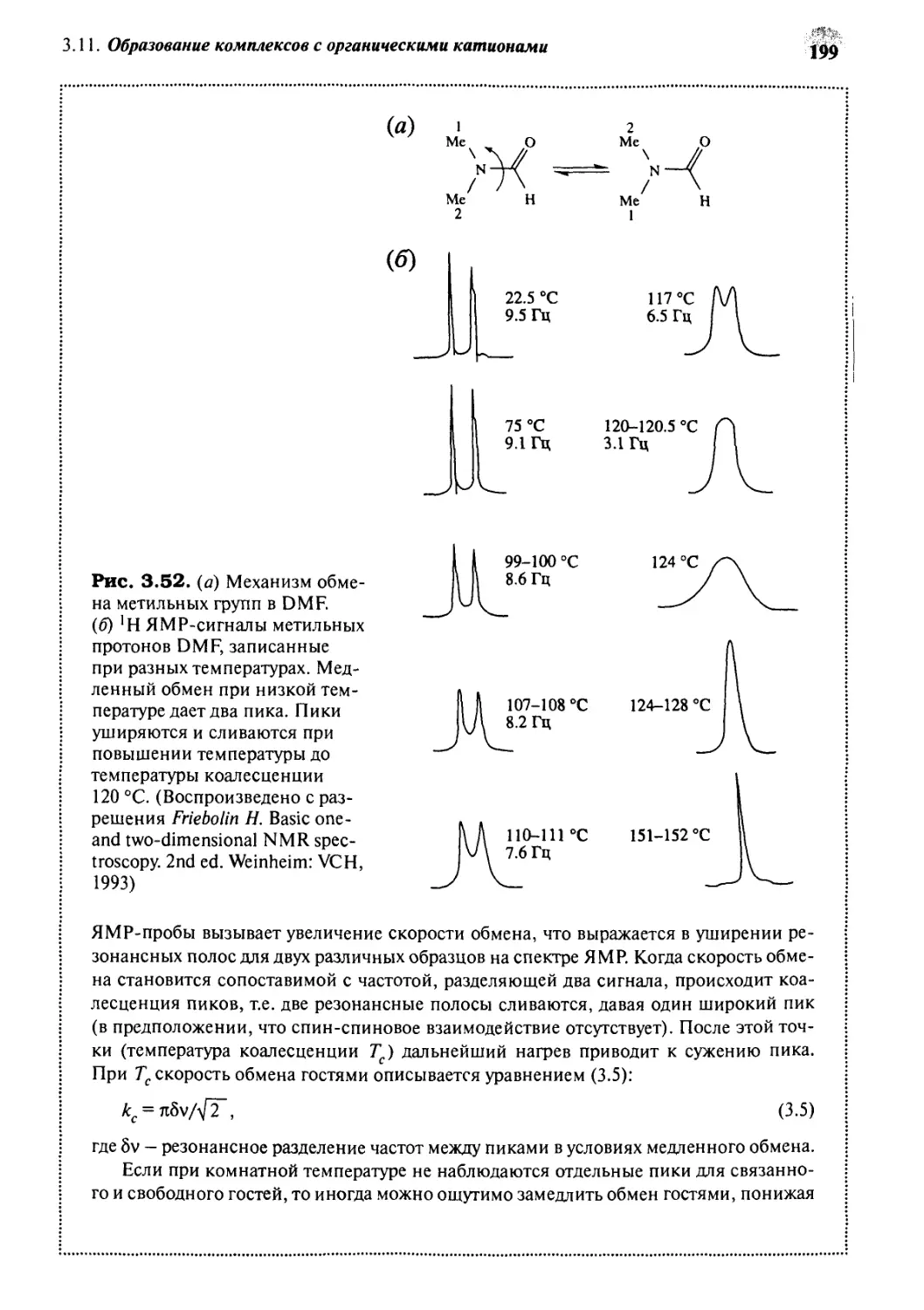
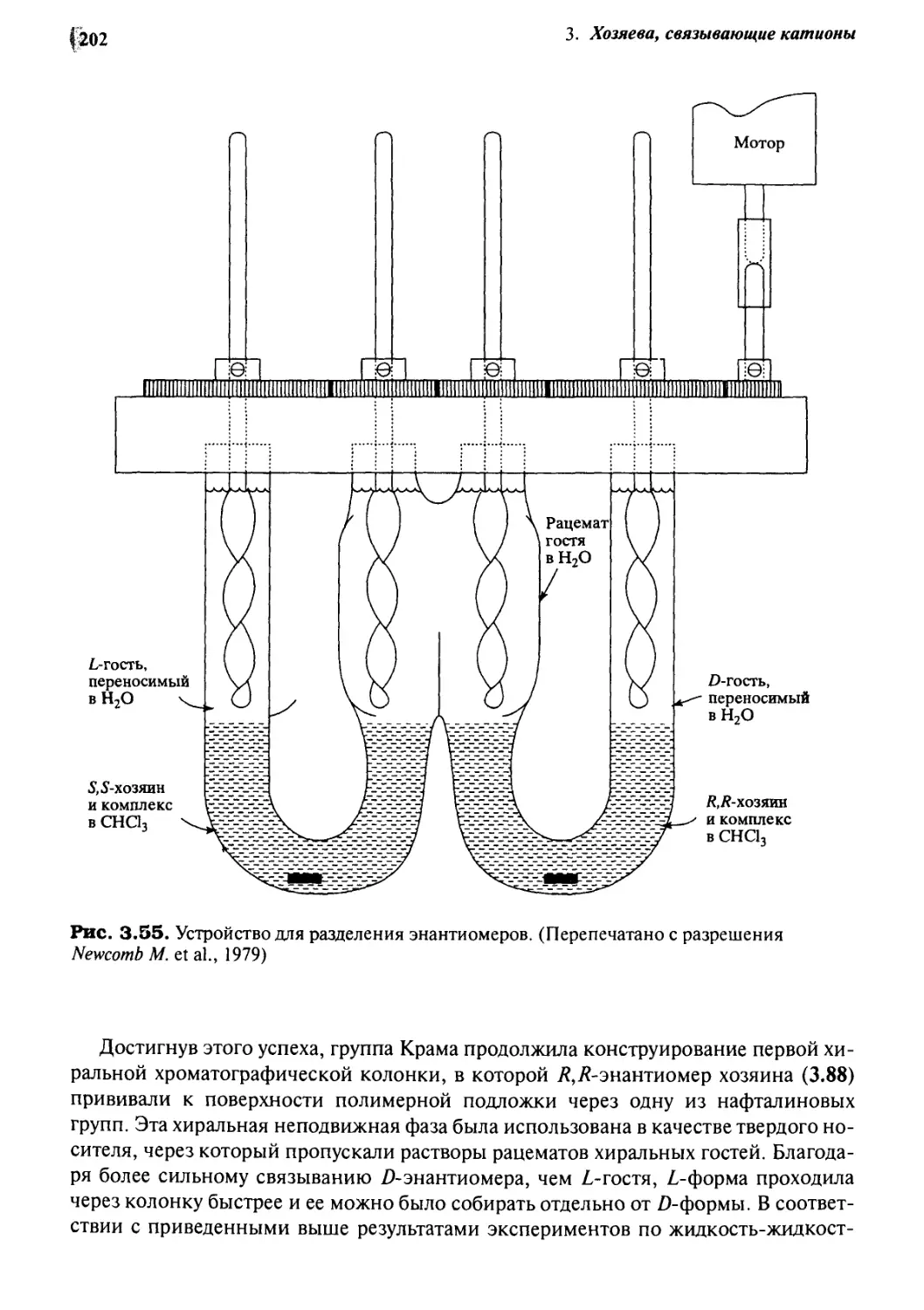
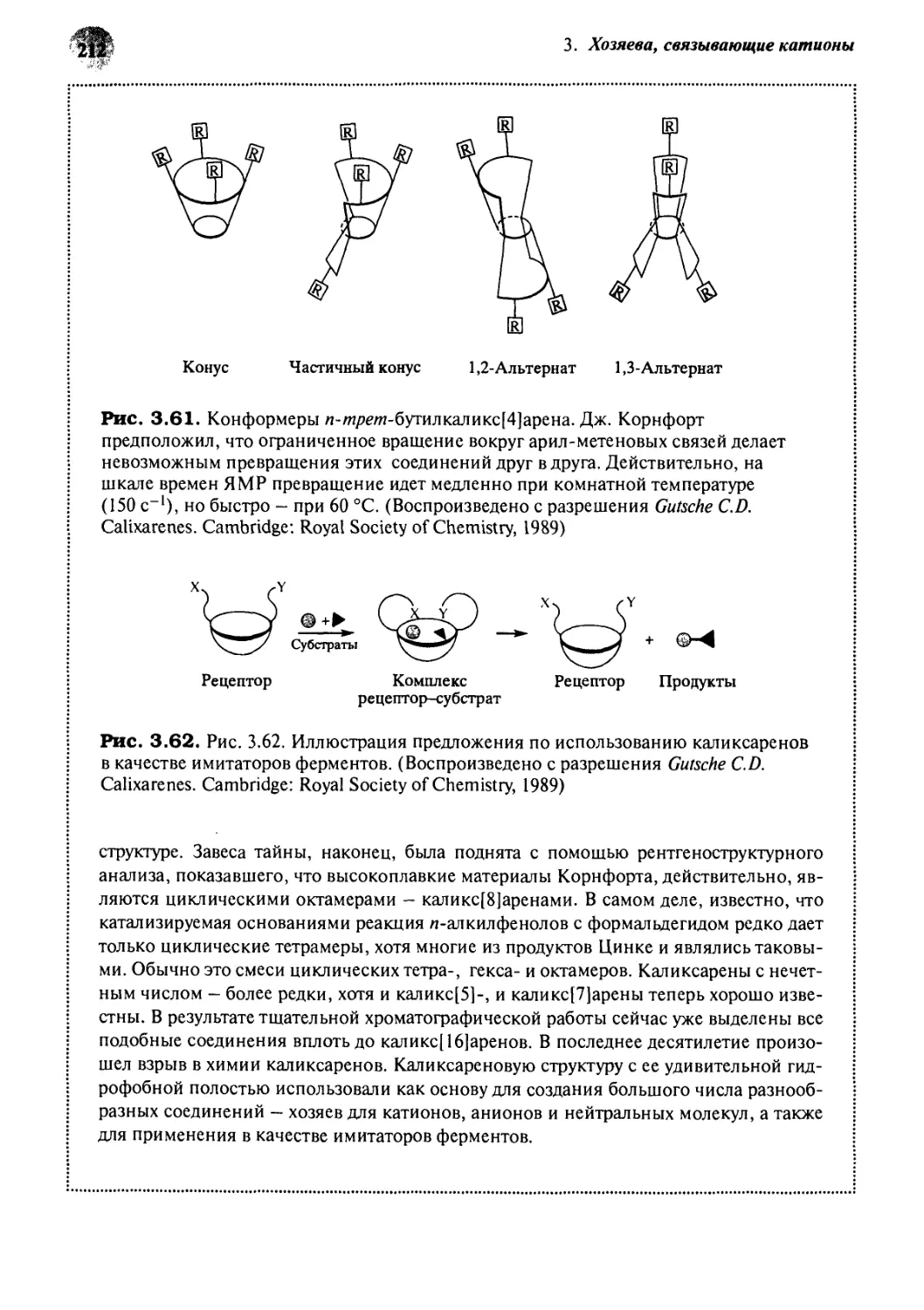
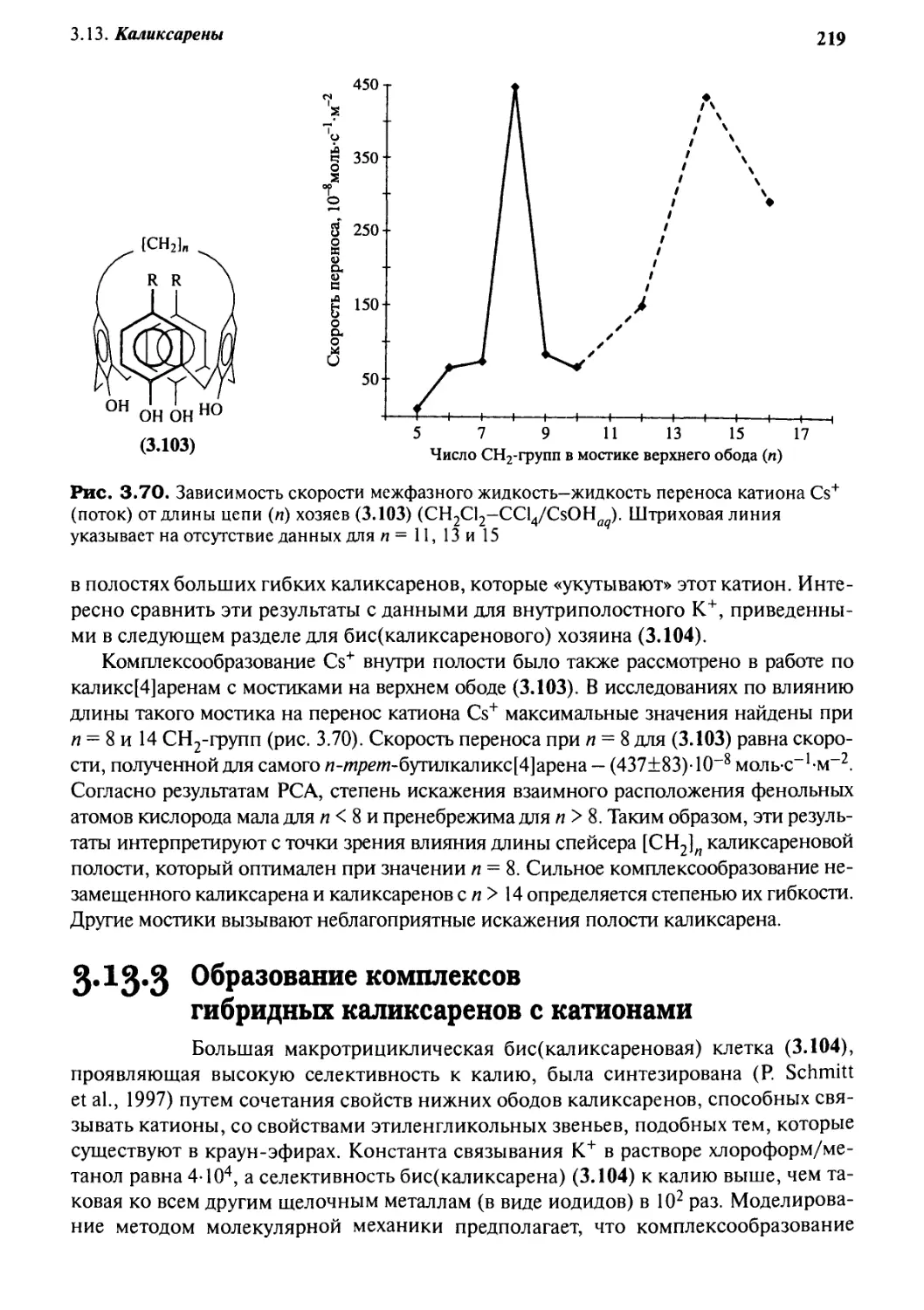
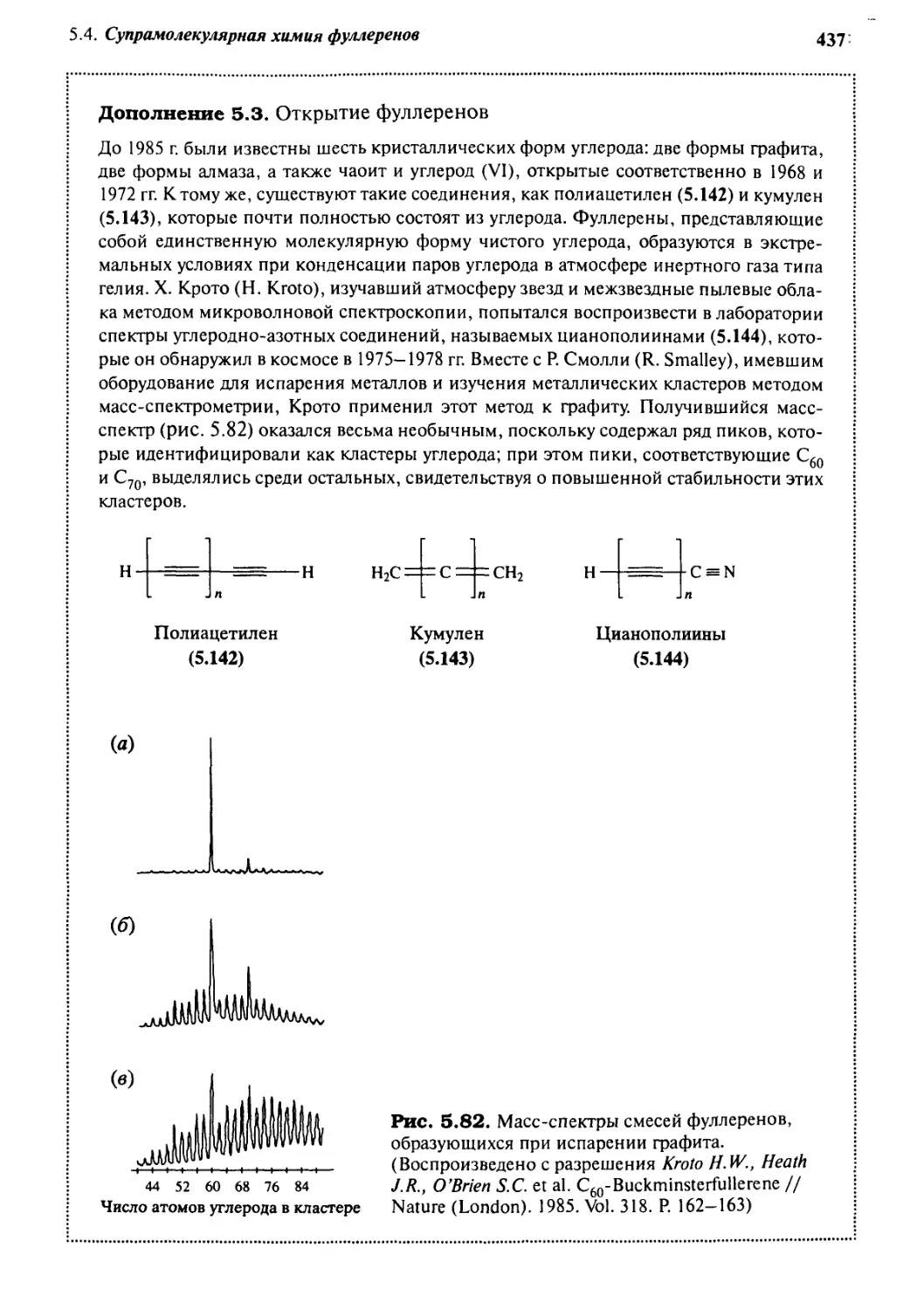
/










































































































































































































































































































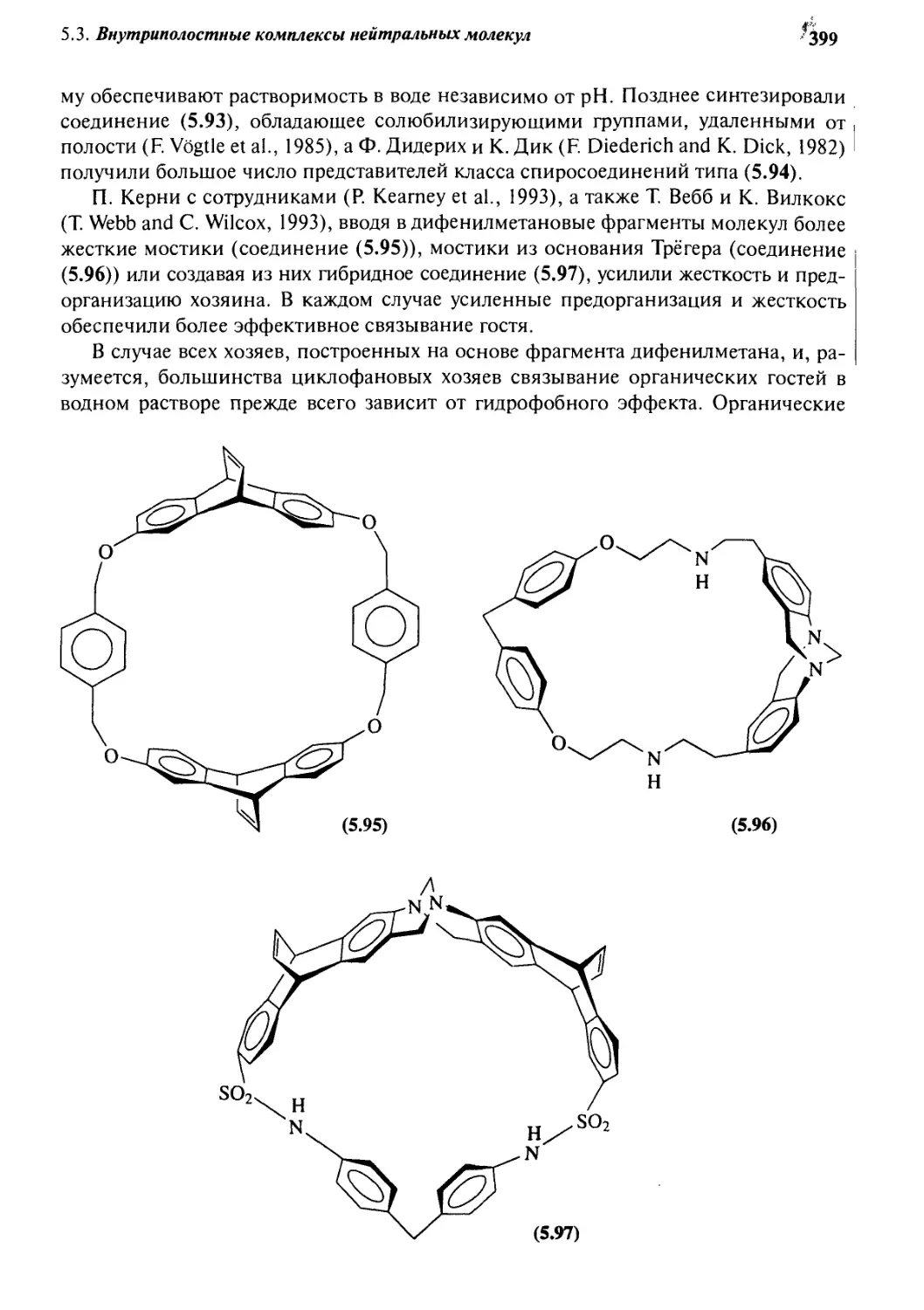

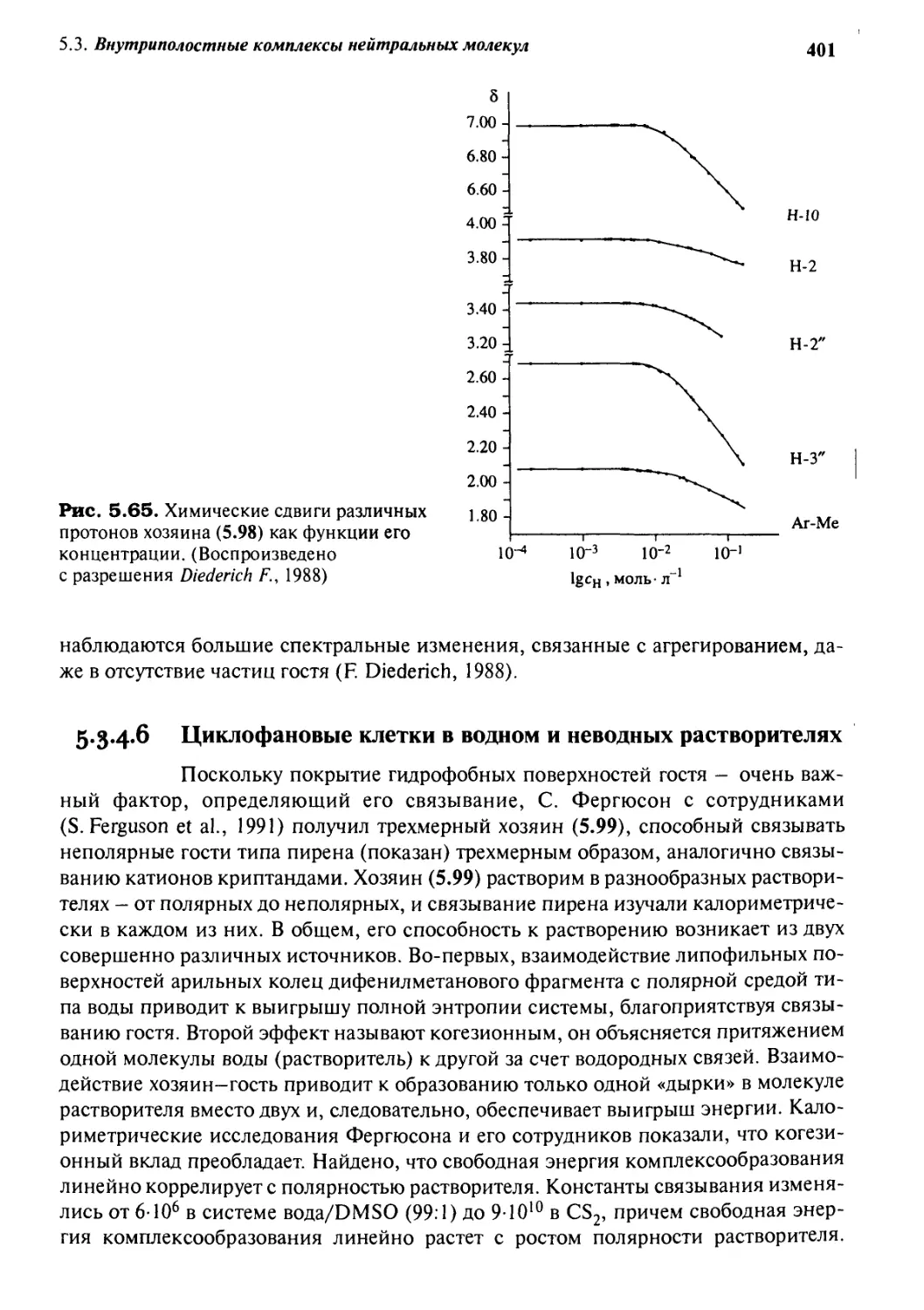
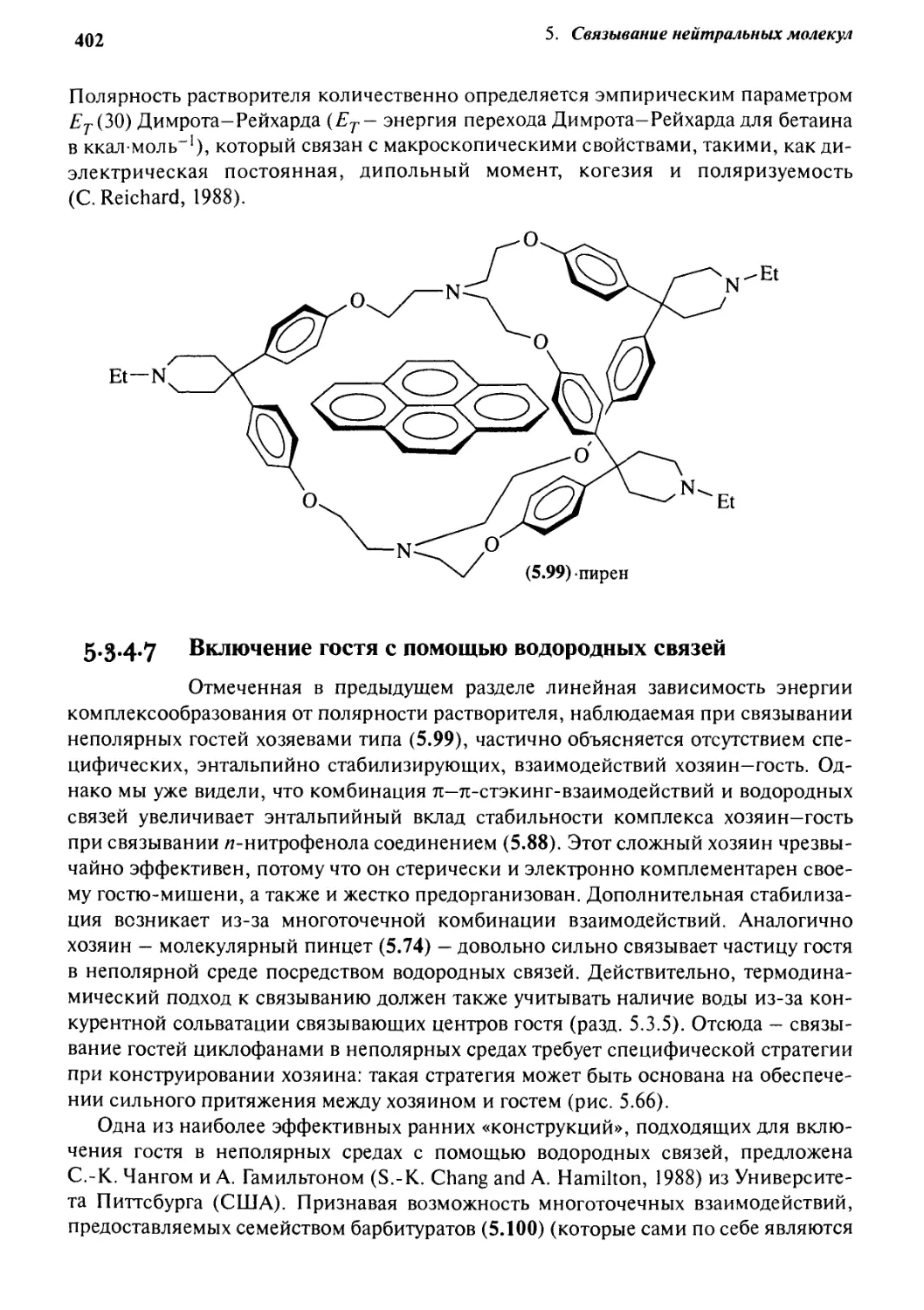




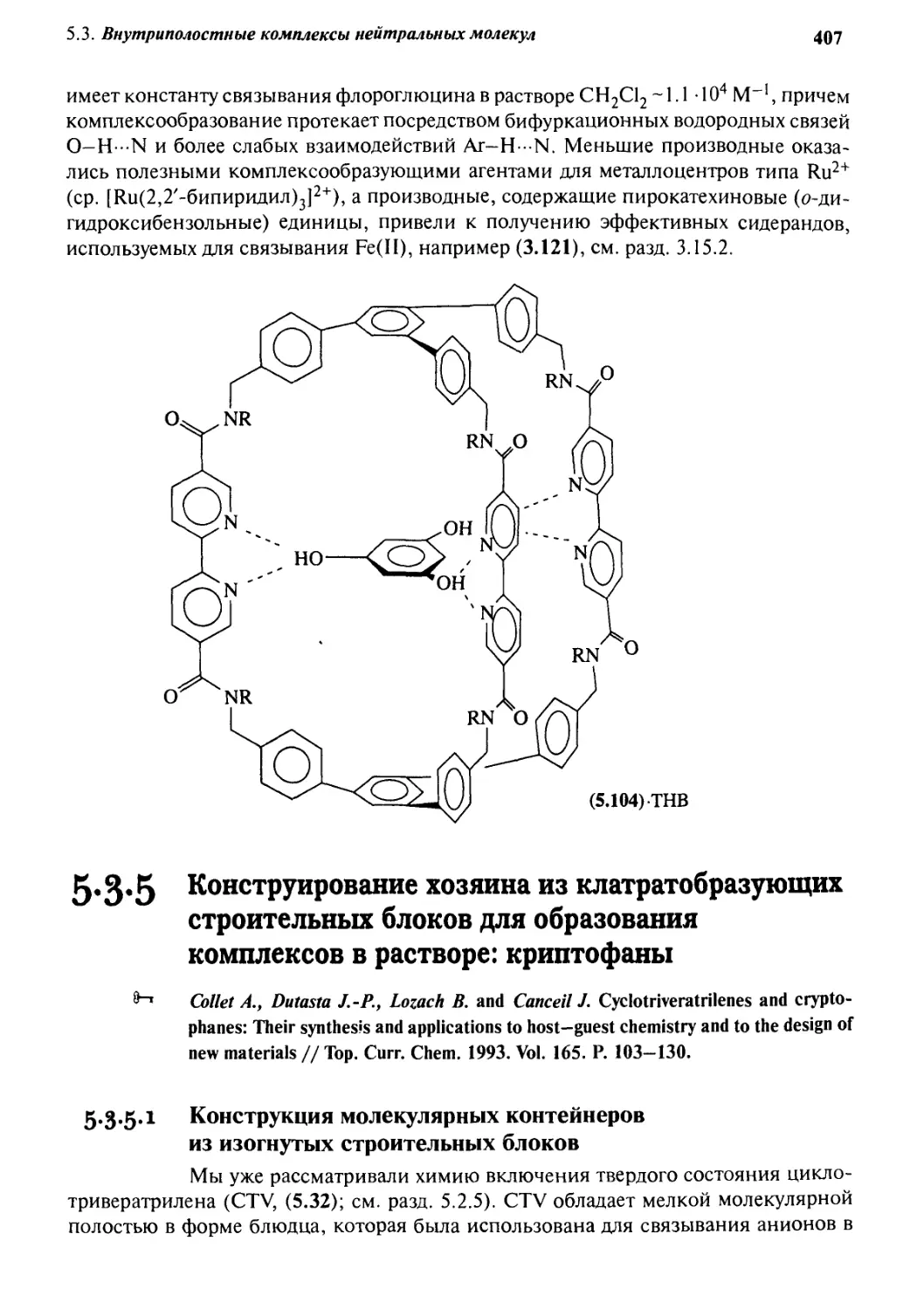
































































Автор: Стид Дж. В. Этвуд Дж. Л.
Теги: органическая химия химия молекулярная биология молекулярная физика
ISBN: 978-5-94628-303-8
Год: 2007




Текст
химия
Jonathan W. Steed
King's College, London
Jerry L. Atwood
University of Missouri, Columbia
John Wiley & Sons, Ш
Chichester • New York • Weinheim • Brisbane • Singapore «Toronto
ДЖ.В.СТИД, Дж.Л. ЭТВУД
химия
Б двух томах
Том 1
Перевод с английского
кандидата химических наук И. Г. Варшавской,
кандидата химических наук Б. И. Харисова,
кандидата химических наук О. В. Белуженко,
кандидата химических наук И. С. Васильченко,
доктора химических наук Ю. А. Алексеева
Под редакцией
академика РАН, профессора А. Ю. Цивадзе,
доктора химических наук, профессора В. В. Арсланова,
доктора химических наук, профессора А. Д. Гарновского
МОСКВА
ИКЦ «АКАДЕМКНИГА».
2007
ББК24.2
С80
Издание осуществлено при поддержке
Российского фонда фундаментальных исследований по проектам
№ 04-03-46004
№ 05-03-46005
Ствд Дж. В., Этвуд Дж. Л.
Супрамолекулярная химия. Пер. с англ.: в 2 т. / Джонатан В. Стид, Джерри Л.
Этвуд. - М. : ИКЦ «Академкнига», 2007. - ISBN 978-5-94628-303-8.
Т. 1. - 2007. - 480 с.: ил. - ISBN 978-5-94628-305-2.
Книга является всеобъемлющим изданием в области супрамолекулярнои химии и по
разностороннему освещению ее фундаментальных и прикладных аспектов, и по ясному и
четкому изложению ее основных концепций, понятий, определений. Большое внимание
уделено состоянию и перспективам развития основных разделов супрамолекулярнои
химии.
Рассмотрены невалентные взаимодействия, создание супрамолекулярных ансамблей,
вопросы современной химии краун-соединений и антикраунов, фуллеренов, геликатов,
дендримеров, супрамолекулярных фото- и биоактивных структур, твердых и жидких
кристаллов, химических сенсоров, нелинейно-оптических материалов и молекулярных
устройств, а также вопросы химии жизненно важных процессов. Предлагаемая книга —
ценное руководство для становления и развития супрамолекулярнои химии в нашей стране.
Эта монография может служить учебником для молодых специалистов.
Для специалистов различных областей, студентов, аспирантов и преподавателей
высших учебных заведений.
ISBN 0-471-98831-6 (англ.)
ISBN 0-471-98791-3
ISBN 978-5-94628-303-8 (рус. общий)
ISBN 978-5-94628-305-2 (т. 1)
© John Wiley & Sons, Ltd, 2000
© ИКЦ «Академкнига», 2007
Оглавление
Предисловие авторов к русскому изданию 15
Предисловие редакторов русского издания 16
Предисловие авторов 17
Благодарности 20
Об авторах 21
Основные сокращения и обозначения 22
Том 1
1 Общие представления и
1.1 Определение и развитие супрамолекулярной химии 27
1.1.1 Что такое супрамолекулярная химия? 27
1.1.2 Химия «хозяин—гость» 28
1.1.3 Развитие представлений 30
1.2 Классификация супрамолекулярных
соединений хозяин-гость 32
1.SJ Рецепторы, координация и аналогия «замок-ключ» 34
1-4 Хелатный и макроциклический эффекты 35
1*5 Предорганизация и комплементарность 40
1.6 Термодинамическая и кинетическая селективность 41
Природа супрамолекулярных взаимодействий 48
1.7.1 Ион-ионные взаимодействия A00-350 кДж-моль) 48
1.7.2 Ион-дипольные взаимодействия E0—200 кДж-моль) 50
1.7.3 Диполь-дипольные взаимодействия E—50 кДжмоль") 51
1.7.4 Водородная связь D-120 кДжмоль) 51
1.7.5 Катион-я-взаимодействия E-80 кДж-моль) 53
1.7.6 п—л-Стэкинг- взаимодействия @—50 кДжмоль) 55
Оглавление
1.7.7 Силы Ван-дер-Ваальса (<5 кДжмоль; переменные) 57
1.7.8 Плотная упаковка в твердом состоянии 58
1.7.9 Гидрофобные эффекты 58
1,8 Супрамолекулярное конструирование хозяина 60
Учебные задания 62
Литература 63
Супрамолекулярная химия жизни 64
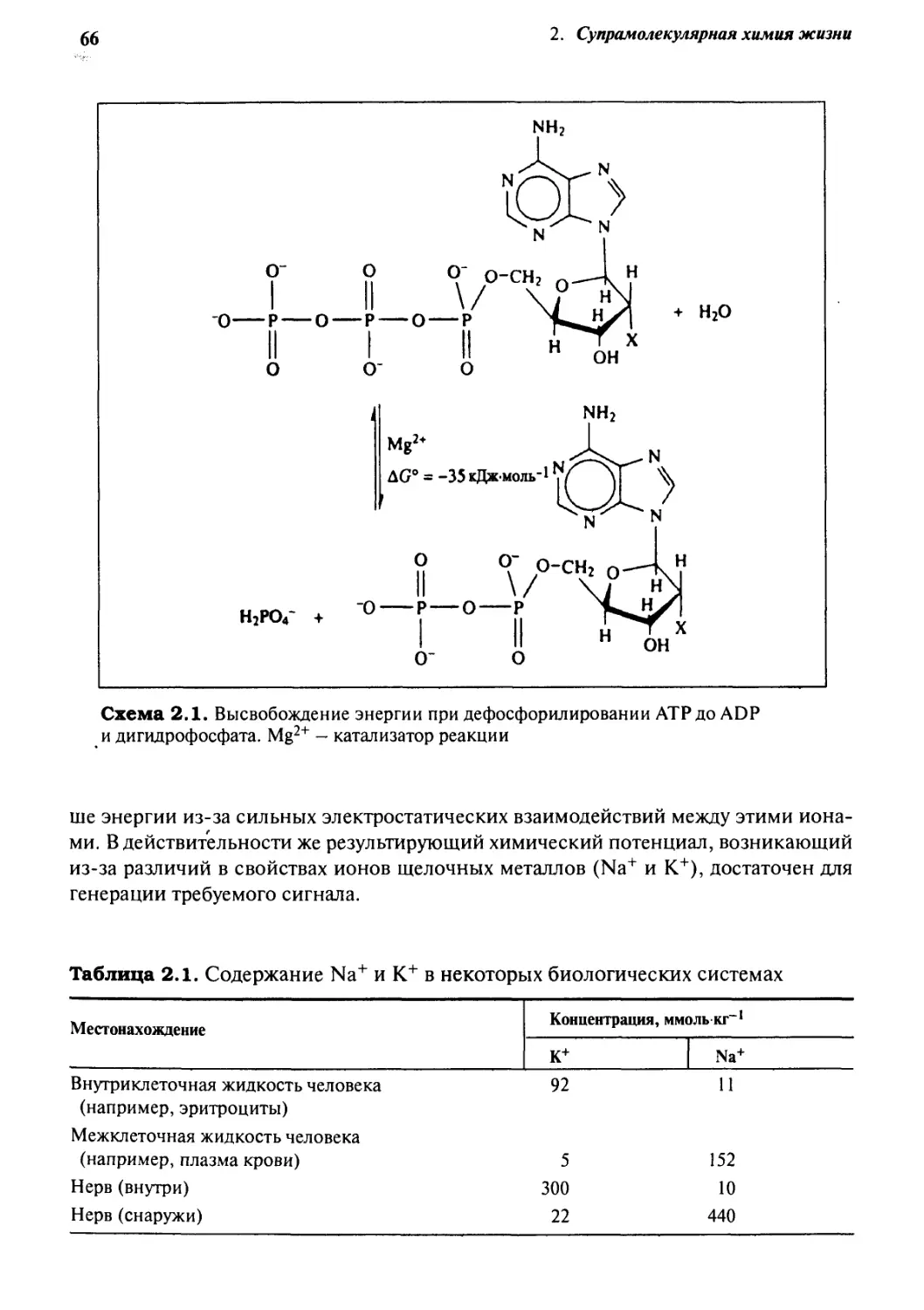
2.1 Катионы щелочных металлов в биохимии 65
2.1.1 Мембранные потенциалы 65
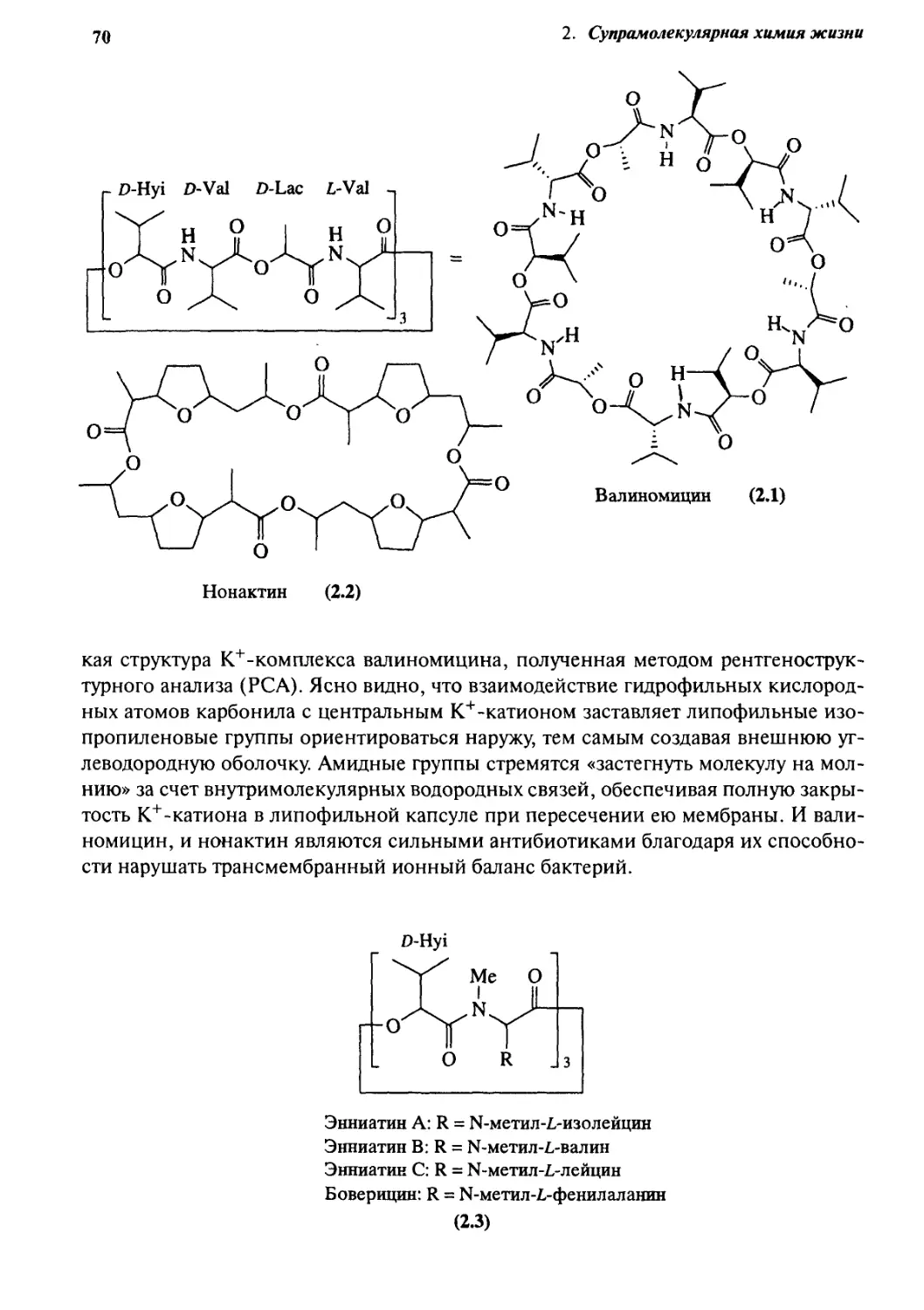
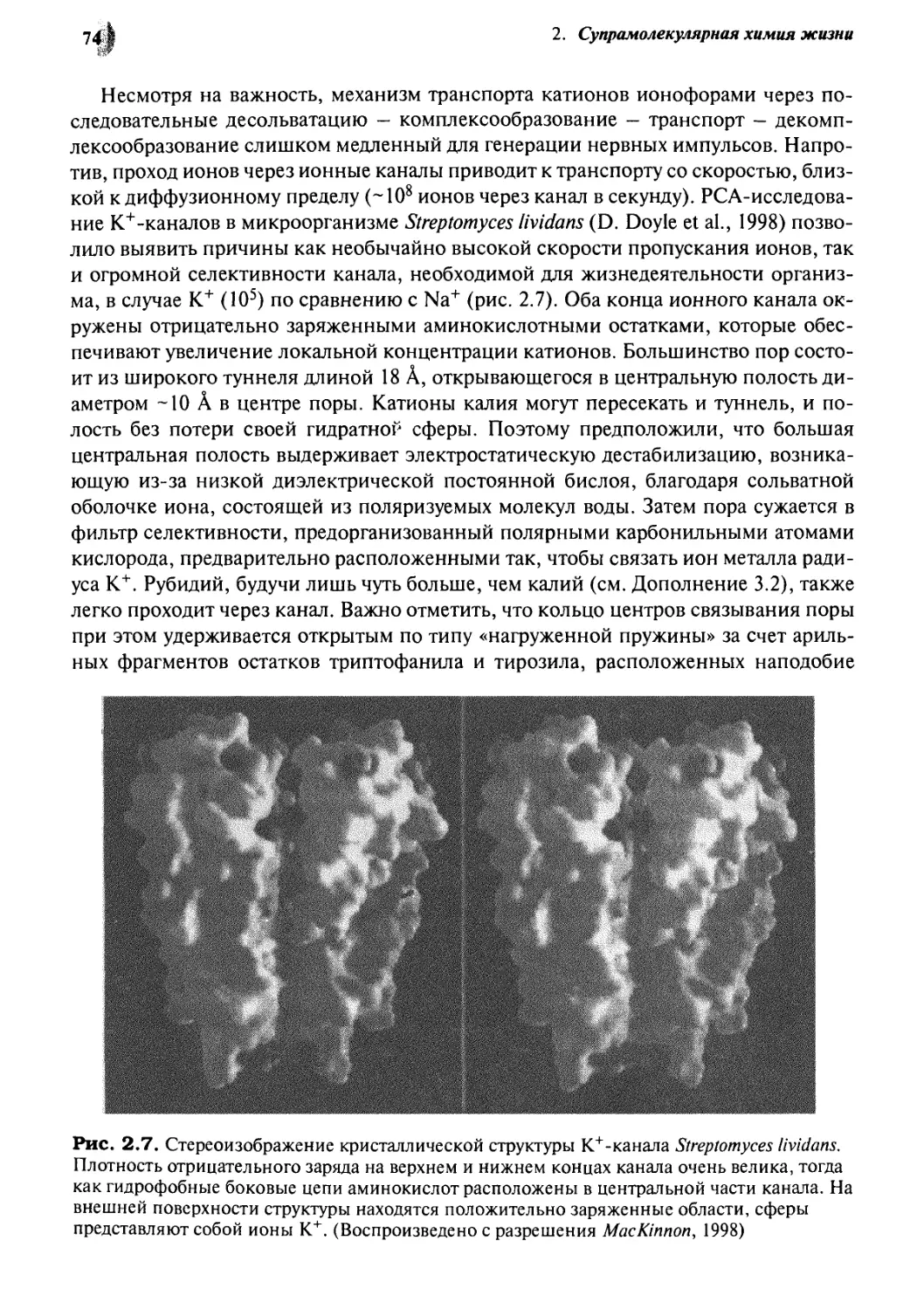
2.1.2 Мембранный транспорт 68
2.1.3 Родопсин: супрамолекулярное фотонное устройство 76
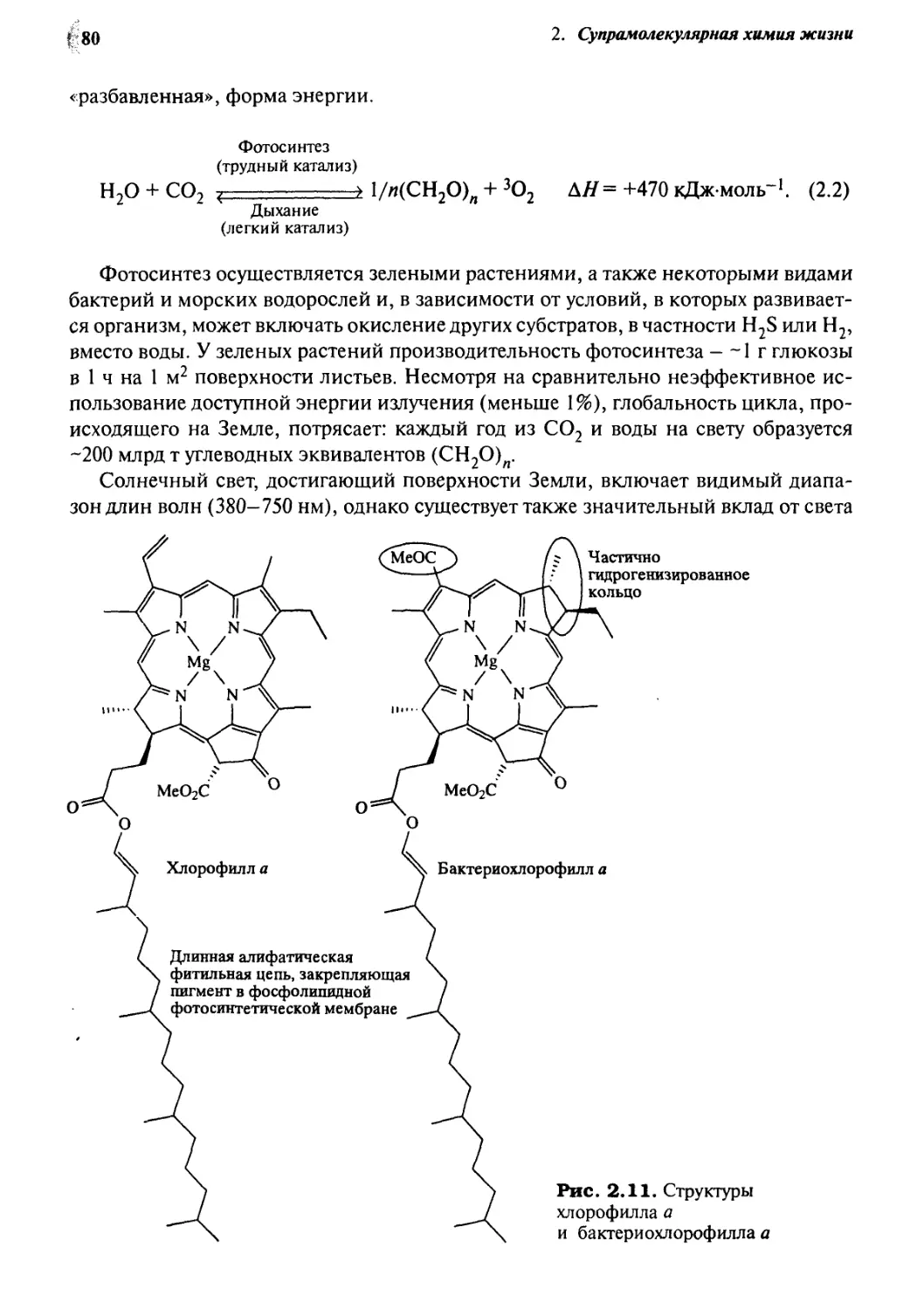
2»% Порфириновые и тетрапиррольные макроциклы 78
2*3 Супрамолекулярные особенности фотосинтеза
в растениях 79
2.3.1 Роль тетрапиррольных комплексов магния 79
2.3.2 Окисление воды до кислорода,
катализированное марганцем 85
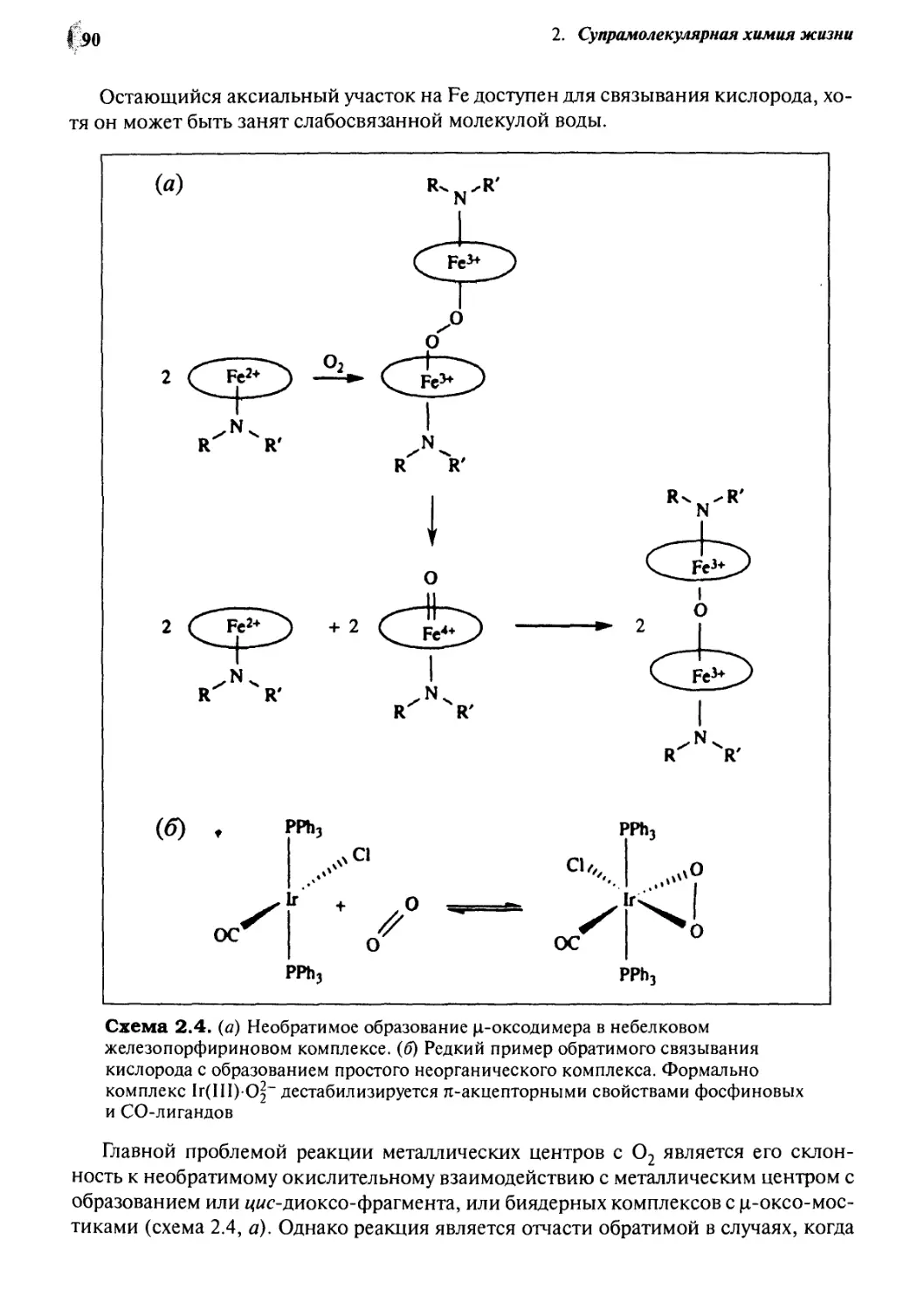
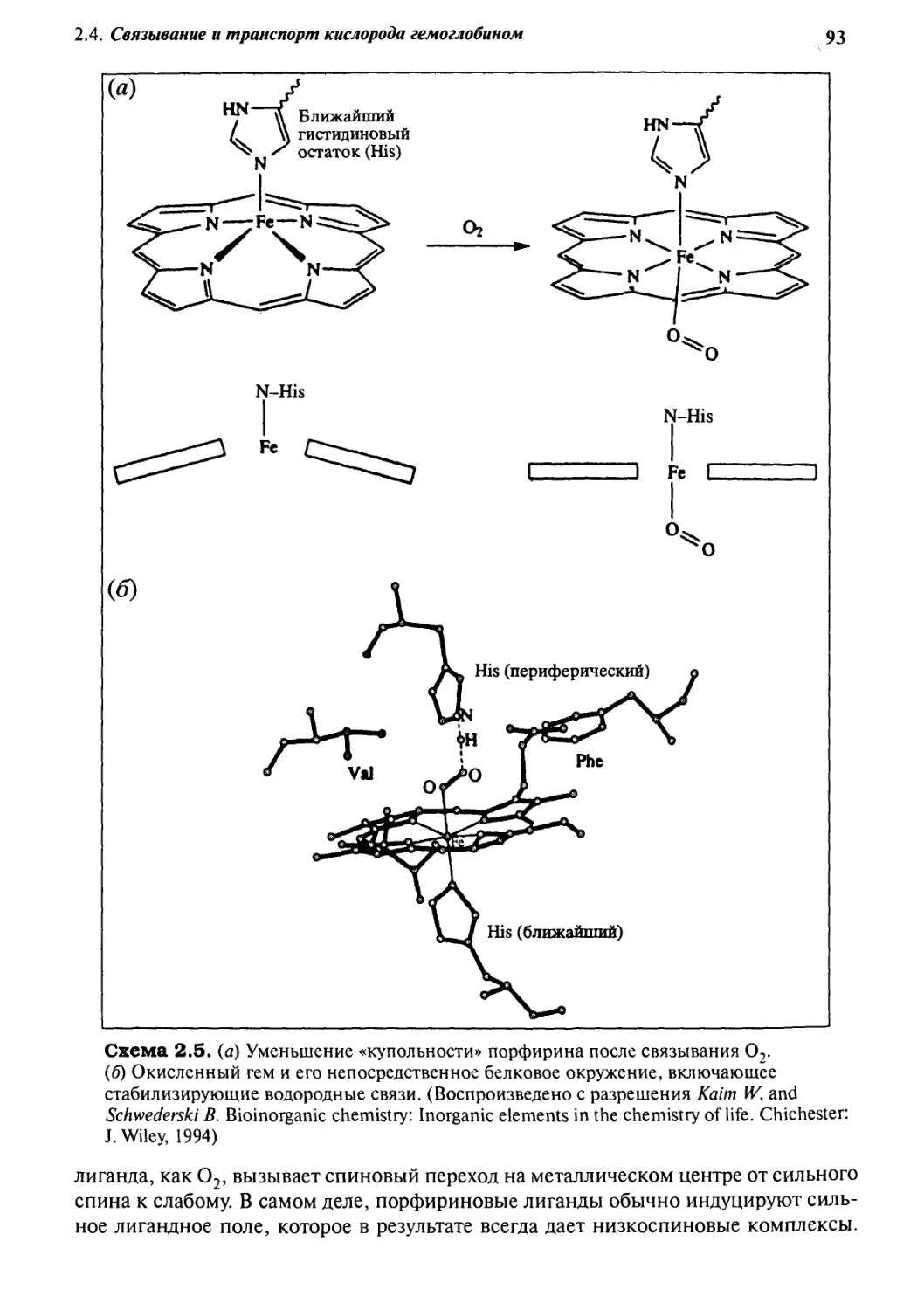
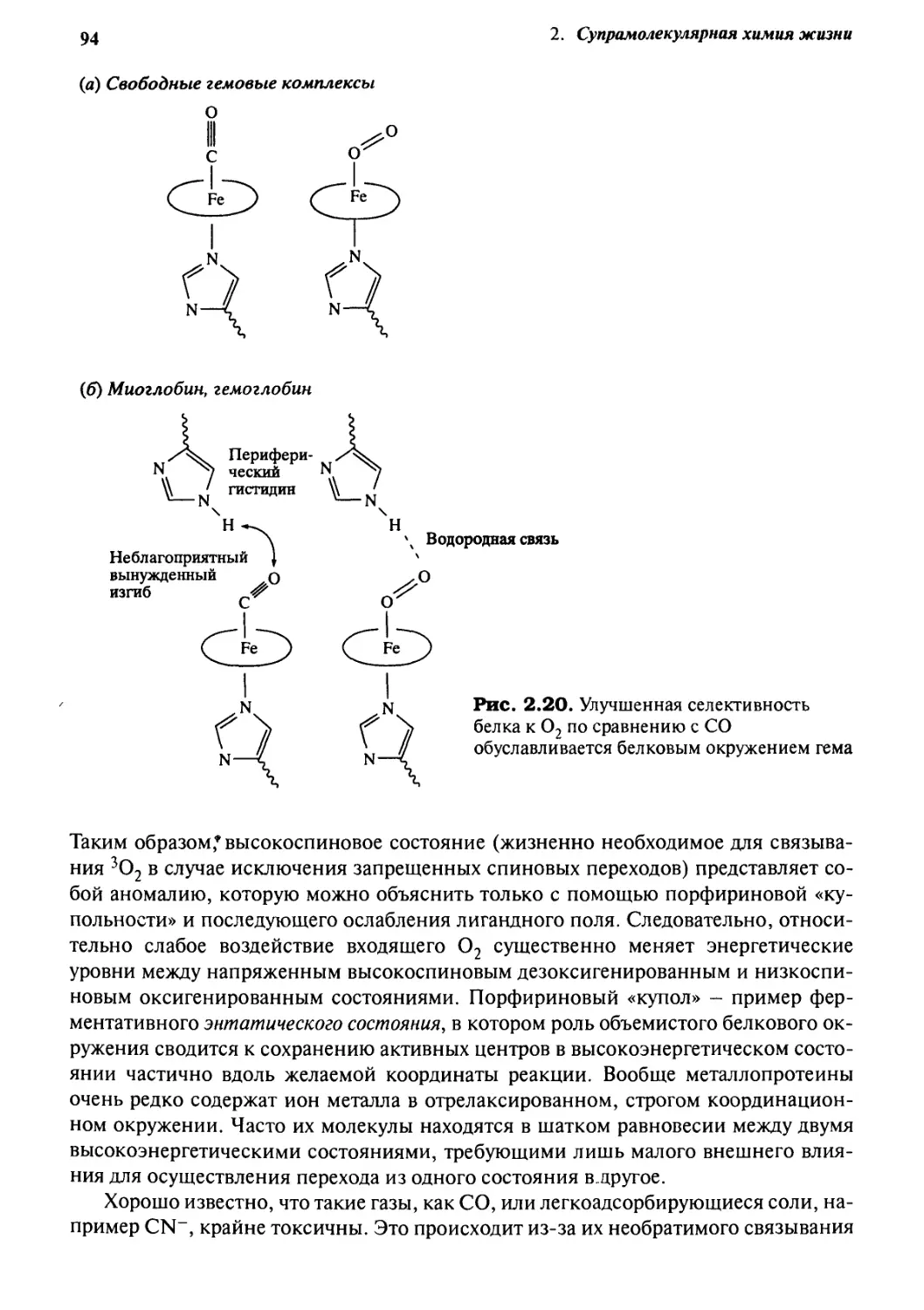
2*4 Связьшание и транспорт кислорода гемоглобином 88
2-5
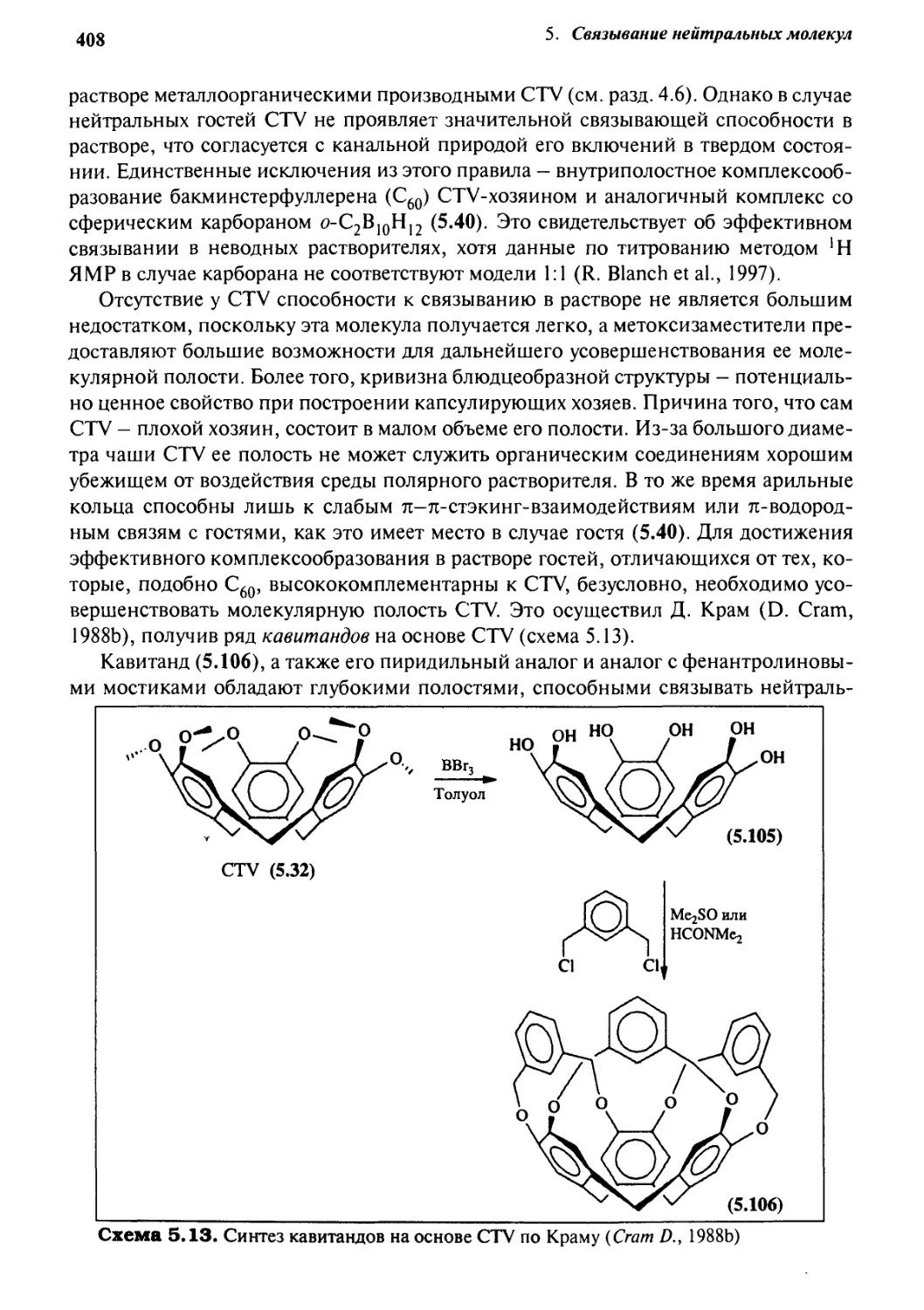
2.6
8.7
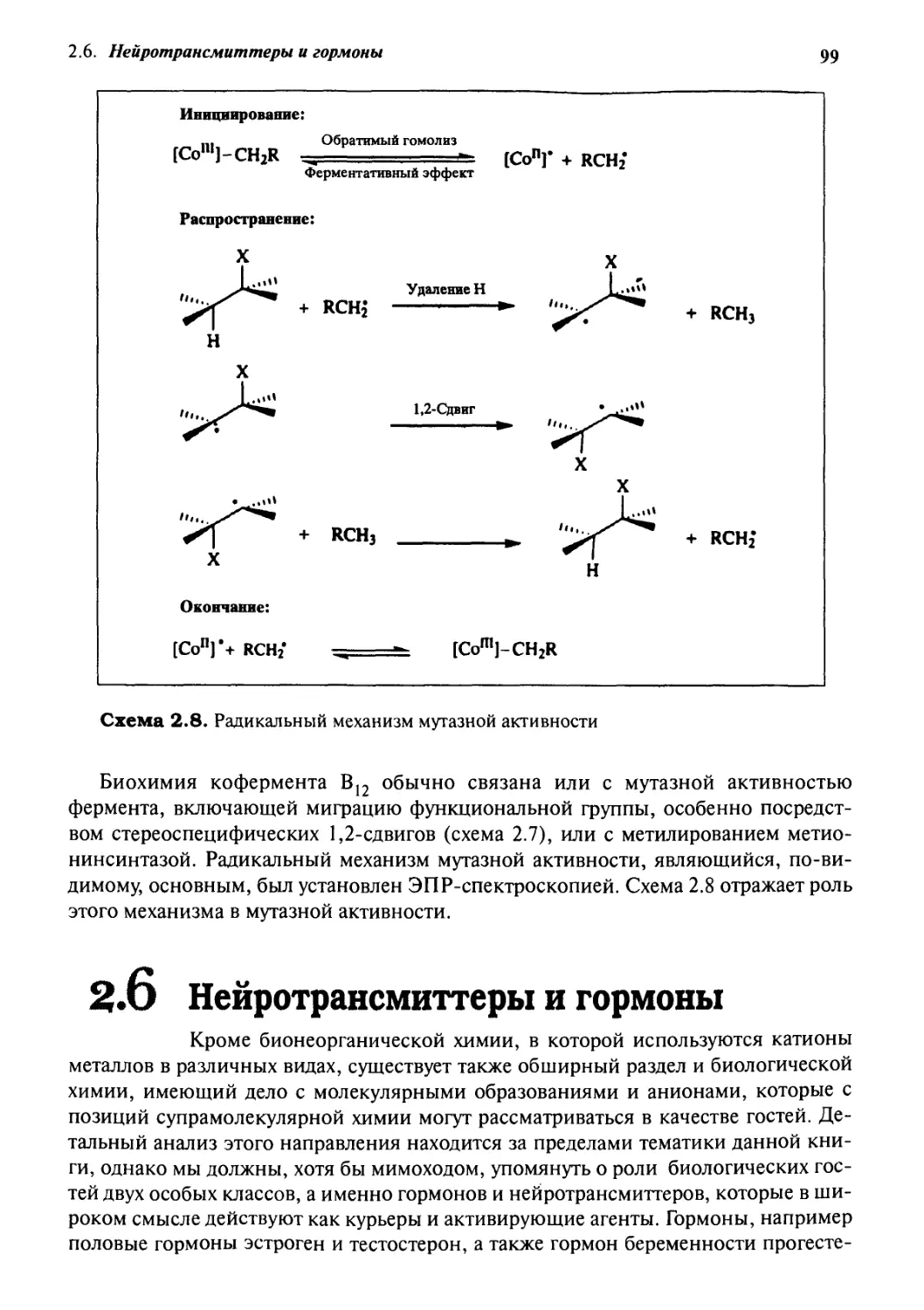
2.8
2-9
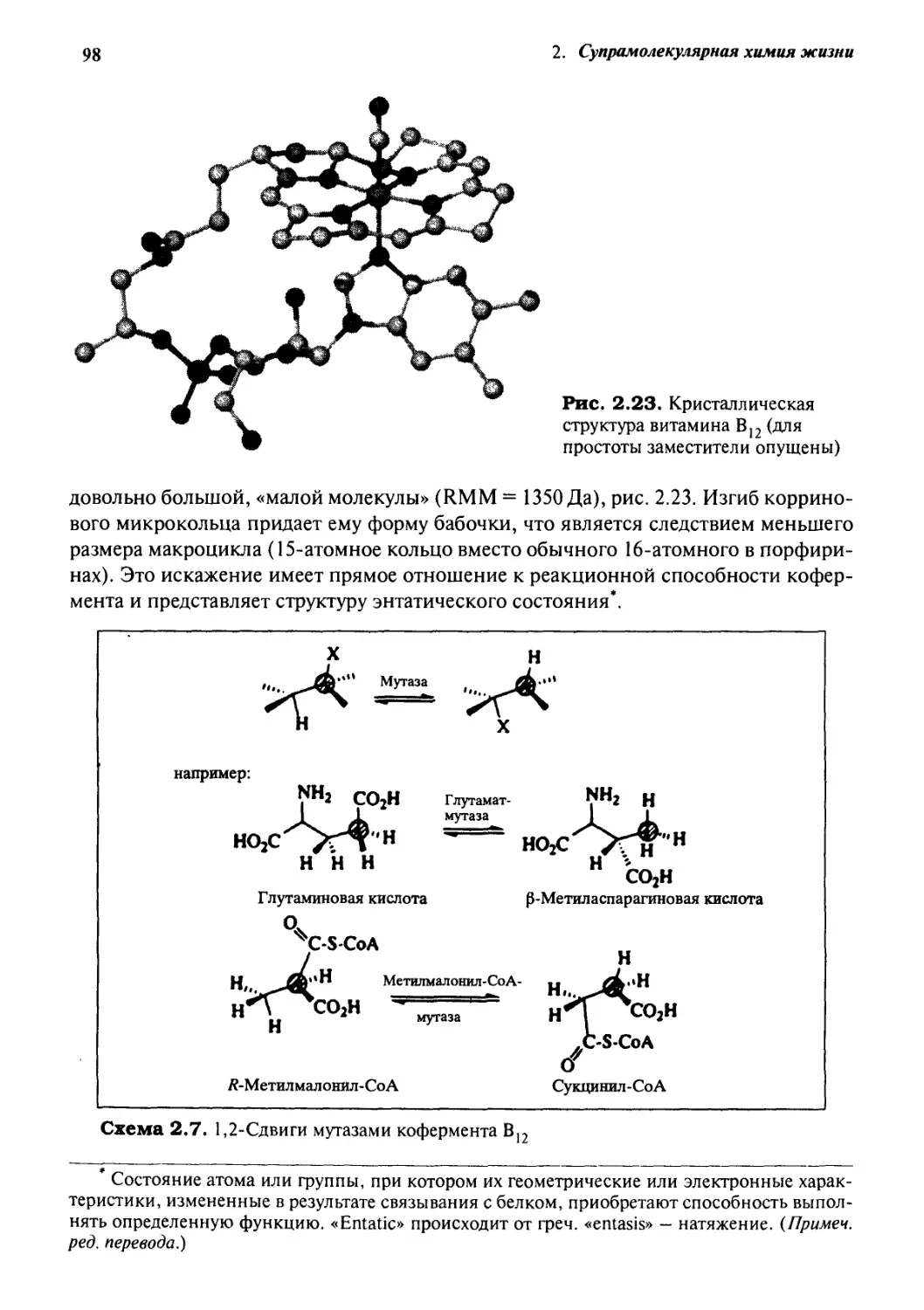
Кофермент В12
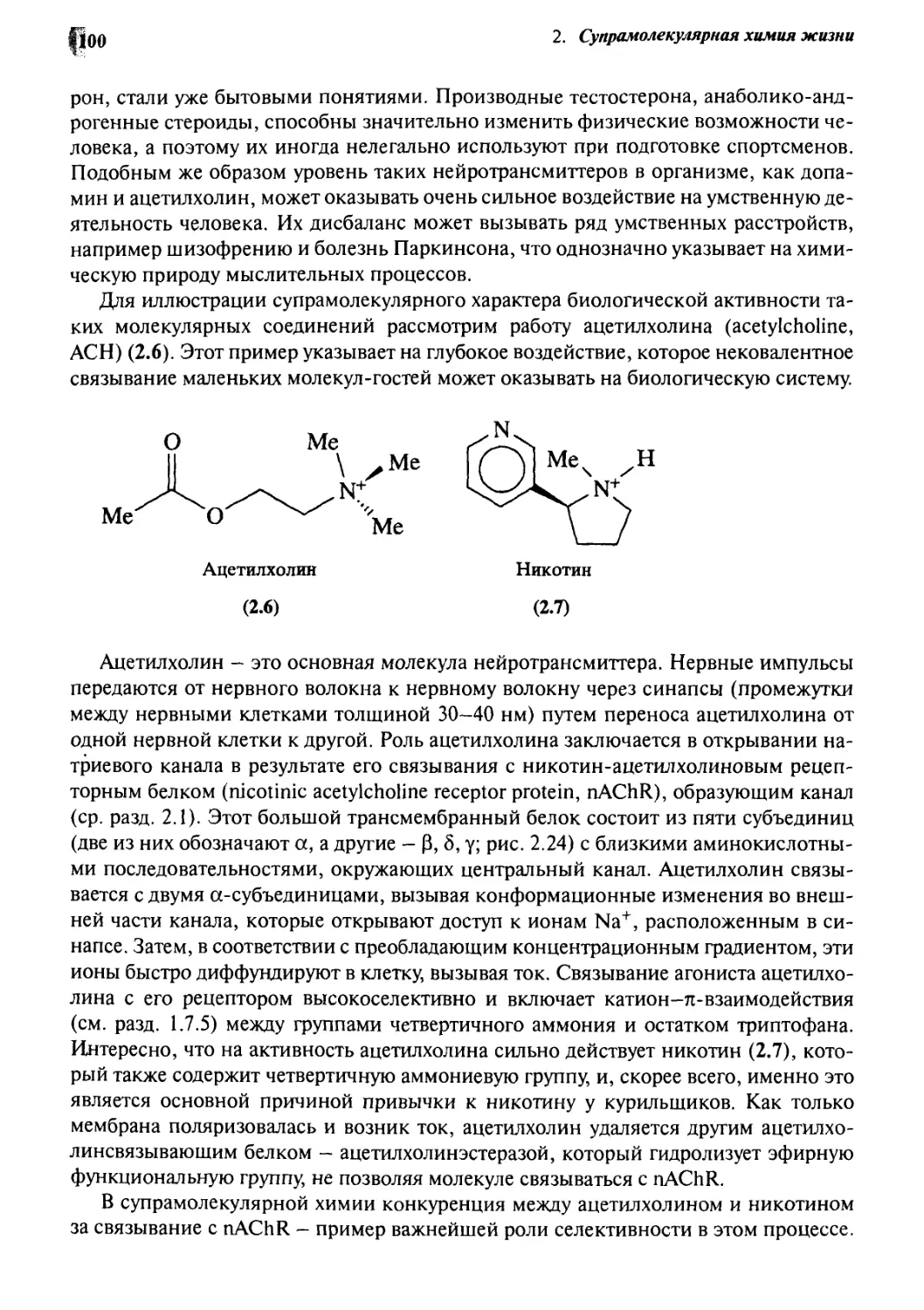
Нейротрансмиттеры и гормоны
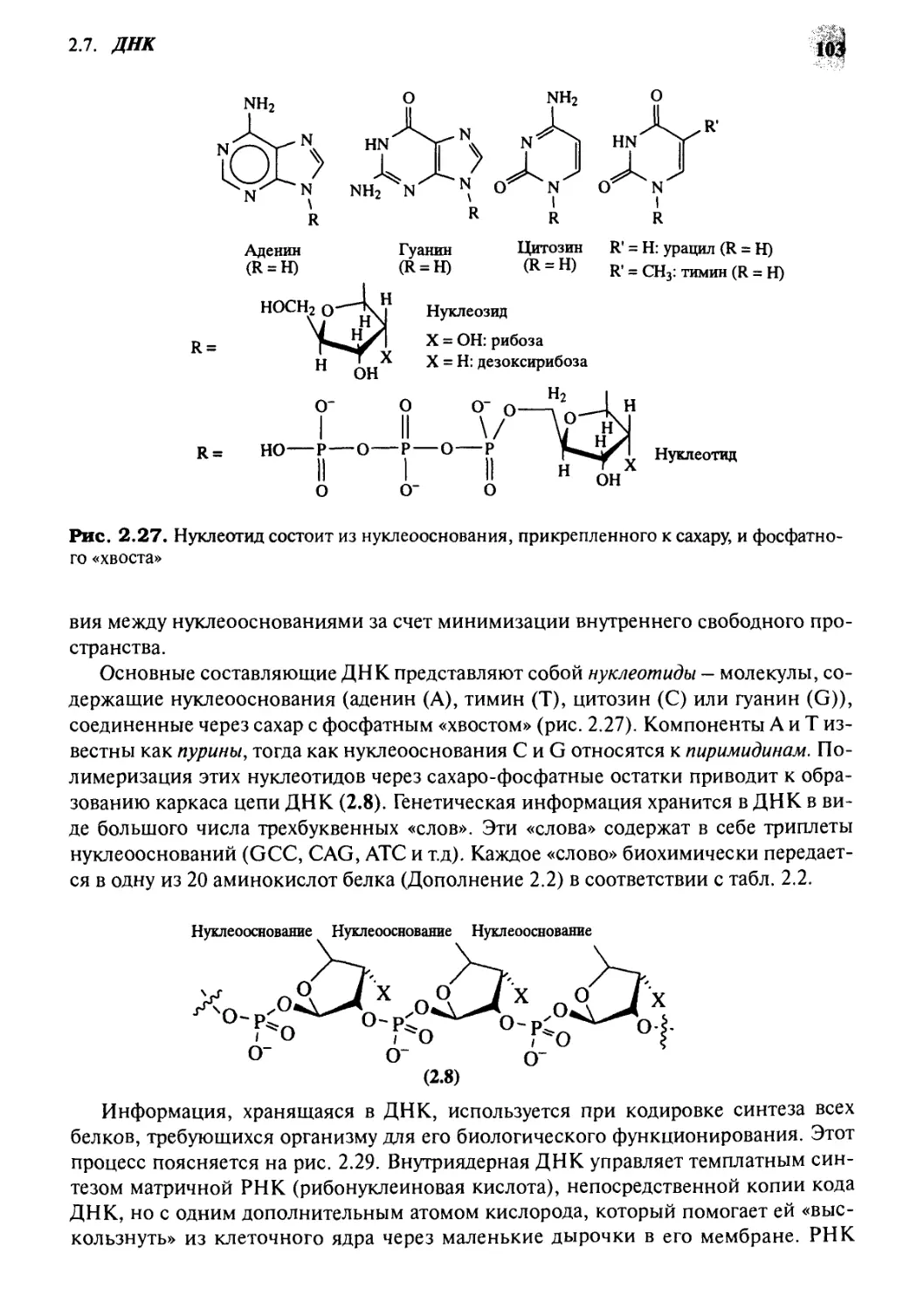
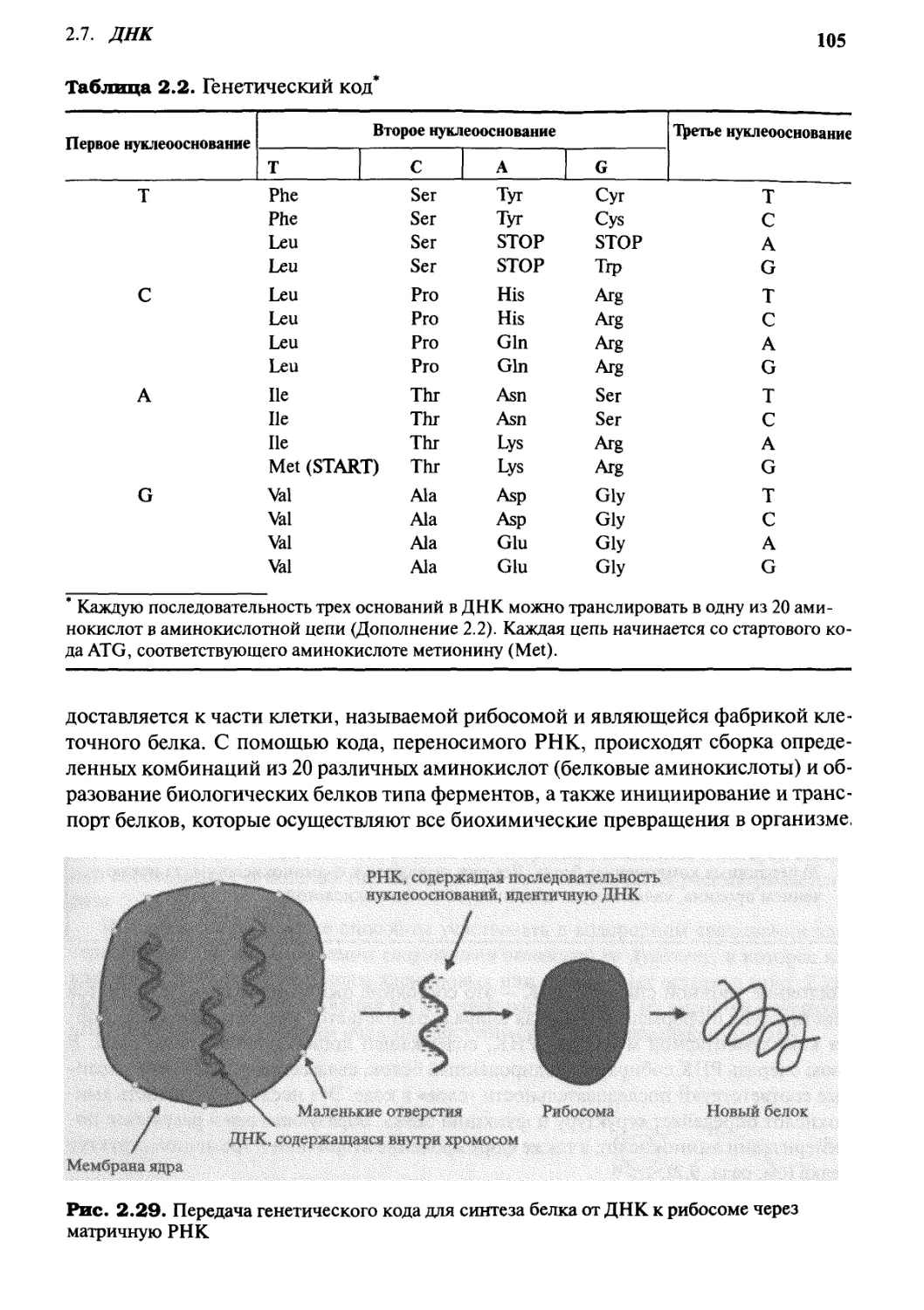
ДНК
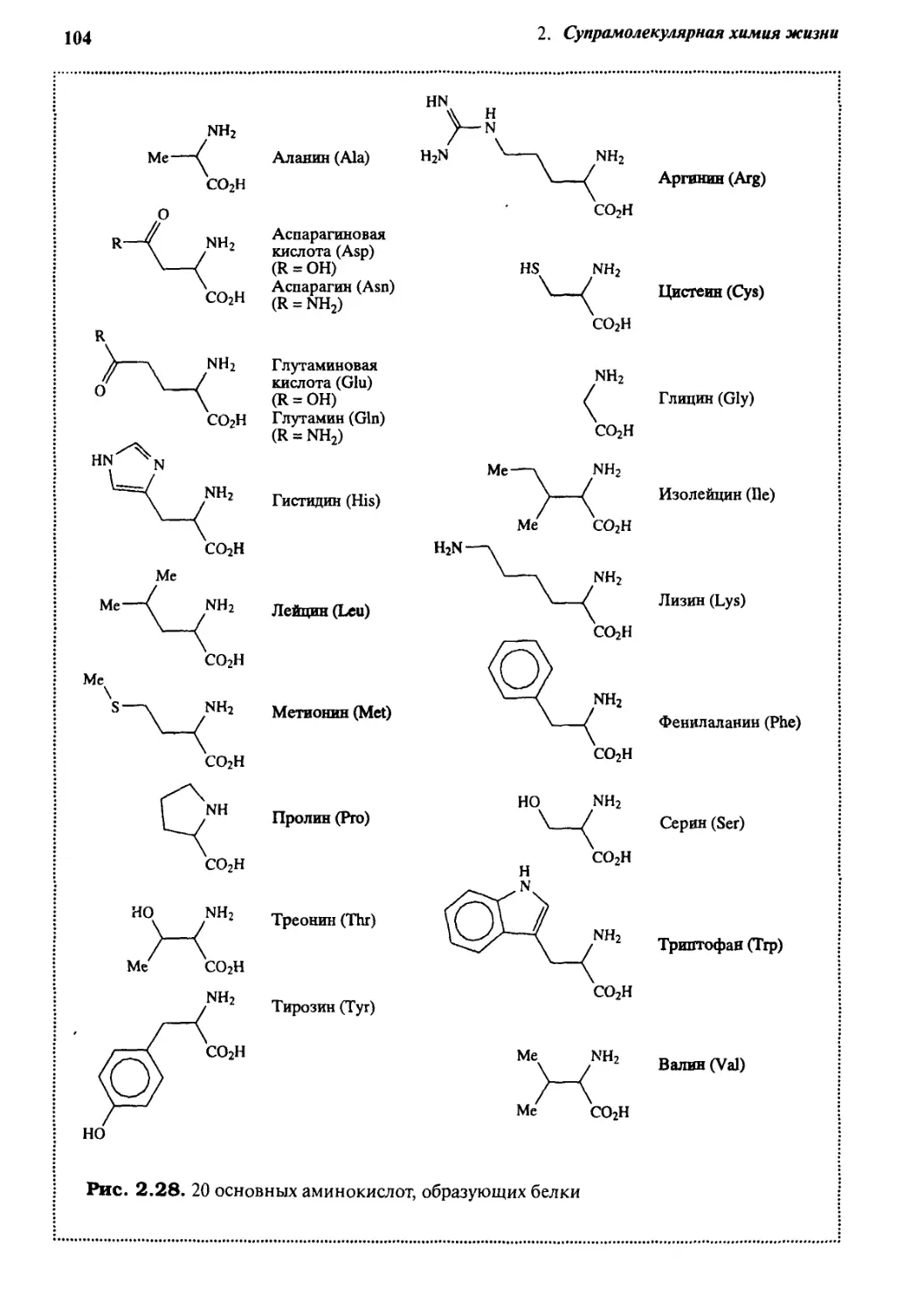
2.7.1 Структура и функции ДНК
2.7.2 Направленный мутагенез
2.7.3 Полимеразная цепная реакция
2.7.4 Связывание с ДНК
Биохимическая самосборка
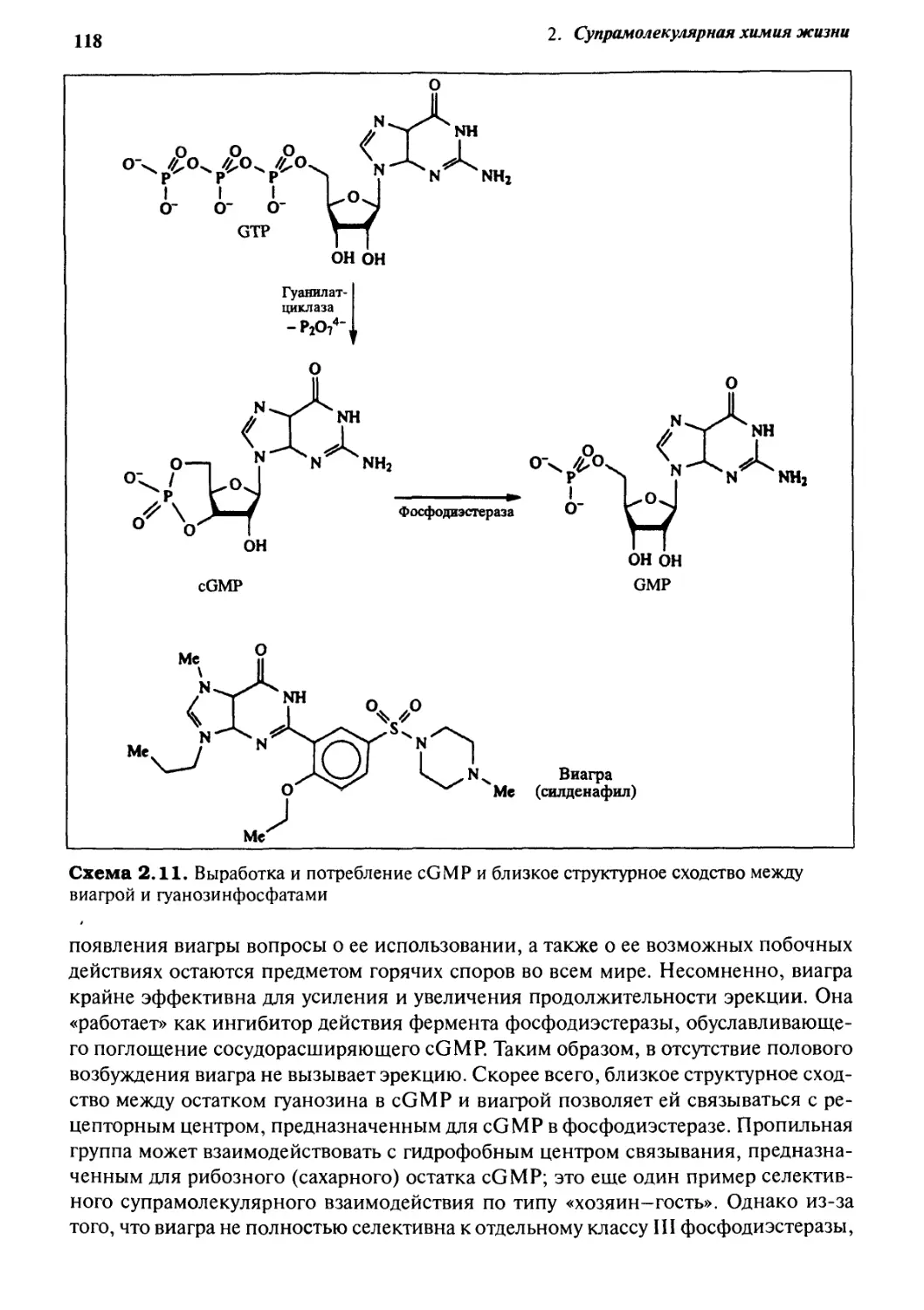
Виагра: по ту сторону газетной шумихи
Учебные задания
Литеоаттоа
96
99
101
101
108
109
ПО
115
117
119
120
Хозяева, связывающие катионы ш
Краун-эфиры 121
Оглавление 7
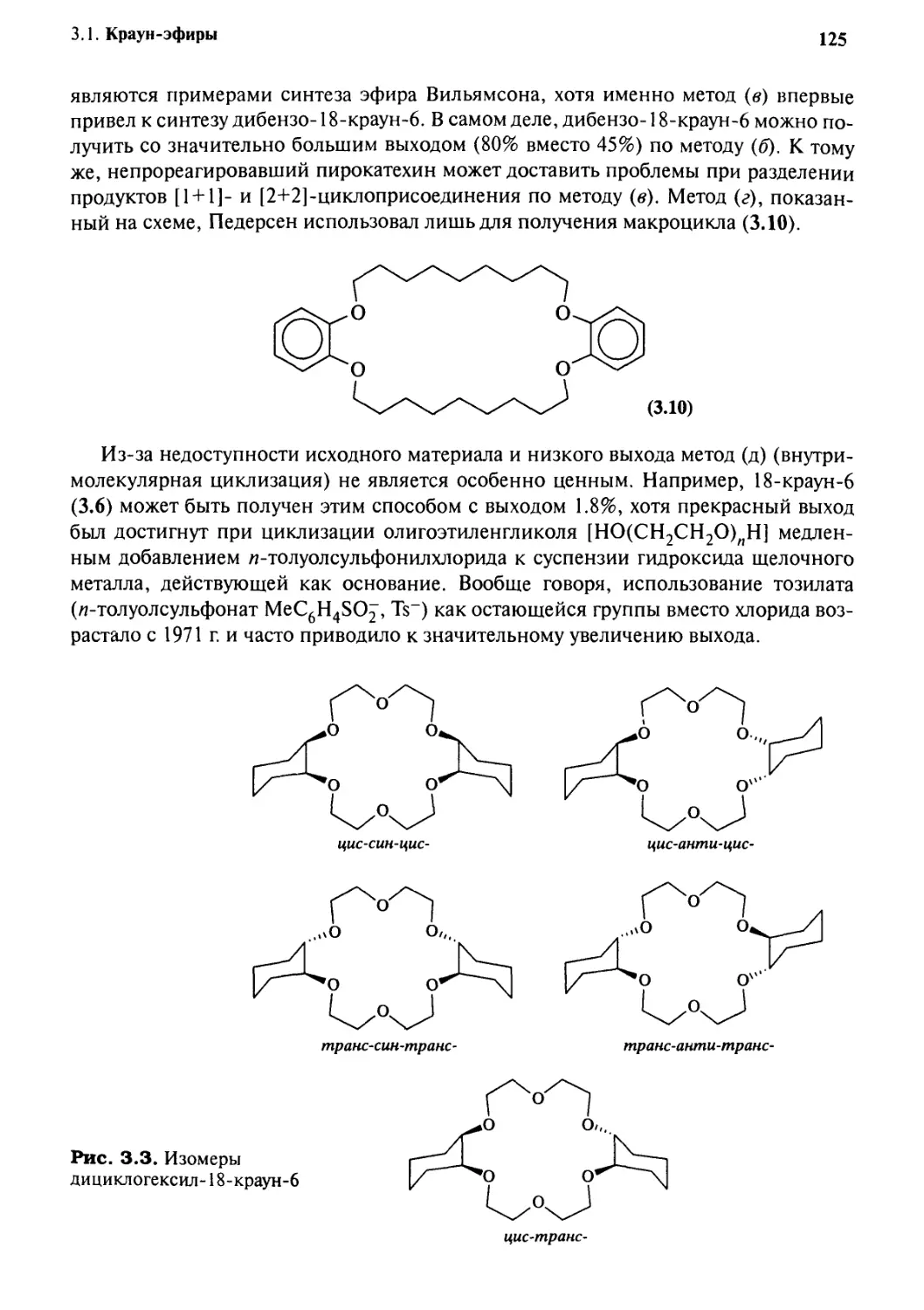
3.1.1 Открытие и область применения 121
3.1.2 Синтез 124
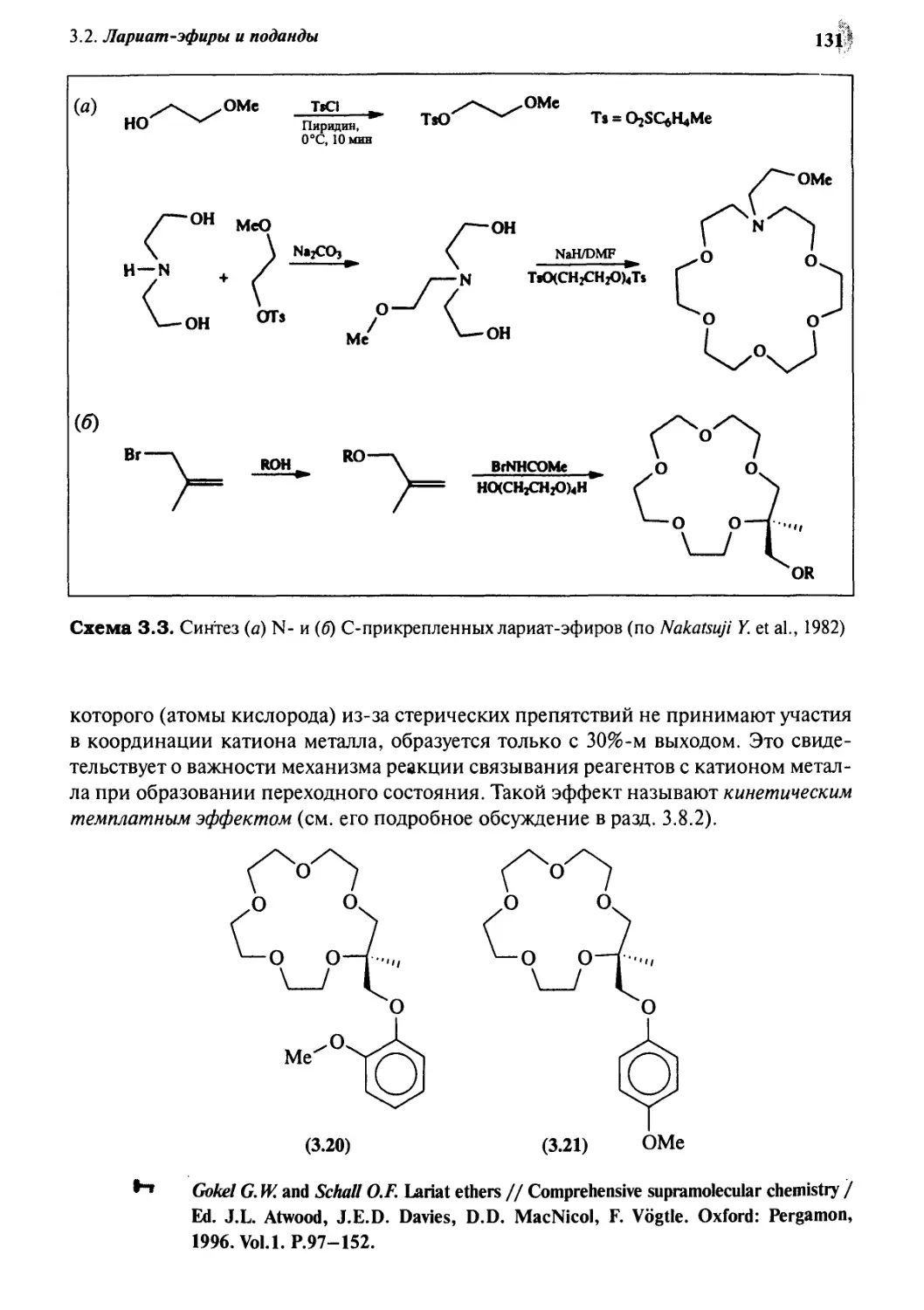
3«2 Лариат-эфиры и поданды 126
3.2.1 Поданды 126
3.2.2 Лариат-эфиры 129
3.2.3 Бибрахиальные лариат-эфиры 132
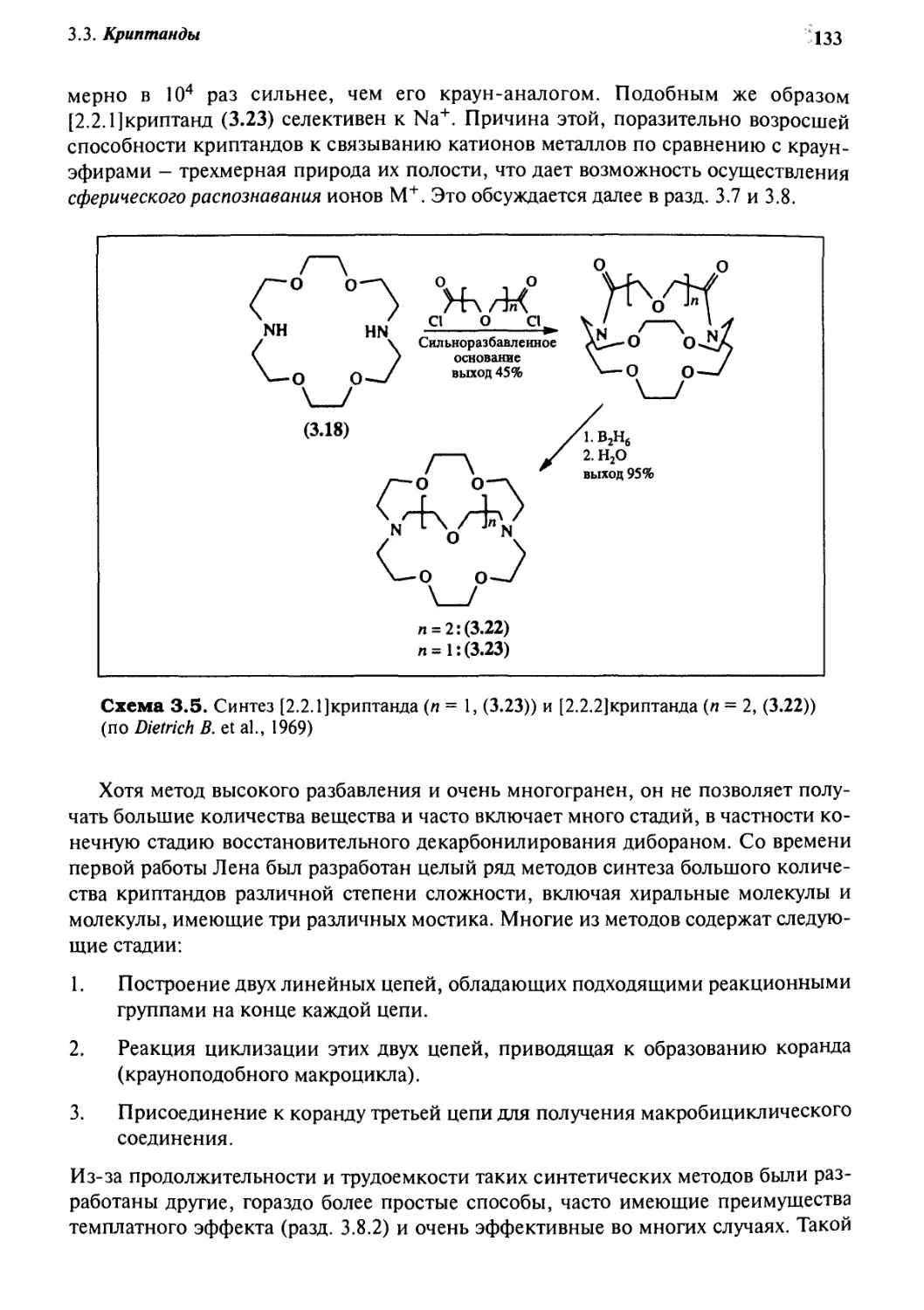
g.g Криптанды 132
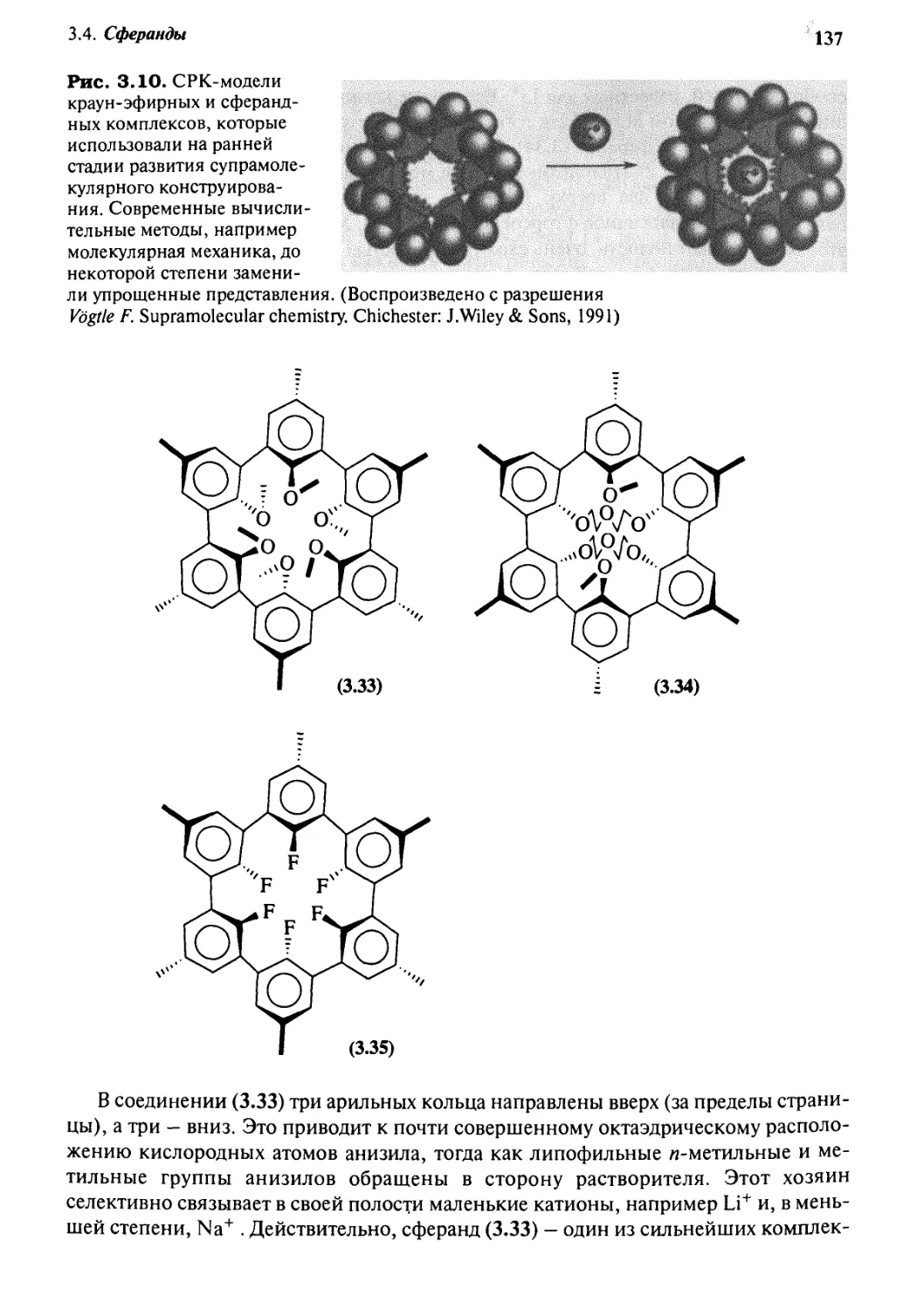
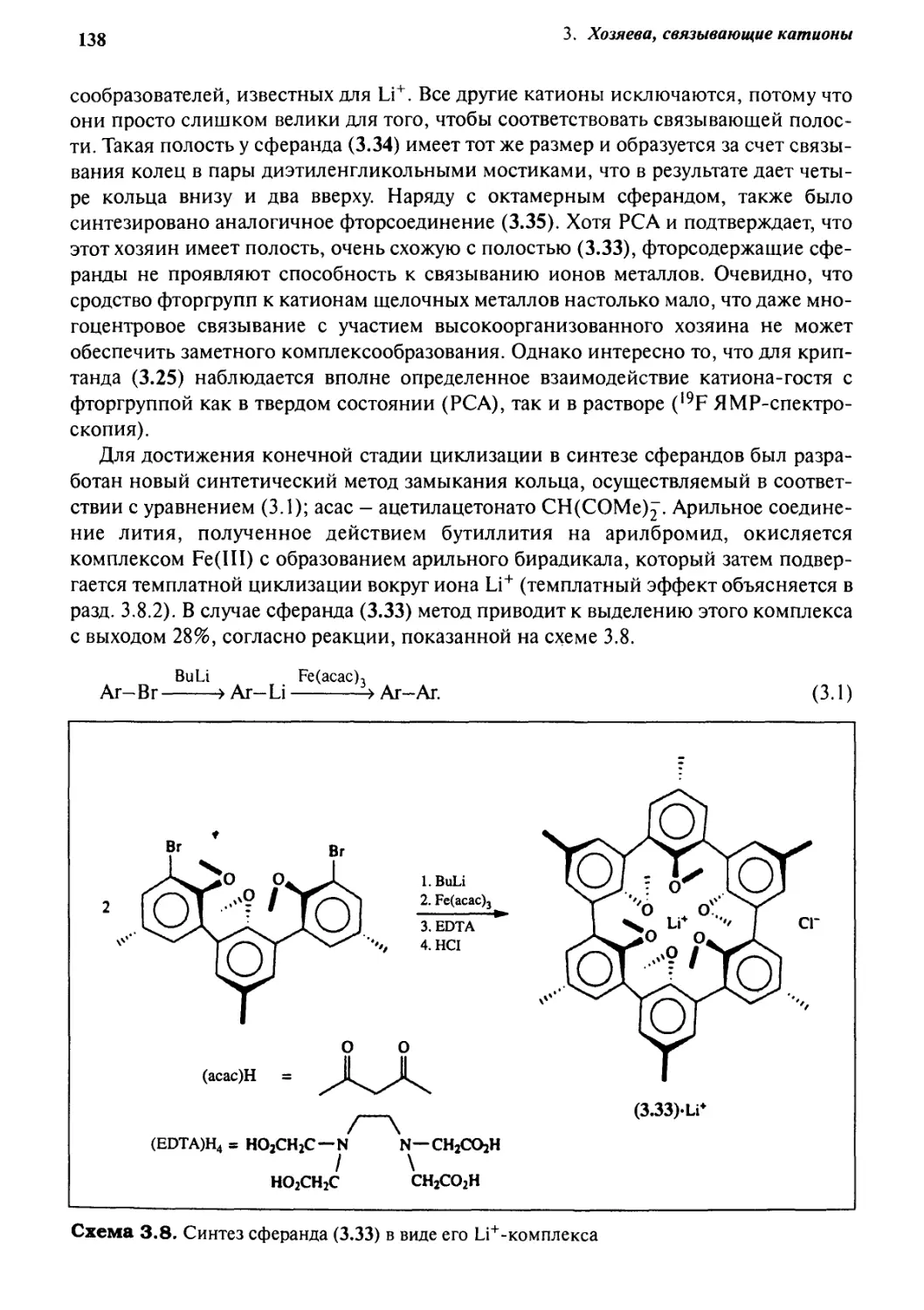
3«4 Сферанды 136
g.g Номенклатура но
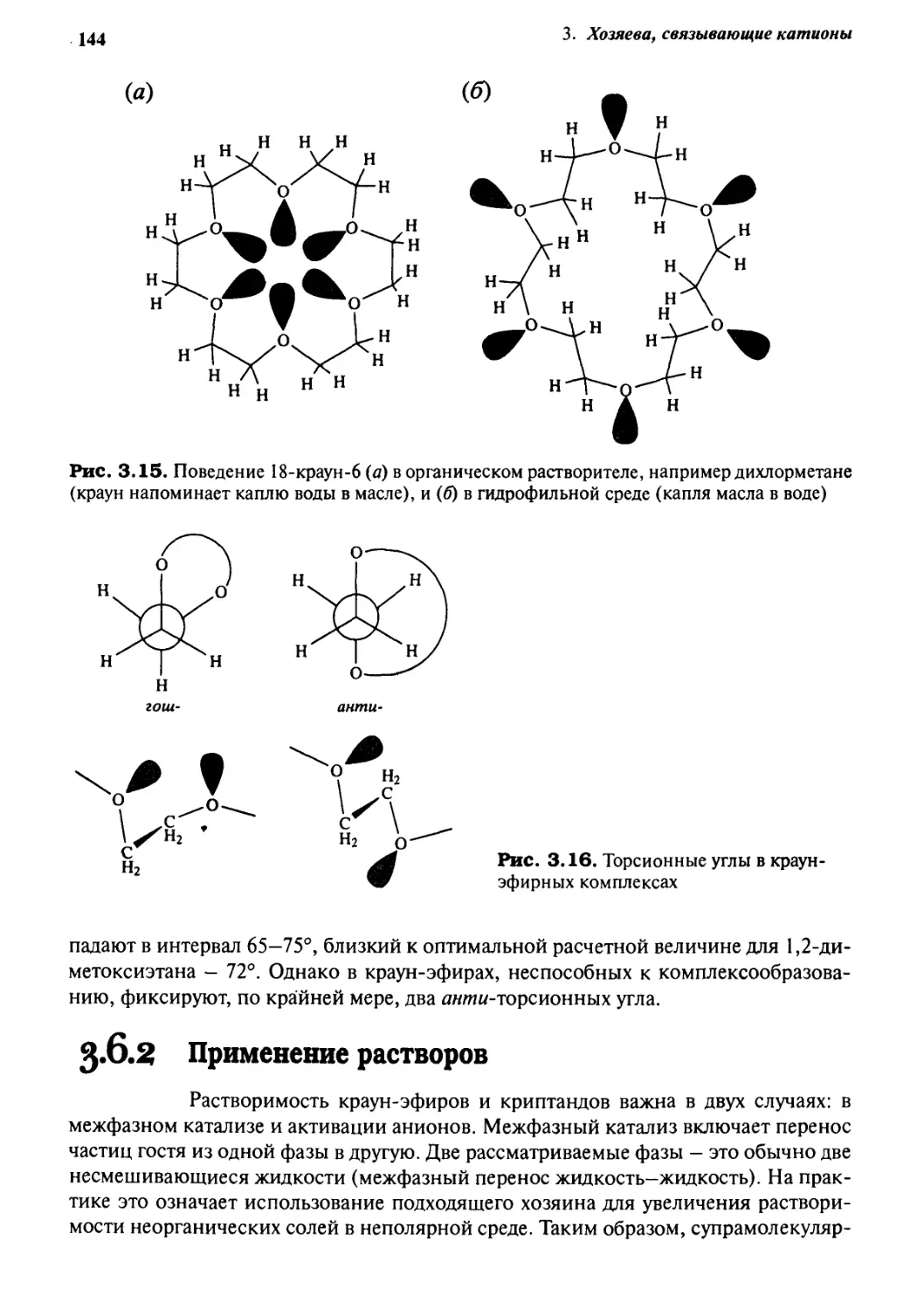
5J.6 Поведение растворов 143
3.6.1 Растворимость 143
3.6.2 Применение растворов 144
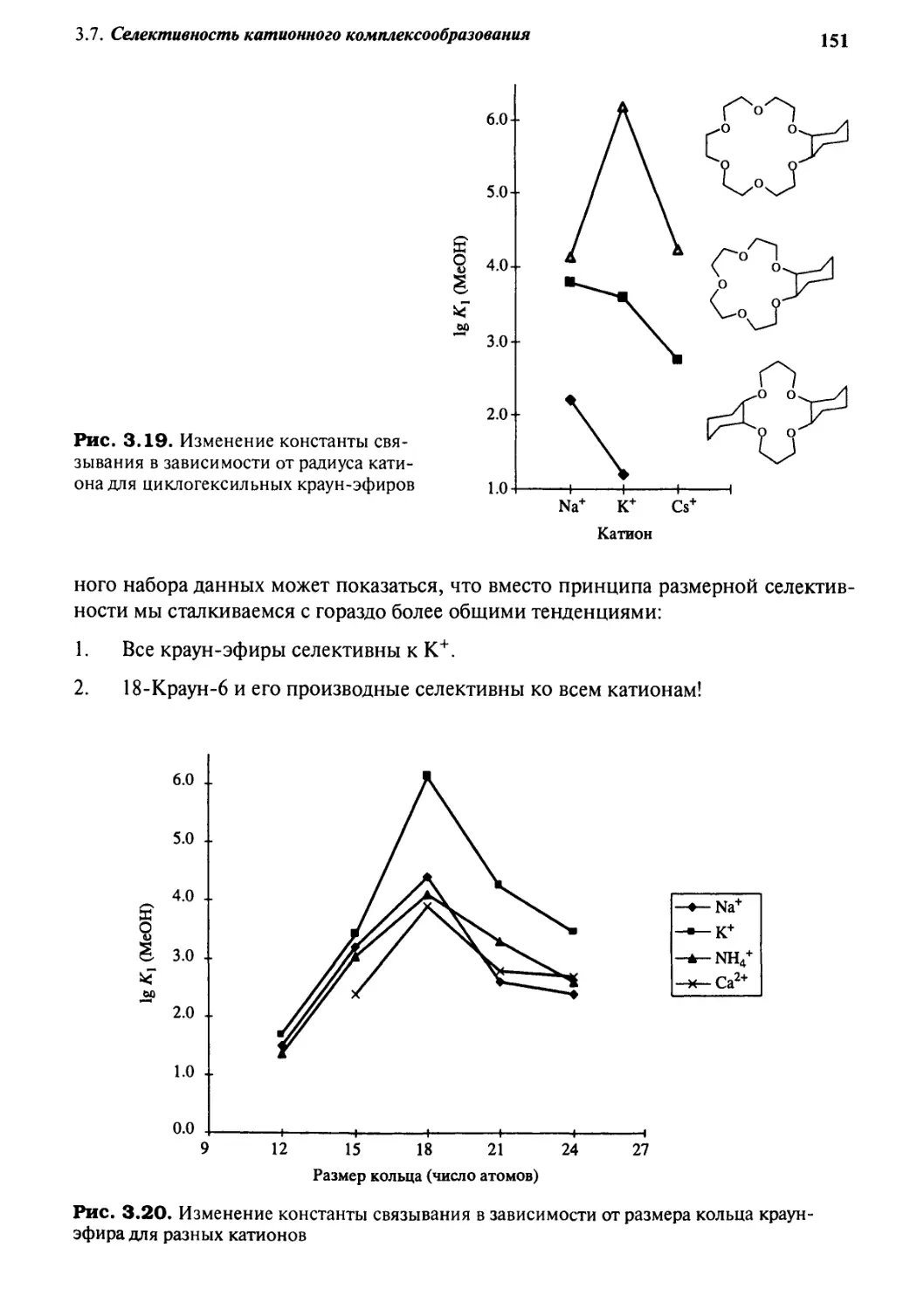
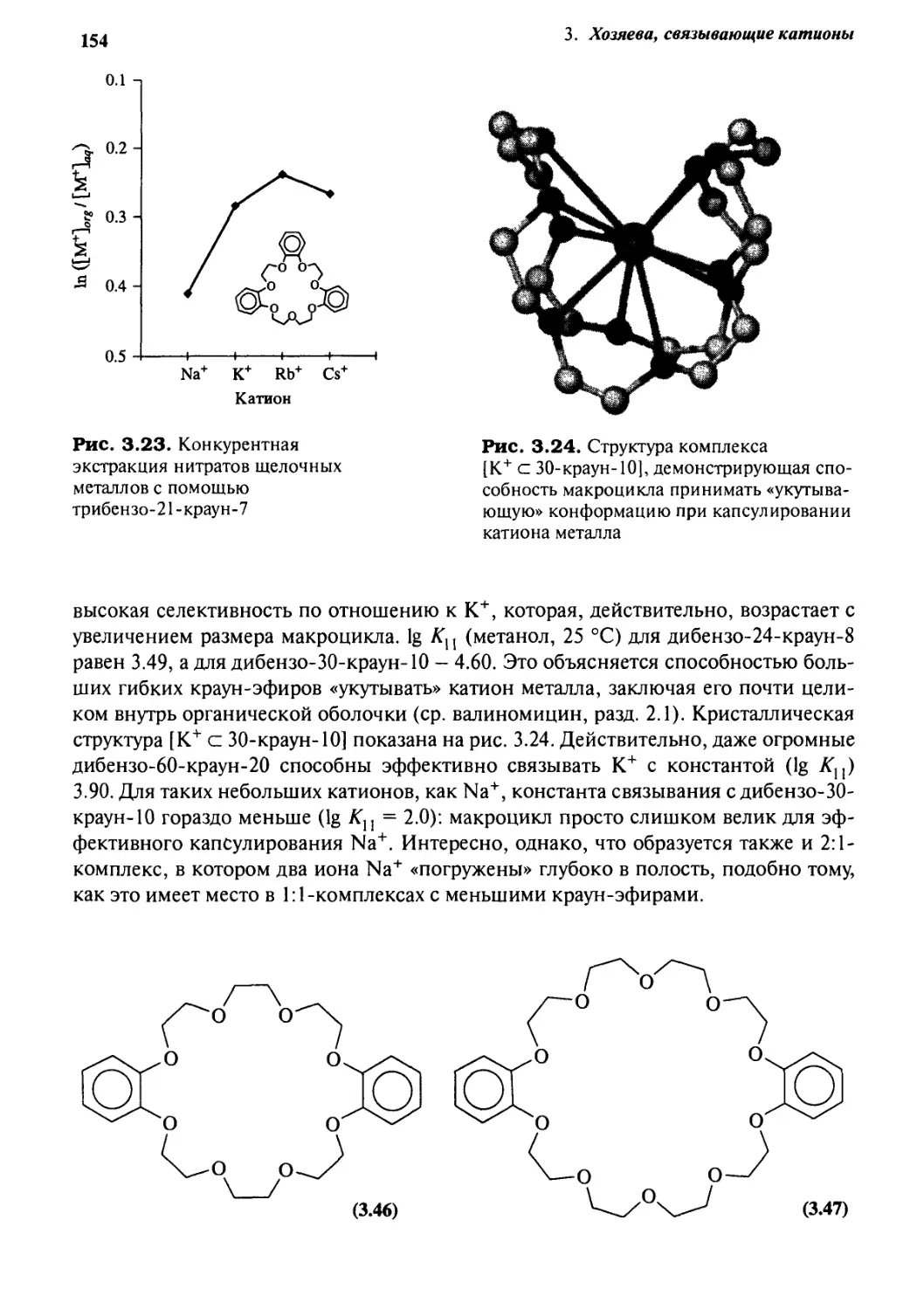
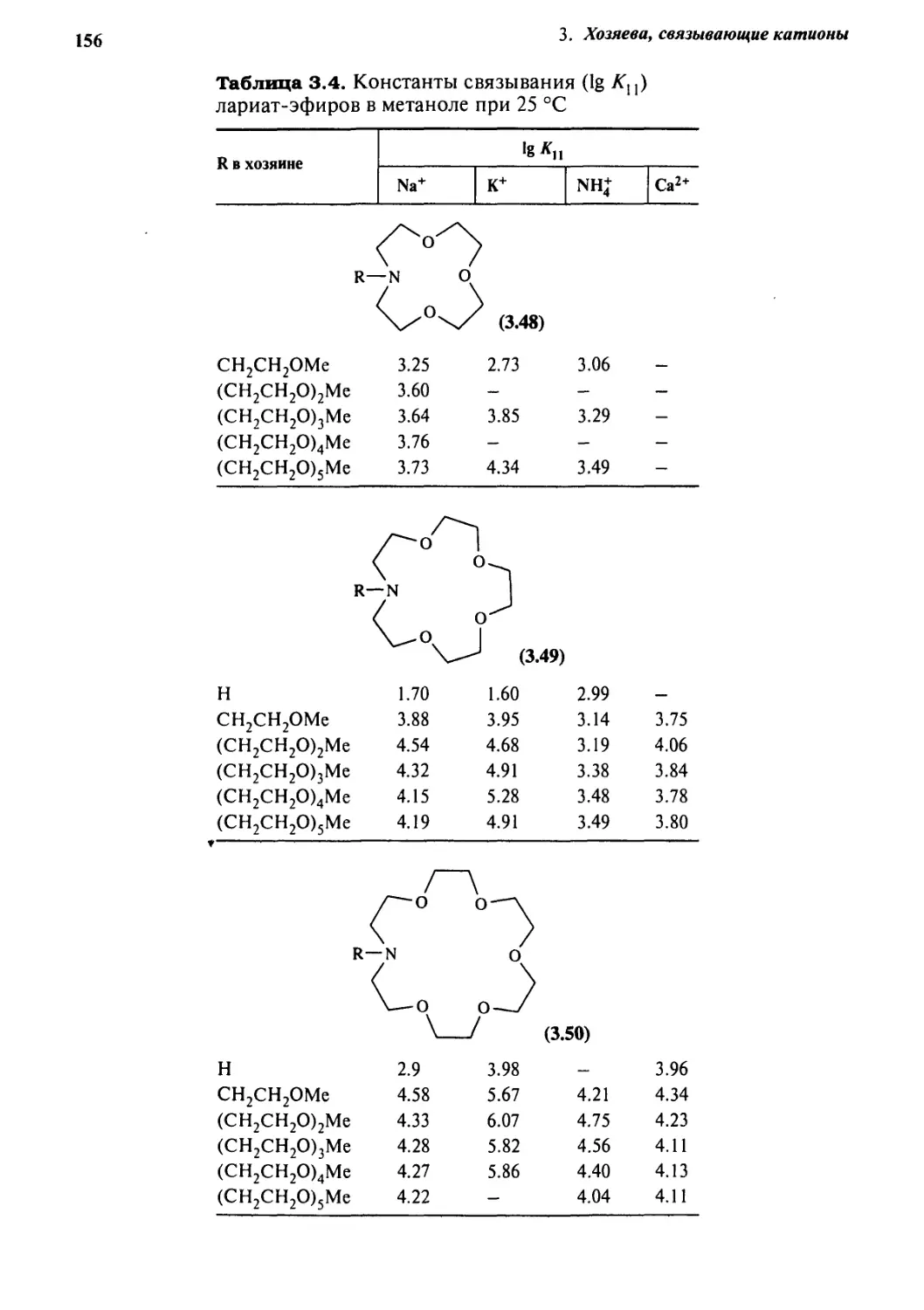
%.J Селективность катионного комплексообразования 149
3.7.1 Общие положения 149
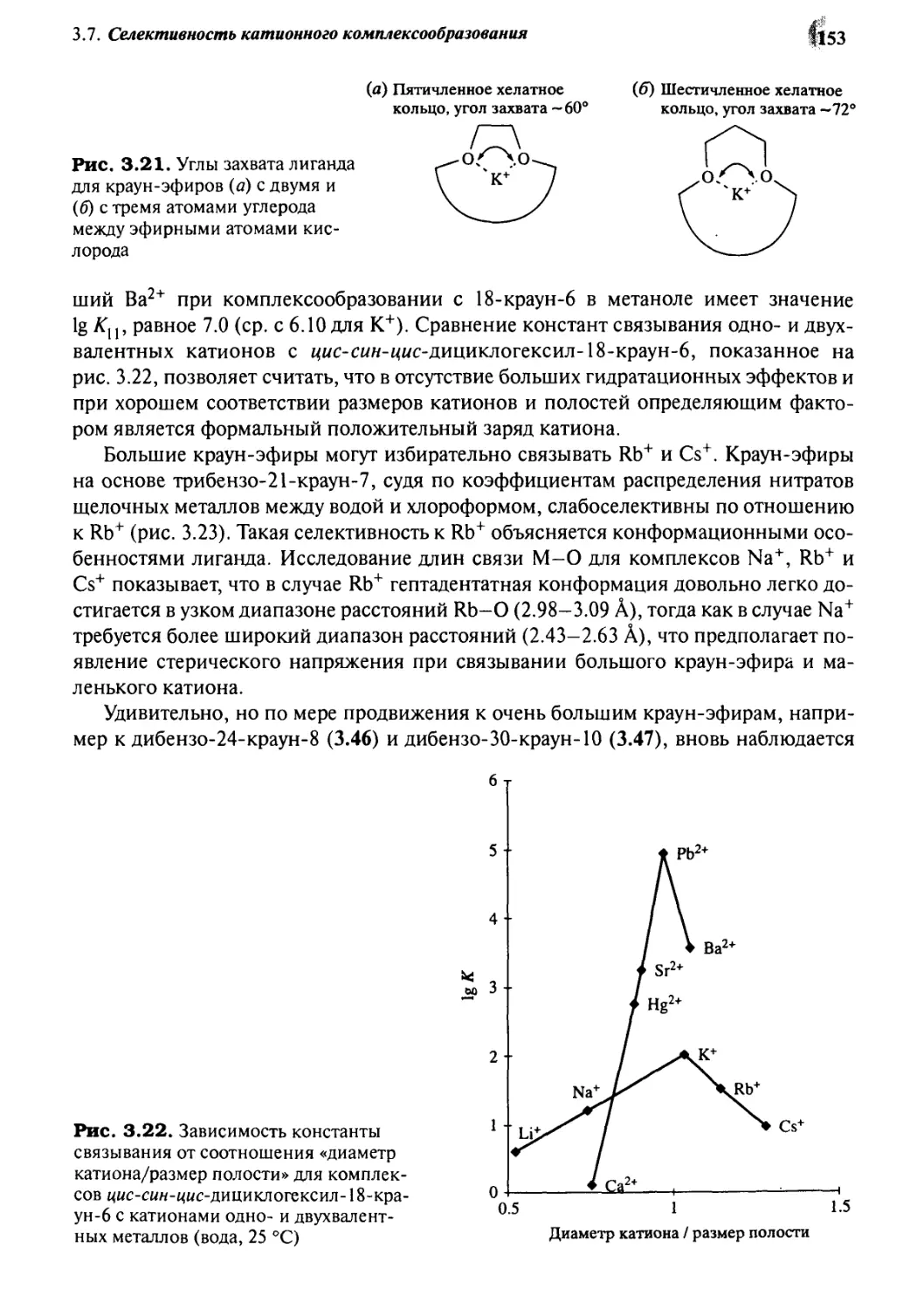
3.7.2 Краун-эфиры 150
3.7.3 Лариат-эфиры 155
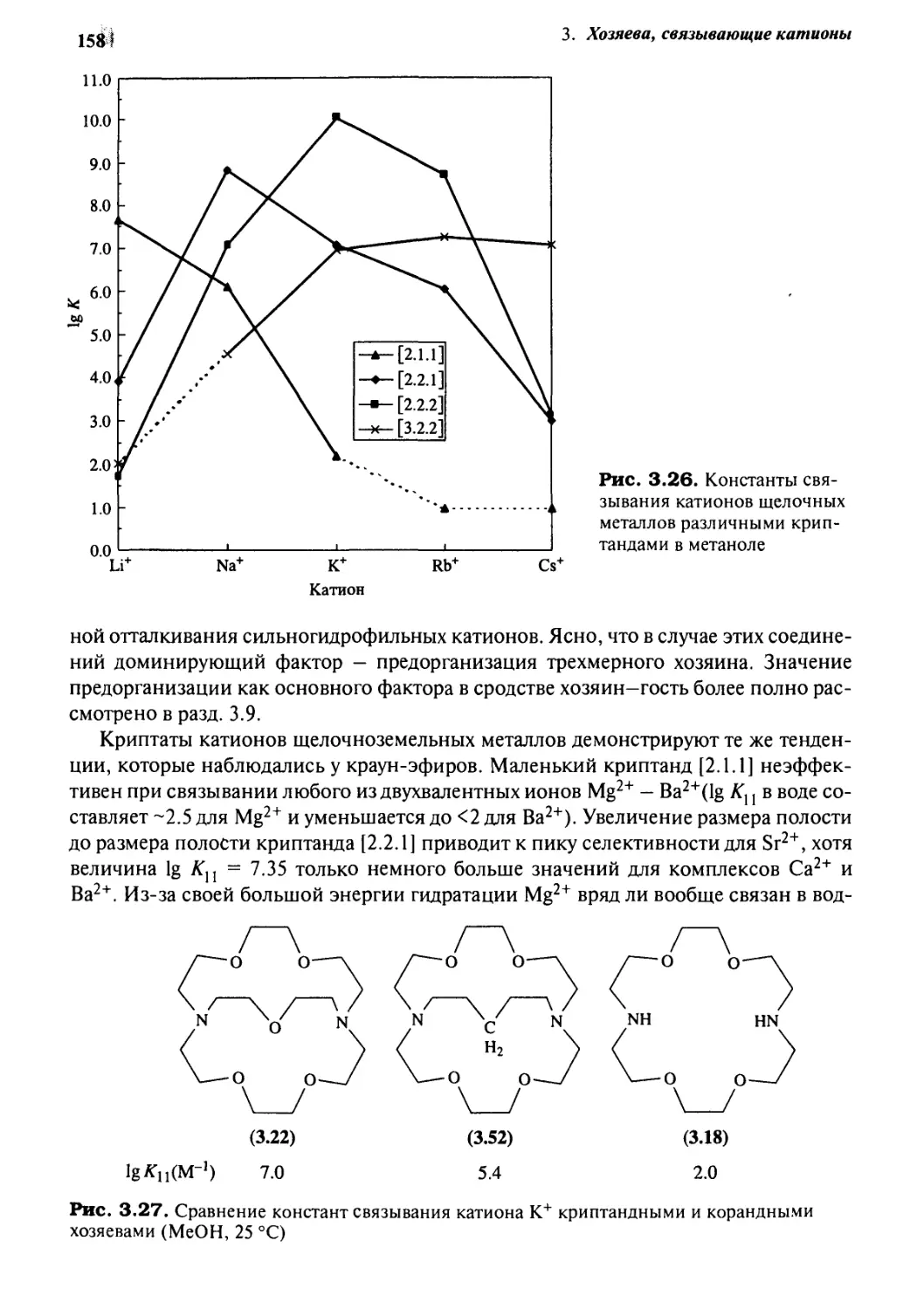
3.7.4 Криптанды 157
3*8 Макроциклический, макробициклический
и темплатный эффекты 159
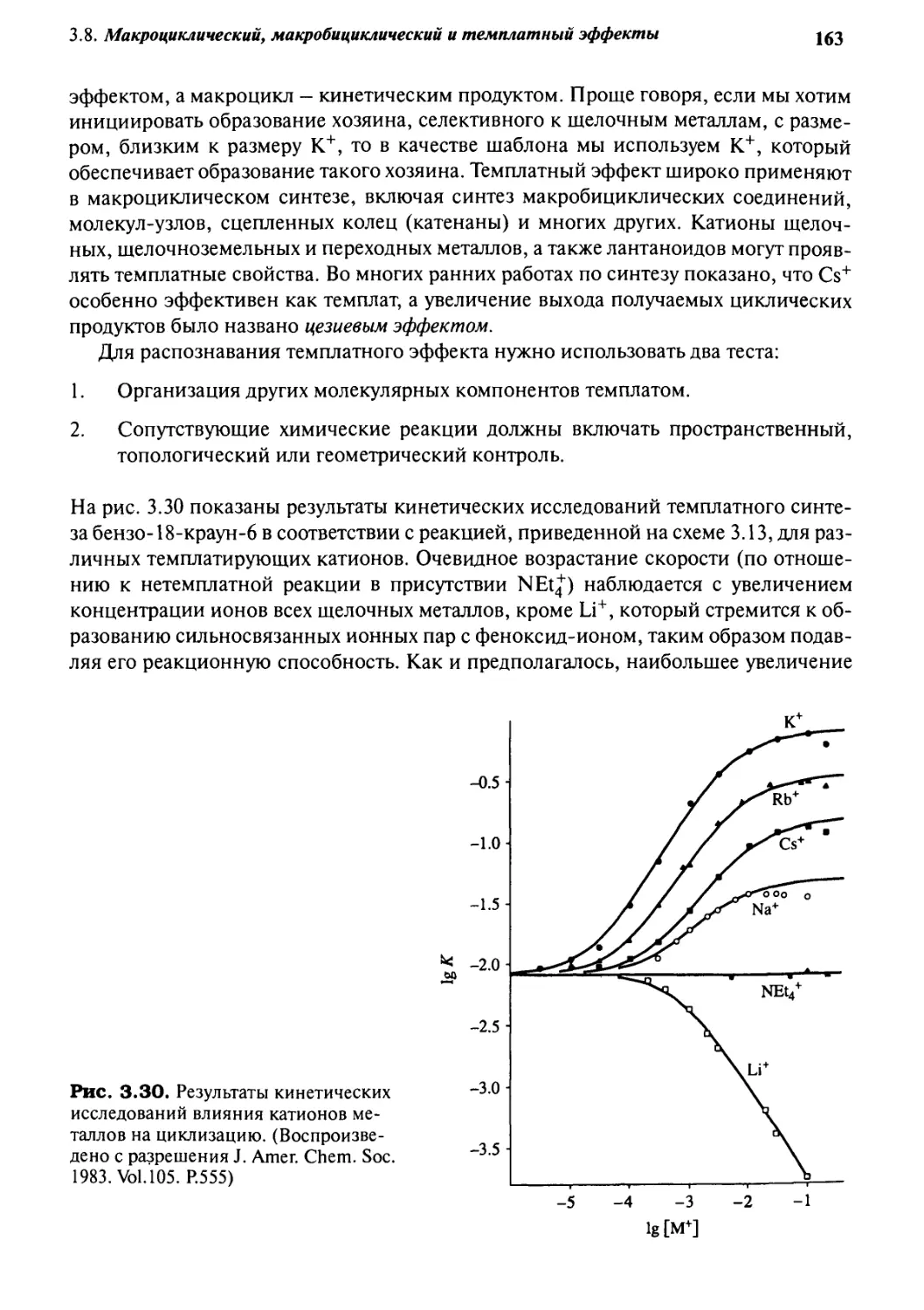
3.8.1 Макроциклический эффект 159
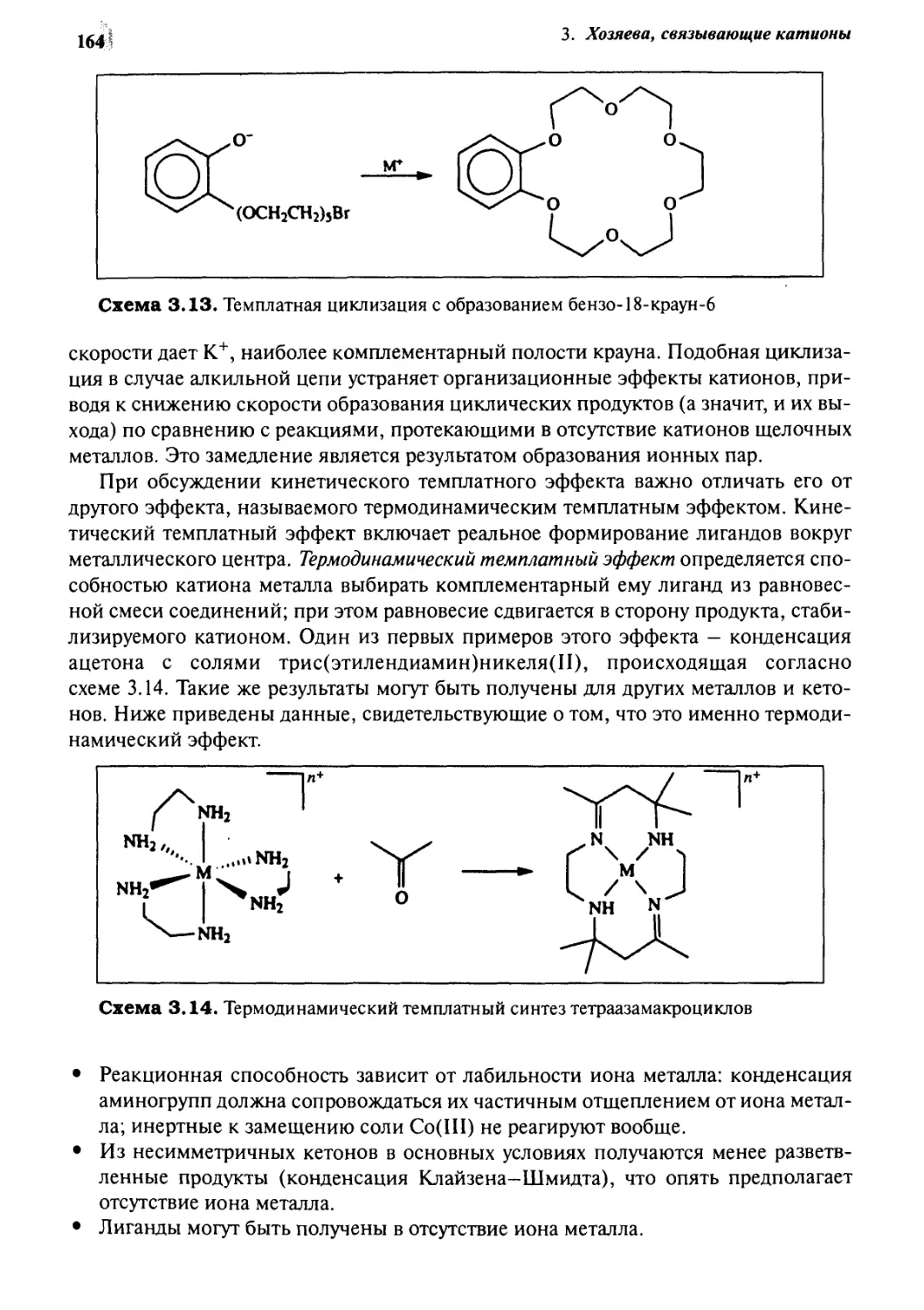
3.8.2 Темплатный эффект 161
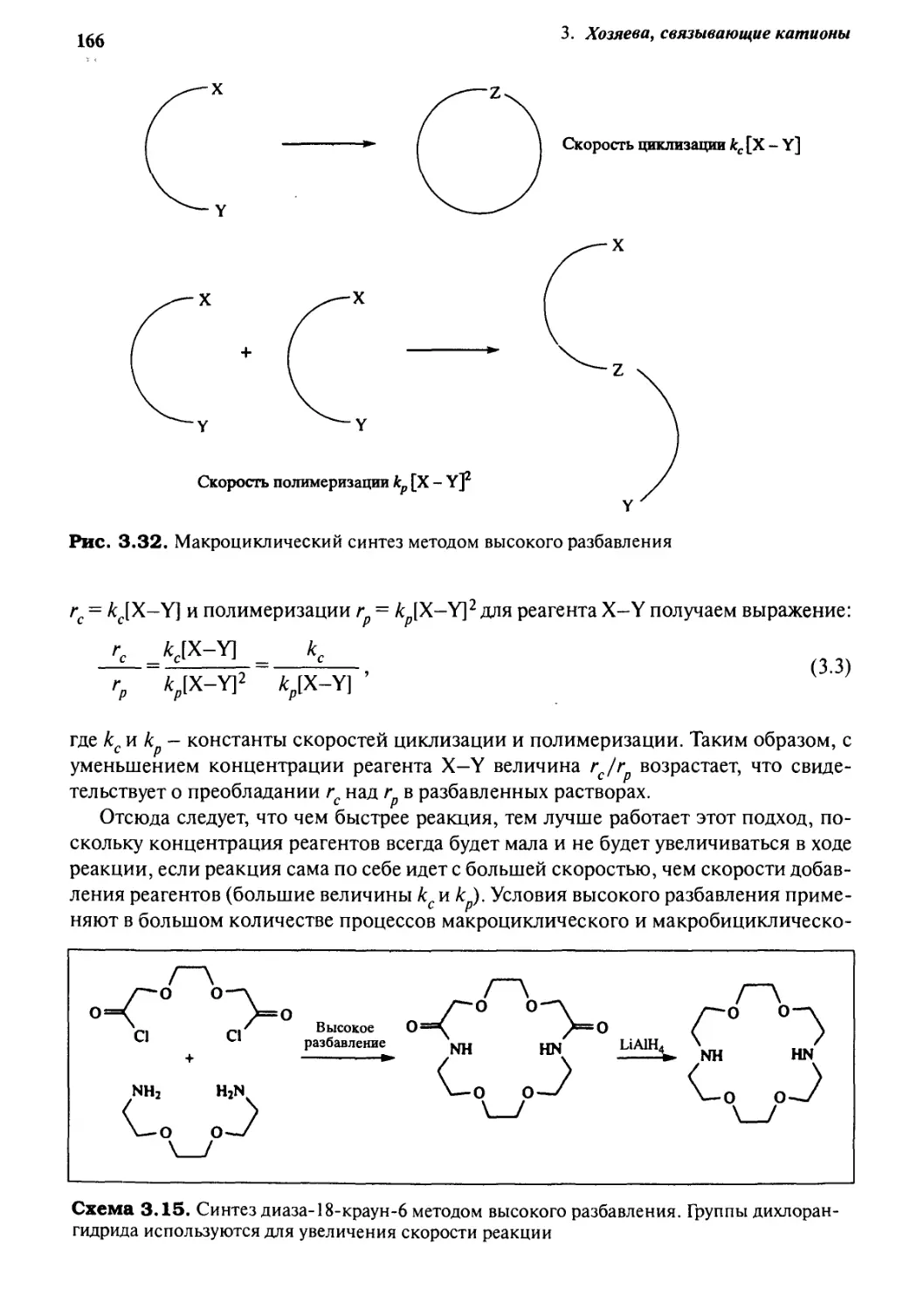
3.8.3 Синтез методом высокого разбавления 165
3.8.4 [2+2]-Циклосоконденсация 167
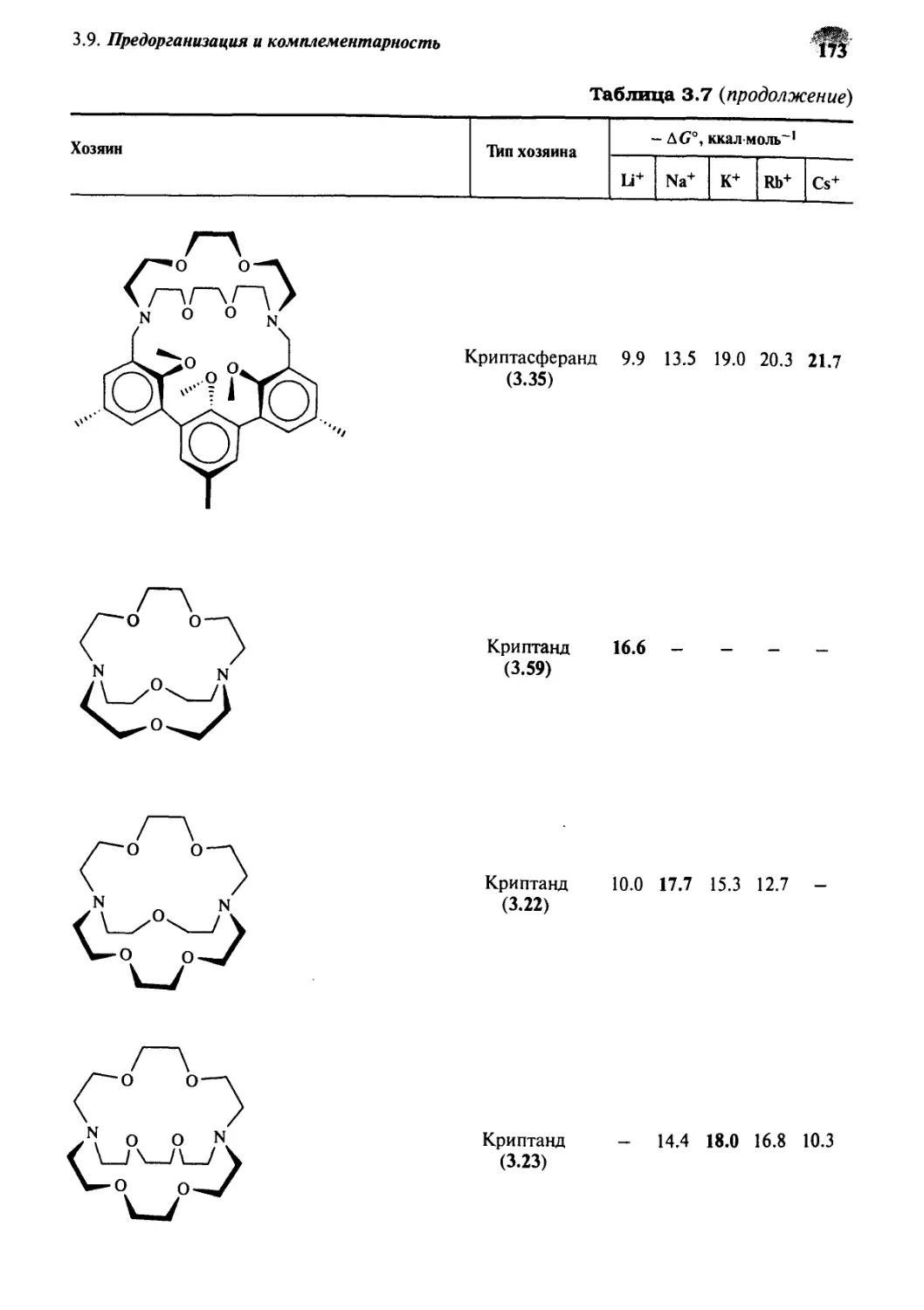
g.Q Предорганизация и комплементарность 169
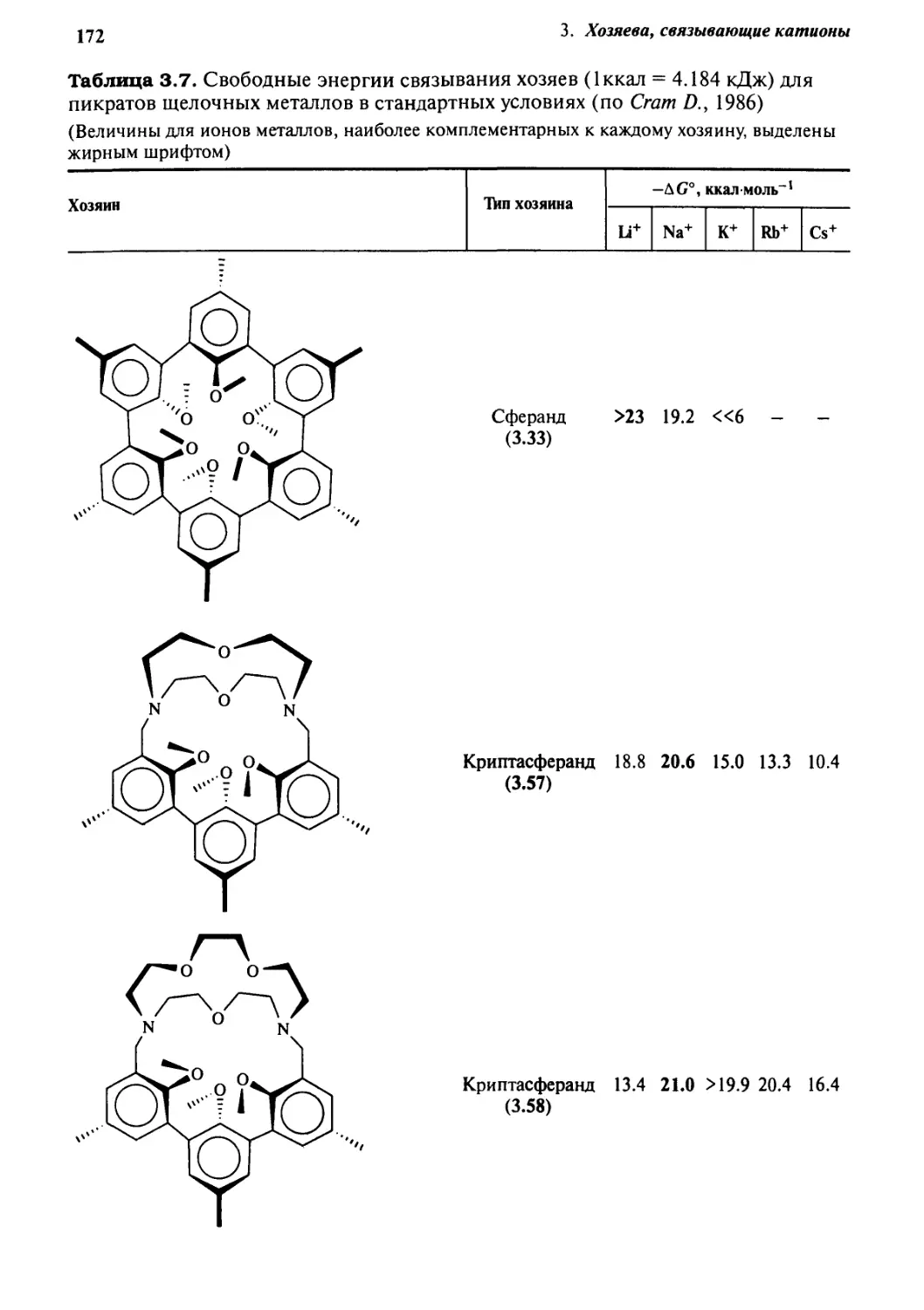
3.9.1 Термодинамические эффекты 169
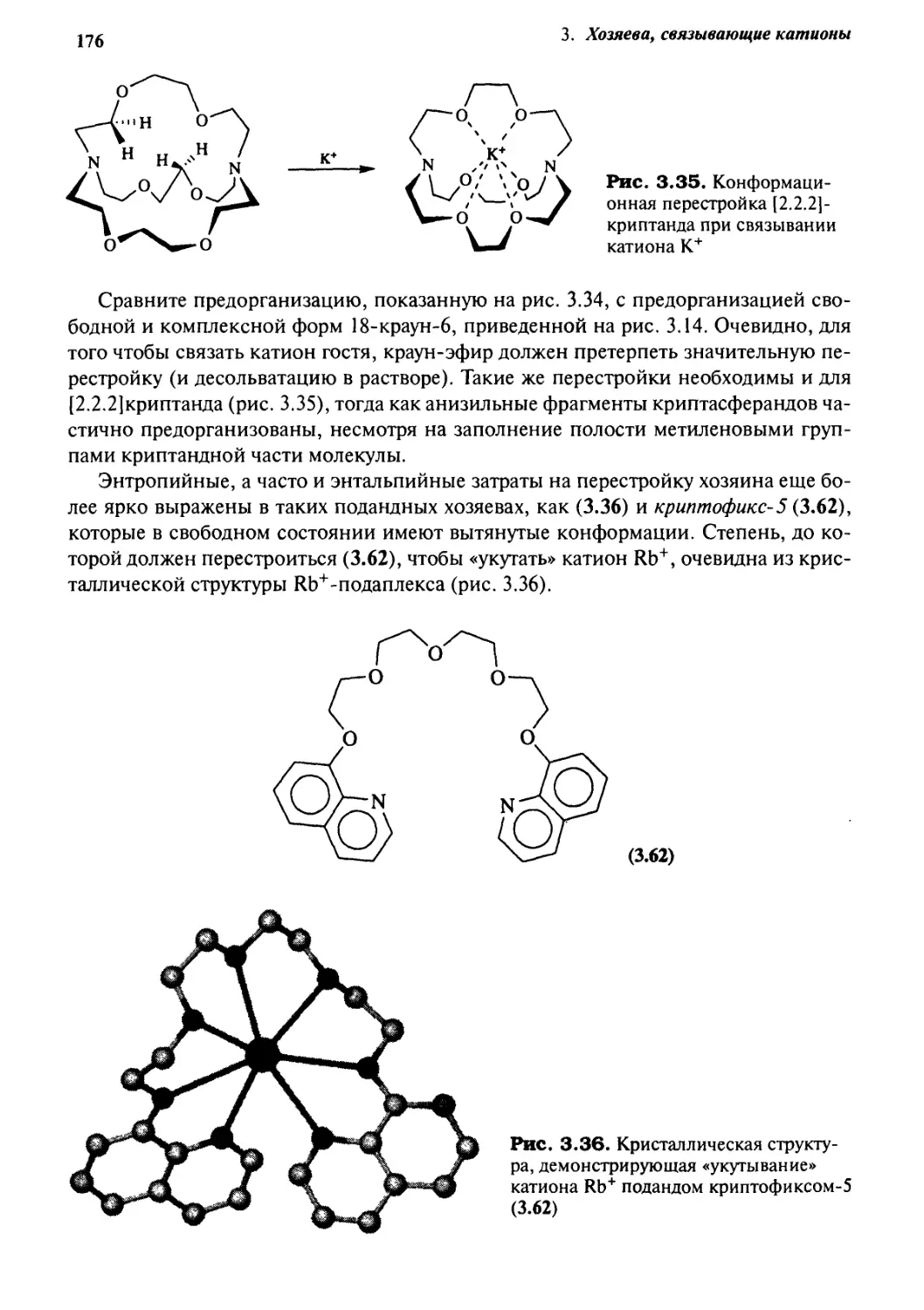
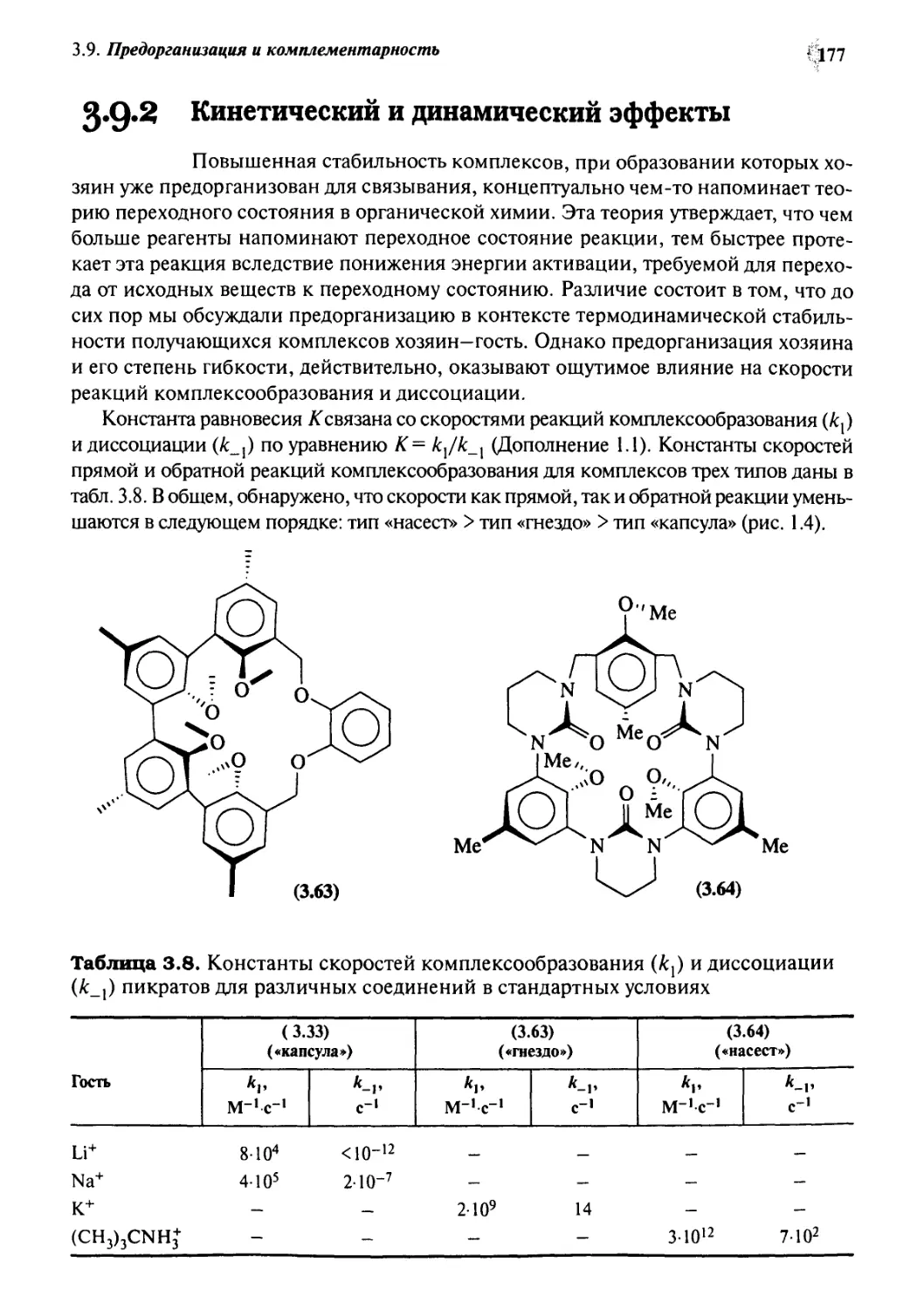
3.9.2 Кинетический и динамический эффекты 177
3.10 Мягкие лиганды для мягких ионов металлов 179
3.10.1 Гетерокраун-эфиры 181
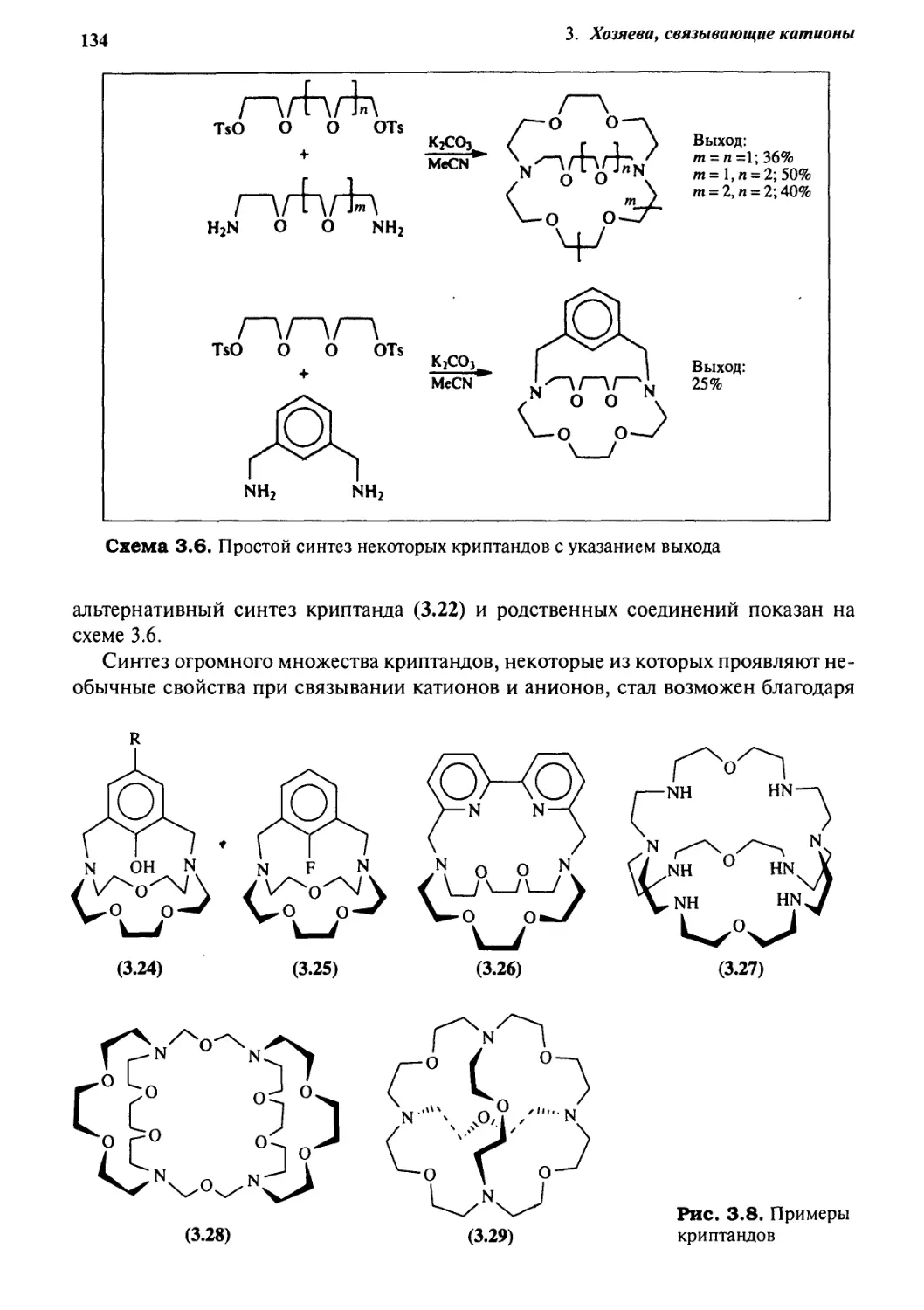
3.10.2 Гетерокриптанды 184
3.10.3 Смешанные криптанды 185
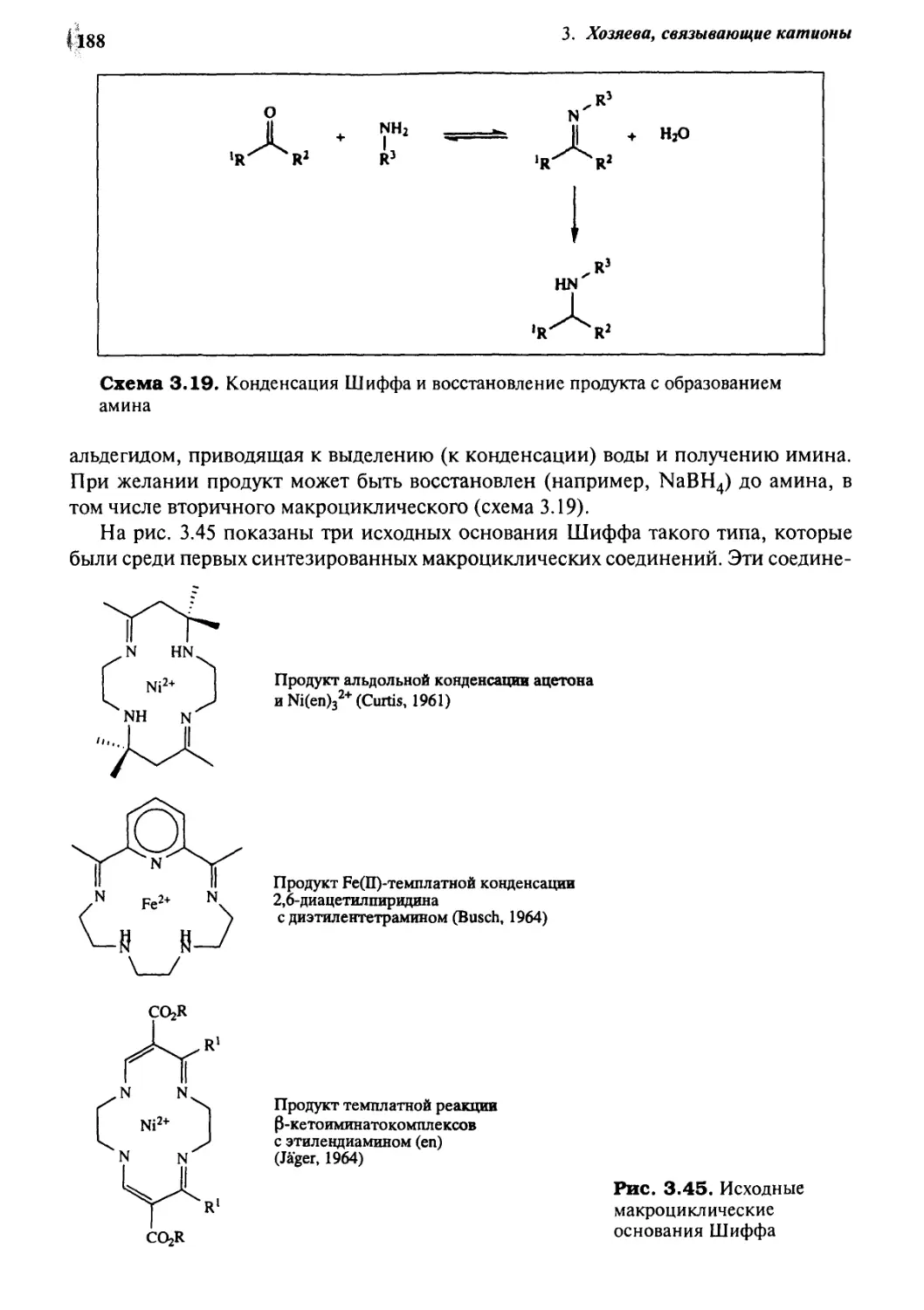
3.10.4 Основания Шиффа 187
3*11 Образование комплексов с органическими катионами 190
3.11.1 Связывание катионов аммония корандами 191
3.11.2 Связывание катионов аммония трехмерными хозяевами 194
3.11.3 Дитопные рецепторы 195
3.11.4 Хиральное распознавание 200
3.11.5 Дифильные рецепторы 203
3.11.6 Частный случай: рецепторы гербицидов 204
й Оглавление
3« 12 Алкалиды и электриды 206
3»*3 Каликсарены 209
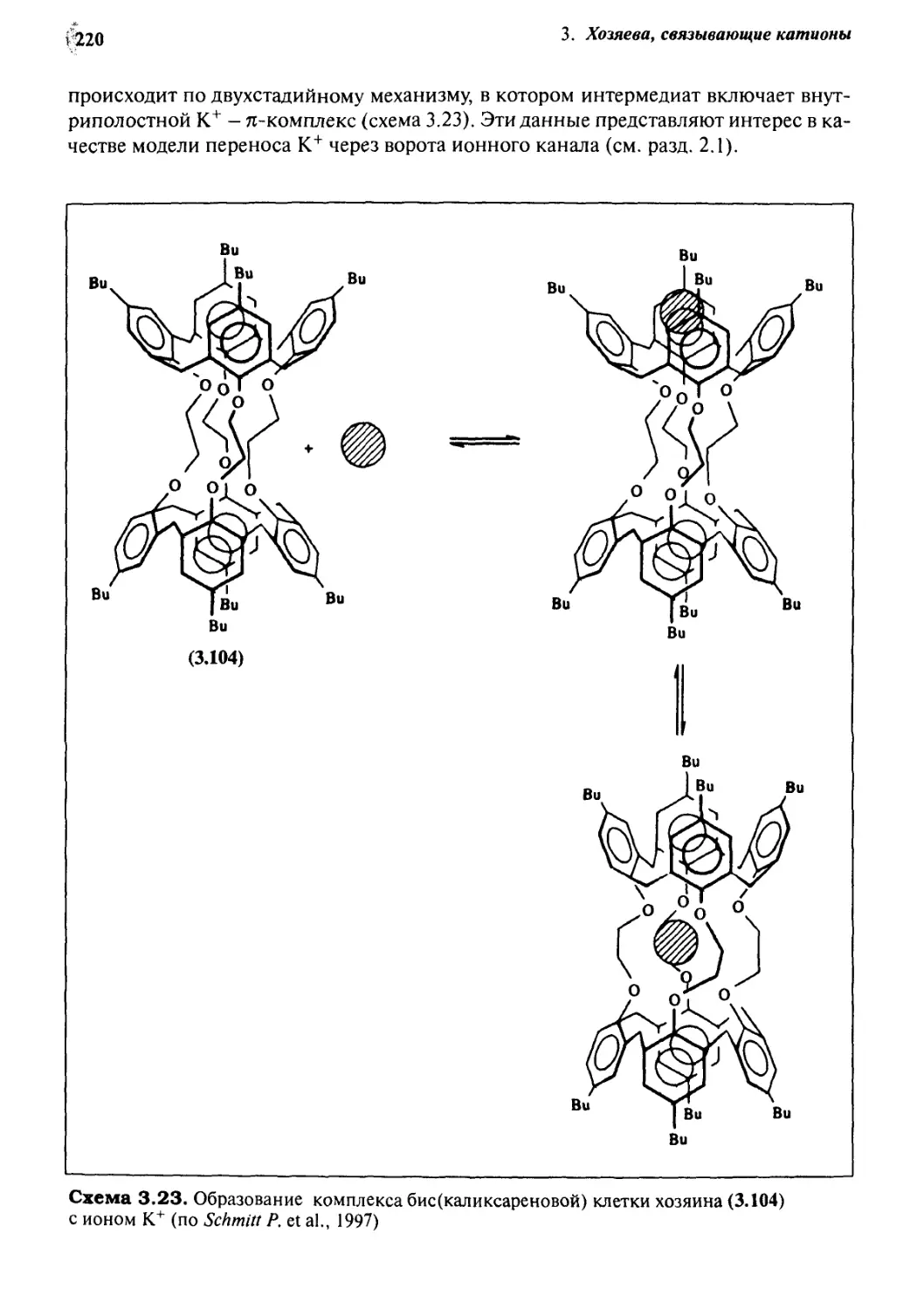
3.13.1 Образование комплексов каликсаренов с катионами 213
3.13.2 Равновесие межфазного переноса 217
3.13.3 Образование комплексов гибридных каликсаренов
с катионами 219
3.I4 Углеродные донорные и л-кислотные лиганды 223
3.14.1 Смешанные С-гетероатомные хозяева 223
3.14.2 Углеводородные хозяева 225
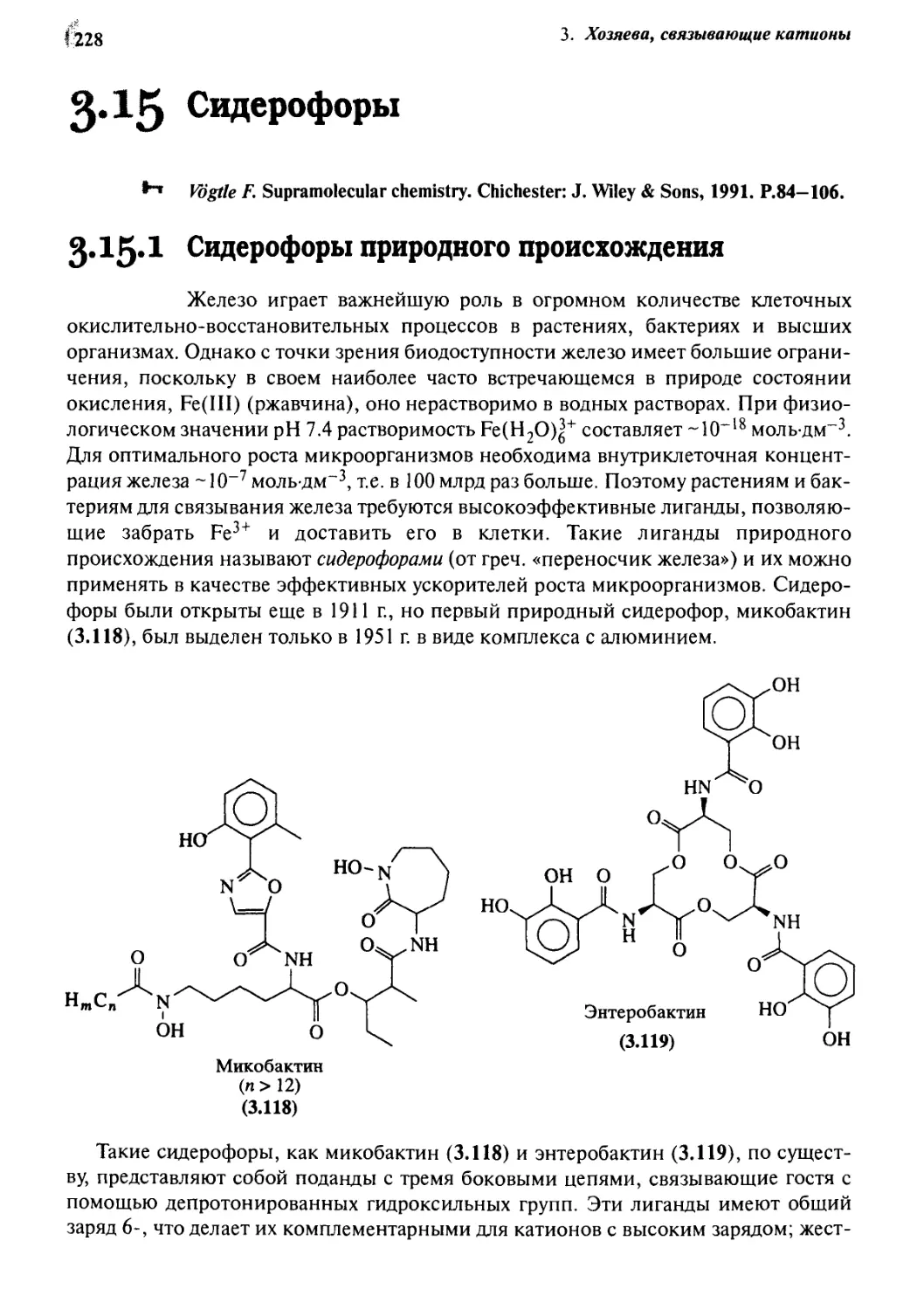
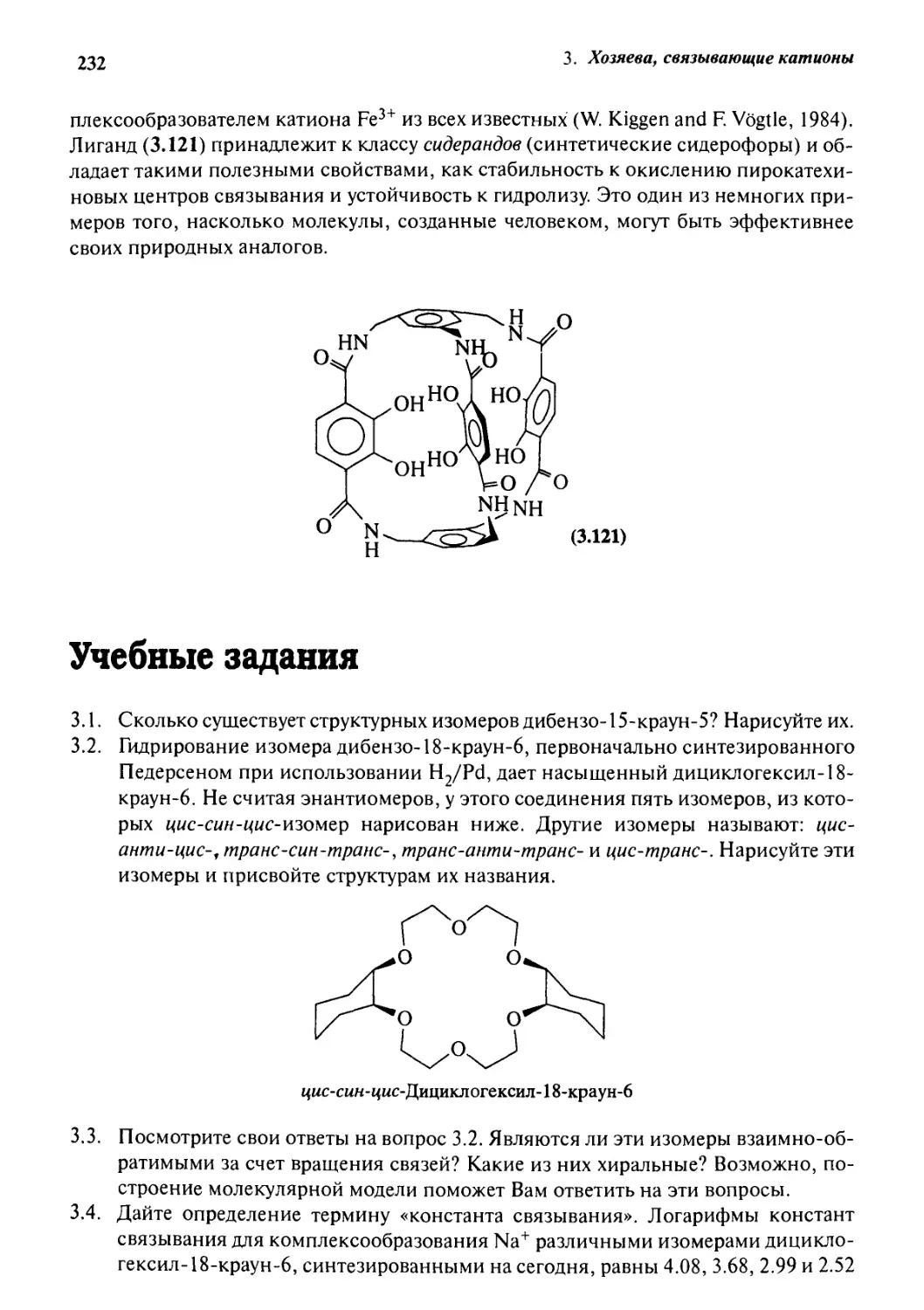
Сидерофоры 228
3.15.1 Сидерофоры природного происхождения 228
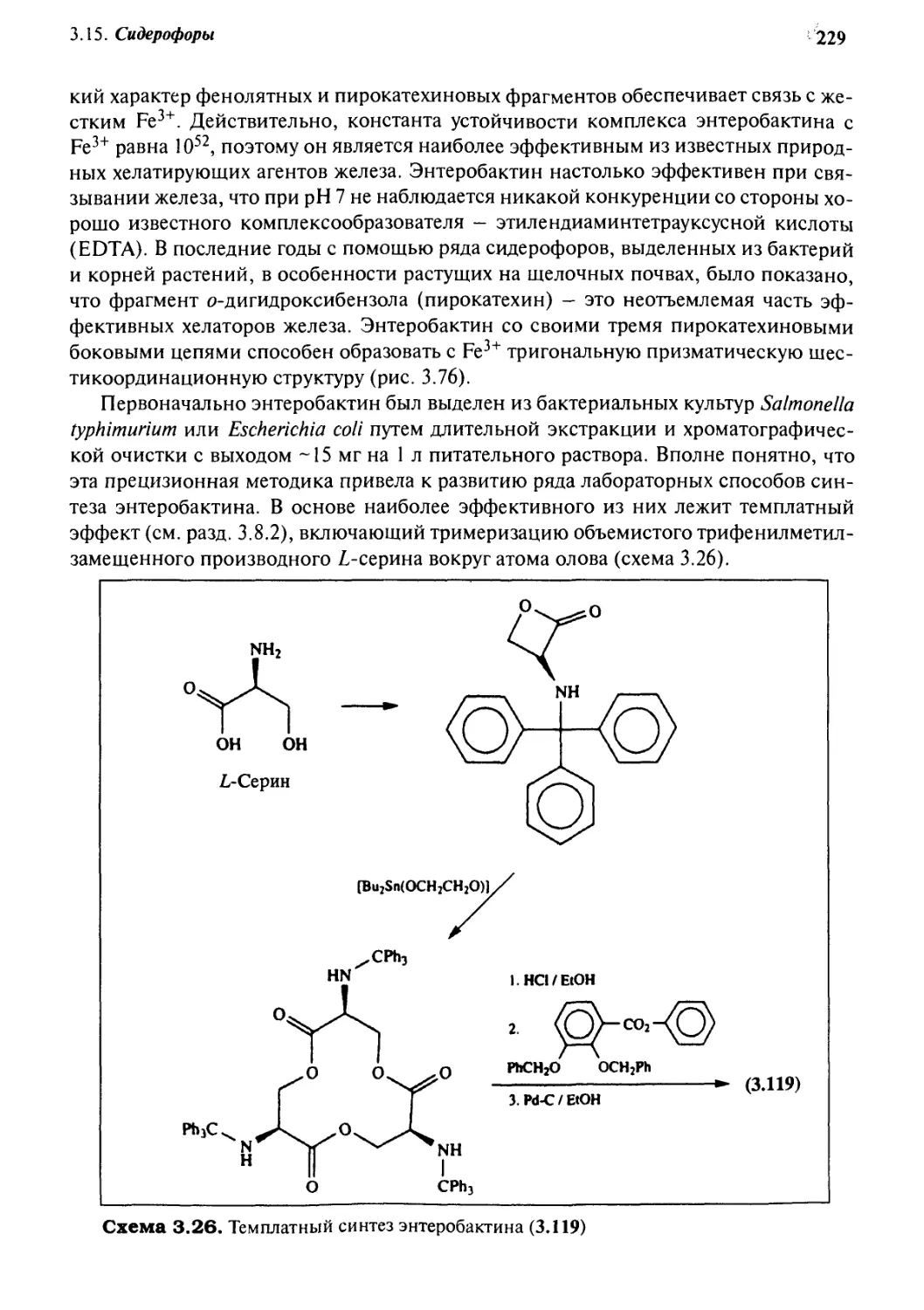
3.15.2 Синтетические системы 230
Учебные задания 232
Мысленный эксперимент 233
Литература 234
4
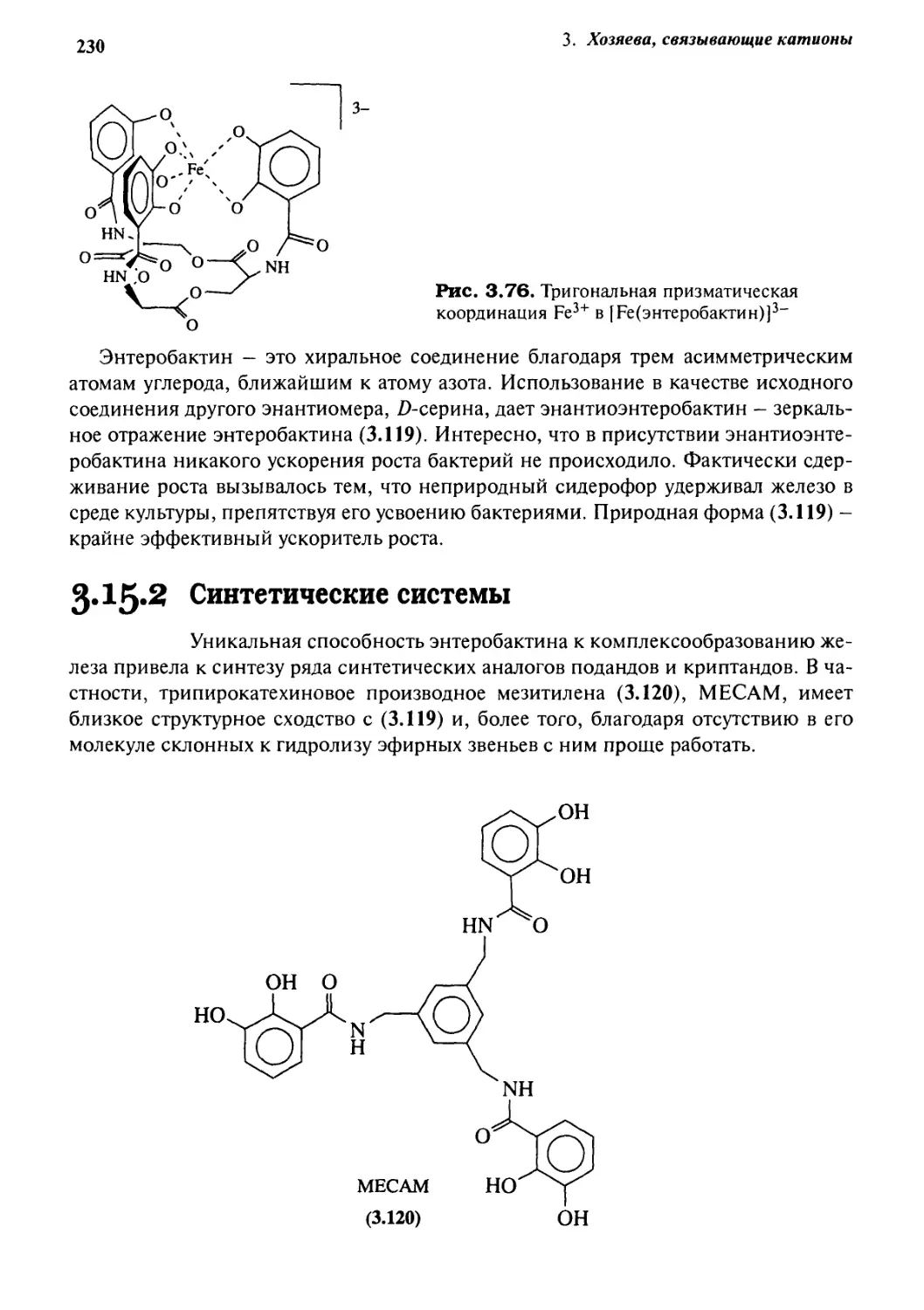
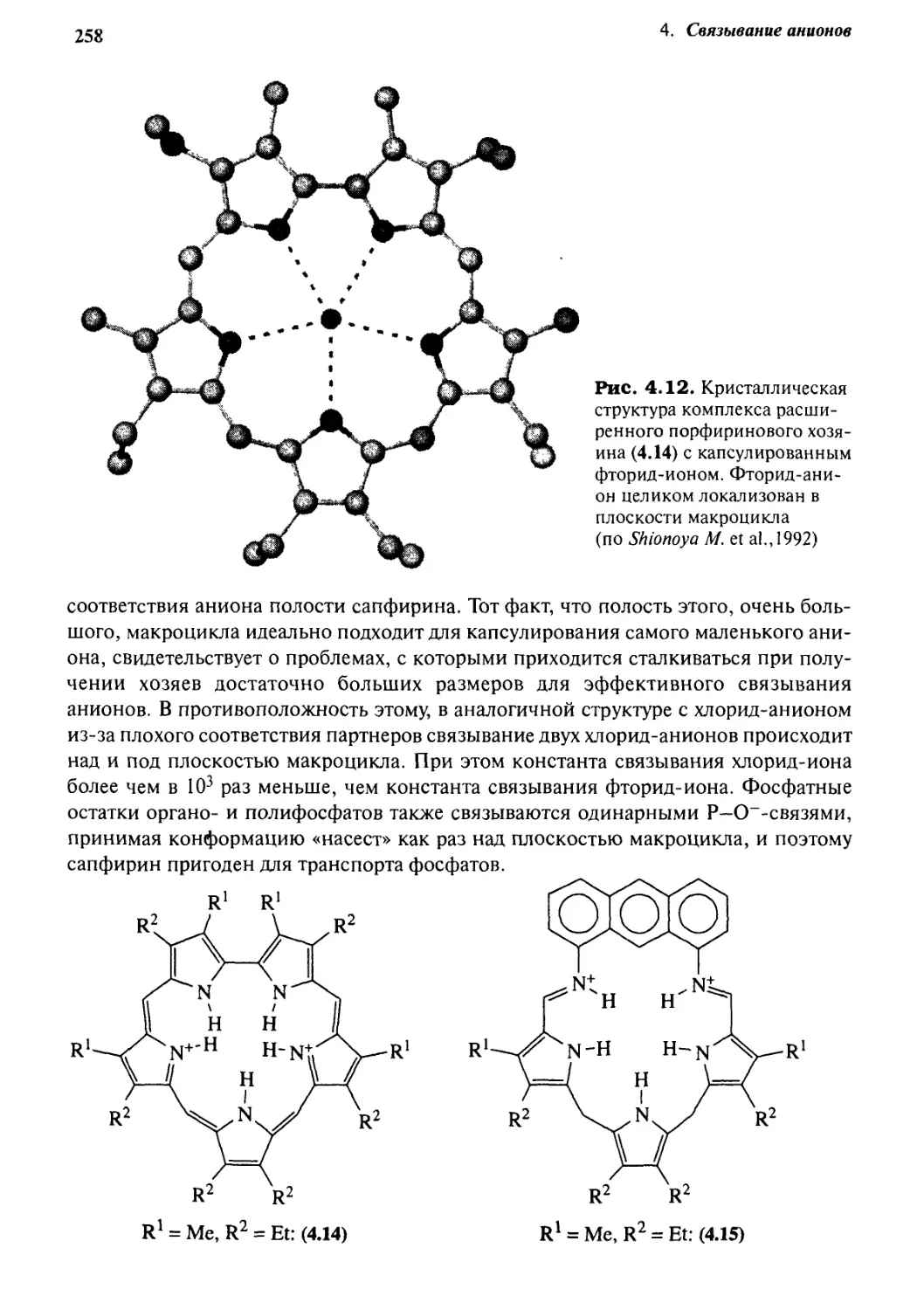
Связывание анионов
237
4* 1 Введение 237
4*2 Биологические рецепторы анионов 240
4.2.1 Белки, связывающие фосфаты и сульфаты 241
4.2.2 Аргинин как центр связывания анионов 242
4.2.3 Карбоксиле птидаза А 243
4«3 Концепции конструирования хозяина для анионов 244
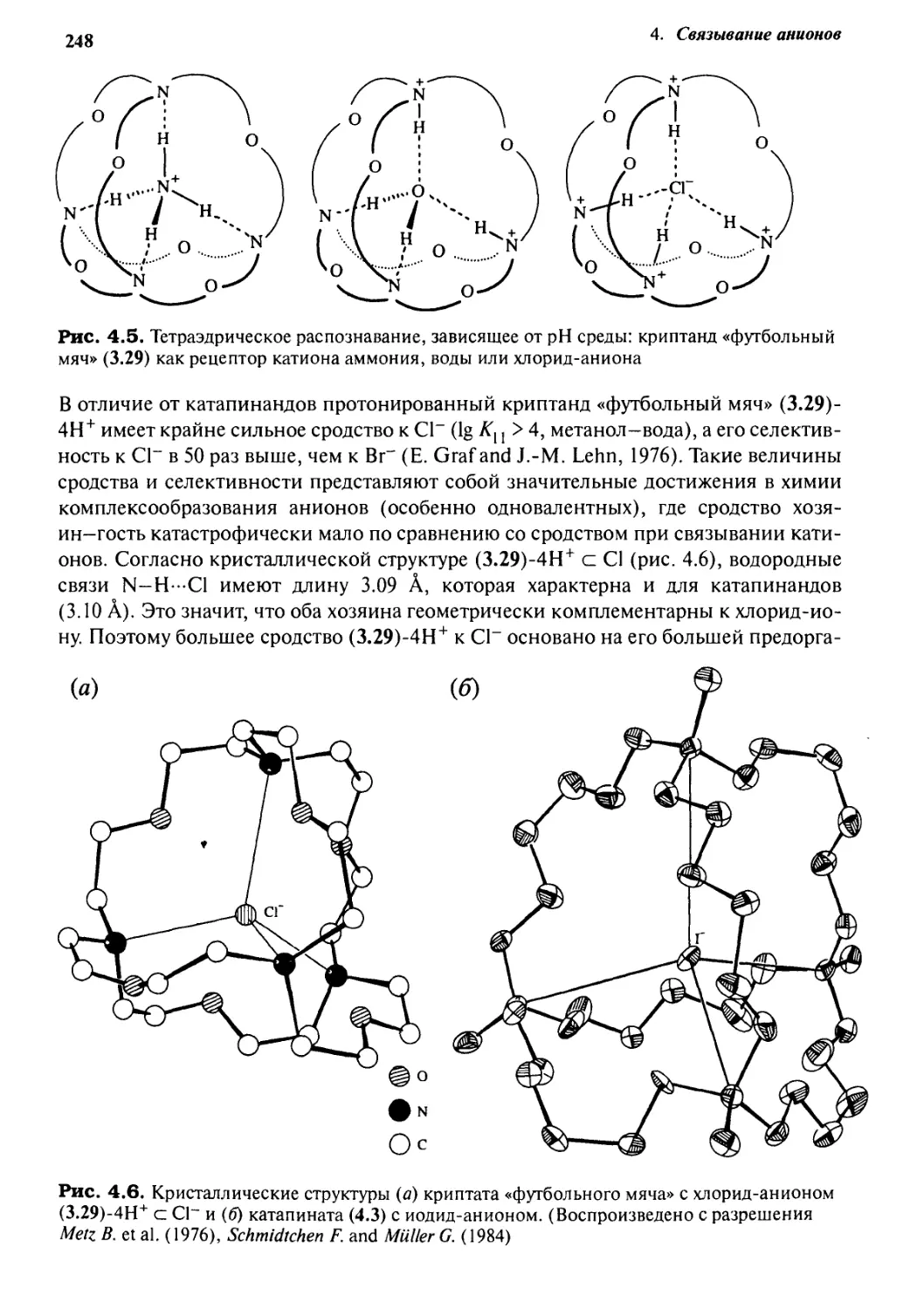
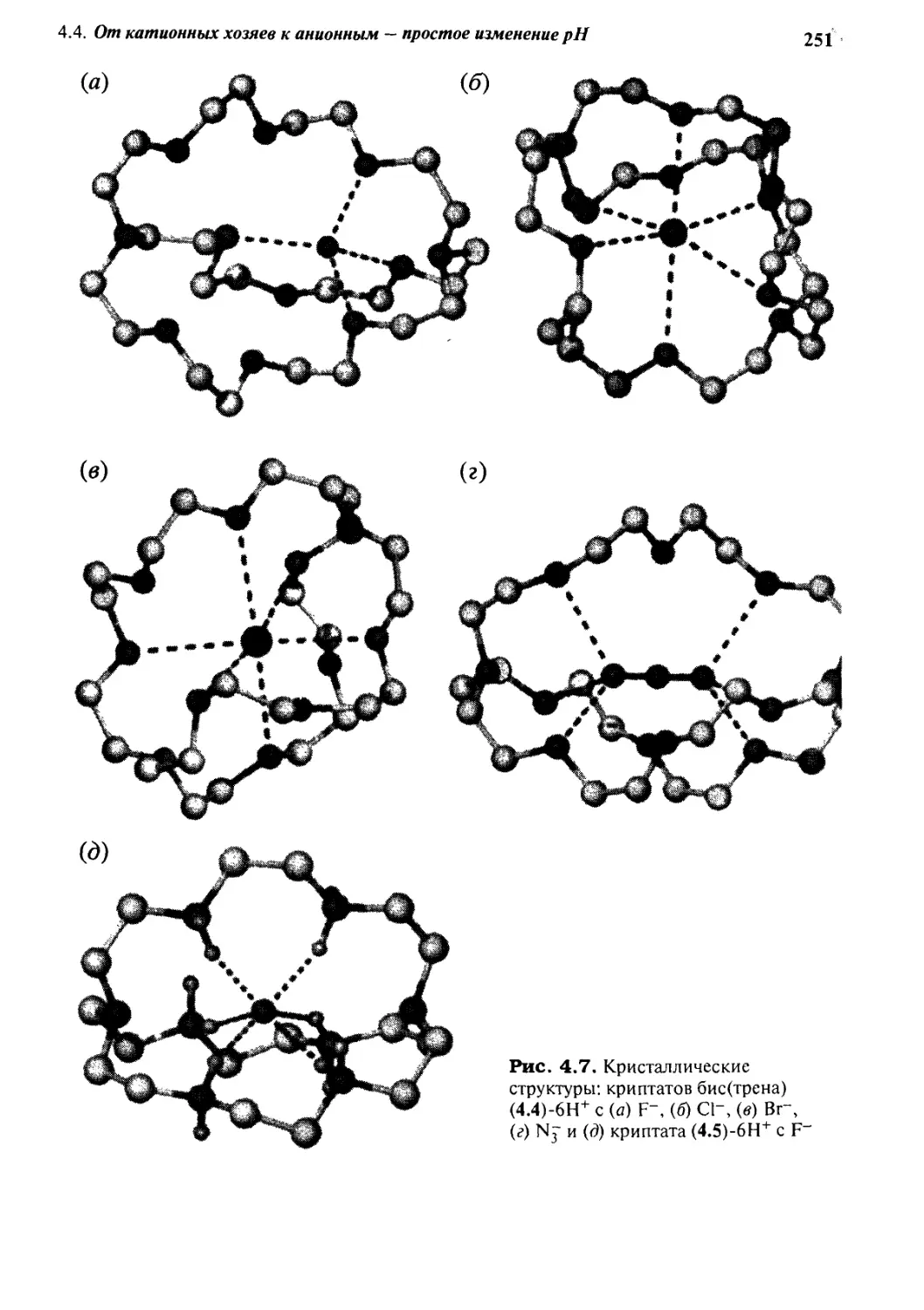
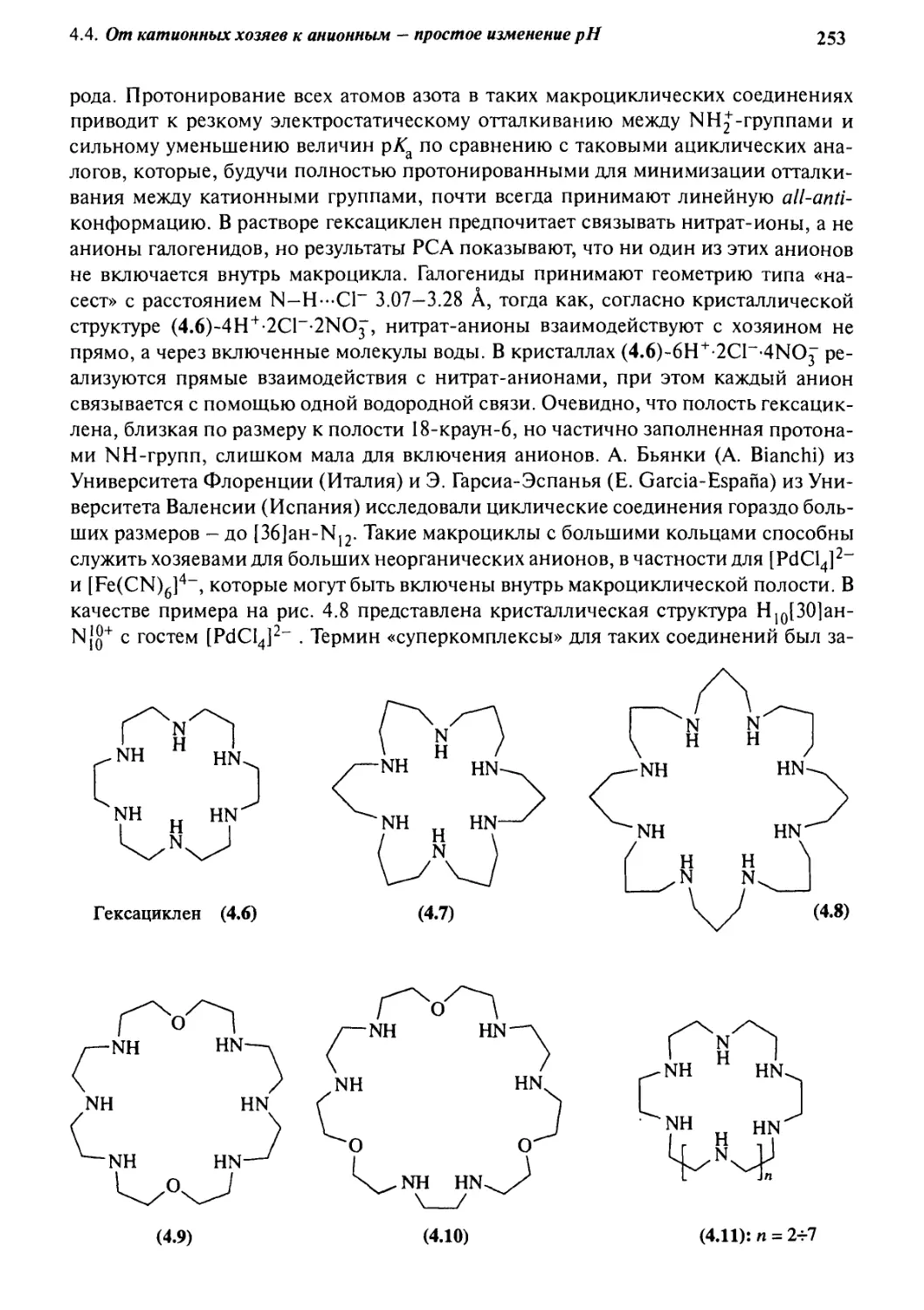
4*4 От катионных хозяев к анионным -
простое изменение рН 247
4.4.1 Тетраэдрические рецепторы 247
4.4.2 Селективность формы 249
4.4.3 Двухмерные хозяева 252
4.4.4 Циклофановые хозяева 260
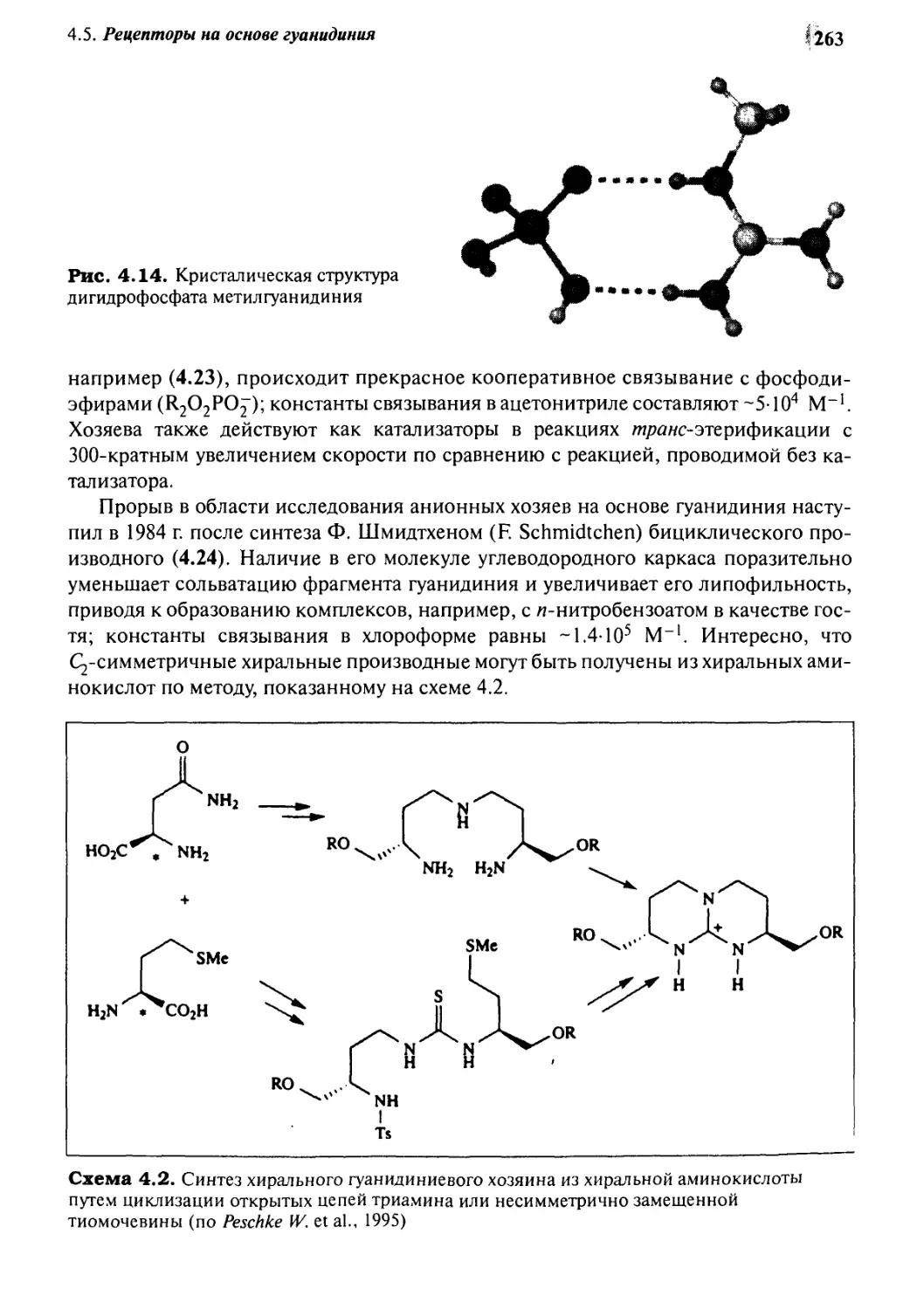
4*5 Рецепторы на основе гуанидиния 262
4*6 Металлоорганические рецепторы 265
4*7 Нейтральные рецепторы 273
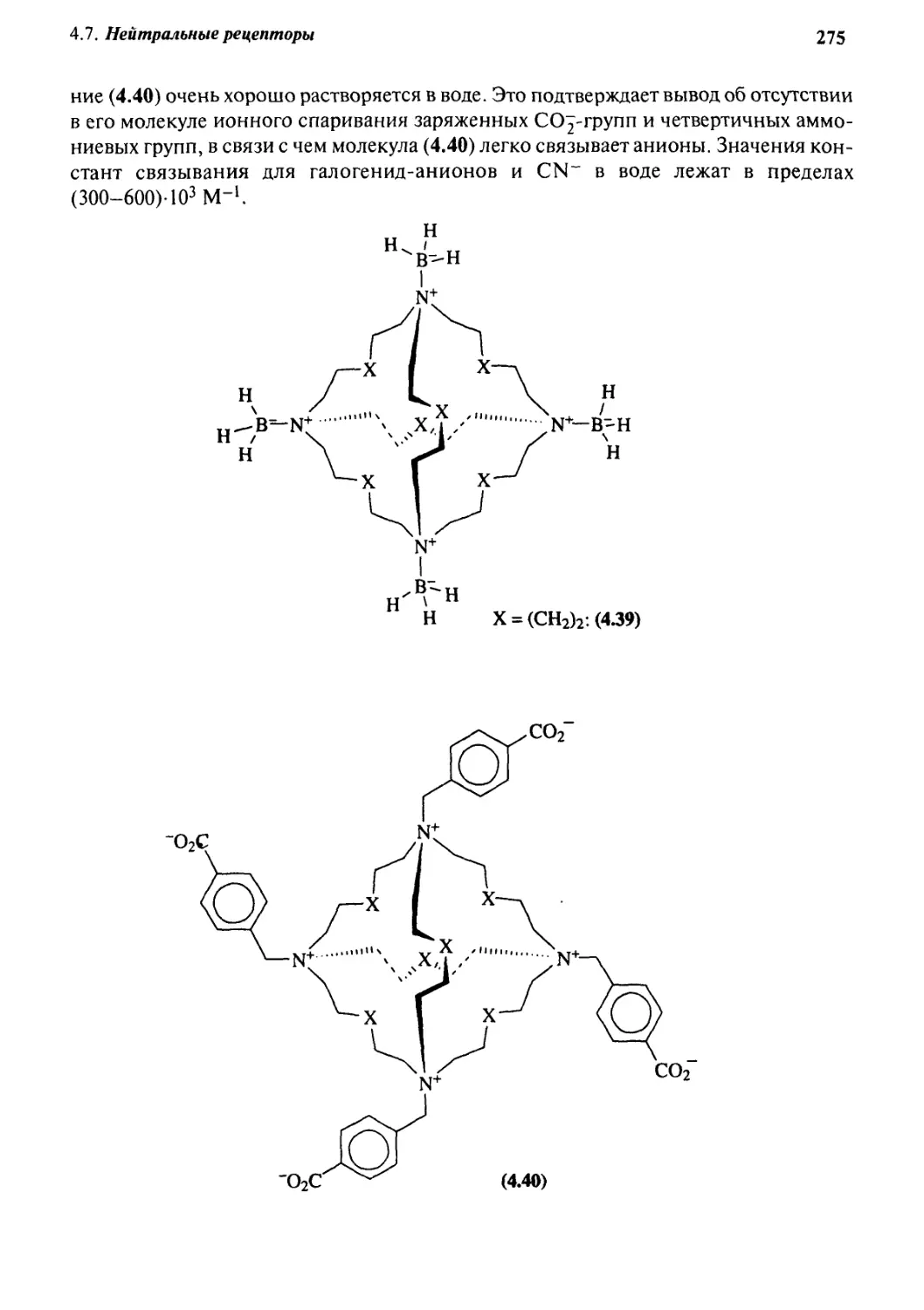
4.7.1 Цвиттер-ионы 274
4.7.2 Хозяева, образующие водородные связи 276
Оглавление
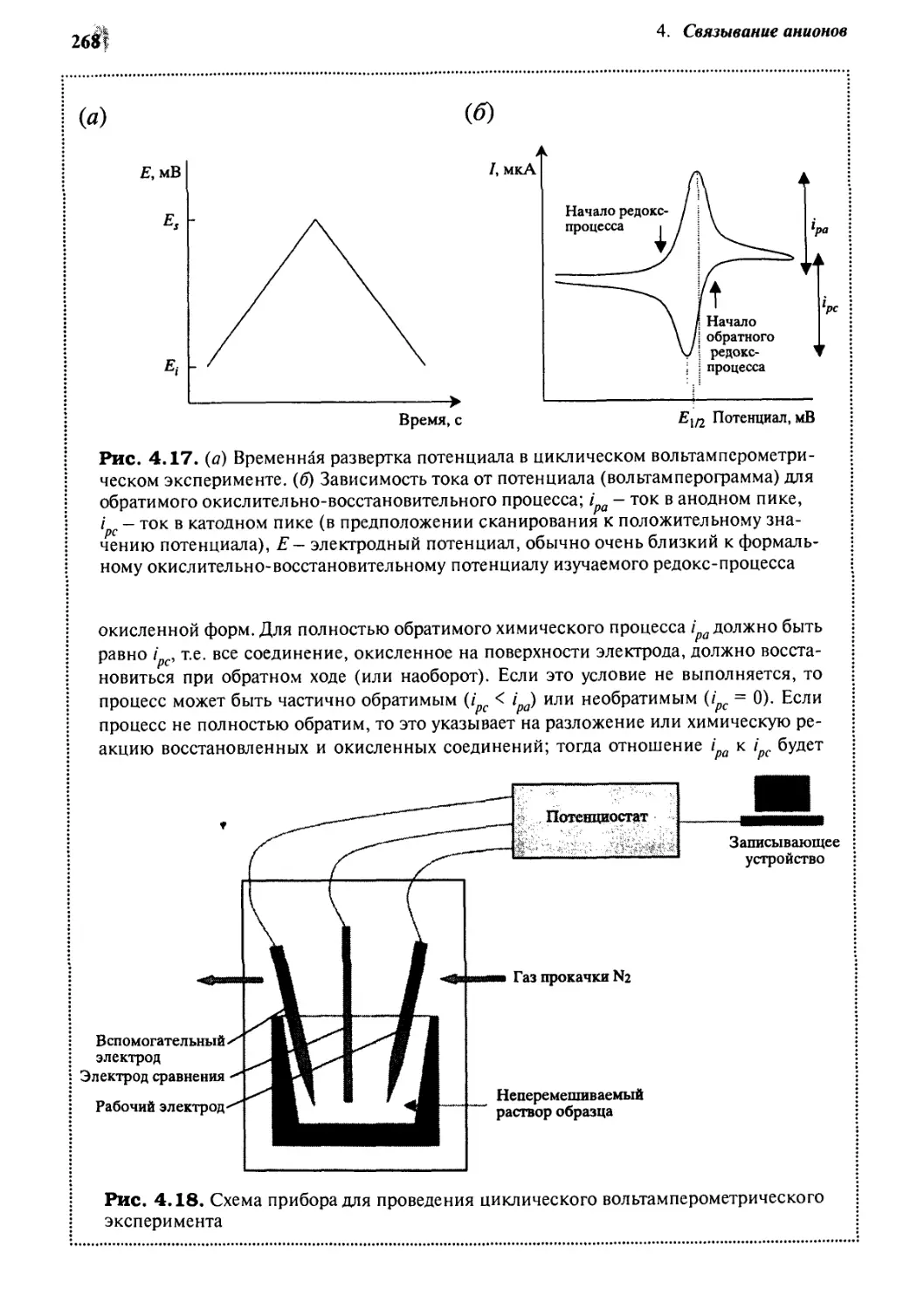
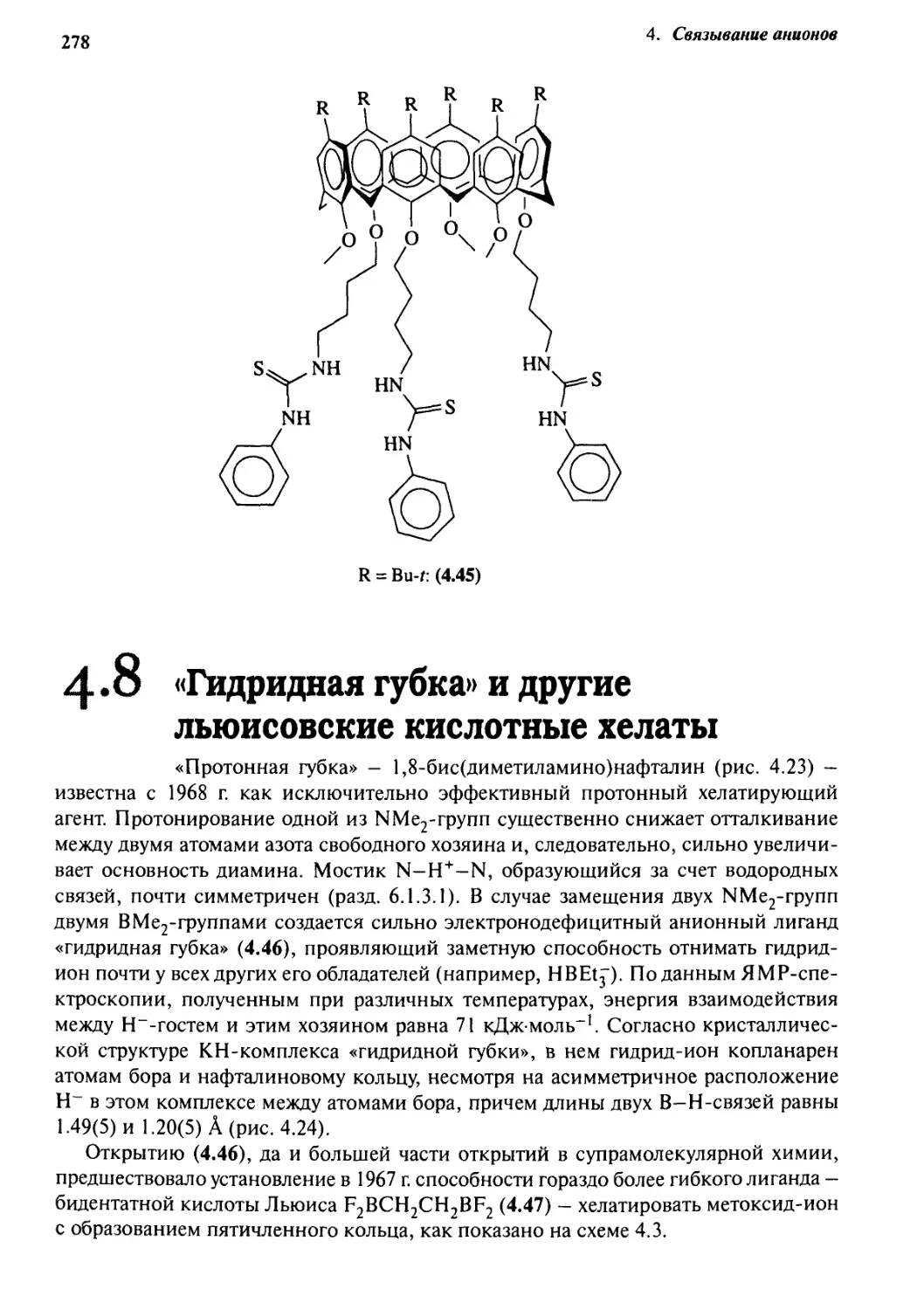
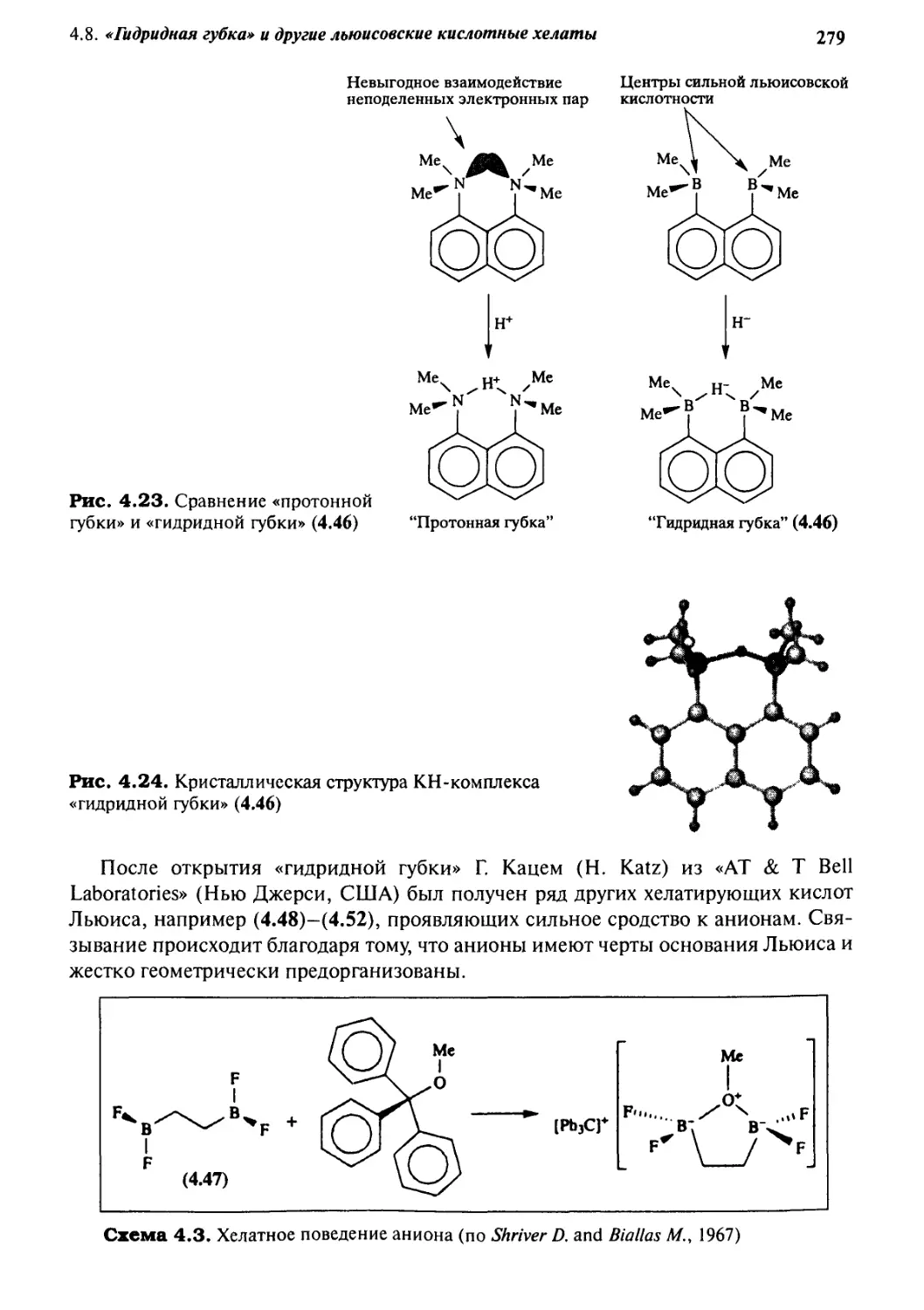
4-8
«Гидридная губка»
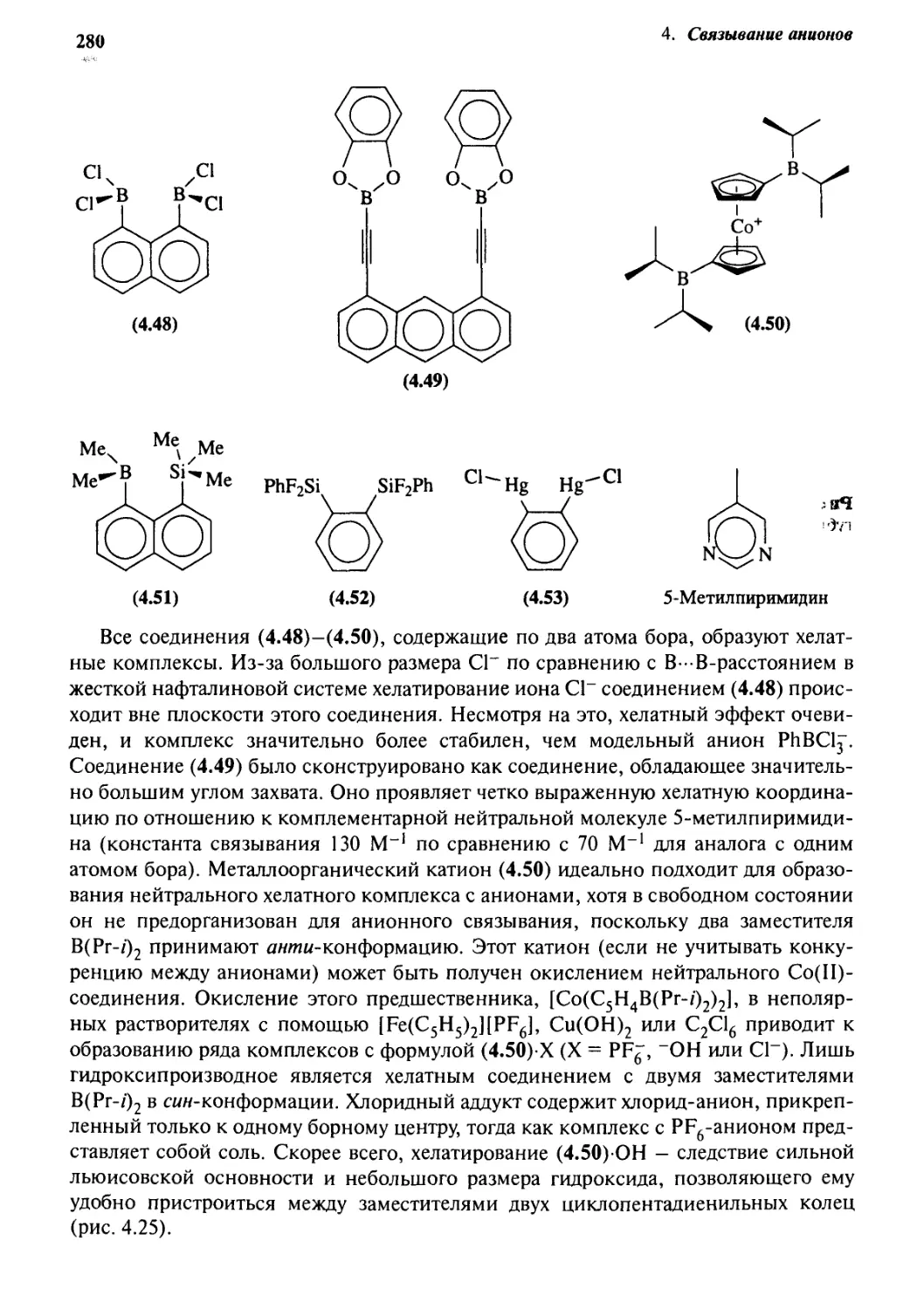
и другие льюисовские кислотные хелаты 278
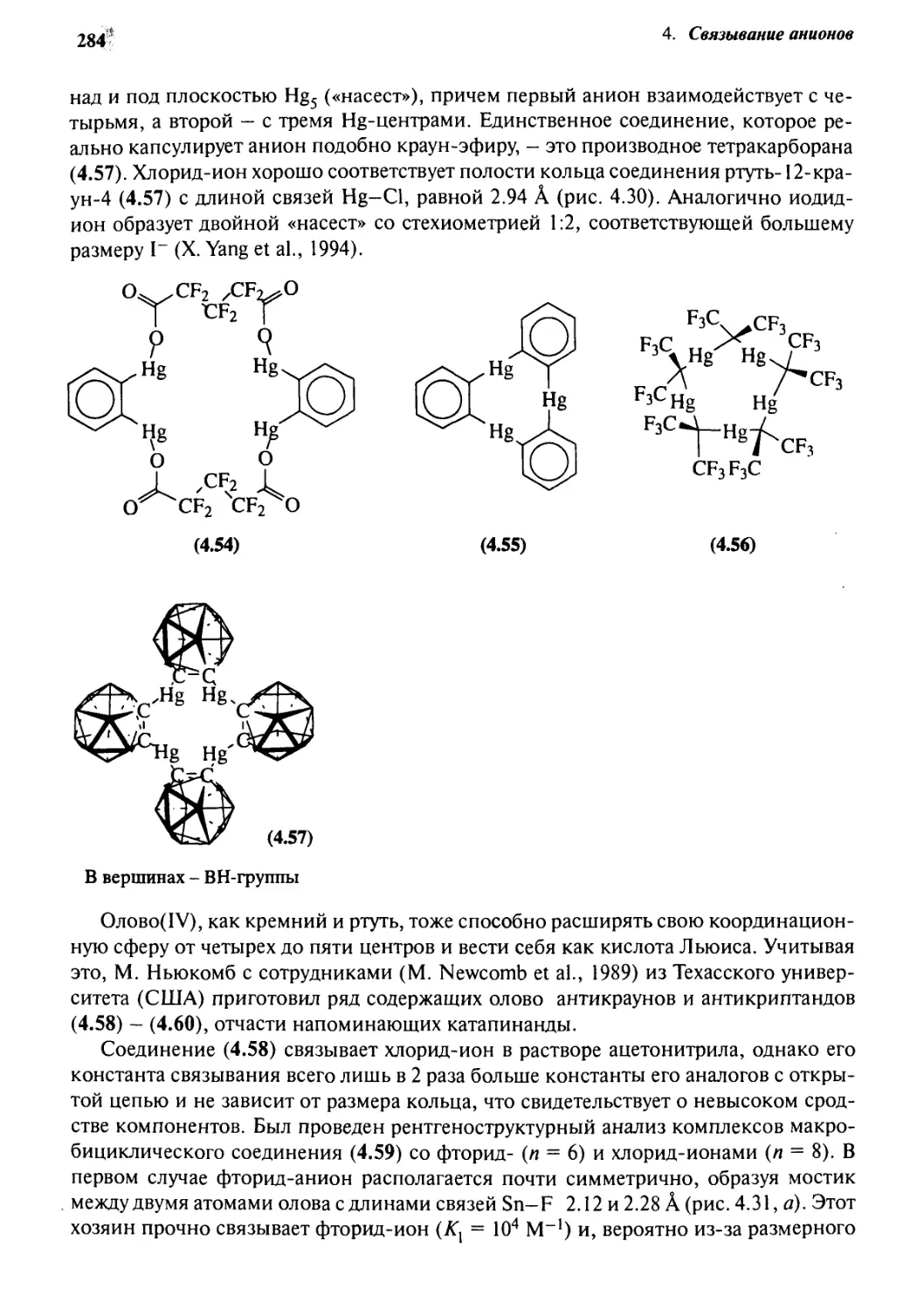
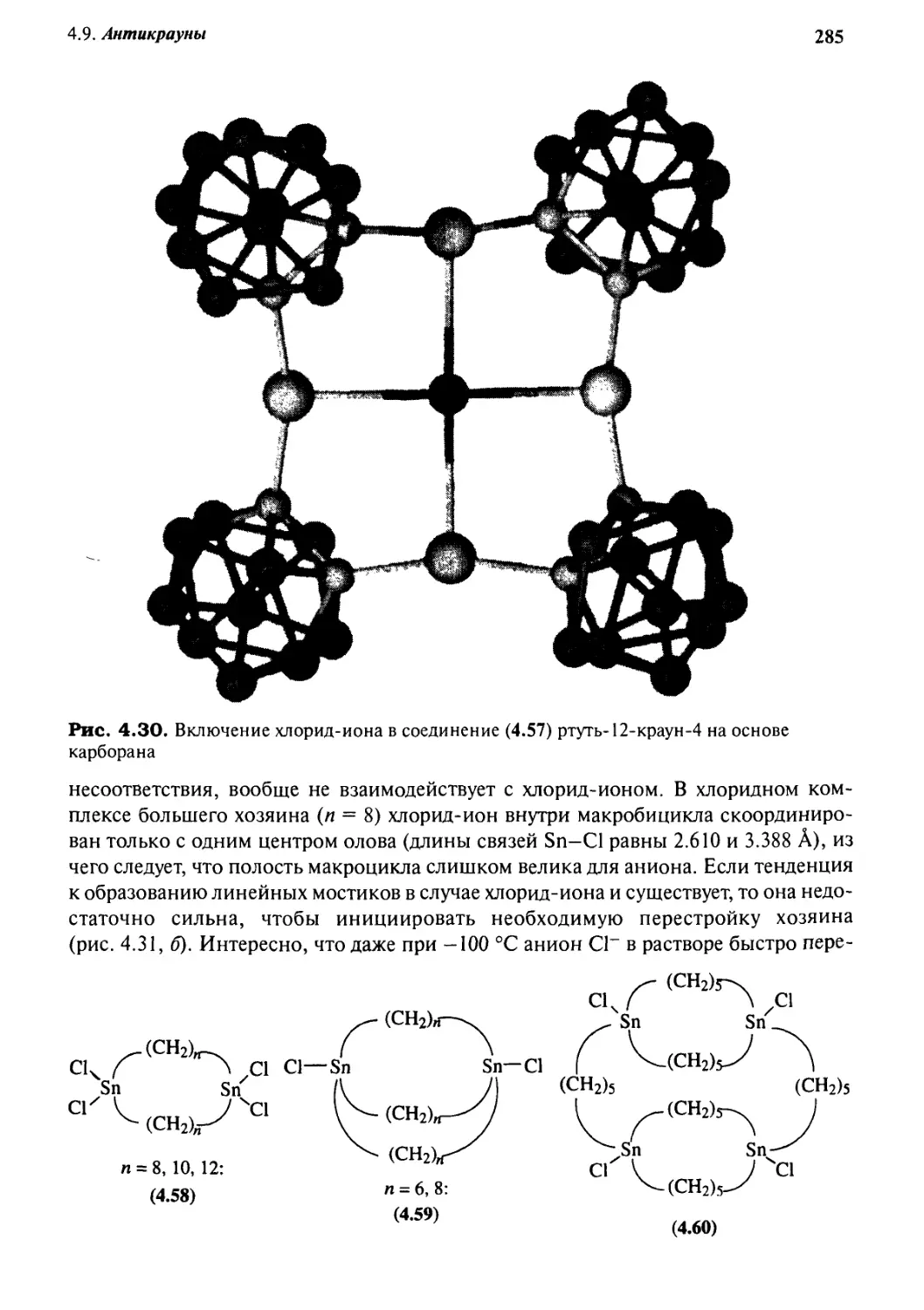
4-9 Антикрауны 282
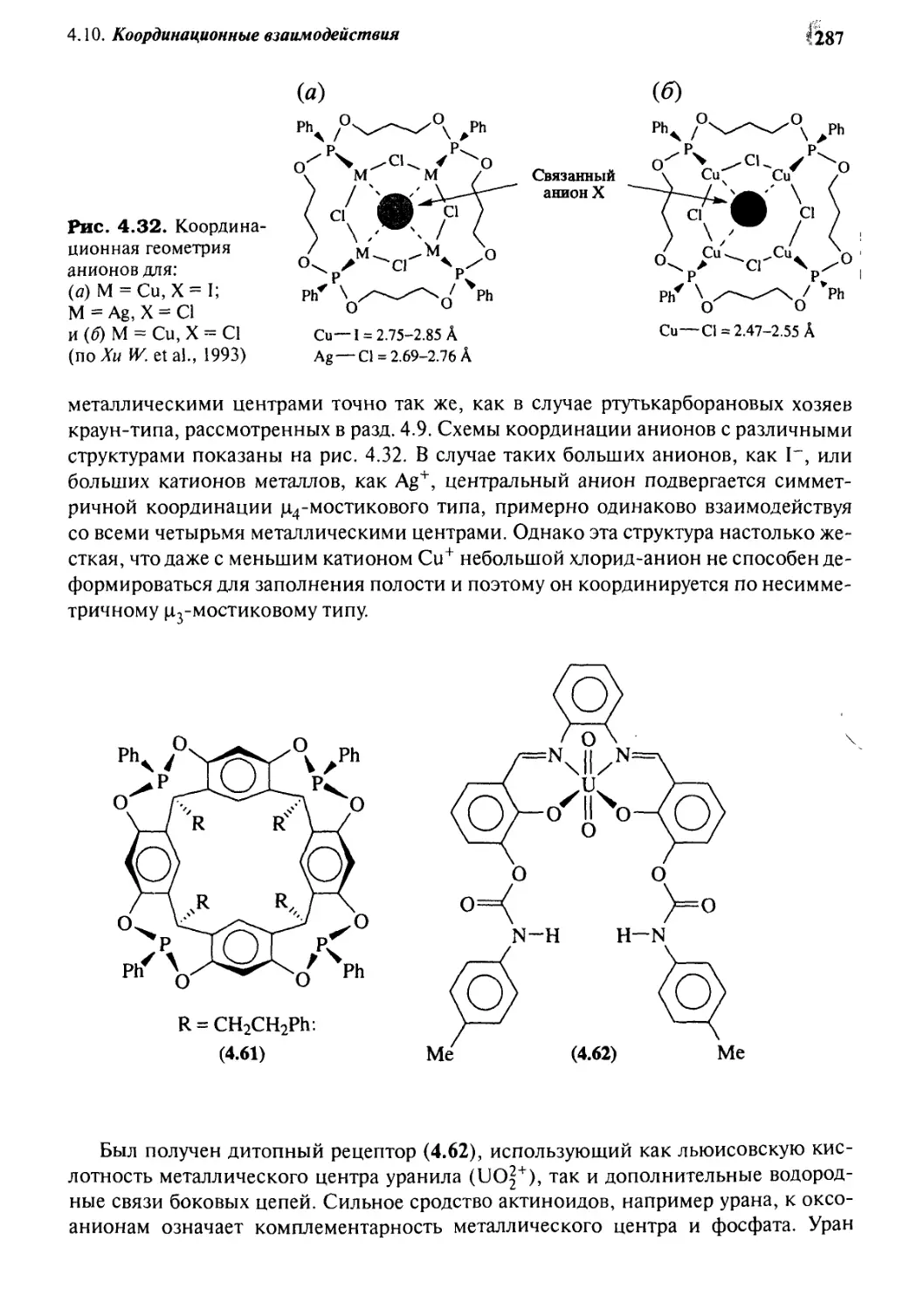
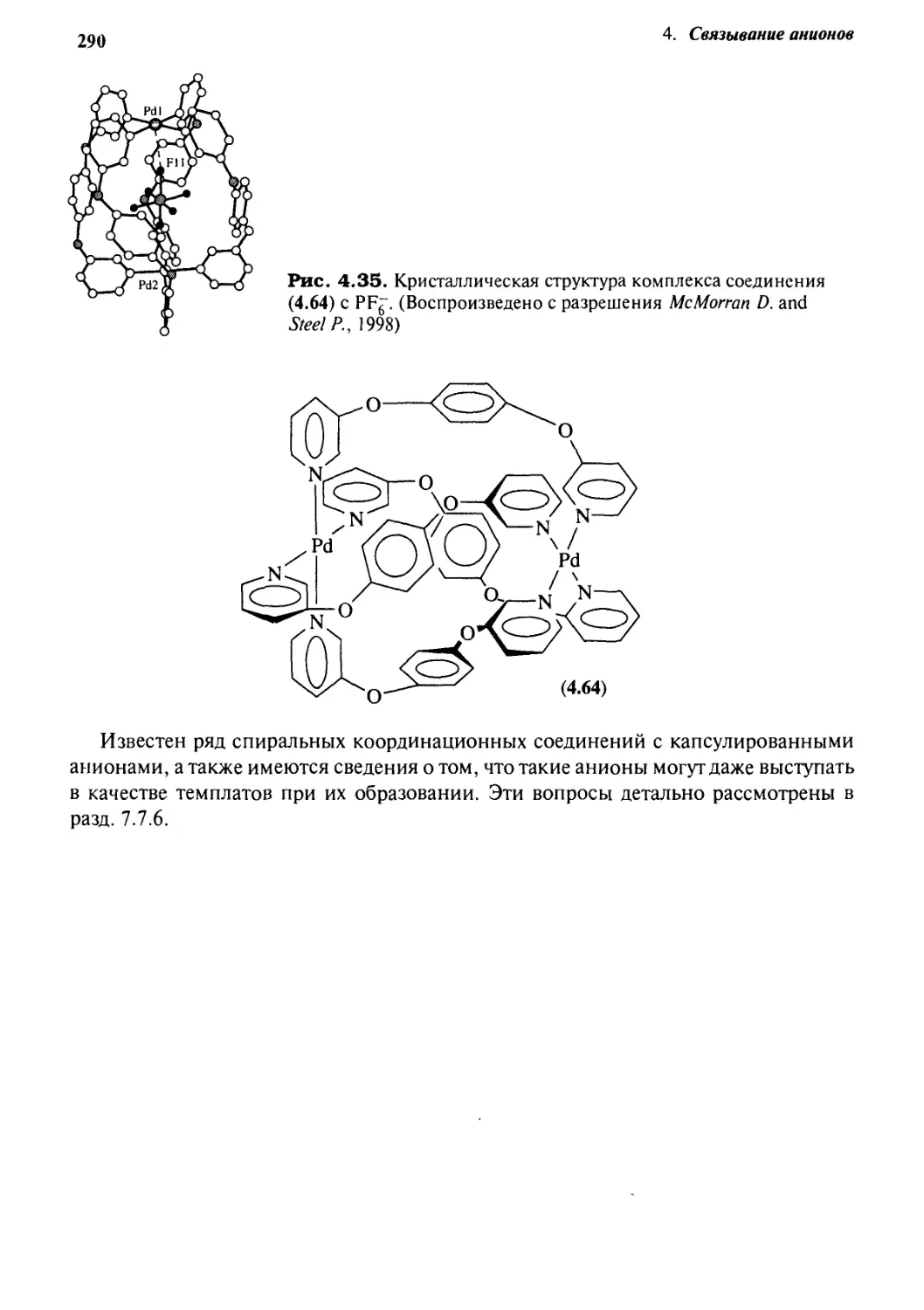
Координационные взаимодействия 286
Учебные задания 291
Мысленный эксперимент 291
Литература 292
295
К Связывание нейтральных молекул
5* 1 Неорганические твердофазные клатратные
соединения 296
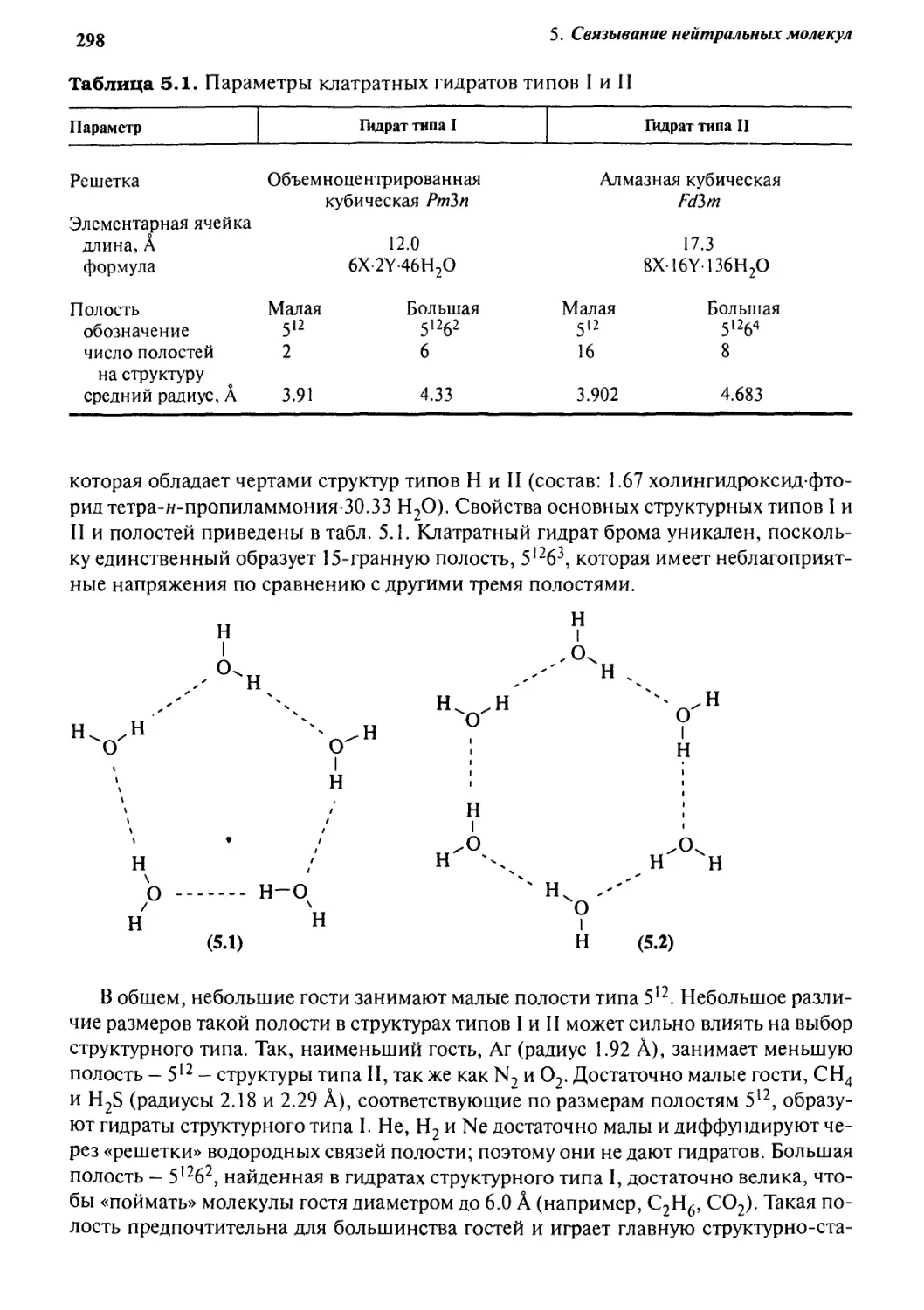
5.1.1 Клатратные гидраты 296
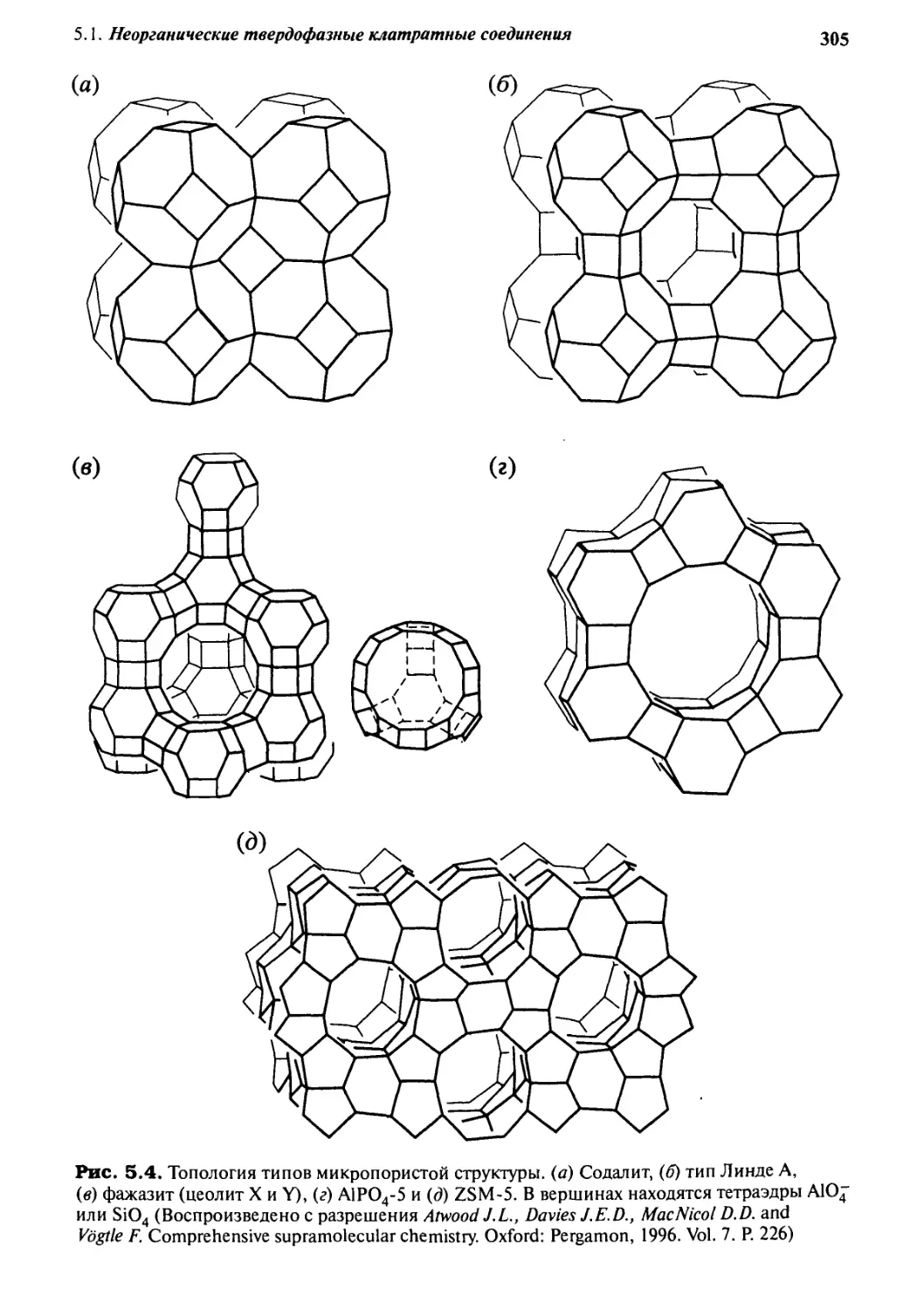
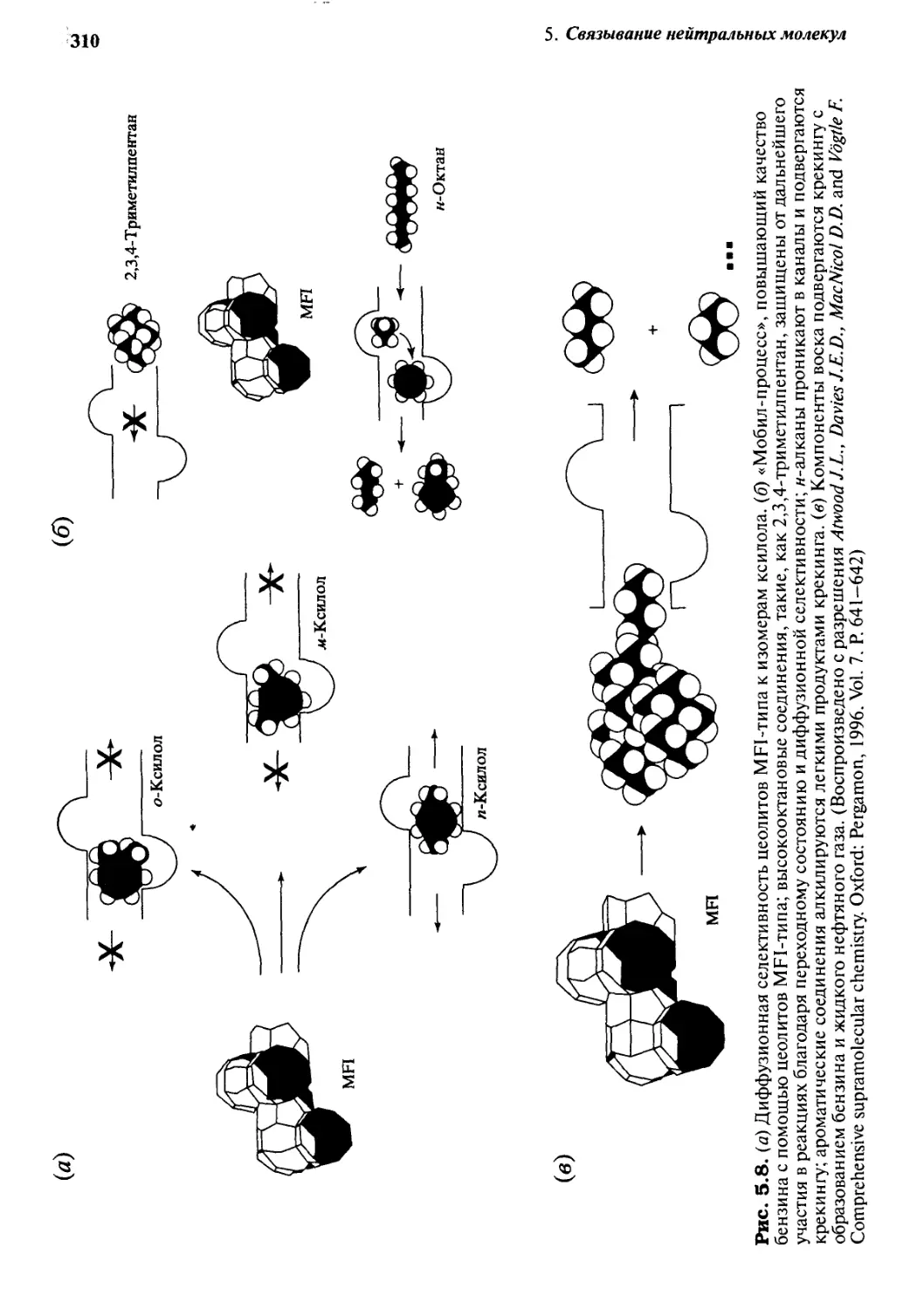
5.1.2 Цеолиты 303
5.1.3 Твердые слоистые материалы и их интеркаляты 311
5.1.4 Клатраты Хофмана и Вернера 317
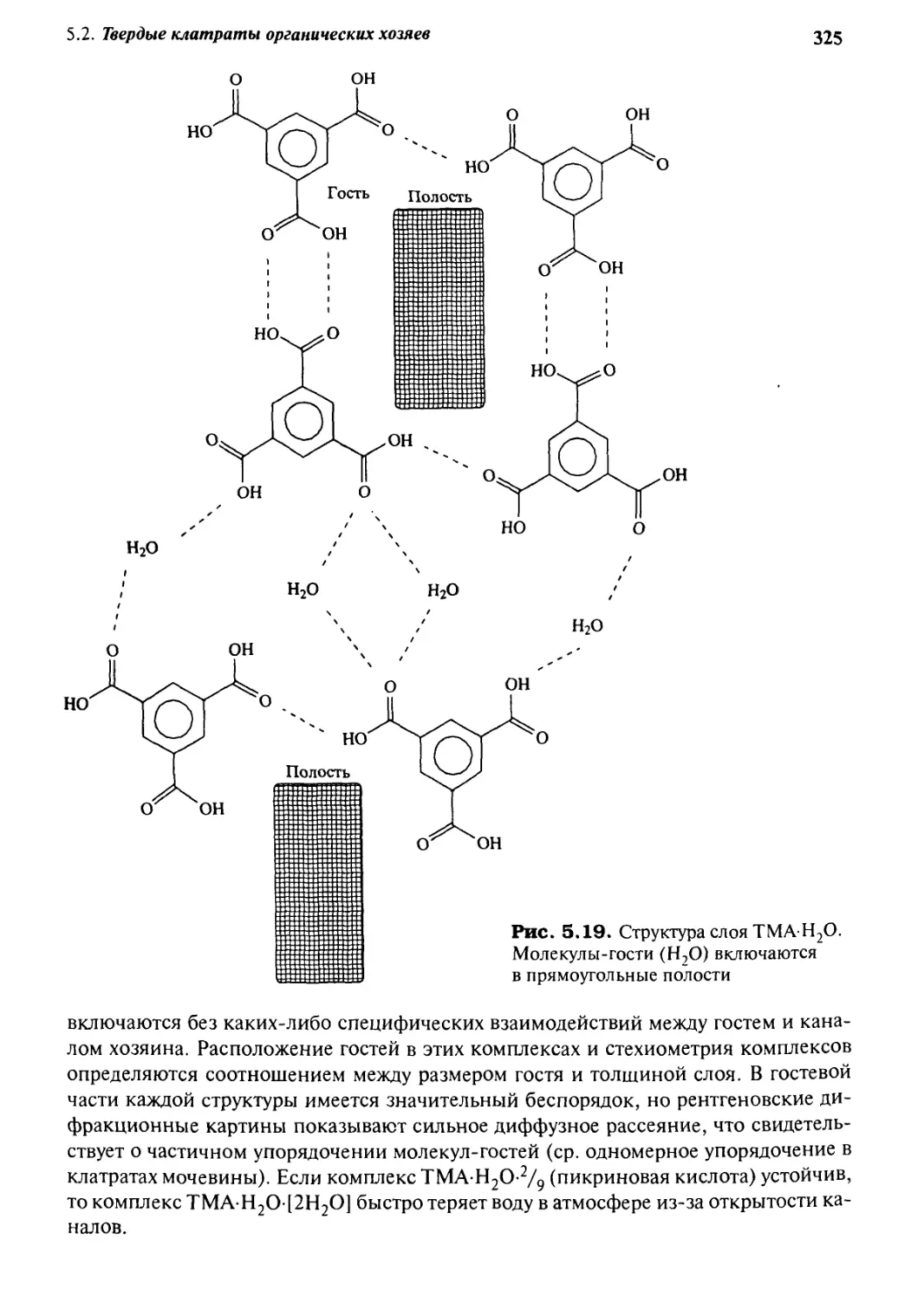
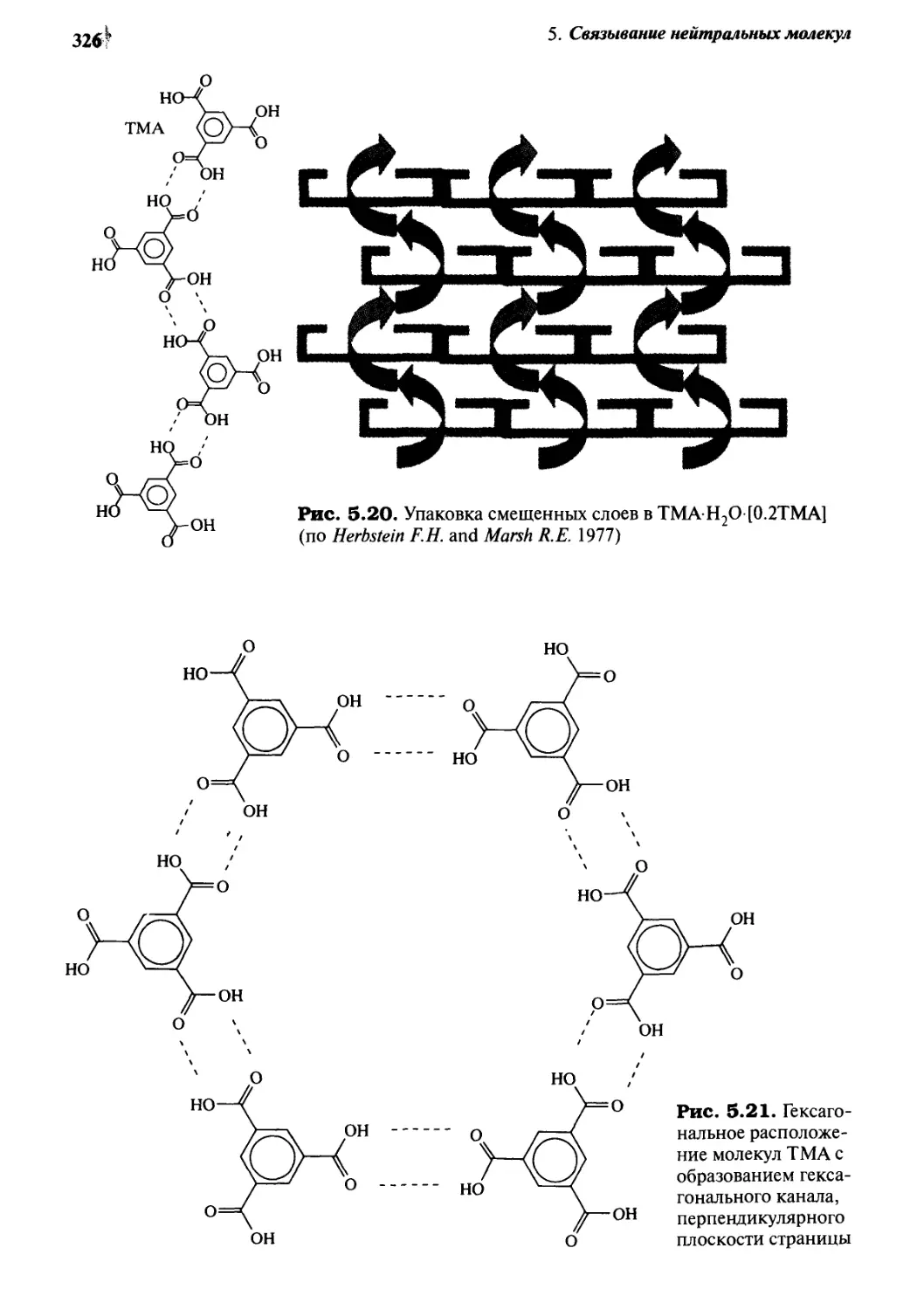
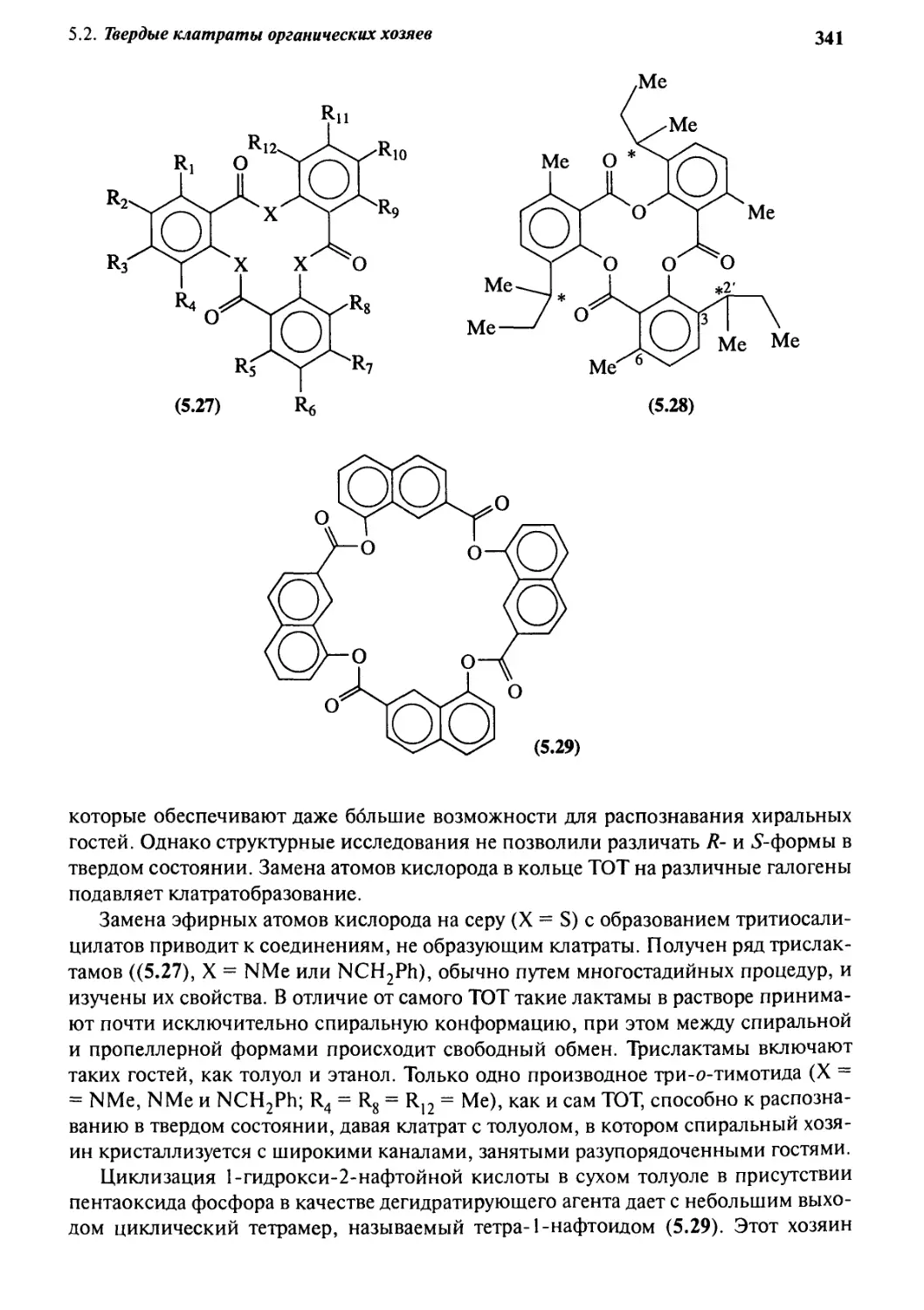
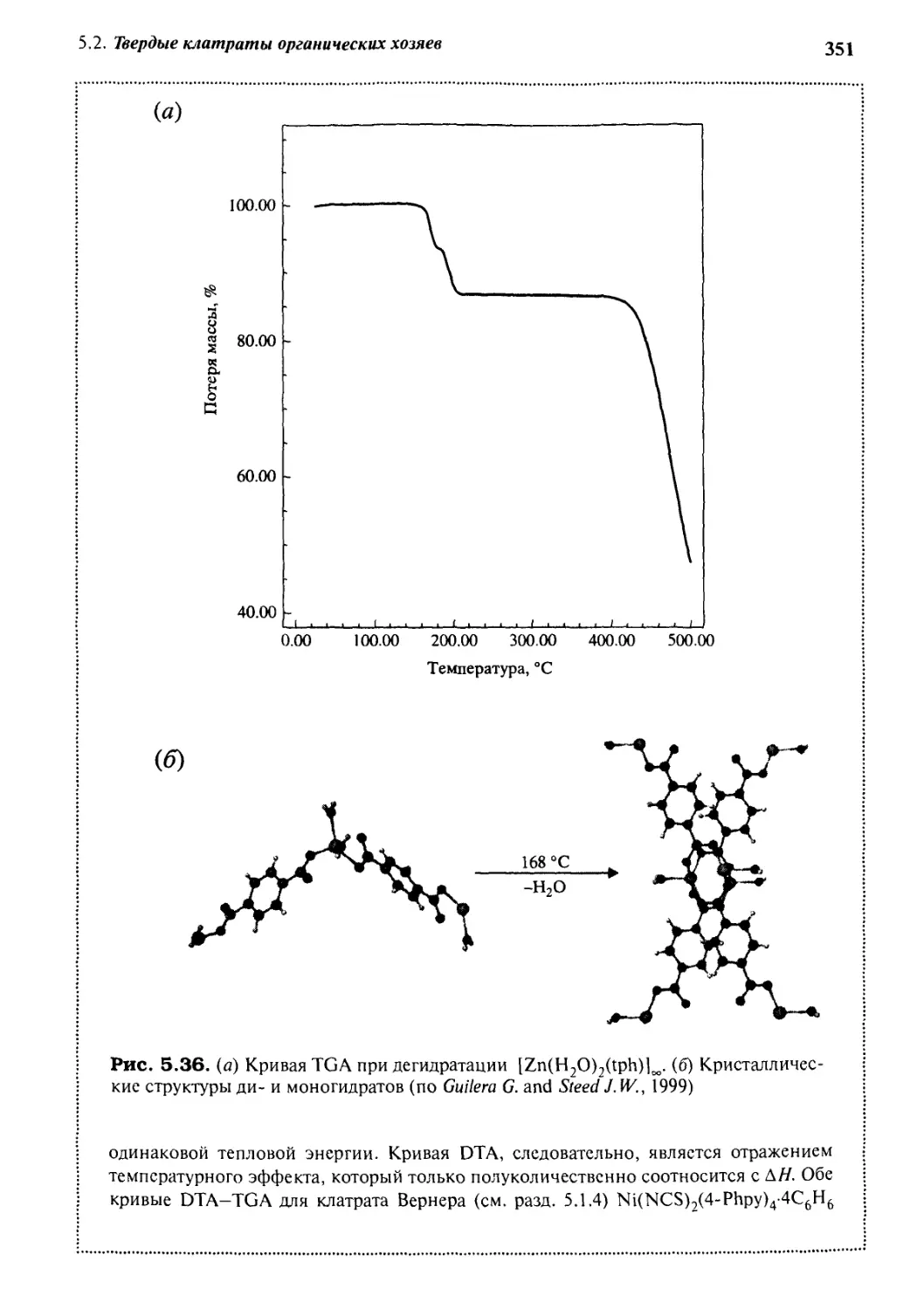
5 • 2 Твердые клатраты органических хозяев з 18
5.2.1 Клатраты мочевины 318
5.2.2 Другие канальные клатраты: клатраты тримезиновой
кислоты, спиральных тубуландов и пергидротрифенилена 324
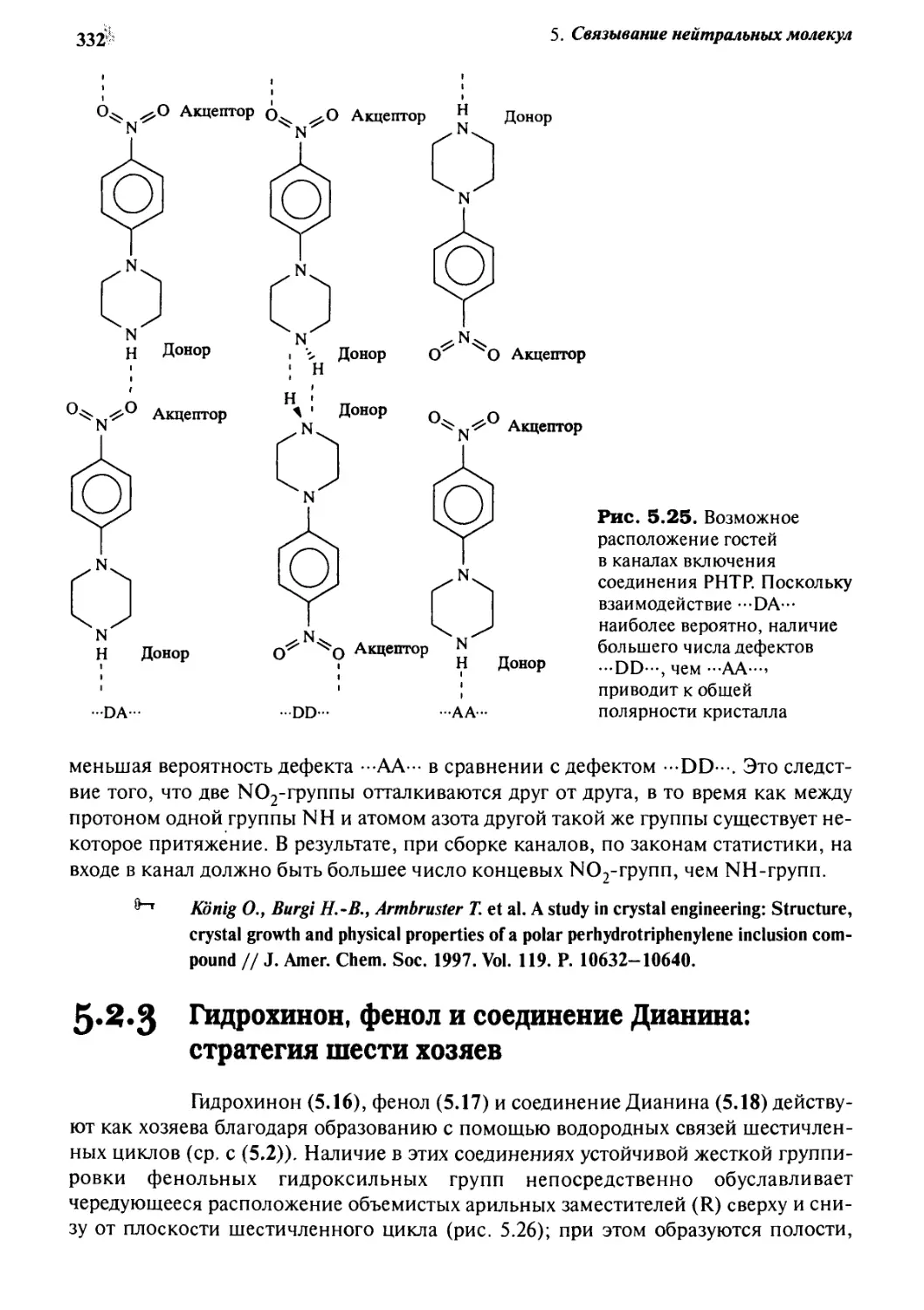
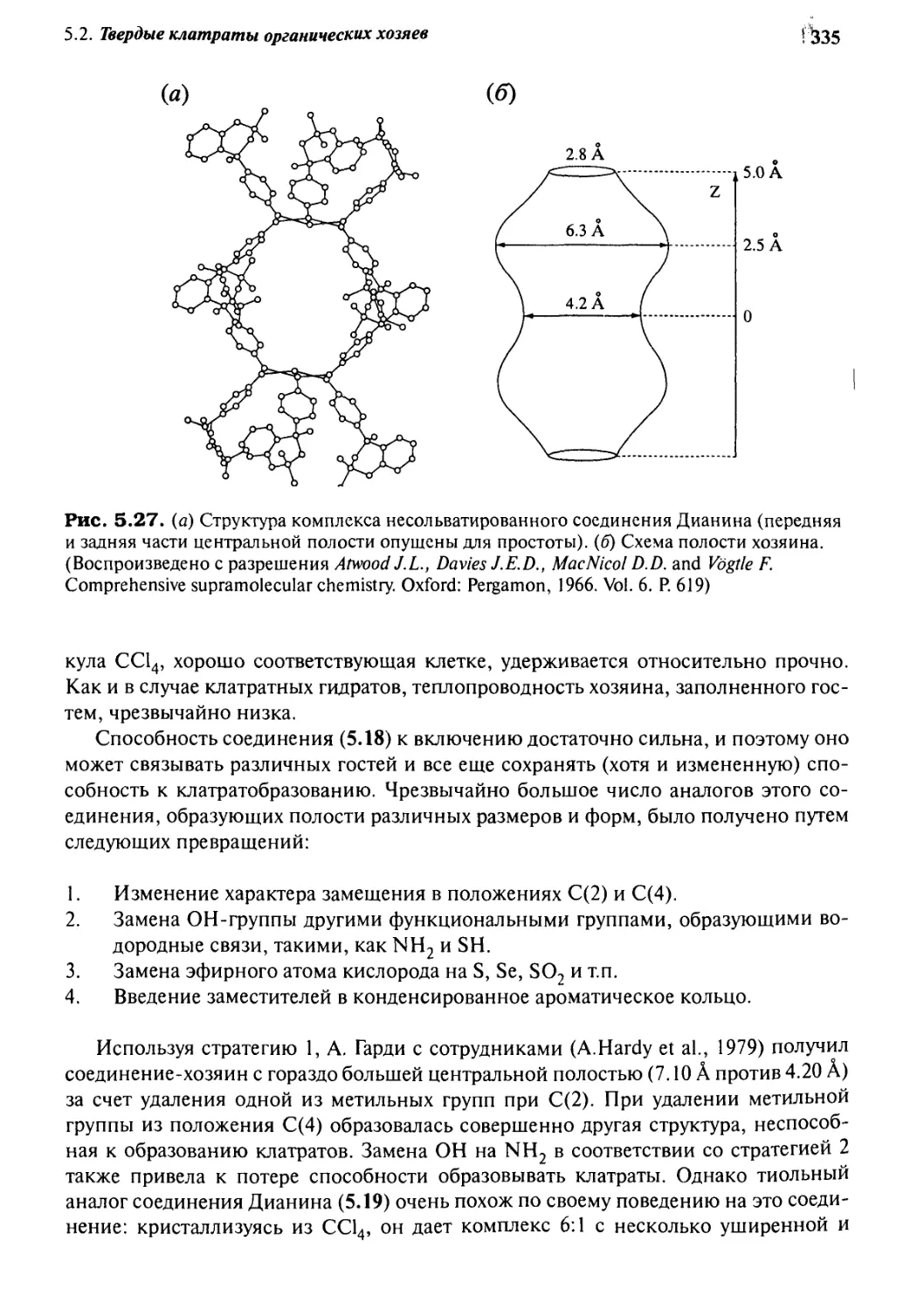
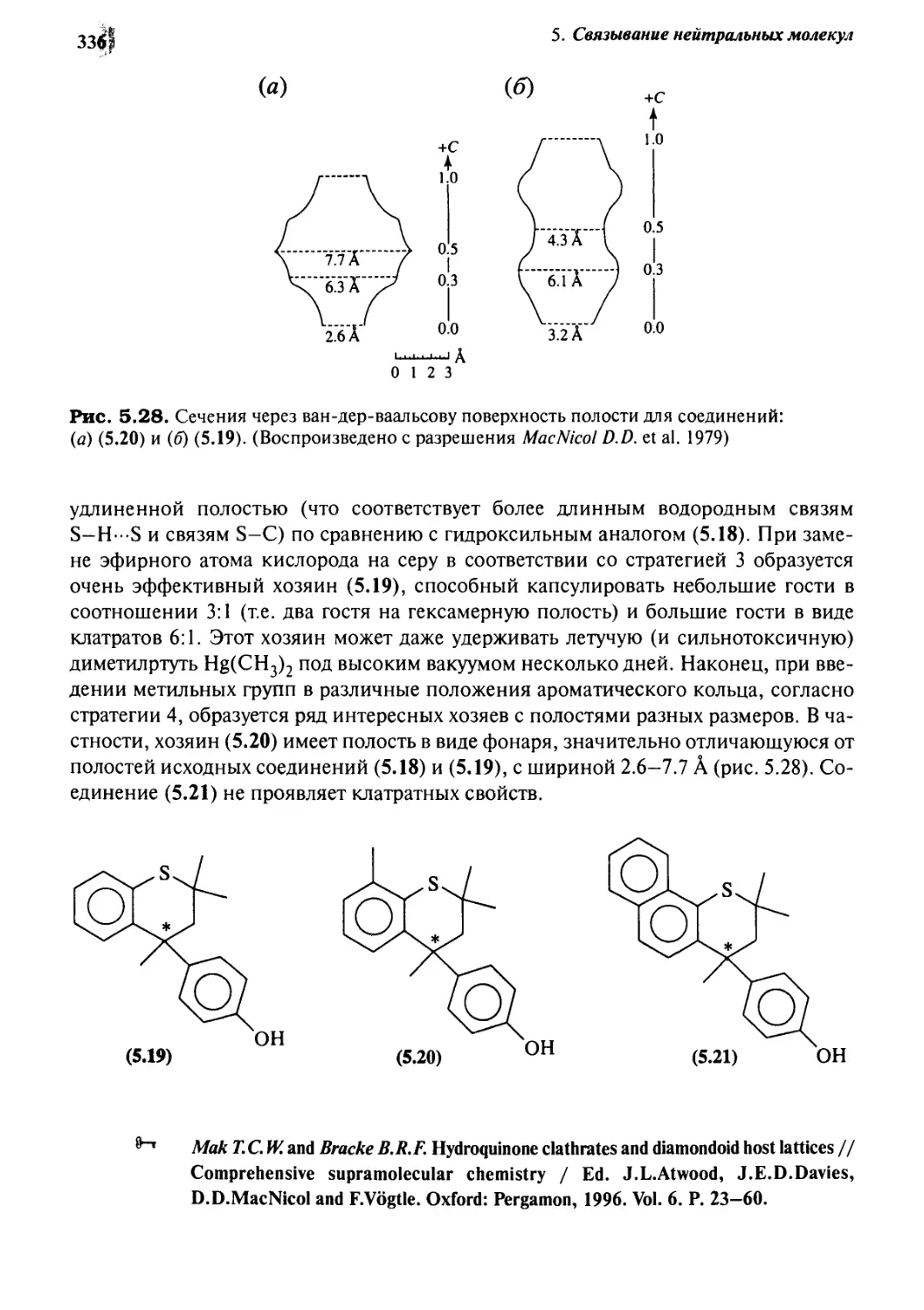
5.2.3 Гидрохинон, фенол и соединение Дианина:
стратегия шести хозяев 332
5.2.4 Три-о-тимотид 337
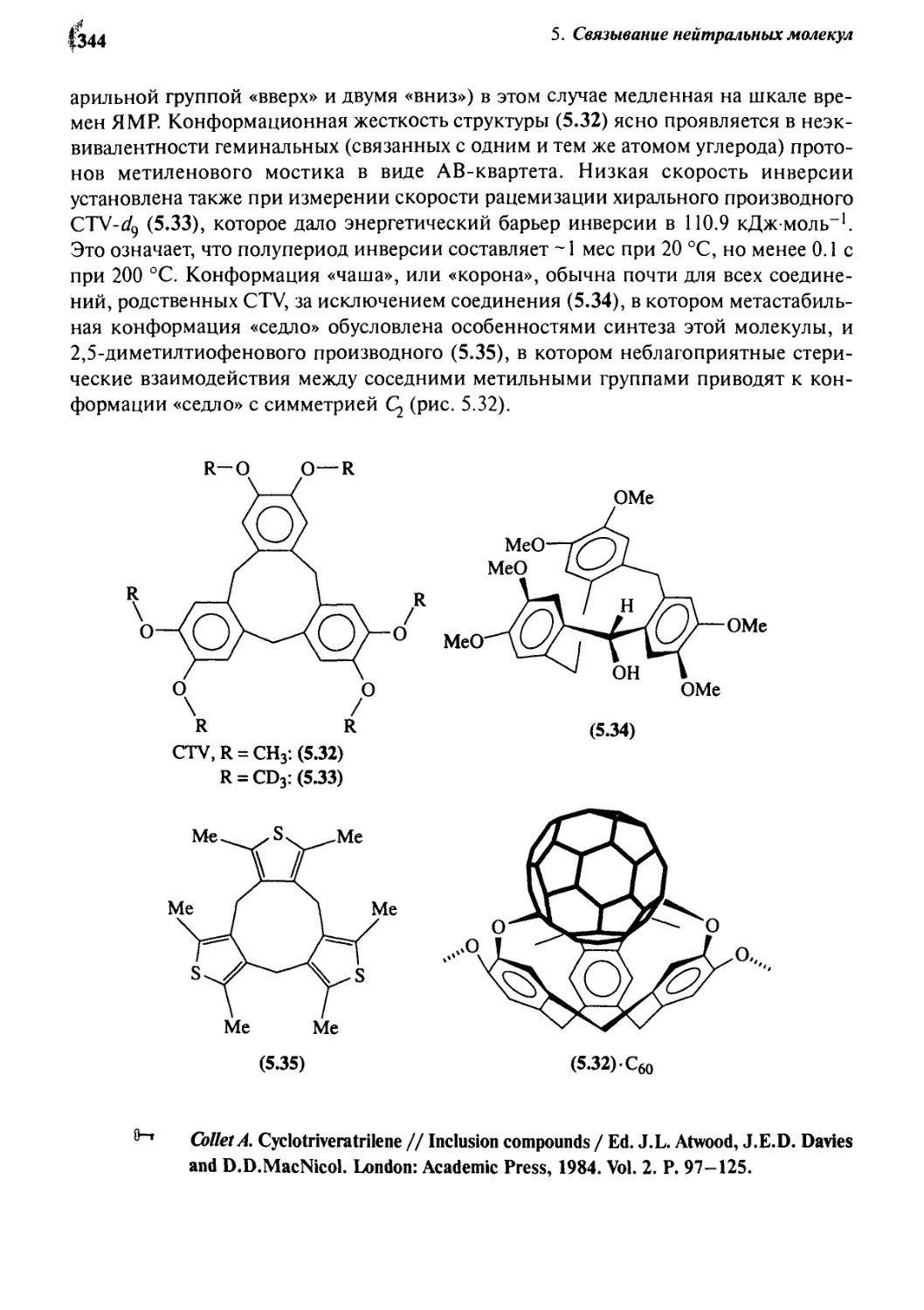
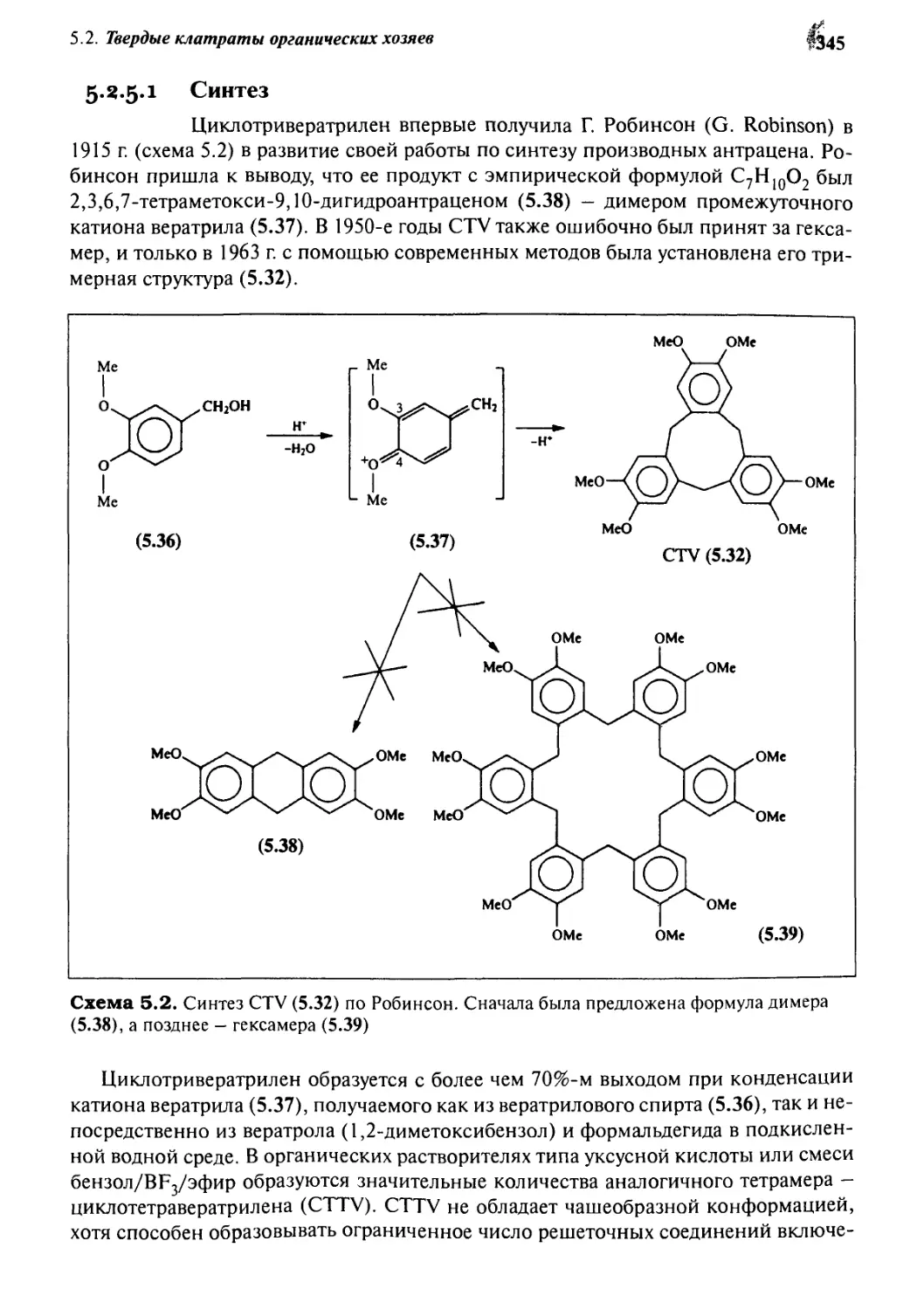
5.2.5 Циклотривератрилен 343
5.2.6 Тетрафенилен 348
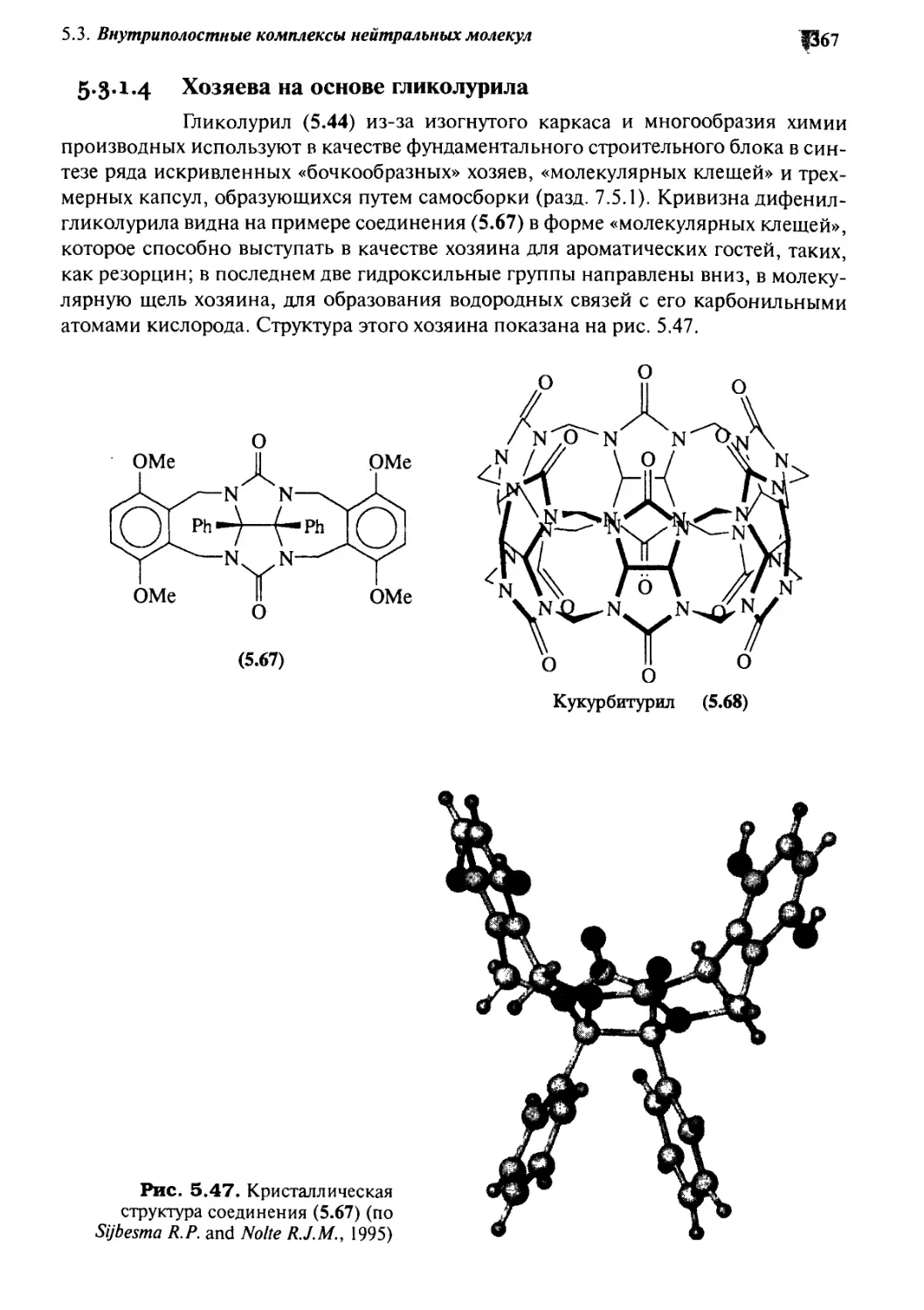
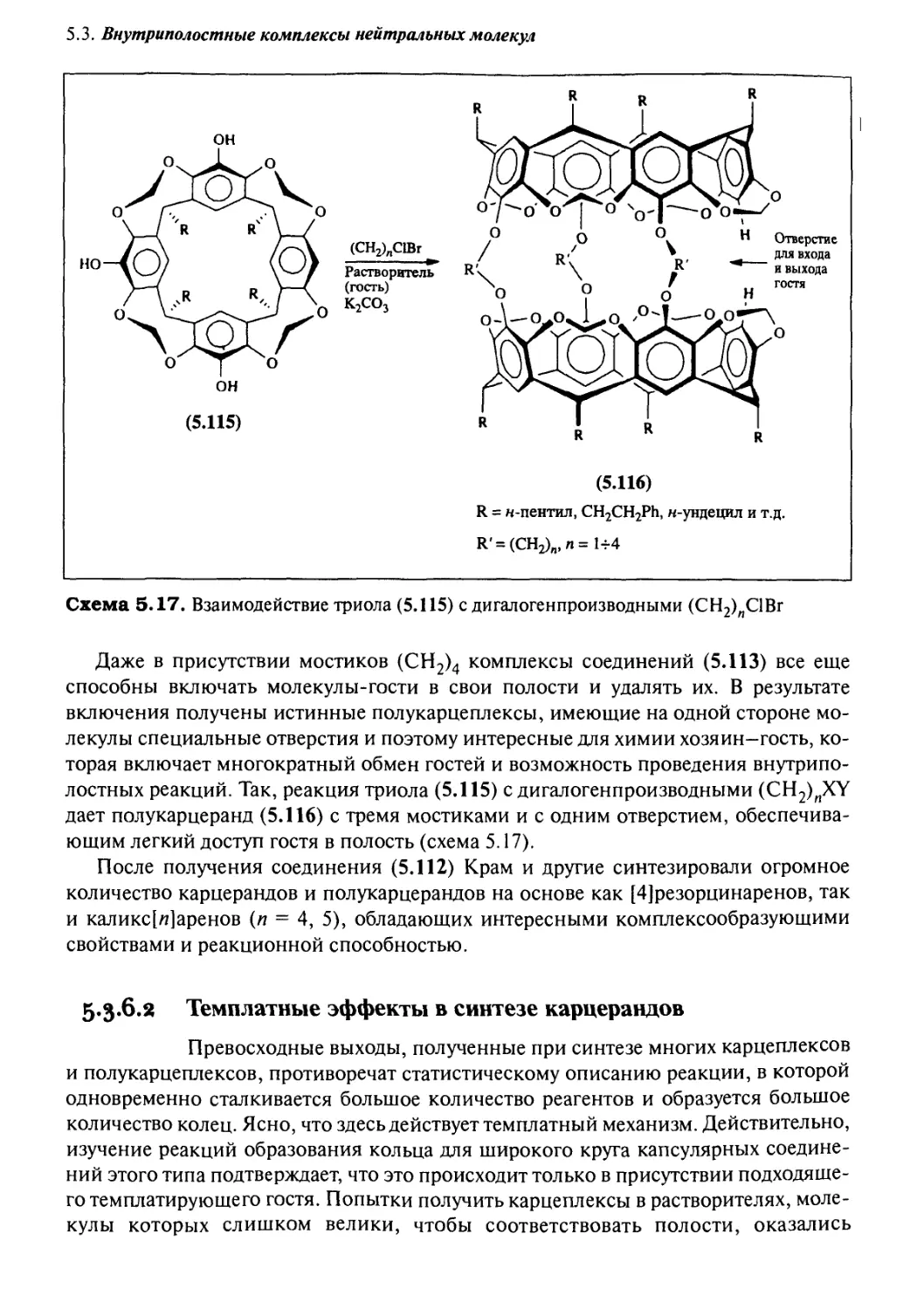
5*3 Внутриполостные комплексы нейтральных молекул:
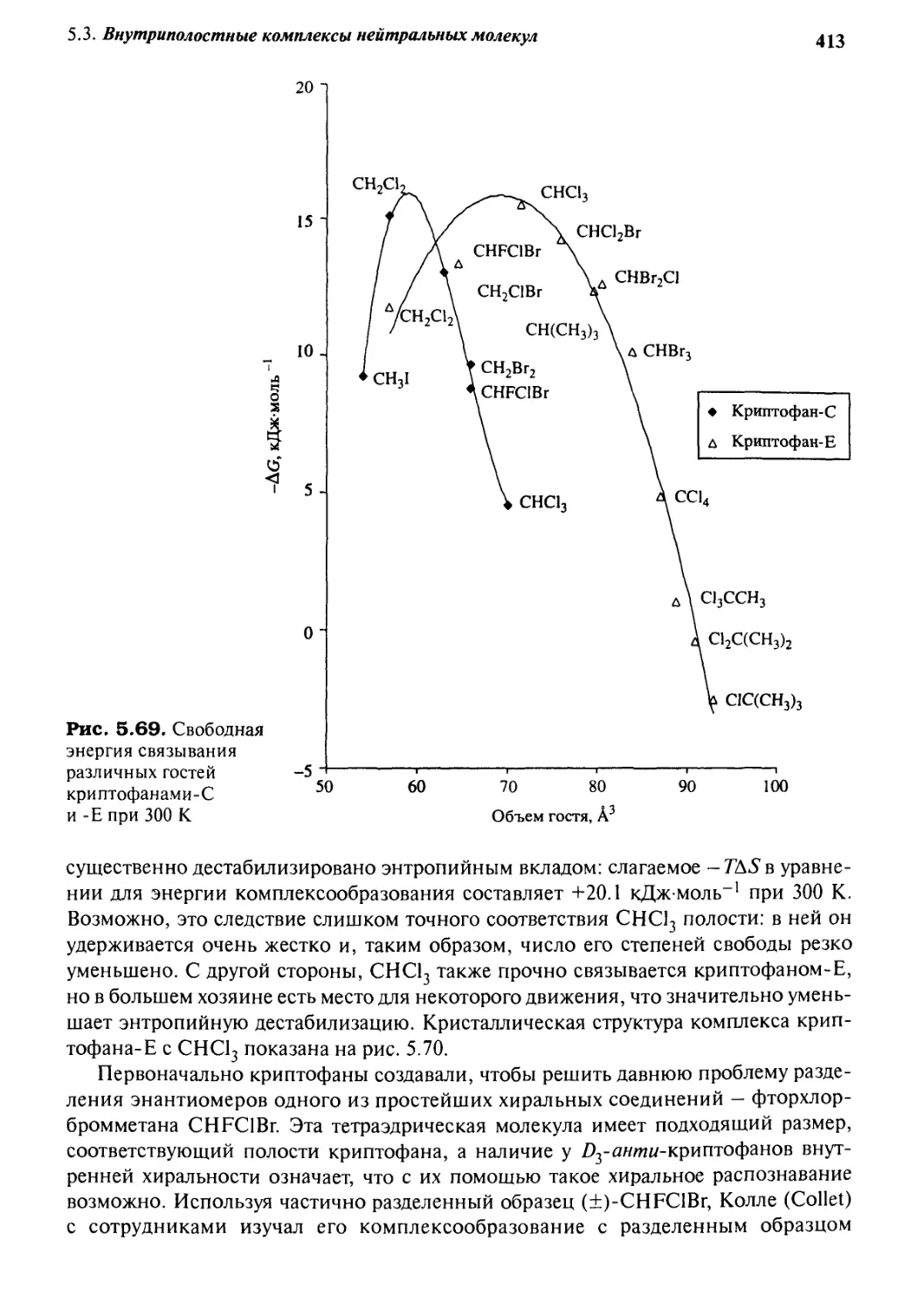
связывание в растворе и в твердом состоянии 353
5.3.1 Внутренняя кривизна хозяев:
связывание гостей кавитандами 354
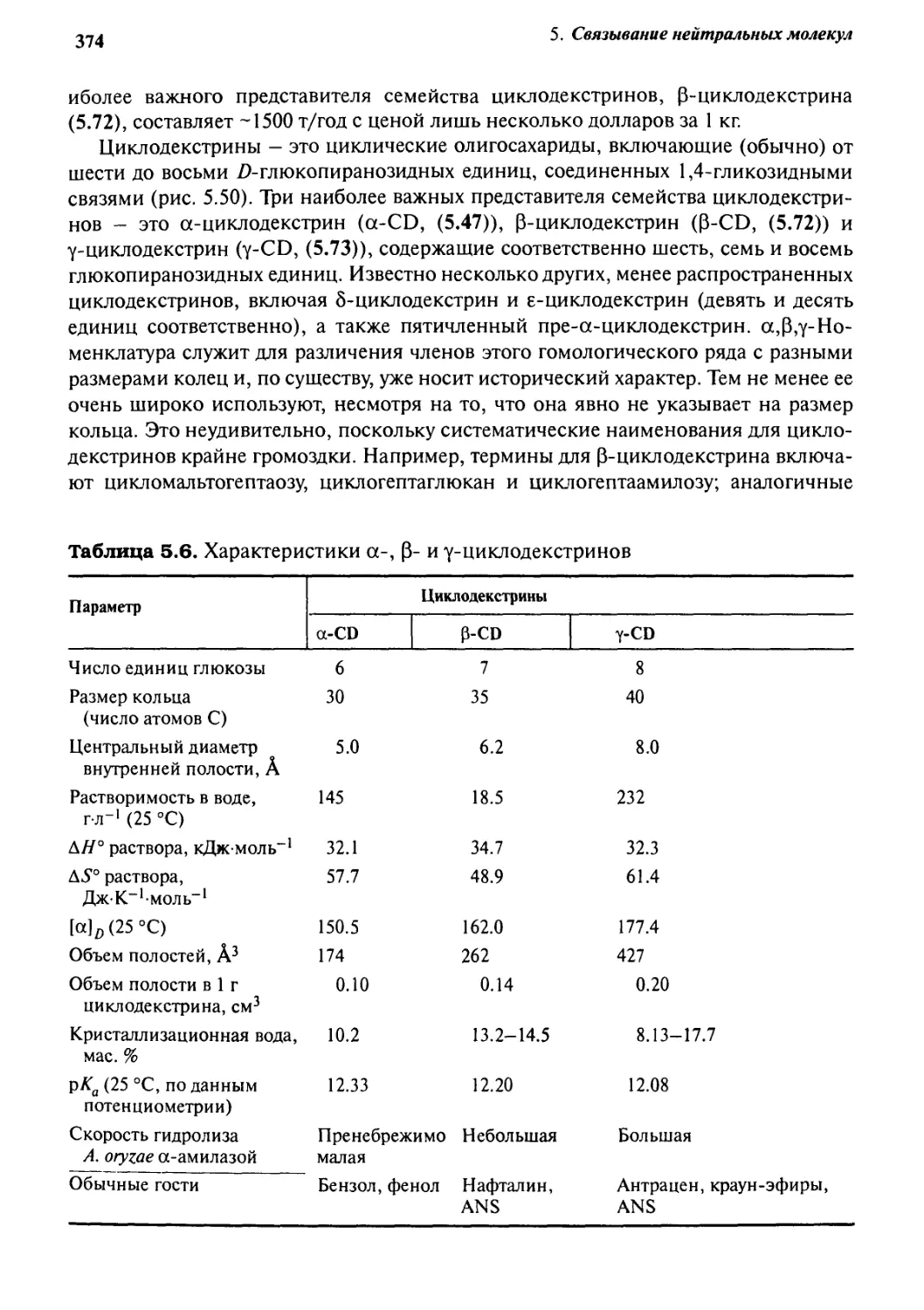
5.3.2 Циклодекстрины 372
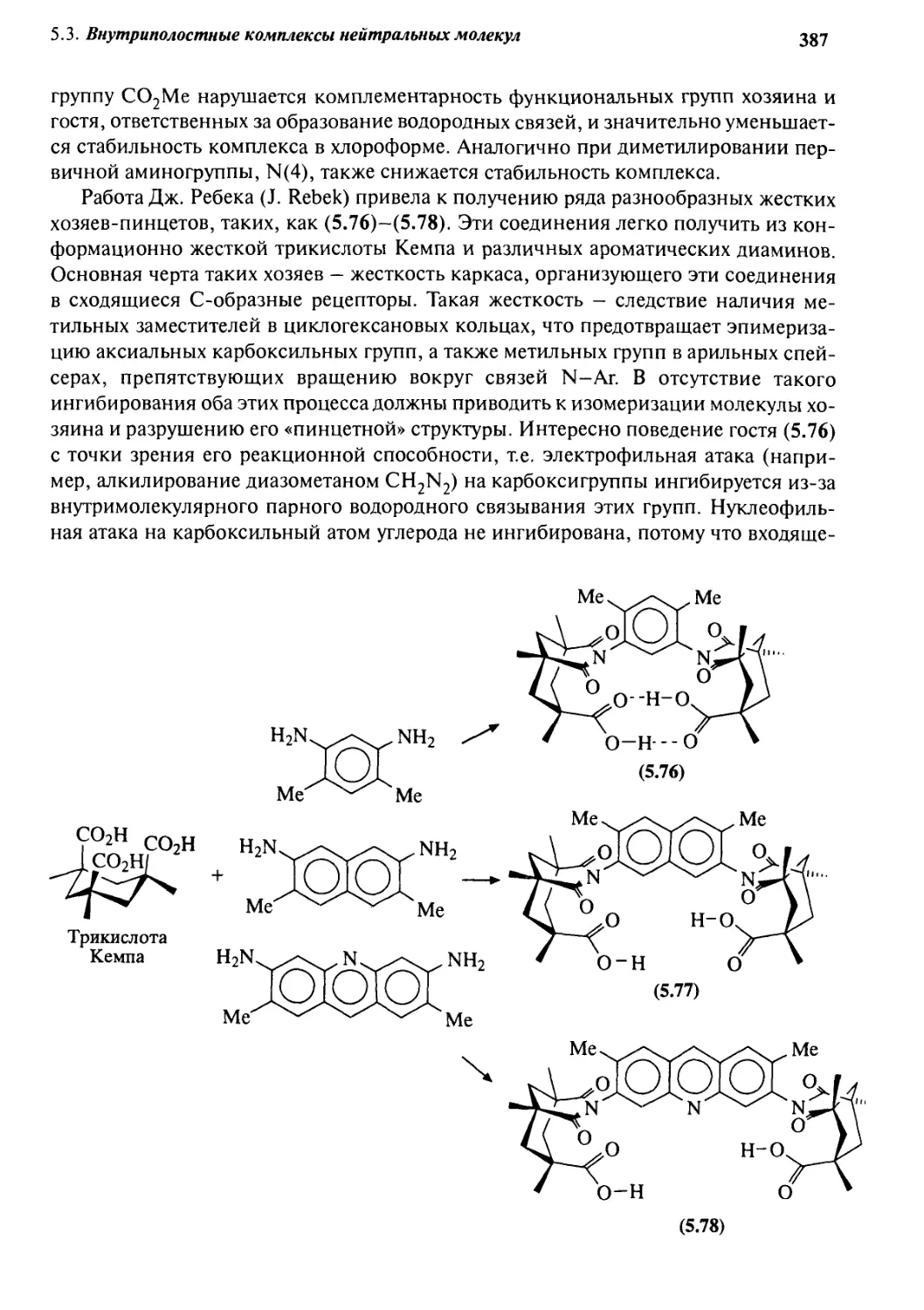
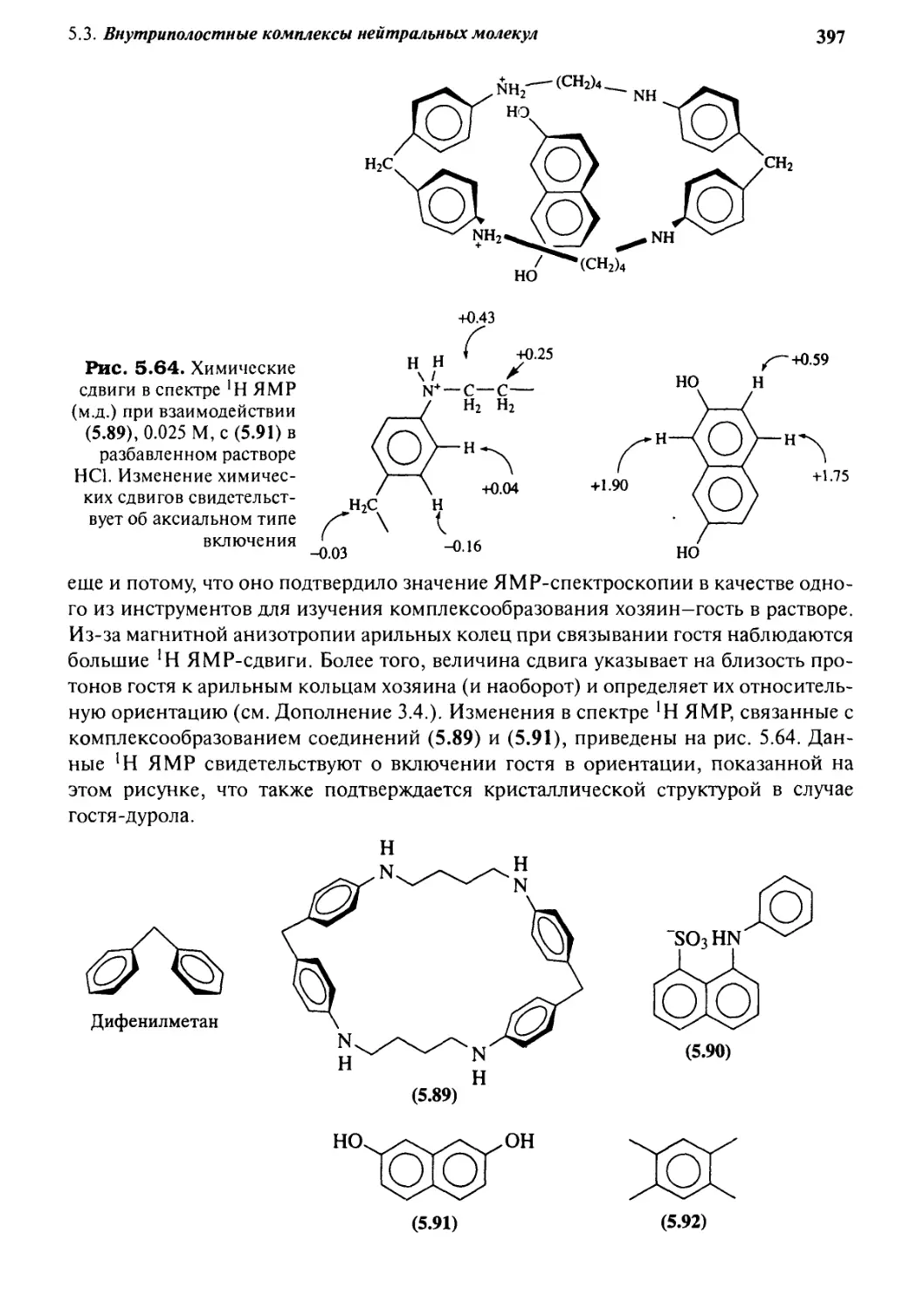
5.3.3 «Молекулярные щели» и «молекулярные пинцеты» 386
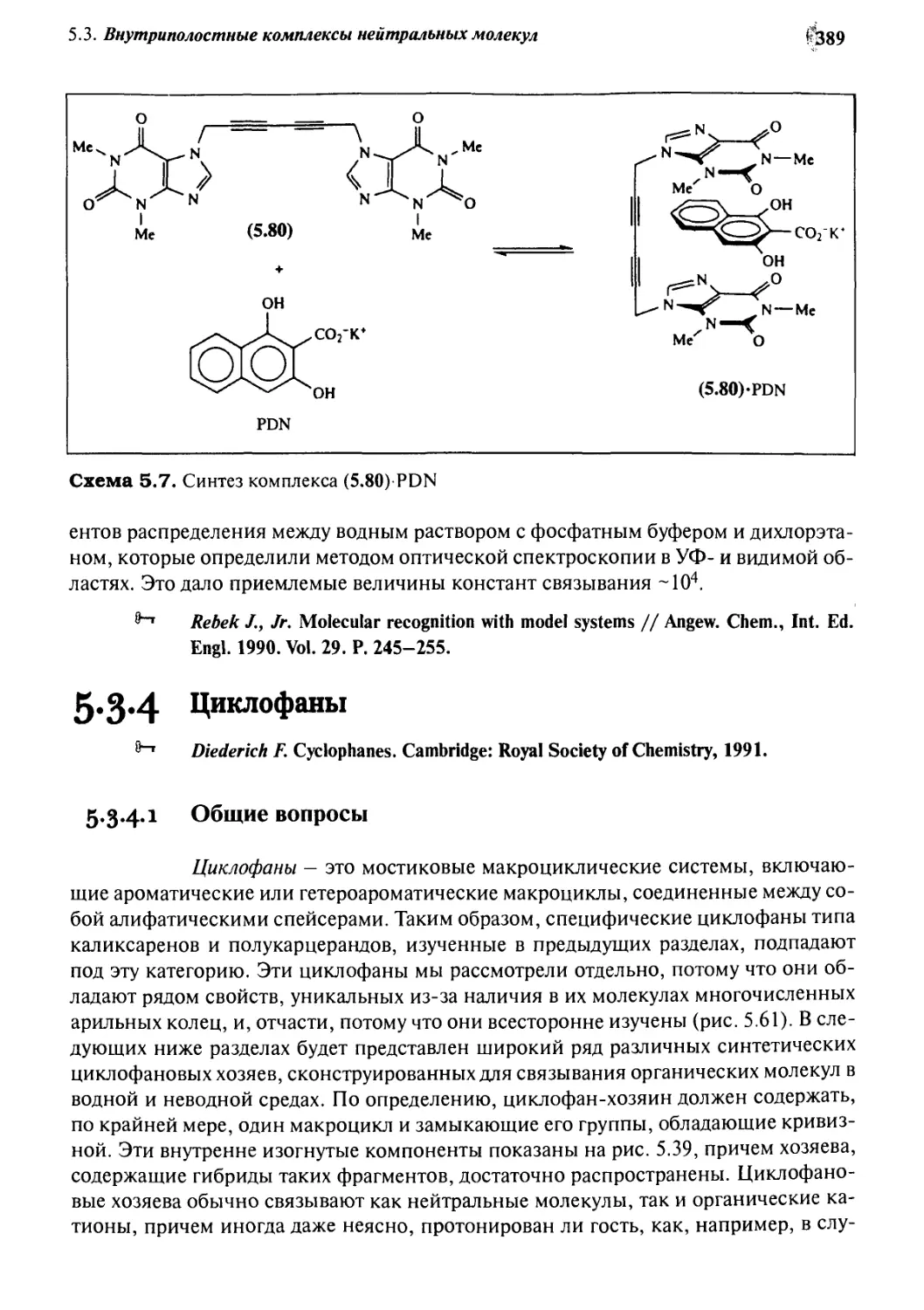
5.3.4 Циклофаны 389
5.3.5 Конструирование хозяина из клатратобразующих
строительных блоков для образования комплексов
в растворе: криптофаны 407
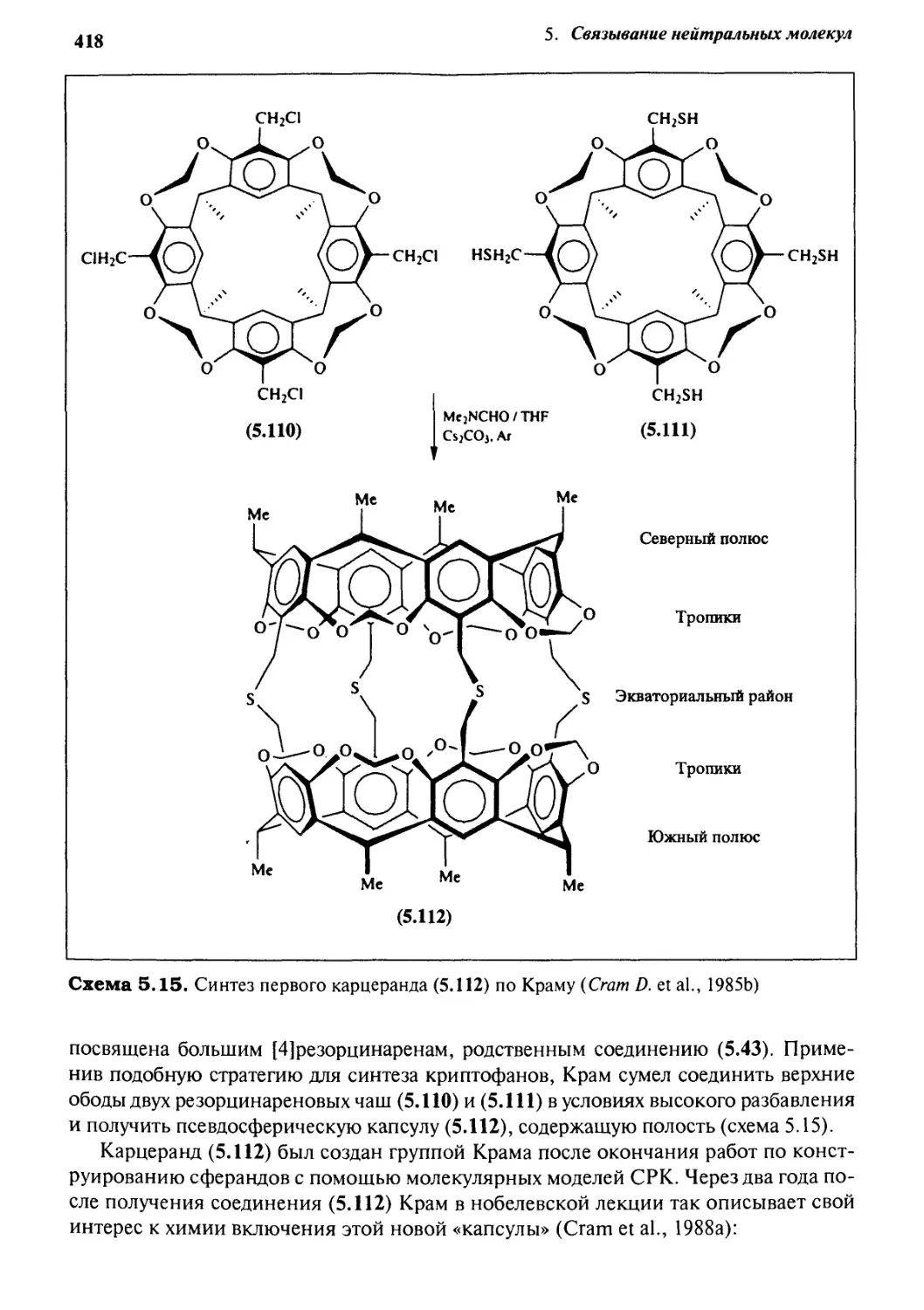
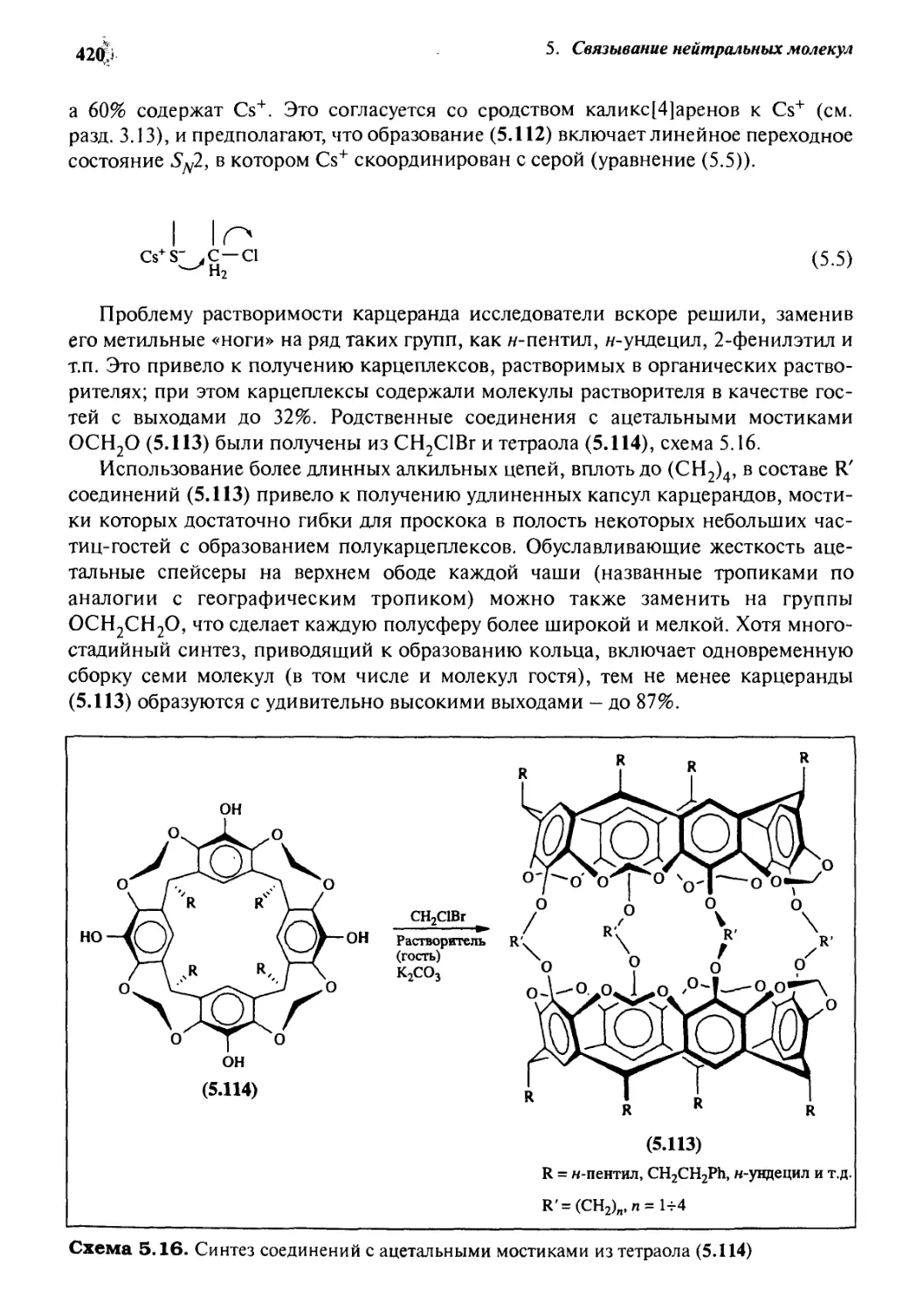
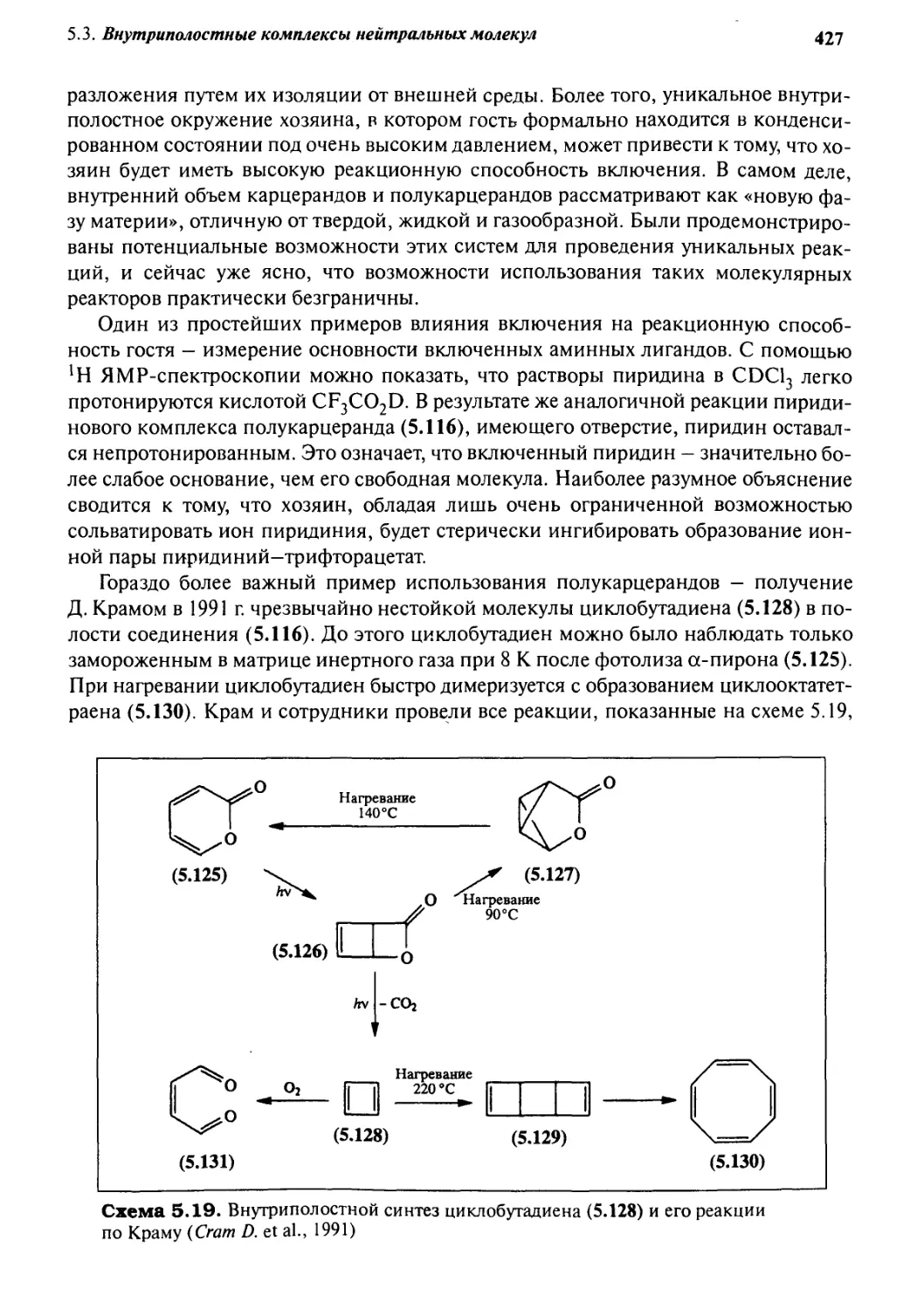
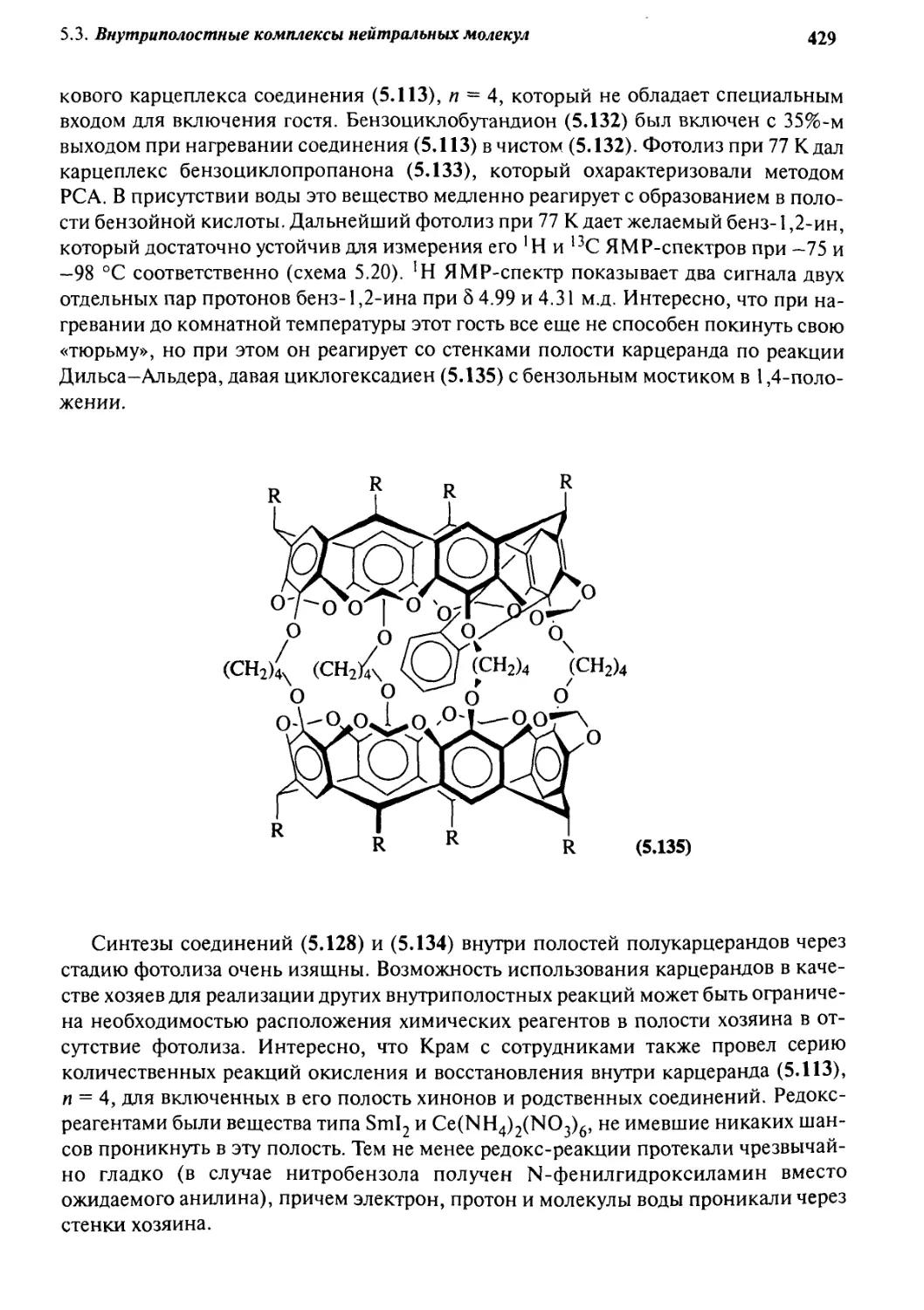
5.3.6 Ковалентные полости: карцеранды и полукарцеранды 417
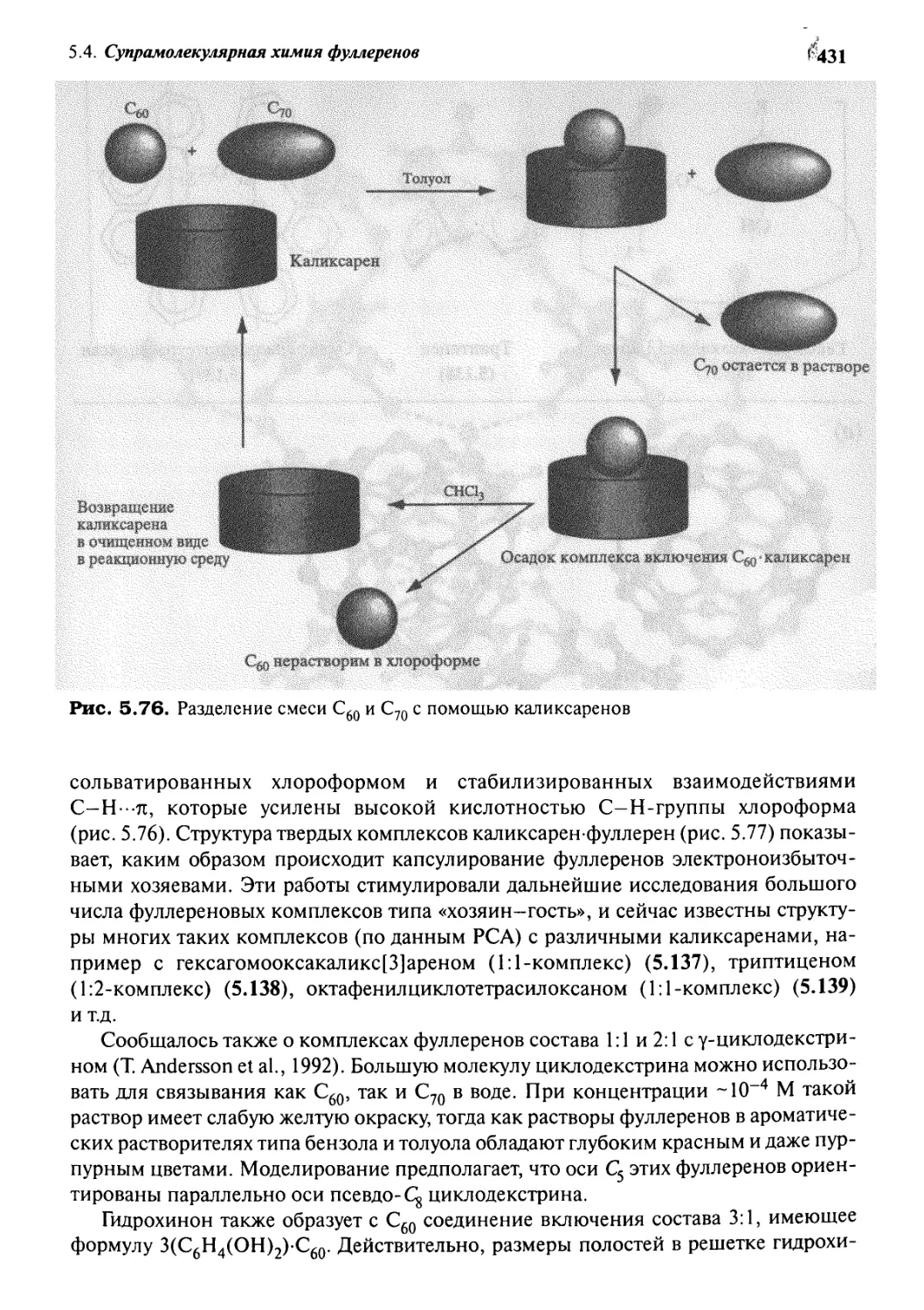
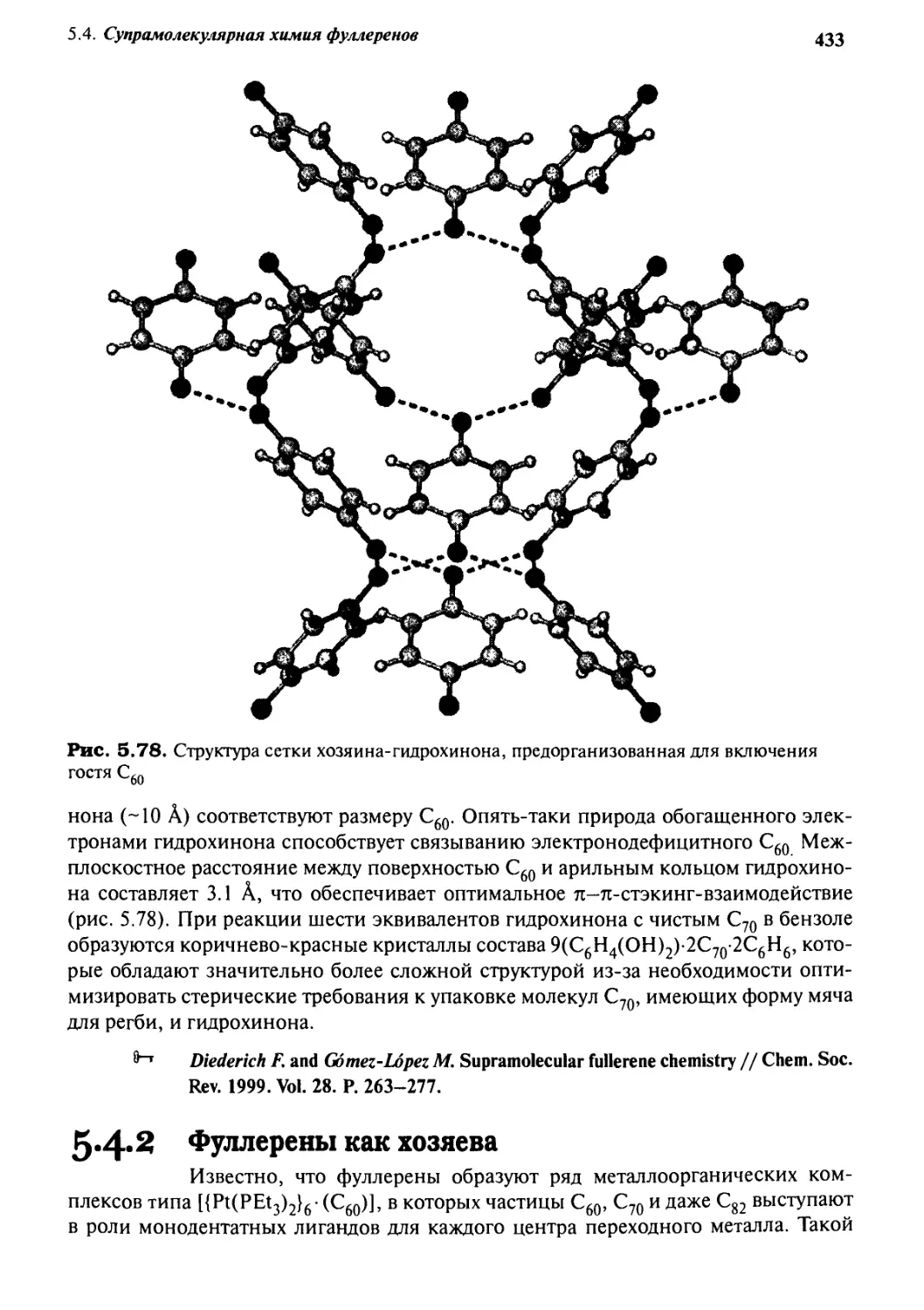
5*4 Супрамолекулярная химия фуллеренов 430
5.4.1 Фуллерены как гости 430
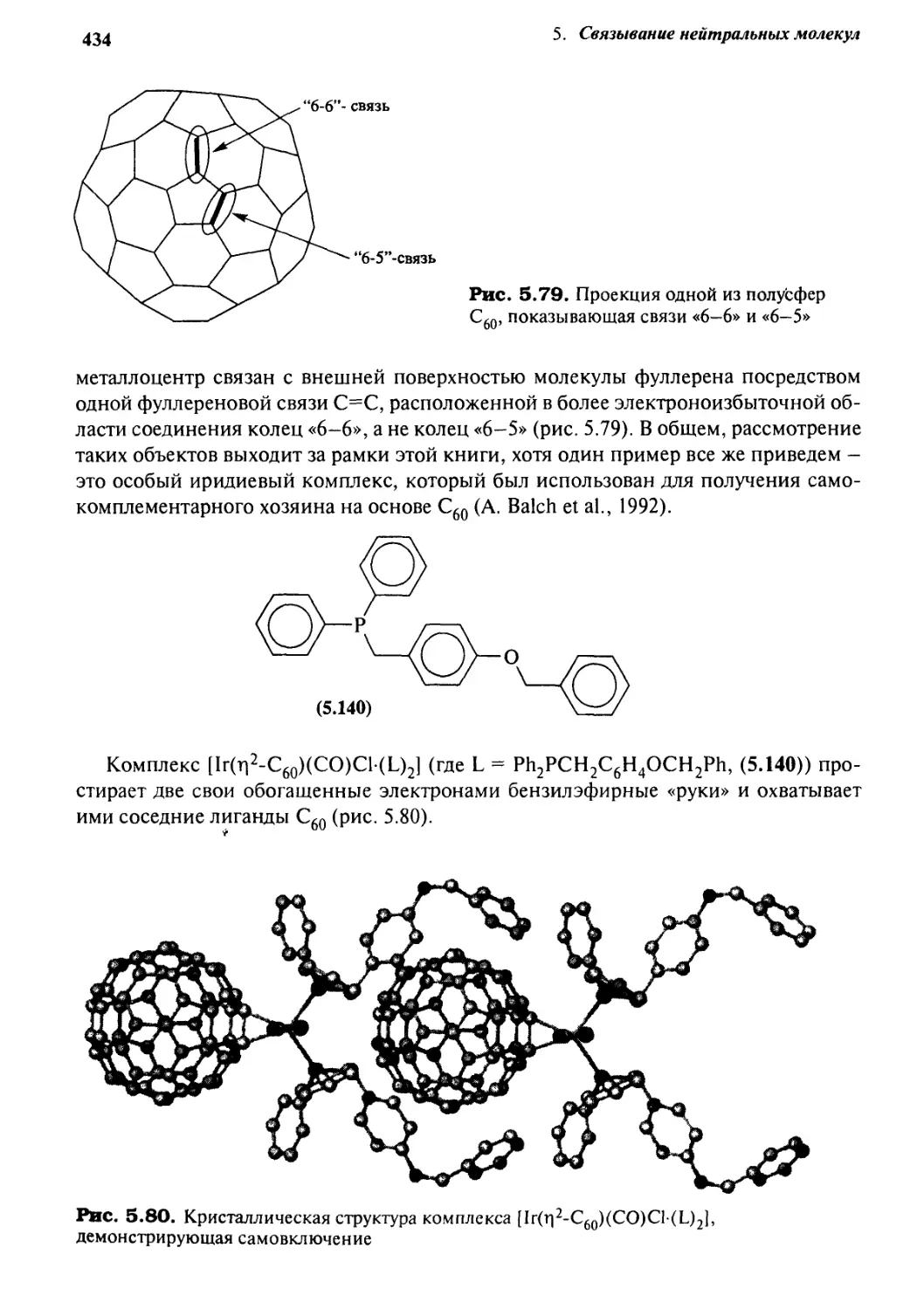
5.4.2 Фуллерены как хозяева 433
IQ Оглавление
5.4.3 Фуллерены как сверхпроводящие соединения
включения 436
Учебные задания 438
Мысленный эксперимент 439
Литература 440
Предметный указатель 444
Том 2
6
6.1
Инженерия кристаллов
Общие вопросы
6.1.1 Введение
6.1.2 Межмолекулярные взаимодействия
6.1.3 Особая роль водородных связей
6.1.4 Анализ набора графов
6.1.5 Рост кристаллов
6.1.6 Разрушение кристаллов
6.1.7 Стратегии дизайна при инженерии кристаллов
Предсказание структуры кристаллов
6.2.1 Расчетные методы
6.2.2 Поавила Эттео
491
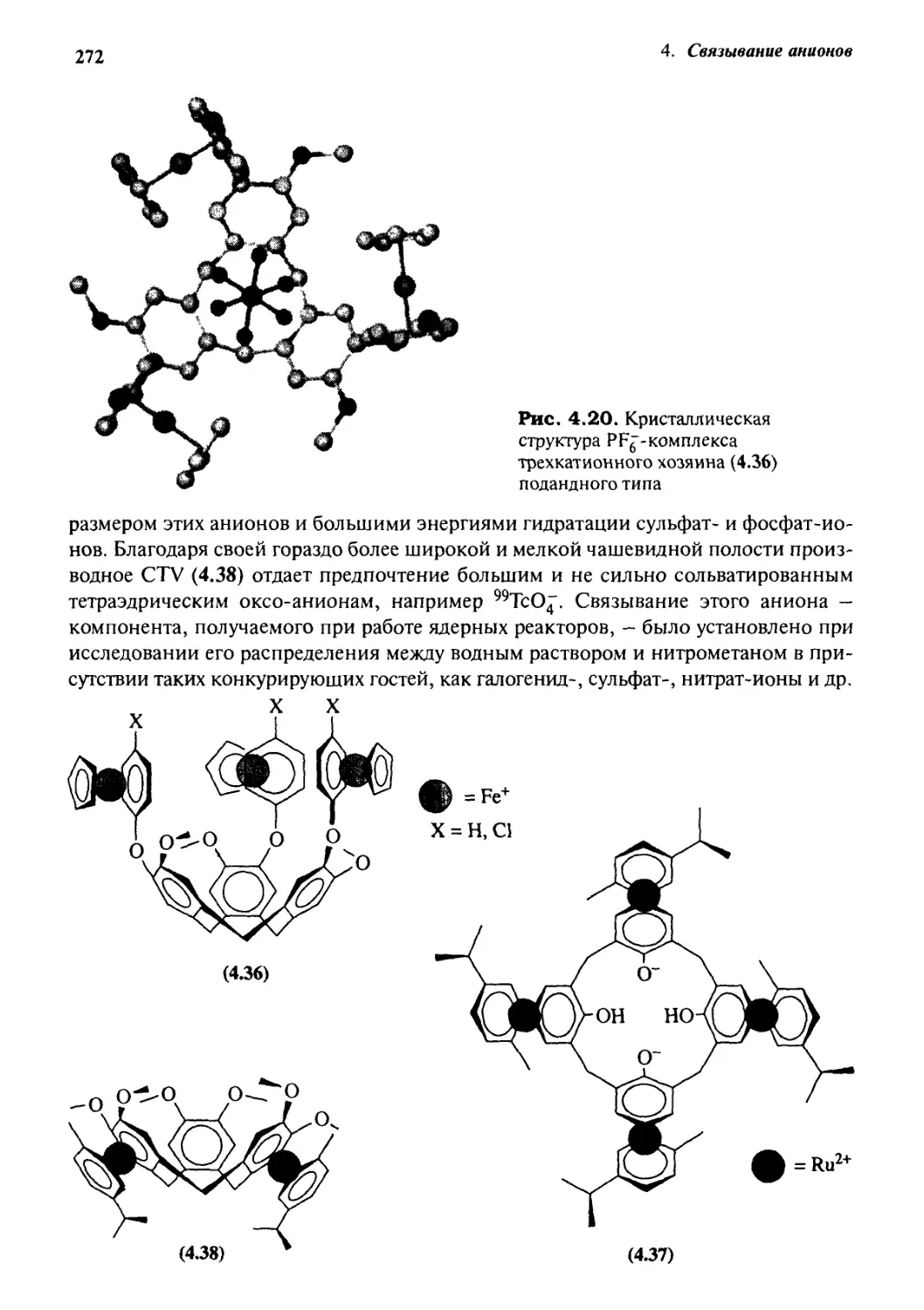
491
491
494
494
501
504
514
515
517
517
521
6.g Кембриджский банк структурных данных 525
6*4 Инженерия кристаллов с алмазоподобными
решетками 529
6. К Инженерия кристаллов с водородными связями 535
6.5.1 Димеры карбоновых кислот 535
6.5.2 Амиды 535
6.5.3 Спирты 536
6.5.4 Смешанные кристаллы спиртов и аминов 540
6.5.5 Смешанные кристаллы аминов с аминами 541
6.6 Водородные связи монооксида углерода 542
Слабые водородные связи 544
6.8 Водородные связи с металлами и гидридами металлов 548
6.8.1 Прямое взаимодействие с металлами 548
6.8.2 Взаимодействие с гидридами металлов 549
6.9 я-я-Стэкинг-взаимодействия 551
Оглавление \ Щ
6.10 Прочие взаимодействия 554
6.11 «Неправильные» формы и «неправильный» подбор 555
6.11.1 Хозяева типа «колесо—ось» 555
6.11.2 «Неправильно» подобранные партнеры 556
6 Л 2 Координационные полимеры 558
Биомиметические (биоподражательные) структуры 562
Смешанные кристаллы: включения типа
«песочные часы» 567
Учебные задания 572
Мысленный эксперимент 574
Литература 574
6.14
у Темплаты и самосборка
576
• 1 Введение 576
7.1.1 Цели и задачи 576
7.1.2 Терминология 578
♦2 Биохимическая самосборка 580
7.2.1 Вирус табачной мозаики 580
7.2.2 Строгая самосборка 581
7.2.3 Самосборка с ковалентной модификацией 583
•3 Самосборка в синтетических системах:
кинетический и термодинамический подходы 584
7.3.1 Темплатные эффекты в синтезе 584
7.3.2 Термодинамическая модель:
самосборка порфириновых комплексов цинка 588
.4 Самосборка координационных соединений 592
7.4.1 Принципы дизайна 593
7.4.2 Супрамолекулярный куб 593
7.4.3 «Молекулярные квадраты» и «молекулярные коробки» 597
7.4.4 Металлические ансамбли 609
7*5 Самосборка закрытых комплексов
с помощью водородных связей 613
7.5.1 «Теннисные мячики» и «мячи для софтбола» 613
7.5.2 Гигантские самособирающиеся капсулы 619
7.5.3 Розетки 625
12
Оглавление
.6 Катенаны и ротаксаны 628
7.6.1 Введение 628
7.6.2 Статистический подход
к синтезу катенанов и ротаксанов 632
7.6.3 Псевдоротаксаны 633
7.6.4 Ротаксаны 636
7.6.5 Катенаны, формируемые с помощью
тс-тс-стэкинг-взаимодействий 637
7.6.6 Принцип «вспомогательной связи» в синтезе катенанов 648
•*7 Геликаты 660
7.7.1 Введение 660
7.7.2 Синтетические аспекты 663
7.7.3 ]4+4] Геликаты 665
7.7.4 [6+6] Геликаты 667
7.7.5 Самораспознавание и позитивная кооперативность 669
7.7.6 Наноциклы 675
7.7.7 Спирали, образованные водородными связями 676
у.8 Молекулярные узлы 679
y.U Каталитические и самовоспроизводящиеся системы 685
Учебные задания 690
Мысленный эксперимент 691
Литература 692
8
Молекулярные устройства 695
8.1 Введение 695
8.1.1 Философия молекулярных устройств 695
8.1.2 Что такое супрамолекулярное устройство? 696
8.2 Супрамолекулярная фотохимия 698
8.2.1 Основы фотохимии 698
8.2.2 Биметаллические системы
и системы со смешанной валентностью 702
8.2.3 Бипиридил и его друзья 704
8.2.4 Фото- и электрохимические устройства
на основе бипиридила 707
8.2.5 Устройства для преобразования света 711
8.2.6 Нековалентно связанные системы 712
Оглавление iljy
8.3 Информация и сигналы: семиохимия 716
8.3.1 Фотохимические сенсоры 717
8.3.2 Флуорофоры 721
8.3.3 Электрохимические сенсоры 724
8.4 Молекулярные электронные устройства:
переключатели, провода и выпрямители 727
8.4.1 Молекулярные провода 727
8.4.2 Молекулярные выпрямители 732
8.4.3 Система 1,2-дитиенилэтена как переключатель 734
8.4.4 Электропереключаемая люминесценция 736
8.4.5 Переключаемое связывание 737
8.4.6 Аллостерические переключатели 739
8*5 Молекулярные машины
на основе катенанов и ротаксанов 740
8.6 Материалы для нелинейной оптики 746
8.6.1 Основы нелинейной оптики 746
8.6.2 Материалы для нелинейной оптики второго порядка 749
8.6.3 Материалы для нелинейной оптики третьего порядка 753
8 Дендримеры 754
8.7.1 Получение и свойства 756
8.7.2 Химия хозяин-гость для дендримеров 759
8.7.3 Фотохимические устройства на основе дендримеров 760
Учебные задания 764
9
Литература 765
Биомиметика 767
. 1 Введение 767
9.1.1 Супрамолекулярная биохимия 767
9.1.2 Характеристики биологических моделей 768
.2 Характеристики ферментов 770
9.2.1 Определение и структура 770
9.2.2 Механизм ферментативного катализа 772
.g Циклодекстрины как имитаторы ферментов 775
9.3.1 Моделирование ферментов
с использованием циклодекстринов как хозяев 776
9.3.2 Циклодекстрины как имитаторы эстераз 778
9.3.3 Функционализированные циклодекстрины 780
14 Оглавление
9*4 Коранды как имитаторы АТРаз 782
Q.g Хозяева, связьшающие катионы,
как имитаторы трансацилаз 786
9.5.1 Хиральные коранды 786
9.5.2 Имитация структуры и функции 789
0.6 Металлобиоцентры 792
9.6.1 Модели гемоцианина 793
9.6.2 Цинксодержащие ферменты 795
Q,^ Аналоги гема 797
9.7.1 Модели связывания и транспорта кислорода 797
9.7.2 Модели цитохромов Р-450 804
Q.8 Модели витамина В12 809
Учебные задания 810
Мысленный эксперимент 811
Литература 811
1О Жидкие поверхности раздела,
жидкие кристаллы
и жидкие клатраты т
ЮЛ Порядок в жидкостях 813
1O.2 Поверхностно-активные вещества
и упорядочение на поверхности раздела 814
lO.g Жидкие кристаллы 820
10.3.1 Природа и структура 820
10.3.2 Дизайн жидкокристаллических материалов 831
10.3.3 Жидкокристаллические полимеры 834
10.3.4 Жидкокристаллические дисплеи 835
1О«4 Жидкие клатраты 837
10.4.1 Открытие и свойства 837
10.4.2 Ион оксония в химии жидких клатратов 842
Учебные задания 845
Литература 846
Предметный указатель 847
Дополнительная литература 883
Предисловие авторов
к русскому изданию
Со времени опубликования в 2000 г. нашей книги «Супрамолекулярная химия»
(Chichester, «J. Wiley & Sons») прошло шесть лет. Мы удовлетворены тем, как эта
книга была принята читателями, и благодарны нашим коллегам во всем мире за
доброжелательные замечания и предложения. За последние пять-шесть лет на полках
книжных магазинов появилось несколько других замечательных книг* по супрамо-
лекулярной химии, в том числе «Законы и методы супрамолекулярной химии»
Х.-Дж. Шнайдера и А. Яцимирского, «Молекулярные устройства и машины:
Путешествие в наномир» В. Бальзани, М. Вентури и А. Креди и пользующийся успехом
учебник П. Бира, Ф. Гэйла и Д. Смита «Супрамолекулярная химия». Мы выпустили
двухтомную «Энциклопедию супрамолекулярной химии»**. Такой уровень
активности свидетельствует о неослабевающем интересе к этой дисциплине и ее
постоянном развитии.
Известие о подготовке русского издания нашей книги «Супрамолекулярная
химия», инициированной директором Института физической химии и электрохимии
им. А.Н. Фрумкина Российской академии наук академиком А.Ю. Цивадзе,
доставило нам особое удовольствие. Для осуществления этого издания ответственный
редактор профессор А.Ю. Цивадзе объединил российских специалистов по
супрамолекулярной химии: ответственных редакторов профессора В.В. Арсланова и
профессора А.Д. Гарновского, а также переводчиков — И.Г. Варшавскую, Б.И. Харисо-
ва, О.В. Белуженко, И.С. Васильченко, Ю.А. Алексеева. Мы выражаем всем им
свою признательность за их тяжелый труд и преданность делу. Понятно, что из-за
незнания русского языка наш вклад в подготовку этого издания пренебрежимо мал!
Нам бы хотелось также выразить свою признательность Российскому фонду
фундаментальных исследований за финансовую поддержку проекта.
Научное содержание настоящего издания практически не отличается от
содержания английского оригинала. Безусловно, со времени опубликования книги
«Супрамолекулярная химия» в этой области был достигнут значительный прогресс,
однако фундаментальные законы и большинство соединений рассмотренных
классов остаются неизменными. Мы верим, что эта книга принесет существенную
пользу как студентам, так и людям, искушенным в этой области химии.
Джонатан В. Стид
(Дарем, Великобритания)
Джерри Л. Этвуд
(Миссури, США)
Октябрь, 2005 г.
Schneider H.-J., Yatsimirsky A. Principles and methods in supramolecular chemistry
(Chichester, Wiley, 1999); Balzani V., Venturi M., Credi A. Molecular devices and machines: A
journey into the nanoworld (Weinheim, Wiley-VCH, 2003); Beer P., Gale Ph., Smith D. Supramolecular
chemistry (Sen "Oxford chemistry primers", 1999).
** Steed J.W., Atwood J.L. Encyclopedia of supramolecular chemistry (M., Dekker, 2004).
Предисловие редакторов
русского издания
С момента выхода в 1998 г. на русском языке блестящей монографии лауреата
Нобелевской премии в области химии Ж.-М. Лена «Супрамолекулярная химия:
Концепции и перспективы»* в переводе Е.В. Болдыревой под редакцией
В.В. Власова и А.А. Варнека (Новосибирск, «Наука», 1998) книги по этой тематике
в России не издавались.
Настоящая монография известных специалистов в области супрамолекулярной
химии Дж.В. Стида и Дж.Л. Этвуда призвана заполнить этот пробел, к сожалению,
затянувшийся по времени. Предлагаемая читателю книга ни в коей мере не заменяет
монографию Ж.-М. Лена, в которой изложены концептуальные проблемы
супрамолекулярной химии. Настоящее издание, как увидит читатель, содержит большой
конкретный материал: множество примеров известных на сегодняшний день соединений
(лиганды и комплексы), образующих супрамолекулярные системы разного уровня
сложности. Оно включает сведения из смежных областей знаний, особенно из
биохимии. Рассмотрены основы методов, наиболее широко применяемых для изучения су-
прамолекулярных структур. Хотя по своему уровню это издание ближе к монографии,
его структура, а также наличие примеров, задач и мысленных экспериментов, которые
позволяют читателю проверить свои знания и закрепить пройденный материал,
приближают книгу к учебнику по супрамолекулярной химии, столь необходимому в
настоящее время студентам, аспирантам, преподавателям вузов и научным работникам.
Как и любая область знания, супрамолекулярная химия использует новые,
часто образные, термины и понятия, которым необходимо находить адекватные для
русского языка слова и выражения. К счастью, нам было легче, чем переводчику и
редакторам книги Ж.-М. Лена, проделавшим большую работу по подбору русских
эквивалентов и выверке терминов. Следует также подчеркнуть, что в настоящем
издании собраны практически все новые лиганды и комплексы, интересные с точки
зрения супрамолекулярной химии, приведены их структурные формулы, что
позволяет рассматривать книгу как справочное пособие по новым, весьма сложным, а
порой и причудливым, в большинстве макроциклическим соединениям.
Настоящая книга требует определенной подготовки и при этом в соответствии с
междисциплинарным характером супрамолекулярной химии предусматривается,
что читатель уже приобрел достаточные знания в области физической химии,
органической химии, химии комплексных соединений и биохимии.
Естественно, шесть лет, прошедшие с момента выхода англоязычного издания
монографии, супрамолекулярная химия не стояла на месте. Однако основные
положения этой области знания, а также метод исследования не претерпели
принципиальных изменений, в то время как круг объектов исследования расширился.
Сейчас супрамолекулярная химия, которая стала в последние годы одной из наиболее
интенсивно развивающихся областей химии, признается в качестве приоритетного
направления и входит в лекционные курсы ведущих российских и зарубежных
вузов. Появление этой книги в России представляется нам весьма своевременным.
А. Ю. Цивадзе, В. В. Лрсланов, А.Д. Гарновский
* Lehn J.-M. Supramolecular chemistry: Concepts and perspectives. Weinheim: VCH, 1995.
Предисловие авторов
Бену и Джошу:
невозможно узнать слишком много.
Супрамолекулярная химия — одна из наиболее популярных, стремительно
развивающихся областей экспериментальной химии. Кто только ею не занимается! Она
привлекает исследователей своей наглядностью и позволяет истолковывать
обычные понятия на молекулярном уровне. По своей природе она крайне
междисциплинарна. Химики, биохимики, биологи,экологи, инженеры, физики, математики и
ученые других специальностей, занимающиеся супрамолекулярной химией,
разрушают традиционные границы своих дисциплин и вместе идут к определенной цели.
Чрезвычайный размах этих исследований и породил среди специалистов шутку о
том, что «в наши дни все вокруг кажется супрамолекулярным». Эта шутка сродни
знаменитой шутке некоторых неоргаников, определяющих свою область как
«химию всех элементов за исключением какого-то там углерода».
Несмотря на огромное число исследователей, работающих в области
супрамолекулярной химии, специалистов, которые, действительно, могут научить ее
премудростям, не так много. В настоящее время большинство практиков в этой области,
включая авторов этой книги, являются самоучками, пополняющими свои знания в
процессе работы. Чтобы помочь своим дипломникам и аспирантам, мы
организовали в наших университетах курсы по супрамолекулярной химии. Оказалось, что
людей, интересующихся этой дисциплиной, множество, а достойного учебника нет.
Его заменяла монография Ф. Фёгтле (Chichester, «J. Wiley & Sons», 1991).
Первоначально идея о создании книги возникла в разговоре между нами летом 1995 г. в
Миссури. В то время курс по супрамолекулярной химии входил в программу лишь
нескольких университетов, но было ясно, что скоро это положение изменится. Мы
даже составили план книги, но дальше этого дело не шло до 1997 г. Благодаря нашим
усилиям и усилиям Э. Слэйда из издательства «Wiley» книга увидела свет летом
1998 г. Мы считаем, что это было лишь общее введение в супрамолекулярную химию
(таким было ее рабочее название), за которым, как мы надеемся, последует много
частных курсов и для дипломников, и для аспирантов по ее отдельным
направлениям. Мы также с удовольствием упоминаем о том, что П. Бир, Ф. Гэйл и Д. Смит
недавно написали учебник по супрамолекулярной химии для начинающих, который,
несомненно, дополнит эту книгу.
При написании своей книги мы очень внимательно отнеслись к ее рабочему
названию, которое раньше содержало слово «введение». В ней мы попытались
упомянуть все ключевые соединения и подробно объяснить терминологию и понятия,
относящиеся к этой области. Мы не старались предложить всеобъемлющий
литературный обзор (это будет сделано в одиннадцатитомной «Comprehensive supramolec-
ular chemistry», соредактором которой является один из нас. К недостаткам нашей
книги можно отнести чрезмерное упрощение текста, но оно было произведено
сознательно, чтобы сделать доступными многие очень запутанные, быстроразвиваю-
lg|| Предисловие авторов
щиеся темы. Мы приносим свои извинения тем ученым, которым, возможно,
кажется, что мы сделали нашу трудную науку более банальной. Мы надеемся, что,
читая эту книгу целиком — от корки до корки (как будто скатываясь с горы на
санках) — или же кусками (ломтиками, удобными для откусывания), читатель сможет
узнать обо всех аспектах или о любом из них этой, многоглавой, как гидра, области
химии.
Супрамолекулярная химия
на мировых веб-сайтах
Современная наука идет вперед семимильными шагами, что особенно верно для
молодой и быстроразвивающейся дисциплины, какой является супрамолекулярная
химия. Поэтому любая более совершенная работа, такая, например, как эта книга,
быстро устаревает, поскольку появляются новые результаты. К счастью, такое быс-
тромодернизирующееся средство массовой информации, как Всемирная паутина,
сейчас настолько развито, что может нивелировать эффекты этой неполноценной
«информационной жизни». Для того чтобы заинтересованный читатель постоянно
совершенствовал свои знания, эта книга дополнена www-сайтом («Supramolecular
chemistry: A homepage»), доступным по адресу http://www.ch.kcl.ac.uk/supramol/text-
book.htm. Этот сайт содержит ряд часто обновляющихся каналов связи с сайтами,
представляющими интерес для специалиста по супрамолекулярной химии, ссылок
на тематические статьи и информацию. Любознательные читатели могут найти
странички авторов и их институтов по адресам http://www.ch.kcl.ac.uk/supramol
(Jonathan Steed), http://www.ch.kcl.ac.uk/ (King's College Chemistry) и
http://www.showme.missouri.edu/chemistry/ (University of Missouri - Chemistry).
Ссылки и библиография
В этой работе всего лишь приоткрывается дверь в очень актуальную и увлекательную
супрамолекулярную химию и она не является всеобъемлющим обзором известной
на сегодняшний день литературы. Поэтому важнейшие работы многих выдающихся
химиков, физиков и биологов — основателей этой ветви науки — рассматриваются на
протяжении всей книги, тогда как литературные ссылки сведены к минимуму.
Самые существенные приведены в конце каждой главы в алфавитном порядке по
основному автору, а в тексте обозначены фамилией основного автора и годом
опубликования работы. Ссылки на наиболее важные и полезные статьи по тематике
данного раздела приводятся в виде ключевых ссылок, помеченных символическим
ключом (Нг) и расположенных в главных разделах. Ключевые ссылки, выбранные в
основном из обзорной литературы, позволяют любознательным студентам получить
доступ к исследовательской литературе по любому подразделу, касающемуся
заинтересовавшего их материала (или являющегося домашним заданием!). Ключевые
ссылки постоянно модернизируются на веб-сайте.
Предисловие авторов \^Л
Учебные задания и мысленные эксперименты
В конце каждой главы приведены задания и мысленные эксперименты по ее
тематике. Задания будут полезны каждому студенту как для закрепления понимания
рассмотренных понятий, так и для подготовки к экзаменам. Ответы к заданиям
даны на веб-сайте. Мысленные эксперименты представляют собой вопросы, ответы
на которые еще не найдены. Они полезны для групповых занятий, где каждый член
группы может выдвигать свои идеи. Эти идеи затем может критически обсуждать
вся группа. Мы надеемся на то, что во многих случаях творческая фантазия
студента превзойдет воображение авторов книги. А это может привести к новым
экспериментальным проектам, способствующим развитию супрамолекулярной химии.
Гиперактивные молекулы
Как правило, супрамолекулы являются большими молекулами, или агрегатами,
которые часто трудно адекватно изобразить на листе бумаги. В соответствии с этим на
веб-сайте этой книги, помимо двухмерных диаграмм, доступны вращающиеся
трехмерные CD) проекции, содержащие ЗО-координаты всех кристаллических
структур, упомянутых в тексте, в формате Brookhaven Database (.PDB). Эти файлы
можно увидеть с помощью просмотровой программы, такой, как RasMol,
написанной Р. Сэйлом из «Glaxo Wellcome», или при помощи веб-программы просмотра,
оборудованной просмотровой головкой Chemscape Chime. Эти сервисные
программы можно бесплатно скачать с приведенных ниже веб-сайтов. На веб-сайте книги
иллюстративный материал разделен по главам: рисункам и соединениям
присвоены соответствующие номера.
Ссылки регулярно обновляются по адресу:
http://www.ch.kcl.ac.uk/supramol/textbook.htm.
Программное обеспечение Chemscape Chime Software для интерактивного
ЗО-просмотра на веб-сайте можно найти по адресу:
http://www.mdli.com/chemscape/chime.
Программное обеспечение RasMol для обзора химических структур доступно по
адресу: http://www.umass.edu/microbio/rasmol/getras.htm.
Джонатан В. Стид (Лондон, Великобритания)
Джерри Л. Этвуд (Колумбия, США)
Благодарности
Мы благодарны многим замечательным студентам, исследователям и
коллегам, которые на протяжении долгих лет работали в наших группах.
Обсуждения с ними помогли нам выкристаллизовать свое видение этой быс-
троразвивающейся области науки. Многие коллеги как в Европе, так и в
США оказали нам неоценимую помощь своими советами и информацией.
В особенности мы признательны Дж. Такеру, М. Хэннону, Дж. Томасу и
Ф. Армитэйджу за обсуждение и конструктивные замечания по рукописи.
Как всегда, мы очень благодарны Л. Барбору за блестящие рентгенострук-
турные исследования, которые облегчили интерпретацию
кристаллографических данных. Э. Слэйд, Д. Бут и С. Карден неустанно трудились в
издательстве «Wiley», для того чтобы довести книгу до должного уровня.
Дж.В. Стад хотел бы поблагодарить Рану за ее терпение и неизменную
поддержку ею этого проекта. Мы даже не представляли себе, как много
времени займет его осуществление!
Об авторах
Джонатан В. Стад родился в Уимблдоне (Лондон,
Великобритания) в 1969 г. В 1993 г. он окончил Университетский
колледж в Лондоне, где получил степени бакалавра и
доктора философии, работая с Д. Точером в области
координационной и металлоорганической химии (синтез
неорганических лекарственных препаратов и разработка новых
темплатных методов синтеза). В 1993 г. за свою докторскую
работу был награжден медалью У. Рамзая. В 1993—1995 гг.,
работая вместе с Дж. Этвудом, он был стипендиатом НАТО
в университетах Алабамы и Миссури, где синтезировал су-
прамолекулярные «хозяева» нового класса для анионов.
Эта область химии — до сих пор часть его научных
интересов. В 1995 г. его назначили преподавателем Королевского колледжа в
Лондоне, где он завоевал научный авторитет в области конструирования кристаллов на
основе сильных и слабых водородных связей и координационных взаимодействий.
В 1999 г. он стал лектором этого колледжа. Доктор Стид — автор более 80 научных
статей, ряда обзоров, глав в книгах и популярных статей. В 1998 г. он был
награжден медалью Р. Мелдола Королевского химического общества.
Джерри Л. Этвуд родился в Спрингфилде (Миссури, США)
в 1942 г. Он учился в Юго-Западном государственном
университете Миссури, где в 1964 г. получил степень
бакалавра. Свою аспирантскую работу он выполнял у Г. Стаки в
Иллинойском университете, где и получил степень
доктора философии в 1968 г. Сразу после этого его назначили
ассистентом профессора в Университете Алабамы, где
позднее он стал ассоциированным профессором A972 г.) и
полным профессором A978 г.). В 1994 г. он получил
должность профессора и кафедру в Университете Миссури
(Колумбия, США). За свою карьеру профессор Этвуд
опубликовал более 500 научных статей. Его научные интересы
охватывают ряд направлений супрамолекулярной химии, включающих самосборку
нековалентных капсул, химию жидких клатратов, конструирование и синтез ани-
онсвязывающих «хозяев», а также взаимодействия «хозяев» в твердом состоянии. В
настоящее время он является ассоциированным редактором журнала «Chemical
Communications». Он отредактировал большое количество основополагающих
работ по супрамолекулярной химии, включая пятитомную серию «Inclusion
compounds» A984 и 1991 гг.) и «Comprehensive supramolecular chemistry» A996 г.).
Профессор Этвуд был награжден премией И. Кристенсена в области
макроциклической химии за 2000-й год.
Основные сокращения
и обозначения
А — поглощение (absorbance)
асас — ацетилацетонато (acetylacetonato)
АСН - ацетилхолин (acetylcholine)
ADP — аденозиндифосфат (adenosine diphosphate)
AmP - аминопиримидин (aminopyrimidine)
AMP - аденозинмонофосфат (adenosine monophosphate)
ANS - 8-анилино-1-нафталинсульфонат (8-anylino-l-naphthalenesulphonate)
Asp - аспартат (aspartate)
ATP - аденозинтрифосфат (adenosine triphosphate)
АТРаза — аденозинтрифосфатаза
ВС — бактериохлорофилл (bacteriochlorophyll)
BEDT-TTF - бис(этилендитио)тетратиафульвален
[bis(ethylenedithio)tetrathiafulvalene]
BiBLE - бибрахиальные лариат-эфиры (bibrachial lariat ethers)
binap - бинафталин (бинафтил) [binapthyl]
bipy - бипиридил (bipyridyl)
biq - 2,2'-бихинолил (biquinolyl)
BP - бактериофеофитин (bacteriopheophytin)
CA - карбоангидраза (carbonic anhydrase)
CAC — критическая концентрация агрегирования
(critical aggregation concentration)
CCD - прибор с зарядовой связью (charge-coupled device)
cGMP - циклический гуанозинмонофосфат (cyclic guanosine monophosphate)
CHEF — флуоресценция, усиленная хелатированием
(chelation-enhanced fluorescence)
CPA - карбоксипептидаза A (carboxypeptidase A)
СРК-модели — модели Кори—Полинга-Колтуна (Corey-Pauling-Koltun models)
CSD - Кембриджский банк структурных данных
(Cambridge crystallographic structural database)
CT - перенос заряда (charge transfer)
CTV- циклотривератрилен (cyclotriveratrylene)
CVA - циклическая вольтамперометрия (cyclic voltammetry)
Cys - цистеин (cysteine)
DC — непосредственное взаимодействие (direct coupling)
Основные сокращения и обозначения 23
de — избыток диастереомера (diastereomeric excess)
Den - дендример (dendrimer)
DMA - диметилацетамид (dimethylacetamide)
DMF - N,N-диметилформамид (N,N-dimethylformamide)
DMSO — диметилсульфоксид (dimethylsulphoxide)
dpp - дипиридинопиразил (dipyridinopirazyl)
dppz - дипиридинофеназил (dipyridinophenazyl)
DSC - дифференциальная сканирующая калориметрия
(differential scanning calorimetry)
DTA - дифференциальный термический анализ (differential thermal analysis)
E — излучение (emittance)
EDRAW — стирание прямого считывания после записи
(erase direct read after write)
EDTA — этилендиаминтетрауксусная кислота (ethenediamine tetraaceticacid)
EFISH — вторая гармоника, индуцированная электрическим полем
(electric-field-induced of second harmonic)
en - этилендиамин (ethylenediamine)
ESFF — расширенное систематическое силовое поле
(extended systematic force field)
ESI — ионизация электрораспылением (electrospray ionisation)
eT - перенос электрона (electron transfer)
ET - перенос энергии (energy transfer)
EXAFS — метод растянутой тонкой структуры рентгеновского поглощения
(extended X-ray absorption fine structure)
FAB - бомбардировка быстрыми атомами (fast atom bombardment)
FAH — ароматические углеводороды с конденсированными кольцами
(fused-ring aromatic hydrocarbons)
GDS — стадия, определяемая гостем (guest-determining step)
GM — генетически модифицированный (genetically modified)
GTP - гуанозинтрифосфат (guanosine triphosphate)
HB - число водородных связей (number of hydrogen bonds)
His — гистидин (histidine)
HOMO — высшая заполненная молекулярная орбиталь
(highest occupied molecular orbital)
HPLC — высокоэффективная жидкостная хромотография
(high pressure liquid chromatography)
HSAB-теория — теория жестких и мягких кислот и оснований
(principle of hard and soft acids and bases)
24 Основные сокращения и обозначения
lh — лед гексагональный
Im — имидазол (imidazole)
IMH — межмолекулярная многоцентровая гетероакцепторная водородная связь
(intermolecular multicentre heteroacceptor hydrogen bond)
IPA — межмолекулярное псевдоагостическое взаимодействие
(intermolecular pseudo-agostic interaction)
ISC — интеркомбинационный переход (intersystem crossing)
ISFET - ион-селективные полевые транзисторы
(ion-selective field-effect transistors)
IT — интервалентный переход (intervalence transition)
LC-переходы — переходы, центрированные на лиганде
(ligand-centered transitions)
LCD - жидкокристаллический дисплей (liquid crystal display)
LFAE — энергия активации поля лигандов (ligand field activation energy)
LFSE — энергия стабилизации поля лигандов (ligand field stabilisation energy)
LUMO - низшая незаполненная молекулярная орбиталь
(lowest unoccupied molecular orbital)
MALDI - лазерная десорбция из матрицы
(matrix-assisted laser desorption ionisation)
МС-переходы — переходы, центрированные на металле (metal-centred transitions)
MLCT — перенос заряда от металла к лиганду (metal-to-ligand charge transfer)
nAChR — никотин-ацетилхолиновый рецепторный белок
(nicotinic acetylcholine receptor protein)
NADH - никотинамидадениндинуклеотид (nicotinamide adenine dinucleotide)
NMP — N-метилпирролидинон
NOE - ядерный эффект Оверхаузера (nuclear Overhauser effect)
NPP — нитрофенилпиперазин (nitrophenylpyperazene)
ODS - октадецилметилкремнезем (octadecylmethylsilica)
OEC - кислородвыделяющий комплекс (oxygen-evolving complex)
Pj - неорганический фосфат
9PA - 9-пропиладенин (9-propyladenine)
PAP - 2,6-адамантан-бисA,1,3,3-тетраметил-7,8-диазациклопентано[1]фенан-
трен-2-илиден)
[2,6-adamantane-bis( 1,1,3,3-tetramethyl-7,8-diazacyclopenta[ 1 ]phenan-
threne-2-ylidene)]
PCR - полимеразная цепная реакция (polymerase chain reaction)
Основные сокращения и обозначения 25
PDN — калиевая соль 1,3-дигидрокси-2-нафтойной кислоты
(potassium salt of l,3-dihydroxy-2-naphthoate)
PDP — дигидрофосфат калия (potassium dihydrogenphosphate)
PET - фотоиндуцированный перенос электрона
(photon-induced electron transfer)
phen - 1,10-фенантролил A,10-phenanthrolyl)
phi - 9,10-фенантренхинондиимин (9,10-phenanthrenequinone diimine)
PHTP - пергидротрифенилен (perhydrotriphenylene)
PNP - «-нитрофенол (p-nitrophenol)
рог - порфирин (porphyrin)
PQ — хиноновая форма пластогидрохинона (quinone form of plastohydroquinone)
PQH2 — пластогидрохинон (гидрохинон) [plastohydroquinone (hydroquinone)]
PS — фотосистема (photosystem)
РТР-аза - протеинтирозинфосфатаза (protein tyrosine phosphatase)
PVP - поливинилпирролидон (polyvinilpyrrolydon)
py - пиридил (pyridyl)
pz - пиразолил (pyrazolyl)
Qa, Qb — хиноны (quinones)
RMM — относительная молекулярная масса (relative molecular mass)
ROMP - метатезисная полимеризация с раскрытием кольца
(ring opening metathesis polymerization)
Ser - серии (serine)
SHB - насыщенные водородными связями (saturated hydrogen-bonded)
смешанные кристаллы
TCNE - тетрацианоэтилен (tetracyanoethylene)
Tf — трифторметансульфонат (трифлат) [trifluoromethane sulphonate]
TGA — термогравиметрический анализ (thermogravimetric analysis)
THB — 1,3,5-тригидроксибензол (флороглюцин) [1,3,5-trihydroxybenzene
(phloroglucinol)]
THF - тетрагидрофуран (tetrahydrofurane)
TNC — закрученная нематическая ячейка (twisted nematic cell)
tph — терефталевая кислота (terephthalic acid)
tpy — трипиридил (tripyridyl)
Ts - л-толуолсульфонат (p-toluenesulphonate)
TSQ - 6-метокси-(8-л-толилсульфонамино)хинолин
[6-methoxy-(8-/?-tolylsulfonamino)quinoline]
WORM - однократная запись, многократное считывание (write once read many)
Основные сокращения и обозначения
ГВГ — генерация второй гармоники
ДНК - дезоксирибонуклеиновая кислота
ИК-область — инфракрасная область
ККМ - критическая концентрация мицеллообразования
ККС - критическая концентрация самосборки
Л/Эф - эффективная молярность
НЛО — нелинейно-оптические (свойства, эффекты)
ПАВ — поверхностно-активные вещества
РНК - рибонуклеиновая кислота
РСА - рентгеноструктурный анализ
УФ-область — ультрафиолетовая область
1 Общие представления
Человечество подразделяется на два огромных класса:
хозяев и гостей.
М. Бирбом (род. 1872). Хозяева и гости
1 ® 1 Определение и развитие
супрамолекулярной химии
1.1.1 Что такое супрамолекулярная химия?
Определение супрамолекулярной химии как «химии молекулярных
ансамблей и межмолекулярных связей» дал один из ее выдающихся основателей
Ж.-М. Лен, лауреат Нобелевской премии, которой он был удостоен в 1987 г. за
работы в этой области. Выражаясь разговорным языком, можно сказать, что это
«химия вне молекулы». Другие определения включают выражения типа: «химия неко-
валентной связи» и «химия за пределами молекулы». Определения этого типа
образно представлены на рис. 1.1, иллюстрирующем связь между молекулярной и
супрамолекулярной химией на примере как структуры, так и функций.
Такие определения, безусловно полезные, по своей природе не являются
всеобъемлющими, и их не стоит принимать слишком буквально. Эти определения
близки определению металлоорганической химии как «химии соединений со
связями металл-углерод». Такой подход сразу же исключает, например, соединение
Уилкинсона RhCl(PPh3K — один из наиболее важных промышленных
катализаторов металлоорганических превращений, известных в этой области. Быстрое
распространение супрамолекулярной химии в последние 15 лет привело к огромному
разнообразию химических систем, полученных как целенаправленно, так и
случайно, которые по происхождению или природе могут претендовать на супрамолеку-
лярность. В частности, специалисты, работающие в области супрамолекулярной
фотохимии, выбрали несколько иное определение супрамолекулярного
соединения: это группа молекулярных компонентов, индивидуальные свойства которых
интегрированы в свойства целого ансамбля (ковалентного или нековалентного).
Так, полностью ковалентную молекулу, содержащую, например, хромофор (свето-
поглощающий фрагмент), спейсер* и редокс-центр, можно рассматривать как су-
прамолекулярную благодаря способностям хромофора и редокс-центра
соответственно поглощать свет или изменять степень окисления, вне зависимости от того,
* Молекулярный фрагмент, соединяющий две одинаковые или разные функциональные
группы молекулы. {Примеч. ред. перевода.)
28
1. Общие представления
Молекулярная химия
Молекулярные предшественники
(прекурсоры)
Супрамолекулярная химия
Ковалентная молекула:
химическая природа,
форма,
редокс-свойства,
зазор HOMO-LUMO,
полярность,
колебание и вращение,
магнетизм,
хиральность
Специфические
характеристики,
функции или свойства:
распознавание,
катализ,
транспорт
Гость
Хозяин
Супрамолекула (комплекс):
степень упорядоченности,
взаимодействия между субъединицами,
симметрия упаковки,
межмолекулярные взаимодействия
Рис. 1.1. Сравнение диапазонов молекулярной и супрамолекулярной химии согласно
Ж.-М. Лену
образуют они часть супра(супер)молекулы или нет (см. гл. 8). Подобным образом
многие недавние работы нацелены на разработку способов синтеза
самоорганизующихся систем для получения больших молекул или молекулярных группировок.
Эти системы часто самоорганизуются с использованием различных
взаимодействий, причем некоторые из них определенно нековалентны (например, водородные
связи), а некоторые содержат значительную ковалентную составляющую
(например, взаимодействия металл—лиганд; см гл 7). Этот сдвиг в акцентах — не более
чем развитие данной области от ее корней, лежащих в химии типа «хозяин—гость»,
до охвата понятий других областей науки. Поэтому при написании книги мы
попытались рассмотреть широкий круг проблем, из которых складывается
супрамолекулярная химия, пытаясь определить как ее современное состояние, так и будущие
направления развития.
1.1.2 Химия «хозяин-гость»
Если мы рассматриваем супрамолекулярную химию в ее простейшем
варианте, предполагая какой-либо тип (нековалентного) связывания или комплек-
сообразования, то сразу должны определить, за счет чего происходит это
связывание. В таком контексте мы обычно рассматриваем молекулу («хозяин»),
связывающую другую молекулу («гость») с образованием комплекса «хозяин-гость», или
супрамолекулы. Обычно хозяин — это большая молекула,, или агрегат, с дыркой по-,
1.1. Определение и развитие супрамолекулярной химии тд$
рядочного размера или полостью в центре. Гостем может служить моноатомный
катион, простой неорганический анион или более сложная молекула, такая, как
гормон, феромон или нейротрансмиттер. Более формально хозяина можно определить
как молекулярное образование со сходящимися центрами связывания (например, до-
норные атомы оснований Льюиса, доноры водородной связи и т.п.). Гостьобладает
расходящимися центрами связывания (например, сферический катион металла
кислоты Льюиса или галогенид-анион как акцептор водородной связи).
Взаимоотношение с образующимся комплексом хозяин—гость Д. Крам определил A986 г.) так:
«Комплексы состоят из двух или более молекул или ионов, связанных вместе в
уникальную структурную систему электростатическими силами, по своей природе иными, чем
чисто ковалентные... Молекулярные комплексы обычно удерживаются водородными
связями, ионными парами, взаимодействиями п-кислота—п-основание, связыванием
металл—лиганд, силами притяжения Ван-дер-Ваальса, перестройкой растворителя и
частично образованными или разорванными ковалентными связями (переходные
состояния)... Высокая структурная организация обычно реализуется только посредством
многочисленных связывающих центров... Высокоструктурированный молекулярный
комплекс образован по крайней мере одним хозяином и одним гостем...
Взаимоотношение хозяин—гость включает комплементарную стереоэлектронную организацию
центров связывания в хозяине и госте... Компонент-хозяин определяется как органическая
молекула (или ион), чьи центры связывания сходятся в комплексе... Компонент-гость —
это любая молекула (или ион), центры связывания которой расходятся в комплексе».
Дальнейшее обобщение может быть достигнуто удалением слова
«органическая» из определения компонента-хозяина, так как в недавних работах описано
множество неорганических хозяев, таких, как цеолиты и полиоксованадаты, а также
смешанные металлоорганические координационные соединения, выполняющие
сходные функции и потому подпадающие под это определение. Связывание
хозяин-гость похоже наловлю мяча рукой. Рука, выполняющая функцию хозяина,
окружает мяч, обеспечивая физический (стерический) барьер, препятствующий
падению (диссоциации). Однако на электронном уровне эта аналогия неправомерна,
так как нет реальных сил притяжения между рукой и мячом. Такая аналогия
годится для введения термина «химия включения» (поскольку мяч включен в руку),
отсюда - включение одной молекулы в другую.
Один из ключевых разделов супрамолекулярной химии хозяин-гость в ее
общем виде относится к стабильности комплекса хозяин—гость в растворе. С одной
стороны, область клатратов, или, в более общем смысле, химия включения,
относится к хозяевам, часто устойчивым только в твердом (кристаллическом)
состоянии, но распадающимся при растворении. В эту категорию попадают газовые
гидраты, клатраты мочевины и многие кристаллические сольваты (см. гл. 5). С другой
стороны, молекулы-хозяева для катионов, например краун-эфиры (коранды),
криптанды и сферанды (см. гл. 3), а также хозяева для нейтральных молекул,
например карцеранды и криптофаны (см. гл. 5), эффективно связывают гостя как в
твердой фазе, так и в растворе. Следует отметить, что существуют и чисто жидко-
фазные системы, в частности жидкие кристаллы и жидкие клатраты, не имеющие
прямых твердофазных аналогов (см. гл. 10).
1зо
1. Общие представления
1.1.3 Развитие представлений
Супрамолекулярная химия считается молодой дисциплиной, которая
берет свое начало в конце 60-х—начале 70-х годов прошлого века. Однако ее
концепции и представления, а также многие простые (и не совсем простые) супрамо-
лекулярные химические системы были известны почти с момента зарождения
современной химии. Ее хронология (хотя, естественно, субъективная и неполная)
представлена в табл. 1.1. Большая часть супрамолекулярной химии обязана
развитию химии макроциклов во второй половине 60-х годов, в частности макроцикли-
ческих лигандов для катионов металлов. Можно идентифицировать четыре
фундаментальные системы A.1)—A.4), полученные в группах Куртиса (N. Curtis),
Буша (W. Busch), Erepa (E.-G. Jager) и Педерсена (С. Pedersen), в трех из которых
были использованы реакции конденсации альдегидов и аминов с образование
иминов (основания Шиффа). Концептуально можно рассматривать эти системы
как аналоги природных макроциклов (ионофоров, гемов, фталоцианинов и др.). К
ним следует добавить циклофаны Д. Крама E0-е годы) и позднее сферанды и кар-
церанды. Отметим также огромный вклад Ж.-М. Лена, получившего криптанды в
конце 60-х годов и с тех пор продолжающего плодотворно работать в этой области.
CO2R
A.1)
(Куртис, 1961)
A.2)
(Буш, 1964)
A.4)
(Педерсен, 1967)
Следует признать, что на сегодня супрамолекулярная химия является одной из
наиболее быстроразвивающихся областей исследований. Ее междисциплинарная
природа привела к широкому сотрудничеству между физиками, специалистами по
компьютерному моделированию, кристаллографами, химиками-неорганиками и
химиками, изучающими твердое состояние, органиками-синтетиками,
биохимиками и биологами. Эстетическая привлекательность супрамолекулярнрых
соединений и непосредственная связь между наглядным представлением, молекулярным
моделированием и поведением хозяев и их комплексов в эксперименте до такой
степени способствовали развитию этой области, что теперь она является
полноправным членом сообщества химических дисциплин. Однако уникальная
междисциплинарная природа супрамолекулярной химии, высокий темп ее развития, а
также усложненность ее объектов и представлений привели к тому, что она отсутствует
АЛ. Определение и развитие супрамолекулярной химии 31
Таблица 1.1. Основные вехи в развитии супрамолекулярной химии
Год
Автор: достижение, открытие
1810 Г. Дэви: открытие гидрата хлора
1823 М. Фарадей: формула гидрата хлора
1841 С. Шафо: изучение интеркалятов графита
1849 Ф. Вёлер: клатратР-гидрохинонН25
1891 А. Вилье и Хебд: соединения включения циклодекстринов
1893 А. Вернер: координационная химия
1894 Э. Фишер: концепция «ключ—замок»
1906 П. Эрлих: введение понятия «рецептор»
1937 К. Вольф: введение термина «супермолекула» для описания организованных
образований, возникающих при объединении координационно насыщенных
частиц (например, димера уксусной кислоты)
1939 Л. Полинг: водородные связи описаны в фундаментальном труде
«The nature of chemical land»
1940 M. Бенген: канальные соединения включения мочевины
1948 Г. Пауэл: рентгенеструктурный анализ соединений включения C-гидрохинона;
введение термина «клатрат» для описания соединений, в которых один
компонент включен в каркас другого
1949 К.Браун и А.Фартинг: синтез [2.2]парациклофана
1953 У. Уотсон и Ф. Крик: структура ДНК
1956 Д. Кроуфут-Ходжкин: рентгеноструктурный анализ витамина В12
1959 Д. Крам: пробный синтез циклофановых комплексов с переносом заряда
(с (NCJC=C(CNJ)
1961 Н. Куртис: первое макроциклическое основание Шиффа из ацетона
и этилендиамина
1964 В. Буш и Э.-Г. Егер: макроциклические основания Шиффа
1967 Ч. Педерсен: синтез краун-эфиров
1968 К. Парк и X. Симмондс: хозяева-катапинанды для анионов
1969 Ж.-М. Лен: синтез первых криптандов
1969 Дж. Этвуд: жидкие клатраты из солей алкилалюминия
1973 Д. Крам: хозяева-сферанды, полученные для проверки важности
предорганизации
1978 Ж.-М. Лен: введение термина «супрамолекулярная химия» и ее определения
«химия молекулярных ансамблей и межмолекулярной связи»
1979 Г. Гокель и М. Окахара: создание лариат-эфиров как подкласса хозяев
1981 Ф. Фёгтле и Э. Вебер: поданды-хозяева и развитие номенклатуры
1987 Присуждение Нобелевской премии по химии Д. Краму, Ж.-М. Лену
и Ч. Педерсену за их работы по супрамолекулярной химии
1996 Дж. Этвуд, Дж. Дэвис, Д. Мак-Никол и Ф. Фёгтле: опубликование книги
«Comprehensive supramolecular chemistry», в которой содержатся результаты
работ почти всех основных групп, а также обобщены достижения
и рассмотрено современное состояние этой области
1996 Присуждение Нобелевской премии по химии X. Крото, Р. Смолли
и Р. Кёрлу за их работы по химии фулеренов
32 1- Общие представления
в основных курсах для студентов. Поэтому цель настоящей книги — создание
сжатого и доступного вводного курса в основы супрамолекулярной химии для
студентов и аспирантов в надежде передать им хотя бы часть того энтузиазма, который
испытывают сами авторы книги к этому удивительному предмету.
1Л2 Классификация супрамолекулярных
соединений хозяин-гость
Одно из первых формальных определений супрамолекулярных
структур хозяин-гость, напоминающих клетку, предложено Г. Пауэлом из
Оксфордского университета в 1948 г. Он ввел термин «клатрат» для определения типа
соединения включения, «в котором два или более компонента объединены без образования
обычного химического соединения - путем полного включения одного набора
молекул в подходящую структуру, образованную другим набором». Начиная
описывать современную химию хозяин—гость, полезно разделить соединения-хозяева на
два главных класса в соответствии с топологической взаимосвязью между хозяином
и гостем. Кавитанды могут быть описаны как хозяева с внутримолекулярными
полостями. Это означает, что способность полости связывать гостя — собственное
молекулярное свойство хозяина и существует как в растворе, так и в твердом
состоянии. Наоборот, клатранды — хозяева с межмолекулярными полостями (такая
полость — это зазор между двумя и более молекулами-хозяевами), существующими
только в кристаллическом или твердом состоянии. Агрегат хозяин—гость, образо-
Молекулярное
включение
Включение
в кристаллическую
решетку
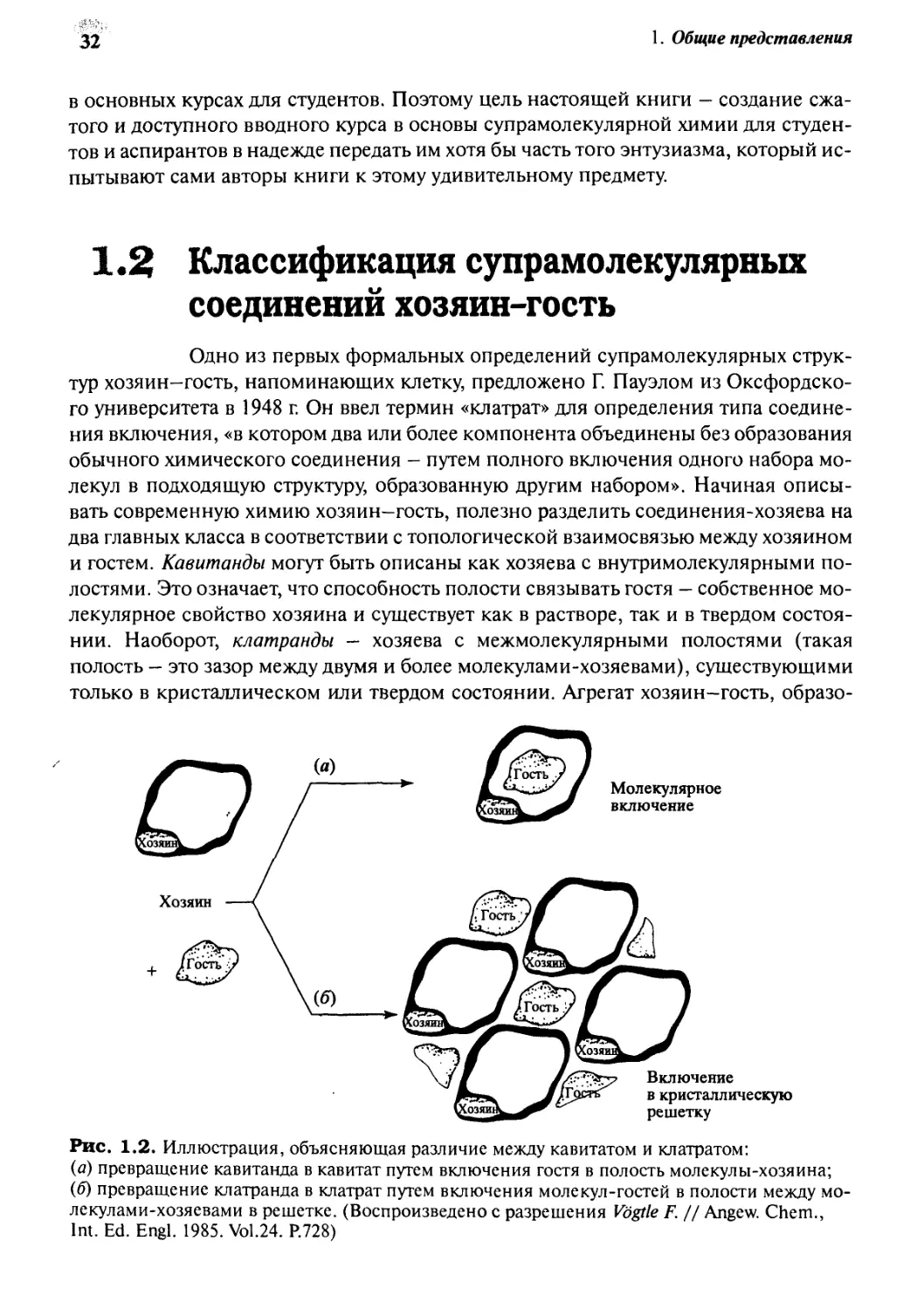
Рис. 1.2. Иллюстрация, объясняющая различие между кавитатом и клатратом:
(а) превращение кавитанда в кавитат путем включения гостя в полость молекулы-хозяина;
(б) превращение клатранда в клатрат путем включения молекул-гостей в полости между
молекулами-хозяевами в решетке. (Воспроизведено с разрешения Vogtle F. // Angew. Chem.,
Int. Ed. Engl. 1985. Vol.24. P.728)
1.2. Классификация супрамолекулярных соединений хозяин—гость
33
Таблица 1.2. Классификация соединений хозяин-гость для нейтральных хозяев
Хозяин
Гость
Взаимодействие
Класс
Пример
Краун-эфир
Сферанд
Катион металла Ион-дипольное
Катион
алкиламмония
Циклодекстрин Органическая
молекула
Водородная
связь
Гидрофобное/
ван-дер-ваальсово
Комплекс
(кавитанд)
Комплекс
(кавитанд)
Кавитат
Вода
Каликсарен
Органическая Ван-дер-ваальсово/ Клатрат
молекула, галоген кристаллическая
и т.д.
Органическая
молекула
Циклотривера- Органическая
трилен (CTV) молекула
упаковка
Ван-дер-ваальсово/ Кавитат
кристаллическая
упаковка
Ван-дер-ваальсово/ Клатрат
кристаллическая
упаковка
[К+сA8-краун-6)]
Сферанд-(СН31ЧН+)
(ос-циклодекстрин) •
• (л-гидроксибензой-
ная кислота)
(Н2ОN(СН4)
(п-трет-бугнл-
каликс[4]арен)-
•(толуол)
(СТУH.5(ацетон)
ванный кавитандом, называют кавитатом, а клатранды образуют клатраты.
Различие между хозяевами этих двух классов схематически представлено на рис. 1.2.
При дальнейшей классификации соединений-хозяев нужно учитывать силы
взаимодействия между хозяином и гостем. Если агрегат хозяин-гость связан
преимущественно электростатическими силами (включая взаимодействия
ион—диполь, диполь-диполь, водородные связи и т.д.), используют термин «комплекс».
Для структур же, удерживаемых менее специфическими, часто более слабыми,
ненаправленными взаимодействиями (например, гидрофобными, ван-дер-ваальсо-
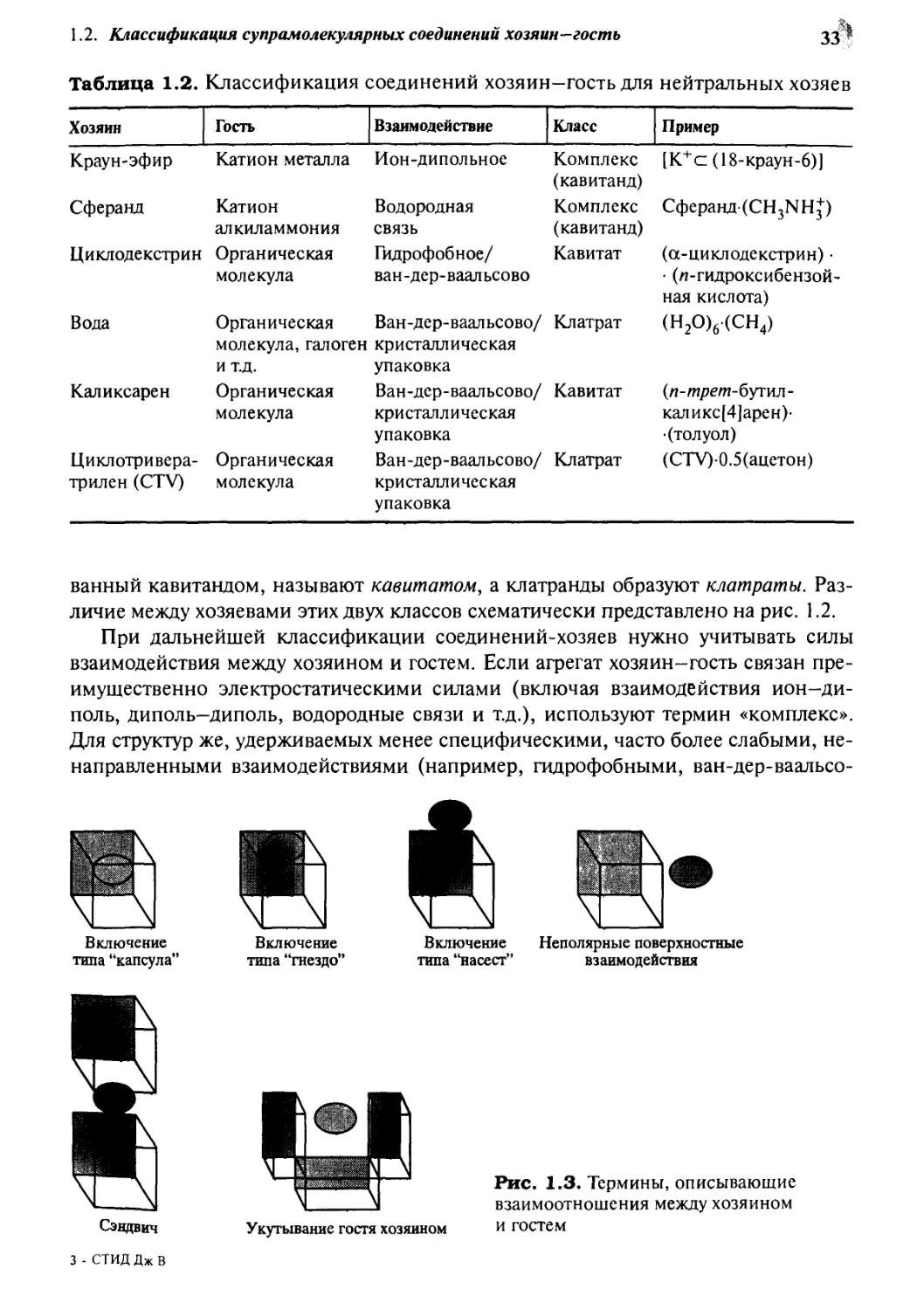
Включение
типа "капсула"
Включение
типа "гнездо"
Включение
типа "насест"
Неполярные поверхностные
взаимодействия
Сэндвич
3 - СТИД Дж В
Укутывание гостя хозяином
Рис. 1.3. Термины, описывающие
взаимоотношения между хозяином
и гостем
34
1. Общие представления
выми или эффектами плотной упаковки), больше подходят термины «кавитаты» и
«клатраты». Некоторые примеры употребления этой номенклатуры показаны в
табл. 1.2. Следует отметить, что в современной литературе наблюдается сильная
тенденция использовать слово «комплекс» для охвата всех этих явлений.
Внутри этой, очень широкой классификации соединений хозяин—гость
существует ряд промежуточных типов; разумеется, точная классификация данного
соединения часто весьма спорна. Иногда дополнительно вводят описательные
термины, заслуживающие внимания (рис. 1.3). Эта номенклатура должна помогать
химику описывать и визуализировать системы, с которыми он работает, и не
должна быть просто ограниченным и жестким набором типов.
Рецепторы, координация
и аналогия «замок-ключ»
2.
Химия типа «хозяин—гость», или «рецептор—субстрат», опирается на
три исторические концепции:
1. Постулат П. Эрлиха A906) о том, что без образования связей молекулы не
могут действовать («corpora поп agunt nisi fixata»; «тела не действуют, если они не
соединены»); так Эрлих ввел понятие «биологический рецептор».
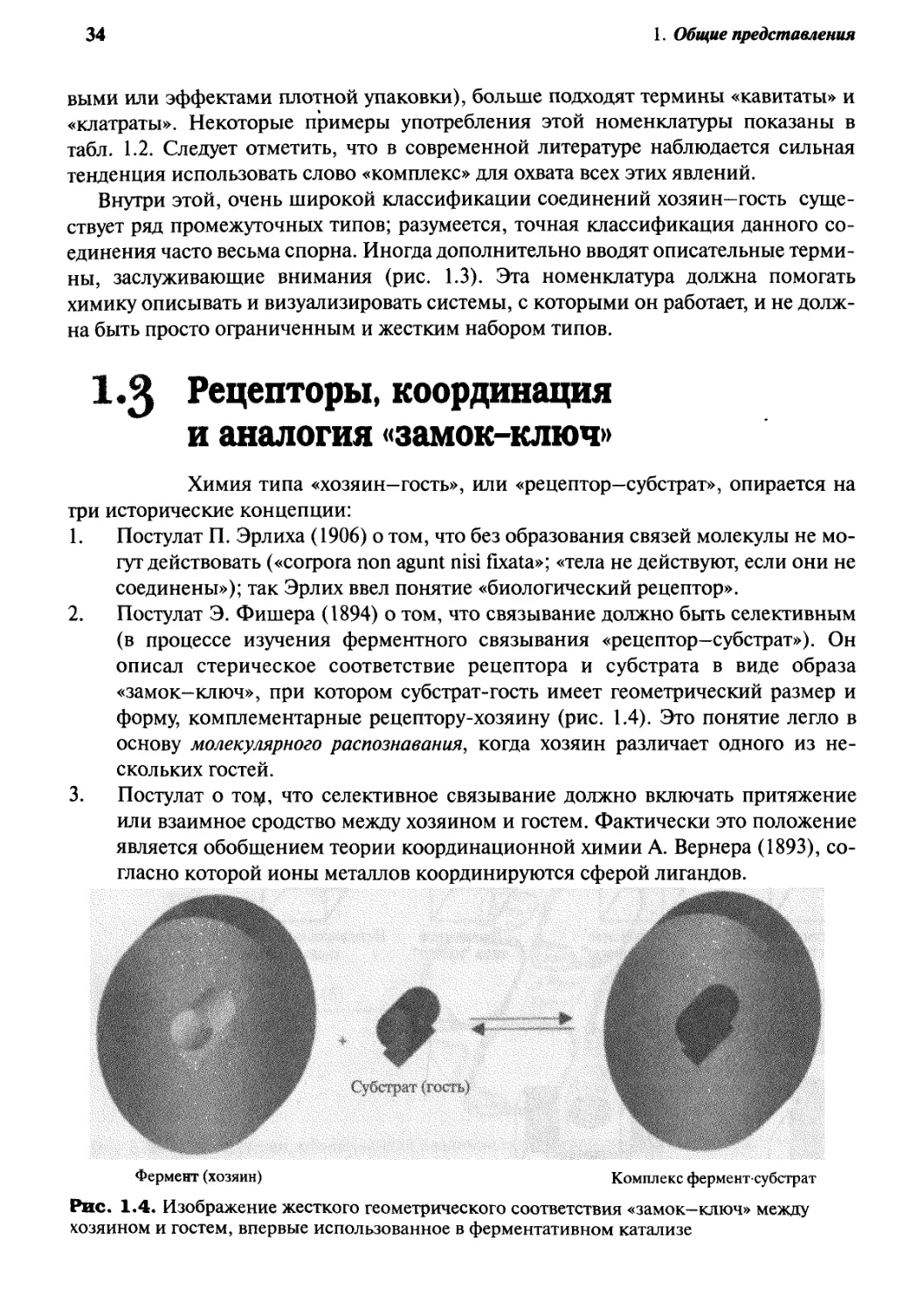
Постулат Э. Фишера A894) о том, что связывание должно быть селективным
(в процессе изучения ферментного связывания «рецептор—субстрат»). Он
описал стерическое соответствие рецептора и субстрата в виде образа
«замок—ключ», при котором субстрат-гость имеет геометрический размер и
форму, комплементарные рецептору-хозяину (рис. 1.4). Это понятие легло в
основу молекулярного распознавания, когда хозяин различает одного из
нескольких гостей.
Постулат о том, что селективное связывание должно включать притяжение
или взаимное сродство между хозяином и гостем. Фактически это положение
является обобщением теории координационной химии А. Вернера A893),
согласно которой ионы металлов координируются сферой лигандов.
3.
Фермент (хозяин)
Комплекс фермент-субстрат
Рис. 1.4. Изображение жесткого геометрического соответствия «замок—ключ» между
хозяином и гостем, впервые использованное в ферментативном катализе
1.4. Хелатный и макроциклический эффекты 35
Эти три концепции возникли, по существу, независимо друг от друга, и прошло
много лет , прежде чем различные дисциплины, давшие им жизнь, слились вместе
и родили междисциплинарную область — супрамолекулярную химию. П. Эрлих,
например, занимался лечением ряда инфекционных заболеваний. В ходе этой
работы он заметил, что краситель метиленовый голубой A.5) имеет удивительное
сродство к некоторым живым клеткам, окрашивая их в интенсивный синий цвет
(его наставник, Р. Кох, использовал метиленовый голубой для открытия бацилл
туберкулеза. «Если окрашиваются только некоторые клетки, — рассуждал Эрлих, —
тогда, может быть, существуют такие красители, которые окрашивают только
носителей болезней и в то же время разрушают их, не атакуя собственные клетки тела?».
В конечном счете в 1910 г. Эрлих разработал лекарство от сифилиса сальварсан
(Salvarsan®) — одно из наиболее эффективных лекарств от этой болезни. В итоге он
стал основателем современной химиотерапии.
СГ
Метиленовый голубой A.5)
Объединению координационной химии, химиотерапии и энзимологии
способствовали успехи в развитии приборной базы и методов синтеза, а также, не в
последнюю очередь, впечатляющие достижения в органическом синтезе,
родившемся как дисциплина в 1828 г., после получения Ф. Вёлером мочевины из цианата
аммония. В ходе развития супрамолекулярной химии громадный прогресс был
достигнут благодаря количественному описанию параметров рецепторов,
обеспечивающих их сродство с гостями. Как будет видно в последующих разделах, образ
«замок—ключ» далее подвергался модифицированию в результате того, что в
супрамолекулярную химию были введены понятия «хелатирование», «предорганизация»,
«комплементарность», «сольватация» и строго определено понятие «молекулярная
форма».
*"» Behr J.P. The lock-and-key principle. The state of the art-100 years on. Chichester:
J.Wiley & Sons, 1994.
1*4 Хелатный и макроциклический эффекты
Многое в конструировании супрамолекулярных молекул-хозяев
касается осуществления суммарных или даже множественных взаимодействий. Это
означает, что можно сконструировать устойчивый комплекс хозяин—гость, используя
нековалентные (часто слабые) взаимодействия, которые способны
стабилизировать комплекс. Небольшое количество стабилизирующей энергии, выделяющееся
1. Общие представления
при одном таком взаимодействии, объединяется с энергией всех других малых
взаимодействий (суммарность), что приводит к значительной энергии связывания, а
следовательно, и к стабильности комплекса. Во многих случаях взаимодействия во
всей системе синергетически сильнее, чем сумма взаимодействий ее компонентов
(множественность). Такая дополнительная стабилизация основана на хелатном и
макроциклическом эффектах.
Хелатный эффект хорошо известен в координационной химии и выражается в
значительно большей устойчивости комплексов бидентатных лигандов (типа
этилендиамина) по сравнению с устойчивостью близких по природе металлокомп-
лексов, содержащих монодентатные лиганды (типа аммиака) A.6). Например, в
реакции, показанной ниже, величина константы равновесия при замене аммиака на
этилендиамин свидетельствует о более высокой (более чем в 108 раз) устойчивости
хелатного комплекса этилендиамина A.7).
[Ni(NH3N]2+ + 3NH2CH2CH2NH2
NH3
i
i
H3N/ __.,
lg*=876
[Ni(NH2CH2CH2NH2K]2+ + 6NH
3.
A.1)
NH3
H2N'
Ni2
NH3
[Ni(NH3N]2+
A.6)
'NH2
2J
[Ni(enK]
A.7)
2+
Особую стабильность хелатных комплексов в растворе можно отнести за счет
как термодинамического, так и кинетического эффекта. Термодинамически
реакция металла с хелатирующим агентом приводит к увеличению числа свободных
частиц (четыре в левой части уравнения A.1) и семь в правой), а следовательно, к
благоприятному энтропийному вкладу (AS0) в общую свободную энергию реакции
(АС0), выражаемую как
А(?° = Д#° - TAS0.
К тому же, рациональное конструирование макроцикла, конформационные и
электростатические вклады которого во взаимодействия лиганд—металл
максимальны, может также обеспечить благоприятную энтальпию реакции.
Энтропийный вклад усиливается также благодаря статистическим причинам,
поскольку для диссоциации хелатного комплекса обе связи металл - донорный
атом должны разрываться одновременно.
Наконец, в образование хелатного комплекса включаются и кинетические
эффекты. Реакция металла с лигандом L протекает со скоростью, близкой к скорости
связывания первого донорного атома хелатирующего лиганда L—L. Связывание
1.4. Хелатный и макроциклический эффекты
37
второго донорного атома молекулы L—L протекает, однако, намного быстрее,
поскольку в своем координированном состоянии он имеет гораздо более высокую
эффективную концентрацию, чем вторая молекула L монодентатного лиганда.
Несмотря на экспериментальное доказательство существования хелатного
эффекта в координационной химии растворов, его природа — предмет многих споров
в литературе. Первая проблема относится к определению понятия «константа
устойчивости»; вторая проблема: ступенчатая константа устойчивости Р12 для
связывания двух монодентатных лигандов имеет другую размерность, чем первая
константа устойчивости бидентатного лиганда, с которой проводится сравнение
(Дополнение 1.1). Поэтому влиянием концентрации растворителя пренебрегают.
Когда это различие учитывается переводом концентраций в мольные доли (т.е.
концентрация в моль-дм'/концентрация растворителя), хелатный эффект почти
исчезает. Более того, измерения устойчивости в газовой фазе также указывают на
небольшую разницу между сравниваемыми хелатными и нехелатными комплексами.
Тем не менее установлено, что, по крайней мере в растворе, хелатные лиганды
всегда почти полностью замещают своих монодентатных аналогов.
В супрамолекулярной химии термодинамическую устойчивость комплекса
хозяин—гость можно повысить действием хелатного эффекта. Донорные атомы
лиганда-хозяина являются центрами связывания (любой природы), а металл — гостем
(который, в действительности, часто является катионом металла, хотя гостями
могут быть также анионы и нейтральные частицы). Действие хелатного эффекта
наблюдается и при связывании металлов подандами (рис. 1.5) — цепочечными хозяе-
Рост
степени
организации
хозяина
Поданд
Коранд
Хелатный эффект
Хелатный и
макроциклический
эффекты
►
Хелатный и макро-
бициклический
эффекты
►
Криптанд
Рис. 1.5. Хелатный, макроциклический и макробициклический эффекты
1. Общие представления
м м
Л Л х' \
Vх \__/
Напряженное Оптимальная Возрастающая гибкость кольца
кольцо геометрия для энтропийно менее благоприятна
больших катионов
Рис. 1.6. Стабилизация, обусловленная хелатным эффектом
вами — с некоторым количеством донорных атомов, расположенных вдоль цепи с
интервалами (разд. 3.2.1).
Стабилизация, обусловленная хелатным эффектом, сильно зависит от размера
хелатного кольца (рис. 1.6). Пятичленные кольца, как в металлокомплексах
этилендиамина, часто наиболее устойчивы, поскольку слабее всего напряжены.
Четырехчленные кольца (например, хелатирующие ацетаты) сильно напряжены, и с
увеличением размера хелатного кольца статистическая вероятность того, что два
донорных атома направлены прямо на металл, постоянно уменьшается. Это
приводит к неблагоприятной энтропии. Однако энергия напряжения хелатного кольца
зависит и от размера катиона металла. Для очень маленьких катионов, таких, как
В3+ и Ве2+, шестичленные хелатные кольца довольно обычны, так как маленький
катион образует связи катион—донор с длинами, близкими тем, которые находят в
ненапряженных шестичленных циклических молекулах типа циклогексана.
Многие комплексы хозяин—гость даже более устойчивы, чем можно было бы
ожидать, учитывая только хелатный эффект. Хозяева в этих образованиях обычно
макроциклические (большое кольцо) лиганды, которые хелатируют своих гостей
посредством определенного числа связывающих центров. Такие соединения
дополнительно стабилизированы макроциклическим эффектом. Этот эффект обусловлен
не только хелатированием гостя многочисленными центрами связывания хозяина,
но и организацией этих центров связывания в пространстве таким образом, что
энергия связывания не расходуется гостем на его «укутывание» хозяином, при этом
из хелатирования извлекается максимальная выгода. Более того, потеря энтальпии,
обусловленная сближением неподеленных электронных пар донорных атомов друг
с другом, компенсируется выигрышем энтальпии еще на стадии синтеза
макроцикла. Макроциклический эффект делает циклических хозяев, таких, как коранды
(например, краун-эфиры), более стабильными (примерно в 104 раз), чем ациклические
поданды с центрами связывания того же типа. Макроциклический эффект впервые
обнаружили Д. Каббинесс (D. Cabbiness) и Д. Маргерум (D. Margerum) в 1970 г. при
изучении комплексов Cu(II) A.8) и A.9). Стабильность обоих соединений
определяется четырьмя хелатирующими донорными атомами. Однако макроцикл
A.8) примерно в 104 раз более устойчив, чем ациклический аналог A.9), что
объясняется дополнительным действием макроциклического эффекта.
1А. Хелатный и макроциклический эффекты 39
Л2+ ~Г
NH HN NH HN
m 1 Г Чм/
/ \ у к / \
NH HN NH HN
М = Zn, Си
A.8) A.9)
Термодинамические измерения аналогичных (неметилированных) комплексов
Zn2+ показали, что стабилизация макроциклическим эффектом имеет как энталь-
пийный, так и энтропийный вклад (табл. 1.3). Энтальпийная составляющая
является результатом того, что макроциклические хозяева часто менее сольватированы,
чем их ациклические аналоги. Это связано с тем, что их поверхность менее
доступна для растворителя. В результате уменьшается число разрывающихся связей
растворитель—лиганд в сравнении с таковым протяженного ациклического хозяина. С
точки зрения энтропии макроциклы конформационно менее гибки и, таким
образом, теряют меньше степеней свободы при комплексообразовании. В сущности,
потеря энтропии, связанная с макроциклом, «оплачена» заранее в ходе его синтеза.
Вообще, роли энтропийного и энтальпииного вкладов меняются в зависимости от
характера изучаемой системы, хотя энтальпия часто доминирует из-за таких
дополнительных факторов, как отталкивание неподеленных электронных пар. Бицикли-
ческие гости — криптанды (разд. 3.3) — оказались во многом по тем же причинам
даже еще более устойчивыми, чем моноциклические — коранды. Это называют
макробициклическим эффектом (см. рис. 1.5). Более подробно макроциклический и
макробициклический эффекты рассмотрены в разд. 3.8.
Таблица 1.3. Термодинамические параметры комплексов Zn2+ A.8)
и A.9), B98К)
Параметр
Комплекс A.8)
Комплекс A.9)
Д#°, кДжмоль
—ТД£°, кДжмоль"
15.34
-61.9
-25.6
11.25
-44.4
-19.8
•"» Hancock R.D. Chelate ring size and metal ion selection // J.Chcm.Ed. 1992. Vol.69.
P.615-621.
1. Общие представления
1.PJ Предорганизация
и комплементарность
Для того чтобы дополнить центры связывания гостя, хозяин должен
иметь центры связывания с подходящими электронными характеристиками
(полярность, эффективность донора или акцептора водородной связи, жесткость
или мягкость и т.д.)- Доноры водородной связи должны соответствовать ее
акцепторам, а кислоты Льюиса — основаниям Льюиса. Более того, эти центры
связывания должны быть размещены в молекуле хозяина таким образом, чтобы обеспечить
их взаимодействие с гостем; при этом молекула-хозяин должна иметь конформа-
цию, необходимую для связывания. Если хозяин удовлетворяет этим требованиям,
то говорят, что он комплементарен гостю.
Если молекула-хозяин не подвергается значительным конформационным
изменениям при связывании гостя, то говорят, что он предорганизюван. Предорганизация
гостя — это основная идея, поскольку она представляет наибольшее (в некоторых
случаях — решающее) увеличение общей свободной энергии комплексообразова-
ния с гостем. Если пренебречь эффектами сольватации, щюцесс связывания гостя
хозяином можно весьма произвольно подразделить на две стадии. Во-первых, это
стадия активации, при которой хозяин конформационно перестраивается, для того
чтобы расположить свои центры связывания наиболее комплементарно по
отношению к гостю, одновременно минимизируя неблагоприятные взаимодействия
между своими разными центрами связывания. Это энергетически невыгодно,
поскольку из-за того, что во время существования комплекса хозяин—гость хозяин должен
оставаться в связывающей конформации, эта энергия никогда не компенсируется.
Во-вторых, за перестройкой следует связывание, энергетически выгодное
вследствие энтальпийно стабилизирующего притяжения между взаимно
комплементарными центрами связывания хозяина и гостя. Полная свободная энергия комплексооб-
разования - это разность между энергией, затраченной на реорганизацию, и
энергией, выделившейся при связывании. Если энергия реорганизации велика,
тогда полная свободная энергия снижается, дестабилизируя комплекс. Если хозяин
предорганизован, то энергия перегруппировки мала.
Естественное следствие предорганизации проявляется в кинетике связывания
гостя. Жестко предорганизованные хозяева могут испытывать значительные
трудности при прохождении через переходное состояние комплексообразования и
таким образом замедлять кинетику связывания гостя. Конформационно подвижные
хозяева способны быстро приспосабливаться к изменяющимся условиям; при этом
и образование комплекса, и его разрушение происходят быстро. Сольватация
усиливает эффекты предорганизации, поскольку сольватационная стабилизация
несвязанного хозяина часто больше, чем в случае, когда хозяин «укутывает» гостя,
эффективно предоставляя ему меньшую площадь поверхности.
Эффекты предорганизации иллюстрируются сравнением предорганизованных
сферандов A.10), разд. 3.4, и конформационно подвижных корандов A.11),
различающихся по сродству к катионам щелочных металлов примерно в 1010 раз.
1.6. Термодинамическая и кинетическая селективность 41
О
О О
О О
О
18-Краун-6
Коранд
Сферанд A.10) A.11)
1.6 Термодинамическая
и кинетическая селективность
_Целью дизайна супрамолекулярного хозяина как в природе
(ферменты, транспортные белки и т.п.), так и в искусственных системах является
достижение селективности — способности отличать одного гостя от другого.
Железосодержащий гемовый транспортный белок — гемоглобин — в крови тонко настроен на
селективное поглощение О2 в присутствии N2, воды и СО2, а также таких
соединений, как СО, который обычно очень прочно связывается железом. Можно легко
оценить сродство гостя к данному рецептору с помощью константы связывания
(К, Дополнение 1.1), которая представляет собой термодинамическую константу
равновесия для процесса связывания Хозяин + Гость <р> (Хозяин-Гость):
К= [Хозяин-Гость]/[Хозяин] [Гость]. A.2)
Термодинамически селективность (S) - это отношение константы связывания
одного гостя к константе связывания другого:
Селективность этого типа достигается легче всего в сочетании с такими
важными понятиями, как аналогия «замок—ключ», «предорганизация» и «комплементар-
ность», а также с детальным знанием взаимодействий хозяин—гость. Однако
существует селективность другого типа, связанная со скоростью превращения
конкурирующих субстратов в ходе реакции. Это кинетическая селективность; она и
является основой для управления такими процессами, как супрамолекулярный
(ферментативный) катализ, обнаружение и подача сигналов гостю. В этом смысле
'42 1. Общие представления
говорят, что система селективна в отношении того гостя, который быстрее всего
превращается, а не того, который прочнее всего связывается. В самом деле, в таких
процессах с временным разрешением большие константы связывания ингибируют
систему вследствие замедления кинетики. Многие биохимические ферменты
селективны кинетически, и изучение их структуры показывает, что они совершенно
комплементарны для желаемого (иногда переходного) состояния гостя в любой
момент и обычно жестко не предорганизуются, так как это должно было бы мешать
быстрому катализу. В искусственных системах достижение разрешенной во
времени селективности (как при конструировании имитаторов ферментов, гл. 9) —
гораздо более трудная задача, так как она требует адаптации хозяина к
изменяющимся потребностям гостя в процессе реакции.
Дополнение 1.1. Константы связывания и их измерение
Термодинамическую стабильность комплекса металл—макроцикл в данном
растворителе (обычно вода или метанол) оценивают константой связывания К (используют
также термины «константа образования», К,, «константа ассоциации», Ка, или
«константа устойчивости», К). Если пренебречь коэффициентом активности, то это
просто константа равновесия реакции (например, между металлом М и хозяином-лиган-
дом в воде).
М(Н2О)«+ + L <z> MLW+ + лН2О A.4)
[MLm+]
К= [М(Н Q)W+][L] (единицы: Дм3-моль-' или М). A.5)
Так, большая константа связывания соответствует высокой равновесной
концентрации связанного металла и, следовательно, более стабильному комплексу
металл—макроцикл. Величины типичных констант связывания для краун-эфиров и
катионов щелочных металлов в воде находятся в интервале 10^—Ю2. В метаноле они
возрастают до 106 для [К+ с A8-краун-6)]. Константа связывания для К+ и
[2.2.2] криптанда составляет ~1010.
Если рассматривать последовательный процесс, включающий связывание более
одного иона металла, то можно измерить две величины К: Кп и КХ2 (например,
связывание двух ионов Na+ и дибензо-30-краун-10).
*..
М(Н2О)«+ + L <=? MLW+ + nW2O A.6)
п
М(Н2О)«+ + MLW+ <=> M2L<2w>+ + лН2О A.7)
[M2LBw>+]
12 [M(H2O)«+][MLW+] ' A *
Тогда ступенчатая константа связывания (Р12) для всего процесса определяется
.6. Термодинамическая и кинетическая селективность
43
как
или, в более общем виде, как
A.9)
[M]m[L]"
A.10)
Величины констант связывания могут изменяться в широких пределах, поэтому
вместо них часто используют lg К, следовательно:
\g$n = \g (Кхх-К{1) = \g Ku +lg Kn. A.11)
Индексы ступенчатой константы связывания указывают, с каким гостем
связывается комплексообразующий партнер: в многостадийном процессе константу
связывания хозяина с первым гостем можно обозначить как Ки, а константу связывания
получившегося комплекса 1:1 со следующим гостем с образованием комплекса 1:2 — как
АГ12 и т.д.
Поскольку константы связывания являются термодинамическими параметрами,
их, согласно уравнению Гиббса, можно выразить через свободную энергию
комплексообразования ДС° = — RT\n К. Таким образом, общее сродство хозяина к гостю в
конкретных условиях (растворитель, температура и т.д.) можно задать в значениях величин
/Гили — ДG°. Значения свободной энергии комплексообразования для комплексов
катионов щелочных металлов колеблются в интервале 20—100 кДжмоль
E—25 ккалмоль, 1 кДж = 4.184 ккал). Большая величина АГ~1010 соответствует
—ДG°, примерно равной 52 кДжмоль A3 ккалмоль). Значения констант
связывания и соответствующей им свободной энергии комплексообразования для некоторых
систем приведены в табл. 1.4.
Таблица 1.4. Константы связывания для некоторых процессов
комплексообразования
Субстрат
Лиганд
Растворитель Кп,М~х
кДжмоль~
Na+
I-
Тетрацианоэтилен
7,7,8,8-Тетрациано-
хинодиметан
Салициловая кислота
Гидрокортизон
сю4-
Гексаметилбензол
Гексаметилбензол
Пирен
Кофеин
Бензоат-ион
Н2О
СС14
СН2С12
СН2С12
Н2О
Н2О
Н2О
Н2О
Метил-/лро«оциннамат Имидазол
л-Гидроксибензойная ос-Циклодекстрин
кислота
Кофеин Кофеин Н2О
Фенол Диметилформамид С6Н6
3.2
1.35
17
0.94
44
2.9
1.0
ИЗО
19
442
_з
-0.8
-7.1
0.0
-9.7
-2.5
0.0
-17.6
-7.1
-15.0
1. Общие представления
Константы связывания можно также определить в терминах констант скоростей
(к) реакций комплексообразования и разрушения комплекса:
*.
M+ + L <==± [M+L] A.12)
K=kx/k_v A.13)
Измерение констант связывания
*-» PolsterJ., Lachmann H. Spectrometric titration. Weinheim: VCH, 1989.
В принципе константы связывания можно определить посредством любой
экспериментальной техники, которая может дать информацию о концентрации комплекса
[M+L] как функции концентрации М+. На практике чаще всего используют
следующие методы.
Потенциометрическое титрование
В случае макроциклов, способных к протонированию (например, криптанды с
основным атомом азота третичной аминогруппы у основания моста), константы протони-
рования (а следовательно, и величины рКа) можно легко определить, используя
рН-электроды для простого титрования кислота—основание. При добавлении
катиона будет изменяться основность макроцикла из-за его конкуренции с Н+ за неподе-
ленную пару азота и, значит, будет меняться вид кривых титрования. Константы
устойчивости реакции комплексообразования металла можно определить анализом
различных равновесий с помощью компьютерной программы подгонки кривых.
Я М Р-титрован ие
Если обмен между комплексом и свободным гостем — медленный процесс в
масштабе времен ЯМР, то константу связывания можно определить в широкой области
концентраций, температур, растворителей и т.д. простым интегрированием сигналов
ЯМР для связанного и свободного хозяина или гостя. Однако большинство
равновесий хозяин—гость устанавливается быстро в масштабе времен ЯМР, и химический
сдвиг, наблюдаемый для данного резонанса (который чувствителен к комплек-
сообразованию), является средневзвешенным между химическими сдвигами
свободной и связанной частиц. В типичном эксперименте ЯМ Р-титрования небольшие
порции гостя добавляют к раствору хозяина известной концентрации в дейтерированном
растворителе, а затем ЯМР-спектр образца анализируют как функцию концентрации
гостя или отношения хозяинггость. Обычно изменение в химическом сдвиге (Д8)
записывают для различных атомных ядер (например, 'Н в ЯМР 'Н) как функцию
влияния связывания гостя на их магнитное окружение. В результате получают два вида
информации. Обнаружение ядра с таким максимальным влиянием может дать
качественную информацию о региоселективности гостя (находится ли гость в
полости хозяина?). Более важным, однако, является вид кривой титрования (зависимость
Д8 от концентрации добавляемого гостя, рис. 1.7), который дает количественную
1.6. Термодинамическая и кинетическая селективность
(а)
0.5-
Концентрация гостя
(б)
Быстрый обмен
Медленный обмен
Концентрация
гостя
I I Г II I I I I I ГП I
б, М.Д.
I I I I III I Г
6, М.Д.
t t
I Ядро, нечувствительное
к комплексообразованию
Чувствительное ядро Аналогичное ядро
свободного лвганда в комплексе
Рис. 1.7. (а) ЯМР-титрование для системы с быстрым установлением равновесия
относительно шкалы времен ЯМР. (б) Схематические ЯМР-спектры равновесных
смесей свободного хозяина, гостя и комплекса хозяин—гость
информацию о константе связывания. Такие кривые титрования часто анализируют
компьютерной подгонкой кривых по методу наименьших квадратов (например, с
помощью программы EQNMR), используя уравнения A.14) для определения
оптимальных величин 8т/7 (химический сдвиг каждого из присутствующих компонентов, где п
и т — цифровые индексы, указывающие, как соотносятся хозяин (Н) и гость (G)) и
$тп (ступенчатая константа связывания).
m=i n=j
8Cfl/c -
A.14)
m=\ Ai=0
>total
Основной аспект таких расчетов — использование правильной стехиометрии (т.е.
выбранного отношения хозяин:гость). В литературе по супрамолекулярной химии
существует устойчивая тенденция подгонять данные к стехиометрии 1:1, при этом
1. Общие представления
Рис. 1.8. График Джоба для
комплекса 1:1 хозяин—гость
0.2 0.4 0.6 0.8
[Хозяин]/([Хозяин] + [Гость])
обычной ошибкой является игнорирование больших агрегатов. Стехиометрия
связывания может быть подтверждена с помощью любого метода титриметрического
анализа, который позволяет определить концентрацию комплекса составлением сепии
растворов с различным соотношением хозяин:гость при их постоянной суммарной
концентрации.
График зависимости концентрации [Комплекс] от выражения [Хозяин]/([Хозяин] +
[Гость]) можно построить, определив изменение концентрации комплекса
хозяин-гость. В случае комплекса 1:1 зависимость такого типа (график Джоба) должна
дать максимум при 0.5 (рис. 1.8).
Флуоресцентное титрование
Эти измерения основаны на пропорциональности интенсивности флуоресценции и
концентрации флуорофора (концентрация флуоресцентных частиц в растворе; часто
это флуоресцентный гость, G). Для комплекса 1:1с хозяином Н и константой
устойчивости Кп = [HG]/[H][G] интенсивность флуоресценции (/^дается выражением
G u A.15)
где kG и ки — коэффициенты пропорциональности для гостя и комплекса 1:1
хозяин—гость соответственно.
В отсутствие хозяина интенсивность флуоресценции (Fo) дается выражением
^0 = *G°Gr A.16)
nieG,= [G] + [HG].
Объединение этих двух соотношений приводит к уравнению A.17), которое
является основой для определения константы устойчивости (/Г,,) большинством
флуоресцентных методов.
1.6. Термодинамическая и кинетическая селективность
Fo \ + Ки[И]
A.17)
Если гость или комплекс хозяин—гость не флуоресцирует, т.е. флуоресценция
«включается» при комплексообразовании или подавляется хозяином, то это
уравнение сильно упрощается. Тогда либо kG, либо ки становится равной нулю. Например,
при kG = к£ и ки = О получаем:
Fo/F= 1 + Ки[И] . A.18)
Зависимость F0/Fot [H], полученная при титровании раствора гостя
хозяином-тушителем, должна быть прямой линией с наклоном Ки. Обычные флуоресцентные
гости, такие, как 8-анилино-1-нафталинсульфонат (8-anilino-l-naphthalene-
sulphonate, ANS A.12)), могут быть использованы для тестирования стабильности и
комплексообразующей способности различных хозяев.
A.12)
ANS
Калориметрическое титрование
Как и другие методы титриметрического анализа, калориметрическое титрование
включает измерение какого-нибудь параметра (в данном случае теплоты, выделяемой
тщательно изолированным образцом) как функции концентрации добавляемого
гостя или хозяина. Изменение теплоты комплексообразования относится
непосредственно к термодинамическим параметрам этого процесса.
Эксперименты по экстракции
Коэффициент распределения (или разделения, Kd) катиона металла между водной
(aq) и органической (org) фазами можно также использовать для оценки
селективности данного макроцикла к ряду металлов в стандартных условиях с участием констант
связывания (К) в следующих процессах (уравнения A.19)—A.22) для пикрата металла
(Пик), воды (aq) и хлороформа, насыщенного водой (org), при 25 °С).
[М+Пик~] + [Гость] = [М+ГостьПик~] , Ки (константасвязывания)
A.19)
[М+]а(? + [Пик~]а(? + [Гость]о^ = [М+-Гость-Пик~] Ке (константа экстракции)
A.20)
48 1- Общие представления
[М+] + [Пик~] = [М+Пик-]0/?, Kd (коэффициент распределения)
A.21)
Концентрацию пикрат-аниона (а следовательно, и М+) определяют из данных по
электронному поглощению C80 нм) для каждого слоя. Предполагается, что хозяин,
по существу, нерастворим в водном слое. Эта методика имеет относительно низкую
точность, но она требует малого времени и годится для мониторинга многих
соединений. Она пригодна для измерения свободной энергии связывания в интервале
20—70 кДжмоль. Величины энергии связывания хозяев выше 70 кДжмоль
определяют при их сопоставлении с данными, полученными для хозяев с известной
энергией связывания.
*-» Connors К.A. Binding constants. Chichester: J. Wiley & Sons, 1987.
Природа
супрамолекулярных взаимодействий
Супрамолекулярная химия имеет дело с нековалентными
связывающими взаимодействиями. Термин «нековалентный» охватывает разнообразные
силы притяжения и отталкивания. Наиболее важные из них рассмотрены ниже с
указанием их приблизительных энергий. При анализе супрамолекулярной системы
очень важно учитывать взаимную «игру» всех этих взаимодействий и эффекты,
относящиеся как к хозяину, так и к гостю, а также к их окружению (например,
сольватация, кристаллическая решетка, газовая фаза и пр.).
Ион-ионные взаимодействия A00-350 кДж моль)
По силе ионная связь сравнима с ковалентным связыванием (энергия
связи 100-350 кДжмоль). Типично ионное твердое тело - хлорид натрия,
имеющий кубическую решетку, в которой каждый ион натрия окружен шестью
анионами хлора (рис. 1.9). Требуется большое воображение, чтобы представить NaCl как
супрамолекулярное соединение, но эта простая ионная решетка, действительно,
иллюстрирует, каким образом катион натрия способен организовать шесть
комплементарных донорных атомов вокруг себя, чтобы максимизировать нековалентные
ион-ионные взаимодействия. Заметим, что в растворе решеточная структура этого
типа распадается из-за эффектов сольватации с образованием частиц, таких, как
лабильный октаэдрический ион Na(H2O>6.
1.7. Природа супрамолекулярных взаимодействий
49
Рис. 1.9. Ионная решетка NaCl
Cl -Na Cl
Na-
Cl-
Na-
£1-
Cl-
■Na
Na
■Cl-
-NaCl-—
Na-
Cl-
-Cl
-Na
—Cl—-
■Na
---Na
-ci-
Na
—Na
—Cl
•Cl
-Na
Гораздо более наглядный пример супрамолекулярных ион-ионных
взаимодействий — взаимодействие хозяина трис(метилен-1,3,5-диазабициклооктан)-2,4,6-
триметилбензола A.13), несущего заряд 3+, с анионами, например с Fe(CN)|~
(рис. 1.10).
Рис. 1.10. Супрамо-
лекулярные
ион-ионные взаимодействия
на примере
комплекса органического
катиона A.13)
с Fe(CNN3~
50
1. Общие представления
(ЫЗ)
1JJ.2 Ион-дипольные взаимодействия
E0-200 кДж моль)
Пример ион-дипольного взаимодействия - связывание иона Na+ с
такой полярной молекулой, как вода. Связывание этого типа наблюдается как в
твердом состояни:1, так и в растворе. Супрамолекулярная аналогия легко
просматривается в структурах комплексов катионов щелочных металлов с макроциклическими
(большое кольцо) простыми эфирами — краун-эфирами (гл.З), в которых эфирные
атомы кислорода играют ту же роль, что и полярные молекулы воды. Неподеленные
электронные пары кислорода притягиваются к положительному заряду катиона.
он2
н2о„, | >л6н2
'".Na+
Н2О
*он2
он2
A.14)
°3
О
О
Na+-KOMimeicc краун-эфира
A.15)
[Ru(bipyK]2+
bipy - 2,2-бипиридил
A.16)
Ион-дипольные взаимодействия включают также координационные связи,
которые в случае взаимодействий неполяризуемых катионов и сильных оснований по
своей природе в основном электростатические. Координационные (дативные)
связи с существенной ковалентной составляющей, как в [Ru(bipyK]2+, также часто
используют в супрамолекулярных ансамблях, и, как будет показано в гл. 7 и 8,
различие между супрамолекулярными и молекулярными частицами может стать весьма
неясным.
1.7. Природа супрамолекулярных взаимодействий §|
1*7*3 Диполь-дипольные взаимодействия
E-50 кДж моль'1)
Взаимная ориентация одного диполя относительно другого может приводить к
значительным притягивающим взаимодействиям благодаря согласованию либо
двух полюсов соседних молекул (тип I), либо одного диполя с другим (тип II),
рис. 1.11. Такое поведение характерно для органических карбонильных соединений
в твердом состоянии и, согласно расчетам, предполагается, что энергия
взаимодействия по типу II равна -20 кДжмоль, что сравнимо с умеренно сильной
водородной связью. Однако точка кипения кетонов, например ацетона E6 °С), говорит об
относительно слабом диполь-дипольном взаимодействии этого типа в растворе.
R'(// 6+
6+ R
RV,, б+ б~
^^.C:=O С
Рис. 1.11. Диполь- A n-
дипольные j? » g_"
взаимодействия R R
в карбонилах т
1«7«4 Водородная связь D-120 кДж моль)
Водородную связь можно рассматривать как особый вид диполь-ди-
польного взаимодействия, в котором атом водорода, присоединенный к
электроотрицательному атому (или группе, отрывающей электрон), притягивается к диполю
соседней молекулы или функциональной группы. Благодаря своей относительно
сильной и направленной природе водородная связь считается «ключевым
взаимодействием в супрамолекулярной химии». Прекрасным примером такого
взаимодействия является образование димеров карбоновой кислоты, которое приводит к
сдвигу v(OH) ИК-частоты валентных колебаний от -3400 до -2500 см; причем этот
сдвиг сопровождается значительным уширением и усилением полосы поглощения.
Типичное расстояние 0-0 при водородном связывании равно 2.50—2.80 А,
хотя взаимодействия на расстоянии свыше 3.0 А тоже могут быть значительными.
Водородная связь с большими атомами, например с хлором, обычно длиннее и
может быть слабее из-за сниженной (по сравнению с малыми атомами)
электроотрицательности большого галогена-акцептора, хотя сила водородных связей сильно
зависит их окружения. В супрамолекулярной химии водородные связи вездесущи. В
частности, водородные связи определяют общую форму многих белков,
распознавание субстратов многочисленными ферментами и структуру двойной спирали
ДНК (рис. 1.12).
Значения длин, сил и геометрий водородных связей могут изменяться в
огромных пределах. Сильная одинарная водородная связь, приходящаяся на молекулу,
1. Общие представления
н
0-Н---0
0---Н-0
Рис. 1.12. Водородные связи при
образовании димеров карбоновой
кислоты и спаривании оснований
в ДНК
Каркас.
N—Н о Каркас
Н гч О Н — N н
Н
Гуанин Цитозин
может быть достаточной, чтобы обусловить структуру твердого состояния и оказать
весомое влияние на растворы и газовую фазу. Более слабая водородная связь
играет роль в стабилизации структуры и может быть важна при многоактовом
взаимодействии. В табл. 1.5 приведены некоторые основные параметры таких связей.
Общепринято, что в случае водородной связи между нейтральными частицами
существует прямая корреляция ее силы (в терминах энергии образования) и
кристаллографически определенного расстояния между донором и акцептором водо-
Таблица 1.5. Свойства водородных связей
Параметры
Водородное взимодействие
среднее
слабое
Взаимодействие А—Н---В
Энергия связи, кДжмоль
Длина связи, A t
Н-В
А-В
Углы, град
Относительный сдвиг
И К-колебаний
(симметричный валентный тип,
см-'), %
Химический сдвиг
'Н ЯМР вдоль поля, м.д.
Примеры
Преимущественно
ковал ентное
60-120
1.2-1.5
2.2-2.5
175-180
25
Преимущественно
электростатическое
16-60
1.5-2.2
2.5-3.2
130-180
10-25
Электростатическое
<12
2.2-3.2
3.2-4.0
90-150
<10
14-22 <14
Газофазные димеры Кислоты
с сильными
кислотами/основаниями
«Протонная губка» Спирты
HF-комплексы Биомолекулы
Второстепенные
компоненты
бифуркационных
связей
Водородные
С—Н-связи
Водородные
О—Н--71-СВЯЗИ
1.7. Природа супрамолекулярных взаимодействий
О" О
И
°\ /° '
К+
/ \
о- о
И
\ /
к+
/ \
О' О
,-оИо- -
°\ /° н
к+
/ \
0" О
и
\—о о-н-
\ /
к+
/ \
О" О
°\ /° н
к+
О" О
И
--о о-н---
\ /
к+
Рис. 1.13. Псевдоводородные
связивКНС2О4
родной связи. Д. Брага с соавторами (D. Braga et al., 1998) показал, что это не
обязательно справедливо в случае ионных соединений. При межионных
взаимодействиях, например в гидрооксалате калия КНС2О4, ab initio расчеты показывают, что
водородные взаимодействия между парами анионов НС2О^" являются
отталкивающими во всех направлениях, т.е. притягивающее взаимодействие типа О—Н---0
отсутствует. Несмотря на это, расстояние 0-0 чрезвычайно мало, что говорит об
очень сильной водородной связи. Это кажущееся противоречие для данной
системы и ряда родственных систем объясняется сильным притяжением катионов К+ к
анионам НС2О^ , превалирующим над анион-анионным отталкиванием (рис. 1.13).
Схожесть с водородной связью в традиционном понимании возникает из-за того,
что при взаимной ориентации в системе, когда О—Н-группа направлена к атомам
кислорода ближайшего аниона цепи, межанионное отталкивание минимально.
В последнее время значительный интерес вызывают водородные
взаимодействия с участием атомов водорода, связанных с атомом углерода, а не
электроотрицательными атомами, такими, как N и О (электроотрицательность: С - 2.55, Н - 2.20,
N — 3.04, О — 3.44). Хотя эти взаимодействия и являются слабейшими на
энергетической шкале водородных связей, присутствие электроотрицательных атомов
около атома углерода может значительно увеличить кислотность С—Н протона, что
приведет к образованию диполя. Взаимодействие метильной группы нитрометана с
пиридильным краун-эфиром, показанное на рис. 1.14, является изящным
примером водородных связей C-H-N и С-Н-О.
*~| Jelfiy G.A. An introduction to hydrogen bonding. Oxford: Oxford University Press,
1997.
1*7*5 Катион-л-взаимодействия E-80 кДж моль)
Хорошо известно, что катионы переходных металлов, например Fe2+,
Pt2+ и других, образуют комплексы с такими олефиновыми и ароматическими
углеводородами, как ферроцен [Fe(C5H5J] и соль Цейзе [PtCl3(C2H4)]~. Связь в
подобных комплексах прочна и никоим образом не может считаться нековалентной,
поскольку она обусловлена частично заполненными d-орбиталями металлов. Даже
такие образования, как Ag+-C6H6, имеют значительную ковалентную составляю-
1. Общие представления
(б)
Рис. 1.14. (а) Водородные связи C-H-N B.21 Л) и C-H--0 B.41 Л, среднее значение)
в комплексе краун-эфира с нитрометаном (по Weber Е. et al., 1986). (б) Схематическая
структура соответствующего краун-эфира
щую. Однако взаимодействие катионов щелочных и щелочноземельных металлов с
двойной связью С=С хотя и является менее ковалентным, «слабым»
взаимодействием, но играет очень важную роль в биологических системах. Например, энергия
взаимодействия К+ и бензола в газовой фазе составляет -80 кДжмоль
(рис. 1.15). Для сравнения, на присоединение К+ к одной молекуле воды
затрачивается только 75 кДжмоль. Причиной лучшей растворимости К+ в воде, чем в
бензоле, является то, что с ионом калия может взаимодействовать много молекул
1.7. Природа супрамолекулярных взаимодействий
Рис. 1.15. Схематическое
изображение катион—71-взаимодействия.
Справа показан квадрупольный
момент бензола, который представлен
в виде двух противоположно
направленных диполей
воды, тогда как только немного объемистых молекул бензола могут устроиться
вокруг него. Взаимодействие неметаллических катионов, например RNH3, с
двойными связями можно считать разновидностью водородной связи Х-Н—п.
•"» Ma J.C. and Dougherty D. The cation-я interaction // Chem.Rev. 1997. Vol.97.
P. 1303-1324.
ji-Ti-Стэкинг-взаимодействия @-50 кДж моль)
Это слабое электростатическое взаимодействие часто происходит
между ароматическими кольцами, когда одно из них относительно богато
электронами, а другое испытывает их недостаток. Существует два основных типа
я-стэкинга: «плоскость к плоскости» и «торец к плоскости», хотя известно и
большое число промежуточных вариантов (рис. 1.16). Взаимодействия при я-упаковке
типа «плоскость к плоскости» отвечают за «скользкость» графита и его смазочные
свойства. Такие же я—я-стэкинг-взаимодействия между арильными кольцами пар
нуклеооснований помогают стабилизировать двойную спираль ДНК.
Взаимодействия типа «торец к плоскости» можно рассматривать как слабые водородные связи
между слегка электронодефицитными атомами водорода одного ароматического
кольца и обогащенным электронами я-облаком другого. Взаимодействия этого
типа обуславливают характерную «ёлочную» упаковку в кристаллических структурах*
ряда малых ароматических углеводородов, включая бензол (рис. 1.17).
Для объяснения разнообразия ориентации, наблюдаемых при л—я-стэкинг-вза-
имодействиях, и для количественного предсказания энергий взаимодействия
Дж. Сандерс (J. Sanders) и К. Хантер (С. Hunter) из университетов Кембриджа и
Шеффилда (Великобритания) предложили простую модель стэкинга, которая учи-
Рис. 1.16. Основные типы л—71-стэкинга.
Отметьте смещение ароматических колец
в типе « плоскость к плоскости» (полное
перекрывание колец приводит к их
отталкиванию)
3.5 А
Торец к плоскости
* Все кристаллические структуры, которые показаны на рисунках, были определены
методом рентгеноструктурного анализа (РСА). (Примеч. ред. перевода.)
56i
1. Общие представления
Рис. 1.17. Кристаллическая
структура бензола с мотивом
«ёлочной» упаковки,
возникающим из-за взаимодействий
«торец к плоскости»
тывает конкуренцию электростатических и ван-дер-ваальсовых сил. Их модель
основана на взаимодействии притяжения Ван-дер-Ваальса (разд. 1.7.7),
пропорциональном площади поверхности контакта двух л-систем. Это притягивающее
взаимодействие преобладает в общей энергии л—я-взаимодействия и может
рассматриваться как притяжение между отрицательно заряженным облаком я-эле-
ктронов одной молекулы и положительно заряженной а-структурой соседней
молекулы. Ориентация двух взаимодействующих молекул друг относительно друга
определяется электростатическим отталкиванием двух отрицательно заряженных
я-систем (рис. 1.18).
Сандерс и Хантер подчеркивают важность взаимодействий между парами
атомов, а не молекул в целом, и, несмотря на то, что их подход был относительно
успешным, природа таких л—л-стэкинг-взаимодействий до сих ггор остается
предметом дискуссий. В частности, в работе К. Вилкокса (С. Wilcox) из Питтсбург-
ского университета (США), затрагивающей эффекты заместителей, утверждается,
что дисперсионные силы Лондона могут играть более важную роль, чем
электростатические взаимодействия (разд. 1.1.7).
*"* Hunter C.A. and Sanders J.K.M. The nature of n—n interactions //
J.Amer.Chem.Soc. 1990. Vol.112. P.5525-5534.
1.7. Природа супрамолекулярных взаимодействий
57
Рис. 1.18.
Взаимодействующие Ti-квадруполи
Притяжение
1«7«7 Силы Ван-дер-Ваальса
(< 5 кДж моль; переменные)
Взаимодействия Ван-дер-Ваальса возникают благодаря поляризации
электронного облака из-за соседства близлежащих ядер, приводящей к слабому
электростатическому притяжению. Эти силы не являются направленными и
поэтому обладают ограниченными возможностями при конструировании конкретных
хозяев для комплексообразования данных гостей. Вообще говоря, ван-дер-ваальсо-
вы взаимодействия обеспечивают основной вклад в притяжение у большинства
«мягких» (поляризуемых) частиц. Именно эти силы обуславливают взаимодействия
между благородными газами. В супрамолекулярной химии они наиболее важны
при образовании соединений включения, в которых небольшие, типично
органические молекулы свободно встраиваются внутрь кристаллических решеток или
молекулярных полостей. Это видно на примере включения толуола внутрь
молекулярной полости макроцикла на основе я-трет-бутилфенола, я-трет-бутилка-
ликс[4]арена (см. разд. 5.3.1.2, рис. 1.19).
Строго говоря, ван-дер-ваальсовы взаимодействия можно подразделить на
дисперсионные (силы Лондона) и обменно-отталкивающие. Дисперсионные силы
являются составляющей притяжения, возникающего благодаря взаимодействию
между флуктуирующими мультиполями (квадруполи, октуполи и др.) соседних
молекул. Притяжение очень быстро уменьшается с расстоянием (зависимость /^6) и
является добавкой к каждой связи, внося вклад в общую энергию взаимодействия.
Обменно-отталкивающие силы определяют форму молекул и при малом
расстоянии уравновешивают дисперсионные силы, убывая по закону г~п.
1. Общие представления
Рис. 1.19. Кристаллическая
структура типичного ван-дер-ваальсового
комплекса включения п-трет-5ути\-
каликс[4]арентолуол (поAndreettiG.
etal., 1979)
1.у.8 Плотная упаковка в твердом состоянии
Плотная упаковка кристалла важна для определения его структуры.
Это подытожено в выражении «природа не терпит пустоты», однако, согласно
теории плотной упаковки Китайгородского, это просто проявление максимизации
благоприятных изотропных взаимодействий Ван-дер-Ваал ьса. Теория
Китайгородского говорит о том, что, по мере того как молекулы усложняются при переходе к
димерам, тримерам, высшим олигомерам и, наконец, к кристаллам, их форма
упрощается. Это означает, что молекула «втискивается» во впадины, образованные ее
соседями, таким образом, что при этом достигается максимальное число
межмолекулярных контактов, подобно популярной компьютерной игре «Тетрис». Известно
очень немного твердотельных структур, имеющих значительную долю «пустого»
пространства. Объекты, содержащие такие пустоты (например, цеолиты; разд.
5.1.2), обладают очень жестким каркасом, способным противостоять «взрыву»,
который может возникнуть из-за огромной разницы давлений между атмосферой и
пустыми порами или каналами кристаллов. Свойства таких материалов могут
оказаться весьма полезными и интересными при катализе и разделении.
*"* Kitaigorodski A.I. Molecular crystals and molecules. New York: Academic Press, 1973.
1.7*9 Гидрофобные эффекты
Гидрофобные взаимодействия, ошибочно принятые за силу, следует
трактовать, однако, как результат выталкивания больших или слабосольватирован-
ных частиц (например, через водородные связи или дипольные взаимодействия) из
полярных растворителей, в основном из воды. Этот эффект проявляется в
несмешиваемости минерального масла и воды. По существу, молекулы воды сильно при-
1.7. Природа супрамолекулярных взаимодействий
§59
VH Ь-н ^н
н
о-н
Сольватированный гость
Сольватированный хозяин
н И," I н
к „or' о~н _^н
Н Комплекс
Рис. 1.20. Гидрофобное связывание органических гостей в водном растворе
тягиваются друг к другу, приводя к агломерации других компонентов системы
(таких, как неполярные молекулы) по мере реализации сильных взаимодействий
внутри раствора. Это напоминает притяжение между двумя органическими
молекулами, хотя между самими органическими молекулами, естественно, действует
ван-дер-ваальсово и п—л-стэкинг-притяжение. При связывании органических
гостей циклодекстриновыми и циклофановыми хозяевами в воде (гл. 5) гидрофобные
эффекты играют решающую роль и могут быть разделены на две энергетические
составляющие: энтальпийную и энтропийную. Энтальпийный гидрофобный эффект
влечет за собой стабилизацию водных молекул, которые, связывая гостя,
освобождают полость хозяина. Из-за того что полости хозяина часто гидрофобны, вода
внутри полости не сильно взаимодействует со стенками хозяина и потому высокоэнер-
гетична. По мере выхода в растворитель происходит стабилизация молекулы воды
за счет ее взаимодействий с такими же молекулами. Энтропийный гидрофобный
эффект возникает благодаря тому, что присутствие двух (часто органических)
молекул в растворе (гость и хозяин) создает две «полости» в структуре воды.
Объединение гостя и хозяина при образовании комплекса приводит к меньшему нарушению
структуры растворителя и, следовательно, к приросту энтропии (приводящему к
уменьшению общей свободной энергии). Процесс схематически показан на
рис. 1.20.
В качестве примера рассмотрим связывание я-ксилольного гостя циклофано-
вым хозяином A.17) (гости этого класса более детально описаны в разд. 5.3.4.5).
Константа связывания в воде равна 9.3-103 М. При 293 К энергия комплексообра-
зования равна -22 кДжмоль и подразделяется на благоприятную, энтальпийную,
Д#° = —31 кДжмоль, и неблагоприятную, энтропийную, T£±.S° - —9 кДжмоль" ,
составляющие стабилизации. Именно вклад энтальпии в гидрофобное связывание
в этом случае доминирует. Этот вклад слишком велик, чтобы быть результатом сил
притяжения между гостем и хозяином (которые испытывают лишь слабые я-я-стэ-
кинг- и ван-дер-ваальсовы взаимодействия), и поэтому он должен включать особые
силы взаимодействия растворитель—растворитель. В случае метанола как
растворителя энтальпийная составляющая уменьшается из-за более слабых взаимодействий
растворитель—растворитель.
60
1. Общие представления
ОМ
О ОМе
п-Ксилол
ОМе
Me
A.17)
Smithrud D.B., Sanford E.M., Chao I. et al. Solvent effects in molecular recognition //
Pure Appl. Chem. 1990. Vol.62. P.2227-2236.
1.8 Супрамолекулярное конструирование
хозяина
Для конструирования такого хозяина, который селективно свяжет
определенного гостя, наряду с концепцией комплементарное™ (соответствие стери-
ческих и электронных состояний хозяина и гостя), используют хелатный и макро-
циклический эффекты и, что является решающим, предорганизацию хозяина.
Четкое определение и тщательный анализ цели — первые шаги в дизайне
(конструирование) хозяина. Этот подход позволяет сразу прийти к заключению о
свойствах нового хозяина. Если гостем должен стать катион металла (гл. 3), то важным
являются его размер (радиус иона), плотность заряда и «жесткость» (Дополнение
3.2). (Например/такие «мягкие» донорные атомы, как сера, удобны для связывания
«мягких» гостей, в частности Hg2+, Ag+ и Pb2+.) При анионном комплексообразо-
вании (гл. 4) эти факторы также влияют на сферические анионы — хлориды,
бромиды и другие, а для несферических гостей в игру вступают такие факторы, как их
форма и заряд, а также донорные характеристики водородной связи. Органические
катионы и анионы могут нуждаться в хозяевах, имеющих и гидрофильные, и
гидрофобные области, в то время как гостям — нейтральным молекулам — может не хватать
особых «рычагов» — полярных групп, которые могут сильно взаимодействовать с
хозяином.
После определения параметров, например размера хозяина, заряда и характера
атомов донора и других, можно начинать творческий процесс сшивки лиганда.
Ключевая концепция этого процесса — организация. Взаимодействия
хозяин-гость происходят через центры связывания. Тип и число этих центров должны
быть такими, чтобы максимально дополнять (быть комплементарными)
характеристики центров связывания гостя (вспомните определение гостя как партнера с
расходящимися центрами связывания), и центры связывания должны, как правило,
1.8. Супрамолекулярное конструирование хозяина 61
располагаться на органическом каркасе хозяина, имеющем подходящий для гостя
размер. Центры связывания должны находиться на некотором расстоянии друг от
друга, чтобы минимизировать взаимное отталкивание, но располагаться таким
образом, чтобы иметь возможность одновременно взаимодействовать с гостем. Чем
больше существует благоприятных взаимодействий, тем лучше. Обычно наиболее
стабильные комплексы получаются в случае предорганизации хозяев (разд. 3.8.1),
т.е. когда при связывании не происходят ни энтропийно, ни энтальпийно
невыгодные перестройки, которые понижали бы полную свободную энергию комплексооб-
разования. Такие хозяева являются идеальными «стоками» для своих гостей, когда
связывание полностью необратимо. Этот вид комплексообразования идеален,
например, для удаления ионов из загрязненной воды. Хозяева, связывающие своих
гостей менее сильно (т.е. существует некое равновесие между связанными и
несвязанными частицами), находят применение как сенсоры и носители, в которых
необходима последовательность событий «связь — обнаружение — выделение» или «связь
— транспорт — выделение».
Природа органического каркаса самого хозяина (лиганд), будь он липофильным
или гидрофильным, играет существенную роль в поведении гостя. Это определяет
растворимость хозяина и его комплексов. Толщина лиганда и легкость доступа к
связывающей полости или трещине влияет как на термодинамическую
стабильность образовавшегося комплекса, так и на кинетику связывания. Введение
боковых групп (например, длинных алкильных цепей) может усилить липофильность
хозяина или стимулировать его взаимодействие с каким-нибудь внешним
комплексом, например с полимерной подложкой или биомолекулой. Такие хозяева
используют для направленного транспорта радиоизотопов к определенным участкам
человеческого тела с целью радиационной диагностики в медицине или разработки
искусственных «имитаторов ферментов» (гл. 9).
Обобщение рассмотренных выше принципов и подходов привело к созданию
области знания, названной обобщенной координационной химией, охватывающей как
супрамолекулярную, так и классическую (вернеровскую) неорганическую
координационную химию.
fr Lehn J.-M. Perspectives in supramolecular chemistry — from molecular recognition
towards self-organisation // Pure Appl. Chem. 1994. Vol. 66. P. 1961-1966.
1. Общие представления
Учебные задания
1.1 Термодинамические параметры реакции Си2* с разными лигандами
приведены ниже (водный раствор, 25 °С). Используйте эти данные для расчета
констант связывания (lg К) комплексов металл-лиганд 1:1. Объясните
наблюдаемую разницу в стабильности.
Лиганд
Д#°, кДж моль
-1
TAS°, кДж моль"
-105
-90.4
7.1
24.3
-76.6
64.0
1.2 Дайте краткое определение термина «супрамолекулярная химия». Объясните
разницу между молекулярными и супрамолекулярными взаимодействиями.
Проиллюстрируйте свой ответ примерами супрамолекулярных
взаимодействий и сравните их важность.
1.3 Изобразите относительную шкалу сил супрамолекулярных взаимодействий,
используя информацию, данную в разд. 1.7. Сопоставьте ее со второй шкалой
важности этих взаимодействий в супрамолекулярном дизайне хозяина для
металлических катионов, принимая во внимание такие факторы, как
направленность, легкость встраивания в каркас хозяина и склонность к усилению
связывания за счет многоцентровых взаимодействий. Изменится ли эта оценка,
если Вам нужно было бы сконструировать хозяина для анионов или
нейтральных молекул? Какие взаимодействия могли бы быть важны при
конструировании хозяина для следующих соединений: метан, бензол, метанол, фенол,
аммоний, С1~, Na+ и Ni2+?
1.4 Используя хронологию, приведенную в табл. 1.1, выскажите предположение о
том, какие открытия наиболее важны для супрамолекулярной химии? Как Вы
думаете, почему понадобилось столько времени, чтобы эта область химии
превратилась в отдельную дисциплину?
Литература
Литература
Andreetti, 1979 Andreetti G.D., Ungaro R., Pochini A. Crystal and molecular structure of
cyclo{quarter[E-/-butyl-2-hydroxy-1,3-phenylene)methylene]}toluene A:1)
c)athrate//J. Chem. Soc, Chem. Commun. 1979. P. 1005.
Braga , 1998 Braga D., Grepioni K, Novoa J.J. Inter-anion O-H—O hydrogen bond like
interactions: the breakdown of the strength-length analogy // Chem. Commun. 1998.
P. 1959.
Cram, 1986 Cram D.J. Preorganisation - from solvents to spherands // Angew. Chem., Int.
Ed. Engl. 1986. Vol.25. P. 1039.
Kim, 1998 Kim E.-I., Paliwal S.t Wilcox C.S. Measurements of molecular electrostatic field
effects in edge-to-face aromatic interactions and СН-л interactions with
implications for protein folding and molecular recognition // J. Amen Chem. Soc.
1998. Vol. 120. P.Ill92.
Weber, 1986 Weber E., Franken $., Puff H., AhrendtJ. Enclave inclusion of nitromethane by a
new crown host — X-ray crystal structure of the inclusion complex and host
selectivity properties // J. Chem. Soc, Chem. Commun. 1986. P.467.
2 Супрамолекулярная
тпмпа •аетпгпж
ХИМИЯ ЖИЗНИ
Природа, что создала нас из четырех стихий,
Воюющих внутри нас за порядок,
Подстегивает дерзновенный ум,
Который души наши побуждает
Познать всю чудную Архитектуру мира
К. Марлоу A564-1593). Завоевания Тамбурлена
Истоки супрамолекулярнои химии лежат в химии живых организмов.
Природа, иногда невероятно сложная, иногда элегантно простая, достигла в этой
области высокой точности и селективности, что дает возможность живым системам
сохранять себя в окружающей среде, питаться, дышать, воспроизводиться и
отвечать на внешние раздражители. Супрамолекулярные хозяева в биологической
химии - это рецепторные участки ферментов, ионов, антител иммунной системы и
ионофоров, а гости — субстраты, ингибиторы, медицинские препараты на основе
кофакторов или антигены. Все такие компоненты проявляют следующие
супрамолекулярные свойства: способность к распознаванию, самосборке,
самоорганизации, а также кинетическую и термодинамическую комплементарность.
Подавляющее большинство этих свойств обусловлено супрамолекулярными
взаимодействиями, такими, как координационные (ион-диполь) связи, водородные связи и п—п-
стэкинг, рассмотренные в разд. 1.7. Поэтому биологические системы — это
совершенные супрамолекулярные системы. Большая часть усилий в области
супрамолекулярнои химии была затрачена на попытки смоделировать или сымитировать
следующие биологические процессы: катализ органических реакций ферментами,
селективный транспорт катионов металлов или молекулярных субстратов, например О2.
Наше понимание биологических систем, являющихся частью этих процессов,
невероятно выросло, но надо честно признать, что созданные стараниями человека
объекты молекулярной и супрамолекулярнои химии еще сильно отстают от своих
биохимических аналогов по масштабу, объему и функциональности. Однако тот факт,
что в природе существует такая богатая и эффективная супрамолекулярная химия,
представляет собой огромный стимул для продолжения поисков еще более
усложненных абиотических (небиологических) аналогов. Это служит также мотивацией
для развития синтетических систем, способных подвергаться превращениям или
обладающих свойствами, не обнаруженными в природе. В этой главе, давая
краткий обзор некоторых, самых важных, биохимических систем, имеющих значение
для специалистов в области супрамолекулярнои химии, мы хотим ознакомить
читателя с наиболее широко распространенными синтетическими системами,
которые более подробно рассмотрены в последующих главах настоящей книги. В этих
главах представлены: пути имитации биологических процессов с помощью
синтетических и модельных систем, методика раскрытия сущности биохимических
2.1. Катионы щелочных металлов в биохимии 65
процессов при изучении супрамолекулярных соединений, а также огромное
разнообразие небиологической супрамолекулярной химии.
•"» Kaim W. and Schwederski В. Bioinorganic chemistry: Inorganic elements in the
chemistry of life. Chichester: J. Wiley, 1994.
2.1 Катионы щелочных металлов
в биохимии
2.1.1 Мембранные потенциалы
Без энергии нет жизни. Растения черпают энергию от Солнца
(фотосинтез), а люди получают ее из пищи (в организме человека пища окисляется до
СО2 и воды). Энергию используют при дыхании - процессе, посредством которого
энергия, содержавшаяся в пище, хранится в виде энергии химической связи АТР
(adenosine triphosphate, аденозинтрифосфат). Строго говоря, АТР имеет заряд иона,
равный 4—, уравновешенный зарядами катионов щелочных и щелочноземельных
металлов. В биологической системе обозначений это часто опускают. АТР способен
долго хранить энергию и доставлять ее в те места, где она нужна для протекания
реакций, идущих с поглощением энергии, таких, как мышечное сокращение.
Энергия высвобождается из АТР с помощью ферментов, называемых АТРазами, среди
которых Ыа+/К+-АТРазы, возможно, наиболее важные. Согласно реакции,
показанной на схеме 2.1, из 1 моль АТР высвобождается 35 кДж энергии. Заметьте, что,
поскольку молекула АТР довольно сложна, в процессе реакции изменяется только
трифосфатный «хвост». Разрыв концевой связи фосфатного эфира Р—О дает анион
дигидрофосфата (Н2РО^~, часто называемый неорганическим фосфатом, Pj) и аде-
нозиндифосфат ADP.
Примерами трансмембранного фермента, т.е. фермента, существующего в фос-
фолипидной мембране (стенка) биологической клетки, являются Ыа+/К+-АТРаз-
ные ферменты. Они переносят катионы щелочных металлов Na+ и К+ с одной
стороны клеточной мембраны на другую, что является частью процесса потребления
АТР. Вопреки градиенту концентрации, фермент эффективно забирает катион Na+
из глубины клетки и переносит его наружу, одновременно доставляя К+ в клетку.
Таким образом, во внутриклеточной жидкости существует высокая концентрация
К+, а снаружи — высокая концентрация Na+ (табл. 2.1).
Такое неоднородное распределение катионов щелочных металлов по обе стороны
мембраны имеет большое значение, так как создает трансмембранный
электрический потенциал, как в батарейке. Эта разность потенциалов используется, среди
прочих процессов, при передаче информации в нервных клетках (рис. 2.1).
Реальная степень разделения заряда через клеточную мембрану очень мала
(число М+-ионов одинаково с каждой стороны мембраны). В принципе, такая разность
потенциалов могла бы установиться при разделении Na+ и С1~ через мембрану.
Однако на такое разделение противоположных ионов потребовалось бы гораздо боль-
5 - СТИД Дж В
66
2. Супрамолекулярная химия жизни
Н20
0
Схема 2.1. Высвобождение энергии при дефосфорилировании АТР до ADP
и дигидрофосфата. Mg2+ — катализатор реакции
ше энергии из-за сильных электростатических взаимодействий между этими
ионами. В действительности же результирующий химический потенциал, возникающий
из-за различий в свойствах ионов щелочных металлов (Na+ и К+), достаточен для
генерации требуемого сигнала.
Таблица 2.1. Содержание
Местонахождение
Na+
иК+
в некоторых биологических
системах
Концентрация, ммоль кг~'
К+
Na+
Внутриклеточная жидкость человека
(например, эритроциты)
Межклеточная жидкость человека
(например, плазма крови)
Нерв (внутри)
Нерв (снаружи)
92
11
5
300
22
152
10
440
2.1. Катионы щелочных металлов в биохимии
67
Изменение
потенциала
Ядро
К+ накапливается,
Na+ исключен,
Na+/K+-ATPa3a
Отток К+,
приток Na+,
канал, зависящий
от потенциала
Ворота
лигандного
канала
Рис. 2.1. Способ передачи нервного импульса. При концентрационных градиентах,
порожденных №+/К+-АТРазой, открытие ионного канала вызывает пассивный отток К+ и
приток Na+, что приводит к небольшому «всплеску» электрического тока (нервный
импульс) и изменению мембранного потенциала. На конце нервной клетки (аксон)
электрический сигнал преобразуется в химический за счет триггерного вброса гормона,
например ацетилхолина (разд. 2.6). В свою очередь, гормон по триггерному механизму
открывает ионный канал с лигандным затвором в аксоне следующего нерва и перезапускает
нервный импульс в виде электрического тока, разрешая протекание пассивного потока К+
и Na+ через следующую мембрану
Поддержание неравновесного градиента ионной концентрации -
наиважнейшее условие использования ионной диффузии такого типа в качестве средства
передачи информации. Из-за того что это состояние относительно нестабильно,
требуется энергия для противодействия природной энтропии, которая увеличивает
обратный поток к равновесию. Это хорошо иллюстрирует модель ионного насоса.
Ионы активно «прокачиваются» через биологическую мембрану навстречу
концентрационному градиенту до тех пор, пока не установится стационарное неравновесное
состояние. Затем происходит пассивный отток ионов в соответствии с
концентрационным градиентом, который регулируется воротами (рис. 2.2).
Тяжелые последствия метаболических расстройств, связанные с нарушением
транспорта катионов щелочных металлов, требуют поддержания точных концент-
Рис. 2.2. Модель ионного насоса. Ионы
металлов активно перекачиваются Na+/K+-
АТРазой (процесс с поглощением энергии) из
участков с низкой концентрацией к участкам с
высокой концентрацией в направлении против
концентрационного градиента до тех пор, пока
не установится динамическое неравновесное
состояние, в котором активная перекачка
уравновешена случайной диффузией. После
активации соответствующим гормоном
открываются ионные каналы с селективными
воротами, позволяя пассивному (а потому и
быстрому) потоку ионов возвращаться назад, к
равновесию, что приводит к протеканию тока
Насос
(активный
транспорт)
Ворота
(пассивный
транспорт)
Пространственная координата
5g 2. Супрамолекулярная химия жизни
рационных градиентов. Например, с одной стороны, избыточное потребление
поваренной соли вызывает повышение кровяного давления. С другой стороны, в
пожилом организме трудно предотвратить выделение очень нестойкого К+ из-за
нарушения проницаемости мембран.
2.1.2 Мембранный транспорт
Итак, как же ионы щелочных металлов переходят из клетки наружу?
Клеточная мембрана состоит из гидрофильных (водорастворимых) фосфатных
«головок», присоединенных к длинному липидному (жирному) «хвосту», и, таким
образом, проявляет свою дифильность (разд. 10.2). В водной среде живого организма
гидрофильные «головки» притягиваются к окружающим молекулам (водородная
связь, дипольные взаимодействия), тогда как органический «хвост» выталкивается
из нее. Это приводит к бислойной организации, при которой органические
составляющие спрятаны от растворителя, а гидрофильные части обращены к нему.
Поэтому все, что должно пройти сквозь стенку клетки, должно суметь преодолеть этот
липофильный (жирорастворимый) участок (рис. 2.3). Катионы натрия и калия не
совсем липофильны. Они не могут эффективно диффундировать сквозь стенку
клетки, если только нечто не делает их липофильными или не создает для них не-
липофильный коридор. Существует два основных возможных механизма такого
пассивного катионного переноса вдоль концентрационного градиента: транспорт
каким-нибудь липофильным переносчиком или же контролируемый проход через
гидрофильный канал в мембране (рис. 2.4).
Транспорт ионов металлов по механизму с участием переносчиков требует ли-
ганда-переносчика, способного и селективно связывать катион металла, и
заслонять его от липофильного участка мембраны. Такие переносчики ионов называют
ионофорами. Среди них наиболее известны природные продукты валиномицин B.1)
и нонактин B.2).
Валиномицин был впервые выделен из бактерии Streptomycesfulvissimus в 1955 г.,
а в 1967 г. было установлено, что он катализирует обмен К+ и Н+ через мембрану
митохондрии внутри клеток по механизму с участием переносчика, не изменяя
концентрацию Na+. Химически валиномицин представляет собой циклический
депсипептид, состоящий из трижды повторяющихся четырех аминокислотных
остатков B.3): L-валина (Val), /)-гидроксиизовалериановой кислоты (Hyi), /)-вали-
на и L-молочной кислоты (Lac). Водородное связывание типа N—Н-О=С как с
эфирными, так и с амидными карбонильными группами играет важную роль в кон-
формации валиномицина (т.е. его предорганизации), которая помогает пептидной
цепи «укутать» катион металла. И валиномицин, и нонактин селективны к К+,
поскольку они способны сворачиваться таким образом, чтобы образовывать почти
октаэдрическую структуру сильными (неполяризуемыми, согласно теории жестких
и мягких кислот и оснований; HSAB-теория; Дополнение 3.2) донорами —
карбонильными атомами кислорода, в точности соответствующую размеру катиона К+.
Катионы Rb+ и Cs+ слишком велики, тогда как для связывания катиона Na+ ионо-
фор не может достаточно сильно сжаться. На рис. 2.5 представлена кристалличес-
2.1. Катионы щелочных металлов в биохимии
Гидрофильная "головка"
Гидрофобные "хвосты"
Рис. 2.3. Упрощенная
схема фосфолипидной
биологической мембраны
(шириной 5—6 нм)
Механизмы ионного транспорта
Внеклеточная жидкость
(а) Механизм с участием
переносчиков
Молекулы
растворителя
Лшгафильная мембрана
Внутриклеточная жидкость
Молекулы
растворителя
(в) Канальный ,
механизм
с воротами
(б) Канальный
механизм
Рис. 2.4.
Механизмы транспорта
ионов через
биологические
мембраны: (а) с участием
переносчиков, (б)
канальный и (в)
канальный с
воротами. Переносчик капсулирует ион щелочного металла, удаляя большинство или все
связанные с ним молекулы воды. После этого переносчик, имеющий липофильную поверхность,
проходит сквозь мембрану и на другом ее конце отдает ион обратно в водный раствор.
Первоначально канал представляет собой гидрофильное отверстие, пронизывающее мембрану.
Ионы могут быстро пересечь канал, не теряя своей сольватной сферы на протяжении почти
всего пути, пока открыты ворота (активированные изменением потенциала или действием
гормонов), и ион может пройти через селективный фильтр (который различает ионы Na+ и
К+). (Воспроизведено с разрешения Kaim W. and Schwederski В. Bioinorganic chemistry:
Inorganic elements in the chemistry of life. Chichester. J.Wiley, 1994)
70
2. Супрамолекулярная химия жизни
О
г D-Hyi D-Val D-Lac L-Val -,
О i u О
Валиномицин B.1)
Нонактин B.2)
кая структура К+-комплекса валиномицина, полученная методом рентгенострук-
турного анализа (РСА). Ясно видно, что взаимодействие гидрофильных
кислородных атомов карбонила с центральным К+-катионом заставляет липофильные изо-
пропиленовые группы ориентироваться наружу, тем самым создавая внешнюю
углеводородную оболочку. Амидные группы стремятся «застегнуть молекулу на
молнию» за счет внутримолекулярных водородных связей, обеспечивая полную
закрытость К+-катиона в липофильной капсуле при пересечении ею мембраны. И
валиномицин, и нонактин являются сильными антибиотиками благодаря их
способности нарушать трансмембранный ионный баланс бактерий.
D-Hyi
Энниатин A: R = >1-метил-/,-изолейцин
Энниатин В: R = М-метил-£-валин
Энниатик С: R = 1Ч-метил-/,-лейцин
Боверицин: R = М-метил-/,-фенилаланин
B.3)
2.1. Катионы щелочных металлов в биохимии у {
Рис. 2.5. Кристаллическая структура
К+-комплекса валиномицина. Краткое
изложение метода РСА приведено
в Дополнении 2.1
Близкими родственниками валиномицина являются энниатины B.3), которые
содержат в 2 раза меньше аминокислотных остатков (Дополнение 2.2 о строении
аминокислоты). Энниатины переносят ионы щелочных и щелочноземельных
металлов, хотя они гораздо менее селективны. Заметьте, что азотные атомы амида
метилированы; это предотвращает возможность водородного связывания.
Дополнение 2.1. Рентгеноструктурный анализ супрамолекулярных
соединений
Первоначальная цель рентгеновской дифракции — получение детальной картины
строения кристалла на атомном уровне, как если бы рассматривали его в
сверхмощный микроскоп. Экспериментальное исследование состоите серии измерений
интенсивности рефлексов от дифракции рентгеновских лучей на небольшом
монокристаллическом образце. В лучшем случае оно приводит к точному определению положения
всех атомов в молекуле и, следовательно, к детальной информации о структуре
молекулы в целом, включая длины и углы связей, а также межмолекулярные контакты.
Помимо этого эксперимент дает представление о тепловом движении в твердом
состоянии. Эти результаты исключительно важны для химиков-супрамолекулярщиков,
так как они содержат непосредственную информацию о межмолекулярных
взаимодействиях, таких, как водородная связь и ион-дипольные взаимодействия, а также о
стерическом соответствии рецептора и субстрата (хозяина и гостя). Однако из-за
того, что РСА основан на дифракции, обусловленной электронной плотностью
кристалла, он не очень подходит для точного обнаружения атомов водорода (каждый из
них имеет только по одному электрону). Это важно учитывать при изучении
водородных связей; для получения точной информации их местоположение определяют
нейтронной дифракцией на монокристалле (см. Дополнение 6.1).
Итак, почему бы нам не взять очень мощный микроскоп и непосредственно не
увидеть атомы? Ответ заключается в том, что атомы слишком малы. Длина волны в
72 2. Супрамолекулярная химия жизни
видимом диапазоне находится между 400 и 700 нм. Для сравнения, обычная длина
связи между атомами (например, углерода) — -0.15 нм, поэтому нам нужно излучение
с гораздо меньшей длиной волны, соизмеримой с межатомными расстояниями, т.е.
рентгеновское излучение. Проблема такого коротковолнового излучения («фазовая
проблема») состоит в том, что невозможно изготовить линзы, достаточно мощные для
его рефокусировки. Ее решают с помощью математической модели структуры на
основе экспериментальных данных и благодаря химической интуиции. Эту модель
используют для расчета дифракционных картин молекул. Результаты расчетов
сравниваете экспериментальными данными и уточняют методом приближения наименьших
квадратов. Эксперимент считается завершенным, когда достигнута наилучшая
согласованность между расчетными и наблюдаемыми данными. Эта согласованность
оценивается по /?-фактору (остаточный фактор) и стандартным неопределенностям
(ошибки, часто называемые стандартными отклонениями) в индивидуальных длинах
и углах связей. При хорошем определении структуры /?-фактор должен быть не более
5%.
Типичный эксперимент
1. Подготовка образца монокристалла (медленное испарение, перекристаллизация
и т.д.)
• Однородный монокристалл без дефектов и трещин.
• Длина кромки 0.1—0.8 мм; идеально сферический или, по крайней мере,
равносторонний.
2. Исследование на оптическом микроскопе (нет ли дефектов).
3. Предварительные рентгеновские снимки (пленки «полароид» или детектор
электронной площади).
• Проверка качества кристалла.
• Определение размеров и симметрии элементарной ячейки (элементарная
ячейка — это основной «кирпичик» кристалла).
4. Запись данных по интенсивности A000—100000 точек в зависимости от размера
молекулы).
5. Разработка приближенной модели структуры.
• Применение метода Паттерсона или прямого метода для определения фазовой
структур.
• Стереохимия молекулы в общих чертах.
• Длины связей с точностью до ±0.1 А A А (Ангстрем) = 10~10 м = 0.1 нм).
6. Уточнение структуры методом наименьших квадратов.
• Оптимизация модели (набор координат и тепловых параметров атомов) для
наилучшего соответствия.
• Длины связей с точностью до ±0.005 А.
• Точная стереохимия.
7. Преобразование координат в таблицы длин и углов связей, представление
структуры в наглядном виде.
2.1. Катионы щелочных металлов в биохимии
73
С увеличением размера группы или ассоциата этот процесс усложняется.
Большинство супрамолекулярных соединений имеют средние размеры и обычно получить
о них точную информацию можно довольно легко. С увеличением размера молекулы
в молекулярном ансамбле число измеряемых данных и параметров, подлежащих
«подгонке», резко возрастает. Часто это сопровождается ростом числа проблем
получения кристаллических образцов хорошего качества. Большие молекулы,
встречающиеся в супрамолекулярной химии, особенно те, которые имеют полости или
сложную форму и плохо соответствуют друг другу, часто включают молекулы
растворителя. Во время РСА-исследования молекулы растворителя могут диффундировать
за пределы кристаллической решетки больших молекул, приводя к уменьшению
кристалличности образца и, следовательно, к уменьшению интенсивности сигнала. Они
также могут передвигаться, приводя к «размазанной» (разупорядоченной) средней
электронной плотности и соответственно к усложнению процесса моделирования.
Даже в отсутствие растворителя плохо упакованные кристаллы дают слабую
дифракцию. РСА-анализ образцов с очень большой молекулярной массой, например белков,
предусматривает затрату больших усилий и требует сбора значительного количества
данных, часто от множества кристаллов. В работе с белками возникают проблемы
рентгеновского повреждения образцов, слабой дифракции и идентификации
различных молекулярных фрагментов. С появлением таких детекторов по площади, как
прибор с зарядовой связью (charge-coupled device, CCD; рентгеновские детекторы,
способные одновременно собирать данные от многих точек; рис. 2.6), необычайно
Рис. 2.6. Современный CCD-дифрактометр.
Обратите внимание на круговой детектор
поверхности, расположенный слева, который
действует как очень чувствительный
эквивалент фотопленки многократного
использования, позволяющий одновременно
получать много данных в разных точках.
(Фотография любезно предоставлена фирмой
«Nonius»)
увеличивших быстроту и чувствительность эксперимента, произошел переворот и в
супрамолекулярной кристаллографии, и в кристаллографии белков. Несмотря на это,
в настоящее время работы по рентгеновской дифракции, в особенности те, в которых
имеют дело с супрамолекулярными соединениями, сольватированными или
содержащими водородные связи, для уменьшения подвижности атомов и предотвращения
диффузионных потерь растворителя обычно выполняют при очень низких
температурах A00-150 К).
*~* AtwoodJ.L. Diffraction studies of supramolecular compounds // Physical supramolecular
chemistry/ Ed. L.Echegoyen, A.E.Kaifer. Amsterdam: Kluwer, 1996. P. 261-272.
*""» Hope H. X-ray crystallography: A fast, first resort analytical tool // Progr. Inorg. Chem. /
Ed. K.D.Kar!in. Chichester: J. Wiley, 1994. Vol.41. P. 1-19.
74<# 2. Супрамолекулярная химия жизни
Несмотря на важность, механизм транспорта катионов ионофорами через
последовательные десольватацию — комплексообразование — транспорт — декомп-
лексообразование слишком медленный для генерации нервных импульсов.
Напротив, проход ионов через ионные каналы приводит к транспорту со скоростью,
близкой к диффузионному пределу (~108 ионов через канал в секунду). РСА-исследова-
ние К+-каналов в микроорганизме Streptomyces lividans (D. Doyle et al., 1998)
позволило выявить причины как необычайно высокой скорости пропускания ионов, так
и огромной селективности канала, необходимой для жизнедеятельности
организма, в случае К+ A05) по сравнению с Na+ (рис. 2.7). Оба конца ионного канала
окружены отрицательно заряженными аминокислотными остатками, которые
обеспечивают увеличение локальной концентрации катионов. Большинство пор
состоит из широкого туннеля длиной 18 А, открывающегося в центральную полость
диаметром -10 А в центре поры. Катионы калия могут пересекать и туннель, и
полость без потери своей гидратноР сферы. Поэтому предположили, что большая
центральная полость выдерживает электростатическую дестабилизацию,
возникающую из-за низкой диэлектрической постоянной бислоя, благодаря сольватной
оболочке иона, состоящей из поляризуемых молекул воды. Затем пора сужается в
фильтр селективности, предорганизованный полярными карбонильными атомами
кислорода, предварительно расположенными так, чтобы связать ион металла
радиуса К+. Рубидий, будучи лишь чуть больше, чем калий (см. Дополнение 3.2), также
легко проходит через канал. Важно отметить, что кольцо центров связывания поры
при этом удерживается открытым по типу «нагруженной пружины» за счет ариль-
ных фрагментов остатков триптофанила и тирозила, расположенных наподобие
Рис. 2.7. Стереоизображение кристаллической структуры К+-канала Streptomyces lividans.
Плотность отрицательного заряда на верхнем и нижнем концах канала очень велика, тогда
как гидрофобные боковые цепи аминокислот расположены в центральной части канала. На
внешней поверхности структуры находятся положительно заряженные области, сферы
представляют собой ионы К+. (Воспроизведено с разрешения MacKinnon, 1998)
2.1. Катионы щелочных металлов в биохимии |- 75
манжеты фильтра селективности (Дополнение 9.1 о структурах аминокислот). В
результате пора не может сжаться до такой степени, чтобы связать Na+. Это
объясняет селективность рассмотренного канала.
Структура Na+/K+-ATPa3bi, фермента, отвечающего за поддержание Na+/K+-
неравновесности в большинстве эукариотических организмов, не известна.
Действительно, фермент трудно получить в чистом виде, поскольку он связан в мембране
и имеет большую молекулярную массу B94 кДа). Известно, что он состоит из двух
пар большого (а) и малого (Р) пептидов, т.е. (офJ. Каждая оф-пара активна. Р-Пеп-
тид — это гликопептид с молекулярной массой -50 000 Да и его роль не ясна; а-пеп-
тид - это пронизывающий мембрану белок с молекулярной массой 100 000 Да,
существующий в различных конформациях в присутствии Na+ (конформация Ej) или
К+ (Е2). Он вовлекается в №+-зависимое фосфорилирование Р-карбоксильной
группы остатка аспарагиновой кислоты, причем этот процесс требует присутствия
Mg2+. Напротив, для потери фосфата нужен К+. Суммарный процесс выглядит
следующим образом:
Mg2+
3Na+c 2К+С + ATP4" + Н2О > 3Na+c + 2К+ + ADP3~ + НРО2.- + Н+ B.1)
(/с - внутриклеточный, ее - внеклеточный).
Механизм энзимного действия АТРазы, без учета ее транспортной функции,
может быть представлен так, как изображено на схеме 2.2.
Фосфорилированный фермент
/
АТР + Е, „ Е,АТР Na*'M8>»- E,PADP - E,P + ADP
Конформационвое
изменение
Р, + Е2К
Р, -Неорганический фосфат
АТР
Дефосфорилирование.катализируемое К+
Конформационное
изменение
Схема 2.2. Механизм ферментативного действия АТРазы
Следовательно, работа системы включает стадию фосфорилирования, сокатали-
зируемую Na+/Mg2+, и стадию дефосфорилирования, катализируемую К+. Каждая
ступень фосфорилирование/дефосфорилирование включает «псевдоротацию» пя-
тикоординационного интермедиата, стабилизированного Mg2+, что приводит к
транспорту катионов щелочных металлов. Способность ферментов переносить
катионы металлов — прямой результат ферментативной реакционной способности
белков. Предполагается, что существует три связывающих центра с большим
сродством к Na+ и два — со сродством к К+. При обработке мембранно-связанного
а-пептида трипсином (который катализирует гидролиз внемембранного белка) и
76
2. Супрамолекулярная химия жизни
(. .-, *Vrt5>? 4<л«;М>^4*;^jy,^'w./
Рис. 2.8. Три спирали, пронизывающие мембрану в Na+/K+-ATPa3e, переносят Na+, тогда
как К+ переносится между ними, чем и объясняется наблюдаемая стехиометрия 3:2
выделении сегментов, пронизывающих мембраны, можно обнаружить четыре
сегмента с 30—40 остатками аминокислоты и один сегмент с 70 остатками. Чтобы
пронзить мембрану, нужно по крайней мере 26 остатков в предположении, что в
сумме существует всего три двойные спирали. Это приводит к образованию трех
цилиндров из двойных спиралей, выступающих в роли ионных каналов, по
которым переносится Na+. При этом К+ транспортируется между этими каналами
(рис. 2.8). Такая^модель согласуется с наблюдаемой стехиометрией 3:2 (Na+:Ka+), a
структура подтверждается данными электронной микроскопии.
2.1 «3 Родопсин:
супрамолекулярное фотонное устройство
Супрамолекулярный транспорт катионов щелочных металлов важен
также в других процессах генерации сигналов, например при стимуляции клеток
палочек и колбочек в сетчатке глаза видимым светом. Клетки палочек лучше
изучены и имеют все черты супрамолекулярного устройства . Структура типичной
палочки показана на рис. 2.9, а. Светособирающая часть клетки палочки содержит
фоточувствительный красноватый пигмент, называемый родопсином (визуально
пурпурный). Он состоит из комплекса белка (опсин) и альдегида витамина А
(каротин), называемого ретиналем. Спектр поглощения родопсина чувствителен к
длинам волн, которые излучаются при низком уровне освещения; при
фотовозбуждении комплекс опсина и ретиналя (азометиновая функция C=NH+) претерпевает
*<мс-АИ/?а«с-изомеризацию. Это вызывает деградацию циклического гуанозинмоно-
2.1. Катионы щелочных металлов в биохимии
(а)
Палочка <
77
Проводящая часть клетки
Слои, содержащие родопсин
Внутренний сегмент,
содержащий митохондрию
Ядро
Основание клетки
(б)
Na+
Гиперполяризация
Рис. 2.9. (а) Светочувствительная клетка палочки сетчатки человека.
Фоточувствительный пигмент родопсин расположен во внешних тонких слоях. Основание
контактирует со зрительным нервом, (б) Функционирование клетки палочки
фосфата (cyclic guanosine monophosphate, cGMP), что необходимо для поддержания
всегда существующего энергопоглощающего потока Na+-HOHOB, включающего в
себя «темновой ток» клетки, и, следовательно, для закрытия трансмембраннных
каналов. Это, в свою очередь, является причиной ощутимой гиперполяризации
внутри клетки и приводит к усилению исходного светового сигнала и генерации
нервного импульса (рис. 2.9, б).
Важность связывания и переноса катионов щелочных металлов для биохимии
очевидна, поэтому большие усилия в супрамолекулярной химии были затрачены на
анализ механизмов природного связывания катионов, транспорта по ионофорному
и канальному типам, а также на разработку искусственных систем, обладающих
такими же селективностью и реакционной способностью. Многое из
перечисленного выше будет рассмотрено в гл. 3.
I 78 2. Супрамолекулярная химия жизни
2,.% Порфириновые и тетрапиррольные
макроциклы
Макроциклические (с большим кольцом) лиганды обычны в
биохимии. Благодаря хелатным и макроциклическим эффектам они способны связывать
даже ионы металлов, инертных к замещению. Это особенно важно при связывании
ионов щелочных металлов с ионофорами — валиномицином и нонактином.
Тетрапиррольные макроциклы также важны в связывании как ионов
переходных, так и ионов непереходных металлов при том огромном разнообразии
ситуаций, в которых требуется сильное размерно-селективное связывание. Тетрапирролы
участвуют в большом числе окислительно-восстановительных реакций благодаря
наличию сопряженного макроциклического каркаса и легкости его
восстановления.
Самые распространенные макроциклические соединения такого типа:
• хлорофиллы, содержащие лабильные в других случаях Mg2+ , важны как
накопители энергии при фотосинтезе;
• кобаламины — активная форма витамина В12, включающая частично
сопряженную корриновую циклическую систему (рис. 2.10, а);
• гем-комплексы, состоящие из центрального атома железа и замещенного пор-
фиринового лиганда, как в Fe-протопорфирине IX (рис. 2.10, б), — активного
центра, связывающего О2 в гемоглобине; гемы обнаружены также во многих
ферментах, например в цитохромах, для которых О2 является субстратом;
• комплекс порфирината никеля(П) — кофермент F450 (рис. 2.10, в) — найден в
организмах, производящих метан; независимость функционирования этих
комплексов от функционирования белков делает их хорошими кандидатами для
катализа «первого часа», т.е. для возникновения жизни.
Тетрапиррольные макроциклы, например гемы и коррины, обладают множеством
замечательных свойств, определяющих важность этих соединений.
• Их плоская (или почти плоская) циклическая система очень стабильна.
• Тетрапиррол может связывать даже высоколабильные ионы металлов, как и те-
традентатные хелатные лиганды, которые после депротонирования несут
единичный (корриновый) или двойной отрицательный заряд. Только в случае
одновременного разрыва всех связей металл—лиганд комплекс может
диссоциировать (ср. хелатный и макроциклический эффекты).
• Обычно макроциклические лиганды вполне селективны к радиусу того иона
металла, с которым они будут связываться (предпочтительнее связываются те,
которые соответствуют размеру полости) В этом отношении тетрапирролы
особенно селективны благодаря жесткости сетки сопряженных двойных связей.
• Большинство тетрапирролов содержит сопряженную я-систему. Порфирины,
имеющие 18 л-электронов во внутреннем 16-членном кольце, подчиняются
правилу Хюккеля для ароматических систем (ароматические системы содержат
4л+2 Ti-электронов; для полностью сопряженных тетрапирролов п — 4). Это не-
2.3. Супрамолекулярные особенности фотосинтеза в растениях
79
(а)
о2с
со2н со2н
Рис. 2.10. Биологические тетрапиррольные макроциклические соединения: (а) коррин,
(б) гем (Fe-протопорфирин IX), (в) кофермент F450
которым образом объясняет термическую стабильность циклической системы и
часто приводит к интенсивному окрашиванию. Поэтому многие тетрапирролсо-
держащие биологические системы называют пигментами. Более того, это
означает, что продукты окисления и восстановления, которые образуются в
некоторых биологических процессах, могут быть вполне стабильными из-за делокали-
зации возникающих зарядов.
• Макроцикл содержит четыре координирующих атома в плоскости и два
доступных участка на октаэдрическом металлическом центре, пригодных для
связывания экстралигандов (субстрат и управляющий лиганд).
• При координации больших ионов металлов, например высокоспинового Fe(II),
плоская структура тетрапирролов может искажаться и принимать
куполообразную форму. Обратимая стабилизация этого высокоспинового состояния
жизненно необходима для функционирования гемоглобина. Напротив, плоские
тетрапиррольные системы расщепляют поле сильных лигандов, а следовательно,
образуют низкоспиновые комплексы.
В следующих разделах мы более подробно рассмотрим некоторые порфириновые и
тетрапиррольные комплексы.
2*3 Супрамолекулярные особенности
фотосинтеза в растениях
2*3*1 Роль тетрапиррольных комплексов магния
Фотосинтез — процесс, с помощью которого органическое вещество
(восстановленный углерод) получается из С02 и воды в ходе трудно идущего (с
поглощением энергии) катализа на свету (всегда доступная «чистая», хотя и довольно
2. Супрамолекулярная химия жизни
;разбавленная», форма энергии.
Фотосинтез
(трудный катализ)
н2о
со
2 ^
Дыхание
(легкий катализ)
± 1/л(СН2О)Л + 3О2 Д# = +470 кДжмоль-1. B.2)
Фотосинтез осуществляется зелеными растениями, а также некоторыми видами
бактерий и морских водорослей и, в зависимости от условий, в которых
развивается организм, может включать окисление других субстратов, в частности H2S или Н2,
вместо воды. У зеленых растений производительность фотосинтеза — ~1 г глюкозы
в 1 ч на 1 м2 поверхности листьев. Несмотря на сравнительно неэффективное
использование доступной энергии излучения (меньше 1%), глобальность цикла,
происходящего на Земле, потрясает: каждый год из СО2 и воды на свету образуется
-200 млрд т углеводных эквивалентов (СН2О)Л.
Солнечный свет, достигающий поверхности Земли, включает видимый
диапазон длин волн C80—750 нм), однако существует также значительный вклад от света
Частично
гидрогенизированное
кольцо
О
Ч\ ХлОроф
Длинная алифатическая
фитильная цепь, закрепляющая
пигмент в фосфолипидной
фотосинтетической мембране
Рис. 2.11. Структуры
хлорофилла а
и бактериохлорофилла а
2.3. Супрамолекулярные особенности фотосинтеза в растениях
81
Рис. 2.12. Складчатая мембрана
фотосинтеза. (Воспроизведено с разрешения
Kaim W. and Schwederski В. Bioinorganic
chemistry: Inorganic elements in the chemistry of life.
Chichester: J. Wiley, 1994)
больших длин волн (и потому меньшей энергии) — примерно до 1000 нм. Для
эффективного фотосинтетического преобразования света в этом диапазоне энергий
требуется множество различных пигментов (световых рецепторов или
хромофоров), каждый из которых чувствителен к определенной части спектра. Эти
пигменты включают хлорофилл а и бактериохлорофилл а (рис. 2.11), так же как и ряд
родственных рецепторов, основанных на тетрапиррольных макроциклах и с гостем, и
без гостя. Пигменты расположены в складчатой фотосинтетической мембране с
большой площадью поверхности, а потому и с большим сечением захвата фотонов
(рис. 2.12).
Хлорофилл содержит полностью сопряженную тетрапиррольную я-систему
A8 я-электронов) с низкоэнергетическим я-л*-переходом. Коэффициент экс-
тинкции велик (~105 М^'см) и на длинноволновом, и на коротковолновом крае
спектра. Комплементарные (дополняющие) цвета — голубой (при
коротковолновом поглощении) и желтый (при длинноволновом поглощении) — комбинируются,
давая характерный зеленый цвет свежих листьев (А.тах 455 и 630 нм). Бактериохло-
рофиллы содержат два частично гидрогенизированных пиррольных кольца, и,
следовательно, их поглощение сдвинуто в сторону больших длин волн, достигая
ближней инфракрасной (ИК-) области. Каротиноиды и молекулы тетрапиррола с
открытой цепью, например фикобилины, дополняют хлорофилльные пигменты
таким образом, что «работает» широкий диапазон поглощения (рис. 2.13). К концу
Рис. 2.13. Спектры поглощения
различных пигментов морских
водорослей и растений: хлорофилл а (—),
ос-каротин (—), фикоцианин (--),
фикоэритрин ( ). (Воспроизведено с
разрешения Kaim W. and Schwederski В.
Bioinorganic chemistry. Inorganic elements
in the chemistry of life. Chichester: J. Wiley,
1994)
и
ощеш
о
Н
л
;
/ / 1 /
/ / у /
1 ! \
,' \
' Ч
а-Каротин
/
\-\ -л
\\ -^ \
1 1
\л •
V A v
\^ • \ ч
400 500 600 700
Синий Зеленый Желтый Красный
Длина волны, нм
2. Супрамолекулярная химия жизни
Рис. 2.14. Пространственно ориентированная
антенная сеть светособирающих пигментов.
(Воспроизведено с разрешения Kaim W. and
Schwederski В. Bioinorganic chemistry: Inorganic
elements in the chemistry oflife. Chichester: J. Wiley,
1994)
каждого периода роста видимые незеленые каротиноидные пигменты изменяют
цвет (что приводит к осенним краскам), поскольку относительно нестабильный
хлорофилл разлагается (например, фикоэритрин, Хтах 455, 510 и 555 нм; фикоциа-
нин, 595 нм).
Из-за того, что скорость поглощения рассеянного солнечного света довольно
низка, большинство (>98%) пигментов представляют собой светособирающие, или
антенные, устройства, поглощающие и передающие энергию к фактически
существующим реакционным центрам. Такая передача поглощенной энергии должна быть
эффективной и пространственно ориентированной. Она становится возможной
благодаря наличию антенной сети пигментов, способной преобразовывать
световую энергию в каскад переносов энергии (рис. 2.14). Существенно то, что перенос
энергии происходит путем перекрывания полос излучения источника и полос
поглощения приемника (рис. 2.15).
Координированный ион Mg2+ в хлорофилле играет роль посредника в
расположении пигментов. Пигменты пространственно закрепляются в определенном
месте с помощью длинной фитильной боковой цепи, глубоко погруженной в
фотосинтетическую мембрану. Однако для жесткой фиксации пигментов два свободных
аксиальных центра октаэдрического Mg2+ связываются с полипептидными боковыми
цепями лигандов, приводя к трехточечному закреплению пигментов и,
следовательно, к четко определенной пространственной ориентации.
Возбуждение
530 нм
! Безызлучательный переход
Л2пс
578 нм
,22 пс
640 нм
,52 пс
660 нм
685 нм
Фикоэритрин Фикоцианин Аллофикоцианин Хлорофилл а
Рис. 2.15. Каскад переносов энергии в светособирающих пигментах морских водорослей
Porphyridium cruetum
2.3. Супрамолекулярные особенности фотосинтеза в растениях 83
Mg2+ в особенности подходит для этой роли по следующим причинам:
• большое распространение в природе (соответствующее этой некаталитической
«объемной» функции);
• недостаток окислительно-восстановительной активности
(окислительно-восстановительная активность металла несовместима с межпигментной передачей
электронов);
• сильная тенденция к гексакоординации;
• подходящие ионные радиусы;
• небольшая константа спаривания спин-орбиталей (в результате
спин-орбитального взаимодействия путем интеркомбинационного перехода образуются долго-
живущие триплетные возбужденные состояния, что приводит к процессам
выделения света и тепла, конкурирующим с химическими реакциями).
РСА-исследование кристаллической структуры этилхлорофиллида (в котором
фитильная группа замещена простым этильным заместителем, рис. 2.16) указывает на
стремление Mg2+ к 5- или 6-координационному окружению. Аксиальный участок
иона Mg2+, связанный с полипиррольным кольцом, координирует молекулу воды,
которая, в свою очередь, водородными связями соединяется с карбонильными
группами соседних молекул, образуя твердый полимер. Это наглядный пример
твердотельной супрамолекулярной организации, но, возможно, что прямое
связывание такого типа не происходит в реальных системах.
Энергия, полученная светособирающими антенными системами, направляется
к центру реакции фотосинтеза, где используется для пространственного разделения
заряда. Другими словами, электрон приводится в возбужденное состояние и участ-
Рис. 2.16. Кристаллическая
структура этилхлорофиллида
pg4 2. Супрамолекулярная химия жизни
Излучательный
переход
Рис. 2.17. Гибель
Основное Внешний Электронно- Внешний фотовозбужденного
состояние донор возбужденное акцептор _
синглетное электрона и образование
состояние положительной «дырки»
вует в реакции химического восстановления до тех пор, пока он не будет иметь
возможность вернуться в основное состояние в результате биохимически
бесполезного излучательного перехода. В простых реакционных центрах, таких, как у
пурпурной бактерии Rhodopseudomonas viridis, возбужденный электрон передается
внешнему акцептору. В более высокоразвитых организмах оставленная «дырка» также
используется для окисления субстрата с участием внешнего донора. В конечном
счете это приводит к получению О2 из воды (рис. 2.17).
Реакционный центр у Rps. viridis располагается в полипротеиновом комплексе,
который пронизывает фотосинтетическую мембрану. Он имеет симметрию почти
С,, причем димер бактериохлорофилла (ВСJ «сидит» на оси симметрии.
Электронное возбуждение этого димера (Р960 - пигмент с длиной волны максимального
поглощения 960 нм) приводит к электронно-возбужденному состоянию. Это
называют первичным разделением заряда. Вслед за этим, еще до того как может произойти
излучательный переход, один энергетически возбужденный электрон переносится
к первичному акцептору — мономерному бактериохлорофиллу (bacteriochlorophyll,
ВС). Восстановленная мономерная молекула ВС, в свою очередь, передает
электрон вторичному акцептору — бактериофеофитину (bacteriopheophytin, BP;
молекула ВС без координированного металлического центра). Это явление известно как
вторичное разделение заряда и оно приводит к пространственному разделению
электрона и его источника (схема 2.3). Благодаря отсутствию металлического центра
молекула легче восстанавливается, поскольку ионная связь Mg2+ с двуханионным
лигандом оставляет лиганду значительную часть электронной плотности.
Действительно, замена Mg2+ протонами приводит к более ковалентному состоянию. В
таком случае электрон передается от анион-радикала ВР к л-хинону, Qa, например
менахинону, восстанавливая его до анион-радикала семихинона. Он, в свою
очередь, восстанавливает более лабильный хинон, Qb, такой, как убихинон, через ше-
стикоординационный центр Fe(II). Qb связан с белком не прочно, но обменивается
с хинонами в «хинонном объединении» мембраны. Возникающий градиент
электронов в конце концов приводит к градиенту Н+ и — как следствие — к
фотосинтетическому фосфорилированию (синтез АТР). В высших организмах происходят
«темновые» реакции, приводящие к восстановлению СО2 (они известны как цикл
Кальвина):
4<r + 4H+ + CO2 -» 1/л(СН2О)„ + Н2О. B.3)
2.3. Супрамолекулярные особенности фотосинтеза в растениях
85
Энергия, эВ
1.40
1.05
0.75
0
(BCJ*BCBPQa
i
буждение
Воз
(В<
i
:)jBCBPQa
(BCV
(ВС):
<ВСJ*
-ИГ2 с /
*
*
ВС" BPQa
1 ~Ю"|2с
f
ВС BP-Qa
1 2 1(Г|Ос
ВС BPQT
ч
' ВС: бактериохлорофилл
ВР: бактериофеофитин
0^,: хинон
Схема 2.3. Разделение зарядов на фотосинтетическом реакционном центре
Rps. viridis
Катион-радикал (ВС)*+ в Rps. viridis, оставшийся после первичного разделения
зарядов, спустя длительное время восстанавливается за счет управляемого потока
электронов через один или более гем-центров цитохромных белков. В высших
организмах окисление субстрата начинается с появления этой «дырки», для чего
необходима дополнительная фотосинтетическая система, включающая катализатор
окисления , содержащий магний (разд. 2.3.2).
Ключевая особенность фотосинтеза — возможность пространственного
переноса заряда подальше от возбужденного состояния реакционного центра, до того как
произойдет обычно высокоэффективная и биохимически бесполезная
рекомбинация. В системах фотосинтеза разделение зарядов происходит примерно в 108 раз
быстрее, чем рекомбинация, что недостижимо в «нормальных» химических
реакциях. Это явление достигается путем пространственного закрепления компонентов в
определенной ориентации относительно друг друга в неполярной области
мембранного белка, что препятствует свободной диффузии и способствует трудноиду-
щей химической реакции.
2.3«2 Окисление воды до кислорода,
катализированное марганцем
В высших растениях существуют две связанные, отдельно
возбуждаемые фотосистемы (photosystems, PS), обозначаемые PS I и PS II. Система PS I
содержит пигмент (Р700), поглощающий при 700 нм, она возбуждается от PS II и в
конечном счете участвует в восстановлении СО2. PS II напоминает более простой
фотосинтетический бактериальный реакционный центр (см. схему 2.3), не
ф%$ 2. Супрамолекулярная химия жизни
содержит поглощающий пигмент с более высокой энергией, Р680. В результате
транспорта электрона в PS II и его использования в синтезе АТР образуется
«дырка», которая затем участвует в окислении. Наличие высоких энергий в системе
означает, что эта «дырка» может быть использована для окисления тирозина до
катион-радикала тирозина (+0.95 В), который, в свою очередь, окисляет гидрохинон
(пластогидрохинон, plastohydroquinone, PQH2) до хиноновой формы (PQ). Реакци-
онноспособный хинон окисляет воду в четырехэлектроннои
окислительно-восстановительной реакции, катализируемой кислородвыделяющим комплексом
(oxygen-evolving complex, OEC), который представляет собой фермент, содержащий
четыре Mn-центра. В этом процессе четыре протона переносятся с внешней стороны
мембраны внутрь ее, создавая градиент протонов. Полная реакция выглядит
следующим образом:
2Н2О + 2PQ + 4Н+Ш/ 4/?V'1MrVaS 3O2 + 2PQH2 + 4Н+ . B.4)
PS II состоит из следующих обязательных элементов (те из них, которые прямо
участвуют в окислении воды, показаны жирным шрифтом):
~200 хлорофиллы-антенны
~50 каротиноиды
1 реакционный центр, Р680
2 хлорофиллы
2 феофитины (первичный акцептор)
2 пластохиноны
2 тирозиновые остатки (первичный донор)
4 Мп-центры
1 Са2+ (до сих пор роль не совсем ясна, но может участвовать
в стабилизации или регулировании структуры)
несколько С1~
1 цитохром bss9
Однако хорошо известно, что ОЕС содержит четыре Mn-центра в одной
субъединице массой 33 кДа. Использование метода растянутой тонкой структуры
рентгеновского поглощения (extended X-ray absorption fine structure, EXAFS) показало,
что расстояния Mn— Mn равны 2.70 и 3.30 А. Это. вероятно, свидетельствует о том,
что оксидные и карбоксилатные мостики соответственно связывают два
металлических центра. Данные подтверждаются на примере большого числа структурных
моделей простых соединений, например B.4). В процессе окисления двух молекул
воды до О2 кластер Мп4 в течение нескольких сотен миллисекунд полностью
проходит через пять различных состояний - от So до S4. Структурные различия между
состояниями малы, что соответствует малой энергии активации; тем не менее за это
2.3. Супрамолекулярные особенности фотосинтеза в растениях
87
время состояние окисления значительно изменяется.
12+
Г \ -А / Л
N—Mn3+ Mn4+-N
NMn MnN
( /I ^(/|\ J
1,4,7-триметил-1,4,7-триазациклононан =
B.4)
На рис. 2.18 представлена последовательность изменений, происходящих в пяти
5-состояниях. Данные EXAFS-спектроскопии подтверждают изменения
окислительных состояний при переходе от SQ к 5t и S2, хотя данные об изменениях при
переходе к ^ противоречивы. Этот переход может включать окисление гистидиново-
го лиганда вместо окисления Mn-центра. Существуют надежные данные ЭПР-спе-
ктроскопии о том, что состояние S2 содержит один неспаренный электрон, а это
согласуется с общим нечетным числом электронов и антиферромагнитным
спариванием.
Очень короткоживущее состояние S4 включает пероксидный лиганд О\~ и один
из центров Mn(IV), который может снова восстановиться до МпAП), давая
нечетное число электронов, наблюдаемое в S2 (у S4 на два электрона меньше, чем у S2,
поэтому если S2 - нечетное, то и 54 должно быть нечетным). Важно, что система
содержит нечетное число неспаренных электронов, поскольку кислород должен
высвобождаться в триплетном 3О2-состоянии, т.е. при четном числе неспаренных
электронов. Соответствие между катализатором и кислородом может приводить к
необратимому связыванию, как в случае реакций большинства переходных
металлов с 3О2. К счастью, изменение спиновой ориентации, требующееся для того,
чтобы эти две системы стали совместимы, формально запрещено, а значит, процесс
является медленным. Подобное «спиновое ингибирование» существует в реакции Н2
Мп4+ Мп3+ Мп4+
/\ /\
н2о он2 но он2
\/ \/
Мп3+ Мп3+ Мп3
Мп3+ Мп4+ Мп4+ Мп4+
/\ /\
но он2 но он
\/ \/
4 ^ Мп4+ Мп4+
Мп4+
/\
о—он
\/
М
Мп3+
2Н,0
/IV
Электроны переносятся к реакционному центру
через катион-радикал тирозина
Н переносится в водную фазу
Рис. 2.18. Изменения в окислительном состоянии марганца при превращении воды в О2
||bg 2. Супрамолекулярная химия жизни
с 02, которая не происходит без катализатора, способствующего разрыву связей,
или инициатора образования радикалов.
Н_Н + Н-Н +0=0 > Н-О-Н + Н-О-Н, B.5)
и и пп пи пп
s=\ s=o
2Н2О + (Mn-Mn)"+ > 3О2 + (Mn-Mn)<"-4>+ + 4H+. B.6)
S=0 S=l/2 5=1 S=3/2
Итак, что же делает марганец таким эффективным катализатором окисления?
Соображения, дающие ответ на этот вопрос, приведены ниже.
• Известно, что свежеобразовавшийся диоксид марганца WlnO2_x- яН2О,
являющийся нестехиометрической системой со смешанной валентностью (IV, III),
действует как хороший гетерогенный катализатор при разложении Н2О2 на О2
и воду.
• Во времена эволюции фотосинтеза (~3-109 лет назад) марганец был легко
биодоступен в морской воде, да и сейчас он в изобилии находится на дне моря в виде
отложений оксидов марганца с содержанием Мп -20%.
• Марганец обладает большим разнообразием стабильных или метастабильных
состояний окисления (II, III, IV, VI, VIII).
• Марганец с лигандами образует лабильные связи.
• Благодаря слабому расщеплению d-орбитали марганец предпочтительнее всего
находится в высокоспиновых состояниях.
При четырехэлектронном окислении воды до О2 полимарганцевая система
действует как резервуар для электронов, накапливая заряд строго контролируемым
образом при физиологически высоком окислительно-восстановительном потенциале, и
как катализатор, не удерживающий 3О2.
В промежутке между высвобождением О2 и связыванием воды ионы С1~ могут
играть существенную роль временных лигандов для Mn-центров. Обычно эту роль
выполняет растворитель, но в данном случае растворитель (вода) является и
субстратом. Вряд ли СГ~ окисляется сам (Е° = +1.36 В для окисления до С12), но если С1~
отсутствует и не предотвращает связывания более чувствительных пептидных
остатков, то они могут разрушаться.
Связывание и транспорт
кислорода гемоглобином
Жизненно необходимой составляющей метаболизма высших
организмов является кислород воздуха. Он метаболически окисляет сахара, такие, как
глюкоза и сахароза, с последующим высвобождением энергии. Энергия этого
управляемого «холодного горения» используется в синтезе АТФ.
С6Н,2О6 + 6О2 > 6СО2 + 6Н2О + энергия.
2.4. Связывание и транспорт кислорода гемоглобином 89
Высшими растениями кислород производится биогенно (разд. 2.3.2) и, скорее
всего, первоначально он был побочным продуктом, появившимся из-за
необходимости замещения фотовозбужденного электрона в фотосинтетическом реакционном
центре хлоропласта. В первобытном океане вода была наиболее мощным
источником электронов. К сожалению, для простейших организмов О2 - крайне реакцион-
носпособный и высокотоксичный газ. Первоначально кислород удалялся из
атмосферы восстановленными ионами таких металлов, как Fe(II) и Мп(Н), но 2 млрд лет
назад , судя по отложениям большого количества осадков оксидов металлов,
содержание О2 в атмосфере начало расти с менее 1 % (как на необитаемых планетах и Луне
сейчас) до 21% (по объему). В результате большинство простейших организмов
должно было бы погибнуть по механизмам гибели радикалов и окисления металло-
ферментов. Выжить смогли только вновь развившиеся аэробные организмы,
появившиеся благодаря избытку этого высокоэнергетического соединения. В настоящее
время выживают только те немногие анаэробные организмы, которые живут в
экологических нишах, например в глубине океан?, куда не может проникнуть
атмосферный кислород, а также те, которые способны развить защитные механизмы от
воздействия О2 и его частично восстановленных радикальных продуктов.
Для того чтобы аэробные организмы могли использовать реакционноспособ-
ный О2 в процессе, противоположном фотосинтезу, необходимо поглотить и
доставить О2 к клеточной митохондрии , где дыхание с «пищей» (т.е. сахарами)
происходит без необратимых реакций и ущерба, наносимого радикалами или
окислителями. Для выполнения этой задачи Природа создала гемоглобин —
замечательный белок, связывающий и транспортирующий кислород. Гемоглобин — тетрамер-
ный белок (RMM = 64.5 кДа), содержащий четыре субъединицы миоглобина
(RMM = 17.8 кДа). Каждая субъединица миоглобина содержит железопорфирино-
вый координационный комплекс, называемый гемом, или Fe-протопорфирином
IX B.5), который аксиально связан с белком посредством координационного
взаимодействия октаэдрического Ре(П)-центра с атомом азота из ближайшего гистиди-
нового остатка белка. Именно способность центра железа обратимо связывать О2 и
является ключом к этой жизненно необходимой биологической системе.
Fe-протопорфирин К
B.5)
НО "О НО
2. Супрамолекулярная химия жизни
Остающийся аксиальный участок на Fe доступен для связывания кислорода,
хотя он может быть занят слабосвязанной молекулой воды.
(а)
PPh,
Cl
ос
PPh,
ОС
PPh,
Ir
PPJb
vO
о
Схема 2.4. (а) Необратимое образование ц-оксодимера в небелковом
железопорфириновом комплексе, (б) Редкий пример обратимого связывания
кислорода с образованием простого неорганического комплекса. Формально
комплекс 1г(Ш)-02~ дестабилизируется п-акцепторными свойствами фосфиновых
и СО-лигандов
Главной проблемой реакции металлических центров с О2 является его
склонность к необратимому окислительному взаимодействию с металлическим центром с
образованием или ^«с-диоксо-фрагмента, или биядерных комплексов с ц-оксо-мос-
тиками (схема 2.4, а). Однако реакция является отчасти обратимой в случаях, когда
2.4. Связывание и транспорт кислорода гемоглобином
Высокий
спин
Низкий
спин
Fe(ID
Октаэдрическое
окружение
Fe(m)
Модель Полинга
Ре-Д
Модель Вайсса
Р.-О-
Рис. 2.19. (а) Описание орбиталей поля кристалла для высоко- и низкоспинового Fe(II) и
Fe(III). (б) Описание поля лиганда согласно моделям Полинга и Вайсса
существует сильная тенденция к образованию более низких окислительных
состояний, например, как в реакции с соединением Васка (схема 2.4, б). Роль гем-центра в
гемоглобине состоит не только в обеспечении обратимости связывания О2, но и в
том, что его комплексообразование и выделение происходят быстро и при
определенных концентрациях. Эти концентрации, или парциальные давления, должны
соответственно быть равны концентрациям, найденным в легких и во внутриклеточной
среде. Более того, связывание О2 должно происходить селективно по отношению к
остальным компонентам атмосферы, таким, например, как вода, N2, СО2, и такому
великолепному для Fe(II) лиганду, как СО. Таким образом, гемоглобин -
превосходный пример функционального и селективного супрамолекулярного рецептора.
До того как связать О2, гем-субъединицы содержат высокоспиновые Fe(II)-ueH-
тры с (/2р4(ер2-конфигурацией валентных электронов , что в результате дает
парамагнитные частицы с четырьмя неспаренными электронами (рис. 2.19, а).
Полагают, что Ре(П)-центр должен располагаться на значительном расстоянии от плоско-
92 2. Супрамолекулярная химия жизни
сти порфиринового макрокольца, в конформации, известной как «купол», и быть
ориентирован в сторону ближайшего гистидинового остатка окружающего белка.
Это следствие того, что высокоспиновое Ре(Н)-состояние имеет больший размер
по сравнению с полостью порфиринового макрокольца (ионный радиус равен 0.78 А).
Связывание О2 (в его основном триплетном состоянии) было предметом
многочисленной полемики вокруг двух противоположных точек зрения: моделей Полинга и
Вайсса. Согласно модели Вайсса (которая сейчас общепринята), связывание О2
сопровождается переносом единичного электрона, что приводит к образованию
дублетного супероксидного лиганда BО2~) и низкоспинового Ре(Ш)-центра.
Меньший ионный радиус Fe(III) (высокоспиновый, 0.65 А; низкоспиновый, 0.55 А)
гораздо лучше соответствует порфирину, т.е. порфириновая «купольность»
отсутствует (схема 2.5). В соответствии с моделью Вайсса РеAП)-центр является
низкоспиновым (t2gM- Это должно приводить к парамагнетизму как от одного остающегося
неспаренного электрона РеAП)-центра, так и от неспаренного электрона
супероксидного лиганда. Действительно, экспериментально было обнаружено, что
окисленная система гема представляет собой диамагнетик (это определенно исключает
высокоспиновый Fe(III)-u,eHTp, у которого было бы пять неспаренных
электронов). Вайсе предположил высокую степень корреляции между неспаренными
электронами низкоспинового Fe(III) и 2О2~, которая приводит к фактическому
диамагнетизму в случае низкого спина. Переход от высокоспинового состояния к
низкоспиновому объясняется возрастанием энергии расщепления в поле кристалла по
мере того, как после связывания О2 Fe-центр движется в направлении к плоскости
гема. Это называют триггерным механизмом. Альтернативное объяснение,
выдвинутое Полингом , предполагает, что спиновый переход происходит таким образом,
что О2 связывается с низкоспиновым центром Fe(II) в синглетной форме 'О2
наиболее удобным синергетическим образом. Оба компонента — диамагнетики, а
следовательно, комплекс в целом — также диамагнетик (рис. 2.19,6). Опять же триггер-
ный механизм объясняет переход от высокого к низкому спину. Модель Вайсса
подкрепляется наличием ИК-полосы валентных колебаний v(O—О) при 1100 см~',
в большей степени соответствующей О2~, чем двойной связи 'О^ которой
отвечает полоса с большим значением волнового числа. Более того, данные ЭПР,
полученные для гема, в котором ионный центр замещен на кобальт при образовании
кобоглобина (дающего систему с одним дополнительным электроном),
подтверждают наличие этого лишнего электрона на кислороде. В результате
получаем формулу СоAП)-О2~, согласующуюся с моделью Вайсса.
О триггерном механизме свидетельствует кристаллическая структура
модельного соединения типа «порфириновая изгородь». Обнаружено, что это соединение
(содержащее на одном аксиальном центре 2-метилимидазольный лиганд в качестве
модели ближайшего гистидинового остатка) в отсутствие кислорода имеет форму
купола, причем ион Fe располагается над плоскостью, образованной четырьмя
азотными атомами порфирина, на расстоянии 0.399 А. Связывание О2 заставляет
Fe перемещаться в положение, которое только на 0.086 А находится вне плоскости
макроцикла. Для облегчения понимания механизма действия гемоглобина был
получен целый ряд модельных соединений на основе порфирина. Эти соединения
рассматриваются в разд. 9.9. Однако удивительно, что связывание такого слабого
2.4. Связывание и транспорт кислорода гемоглобином
93
(а)
f Ближайший
\ гистидиновый
, S остаток (His)
N
О
N-His
N-His
Fe С
(б)
Схема 2.5. (а) Уменьшение «купольности» порфирина после связывания О2.
(б) Окисленный гем и его непосредственное белковое окружение, включающее
стабилизирующие водородные связи. (Воспроизведено с разрешения Kaim W. and
Schwederski В. Bioinorganic chemistry: Inorganic elements in the chemistry of life. Chichester:
J.Wiley, 1994)
лиганда, как О2, вызывает спиновый переход на металлическом центре от сильного
спина к слабому. В самом деле, порфириновые лиганды обычно индуцируют
сильное лигандное поле, которое в результате всегда дает низкоспиновые комплексы.
94
2. Супрамолекулярная химия жизни
(а) Свободные гемовые комплексы
О
(б) Миоглобин, гемоглобин
Перифери-
й
гистидин
Неблагоприятный
вынужденный о
изгиб •#
ч Водородная связь
О
Рис. 2.20. Улучшенная селективность
белка к 02 по сравнению с СО
обуславливается белковым окружением гема
Таким образом*высокоспиновое состояние (жизненно необходимое для
связывания 3О2 в случае исключения запрещенных спиновых переходов) представляет
собой аномалию, которую можно объяснить только с помощью порфириновой «ку-
польности» и последующего ослабления лигандного поля. Следовательно,
относительно слабое воздействие входящего О2 существенно меняет энергетические
уровни между напряженным высокоспиновым дезоксигенированным и
низкоспиновым оксигенированным состояниями. Порфириновый «купол» - пример
ферментативного энтатического состояния, в котором роль объемистого белкового
окружения сводится к сохранению активных центров в высокоэнергетическом
состоянии частично вдоль желаемой координаты реакции. Вообще металлопротеины
очень редко содержат ион металла в отрелаксированном, строгом
координационном окружении. Часто их молекулы находятся в шатком равновесии между двумя
высокоэнергетическими состояниями, требующими лишь малого внешнего
влияния для осуществления перехода из одного состояния вдругое.
Хорошо известно, что такие газы, как СО, или легкоадсорбирующиеся соли,
например CN~, крайне токсичны. Это происходит из-за их необратимого связывания
2.4. Связывание и транспорт кислорода гемоглобином
95
Рис. 2.21. Кривые насыщения
кислородом миоглобина и
гемоглобина и различные величины
рН. (Воспроизведено с разрешения
Kaim W. and Schwederski В.
Bioinorganic chemistry: Inorganic
elements in the chemistry of life.
Chichester: J.Wiley, 1994)
20 40 60 80 100 120 140
Парциальное давление О2, мм рт. ст.
с Fe в гемоглобине, что препятствует переносу кислорода и вызывает удушье. В
частности, СО — гораздо лучший л-акцептор, чем О2, и поэтому связан гораздо
сильнее. Как и ожидалось, сродство модельных систем гема, не содержащих белков, к
СО намного больше, чем к О2: Ксо/Ко = 25000. Однако в гемоглобине это
соотношение снижается до более благоприятного значения, 200, позволяя человеку
выдерживать небольшие дозы СО. Это явление объясняется тем, что белок
ограничивает доступ к связывающему центру железа, так что геометрия связывающего
«кармана» более приспособлена к изогнутому виду связывания О2, чем к линейной
форме СО (рис. 2.20). В окисленном геме молекула О2 связана только через один атом
О при угле Fe-0-O -120° (расстояние 0-0 - 1.89 А) вследствие наличия неподе-
ленной пары на связывающем атоме О. Взаимодействие гем—О2 стабилизируется
водородной связью несвязанного О2 с периферической гистидиновой
функциональной NH-группой.
В заключение рассмотрим вопрос, касающийся встраивания четырех
субъединиц миоглобина в белок гемоглобина. Ясно, что миоглобин сам по себе вполне
способен к поглощению и переносу О2, так почему же ему необходимо встраиваться в
белок? Ответ кроется во взаимодействиях четырех субъединиц миоглобина в
гемоглобине. Эти взаимодействия приводят к эффекту синергетического связывания и
высвобождения О2, что обеспечивает необходимые парциальные давления О2 в
легких и мышцах. Поведение миоглобина и гемоглобина (рис. 2.21) показывает, что
без этих кооперативных взаимодействий миоглобин не был бы способен
высвобождать О2 в кислороддефицитных тканях, таких, как мышцы, где он необходим.
Такое кооперативное поведение объясняет усиление связывания на одном
участке гема, если другой насыщен кислородом, и, наоборот, если один гем-центр
теряет свой О2, то это вызывает уменьшение сродства к кислороду на других центрах
железа. Влияние связывания субстрата с одним центром на связывание с другим
центром такого же типа, находящимся на значительном расстоянии от первого,
называет аллостерическим эффектом. Для него характерен сигмавидный профиль
связывания (ср. разд. 7.7.5). Результатом является тип поведения «все или ничего», в
том смысле, что как только содержание О2 становится достаточно высоким, для
того чтобы вызвать его связывание на одном центре (например, в легких), все
участки быстро насыщаются. После высвобождения О2 в мышцах диссоциация ком-
96!
2. Супрамолекулярная химия жизни
плексов быстро происходит на всех четырех участках после его выделения на любом
из них. Механизм этого аллостерического эффекта был описан моделью
«нагруженная пружина», в которой гемоглобин изменяется между двумя состояниями,
названными напряженным (Т) и релаксированным (R). Следовательно, каждый
гем может быть оксигенированным и дезоксигенированным и может находиться в
Т- или /?-состоянии. Связывание О2 с купольной деоксигенированной Г-формой
вызывает уменьшение искажения структуры и приводит к образованию
напряженной оксигенированной Г-формы. Напряжение уменьшается за счет движения
соседних гистидиновых остатков. Это движение передается всем субъединицам миог-
лобина, что стимулирует связывание О2.
2-5 Кофермент В12
Другой класс химически интересных соединений на основе тетрапир-
рольных макроциклов составляют кофермент В12 и его производные, например
витамин В|2. Структуры этих соединений показаны на рис. 2.22. Все они — комплексы
Со(Ш) и коррина, который выступает в качестве лиганда, несущего единичный
отрицательный заряд. Заряд, остающийся на кобальте, уравновешивается сложным
фосфатным остатком и заместителем X, который представляет собой
ферментативный «деловой край» молекулы. Для кофермента в активной форме таким замести-
h2noc
conh2
H2NOC
CONH2
CONH2
X = СН3: метилкобаламин
X = CN: цианокобаламин (витамин В^)
но он
-дезоксиаденозилкобаламин
(кофермент В12)
СН2ОН
Рис. 2.22. Кофермент В12 и его производные
NH2
2.5. Кофермент BJ2 97
телем является карбанион. Поэтому кофермент В12 и его производные —
единственные металлоорганические соединения естественного происхождения.
Примечательно, что при физиологических условиях (рН 7, окисленный водный раствор)
связь Со-С очень стабильна.
Для понимания биохимической роли кофермента В,2 сначала необходимо дать
определение понятию «кофермент». Кофермент — одна из составных частей
биологической каталитической системы. Для функционирования такой системы
требуются щфермент, апофермент и субстрат. Химическая реакция происходит между ко-
ферментом и субстратом, а оба они связываются апоферментом. Объединенные
апофермент и кофермент называют голофврмвнтом (схема 2.6). Такой модульный
подход позволяет коферменту В,2 участвовать в целом ряде биологических реакций
с различными апоферментами. Эти реакции могут включать гомолиз связи Со—С,
приводящий к образованию алкильного радикала, который вызывает перестройку
структуры, а также к редокс-реакции путем восстановления до Со(Н) и СоA) и ал-
килирования. Тип реакции определяется природой кофермента, тогда как природа
апофермента отвечает за селективность реакции по отношению к субстрату и за ре-
гиоспецифичность.
Из-за малой биодоступности кобальта соединения класса В12 требуются в очень
малых количествах и используются в основном микроорганизмами, которые также
сами могут их синтезировать. Однако у млекопитающих мутазная система метилма-
лонил-СоА, зависящая от В12, является исключительно важной. Она необходима
для аминокислотного метаболизма в печени, а ее отсутствие из-за генетических
дефектов смертельно. В самом деле, еще в 20-х годах прошлого века было замечено,
что смертельно опасную пернициозную анемию можно вылечить экстрактами из
печени животных. В этих экстрактах было обнаружено присутствие кобальта, что и
привело к большой работе по выделению этого необычного кобальтсодержащего
соединения, концентрация которого в крови -0.01 мг-л. Благодаря
использованию хроматографических методов и большой настойчивости исследователей циа-
нокобаламин наконец был выделен в чистом виде в 1948 г. Причем CN~-rpynna -
это не часть активной формы комплекса, а артефакт процедуры выделения.
Однако до сих пор цианпроизводное используется в терапии. Новое соединение было
названо витамином В12. В 1964 г. Д. Кроуфут-Ходжкин была удостоена
Нобелевской премии в области химии за определение кристаллической структуры этой,
Особые черты субстракта (селективность)
и скорость реакции обуславливаются белком
с большой молекулярной массой
I
Кофермент + апофермент *- голофермент
Полностью функциональный
фермент
Тип реакции обуславливается
низкой молекулярной массой
Схема 2.6. Модульная структура голофермента
7- СТИДДж.В
98
2. Супрамолекулярная химия жизни
Рис. 2.23. Кристаллическая
структура витамина В]2 (для
простоты заместители опущены)
довольно большой, «малой молекулы» (RMM = 1350 Да), рис. 2.23. Изгиб коррино-
вого микрокольца придает ему форму бабочки, что является следствием меньшего
размера макроцикла A5-атомное кольцо вместо обычного 16-атомного в порфири-
нах). Это искажение имеет прямое отношение к реакционной способности кофер-
мента и представляет структуру энтатического состояния*.
например:
nh2 со2н
Глутамат-
мутаза
H
н н н
Глутаминовая кислота
н
СО2Н
р"-Метиласпарагиновая кислота
^
C-S-CoA
i
иН Метвлмалонил-СоА-
Л-Метилмалонил-СоА
:-s-coa
Сукцинил-СоА
Схема 2.7. 1,2-Сдвиги мутазами кофермента В,
Состояние атома или группы, при котором их геометрические или электронные
характеристики, измененные в результате связывания с белком, приобретают способность
выполнять определенную функцию. «Entatic» происходит от греч. «entasis» — натяжение. {Примеч.
ред. перевода.)
2.6. Нейротрансмиттеры и гормоны
99
Инициирование:
п| Обратимый гомолиз
[Co'"]-CH2R ==: [Со11]' + RCH2*
Ферментативный эффект
Распространение:
X
+ RCHj*
Удаление Н
1,2-Сдвиг
RCH3
+ RCH3
X
Окончание:
[Соп]*+ RCH2*
RCH2
[Com]-CH2R
Схема 2.8. Радикальный механизм мутазной активности
Биохимия кофермента В12 обычно связана или с мутазной активностью
фермента, включающей миграцию функциональной группы, особенно
посредством стереоспецифических 1,2-сдвигов (схема 2.7), или с метилированием метио-
нинсинтазой. Радикальный механизм мутазной активности, являющийся,
по-видимому, основным, был установлен ЭПР-спектроскопией. Схема 2.8 отражает роль
этого механизма в мутазной активности.
2.6 Нейротрансмиттеры и гормоны
Кроме бионеорганической химии, в которой используются катионы
металлов в различных видах, существует также обширный раздел и биологической
химии, имеющий дело с молекулярными образованиями и анионами, которые с
позиций супрамолекулярнои химии могут рассматриваться в качестве гостей.
Детальный анализ этого направления находится за пределами тематики данной
книги, однако мы должны, хотя бы мимоходом, упомянуть о роли биологических
гостей двух особых классов, а именно гормонов и нейротрансмиттеров, которые в
широком смысле действуют как курьеры и активирующие агенты. Гормоны, например
половые гормоны эстроген и тестостерон, а также гормон беременности прогесте-
Mqq 2. Супрамолекулярная химия жизни
рон, стали уже бытовыми понятиями. Производные тестостерона, анаболико-анд-
рогенные стероиды, способны значительно изменить физические возможности
человека, а поэтому их иногда нелегально используют при подготовке спортсменов.
Подобным же образом уровень таких нейротрансмиттеров в организме, как допа-
мин и ацетилхолин, может оказывать очень сильное воздействие на умственную
деятельность человека. Их дисбаланс может вызывать ряд умственных расстройств,
например шизофрению и болезнь Паркинсона, что однозначно указывает на
химическую природу мыслительных процессов.
Для иллюстрации супрамолекулярного характера биологической активности
таких молекулярных соединений рассмотрим работу ацетилхолина (acetylcholine,
АСН) B.6). Этот пример указывает на глубокое воздействие, которое нековалентное
связывание маленьких молекул-гостей может оказывать на биологическую систему.
О
Ацетилхолин
B.6)
Ацетилхолин — это основная молекула нейротрансмиттера. Нервные импульсы
передаются от нервного волокна к нервному волокну через синапсы (промежутки
между нервными клетками толщиной 30—40 нм) путем переноса ацетилхолина от
одной нервной клетки к другой. Роль ацетилхолина заключается в открывании
натриевого канала в результате его связывания с никотин-ацетилхолиновым рецеп-
торным белком (nicotinic acetylcholine receptor protein, nAChR), образующим канал
(ср. разд. 2.1). Этот большой трансмембранный белок состоит из пяти субъединиц
(две из них обозначают а, а другие — 3, 5, у; рис. 2.24) с близкими
аминокислотными последовательностями, окружающих центральный канал. Ацетилхолин
связывается с двумя а-субъединицами, вызывая конформационные изменения во
внешней части канала, которые открывают доступ к ионам Na+, расположенным в
синапсе. Затем, в соответствии с преобладающим концентрационным градиентом, эти
ионы быстро диффундируют в клетку, вызывая ток. Связывание агониста
ацетилхолина с его рецептором высокоселективно и включает катион-л-взаимодействия
(см. разд. 1.7.5) между группами четвертичного аммония и остатком триптофана.
Интересно, что на активность ацетилхолина сильно действует никотин B.7),
который также содержит четвертичную аммониевую группу, и, скорее всего, именно это
является основной причиной привычки к никотину у курильщиков. Как только
мембрана поляризовалась и возник ток, ацетилхолин удаляется другим ацетилхо-
линсвязывающим белком - ацетилхолинэстеразой, который гидролизует эфирную
функциональную группу, не позволяя молекуле связываться с nAChR.
В супрамолекулярной химии конкуренция между ацетилхолином и никотином
за связывание с nAChR — пример важнейшей роли селективности в этом процессе.
2.7. ДНК
101
70 А
зо А
120 А
Рис. 2.24. Никотин-
ацетилхолиновый
рецептор (nAChR)
Из-за недостаточно высокой селективности nAChR может также связываться и с
мешающим никотином. Тот факт, что никотин, подобно ацетилхолину, содержит
четвертичную аммониевую группу, которая может участвовать в его связывании,
делает очень важной зависимость биологических процессов от кажущихся слабыми
супрамолекулярных взаимодействий.
*"* Dougherty D.A. Is the brain ready for physical organic chemistry? //
J.Phys.Org.Chem. 1998. Vol. 11. P.334-340.
2.7 днк
•""* Calladine C.R. and Drew H.R. Understanding DNA: The molecule and how it works.
2nd ed. New York: Academic Press, 1997.
Структура и функции ДНК
ДНК (дезоксирибонуклеиновая кислота) хорошо известна как
молекула, несущая в себе всю генетическую информацию, необходимую для построения и
функционирования живого организма. Ядра клеток всех эукариотических
организмов содержат ДНК, а каждая клетка - генетический код, необходимый для сборки
всего организма в целом, — весьма примечательное удвоение информации. Объем
содержащейся информации определяет необычайно большую длину цепи ДНК.
Суммарная длина молекул ДНК в каждой клетке достигает 3 см. То, что такая
большая молекула помещается внутри клетки шириной ~10~5 м, объясняется тем, что
молекула ДНК очень тонкая, ее диаметр всего 210~9 м. В обычном состоянии по-
1102
2. Супрамолекулярная химия жизни
коя ДНК имеет структуру двойной
спирали, состоящей из двух идентичных
цепей. Цепи удерживаются между собой
водородными связями и л—л-стэкинг-взаи-
модействиями (рис. 2.25). Реальная (хотя
Малая бороздка ^-"^^^ и идеализированная) структура ДНК
показана на рис. 2.26. Полагают, что
движущей силой такой организации является
то, что структура двойной спирали
позволяет гидрофобным участкам молекулы
(нуклеооснованиям) избежать контакта с
водой за счет их расположения внутри
Большая бороздка спирали. Структура спирали дает
возможность оптимизировать взаимодейст-
Рис. 2.25. Двойная спираль ДНК.
Штриховыми линиями обозначены
водородные связи между парами оснований
Рис. 2.26. Идеализированное изображение трех наиболее распространенных форм ДНК:
В, А и Z. Структуры В и А — правовращающие спирали с 10 и 11 фосфатными группами на
виток соответственно. Структура Z — левовращающая спираль с 12 фосфатными группами
на виток. (Воспроизведено с разрешения Calladine C.R. and Drew H.R. Understanding DNA:
The molecule and how it works. 2nd ed. New York: Academic Press, 1997)
2.7. ДНК
Аденин
(R = H)
R =
R =
Гуанин Цитозин R' = Н: урацил (R = Н)
(R = H) (R = H) R. = Cf
Нуклеозид
Н^\ Х = ОН:рибоза
X X = Н: дезоксирибоза
ОН
О" О
но-
-р—о-
II
о
-р—о-
I
О"
H ' X
H OH
Нуклеотид
Рис. 2.27. Нуклеотид состоит из нуклеооснования, прикрепленного к сахару, и
фосфатного «хвоста»
вия между нуклеооснованиями за счет минимизации внутреннего свободного
пространства.
Основные составляющие ДНК представляют собой нуклеотиды — молекулы,
содержащие нуклеооснования (аденин (А), тимин (Т), цитозин (С) или гуанин (G)),
соединенные через сахар с фосфатным «хвостом» (рис. 2.27). Компоненты А и Т
известны как пурины, тогда как нуклеооснования С и G относятся к пиримидином.
Полимеризация этих нуклеотидов через сахаро-фосфатные остатки приводит к
образованию каркаса цепи ДНК B.8). Генетическая информация хранится в ДНК в
виде большого числа трехбуквенных «слов». Эти «слова» содержат в себе триплеты
нуклеооснований (GCC, CAG, АТС и т.д). Каждое «слово» биохимически
передается в одну из 20 аминокислот белка (Дополнение 2.2) в соответствии с табл. 2.2.
Нуклеооснование Нуклеооснование Нуклеооснование
Информация, хранящаяся в ДНК, используется при кодировке синтеза всех
белков, требующихся организму для его биологического функционирования. Этот
процесс поясняется на рис. 2.29. Внутриядерная ДНК управляет темплатным
синтезом матричной РНК (рибонуклеиновая кислота), непосредственной копии кода
ДНК, но с одним дополнительным атомом кислорода, который помогает ей
«выскользнуть» из клеточного ядра через маленькие дырочки в его мембране. РНК
104
2. Супрамолекулярная химия жизни
Me { Алании (Ala) H2N
со2н
о
s ff NH Аспарагиновая
< \ /2 кислота (Asp)
N ( (R = OH)
\ Аспарагин (Asn)
CO2H (R _ j^^
NH2 Глутаминовая
кислота(Glu)
(R = OH)
CO2H Глутамин (Gin)
(R = NH2)
со2н
Пролин (Pro)
Треонин (Thr)
Тирозин (Туг)
HS
Me
Me
NH2
CO2H
NH2
CO2H
CO2H
Аргинин (Arg)
Цистеин (Cys)
Глицин (Gly)
Изолейцин(Пе)
Лизин (Lys)
Фенилаланин (Phe)
Серии (Ser)
Триптофан (Тф)
Валин (Val)
CO2H
Рис. 2.28. 20 основных аминокислот, образующих белки
2.7. ДНК
105
Таблица 2.2. Генетический код*
Первое нуклеооснование
G
Второе нуклеооснование
Третье нуклеооснование
Phe
Phe
Leu
Leu
Leu
Leu
Leu
Leu
He
He
He
Met (START)
Val
Val
Val
Val
Ser
Ser
Ser
Ser
Pro
Pro
Pro
Pro
Thr
Thr
Thr
Thr
Ala
Ala
Ala
Ala
Tyr
Тут
STOP
STOP
His
His
Gin
Gin
Asn
Asn
Lys
Lys
Asp
Asp
Glu
Glu
Cyr
Cys
STOP
Trp
Arg
Arg
Arg
Arg
Ser
Ser
Arg
Arg
Gly
Gly
Gly
Gly
T
С
A
G
T
С
A
G
T
С
A
G
T
С
A
G
* Каждую последовательность трех оснований в ДНК можно транслировать в одну из 20
аминокислот в аминокислотной цепи (Дополнение 2.2). Каждая цепь начинается со стартового
кода ATG, соответствующего аминокислоте метионину (Met).
доставляется к части клетки, называемой рибосомой и являющейся фабрикой
клеточного белка. С помощью кода, переносимого РНК, происходят сборка
определенных комбинаций из 20 различных аминокислот (белковые аминокислоты) и
образование биологических белков типа ферментов, а также инициирование и
транспорт белков, которые осуществляют все биохимические превращения в организме,
РНК, содержащая послецокательность
ДНК
*&$
$ЙШШ$у* £$3^
Маленькие отверстия
ДНК, шдсра»лад>яс« йнугпп хроьичом
Мембрана
Рис. 2.29. Передача генетического кода для синтеза белка от ДНК к рибосоме через
матричную РНК
2. Супрамолекулярная химия жизни
Дополнение 2.2. Аминокислоты белка
Аминокислоты белка — основные «строительные блоки» всех пептидных
(аминокислотных полимеров и олигомеров) и белковых цепей (белок — большой полипептид),
которые составляют каркас структуры огромного числа биологических молекул. Цепи
состоят из пептидных звеньев (амидные связи), образующихся в результате
межмолекулярной конденсации между карбоксильной и аминогруппами аминокислот (схема 2.9).
NH,
R-( +R
CO2H
nh2
CO2H
H2N,
R' ^
) R
Д ♦ H20.
^""^ Пептидное звено
COjH
Схема 2.9. Реакция конденсации между карбокси- и аминогруппами
аминокислот
Группы R, называемые боковыми цепями, определяют состав аминокислоты.
Аминокислоты являются хиральными молекулами, в высших организмах все
множество белков принадлежит к L-cx-аминокислотам. В бактериях и грибах
обнаружено гораздо большее разнообразие аминокислот. Аминокислоты синтезируются
исключительно с помощью ферментов, но образование пептидов и белков — более
сложный процесс, требующий участия как транспортной РНК, так и матричной РНК.
В высших организмах этот процесс протекает в пределах особой части клетки
(рибосомы) путем расщепления предшественников белка, что ограничивает диапазон
доступных аминокислот. В низших организмах пептиды возникают в нерибосомных
ферментативных процессах, которые менее специфичны. На рис. 2.28 представлены
20 первичных компонентов белка. Все они относятся к а-аминокислотам, за
исключением пролина, являющегося циклической а-иминокислотой.
Состояние двойной спирали ДНК — это состояние ожидания. Когда считывается
генетическая информация, двойная спираль расплетается и используется для
сборки комплементарной молекулы РНК, содержащей копию генетического кода. В
свою очередь РНК собирает закодированный белок, связывая аминокислоты,
которые соответствуют последовательности «слов» в коде. Эта последовательность
аминокислот определяет структуру и функцию белка, образующегося в результате
полимеризации аминокислот, а также формирование вторичной и третичной структур
белка (см. разд. 9.2).
Внутри двойной спирали ДНК существуют пары оснований специфического
типа, соединенные водородными связями. Это пары оснований Уотсона—Крика
2.7. ДНК 107
^ 10.85 А (Ф 10.85 А
^ ».
HN 2.84 А I
N-H--- Оч dR
dR ,N=\ 2.92 А Г~\ dR
SN-( N-H-- N
^N Ъ--- H-N Me "N N-H--- О
2.84 А Чн и 2.82 A
dR - дезоксирибоза
G-C A-T
Рис. 2.30. Спаривание оснований {а) С—G и (б) А—Т по Уотсону и Крику
(в честь Дж. Уотсона и Ф. Крика, впервые описавших структуру ДНК в 1952 г.)
Комплементарность пар оснований обеспечивается особыми водородными
связями или между аденином и тимином, или между гуанином и цитозином (в РНК ти-
мин заменен родственным ему урацилом). Никаких других комбинаций не
существует, и именно эта способность нуклеиновой кислоты к распознаванию своего
комплементарного партнера составляет основу для работы ДНК.
Спаривание оснований А—Т и C—G с помощью водородных связей показано т
рис. 2.30. В обоих случаях за взаимное распознавание комплементарных оснований
ответственна более чем одна водородная связь, причем геометрия такова, что обе
пары имеют одну и ту же длину. Длины водородной связи 2.8—2.9 А типичны для
водородных связей средней силы N—H--O/N. Спаривание оснований и п—л-стэкин1
в сборке двойной спирали ДНК - великолепный пример супрамолекулярной
самоорганизации. Именно это обеспечивает репликацию ДНК, так же как и передач)
закодированной информации на РНК. Самосборка имеет все признаки каскадногс
процесса, в котором сумма вкладов от образования каждой последующей пары
оснований постепенно превышает уменьшение энтропии, связанное с потерей
степеней свободы двух отдельных цепей. Детально самосборка ДНК рассматривается е
разд. 7.2.
Нуклеооснования также способны участвовать в водородном связывании
альтернативного типа, называемом спариванием оснований по Хугстену, в которое
вовлекаются пятичленные кольца адениновых или гуаниновых остатков (рис. 2.31)
Центры водородного связывания по Хугстену расположены на внешней
поверхности молекулы ДНК и, таким образом, доступны агентам, связывающим ДНК.
Рис. 2.31. Спаривание оснований аденина
и тимина по Хугстену
108
2. Супрамолекулярная химия жизни
Направленный мутагенез
Нобелевскую премию 1993-го года в области химии за развитие
направленного мутагенеза и полимеразной цепной реакции — двух методов
манипуляций с ДНК — разделили М. Смит (М. Smith) из Университета Британской
Колумбии (Канада) и К. Муллис (К. Mullis; Jla Хойа, Калифорния, США). Направленный
мутагенез позволяет перепрограммировать генетический код ДНК для получения
разновидностей белков с новыми свойствами. Этот метод применяют в белковой
инженерии антител для атаки на особые области, такие, как опухолевые клетки, а
также для получения более стабильных белков в практических целях и лечения
наследственных болезней, например кистозных фиброзов. Метод также можно
использовать в генной инженерии сельскохозяйственных культур для получения
быстрорастущих и устойчивых к болезням сортов. Эта проблема стала центром
недавней полемики по генетически модифицированным (genetically modified, GM)
продуктам, так как в средствах массовой информации появились сведения о том, что
модифицированные белки, обладая непредсказуемыми свойствами, в перспективе
смогут заменить природные продукты.
Синтетический олигонуклеотид
с измененным генетическим кодом
-AAA-GGG- (GGG вместо GAG)
-1ТТТ-СТС-1
ДНК вируса
ДНК-полимераза
- AAA-GGG-
Молекула ДНК с обычным кодом
^AAA-GAC
Молекула ДНК
с мутантным кодом
-AAA-GGG-
Размножение в бактерии
Лейцин
Обычный белок
Рис. 2.32. Направленный мутагенез
Мутантный белок
Обмен пролина
на ле£
2.7. ДНК 109
Процесс направленного мутагенеза включает сборку олигонуклеотидных
фрагментов, обеспечивающих желаемую мутацию. Мутация происходит путем
изменения одного из «слов» генетического кода; например, замена САС на GAC вызывает
замену гистидинового остатка аминокислоты на аспаргиновую кислоту. Мутант-
ный олигонуклеотид вводится в сегмент вирусной ДНК, с которым он связывается
в том же месте, что и немутантный аналог. Это происходит потому, что комплемен-
тарность между цепями может выдержать небольшое количество несоответствий
пар оснований. Мутантный олигонуклеотид встраивается в копию исходной ДНК с
помощью фермента ДНК-полимеразы, которая формирует сахаро-фосфатный
каркас, связывая вместе фрагменты нуклеотидов. Затем полученный мутант ДНК
вводится в подходящий организм бактериального хозяина, в котором он
размножается, производя свои копии, и, когда их наберется достаточное количество,
синтезирует заданный мутантный белок. Как случайные события такие мутации
происходят в природе постоянно и согласуются с теорией эволюции. Большинство
природных мутаций губительно для организма, но управляемый мутагенез может быть
весьма полезным. Схема этого процесса дана на рис. 2.32.
2.7.3 Полимеразная цепная реакция
Полимеразная цепная реакция (polymerase chain reaction, PCR) —
другая крайне важная недавняя разработка в химии ДНК. PCR-дает возможность
синтезировать отдельные участки ДНК или фрагмент олигонуклеотида, начиная с
совсем маленького образца, в больших количествах. Эту методику широко
используют в криминалистике, когда из крови или ткани человека (следы) может быть
выделено такое количество ДНК, которого достаточно для проведения анализа.
Процесс PCR жизненно важен для проекта HUGO (Human Genom - Геном
Человека), в результате работы над которым в 2000 г. был определен каждый отдельный
код ДНК в генетическом материале человека. Этот процесс также предоставляет
уникальные возможности, например, для выделения больших количеств ДНК
давно исчезнувших видов животных из их окаменелых останков. Это вдохновило
создателей научной фантастики на «возрождение» динозавров в фильмах «Парк
Юрского периода» и «Затерянный мир», хотя при современной технологии
возрождение вымерших особей невозможно.
PCR состоит в синтезе двух коротких олигонуклеотидов, точно связывающихся
с противоположными цепями ДНК, которые будут копироваться. Это позволяет
ферменту ДНК-полимеразе начать сборку копий двух цепей, приводящую к
получению двух новых цепей ДНК. Нагревание образца вызывает расплетение двойных
спиралей и дает четыре новые цепи, которые могут использоваться в качестве тем-
платов для образования еще четырех цепей. Процедура повторяется от 20 до 60 раз
в течение нескольких часов или вручную, или обычно с помощью автоматического
устройства. В результате это дает 220—260 A06—1019) копий исходного фрагмента
(рис. 2.33).
IK
2. Супрамолекулярная химия жизни
Повтор 20-60 раз
Рис. 2.33. Схема полимеразной цепной
реакции (PCR)
Много миллионов копий ДНК
Связывание с ДНК
Некоторые разновидности рака очень восприимчивы к обработке
платиновыми комплексами, которые связывают ДНК опухоли. Почти все активные
антиопухолевые агенты содержат Pt(II) в плоскоквадратной геометрии с двумя цис-
первичными или ^wc-вторичными аминогруппами и двумя более слабо связанными
лигандами, такими, как хлорид или карбоксилаты. Родственные октаэдрические
комплексы Pt(IV) с дополнительными аксиальными лигандами также очень
активны, и, вероятно, in vivo (в реальной живой системе) они могут восстанавливаться до
комплексов Pt(II) с потерей аксиальных лигандов.
Два наиболее важных противораковых препарата на основе платины — это цис-
платин B.9) и карбоплатин B.10). Цисплатин эффективен при опухолях яичек,
карциноме яичников и других типах рака, но относительно неактивен в случае
рака груди и легких; это очень токсичное вещество. Побочные эффекты включают
потерю слуха в высокочастотном диапазоне, невропатию и тошноту. Он может
вызвать серьезное нарушение функции почек. Его действие можно свести к
минимуму, употребляя большое количество воды, что обеспечивает быстрое вымывание
неиспользованного лекарства из организма. Карбоплатин — более новое и менее ток-
2.7. ДНК
111
сичное лекарство. Примечательно, что оно не приводит к потере слуха в
высокочастотном диапазоне.
I3N^ 4C1
Цисплатин
B.9)
H3N.
H3N
О
//
o-c
w
о
Карбоплатин
B.10)
По-видимому, способ действия многих неорганических противораковых
препаратов заключается в их способности связываться с ДНК. Остатки гуанина,
находящиеся на поверхности большой бороздки ДНК некоторых типов, являются главной
мишенью для платины в теле человека. Лекарство напрямую вводят путем
инъекции (во избежание гидролиза кислотой желудочного сока). Значительная его часть
связывается с белками плазмы и затем быстро вымывается через почки.
Оставшаяся часть переносится кровью и проходит через клеточную мембрану в нейтральной
форме. Хлоридные B.9) или карбоксильные B.10) лиганды теряются за счет более
низкой внутриклеточной концентрации С1~, образуя [Pt(NH3JCl(H2O)]+;
положительный заряд этого катиона обеспечивает его притяжение к отрицательно
заряженной ДНК, в конечном счете приводя к связыванию группы (NH3JPt2+ с ДНК.
Возможно, образование такого мостика вызывает разрыв связей между парами
оснований, удерживающих вместе цепи двойной спирали ДНК (рис. 2.34). Это, в
свою очередь, вызывает «изгиб» цепи ДНК, которая теперь не распознается
«ремонтными» ферментами ДНК в клетках опухоли. транс-[РхС12(^Н^J] не может об-
p (I
dR dR
Рис. 2.34. Связывание платины с ДНК
112
2. Супрамолекулярная химия жизни
Рис. 2.35. Кристаллическая структура
«i/c-Pt(NH3)£4d(pGpG)J (no Sherman S.
etal., 1989)
разовать сшивку ДНК той же геометрии, что объясняет отсутствие активности у
этого соединения.
Моделью взаимодействия ДНК-Pt является комплекс Pt(NH3J+ и гуанозинди-
нуклеотида d(pGpG), содержащего два основания гуанина, связанных сахаро-фос-
фатным каркасом. На рис. 2.35 показана кристаллическая структура этого
соединения, из которой видно взаимодействие платины с соседними гуаниновыми NG)-
донорами. Водородные связи между аммониевыми лигандами и кислородным
атомом фосфата дополнительно стабилизируют этот комплекс.
Связывание с переходными металлами с помощью координационных
взаимодействий — далеко не единственный способ атаки на молекулу ДНК. Известно
множество координационных соединений, называемых интеркаляторами, которые
способны связываться с ДНК исключительно путем комбинации
электростатического и 71—Ti-стэкинг-взаимодействий. Недавно показали, что плоскоквадратные
комплексы Pt(II), содержащие плоские ароматические гетероциклические лиган-
ды, например 2,2'-бипиридил, способны связываться с ДНК путем частичного
' :Ru N
dppz
B.12)
D-[Ru(phenK]2+
B.11)
Me
[Rh(/?,/?-Me2trien)(phi)]3+
B.13)
2.7. ДНК
113
включения (интеркаляция) ароматического лиганда между нуклеооснованиями,
взаимодействующими по типу п—я-стэкинга, со стороны большой бороздки.
Подобная интеркаляция установлена для хиральных октаэдрических комплексов,
например для [Ru(phenK]2+ B.11), где phen - 1,10-фенантролил, и, что важно, для
правовращающих D-изомеров в правозакрученную спираль ДНК она предпочтительна.
Сродство интеркаляторов к ДНК может быть заметно усилено при увеличении
площади поверхности интеркалирующих лигандов; так, величины констант
связывания аналогичных комплексов с большой площадью поверхности dppz-лиганда
B.12) превышают 106 М. Любую неоднозначность в природе связывания
интеркаляторов с ДНК недавно исключила Ж. Бартон (J. Barton) из Калифорнийского
технологического института (США), определившая кристаллическую структуру
комплекса другого интеркалятора, [Rh(/?,/?-Me2trien)(phi)]3+ B.13), связанного с
центральной 5'-ТОСА-3'-областью олигонуклеотида, содержащей восемь пар
оснований. Из структуры ясно видно, что phi-лиганд, интеркалированный в
область данного нуклеооснования (рис. 2.36), участвует в формировании нескольких
водородных связей между аксиальными группами аминного лиганда и соседними
Рис. 2.36. Два вида кристаллической структуры комплекса [Rh(/?,/?-Me2trien)(phi)]3+
B.13), связанного внутри олигонуклеотида с восемью парами оснований. (Воспроизведено
с разрешения Erkkila K.E., Odom D.T., Barton J.K. Recognition and reaction of metallointercala-
tors with DNA // Chem.Rev. 1999. Vol.99. P.2777-2795)
114
2. Супрамолекулярная химия жизни
гуаниновыми остатками. Примечательно, что структура, включающая водородные
связи, была предсказана исследователями из группы Бартон; эта структура
образовалась в результате успешной атаки сконструированного ими интеркалятора на
область 5'-TGCA-3'. Константа связывания, соответствующая этому элегантному
образцу молекулярного конструирования, очень велика (9-107 М~'). Пока еще
применение интеркалирующих агентов для ДНК находится в стадии младенчества.
Однако такой подход предполагает возможное введение флуоресцентных меток и для
диагностирования повреждений, и для их залечивания, а также для увеличения
чувствительности комплексов к окислительному и фотолитическому расщеплению.
Целый ряд хорошо известных полностью органических пептидов также
связывается с ДНК. Антибиотики, например ионофор нонактин, так же как и природные
продукты нетропсин и дистамицин А, способны связываться в пределах большой
бороздки ДНК в определенной последовательности с образованием множества
водородных связей. Пептиды имеют выраженную серповидную форму и лежат в
пределах последовательностей, обогащенных AT, которые образуют водородные связи
с атомами NC) аденина и 0B) тимина, рис. 2.37. Свободные энергии связывания
равны ~53 кДжмоль. Синтетическая податливость каркаса пептидов в
антибиотиках этого типа позволяет легко изменять их структуру, благодаря чему были
получены многочисленные синтетические производные пептидов.
Большая часть исследований в области супрамолекулярной химии посвящена
поиску современных противораковых препаратов. Наиболее перспективное
Рис. 2.37. Водородное связывание
нетропсина с 5'-ААТТ-3'-областью
ДНК
2.8. Биохимическая самосборка j J5
направление таких исследований — вероятно, поиск агентов, связывающих ДНК с
помощью координационных взаимодействий и/или комбинированных
водородных взаимодействий по Хугстену и 71—71-стэкинга (интеркаляция ДНК).
•""» Erkkila K.E., Odom D.T., Barton J.K. Recognition and reaction of metallointercala-
tors with DNA// Chem. Rev. 1999. Vol.99. P.2777-2795.
2.8 Биохимическая самосборка
Очевидно, что структура двойной спирали ДНК и способность ее
молекулы развертываться для воспроизводства своих копий (репликация ДНК) и
транскрипции РНК тесно связаны с супрамолекулярными взаимодействиями,
удерживающими две цепи нуклеотида вместе. Именно информация,
закодированная в отдельных нуклеооснованиях заставляет молекулу формировать двойную
спираль. Это пример гораздо более общего явления самосборки, которое
невероятно важно в современной супрамолекулярной химии. В очень общем виде
самособирающиеся системы могут содержать два или более элемента, которые могут
обратимо объединяться таким образом, что получающийся агрегат будет представлять
собой наиболее термодинамически устойчивую структуру, присущую системе в
данных условиях. Строго говоря, вся информация, необходимая для образования
конечного соединения, должна быть заключена (закодирована) в реагирующих
компонентах. Именно эта термодинамическая стабильность в сочетании с
возможностью исправления ошибок благодаря обратимости равновесного процесса отвечает
за структуру многих биологических систем и молекулярных устройств. Примеры
такого процесса - самосборка ДНК и трансляция матричной РНК в белок в
рибосоме, а также самосборка вирусов (которые часто обладают потрясающе красивой
симметричной структурой, формирующейся в результате самосборки одинаковых
субъединиц), сворачивание ферментов и белков, накопление света в фикобилисо-
ме, фотоиндуцированная прокачка ионов через мембраны с помощью бактериоро-
допсина. Следует, однако, заметить, что живые организмы в целом далеки от
равновесия и требуют постоянной подпитки энергией для поддержания гомеостаза.
В качестве примера самосборки белков рассмотрим самосборку рибонуклеазы,
содержащую 124 аминокислотных остатка и четыре дисульфидные (—S—S—) связи.
Восстановление этих связей дает восстановленный белок, содержащий восемь SH-
групп, т.е. полностью денатурированный (ферментативно неактивный) продукт.
При окислении этого белка в присутствии мочевины (которая действует как
денатурирующий агент благодаря своей способности к водородному связыванию)
образуется смесь из спутанных (неправильно сложенных) белков, содержащих
большинство (если не все) из 105 возможных комбинаций дисульфидных связей.
Добавление C-меркаптоэтанола вызывает медленный разрыв дисульфидных связей и
переход к равновесному процессу; если удалить мочевину и добавить C-меркаптоэта-
нол, то белки распутываются, давая нативную рибонуклеазу с полной
ферментативной активностью (рис. 2.38). Отсюда следует, что нативный белок представляет
2. Супрамолекулярная химия жизни
Нативный белок
Повторное свертывание
Нативный белок Спутанный белок
Рис. 2.38. Денатурирование и повторная самосборка рибонуклеазы. Правильное
свертывание происходит только тогда, когда образование дисульфидной связи становится
обратимым при добавлении Р-меркаптоэтанола (RSH), позволяющего системе достичь
равновесия. (Воспроизведено с разрешения Lindsey J S. Self-assembly in synthetic routes to
molecular devices. Biological principles and chemical perspectives: A review // New J. Chem. 1991.
Vol.15. P. 153-180)
собой структуру с наименьшей свободной энергией Гиббса и что вся информация,
необходимая для сборки белка, закодирована в последовательности аминокислот.
Рассмотрим железонакапливающий белок ферритин в качестве примера
кодирования информации. Этот белок содержит 24 идентичные субъединицы, которые
вместе образуют почти сферическую оболочку октаэдрической симметрии. Почему
же 24 субъединицы и почему октаэдрическая симметрия? Исследование отдельных
субъединиц показывает, что в зависимости от распределения их центров
связывания могут быть реализованы два способа агрегации. Один из них включает
взаимодействие по типу «замок—ключ» каждой субъединицы со своим соседом под углом
90°, что дает агрегат из четырех субъединиц с симметрией четвертого порядка.
Другой же вовлекает во взаимодействие по типу «замок—ключ» три субъединицы,
расположенные под углом 60° друг к другу, что дает соединение с симметрией третьего
порядка. Решающим поэтому является то, что если белок должен одновременно
насытить все свои центры связывания, то получающийся агрегат должен обладать
осями симметрии как третьего, так и четвертого порядка. Это может быть
достигнуто единственным путем — образованием кластера из 24 субъединиц с
октаэдрической симметрией (схема 2.10).
Другой пример биохимической самосборки — димеризация протеазы вируса
иммунодефицита человека 1 (HIV-1) - фермента, определяющего сборку и
инфекционную активность вируса СПИД, а также протеазы икосаэдрического римовируса —
причины обычной простуды. Однако, вероятно, наиболее известным и важным
примером биохимической самосборки помимо ДНК является вирус табачной
2.9. Виагра: по ту сторону газетной шумихи j j<7
Схема 2.10. Изображения структуры Н
цепочечного ферритина человека с осями
симметрии четвертого и третьего порядка (левое
и правое соответственно). Взаимодействия
вокруг оси четвертого порядка происходят под
углом 90° и приводят ктетрамерам. Белковые
тримеры, расположенные под углом 60° друг к
другу, имеют ось третьего порядка.
(Воспроизведено с разрешения Caulder D.L. and
Raymond K.N. // Ace. Chem. Res. 1999. Vol. 32.
P. 975)
мозаики. Этот механизм, который вводит нас в быстроразвивающуюся область
синтетической самосборки и ее возможного применения при формировании
молекулярных машин и устройств, рассмотрен в гл. 7.
•""* Lindsey J.S. Self-assembly in synthetic routes to molecular devices. Biological
principles and chemical perspectives: A review // New J. Chem. 1991. Vol.15. P.153-180.
2*9 Виагра: по ту сторону
газетной шумихи
Один из последних примеров в области биохимии, имеющий супрамо-
лекулярное происхождение, — реакция мужчины на половое возбуждение.
Нарушения этой реакции успешно лечат с помощью абиотических лекарств. Фактически
такая реакция на половое возбуждение, приводящая к эрекции, — результат
выработки и связывания одной маленькой, быстро диффундирующей молекулы оксида
азота (N0). При половом возбуждении окончания аксонов парасимпатических
нервов (часть автономной нервной системы, работающая по механизму
бессознательных реакций тела) высвобождают N0. Затем N0 быстро диффундирует в
клетки гладкой мускулатуры, окружающей главную артерию, ведущую в пенис,
связывается ферментом гуанилатциклазой и активирует его. Роль этого фермента
состоит в связывании гуанозинтрифосфата (GTP, ср. с АТР) и превращении его в
циклический гуанозинмонофосфат (cGMP). Последний вызывает релаксацию клеток
гладкой мускулатуры, что приводит к увеличению притока крови в пенис и - как
следствие — к его распуханию. После эякуляции и завершения полового
возбуждения выработка N0 и cGMP прекращается, а остающийся cGMP удаляется с
помощью фермента фосфодиэстеразы, который превращает cGMP в его нециклический
аналог GMP (схема 2.11). Неправильное функционирование этого механизма
(например, в результате побочного действия лекарства) вызывает дисфункцию
эрекции — расстройство, поражающее -20—30 млн мужчин только в США.
В марте 1998 г. Управление по санитарному надзору за качеством пищевых
продуктов и медикаментов США одобрило препарат виагра (Viagra®, сидденафилцит-
рат), производства «Pfizer Inc.», для лечения дисфункции эрекции. С момента
118
2. Супрамолекулярная химия жизни
N..
Т Т Т \°J
GTP y-f
он он
Гуанилат-
циклаза
-Р2О74",
f
О
<^х
он
cGMP
м. ?
Me
О
II
N NH2
Фосфодиэстераза
Ч-" ^Ме
0
/Y^NH
O"v ^°\ N^v. ^Ч
Р ^ 7 N NH2
он он
GMP
Виагра
(силденафил)
Схема 2.11. Выработка и потребление cGMP и близкое структурное сходство между
виагрой и гуанозинфосфатами
появления виагры вопросы о ее использовании, а также о ее возможных побочных
действиях остаются предметом горячих споров во всем мире. Несомненно, виагра
крайне эффективна для усиления и увеличения продолжительности эрекции. Она
«работает» как ингибитор действия фермента фосфодиэстеразы,
обуславливающего поглощение сосудорасширяющего cGMP. Таким образом, в отсутствие полового
возбуждения виагра не вызывает эрекцию. Скорее всего, близкое структурное
сходство между остатком гуанозина в cGMP и виагрой позволяет ей связываться с ре-
цепторным центром, предназначенным для cGMP в фосфодиэстеразе. Пропильная
группа может взаимодействовать с гидрофобным центром связывания,
предназначенным для рибозного (сахарного) остатка cGMP; это еще один пример
селективного супрамолекулярного взаимодействия по типу «хозяин—гость». Однако из-за
того, что виагра не полностью селективна к отдельному классу III фосфодиэстеразы,
Учебные задания
119
являющейся ее целью, она может вызывать некоторые временные побочные
эффекты: изменение зрения, особенно цветового восприятия, головную боль, боли в
области таза, покраснение лица, носовые кровотечения и понос. Кроме этого, еще
более серьезен риск при возможном взаимодействии виагры с другими
органическими NO-содержащими препаратами, которые больные могут принимать для
поддержания сердечной деятельности, снижения давления и т.д. Органические
нитраты, например нитроглицерин, изосорбитмононитрат и пентаэритритолтетранитрат,
действуют как сосудорасширяющие препараты, повышающие уровень N0,
который, в свою очередь, повышает уровень cGMP. Синергетическое взаимодействие
виагры с нитратными препаратами может привести к крайне высоким уровням
cGMP и катастрофическому падению кровяного давления. Симптомы колеблются
от головокружения и бреда до обморока и даже сердечного приступа. Проблемы
такого рода не были замечены в случае других сосудорасширяющих препаратов,
действующих без участия N0.
Учебные задания
2.1. Используя информацию, приведенную в табл. 2.1, рассчитайте разность
потенциалов (в мВ) через нервную клетку.
2.2. Рассчитайте энергию, необходимую для удержания ионов Na+ и С1~ на
расстоянии 5 нм друг от друга в 1.0-10 М NaCl. Что является аналогом
электростатической потенциальной энергии между 1 моль Na+ и С1~, расположенных на
расстоянии 1000 км друг от друга? (Электростатическая энергия - qvq2/4nE0r,
единичный заряд - 1.602-10~19 Кул; е0 = 8.854-Ю-12 О-мЧ)
2.3. Используя табл. 2.2, укажите последовательность аминокислот, закодированную
следующей последовательностью нуклеооснований, начиная с основания 1:
ATGTTCCATAGCAAGTAG
123
Повторите процесс, начиная с оснований 2 и 3. Заметьте, что эта
последовательность аминокислот решающим образом зависит от исходной точки или
«системы координат чтения». Добавление одного нуклеооснования
где-нибудь в середине последовательности полностью изменяет код. Это известно
как «мутационный сдвиг системы координат». Попытайтесь ввести С после
ATG-START-кода и повторите сборку белка.
2.4. Обсудите значение биологической трансмембранной разности потенциалов.
Как создаются и поддерживаются такие разности потенциалов?
2.5. Ежедневные рекомендуемые в США диетические нормы (РДН) для ряда
различных металлов приведены ниже. Объясните эти различия на основе
краткого анализа функций этих элементов в биохимии человека.
120
2. Супрамолекулярная химия жизни
Элемент
К
Na
Са
Mg
Fe
Со
РДН, мг день
2000-5500
1100-3300
800-1200
300-400
10-20
0.2
Литература
Caulder, 1999 Caulder D.L, Raymond K.N. Supermolecules by design // Ace. Chem. Res.
1999. Vol.32. P.975.
Doyle, 1998 Doyle D.A., CabralJ.M., Pfuetzner R.A. et al. The structure of the potassium
channel: Molecular basis of K+ conduction and selectivity// Science. 1998,
Vol.280. P.69.
Glusker, 1985 GluskerJ.R, Trueblood K.N. Crystal structure analysis. 2nd ed. Oxford: Oxford
University Press, 1985.
Kobuke, 1997 KobukeY. Artificial ion channels // Advances in supramolecular chemistry / Ed.
G.W. Gokel. Greenwich: JAI Press, 1997. Vol.4. P. 163-210.
Sherman, 1989 Sherman S.E., Gibson D., WangA.H.J., Lippard S.J. Crystal and molecular-
structure of m-[Pt(NH3J(d(pgpg))] the principal adduct formed by cis-
diamminedichloroplatinum(II) with DNA// J. Amer. Chem. Soc. 1989.
Vol.110. P.7368.
3
Хозяева,
связывающие катионы
Коня пришпорил запоздалый путник,
Спеша к ночлегу.
Мы otce, смешавшись с остальными,
Сыграем роль смиренную хозяев.
В. Шекспир A564-1616). Макбет
Благодаря открытию таких природных ионофоров, как валиномицин,
нонактин и энниатины (гл. 2), химики-супрамолекулярщики получили мощный
импульс для синтеза искусственных имитаторов ионофоров и моделирования
соединений, способных селективно связывать и переносить катионы не только
щелочных металлов, но также большинства s-, p-, d- и/-металлов и неметаллические
катионы, например NHJ и органические аммониевые. Особенно сложной задачей
оказалось энантиоспецифическое связывание хиральных молекул, в частности
протонированных аминокислот. Химия катионного комплексообразования
положила начало целой области молекулярного распознавания (сфера химических
исследований, включающая селективное связывание хозяевами, геометрическая и
электронная структура которых запрограммирована для комплексообразования с
данным гостем). Было синтезировано большое количество разнообразных лиган-
дов (хозяева для ионов металлов), проявляющих и удивительную селективность, и
«полезную» реакционную способность. В этой главе мы рассмотрим некоторые
наиболее распространенные классы лигандов и используем их в качестве примеров
при обсуждении вопросов молекулярного распознавания. Затем будет рассмотрено
применение этих понятий в более сложных или специализированных системах.
*"* Lehn J.-M. Supramolecular chemistry — scope and perspectives molecules,
supermolecules, and molecular devices //Angew. Chem., Int. Ed. Engl. 1988, Vol.27,
N1. P.89-112.
*"* Cram D.J. The design of molecular hosts, guests and their complexes (Nobel Lecture)//
Angew. Chem., Int. Ed. Engl. 1988. Vol.27, N8. P. 1009-1020.
*"» Pedersen C.J. The discovery of crown ethers (Nobel Lecture) //Angew. Chem., Int.
Ed. Engl. 1988. Vol.27, N8. P.1021-1027.
1 Краун-эфиры
•"» Gokel G. W. Crown ethers. Cambridge: Royal Society of Chemistry, 1990.
3.1.1 Открытие и область применения
Краун-эфиры относят к числу простейших и привлекательных макро-
циклических (с большим кольцом) лигандов, в супрамолекулярной химии они -
122
3. Хозяева, связывающие катионы
вездесущие хозяева и для катионов, и для нейтральных молекул (гл. 5). Краун-эфи-
ры представляют собой циклические соединения, содержащие эфирные атомы
кислорода, связанные с органическими спейсерами, обычно —СН2СН2-группами.
В то время как способность связывать металлы у однодентатных эфиров, например
у обычного растворителя - диэтилового эфира, очень низка, у краун-эфиров,
благодаря хелатным и макроциклическим эффектам, она несравненно выше (разд. 1.4).
За открытие краун-эфиров в 1967 г. Ч. Педерсену, химику компании «American
du Pont de Nemours» , была присуждена Нобелевская премия в области химии за
1987-й год. Интересно, однако, что первый краун-эфир, дибензо-18-краун-6 C.4),
Педерсен синтезировал случайно. Пытаясь осуществить синтез линейного диола
C.3), который, как он надеялся, мог бы выступить в качестве лиганда для
каталитического иона ванадила, Педерсен провел реакцию, показанную на схеме 3.1.
Исходным веществом являлось производное пирокатехина A,2-дигидроксибензола)
C.1), в котором одна из гидроксильных групп для подавления ее химической
активности защищена тетрагидропираном. Педерсен не знал, что его исходное вещество
слегка загрязнено свободным пирокатехином C.2)*. Полученный продукт оказался
смесью ожидаемого соединения C.3) и малого количества дибензо-18-краун-6 с
выходом всего лишь 0.4%. Надо отдать должное искусству Педерсена, сумевшего
выделить и изучить свойства побочного продукта, полученного в столь малом
количестве (см. С. Pedersen, 1967).
Схема 3.1. Синтез первого краун-эфира, дибензо-18-краун-6 (по Pedersen С, 1967)
Педерсен заинтересовался соединением C.4) благодаря его необычному
поведению в растворах и высокой степени кристалличности (это, скорее, указывало на
низкомолекулярную, а не на полимерную природу соединения). Вещество умерен-
* Это не соответствует действительности. Ч. Педерсен знал, что исходное вещество
содержит 10% пирокатехина, и предполагал очистить смесь продуктов реакции от него в конце
процесса. См.: Педерсен Ч.Дж. Открытие краун-эфиров. Крам Д.Дж. Получение
молекулярных комплексов типа «хозяин—гость»: Нобелевские лекции. М.: Знание, 1989. С. 6 (Новое в
жизни, науке, технике. Сер. «Химия», № 1). (Примеч. ред. перевода.)
3.1. Краун-эфиры
123
Рис. 3.1. Пространственная модель комплекса
18-краун-6 C.6) и иона К+
но растворялось в метаноле, но при добавлении солей щелочных металлов его
растворимость значительно возрастала. Вскоре Педерсен синтезировал это вещество с
гораздо большим выходом. Он установил, что вещество растворяет неорганические
соли, например КМпО4, в органических растворителях типа бензола, придавая ему
пурпурную окраску («пурпурный бензол»). Педерсен также обнаружил способность
краун-эфира растворять сами щелочные металлы с образованием синих растворов,
которые теперь известны как алкалидные и электридные соли (разд. 3.12). В конце
концов он пришел к заключению о том, что «...ион калия упал в полость в центре
молекулы» (рис. 3.1 >. Для того времени это было очень смелое, если не
фантастическое, утверждение. Скоро события доказали его правоту. Этот первоначальный
результат быстро привел к синтезу родственных соединений (рис. 3.2), которым
С
о о-
\ ' 15-Краун-5,
C.5) комплементарный Na+
С 3
о
C.8)
о
Дициклогексил-18-краун-6,
конформационно более жесткий
18-Краун-б,
C.6) комплементарный К+
С
■о
о-
C.7)
21-Краун-7,
комплементарный Cs+
C.9)
Дибензо-30-краун-10,
связывает два иона Na+
Рис. 3.2. Некоторые наиболее распространенные краун-эфиры
124
3. Хозяева, связывающие катионы
Педерсен дал название «краун-эфиры» из-за короноподобной формы капсулярно-
го комплекса соединения C.4) и К+.
С первых работ Педерсена краун-эфиры стали очень популярными в химии ка-
тионного комплексообразования. Способность к связыванию и почти бесконечная
синтетическая «плодовитость» таких эфиров привели к получению огромного
количества их производных и аналогов с различной селективностью по отношению к
разнообразным гостям. Сразу же стала очевидной аналогия между способностью
C.4) и ионофорных металлокомплексов, таких, как валиномицин и нонактин, к
связыванию К+ (см. разд. 2.1). Поэтому краун-эфиры начали широко использовать
в исследованиях, моделирующих поведение ионофоров.
L.2 Синтез
В своей оригинальной работе Педерсен описал всего шесть различных
методов получения краун-эфиров. Они до сих пор составляют основу для синтеза
большинства краун-эфиров (схема 3.2). Значительная часть новых краун-эфиров
получена или методом (а), или методом (б). Все методы, показанные на этой схеме,
(а)
ОН
он
CI— R— С\
2NaOH
С1-Т-С1
2NaOH
(е)
Схема 3.2. Методы синтеза краун-эфиров
3.1. Краун-эфиры
125
являются примерами синтеза эфира Вильямсона, хотя именно метод (в) впервые
привел к синтезу дибензо-18-краун-6. В самом деле, дибензо-18-краун-6 можно
получить со значительно большим выходом (80% вместо 45%) по методу (б). К тому
же, непрореагировавшии пирокатехин может доставить проблемы при разделении
продуктов [1 + 1]- и [2+2]-циклоприсоединения по методу (в). Метод (г),
показанный на схеме, Педерсен использовал лишь для получения макроцикла C.10).
C.10)
Из-за недоступности исходного материала и низкого выхода метод (д)
(внутримолекулярная циклизация) не является особенно ценным. Например, 18-краун-6
C.6) может быть получен этим способом с выходом 1.8%, хотя прекрасный выход
был достигнут при циклизации олигоэтиленгликоля [НО(СН2СН2О)лН]
медленным добавлением «-толуолсульфонилхлорида к суспензии гидроксида щелочного
металла, действующей как основание. Вообще говоря, использование тозилата
(я-толуолсульфонат MeC6H4SO2~, Ts~) как остающейся группы вместо хлорида
возрастало с 1971 г. и часто приводило к значительному увеличению выхода.
цис-син-цис-
О
транс-син-транс-
ov>1
цис-анти-цис-
о о4'
о.
транс-анти-транс-
Рис. 3.3. Изомеры
дициклогексил-18-краун-6
О/,.
О
цис-транс-
|||26 3. Хозяева, связывающие катионы
Для получения насыщенных циклогексанов из соответствующих аренов
используется метод (е) каталитического восстановления. В принципе, в случае дибензо-
18-краун-6 это может привести к образованию пяти изомеров дициклогексил-18-
краун-6 C.8), из которых выделены только первые четыре (рис. 3.3). Различная
конфигурация этих соединений (которые не взаимопревращаемы) приводит к
широкому изменению их связывающей способности. Например, в метаноле их
сродство к Na+ варьируется в пределах 1.5 порядков, причем наиболее эффективен цис-
син-цис-изомер.
*"* Parker D.A. Macrocycle synthesis: A practical approach. Oxford: Oxford University
Press, 1996.
Лариат-эфиры и поданды
3-2.1 Поданды
Подандами называют ациклические хозяева с центрами связывания в
виде «подвесок». Простейшие поданды — это ациклические аналоги краун-эфиров;
например, диметиловый эфир пентаэтиленгликоля C.11) подобен 18-краун-6 или
его диольному аналогу C.12).
О О
О О
он но
C.11) C.12)
В результате неблагоприятных энтропийного и энтальпийного эффектов (разд.
3.8) поданды-хозяева обычно проявляют меньшее сродство к катионам, чем их
циклические аналоги, но, подобно краун-эфирам, могут принимать аналогичную
«укутывающую» конформацию в присутствии подходящих катионов металлов,
например катионов лантаноидов (рис. 3.4). Однако предельно высокая гибкость
подандов-хозяев позволяет им участвовать в многоцентровом мостиковом и
спиральном связывании, которое не может быть реализовано в случае краун-эфиров
(рис. 3.5).
Еще в 1972 г. группа Симона (Simon) из Федерального технологического
института в Цюрихе (Швейцария) сообщила о подандах — переносчиках Са2+, которые
ведут себя подобно трансмембранным ионофорам. Действие этих хозяев
(например, C.13)) основано на связывающей способности полярных амидных групп, а
также на том, что гибкие длинные спейсеры эфирных групп способны изгибаться
назад и одновременно координировать катион металла. Алифатические цепи также
3.2. Лариат-эфиры и поданды
127
Рис. 3.4. Кристаллическая структура
подандного Еи(Ш)-комплекса
[Еи(Н2ОKC.12)]3+
С1
С1
С1
\
Hg
'с
Hg
С1
он
но
п\ но
НО о Cd-
Он
Рис. 3.5. Чередующиеся координационные формы простого подандного
тригидроксиэтиленгликоля
4+
вносят большой вклад в липофильность соединения, а следовательно, в его
способность к мембранному переносу.
О
Me.
N ^^ ^ ^ \^ -^ OEt ^\ ,
О
О О-
о
о
:оо сап
OEt
Me
C.14)
C.13)
§128
3. Хозяева, связывающие катионы
Термин «поданд» ввели Ф. Фёгтле и Э. Вебер (F. Vogtle and E. Weber, 1979), чья
ранняя работа была посвящена подандам с концевыми хинолиновыми группами,
например C.14). Такие поданды образуют кристаллические соединения с целым
рядом солей щелочных металлов, а также с переходными металлами и даже уранил-
нитратом иО2(МОзJ-6Н2О. Как часть своей работы по подандам эти
исследователи сформулировали концепцию жесткой концевой группы. Одна из проблем —
высокая гибкость подандов, позволяющая им принимать несвязывающие открытые
конформации. Однако если поданд заканчивается жесткой функциональной
группой (например, арилом, сложным эфиром, амидом), то связывание усиливается за
счет дополнительной степени организации, придаваемой поданду-хозяину
концевой группой. Хороший пример этой концепции — соединение C.15), где
сопряженные фрагменты бензойной кислоты обеспечивают жесткую, плоскую и частично
предорганизованную связывающую щель в сочетании с коротким диэтиленгли-
кольным мостиком. Хозяин с двумя жесткими концевыми хинонмоноиминными
группами C.16) был изучен в качестве возможного ионофора для обнаружения
катионов по изменениям в спектре поглощения в УФ-видимом диапазоне {хромоионофор).
Этот цвиттер-ионный поданд, осуществляющий связывание через два анионных
кислородных донора, вызывает большие спектральные изменения в присутствии
двухвалентных катионов. Такое катион-сенсорное применение обсуждается в гл. 8.
О
C.15)
N+-Me
C.16)
Эти представления можно использовать для триподальных молекул, т.е.
подандов с тремя гибкими цепочками, например для C.17), которые способны к более
полному капсулированию гостей. Здесь опять-таки применима концепция жесткой
концевой группы, поскольку триподальные хозяева часто слишком гибки
вследствие инверсии атомов азота, стоящих в голове мостика.
3.2. Лариат-эфиры и поданды
129
МеО
ОМе
C.17)
*"* Gokel G. and Murillo О. Podands // Comprehensive supramolecular chemistry / Ed.
J.L. Atwood, J.E.D. Davies. D.D. MacNicol, F. Vogtle. Oxford: Pergamon, 1996.
Vol. 1. P. 1-33.
3.2.2 Лариат-эфиры
Термин лариат-эфир относится к краун-эфиру или похожему макро-
циклическому производному с одним или более дополнительных фрагментов,
введенных для увеличения его способности к катионному комплексообразованию
путем создания при связывании подобия трехмерной структуры (ср. C.17)). Эти
Рис. 3.6. Константы
связывания (lg К) при
образовании комплексов К+
с простым подандом, кра-
ун- и лариат-эфирами.
Количественно
А'определяют как константу
равновесия реакции
М+ + Хозяин <=»(М-Хозя-
ин)+ (см. Дополнение 1.1)
9 - СТИД Дж В
О
С Э С
о
18-Краун-6
C.6)
lg К 6.08
о
NH
HN
о
\gK
C.18)
2.04
-О
о
О
О
о
'Ме
Me
Диметиловый эфир
пентаэтиленгликоля (EG5)
C.11)
2.3
ГЛ
о с
Me-N
о
о
C.19)
4.8
Me
13ф i 3. Хозяева, связывающие катионы
соединения можно считать макроциклами краун-типа с подандной боковой цепью.
(Название «лариат-эфиры» происходит от испанского «la reata», что означает
«веревка»; при этом макроцикл рассматривают как петлю лассо, а подандную боковую
цепь - как веревку, за которую держат лассо.) Таким образом, эти соединения
сочетают высокую жесткость и предорганизацию макроциклических соединений с
дополнительной стабильностью и гибкостью подандного комплексообразования,
ведущей к быстрой кинетике катионного связывания. Способности к связыванию К+
простым подандом, краун- и лариат-эфирами сопоставлены на рис. 3.6. Ясно, что
18-краун-6 C.6) — гораздо более эффективный лиганд для К+, чем поданд EG5
C.11) (К+-комплекс первого из них почти на 4 порядка стабильнее). На первый
взгляд кажется, что связывающая способность лариат-эфира C.19) находится где-
то между таковыми циклического и нециклического соединений. Однако сродство
к К+ регулируется также характером донорных атомов. Сравнение азакраун-эфира
C.18) и лариат-эфира C.19) наводит на мысль о том, что именно за счет
трехмерного связывания C.19), рис. 3.7, наиболее эффективен, хотя большая аминная
основность у C.18) также усиливает константу связывания (ср. М,М'-ди-«-бутилдиаза-
18-краун-6, lg K= 3.8). Важным соображением является и число донорных атомов
(восемь вместо шести).
В соединении C.19) атом азота третичной аминогруппы - это место, где
боковая цепь поданда прикреплена к краун-эфирному кольцу (N-прикрепление).
Однако лариат-эфиры можно синтезировать, используя углеродный атом как место
прикрепления (С-прикрепление), подобно C.20). На схеме 3.3 представлены примеры
синтеза соединений обоих типов.
Интересно, что тогда как C.20), o/wio-изомер, можно легко получить с выходом,
превышающим 70%, аналогичный яя/хз-изомер, C.21), два донора боковой цепи
о б
Me—N N \ - Me—N
v_~o О—/ О
Me
М\/~Л
о о
_j ^P* N
Me
•О О
< W ) I
у—-о о—У о
Рис. 3.7. Образование комплекса катиона металла с лариат-эфиром
3.2. Лариат-эфиры и поданды
13Й
(а)
,ОМс Т«С1
но
он
МеО
Пиридин,
0°С, 10 мин
NtjCOj
(б)
Вг
ROH
RO
TsO'
-О Me
= O2SC6H4Me
V-OH ОГ. /° (
ОН
ОН
NaH/DMF
№ С
ОМе
BrNHCOMe
НО(СН]СН^)LН
О
Схема 3.3. Синтез (a) N- и (б) С-прикрепленныхлариат-эфиров (по Nakatsuji Y. et al., 1982)
которого (атомы кислорода) из-за стерических препятствий не принимают участия
в координации катиона металла, образуется только с 30%-м выходом. Это
свидетельствует о важности механизма реакции связывания реагентов с катионом
металла при образовании переходного состояния. Такой эффект называют кинетическим
темплатным эффектом (см. его подробное обсуждение в разд. 3.8.2).
Me
C.20)
C.21) ОМе
Gokel G. W. and Schall O.F. Lariat ethers // Comprehensive supramolecular chemistry /
Ed. J.L. Atwood, J.E.D. Davies, D.D. MacNicol, F. Vogtle. Oxford: Pergamon,
1996. Vol.1. P.97-152.
J32 3. Хозяева, связывающие катионы
3-2-3 Бибрахиальные лариат-эфиры
Можно легко распространить концепцию лариат-эфиров на
получение бибрахиальныхлариат-эфиров (bibracchial lariat ethers, BiBLE) — хозяев, которые
имеют две подандные боковые цепи. Благодаря этому достигается характерный для
трехмерного связывания более полный охват поверхности гостя — иона металла.
В. Гатто и другие (V. Gatto et al., 1986) получили большое количество N-прикреплен-
ных BiBLE-лигандов с помощью удобного, хотя и малоэффективного метода, в
общих чертах показанного на схеме 3.4.
г~\
о о—у
> R
OMe
•О О
(* - место прикрепления)
Схема 3.4. Одновременное образование четырех связей при получении BiBLE-
лигандов
«3* Криптанды
Вскоре после работы Ч. Педерсена, Ж.-М. Лен, тогда молодой
исследователь из Страсбургского университета (Франция), предпринял попытку
сконструировать трехмерные аналоги краун-эфиров. Он предвидел, что ионы металлов
могут быть полностью капсулированы внутри крауноподобного хозяина. Это
должно было приводить к последующему увеличению катионной селективности
хозяина и усилению ионофороподобных транспортных свойств. Учитывая эти факторы и
используя метод высокого разбавления (разд. 3.8.3), Лен синтезировал бицикличе-
ские криптанды (названные так из-за их способности сферически окружать, как бы
«погребать в склепе» ионы металлов, от греч. «kruptos», означающего «скрытый») и
огромное количество родственных соединений, причем большинство из них — с
очень большим выходом (схема 3.5). Первый и наиболее важный представитель
этого ряда - [2.2.2]криптанд C.22), который продается под торговым названием
криптофикс (Kryptofix®). Поскольку этот хозяин имеет тот же размер, что и 18-кра-
ун-6, он также проявляет большую селективность к К+ по сравнению с другими
щелочными металлами. При этом связывание К+ [2.2.2]криптандом в метаноле при-
3.3. Криптанды 133
мерно в 104 раз сильнее, чем его краун-аналогом. Подобным же образом
[2.2.1]криптанд C.23) селективен к Na+. Причина этой, поразительно возросшей
способности криптандов к связыванию катионов металлов по сравнению с краун-
эфирами - трехмерная природа их полости, что дает возможность осуществления
сферического распознавания ионов М+. Это обсуждается далее в разд. 3.7 и 3.8.
о о-
гч г»
NH HN
/ \ Сильноразбавленное
\ / основание
V-0 O-/ выход 45% V_O О
C.18) ХВ2н6
I . / 2.H2O
J Чо_-. выход 95%
N L V JnN
>—о о-У
и0'
n = 2:C.22)
n = l:C.23)
Схема 3.5. Синтез [2.2.1]криптанда (я = 1, C.23)) и [2.2.2]криптанда (п = 2, C.22))
(по Dietrich В. et al., 1969)
Хотя метод высокого разбавления и очень многогранен, он не позволяет
получать большие количества вещества и часто включает много стадий, в частности
конечную стадию восстановительного декарбонилирования дибораном. Со времени
первой работы Лена был разработан целый ряд методов синтеза большого
количества криптандов различной степени сложности, включая хиральные молекулы и
молекулы, имеющие три различных мостика. Многие из методов содержат
следующие стадии:
1. Построение двух линейных цепей, обладающих подходящими реакционными
группами на конце каждой цепи.
2. Реакция циклизации этих двух цепей, приводящая к образованию коранда
(крауноподобного макроцикла).
3. Присоединение к коранду третьей цепи для получения макробициклического
соединения.
Из-за продолжительности и трудоемкости таких синтетических методов были
разработаны другие, гораздо более простые способы, часто имеющие преимущества
темплатного эффекта (разд. 3.8.2) и очень эффективные во многих случаях. Такой
134
3. Хозяева, связывающие катионы
TsO О О OTs
MeCN
гл»
H2N О О NH2
глглгл
TsO О О OTs
MeCN
NH2
NH,
ГЛ
о о—v
( ° ° Л
\—О О—-У
\ Выход:
- / т = л=1;36%
т=1,п = 2;50%
m = 2,n = 2;40%
Выход:
25%
Схема 3.6. Простой синтез некоторых криптандов с указанием выхода
альтернативный синтез криптанда C.22) и родственных соединений показан на
схеме 3.6.
Синтез огромного множества криптандов, некоторые из которых проявляют
необычные свойства при связывании катионов и анионов, стал возможен благодаря
A.J9
C.24)
N ОН N( N F N
C.25)
NH
HN-
C.27)
C.28)
О
о
|Л\ O/i /'""N
0H
N
C.29)
Рис. З.8. Примеры
криптандов
3.3. Криптанды
синтетической разносторонности азакраунов (это краун-эфиры, в которых часть
атомов кислорода заменена на функциональные группы NH); см., например,
C.18). Некоторые примеры таких криптандов показаны на рис. 3.8.
Совершенно новый класс криптандов — сепулькраты — получают в две стадии. На
первой стадии в результате реакции, темплатируемой Со(Ш), образуются трис(хе-
латные) комплексы бидентатных лигандов, содержащих «венчающие» группы.
Первый пример, о котором сообщили Д. Бостон и Н. Роуз (D. Boston and N. Rose) в 1968 г.,
в качестве второй стадии включал реакцию трис(диметилглиоксимато)кобальта(Ш)
с эфиратом трифторида бора, представляющим собой сильную кислоту Льюиса
(схема 3.7). Крайняя инертность иона Со(Ш) (Дополнение 7.2) означает, что крип-
татный комплекс, весьма вероятно, образуется при этом благодаря способности
иона металла удерживать лиганд в ориентации, подобной геометрии конечного
продукта. Работа А. Саргесона (A. Sargeson) из Австралийского национального
университета привела к получению ряда родственных комплексов — производных
ООН
6НСНО
2NH,
Li,CO3
Сепулькрат
C.30)
Саркофагины
R=NO2,NH2.CH2ClHT.fl.
C.31)
i , C.32)
M = Со(НП.
Схема З.7. Сепулькраты и саркофагины
3. Хозяева, связывающие катионы
Рис. 3.9. Кристаллическая
структура Pt(IV)-KOMimeKca
саркофагина C.32) (по Brown К.
etal., 1996)
этилендиамина. Саргесон придумал названия «сепулькраты» для соединений
металла, «увенчанных» атомами азота, например C.30), и «саркофагины» для
аналогичных соединений с углеродными мостиками (саркофагины названы так, потому что
эти металлические комплексы очень стабильны и нереакционноспособны).
Последние работы привели к удивительному регио- и стереоселективному
получению таких саркофагинов Со(Ш) и Pt(IV), как C.32), по методу конденсации
оснований Шиффа. Макроциклические основания Шиффа описываются в разд. 3.10.4.
Кристаллическая структура одного из таких соединений показана на рис. 3.9.
•"* Lehn J.-M. Supramolecular chemistry: Concepts and perspectives. Weinheim: VCH,
1995.
Сферанды
Нобелевскую премию по химии A987 г.) с Ч. Педерсеном и Ж.-М.
Леном разделил третий химик-супрамолекулярщик, Д. Крам (D. Kram), за
исследования в области супрамолекулярной химии макроциклических катионных хозяев
нового класса — сферандов.
Крам осознавал, что в отличие от относительно гибких молекул краун-эфиров и
даже криптандов в растворе жесткая молекула гипотетического хозяина, имеющего
донорные центры, обращенные в сторону центральной связывающей полости еще
до введения в раствор катиона металла, должна проявлять сильное связывание и
превосходную катионную селективность. Используя пространственные
молекулярные модели (называемые моделями Кори—Полинга—Колтуна (Corey-
Pauling— Koltun), или СРК), рис. 3.10, Крам и сотрудники сконструировали жесткие
трехмерные сферанды C.33) и C.34), атомы кислорода в которых, предорганизо-
ванные в октаэдрическом порядке, готовы принять ион металла.
3.4. Сферанды
137
Рис. 3.10. СРК-модели
краун-эфирных и сферанд-
ных комплексов, которые
использовали на ранней
стадии развития супрамоле-
кулярного
конструирования. Современные
вычислительные методы, например
молекулярная механика, до
некоторой степени
заменили упрощенные представления. (Воспроизведено с разрешения
Vogtle F. Supramolecular chemistry. Chichester: J.Wiley & Sons, 1991)
C.33)
C.34)
C.35)
В соединении C.33) три арильных кольца направлены вверх (за пределы
страницы), а три — вниз. Это приводит к почти совершенному октаэдрическому
расположению кислородных атомов анизила, тогда как липофильные л-метильные и ме-
тильные группы анизилов обращены в сторону растворителя. Этот хозяин
селективно связывает в своей полости маленькие катионы, например Li+ и, в
меньшей степени, Na+ . Действительно, сферанд C.33) - один из сильнейших комллек-
138
3. Хозяева, связывающие катионы
сообразователей, известных для Li+. Все другие катионы исключаются, потому что
они просто слишком велики для того, чтобы соответствовать связывающей
полости. Такая полость у сферанда C.34) имеет тот же размер и образуется за счет
связывания колец в пары диэтиленгликольными мостиками, что в результате дает
четыре кольца внизу и два вверху. Наряду с октамерным сферандом, также было
синтезировано аналогичное фторсоединение C.35). Хотя РСА и подтверждает, что
этот хозяин имеет полость, очень схожую с полостью C.33), фторсодержащие сфе-
ранды не проявляют способность к связыванию ионов металлов. Очевидно, что
сродство фторгрупп к катионам щелочных металлов настолько мало, что даже
многоцентровое связывание с участием высокоорганизованного хозяина не может
обеспечить заметного комплексообразования. Однако интересно то, что для крип-
танда C.25) наблюдается вполне определенное взаимодействие катиона-гостя с
фторгруппой как в твердом состоянии (РСА), так и в растворе A9F ЯМР-спектро-
скопия).
Для достижения конечной стадии циклизации в синтезе сферандов был
разработан новый синтетический метод замыкания кольца, осуществляемый в
соответствии с уравнением C.1); асас - ацетилацетонато СН(СОМе)^". Арильное
соединение лития, полученное действием бутиллития на арилбромид, окисляется
комплексом Fe(III) с образованием арильного бирадикала, который затем
подвергается темплатной циклизации вокруг иона Li+ (темплатный эффект объясняется в
разд. 3.8.2). В случае сферанда C.33) метод приводит к выделению этого комплекса
с выходом 28%, согласно реакции, показанной на схеме 3.8.
Аг-Вг
BuLi
Ar-Li
Fe(acac),
Аг—At.
C.1)
l.BuLi
2. Fe(acac)j
3. EDTA
4.HCI
(acac)H =
(EDTA)H4 * HO2CH2C—N N—CH2COjH
CH2COjH
C.33).Li*
Схема З.8. Синтез сферанда C.33) в виде его и+-комплекса
3.4. Сферанды
139
Для того чтобы определить важность жесткой предорганизации циклического
хозяина, были получены ациклические подандные хозяева, аналогичные
соединению C.33). Сравнение близких родственников - поданда C.36) (обладающего
более чем 10 000 возможных конформаций, из которых только две могут связывать
катионы по сходящемуся типу) и сферанда C.33) (имеющего только одну замкнутую
конформацию) - показывает, что сферанд связывает Li+ более чем в 1012 раз
эффективнее. Это подчеркивает важность статистических (энтропийных) эффектов в
конструировании хозяина.
.Me
Me
.Me
ололсгпгсош
Me
Me
Me
Me
C.36)
Сочетание краун-эфиров (или, вообще, корандов), криптандов, сферандов и по-
дандов привело к появлению большого числа гибридных хозяев, например крипта-
сферандов и полусферандов, многие из которых проявляют все полезные черты
соединений-родителей (рис. 3.11).
Полусферанд
C.37)
Криптасферанд
C.38)
Рис. 3.11. Гибридные хозяева для
катионов-гостей
Полусферанд
C.39)
140
3. Хозяева, связывающие катионы
Maverick E. and Cram D.J. Spherands: Hosts preorganised for binding cations //
Comprehensive supramolecular chemistry / Ed. J.L. Atwood, J.E.D. Davies,
D.D. MacNicol, F. Vogtle. Oxford: Pergamon, 1996. Vol.1. P.213-243.
Номенклатура
Наименования краун-эфиров относительно просты, хотя, по
историческим причинам, они недвусмысленны только тогда, когда имеется в виду
использование исключительно этиленгликольных (—ОСН2СН2О—) звеньев; в более
сложных случаях название обычно сопровождают изображением.
1. Первое число в названии краун-эфира означает число атомов в кольце (в
англоязычной литературе обычно дается в квадратных скобках).
2. Второе число показывает число атомов кислорода (или другого донора).
3. Заместители обозначены префиксами: бензо-, дициклогексано- и т.д.
Например, дибензо-18-краун-6 C.4) - это краун-эфир с 18-членным макроцикли-
ческим кольцом, содержащий шесть атомов кислорода и два бензольных
заместителя. Более обобщенная номенклатура для таких нейтральных органических лиган-
дов была разработана Фёгтле и Вебером (F. Vogtle and E. Weber, 1979), а позже
модифицирована Крамом A986). По этой номенклатуре любую моноциклическую
систему, в частности краун-эфир, называют корандом (первоначально - коронан-
дом). Молекулы с разомкнутой цепью, например C.3) и C.11), именуют подандами;
би- или олигоциклические системы называют криптандами (рис. 3.12); жестким
системам на основе я-метиланизола дано имя сферанды. В целом, для чисто
кислородных донорных лигандов исторически сложившаяся номенклатура краун-соедине-
ний сохранена.
Открытая цепь
Циклический лиганд
Поданд Коранд
D - донорный атом D = О, краун-эфир
Рис. 3.12. Классы ациклических и циклических лигандов
Криптанд
В - атом в голове мостика
3.5. Номенклатура
СИ КО
Me Me
C.11)
C.42)
NH О
О HN
N
NH 0-/
C.43)
C.22)
Рис. 3.13. Поданды C.11), C.40), коранды C.41)—C.43) и криптанд C.22), родственные
18-краун-6. Соединения C.41) и C.42) — члены гибридного корандно-подандного
семейства, называемого лариат-эфирами. Лариат-эфиры с двумя подандными боковыми цепями
C.42) называют BiBLE (бибрахиальными лариат-эфирами)
На рис. 3.13 показаны примеры подандов, корандов и криптандов. Согласно
этой системе, все краун-эфиры принадлежат к общему классу корандов. Название
«18-краун-6» C.6) превращается в название «18(О626коранд-6)>>; это означает
18-членное моноциклическое соединение (т.е. коранд), содержащее шесть донор-
ных атомов кислорода, соединенных шестью спейсерами, каждый из которых
имеет два атома углерода. Иногда приводят сокращенное название <<[18]ан-О6».
Точно так же азакоранд C.43) называют 18(О3^26коранд-6). Аналогична и
номенклатура, общепринятая в литературе особенно для корандов, содержащих
многочисленные донорные атомы азота или серы. В соответствии с ней коранд
рассматривают как большой замещенный циклоалкан; отсюда и употребление суффикса
«-ан» наряду с числами, означающими размер кольца и количество донорных
атомов. Таким образом, 18(М626коранд-6) C.44) сокращенно называют «[18]aH-N6» a
его серный гомолог — <<[18]aH-S6». В литературе также приняты другие названия,
часто описательные. Соединение C.44) называют и гексацикленом (из-за его
сходства с этилендиамином, часто сокращенно обозначаемым «en»). Соединение C.41) -
член семейства лариат-эфиров (лассо-эфиров), у которых кольцо коранда (крауна)
представляет собой петлю лассо, а подандная боковая цепь — веревку. Азакоранд-
ные аналоги краун-эфиров, например C.45), у которых эфирные атомы кислорода
полностью заменены NH-группами, получали и изучали отдельно как лиганды для
переходных металлов, а потому они часто имеют вненоменклатурные
(тривиальные) названия. Так, коранд C.45) называют цикламом. Его могут также называть
142 3. Хозяева, связывающие катионы
«тетрааза-14-краун-4», или, более систематично, «14(М42232-коранд-4)».
Тривиальные названия, например сепулькраты и саркофагины, C.30)—C.32), присвоены
этим соединениям по аналогии с криптандами. (Путь проникновения в литературу
новых наименований до некоторой степени случаен. Взрывоопасный макроцикл
Se4N4 был назван фетодигитогеником; термин, придуманный Вулинсом и другими
(Woolins et al.) из Королевского колледжа (Лондон, Великобритания), по существу,
подразумевает, что это соединение при взрыве отрывает пальцы !).
N
NH
й ' '
H HN NH NH
NH HN. ^NH NH
^NH „ ЮГ HN HN
N
C.44) \^ C.45)
Для криптандов, имеющих, скорее, сферическую, чем крауноподобную
структуру, используют несколько другую номенклатуру. При этом в названии каждого
хозяина указывают число донорных атомов в каждом из мостиков между атомами в
головах этих мостиков. Таким образом, соединения C.22) и C.23) называют
соответственно <<[2.2.2]криптанд» и <<[2.2.1]криптанд». Было синтезировано большое
количество криптандов с мостиками, которые начинаются у атомов углерода или
атомов азота и содержат атомы серы, азота или другие донорные атомы и фрагменты
(например, криптанд типа «футбольный мяч» C.29)). Номенклатура этих
соединений часто очень привлекательна, но не строга.
Все эти лиганды были синтезированы для связывания гостей — катионов
металлов, а поэтому номенклатуры для свободных (не содержащих металл) лигандов и их
металлокомплексов должны различаться. Этого достигают, используя окончание
«-ан&» при обозначении свободного лиганда и окончание «-am» при обозначении
его металлокомплекса. Таким образом, металлокомплексы подандов называют
податами, комплексы корандов — коратами, а криптандов — криптатами. В
названия этих комплексов включают математический символ «с», означающий
принадлежность к системе. Так, коранд 18(О626-коранд-6) C.6) образует корат
[К+ с 18(О626коранд-6)], показанный на рис. 3.1.
Однако эта общепринятая номенклатура вводит в заблуждение, поскольку
суффикс «-am» обычно используют и для анионов (ср. ацетат, сульфат), тогда как
многие комплексы хозяин—гость относят к катионам. Было предложено заменить
«-am» на суффикс «-плеко, что дает кораплекс, сфераплекс и т.д. (Cram D., 1986). Во
избежание путаницы, насколько это возможно, в этой книге будут использоваться
наиболее общепринятые термины. Несмотря на большое разнообразие
используемых терминов, в большинстве случаев они очень наглядны и достаточно понятны.
3.6. Поведение растворов 143
Поведение растворов
.6.1 Растворимость
Краун-эфиры, например 18-краун-6, примечательны тем, что в
отсутствие гостей они растворимы в разнообразных растворителях — от воды до алканов.
Действительно, 18-краун-6 имеет нулевое значение на шкале липофильности,
основанной на распределении растворяемых веществ между октаном и водой. Это
указывает на превосходную сбалансированность гидрофильности и
липофильности молекулы краун-эфира. Сравнение кристаллических структур свободного
хозяина и его комплекса с К+ (рис. 3.14) позволяет нам понять причину такого
поведения. В К+-комплексе все атомы кислорода направлены внутрь (вспомните
определение хозяина как химического соединения со сходящимися центрами
связывания в комплексе), что приводит к полной гидрофобности его внешней
поверхности, как и в случае мембранного транспорта с участием ионофоров, например ва-
линомицина (разд. 2.1). Однако в свободном лиганде два кислородных атома
направлены наружу, что придает внешней поверхности большую полярность. В
действительности такие коранды, как 18-краун-6, крайне гибки в растворе.
Поэтому они имеют либо полностью гидрофильную внешнюю поверхность (и таким
образом маскируют свой липофильный этиленовый каркас), либо полностью
гидрофобную внешнюю поверхность с неподеленными электронными парами
кислорода, направленными внутрь (и таким образом предорганизуются (разд. 3.9)
для катионного комплексообразования), рис. 3.15. Криптанды проявляют такую же
гибкость. Сферанды, однако, гораздо более жестки.
Конформационный критерий связывания катиона металла большинством
хозяев, основанных на этиленгликоле (—ОСН2СН2О—), можно вывести на основании
величин торсионных углов О-СН2-СН2-О (рис. 3.16). Для того чтобы связать
гостя, все углы должны быть гош-торсионными (т.е быть меньше 90°). Обычно они по-
Рис. 3.14. Сравнение кристаллических структур (а) свободного дициклогексил-
18-краун-6 и (б) калиевого комплекса его близкого «родственника» — 18-краун-6
144
3. Хозяева, связывающие катионы
(а)
н н н
н н
Рис. 3.15. Поведение 18-краун-6 (а) в органическом растворителе, например дихлорметане
(краун напоминает каплю воды в масле), и (б) в гидрофильной среде (капля масла в воде)
О
н
гош-
о
и-
н2
У
н2
с
о-
Рис. 3.16. Торсионные углы в краун-
эфирных комплексах
падают в интервал 65—75°, близкий к оптимальной расчетной величине для 1,2-ди-
метоксиэтана - 72°. Однако в краун-эфирах, неспособных к комплексообразова-
нию, фиксируют, по крайней мере, два аняш-торсионных угла.
\ Применение растворов
Растворимость краун-эфиров и криптандов важна в двух случаях: в
межфазном катализе и активации анионов. Межфазный катализ включает перенос
частиц гостя из одной фазы в другую. Две рассматриваемые фазы — это обычно две
несмешивающиеся жидкости (межфазный перенос жидкость—жидкость). На
практике это означает использование подходящего хозяина для увеличения
растворимости неорганических солей в неполярной среде. Таким образом, супрамолекуляр-
3.6. Поведение растворов
Й45
ные частицы хозяина катализируют реакцию между гидрофильной неорганической
солью и липофильным органическим субстратом, допуская гомогенное смешение
реагентов в органической фазе.
Большие липофильные комплексы катион металла - макроцикл,
легкорастворимые в неполярных растворителях, например в бензоле, толуоле и галогеналканах
образуются при связывании макроциклическими полиэфирами таких
неорганических катионов, как ионы щелочных и щелочноземельных металлов. Для
сохранения баланса заряда катионный комплекс принимает ассоциированный противо-
анион. В такой несмешивающейся двухфазной системе, как смесь хлороформа и
воды, анион неизбежно втягивается в органическую фазу при пересечении фазовой
границы катионным комплексом. Этот принцип можно проиллюстрировать
добавлением раствора 18-краун-6 в хлороформе к водному раствору пикрата калия B,4,6-
тринитрофенолят калия). После перемешивания прилегающая (находящаяся в
физическом контакте) фаза хлороформа приобретает желтый цвет пикрат-аниона
(рис. 3.17).
Находясь в органической фазе, реакционноспособные анионы будут, скорее,
участвовать в гомогенной химической реакции с веществами в органической фазе,
а не на границе фаз или поверхности раздела. Это приводит к огромному
увеличению скорости реакции и несоответствию между прекрасным (количественным)
выходом и отсутствием какой-либо заметной реакции вообще. Поскольку требуется
Рис. 3.17.
Межфазный катализ
с помощью краун-
эфира
Водная
фаза
18-Краун-б
Фаза
СНС13
i4g 3. Хозяева, связывающие катионы
лишь малое количество краун-эфира или криптанда, это истинно каталитический
процесс. Как только произошла реакция, краун может вернуться к поверхности
раздела органическое вещество - вода, чтобы захватить следующий реагент и
отдать неорганические побочные продукты. Таким образом, общая равновесная
концентрация неорганической соли в органической фазе не обязательно должна быть
высокой, но тот факт, что анион (например, МпО^) постоянно участвует в реакции,
вызывает промотируемую крауном диффузию свежей порции соли через
поверхность раздела в соответствии с концентрационным градиентом (концентрация
неорганической соли в органическом слое всегда ниже, чем в водной фазе).
Другим интересным аспектом реакционного катализа макроциклическими ка-
тионными хозяевами является эффект «обнаженного» аниона. В обычной анионной
реакции, например в реакции нуклеофильного замещения, анионный нуклеофил
теряет активность за счет координации или ионного спаривания с катионом (его
противоион). Таким образом, энергия активации реакции возрастает на величину
энергии, необходимой для разрыва анион-катионной связи. Сильное физическое
разделение аниона и катиона происходит из-за того, что катион изолирован внутри
липофильной оболочки в присутствии краун-эфира или, в особенности,
криптанда. В результате анион почти не взаимодействует с катионом. Этот эффект
приводит к снижению энергии активации реакции, поскольку в этом случае нет
необходимости учитывать вклад анион-катионного взаимодействия.
В принципе, макроциклические комплексообразующие агенты могут быть
использованы для усиления любой реакции, в которой участвуют ионы, ионные ин-
термедиаты или сильнополярные соединения. Важные примеры таких реакций:
• нуклеофильное замещение;
• реакции с карбанионами;
• образование С-С-связи (например, конденсация Дарзана или Кнёвенагеля);
• присоединение;
• элиминирование;
• генерация карбена;
• восстановление (особенно с электридными солями, разд. 3.12);
• перегруппировки (Фаворского, Коупа).
Приведенные далее реакции показывают, как можно использовать краун-эфиры и
криптанды в межфазном катализе и при анионной активации.
3-6.2.1 Окисление органических субстратов
под действием КМпО4
Добавление липофильных краун-эфиров к неорганическим солям,
например к перманганату калия, дихромату калия и другим, обычно нерастворимым
в неполярных растворителях, позволяет им благополучно окислять органические
субстраты с количественным выходом (схема 3.9). В отсутствие макроциклов выход
крайне низок.
3.6. Поведение растворов
о
КМлО4
дициклогексил-18-краун-6
, ».
Бензол
КМпО4
ОН 18-краун-6
сн2а2
Схема 3.9. Перенос обычно нерастворимого иона МпО^" в органический
растворитель с помощью комплексов (К+ с краун-эфир), приводящий
к количественному окислению органических субстратов
Реакция солей калия с бензилхлоридом в ацетонитриле
Обычная реакция анионов А~ солей калия с бензильными
производными показана на схеме 3.10.
Схема 3.10. Реакция солей калия с бензилхлоридом в ацетонитриле (X = С1;
А — анион, например ацетат)
Из табл. 3.1 видно, что скорость этого простого нуклеофильного замещения
резко увеличивается в результате участия краун-эфира, который способствует
отделению катиона К+ от сравнительно сильно координирующего его ацетат-аниона и
таким образом обеспечивает аниону возможность выступать в качестве нуклеофила.
Таблица 3.2 показывает, что в протолитических средах , где связывание краун-эфи-
рами проявляется слабее, наблюдается большая разница в скоростях реакций
различных анионов в соответствии с их способностью к комплексообразованию К+.
Так, мягкий 1~ вдобавок к своей малой растворимости слабо связан с К+ и
реагирует быстро. В то же время фтор образует с К+ прочную пару, хорошо растворим и
реагирует медленно. В органических растворителях ион К+ гораздо сильнее
связывается краун-эфиром и в результате все анионы реагируют с почти одинаковыми
скоростями. В самом деле меньший, более подвижный F~ со своей высокой
плотностью отрицательного заряда более нуклеофилен, чем 1~.
148
3. Хозяева, связывающие катионы
Таблица 3.1. Зависимость периода полупревращения
от наличия краун-эфира
(А = СН3СО2-, X = С1)
Краун-эфир
Отсутствует
18-Краун-6
Дибензо-18-краун-6
Дициклогексил-18-краун-6
Период полупревращения, ч
685
3.5
9.5
1.5
Таблица 3.2. Нуклеофильность различных анионов (А )
в присутствии 18-краун-6
(X — л-толуолсульфонат)
Нуклеофил
N3-
CH3COj
CN"
Вг-
С1-
I-
F-
SCN"
Относительная
скорость
' 10.0
9.6
2.4
1.3
1.3
1.0
1.4
0.3
Относительная скорость
в протолитической среде
100
5
1250
80
10
1000
1
625
Образование «обнаженного» карбидного лиганда
Индивидуальные атомы углерода - весьма необычные лиганды для
переходных металлов. Синтез молибденового комплекса, содержащего один анион-
радикал С'~, который не образует комплексы ни с одним из других металлов или
со
Mo
R2N
NR2
l.Na/Hg(THF)
2. t-BuC(O)Cl
т
3. Na(THF)
NR2
R2N,
Mo
NR2
'--NR2
"'NR2
KCHjPh
C"
A
V
Бензо-15-краун-5
или [2.2.2] криптанд
R2N
R2N
NR2
NR2
Схема 3.11. Образование «обнаженного» карбидного комплекса путем связывания
К+ бензо- 15-краун-5 или [2.2.2]криптандом
3.7. Селективность катионного комплексообразования
Рис. 3.18.
Кристаллическая структура
комплекса карбида
молибдена.
Координирующий ион К+
связан с помощью
бензо-15-краун-5
функциональных групп, был проведен путем удаления противоионов К+ с
помощью бензо-15-краун-5 или [2.2.2]криптанда по схеме 3.11 (J. Peters et al., 1997).
Кристаллическая структура продукта ясно указывает на связывание катиона К+
(рис. 3.18).
Селективность
катионного комплексообразования
Общие положения
Термодинамическая селективность данного хозяина к отдельному
катиону представляет собой соотношение между сродством хозяина к данному
металлу (например, к К+) и его сродством к катионам другого гостя. Основой
молекулярного распознавания является сильное, но селективное связывание. Так,
«успешный» хозяин проявляет сильное сродство именно к отдельно взятому гостю
и гораздо меньшее сродство (измеренное на основании констант связывания, см.
Дополнение 1.1) к другим катионам. Конструирование синтетического хозяина,
высокоселективного к данному катиону, - очень сложная задача, из-за того что
селективность определяется огромным количеством факторов. Самые важные из этих
факторов:
• соответствие между размерами катиона и полости хозяина;
• электростатический заряд;
• растворитель (полярность, водородное связывание и способность к
координации);
• степень предорганизации хозяина;
• энтальпийный и энтропийный вклады во взаимодействие катион—хозяин;
• свободные энергии сольватации катиона и хозяина;
• природа противоаниона и его взаимодействия с растворителем и катионом;
• кинетика катионного связывания;
• размер хелатного кольца.
J5Q 3. Хозяева, связывающие катионы
Среди перечисленных факторов важным и легкоучитываемым, но не
обязательно главным фактором является размерное соответствие (ср. селективность валино-
мицина по отношению к К+, см. разд. 2.1). Наряду с менее значимыми факторами,
например с размером хелатного кольца (R. Hancock, 1992), сольватация и предорга-
низация также играют важную роль. Перед тем как подробно обсуждать эти
эффекты, необходимо определить меру сродства отдельного хозяина к каждому катионно-
му гостю, чтобы иметь возможность количественно сравнивать связывание гостя
тем или иным хозяином. Термодинамические факторы, определяющие
способность к катионному комплексообразованию, обычно входят в константу
связывания (А), которая, по существу, представляет собой константу равновесия реакции
комплексообразования. Таким образом, селективность хозяина может быть
выражена в виде отношения одной константы связывания к другой. Способы
определения констант связывания, наряду с некоторыми наиболее общепринятыми
способами их измерения, приведены в Дополнении 1.1. Заметьте, что существует важное
различие между термодинамической стабильностью комплекса (статическая
стабильность комплекса) и кинетикой его поведения в процессе реакций
образования и диссоциации комплекса (динамическая стабильность комплекса), разд. 3.9.2.
3-7-2 Краун-эфиры
Изменение констант связывания данного макроцикла с катионами
I группы для крайне гибких макроциклов типа краун-эфиров часто представляет
собой плато селективности. Это означает, что константы связывания не проявляют
тенденцию к плавному увеличению или уменьшению в ряду таких гостей, как
катионы щелочных металлов, а имеют близкие значения для ионов нескольких
металлов. Например, константы связывания для малых катионов металлов могут быть
одними и теми же и могут уменьшаться по мере роста радиуса катиона, что
приводит к слабому различию между Na+ и К+. И наоборот, константы связывания могут
увеличиваться с ростом радиуса катиона, давая плато для Rb+ и Cs+. Это может быть
связано с двухмерной природой комплексообразования, что позволяет краун-ли-
гандам приспосабливаться к большому разнообразию катионов металлов
независимо (до некоторой степени) от их ионного радиуса. Итак, насколько важно
соответствие радиуса катиона радиусу полости хозяина в столь простых лигандах, как
краун-эфиры? На рис. 3.19 и 3.20 показано изменение константы связывания в
зависимости от радиуса катиона для краун-эфиров с различным размером кольца. В
табл. 3.3 сопоставлены диаметры катионов металлов и размеры полостей краун-
эфиров, найденные с помощью молекулярных моделей СРК. Для сравнения
приведены диаметры катионов других металлов.
Плато селективности для больших краун-эфиров, возникающее благодаря их
крайне высокой гибкости, показано на рис. 3.20. Ясно также, что К+ гораздо
сильнее других катионов связан с 18-членными макроциклами; это отвечает
представлениям о соответствии размеров катиона и полости. Ряд других особенностей
зависимостей рис. 3.19 и 3.20 не так легко объяснить. Действительно, 18-краун-6 и его
производные сильнее других хозяев связывают все катионы. Более того, из всех
катионов металлов именно К+ связан наиболее сильно. В пределах этого ограничен-
3.7. Селективность катионного комплексообразования
151
Рис. 3.19. Изменение константы
связывания в зависимости от радиуса
катиона для циклогексильных краун-эфиров
1.0
Na+ K+ Cs+
Катион
ного набора данных может показаться, что вместо принципа размерной
селективности мы сталкиваемся с гораздо более общими тенденциями:
1. Все краун-эфиры селективны к К+.
2. 18-Краун-6 и его производные селективны ко всем катионам!
б.о .
5.0 -
4.0
3.0 •
2.0
1.0 ..
0.0
12 15 18 21 24
Размер кольца (число атомов)
27
Рис. 3.20. Изменение константы связывания в зависимости от размера кольца краун-
эфира для разных катионов
2 3. Хозяева, связывающие катионы
Таблица 3.3. Сравнение диаметров полостей различных краун-эфиров
с диаметрами катионов металлов
Катион
Диаметр, А Краун-эфир Диаметр полости, А
Li+
Na+
К+
Cs+
Cu+
Ag+
Mg2+
Ca2+
La3+
Lu3+
Zi4*
1.36
1.90
2.66
3.38
1.92
2.52
1.44
2.20
2.34
2.00
1.72
12-Краун-4
15-Краун-5
18-Краун-6
21-Краун-7
-
-
-
-
—
—
-
1.20-1.50
1.70-2.20
2.60-3.20
3.40-4.30
-
-
-
-
-
—
-
Если бы это было верно для всех супрамолекулярных систем, то перспективы
управляемой селективности были бы, действительно, мрачными. Для объяснения
этих явлений мы должны посмотреть на взаимодействие всех факторов,
перечисленных в начале разд. 3.7. Наиболее важные из них приведены ниже.
• Соответствие размеров полости хозяина и гостя-катиона. Действительно,
гибкий 18-краун-6 имеет достаточно хорошее размерное соответствие со всеми ка-
тионными гостями, хотя и оптимален для К+.
• Число донорных атомов. Как правило, супрамолекулярные взаимодействия
аддитивны; следовательно, можно ожидать, что связывание больших краун-эфиров с
катионами металлов будет наиболее сильным до тех пор, пока все донорные
атомы не выстроятся вокруг металла. Это вносит вклад в плато селективности,
которое мы видим для большинства катионов в правой части рис. 3.20.
• Сольватация катионов и лигандов. Свободная энергия сольватации возрастает в
следующем ряду: K+<Na+<Ca+. Поэтому, для того чтобы десольватировать
(частично) К+ , что необходимо для его связывания, требуется наименьшая энергия.
• Размер хелатного кольца (угол захвата лиганда). В комплексных соединениях
хозяев, содержащих этенильные (двууглеродные) мостики, формируются пяти-
членные хелатные кольца (рис. 3.21). Малый угол захвата этилендиоксидной
группы подходит для катиона большего размера, например для К+. Ионы
металлов меньшего размера, особенно Li+, не так хорошо приспособлены для
обеспечения контакта с соседними донорными атомами и поэтому не образуют
столь прочные комплексы. Это подтверждается большим сродством дицикло-
гексил-14-краун-4 к Na+, рис. 3.19 (ср. 12-краун-4, рис. 3.20).
Заряд катиона - другой важный фактор, не отраженный на рис. 3.20. Ясно, что Са2+
лишь слабо связывается с большим числом краун-эфиров. Однако это в основном
определяется его малым размером и большой энергией гидратации. Гораздо боль-
3.7. Селективность катионного комплексообразования
153
(а) Пятичленное хелатное
кольцо, угол захвата ~ 60°
(б) Шестичленное хелатное
кольцо, угол захвата -72°
Рис. 3.21. Углы захвата лиганда
для краун-эфиров (а) с двумя и
(б) с тремя атомами углерода
между эфирными атомами
кислорода
ший Ва2+ при комплексообразовании с 18-краун-6 в метаноле имеет значение
lg Ku, равное 7.0 (ср. с 6.10 для К+). Сравнение констант связывания одно- и
двухвалентных катионов с ^мс-сми-г/мс-дициклогексил-18-краун-6, показанное на
рис. 3.22, позволяет считать, что в отсутствие больших гидратационных эффектов и
при хорошем соответствии размеров катионов и полостей определяющим
фактором является формальный положительный заряд катиона.
Большие краун-эфиры могут избирательно связывать Rb+ и Cs+. Краун-эфиры
на основе трибензо-21-краун-7, судя по коэффициентам распределения нитратов
щелочных металлов между водой и хлороформом, слабоселективны по отношению
к Rb+ (рис. 3.23). Такая селективность к Rb+ объясняется конформационными
особенностями лиганда. Исследование длин связи М—О для комплексов Na+, Rb+ и
Cs+ показывает, что в случае Rb+ гептадентатная конформация довольно легко
достигается в узком диапазоне расстояний Rb-0 B.98-3.09 А), тогда как в случае Na+
требуется более широкий диапазон расстояний B.43-2.63 А), что предполагает
появление стерического напряжения при связывании большого краун-эфира и
маленького катиона.
Удивительно, но по мере продвижения к очень большим краун-эфирам,
например к дибензо-24-краун-8 C.46) и дибензо-30-краун-10 C.47), вновь наблюдается
РЬ2+
Рис. 3.22. Зависимость константы
связывания от соотношения «диаметр
катиона/размер полости» для
комплексов цис-син-цис-тшклогексил-18-кра-
ун-6 с катионами одно- и
двухвалентных металлов (вода, 25 °С)
0.5 1 1.5
Диаметр катиона / размер полости
154
3. Хозяева, связывающие катионы
I
0.1 -|
0.2-
о.зЧ
0.4-
0.5
Na+ K+ Rb+ Cs+
Катион
Рис. 3.23. Конкурентная
экстракция нитратов щелочных
металлов с помощью
трибензо-21-краун-7
Рис. 3.24. Структура комплекса
[К+ с 30-краун-10], демонстрирующая
способность макроцикла принимать
«укутывающую» конформацию при капсулировании
катиона металла
высокая селективность по отношению к К+, которая, действительно, возрастает с
увеличением размера макроцикла, lg Ku (метанол, 25 °С) для дибензо-24-краун-8
равен 3.49, а для дибензо-30-краун-10 - 4.60. Это объясняется способностью
больших гибких краун-эфиров «укутывать» катион металла, заключая его почти
целиком внутрь органической оболочки (ср. валиномицин, разд. 2.1). Кристаллическая
структура [К+ с 30-краун-10] показана на рис. 3.24. Действительно, даже огромные
дибензо-60-краун-20 способны эффективно связывать К+ с константой (lg A',,)
3.90. Для таких небольших катионов, как Na+, константа связывания с дибензо-30-
краун-10 гораздо меньше (lg Ku = 2.0): макроцикл просто слишком велик для
эффективного капсулирования Na+. Интересно, однако, что образуется также и 2:1-
комплекс, в котором два иона Na+ «погружены» глубоко в полость, подобно тому,
как это имеет место в 1:1-комплексах с меньшими краун-эфирами.
C.46)
C.47)
3.7. Селективность катионного комплексообразования 155
3-7*3 Лариат-эфиры
Лариат-эфиры состоят из кольца краун-эфирного типа с подандной
боковой цепью, предназначенной для придания некоторой трехмерности катион-
ному комплексообразованию. При этом сохраняется высокая скорость
комплексообразования, ожидаемая для крайне гибких корандных систем (см. разд. 3.2.2).
Таким образом они сочетают свойства подандов, корандов и криптандов. Константы
связывания лариат-эфиров с N-прикрепленной полиэфирной боковой цепью для
хозяев с различной длиной боковой цепи приведены в табл. 3.4. N-Прикрепленные
макроциклы способны более точно направить свою боковую цепь к гостю, и,
поскольку атом, к которому прикреплена боковая цепь, смотрит в сторону катиона
металла, связывание в этих системах более сильное, чем в их С-прикрепленных
аналогах. В табл. 3.4 показана эффективность лариат-эфира (связывание и боковой
цепью, и кольцом ) во всех случаях по сравнению со случаями, где R = Н. Это
особенно поразительно для BiBLE-лигандов типа C.51), где даже в случае
моноэфирных цепей (R = СН2СН2ОМе) константа связывания для К+ в 104 раз выше, чем для
корандного аналога. Удлинение боковой цепи приводит к очень незначительному
усилению связывания Na+ соединениями с малым кольцом типа C.48). Для
производных 15-краун-5, имеющих до двух донорных атомов в боковой цепи, это также
справедливо, но в таком случае сродство постоянно, lg Кх, = 4.2, для боковых цепей
с числом донорных атомов до пяти. Эта тенденция может также наблюдаться в
случае К+-комплексов типа C.49) и C.50), где связывание К+ достигает максимума
при lg Ku ~ 6 для восьми донорных атомов (коранд + боковая цепь), причем
делается вывод о том, что дальние донорные атомы являются «неэффективными»
донорами, т.е. они не способны стерически координировать катионы металла. Как и в
случае металлов, катион аммония тоже может соединяться с лигандами типа краун-
эфиров, причем связывание происходит через водородные связи N—Н+--О и
N-H+-N (разд. 3.11). В случае лариат-эфиров результаты по связыванию NH^
поразительны, поскольку оказалось, что они практически одинаковы для всех
рецепторов типа C.48) и C.49), но почти на порядок возрастают для хозяев типа C.50).
Такой факт можно объяснить с помощью молекулярных моделей,
предсказывающих связывание NH^ по триподальному способу водородного связывания трех
атомов водорода с двумя донорными атомами корандного кольца и одним атомом
боковой цепи. Это единственная стерическая возможность для четвертого атома
водорода группы NH4 образовать водородную связь в производных 18-краун-6 и,
таким образом, усилить связывание (рис. 3.25).
Рис. 3.25. Стерические
ограничения на водородное связывание
катиона NH| лариат-эфирами
Стерическое несоответствие
156
3. Хозяева, связывающие катионы
Таблица 3.4. Константы связывания (Ig Ku)
лариат-эфиров в метаноле при 25 °С
R в хозяине
■8*11
Na+
К+
NH+
Са2+
R—N
C.48)
СН2СН2ОМе
(СН2СН2ОJМе
(СН2СН2ОKМе
(СН2СН2ОLМе
(СН2СН2ОMМе
3.25
3.60
3.64
3.76
3.73
2.73
-
3.85
-
4.34
3
3
3
.06
-
.29
-
.49
R—N
О
C.49)
н
СН2СН2ОМе
(СН2СН2ОJМе
(СН2СН2ОKМе
(СН2СН2ОLМе
(СН2СН2ОMМе
1.70
3.88
4.54
4.32
4.15
4.19
1.60
3.95
4.68
4.91
5.28
4.91
гл
/—о
о—л
2.99
3.14
3.19
3.38
3.48
3.49
—
3.75
4.06
3.84
3.78
3.80
R—N
н
СН2СН2ОМе
(СН2СН2ОJМе
(СН2СН2ОKМе
(СН2СН2ОLМе
(СН2СН2ОMМе
>—о о—t
V_7
2.9
4.58
4.33
4.28
4.27
4.22
3.98
5.67
6.07
5.82
5.86
-
C.50)
—
4.21
4.75
4.56
4.40
4.04
3.96
4.34
4.23
4.11
4.13
4.11
3.7. Селективность катионного комплексообразования
Таблица 3.4 {окончание)
157
R в хозяине
Na+
NH+
Са2+
/—О О v
R—N
N —R
C.51)
н
СН2СН2ОМе
CH2CO2Et
1.5
4.75
5.51
1.8
5.46
5.78
—
4.48
6.78
3*7*4 Криптанды
Трехмерные криптанды, в противоположность краун-эфирам,
проявляют предельно высокую селективность. Их макробициклические полости более
жестки и ограничены, потому они не способны ни достаточно сжиматься для
связывания с катионами, слишком малыми для такой полости, ни расширяться,
приспосабливаясь к радиусам, большим, чем требуется для оптимального размерного
соответствия. Более того, трехмерная полость криптанда гораздо больше предорга-
низована для связывания катиона, в том смысле, что энтропийные, а часто и эн-
тальпийные затраты на конформационную перестройку, которая должна
происходить, для того чтобы комплекс принял оптимальную геометрию, здесь менее
значительны. Так, константа связывания для криптанда [2.2.2] максимальна в
случае солей К+ в метаноле (lg Ки = 10.0), тогда как меньший криптанд — [2.2.1] —
селективен к Na+(lg Кп = 9.0), а криптанд [2.1.1] - к Li+(lg Ku = 7.6). Для криптанда
[3.2.2] К+/Ыа+-селективность равна ~ 103, а для 18-краун-6 в тех же условиях эта
величина меньше 102 (рис. 3.26).
Поразительный пример влияния предорганизационных эффектов,
увеличивающих жесткость трехмерной полости криптандов, — данные, приведенные на
рис. 3.27. Ясно, что даже если размерное соответствие и не идеально, то трехмерная
полость и семь донорных атомов криптанда [2.2.1] C.22) очень сильно связывают
катион К+; его константа связывания примерно на 5 порядков выше, чем таковая
егодиаза-18-краун-6-аналога C.18), имеющего только шесть центров связывания и
двухмерную полость. Примечательно, однако, что криптанд C.52) с шестью донор-
ными атомами, как у C.18), и углеводородным мостиком, связывает К+ более чем в
103 раз сильнее, несмотря на то, что его липофильный мостик может быть причи-
15Й
11.0
10.0
9.0
8.0
7.0
6.0
3. Хозяева, связывающие катионы
5.0
4.0
3.0
2.0}
1.0
0.0
/ / • \
' / ■• \
/-••■ \
/•■■ \
—а—[2лл;
-♦-[2.2. Г
-»- [2.2.2;
-*- [3.2.2;
т
Рис. 3.26. Константы
связывания катионов щелочных
металлов различными крип-
тандами в метаноле
Na+
К+
Катион
Rb+
Cs+
ной отталкивания сильногидрофильных катионов. Ясно, что в случае этих
соединений доминирующий фактор — предорганизация трехмерного хозяина. Значение
предорганизации как основного фактора в сродстве хозяин—гость более полно
рассмотрено в разд. 3.9.
Криптаты катионов щелочноземельных металлов демонстрируют те же
тенденции, которые наблюдались у краун-эфиров. Маленький криптанд [2.1.1]
неэффективен при связывании любого из двухвалентных ионов Mg2+ - Ba2+(lg К] j в воде
составляет -2.5 для Mg2+ и уменьшается до <2 для Ва2+). Увеличение размера полости
до размера полости криптанда [2.2.1] приводит к пику селективности для Sr2+, хотя
величина lg Kn = 7.35 только немного больше значений для комплексов Са2+ и
Ва2+. Из-за своей большой энергии гидратации Mg2+ вряд ли вообще связан в вод-
NH
О О
О О
HN
C.18)
2.0
Рис. 3.27. Сравнение констант связывания катиона К+ криптандными и корандными
хозяевами (МеОН, 25 °С)
3.8. Макроциклический, макробициклический и темплатный эффекты 159
ном растворе. Криптанд [2.2.2] — крайне эффективный лиганд для Ва2+. Влияние
двойного положительного заряда щелочноземельного металла хорошо видно при
сравнении значений логарифма констант связывания для Ва2+ (9.5) и К+ (lg Кх х = 5.4)
в воде. Катион стронция связывается значительно слабее (lg Kn = 8.0), а
стабильность продолжает уменьшаться до величин lg К, равных 4.4 и < 2, для Са2+ и Mg2+
соответственно.
Макроциклический,
макробициклический и темплатный
эффекты
3*8.1 Макроциклический эффект
Взаимодействие К+ со слабым монодентатным основанием, например
с диметиловым эфиром (как показывает измерение его константы связывания),
пренебрежимо мало. Однако мы видели, что эфирные макроциклы проявляют к
катионам щелочных металлов значительное сродство. Такое различие в поведении
можно объяснить хелатным и макроциклическим эффектами, обычно
наблюдаемыми в координационной химии (разд. 1.4). Исходя из энтальпийного,
энтропийного и статистического подходов, объединение шести донорных атомов в таком ли-
ганде, как 18-краун-6, сильно увеличивает стабильность [К+ с 18-краун-6] по
сравнению с [К(ОМе2N]+. В этих системах роль макроциклического эффекта
очевидна. Сравните стабильность [К+ с C.6)] и [К+ с C.11)], рис. 3.28. Хотя оба эти
соединения используют хелатную стабилизацию, тем не менее в растворе метанола
макроциклические комплексы типа [К+ с C.6)] примерно в 104 раз более
стабильны. Такая предельно высокая стабилизация — следствие макроциклического
эффекта. В результате макробициклического эффекта комплекс [2.2.2]криптанда C.22)
еще более стабилен.
оо— • °
о
о
о о
Me Me
[К+-C.11)] [К+-C.6)] [К+-C.22)]
lgK(M~l; MeOH, 25°C) 2.0 6.1 10.0
Рис. 3.28. Стабильность подандного (ациклического), краун-эфирного
(макроциклического) и криптандного (макробициклического) комплексов катиона К+
3. Хозяева, связывающие катионы
Таблица 3.5. Термодинамические вклады в стабильность
подандного и краун-эфирного комплексов К+
Комплекс
ДС°, Джмоль
Д#°, Джмоль"
&S°, Джмоль"
[К+сC.11)] -11368 -36400 -84
[К+сC.6)] -34 842 -56 000 -71
Для объяснения макроциклического и макробициклического эффектов мы
должны, согласно уравнению Гиббса, разделить энтальпийный и энтропийный
вклады в стабильность таких комплексов, как [К+ с C.11)] и [К+ с C.6)]:
ДС° = -RT\n К = ЛЯ° - TAS0. C.2)
При 298 К мы получим значения этих параметров, приведенные в табл. 3.5. Из
этих данных ясно, что и энтальпийный, и энтропийный вклады способствуют
стабильности макроциклического комплекса. В данном случае стабильность
определяется энтальпийным вкладом, хотя в другом случае более значимым может быть
второе слагаемое.
Поданд C.11) за счет вытянутой линейной конформации способен сводить к
минимуму неблагоприятное изменение энтальпии, вызываемое отталкиванием
между неподеленными электронными парами кислорода. Связывание с
металлическим центром влечет за собой конформационную перестройку и поэтому дает
дополнительный неблагоприятный вклад в изменение энтальпии, обусловленное
отталкиванием сближенных неподеленных электронных пар атомов кислорода в
связывающей конформации. В случае циклического лиганда C.6) этот
неблагоприятный энтальпийный вклад в общую свободную энергию связывания катиона
металла лигандом энергетически уже «оплачен» во время синтеза. Этот лиганд более
предорганизован для связывания катиона металла и в нем существуют
неблагоприятные взаимодействия между неподеленными электронными парами атомов
кислорода независимо оттого, связывает ли он катион металла или нет (рис. 3.29).
Поэтому отталкивание между неподеленными электронными парами в нем не влияет
на свободную энергию процесса комплексообразования катиона металла. Конечно,
в первую очередь, это делает синтез собственно макроцикла более трудным,
приводя к необходимости использовать специальный метод синтеза (разд. 3.8.3). Для
таких макробициклических соединений, как криптанды, степень предорганизации
еще более явно выражена, что и приводит к макробициклическому эффекту.
Таблица 3.6. Соотношение между стабильностью
и стабилизирующим эффектом комплексов типа
«хозяин-гость»
Тип комплекса
*(МеОН)
Эффект
Подат
Корат
Криптат
102-
104-
106-
ю4
106
ю9
Хслатный
Макроциклический
Макробициклический
3.8. Макроциклический, макробициклический и темплатный эффекты
Отталкивание "неподеленная электронная пара - неподеленная электронная пара"
Очень слабое отталкивание
Рис. 3.29. Объяснение макроциклического эффекта с позиций энтальпийного вклада
Следует заметить, что 18-краун-6 слабее сольватирован, чем свободный C.11),
из-за ограниченного объема макроциклической полости. Ациклический полиэфир
C.11) способен также принимать вытянутую конформацию, предоставляя
максимально возможную площадь поверхности для взаимодействия с молекулами
растворителя. Поэтому при связывании катиона для десольватации неподеленных
электронных пар атомов кислорода поданда требуется большая энергия, чем в случае
макроцикла, и поданды перестраиваются с образованием более упорядоченной
конформации. Это приводит к неблагоприятным энтропийным эффектам.
Количественное соотношение между хелатным, макроциклическим и макробицикличес-
ким эффектами представлено в табл. 3.6.
3*8*2 Темплатный эффект
При рассмотрении синтеза дибензо-18-краун-6 (схема 3.1) возникает
предположение о том, что возможную побочную реакцию, приводящую к
образованию краун-эфиров, могли вообще не обнаружить. Макроцикл дибензо-18-краун-6,
который был получен случайно и положил начало современной супрамолекуляр-
ной химии, мог бы и не образоваться, если бы реакция пошла по другому пути - по
пути, ведущему к полимерным (поликонденсатным) продуктам. Возможные
направления реакций, идущих при синтезе 18-краун-6, приведены на схеме 3.12.
Макроциклические краун-эфиры являются основными продуктами вовсе не из-
за того, что они наиболее термодинамически стабильны. Действительно, не
существует связи между тем, что 18-членные крауны селективны к К+ и что в их синтезе
чаще всего используется карбонат калия (или КОН). В самом деле, замена К2СО3 на
11 - СТИД Дж.В
162
3. Хозяева, связывающие катионы
Схема 3.12. Возможные пути образования циклических и ациклических продуктов при
синтезе 18-краун-6 C.6)
органическое основание, например триэтиламин, приводит к образованию
преимущественно полимерного продукта. Принципиальная разница в поведении
оснований этих двух классов лежит в способности иона К+ таким образом
организовывать вокруг себя реагирующие вещества, что получается интермедиат (возможно,
напоминающий C.53)), который предорганизован для образования циклического
продукта. Функциональные группы ОН и С1 вступают в непосредственный контакт
за счет координации с катионом калия (через хелатныи эффект) и происходит
быстрая циклизация. Органическое основание не способно вызвать образование
этого интермедиата, и реакция идет по межмолекулярному, а не внутримолекулярному
пути. Полагают, что ион К+ - темплат для реакции, и образование макроцикличе-
ских соединений таким способом называют темплатным эффектом или, более
строго, кинетическим темплатным эффектом. В действительности, это
разновидность катализа, в котором катион металла действует как стабилизатор
циклического интермедиата, за счет чего резко увеличивается скорость образования
циклического продукта. Таким образом, темплатный эффект является кинетическим
3.8. Макроциклический, макробициклический и темплатный эффекты
163
эффектом, а макроцикл — кинетическим продуктом. Проще говоря, если мы хотим
инициировать образование хозяина, селективного к щелочным металлам, с
размером, близким к размеру К+, то в качестве шаблона мы используем К+, который
обеспечивает образование такого хозяина. Темплатный эффект широко применяют
в макроциклическом синтезе, включая синтез макробициклических соединений,
молекул-узлов, сцепленных колец (катенаны) и многих других. Катионы
щелочных, щелочноземельных и переходных металлов, а также лантаноидов могут
проявлять темплатные свойства. Во многих ранних работах по синтезу показано, что Cs+
особенно эффективен как темплат, а увеличение выхода получаемых циклических
продуктов было названо цезиевым эффектом.
Для распознавания темплатного эффекта нужно использовать два теста:
1. Организация других молекулярных компонентов темплатом.
2. Сопутствующие химические реакции должны включать пространственный,
топологический или геометрический контроль.
На рис. 3.30 показаны результаты кинетических исследований темплатного
синтеза бензо-18-краун-6 в соответствии с реакцией, приведенной на схеме 3.13, для
различных темплатирующих катионов. Очевидное возрастание скорости (по
отношению к нетемплатной реакции в присутствии NEt^") наблюдается с увеличением
концентрации ионов всех щелочных металлов, кроме Li+, который стремится к
образованию сильносвязанных ионных пар с феноксид-ионом, таким образом
подавляя его реакционную способность. Как и предполагалось, наибольшее увеличение
Рис. 3.30. Результаты кинетических
исследований влияния катионов
металлов на циклизацию.
(Воспроизведено с разрешения J. Amer. Chem. Soc.
1983. Vol.105. P.555)
-0.5-
-1.0
-1.5-
-2.0-
-2.5-
-3.0-
-3.5-
NEt4+
-5 -4 -3 -2 -1
3. Хозяева, связывающие катионы
Схема 3.13. Темплатная циклизация с образованием бензо-18-краун-6
скорости дает К+, наиболее комплементарный полости крауна. Подобная
циклизация в случае алкильной цепи устраняет организационные эффекты катионов,
приводя к снижению скорости образования циклических продуктов (а значит, и их
выхода) по сравнению с реакциями, протекающими в отсутствие катионов щелочных
металлов. Это замедление является результатом образования ионных пар.
При обсуждении кинетического темплатного эффекта важно отличать его от
другого эффекта, называемого термодинамическим темплатным эффектом.
Кинетический темплатный эффект включает реальное формирование лигандов вокруг
металлического центра. Термодинамический темплатный эффект определяется
способностью катиона металла выбирать комплементарный ему лиганд из
равновесной смеси соединений; при этом равновесие сдвигается в сторону продукта,
стабилизируемого катионом. Один из первых примеров этого эффекта - конденсация
ацетона с солями трис(этилендиамин)никеля(П), происходящая согласно
схеме 3.14. Такие же результаты могут быть получены для других металлов и кето-
нов. Ниже приведены данные, свидетельствующие о том, что это именно
термодинамический эффект.
NH2
NH2
~T -^n"*
NH2/, \ ^ .N NH
"•-. I .„nNH2 >^ { \ / ^
•NH2
Схема 3.14. Термодинамический темплатный синтез тетраазамакроциклов
Реакционная способность зависит от лабильности иона металла: конденсация
аминогрупп должна сопровождаться их частичным отщеплением от иона
металла; инертные к замещению соли Со(Ш) не реагируют вообще.
Из несимметричных кетонов в основных условиях получаются менее
разветвленные продукты (конденсация Клайзена—Шмидта), что опять предполагает
отсутствие иона металла.
Лиганды могут быть получены в отсутствие иона металла.
3.8. Макроциклический, макробициклический и темплатный эффекты
i|65
Более необычные темплатные эффекты, например те, которые затрагивают синтез
катенана и ротаксана, а также начало «самовоспроизведения», обсуждаются в гл. 7.
•""» Busch D.H. and Stephenson N.A. Molecular organisation, portal to supramolecular
chemistry // Coord. Chem. Rev. 1990. Vol.100. P. 119-154.
3.8.3 Синтез методом высокого разбавления
Синтез макроциклических лигандов сильно затруднен без
подходящего темплата, поэтому необходимо использовать условия высокого разбавления. Под
термином «высокое разбавление» мы подразумеваем использование малых
количеств реагирующих веществ в большом объеме растворителя. Типичный аппарат
для выполнения такой процедуры показан на рис. 3.31. Реагирующие вещества
очень медленно капают из двух резервуаров (это может происходить автоматически
с помощью электронно-управляемого шприцевого насоса) и смешивают в большой
круглодонной колбе.
Разумность этого подхода заключается в том, что в разбавленных растворах
образование циклического продукта путем внутримолекулярной реакции (т.е. один
конец молекулы замыкается на себя) - более вероятный, а значит, и более быстрый
процесс, чем образование полимера, требующее столкновений между двумя
реагентами (межмолекулярная реакция), рис. 3.32. При сравнении скоростей циклизации
Рис. 3.31. Аппарат,
предназначенный для
достижения высокого разбавления в
реакциях макроциклизации.
(Перепечатано с разрешения
AtwoodJ.L., Davies J.E.D.,
MacNicol D.D., Vogtle F.
Comprehensive
supramolecular chemistry. Oxford:
Pergamon, 1996)
Высокоскоростной мотор C000 об-мюГ1)
i^~ Трубка с осушителем
Прецизионная капельница
Стальной стержень, покрытый
тефлоном
Круглодонная колба
Тефлоновая лопасть
166
3. Хозяева, связывающие катионы
Скорость циклизации кс [X - Y]
Скорость полимеризации кр [X - Y]2
Рис. 3.32. Макроциклический синтез методом высокого разбавления
гс = кс[Х-У] и полимеризации г = кJX-Y]2 для реагента X-Y получаем выражение:
кс[Х-У]
кр[Х-У]2
C.3)
где кс и к — константы скоростей циклизации и полимеризации. Таким образом, с
уменьшением концентрации реагента X-Y величина гс/г возрастает, что
свидетельствует о преобладании гс над гр в разбавленных растворах.
Отсюда следует, что чем быстрее реакция, тем лучше работает этот подход,
поскольку концентрация реагентов всегда будет мала и не будет увеличиваться в ходе
реакции, если реакция сама по себе идет с большей скоростью, чем скорости
добавления реагентов (большие величины кс и к). Условия высокого разбавления
применяют в большом количестве процессов макроциклического и макробициклическо-
ГЛ
У—О О—\
г~\
С1
NH2
CI
Высокое О
разбавление ^п\
< >
\—О О—/
Схема 3.15. Синтез диаза-18-краун-6 методом высокого разбавления. Группы дихлоран-
гидрида используются для увеличения скорости реакции
.3-8. Макроциклический, макробициклический и темплатный эффекты
167
го синтеза, в том числе и при получении исходных соединений для криптандов
(схема 3.5). В частности, реакция амина с дихлорангидридом происходит
достаточно быстро из-за отрыва электрона и резонансной стабилизации карбонильных
групп. Использование дихлорангидрида в синтезе методом высокого разбавления
простого азакраун-эфира показано на схеме 3.15.
*"» Illuminati G. and Mandolini L. Ring closure reactions of biftinctional chain molecules
//Ace. Chem. Res. 1981. Vol.14. P.95-102.
3.8.4 [2+2]-Циклосоконденсация
Большое разнообразие весьма интересных криптандов, в особенности
содержащих большие полости, было синтезировано сочетанием внутри- и
межмолекулярных реакций. Наиболее доступная из них — реакция [2 + 2]-циклосоконденса-
ции при синтезе краун-эфира или криптанда, в которой, вместо [1 + 1]-реакции,
показанной на схеме 3.15, две пары реагентов сталкиваются, часто случайно, с
образованием макробицикла или макротрицикла. Реакция (в) схемы 3.2 — это
пример [2 + 2]-циклоконденсации краун-эфиров. В случае синтеза криптандов такая
циклосоконденсация схематически представлена на схеме 3.16. Обычно ситуации, в
которых условия реакции (скорость и концентрация) создают равновесие между
полимеризацией и циклизацией, или же обстоятельства, при которых продукт [1 + 1]-
циклизации стерически напряжен или его образование неблагоприятно по какой-
либо другой причине, приводят к протеканию реакции [2 + 2]-циклосоконденсации.
На схеме 3.17 (С. Hall et al., 1997) представлен прекрасный пример химии
такого рода, изначально создававшейся для синтеза [1 + 1]-продукта C.54). Предполага-
Схема 3.16. Схематическое представление [1 + 1]- и [2+2]-циклосоконденсациидля
получения криптандов
168
3. Хозяева, связывающие катионы
C.56)
Схема 3.17. Одновременная [1 + 1]-, [2+2]- и [3+3]-циклосоконденсация
для получения криптандов на основе бипиридила
3.9. Предорганизация и комплементарность 169
Рис. 3.33. Кристаллическая
структура 2:2-комплекса
криптанда C.55) с NaC104
{no Hall С. etal., 1997)
лось, что при связывании редокс-активного бис(циклопентадиенил)железа(П)
(ферроценил) с макроциклом бис(бипиридила) получится редокс-чувствительный
и редокс-переключаемый криптанд, способный связывать и обнаруживать такие
катионы, как Na+ и Еи3+. Действительно, с помощью хроматографии были
выделены продукты [1 + 1]-, [2+2]- и [3+3]-циклосоконденсации. Как показывает
кристаллическая структура (рис. 3.33) 2:2-комплекса соединения C.55) с NaC104, этот
криптанд способен капсулировать два катиона Na+ внутри гидрофобной арильной
полости.
3*9 Предорганизация
и комплементарность
•""» Cram D.J. Preorganisation - from solvents to spherands // Angew. Chem., Int. Ed.
Engl. 1986. Vol.25. P. 1039-1057.
3*9*1 Термодинамические эффекты
Как мы уже видели в разд. 3.8.2, простая предорганизация такого
хозяина, как краун-эфир, может резко усилить его способность к связыванию, если
принять меры против нежелательных взаимодействий при синтезе молекулы.
Общая концепция предорганизации заключается в заблаговременном (до связывания
гостя) конструировании хозяина в точном соответствии со стерическими и
электронными требованиями гостя. Подходящие друг другу хозяева и гости называют
комплементарными. В стерическом смысле хозяин, не будучи ни слишком
большим, ни слишком маленьким, должен соответствовать гостю. В электронном
смысле хозяин должен предоставлять свои центры связывания противоположного
электростатического заряда или диполи соответствующим центрам или диполям гостя
(например, хозяева с основными центрами Льюиса - гостям с кислотными
центрами Льюиса; доноры водородной связи - акцепторам водородной связи). Принцип
170
3. Хозяева, связывающие катионы
комплементарное™ был сформулирован Д. Крамом: «Для образования комплекса
хозяева должны иметь центры связывания, которые кооперативно контактируют и
притягивают центры связывания гостей, не вызывая при этом сильное несвязыва-
юшее отталкивание». Отдельные взаимодействия между хозяевами и гостями
(см. разд. 1.7) относительно слабы — гораздо слабее ковалентной связи, но именно
благодаря комплементарным взаимодействиям многочисленных пар центров
связывания достигается сильное селективное комплексообразование. Константа
связывания (или свободная энергия комплексообразования) представляет собой
количественную меру комплементарности при данном наборе условий.
В рамках этой концепции очень важна роль растворителя. Часто
взаимодействия между растворителем и гостем, а также между растворителем и хозяином
имеют тот же тип, что и между гостем и хозяином. Для того чтобы связать гостя, и
хозяин, и гость должны лишиться своих сольватных оболочек. Таким образом,
свободная энергия комплексообразования хозяин—гость включает энтальпийный и
энтропийный приросты энергии, являющиеся результатом благоприятных
взаимодействий хозяин—гость и возрастания числа свободных частиц, причем энтальпий-
ная потеря отдесольватации хозяина и гостя имеет меньшую величину (схема 3.18).
Вообще говоря, этот процесс благоприятен только благодаря высокой степени
1. Сольватированный хозяин
(гидрофобные
и ван-дер-ваальсовы
взаимодействия)
2. Десольватированный хозяин
и свободный растворитель.
Энталышйно затратный,
энтропийно выгодный процесс
. •
Конформационная
перестройка хозяина.
Энтальпийно и энтропийно
затратный процесс.
Не обязателен для
предорганизованного
хозяина
6. Комплексообразование. ^ 1 ' ф
Энтальпийно очень * ф
Взаимодействия между
комплексом и растворителем
могут включать как
гидрофобные
и ван-дер-ваальсовы
взаимодействия, так
и координационные эффекты
выгодный процесс
7. Растворение комплекса.
Энталышйно выгодный,
энтропийно затратный >
процесс
4. Сольватированный
гость (координационный
комплекс)
Катион металла (
Молекула
растворителя.
Полярная "головка". (
Гидрофобный "хвост"
Фрагмент хозяина
5. Десольватация гостя.
Энтальпийно затратный,
энтропийно выгодный
процесс
\ 8. Остаточный растворитель
не включается во взаимодействие.
Энтропия возрастает
Схема 3.18. Процесс комплексообразования хозяин—гость
3.9. Предорганизация и комплементарность
171
организации хозяина (комплементарно гостю) и ее отсутствию у растворителя.
Если хозяин предорганизован (т.е. существует небольшая разница между
минимальными энергиями конформации свободного и связанного хозяев), то связывание
особенно благоприятно, поскольку тогда не возникают дополнительные
энергетические потери из-за конформационной перестройки хозяина, необходимой для
создания оптимальной связывающей конформации.
В табл. 3.7 приведены свободные энергии связывания целого ряда хозяев для пи-
кратов щелочных металлов. Как можно заметить из этого небольшого набора
данных, семейства соединений в соответствии с величинами — Д(?°, с которой они
связывают наиболее комплементарные гости, в основном располагаются в следующем
порядке: сферанды > криптасферанды > криптанды > полусферанды > коранды >
Рис. 3.34. Кристаллические структуры
(а) свободного сферанда C.33) и (б) его
комплекса с Li+ (по Trueblood К. et al.,
1981)
172
3. Хозяева, связывающие катионы
Таблица 3.7. Свободные энергии связывания хозяев Aккал = 4.184 кДж) для
пикратов щелочных металлов в стандартных условиях (по Cram D., 1986)
(Величины для ионов металлов, наиболее комплементарных к каждому хозяину, выделены
жирным шрифтом)
Хозяин
Тип хозяина
—ДС°, ккал моль
Na"
CsH
r\
Сферанд >23 19.2 «6
C.33)
Криптасферанд 18.8 20.6 15.0 13.3 10.4
C.57)
Криптасферанд 13.4 21.0 >19.9 20.4 16.4
C.58)
3.9. Предорганизация и комплементарность
Хозяин
Тип хозяина
Таблица 3.
и+
Na+
7
(продолжение)
, ккалмоль
к+
Rb+
Cs+
ГУ
Криптасферанд 9.9 13.5 19.0 20.3 21.7
C.35)
ГЛ
О О
Криптанд 16.6 - - -
C.59)
О О
Криптанд 10.0 17.7 15.3 12.7
C.22)
О О
N О О N
V_J\ 1\ /
Криптанд
C.23)
- 14.4 18.0 16.8 10.3
174
3. Хозяева, связывающие катионы
Таблица 3.7 (продолжение)
Хозяин
Тип хозяина
—ДG°, ккал моль"
Na"
Полусферанд 7.0 12.2 11.9 10.4 9.0
' C.34)
Полусферанд 7.2 13.5 10.7 8.4 7.1
C.60)
Полусферанд 6.5 7.1 11.6 11.4 10.8
C.39)
3.9. Предорганизация и комплементарность
Таблица 3.7 {окончание)
Хозяин
Тип хозяина
—AG°, ккалмоль"
U+ Na+ K+ Rb+ Cs
Коранд 6.3 8.4 11.4 9.9 8.5
(лариат-эфир)
C.61)
Поданд <6 <6 <6 <6 <6
C.36)
поданды > растворители. Разница величин -AG0 для Li+, связанного сферандом
C.33) и подандом C.36), равная 17 ккал-моль G1 кДж-моль), соответствует
разнице констант связывания К более чем 1012. При уменьшении порядка пик
селективности уступает место плато селективности до того момента, пока для поданда
C.36) вообще не будет наблюдаться никакой селективности. На свободные энергии
связывания, приведенные в табл. 3.7, значительное влияние оказывают количество
и природа донорных атомов, находящихся в пределах от шести в сферанде C.33) до
девяти в криптасферанде C.35). По мере того, как энергия каждого
координационного взаимодействия вносит вклад в связывание, можно ожидать аддитивного
увеличения стабильности с ростом числа центров связывания. Однако над этой
тенденцией явно преобладает другой фактор — степень предорганизации.
Сравнение кристаллических структур сферанда C.33) и его очень стабильного
сфераплекса C.33) • Li+ показывает, что в обоих случаях хозяин имеет одну и ту же
конформацию (рис. 3.34). Хозяин C.33) полностью предорганизован для
связывания металла, и на комплексообразование ему не требуется никаких энтальпийных
затрат. Более того, в свободном хозяине шесть анизильных атомов кислорода
глубоко погружены внутрь несольватированной полости, образованной гидрофобным
каркасом хозяина. Таким образом, при комплексообразовании катиона металла не
существует стадии десольватации этих донорных атомов.
176
3. Хозяева, связывающие катионы
к+
гл
о о
Рис. 3.35. Конформаци-
онная перестройка [2.2.2]-
криптанда при связывании
катиона К+
Сравните предорганизацию, показанную на рис. 3.34, с предорганизацией
свободной и комплексной форм 18-краун-6, приведенной на рис. 3.14. Очевидно, для
того чтобы связать катион гостя, краун-эфир должен претерпеть значительную
перестройку (и десольватацию в растворе). Такие же перестройки необходимы и для
[2.2.2]криптанда (рис. 3.35), тогда как анизильные фрагменты криптасферандов
частично предорганизованы, несмотря на заполнение полости метиленовыми
группами криптандной части молекулы.
Энтропийные, а часто и энтальпииные затраты на перестройку хозяина еще
более ярко выражены в таких подандных хозяевах, как C.36) и криптофикс- 5 C.62),
которые в свободном состоянии имеют вытянутые конформации. Степень, до
которой должен перестроиться C.62), чтобы «укутать» катион Rb+, очевидна из
кристаллической структуры КЬ+-подаплекса (рис. 3.36).
C.62)
Рис. 3.36. Кристаллическая
структура, демонстрирующая «укутывание»
катиона Rb+ подандом криптофиксом-5
C.62)
3.9. Предорганизация и комплементарностъ |%77
3*9*3 Кинетический и динамический эффекты
Повышенная стабильность комплексов, при образовании которых
хозяин уже предорганизован для связывания, концептуально чем-то напоминает
теорию переходного состояния в органической химии. Эта теория утверждает, что чем
больше реагенты напоминают переходное состояние реакции, тем быстрее
протекает эта реакция вследствие понижения энергии активации, требуемой для
перехода от исходных веществ к переходному состоянию. Различие состоит в том, что до
сих пор мы обсуждали предорганизацию в контексте термодинамической
стабильности получающихся комплексов хозяин—гость. Однако предорганизация хозяина
и его степень гибкости, действительно, оказывают ощутимое влияние на скорости
реакций комплексообразования и диссоциации.
Константа равновесия /^связана со скоростями реакций комплексообразования (к{)
и диссоциации (&_,) по уравнению К= kx/k_x (Дополнение 1.1). Константы скоростей
прямой и обратной реакций комплексообразования для комплексов трех типов даны в
табл. 3.8. В общем, обнаружено, что скорости как прямой, так и обратной реакции
уменьшаются в следующем порядке: тип «насест» > тип «гнездо» > тип «капсула» (рис. 1.4).
C.63)
Таблица 3.8. Константы скоростей комплексообразования (кх) и диссоциации
(&_,) пикратов для различных соединений в стандартных условиях
Гость
C.33)
(«капсула»)
М-'"с-
*-р
С
C.63)
(«гнездо»)
М-'-с
с
C.64)
(«насест»)
М-'с
*-р
С
Na+
К+
(CH3KCNH3+
8104
4105
2-Ю
-7
2109
14
3-Ю12
7102
173 3. Хозяева, связывающие катионы
Рис. 3.37. Кристаллическая
структура /wpe/w-бутиламмониевого
комплекса полусферанда C.64).
(Воспроизведено с разрешения
Cram D., 1986)
В комплексе типа «насест», где гость остается на верхушке связывающей
поверхности хозяина, как в mpem-бутиламмониевом комплексе полусферанда C.64)
(рис. 3.37), разрыв сольватной оболочки катиона при связывании относительно
невелик. Разрыв возрастает при переходе к комплексу типа «гнездо», в котором гость
(катион) частично погружен в полость хозяина, и максимален в капсулярных
комплексах. Таким образом, свободный катион в комплексах типа «насест» более всего
напоминает переходное состояние комплексообразования, которое приводит к
быстрой реакции. Исследование СРК-моделей сфераплексов соединения C.33)
указывает на то, что в переходном состоянии комплексообразования (при вхождении
гостя в полость) существует пространство только для одной молекулы
растворителя (или, возможно, фенольного кислорода пикрат-аниона) и катиона,
окруженного углеводородным «пузырем», образованным предорганизованной полостью
хозяина и метальными группами его анизильных фрагментов. Таким образом, для
комплексообразования с участием C.33) следует ожидать замедленной кинетики.
Это связано с тем, что катион должен почти полностью десольватироваться
(процесс энтальпийно невыгодный, ведущий к высокоэнергетическому переходному
состоянию), прежде чем он сможет восполнить энтальпию за счет
комплексообразования. Фактически это общее явление: чем жестче предорганизован хозяин для
катионного связывания, тем медленнее кинетика процесса. Более гибкие хозяева,
например коранды и особенно поданды, должны быть способны к более плавному
переходу от сольватации к комплексообразованию без прохождения через
нестабильные десольватированные промежуточные состояния.
Из табл. 3.8 видно, что все скорости комплексообразования очень велики и их
нельзя определить даже с помощью 'Н ЯМР-спектроскопии (действительно,
реакции происходят сразу после смешения). Для более гибких хозяев скорости
диссоциации тоже велики, но они доступны для 1Н ЯМР, в то время как для сфераплексов
обратная реакция при 25 °С протекает очень медленно.
При использовании супрамолекулярных катионных хозяев большое значение
имеет связь между стабильностью комплекса и кинетикой катионного обмена. На
основании этих свойств можно установить различие между рецепторами катионов
(медленная кинетика, большие константы стабильности) и переносчиками катионов
(быстрая кинетика, низкая стабильность). Выше (разд. 3.6) мы видели, как быстрая
кинетика обмена делает переносчиков катионов крайне полезными, например, в
таких приложениях, как межфазный катализ.
3.10. Мягкие лиганды для мягких ионов металлов \ 791
Мягкие лиганды
для мягких ионов металлов
До сих пор большинство рассмотренных нами комплексов ионов
металлов были комплексами жестких неполяризуемых ионов, например катионов ше-
лочных металлов. Связывание в этих комплексах имеет в основном
электростатическую природу, причем донорные атомы хозяина располагаются вокруг сферы
катиона таким образом, чтобы минимизировать стерическое отталкивание внутри
хозяина. Это связывание почти не ковалентно и не имеет преимущественного
направления, а получающаяся координационная геометрия иона металла и его
координационное число сильно зависят от конформационных предпочтений хозяина
(Дополнение 3.1). Как правило, лиганды, эффективные при связывании жестких
катионов металлов, проявляют только ограниченное сродство к более мягким
ионам металлов, например к Ag+, и ионам многих переходных металлов с низкими
состояниями окисления, особенно из второго и третьего рядов d-элементов.
Дополнение 3.1. Селективное связывание катионов щелочных металлов
Свойства катионов щелочных металлов:
• Жесткие неполяризуемые сферы (Дополнение 3.2).
• Небольшое предпочтение определенной координационной геометрии.
• Относительно высокие свободные энергии гидратации.
• Сродство к сильнозаряженным, неполяризуемым основаниям.
Эти особенности создают трудности при конструировании приемлемых лигандов,
которые могли бы вытеснять сольватационную воду и связываться прочно и
селективно со свободно диффундирующими катионами. Единственный базовый
параметр, определяющий селективность хозяев по отношению к катионам щелочных
металлов, — это размер последних (рис. 3.38).
v* Rb+
Радиус иона, А
0.60 о.97
1.33 1 4»
148 1.67
Рис. 3.38. Размер катионов щелочных металлов
3. Хозяева, связывающие катионы
Дополнение 3.2. Теория жестких и мягких кислот и оснований
Ионы металлов можно разделить на два класса:
1. Ионы, у которых стабильность галогенных комплексов и сила связи
металл—галоген меняются в следующем ряду. F > С1 > Вг > I; эта
последовательность сохраняется для групп из 16 и 15 донорных атомов. Их называют
акцепторами класса «а». Примерами являются Al3+, Ti4+ и большинство ионов s- и
/^-элементов, Mn3+, Fe3+, Co3+, а также некоторые другие ионы переходных
металлов в высоких состояниях окисления.
2. Ионы, у которых стабильность меняется в ряду: F < С1 < Вг < 1, О < S~Se~Te и
N < Р > As > Sb (комплексы с Р-донорами почти всегда более стабильны, чем
комплексы на основе As и Sb). Большинство ионов металлов класса «Ь»
принадлежат переходным металлам, например Cu(I), Ag(I), Au(I), Pd(II) и
Pt(II). Ионы класса «Ь» стремятся иметь большое число ^-электронов на
внешней оболочке. Размер таких ионов может быть признан еще одним фактором,
определяющим «Ь»-характер. Это происходит потому, что ионы металлов
легче поляризуются с ростом их размера и уменьшением проникновения
валентного электрона.
Система классов «а» и «Ь» была обобщена в теории жестких и мягких кислот и
оснований (principle of hard and soft acids and bases, HSAB). В данном контексте слово
«основание» означает основание Льюиса, т.е. донор электрона (обычно лиганд в
координационном комплексе), а слово «кислота» относится к акцептору электрона (обычно
металл). Теория HSAB гласит, что жесткие кислоты предпочитают координироваться с
жесткими основаниями, а мягкие кислоты предпочитают мягкие основания. Ионы
металлов класса «а» — жесткие кислоты, тогда как в класс «Ь» входят мягкие кислоты.
Жесткие кислоты Жесткие основания
Большой положительный заряд Высокая электроотрицательность
Низкая поляризуемость Трудность окисления
Маленький размер (Н+, А13+) Низкая поляризуемость (F~)
Мягкие кислоты Мягкие основания
Небольшой положительный заряд Низкая электроотрицательность
Высокая поляризуемость Легкость окисления
Большой размер (Ag+) Высокая поляризуемость
Большой отрицательный заряд (Н~)
Реакция C.4) — интересный пример теории HSAB.
Lil + CsF^LiF + CsI. C.4)
Как предсказывает теория HSAB, эта реакция протекает слева направо, хотя это
противоречит определению электроотрицательности по Полингу, согласно которому
большая энергия связи в соединении соответствует большей разнице величин
электроотрицательности взаимодействующих ионов.
*""» Pearson R.G. Chemical hardness. New York: Wiley-VCH, 1997.
ЗЛО. Мягкие лиганды для мягких ионов металлов
181
- Ю. 1 Гетерокраун-эфиры
Введение одного или более мягких донорных атомов (например, N, S и
т.д.) приводит к поразительным изменениям как в связывающей способности лиган-
да, так и в геометрии получающегося комплекса (Дополнение 3.2). Резкое усиление
связывания мягкого Ag+ (и соответственно падение сродства к К+) после замены
эфирных кислородных доноров на S или N показано в табл. 3.9. Ясно, что мягкий
Ag+ имеет сильное сродство к S-донорам, причем связывание увеличивается в
последовательности: NH, S > О, в то время как для промежуточного РЬ2+ имеет место
другая последовательность: NH > О > S. Напротив, константы связывания жестких
катионов, таких, как К+, Т1+ и Ва2+, уменьшаются с уменьшением
электроотрицательности донорных атомов: О > NH > S. Это разумно с точки зрения катион-ди-
польных взаимодействий, лежащих в основе связывания, однако очень сильное
снижение при переходе от О к N (например, от 18-краун-6 к C.18)) с учетом близкого
сродства К+ к N- и О-основаниям в газовой фазе просто поразительно. Ясно, что в
этом случае благодаря донорнои способности водородной связи NH-группы сольва-
тационные и конформационные эффекты должны играть большую роль.
Поразительные различия кристаллических структур, показанных на рис. 3.39, -
Рис. 3.39.
Кристаллические структуры
нитратов Ag+-KOM-
плексов(а) 18-краун-6
C.6) и (б) его тиаана-
лога C.66)
Таблица 3.9. Сравнительные константы связывания (lg А"п) жестких и мягких
ионов металлов разными лигандами
К+ (метанол)
К+ (вода)
Ag+ (метанол)
Ag+ (вода)
ТГ (вода)
Ва2+(вода)
РЬ2+ (вода)
18-краун-6
6.10
2.10
4.58
1.60
2.27
3.78
4.27
C.65)
1.15
-
-
4.34
0.93
—
3.13
C.18)
2.04
<1
-
7.80
1.1
2.51
6.9
Лиганд
15-краун-5
_
0.74
—
0.94
1.23
—
1.85
C.67)
_
-
-
5.0
0.8
—
1.65
C.68)
_
1.0
-
5.85
-
1.0
5.85
C.69)
_
-
-
8.95
-
—
5.67
ie#| 3. Хозяева, связывающие катионы
это следствие различий между ненаправленным электростатическим связыванием
в [Ag+ с 18-краун-6] C.6) и дативной ковалентной связью в его тиааналоге
[Ag+ с [18JaH-S6] C.66). Кольцо крауна в [Ag+ с 18-краун-6] имеет слегка
искаженную плоскую конформацию и занимает в комплексе экваториальное положение,
при этом вода и нитрат-анион находятся в аксиальных положениях. В то же время
комплекс [Ag+ с [18]ан-56]имеет более узнаваемую октаэдрическую
координационную геометрию, подобную геометрии обычных координационных комплексов
Вернера. Лиганд C.66) также связывает мягкие ионы переходного металла,
например Pd2+. В этом случае краун достаточно гибок, для того чтобы с помощью
четырех коротких Pd-S-связей B.31 А) и двух, гораздо более длинных, аксиальных
связей C.27 А) приспособиться к квадратной плоской координации, предпочитаемой
ионом палладия.
N
О О^ ^6 Н 6
о о о н ? s
C.65) C.18) C.66)
s' > < No
°0 °j с ь с ">
О О—7 v—NH HN—' N—S S—'
\~J \_J V_V
C.67) C.68) C.69)
Краун-эфиры также не образуют сильных связей с катионами переходных
металлов. В самом деле, из-за своих малых ионных радиусов эти катионы часто
совсем не связываются с большими краун-эфирами, например с 18-краун-6, а вместо
этого их ионы с шестью молекулами воды за счет водородных связей образуют
вторую координационную сферу комплекса (рис. 3.40, а). Это согласуется с
исследованиями Дж. Кристенсена и других (J. Christensen et al., 1971), которые
предположили, что в случае щелочных металлов отношение диаметра катиона к размеру
полости хозяина в пределах 0.75—0.90 А благоприятно для прямого связывания
ион—краун-эфир. Так, для 18-краун-6 (диаметр полости 2.6—3.2 А в зависимости от
конформации) в случае Na+, K+ и Cs+ получены отношения 0.61—0.75, 0.83—1.02 и
1.03—1.27 соответственно, что совпадает с наблюдаемыми величинами
селективности. Точно так же в случае 15-краун-5 (диаметр полости 1.7-2.2 А) для Ыа+получе-
но отношение 0.89—1.15, что отражает размерное соответствие краун-эфира этому
иону. Ионы же большинства двухвалентных переходных металлов, например Со2+,
Ni2+ и Zn2+, слишком малы, чтобы быть заключенными внутри 18-краун-6, зато
3.10. Мягкие лиганды для мягких ионов металлов
~Н20 0Н2
Н20 —'nj2+—0H2
_,-Н20 \н2
(д)
а
Со*
Рис. 3.40. Продукты реакций дикатионов переходных металлов с 18-краун-6 (а, 6),
15-краун-5 (в) и 12-краун-4 (г, д) в водном или спиртовом растворах
они уютно устраиваются в 15-краун-5. В этих системах и аксиальные, и
экваториальные лиганды с множественными водородными связями образуют
высококристаллические супрамолекулярные полимеры (обычно их молекулы удерживаются
вместе водородными связями), которые для ряда солей М2+(С1О4J (М = Со, Ni, Си,
Zn) были выделены в твердом состоянии. Однако в водных растворах водородные
связи легко разрушаются, что приводит к образованию высокоподвижных (быстро-
обменивающихся) систем. В случае Си2+ может быть выделено второе соединение,
в образовании которого участвует первая координационная сфера макроцикла
(рис. 3.40, б). Меньшие краун-эфиры, например 15-краун-5 и 12-краун-4, будут
связывать катионы переходных металлов (рис. 3.40, в и г). В случае 15-краун-5 обычно
катион металла образует семь координационных связей: пять — с краун-эфиром и
j£4 3. Хозяева, связывающие катионы
две - с аксильными лигандами Н20. В случае 12-краун-4 продукты сильно зависят
от природы противоаниона и катиона металла: при этом выделены комплексы 2:1
(сэндвич) и 1:1 (типа «насест»), а также полимеры, содержащие ионы металлов с
шестью молекулами воды, которые наблюдали для 18-краун-6 (J. Steed et al., 1998).
3.Ю.2 Гетерокриптанды
Замена в криптандах кислорода на донорные функциональные NH-
или S-группы резко меняет поведение этих гетерокриптандов как комплексообра-
зователей при сохранении размеров О-криптандов. Таким образом, включение двух
NMe-rpynn в [2.2.2]азакриптанд C.22) приводит к образованию криптанда C.71),
который даже в воде имеет константу связывания для Ag+ (lg К), равную 11.5.
Соответствующая величина для К+ равна всего лишь 2.7 (по сравнению с 5.4 для
C.22)), тогда как Т1+ дает промежуточную величину lg К, равную 5.5. Сродство
ряда гетерокриптандов к некоторым представителям ионов металлов приведено в
табл. 3.10.
Ряд, включающий соединения C.22) и C.70)—C.72), особенно интересен,
поскольку константа связывания плавно изменяется, по мере того как атомы
кислорода последовательно заменяются на атомы азота. С одной стороны, возросшее
сродство к Ag+ подтверждает факт сильного взаимодействия Ag+—N, которое
частично осуществляется за счет ковалентной связи. С другой стороны, К+
связывается исключительно по ионному механизму и гораздо больше стабилизирован
кислородными донорами. Ясно, что Т1+ ведет себя подобно К+, т.е. его стабильность
падает с ростом числа атомов азота. Однако более высокая поляризуемость иона Т1+
подтверждается величинами констант связывания и приводит к их более
постепенному изменению. Сравнение данных по связыванию Ag+ для большего C.72) и
меньшего C.73) хозяев показывает, что, несмотря на то, что у соединения C.72)
шесть N-донорных атомов вместо четырех, сродство Ag+ к C.73) — аналогу [2.1.1]-
криптанда — почти такое же большое, как к большему хозяину C.72). Это
приписывали более близкому размерному соответствию Ag+ и C.73). В общем, очевидно,
Таблица 3.10. Константы связывания катионов Ag+, K+ и Т1+ разными
криптандами
Хозяин
•8*1.
Ag+
Растворитель
C.22)
C.70)
C.71)
C.72)
C.73)
C.74)
9.6
12.22
10.8
11.5
13.0
12.7
12.39
5.4
10.49
4.2
2.7
1.7
-
6.92
6.3
-
6.3
5.5
4.1
3.9
н2о
МеС
н2о
Н2О
н2о
Н2О
МеС
3.10. Мягкие лиганды для мягких ионов металлов 185
что Ag+ проявляет исключительное сродство к большинству криптандов, в том
числе и с небольшим числом атомов азота, например к C.74), даже в таком, способном
к конкуренции растворителе, как вода. В целом это можно объяснить более
инертным характером Ag+ по сравнению с К+, ковалентностью Ag—N-взаимодействий и
более низкой свободной энергией сольватации Ag+ в сочетании с пластичностью
его координационной сферы.
-N N
Me ^ ' Me
C.72) C.73) C.74)
Смешанные криптанды
Сочетание жестких и мягких донорных атомов в одном рецепторе
позволяет либо связывать порознь разнообразные ионы металлов, либо, что более
интересно, одновременно связывать мягкие и жесткие ионы металлов. Например,
смешанные 5,Ы,О-криптанды (рис. 3.41) способны связывать и мягкие катионы,
например Rh+, и катионы щелочных металлов, являющиеся кислотами Льюиса,
образуя криптаты, содержащие и редокс-активные металлические центры, и
кислотные центры Льюиса. Это может представлять интерес для супрамолекулярного
связывания и активации малых органических субстратов, в частности СО.
Присутствие СО-лиганда в C.75) указывает на возможную модель такого процесса,
в котором СО связывается с переходным металлом, имеющим низкое состояние
окисления (Дополнение 3.3) в стабильном синергетическом варианте, и
активируется путем координации с катионом щелочного металла.
186
3. Хозяева, связывающие катионы
C.75)
Катион щелочного металла
Рис. 3.41. Субстрат СО расположен
между редокс-активным переходным
металлом и катионом щелочного металла
(кислота Льюиса) в криптанде с жестким
и мягким донорными центрами
Дополнение 3.3. Синергетический эффект
Стабилизация комплексов с низким состоянием окисления обычно осуществляется
такими л-кислотными лигандами, как СО, PR3 (фосфины), алкены и др. Термин
«кислота» здесь относится к кислотности Льюиса, т.е. к способности принимать
электроны. Связывание л-кислотных лигандов происходит синергетическим путем,
включающим два компонента:
1. Перенос электронной плотности от лиганда на незаполненные р- или s-орби-
тали металла с образованием а-связи (т.е. максимум электронной плотности
расположен вдоль линии, соединяющей два атомных центра , рис. 3.42).
Незаполненная р-орбиталь металла
Молекулярная сг-орбиталь, заполненная
лигандом (неподеленная электронная пара)
Донорное связывание L-M
Рис. 3.42. Перенос электронной плотности от лиганда к незаполненным р- или
5-орбиталям металла
Заполненная J-орбиталь металла
О
Незаполненная я*-орбиталь лиганда
Рис. 3.43. Обратное связывание металл—лиганд
2. Обратное связывание металл—лиганд (рис. 3.43). Перенос электронной пгют-
ности от заполненной </-орбитали металла на незаполненную антисвязываю-
щую орбиталь лиганда/?- или d-rvina. Обратное связывание происходит в л-ва-
3.10. Мягкие лиганды для мягких ионов металлов
рианте, при котором два максимума электронной плотности связи металл—ли-
ганд расположены по разные стороны связи. Это усиливает М—L-связь, но
ослабляет связи внутри самого лиганда (например, С-О-связь в СО), так как
обратный перенос происходит на антисвязывающую орбиталь лиганда, что
уменьшает общий порядок связи.
Обратное связывание M-L
Заполненная J-орбиталь металла Незаполненная я*-орбиталь лиганда
Заполненная л*-связывающая орбиталь лиганда,
образующая донорную О-связь с незаполненной
р-орбиталью металла
Рис. 3.44. Модель связывания Д. Чатта
Об этих двух компонентах, усиливающих друг друга, говорят как о «синергетичес-
ких». Эта модель связывания соответствует принципу электроотрицательности
Полинга (который не допускает скопления заряда на каком-либо отдельном атоме),
потому что заряд, отданный неподеленной электронной парой л-кислотного лиганда,
возвращается к нему путем обратного связывания. В случае алкеновых соединений
эта модель известна как модель Д. Чатта (рис. 3.44):
1. Прямой перенос электрона с л-связывающей орбитали алкена на
незаполненную /ьорбиталь металла.
2. Обратный перенос электрона с заполненной ^-орбитали металла на
антисвязывающую орбиталь алкена.
В связи с этим в таких алкенах, как этен, наблюдаются следующие эффекты.
• Ослабление С=С-связи с последующим переносом электрона на л*-орбиталь.
Длина С=С-связи в типичном синергетическом комплексе [PtCl3(C2H4)]~
равна 1.37 А (ср. со свободным этеном, 1.34 А).
• Снижение частоты валентных колебаний. В приведенном выше примере
v(C=C) равна 1520 см (ср. с v(C=C) в свободном этене, 1623 см).
• Смещение Н-атомов на 16° от металлического центра по сравнению с их
положением в плоском этене из-за частичной регибридизации 5/?3-атомов углерода.
3.Ю.4 Основания Шиффа
Макроциклические и макробициклические основания Шиффа
необычайно распространены и играют очень важную роль в координационной химии
макроциклов, особенно в химии переходных металлов, начиная с самых истоков
супрамолекулярной химии. Реакция конденсации Шиффа — это реакция амина с
3. Хозяева, связывающие катионы
Схема 3.19. Конденсация Шиффа и восстановление продукта с образованием
амина
альдегидом, приводящая к выделению (к конденсации) воды и получению имина.
При желании продукт может быть восстановлен (например, NaBH4) до амина, в
том числе вторичного макроциклического (схема 3.19).
На рис. 3.45 показаны три исходных основания Шиффа такого типа, которые
были среди первых синтезированных макроциклических соединений. Эти соедине-
COjR
N N
Ni2+
N N
Продукт альдольной конденсации ацетона
и Ni(enK2+ (Curtis, 1961)
Продукт Ре(П)-темплатной конденсации
2,6-диацетилпириднна
с диэтилентетрамином (Busch, 1964)
Продукт темплатной реакция
Р-кетоиминатокомплексов
с этилендиамином (en)
(Jager, 1964)
COjR
Рис. 3.45. Исходные
макроциклические
основания Шиффа
ЗЛО. Мягкие лиганды для мягких ионов металлов
ния обычно получают методом термодинамического темплатирования, так как
если в процессе реакции вода не удаляется, то конденсация будет обратимой и не
позволит выделить наиболее стабильные соединения металл—продукт (см. разд. 3.8.2).
Темплатные ионы металлов можно использовать для управления размером
образующегося макроцикла. Так, малые катионы Mn2+, Fe2+ и Mg2+ могут приводить к
образованию 1:1-продуктов конденсации, как показано в схеме 3.20, а, в то время
как, например, катионы Ва2+ и РЬ2+ способствуют получению больших B:2-)
макроциклов (схема 3.20, б). Однако неудобство темплатной методики заключается в
том, что если требуется макроциклическое соединение, не содержащее металл
(например, для дальнейшего комплексообразования или как участник более сложного
синтеза), то темплатный катион, например Ni2+, нужно удалить. Для выполнения
этой процедуры был разработан ряд способов, среди которых самые
распространенные — экстракция макроциклического соединения в органический
растворитель (для таких слабокоординированных ионов, как катионы щелочных и
щелочноземельных металлов) или замена макроцикла очень сильными координирующими
лигандами, например CN~ (схема 3.21).
Однако, несмотря на эти недостатки, реакции конденсации Шиффа
исключительно многообразны и идут с высокими выходами; их применяют для получения
огромного множества металломакроциклических и -макробициклических соедине-
Схема 3.20. (а) Темплатная 1:1-циклоконденсация с участием катионов металлов
небольшого размера и (б) получение больших макроциклов с помощью больших темплатов
190
3. Хозяева, связывающие катионы
Схема 3.21. Удаление темплатного катиона Ni2+ в синтезе циклама A4(Ы43222коранд-4))
ний и для аналогов подандов с открытой цепью. Действительно, переходные
металлы взаимодействуют с азотом имина особенно сильно благодаря обратному
связыванию с антисвязывающей 71*-орбиталью C=N.
Большое количество реакций конденсации по типу конденсации Шиффа
обнаружено в химии макроциклов полипиррола. Подобная темплатная циклизация
лежит в основе синтеза фталоцианинов. Например, в синтезе суперфталоцианина
C.76), в котором пять повторяющихся блоков организованы вокруг пентагональ-
ного бипирамидального ядра UO^" вместо четырех блоков у более традиционного
фталоцианинового комплекса C.77), необходимо применять большой темплат.
C.76)
3*11 Образование комплексов
с органическими катионами
До сих пор мы обсуждали только комплексообразование
металлических катионов, стабильность которых обусловлена, в первую очередь,
кооперативными ион-дипольными взаимодействиями или взаимодействиями типа
кислота/основание Льюиса. Однако неметаллические катионы тоже могут
3.11. Образование комплексов с органическими катионами
Рис. 3.46. Тетраэдрическое распознавание катиона
NH^" криптандом «футбольный мяч» C.29)
взаимодействовать с корандами и криптандами, как и происходит в случае тетра-
эдрического распознавания аммониевого катиона (NH4), криптандом
«футбольный мяч» C.29), рис. 3.46. В то время как это распознавание происходит только с
помощью водородной связи (типа N+—H--N), воздействие на ион аммония таково,
что величина рКа увеличивается на 6 единиц; таким образом, депротонирование
криптата в 1 млн раз менее вероятно, чем депротонирование его некомплексного
аналога.
Значительный интерес представляет также связывание органических катионов,
в частности производных аммония, с помощью водородной связи, а также за счет
электростатических взаимодействий, так как эти, более сложные, катионы могут
быть хиральными. Таким образом, возникает вопрос: может ли хиральный хозяин
отличить один энантиомер от другого!
» 11.1 Связывание катионов аммония корандами
18-Краун-6 и его N-гетероаналоги связывают катионы аммония и ал-
киламмония с помощью трех водородных связей N+—H-O, давая комплекс типа
«насест» (рис. 3.47), хотя макроцикл с чисто кислородными донорами связывает К+
сильнее, что вызвано относительно жестким характером эфирных атомов
кислорода. Аналогичный триазакоранд 18(O3N326Kopann-6) C.43) образует стерически
почти идентичную триаду водородных связей N+—H---N и проявляет селективность
к ионам ал кил аммония по сравнению с К+, что может быть в одинаковой степени
результатом уменьшения сродства к К+ и увеличения сродства к производным
аммония.
В табл. 3.11 даны свободные энергии комплексообразования для различных
ионов аммония с тремя корандами-рецепторами (хозяева). Сразу же очевидно, что
^ связан сильнее, чем любой из его алкильных аналогов, а метиламмоний все-
Рис. 3.47. Геометрия
типа «насест» в комплексах
катиона алкиламмония с
азакорандами типа C.43),
стабилизированных
водородными связями, которые
усиливают электрический
заряд
A R
,Ме
^ ч н'
Водородная связь О Н О
Ион-дипольное взаимодействие
192
3. Хозяева, связывающие катионы
Таблица 3.11. Свободные энергии комплексообразова-
ния корандных хозяев с ионами аммония
A ккал = 4.184 кДж)
Хозяин
C.78)
C.79)
C.80)
-AG0, ккалмоль
NH+
10.5
9.5
8.9
CH3NH+
9.0
7.5
6.9
(CH3KCNH+
8.3
6.9
6.4
гда связан сильнее, чем mpe/w-бутиламмоний, причем особенно большая разница
существует между комплексами NHJ и CH3NH^" с C.79) и C.80) — 2 ккал-моль.
Это наводит на мысль о важности стерических эффектов для всех хозяев. В
частности, у алкиламмониевых комплексов C.80) найдены низкие свободные энергии
связывания, что объясняют неблагоприятными стерическими взаимодействиями
между метильными группами хозяина и гостя. Согласно кристаллической
структуре комплекса C.80)-(CH3KCNH^, эти метильные группы соприкасаются друг с
другом в положениях, соответствующих положениям часовой стрелки — 7 и 12 ч
(рис. 3.48).
Г~\
О О—\
N О
C.79)
C.78)
C.80)
Рис. 3.48. Кристаллическая структура
комплекса C.80) (CH3KCNH+,
демонстрирующая неблагоприятные
стерические взаимодействия между
метильными группами хозяина и гостя.
(Воспроизведено с разрешения Cram D., 1986)
3.11. Образование комплексов с органическими катионами
Латеральное распознавание
193
Центральное распознавание
Рис. 3.49. Латеральное
и центральное распознавание
X = С02": C.81а)
X = CONYY': C.816)
При введении в молекулу 18-краун-6 отрицательно заряженных групп,
например карбоксигрупп, связывание катиона алкиламмония усиливается за счет
укрепления сетки водородных связей электростатическим притяжением между NH-^-oc-
татком и анионным С(О)—О~-фрагментом. Такие рецепторы, как C.81а), сильно
связываются с первичными солями аммония и, действительно, более селективны к
ним по сравнению со вторичными и третичными солями. Это явление называют
центральным распознаванием (рис. 3.49), так как оно включает селективность,
возникающую благодаря центральному ядру корандного макроцикла, и позволяет
распознавать биологически важные первичные соли аммония, например норадрена-
лин и норэфедрин на фоне их N-метильных аналогов. Изменение боковых цепей X
приводит к появлению ряда таких рецепторов, которые могут быть настроены на
селективное связывание именно первичных ионов аммония благодаря
специфическим взаимодействиям (водородные связи, электростатические, п—л-стэкинг-,
ван-дер-ваальсовы взаимодействия и т.д.) с заместителями в группе R гостя. Этот
вид связывания называют латеральным распознаванием (см. рис. 3.49), которое в
хорошо сконструированном хозяине комплементарно центральному распознаванию.
В случае же вторичных ионов аммония R2NH2 триподальный мотив типа
«насест», наблюдаемый выше, не возможен. Однако для таких малых корандов, как
12(N2O224KopaHfl-4) (диаза-12-краун-4), наблюдается связывание через две
водородные связи N+-H--N.
Увеличение размера рецептора способствует комплексообразованию больших
катионов аммония, например ионов гуанидиния (С(ЫН2)з") и имидазолия
(N2C3H^). Следует заметить, что в 18-членных корандах центральный
связывающий участок способен принять симметрию третьего порядка, комплементарную
симметрии псевдотретьего порядка RNHJ-группы. Таким образом, рецептор
комплементарен гостю по симметрии. Точно так же большие хозяева — соединения на
основе 27-краун-9, в частности C.82), - комплементарны гуанидинию, имеющему
симметрию третьего порядка.
13 - СТИДДж В
3. Хозяева, связывающие катионы
H H
i i
СК N -NL
-н Х+ н
н н
о
RC R
R = СО2" C.82)
g. 11.2 Связывание катионов аммония
трехмерными хозяевами
Как и в случае связывания катионов металлов, трехмерные бицикли-
ческие хозяева связывают катионы алкиламмония сильнее, чем их корандные
аналоги. Свободные энергии связывания ряда трехмерных хозяев с гостями — ионами
аммония и алкиламмония - представлены в табл. 3.12. В частности, очень сильное
комплексообразование криптасферанда C.38) с NH| свидетельствует о капсуляр-
ном типе его комплекса с ориентацией протонов катиона NH^ по направлению к
атомам азота криптанднои части молекулы и к анизильным атомам кислорода. В
соответствии с этим кристаллические структуры К+-и С8+-криптасфераплексов
соединения C.38) с близкими свободными энергиями связывания также имеют кап-
сулярную природу. Гораздо меньшие свободные энергии полусфераплексов
соединения C.83) не предполагают их капсулярность; действительно, их молекулярные
СРК-модели свидетельствуют о напряженной структуре типа «гнездо». Еще более
низким свободным энергиям образования 1^Н;}"-комплексов соединений C.84) и
C.85) соответствуют модели типа «насест».
Таблица 3.12. Свободные энергии комплексообразова-
ния рецепторов сферандного типа с ионами аммония
(по Cram D., 1986)
Хозяин
C.38)
C.64)
C.83)
C.84)
C.33)
C.85)
NH+
20.2
14.4
12.7
9.2
9.9
7.0
-AG0, ккалмоль
CH3NH+
_
14.4
11.9
8.4
8.2
7.4
(CH^CNH^
_
13.2
11.7
8.3
7.7
8.6
3.11. Образование комплексов с органическими катионами
195
■•''/ °
C.83)
C.84)
C.85)
3.II.3 Дитопные рецепторы
Дитопные рецепторы — это рецепторы, обладающие двумя центрами
связывания гостей. Хозяева с тремя и более центрами связывания называют
политопными рецепторами. По существу, сродство дитопного хозяина к
бифункциональному гостю должно быть больше (по крайней мере в 2 раза), чем сродство мо-
нотопного аналога, из-за того, что происходят два или более случаев
распознавания. Кроме того, эффективность селективного молекулярного распознавания
можно сильно увеличить за счет размещения и разнесения центров связывания
рецептора. При этом дитопные рецепторы могут проявлять макрохелатный эффект,
при котором многоточечное (например, макроциклическое) связывание более чем
на одном участке (например, в кольце коранда) больше суммы отдельных
взаимодействий.
В 1980 г. Лен синтезировал дитопные хозяева C.86), способные к линейному
молекулярному распознаванию катионов диаммония в соответствии с длиной спей-
серной группы гостя. 'Н ЯМР-измерения показывают, что меньший хозяин C.86а)
проявляет меньшую селективность к гостям NH^CH^NH^. Однако при
связывании гостей с длиной цепи соответственно п = 5, 7 и 10 (рис. 3.50) хозяева C.866),
C.86в) и C.86г) вызывают максимальные изменения химического сдвига протонов
цепи гостя по сравнению с аналогами, имеющими как более длинные, так и более
короткие цепи. При нахождении гостя внутри молекулярной полости ожидаются
3. Хозяева, связывающие катионы
7 9 11
Длина цепи (я)
Рис. 3.50. Зависимость
наибольшего сдвига в сторону сильного поля
(Дб) от длины цепи гостя (п) для
хозяев C.86а)-C.86г)
большие изменения химического сдвига, поскольку в этом случае алифатическая
цепь гостя попадает в область экранирования кольцевого тока, генерируемого
ароматическими спейсерными группами R (Дополнение 3.4).
С0 --°-л
NtH ^
1^
OHOHQ
C.86)
Особенно интересные результаты были получены при измерении времен
релаксации ЯМР хозяина и гостя. Эти результаты показали, что у стерически
комплементарных пар хозяин—гость очень сходные движения молекул в растворе (сильное
динамическое спаривание) и они значительно более медленные, чем у свободного
хозяина. Как только соответствие хозяин—гость становится менее
комплементарным, движения становятся анизодинамическими, т.е. движения гостя ускоряются,
а движения хозяина остаются почти неизменными. В случаях особенно плохого
соответствия (например, C.86г), п = 8) движения хозяина еще более замедляются ,
тогда как скорость движений гостя увеличивается. Это объясняется тем, что через
одну группу гостя NH^ и одно корандное кольцо хозяина осуществляется
связывание, внешнее по отношению к полости второго кольца. При этом молекулярная
масса хозяина заметно увеличивается, замедляя его движение. Гость, находящийся
во внешней полости, обладает свободой движений.
3.11. Образование комплексов с органическими катионами
Й97
Дополнение 3.4. ЯМР-спектроскопия супрамолекулярных соединений
Обсуждение основ метода ЯМР находится за пределами тематики этой книги, их
можно найти в ключевых ссылках. Однако особый интерес для специалистов в
области супрамолекулярной химии представляют один-два аспекта ЯМР, в том числе и
необходимость иметь методику для определения свойств новых химических
соединений. В Дополнении 1.1 мы уже познакомились с методикой ЯМР-титрования для
измерения констант связывания хозяин—гость в разбавленных растворах. Однако
ЯМР-спектроскопия может предоставить гораздо большую информацию, чем
величины констант связывания.
*"* Friebolin H. Basic one- and two-dimensional NMR spectroscopy. 2nd ed. Weinheim:
VCH, 1993.
Эффекты кольцевого тока ароматического кольца
Даже при простом сравнении спектров гостя в присутствии и в отсутствие хозяина
можно обнаружить некоторые весьма необычные черты, в особенности в тех случаях,
когда или хозяин, или гость содержит ароматическое кольцо. В производных бензола
или в таких высших ароматических соединениях, как порфирин, делокализованные
я-электроны в присутствии магнитного поля представляют собой циркулирующий
заряд, который, согласно электромагнитной теории, создает магнитное поле,
противоположное полю, приложенному извне, BQ. Это приводит к эффективному дезэкра-
нированию С—Н-протонов бензольных соединений, поскольку эти протоны
размещаются в области индуцированного поля, совпадающего по направлению с BQ.
Однако гость, находясь внутри ароматического хозяина (обычно циклофан, гл. 5),
часто располагается прямо над арильным кольцом в области экранирования, что
вызывает очень большие изменения химического сдвига в сторону более высокого поля
(т.е. меньшего химического сдвига, рис. 3.51. Степень, до какой ядра различных
гостей подверглись воздействию, может дать качественную информацию об
относительной ориентации хозяина и гостя, а также о региоселективности их связывания.
Область экранирования
Beffect = Bo~Bi
Индуцированное поле, В{
Область дезэкранирования
В effect = B0 + &i
Рис. 3.51. Эффекты кольцевого тока ароматического кольца
198 3. Хозяева, связывающие катионы
Ядерный эффект Оверхаузера
Больше количественной информации о соседстве хозяин—гость можно получить,
используя ядерный эффект Оверхаузера (nuclear Overhauser effect, NOE). NOE
заключается в насыщении спина одного ядра путем непрерывного облучения и мониторинге
возникающего усиления сигнала ЯМР-резонанса близлежащих атомов. Идея состоит
в том, что облучение одного ядра вызывает его возбуждение до неравновесного
распределения спиновых состояний. Релаксация этого возбужденного состояния
происходит путем диполь-дипольного спин-решеточного переноса избытка энергии и
приводит к усилению интенсивности сигналов от ядер, физически близких к
облученному ядру, независимо оттого, связаны ли они на самом деле или нет.
Исходная интенсивность NOE может усиливаться в пределах от 0 до 200%, и поэтому
эффект NOE — хороший индикатор пространственной близости ядер. Например, ядра
На и Нь полуклатратного соединения C.87) очень близки друг к другу. Если
резонансный уровень На насыщен, то интенсивность сигнала от Hh возрастает на 45%, тогда
как более удаленные протоны не испытывают существенного воздействия. С
помощью этого эффекта можно с достаточной степенью уверенности описать
стереохимию молекулы. Пример для истинной системы хозяин—гость описан в разд. 3.13.1.
C.87)
Спин-спиновое взаимодействие
В ЯМР спин-спиновое взаимодействие — результат влияния близлежащих ядер на
эффективное магнитное поле отдельного ядра. Вообще эти взаимодействия очень
полезны с точки зрения той информации, которую они дают о связи. Из-за того, что в
супрамолекулярных соединениях частицы хозяина и гостя не связаны между собой
ковалентно, не следует ожидать реализации спин-спиновых взаимодействий между
ними. Однако если взаимодействие между хозяином и гостем приводит к обобщению
электронной плотности, то может наблюдаться расщепление сигналов хозяина в
результате спин-спинового взаимодействия с гостем и наоборот. Это особенно заметно
у ядер, испытывающих сильное взаимодействие, например у 19F.
Динамика обмена гостями
В случаях, когда энергия связывания гостя имеет подходящее значение
(-20—100 кДжмоль), с помощью метода коалесценции можно наблюдать обмен
между свободным и связанным гостями. Если в соответствии со шкалой времен ЯМР
обмен гостями кинетически замедлен (время жизни комплекса > ~10~2—10~3 с), то
для связанного и свободного гостей наблюдаются отдельные пики. Рост температуры
3.11. Образование комплексов с органическими катионами
Рис. 3.52. (а) Механизм
обмена метальных групп в DMF.
(б) 'Н ЯМР-сигналы метальных
протонов DMF, записанные
при разных температурах.
Медленный обмен при низкой
температуре дает два пика. Пики
уширяются и сливаются при
повышении температуры до
температуры коалесценции
120 °С. (Воспроизведено с
разрешения Friebolin H. Basic one-
and two-dimensional NMR spec-
troscopy. 2nd ed. Weinheim: VCH,
1993)
(a)
Me ^ О
Ж
Me
2
2
Me
Me
1
F)
22.5 °C
9.5 Гц
99-100 °C
8.6 Гц
117°C
6.5 Гц
120-120.5 °C
3.1Гц
124 °C
107-108°C
8.2 Гц
110-111
7.6 Гц
124-128 °C
151-152°C
ЯМР-пробы вызывает увеличение скорости обмена, что выражается в уширении
резонансных полос для двух различных образцов на спектре ЯМ Р. Когда скорость
обмена становится сопоставимой с частотой, разделяющей два сигнала, происходит коа-
лесценция пиков, т.е. две резонансные полосы сливаются, давая один широкий пик
(в предположении, что спин-спиновое взаимодействие отсутствует). После этой
точки (температура коалесценции Тс) дальнейший нагрев приводит к сужению пика.
При Тс скорость обмена гостями описывается уравнением C.5):
kc = n5v/{T, C.5)
где 8v - резонансное разделение частот между пиками в условиях медленного обмена.
Если при комнатной температуре не наблюдаются отдельные пики для
связанного и свободного гостей, то иногда можно ощутимо замедлить обмен гостями, понижая
200
3. Хозяева, связывающие катионы
температуру ЯМР-пробы. Температуры до —90 °С можно достичь в обычных дейтери-
рованных растворителях, например в CD2C12 или CO(CD3J, а в особых случаях еще
более низкие температуры могут быть получены при использовании фреонов,
например CF2C12.
Если измерить скорость обмена гостями, то по уравнению Эйринга C.6) можно
определить свободную энергию активации комплексообразования AG*.
kc = xkBTe-AG'/RT/h, C.6)
где х — коэффициент пропускания (обычно равный единице), къ — константа Больц-
мана A.3805-10"3 ДжК), Т - температура (в градусах Кельвина), И - постоянная
Планка F.625-10~34 Дж-с).
На рис. 3.52 в качестве очень простого приложения этого метода рассмотрена ме-
тильная область ЯМР-спектра Ы,Ы-диметилформамида (DMF) при различных
температурах. Коалесценция двух метильных групп вызвана постепенным ростом
скорости вращения вокруг С—N-связи, которая отчасти имеет характер двойной связи. Это
как раз может соответствовать легкому сдвигу DMF-гостя внутри несимметричного
карцерандного хозяина (разд. 5.3.6.3). В этом случае температура коалесценции равна
120 °С C93 К), а разница между частотами линий составляет 9.5 Гц (что соответствует
очень малой разнице химических сдвигов при 60 МГц); следовательно, используя
уравнение C.5), мы получим кс = 21 с. Подстановка этого значения в уравнение C.6)
дает AGj93 = 87.5 кДж-моль. Обычно ошибка равна ±1 кДж-моль.
3» 11 »4 Хиральное распознавание
Энантиоспецифическое связывание рецептор—субстрат является
крайне важным в биохимических системах. В результате этого большой интерес
представляют как использование хиральных супрамолекулярных соединений в
качестве имитаторов и моделей ферментов (гл. 9), так и абиотический хиральный
катализ, который в итоге может, например, найти применение в синтезе или
разделении хиральных фармацевтических препаратов. Еще в 1978 г. С. Пикок и другие
(S. Peacock et al.) сконструировали и получили хиральный коранд C.88). Несмотря
на то, что C.88) не имеет асимметрических атомов углерода, он хирален благодаря
взаимной ориентации двух бинафталиновых (binap) фрагментов. Наличие двух
метильных заместителей препятствует вращению вокруг связи между двумя
нафталиновыми группами елевой стороны хозяина, приводя к скрученной (твист-)конфор-
мации, называемой хиральным барьером (рис. 3.53). Строение комплекса Д,/?-C.88)
таково, что он обладает осью симметрии С,, проходящей горизонтально через две
связи между парами нафталиновых остатков. Это делает обе связывающие
поверхности эквивалентными (т.е. не различающимися по отношению к триподальным
гостям типа «насест»). Следовательно, хиральное распознавание должно
происходить независимо от того, какой стороной хозяин связывает гостя.
Способность соединения C.88) к хиральному распознаванию оценивали в
экспериментах по жидкость-жидкостной двухфазной экстракции рацемических
3.11. Образование комплексов с органическими катионами
201
Хиральный барьер
C.88)
Рис. 3.53. Пример хирального барьера
смесей аминокислотных и аминоэфирных гостей. Крам и сотрудники обнаружили,
что в каждом случае эти эксперименты приводили к селективной экстракции
£>-энантиомеров. Количественной характеристикой процесса служил фактор
хирального распознавания (отношение концентрации £-энантиомера к
концентрации L-энантиомера, экстрагированных в органическую фазу), который изменялся
от большого значения, 31, для C6H5CH(CO2Me)NH^ до малого, 2.3, для
C6H5CH(CO2H)NH^" (протонированная форма фенилглицина — синтетической
аминокислоты). Хиральное распознавание основано на неблагоприятных стериче-
ских взаимодействиях между объемистыми заместителями гостя и выступающими
метальными заместителями хозяина (рис. 3.54).
Для последующего исследования хиральной способности хозяев, Я,Я-C.88) и
его З^-энантиомера, к распознаванию было создано устройство для разделения
энантиомеров, изображенное на рис. 3.55. Основой прибора является W-образная
трубка с раствором хозяина в хлороформе на дне. З^-Энантиомер находится в
левой части трубки, а /?,/?-энантиомер — в правой. Центральный резервуар содержит
водный раствор рацемата гостя, который перед началом эксперимента
механически перемешивается. На каждой из фазовых границ вода—хлороформ, левой и
правой, происходит энантиоселективное связывание; при этом D-гость переносится из
хлороформа в водную фазу в правой части трубки, а /,-гость — в левой части.
Градиент концентрации поддерживают, непрерывно удаляя 86-90% энантиомерно
чистых гостей из водных слоев обеих частей трубки, одновременно добавляя в
центральный резервуар свежий рацемат.
,k
Рис. 3.54. Селективное связывание £-энантиомера катиона фенилглицината корандом
C.88). В L-энантиомере карбоксигруппа гостя находится в близком соседстве с метальным
заместителем хозяина
3. Хозяева, связывающие катионы
L-гость,
переносимый
вН2О
5,5-хозяин
и комплекс
всна3
£>-гость,
переносимый
вН2О
Й.Л-хозяин
и комплекс
в СНС13
Рис. 3.55. Устройство для разделения энантиомеров. (Перепечатано с разрешения
Newcomb M. et al., 1979)
Достигнув этого успеха, группа Крама продолжила конструирование первой хи-
ральной хроматографической колонки, в которой /?,/?-энантиомер хозяина C.88)
прививали к поверхности полимерной подложки через одну из нафталиновых
групп. Эта хиральная неподвижная фаза была использована в качестве твердого
носителя, через который пропускали растворы рацематов хиральных гостей.
Благодаря более сильному связыванию £-энантиомера, чем L-гостя, L-форма проходила
через колонку быстрее и ее можно было собирать отдельно от /)-формы. В
соответствии с приведенными выше результатами экспериментов по жидкость-жидкост-
3.11. Образование комплексов с органическими катионами ШОЗ
ной экстракции факторы разделения, полученные таким способом, изменялись в
интервале от 26 до 1.4.
Сейчас эту технику широко используют для оценки энантиомерных избытков в
органических реакциях и разделения малых количеств энантиомеров. Хиральные
коранды также весьма полезны при оценке оптической чистоты аминокислот, хотя
цена современных хиральных колонок для HPLC (высокоэффективная жидкостная
хроматография, high pressure liquid chromatography) может превышать 1500 дол.
США.
3« 11 «5 Дифильные рецепторы
Рецептор, совмещающий две или более функции распознавания гостя
и тем самым обеспечивающий синергетическое усиление связывания, называют
дифильным'. Дифильные рецепторы, в молекулах которых сочетаются полярные
или заряженные участки с гидрофобным остатком (этот остаток защищает
полярные участки от сольватации и усиливает электростатическое притяжение), носят
название спелеанды, а их криптатные комплексы - спелеаты. Один такой спелеат,
C.89), содержит корандную 18-членную полярную связывающую группу N3O3 и
гидрофобную полость производного циклотривератрилена (CTV). Обычно
макроцикл CTV осуществляет включение нейтральных гостей (разд. 5.2.5), но в данном
случае он обеспечивает гидрофобное распознавание метильной группы катиона
CH3NH^, тогда как фрагмент коранда образует водородные связи с триподальной
группой NH} (J. Canceill et al., 1982).
0@H
МеО
СО,
< Н'Гн Me V J
Ч/^Ь" ) = о. о
о.
О"
0\/ N
К S
C.89)
СО,
C.90)
CO,
СО,
В состав другого спелеата, C.90), (М. Dhaenens et al., 1984) входит коранд
большей величины, содержащий кислородные донорные центры, подандоподобные
анионные карбоксигруппы и гидрофобную область, образованную четырьмя
центральными арильными кольцами. Здесь налицо концептуальное сходство с
соединением C.81). В воде, при нейтральном рН, C.90) образует комплексы со многими
органическими солями аммония. Тот факт, что гидрофобная связывающая полость
* В последние годы в русскоязычной научной литературе часто вместо термина «дифиль-
ный» неправомерно используют термин «амфифильный» — англ. «amphiphilic». (Примеч. ред.
перевода.)
204
3. Хозяева, связывающие катионы
находится в области экранирования ароматических колец хозяина, приводит к
большому сдвигу 'Н ЯМР-линий гостя в сторону возрастания поля. В частности,
C.90) — эффективный рецептор для ацетилхолинового нейротрансмиттера.
Интерес к этому гостю возникает в связи с естественным желанием как можно больше
узнать о передаче импульсов путем моделирования взаимодействия
нейротрансмиттера с природным нейрорецептором. Спелеат C.90), подобно более
простому аналогу — дибензо[30]краун-10, тоже связывает дикатион N,N '-диметил-4,4'-
бипиридила (метилвиологен) с константой связывания, превышающей 105 М.
Частный случай: рецепторы гербицидов
Благодаря тому, что редокс-активные ионы аммония дикват C.91) и
паракват (метилвиологен) C.92) являются переносчиками электронов, они
представляют особый интерес как гости, используемые для электронного транспорта,
накопления фотохимической энергии и практического применения в качестве
гербицидов.
C.91)
NH3
I
Me C.92)
C.93)
Мы уже видели (см. разд. 3.7.2), как конформационно гибкий дибензо-30-кра-
ун-10 C.47) может «укутывать» такой ион, как К+, образуя 1:1-комплекс. В случае
диквата C.91) также наблюдается U-образная «укутывающая» конформация,
позволяющая крауну выступать в качестве дифильного рецептора, эфирные атомы
кислорода которого участвуют в ион-дипольных взаимодействиях с четвертичными
атомами азота диквата, а также в донорно-акцепторных я-я-стэкинг-взаимодейст-
виях, в которые вовлекаются электроноизбыточные бензольные заместители C.47)
и электронодефицитные ароматические кольца диквата. Слабые водородные связи
С—Н---О, в которых участвуют углеводородные группы, ближайшие к заряженным
атомам азота, заметно стабилизируют комплекс. Интересно, что весьма похожий
комплекс дибензо-30-краун-10 C.47) образует и с катионом C.93), в котором
водородные связи N—H---0 от амминных лигандов усиливают ион-дипольную и п—п-
стэкинг-стабилизацию. В самом деле, краун-эфир действует как лиганд второй
сферы для Р1(П)-центра, почти так же, как в случае, приведенном на рис. 3.40, а.
Кристаллическая структура этого РЦЩ-комплекса представлена на рис. 3.56.
Новая конформация C.47), показанная на рис. 3.56, привела к синтезу более
предорганизованных криптандных хозяев-комплексообразователей со сходными
3.11. Образование комплексов с органическими катионами
205
Рис. 3.56. Кристаллическая структура
комплекса катиона бипиридилдиаммин-
платины(П) C.93) с корандом C.47)
свойствами. Хозяин C.94) - производное краун-эфира C.47), полностью предорга-
низованный для связывания диквата, образуется благодаря встраиванию в C.47)
третьего, пирокатехинового, мостика. За счет предорганизации (рис. 3.57) и
трехмерной криптандной природы соединения C.94) его константа связывания для
диквата (ацетон, 25 °С), превышает 9105 М; для C.47) эта константа составляет
1.7-104 М-1.
C.94)
C.95)
/f\ /^\ /\
О 0 0 0
C.96)
В противоположность диквату, паракват C.92) из-за большего расстояния
между двумя его 1^+-центрами, не комплементарен таким хозяевам, как C.45) и C.94),
2Q5 3. Хозяева, связывающие катионы
Рис. 3.57. Кристаллические структуры
свободного C.94) и его комплекса с
дикватом, демонстрирующие
предорганизацию хозяина при связывании дик-
вата. (Воспроизведено с разрешения
AllwoodB.L, Kohnke F.H., StoddartJ.F.,
Williams D.J. A macrobicyclic receptor,
molecule for the diquat dication // Angew.
Chem., Int. Ed.Engl. 1985. Vol.24. P.581)
и не связывается ими. Однако его связывают краун-эфиры C.95) и C.96). В первом
случае образуется красный 1:1-комплекс (К= 730 М, ацетон, 25 °С).
Интенсивный цвет возникает из-за поглощения при 436 нм за счет переноса заряда. Данные
РСА-исследования вновь указывают на предорганизацию хозяина C.95) для гостя
C.92). В случае хозяина C.96) образуется 2:1-комплекс с катионами гостя как
внутри, так и снаружи кольца хозяина, вовлеченными в л-я-стэкинг-взаимодействия
при переносе заряда. Химия переноса заряда была широко использована при
конструировании сцепленных молекул, называемых катенанами, в которых одно
кольцо корандного типа продевается через другое, давая супрамолекулярные
соединения, которые могут быть разъединены только путем разрыва ковалентных связей.
Синтез катенанов и соответствующая химия описаны в гл. 7.
*"» Colquhoun #., Stoddart К, Williams D. Chemistry beyond the molecule // New
Scientist. 1986. May 1. P.44-48.
Алкалиды и электриды
Еще в 1969 г. Ч. Педерсен был заинтригован интенсивно синим
цветом, наблюдавшимся при растворении малых количеств металлического натрия и
калия в координирующих органических растворителях в присутствии краун-
эфиров. На самом деле началом истории химии растворения щелочных металлов
(в противоположность катионам металлов) можно считать заметку, которую Г.
Дэви (Н. Davy) сделал в записной книжке в 1808 г., касающуюся синего или
бронзового цвета растворов калия в жидком аммиаке. Синий цвет объясняют присутствием
в растворе сольватированной формы свободных электронов. Его также наблюдают
при растворении металлического натрия в жидком аммиаке: такой раствор
является реагентом для восстановления аренов растворяющимися металлами, например,
для их селективного восстановления до 1,4-диенов (восстановление по Бёрчу). Для
этих целей сейчас широко используют растворы щелочных металлов в эфирных
растворителях в присутствии краун-эфиров и криптандов. Однако только в 1983 г.,
Дж. Дай (J. Dye) и его сотрудники из Мичиганского университета (США), впервые
получив кристаллическую структуру электридной соли (катион с электроном в
качестве противоиона), охарактеризовали это удивительное соединение. Было показано,
что высокочувствительное к воздуху и влаге вещество [CsA8-KpayH-6J]+e~ содержит
3.12. Алкалиды и электриды
(а)
(б)
Рис. 3.58. (а) Кристаллическая
структура [CsA8-KpayH-6J]+ e~,
первого электридного
комплекса, (б) полости и каналы
структуры в плоскости а-Ь.
(Перепечатано с разрешения AtwoodJ.L.,
Davies J.E.D., MacNicol D.D.,
Vogtle F. Comprehensive supra-
molecular chemistry. Oxford:
Pergamon, 1996. Vol.1. P.500)
катион Cs+, расположенный между двумя 18-краун-6-хозяевами. Электрон попадает
в ловушку внутри почти сферических полостей решетки диаметром -2.4 А с
наименьшими расстояниями между электронами 8.68 А. Из-за этого большого
расстояния твердое тело не является электропроводником (проводимость
-Ю-10 Ом-'см при 200К, рис. 3.58).
Поскольку РСА-исследования не позволяют наблюдать отдельный электрон
(см. Дополнение 2.1), то остается вопрос о возможности его расположения на таком
расстоянии от катиона Cs+. Если это, действительно, так, то мы имеем дело с
предельным случаем эффекта «обнаженного» аниона (см. разд. 3.6.2). Согласно
альтернативному объяснению, электрон локализуется на катионе Cs+, что также
соответствует наблюдаемой низкой проводимости. Однако почти изоструктурные аналоги
соединения [CsA8-KpayH-6J]+-e~ - содид (Na~) и калид (Кг) - представляют
собой убедительное свидетельство разделения катиона и электрона. В них анионы
щелочных металлов располагаются в тех же локализованных полостях, что и в их
электридных аналогах.
203 3. Хозяева, связывающие катионы
Рис. 3.59. Канальная структура криптандного
электрида [К([2.2.2]криптанд)]+е~.
(Перепечатано с разрешения Atwood J.L., Davies J.E.D.,
MacNicol D.D., Vogtle F. Comprehensive supramole-
cular chemistry. Oxford: Pergamon, 1996. Vol.1.
P.505)
Анионы щелочных металлов и электридные комплексы образуются при
растворении и кристаллизации щелочного металла и краун-эфира или криптанда в
неполярных, но координирующих растворителях, таких, как тетрагидрофуран (THF)
или диметиловый эфир. Из-за крайней чувствительности продукта единственным
условием его получения является строжайшее исключение воздуха, влаги,
протонных растворителей и любого рода загрязнений или примесей. Содид
[№([2.2.2]криптанд)]+^а~ стабилен при комнатной температуре несколько часов,
тогда как электрид [Ы([2.2.2]криптанд)]+-е~ быстро разлагается при температуре
выше 230 К.
Более интересный электрид, [К([2.2.2]криптанд)]+-е~, образуется в ходе реакции
[2.2.2]криптанда с металлическим калием в диэтиловом эфире при —50 °С. На
основании кристаллической структуры этого соединения можно сделать вывод о том,
что между катионами [К([2.2.2]криптанд)]+ располагаются большие гантелеобраз-
ные полости, в которых электронные пары находятся на расстоянии друг от друга
5.3 А. Каждая пара полостей соединена с соседними полостями каналами длиной
7.8 и 8.4 А (рис. 3.59). Были получены структуры алкалидных анионных аналогов
[К([2.2.2]криптанд)]+М~ (J. Dye, 1990) и было показано, что анионы щелочных
металлов также занимают полости, предназначенные для электронов, с небольшим
расстоянием Кг-К~, равным 4.90 А, которое увеличивается до 6.38 А в цезидном
(Cs~) аналоге, что предполагает существование удивительных димеров Ы\~.
Благодаря такой природе своих полостей [К([2.2.2]криптанд)]+е~ имеет значительную
(хотя и не металлическую) мольную проводимость, -10 Ом-см, гораздо
большую, чем у его выделенного аналога [CsA8-KpayH-6J]+-e~. Это объясняет влияние
топологической структуры катиона-хозяина и его кристаллического каркаса на
электрические и магнитные свойства электронных гостей. Способность макроцик-
лических хозяев изолировать катионы щелочных металлов внутри глубокой липо-
фильной оболочки, таким образом создавая стерический барьер для рекомбинации
катиона и электрона, составляет основу для формирования этих интересных
соединений. Полная свободная энергия комплексообразования соединения органичес-
3.13. Каликсарены 209?^
кий хозяин — катион щелочного металла должна превышать энергию ионизации (к
счастью, низкую) атома этого металла.
•""» Dye J.L., Wagner M.J., Overney G. et al. Cavities and channels in electrides //
J. Amer. Chem. Soc. 1996. Vol.118. P.7329-7336.
3-13 Каликсарены
*""» Gutsche CD. Calixarenes. Cambridge: Royal Society of Chemistry, 1989.
•""» Gutsche CD. Calixarenes 2. Cambridge: Royal Society of Chemistry, 1997.
Каликсарены представляют собой множество макроциклических
соединений, образующихся при конденсации пара-замещенного фенола (например,
я-трет-бутилфенола) с формальдегидом. Наличие в них мостиковых
ароматических колец позволяет формально отнести их к семейству циклофанов (разд. 5.3.4). По
номенклатуре циклофанов их называют замещенными [1.1.1]метациклофанами.
Из-за сходства чашеобразной конформации небольших каликсаренов с греческой
вазой, которая носит название чашевидный кратер (рис. 3.60), К. Гутше (С. Gutsche) из
Вашингтонского университета (США) предложил называть их каликсаренами.
Число в квадратных скобках означает число фенольных остатков. Таким образом,
наиболее распространенный циклический тетрамер с пара-трет-Ъутшьнымн
заместителями называют я-трет-бутилкаликс[4]ареном C.97). Если сравнить это название
с систематическим названием соединения C.97) в «Chemical Abstracts»:
[19.3.1.13-719>13115'19]октакоза-1B5),3,5,7B8),9,11,13B7),15,17,19B6),21,23-додекан,
то легко понять, почему упрощенная номенклатура получила такое широкое
распространение. Обзор истории развития химии каликсаренов дан в Дополнении 3.5.
Рис. 3.60. л-/и/?е/и-Бутилкаликс[4]арен C.97)
по форме напоминает вазу «чашевидный кратер» 1 C.97)
2IQ 3. Хозяева, связывающие катионы
Дополнение 3.5. История развития химии каликсаренов
Еще в 1872 г. была проведена реакция между фенолом и формальдегидом, которая
привела к образованию различных смоло- и цементоподобных материалов, однако с
помощью примитивных методов исследования того времени (в основном элементный
анализ) определить состав продуктов не удалось. В 1902 г. Л. Бакеланд (L. Bakeland),
одаренный и, к тому же, весьма состоятельный химик, обратил внимание на эту
реакцию в надежде найти коммерческое применение получающейся при этом твердой и
труднообрабатываемой фенол-формальдегидной смоле. Проводя реакцию в
присутствии тщательно контролируемых количеств основания, Бакеланд сумел получить
гораздо более гомогенный и привлекательный материал, названный им бакелитом. Как
предполагалось, этот материал представлял собой полимер, состоящий из сильно
сшитых фенольных фрагментов, соединенных —СН2- и — С Н2ОСН2-мостикам и как в
орто-, так и в ядро-положениях относительно фенольных гидроксильных групп в
исходных фенолах. 18 февраля 1907 г. Бакеланд запатентовал свой процесс, открыв
таким образом эпоху синтетических пластмасс. Начался бурный рост использования
бакелитовых пластмасс: формованные бакелитовые изделия широко применяли чуть ли
не во всех отраслях промышленности, пока совсем недавно их не заменили новые
материалы — продукты нефтехимического синтеза. Однако, несмотря на огромный
объем исследований по бакелитовым материалам, часто их точный химический состав до
сих пор остается неизвестным.
В 1942 г. А. Цинке (A. Zinke) из Университета Граца (Австрия) при изучении
процесса получения бакелита решил упростить реакцию и провел конденсацию пара-за-
мещенных фенолов, в частности п~трет-бугшфенола, с формальдегидом. Цинке
удалось выделить кристаллический продукт, состав которого соответствовал
эмпирической формуле СПН]4О. Основываясь на том, что замещенный фенол мог
реагировать только в о/шо-положении (гидроксильная группа индуцирует реакции в
орто- и лора-положения), и учитывая современные литературные данные, он
предложил циклическую тетрамерную структуру продукта C.97). Однако криоскопические
измерения молекулярной массы ацетатного производного показали, что в
действительности продукт обладал гораздо большей молекулярной массой. Это объяснили
образованием смесей соединений, и только продукт, полученный позже из л-1,1,3,3-
тетраметилбутилфенола, имел молекулярную массу, полностью соответствовавшую
формуле тетрамера. Следующее доказательство тетрамерной структуры было
получено в 1956 г. Б. Хайесом и Р. Хантером (В. Hayes and R. Hunter), специалистами из
Отделения исследования и развития фирмы «Bakelite Ltd», осуществившими
ступенчатый синтез /шро-метильного производного. Они провели завершающую стадию
циклизации (схема 3.22) в условиях высокого разбавления и получили продукт C.98)
с неустановленным, но, предположительно, низким выходом.
В 1955 г. британский химик Дж. Корнфорт (J. Cornforth), интересовавшийся
циклическими структурами как возможными веществами, замедляющими развитие
туберкулеза, повторил синтез производных Цинке. Проведя реакции с участием трет-
бутил- и 1,1,3,3-тетраметилбутилпроизводных, он сумел выделить по два продукта с
близкими, но все же различными температурами плавления. Результаты
предварительного РСА и определения молекулярной массы заставили Корнфорта согласиться
с Цинке и отнести все четыре соединения к тетрамерным структурам. На основании
анализа молекулярных моделей СРК он предположил, что два соединения каждого
3.13. Каликсарены
211
Me
Me
Me
Me
он
он
он
OH
Высокое
разбавление
Me
C.98)
Схема 3.22. Стадия циклизации в синтезе л-метилкаликс[4]арена (по Hayes В.
and Hunter R., 1956)
типа наблюдаются из-за офаниченного вращения вокруг связей, соединяющих фе-
нольные ядра с метеновыми мостиками. Это приводит к образованию диастереоме-
ров. В настоящее время эти диастереомеры известны как конический, частично
конический, 1,2- и 1,3-чередующиеся конформеры (альтернаты, рис. 3.61). Из них
конический конформер, несомненно, наиболее распространен; он стабилизирован
циклической сеткой внутримолекулярных водородных связей между гидроксильны-
ми фуппами на нижнем «ободе» молекулы. В действительности данные 'Н ЯМР-спе-
ктроскопии показывают, что молекулярные модели значительно завышают барьеры
вращения.
Только в 1972 г. К. Гутше (С. Gutsche), надеясь получить ряд веществ, содержащих
полости, подходящие для конструирования имитаторов ферментов, особенно при
использовании корзиноподобной конической конформации (рис. 3.62), возродил химию
этих циклических олигофенольных продуктов. После того как Гутше убедился в том,
что синтезировал тетрамерные соединения, он обнаружил литературные данные с
неоднозначной трактовкой этой проблемы. Это заставило его детально ознакомиться с
результатами по синтезу каликсаренов. Тщательная перекристаллизация неочищенных
продуктов конденсации привела к получению вещества с острым пиком плавления и
осмометрической молекулярной массой 1330 Да, что соответствовало циклическому
октамеру. Масс-спектр триметилсилилпроизводного также говорил об октамерной
3. Хозяева, связывающие катионы
Конус
Частичный конус
1,2-Альтернат 1,3-Альтернат
Рис. 3.61. Конформеры л-т/?ет-бутилкаликс[4]арена. Дж. Корнфорт
предположил, что ограниченное вращение вокруг арил-метеновых связей делает
невозможным превращения этих соединений друг в друга. Действительно, на
шкале времен ЯМР превращение идет медленно при комнатной температуре
A50 с), но быстро - при 60 °С. (Воспроизведено с разрешения Gutsche CD.
Calixarenes. Cambridge: Royal Society of Chemistry, 1989)
Y—V ©+►
N^ -^f *-
\^^^y Субстраты
Рецептор
Комплекс
рецептор-субстрат
Рецептор Продукты
Рис. 3.62. Рис. 3.62. Иллюстрация предложения по использованию каликсаренов
в качестве имитаторов ферментов. (Воспроизведено с разрешения Gutsche CD.
Calixarenes. Cambridge: Royal Society of Chemistry, 1989)
структуре. Завеса тайны, наконец, была поднята с помощью рентгеноструктурного
анализа, показавшего, что высокоплавкие материалы Корнфорта, действительно,
являются циклическими октамерами — каликс[8]аренами. В самом деле, известно, что
катализируемая основаниями реакция л-ал кил фенолов с формальдегидом редко дает
только циклические тетрамеры, хотя многие из продуктов Цинке и являлись
таковыми. Обычно это смеси циклических тетра-, гекса- и октамеров. Каликсарены с
нечетным числом — более редки, хотя и каликс[5]-, и каликс[7]арены теперь хорошо
известны. В результате тщательной хроматофафи ческой работы сейчас уже выделены все
подобные соединения вплоть до каликс[16]аренов. В последнее десятилетие
произошел взрыв в химии каликсаренов. Каликсареновую структуру с ее удивительной
гидрофобной полостью использовали как основу для создания большого числа
разнообразных соединений — хозяев для катионов, анионов и нейтральных молекул, а также
для применения в качестве имитаторов ферментов.
3.13. Каликсарены
13*1 Образование комплексов
каликсаренов с катионами
Каликсарены обладают крайне многообразными каркасами и, как мы
увидим в гл. 4 и 5, в зависимости от степени функционализации, могут служить
хозяевами для катионов, анионов и нейтральных молекул. Фенольные атомы
кислорода на нижнем ободе каликсарена (рис. 3.63) в случае его использования как
хозяина для катионов могут действовать так же, как анизильные остатки сферандов,
или в исходной гидроксильной форме, или в виде алкоксипроизводных.Такой
тип поведения наблюдался для метилового эфира исходного я-трет-бутилка-
ликс[4]арена C.100) в реакции со смесью бензоата натрия, одного эквивалента
воды и триметилалюминия. Получающийся комплекс содержит катион Na+,
координированный всеми четырьмя атомами кислорода каликсаренового тетраэфира.
Наружная сторона катиона Na+ связана по типу «гнездо» с метилоксиалюминат-
анионом, образующимся из других компонентов реакционной смеси (рис. 3.64).
Заместители верхнего обода
Рис. 3.63. Строение
каликс[4]арена в конической
конформации
Заместители нижнего обода
Сферандоподобный центр
связывания катионного гостя
Рис. 3.64. Кристаллическая структура
Ыа+-комплекса тетраметилового эфира
и-трет-бутилкаликс[4]арена C.100)
(по Bott S.etaL, 1986)
2iл 3. Хозяева, связывающие катионы
Интересно, что гидрофобная полость каликсарена также содержит молекулу гостя
- толуола. Таким образом, хозяин одновременно служит рецептором и для
катионов, и для нейтральных молекул.
R = H,Y = Me:C.99)
R = f-Bu, Y = Me: C.100)
R = H, Y = и-Рг: C.101)
Хотя из рис. 3.64 ясно, что связывание Na+ с помощью C.100) происходит в
конической конформации, для этого метилового эфира она уже не является наиболее
стабильной конформацией в растворе, поскольку на нижнем ободе каликсарена не
существует сетки водородных связей, стабилизирующих конический конформер.
Действительно, изомерия каликсаренов C.99) и C.100) сильно зависит от природы
растворителя, и в равновесии могут присутствовать все изомеры. Например, C.99)
существует в растворе CDC13/CD3CN A0:1 по объему) при —50 °С в виде смеси
частично конического и конического изомеров в соотношении 69:31. Вообще, с
ростом полярности растворителя доля конического изомера заметно возрастает. Это
согласуется с тем, что конический изомер - более полярное соединение, чем
частично конический, поскольку в его молекуле все атомы кислорода находятся с
одной стороны. Таким образом, в неполярных растворителях преобладает частично
коническая форма, которая не предорганизована для связывания М+ нижним
ободом. Спектры 'Н ЯМР, соответствующие как коническим, так и частично
коническим М+-комплексам C.100), наблюдаются при добавлении МС1О4 (М+ = Li+, Na+)
к C.100) в растворе хлороформ/метанол D:1). Однако, когда М+ = К+ или Ag+,
происходят большие изменения химического сдвига, обусловленные наличием
частично конического изомера и 1,3-альтерната. Это результат связывания более жестких
оксофильных Li+ и Na+ кислородными атомами нижнего обода, которые до
некоторой степени предорганизованы для сходящегося комплексообразования М+ с
коническим и частично коническим изомерами. Катионы Ag+ и К+ связываются с
атомами кислорода и участвуют в катион—я-взаимодействиях с ароматическими
кольцами (рис. 3.65). Добавление катионов алкиламмония RCHjNH^ приводит к
связыванию в конической конформации. Это может быть следствием или кати-
он-я-взаимодействий с ароматическими кольцами хозяина, или гидрофобного
включения органического «хвоста» гостя.
Работа с N-метилпиридинийиодидом ясно указывает на то, что после
добавления этого гостя процесс комплексообразования способен вызвать конформацион-
ную перестройку с возрастанием доли конической конформации C.99) от 31 до
67%. Замена метильного заместителя на //-пропильный полностью препятствует
конформационной перестройке, приводя к жестким стабильным конформерам.
Геометрию комплекса н-пропилэфирного аналога соединения C.99) - C.101) -
с N-метилпиридинийиодидом изучали методом NOE ЯМР-спектроскопии.
3.13. Каликсарены
215
О О °ч
/ \ Me
Me Me
Рис. 3.65. Конформационные аспекты
включения катиона в л-т/?ет-бутилкаликс[4]арен
C.100) (wpew-бутильные заместители для
простоты опушены)
'Н ЯМР-спектроскопия этого типа исследует передачу спинового возбуждения от
данного протона к соседним (см. Дополнение 3.4). Поскольку это
пространственный эффект, то степень такой передачи, измеряемая по усилению интенсивности
пиков ЯМР, — надежная мера пространственной близости одного водородного
ядра (например, ядра хозяина) к другому (например, к ядру гостя). Результаты
(рис. 3.66) свидетельствуют о взаимодействии атома азота катиона пиридиния с
я-электронной плотностью ароматических колец каликсарена.
С помощью РСА (A. Ikeda and S. Shinkai, 1994) было четко установлено, что ком-
плексообразование Ag+ происходит с участием ароматических колец каликсарена в
0.126
Рис. 3.66. Интенсивность пиков NOE
при возбуждении jwe/яа-арильных протонов
полости каликсарена
1.941
0.388
Рг
0.130
216
3. Хозяева, связывающие катионы
Рис. 3.67. Кристаллическая структура
каликс[4]арена C.101)
случае как частично конической, так и конической конформации неподвижных те-
тра-н-пропиловых эфиров. В обоих случаях катион Ag+ зажат между двумя
кольцами, расположенными друг напротив друга, в отличие от соседних каликсареновых
колец, которые почти перпендикулярны друг другу, что приводит к искаженной
конической конформации. Наружная поверхность катиона в обоих случаях
координируется с трифлат-анионом. В случае частично конической конформации ион Ag+
также взаимодействует с одним из анизильных атомов кислорода, изменившим
свое положение по отношению к трем другим на нижнем ободе (рис. 3.67).
Параллельность колец, наблюдаемая в обеих кристаллических структурах,
достигается и в 1,3-альтернате. Кроме того, ЯМР-спектроскопией было установлено,
что этот изомер связывает катионы К+ и Ag+. В таком случае обе стороны
молекулы хозяина эквивалентны и наблюдается интересный, очень быстрый эффект тун-
нелирования катиона с температурой коалесценции ~-70 °С, который состоит в
проскоке иона Ag+ через кольцо каликсарена (рис. 3.68). Этот эффект для ионов
щелочных металлов в замещенном каликскрауне C.102) был медленным даже при
Рг
1. Быстрый
внутримолекулярный
катионный обмен
2. Медленный
межмолекулярный
катионный обмен
Рг
Рис. 3.68. Туннелирование Ag+ . Этот быстрый внутримолекулярный эффект можно
выделить из межмолекулярных процессов методом 'Н ЯМР
3.13. Калике арены jn
комнатной температуре. Интегрирование пиков свободного хозяина и его
комплексов дает следующие величины lg Ku (при 25 °С): 3.8 для К+ и 2.9 для Rb+.
Включение второго катиона К+ , приводящее Klg Ки = 2.7, стало возможным
благодаря присутствию двух крауноподобных петель. Хозяева были способны
связывать NH|, но для замещенных солей алкиламмония никакого сродства не
наблюдалось, что отражает капсулярную геометрию комплекса.
•""* Roundhill D.M. Metal complexes of calixarenes // Prog. Inorg. Chem. 1995. Vol.43.
P.533-607.
*""* Ikeda A. and Shinkai S. Novel cavity design using calix[w]arene skeletons: Towards
molecular recognition and metal binding // Chem. Rev. 1997. Vol.97. P.1713-1734.
3.13.2 Равновесие межфазного переноса
Исходные л-трет-бутилкаликс[л]арены (п = 4,6,8) почти полностью
нерастворимы в воде. Однако благодаря своему сходству с краун-эфирами и сфе-
рандами они представляют интерес для межфазного катализа (разд. 3.6.2). В
табл. 3.13 приведена селективность каликсарена C.97), а также его гекса- и окта-
мерных гомологов к экстракции катионов различных металлов из гидроксидов в
органическую фазу — хлороформ. К счастью, в водных растворах гидроксидов ка-
ликсарены достаточно хорошо растворимы для осуществления межфазного
катализа и депротонирования одной из фенольных гидроксильных групп. Напротив,
18-краун-6 проявляет наибольшую эффективность в нейтральных растворах.
В табл. 3.13 показано, что все каликсарены очень хороши для транспортировки
Cs+, но обладают малым сродством к катионам других металлов, включая Ва2+,
имеющий большой заряд. В этой таблице приведены также приблизительные
диаметры полостей между атомами кислорода нижнего обода каликсарена; эти
полости, по аналогии с рис. 3.64, могут служить основными центрами связывания
катионов металлов. Фактически ни один из катионов металлов по размеру не
соответствует диаметру полости, хотя, по крайней мере в случае метилового эфира,
218
3. Хозяева, связывающие катионы
Таблица 3.13. Селективный межфазный перенос катионов
металлов каликсаренами из водных растворов гидроксидов
(Приведены значения скорости катиониого переноса (поток) через фазовую
границу (мольс~'м~2108)
Гидроксид
Диаметр катиона
п -трет- Бутилкаликс[ л] арен
л = 4, d= 1.0
л = 6, d=2A
л = 8, rf=4.8
LiOH
NaOH
КОН
RbOH
CsOH
Ba(OHJ
Примечание.
1.52
2.04
2.76
3.04
3.40
2.70
—
2
<0.7
6
260
1.6
10
22
13
71
810
3.2
d — диаметр полости нижнего обода каликсарена (в А).
2
9
10
340
996
-
ясно, что каликсарен достаточно гибок для включения Na+. Тогда в чем же
причина особой селективности каликсаренов к относительно мягкому, поляризуемому
Cs+? Хотя катион цезия и слишком велик для связывания внутри квадрата из
четырех кислородных атомов каликс[4]арена, кристаллическая структура Cs+-Ka-
ликс[4]аренового моноанионного комплекса (рис. 3.69), лишенного бутильных
заместителей, показывает, что этот катион прекрасно соответствует полости
каликсарена, стабилизированной сильными катион—я-взаимодействиями.
Большой катион щелочного металла в этом комплексе расположен симметрично внутри
чашеобразной полости каликсарена с Cs-C-расстояниями в интервале 3.55—4.12 А.
Эта работа была опубликована под названием «Цезий в каликсареновой чаше:
координационная головоломка (J. Harrowfield et al., 1991). Очевидно, что большой
мягкий катион Cs+ и стерически, и электронно подходит для капсулирования
Рис. 3.69. Связывание катиона Cs+
ароматическими кольцами
каликс[4]арена и атомом азота MeCN
3.13. Каликсарены
219
[СН2]
450т
§ 35°
i
8 250 ■
150
50 ■
ОН
ОН ОН
C.103)
Л
Н 1 1 1-
5 7 9 И 13 15 17
Число СН2-групп в мостике верхнего обода (и)
Рис. 3.70. Зависимость скорости межфазного жидкость—жидкость переноса катиона Cs+
(поток) от длины цепи (п) хозяев C.103) (CH2Cl2-CCl4/Cs0Ha(?). Штриховая линия
указывает на отсутствие данных для п — 11, 13 и 15
в полостях больших гибких каликсаренов, которые «укутывают» этот катион.
Интересно сравнить эти результаты с данными для внутриполостного К+,
приведенными в следующем разделе для бис(каликсаренового) хозяина C.104).
Комплексообразование Cs+ внутри полости было также рассмотрено в работе по
кал икс [4] аренам с мостиками на верхнем ободе C.103). В исследованиях по влиянию
длины такого мостика на перенос катиона Cs+ максимальные значения найдены при
п — 8 и 14 СН2-групп (рис. 3.70). Скорость переноса при п = 8 для C.103) равна
скорости, полученной для самого я-трет-бутилкаликс[4]арена — D37±83I0~8 моль-с'-м.
Согласно результатам РСА, степень искажения взаимного расположения фенольных
атомов кислорода мала для я < 8 и пренебрежима для п > 8. Таким образом, эти
результаты интерпретируют с точки зрения влияния длины спейсера [CH2]W каликсареновой
полости, который оптимален при значении п — 8. Сильное комплексообразование
незамещенного каликсарена и каликсаренов с п > 14 определяется степенью их гибкости.
Другие мостики вызывают неблагоприятные искажения полости каликсарена.
3* * 3*3 Образование комплексов
гибридных каликсаренов с катионами
Большая макротрициклическая бис(каликсареновая) клетка C.104),
проявляющая высокую селективность к калию, была синтезирована (P. Schmitt
et al., 1997) путем сочетания свойств нижних ободов каликсаренов, способных
связывать катионы, со свойствами этиленгликольных звеньев, подобных тем, которые
существуют в краун-эфирах. Константа связывания К+ в растворе
хлороформ/метанол равна 4104, а селективность бис(каликсарена) C.104) к калию выше, чем
таковая ко всем другим щелочным металлам (в виде иодидов) в 102 раз.
Моделирование методом молекулярной механики предполагает, что комплексообразование
3. Хозяева, связывающие катионы
происходит по двухстадийному механизму, в котором интермедиат включает внут-
риполостной К+ — я-комплекс (схема 3.23). Эти данные представляют интерес в
качестве модели переноса К+ через ворота ионного канала (см. разд. 2.1).
Ви
Ви
C.104)
Ви
Ви
Ви Ви
Ви Ви
Ви
Схема 3.23. Образование комплекса бис(каликсареновой) клетки хозяина C.104)
с ионом К+ (по Schmitt P. et al., 1997)
3.13. Каликсарены
111
Для большого бис(каликс)крауна C.105) могут наблюдаться два динамических
процесса. Межмолекулярное равновесие ассоциация/диссоциация, при котором
комплекс находится в равновесии со свободным лигандом и катионом металла, - это
медленное событие на шкале времен Я MR В самом комплексе происходит быстрое
внутримолекулярное колебание катиона металла от одного центра связывания к
другому (схема 3.24). Соединение C.105) также представляет интерес из-за того,
что механизм этого быстрого внутримолекулярного челночного движения катиона
может иметь важное значение по аналогии с движением катионов металлов через
трансмембранные ионные каналы (разд. 2.1).
Кроме ранее упомянутых хозяев были синтезированы и молекулы других
гибридных хозяев, сочетающие преимущества жесткого, легкодоступного каликсаре-
нового каркаса со связывающими способностями криптандов и сферандов. В
экспериментах по экстракции растворов пикратов щелочных металлов получены
следующие энергии связывания (—АС/0, ккалмоль, ±0.2 ) для каликссферанда
C.106): Na+, 13.6; К+, 14.0; Rb+, 12.0; Cs+, 9.8. Скорости комплексообразования
свидетельствуют о связывании катиона металла в капсулированном виде,
поскольку комплексы с Rb+ и Cs+ образуются менее чем за 6 ч, тогда как для
комплексообразования с Na+ требуется более 48 ч, причем предполагается, что перед
связыванием происходит полная десольватация катионов, так как свободная энергия
С ' °: з
1. Быстрый внутримолекулярный
катионный обмен
2. Медленный межмолекулярный
катионный обмен
|Ви
Ви C.105>К+
Схема 3.24. Внутри- и межмолекулярные процессы обмена катионом металла
для бис(каликс)крауна C.105) (по IkedaA. and Shinkai S., 1994)
1^22 3. Хозяева, связывающие катионы
гидратации Na+ больше, чем таковые Rb+ и Cs+. Соединения типа каликскрауна
C.107) — эффективные ионофоры для больших катионов щелочных металлов;
величина —ДС° равна 11.3; 10.6 и 7.8 ккалмоль для К+, Rb+ и Cs+ соответственно.
Фактор селективности лиганда (отношение констант связывания) достигает -2000
для пары K+/Na+. Напротив, А. Икеда и С. Шинкай (A. Ikeda and S. Shinkai, 1994)
разработали ионофор меньшего размера C.108), который ввели в ионоселективные
электроды. Как для конического, так и для частично конического изомера эти
устройства имеют высокую селективность по отношению к паре K+/Na+ с фактором
селективности до 1053.
Ви
C.106)
C.107)
C.108)
Интересный прототип молекулярного устройства, подобного шприцу, был
получен на основе несимметричного каликсарена C.109). Сродство Ag+ к донорному
атому азота крауна таково, что значение \g Ки в растворе CD2C12/CD3OD
составило 9.78, причем катион Ag+ располагается в части молекулы, относящейся к крауну.
В результате протонирования основного атома азота (рис. 3.71) связывание Ag+
C.109)
Рис. 3.71. «Перекачка»
катиона металла
«молекулярным
шприцем»
3.14. Углеродные донорные и п-кислотные лиганды 223
крауном «выключается» и происходит «перекачка» катиона из кольца каликсарена
к нижнему центру связывания. При депротонировании катион Ag+ возвращается
обратно.
Углеродные донорные
и я-кислотные лиганды
Комплексообразование мягких ионов металлов не ограничивается ге-
тероатомными донорами. Такие ионы, богатые электронами и имеющие большие
несжатые орбитали, проявляют весьма интересные особенности при комплексооб-
разовании с я-кислотными лигандами. Многие переходные металлы образуют ко-
валентные комплексы с алкеновыми, алкиновыми и арильными лигандами,
стабилизированные синергетическим обратным связыванием (см. Дополнение 3.3).
Нековалентное катион—л-взаимодействие (см. разд. 1.7.5) играет значительную
роль в качестве «супрамолекулярной силы» даже в отсутствие ковалентного синер-
гетического связывания. Мы уже видели, какую важную роль играют катион—
Ti-взаимодействия в комплексообразовании Ag+ и больших катионов щелочных
металлов карбоциклами каликсаренов (см. разд. 3.13). Как более общее явление этот
вид комплексообразования будет рассмотрен в следующем разделе.
3* 14* 1 Смешанные С-гетероатомные хозяева
Аллильный лариат-эфир C.110), см. разд. 3.2, образует комплексы и с
К+, и с Ag+ (которые имеют близкие ионные радиусы, см. табл. 3.3). Можно
ожидать, что в случае К+-комплекса катион калия слишком велик, для того чтобы
удобно расположиться внутри кольца аза-15-краун-5, и поэтому он лежит несколько
выше плоскости донорных атомов. Наружная сторона атома металла занята
анионом PFg". Подобная координация с крауном характерна и для Ag+. Однако этот
катион металла своей наружной стороной связывается не с анионом, а с аллильной
боковой цепью близлежащей молекулы, что приводит к образованию твердого
полимера (рис. 3.72).
N f О О
(ЗЛЮ) C.111) C.112) \ 1
R = 2-NO2, 4-NO2, 2-CN, 4-CN, 2-C1, 4-C1, H, 2-OMe, 4-OMe
24
3. Хозяева, связывающие катионы
E)
Рис. 3.72. Контраст между связыванием
(а) жесткого катиона К+ и (б) мягкого Ag+
(соли PFg") лариат-эфиром C.110).
Расстояния Ag—С равны 2.36-2.39 А
Изучение селективности лариат-аза-15-краун-5 C.111) и производных лигандов
BiBLE (бибрахиальный лариат-эфир) диаза-18-краун-6 C.112), содержащих по-
разному замещенные ароматические кольца, показало, что стабильность Ag+-KOM-
плекса уменьшается с ростом способности заместителей R оттягивать электроны,
которая измеряется константой индуктивности Тафта — а0 (параметр,
характеризующий способность яярд-заместителей производных бензойной кислоты оттягивать
электроны и оцениваемый на основании значений рКа кислоты) (D. Gustowski et
al., 1987). Константы связывания катионов серебра (lg К) находятся в пределах от
2.47 (R = o-NO2) до 4.22 (R = о-ОМе) для лигандов типа C.111) и от 3.17 до 6.30 для
соединения C.112) с теми же заместителями. Этот результат объясняется как нераз-
рушающими связи индуктивными воздействиями на электронную плотность атома
азота, так и Ag+— тх-взаимодействиями в растворе. Корреляция lg К: а0 значительно
3.14. Углеродные донорные и п-кислотные лиганды
225*
C.113)
■^
Капсулирование
Триподальное хелатирование
Рис. 3.73. Способы координации коелентерандов
менее выражена для Li+ и Na+, что соответствует их более слабому взаимодействию
с азотом и малому вкладу в катион—я-взаимодействия.
Взаимодействия между Ag+ и алкенами в растворе относительно слабы.
Катионы переходных металлов с низкой степенью окисления и такие нейтральные
атомы, как Cr@), Mn(I), Fe(II), Ru(II) и Rh(III), часто участвуют в более сильных
взаимодействиях, образуя полностью ковалентные я-ареновые и я-алкеновые
комплексы. Хотя большинство этих удивительных соединений находится за
пределами тематики этой книги, следует заметить, что взаимодействия такого типа
начинают использовать при разработке геометрически и электронно селективных кати-
онных хозяев, обладающих очень высокой способностью к связыванию благодаря
образованию комплексов со значительной степенью ковалентности. В частности,
были получены представители нового класса трис(пиразолил)- и пиридил-лиган-
дов, например C.113), называемые коелентерандами (от греч. «пустой живот»;
С. Hartshorn and P. Steel, 1996).
Коелентеранды могут координироваться с катионами металлов двумя
способами (рис. 3.73). Триподальная хелатная форма напоминает обычные лиганды, такие,
как трис(пиразолил)бораты и их трис(пиразолил)метановые аналоги. Капсулиро-
ванная форма является редкой, но реакция Ru(II)-димeтилcyльфoкcиднoгo
(DMSO) предшественника - [RuQ2(DMSOL] - с C.113) приводит к получению
полностью капсулированного комплекса при наличии в качестве противоаниона
ZnCl4 . В этом случае кристаллическая структура показывает, что катион Ru(II)
глубоко внедрен внутрь «пустого живота» лиганда с довольно коротким расстоянием
от Ru до центра ароматического кольца — ~ 1.58 А (по сравнению с нормальными
значениями для металлоорганических комплексов ~ 1.67-1.70 А), а это
предполагает хелатное усиление связи Ru-C. Данные 13С ЯМР-спектроскопии говорят о
стабильности комплекса в растворе в течение долгого времени.
Углеводородные хозяева
Наличие в молекуле хозяина донорных гетероатомов не является
существенным для катион-я-комплексообразования; действительно, был получен
ряд чисто углеводородных лигандов типа я—С-донор. Лиганд C.114) синтезирова-
15- СТИДДж В
4226 3' Хозяева, связывающие катионы
TiClj-Zn-Cu
Диметоксиэтан
^.
48 ч
кипячение с обратным
холодильником
C.114)
Схема 3.25. Синтез ненасыщенного углеводородного макроцикла
ли с 90%-м выходом циклизацией дикетонового предшественника в условиях
высокого разбавления, примененного для минимизации образования полимерных
побочных продуктов (схема 3.25).
Согласно РСА-данным, расстояние между двумя обращенными друг к другу
парами двойных связей в этом лиганде равно 5.11 А, что свидетельствует о
прекрасном соответствии его полости размеру катиона Ag+, который обычно образует
связи с алкенами при расстояниях Ag—С в интервале 2.4-2.6 А. Соответственно
реакция между C.114) и Ag[CF3SO3] (CF3SO3", трифторметансульфонат-анион, или
трифлат-анион, - это обычный, легкодоступный анион, который слабо
координирует центры металла) привела к получению комплекса [Ag+ с C.114)] с хорошим
выходом, рис. 3.74. Кристаллическая структура этого комплекса позволила
установить плоскоквадратную координационную геометрию Ag+, необычную для ионов
металлов ^/10-типа. Примечательно, что 13С ЯМР-спектр этого комплекса
указывает на сильное взаимодействие; оказалось, что комплекс очень устойчив на воздухе,
а также к действию света и тепла, т.е. в условиях, обычно разрушающих Ag-тс-ком-
плексы.
Были получены аналогичные ди- и тримерные соединения, но лишь тетрамер-
ный макроцикл C.114) достаточно велик, для того чтобы включить катион металла
в свою полость. Подобные соединения были также обнаружены для хозяев,
состоящих из арильных колец. В частности, [2.2]парациклофан C.115), [3.3]парацикло-
фан C.116) и [2.2.2]парациклофан C.117) образуют интересные металлоорганичес-
кие комплексы с рядом металлов, имеющих низкие состояния окисления. Более
подробно номенклатура и химия циклофанов обсуждается в разд. 5.3.4.
Рис. 3.74.
Стереоизображение
высокосимметричного Ag+-KOMrmeKca
лиганда C.114).
(Воспроизведено с разрешения
VogtleF., 1991)
3.14. Углеродные донорные и п-кислотные лиганды
111
C.115)
C.116)
C.117)
Соединения C.115) и C.116) реагируют с металлическим хромом в паровой
фазе в соответствии с методом синтеза, называемым «атом металла — пар лиганда», в
котором испаренные при высокой температуре атомы металла и газообразные
лиганды совместно конденсируются на поверхности, охлаждаемой жидким азотом
или жидким гелием, а затем постепенно нагреваются до тех пор, пока не
произойдет реакция. Образующиеся комплексы содержат Сг°, капсул ированный (зажат как
в сэндвиче) между обращенными друг к другу ароматическими кольцами. Эти
комплексы слишком реакционноспособны, чтобы кристаллизоваться, но при их
окислении иодом до аналогичных Сг+-комплексов удалось получить кристаллы обоих
соединений (рис. 3.75).
НF)
НG1)
НG2)
С(б')
Н(81)
НE')
Н(91)
Рис. 3.75. Кристаллическая структура Сг+-комплскса [3.3]парациклофана C.116).
(Воспроизведено с разрешения Вепп R., Blank N.E., Haenel M.W. et al. Structural investigations on
(rI2-[3.3]paracyclophane) chromium(O) and its cation (ri12-[3.3]paracyclophane) chromium(I) tri-
iodide // Angew. Chem., Int. Ed. Engl. 1980. Vol.19. P.44)
1228
3. Хозяева, связывающие катионы
3*15 Сидерофоры
*~» Vogtle F. Supramolecular chemistry. Chichester: J. Wiley & Sons, 1991. P.84-106.
3.15* 1 Сидерофоры природного происхождения
Железо играет важнейшую роль в огромном количестве клеточных
окислительно-восстановительных процессов в растениях, бактериях и высших
организмах. Однако с точки зрения биодоступности железо имеет большие
ограничения, поскольку в своем наиболее часто встречающемся в природе состоянии
окисления, Fe(III) (ржавчина), оно нерастворимо в водных растворах. При физио-
логическом значении рН 7.4 растворимость Fe(H2O)^+ составляет ~10'18 моль-дм~3.
Для оптимального роста микроорганизмов необходима внутриклеточная
концентрация железа ~ 10~7 моль-дм, т.е. в 100 млрд раз больше. Поэтому растениям и
бактериям для связывания железа требуются высокоэффективные лиганды,
позволяющие забрать Fe3+ и доставить его в клетки. Такие лиганды природного
происхождения называют сидерофорами (от греч. «переносчик железа») и их можно
применять в качестве эффективных ускорителей роста микроорганизмов.
Сидерофоры были открыты еще в 1911 г., но первый природный сидерофор, микобактин
C.118), был выделен только в 1951 г. в виде комплекса с алюминием.
О
Л
w
HO-n )
oV
он о
но.
O^NH Uy"
он о 1\
Микобактин
°" Id
Энтеробактин НО
C.119) ОН
C.118)
Такие сидерофоры, как микобактин C.118) и энтеробактин C.119), по
существу, представляют собой поданды с тремя боковыми цепями, связывающие гостя с
помощью депротонированных гидроксильных групп. Эти лиганды имеют общий
заряд 6-, что делает их комплементарными для катионов с высоким зарядом; жест-
3.15. Сидерофоры
229
кий характер фенолятных и пирокатехиновых фрагментов обеспечивает связь с
жестким Fe3+. Действительно, константа устойчивости комплекса энтеробактина с
Fe3+ равна 1052, поэтому он является наиболее эффективным из известных
природных хелатирующих агентов железа. Энтеробактин настолько эффективен при
связывании железа, что при рН 7 не наблюдается никакой конкуренции со стороны
хорошо известного комплексообразователя — этилендиаминтетрауксусной кислоты
(EDTA). В последние годы с помощью ряда сидерофоров, выделенных из бактерий
и корней растений, в особенности растущих на щелочных почвах, было показано,
что фрагмент о-дигидроксибензола (пирокатехин) - это неотъемлемая часть
эффективных хелаторов железа. Энтеробактин со своими тремя пирокатехиновыми
боковыми цепями способен образовать с Fe3+ тригональную призматическую шес-
тикоординационную структуру (рис. 3.76).
Первоначально энтеробактин был выделен из бактериальных культур Salmonella
typhimurium или Escherichia coli путем длительной экстракции и хроматографичес-
кой очистки с выходом -15 мг на 1 л питательного раствора. Вполне понятно, что
эта прецизионная методика привела к развитию ряда лабораторных способов
синтеза энтеробактина. В основе наиболее эффективного из них лежит темплатный
эффект (см. разд. 3.8.2), включающий тримеризацию объемистого трифенилметил-
замещенного производного L-серина вокруг атома олова (схема 3.26).
— C.119)
PhjC
О
Схема 3.26. Темплатный синтез энтеробактина C.119)
230
3. Хозяева, связывающие катионы
0=^-0 о-<
HN О V
Ч .о—'
Рис. 3.76. Три тональная призматическая
координация Fe3+ в [Ре(энтеробактин)]3"
Энтеробактин — это хиральное соединение благодаря трем асимметрическим
атомам углерода, ближайшим к атому азота. Использование в качестве исходного
соединения другого энантиомера, /)-серина, дает энантиоэнтеробактин —
зеркальное отражение энтеробактина C.119). Интересно, что в присутствии энантиоэнте-
робактина никакого ускорения роста бактерий не происходило. Фактически
сдерживание роста вызывалось тем, что неприродный сидерофор удерживал железо в
среде культуры, препятствуя его усвоению бактериями. Природная форма C.119) -
крайне эффективный ускоритель роста.
Синтетические системы
Уникальная способность энтеробактина к комплексообразованию
железа привела к синтезу ряда синтетических аналогов подандов и криптандов. В
частности, трипирокатехиновое производное мезитилена C.120), МЕСАМ, имеет
близкое структурное сходство с C.119) и, более того, благодаря отсутствию в его
молекуле склонных к гидролизу эфирных звеньев с ним проще работать.
МЕСАМ
C.120)
3.15. Сидерофоры |^31
Как и ожидалось, синтетический хозяин C.120), имеющий тесное родство с
C.119), — также сильный комплексообразователь для железа. Однако константа
связывания Fe3+ этим соединением не превышает 1046, т.е. она в несколько
миллионов раз ниже, чем у самого энтеробактина, что сразу наводит на мысль об
огромной роли структурного фактора в объяснении исключительно эффективной ком-
плексообразующей способности энтеробактина. По-видимому, меньшая
мезитильная спейсерная группа в МЕСАМ приводит к более напряженной
геометрии комплекса. Комплекс [Fe(MECAM)]3" был испытан в культуре E.coli; несмотря
на его меньшее, чем у C.119), сродство к Fe3+, он способен связываться с системой j
рецепторов энтеробактина E.coli и ускорять рост организмов.
Пирокатехин — всего лишь слабая кислота, и в виде аниона с зарядом 6- — это
сопряженное основание (гость), которое восприимчиво к протонированию, что
приводит к существенному влиянию рН на его комплексообразующую способность
по отношению к Fe3+. Например, в слабокислых растворах (рН 5) существует
сильная конкуренция за Fe3+ со стороны EDTA (более сильная кислота в протонирован-
ной форме), что можно обнаружить с помощью УФ-спектроскопии в видимой
области. Чтобы иметь возможность надежно определять комплексообразующую
способность сидерофоров по отношению к металлам, для этих лигандов ввели
параметр рМ. Величина рМпо аналогии с рН определяется следующим соотношением:
pM=-lg[Fe(H2OK+]. C.7)
Таким образом, большая положительная величина рМ соответствует низкой
равновесной концентрации катиона Fe3+ в воде и, следовательно, свидетельствует о
высокой комплексообразующей способности лиганда. Важно, чтобы величину рМ
измеряли в стандартных условиях в водных растворах при концентрации
Fe3+ 1 мкМ, концентрации лиганда 10 мкМ и рН 7.4. Величины рМдля некоторых
лигандов даны в табл. 3.14.
Таблица 3.14. Величины рМ
для хелатных Ре3+-лигандов
Лиганд
Энтеробактин 35.5 52
МЕСАМ 29.1 46
EDTA 12.2
Поданды энтеробактин C.119) и его синтетический аналог C.120) чрезвычайно
эффективны в связывании железа, хотя подандные хозяева, как правило, менее
эффективны, чем их циклические аналоги (см. разд. 3.2.1). Поэтому были
синтезированы макробициклический лиганд C.121) и целый ряд родственных ему хозяев со
спейсерами разного размера (например, трифенилметановые чаши, бипиридиль-
ные центры связывания). Этот лиганд, в соответствии со своей жесткой предорга-
низованной структурой C.121), имеет огромную константу связывания для Fe3+ в
физиологических условиях - ~1059, что делает его наиболее эффективным ком-
232
3. Хозяева, связывающие катионы
плексообразователем катиона Fe3+ из всех известных (W. Kiggen and F. Vogtle, 1984).
Лиганд C.121) принадлежит к классу сидерандов (синтетические сидерофоры) и
обладает такими полезными свойствами, как стабильность к окислению пирокатехи-
новых центров связывания и устойчивость к гидролизу. Это один из немногих
примеров того, насколько молекулы, созданные человеком, могут быть эффективнее
своих природных аналогов.
C.121)
Учебные задания
3.1. Сколько существует структурных изомеров дибензо-15-краун-5? Нарисуйте их.
3.2. Гидрирование изомера дибензо-18-краун-6, первоначально синтезированного
Педерсеном при использовании H2/Pd, дает насыщенный дициклогексил-18-
краун-6. Не считая энантиомеров, у этого соединения пять изомеров, из
которых цис-син-цис-изомер нарисован ниже. Другие изомеры называют: цис-
анти-цис-1 транс-син-транс-, транс-анти-транс- и цис-транс-. Нарисуйте эти
изомеры и присвойте структурам их названия.
цис-син-цис- Дицикл огексил-18-краун-6
3.3. Посмотрите свои ответы на вопрос 3.2. Являются ли эти изомеры
взаимно-обратимыми за счет вращения связей? Какие из них хиральные? Возможно,
построение молекулярной модели поможет Вам ответить на эти вопросы.
3.4. Дайте определение термину «константа связывания». Логарифмы констант
связывания для комплексообразования Na+ различными изомерами дицикло-
гексил-18-краун-6, синтезированными на сегодня, равны 4.08, 3.68, 2.99 и 2.52
Учебные задания
233
для цис-син-цис-, цис-анти-цис-, транс-син-транс- и транс-анти-траис-шо-
меров соответственно (МеОН, 25 °С). Что, по-вашему, служит причиной столь
большой разницы? Соответствующая константа связывания для 18-краун-6
равна 4.32. Как это соотносится с Вашими представлениями?
3.5. Логарифмы констант связывания К+ (М, МеОН, 25 °С) для трех хозяев
приведены ниже. Объясните такую большую разницу.
lg /Cx(K+) 9.0 5.4 2.0
3.6. Дайте краткие объяснения следующим понятиям. Воспользуйтесь для этого
материалом гл. 1 и 3: (а) темплатный эффект; (б) хелатный и макроцикличес-
кий эффекты; (в) метод высокого разбавления в макроциклическом синтезе;
(г) предорганизация и комплементарность.
3.7. Сопоставьте свойства молекул-хозяев следующих классов. Включите в Ваш
ответ информацию о селективности, растворимости и кинетике связывания:
(а) ионофоры природного происхождения; (б) поданды; (в) коранды;
(г) лариат-эфиры; (д) криптанды; (е) каликсарены; (ж) сферанды.
Информацию можно представить в виде таблицы.
3.8. Исходя из своего ответа на вопрос 3.6 или сведений другого источника,
предложите искусственные системы, имитирующие (а) некоторые из свойств
природных ионофоров и (б) особенности трансмембранных ионных каналов.
Мысленный эксперимент
Ниже показан ряд структурных субъединиц. Основываясь на прочитанной Вами
главе, предложите структурные модификации или соответствующие
функциональные группы, которые можно добавить, для того чтобы хозяин каждого типа мог
служить хозяином (а) для катионов металлов и (б) для ионов органических
производных аммония. Используя понятия «предорганизация», «сольватация», «энталь-
пийный и энтропийный эффекты», а также «комплементарность», расположите в
ряд Ваши хозяева в соответствии с их селективностью по отношению к гостям
следующих серий:
(а) Na+, K+, Rb+, Cs+;
(б) RNH3+ (R = Me, Et, /-Bu, PhC(COj));
(в) NH+R'NH3+ (R' = CH2, C2H4, o-C6H4, />-C6H4).
234
3. Хозяева, связывающие катионы
В каждом случае рассмотрите, будет ли комплексообразование гостя капсулярного
типа, типа «гнездо» или «насест», а может быть, и какого-нибудь другого типа.
Используйте эту информацию для качественной оценки скорости образования и
диссоциации комплекса в каждом случае.
R
Дифенилметан
он он он
Калике [4]арен
^7 Л
CTV
X
X
X
X = О, NH, S
но
он
но он
а-Циклодекстрин
Литература
Boston, 1968 Boston D.R., Rose N.J. Encapsulation reaction: Synthesis of clathro chelate 1,8-
ta(fluoroboro)-2,7,9,14,15,20-hexaoxa-3,6,10,13,16,19-hexaaza-
4,5,11,12,17,18-hexamethylbicyclo[6.6.6]eicosa-3,5,10,12,16,18-hexa-
enecobalt(III) ion//J.Amer.Chem.Soc.l968.Vol.90. P.6859.
Bott, 1986 Bott S.G., Coleman A. W., Atwood J.L. Inclusion of both cation and neutral
molecule by calixarene - structure of the [para-terf-butylmethoxycalix[4]arene-sodi-
um-toluene]+cation//J. Amer. Chem. Soc. 1986. Vol.108. P. 1709.
Brown, 1996 Brown K.N., Geue R.J., Hambley T. W. et al. Stereospecific template synthesis of
a new class of cage complexes: An example of self assembly// Chem. Commun.
1996. P.567.
Литература
235
Canceill, 1982 CanceillJ., ColettA., GabardJ. et al. Speleands — macropolycyclic receptor cage;
based on binding and shaping subunits — synthesis and properties of macrocy-
cle—cyclotriveratrylene combinations // Helv. Chim. Acta. 1982. Vol.65. P. 1984.
Christensten, 1971 Christensten J.J., Hill J. 0., Izatt R.M. Ion binding by synthetic macrocyclic
compounds//Science. 1971. Vol.174. P.459.
Cram, 1986 Cram D.J. Preorganisation — from solvents to spherands // Angew. Chem., Int.
Ed. Engl. 1986. Vol.25. P. 1039.
Dhaenens, 1984 Dhaenens M., Lacombe L, Lehn J.-M., Vigneron J.-P. Binding of acetylcholine
and other molecular cations by a macrocyclic receptor molecule of speleand type
//J. Chem. Soc, Chem. Commun. 1984. P. 1097.
Dietrich, 1969 Dietrich В., Lehn J.-M., Sauvage J.-P. Diaza-polyoxa-macrocycles et macrobi-
cycles//Tetrahedron Lett. 1969. P.2885.
Dye, 1990 Dye J.L. Electrides — ionic salts with electrons as the anions // Science. 1990.
Vol.247. P.663.
Gatto, 1986 Gatto V.J., Arnold K.A., Viscariello A.M. et al. Novel synthetic access to 15-mem-
bered and 18-membered ring diaza-bibracchial lariat ethers (bibles) and a study
of sidearm-macroring cooperativity in cation binding // Tetrahedron Lett. 1986.
Vol.27. P.327.
Geue, 1984 Geue R.J., Hambley T. W., Harrow/field J.M. et al. Metal-ion encapsulation -
cobalt cages derived from polyamines, formaldehide, and nitromethane //
J. Amer. Chem. Soc. 1984. Vol.106. P.5478.
Gustowski, 1987 Gustowski D.A., Gatto V.J., Mallen J. et al. Direct correlation of cation binding
strengths to Hammett parameters in substituted N-benzylaza-15-crown-5 lariat
ether and N,N '-dibenzyl-4,13-diaza-18-crown-6 BiBLE derivatives // J.Amer.
Chem. Soc. 1987. Vol.52. P.5172.
Hall, 1997 Hall CD., Truong T.-K.-U., Tucker J.H.R., Steed J. W. Sodium springs another
surprise // Chem. Commun. 1997. P.2195.
Hancock, 1992 Hancock R.D. Chelate ring size and metal-ion selection - the basis of selectivity
for metal-ions in open-chain ligands and macrocycles // J. Chem. Ed. 1992.
Vol.69. P.615.
Harrowfield, 1991 Harrowfield J.M., Ogden M.I., Richmond W.R., White A.H. Calixarene-cupped
cesium — a coordination conundrum //J. Chem. Soc, Chem. Commun. 1991.
P.I 159.
Hartshorn, 1996 Hartshorn СМ., Steel P.J. Coelenterands: A new class of metal-encapsulating
ligands//Angew. Chem., Int. Ed. Engl. 1996. Vol.35, N.22. P.2655-2657.
Ikeda, 1994 IkedaA., Shinkai S. On the origin of high ionophoricity of 1,3-alternate
calix[4]arenes - л-donor participation in complcxation of cations and evidence
236 3. Хозяева, связывающие катионы
for metal-tunneling through the calix[4]arene cavity // J. Amer. Chem. Soc.
1994. Vol.116. P.3102.
Kiggen, 1984 Kiggen W., Vogtle F. Functionalized, oligocyclic cavities - a novel siderophore //
Angew. Chem., Int. Ed. Engl. 1984. Vol.23. P.714.
Nakatsuji, 1982 Nakatsuji Y., Nakamura Т., Okahara M. et al. The effect of sidearm heteroatoms
and aquaternary methyl-group at the pivot position in lariat ethers //
Tetrahedron Lett. 1982. P. 1352.
Newcomb, 1979 Newcomb M., Toner J.L., Helgeson R.C., Cram D.J. Host-guest complexation. 20.
Chiral recognition in transport as a molecular basis for a catalytic resolving
machine//J. Amer. Chem. Soc. 1979. Vol.101. P.4941.
Peacock, 1978 Peacock S.C., Domeier L.A., Gaeta F.C.A. et al. Host-guest complexation. 13.
High chiral recognition of amino esters by dilocular hosts containing extended
steric barriers // J. Amer. Chem. Soc. 1978. Vol.100. P.8190.
Pedersen, 1967 Pedersen C.J. Cyclic polyethers and their complexes with metal salts // J. Amer.
Soc. 1967. Vol.89. P.7017.
Peters, 1997 Peters J.C., Odom A.L., Cummins С.С A terminal molybdenum carbide prepared
by methylidyne deprotonation // Chem. Commun. 1997. P.1995.
Schmitt, 1997 Schmitt P., Beer P.D., Drew M.G.B., Sheen P.D. Calixl4]tube: A tubular receptor
with remarkable potassium ion selectivity//Angew. Chem., Int. Ed. Engl. 1997.
Vol.103. P. 1840.
Steed, 1998 Steed J. W., McCool B.J., Junk P.C. Hydrogen bonded polymers and oligomers
from metal salts and 18-crown-6//J.Chem. Soc, Dalton Trans. 1998. P.3417.
Trueblood, 1981 Trueblood K.N., Knobler СВ., Maverick E. et al. Spherands, the 1st ligand
systems fully organized during synthesis rather than during complexation //
J. Amer. Chem. Soc. 1981. Vol.103. P.5594.
Vogtle, 1979 Vogtle F., Weber E. Multidentate acyclic neutral ligands and their complexation //
Angew. Chem., Int. Ed. Engl. 1979. Vol.18. P.753.
Vogtle, 1991 Vogtle F. Supramolecular chemistry. Chichester: J .Wiley & Sons, 1991.
4
Связывание анионов
Человеку следует с осторожностью
приступать к расходам,
которые, однажды начавшись,
продолжатся.
Ф. Бэкон A561-1626). Эссе (о расходах), 1625 г.
•""» Bianchi A., Bowman-James К., Garcia-Espana E. Supramolecular chemistry
of anions. New York: Wiley. 1997.
Введение
Отправной точкой в области нековалентной координационной химии
анионов можно считать сообщение К. Парка (С. Park) и X. Симмондса
(Н. Simmonds) из компании «Du Pont de Nemours Company», 1968 г., посвященное
комплексообразованию галогенидов с рядом макробициклических хозяев,
называемых катапинандами D.1). Катапинанды (от греч. «katapino», означающего
«заглатывать», или «поглощать») при протонировании атомов азота, находящихся в
голове мостика, способны связывать галогенид-ионы внутри своей
макробициклической полости. Такое капсулирование, позже подтвержденное РСА
(рис. 4.1), — первый пример анионного связывания макроциклическим хозяином;
оно на несколько лет предвосхитило открытие криптатов (см. разд. 3.3).
Упомянутое выше сообщение Парка и Симмондса, представленное в редакцию 13 ноября
1967 г., стало важным вкладом лаборатории компании «Du Pont» в тогда еще
неизвестную область супрамолекулярной химии. Семью месяцами ранее, 13 апреля,
Ч. Педерсен представил свою основополагающую работу по связыванию катионов
с помощью дибензо-18-краун-6, которая положила начало современной
супрамолекулярной химии.
Н—Nt N+—H
\п
in
D.1), п = 0: D.1а)
п = 2: D.1в)
D.16): 1,11-диазабицикло[9.9.9]нонакозан
238 4. Связывание анионов
Рис. 4.1. Минимизированная структура
комплекса хлорид-иона с катапинандом D.1а)
Несмотря на довольно раннее открытие катапинандов, нековалентная
координационная химия анионов развивалась относительно медленно по сравнению с
химией хозяев для катионов и даже нейтральных молекул. Хотя, как правило,
поведение анионных хозяев подчиняется тем же общим закономерностям, которые
характерны для катионных хозяев (прежде всего, предорганизация, комплементар-
ность и сольватация) и выражаются константами связывания и селективностью
хозяев к катионным гостям, использование этих представлений может быть
затруднено из-за некоторых свойств анионов.
• Поскольку анионы довольно велики, для них требуются рецепторы значительно
большего размера, чем для катионов. Например, ионный радиус одного из
самых маленьких анионов, F~, сравним с ионным радиусом К+ A.33 и 1.38 А).
Другие анионные радиусы приведены в табл. 4.1.
• Даже у простых неорганических анионов существует большое разнообразие
формы и геометрии, например, сферическая (галогениды), линейная (SCN~,
Щ), плоская (NO^~, PtCl^"), тетраэдрическая (РО^~, SO^"), октаэдрическая
(PF^, Fe(CN)|~), а в случае биологически важных олигофосфатных анионов
формы еще более сложны.
• У анионов свободные энергии сольватации больше, чем у катионов того же
размера; значит, хозяева анионов должны более эффективно конкурировать
с окружающей средой, например, AGhydration(F~) = -465 кДж-моль,
A^hydration(^+) = ~295 кДж-моль. Свободные энергии сольватации других
анионов и катионов приведены в табл. 4.1.
• Многие анионы существуют в узких пределах рН. Это может создавать
трудности, особенно в случае рецепторов на основе полиаммониевых солей, где хозяин
может быть не полностью протонирован как раз в той области рН, в которой
анион присутствует в нужном виде.
• Обычно анионы координационно насыщены и поэтому связываются только
посредством слабых взаимодействий, таких, как водородная связь и ван-дер-ва-
альсовы взаимодействия.
4.1. Введение
1239
Таблица 4.1. Свойства некоторых наиболее
распространенных анионов и катионов
Ион
F-
CI-
Вг~
I-
сю4-
NO3"
со2-
so42-
poJ-
н2ро4-
PdCl2"
Na~
Cs~
Li+
Na+
K+
Cs+
Ca2+
Zn2+
Al3+
La3+
NH4+
Радиус, А
1.33
1.81
1.95
2.16
1.50
1.79
1.78
2.30
2.38
2.00
3.19
2.2
3.5
0.69
1.02
1.38
1.70
1.00
0.75
0.53
1.05
1.48
-465
-340
-315
-275
-430
-300
-1315
-1080
-2725
-465
-695
Нет данных
Нет данных
-475
-365
-295
-250
-505
-1955
-4525
-3145
-285
Р*а B98 К)
3.3
Низкий
Низкий
Низкий
-
-1.4
6.4, 10.3
Низкий, 2.0
2.1,6.2, 12.4
2.1,6.2, 12.4
-
-
—
—
—
—
—
—
—
—
—
9.3
Таким образом, количественное описание селективного связывания анионных
гостей представляет до некоторой степени более сложную задачу, чем описание
связывания катионов металлов, даже если качественно в обоих случаях можно
использовать одни и те же концепции и идеи супрамолекулярной химии хозяин—гость.
Развитие нековалентной координационной химии анионов продолжалось, хотя
и с перерывами, в 1970-е и в начале 1980-х годов, когда группами Ф. Шмидтхена
(F. Schmidtchen) из Мюнхенского технического университета (Германия) и
Ж.-М. Лена (J.-M. Lehn) из Института Л. Пастера (Париж, Франция) были
синтезированы некоторые принципиально важные хозяева (в основном криптандного
типа, аналогичные катапинандам). И только в конце 1980-х годов, когда новое
поколение химиков обратилось к этой, почти неизвестной, области, комплексообра-
зование анионов стало популярной темой. Даже в вышедшей в 1996 г. книге
«Comprehensive supramolecular chemistry» (J. Atwood et al.) всего лишь одна глава
полностью посвящена связыванию анионов (т. 2, с. 519—552). В 1992 г. Лен,
возможно, с некоторой долей предвидения, назвал область супрамолекулярной координа-
240
4. Связывание анионов
ционной химии анионов полноправным членом супрамолекулярной химии. Его
правота полностью подтвердилась после опубликования первой книги по
координационной химии анионов (A. Bianchi et al., 1997) и большого обзора Ф. Шмидтхена и
М. Бергера (F. Schmidtchen and M. Berger, 1997). Действительно, исследования в
области химии комплексообразования анионов привели к появлению большого
количества разнообразных молекул хозяев. Мастерство химиков проявилось как при
конструировании и синтезе этих соединений, очень различных по строению, так и
при разработке способов связывания анионов. За последние годы в журнале
«Chemical Communications» опубликованы четыре работы групп Этвуда (J. Atwood,
1996), Вира (P. Beer, 1996), Рейнхоудта (D. Reinhoudt; M. Antonisse et al., 1998) и Сес-
слера (J. Sessler; P. Gale et ah, 1998) об аспектах химии анионного связывания.
*"» Schmidtchen F.P. and Berger M. Artificial organic host molecules for anions //
Chem.Rev. 1997. Vol. 97. P. 1609-1646.
Биологические рецепторы анионов
Анионами являются 70—75% ферментативных субстратов и
кофакторов; очень часто это фосфатные остатки (как в АТР и ADP) или неорганические
фосфаты. Такие анионы, как сульфаты и карбоксилаты, тоже характерны для
биохимических систем. Хлорид-анион — главный внеклеточный анион; предполагают,
что функционирование именно хлоридных транспортных каналов обуславливает
циститный фиброз. Прежде чем приступить к обсуждению конструирования и
получения искусственных анионных хозяев, следует обратить внимание на способ,
которым природа осуществляет комплексообразование и транспорт анионов. При
биохимическом связывании анионов ферментные или белковые хозяева всегда
являются частью функционирующей биологической системы (например, биокатализ
и транспорт). При этом природные системы связывания анионов должны не
только иметь большое сродство к аниону-мишени, но и малое сродство ко всем другим
компонентам, присутствующим в клетке или во внеклеточной жидкости
(термодинамическая селективность). Кроме того, они должны быстро и в нужный момент
связывать и высвобождать свои субстраты (кинетическая селективность, см.
разд. 1.6). В результате белки, связывающие анионы, не стремятся к образованию
жестко предорганизованных макроциклических или макробициклических систем,
а формируют гораздо более гибкие, нитевидные, образования, способные в
результате взаимодействий принимать анионсвязывающие конформации. Большое число
взаимодействий белок—анион (стабилизирующий энтальпийный фактор)
компенсирует недостаточную предорганизацию.
*"» Mangani S. and Ferraroni M. Natural anion receptors: Anion recognition by proteins //
Supramolecular chemistry of anions / Ed. A. Bianchi, K. Bowman-James., E. Garcia -
Espafta. New York: Wiley-VCH. 1997. P.63-78.
4.2. Биологические рецепторы анионов
241
Белки, связывающие фосфаты и сульфаты
В работе Ф. Куиоко (F. Quiocho) из Университета в Раисе (Техас, США)
проведены кристаллографические исследования двух бактериальных периплазма-
тических анионтранспортных белков, названных фосфатсвязывающими (РВР) и
сульфатсвязывающими (SBP) белками (Quiocho, 1990). Их функция состоит в
устойчивом связывании аниона сразу после того, как он в результате пассивной
диффузии пересечет мембрану клетки бактерии. Структуры этих двух белков очень
близки. В каждом случае анионы связываются внутри щели глубиной ~8 А,
образующейся при пересечении двух глобулярных областей белка. Основная разница
между двумя структурами, приводящая к практически полной селективности по от-i
ношению к соответствующим субстратам (фактор селективности составляет ~104),i
заключается в оптимальной организации остатков, участвующих в образовании во-i
дородных связей у связывающего центра белка. В случае РВР белок реагирует на то,
что как НРО4-, так и Н2РС>4 могут служить и донорами, и акцепторами при
образовании водородной связи. Все данные по кристаллической структуре этого белка
дают четкую картину связанного НРО^", удерживаемого в общей сложности 12
водородными связями с расстояниями N/O--0 от 2.62 до 2.92 А. В семи из них
участвуют NH-группы основной цепи белка или боковой цепи аргинина, в четырех —
ОН-группы (две сериновые и две треониновые) и в одной — атом кислорода кар-
боксилат-аниона (Asp56), который действует как акцептор водородной связи.
Именно этот акцептор водородной связи обеспечивает селективность
распознавания белка, она отсутствует в SBP. Сульфат-анион в структуре SBP удерживается се-
Рис. 4.2. (а)
Стереоизображение взаимодействий
SBP-сульфат в Salmonella
typhimurium.
(Воспроизведено с разрешения
Pflugrath J. W. and Quiocho
F.A. The 2-A resolution
structure of the sulfate-bind-
ing protein involved in active
transport in Salmonella
typhimurium // J.Mol. Biol.
1988. Vol. 200. P. 175).
(б) Структуры серина, цис-
теина, аланина и глицина
(б)
НО "" СО2Н HS ~ СО2Н
Серии Цистеин
NH2 NH2
СО2Н
Глицин
Me CO2H
Алании
242* 4. Связывание анионов
мью водородными связями от NH-групп каркаса белка, ОН-групп серина и NH-
групп триптофана; они являются донорами водородной связи (рис. 4.2). Сродство
белка к сульфату уменьшается при замене донора водородной связи — серина 130 —
на цистеин (SH вместо ОН), аланин (Me) или глицин (без заместителя) с помощью
направленного мутагенеза. В результате неблагоприятных стерических
взаимодействий сродство в случае цистеина снижается в 3200 раз. Две другие замены,
приводящие к потере одной из семи водородных связей, уменьшают сродство
соответственно в 100 и 15 раз, что равно потере водородной связи с энергией
~7 кДжмоль.
Обзор Кембриджского банка структурных данных также был посвящен
биологическому распознаванию фосфата и сульфата (разд. 6.3). Из этого исследования
следует, что средний угол водородной связи S=O-H, равный 127.9°, примерно на 9°
больше, чем при аналогичном взаимодействии с фосфатом. Близкие значения
углов наблюдаются для водородных связей сульфонильной группы, причем в этом
случае реализуется почти заслоненная конформация. Напротив, фосфорильные
аналоги предпочитают взаимодействия под острыми углами.
> Аргинин как центр связывания анионов
Аргининовый остаток, содержащий гуанидиновую группу, особенно
важен среди белков и ферментов, связывающих анионы. Гуанидиний, протониро-
ванная форма гуанидина, является прекрасным центром связывания анионов,
поскольку он остается протонированным в очень широком диапазоне рН (рКа 13.5 для
исходного CN3H6) и может участвовать в создании двух водородных связей с кар-
боксилатами, фосфатами, сульфатами и др. (рис. 4.3).
Важные аргининсодержащие биологические системы включают супероксидди-
смутазу (Си—Zn-фермент, катализирующий превращение супероксида (О^") в пе-
роксид водорода и молекулярный кислород) и цитратсинтазу
Кристаллическая структура другой системы на основе аргинина — Yersinia про-
теинтирозинфосфатазы (РТР-азы) — была определена с разрешением в 2.5 А в
присутствии вольфрамат-аниона. Протеинтирозинфосфатазы, наряду с киназами,
регулируют уровни фосфорилирования, что необходимо для роста клеток, передачи и
дифференциации сигналов. Yersinia - бактерия, вызывающая бубонную чуму, —
выделяет РТР-азу, подавляющую иммунные клетки хозяина. Структура белка
демонстрирует, каким образом гость WO|~ располагается внутри сетки, состоящей из 12
Аргинин I у
н н
Гуанидиновая группа ; ; Рис. 4.3. Аминокислота
аргинин и возможный
способ его связывания
с карбоксилат-анионами
4.2. Биологические рецепторы анионов 243
водородных связей длиной от 2.7 до 3.4 А. Примечательно, что четыре из них
возникают благодаря гуанидиниевым фрагментам остатков аргинина внутри
фермента; это указывает на важность таких функциональных фрагментов для
биологических систем. Вольфрамат часто используют как модель для других тетраэдрических
анионов, а процесс растворения белковых структур облегчается благодаря
присутствию такого тяжелого атома, как вольфрам. Структура вольфраматного комплекса
отличается от структуры нативного белка тем, что петля, состоящая из 10 остатков,
сместилась в среднем на 3.3 А для капсулирования связываемого аниона. Если
такие же конформационные изменения наблюдаются при связывании фосфотирози-
нового субстрата, то подобные структуры дают четкое представление о механизме
переноса протона при гидролизе, сопровождающем комплексообразование
фермент—субстрат.
4-2.3 Карбоксипептидаза А
Карбоксипептидаза А (СРА) — еще один фермент, использующий ар-
гининовый остаток для связывания анионного субстрата. Роль этого цинксодержа-
щего фермента состоит в гидролитическом расщеплении концевой пептидной или
эфирной связи на карбоксилатном конце полипептидных или эфирных субстратов,
имеющих боковые C-ароматические цепи на последнем остатке. Связывание кар-
боксилат-анионного участка субстрата осуществляется положительно заряженным
аргининовым остатком (Argl45), который образует «солевой мостик» из двух
одинаковых водородных связей (см. рис. 4.3). Существуют также дополнительные
водородные связи с другими остатками, содержащими тирозин (Туг248). В целом это
дает две водородные связи на один кислород карбоксилата. Хороший пример
индуцированного соответствия — участие тирозина, поскольку после связывания
субстрата этот остаток сдвигается со своего места в нативном ферменте на 14 А. (З-Аро-
матическое кольцо субстрата связывается внутри точно соответствующего
Туг 248 ТУГ 248
Рис. 4.4. Стереоизображение связывания L-фениллактата с карбоксипептидазой А (СРА).
(Воспроизведено с разрешения Bianchi A., Bowman-James К., Garcia-Espana E. //
Supramolecular chemistry of anions. New York: Wiley-VCH, 1997. P.65)
244 4. Связывание анионов
гидрофобного кармана, что обеспечивает необходимую селективность ферментов.
Кристаллическая структура активного центра фермента со связанным /,-фениллак-
татом (который действует как ингибитор ферментативной функции) показана на
рис. 4.4. Другие участки, расположенные ближе к поверхности фермента, также
играют роль при связывании полимерных субстратов.
4.3 Концепции конструирования
хозяина для анионов
В гл. 1 было дано определение понятия «хозяин» как соединения со
сходящимися центрами связывания, а понятия «гость» — как соединения с
расходящимися центрами связывания. Очень важно, чтобы хозяин мог отличать одного
гостя от другого. Эти определения полезны и в случае координационной химии
анионов, но по двум причинам мы попадаем в затруднительное положение.
Во-первых, большой размер и высокая поляризуемость анионов предполагают при их
связывании значительную роль ненаправленных взаимодействий, например
дисперсионных. Во-вторых, из-за своего отрицательного заряда анион неизбежно
испытывает электростатическое притяжение к большинству молекулярных
образований, даже не заряженных положительно, а просто благодаря разности заряда
между отрицательно заряженным анионом и электронейтральной молекулой. Это
означает, что дисперсионные и электростатические силы, являясь полностью
ненаправленными, вносят значительный вклад в связывание анионов. Поэтому хозяин
в определенном смысле представляет собой сплошной центр связывания, хотя
некоторые его участки будут взаимодействовать с анионом сильнее, чем другие.
Вторая проблема касается определения центров связывания на самом анионе.
Координационно насыщенная природа анионов и недостаток (за исключением таких
анионов, как Н2РО^) функциональных групп, способных к образованию
водородных связей, означают, что, например, галогениды ведут себя как сферические
заряды и обладают очень гибкой координационной геометрией, не имеющей
специфических центров связывания.
Обобщенное определение хозяина как конвергентного соединения (со
сходящимися центрами связывания) позволяет нам исключить катионы металлов из
числа анионных хозяев, даже если они и проявляют большую селективность в своих
взаимодействиях с анионами (ср. HSAB-теория, см. Дополнение 3.3). Это связано с
тем, что, как правило, на металл приходится по одному центру связывания
отдельного аниона и центры связывания ионов металлов являются расходящимися (никто
бы всерьез не предложил ион Na+ в NaCl в качестве хозяина для связывания
анионов, хотя Na+ и селективен к С1~ в большей степени, чем к 1~, а в кубической
решетке NaCl ион С1~ октаэдрически окружен шестью ионами Na+, рис. 1.9). Ясно,
однако, что бициклические катапинанды D.1) могут быть классифицированы как
хозяева анионов. Они связывают свои анионные гости с помощью двух
сходящихся водородных связей N+—H ••• Х~ (центров связывания) и проявляют размерную
селективность в соответствии с величиной хозяина (определяемой длиной цепи п).
4.3. Концепции конструирования хозяина для анионов 245
Мостиковый [8.8.8]катапинанд D.1а) достаточно слабо связывает анионы;
аналогичный [9.9.9]-хозяин D.16) имеет константу связывания ~102 М для С1~ в
растворителе вода/трифторуксусная кислота с умеренной селективностью к хлоридам
по сравнению с бромидами (примерно в 8 раз), тогда как мостиковый 110.10.10]ка-
тапинанд D.1в) связывает С1~, Вг~ и I" с низкой селективностью. Однако катапи-
нанды не предорганизованы и претерпевают основное конформационное
изменение уже после связывания аниона, что соответствует перестройке от
ом/,ом/-конформера к ш,ш-конформеру (схема 4.1). В действительности, это
процесс того же типа, что и процесс, происходящий с краун-эфирами после
связывания катионов в водном растворе.
N*—Н
out,out- in,m-
Схема 4.1. Конформационное изменение катапинандов при связывании аниона
С учетом этих данных важно знать, насколько бициклическая природа
катапинандов содействует их связыванию с анионами. Помогает ли трехмерная структура
хозяина организовать два центра связывания или же всю ответственность за
организацию берет на себя анион С1~? Это равносильно вопросу о важности предорга-
низации в химии комплексообразования анионов. Низкие константы связывания
катапинандов свидетельствуют о ее неоспоримой важности и о необходимости ее
учитывать при конструировании хозяина, как и в случае хозяев для связывания
катионов.
При конструировании анионсвязывающих хозяев, включающем предорганиза-
цию, необходимо принимать во внимание и комплементарность. Конструирование
комплементарных центров анионного связывания должно учитывать некоторые
фундаментальные характеристики анионов.
Отрицательный заряд. Отрицательный заряд предполагает, что связывать
анионы будут как нейтральные, так и, в особенности, положительно заряженные
хозяева. К сожалению, электростатические взаимодействия не являются
направленными, а поэтому все анионы будут электростатически притягиваться к хозяину,
образуя в растворе сольватированную ионную пару. С ростом заряда анионов
следует ожидать увеличения сродства. Для данного положительно заряженного хозяина
следует учитывать конкуренцию со стороны противоанионов, т.е. на самом деле
любая из наблюдаемых констант связывания будет представлять собой фактор
относительной селективности связывания одного аниона по сравнению с другим.
*>246 ^ Связывание анионов
Основность Льюиса. Подавляющее большинство анионов является основаниями
Льюиса, но есть и некоторые исключения: анионы, у которых нет неподеленных
электронных пар (например, А1Н^", В(С6Н5)^, клозо-Вп^\2~), или анионы,
являющиеся очень слабыми основаниями (например, B(C6F5)^). Это свидетельствует о
том, что хозяева, содержащие кислотные атомы Льюиса, такие, как органические
соединения бора, ртути, олова или катионы металлов, могут быть основой для
создания подходящего хозяина за счет образования координационных связей. Это
привело к появлению хозяев-антикраунов, напоминающих краун-эфиры, у
которых кислород основания Льюиса заменен кислотными центрами связывания
Льюиса (разд. 4.9). Хороший фундамент для конструирования селективных хозяев
создает сильная направленность льюисовских координационных взаимодействий
кислота—основание. Благодаря основности Льюиса анионы могут выступать в
качестве акцепторов водородной связи, так же как в случае связывания галогенида ка-
тапинандами, при котором усиленные зарядами водородные связи N—H--C1
обеспечивают основной вклад в комплексообразующую способность хозяев.
Высокая поляризуемость. За счет высокой поляризуемости анионов возникают
значительные ван-дер-ваальсовы взаимодействия. Будучи ненаправленными, они
относятся ко всей площади поверхности контакта хозяина и аниона; таким
образом, трехмерное капсулирование данного аниона должно усилить связывание всех
анионов, соответствующих хозяину.
Сольватация. Анионы в основном имеют большие энергии сольватации, а
поэтому влияние среды, в которой выполняют эксперименты по комплексообразова-
нию анионов, на измеряемые константы связывания будет еще большим, чем при
связывании катионов. Константы связывания для одновалентных неорганических
анионов в воде имеют значения ~102—103. Это свидетельствует о прочном
связывании, тогда как из-за отсутствия у анионов склонности к сольватации прочная связь
гораздо легче образуется в неполярных растворителях, например в хлороформе.
Полагают, что константы связывания анионов возрастают в следующем ряду
растворителей: Н2О < DMSO < MeCN < СНС13 < СС14, хотя интересные
компенсационные энтальпийно-энтропийные эффекты могут наблюдаться при
комплексообразовании гостей всех видов (см., например, разд. 5.3.3).
Координационное число и геометрия связанного аниона - также потенциально
важные параметры. При поисках комплементарного хозяина для катионов
металлов надо учитывать, что связывание металлов гораздо сильнее в тех случаях, когда
координационное число и геометрия металла соответствуют этим же параметрам
лиганда (октаэдрическая, тетраэдрическая, плоскоквадратная и т.д.). Эта важная
особенность не так существенна в случае катионов щелочных металлов, которые не
имеют четких координационных предпочтений. В кристаллическом NaCl хлорид-
анион находится в регулярном шестикоординационном октаэдрическом
окружении. Напротив, хлорид-анион, капсулированный катапинандами, можно считать
двухкоординационным, поскольку он занят в образовании двух водородных связей
N-H-C1. В действительности, существует много разнообразных видов
координационной геометрии анионов, причем число взаимодействий, на которые способен
анион, возрастает с увеличением его размера. Синтетические хозяева галогенидов
могут участвовать в образовании двух-шести водородных связей в ациклических и
4.4. От катионных хозяев к анионным — простое изменение рН 247
циклических структурах, тогда как большие многоатомные анионы могут давать до
трех водородных связей в расчете на один концевой атом. Таким образом,
структура РВР-белка с его двенадцатью водородными связями, по три на каждый атом
кислорода в фосфате, — это прекрасный пример максимизации координационного
числа для получения наибольшей стабилизации.
4*4
катионных хозяев к анионным -
простое изменение рН
Dietrich В, Design of anion receptors: Applications // Pure Appl. Chem. 1993.
Vol.65. P. 1457-1464.
4*4-1 Тетраэдрические рецепторы
Наиболее очевидный путь, обеспечивающий связывание анионов,
состоит в сочетании электростатического притяжения к положительно заряженному
хозяину с анионным характером основания Льюиса, способного выступать в
качестве акцептора водородной связи. Именно эта стратегия привела Парка и Сим-
мондса A968) к подлинному успеху при работе с катапинандами. Существует
большое число разнообразных криптандов, способных связывать ионы металлов
благодаря тому, что их третичные атомы азота, находящиеся в голове мостика, и
вторичные аминогруппы цепей представляют собой основания Льюиса. Простое
изменение рН раствора должно приводить к протонированию этих аминогрупп в
соответствии с величинами их относительной основности и образованию катапи-
нандоподобного хозяина, который должен связывать катионы. Наиболее важны
следующие факторы: криптанд должен быть достаточно большим, чтобы иметь
возможность включить анион (т.е. таким же, как [2.2.2]криптанд или больше); между
протонированными атомами азота не должно существовать сильного
отталкивания, которое препятствовало бы схождению центров связывания хозяина, и
основность не должна быть такой низкой, чтобы протонирование вообще исключалось.
Прекрасная иллюстрация этого простого подхода - макротрициклический
криптанд C.29), состоящий из четырех колец триаза-18-краун-6. Соединению C.29)
дали название «футбольный мяч» («soccer ball») из-за почти совершенной
сферической формы его молекулы. Наличие четырех атомов азота в головах мостиков этой
молекулы позволяет ее рассматривать в качестве весьма характерного
представителя тетраэдрических рецепторов. Этот сферанд, в дополнение к свойствам
основания Льюиса, позволяющим ему прочно связываться катионами, в своей тетрапро-
тонированной форме способен также связывать такие анионы, как С1~, за счет
четырех водородных связей, усиленных электростатическими взаимодействиями с
эфирными атомами кислорода. Таким образом, его способность к связыванию
зависит от рН среды: нейтральный хозяин может связывать катионы, особенно
аммоний NH^; в дипротонированной форме он связывает нейтральные молекулы,
например воду, а в тетрапротонированной форме — такие анионы, как С1~ (рис. 4.5).
248
4. Связывание анионов
N-Г У^Н.
;н. о
N'
Рис. 4.5. Тетраэдрическое распознавание, зависящее от рН среды: криптанд «футбольный
мяч» C.29) как рецептор катиона аммония, воды или хлорид-аниона
В отличие от катапинандов протонированный криптанд «футбольный мяч» C.29)-
4Н+ имеет крайне сильное сродство к Cl~ (lg K{, > 4, метанол—вода), а его
селективность к С1~ в 50 раз выше, чем к Br~ (E. Graf and J.-M. Lehn, 1976). Такие величины
сродства и селективности представляют собой значительные достижения в химии
комплексообразования анионов (особенно одновалентных), где сродство
хозяин—гость катастрофически мало по сравнению со сродством при связывании
катионов. Согласно кристаллической структуре C.29)-4Н+ с С1 (рис. 4.6), водородные
связи N—H---C1 имеют длину 3.09 А, которая характерна и для катапинандов
C.10 А). Это значит, что оба хозяина геометрически комплементарны к
хлорид-иону. Поэтому большее сродство C.29)-4Н+ к С1~ основано на его большей предорга-
(а)
Рис. 4.6. Кристаллические структуры (а) криптата «футбольного мяча» с хлорид-анионом
C.29)-4Н+ с С1~ и (б) катапината D.3) с иодид-анионом. (Воспроизведено с разрешения
Metz В. et al. A976), Schmidtchen F. and Muller G. A984)
4.4. От катионных хозяев к анионным — простое изменение рН
249
низации, большем положительном заряде и большем числе центров связывания.
Меньшее сродство этого криптанда к Вг~ можно объяснить отсутствием
соответствия между более длинной, чем в случае С1~, водородной связью N—Н -Вг и
размером полости.
N
О / О
N"'x\ .A.?y""N
о \ о
L.VJ
Me-NT"
C.29)
N+-Me
X = 0.D.2)
Х = (СН2J:D.3)
Очевидно, что криптат «футбольного мяча» прекрасно соответствует своему
аниону-мишени с точки зрения предорганизации, размера, заряда и образования
водородных связей. Однако если сравнивать такие хозяева, как «футбольный мяч»
и катапинанд, то не совсем ясно, какой из этих факторов наиболее важен при
определении сродства к анионам и селективности. Еще в 1977 г. Ф. Шмидтхен
(F. Schmidtchen), синтезировав N-метиловый аналог криптанда C.29) -
соединение D.2) — и родственное ему соединение D.3) с углеводородными мостиками,
получил определенные данные о роли водородных связей в этих системах. В обоих
случаях при наличии в молекуле метильной группы хозяин вынужден принимать
конформацию с обращенными наружу заместителями (в противном случае метиль-
ные группы находились бы в непосредственной близости друг к другу в центре
молекулы). Это приводит к тому, что тетракатионная полость соединений D.2) и D.3)
гораздо больше, чем полость у C.29)-4Н+, в результате чего константы связывания
бромид- и иодид-анионов имеют наиболее высокие значения (lg К— 2.25 в воде).
Согласно кристаллической структуре иодидного комплекса, показанной на
рис. 4.6, б, иодид-анион расположен в центре тетраэдрической макроциклической
клетки на расстоянии -4.54 А от катионных азотных центров.
4*4*2 Селективность формы
В случае тетраэдрических рецепторов, рассмотренных в разд. 4.4.1,
координационную геометрию галогенидов (определенную на основе измерений их
кратчайших межмолекулярных расстояний или длин водородных связей) можно
считать тетраэдрической. В предыдущих разделах уже говорилось о том, что
величины координационных чисел галогенидов колеблются от двух до шести. Итак,
существует ли какое-либо доказательство предпочтительной координационной гео-
25«
4. Связывание анионов
метрии или координационного окружения галогенидов, навязанных им предорга-
низацией и топологией хозяина? Глубже вникнуть в эту проблему и понять роль
других факторов, влияющих на селективность связывания анионов, помог анализ
особенностей связывания цилиндрического макробицикла бис(трена) D.4) и его
меньшего аналога D.5). На рис. 4.7 показаны кристаллические структуры крипта-
тов гексапротонированного бис(трена) D.4)-6Н+ с анионами F~, С1~, Вг~ и N^"
(азид), а также криптата D.5)-6Н+ с F'-анионом. Отметим, что в каждом случае
третичный атом азота в голове мостика не протонирован, потому что его
основность очень сильно снижена из-за его близости к протонированным вторичным
атомам азота.
Бис(трен) D.4) D.5)
Константы связывания галогенидов с протонированным бис(треном) в
растворе (lg К) равны 4.19, 3.0, 2.6 и 2.15 для F~, С1~, Вг~ и 1~ соответственно. Как и
фторид-ион, азид-ион тоже связывается очень прочно (lg К— 4.30). Удивительно,
что последовательность изменения констант связывания галогенидов
соответствует последовательности изменения их энергий гидратации, т.е. наиболее сильно
сольватированный фторид-ион связан прочнее всех остальных. Это кажется еще
более удивительным, так как, согласно кристаллической структуре криптата
бис(трена) (рис. 4.7, a), F~-hoh плохо соответствует длинной цилиндрической
полости бис(трена). Анион F~ находится приблизительно в тетраэдрическом
окружении и смещен к одной стороне макроцикла со средней длиной N—H-F водородных
связей 2.72 А. Более крупные хлорид- и бромид-ионы (рис. 4.7, бив) расположены
в полости гораздо более симметрично, взаимодействуя с шестью NH-группами со
средними длинами связей N-H--C1 3.19-3.39 А и N-H-Br 3.33-3.47 А. Следует
отметить, что эти связи гораздо больше оптимальных связей N—H-C1, которые в
случае D.16) и C.29)-6Н+ равны ~3.10 А. В соответствии с этим хлорид-, бромид- и
иодид-ионы связываются гораздо слабее, чем фторид-ион. С точки зрения
топологического соответствия и соответствия формы цилиндрической полости лучше
всего подходит цилиндрический азид-анион (рис. 4.7, г), связывающийся наиболее
прочно. Связи N—H--NJ имеют оптимальную длину в узком интервале значений
2.81—3.02 А, что свидетельствует о симметричном связывании, возникающем в
результате высокой комплементарности. По селективности к бис(трену) D.4)
одновалентные анионы располагаются в следующем порядке: СЮ^, 1~, Вг~, СГ <
< СН3СО^" < НСО^~ < N0^, N0^ < F~, Щ. Эта последовательность не коррелирует
с размерами анионов, значениями энергии гидратации или с таким, менее
очевидным рядом, как ряд Хофмейстера, в котором анионы группируются в соответствии
4.4. От катионных хозяев к анионным — простое изменение рН
251
(а)
(в)
(г)
Рис. 4.7. Кристаллические
структуры: криптатов бис(трена)
D.4)-6Н+ с (a) F", (б) С1-, (в) Br~,
(г) Nj и ((?) криптата D.5)-6Н+ с F"
252
4. Связывание анионов
с их способностью «высаливать» белки из воды. Ряд Хофмейстера, или лиотропный
ряд, впервые описанный в 1888 г., представляет собой следующую
последовательность анионов: С1О^ > Г > SCN" > NO3~ > ClOj > Вг > С1~ » F", IOj > СН3СО^,
СО3~ > НРО^", SO!" > цитрат3", где в левой части представлены анионы,
растворяющие и денатурирующие белки в концентрированных солевых растворах, а в
правой — анионы, осаждающие белки. Полагают, что этот ряд характеризует
упорядочение водного растворителя, индуцируемое различными соединениями, а также
соответствует энергии гидратации анионов (W. Jencks, 1969). Действительно,
связывание с бис(треном) D.4) складывается из электростатической составляющей,
определяющей более прочную связь анионов, имеющих относительно большую
плотность заряда, например F~, и топологической комплементарности (т.е. форма, а не
размер), благоприятной для азид-иона (который также обладает высокой
плотностью заряда). Электростатический фактор становится заметным при рассмотрении
сродства таких многозарядных ионов, как Р2С>7~ (lg K= 10.30), АТР4" и ADP3~. Все
эти анионы связываются очень прочно, причем частицы с зарядом 4— связываются
сильнее, чем частицы с зарядом 3-. При сочетании всех этих факторов (компле-
ментарность размера и формы, сильные электростатические взаимодействия и
многочисленные водородные связи) наблюдается чрезвычайно сильное
связывание. Маленький хозяин D.5)-6Н+ в высшей степени подходит для фторид-иона
(рис. 4.7, д). При его связывании реализуется искаженная тригональная
призматическая геометрия с расстояниями N—H---F" в пределах 2.76—2.86 А, константа
связывания в воде (lg К) составляет 11.2 (В. Dietrich et al., 1996).
*"** Dietrich В., Guilhem /., Lehn J.-M. et al. Molecular recognition in anion
coordination chemistry // Helv. Chim. Acta. 1984. Vol.67. P.91-104.
4-4*3 Двухмерные хозяева
Координация анионов в водных растворах была изучена для большого
семейства макроциклов азакорандного типа, таких, как D.6)—D.11),
представляющих собой азотсодержащие аналоги краун-эфиров. Константы связывания и про-
тонирования легко определяют потенциометрическим титрованием (см.
Дополнение 3.1). Часто приходится иметь дело с целым рядом соединений,
протонированных в различной степени, а это означает, что результаты
экспериментов по связыванию анионов нужно рассматривать как сумму вкладов от нескольких
равновесий в растворах. Поэтому при обработке данных титрования могут быть
допущены большие ошибки. Поскольку вода является сильнорастворяющей средой с
большой диэлектрической постоянной, то в основном имеют значение только
взаимодействия между многозарядными частицами; при этом по мере увеличения
заряда образуются термодинамически чрезвычайно стабильные комплексы.
Потенциометрическое титрование показало, что гексациклен D.6), [18]aH-N6,
являющийся азааналогом 18-краун-6, в гексапротонированной форме
представляет собой очень сильную двухосновную кислоту. Для большинства практических
целей во всех растворах, кроме очень кислотных, гексациклен следует рассматривать
как тетрапротонированный макроцикл D.6-4Н+). Это общее наблюдение для
макроциклов типа D.11), в которых атомы азота разделены только двумя атомами угле-
4.4. От катионных хозяев к анионным — простое изменение рН 253
рода. Протонирование всех атомов азота в таких макроциклических соединениях
приводит к резкому электростатическому отталкиванию между ИН^-группами и
сильному уменьшению величин рКа по сравнению с таковыми ациклических
аналогов, которые, будучи полностью протонированными для минимизации
отталкивания между катионными группами, почти всегда принимают линейную all-anti-
конформацию. В растворе гексациклен предпочитает связывать нитрат-ионы, а не
анионы галогенидов, но результаты РСА показывают, что ни один из этих анионов
не включается внутрь макроцикла. Галогениды принимают геометрию типа
«насест» с расстоянием N—Н-С1~ 3.07—3.28 А, тогда как, согласно кристаллической
структуре D.6)-4H+-2Cl~-2NOj, нитрат-анионы взаимодействуют с хозяином не
прямо, а через включенные молекулы воды. В кристаллах D.6)-6H+-2Cl~~4NO^
реализуются прямые взаимодействия с нитрат-анионами, при этом каждый анион
связывается с помощью одной водородной связи. Очевидно, что полость гексацик-
лена, близкая по размеру к полости 18-краун-6, но частично заполненная
протонами NH-групп, слишком мала для включения анионов. А. Бьянки (A. Bianchi) из
Университета Флоренции (Италия) и Э. Гарсиа-Эспанья (Е. Garcia-Espana) из
Университета Валенсии (Испания) исследовали циклические соединения гораздо
больших размеров - до [З6]ан-Ы|2. Такие макроциклы с большими кольцами способны
служить хозяевами для больших неорганических анионов, в частности для [PdCl4]2~
и [Fe(CNN]4~, которые могут быть включены внутрь макроциклической полости. В
качестве примера на рис. 4.8 представлена кристаллическая структура Н10[30]ан-
N{[j+ с гостем [PdCl4]2~ . Термин «суперкомплексы» для таких соединений был за-
■ в > v " н
ttrS x—NH HN-^ /—NH HN^
H H
.N N.
Гексациклен D.6) D.7) \ / D.8)
NH HN ^-NH HN
Н J NH „ HN
NH HN-, / "" \ [ N
NH
NH HN
NH
/ ^O n-^
NH HN
О
1
NH HN^
D.9) D.10) D.11): n = 2ч-7
254
4. Связывание анионов
Рис. 4.8. Кристаллическая
структура [РAС14]2~-комплекса
соединения H^pOlaH-NJj^
(по BenciniA. eta!., 1990)
имствован из химии металлокомплексов. Элегантность и простота этой структуры
не должны вводить в заблуждение: этот макроциклический амин из-за большого
числа различных протонированных состояний отличается сложностью поведения в
растворе.
Те же исследователи изучили комплексообразование [Fe(CNN]4~ с рядом мак-
роциклических азакорандов. Кривые распределения (состав раствора как функция
рН) для этого аниона и его комплексов с [27]aH-N9 показаны на рис.4.9. Как видно
из этих зависимостей, в растворе присутствуют все соединения — от тетрапротони-
рованных до октапротонированных циклов; действительно, даже при рН 9 и выше
макроциклы могут сохранять четыре протона. Анион гексацианоферрата(П)
представляет интерес, потому что помимо потенциометрии, мониторинг его
связывания можно проводить с помощью циклической вольтамперометрии (Дополнение
4.1). В этих соединениях редокс-пара Fe(II)/Fe(III) обратима и чувствительна ко
второй координационной сфере атома железа. На рис. 4.9 показан сдвиг редокс-
(окислительно-восстановительного) потенциала как функция рН. Из рисунка
следует, что с возрастанием степени протонирования хозяина окисление аниона стано-
100 Н
50 -
Fe(CN)£
H7LlFe(CNN3+ H5LlFe(CNN+j
. eH6LlFe(CNN2+
Рис. 4.9. Кривые распределения ( )
для системы H+/[Fe(CNN]47[27]aH-N9
и зависимость редокс-потенииала
Fe(II)/Fc(III)(£-1/2)C)OTpH.
(Воспроизведено с разрешения BianchiA.
et al. // Inorg. Chem. 1987. Vol.26. P.3902)
4.4. От катионных хозяев к анионным — простое изменениерН 255
Таблица 4.2. Константы связывания (lg К) для различных стадий образования
суперкомплексов азакорандов D.11) {п = 3 -*- 5) с [Fe(CNN]4~ в соответствии
с приведенными реакциями
Реакция
л=3 л = 4 л=5
■ = [H4D.11)Fc(CN6)] 4.06 3.69 3.61
■ = [H5D.11)Fe(CN6)]+ 5.63 4.78 4.66
[Fe(CNN]4-=[H6D.11)Fe(CN6)]2+ 7.60 6.23 5.72
]4~= [H7D.11)Fe(CN6)]3+ 9.33 7.92 6.93
|4+ - 9.03 8.07
вится все более трудным. Отнимать электрон у комплексов хозяин—гость с
большим положительным зарядом становится труднее. Кроме того, потенциал растет не
монотонно, а ступенчато в соответствии с пиками концентрации различных
соединений в растворе. В табл. 4.2 представлены константы для различных стадий
образования суперкомплексов азакорандов D.11) в растворе.
Во-первых, таблица показывает, что в водном растворе константы связывания
для всех образующихся комплексов очень велики (по сравнению с комплексами
одновалентных анионов), что объясняется большими зарядами взаимодействующих
соединений. Во-вторых, с ростом заряда макроцикла явно увеличивается и
стабильность этих комплексов, что соответствует электростатической природе
взаимодействий. Наконец, следует также отметить, что наиболее стабильные
комплексы образуются при участии наименьшего макроцикла, у которого плотность
положительного заряда выше (меньшее влияние непротонированных NH-rpynn).
Взаимодействия хозяин-анион N+—Н- NCFe~ относятся к типу взаимодействий,
усиленных зарядом, и поэтому если у макроциклов меньше четырех протонов —
того минимума, который необходим для нейтрализации заряда, то комплексы вообще
не образуются.
Для того чтобы исключить проблемы, связанные с поведением полностью
протонированных макроциклов, например гексацилена и его высших аналогов,
имеющих две СН2-группы между атомами азота, было получено большое количество
других соединений. Введение пропильных спейсеров между протежированными
атомами азота в макроциклах типа D.7) и D.8) позволяет развести эти атомы на
большее расстояние. В менее конформационно гибких макроциклах D.9) и D.10)
содержатся эфирные атомы кислорода, необходимые для разделения
положительных зарядов. Эта стратегия оказалась успешной в том смысле, что позволила
получить для всех полностью протонированных лигандов значения рКа выше 7.
Зависимость р/С, макроцикла от длины спейсера для ряда [4«]aH-N4
D.12) показана в табл. 4.3. Тип анионного связывания для D.7)—D.10) подобен
типу, наблюдаемому для связывания соединения D.11), только с ростом заряда
аниона их константы связывания увеличиваются. Для одинаково заряженных анионов
наблюдаются структурные эффекты: анионы с симметрией, соответствующей
симметрии макроциклов, связываются с макроциклом более прочно (например, ось
25$
4. Связывание анионов
Таблица 4.3. Величины рА^ для макроциклов типа Hw[4«]aH-N^+ D.12)
(п = 2 -5- 4; т = 3, 4) в зависимости от длины спейсера
Соединение
D.12а)
D.126)
D.12в)
л
2
3
4
Р*«з
1.7
6.9
10.6
Р*«4
<1
5.4
8.9
н н
сн2
п
сн,
Н2 \п D.12)
симметрии третьего порядка у SO| с D.10)-6Н+, ось симметрии четвертого
порядка у скварат-аниона С4С>4~ с D.8)-8Н+). Кроме того, большие анионы прочнее
связываются с большим макроциклом D.8)-8Н+. Интересно, что константы
связывания для таких многозарядных анионов, как АТР, значительно выше, чем для
аналогичных ациклических соединений, например спермина
H2N(CH2KNH(CH2LNH(CH2KNH2. Это указывает на ощутимый макроцикличес-
кий эффект анионного связывания, подобный эффекту, обнаруженному для краун-
эфиров в роли катионных хозяев. Способность АТР и ADP к связыванию с такими
макроциклами, как D.9), стала основой для синтеза АТР (разд. 9.4).
Способность моноциклических хозяев типа азакраунов, к распознаванию
анионов по размеру и форме была изучена при получении двух связывающих доменных
систем D.13). Предполагалось, что дитопные хозяева D.13), обладающие полостью,
которая меняет размер в зависимости от длины спейсера (СН2)Л, должны проявлять
селективность к а,ш-дикарбоновым кислотам ~O2C(CH2)WCO2~ в соответствии с их
длиной (определяется длиной (СН2)т-группы), рис. 4.10. Те дианионные гости,
которые в наибольшей степени соответствуют полости, должны связываться сильнее
всего. Зависимость констант связывания соединений D.13) с п = 7 и 10 от длины
гостя, т, показана на рис. 4.11. Кривые имеют умеренные пики селективности,
означающие размерное соответствие хозяина и гостя, несмотря на относительную
гибкость обоих партнеров.
D.13)- а,ш-дикарбоновая
кислота: п = 7, 10
Представители еще одного интересного класса рецепторов (хозяева) анионов,
содержащих протонированные атомы азота, - расширенные порфириновые мак-
4.4. От кат ионных хозяев к анионным — простое изменение рН
257
Рис. 4.10. Распознавание
моноциклическими хозяевами
D.13) а,ы-дикарбоновых кислот
в соответствии с их длиной
роциклы, например D.14), сапфирин, и D.15). Тетрапиррольные порфириновые
макроциклы - превосходные хозяева для таких катионов металлов, как Fe2+ и Mg2+
(например, гемоглобин и хлорофилл, разд. 2.3, 2.4); однако размеры их полостей
слишком малы для включения анионов. Напротив, расширенные порфирины,
например D.14), содержащие пять или более пиррольных остатков, имеют жесткую
макроциклическую полость диаметром ~5.5 А, в которой NH-группы способны
связывать анионы. Дипротонированный сапфирин D.14) образует со
фторид-ионом в растворе метанола весьма стабильный комплекс (lg K= 5.0).
Кристаллическая структура этого комплекса (рис. 4.12) с пятью короткими симметричными
связями N-H-F длиной в интервале 2.697-2.788 А отражает высокую степень
Рис. 4.11.
Зависимость констант
связывания хозяев
D.13) от длины {т)
спейсера дикарбок-
си-гостя
4.5 -
4 -
3.5 -
3 -
2.5
«=10
3 4 5
Длина цепи (т)
17 - СТИДЛа В
258
4. Связывание анионов
Рис. 4.12. Кристаллическая
структура комплекса
расширенного порфиринового
хозяина D.14) с капсулированным
фторид-ионом.
Фторид-анион целиком локализован в
плоскости макроцикла
(по Shionoya M. et aL, 1992)
соответствия аниона полости сапфирина. Тот факт, что полость этого, очень
большого, макроцикла идеально подходит для капсулирования самого маленького
аниона, свидетельствует о проблемах, с которыми приходится сталкиваться при
получении хозяев достаточно больших размеров для эффективного связывания
анионов. В противоположность этому, в аналогичной структуре с хлорид-анионом
из-за плохого соответствия партнеров связывание двух хлорид-анионов происходит
над и под плоскостью макроцикла. При этом константа связывания хлорид-иона
более чем в Ю3 раз меньше, чем константа связывания фторид-иона. Фосфатные
остатки органо- и полифосфатов также связываются одинарными Р—О~-связями,
принимая конформацию «насест» как раз над плоскостью макроцикла, и поэтому
сапфирин пригоден для транспорта фосфатов.
R1 R1
R2
R1 R1
R
R2 R2
R1 = Me, R2 = Et: D.14)
V-R1
R2
R1 = Me, R2 = Et: D.15)
4.4. От катионных хозяев к анионным — простое изменение рН 1259
Для получения хлоридселективных макроциклов на основе полипиррола были
сконструированы неароматические расширенные порфирины, обладающие
гораздо большей полостью, чем традиционные порфирины. Так, соединение D.15), по
данным РСА, хорошо подходит для связывания хлорид-иона четырьмя
водородными связями N—H---C1" длиной 3.11—3.25 А. Предполагают, что комплекс
хозяин-хлорид-ион образуется в газовой фазе и его можно обнаружить масс-спектро-
скопией с бомбардировкой быстрыми атомами (fast atom bombardment, FAB).
Константы связывания в растворе СН2С12 (являющемся гораздо менее полярным,
растворителем, чем требуется в соответствии с низкой растворимостью
макроцикла в более полярной среде) равны 2-Ю5 М для С1~ и 1.4-104 М для F".
Расширенные порфириновые системы были также изучены в экспериментах по переносу
анионов из одного водного слоя в другой через жидкую мембрану из СН2С12
(рис. 4.13). Показано, что D.15) — гораздо лучший переносчик фторид-иона, чем
D.14), но С1~-ион оно не переносит столь же эффективно. Это согласуется с более
сильным связыванием иона С1~ соединением D.15) и более сильным связыванием
иона F~ соединением D.14). При этом после переноса анионов комплексы
неохотно расстаются с ним (ср. с рецепторами и переносчиками катионов, см. разд. 3.9.2).
Исходный поток анионов, Ф (определяемый как число молей анионов,
переносимое в единицу времени), равен 5.03 мкмоль-ч для F~ и 1.56 мкмоль-ч для С1~.
Далее обнаружили, что при введении СГ~ перенос F~ замедляется. В отсутствие
Водный слой
NaOH
рН12
Рис. 4.13. Одновременный перенос
(симпорт) Н+ и X" через жидкую
СН2С12-мембрану (X = F, C1)
СН2С12
260
4. Связывание анионов
макроцикла перенос практически не наблюдали. Была также отмечена постепенная
нейтрализация рН с обеих сторон жидкой мембраны, что согласуется с механизмом
одновременного переноса (симпорт), согласно которому Н+ и галогенид-анионы
переносятся одновременно и в одном направлении (ср. с переносом Na+ и К+ через
клеточные мембраны (антипорт), см. разд. 2.1).
*""* Boudon S., Decian A., Fischer /. et al. Structural and anion coordination features of
macrocyclic polyammonium cations in the solid, solution and computational phases //
J. Coord. Chem. 1991. Vol. 23. P. 113-135.
4*4*4 Циклофановые хозяева
В качестве анионсвязывающих хозяев с большим успехом применяли
множество циклофанов (соединения, содержащие ароматические кольца) с прото-
нированными аминогруппами. В таких лигандах в качестве спейсера обычно
используют фрагмент дифенилметана. В циклофановых хозяевах, предназначенных
для связывания как анионов, так и нейтральных молекул, эти фрагменты
необходимы для придания кривизны и увеличения размера полости хозяина при
сохранении его жесткости. Макроцикл D.16) связывает анионы, например 8-анилино-
1-нафталинсульфонат (ANS) D.17), который служит флуоресцентным зондом для
спектрофотометрического определения сродства хозяин—AN S (кривизна хозяина,
в том числе D.16) и ряда других лигандов циклофанового типа, обсуждается в
разд. 5.3.4.5). Связывание ANS происходит за счет гидрофобных и я-я-стэкинг-
взаимодействий, а также электростатических взаимодействий и водородных
связей, что характерно для нейтральных молекул, прочно удерживаемых такими
хозяевами.
D.16)
D.17)
Бициклический лиганд D.18) является эффективным хозяином для терефталат-
дианиона. Согласно кристаллической структуре этого комплекса, анион полностью
капсулируется бициклической системой хозяина и удерживается в ней благодаря
водородным связям N—Н---О, а также гидрофобным, ван-дер-ваальсовым и п—п-
стэкинг-взаимодействиям. После комплексообразования с терефталат-дианионом
два атома азота в головах мостиков молекулы хозяина смещаются по направлению
к гостю примерно на 1.8 А, тем самым обеспечивая необходимое соответствие.
4.4. От катионных хозяев к анионным — простое изменение рН 14б1
Нуклеотиды также связываются этим хозяином с lg K= 4 + 5. (Нуклеотиды
содержат анионный «хвост» фосфата, связанный с сахарным остатком, который, в свою
очередь, прикреплен к нуклеооснованию. Если последнее — аденин, то это адено-
зинтрифосфат (АТР4".) При отсутствии фосфатного «хвоста» соединение
«основание + сахар» называют нуклеозидом). Другие бициклические циклофановые
хозяева, например D.19) и D.20), связывают анионных гостей, уже будучи
протонированными. В частности, рН-титрование и данные ЯМР показали, что при
образовании 1:1-комплексов соединением D.20) с рядом анионов величина lg
А"находится в пределах 2.5-6.0 для одно- и двухвалентных анионов. Удивительно,
однако, что, согласно кристаллической структуре этого соединения (например, его
нитрата), анионы располагаются с внешней стороны полости, несмотря на то, что
даже в растворе кинетика обмена хозяин—гость указывает на их капсулирование.
Отсюда следует, что сравнение кристаллографических данных для твердого
состояния и результатов, полученных в растворе, необходимо проводить с большой
осторожностью.
D.18) • терефталат2
NH
D.20)
262
4. Связывание анионов
Рецепторы на основе гуанидиния
Ион гуанидиния часто используют при конструировании хозяев для
комплексообразования анионов. Интерес к нему был подогрет тем, что гуаниди-
ний-ион, как правило, входит в состав аргининовых остатков в природных анион-
связывающих хозяевах (разд. 4.2.2). Ион гуанидиния имеет рКа 13.5; следовательно,
он протонирован, а потому положительно заряжен и является эффективным
донором водородной связи в широком интервале рН. В твердом состоянии метилгуани-
диний дает с дигидрофосфатом 1:1-комплекс, что говорит о бидентатном типе его
координации с образованием водородных связей (рис. 4.14). Пытаясь использовать
преимущество такого поведения макроциклического хозяина, Б. Дитрих и другие
(В. Dietrich et al., 1979) получили макромоноциклы D.21) и D.22). Заряд 3+ и
симметрия третьего порядка соединения D.21), вероятно, свидетельствуют о том, что
этот хозяин должен прочно связывать, в частности, анион РО^~. Из-за низкой
брёнстедовской кислотности протонированных фрагментов гуанидиния хозяину
следовало бы оставаться протонированным в условиях сильной основности,
требующейся для существования РО^~~. Удивительно, но величины lg AT для макроциклов
D.21) и D.22) равны всего лишь 2.4 и 1.7 соответственно. Первоначально
предполагалось, что это результат низкой плотности заряда фрагмента гуанидиния, а значит,
и слабых электростатических взаимодействий, хотя это не совсем понятно, если
принять во внимание его распространенность в природе. Может быть, макроциклы
слишком малы, чтобы принять большие фосфатные анионы, и недостаточно гибки,
чтобы направить свои гуанидиниевые фрагменты к связанному аниону,
находящемуся в геометрии «насест». Возможно, прочному связыванию вредит и большая
степень сольватации иона гуанидиния в полярных растворителях.
н
D.21)
D.22)
Несмотря на эти, не слишком оптимистические, результаты, работа по
созданию хозяев на основе гуанидиния проводилась весьма активно. Большинство
новых систем — ациклические аналоги подандных хозяев для катионов. Эти
соединения сочетают легкость синтеза (не требуется высокого разбавления) с быстрой
кинетикой комплексообразования/диссоциации, характерной для биологических
систем благодаря предорганизации. В случае простых соединений бис(гуанидиния),
4.5. Рецепторы на основе гуанидиния
1263
Рис. 4.14. Кристалическая структура
д и гидрофосфата метилгуанидиния
например D.23), происходит прекрасное кооперативное связывание с фосфоди-
эфирами (R2O2PO^); константы связывания в ацетонитриле составляют -5-104 М.
Хозяева также действуют как катализаторы в реакциях тря«с-этерификации с
300-кратным увеличением скорости по сравнению с реакцией, проводимой без
катализатора.
Прорыв в области исследования анионных хозяев на основе гуанидиния
наступил в 1984 г. после синтеза Ф. Шмидтхеном (F. Schmidtchen) бициклического
производного D.24). Наличие в его молекуле углеводородного каркаса поразительно
уменьшает сольватацию фрагмента гуанидиния и увеличивает его липофильность,
приводя к образованию комплексов, например, с л-нитробензоатом в качестве
гостя; константы связывания в хлороформе равны -1.4-105 М~1. Интересно, что
С^-симметричные хиральные производные могут быть получены из хиральных
аминокислот по методу, показанному на схеме 4.2.
Схема 4.2. Синтез хирального гуанидиниевого хозяина из хиральной аминокислоты
путем циклизации открытых цепей триамина или несимметрично замещенной
тиомочевины (по Peschke W. et al., 1995)
264
4. Связывание анионов
Н
Н
Н
Н Н
D.24)
D.23)
Хиральное производное D.25) способно экстрагировать N-ацетил триптофан
(производное аминокислоты) из водного раствора рацемата в хлороформ со
скромным 17%-м избытком диастереомера (diastereomeric excess, de). Связывание
происходит благодаря образованию водородных связей и п—я-стэкингу гуанидиний-иона
соединения D.25) с ацетил-анионом. При искусном молекулярном
конструировании один из нафталиновых остатков соединения D.25) был заменен на краун с
образованием производного аза-18-краун-6 D.26), что обеспечило дитопное
распознавание и карбокси-, и NH^-группы у таких немодифицированных аминокислот, как
триптофан D.27) и фенилаланин D.28). В этом случае для L-изомеров (A. Galan
et al., 1992) были получены значения de, равные 80%. Работы по молекулярному
моделированию свидетельствуют о том, что вклад группы гуанидиния составляет
примерно половину от общей энергии связывания: одну треть дает связывание ^
группы краун-эфиром, а остальное — это вклад тг—тг-стэкинг-взаимодействий.
О
D.25)
D.26)
4.6. Металлоорганические рецепторы 265
Бициклические производные гуанидиния были также встроены в рецепторы,
способные распознавать тетраэдрические оксо-анионы, например полифосфатные
остатки важных биологических молекул. Гибкость рецептора D.29) позволяет двум
гуанидиниевым фрагментам перемещаться с сохранением взаимно
перпендикулярной ориентации, что обеспечивает более эффективное связывание полифосфатов
даже в воде.
о
•""» Наппоп C.L. and Anslyn E. V. The guanidinium group: Its biological role; synthetic
analogs // Bioorganic chemistry frontiers / Ed. H. Dugas, F.P. Schmidtchen.
Heidelberg: Springer, 1993. P. 193.
4*6 Металлоорганические рецепторы
В 1989 г. П. Бир с сотрудниками из Оксфордского университета
(Великобритания) получил бис(кобальтоцениевый) рецептор D.30). Этот металлоорга-
нический макроцикл, имеющий два положительных заряда на центрах Со(Ш),
способен связывать анионы исключительно за счет электростатических
взаимодействий. Бир и сотрудники быстро поняли, что значительно больших
сродства к аниону и селективности можно достичь при дополнительном введении
функциональных групп, способных образовывать с анионом водородные связи.
Тогда этот фактор, наряду с положительно заряженным металлом, будет
увеличивать сродство к аниону. Эти представления были использованы ими при синтезе
ряда амидсодержащих хозяев, таких, как бис(амид) D.31). Действительно, согласно
кристаллической структуре этого соединения, оно может взаимодействовать с
анионами Вг~ с помощью водородных связей N-H-Вг и С-Н-Вг~ (рис. 4.15). Этому
соответствуют большие сдвиги протонов NH-групп в сторону уменьшения поля
(несколько миллионных долей), установленные ]Н ЯМР-спектроскопией при
титровании различными анионами в диполярных апротонных растворителях,
например DMSO и MeCN. Использование апротонных растворителей важно, поскольку
кислый протон NH-группы обменивается на дейтерий, что ведет к потере ЯМР-
сигнала от этого ядра в D2O. Циклическая вольтамперометрия (Дополнение 4.1),
наряду с !Н ЯМР, также является надежным «инструментом» для наблюдения за
4. Связывание анионов
Рис. 4.15. Кристаллическая
структура бромидного
комплекса бис(амида) D.31)
связыванием гостя: сдвиг редокс-потенциала пары Со(Ш)/Со(И) обусловлен
близостью анионов-гостей, которые образуют вторую координационную сферу
металла. Найдено, что в присутствии С1~ сдвиг потенциала (АЕу2) Для редокс-пары
Со(Ш)/Со(П) в бис(амиде) D.31) составляет 85 мВ (рис. 4.16), тогда как в
присутствии Н2РО^ — 240 мВ. Таким образом, фрагмент кобальтоцения непосредственно
способствует связыванию аниона благодаря положительному заряду и кислотности
150т
100
50-
-50
-100
(а)
0.5 0 -0.5 -1 -1.5 -2
Е,В (относительно Ag/Ag+)
Рис. 4.16. Циклическая вольтамперограмма
для D.31): (а) в отсутствие и (б) в
присутствии С1~. (Воспроизведено с разрешения
Beer P.D. Transition-metal receptor systems for
the selective recognition and sensing of anionic
guest species//Ace. Chem. Res. 1998. Vol. 31.
P. 71-80)
4.6. Металлоорганические рецепторы
Льюиса, а также позволяет провести электрохимическое определение процесса
анионного связывания.
О
Y V
0
D.30)
Y6 «V
D.31)
о
о о
= (CH2J,(CH2K,(CH2L,
дифенилметан: D.32)
ОМе
ОМе
Дополнение 4.1. Циклическая вольтамперометрия супрамолекулярных
соединений
Циклическая вольтамперометрия — это электрохимический метод (мониторинг
процессов, происходящих вблизи поверхности электрода), в котором измеряют
зависимость величины тока от приложенного напряжения. Потенциал увеличивается с
постоянной скоростью (обычно 10—500 мВс) от исходной величины £, (измеряемой
относительно насыщенного каломельного электрода сравнения или потенциала пары
Fe(II)/Fe(III) в ферроцене) до максимальной величины Es, а затем возвращается к
исходной величине (рис. 4.17, а). Любые обратимые окислительно-восстановительные
процессы, происходящие при изменении потенциала в прямом и обратном
направлениях, дают характерную вольтамперограмму наподобие показанной на рис. 4.17, б.
Экспериментальная установка изображена на рис. 4.18. Следует отметить, что раствор
не перемешивается во время измерения, а поэтому восстанавливается или окисляется
только то соединение, которое находится у поверхности электрода. Это позволяет
многократно повторять эксперимент, по мере того как свежие реагенты
диффундируют к электроду из объема раствора.
С помощью циклической вольтамперометрии можно получить информацию о ре-
докс-активности соединения, о стабильности и доступности его восстановленной и
26Й
4. Связывание анионов
(а)
(б)
Е, мВ
Е.
/, мкА
Начало редокс-
процесса
Время, с
El/2 Потенциал, мВ
Рис. 4.17. (а) Временная развертка потенциала в циклическом вольтамперометри-
ческом эксперименте, (б) Зависимость тока от потенциала (вольтамперограмма) для
обратимого окислительно-восстановительного процесса; / — ток в анодном пике,
/ с — ток в катодном пике (в предположении сканирования к положительному
значению потенциала), Е- электродный потенциал, обычно очень близкий к
формальному окислительно-восстановительному потенциалу изучаемого редокс-процесса
окисленной форм. Для полностью обратимого химического процесса /^должно быть
равно /" т.е. все соединение, окисленное на поверхности электрода, должно
восстановиться при обратном ходе (или наоборот). Если это условие не выполняется, то
процесс может быть частично обратимым (/ < / ) или необратимым (/ = 0). Если
рс ра'
•рс
процесс не полностью обратим, то это указывает на разложение или химическую
реакцию восстановленных и окисленных соединений; тогда отношение ipa к ipc будет
Вспомогательный У
электрод
Электрод сравнения
Рабочий электрод'
Потенциостат
Записывающее
устройство
Газ прокачки N2
Неперемешиваемый
раствор образца
Рис. 4.18. Схема прибора для проведения циклического вольтамперометрического
эксперимента
4.6. Me таллоорганические рецепторы 269
сильно зависеть от скорости сканирования потенциала, поскольку обратный ток
связан со временем жизни соединения, образующегося в ходе
окислительно-восстановительных процессов. Следует отметить, что химически обратимые процессы (в том
смысле, что и восстановленные, и окисленные соединения стабильны) могут и не
быть электрохимически обратимыми (термин, относящийся к относительным
скоростям прямого и обратного переноса электронов). Характерная черта
электрохимически обратимых процессов — расстояние между пиками потенциалов прямого и
обратного процессов, равное точно 59 мВ.
С точки зрения специалиста в области супрамолекулярной химии более важным
является сильное влияние электронного окружения редокс-центра на окислительно-
восстановительный потенциал Е. Так, в эксперименте по связыванию анионов
большая константа связывания приводит к тому, что заряженный анионный гость в
среднем будет достаточно долго находиться вблизи редокс-центра (например, в случае
металлоцениевого хозяина как ионная пара в его второй координационной сфере).
Это сильно изменяет окислительно-восстановительный потенциал металлического
центра, затрудняя его восстановление и/или облегчая его окисление. Таким образом,
типичный сдвиг потенциала Д/Г, равный 0—250 мВ, возникает из-за связывания.
Цилическая вольтамперометрия может также быть использована для оценки
взаимного сродства восстановленных и окисленных хозяев и гостей методом титрования.
Например, рассмотрим связывание анионного гостя (G) с хозяином (Н), содержащим
Cu(II) или Cu(I). Титрование хозяина Н пробой гостя G и запись циклической вольт-
амперограммы после каждого добавления дают кривую титрования £",/2 в
зависимости от концентрации гостя G, которую можно проанализировать, используя уравнение
D.1).
RT
+ In
1«F 1 + *bu(ii)[G]
D.1)
где R — газовая постоянная; Т— абсолютная температура; п — число электронов,
участвующих в редокс-процессе (в данном случае — один); F — постоянная Фарадея;
^Cu(i) и ^Cu(ii) ~ константы связывания G с Н для форм последнего с Cu(I) и Cu(II)
соответственно. Гораздо более простой способ состоит в записи величины Е^2 Д° и
после добавления большого избытка гостя (G) к раствору хозяина (Н) и
использовании уравнения D.2). Задача упрощается, если при сильном связывании в растворе
будут наблюдаться отдельные редокс-пики и для связанных, и для несвязанных
соединений, как при ЯМР-титровании (ср. Дополнение 1.1).
RT
Em(H) = £1/2(HG) + In
1/2
nF
К.
red
D.2)
•""» Kaifer A.E. Electrochemical techniques // Comprehensive supramolecular chemistry /
Ed. J.L. Atwood, J.E.D. Davies, D.D. MacNicol, F. Vogtle. Oxford: Pergamon, 1996.
P. 499-535.
4. Связывание анионов
Бир (P. Beer) и сотрудники, используя сочетание ЯМР-спектроскопии и
электрохимии для изучения связывания анионов, продолжили работу по
конструированию бис(кобальтоцениевых) хозяев D.32) и макроцикла D.33). Соединения D.32)
связывают галогенид-анионы, однако при увеличении длины спейсерной группы R
эффективность связывания резко снижается, что подтверждает роль двух
NH-групп в хелатировании анионов (табл. 4.4).
При сопоставлении свойств циклического рецептора D.33) и его ациклического
аналога D.34) было установлено влияние макроциклического эффекта на связывание
хлорид-аниона в DMSO: D.33) имеет константу 250 М, а D.34) - только 20 М~'.
Таблица 4.4. Константы связывания
галогенид-анионов (MeCN) в
зависимости от длины спейсера в
соединении D.32)
Спейсер R
-(СН2J-
-(СН2K-
-(СН2L-
Константа связывания К, М~'
С1-
2500
1300
280
Вг"
330
270
260
I-
450
275
100
Как и в случае катионсвязывающих хозяев, каликсареновые структуры (см. разд.
3.13) могут быть использованы для формирования анионсвязывающих фрагментов.
При введении фрагмента бис(амида) кобальтоцения в верхний обод каликс[4]аре-
на, содержащего л-толуолсульфонатные группы для увеличения растворимости,
образуется жесткий хозяин D.35), в котором расположение двух амидных NH-
групп идеально подходит для комплексообразования карбоксилат-аниона.
Константа связывания ацетат-аниона в растворе DMSO равна 41 520 М~1, для
сравнения константа связывания хлорид-иона только 70 М~'. Кристаллическая структура
хлоридного комплекса с водородными связями N—H---C1" показана на рис. 4.19.
Бис(амид) кобальтоцения
Каликсарен
D.35)
4.6. Металлоорганические рецепторы
Рис. 4.19. Кристаллическая
структура хлоридного комплекса каликса-
ренового хозяина D.35)
Работа К. Хольмана и других (К. Holman et al., 1998) привела к получению
родственного трехкатионного хозяина D.36) на основе макроциклического циклотри-
вератрилена (CTV, разд. 5.2.5), имеющего глубокий карман для связывания
анионов, окруженный тремя металлическими центрами. Анионный гость PFg~ очень
точно соответствует полости, стабилизированной С—H-'F-взаимодействиями,
которые регистрируются 'Н ЯМР-спектроскопией. Кристаллическая структура этого
комплекса приведена на рис. 4.20.
При получении D.36) использовали производные рутенийкаликс[4]арена D.37)
и CTV D.38), которые образуют достаточно простые соединения, связывающие
анионы за счет электростатических взаимодействий, и, обладая размерной
селективностью, способны распознавать анионы даже в таком сильном
координирующем растворителе, как вода (М. StafTilani et a!., 1997). Если размер аниона точно
соответствует размеру макроциклической полости, то между ним и окружающими его
катионами металлов действуют электростатические силы. Наличие в комплексах
D.37) и D.38) 1-метил-4-изопропилбензольных заместителей увеличивает их
растворимость в органической среде. Каликс[4]арен, содержащий четыре атома
металла, имеет константу связывания в воде для С1~, равную 551 М, по сравнению с
133, 51 и 49 М для Вг~, 1~ и NOj соответственно. Ацетат-ион, сульфат-ион и
Н2РО^ вообще не поддаются комплексообразованию, что согласуется с большим
272
4. Связывание анионов
Рис. 4.20. Кристаллическая
структура PF^"-комплекса
трехкатионного хозяина D.36)
подандного типа
размером этих анионов и большими энергиями гидратации сульфат- и
фосфат-ионов. Благодаря своей гораздо более широкой и мелкой чашевидной полости
производное CTV D.38) отдает предпочтение большим и не сильно сольватированным
тетраэдрическим оксо-анионам, например "ТсО^. Связывание этого аниона —
компонента, получаемого при работе ядерных реакторов, — было установлено при
исследовании его распределения между водным раствором и нитрометаном в
присутствии таких конкурирующих гостей, как галогенид-, сульфат-, нитрат-ионы и др.
X X
= Ru2+
D.38)
D.37)
4.7. Нейтральные рецепторы 273
*~* Beer P.D. Transition-metal receptor systems for the selective recognition and sensing
of anionic guest species //Ace. Chem. Res. 1998. Vol.31. P.71-80.
*"* Atwood J.L., Holman К. Т., Steed J. W. Laying traps for elusive prey: Recent advances
in the non-covalent binding of anions // Chem. Commun. 1996. P.1401-1407.
Нейтральные рецепторы
Все хозяева, рассмотренные в разд. 4.4 и 4.5, обладают формальным
положительным зарядом, который способствует их комплексообразованию с
анионами за счет ненаправленных электростатических взаимодействий. Несмотря на
прочное связывание заряженными хозяевами, катионы, используемые для
связывания анионов, имеют два существенных недостатка. Ненаправленность
электростатических сил означает, что с некоторой степенью прочности связываются все
анионы. Понятно, что это сильно снижает селективность по отношению к
анионам. Вклад в селективность, являющийся результатом таких дополнительных
взаимодействий, как водородные связи, размерное соответствие и т.д., часто очень
невелик и не может конкурировать со вкладом электростатических взаимодействий.
Не обладая селективностью, каждый катионный хозяин должен иметь противоани-
он, встраивающийся во время синтеза для соблюдения требования
электронейтральности. Эти противоанионы - обычно помеха при связывании аниона-цели, а
наблюдаемые константы связывания в большей степени представляют собой
относительное сродство к одному аниону по сравнению со сродством к другому, чем
абсолютное сродство хозяина и гостя. Такая конкуренция наглядно подтверждается
данными *Н ЯМР-титрования металлоорганического хозяина D.37). Соединение
D.37), имеющее заряд 6+, было синтезировано в виде соли гексакистрифторметан-
сульфоната (CF^SOj). Титрование с помощью NaBr в водном растворе дает гладкую
кривую связывания, соответствующую замещению анионов CF3SOj на Вг~ внутри
и вне полости. Однако если эксперимент проводят с добавлением NaCF3SO3 для
поддержания постоянной ионной силы ( общая концентрация соли), то из-за
большого избытка CF-jSOj связывание Вг~ резко тормозится в начале эксперимента.
При сдвиге равновесия в сторону Вг~ начинается его связывание, но торможение
отмечается во время всего опыта до тех пор, пока концентрация добавленного
CFjSOj не станет равной нулю (рис. 4.21).
Заряженные хозяева, обладающие селективностью, создать трудно и из-за
ненаправленного электростатического связывания, и из-за конкуренции между
анионами. Это причина интенсивных исследований по синтезу нейтральных анионсвязы-
вающих соединений. Необходимость проведения таких исследований базировалась
также на осознании того, что нейтральные хозяева (по сравнению с заряженными)
должны быть гораздо более эффективными переносчиками благодаря своей липо-
фильной природе, как в биологических системах.
274 4. Связывание анионов
Постоянная ионная сила
] + [CF3SO3-] = 0.1M
Рис. 4.21. Данные 'Н
ЯМР-титрования хозяина
' ' ' ' ' ' D.37) с помощью Вг~ и
0.0? 0.04 0.06 0.08 0.1 0.12 смеСи Вг и CFjSOj при
[ВГ], М постоянной ионной силе
Antonisse M.M.G. and Reinhoudt D.N. Neutral anion receptors, design and
application //Chem. Commun. 1998. P.443-448.
Цвиттер-ионы
Цвиттер-ион — это нейтральная молекула, содержащая как
положительный, так и отрицательный заряд (например, аминокислоты фенилаланин и
триптофан, в которых кислотная группа СО2Н является достаточно кислой для
протонирования- своей собственной 1ЧН2-группы). Большинство биологических
белков и ферментов, связывающих анионы, представляют собой цвиттер-ионы с
положительно заряженными участками, на которых происходит связывание
анионов по соседству с отрицательно заряженными карбоксигруппами,
обеспечивающими суммарную электронейтральность, которая, в свою очередь, улучшает
растворимость белков в мебранах. Шмидтхен и другие (Schmidtchen et al.) в простом
эксперименте по синтезу молекул нейтрального хозяина провели реакцию трицик-
лического тетрамина C.32) с бораном (В2Н6) в тетрагидрофуране (THF) и
получили тетракис(ВН3)-аддукт D.39). Несмотря на то, что в целом молекула
электронейтральна, атомы азота хозяина все же имеют значительный положительный заряд
из-за полярных связей +N—В~, поле которых направляет анионы в полость таким
же образом, как и в случае метилированного хозяина D.3). Хозяин D.40) связывает
в своей полости целый ряд анионов и способен переносить их в раствор
хлороформа. В подражание биологическим системам цвиттер-ион D.40) был получен тем же
способом, что и соединение D.39). Жесткие арильные спейсеры препятствуют
проникновению карбоксилатных фрагментов молекулы в полость. Поэтому соедине-
4.7. Нейтральные рецепторы
275
ние D.40) очень хорошо растворяется в воде. Это подтверждает вывод об отсутствии
в его молекуле ионного спаривания заряженных СО^-групп и четвертичных
аммониевых групп, в связи с чем молекула D.40) легко связывает анионы. Значения
констант связывания для галогенид-анионов и CN~ в воде лежат в пределах
C00-600)- КЯМ.
Н
Н /
н
в—кг
н
/
• N+-B-H
н
= (СН2J:D.39)
СЬС
D.40)
276fi
4. Связывание анионов
4*7*2 Хозяева, образующие водородные связи
Нейтральные хозяева для связывания анионов образуют водородные
связи, которые обладают достаточными прочностью и направленностью.
Взаимодействия этого типа могут быть реализованы для большого числа различных
соединений, в которых водородные связи обеспечивают необходимый уровень предорга-
низации. По аналогии с хозяевами на основе гуанидиния, на базе мочевины был
получен жесткий гость с молекулярной щелью D.41), способный хелатировать кар-
боксилат-, фосфат-, сульфонат-анионы и анионы подобной формы. Установлено,
что сродство к анионам зависит от основности по Брёнстеду и заряда гостя, при
этом чем заряд больше, тем лучше, хотя изменения и не были очень большими.
Хозяева D.42) и D.43) получены на основе трена - фрагмента 2,2',2"-трис(ами-
ноэтил)амина, содержащего три N—Н-связи, сходящиеся к центру связывания. В
случае D.42) стэкинг-взаимодействия анионов с нафталиновыми кольцами
способствуют их связыванию. В случае соединения D.43) три ферроценовых
фрагмента действуют как редокс-зонды для обнаружения анионного связывания и как
стабилизаторы благодаря льюисовской кислотности металла и взаимодействиям
С—Н-анион. Несмотря на свою гибкость, этот лиганд связывает Н2РО^ в ацетони-
триле со значительной константой связывания 1.4-104 М. Каликс[4]пиррол D.44),
названный так из-за сходства с каликс[4]аренами, обладает лишь небольшой
полостью, недостаточной для капсулирования любых анионов, за исключением самых
малых. Несмотря на это, D.44) может образовать четыре водородные связи с
анионами, принимающими положения типа «насест» на том месте, которое у каликса-
рена называют нижним ободом (P. Gale et al., 1998). Это соединение представляет
особый интерес, поскольку оно легко получается в одну стадию конденсацией
пиррола и формальдегида. Было показано, что оно связывает фторид-ион в растворе
СН2С12 с константой связывания 1.7-104 М и с селективностью по отношению к
хлорид-иону, равной 50 (рис. 4.22). Интересно, что свободный макроцикл имеет
(а)
Рис. 4.22. Структуры (а) хлоридного комплекса каликс[4]пиррола D.44) и (б) фторидного
комплекса его циклогексильного производного, демонстрирующие лучшее соответствие
р--аниона предорганизации хозяина
4.7. Нейтральные рецепторы
277
D.41)
V-NH
7
D.42)
D.43)
D.44)
конформацию 1,3-альтерната, а значит, комплексообразование с анионом должно
вызывать конформационную перестройку.
При конструировании хозяев этого типа воображение химика ограничено
только его или ее способностями к синтезу. Известно огромное количество гибридных
хозяев, образующих водородные связи или различные макроциклические и макро-
бициклические структуры. Хороший пример - каликс[6]ареновое производное
тиомочевины D.45), в котором большая полость каликсарена использована для
пространственной организации трех фрагментов тиомочевины в структуре с симметрией
третьего порядка. Этот хозяин достаточно хорошо связывает галогенид-анионы в
хлороформе и исключительно эффективен при комплексообразовании 1,3,5-три-
карбоксибензола (трианион тримезиновой кислоты), имеющего симметрию
третьего порядка, с константой связывания в хлороформе 2-Ю5 М~*. В 10—100 раз более
слабое связывание 1,2,4- и 1,2,3-замещенных изомеров (хозяева), несмотря на
гибкость их трех боковых цепей, подтверждает значение топологической
(обусловленной симметрией) комплементарности для связывания анионов.
278
4. Связывание анионов
R
R = Bu-f: D.45)
«Гидридная губка» и другие
льюисовские кислотные хелаты
«Протонная губка» - 1,8-бис(диметиламино)нафталин (рис. 4.23) -
известна с 1968 г. как исключительно эффективный протонный хелатирующий
агент. Протонирование одной из NMe2-rpynn существенно снижает отталкивание
между двумя атомами азота свободного хозяина и, следовательно, сильно
увеличивает основность диамина. Мостик N-H+-N, образующийся за счет водородных
связей, почти симметричен (разд. 6.1.3.1). В случае замещения двух NMe2-rpynn
двумя ВМе2-группами создается сильно электронодефицитный анионный лиганд
«гидридная губка» D.46), проявляющий заметную способность отнимать гидрид-
ион почти у всех других его обладателей (например, HBEt0. Поданным ЯМР-спе-
ктроскопии, полученным при различных температурах, энергия взаимодействия
между Н~-гостем и этим хозяином равна 71 кДжмоль. Согласно
кристаллической структуре КН-комплекса «гидридной губки», в нем гидрид-ион копланарен
атомам бора и нафталиновому кольцу, несмотря на асимметричное расположение
Н~ в этом комплексе между атомами бора, причем длины двух В—Н-связей равны
1.49E) и 1.20E) А (рис. 4.24).
Открытию D.46), да и большей части открытий в супрамолекулярной химии,
предшествовало установление в 1967 г. способности гораздо более гибкого лиганда —
бидентатной кислоты Льюиса F2BCH2CH2BF2 D.47) - хелатировать метоксид-ион
с образованием пятичленного кольца, как показано на схеме 4.3.
4.8. «Гидридная губка» и другие льюисовские кислотные хелаты
279
Невыгодное взаимодействие Центры сильной льюисовской
неподеленных электронных пар кислотности
Me
Рис. 4.23. Сравнение «протонной
губки» и «гидридной губки» D.46)
н-
"Протонная губка"
"Гидридная губка" D.46)
Рис. 4.24. Кристаллическая структура КН-комплекса
«гидридной губки» D.46)
После открытия «гидридной губки» Г. Кацем (Н. Katz) из «AT & Т Bell
Laboratories» (Нью Джерси, США) был получен ряд других хелатирующих кислот
Льюиса, например D.48)-D.52), проявляющих сильное сродство к анионам.
Связывание происходит благодаря тому, что анионы имеют черты основания Льюиса и
жестко геометрически предорганизованы.
Схема 4.3. Хелатное поведение аниона (по Shriver D. and Biallas M., 1967)
280
4. Связывание анионов
D.48)
V0 V0
D.49)
D.50)
D.51)
PhF2Si
SiFoPh
Cl-
-Cl
■■иЧ
D.52)
D.53)
5-Метилпиримидин
Все соединения D.48)-D.50), содержащие по два атома бора, образуют хелат-
ные комплексы. Из-за большого размера С1~~ по сравнению с В-В-расстоянием в
жесткой нафталиновой системе хелатирование иона СГ соединением D.48)
происходит вне плоскости этого соединения. Несмотря на это, хелатный эффект
очевиден, и комплекс значительно более стабилен, чем модельный анион PhBCl^".
Соединение D.49) было сконструировано как соединение, обладающее
значительно большим углом захвата. Оно проявляет четко выраженную хелатную
координацию по отношению к комплементарной нейтральной молекуле 5-метилпиримиди-
на (константа связывания 130 М~' по сравнению с 70 М для аналога с одним
атомом бора). Металлоорганический катион D.50) идеально подходит для
образования нейтрального хелатного комплекса с анионами, хотя в свободном состоянии
он не предорганизован для анионного связывания, поскольку два заместителя
В(Рг-/J принимают яшш-конформацию. Этот катион (если не учитывать
конкуренцию между анионами) может быть получен окислением нейтрального Со(П)-
соединения. Окисление этого предшественника, [Со(С5Н4В(Рг-/JJ], в
неполярных растворителях с помощью [Fe(C5H5J][PF6], Cu(OHJ или С2С16 приводит к
образованию ряда комплексов с формулой D.50)Х (X = PF^, ~OH или С1~). Лишь
гидроксипроизводное является хелатным соединением с двумя заместителями
В(Рг-/J в о/«-конформации. Хлоридный аддукт содержит хлорид-анион,
прикрепленный только к одному борному центру, тогда как комплекс с РР6-анионом
представляет собой соль. Скорее всего, хелатирование D.50)ОН - следствие сильной
льюисовской основности и небольшого размера гидроксида, позволяющего ему
удобно пристроиться между заместителями двух циклопентадиенильных колец
(рис. 4.25).
4.8. «Гидридная губка» и другие льюисовские кислотные хелаты
281
Рис. 4.25. Кристаллическая структура D.50)ОН,
демонстрирующая симметричное хелатирование
гидроксид-аниона (по Herberich G. et al., 1996)
Соединения D.51) и D.52) формально содержат четырехвалентный кремний, не
являющийся электронодефицитным. Однако кремний — сильная кислота Льюиса и
при реакции с F~ образует пятикоординированные соединения. В случае D.51)
фторид-анион локализован главным образом на атоме бора, хотя и проявляет
динамические свойства, включающие перескок от бора к кремнию. Аддукт соединения
D.52) и KF содержит два пятикоординированных атома кремния, хелатирующих
анион F~ (рис. 4.26).
Как и в случае формально насыщенных соединений кремния, соединение
D.53), содержащее два атома ртути, способно легко расширять свою
координационную сферу и образовывать аддукт с галогенид-ионами. Результаты 199Hg ЯМР-
спектроскопии свидетельствуют о том, что в растворе образуется 1:1-комплекс с
хлорид-ионом, в котором два Hg-центра хелатируют анион подобно хозяевам типа
«гидридной губки». Однако кристаллическая структура этого соединения
соответствует 2:1-аддукту, в котором один дополнительный хлорид-ион связан сразу двумя
хозяевами, причем длина ковалентных связей Hg-Cl равна 2.93 А, а длина
вторичных связей - 3.17 А (рис. 4.27).
Рис. 4.26. Кристаллическая структура
KF-аддукта соединения D.52). Длины
Si—F-мостиков равны 1.898 и 2.065 А по
сравнению с длинами концевых связей
Si-F 1.60-1.65 А
4. Связывание анионов
Рис. 4.27. Кристаллическая
структура 2:1-аддукта
о-фенилендиртутного соединения
D.53) с С1-
»-t Каи Н.Е. Hydride sponge // J. Org. Chem. 1985. Vol. 50. P. 5027.
Антикрауны
Бидентатное координационное поведение таких хелатирующих лиган-
дов на основе кислот Льюиса, как D.46)—D.53), предполагает, что очень сильное
анионное связывание может быть достигнуто, когда группы, представляющие
собой кислоты Льюиса, являются частью кольца макроцикла. В противоположность
основаниям Льюиса в обычных краун-эфирах, аналог краун-эфира, собранный из
электроноакцепторных групп, может быть представлен как «антикраун» из-за его
альтернативных комплексообразующих свойств, т.е. из-за его сродства к анионам —
основаниям Льюиса, а не к кислотам Льюиса, например к катионам щелочных
металлов. Будучи частью системы макроциклического кольца, антикрауны должны
повышать стабильность комплекса за счет макроциклического и хелатного
эффектов, а также более строгой предорганизации.
Антикраун D.54), содержащий четыре атома металла, получили по реакции
оксо-мости кового аналога о-фенилендиртутного фрагмента D.53) с перфторглута-
ровой кислотой. Встраивание перфторалкильной цепи представляется несколько
необычным, тем не менее это единственный «ртутный цикл», который удалось
синтезировать после многочисленных попыток с целым рядом фторированных и не-
фторированных а,ш-дикарбоновых кислот, причем большинство других реакций
приводило к образованию полимеров, не поддающихся обработке. Возможно, что,
в отличие от таких темплатирующих агентов, как щелочные металлы, в синтезе кра-
ун-эфиров для большинства рассматриваемых здесь реакций не найден
подходящий «антитемплат». Согласно кристаллической структуре D.54), макроцикл
связывает THF как гостя, который координируется с ртутным участком, имеющим
свойства кислоты Льюиса (рис. 4.28). До сих пор неизвестно, выполняют ли эти
молекулы THF определенные функции при образовании антикрауна, получаемого с
81%-м выходом. Пока нет никаких данных о поведении D.54) при комплексообра-
зовании анионов.
4.9. Антикрауны
283
А1
Рис. 4.28. Кристаллическая структура комплекса соединения D.54) с THF
Вслед за D.54) был получен ряд
других циклических ртутьсодержа-
щих краун-соединений, которые
ведут себя подобным образом при
комплексообразовании с галоге-
нид-ионами. Соединение D.55)
дает 1:1-полимер с бромид-анионом в
твердом состоянии, в котором
анионы Вг~ располагаются над
плоскостью Hg3 (структура типа «насест»).
Длина связи Hg—Вг, равная
3.07-3.39 А, значительно больше
длины обычной ковалентной связи
Hg-Br (-2.54 А), рис. 4.29.
Аналогичный хлоридный комплекс имеет
стехиометрию 3:2, что предполагает
трехпалубный сэндвич типа
[D.55)-С1-D.55)-С1-D.55)]2-.
Комплекс соединения D.56) с
хлорид-ионом имеет 1
^-стехиометрию хозяин—гость. В этом
комплексе два хлорид-аниона расположены
Рис. 4.29. Сэндвичевый полимер комплекса
соединения D.55) с Вг~. (Воспроизведено с
разрешения Shur V. et al., 1991)
В"
©Hg
0Br
° С
2S4'
4. Связывание анионов
над и под плоскостью Hg5 («насест»), причем первый анион взаимодействует с
четырьмя, а второй — с тремя Hg-центрами. Единственное соединение, которое
реально капсулирует анион подобно краун-эфиру, - это производное тетракарборана
D.57). Хлорид-ион хорошо соответствует полости кольца соединения ртуть- 12-кра-
уН_4 D.57) с длиной связей Hg-Cl, равной 2.94 А (рис. 4.30). Аналогично иодид-
ион образует двойной «насест» со стехиометрией 1:2, соответствующей большему
размеру I" (X. Yang et al., 1994).
О
Л
Y *£ i
? я
Hg Hg
яр
о о
D.54)
D.55)
CF,
Hg
1
CF3F3C
D.56)
D.57)
В вершинах - ВН-группы
ОловоAУ), как кремний и ртуть, тоже способно расширять свою
координационную сферу от четырех до пяти центров и вести себя как кислота Льюиса. Учитывая
это, М. Ньюкомб с сотрудниками (М. Newcomb et al., 1989) из Техасского
университета (США) приготовил ряд содержащих олово антикраунов и антикриптандов
D.58) — D.60), отчасти напоминающих катапинанды.
Соединение D.58) связывает хлорид-ион в растворе ацетонитрила, однако его
константа связывания всего лишь в 2 раза больше константы его аналогов с
открытой цепью и не зависит от размера кольца, что свидетельствует о невысоком
сродстве компонентов. Был проведен рентгеноструктурный анализ комплексов макро-
бициклического соединения D.59) со фторид- (п = 6) и хлорид-ионами (п = 8). В
первом случае фторид-анион располагается почти симметрично, образуя мостик
между двумя атомами олова с длинами связей Sn-F 2.12 и 2.28 А (рис. 4.31, а). Этот
хозяин прочно связывает фторид-ион (A"t = 104 М) и, вероятно из-за размерного
4.9. Антикрауны
285
Рис. 4.30. Включение хлорид-иона в соединение D.57) ртуть- 12-краун-4 на основе
карборана
несоответствия, вообще не взаимодействует с хлорид-ионом. В хлоридном
комплексе большего хозяина (п = 8) хлорид-ион внутри макробицикла
скоординирован только с одним центром олова (длины связей Sn-Cl равны 2.610 и 3.388 А), из
чего следует, что полость макроцикла слишком велика для аниона. Если тенденция
к образованию линейных мостиков в случае хлорид-иона и существует, то она
недостаточно сильна, чтобы инициировать необходимую перестройку хозяина
(рис. 4.31, б). Интересно, что даже при -100 °С анион С1~ в растворе быстро пере-
(СН2)
С1Ч ( Л С1 С1—Sn
сг
Sn
Sn
8,10, 12:
D.58)
(СН2>
(СН2),
(СН2
п = 6,8
D.59)
С1
\ ,С1
Sn
Sn
Sn—Cl
(СН2M
(СН2M
Sn
J "
Cl
D.60)
4. Связывание анионов
Рис. 4.31. Кристаллические структуры комплексов: (а) хозяина D.59), п = 6, с F~
и (б) хозяина D.59), п = 8, с С1~ ( по Newcomb M. et al., 1989)
скакивает от одного металлического центра к другому и обратно. Константа
связывания очень мала и сравнима только с константой связывания единичного Sn-цен-
тра в ациклических системах.
Сродство хлорид-иона к хозяину D.60), имеющему большее число атомов Sn и
более высокую степень организации хозяина, резко возрастает, и константа
связывания в хлороформе достигает 500 М~!. Несмотря на то, что в общем
взаимодействия хлорид-иона с макроциклами на основе олова не кажутся особенно сильными,
нужно учитывать то, что эти соединения, подобно катапинандам, содержат только
алкильные спейсеры. По-видимому, существует большой простор для встраивания
большего числа центров связывания в молекулу хозяина и увеличения ее
жесткости, что вполне может привести к резкому улучшению характеристик хозяина.
•""* Katz H.E. Recent advances in multidentate anion complexation // Inclusion
compounds / Ed. J.L. Atwood, J.E.D. Davies, D.D. MacNicol. Oxford: Oxford
University Press, 1991. Vol. 4. P. 391-405.
*""* WuestJ.D. Multiple coordination and activation of Lewis bases by multidentate Lewis
acids // Ace. Chem. Res. 1999. Vol. 32. P. 81-89.
4.1O Координационные взаимодействия
Катионы других металлов (кислоты Льюиса), введенные в различные
органические структуры, могут быть использованы для связывания анионов точно
так же, как и антикрауны. Лиганд D.61) легко связывает одновалентные металлы
М = Cu(I), Ag(I), Au(I), используемые для чеканки монет, и дает тетраметалличес-
кие комплексы, в которых верхний обод чаши каликс[4]резорцинарена
(разд. 5.3.1.2) декорирован кольцом М4С14 (М = Си, Ag; комплекс с АиA) не
образует мостиков с хлорид-ионом). Это приводит к образованию центральной, крайне
жесткой, полости с четко определенными размерами, в которую анионы могут быть
встроены благодаря дальнодействующим латеральным взаимодействиям с двумя
4.10. Координационные взаимодействия
1287
Рис. 4.32.
Координационная геометрия
анионов для:
(o)M = Cu,X=I;
М = Ag, X = С1
и (б) М = Си, X = С1
(поХи W.etal., 1993)
{б)
Phy
Связанный \ Cu^
анион X "" Т——-/*..
( С1
> ),
Ph'
Ph
Си—I = 2.75-2.85 А
Ag—Cl = 2.69-2.76 А
= 2.47-2.55 А
металлическими центрами точно так же, как в случае ртутькарборановых хозяев
краун-типа, рассмотренных в разд. 4.9. Схемы координации анионов с различными
структурами показаны на рис. 4.32. В случае таких больших анионов, как 1~, или
больших катионов металлов, как Ag+, центральный анион подвергается
симметричной координации ц4-мостикового типа, примерно одинаково взаимодействуя
со всеми четырьмя металлическими центрами. Однако эта структура настолько
жесткая, что даже с меньшим катионом Си+ небольшой хлорид-анион не способен
деформироваться для заполнения полости и поэтому он координируется по
несимметричному Hj-мостиковому типу.
Ph
Ph
О
R = CH2CH2Ph.
D.61)
D.62)
Был получен дитопный рецептор D.62), использующий как льюисовскую
кислотность металлического центра уранила (UO^+), так и дополнительные
водородные связи боковых цепей. Сильное сродство актиноидов, например урана, к оксо-
анионам означает комплементарность металлического центра и фосфата. Уран
288
4. Связывание анионов
Рис. 4.33. Кристаллическая
структура 1:2-комплекса соединения
D.62) с
Непринимает пентагональную бипирамидальную координационную
геометрию, что означает наличие вакантного координационного центра для связывания
анионов. Анионы фосфата в комплексе могут также образовывать водородные
связи как с NH-группами, так и с кислородными атомами арильных эфиров. В
результате с анионом Н2РО^ возникает 1:2-комплекс хозяин—гость (рис. 4.33), причем
одна фосфатная группа прочно связывается с металлическим центром, а другая —
закрепляется на. первой с помощью водородных связей. Если блокирующие пара-
толильные группы в исходном рецепторе отсутствуют, то образуется похожий 2:2-
комплекс, в котором вторая фосфатная группа связана со следующей молекулой
хозяина.
Наконец, в двух интересных сообщениях было рассмотрено трехмерное ком-
плексообразование с участием фторсодержащих анионов BF^ и PF^~, которые
обычно считают неспособными к заметной координации и к сильным
взаимодействиям.
Работа С. Манна и других (S. Mann et al., 1996), использовавших октаэдрические
«строительные блоки» Fe(II), содержащие трифосфиновый лиганд на внешней
стороне блока, завершилась получением самособирающегося тетраэдрического
комплекса [Fe{MeC(CH2PPh2K}4(NCC2H2CNN с BF4][BF4]7 D.63) с линейными мос-
тиковыми лигандами NC—CH=CH—CN. Вполне возможно, что образование
тетраэдрического тетраметаллического комплекса (вместо шести блоков Fe(II),
образующих октаэдр, или пяти, образующих тетраэдр; разд. 7.4) обусловлено
соответствием симметрии тетраэдрического хозяина и тетраэдрического анионного гостя.
4.10. Координационные взаимодействия 289
Рис. 4.34.
Кристаллическая структура
комплекса D.63) со
связанным BF^~ (для простоты
фенильные группы
опущены)
Кристаллическая структура D.63) представлена на рис. 4.34. Кратчайшие
расстояния Fe-F равны 4.76—5.55 А, и при комнатной температуре капсулированный
анион BF^ быстро, на шкале времен 19F ЯМР, обменивается с анионами,
расположенными вне полости. Примечательно, что при -30 °С наблюдаются четкие сигналы
для связанных и свободных анионов в ожидаемом соотношении 1:7
E = -202.5 и-197.0 м.д.).
Еще более примечателен, с точки зрения прочности связывания, комплекс PF^~
и соединения, содержащего два атома Pd(II) D.64). Хозяин D.64) интересен сам по
себе, поскольку представляет собой редкий пример квадрупольного скрученного
геликата (разд. 7.7). Симметрия четвертого порядка хозяина прекрасно
соответствует симметрии четвертого порядка октаэдрического аниона PF^, который
располагается вдоль оси Pd-"Pd. При этом длина связей Pd--F равна 2.79 и 2.91 А, что
предполагает наличие значительных и очень необычных координационных
взаимодействий с квадратными плоскими металлическими центрами (рис. 4.35).
19F ЯМР-спектр тетракатионного хозяина в присутствии четырех противоанионов
PF^, полученный при комнатной температуре, содержит два дублета в
соотношении 1:3. Дублеты проявляются из-за взаимодействия с31P (спиновое квантовое
число равно 1/2, 100%-й избыток); поэтому обмен между одним капсулированным
анионом и тремя анионами, находящимися вне полости, при комнатной температуре
происходит медленно, как в случае BF^~ при низкой температуре. Однако
взаимодействия Pd-F недостаточно сильны, для того чтобы остановить быстрый поворот
аниона в пределах полости хозяина (D. McMorran and P. Steel, 1998).
19- СТИДДжВ
290
4. Связывание анионов
Рис. 4.35. Кристаллическая структура комплекса соединения
D.64) с PF^. (Воспроизведено с разрешения МсМопап D. and
Steel P., 1998)
D.64)
Известен ряд спиральных координационных соединений с капсулированными
анионами, а также имеются сведения о том, что такие анионы могут даже выступать
в качестве темплатов при их образовании. Эти вопросы детально рассмотрены в
разд. 7.7.6.
Учебные задания 91
Учебные задания
4.1. Согласно табл. 4.1, свободная энергия гидратации анионов уменьшается с
ростом их радиусов. Несмотря на это, самые маленькие, наиболее сильно
сольватированные анионы связываются большинством хозяев прочнее
других. Объясните это наблюдение.
4.2. Наибольшие константы связывания для гостей любого типа часто
наблюдаются у жестких хозяев и хозяев, предорганизованных для связывания гостя.
Объясните, почему эти особенности не проявляются при связывании анионов
белками и ферментами (разд. 4.2). Как это соотносится с применением [24]ан-
N6O2 D.9) в качестве искусственного фермента АТРазы (разд. 9.4)?
4.3. Анионы связываются такими катапинандными хозяевами, как диазабицик-
ло[9.9.9]нонакозан, гораздо слабее, чем родственным азакриптандом бис(тре-
ном) D.4). Какое отношение это наблюдение имеет к явлению in- и ои/-изоме-
ризации?
4.4. Предполагая полностью жесткое тетраэдрическое расположение атомов азота
в голове мостика у протонированного «футбольного» криптанда C.29) с
расстояниями N--N между несвязанными атомами азота 4.65 А (см. рис. 4.5),
подсчитайте, насколько стерически комплементарен этот хозяин для NH^
(непротонированный хозяин), Н2О (дипротонированный) и галогенид-ионов
С1~, Вг~, 1~ (тетрапротонированный). Объясните качественно, как эта ком-
плементарность может измениться в реальном, до некоторой степени гибком,
хозяине. (Примите длины О—Н- и N—Н-связей равными 1.00 А, а водородные
связи — линейными и что при оптимальной длине они на 0.5 А короче, чем
ван-дер-ваальсово расстояние между водородом и акцептором. Радиусы Ван-
дер-Ваальса: 1.10 А для Н, 1.40 А для О, 1.50 А для N, 1.80 А для С1, 1.95 А для
Вг и 2.15 А для I.)
4.5. Объясните, почему тетрапиррольные макроциклы в биологических системах
выступают в роли хозяев, прочно связывающих катионы (гл. 2), тогда как
расширенные порфирины, например D.14), больше подходят для связывания
анионов.
4.6. Предложите экспериментальные методики для определения сродства хозяина
и отдельно взятого аниона в растворе. Какие из этих методик могут подойти
для: (а) поданда D.43), (б) хозяев, содержащих два атома бора, таких, как «ги-
дридная губка» D.46), и (в) цвиттер-ионов, например D.39).
Мысленный эксперимент
Перечислите сходства и различия при конструировании селективных супрамолеку-
лярных хозяев для анионов и катионов. Что наиболее важно в каждом случае? На
основании этих соображений предположите, каковы причины более медленного
развития супрамолекулярной химии анионов, чем химии катионов.
292
4. Связывание анионов
Величины рКа для Н3РО4 равны 2.1, 6.2 и 12.4. Исходя из того, что Вы
прочитали в этой главе или в других источниках, предложите возможные типы хозяев,
которые могут быть селективными к анионам Н2РО^, НРО|~ и РО^~. Чем
необходимо руководствоваться при конструировании хозяина?
Литература
Antonisse, 1998
Atwood, 1996
Beer, 1996
Bell, 1975
Bianchi, 1997
Bencini, 1990
Dietrich, 1996
Dietrich, 1979
Galdn, 1992
Gale, 1998
Graf, 1976
Antonisse M.M.G, Reinhoudt D.N. Neutral anion receptors: design and application
// Chem. Commun. 1998. P.443-448.
Atwood J.L., Holman K.T., Steed J.W. Laying traps for elusive pray: Recent
advances in non-covalent binding of anions // Chem. Commun. 1996. P. 1401.
Beer P.D. Anion selective recognition and optical/electrochemical sensing by
novel transition-metal receptor systems // Chem. Commun. 1996. P.689-696.
Bell R.A., Christoph G.G., Fonczek F.R., Marsh R.E. The cation H13O^: A short,
symmetric hydrogen bond//Science. 1975. Vol.190. P.151.
Bianchi A., Bowman-James K., Garcia-Espana E. // Supramolecular chemistry of
anions. New York: Wiley-VCH, 1997.
Bencini A., Bianchi A., Dapporto P. et al. (PdCl4J~ inclusion into the deca-
charged polyammonium receptor (H10[30]aneN10I0+ ([30]aneN10 =
1,4,7,10,13,16,19,22,25,28-deca-azacyclotriacontane) //J. Chem. Soc, Chem.
Commun. 1990. P.753.
Dietrich В., Dilworth В., Lehn J.-M. et al. Anion cryptates: Synthesis, crystal
structures, and complexation constants of fluoride and chloride inclusion complexes of
• polyammonium macrobicyclic ligands// Helv. Chem. Acta. 1996. Vol. 79. P. 569.
Dietrich В., Fyles D.L., Fyles T.M., Lehn J.-M. Anion coordination chemistry:
Polyguanidinium salts as anion complexones // Helv. Chem. Acta. 1979. Vol.62.
P.2763.
Galdn A., Andreu D., Echavarren A.M. et al. A receptor for the enantioselective
recognition of phenylalanine and tryptophan under neutral conditions // J. Amer.
Chem. Soc. 1992. Vol.114. P.1511.
GaleP.A., SesslerJ.L, Krdl V. Calixpirroles//Chem. Commun. 1998. P.I.
Graf E., Lehn J.-M. Anion cryptates — highly stable and selective macrotricyclic
anion inclusion complexes // J. Amer. Chem. Soc. 1976. Vol.98. P.6403.
Herberich, 1996 Herberich G.E., Fisher A., Wiebelhaus D. Bis(boryl)metallocenes.l. l,l'-bis(diiso-
propylboryl)cobaltocenium cation: A novel anion-binding ligand //
Organometallics. 1996. Vol.15. P.106.
Литература
Holman, 1998
Jencks, 1969
Mann, 1996
Holman K.T., Orr G.W., Steed J.W, Atwood J.L. Deep cavity [CpFe(arene)]+
derivatized cyclotriveratrylenes as anion hosts // Chem. Commun. 1998. P.2109.
Jencks W.P. Catalysis in chemistry and enzymology. New York: McGraw Hill,
1969. P.351-364.
Mann S., Huttner G., Zsolnai L, Heinze K. Supramolecular host-guest compounds
with tripod-metal templates as building blocks at the corners // Angew. Chem.,
Int. Ed. Engl. 1996. Vol.35. P.2808.
McMorran 1998 McMorran D.A., Steel P.J. The first coordinatively saturated, quadruply stranded ■
helicate and its encapsulation of a hexafluorophosphate anion // Angew. Chem.,
Int. Ed. Engl. 1998. Vol.37. P.3295.
Metz, 1976 Metz В., Rosalsky J.M., Weiss R. [3]Cryptates: X-ray crystal structure of the
chloride and ammonium ion complexes of a spheroidal macrotricyclic ligand //
J. Chem. Soc, Chem. Commun. 1976. P.533.
Newcomb, 1989 Newcomb M., Homer J.H., Blanda M.T., Squattrito P.J. Macrocycles containing
tin — solid complexes of anions encrypted in macrobicyclic Lewis acidic hosts //
J. Amer. Chem. Soc. 1989. Vol.111. P.6294.
Peschke, 1995 Peschke W., Schiessl P., Schmidtchen F.P. et al. Building-blocks for artificial anion
receptors - derivatives of chiral bicyclic guanidines //J. Org. Chem. 1995. Vol.60.
P. 1039.
Quiocho, 1990 Quiocho FA. Atomic structures of periplasmic binding-proteins and the high-
affinity active-transport systems in bacteria // Phil. Trans. R. Soc. Lond. B. 1990.
Vol.326. P.341.
Schmidtchen, 1997 Schmidtchen F.P., Berger M. Artificial organic host molecules for anions // Chem.
Rev. 1997. Vol.97. P. 1609.
Schmidtchen, 1984 Schmidtchen F.P., Miiller G. Anion inclusion without auxiliary hydrogen bonds:
X-ray structure of the iodide cryptate of a macrotricyclic tetra-quaternary
ammonium receptor//J. Chem. Soc, Chem. Commun. 1984. P.I 115.
Shionoya, 1992 Shionoya M., Furuta H., Lynch V. et al. Diprotonated sapphyrin - a fluoride
selective halide anion receptor // J. Amer. Chem. Soc. 1992. Vol.114. P.5714.
Shriver, 1967 Shriver D.F., Biallas M.J. Observation of the chelate effect with a bidentate Lewis
acid, F2BCH2CH2BF2//J. Amer. Chem. Soc. 1967. Vol.89. P.1078.
Shur, 1991 Shur V.B., Tikhonova LA., Yanovsky A.I. et al. Crown compounds for anions -
unusual complex of trimeric perfluoro-o-phenylenemercury with the bromide
anion having a polydecker sandwich structure // J. Organomet. Chem. 1991.
Vol.418. P.C29.
Simmonds,1968 Simmonds H.E., Park C.H. Macrobicyclic amines. I. Out-in isomerism of 1 ,
diazabicyclo[£./.m]alkanes// J. Amer. Chem. Soc. 1968. Vol.90. P.2428.
4. Связывание анионов
Simmonds,1968 Simmonds H.E., Park C.H. Macrobicyclic amines. II. Out-out <-> in-in prototropy
in l,(£+2)-diazabicyclo[£./.m]alkaneammonium ions // J. Amer. Chem. Soc.
1968. Vol.90. P.2429.
Simmonds,1968 Simmonds H.E., Park C.H. Macrobicyclic amines . III. Encapsulation of halide
ions by in-in-l,(fc+2)-diazabicyclo[/c./.m]alkaneammonium ions // J. Amer.
Chem. Soc. 1968. Vol.90. P.2431.
Staffilani, 1997 Staffilani M., Hancock K.S.B., Steed J. W. et al. Anion binding within the cavity of
л-metalated calixarenes // J. Amer. Chem. Soc. 1997. Vol.119. P.6324.
Xu, 1993 Xu W., VittalJ.J., Puddephatt R.J. Selective anion inclusion in calix[4]arene
complexes driven by face-bridging u.4-halide binding // J. Amer. Chem.Soc. 1993.
Vol.115. P.6456.
Yang, 1994 Yang X.G., Knobler C.B., Zheng Z.P., Hawthorne M.F. Host-guest chemistry of a
new class of macrocyclic multidentate Lewis-acids comprised of carborane-sup-
ported electrophilic mercury centers // J. Amer. Chem. Soc. 1994. Vol.116.
P.7142.
5
Связывание
нейтральных молекул
Каменные стены — еще не тюрьма,
как и железные прутья — еще не клетка.
Р. Лавлас A618-1658). КАльтее, из тюрьмы
Связывание нейтральной (обычно органической) молекулы может
происходить путем ее физического включения в твердофазный каркас (клатрат)
либо в полость растворенной частицы (кавитанд). Взаимодействие между гостем и
каркасом хозяина может быть или очень слабым (например, ван-дер-ваальсово
взаимодействие), или достаточно сильным (например, водородные связи). Однако, в
отличие от заряженных частиц (см. гл. 3 и 4), нейтральные молекулы не
связываются постоянными электростатическими силами, а также не подвержены
координационным взаимодействиям, и, следовательно, их связывание может быть более
слабым. Это, в сочетании с их часто намного большими размерами по сравнению с
катионами металлов и даже с простыми молекулярными анионами, обусловило
получение чрезвычайно разнообразных хозяев или кристаллических соединений,
способных связывать нейтральные молекулы.
Образование твердофазных клатратов, или соединений типа «клетка» (разд. 1.2),
известно со времени открытия Г. Дэви в 1810 г. клатратного хлоргидрата —
соединения включения газа С12 и льда. Как и для решения многих других вопросов супра-
молекулярной химии, для полного понимания поведения этих соединений
включения необходимо было, чтобы появилась современная техника РСА (Дополнение
2.1). Только когда стали осознавать, что твердофазные клатраты, по существу,
включают физическое «пленение» молекулы гостя либо полостью хозяина, либо чаще
пустотами в кристаллической решетке хозяина, современная супрамолекулярная
химия обратилась к конструированию молекулярных хозяев, способных лишать
свободы нейтральных гостей в растворе и даже осуществлять химические реакции
внутри матрицы или полости хозяина. Несмотря на часто слабые силы притяжения
и, следовательно, низкие константы связывания, химия включения нейтральных
частиц имеет множество важных применений. Это, например, разделение смесей
близкородственных соединений и энантиомеров, хранение газов и токсичных
веществ, стабилизация реакционноспособных соединений, медленное выделение
лекарств при физиологических условиях и контроль за маршрутами реакций
посредством включения таких частиц в молекулярные реакторы или каналы (топохимия).
Настоящая глава начинается с рассмотрения «традиционных» клатратобразующих
соединений, а затем в ней прослеживается развитие этой области в сторону более
усложненного конструирования клатратов и химии включения в растворах.
29|Ц 5. Связывание нейтральных молекул
КЛ Неорганические твердофазные
клатратные соединения
5-1.1 Клатратные гидраты
•""» Englezos P. Clathrate hydrates // Industrial and engineering chemistry research.
1993. Vol. 32. P. 1251-1274.
5.1.1.1 Образование
Многие относительно неполярные частицы, не образующие
водородных связей с водой, способны давать клатратные гидраты. Первым гидратным
соединением было твердое соединение формулы С12-10Н2О, образующееся при
охлаждении водного раствора хлора ниже 9 °С и открытое Г. Дэви (Н. Davy) в 1810 г.
Формулу этого соединения подтвердил М. Фарадей (М. Faraday) в 1823 г. Позднее
было получено большое число гидратов с такими гостями, как SO2 (de la Rive, 1829),
Br (Alexeyeff, 1876), алканы, в том числе метан (Villard, 1888), благородные газы
(Forcrand, 1923), H2S, CS2, N2O, и многими другими. Во всех этих случаях гидрат-
образующий гость в матрице хозяина-воды заключается в пустоты
кристаллической решетки кристалла воды (лёд) и не вовлекается в какое-либо сильное
взаимодействие (водородное связывание, диполь-дипольное взаимодействие и т.п.).
Соединения, подобные спиртам, кислотам и аммиаку, которые способны к таким
сильным взаимодействиям, обычно не образуют гидратов, хотя известно несколько
примеров, когда анионы (например, F~) включаются в каркас хозяина, не вызывая
его разрушения. Процессы включения этого типа — пример клатрации; они сильно
отличаются от образования стехиометрических гидратов солей металлов, таких, как
NiCl2-6H2O, который, действительно, содержитоктаэдрический координационный
комплекс Ni(HjO)|+.
Твердые клатратные гидраты получаются при определенных температуре и
давлении. Одним из их наиболее важных и замечательных свойств является то, что
зачастую их точки плавления заметно выше, чем точки плавления чистого льда
(символ \h, означающий «ice-hexagonal» — лед гексагональный). Действительно,
некоторые известные газовые гидраты устойчивы вплоть до 31.5 °С. Это свойство
отметил Дэви, который так описал свой гидрат хлора (названный затем оксимури-
евым газом):
«В книгах по химии часто утверждается, что оксимуриевый газ способен
конденсироваться и кристаллизоваться при низкой температуре; с помощью нескольких
экспериментов я обнаружил, что это не так. Раствор оксимуриевого газа в воде замерзает
легче, чем чистая вода, но чистый газ, высушенный муриатом извести [безводный
хлорид кальция!, не подвергается никакому изменению при температуре на 40° ниже нуля
по Фаренгейту».
Столь высокая степень термостабильности клатратных гидратов в настоящее
время - серьезная проблема в газовой промышленности. Транспорт природного га-
5.1. Неорганические твердофазные клатратные соединения
297
за (в основном метана) по трубопроводам от месторождений в районах вечной
мерзлоты или низких температур морского дна (например, Аляска, Сибирь,
Северное море) в места использования затрудняется пробками, вызванными
образованием таких гидратов. К тому же, их образование в буровых установках также является
большой проблемой. На ее решение ежегодно тратятся миллионы долларов с
применением таких приемов, как высушивание и нагревание газа, а также добавление
больших количеств кинетических и термодинамических ингибиторов гидратобра-
зования (Дополнение 5.1).
5-1.1.2 Структуры
В обычном льду, \h, нет никаких полостей, способных включать
молекулы-гости. Однако в присутствии гидратобразующих частиц во льду происходит
темплатная реакция, когда вокруг гостя возникают полиэдрические полости,
соответствующие его размерам. Эти полости состоят исключительно из
конденсированных пяти- и шестичленных колец с водородными связями E.1) и E.2). Их
обозначают верхними индексами согласно числу колец каждого типа, присутствующих в
клетке (рис. 5.1). Почти все клатратные гидраты имеют одну из двух основных
структур, которые обозначают как «тип I» и «тип II». Клатраты типа I состоят из
двух полостей 512 и шести полостей 51262, а гораздо большая элементарная ячейка —
клатраты типа II (кристаллографическая элементарная ячейка — это основная
структурная повторяющаяся единица, или строительный блок кристалла) —
состоит из шестнадцати полостей 512 и восьми 512 б4. Среди более чем 100 клатратобразу-
ющих соединений существуют только шесть исключений из двух основных типов.
Это гидраты брома, диметилового эфира, этанола, аммониевых и алкиламинных
солей (структуры низкой симметрии типов III—VII), а также молекулы размера ме-
тилциклогексана или большего размера (структура типа Н;
J. Ripmeester , 1987). Позднее К. Юдашин и Дж. Рипмистер (К. Udachin and
J. Ripmeester, 1999) сообщили также о новой необычной «комплексной» структуре,
Рис. 5.1. Структура льда AА) и полости трех
типов, найденные в клатратных гидратах типа I и
II. (а) Пентагональный додекаэдр;
(б) тетракайдекаэдр и (в) гексакайдекаэдр.
Каждая вершина означает молекулу воды;
соединительные линии — водородные связи
(аM12 (бM1262 (вM1264
298 5. Связывание нейтральных молекул
Таблица 5.1. Параметры клатратных гидратов типов I и II
Параметр
Решетка
Элементарная ячейка
длина, А
формула
Полость
обозначение
число полостей
на структуру
средний радиус, А
Гидрат типа I
Объемноцентрированная
кубическая РтЗп
12.0
6X2Y46H2O
Малая
512
2
3.91
Большая
51262
6
4.33
Гидрат типа II
Алмазная кубическая
Fd3m
17.3
8X16Y136H2O
Малая
512
16
3.902
Большая
51264
8
4.683
которая обладает чертами структур типов Н и II (состав: 1.67 холингидроксидфто-
рид тетра-н-пропиламмония-30.33 Н2О). Свойства основных структурных типов I и
II и полостей приведены в табл. 5.1. Клатратный гидрат брома уникален,
поскольку единственный образует 15-гранную полость, 51263, которая имеет
неблагоприятные напряжения по сравнению с другими тремя полостями.
Н
I
О,
н
I
о.
н
н
н.
н
н.
*0'
н
О'
I
н
н
о'
I
н
н
н
о -- н-о
н н
E.1)
н
н
I
,0
н.
н
н'
E.2)
В общем, небольшие гости занимают малые полости типа 512. Небольшое
различие размеров такой полости в структурах типов I и II может сильно влиять на выбор
структурного типа. Так, наименьший гость, Аг (радиус 1.92 А), занимает меньшую
полость — 512 - структуры типа II, так же как N2 и О2. Достаточно малые гости, СН4
и H2S (радиусы 2.18 и 2.29 А), соответствующие по размерам полостям 512,
образуют гидраты структурного типа I. Не, Н2 и Ne достаточно малы и диффундируют
через «решетки» водородных связей полости; поэтому они не дают гидратов. Большая
полость — 51262, найденная в гидратах структурного типа I, достаточно велика,
чтобы «поймать» молекулы гостя диаметром до 6.0 А (например, С2Н6, СО2). Такая
полость предпочтительна для большинства гостей и играет главную структурно-ста-
5.1. Неорганические твердофазные клатратные соединения iЪэд
билизирующую роль почти во всех гидратах структурного типа I. Самая большая
полость - 51264 — способна «улавливать» молекулы гостя диаметром до 6.6 А, такие,
как С3Н8 и /50-С4Н1О. Даже «-бутан может быть «пойман» клатратным гидратом в
ограниченном интервале температур и давлений, если небольшие
«вспомогательные» гости могут заполнять полости 512. Интервал диаметров молекул гостей и их
влияние на структурный тип клатратных гидратов представлены на рис. 5.2.
На рисунке приведены также «идеальные» гидратные числа (т.е. число молекул
воды на молекулу гостя в предположении, что заполнены все подходящие полости).
Однако почти во всех случаях некоторые полости остаются пустыми, и реальные
гидраты содержат больше воды, чем идеальные. Например, нашли, что клатратный
гидрат хлора имеет формулу С12-10Н2О, в то время как его идеальная формула
содержит 7 2/3 молекул воды для структуры типа I, когда заняты только полости 51262.
ЗА
4А
5А
бА
7А
Рис. 5.2. Сравнение диаметров
гостей и размеров полостей,
занятых, как в простых гидратах
8А
Гость
-Аг
-Кг
-N2
-02 *
-сн4
-Хе; H2S
Гидраты
отсутствуют
Н2О
Н2О
-СО,
-cyclo-C,H6
Н2О
-(СН2KО
-С3Н8
— i'jo-C4H10
17 Н2О
, Гидраты
4 |0 отсутствуют
Полости
и их структурные типы
£> Структурный
тип П
Структурный
ТИП1
Структурный
ТИП1
E12бЛ
Структурный
тип П
'■jOO 5. Связывание нейтральных молекул
5.1.1.3 Свойства
Поскольку вода в структурах клатратных гидратов обычно составляет
более 85%, имеет смысл обсудить влияние молекул-гостей на физические свойства
гидратов в сравнении со льдом. Данные измерения ширины линий твердотельного
1Н ЯМР указывают на то, что при температурах ниже 50К структуры гидратов,
подобно льду, являются прочными. Выше этой температуры индивидуальные
молекулы воды внутри гидратных структур начинают подвергаться переориентации и
трансляционной диффузии. При 273К скорость диффузии более чем в 100 раз
превышает таковую во льду. Переориентация фиксированного центра происходит
каждые 10 мкс в сравнении с 21 мкс во льду. Столь быстрая диффузия влияет также на
объемную диэлектрическую постоянную гидратов, которая составляет примерно
(а) .... у.
Рис. 5.3. Кристаллофафические
структуры клатратных гидратов
типа I (а) и типа II (б). Сферами
обозначены молекулы гостя
5.1. Неорганические твердофазные клатратные соединения 301
половину от этого параметра для льда. Однако, судя по данным дальнего ИК-спек-
тра (низкие волновые числа), прочность водородных связей в молекуле гидратов
почти такая же, как и во льду. В самом деле, длины водородных связей в этих
соединениях, определенные методом РСА (рис. 5.3), только на 1% превышают длины в
недеформированных полостях 512 льда \h.
Другое заметное отличие гидратов от льда AЛ) — их теплопроводность, которая у
1Л почти в 5 раз выше. Предположили, что такое различие - следствие фононного
рассеяния, происходящего из-за ангармонических колебаний и вращений
молекулы-гостя. Это, возможно, связано с тем фактом, что коэффициент теплового
расширения гидратных структур обычно значительно превышает таковой для льда.
5.1.1.4 Применение
Если пока образование гидрата природного газа — метана — ведет к
удорожанию добычи нефти, то по мере истощения запасов природного
ископаемого топлива гидраты метана, возможно, станут его ценным источником. Расчет
плотности метана внутри его гидрата СН4-6Н2О показывает, что этот гидрат
соответствует сильносжатой форме газа (в гидрате содержится в 184 раза больше метана, чем
в эквивалентном объеме этого газа при нормальных температуре и давлении),
вопреки его стабильности при сравнительно низких давлениях. Энергия решетки
гидрата метана такова, что для его разложения потребовалось бы только -10%
энергии, выделяющейся при сжигании метана. Это убедительно иллюстрируется тем
фактом, что гидрат метана поддерживает горение.
Последние оценки мировых запасов гидратов природного газа на Земле дают
~5-1013 м3, преимущественно для районов вечной мерзлоты Аляски и Сибири, а
остальные E—25)-1015 м3 гидратов находятся в океане, в частности в той его части, где
он омывает берега Центральной Америки. Это примерно вдвое больше запасов
ископаемого топлива других видов. Понятно, что значение этого громадного
богатства будет возрастать по мере истощения запасов ископаемого топлива.
Действительно, большие залежи гидратов природного газа в России (Мессоянское
месторождение, полуостров Ямал) разрабатывают для получения природного газа с
1971 г.
Газовые (клатратные) гидраты применяют для хранения и разделения газов. Так,
в 1962 г. Бардхан (Bardhun) показал, что такие гидраты можно использовать для
обессоливания морской воды. Кроме того, гидраты можно применять для хранения
и транспортировки метана без использования контейнеров высокого давления.
Дополнение 5.1. Клатратные гидраты в газовой промышленности
С ростом нефтяной и газовой промышленности США еще в 1934 г. стало понятно, что
наличие пробок в газопроводах при температурах, когда обычная вода не замерзает,
вызвано образованием клатратных гидратов. Идеальные условия для образования
гидратов — наличие воды и углеводородов при низкой температуре и зачастую
повышенном давлении. В настоящее время это явление понимают гораздо лучше, однако
3Q2 5. Связывание нейтральных молекул
образование клатратных гидратов продолжает оставаться главной проблемой и
предметом очень активных исследований в этой отрасли промышленности, так как
условия бурения и транспортировки становятся все более экстремальными.
Залежи газов, извлекаемых из природных резервуаров, насыщены водой.
Поскольку газ расширяется в сепараторах или в скважинах, его температура понижается
и в результате получается твердый гидрат, блокирующий трубопроводы и
оборудование. При образовании гидрата из нефти уходят ее, более легкие, менее вязкие
фракции, в результате чего остаются тяжелые фракции нефти и битумные пески. В самом
деле, разложение гидратов in situ может облегчить разработку таких залежей
благодаря пониженной вязкости сырой нефти за счет повторного растворения
низкомолекулярных углеводородов.
В нефтяной и газовой промышленности разработан ряд методов борьбы с гидрат-
образованием, включающих предварительную обработку газа и использование
специального оборудования, а также усовершенствованы сами процессы, чтобы
минимизировать условия гидратобразования и последствия отложения гидратов. Действительно,
согласно оценкам, 5—8% от общей стоимости процесса направлены на решение
проблемы гидратов (-500 млн долларов ежегодно). Наиболее важные методы:
• высушивание природного газа;
• нагревание газа до температур выше температуры плавления гидрата;
• влияние на фазовую диаграмму гидратобразования путем введения в скважину
больших количеств ингибиторов, таких, как метанол, гликоль или растворы
солей, хотя многие из них могут вызывать коррозию (термодинамические
ингибиторы);
• уменьшение скорости гидратобразования путем добавления небольших количеств
органических полимеров, которые замедляют нуклеацию гидратных кристаллов
(кинетические ингибиторы), например PVP (поливинилпирролидон, E.3)).
PVP E.3)
Помимо постэкстракционных проблем наличие газогидратного слоя в земле
может также иметь катастрофические последствия при бурении. Известно, что
образование при этом газовых гидратов и продуктов их разложения из-за внезапных
изменений давления или температуры вызывало повреждение стенок аппарата,
неконтролируемые выбросы газа, а также утечку, воспламенение и просачивание газа
через кожух. Поэтому, чтобы установить, как гидраты распределяются в породе,
определить их объем, температуру в скважине, давление в порах, пористость и
проницаемость пород, проводят обширные геологические исследования. Особенно большие
слои природных гидратов могут находиться в осадочных породах либо ниже слоя
вечной мерзлоты, либо под океанским дном. Обычно принимают меры для
предотвращения неконтролируемого разложения гидратов.
*** Sloan E.D., Jr. Clathrate hydrates of natural gases. New York: Dekker, 1996.
5.1. Неорганические твердофазные клатратные соединения 303
5* 1*2 Цеолиты
5-1.2.1 Состав и структура
Цеолиты — это пористые алюмосиликаты, в которых обычно
анионный каркас сбалансирован катионами, как правило, расположенными внутри
твердых полостей или каналов, но их не заполняют. Общая формула цеолитов,
согласно определению ИЮПАК, состоит из трех частей:
Si^Cy (*Н2О,
Катионы А,В и С Каркас Включенные гости
Каждому цеолиту присваивают также трехбуквенный структурный код,
описывающий топологию его каркаса (связность, размерность каналов и т.п.). Примеры
приведены в табл. 5.2, а наиболее распространенные структуры показаны на рис. 5.4.
Как правило, цеолиты — регулярные кристаллические соединения, хотя для них
обычны и дефекты, такие, как немостиковые атомы кислорода, вакантные участки
или большие поры, которые вносят свой вклад в реакционную способность этих
материалов. Кремний — основной элемент цеолитного каркаса, алюминий
содержится в каркасе в виде аниона АЮ^, который легче всего замещается нейтральными
участками SiO4. В молекулу цеолита могут быть включены и другие соединения —
ТО4 (Т— тетраэдрический центр, например Ge, Ga, P, As и т.п.).
Известно, что соотношение Si/Al изменяется от единицы до бесконечности, это
соответствует минимальному требованию отсутствия связей А1—О—А1 в любом
месте структуры; стабильны только связи А1—О—Si и Si—О—Si. На основании
соотношения Si/Al цеолиты обычно подразделяют на две большие категории:
1. Цеолиты с низким или средним соотношением Si/Al (< 5).
2. Цеолиты с высоким соотношением Si/Al (> 5).
Цеолиты с очень высоким соотношением Si/Al (стремящимся к бесконечности)
называют полностью кремниевыми молекулярными ситами, цеосилами или поро-
силами. В присутствии алюминия поры могут быть заполнены некаркасными
катионами: катионами щелочных и щелочноземельных металлов либо органическими
ионами тетраалкил- или тетраариламмония. В порах могут также содержаться
органические молекулы или молекулы растворителя и воды в зависимости от условий
синтеза.
Роль цеолитов и цеолитных материалов в промышленности трудно
переоценить. Их используют для решения широкого круга проблем катализа и разделения,
в частности в нефтехимической промышленности. Основные области их
применения: адсорбционное разделение углеводородов, очистка газов и жидкостей, а также
каталитический крекинг длинноцепных углеводородов, осуществляемый для
получения более ценных короткоцепных гомологов. Цеолиты используют и как
детергенты (умягчение воды) в ионном обмене, и как молекулярные сита для
обезвоживания органических растворителей.
Таблица 5.2. Характеристики топологии каркасов некоторых распространенных цеолитов
Код структурного
Цеолит
формула
Состав
каркаса
Системы
каналов
Окно поры,
диаметр
Тип ячейки
Примечаниия
AFI
FAU
LTA
MEL
MFI
SOD
AIPO.-5
Фожазит
Цеолит
Линде А
ZSM-11
ZSM-5
Содалит
А112Р12О48
■•
M29[Al58Si134O384]-240 H20
(М = Na2, Ca, Mg)
{Na12[Al12Si12O48]-27H2O}8
NajA^S^ ЯО192]16Н2О
(л < 16)
Na/,[4Si96-«O.92]16H2O
(я < 27)
Na6[AJ6Si6024]-2NaCl
На основе
А1РО4
Алюмо-
силикатный
Высококремниевый
На основе
А1РО4
Алюмоси-
ликатный
Высококремниевый
На основе
А1РО4
На основе
GaPO4
Высококремниевый
Высококремниевый
Множество
комбинаций
Al, Si, P, Ga,
Be, As и Zn
ID
3D
3D
3D
3D
_
12-членный
цикл
12-членный
цикл
7.4 А
8-членный
цикл
4.1 А
Эллиптический 10-член-
ный цикл,
5.5 А (среднее)
Эллиптический 10-член-
ный цикл,
5.5 А (среднее)
Только
6-членный
цикл, 2.8 А
Отсутствует
/аи
sod
abR
sod
а
—
_
Отсутствует
Отсутствует
sod
—
Складчатые
слои содалитовых
ячеек, сложенные
по типу ABC
—
-
—
-
—
_
Прямые каналы
Один прямой и один
зигзагообразный
каналы
Шесть циклов,
сложенных по типу
ABC
О"
1
а
а
О"
§
5.1. Неорганические твердофазные клатратные соединения
(а) (б)
305
Рис. 5.4. Топология типов микропористой структуры, (а) Содалит, (б) тип Линде А,
(в) фажазит (цеолит X и Y), (г) А1РО4-5 и (д) ZSM-5. В вершинах находятся тетраэдры
или SiO4 (Воспроизведено с разрешения AtwoodJ.L, Davies J.E.D., MacNicol D.D. and
Vdgtle F. Comprehensive supramolecular chemistry. Oxford: Pergamon, 1996. Vol. 7. P. 226)
306
5. Связывание нейтральных молекул
Известно около 60 природных цеолитов, например, бикитаит
Li2[Al-,Si40J]-2H20, геландит Ca4fAl8Si28O72]-24H2O и фожазит
(Na2,Ca,MgJ9[Al58SiI34O384l'240H2O. Однако многие из наиболее важных цеолитов,
например ZSM-5, применяемый в нефтехимической промышленности при
производстве бензина, получают синтетически. Благодаря недавним темплатным
синтезам с использованием поверхностно-активных веществ стали доступными очень
интересные мезопористые материалы, такие, как МСМ-41 и МСМ-48, которые
имеют намного большие поры, чем традиционные микропористые материалы.
(ZSM и МСМ означают «Zeolite Socony Mobil» и «Mobil Catalytic Material»
соответственно. Эти обозначения относятся к большому ряду обозначений, принятых для
определенных материалов, в особенности материалов промышленного значения,
которые исторически сложились раньше, но все еще используются.) Многие
полезные свойства цеолитов и их химия объясняются наличием в их структурах каналов
и полостей, которые включают катионы металлов (уравновешивающие заряд
анионного каркаса), воду и множество других гостей. Ценность цеолитов заключается
в том, что алюмосиликатные полости достаточно прочны, для того чтобы гости
могли проникать в каналы и покидать их, не разрушая структуру хозяина. Вот
почему цеолиты используют как молекулярные сита, разделяющие катионных и
молекулярных гостей по размеру или по адсорбционной селективности, а также как
реакционные сосуды для высокоселективных реакций внутри каналов и полостей.
Химия включения цеолитов определяется размерами каналов и пор, а также
размером входов-окон, через которые осуществляется доступ к этим порам. В случае
содалита 0-ячейки (рис. 5.5) доступны только через четырех- и шестичленные
циклы (кольца), которые недостаточно велики для проникновения подавляющего
большинства гостей. Напротив, топология Линде типа А, хотя и основана на сода-
литовых ячейках, но дополнительно содержит двойные четырехчленные спейсеры.
В результате получаются а-ячейки, с окнами из восьми циклов, образующих
каналы с трехмерной структурой. Следуя дальше, приходим к типу фожазита, когда
ячейки содалита группируются тетраэдрически, точно так же, как атомы углерода в
алмазе (разд. 6.4), связываясь двойными шестичленными циклами. В результате по-
Двойной четырехчленный
цикл (dAR)
Двойной шестичленный
цикл (d6R)
Р-Ячейка содалита
(sod)
а-Ячейка
Рис. 5.5. Структуры цеолитных ячеек
5.1. Неорганические твердофазные клатратные соединения
307
Рис. 5.6. Прямые и зигзагообразные каналы в ZSM-5 чш
лучается ячейка фожазита (fau), представляющая собой трехмерную систему
каналов с окнами из 12-членных циклов. Такой каркас очень пористый и идеален для
целей катализа, основанного на химии включения.
В противоположность топологиям SOD, LTA и FAU, структуры ZSM-5 и ZSM-11
основаны не на мотиве содалита. Это сложные структуры с каналами, имеющими
окна из 10-членных циклов, в основе которых лежит мотив «оболочка» из шести-
членных циклов; в такой оболочке стенки канала состоят из конденсированных
шестичленных колец. Единственное различие между этими двумя структурами -
наличие центра инверсии в ZSM-5 и зеркальной плоскости в ZSM-11. В результате
в ZSM-5 существуют один прямой и один зигзагообразный каналы (рис. 5.6) и
только прямые каналы в ZSM-11. Тип AFI, представленный цеолитом А1РО.-5, также
основан на каналах. В чисто алюмофосфатных цеолитах компоненты Аг+ и РО|~
строго чередуются, образуя нейтральный каркас, в котором имеются только циклы,
содержащие четное число членов. Система пор представляет собой одномерный
канал с окнами из 12-членных циклов.
Цеолиты, приведенные в табл. 5.2, — это примеры микропористых материалов,
названных так из-за относительно малых размеров их пор. В 1992 г. сотрудники
фирмы «Мобил» сообщили о семействе цеолитов M41S, членами которого
являются МСМ-41 и МСМ-48. Эти материалы получены с участием
поверхностно-активных молекул, образующих мицеллы в растворе (разд. 9.2), которые темплатируют
образование очень больших пор (мезопоры). Цеолит МСМ-41 имеет одномерную
гексагональную организацию открытых каналов с размерами 15-100 А, которые
можно наблюдать с помощью просвечивающей электронной микроскопии.
МСМ-48 имеет поры диаметром ~30 А с трехмерной кубической организацией.
5-1.2.2 Синтез
Для приготовления цеолитов точно заданного структурного типа
следует использовать темплатные материалы, определяющие распределение пор по
размерам. Считается, что общий механизм образования цеолитов включает
постепенное замещение гидратной воды вокруг катиона-темплата силикатными или
алюмосиликатными единицами. Таким образом, размер поры определяется
размером катиона, темплатирующего метастабильный каркас. Некоторые примеры
катионов и типов цеолитов, образовавшихся в результате темплатирования, приведены
в табл. 5.3. Необходимо также контролировать и ряд других факторов, таких, как
кинетика образования кристаллов и соотношение Si/Al. Поэтому синтез цеолитов
308
5. Связывание нейтральных молекул
Таблица 5.3. Катион ы-темплаты и типы образующихся цеолитов
Катионы
Тип цеолита
Na+
Na+ + NMe4+
Na+ + NPr|
Na+ + бензилтрифениламмоний
Na+ + 15-краун-5
CwH2/|+1Me3N+(/i==8+16)
Содалит
Фожазит, содалит, цеолит Линде А
ZSM-5
ZSM-11
Высококремниевый фожазит
МСМ-41
обычно проводят в твердом геле, куда частицы - «строительные блоки» каркаса —
непрерывно поступают при контролируемой скорости за счет непрерывного
растворения. Общая схема синтеза представлена на рис. 5.7.
Исходная смесь
(обычно гель)
Материалы на основе SiO4: pH 10-14
Материалы на основе РО4: рН 3-10
Растворение: + ОН"
Раствор, содержащий материалы для:
1. построения каркаса, например силикаты (ОН^
2. заполнения микропор, например катионы, вода,
амины, соли
-ОН-
Заполнение микропор
при поликонденсации
Рис. 5.7. Схематическая диаграмма,
иллюстрирующая синтез цеолитов
в присутствии аниона ОН~ в водной
фазе
5.1. Неорганические твердофазные клатратные соединения 309
Для получения заданного соотношения Si/Al критическим является контроль
рН. По мере уменьшения* рН способность силиката к конденсации уменьшается
вследствие уменьшения количества частиц Si—О~ по сравнению с количеством
Si—ОН. Анионная форма необходима для осуществления первоначальной нуклео-
фильной атаки. Напротив, скорость конденсации А1(ОН)^~ остается постоянной, и
потому цеолиты, обогащенные алюминием, кристаллизуются предпочтительно при
высоких рН и наоборот. Синтез цеолитов зависит также от большого числа
экспериментальных параметров, включая концентрации и степень пересыщения,
источник материала каркаса, растворитель (иногда используют спирты или гликоли),
скорость растворения геля, созревание, добавление затравочных кристаллов,
температуру, время перемешивания и давление. Идеальные параметры такого синтеза
достаточно точно определены экспериментально, и цеолиты можно легко
приготовить в больших количествах.
5-1-2-3 MFI-цеолиты в нефтяной промышленности
Благодаря своим адсорбционным свойствам, зависящим от формы
пор, а также другим физико-химическим свойствам, канальные цеолиты MFI-ти-
па, к которому принадлежит цеолит ZSM-5, очень важны в нефтяной
промышленности. Наиболее известный пример - селективный синтез и диффузия «-ксилола
через ZSM-5 в отличие от орто- и л/е/гш-изомеров. Благодаря «сверхкислым»
участкам в порах такие кальцинированные** цеолиты, как ZSM-5, могут способствовать
значительным преобразованиям обычно нереакционноспособных органических
молекул. Отрицательный заряд каркаса кальцинированных цеолитов уравновешен
протонами, имеющимися или на дефектных участках, или на мостиковых
кислородных атомах. В пустой полости цеолита протон находится в несольватированном
состоянии и поэтому крайне активен. В результате даже очень слабые основания,
например ароматические углеводороды, «-алканы и воски, проходя через каналы
цеолита, протонируются и образуют реакционноспособные карбкатионы, которые
могут быстро перегруппироваться, образуя смесь продуктов. Синтез ксилолов
внутри полости осуществляют по реакции толуола с метанолом. Кислотность цеолита
приводит к электрофильному ароматическому замещению в арильном кольце,
благодаря чему образуется смесь о, м- и л-ксилолов, находящаяся в цеолите в
состоянии равновесия. Решающим является тот факт, что только пара-изомер способен
быстро диффундировать через канал цеолита именно благодаря своей линейной
нитевидной форме. Более объемистые орто- и .ие/гш-изомеры гораздо менее
подвижны внутри цеолита, а потому более склонны к обратной изомеризации с
образованием дополнительного статистического количества «-ксилола, который вновь
диффундирует наружу. Таким образом, такие цеолиты, как ZSM-5, проявляют
сильную «оря-селективность. Действительно, пара-изомер диффундирует в 14 раз
быстрее ор/яо-изомера и в 100 раз быстрее л/е/гш-изомера (рис. 5.8, а).
* В тексте: «увеличения», что ошибочно. (Примеч. ред. перевода.)
** Кальцинация — прокаливание (или обжиг) веществ, производимое с целью их
окисления или разложения. (Примеч. ред. перевода.)
(а)
ми
(б)
2,3,4-Триметилпентан
MFI
и-Октан
w-Ксилол
(в)
МИ
Рис. 5.8. (а) Диффузионная селективность цеолитов MFI-типа к изомерам ксилола, (б) «Мобил-процесс», повышающий качество
бензина с помощью цеолитов MFI-типа; высокооктановые соединения, такие, как 2,3,4-триметилпентан, защищены от дальнейшего
участия в реакциях благодаря переходному состоянию и диффузионной селективности; н-алканы проникают в каналы и подвергаются
крекингу; ароматические соединения алкилируются легкими продуктами крекинга, (в) Компоненты воска подвергаются крекингу с
образованием бензина и жидкого нефтяного газа. (Воспроизведено с разрешения Atwood J.L., Davies J.E.D., MacNicol D.D. and Vogtle F.
Comprehensive supramolecular chemistry. Oxford: Pergamon, 1996. Vol. 7. P. 641—642)
f
!
1
5.1. Неорганические твердофазные клатратные соединения .; 311
Высокая кислотность цеолитов также имеет решающее значение при
производстве бензина по «Мобил-процессу». Из-за более низких октановых чисел линейные
«-алканы в бензине более нежелательны по сравнению со своими разветвленными
изомерами. Необходимость разделять линейные и разветвленные соединения
приводит к повышению стоимости бензина. Значительно выгоднее превращать
линейные изомеры в разветвленные. Такие сильноразветвленные алканы, как 2,3,4-три-
метилпентан, очень медленно диффундируют в каналах ZSM-5. Более того, даже
если они находятся внутри цеолита, изомеризация алкана включает передачу
гидрид-иона катионному участку цеолита. Переходное состояние в этой реакции очень
объемисто, в результате чего только линейные алканы способны участвовать в
реакции. Это явление называют селективностью переходного состояния. С одной
стороны, и селективность переходного состояния, и диффузионная селективность
приводят к тому, что ценные разветвленные углеводороды не изменяются под
действием цеолита. С другой стороны, линейные соединения, например «-октан,
быстро диффундируют в цеолит и реагируют с кислотными участками, что вызывает их
каталитический крекинг на более легкие фракции, без труда отделяемые от смеси.
Ароматические соединения алкилируются продуктами крекинга и входят в состав
получаемого бензина, что приводит к небольшим потерям объема. Поскольку
бензол канцерогенен и обладает высокой реакционной способностью, то желательно
уменьшить соотношение бензол.толуол в продукте (рис. 5.8, б).
Цеолиты большего размера, типа FAU, применяют для крекинга восков и алка-
нов с длинной цепью, которые из-за своей высокой вязкости не имеют большой
ценности. Продукты этого процесса — бензин и жидкий газ, последующая
обработка которого цеолитами типа MFI детально показана на рис. 5.8, в.
5« 1*3 Твердые слоистые материалы и их интеркаляты
5.1.3.1 Введение
В группу твердых слоистых материалов включают графит, катионные и
анионные глинистые минералы, фосфаты и фосфонаты металлов, а также ряд
других неорганических и координационных соединений. Первое свидетельство их
существования — по-видимому, производство фарфора в Китае в 600—700 гг. н.э.
Фарфор получают путем интеркаляции*, т.е. включения, ионов щелочных металлов в
структуры природных слоистых минералов, например в структуры полевого шпата
или каолина. Твердый слоистый материал состоит из двухмерных слоев, в каждом
из которых компоненты взаимодействуют ковалентно (или прочно связаны другим
способом), а взаимодействия между слоями слабые, обычно типа взаимодействий
Ван-дер-Ваальса. Некоторые характеристики твердых слоистых материалов
представлены на рис. 5.9, а примеры слоистых материалов различных классов даны в
табл. 5.4.
* Обратная реакция внедрения атомов металла или нейтральных молекул в межслоевое
пространство кристаллических веществ со слоистым типом структуры. (Примеч. ред.
перевода.)
312
5. Связывание нейтральных молекул
Межслоевая
область
Высота галереи df
Период повторяемости d
(расстояние между
центрами слоев)
Толщина слоя
Интеркалированные гости
Рис. 5.9. Характеристики твердых слоистых материалов
Слоистая структура делает эти материалы очень интересными с точки зрения
взаимодействия хозяин—гость, поскольку ионные или молекулярные частицы гостя
могут встраиваться между соседними слоями хозяина, вызывая их расширение или
набухание. Интеркаляция гостя обычно обратима. Важной характеристикой
твердых слоистых материалов является то, что, как и цеолиты, они могут сохранять свою
слоистую структуру хозяев и на стадии интеркаляции, и на стадии деинтеркаляции.
Однако, в отличие от цеолитов, интеркалирующие слои хозяина подвижны и могут
Таблица 5.4. Классы твердых слоистых материалов
Слоистый материал
Формула
1. Незаряженные слои
(а) Изоляторы
Глины
каолинит, дикит
серпентин
Цианид никеля
(б) Электропроводящие слои
Графит
Дихалькогениды переходных металлов
Оксифосфаты металловAУ)
2. Заряженные слои
(а) Анионные слои
Глины
монтмориллонит
сапонит
вермикулит
мусковит
(З-Оксид алюминия-натрия
Оксиды переходных и щелочных металлов
(б) Положительно заряженные слои
Гидротальцит
Al2Si205@HL
Mg3Si205@HL
Ni(CNJ
МХ2 (М = Ti, Zr, Hf, V, Nb, Та, Mo, W;
X = S, Se, Те)
MOPO4 (M = V, Nb, Та)
Nax(Al2.xMgx)(Si4O10)(OHJ
Cax/2Mg3(AlxSi4.xO10)(OHJ
(Na, Ca)x(Mg3.xLixSi4O10)(OHJ
KAl2(AlSi3O10)(OHJ
NaAl,,O17
M'XO2 (M1 - щелочной металл; X = Ti, V,
Cr, Mn, Fe, Co, Ni)
[Mg6Al2(OHN]CO34H2O
5.1. Неорганические твердофазные клатратные соединения
313
изгибаться, обеспечивая частичное включение гостя в определенные зоны.
Слоистые интеркалированные соединения обычно образуют каскады структур,
в которых номер каскада представляет собой отношение числа слоев хозяина к
числу слоев гостя. Так, комплекс каскада 1 состоит из чередующихся слоев хозяина и
гостя. Комплекс каскада 2 содержит два слоя хозяина на один слой гостя и т.д.
Встречаются также и дробные каскады, относящиеся к интермедиатам, например,
при превращении соединения каскада 2 в соединение каскада 1. В классическом
варианте превращение такого рода через интермедиат дробного каскада потребовало
бы удаления всех гостей из некоторых слоев, схлопывания структуры до исходного
состояния, свободного от гостей, с периодом повторяемости d и повторного
заселения других слоев. Такая модель не согласуется с наблюдаемыми легкими
взаимопревращениями и поэтому ее реализация маловероятна. Вот почему для интерка-
лятной каскадности была предложена неклассическая модель — модель
Даумаса—Геролда, которая допускает изменение плотности гостей внутри слоя и тот
факт, что незанятые области в одном слое располагаются параллельно занятым
областям в соседних слоях. При этом обеспечиваются минимальное искажение всей
структуры и максимальное электростатическое притяжение. Разница между этими
двумя картинами интеркаляции показана на рис. 5.10; картина превращения интер-
Классическая модель
Модель Даумаса-Геролда
шш мтт
Каскад 2
(Bfttt
Каскад 3
О С fr *О
Рис. 5.10. Классическая модель и модель Даумаса—Геролда каскадности в интеркалятных
соединениях. Номера каскадов означают отношение числа слоев хозяина к числу слоев гостя
314
5. Связывание нейтральных молекул
РОСС
Каскад 2
Каскад 4/3
1
tfttttt
Рис. 5.11. Схематическое представление
превращения интеркалята каскада 2
в интеркалят каскада 1 с промежуточным
каскадом 4/з по меРе интеркаляции
дополнительных гостей
Каскад 1
калята каскада 2 в интеркалят каскада 1 по Даумасу-Геролду представлена на
рис. 5.11.
Исторически химия слоистых интеркалятов началась в 1840 г. с сообщения, что
графит способен интеркалировать серную кислоту между соседними слоями своей
«сетки». Серьезный интерес к интеркалятам возник только после 1960-х годов,
когда осознали, что интеркаляция гостя может значительно изменить химические,
каталитические, электронные и оптические свойства хозяина. Это особенно верно,
если свойства хозяина зависят от свойств его слоистой структуры.
Например, графит можно применять в качестве «сухой» низкотемпературной
смазки благодаря той легкости, с которой один его углеродный слой скользит по
другому (Вы, возможно, ощущали его «скользкость», например, при пользовании
мягким карандашом, грифель которого представляет собой графит с низким
содержанием глины). Интересно, что смазывающие свойства графита сильно зависят от
наличия в нем интеркалированного кислорода. В отсутствие кислорода,
действующего наподобие молекулярного мяча, графит значительно менее скользкий. Это
оказалось серьезной проблемой для применения графитовых смазок в космической
программе. Другие интеркалирующие материалы, в частности глины, используют в
качестве ионообменников и для катионов, и для анионов. Самая важная область
применения интеркалятов — их использование в твердотельных электрохимических
5.1. Неорганические твердофазные клатратные соединения 315
устройствах и при гетерогенном катализе. Как графит, так и слоистые халькогениды
металлов интеркалируют щелочные металлы; интеркаляты применяют как
электроды в твердотельных батареях для сотовых телефонов. TiS2 (хозяин), интеркалирую-
щий литий (гость), нашел коммерческое применение в батареях с высокой
плотностью энергии благодаря их высокой надежности — для кардиостимуляторов.
В нефтехимическом синтезе для осуществления каталитических превращений
до открытия цеолитов интенсивно использовали глины. Возрождается интерес к
столбчатым глинам с порами более 1 нм из-за неспособности цеолитов с
небольшими порами к крекингу очень тяжелых фракций сырой нефти. Внутри глинистых
минералов также могут протекать многие каталитические превращения
органических соединений. Более того, глины могут сорбировать нейтральные и заряженные
органические частицы; например, смектитовые глины применяют для
обесцвечивания пищевых масел, осветления алкогольных напитков и удаления кислотных
примесей из поливинилхлорида. Детальное рассмотрение этих областей выходит за
рамки настоящей книги, и в качестве типичного примера мы рассмотрим только
графитовые интеркаляты. Более полное обсуждение и наиболее важные работы
можно найти в ключевой ссылке к этому разделу.
8-7 O'Hare D. Inorganic intercalation compounds // Inorganic materials / Ed.
D.W.Bruce and D.O'Hare. Chichester: J.Wiley and Sons, 1996. P. 171-254.
5.1.3.2 Графитовые интеркаляты
Структура графита E.4) - аллотропа очень чистого углерода — это
бесконечный слой, состоящий только из шестичленных колец с 5/?2-гибридизованны-
ми атомами углерода. В его структуре слои уложены друг на друга на расстоянии
-3.35 А с максимальными п—я-стэкинг-взаимодействиями С---С (разд. 6.9). Чистый
графит — это полуметалл с заполненной валентной я-зоной, за которой сразу
следует пустая зона л*-проводимости без запрещенной зоны, характерной для
полупроводников. В результате п—п-взаимодействий происходит слабое перекрывание
валентной зоны и зоны проводимости и, следовательно, имеется ненулевая
плотность состояний на уровне Ферми прямо между этими двумя зонами. В терминах
электронных свойств, согласно своему значению электроотрицательности по По-
лингу, равному 2.5, углерод находится в середине 2-го периода Периодической
таблицы. Это означает, что атом углерода может и легко терять, и легко приобретать
электрон в зависимости от электроноакцепторнои или электронодонорнои
природы молекул-гостей, способных оказаться между слоями. Фактически графит
образует интеркаляты с атомами металлов; в таких интеркалятах металл
восстанавливает графит, а фторид-анион окисляет его. Типичные комплексы металлов включают
LiC6 и МС8 (М = К, Rb, Cs, Ca, Sr, Ba, Sm, Eu, Yb). Фториды металлов, дающие
комплексы с фторид-анионом, и их энтальпии восстановления приведены на рис.
5.12. Ясно, что только те частицы, энтальпия восстановления которых более
отрицательна, чем —502 кДж-моль, могут образовывать полные интеркаляты каскада
1. Материалы с частично заполненными каскадами более высоких уровней
получаются при значениях энтальпии восстановления ниже —440 кДжмоль.
5. Связывание нейтральных молекул
Графит E.4)
Интересно, что когда КС8 с плоской 2х2-структурой E.5) помещают в
атмосферу СО или СО2, гость постепенно исчезает, последовательно образуя фазы КС24,
КС36 и КС48. Это свидетельствует в пользу модели Даумаса—Геролда (см. разд.
5.1.3.1).
GeF4
О
WF6
о
5
О
BF,
SiF4
^ReF6 ^OsF(
+V2F2 =
pF AsF5 SbF5
PtF,
SbF5
«r + MF6 = M
e~ + 3/2MF5 =
PF5
-^гС^ + HF = MF^.] + HC1
+i/2Cl2 = MFy"+1 + MF^jCl
BF3OOpFsWGeF4
1 111 1 1
-300 -400 -500 -600 -700 -800 C~~J Реакция отсутствует
АЯОB98К),кДжмоль-1
^Щ Частичная интеркаляция
Интеркаляция по каскаду 1
Рис. 5.12. Энтальпии восстановления (кДжмоль) указанных реакций и степень интер-
каляции фторид-анионов графитом
5.1. Неорганические твердофазные клатратные соединения
317
E.5)
Графит также легко образует интеркаляты с Вг2 и с IBr, IC1, но не со F2, Cl2, 12
или с натрием. Структура интеркалята с Вг2 содержит недиссоциированные
молекулы Вг2, каждый атом которых располагается над шестичленным графитовым
кольцом. Возможно, что длины связи Вг—Вг, а также связей I—Вг и I-C1 B.27, 2.49
и 2.40 А соответственно) хорошо подходят для размещения между
шестиугольниками, a F2, Cl2 и 12 (длины связей 1.41, 1.99 и 2.67 А соответственно) или слишком
малы, или слишком велики. В случае натрия вероятно, что радиус его атома слишком
велик для эффективного образования NaC6, но слишком мал для возникновения
структуры МС8, обычной для ряда других металлов.
5* 1 «4 Клатраты Хофмана и Вернера
Комплексы включения (клатраты) типа комплексов Хофмана и
Вернера образуются в пустотах решеток ряда неорганических координационных
соединений.
Соединения включения Хофмана имеют общую формулу M(NH3JM'(CNL-2G
(где М — переходный металл 4-го периода Периодической системы, М = Mn—Zn
или Cd; М' = Ni, Pd, Pt, a G — небольшая ароматическая молекула). Твердофазная
структура этих образований состоит из полимерного слоя, в котором лиганды CN~
- это мостики между металлами М', обладающими плоскоквадратной
координацией и экваториальными центрами переходных металлов М с октаэдрической
координацией. В результате лиганды NH3 выступают вверх и вниз из этого слоя, образуя
в решетке полости, подходящие для включения небольших ароматических
молекул, например бензола или тиофена. Структура бензольного клатрата Хофмана
Ni(NH3JNi(CNL-2C6H6 показана на рис. 5.13.
Клатраты Вернера образуют многие вернеровские координационные металло-
комплексы типа МХ2А4 (где М - переходный металл 4-го периода, Cr-Zn, Cd или
Hg; X = NCS-, NC0~, CN~, N0^, NOj, Cl", Br~, I~; A - нейтральный замещенный
пиридин). Как и в клатратах Хофмана, в клатратах Вернера гостями являются не-
318
5. Связывание нейтральных молекул
(>«офо#-6
Рис. 5.13. Структура
бензольного клатрата Хофмана
Ni(NH3JNi(CNL-2C6H6
большие ароматические молекулы, хотя хозяин — не полимер. В этом случае
пустоты решеток возникают из-за присутствия широких плоских пиридильных лиган-
дов. Клатраты Вернера используют для разделения орто-, мета- и пара-изомеров
дизамещенных бензолов хроматографическими методами (J.Lipkowski et al., 1979).
В общем, такие неорганические клатратные комплексы могут образовывать многие
координационные соединения, а не только металлокомплексы типа МХ2Аг4. Их
образование происходит всегда, когда комплекс не может эффективно упаковаться в
твердом состоянии (прежде всего из-за формы), причем в кристаллизационной
среде имеется достаточно подходящая молекула растворителя или другая молекула,
которая может действовать как гость. Клатраты Вернера — стабильные и удобные
модельные системы для систематического изучения, однако в приложении к
процессам разделения они не могут конкурировать с цеолитами (см. разд. 5.1.2).
9-т
Lipkowski J. Werner clathrates // Comprehensive supramolecular chemistry / Ed.
J.L.Atwood, J.E.D.Davies, D.D.MacMcol and F.Vogtle. Oxford: Pergamon, 1996.
Vol. 6. P. 691-714.
5-2.1
9-т
Твердые клатраты
органических хозяев
Клатраты мочевины
Harris K.D.M. Investigating the structure and dynamics of a family of organic solids —
the alkane urea inclusion compounds // J. Solid State Chem. 1993. Vol. 106. P. 83.
5.2.1.1 Структура
Простая органическая молекула, мочевина E.6), как и ее
серосодержащий аналог, тиомочевина E.7), образуют твердые клатраты с длинноцепными
углеводородами типа «-алканов. Мочевина способна действовать как хозяин благодаря
сильным межмолекулярным водородным связям между кислотными протонами
NH2-rpynn и атомами кислорода или серы соседних молекул. В результате образу-
5.2. Твердые клатраты органических хозяев
19
0 2 4 6 8 ЮА
1 i i i i i
Рис. 5.14. (а) Стереоизображение одного канала мочевины как хозяина. Хиральные
ленты образованы за счет водородных связей анти-N—Н---0 и объединены водородными
связями син-N—Н---О. (Воспроизведено с разрешения Hollingsworth M.D., Brown M.E.,
HillierA.C. et al. Superstructure control in the crystal growth and ordering of urea inclusion
compounds // Science. 1996. Vol. 273. P. 355). (б) Размеры ван-дер-ваальсова поперечного сечения
полости в канале мочевины в сравнении с размерами «-октана (слева), бензола (вверху),
3-метилгептана (справа) и 2,2,4-триметилоктана (внизу)
ется хиральная спиральная полая трубка из молекул мочевины с минимальным
ван-дер-ваальсовым диаметром 5.5—5.8 А, в которую могут войти гости с малым
поперечным сечением. Такое спиральное расположение позволяет каждой молекуле
мочевины образовывать водородные связи со всеми ее NH-протонами,
максимизируя межмолекулярные взаимодействия. Это означает, что каждый атом кислорода
(или серы) должен принять всего четыре водородные связи. Сходные, но большие
каналы образует тиомочевина, что позволяет включать разветвленные
углеводороды, но не небольшие w-алканы (рис. 5.14).
О Q
--А-»
н
N
I
Н
Мочевина
E.6)
N N
I I
Н Н
Тиомочевина
E.7)
Поскольку в молекулах мочевины нет никаких «резервных» водородных связей,
они предоставляют встроившимся гостям лишь однородную поверхность, что
приводит к слабому связыванию, часто в нестехиометрических соединениях; при этом
могут происходить значительные трансляционные и колебательные движения гостя
в твердом состоянии. Так, независимо от индивидуальности гостя, сохраняется
гексагональная симметрия решетки. Удаление гостя (например, внешним вакуумом)
приводит к немедленному разрушению каналов и образованию тетрагональной
мочевины, неспособной к включению и имеющей совершенно другую структуру.
32ij| 5. Связывание нейтральных молекул
5.2.1.2 Гости
Одна из главных причин интереса к соединениям включения
мочевины состоит в их возможном применении для разделения линейных и разветвленных
углеводородов в нефтехимической промышленности. Поскольку диаметр канала
лишь ненамного превышает ван-дер-ваальсов диаметр сечения линейной
углеводородной цепи, для осуществления реакции включения допустимо лишь
незначительное разветвление. Возможность мочевины образовывать данное соединение
включения можно просто оценить, сравнивая размер канала с диаметром
соответствующего гостя. Например, найдено, что молекула бензола шире, чем канал, и
включение происходит с трудом (рис. 5.14, 6). Интересно, однако, что увеличение
длины цепи допускает наличие больших, более разветвленных концевых групп.
Так, 1-фенилоктан в качестве гостя не образует соединения включения, а намного
более длинный 1-фенилэйкозан дает его. Измерение равновесного давления пара
показало, что энтальпия комплексообразования возрастает примерно на
10.0 кДж-моль при увеличении линейной цепи на одну СН2-группу.
Следовательно, дополнительная стабилизация, достигаемая при комплексообразовании более
длинного гостя, перевешивает неблагоприятные стерические взаимодействия с
концевой фенильной группой. В общем алкановые молекулы-гости проявляют
одномерное упорядочение, располагаясь вдоль канала мочевины, но структура в
одном канале никак не соотносится со структурой в соседних каналах. Об этом
свидетельствует диффузное рассеяние, наблюдаемое при дифракции рентгеновских
лучей в образце.
5.2.1.3 Соразмерные и несоразмерные структуры
Интересной и важной проблемой соединений включения мочевины
является соотношение между кристаллографическими повторяющимися
расстояниями хозяина и гостя, параллельными оси канала (кристаллографическая ось с).
Под «повторяющимся расстоянием» мы подразумеваем расстояние, которое
воображаемый наблюдатель должен пройти в кристалле в определенном направлении
до окружения (в смысле окружающих молекул), идентичного первоначальному.
Расположение хозяина повторяется через каждые 11.02 А - это величина,
обозначаемая сИ (с означает тот факт, что канал расположен по направлению
кристаллографической оси с элементарной ячейки). Соразмерная структура - это структура,
в которой гость повторяется через расстояние с , такое же, как и у хозяина, или
через расстояние, кратное размеру хозяина (рис. 5.15). Для соразмерной структуры
Рис. 5.15. Повторяющиеся
расстояния хозяина (ch)
и гостя (с) в соединениях
включения мочевины не
обязательно одинаковы
5.2. Твердые клатраты органических хозяев 321
справедливо соотношение pch ~ qcg, когда р и q — небольшие целые числа. В
несоразмерных структурах такого простого соотношения не существует.
5.2.1.4 Изучение фактов: алканоны и алкандионы
Влияние упорядочения при образовании соединения включения на
поведение гостя лучше всего видно на примере длинноцепных алканонов E.8) и
алкандионов E.9) (М. Hollingsworth and M. Brown, 1995).
О О
О
и=1:(в)
E.8) л = 2.-(б) E-9)
п = 3: (в)
Изучение более чем 100 таких гостей показало, что, в отличие от алкановых
аналогов, многие из них при дифракции рентгеновских лучей обнаруживают
значительное трехмерное упорядочение в противоположность расположению только
вдоль оси канала. Это означает межканальные взаимодействия между гостями и их
регулярную упаковку «голова к голове» или «голова к хвосту» вдоль оси канала.
Однако расчеты показывают, что диполь-дипольные взаимодействия типа
С8+=О8~-•С8+=О8~ слишком слабы, чтобы влиять на межканальное упорядочение,
и, действительно, в некоторых случаях (например, в случае соединений E.86) и
E.8в)) дифракция рентгеновских лучей и твердотельная 13С ЯМР-спектроскопия
показали только одномерное упорядочение. Ключом к пониманию этой проблемы
являются соразмерные структуры, образованные соединениями E.9а)—E.9в).
Разница между соразмерной фазой алкандиона и несоразмерной фазой алканона
четко показана на рентгеновской дифракционной картине комплексов мочевины с
E.8в), рис. 5.16, а, и с E.9в), рис. 5.16, б. В первом случае присутствие
неупорядоченного гостя подтверждается только наличием диффузных линий в слоях,
обозначенных индексом «g». Напротив, четкая дифракционная картина с Ъс = 4ch = 44.0 А
ясно видна для соразмерного комплекса на рис. 5.16, б.
Происхождение этих эффектов было окончательно установлено путем
определения полной кристаллической структуры методом РСА, которое показало, что
между определенными NH2-rpynnaMH мочевины и карбонильными атомами
кислорода гостя образуются водородные связи (рис. 5.17). Фактически в структурах
всех комплексов алканонов и алкандионов соответстветствующим образом
расположенные молекулы хозяина способны выходить из плоскости стенки канала,
предоставляя одну из своих NH2-rpynn для образования водородной связи с гостем. В
случае алкандионов E.9) силы двух водородных связей достаточны для
формирования трехмерно упорядоченной соразмерной структуры при комнатной
температуре. Для гостей типа E.8), таких, как деканон-2, образующих несоразмерные
структуры, охлаждение до —85 °С также приводит к подобным соразмерным структурам.
21 - СТИД ДжВ
322
(а)
5. Связывание нейтральных молекул
(б)
Рис. 5.16. Рентгеновская дифракционная
картина: (а) для комплекса деканон-2-мочевина с
осциллирующими каналами, показывающая
дифракцию решетки хозяина (Л) и диффузные
полосы в области дифракции решетки гостя (g);
(б) для комплекса декандион-2,9мочевина,
показывающая соразмерное соотношение
Ъс = 4ch — 44.0 А. (Воспроизведено с разрешения
Hollingsworth M.D., Brown M.E., HillierA.C. et al.
Superstructure control in the crystal growth and
ordering of urea inclusion compounds // Science.
1996. Vol. 273. P. 1355)
Рис. 5.17. Стереоизображение вдоль оси канала комплекса декандион-2,9мочевина,
показывающее образование водородных связей между карбонильными группами гостя и
«выступающими» донорными атомами мочевины. (Воспроизведено с разрешения Atwood J.L.,
Davies J.E.D., MacNicol D.D. and Vdgtle F. Comprehensive supramolecular chemistry. Oxford:
Pergamon, 1996. Vol. 6. P. 199)
5.2. Твердые клатраты органических хозяев
1323
@ 10)
Рис. 5.18. Необратимая
фазовая переориентация
в комплексе ундеканди-
он-2,10мочевина (а),
индуцированная
напряжением, (б) Схема кристалла до
приложения напряжения:
стрелки — это
карбонильные диполи гостей в
одном слое, (в) Кристалл под
однородным
напряжением, приложенным слева в
нижней части картинки,
(г) Схема напряженного
кристалла.
(Воспроизведено с разрешения
Hollingsworth M.D. and
Brown M.E. Stress-induced
domain reorientation in urea
inclusion compounds //
Nature. 1995.
Vol. 376. P. 323)
Точная ориентация молекул, связанных с молекулами мочевины водородными
связями, решающим образом зависит от длины цепи гостя. Случай ундекандиона-
2,10 особенно интересен. Для дионов с нечетным числом атомов углерода в цепи
две карбонильные группы находятся по одну сторону молекулы в вытянутой
д//-дл?/-конформации цепи. В результате образуется иная соразмерная структура с
2cg = 3ch = 33.0 А, в которой вследствие водородного связывания канал сужается
только с одной стороны. Такая структура с пониженной симметрией проявляет
значительные ферроупругие свойства. Небольшие механические напряжения могут
переориентировать объемные домены внутри кристалла мочевины в соответствии с
согласованным поворотом групп гостей на 60° в соседних каналах. Это приводит к
образованию четких параллельных полосок, видимых при рассмотрении кристалла
в поляризованном свете (рис. 5.18). Если приложенное напряжение достаточно
слабо, переориентированный домен возвращается к первоначальной ориентации за
несколько секунд. Более сильные напряжения приводят к постоянным
изменениям ориентации, хотя даже такое воздействие можно нивелировать вращением
кристалла на 60° с повторным приложением давления.
5.2.1.5 Полимеризация при включении
Более широкие каналы в молекуле тиомочевины предполагают
интересное использование ее соединений включения для проведения химических
реакций в матрице хозяина. Следует ожидать, что строгие стерические ограничения
хозяина повлекут за собой высокую стерео- и региоселективность реакций, что
трудно контролировать в обычных условиях. Поскольку соединения мочевины
224 5. Связывание нейтральных молекул
имеют канальную структуру, предполагают, что они будут пригодны для
селективного образования длинных линейных полимеров. В частности, еще в 1960 г. Браун
и Байт (Braun and White) сообщили о полимеризации бутадиена-2,3, 2,3-дихлорбу-
тадиена-2,3, циклогексадиена и родственных им соединений при включении их в
канал мочевины. В большинстве случаев были выделены кристаллические
полимеры с высокими температурами плавления, что свидетельствует о длинноцепных
соединениях с узким интервалом молекулярных масс, которые, безусловно,
образуются в строго контролируемых условиях.
5*2*2 Другие канальные клатраты:
клатраты тримезиновой кислоты,
спиральных тубуландов и пергидротрифенилена
5.2.2.1 Тримезиновая кислота
Тримезиновая, или бензол-1,3,5-трикарбоновая, кислота (ТМА,
E.10)) способна образовывать ряд комплексов за счет водородного связывания
остатков карбоновой кислоты или ионизации (солевые комплексы). Однако особый
интерес представляют сетчатые структуры моногидрата тримезиновой кислоты
(ТМАН2О). Кристаллизация соединения E.10) из воды без строгого контроля
температуры дает кристаллогидраты двух типов, ТМА-2Н2О и ТМА-5Д Н2О, а также
кристаллы несольватированной ТМА. Добавление пикриновой кислоты B,4,6-три-
нитрофенол) к этой системе приводит к желтым иглам состава ТМАН2О2/9(ПИК~
риновая кислота). Удивительно, что, несмотря на различные составы, структуры
этих трех гидратов весьма близки.
О E.10)
В каждом случае прямоугольная элементарная ячейка состоит из четырех
молекул ТМА, образующих в структуре хозяина двухмерную прямоугольную полость.
Каждая ячейка соединена с соседними прямоугольными ячейками, лежащими в
той же плоскости, водородными связями с участием молекул воды. Стехиометрия
всего сетчатого слоя хозяина отвечает формуле ТМА Н9О, что, следовательно,
оправдывает термин «моногидрат» (рис. 5.19).
Как в ТМАН2О-2/9(пикРиновая кислота)> так и в ТМА-2Н2О (точнее,
ТМАН2О[2Н2О], где в квадратных скобках указаны частицы гостя) моногидрат-
ные слои располагаются непосредственно друг над другом, образуя бесконечные
прямоугольные полости, в которые молекулы гостей (вода или пикриновая кислота)
S.2. Твердые клатраты органических хозяев
О ОН
325
Н2О
он
Н2О
н2о
Н2О
аг»-.
о
О' ОН
о
Рис. 5.19. Структура слоя ТМАН2О.
Молекулы-гости (Н7О) включаются
в прямоугольные полости
включаются без каких-либо специфических взаимодействий между гостем и
каналом хозяина. Расположение гостей в этих комплексах и стехиометрия комплексов
определяются соотношением между размером гостя и толщиной слоя. В гостевой
части каждой структуры имеется значительный беспорядок, но рентгеновские
дифракционные картины показывают сильное диффузное рассеяние, что
свидетельствует о частичном упорядочении молекул-гостей (ср. одномерное упорядочение в
клатратах мочевины). Если комплекс ТМА-Н2О-2/9 (пикриновая кислота) устойчив,
то комплекс ТМАН2О[2Н2О] быстро теряет воду в атмосфере из-за открытости
каналов.
32$?
5. Связывание нейтральных молекул
ТМА
Рис. 5.20. Упаковка смещенных слоев в ТМАН20 [0.2ТМА]
(по Herbstein F.H and Marsh R.E. 1977)
О
О
Рис. 5.21.
Гексагональное
расположение молекул ТМА с
образованием
гексагонального канала,
перпендикулярного
плоскости страницы
5.2. Твердые клатраты органических хозяев 327
Рис. 5.22.
Стереоизображение комплекса 2ТМА[н-тет-
радекан] при рассмотрении
вдоль кристаллографической
оси с. Молекулы н-тетрадека-
на очень разупорядочены.
Структура хозяина
повторяется через каждые три слоя.
(Воспроизведено с
разрешения Herbstein F.H. et al., 1987)
Структура гидратов ТМА-5'/6Н2О основана на той же прямоугольной сетке
хозяина. Необычную стехиометрию можно объяснить, если написать эту формулу в
виде 6ТМА-5Н2О или 5ТМА-5Н2О ТМА, или, по аналогии со структурой комплекса
пикриновой кислоты, как ТМАН2О[0.2ТМА]. Фактически каркас моногидратно-
го хозяина содержит бесконечную зигзагообразную цепочку ТМА-гостей,
удерживаемых водородными связями, что приводит к смещению одного слоя
относительно другого с образованием зигзагообразных каналов (рис. 5.20).
Эта структура идеально упорядочена и устойчива, в ней нет значительного
взаимодействия между цепями ТМА-гостей и слоями хозяина, перпендикулярными им.
Интересно, что химия включения ТМА не ограничивается образованием
прямоугольных каналов. В присутствии больших частиц-гостей типа линейных и
разветвленных длинноцепных углеводородов, длинноцепных спиртов и т.п. образуются
гексагональные каналы хозяев, состоящие из повторяющихся двухмерных ТМА-
мотивов типа «мелкая проволочная сетка» («chicken wire», рис. 5.21). Возникшие
каналы заполняются молекулами-гостями и могут занимать два, три или даже пять
слоев хозяина, прежде чем эта структура снова повторится. Структура комплекса
2ТМА-[«-тетрадекан] показана на рис. 5.22.
fr Herbstein F.H. Structural parsimony and structural variety among inclusion
complexes // Top. Curr. Chem. 1987. Vol. 140. P. 107-139.
5.2.3.2 Спиральные тубуланды
К структурам, родственным хозяевам на основе мочевины и тиомоче-
вины (см. разд. 5.2.1), относят спиральные тубуланды - канальные структуры,
образовавшиеся из большого числа циклических диолов типа E.11)—E.13). Структуры
канального типа получаются в результате кристаллизации соединения E.11) в
присутствии различных растворителей или других потенциальных моле кул-гостей. В
этих структурах стенки тригональных каналов (рис. 5.23) включают две спиральные
нити молекул диола, имеющие благодаря водородным связям общий
«позвоночный столб» винтовой симметрии третьего порядка C,; поворот на 120°
сопровождается трансляцией вдоль оси вращения).
5. Связывание нейтральных молекул
НО/,.
E.11)
E.13)
Такая канальная структура может содержать множество частиц-гостей,
способных устраиваться внутри доступного пространства, за исключением фенолов и
других соединений, участвующих в образовании сильных водородных связей. За счет
этих взаимодействий такие соединения сокристаллизуются с хозяевами, давая
структуру без каналов. В трубчатых структурах соотношение диолтость обычно
составляет 3:1 (с гостями типа ацетонитрила, тиофена, диоксана и т.п.), но известен
также ряд нестехиометрических соединений включения, состав которых зависит от
соотношения длины гостя и длины повторяющейся канальной единицы E.11K.
Несмотря на сильные водородные связи, стенки каналов обладают значительной
гибкостью, и в присутствии различных гостей размеры элементарной ячейки,
перпендикулярные направлению канала, могут варьироваться в пределах 5% A1.89-12.47 А,
ацетонитрил — диоксан) по мере того, как структура изгибается для включения все
больших гостей.
Рис. 5.23. Пространственная модель структуры тригонального канала соединения E.11) с
треугольным поперечным сечением и длиной ребра 6.30 А
5.2. Твердые клатраты органических хозяев 319
Мотив алициклического диола E.11) может структурно значительно меняться с
образованием каналов, имеющих сильно различающиеся размеры (например, в
соединениях E.12) и E.13)). В широком смысле семейство спиральных тубуландов
распадается на две категории: хозяева с узкими каналами, способными
существовать без гостей, и хозяева с гораздо большими каналами, которые сжимаются при
удалении частиц-гостей. Меньшие каналы имеют незаслоненную площадь
поперечного сечения (это площадь канала, видимая при рассмотрении вдоль его оси, т.е.
минимальная площадь канала при наличии в нем извилин), равную примерно
1.2-9.9 А2, а большие каналы - 19.8-34.0 А2.
9-1 Bishop R. and Dance I.G. New types of helical canal inclusion networks // Top. Curr.
Chem. 1988. Vol. 149. P. 137.
5.3.2.3 Пергидротрифенилен
Пергидротрифенилен (РНТР, E.14)) - хиральное насыщенное
органическое соединение симметрии D^, которое в твердом состоянии образует
гексагонально расположенные стопки (наподобие пчелиных сот), содержащие каналы,
находящиеся друг от друга на расстоянии 15 А, в которые включаются молекулы-
гости. Гости — это обычно длинные линейные молекулы, например молекулы
углеводородов, спиртов, сложных эфиров, простых эфиров и кислот, размеры которых
соответствуют размерам длинных каналов, однако разветвленные и объемистые
гости также могут включаться в каналы. Взаимодействия хозяин—гость (типа ван-
дер-ваальсовых) минимальны. Из-за отсутствия в молекуле углеводорода
функциональных групп молекулы хозяина и гостя часто показывают низкую степень
порядка и несоразмерность или одномерно упорядоченную упаковку, как в соединениях
включения мочевины (см. разд. 5.2.1). Это дает размер элементарной канальной
ячейки для гостя (с), который кратен размеру единичной ячейки хозяина (ch),
зависящему от длины гостя. РНТР представляет особый интерес из-за его
способности включать гиперполяризуемые частицы-гости с образованием кристаллов,
обладающих полярностью. В результате образуются РНТР-соединения, имеющие
интересные нелинейно-оптические и электрооптические свойства (разд. 8.6).
Кажущийся кристаллографический беспорядок, возникающий вследствие
одномерного упорядочения гостей, приводит к значительным структурным проблемам при
характеристике соединений включения РНТР. Только в 1997 г. Г.-Б. Бюрги
(Н.-В. Burgi) из Бернского университета (Швейцария) удалось дать полную
структурную характеристику сильнополярного соединения включения РНТР. Изученное
вещество представляло собой соединение включения (±)-РНТР с 1-D'-нитрофе-
нил)пиперазином (NPP, E.15)) формулы [(±)-PHTP]5[NPP], с помощью которого
осуществлены генерация второй гармоники, электрооптический и
пироэлектрический эффекты, возникающие из-за полярности кристалла. Соотношение гостыхо-
зяин, равное 5:1, — это прямое следствие относительных длин повторяющихся
единиц хозяина (ch = 4.73 А) и гостя (с = 5 ch).
330
5. Связывание нейтральных молекул
РНТР E.14)
NPP E.15)
Проекция структуры этого комплекса хозяин-гость вдоль оси канала показана
на рис. 5.24, а, а расположение гостей - на рис. 5.24, б.
Ключевой вопрос о структуре комплекса [PHTP]5[NPP] связан с причиной
нелинейной поляризации его кристалла. Происхождение таких свойств можно легко
объяснить наличием полярной стопки молекул гостей или хозяев, но остается
вопрос: каким образом и почему стопки этих молекул образуют нецентросимметрич-
ные структуры.
Объемная полярность кристалла могла бы возникнуть при спонтанном
разделении (+)- и (—)-форм хозяина на дискретные энантиомерные кристаллы (разд.
6.1.5.3). Однако кристаллическая решетка хозяина центросимметрична, и
существует беспорядок между (+)-РНТР и (-)-РНТР в одном и том же кристалле. При
детальном анализе коротких упаковочных контактов в хозяине обнаруживается, что
образуются индивидуально полярные гомохиральные стопки (т.е. только один
энантиомер в каждой стопке) либо (+)-, либо (-)-формы. В кристалле эти стопки
распределяются беспорядочно. Ван-дер-ваальсова энергия взаимодействия между
парами форм (+)-(+), (-)-(-) или (+)-(-) примерно одинакова, и поэтому
беспорядочное распределение неудивительно. Важно, что решетка хозяина в среднем не-
полярна в результате погашения (+)-стопок равным числом (—)-стопок.
Следовательно, полярность должна обеспечиваться молекулами-гостями. Опять-таки легко
представить себе полярную цепь молекул-гостей, расположенных в
последовательности донор-акцептор—донор—акцептор (-DADA--), с учетом донорной
способности NH-группы и акцепторной способности NO2-rpynnbi при образовании
водородной связи. Напротив, взаимодействия D—D и А—А, когда молекула в цепи
переворачивается, должны быть неблагоприятными. Поэтому цепи DADA должны
быть индивидуально полярными, но, поскольку эти цепи отделены друг от друга на
15 А, трудно представить себе, чтобы кооперативные взаимодействия гость—гость
между каналами приводили бы к ориентации всех цепей в одном направлении.
Таким образом, и межцепные взаимодействия должны вызывать погашение
полярности.
Изучение размеров каналов показало, что молекула NPP не может
перевернуться, если трехмерный канал полностью сформировался, и, таким образом,
ориентация цепей NPP должна задаваться в ходе роста кристалла. Если кристалл имеет
объемную полярность, то это должно означать наличие движущей силы, способной
5.2. Твердые клатршпы органических хозяев
3311
F)
Рис. 5.24. (а) Проекция структуры
комплекса [(±)-РНТР]5 [NPP] по оси
канала, (б) Схематическое изображение
каналов с молекулами-гостями NPP.
Гости взаимодействуют посредством
водородных связей N—H—O2N
и упорядочены внутри каждого канала.
Положения гостей в соседних каналах
различаются целочисленными
множителями с /5. (Воспроизведено с
разрешения Konig О., Burgi H.-B.,
Armbruster T. et al. A study in crystal
engineering: Structure, crystal growth and
physical properties of a polar perhydrotripheny-
lene inclusion compound // J. Amer. Chem.
Soc. 1997. Vol. 119. P. 10632-10640)
f
\
расположить более чем 50% цепей в одном направлении, т.е. такой движущей силы
которая располагает, например, группу NO2 у входа в растущий канал чаще, чел
противоположный конец молекулы-гостя. Фактически общая полярность — это ре
зультат разной вероятности различных взаимодействий гость-гость: •••DA—
-DD- и -АА- (рис. 5.25). Комплементарный мотив -DADA-, разумеется, наи
более благоприятен, так как образует индивидуальные полярные цепи. Данные по
казывают, что эти цепи тянутся примерно на 100 А до возникновения «ошибки»
когда включаются дефекты —DD— (например, N—Н—Н—N) или —АА— (например
NO2-O2N), которые влияют на обращение полярности цепи. Ключевой момент -
332'-
5. Связывание нейтральных молекул
АкЧепт°Р
i
Акцептор н Донор
Q
N
Н Донор
Акцептор
Донор О О Акцептор
Донор о о
^^ Акцептор
Н Донор
•DA-
Акцептор N
7 Н Донор
DD-
•А А-
Рис. 5.25. Возможное
расположение гостей
в каналах включения
соединения РНТР. Поскольку
взаимодействие •••DA---
наиболее вероятно, наличие
большего числа дефектов
•DD-.чем "АА-,
приводит к обшей
полярности кристалла
меньшая вероятность дефекта •••АА-- в сравнении с дефектом •-DD-••. Это
следствие того, что две 1ЧО2-группы отталкиваются друг от друга, в то время как между
протоном одной группы NH и атомом азота другой такой же группы существует
некоторое притяжение. В результате, при сборке каналов, по законам статистики, на
входе в канал должно быть большее число концевых NO2-rpynn, чем NH-rpynn.
8-1 Konig О., Burgi H.-B., Armbruster T. et al. A study in crystal engineering: Structure,
crystal growth and physical properties of a polar perhydrotriphenylene inclusion
compound // J. Amer. Chem. Soc. 1997. Vol. 119. P. 10632-10640.
5»2«3 Гидрохинон, фенол и соединение Дианина:
стратегия шести хозяев
Гидрохинон E.16), фенол E.17) и соединение Дианина E.18)
действуют как хозяева благодаря образованию с помощью водородных связей шестичлен-
ных циклов (ср. с E.2)). Наличие в этих соединениях устойчивой жесткой
группировки фенольных гидроксильных групп непосредственно обуславливает
чередующееся расположение объемистых арильных заместителей (R) сверху и
снизу от плоскости шестичленного цикла (рис. 5.26); при этом образуются полости,
5.2. Твердые клатраты органических хозяев
333
выстланные арильными остатками. В кристалле все эти полости вместе составляют
закрытую клетку, в которой могут разместиться разнообразные молекулярные гости.
E.17)
E.18)
В твердом состоянии гидрохинон существует в трех формах, обозначаемых как
а-, C- и у-формы, из которых только C-форма образует клатраты. Клатраты C-гид-
рохинона имеют общую формулу 3C6H4(OHJ-jcG, где G - частицы гостя, ах-
фактор занятости центра, зависящий от условий реакции. При полной занятости
каждой полости х = 1, но для частиц многих гостей х может быть меньше единицы
(например, G = Хе, х = 0.866; G = H2S, x — 0.874); при этом устойчивые клатраты
все же образуются. Размеры шестичленных циклов и полостей могут значительно
изменяться в соответствии с размерами гостя. Так, небольшие сферические гости,
такие, как Хе и H2S, образуют клетки с высокой симметрией /?3 (клатрат типа I).
В случае метанола, SO2 и НС1 симметрия понижается до R3 с потерей центра
симметрии и образованием клатрата типа II, в котором длина клеток увеличена для
приема более протяженного гостя. Известен также один пример клатрата типа III
(гость — MeCN), в котором наблюдаются три различные ориентации гостя
(симметрия РЗ). Бакминстерфуллерен также образует высокосимметричную структуру
КЗт, напоминающую металлический полоний (разд. 5.4).
Соединение Дианина E.18) названо в честь русского химика А.П.Дианина,
который впервые выделил его в 1914 г. при конденсации фенола с оксидом мезитила
и случайно получил из него несколько клатратов хозяина с обычными
растворителями. Соединение Дианина — хиральная молекула (асимметрический атом
углерода обозначен звездочкой), в твердом состоянии оно имеет геометрию с одним энан-
тиомером «вверху» и с другим «внизу» (рис. 5.27, а). Действительно, чистый
S-энантиомер совсем не образует клатратов. Среди фенолятных хозяев соединение
Дианина — одно из наиболее «всеядных». Оно помимо широкого круга
органических и неорганических гостей включает аргон, глицерин, небольшие углеводы и, что
особенно важно, SF6, широко используемый как газ-изолятор в
электротехнической промышленности. Предполагают, что соединение E.18) можно применять для
хранения и транспорта летучих веществ. Соединение Дианина включает также
многие растворители и родственные молекулы. Для большинства клатратов,
включающих ароматические гости, соотношение хозяинтость составляет 6:1, что
согласуется с их гексамерной структурой. Молекулы растворителя большего размера -
типа бутанола, этанола или ацетона — образуют аддукты 3:1, в которых две
молекулы растворителя занимают гексамерные полости хозяина, тогда как метанол дает
аддукт 2:1. Лишь пиперидин образует аддукт 1:1. Структура комплекса несольвати-
334
5. Связывание нейтральных молекул
Рис. 5.26. (а) Схема,
показывающая клеточную структуру
гидрохинона, (б) Структура
решеточной полости в C-гидро-
хиноне с гостем-метилизоциа-
нидом
рованного соединения Дианина, имеющая гексагональную группировку хозяина, и
схема его полости приведены на рис. 5.27, где также указаны размеры
образующейся полости.
С одной стороны, изучение динамики включенных молекул показывает, что
небольшие молекулы, например этанол, (две на клетку) в твердом состоянии быстро
двигаются, как в жидкости, даже при низкой температуре. С другой стороны, моле-
5.2. Твердые клатраты органических хозяев
(а) (б)
2.8 А
5.0 А
2.5 А
Рис. 5.27. (а) Структура комплекса несольватированного соединения Дианина (передняя
и задняя части центральной полости опушены для простоты), (б) Схема полости хозяина.
(Воспроизведено с разрешения AtwoodJ.L., DaviesJ.E.D., MacNicol D.D. and Vbgtle F.
Comprehensive supramolecular chemistry. Oxford: Pergamon, 1966. Vol. 6. P. 619)
кула СС14, хорошо соответствующая клетке, удерживается относительно прочно.
Как и в случае клатратных гидратов, теплопроводность хозяина, заполненного
гостем, чрезвычайно низка.
Способность соединения E.18) к включению достаточно сильна, и поэтому оно
может связывать различных гостей и все еще сохранять (хотя и измененную)
способность к клатратобразованию. Чрезвычайно большое число аналогов этого
соединения, образующих полости различных размеров и форм, было получено путем
следующих превращений:
1. Изменение характера замещения в положениях СB) и СD).
2. Замена ОН-группы другими функциональными группами, образующими
водородные связи, такими, как NH2 и SH.
3. Замена эфирного атома кислорода на S, Se, SO2 и т.п.
4. Введение заместителей в конденсированное ароматическое кольцо.
Используя стратегию 1, А. Гарди с сотрудниками (A.Hardy et al., 1979) получил
соединение-хозяин с гораздо большей центральной полостью G.10 А против 4.20 А)
за счет удаления одной из метальных групп при СB). При удалении метильной
группы из положения СD) образовалась совершенно другая структура,
неспособная к образованию клатратов. Замена ОН на NH2 в соответствии со стратегией 2
также привела к потере способности образовывать клатраты. Однако тиольный
аналог соединения Дианина E.19) очень похож по своему поведению на это
соединение: кристаллизуясь из СС14, он дает комплекс 6:1 с несколько уширенной и
ззЙ
5. Связывание нейтральных молекул
(а)
(б)
ТзХ
"бТА"
"Ш"
+с
1.0
0.5
0.3
0.0
Рис. 5.28. Сечения через ван-дер-ваальсову поверхность полости для соединений:
(а) E.20) и (б) E.19). (Воспроизведено с разрешения MacNicol D.D. et al. 1979)
удлиненной полостью (что соответствует более длинным водородным связям
S—H--S и связям S—С) по сравнению с гидроксильным аналогом E.18). При
замене эфирного атома кислорода на серу в соответствии со стратегией 3 образуется
очень эффективный хозяин E.19), способный капсулировать небольшие гости в
соотношении 3:1 (т.е. два гостя на гексамерную полость) и большие гости в виде
клатратов 6:1. Этот хозяин может даже удерживать летучую (и сильнотоксичную)
диметилртуть Hg(CH3J под высоким вакуумом несколько дней. Наконец, при
введении метальных групп в различные положения ароматического кольца, согласно
стратегии 4, образуется ряд интересных хозяев с полостями разных размеров. В
частности, хозяин E.20) имеет полость в виде фонаря, значительно отличающуюся от
полостей исходных соединений E.18) и E.19), с шириной 2.6-7.7 А (рис. 5.28).
Соединение E.21) не проявляет клатратных свойств.
E.19)
E.20)
9-т
Мак Т.С. W. and Bracke B.R.F. Hydroquinone clathrates and diamondoid host lattices //
Comprehensive supramolecular chemistry / Ed. J.L.Atwood, J.E.D.Davies,
D.D.MacNicol and F.Vogtle. Oxford: Pergamon, 1996. Vol. 6. P. 23-60.
5.2. Твердые клатраты органических хозяев
337
5» 2 «4 Три-о-тимотид
8-т
Arad-Yellin R., Green B.S., Knossow M. and Tsoucaris G. Enantiomeric selectivity of
host lattices // Inclusion compounds / Ed. J.L.Atwood, J.E.D.Davies and
D.D.MacNicol. New York: Academic Press, 1984. Vol. 3. P. 278-284.
5.2.4.1 Соединения включения
Известно, что три-о-тимотид (ТОТ, E.22)) образует очень широкий
круг твердых соединений включения, из которых к настоящему времени более 140
уже выделено. Замечательная черта химии хозяин-гость соединения E.22) —
большое число симметрии кристаллов, принимаемых его клатратами. К настоящему
времени идентифицированы, по крайней мере, 12 способов кристаллической
упаковки. Причина такого чрезвычайного разнообразия пока неизвестна, но,
по-видимому, это следствие способности ТОТ капсулировать огромное количество гостей,
различающихся по размерам и форме, включая гостей с линейным, плоским или
шарообразным каркасом. Только линейные и сильноразветвленные гости
эффективно не капсулируются. Например, настолько различные гости, как9-метилантра-
цен E.23) и пентакарбонил-4-аминопиридилвольфрам E.24), капсулируются
легко, а тра«с-2-бромциклогексанол-1 E.25) не образует клатрата. С небольшими
гостями типа метанола клатраты также образуются, но с некоторым трудом, а при
образовании клатрата с 12 90% полостей ТОТ не содержат гостей.
i-Pr
Me
E.23)
ТОТ E.22)
H2N
со
W.
ОС
E.24)
СО
он
.со
со
338 5. Связывание нейтральных молекул
Результаты 'Н ЯМР указывают, что ТОТ в растворе существует в двух
равновесных конформациях (каждая из них присутствует в виде двух энантиомерных форм):
в более предпочтительной форме пропеллера, похожей на гребной винт корабля, и
в менее распространенной «спиральной» форме, у которой одна группа С=О
ориентирована в противоположном направлении по сравнению с двумя другими.
Только хиральная «пропеллерная» форма ТОТ, наблюдаемая в кристаллическом
состоянии, имеет (-)-(Л/)-конформацию (или энантиомерную (+)-(/г)-конформацию
противоположной спиральности, обсуждение спиральности см. разд. 7.7.1),
показанную на рис. 5.29. Растворение хирального кристаллического образца и
измерение его оптического вращения как функции времени по мере рацемизации образца
(равновесие между (—)-(Л/)- и (+)-(/3)-конформациями) позволяют измерить
оптическое вращение чистого энантиомера. Экстраполяция графика к нулевому
времени дает большое значение оптического вращения: [ос]^ = 88.75 ± 0.16° в СНС13.
В результате кристаллизации обычной формы клатрата ТОТ образуются
дискретные полости симметрии С^ в хиральной пространственной группе Fhx2\. Это
означает, что при кристаллизации формы (-)-(А/) и (+)-(Р) спонтанно
разделяются. Хиральность кристаллической формы допускает некоторую дифференциацию
между хиральными гостями. Полость решетки ТОТ, содержащая молекулу гостя,
образована всего восемью молекулами ТОТ-хозяев, сгруппированными в пары,
связанные осями вращения Cj. Поверхность полости выстлана атомами водорода и
двумя карбонильными атомами кислорода. Карбонильные заместители молекул
ТОТ обычно не принимают участия в водородных связях между хозяином и гостем
(единственное исключение — 2-гидроксициклопентенон-1, который образует
сильную водородную связь с С=О-группой), а взаимодействия хозяин-гость являются
ван-дер-ваальсовыми. Объем полости и, следовательно, параметры элементарной
ячейки ТОТ-хозяина показывают примерно линейную корреляцию с
молекулярным объемом гостя. Такие клатраты очень термостабильны, несмотря на то, что
факторы занятости полости обычно находятся между 0.65 и 0.75, т.е. 25—35% поло-
Рис. 5.29. Кристаллическая структура
(—)-(Л/)-конформации ТОТ
5.2. Твердые клатраты органических хозяев 339
(а)
(б)
Рис. 5.30. Структуры кла- \
тратов (а) транс-диметил- \
оксирана и (б) 2-бромбутана
с три-о-тимотидом.
(Воспроизведено с разрешения
Atwood J.L., Davies J.E.D.,
MacNicol D.D. and Vogtle F.
Comprehensive supramolecu-
lar chemistry. Oxford:
Pergamon, 1996. Vol. 6)
стей (соотношение хозяин:полость составляет 2:1) пусты, что зависит от характера
гостя и условий кристаллизации. Это удивительно, так как термостабильность
обычно не связана с долей незанятых пустот решетки. Структуры таких клатратов
т/?я«с-диметилоксирана и 2-бромбутана показаны на рис. 5.30. ТОТ включает эти
хиральные гости с энантиомерным избытком 47 и 35% соответственно, что
указывает на значительную, но не подавляющую хиральную селективность при
связывании гостя.
Помимо полостных структур ТОТ также может образовывать канальные
клатраты (симметрии пространственных групп P6V P62 или РЗу) в присутствии длинных
линейных молекул-гостей типа Ме(СН2)иХ (X = ОН, Br, I; n = 4-^7, 16, 18).
Соотношения хозяин:гость определяются неточно и зависят от длины гостя, а структуры
несоразмерны, подобно клатратам мочевины (разд. 5.2.1). Каналы хозяина
возникают при объединении 12 молекул ТОТ с образованием повторяющейся единицы
канала. Две последовательности из шести молекул дают структуру двойной
спирали (рис. 5.31).
340
5. Связывание нейтральных молекул
Рис. 5.31. Стереоизображение вдоль кристаллографической оси с, показывающее центры
12 молекул ТОТ, составляющих канал хозяина. Оси а и b элементарной ячейки
представлены штриховыми линиями. (Воспроизведено с разрешения AtwoodJ.L., DaviesJ.E.D.,
MacNicol D.D. and Vogtle F. Comprehensive supramolecular chemistry. Oxford: Pergamon, 1996.
Vol. 6. P. 247)
5.2.4.2 Синтез
Ранняя работа по клатратам ТОТ при непосредственном получении
ТОТ E.22) из о-тимотовой кислоты E.26) была затруднена из-за относительно
низкого выхода хозяина вследствие образования линейного димера и других открыто-
цепных продуктов конденсации, возникающих при кислотно-каталитическом де-
карбоксилировании кислоты E.26). В более поздней работе хозяин E.22) был
получен с выходом 84% тримеризацией кислоты E.26) при 100 °С в чистом РОС13
(J. Gnaim et al., 1991). Хозяин E.22) выделен путем десольватации при пониженном
давлении и повышенной температуре.
СО2Н
он
E.26)
5-2-4-3 Производные три-0-тимотида и родственные хозяева
Каркас ТОТ предоставляет большое число возможностей для
структурного модифицирования, включая изменения R-групп и сложноэфирных связей
в структуре E.27). Однако небольшие изменения в структуре могут очень сильно
влиять на клатратобразующую способность хозяина. Даже простая замена метиль-
ного и изопропильного заместителей в ТОТ с образованием три-о-карвакротида
приводит к тому, что это соединение полностью теряет способность формировать
клатрат. Напротив, трис-3-B'-бутил)-6-метилсалицилат E.28) образует свыше 30
клатратных соединений, все полостного типа. Этот хозяин интересен тем, что он
имеет хиральные заместители в ароматических кольцах (отмеченные звездочкой),
5.2. Твердые клатраты органических хозяев
341
Me
Me Me
E.28)
О
E.29)
которые обеспечивают даже большие возможности для распознавания хиральных
гостей. Однако структурные исследования не позволили различать R- и 5-формы в
твердом состоянии. Замена атомов кислорода в кольце ТОТ на различные галогены
подавляет клатратобразование.
Замена эфирных атомов кислорода на серу (X = S) с образованием тритиосали-
цилатов приводит к соединениям, не образующим клатраты. Получен ряд трислак-
тамов (E.27), X = NMe или NCH2Ph), обычно путем многостадийных процедур, и
изучены их свойства. В отличие от самого ТОТ такие лактамы в растворе
принимают почти исключительно спиральную конформацию, при этом между спиральной
и пропеллерной формами происходит свободный обмен. Трислактамы включают
таких гостей, как толуол и этанол. Только одно производное три-о-тимотида (X =
= NMe, NMe и NCH2Ph; R4 = R8 = R,2 = Me), как и сам ТОТ, способно к
распознаванию в твердом состоянии, давая клатрат с толуолом, в котором спиральный
хозяин кристаллизуется с широкими каналами, занятыми разупорядоченными гостями.
Циклизация 1-гидрокси-2-нафтойной кислоты в сухом толуоле в присутствии
пентаоксида фосфора в качестве дегидратирующего агента дает с небольшим
выходом циклический тетрамер, называемый тетра-1-нафтоидом E.29). Этот хозяин
342 5. Связывание нейтральных молекул
образует ряд клатратов 1:2 с небольшими молекулами типа СНС13 и бензола, очень
неустойчивых к десольватации. Комплексы гостей — 2-броммасляной кислоты и
нафталина — значительно более устойчивы. Известен также комплекс с переносом
заряда (гость - тетрацианоэтилен, TCNE), в котором электронодефицитный TCNE
увеличивает свою электронную плотность за счет электроноизбыточных арильных
колец макроцикла E.29).
5.2.4*4 Применение
Три-о-тимотид интенсивно используют в твердофазных реакциях,
когда молекулы-гости, захваченные решеткой ТОТ, либо участвуют в фотолитических
реакциях, либо взаимодействуют с небольшими молекулами, такими, как О2, НС1 и
НВг. Эти молекулы могут проникать между молекулами ТОТ через твердую
матрицу по узким каналам, имеющим форму замочной скважины. Реакции проводят с
мелкодисперсным порошком ТОТ для повышения скорости реакции (за счет
увеличения площади поверхности и меньших размеров частиц, что ускоряет их
проникновение) в растворителе, в котором данный клатрат совсем не растворяется.
Преимущества таких твердофазных синтезов — высокая специфичность наряду с
регио- и энантиоселективностью. Эти реакции аналогичны реакциям включения,
проводимым на цеолитных подложках (см. разд. 5.1.2), но они отличаются тем, что
полость ТОТ хиральна и не обладает активными центрами, способными
участвовать в конкурентных реакциях. В качестве примера приведем фотоокисление про-
хирального (.2Г)-метоксибутена-2 E.30) синглетным кислородом, которое дает
рацемический аллильный гидропероксид E.31) без какой-либо энантиоселективности.
Однако соединение E.30) образует с ТОТ клеточный клатрат, спонтанно
разделяющийся на кристаллы (+)-(Р)- и (-)-(Л/)-типов. Облучение разделенных кристаллов
в присутствии О2 и фотосенсибилизатора приводит к реакции включения, причем
снова образуется продукт E.31) в твердом состоянии. Растворение кристаллов
этого продукта и определение оптического вращения образца после рацемизации
хозяина ТОТ дают удельное вращение, показанное на схеме 5.1. Это свидетельствует
(-)-(АО-ТОТ- E.30)
но
о
н ОМе
(♦H/VTOT- E.30) E.31)
Схема 5.1. Перенос хиральности с помощью реакций включения (по Gerdil R. et al.
1984)
5.2. Твердые клатраты органических хозяев 343
о переносе хиральности решетки хозяина ТОТ на продукт фотоокисления. При
этом важно дождаться полной рацемизации ТОТ в растворе, чтобы быть уверенным
в том, что измеренное оптическое вращение (а следовательно, и энантиомерный
избыток) не обусловлено самим хозяином ТОТ.
Помимо реакций включения ТОТ используется для разделения энантиомеров
при энантиоселективном комплексообразовании. Энантиомерные избытки (ее)
гостей внутри монокристалла клатрата ТОТ, растущего из раствора рацемического
гостя, находятся обычно в интервале 2-60% у клатратов полостного типа. Такие
избытки не пригодны для промышленных целей, требующих, по крайней мере, 80%-го
избытка после одной перекристаллизации. Тем не менее величины ее можно
улучшить «умножением хиральности». Если в раствор рацемата внести затравочные
кристаллы, содержащие некоторый избыток одного энантиомера, то растущие
кристаллы имеют величины ее более 90%.
5*2*5 Циклотривератрилен
Циклотривератрилен (CTV, E.32)) — не только весьма гибкий хозяин,
но и основной «строительный блок», имеющий форму чаши или мелкого блюдца)
для конструирования целого ряда хозяев, способных связывать нейтральные
молекулы и даже анионы как в растворе, так и в твердом состоянии (разд. 5.3.3 и 4.6).
«Полезность» CTV обусловлена тем, что предпочтительная конформация
центрального девятичленного кольца (производное циклононатриена) — это конформация
«корона», которая вынуждает все три арильных цикла располагаться в одном
направлении. Температурные исследования методом 'Н ЯМР показали, что кон-
формационная инверсия (через промежуточную конформацию «седло» с одной
(б)
Рис. 5.32. (а) Чашеобразная конформация CTV E.32) и (б) седловидная конформация
родственного ему E.35), возникающая из-за неблагоприятных стерических взаимодействий
между метильными заместителями
t344
5. Связывание нейтральных молекул
арильной группой «вверх» и двумя «вниз») в этом случае медленная на шкале
времен Я MR Конформационная жесткость структуры E.32) ясно проявляется в
неэквивалентности геминальных (связанных с одним и тем же атомом углерода)
протонов метиленового мостика в виде АВ-квартета. Низкая скорость инверсии
установлена также при измерении скорости рацемизации хирального производного
CTV-J9 E.33), которое дало энергетический барьер инверсии в 110.9 кДжмоль.
Это означает, что полупериод инверсии составляет ~1 мес при 20 °С, но менее 0.1 с
при 200 °С. Конформация «чаша», или «корона», обычна почти для всех
соединений, родственных CTV, за исключением соединения E.34), в котором метастабиль-
ная конформация «седло» обусловлена особенностями синтеза этой молекулы, и
2,5-диметилтиофенового производного E.35), в котором неблагоприятные стери-
ческие взаимодействия между соседними метальными группами приводят к кон-
формации «седло» с симметрией С^ (рис. 5.32).
R-0
О—R
R
CTV, R = СН3: E.32)
R = CD3: E.33)
Me
Me
ОМе
MeO
MeO
MeO
ОМе
ОМе
E.34)
E.32). C60
(Hr
Collet A. Cyclotriveratrilene // Inclusion compounds / Ed. J.L. Atwood, J.E.D. Davies
and D.D.MacNicol. London: Academic Press, 1984. Vol. 2. P. 97-125.
5.2. Твердые клатраты органических хозяев
L5
5.3.5*1 Синтез
Циклотривератрилен впервые получила Г. Робинсон (G. Robinson) в
1915 г. (схема 5.2) в развитие своей работы по синтезу производных антрацена.
Робинсон пришла к выводу, что ее продукт с эмпирической формулой С7Н10О2 был
2,3,6,7-тетраметокси-9,10-дигидроантраценом E.38) - димером промежуточного
катиона вератрила E.37). В 1950-е годы CTV также ошибочно был принят за гекса-
мер, и только в 1963 г. с помощью современных методов была установлена его три-
мерная структура E.32).
МеО ОМе
Me
СН2
+0^4
Me
-Н*
МеО
E.36)
E.37)
МеО ОМе
CTV E.32)
ОМе ОМе
ОМе
MeO
ОМе
ОМе
ОМе ОМе E.39)
Схема 5.2. Синтез CTV E.32) по Робинсон. Сначала была предложена формула димера
E.38), а позднее — гексамера E.39)
Циклотривератрилен образуется с более чем 70%-м выходом при конденсации
катиона вератрила E.37), получаемого как из вератрилового спирта E.36), так и
непосредственно из вератрола A,2-диметоксибензол) и формальдегида в
подкисленной водной среде. В органических растворителях типа уксусной кислоты или смеси
бензол/ВР3/эфир образуются значительные количества аналогичного тетрамера —
циклотетравератрилена (CTTV). CTTV не обладает чашеобразной конформацией,
хотя способен образовывать ограниченное число решеточных соединений включе-
346 5. Связывание нейтральных молекул
ния, использующих водородные связи метоксизаместителей (H.Zhang et al., 1995).
В реакциях циклизации, показанных на схеме 5.2, возможна замена на другие
функциональные группы метоксигрупп исходного соединения E.36) в положениях
3 и 4. Существенно, однако, что положение 4 должно быть занято электронодонор-
ной группой, чтобы стабилизировать положительный заряд промежуточного
катиона вератрила E.37) Легкое образование циклического тримера E.32) (в
противоположность высшим олигомерам и полимерам) — интересная особенность этой
реакции, причем не ясно, результат ли это термодинамического равновесия
(возможно, вследствие нерастворимости продукта) или кинетического темплатного
эффекта.
5.^.5-2 Соединения включения
Относительно давно было известно, что CTV образует твердые
соединения включения с некоторыми небольшими нейтральными молекулами, такими,
как бензол, толуол, вода, ацетон и CS2. Согласно корреляции между ИК-спектро-
скопическими данными и кристаллографическими параметрами элементарной
ячейки, клатраты CTV разделяются на две четкие твердые фазы, обозначаемые как
а- и C-фазы, с небольшими различиями в свойствах. Данные РСА показали, что, по
крайней мере, в а-фазе молекулы CTV расположены одна на другой, как в стопке
блюдец, давая характерный размер элементарной ячейки вдоль
кристаллографической оси b (9.6-9.7 Адляа-фазы; 8.1—8.3 А-для (З-фазы). Именно это характерное
«самовключение» молекул CTV делает, казалось бы, привлекательную блюдцеоб-
разную форму полости неприемлемой для включения гостя, и все соединения
включения CTV, выделенные вплоть до 1994 г., содержат частицы гостей только в
каналах между стопками молекул CTV. Определение структуры двух комплексов
C-фазы (J. Steed et al., 1996) позволило окончательно выяснить причину различий
между соединениями этих двух типов. Найдено, что различия в их
спектроскопических параметрах возникают вследствие поворота одной из метоксигрупп в ответ на
водородное связывание гостя. На языке современного конструирования
кристаллов (гл. 6) CTV — акцептор водородной связи с небольшой донорной способностью.
В присутствии гостей - доноров водородной связи (даже таких слабых кислот, как
бензол) - протоны гостя образуют водородные связи с атомами кислорода
метоксигрупп. Однако если гость не способен выступать в качестве такой кислоты, то
молекула CTV искажается, чтобы предоставить протоны метоксигруппы (слабые
доноры водородной связи) для взаимодействия с гостем-акцептором.
В течение года с момента завершения классификации этих двух фаз Ибрагимов
(Ibragimov et al.) с сотрудниками охарактеризовал третью фазу, названную у-фазой.
Единственный, до сих пор известный пример - клатрат (CTVLMe2CO,
содержащий молекулу ацетона, расположенную между двумя чашами CTV «лицом к лицу»
для создания полости. Как и в клатратах (З-фазы, взаимодействие хозяин—гость
включает водородные связи ОСН3-ОСМе2 (рис. 5.33).
Согласно структуре твердого комплекса (CTVLMe2CO, в ней осуществляется
внутриполостное включение; этот комплекс представляет собой нековалентный
аналог капсулярных криптофанов на основе CTV (разд. 5.3.3). Комплекс является
5.2. Твердые клатраты органических хозяев
347
Рис. 5.33. Стереоизображение
кристаллической структуры
клатратау-фазы (СТУLаиетон
(гость — ацетон — разупорядочен)
единственным из известных соединений CTV, содержащим растворитель. Тем не
менее для более необычных сферических гостей, таких, как бакминстерфуллерен
(С60) и 1,2-дикарбадодекаборанA2) E.40), внутриполостные соединения известны
с 1994 г. В случае С60 имеет место как стерическая, так и электронная комплемен-
тарность между хозяином и гостем. Радиус кривизны широкой полости хозяина
CTV близок к радиусу большой сферы гостя фуллерена. К тому же, арильные
кольца CTV очень богаты электронами вследствие электронодонорных свойств меток-
сизаместителей, что дополняет электронодефицитную природу соединения С60 и
способствует образованию в растворе комплексов с переносом заряда. За
взаимодействием в растворе можно проследить с помощью спектроскопии в УФ-видимой
области. Факт внутриполостного включения в твердом состоянии был подтвержден
и для комплекса карборана, и для комплекса фуллерена методом РСА. В случае
гостя С60 его шестичленное кольцо располагается прямо над осью вращения С3
хозяина, приводя к хорошему (по симметрии) соответствию. В случае соединения E.40)
его комплекс с CTV стабилизирован взаимодействиями С-Н •••п с расстояниями
2.18 и 2.56 А между кислотными С—Н-протонами карборана и кольцами CTV.
Важно, что значительно более электроноизбыточные атомы водорода связи В—Н
отталкиваются арильными кольцами CTV (самое короткое расстояние В-Н--я
составляет 2.77 А, рис. 5.34).
В вершинах - ВН:
E.40)
Rn = Me, C1, 0-С12, m-Me2, 1,3,5-Me3:
E.41)
Помимо этих комплементарных соединений электроноизбыточная природа ве-
ратрильных колец CTV способствует образованию ряда необычных комплексов
включения E.41) с катионами циклопентадиениларешкелеза(П) (К. Holman et al.,
348
Ь. Связывание нейтральных молекул
Рис. 5.34. Кристаллическая структура
полостного комплекса включения
1,2-дикарбадодекаборанаA2) с CTV
1997). В общем, меньшее кольцо С5Н5 располагается внутри полости CTV, а его
кислотные протоны образуют водородные связи С-Н---Я с электроноизбыточными
арильными фрагментами CTV. При расположении «лицом к лицу» кольца С5Н5 и
одного из арильных остатков CTV образуется комплекс с переносом заряда. Это
приводит к формированию нецентросимметричных двухмерных полярных
каркасов в твердом состоянии.
5*2.6 Тетрафенилен
Тетрафенилен E.42) в основном состоянии принимает седловидную
конформацию D2d, и именно такая необычная форма обуславливает его клатратоб-
разующую способность. Этот хозяин дает большой ряд клатратов общей формулы
E.42JG, где гость (G) может быть нейтральной молекулой размером от дихлорме-
тана до циклогексана. Такие клатраты имеют тетрагональную пространственную
группу РА2/п, а молекулы гостей расположены в пространстве между молекулами
тетрафенилена, которое в предельном случае (включение СС14) представляет собой
сферу диаметром -7.3 А. Включение гостя соответствует принципу эффективной
упаковки, а взаимодействия хозяин-гость относятся к типу ван-дер-ваальсовых.
Конформация тетрафенилена отчасти гибкая и потому легко приспосабливается к
предпочтительной ориентации частиц-гостей. Кристаллическая структура клатрата
четыреххлористого углерода показана на рис. 5.35.
E.42)
1вероые клатраты органических хозяев
349
Рис. 5.35. Кристаллическая структура клатрата СС14 с тетрафениленом
Дополнение 5.2. Термогравиметрический анализ и дифференциальная
сканирующая калориметрия при изучении соединений включения
Тот факт, что молекулы-гости могут включаться в полости хозяев или захватываться
их решетками в твердом состоянии, автоматически предполагает, что это включение
должно быть обеспечено энергией. Для растворов о термодинамике включения
можно судить, измеряя константы связывания (Дополнение 1.1). В твердом состоянии
обычно не существует равновесия ассоциация—диссоциация; поэтому для оценки и
устойчивости, и стехиометрии соединений включения надо использовать другие
методы. Наиболее распространены термофавиметрический анализ (TGA) и
дифференциальная сканирующая калориметрия (DSC). Оба метода довольно быстры (от минут
до нескольких часов), требуют лишь незначительные количества образца
исследуемого вещества (-10 мг) и их можно выполнять на лабораторном оборудовании
стоимостью 20000—30000 долларов (это относительно недорого по сравнению со стоимостью
оборудования для ЯМР и РСА).
350 5. Связывание нейтральных молекул
Термогравиметрический анализ
В TGA-эксперименте масса твердого образца записывается как функция постоянно
возрастающей температуры. В ходе эксперимента масса образца выражается в
процентах от начальной массы, причем на термограммах присутствует одно плато или
несколько, разделенных наклонными участками, соответствующими потере молекул-
гостей при различных температурах. Если известны молекулярные массы хозяина и
гостя, то можно получить соотношение хозяинтость при сравнении расчетной и
наблюдаемой потери массы для различных стехиометрических соотношений.
Температура, при которой теряется гость, некоторым образом указывает на стабильность
комплекса хозяин-гость, хотя точная температура, при которой происходит потеря гостя,
зависит от скорости нагревания. Кривая TGA также часто содержит наклонные
участки, возникающие из-за разложения хозяина при повышенных температурах. TGA-
эксперимент проводят в струе газа (обычно N2) для удаления продуктов деструкции,
которые можно направить в другой анализатор, например, такой, как
ЯК-спектрометр (TGA-IR), чтобы дополнить полученные данные характеристикой
выделяющихся соединений.
ЗаписьTGA при дегидратации координационного 7п(И)-полимера [Zn(H20J(tph)]oo
(H7tph — терефталевая кислота и-С6Н4(СО2НJ) показана на рис. 5.36. Потеря
координационной воды происходит в два четких этапа при 168 и 192 °С, соответствуя
потере массы 6.8% в каждом случае. Предполагается, что потеря первой молекулы воды
соответствует превращению линейного зигзагообразного полимера на основе
искаженного тетраэдра Zn2+ в полимостиковую структуру, включающую тригонально-би-
пирамидальные центры цинка.
Дифференциальная сканирующая калориметрия
DSC-метод включает измерение разницы в энергетических потребностях образца и
эталона при одинаковой температуре в процессе повышения или понижения
температуры со скоростью в несколько К/мин. Это классический калориметрический
метод, имеющий дело с разностями значений энергии. Преимуществом DSC в
сравнении с TGA является то, что он чувствителен к фазовым изменениям, которые не
приводят к изменению массы (например, плавление), а интегрирование площади
пика дает количественную меру изменения энтальпии, АЯ, в изучаемом процессе. Так,
если образец претерпевает фазовый переход с поглощением теплоты (например,
плавление), то к камере образца требуется подводить больше энергии по сравнению с
энергией, необходимой для эталона (имеющего близкую массу, но не
претерпевающего фазового перехода). В результате на кривой DSC появляется положительный пик.
Таким же образом экзотермические процессы дают отрицательные пики, а плоский
участок означает, что различие между поведением образца и эталона отсутствует.
Методы TGA и DSC часто используют совместно, чтобы различать перекрывающиеся
термические события типа фазовых переходов и разложения.
Кривая DSC для [Zn(H20J(tph)]oo, показанная на рис. 5.37, свидетельствует о том,
что как потеря воды образцом, так и его разложение, происходящее при более
высокой температуре, представляют собой эндотермические процессы.
Тесно связан с методом DSC гораздо более старый метод - дифференциальный
термический анализ (DTA). DTA работает на более простом принципе измерения
(с помощью термопары) разности температур образца и эталона при подводе к ним
5.2. Твердые клатраты органических хозяев
351
100.00
I 80.00
60.00
40.00 -
_i i_
0.00
100.00
200.00 300.00
Температура, °С
400.00 500.00
(б)
168 °С
-н,о
Рис. 5.36. (а) Кривая TGA при дегидратации [Zn(H2OJ(tph)lTO. (б)
Кристаллические структуры ди- и моногидратов (по Guilera G. and Steed J. W., 1999)
одинаковой тепловой энергии. Кривая DTA, следовательно, является отражением
температурного эффекта, который только полуколичественно соотносится с ДЯ. Обе
кривые DTA-TGA для клатрата Вернера (см. разд. 5.1.4) Ni(NCSJD-PhpyL-4C6H6
352
5. Связывание нейтральных молекул
D-Phpy — 4-фенилпиридин) представлены на рис. 5.38. Кривая DTA показывает, что !
все три наблюдаемых термических события эндотермичны. Первое связано с потерей ;
; гостя-бензола, а второе и третье относятся к потере координированных лигандов \
\ 4-Phpy. Отметим высокую температуру (~350 °С), требующуюся для удаления клатрат- ;
ного бензола, что указывает на термическую стабильность семейства клатратов I
Вернера. !
1 Е, мВт Температура, °С 1
| 0.00
1 -10.00
-
i . . , . i , . , , i . . . , i
500.00 1
400.00 1
300.00 |
200.00 1
100.00 |
о.оо !
| 0.00 10.00 20.00 30.00 |
; Время, мин |
1 Рис. 5.37. Кривая ОБСдля [2п(Н2ОJAрп)]те
5.3. Внутриполостные комплексы нейтральных молекул
353
400 500
Температура, К
Рис. 5.38. Кривые DTA — ТвАдля Ni(NCSJD-PhpyL-4C6H6 показывают
эндотермические потери вначале бензола, а затем 4-фенилпиридильных лигандов,
происходящие в две четкие стадии. (Воспроизведено с разрешения Lavelle L. et al,.
1989)
Внутриполостные комплексы
нейтральных молекул: связывание
в растворе и в твердом состоянии
В разд. 5.2 мы имели дело лишь с кристаллическими клатратными
образованиями. Эти соединения существуют только в твердом состоянии, поскольку
полости, содержащие частицы-гости, относятся к межмолекулярному типу. Иными
словами, такая полость — непосредственный результат неэффективного
заполнения пространства, когда молекулы-хозяева упаковываются вместе в
кристаллической форме (см. рис. 1.3). Для комплексообразования нейтрального гостя как в
растворе, так и в твердом состоянии необходимо, чтобы хозяин обладал внутренней
полостью или создавал ее путем организации либо самосборки (гл. 7) нескольких
компонентов в растворе. В этом разделе мы рассмотрим разнообразные
индивидуальные молекулы-хозяева, обладающие внутренней кривизной, позволяющей им
включать частицы гостя как в капсулы (см. рис. 1.3). Так же как в случае
комплексообразования катионов и анионов, в случае нейтральных гостей можно измерить
константы связывания в растворе (см. Дополнение 1.1); и эта информация будет
полезной для выявления связи структура—функция, знание которой необходимо
при конструировании хозяев.
23 - СТИДДж.В
354 5. Связывание нейтральных молекул
5*3* 1 Внутренняя кривизна хозяев:
связывание гостей кавитандами
5-3» 1-1 «Строительные блоки»
Индивидуальные молекулы-хозяева, обладающие внутренней
полостью как в твердом состоянии, так и в растворе, называют кавитандами. Кавитанд
определяют как молекулярный контейнер с вынужденно вогнутой поверхностью.
Обычно его молекулярная полость открыта с одного конца. Включение
частиц-гостей в кавитанд приводит к образованию кавитата (или, что то же, кавиплекса).
Молекулы или фрагменты хозяев, обладающие внутренней кривизной (т.е. структурно
изогнутые, или искривленные), можно использовать для связывания
молекул-гостей как в растворах, так и в твердом состоянии, поскольку растворение хозяина не
приводит к исчезновению его полости. Внутренне изогнутые молекулярные блоки
для «строительства» хозяев, синтез которых сравнительно сложен, достаточно
необычны. Хозяева-кавитанды и большое число родственных им соединений можно
разделить на отдельные семейства. Такие семейства можно группировать согласно
типам «строительных блоков», использованных для придания молекуле хозяина
кривизны и, следовательно, для получения связывающей полости. Ряд примеров
синтетически доступных кавитандов и их изогнутых предшественников,
применяемых для захвата нейтральных частиц-гостей, показан на рис. 5.39.
Помимо вынужденно искривленных соединений существует обширный круг
хозяев, которые в широком смысле можно назвать циклофанами. Молекулы
последних содержат мостиковые арильные кольца, используемые в качестве стенок
контейнера, а также разнообразные гибкие или угловые спейсеры, позволяющие
хозяину замыкаться (т.е. образовывать цикл).
В основном большинство кавитандов связывает строго нейтральные
неполярные органические молекулы в неполярных растворителях относительно слабо из-за
отсутствия значительного выигрыша в энтальпии от сильных взаимодействий
хозяин—гость. Во многих комплексах наблюдается связывание полярных или
заряженных органических гостей, причем особенно распространено связывание катионов
алкиламмония. Такое связывание обычно осуществляется за счет диполь-диполь-
ных взаимодействий или водородных связей, а часто и с участием заряда
(ион-диполь). Гидрофобные участки гостя связываются гидрофобными участками хозяина.
В воде связывание органического гостя сильно возрастает из-за гидрофобных
эффектов. В следующих разделах кратко рассмотрена химия внутриполостного
включения для некоторых важных хозяев.
5-3л-2 Каликсарены и резорцинарены
Синтез и номенклатура каликсаренов* типа C.97) уже были кратко
рассмотрены (см. разд. 3.13) в контексте их широкого использования в качестве
хозяев, обычно связывающих катионы металлов через свои фенольные атомы кисло-
Введенис в химию каликсаренов и история их развития представлены в Дополнении 3.5.
5.3. Внутриполостные комплексы нейтральных молекул
fl55
НО
,он
он но. он
Каликс[4]резорцинарен
E.43)
Гликолурил
E.44)
R = СН,, CH2CH2Ph и т.д.
Каликс[4]арен R = Н, СН3, /-Bu
C.93)
ОН
ОН
а-Циклодекстрин
E.47)
Предшественник конкина
E.45)
Основание Трёгера
E.46)
Трифенилметан
R = Н, Me
E.48)
Дифенилметан
E.49)
Рис. 5.39. Ряд изогнутых «строительных блоков» и их структурные типы
356
5. Связывание нейтральных молекул
НС1 C7%), 1ГСН0
80 °С, 16 ч, 75%
R1 = Н, ОН, А1к
R2 = Alk,Ar
НО
ОН
Схема 5.3. Типичный синтез [4]резорцинаренов
рода. Родственные им резорцинарены (или каликсрезорцинарены), такие, как
E.43), получают подобным образом при конденсации резорцина C-гидроксифе-
нол) с альдегидами, как показано на схеме 5.3. В этом случае реакцию проводят в
условиях кислотного катализа, причем с самим формальдегидом не работают из-за
реакций полимеризации, протекающих в положение 2. Высокой эффективностью
обладают многие другие альдегиды, но обычно используют ацетальдегид (дающий
метальные «ножки» чаше резорцинарена) или 2-фенилэтаналь (обеспечивающий
повышенную растворимость продукта в органических растворителях). И ка-
ликс[4]арены, и [4]резорцинарены в их наиболее устойчивых формах имеют
чашеобразную конформацию. В случае [4]резорцинаренов она шире и мельче из-за
присутствия в верхнем ободе структуры гидроксизаместителей, которые стабилизируют
эту чашу внутримолекулярными водородными связями. Как каликсарены, так и
резорцинарены, несущие небольшие заместители, обладают относительной конфор-
мационной гибкостью и могут принимать частично конические, 1,2- и 1,3-череду-
ющиеся (альтернантные) конформации (рис. 3.61). Действительно, резорцинарены
значительно более конформационно гибки: свободная энергия активации AG* кон-
формационного превращения составляет 18.4 кДж-моль для [4]резорцинаренов с
R1 = «-гексил и R2 = Н. Для каликс[4]аренов в CDC13 величины этого параметра
лежат в интервале 63—67 кДжмоль"", причем у этих соединений
внутримолекулярные водородные связи в нижнем ободе цикла значительно сильнее аналогичных
взаимодействий в верхнем ободе соединений типа E.43), рис. 5.40. Однако в обоих
случаях только коническая конформация способна прочно связывать органические
молекулы-гости. Типичная твердофазная структура комплекса конического
я-т/?ети-бутилкаликс[4]арена C.97) с толуолом, показана на рис. 1.19 (заметьте, что
именно метильная группа толуола входит в полость каликсарена).
Внутриполостное включение разнообразных ароматических молекул-гостей
с образованием комплексов состава 1:1 наблюдали для многочисленных ка-
ликс[я]аренов. В случае 1:1-комплекса производного я-изопропилкаликс[4]арена
с я-ксилолом в твердом состоянии нагревание вызывает потерю половины
молекул-гостей и образование полостной структуры 2:1, в которой каждый из двух ме-
тильных заместителей гостя дает комплекс с соседними каликсаренами. Подобная
5.3. Внутриполостные комплексы нейтральных молекул
H-Q
0-Н
Рис. 5.40. Внутримолекулярные водородные связи нижнего и верхнего ободов
в каликс[4]аренах и [4]резорцинаренах, стабилизирующие конические конформации
организация наблюдается и у комплекса калике[4]арена C.97) с анизолом, хотя
молекулы-гости анизола сильно разупорядочены. Аналогичное капсулирующее
поведение характерно также для Мо(У1)-производного тетрааниона соединения C.97)
[МоО(я-Аи/?ет-бутилкаликс[4]арен-4Н)], которое образует удивительный
твердофазный комплекс со свободным C.97), молекулой воды и нитробензолом, рис. 5.41
(F. Corazza et al., 1990). В каждом случае комплексы обычно стабилизированы
взаимодействиями CH3-t между алкильными заместителями верхнего обода хозяина
и арильным кольцом гостя на расстоянии ~3 А.
Многочисленные комплексы включения в твердом состоянии с
ароматическими и алифатическими гостями (галогеналканы, ацетон, DMF, DMSO и т.д.),
образованные как каликс[4]аренами, так и [4]резорцинаренами, обычно
стабилизированы слабыми взаимодействиями типа С—Н--71. Каликс[5]арены также обладают
четко определенной полостью и образуют аналогичные комплексы, в то время как
высшие каликсарены типа каликс[6]арена и каликс[8]арена (все каликсарены,
вплоть до калике[16]арена, получены и выделены хроматографически группой
Рис. 5.41. Капсула комплекса [МоО(л-/ярет-бутилкаликс[4]арен-4Н)]-
• [л-т/?ет-бутилкаликс[4]арен] H2ONO2Ph (по Corazza F. et al., 1990)
но он он он
358
5. Связывание нейтральных молекул
К.Гутше (С. Gutsche)) дают намного менее определенные полости и не склонны к
образованию таких четких структур типа «гнездо» или «капсула». Единственное
исключение из этого — встраивание фуллеренов в полости каликсаренов, так же как и
в полости CTV (разд. 5.4.1). Тем не менее, несмотря на такие интересные
результаты, имеется мало свидетельств связывания каликсаренов с большинством
нейтральных молекул в неводных растворах. Полости каликсаренов относительно малы
и слишком конформационно гибки, чтобы можно было ожидать от них
значительную сольватофобную защиту, а отсутствие при этом сильных взаимодействий
хозяин—гость означает малую энтальпийную стабилизацию таких комплексов.
Исключение составляет комплексообразование аминов с алкилкаликс[4]аренами, дающее
константы связывания в CD3CN ~104 М. Считается, что оно протекает по двух-
стадийному механизму, включая первоначальное протонирование амина одной из
кислотных фенольных групп каликсарена с последующим связыванием катиона
гостя образовавшимся анионом каликсарена. Таким образом, этот комплекс
значительно стабилизирован ионными взаимодействиями. Даже нейтральные каликса-
рены могут давать устойчивые комплексы с рядом органических аммониевых
катионов в растворе. Например, катион N-метилпиридиния связывается
метоксипроизводными ряда каликс[я]аренов (п = 4, 6 и 8) в смеси CDC13—CD3CN
A0:1) с энергиями взаимодействия 4.3-5.7 кДжмоль. Вероятно, что такие
катионы аммония взаимодействуют с нижним полярным ободом каликсарена, а не с его
гидрофобной полостью.
В водных растворах водорастворимые каликсарены имеют возможность гораздо
сильнее связывать органические гости, чем в липофильных средах, из-за
гидрофобного эффекта (см. разд. 1.7.9). К сожалению, я-алкилкаликсарены нерастворимы в
воде и для использования в водных растворах их нужно химически
модифицировать. Проще всего это достигается сульфированием заместителей верхнего обода с
образованием ряда я-сульфокаликс[4]аренов E.50а-д), обычно в виде натриевых
солей, очень хорошо растворимых в воде.
SChNa
п = 4: E.50а)
п = 5: E.506)
п = 6: E.50в)
п = 7: E.50г)
п = 8: E.50д)
E.51)
Соединение E.50а) в водном растворе оказалось чрезвычайно кислым, что
согласуется с протонированием гостей-аминов соединением C.97) и величиной рКа
~3.3 для первого фенокси-протона в сравнении с рКа > 11 для трех остальных фе-
нокси-протонов. Обычно считается, что это следствие стабилизации фенокси-ани-
она — продукта депротонирования — внутримолекулярными водородными связями.
Соединение E.50а) образует в водном растворе прочные комплексы с
органическими катионами, например, константа связывания с катионом адамантилтриметил-
5.3. Внутриполостные комплексы нейтральных молекул
(«) (б)
359
15.0 А
Рис. 5.42. Сравнение кристаллической структуры (а) л-сульфокаликс[4]арена со
структурой (б) природного глинистого минерала натрийвермикулита. (Воспроизведено с разрешет
ния Atwood J.L. Cation complexation by calixarenes // Cation binding by macrocycles / Ed.
J.Inoue and G.Gokel. New York: Dekker, 1991)
аммония E.51) составляет 21 000 M. Это соединение также способно связывать
нейтральные молекулы типа толуола, хотя константа связывания при этом
составляет только 7.0 М. В твердом состоянии все комплексы соединений E.50а-в)
образуют двухслойные структуры, в которых гидрофобный участок каликсарена
чередуется с гидрофильными слоями, включающими молекулы воды, ионы Na+ и
каликсареновые сульфогруппы. Такие структуры сильно напоминают природные
глинистые минералы, рис. 5.42 (см. также разд. 5.1.3). Еще более примечательно,
что в твердом состоянии каликсарен E.50а) прочно связывает молекулу воды,
которая внедряется в его полость. Эта структура - первое свидетельство образования
водородных связей О-И-п между молекулами воды и арильными кольцами; такое
явление может объяснить стабильность третичной структуры белка (J. Atwood et al.,
1991). Внутриполостная молекула воды предоставляет оба своих протона для
образования водородных связей с противоположными поверхностями полости
каликсарена, при этом О-Н-я-расстояния равны 2.38 и 2.50 А (рис. 5.43). Расчеты ab initio
взаимодействия воды с бензолом дают энергию взаимодействия 14.2 кДж-мольг1.
W10
Рис. 5.43. Включение воды (W)
водорастворимым л-сульфокаликс[4)ареном E.50а).
(Воспроизведено с разрешения
Atwood J.L. et al., 1991)
360
5. Связывание нейтральных молекул
Поведение каликсаренов с другими гидрофильными заместителями (например,
с аминометилом и карбоксиэтилом) типа соединений E.52) и E.53) исследовали
соответственно в разбавленных кислоте или основании. При изучении комплексооб-
разования этих каликсаренов с ароматическими соединениями типа дурола и
нафталина определили, что константа связывания лежит в интервале 6-102—1.5-104 М.
При изучении комплексообразования производного E.54), имеющего большую
полость, с рядом ароматических гостей - от дурола до хризена - нашли линейную
корреляцию констант связывания с числом ароматических колец в молекуле гостя,
причем константа связывания с хризеном, молекула которого состоит из четырех
арильных колец, равна 7.0-104 М.
п = 4, 6, 8:
E.52)
и =4, 6,8:
E.53)
*= H: E.55а)
Me : E.556)
ОН: E.55в)
Были также получены сульфированные 14]резорцинарены и изучена их
способность связывать сахара и циклогексанолы в водных растворах. Эти исследования
были начаты после того, как обнаружили, что благодаря однослойным ансамблям
алкилпроизводных [4]резорцинаренов можно определять рибозу при
концентрациях до 4-10~5 М на поверхностях раздела водный раствор - электрод. Константы
связывания для хозяев E.55а-в) представлены в табл. 5.5. Первичным является
взаимодействие органических фрагментов гостя с неполярным участком хозяина в
отсутствие водородных связей между хозяином и гостем. Это согласуется с
данными об отсутствии связывания для липофильных резорцинареновых хозяев в
органических средах, когда основная движущая сила комплексообразования — это водо-
5.3. Внутриполостные комплексы нейтральных молекул
361
Таблица 5.5. Константы связывания (К, М) сульфированными
резорцинаренами (хозяева) различных спиртов (гости)
Гость
формула
номер
Хозяин
E.55а)
E.556)
E.55b)
/-BuOH
E.56)
E.57)
E.58)
E.59)
4.2
30
29
16
19
200
180
125
24
82
160
64
E.60)
E.61)
E.62)
14
1.8
80
6.0
80
8.4
родные связи. Оба производных, E.556) и E.55в), связывают гостей более прочно,
чем соединение E.55а), несмотря на различную полярность их заместителей и
степень, с которой они подают электроны в арильные кольца. Кажется, что такие
относительно гидрофобные гости, как E.57), предпочитают хозяина E.556) хозяину
E.55в), в то время как обратное справедливо для гидрофильного гостя E.61).
Сильногидрофильный гость E.62) не связывается вовсе. Итак, рассмотренные
результаты свидетельствуют о значительных взаимодействиях С-Н-л между гостем и эле-
ктроноизбыточным арильным кольцом хозяина (оба заместителя — Me и ОН —
являются электронодонорными).
В то время как исходные каликсарены и резорцинарены обычно не обладают
большим сродством к органическим молекулам в растворе, их связывающую
способность можно существенно увеличить, усовершенствовав их полости. Синтез
более глубоких полостей имеет двойственный эффект: такая полость увеличивает
степень экранирования гостя от растворителя, но это часто приводит к увеличению
жесткости хозяина, что увеличивает степень его предорганизации и препятствует
схлопыванию полости.
362
5. Связывание нейтральных молекул
Крам и сотрудники (D. Cram et al., 1985a) получили ряд полимостиковых [4]ре-
зорцинаренов типа E.63) и E.64), которые проявляют низкую конформационную
гибкость в растворе. Заместители X - перспективные функциональные группы,
пригодные даже для дальнейшей функционализации этих соединений (разд. 5.4).
Наличие жесткой молекулярной полости в соединениях E.63) и E.64) — причина
того, что эти соединения неизменно кристаллизуются с внутриполостным гостем.
Группа Крама методом РСА изучила комплексы этих веществ с такими гостями, как
SO2, CS2, Me3C=CH, MeCN и СН2С12.
О
О
О
О
о
о
E.63)
(а): X = Н, R = СН2
(б): X = Br, R = СН2
(в): X = CH2SH, R = СН2
E.64)
(г): X = СН2С1, R = СН2
(д): X = Н, R = SiMe2
Кавитанд E.64) обладает полостью чашеобразной формы, достаточно большой,
чтобы включить Me2NCHO в твердом состоянии. Наиболее эффективный хозяин в
растворе - диметилсилильное производное этого кавитанда E.63д), имеющее
длинную узкую полость, подходящую для включения линейных гостей, подобных
указанным выше. Соединение E.63д) легко получают из каликс[4]резорцинарена
E.43), обрабатывая его SiMe2Cl2. Кристаллическая структура комплекса
E.63fl)CS2, полученная методом РСА, показана на рис. 5.44, где ясно видна четкая
подгонка этого линейного гостя, втиснутого в полость хозяина через узкое
отверстие, образованное метальными заместителями хозяина. В неполярных
растворителях (CDC13, C6D6) свободная энергия комплексообразования (—ДGО) составляет
~8 кДж-моль.
Каликсарены также можно превратить в растворимые хозяева, модифицируя их
верхний обод. Легкая доступность каркаса каликсаренов позволила синтезировать
ряд хозяев, в которых каликсарен используют в качестве спейсера и жесткого трех-
5.3. Внутриполостные комплексы нейтральных молекул
363
Рис. 5.44. Кристаллическая структура комплекса
E.63fl)CS2. (Воспроизведено с разрешения
Cram D. etal., 1988a)
мерного «якоря», а также молекулярной платформы для введения других
связывающих групп. Это аналогично химии каликскраунов, описанной в разд. 3.13. Один
из самых наглядных примеров такого подхода — введение 2,4-диаминотриазиновых
групп в два противоположных арильных фрагмента каликсарена с образованием
рецептора E.65). Введение в нижний обод каликсаренов объемистых алкилэфир-
ных цепей вынуждает два триазиновых остатка расположиться параллельно друг к
другу. В такой предорганизованной конформации они выступают в качестве
эффективного рецептора барбитуратов (J.-D. van Loon et al., 1992). Примечательно,
что родственные хиральные производные соединения E.65) образуют как в
растворе, так и в твердом состоянии хиральные спиральные молекулярные капсулы,
включающие три бифункциональные молекулы каликсарена и шесть гостей —
молекул производных 5,5'-диэтилбарбитуровой кислоты (рис. 5.45). Как видно,
наблюдается спонтанное нековалентное хиральное распознавание (L. Prins et al.,
1999).
A...-H T. Н--.Л,
E.65)-барбитуровая кислота:
R = CH2CH2OEt
364
5. Связывание нейтральных молекул
Рис. 5.45. Структура
хирального девятимолеку-
лярного ансамбля,
образованного тремя молекулами
типа E.65) и шестью
молекулами производных ди-
этилбарбитуровой
кислоты. (Воспроизведено
с разрешения Prins L.J.
etal., 1999)
Bohmer V. Calixarenes: Macrocycles with (almost) unlimited possibilities // Angew.
Chem., Int. Ed. Engl. 1995. Vol. 34. P. 713-745.
5.3.1.3 Динамика обмена гостей в кавитатах
Для изученных до сих пор комплексов катионов (гл. 3), анионов и
нейтральных частиц общим является то, что в них гость (ион или молекула) полностью
заключен, по существу, в закрытую полость хозяина. Такие комплексы имеют
большие константы связывания и для них характерен медленный обмен гостя с гостями
внешней среды. Это было четко установлено при сравнении связывания катионов
щелочных металлов двухмерными краун-эфирами и трехмерными сферандами. В
разд. 5.3.3 и 5.3.4 мы увидим, что эти эффекты проявляются и при связывании
нейтральных молекул каликсаренами и резорцинаренами, а также сферическими полу-
карцерандами и, в особенности, карцерандами, в которых обмен гостей
практически отсутствует. В случае нейтральных молекул причиной прочного связывания
внутри трехмерных хозяев, несомненно, является стерическое «заточение» гостя, а
не энтальпийно благоприятное притяжение. Рассмотрим возможный механизм
входа гостя в «открытые» контейнерные молекулы (кавитанды) типа каликс[4]аре-
на C.97) и его выхода из них. Гость, имеющий размер и форму, соответствующие
таковым чашеобразной полости каликсарена, в процессе обмена должен входить и
выходить через открытую верхнюю часть молекулы хозяина. Поскольку данный
гость соответствует полости хозяина, совершенно очевидно, что в ней нет места для
одновременного входа и выхода еще одного гостя. По другому варианту механизм
обмена гостя должен быть раздельным: первый гость должен оставить полость,
прежде чем в нее сможет войти второй. Это подразумевает, что существует какая-то
5.3. Внутри полостные комплексы нейтральных молекул
365
промежуточная стадия с «пустым» хозяином, т.е. когда в хозяине имеется вакуум.
Энергетически это крайне неблагоприятная ситуация («природа не терпит
пустоты»), и, следовательно, энергия активации, требуемая для такого обмена гостей,
должна быть исключительно высокой, а отсюда кинетика такого обмена должна
быть медленной. Фактически же нам известно, что в случае открытых гибких
хозяев типа C.97) обмен гостей (с другими гостями и с растворителем) является
чрезвычайно быстрым при комнатной температуре. Итак, как можно объяснить это явное
противоречие? Ответ лежит в способности молекулы хозяина временно искажаться
для уменьшения объема пустой полости. Искажение хозяина согласовано с
выходом гостя из полости хозяина, и, следовательно, переходное состояние при обмене
гостя не слишком энергетически невыгодно, так как является следствием
незначительной деформации связей. При входе нового гостя хозяин, разумеется, может
вернуться к своей первоначальной конформации. В случае каликсаренов это
искажение принимает форму «тиски» с переходом от симметрии C4v к Cjv (схема 5.4).
Эта особенность становится решающей в случае жесткого хозяина или когда
такое конформационное изменение связано со значительной энтальпийной
дестабилизацией. Интересно, однако, обсудить вопрос о скоростях обмена гостей в
теоретически полностью жестком хозяине. Станет ли обмен гостей невозможным из-за
образования вакуума? Такое явление сродни попытке вылить молоко из бутылки
при ее переворачивании. Молоко не способно хлынуть наружу под влиянием силы
Искажение полости в виде
"тисков" для минимизации
свободного объема
пустой полости
Гость
Реконструкция
комплекса - форма
полости хозяина
восстанавливается
Схема 5.4. Объем полости уменьшается при диссоциации комплекса для гибких
хозяев
366
5. Связывание нейтральных молекул
Жесткая полость
из сетки водородных
связей
Чаша резорцинарена
Рис. 5.46. Форма полости типа «бокал для виски» молекулы соединения E.66)
тяжести из-за вакуума, который при этом возникает. Воздух не может заполнить
вакуум, поскольку на его пути находится молоко. Результат — (в лучшем случае)
турбулентный медленный выход молока.
Для проверки этой теории был спроектирован жесткий хозяин (D. Rudkevich et
al., 1997). В основе структуры молекулы такого хозяина (соединение E.66)) лежит
структура каликс[4]резорцинарена E.43), верхний обод которого представляет
собой жесткую сетку из доноров и акцепторов водородной связи. Эти фрагменты
образуют глубокую открытую полость симметрии C4v, по форме несколько
напоминающую бокал для виски (рис. 5.46). Хотя этот хозяин способен принимать
конформации симметрии C4v и C,v, но наличие сетки водородных связей,
содержащей четыре циклических фрагмента, благоприятствует конформации C4v.
Большие молекулы-гости типа адамантана плотно входят в этот «молекулярный
бокал» и дают сигнал 'Н ЯМР при -1 м.д.; это согласуется с экранирующим
влиянием, ожидаемым от ароматических колец хозяина. Чрезвычайно важно, что
наблюдаются отдельные сигналы для связанного и свободного адамантановых гостей.
Это указывает на медленный обмен гостя на шкале времен ЯМР. Такой результат
трудно было ожидать, поскольку полость открыта и не содержит препятствий.
Однако эти данные можно объяснить тем, что удаление гостя или растворителя из
полости сопровождается образованием вакуума. Действительно, обмен гостя
протекает медленно из-за необходимости «распрямления» водородно-связанных
заместителей, составляющих верхний обод, подобно раскрытию цветка, что
сопровождается разрушением сетки водородных связей. Измерение динамического ЯМР
указывает на значительный активационный барьер G1 кДжмоль) процесса
обмена гостя, причем считается, что такой энергетический барьер непосредственно
связан с потерей энтальпийной стабильности из-за разрушения водородных связей.
Учитывая эти результаты, предполагают, что для всех открытых кавитандных
хозяев связывание нейтрального органического гостя в растворе можно кардинально
усилить конструированием жестких вогнутых поверхностей с единственным,
относительно узким, отверстием для входа и выхода.
5.3. Внутриполостные комплексы нейтральных молекул
5-3-1 -4 Хозяева на основе гликолурила
Гликолурил E.44) из-за изогнутого каркаса и многообразия химии
производных используют в качестве фундаментального строительного блока в
синтезе ряда искривленных «бочкообразных» хозяев, «молекулярных клещей» и
трехмерных капсул, образующихся путем самосборки (разд. 7.5.1). Кривизна дифенил-
гликолурила видна на примере соединения E.67) в форме «молекулярных клещей»,
которое способно выступать в качестве хозяина для ароматических гостей, таких,
как резорцин; в последнем две гидроксильные группы направлены вниз, в
молекулярную щель хозяина, для образования водородных связей с его карбонильными
атомами кислорода. Структура этого хозяина показана на рис. 5.47.
ОМе
W //
\ Y !
Кукурбитурил E.68)
Рис. 5.47. Кристаллическая
структура соединения E.67) (по
Sijbesma R.P. and Nolte R.J.M., 1995)
здо 5. Связывание нейтральных молекул
Наиболее известный хозяин на основе гликолурила — кукурбитурил E.68). Ку-
курбитурил назван так из-за сходства его бочкообразной молекулы с тыквой
(семейство Cucurbitaceae), в особенности с фонарем из тыквы Cucurbita pero. Соединение
E.68) известно с 1905 г., однако его полная характеристика была дана только в
работе У. Мока (W. Mock) из Чикагского университета (США), который исследовал его
методом РСА и другими методами в начале 1980-х годов. Это соединение легко
получить конденсацией гликолурила с формальдегидом. Сначала образуется
соединение, которое до сих пор мало изучено и, возможно, является поперечно-сшитым
полимером аминного типа. Это вещество очень неактивно и малорастворимо. Беренд
(Behrend), выполнивший в 1905 г. первую работу по этой тематике, обработал его
горячей концентрированной серной кислотой, в которой это соединение медленно
растворилось. Полученный раствор разбавили холодной водой с последующим
кипячением. Выделенный в конце концов кристаллический осадок, как показали
более поздние исследования, представлял собой соединение E.68), схема 5.5.
x
HN NH l.KoHa-HjSO,
МН20,НС1 Полимешшй 110°С т, Нагревание
CH o _i ^ полимерный ^ Раствор — Кукурбитурил
осадок 2.И,О.0-10С rs й8^
Y
HNV ^NH (избыток) E<68)
о
E.44)
Схема 5.5. Синтез кукурбитурила E.68)
Считается, что этот удивительный гексамер гладко образуется при темплатной
реакции (см. разд. 3.8.2), в процессе которой ионы оксония (возможно, Н3О+)
связываются водородным^ связями с карбонильными атомами кислорода верхней и нижней
частей этой молекулы. При этой реакции не обнаружили никаких других олигомеров
гликолурила, хотя А. Флинн и сотрудники (A. Flinn et al., 1992) смогли получить из ди-
метилгликолурила в умеренно кислых условиях родственный пентамер, названный
декаметилкукурбит[5]урилом. Совсем недавно B000 г.) группе Кима (Kim) удалось
видоизменить первоначальные условия реакции и синтезировать кукурбит[л]урилы, где
п = 5, 7, 8 и 9. Эти новые большие «бочки» предоставляют интересные возможности
для капсулирования больших гостей и проведения реакций включения.
Установили, что кукурбитурил не только очень устойчив химически, но и образует
ряд кристаллических комплексов с солями металлов и красителями. Исследования
показали, что эта молекула — эффективный хозяин для протонированных
полиаминов и ионов металлов как в водных растворах, так и в твердом состоянии, но не для
нейтральных органических молекул. Такая химия включения обусловлена наличием в
молекуле кукурбитурила бочкообразной полости глубиной ~6 А и координирующей
способности кольца из карбонильных атомов кислорода. Пентамерный диметилку-
курбит[5]урил имеет более узкую полость и небольшое число соединений включения.
5.3. Внутриполостные комплексы нейтральных молекул 369
н-алкилендиаммониевые ионы
н-алкиламмониевые ионы
6 -
5 -
4 "
3 -
2 ' ■ " ^ ^ Рис. 5.48. Зависимость константы
связывания от длины цепи для хозяина — ку-
курбитурила — и гостей — протонирован-
| ных аминов NH2(CH2)wMe и диаминов
'о 1 2 3 4 5 6 7 8 9 10 NH2(CH2),NH2 - в HCO2H-D2O
Длина цепи (л)
Поскольку кукурбитурил не обладает ароматическими кольцами, следует
ожидать, что сдвиги в спектре ЯМР гостя, индуцированные комплексообразованием,
должны быть меньше по сравнению со сдвигами, наблюдаемыми в производных,
содержащих бензольные кольца (ср. Дополнение 3.4). В реальности, однако,
алифатический каркас этого соединения также приводит к значительному
экранирующему эффекту. Спектр 'Н ЯМР 1,5-диаминопентана в растворе HCO2H-D2O,
имеющий два мультиплета при 5 3.17 и 1.77 м.д. в отсутствие соединения E.68),
обнаруживает два новых сигнала при 8 2.73 и 0.77 м.д. взамен сигналов свободного
гостя при наличии кукурбитурил а. В конце концов, образуется устойчивый 1:1 -ад-
дукт хозяин—гость, в котором этот амин (дважды протонированный муравьиной
кислотой) втиснут в полость хозяина. Обмен гостя является медленным на шкале
времен 'Н ЯМР. В ряду гостей-диаминов ЫН2(СН2)ЯЫН2 (п = НЮ) в той же среде
найдено, что максимальные константы связывания наблюдаются при п = 5 и 6, это
соответствует гидрофобному включению алкильной цепи при возможности
образования водородных связей между группами NH^" и карбонильными атомами
кислорода (рис. 5.48). Заметьте, что имеется соответствие по симметрии между
(^-симметрией аммониевого заместителя и С6-симметрией соединения E.68), позволяющее
образовать три бифуркационные усиленные зарядом водородные связи N+—H---O.
8-1 Mock W.L. Cucurbituril // Comprehensive supramolecular chemistry / Ed.
J.L.Atwood, J.E.D.Davies, D.D.MacNicol and F.Vogtle. Oxford: Pergamon, 1996.
Vol. 2. P. 477-493.
* См. также обзор: Герасько О.А., Самсоненко Д.Г., Федин В.П. Супрамолекулярная химия
кукурбитурилов // Успехи химии. 2002. Т. 71, № 9. С. 840-861. {Примеч. переводчика.)
370
5. Связывание нейтральных молекул
5.3*1.5 Хозяева на основе конкинов
В середине 1980-х Дж. Стоддарт и Ф. Конке (J. Stoddart and F. Kohnke),
исследователи из Университета Шеффилда (Великобритания), синтезировали
другой важный строительный блок - E.45), образующийся при реакции 1,2,4,5-тетра-
бромбензола с фураном (схема 5.6).
Конкин E.69), названный так в честь своего создателя, — очень жесткая
структура, которая обладает небольшой эллиптической полостью, непригодной для
включения молекул гостей. Однако это соединение можно дезоксигенировать с
образованием дидезоксиконкина E.70), имеющего более квадратную полость, как раз
подходящую по размеру для включения единственной молекулы воды (рис. 5.49).
Предшественник
конкина
E.45)
Конкин E.69)
Схема 5.6. Синтез конкина путем ряда реакций присоединения
по Дильсу—Альдеру
5.3. Внутриполостные комплексы нейтральных молекул
371
Рис. 5.49. Кристаллические структуры (а) конкина и (б) дидезоксиконкина с включенной
в решетку молекулой воды
Этот пример проливает свет на чрезвычайную трудность получения молекулярных
полостных соединений даже с маленькой полостью, причем трудность возрастает с
увеличением ее размера . При этом исследователю приходится проявлять большую
изобретательность.
Н
Н
н \СУ1 ;\—у н
Дидезоксиконкин E.70)
Стоддарт и сотрудники расширили этот общий подход и предложили
многоцелевой молекулярный конструктор, которому они дали название «молекулярный
LEGO» по аналогии с детским конструктором, содержащим строительные блоки.
Это позволило исследователям получить циклические структуры типа конкина
различных размеров, молекулярные «волны» (образующиеся по ретрореакциям
Дильса—Альдера) и даже полостное соединение E.71), названное ими тринакреном
(Тринакрия - старинное название Сицилии, родины Ф. Конке).
5. Связывание нейтральных молекул
° V7 °
Тринакрен E.71)
Mathias J.P. and Stoddart J.F. Constructing a molecular LEGO set //
Chem.Soc.Rev. 1992. Vol. 21. P. 215-225.
5*3*2 Циклодекстрины
8-* Sze/f/f /. Introduction and overview of cyclodextrin chemistry // Chem.Rev. 1998.
Vol. 98. P. 1743-1753.
5.3.3.1 Свойства
Химия каликсаренов и кавитандов (разд. 5.3.1) ясно демонстрирует,
что, по мере увеличения жесткости молекулярной полости (и, следовательно, ее
предорганизации) и ее глубины, способность полости связывать органические
гости в растворах возрастает. Как мы видели на примерах хозяев, полученных на
основе гликолурила (см. разд. 5.3.1.4 и рис. 5.39), использование связанных
ароматических колец (циклофаны) — не единственный путь создания жесткой структуры
хозяина. Действительно, обычные, наиболее изученные и самые дешевые,
коммерчески доступные хозяева — циклодекстрины - полностью насыщены и их
жесткость обеспечивается как внутримолекулярными водородными связями, так и
радиусом кривизны их полости. Циклодекстрины относятся к классу чрезвычайно
важных соединений-хозяев, которые широко используют в пищевой,
косметической и фармацевтической отраслях промышленности, обычно в качестве агентов для
медленного высвобождения и доставки веществ. Они также чрезвычайно важны
как имитаторы ферментов (см. разд. 3.9). Одно из их преимуществ — абсолютная
нетоксичность в широком диапазоне дозировки. Они настолько важны, что им
посвящен целый том 11-томного издания «Comprehensive supramolecular chemistry
(J. Atwood et al. Oxford, «Pergamon», 1996) - том З. Промышленное производство на-
5.3. Внутриполостные комплексы нейтральных молекул
{а)
373
ОН
(б)
а-Циклодекстрин
E.47)
Р-Циклодекстрин
E.72)
ОН
он
у-Циклодекстрин
E.73)
6 ОН
1,4-Гликозидная связь
Рис. 5.50. (а) Три наиболее важных циклодекстрина. (б) 1,4-Гликозидная связь,
соединяющая соседние D-глкжопиранозидные единицы
374
5. Связывание нейтральных молекул
иболее важного представителя семейства циклодекстринов, C-циклодекстрина
E.72), составляет -1500 т/год с ценой лишь несколько долларов за 1 кг.
Циклодекстрины — это циклические олигосахариды, включающие (обычно) от
шести до восьми /)-глюкопиранозидных единиц, соединенных 1,4-гликозидными
связями (рис. 5.50). Три наиболее важных представителя семейства
циклодекстринов - это а-циклодекстрин (a-CD, E.47)), Р-циклодекстрин (P-CD, E.72)) и
у-циклодекстрин (y-CD, E.73)), содержащие соответственно шесть, семь и восемь
глюкопиранозидных единиц. Известно несколько других, менее распространенных
циклодекстринов, включая 8-циклодекстрин и е-циклодекстрин (девять и десять
единиц соответственно), а также пятичленный npe-a-циклодекстрин. а,Р,у-Но-
менклатура служит для различения членов этого гомологического ряда с разными
размерами колец и, по существу, уже носит исторический характер. Тем не менее ее
очень широко используют, несмотря на то, что она явно не указывает на размер
кольца. Это неудивительно, поскольку систематические наименования для
циклодекстринов крайне громоздки. Например, термины для Р-циклодекстрина
включают цикломальтогептаозу, циклогептаглюкан и циклогептаамилозу; аналогичные
Таблица 5.6. Характеристики а-, Р- и у-циклодекстринов
Параметр
Число единиц глюкозы
Размер кольца
(число атомов С)
Центральный диаметр
внутренней полости, А
Растворимость в воде,
г-л-' B5 °С)
Д#°раствора, кДжмоль
AS° раствора,
Дж-Кг1-мол Б
[abB5°Q
Объем полостей, А3
Объем полости в 1 г
циклодекстрина, см3
Кристаллизационная вода,
мае. %
рКа B5 °С, по данным
потенциометрии)
Скорость гидролиза
A. oryzae a-амилазой
Обычные гости
Циклодекстрины
a-CD
6
30
5.0
145
32.1
57.7
150.5
174
0.10
10.2
12.33
Пренебрежимо
малая
Бензол, фенол
P-CD
7
35
6.2
18.5
34.7
48.9
162.0
262
0.14
13.2-14.5
12.20
Небольшая
Нафталин,
ANS
Y-CD
8
40
8.0
232
32.3
61.4
177.4
427
0.20
8.13-17.7
12.08
Большая
Антрацен, краун-эфиры,
ANS
5.3. Внутриполостные комплексы нейтральных молекул
375
термины существуют для других членов этого семейства. Наименование «цикло-
декстрин» происходит от слова «декстроза» — старинного синонима слова
«глюкоза». Известны также другие циклические олигосахариды, производные маннозы и
галактозы, но они гораздо менее изучены.
Форму циклодекстрина часто представляют в виде конусообразного тора или
усеченной воронки и, подобно верхнему и нижнему ободам каликсаренов, в цикло-
декстринах имеются две различные торцевые поверхности: узкая и широкая. Узкая
поверхность — это узкий конец тора, включающий первичные гидроксильные
группы. Более широкая поверхность содержит группы СНОН. Шестичленные /)-глюко-
пиранозидные кольца соединены так, что все их поверхности направлены к
центральной гидрофобной полости различных размеров. Именно наличие этой полости
в сочетании с растворимостью в воде, обусловленной гидрофильными спиртовыми
группами, придает циклодекстринам уникальную способность к комплексообразо-
ванию в водном растворе. Функциональная схема строения циклодекстринов
наряду с размерами полостей показана на рис. 5.51. Важнейшие параметры приведены в
табл. 5.6.
В целом, физические и химические свойства циклодекстринов относительно
равномерно изменяются в ряду от а- до y-CD, хотя характеристики очень больших
циклодекстринов типа 8-CD начинают отклоняться от характеристик этого ряда,
потому что они не такие жесткие, как хозяева меньших размеров, и - как следствие
— проявляют намного меньшую способность связывать гости. Однако монотонное
изменение свойств нарушается при рассмотрении растворимости циклодекстринов
в воде (см. табл. 5.6), которая значительно меньше у нечетного члена ряда C-CD,
Гидрофобная полость
Вторичные гидроксильные группы
Первичные гидроксильные группы
13.70 А
15.30 А.
16.90 А
7.80 А
a-CD P-CD
Рис. 5.51. Строение циклодекстринов
y-CD
376
5. Связывание нейтральных молекул
чем у меньшего (а-) и большего (у-) аналогов (данные рефрактометрии).
Растворимость нечетного члена ряда 6-CD также меньше, чем растворимость a-CD и y-CD,
хотя это отклонение не столь велико, как в предыдущем случае. Такое поведение
P-CD имеет определенное значение при его использовании в качестве хозяина в
растворе. Предложено несколько интерпретаций такого поведения циклодекстри-
нов. Определение энтальпии и энтропии гидратации циклодекстринов показало,
что оба параметра менее благоприятны для P-CD, чем для его аналогов большего и
меньшего размеров. Это объясняется нарушением водородных связей структуры
воды агрегированным P-CD. Кроме того, циклодекстрины с симметрией С6 и Q
более совместимы с агрегатами растворителя, имеющими четное число доноров и
акцепторов водородной связи. Предположили также, что внутримолекулярные
водородные связи на широкой поверхности P-CD ограничивают его взаимодействие с
растворителем - водой. В a-CD аналогичная сетка водородных связей неполная
из-за напряжения, тогда как молекула y-CD более гибкая. Действительно,
метилирование вторичных гидроксильных групп увеличивает растворимость P-CD.
Все циклодекстрины кристаллизуются из воды в виде гидратов, содержащих
различное количество воды, в зависимости от условий кристаллизации. В каждом
случае полость циклодекстрина заполнена молекулами воды с относительно
высокой энергией вследствие ограниченности их взаимодействий со стенками полости.
Предполагается, что в растворе большой вклад в комплексообразование частиц-
гостей вносит выталкивание этих высокоэнергетических молекул воды из полости,
являющееся следствием общего гидрофобного эффекта. Скорее всего, этот вклад
сводится к уменьшению влияния неблагоприятного процесса (десольватация
хозяина), но сам по себе не является реальной движущей силой, как мы увидим позже.
Распределение воды в различных формах кристаллогидратов циклодекстринов
представлено в табл. 5.7. Отметим, что для больших циклодекстринов объем
полости в твердом состоянии ограничен из-за включения остатков глюкозы соседних
хозяев. Каждый циклодекстрин кристаллизуется в разнообразных формах (которые
Таблица 5.7. Распределение воды в твердых гидратах циклодекстринов
п
Распределение
Вне полости
В ПОЛОСТИ
a-CD
6A)
6A1)
7.57A11)
2A)
1A)
2.57A11)
P-CD
12A)
11A1)
-
7.3
6.3 (II)
-
Число молекул водь
Y-CD
19
14.1
15.7
7.1 из 14.1
Две единицы
глюкозы также
занимают полость
-
6-CD
13.75
—
-
Две единицы глюкозы
занимают полость; вода
распределена в открытом
пространстве на
первичной поверхности
—
-
5.3. Внутриполостные комплексы нейтральных молекул
377
Рис. 5.52. Две проекции
кристаллической структуры формы I
гидрата a-CD
обозначены римскими цифрами для надежно установленных случаев). Вообще,
структуры самих циклодекстринов-хозяев сильно не изменяются от одной формы к
другой, и вся разница заключается в числе молекул воды. В формах I и III a-CD
принимает эллиптически искаженный неправильный вид, причем две единицы
глюкозы повернуты относительно их гликозидной связи таким образом, что
стороны с первичными гидроксильными группами направлены внутрь полости; при
этом ее объем уменьшается. Гидратная форма II более правильна. Обе формы P-CD
имеют похожую геометрию хозяина, которая более правильна, чем геометрия
a-CD; занятость некоторых из разупорядоченных участков воды больше в форме I.
Структура формы I гидрата a-CD показана на рис. 5.52. Структура гидрата y-CD
очень напоминает таковую для гидрата P-CD, причем снова молекулы воды
организованы беспорядочно. Оба хозяина содержат последовательность
внутримолекулярных водородных связей на узкой поверхности с расстояниями 0-0 в интервале
2.70—3.00 А. Искаженная структура гидрата 8-CD имеет форму «лодка» и образует
меньшую полость.
..А 5. Связывание нейтральных молекул
§.§.2.2 Синтез
В результате фотосинтеза в растениях образуются два главных
продукта: крахмал и целлюлоза. Целлюлоза - нерастворимый, упругий,
структурообразующий компонент клеток. Напротив, крахмал, по существу, - хранилище энергии.
Он легко растворяется и превращается в биохимически полезные формы, и именно
из него получают циклодекстрины. Крахмал включает два полимера глюкозы:
линейный (амилоза), состоящий из сотен и даже тысяч остатков D-глюкопиранозида,
и разветвленный (амилопектин). Амилоза содержит только 1,4-гликозидные связи,
а разветвленный амилопектин имеет также 1,6-связи. Разложение крахмала под
действием ферментов в присутствии воды приводит к декстринам, образующимся в
результате гидролиза гликозидных связей. Декстрины используют в пищевой,
текстильной и бумажной промышленности, а также при производстве хлеба, пива и
т.п. Если декстрины разложить ферментом глюкозилтрансферазой, то первичный
продукт расщепления цепи - линейный олигосахарид — подвергается
внутримолекулярной циклизации без участия воды, образуя циклодекстрин. Индивидуальные
циклодекстрины выделяют из смеси комплексообразованием с неполярными
гостями типа толуола (рис. 5.53), что приводит к резкому снижению растворимости.
Чистота промышленно производимых циклодекстринов обычно превышает 99%.
5'3'2*3 Соединения включения
Обычно взаимодействие циклодекстрина с неполярной
молекулой-гостем приводит к образованию молекулярных соединений включения состава 1:1, в
которых гость «встроен» в полость циклодекстрина. Включение — термодинамически
равновесный процесс с константой связывания К, даваемой обычным соотношением:
E.1)
[CDG]
E2)
Обычны также равновесные процессы более высокого порядка, в которые
входит образование комплексов 1:2 или кратных агрегатов с участием более одного
циклодекстрина, часто существующих одновременно. Движущую силу включения
гостя составляют вклады, степень важности которых еще обсуждается. Эта
проблема подробно освещена в ключевой ссылке данного раздела.
Особо важными факторами являются:
• стерическое соответствие;
• высвобождение высокоэнергетической воды;
• гидрофобные эффекты;
• взаимодействия Ван-дер-Ваальса;
• дисперсионные силы;
• диполь-дипольные взаимодействия;
• взаимодействия с переносом заряда;
• электростатические взаимодействия;
• водородные связи.
Глюкозилтрансфераза
Крахмал
Смесь после разложения
I
I
Комплексы циклодекстрин-гость
Рис. 5.53. Вьщеление циклодекстринов при разложении крахмала глюкозилтрансферазой
2gn 5. Связывание нейтральных молекул
В табл. 5.6 показано, что в основном размер приспосабливающегося гостя
увеличивается по мере роста размера полости хозяина — циклодекстрина. Однако
структуры хозяина обладают некоторой гибкостью, и поэтому соответствие по
размеру не является строгим критерием. Высвобождение высокоэнергетической воды
и гидрофобный эффект (см. разд. 1.7.9) включают два энергетических компонента:
выигрыш в энтальпии вследствие воссоединения высокоэнергетических молекул
растворителя с его основной массой и выигрыш в энтропии, поскольку две дырки в
растворителе (хозяин и гость) становятся одной (комплекс). Классический
гидрофобный эффект дает явно отрицательную величину AS°, но это не всегда
справедливо для комплексов циклодекстринов, так как, в действительности, полость не
является неполярной. Она имеет ряд функциональных групп, и ее лучше
рассматривать как полуполярную. Тем не менее и неклассические гидрофобные
эффекты могут стать чрезвычайно важными. Наряду со стабилизацией за счет
эффектов соответствия по размеру и сольватации, твердые комплексы и комплексы в
растворе часто дополнительно стабилизированы энтальпийно благоприятными
взаимодействиями, такими, как водородные связи между гостем и первичными или
вторичными гидроксильными группами циклодекстрина и дипольные
взаимодействия. К. Коннорс (К. Connors, ключевая ссылка этого раздела) разработал
обобщенную модель комплекообразования циклодекстринов в растворе, которая дает
общую свободную энергию комплексообразования АGС , включающую
следующие члены:
AGcomp = ЩтгазЫ + Що/v + ^genm' <5'3)
Член &Gjntrasoj описывает взаимодействия хозяин—гость и должен иметь
благоприятный или, в худшем случае, нулевой вклад. Он включает вклады водородных
связей, дипольных взаимодействий и сил Ван-дер-Ваальса. AGyo/v описывает
эффекты растворителя (гидрофобный, перегруппировка растворителя). Он может
быть благоприятным или неблагоприятным, но обычно неблагоприятен. Этот член
представляет собой разность между свободной энергией сольватации комплекса и
суммой свободных энергий сольватации индивидуальных хозяина и гостя. Если
вода в свободных циклодекстринах-хозяевах имеет высокую энергию, тогда AG5O/v
будет стремиться стать более благоприятным (или менее неблагоприятным). ДС' т —
общий эффект среды, который связан с созданием полости в растворителе при
растворении вещества. Этот член зависит от площади поверхности полости и
поверхностного натяжения раствора. Смысл этой зависимости состоит в том, что
эффективное комплексообразование должно вызываться благоприятными вкладами
^imrasoi и ^genm- Константы связывания заряженных и незаряженных гостей
а-циклодекстрином показаны на рис. 5.54. Ясно, что 4-нитрофенолят-анион
связан значительно сильнее, чем аналогичное нейтральное соединение. Гидрофобный
нитрофенильный заместитель этого гостя включен в полость циклодекстрина, а
остаток фенолята расположен у широкой поверхности вторичных гидроксилов.
Таким образом, гидрофобные взаимодействия одинаковы для двух следующих гостей
(ср. константы связывания производных диметилнитрофенола: объемистое 3,5-ди-
метилпроизводное не вгоняется в узкую область первичных гидроксилов циклодек-
5.3. Внутриполостные комплексы нейтральных молекул
381
ОН
Широкая
поверхнос
Узкая
поверхнос
lg AT 1.78 3.07 ~0 ~0
Рис. 5.54. Гости и их константы связывания с а-циклодекстрином в воде
стрина, в то время как для 2,6-диметильного аналога, в котором метальные
заместители расположены у более широкой поверхности вторичных гидроксилов,
наблюдается прочное связывание. Дополнительная стабилизация достигается за счет
индуцированных диполь-дипольных взаимодействий и сил Ван-дер-Ваальса,
возникающих вследствие повышенной поляризуемости аниона, в котором
отрицательный заряд делокализован по всей молекуле. Это противоречит результатам по
бензойной кислоте и ее сопряженному основанию, для которых анион связан
слабее, чем кислота, поскольку центр ионизации этого гостя (т.е. карбоксилатный
остаток) является также центром связывания.
Найдено, что при комплексообразовании алифатических гостей типа
Ме(СН2)лХ связывание значительно усиливается, если X - полярная группа,
например СООН или ОН, а не Me. Это опять-таки объясняется значительным энталь-
пийным выигрышем от таких взаимодействий хозяин—гость, как диполь-диполь-
ное взаимодействие или водородные связи с ободом молекулы хозяина.
Интересно, что факт обнаружения комплексообразования в растворе, например
методами ЯМР, кругового дихроизма или по каталитическому эффекту, не
обязательно означает возможность выделения кристаллических или твердых
комплексов. В твердом состоянии гидрофобное отталкивание между гостем и
растворителем не имеет места, и, таким образом, в отсутствие значительного притяжения
может сильно уменьшиться движущая сила комплексообразования. Иногда
комплексы, устойчивые в растворе, кристаллизуются в виде тонкой дисперсии
отдельных кристаллов хозяина и гостя. Тем не менее было выделено большое число
устойчивых кристаллических комплексов включения, и, даже когда отсутствуют
значительные благоприятные взаимодействия хозяин—гость, твердофазное ком-
плексообразование может быть вызвано необходимостью избежать образования
пустого пространства в кристаллической структуре. Наличие гостей также
необходимо, поскольку тор циклодекстрина не может схлопываться сам по себе.
382i
5. Связывание нейтральных молекул
В общем, комплексы циклодекстринов в твердом состоянии можно разделить на
три обширные категории — канальные, клеточные и слоистые — в зависимости от
ориентации циклодекстриновых остатков и связей между соседними полостями.
Далее их подразделяют в зависимости от относительной ориентации поверхностей
первичных и вторичных гидроксилов. Возможные способы упаковки представлены
на рис. 5.55.
Структуры клеточного типа наблюдаются, когда молекула гостя достаточно
мала для полного включения в полость циклодекстрина. В результате молекулы цик-
лодекстрина организуются по типу «елочка». Полость oc-CD достаточно велика для
захвата молекулы бензола, имеющей длинную ось, параллельную оси симметрии
псевдо-С6, а также для захвата малых органических молекул, например метанола,
пропанола-1 и 3-иодпропановой кислоты; и все такие комплексы кристаллизуются
по клеточному типу. Размер полости C-CD также подходит для включения
молекулы бензола с его длинной осью, перпендикулярной оси симметрии псевдо-С^.
C-CD также образует комплексы клеточного типа с небольшими гостями наподо-
(а)
(в)
(б)
Рис. 5.55. Схематическое
представление упаковки
циклодекстриновых структур, (я)
Канальный тип «голова к голове»;
(б) канальный тип «голова
к хвосту»; (в) клеточный тип;
(г) слоистый тип; (д) слоистый
тип, состоящий из димеров
C-CD. (Воспроизведено с
разрешения AtwoodJ.L., DaviesJ.E.D.,
MacNicol D.D. and Vdgtle F.
Comprehensive supramolecular
chemistry. Oxford: Pergamon,
1996. Vol. 3. P. 287)
5.3. Внутриполостные комплексы нейтральных молекул
383
Рис. 5.56. Клеточная
организация в комплексе
P-CD-бензиловый спирт
{по Harata К. etal., 1985)
бие бензилового спирта (рис. 5.56). Нет
свидетельств, что C-CD образуют соединения
клеточного типа, за исключением комплекса с водой.
Это следствие включения остатков глюкозы
соседних молекул по широкой поверхности, что
значительно сокращает объем полости при
клеточной организации.
С большими молекулами-гостями, не
соответствующими полости, все три главных цикло-
декстрина образуют канальные структуры, в
которых циклодекстриновые полости
выстраиваются в ряд, давая одну протяженную
гидрофобную полость. В эту полость можно ввести
гости так же, как и в клатраты мочевины (см.
разд. 5.2.1). Такие структуры наблюдали для
комплексов oc-CD с индикатором метиловым
оранжевым, в которых гость простирается на три
циклодекстриновые полости, и комплекса с
ферроценом (рис. 5.57), а также для комплекса y-CD
с гораздо большим [NaA2-KpayH-4J]+ (рис. 5.58).
Включение катиона Na+ в две молекулы краун-
эфиров, которые, в свою очередь, включены в
пару циклодекстриновых полостей, назвали
комплексом «русская кукла» по аналогии с тра-
Рис. 5.57. Канальная структура комплекса a-CD
с ферроценом
5. Связывание нейтральных молекул
Рис. 5.58. Канальная структура
комплекса y-CD с [ЫаA2-краун-4J]+
типа «русской куклы» («матрешка»)
диционными деревянными русскими куклами «матрешками», когда одна кукла
вкладывается в другую. Присутствие противоиона хлорид-аниона делает
необходимым чередование гостя (металл-краун), капсулированного парой y-CD с еще
одной молекулой yCD, который включает в себя молекулу свободного 12-краун-4.
Тогда общая стехиометрия такова: [(y-CDK с №A2-краун-4J-С112-краун-4].
^~Т Connors К.A. The stability of cyclodextrin complexes in solution // Chem. Rev. 1997.
Vol. 97. P. 1325-1357.
5.3.2.4 Промышленное применение
Циклодекстрины находят широкое применение в ряде отраслей про-
мышленности. Рынок циклодекстринов растет благодаря их уникальным
свойствам по включению гостей и кинетике диссоциации комплексов наряду с их
стабильностью, нетоксичностью и относительной дешевизной. Основные области
интереса к ним представлены в виде распределения резюме статей по циклодекст-
ринам, опубликованным в 1996 г. в «Cyclodextrin News» (рис. 5.59).
Распределение исследований, приведенное на этом рисунке, не включает,
однако, самых последних данных о применении циклодекстринов. Около 80-90%
полученных в промышленности циклодекстринов (в основном C-CD) расходуется в
пищевой промышленности, где использование циклодекстринов предпочтительно
из-за их высокотемпературной стабильности в процессах переработки пищевых
продуктов. Комплексообразование циклодекстринами дорогих душистых масел и
специй, приготовленных из яблок, цитрусовых, корицы, чеснока, имбиря, мяты и
тимьяна, значительно уменьшает их количества, необходимые для достижения
запаха определенной силы при добавлении к пище. Комплексообразование делает
5.3. Внутриполостные комплексы нейтральных молекул 385
Химия комплексов
Фармацевтика 24% циклодекстрина 22%
Пища _^^__„ ,™__^^^ _^__^^^_^^^^^^_
и косметика 7% |^^ррни->^ ^^^^| Химия, энзимология,
биологические эффекты 16%
Пестициды 1%
Химические Аналитическая химия 19%
и биохимические
процессы 11%
Рис. 5.59. Распределение 1706 исследовательских работ по химии циклодекстринов и их
применению, опубликованных в «Cyclodextrin News» в 1996 г.
такие ароматизаторы более устойчивыми к окислению, фотохимической
деструкции, термическому разложению и уменьшает их летучесть. Приведем лишь один
пример. Количество луковой приправы для пикантного овечьего сыра можно
снизить от 550 г (на 100 порций) до 10 г, если эта специя связана в виде комплекса с
Р-циклодекстрином. Подобная стабилизация достигается в случае пищевых
красителей и пигментов, которые могут разлагаться фотолитически или при изменении
рН. Привлекательность циклодекстринов для производителей пищевых продуктов
возрастает еще более, если учесть преимущества переработки и взвешивания (сухой
порошок в противоположность летучему маслу), которые снижают цены при
упаковке и хранении, а также вероятность микробного загрязнения.
Кроме использования в пищевой промышленности циклодекстрины также
применяют в фармацевтической промышленности в качестве систем доставки
лекарств. Как и в пищевой промышленности, циклодекстрины могут служить
защитными агентами, которые предотвращают преждевременный метаболизм лекарств и
допускают таким образом, например, оральное, а не внутривенное введение. Они
также могут изменять растворимость лекарства и особенности его биологического
транспорта, увеличивая специфичность к мишени. Циклодекстрины могут также
увеличивать растворимость слаборастворимых лекарств, что исключает
необходимость химического модифицирования самого лекарства введением гидрофильных
остатков, способных вмешиваться в мембранный транспорт этого лекарства.
Известно, наконец, что комплексообразование циклодекстринами уменьшает местное
раздражение или повреждения, вызванные лекарством, а также маскирует
неприятные или горькие привкусы.
Другая большая область применения циклодекстринов - аналитическая химия,
в особенности хроматографические методы, такие, как тонкослойная и газовая
хроматография, капиллярный электрофорез и высокоэффективная жидкостная
хроматография (high-pressure liquid chromatography, HPLC). И снова циклодекстрины
выполняют роль комплексообразователей для изучаемых гостей-аналитов. Наличие
циклодекстринов (и их производных) либо в качестве добавок к подвижной фазе,
либо в качестве добавок, химически связанных со страционарной фазой, может
повысить эффективность разделения и скорость анализа, а также привести к
разделению родственных соединений и изомеров, в частности энантиомеров. HPLC-разде-
25 - С1ИЛ Лм И
5. Связывание нейтральных молекул
ление энантиомеров с использованием хиральных колонок обычно чрезвычайно
дорого из-за высокой стоимости стационарной фазы A000—2000 долларов за
колонку). Напротив, применение значительно более дешевой, ахиральной, колонки
(с октадецилдиметилсиликагелем, ODS) с добавлением (З-циклодекстрина к
подвижной фазе позволяет проводить, например, хиральное разделение барбитуратов
в человеческой сыворотке (S. Eto et al., 1992).
5*3*3 "Молекулярные щели» и «молекулярные пинцеты»
«Молекулярный пинцет» представляет собой рецептор простого вида,
в котором два домена («лапки» пинцета), связывающих гостя, расположены (в
более или менее предорганизованном состоянии) с каждой стороны от
предполагаемого центра связывания хозяина. Этими лапками хозяин удерживает гостя.
Обычно для удержания лапок хозяина на требуемом расстоянии друг от друга в качестве
жестких спейсеров используют арильные кольца, которые при связывании гостя
служат мостиком. Дж. Адриан и К. Вилкокс (J. Adrian and С. Wilcox, 1991)
получили молекулярные пинцеты E.74), используя основание Трёгера для достижения
необходимой молекулярной кривизны. Хозяин E.74) способен связывать 2-аминопи-
римидин (AmP) с образованием комплементарного комплекса E.74)АтР состава
1:1. В сухом хлороформе константы связывания достигают величины 2.4-104 М~',
хотя с ростом температуры они значительно уменьшаются. Интересно, что
насыщение хлороформа водой не приводит к значительному росту сродства
хозяин—гость, что следует из измерения величины общей свободной энергии комплек-
сообразования. Это обусловлено тем, что конкуренция воды (которая уменьшает
энтальпийныи вклад в связывание) за центры связывания компенсируется более
благоприятным энтропийным вкладом, обусловленным высвобождением воды при
образовании этого комплекса (ср. гидрофобный эффект в чистой воде).
R V R
Me2N
E.74) AmP
E.75)-9Р А
Концепция молекулярных пинцетов была распространена С. Циммерманом
(S. Zimmerman, 1993) на двойное распознавание гостей: с помощью п—тх-стэкинга,
хозяев-пинцетов и водородных связей с ядром лиганда. Хозяин E.75) связывает
9-алкиладенины, например 9-пропиладенин (9РА), при этом константы связывания
достигают 1.2-105 М. При замене группы СО2Н на аналогичную сложноэфирную
5.3. Внутриполостные комплексы нейтральных молекул
387
группу СО2Ме нарушается комплементарность функциональных групп хозяина и
гостя, ответственных за образование водородных связей, и значительно
уменьшается стабильность комплекса в хлороформе. Аналогично при диметилировании
первичной аминогруппы, ND), также снижается стабильность комплекса.
Работа Дж. Ребека (J. Rebek) привела к получению ряда разнообразных жестких
хозяев-пинцетов, таких, как E.76)—E.78). Эти соединения легко получить из кон-
формационно жесткой трикислоты Кемпа и различных ароматических диаминов.
Основная черта таких хозяев - жесткость каркаса, организующего эти соединения
в сходящиеся С-образные рецепторы. Такая жесткость — следствие наличия ме-
тильных заместителей в циклогексановых кольцах, что предотвращает эпимериза-
цию аксиальных карбоксильных групп, а также метальных групп в арильных спей-
серах, препятствующих вращению вокруг связей N-Ar. В отсутствие такого
ингибирования оба этих процесса должны приводить к изомеризации молекулы
хозяина и разрушению его «пинцетной» структуры. Интересно поведение гостя E.76)
с точки зрения его реакционной способности, т.е. электрофильная атака
(например, алкилирование диазометаном CH2N2) на карбоксигруппы ингибируется из-за
внутримолекулярного парного водородного связывания этих групп. Нуклеофиль-
ная атака на карбоксильный атом углерода не ингибирована, потому что входяще-
Трикислота
Кемпа
E.78)
388
5. Связывание нейтральных молекул
Нуклеофил
Электрофил
Рис. 5.60. Траектории нуклеофильной
и электрофильной атак на пинцет E.76)
му нуклеофилу не нужно втискиваться между двумя лапками пинцета, чтобы
прореагировать (рис. 5.60).
В больших пинцетах E.77) и E.78) отсутствуют внутримолекулярные
водородные связи и лапки пинцетов контактируют друг с другом только через молекулы
растворителя. Такая щель, следовательно, доступна для связывания субстрата.
Исследование этих систем направлено на связывание биологически значимых гостей,
чтобы использовать их в качестве моделей для биологического (ферментативного)
катализа. В частности, родственный имидный хозяин-пинцет E.79) способен
экстрагировать аденин и даже углеводы типа аденозина и дезоксиаденозина из
водного раствора и транспортировать их через жидкие органические мембраны.
E.79)-аденин
Гораздо более ранняя работа К. Чена и X. Витлока (С. Chen and H. Whitlock,
1978) привела к получению молекулярного пинцета E.80), использующего
комбинацию п—71-стэкинг- и ион-дипольных взаимодействий для связывания гостя.
Работа Витлока была инициирована данными об ингибировании гидролиза аспирина
кофеином, полученными в 1970 г. Это предполагало, что кофеин связывает
лекарство. Виток ожидал, что такое связывание лучше всего изучать на примере более
простых систем, в частности хозяина типа соединения E.80) и гостей типа
калиевой соли 1,3-дигидрокси-2-нафтойной кислоты (potassium salt of l,3-dihydroxy-2-
naphthoate, PDN). Расстояние между двумя лапками теофиллиновых пинцетов в
E.80) довольно жестко поддерживается шестиуглеродным дииновым спейсером,
допускающим некоторое вращение метиленовых 5/?3-звеньев (схема 5.7). Комплекс
E.80) PDN практически нерастворим, и его невозможно изучить методом ЯМР-
спектроскопии. Поэтому константу связывания измерили с помощью коэффици-
5.3. Внутриполостные комплексы нейтральных молекул
PDN
E.80)-PDN
Схема 5.7. Синтез комплекса E.80) PDN
ентов распределения между водным раствором с фосфатным буфером и
дихлорэтаном, которые определили методом оптической спектроскопии в УФ- и видимой
областях. Это дало приемлемые величины констант связывания ~104.
^ Rebek /., Jr. Molecular recognition with model systems // Angew. Chem., Int. Ed.
Engl. 1990. Vol. 29. P. 245-255.
5*3*4 Циклофаны
^ Diederich F. Cyclophanes. Cambridge: Royal Society of Chemistry, 1991.
5.3.4.1 Общие вопросы
Циклофаны — это мостиковые макроциклические системы,
включающие ароматические или гетероароматические макроциклы, соединенные между
собой алифатическими спейсерами. Таким образом, специфические циклофаны типа
каликсаренов и полукарцерандов, изученные в предыдущих разделах, подпадают
под эту категорию. Эти циклофаны мы рассмотрели отдельно, потому что они
обладают рядом свойств, уникальных из-за наличия в их молекулах многочисленных
арильных колец, и, отчасти, потому что они всесторонне изучены (рис. 5.61). В
следующих ниже разделах будет представлен широкий ряд различных синтетических
циклофановых хозяев, сконструированных для связывания органических молекул в
водной и неводной средах. По определению, циклофан-хозяин должен содержать,
по крайней мере, один макроцикл и замыкающие его группы, обладающие
кривизной. Эти внутренне изогнутые компоненты показаны на рис. 5.39, причем хозяева,
содержащие гибриды таких фрагментов, достаточно распространены. Циклофано-
вые хозяева обычно связывают как нейтральные молекулы, так и органические
катионы, причем иногда даже неясно, протонирован ли гость, как, например, в слу-
390
5. Связывание нейтральных молекул
Рис. 5.61. Иллюстрация из журнала «Berichte der Durstigen Chemischen Gescllschafl»
(Берлин, 1886). (а) Круг из обезьян, ухватившихся друг за друга, изображающий структуру
бензола Ф. Кекуле, и (б) шутливый «таутомер» бензола, в котором обезьянам позволено
использовать для связывания хвосты вместо ног. Такому эффекту дали название «хвостовое
остаточное сродство»
чае кислоты-хозяина, образующей с ним водородные связи, или нет. Циклофано-
вые хозяева могут содержать или не содержать молекулярную полость, и в
некоторых случаях такие полости не нужны для достижения высокого сродства к
связыванию, если связывающие центры в молекуле хозяина, обладающие
стереоэлектроннои (т.е. стерическои и электронной) комплементарностью к гостю,
расположены надлежащим образом (предорганизованы). Это может приводить к
связыванию по типу «капсула» или «гнездо» или даже к неполярным
поверхностным взаимодействиям, последние часто ведут к агрегированию (см. рис. 1.4). Тем не
менее встречаются и полости, когда прирост энтропии и энтальпии обусловлен
сферическим или трехмерным капсулированием гостя хозяином, имеющим
сходящиеся связывающие центры.
5-3-4-2 Номенклатура
Если алифатические мостики циклофанов содержат п атомов
углерода, то это число заключают в квадратные скобки перед наименованием «циклофан»
наряду с обозначением (орто-, мета- или пара-), указывающим на характер
замещения в арильном кольце (кольцах). Количество цифр в квадратных скобках означает
число таких мостиков. Так, соединение E.81), впервые полученное независимо
Брауном и Фартингом (С. Brown and A. Farthing, 1949), а также Крамом и Стейнбер-
гом (D. Cram and H. Steinberg, 1951), называют [2.2]парациклофаном. Ка-
ликс[4]арены типа C.97) формально представляют собой замещенные [1.1.1.1]ме-
5.3. Внутриполостные комплексы нейтральных молекул 391
тациклофаны, а циклотривератрилен E.32) — гексаметокси[ 1.1.1 ]ортоциклофан.
Другие примеры показаны ниже.
[2.2]Парациклофан [3.3]Парациклофан [2.2.2]Парациклофан
E.81) E.82) E.83)
5.34-3
Синтез циклофанов обязательно включает образование колец средних
и больших размеров и, следовательно, всегда проблематичен из-за конкурирующих
реакций олигомеризации и полимеризации. Детальное обсуждение
многочисленных методов получения циклофанов выходит за пределы данной книги, но превос-!
ходный обзор можно найти в ключевой ссылке этого раздела. Уместно, однако,
привести обычные методы и особые «хитрости» в этой продвинутой области
органического синтеза. Несколько важных реакций синтеза циклофанов (и
макроциклов вообще) перечислены ниже:
1. Нуклеофильное замещение, включая реакции с участием серы и реакции с ее
удалением. Сера имеет гораздо большую реакционную способность, чем
кислород, вследствие повышенной кислотности группы SH. Промежуточные тио-
эфиры можно окислить в соответствующие S=O- или 5О2-производные,
пиролиз которых приводит к удалению серы с образованием соединений с
С—С-связью, таких, как E.84), схема 5.8.
2. Конденсация аминов с альдегидами. Реакцию диаминов с диальдегидами, иногда
называемую реакцией Шиффа, широко используют при получении макро-
циклических лигандов для катионов металлов. Восстановление
образовавшейся иминогруппы (C=N) боргидридом натрия дает соответствующий
алифатический амин (см. разд. 3.10.4).
3. Превращение со-гидроксикислот в лактоны по реакции Митсонобу (схема 5.9).
Размер кольца и степень олигомеризации зависят от температуры и характера
добавок.
4. Реакции образования углерод-углеродной связи типа реакции Вюрца и
1,6-элиминирование, например, синтез [2.2]парациклофана (схема 5.10).
5. Образование амидов по реакции аминов с хлор ангидридом кислоты, как в
синтезах разнообразных криптандов (схема 3.5).
6. Катализируемое медью сочетание алкинов (схема 5.11).
92
5. Связывание нейтральных молекул
Вг
Na2S
Вг
1. Окисление
2. Пиролиз
E.84)
Схема 5.8. Синтез макроциклов с удалением серы
При синтезе циклофанов наиболее важны темплатные эффекты и применение
высокого разбавления (см. разд. 3.8.2 и 3.8.3; перспективные темплатные эффекты
при самосборке обсуждаются в гл. 7). Высокого разбавления можно достигнуть
даже при гетерогенном разбавлении, когда либо продукты, либо один из реагентов
нерастворимы в реакционной среде. Разбавление при этом достигается путем
протекания реакций только на границах раздела фаз или удаления продукта осаждением.
Синтезу циклофанов способствуют также эффект цезия и принцип жесткой
концевой группы. Эффект цезия состоит в значительном увеличении выхода при ис-
pph'
Лн
Н О
О
О— PPhj"
О.
►V>
■ О = PPh3
Схема 5.9. Превращение со-гидроксикислот в лактоны по реакции Митсонобу
внутриполостнык комплекс
скул
(а)
(СН2)Л
Na
(СН2)„
СН;ВГ
(б)
NMe3
Нагревание
+ Полимер
+ Полимер
Схема 5.10. Синтез простого циклофана (а) по реакции Вюрца
и (б) 1,6-элиминированием
Н CuCi/CuClj
E.85)
Схема 5.11. Катализируемое медью сочетание алкинов
5- Связывание нейтральных молекул
пользовании CsF, Cs2CO3 или CsOH в качестве оснований в реакциях циклизации.
Возможно, что это проявление кинетического темплатного эффекта. Согласно
принципу жесткой группы, по мере укорочения длины цепи исходного соединения
и, следовательно, уменьшения числа степеней свободы реакционной системы
скорость циклизации возрастает. Это просто статистический эффект: чем больше
число возможных конформаций реагента, тем менее вероятна конформация,
подходящая для образования кольца. При введении жестких групп, таких, как арильные
кольца, алкины и т.п., выход циклических продуктов должен увеличиваться. Это
похоже на синтетическую аналогию принципа предорганизации, которая также
проявляется в принципе жесткой концевой группы при комплексообразовании с
участием подандов (см. разд. 3.2.1).
Я"* Breitenbach /., Boosfield /. and Vogtle F. Some general synthetic strategies towards
macrocyclic systems // Comprehensive supramolecular chemistry / Ed. J.L.Atwood,
J.E.D.Davies, D.D.MacNicol and F.Vogtle. Oxford: Pergamon, 1996. Vol. 2.
P. 29-67.
5.3.4.4 От пинцетов к циклофанам
Черты, свойственные всем успешным молекулярным
пинцетам-хозяевам, рассмотренным в разд. 5.3.3, — это достаточно высокая степень структурной
жесткости в сочетании с многоточечным связыванием. Эти черты обычно
выражаются в виде синергетических эффектов 71—Ti-стэкинга и водородного связывания и
приводят к капсулированию гостя. При рассмотрении химии катионсвязываюших
хозяев (см. гл. 3) становится ясной взаимосвязь их жесткой предорганизации и
высоких констант связывания, причем сродство к связыванию возрастает в ряду: по-
данды, коранды, криптанды и сферанды. Эффективность молекулярных пинцетов
можно объяснить ростом подандного связывания в соответствии с принципом
жесткой концевой группы Фёгтле и Вебера (см. разд. 3.2.1), которая увеличивает
степень предорганизации. Ясно, однако, что при переходе от подандоподобных
пинцетов к истинным циклофанам, в которых связывающая полость является частью
всей циклической структуры, следует ожидать дальнейшего усиления связывания
(благодаря дополнительной предорганизации хозяина) и выигрыша в энтропии
(обусловлен снижением степени конформационной свободы хозяина).
Пользуясь понятием «пинцет» применительно к соединению E.80), Э. Джарви
и X. Витлок (Е. Jarvi and H. Whitlock) в 1980 г. получили [8.8]парациклофан E.86),
содержащий два дииновых мостика. Многоточечное связывание осуществляется
путем ион-парного взаимодействия между карбоксилат-анионом и
положительным зарядом 2-нафталинметилентриэтиламмониевого катиона-гостя. Тем не менее
константа связывания этой, очевидно комплементарной, пары составляет лишь
55 М~'. Изучение молекулярных моделей показывает, что такая полость слишком
мала для гостя и не подходит ему. Более того, может существовать и открытая
конформация, не подходящая для большинства л—я-стэкинг-взаимодействий. Для
решения этой проблемы был сконструирован ряд других хозяев, типа E.87), с
большими полостями, которые в некоторых случаях также включали дополнительные
5.3. Внутриполостные комплексы нейтральных молекул
395
предорганизуюшие мостики и акцепторные центры водородного связывания. К
сожалению, константы связывания соединения E.87) в СНСЦ с различными гостями
оказались также низкими. В этом случае полость слишком велика для
эффективного связывания гостя, что приводит к нарушениям двойного п—л-стэкинга. Именно
из-за таких очевидных неудач Витлок признал важность основных взаимодействий
в этой системе и комплементарности хозяин—гость.
В качестве компромисса был сконструирован хозяин E.88). В этом соединении
использованы нафталиновый спейсер и пиридильный остаток, участвующий в
образовании водородной связи с гостем. Один из дииновых мостиков соединений
типа E.86) также заменен более коротким л-ксилильным спейсером для создания
дополнительных л—71-стэкинг-взаимодействий типа «торец к плоскости»
(водородные связи С—H--7I). Константа связывания этого хозяина с
«-нитрофенолом (PNP) уже значительно больше: 9.6-104 М~' в растворе CD2C12. Внутриполост-
ное комплексообразование доказано благодаря анализу химических сдвигов для
гостя в спектре 'Н ЯМР и данных РСА (рис. 5.62).
NEt3
E.86) • гость:
X = СО2"
О
NMe2
О
■О
E.87)
E.88) PNP
396
5. Связывание нейтральных молекул
Рис. 5.62. Кристаллическая
структура л-нитрофенольного
комплекса соединения E.88).
Обратите внимание на водородные
связи С—Н-я и OH---N, а также на
я—я-стэкинг-взаимодействия
{Whitlock B.J. and Whitlock НЖ,
1990)
5-34-5 Фрагмент дифенилметана
Одна из наиболее важных попыток найти синтетические хозяева,
способные связывать органические молекулы в растворах (в частности, водных), —
получение К. Одашимой с сотрудниками (К. Odashima et al., 1980) циклофана E.89) —
это 1,6,20,25-тетрааза[6.1.6.1 ]парациклофан. В соединение E.89) для создания
кривизны на двух сторонах почти прямоугольной макроциклической полости введен
фрагмент дифенилметана. При этом два таких фрагмента соединены гибкими
спейсерами на основе алифатических аминов. При рН < 2 хозяин E.89)
растворяется в воде вследствие протонирования атомов азота. Значительные сдвиги
отмечены в спектре 'Н ЯМР этого соединения в присутствии ряда органических молекул,
в частности молекул ароматических гостей, таких, как 8-анилино-1 -нафталинсульфонат
(ANS, E.90)), за связыванием которого легко проследить с помощью
флуоресцентной спектроскопии, и 2,7-дигидроксинафталин E.91). Такие взаимодействия до
1980 г. наблюдали и в других системах, но главное достижение при изучении
соединения E.89) сводилось к следующему: при добавлении дурола A,2,4,5-тетраметил-
бензол, E.92)) к раствору этого хозяина образуется кристаллическое вещество.
Кристаллическая структура комплекса E.89)-С6Н2Ме4 (РСА) показана на рис. 5.63.
Эта структура - первое определенное свидетельство включения органического
гостя в центр макроциклической полости как раз таким образом, как можно было
ожидать на основании простой молекулярной модели. Следовательно, стало
возможным сконструировать полностью искусственную полость, способную
связывать органические гости в воде. Молекула дурола связана с E.89) посредством п—п-
стэкинг-взаимодействий типа «торец к плоскости» и «плоскость к плоскости»,
причем наименьшее расстояние С--С между метильной группой гостя-дурола и
арильным углеродным атомом хозяина составляет 3.59 А. Соединение E.89) важно
Рис. 5.63. Кристаллическая
структура 1:1-комплекса
E.89)С6Н2Ме4, показывающая
внутри полостное связывание
дурола
5.3. Внутриполостные комплексы нейтральных молекул
397
(СН2L.
Рис. 5.64. Химические
сдвиги в спектре *Н ЯМР
(м.д.) при взаимодействии
E.89), 0.025 М, с E.91) в
разбавленном растворе
НС1. Изменение
химических сдвигов
свидетельствует об аксиальном типе
включения
/ ^^
н2с ^^^
+0.43
н н '
\ /
N+ —С—
/ н2
но
Н7^(сн!L
+0.25
/
с—
н2
^-+0.59
+0.04
+1.90
-0.16
+1.75
еще и потому, что оно подтвердило значение ЯМР-спектроскопии в качестве
одного из инструментов для изучения комплексообразования хозяин—гость в растворе.
Из-за магнитной анизотропии арильных колец при связывании гостя наблюдаются
большие *Н ЯМР-сдвиги. Более того, величина сдвига указывает на близость
протонов гостя к арильным кольцам хозяина (и наоборот) и определяет их
относительную ориентацию (см. Дополнение 3.4.). Изменения в спектре 'Н ЯМР, связанные с
комплексообразованием соединений E.89) и E.91), приведены на рис. 5.64.
Данные 'Н ЯМР свидетельствуют о включении гостя в ориентации, показанной на
этом рисунке, что также подтверждается кристаллической структурой в случае
гостя-дурола.
Н
N.
Дифенилметан
E.89)
о©
E.91)
391
5. Связывание нейтральных молекул
Будучи концептуально крайне важным шагом вперед, синтез хозяина E.89)
предопределил работы многих научных групп по созданию гигантского числа новых и
усовершенствованных систем на основе дифенилметана. Особые проблемы,
связанные с соединением E.89), включали:
1. Необходимость работать в сильнокислых растворах. Растворимость при
нейтральном рН могла бы быть полезной при изучении физиологических систем.
2. Близость полярных аминогрупп, необходимых для растворимости в воде и
работы связывающего центра гостя, затрудняет разделение эффектов: ионного
спаривания (солевые мостики) и гидрофобного связывания. Расположение
этих полярных групп вдали от связывающего центра могло бы улучшить
понимание того, каковы вклады этих эффектов.
3. Полиметиленовые звенья между двумя дифенилметановыми фрагментами
весьма гибки, что снижает предорганизацию хозяина. Более жесткие спейсе-
ры могли бы обеспечить более высокую селективность.
4. Хозяин E.89) ахирален. Введение хиральности могло бы создать условия для
хирального распознавания и асимметрического катализа.
Растворимость E.89) в воде при нейтральном рН была легко достигнута перал-
килированием NH-групп с образованием групп NMet, которые заряжены и пото-
СО
Ме,
лМе
E.94)
5.3. Внутриполостные комплексы нейтральных молекул
му обеспечивают растворимость в воде независимо от рН. Позднее синтезировали
соединение E.93), обладающее солюбилизирующими группами, удаленными от
полости (F. Vogtle et al., 1985), а Ф. Дидерих и К. Дик (F. Diederich and К. Dick, 1982)
получили большое число представителей класса спиросоединении типа E.94).
П. Керни с сотрудниками (P. Kearney et al., 1993), а также Т. Вебб и К. Вилкокс
(Т. Webb and С. Wilcox, 1993), вводя в дифенилметановые фрагменты молекул более
жесткие мостики (соединение E.95)), мостики из основания Трёгера (соединение
E.96)) или создавая из них гибридное соединение E.97), усилили жесткость и пред-
организацию хозяина. В каждом случае усиленные предорганизация и жесткость
обеспечили более эффективное связывание гостя.
В случае всех хозяев, построенных на основе фрагмента дифенилметана, и,
разумеется, большинства циклофановых хозяев связывание органических гостей в
водном растворе прежде всего зависит от гидрофобного эффекта. Органические
О
E.95)
E.96)
SO2
E.97)
40(|| 5. Связывание нейтральных молекул
молекулы, как правило, нерастворимы в воде и ищут убежища в неполярной
полости хозяина, если она стерически соответствует размеру и форме гостя. Константы
связывания изменяются в том же порядке, что и липофильность гостей,
измеряемая коэффициентом распределения гостя в системе октанол—вода. Например,
производное соединения E.94) связывает перилен со свободной энергией связывания
-40.2 кДж-моль~', соответствующей К~ 107. Найдено, что константы связывания
этого хозяина с рядом гостей изменяются в последовательности, обратной
изменению растворимости гостя в воде. Помимо гидрофобных эффектов константы
связывания обычно возрастают при увеличении площади поверхности гостя,
покрываемой хозяином, и можно считать, что энергии связывания содержат аддитивный
ряд вкладов гидрофобных взаимодействий. Связывание алифатических гостей
изучено меньше, но имеющиеся данные указывают, что в этих случаях связывание, в
общем, более слабое. Иногда наблюдается более сильное связывание алкиламмо-
ниевых катионов, причем константы связывания возрастают за счет катион—я-вза-
имодействий.
Следует, однако, соблюдать осторожность при оценке констант связывания в
водном растворе, поскольку связывание гостя может значительно конкурировать с
агрегированием хозяина (т.е. со связыванием одного хозяина другим). Сами
молекулы-хозяева обладают липофильными поверхностями и при определенных
критических концентрациях (называемых критическими концентрациями
агрегирования, critical aggregation concentration, CAC) будут агрегироваться с образованием
мицеллоподобных частиц, обладающих высокой молекулярной массой и плохо
определенной взаимной ориентацией (подобно микроскопическим шарикам масла в
воде). Такое агрегирование существенно влияет на ассоциацию хозяин—гость.
Истинные величины С АС могут лежать в интервале 10~5—10 М. Зависимость
химических сдвигов различных протонов в спектре !Н ЯМР типичного хозяина E.98) от
концентрации показана на рис. 5.65. Ясно, что ниже значения САС, равного ~ 10~2 М,
химический сдвиг не зависит от концентрации, и это наилучший
концентрационный интервал для измерения связывания гостя. При более высоких концентрациях
Me, ч Me
MeN Me E.98)
5.3. Внутриполостные комплексы нейтральных молекул
401
Рис. 5.65. Химические сдвиги различных
протонов хозяина E.98) как функции его
концентрации. (Воспроизведено
с разрешения Diederich F., 1988)
5
7 00
6.80-
6.60-
4.00-
3.80-
3.40-
3.20 :
2.60-
2.40-
2.20-
2.00-
1.80-
\
\
10 1(Г3 10 10"'
lgcH , моль- л 1
Н-10
Н-2
Н-2"
Н-3"
Аг-Ме
наблюдаются большие спектральные изменения, связанные с агрегированием,
даже в отсутствие частиц гостя (F. Diederich, 1988).
5-3-4-6 Циклофановые клетки в водном и неводных растворителях
Поскольку покрытие гидрофобных поверхностей гостя - очень
важный фактор, определяющий его связывание, С. Фергюсон с сотрудниками
(S.Ferguson et al., 1991) получил трехмерный хозяин E.99), способный связывать
неполярные гости типа пирена (показан) трехмерным образом, аналогично
связыванию катионов криптандами. Хозяин E.99) растворим в разнообразных
растворителях — от полярных до неполярных, и связывание пирена изучали
калориметрически в каждом из них. В общем, его способность к растворению возникает из двух
совершенно различных источников. Во-первых, взаимодействие липофильных
поверхностей арильных колец дифенилметанового фрагмента с полярной средой
типа воды приводит к выигрышу полной энтропии системы, благоприятствуя
связыванию гостя. Второй эффект называют когезионным, он объясняется притяжением
одной молекулы воды (растворитель) к другой за счет водородных связей.
Взаимодействие хозяин—гость приводит к образованию только одной «дырки» в молекуле
растворителя вместо двух и, следовательно, обеспечивает выигрыш энергии.
Калориметрические исследования Фергюсона и его сотрудников показали, что когези-
онный вклад преобладает. Найдено, что свободная энергия комплексообразования
линейно коррелирует с полярностью растворителя. Константы связывания
изменялись от 6-Ю6 в системе вода/DMSO (99:1) до 9-Ю10 в CS2, причем свободная
энергия комплексообразования линейно растет с ростом полярности растворителя.
402
5. Связывание нейтральных молекул
Полярность растворителя количественно определяется эмпирическим параметром
/^-(ЗО) Димрота—Рейхарда (ЕТ— энергия перехода Димрота—Рейхарда для бетаина
в ккалмоль), который связан с макроскопическими свойствами, такими, как
диэлектрическая постоянная, дипольный момент, когезия и поляризуемость
(С. Reichard, 1988).
Et—N
•Et
Et
E.99) пирен
5.3.4.7 Включение гостя с помощью водородных связей
Отмеченная в предыдущем разделе линейная зависимость энергии
комплексообразования от полярности растворителя, наблюдаемая при связывании
неполярных гостей хозяевами типа E.99), частично объясняется отсутствием
специфических, энтальпийно стабилизирующих, взаимодействий хозяин—гость.
Однако мы уже видели, что комбинация п—л-стэкинг-взаимодействий и водородных
связей увеличивает энтальпийный вклад стабильности комплекса хозяин—гость
при связывании л-нитрофенола соединением E.88). Этот сложный хозяин
чрезвычайно эффективен, потому что он стерически и электронно комплементарен
своему гостю-мишени, а также и жестко предорганизован. Дополнительная
стабилизация возникает из-за многоточечной комбинации взаимодействий. Аналогично
хозяин — молекулярный пинцет E.74) — довольно сильно связывает частицу гостя
в неполярной среде посредством водородных связей. Действительно,
термодинамический подход к связыванию должен также учитывать наличие воды из-за
конкурентной сольватации связывающих центров гостя (разд. 5.3.5). Отсюда -
связывание гостей циклофанами в неполярных средах требует специфической стратегии
при конструировании хозяина: такая стратегия может быть основана на
обеспечении сильного притяжения между хозяином и гостем (рис. 5.66).
Одна из наиболее эффективных ранних «конструкций», подходящих для
включения гостя в неполярных средах с помощью водородных связей, предложена
С.-К. Чангом и А. Гамильтоном (S.-K. Chang and A. Hamilton, 1988) из
Университета Питтсбурга (США). Признавая возможность многоточечных взаимодействий,
предоставляемых семейством барбитуратов E.100) (которые сами по себе являются
5.3. Внутриполостные комплексы нейтральных молекул
403
Хо5йШ1 без полярного или способного
1С образованию водородных связей центра
Гость 6ei полярного или способного
к. образованию» водородных связей центра
Полярный ^
растворитель \ (б)
(8)
Неполярный
растворитель
Хозяин с полярным или способным Гость с полярным или спосооным
к образованию водородных связей центром к образованию водородных связей центром
Рис. 5.66. Связывание полярного и неполярного гостей в полярных и неполярных средах
хозяевами различных типов, (а) Гидрофобная полость хозяина, связывающая неполярный
гость: небольшой выигрыш энтальпии вследствие легкой десольватации и ван-дер-
ваальсовых притяжений; большой выигрыш энтропии вследствие гидрофобного эффекта,
(б) Полость хозяина с полярными центрами, связывающая неполярный гость: менее
устойчивый комплекс вследствие неблагоприятной десольватации функциональных групп
гостя, (в) Полость хозяина с полярными центрами, связывающая полярный гость: более
устойчивый комплекс, так как неблагоприятная десольватация функциональных групп
хозяина уравновешивается его взаимодействием с гостем, (г) Полость хозяина с полярными
центрами, связывающая полярный гость в неполярном растворителе: устойчивый комплекс
вследствие благоприятных взаимодействий хозяин—гость; сольватофобная стабилизация
отсутствует
интересными гостями и находят широкое клиническое применение в качестве се-
дативов и антиконвульсантов), эти исследователи сконструировали
комплементарный макроциклический хозяин E.101) и его ациклический аналог E.102); ср. с
комплексом каликсарена E.65). Результаты были чрезвычайно обнадеживающими.
Изучение комплексообразования E.101) с E.100) в растворе хлороформа методом
ЯМР ясно показало, что барбитал E.1006) (К= 1.37-106 М) связывается более
селективно по сравнению с более объемистым E.100г) (К = 6.80-102 М~'). При этом
была выявлена большая эффективность предорганизованного макроциклического
хозяина E.101) по сравнению с ациклическим E.102) (барбитал, К= 2.08-104 М").
Близкие результаты получены и для родственных соединению E.102) хозяев и ди-
карбоновых кислот в качестве гостей, причем сила связывания зависит от длины
404%
5. Связывание нейтральных молекул
молекулы дикарбоновой кислоты. Эта концепция использована при разработке
прототипа редокс-сенсора для дикарбоновых кислот (разд. 8.3.3).
(Г N
о
E.102):
E.100). E.101)
(a): R1 = R2 = R3 = Н R3 = СО(СН2JМе
F):X = R2 = Et,R3 = H
(в): X = Et, R2 = Ph, R3 = Н
(r):X = Et,R2 = Ph,R3 = Me
В работе К. Хантера (С. Hunter, 1991) для связывания акцептора водородной
связи - л-бензохинона - привлечены множественные водородные связи и
множественные л—л-стэкинг-взаимодействия, что привело к конструированию и синтезу
рецептора E.101), включающего комплементарный набор из четырех доноров
водородных связей (NH-группы, по одной на каждую неподеленную электронную
пару хинона) и дифенилметановые тг-поверхности (схема 5.12). Получение
соединения E.103) осложнено образованием с большими выходами аналогичных
циклических тримера, тетрамера и даже [2]катенана, в котором две молекулы
E.103) сцеплены механически (разд. 7.5). Однако применение постадийного
синтеза позволило выделить это макроциклическое соединение с приемлемым выходом.
Константы связывания л-бензохинона хозяевами E.103) приведены в табл. 5.8.
Наблюдалось сильное связывание гостя-хинона, что, например, следует из
сдвигов амидных протонов в спектре [Н ЯМР вплоть до 2.5 м.д. и характерно для
водородного связывания хинона. Хиноны с большим числом заместителей типа те-
траметилхинона, которые не соответствуют полости хозяина, вообще не
связываются. Типичный потенциал восстановления хинона также сдвигается в
положительную сторону примерно на 200 мВ, отражая сильный поляризующий эффект
амидных NH-связей. Наиболее вероятный способ связывания этого хинона
схематически показан на рис. 5.67.
5.3. Внутриполостные комплексы нейтральных молекул
405
NEt3, CH2C12
Высокое
разбавление
E.103)
л-л
л-л.
Л—Л
Н-СВЯЗЬ ,f
Н-связь хЧ
vН-связь
'Н-связь
л-л
л-л
л-л
Схема 5.12. Синтез рецептора хинонов E.103) [X,Y = CH или N, Hunter С, 1991:
R = N+Me2Cl-, Alott С. et al., 1998]
Таблица 5.8. Константы связывания
гостя-хинона хозяевами E.103) в CDC13
Хозяин E.103)
К, М-1
X=Y=CH
X = N,Y=CH
X = CH,Y = N
1200
1800
230
В более поздней работе К. Алотт с сотрудниками (С. Allot et al., 1998) также
показал, что хозяин E.103), R = N+Me2Cl~, способный образовывать водородные
связи, может быть использован для комплексообразования органических гостей
406
5. Связывание нейтральных молекул
О
о
О
Рис. 5.67. Связывание
бензохинона хозяином E.103)
(R = СН2, заместители для
простоты опущены)
в воде (ср. гидрофобное распознавание для других соединений на основе дифенил-
метана). Оказалось, что константа связывания для и-хинона в воде составляет
менее 5 М~', а в хлороформе — 230 М~'. Тем не менее увеличение в молекуле гостя
числа групп, способных давать водородные связи, привело к значительному росту
их констант связывания, измеренных как в воде, так и в хлороформе (табл. 5.9).
Таблица 5.9. Константы связывания гостей хозяевами E.103)
(R = N+Me2Cl", no Aloft С. et al., 1998)
Гость
К, М-1 (вода)
К, М (хлороформ)
<5
94
760
230
850
Подход, использующий представление о водородных связях, был также развит
для комплексообразования доноров таких связей типа 1,3,5-тригидроксибензола
(ТНВ, флороглюцин). Основываясь на реакции конденсации хлорангидридов
кислот с аминами, К. Зеел и Ф. Фёгтле (С. Seel and F. Vogtle, 1992) получили ряд
огромных цилиндрических циклофанов типа E.104) с тремя мостиками. Хозяин E.104)
5.3. Внутриполостные комплексы нейтральных молекул
407
имеет константу связывания флороглюцина в растворе СН2С12 ~ 1.1 • 104 М, причем
комплексообразование протекает посредством бифуркационных водородных связей
О—H---N и более слабых взаимодействий Ar—H--N. Меньшие производные
оказались полезными комплексообразующими агентами для металлоцентров типа Ru2+
(ср. [ЫиB,2'-бипиридилK]2+), а производные, содержащие пирокатехиновые (о-ди-
гидроксибензольные) единицы, привели к получению эффективных сидерандов,
используемых для связывания Fe(II), например C.121), см. разд. 3.15.2.
E.104) ТНВ
5*3*5 Конструирование хозяина из клатратобразующих
строительных блоков для образования
комплексов в растворе: криптофаны
9-1 Collet A., Dutasta J.-R, Lozach В. and CanceilJ. Cyclotriveratrilenes and crypto-
phanes: Their synthesis and applications to host-guest chemistry and to the design of
new materials // Top. Curr. Chem. 1993. Vol. 165. P. 103-130.
5.3.5.1 Конструкция молекулярных контейнеров
из изогнутых строительных блоков
Мы уже рассматривали химию включения твердого состояния цикло-
тривератрилена (CTV, E.32); см. разд. 5.2.5). CTV обладает мелкой молекулярной
полостью в форме блюдца, которая была использована для связывания анионов в
408
5. Связывание нейтральных молекул
растворе металлоорганическими производными CTV (см. разд. 4.6). Однако в случае
нейтральных гостей CTV не проявляет значительной связывающей способности в
растворе, что согласуется с канальной природой его включений в твердом
состоянии. Единственные исключения из этого правила — внутриполостное комплексооб-
разование бакминстерфуллерена (С60) CTV-хозяином и аналогичный комплекс со
сферическим карбораном о-С2В10Н12 E.40). Это свидетельствует об эффективном
связывании в неводных растворителях, хотя данные по титрованию методом 'Н
ЯМР в случае карборана не соответствуют модели 1:1 (R. Blanch et al., 1997).
Отсутствие у CTV способности к связыванию в растворе не является большим
недостатком, поскольку эта молекула получается легко, а метоксизаместители
предоставляют большие возможности для дальнейшего усовершенствования ее
молекулярной полости. Более того, кривизна блюдцеобразной структуры —
потенциально ценное свойство при построении капсулирующих хозяев. Причина того, что сам
CTV — плохой хозяин, состоит в малом объеме его полости. Из-за большого
диаметра чаши CTV ее полость не может служить органическим соединениям хорошим
убежищем от воздействия среды полярного растворителя. В то же время арильные
кольца способны лишь к слабым п—л-стэкинг-взаимодействиям или л-водород-
ным связям с гостями, как это имеет место в случае гостя E.40). Для достижения
эффективного комплексообразования в растворе гостей, отличающихся от тех,
которые, подобно С60, высококомплементарны к CTV, безусловно, необходимо
усовершенствовать молекулярную полость CTV. Это осуществил Д. Крам (D. Cram,
1988b), получив ряд кавитандов на основе CTV (схема 5.13).
Кавитанд E.106), а также его пиридильный аналог и аналог с фенантролиновы-
ми мостиками обладают глубокими полостями, способными связывать нейтраль-
E.105)
CTV E.32)
Me2SO или
HCONMej
E.106)
Схема 5.13. Синтез кавитандов на основе CTV по Краму {Cram D., 1988b)
5.3. Внутриполостные комплексы нейтральных молекул 409
ные молекулы типа СН2С12 как в растворе, так и в твердом состоянии. В свободном
E.106) л/-ксилильные мостики очень гибки, однако эта гибкость значительно
снижается при образовании комплекса с гостем. Молекулярно-механические расчеты
предполагают, что кавитат E.106)СН2С12 устойчивее свободного хозяина на
57.3 кДжмоль", хотя эту большую величину нельзя сопоставлять с константой
связывания в растворе, поскольку расчет не принимает во внимание влияния среды.
Ясно, что углубление полости CTV с образованием кавитандов
высокоэффективно для усиления комплексообразующих свойств соединений на основе CTV в
растворе. Тем не менее в случае криптандов мы видели (см. гл. 3), что сродство
хозяин—гость возрастает еще более при наличии у криптанда закрытой трехмерной
полости, которая может полностью капсулировать частицу гостя. Эта трехмерность
обеспечивает предорганизацию, защищает гостя от среды растворителя и
замедляет кинетику обмена гостя. А. Колле (A. Collet) получил ряд трехмерных хозяев на
основе CTV, в которых две полости типа CTV обращены друг к другу и ковалентно
связаны мостиками -(СИ2)п-, -СН2-СН=СН-СН2- или -СН2-ОС-СН2-
(п = 3+8). Аналоги с арильными мостиками были также получены путем
перекрывания мостиками подандных анионсвязывающих хозяев типа D.36) на основе
CpFe+ (см. разд. 4.6). Эти капсулярные хозяева назвали криптофанами по аналогии
с криптандами (окончание «-фан» означает, что они принадлежат к классу [1.1.1 ]ор-
тоциклофанов; см. разд. 5.3.4.2 о номенклатуре циклофанов). Криптофаны
существуют в двух разновидностях с анти-EА01) и о/«-E.108)ориентациями
заместителей двух полусфер, обычно имеющих симметрию Dyanmu- или С^-син-. Названия
криптофанам присваивали по мере их открытия. Так, криптофан-А и криптофан-В —
это анти- и о/«-производные с мостиками X = -(СН2J-, а криптофаны-Е и -F с
мостиками X = -(СН2K-. В криптофанах-С и -D имеются мостики -(СН2J-, как
и криптофанах-А и -В, но отсутствуют ОМе-группы на полусферах. Геометрия
«поверхность к поверхности» характерна и для недавно описанных соединений
включения самого CTV в у-фазе (разд. 5.2.5.2).
Криптофаны синтезируют двумя методами. В начальных стадиях первого из них
используют ковалентный темплатный эффект, когда одна чаша производного CTV
предорганизует циклизацию второй в условиях высокого разбавления (схема 5.14, а).
Схема 5.14. Методы синтеза криптофанов. (Воспроизведено с разрешения Collet А.
et al. Cyclotriveratrylenes and cryptophanes: Their synthesis and applications to host-guest
chemistry and to the design of new materials //Top. Curr. Chem. 1993. Vol. 165. P. 103)
410
5. Связывание нейтральных молекул
Dyaumu- E.107) Сжсш- E.108)
X = -(СН2)„-, -СН2-СН=СН-СН2-, алкин, С6Н4
НО2С
СО2Н
НО2С
E.109)
Недавно разработан более прямой метод, когда криптофаны образуются
непосредственно в процессе двойной тримеризации (схема 5.14, б).
Выходы различных криптофанов, полученных по этим методам, показаны в
табл. 5.10. Ясно, что прямой метод приводит всегда к более низким выходам,
вероятно, потому, что он основан на тройном замыкании больших циклов как части
трехкомпонентной самосборки без значительного темплатного содействия.
Конкуренцию получению продукта по этому методу составляет образование полимеров.
Тем не менее этот метод не требует условий высокого разбавления (см. разд. 3.8.3) и
поэтому позволяет получать большее количество продукта за то же время. К тому
же, он гораздо более прямой. Более интересно, что метод (б) приводит к совсем
другому шшш/син-соотношению, нежели темплатный метод {а). В методе (а) анти/син-
соотношение смещено в сторону ^www-конфигураций для мостиков с четным чис-
5.3. Внутриполостные комплексы нейтральных молекул
411
Таблица 5.10. Выходы (%) криптофанов, полученных методами,
показанными на схеме 5.14
X в мостике О—X—О
-(СН2J-
-(СН2K-
-(СН2L-
-(СН2M-
-(СН2N-
-(СН2O-
—(СН2)8—
-СН2-СН=СН-СН2- (£-)
-СН2-СН=СН-СН2- (Z-)
-сн2-а=с-сн2-
Темплатный метод (а)
анти-
80
27
-
21
18
-
60
34
25
43
син-
0
50
-
43
9
-
20
5
50
20
Прямой метод (Ь)
анти-
5
17
8
12
8
5
0
5
9
0
син-
0
3
2
6
2
1
0
1
8
0
лом атомов углерода и в сторону сын-конфигураций для мостиков с нечетным
числом. Поскольку изменение конфигурации двойной связи от цис- к транс- имеет тот
же эффект, то предполагается, что мостики определяют предпочтительную
ориентацию в предшественнике. Прямой подход всегда дает в основном анти-изомер.
Вероятно, это следствие того, что в методе (б) две части производного CTV цикли-
зуются согласованно, причем оба кольца образуются одновременно.
5-3-5-2 Образование комплексов с галогенуглеводородами
Криптофаны, в соответствии с их капсулирующей природой,
обладают очень высокой комплексообразующей способностью для небольших тетраэдри-
ческих молекул, таких, как метан и его галогенпроизводные. Спектр 1Н ЯМР крип-
тофана-Е в присутствии двух эквивалентов СНС13 показан на рис. 5.68. Спектр
записан в дейтерированном 1,1,2,2-тетрахлорэтане, молекула которого слишком
велика, чтобы соответствовать полости криптофана-Е. Резкие пики связанных и
внеполостных молекул СНС13 легко наблюдаются при комнатной температуре. По
мере повышения температуры до 340 К они постепенно уширяются, поскольку
обмен гостя становится быстрым на шкале времен ЯМР. Высокопольный химический
сдвиг включенного гостя - характерное следствие экранирующих эффектов ариль-
ных колец. Наблюдаемый медленный обмен согласуется с необходимостью для
гостя протиснуться сквозь «окна» хозяина. Анализ формы линий этого хозяина дает
энергии активации обмена гостя в интервале 54-63 кДж-моль. При 300 К
константа связывания составляет 470 М~', т.е. является довольно высокой для слабых
сил связывания, если учесть отсутствие гидрофобных эффектов в среде
неполярного растворителя. Это следует также из большой величины константы связывания —
7700 М~' — хлороформа в воде родственным соединению E.108) водорастворимым
хозяином E.109), в котором заместители ОМе заменены гидрофильными группами
осн2со2н.
412
5. Связывание нейтральных молекул
н,со -, ббсн, хД
S ОСИ, | Г
о Т \ ? ? осн,
сна
Растворитель
_л
J* fj
340 К
СНС1,
300 К
7.5
7.0
6.5
6.0
5.5
5.0
4.5
4.0
3.5
3.0
2.5
2.0
Рис. 5.68. Спектр 'Н ЯМР криптофана-Е (E.107), X = (СН2K) в присутствии двух
эквивалентов СНС13; растворитель (CDC12J- (Воспроизведено с разрешения CanceillJ.
etal., 1986)
Свободные энергии комплексообразования различных нейтральных гостей
криптофанами-С и -Е приведены на рис. 5.69. Как можно было ожидать исходя из
их трехмерной природы, эти хозяева показывают пики селективности с хорошей
корреляцией между размером полости хозяина и ван-дер-ваальсовым объемом
гостя. Так, меньший криптофан, криптофан-С с мостиками —(СН2J—, явно
селективно связывает меньшие гости типа СН2С12, а больший гость СНС13 связывается
более прочно криптофаном-Е, который имеет мостики —(СН2K. Однако это простое
представление о соответствии размера гостя размеру полости хозяина терпит
неудачу, если свободные энергии комплексообразования рассматривать с учетом энталь-
пийного и энтропийного вкладов. Вклады можно разделить с помощью графика
Вант-Гоффа (зависимость lg К от \/Т). Параметры связывания СН2С12 и СНС13
криптофаном-С и большим криптофаном-Е приведены в табл. 5.11.
Тот факт, что меньший криптофан — криптофан-С - селективен к СН2С12, не
определяется исключительно соответствием малого размера полости хозяина и
размера гостя. Энтальпия взаимодействия СНС13 с этим хозяином имеет гораздо
большую величину, несмотря на его (хозяина) малый размер. Однако связывание СНС13
Таблица 5.11. Термодинамические параметры связывания гостя криптофанами
в (CDC12J при 300 К
Хозяин
Криптофан-С
Криптофан- Е
Гость
СН2С12
СНС13
СН2С12
СНС13
ДС°, кДж моль
-15.1
-6.7
-11.7
-15.5
ЛЯ, кДжмоль
-16.3
-26.8
+4.2
-25.1
Д5, Дж К-'моль
_4
-67
+25
-29
5.3. Внутриполостные комплексы нейтральных молекул
20
413
15 "
10.
5 -
о-
Рис. 5.69. Свободная
энергия связывания
различных гостей
криптофана.ми-С
и -Е при 300 К
-5
СН2С1
СНС12Вг
д СНВг2С1
Д СНВг3
♦ Кргаггофан-С
д Криптофан-Е
СС14
С]3ССН3
С12С(СН3J
С1С(СН3K
50
60
70 80
Объем гостя, А3
90
100
существенно дестабилизировано энтропийным вкладом: слагаемое — 7Д5в
уравнении для энергии комплексообразования составляет +20.1 кДжмоль при 300 К.
Возможно, это следствие слишком точного соответствия СНС13 полости: в ней он
удерживается очень жестко и, таким образом, число его степеней свободы резко
уменьшено. С другой стороны, СНС13 также прочно связывается криптофаном-Е,
но в большем хозяине есть место для некоторого движения, что значительно
уменьшает энтропийную дестабилизацию. Кристаллическая структура комплекса крип-
тофана-Е с СНС13 показана на рис. 5.70.
Первоначально криптофаны создавали, чтобы решить давнюю проблему
разделения энантиомеров одного из простейших хиральных соединений — фторхлор-
бромметана CHFClBr. Эта тетраэдрическая молекула имеет подходящий размер,
соответствующий полости криптофана, а наличие у /K-ш/яш-криптофанов ВНУТ"
ренней хиральности означает, что с их помощью такое хиральное распознавание
возможно. Используя частично разделенный образец (±)-CHFClBr, Колле (Collet)
с сотрудниками изучал его комплексообразование с разделенным образцом
л\л . 5. Связывание нейтральных молекул
Рис. 5.70. Кристаллическая
структура комплекса криптофана-Е
с СНС13
(+)-криптофана-С. Отдельные пики наблюдали для сигналов гостя двух диастерео-
мерных комплексов хозяин—гость: (+)-хозяин—(+)-гость и (+)-хозяин—(—)-гость.
Интегрирование показало, что образец имел 4.5%-й энантиомерный избыток, а это
в сочетании с данными кругового дихроизма дало оптическое вращение гостя
(а)^5 = 1.6±0.5 — информация, которую искали целое столетие.
5*3*5-3 Конкуренция с растворителем
Первые исследования комплексообразующей способности криптофа-
нов были проведены в растворителе CDC13 с родственными гостями типа СН2С12,
при этом получили чрезвычайно малые константы связывания — ~1—2 М.
Отсюда следовал вывод о существовании сильного эффекта сольватации при капсулиро-
вании гостя. В случае связывания гостя в воде рост эффективности комплексообра-
зования обусловлен гидрофобными эффектами, возникающими в результате
образования водородных связей вода—вода. Однако если растворитель является
эффективным конкурентом для включения в полость хозяина, то константы
связывания сильно уменьшаются, как в случае связывания СН2С12 в CDC13. Этого следует
ожидать из-за высокого сродства криптофановых хозяев - криптофанов-С и -Е — к
обоим этим соединениям (см. рис. 5.69), что приводит к получению константы
равновесия, являющейся концентрационно-взвешенной мерой относительного
сродства хозяина к гостю и к растворителю. Так, константа комплексообразования
А'дается уравнением E.4), рис. 5.71:
K=KU(\+KS[S)), E.4)
где Ки - константа связывания гостя, Ks - константа равновесия при связывании
растворителя, [S] — мольная концентрация (активность) растворителя.
Ясно, что К~ Кп только когда A^[S] << 1. Вообще для органических
растворителей [S] = 10 М и, следовательно, /^должна быть меньше 10~2. В случае криптофа-
5.3. Внутриполостные комплексы нейтральных молекул 415
*<£ # О
*А*
Хозяин
Гость
Растворитель
▼ Рис. 5.71. Равновесие
конкуренции растворителя
на-С константа комплексообразования АГдля СН2С12 в CDC13 равна 2.6 дм3-моль~'.
Для CDC13 при комнатной температуре [S] = 12.4 моль-дм. Измерения в
неконкурирующих растворителях типа (CDC12J показывают, что CDC13 связывается крип-
тофанами с Ks = 10 дм3-моль~'. Применение уравнения E.4) для СН2С12 дает Кп,
равную -325 М. Ясно, что эффект среды имеет первостепенное значение.
5.3.5*4 Образование комплексов с ионами алкиламмония
Одна из наиболее замечательных черт криптофанов — их способность
очень эффективно связывать органические аммониевые катионы в неполярных
средах. Тетраметиламмоний-катион (NMe^) связывается криптофаном-Е с
константой 250 000 дм3-моль~' в (CDC12J. Напротив, нейтральный гость СС1Ме3
близкого размера связывается с трудом. Ясно, что в случае катионного гостя связывание
определяется другими, более важными причинами. Сильное связывание
свойственно также ряду других алкиламмониевых катионов, причем константа
комплексообразования уменьшается с увеличением размера алкильных групп. Сильное
связывание такого катиона может быть следствием его намного более гидрофильной
природы. Если полость хозяина представляет собой более полярную «сольватную
оболочку», чем весьма неполярный растворитель, то энтальпийный вклад будет
благоприятным. Заметим, что, в противоположность соединениям типа D.63),
близкие им по величине анионы типа BF7 не связываются вообще. Интересно
jjg 5. Связывание нейтральных молекул
также сравнить способность криптофанов связывать катионы с таковой для спеле-
андов (см. разд. 3.11.5), молекулы которых также содержат каркас CTV. Однако в
последнем случае взаимодействия хозяин—гость определяются направленными
водородные связями.
5-3-5-5 Образование комплексов с метаном
А. Колле и сотрудники (Collet et al.) изучили также комплексообразу-
ющую способность малого криптофана — криптофана-А — по отношению к метану.
Метан, со своей небольшой молекулой, имеет важное значение не только из-за
того, что он входит в состав природного газа угольных и нефтяных месторождений.
Метан чрезвычайно трудно связать, поскольку он высокосимметричен, незаряжен,
неполярен и способен взаимодействовать со своим окружением только
посредством слабых сил Ван-дер-Ваальса. Эти сложности усугубляются его чрезвычайно
высокой летучестью, что создает трудности при изучении комплексообразования
метана в растворе. Установлено, что криптофан-А высокоэффективен при
связывании метана, причем свободные энергии комплексообразования при 300 К
сравнимы с энергиями связывания СНС13 тем же хозяином, несмотря на
значительное различие их молекулярных объемов. Связывание метана в (CDC12J
сопровождается благоприятным изменением как энтальпийного, так и энтропийного
вкладов: Д#° = -6.7 кДж-моль и А5 = +17 Дж-Кг'моль. Константа
связывания составляет 130 М~', а капсулированный метан имеет достаточно большое
время полужизни - 9 мс - при 199 К. Это означает, что в спектре 'Н ЯМР при такой
температуре можно наблюдать разделенные пики свободного и связанного гостей.
Свободный метан резонирует при ~0.2 м.д., а связанная молекула СН4 дает сигнал
при -4.4. м.д. При 298 К наблюдается усредненный сигнал. Молекулярное
моделирование этой структуры показывает, что среднее расстояние Сгость---Схозяин
составляет ~4.5 Л, т.е. оно на 20% больше, чем сумма ван-дер-ваальсовых радиусов. На
этом расстоянии силы притяжения в потенциале притяжения Ван-дер-Ваальса
достигают максимального значения, что может быть единственной причиной
экстраординарной стабильности этого комплекса.
Соотношение между размерами полости и гостя в терминах их молекулярных
объемов можно выразить фактором занятости полости (р). Величина р = 1
представляет целиком заполненную полость. Факторы занятости для различных крип-
тофановых комплексов приведены в табл. 5.12.
Таблица 5.12. Факторы занятости (р) для криптофановых комплексов*
Гость
сн4
сн2а2
СН2Вг2
СНС13
* Объемы
Криптофан-А
0.35
0.70
0.80
0,89
полостей для криптофанов-А и
Криптофан-С
0.70
0.89
-С равны 81.5 h
Криптофан-Е
0.65
0.81
i, a для криптофана-Е - 89.0 А.
5.3. Внутриполостные комплексы нейтральных молекул 417
Ясно, что метан гораздо менее соответствует полости криптофана, чем галоген-
алканы. Интересно, однако, сравнить полученную величину р @.35) с параметрами
ряда других «состояний материи» метана. Газообразный метан, например, имеет
р = 0.77-10~3 при 1 атм и 298 К. В критической точке D5.6 атм, 190.6 К,
температура и давление, при которых нет различий между жидкостью и газом) эта величина
падает до 0.17, а твердый метан при 0 К имеет р = 0.67. Тогда в терминах занятости
состояние внутри полостного окружения метана близко к сверхкритической
жидкости. При макроскопическом рассмотрении одна молекула метана в полости крип-
тофана-А эквивалентна одному молю метана в 49 мл при 610 атм и 298 К. Напротив,
в случае СНС13 фактор занятости р = 0.89 соответствует очень плотноупакованно-
му кристаллу и, действительно, изменения энтропии и энтальпии при связывании
СНС13 подобны изменениям этих параметров при образовании кристаллических
органических соединений.
5*3*6
Ковалентные полости:
карцеранды и полукарцеранды
Я Jasat A. and Sherman J.C. Carceplexes and hemicarceplexes // Cbem. Rev. 1999.
Vol. 99. P. 931-967.
5.3.6.1 Определения и синтез
Карцеранд — это закрытый молекулярный контейнер, или капсула без
отверстий значительного размера, через которые гости могли бы входить или
выходить. Частица гостя , следовательно, постоянно удерживается («посажена в
карцер») во внутреннем объеме карцеранда, если не произойдет разрыва ковалентной
связи в хозяине. Карцеранд, который содержит гость, т.е. образует комплекс
хозяин-гость, носит название карцеплекс. Аналоги карцерандов, имеющие
молекулярные контейнеры, которые допускают вход и выход гостя с конечным активацион-
ным барьером, называют полукарцерандами. В присутствии частиц гостя
полукарцеранды образуют полукарцеплексы. Хорошими примерами полукарцеран-
дов являются криптофаны (см. разд. 5.3.5), которые способны обратимо
захватывать относительно малые гости, например метан, галогеналканы и т.п. Интерес к
этому виду химии трехмерного включения в основном обусловлен возможностью
стабилизации реакционноспособных частиц гостя внутри полости хозяина,
доставки лекарств, внутриполостного катализа. Реализация этих процессов требует, чтобы
полукарцеранды были способны селективно связывать и выталкивать частицы
гостя в ответ на внешние условия. Сами карцеранды не могут быть использованы в
этих процессах, хотя можно предвидеть их применение в молекулярной
электронике и молекулярных устройствах.
Криптофаны — очень перспективные хозяева для связывания отдельных
молекул гостя, однако объем их полостей (80-90 А3) слишком мал для одновременного
связывания двух частиц гостя, что исключает возможность протекания внутрипо-
лостных реакций и катализа. Работа Д. Крама с сотр. (D. Cram et al., 1985b) была
27 - СТИД Дж.В
418
5. Связывание нейтральных молекул
С1Н2С
CH2SH
E.110)
Me2NCHO / THF
CsjCO3.Ar
E.111)
Северный полюс
Тропики
S Экваториальный район
Тропики
Южный полюс
Me
E.112)
Схема 5.15. Синтез первого карцеранда E.112) по Краму (Cram D. et al., 1985b)
посвящена большим [4]резорцинаренам, родственным соединению E.43).
Применив подобную стратегию для синтеза криптофанов, Крам сумел соединить верхние
ободы двух резорцинареновых чаш E.110) и E.111) в условиях высокого разбавления
и получить псевдосферическую капсулу E.112), содержащую полость (схема 5.15).
Карцеранд E.112) был создан группой Крама после окончания работ по
конструированию сферандов с помощью молекулярных моделей СРК. Через два года
после получения соединения E.112) Крам в нобелевской лекции так описывает свой
интерес к химии включения этой новой «капсулы» (Cram et al., 1988a):
5.3. Внутриполостные комплексы нейтральных молекул
419
«Сначала нужно было ответить на следующий вопрос: какое соединение-гость могла бы
захватить капсула при замыкании? Это все равно, что спросить: может ли котел с
тушенкой, содержащий две суповые миски, сомкнутые ободок к ободку под
поверхностью котла, вместить еще сколько-то тушенки. Ответ был таков: соединение E.112,)
"содержало "все компоненты среды, присутствовавшие при образовании этой капсулы».
Продукт реакции (см. схему 5.15) очень плохо растворялся во всех
растворителях, и поэтому его исследование потребовало много усилий. Примеси и непрореа-
гировавшие исходные соединения экстрагировали, обрабатывая смесь веществ
наиболее сильными растворителями разных типов (полярный, неполярный,
образующий водородные связи, полярный апротонный и т.д.). Оставшуюся смесь
(различные карцеплексы, т.е. комплексы хозяин—гость соединения E.112))
подвергли элементному анализу на С, Н, S, О, N, Cl, Cs и определили, что каждый из
этих элементов содержится в нем в различных количествах при стехиометрическом
соотношении Cs и С1. По характеристической частоте связи С=О в ИК-спектре
установили, что в твердой смеси присутствует растворитель реакции — Me2NCHO
(DMF). Масс-спектрометрический анализ этой смеси (бомбардировка быстрыми
атомами) позволил обнаружить наличие всех карцеплексов, показанных на
рис. 5.72. Никаких других пиков, в том числе соответствующих молекулярным
массам, большим, чем у самого тяжелого карцеплекса, не обнаружено. Ясно, что как
только кольцо замкнулось, любые частицы, включая аргон, присутствующие во
внутренней области образовавшегося карцеранда, не способны его покинуть.
Крам и сотрудники обнаружили, что тщательное высушивание гидратирован-
ных комплексов с последующим кипячением в D2O привело к замене
значительного количества молекул Н2О на молекулы их дейтерированного аналога.
Предположили, что отверстия на сторонах молекул этого карцеранда достаточно велики,
чтобы небольшие молекулы типа молекул воды могли диффундировать сквозь
стенки полости, хотя возможен также и обмен ионов Н+ на D+. Данные
элементного анализа показали, что ~5% смеси представляют собой свободный карцеранд,
Cs+
сг сг сг сг
Рис. 5.72. Карцеплексы, образующиеся при получении соединения E.112)
42(f). 5. Связывание нейтральных молекул
а 60% содержат Cs+. Это согласуется со сродством каликс[4]аренов к Cs+ (см.
разд. 3.13), и предполагают, что образование E.112) включает линейное переходное
состояние SN2, в котором Cs+ скоординирован с серой (уравнение E.5)).
Cs+S"
,с—ci
н2
E.5)
Проблему растворимости карцеранда исследователи вскоре решили, заменив
его метальные «ноги» на ряд таких групп, как //-пентил, //-ундецил, 2-фенилэтил и
т.п. Это привело к получению карцеплексов, растворимых в органических
растворителях; при этом карцеплексы содержали молекулы растворителя в качестве
гостей с выходами до 32%. Родственные соединения с ацетальными мостиками
ОСН2О E.113) были получены из СН2С1Вг и тетраола E.114), схема 5.16.
Использование более длинных алкильных цепей, вплоть до (СН2L, в составе R'
соединений E.113) привело к получению удлиненных капсул карцерандов,
мостики которых достаточно гибки для проскока в полость некоторых небольших
частиц-гостей с образованием полукарцеплексов. Обуславливающие жесткость аце-
тальные спейсеры на верхнем ободе каждой чаши (названные тропиками по
аналогии с географическим тропиком) можно также заменить на группы
ОСН2СН2О, что сделает каждую полусферу более широкой и мелкой. Хотя
многостадийный синтез, приводящий к образованию кольца, включает одновременную
сборку семи молекул (в том числе и молекул гостя), тем не менее карцеранды
E.113) образуются с удивительно высокими выходами — до 87%.
\1 'R
ОН
R4
ОС
ОН
E.114)
о
СН2С1Вг
Растворитель
(гость)
К2СО3
R
О
*\
м
R
R =
R' =
R
1
/-«
sfe
0
»/ V
Rv R'
R
н-пентил,
(СН2)„,п
R
E.113)
°"Г/
О
\
R'
¥
R
CH2CH2Ph, н-ундецил и т.д.
= 1-1-4
Схема 5.16. Синтез соединений с ацетальными мостиками из тетраола E.114)
5.3. Внутриполостные комплексы нейтральных молекул
он
E.115)
Растворитель R\.
(гость) ^n
о к2со3
E.116)
R = w-пентил, CH2CH2Ph, м-ундецил и т.д.
Схема 5.17. Взаимодействие триола E.115) с дигалогенпроизводными (СН2)„С1Вг
Даже в присутствии мостиков (СН2L комплексы соединений E.113) все еще
способны включать молекулы-гости в свои полости и удалять их. В результате
включения получены истинные полукарцеплексы, имеющие на одной стороне
молекулы специальные отверстия и поэтому интересные для химии хозяин—гость,
которая включает многократный обмен гостей и возможность проведения внутрипо-
лостных реакций. Так, реакция триола E.115) с дигалогенпроизводными (СН2)„ХУ
дает полукарцеранд E.116) с тремя мостиками и с одним отверстием,
обеспечивающим легкий доступ гостя в полость (схема 5.17).
После получения соединения E.112) Крам и другие синтезировали огромное
количество карцерандов и полукарцерандов на основе как [4]резорцинаренов, так
и каликс[я]аренов (п = 4, 5), обладающих интересными комплексообразующими
свойствами и реакционной способностью.
5-3-6-2 Темплатные эффекты в синтезе карцерандов
Превосходные выходы, полученные при синтезе многих карцеплексов
и полукарцеплексов, противоречат статистическому описанию реакции, в которой
одновременно сталкивается большое количество реагентов и образуется большое
количество колец. Ясно, что здесь действует темплатный механизм. Действительно,
изучение реакций образования кольца для широкого круга капсулярных
соединений этого типа подтверждает, что это происходит только в присутствии
подходящего темплатирующего гостя. Попытки получить карцеплексы в растворителях,
молекулы которых слишком велики, чтобы соответствовать полости, оказались
422 5. Связывание нейтральных молекул
безуспешными. Темплатирующая способность огромного числа потенциальных
гостей определена количественно путем измерения темплатных соотношений при
синтезе карцеранда E.113); п = 1, R = CH2CH2Ph. Чтобы оценить этот параметр, в
качестве стандарта (приняли за единицу) использовали N-метилпирролидинон
(NMP, E.117)), чрезвычайно плохой растворитель для такого синтеза, а сам синтез
проводили в присутствии двух конкурирующих потенциальных гостей, которые в
равных количествах вводили в реакционную смесь в растворе NMP. Это приводило
к образованию двух разных карцеплексов соединения E.113) с каждым из
конкурирующих гостей. Анализ реакционной смеси 'Н ЯМР-спектроскопией
(интегрирование сигналов от гостей) позволил рассчитать соотношение между ними.
Предпочтительное образование одного карцеплекса перед другим предполагает, что гость
внутри первого хозяина способен лучше стабилизировать интермедиат и
обеспечивать замыкание карцеранда. Эту стадию реакции называют стадией, определяемой
гостем (guest-determining step, GDS), поскольку гость уже не может выйти из
карцеплекса после этой стадии. Найдено, что величины темплатных соотношений
могут достигать 106 в случае пиразина E.118), оказавшегося наилучшим темплатом.
Высокое темплатное соотношение в случае пиразина хорошо коррелирует с
выходом реакции G5%), однако существуют некоторые исключения из этого правила.
Найдено также, что это темплатное соотношение согласуется с константами
относительной устойчивости карцеплексов (Кге1). Это означает, что гости аналогичным
образом влияют как на полную свободную энергию комплекса, так и на энергию
активации переходного состояния. Некоторые значения темплатных соотношений
и Кге1 приведены в табл. 5.13, анализ которой позволяет сделать заключение о
важности размера и формы гостя. Несоответствие данных этой таблицы для
родственных 1,3-E.122)- и 1,4-E.119)диоксанов свидетельствует о наличии в системе
специфических направленных эффектов. Последние могут быть связаны с
нековалентными взаимодействиями между гостем и стенками полости по мере ее
образования. Таким образом, гости, которые могут обеспечить наиболее интенсив-
Таблица 5.13. Темплатные соотношения при
образовании комплексов E.113) (п = 1, R = CH2CH2Ph) с рядом
растворителей-гостей и относительная устойчивость
образующихся карцеплексов в нитробензоле-^ (по
Chapman /?. et al., 1998).
(Величины нормализованы по [NMP] = 1)
Гость
Пиразин E.118)
1,4-Диоксан E.119)
Пиридин E.120)
Бензол E.121)
1,3-Диоксан E.122)
Диметилацетамид E.123)
NMP E.117)
Темплатное соотношение
1 000 000
290 000
34 000
2 400
200
20
1
980 000
240 000
7 100
540
140
8.9
1
5.3. Внутриполостные комплексы нейтральных молекул 423
ные ван-дер-ваальсовы и С—Н--тт-взаимодействия с полостью, являются наиболее
эффективными темплатами.
., о © © о х
E.117) E.118) E.119) E.120) E.121) E.122) E.123)
(V
5-3-6-3 Образование комплексов с карцерандами
Связывание гостей хозяевами типа карцерандов определяется
скоростью обмена гостя с внешней средой. Истинные карцеплексы, в сущности, не
способны к обмену гостей. Медленный обмен гостя (от часов до дней) характерен для по-
лукарцеплексов, в то время как связывание очень малых молекул, типа О2, которые
быстро обмениваются (по шкале времен 'Н ЯМР), строго говоря, свойственно кар-
цеплексам, а не полукарцеплексам. Как и в случае криптофанов, комплексообразую-
щие свойства карцерандов легко определяют благодаря спектроскопии !Н ЯМР.
Магнитная анизотропия арильных колец, образующих стенки полости, приводит к
большим изменениям химических сдвигов (обычно высокопольным) для резонан-
сов, приписываемых частицам гостя. По мере более глубокого проникновения
гостя в полость, этот эффект проявляется сильнее и величины Л8 (= Ьггее — ЬеМ £
в интервале 2—4.5 м.д. обычны и зависят от степени проникновения гостя в чаши
резорцинарена. ИК-спектры захваченных частиц также дают значительные
отклики. В случае карцеплексов, образованных карцерандом E.113) и диметилформами-
дом (DMF) или диметилацетамидом (DMA, E.123)), найдено, что
характеристические частоты валентных колебаний связи С=О находятся в промежутке между
обычно наблюдаемыми частотами для жидкой и газообразной фаз чистых молекул
(ср. метан внутри криптофана-А, разд. 5.3.5).
Кристаллическая структура пиразинового карцеплекса соединения E.113)
(РСА) показана на рис. 5.73. Пиразин имеет одно из самых больших известных тем-
платных соотношений, что предполагает благоприятные взаимодействия
хозяин—гость. Это подтверждается тем, что атомы азота гостя участвуют в образовании
водородных связей С—H-N с метиленовыми мостиками хозяина. Кроме того,
реализуются взаимодействия С—Н 1 между другими метиленовыми мостиками
хозяина и л-системой пиразина, а также между пиразиновыми протонами и арильными
кольцами хозяина. Такие взаимодействия приводят к замедлению внутриполостно-
го вращения пиразина вокруг оси N-N. В асимметричном хозяине, молекула
которого содержит внеполостные метальные заместители на одной чаше и 2-фенил-
этильные группы — на другой, такое вращение имеет большой энергетический
барьер - 80 кДжмоль. Оно оказалось достаточно медленным, чтобы можно было
различить взаимодействие между протонами На и Иь на шкале времен ЯМР
(рис. 5.74). Анализ структуры пиразинового комплекса также показал превосходное
соответствие пиразинового гостя хозяину. В этом карцеплексе две чаши хозяина
S. Связывание нейтральных молекул
Рис. 5.73. Стереоизображение кристаллической структуры пиразинового карцеплекса
соединения E.113). (Воспроизведено с разрешения FraserJ.R. et al., 1995)
хотя и повернуты одна относительно другой на 21° вокруг оси псевдо-С4, но, по
существу, они остаются параллельными, что обеспечивает полное сопряжение
атомов, находящихся между чашами с ароматическими кольцами. Напротив, в
структуре с DMA-производным чаши отклонены на 5.2° от параллельного расположения
из-за большего объема гостя.
ОсьС4
Ph
Рис. 5.74. Геометрия включения пиразина
в асимметричный карцеранд
5.3. Внутриполостные комплексы нейтральных молекул
425
5.3*6.4 Карцерия
Изучая в качестве хозяев карцеранды, исследователи не
ограничивались производными резорцинарена. П. Тиммерман с сотрудниками (P. Timmerman
et al., 1994) получил смешанный хозяин [4]резорцинаренкаликс[4]арен E.124) при
сочетании «лицом к лицу» двух разных чашеобразных макроциклических полусфер
(схема 5.18). Если внутримолекулярные полости в резорцинареновых карцерандах,
по существу, шарообразны или имеют форму удлиненных цилиндров с
искривленными концами, то полость в соединении E.124), скорее, яйцеобразна, т.е. чрезвы-
с,,н
с.,н
СцН23
С„Н23 E.124)
Схема 5.18. Синтез яйцеобразного карцеранда E.124)
426
5. Связывание нейтральных молекул
Ось С а
Каликс[4]арен
[4]Резорцинарен
Рис. 5.75. Карцерия в комплексах DMA с гибридными карцерандами (по Timmerman P.
etal., 1994)
чайно асимметрична, причем более широкая и более мелкая резорцинареновая
полость образует основание яйца, а более узкая и более глубокая полость
калике [4] арена формирует его вершину.
Карцеранд E.124) получают в разнообразных растворителях, таких, как DMF,
DMA, DMSO и этилметилсульфоксид, при 80 °С с количественным выходом.
Однако в больших растворителях-гостях (слишком больших, чтобы они могли
соответствовать полости) его выход значительно снижается. В частности, 1,5-диметил-
пирролидинон-2 дает только 5%-й выход этого соединения. В случае маленьких
гостей, например DMF, вращение вокруг оси типа Cj осуществляется быстро, и
поэтому наблюдается только один продукт. Однако большие гости типа DMA
вращаются с трудом, их вращательный барьер достигает 53-73 кДжмоль, что приводит
к получению двух ориентационных изомеров — это стереоизомерия нового типа.
Д. Рейнхоудт с сотрудниками (D. Reinhoudt et al.) для описания этого явления
предложил термин «карцероизомерия» (или, для краткости, «карцерия»), рис. 5.75.
5-3-6.5 Реакции включения
Совсем не завидую я
Ни пленительной пустоте благородной страсти,
Ни коноплянке, рожденной в клетке,
Которая никогда не узнает о летних рощах.
А. Теннисон A809-1892). В память
Прочная, хорошо экранированная полость, обнаруженная в полукар-
церандах, предопределяет большие возможности использования этих хозяев в
качестве микрореакторов, защищающих реагирующие частицы от бимолекулярного
S.3. Внутриполостные комплексы нейтральных молекул
411
разложения путем их изоляции от внешней среды. Более того, уникальное внутри-
полостное окружение хозяина, в котором гость формально находится в
конденсированном состоянии под очень высоким давлением, может привести к тому, что
хозяин будет иметь высокую реакционную способность включения. В самом деле,
внутренний объем карцерандов и полукарцерандов рассматривают как «новую
фазу материи», отличную от твердой, жидкой и газообразной. Были
продемонстрированы потенциальные возможности этих систем для проведения уникальных
реакций, и сейчас уже ясно, что возможности использования таких молекулярных
реакторов практически безграничны.
Один из простейших примеров влияния включения на реакционную
способность гостя — измерение основности включенных аминных лигандов. С помощью
*Н ЯМР-спектроскопии можно показать, что растворы пиридина в CDC13 легко
протонируются кислотой CF3CO2D. В результате же аналогичной реакции
пиридинового комплекса полукарцеранда E.116), имеющего отверстие, пиридин
оставался непротонированным. Это означает, что включенный пиридин — значительно
более слабое основание, чем его свободная молекула. Наиболее разумное объяснение
сводится к тому, что хозяин, обладая лишь очень ограниченной возможностью
сольватировать ион пиридиния, будет стерически ингибировать образование
ионной пары пиридиний—трифторацетат.
Гораздо более важный пример использования полукарцерандов — получение
Д. Крамом в 1991 г. чрезвычайно нестойкой молекулы циклобутадиена E.128) в
полости соединения E.116). До этого циклобутадиен можно было наблюдать только
замороженным в матрице инертного газа при 8 К после фотолиза а-пирона E.125).
При нагревании циклобутадиен быстро димеризуется с образованием циклооктатет-
раена E.130). Крам и сотрудники провели все реакции, показанные на схеме 5.19,
E.131)
а Нагревание
140°С
E.125) \
О
О
E.127)
О Нагревание
90°С
E.126)
О
-СО,
Нагревание
220 °С
E.128)
E.129)
E.130)
Схема 5.19. Внутриполостной синтез циклобутадиена E.128) и его реакции
по Краму (Cram Z). et al., 1991)
428
5. Связывание нейтральных молекул
внутри полости соединения E.116). Тщательно контролируемые условия
позволили им осуществить циклизацию при фотолизе а-пирона (который связывался при
кипячении со свободным хозяином) с образованием лактама E.126), получить
перегруппировкой лактам E.127) и снова вернуться к ос-пирону. Полукарцеплекс
гостя E.126) оказался устойчивым в течение двух недель в твердом состоянии при
комнатной температуре. Однако наиболее примечательно, что облучение этого
полукарцеплекса 75-ваттной ксеноновой дуговой лампой в течение 30 мин при
комнатной температуре привело в результате включения к образованию циклобута-
диена E.128). Этот новый гость в спектре 'Н ЯМР давал сигнал при 8 2.27 м.д. и был
очень устойчивым, находясь под защитой полукарцеранда. СО2, образующийся как
побочный продукт, выбрасывался из полости. По аналогии с высокопольным
сдвигом в спектре включенного бензола Крам рассчитал, что химический сдвиг
свободного циклобутадиена должен составлять 5.2—5.7 м.д. Наблюдение резких сигналов
подтверждает, что циклобутадиен находится в основном синглетном состоянии.
Реакционную способность включенного циклобутадиена исследовали методом
ЯМР, нагревая образец до 220 °С в течение 5 мин. При этом образовался свободный
циклооктатетраен E.130), очевидно, в результате выброса циклобутадиена E.128)
из полости и его последующей димеризации через интермедиат E.129). Реакция
E.128) с О2, который способен проникать в полость полукарцеранда E.116), дала
включенный в эту полость малеиновый ангидрид E.131).
Некоторое время «укрощение» циклобутадиена оставалось единственным
большим достижением в применении контейнерных молекул-хозяев для стабилизации
реакционноспособных частиц. Однако появились данные (R. Warmuth, 1997) о
стабилизации еще более химически активного гостя - бенз-1,2-ина E.134).
Циклобутадиен неустойчив из-за трудностей при реализации углов в 90° между гибридными
5/>2-орбиталями, которые имеют оптимум при 120°. Напротив, бенз-1,2-ин
вынужден принимать углы в 120°, составленные .sp-орбиталями, для которых
оптимальный угол равен 180°. Стабилизация бенз-1,2-ина достигалась внутри четырехмости-
,0
hi
77К
со2н
Н2О
E.132)
E.133)
Бенз-1,2-ин E.134)
Схема 5.20. Внутриполостной синтез бенз-1,2-ина E.134)
S.3. Внутриполостные комплексы нейтральных молекул
429
кового карцеплекса соединения E.113), п — 4, который не обладает специальным
входом для включения гостя. Бензоциклобутандион E.132) был включен с 35%-м
выходом при нагревании соединения E.113) в чистом E.132). Фотолиз при 77 К дал
карцеплекс бензоциклопропанона E.133), который охарактеризовали методом
РСА. В присутствии воды это вещество медленно реагирует с образованием в
полости бензойной кислоты. Дальнейший фотолиз при 77 К дает желаемый бенз-1,2-ин,
который достаточно устойчив для измерения его 'Н и 13С ЯМР-спектров при -75 и
—98 °С соответственно (схема 5.20). 'Н ЯМР-спектр показывает два сигнала двух
отдельных пар протонов бенз-1,2-ина при 5 4.99 и 4.31 м.д. Интересно, что при
нагревании до комнатной температуры этот гость все еще не способен покинуть свою
«тюрьму», но при этом он реагирует со стенками полости карцеранда по реакции
Дильса—Альдера, давая циклогексадиен E.135) с бензольным мостиком в 1,4-поло-
жении.
О
(СН2L
R
E.135)
Синтезы соединений E.128) и E.134) внутри полостей полукарцерандов через
стадию фотолиза очень изящны. Возможность использования карцерандов в
качестве хозяев для реализации других внутриполостных реакций может быть
ограничена необходимостью расположения химических реагентов в полости хозяина в
отсутствие фотолиза. Интересно, что Крам с сотрудниками также провел серию
количественных реакций окисления и восстановления внутри карцеранда E.113),
п = 4, для включенных в его полость хинонов и родственных соединений. Редокс-
реагентами были вещества типа Sml2 и Ce(NH4J(NO3N, не имевшие никаких
шансов проникнуть в эту полость. Тем не менее редокс-реакции протекали
чрезвычайно гладко (в случае нитробензола получен N-фенилгидроксиламин вместо
ожидаемого анилина), причем электрон, протон и молекулы воды проникали через
стенки хозяина.
430 5. Связывание нейтральных молекул
Супрамолекулярная химия фуллеренов
Открытие в 1985 г. фуллеренов — замкнутых трехмерных молекул,
состоящих исключительно из углерода, — одно из самых замечательных открытий в
химии за последние десятилетия. В 1996 г. оно принесло Нобелевскую премию по
химии Р. Керлу-младшему, Р. Смолли (R. Curl (Jr.), R. Smalley) из Университета
Раиса в Хьюстоне (США) и X. Крото (Н. Kroto) из Университета Суссекса
(Великобритания) за исследования в этой области (Дополнение 5.3). Фуллерены, в
особенности бакминстерфуллерен (С60, «бакибол», «футбольный мяч») и С70, чрезвычайно
популярны во многих областях химии и, не в последнюю очередь, в супрамолеку-
лярной химии. Фуллерены могут быть как хозяевами, так и гостями. В качестве
хозяев они проявляют способность к интеркаляции, в какой-то степени подобно
графиту, и могут включать в свою закрытую полость такие молекулы, как, например,
молекулы гелия и металлов. В качестве гостей они представляют собой большой
электронодефицитный темплат, способный образовывать с донорами электронов
типа ферроцена и вогнутого BEDT-TTF (бис(этилендитио)тетратиафульвален
E.136)) ряд соединений с переносом заряда, а также соединения включения ван-
дер-ваальсова типа, особенно в твердом состоянии. Первые сообщения о супрамо-
лекулярной химии соединения С60 как хозяина появились в начале 1990-х годов, и
до сих пор исследования в этой области очень актуальны. В последующих разделах
будут рассмотрены лишь некоторые свойства этих соединений.
BEDT-TTF E.136)
*■* Kroto H.W., Heath J.X., O'Brien S.C. et al. C60-Buckminsterfiillerene // Nature
(London). 1985. Vol. 318. P. 162-163.
5* 4 • 1 Фуллерены как гости
Мы уже знаем о способности фуллеренов С60 образовывать
соединения включения с электроноизбыточным чашеобразным макроциклом CTV (см.
разд. 5.2.5.2). Очевидно, что фуллерены должны давать соединения включения с
ароматическими макроциклами. Действительно, каликсарены, такие, как
л-трет-бутилкаликс[8]арен и л-т/>ет-бутилкаликс[6]арен, можно использовать
для выделения фуллеренов С60 и С70 из сложной сажевой смеси, образующейся при
испарении графита в угольной дуге. В основе этого метода лежит способность
больших каликсаренов селективно связывать фуллерены в растворе толуола, в
результате чего образуются кристаллические 1:1-комплексы п-трет-Ьутилка-
ликс[8]арен-С60, а также 1:2-комплексы л-трет-бутилкаликс[6]арен(С60J и
л-трет-бутилкаликс[6]арен-(С70J (J. Atwood et al., 1994; Т. Suzuki et al., 1994).
Обработка этих твердых комплексов включения хлороформом приводит к разделению
хозяев и гостей, давая осадки очищенных фуллеренов и растворы каликсаренов,
5.4. Супрамолекулярная химия фуллеренов
431
Толуол
Каликсарен
С-70 остается в растворе
Возвращение
каликсарена
в очищенном виде
в реакционную среду
CHCI,
Осадок комплекса включения Сзд-калаксарен
С60 нерастворам в хлороформе
Рис. 5.76. Разделение смеси С60 и С70 с помощью каликсаренов
сольватированных хлороформом и стабилизированных взаимодействиями
С—Н--71, которые усилены высокой кислотностью С—Н-группы хлороформа
(рис. 5.76). Структура твердых комплексов каликсарен-фуллерен (рис. 5.77)
показывает, каким образом происходит капсулирование фуллеренов электроноизбыточ-
ными хозяевами. Эти работы стимулировали дальнейшие исследования большого
числа фуллереновых комплексов типа «хозяин—гость», и сейчас известны
структуры многих таких комплексов (по данным РСА) с различными каликсаренами,
например с гексагомооксакаликс[3]ареном A:1-комплекс) E.137), триптиценом
A:2-комплекс) E.138), октафенилциклотетрасилокеаном A:1-комплекс) E.139)
и т.д.
Сообщалось также о комплексах фуллеренов состава 1:1 и 2:1 с у-циклодекстри-
ном (Т. Andersson et al., 1992). Большую молекулу циклодекстрина можно
использовать для связывания как С60, так и С70 в воде. При концентрации ~10~4 М такой
раствор имеет слабую желтую окраску, тогда как растворы фуллеренов в
ароматических растворителях типа бензола и толуола обладают глубоким красным и даже
пурпурным цветами. Моделирование предполагает, что оси С5 этих фуллеренов
ориентированы параллельно оси псевдо-Q циклодекстрина.
Гидрохинон также образует с С60 соединение включения состава 3:1, имеющее
формулу 3(С6Н4(ОНJ)С60. Действительно, размеры полостей в решетке гидрохи-
432
5. Связывание нейтральных молекул
Гексагомооксакаликс[3]арен
E.137)
Триптицен Октафенилциклотетрасилоксан
E.138) E.139)
Рис. 5.77.
Кристаллические структуры:
(а) каликс[6]арен(С60J
и (б) каликс[6]арен(С70J
(AtwoodJ.L. etal., 1998)
5.4. Супрамолекулярная химия фуллеренов
433
Рис. 5.78. Структура сетки хозяина-гидрохинона, предорганизованная для включения
гостя С
60
нона (—10 А) соответствуют размеру С60. Опять-таки природа обогащенного
электронами гидрохинона способствует связыванию электронодефицитного С60
Межплоскостное расстояние между поверхностью С60 и арильным кольцом
гидрохинона составляет 3.1 А, что обеспечивает оптимальное п—тг-стэкинг-взаимодействие
(рис. 5.78). При реакции шести эквивалентов гидрохинона с чистым С70 в бензоле
образуются коричнево-красные кристаллы состава 9(С6Н4(ОНJ)-2С70-2С6Н6,
которые обладают значительно более сложной структурой из-за необходимости
оптимизировать стерические требования к упаковке молекул С70, имеющих форму мяча
для регби, и гидрохинона.
9-1 Diederich F. and Gomez-Lopez M. Supramolecular fullerene chemistry // Chem. Soc.
Rev. 1999. Vol. 28. P. 263-277.
5*4-2 Фуллерены как хозяева
Известно, что фуллерены образуют ряд металлоорганических
комплексов типа [{Pt(PEt3J}6- (С60)], в которых частицы С60, С70 и даже С82 выступают
в роли монодентатных лигандов для каждого центра переходного металла. Такой
434
5. Связывание нейтральных молекул
6-6"- связь
-5"-связь
Рис. 5.79. Проекция одной из полусфер
С60, показывающая связи «6—6» и «6—5»
металлоцентр связан с внешней поверхностью молекулы фуллерена посредством
одной фуллереновой связи С=С, расположенной в более электроноизбыточнои
области соединения колец «6—6», а не колец «6-5» (рис. 5.79). В общем, рассмотрение
таких объектов выходит за рамки этой книги, хотя один пример все же приведем —
это особый иридиевый комплекс, который был использован для получения
самокомплементарного хозяина на основе С60 (A. Balch et al., 1992).
E.140)
Комплекс [Ir(ri2-C60)(CO)Cl-(LJ] (где L = Ph2PCH2C6H4OCH2Ph, E.140))
простирает две свои обогащенные электронами бензилэфирные «руки» и охватывает
ими соседние лиганды С60 (рис. 5.80).
Рис. 5.80. Кристаллическая структура комплекса [Ir(r|2-C60)(CO)Cl-(LJ]
демонстрирующая самовключение
5.4. Супрамолекулярная химия фуллеренов
435^
Недавно выдающаяся работа Ф. Дидериха (F. Diederich, 1988) из Федерального
технологического института в Цюрихе привела к созданию редокс-чувствительно-
го сенсорного устройства E.141) на основе бакминстерфуллерена. Этот новый
сенсор, полученный с выходами до 50%, содержит полость дибензо-18-краун-6 вблизи
поверхности фуллерена. Связывание К+ внутри этой краун-эфирной полости
приводит к относительно большому анодному сдвигу — на 90 мВ — на первой стадии
восстановления фуллерена (С60 —> С6*0~). Близость катиона калия к фуллерену
значительно облегчает восстановление последнего до моноаниона. Связывание К+ в
полости крауна вблизи внешней поверхности фуллерена подтверждено данными
РСА.
E.141)
Возможность связывания катионов металлов и нейтральных атомов внутри
полостей фуллеренов с образованием «захваченного» атома металла вызывает
активный интерес у специалистов в области супрамолекулярной химии. Полость в С60
имеет сечение ~7 А в диаметре, т.е. достаточно велика для включения, по крайней
мере, одноатомных гостей. Большие гЪуллерены, такие, как С70 и С82, вероятно,
могут включать более одного атома или даже небольшие молекулы-гости. Ясно, что
закрытая структура молекул типа С60, содержащая пяти- и шестичленные кольца,
не имеет отверстия, достаточно большого для проникновения хотя бы одного
атома в нормальных условиях химической реакции, и, следовательно, нужны
альтернативные стратегии. Фактически соединения включения фуллеренов,
обозначаемые как М@СХ (где М - частица гостя, обычно атом металла, аСх- фуллерен,
х = 60, 70, 74, 82 и т.д.), были получены либо в результате «заточения» металла в
клетке, образующейся в ходе синтеза фуллерена в присутствии паров данного
металла, либо при высокоэнергетических бимолекулярных столкновениях. Почти
сразу после открытия фуллеренов Р. Смолли (R. Smalley) удалось получить
комплексы М@С60 (М = La, Ni, Na, К, Rb, Cs), а также комплексы лантана с С70, С74 и
С82. О включении атома металла в полость фуллерена свидетельствует тот факт, что
синтезированные комплексы, обычно содержащие металлы — сильные
восстановители, инертны к О2, NH3 и воде. Металл защищен окружающей его клеткой
фуллерена. Были выделены макроскопические количества La@C82, и методом
рентгеновской фотоэлектронной спектроскопии определено наличие иона La3+ (наиболее
436 5. Связывание нейтральных молекул
устойчивое состояние окисления лантана), аниона С|^~, до которого фуллерен
восстановлен металлом (фуллерены имеют очень важные электрохимические
свойства, и C|jсчитается особенно устойчивым), и «свободного» электрона, также
включенного в полость. Таким образом, это соединение является весьма необычным
электридом (ср. разд. 3.12). Были также получены биметаллические комплексы
М2@С80 (М = La, Y) и высказано предположение, о том, что они могут оказаться
сверхпроводящими.
*Ъг Schwartz H. C60-Fullerene - a playground for chemical manipulations on curved
surfaces and in cavities // Angew. Chem., Int. Ed. Engl. 1992. Vol. 31. P. 293-298.
5.4*3 Фуллерены как сверхпроводящие
соединения включения
Какими бы ни оказались при дальнейших исследованиях свойства эн-
доэдральных* металлофуллеренов, уже сейчас получен ряд сверхпроводящих фул-
лереновых интеркалятов с ионами щелочных металлов, проявляющих
сверхпроводимость при температуре до 33 К (для Cs2RbC60). Этот результат не особенно
удивляет, если сравнить его с данными для неорганических сверхпроводников
керамического типа (например, оксиды иттрия-бария-меди), которые обладают
сверхпроводимостью выше температуры жидкого азота (до 123 К). Подобно
графиту, фуллерены легко восстанавливаются щелочными металлами, давая соединения
типа Мх-С60 (М = Na, К, Rb, Cs, Ва, Са и т.д.; х = 1-М, 6), из которых наиболее
известны К3С60 и К6-С60, имеющие гранецентрированную (fee) и объемноцентриро-
ванную {bee) кубические структуры соответственно (рис. 5.81). Эти соединения
получены реакцией фуллерена С60 с таким восстановителем, как NaBH4, и можно
считать, что их молекулы содержат анионы фуллерена и катионы щелочных
металлов, занимающих как тетраэдрические, так и октаэдрические центры внутри фул-
лереновой решетки.
8-1 Okino F. and Touhara H. Graphite and fullerene intercalation compounds //
Comprehensive supramolecular chemistry / Ed. J.L.Atwood, J.E.D.Davies,
D.D.MacNicol and F.Vogtle. Oxford: Pergamon, 1996. Vol. 7. P. 25-76.
Гранецентрированная Объемноцентрированная рис g 81 c фуллереновых
кубическая кубическая ., -, „ >.
(fee) (bee) интеркалятов K3C60 и К6С60
* В эндоэдральных соединениях атом металла расположен внутри, а в экзоэдральных —
снаружи углеродной сферы. (Примеч. ред. перевода.)
5.4. Супрамолекулярная химия фуллеренов
437:
Дополнение 5.3. Открытие фуллеренов
До 1985 г. были известны шесть кристаллических форм углерода: две формы фафита,
две формы алмаза, а также чаоит и углерод (VI), открытые соответственно в 1968 и
1972 гг. К тому же, существуют такие соединения, как полиацетилен E.142) и кумулен
E.143), которые почти полностью состоят из углерода. Фуллерены, представляющие
собой единственную молекулярную форму чистого углерода, образуются в
экстремальных условиях при конденсации паров углерода в атмосфере инертного газа типа
гелия. X. Крото (Н. Kroto), изучавший атмосферу звезд и межзвездные пылевые
облака методом микроволновой спектроскопии, попытался воспроизвести в лаборатории
спектры углеродно-азотных соединений, называемых цианополиинами E.144),
которые он обнаружил в космосе в 1975—1978 гг. Вместе с Р. Смолли (R. Smalley), имевшим
оборудование для испарения металлов и изучения металлических кластеров методом
масс-спектрометрии, Крото применил этот метод к фафиту. Получившийся масс-
спектр (рис. 5.82) оказался весьма необычным, поскольку содержал ряд пиков,
которые идентифицировали как кластеры углерода; при этом пики, соответствующие С6о
и С70, выделялись среди остальных, свидетельствуя о повышенной стабильности этих
кластеров.
Полиацетилен
E.142)
Кумулен
E.143)
Цианополиины
E.144)
(б)
(в)
44 52 60 68 76 84
Число атомов углерода в кластере
Рис. 5.82. Масс-спектры смесей фуллеренов,
образующихся при испарении фафита.
(Воспроизведено с разрешения Kroto H.W., Heath
J.R., O'Brien S.C. et al. C60-Buckminsterfullerene //
Nature (London). 1985. Vol. 318. P. 162-163)
5. Связывание нейтральных молекул
Рис. 5.83. Первая модель соединения С6о,
построенная Р. Смолли из бумажных
шестиугольников и пятиугольников.
(Воспроизведено с разрешения Kroto H. W.
С60: Buckminster fullerene. The celestial sphere that
fell to earth // Angew. Chem., Int. Ed. Engl. 1992.
Vol. 31. P. 111-129)
Вначале исследователи не могли объяснить такое высокое содержание кластера с
отношением масса/заряд 720, соответствующим соединению С60. Это удалось сделать
только после того, как они предположили, что аГрегат представляет собой закрытый
кластер, причем команда исследователей потратила целый вечер, пытаясь сложить
такой кластер из шестиугольных кусочков бумаги, а также скрепить зерна набухшего
гороха зубочистками. К счастью, с помощью модели «звездного свода», которую
предложил Крото, исследователи осознали необходимость использования также и
пятиугольных граней. В конце концов, из 12 пятиугольников и 20 шестиугольников
им удалось собрать элегантное сферическое твердое тело с 60 вершинами (усеченный
икосаэдр), соответствующее структуре соединения С60 (рис. 5.83). Эта молекула
имеет точно такую же структуру, как звездный свод Крото или европейский футбольный
мяч, или геодезические купола архитектора Р. Бакминстер Фуллера. В его честь эта
новая молекула была названа бакминстерфуллереном, и немедленно была написана
статья. Родственная молекула — С70 — имеет эллипсоидную форму с теми же
полусферами по краям, что и у С6о, но с дополнительным рядом шестиугольников в
срединной части. Фактически, чтобы составить любую твердую замкнутую фигуру,
требуются еще и 12 пятиугольников, потому что из одних только шестиугольников это сделать
невозможно (геометрический факт, известный как закон Эйлера).
Открытие фуллеренов было встречено с энтузиазмом и враждебностью. Эти
молекулы присутствовали только в масс-спектрометре, и некоторые исследователи
сомневались в их существовании. Окончательное доказательство появилось в 1990 г., после
выделения макроскопических количеств С60. Это позволило получить 13С ЯМР-
спектр этого соединения, в котором, как и предсказывалось, он был представлен
одной линией (при 8 143 м.д.). Все атомы углерода в С6о эквивалентны.
9-1 Kroto H.W. C60: Buckminster fullerene. The celestial sphere that fell
to earth // Angew. Chem., Int. Ed. Engl. 1992. Vol. 31. P. 111-129.
Учебные задания
5.1. Константа связывания криптофана-А с метаном составляет 130 М в
(СНС12J, растворителе, который не связывается полостью. Рассчитайте
константу связывания в СНС13, если Лдля этого растворителя составляет 10 М,
а плотность жидкого хлороформа равна 1.48 г-см~3 при 25 °С.
Учебные задания
439
5.2. Большой карцеранд имеет полость объемом 120 А3. Если молекулярный
объем метана составляет 28.5 А3, вычислите фактор занятости р для метанового
1:1-карцеплекса этого хозяина. Какой объем хозяина получится при факторе
занятости 0.67, эквивалентном твердому метану? Рассчитайте воображаемое
давление одной молекулы метана в полости такого размера. Считаете ли Вы
образование такого карцеплекса вероятным?
5.3. Предполагая, что полости 512 в структуре I клатратных гидратов имеют форму
сферы (радиус 3.91 А), рассчитайте фактор занятости и воображаемое
давление молекулы метана, заключенной в такую полость. Сравните эти данные с
полостью 51262 (радиус 4.33 А). Какую полость метан займет предпочтительно?
5.4. Объясните, почему карцерия не наблюдается для небольших карцерандов,
основанных только на [4]резорцинаренах типа E.115).
5.5. Объясните мотивы введения метильных заместителей в молекулярные
пинцеты E.76)—E.78) с точки зрения принципа предорганизации, рассмотренного в
гл. 1.
5.6. Назовите, наряду с соединениями E.84) и E.89), следующие циклофаны.
5.7. Предложите возможный путь синтеза [4]резорцинарена E.43).
5.8. Рассчитайте потерю массы (измеренную методом TGA, в процентах)
соединений включения 1:1, 2:1 и 3:1 тетрафенилена С24Н16, мочевины CO(NH2J и
А7-т/?ет-бутилкаликс[4]арена С44Н56О4 со следующими гостями: вода, СНС13,
бензол, С60. Насколько полезным мог бы быть метод TGA при изучении химии
хозяин—гость каждого из этих комплексов?
Мысленный эксперимент
Ознакомившись с разд. 5.3.6.1, оцените значения темплатного соотношения при
синтезе соединения E.113) в NMP в присутствии приведенных ниже гостей (см.
табл. 5.13). Рассмотрите влияние размера и формы молекулы, а также
взаимодействий хозяин-гость типа водородных связей, л-л-стэкинг-взаимодействий и т.п.
о о t
440
5. Связывание нейтральных молекул
Литература
Adrian, Jr., 1991 Adrian J.C., Jr, Wilcox C.S. Chemistry of synthetic receptors and functional-group
arrays. 15. Effects of added water on thermodynamic aspects of hydrogen-bond-based
molecular recognition in chloroform // J.Amer. Chem. Soc. 1991. Vol. 113. P. 678.
Allott, 1988 Allott C, Adams H., Bernad P.L., Jr. et al. Hydrogen-bond recognition of cyclic
dipeptides in water// Chem. Commun. 1998. P. 2449.
Andersson, 1992 Andersson Т., Nilsson K., Sundahl M. et al. C60 embedded in y-cyclodextrin - a
water-soluble fullerene // J.Chem. Soc, Chem. Commun. 1992. P. 604.
Andretti, 1979 Andretti G.D., Ungaro R., PochiniA. Crystal and molecular structure of cyclo{quar-
ter[E-/-butyl-2-hydroxy-I,3-phenylene)methyiene]} toluene A:1) clatrate //
J.Chem. Soc, Chem. Commun., 1979. P. 1005.
Atwood, 1991 Atwood J.L., Hamada F, Robinson K.D. et al. X-ray diffraction evidence for
aromatic pi hydrogen-bonding to water// Nature. 1991. Vol. 349. P. 683.
Atwood, 1994 Atwood J.L., Koutsantonis G.A., Raston C.L. Purification of C60 and C70 by
selective complexation with calixarenes // Nature. 1994. Vol. 368. P. 229.
Atwood, 1998 Atwood J.L., Barbour L.J., Raston C.L., Sudria I.B.N. C60 and C70 compounds in
the pincerlike jaws of calyx[6]arene // Angew. Chem., Int. Ed. Engl.. 1998.
Vol.37. P. 981.
Balch, 1992 Balch A.L., Catalano V.J., LeeJ.W., Olmstead M.M. Supramolecular aggregation
of an (Л2-С60) iridium complex involving phenyl chelation of the fullerene //
J. Amer. Chem. Soc. 1992. Vol. 114. P. 5455.
Blanch, 1997 Blanch R.J., Williams M., Fallon G.D. et al. Supramolecular complexation of 1,2-
dicarbadodecaboraneA2)//Angew. Chem., Int. Ed. Engl. 1997. Vol. 36. P. 504.
Brown, 1949 Brown C.J., Farthing A.C. Preparation and structure of di-/?-xylylene // Nature
(London). 1949. Vol. 164. P. 915.
Canceill, 1986 Canceill J., Lacombe L., Collet A. New cryptophane forming unusually stable
inclusion complexes with neutral guests in a lipophilic solvent // J. Amer. Chem.
Soc. 1986. Vol. 108. P. 4230.
Chang, 1988 Chang S.-K., Hamilton A.D. Molecular recognition of biologically interesting
substrates — synthesis of artificial receptor for barbiturates employing 6 hydrogen-
bonds//J. Amer. Chem. Soc. 1988. Vol. 110. P. 1318.
Chapman, 1998 Chapman R.G., Olovsson G., Trotter J., Sherman J.C. Crystal structure and
thermodynamics of reversible molecular capsules // J. Amer. Chem. Soc. 1998. Vol.
120. P. 6252.
Chen, 1978 Chen C.W., Whitlock H.W. Molecular tweezers. A simple model of bifunctional
intercalation //J. Amer. Chem. Soc. 1978. Vol. 100. P. 4921.
Corazza, 1990 Corazza F., Floriani C, Chiesi- Villa A., Guastini С The oxo-molybdenum(VI)
group over a calixarene-oxo surface — calyx[4]arene complexing inorganic and
organic functionalities//J.Chem. Soc, Chem. Commun. 1990. P. 640.
Cram, 1951 Cram D.J., Steinberg H. Macro rings. I. Preparations and spectra of the paracyclo-
phanes// J.Amer. Chem., Soc. 1951. Vol. 73. P. 5691.
Cram, 1985a Cram D.J., Stewart K.D., Goldberg I., Trueblood K.N. Complementary solutes
enter nonpolar preorganized cavities in lipophilic noncomplementary media //
J. Amer. Chem. Soc 1985a. Vol. 107. P. 2574.
Cram, 1985b Cram D.J., Karbach S., Kim Y.H. et al. Shell closure of 2 cavitands forms
carcerand complexes with components of the medium as permanent guests //
J. Amer. Chem. Soc. 1985b. Vol. 107. P. 2575.
Литература
441
Cram, 1988a Cram D.J. The design of molecular hosts, guests, and their complexes (Nobel
Lecture) //Angew. Chem., Int. Ed. Engl. 1988a. Vol. 27. P. 1009.
Cram, 1988b Cram D.J., Weiss J., Hegeson R.C. et al. Design, synthesis, and comparison of
crystal, solution, and calculated structures within a new family of cavitands //
J.Chem., Soc, Chem. Commun. 1988b. P. 407.
Cram, 1991 Cram D.J., Tanner M.E., Thomas R. The taming of cyclobutadiene // Angew.
Chem., Int. Ed. Engl. 1991. Vol. 30. P. 1024.
Diederich, 1982 Diederich F, Dick K. New water-soluble macrocycles of the paracyclophane-type
aggregation behavior and host-guest-interaction with hydrophobic substrates //
Tetrahedron Lett. 1982. Vol. 23. P. 3167.
Diederich, 1988 Diederich F. Complexation of neutral molecules by cyclophane hosts // Angew.
Chem., Int. Ed. Engl. 1988. Vol. 27. P. 362.
Eto, 1992 Eto S., Noda H., Noda E. Chiral separation of barbiturates and hydantoins by
reversed-phase high-performance liquid-chromatography using a 25 or 50 mm
short ODS cartridge column via C-cyclodextrin inclusion complexes //
J. Chromatogr. 1992. Vol. 579. P. 253.
Ferguson, 1991 Ferguson S.B., Sanford E.M., Seward E.M., Diederich F. Cyclophane arene
inclusion complexation in protic solvents — solvent effects versus electron-donor
acceptor interactions//J.Amer. Chem. Soc. 1991. Vol. 113. P. 5410.
Flinn, 1992 Flinn A., Hough G.C., Stoddart J.F., Williams D.J. Decamethylcucurbit[5]uril //
Angew. Chem., Int. Ed. Engl. 1992. Vol. 31. P. 1475.
Fraser, 1995 FraserJ.R., Borecka В., Trotter J., Sherman J.C. An asymmetric carceplex and new
crystal-structure yield information regarding a 1-million-fold template effect //
J. Org. Chem. 1995. Vol. 60. P. 1207.
Gerdil, 1984 Gerdil R., Barchietto G, Jejford C.W. Heterogeneous chirality transfer on pho-
tooxygenation // J. Amer. Chem. Soc. 1984. Vol. 106. P. 8004.
Gnaim, 1991 Gnaim J.M., Green B.S., Arad-Yellin R., Keehn P.M. Improved preparation of the
clathrate host compound tri-orto-thymotide and related trisalicylide derivatives//
J. Org. Chem. 1991. Vol. 56. P. 4525.
Guilera, 1999 Guilera G, Steed J. W. Topological control in coordination polymers by noncova-
lent forces//Chem. Commun. 1999. P. 1563.
Harata, 1985 Harata K., Ukema K., Otagiri M., Hirayama F. The structure of the cyclodextrin
complex. 18. Crystal-structure of beta-cyclodextrin benzyl alcohol A-1) complex
pentahydrate // Bull. Chem. Soc. Jpn. 1985. Vol. 58. P. 1234.
Hardy, 1979 Hardy A.DM., McKendrick J.J., MacNicol D.D. Alternation of cage geometry by
systematic structural modification of a clathrate host molecule // J. Chem. Soc,
Perkin Trans. 2. 1979. P. 1072.
Hare, 1992 Hare J.P., Kroto H. W. A postbuckminsterfullerene view of carbon in the galaxy //
Ace. Chem., Res. 1992. Vol. 25. P. 106.
Herbstein, 1977 Herbstein F.H., Marsh R.E. The crystal structure of trimesic acid, its hydrates and
complexes. II. Trimesic acid monohydrate 2/9 picric acid and trimesic acid 5/6
hydrate // Acta Crystallogr. B. 1977. Vol. 33. P. 2358.
Herbstein, 1987 Herbstein F.H., Kapon M., ReisnerG. Catenated and noncatenated inclusion
complexes of trimesic acid//J. Inclusion Phenom., 1987. Vol. 5. P. 211.
Hollingsworth, 1995 Hollingsworth M.D., Brown M.E. Stress-induced domain reorientation in urea
inclusion-compounds // Nature. 1995. Vol. 376. P. 323.
Holman, 1997 Holman K.T., Steed J.W., Atwood J.L. Intra-cavity inclusion of [CpFe"(arene)]+
guests by cyclotriveratrilene// Angew. Chem., Int. Ed. Engl. 1997. Vol. 36. P. 1736.
Hunter, 1991 Hunter C.A. Molecular recognition of para-benzoquinone by a macrocyclic host //
J.Chem. Soc, Chem. Commun. 1991. P. 749.
442
5. Связывание нейтральных молекул
Jarvi, 1980 Jarvi E.T., Whitlock H.W. Synthesis and characterisation of 1,8,15,22-
tetraoxa[8,8]paracyclophane-3,5,17,19-tetrayne-10,25-dicarboxylic acid, a
novel, water-soluble and donut shaped molecule // J. Amer. Chem. Soc. 1980.
Vol. 102. P. 657.
Kearney, 1993 Kearney P.C., Mizoue L.S., Kumph R.A. et al. Molecular recognition in aqueous-
media — new binding-studies provide further insights into the cation-я interaction
and related phenomena//J. Amer. Chem. Soc, 1993. Vol. 115. P. 9907.
Lavelle, 1989 Lavelle L., Nassimbeni L.R., Niven M.L., Taylor M.W. Studies in Werner
clathrates. 2. Structure and thermal-analysis of bis(isothiocyanato)tetrakis-
D-phenylpyridine)nickel(II) benzene clathrate A-4) //Acta Crystallogr. С 1989.
Vol. 45. P. 591.
LePree, 1994 LePree J.M., Mulski M.J., Connors K.A. Solvent effects on chemical processes. 6.
The phenomenological model applied to the solubility of naphthalene and
4-nitroaniline in binary aqueous-organic solvent mixtures // J. Chem. Soc,
Perkin Trans. 2. 1994. P. 1491.
Lipkowski, 1979 LipkowskiJ., Sybilska D., Pawlowska M. Experimental study of selectivity and
column efficiency in clathrate chromatography using Werner complexes as clathrate
host components. I. Relationship between selectivity of Ni(NCSJD-methylpyri-
dineL • guest clathrate sorbents and composition of the mobile phase //
J. Chromatogr. 1979. Vol. 177. P. 1.
Loon van, 1992 LoonJ.-D. van, HeidaJ.F., Verboqm W., Reinhoudt D.N. Calyx[4]arenes as building-
blocks for molecular receptors // Rec. Trav. Chim. Pays-Bas. 1992. Vol. 111. P. 353.
Odashima, 1980 Odashima K., IitaiA., Iitaka Y., Koga K. Host-guest complex formation between a
water soluble polyparacyclophane and a hydrophobic guest molecule // J. Amer.
Chem. Soc. 1980. Vol. 102. P. 2504.
Prins, 1999 Prins L.J., Huskens J., de Jong F. et al. Complete asymmetric induction of
supramolecular chirality in a hydrogen-bonded assembly // Nature, 1999. Vol.
398. P. 498.
Reichard, 1988 Reichard С Solvents and solvent effects in organic chemistry. 2nd ed. Weinheim:
VCH, 1988.
Ripmeester, 1987 RipmeesterJ.A. A new clathrate hydrate structure // Nature. 1987. Vol. 325. P. 135.
Rudkevich, 1997 Rudkevich DM., Hilmersson G., Rebek J., Jr. Intramolecular hydrogen bonding
controls the exchange rates of guests in a cavitand // J. Amer. Chem. Soc. 1997.
Vol. 119. P. 9911.
Seel, 1992 Seel C, Vogtle F. Molecules with large cavities in supramolecular chemistry //
Angew. Chem., Int. Ed. Engl. 1992. Vol. 31E). P. 528-549.
Sijbesma, 1995 Sijbesma R.P., Nolle R.J.M. Molecular clips and cages derived from glycoluril //
Top. Curr. Chem. 1995. Vol. 175. P. 25.
Steed, 1996 Steed J.W., Zhang H., Atwood J.L. Inclusion chemistry of cycloveratrylene and
cyclotricatechylene // Supramol. Chem. 1996. Vol. 7. P. 37.
Suzuki, 1994 Suzuki Т., Nakashima K., Shinkai S. Very convenient and efficient purification
method for fullerene (C-60) with 5,11,17,23,29,35,41,47-octa-rm-
butylcalix[8]arene-49,50,51,52,53,54,55,56-octol//Chem. Lett. 1994. P. 699.
Timmerman, 1994 Timmerman P., Verboom W., van Veggel F.C.J.M. et al. An organic molecule with
rigid cavity of nanosize dimentions // Angew. Chem., Int. Ed. Engl. 1994. Vol. 33.
P. 1292.
Udachin, 1999 Udachin K.A., Ripmeester J.A. A complex clathrate hydrate structure showing
bimodal guest hydration // Nature, 1999. Vol. 397. P. 420.
Литература
443
Vbgtle, 1985 Vogtle F., Men Т., Wirtz H. Large carbocyclic cavities for the selective inclusion of
organic guest molecules in aqueous-solution // Angew. Chem., Int. Ed. Engl.
1985. Vol. 24. P. 221.
Warmuth, 1997 Warmuth R. o-Benzyne: strained alkyne or cumulene? — NMR characterisation in
a molecular container//Angew. Chem. Int. Ed. Engl. 1997. Vol. 36. P. 1347.
Webb, 1993 Webb Т.Н., Wikox C.S.. Enantioselective and diastereoselective molecular
recognition of neutral molecules// Chem. Soc. Rev. 1993. P. 383.
Whitlock, 1990 Whitlock B.J., Whitlock H.W. Concave functionality - design criteria for non-
aqueous binding-sites // J. Amer. Chem. Soc. 1990. Vol. 112. P. 3910.
Zhang, 1995 Zhang H., Steed J. W., AtwoodJ.L. Inclusion chemistry of cyclotetraveratrylene //
Supramol. Chem. 1995. Vol. 4. P. 185.
Zimmerman, 1993 Zimmerman S.C. Rigid molecular tweezers as hosts for the complexation of
neutral guests//Top. Curr. Chem. 1993. Vol. 165. P. 71.
Предметный указатель
Автокатализ 687
Агонист 100
Агостическое взаимодействие 548
Аденозиндифосфат (ADP) 65, 66,
240, 252, 256, 782, 784, 785
Аденозинмонофосфат (AMP) 779,
782, 784
Аденозинтрифосфат (АТР) 65, 66,
240, 252, 256, 778, 779,
782-786
депротонированная форма
784
переходное состояние,
реакция дезаминирования 773
свойства 65, 66
синтез 84, 86, 785, 786
ЯМР-спектр 31Р
Азакоранды
аналоги 141
антрацензамещенный 719
макроциклы 252—260
Азакрауны 129-135
Азид-анион, криптаты бис(трена)
250
Азобензол, переключаемое
связывание 737-739
Активный центр 770
Алкалиды 206-209
Алкиламмония катионы
свободные энергии
связывания 194
связывание криптофанами
191,415
Алмазоподобные решетки
529-534
Аминные лиганды 204
Аминокислоты белков 104—106,
метаболизм, метилмалонил-
СоА-мутаза 97, 98
/)-Аминоэфиры, трансацилиро-
вание 788
Аммония катионы 155—157,
191-194
связывание азакорандами,
геометрия типа «насест» 191 — 193
связывание корандами
191-193
связывание лариат-эфирами,
стерические ограничения 155
связывание трехмерными
хозяевами 194
Анализ набора графов,
водородные связи 501—504
Аналоги гема
биологические имитаторы
797-808
модели, не содержащие железа
807, 808
модели связывания и
транспорта кислорода 797—803
модели цитохромов Р-450
804-808
Анаэробные организмы 89
8-Анилино-1 -нафталинсульфонат
(ANS) 47, 260, 396
Анион BF^, кристаллическая
структура комплекса 289
Анионы
нуклеофильность 146
свойства 238, 239
Предметный указатель
Антенное устройство светоакку-
мулирующее 82,83,760,761,
хлорофилл а 80-82
каскад переносов энергии 82
Антикрауны 246, 282-286
комплекс ртуть- 12-краун-4
на основе карборана с С1~ 285
сэндвичный полимер
комплекса с Вг~ 283
Антрацен, система
краун—антрацен 719
Апоферменты 97, 809
Аргинин 242,
«солевой мостик», 243
Ароматические кольца
п—тг-взаимодействия 55, 56
эффекты кольцевого тока 197
Ароматические системы, правило
Хюккеля 78, 79
Ароматические углеводороды
с конденсированными
кольцами (FAH) 551
Архимедовы тела 607, 621
Асимметрическая ячейка 513, 556
Атропоизомеры 800
Ацетилхолин (АСН) 100, 101
Аэробные организмы 89
АТРазы 65, 75, 76
имитация корандами 782—786
свойства 65
транспортное действие 65—69
ферментативное действие 75
Na+/K+-ATPa3a 65-68, 75-77,
782
Бакелит 210
Бакминстерфуллерен 347,
430-438, 619-621, см. также
Фуллерены
редокс-чувствительное
устройство 435
темплат 619
Бактериальные анионтранспорт-
ные белки 241, 242
белки, связывающие сульфаты
241,242
белки, связывающие фосфаты
241
Бактериофеофетин 84
Бактериохлорофилл а 80, 81
Барбитураты 363, 364, 402-404
Бенгальский розовый,
ассоциация дендример-DAB 760
Бенз-1,2-ин 428,429
Бензол
4,4 '-диметоксиазоксибензол
826
квадрупольный момент,
катион—тс-взаимодействия 53—55
кристаллическая структура 56
разделение смесей с помощью
жидких клатратов 841
п—тг-стэкинг-взаимодействия,
55,56
6:1-цикламер циклогександи-
онбензол 495
я-Бензохинон, связывание
404-406
Бибрахиальные лариат-эфиры
(BiBLE), 132, 141,224,
одновременное образование
четырех связей 132
Бидентатные лиганды 36, 135
Биметаллические системы,
фотохимия 702-704
Биологические модели,
структурная и функциональная
768-770
446
Предметный указатель
Биологические системы 64— 120
гемоглобин, связывание
и транспорт кислорода 88—96
ДНК 101-115
катионы щелочных металлов
65-76
кофермент (витамин) В12
96-99, 809, 810, см. Цианоко-
баламин
нейротрансмиттеры и гормоны
99-101
порфириновые и тетрапир-
рольные макроциклы 78, 79
фотосинтез 79-88
Биомиметика 767—810
Биомиметические структуры
562-567
Биоминералы 562—564
Биохимическая самосборка
115-119,580-584
вирус табачной мозаики 580,
581
ковалентная модификация
583,^584
строгая самосборка 581—583
2,2'-Бипиридил (bipy) 50, 112,
660,704-710,712,761,763
4,4'-Бипиридил (bipy) 593
Бислои 816, 817, 819, см. также
Мембраны биологических
клеток
Бис(трен) 250-252
Благородные газы 57
Большие молекулы, см. также
Супрамолекулярные частицы
696, 697
Бомбикол 768
л-т/?ет-Бутилкаликс[4]арен 57, 58
катализаторы межфазного
переноса 217-219
комплекс тетраэтилового
эфира с Na+ 213
конформеры 212
растворимость 217
Валиномицин 70, 71
Взаимодействия Ван-дер-Ваал ьса
57,246,418
дисперсионные (силы
Лондона) 57
обменно-оттал кивающие 57
Взаимодействия, типы
агостические 548
водородные связи 51—53,
494-501
с гидридами металлов
549-551
с лариат-эфирами, стеричес-
кие ограничения 157
с металлами 548, 549
диполь-дипольные 51,321
ион-дипольные 50
ион-ионные 48, 49
катион—тс- 53—55
координационные 286—290
координационно-клатратные
516
межмолекулярные 494, см.
также Взаимодействия Ван-
дер-Ваальса
немолекулярные 48—60
я—тг-стэкинг- 55—57,
катенаны 637—648
супрамолекулярные
взаимодействия 48—60
Виагра 117-119
Витамин В12 96-99, 809, 810, см.
Цианокобаламин
Предметный указатель
447
модели, биологические
имитаторы 809,810
производные 96
Вода
декамер 605, 606
кислотность 795
окисление до кислорода,
катализированное марганцем
85-88
равновесие, описывающее
изменение жесткости воды
564
Водородные связи 51,183,276,
277, 402-407, 495-504,
535-551,813,832
амиды 535, 536
анализ набора графов 501—594
бифуркационные 499
в ДНК 51-53, 107
в молекулярных устройствах
711,712
включение гостя, циклофан
402-407
димеры карбоновых кислот
52, 535
жидкие кристаллы 832
закрытые комплексы,
самосборка 613-628
инженерия кристаллов
535-542
комплекс краун-эфира с нит-
рометаном 54
мотивы «ограда курятника» и
«лестница» 540, 541
мотивы распознавания 724,
725
нейтральные хозяева 276, 277
нетропсин 114
определение мотива 502
свойства 52
с металлами и гидридами
металлов 548—551
взаимодействие с гидридами
металлов 549—551
прямое взаимодействие
с металлами 548, 549
с монооксидом углерода
542-544
сильные 51-53, 498
системы М-Н--Н-Х,
параметры 549-551
слабые 51-53,500,501,
544-547
смешанные кристаллы аминов
с аминами 541, 542
смешанные кристаллы
спиртов и аминов 540, 541
спирали 676—679
спаривание оснований по Уот-
сону и Крику 52, 106, 167
спаривание оснований по Хуг-
стену 107
спирты 540
средние 51-53,498-500
темплатные эффекты 586
ферроценильный редокс-ак-
тивный хозяин 724
фосфаты и сульфаты 241, 242
КНС2О4 53
C-H-N 525, 526
С-Н-0 495, 543
О-Н-0 495,525
Х-Н-Х 495,496
Вольтамперометрия циклическая
267-269
Вольфрамат, модель для тетраэд-
рических анионов 243
Ворота ионного канала 69, 220
Времена релаксации ЯМР
хозяина и гостя 196
448
Предметный указатель
Вспомогательная связь 648—659
Вторая гармоника,
индуцированная электрическим полем
(EFISH) 750
Галогенид-анионы
константы связывания 270
координационные числа 249,
250
Галогенуглеводороды, связывание
криптофанами 411—414
Гексафторид серы 333
Гексациклен 141, 142, 252, 253
Геликанд двухдоменный,
потенциальные комплексы 662
Геликаты 289,660-679,
[4 + 4]геликаты 665-667,
кристаллическая структура
666
[6 + 6]геликаты 667-669,
кристаллическая структура
669
наноциклы 675
номенклатура 660
полиядерные 683, 684
самораспознавание и
позитивная кооперативность 669—674
синтетические аспекты 663,
664
спирали, образованные
водородными связями 676—679
темплаты и самосборка
660-669
Гем 78,89
комплексы 78
модели Полинга и Вайсса 91,
92
тетрапиррольные макроциклы
78,79
Гемоглобин 89
аллостерический эффект 95
кооперативность, управляемая
белком 804
миоглобин 95
насыщение кислородом 95
связывание и транспорт
кислорода 88—96
сродство к кислороду, влияние
аксиальных лигандов 804
структура 89
Гемоцианин, модели,
биологические имитаторы 793—795
Генерация второй гармоники
(ГВГ) 746
Генетически модифицированные
(GM) продукты 108
Генетический код 105, 106, 108
Генная инженерия 108,109
Геометрия типа
«гнездо» 93, 177, 194,213,262
«капсула» 33, 177
«насест» 33, 177, 178, 191, 194,
262,264,283,358,615
Гербициды 204-206
Гетерогенное разбавление 392
Гетерокраун-эфиры 181—184
Гетерокриптанды 184, 185,
константы связывания катионов
Ag+, К+иТ1+ 184
Гибридные хозяева катионов 139
Гигантские самособирающиеся
капсулы 619—625
«Гидридная губка» 278, 279
другие льюисовские
кислотные хелаты 278-281
«протонная губка» 279
хелатное поведение аниона
279
Гидриды металлов 549—551
Предметный указатель
449
Гидроксиапатит, биоосаждение
565
Гидрофобные эффекты 58—60,
354,399,400,406,816
связывание органических
гостей в водном растворе 59
энтальпийные и энтропийные
59
Гидрохинон 332—336, 433
структура полости 334
фенол и соединение Дианина,
стратегия шести хозяев
332-336
Гиперполяризуемость 736
Гликолурил 367—369, 615—618
Глины 311-315,359
Глиоксалазы, их имитация 781
а-Глицин 503, 506-508
кристаллическая упаковка 506
наборы графов 503
пирамидальные кристаллы,
монослои МБ-пальмитоил-
(ад-лизина 508
Голоферменты 97
Гомохиральные стопки 329, 330
Гормоны 99, 100
Гости 320
динамика обмена гостями
198-200
связывание в полярных и
неполярных средах 403
стадия, определяемая гостем
(GDS) 422
темплатирование 603
Гость, определение 28, 29, 244
График Вант-Гоффа 412
График Джоба 46
Графит 312,314-317
Гуанидиния катион 193, 242,
262-265
рецепторы на основе
гуанидиния 262-265
комплекс метилгуанидиния
с дигидрофосфатом 262,
кристаллическая
структура 263
хиральный хозяин, синтез
263
центр связывания анионов
242
связывание 193
Гуанозинтрифосфат (GTP)
117-119
Дансиламида производные,
сенсоры на цинк, флуорофоры
721-723
Дендримеры 754-763
дивергентный синтез 756, 757
итерационное получение 754,
755
конвергентный синтез 756—758
фотохимические устройства
на основе дендримеров
760-763
химия хозяин—гость для
дендримеров 759, 760
Диаграмма Скэтчарда 672, 673
1,4-Диаминобутил (DAB) 760
Диаммония катионы,
распознавание 193, 195
Диарилмочевины 521,522
Диастереомерия 631, 652,
тройной узел 631
Диатомеи, микроскелет 565
1,2-Дигидроксибензол 673
Дигидрофосфат калия 529
а,со-Дикарбоновые кислоты
гости 256
димеризация 532, 535
29- СТИДДж.В
450
Предметный указатель
распознавание в соответствии
с длиной 257
Дикват, связывание 204, 205, 634
Димеры типа «мяч для софтбола»
615-618
капсулирование гостя 616, 617
присоединение по Дильсу-Аль-
деру, катализируемое
хозяином 617
самосборка 615—618
Димеры типа «теннисный мячик»
613-616
кристаллическая структура 614
самосборка 613—616
связывание гостя, константы
616
Диметилртуть 336
Д и метил формам ид (DM F) 423
механизм обмена метальных
групп 199
Динамика обмена гостями 198
Динамическое спаривание
хозяин—гость 196
Дисклинации 825
Дисперсионные взаимодействия
при анионном связывании
56,244 '
Дистамицин А 114
Дитопные рецепторы 195,196,
256, 257, 287
линейное молекулярное
распознавание катионов
диаммония 196
Дифенилгликолурил 613—618
Дифенилметан 260, 396-401
Дифильные рецепторы 68, 203,
204, 815, 816, еж
Поверхностно-активные вещества
Дифрактометр, прибор
с зарядовой связью (CCD) 73
Дифракция нейтронов 497, 544
Дифференциальная
сканирующая калориметрия (DSC) ,
соединения включения
349-353
Диффузионное рентгеновское
рассеяние 320, 325
ДНК 101-115
водородные связи 51
двойная спираль 51, 102
интеркаляция 112—115
направленный мутагенез 108,
109
спаривание оснований 52, 106,
107, 578
самосборка 581—583
связывание 110-115
структура и функции 101 — 107
полимеразная цепная реакция
(PCR) 109, ПО
чувствительность комплексов
114
Домены связывающие 660
Железа катионы Fe2+ и Fe3+
89-96, 228-232, см. также
Гем, Гемоглобин, Миоглобин,
Порфирины
гексацианоферрат(П)-анион
Fe(CNN]4~, суперкомплексы
253, 254
железосодержащие лиганды,
см. Сидерофоры
оксиды как биоминералы 563
хелатные Ре3+-лиганды,
величины рМ 231
Жесткие ионы металлов 179
Жидкие клатраты 837—845
ион оксония в химии жидких
клатратов 842—845
Предметный указатель
451
открытие и свойства 837—841
Жидкие кристаллы 677, 814,
820-837
дизайн 831-833
жидкокристаллические
дисплеи (LCD) 835-837,
закрученные нематические ячейки
(TNC) 836
жидкокристаллические
полимеры 834,835
классификация 823, 824
применение 835—837
природа и структура 820—831
Жидкие фазы 813—846,
поверхностно-активные вещества и
упорядочение на поверхности
раздела 814-820
Жидкокристаллические фазы
голубые 829
дискотические 824,831
лиотропные 824, 830
нематические 824,826,828,
830,834
смектические 824—827, 829,
834
столбчатая 830, 831
сэндиковая 834
трубчатые 823, 825
холестерические 824, 825, 828,
829, 834,835
Жидкостно-мозаичная модель
биологической клетки 819,
820
Жуки, окраска 835
Закрученные нематические
ячейки (TNC) 836
Закрытые комплексы 613—628
гигантские самособирающиеся
капсулы 619—625
димеры типа «теннисный
мячик» и «мяч для софтбола»
613-618
розетки 625-628
самосборка с помощью
водородных связей 613—618
«Замок—ключ», модель Фишера,
аналогия 34, 35, 767, 768
Змеи,спиральность 661
Изменение жесткости воды 564
Изменение pKfl макроцикла 255
Изоляция центра при росте денд-
римера 759
Изомерия, обусловленная
растяжением связей 570—572
Изомеры электронные 702, 709
Имидазолия катион, комплексо-
образование 193
Имитаторы трансацилаз
786-789
Индукция хиральности в
кристаллах 508-510
Индуцированное соответствие
243, 603, 768, 773, 774, см.
«Замок—ключ» 768
Инертные комплексы 600—602
Инженерия (конструирование)
кристаллов 491—573
алмазоподобные решетки
529-534
биомиметические структуры
562-567
водородные связи 494—501,
с монооксидом углерода
542-544
дифракция нейтронов от
монокристалла 497
история 491—494
45Й
Предметный указатель
Кембриджский банк
структурных данных 525—528
координационные полимеры
558-561
межмолекулярные
взаимодействия 494
«неправильно» подобранные
партнеры 556, 557
определение 493
предсказание структуры
кристаллов 517—524
прочие взаимодействия 554,
555
разрушение кристалла 514,
515
самосборка 491,492
синтез 492, 493
соединения включения типа
«песочные часы» 567—570
стратегии дизайна 515—517
71—71-стэкинг-взаимодействия
553, 554
хозяева типа «колесо—ось»
555,556
Инсулин 583, 584, 648
Интеркаляторы 112, сродство
кДНКПЗ
Интеркаляты 311-317,
классическая модель и модель Дау-
маса- Геролда 313,314
Информация и сигналы 716-727
нервный импульс 67
семиохимия 716-727
флуорофоры 721-724
фотохимические сенсоры
717-721
электрохимические сенсоры
724-727
Иодид-анион, связывание тетра-
эдрическими рецепторами
248
Ион Ru(II)-Ru(III) Крейтца-
Таубе 702
Ионизация электрораспылением
(ESI) 756
Ионная сила 273, 274
Ионные каналы 67—69
Ионофоры 68, 69
трансмембранные 126, 127
транспортные свойства 132
хромоионофор 128
Ион-селективные полевые
транзисторы (ISFET) 725
Кавиплекс 354
Кавитанды 32, 354-386, 408
определение 32,354
синтез на основе цикловера-
трилена (CTV) по Краму 408
строительные блоки 308, 343,
354, 355
Кавитаты 33,354
динамика обмена гостей
364-366
определение 32, 354
Калид 207
Калия катион К+
биохимия 65—76
мембранный транспорт 69—76
связывание, константы
172-175
гибидными каликсаренами
219-223
лариат-эфирами 155
содержание в биологических
системах 66
темплатный эффект для мак-
роциклических соединений
161, 162, 164
флуоресцентный сенсор 719
Предметный указатель
453
Калия соли
реакция с бензилхлоридом
в ацетонитриле 147,148
сульфат калия,
кристаллические формы 570
Каликсарены 209-223, 271,
354-364,431,432
я-трет-бутилкаликсарены
57, 58, 212, 217, 218, конфор-
меры 212
имитаторы ферментов 212
история 210—212
каликс[4]арен, структура
234
каликс[5]арен 212, 724,
электрохимический сенсор 724
каликс[4]резорцинарен 619
кристаллические структуры
комплексов 432
я-метилкаликс[4]арен, синтез
по Хайесу и Хантеру 210
определение комплекса 33, 34
равновесие межфазного
переноса катионов 217—219
внутриполостное
связывание Cs+ 218, 219
связывание катионов
металлов 218
разделение смеси С60 и С70
430
связывание анионов 270, 272,
255, 256
связывание катионов 213—217
конформационные аспекты
включения 215
туннелирование Ag+ 216
Ag+—Ti-комплекс 216
связывание катионов
гибридными каликсаренами
219-223
макротрициклическая
бис(каликсареновая
клетка) 219-221
«перекачка» металла
«молекулярным шприцем»
222
процессы обмена катионом
металла, внутри-
и межмолекулярные 221
связывание К+ 219-222
Каликс[4]пиррол 276
Калориметрическое титрование
47
Кальция катион Са2+
биоминералы 563—565
переносчик Са2+, поданды
126
сенсоры, флуорофоры 721
Карбид молибдена 149
Карбоангидраза (СА) 721,795
Карбоновые кислоты
димеризация 535, 536
водородные связи 536
нуль-, одно- и двухмерные
сетки 532
кристаллические структуры
531-533,535,536
продукты совместной
кристаллизации с 2-аминопириди-
ном, правила Эттер,
предсказание структуры 523
Карбоксипептидаза А, рецепторы
анионов 243, 244
Карбонилы, диполь-дипольное
взаимодействие 51
Карбоплатин 110,111
Каровиологены 727, 728
Карцеплексы 417, 419, 423-426,
полукарцеплексы 417,420,
421
454
Предметный указатель
Карцеранды 417-423
карцерия 425, 426
определение и синтез
417-421
полукарцеранды 417,421,
617,619
реакции включения 426—429
связывание 423, 424
темплатные эффекты в синтезе
карцерандов 421-423
Каскадные молекулы, см. Денд-
римеры
Каталановы тела 621
Катализ краун-эфирами 145,146
Катализ супрамолекулярный
685-690
Катализируемые и некатализиру-
емые реакции 773
Каталитические и
самовоспроизводящиеся системы 685—690
Каталитический крекинг 311
Катапинанды 237-240, 244, 245,
286
определение 237
связывание анионов 245
Катапинаты 248, 249
Катенаны (катенанды) 163, 206,
404, 598, 628-659
бис([2]катенан), синтез 647,
648
динамические свойства 638,
639
[2]катенан 629-632,637,641,
643, 647
вспомогательная связь,
трилистный узел 648-650,
680
выход 640
кристаллические структуры
639, 658
направленный синтез
649-654
темплатный синтез 638
электрохимически
контролируемые
перемещения циклов 745
ЯМР-спектр !Н 656
[3]катенан 629,641-643
изомеры 649—651
синтез 648—651
синтез под высоким
давлением 642
[4]катенан 641,643,645
[5]катенан, первый синтез 641,
643-646
кинетические и
термодинамические эффекты 587
макроциклизация в условиях
высокого разбавления 653,
655
молекулярные машины
740-745
номенклатура 629
олимпиадан 645,646
определение 628, 629
принцип вспомогательной
связи в синтезе катенанов
648-659
самосборка 598
синтез 630,632,648-659
статистический подход 632,
633
стерически управляемая
циклизация 651
71—Ti-стэкинг-взаимодействия
637-639
топологическая изомерия 631
трансляционная изомерия
743
хиральность 631
Предметный указатель
455
Катенаты 628, 629, 655-659
кристаллическая структура
658
синтез 656—659
Катион бипиридилдиамминпла-
тины(П), кристаллическая
структура 205
Катион-тг-взаимодействия
53-55, 100,214,218,224,839,
845, схематическое
изображение, квадрупольный момент
бензола 55
Катионы, свойства 239
Катионы металлов (кислоты
Льюиса), координационные
взаимодействия 286—290
Катионы щелочных металлов
65-76, 179
константы связывания 181
мембранные потенциалы
65-68
мембранный транспорт
68-76
родопсин 76, 77
размер и свойства 179
Кинетика
связывание катионов 130,177
связывание нейтрального
гостя 364-366, 422
Кинетическая селективность
41-48, 149-159
Кинетическая стабильность
комплекса 600
Кинетический темплатный
эффект 131, см. Темплатный
эффект
Кислород 88-96, 793-795,
797-804
аэробные и анаэробные
организмы 88,89
модели связывания и
транспорта, аналоги гема, порфи-
рины 797-804
связывание и транспорт
гемоглобином 88—96
Кислотность С-Н-кислот,
корреляция с длиной водородной
связи C-H--0 545
71-Кислоты 186,187,223-227
Классификация соединений
хозяин-гость 32-34
кавитат и клатрат 32, 33
нейтральные хозяева 33
пространственное
соотношение между гостем и хозяином
33
Клатранды, определение 32
Клатратный хлоргидрат 31,295,
296
Клатраты 32, 34, 295,
применение 295
Клатраты жидкие 837—845
Клатраты мочевины 318—324
алканоны и алкандионы
321-323
гости 320
полимеризация при
включении 323
соразмерные и несоразмерные
структуры 318—324
структура 318, 319
Клатраты неорганические
твердофазные 296-318
история 296
клатратные гидраты 296—302
образование 296, 297,
проблемы в газовой
промышленности 301, 302
применение 301, 302
свойства 300, 301
Предметный указатель
сравнение размеров гостей
и полостей, занятых, как
в простых гидратах 299
структуры 297—299
клатраты Хофмана и Вернера
317,318
твердые слоистые материалы
и интеркаляты 311—317
графитовые интеркаляты
315-317
классическая модель
и модель Даумаса—Геролда
313,314
характеристики и классы
311,312
цеолиты 303-311
Клатраты твердых органических
хозяев 318—353
гидрохинон, фенол и
соединение Дианина 332—336
канальные клатраты 324—332
клатраты мочевины 318—324
пергидротрифенилен 329—332
спиральные тубуланды
327-329
тетрафенилен 348, 349
тримезиновая кислота (ТМА)
324-327
гексагональное
расположение молекул 326
структура слоя моногидрата
324,325
три-о-тимотид 337-343
циклотривератрилен (CTV)
343-348
«Книга кельтов» 679
Кобаламины 78, 96, 97
тетрапиррольные макроциклы
78
цианокобаламин, см.
Витамин В,2, модели,
биологические имитаторы 809, 810
Кобальтоцений 266
Кобальтпорфириновый комплекс
797, 798
Ковалентные связи
деформационное
укорачивание 496
самосборка с ковалентной
модификацией 583,584
Коелентеранды, определение 225
Комплекс карбида молибдена
148, 149
Комплекс кислородвыделяющий
(ОЕС) 85-88
Комплекс «матрешка» 384
Комплекс переходного металла,
молекулярно-орбитальное
описание 700
Комплекс PF^~, трехкатионный
хозяин подандного типа 272
Комплексы из лигандов типа
«жесткий стержень» 609
Комплексы меди 39
Комплексы типа «рейки»,
«лестницы» и «решетки» 609—612
Комплементарность 40,
169-176, 193
определение 170
предорганизация 169—176
связывание хозяин—гость
40-41
Комплементарные
(дополняющие) цвета 81
Конденсация аминов с
альдегидами 391
«Коническая поправка» 499, 500
Конкин 370-372
Константы ассоциации, см.
Константы связывания
Предметный указатель
45',
Константы образования, см.
Константы связывания
Константы связывания 42—48
галогенид-анионы,
зависимость от длины спейсера 270
гетерокриптанды 184
двухмерные хозяева,
связывание анионов 256,257
жесткие ионы металлов 181
измерение 42—48
катионы
Ag+ 184
Ва2+ 158, 181
К+181, 184
РЬ2+ 181
Т1+ 181, 184
краун-эфиры 129, 151
криптанды 158,184
лариат-эфиры 129, 156, 157
мягкие ионы металлов 181
образование суперкомплексов
255
поданды 129
связывание 43
«Конструирование вниз» 577
Конструирование хозяина 60, 61
Конструирование хозяина для
анионов 244—247
антикрауны 282—286
биологические рецепторы
анионов 240—244
«гидридная губка» и другие
льюисовские кислотные хела-
ты 278-282
константы связывания,
длина спейсера
дикарбокси-гостя 256, 257
[Рс1С14]2~-комплекс
соединения H10[30]aH-N{§+
254
расширенный порфирино-
вый хозяин с капсулиро-
ванным фторид-ионом 258
суперкомплексы азакоран-
дов 255
изменение рН 247-261
двухмерные хозяева
252-260
селективность формы
249-252
тетраэдрические рецепторы
247-249, криптанд
«футбольный мяч» и его
криптат с хлорид-анионом
249
циклофановые хозяева
260,261
концепции 244—247
координационные
взаимодействия 286-290
металлоорганические
рецепторы 265-273
нейтральные рецепторы 273,
274
основность Льюиса анионов
246
рецепторы на основе гуаниди-
ния 262—265
Конформации каликсаренов
1,2- и 1,3-альтернаты
(чередующиеся) 211,212,214,216,
277
коническая (конус) 212,214,
216
частично коническая
(частичный конус) 212,214,216
Концентрационный градиент
67,68
Концепция жесткой концевой
группы 128, 392
458
Предметный указатель
Кооперативность (модель),
влияние аксиальных лигандов
на сродство к О2 803, 804
Кооперативность
позитивная 669—674
негативная 673
Координационная геометрия
анионов 287
Координационная химия 34, 35
«Координационно-клатратный»
подход Вебера 516
Координационное число 246, 247
Координационные
взаимодействия 286-290
комплекс с Н2РО^~ 288
координационная геометрия
анионов 287
Координационные полимеры,
инженерия кристаллов
558-562
Координационные (дативные)
связи 50
Координационные соединения
металлические ансамбли
609-612
«молекулярные квадраты»
и «молекулярные коробки»
597-608
принципы дизайна 593
самосборка 592-612
супрамолекулярный куб
593-596
Коранды 40, 140
донорные центры кислорода
203
как имитаторы АТРаз
782-786
макроциклический эффект 38
модель активности АТРаз 782
номенклатура 140—142
определение 140
структуры 41, 141
хиральные 786—789
хиральный барьер 200
хозяева, свободные энергии
связывания ионов аммония
192
хозяева, связывающие
катионы, как имитаторы трансаци-
лаз 786-791
Кораплекс 142
Кораты 142
«Королевский разрез» 661,662
Корреляция силы и длины
водородной связи 52
Корриновое кольцо, тетрапир-
рольные макроциклы 78, 79,
96, 809
Коферменты 96,97,781
В12 96,97
модели, биологические
имитаторы 809,810
производные 96, 97
F450, тетрапиррольные
макроциклы 78, 79
Краун-эфиры 121—126
аза-18-краун-6, комплекс с ни-
трометаном 54
азотсодержащие аналоги 252
ациклические аналоги (подан-
ды) 126
бензо-15-краун-5 148, 149
бензо-18-краун-6, темплатный
синтез 163
в химии жидких клатратов
842-845
гетерокраун-эфиры 181 — 184
диаза-18-краун-6, синтез
методом высокого разбавления
157, 166
Предметный указатель
459
диаметры полостей, сравнение
с диаметрами катионов
металлов 152
дибензо-18-краун-6 161, синтез
122, 125
дибензо-24-краун-8 153, 154,
843, 844
дибензо-30-краун-10 123,153,
154, паракват (метилвиоло-
ген) 204
дицикл огексил -14-краун -4
152
дицикл огексил-18-краун-6
123, изомеры 125, 126, 143
изменение константы
связывания
как функция радиуса
катиона металла 151
как функция размера
кольца краун-эфира 151
капсулирование катиона
металла макроциклом 154
12-краун-4 183
15-краун-5 123, 183
18-краун-6 122-126, 141, 143,
148, 150, 151, 153, 160, 162,
183
в органическом
растворителе и гидрофильной среде
144
ион оксония 842—844
комплекс с К+ 143, 160
комплекс с Ag+ 182
21-краун-7 123,843,844
30-краун-10 154
межфазный катализ 144, 145
модели Кори—Полин га—
Колтуна (СРК-модели) 136,
137, 194
номенклатура 140-142
окисление органических
субстратов КМпО4 146, 147
определение 122,
классификация комплексов 33
открытие 121 — 124
полимерные продукты
161-163
применение 124
растворимость 143, 144
ртуть-12-краун-4 на основе
карборана 285
связывание иона оксония
842-845
связывание катионов, плато
селективности 150, 151
связывание нитрометана 54
селективность связывания
катионов 149—154
синтез 122, 124-126, 161, 162
система краун—антрацен 719
скорость реакции 148
структуры 123
темплатный эффект 161,162,
164
торсионные углы 144
триаза-18-краун-6 247
трибензо-21-краун-7 154
угол захвата лиганда 152,153
Крахмал, получение цикл оде кст-
ринов 378, 379
Криоскопия 210
Криптанды 132-136, 157-159,
173, 176, 248, 249
гетерокриптанды, константы
связывания Ag+, K+ и Т1+ 184
константы связывания
катионов
щелочноземельных металлов 158
катионов щелочных
металлов 158
F0
Предметный указатель
криптофикс 132
макробициклический эффект
159
макротрициклические крип-
танды 247—249
макроциклический эффект
159
номенклатура 140—142
определение 140
переключаемое связывание
737
селективность связывания
катионов 157-159
сепулькраты и саркофагины
135, 136
синтез 132-136; [1 + 1]-,
[2+2]- и [3+3]-циклосокон-
денсация 167-169
спелеанды 203
структуры 134, 141
«футбольный мяч» 134,191,
248, 249
Криптасферанды 139, 194
Криптаты 142, 158, 160,738
бис(трена) 250,251
катионов щелочноземельных
металлов 158
смешанные, синергетический
эффект 185-187
спелеаты 203
«футбольного мяча» 248, 249
Криптофаны 407-417
комплексы с галогенуглеводо-
родами 411-414
комплексы с катионами
алкиламмония 415,416
комплексы с метаном 416,
417
конкуренция с растворителем
414,415
конструкция молекулярных
контейнеров из изогнутых
строительных блоков 407—411
синтез 409
Кристаллы, плотная упаковка
в твердом состоянии 58, см.
также Жидкие кристаллы
Критическая концентрация
агрегирования (САС) 400
Критическая концентрация ми-
целлообразования (ККМ)
818
Критическая концентрация
самосборки (ККС) 590
«-Ксилол, связывание циклофа-
новым хозяином 59
Кубен 531
Кубооктаэдры 607, 608
Кукурбитурил 367, 368
«Купольность» 79, 92-94, 798,
799
Лабильные комплексы 600—602
Лазерная десорбция из матрицы
(MALDI) 757
Лактаноидные я-сульфока-
ликс[4]ареновые ансамбли
623, 624
Лантаноидов катионы 126,163
Лантаноиды, светопреобразую-
щие устройства 712
Лариат-эфиры 129—131, 141,
155-157,223,224
бибрахиальные лариат-эфиры
(BiBLE) 132,141,155,224
водородное связывание NHJ,
стерические ограничения
155
комплекс с катионом металла
130
Предметный указатель
461
комплексы с К+ и Ag+ 223,
224
константы связывания 129,
156, 157
селективность связывания
катионов 149-159
N- и С-прикрепленные,
синтез 131
Ленты 625-627
Лёд
гексагональный AЛ) 296, 297,
300,301,606
кубический AС) 606
Лиганды моно- и бидентатные
36,37
Лиганды, связывающие железо,
см. Сидерофоры
Лиганды типа «жесткий
стержень» 609
Лиотропные жидкие кристаллы
818,822
Лиотропные фазы 823, 824, 830
Липофильность 61, 143
Лития катион Li+
арильные соединения 138
электрид 208
Локализованные валентности
702
Льюисовские кислотные хелаты
278-282
Люминесценция 698, 699, 736,
737
светопреобразующие
устройства 711,712
тушители 713
Магния катион Mg2+
в хлорофиллах 79—88
транспорт щелочных металлов
75,76
Макробициклический эффект
37-39, 159-161,250,251
Макротрициклическая бис(ка-
ликсареновая) клетка,
связывание катионов гибридными
каликсаренами 219—222
Макротрициклические криптан-
ды 247-249
Макроциклические азакоранды,
суперкомплексы 254, 255
Макроциклические лиганды 78,
79
ковалентный темплатный
синтез 586
межфазный катализ, эффект
«обнаженного» катиона 146
синтез методом высокого
разбавления 165—167
синтез с удалением серы 392
Макроциклический эффект
35-39,78, 159-161,282
Масс-спектрометрия 419, 437
дендримеры 756—758
катенаны 645, 646, 649, 656, 657
Материалы для нелинейной
оптики 746-754
второго порядка 749—753
генерация второй
гармоники (ГВГ) 745
переключаемое устройство
753
полиены типа «тяни—
толкай» 736,750,751
молекулярная поляризация
индуцированная 747
общая 747, 748
основы нелинейной оптики
746-749
коэффициент
гиперполяризуемости 748,749
462
Предметный указатель
поляризуемость 746, 747,
749
третьего порядка 753, 754
Машина, определение 695
Межмолекулярные
взаимодействия, инженерия кристаллов
494
Межмолекулярные
многоцентровые гетероакцепторные
водородные связи 549
Межфазный перенос 144,178,
217-219,259
Мезогены 820-831,834,835
Мезоморфные
(жидкокристаллические) соединения 820
Мезопоры 307
Мезофазы 822, 830
Меламин 625, структура
комплекса меламинциануровая
кислота 625-628
Мембранные потенциалы
65-68
Мембранный транспорт ионов
68-76, 127,273,388
канальный механизм 69
канальный механизм с
воротами 69
механизм с участием
переносчиков 69
Мембраны биологических клеток
68-70,814,819
Металлобиоцентры 792, 793
биологические имитаторы
792-796
модели гемоцианина 793-795
модели для имитации,
подтверждающие и
спекулятивные 792
Металлоорганические рецепторы
265-273
комплекс бис(амида) с Вг~,
кристаллическая структура
265, 266
комплекс каликсарена с С1~
271
комплекс поданда с PF^ 272
циклическая вольтамперомет-
рия 267-269
Металлопротеины 792
Металлы,
см. также Щелочные металлы
биметаллические системы
702-704
водородные связи с металлами
и их гидридами 548—551
катионы, равновесие
межфазного переноса 217—219
комплекс с лариат-эфиром
130
комплексы катион
металла—макроцикл 144—149
конструирование хозяина
60-61
координационные центры
792
металлические ансамбли
609-612
«молекулярные квадраты»
и «молекулярные коробки»,
самосборка 597—608
октаэдрический ион, диаграмма
кристаллического поля 601
перенос заряда от металла
клиганду(МЬСТ) 701
переходы, центрированные на
металле (М С) 701
селективность связывания
катионов 149-159
синтез «атом металла — пар
лиганда» 227
Предметный указатель
463
системы со смешанной
валентностью 702-704
суперкомплексы макроцикли-
ческих соединений 253
Метан 416,417
клатратный гидрат 299—302
связывание криптофанами 416
Метациклофаны 390,391
Метилвиологен, см. Паракват
Метиленовый голубой, структура
35, 807, 808
Метилимидазол (Melm),
структурная модель гема 798, 799,
801-804
Метилмалонил-СоА-мутаза,
аминокислотный метаболизм 99
Метод атом-атомных
потенциалов 518-520
Метод высокого разбавления
165-167,392,409,418
аппарат 165-167,392,409,418
Метод коалесценции 199, 200
Метод «молекулярного замка»
600
Метод растянутой тонкой
структуры рентгеновского
поглощения (EXAFS) 86, 87, 792,
793
Метоксид-ион 278
Микобактин 228
Миоглобин 89, 94-96
в гемоглобине 95
насыщение кислородом 95
Мицеллы 307,400,817-819,830,
обратные 818
«Мобил-процесс» 310,311
Модели Кори—Полинга—Колтуна
(СРК-модели) 136, 137, 178,
194,210,418,789
Модель Вайсса 92
Модель ионного насоса 67
Модель Полинга 92
Модель связывания Чатта 187
«Молекулярная карусель» 643
«Молекулярный поезд» 643
«Молекулярный челнок» 740,741
«Молекулярный шприц» 222, 223
Молекулярный конструктор
LEGO 371
Молекулярные выпрямители
732-734
«Молекулярные квадраты» и
«молекулярные коробки»
597-608
Молекулярные орбитали (МО)
701
«Молекулярные пинцеты»
386-389
Молекулярные провода 727—731
Молекулярные сита 306
Молекулярные узлы 652,
679-685
многократно сцепленные
соединения 683—685
пятилистный 683, 684
сборка 679-684
трилистный 652, 679, 680,
682, 683
Молекулярные устройства
695-763
дендримеры 754—763
информация и сигналы
(семиохимия) 716—726
материалы для нелинейной
оптики 746—754
молекулярные машины на
основе катенанов и ротаксанов
740-746
определение 695
философия 695
«464
Предметный указатель
фотохимия 698—716
«Молекулярные щели» 386—389
Молекулярные электронные
переключатели 734—740
аллостерические
переключатели 739,740
переключаемое связывание
737-739
система 1,2-дитиенилэтена как
переключатель 734—736
электропереключаемая
люминесценция 736, 737
Монодисперсность 756
Монооксид углерода
водородные связи 542—544
связывание в гемоглобине 94,
802
Монослой ПАВ 816,817
Морфосинтез 565
Мотив «ёлочка», см. Упаковка
молекул по типу «ёлочка»
Мочевина 113,318-324, ферро-
упругие свойства комплекса
323
Мыла 815,816,824
Мягкие ионы металлов 179
Мягкие лиганды для мягких
ионов металлов 179-190
гетерокраун-эфиры 181-184
гетерокриптанды 184, 185
катионы щелочных металлов
179
основания Шиффа 187-190
смешанные криптанды
185-187
теория жестких и мягких
кислот и оснований (теория
HSAB) 180
М ЕСАМ (комплексообразователь
железа) 230, 231, структура 230
Наноразмерная инженерия, см.
Молекулярные устройства
Направленный мутагенез 108,
109
Насыщенные водородными
связями (SHB) структуры 540
Натрия катион Na+
биохимия 65—77
содержание в биологических
системах 67
содид 207
транспорт, биологическая
мембрана 68, 69, 75, 76
Нейротрансмиттеры 99—101
Нейтральные молекулы,
связывание 295-438
внутриполостные комплексы
353-429
неорганические твердофазные
клатратные соединения
296-318
твердые клатраты
органических хозяев 318—353
фуллерены 430—438
Нейтральные рецепторы
273-277
анионсвязывающие хозяева
273
хозяева, образующие
водородные связи 276, 277
цвиттер-ионы 274, 275
Нековалентная координационная
химия анионов 237—240
Нековалентные взаимодействия
48-60
Нелинейно-оптические (НЛО)
свойства 330, 736, 746-754
Нервный импульс 67, 100
Нервные клетки, передача
информации 67
Предметный указатель
465
Несоразмерные структуры 320,
321,329,339
Нестехиометрические
соединения 319
Нетропсин, водородные связи
114
Никотин 100, 101
Никотинамидадениндинуклеотид
(NADH) 805,807
Никотин-ацетилхолиновый ре-
цепторный белок (nAChR)
100, 101
Нитроанилины 522
Нонактин 70, 114
Нуклеиновые кислоты,
самосборка 581-583
Нуклеозиды 103, определение 261
Нуклеооснования 103
Нуклеотидные полифосфаты
782-786
Нуклеотиды, определение 103,
261
Нуклеофил анионный 146
Нуклеофильное замещение 146,
391
Обобщенная координационная
химия 61
Образование центров
кристаллизации (нуклеация) 504, 505,
508-510
Однократная запись,
многократное считывание (WORM) 736
Оксид азота 117-119
Оксид кремния как биоминерал
563
Олимпиадан 645, 646
Оптическое вращение энантио-
мера 338
Органические гости,
гидрофобное связывание 59
Ортоциклофаны 391
Основание Трёгера 386, 399
Основания Шиффа 30,187-190
жидкие кристаллы 832
конденсация 30,187-189
макроциклические 136,188,793
оксигемоцианин 794
1:1 -циклоконденсация 189
«Офис» 609-612
кристаллическая структура 610
«полы» и «стены» 609, 611
Паракват(метилвиологен) 204,
205, структура 634
Параметр ЕТ C0) Димрота-Рей-
харда 402
Параметр Тафта (константа
индуктивности) 224
Парациклофаны 226, 227, 260,
261,390,391,394,401,402
Пептидные звенья 106
Пергидротрифенилен (РНТР)
329-332
Переключаемое связывание
737—739, см. также
Молекулярные электронные
переключатели
Переключатель
нелинейно-оптический 753
хромофорный молекулярный
744
Переключение 734—740
Перенос заряда от металла к ли-
ганду(МЬСТO01
Перенос электрона (еТ) 709—711
нековалентно связанные
системы 712—716
энергетические уровни 709
1466
Предметный указатель
Перенос энергии (ЕТ) 700, 712
фотосинтез 82-84
фотохимия 698—702
Переносчики катионов 178
Переносчики электронов 204
Переходные металлы
моноядерный комплекс,
диаграмма энергетических
уровней 701
реакции дикатионов 182, 183
редокс-активный металл, СО-
субстрат 185, 186
связывание 182, 225
Периферическое уплотнение 626
Пероксид водорода 242
Пигменты светособирающие 81,82
Пикраты щелочных металлов
константы скоростей
связывания и диссоциации 177
свободные энергии
связывания 169-175
экстракция 47, 48
Пиразин 423,424
Пиримидины 103
Пирокатехин, свойства 231
Платины катион Pt2+
катион бипиридилдиаммин-
платины(П) 204, 205
лекарства ПО, 111
связывание ДНК 110-112
Плато селективности 150,175
Платоновы тела 620, 621
Пленка Ленгмюра—Блоджетт 750
Поверхностно-активные
вещества (ПАВ) 814-819
классы 815,816
упорядочение на поверхности
раздела 814-820
Поверхностные весы Ленгмюра
816
Поданды 37, 126-129
комплекс с Eu(III) 127
комплекс с К+ 160
константы связывания 129
концепция жесткой концевой
группы 128
макроциклический эффект 38
определение 140
переносчики Са2+ 126
структуры 141
термодинамические вклады
в стабильность 160
трансмембранные ионофоры
126
подандный тригидроксиэти-
ленгликоль 127
«укутывание» катиона Rb+
криптофиксом-5 176
хелатный эффект 37
Податы, определение 142
Полиены типа «тяни—толкай»
750,751
Полимеразная цепная реакция
(PCR) 109,110
Полимеризация при включении
323
Полимерные продукты 161,162,
166
Полиморфизм 518
Политопные рецепторы 195—200
Полости 32
Полукарцеранды 417,421,617,619
Полупроводник TiO2 715
Полусферанды 139, 790
Поляризационная оптическая
микроскопия с
нагревательным столиком 820,821, 827
Поляризованный свет 822
Полярное и неполярное
связывание гостей 403
Предметный указатель
467
Полярность кристалла 330
Пористые координационные
полимеры 558—562,
металлические 4,4 '-бипиридильные
пористые сетки 561
Поросилы 303
Порфирины 78, 79, 89-94,
256-259, 588-592, 797-804
аналоги гема, модели
связывания и транспорта кислорода
797-804
атропоизомеры 800
«карманные» 800
комплекс с капсулированным
F- 258
комплексы цинка, самосборка
588-592
«корзинка для пикника» 800
«купольность» 92—94
лиганды 92
«огороженные» 92, 800—802
перенос электронов 713
расширенные 256—259
семиохимия 716
экранирование железопорфири-
нового ядра дендримером 759
Потенциометрическое титрование
44, 252
Правила Эттер 521-524
диарилмочевины 522
нитроанилины 522
продукты кристаллизации кар-
боновых кислот с 2-аминопи-
римидином 523
продукты кристаллизации
оснований нуклеотидов 523
Правило Хюккеля,
ароматические системы 78, 79
Предорганизация 40, 158,
169-178,206,245,282,626
динамический эффект 177,
178
кинетический эффект 177,178
связывание хозяин—гость 40,
169-176
термодинамические эффекты
169-176
Предсказание структуры
кристаллов 517-524
расчетные методы 517-520
экспериментальные методы
519
Преобразование света 711,712
Преобразование сигналов 67, см.
также Информация
и сигналы
Призмы 621
Применение растворов 144—148
Принцип Франка-Кондона 699
Присоединение по Дильсу—Аль-
деру 617
синтез конкина 370
катализируемое «мячом для
софтбола» 618
Пространственные группы 513
Протеинтирозинфосфатаза
(РТР-аза) 242
Противораковые препараты
карбоплатин 110,111
цисплатин ПО, 111
Протоклетка, происхождение
жизни 819
«Протонная губка» 278, 279
Процессы, индуцированные
светом, см. Фотохимия
Псевдоводородные связи 53
Псевдоротаксаны 629—631,
633-635, 743, хромофорный
молекулярный
переключатель 744
Предметный указатель
Пурины 103
Пурпурный бензол 123
Разделение (рекомбинация)
зарядов 85,742,743
Разрушение кристаллов 514,515
Распознавание ионов 200—203
латеральное 193
молекулярное 34,149, 195,
696,716
сферическое 133
хиральное 200-203, 264, 341
центральное 193
Распространение спаривания
оснований 582
Рассеяние рентгеновских лучей
малоугловое 827
Растворители, взаимодействия
хозяин—гость 170
Расширенное систематическое
силовое поле (ESFF) 607
Реакции включения 426—429,
водородные связи 402—407
Реакции типа реакции Вюрца 391
Реакция Митсонобу 391,392
Резорцинарены 354-364, 418, 622
каликс[4]резорцинарен 355,
усеченный куб 621, 622
[4]резорцинаренкаликс[4]арен
425
Рентгеноструктурный анализ
(РСА) 55,71-73,206,321,
322, 606
Рецептор хинона, синтез 405
Рецепторы анионов биологические
240-244
аргинин как центр связывания
анионов 242, 243
белки, связывающие фосфаты
и сульфаты 241, 242
карбоксипептидаза А 243,244
Рецепторы гербицидов 204—206
Рецепторы дитопные и политоп-
ные 195
Рецепторы, координация и
модель «замок—ключ» 34, 35,
767, 768
Рибонуклеаза , самосборка 115
Рибонуклеиновая кислота (РНК)
103, 105-107
Рибосома 105
Родопсин 76, 77
Розетки 625-628
Рост кристаллов 504—513
образование центров
кристаллизации (нуклеация) 504,
505, 508
операции трансляционной
симметрии 512
рост на поверхности раздела
воздух-жидкость 505—508
симметрия молекул и
кристаллов 511
системы Шёнфлиса и
Германа—Могена 511
хиральность, эффект Адама
508-510
Ротаксаны 628—630, 632, 633,
636, 637, 652
кинетический и
термодинамический темплатные эффекты
586
молекулярные машины
740-743
состояние с разделенными
зарядами 743
«молекулярный челнок»
740, 741
номенклатура 628
определение 628
Предметный указатель
469
[2]ротаксан, синтез
статистическим методом
проскальзывания 652
синтез 630, 632, 636, 637
подход на основе
самосборки (защелкивание,
продевание,
проскальзывание) 632, 636, 637
статистический подход
632, 636,652
Рутения катионы Ru2+ и Ru3+
593-596, 702, 705-709
22-ядерный металлокомплекс
708
[Ru(bipyJCl2] 708
[Ru(bipyK]2+ 702, 705,
спектры поглощения и
фосфоресценции 705
Ru(II)—Ru(III), ион Крейт-
ца-Таубе 702
Ряд Хофмейстера 250, 252
Сальварсан 35
Самоингибирование 618
Самовоспроизводство
(репликация) 578,685-690
Самоорганизация 580, 677, 678
Самораспознавание 669—673,
677
Самосборка 115-117, 578-581
биохимическая 115—117,
580-584
в синтетических системах
584-592
геликаты 660—679
ДНК 105-107,115,581-583
закрытые комплексы,
водородные связи 613—628
гигантские
самособирающиеся капсулы 619-625
розетки 625—628
«теннисные мячики»
и «мячи для софтбола»
613-618
капсулярные и полимерные
самособирающиеся
структуры 594
каталитические и
самовоспроизводящиеся системы
685-690
катенаны и ротаксаны
628-659
координационные соединения
592-612
металлические ансамбли
609-612
«молекулярные квадраты»
и «молекулярные
коробки» 597-608
принципы дизайна 593
супрамолекулярный
куб 593-596
кристалл 491, 492
молекулярная 579
молекулярные узлы 679—685
определение 115
с ковалентной модификацией
583, 584
модификация
предшественника 583
обработка после сборки 583
синтетические системы
кинетический и
термодинамический темплатные
эффекты в синтезе
584-592
термодинамическая модель
587-592, 626, порфирино-
вые комплексы цинка
588-592
470
Предметный указатель
строгая 581-583
супрамолекулярная 579
терминология 578—580
цели и задачи 576—578
Самособирающиеся
многоэтажные структуры 609—612
Сапфирин 256
Саркофагины, Со(Ш) и Pt(IV)
135, 136, 142
Светопреобразующие устройства
711,712
Сворачивание дендримеров 758
Связывание анионов 237—292
биологические рецепторы
240-244
конструирование хозяина 60,
61
Связывание катионов 121—232
заряд катиона 152
факторы, определяющие
селективность хозяина 149,152
Связывание нейтральных
молекул 295-438
Связывание органических
катионов 190-206
дитопные рецепторы 195-200
эффекты кольцевого тока
ароматического кольца
197
ЯМР-спектроскопия 197
дифильные рецепторы 203,
204
рецепторы гербицидов
204-206
связывание катионов аммония
корандами 191-194
связывание катионов аммония
трехмерными хозяевами 194
хиральное распознавание
200-203
устройство для разделения
энантиомеров 202
хиральный барьер 200, 201
Селективность
кинетическая и
термодинамическая 41-48, 149-159, 240,
768, 787
связывание анионов 244—252,
273
связывание катионов
149-154, 170
связывание циклодекстринами
380-384
переходного состояния 311
формы 249-252
Семиохимия 716—727, см.
Информация и сигналы
Сенсоры 60, 716-727
флуорофоры 721—724
фотохимические 717—721
химические 717
электрохимические 724—727
PET 719
Сепулькраты, Со(Ш) 135, 136, 142
Серебра катион Ag+
комплекс 18-краун-6 с Ag+ 181
комплекс углеводородного ли-
ганда с Ag+ 226
туннелирование Ag+,
связывание каликсаренами 216
ЯМР-спектроскопия 109Ag
609
Сигмаидальная зависимость
скорости реакции от времени
688, 690
Сидеранды 231,232
Сидерофоры 228—232
природные 228—230
синтетические системы
230-232
Предметный указатель
47 IP
МЕСАМ 230
темплатный синтез энтеробак-
тина 229
Симметрия молекул и кристаллов
511-513
Симпорт (перенос) Н+ и С1~-ани-
онов 259
Синапс 100
«Синтез вверх» 577
Синергетическое связывание
185-187,223,494
Системы со смешанной
валентностью, фотохимия 702—704
Слой Гуи-Чепмена 817,818
Слой Штерна 817,818
Соединение Дианина 332—336
Соединения включения
дифференциальная
сканирующая калориметрия (DSC)
349-353
клатраты Хофмана и Вернера
317,318,559
конформационные аспекты,
включение катиона в калик-
сарен 215
кристаллическое состояние 33
суперкомплексы металлов 253
термогравиметрический
анализ (TGA) 349-353
типа «песочные часы»
(смешанные кристаллы) 567-569
три-о-тимотид 337-340
цикл оде кстрины 378, 384
циклотривератрилен (CTV)
346-348
циклофаны 402—407
Соединения (комплексы)
хозяин—гость
геометрия типа «гнездо»,
«капсула», «насест» 177
график Джоба 46
классификация 32—34
конструирование хозяина 60, 61
определение 28, 29
предорганизация и компле-
ментарность 40,41,169,170
связывание 170, константы
43-48
термодинамическая
устойчивость 37
Соль Цейзе 53
Сольватация гостей и хозяев
246,414,415
конкуренция гостя и
растворителя 414,415
свободная энергия 152, 238
Соответствие размеров полости
хозяина и гостя-катиона
149, 150, 152,394
Сочетание алкинов,
катализируемое медью 391, 393
Спаривание оснований в ДНК
52, 106, 107, 578
по Уотсону и Крику 106, 107
по Хугстену 107
Спейсер 27
Спелеанды 203
Спелеаты 203
«Спиновое ингибирование» 87
Спирали 319, 327-329, 337-340,
660-663, 676-679, 828
Спиральность М- и Р- 338,661
Спиральные тубуланды 327—329
Спирты 536, 538, структура 538
Стабилизация, обусловленная хе-
латным эффектом 38
Стадия, определяемая гостем
(GDS) 422
Статистический синтез катенанов
и ротаксанов 633
47Й
Предметный указатель
Статическая стабильность
комплекса 150
Стерическое соответствие
хозяин-гость 34
Стехиометрия 45
Стирание прямого считывания
после записи (EDRAW) 736
Структуры из лигандов типа
«жесткий стержень» 609
Сульфаты
водородные связи 241, 242
как биоминералы 564
Супервюрцит, структура 541
Суперкомплексы азакорандов
с анионами 253
азакоранды с большими
кольцами 253
константы связывания 255
Супероксиддисмутаза 242
Супрамолекулярная фотохимия
698-716
Супрамолекулярная химия
история 30-35, 206,239
классификация соединений
хозяин—гость 32—34
комплементарность 40
конструирование хозяина 60,
61
координация 34,35
определение 27-29
предорганизация 40
природа взаимодействий
48-60
развитие представлений 30-33
рецепторы 34, 35
термодинамическая и
кинетическая селективность 41 —48
хелатный и макроциклический
эффекты 35-39
химия хозяин-гость 28, 29
Супрамолекулярное устройство,
определение 696
Супрамолекулярные
взаимодействия, типы 48—60
Супрамолекулярные частицы 27
большие молекулы 696, 697
история 34, 35, 206, 237
конструирование хозяина 60,
61
определение 696
фотохимические и
электрохимические критерии 697
Супрамолекулярный куб 593—596
компоненты 595
самосборка 593—596
Сферанды 40, 136-140
как хозяева для
катионов-гостей 139
классификация соединений
хозяин—гость 33
комплекс с Li+ 138,175
модели
Кори—Полинга—Колтуна (СРК-модели) 136,137,
418
синтез, циклизация 138
структура гибридных хозяев
41
Сфераплекс 142
Таллия катион Т1+ 181,184
Твердофазные реакции 342
Тектоны (строительные блоки)
491,515,516,529-531,535,
определение 578, 579
Темплатирование 578,585
Темплатированный рост
кристаллов 505-508
Темплатирующие гости 604, 616
Темплатная биоминерализация
564
Предметный указатель
:f473
Темплатный эффект 131,133,
161-165, 189, 190,229,578,
616,656
в синтезе 584—587
бензо-18-краун-6 163, 164
[2]катената 656
карцерандов 421-423
тетраазамакроциклов 164
ионы металлов, влияние
на циклизацию 163
кинетический 164
синтез методом высокого
разбавления 165—167
термодинамический 164
удаление темплатного катиона
Ni2+ 190
циклические и ациклические
продукты 162
1:1 -циклоконденсация 189
[2 + 2]-циклосоконденсация
167-169
Темплаты 576—690, см. также
Самосборка
в синтезе 584—592
геликаты 660—679
закрытые комплексы,
водородные связи 613-628
каталитические и
самовоспроизводящиеся системы
685-690
катенаны и ротаксаны
628-695
координационные соединения
592-613
молекулярные узлы 679—684
терминология 578—580
цели и задачи 576—578
Теория жестких и мягких кислот
и оснований (HSAB) 180,
244
Термогравиметрический анализ
(TGA), соединения
включения 349-353
Термодинамическая
селективность 41, 240
Термодинамическая стабильность
комплекса 600-602
Термотропные жидкие кристаллы
822, 823, трубчатые и диско-
тические 823,824
Тетрагидрофуран (THF) 208,
274, 282, 283
Тетрапиррольные макроциклы
78,79
корриновое кольцо 78, 79, 98,
809
комплексы магния 79—85
Тетраол, синтез соединений
с ацетальными мостиками
420
Тетрафенилен 348, 349
л-Толуолсульфонат (Ts) 125
Топологические диастереомеры
и энантиомеры 631,652
Топологический захват 760
Топохимия 295, 493
Торсионные углы в комплексах
краун-эфиров 143, 144
Трансаминирование циклодекст-
рином и пиридоксаминовым
коферментом 782
Трансацилазы 787
/)-аминоэфиры 788
хиральные коранды 786, 787
хозяева, связывающие
катионы 786-791
Трансляционная изомерия 647,
651,652
1,3,5-Тригидроксибензол (ТНВ,
флороглюцин) 406
474
Предметный указатель
Григидроксиэтиленгликоль
подандный 127
Грикислота Кемпа 387
Грилистный узел 631, 652, 680,
682, 683
базовый 680
диастереомерия 631
Гримезиновая кислота (ТМА)
324-327, трианион 277
Грис(пиразолил)бораты 225
Гри-о-тимотид (ТОТ) 337-343
применение 342, 343
производные и родственные
хозяева 340
синтез 340
соединения включения
337-340
спонтанное разделение кон-
формаций 338
Грифлат-анион 226
1,3,5-Трицианобензол 500,501
Тройной комплекс 685, 686
Губулярные мезофазы 830
Тушители 713,719
люминесценция 713
флуоресценция^ 19
Углеводородные хозяева 225-227
Углерод (углеводные
эквиваленты), фотосинтетическое
получение 79, 80
Углеродные донорные и л-кис-
лотные лиганды 223-227
смешанные С-гетероатомные
хозяева 223-225
углеводородные хозяева
225-227
комплекс с Ag+ 226
комплекс парациклофана
сСг+ 227
Угол захвата лиганда 152,153,
280,705
Упаковка молекул по типу
«ёлочка» 56, 382, 552, 553, 826
Уравнение Гиббса 160
Уравнение Эйринга 200
Уранилнитрат 128
Уранилхлорид,гидрат 556
Урацил 676-679
Уреаза из бобов 792
Фактор занятости 416,617
Фенол 332, 333
Ферментативный катализ 34
Ферменты 767-796, см. также
Коферменты
активные центры 770, 790
апофермент 97, 809
голофермент 97
имитаторы
каталитические и
самовоспроизводящиеся системы
685-690
структурный 789
циклодекстрины
(как хозяева) 775—782
частичный 789
индуцированное соответствие
768, 769, 773, 774
искусственный 780
история 792
металлобиоцентры 792—796
модели 767-770, 792, 793
определение 770—772
перенос протона 243
размер молекул 775
свойства 768, 769
серинпротеазные 776
структура 770—772
трансмембранные 65
Предметный указатель
ферментативный катализ 34,
763, 768
механизм 772—775
модель «замок—ключ» 34,
767-769
характеристики 770—775
энтатическое состояние 94
Феромоны 768
Ферритин,самосборка 116
Ферроцен 53, 430
комплекс с ос-циклодекстри-
ном(а-СО) 383
полиморфные формы 553
Фетодигитогеник 142
Фильтр селективности 74, 75
Фильтрация ошибки 673
Флуоресцентное титрование 46
Флуоресценция
определение 698—700
система краун—антрацен
719-721
Флуоресценция, усиленная хела-
тированием (CHEF) 722, 723
Флуорофоры, семиохимия
721-724
Формамид 536, структура 537
Фосфаты, водородные связи
241,242
Фосфолипидная везикула,
молекулярная проводимость 728,
729
Фосфолипиды 815, мембрана
69,70
Фосфоресценция, определение
700
Фотовозбуждение, последующие
излучательные явления
698-702
Фото- и электропереключаемая
система 735, 736
Фотоионный сигнал 737,
переключаемое связывание
737-739
Фотосинтез 79, 378, 709
окисление воды до кислорода,
катализированное марганцем
перенос энергии (ЕТ) 700
реакционный центр 85
тетрапиррольные комплексы
магния 79-85
фотосинтетическая мембрана
81
Фотосистемы (PS) I и II 85, 86
Фотохимические устройства
на основе дендримеров
760-763
сенсоры, семиохимия 716—727
Фотохимия 698-704
биметаллические системы
и системы со смешанной
валентностью 702—704
бипиридил, трипиридил
и 1,10-фенантролин 704-707
критерии для отнесения
частиц 697
накопление энергии 204
основы 698-702
светопреобразующие
устройства 711,712
системы с нековалентными
связями 712—715
фотохимические устройства
на основе дендримеров
760-763
фото- и электрохимические
устройства на основе бипири-
дила 707-711
Фталоцианины, суперфталоциа-
нин 190
476
Предметный указатель
Фторхлорбромметан 413
Фуллерены 358, 430-438, 579
бакминстерфуллерен 347,
430-438,619-621
как гости 430—433
как сверхпроводящие
соединения включения 436
как хозяева 433—436
открытие 437, 438
редокс-чувствительное
сенсорное устройство на основе бак-
минстерфуллерена 435
самосборка 579
структура 621
Фумаровая кислота и ее комплекс
535, структуры 536
«Хвостовое остаточное сродство»
390
Хелатирование 280,281
Хелатирующие агенты 278—282
Хелатное кольцо 38, 149, 152, 153
размер (угол захвата лиганда)
152, 153,280
энергия напряжения 38
Хелатный эффект 35—39
анионы 270, 279, 280
стабилизация 38
Хелаты 280, кристаллические
структуры 280,281
Химически модифицированные
ион-селективные полевые
транзисторы (CHEMFET)
726, 727
Химия растворения щелочных
металлов, история 206
Химия хозяин-гость 28, 29
Химотрипсин 789, 790,
активный центр 790
Хиральная хроматографическая
колонка 202
Хиральность 631
двойные геликаты 663
двухдоменный геликанд,
комплексы 662
индукция 508—510
коранды 786-789
трилистный узел 682
умножение 343
Хиральные капсулы 363
Хиральный барьер 200, 201,
определение 200
Хиральный сдвигающий реагент
663
Хлорид-анион, связывание тетра-
эдрическими рецепторами 248
Хлорофиллы 78, 80—85
спектр поглощения 81
тетрапиррольные макроциклы
78
этилхлорофиллид,
кристаллическая структура 83
Хозяева кремнийсодержащие,
связывающие анион 281
Хозяева на основе гликолурила
367-369
Хозяева на основе конкинов
370-372
Хозяева, связывающие катионы
121-232
алкалиды и электриды
206-209
бибрахиальные лариат-эфиры
(BiBLE) 132, 141, 155,
224
гибридные 139, 171-174
имитаторы трансацилаз
786-791
каликсарены 209—223
Предметный указатель
411
комплексы с катионами
металлов 149-159
комплексы с органическими
катионами 190—206
дитопные рецепторы
195-200
коранды 191—193
трехмерные хозяева 194, 195
коранды 160, 175, 178
краун-эфиры 121—126,
143-154, 159-164, 166, 167,
169
криптанды 132—136, 157—160,
167-169, 171, 173, 176
лариат-эфиры 129—131,
155-157,223,224
мягкие лиганды для мягких
ионов металлов 179— 190
номенклатура 140—142
поданды 126-129, 159-161,
175, 178,272
сидерофоры 228—232
сферанды 136-140, 171, 172,
175
углеродные донорные и л-кис-
лотные лиганды 223—227
Хозяева типа «колесо—ось» 555,
556
Хозяин, определение 28, 29,
244
Хрома катион Сг+
парациклофаны, комплекс
сСг+ 227
Сг(СОN, разрушение
кристаллической структуры 514
Хромионофор 128
Хромофоры 27, 698, 715
Хромофорное молекулярное
переключение, псевдоротак-
саны 743, 744
Цвиттер-ионы, нейтральные
рецепторы 274, 275
Цезиевый эффект 163, 392
Целлюлоза, получение циклодек-
стринов 378
Центры связывания хозяина
и гостя, сходящиеся и
расходящиеся 29
Цеолиты 303-311
синтез 307-309
катионы-темплаты и типы
образующихся цеолитов
308
состав и структура 303—307
зигзагообразные каналы
bZSM-5 307
топология каркаса
структуры 304, 305
MFI-типа в нефтяной
промышленности 309—311
Цеосилы 303
Цианокобаламин, см.
Витамин В12
Циануровая кислота 625
структура комплекса мела-
минциануровая кислота
625-628
Циклобутадиен, внутриполост-
ной синтез 427, 428
Циклам 190
Циклическая вольтамперометрия
254,267-269,640,641,658
Циклические диолы 327—329
Циклический 3',5'-аденозинмо-
нофосфат (сАМТ), гидролиз
фосфодиэстеразой 778, 779
Циклодекстрины 372—386,431
ацетилирование 777
как имитаторы ферментов
775-782,809,810
478
Предметный указатель
как имитаторы глиоксалаз
781
как имитаторы эстераз
778-780
как катализаторы гидролиза
сложных эфиров 778, 779
как хозяева 776—778
функционализированные
780-782
определение класса комплекса
33
применение 384-386
продевание каровиологенов
сквозь полости циклодекст-
ринов 728,729
свойства 372-377
синтез 378
соединения включения
378-384
структуры 234, 373
упаковка 382
Циклосоконденсация 167—169
комплекс криптанда с NaC104,
кристаллическая структура
169
криптанды 167
криптанды на основе бипири-
дила 168
Циклотетравератрилен (CTTV) 345
Циклотривератрилен (CTV) 33,
203,271,272,343-348,
407-417,430
определение 33
синтез 345, 346
соединения включения
346-348
фазы 346
Циклофаны 59, 209, 226, 227,
260,261,389-407,390,391,
см. также Каликсарены
включение гостя с помощью
водородных связей 402-407
дифенилметан 396—401
изменение рН 261
клетки в водном и неводных
растворителях 401, 402
концепция пинцета 394—396
номенклатура 390,391
определение 389
связывание я-ксилола 59, 60
синтез 391-394
супрамолекулярная
самосборка 598
циклические вольтамперо-
граммы 641
Цинкин 721,722
Цинковые сенсоры 721—723,
флуорофоры 721—724
Цинкпорфириновые комплексы
588-592,713, 714, самосборка
588-592
Цинксодержащие ферменты
721,795,796
Цисплатин 110, 111
Цитохромы Р-450 804-808
катализ реакций 804, 808
каталитический цикл оксиге-
назной активности 805
микрореактор, моделирующий
работу цитохрома 807
модели 804-808
циклодекстрины как
имитаторы цитохрома 808
Цитратсинтаза 242
Число А/А в жидких клатратах
838
Частичный имитатор фермента
789
Предметный указатель
479
Шампуни 815
Шлирен-текстура
823,824
Экзотермическая реакция,
изменение свободной энергии
601
Электриды 206—209, 436,
первый электридный комплекс
207
Электропереключающаяся
люминесценция 736, 737
1,6-Элиминирование 391
Энтатическое состояние 94
Энтеробактин 229, 230, тем-
платный синтез 229
Энтропийные и энтальпийные
вклады хелатного и макро-
циклического эффектов
35-39
Эстеразы 778-780
т/?яяс-Этерификация 263
Эфир Вильямсона, синтез 125
Эффект Адама, см. Индукция хи-
ральности в кристаллах
Эффект «обнаженного» аниона
146, 207
Эффективная концентрация
гостя в полости хозяина 618
Эффективная молярность 590,
773
Эффективность самосборки 591,
592
Эффекты кольцевого тока
ароматического кольца 197
Ядерный эффект Оверхаузера
(NOE) 198,214,215
ЯМР-спектроскопия 138,
197-200, 265, 343, 396, 397
аденозинтрифосфат C1Р) 784
дитопные рецепторы ('Н) 195
каликсарены (!Н) 211
катенаны ('Ни 13С) 649
криптанды A9F) 138
металлические ансамбли
A09Ag) 609
ртутьсодержащий хозяин
A99Hg) 281
спин-спиновое взамодействие
198
тетракатионный хозяин A9F)
289
ЯМР-титрование 44—46
Научное издание
Стид Джонатан В., Джерри Л. Этвуд
СУПРАМОЛЕКУЛЯРНАЯ ХИМИЯ
Том 1
Редактор М.Л. Франк
Художник И.А. Слюсарев
Дизайнер И.М. Кадченко
Компьютерный дизайн и верстка О.Д. Эшлиман
ИД №04284 от 15.03.2001.
Подписано в печать 01.11.2006. Формат 70x100/16. Гарнитура NewtonC.
Печать офсетная. Печ. л. 30. Тираж 400 экз. Тип. зак. 2706.
Издательско-книготорговый центр «Академкнига»
117997, Москва, Профсоюзная ул., 90
По вопросам поставок обращаться
в отдел реализации ИКЦ «Академкнига»
Тел./факс: D95) 334- 73-18
e-mail: bookreal@maik.ru, web-site: http://www.akademkniga.com
Отпечатано с готовых диапозитивов в
ОАО «Ивановская областная типография»
153008, г. Иваново, ул. Типографская, 6.
E-mail: 091-018@adminet.ivanovo.ru
Special than fe; for
ЮШ-scan
'Revenae ~ editS^ encodina
химия
Jonathan W. Steed, King s College, London
and
«Jerry L Atwood, Untversity of Missoun, Columoia
Супрамолекулярная химия ~ это междисциплинарная
область, которая до сих пор активно сопротивляется
всем попыткам очертить ее рамки или дать
достаточно полное ее определение. Книга
Супрамолекулярная химия - это на сегодняшний день первый в
мировой литературе учебник, охватывающий
практически все разделы супрамолекулярнои химии. В ней
рассказывается обо всем, что студент должен знать
для успешного начала своей научной деятельности в
этой области. Предполагая, что читатель уже
обладает определенным уровнем подготовки, авторы
включили в книгу и теоретические вопросы относящиеся
к рассматриваемой области и сведения о наиболее
важных экспериментальных методах исследования
супрамолекулярных систем. Значительное внимание
уделено истокам супрамолекулярнои химии ее
объектам а также вопросам терминологии. Книга
содержит большое число примеров супрамолекулярных
соединении, вопросы и задачи для повторения
материала каждой главы.
Supramolecular Chemistry
Is the iirst integrated supramolecular book written specifically for students
Is clearly structured and lucidly written with many examples,
applications and problems drawn from this diverse discipline.
Features a web site with additional information and the latest developments:
www.ch.kcl.ac.uk/supramol/textoook.htm
ISBN 978-5-94628-303-8
>BN 0-471-98831-6
785946 283038"
ИКЦ «АКАДЕМКНИГА»