 5,0
(CeH5)3SiH 4,57 10,0
(С6Н5)2С = СН2 4,60 5,0
Дициклопентадиен (олефиновые Н, циклопентено- 4,60 ш 10,0
вое кольцо)
Метилрицинолеат (олефиновые Н) 4,62 10,0
Лимонен (олефиновые Н, кольцо) 4,65 10,0
СН2С12 4,67 5,0
Метиллинолеат (олефиновые Н) 4,68 10,0
1-Метилциклогексен (олефиновые Н) 4,70 3 5,0
Метилолеат (олефиновые Н) 4,72 10,0
СвН5ССН3 = снн 4,72 НК 5,0
Нойол (олефиновые Н) 4,73 щ 10,0
Фталид (бензильный Н) 4,74 5,0
СвН5СНВгСН2СС13 4,74 м 5,0
СН = СССНз = СН2 4,74 м 6,0
(СН3)2С = СНСН3 4,79 97,0д
СН2С1Вг 4,84 5,0
а-Пинен (олефиновые Н) 4,88 3 10,0 .
Сквален (олефиновые Н) 4y9F 10,0
Цитронеллаль (олефиновые Н) 4,97 10,0
С6Н5ССН3 =;СНН 4,98 нк . 5,0
419
Продолжение приложения I
Соединение а Г6, м. д. Мульти-плетность сигнала в Концентрация в CCI4 г, %
С1СН2ССН3 = снн 4,98 нк 5,0
Гераниол (олефиновые Н) -5,00 Ш 10,0
Триоксан 5,00 4,0
СН2СП 5,01 5,0
НС(ОС2Н5)3 5,04 5,0
СН2Вг2 5,06 5 0
СН2 = СНССНз = СН2 5,06 10,0
С1СН2ССН3 = СНН 5,10 нк 5,0
(СбН5)2^1Н2 5,14 5 0
(CF3CH2O)2CH2 5,18 2 0
Диформа ль пентаэритрита 5,29 5 0
Лимонен (олефиновые Н) 5,34 10,0
Изобутилен (олефиновые Н) 5,399 5,0
СН3СО2ССНз = СН2 5,40 5 0
«-C3F7CH2OOCC2H5 5,44 3 5 0
Дигидропиран (3) 5,46 2, м 10,0
C6HSCH2C1 5,50 5 0
n-GeH4(CH2Cl)2 5,50 4,0
(С6Н5СН2)2О 5,51 5 0
(СН3О)2СН2 5,51 5 0
(CH3)2CHNO2 5,56 7 6 0
C6HsCH2Br 5,57 5,0
п-СбН4(СН2Вг)2 5,59 4 0
С6Н5СН2ОН 5,60 6,0
СР3СО2СН2СНз 5,61 3 5 0
Циклопентилбромид (1) 5,61 м 10,0
(С1СН2)2ССН2ОСН2 5,64 10 0
(4-СН3С6Н4СН2)2О 5,64 5 0
5,65 2, ш 10,0
«-С3Ь7СО2СН3 • 5,70 5,0 10 0
СР3СО2СН2СзН7 5,71 3
у-Бутиролактон (СН2О) 5,72 3 5,0
CH3NO2 5,722 2,0
CH3F 5,74 ж 2
(СН3)2СН1 5,76 7 10 0
Р-Пропиолактон (СН2О) 5,78 3 5 0
Диэтилфумарат (СН2О) 5,79 4 5,0 3,0 .10,0 5,0 5,0
Этиленкарбонат 5 80
(СН3)2СНВг С1СН2РОС12 Диэтилмалеат C6HsSiH3 (СН2О) 5^80 5,80 5,81 2 2 4
(/г-СНзСбН4)2СНСНз НС = ССН2ОН С1СН2С = ССН2С1 5,81 5,81 5,82 5,84 5,85 5,85 5,87 4 2 5,0; 10,0 5,0 3,0
(С2Н5)2С(СО2СН2СНз)2 5,0 5,0 5,0 10,0 ,
СН2 = ССН3СО2СН2СН2
(СН3)2СНС1 7
27*
420
Продолжение приложения I
— _ б Мульти- Концентра-
Соединение а т , м. д. плетиость сигнала в ция в CCI4 г, %
Циклогексилбромид Д2-Циклогексенол НС = ССН2С1 (1) (1) 5,89 5,89 5,91 М Ш 10,0 10,0 6,0
Вг(СН2)ю СО2СН2СН3 C1CH2CN 5,92 5,93 4 20,0 2,0
6-Валеролактон (СН2О) Ферроцен СН3СО2СН2СН3 5,94 5,95 5,95 ш 4 10,0 4,0 5,0
3,3,5-Тр иметилциклогексанол (1) 5,95 10,0 10,0
С1СН2СН = СНСН2С1 5,96 2
2-Бромоктан н-С3Н7СН2ОН (2) 5,97 5,98 м 3 10,0 3,0; 6,0
СНС12СН2С1 6,03 3 10,0
Гераниол (СН2О) CF3CO2CH3 6,03 6,04 2 10,0 3,0
СН2 = ССН3СН2С1 6,04 м 5,0
Этилентритиокарбонат 6,06 5,0
Диметилсульфат 6,06 ж 2,0 10,0
(CF3CH2O)2CH2 h-C3F7CH2C1 6,07 6,07 3
СНС12СО2СН3 6,08 5,0
п-СбН^ОСНгСНзЬ (С6Н5)2СН2 6,08 6,08 4 5,0 5,0
(С1СН2)2ССН2ОСН2 Дигидропиран 2-Хлорбутан ' СН212 (6) (2) 6,09 6,10 6,10 , 6,10 10,0 10,0 5,0 5,0
С6Н5СО2СН3 6,10 5,0
СН3СН = СНСН2ОН 6,10 ш 5,0
Циклогексилхлорид (1) 6,11 ш 10,0 5,0
4-СН3ОС6Н4СНО 6,143
СН2 = СНСН2Вг 6,15 2 5,0
CF3CH2OH 6,15 4 3,0
4-СН3ОС6Н4СО2СН3 6,173 5,0
СН s ССН2Вг 6,18 5,0
Диметилфтал ат (СН3) 6,18 5,0
Флуорен (4-СН3СвН4)2СН2 4 <СН2) 6,19 6,19 5,0 6,0
Диметилкарбонат 6,23 ж 5,0
ВгСН2СО2СН3 6,24
(CF3)2CHCF2OCH3 (С1СН2)3ССН2ОН (СН3О)2РНО СвН6ОСН3 6,24 6,25 6,25 6,266 2 10,0 6,0 5,0 6,0
4-СН3ОСвН4СН3 6,293 5,0
СвН5СНВгСН2СС13 6,30 2 5,0
ВгСН2СО2СН3 6,30 5,0
mpm-2-Метилциклогексанол (СНОН) 6,30 ш 10,0
421
Продолжение приложения I
. б Мульти- Концентра-
„ а Соединение м. д. плетность ция в ССЦ г,
сигнала %
CHO2CH3 6,31 10,0
Борнеол (СНОН) 6,31 3 10,0
С1СН2СН2С1 6,31 2,0
СН3СО2СН3 6,35 ж
CH3SO2C1 6,36 4,0
(С1СН2)зССН2ОН 6,36 6,0
Тетрагидрофуран (2) 6,37 3 6,0
ВгСН2СН2Вг 6,369 5,0
Диформаль пентаэритрита 6,38 5,0
Метилолеат, метиллинолеат 6,38 10,0
Метилрицинолеат 6,39 10,0
CH3CH2NCS 6,39 4 5,0
СН3СН2ОН 6,41 4 3,0; 6,0
K-C3F7CH2I 6,42 3 10,0
ВгСН2СН2СН2Вг 6,42 3 5,0
N-Метилморфолин (СН2О) 6,42 3 5,0
Диметилсульфит 6,42 ж
Диоксан 6,43 2,0
(СН2СН2С1)2 6,43 3 5,0
Морфолин (СН2О) 6,43 3 6,0; 2,0
Тетрагидропиран (СН2О) 6,44 м 10,0
cf3ch2i 6,44 4 5,0
(СНзОС2Н4ОС2Н4)2О 6,44 5,0
Тетраметилсиликат 6,44 ж
С(СН2С1)4 6,46 5,0
4-тргт-Бутилциклогексанол (1) 6,46 м 10,0
Триметилборат* 6,47 ж
Нопол (СН2О) 6,49 , 3 10,0
НС(ОСН2СН3)з 6,50 4 5,0
(З-Пропиолактон (СН2СО) 6,52 3 5,0.
(СН3ОС2Н4ОС2Н4)2О 6,52 5,0
Норборнадиен (СН) 6,53 м 10,0
(СН3ОС2Н4)2О 6,54 5,0
н-С3Н7СН2С1 6,54 3 10,0
С2Н5СН2С1 6,55 3 5,0
(СН2СН2Вг)2 6,58 3 5,0
(CH3CH2)4N+I- (в CF3CO2H) - 6,60 4 (5,0)
СН3ОН 6,622 1,0; 2,0
6,63 3,6; 97 Д
Пирролидон (CH2N) 6,63 3 6,0
СН3СН2Вг 6,64 4 5,0
С2Н5СН2Вг 6,64 3 5,0
(СН3СН2)2О 6,64 4 5,0
CF3C(OCH3)3 6,64 3,0
(СНз)2СНСН2С1 6,65 2 10,0
ВгСН2(СН2)вСО2С2Н5 6,65 3 20,0
«-С7Н15СН2Вг 6,65 3 10,0
Аценафтен (СН2) 6,66 5,0
Н-СЗН7СН2ВГ 6,66 3 10,0
422
Продолжение приложения I
Соединение а Гб, м. д. Мульти-плетность сигнала в Концентрация в CCI4 г, %
(CH3)4N+I- (в CF3CO2H) 6,67 (1,0)
C6H5CH2N(CH3)2 6,68 5,0
Димето кситетрагл икол ь (СН3) 6,69 5,0
Д иметоксидигл икол ь (СН3) 6,70 5,0
h-C3F7CON(CH2CH3)2 6,72 4 5,0
ch3ch2so2f 6,72 4, 2 5,0
(СН3О)2СН2 6,72 5,0
ch3so2f 6,73 2 2,0
(СН3)2СНСН2Вг 6,74 2 5,0
(СН3)2О 6,76 ж
с2н5сн21 6,80 3 10,0
н-С3Н7СН21 6,81 3 10,0
СН3СН21 6,85 4 5,0
ch3so2cf3 6,87 5,0
е-Капролактам (CH2N) 6,88 м 6,0
h-C3F7CON(CH3)2 6,90 2 5,0
(СН3)2СНСН21 6,93 2 10,0
СН3С1 6,95 ж
2,2-Парациклофан (СН2) 6,95 е
4-(CH3)2NCeH4CHO 6,951 5,0
CF3SO2NHCH3 6,96 2 5,0
QB,С = сн 7,07 3,0
CH2CH2SO2CH2CH2 7,08 3 5,0
C6H5N(CH3)2 7,096 5,0
Циклопейтадиен (СН2) 7,10 6,0
СбНйСНгСНгСеНб 7,129 5,0
(CH3)2CHNH2 7,13 м 3,0; 6,0
С6Н5СН(СН3)2 - 7,13 м 5,0 '
(CH3)2NCHO 7,15 2 5,0
Морфолин (CH2N) 7,17 3 2,0; 6,0
Ацетилбромид 7,19 ж
Дициклопентадиен (кольцо Се, протон СН в голове 7,20 10,0
моста)
Норборнен (аллильный Н) 7,22 2,0
Пирролидин (CH2N) 7,26 м 3,0
(СНСН2О)2 7,28 м 15,0
СНзСС1з 7,28 3,0
НС = СССНз = сн2 7,29 6,0
Тетралин (бензильный Н) 7,30 м 5,0
Пиперидин (CH2N) 7,31 м 3,0
1-С10Н7СОСН3 7,32 5,0
СН3Вг 7,32 ж
СН3СНСН2О 7,34 м 10,0
Ацетилхлорид 7,34 .3,0
(СНСН2О)2 7,37 м 15,0
h-C3H7CH2NH2 7,37 3 6,0
CH3SCN 7,37 3,0
4-ВгС6Н4СОСН3 7,38 5,0
423
Продолжение приложения I
Соединение а тб, М. д. Мультиплети ость сигнала в Концентрация в ССЦ г, %
С6Н5СН2СН3 7,382 4 5,0
(CH3)3N-BH3 7,40 5,0
[(CH3)2N]3PO 7,40 2,0
С6Н5СОСН3 7,45 3,0
2-C4H3SCOCH3 7,46 5,0
(CH3)2SO 7,50 2,0
4-FO2SCeH4CH3 7,508 5,0
CH3CH2SH 7,56 4 3,0
(CH3CH2)3N 7,58 4 5,0
Циклогептанон (СН2СО) 7,58 ш 12,0
W-C3F7COCH3 7,59 5,0
НС = CCH2C1 7,60 3,0; 6,0
(CH3CH2)2CO 7,61 4 5,0
(Ca^C^S 7,61 3 5,0
ВгСН2СН2СН2Вг 7,64 5 5,0
с6н5сн3 7,663 2,0
ch3nnh2 7,67 2,0; 6,0
CH3COSH 7,67 2,0; 6,0
НС ЗЕ ССН2Вг 7,67 5,0
НС = ССН2ОН 7,67 3,0; 6,0
4-СН3СО2С6Н4СН3 7,682 5,0
3,5-Вг2С6Н3СН3 7,682 5,0
е-Капролактам (СН2СО) 7,69 ш 6,0
у- Бутиролактон (СН2СО) 7,69 3 5,0
Ци клооктатр иен -1,3,5 (СН2) 7,69 е
С6Н5СН = СНСОСНз 7,69 5,0
(4-СН3СвН4СН2)2О 7,69 5,0
4-FC6H4CH3 7,700 5,0
п-НОСеН4СН3 7,70 ' 3,0
СН3СНСН2О 7,71 м 10,0
N-Метилморфолин (CH2N) 7,72 3 5,0
л4-СвН4(СН3)2 7,726 4,0
П-С6Н4(СН3)2 7,727 5,0
4-СН3С6Н4СН(СН3)2 7,73 3,0
б-Валеролактон (СН2СО) 7,73 ш 10,0
(4-СН3СвН4)2СН2 7,73 6
4-СН3ОС6Н4СН3 7,748 5,0
Циклогексанон (СН2СО) 7,75 м 6,0
2,5-Д иметилфуран 7,76 5,0
О-СбН4(СНз)2 7,769 5,0
1-Фенилциклогексанол (2) 7,77 ш 10,0
Диацетил 7,77 2,0
2-Пирролидон (3) 7,77 3 6,0
Мезитилен (СН3) 7,778 5,0
Нопол (аллильная СН2-группа) ~ 7,78 ш 10,0
CH3CH2CO2CH2C3F7 7,78 4 5,0
Вг(СН2)9СН2СО2С2Н5 7,79 3 20,0
N-Метилморфол ин 7,80 5,0
Уксусный ангидрид 7,803 3,0
424
Продолжение приложения I
Соединение а г6, м. д. Мульти-плетность сигнала в Концентрация в CCI4 Г» %
Норборнан (СН) 7,81 5,0
Цитронелл аль (СН2СО) 7,81 М 10,0
4-СН3СО2СвН4СНз 7,815 5,0
Метилол еат (СН2СО) 7,83 Ш 10,0
СвН5СН2М(СН3)2 7,83 5,0
СН3СНО 7,83 5,0
(2H2CH2SO2CH2CH2 7,84 5,0
Циклогексадиен-1,3 (СН2) 7,84 е
Метил иодид 7,843 2,0
СвН5ССН3 = СН2 7,86 5,0
2-Пирролидон (4) 7,86 м 6,0
Дурол (СН3) 7,862 5,0
Триметиламин 7,876 2,0
CH3CONCH2CH2 7,88 10,0
(СН2 = СНСН2)2 7,88 2 20,0
Ацетон 7,914 3,0
(CH3)2S 7,92 2,0
у-Бутиролактон (Р-Н) 7,92 м 5,0
СН3СО2ССН3 = СН2 7,93 5,0
СвН5СН2СН2СОСН3 7,93 5,0
(CF3)2CHCF2OCH3 7,93 м 97,0 Д
Метил рицинолеат (СН2СО) 7,93 3 10,0
Уксусная кислота 7,933 6,0
(СН3)2С = СНСОСНз (СН3СО и СН3 цис относи- 7,94 5,0
тельно СО) CH3CO2CH2C3F7 7,94 5,0
Сквален (СН2СН = С) 7,96 2 10,0 .
Камфора (СН2СО) ' 7,96 м 10,0
(ВгСН2СН2)2 7,97 3 5,0
СН2 = CCH3CN 7,97 5,0
СНзСН = СНСНО 7,98 2 5,0; 25.
Циклопентанон (2, 3) 7,98 6,0
Д2-Циклогексенол (4) 7,98 ш 10,0
СН3СО2СН3 7,99 ж
Сквален (СН2ССНз = С) 8,01 10,0
а-Фелландрен (аллильная СН2-группа) 8,01 ш 10,0
Ацетонитрил 8,029 3,0
Лнмонен (аллильная СН2-группа) 8,03 м 10,0
(С1СН2СН2)2 8,04 3 5,0
Циклогексен (аллильная СН2-группа) 8,04 м 10,0
Декадиен-1,9 (аллильная СН2-группа) 8,05 ш 5,0
СН3СО2С2Н5 8,05 5,0
Циклопентилбромид (2, 3) 8,05 м 10,0
Норборнадиен (СН2) 8,05 3 10,0
н-С,Р15ССН3 = СН2 8,06 10,0
СН2 = ССН3СО2С2Н5 8,06 5,0
а-Терпинеол (аллильная СН2-группа) 8,06 ш 10,0
Циклогексилбромид (2) 8,07 ш 10,0
Дигидропиран (4) 8,07 м 10,0
425
Продолжение приложения I
Соединение а г6, М. Д. Мульти-плетность сигнала в Концентрация в ССЦ г, %
СН3СН = СНСО2Н 8,07 2 6,0
СвН5СОС(СО2Н)СН2СН2 (в CF3CO2H) 8,08 (5,0>
СНзСО2ССН3 = СН2 8,09 5,0
СН2 = ССН3СН2С(СН3)з 8,09 7,0'
Терпинилацетат (СН2С — О) 8,10 ш 10,0-
СН2 = ССНзС = СН 8,11 6,0
СН3СН2СН2Вг 8,11 ш 5,0
Цитронеллаль (аллильная СН2-группа) 8,11 м 10,0
СН3СО2ССН3 = СН2 8,11 5,0
Терпинилацетат 8,12 10,0
(СН3)2СН1 8,12 10,0
Циклогексилхлорид (2) 8,13 ш 10,0
.СН2 = ССН3СН2С1 8,13 5,0
Циклогексанон (3, 4) 8,13 ш 6,0
(СН3)2С = СНСОСНз (СН3 транс относительно СО) 8,12 2 5,0
СН3СН2СН21 8,14 ш 10,0
СН3СН21 8,14 3 5,0
1 -Метилциклогексен (3, 6) 8,14 м 10,0
СН2 = ССНзСН = СН2 8,16 10,0
(СН3СН2)2С(СО2С2Н5)2 8,16 4 5,0
СН3СН2СН2С1 8,17 ш 5,0
(СН3)2С = NOH 8,19 ж
4-трет-Бутилциклогексанол (2) 8,19 10,0
(СН3)2СНСН2Вг 8,20 м 97,0 Д’-
Тетрагидрофуран (3) 8,21 3 6,0
Адамантан (СН2) 8,218 2,0
Тетралин (р-Н) 8,22 ш 5,0
СН2 = ССН3СН2С(СН3)з 8,23 7,0
(СНз)зСВг 8,24 2,0
1 -Метилциклобутилхлор ид 8,28 8,6
Цинеол (СН2С — О) 8,28 ш 10,0
Лимоиен (СН3С = СН2) 8,28 10,0
(СНзЦСНВг 8,29 2 10,0
1 - Фен илциклогекса нол (3, 4) 8,29 м 10,0
СН3СНВГбН13 8,29 2 10,0
Камфора (4, 6?) 8,29 10,0
Изобутилен (СНз) 8,299 5,0
СН3СН2СН2СН2Вг 8,30 м 10,0
2-Фенилбутан (3-СН2) 8,30 ш 10,0
СН3СН = СНСН2ОН 8,31 ш 5,0'
а-Фелландрен (аллильная СНз-группа^ 8,32 м 10,0
СН3СНС1СН2СН3 8,32 м 5,0
СН3СН2СН2СН21 8,33 м 10,0
СС12СН2СНСН = СН2 8,33 м 5,0
е-Капролактам (4, 5) 8,33 ш 6,0
Циклогептанон (3, 4) 8,34 ш 12,0
СН3СН2СН2СН2С1 8,34 м 10,0
а-Терпинеол (аллильная СН3-группа) 8,35 10,0
СН3СН2Вг 8,35 3 5,0
426
Продолжение приложения I
Соединение а Г6, М. д. Мульти-плетность сигнала в Концентрация в ССЦ Г, %
Лимонен (кольцевая аллильная СН3 группа) 8,36 10,0
(СН3)2С = СНСНз 8,37 6,0
а-Пинен (аллильная СН3-группа) 8,37 М 10,0
CF3CO2CH2(CH2)2CH3 8,37 Ш 10,0
Сквален (аллильная СН3-группа) 8,37 10,0
Гераниол (аллильная СН3-группа) 8,38 Ш 10,0
Пирролидин (3) 8,38 3 3,0; 6,0
д-Валеролактон (₽, Y) 8,38 ш 10,0
1-Метилциклогексен 8,40 10,0
1-Метилциклогексен (4, 5) 8,40 10,0
Цитронеллаль (аллильная СН3-группа) 8,40 10,0
Линалоол (аллильная СН3-группа) 8,41 ш 10,0
Борнеол (СН2) 8,41 ш 10,0
(СНз)зСС! 8,42 2,0
Тетрагидропиран (3, 4) 8,42 ш 5,0
(СН3)2СНСН2С1 8,42 м 10,0
(CH3)2CHNO2 8,43 2 6,0
(CH3CH2CH2)2S 8,44 м 5,0
CH3CH2SO2F 8,44 3 5,0
(CH3)2CHCH2I 8,44 ш 10,0
C6H5C(CH3)2OOH 8,44 6,0
(СНз)зСН ~ 8,44 м 10,0
Циклогексил хлорид (3, 4) 8,45 ш 10,0
Циклогексилбромид (3, 4) 8,46 ш 10,0
Метилциклогексан (СН2) 8,46 10,0
(СН3)2СНС1 8,46 2 10,0
Циклогептан 8,470 2,0
(4-СНзСвН4)2СНСНз ,8,47 2 5,0
Камфен (СН2) 8,48 ш 10,0
Циклопентан 8,492 2,0
Камфора (5) 8,51 10,0
СС12СН2ССН3СН = сн2 8,51 5,0
Пиперидин (3, 4) 8,51 ш 3,0; 6,0
транс-2-Метилциклогексанол (СН2) 8,52 ш 10,0
1 1 ch2ch2nh 8,52 3,0; 6,0
цисло-СеНиСРз (СН2) 8,52 ш 5,0
СН3СНС1СН2СН3 8,53 2 10,0
1,2-Бензциклооктен-1 (4, 5) 8,54 е
4-трет-Бутилциклогексанол (3, 4) 8,56 ш 10,0
Циклогексан 8,564 1,0
CF3CO2CH2CH3 8,59 3 5,0
(CH3CH2)4N+I- (в CF3CO2H) 8,59 3 (5,0)
Декалин 8,59 ш 10,0
ch3ch2ncs 8,60 3 5,0
(CH3)3CSH 8,61 3,0
C3F7C(CH3)2OH 8,63 6,0
Терпинилацетат (СН3) 8,63 10,0
Дициклопентадиен (мостикова я СН2-группа) 8,64 м 10,0
427
Продолжение приложения I
Соединение а Г6, м. д. Мультиплетность сигнала в Концентрация в CCI4 г, %
л-С6Н4(ОСН2СН3)2 8,64 3 5,0
CH3(CH2)4CH2F 8,65 ш 10,0
СН2 = ССН3СО2СН2СН3 8,65 3 5,0
ВгСН2(СН2)8СН2СО2С2Н5 8,66 10,0
Дигидрокамфен (СН2) 8,66 ш 10,0
3,3,5-Триметилциклогексанол (СН2) 8,67 ш 10,0
Аскаридол (СН3С — О) 8,68 5,0
СбН5С(СН3)3 8,681 4,0
CH3(CH2)2CH2NH2 8,69 ш 6,0
CH3CH2SH 8,69 3 3,0; 6,0
Зтилмалеат (СН3) 8,69 3 5,0
Зтилфу Марат (СН3) 8,69 3 5,0
Метилрицииолеат (СН2) 8,69 10,0
1 -Метилциклобутанол 8,70 3,0; 6,0
Нопол (одна СН3-группа) 8,70 10,0
(СН2 = СНСН2СН2СН2)2 8,70 5,0
СН3(СН2)4СН2СНВгСН3 8,71 10,0
СН;1СНСН2О 8,72 2 10,0
СН3(СН2)5СН2СН2Вг 8,72 10,0
Метилолеат (неаллильная СН2-группа) 8,72 ш 10,0
Норборнан (СН2) 8,74 ш 5,0
а-Пинен [(СН3)2С, только одна СН3-группа] 8,74 10,0
(С2Н5)2С(СО2СН2СН3)2 8,75 3 5,0
н-Гексан (СН2) 8,75 7,0
н-Гептан (СН2) 8,75 10,0
н-Гексадекан (СН2) 8,75 5,0
Вг(СН2)10СО2СН2СН3 8,75 3 20,0
(СН3)3ССО2Н 8,77 10,0
С6Н5СН(СН3)2 8,77 2 5,0
2-Фенилбутан (1-СНз) 8,77 2 10,0
(СН3)3СОН 8,78 6,0; 3,0
СН3СО2СН2СН3 8,79 3 5,0
Линалоол (СН3С - О) 8,79 10,0
1,3,5-[(СН3)2СН]3СвН3 8,80 2 10,0
с6н5сн2сн3 8,80 3 5,0
h-C3F7CON(CH2CH3)2 8,80 3 5,0
4-СН3СвН4СН(СН3)2 8,80 2 2,0
(СН3)3СООН 8,81 6,0
СН(ОСН2СН3)3 8,81 3 5,0
Цинеол [(СН3)2С - О] 8,83 10,0
СН3СН2ОН 8,83 3 6,0; 3,0
(СН3СН2)2О 8,84 3 5,0
[(СН3)зС]2О2 8,84 2,0
CH3CH2CO2CH2C3F7 8,85 3 5,0
а-Терпинеол [(СН3)2С - О] 8,87 10,0
(СН3)2СНСНО 8,88 2 5,0
3,3,5-Триметилциклогексанол (3-СН з, только одна 8,89 6,0
группа)
428
Продолжение приложения I
Соединение а г6, м. д. Мульти-плетность сигнала в Концентрация В CCI4 г, %
(CH3)3CNH2 8,92 6,0; 3,0
СН3СН2СН2С1 8,96 3 5,0
СНЗСН2СН2ВГ 8,96 3 5,0
СН3СН2СН21 8,96 3 5,0
(СН3СН2)2СО 8,96 3 5,0
(СН3)2СНСН2Вг 8,96 2 5,0
транс-2-Метилциклогексанол 8,97 2 10,0
(СН3)2СНСН2С1 8,99 2 10,0
(CH3)2CHNH2 8,99 2 6,0; 3,0
Камфен (СН3) 8,99 10,0
(CH3CH2CH2)2S 9,00 3 5,0
Фенхон (СНз) 9,00 10,0
Аскаридол [(СНЭ)2СН] 9,02 2 5,0
Цинеол (1-СНз) 9,02 10,0
(СН3)2СНСН21 9,02 2 10,0
(СН3)3ССН2СН(СН3)2 9,02 м 10,0
СН3СНС1СН2СН3 9,02 3 10,0
Камфора (1-СНэ) 9,03 10,0
CH3(CH2)3O2CCF3 9,03 3 10,0
(CH3CH2)3SiF 9,03 м 5,0
СН3(СН2)3С1 9,04 3 10,0
СН3(СН2)3Вг 9,04 3 10,0
СН3(СН2)31 9,04 3 10,0
Цитронеллаль (СНЭСН) 9,04 2 10,0
(CH3CH2)3N 9,05 3 5,0
(СН3)2СНСН2СН3 9,06 2 10,0
(СН3)4С 9,06 ж
CH3(CH2)3NH2 ,9,07 3 6,0
Метилциклогексан 9,08 10,0
(СН3)3ССН2ССНз = СН2 9,08 7,0
CH3(CH2)5F 9,10 3 10,0
н-Гексан (СНз) 9,10 3 7,0
н-Гептан (СНз) 9,11 3 10,0
и-Гексадекан (СНз) 9,П 3 5,0
Дигидрокамфен [(СН3)2С, только одна СНэ-группа] 9,П 10,0
Цитронеллол (СНЭСН) 9,11 2 2,0
СН3(СН2)5СНВгСН3 9,11 3 10,0
(СНз)зСН 9,112 2 10,0
а-Фелландрен [(СНЭ)2СН) 9,12 2 10,0
3,3,5-Трнметнлциклогексанол (3-СН3, только одна 9,12 10,0
группа)
3,3,5-Трнметилциклогексанол (5-СН3) 9,12 2 10,0
СН3(СН2)7Вг 9,12 3 10,0
Метнлолеат (концевая СН3-группа) 9,12 3 10,0
Метилрицинолеат (концевая СН3-группа) 9,12 3 10,0
Метиллннолеат (концевая СН3-группа) 9,12 3 10,0
(CH3CH2)3SiF 9,13 м 5,0
(СН3)3ССН2СН(СН3)2 9,14 м 10,0
Нопол (только одна СН3-группа) 9,14 10,0
429
Продолжение приложения I
Соединение а Г6, м. д. Мульти-плетиость сигнала в Концентрация в CCI4 Г» %
Камфора [(СН3)2С] 9,14 10,0
Борнеол [(СН3)2С?] 9,14 10,0
(СН3)2СНСН2СН3 9,14 3 10,0
4-трет-Бутилциклогексанол (СН3) 9,15 10,0
а-Пинен [(СН3)2С, только одна СН3-группа] 9,16 10,0
Борнеол (1-СН3?) 9,19 10,0
2-Фенилбутан (4) 9,20 м 10,0
(СН3СН2) 2С(СО2С2Н5)2 9,21 3 5,0
Дигидрокамфен (СН3СН) 9,21 2 10,0
Дигидрокамфен [(СН3)2С, только одна СН3-группа] 9,23 10,0
Циклопропан 9,778 5,0
[(CH3)2SiO]4 9,91 2,0
(CH3)4Sn 9,94 1,0
Тетраметилсилаи (по определению) 10,000 1,0; 5,0
а Приведенные данные относятся к протонам, выделенным в формуле жирным шрифтом или указанным цифрой в скобках.
б. ,n non 106 ('’набл— VTMC) ппп 106 (НТМС~ Ннабл) , , „
v т = 10,000 ----------------------- 10,000 ---------—----------- (м. д.). Увеличение зиаче-
VTMC НТМС
иий т соответствует возрастанию экранирования. Стандартные отклонения равны ± 0,024 м. д. для значений т с двумя цифрами после запятой и ± 0,003 м.д. для значений т с тремя цифрами после запятой.
Если особо не оговорено, резонансные сигналы представляют собой синглеты. Цифры и буквы означают: 2 — дублет; 3 — триплет; 4 — квартет; 5 — квинтет и т. д.; м — мультиплет (слабо разрешенный); ш — широкий или плохо разрешенный мультиплет; нк — половина «неэквивалентного квартета», проанализированного и отнесенного как указано в тексте [Hahn, Maxwell, Phys. Rev., 88 , 10 70 (1952)]. Отметим, что в таких случаях истинные т-значения дает «центр тяжести» несимметричной пары сигналов.
г Для жидкостей концентрация указывается в объемных процентах, для твердых веществ — в е/100 мл, причем растворитель представляет собой смесь 99 об.% CCI4 и 1 об,% 81(СНз)4. В случае необходимости изучались сильно охлажденные растворы. Скобки указывают на использование CF3COOH вместо CCI4.
д В чистую жидкость добавлялся чистый тетраметилсилаи; ССЦ'не использовался.
е По данным Уо и Фессендена [J. Am. Chem. Soc., 79,846 (1957)]. т-Зиачеиия были получены прибавлением 5,24 м. д. к химическому сдвигу сигнала: т = б + 5,24 (м. д.). Возможная ошибка (стандартное отклонение) составила ± 0,033 м.д. (для 8 измерений).
ж По данным Оллреда и Рохова [J. Am. Chem. Soc., 79, 5361 (195 7)]; т = б + 5,21 [(м. д.); возможная ошибка (стандартное отклонение) составила ± 0,036 м. д. (для 9 измерений).
ПРИЛОЖЕНИЕ II
Диаграммы протонных химических сдвигов и констант спин-спинового взаимодействия
Диаграммы т-значений для СН2 и СН3-групп, приведенные на рис.
11,1 — II.5, составили К- Нукада и сотр. [Anal Chem., 35, 1892 (1963)].
Диаграммы спектральных параметров для олефиновых соединений, приведенные на рис. II.6 — II.7 и в табл. II.1, составлены Штелингом.
— 1 с 5 7 9
СН3-сн = СНг=: СН $ СНг= -СН = О
Уисло\ . сюъекпюу
298 8
361
97 11 51
80
Рис. II. 1
43 k
Продолжение приложения II
г—>- 6 7 8 9 10 Мин. Макс. числа объем
1 2 3 4 СН38К СН3С< СН3С= СН3Й= ch3s-сн3-© CH3N< сн3о- CH3X 11 1 1 1 1 1 1 1 1 1 1 II I 1 II 1 1 1 1 1 1 1 1 1 1 1 1 ' - —L 1 1 1 1 1 1 1 1 1 9,43 8.12 7,88 7,32 7Л2 7,24 6,90 5,98 5,72 10,0 9,23 8,17 8,41 7,98 7,86 7,88 6,76 7,84 5 146 5 58 3 20 18 33 10
— L——
l
_!LL_ 1 1 1 1 1 1 1 i 1 1 1 1 1 1 1111. 1 1 1 1 1 1 1 1 1 1 1II1 1 1 1 1
г—»- 6 7 & 9 10 298
Рис. II.2
Г 6 7 8 5 Мин. Макс. ^иСЙО ооьек Off!
1 \|/ \/ <>,11 О Z О уо <Л X 1 i II Т 1 1 1 -о- -о 1(0 1 X О —ГТ 1 1 1 1 1 1 1 1 1 I I J i 1 1 1 1 1 1 1 11 1 1 1 1 1 X _L j 1 1 L 8,90 8,77 8,77 8,68 8,56 8,47 8,12 9,21 9,05 8,96 8,80 9,02 8,77 8,51 51 14 4 6 33 5 11
2 1 1 о ! ° 1 “A I । i А ?’> ? । ii-?' и о о о 1 о о 1\ /1\ 1 | л 1 7,86 7,82 7,89 7,59 7,69 7,32 8,41 8,05 8,03 8,05 7,94 7,55 25 31 8 10 3 5
3 CHjN-C^ -с=о J— 7,66 6,90 6,95 7,88 7,29 7,26 5 4 5
4 CHjO-CS -о -С=0 Т т t 1 1 У f f f t t t t t T I f 1 t 1 И 11 6,53 6,14 6,04 6.76 6,39 6,43 11 6 12
Т *- 6 7 8 9
Р и с. II.3
432
Продолжение приложения II
г 5 t 7 8 9 10 Mun. Макс. 4ucn<f объектов
У V tl III I М/ V 1 II 1. V II 1/ II 1 III 1 Л L V t III и II О и , 1 1 I 1 и <_ 1 <Л 0-0 О <Л О Z W <Л-О tO Z 2 U Z-О <Л О к- О Г — СО Z Щ и Z Q-O.-Z и. СО О О О О О, СО 10i о о Jft Jfl.bl Ы Jr. Jj. !Nh4 Jj. £ к, * ±« ±ч±~ -S'. £ Iм ±« iNi- и оиооиОи oooooooC>o<jooo<Jooo<J<J<Jlr>tT,<7>'7'¥'r><?<?<T,'7,'7!YV 6 6 <j 6 о А 6 6-6-6 6 6 6 6 6 А Л-1-6 6 А 6 6 А-6 S-6-о о о о (Ji(^)oq-o(AjcS<q об л\ /|\ /к /|\ л w л\ m и Н л\ и /л И' hi и и л' v in у и и 11 hi Л' 1 111 11 I I I 11 i 1 i 1 111111111 I 1 1 1 1 1 1 1 I 1 9,03 7,97 7,58 7,20 7,03 6,66 6,40 6,80 6,75 6,08 6,37 6,21 6,39 6,31 6,12 6,03 5,97 5,92 6,16 5,52 6,04 5,95 5,57 5,63 5,87 5,42 5,63 5,57 5,40 5,18 5,04 4,93 4,66 4,49 9,49 9,02 8,14 7,87 7Л? 7,38 7,72 6,93 6,98 7,30 6,74 7,07 6,66 6,65 6,76 6,55 6,55 6,59 6,31 6,64 6,19 6,18 6,68 $30 5,91 6,08 5,72 5,68 5,55 5,59 5,99 5,83 5,66 4,65 4 69 25 15 9 9 38 4 3 7 8 6 2 7 4 5 4 2 2 42 5 1 2 11 6 2 1 10 1 2 1 4 4 5 10 6 18 1 1 1 2
r
!'
X I
I
I l__ r
t
X I I
J. 1 , 1 I L
1 X X 1
I I X < г 1 1 ' 1 , 1 1 —1 1 —1—LJ—LL L.l t i I I I I I I I I I J i i i i i i > i i 1 i I i i t i i 1 1 1 1 1 1 1 1 1
Т 5 5 7 8 3 10 361
Рис. II.4
433
Продолжение приложения II
Т 5 6 7 8 9 Мии. Мша Чисм*
1 1 jc-cH^c-ce -X -0 1 11 11 | 1 1 1 1 I II 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 тт 8,46 8,38 7,97 7,98 9,02 8,67 8,34 8,21 11 3 11 3
2 $С-СНгС=С< -6=0 «4 7,88 7,58 8,14 7,93 11 13
3 М/ п 1 1 1 1 1? Х-Ж-Х Ж Ж 2-6 ? - по 1 - 7,47 7,37 7,26 6,62 6,72 7,70 7,55 7,51 7,00 6,88 6 4 6 4 5
4 7С-С-СНгС=С< 0=^-СНгС=0 1 X 7,27 6,08 7,30 6,75 2 5
5 SCCHfO-C$ -С=С -© -6=0 —1 11 1 6,37 6,33 5,84 5,67 8,64 6,64 6,12 5,98 7 3 5 12
6 D-CH2-N<£| -О —с=о 1 6,18 5,57 6,68 5,92 5 5 1
7 SCCH^-O-C? -О 5,79 5,04 5,98 5,37 3 6
8 O-CHj-0-C^ -6=0 J ' LLLLL 1 11111-111 11)111111 1 1 1 1 1 1 1 1 1 111111111 1 1 5,48 4,90 4,66 5,66 5,04 5,03 8 2 6
г * 5 6 18 9
Рис. II.5
28—48»
25
11
7,0 т 0,2 03 0,4 0,5 0,6 0,7 ОД 0,9 8,0 0,1 0,2 ОД 0,4 0,5 0,6 0,7 0g 0,9 0.0 0,1 0,2 0,3
---1--1---1--1--1--1---1--1--1--1--7^—1---1--1--1---1--1--1--1---Г" I ~|----
7<н3
-Шснэ I п>2
31
11
10
2
5
3
/з-сн3
Все группы -СН2СН3 u - CH(CH3)R
GHz=CHC (сНэ^й
□ цис-1 Зне ццс-R
у-сн2-
/3 -CH2-
р -сн:
41
10
7 б
12
6
*-сн3
30
11
6
3
6
5,8
Число объекта i
-CfcHj^R siu-u тризамещенныхзпиеиах
нецис-R uuc-R
0Нг=С(сНз)й транс-R СН=СНСН3 Zjuc-RCH’CHCHj R,C=C(CH$)R,nuc и транс RCH=c(cH3)R й2С=СНСНз
сС-СН2-
Все группы -СН2СН3
СН2=СН-СНг (СНД, СН3 п>1 трапа -КСН=СН-СН2(СН2\,СН3 п»1
Всегдут>и-СН2СК3
сС-СН'
ццс-R
7J) V <J2 0,3 0,4 0,5 0,6 0,7 0,8 0,3 8,0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,3 9,0 01 0,2 0,3 V-злаченая---------*-
6
3
Рис. II.6. Химические сдвиги неолефиновых протонов в моноолефиновых углеводородах.
Диаграмма может быть использована для чистых жидкостей, разбавленных растворов в СС14 или для смесей олефинов.
Р и с. II.7. Химические сдвиги олефиновых протонов в моноолефиновы’с углеводородах (изученных в виде чистых жидкостей).
435
Таблица П.1
КОНСТАНТЫ СПИН-СПИНОВОГО ВЗАИМОДЕЙСТВИЯ в МОНООЛЕФИНОВЫХ УГЛЕВОДОРОДАХ
Число исследованных соединений Структура соединения а Константа J, гц
л и II и к •^й-нС=16’6—17>4 '
н/ \н •/н-н = 9- 6-11,1 U
•/н-н = °>1—2-1
3 X л и II и Е к ^н-нС= 15,5-15,7
4 Нч /Н >с=с< /'ib-H-11’0—12-3
я \ я
.СН3 zCH2R zCHR2 J = 6,1-7,1
3, 6, 1 = С< , =С< , =с< Н \н
4 1 /СНз i RCHa = C< 1 ХНЬ •61а-СНз + 7нй-СНз = 4 ’7 — 5,6
! цис или транс
8 СНз>с = с/ Жн3=-(1-2-1,5)
/ \н
8 сн3ч ,н /с=с\ СНз = - (°’8-1 •4)
6 1 1 rch2-c = ch 7н-сн2= - (°,8- 1,4)
цис или транс
8 х /CH(CH3)R \с=с/ •7Н-СНз = 6-7±°’2В
7 . .СНо-СНз / = \ Ан2-СНз = 6,8+0,2; 7,8+0,2 г
5 , ,С —СН2СН3 /с=с\ 7СН2-СНз = 6,2+0,2; 7,6+0,1 г
а R — только алкильные группы. *
б Данные взяты из работы BrOgel W., Elektrochem., 64, 1 12 1 (1960).
R Расстояния .между компонентами возмущенного дублета СНз-группы.
г Расстояния между центральным и слабопольным, а также центральным и сильнопольным сигналами соответственно в возмущенном триплете СНз-группы. Расстояния измерены между наиболее интенсивными линиями (при разрешении около 0„4 гц).
28*
ПРИЛОЖЕНИЕ III
Химические сдвиги протонов некоторых диазосоединений (Ledwith А., неопубликованные данные)
Диазосоединение Растворитель Т-Значение, м. д. Мультиплетность сигнала
ch2n2 CC12FCF2C1 6,92
ch3chn2 3 а cci2fcf2ci 6,78
8,30 Дублет
ch2=chchn2 V 3 а СС14 5,42 Мультиплет
6,72 Квартет
8,93 Триплет
\2СЙСООС2Н5 а Чистая жидкость 5,04
n2chcooch3 а Чистая жидкость 5,04
сн=сн, 1 >cn2 СН = СН/ 3 а СС14 3,3$ 4,14 Квартет Квартет
c6h5chn2 СС14 5,42
3,10 Мультиплет
Чистая жидкость 5,47
3,06 Мультиплет
С8Н5СНО а СС14 0,01
C6H5CH = NNH2 а СС14 4,71
(C6H5)2CN2 СН2С12 2,74 Синглет
n-CH3OC6H4)2CN2 СН2С12 3,06 Квартет
6,32
9-Диазофлуорен СН2С12 2,70 Мультиплет
< 2,20 Мультиплет
ПРИЛОЖЕНИЕ IV
Спектральные характеристики sopmo и sMema влияния заместителей на экранирование бензольных протонов. Таблица составлена Г. У. Смитом на основании материалов, представленных указанными в таблице авторами (См. разд. 10.12.4)
Заместитель sopmo , M. Д. sterna м д_
Смит Диль Тирс Фирма «Varian» а Брей и Лоусон Смит Диль Тирс Фирма «Varian» а Брей и Лоусон
F 0,23 (5) 6 0,25 0,18 (1) 0,30 (2) 0,04 0,01 0,01 0,09
С1 -0,07 (9) -0,05 -0,07 (3), 0 (4) -0,05 0,06 0,06 0,06 0,11 0,06
-Зв
Вг -0,18 (8) -0,22 -0,24 (2), -0,10 (2) -0,11 0,13 0,11 0,08 0,17 0,08
— 4B
I -0,38 (5) -0,41 -0,39 (1) -0,33 (1) -0,22 0,23 0,22 0,20 0,21 0,19
no2 -0,96 (6) -0,98 -0,96 (2) -0,91 (1) -0,93 -0,18 -0,21 -0,23 -0,13 -0,25
nh2hci -0,10 (2) a -0,17 а
-1,45a -0,47 а
—n = nbf4 ИЛИ ИЛИ
— 1,25 a -0,67а
nh2 0,725 (4) 0,68 0,69 (1) 0,78 0,24 0,22 0,34 0,17
N(CH3)2 0,80 (1) a 0,80 (1) 0,82 (2) 0,21 а 0,20 0,22
cf2h
NH —N = C—CH2
\
COOC2H5 -0,53 (1) -1,12
NHCOCHg -0,22 (2) 0,04
OH 0,56 (4) a 0,50 0,58 0,12 а 0,16 0,14
OCH3r 0,44 (5) 0,42 0,46 (4) 0,50 0,12 0,10 0,09 0,03
OC2H5 0,45 (3) 0,43 (1) 0,46 (1) 0,15 0,13 0,23
437
Заместитель s°Pm°, M. Д
Смит Диль Тирс
ососн3 so2f SO2C1 SO2NH2 SOgCnH2n+j SO3Na SH COOH -0.80 (1) -0,60 (l)a -0,62 (4) От — 0,27 a до —0,55 (2)8 -0,75 (1) a 0,21 (1) -0,76 (1) -0,85 (1)
COOCH3 COOC4H9 COOC2H4N (C2H6) 2 CHO COCH3 COCH2Br coci -0,73 (l)a -0,79 (1) -0,61 (2) -0,70 (3) -0,88 (2) -0,74 -0,80 (1) -0,62 (4) -0,68 (1)
CN -0,35 (2) -0,35 -0,32 в
CH3 CH2CH3 CH(CH3)2 C(CH3)3 С(СНз)2СН2С(СНз)з CH2C1 CH2Br CHBrCH2CCl3 CF, 0,17 (9) 0,H (4) 0,17 0,15 (3), 12в 0,10 е 0,10 (1), 10В 0,03в -0,06 В -0,05 в -0,14в -0,38 (1), —29 в
Продолжение приложения IV
sMemat м Д-
Фирма «Varian» а Брей и Лоусон Смит Диль Тирс Фирма «Varian» а Брей и Лоусон
0,02 -0,32
-0,33 -0,17 а -0,27
-0,27 От 0,05 а ДО —0,31 а
—0,13 (1) -0,01
-0,88 (1) —0,20а -0,09 а -0,21 -0,10 -0,06 -0,10
-0,75 (1) -0,03 -0,32
-0,57 (3) -0,52 -0,21 -0,18 -0,14
-0,67 (2), -0,10 -0,17 -0,05
-59 В
-0,65 (1) -0,24 -0,15 -0,17 -0,13 -0,11 -0,17
0,15 (3) 0,23 0,14 0,13 о,п 0,11 0,09
0,20 0,05
0,10 (1), 0,13 0,08 0,07 0,09 0,03
7в
0,0 (1) -0,05 (1) -0,06(1) 0,01
-0,30 (1) -0,18 -0,10
h-C3F7
C3H6Br
CH2C(CH3)2C1
СН2ОН
СН(СН3)ОН
о
-СН—сн2
С2Н4ОСОСН3
CH2N(CH3)2
CH(CH3)NH2
CH2N(CH2CN)2
CH2N(CH2<p)CH2N(CH2<p)2 c2h4nhcoch3
ch2—
CH(<p)COCH3
CH(<p)NCCr(CO)5
C(CH3)2COOH
-12 B
-0,31 в
0,05 в 0,10 в 0,05 в 0,04 в 0,06 (1) -0,06 (1) 0,11
0,04в
0,02 в 0,03 в 0,02 в 0,07 в 0,07 в 0,07 в
0,08 в 0,07 в
— 0,10 в
439
Заместитель S°Pm0, м. д.
Смит Диль Тирс
СзЩСОСНз -сн=снсн3 0,11 в
ссн3=сн2 -0,08 в
СН = СН-СН = СНф 0,01 в
СН = СНСНО — 0,24 в
СН = СНСОСНз -0,14»
С(ф)=СН2 -0,02 в
СН = СНВг -0,01 в
СН = СН-СН = СНф 0,04»
\e(CO)f
Н . . . .0 0 II -С = СН-СОС2Н5
Ферроцен
С=СН -0,11 в
SiH3 1 — 0,15 в
а Данные получены только для полярных растворителей.
б Цифры в скобках указывают число изученных спектров.
в Для монозамещенных бензолов.
г Изучен ргд метоксибензолов [454],
<р — фенильная группа.
Продолженйе приложения IV
sMemat м. д
Фирма «Varian» а Брей и Лоусон Смит Диль Тирс Фирма <Varian> а Брей и Лоусон
-0,03 (1) 0,08
-0,78 (1) -0,09
-0,12 (1) 0,10 ,
ПРИЛОЖЕНИЕ V
Таблица химических сдвигов Р31
[Van Waser, Callis, Shoolery, Jones, J. Am. Chem. Soc., 78, 5715 (1956)]
Название Формула Агрегатное состояние Химический сдвиг относительно 85%-ной ортофосфорной кислоты, м. д.
А. Соединения трехвалентиого фосфора
Этилдифторфосфин c2h6pf2 Жидкость +зо±з
T риметилфосфит Р (ОСН3)3 » -141 + 1
Триэтилфосфит Р(ОС2Н6)3 » -139±1
Т риизопропилфосфит Р[ОСН(СН3)2]3 » -138+1
Т рибути лфосфит Р(ОС4Н9)3 » -139+1
Т ри-(Р-хлорэтил)фосфит Р[О(СН2)2С1)]3 » -139+1
Три-(2-этилгексил)фосфит Р(ОС8Н17)3 » -140+1
Трифеиилфссфит Р(ОС6Н5)з » -128+1
Этилхлорфторфосфии C2H5PC1F » +20+2
Этилфторизопропоксифосфии C2H5P[OCH(CH3)2]F » —29±3
Трихлорид фосфора РС13 » -220± 1
Г ексаметилтриамидофосфит P[N(CH3)213 » -122+2
Гексаэтилтриамидофосфит P[N(C2H5)2]3 » -118+3
Дихлорфеиилфосфин свн5РС12 » -166+1
Трибромид фосфора РВгз » -229+1
Демитиламидодиметилфосфин (CH3)2NP(CH3)2 » -39+1
Т рифенилфосфин Р(С6Н5)з Раствор в эфире +8±1
Триметилфосфин Р(СН3)з Жидкость +62±1
Диметилфосфин НР(СНз)2 +98,5+1
Монометилфосфин Н2РСН3 » + 163,5±1
Продолжение приложения V
Название Формула Агрегатное состояние Химический сдвиг относительно 85%-иой ортофос-форной кислоты, м. д.
Фосфин Полуторасернистое производное фосфора РН3 P(S)3 в P4S3 Жидкость при —90° Раствор в CS2 +238+1 — 71 + 1
Б. Изогнутые связи
442
Элементарный фосфор Р4 Твердый при —90 Широкий сигнал
Полуторасериистый фосфор SP(P)2 в P4S3 Раствор в CS2 в очень силь- ном поле + 120zbl
Органические фосфаты. В. Соединения пятивалентного фосфора
Три-о-толилфосфат / /СНз^ Жидкость + 17±0,3
\ \ '/-О / рО X X / J 3
Т ри-л4-тол и лфосфат /снз\ Жидкость +17±0,5
1 / \-о)ро
Т ри-п-толилфосфат (сн3/ о) РО Раствор в бензоле + 16+0,5
Трикрезилфосфат [(СН3)(ОН)СвН3оКРО Жидкость + 18+1
Трифеиилфосфат (СвН5О)3РО Раствор в эфире +18±0,5
ч-Хлорфеиилдифеиилфосфат С1СвН4О(С6Н5О)2РО Жидкость + 17,5±0,5
Бутилдифеиилфосфат С4Н9О(С6Н5О)2РО » + 12+1
2-Этилгексилдифеиилфосфат С3Н17О(С6Н5О)2РО » + 12+1
Этилдифеиилфосфат С2Н5О(С6Н5О)2РО » + 12+1
Дибутилфенилфосфат (С4Н9О)2С6Н5ОРО » +4±1
Дибутилфосфат (С4Н9О)2НОРО Жидкость 0+0,5
Триэтилфосфат (С2Н5О)3РО » +1+1
Три-р-хлорэтилфосфат (С1С2Н4)3РО » +2+1
Три-н-бутилфосфат (н-С4Н9О)3РО » -1+0,5
Трибутилфосфат (С4Н9О)3РО » + 1+0,5
Амилоктилоксифосфат (С5НнО)(С8Н17О)НОРО » +1+0,5
Неорганические ортофосфаты
Тризамещенные ортофосфаты _ . , Н 1 Ортофосфаты с -р- = -у PO?- + 3Na+ (или ЗК+) РО4Н2- Раствор в воде То же -5 0
Ортофосфорная кислота (85%) (НО)зРО Жидкость (водн.) 0
Водный раствор (1 : 1) 85%-ной Н3РО4 (НО)зРО Раствор в воде — 1 дЬ 1
Первичный кислый фосфат натрия NaH2PO4 То же 0+1
Вторичный кислый фосфат натрия Na2HPO4 » » +3±1
Фосфат натрия Na3PO4 » » -5+1
Первичный кислый фосфат калия КН2РО4 » » -1+1
Вторичный кислый фосфат калия К2НРО4 » » -1±1
Фосфат калия К3РО4 » » -6±1
Первичный кислый фосфат аммония NH4H2PO4 » » -1 + 1
Вторичный кислый фосфат аммония (NH4)2HPO4 » » — 1±1
Фосфорномолибденовая кислота » » +5±1
Конденсированные фосфаты
Концевые группы, дважды замещенные
— OPO|- + 2Na+ (или 2К+)
Раствор в воде
+5±1
Продолжение приложения V
Название Формула Агрегатное состояние Химический сдвиг относительно 85%-ной ортофосфорной кислоты, м. д.
TZ Н 1 Концевые группы с р- — -ОРО3Н- Раствор в воде + 10±1
Пирофосфат натрия О3РОРО|- То же +6±1
Кислый пирофосфат натрия НО3РОРО3Н2- » » + 10+1
Кислый пирофосфат калия НО3РОРО3Н2- » » +9±1
Пирофосфат аммония О3РОРО|- » » -|-8+1
Срединные группы, короткие цепи — OPOjO — » » +18±1
Срединные группы, длинные цепи и кольца — OPOjO — » » +21±1
Триполифосфат натрия P3O+ Раствор в воде (пересыщенный) Г +4+11 1+18+1/
Триполифосфат калия P3O+ Раствор в воде Г +6+11 1+19+1J
Триполифосфат аммония Psoto То же Г +8+11 1+22±1 !
Натриевое фосфатное стекло п^4 Р/Яз » » Г +3+!| 1+18+1J
п = 5,4 2“О3РОРО2О . ,ОРО2ОРО|- » » +5+1, +21±1
п = 6,8 2~О3РОРО2О . . ОРО2ОРО|- » » Два пика
п=14 2-ОзРОРОгО . ,ОРО2ОРО|- » » +6+1, +20+1
п = 25 2-О3РОРО2О . .ОРО2ОРО|- » » +5±1, +19±1
п = 80 2-О3РОРО2О . .ОРО2ОРО|- » » + 19±1
п = 230 2-О3РОРО^О . . .OPf+OPO*- » » +21 + 1
Триметафосфат натрия (NaPO3)3, циклическая структура » » +21 + 1
Тетраметафосфат натрия (NaPO3)4, циклическая структура » » +21 + 1
Тетраполифосфат аммония (NH4)eP4O13 Раствор в воде Г +9+11 1+22+1/
Ультрафосфат натрия Na2O/P2O5 = 0,82 Na20/P205=-0,50 Na2O/P2O5 = 0,42 Теоретически сильно разветвлен Нагрет до размягчения | +31 +31 +30
Н2О-Р2О5 (азеотроп) Ультрафосфат натрия Na2O/P2O5 = 0,96 Тетрагидрат изогипофосфата натрия Г ексафенил-бис- (диэтиленгликоль)тетрафосфат Пентафен ил-бис- (диэтиленгликоль) трифосфат Теоретически сильно разветвлен По-видимому, не разветвлен Na2O3POPHO2Na ((СвН6О)2ОРОС2Н4О — — С2Н4ОР(ОСвН5)2О —)2 (С6Н5О) 2ОРОС2Н4О - С2Н4ОР (ОС6Н5) 2О - — С2Н4ОС2Н4ОрО(ОС6Н5) 2 Нагрет до размягчения Через 30 мин после растворения в воде Раствор в воде Жидкость > +34 + 14 + 1+0,5 +3±0,5 Г +7,5+П 1+12,5+1 J / +7±П 1+13+1/
Продукт реакции пятиокиси фосфора с диэтиловым эфиром Вязкая жидкость (+14+1-] +29+1 > I+41 + 1J
Органические фосфиты
Диметилфбсфит (СН3О)2НРО Жидкость -11+1
Днэтилфосфит (С2Н5О)2НРО » — 8+1
Дибутилфосфит (С4Н9О)2НРО » -8+1
Бис- (2-этилгексил) фосфит (С3Н17О)2НРО » -7+1
Продолжение приложения V
Название Формула Агрегатное состояние Химический сдвиг относительно 85%-ной ортофос-форной кислоты, м. Д.
Неорганические фосфиты
Фосфорная кислота (НО)2НРО 30%-ный раствор вводе -5+2
Фосфорная кислота (НО)2НРО 13,1 М водный раствор, приготовленный из кристаллической кислоты — 8+2
Первичный фосфит натрия NaO(HO)HPO Раствор в воде —4±2
Вторичный фосфит натрия (NaO)2HPO То же — 4±2
Молекулы с низкой степенью окисления фосфора
Гипофосфат натрия HO(NaO)POPO(ONa)OH Раствор в воде —9±1
Гипофосфорная кислота Н2РО(ОН) 50%-ный водный раствор —13±2
Гипофосфит кальция Н2РО(ОСа) Раствор в воде —8±2
Гипофосфит натрия H2PO(ONa) То же -8+2
Гипофосфит калия Н2РО(ОК) » » — 6±2
Азотзамещенные соединения
446
Первичный фосфорамидат натрия NaOPO(OH)NH2 Раствор в воде -3+1
Диймидофосфорная кислота HOPO(NH2)2 То же +3,5 (?)
Натриевая соль тримерной азотистофосфорной (NaPO2NH)3, циклическая структура » » +1±2
КИСЛОТЫ
Г ексаметилфосфорамид ((CH3)2N)3PO Жидкость -27+1
- 26,5±0,5
Цекса-н-бутилфосфорамид ((h-C4Hb)2N)3PO » - 23+1
Этклтетраметилфосфордиамидат C2H5OPO(N(CH3)2)2 » -18+1
Октаметилпирофосфорамид ((CH3)2N)4P2O3 Жидкость -12+1 -11 + 1
Тример хлорида фосфонитрила Полимер хлорида фосфонитрила Дифенилдодецилфосфорамид н-Бутил-трис-(диметиламидо)фосфонийбромид (PNC12)3, циклическая структура (PNC12)~, цепочечная структура (C6HEO)2PO(NHR) [h-C4H9P(N(CH3)2)3]+Bt_ Раствор в бензоле То же » » Жидкость -19+1 -20+1, +7+1 -1±1 — 62±1
Хлорбензил-трис-(диметиламидо)фосфоний хлорид [C1<J ^CH2P(N(CH3)2)3]+C1- Раствор в спирте -55,5+1
ДйхЛорфтормети л-трис-(диметиламидо) фосфоний-. Хлорид [C12FC^P(N(CH3)2)3]+C1- То же -44+1
Другие производные пятивалентного фосфора
Т риэтилселенофосфат (C2H6O)3PSe Жидкость -71 + 1
Дихлорид хлорметилфосфиновой кислоты С1СН2РОС12 -38+2
Соединения с связью С — Р
фенилфосфоновая кислота С6Н6РО(ОН)2 Раствор в ацетоне -17,5±1
n-Хлорфенилфосфоновая кислота С1С6Н4РО(ОН)2 То же -14+1
n-Этилбензилфосфоновая кислота (С2Н5СеН4СН2)РО(ОН)2 » » —26+2
З-Бромпропилфосфоновая кислота (ВгС3Н8)РО(ОН)2 » » -30+2
н-Бутилфосфоновая кислота (С4Н9)РО(ОН)2 » » -32,5±1
м-Пентилфосфоновая кислота (С5Нн)РО(ОН)2 Раствор в воде -33+1
|3-Стирилфосфоновая кислота С6Н5СН = СНРО(ОН)2 Раствор в ацетоне -18+1
Дибутилэтилфосфонат С2Н5РО(ОС4Н9)2 Жидкость -31+2
Дибутилбутилфосфонат С4Н9РО(ОС4Н9)2 » -32+1
Днбутилнонилфосфонат С9Н19РО(ОС4Н9)2 » -31 + 1
Продолжение приложения V
•448
Название Формула Агрегатное состояние Химический сдвиг относительно 85%-иой ортофос-форной кислоты, м. д.
Ди-н-бутилдецилфосфонат СюН21РО (ОС4Н9) 2 Жидкость - 32+1
Диэтилметилфосфонат СН3РО(ОС2Н5)2 » -30±1
Диэтил этилфосфонат С2Н5РО(ОС2Н5)2 » -32,5+1
бис- (2-Этилгексил)-2-этилгексилфосфонат С8Н17РО(ОС8Н17)2 » — 32±1
Децилфосфонат натрия С10Н21РО (ONa) 2 Раствор в воде —27+2
бнс-(Р-Хлорэтил)винилфосфонат СН2=СНРО(ОС2Н4С1)2 Жидкость -22+1
Дибутиламилнафтилфосфонат С5НЦ — С10Н6РО(ОС4Н9)2 » -26,5±1
бис-(2-Бутоксиэтил)фенилфосфонат С6Н5РО(ОС2Н4ОС4Н9)2 » -19±0,5
Диэтилтрихлорметилфосфонат С13СРО(ОС2Н5)2 » -6,5+0,5
Диэтилацетилфосфонат СН3СОРО(ОС2Н5)2 » +2±1
Диэтилбензилтиометилфосфонат CeH5CH2SCH2PO(OC2H5)2 » -24+1
Диэтилхлорметилфосфонат С1СН2РО(ОС2Н5)2 » -20+2
О
Диэтил бензоилфосфонат - с- РО(ОС2Н5)2 » +2±1
Диэтилдиметилкарбамилфосфонат (CH3)2NCPO(OC2H5)2 » 0+0,5
2,4, §-трис-( Д иэтилфосфонил) -1,3,5-триазин N3C3(PO(OC2H5)2)3 » 0+1
Фенилфосфонистая кислота CgH5PO(H)(OH) Раствор в воде -23+1
С6Н5ОР(Н) (ОН) Раствор в ацетоне -20+1
2-Метил-4-диметилминофенилфосфонит натрия ((CH3)2N)(CH3)CgH3PO(H)ONa Раствор в воде -20+5
Этил-бнс-(п-хлорфенил)-втор-фосфонат (С1С8Н4)2РО(ОС2Н5) Жидкость -27±1
Окись трис-( п-хлорфенил)фосфина (ClCgH4)3PO Раствор в бензоле -23+1
Производные кислорода и серы
Сульфоксид фосфора P4S40g Раствор в CS2 -16+1
Оксихлорид фосфора РОС13 Жидкость -4+1
Окись дихлорфенилфосфина CgH5POCl2 » -34+1
1/429-489
]
Сульфид дихлорфеиилфосфина CeH5PSCl2 Жидкость - 80±1
Три-п-толилтиофосфат (CH3CgH4O) gPS Раствор в бензоле —53,5±1
О, О-Д иметил-Б-пропилфосфортиоат (CH3O)2PO(SC3H7) Жидкость - 31±1
О, О-Д иэти л-S-n-x лорфенилфосфортиоат (C2H5O)2PO(SCeH4Cl) » - 21+1
О, О-Д иэтил-О- (п-нитрофенил)фосфортиоат (C2H5O)2PS(OCeH4NO2) » —42+1
О,О-Диэтил-5-фенилфосфортиоат (C2H5O)2PO(SC6H5) » —22+1
О-Этил-S- (n-хлорфеиил) фени лфосфортиоат CeH5PO(OC2H5)(SCgH4Cl) » - 39±1
Диэтилтиофосфат натрия (C2H5O)2PSONa Раствор в спирте - 57+1
О, О-Д иизопропил-5-(п-хло рфенил) пероксифосфор- (u30-C3H70)2PS(SS<f ^>C1) Жидкость —74+2
тритиоат
Пентахлорид фосфора PC15 Раствор в CS2 +80+2
Соединение Химический сдвиг Литературная ссылка в гл. 12 Соединение Химический сдвиг Литературная ссылка в гл. 12
(С4Н9О)2Р(О)С4Н9 (С2Н5О)2Р(О)СН2СООС2Н5 (С2Н5О)2Р(О)СН2СОСН3 (C2H5O)2P(O)CH2CON(C2H5)2 (С2Н5О) 2Р (О) CH2CON (С3Н7) 2 (С2Н5О)2Р(О)ОС(СН3) = сн2 (С2Н5О)2Р(О)ОС(ОС2Н5) = СС12 (С2Н5О)3Р(О) С2Н5Р(О)С12 С6Н5Р(О)С12 сн3РС12 -30,8 -19,5 -19,9 -21,3 -22,0 +7,2 +7,5 +0,9 -53,0 -34,5 -191,2 248 248 248 248 248 248 248 248 . 146 ' 146 146 С6Н5РС12 C2H5P(S)C12 C6H5P(S)C12 -161,6 -94,3 -74,8 146 146 146
Циклические фосфоннтрилы
P3N3C14(SC2H5)2 PS(C2H5)2 —51,7 РС12 -17,7 (7рр —4,8 ац) 169
449
Циклические фосфоннтрнлы
P4N4C14(SC2H5)4 P(SC2H5)2 -28,6 169
PC12 +9,7
(Jpp =11,8 гц)
P3N3C10 -19,7 169
P3N3(SC2H5)0 -45,7 169
p4n4ci8 +7,4 169
P3N3C14(SC0Hs)2 P(SC0H5)2 -46,8 169
PC12 -18,6
(Jpp = 4,2 гц)
Br3PS + 111,8 4
ВгзРО + 102,9 4
NaH2PO4 (води) + 1,3 4
(C2H5O)3PO +0,9 4
H3PO4 0,0 4
[(CH3)2N]3PO -23,4 4
(C2H5)3PSe -45,8 4
(CgH^O -48,3 4
(C2H5)3PS -54,5 4
(C2H5S)3PO -61,3 4
Продолжение приложения V
450
Циклические фосфоннтрнлы
(C2H5O)3PS -68,1 4
[(C2H6)2N]3PS -77,8 4
(C2H6S)3PS -92,9 4
(C2H5S)3P -115,6 4
(C3H7S)3P -118,2 4
[(C2H5)2N]3P -118,2 4
(C0H6O)3P -126,8 4
(C2H5O)3P -136,9 4
(изо-С3Н70)3Р -136,9 4
PCI7 (твердый PC15) +281 162, 163
PClt -96 162, 163
PFl + 148 162, 163
CH3PS(OC2H5)2 -94,9 247
CH3(CH2C1)POC1 -59,2 246
CH3(CH2C1)PSC1 -84,9 246
CH2C1PSC12 -72,8 246
OPC1(OC0H5)2 +6,2 176
PC15 +80,0 162, 163
OPC12(OC0H6) -1,5 143
Соединение Химический сдвиг, м. д. Коистаита взаимодействия гц Ссылка в гл. 12
\ ZN+/X X = Y = C1 -19
\pz р X = Y = N(CH3)2 -25 251
Y 11 J>Y X==C1; Y = N(CH3)2 -21
N N \z X = Y = NH(CH3) —24
p
x\
Cl 1 Pa 4-14
[C13PB = N - PA = N - PBCl3]PcCle Pb -12,5 J РАРв — 45,3 177
il Pc 4-305,0
Cl Cl 1 1 Pa +20 /РдРХ = 29 ’
C13PX—-N —PA = N —PB = O Pb + 13,4 177
ci c!i Px -7,1 7раРв = 26,7
Cl
C13Pa = N-Pb = O 1 Pa Pb +0,1 + 14,2 ^РАРВ= I5’0 250
Cl
Cl 1 Pa ’29,6 ^рарх = 28’7
C13PX= N —PA—N = PXC13 II N Px + 1,9 *^РДРр~31,0 177
CIPpCi II о PP + 13,6
Химические СДВИГИ Р81 Для 140 Соединений фосфора приведены в литературной ссылке [302] главы 12,
Предметный указатель
Азот, ядерные свойства 343
Азотная кислота, химические сдвиги О17 352, 355
Азулен, химические сдвиги С13 314
Азулиний-ион, спектры ЯМР-Н1 108
Акриловая кислота, химический сдвиг карбонильной группы 313, 320
Акролеин, химический сдвиг О17 355
Алкалоиды, спектры ЯМР-Н1 134
Алканы 6
химические сдвиги 6
— — корреляция с электроотрицательностью 10, 12, 14
Алкены, химические сдвиги протонов 46, 57 Алкилхинолины
константы спин-спинового взаимодействия 128
химические сдвиги протонов 127
Алкильные соединения ртути, константы спин-спинового взаимодействия Н1 —
Hg199 27
Аллены, константы спин-спинового взаимодействия С13—Н 334, 341
Альдегид
анисовый, химические сдвиги формильных протонов 96, 97
кротоновый, 7С13_Н341
масляный, химический сдвиг О17 355 мезитиловый, химический сдвиг формильных протонов 97
1-нафтойный, химический сдвиг формильных протонов 97
пропионовый, химический сдвиг О17 355
салициловый, химические сдвиги протонов 97, 143
толуиловый, химический сдвиг формильных протонов 97
Альдопентапиранозы ацетилированные, спектры ЯМР-Н1 40
Алюминий, химические сдвиги соединений 398
Амиды N-замещенные, NH-группа в спектрах ЯМР-Н1 349
Аминокислоты, химические сдвиги 140
Аминопиридин, константы спин-спинового взаимодействия Н1—Н1 124
Аминофенетол, химические сдвиги протонов 96
2-Аминофуран, химические сдвиги протонов 114
Аммиак, ассоциативный химический сдвиг 145
Андростерон, химические сдвиги 33, 138 n-Анизилдифенилкарбинол, химические сдвиги протонов 105
Анизол, химические сдвиги 95, 105, 318
Анизола замещенные, химические сдвиги 95
Аннулены, спектр ЯМР-Н1 77
Аценафтилен, химические сдвиги С13 314
Ацеталь, химические сдвиги О17 355 Ацетальдегид константа дальнего спин-спинового взаимодействия С13 — Н 339 химические сдвиги С13 321
-----О17 355
Ацетамид, влияние температуры на сигнал протона NH-группы 146
Ацетат калия, химические сдвиги С13 320
Ацетилацетон, химические сдвиги
С13 321 О17 352, 355
Ацетилены 77, 313, 341
константы спин-спинового взаимодействия 80
химические сдвиги 77
— — влияние растворителей 81, 170, 173
2-Ацетилпиррол, химические сдвиги протонов 120
Ацетоксихолестанона производные, константы спин-спинового взаимодействия 42
Ацетон, химические сдвиги
С13 321
О17 351, 352, 355
Ацетонитрил, константа дальнего спин-спинового взаимодействия С13 — Н 339
Ацетоуксусный эфир, химические сдвиги О17 352, 355
Ацетофенон константа дальнего спин-спинового взаимодействия С13 — Н 339
химические сдвиги С13 321
Бензальдегиды замещенные, химические сдвиги формильных протонов 96
лг Бензилоксибензальдегид, химический
сдвиг формильного протона 96
Бензоилацетилен, влияние растворителей на химические сдвиги протонов 171
Бензол
влияние растворителей на спектры протонного резонанса 81
галогензамещенные 92, 334, 335
диза'мещенные 85
константы спин-спинового взаимодействия С13 — Н 323, 324
монозамещенные 82
сателлиты С13 — Н в спектрах протонного резонанса 336
тризамещенные 99
Бензотрифториды замещенные, химические сдвиги F19 225
3,4-Бензпирен, химические сдвиги протонов 107
Бор, ядерные свойства 285
Бораны замещенные, спектры ЯМР-В11 298
Боргидрид алюминия, спектры ЯМР-Н1 и ЯМР-В11 300
Боргидрид натрия, спектр ЯМР-Н1 300 jH-Бромбензальдегид, химические сдвиги 96
Бромистый винил, химические сдвиги протонов 52
З-Бромпиридин, константы спин-спинового взаимодействия Н1 — Н1 125
Бутандиол-2,3-циклокарбонат, химические сдвиги протонов 28
Бутаны
константы спин-спинового взаимодействия Н1 — Н1 28
химические сдвиги 28
Предметный указатель
453
Бутен-1 52, 71
константы спин-спинового взаимодействия 59
химические сдвиги 59
/пре/п-Бутилбензол, химические сдвиги протонов 105
Бутилпиррол, химические сдвиги протонов
176
n-mprm-Бутилфенилдифенилхлорметан, химические сдвиги протонов 105
Бутиролактон, химические сдвиги С13 321
Винилацетат, химические сдвиги С13 320 Винилиденкарбонат
константа спин-спинового взаимодействия С13 — Н 334
химические сдвиги протонов 334
Виниллитий, спектр ЯМР-Н1 75
Винилметиловый эфир, химические сдвиги протонов 52
Виниловые эфиры
константы спин-спинового взаимодействия Н1 — Н1 56
химические сдвиги протонов 56
Винилформиат, константы дальнего взаимодействия Н1—Н1 73
Винилцианид, химические сдвиги поотонов
52, 53, 67
Вода, химический сдвиг О17 351, 352
Водород, связанный с атомами
азота 145
бор а 146
германия 149
кислорода 143
кремния 147
олова 149
переходных металлов 150
свинца 149
серы 147
Галогенопроизводные метилацетилена, химические сдвиги протонов 79
Галогены, химические сдвиги и константы спин-спинового взаимодействия 399
Гаммета о-константы 219, 220
корреляция с химическим сдвигом F19 219
уравнения 219
Гексаметилбензол, химические сдвиги
С‘3 315
Н1 107
Гексафторид серы 248 алкильные замещенные, химические сдвиги F19 250
дизамещенные производные, константы спин-спинового взаимодействия F19 _ pis 252
монозамещенные производные, химические сдвиги F19 250
перфторалкильные производные, химические сдвиги F19 251
Гексаэтилбензол, химические сдвиги протонов 107
Гексен-1
константы спин-спинового взаимодействия 59
химические сдвиги 59
30-489
Гетероциклические соединения 111 влияние растворителей на спектры протонного резонанса 127
фторированные, химические сдвиги F19 244—246
Гипофторит пентафторида серы, спектр ЯМР-F19 255
Двойной резонанс, применение для снятия квадрупольного уширения ядер N14 348 Дейтерий, спин-спиновое взаимодействие
Н — D 396
Декалины, влияние конформаций на спектр ЯМР-Н1
Диалкилфосфонаты щелочных металлов, химические сдвиги Р31 378
Диамагнитная анизотропия связей С — Н и С — С 15
Диаммиакат диборана, спектр ЯМР-В11 299
Диацетат этиленгликоля, химические сдвиги О17 355
Диацетоксибутан, химические сдвиги протонов 28
Дибромбензол, константы спин-спинового взаимодействия 102, 103
Дибромбутан, химические сдвиги протонов 28
Дивиниловый эфир, химические сдвиги протонов 52
Дигидродекафторциклогексаны, химические сдвиги F19 242
Дигидроперфторциклогексаны, исследование спектров ЯМР-F19 240
2,3-Дизамещенные пропены
константы спин-спинового взаимодействия Н1—Н1 66
химические сдвиги протонов 66
Дииодбензол, константы спин-спинового взаимодействия 102, .103
м-Диметил аминобензальдегид, химические сдвиги 96
Диметиламм’онийхлорид, влияние температуры на сигнал протона NH-группы 146 9,10-Диметилантрацен, химические сдвиги протонов 107
9,10-Диметил-1,2-бензантрацен, химические сдвиги 106, 107
3,3-Диметилбутен-1, химические сдвиги 52
Диметилкарбонат, химические сдвиги С13 320
Диметилмуконат, константы спин-спинового взаимодействия Н1 — Н1 71
Диметиловый эфир
ацетилендикарбоновой кислоты, химические сдвиги С13 320
малоновой кислоты, химический сдвиг О17 355
этиленгликоля, химический сдвиг О17 355
янтарной кислоты, химический сдвиг О17 355
Диметилсульфоксид, химический сдвиг О17 355
Диметилфенолы
константы спин-спинового взаимодействия С13 — Н 318
химические сдвиги С13 317, 318
Диметилформамид, химический сдвиг С13 321
454
Предметный указатель
2,5-Диметилфураны, химические сдвиги протонов 113, 115, 120
Диметилциклогексан, ширина резонансных сигналов в спектрах ЯМР-Н1 44
3,5-Диметоксибензальдегид, химические сдвиги 95, 96
Диметоксибензол, химические сдвиги
С13 317
Н1 95
Диметоксиметан, химический сдвиг О17 355
Динитробензол, константы спин-спинового взаимодействия 102
Диоксан
константа спин-спинового взаимодействия С13 — Н 341
химические сдвиги О17 355
Дифенилбутан, химический сдвиг протонов 28
Дифенилы
спектры ЯМР-Н1 99
химические сдвиги С13 314, 317
Дифосфин, анализ спектра ЯМР-Н1 380
Дифосфит-ион, анализ спектра ЯМР-Р31 376
Дифтор дихлорэтилен
константы спин-спинового взаимодействия 236
химические сдвиги F19 236
1,2-Дифторэтилен >
константы спин-спинового взаимодействия 232
химические сдвиги F19 232
2,3-Дифурандикарбоновая кислота, константы спин-спинового взаимодействия Н1 — Н1 114
3,4-Дихлорбензальдегид, химические сдвиги 96
Дихлорбутан, химические сдвиги протонов 28
2,3-Дихлоргексафторбутан
константы спин-спинового взаимодействия 236
химические сдвиги F19 236
1,3-Дихлорпропены, константа спин-спинового взаимодействия Н1 — Н1 71
Диэтилкарбонат, химические сдвиги
С13 320
О17 355
Диэтилмалеат, константы спин-спинового взаимодействия С13 — Н 338
Диэтиловый эфир
химический сдвиг О17 355
— — — малоновой кислоты 355
— — — щавелевой кислоты 355
— — — янтарной кислоты 355
Диэтилфумарат, химический сдвиг О17 355
Диэтоксибензол, химические сдвиги протонов 95
Диэтоксиметан, химический сдвиг О17 355
Дурол
константы спин-спинового взаимодействия С13 — Н 315
химические сдвиги С13 315
Жирные кислоты, химические сдвиги протонов 139
Изобутиламин, ассоциативный химический сдвиг 145
Изобутилен, константа спин-спинового взаимодействия С13 — Н 341
Изогипофосфат-ион, спектр ЯМР-Р31 37&
Изопропенилацетат, химический сдвиг С13 320
Изопропильная группа, химические сдвиги протонов 56
Изофталоилхлорид, константы спин-спинового взаимодействия Н1 — Н1 102
Инден
константы спин-спинового взаимодействия 100
химические сдвиги протонов 100
Индолы, спектр ЯМР-Н1 122
лшо-Инозитолгексаацетат, внутренний химический сдвиг 33
Ион BFf, спектр
ЯМР-В11 300
ЯМР-F19 261
З-Карбметоксипиррол, химические сдвиги протонов 120
Карбодиимиды, спектр ЯМР-N14 350
Карбонат, константа спин-спинового взаимодействия С13 — Н 341
Карплуса уравнение 16
Кафестол, химические сдвиги протонов;
Квадрупольная релаксация ядер N14 347
Кислород-17, химические сдвиги 351
Кобальт-59, 384
резонансные частоты 385
электронные максимумы поглощения 385
Комплексные соединения, химические сдвиги 151, 267, 268
Константы спин-спинового взаимодействия алкильных производных, содержащих магнитные ядра 25 даль'него ацетиленов 78 — бензолов замещенных 223, 224 — экзо-бицикло- [2,1]-гексанола-5, 21 — бутанов 2,3-дизамещенных 28 — 1,2-дибром-2-фенилпропана 20 — знаки 19 — — 1-хлорбутадиена-1,2 48 — 2-эн5о-3-эн5о-камфандиола-2,3 22 — кольцевых протонов ароматических молекул 102 — насыщенных систем 20 — изобутенов 69 — моноолефинов 435 — моноциклических ароматических соединений 118 — метилацетилена 342 — олефинов 47, 61, 62 71 — пиридинов 118 — — 3-замещенных 126 — — метилзамещенных 126 — пирролов 117, 118 — пропена-1 59 — пропенов 69 — тиазолов 133 — тиофенов 118, 132 — фуранов 112, 113, 118 — хинолинов алкилзамещенных 128 В41 — F19 260
Предметный указатель
455
В44 —Н4 289, 291, 292
С42 — С13 342
С13 — F49, аддитивные соотношения 327
— — вклады заместителей 329
— — дихлорфторметана 322
— — знак относительно 7С13Н1 322
— — — — ^С13—CF 235
— — связь с изотопным химическим сдвигом (С13 — С12) 333
— — фторалканов 329
— — фторорганических соединений 276
С13 — Н1 341
— — аддитивные соотношения 327
— — — — для $р2-гибридизованных углеродов 331
— — альтернантных углеводородов 317
— — анизола 318
— — бензолов симметричнозамещен-ных 335
— — взаимосвязь с /дц и 322
— — вклады заместителей 327
— — дальнего взаимодействия 337 — 339
— — дихлорметана 322
— — зависимость от гибридизации углерода 323, 324
— — — от длины связи СН в метанах 327
— — замещенных альдегидов 332
— — — метанов 325—327, 331
— — знак относительно 7С13_р1() 322
— — корреляция с протонными химическими сдвигами 333
— — метилбензолов 315
— — метилфенола 318
— — насыщенных молекул 329
— — связь с электроотрицательностью заместителя 325
— — соединений, обогащенных изотопом С13 340
— — — содержащих формильную группу 326
— — сравнение с рассчитанными 329, 330, 331
— — фенола 318
р19 _ р19
— — влияние температуры 199
— — гипофторитов 264
— — перфторалкановых производных 273
— — перфторалкилов металлов 218
— — перфторалкильных производных гексафторида серы 252
— — — — дифторида серы 255
— — — — тетрафторида серы 255
— — перфторацилов металлов 218
— — перфторвинильных соединений 227
— — пропанов замещенных 209
— — соединений типа RPF2 276
----------R2PF3 276
— — фторалканов, знаки относительно J ц____р 210
— — фтор алкенов 228—231
— — — относительные знаки констант 234
F19 — Fl9 фторалкилов ртути 219 — — фторметанов 205 — — этанов замещенных 209 F49 — Н1 соединений типа R4PF, и R2PF3 276 . 2
--фторалкенов 230, 231, 235 -----1-фтор-2-(перфторизопропил)-этилена 233
-----1-фторпропена 233
-----2-хлоргептафторбутена-2 232
F19 — N44 изомеров N2F2 263
F49 — Si29 3 5 0
Н1 — D 396
Hl — Hl бензола 118
-----бутена-1 59
— — вицинальные, зависимость от двугранного угла 16
-------взаимосвязь с 7С13_Н в этиленах 332
— — корреляция с валентными углами 48
----винильных соединений 48—50 — — — корреляция с электроотрицательностью заместителя 49
— — гексена-1 59
— — геминальные 16
-------взаимосвязь с 7С13_Н в этиленах 332
Н Hg499 алкильных соединений ртути 27
---- ртутьорганических соединений типа CH3ORHgX 402
— — трихлормеркурфурана 403
Н1 — N44 дальние изонитрилов 350
Н4 — О17 воды 357
Н4 — Р34 369, 371
-----диалкилфосфонатов 379
— — знаки 370
-----триалкилфбсфатов 371
----- фосфорорганических эфиров 372
Н4 — Pt493 401
-----гидрида платины 401
Н4 — Si29 358-360
Н1 — Sn449 метильных соединений оло-вз 393
— — станнанов 149, 391
----- тетравинилолова 392
Н4 — Т1206 винилталлия 75, 395
— триалкилов таллия 305 psi _ pi9 266, 370
----перфторалкильных производных фосфора 274
~276 Соединений типа R2PF3 и R3PF2 -----фторидов фосфора 274 Р34 - Р34 369, 384 — — знаки 370
-----тетраполифосфат-иона 377
Константы экранирования 164 вклад слабых вандерваальсовых взаимодействий 165 — среды 164 — электрического поля 165 неидеального газа 177, 178 Контактные сдвиги 151 аминотропониминатов 152
Конфигурации полимеров 157
Конформации Замещенных этанов 210
30*
456
Предметный указатель
Крезол
константы спин-спинового взаимодей-
ствия 317, 318
химические сдвиги С13 318
Кремний, химические сдвиги 357
Кротонитрил
константы спин-спинового взаимодействия С13 — Н1 341
химические сдвиги 67
/пракс-Кротоновый альдегид, константы спин-спинового взаимодействия Н1 — Н1 73
Ксилол
константы спин-спинового взаимодействия 314, 315
химические сдвиги С13 315
Ксилолы, химические сдвиги протонов 93
Ликонен, химические сдвиги протонов 77
Лимонин, химические сдвиги протонов 113
Лунакрин, спектр ЯМР-Н1 134
Лунин, спектр ЯМР-Н1 135
Магнитные моменты ядер 405
Малиновского инкременты 327—329,
см. также Константы спин-спинового взаимодействия С13 — F19 и С13 — Н1, аддитивные соотношения
Маллера уравнение, расчет числа несбалансированных электронов 365
Малоновый эфир, химический сдвиг О17 352
Медь-63 401
применение ЯМР для определения скорости реакции 401
Мезитилен константы спин-спинового взаимодействия 315, 324
химические сдвиги С13 314, 315
а-Метакрилонитрил, химические сдвиги протонов 67
Метанол, химические сдвиги О17 352, 355
Метиламмонийхлорид, влияние температуры на сигнал протона NH-группы 146 Метилацетат константы дальнего спин-спинового взаимодействия С13 — Н1 339
химические сдвиги С13 321
-----О17 339
Метилацетилен, константы спин-спинового взаимодействия С13 — Н1 323, 341
Метилбензоат, химические сдвиги протонов 320
З-Метилбутен-1, химические сдвиги 52 а-Метилвинилметиловый эфир, константа спин-спинового взаимодействия С13 — Н 341
Метилгалогениды 6 химические сдвиги протонов 6 — — — зависимость от электроотрицательности 8, 9
Метилдекалол, химические сдвиги протонов 45
1-Метил-2,2-дифторциклопропан, химические сдвиги протонов 237
Метилимидазол, константы спин-спинового взаимодействия С13—Н1 341
Метиловый эфир р,р-диметилакриловой кислоты, химические сдвиги протонов 70
Метиловый эфир метакриловой кислоты, химические сдвиги протонов 70
Метил-о-метоксибензоат, химические сдвиги протонов 95
Метил-м-метоксибензоат, химические сдвиги протонов 95
Метилолеат, химический сдвиг С13 321
Метилпиридин, константы спин-спинового взаимодействия Н1 — Н1 124
4-Метилпиридин, химические сдвиги протонов 176
Метилпиррол, химические сдвиги протонов 120
Метилпропионат, химические сдвиги О17 355
Метилсалицилат, химические сдвиги
С13 321
Н1 143
Метилфенетол, химические сдвиги протонов 96
Метилформиат, химические сдвиги О17 355
Метилфторсилан RxSiF4_x, зависимость химических сдвигов F19 от числа алкильных заместителей 247
2-Метилфуран, химические сдвиги протонов 113—115, 120
Метильная группа
константы спин-спинового взаимодействия 56
химические сдвиги протонов 56
Метилэтилкетон, химические сдвиги О17 352, 355
n-Метоксибензиловый спирт, химические сдвиги протонов 95
Метоксибензоат, химические сдвиги протонов 95
Метоксибензойная кислота, химические сдвиги протонов 95
Монодейтробензол, константы спин-спинового взаимодействия 396
Мочевина, химические сдвиги О17 355
Муравьиная кислота, химические сдвиги
С13 320
О17 351, 352, 355
Мышьяк-75, ширина сигнала 401
Натрий-23, структура сигналов 397
Натрия ацетат, константы дальнего спин-спинового взаимодействия С13—Н 339
Нафталины
фторзамещенные 225
химические сдвиги С13 314, 315
а,|3-Ненасыщенные эфиры, химические сдвиги протонов 70
Ниобий-93, спектр ЯМР иона NbFe 405
Нитрит натрия, химический сдвиг О17 351
Нитробензол, химические сдвиги О17 355
Нитрозилфторид, химический сдвиг F19 262
Нитрофенетол, химические сдвиги протонов 96
Нитрофуран, химические сдвиги 114, 115
Нитроэтан, химические сдвиги О17 351
Нонанон-2, химический сдвиг С13 321
19-Нортестерон, химические сдвиги протонов 138
Обмен, равновесные процессы с участием кобальта 387 фосфора 382
Предметный указатель
457
Окись мезитила, химические сдвиги О17 355
Па-Оксиаллопрегнан-3,20-дион, химические сдвиги протонов 138
а-Оксикислоты, химические сдвиги С13 304, 320
1 la-Оксипрегнан-З,20-дион, химические сдвиги протонов 138
Оксипрогестерон
внутренний химический сдвиг 33
химические сдвиги протонов 138
Оксихлориды металлов, ядерное квадру-польное расщепление 400
Октаметилциклотетрасилоксан, химические сдвиги О17 351
Олефины
алкилзамещенные 62
дальнее спин-спиновое взаимодействие 72
дизамещенные 63
типичные константы спин-спинового взаимодействия 47
тризамещенные 68
Олово, ядерные свойства 387
Олово IV, химические сдвиги Sn119 смешанных галогенидов 390
Паральдегид константы спин-спинового взаимодействия С13—Н 341 химический сдвиг О17 355
Пентаборан-9, спектр ЯМР-В11 295
Пентаборан-11, спектр ЯМР-В11 295
Пентакарбонил железа, химические сдвиги С13 322
Пентаметилбензол, химические сдвиги протонов 107
Пентафторид брома, спектр ЯМР-F19 258
Пентафторид иода, спектр ЯМР-F19 258
Пентафторид сурьмы, спектр ЯМР-F19 247
Пептиды, химические сдвиги протонов 140
Перекись водорода, химический сдвиг О17 351
Перфторбензол, химические сдвиги F19 243 Перфторвинильные производные константы спин-спинового взаимодействия F19 — F19 227 химические сдвиги F19 227
Перфтордекалины, спектр ЯМР-F19 243
Перфтордекановая кислота, химические сдвиги F19 270
Перфторметилциклогексан, спектр ЯМР-F19 240
Перфторпиперидин, химические сдвиги F19 245
Перфторциклогексан, спектр ЯМР-F19 240
Перфторциклогексан диены, химические
сдвиги F19 243
Перфторциклогексен, химический сдвиг F19 243
Перхлорилфторид FC1O3, спектр F19 259
Пиколин, константы спин-спинового взаимодействия Н1 — Н1 126
Пиперональ, химические сдвиги протоновЭб Пирен константа спин-спинового взаимодействия С13 — Н 317
химический сдвиг С13 317
----- Н1 107
Пиридин
константы спин-спинового взаимодей-
ствия Н1 — Н1 122, 125, 126
метилзамещенные, константы спин-спинового взаимодействия 126
спектр ЯМР-Н1 123
химические сдвиги атома азота 345
— — протонов монозамещенных производных 124
Пиридиния ион, химический сдвиг N14 344
Пиррол
влияние температуры на сигнал протона NH-группы 146
дизамещенные, константы спин-спинового взаимодействия С13 — Н 341
спектры ЯМР-Н1 177
химические сдвиги протонов 176
2-Пирролальдегид, химические сдвиги протонов 120
Пирролидин, влияние температуры на сигнал протона NH-группы 146
Платина-195, спин-спиновое взаимодей-
ствие Pt — Н 401
Полиакриламид, влияние сегментного движения на ширину резонансных сигналов 155
Поливинилиденфторид, химические сдвиги F19 270
Поливинилфторид, химические сдвиги F19 270
Полимеры
определение молекулярной структуры 156
— пространственной конфигурации 158
— — — с использованием дейтерирования 160
применение двойного резонанса 159
— спектроскопии ЯМР высокого разрешения 159-
сегментное движение 154
фторсодержащие, химический сдвиг F19 269
Полнметакриламид, влияние сегментного движения на ширину резонансных сигналов 155
Поли(2-метил-1,1,4,4-тетрафторбутадиен), химический сдвиг F19 270
Политрифторхлорэтилен, химический сдвиг F19 270
Порфирины, спектр ЯМР-Н1 121
А4,17 <29'-Прегнадиен-11|3,21-диол-3-он, хи-
мический сдвиг протонов 137, 138
Пропанол, химический сдвиг О17 355
Пропаргилхлорид, химические сдвиги протонов 171
Пропен, спектр протонного резонанса 58
Пропенилбензол, константы спин-спинового взаимодействия Н1 — Н1 71
Пропены 2-замещенные, химические сдвиги протонов 63
к-Пропилформиат, химические сдвиги О17 355
Пропионовая кислота, химические сдвиги О17 352, 355
Пропионовый альдегид, химические сдвиги О17 352, 355
Птерокарпин, спектр ЯМР-Н1 139
458
Предметный указатель
Резонанс ядер, экспериментальные методы наблюдения
С13 202
Р31 361, 363
Si29 3 5 7
Ртуть-199, константа спин-спинового взаимодействия Н — Hg 401
Рубидий-87, химические сдвиги галогенидов 397
Сателлиты, обусловленные взаимодействием С13 — Н 235
Свинец-207, химические сдвиги соединений 404
Селеи-77, химические сдвиги H2SeO3, H2SeO4 401
Серная кислота, химический сдвиг О17 355
Сополимеры
бутадиен—изопрен, спектры ЯМР-Н1 156
поли-(п- 1)-метилалкены-1, химические сдвиги протонов 157
Сопряженные диены, константа спин-спи-иового взаимодействия Н—Н 74
Спектр ЯМР на ядрах
В11 294
— гексаборана 297
— диборанов 294
• — метилборанов 295
— моноиодидпентаборана-9 298, 299
— пентаборана-9 295, 297
— пентаборана-11 297
С13
— азулена 314
— анизола 317
— ароматических соединений 314
— аценафтилена 314
— бензола 314
— гексаметилбензола 314
— диметилфенола 317
— о-диметоксибензола 319
— дифенила 314
— дурола 314
— 2,4,6-коллидина 320
— крезола 317
— ксилола 314
— 3,5-лутидина 320
— мезитилена 314
— нафталина 314
— олефинов 312
— пирена 314
— пиридина 306
— толуола 314
— 2,6,8-триметилазулена 314
— уксусной кислоты 305, 306
— фенола 317
— флуорантена 314
— этилового спирта 306
pis
— 1,1-диметил-2,2,3,3-тетрафторцик-лобутана 237
— перфтордекалииа 244
• — перфторметилциклогексана 241
— перфторпиперидина 244
— смешанных галогенидов бора 260
— 1-фтор-2-(перфторизопропил)-этиле-нов 234
Н1
— азулена 108
— азулиний-иона 108
— антрацена 103
— ароматических соединений, влияние растворителей 173
— ацетиленовых соединений, влияние растворителей 170
— ацетилированных альдопентапираноз 40
— бромистого винила 46
— гетероциклических соединений 127
— диборанов 294
— 9,10-диметил-1,2-бензантрацеиа 106
— 1а,зр-диметокси-2а-ацетоксици-клогексана 38, 40
— 2,4-дихлорбензальдегида 98
— 1,1-дихлор-2-метоксициклопропана
30
— диэтилртути
— индена 100
— индола 123
— 3-кетоаллопрегнана 136
— лунакрина 134
— Лунина 135
— метилглутаровой кислоты 142
— цис-метилкротоната 72
— метилянтарной кислоты 142
— нафталина 103
— ненасыщенных гетероциклических соединений 176
— пиридина 349
— пиррола 116
— — влияние растворителей 177
— поливинилхлорида 159
— полиметилметакрилата 158
— полипропилена 160
— полистирола 155, 161
— порфирина 121
— пропаргилбромида 79
— пропена-1 58
— птерокарпина 141
— о-терфенила 99
— тетра-о-ацетил-1,4-дидезокси-1,4-ди-нитронеоинозитола 41
— 1,1,4,4-тетраметилциклогексил-цис-2,6-диацетата 37
— тетраэтилсвинца 24
— циклогексанов 38
— — дизамещенных 43
— NH-содержащих молекул, влияние температуры 146
Hg199 диметилртути 405
N«
— аммиака 348
— аммоний-иона 344
— влияние квадрупольного момента ядра азота 347
— четвертичных аммониевых ионов 348
Nb93 иона ниобия NbFj 405
О17 воды 357
Р31 дифосфит-иона 376
— изогипофосфат-иона 376
— тетраполифосфита аммония 378
— фосфат-ионов 380
— фосфонитрилов 382
Sn119
— смеси галогенидов олова 389, 390
— тетраметилолова 392
Т1205 расплава TI2CI4
Предметный указатель
459
Стероиды, химические сдвиги
метильных групп С(18) и С(19) 137 протонов в ряду гидроксильных производных 138
Стирол, отнесение сигналов 55
Сукцинимид, влияние температуры на сигнал протона NH-группы 146
Сурьма-121, константа спин-спинового вза-
имодействия Sb — F 401
Таллий, ядерные свойства 392
Тафта
<т-константы 220
— корреляция с химическими сдвигами F19 220
уравнения 220
Тетраборан, спектр ЯМР-В11 295
Тетравинилолово, константы спин-спинового взаимодействия Sn119 — Н 392
1,2,5,6-Тетрагидропиридин, спектр протонного резонанса 126
Тетрагидрофуран, химический сдвиг О17 355
2,2,4,5-Тетраметилдиоксолан
константы спин-спинового взаимодействия Н1 — Н1 28
химические сдвиги протонов 28
Тетраметилортосиликат, химический сдвиг О17 351
Тетрафторид серы, химические сдвиги F19 253
перфторалкильные производные 254
Тетрафторциклобутаны, спектр ЯМР-F19 237
Тиазолы, химические сдвиги протонов 133
Тиглиновый альдегид, константы спин-спинового взаимодействия Н1 — Н1 73
Тиокарбоновые кислоты, химические сдвиги F19 256
Тионилтетрафторид, химические сдвиги F19 255
2-Тиофенальдегид, химические сдвиги протонов 120
Тиофены
константы спин-спинового взаимодействия Н1 — С13 334, 341
--------Н1 — Н1 132
химические сдвиги протонов 120, 131, 176
л-Толилдифенилкарбинол, химические сдвиги протонов 105
л-Толилфенилкарбинол, химические сдвиги протонов 105
Толуол
константы спин-спинового взаимодействия Н1 — С13 315, 339, 341
химические сдвиги протонов 95, 105
Триалкилфосфаты, константы спин-спинового взаимодействия Р31 — Н1 371
Триарилкарбониевые ионы, химические сдвиги протонов 105
2,6,8-Триметилазулен, спектр ЯМР-С13 314
2,4,6-Триметилацетофенон, химический сдвиг С13 321
2,4,6-Триметилпиридин, константы спин-спинового взаимодействия ^'зн 335
Триметилфосфит, спектр ЯМР-Р31 362
Триоксан, константа спин-спинового взаимодействия С13 — Н 341
Три-п-толилкарбинол, химические сдвиги протонов 105
Трифенилкарбинол, химические сдвиги протонов 105
Трифосфонитрил, спектр ЯМР-F19 266
Трифторид брома, спектр ЯМР-F19 258
Трифторид хлора, спектр ЯМР-F19 256
Трифторуксусная кислота, спектр межъядерного двойного резонанса С13 203
Трифторхлорметан, внутренний эталон при измерении химических сдвигов F19 194 Тропилий-катион, химические сдвиги протонов 109, НО
Углеводы ацетилированные, конфигурационные эффекты 38
Уксусная кислота, химические сдвиги О17
351, 352, 355
Уксусный ангидрид, химические сдвиги С13 320
О17 355
Фенантрен, химические сдвиги С13 317
Фенетолы, химические сдвиги протонов 96
Фенилацетилен, влияние растворителей на химические сдвиги протонов 171
1-Фенил-2,2,3,3-тетрафторциклобутан, химические сдвиги F19 237
Фенол, химические сдвиги
Н1 143
С13 317, 318
Ферроцен, химические сдвиги С13 341
Флуорантен, спектр ЯМР-С13 314
Формамид
влияние температуры на сигнал протона NH-группы 146
химические сдвиги О17 351, 355
Формиат натрия, химический сдвиг О17 351
Фосфаты конденсированные, химические сдвиги Р31 379
Фосфиты, химические сдвиги Р31 374
Фосфонитрилгалогениды, химические сдвиги Р31 383
Фосфонитрилы нециклические, химические сдвиги Р31 383
Фосфор 361
количественный анализ соединений 382
структура тиокислоты 380
Фосфора пентахлорид, спектр ЯМР-Р31 380
Фосфорных кислот анионы, спектр ЯМР-Р31 377
Фталоилхлорид, константы спин-спинового взаимодействия Н1 — Н1 103
Фтор 193, 196
Фторалканы
знаки констант спин-спинового взаимодействия 209
химические сдвиги 216
Фторалкильные карбоновые кислоты, химические сдвиги F19 256
Фторацетилены, химические сдвиги F19 236
Фторбензолы
индуктивный эффект 219
резонансный (сопряжения) эффект 219
стерический эффект 219
Фториды бинарные, химические сдвиги F19 203
460
Предметный указатель
Фтористый винил, химические сдвиги протонов 52
9а-Фторкортикостеронацетат, химические сдвиги протонов 138
1-Фторпропен
константы спин-спинового взаимодействия F19 — Н1 233
химические сдвиги F19 и Н1 233
Фторсульфонат фтора, спектр ЯМР-F19 255
Фторхлоралканы
константы спин-спинового взаимодействия 205
химические сдвиги F19 205, 206
2-Фтор-3-хлорпропен-2
константы спин-спинового взаимодействия 231
химические сдвиги 231
Фуранакролеин, химические сдвиги 115
Фураикарбоновые кислоты
константы спин-спинового взаимодействия Н1 — Н1 114
химические сдвиги протонов 113
Фураны
влияние заместителей на химические сдвиги кольцевых протонов 115
константы спин-спинового взаимодействия 112—115
химические сдвиги протонов 112, 176
---С13 341
---О17 355
2-Фуроилхлорид, химические сдвиги протонов 114, 115
Фурфурол
химические сдвиги
Н1 113—115
О17 355
2-Фуротрифторметилкетон, химические сдвиги протонов 115
Фурфуриламин, химические сдвиги протонов 115
Химические сдвиги ядер
азота-14 344, 345
— аммиака и родственных соединений 345, 346
— изомеров N2F2 256
— • нитратов 347
— нитритов 347
— нитросоединений 347
алюминия-27 398
бора-11 287, 289, 290
— гидридов 291
— тригалогенидов 288, 291
— тригональных соединений 299
вклады от различных заместителей и групп 162
кислорода-17
— влияние парамагнитных ионов 356
— карбонилов металлов 356
— органических молекул 352, 355
— связанного с непереходными элементами 353
— — с переходными металлами 352
кобальта-59, влияние растворителей 386
----температуры 386
— связь с длинами волн низкочастот-
ного максимума поглощения 386 кремния-29 358, 359
олова-119 388, 389
— влияние растворителя 388
— смешанных галогенидов олова 391, 390
протонов алкилзамещенных олефинов 62
— алкильных производных углерода, кремния и олова 23
— — соединений 11, 12
— аминокислот 140
— анизола и родственных соединений 95
— ацетиленов 78
— ацетонитрила в различных растворителях 167, 169
— бензоилацетата 171
— бензолов галогензамещенных 92
— — дизамещенных 85, 87, 174
— — монозамещенных 82
— — тризамещенных 69
— бензольного кольца (приложение IV) 438
— бутена-1 59
— виниловых эфиров 56
— винильных производных металлов 74
— — протонов, аномальные 53
— — — в зависимости от электроотрицательности заместителей 52
— внутренние, в металлалкилах 13
— гексена-1 59
— гетероциклических соединений 176
— диаграммы (приложение II) 430
— 2,3-дизамещенных бутанов 28
— — пропенов 66
— 9,10-диметил-1,2-бензантрацена
106
— зависимость от JC13_н 333
— изонитрилов 350
— колец некоторых гетероциклических соединений 176
— кольца пара-дизамещенных бензо; лов. 174
— кислот ненасыщенных 71
— магноцена 110
— метана в различных растворителях 166, 169
— — зависимость от теплоты испарения растворителя 168
— метильных групп стероидов 137
— — производных 79
— — — элементов группы IVB 23
— органических соединений (приложение I) 415
— осмоцена НО
— пептидов 140
— пирролов 119, 120
— пропаргилхлорида 171
— пропена-1 55, 59, 67
— рутеноцена 110
— связанных с азотом 145, 146
— — с бором 146
— — с германием 149
— — с кремнием 147
— — с оловом 149
— — с свинцом 149
— — с серой 147
— ОН-систем при бесконечном разбавлении 143
— солей тропилий-катиона 110
Предметный указатель
461
протонов ОН-систем циклопентадиенил-аниона 110
— сополимеров бутадиена и изопрена 157
— тиазола 133
— тиофена 120
— — замещенных 131
— триарилкарбониевых ионов НО
— фенетола и родственных соединений 95
— фенилацетилена 171
— фенолов 90
— ферроценов 110
— формильных альдегидов полициклических 97
— — бензальдегидов 96
— фурана 112, 120
— — метилзамещенных 112
— хинолинов 127, 129
— — алкилзамещенных 128
— — замещенных 129
— хлористого винила 57
— циклоалканов 29
— циклоалканонов 29
— циклоалкенов 29
— циклогексанов монозамещенных 33, 42
— эмпирическая оценка 161
— эпоксидов 32
— этиленов 333
— эфиров а, ^-ненасыщенных 70
ртути-199 402, 404
свинца-207 404
таллия-205 394, 404
углерода-13
— алканов 304
— алкенов 312
— альтернантных углеводородов 317
— анизола 318
— ацетиленов 313
— бензолов монозамещенных 315
— двуокиси углерода 321
— зависимость от числа атомов хлора
в хлорбромметанах 311
— — — заместителей X в ряду СНхХ4_х 311
— — от электроотрицательности X
в ряду СН3Х 309
-------------СН3СН2Х 310
— углерода в бутил-, изопропил-
и этилпроизводных 309
— изотопный (С13 —С12) 334
— карбонилов железа 322
— — никеля 322
— карбонильных групп 320
— метилбензолов 315
— метилгалогенидов 304
— метилфенолов 318
— метильных групп 308
— окиси углерода 321
— соединений типа СН3Х 307
— фенола 318
— хлорбромметанов 308
фосфора-31 362—364, 442
— дифосфинов замещенных 381
— конденсированных фосфатов 379
— корреляция с числом несбаланси-
рованных электронов 365
— органических фосфитов 374
— пентахлорида фосфора 380
фосфора-31 пятивалентного, изменение при замене одного органического лиганда на другой 368
— — тиокислоты 380
— связь с числом несбалансированных р-электронов 365
— трехвалентного, изменения при замене галогена или органического радикала 366
— — сравнение с рассчитанными 367
— фосфитов 378
— фосфонатов 378
— циклических соединений 381
фтора-19 271
— алкилселенидов 278
— алкоксипроизводных перфторизобутилена 272
— бинарных фторидов 203
— галогензамещенных пропенов 229
— — этанов 208
— гипофторитов 264
— дигидродекафторциклогексанов 242
— изомеров N2F2 263
— монозамещенных бензолов 220
— — — зависимость от а-констант
221
— перфторалкановых производных 273
— перфторалкильных соединений металлов 218
— — производных SF2 и SF4 254
— — — фосфора 274
— перфторацильных соединений металлов 218
— перфторбензолов С6Н5Х монозамещенных 226
— перфторвинильных соединений 227
— перфторциклогексана 24
— производных гексафторида серы
250, 251
— силилселенидов 278
— соединений, содержащих перфторметильные группы 274
— — —' связи В — F 260, 261
-----------N — F 262
-----------Р — F 264
— тетрафторциклобутанов 238
— фтора F2 196
— фторалканов 216
— фторалкенов 228
— фторалкилов ртути 219
— фторалкильных карбоновых кислот 256
225-
272
276
— фторбензолов полизамещенных — фторидов фосфора 275
— фторированных углеводородов — фторорганических соединений
—• i-фторпропена 233
— фторуглеводородов, содержащих азот 211, 212
— фторуглеродных групп в полимерах 269
— фторхлоралканов 206
— фторциклогексанов 243
— • фторэтанов 207, 273
— 2-хлоргептафторбутена-2 232
хлора-35 хлоридов жидких 399
цезия-133 галогенидов твердых 397
Хинолины, химические сдвиги протонов 127
Хлор, химические сдвиги С135 и С137 399,.
400
462
Предметный указатель
1-Хлорбутадиен-1,2, знаки констант спин-спинового взаимодействия 48
2-Хлоргептафторбутен-2, константы спин-
спинового взаимодействия 232
Хлориды металлов, ядерное квадрупольное расщепление 400
Хлористый винил
константы спин-спинового взаимодей-
ствия Н1 — С13 333
--------Н1 — Н1 57
химические сдвиги 52, 57
З-Хлормеркурфуран, константы спин-спинового взаимодействия Н1 — Hg 403
Хлорная кислота, химический сдвиг О17 355
Хлорпиридин, константы спин-спинового взаимодействия Н1 — Н1 124
Хлорпропены, рассчитанные и наблюдаемые химические сдвиги 67
Хлорэтилены, химические сдвиги протонов 57
Цезий-133, химические сдвиги галогенидов 397
Цианимиды, химические сдвиги N1' 350
Цианистый калий, химический сдвиг С13 204
Цианопиридин, константы спин-спинового взаимодействия 124
Цианпропены, рассчитанные и наблюдаемые химические сдвиги 67
Циклические соединения фторированные, исследование структуры методом ЯМР 236
Циклоалканы, химический сдвиг С13 204
Циклогександиацетат, ширина резонансных сигналов в спектрах ЯМР-Н1 44
Циклогександибензоат, ширина резонансных сигналов в спектрах ЯМР-Н1 44 Циклогександиолы, ширина резонансных сигналов в спектрах ЯМР-Н1 44
Циклогексанон, химические сдвиги С13 321
О17 355
Циклогексаны
дизамещенные, определение стереохимии из спектров ЯМР-Н1 43
константы спин-спинового взаимодействия С13 — Н 324, 341
монозамещенные, спектр ЯМР-Н1 42, 56
фторированные 240 1
химический сдвиг внутренний 33
Циклогексен, константа спин-спинового взаимодействия С13 — Н 323
Циклопентадиенил-анион, химические сдвиги протонов 109, ПО
Циклопентадиенилы, спектры ЯМР-Н1 76
Циклопентан, константа спин-спинового взаимодействия С13 — Н 324
Циклопентанон, химический сдвиг С13 321
О17 355
Циклопропаны
константы спин-спинового взаимодействия С13 — Н 324
--------Н1 — Н1 31
фторированные, спектр ЯМР-F18 236
химические сдвиги протонов 31
Щелочные металлы, химические сдвиги соединений 397
Экранирование
анизотропными группами 5
водорода 15 диамагнитное 5 дифференциальное дизамещенных олефинов 63
кобальта-59 385
протонов бензольного кольца 83, 84
ртути 403
фосфора-31 364
— вклады различных групп 366
фтора-19, влияние растворителей 195
Электроотрицательность заместителя корреляция с 7HiHi в винильных производных 49—51
— с химическими сдвигами протонов алкильных соединений 10, 12
— — — — протонов в метилгалогенидах 8, 9
— — — внутренними в металлалки-лах 14
Эпиандростерон, химические сдвиги протонов 33, 138
Эпихлоргидрин, химические сдвиги О17 355
Этанол, химические сдвиги О17 352, 355
Этаны замещенные
константы спин-спинового взаимодействия F18 — F18 208
F18 — Н1 208
Н1 — Н1 18
конформация 211
химические сдвиги F18 208
Этиламмоний хлорид, влияние температуры на сигнал протона NH-группы 146
Этилат таллия, рпектр ЯМР-Т1 396
Этилацетат, химические сдвиги О17 352, 355
2-Этилгексильная группа, химические сдвиги протонов 56
Этиленгликоль, химические сдвиги О17 355
Этилендиамин, комплекс с никелем 152
Этилены галогензамещенные, химические сдвиги протонов 333
Этиллактат, химические сдвиги С13 321
Этилнитрат, химические сдвиги О17 355
Этилпропионат, химические сдвиги О17 355
Этилформиат, химические сдвиги О17 355
Этилфторсиланы RxSiF4_x, зависимость химических сдвигов F18 от числа алкильных заместителей 247
Этил-2-фуроилацетат, химические сдвиги протонов 114, 115
Этилхлоргидрин, химические сдвиги О17 355
Этилхлорформиат, химические сдвиги О17 355
Эфиры
константы спин-спинового взаимодействия Н1 — Н1 56
х имические сдвиги протонов 56 О17 352
Содержание
Глава 10. КОРРЕЛЯЦИИ СПЕКТРОВ ЯМР-Н1 СО СТРОЕНИЕМ МОЛЕКУЛ 5
Алканы ................................................................. 6
10.1. Химические сдвиги производных алканов .......................... 6
10.1.1. Корреляция протонных химических сдвигов метильных производных СН3Х с электроотрицательностью заместителей 6
10.1.2. Корреляция химических сдвигов с электроотрицательностью заместителей в этильных производных............................... 10
10.1.3. Пропильные и изопропильные производные.................. 14
10.1.4. Вклады диамагнитной анизотропии связей С— Н и С — С в экранирование ядер водорода ................................... 15
10.2. Константы спин-спинового взаимодействия производных алканов 15
10.2.1. Константы геминального взаимодействия протонов у насыщенного атома углерода.............................................. 16
10.2.2. Константы вицинального взаимодействия протонов.......... 16
10.2.3. Знаки констант взаимодействия протонов.................. 19
10.2.4. Константы дальнего взаимодействия протонов в насыщенных системах ........................................................ 20
10.3. Другие алкильные соединения.......................’............ 22
10.3.1. Метильные производные элементов группы IVB.............. 22
10.3.2. Алкильные производные элементов, имеющих ядерные магнитные моменты 24
10.3.3. Другие алкильные производные металлов .................. 26
10.3.4. 2,3-Дизамещенные бутаны................................. 27
10.3.5. Циклопропан и его производные........................... 28
10.3.6. Производные циклогексана................................ 33
Алкены................................................................. 46
10.4. Винильные производные СН2 = СНХ................................ 46
10.4.1. Корреляция между константами геминального взаимодействия /Уц и валентными углами Н — С — Н.......................... 47
10.4.2. Константы вицинального взаимодействия в алкенах .... 48
10.4.3. Корреляция между константами взаимодействия и электроотрицательностями заместителей в винильных производных 49
10.4.4. Изменения химических сдвигов в винильных группах в зависимости от электроотрицательности заместителей................... 52
10.4.5. Корреляция химических сдвигов в винильных системах с а-константами Гаммета.......................................... 52
10.4.6. Корреляция химических сдвигов с групповыми дипольными моментами в винильных системах................................... 53
10.4.7. Аномальные химические сдвиги протонов в винильных
системах ................................................ 53
10.4.8. Хлористый винил и хлорэтилены........................... 57
464
Содержание
10.4.9. Пропен-1, бутен-1 и гексен-1............................. 57
10.4.10. Константы спин-спинового взаимодействия в алкенах-1 .... 60
10.5. Дизамещенные олефины............................................ 51
10.5.1. цис-ъ транс-Дизамещенные олефины.......................... 63
10.5.2. 2-Замещенные пропены...................................... 63
10.5.3. 2,3-Дизамещенные пропены.................................. 65
10.5.4. Влияние метильных групп на спектры протонного резонанса
производных этилена...................................... 66
10.6. Тризамещенные олефины........................................... 68
10.6.1. 1-Замещенные изобутены.................................... 68
10.6.2. а,р-Ненасыщенные эфиры ................................ 69
10.7. Дальнее спин-спиновое взаимодействие через олефиновые системы 72
10.8. Другие олефины.................................................. 74
10.8.1. Сопряженные диены......................................... 74
10.8.2. Олефиновые пройзводные металлов........................... 74
10.8.3. Метильные группы в сопряженных полиенах................... 76
10.8.4. Кольцевые токи в аннуленах................................ 77
Ацетилены............................................................... 77
10.9. Ацетилен и родственные молекулы................................. 77
10.9.1. Галогенопроизводные метилацетилена ХСН2С = СН .... 79
10.10. Константы спин-спинового взаимодействия в ацетиленах........... 80
10.11. Влияние растворителей на химические сдвиги протонов в ацетиленах 81
Ароматические молекулы.................................................. 81
10.12. Производные бензола............................................ 81
10.12.1. Влияние растворителей на спектры протонного резонанса ароматических соединений........................................ 81
10.12.2. Монозамещенные бензолы................................... 82
10.12.3. Корреляция химических сдвигов протонов с а-константами Гаммета в монозамещенных бензолах............................... 84
10.12.4. Дизамещенные производные бензола......................... 85
10.12.5. Галогензамещенные бензолы............,................... 91
10.12.6. Замещенные ксилолы....................................... 93
10.12.7. Оксибензолы.............................................. 94
10.12.8. Ароматические алкоксисоединения.......................... 94
10.12.9. Ароматические альдегиды.................................. 96
10.12.10. Тризамещенные бензолы................................... 99
10.12.11. Дифенилы................................................ 99
10.12.12. Инден.................................................. 100
10.12.13. Кольцевые токи в ароматических молекулах............... 100
10.13. Полициклические ароматические молекулы......................... 101
10.14. Спин-спиновое взаимодействие в ароматических молекулах .... 101
10.15. Ароматические ионы............................................. 104
10.15.1. Триарилкарбониевые ионы................................. 104
10.15.2. Карбониевый ион 9,10-диметил-1,2-бензантрацена и другие ароматические карбониевые ионы................................. 105
10.15.3. Ион азулиния и родственные системы...................... 108
10.15.4. Циклопентадиенил-анион С5Нг и тропилий-катион С7Н+ 109
Гетероциклические соединения.......................................... 111
10.16. Фуран и его производные....................................... 111
10.17. Пиррол и его производные ...................................... 116
10.17.1. Химические сдвиги в пирролах............................ 118
10.17.2. Порфирины............................................... 121
10.17.3. Индолы ................................................. 122
10.18. Пиридин и родственные молекулы................................. 122
Содержание 465
10.19. Влияние растворителей на спектры протонного резонанса ненасыщенных гетероциклических соединений.............................. 127
10.20. Хинолины..................................................... 127
10.21. Тиофен и его замещенные...................................... 130
10.22. Тиазол и его метильные производные........................... 133
Сложные органические соединения....................................... 133
10.23. Алкалоиды.................................................... 134
10.24. Стероиды .................................................... 135
10.25. Жирные кислоты............................................... 139
10.26. Аминокислоты и пептиды....................................... 139
10.27. Другие исследования.......................................... 139
10.28. Определение С-метильных групп методом ЯМР.................... 141
Соединения, в которых водород связан с атомами, отличными от углерода 143
10.29. Водород, связанный с атомами кислорода....................... 143
10.30. Водород, связанный с атомами азота........................... 145
10.31. Водород, связанный с атомами бора............................ 146
10.32. Водород, связанный с атомами серы............................ 147
10.33. Водород, связанный с атомами кремния......................... 147
10.34. Водород, связанный с атомами германия, олова и свинца........ 148
10.35. Водород, связанный с атомами переходных металлов............. 150
Другие исследования................................................... 151
10.36. Контактные химические сдвиги в парамагнитных частицах........ 151
10.37. Исследования растворов полимеров............................. 153
10.37.1. Сегментное движение в полимерах....................... 154
10.37.2. Определение молекулярной структуры полимеров.......... 156
10.37.3. Определение пространственной конфигурации полимеров 158
10.37.4. Применение двойного резонанса для определения строения полимеров ..................................................... 159
10.37.5. Использование дейтерирования при определении стереорегулярности полимеров............................................. 160
10.38. Эмпирическая оценка химических сдвигов ...................... 161
10.39. Влияние среды в жидкостях.................................... 164
10.39.1. Влияние растворителей на спектры протонного резонанса ацетиленовых соединений............................................ 170
10.39.2. Влияние растворителей на спектры протонного резонанса ароматических соединений ....................................... 173
10.39.3. Влияние растворителей на спектры протонного резонанса ненасыщенных гетероциклических соединений............... 176
10.40. Влияние среды в газах........................................ 177
Литература .............................................................. 180
Глава И. ИССЛЕДОВАНИЕ ЯДЕРНОГО МАГНИТНОГО РЕЗОНАНСА ФТОРА 193
11.1. Введение...................................................... 193
11.2. Химические сдвиги ядер фтора................................ 196
11.3. Константы спин-спинового взаимодействия F19................... 197
11.4. Бинарные соединения фтора..................................... 202
11.5. Галогенопроизводные фторуглеродов . .......................... 205
11.6. Замещенные фторалканы ........................................ 207
11.6.1. Относительные знаки констант спин-спинового взаимодействия F19 во фторалканах........................................ 209
11.6.2. Данные, полученные при исследовании конформации замещенных этанов................................................... 210
11.7. Фторуглеводородиые соединения, содержащие азот................ 211
11.8. Другие производные фторуглеводородов.......................... 213
466
Содержание
11.9. Перфторалкильные и перфторацильные соединения металлов ... 217
11.10. Фторированные ароматические соединения....................... 219
11.10.1. Химические сдвиги F19 в замещенных фторбензолах....... 219
11.10.2. Спин-спиновое взаимодействие во фторированных ароматических соединениях.............................................. 222
11.10.3. Замещенные бензотрифториды............................ 225
11.10.4. Полизамещенные производные фторбензолов............... 225
11.10.5. Фторзамещенные нафталины.............................. 226
11.11. Фторалкены................................................... 226
11.11.1. Перфторвинильные производные.......................... 226
11.11.2. Другие фторалкены..................................... 229
11.11.3. Константы взаимодействия для фторалкенов ............. 229
11.11.4. Относительные знаки констант взаимодействия F19 во фтор-алкенах......................................................... 234
11.11.5. Сведения, получаемые при исследовании сателлитов, обусловленных взаимодействием С13 — F.................................. 235
11.12. Фторацетилены................................................ 236
11.13. Фторированные циклические соединения......................... 236
11.13.1. Производные циклопропана.............................. 236
11.13.2. Моно- и дизамещенные производные тетрафторциклобутана 237
11.13.3. Фторированные циклогексаны............................ 246
11.13.4. Фторированные гетероциклические соединения............ 244
11.14. Метил- и этилфторсиланы RxSiF4_x............................. 247
11.15. Пентафторид сурьмы........................................... 247
11.16. Соединения, содержащие связи F — S.......................... 248
11.16.1. Гексафторид серы и его монозамещенные производные . . . 248
11.16.2. Дизамещенные производные гексафторида серы............ 252
11.16.3. Тетрафторид серы...................................... 253
11.16.4. Перфторалкнльные производные SF4...................... 254
11.17. Другие фторсодержащие соединения серы........................ 255
11.18. Соединения фтора с другими галогенами........................ 256
11.18.1. Трифторид хлора....................................... 256
11.18.2. Трифторид брома....................................... 257
11.18.3. Пентафториды иода и брома.......................... 257
11.18.4. Перхлорилфторид FC1O3 и родственные соединения .... 259
11.19. Соединения, содержащие связи В — F........................... 259
11.19.1. Трифторид бора и родственные соединения............... 259
11.19.2. Ион BFj............................................... 261
11.20. Соединения, содержащие связи N — F.......................... 262’
11.21. Соединения, содержащие связи О— F............................ 264
11.22. Соединения, содержащие связи Р — F........................... 264
11.23. Комплексные соединения ...................................... 267
11.23.1. Комплексы трнфторида бора............................. 267
11.23.2. Комплексы тетрафторидов металлов...................... 267
11.23.3. Другие комплексы фторидов металлов.................... 268
11.24. Фторсодержащие полимеры...................................... 269
11.25. Другие исследования.......................................... 272
Литература .............................................................. 278
Глава 12. СПЕКТРЫ ЯМР ДРУГИХ ЯДЕР (КРОМЕ ВОДОРОДА И ФТОРА) 284
12.1. Бор........................................................... 285
12.1.1. Химические сдвиги В11.................................. 286
12.1.2. Спин-спиновое взаимодействие . ........................ 291
12.1.3. Гидриды бора........................................... 291
Содержа н и е 467
12.1.4. Боргидриды................................................ 300
12.1.5. Другие исследования резонансов В11...................... 300
12.2. Углерод......................................................... 301
12.2.1. Экспериментальные методы измерения резонанса на ядрах С13 302
12.2.2. Химические сдвиги С13.................................... 305
12.2.3. Корреляция химических сдвигов С13 с электроотрицательностью групп, связанных с атомами углерода...................... 307
12.2.4. Метил- и этилпроизводные элементов IV группы............ 312
12.2.5. Алкены................................................... 312
12.2.6. Ацетилены................................................ 313
12.2.7. Ароматические соединения................................. 313
12.2.8. Пиридин и замещенные пиридины............................ 319
12.2.9. Химические сдвиги С13 карбонильных соединений............ 320
12.2.10. Другие исследования ЯМР-С13 ............................ 321
12.2.11 Спич-спиновое взаимодействие между ядрами Н1 и С13 . . . 322
12.2.12. Аддитивные соотношения для констант спин-спинового взаимодействия Н1 — С13 и F19 — С13................................. 327
12.2.13. Аддитивные соотношения для констант /д1з_н атомов углерода с хр2-гибридизацией........................................ 331
12.2.14. Взаимосвязь /С1з_н с и Удд для этиленов................. 332’
12.2.15. Корреляция констант спин-спинового взаимодействияН1 — С13 с химическими сдвигами протонов................................. 333
12.2.16. Константы спин-спинового взаимодействия С13 — F19 .... 334
12.2.17. Сателлиты С13 — Н в протонных спектрах симметрично замещенных бензолов................................................ 335-
12.2.18. Константы дальнего спин-спинового взаимодействия С13 — С — Н1 ................................................... 336
12.2.19. Константы спин-спинового взаимодействия С13 — С13 . . . 342
12.2.20. Различные константы спин-спинового взаимодействия С13 — Н1........................................................ 342
12.3. Азот............................................................ 343
12.3.1. Химические сдвиги N14.....................•.............. 344
12.3.2. Эффекты в спектрах ЯМР, обусловленные квадрупольным моментом ядра азота......................’...................... 347
12.3.3. Константы дальнего спин-спинового взаимодействия Н1 — N14 349
12.3.4. Определение строения молекул с помощью ЯМР-N14 . . . 350
12.4. Кислород...................................................... 351
12.4.1. Химические сдвиги О17.................................... 351
12.4.2. . Влияние парамагнитных ионов на химические сдвиги О17 356
12.4.3. Спин-спиновое взаимодействие Н1 — О17 в спектре воды . . . 357
12.5. Кремний......................................................... 357
12.5.1. Химические сдвиги Si29 .................................. 359
12.5.2. Спин-спиновое воздействие ядра Si29 ..................... 360
12.6. Фосфор ......................................................... 361
12.6.1. Химические сдвиги Р31.................................... 362
12.6.2. Спин-спиновое взаимодействие ядер Р31.................... 369
12.6.3. Применение ЯМР-Р31 для структурных исследований .... 373
12.6.4. Количественный анализ................................... 382
12.7. Кобальт........................................................ 384
12.7.1. Химические сдвиги Со59 ................................. 38 4
12.7.2. Исследование равновесных процессов с участием кебальта 387
12.8. Олово ......................................................... 387
12.8.1. Химические сдвиги Sn119.................................. 388
12.8.2. Смешанные галогениды Sn(IV)............................. 389
12.8.3. Константы спин-спинового взаимодействия Sn119........... 391
468
Содержание
12.9. Таллий ............................................................. 392
12.9.1. Химические сдвиги ЯМР-Т1205 ................................ 393
12.9.2. Спин-спиновое взаимодействие Т1205 — Н1 .................... 395
12.9.3. Структура этилата таллия ТЮС2Н5 ............................ 396
12.10. Другие ядра ....................................................... 396
12.10.1. Дейтерий................................................... 396
12.10.2. Щелочные металлы........................................... 397
12.10.3. Алюминий А127 ............................................. 398
12.10.4. Галогены ...................................................399
12.10.5. Медь Си63 ................................................. 401
12.10.6. Мышьяк As75 ............................................... 401
12.10.7. Селен Se77 ................................................ 401
12.10.8. Сурьма Sb121 .............................................. 401
12.10.9. Платина Pt195 401
12.10.10. Ртуть Hg199............................................... 401
12.10.11- Свинец РЬ207 ............................................. 404
12.10.12. Ниобий Nb93 .............................................. 405
12.10.13. Магнитные моменты ядер ................................... 405
Литература .................................................................... 405
Приложения....................................................................... 413
Приложение I.................................................... 415
Приложение II.................................................... 430
Приложение III.................................................... 436
Приложение IV.................................................... 437
Приложение V.................................................... 441
Предметный указатель........................................................... 452
Дж. Эмсли, Дж. Финей, JI. Сатклиф СПЕКТРОСКОПИЯ ЯДЕРНОГО МАГНИТНОГО РЕЗОНАНСА ВЫСОКОГО РАЗРЕШЕНИЯ
том 2
Редактор И. С. Беленькая
Художник Л. Г. Ларский Художественный редактор Н. А. Фильчагина Технический редактор Е. С. Потапенкова Корректор Л. А. Брычкова
Сдано в производство 4/IX 1968 г. Подписано к печати 24/11 1969 г. Бумага № 1 70 X 108’/ie= 14,63 бум. л. усл. печ. л., 40,95. Уч.-изд. л. 40,88. Изд. № 3/4192. Цена 4 р. 28 к.
Зак. 489. Темплаи изд-ва «Мир» 1968 г. пор. № 124.
ИЗДАТЕЛЬСТВО «МИР» Москва, 1-й Рижский пер., 2 Московская типография № 16 Главполиграфпрома Комитета по печати при Совете Министров СССР
Москва, Трехпрудиый пер., 9