/
































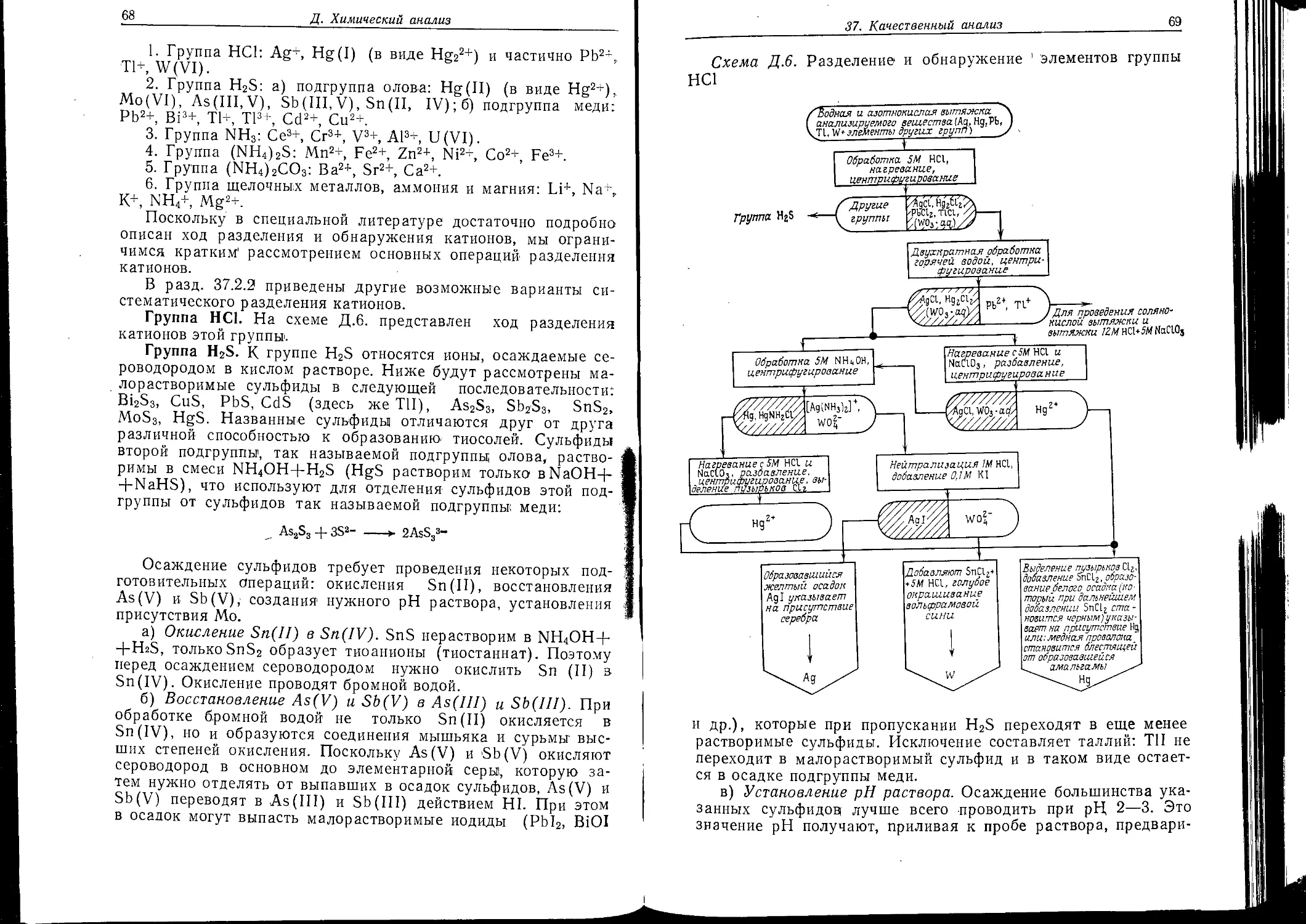






























































































































































































































































































Текст
ANOROANIKUM
Lehr- und Praktikumsbuch der anorganischen Chemie mit einer Einfuhrung in die physikalische Chemie
Vom Autorenkollektiv:
G. Blumenthal, S. Engels, I. Fitz, W. Haberditzl, K.-H. Heckner, G. Henrion, R. Landsberg, W. Schmidt, G. Scholz, P. Starke, I. Wilke, К.-Th. Wilke
Herausgegeben von LOTHAR KOLDITZ
10. iiberarbeitete Auflage in 2 Teilen
Teil 2
VEB Deutscher Verlag der Wissenschaften Berlin 1984
ЛНСЖНИйМ
В двух томах
Том 2
ХИМИЧЕСКИЙ АНАЛИЗ
ОБОРУДОВАНИЕ ЛАБОРАТОРИЙ И ПРАКТИКУМ ПО ПРЕПАРАТИВНОЙ ХИМИИ
Редактор Л. Кольдиц
Перевод с немецкого канд. хим. наук Л. Б. Кузнецовой под редакцией д-ра хим. наук, проф. О. М. Петрухина
МОСКВА «МИР» 1984
ББК 24.4
А 69
УДК 543 + 542
Блументаль Г., Энгельс 3., Фиц И., Хабердитцль В., Хекнер К.-Х., Хенрион Г., Ландсберг Р., Шмидт В., Шольц Г., Штарке П., Вильке И., Вильке К.-Т.
Анорганикум: В 2-х т. Т. 2. Пер с нем./Под ред. А 69 Л. Кольдица. — М.: Мир, 1984. 632 с., ил.
В учебном пособии, составленном авторами из ГДР н выдержавшем 10 изданий, излагается курс неорганической химии с основами физической и аналитической ХИМИН.
Том 2 посвящен классическому химическому анализу неорганических веществ и инструментальным аналитическим методам, включая электрохимические, спект» роскопическне, радиохимические и термические. Рассматриваются аналитическая химия микроколичеств веществ н способы оценки результатов анализа. Приводится лабораторный практикум по препаративному неорганическому синтезу.
* Для студентов и преподавателей высших и средних химических и химнко-
технологических учебных заведений.
1802000000—346 --------------95—84, ч. 1
041(41)—84
ББК 24.4
543
Редакция литературы по химии
УВАЖАЕМЫЙ ЧИТАТЕЛЬ!
Ваши замечания о содержании книги, ее оформлении качестве перевода и другие просим присылать по адресу: 129820, Москва, И-140, ГСП 1-й Рижский пер., д. 2, издательство «Мир».
© VEB, Dentscher Verlag det Wis-senschaften, Berlin, 1984
© Перевод на русский язык, «Мир», 1984
ПРЕДИСЛОВИЕ РЕДАКТОРА ПЕРЕВОДА
Исторически для получения информации о качественном и количественном составе вещества прежде всего использовали химические методы, т.е. методы, основанные на получении в результате химической реакции того или иного соединения, обладающего определенными аналитическими свойствами. Эта ситуация закреплена в самом названии «аналитическая химия». Поэтому классические методы аналитической химии, особенно в той части, которая касается анализа неорганических веществ, опираются прежде всего на неорганическую химию* как более общую дисциплину. Кроме того, нужно учесть следующее. Преподавание аналитической химии в высшей школе имеет помимо конечной главной цели — обучение основам аналитической химии-—также задачу научить химическому мышлению. Распространено мнение (и оно вполне справедливо), что аналитическая химия представляет собой идеальное средство для достижения этой, второй цели, иначе говоря, аналитическая химия естественно входит в структуру общехимических дисциплин вуза. Поэтому, как правило, курс классической аналитической химии, представляющий по существу неорганическую аналитическую химию, излагается в, вузах сразу же после неорганической химии, а иногда совмещается с ней в единый курс. Именно* для такого вузовского курса и написан двухтомный учебник «Анорганикум», изданный в ГДР.
В книге весьма подробно изложены качественный и количественный анализы, основанные на химических реакциях, т. е. на реакциях осаждения, протолитических, окисления — восстановления, комплексообразования. Приведено большое число- расчетов и примеров.
Следует, однако, отметить, что аналитическая химия представляет собой чрезвычайно динамичную дисциплину, использующую достижения многих областей науки и техники; для ре
6 Предисловие редактора перевода
шения своих задач она интенсивно развивает различные методы и использует любые принципы, но в большей степени физические и физико-химические. Последние рассматриваются в дисциплинах, с которыми студенты знакомятся на старших курсах. Поэтому в СССР, как правило, курс физических и физико-химических методов анализа преподается на старших курсах. Соотношение между различными методами аналитической химии меняется как в зависимости1 от 'Времени и места, так и в зависимости от объекта анализа, и при составлении вузовской программы приходится сталкиваться с задачей выбора тех или иных методов хотя бы потому, что! вся совокупность не может войти ни в одну из программ. Эту задачу можно, естественно, решить по-разному. В данном учебнике очень подробно рассмотрены электрохимические и ряд спектроскопических методов анализа. Интересно подчеркнуть, что едва ли не впервые в учебнике такого типа введена глава, посвященная аналитической химии микроколичеств веществ, что отражает, помимо прочего, возросшее внимание к экологическим проблемам— проблемам, к которым химики-аналитики имеют самое непосредственное отношение.
Мы надеемся, что книга будет эффективно использоваться преподавателями вузов, студентами и сотрудниками аналитических лабораторий.
О. Петрухин
Д. ХИМИЧЕСКИЙ АНАЛИЗ
37. КАЧЕСТВЕННЫЙ АНАЛИЗ
37.1. Общая часть
37.1.1. Историческое развитие и задачи
качественного анализа
По сложившейся традиции принято различать качественный и количественный анализы. С помощью качественного анализа устанавливают, какие элементы, молекулы илй ионы входят в состав вещества. Количественный анализ позволяет определить содержание компонентов в веществе после идентификации их методами качественного анализа. Это различие между качественным и количественным анализом, кажущееся таким простым, в действительности проблематично. При проведении анализа по существующим в настоящее время методикам в любом веществе возможно обнаружить большое количество элементов, й том1 числе и таких, присутствие которых, не предполагалось. Содержание этих элементов может быть на несколько порядков меньше содержания основных компонентов. Поэтому, когда аналитик утверждает, что в веществе А содержится элемент В, то это высказывание имеет смысл только в том случае, если указан порог чувствительности (см. прим, на с. 434) реакции обнаружения. Отсюда следует вывод, что к реакциям, применяемым в качественном анализе, также необходим количественный подход.
Как и другие отрасли химии, качественный анализ находится в постоянном развитии. В XVIII в. значительно возросло число известных элементов и соединений и их! реакций, в связи с чем появилась необходимость систематизации. Шведский ученый Т. Бергман разделил элементы! на группы на основе различного отношения их ионов, к действию сероводорода. Отдельные ионы он идентифицировал, используя специфические реакции. Этот метод получил дальнейшее развитие в XIX в. Значительный вклад в развитие метода внесли К. Р. Фрезениус и Ф. Тредвелл. В итоге возник надежный «классический» метод разделения. Количество анализируемого вещества в этом методе, называемом макроанализом, составляет 0,5—5,0 г.
Для анализа небольших количеств веществ в конце XIX в. был разработан микроанализ, в котором вместо пробирок .применяют предметные стекла и рабочее место оборудуют микроскопом. Исследователь получает кристаллы вещества и идентифицирует его по форме образовавшихся кристаллов. Осново-положнико'м этого метода является Беренс. Масса исследуемой
8
Д. Химический анализ
пробы может достигать 1 мг; следовательно, в микрометоде работают с количествами веществ совершенно другого! порядка, чем в макрометоде. Еще одно отличие микрометода — то, что в нем невозможно использовать принцип систематического хода анализа, так как кристаллы различных веществ почти невозможно разделить под микроскопом. Удалось найти большое число реакций, благодаря которым можно идентифицировать компоненты1 непосредственно из их смеси без предварительного разделения. Реакции образования кристаллов оказались не единственными реакциями, пригодными для идентификации. С развитием органической химии появились аналитические реагенты, образующие ярко окрашенные соединения с неорганическими ионами. В этом случае для (проведения исследования достаточно одной капли раствора, отсюда и название метода — капельный анализ. Начиная с 1920 г. этот метод разрабатывали Ф. Файгль и Н. А. Тананаев.
Для устранения недостатков, присущих микрометоду, а также затруднений, связанных с техникой работы, предложен полумикрометод анализа (полумикроанализ), в котором работают с количествами веществ 20—200 мг и который является очень гибким по своим приемам. Выдающийся вклад в разработку этого метода внес шведский химик А. Шоландер. Полумикрометод анализа чаще всего применяется при обучении студентов.
Студенты изучают также дробный и систематический методы анализа, при этом основное внимание уделяется методам разделения. Систематический ход анализа, основанный на использовании различной основности, растворимости и других свойств веществ, дает возможность наиболее полно на практике изучить эти свойства.
Реакции идентификации веществ широко применяют в заводских и научно-исследовательских лабораториях, что обусловлено быстротой выполнения анализа. Основной компонент пробы чаще всего известен, поэтому заранее можно предвидеть, будет ли он мешать идентификации других компонентов пробы. Существенный вклад в развитие этой области анализа внес Г. Шар ло.
37.1.2. Общие реакции в качественном анализе
В зависимости от природы анализируемой смеси для ее анализа можно применить различные общие реакции, на основании критической оценки которых1 можно сделать важные выводы о результатах анализа. Различают групповые, селективные и специфические реакции.
Групповые реакции. Групповые реактивы одинаково реагируют с ионами одной группы. Применение групповых реакти-
37. Качественный анализ
9
bob и правильная оценка результатов их действия оказывает большое влияние на ход анализа. При этом, чтобы сделать вывод о наличии и отсутствии определенной группы ионов, можно использовать сравнительно небольшое число реактивов. Положительный эффект, наблюдаемый при добавлении группового реактива после выделения ионов определенной группы из раствора, указывает на то,, что! в растворе еще содержатся ионы данной группы. Групповыми реактивами являются, например, H2S, раствор К1з, раствор AgNOa.
Селективные реакции — реакции, дающие сходный аналитический эффект с ограниченным числом ионов. Если эти ионы относятся к различным аналитическим группам, селективную реакцию можно рассматривать как реакцию обнаружения при условии четкого разделения ионов. Необходимо точно соблюдать условия проведения реакции, так как селективность реакции зависит от pH, температуры и концентрации реагентов.
Специфические реакции. Реакция является специфической, если при действии реактива в определенных условиях однозначно идентифицируют только, один элемент или ион. Специфические реакции должны быть легко выполнимыми и наглядными. Если реакция ненадежна,, то применять ее не следует. До сих пор немного предложено так называемых «идеально специфичных» реакций. Благодаря применению органических реагентов число, таких реакций постоянно1 растет. Типично специфическим реагентом, давно применяемым для обнаружения никеля, является диметилглиоксим, который с ионами никеля(П) образует малиново-красный характерный осадок.
Групповые или селективные реагенты можно использовать как специфические при1 условии предварительного разделения ионов. Например, кадмий можно специфично обнаружить в виде сульфида кадмия при взаимодействии с сероводородом.
37.1.3. Чувствительность аналитических реакций
Аналитические реакции должны быть чувствительными, т. е. давать возможность четко и надежно идентифицировать небольшие количества компонентов. С помощью очень чувствительных реакций и при тщательном разделении ионов можно обнаружить! в природных минералах и рудах большое число элементов. С другой стороны, чувствительность имеет определенные границы. Для качественного анализа оптимальным является применение специфических реакций с примерно равной чувствительностью. Это, к сожалению, недостижимо не только в настоящее время, но и в ближайшем будущем. Поэтому для каждой аналитически важной реакции указывают чувствительность.
10
Д. Химический анализ
Согласно теории, образование следовых количеств осадка в растворе происходит в том- случае, когда произведение концентраций ионов1 реагирующих веществ достигает значения произведения растворимости продукта реакции. Однако такие 'количества осадка еще неразличимы), так как первые стадии осаждения— этд образование зародышей и коллоидообразование. Только вслед за этим наблюдаются видимые изменения в растворе, характеризующие чувствительность данной реакции.
При проведении цветных реакций обнаружения концентрации реагирующих веществ должны достигать таких значений, чтобы глаз мой воспринять изменение окраски. В, зависимости от длины волны неа-бсорбированной составляющей света интенсивность окраски различна. Таким о'бразом, чувствительность цветной реакции зависит также от окраски продукта реакции. Как и .в случае реакции -осаждения, для обнаружения эффекта химической, реакции в, растворе должны находиться достаточно большие количества компонентов. Поэтому чувствительность реакции невозможно рассчитать, ее нужно определять экспериментальным путем. Величина теоретической’ чувствительности выше действительной.
Чувствительность аналитической реакции зависит от чувствительности наших органов1 чувств, применяемых посуды и инструментов (пробирок, предметных стекол, луп, микроскопов), условий реакции (pH, температуры, концентрации), присутствия посторонних веществ. Так как чувствительность определяют визуально, она зависит и от субъективных свойств каждого наблюдателя, поэтому разные авторы приводят различающиеся данные по чувствительности реакций. Известны следующие способы выражения чувствительности реакций.
Абсолютная чувствительность или открываемый минимум ’(ОМ). Дс1 Файглю, это наименьшее количество элемента в ’микрограммах (раньше в у), которое можно обнаружить в данном объеме с помощью соответствующей реакции. Этот способ выражения чувствительности в настоящее время уже не имеет большого значения.
Файгль унифицировал обозначения ОМ следующим образом: 5 [А]°л3. Перед скобками указывается количество вещества в микрограммах, которое еще можно надежно определить в объеме раствора (см3), записанном в показателе степени. Буква в скобках обозначает способ проведения реакции: [А]—капельная на капельной пластинке; [В] — капельная на фильтровальной бумаге; [С] — реакция в пробирке для полумикроанализа; [D]—реакция в пробирке для макроанализа; [Н]—образование кристаллов под микроскопом; [ И — другие методы.
Предельная концентрация (ПК). По Гану, это предельное разбавление раствора, при котором еще может быть обнаружен соответствующий ион или элемент. ПК выражают как отношение массы определяемого иона к общей массе раствори
37. Качественный анализ
11
теля. 'При этом количество обнаруживаемого иона составляет примерно 1 г, поэтому ПК записывают как частное, например 1:100 000. Так, ПК= 1:50 000 означает, что 1 г иона можно обнаружить в 50 кг растворителя. Как и в случае ,ОМ, здесь имеет значение объем, применяемый .для анализа. В 1 см3 раствора данный ион можно не обнаружить, в то время как в 1 дм3 раствора такой же концентрации окрашивание или осадок уже можно видеть. Аналитическую идентификацию ионов, в зависимости от метода, можно 'проводить в различных объемах. В пределах одного метода (макро-, полумикро-, капельный анализ) объемы! могут быть постоянными.
Принимают следующие стандартные объемы V:
макрореакции, проводимые в пробирках (15X1,5 см) полумикрореакции, проводимые в пробирках (8X1 см) реакции на капельной пластинке реакции на фильтровальной бумаге
реакции под микроскопом
V=5 см3
V=1 см3
У=0,03 см3
V=0,03 см3
К=0,01 см3
При соблюдении указанных объемов пробы чувствительность однозначно Определяется величиной щредельной концентрации, так как из1 прописи анализа! объем известен. Целесообразно выражать ПК в виде числа й определенной степени, так как часто приходится иметь дело с большими величинами. Тогда
1 : 50 000= 1 : 5-104= 10*4’7
1 : 100 000= 1 : 105= 10’5
Эту величину можно выразить в виде отрицательного десятичного логарифма аналогично pH или рК. Тогда ее обозначают как рл. Если ПК=10~4'7, то рс=4,7.
Чувствительность большинства аналитических реакций характеризуется ро 5—6. Верхний порог чувствительности, используемый для аналитических целей, соответствует pz> = 8, нижний — ро=3. При pz>=3 реакция нечувствительна. Верхний порог чувствительности определяют по эффективности обнаружения данного иона в присутствии следовых количеств фоновых элементов, концентрации которых различны для разных элементов. Высокочувствительные реакции обнаружения, за исключением анализа следовых количеств, не применяются на практике. Например, спектральное обнаружение натрия можно лишь условно использовать для обычных аналитических определений, так как уже 0,001 мкг Na+ окрашивает пламя в желтый цвет.
В присутствии больших концентраций посторонних ионов общая чувствительность снижается. Это указывают при описании реакций обнаружения. При изменяющемся значении рь нужно указывать отношение концентраций мешающих ионов к концентрации обнаруживаемого. Это отношение называют предельным отношением.
37.1.4. Органические реагенты в анализе
Русский ученый Л. А. Чугаев в 1905 г. опубликовал работу, в которой описал реакцию взаимодействия «-диоксимов с ионами никеля. Он обнаружил, что при взаимодействии соли ни-
12
Д. Химический анализ
келй с диметилглиоксимом в аммиачной или уксуснокислой среде образуется ярко-красный осадок. Это была одна из первых реакций с органическими реагентами, примененная в аналитических целях. С тех пор постоянно появлялись все новые органические реагенты, успешно используемые как в качественном, так и в количественном анализе.
Органические реагенты) по сравнению с неорганическими обладают рядом преимуществ.
а) Многие органические ,реагенты являются более селективными, отчасти также и более специфичными. При умелой работе часто отпадает необходимость в проведении разделения з пределах одной группы ионов.
б) Реакции с органическими реагентами более чувствительны. Анализ следовых количеств веществ в технических продуктах успешно проводят капельным методом с применением преимущественно органических реагентов. При использовании органических реагентов в обычном анализе необходимо проводить холостую1 пробу.
в) Большая чувствительность реакций органических реагентов связана с образованием» интенсивной характерной окраски. Наряду *с образованием окрашенных осадков большое значение имеет и окраска растворов. По интенсивности окраски можно судить о содержании компонента в пробе, для этого1 часто, применяют колориметрию и спектрофотометрию.
г) Произведения растворимости малорастворимых осадков продуктов реакции, как правило, очень малы. С этим связана большая чувствительность реакций обнаружения. Органические реагенты чаще всего нельзя применять для отделения одного иона от других, так как избыток реагента мешает проведению дальнейших реакций и может быть удален только в ходе длительных и тщательно проводимых операций.
д) Большинство комплексных соединений с органическими реагентами и красителями хорошо1 растворяются в не смешивающихся с водой органических растворителях. Поэтому, применяя экстракцию, можно повысить чувствительность и специфичность реакции.
Как видим, органические реагенты имеют явные преимущества перед неорганическими, но при использовании их в химическом практикуме необходимо учесть следующее: а) так как на этой стадии обучения студент еще не изучал органическую химию, он не в состоянии в полном! объеме теоретически осмыслить протекание этих реакций; б)' высокая специфичность и простота применения, связанные с большой чувствительностью определения^ часто приводят к неверному использованию органических реагентов в ходе анализа. Следствием этого является невозможность последующего разделения, изменение
37. Качественный анализ
13
реакционной способности элементов, не говоря уже о получении неверных результатов анализа.
Примерно 25 органических соединений, перечисленных в разд. 37.2 наряду с неорганическими реагентами, — это лишь небольшая) часть применяемых в настоящее время реагентов. Реакции с этими органическими реагентами необходимо использовать в обучении.
Поскольку во многих случаях органические реагенты- образуют устойчивые комплексы с ионами металлов, их применяют преимущественно для обнаружения катионов. Для анионов известно .небольшое число чувствительных органических реагентов, поэтому идентификацию анионов лучше проводить с помощью классических реакций с' неорганическими веществами.
Теория комплексообразования, а также строение и типы комплексных соединений были рассмотрены в разд. 6.52, 33.3 и 35.6.
По аналитическим свойствам органические реагенты можно разделить на три большие группы.
I. Органические реагенты, образующие окрашенные внутри-комплексные соединения с определяемыми ионами. В соединениях этого типа ион металла замещает водород кислотной органической группы (—ОН, —SH, —СООН, —NH, =NOH) и, кроме того, образует с органическим реагентом координационные связи. Координирующийся атом функциональной группы реагента должен иметь по меньшей мере одну свободную пару электронов например:
\с=О /С=8 —N=O =N—ОН
В этом случае при комплексообразовании образуется очень устойчивый цикл, состоящий из пяти или шести атомов (см. т. I, разд. А). Как правило, орбитали органических молекул и ионов металлов перекрываются, в результате чего1 ионность связи между кислотной органической группой и ионом металла снижается. Поэтому обычного различия между основными и побочными валентностями в молекуле комплекса нет. Применяемое еще различие в написании формул (черта и шт.рих-линия) дает представление о степени окисления металла, но не отражает соотношение связей.
Природа связей определяет интенсивность окраски комплексных соединений и их растворимость в органических растворителях. Поскольку металл координационно насыщен и уже не обладает свойствами иона, гидратация невозможна. Из этого следует, что соединения этой группы крайне плохо растворимы в воде. Ниже приведены примеры) органических реаген
14
Д, Химический анализ
тов, образующих с ионами металлов внутрикомплексные соединения.
8-Оксихинрлин ((оксин)
Образует со многими металлами внутрикомплексные соединения с пятичленными циклами. Важным для аналитических целей является оксихинолинат магния, представляющий собой желтое кристаллическое вещество:
он
-4- Mg2+(aq)
Н20
Реакция 8-оксихинолина с Mg2+ не является специфичной, ионы многих тяжелых металлов образуют аналогичные осадки.
Диметилглиоксим
CH3-C=N-OH
ch3-4=n-oh
СН,- C=N-ОН I CH,-C=N — О
3 I
I I
СН3—C=N-.. /N=C~CH3
I /N< I
CH3-C=N N=C—CH3
3 I I
Реагент специфичен на ионы никеля, В солянокислой среде образует желтый осадок с ионами палладия(П). С ионами железа (II) в аммиачном растворе образуется темно-красное
окрашивание.
а-Нитрозо-$-нафтол
Чувствительный реагент на кобальт.
Рубеановодородная кислота (диамид тиощавелевой кислоты)
S = С — NHZ
S = С — NHZ
Применяют для чувствительного обнаружения меди.
II . Органические реагенты, образующие окрашенные лаки. Лаки образуются при адсорбции окрашенных хелатообразующих реагентов или красителей поверхностью гидроксидов и оксидов. Образование лаков происходит при совместном осаждении гидроксида металла с водорастворимыми красителями. Оно наиболее характерно для оксиантрахинонов, так как эти
37. Качественный анализ
15
лиганды по стерическим .причинам) занимают не все места в координационной сфере ионов металлов, благодаря чему сохраняется полимерная структура гидроксида и он не переходит в раствор.
Важным (Представителем этого класса красителей является ализарин S, 1,2-диоксиантрахинон-З-сульфоновая кислота, образующий с А13+ в уксуснокислой среде лак, окрашенный в кирпично-красный цвет:
НО - Al-он о' 'о
О
Соединение ализарина S с алюминием
Ализарин S образует также окрашенный лак с цирконием, который в отличие от лаков других катионов устойчив в среде 1Г М НС1. Поэтому ализарин 5 является специфическим реагентом на Zr.
Магнезон :(4-нитро<бензол-1-азорезорцинол) дает характерно окрашенный лак с Mg(OH)2; хинализарин (1,2,5,8-тетраокси-антрахинон)—с Ве(ОН)2; морин (3,5,7,2',4'-пентаоксифла-вон) — с А1 (ОН)з.
II I. Органические реагенты, синтезируемые в результате реакции взаимодействия органических веществ с идентифицируемым ионом. Число таких реакций, естественно, гораздо меньше реакций предыдущих групп. Сюда относится, например, реакция- синтеза индиго как реакция обнаружения ацетат-ио-нов. ,При сухой (перегонке ацетата кальция образуется ацетон:
СН3
Са(СН3СОО), -+ СаСО3 + )С=О сн/
Ацетон затем конденсируется с о-нитробензальдегидом с образованием индиго.
Реакцией диазотирования сульфаниловой кислоты (реакция Лунге I) и азосочетания образующегося диазония с ароматическими аминами можно обнаружить нитрит-ионы. Наиболее часто в реакциях азосочетания применяют 1-аминонафталин (реакция Лунге II). Образуется красный азокраситель:
16
Д. Химический анализ
Бром реагирует с флуоресцеином с образованием красного красителя эозина, В этом .случае происходит бромирование ароматического ядра:
Вг Вг
Для аналитических целей можно использовать образование и разрушение красителей в ходе реакций окисления-восстановления. Эти реакции не очень специфичны, поэтому в основном их применяют для обнаружения окислителей и восстановителей. Реагентом на окислители, такие, как HNO3, Cr(VI), HNO2, является дифениламин. В результате реакции образуется окрашенный в голубой цвет имин:
Сходную реакцию дает бензидин:
Эффект обесцвечивания красителя при восстановлении используют при обнаружении сульфит-ионов с малахитовым зеленым или фуксином. В этом случае, наоборот, хиноидная система переходит в бензоидную, например в случае малахитового зеленого:
' Реже для целей идентификации применяют образование малорастворимых солей. Например, в последние годы для обнаружения калия? все чаще используют реакцию с тетрафенил-боратом натрия:
37. Качественный анализ 1/
Этот реагент образует малорастворимые соли также с ионами аммония, рубидия и цезия. Реакция очень чувствительна. Малорастворимым соединением нитрат-ионов является нитрат нитрона. В основном его применяют в гравиметрическом анализе.
37.1.5. Техника полумикроанализа
Классический макрометод химического анализа связан с большими затратами времени и средств! и, кроме того, со значительным расходом реактивов. Оборудование дорого стоит и требует много места, а обильные осадки зачастую являются причиной неточной работы,. Полумикрометод анализа лишен недостатков макро- и микрометодов. В полумикрометоде используют сравнительно простое оборудование, техникой метода, легко овладеть. Жидкости в основном дозируют по каплям. Так как объем' капли зависит от поверхностного натяжения, величина которого примерно одинакова для воды и разбавленных растворов, по каплям можно дозировать равные объемы. Это дает возможность проводить полуколичественные определения и постоянно знать примерную концентрацию иона в добавляемом, растворе. Для упрощения полуколичественных определений используют молярные растворы реагентов.
Отдельные рабочие операции иногда можно варьировать. В процессе работы каждый учащийся накапливает опыт н может затем!, сам отладить технику эксперимента*. Решающими критериями работы являются ее быстрота, чистота и безупречность.
37.1.5.1. Посуда и реактивы
Растворы реагентов хранят в полумикросклянках вместимостью 30 см13. Склянки обычно снабжены, крышками с резиновыми всасывающими баллончиками для пипеток. Для концентрированных кислот и агрессивных растворов, таких, как бромная вода, применяют склянки с пипетками ,на шлифах. (Не путать шлифы при очистке!) Для хранения растворов, чувствительных к действию света, применяют склянки из коричневого стекла. Этикетки на склянках должны быть по возможности написаны чертежной тушью и защищены, например, прозрачной полимерной пленкой. В ГДР используют стандартные склянки формы А (с навинчивающейся крышкой) и формы В (с коническим шлифом NS 12,5) по стандарту TGL** 40-313.
* Более подробные сведения о технике химического анализа читатель, может найти в книге: Коростелев П. П. Лабораторная техника химического» ЭНаДиза' — М"' Химия, 1981. — Прим, перев.
Технические нормы и методики испытаний (ГДР). — Прим, перев.
2—1880
Таблица Д.1. Основные растворы реактивов
№ Реактив Количество вещества (г или см3) иа 1 дм3 раствора; способ приготовления
1 Ализарин S, 1% 200 г NH4NO3 и 10 г ализарина отдельно растворяют в небольшом количестве воды, растворы объединяют и доливают водой до 700 см3. Затем добавляют 100 см3 СН3СООН и медленно вносят в раствор 400 см3 2 М NH4OH. Доливают водой до 1 дм3 и через 24 ч фильтруют
2 Гидроксид аммония, 13 М NH3 из стального баллона пропускают в воду до получения раствора с пл. 0,91 при 20 °C
3 Гидроксид аммония, 5 М 384 см3 13 М NH4OH
4 Ацетат аммония, 5 М 385 г NH4CH3COO
5 Хлорид аммония, 5 М 267 г NH4CI
6 Гидросульфид аммония, 1 Ма 225 см3 1 М NH4OH охлаждают до 0 °C. Пропускают через раствор H2S до насыщения и добавляют к раствору 25 см3 1 М NH4OH. Раствор малоустойчив
7 Молибдат аммония, 0,5 М 100 г (NH4)2MoO4+200 г NH4NO3 + 7O см3 13 М NH4OH
8 Гидрофосфат аммония, 2,5 М 330 г (NH4)2HPO4
9 Хлорид бария, 0,5 М 122 г ВаС12-2Н2О
10 Гидроксид бария, насыщенный раствор3’ 6 39 г Ва(ОН)2-2Н2О
11 Ацетат свинца, 1 М 380 г РЬ(СН3СОО)2-ЗН2О
12 Бромная вода, насыщенный раствор3’ 6 15 см3 Вг2
13 Ацетат кадмия, 0,5 М 116 г Cd(CH3COO)2
14 Ацетат кальция, 2 М ИЗ г СаО осторожно смешивают с 180 см3 СН3СООН и несколько раз размешивают с активированным углем. Отсасывают и доливают водой до 1 дм3
15 Уксусная кислота, 5 М (2) 296 см3 СН3СООН
16 Иодоводородная кислота, 1 Ма’6 96 см3 67%-ной HI
17 Цианид калия, '1 М 65,1 г KCN
18 Бромид калия, 0,2 М 24 г КВг
19 Хромат калия, 0,5 М 97 г К2СгО4
Продолжение
№ Реактив Количество вещества (г или см3) иа 1 дм3 раствора; способ приготовления
20 Иодид калия, 0,1 М 16,6 г KI
21 Иодид калия, 1 М 166 Г KI
22 Трииодид калия, 0,05 Мв 8,8 г KI растворяют в небольшом количестве воды +12,7 г 12
23 Карбонат калия, 2,5 М 436 г К2СО5-2Н2О
24 Нитрит калия, 5 М 426 г KNO2
25 Перманганат калия, 0,02 Ма 3,16 г КМпО4
26 Хлорат натрия, 5 М 532 г NaClO3
27 Карбонат натрия, 2 М 572 г Na2CO3-10H2O
28 Гидросульфид натрия, 1 Ма 900 см3 1 М NaOH охлаждают до 0°С. Через раствор пропускают H2S до насыщения н добавляют 100 см3 1 М NaOH. Раствор малоустойчив
29 Гидроксид натрия, 5 М 200 г NaOH
30 Азотная кислота, 14,5 М® Конц. HNO3 (пл. 1,40 при 15 °C)
31 Азотная кислота, 2,5 М8 172 см3 14,5 М HNO3
32 Соляная кислота, 12 Мб Конц. НС1 (пл. 1,19 при 15 °C)
33 Соляная кислота, 5 М® 416 см3 И2 М НС1
34 Серная кислота, 18 М® Конц. H2SO4 (пл. 1,84 при 15 °C)
35 Серная кислота, 2,5 М8 122 см3 18 М H2SO4 добавляют (осторожно!) к 878 см3 воды
36 Нитрат стронция, насыщенный раствор 720 г Sr(NO3)2-4H2O
37 Нитрат серебра, 5%а 50 г AgNO3
38 Нитрат серебра, 0,5 Ма 85 г AgNO3
39 Тетрахлорид углерода6
40 Пероксид водорода, 2,5 Ма 283 см3 30%-ного Н2О2
а Склянка из коричневого стекла. 6 Скляика с пришлифованной пробкой.
2*
Таблица Д.2. Растворы реактивов специального назначения
№ Реактив Количество вещества (г или см3) иа 250 см3 раствора, способ приго* товления
1 Алюминон, 0,2% 0,5 г растворяют в 25 см3 воды, разбавляют до 250 см3
2 н-Амиловый спирт
3 Ацетат беизидниа, 0,1 %а 0,25 г бензидина растворяют в 25 см3 СНзСООН, разбавляют до
250 см3
4 Ацетат бензидина, 1%а 2,5 г бензидина растворяют до 25 см3 СНзСООН, разбавляют до 250 см3
5 Бензол®
6 Тетрародаиомеркурат(И) аммония 75 г HgCl2+83 г NH4SCN
7 Диметилглиокснм, 1 % 2,5 г растворяют в 25 см3 абсолютного этанола, доливают этанолом
8 Дифенилкарбазид, 1%а 2,5 г растворяют в 25 см3 абсолютного этанола, доливают этанолом
9 Формалин, 35% а
10 Раствор индиго, 0,02 М 1,6 г медленно вносят в 10,4 г дымящей H2SO4 через 2 сут вливают в 20-кратное количество Н2О, фильтруют
11 Йодноватая кислота, 0,5 М 22 г НЮз
12 Гексацианоферрат(П) калия, 1 М 105 г K4[Fe(CN)6]-3H2O
13 Иодат калия, насыщенный раствор 20 г КЮз
14 Магнезиальная смесь 14 г MgCl2-6H2O+35 г NH4CI растворяют в Н2О, добавляют 31 см3 13 М NH4OH, доливают водой до 250 см3
15 Магнийуранилацетат 13 г UO2(CH3COO)2-2H2O растворяют в 8 г СНзСООН и 125 см3 воды. 41 г Mg(CH3COO)2-•4Н2О растворяют в 13 г СНзСООН н 1125 см3 воды. Растворы смешивают. Вносят кристалл NaCl. Через 24 ч фильтруют
16 Магнезон, 0,2% 0,5 г растворяют в 2 и. NaOH и доливают 2 н. NaOH до 250 см3
17 Малахитовый зеленый, 0,005%а ОД! г растворяют в СН3СООН, доливают водой до 250 см3
18 Сульфат марганца, насыщенный раствор 165 г MnSO4-4H2O
19, Метанол
20 Метиловый оранжевый 0,1 %а 0,25 г
20
37. Качественный анализ
Z1
Продолжение
№ Реактив Количество вещества (г или см3) на 250 см3 раствора, способ приготовления
21 Смешанный индикатор® 0,31 г метилового оранжевого
22 Морин, 2%а смешивают с 0,21 г метилового синего, доливают метанолом до 250 см3 5 г растворяют в метаноле
23 а-Нафтиламин, 0,3% 0,75 г растворяют в 83 см1
24 Реактив Несслера® СНзСООН, доливают водой до 250 см3 13 г KI растворяют в 13 см3 во-
ды, добавляют насыщенный раствор HgCl2 (6 г/100 см3) до образования устойчивого осадка Hgl2; добавляют 50 см 6 М NaOH и доливают водой до
25 Нейтральный красный, 0,1% 250 см3. Дают отстояться и прозрачный раствор сливают в темную склянку 0,25 г растворяют в 250 см3
26 Хлорная кислота, 70%® 60%-ного этанола
27 клорная кислота, 3 М 65 г 70%-ной НС1О4
28 Фенолфталеин, 1%® 2,5 г растворяют в 70%-ном эта-
29 Фосфорная кислота, 85%® иоле
30 Сульфаниловая кислота, 1% 2,5 г растворяют в 75 см3
31 Оксисульфат титана, 0,05 М СНзСООН, доливают водой до 250 см3 2 г ТЮ2 сплавляют с 15-кратным
32 Нитрат висмута, 0,2 М количеством K2S2O7, расплав растворяют в 2,5 М H2SO4, доливают водой до 250 см3 24 г В1(НОз)з-5Н2О растворяют в
33 Оксихлорид циркония, 0,5 М 8 см3 '14,5 М HNO3, доливают водой до 250 см3 40 г ZrOCl2-8H2O
a Склянка из коричневого стекла, в Склянка с пришлифованной пробкой.
Склянки с реактивами размещают в деревянном штативе, выполненном в виде лесенки* (рис. Д.1), благодаря чему легко доставать пипетки из склянок. Кроме того, растворы в штативе хорошо видны и расположены в определенном порядке. Склянки с концентрированными кислотами должны находить
* Так называемая горка. — Прим, перев.
22
Д, Химический анализ
ся на нижней ступеньке штатива, иначе падающие с них капли могут (повредить' этикетки и пипетки других склянок. Основные растворы реактивов, которые должны находиться в штативе, перечислены в табл. Д.1.
Так как в штативе может находиться не больше 40 склянок с реактивами, то редко применяемые реактивы - размещают отдельно — в одном1 или нескольких особых штативах (так называемых штативах для реактивов специального назначения), а реактивы общего .пользования размещают в центре лаборатории. Перечень реактивов специального назначения приведен в табл. Д.2.
На рабочем месте всегда надо иметь запас всасывающих баллончиков для пипеток и навинчивающихся крышек с резиновыми прокладками для быстрой замены разрушенных или поврежденных частей склянок.
Твердые реагенты хранят в широкогорлых склянках для порошкообразных веществ вместимостью 10 см3. Для удобства их ставят а гнезда специального штатива (рис. Д.2). Этикетки на склянках для реактивов специального назначения такие же, как на склянках с растворами реактивов. Перечень твердых реактивов, которые должны быть на рабочем; месте, а также твердых реактивов специального назначения, приведен в табл. Д.З.
Таблица Д.З. Твердые реактивы
Основные реактивы
Реактивы специального назначения
1. Ацетат аммония
2. Карбонат аммония
3. Хлорид аммония
4. Тетраборат аммония
5. Сплав Деварда
6. Хлорид натрия
7. Соль Мора
8. Гидросульфат калия
9. Нитрат калия
10. Карбонат натрия, безводный
1<1. Гексанитрокобальтат натрия
12. Гидрокарбонат натрия
13. Гидроксид натрия
14. Нитропруссид натрия
15. Цинковая пыль
1. Амидосерная кислота
2. Дифенилкарбазид
3. Этилксантогенат калия
4. Гексацианоферрат(Ш) калия
5. о-Нитробензальдегид
6. >8-Оксихинолин
7. Диоксид кремния
8. Тиомочевина
Аналогично макрометоду аналитические реакции в полумикрометоде проводят в основном в пробирках. Стандартные пробирки для проведения полумикроанализа в соответствии с TGL 40-315 имеют размеры 9,5X751 мм! и сделаны из стекла марки 490 (TGL 7209) (рис. Д.З).
Рис. Д.1. Штатив для склянок с растворами реактивов.
Рис. Д.2. Специальный штатив с гнездами для склянок с твердыми реактивами.
Рис. Д.З. Пробирка для полумикроанализа.
Рис. Д.4. Центрифужная пробирка.
Рис. Д.5. Стакан со сферическим дном, применяемый в полумикроанализе для кипячения растворов.
24
Д. Химический анализ
Для нагревания твердых веществ применяют трубочки для прокаливания 5Х(30—50) мм, изготовленные из термостойких сортов йенского стекла, которые после употребления выбрасывают.
Реакции, заканчивающиеся разделением фаз, проводят в центрифужных пробирках различных типов. Типы пробирок сходны с применяемыми для полумикроанализа и регламентируются стандартом, i(TGL! 40-317). Центрифужные пробирки для полумикроанализа 'вмещают 3 см3 раствора (рис. Д.4).
При работе с несколько большими объемами жидкостей, например при осаждении сульфидов, применяют конические стаканы вместимостью 15 см3 (стандарт TGL 40-316), которые также изготовляют из йенского стекла, применяемого для изготовления толстостенных сосудов. Дно их должно быть закруглено для удобства удаления и промывания осадка. Толстостенные стеклянные сосуды! нельзя нагревать на открытом пламени (почему?).
Максимальные объемы растворов, которые применяют в полумикроанализе, не превышают 10—15 см3. Для нагревания и упаривания таких объемов жидкостей; а также удаления летучих компонентов применяют полумикростаканы со сферическим дном вместимостью 25 см3 (рис. Д.5). Их можно нагревать на открытом пламени.
Для поддержания чистоты и порядка на рабочем месте всю посуду следует располагать продуманно, на строго определенных местах. Обычные и центрифужные пробирки должны на
37. Качественный анализ
ходиться в деревянных или пластмассовых штативах (рис. Д.6).
Для размещения стаканов со сферическим1 дном применяют простые деревянные колодки с тремя'отверстиями (рис. Д.7).
37.1.5.2. Приемы перенесения жидкостей
В полумикрометоде работают с малыми объемами жидкостей, поэтому жидкости нецелесообразно переливать из одного сосуда в другой, так как это сказывается на чистоте работы' и при этом нельзя точно отмерить количество жидкости. Для этих целей применяют капельные пипетки. На рис. Д.8 показаны капельные пипетки двух типов. Пипетки с длинным капилляром (стандарт TGL 40-314,А) применяют для отделения раствора от осадка и для добавления растворов реактивов. Пипетки с коротким капилляром (стандарт TGL 40-314, В) применяют для перенесения жидкостей в сосуды большого размера— стаканы для кипячения или большие центрифужные пробирки. При работе с полумикропробирками применяют тонкие
Рис. Д.8. Пипетки для полумикроана- Рис. Д.9. Штатив для капельных пилиза; а — с длинным капилляром, петок.
б — с коротким капилляром.
Длинные пипетки. Всасывающий баллончик пипетки держат большим и указательным пальцами, одновременно придерживая пипетку* ладонью. Раствор реактива из пипетки добавляют по каплям (дозируют).
Для обеспечения непрерывной работы рекомендуется иметь Достаточно большой запас чистых, сухих пипеток. Использованные пипетки помещают в склянку с дистиллированной водой таким образом, чтобы они были заполнены водой. В конце рабочего дня пипетки очищают, ополаскивают дистиллированной водой и высушивают в сушильном шкафу. Баллончики
26
Д. Хим.ический^нали3-
для всасывания делаются* из мягкой резины, поэтому при очи стке с ними надо обращаться остоРожн°" в коем случае нельзя использовать для очистки хромовую смесь или кон центрированные кислоты! Достаточно промыть теплой водой Загрязненные, старые, потрескавшиеся баллончики нужно заменять новыми.
На рабочем месте1 капельные пипетки хранят в специальном штативе (рис. Д.9) в горизонтальном положении, их нельзя класть на стол.
37.1.5.3. Приемы перенесения тв?Р^ых веЩеств
Твердые реактивы или анализируем?'10 смесь отбирают при помощи ,полумикрошпателя. При этом применяют обычные зубоврачебные или маленькие никелевые шпатели (рис. Д.10). Шпатель по ширине должен входить в полумикропробирку. Материалом для изготовления шпателей служит нержавеющая или хромированная сталь либо никель.
Рис. Д.10. Шпатели-
Избыток вещества со шпателя ссыпают; шпатель после употребления хорошо протирают чисто® ТРЯПКОИ или фильтровальной -бумагой. Его также можно хранить в штативе для капельных пипеток.
37.1.5.4. Нагревание
В качественном анализе часто приходится производить нагревание для полноты образования осаДка> упаривания, прокаливания и плавления. Наиболее важным источником тепла является известная горелка Бунзена. Она Д°лжна быть снабжена насадкой для предотвращения пр°скока пламени.
В пламени бунзёновской горелки нельзя нагревать узкие пробирки для полумикроанализа, так как содержимое пробирки при этом выкипает (а также задерживается кипение.). Эти пробирки нагревают на водяной бане-
Баня для полумикроанализа — это обычный широким стакан вместимостью 400 см3, изготовЛеннь111 J13 йенского стекла, с соответствующей вставкой (рис. Д-11)- вставке имеются отверстия для полумикропробирок, центрифужных пробирок и стаканчиков для кипячения жидкосте®- Ур°вень дистиллированной воды в стакане должен находиться примерно на 1
37. Качественный анализ
27
2 см ниже поверхности вставки с отверстиями. В воду помещают кипятильники и в ходе аналитических работ (постоянно (поддерживают небольшое кипение воды.
В стакане со сферическим, дном, предназначенном для кипячения жидкостей, можно проводить упаривание, нагревая стакан в слабосветящемся, подвижном пламени бунзеновской
горелки. Таким способом можно упарить 5 см3 жидкости за несколько минут.
Особой осторожности требует выпаривание жидкости на предметном стекле. При этом можно использовать небольшое пламя бунзеновской горелки, двигая взад и вперед зажатое пинцетом или зажимом предметное стекло над пламенем горелки на высоте 20—30 см. Вместо горелки лучше применять для нагревания инфракрасные лампы, освещающие предметное стекло сверху. Ни в коем случае
Рис. Д.11. Водяная баня для полу-
нельзя выпаривать раствор на микроанализа.
стекле досуха. Его нужно
лишь слегка нагревать. Для образования кристаллов предмет-
ное стекло затем переносят в место, защищенное от пыли.
Нагревание в калильной трубочке или с 18 М H2SO4, а также переведение вещества в .растворимое состояние сплавлением проводят на несветящемся .пламени бунзеновской горелки или горелки с насадкой.
37.1.5.5. Работа с газами
В полумикроанализе некоторые анионы обнаруживают по образованию газообразных продуктов реакции. (Важную роль в анализе катионов еще и в настоящее время играет сероводород, применяемый в качестве группового реактива. В ходе некоторых! аналитических операций необходимо применять слабый поток воздуха. Для проведения этих работ существуют приборы, принцип действия которых описан ниже.
Прибор для исследования газов, применяемый в полумикроанализе. Несколько миллиграммов анализируемого вещества помещают в прибор для получения газа (стандарт TGL 40 320) (рис. Д.12) и насаживают согнутую под углом трубку на штуцер прибора, снабженный коротким резиновым шлангом. К этой трубке подсоединяют пробирку для полумикроанализа,
28
Д. Химический анализ
служащую приемником газа. Пробирку предварительно заполняют необходимым реагентом. Затем в длинную вертикально укрепленную трубку засасывают несколько капель жидкости, применяемой для получения газа. Вертикальную трубку закрывают сверху дальнем и так вносят ее в прибор для получения газа. С помощью груши жидкость из трубки выдавливают в реакционный объем. Когда палец опускают, давление выравнивается, после чего верхнее отверстие вертикальной трубки опять закрывают пальцем. Образующийся газ с помощью груши выдавливают в приемник. Этот процесс при необходимости повторяют.
К вертикальной трубке можно при необходимости подключить трубку для подвода промывающего газа.
Обнаружение газа проводят в «газовой микрокамере» (рис. Д.13). Ее применяют, если газ можно идентифицировать-кристаллографически, а также для обнаружения аммиака. Газовую микрокамеру можно изготовить самим. Из толстой стеклянной трубки диаметром 40—15 мм вырезают кольцо высотой 8—10 мм и шлифуют его с обеих! сторон для получения; ровных поверхностей. Два предметных стекла или одно предметное стекло и одно большое покровное стекло образуют дно и крышку «газовой микрокамерьв». На нижнее предметное стекло наносят каплю пробы и каплю реактива, затем ставят на предметное стекло стеклянное кольцо таким образом, чтобы капли находились внутри кольца, и накрывают его покровным. стеклом, на котором находится капля реактива для обнаружения газа. Дают некоторое время постоять и наблюдаюг эффект реакции.
Получение сероводорода. Сероводород, необходимый для< осаждения, в основном получают при взаимодействии FeS с НС1. Для (этой цели в качественном! полумикроанализе применяют не аппарат Киппа, а полумикропрйбор для получения: газа (рис. Д.14).
Устройство прибора и принцип его действия понятны из рисунка. Этот прибор можно изготовить самим. Трубку, заполненную кольцами Рашита и сульфидом железа, делают из пробирки для микроанализа; в дне пробирки проделывают несколько отверстий диаметром 1 ,мм. Кислоты находятся в нижней части промывной склянки.
Ф. Зеель предложил простой метод для получения чистого H2S. При нагревании смеси серы и парафина образуется сероводород с выходом ~98%:
С„Н2П+2 + (я + 1) S -> пС + (я + 1) H2S
Сырье для получения газа имеется в продаже. Установка проста, и состоит из пробирки, снабженной пробкой с отверстием, и газоотводной трубки. Над палочками парафина помеща-
Рис. Д.12. Прибор для идентификации газов в полумикроанализе.
Рис. Д.13. Газовая микрокамера.
Рис. Д.16. Воздушная камера.
Рис. Д.14. Прибор для получения газов в полумикроанализе.
Рис. Д.15. Прибор для получения H2S (по Ф. Зеелю).
30
Д. Химический, анализ
ют ватный тампон, который задерживает уносимые газом твердые частицы (рис. Д.15). При нагревании до 170 °C и выше выделяется сероводород. Как только нагревание (прекращают, прекращается и выделение сероводорода. Изменяя температуру, можно регулировать поток газа.
Работа! с воздухом. Для получения потока воздуха применяют обычные резиновые пузыри, имеющиеся в продаже. Для создания равномерной скорости потока используют круглодонную колбу вместимостью 750 см3 в качестве воздушного резервуара. Шланг, отходящий от резервуара, закрывают винтовым зажимом для регулирования потока воздуха (рис. Д. 16).
Для обнаружения СО2 в прибор для исследования газов необходима подавать воздух, свободный от ,СО2. Удаление СО2 осуществляют, пропуская воздух через трубку с натронной известью или через промывную склянку, заполненную’ 30%-ным раствором КОН, которую1 устанавливают; между резервуаром для воздуха и прибором для исследования выделяющегося газа.
Выпаривание в стаканчике для кипячения и удаление растворенного хлора или иода можно значительно ускорить, пропуская через раствор поток воздуха с помощью газоподводя-щей трубки. Так, например, после растворения вещества в хлорной воде перед пропусканием1 H2S можно удалить хлор продуванием.
Воздух используют и для проведения фильтрования под давлением по Барберу.
37.1.5.6. Разделение фаз
Осадки отделяют от раствора в основном центрифугированием. В лабораторном практикуме применяют электрические лабораторные центрифуги со скоростью вращения 4000 оборотов в минуту, в которых одновременно можно разместить шесть пробирок. В угловых центрифугах гильзы жестко закреплены под постоянным углом К оси вращения. -Радиальная центрифуга имеет гильзы, -которые при центрифугировании принимают горизонтальное положение. Благодаря жестко укрепленным гильзам угловая центрифуга1 удобнее и надежнее в обращении, однако осадок собирается не на дне пробирки, что является недостатком при работе с небольшими осадками.
Электрическая центрифуга требует очень тщательного обслуживания. Гильзы- центрифуги должны быть точно тарированы на весах. Противовесом пробирке с центрифугируемым веществом служит такая же ,центрифужная пробирка, заполненная водой. Пробирки уравновешивают на весах, добавляя воду из капельной пипетки. При этом объемы жидкости в Пробирках не обязательно должны быть равными, так как надо учитывать различие масс-самих пробирок.
37. Качественный анализ
При работе с небольшими центрифужными пробирками гильзы следует снабжать предохранительными устройствами. При работе б большими пробирками в гильзу кладут ватный там-пом (гильзы центрифуги уравновешены). Быстро центрифугирующиеся осадки, которые» выделяются уже при небольшом числе оборотов, можно отделять в .пробирках для полумикроанализа. Пробирку вставляют в большую центрифужную пробирку с ватным тампоном (рис. Д.17), откуда ее можно, вынуть пинцетом.
Скорость вращения увеличивают постепенно; после отключения центрифуге дают возможность остановиться. При быстром включении пробирки разбиваются и наносят повреждения центрифуге; при быстром торможении осадок снова взмучивается. Центрифугирование проводят в течение примерно одной-двух минут. Если осадок отделен полностью, жидкость, находящуюся над осадком, осторожно отбирают длинной пипеткой (рис. Д.18). Промывную жидкость добавляют пипеткой с коротким капилляром и осадок взмучивают маленькой мешалкой, которая представляет собой тонкую стеклянную палочку с оплавленым плоским концом (рис. Д.19). Затем .центрифугирование повторяют и т. д.
Рис. Д.17. Центри- Рис. Д.18. Способ от- Рис. Д.19. Па-Фужная пробирка сасывания жидкости, дочка для пере-вместимостью 15 см3 с находящейся над мешивания. ватной прокладкой осадком, пипеткой с
Лля центрифугирова- длинным капилляром.
ния пробирок.
Рис. Д.20. Установка для фильтрования под давлением по Барберу.
32
Д. Химический аналиэ
37. Качественный анализ
33
Если осадок нельзя отцентрифугировать, можно применить фильтрование под давлением по Барберу. Фильтрование проводят через вату, находящуюся в трубочке для фильтрования. Давлением воздуха фильтруемую жидкость продавливают через фильтр в пробирку. Устройство установки и способ работы понятны из рис. Д.20.
37.1.5.7. Работа с микроскопом
В качественном анализе нельзя обойтись без идентификации кристаллических веществ под микроскопом. Для аналитических целей используют обычный микроскоп. Необходимо увеличение трех размеров: 40Х, 80Х> 200Х- При большом рас- j стоянии до рассматриваемого объекта нужны слабые объек-! тивы и сильные окуляры. Использование конденсора и ново-! ротного столика не обязательно. Микроскоп устанавливают на специальном столе; в нерабочее время его надо хранить в i специальном ящике или покрыть полимерной пленкой для защиты, от агрессивных лабораторных паров.
При исследовании веществ под микроскопом сначала применяют самое малое увеличение, так как при большем увели-, чении ничего не видно. Объектив всегда перемещают снизу вверх, иначе чувствительный объектив может попасть в жидкость, часто являющуюся агрессивной. Кристаллы получают на предметном стекле. Каплю анализируемой пробы наносят на предметное стекло рядом с каплей реактива, затем обе капли соединяют с помощью платиновой проволоки или стеклянной палочки. Кристаллы должны расти медленно, из разбавленных растворов. Для этого обычно раствор с кристаллами оставляют стоять при комнатной температуре, затем очень медленно выпаривают жидкость. Следует отметить, что кристаллы одного и того же вещества могут выглядеть по-разному в зависимости от условий образования, концентрации, скорости выделения, присутствия посторонних солей и значения pH. Доваренная соль, например, кристаллизуется из воды в виде кубических кристаллов. В присутствии мочевины образуются Октаэдры, а кубы не формируются. Лучше всего сравнивать форму изучаемых кристаллов с полученными при тех же условиях кристаллами соответствующего чистого вещества.
37.1.5.8. Капельные реакции
Капельные реакции проводят по завершении стадий разделения, т. е. в том случае, когда анализируемое вещество даль-toe уже не обрабатывают. Подготавливают пластинку из белого или черного фарфора или наполовину черную, а наполовину белую. Капельные пластины из йенского стекла прозрач
ны. Подкладывая листы бумаги определенного цвета, можно так подобрать цвет подложки, чтобы осадок был ясно виден.
Реактивы и раствор пробы наносят на капельную пластинку с помощью пипетки. Капли должны падать свободно, чтобы не загрязняться. При образовании паров или газов реактивы сначала наносят по каплям на стеклянную палочку, а затем 'вносят в углубление капельной пластинки.
К капельным реакциям относятся также реакции, проводимые на бумаге (подробно см. разд. 37.3.2.1).
37.1.5.9. Ионный обмен
Некоторые анионы, например фосфат, борат и оксалат, мешают разделению катионов в качественном анализе. Для удаления этих анионов известно много методов. (В полумикроанализе часто используют ионный обмен. Теория и практика этого метода рассмотрены в разд.
38.3.7.1. Применяемые в полумикроанализе ионообменные колонки показаны на рис. Д.21.
Для катионного обмена применяют смолу типа KPS 200 (катионит КУ-2), для анионного обмена — смолу типа SBW (анионит АВ-17). Ионообменную колонку, закрытую внизу тампоном из стекловаты, готовят к работе следующим способом. Сначала ее заполняют дистиллированной водой и затем порциями вносят в нее катионит, набухавший в течение полусуток в 5М НС1. Сверху смолу покрывают слоем стекловаты. Пропускают несколько раз по 5 см3 5 М НС1 через колонку и промывают затем водой до нейтральной реакции элюата.
В нерабочем состоянии колонку заполняют 5 М НС1; после промывания водой до нейтральной реакции колонку снова можно применять для ионного обмена.
Максимальная катионообменная емкость ионита при диаметре слоя 7 (мм и длине колонки ЮО мм составляет 4 ммоль (при проведении1 ионного обмена в нейтральной среде) и 3 ммоль Дв растворе 0,5 ,М НС1). Скорость прохождения 1 — 3—1880
обмен. Теория и практика это-
Рис. Д.21. Ионообменная колонка для полумикроанализа.
34
Д. Химический анализ
2 см3/мин, что соответствует 1—2 каплям в секунду. Поскольку смола в кислой среде .«сморщивается», скорость прохождения может изменяться. При длительном^ применении смола в колонке уплотняется; ее нужно заменять, но не ранее чем после шестикратного использования. При работе со смолами (даже марки ч. д. а.) необходимо (Проводить испытание смолы на наличие загрязнений. Прежде всего нужно обратить внимание на присутствие ионов кальция. При обнаружении ионов, кальция смолу следует промыть разбавленным аммиачным раствором комплексона.
37.2. Практическая часть
37.2.1. Ход качественного химического анализа
Смесь веществ1 для качественного анализа может иметь различное происхождение. Речь может идти об исследовании искусственной, специально для анализа приготовленной, смеси или об анализе природного или технического продукта.
В состав искусственных проб, полученных смешиванием веществ, могут входить примерно в равных количествах отдельные компоненты, число которых часто довольно велико. Технические и природные продукты содержат относительно малое число основных компонентов, но число элементов, присутствующих в следовых количествах, может быть довольно большим. Некоторые ионы можно исключить сразу. При анализе технических продуктов и минералов трудность состоит в 'определении следовых количеств примесей в присутствии основных компонентов. В этом' случае нужно применять другую технику работы, чем при анализе пробы, полученной смешиванием веществ.
В процессе развития аналитической химии была разработана определенная техника качественного анализа. Каждый аккуратно работающий аналитик использует эту технику, так как она гарантирует получение надежных результатов наиболее быстрым способом. Однако это не означает, что нужно слепо воспроизводить все прописи анализа и процессы разделения, Каждую операцию нужно хорошо продумать и делать необходимые выводы из результатов опытов. Качественный анализ включает следующие этапы: а) отбор пробы; б) описание' внешнего вида1 пробы; в) предварительные испытания (мюкрым или сухим путем); г) растворение пробы; д) обнаружение анионов; е) обнаружение катионов; ж) анализ нерас-творенного остатка.
В этой главе основное внимание будет уделено систематическому .ходу анализа искусственных проб, полученных смешением веществ.
37. Качественный анализ
35
37.2.1.1. Отбор пробы
Первой операцией в ходе качественного анализа технического или (Природного продукта является отбор пробы; эту операцию необходимо проводить очень тщательно. В принципе для отбора пробы можно1 лрименять методы, описанные в количественном анализе. Часто операция отбора пробы упрощается, так как получение точных количественных соотношений в данном случае необязательно.
Очень важно тщательно измельчать пробу, поскольку проба при этом хорошо перемешивается и быстрее растворяется. Лучше затратить1 15 мин на измельчение пробы, чем тратить время на многочасовое растворение неизмельченного вещества. Необходимо следить за тем, чтобы при измельчении вещество не загрязнялось пылью. Пробы можно измельчать в агатовой ступке.
Учебную пробу для анализа, полученную смешением веществ, также нужно тщательно измельчить в ступке и (перемешать, так как в негомогенной пробе можно не обнаружить всех компонентов. После отбора пробы анализируемого вещества или после приготовления искусственной пробы ее сразу изолируют от доступа воздуха во» избежание загрязнения (так, вещества сильноосновного характера поглощают из воздуха СО2 и другие компоненты кислотного характера).
Необходимо составить точный отчет, включающий следующие сведения: источник пробы, ее количество, внешний вид, время ютбора пробы.
37.2.1.2. Предварительные испытания
Предварительные испытание проводят с помощью 'простых Вспомогательных средств. Они требуют очень небольших затрат времени и тем не менее в гораздо большей степени, чем любая другая операция химического анализа, выявляют способности химика внимательно наблюдать и делать из наблюдений правильные выводы. Предварительные испытания дают ценные сведения о примерном составе анализируемого вещества, и хотя не могут заменить идентификацию' в ходе разделения, но позволяют модифицировать и упрюстить ход анализа. Ионы, .определяемые в предварительных испытаниях, Должны быть надежно идентифицированы химическими методами.
Важно установить, какие ионы в пробе отсутствуют. Предварительные испытания1 в основном проводят с твердыми исходными веществами, растворы выпаривают. Целесообразно установить, содержатся ли при этом в парах летучие неорга-3*
36
Д. Химический анализ
нические вещества (AsCU, HgCl2 и т. д.) или продукты разложения (например, НС1).
Проба на восстановление в пламени при помощи паяльной трубки.' Эта проба основана! на восстановлении расплавов солей углем и веществами в восстановительной части пламени газовой горелки (например СО, Н2, СН4, радикалами). Для защиты восстановленных веществ используют добавку соды, которая является основным флюсом.
Для работы необходимы паяльная трубка, кусок липового или тополиного древесного угля, безводный |Na2CO3 (ч.д. а.) и горелка.
Обычные реакции можно проводить, применяя горелку Бунзена без подачи в нее воздуха. Светящееся пламя газовой горелки нельзя применять при идентификации серуоодержащих соединений, так как в светильном газе могут быть соединения серы. В этих случаях используют пламя обычной спиртовки, применяемой в стоматологической практике.
Для ’проведения реакции в куске древесного угля просверливают круглое углубление диаметром ^0,5 мм. Тонкоизмель-ченное (порошок)* обезвоженное вещество на кончике шпателя переносят на часовое стекло и тщательно смешивают с двойным количеством измельченной в порошок безводной соды. Долученной смесью заполняют углубление в древесном угле, смачивают смесь каплей дистиллированной воды и уплотняют. Затем смесь сплавляют в восстановительном-, пламени с применением! паяльной трубки. При этом, происходят реакции, в результате которых большинство солей и оксидов восстанавливаются' с образованием металлов. Уголь впитывает избыток расплавленной соды и образовавшиеся1 соли натрия. В зависимости от температуры плавления металлы могут образовываться в виде сплавленных! металлических зерен (РЬ, Sn) или несплавленных металлических блесток (Fe). Легкоплавкие или легкоокисляющиеся металлы частично переходят в парообразное состояние (Zn, Cd). За пределами восстановительной зоны пламени пары металла снова окисляются и оксиды осаждаются на угле по краям углубления.
Для получения восстановительного пламени на светящееся пламя дуют сбоку, тем самым сгибая его. Для успешного проведения реакции необходимо непрерывно дуть на пламя, иначе исследуемое вещество, взятое в небольшом количестве, сразу же охлаждается и не вступает в реакцию. Необходимо no-j упражняться для овладения техникой эксперимента. На пламя дуют ртоми при этом дышат через нос. 1
Так же как в пламени горелки Бунзена, в пламени, полу-1 ченном с применением паяльной трубки, различают восстановительную и окислительную зоны. Пламена имеют примерно одинаковое расположение окислительной и восстановительной
37. Качественный анализ
37
Восстановительная зона
Окислительная зона
Рис. Д.22. Способ получения пламени для восстановления с применением паяльной трубки.
зон. На рис. Д.22 изображено пламя, полученное при помощи паяльной трубки и обозначены важные для анализа зоны.
При поддувании необходимо следить за тем, чтобы язычок внутреннего конуса пламени касался внесенного в пламя древесного угля. Примерно через 2—3 мин реакция завершается и расплав впиты-
вается углем; его можно охладить и остаток обработать на часовом стекле теплой водой. Образовавшийся металл можно затем растворить в 1—3 к. 14,5 М. HNO3 и идентифицировать химическим путем. Не рекомендуется проводить полное разделение компонентов пробы из этого небольшого объема раствора. Следует провести лишь некоторые реакции обнаружения.
В табл. Д.4 указаны некоторые типичные признаки металлических зерен и налетов оксидов металлов.
Для ускорения процессов восстановления и сплавления можно вместо соды применять оксалат или цианид калия. Это способствует лучшему оплавлению металлов.
Для обнаружения серы, селена и теллура служит Любые серусодержащие соединения можно восстановить нии с содой с образованием сульфидной серы. Расплав
Таблица Д.4. Поведение солей металлов при восстановлении в пламени с применением паяльной трубки
гепариновая проба. углем при нагрева-помещают на глад-
Не восстанавливаются соединения Образуют зерна или блестки металлов Дают налет оксида без образования зерен металлов
без налета оксида С налетом оксида
Mg, Са, Sr, Ва Sn — белый, пластичный РЬ — желтый налет As — белый
А1, Мп, Cr, V Ag — белый, пластичный Bi — желтый налет Zn — белый, при нагревании желтый
Си — желтые блестки Au — желтый, пластичный Fe —серые блестки Со — серые блестки Ni — серые блестки Мо — желтый налет при нагревании; белый налет на холоду Sb — белый налет на холоду Cd — коричневый
38
Д. Химический анализ
кий лист серебра (серебряную монету), смачивают несколькими каплями
воды и дробят. Примерно через пять минут после смачивания на листе появляется черное пятно сульфида серебра. Для проведения реакции нельзя использовать сетевой газ. Необходимо каждый раз проводить холостой опыт, так
как соединения серы могут загрязнить древесный уголь.
Определение лучше проводить полумикрометодом; простейшим вариантом является восстановление на обугленных содовых палочках по Бунзену. Недостатком этого способа является отсутствие налетов оксидов. Для проведения реакции необходима сода и деревянные палочки длиной 20—30 см и толщиной 3,5 мм, а также горелка Бунзена^ Конец палочки окунают в пасту, полученную смешиванием соды с водой, и нагревают в пламени (операции повторяют несколько раз). Концом полученной таким образом сдовой палочки
прикасаются к сухому анализируемому веществу и восстанавливают его в пламени, при этом сода предотвращает горение самой палочки. Обугленный конец
вносят в воду, смывают с него уголь и иследуют металлы, как описано выше (восстановление на угле при помощи паяльной трубки).
Окрашивание пламени и изучение спектров. При действии высоких температур электроны в атоме возбуждаются и переходят на более высокий энергетический уровень. Дри переходе электронов на прежний .энергетический уровень излучается свет определенной длины волны. Для каждого элемента существует характеристическая длина волны. Под действием сравнительно низкой температуры газового пламени излучают свет лишь немногие элементы. К ним относятся щелочные, щелочноземельные, а также некоторые тяжелые металлы. Температура возбуждения зависит и от присутствующих анионов. Сульфаты щелочноземельных металлов в пламени практически не излучают света. Для испытаний на окрашивание пламени лучше всего применять ^хлориды. Поскольку следовые количества натрия практически невозможно устранить, окрашивание пламени соединениями натрия часто маскирует окрашивание других элементов. Дерекрывание окрасок наблюдается также при одновременном присутствии нескольких элементов. В этих случаях лучше применять простейший спектроскоп.
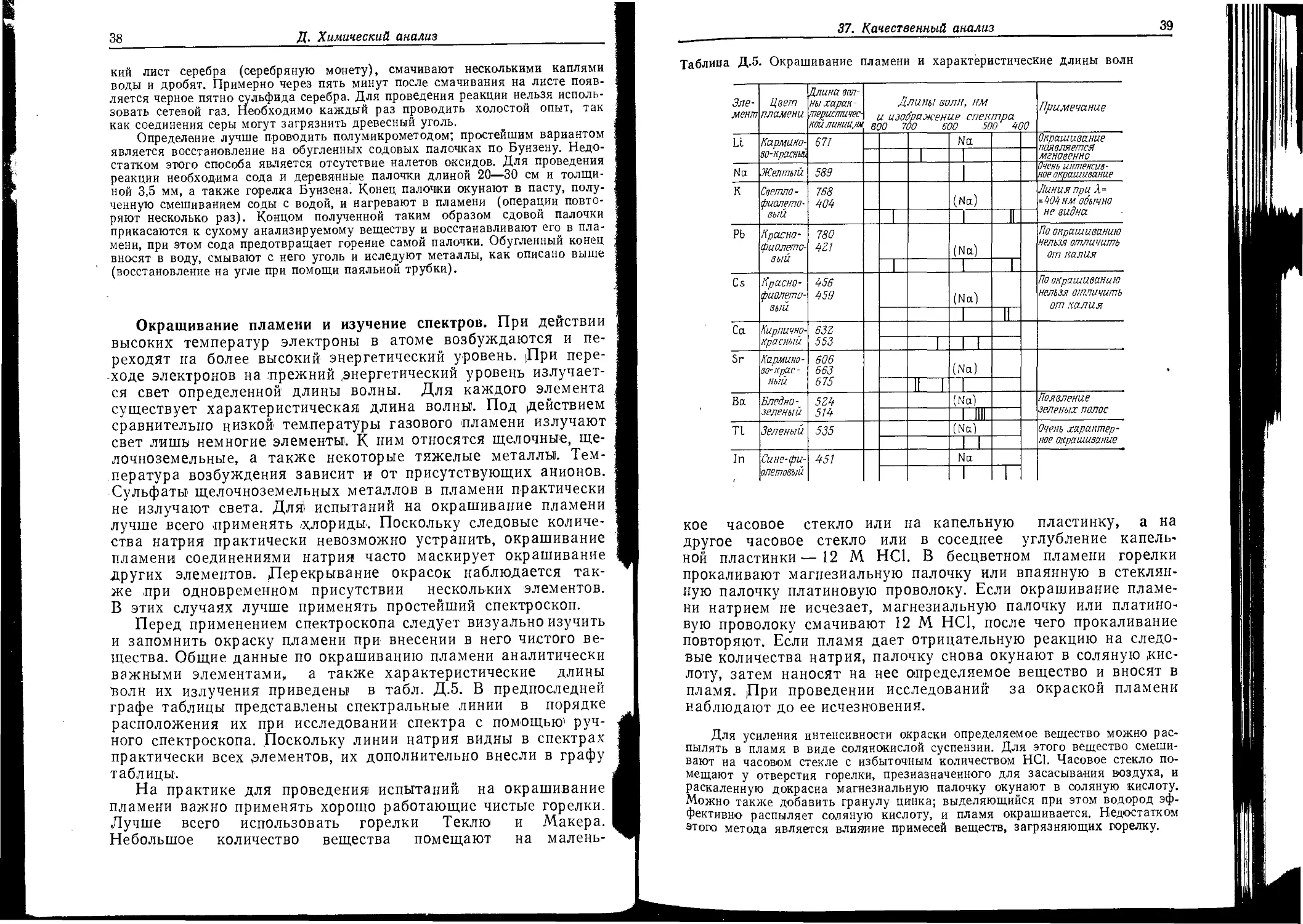
Перед применением спектроскопа следует визуально изучить и запомнить окраску пламени при внесении в него чистого вещества. Общие данные по окрашиванию пламени аналитически важными элементами, а также характеристические длины Волн их излучения приведены в табл. Д.5. В предпоследней графе таблицы представлены спектральные линии в порядке расположения их при исследовании спектра с помощью'1 ручного спектроскопа. Поскольку линии натрия видны в спектрах практически всех элементов, их дополнительно внесли в графу таблицы.
На практике для проведения испытаний на окрашивание пламени важно применять хорошо работающие чистые горелки. Лучше всего использовать горелки Теклю и Макера. Небольшое количество вещества помещают на малень-
37. Качественный анализ
39
Таблица Д.5. Окрашивание пламени и характеристические длины волн
Элемент Цвет пламени Илина волны харак-теристичес кой линии,.нм Длины волн, нм и изображение спектра 800 700 000 500 400 Примечание
Li Карминово-красный 671 Na Окрашивание появляется мгновенно
1 1
Nd Желтый 589 1 Очень интенсивное окрашивание
к Светло-фиолетовый 768 404 (Na) Линия при Л= -404 нм обычно не видна
1 1 II
РЬ Красно-фиолетовый 780 4Z1 (Na) Ко окрашиванию нельзя отличить от калия
т 1 “T
Cs Красно-фиолетовый 456 459 (Na) По окрашиванию нельзя отличить от калия
1 T
Са Кирпичнокрасный. 63Z 553
1 I |
Sr Кармино-во-крас -ный 606 663 675 (Na)
II 1 1
Ва Бледно-зеленый 5Z4 514 (Na) Появление зеленых полос
1 Illi
П Зеленый 535 (Na) Очень характерное окрашивание
1 1
In Сине-фиолетовый 451 Na
1 1
кое часовое стекло или на капельную пластинку, а на другое часовое стекло или в соседнее углубление капельной пластинки—12 М НС1. В бесцветном пламени горелки прокаливают магнезиальную палочку или впаянную в стеклянную палочку платиновую проволоку. Если окрашивание пламени натрием не исчезает, магнезиальную палочку или платиновую проволоку смачивают 12 М НС1, после чего прокаливание повторяют. Если пламя дает отрицательную реакцию на следовые количества натрия, палочку снова окунают в соляную цис-лоту, затем наносят на нее определяемое вещество и вносят в пламя. Дри проведении исследований за окраской пламени наблюдают до ее исчезновения.
Для усиления интенсивности окраски определяемое вещество можно распылять в пламя в виде солянокислой суспензии. Для этого вещество смешивают на часовом стекле с избыточным количеством НС1. Часовое стекло помещают у отверстия горелки, презназначенного для засасывания воздуха, и раскаленную докрасна магнезиальную палочку окунают в соляную кислоту. Можно также добавить гранулу цинка; выделяющийся при этом водород эффективно распыляет соляную кислоту, и пламя окрашивается. Недостатком этого метода является влияние примесей веществ, загрязняющих горелку.
40
Д. Химический анализ
Для надежной идентификации веществ следует применять ручной спектроскоп. Хорошие результаты' получаются при работу а ручным спектроскопом, выпускаемым предприятием Карл Цейс (Иена), снабженным шкалой длин волн и призмой сравнения. На рис. Д.23 приведена оптическая схема ручного спектроскопа.
Свет от пламени горелки попадает на входную щель 1 и затем через фокусирующую линзу 3 на состоящую из трех частей преломляющую призму 4. Спектр рассматривают через окуляр 5. В тубусе размещена шкала длин волн 7, которая через систему линз и зеркало проецируется на спектр. Поло-
Рис. Д.23. Оптическая схема ручного спектроскопа.
/ — щель; 2 — защитное стекло; 3 — ахроматическая линза; 4 — преломляющая призма; 5 — окуляр; 6 — отражательная призма; 7 — шкала длин волн; 8 — винт для юстировки.
жение шкалы можно менять с помощью юстировочного винта 8. Перед работой шкалу прибора градуируют по спектру пламени натрия (Л=589,3 нм). Для того чтобы шкалу было видно и при работе с пламенем, короткую трубку снабжают небольшим приспособлением для освещения. При этом достаточно применить трансформатор и лампочку накаливания (6 В, железнодорожная модель). Подробности конструкции можно видеть на рис. Д.24.
Для удобства обслуживания ручной спектроскоп лучше укрепить в штативе. При этом у работающего со спектроскопом высвобождаются руки- и он не сможет уже неосторожным движением переместить спектроскоп слишком близко к пламени горелки. Расстояние до пламени должно .быть ~8 см/ Ширину щели можно регулировать о помощью винта с накатанной головкой. Оптимальную ширину щели можно определить только на практике. При работе со слишком широкой щелью получают широкие, нечеткие линии, частично ’перекрывающие другие слабые линии,. Хотя слишком узкая щель и дает возможность получить четкие линии, но она пропускает слишком мало света, поэтому наряду с другими могут не появиться и аналитически важные линии.
Спектральный анализ и пробы на окрашивание1 пламени лучше всего проводить в полностью затемненном помещении.
37. Качественный анализ
41
Если это практически невозможно осуществить в аналитической лаборатории, нужно загородить прибор и горелку от прямых солнечных лучей.
Элементы, обнаруженные в( исходном веществе спектральным методом, необходимо идентифицировать также химическим путем. С другой стороны, осадки, полученные при разделении и химической идентификации веществ, можно дополнительно исследовать спектральным методом. При исследовании сульфатов, фосфатов или силикатов щелочноземельных метал-
Рис. Д.24. Схема осветительного приспособления для ручного спектроскопа.
лов пробу предварительно восстанавливают, нагревая ее во внутреннем конусе пламени, и только затем смачивают НС1, При идентификации бора в пламени бораты переводят в бор-нометиловый эфир или в BF3.
К особым случаям предварительных испытаний с помощью спектрального анализа относится идентификация редких металлов. При рассматривании в ручном спектроскопе спектра редких металлов, образующегося при прохождении через них солнечного света, видны четкие полосы поглощения.
Получение перлов веществ с тетраборатом натрия (бурой) или с фосфорной солью. Расплавленный NaNH4HPO4, так называемая фосфорная соль, при добавлении аммиака и воды превращается в полимерный стекловидный полифосфат натрия:
nNaNH4HPO4 ---> (NaPO3)n-H2O + nNH3 + (n — 1) H2O
Тетраборат натрия (Na2B4O7-ЮН2О также образует стекловидные соединения! i При нагревании до 350—400 °C он теряет кристаллизационную! воду и вспучивается, а 'при 870 °C плавится с образованием стекловидной массы.
Как полифосфат, так и тетраборат натрия могут растворять оксиды металлов. В случае оксидоц тяжелых металлов при этом появляется характерная окраска. В настоящее время считается, что получается коллоидный раствор оксида металла
42
Д. Химический анализ
в расплаве. В аналитической химии ;эту реакцию применяют в предварительных испытаниях на содержание тяжелых металлов для получения перлов. Однако применение этой пробы ограничено, так как в, случае смеси металлов получается нехарактерная окраска. Чувствительность, реакции различна для разных элементов, поэтому можно сделать некоторые выводы о составе смеси. Так, марганец, кобальт и ванадий можно надежно определить, а реакции обнаружения хрома, титана, молибдена и вольфрама нечувствительны.
Для проведения реакции нагревают магнезиальную палочку и окунают ее в соль. Прилипшую соль медленно, плавят до получения перла. Перл слегка охлаждают, добавляют к нему небольшое количество твердого анализируемого вещества и сплавляют в течение нескольких минут в окислительном пламени горелки. Окраску горячего и холодного перла записывают. Затем! сплавление повторяют в восстановительном пламени. При этом перльг нужно охлаждать во внутреннем, конусе пламени. Затем- их быстро проносят через окислительную, зону пламени. Если перл слабо окрашен, сплавление повторяют с большим, количеством вещества. Нельзя с самого начала брать слишком много анализируемого вещества, так как избыток оксида окрашивает перл в черный цвет.
Окраски перлов буры и фосфорной соли идентичны. Исключение составляет кремневая кислота, которая в перле фосфорной соли образует так называемый кремниевый каркас, что не наблюдается для перлов буры. Многие оксиды, как, например, SnO2, нерастворимы) в перлах и, так же как S1O2, вы-
Таблица Д.6. Окраска перлов с тетраборатом натрия (бурой) или «фосфорной солью»а
В окислительном пламени В восстановительном пламени
Элемент горячий перл холодный перл горячий перл холодный перл
Ni Желтый Коричневый Серый Серый
Fe » » Желто-красный Зеленый Зеленый
V » » Коричневый » » » »
и » » Желто-зеленый » » » »
Sb Слабо-желтый Бесцветный Серый Серый
Bi » » » » » » » »
Си Зеленый Синий Бесцветный Красный
Cu+Sn Cr Красный Желто-зеленый Красный Желто-зеленый Зеленый Зеленый
Co Синий Синий Синий Синий
Мп Фиолетовый Фиолетовый Бесцветный Бесцветный
а Перлы Ag, Pb, Bi, Sb, Sn, Ni, Cd, Zn в восстановительном пламени имеют окраску от серой до черной (восстановление до металлов).
37. Качественный анализ
43
зывают их помутнение. Выбор солей для проведения идентификации не имеет значения.
В табл. Д.6 указаны характерные окраски перлов.
Нагревание в калильной трубочке. Толстостенную калильную трубочку изготовляют из йенского стекла. Она имеет длину 5—6 см и внутренний диаметр 5—6 мм.
Тонкоизмельченную в порошок пробу вносят в сухую трубочку так, чтобьп вещество не пристало к стенкам. Трубочку осторожно начинают нагревать на небольшом пламени горелки, подняв ее на 1—3 см над пламенем, и затем усиливают величину пламени и степень нагрева до красного каления трубочки. Растворители заранее испаряют, влажные вещества осторожно высушивают и воду из трубки удаляют. При мед-ленном; повышении температуры вплоть до красного каления могут наблюдаться различные явления, позволяющие сделать определенные выводы:
Наблюдения Возможно присутствуют
Не происходит никаких изменений Оксиды многих тяжелых металлов, оксиды и сульфаты щелочноземельных металлов, силикаты
Вздутие (вспучивание) вещества Образование возгонов Выделение газов Бораты, фосфаты, содержащие воду карбонаты щелочных металлов, квасцы См. табл. Д.7 См. табл. Д.8
Если при проведении гепариновой пробы (см. выше) обнаружена сера, то анализируемой вещество сплавляют затем с содой в калильной трубочке. Если при положительном эффекте гепариновой пробы проба в калильной трубочке дает отрицательный результат, это означает, что в исследуемом веществе присутствует только сульфатная' сера. В противном^ случае обнаруженная сера не является сульфатной.
Таблица Д.7. Возгоны, образующиеся при нагревании веществ в калильной трубке
Цвет Возгон
Белый Соли аммония, соли ртути, AS2O3, ЗЬгОз, А1С1з, Z11CI2, CdCh, MgClz, S11CI2, PbCk
Серый Hg, HgS, I2 (фиолетовые пары), As, Se
Желтый As2S3, S, Hgl2 (при потирании стеклянной палочкой красный), Pbl2, FeCl3
Красный Sb2S3, CrCla
44
Д. Химический анализ
Таблица Д.8. Газы, выделяющиеся при нагревании веществ в калильной трубке
Цвет Запах Другие свойства Газ Исходные вещества
Бесцветный Нет Воспламеняет тлеющую лучинуа О2 Пероксиды, хлораты, нитраты, HgO, MnO2, PbO2
Бесцветный Нет Вызывает помутнение баритовой воды со2 Карбонаты
Бесцветный Резкий Вызывает помутнение баритовой воды, обесцвечивает иодкрах-мальную бумагу so2 Сульфиды, сульфиты, тиосульфаты, сульфаты некоторых тяжелых металлов
Бесцветный Резкий Окрашивает в синий цвет красную лакмусовую бумагу, образует «туман» с НС! NH3 Соли аммония
Бесцветный Резкий Вызывает помутнение раствора AgNO3, образует «туман» с NH3 НС! Хлориды
Зеленовато-желтый Резкий Вызывает посинение крахмальной бумаги, пропитанной раствором KI Cl2 Хлориды + окислители, гипохлориты
Коричневый Удушливый Вызывает посинение крахмальной бумаги, пропитанной раствором KI Br2 Бромиды + окислители
Фиолетовый Удушливый Вызывает посинение крахмальной бумаги, пропитанной раствором KI I2 Йодиды + окислители
Коричневый Удушливый Вызывает посинение крахмальной бумаги, пропитанной раствором KI no2 Нитриты, нитраты тяжелых металлов
Бесцветный «Тухлых яиц» Вызывает почернение бумаги, пропитанной РЬ(СН3СОО)2 H2S Летучие сульфиды, тиосульфаты (с одновременным выделением SO2)
а Кислород обычно смешай с другими газами, в связи с чем эта малочувствительная проба часто дает отрицательный результат.
Нагревание с серной кислотой. Анализируемую пробу помещают в пробирку, добавляют к, ней примерно ,1 см3 2,5 М. H2SO4 и наблюдают за выделением газа (табл. Д.9). После прекращения выделения! газа пробирку! медленно нагревают на водяной бане. В лабораторном журнале отмечают раствори-
37. Качественный анализ
Таблица Д.9. Газы, выделяющиеся при нагревании веществ с 2,5 М H2SO4
Цвет Газ Исходное вещество Свойства
Бесцветный Бесцветный Коричневый Бесцветный Зеленовато-жел.тый со2 so2 no2 о2 С12 Карбонаты Сульфиты, тиосульфаты Нитриты Пероксиды Гипохлориты См. табл. Д.8
Бесцветный H2S Сульфиды Вызывает почернение бумаги, пропитанной РЬ(СН3СОО)2, имеет запах «тухлых яиц»
Бесцветный Бесцветный HCN н2 Цианиды Металлы Запах горького миндаля (осторожно, яд!)
мость вещества, так как1 из этого можно сделать выводы' о присутствии или отсутствии некоторых ионов. Пробирку с пробой не нужно выбрасывать, а следует использовать для проведения других реакций идентификации.
При нагревании с 18 М H2SO4 необходимо работать с полумикроколичествами веществ, иначе реакция протекает очень бурно, иногда даже со взрывом (выделение С1О2 из хлоратов, •Mn2O7 из перманганатов). Обязательно работать в защитных очках и под тягой! Кислоту добавляют по каплям с помощью специальной пипетки во избежание загрязнения содержимого склянки. При этом1 выделяются в основном, те же газы {табл. Д.10), что и при действии разбавленной серной кислоты. ,Поскольку конц. H2SO4 обладает обезвоживающей и Окислительной способностью, наблюдаются и другие явления.
Если получают прозрачный раствор, одну каплю его осторожно вносят в пробирку с водой. .Образование осадка указывает на присутствие щелочноземельных металлов! или свинца, так как сульфаты этих) элементов растворимы в конц. H2SO4, но при разбавлении снова выпадают в осадок (почему?)! Обращают внимание на изменение окраски серной кислоты; молибден дает голубое окрашивание, селен — зеленое, а теллур — красное.
1 Наряду с указанными) предварительными испытаниями можно провести также реакции предварительного обнаружения различных ионов. Ниже описаны важнейшие реакции, применяемые в полумикроанализе. Следует отметить, что трудно провести четкое различие между реакциями, используемыми Для предварительных испытаний, и реакциями идентификации.
Сплавление с металлическим натрием. Расплавленный, натрий применяют для проведения предварительных испытаний
46
Д. Химический анализ
Таблица Д.10. Газы, выделяющиеся при нагревании веществ с 18 М H2SO<
Цвет Г аз Исходное вещество Свойства
Бесцветный so2 Серная кислота в при- См. табл. Д.8
Бесцветный HF сутствии восстановителей (металлы, сульфиды, роданиды и т. д.) Фториды, гексафторсили- H2SO4 не смачивает стек-
Бесцветный Зеленовато- НС1 Cl2 каты Хлориды Хлориды+окислители ло; пузырьки газообразного HF образуют маслянистое вещество на стеклянных стенках пробирок (проба на травление стекла); пробирки необходимо постоянно менять } См. табл. Д.8
желтый Желтый сю2 Хлораты Резкий запах, взрывает-
Коричневый Фиолетовый Вг2 Ь Бромиды Йодиды ся с треском (осторожно!) См. табл. Д.8
Коричневый Красно-коричневый Фиолетовый no2 СгОгС12 Нитраты Хлориды-(-хроматы При смешивании с NO2
Мп2О? Перманганаты может взорваться (осторожно!)
на серу и серусодержащие анионы1. Анализируемую смесь на кончике? шпателя вносят в пробирку с кусочком натрия (очищенного от следов бензола и керосина) и осторожно нагревают до получения расплава {защитные очки!). В присутствии серы или серусодержащих анионов реакцию проводят до образования Na2S. После быстрого охлаждения смесь выщелачивают водой. На центрифугат действуют Na2[Fe(CN)5NO].
При сплавлении с натрием из фосфорсодержащих соединений выделяется также РНз, имеющий характерный запах ^осторожно, ядовит!). Проба не очень характерна. При содержании в пробе мышьяка выделяется AsH3.
Сплавление с натрием можно применять такжё как предварительную пробу на Ti, V, Мо и W. После подкисления центрифугата можно обнаружить Ti3+ по фиолетовому окрашиванию. У34* по зеленому окрашиванию, молибден — по образо
37. Качественный анализ*н
ванию молибденовой сини, вольфрам — по образованию вольфрамовой сини.
Окислительное сплавление (предварительные испытания на Мп и Сг). Небольшое количество анализируемой смеси и смеси безводных Na2CO3 и KNO3 ,(1 :4) сплавляют в углублении магнезиальной палочки. Зеленый или сине-зеленый цвет расплава указывает на присутствие марганца, желтый цвет—на присутствие хрома. (Какие соединения марганца и хрома ^обусловливают соответствующие окраски расплава?)
При растворении расплава в воде и подкислении НС1 получается фиолетовый раствор и выпадает коричневый осадок MnO2-aq (уравнение реакции?).
Предварительные испытания на As и Sb. К анализируемой пробе добавляют гранулы! цинка и 1 см3 2,5 М H2SC>4. Пробирку со смесью закрывают пробкой с просверленным отверстием, через которое вставлена маленькая воронка. В воронку помещают пропитанный раствором! ацетата свинца и хорошо выжатый ватный тампон. Воронку накрывают часовым стеклом, к нижней стороне которого приклеен кусочек фильтровальной бумаги, смоченный 0,5 М раствором AgNO3. Пробирку нагревают на водяной бане в течение примерно 5 мин и наблюдают за явлениями, происходящими на фильтровальной бумаге. Серое, черное или желтоватое пятно Ag3M-3AgNO3 указывает •на присутствие As или Sb. Образующийся сероводород улавливается ацетатом свинца (проба Гутцайта).
Проба на олово (возникновение свечения). К анализируемой смеси (~5 мг), находящейся: в фарфоровой чашечке, добавляют 12 М. НС1 и маленькую гранулу цинка. Через несколько минут в смесь окунают наполненную раствором KMnOd или водой макропробирку и затем вносят ее в бесцветное пламя горелки Бунзена. Светло-голубая люминесценция, появляющаяся на наружных стенках пробирки, указывает на присутствие олова. Свечение вызвано хлоридом олова (II). Следовые количества Sn(IV) восстанавливаются цинком. Проба очень чувствительна.
Предварительные испытания на Мо и W. Эти испытания необходимы для правильного выбора хода разделения катионов (в присутствии молибдена модифицируют операцию второго осаждения сероводородом (разд. 37.2.1.8, п. в), в присутствии вольфрама исключают водную вытяжку). Обнаружение молибдена и вольфрама проводят из содовой вытяжки, в которой они находятся (в виде ионов МоО42- и WO42-. Нерастворимые соединения вольфрама, входящие в состав минералов, и прокаленный WO3 переводят в вольфраматы сплавлением со Щелочами.
а) Проба на вольфрам с SnCl2. К двум каплям содовой вытяжки добавляют 5 к. смеси 1 М SnC12-|-12 М. НС1. В при
48
Д. Химический анализ
сутствии вольфрама образуется осадок вольфрамовой сини (см. также разд. 36.12.3).
б) Проба с KSCN и SnCl2 на вольфрам и молибден (см. также разд. 36.12.2). Каплю 5 М НС1 помещают на фильтровальную бумагу и добавляют к ней 1 к. подкисленной кодовой вытяжки. При этом образуется H2WO4, окрашенная в желтый цвет .(мешает ЗОЛ). Затем добавляют 1 М KSCN и 0,5 М. SnCl2 в 3 М. НС1. В присутствии вольфрама образуется синее окрашивание (вольфрамовая синь), а в присутствии молибдена— красное((Кз[А1о(5СК)б]. Окраска исчезает при действии 12 М НС1. Л
’в) Проба на молибден с етилксантогенатом калия. (На капельную пластинку помещают нейтрализованную почти до pH 7 содовую вытяжку, добавляют к ней крупинку' этилксан-тогената калия и несколько капель 5 М. НС!. В присутствии молибдена образуется комплекс, имеющий окраску от розовой до фиолетовой:
MoO3-2S = C
zsh
XOCaHs
Предварительные испытания на V и U (cм^ также разд. 36.13). Сернокислую вытяжку исходного вещества смешивают с 1 к. 2,5 М.1 Н2О2. В присутствии ванадия появляется краснокоричневое .окрашивание, вызванное образованием ионов пероксованадия (V) [VO2]3+. В присутствии избытка; Н2О2 образуются желтые трипероксованадат-ионы [VO2(O2)2]3~, что вызывает частичное обесцвечивание раствора. При добавлении к сернокислому анализируемому раствору 5 М раствора NaOH до сильно щелочной реакции образуется бесцветный ванадат, а в присутствии U(VI)—окрашенные в оранжевый цвет ди-пероксоуранат-ионы <[UO2(O2)2]2~. Реакцию получения пероксоураната лучше проводить при нагревании и в присутствии избытка Н2О2. Если при нагревании раствора на водяной бане в течение 5 мин не появляется желтого или оранжевого окрашивания, это означает, что уран отсутствует. Окрашенные в желтый цвет ионы СгО42~ мешают проведению реакции.
Предварительные испытания на бораты (см. разд. 35.10.2, опыт 8). Анализируемую пробу смешивают с СаЁ2 и KHSO4, смесь наносят на магнезиальную палочку и вносят палочку во внешний край пламени горелки Бунзена. Образующийся BF3 окрашивает пламя в .зеленый цвет.
Растирание с KHSO4. При растирании анализируемой пробы с KHSO4 в ступке (анализируемая проба -(-четырехкратное количество KHSO4+1 к. водьп) могут образоваться: СН3СООН из ацетатов, H2S из растворимых сульфидов, SO2 из сульфидов и тиосульфатов, HCN из цианидов и HSCN из роданидов.
37. Качественный анализ 4tf
Каждое из этих веществ имеет характерный запах. Из этого следует, что по запаху нельзя однозначно определить вещество, и, следовательно, значение этой пробью невелико. Проба на ацетат-ионы растиранием с KHSO4 является уже не предварительным испытанием, а реакцией -идентификации. Необходимо позаботиться' об удалении мешающих обнаружению ионов, что можно осуществить действием Ag2CrO4) или соответственно Ag2SO4, и КМпО4 (образование продуктов обмена и окисление).
37.2.1.3. Растворение пробы
Для качественного анализа независимо от способа его проведения пробу необходимо растворить. Для рационального .проведения анализа этот раствор должен обладать определенными свойствами. Желательно иметь разбавленный солянокислый раствор анализируемого вещества, не содержащий мешающих ходу анализа анионов. Присутствующие в пробе органические компоненты необходимо удалить экстракцией, прокаливанием или действием сильных окислителей, например минеральных кислот.
Для растворения вещества можно в основном предложить два способа) 1. Путем предварительных испытаний подбирают растворитель, в котором можно полностью растворить пробу. Затем применяют его для растворения вещества в ходе анализа. Таким образом, получают раствор, в котором проводят разделение ионов. 2. Применяя растворители селективного действия, цолучают несколько вытяжек, которые затем исследуют. При этом можно добиться количественного извлечения определенных ионов, а также в зависимости от условий избежать образования малорастворимого остатка. Например, если смесь MgSO4 и ВаСО3 растворять в 5 М НС1, то образуется малорастворимый BaSO4. Если же обработать смесь водой, то MgSO4 перейдет в1 раствор, после чего ВаСО3 можно растворить в 5 М HCI.
Селективными растворителями могут быть: Н2О, 5 М НСТ, 12 М НСЦ-5 М NaC103, ,5 М NaOH-]-l М KCN, 5 М NaOH, 2,5 М К2СО3. Все растворители применяют при нагревании.
Растворение в воде. Обработку анализируемой смеси водой проводят при перемешивании и нагревании на водяной бане. Растворимость вещества в воде проверяют после отделения не-растворившегося остатка центрифугированием, выпаривая каплю центрифугата на (предметном стекле. Если вещество растворилось хотя бы частично, водную вытяжку повторяют до полного его растворения.
В каждом случае определяют pH водной вытяжки. При РН^>7 вытяжку подкисляют 2,5 М HNO3. Следует обратить вни-4—1880
50 Д. Химический анализ
мание на возможное помутнение водной вытяжки при протека-1 нии реакций протолиза (например, А1(СН3СОО)3 или BiCl3-]-| Ч-Н2О). В этом случае подкисление 2,5 М. HNO3 дает прозрач-Ч ный раствор.
Нецелесообразно проводить водную вытяжку в присутствии вольфрама (см. разд. 37.2.1.2); в этом случае анализируемую пробу нужно обработать 2,5 М HNO3 (большая часть 'Вольфрама в виде WO3-H2O останется в нерастворившемся остатке).
Растворение в 5 М НС1. Остаток, полученный после водной вытяжки, обрабатывают при нагревании 5 М НС1, разбавляют водой и центрифугируют в горячем состоянии (РЬС12 только при нагревании хорошо растворяется в 5 М НС1).
При обработке анализируемой смеси 5 М НС1 может происходить выделение газов: С02 (из карбонатов), H2S (из сульфидов, разлагающихся при действии кислот), SO2 (из сульфатов, тиосульфатов), HCN (из цианидов). Могут образоваться также С12 (в присутствии ,МпО2, РЬО2 и других сильных окислителей) и Н2 (в присутствии металлов).
Растворение в смеси 12 М HCl-j-5 М NaC103. При проведении полумикроанализа для растворения анализируемого вещества вместо HNO3 или царской водки применяют смесь 12 М НС1 с 5 М NaC103. В этом случае отпадает необходимость длительного удаления NO3~ (нитрат-ионы мешают разделению катионов, почему?). Окислителем в данном случае является хлор в момент выделения. В дальнейшем при идентификации ионов .щелочных металлов следует учесть, что в раствор введены ионы Na+.
При действии1 указанной смеси в раствор прежде всего переходят растворимые в кислотах сульфиды .(окисляясь при атом в сульфаты). Соли Hg(I) также растворяются при окислении. Остаток, полученный после обработки анализируемого вещества 5 М НС1, обрабатывают смесью 12 М HC1-J-5 М МаС1О3 сначала на холоду, а затем' при (нагревании. По окончании реакции С12 удаляют продуванием и горячий раствор с остатком центрифугируют. В нерастворившемся остатке, промытом водой, могут находиться: сульфаты щелочноземельных металлов, и свинца, галогениды серебра, шпинели, силикаты, прокаленные оксиды, например SnO2, Sb2O4, А12О3, MgO, Ге2О3, Cr2O3, WO3, МоО3 (небольшое количество).
Растворение в смеси 5 М NaOH + 1 М KCN. Водную и солянокислую вытяжки, а также вытяжку, полученную при обработке вещества смесью 12 М НС1 + 5 М NaC103, можно объединить для дальнейшего проведения анализа. Вытяжку, полученную после обработки анализируемого вещества смесью NaOH+KCN, нужно исследовать отдельно (почему?). Остаток, полученный после растворения в смеси 12 М НС!+5 М NaC103, при нагревании обрабатывают смесью 5 М NaOH+ 1 М KCN. При этом в раствор переходят гало
3/. качественный ипилиа
гениды серебра в виде [Ag(CN)2]_, PbSO4 в виде [Pb(OH)i]2~, W03 и МоО3 в виде WO42~ или МоО42~ соответственно. После отделения серебра и свинца действием 1 М NaHS можно идентифицировать вольфрам и молибден.
Если анализируемая смесь все еще полностью не растворилась, остаток нужно перевести в растворимое состояние сплавлением.
При растворении анализируемой пробы, конечно, совершенно не обязательно использовать предложенную последовательность операций, здесь приведен лишь один из многих возможных вариантов. Исходя из предварительных испытаний, часто можно1 выбрать другие способы растворения пробы или упростить эту операцию. Например, в отсутствие серебра пробу можно растворить в 5 М NaOH. Сульфаты щелочноземельных металлов переводят в карбонаты кипячением остатка, полученного после обработки анализируемого вещества смесью NaOH + KCN, с 2,5 М К2СО3. В присутствии силикатов идентификацию ионов щелочноземельных металлов в остатке целесообразно проводить только после его сплавления с Na2CO3, так как образуются силикаты щелочноземельных металлов. Отрицательный результат пробы иа кальций, стронций и барий после переведения соединений в растворимое состояние действием 2,5 М КгСО3 также еще не указывает на отсутствие щелочноземельных металлов в нерастворившемся остатке.
37.2.1.4. Переведение веществ в растворимое состояние сплавлением
Вещества, нерастворимые в водных растворах кислот или щелочей, переводят в растворимое состояние сплавлением с плавнями. Для этого труднорастворимый остаток нужно хорошо высушить <и тонко измельчить в порошок. Затем его тщательно смешивают с соответствующим количеством плавня. Материал сосуда для сплавления не должен разрушаться под действием расплава. Если это произошло, примеси, 'перешедшие из него в раствор, не должны мешать идентификации, ионов.
Сплавление с карбонатами лучше всего проводить в платиновых сосудах. Перед их применением, обязательно следует ознакомиться с указаниями по работе с платиновыми сосуда-
Рис. Д.25. Платиновая чашечка.
'ми. Для' полумикроанализа целесообразно применять платиновые чашечки (рис. Д.25). Можно также использовать маленькие платиновые крышки для тиглей.
' Платиновые тигли непригодны для оплавления проб' с КОН. В (Этом случае используют маленькие серебряные или никелевые тигли или крышки для тиглей. При сплавлении веществ со щелочными плавнями в фарфоровом тигле нужно проводить холостой опыт, так как в этом случае невозможно избежать разрушения глазури.
' Сплавление с гидросульфатом калия можно проводить также в полумикропробирке из йенского стекла, так как плавле-
4*
52
Д. Химический анализ
ние в этом случае происходит при — 300 °C. В нерастворенном Остатке могут находиться: SiO2 и некоторые силикаты; А120з, Fe2O3, Cr2O3, TiO2, СеО2; шпинели, такие, как Fe3O4 и Cr2FeO4; SnO2, ,Sb2O4; SrSO4, BaSO4 и частично CaSO4.
Выбор плавня зависит от природы анализируемой пробы и от результатов предварительных испытаний. В полумикроанализе применяют следующий ход сплавления.
1. Сплавление с K2S2O7. При сплавлении с K2S2O7 в растворимое состояние переходят прокаленные оксиды, практически полностью толЫко TiO2 (см. разд. 36.10) и СеО2. Такие Соединения, как шпинели, А12О3 (природный корунд), Sb2O4, прокаленный при высокой температуре Сг2О3 растворяются 'обычно лишь частично. SnO2 не вступает в реакцию. Ti, Се, AU Fe,t Сг и Мп после сплавления переходят в растворимые Сульфаты.
Перед идентификацией этих элементов нужно удалить Sn и Sb, пропуская в сернокислый раствор H2S.
Для проведения сплавления с K2S2O7 предварительно высушенный остаток, полученный после испытаний пробы на растворимость, смешивают примерно с 150 мг измельченного в тончайший порошок K2S2O7 в платиновой чашке. Смесь нагревают на небольшом пламени горелки (расплав при этом должен оставаться прозрачным и не пузыриться). Затем расплав быстро охлаждают, опустив платиновую чашечку ненадолго в холодную воду. Полученный плав измельчают и обрабатывают минимальным количеством 18 М H2SO4 и воды. Перед центрифугированием раствор разбавляют 2,5 М H2SO4 до объема 3 см3.
2. Сплавление с Na2CO3 или со смесью ,NasCO3-]-KNO3. В остатке, полученном после сплавления пробы с пиросульфатом калия, могут находиться некоторые из упомянутых выше оксидов, не полностью перешедшие в раствор, и SnO2, а также сульфаты щелочноземельных металлов и силикаты.
В ходе сплавления с содой сухой остаток, предварительно отмытый от SOi1 2-, смешивают с восьми- или десятикратным количеством Na2CO3 и сплавляют смесь в платиновой чашечке. Затем плав обрабатывают водой и центрифугируют. При этом обходятся без обычного добавления К2СО3 (применяемого для снижения температуры плавления). Сплавление больших количеств в полумикроанализе успешно проводят при температуре пламени горелки Бунзена.
1 Для обнаружения щелочноземельных элементов в остатке, .полученном после сплавления с Na2CO3, в части центрифугата проводят пробу на содержание SO42~. Положительный рффект пробы указывает на присутствие сульфатов щелочноземельных металлов. (Отрицательный результат пробы на ,SO42~ не обязательно указывает на отсутствие щелочноземельных элементов в остатке.) В содовом расплаве кальций, содержащийся в
силикате, может также образовать карбонат.
37. Качественный анализ
53
Наряду со SnO2 и остатками других тугоплавких оксидов (Sb2O4, СГ2О3, СггРеО4) в остатке после сплавления с содой щелочноземельные металлы содержатся в, виде карбонатов, которые .можно* разделить после вымывания 5 М раствором НС1. Необходимо только подкислить центрифугат после отделения его от остатка (почему?).
В центрифугате нужно провести пробы на силикат-ионы, А13+, Fe3+ и Mg2+.
Перед идентификацией катионов силикаты необходимо отделить в виде нерастворимого S1O2; для этого к центрифугату добавляют 12 М НС1, раствор выпаривают досуха, и эти операции повторяют. После обработки 12 М НС1 и нагревания на водяной бане раствор с осадком центрифугируют. В растворе находятся катионы, в остатке SiOs.
В присутствии Сг2О3, Обнаруженного по зеленой окраске остатка пробы после сплавления с пиросульфатом калия, вместо сплавления с Na2CO3 лучше -применять сплавление со смесью Na2CO3+KNO3 (Na2CO3: KNO3 = 5 : 1). При этом образуется хромат. Аналогично можно перевести в растворимое состояние Cr2FeO4. Вместо KNO3 можно также использовать ’Na2O2 (осторожно!). Это окислительное сплавление проводят 'в никелевом тигле.
Остаток, полученный после сплавления с содой, также можно сплавлять со смесью Na2CO3+KNO3.
3. Сплавление со смесью Aa2CO3+S (плавень Фрайбергера). Для переведения в растворимое состояние SnOs и Sb2O4, их сплавляют с плавнем Фрайбергера. Для этого используют остаток, полученный после сплавления с содой или со смесью Na2CO3+KNO3.
Нерастворимый остаток помещают в фарфоровый тигель (почему не платиновый?) и смешивают с шести-семикратным количеством тщательно перемешанной смеси NaaCOs и серы (Na2CO3: S=1 : 1). Полученную смесь покрывают слоем смеси соды с серой. Нагревают смесь сначала при медленном повышении температуры, а через 15 мин включают сильный нагрев. После охлаждения содержимое тигля обрабатывают холодной водой.
В растворе находятся тиостаннат и тиоантимонат, при подкислении в осадок выпадают сульфиды.
4. Сплавление с HF применяют для обнаружения щелочных металлов в силикатах, что невозможно при сплавлении с содой. (Присутствие больших количеств натрия затрудняет или делает практически невозможным обнаружение калия и лития.)
Для сплавления с HF берут небольшое количество исходного вещества. Сплавление нужно проводить очень осторожно, работать только в защитных очках и в резиновых перчатках!
54
Д. Химический анализ
Пробу смешивают с 5 к. 40 %-ной HF и 5 к. 18 М H2SO4 (в платиновом тигле; не применять стеклянную посуду!). Затем смесь осторожно нагревают в платиновой чашечке на небольшом пламени горелки. После охлаждения и повторного добавления 1 к. HF эту операцию повторяют. Смесь нагревают затем до образования дыма (весь кремний должен улетучиться в виде SiF* или соответственно H2SiF6) и разбавляют.
В растворе можно идентифицировать ионы щелочных металлов.
37.2.1.5. Групповые реакции на анионы
С помощью групповых реакций' получают довольно точные сведения о присутствии или отсутствии многих ионов. При этом обычно применяют реакции осаждения или окисления-восстановления, характерные для ионов определенной группы. Групповые реакции имеют особенно большое значение в анализе анионов, поскольку практически не существует систематического хода разделения анионов.
Описанные в этом .разделе групповые реакции проводят в пробирках с 3—5 каплями предварительно подкисленной содовой вытяжки. Можно также использовать для проведения реакций и другие вытяжки, содержащие анионы, вводя необходимые изменения в ход анализа. Здесь следует еще раз подчеркнуть, что ход анализа, разделения, идентификации и т. д. не является чем-то раз и навсегда установленным, возможны другие варианты проведения анализа. При отрицательном эффекте при действии группового реактива можно надежно говорить об отсутствии в растворе соответствующих анионов, в то время как положительный эффект свидетельствует лишь о возможности присутствия данного аниона. Окончательное обнаружение ионов всегда проводят с помощью реакций идентификации.
Катионы, мешающие обнаружению анионов и проведению групповых реакций на них, должны быть отделены1. Эту операцию осуществляют, применяя содовую вытяжку. Примерно 50—100 мг анализируемой смеси, Предварительно очищенной от (Элементарной серы экстракцией ее сероуглеродом, смешивают с 5 см3 1 М ЫагСОз.
Раствор соды для проведения содовой вытяжки получают, растворяя NaaCOa (ч.д.а.), степень чистоты которого проверена. Полученный раствор нагревают в течение 15 мин на водяной бане, после чего центрифугируют горячим. Остаток промывают небольшим количеством горячей воды или горячего раствора соды, промывные воды объединяют с центрифугатом.
В растворе содовой вытяжки можно проводить обнаружение анионов, при этом основная часть мешающих обнаружению катионов находится в нерастворившемся остатке bi виде карбонатов или гидроксидов. Этот остаток часто исследуют на
Схема Д.1. Групповые реакции на анионы
56
Д. Химический анализ
'присутствие катионов щелочноземельных металлов и некоторых! 'анионов. |
В содовой вытяжке идентифицируют также катионы, нахо-’ дящиеся в ней в виде анионов (типа1 CrO42 \ MnO4~, AsO43~,: .[А1(ОН)4]~ [Sn(OH)6]2-). j
На схеме Д.1 представлен ход обнаружения анионов прй1 действии групповых реактивов. ,i
37.2.1.6. Обнаружение анионов
Не существует систематического метода разделения анионов, попытки его создания до сих пор были малоуспешными. Анионы можно обнаруживать либо из исходного вещества, либо из содовой вытяжки.
Обнаружение анионов из исходного вещества
На схеме Д.2 представлен? ход анализа смеси анионов NO3-, СО32-, SiO42-, ,ВО33~, СН3СОО“ и CN~ непосредственно из исходного вещества.
Примечания: а Пробу на травление стекла нужно проводить в пробирке, не бывшей в употреблении.
6 Проба однозначно указывает только на отсутствие нитратов и не очень чувствительна.
в Обнаружению карбонат-ионов мешают CN~, F~, SO32-, S2O32~. В присутствии CN~ нельзя проводить обнаружение небольших количеств СО2 (почему?). Добавлением HgCl2 связывают цианид в недиссоцнированный Hg(CN)2. Фториды маскируют раствором ZrOCl2; SO32~ и S2O32- окисляют пероксидом водорода.
г При обнаружении силикат-ионов в присутствии боратов в результате гидролиза BF3 на влажной бумаге образуется белое пятно Н3ВО3. В отличие от SiO2-aq Н3ВО3 растворима в горячей воде. Ни в коем случае нельзя применять избыток CaF2, так как при этом образуется устойчивая H2SiF6.
д Реакция непригодна для боросиликатов.
е Обнаружение ацетатов из исходного вещества проводят реакцией с о-нитробензальдегидом (ср. разд. 37.1.4): смесь анализируемой пробы с небольшим количеством СаСО3 нагревают в калильной трубочке. При этом образуется ацетон, который в щелочной среде реагирует с о-нитробензальдеги-дом с образованием индиго (синтез индиго по Байеру). При этом калильную трубочку покрывают маленькой полоской фильтровальной бумаги, смоченной раствором о-нитробензальдепида в 2 М NaOH. При добавлении капли 2 М НС1 возникающая вначале зеленая окраска переходит в чисто синюю.
ж Выпавший осадок AgCN после отделения центрифугированием и растворения в 14,5 М HNO3 идентифицируют микрокристаллоскопическим методом. Раствор, полностью освобожденный от HCN, помещают в установку для получения газов и используют для обнаружения галогенидов и ионов SCN-. В присутствии Hg2+ образуется очень слабо диссоциированный Hg(CN)2> мешающий обнаружению цианидов. Применяя большой избыток С1~ (насыщение раствора хлоридом натрия), можно получить существенное увеличение концентрации CN-, как это следует из уравнения реакции:
Hg(CN)3 4- 4С1- ^==t [HgCl4p~ + 2CN-
Схема Д.2. Идентификация анионов из исходного вещества
58
Д, Химический анализ
Обнаружение и идентификация анионов из содовой вытяжки
I. CN-, SCN-, F", С1-, Вг- I-.
Цианид-ион обнаруживают по реакции образования Fe(SCN)3 или берлинской лазури. В первом случае к пробе содовой вытяжки добавляют так называемый желтый сульфид аммония, досуха упаривают пробу при слабом нагревании, смачивают 5 М НС1, снова нагревают (для удаления H2S) и смешивают с 0,5 М FeCl3. В присутствии ионов CN~ образуется красный Fe(SCN)3 (см. также 36.15.1). Эту реакцию нужно проводить только в отсутствие SCN-.
Во втором случае смесь пробы содовой вытяжки со свежеприготовленным раствором FeSO4 выпаривают почти досуха и смешивают с 5 М НС1 и 0,5 М FeCls. Образование зеленого окрашивания или осадка берлинской лазури указывает на присутствие CN~ (ср. разд. 36.15.3).
Для обнаружения роданид-иона пробу содовой вытяжки подкисляют 5 М НС1 и смешивают с избытком 0,5 М FeCl3. Красное окрашивание Fe(SCN)3 свидетельствует о присутствии SCN~.
Следует помнить, что ион SCN- может образоваться из CNJ и S2~ или S2O32 в процессе получения содовой вытяжки. Перед идентификацией SCN- необходимо разрушить NO2~, добавляя насыщенный раствор NH2SO3H.
Фторид-иой обнаруживают следующим способом. Пробу содовой вытяжки подкисляют 5 М СН3СООН и смешивают с 2 М Са(СН3СОО)2. Нагревают 5 ' мин на водяной бане. Осадок CaF2 отделяют центрифугированием. С осадком1 CaF2 можно проделать следующие реакции: а) травление стекла;' б) обесцвечивание ализаринциркониевого лака.
Красный ализаринциркониевый лак образуется при добавлении 1 к. 0,5 М ZrOCl2 и 2 к. 1%-ного раствора ализарина S к 10 к. 5 М НС1. При добавлении по каплям полученного солянокислого раствора к осадку CaFa образуется бесцветный очень устойчивый комплекс [ZrFe]2-. (Наряду с ним образуются и другие комплексы, например [ZrF5(OH)]2~, или npoj дукты конденсации.) Появление желтой окраски, свойственной ализарину S, обусловлено присутствием F~. 1
II. Разделение и обнаружение хлоридов, бромидов и иоди| дов. I
Обнаружение хлорид-, бромид- и иодид-ионов можно про! водить как в растворе, не содержащем CN-, оставшемся а приборе для обнаружения газов, так и, в отсутствие CN-] в центрифугате после осаждения CaF2. Ионы CN~ можно уда] лить, просасывая через раствор воздух с помощью резиновой груши (осторожно HCN1). ’
Схема Д.З*. Разделение и обнаружение Cl", Br , J
Na*, NO3, HjO'
'AgCl, AgBr, Agl>
Центрифугирование, промывание, двухкратная обработка 13 М NHijOH, центрифугирование
___________"___________
Нагревание с Zn+2,5M H,SOi до прекращения выделения н2, центрифугированы е
Подкисление 2,5М H2SO4. центрифугирование
Слабое нагревание со свежеприготовленным насьиц. кони, раствором (МЩ)2СО3, центрифугирование
Цобакпение 0,2 М КВг Выпадает желтоватый осадок AgBr (в присутствии СГ)
______у_______
Восстановление Zn*Z,5M Н250^, центрифугирование. Центрифугат смеиш-вссют с добавляют по каплям хлорную воду; в присутствии or* коричневая окраска
4 s'
_________У_________
Смешивание с CClq, добавление 2-5к. 5М КМ02 рли хлорной воды6, встряхивание; в присутствии I ~ слой CClif окрашивается в фиолетовый цвет
I"
Здесь и далее заштрихованные части относятся к осадкам. — Прим. ред.
60
Д. Химический анализ
На схеме ,Д.З приведен ход разделения и идентификации С1~, Вг~ и В в отсутствие ,S2-, S2O32- и SCN-.
Примечания. а В присутствии S2-, S2O32-, SON- образуется черный осадок Ag2S. Осадок затем кипятят с 14,5 М HNO3 до полного окисления Ag2S. При этом также растворяется основная часть AgSCN. Оставшиеся галогениды серебра отделяют центрифугированием и промывают осадок водой. AgCl и AgBr можно затем извлечь раствором аммиака.
Для обнаружения хлорид- и бромид-ионов аммиачный раствор подкисляют 2,5 М H2SO4. Выпадающий в присутствии роданидов осадок AgSCN необходимо разрушить. Для этого осадок напревают в фарфоровом тигле до темно-красного каления, причем AgSCN количественно разлагается с образованием Ag2S, S, CS2, (CN)2 и Na. К темному осадку в тигле добавляют кусочек цинка и 2,5 М H2SO4. Из этого раствора снова можно осадить С1~ и Вг- действием 0,5 М AgNO3.
6 Важно добавлять хлорную воду по каплям, так как избыток С12 приводит к образованию 1С13 и окислению 12 в 1О3-. В обоих случаях происходит обесцвечивание раствора. При правильном проведении реакции иодид обнаруживают по выделению иода и фиолетовому окрашиванию слоя СС14. Коричневое окрашивание слоя СС14 вызвано образованием брома и указывает на неполноту извлечения галогенидов серебра раствором аммиака.
Особые случаи обнаружения С1~, Вг-, I- и SCN-:
в Оксигалогениды (например, BiOI) после содовой вытяжки могут находиться в остатке. Поэтому их обнаруживают в растворе, полученном при обработке остатка или анализируемой смеси кислотой (2,5 М H2SO4).
г В присутствии Ag+ в остатке содовой вытяжки находятся AgCl, AgBr, Agl и AgSCN. Остаток тщательно обрабатывают 13 М NH4OH. Раствор, в который переходят С1~, Вг- и /SCN~, отделяют центрифугированием, подкисляют центрифугат 2,5 М HNO3 и дальше поступают, как описано выше.
В остатке, полученном после обработки аммиаком, содержится Agl. Нагреванием с 18 М H2SO4 окисляют I- в 12. При проведении этой реакции в установке для идентификации газов можно обнаружить иод по фиолетовой окраске слоя СС14.
III. Обнаружение S2-, S2O32-, SO42-, SO32-
Из отдельной пробы содовой вытяжки проводят идентификацию сульфидов, сульфитов, сульфатов и тиосульфатов. Предлагаемый ход разделения и обнаружения этих ионов представлен на схеме Д.4.
Примечания. а Обнаружение тиосульфат-нонов см. в разд, 35.6.4.2.
6 Обнаружение сульфидов в присутствии Ag+ и Hg2+. Ag2S и HgS нерастворимы в кислотах, не являющихся окислител'ями, поэтому остаются в остатке содовой вытяжки. Обнаружение можно провести в установке для идентификации газов при обработке остатка 12 М НС1 и добавлении металлического цинка.
Б Сульфаты щелочноземельных металлов (особенно BaSO4 и SrSO4) могут оставаться в остатке содовой вытяжки. В этом случае сплавляют остаток с содой (см. разд. 37.2.1.4).
IV. Обнаружение NO2~, NO3-
В то время как нитрит-ионы можно однозначно обнаружить в содовой вытяжке, нитраты в виде оксонитратов (например, BiONO3) находятся в остатке, поэтому отрицательная реакция
Схема Д. 4. Разделение и обнаружение S2 , SO32 , SO42 и s2o32-
SOj', 50|',
Добавление 5М НС1
Нейтрализация 5М ОН3СООН по нейтральному красному, нагревание, снова нейтрализация, добавление Зк.
5М ЧНфН+насыщ. раствор
Sr(W03)2, нагревание, центрифугирование
Добавление Юк. Н20 и
7 к. HCL, центрифугирование
обработка. 5М НС1. центрифугирование
Обнаружение S2 То запаху Нг5 или по почернению бумаги, омоченной ₽Ь(СН3С00)2
а)Смеишвание осадка elM НС1, я?-| давление 0,5М ВаС12; выпадающий осадок BctSOi, (помутнение) указывает на присутствие SC4; б'растворение остатка в 2М Na2C0. обнаружение дейсгт ^'^вием BtiClz ж'
Нейтрализация 1,1М NaDH, добавление по каплям 0,0051 раствора милахи -елового зеленого-, обесцвечивание ЗО-ЗОка-пель -указывает на присутствие 503
_____________/
а'Добавление Юк 5М НС1; выделение
S и запах 302 указывает на присутствие 5г03’; б) добавление 2,5:w HN03 ♦ 0,5№ AgNOy, изменение цвета бсадка указывает на пршупн ствие SzOf J
62
Д. Химический анализ
на нитрат-ионы из содовой вытяжки еще не доказывает их отсутствия. В этом! случае обнаружение нитрат-ионов проводят в растворе, полученном при обработке на холоду исходного вещества или остатка содовой вытяжки 2,5 М раствором H2SO4. Обнаружению нитрит- и нитрат-ионов, по реакции с FeSO4 мешают Вг- I-, [Fe(CN)6]3-, [Fe(CN)6]4-, SON-, S2-, S2O32-, SO32- и СГ2О72-, которые удаляют в виде малорастворимых солей серебра. Для этого раствор нейтрализуют 5 М. СН3СООН, прибавляют ’к нему содовую вытяжку, смешанную с одной каплей 2 М. ЫагСОз,, и встряхивают с твердым Ag2CO3 или смешивают с насыщенным раствором Ag2SO4. Осадок отделяют центрифугированием и для осаждения SO32- к центрифуга-ту добавляют 0,5 М. ВаС12. После повторного центрифугирования проводят реакции обнаружения.
Нитрит-ион
а) Реакция с FeSO4 (уравнение реакции см. разд. 35.7.3.1, опыт 21). На капельной пластинке смешивают 1 к. раствора пробы с 1 к. 2,5 М H2SO4. При внесении кристаллика FeSO4X Х7Н2О, промытого 2,5 М H2SO4, в присутствии ионов NO2~ образуется [Fe(NO) (НгО)5]2+ темно-коричневого цвета.
б) Обнаружение по Лунге. Эту очень чувствительную и специфичную реакцию на нитриты проводят на капельной пластинке. Каплю анализируемой пробы, подкисленной 5 М СН3СООН, смешивают с 1 к. 1%-ного раствора сульфаниловой кислоты и 1 к. 0,3% раствора 1-нафтиламина. В результате диазотирования и азосочетания образуется красный азо^ краситель (подробнее см. учебники по органической химиии
в) Реакция с тиомочевиной: I
SC(NH2)2 + HNO2 ->- N2 + Н2О + Н3О+ + SCN- I
В пробу содовой вытяжки, подкисленную 5 М СН3СООМ вносят крупинку тиомочевины. После кратковременного наВ гревания добавляют 1 к. 0,5 М FeCl3. Возникновение красногВ окрашивания Fe(SGN)3 указывает на присутствие NO2-.. НитВ раты не мешают определению. I
Нитрат-ион В
Надежное обнаружение нитрат-ионов возможно только В отсутствие нитрит-ионов, так как для обоих ионов характерВ ны одни и те же реакции. На большей по сравнению с нитратгВ ми реакционной способности нитритов основан ряд методов и| удаления. В качественном анализе используют разложение, нитритов действием амидосерной кислоты (HSO3—NH2) и азида натрия (NaN3).
а) Реакция с FeSO4. Эта реакция, так же как и в случае нитритов, основана на образовании (Fe(NO) (Н2О)5]2+. В от
37. Качественный анализ
63
личие от HNO2 HNO3 окисляет Fe2+ в Fe3+ только при высокой концентрации Н3О+:
4Н3О+ + NO3" + 3Fe2+-► NO + 3Fe3+ + 6H2O
Для разложения нитритов целесообразно применять амидосер-ную кислоту. При использовании NaN3 в присутствии его избытка может образоваться окрашенный в красный1 цвет азид железа, окраска которого мешает обнаружению NO3_.
Раствор анализируемой пробы вносят по каплям в свежеприготовленный насыщенный на холоду раствор амидосерной кислоты (почему не наоборот?). Через небольшой промежуток времени раствор слабо нагревают на водяной бане до прекращения! выделения N2. После охлаждения несколько капель раствора, не содержащего NO2_, в пробирке смешивают с примерено равным объемом, насыщенного на холоду раствора/ FeSO4 (свежеприготовленного) и добавляют 1 к. 2,5 М H2SO4. Раствор затем осторожно перемешивают с 18 М H2SO4. На гра-'нице слоев в присутствии нитрат-ионов образуется фиолетовое или коричневое (при больших количествах NO3_) кольцо.
При проведении этой реакции на капельной пластинке с твердым сульфатом железа(II) на кристалле FeSO4 и в месте соприкосновения раствора с H2SO4 образуется [Fe(NO) (Н2О)б]2+, окрашенный в коричневый цвет.
б) Реакция восстановления до нитритов с последующим обнаружением по Лунге. Перед проведением этой реакции рекомендуют разложить нитрит действием NaN3, тщательно удалив избыток азида. Амидосерную кислоту в данном методе не применяют, так как избыток ее трудно удалить, а оставшаяся кислота может в дальнейшем разрушить нитрит, образующийся из нитрата.
1 К раствору пробы добавляют немного твердого NaN3, подкисляют 5 М. СН3СООН и нагревают на водяной бане до прекращения выделения азота. В конце раствор упаривают на открытом пламени горелки до половины его объема. (Осторожно! Работать под тягой в защитных очках, так как выделяющийся 'HN3 сильно ядовит.) Пробу, из которой удален NO2-^ .смешивают с несколькими кусочками цинка (предварительно •промытого 5 М СН3СООН) для восстановления NO3_ в NO2~ и с 5 М СН3СООН. Обнаружение по Лунге проводят, как описано выше.
V. Обнаружение РО43-, С2О42-
Оба иона идентифицируют из отдельных проб содовой вы-Дяжки.
Фосфат-ион
Отрицательный результат пробы на фосфат-ионы в содовой вытяжке однозначно доказывает их отсутствие. Обнаружению
64
Д. Химический анализ
фосфат-ионов из содовой вытяжки мешают арсенат-ионы. НаИ дежное обнаружение фосфат-ионов можно провести только посИ ле количественного отделения элементов группы сероводороде в ходе разделения катионов. Н
а) Обнаружение в виде фосфоромолибдата (см. такжЛ разд. 36.Г2.2). 0,5 М раствор молибдата аммония смешиваю» с равным объемом 14,5 М HNO3, достаточным для растворение выпадающей в осадок молибденовой кислоты. К полученном» раствору добавляют несколько капель содовой вытяжки, подИ кисленной 2,5 М. HNO3 (или содержащий анионы раствор, поИ лученный в результате ионного обмена, или азотнокислый расИ твор, полученный после обработки исходного вещества азотное кислотой). Образующийся примерно через 3 мин на хоМ лоду желтый осадок додекамолибдатофосфата аммония! ,(NH4)3[P(Mo12O40]-6Н2О указывает на присутствие фосфат-ио-1 НОВ. I
Неоднократно упоминавшееся отличие фосфатов от арсенатов, заключаю-1 щееся в том, что арсенатомолибдат в этих условиях дает желтое окрашива-J ние раствора и выпадает в осадок только при кипячении раствора, очени ненадежно. И
Растворимые силикаты также могут мешать обнаружению фосфатов (желтое окрашивание раствора), но они не образуют малорастворимые осадИ ки кремнемолибдатов. Большие количества фторидов и хлоридов могут меИ шать определению (снижение чувствительности обнаружения), образуя фтоИ ро- и хлорокомплексы молибдена. Ч|
б) Реакция образования MgNH4P04-6H2O (см. также! разд. 35.7.3.3). |
Чтобы обнаружить, содержится ли в осадке наряду с фос-1 форомолибдатом арсеномолибдат, осадок растворяют в 5 М ] NH4OH и смешивают со свежеприготовленным раствором маг- 1 незиальной смеси. При этом выпадает белый осадок | MgNH4PO4-6H2O или соответственно MgNH4AsO4- 6Н2О. После! его промывания проводят обнаружение фосфат- и арсенат-ио- 1 нов действием 0,5 М AgNO3. Образуется желтый Ag3PO4 или 1 шоколадно-коричневый Ag3AsO4. |
Оксалат-ион )
Оксалат-ионы мешают при разделении катионов, поэтому | наряду с фосфат- и борат-ионами их нужно предварительно ,| удалить с помощью* ионного обмена. Для разрушения оксала- | тов рекомендуют быстрый метод окислительного разложения 1 оксалата действием HNO3, Н2О2 или NaC103. ]
Реакцию обнаружения оксалат-ионов в содовой вытяжке J проводят следующим образом: пробу, подкисленную 5 М| <СН3СООН, смешивают с 0,05 М К1з Д° получения желтой i окраски раствора, указывающей на присутствие избытка Ъч '(окисление SO32~ в SO42~). Затем осаждают оксалат дейст-1
37. Качественный анализ
65
вием 2 М Са(СН3СОО)2. После тщательного промывания осадок растворяют в 18 М H2SO4 и раствор переносят в установку для идентификации газов (в сосуд для получения газа). <В вертикальной трубке установки находится 0,02 М КМпО4, в приемнике — насыщенный раствор Ва(ОН)2. В сосуд для получения газа по каплям добавляют раствор КМпО4. В присутствии щавелевой кислоты КМпО4 обесцвечивается, а из С2О42~ образуется СО2: Прй окислении всего количества С2О42- (что определяют по прекращению обесцвечивания КМпО4) СО2 переводят в приемник потоком воздуха (очищенного от СО2 путем пропускания! через трубку с натронной известью). В приемнике образуется белый осадок ВаСОз, указывающий на присутствие оксалат-ионов в пробе.
VI. Обнаружение [Fe(CN)6]4- и [Fe(CN)6]3-
Присутствие гексацианоферратов значительно усложняет ход анализа. Такие малорастворимые соли, как Cu2[Fe(CN)6], при обычных условиях почти не вступают в реакцию обмена с 2 М Na2CO3 и поэтому остаются в остатке содовой вытяжки. Для обнаружения гексацианоферрат-ионов часть анализируемой смеси, предназначенную для обнаружения анионов, выдерживают -~5 мин с 5 М NaOH, добавляют равное количество 2 М Na2CO3 и повторно нагревают в течение 5 мин на водяной бане. После отделения остатка центрифугированием раствор снова нагревают с 2 М Na2CO3 на водяной бане. Объединенные центрифугаты используют для обнаружения анионов.
Г ексацианоферрат( 11 )-ион
Каплю содовой вытяжки подкисляют 5 М НС1 и смешивают с 1 к. 0,5 М FeCl3. В присутствии гексацианоферрата(II) образуется берлинская лазурь NaFe[Fe(CN)e] или Fe4[Fe(CN)6]3- В присутствии гексацианоферрата(III) появляется коричневое окрашивание раствора.
Гексацианоферрат( 111 )-ион
Каплю содовой вытяжки подкисляют 5 М НС1 и добавляют к ней кристалл соли Fe2+ (соли Мора), не содержащей Fe3+. Темно-синее окрашивание (берлинская лазурь) указывает на присутствие гексацианоферрата (III). Происходит окисление Fe2+ в Fe3+ и восстановление [Fe(CN)6]3- в [Fe(CN)6]4-. Гек-сацианоферрат(П) образует при этом беловатый или светло-голубой осадок, который на воздухе быстро становится синим, но он не мешает обнаружению [Fe(CN)6]3- (см. разд. 36.15.3).
Устранение мешающего влияния гексацианоферратов при проведении реакций обнаружения анионов и катионов. При обнаружении галогенидов осаждают мешающие гексацианоферраты из нейтрализованной содовой вытяжки, 5—1880
66
Д. Химический анализ
из которой удален СО2 действием 0,5 М Cd(CH3COO)2. В центрнфугате проводят разделение и обнаружение анионов обычным способом.
При обнаружении NO2~ и NO3~ по реакции с Fe2+ гексацианоферраты осаждают из слабосернокислого раствора действием насыщенного раствора
Обнаружение цианидов в присутствии гексацнаноферратов проводят, как указано на схеме Д.2. В этих условиях гексацианоферраты не мешают их обнаружению.
Для разделения н обнаружения катионов в присутствии гексацианоферратов анализируемую смесь выпаривают досуха в маленькой свинцовой чашке с 18 М H2SO4 при добавлении кристаллика (NH4)2S2O8. Следует учесть, что при этом вводится свинец, который затем можно обнаружить.
Если в предварительных испытаниях были обнаружены ионы ртути и мышьяка, в растворе, полученном после обработки анализируемой смеси растворами 12 М НС1 и 5 М INaC103, нужно идентифицировать эти элементы.
Если присутствуют гексацианоферраты, то нельзя проводить определение ионов железа в ходе систематического анализа катионов, так как находящиеся в пробе гексацианоферраты разлагаются при действии кислот. Поэтому определение железа проводят в остатке, полученном после нагревания анализируемой пробы в течение 5 мин с 2 М Na2CO3 на водяной бане. При этом железо, не связанное в комплекс, осаждается в виде гидроксида.
VII. Обнаружение пероксидов
Пероксиды лучше всего идентифицировать в растворе, полученном при обработке анализируемого вещества 5 М СН3СООН на холоду, так как в процессе содовой вытяжки пероксиды обычно разрушаются. Уксуснокислую вытяжку смешивают с 2,5 М H2SO4 и 0,05 М TiOSO4. В присутствии О22- образуются катионы пероксотитанила [TiO2]2+, окрашивающие раствор в оранжевый цвет.
Фториды мешают проведению этой реакции (почему?).
37.2.1.7. Групповые реакции на катионы
Групповые реакции имеют большое значение в анализе катионов, так как с их Помощью можно сделать важные выводы о составе анализируемой смеси. Известно большое число групповых реактивов! на катионы; кроме того, комбинируя групповые реактивы, меняя последовательность их применения, pH, температуру и другие условия реакций, можно получить практически полную информацию* о присутствии различных ионов и 'благодаря этому наметить четкий ход последующего разделения катионов.
Групповые реакции можно проводить в растворах, полученных объединением* различных вытяжек. Но, конечно, большие преимущества дает исследование отдельных вытяжек. Групповые реактивы можно добавлять последовательно к анализируемой смеси (т. е. отделить осадок, полученный при действии одного группового реактива, а на центрифугат подействовать следующим групповым' реактивом)* или к отдельным порциям анализируемой смеси. Поскольку существует много различных
37. Качественный анализ
67
вариантов анализа смеси катионов и выбор конкретного хода анализа проводит сам экспериментатор, нельзя дать общую, пригодную во всех случаях схему анализа. При применении групповых реактивов нужно помнить о том, что ионы, мешающие друг другу при проведении реакций, находятся в одном растворе. Одна из возможных схем разделения катионов действием групповых реактивов приведена ниже (схема Д.5).
Схема Д.5. Групповые реакции на катионы
37.2.1.8. Разделение и обнаружение катионов
Приведенные схемы систематического хода разделения катионов в основном соответствуют классическому анализу по Фрезениусу. При этом катионы металлов1 делят на следующие 'группы:
5*
68
Д. Химический анализ
1. Группа НС1: Ag+, Hg(I) (в виде Hg22+) и частично РЬ2+, Т1+, W(VI).
2. Группа H2S: а) подгруппа олова: Hg(II) (в виде Hg2+). Mo(VI), As(III.V), Sb (III, V), Sn (II, IV); б) подгруппа меди: Pb2+, Bi3+ T1+ TP+, Cd2+, Cu2+.
3. Группа NH3: Ce3+, Cr3+, V3+, Al3+, U(VI).
4. Группа (NH4)2S: Mn2+, Fe2+, Zn2+, Ni2+, Co2+, Fe3+.
5. Группа (NH4)zCO3: Ba2+, Sr2+, Ca2+.
6. Группа щелочных металлов, аммония и магния: Li+, Na+, к+, nh4+, Mg2+
Поскольку в специальной литературе достаточно подробно описан ход разделения и обнаружения катионов, мы ограничимся кратким1 рассмотрением основных операций разделения катионов.
В разд. 37.2.2 приведены другие возможные варианты систематического разделения катионов.
Группа НС1. На схеме Д.6, представлен ход разделения катионов этой группы:
Группа H2S. К группе H2S относятся ионы, осаждаемые сероводородом в кислом растворе. Ниже будут рассмотрены малорастворимые сульфиды в следующей последовательности: Bi2S3, CuS, PbS, CdS (здесь же TH), As2S3, Sb2S3, SnS2, MoS3, HgS. Названные сульфиды отличаются друг от друга различной способностью к образованию' тиосолей. Сульфиды второй подгруппы, так называемой подгруппы олова, растворимы в смеси NH4OH+H2S (HgS растворим только1 BNaOH-(-+NaHS), что используют для отделения сульфидов этой подгруппы от сульфидов так называемой подгруппы! меди:
As2S3 + 3S2- ► 2AsS33-
Осаждение сульфидов требует проведения некоторых подготовительных операций: окисления Sn(II), восстановления As(V) и Sb(V), создания- нужного pH раствора, установления присутствия Мо.
а) Окисление Sn(II) в Sn(IV). SnS нерастворим в NH4OH + +H2S, только SnS2 образует тиоанионы (тиостаннат). Поэтому перед осаждением сероводородом нужно окислить Sn (II) в Sn(IV). Окисление проводят бромной водой.
б) Восстановление As(IZ) и Sb(V) в As(III) и Sb(III). При обработке бромной водой не только Sn (II) окисляется в Sn(IV), но и образуются соединения мышьяка и сурьмы высших степеней окисления. Поскольку As(V) и Sb(V) окисляют сероводород в основном до элементарной серы, которую затем нужно отделять от выпавших в осадок сульфидов, As(V) и Sb(V) переводят в As(III) и Sb (III) действием HI. При этом в осадок могут выпасть мадорастворимые иодиды (РЫ2, BiOI
37. Качественный анализ
69
Схема Д.6. Разделение и обнаружение 1 элементов группы НС1
/Годная и азотнокислая вытяжка ( анализируемого вешества (Ад, На,Р \T\,Vi*элементы других групп}
t
Обработка. SM НС1, нагревание, центрифугирование
Другие группы
z^g'cl, 'РЬС12, Tin,.
Двухкратная обработка горячей водой, центрифугирование
igCl, Hg2Clzz
РЬ^+ TV** ) >-
' /Для проведения соляно-----------''кислой, вытяжки и
।вытяжки 1ZM HCl*5MNaCl0j
Обработка 5М ЫЩОН, ц енгприфугирование
Нагревание сбМ НС1 и NtiClOj, разбавление, центрифугирование
Z/ZZZ/,
jCl, WOj • ас/z
Z////A
I
Образовавшийся желтый осадок Agl указывает на присутствие серебра
Добавляют $пС12+ з5М НС1, голубое окрашивание вольфрамовой сини
Выделение пузырьков С1г, добавление SnCl2, образование белого осадка (который при дальнейшем добавлении 5пС1г ста -ловится черным)указывает на присутствие Нд, или:медная проволока становится блестящей от образовавшейся
и др.), которые при пропускании H2S переходят в еще менее растворимые сульфиды. Исключение составляет таллий: ТП не переходит в малорастворимый сульфид и в таком виде остается в осадке подгруппы меди.
в) Установление pH раствора. Осаждение большинства указанных сульфидов! лучше всего проводить при pH, 2—3. Это значение pH получают, приливая к пробе раствора, предвари
70
Д. Химический анализ
тельно доведенной до объема 3 см3, 5 М NaOH или 5 М НС1. Некоторые сульфиды, такие, кай PbS, CdS, M0S3 и частично также В125з, количественно выпадают в осадок только при более высоких значениях pH (pH 5). Поэтому осаждение сульфидов проводят при двух значениях pH (первое и второе осаждение сероводородом).
г) Предварительные испытания на Мо (см. разд. 37.2.1.2). Для правильного проведения второго осаждения сероводородом необходимо предварительно провести пробу на присутствие молибдена в растворе. При действии сероводорода M0S3 выпадает в осадок очень медленно и лишь при нагревании. Для ускорения его осаждения и количественного отделения второе осаждение сероводородом нужно проводить под давлением. Для этого центрифугат и промывные воды после первого' осаждения сероводородом помещают в ампулу вместимостью 5 см3 и смешивают с тиоуксусной кислотой, избытка которой следует избегать. В случае присутствия в растворе трех элементов (Мо, Pb, Cd) для анализа достаточно иметь 0,2 см3 раствора. Ампулу осторожно запаивают и, обернув тряпкой, нагревают на водяной бане. Нагревание нужно проводить очень осторожно, под закрытой тягой, работать в1 защитных очках. Как видно из уравнения реакции, образуется H2S, осаждающий M0S3 и другие сульфиды:
СН3С + Н20 -*СН3С + Нг5 ОН ^он
Осаждение заканчивается через 5 мин. Конец обернутой тряпкой ампулы нагревают в пламени горелки Бунзена, при этом находившийся под давлением H2S выделяется через размягченное стекло. Верхнюю1 часть ампулы теперь можно отделить, отрезав ее ножом для резки стекла.
В отсутствие молибдена катионы осаждают, как описано выше, пропуская через раствор H2S. Осадок отделяют центрифугированием, промывают и объединяют с осадком, полученным после первого осаждения' сероводородом. Из центрифуга-та и промывных вод удаляют H2S и упаривают их до объема 5 см3. Этот раствор подвергают ионному обмену или, в отсутствие РО43- и ВОз3-, подготавливают для осаждения действием NH3.
Разделение катионов подгруппы олова и подгруппы меди.
На схеме Д.7 приведен ход разделения ионов подгрупп меди и олова, а также разделения1 и обнаружения элементов подгруппы олова; на схеме Д.8 показан ход разделения и обнаружения ионов подгруппы меди.
Схема Д.7. Разделение элементов подгруппы меди и подгруппы олова. Обнаружение элементов подгруппы олова.
Подкисление ZfiM HzSOq, удаление H2S кипячением
Добавление 5М №гОН+/ЛТ NaHS, нагревание (двухкратное}
-TuS, PbS, /Bi2S3, CcLS,
Добавление 1ZM НС1, нагревание
Добавление гранулированного и инка
Проба на окра -шивание пламени или добавление
Hgci2 Образование осадка Нд2С1,> Нд (черный]
Растворение в 1ZM HCI+JAf NttClO? (NH4)2fWH25, образование SbzS5 или 5bzSj
Растворение в М.5М HNOj; недальние количество раствора добавляют к смеси 0,5М (МН4)2МоОч* 74.5М HN03, нагревание, появление жел -того опрашивания
Обнаружение Нд (см схему Д.5}
Псантогенат-ная проба
As
нд
Мо
Схема Д.8. Разделение и обнаружение элементов подгруппы меди.
Отделение от подгруппы
Цобавление 2,5М Hj02+ 13М HzSO 4, разбавление до Z,5M HjSOi,
Добавление 7MNO.H5 и нагревание
При добавлении ШЫа.Н5+г,5МНД образование черного осадка
Cu2S
Си
Растворение в 5М НС1, выпаривание досуха на предметном сте-кле+1к. 5М НС1* tin. соли Рейнене +1 кристалл тиомочевины
Бледнорозовые т-сталлы стержневидной формы
Зеленая линия в спектроскопе или образование черного Т1Н2 при добавлении Im KI
Т1
Растворение в ДОВДИНД разбавление, образование осадка Bi(0H)3, при действии ИНз+5М МаОН*2к. 0,5М SnClz образование черного осадка Bi (металл)
Bi
Растворение в I 5ММаОН+2,5/И Нг0г1 +Нг0Чк.Й5М К# +5Й CHjCOOH, обра-1 зование PbCrOi, I желтого цвета j
37. Качественный анализ
73
Если в анализируемой пробе присутствуют РО43- и ВО33~, то центрифу-Гат, полученный после второго осаждения сероводородом, перед дальнейшим анализом нужно подвергнуть ионному обмену. В аммиачном растворе почти все катионы образуют осадки с РО43- и ВО33-, что усложняет последующий систематический ход анализа катионов.
Техника проведения ионного обмена описана в разд. 37.1.5.9. Упаренный слабокислый раствор пропускают через колонку с катионитом. После того как весь раствор поступит в колонку, ее промывают четыре раза водой порциями по 3 см3. Вытекающий из колонки раствор (элюат) исследуют на содержание РО43~ и ВО,3-. Катионы элюируют примерно 5 см3 5 М НС1 (в присутствии трудноэлюируемых ионов бария необходимо пропустить через колонку 10 см3 5 М НС1). Затем колонку промывают водой и элюат упаривают для удаления большого избытка НС1.
Группа NH3. На схеме Д.9 приведен ход разделения и обнаружения элементов группы NH3.
Группа (NH4)2S. На схеме Д.10 приведен ход разделения и обнаружения элементов группы сульфида аммония.
Группа (NH4)2CO3. Перед осаждением катионов щелочноземельных металлов действием раствора (NH4)2CO3 из анализируемой пробы необходимо удалить основную часть NH4+. (В присутствии большого количества NH4+ может не осесть СаСО3). Кроме того, в присутствии ванадия, молибдена или вольфрама часто, необходимо отделять остаточные количества тиосолей этих элементов в ходе анализа.
Для получения бесцветного раствора, не содержащего! ванадия, молибдена или вольфрама, анализируемый раствор подкисляют 5 М НС! и удаляют выделяющийся H2S. В присутствии тиосолей указанных элементов в осадок выпадают соответствующие сульфиды. Если в растворе еще остались тиосоли, то после центрифугирования к центрифугату можно, добавить 5 М NH4OH- и несколько капель 2,5 М. Н2О2 и прокипятить (избыток Н2О2 полностью разлагают, почему?). Затем, после подкисления 5 М НС1, сульфиды мешающих ионов осаждают действием H2S. Из центрифугата удаляют H2S кипячением, а избыток NH4+ —добавлением 14,5 М HNO3. К остатку, смешанному с 1 к. 5 М НС1, добавляют воду.
В присутствии ионов магния осадок, полученный при действии раствора (NH4)2CO3, нужно переосадить для отделения соосажденного гидроксикарбоната магния. На этом этапе анализа проводят предварительные испытания на магний. Для этого смешивают каплю солянокислого раствора с несколькими каплями 0,02%-ного раствора магнезона. После добавления 5М NaOH происходит взаимодействие Mg(OH)2 с магнезоном с образованием лака василькового цвета.
На схеме Д.11 представлен ход разделения и обнаружения элементов группы карбоната аммония.
Группа щелочных металлов, аммония, магния. В присутствии Li+ (спектральная проба) в центрифугате, полученном после осаждения карбонатов щелочноземельных металлов,
Схема Д.9. Разделение и обнаружение элементов группы NH3.
&и. схему Д. 10
Нейтрализация 14,5 HN03, добавление 5М СН3 CD0H +
IM Pb (CH3CD0)z
5М CH3CODH+
+ Д/М КцГРе(СМ)6], образование K2U02[Fe(CN)6] красновато-коричневого
цвета
Растворение в 1МК\.* 0,1%-ный раствор ализари-/rtZ+57W NH4OH светлокрасный лак, нерастворимый в разбавленной уксусной кислоте
Перемешивание с амиловым спиртом*?,5М Н202, встряхивание -* красновато-коричневый цвет водной фазы
Перемешивани е с амиловым спиртом* ?,5М.
Н202, встряхивание -* голубое окрашивание слоя амилового спирта
Растворение в 14,5М HNO3+ РЪ02, нагревание, желтое окрашивание -» Се; можно обесцветить действием Na3AsO3+ *0,1М KI, восстановление Се!у-Се"
U
Al
Схема Д.10. Разделение и обнаружение элементов группы (NH4)2S.
Центршругат группы NH3
Пропускание H2S, добавление 5М NH^OH, нагревание
Группа. fNH^zCOj
^NiS, CoS//^
Растворение в Ve,5 М HN03, добав-
ление 5М Na.CI.O3, двухкратное выпаривание досуха, растворение в воде
Буферирование добавлением 5М NHijCl + 5M NHjjOH
Нейтрализация 5М СН3С00Н, пропускание Hj,5
I ________
Нейтрализация 5М NH4OH+KSCN+ * амиловый спирт, встряхивание
Подавление 5М NH^OH+ftWtz-метилглиотим: красные иглы
Растворение в, 2,5 М НЫ03. добавление (ИМ Фр)5].
KzZn[Fe(CN)6] белый
'г
Растворение в 5М НС1, добавив Hue IM NHitSCN,
Fe(SCNl3 красный
Окислительное сплавление, зеленый
Co(SCN)2 голубой
''
Со
Ni
Zn
Схема Д.11.
(NH4) 2СО3.
Разделение и обнаружение элементов группы
Дентрисругит группы (NHi^jS
Ъцрерирование добавлением 5М NH4CI+ 5М МНгДЬЬнасб/-щенный раствор (NH^COj
Растворение в SM CH3C00H,tfus0ep«po-вание добавлением 5М NH^Cl, добавление К2Сг0г,
Растворение в 5М НС1
Добавление 2,5 М HzS0z,; иглы CtxSO^ (люкраскоп'}
Добавление гипсовой воды, выпадает осадок Sr30ц; или выпаривание 1к. раствора* * 1к. Н?0*
* 1к раствора Н103 (микроскоп}:
Sr(I03W
37. Качественный анализ
77
Mg2+ необходимо отделить, в отсутствие Li+ можно раздельно определить Mg2+, Na+- и К+. В то же время проводить идентификацию щелочных металлов в центрифугате после осаждения группы карбоната аммония не всегда целесообразно, часто даже невозможно. Так, например, в вытяжке, полученной после обработки анализируемого вещества смесью 12 М НС1+5 М NaC103, нельзя определять Na+. Поэтому обычно готовят вод-нук1 или слабосернокислую вытяжку исходного вещества и в ией определяют Na+ и К+ (обнаружение щелочных металлов в труднорастворимых силикатах см. в разд. 37.2.1.4).
Обнаружение магния. В присутствии ионов Li+ магний отделяют в виде оксонитрата или MgO. Для этого дважды выпаривают досуха центрифугат, полученный после осаждения раствором (ПН4)2СОз:: первый раз со смесью 14,5 М HNO3+ Д-12 М. НС1 и второй-—с 14,5 М HNO3. При этом из полученного остатка не должны выделяться газообразные нитрозосоединения и соли аммония. Затем к осадку, измельченному при помощи стеклянной палочки, добавляют воду. Раствор упаривают почти наполовину, переносят в центрифужную пробирку и центрифугируют. Остаток представляет собой оксонитрат магния, его промывают и растворяют в 5 М НС1.
В отсутствие Li+ в анализируемой смеси отделение магния не проводят. Ионы аммония в любом случае нужно удалять выпариванием, как указано выше; остаток от выпаривания можно затем растворить в 5 М ;НС1.
Обнаружение магния проводят из полученного солянокислого раствора, как указано ниже: после добавления к раствору 1—2 капель 2,5 М (NH4)2HPO4 и 5 М NH4OH (раствор должен иметь слабощелочную реакцию) - при нагревании на водяной бане осаждают MgNH4PO4-6H2O в виде белого мелкокристаллического осадка. Эта реакция иногда протекает медленно, так что осадок выпадает только через некоторое время. Под микроскопом видны кристаллы характерной формы.
При слишком быстром росте кристаллы имеют не характерную или не ярко выраженную форму. В этом случае осадок можно еще раз растворить в минимально возможном количестве 5 М НС1, каплю полученного раствора поместить на предметное стекло и положить его этой стороной на чашечку, наполненную 13 М раствором NH4OH. Образующиеся при этом кристаллы имеют характерную форму.
Mg2+ можно обнаружить также реакцией с магнезоном в растворе, полученном при растворении MgNH4PO4, как описано в разд. 37.2.1.8.
Обнаружение натрия. Обнаружение ионов Na+ и других Щелочных металлов проводят в центрифугате, полученном пос-ле осаждения магния (оксонитрата магния), или в водной вытяжке исходного анализируемого вещества.
78
Д. Химический анализ
а) Обнаружение по реакции образования NaMg(UO2)s. (СН3СОО)э-9Н2О. На предметном стекле соединяют каплю исследуемого раствора с каплей- магнийуранилацетата. Образуются желтоватые, прозрачные ромбоэдрические кристаллы.
б) Обнаружение по реакции образования двойного сульфата натрия-висмута.
Упаренную на предметном стекле каплю анализируемого раствора нагревают с каплей 2,5 М H2SO4 до образования тумана H2SO4. Смешивают затем1 с 1 к. Н2О и 1 к. 0,2 М Bi (NO3) 3. При этом в осадок не должен выпасть оксинитрат висмута; 'если же это произошло, осадок растворяют в капле 2,5 М. HNO3 или H2SO4. При медленном высушивании при комнатной температуре под микроскопом можно наблюдать образование 'кристаллов двойной соли 3Na2SO4-2Bi2(SO4)3-2H2O в форме 'призм (палочек).
1 Образование шестиугольных пластинок и звездочек указывает на присутствий К+ в анализируемом растворе. Ионы аммония мешают проведению1 реакции.
Обнаружение калия.
а) Реакция образования K2SO4-Bi2(SO4)3-2H2O.
Реакцию проводят так же, кай указано для обнаружения натрия.
б)' Осаждение в виде гексанитрокобальтата. Образование желтого осадка при смешивании раствора пробы (предварительно слабо подкисленного 5 М СН3СООН) со свежеприготовленным насыщенным1 раствором Na3[Co(NO2)6] указывает на присутствие К+. Образуется1 соединение KnNa3_n[Co(NO2)6]^ •где п^З. Эту реакцию нельзя проводить в предварительных, испытаниях' в водной или уксуснокислой вытяжке анализируемого вещества, так как ее проведению мешают очень многие катионы. При анализе смеси катионов щелочных металлов, аммония и магния мешают только NH4+ ;(образуется похожий по-внешнему виду осадок). В присутствии NH4+ осаждение проводят избытком раствора Na3[Co(NO2)e] при нагревании на. водяной бане. При этом комплекс разрушается и одновременно происходит окисление NH4+. Раствор охлаждают и снова, проверяют на К+.
в) Обнаружение реакцией с НСЮ4. При смешивании на предметном стекле капли анализируемого раствора с 1 к. 3 М НСЮ4 образуются (иногда после кратковременного нагревания) характерные ромбические кристаллы КСЮ4.
Обнаружение лития. Надежным и чувствительным способом обнаружения лития1 является спектральный метод.
’ Обнаружение NH^. Так как соли аммония часто применяют в ходе разделения, обнаружение ионов NH4+ проводят из отдельной пробы исходного вещества.
37. Качественный анализ
79
а) Проба на запах. В маленькой ступке растирают небольшое количество вещества с влажными гранулами NaOH или КОН. Характерный запах является достаточным признаком присутствия NH4+. Можно смешать вещество с пластинками гидроксида натрия также в бюксе. При этом приклеенная к внутренней стороне крышки бюкса влажная красная, лакму- , совая бумага окрашивается в синий цвет, а капля реактива Несслера становится коричневой.
Вопросы- и задания
1. Почему в полумикроанализе анализируемую пробу обрабатывают не царской водкой, а смесью 12 М НС1 + 5 М NaC103?
2. Напишите уравнения реакций, происходящих при действии смеси 12 М НС1 + 5 М NaClOs на анализируемую пробу.
3. Какая реакция протекает при удалении солен аммония (например, из фильтрата после осаждения группы (NH4)2S) действием HNO3? Напишите уравнение реакции. К какому типу относится данная реакция?
4. Почему сплавление с пиросульфитом нельзя проводить при слишком высокой температуре?
5. Почему вытяжку, полученную при обработке вещества смесью NaOH + + KCN, нельзя объединять с другими вытяжками (водной, солянокислой или вытяжкой, полученной при обработке смесью 12 М НС1 + 5 М NaC103)?
6. Вещество голубого цвета при нагревании в калильной трубочке обесцветилось, а при добавлении концентрированной НС1 окрасилось в зеленый цвет. Солянокислый раствор при разбавлении окрасился в голубой цвет. О каком соединении идет речь?
7. Белое вещество, нерастворимое в воде, растворимо в разбавленном растворе аммиака. При добавлении к этому раствору NaOH выпадает чернокоричневый осадок. Для какого соединения характерны такие реакции?
8. При действии какого реактива раствор Fea(SO4)3 окрашивается в желтый цвет?
9. При помощи каких реакций можно установить, что за соединение находится в осадке: MgNH4AsO4-6H2O или MgNH4PO4-6H2O?
10. Пробу каждого из перечисленных веществ: NaF, NaCl, NaBr, Nal, NaCN и NaSCN напрели с 18 M H2SO4. Напишите соответствующие уравнения реакций.
И. Объясните, почему Ag3[Fe(CN)6] растворяется в 5 М NH4OH, a Ag4[Fe(CN)6]— нет.
12. Почему сульфиды группы сероводорода выпадают в осадок при других значениях pH, чем сульфиды группы (NH4)2S?
13. Объясните, почему осадок сульфидов группы сероводорода промывают слегка подкисленным НС1 раствором сероводородной воды, а ие дистиллированной водой.
14. Почему сульфиды легче осаждаются из растворов арсенитов, чем из растворов арсенатов?
15. В подгруппе меди свинец отделяют от висмута действием 18 М H2SOt я Н2О2. Объясните, какие реакции протекают: а) если ие разбавлять серную кислоту; б) если применить сильно разбавленную серную кислоту.
16. Объясните реакцию, происходящую между AgNO3 и Na2S2O3. Напишите ее уравнение.
17. Почему реакция разложения нитритов производными аммиака является реакцией окисления — восстановления?
18. Почему HgS не растворяется в (NH4)2S, но растворим в NaOH+ NaHS?
80
Д. Химический анализ
19. Какая реакция протекает при добавлении CN- к Си2+ (при разделении смеси Cu2+ + Cd2+)?
20. Какие реакции происходят при добавлении к А1(ОН)3, Сг(ОН)з, VO(OH)2 и (NH^aUaO? смеси NaOH + HaOa? Напишите уравнения реакций, укажите типы реакций.
21. Почему при пропускании H2S в раствор [Cu(ON)4]2-+[Cd(CN)<]2- не осаждается Cu2S? При каких условиях можно Си+ осадить в виде Cu2S из указанного раствора?
22. Почему при добавлении фторида к ализаринциркониевому лаку ализарин выделяется в свободном состоянии?
23. Почему AgCl растворяется в насыщенном растворе карбоната аммония, a AgBr и Agl нерастворимы? Напишите уравнения реакций. К какому типу относятся данные реакции?
24. На каком этапе разделения катионов применяют насыщенный раствор карбоната аммония? Почему этот раствор должен быть свежеприготовленным и почему при применении его нагревают до 50 °C?
37.2.2. Другие способы разделения катионов
В ходе разработки качественного анализа были предложены разнообразные методы разделения, целью большинства которых являлась замена газообразного H2S другими реагентами или устранение операции осаждения ионов в виде сульфидов-Однако классический метод разделения по Фрезениусу все еще применяется и в настоящее время.
37.2.2.1. Методы разделения без применения газообразного H2S
Эта группа методов основана на гомогенном осаждении. К достоинствам методов наряду с устранением ядовитого сероводорода относится хорошая фильтруемость и легкость центрифугирования осадков сульфидов. Кроме того, при использовании в качестве осадителей твердых сульфидов или других' серусо-держащих соединений, которые в кислом растворе протолизу-ются с образованием) ионов HS~ или гидролизуются, можно-регулировать количество осадителя.
При использовании Na2S в качестве осадителя последовательность разделения катионов такая же, как в сероводородном методе. При действии Na2S, Na2CO3 и NaOH катионы отделяют в виде нерастворимых в щелочах сульфидов, карбонатов и гидроксидов соответственно. В растворе остаются1 тиосоли, ионы алюминия и бериллия. Дальнейшее разделение проводят, обрабатывая осадой и раствор соляной кислотой. Недостатком метода является выделение больших количеств Н2& при избытке кислоты.
37. Качественный анализ
81
В качестве осадителей применяют также Р255„ Na2S2O3 и (NH4)2$, органические серусодержащие соединения, например-тиоацетамид:
СН3С +2Н20-*СН3С^ + NHX + HS“ NH2 ОН
При этом и фильтрату, полученному после осаждения группы НС1, добавляют избыток 2%-ного раствора тиоацетамида и нагревают смесь в течение ~5 мин до 100 °C. Затем pH раствора доводят до 0,5. При дальнейшем нагревании выпадает осадок сульфидов. В фильтрате группы H2S избыток тиоацетамида разрушают действием бромной воды и HNO3. После отфильтровывания выделившейся серы осаждают Al, Fe и Сг действием NH4OH. Фильтрат группы NH3 подкисляют HCU и снова добавляют к нему тиоацетамид. Раствор нагревают на водяной бане в течение 5* мин и добавляют к нему раствор аммиака. После повторного нагревания выпадают сульфиды Со, Мп, Ni и Zn. В фильтрате после разрушения тиоацетамида проводят обнаружение ионов щелочных и щелочноземельных металлов.
В качестве органических серусодержащих осадителей применяют и такие, как
тиоуксусная кислота
тиоформамид
этилксантогенат калия
/8
нс<с
NHa
/ОС2Н5 s=c<
SK
37.2.2.2- Бессероводородные методы разделения
Из большого числа опубликованных методов можно остановиться лишь на некоторых.
1 Осаждают и отделяют серебро, свинец и ртуть в виде хлоридов; затем.' отделяют щелочноземельные металлы в виде сульфатов. После этого действием металлического цинка на кислый раствор выделяют Cu, Cd, Bi, As, Sb и Sn. Осажденные металлы растворяют в кислоте и идентифицируют с помощью селективных реакций.
6—1880
82
Д. Химический анализ
2. Катионы металлов делят на небольшие группы по сле^ дующей схеме (р—р-раствор):
р-р
раствор 4* HNO3 ----►
Stjsn
р-р
+ НС1 -----►
Ag, Pb, Hg
р-р
4- NH3------>
Fe, Cr, Al, Bi
+ H2O2
Mn
+ (NH4)2co3------>
Ba, Sr, Ca
+ Ba(OH)2
K, Na
Mg, Ni, Co, Zn, Cu.Cd
Недостатком этого метода является необходимость предвари' тельного удаления органических веществ и неполнота некО' торых разделений.
3. Катионы делят на пять групп: а) группа хлоридов б) группа основных бензоатов (bi осадок выпадают гидрокси ды трех- и четырехзарядных ионов металлов); в) группа фто ридов; г) группа катионов неамфотерных: металлов; д) труп па катионов амфотерных металлов.
При этом происходит количественное разделение катионов
37.2.2.3. Метод экспресс-анализа по Шарло
Метод/ Шарло — это современный метод экспресс-анализа ка тионов, в котором число операций разделения сведено к ми нимуму. В нем применяют реакции идентификации, отличаю щиеся высокой селективностью, которые дают возможность последовательно идентифицировать катионы из отдельных проб раствора.
Для проведения анализа используют солянокислый рас твор анализируемого вещества, из которого удалены Ag+ и Hg(I). Раствор не должен содержать F~ и СгОД-. Для от деления SiO2 раствор дважды выпаривают досуха с соляной кислотой. При этом мешающие кислоты разлагаются, в ана лизируемом растворе должны находиться только Cl-, SO42-РО43' и ВОз3". Мышьяк, сурьму и ртуть нужно обнаружить в исходном растворе, так как их хлориды летучи. Реакцией с метиленовым голубым обнаруживают Sn(II) и Fe(II). Эти ио ны нужно окислить бромной водой и избыток брома удалить кипячением.
37. Качественный анализ
8а
Для получения информации о качественном составе анализируемого раствора проводят ряд предварительных реакций с групповыми реактивами:
1 а) добавляют NH3, затем Na2S и наблюдают окраску образующихся гидроксидов и сульфидов, по которой делают определенные выводы;
б) добавляют раствор купферона в 1,5 М НС1; если образуется осадок, то возможно присутствие Sn, Zr, Fe, V, Мо, W,. Ti, Bi;
в) добавляют раствор бензоиноксима в 4 М НС1; в присутствии Мо, W и V образуется осадок;
г) добавляют раствор (NH4)2SO4 в 2 М НС1; образующийся осадок указывает на присутствие Sr и Ва или больших количеств Са и РЬ.
В исходном растворе проводят обнаружение катионов^ дробным методом с помощью капельных реакций, выполняемых на капельной пластинкё или на фильтровальной бумаге. Мешающее влияние отдельных ионов обычно устраняют маскировкой с помощью ЭДТА.
Метод Шарло можно дополнить качественным анализом в среде ледяной уксусной кислоты. Можно использовать чистую безводную ледяную уксусную кислоту, уксусную кислоту с максимальным содержанием воды (2%) или ледяную уксусную кислоту с добавлением 10% уксусного ангидрида.
Для проведения анализа готовят солянокислый раствор анализируемого вещества в воде. При этом могут образоваться осадки AgCl и РЬС12. К фильтрату добавляют NH4C1 и выпаривают досуха. К остатку, полученному после выпаривания, добавляют смесь ледяной уксусной кислоты и ацетил-хлорида (1:1) и кипятят 15 мин. В остатке при этом остаются CuCl2, №С12г (NH4)2[SnCl4], CdCl2, Т1С1 и алюминий, образующий многоядерный ацето-комплекс. Компоненты остатка идентифицируют без дальнейшего разделения, применяя селективные реакции на капельной пластинке. В уксуснокислый раствор вносят магниевые стружки. При этом в виде металлов выделяются Hg, As, Sb и Bi. Их отделяют центрифугированием и смешивают с раствором КВгОз, приготовленным в небольшом количестве ледяной уксусной кислоты; образуются броматные комплексы. Металлы, не восстанавливающиеся магнием, находятся в центрифугате в виде хлоридных комплексов: [СоСЦ]2-, [FeCh]2”, [MnCk]2-, [ZnCk]2-, [А1С16]3~ и [СгС16]3-. Эти ионы также идентифицируют дробным методом, проводя капельные реакции на капельной пластинке.
Г. Аккерман описал метод обнаружения катионов, также основанный на применении селективных реакций обнаружения (особенно капельных и микрокристаллоскопических).
37.2.2.4. Применение дитизона в качественном анализе
Дитизон можно применять: в микро- и макроанализе как аналитический групповой реагент. При этом: с помощью немногих реакций можно сделать выводы о присутствии или отсутствии различных групп и тем самым быстро получить информацию о составе анализируемой смеси.
6*
84
Д. Химический анализ
, Для этого готовят четыре раствора анализируемого вещества, которые затем обрабатывают дитизоном. Из раствора, полученного растворением анализируемого вещества в минеральных кислотах, дитизоном можно извлечь Pd, Au, Hg, Ag и Си. В уксуснокислых растворах с дитизоном реагируют Pd, Zn, Со, Ni, а также Cd и Sn при относительно большом их содержании в растворе. В щелочном растворе в реакцию с дитизоном вступают Ag, Hg, Pd, Au, Cu, Co, Ni и Cd. В растворе, содержащем, цианиды, с дитизоном реагируют Pb, Bi, TI, Sn.
Для проведения групповых реакций в делительную воронку помещают несколько кубических сантиметров исследуемого раствора при соответствующих условиях (pH, присутствие маскирующих веществ, и т. д.) и встряхивают о раствором реактива. Отрицательный эффект реакции свидетельствует об отсутствии в растворе ионов металлов, относящихся к данной группе. При изменении окраски раствора дитизона можно уже по окраске экстракта или образующихся хлопьев сделать вывод о присутствии в растворе определенного иона металла. Дополнительные реакции обнаружения ионов можно провести в органическом экстракте или в водной фазе. Кроме того, ионы металлов, находящиеся в результате в экстрактах различных групп, после разрушения дитизо.на можно идентифицировать другими способами, например по образованию кристаллов.
37.2.3. Экспресс-анализ
При! проведении практикума по качественному неорганическому анализу успешно применяют такую форму обучения, как экспресс-анализ, под которым понимают идентификацию нескольких простых веществ в течение часа.
Хотя химику в его практической деятельности вряд ли придется решать подобную задачу, экспресс-анализ нельзя рассматривать как устаревшую, бесполезную форму обучения. В процессе проведения экспресс-анализа развивается наблюдательность, привлекается знание химических свойств веществ, умение делать быстрые и точные выводы, приобретаются навыки техники работы в лаборатории. Развитие современных физических и физико-химических методов анализа1 не снижает ценности этих навыков.
Нельзя привести/ общую, пригодную для всех случаев схему экспресс-анализа. Для проведения работы необходима безупречная подготовка рабочего места с рациональным расположением оборудования и реактивов и легким доступом к ним. На рабочем месте наряду с обычными и центрифужными пробирками, а также несколькими капельными пипетками должны находиться две горелки, магнезиальные палочки, ручной спектроскоп, центрифуга, капельная пластинка и стеклянные
37. Качественный анализ
85
палочки и, кроме того, универсальная индикаторная бумага для измерения pH и кипящая водяная баня. В рабочем состоянии должны находиться основный чистые реактивы, придем особенно важны групповые реактивы. Растворы сероводородной воды и хлорной воды должны быть свежеприготовленными.
Можно дать некоторые рекомендации по проведению экспресс-анализа. Анализ начинают с растворения вещества и получения различных вытяжек (см. разд. 37.2.1.3). Если вещество растворяется в воде, определяют pH раствора. В то время как вещество с растворителем при необходимости нагревается на водяной бане (чаще перемешивать!), проводят предварительные испытания.
Важнейшей операцией экспресс-анализа является выполнение групповых реакций (см. разд. 37.2.1.5 и 37.2.1.7). Применяя логически обоснованную последовательность действия групповых реактивов, неизвестное вещество обычно можно идентифицировать за короткий промежуток времени.
37.3. Применение физико-химических методов
в качественном анализе
37.3.1. Хроматографические методы
Среди методов разделения веществ важное место занимают хроматографические методы, которые в последние годы находят все большее применение в аналитической химии. Хроматографию на бумаге и в тонких слоях применяют в качественном анализе чаще, чем колоночную. Хотя основной областью применения хроматографии является органическая химия, в хроматографии неорганических! веществ также достигнуты определенные успехи, о чем можно судить по постоянно растущему числу публикаций на эту тему.
Теория хроматографических методов анализа и техника их выполнения изложена в разд. 38.3.6.
37.3.1.1. Разделение и идентификация катионов методом бумажной хроматографии
Число работ, относящихся к этой области хроматографического анализа, велико и постоянно растет. Мы приведем некоторые типичные примеры.
Обнаружение щелочных и щелочноземельных металлов. Для разделения щелочных и щелочноземельных металлов готовят Ю%-ный раствор пробы в 2 М НС1, не содержащий ионов других аналитических групп. При этом применяют хроматографиче
86
Д. Химический анализ
скую бумагу марки FN 11 в листах размером 25x30 см*. Линия старта находится на расстоянии 2,5 см от нижнего края бумаги.
Исследуемые растворы наносят на бумагу в виде точек е интервалом 3 см. Подвижной фазой является смесь 80 см3 метанола с 10 см3 воды и 10 см3 12,5 М НС1. Хроматограмма развивается при 20 °C в течение ~ 3 ч; подвижная фаза при этом проходит расстояние около 20 см. На линию старта наносят последовательно: 1%-ный раствор смеси KC1+NH4C1, служащий контрольной пробой; пробу неразбавленного анализируемого раствора; пробу анализируемого раствора, разбавленного в отношении 1:10; вторую контрольную пробу-—смесь NaCI+ 4-LiCl, и затем снова две пробы анализируемого раствора. Далее наносят 0,2%-ный раствор смеси солей щелочноземельных металлов и две пробы анализируемого раствора.
Бумагу сушат в течение 30 мин при комнатной температуре, скручивают в цилиндр и вставляют в сосуд с подвижной фазой. После того как подвижная фаза пройдет расстояние 20 см, бумагу сушат на воздухе (сушилкой) до исчезновения едкого запаха газообразного НС1. Бумагу разрезают на три части. Часть, на которую был нанесен стандартный раствор смеси солей К+ и NH4+, окунают в 10%-ный раствор гексанитрокобальтата натрия. Бумагу слегка подсушивают и вымывают избыток реактива водой. При этом на бумаге появляются желтые пятна К+ и NH4+ (для NH4+ 7?/ = 43).
Часть бумаги с нанесенным раствором Na+ и Li+ обрызгивают насыщенным раствором цинкуранилацетата в 5%-ной СНзСООН. При облучении ультрафиолетовым светом появляется флуоресцирующее зеленое пятно, которое сравнивают с пятном контрольной пробы (для Li+ 7?/ = 0,74, для Na+ 7?/ = 0,31).
Третью часть хроматограммы обрызгивают 1%-ным щелочным раствором 8-оксихинолина и высушивают. Затем сильно смачивают хроматограмму концентрированным раствором аммиака и тот час же подвергают влажную бумагу ультрафиолетовому облучению. В присутствии бария и стронция возникает бирюзовая флуоресценция (для Ва2+ 7?f = 0,ll; для Sr24* /?f = 0,26), в присутствии кальция — желто-зеленая (J?f = 0,47).
Быстрый анализ сталей (Аккерман, 1963). Легирующими элементами в сталях являются Al, Cr, Ni, V, Мп, Ti, Си, Со и Мо. Подвижной фазой служит смесь 25% НС1 (пл. 1,19)+ 30% н-бутанола+45% уксусноэтилового эфира (проценты по объему). Ионы железа перемещаются вместе с фронтом растворителя и поэтому не мешают определению других компонентов.
Для обнаружения всех компонентов необходимо получить
* В СССР выпускают несколько сортов хроматографической бумаги, различающейся по плотности: № 1 и 2 — «быстрая», № 3 и 4 — «медленная».— Прим, перед.
37. Качественный анализ
87
две хроматограммы. На первой хроматограмме обнаруживают хром после его окисления гипобромитом в виде окрашенного в желтый цвет хромата. Идентификацию Мп, Ni, Си, Со проводят с помощью салицилальдоксима и рубеановодородной кислоты. Вторую хроматограмму делят на части. В верхней части идентифицируют молибден действием KSCN и SnCU. При опрыскивании оставшейся части хроматограммы смесью ализарина 8 и хромотроповой кислоты обнаруживают титан, ванадий и алюминий.
Выполнение анализа
Сталь растворяют в царской водке, выпаривают досуха и добавляют 12 М НС! до получения 10%-ного раствора. По 5 мм3 полученного таким образом раствора наносят на две полоски хроматографической бумаги (WF 11, FN 11) длиной по 22 см и получают восходящие хроматограммы. После окончания разделения (в течение 3 ч) полосы бумаги высушивают до исчезновения запаха НС1.
Полосу 1 опрыскивают 2 М NaOH и подвешивают в цилиндре с парами брома. После высушивания ее обрабатывают раствором 1 г рубеановодородной кислоты и 0,5 г салицилальдоксима в 100 см3 этанола и парами аммиака.
Полосу 2 разрезают, отступив на 1 см ниже линии фронта. Верхнюю часть опрыскивают раствором 2,5 г SnCl2 и 5 г KSCN в 100 см3 НС1 (1 : 1).
Таблица Д.11. Идентификация компонентов стали, разделенных методом бумажной хроматографии
Элемент Величина R; Полоса 1 Полоса 2
Мо 0,85 Красный (верхняя часть)
Со 0,6 Коричневый —-
Си 0,55 Оливковый —
Ti 0,35 — Красный (нижняя часть)
Мп 0,25 Коричневый —
V 0,2 — Оранжевый (нижняя
часть)
Ni 0,07 Сине-фиолетовый —
Сг 0,07 Желтый —
А! 0,0 — Розовый (нижняя часть)
Нижнюю часть хроматограммы опрыскивают растворогм 0,1 г ализарина S и 1 г хромотроповой кислоты в 100 см3 50%-ного этанола и обрабатывают парами НС1. В табл. Д.11, представлены значения Rs и указаны соответствующие окраски пятен.
37.3.1.2. Тонкослойная хроматография
Основными областями применения тонкослойной хроматографии, так же как и бумажной, являются органическая химия и биохимия. Но в последние годы этот метод все более широко внедряется в практику аналитической химии. Его можно применять и для разделения неорганических веществ. При этом тонкослойная хроматография имеет ряд преимуществ перед бу
88
Д. Химический анализ
мажной; важнейшие из них — возможность четкого разделения ионов, высокая чувствительность и быстрота анализа. Если в. бумажной хроматографии разделение проводят в течение нескольких часов, то при выборе соответствующего адсорбента в. методе тонкослойной хроматографии разделение заканчивают за несколько минут. Однако бумажная хроматография, также применяемая в настоящее время, имеет некоторые преимущества при разделении гидрофильных веществ.
Сущность метода. На стеклянную пластинку наносят слой адсорбента толщиной 250 мкм (кизельгура G, порошкообразной целлюлозы, оксида алюминия). При этом лучше использовать имеющиеся в продаже пленки. Оправдало себя применение выпускаемых в ЧССР специальных пластинок (силуфолов), представляющих собой алюминиевую фольгу, покрытую слоем, силикагеля. На пластинку на расстоянии 1,5 см от нижнего-края наносят с помощью микропипетки анализируемые растворы. После испарения растворителя пластинки ставят в специальную разделительную камеру, заполненную подвижным растворителем на высоту примерно 0,5 см. Пространство камеры должно быть насыщено парами растворителя. При получении восходящей хроматограммы подвижная фаза движется от линии старта вверх. По мере ее развития появляются пятна, характерные для определенных веществ, так как компоненты смеси движутся с различной скоростью. В основе разделения лежат адсорбционные процессы.
Когда фронт растворителя переместится на определенное-расстояние (обычно 10 см), пластинку вынимают из камеры и высушивают. Разделенные вещества можно идентифицировать, различными методами. Мерой скорости передвижения веществ-, в бумажной хроматографии является Rf — относительная величина, зависящая от условий определения. Хроматограмма дает-лишь информацию о распределении пятен, так называемый рисунок пятен.
Оборудование и техника работы. Для проведения простых: исследований необходимы:
а) силуфольные пластинки производства ЧССР размером 10X20 см или 5x20 см, которые очищают путем хроматографирования на них чистого подвижного растворителя и сушат в сушильном шкафу при ПО °C в течение 30 мин; б) угловая разделительная камера из стекла с пришлифованной крышкой (для насыщения пространства камеры парами подвижного растворителя стенки камеры выкладывают фильтровальной бумагой, смоченной этим растворителем); в) сушильный шкаф для высушивания пластинок; г) эксикатор для хранения пластинок; д) сушилка; е) пульверизатор.
Разделяемую смесь, как и в бумажной хроматографии, с. помощью микропипетки наносят на линию старта на расстоя
37. Качественный анализ
89
нии 1,5—2 см от края пластинки. Диаметр наносимых пятен должен быть небольшим. Пятно высушивают с помощью сушилки. Пластинки затем вносят в разделительную камеру, насыщенную парами подвижного растворителя и заполненную растворителем на высоту 0,5—1 см. Расстояние, пройденное фронтом растворителя, должно составлять примерно 100 мм. По достижении растворителем линии финиша пластинку вынимают из камеры. Отмечают линию фронта растворителя и высушивают хроматограмму при комнатной температуре в сушильном шкафу или с помощью сушилки. Идентификацию разделенных катионов проводят или по их собственной окраске под действием ультрафиолетового облучения, или при обрызгивании специальными реактивами для идентификации.
На готовой хроматограмме пятна очерчивают карандашом, отмечают центр пятна и замеряют расстояние от него до линии старта. В тонкослойной хроматографии неорганических веществ невозможно указать постоянные значения Rf, так как отношение расстояния, пройденного ионом, к расстоянию, пройденному фронтом растворителя, зависит от влажности сорбента. Поэтому можно говорить только о последовательности расположения характерных пятен отдельных ионов. Целесообразно одновременно нанести на бумагу эталонные растворы соединений известных ионов, а также получать хроматограммы для различных концентраций анализируемых веществ.
Разделение катионов погруппы меди. Для разделения сульфидов подгруппы меди, растворенных в 6 М HNOs, применяют смесь 100 см3 бутанола, 20 см3 1,5 М. НС1 и 0,5 см3 ацетилаце-тона. Ацетилацето'н, образующий в этих условиях неустойчивые комплексы с определенными ионами, применяется для уменьшения образования так называемых «хвостов» на хроматограмме. Последовательность разделения ионов:
сиг+ < ръг+ < сй.г+ < Bt3+ < Hgz+
Значение fy
В качестве сорбента применяют силикагель GT. В силикагеле может содержаться железо, которое необходимо предварительно удалить. Пластинки помещают в камеру со смесью СН3ОН+ 4-НС1 (1:1) и обрабатывают подвижным растворителем. Железо перемещается вместе с фронтом растворителя и таким образом вымывается.
В качестве эталонных образцов берут по 0,002 см3 0,1 М. растворов следующих солей: Н§(МОз)2-2Н2О, Сс1(СНзСОО)2Х Х2Н2О, BiONOs, Pb(NO3)2, Cu(CH3COO)2-H2O.
Идентификацию ионов после их разделения проводят, опрыскивая пластинку 2%-ным раствором КВ Пластинку высуши-
90
Д. Химический анализ
Таблица Д.12. Идентификация ионов подгруппы меди методом тонкослойной хроматографии
Ион Цвет при проявлении, раствором KI Цвет при обработке H2S
Hg2+ Красный Коричнево-черный
Bi3+ Коричнево-желтый Коричнево-черный
Cd2+ — Желтый
Pb2+ Желто-коричневый Коричневый
Cu2+ Коричневый Темно-коричневый
вают; обрабатывают парами NH3 ненную сероводородом. Окраски, ведены в табл. Д.12.
и помещают в камеру, запол-характерные для ионов, при-
37.3.2. Микроанализ на фильтровальной бумаге
37.3.2.1. Капельный анализ
Применение органических реагентов в качественном анализе привело к развитию нового метода — капельного анализа. Благодаря интенсивной окраске, возникающей при взаимодействии
ионов с органическими реагентами, анализ можно проводить капле раствора. Начиная с 1920 г. разработкой и совершенствованием капельного анализа занимался Ф. Файгль. Результаты своих исследований он обобщил в вышедшей в 1954 г. монографии*. Существенный вклад в эту область анализа внес также Н. А. Тананаев.
Для проведения капельного анализа применяют округлые бумажные фильтры, используемые в количественном анализе, поскольку обычная фильтровальная бумага содержит большое количество посторонних ионов. Лучше всего брать фильтры со средней величиной пор или фильтры из плотной бумаги. Раствор анализируемой пробы отбирают капилляром, концом которого касаются затем фильтровальной бумаги. Бумага впитывает из капилляра необходимое количество анализируемого» раствора. Аналогично на бумагу наносят раствор реактива* Не рекомендуется добавлять растворы по каплям из пипеток,, так как может образоваться пятно неправильной формы из-за избытка реактива. Жидкость распространяется концентрическими кругами, при этом образуются кольцеобразные зоны раз
* Есть русский перевод шестого переработанного издания: Файгль Ф., Ангер В. Капельный анализ неорганических веществ. Пер. с англ. — М.: Мир, 1976. — Прим. ред.
37. Качественный анализ
91
деляемых компонентов. Иногда этим методом можно разделить довольно большое число компонентов. Часто разделение происходит только после добавления соответствующих реактивов. При этом в центре пятна могут остаться малорастворимые гидроксиды, в то время как растворимые соединения переместятся к краю фильтра, где их можно идентифицировать.
Для увеличения порога чувствительности реакции целесообразно применять фильтровальную бумагу, пропитанную раствором реактива. Например, А13+ идентифицируют на бумаге, смоченной спиртовым раствором ализарина. Для этого на высушенную бумагу помещают пробу вещества, обрабатывают ее парами аммиака и обрызгивают уксусной кислотой. Красное пятно указывает на присутствие алюминия. Лучший эффект получают при применении бумаги, импрегнированной реагентами.
Из-за явлений коллоидообразования важное значение имеет порядок нанесения растворов на бумагу; при проведении этой операции надо строго следовать методике анализа.
В определенных случаях возможно проводить разделение веществ на бумаге, используя различное отношение их к действию осадителей и растворителей. Например, на смесь Sb3+ и Bi3+ действуют полисульфидом аммония и осадок обрабатывают избытком реактива. Bi2S3 осаждается в виде черного пятна в центре фильтровальной бумаги, a Sb3+ переходит в раствор в виде тиоантимонита, который перемещается из центра и разрушается с образованием оранжево-красного Sb2S3, располагающегося в виде кольца вокруг черного пятна.
В ходе качественного полумикроанализа для идентификации разделенных ионов можно использовать капельные реакции. Примерами могут служить проба на W и Мо (действие KSCN+,SnCl2) и идентификация Sn2+ (действие фосфоромо-либдата аммония).
Многие реакции идентификации можно проводить на капельных пластинках. При этом можно идентифицировать даже сильно агрессивные вещества. К недостаткам такого способа работы относятся необходимость работы с большими количествами веществ, а также отсутствие капиллярного эффекта, возникающего при выполнении реакции на бумаге.
37.3.2.2. Электрография
Для проведения качественного анализа металлов или проб, обладающих электропроводностью, существует метод неразрушающего анализа, называемый электрографией.
Шлифуют небольшую часть поверхности пробы металла. Затем помещают на него кусочек фильтровальной бумаги, смочен
92
Д. Химический анализ
ный раствором электролита (K.NO3, NaCl). Гибкий платиновый или алюминиевый штифт подсоединяют к отрицательному полюсу источника постоянного тока (аккумулятора, батарейки карманного фонарика) и прижимают к влажной фильтровальной бумаге (рис, Д.26). Пробу соединяют с положительным полюсом источника тока. Компоненты пробы, растворяющиеся на аноде, переходят на фильтровальную бумагу и могут быть идентифицированы на ней обычными капельными реакциями.
Рис. Д.26. Схема установки для электрографического анализа.
/ — катод из листового алюминия; 2 — бумага; 3 — проба; 4 — анод; А — амперметр; V — вольтметр.
Для более точного контроля силы тока можно включить в цепь миллиамперметр и регулирующее сопротивление.
В течение небольшого времени (обычно бывает достаточно нескольких секунд) пропускают ток (силой примерно 10— 50 мА). Затем с помощью соответствующей реакции идентифицируют элемент на фильтровальной бумаге. Лучшие результаты получают, применяя бумагу, смоченную раствором реактива. В этом случае характерная для данного элемента окраска возникает практически мгновенно. При использовании органических реагентов в анализируемый раствор необходимо добавлять электролиты.
В качестве примера можно привести обнаружение кобальта в присутствии никеля и железа. Бумагу смачивают 25%-ным раствором NH4SCN, подкисленным СНзСООН, и пропускают ток при напряжении 2—4 В в течение 15 40 с. Фильтр обрабатывают парами ацетона. В присутствии кобальта появляется голубое окрашивание. Затем фильтровальную бумагу обрызгивают раствором диметилглиоксима и обрабатывают парами аммиака. В присутствии никеля и железа возникает красное окрашивание. Красная окраска, обуслов
37. Качественный анализ
93
ленная присутствием ионов железа, исчезает при действии концентрированного раствора фторида калия. Если нужно обнаружить только никель в присутствии больших количеств железа, то бумагу вначале смачивают раствором KF.
Полуколичественные результаты можно получить при проведении анализа при постоянной силе тока и постоянном времени. Для этого готовят стандартные образцы бумаги, на которые наносят пробы известной концентрации.
Поскольку на аноде растворяются лишь микрограммовые количества веществ, внешняя поверхность пробы практически не разрушается. Поэтому электрографию можно применять для анализа изделий из пластмасс. Этот метод также дает возможность установить распределение легирующих элементов на поверхности металлов. Благодаря простоте выполнения и незначительным аппаратурным затратам электрографию используют в металлургической промышленности для быстрого решения аналитических задач, например для сортировки и классификации неизвестных образцов легированных сталей. С помощью этого метода можно определять также состав деталей из мед-но-никелевых сплавов и нержавеющих сталей, доступ к которым затруднен. Для этих целей применяют выпускаемые промышленностью переносные приборы, снабженные портативной капсулой с электрографическим устройством для проведения анализа. При использовании вместо фильтровальной бумаги желатиновых пластинок, импрегнированных электролитами, на них появляется так называемый химический «отпечаток» поверхности металла. После соответствующей обработки растворами реактивов можно наблюдать под микроскопом распределение компонентов на поверхности металла.
37.3.2.3. Метод кольцевой печи
В 1954 гг. Вейс опубликовал работу, посвященную новому модифицированному способу проведения капельного анализа. Используя простую установку, он смог эффективно разделить микрограммовые количества неорганических веществ.
Основным оборудованием этого метода является кольцевая печь (рис. Д.27). Она состоит из цилиндрического блока с электрообогревом высотой около 35 мм и диаметром 55 мм.. В центре блока находится отверстие диаметром 22 мм. Алюминиевый блок снабжен электронагревателем с регулятором температуры. Температуру печи контролируют ртутным термометром. Печь установлена на штативе. Отверстие снизу освещают лампой накаливания. Над кольцевой печью укреплена капиллярная пипетка, которую можно поворачивать в двух направлениях. Пипетка отцентрована таким образом, чтобы она размещалась вертикально над центром отверстия. При работе
94
Д. Химический анализ
с водными растворами температуру поверхности кольцевой печи регулируют в пределах 105—110 °C.
Принцип действия кольцевой печи можно пояснить на следующем примере. В центр фильтровальной бумаги со средним размером пор, применяемой в количественном анализе, вносят
каплю 10~4 М раствора FeCl3. Бумагу кладут на кольцевую печь, нагретую до 105 °C, таким образом, чтобы пятно раствора находилось точно под капиллярной пипеткой. Капиллярную пипетку заполняют 0,05 М раствором НС1, прикоснувшись пипеткой к поверхности раствора кислоты. Как только заполненная пипетка с помощью держателя касается центра фильтровальной бумаги, бумага впитывает соляную кислоту, которая вымывает пятно FeCl3 к краю фильтровальной бумаги. В ходе перемещения жидкости к краю нагретой кольцевой печи раство-
37. Качественный анализ
95.
Рис. Д.28. Схема газогенератора для метода кольцевой печи.
1 — место подсоединения водоструйного насоса; 2 — бумажный фильтр с пятном; 3 — фильтр из ваты.
Рис. Д.29. Капиллярная пипетка для нанесения проб.
Рис. Д.30. Держатель для вырезанного из фильтра-круга.
ритель испаряется, а пятно остается в виде зоны диаметром 22 мм. Растворенное вещество (FeCl3), вымываемое соляной кислотой (объем примерно 10 пипеток), движется к периферии пятна и размещается там в виде узкой кольцевой зоны. При малом внутреннем диаметре пипетки процесс вымывания саморегулируется, так как из пипетки может вытечь только такое количество жидкости, которое адсорбирует бумага. На весь процесс вымывания затрачивают 1—2 мин. Бумагу снимают с печи, сушат специальной сушилкой и обрызгивают 1%-ным водным раствором K4[Fe(CN)6], Узкое голубое кольцо диаметром 22 мм свидетельствует о том, что все ионы железа вымыты из центра к краю фильтровальной бумаги.
Концентрация вещества в кольце на единицу поверхности в 3—10 раз больше, чем в исходном растворе. Если к периферии перемещаются пятна ионов нескольких веществ, высушенную-бумагу можно разрезать на несколько секторов. Поскольку не-
S6
Д. Химический анализ
Рис. Д.31. Способ вымывания растворенного осадка в кольцо.
I I
А
^-5
Рис. Д.32. Способ вымывания вещества из кольца в центр.
1 — стеклянное кольцо с внутренним диаметром 55 мм; 2 — стеклянное кольцо с наружным диаметром 53 мм; 3 — кольцевая зона; 4 — растворитель; 5 — стеклянная пластинка с отверстием 25 мм; 6 — асбестовая пластинка с отверстием 10 мм; 7 — стеклянная трубка диаметром 10 мм, длиной 170 мм для подвода тепла; в—-пламя микрогорелки.
раствором НС1 ионы железа и никеля мываний Fe и Ni концентрируются в к -лом CuS и кольцевой зоной железа и
риметр кольца равен 77 мм, пятно можно разрезать максимально на десять секторов, т. е. одновременно можно проводить обнаружение десяти ионов при их совместном присутствии.
Кольцевую печь можно использовать также для разделения ионов. Для этого один из компонентов смеси должен образовывать осадок на фильтровальной бумаге. Растворимые компоненты при этом вымывают в кольцевые зоны.
Осадки лучше всего получать, действуя газообразными реагентами. Вейс применял для этого установку для получения газов, изображенную на рис. Д.28. Принцип ее действия ясен из рисунка. Сероводород при этом получают взаимодействием ZnS с H2SO4 (1:1) или AI2S3 с водой.
Для нанесения капли пробы применяют специальные капиллярные пипетки, благодаря которым на бумагу попадают одинаковые по величине объемы пробы (рис. Д.29). Капиллярные пипетки заполняют, поднося их к поверхности раствора анализируемой пробы. Обычно их объем равен 1,5 мм3.
Пример аналитического разделения. Требуется разделить смесь меди, железа и никеля. Каплю слабосолянокислого раствора пробы вносят в центр круглого фильтра. Фильтр обрабатывают сероводородом в установке для получения газов. При этом CuS выпадает в осадок. Фильтр переносят на нагреваемую кольцевую печь и вымывают 0,05 М к периферии пятна. После 5—10 про-мьцевой зоне. Между внутренним пят-никеля находится «нейтральная зона»,
37. Качественный анализ
97
не содержащая ионов металла. Пятно CuS вырезают из фильтра с помощью сверла для пробок диаметром 12 мм. Оставшуюся часть фильтра делят на два сектора и идентифицируют железо и никель реакциями с Кл[Ге(СМ)б] и ди-метилглиоксимом. Вырезанный участок фильтра с пятном CuS переносят в специальный стеклянный держатель (рис. Д. 30). Медь можно идентифицировать окислением CuS парами брома с последующим обрызгиванием раствором а-бензоиноксима.
Если в вырезанном участке фильтра находятся осадки нескольких ионов, осадки растворяют, вырезанный участок помещают на середину круглого фильтра и смачивают каплей воды. Оба фильтра переносят на кольцевую печь. Применяя соответствующие растворители, вещества вымывают в кольцевую зону, где их идентифицируют обычными реакциями. Способ вымывания показан на рис. Д.31.
Для проведения большого числа разделений растворенные соединения необходимо перемещать из кольцевой зоны в центр фильтра, где растворитель должен испариться. Соответствующее приспособление показано на рис. Д.32. Края фильтров прижаты стеклянным кольцом с канавкой, заполненной растворителем. Растворитель испаряется с нагретой середины фильтра, и ионы растворенных веществ концентрируются в центре пятна.
По описанной методике можно быстро провести полный анализ микрограммовых количеств веществ. Вейс предложил схему разделения четырнадцати катионов в пробе раствора объемом 1,5 мм3. Продолжительность определения менее 15 мин.
Применение метода кольцевой печи для полуколичественного анализа. В методе кольцевой печи можно поддерживать постоянные значения всех параметров (температуры, концентрации), что дает возможность проводить по-луколичественный анализ веществ, сравнивая интенсивность получаемых окрасок с окраской стандартной кольцевой шкалы.
Для получения стандартной шкалы 1, 2, 4, 6, 8 и 10 капель стандартного раствора с известным содержанием компонентов (примерно 0,1 мг/см3) последовательно вымывают в кольцевую зону и проявляют при одинаковых условиях. Затем можно получить кольцевую зону определяемого вещества из нескольких капель его раствора и визуально сравнить ее окраску с окраской стандартных колец. Сравнивая число капель раствора, необходимых для получения окрашенного кольца, можно определить неизвестные концентрации веществ в растворе. Ошибка определения не превышает 10%. Метод упрощается при использовании стандартной универсальной шкалы. Упрощенный метод можно применять в том случае, если определяемый ион количественно осаждается в виде сульфида, имеющего определенный стехиометрический состав. При этом должна существовать возможность количественного перевода сульфида металла в сульфид серебра.
Стандартную шкалу сульфида серебра готовят из растворов соединений серебра различных известных концентраций. Сульфид определяемого металла переводят в кольцевую зону, в которой образуется Ag2S, и интенсивность возникающей окраски сравнивают со шкалой. Применяя факторы пересчета, можно рассчитать содержание определяемого металла. Метод кольцевой печи в сочетании с электрографией применяют для быстрого полуколичественного определения продуктов металлургических производств.
7—1880
38 Д.Химический анализ
38. ОСНОВЫ КОЛИЧЕСТВЕННОГО
ХИМИЧЕСКОГО АНАЛИЗА
38.1. Общие положения
Целью количественного анализа является определение содержания (количества) компонента в веществе. Обычно в анализируемом веществе присутствуют другие, часто мешающие определению компоненты, которые необходимо удалить' или перевести в такую форму, в которой они не мешают ходу анализа. Поэтому перед количественным определением обычно необходимо провести ряд операций, правильное выполнение которых часто оказывает большее влияние на результаты, чем сам метод анализа.
Вначале изучают литературу, в результате чего составляют точный план работы. Затем нужно приготовить и испытать необходимые реактивы. Как известно, дЛя успешной работы нужен хороший инструмент, этот принцип в значительной степени верен и для количественного анализа. Важным этапом является идентификация реактивов и проверка их чистоты. Идентификация дает ответ на вопрос, является лй покупной препарат, например, щавелевой кислоты, действительно С2Н2О4, а' не СгИгО^НгО или оксалатом натрия. Определение чистоты реактива помогает установить, какие примеси содержатся в препарате. Эти испытания должны стать привычными для каждого' студента-химика. Отговорки, что на это затрачивается слишком много времени, опровергаются практикой. Об этом свидетельствуют неудачные анализы, испорченные реактивы или даже взрывы из-за неправильно примененных реактивов.
Следует указать и на другие источники ошибок. Концентрированные растворы аммиака, которые долгое время хранили в стеклянных бутылях, содержат большие количества силикатов и карбонатов, что может привести к ошибкам, например, при определении фосфатов в виде магнийаммонийфосфата или при отделении группы сульфида аммония от группы щелочноземельных металлов. Поэтому целесообразно раствор аммиака готовить перед проведением анализа, пропуская аммиак в охлаждаемую льдом дистиллированную воду, и этот раствор хранить в полиэтиленовой бутыли. Такие же загрязнения характерны для растворов щелочей. Следует упомянуть, что азотная кислота, долгое время хранящаяся в лаборатории, может содержать хлориды. Необходимо обращать внимание на то, чтобы была известна формула вещества, применяемого в качестве реактива. Ц<ак правило, в лаборатории редко имеются в рас-поряженишнеоткрытые банки с реактивом в фабричной упаковке. снабженные этикеткой. Неверные - или неполные этикетки
38. Основы количественного химического анализа 99,
на.склянках с..реактивами,..в, которые реактивы отсыпаны .из, больших банок, могут легко привести к ошибкам .при пров'е.-., дении анализа. Например, часто путают дигидрофосфат натрия (NaH2PO4), гидрофосфат натрия (Na2HPO4) и частично также, фосфат натрия (Na3PO4). Часто важно знать содержание кри-. сталлизационной воды в соединении, так как только при этом! можно приготовить раствор нужной . концентрации.. В. со^ыц--тельных случаях необходимо определить содержание воды. Органические растворители, если они. не. находятся в.фабричцы^. упаковках, необходимо перегонять. .Следует также помнить том, что при проведении некоторых определений может мешать диоксид." углерода, находящийся в. дистиллированной воде, н<ь, пример, "при, титровании с фенолфталеином., Егр удаляют, квдяп че-нием. дистиллированной воды. . . >. • ,
, Все сказанное р. реактивах относится и к рабочей аппара-. туре. С такими дорогостоящими приборами,, как фотомртры,, потенциометры и т. д., обычно обращаются аккуратно,. но при работе со стаканами, мешалками, ершиками, фильтрами из пористого стекла об аккуратности нередко забывают.
Количественное вымывание осадков из стаканов, поцарапанных стеклянными палочками, обычно очень, затруднено. Образование царапин можно . предотвратить, используя электрические мешалки. При фильтровании растворов после осаждения можно применять мешалки, представляющие собой стеклянные пдлочки, конец которых на длину 3 см согнут под углом 45°. Естественно, стеклянные стаканы нельзя внутри тереть песком.
Каждый студент кроме. большой промывалки вместимостью 1-дм3 должсщ;иметь.,еще две или три небольшие промывалки вместимостью по, 10.0 .см?,. £ помощью этих, промывалок проще и удобнее процедить вымывание осадков и ополаскивание стаканов. Кроме-того, онц дают возможность промывать осадки теплой водой,, разбавленными,.кислотами или основаниями и другими растворами, что сложно: при .использовании .больших промывадок. , г : . . .
: После подготовки необходимых реактивов и .приборов приступают, к анализу стандартных образцов. Это значит, что перед определением; неизвестного содержания вещества в анализируемой пробе определяют содержание этого вещества в соединении с точно- известным его* количеством. Пусть, например, нужно провести гравиметрическое определение содержания хлорида в растворе поваренной соли. Анализ стандартных образцов в.этом случае заключается в.тщщ.что проводят нечетное число гравиметрических определений (обычно три) хлорида в раство-* Ре, содержащем точно взвешенное количество вещества, соответствующего формуле NaQL;-Совпадение результатов анализа стандартных .образцов, междуд.собой ^«воспроизводимость»), а также совпадение: этих результатов щдеоретическщ;рдесчцтан-7*
100
Д. Химический анализ
ными данными («точность») показывают студенту, в какой ме- I ре он овладел методом. I
В настоящее время анализы стандартных образцов не ис-® пользуют при обучении студентов. Но они являются очень важ-:Я ными, поскольку вырабатывают определенный стиль работы, * В ходе анализа стандартных образцов студент знакомится с влиянием различных факторов на точность результатов определений. Именно здесь он выбирает подходящий метод анализа. Методологически более правильно выполнить небольшое число количественных определений, но с предварительным анализом стандартных образцов, чем большое количество определений 4 без него. Опыты надо повторять до получения сходящихся ре- ж зультатов серии определений. Если нет веществ, точно соответ- и ствующих формуле, содержание компонента в стандартном об- Ц разце определяют, применяя надежный метод анализа. Если и при анализе стандартных образцов постоянно или периодически» получают неверные результаты, то это указывает на необходи-Я мость использования другого метода анализа. «
Анализ стандартных образцов нужно проводить в условиях,” аналогичных условиям последующих определений (концентрация веществ, присутствие мешающих определению ионов, температура осаждения и др.). Конечно, анализ сложных соединений, таких, как сплавы или минералы, нецелесообразно полностью воспроизводить на искусственных смесях. Но основные операции разделения, выделения и определения компонентов нужно отработать на стандартных образцах.
Студент должен подробно описывать весь ход экспериментальной работы от начала до конца. Отчет о работе следует составлять таким образом, чтобы посторонний человек смог без слишком больших затруднений продолжить эту работу. В отчете должно быть отражено не только, что проделано, но и почему это было сделано и какие результаты при этом получены. Все наблюдения должны быть записаны точно с указанием даты и времени проведения опыта. Должно стать правилом, что в конце каждого отчета приводится оценка ошибок эксперимента и даже их расчет, чтобы студент мог критически оценить результаты анализа и эту оценку выразить количественно.
В связи со все более широким проникновением микроэлектроники в аналитическую практику и использованием в лабораториях все более сложных и эффективно действующих аналитических приборов и систем иногда недооценивают значение точных химических методов анализа. Студент-химик должен как можно раньше понять, что во всех современных физических методах серийных анализов, например основанных на использовании явлений атомной абсорбции, УФ-эмиссии или рентгено-флуоресценции, необходимо применять градуировку, которую
38. Основы количественного химического анализа
ЮГ
проводят по стандартным образцам, химический состав которых обычно устанавливают химическими методами анализа* тщательно и многократно повторяя определения.
Правильная и рациональная оценка результатов анализа, как и вся аналитическая химия в целом, основана на использовании законов стехиометрии. Студент должен основательно усвоить этот материал и поупражняться в расчетах.
38.2. Основы практики количественного
химического анализа
38.2.1. Взвешивание
Важнейшей частью аналитических весов является двухплечевое коромысло. Примем, что оно абсолютно жесткое и может вращаться вокруг горизонтальной оси. На одно плечо коромысла действует сила тяжести взвешиваемого тела. Момент вращения при этом должен компенсироваться встречным моментом одинаковой величины. Это достигается следующими приемами: а) нагружают такой же массой второе плечо коромысла равноплечих весов; б) при неравенстве масс изменяют длину плеча коромысла, применяя подвижную гирьку (на аналитических весах — с помощью рейтера); в) центр тяжести коромысла располагают ниже оси вращения. При отклонении возникает встречный вращающий момент, пропорциональный синусу угла отклонения (рычажные весы, почтовые весы). Все три приема используют при взвешивании на аналитических весах.
Полная компенсация эталонами определенной массы (гирьками) требует слишком больших затрат времени и применения разновесов с массой гирек до 0,1 мг или даже меньше. Поэтому гирьки, находящиеся в разновесе, используют до самой маленькой (способ а) и затем, в случае недемпфированных аналитических весов, проводят компенсацию, передвигая рейтер (обычно массой 10 мг) по градуированному коромыслу весов (способ б). Наконец, по отклонению стрелки от нуля (способ в) можно определить разность масс и прибавить это значение к полученному результату или вычесть из него, если известна чувствительность весов.
Чувствительность Е аналитических весов определяется числом делений шкалы Да, на которые отклоняется стрелка весов под действием массы Д/тг (обычно 1 мг):
E = \a/&m (1)
Чувствительность весов несколько зависит от их нагрузки. Эта зависимость показана на рис. Д.ЗЗ. Таким образом, при точном взвешивании необходимо учитывать изменение чувствительности весов при различной нагрузке.
10?
Д. Химич^ский анализ
. Рис. Д,33. Зависимость, чувствительности весов qi .нагрузки.
Для выявления сути этой зависимости рассмотрим схему, приведенную, на рис. Д.34, .на которой в увеличенном масштабе показан небольшой изгиб коромысла, возникающий при нагрузке.
В состоянии равновесия сумма моментов вращения равна нулю. Поскольку момент вращения зависит от силы и расстояния от места действия силы
Рис. Д.34. Схема изгиба коромысла весов.
Pt, Рг, Рз— точки вращения (опоры); Л1Н-Аллг определяемая неизвестная масса; М — масса эталонов (гирек); ср — угол отклонения, вызываемого перегрузкой Ат; а — угол изгиба коромысла под действием нагрузки; £ — положение центра тяжести коромысла перед отклонением; £'— положение центра тяжести коромысла после отклонения на угол ср; s — расстояние от центра тяжести коромысла до точки вращения Pi; / — длина плечей коромысла; Мв — масса коромысла.
до точки вращения и это расстояние (Л) на левом плече определяют как A — I cos(a + <p), возникает момент вращения 0:
0/ — Ы (М + Xm) cos (a + ср) (2)
где 6 — гравитационная постоянная. На правое плечо действуют два вращающих момента:
0Г1 = ЫМ cos (a — ср) и 0Г2 — bsMB sin <р (3), (4)
Уравнения (3) и (2) аналогичны. Уравнение (4) объясняется тем, что массу коромысла тИв можно считать сосредоточенной в точке S'. Тогда сила равна ЬМв и расстояние s sin ср.
38. Основы количественного1 Хймического анализа
.. ЮЗ
В состоянии равновесия:'- и ’
Ы (М + Am) cos (а + <р) — ЫМ cos (а — ср) — bsMrj sin ср = О
Применяя правило суммирования (cos (ср± а) = cos ср cos a =Fsin ср sin а) и разделив уравнение наЬ и на sin ср, получим
/Amcosa <
tg <Р — i (2М + Am) sin а ( '
Поскольку при небольшом отклонении стрелки весов tgcp«Aa/?z, (lz— длина стрелки весов), из уравнения (1) получим выражение для чувствительности равноплечих весов в зависимости от величины нагрузки:
ZzZcosa
£ = Mgs + Z (2Л4 + Дт) sin а ( '
Из этого выражения можно сделать следующие выводы: чувствительность равноплечих весов тем больше, чем меньше масса Мв коромысла весов, расстояние s между точкой действия силы тяжести и точкой вращения коромысла весов, угол отклонения ср и нагрузка (2Л4+Дт) на коромысло, и чем больше длина стрелки весов lz-
Только в идеальном случае, при условии, что а=0 независимо от нагрузки на коромысло, чувствительность не зависела бы от нагрузки, поскольку из уравнения (6) следует, что
Е я lzl/M5s (7)
При увеличении I числитель в уравнении (6) растет несколько быстрее знаменателя, следовательно, чувствительность, казалось бы, должна возрастать. В соответствии с уравнением (7) она стала бы даже прямо пропорциональна I. Но если вспомнить, что при изменении длины Z масса коромысла Мв должна возрастать, чтобы прочность не изменялась (маленькое значение а), то очевидно, что с увеличением I чувствительность не увеличивается, а даже уменьшается. Поэтому в настоящее время оптимальной считают среднюю длину коромысла.
Поскольку в чувствительных весах масса Мд коромысла, а также угол изгиба а, вызванный нагрузкой на коромысло, должны быть небольшими, коромысло весов изготавливают из наиболее легких и высокопрочных материалов. Поскольку прочность стержня на изгиб пропорциональна его ширине и высоте (в третьей степени), нужно применять коромысла высокие, узкие и с прорезями.
Условие а=0 соблюдается только для определенной средней нагрузки на коромысло, что связано с типом весов. Тип весов целесообразно выбрать таким образом, чтобы они позволяли взвешивать обычно используемые в работе массы веществ. При этой нагрузке чувствительность весов должна быть максимальной. Величину s из уравнения (7) можно изменить с помощью регулировочного винта на коромысле весов, что приводит к изменению чувствительности весов.
При очень точных взвешиваниях необходимо помнить о том, что оба плеча коромысла весов не абсолютно равны (допуск при изготовлении). Эту небольшую погрешность можно устранить, используя метод замещения Борда или метод двойного взвешивания Ф. Гаусса. Описание методов можно найти в литературе*.
* Рудо Н. М. Лабораторные весы и точное взвешивание. — М.: Стап-Даптиздат, 1963. — Прим, персе.
104
Д. Химический анализ
С недавнего времени для аналитических целей начали широко применять одночашечные весы, позволяющие быстрее и точнее проводить взвешивание, чем на двухчашечных весах. Одночашечные весы являются неравноплечими и работают в основном по принципу замещения (рис. Д.35). Они отличаются тем, что при ненагруженной чашке весов коромысло на этом же
Рис. Д.35. Принципиальная схема одночашечных неравноплечих аналитических весов.
JWi — масса поднимаемых гирек; Мг —масса противовеса, жестко укрепленного на коро^ мысле.
конце нагружено помещенными на стержне гирями, которые можно выборочно поднимать специальным механизмом. Этот набор гирь уравновешен противовесом, жестко укрепленным на другом конце коромысла. Когда на чашку весов помещают взвешиваемый предмет, после разарретирования весов равновесие коромысла под действием результирующего вращающего момента нарушается. Для уравновешивания гири поднимают с коромысла до тех пор, пока оно не вернется в положение, близкое к исходному. Масса поднятых гирь, а также остаточный вращающий момент определяют по цифровому устройству. Такой принцип действия дает возможность проводить взвешива
38. Основы количественного химического анализа
105
ния при одной и той же нагрузке коромысла и призм весов независимо от массы взвешиваемого предмета, что устраняет зависимость чувствительности весов от нагрузки, характерную для равноплечих двухчашечных весов. Кроме того, в неравноплечих одночашечных весах исключается погрешность, связанная с практически всегда существующей разницей плеч в равноплечих весах. В одночашечных неравноплечих весах делают две призмы (вместо трех в случае равноплечих), что является еще одним конструктивным достоинством.
Обычно одночашечные весы снабжены магнитным демпферным устройством и имеют очень небольшое время колебаний. Дальнейшим конструктивным улучшением весов является применение встроенного специального устройства, которое сразу после разарретирования весов дает возможность определить примерную массу взвешиваемого предмета, облегчая тем самым подбор гирь. В некоторых типах современных весов вращающий момент компенсируют не механическим путем, снимая гири, а силой электромагнитного поля, регулируемого электронными устройствами. Электронные весы снабжены цифровыми устройствами и могут быть подключены к вычислительным машинам, печатающим устройствам и самописцам.
Поправка на взвешивание в воздухе. Негласно принимаемое допущение о том, что весы практически мгновенно реагируют на массу взвешиваемого предмета, верно только при взвешивании в вакууме или при равных объемах сравниваемых масс. Как в жидкости, так и в воздухе (хотя и в меньшей степени), все вещества испытывают влияние выталкивающей силы, пропорциональной объему вещества, которое необходимо учитывать, когда массу нужно определить с возможно большей точностью.
Для расчета выталкивающей силы обозначим через Мд силу воздействия воздуха на неизвестную массу М вещества (по сравнению с вакуумом), а через Gl— силу воздействия воздуха на массу гирек, необходимых для компенсации массы М. Тогда
Ml = Gl (8)
Выталкивающая сила воздуха определяется произведением объема массы М на плотность воздуха (Умрд). Аналогичные рассуждения можно применить для Gl, при этом уравнение (8) примет вид
M-VmPl = G-VcPl или M = G4-Pl(Vai-Vg) (9),(10) где G — масса гирек в вакууме.
Поскольку объем любого вещества равен частному от деления его массы на плотность, получим
M = G-tpLf—-----— 'j (11).
1 L ( Рм Pg ) ' ’
где pL — плотность воздуха (pl=0,001293 г/см3); рм—плот-
106
Д, Химический анализ
ность материала вещества массой. М', р0 — плотность гирек (при измерении ро = 8,4 г/см3).
В уравнении (11) величину- М в Скобках можно считать примерно равной G, поскольку разйость очень'невелика:и величина дроби в значительно большей степени зависит от сильно различающихся значений плотности. Тогда получим
M = G+Gp^-^ (12)
Оцените на примерах ошибку, возникающую без введения поправки на взвешивание в воздухе!
Выполнение взвешивания. От воздействия потока воздуха весы защищают стеклянным футляром. Во избежание конвекционных потоков воздуха при взвешивании температура объекта не должна отличаться от температуры внутри футляра; следует также избегать тепловых излучений. Поэтому взвешиваемые вещества, помещаемые на чашки весов через боковые дверцы, предварительно должны приобрести температуру весов (тигли после прокаливания выдерживают в эксикаторе рядом с весами до выравнивания температур). Изменение нагрузок на чашках весов нужно проводить на арретированных весах во избежание повреждений из-за вибраций крайне чувствительных опорных призм. По этой же причине открывать арретир нужно очень осторожно.
Вопросы, и задания
1. Объясните ход кривой на рнс. Д.ЗЗ. . и
2. Рассчитайте ошибку при взвешивании 10 г алюминия (Pai=2,7 г/см3) с применением гирек (рм = 8,4 г/см3) без учета поправки на взвешивание в воздухе. ' ' •1 ‘'1 1 '
" 3. Найдите в литературе сведения о калибровке гирек.
' ‘ - 1 5 • • . 1 . __
38.2.2. Гравиметрия Й ]
38.2.2.1.' • Общие полайфиЯя г-
Сущность гравиметрии состоит в переводе определяемого'йёще-стца в малорастворимое соединение известного состава, очистке осадка от загрязнений и взвешивания. По массе осадка и его формуле можно рассчитать количество определяемого вещества.
Реакции, применяемые в гравиметрических определениях, должны удовлетворять следующим требованиям: а) осадок должен быть достаточно малорастворимым; б) осадитель должен быть специфичным для определяемого компонента, т. е. це соосаждать другие компоненты раствора, которые затем
38. Основы количественного -химического анализа
'167
нельзя , будет удалить из осадка; в) осадок должен быстро и легко фильтроваться; г) необходимо, чтобы ^осадок можно было воспроизводимо переводить в устойчивую гравиметрическую форму определенного состава; д) гравиметрический фактор для определяемого вещества должен быть как можно меньшим. Первые три условия не требуют разъяснений. Четвертое определяется тем, что обычно форма осаждения непригодна для взвешивания, поскольку или осадок нестабилен, или его состав зависит от условий осаждения и часто неточен. Поэтому перед взвешиванием форму осаждения необходимо перевести в устойчивое соединение определенного состава — гравиметрическую форму.
Пример. Для гравиметрического определения железа его осаждают в виде гидроксида железа(Ш) Fe(OH)3-aq. Этот объемистый осадок красно-корпч-иевого цвета содержит большие и непостоянные количества воды и адсорбирует большие количества посторонних нонов, особенно хлоридов. Он не пригоден для прямого взвешивания. Для перевода в гравиметрическую форму осадок фильтруют, тщательно промывают, сушат и прокаливают при 800 °C. Промыванием удаляют основную часть адсорбированного хлорида. Остаток хлорида испаряют прн прокаливании. При температуре прокаливания гидроксид железа(Ш) переводят в оксид железа(Ш) РегОз, пригодный для взве-шиваиия:
2Fe(OH)3--► Fe2O3 + ЗН2О (13)
Другие Примеры перевода осаждаемой формы в гравиметрическую форму:
Sb2S3 --► Sb2O3; КС1О3 —-► КС1; СаС2О4 --► СаО (14)
Если гравиметрическая форма имеет состав АаВв, а ее взвешенная масса mw, то массу определяемого вещества рассчитывают по формуле
т.д = mwvAMA/Mw (15)
где МА, М«г — молярные массы А и гравиметрической формы АаВв соответственно; va— отношение стехиометрических коэффициентов. Произведение vaMa/Mw называют гравиметрическим фактором /; для многих веществ составления таблицы этих факторов.
!Еслй-состав осадка- при точно соблюдаемый условиях осаждения постоянен, но точно не известен, то,1 проанализировав ряд стандартных образцов, по уравйейийгопределяют так порываемый эмпирический, фактор., При использовании эмпирического фактора правильные рёзультат'ы получают только при особенно тщательном соблюдении методики рнализа.
Обычно иуЖно знать не абсолютное количество /па, а долю р вещества' А в массе навески тЕ веществу Е, взятой для анализа. Эту долю определяют по уравнению '
. а' я ’ р- !Па —
108
Д. Химический анализ
В соответствии с законом распространения ошибок относительная ошибка Fr доли р равна
<17>
При логарифмировании до четвертого знака после запятой величина (df/f)2 пренебрежимо мала, так что можно записать
F (18)
г I/ \ W ) 1 \ тЕ J '
Если из уравнения (16) выразить mw и тЕ через тА и учесть, что ошибки взвешивания dmE и dmw примерно равны, так что dmE~dmw=dm, то получим
: У>.„.‘L ^“т/(£)>+«. , . <19>
Из уравнения (19) вытекают условия, выполнение которых необходимо для получения точных результатов гравиметрического .•определения: а) ошибка взвешивания dm должна быть малой;
•б), масса тА взвешиваемого элемента А (в виде навески тЕ и массы весовой формы mw) должна быть как можно большей;
; в)- фактор f и доля р должны быть небольшими.
Значение величины фактора f следует пояснить на нескольких примерах. . При электрогравиметрическом определении металлов f имеет неудовлетворительно большую величину, часто равную единице или даже больше, как, например, при определении содержания оксида никеля в веществе при электролитическом выделении никеля, где = 1,2725. При определении
фосфата в виде молибдатофосфата эмпирический фактор (по Финкенеру) имеет хорошую величину: /=0,01639. Осаждением сульфата бария лучше определять содержание в веществе серы (/=0,13738), чем бария (/=0,58843).
В некоторых случаях компоненты смеси можно определять без их предварительного отделения. Например, анализ смеси КС1 и КВт можно проводить следующим методом: смесь взвеН шивают (mJ, растворяют в воде и к раствору добавляют нитрат серебра. Получают суммарную массу галогенидов т2. Мас-! су КВ г определяют из уравнения
mKBr= 5,58m!—2,90m2
(20)
Факторы mi и т2 в уравнении (20) имеют довольно значительную величину, так что ошибки взвешивания т\ и т2 возрастают до больших значений и возникает относительно большая ошибка определения тквг- Поэтому косвенные методы анализа отличаются значительно меньшей точностью, чем прямые, и по возможности их следует избегать.
38. Основы количественного химического анализа
109
.38.2.2.2. Основные операции гравиметрии
К основным операциям гравиметрического анализа относятся: осаждение, фильтрование, промывание, высушивание, озоление, прокаливание и взвешивание.
Фильтрование и промывание. После осаждения осадка его .нужно количественно отделить от маточного раствора. В гравиметрии это обычно осуществляют фильтрованием. В общем для фильтрования применяют фильтры с различной величиной пор и известным небольшим содержанием золы, выпускаемые промышленностью. Фильтры выбирают в зависимости от свойств осадка. Для крупнокристаллических и гелеобразных осадков, таких, например, как гидроксид алюминия, применяют крупнопористые фильтры, для мелкокристаллических, .например сульфата бария, — мелкопористые плотные фильтру. Фильтры при этом помещают в воронки таким образом, чтобы-трубка воронки была постоянно заполнена жидкостью. Правильное размещение фильтра в воронке и выбор фильтра часто являются решающими условиями продолжительности гравиметрического .анализа.
В ряде случаев вместо бумажных фильтров применяют стеклянные нутч-фильтры (фритты) из спекшегося пористого стекла . (применимы до 450 °C) или фарфоровые фильтрующие тигли ‘(применимы до 1000°C). Эти типы фильтров особенно рекомендуют применять, если при промывании осадка на фильтре он может восстановиться.
Чтобы получить наиболее чистый осадок, его нужно тщательно отмыть от маточного раствора. Нельзя проводить промывание произвольно выбранным большим количеством воды, поскольку значительная часть осадка при этом может раствориться (например, растворимость BaSCU при 20 °C равна 0,2 мг/100 г воды). Обычно промывают отмеренным объемом промывной жидкости ('"'50 см3) как можно меньшими порциями. Это требование обусловлено тем, что после фильтрования на осадке может еще находиться vm см3 маточного раствора с концентрацией посторонних ионов Со. При промывании промывной жидкостью объемом vw см3 концентрация посторонних ионов ci после первого промывания равна
При повторном промывании таким же объемом жидкости и задерживании vm см3 маточного раствора на осадке концентрация посторонних ионов с2 составляет
;с2 = с° <22>
vm \ -j- Vw ]
1 10- ——— ’„Ji—Дд( -:—-— ------------------------------— —
После п промыванйй'хо'нЙ^ЙТрациЙгс^райнаи'0'"‘' .........
’•!!:.'!>l?l i:.:n.„;li; : ;i!UHlii(31ll ШШНПК . -b J
.люпит-п ..шшшшг Vm-hUfe J;)fflna<ttr.<r.-.no „дао ' q
„ i /шшшш.с: : i
Ь(}ли степень вымывания обозаа^и^.^к/р^^у^иди i
-ittiHt- £ ' _ / Pm'ir .‘V*- : лшйП.лдао; ,щ
."НИШ £ ( ад-ф-Ц»-/ИГ': . ШР’Щ:: ГЛ'. Hl! ' 3
II!!! I bi''!''!:1 ’ ; :: .; lb) • nihl'Uitn г!;1:Ш(Пг’ ; 1 |
. . 'Уравнение (24) показывает, что многократное промывание! .небольшими объемами промывнойл жидкости действеннее, чем! однократное промывание большим.объемом. 1
Пример. На .осадке мог задержаться 1 см3 маточного раствора. В первом! опыте на однократное промывание осадка затрачено 10 см3 промывной жид-1 кости, во втором‘опыте промывание проводили 10 раз порциями по 1 см*' промывной жидкости. В первом опыте <р = 0,1, во втором опыте <p»0,001J Таким образом, промывание во втором опыте в 100 раз эффективнее, чем<в| первом. ’
Лучше, чем на фильтре, осадки можно промыть декантацией в стакане или в центрифужной пробирке. Обычно осадки промывают не чистой водой, а разбавленными растворами электролитов, содержащих ионы, одноименные с осадком, и улетучивающихся при нагревании (соли аммония, НС! и др.), j Добавка электролита к промывной жидкости уменьшает рас- я творимость промываемого осадка и тем самым пептизацию.; Я
Высушивание, озоление, прокаливание. После отфильтровы- Ф вания осадка бумажный фильтр перед взвешиванием нужно сжечь. Для этого влажный осадок с фильтром складывают и .озоляют в наклонно поставленном тигле .(для лучшей циркуля-.ции кислорода) на небольшом пламени бунзеновской горелки. Температура озоления должна быть, как можно, меньше во избежание восстановления осадка углем фильтра. .После озоле-,ния .тигель ,с осадком прокаливают при температуре, указанной в . методике анализа. Необходимо помнить,., что в раскаленный платиновый тигель очень легко может диффундировать водород,. что. может, привести к восстановлению даже , устойчивых соединений. Этому способствуют тацже реакции,, вздимодейдт- л вия продукта восстановления с платиной или его сплавление М ней. Такое поведение характерно для соединений всех тяжелыяИ м'еталлов, а также силикатов, которые могут восстанавливатьс^И с образованием элементарного кремния, образующего нпзкс^И плавкий силицид'платины. При: этом можно -легко раЗрушит^И дорогие платиновые - сосуды. Поэтому легко восетай'аВливай^И щиеся соединения нужно прокаливать'вг фарфоровых; Тиглях электрических печах.
Операция озоления фильтра: не нужна, если использую^И фарфоровый фильтрующий тигель. Этот фильтр удобен в р^И
38. Основы количественного химического анализа
1:11
боте. Перед употреблением необходимо- убедиться в том,' что фритта прочно вставлена в тигель и не перемещается в нем.
Некоторые осадки можно отсасывать- на стеклянных порис? тых пластинках и затем отмывать от захваченной осадком воды спиртом, эфиром или ацетоном (например, магнийаммоний-фосфат или аммониймолибдатофосфат захватывают до 6 моль воды). Иногда подобные осадки окончательно высушивают в вакуумированном эксикаторе; при этом необходимо предварительно в течение достаточно .долгого времени просасывать через фильтр и осадок воздух, так как оставшиеся в дне фильтрую? щегося тигля следы растворителя при последующем вакуумировании могут привести к разбрызгиванию-вещества. .
38.2.3. Титриметрический (объемный) анализ
Л1' , 'illr
38.2.3.1. Общие положения, ^триметрии лч;
'.ni;1 .-ш: i-ш.-
Реакции, применяемые в объемном анализе, должны удовлетворять следующим требованиям: протекать быстро и количественно; быть стехиометрически однозначными; должна существовать возможность определения конца протекания реакции.
Сущность титриметрических методов анализа состоит в том, что к анализируемому раствору известной концентрации добавляют титрант до тех пор, пока определенная система индикации не укажет на конец протекания реакции. Для установления конечной точки могут быть применены колориметрические индикаторы, изменение окраски которых наблюдают визуально, а также физико-химические методы. В большинстве случаев измеряют объем израсходованного титранта. Все методы, основан-? ные на использовании указанных принципов, относятся к обън емкому анализу/ - ' чп)
Основным достоинством титриметрии по сравнению::С гравиз. метрией-является быстрота. Кроме того, можно1 определять ве-н щества, которые не образуют малорастворимых соединений или;' Для которых нельзя получить гравиметрическую- форму,' удовлетворяющую предъявляемым к . ней требованиям. с*С другой стороны, при сравнимых затратах времени методы;,-титриметри-ческого анализа менее точны, чем гравиметрические, . так. как массу -вещества можно определить точнее, чем объем. Однако с учетом всех источников ошибок в некоторых методах титрования можно получить точность, соответствующую наибольшей точности определения известных атомных масс. ;
Расход титранта можно определить также взвешиванием. Этот метод называется гравиметрическим титрованиемс.н'
112
Ц. Химический анализ
38.2.3.2. Основные операции титриметрии
Измерение объемов мерной посудой. К мерной посуде, применяемой в титриметрическом анализе, относятся: простые пипетки, градуированные пипетки, бюретки и мерные колбы. На мерной посуде имеются кольцевые метки, которые с известной ошибкой ограничивают определенный объем.
Посуду для титриметрии перед употреблением надо тща- тельно очистить. При долгом стоянии на стенках стеклянной' посуды образуется жировая пленка, что может привести к су- = щественным ошибкам при выполнении анализа. Для очистки мерной посуды в объемном анализе применяют смесь бихромата калия с концентрированной азотной кислотой. Смесь бихромата калия с серной кислотой применять не рекомендуется, поскольку она действует как эффективный окислитель только в нагретом состоянии. Кроме того, смесь бихромата с серной кис-, лотой гигроскопична и при разбавлении быстро теряет свою* эффективность. Стеклянное оборудование можно быстро очистить щелочным раствором перманганата калия и концентрированной соляной кислотой (защищать глаза!). Очищаемое оборудование оставляют стоять со щелочным раствором перманганата калия в течение примерно 15 мин. (Бюретки с пришлифованными кранами или мерные колбы с пришлифованными пробками нельзя оставлять на ночь заполненными щелочным, раствором перманганата, так как при этом краны или соответственно пробки заклинивает.) После сливания раствора перманганата сосуд заполняют конц. НС1 (без промежуточного, ополаскивания водой) и снова оставляют стоять примерно 15 мин (под тягой!); затем раствор сливают и посуду ополаскивают водой, последний раз — дистиллированной.
Высушивание сосудов можно проводить многократно (не менее пяти раз) ополаскивая сосуд маленькими порциями измеряемого раствора. Перед каждым ополаскиванием дают полностью стечь каплям. Если измеряемый раствор нельзя расходовать, пипетки или бюретки лучше сушить, просасывая через них воздух, причем входное отверстие при этом закрывают кусочком фильтровальной бумаги, чтобы уберечь кончик.
Мерную посуду ни в коем случае нельзя сушить нагреванием, например, в сушильном шкафу (почему?).
Градуировка. Обычно истинный объем мерной посуды в большей или меньшей степени отличается от указанного на ней номинального объема. Для получения высокой точности нужно измерить вместимость посуды до метки или заново определить положение метки в соответствии с номинальным объемом. Эту операцию называют градуировкой.
Мерную посуду, применяемую в титриметрическом анализе, в настоящее время градуируют в кубических сантиметрах
38. Основы, количественного химического анализа
ПФ
(см3), иногда еще в миллилитрах (мл), при этом в соответствии с интернациональной системой единиц (СИ), принятой в 1960 г., 1 л = 1 дм3=1-10~3 м3.
При градуировке сосуда его заполняют жидкостью (ртутью, водой) точно известной плотности до определенного уровня, взвешиванием определяют массу жидкости и рассчитывают занимаемый ею объем при температуре градуировки t. Обычно определяют массу жидкости, занимающую определенный объем при температуре 20 °C — стандартной температуре градуировки. Таким образом нужно рассчитать массу жидкости, взвешенную при температуре t, которая занимает нужный объем при 20 °C.
Пример. Градуировка колбы вместимостью 1000 см3 при заполнении ее водой. При уравновешивании 1000 см3 воды на рычажных весах при температуре t на воздухе с помощью гирек mM = mw, где mw и тм— массы воды и гири соответственно в воздухе. Значение mw меньше, чем указанная на гире номинальная масса тм, у, определенная в вакууме, вследствие выталкивающей силы воздуха.
Если массу в воздухе выразить через массу в вакууме (тм. v или mv, и), получим
«aiv ,„еч
рь = mw,V — Рй7 рд (25)
Поскольку (тму/рм')рс<^тму, для простоты расчета с достаточным приближением можем в этом выражении заменить тм, у (массу гири в вакууме) на mw, у (массу воды), т. е. принять тм, v^mw, v. Кроме того, mw, у — это масса 1000 см3 воды, поэтому
mM,v ~ mw\v — Pw ЮОО (26)
Подставив выражение (26) в уравнение (25), получим
mM,V — (lOOOpnz/pAi) PL = Ю00 (paz — рд) (27)
Откуда mM,v = 1000 [ру — р/, (1 — Рщ/Рм)] (28)
Поскольку масса воды, уравновешенная номинальной массой тм, у гирн в воздухе при температуре t, занимает при этой температуре объем 1000 см3, а количество воды т, занимающее при 20 °C объем 1000 см3, при температуре t имеет объем щ, то
т = f/^M.v/lOOO (29>
Нужно также учесть зависимость изменения объема мерной колбы от температуры по уравнению
vt = 1000 + ₽ (1 — 20) (30)
где Р —коэффициент объемного расширения стекла.
Подстановка в уравнение (29) уравнений (28) и (30) окончательно дает
т = [рУ — PL (1 — Рщ-'/Рлт)] [Ю00 + Р (t — 20)] (31)
где т— масса воды, взвешенная прн температуре t в воздухе н занимающая при 20 °C объем 1000 см3.
8—1880
114
Д. Химическийанализу,-. ьу.у
.Точное определение объема и нанесение: новой .метки обычно ре составляет труда. Для этого несколько.раз определяют •объем градуируемого сосуда до метки, взвешивая сосуд, заполненный водой иди ртутью, с последующим расчетом (Действительного объема. ..-(Студент должен- уметы вывести-соответствующую формулу для расчета!) .ъг.д.щ • т .;
Аналогично градуируют бюретки. С этой целью!бюретку, вместимостью, например, 50 см3, заполняютдводой и мениск-ус!-тганавливают на 0. Дают воде медленно .'стекать в. сукой* взве- ц;.-.. .-.т.-"- ысн 11
” - <Рис. Д.36. Градуировочная кривая бюретки;-'-'-
• ГГГИ11 -il.VC
пленный стаканчик до тех пор, пока мениск жидкости не установится на делении 5 см3. Воду в стаканчике взвешивают. Из нечетного числа таких определений (обычно 3 или 5) рассчитывают среднее значение и пересчитывают его на температуру 20°C. Затем взвешивают объемы воды от 0 до 10 см3, от 0 до 15 см3 и г. д. В прямоугольной системекоординат строят график зависимости отклонений Av между найденным и теоретическим (т. е. тем, что обозначено на бюретке) значениями в зависимости от теоретического значения. Пример градуировочной кривой представлен на рис. ’Д.ЗбД’Такая кривая является важ-нейшейлхарактеристикой бюрет-кида; .- ичк-м’ а :
При градуировке пипеток с одной меткой измеряемую жидкость засасывают в пипетку вцтше метки. Кончик пипетки осторожно осушают мягким фильтром. Дают мениску жидкости медленно опуститься до метки...-Кончиком пипетки, в вертикальном положении прикасаются к стенке наклонно стоящего стакана, дают стечь жидкости и. выдерживают пипетку в этом положении еще в течение 15 с?-Таким образом измеряют действительней объем пипетки..,.^рдко§ть..и^.д1ццетки, нельзя _выду-вать или вытряхивать. Д. гд;5 jy
4 дян
. о:
38^0сновы^т'личёств^ио1Х>2^мйческога-лналим-------——-Н5»
. ;Ошибки титрования. При проведении т'иТррйания могут возникать следующие ошибки':- а1) градуировки/’’б) Аемйёратурц^я;. в) отсчета; т) вытекания; д) см’ачи’ваемости; ,!ё) капельная/ .! Ошибка градуировки-возникает в- том случае, кЬГдй отметка на посуде не соответствует истинному значению.’ Эту ошибку: можно устранить градуировкой. Если мерную ' посуду применяют при температуре, отличной от температуры градуировки, то нужно вносить соответствующую температурную поправку. Ошибка отсчета может быть вызвана параллаксом и нечетким определением уровня окрашенных в темный цв^т' жидкостей (например, растворов КМпОд, КДз и т. п.). Ошибка вытекания может возникнуть, если при выливании жидкости номерного сосуда (пипетки, бюретки) не соблюдают времени,, определенного при градуировке (ровно 15 с), или при пипетй-ровании вязких жидкостей. Ошибка вытекания обычно имеет знак минус. При выдувании или вытряхивании жидкости из-пипетки ошибка может иметь также и плюсовое значение. Ошибка смачиваемости обусловлена жировой пленкой, находящейся на внутренних стенках мерной посуды, поэтому при проведении объемных определений необходимо следить за чистотой посуды..
В то время как все рассмотренные ошибки практически могут быть устранены, капельной ошибки избежать нельзя. Она* возникает оттого, что последняя капля титранта, необходимая для изменения окраски индикатора, является избыточной, т.. е. титранта вносят больше, чем это нужно для достижения точкиг эквивалентности.
38.2.3.3. . Основные операции гравиметрического титрования:
В методе гравиметрического титрования измеряют не объемны а массы растворов. Например, для определения количества титранта, затраченного на титрование, бюретку для гравимэдр.-р-ческого титрования взвешивают до и после титрования.. Аналогично поступают-ДЙя определения количеств жидкостей^ Таранты готовят растворением реагента в 1 кг1 растворителй/ а в 1 дм3 раствора. , .. ’
- ВаЖйейиТим 'бборудоваЖУм для; прЬвё^йпГй- гравймет^ичр-скотО:<!кйТрЬва,Ййй':1являётся!<'Грайиметричё'скйЯ’/(1 пййётка' '/сна'б-'.жеийй’я >вйй¥б'вЖ1 йёха'МйШм^дл'А ‘доЗЙрАв’а'й'Йя 'ЬЧейь кйалых объемов и применяемая так' жё; кйк' Ьктрётка.’/'П^кйцйй’действия гравиМ'ёТрййёской 'пййё'тктЕтот1 Нйпет-
’Кё’НЛй 1йедиЦЙйеком;'Щпрйце.1“ гл--п. .
Гравиметрическое титрование имеет ряД1 ДоСТОиНств, ‘Однй'м Из ййх являёГся йёнужнЬсть|трРдуирЬвкЙ.,/‘ЙоскоЛк'ку'' измеряют1 чм-аСсу:,раствордв; из’мёнбния' Т'емпеба'тУры ‘'не оказывают й:ЛйЯййя‘: на1 •-результаты тйтрбйаний. Дакее, грквйЖт^иАёсйие пипетки после очистки можно сушить при высоких темпер'Рт’у-
8*
116
Д. Химический анализ
$>ах (120°C). Незначительные их загрязнения, например жировая пленка, не сказываются на результатах определения в отличие от объемных методов анализа. Поскольку в этом методе не нужна смазка для шлифов, капилляр бюретки не забивается ею в отличие от бюреток с кранами.
Кроме того, в этом методе не существует ошибки, связанной с выливанием. Можно пипетировать вязкие растворы или использовать их в качестве титрантов. Так как в процессе титрования кончик капилляра опущен в титруемое вещество, а титрант дозируют винтовым механизмом, подача титранта происходит практически непрерывно, чем устраняется капельная ошибка. Отпадает трудность фиксирования положения мениска вследствие параллакса или в случае непрозрачных или окрашенных растворов.
Воспроизводимость результатов гравиметрического титрования по сравнению с обычным титрованием на порядок выше. Бюретки для гравиметрического титрования проще заполнять, чем обычные бюретки, что особенно важно при работе с микроколичествами. •;
Основным недостатком, метода является необходимость -большого количества взвешиваний. Однако, применяя современные полуавтоматические весы, можно существенно сократить затраты времени. В этом случае гравиметрическое титрование дает даже выигрыш во времени по сравнению с обычным! титрованием. ' |
38.2.3.4. Т итранты |
Для приготовления раствора титранта взвешивают определенное количество вещества, растворяют в мерной колбе и доливают до определенного объема растворителем, обычно водой. Идеальный титрант должен удовлетворять следующим требованиям:
1. Должна существовать возможность простого и воспроизводимого приготовления. Состав титранта должен точно соответствовать формуле.
2. Раствор должен быть устойчивым и не реагировать с компонентами воздуха, т. е. должен быть нечувствительным к действию кислорода, негигроскопичным, не поглощать диоксид углерода и не разлагаться на свету.
3. Эквивалентная масса его должна быть большой.
4. Титр раствора должен оставаться постоянным в течение длительного времени.
Поскольку сравнительно немногие химические соединения полностью удовлетворяют всем перечисленным требованиям, выбор веществ для приготовления растворов титрантов ограничен.
38. Основы количественного химического анализа
117
Если вещество, применяемое для приготовления титранта, удовлетворяет указанным требованиям, раствор готовят по точной навеске вещества. При более высоких требованиях к точности нужно применять мерную колбу, градуированную при 20 °C, и воду, температура которой также доведена до 20 °C. Идеальными веществами для приготовления растворов титрантов являются, например, хлорид натрия, бихромат калия, бромат калия, оксалат натрия. Если вещество удовлетворяет не всем перечисленным выше требованиям, то для приготовления раствора отвешивают приблизительное количество вещества, а затем устанавливают титр раствора.
Установку татра раствора (стандартизацию) проводят следующим образом: определенное количество соединения, состав которого точно соответствует его формуле, — первичное «стандартное вещество-— растворяют в воде и титруют раствором, титр которого надо установить. На основании уравнения реакции по навеске первичного стандартного вещества и расходу титранта можно- рассчитать титр растворит
Первичное стандартное вещество должно в основном удовлетворять требованиям, перечисленным выше в пп. 1, 2 и 3. Соблюдать условие 4 нет необходимости, так как водный раствор первичного стандартного вещества титруют сразу после приготовления. -Первичное стандартное вещество не обязательно должно- растворяться в воде (например, оксид ртути имеет преимущества при установке титра растворов кислот). В общем идеальное вещество, пригодное в качестве титранта, можно применять также и как первичное стандартное вещество, но первичное стандартное вещество не всегда можно применять для приготовления раствора титранта.
Концентрацию раствора титранта можно выразить по-разному. Из практических соображений в настоящее время ее выражают почти исключительно в виде нормальности.
Например, однонормальный раствор кислоты (сокращенно 1 н.) содержит 1 моль эквивалентов кислоты в 1000 см3 раствора, т. е. концентрация раствора, выраженная в эквивалентах, составляет сэ=1 моль/дм3. Соответственно концентрация децинормального раствора (0,1 н.), выраженная в эквивалентах, составит сэ = 0,1 моль/дм3. Эквивалентная концентрация сэ и концентрация с связаны между собой через стехиометрический коэффициент z:
c = c3lz (31а)
Стехиометрический коэффициент вещества не является его константой, а зависит от типа реакции. Многие вещества, применяемые для приготовления титрантов, в зависимости от условий реакции изменяют свои химические свойства и могут поэтому иметь различные стехиометрические коэффициенты.
118 . Д. Химический анализ
,.Пример. Для фосфорной кислоты при титровании её с индикатором мети] ловим красным z=l, а при использовании в качестве индикатора фенолфта] леина .2=2. Йодноватая кислота НЮ3 может реагировать как одноосновна-я кислота (г=1), а также как окислитель с присоединением шести электронов] (2*=6). . |
: Из сказанного ясно, что указание на то, что имеется 1 н] раствор кислоты, не всегда однозначно дает возможность уз] нать содержание кислоты в этом растворе и понять, как при] готовить раствор, соответствующий этой концентрации. Во из] бежание разночтений концентрацию титранта следует выра] жать в виде частного в соответствии с уравнением (31а), на] пример Ch2so4 =0,1/2 моль/дм3. Из такой записи следуют одно] значные выводы: а) для приготовления раствора нужно раство] рить 0,05 моль H2SO4 в 1000 см3 воды; б) эквивалентная кон] центрация раствора кислоты сэ = 0,1 моль/дм3, т. е. раствор де] цинормальный (0,1 н.); в) стехиометрический коэффициент серной кислоты в предполагаемом титровании z=2. Следует помнить, что концентрация растворов титрантов в объемном анализе зависит также от температуры (массовая единица] моль/дм3!).
Применение растворов нормальной концентрации при титро] вании имеет то преимущество, что стехиометрические соотно] шения компонентов в этом случае отчетливо видны. Например,) для титрования какого-либо объема 1 н. кислоты необходим равный объем 1 н. раствора щелочи независимо от типа кис] лоты или основания. Это основано на том, что одинаковые объемы эквинормальных кислот или щелочей содержат одинаковые количества протонов (в виде ионов Н3О+) или ионов гидроксила соответственно. При расчетах- используют ^следующие соотношения: U..i ,Е ...3.. ' J.U ’I.
к". , - лнгг.-..i ; Ингzciiii.xr .щин с. • • п I
^reop^teoji™ ^пЩктспракт >
' ЩЭШ п:-пт:и:( ПЛИЛжЩГНЩШШ.:.: .шллпщ;. -
где индексы теор лИ.щрйКТ^-од^д^еЖйЯеоретическдй^'Л’рактцче-ЙКИЙ роответствецщд.;.,-.п;,:< -.ж( шш.щщшшо: .•
щщПрц-.дрбавлщищ в-процессе титррранищ.найдэдда^е- объема тртрднтд; г/рраи, . дсщцентрацией ।спряжв;.г-нд^одя;гп;,йб<ье1м.:у'теЙр. рдртдора.й;крнде.вдр.здийй^геор. Пдаундот 'ЛНЕ7 ;<
• кг л: inn:/? аднг.г. щшкщ
V тсор V практ: -.щадмШЩГЖО; Н!. л*
Далее из уравнения (32) следует; что
щ: -и ^?ifepp.i'.oxo: ншщ-лшг:
чг пип’. ;к-лиы. ; .шт.ш.
Тогда г.: :1)*п:з1ш.с i .ап лыс :г. яшгкшптгщщ н...
•.Л ’,О‘ I ВГ.-ЩЩ-Щ !M;"#r*W-Ur- lh.£.y.I!!l!'m.’.I Hilb'.iИ
.•:71!ад111(0.1фА.тв9йй?>ед^щ?л»гй‘’1~'^йаит.щ1м .. >
38. Основы количественного химического анализа
119
Частное / = Утеор/ппра-кт называется титриметрическим фактором. Это безразмерная величина. Для раствора титранта с кб'йЦент-рацией, равной Стеор, /=1,0000. На практике для. .. расчетов пользуются произведением fcTeop.
В качестве примера рассмотрим установку характеристик 0,1 н. раствора серной кислоты по первичному стандартному веществу — карбонату натрия.
Взвешивают 245,6 мг, 250,3 мг и 249,8 мг карбоната натрия. Раствор серной кислоты ~0,1 н. готовят разбавлением конц. H2SO4. Этой кислотой титруют пробы Па2СО3. При этом на титрование затрачивается 45,70 см3; 46,70 см3; 46,55 см3 H2SO4 с концентрацией спракт. Рассчитанные объемы раствора кислоты (Щеор) составляют 46,34 см3; 47,22 см3; 47,13 см3 соответственно. Таким образом, объем кислоты, затраченный на титрование, меньше рассчитанного (теоретического). Это означает, что раствор серной кислоты имеет концентрацию больше чем 0,1 н. Истинную концентрацию раствора кислоты спракт получают из уравнения (33). Среднее значение из полученных результатов Спракт — 0,050245 моль/дм3 = 0,10049 н.
Раствор этого титранта можно разбавить рассчитанным объемом воды для получения раствора с концентрацией 0,05000 моль/дм3. Отклонение концентрации раствора применяемого титранта от необходимого значения определяют по уравнениям (35) и (36). Титриметрический фактор в приведенном примере будет равен 1,0049.
Стандартизацию раствора титранта надо проводить очень тщательно, поскольку ошибка стандартизации явится причиной ошибок всех последующих титриметрических определений.
Рекомендуем снабжать посуду с титрованными растворами этикеткой с указанием градуировочной кривой, формулы, концентрации, титриметрического фактора, его десятичного логарифма и дату приготовления раствора.
, - - : ' \ 'I
' ' Р" lb
38.2.3.5. . У становление конечной точки титрования v .И4-Д«.’
Способы установления конечной точки титрования имеютг важное значение, так как часто оказывают решающее влияние иа точность титрования. Для этого в принципе можно применять все химические и физические методы; позволяющие измерять один из параметров, характерных для данной реакции. В зависимости от вида этих параметров различают следующие спосо-
бы индикации: .
Параметр -- ищ
Концентрация Н3О+-ионов ,
Концентрации других анионов или катионов . . .
Электрический заряд поверхности осадка
Окислительно-восстановительный потенциал
Электропроводность
Электрическая емкость, индуктивность - . г’..
Способ индикации
Кислотно-основные индикаторы
Осадительные индикаторы, комплексометрические индикаторы
Адсорбционные индикаторы
Редокс-индикаторы
Кондуктометрический
Высокочастотное титрование
120
Д. Химический анализ
Диффузионный ток
Оптическая плотность раствора
Температурные изменения, обусловленные энтальпией реакции
Амперометрический, Фотометрический Термометрический
Важной задачей аналитической химии является нахождение новых методов установления конца титрования, поскольку с этим связано расширение типов реакций, применяемых в объемном анализе. Тенденция развития направлена в сторону физических методов индикации, которые в отличие от химических не вносят изменений в аналитическую систему и тем самым обусловливают принципиально большую точность индикации. Кроме того, это способствует автоматизации титриметрических определений, что имеет большое значение для химической промышленности. Однако наиболее пригодны для автоматизации методы, не связанные с измерением объемов, например метод меченых атомов, измерение УФ- и ИК-поглощения, УФ- и рентгеноэмиссионный спектральный анализ.
38.3.
38.3.1.
Теоретические основы количественного
химического анализа
Кислотно-основные равновесия
38.3.1.1. Общие вопросы теории
Расчеты кислотно-основных равновесий включают вычисление pH, поскольку, зная величину pH, можно рассчитать равновесные концентрации всех компонентов раствора. Общий метод расчета равновесных концентраций основан на том, что выражают все условия равновесия в виде неизвестных и затем решают систему уравнений.
Простейшим примером является реакция взаимодействия слабой одноосновной кислоты НХ с водой. При этом происходят следующие реакции:
НХ + Н2О Н3О+4-Х~ (37)
Н2О + Н2О Н3О+4- ОН- (38)
НХ + ЗН2О 2Н3О++ X- + ОН- (39)
Исходя из уравнений реакций, можно рассчитать следующие равновесные молярные концентрации: недиссоциированной кислоты Снх, ионов водорода сНзо+> анионов кислоты сх", ионов гидроксила Сон". Для расчета можно составить систему четырех уравнений с четырьмя неизвестными.
1. Уравнение, основанное на применении закона действую
38. Основы количественного химического анализа
121
щих масс к диссоциации слабой кислоты (реакция (37)):
сн3о+сх_/снх — Ks (40)
где Ks — константа кислотности слабой кислоты НХ.
2. Выражение для ионного произведения воды в соответствии с реакцией (38):
сн3о+сон~ — Kw (41)
где Kw — константа автопротолиза воды.
3. Уравнение, полученное исходя из того„ что в водных растворах протоны не могут находиться в свободном состоянии и число протонов, отданное одноосновными кислотами, равно количеству протонов, принятых однокислотными основаниями. Иначе говоря, суммарные концентрации всех образовавшихся одноосновных кислот и однокислотных оснований этой системы равны между собой. Это отражено в правой части реакции (39), откуда следует
сн3о+ = сон Г сх~ (42)
4. Уравнение, основанное на применении закона сохранения масс к кислоте или основанию соответственно:
Со = снх+ сх~ (43)
где Со — общая концентрация кислоты или основания. Общая концентрация постоянна при постоянном объеме раствора; это условие обычно хорошо соответствует практике (например, при титровании). Концентрацию Со можно представить также как количество чистой кислоты или соответственно основания (в молях), деленное на конечный объем раствора. Сочетанием уравнений (40) — (43) получают.
сн3о+сх- _ сн3о+ (сн3о+ — срн~) сн3о+ (сн3о+ — Kit/ch3o+) _ „ , Со — сх~ Со — (сн3о+ — сон~) Со — (cHgo+— Xir/cH3o+) s
с3н3о+ ^5с2ц3о+ — (Кц?+KsC0) сн3о+ — KwKs — 0 (45)
Уравнение (45) можно применять для построения графической зависимости pH от константы кислотности Ks и общей концентрации Со. Для этого уравнение (45) логарифмируют (в соответствии с известным соотношением х=101г%) и получают:
Со = 10м ~2рН 4-1 О’ н—10рК^р/<®' — 10рН~₽к®' (46)
При построении зависимости значений pH одноосновных кислот различной силы, т. е. при разных pKs, от соответствующей общей концентрации Со получают кривые, изображенные в нижней части рис. Д.37. При замене в уравнении (46) pH на рОН и pKs на pKs (отрицательный десятичный логарифм константы основности основания) получают кривые Со/pH для ос
122
Д. Химический анализ
нований (верхняя часть рис. Д.37). Кривые кислот и оснований с одинаковыми значениями рД зеркально симметричны относительно прямой рН = 7. Все кривые заканчиваются в точке (Со = О, рН = 7), и с увеличением Со наблюдается образование постоянно нисходящих или соответственно восходящих кривых..
По ходу кривых рис. Д.37 можно предсказать кислотно-ос-
Рис. Д. 37. Диаграмма pH — Со (Со — общая концентрация).
новное поведение водных растворов кислот и оснований: уже при небольшом содержании кислоты или основания в чистом воде происходит резкое изменение pH, которое тем больше, чем сильнее кислота или основание. Напротив, при изменении концентрации концентрированных кислот или оснований наблюдав ется незначительное изменение pH. По рис. Д.37 нельзя точно» определить изменения pH для небольших общих концентраций, поэтому для этой области концентраций строят логарифмическую кривую, представленную на рис. Д.38. По данным кривой рис. Д.38 можно непосредственно или после интерполяции определить pH растворов одноосновных кислот и однокислотных оснований в широкой области концентраций.
38. Основы количественного, химического анализа 123
. , Пример, В.0,1 М растврре .хдориущй'кислоты pH равдр 1,55 (рЛ'5=2,0). Для соответствующего ' сопряженного основания,—хлорит-ионов. (,С1О$~ \;Ki,=рК.'х—рЛ\= 14—2=12, откуда следует, что 0,1 М раствор хлорита натрия имеет pH-7,5. ; " ''
.и(. ' •
.38.3.1.2. - Логарифмические pH-диаграммы одноосновныхJs.,
• ' ' кислот и однокислотных оснований
Хотя кривые, приведенные на рис.. Д.37 и Д.38, дают возможность определить-зависимость между общей концентрацией растворов чистых одноосновных кислот (или оснований) и ве-
Рис. Д.38. Логарифмическая диаграмма (1g Со—pH).
личиной pH, по ним нельзя определить равновесные концентрации кислоты и аниона (или основания и катиона). Но для оценки кислотно-основного равновесия необходимо знать именно эти величины. Такую возможность дают так называемые логарифмические pH-диаграммы. Для их построения откладывают на оси абсцисс значения pH, а в области отрицательных значений оси ординат — значения десятичных логарифмов концентраций (в одинаковом масштабе). На рис. Д.39 изображена логарифмическая диаграмма, построенная для одноосновной кислоты СбН5СООН с константой диссоциации /С = 6,46 моль/дм3 (pAs = 4,19) и общей концентрацией Со=1О-2 моль/дм3. На примере диаграммы, изображенной на рис. Д.39, можно дать методику применения логарифмической диаграммы: общую концентрацию Со определяют в точке, в которой ветвь кривой НХ (линия кислоты), параллельная оси pH, или соответственно кривой Х“ (линия основания) пересекает ось ординат, так что в этой точке пересечения: lgC = lgC0. Силу кислоты, характеризующуюся константой диссоциации Ks, определяют по уравнению рН = рД5, беря значение pH на параллельной ветви кривой НХ, начиная с которого (при одном и том же масштабе по осям
124
Д. Химический анализ
абсцисс и ординат) линия кислоты отклоняется под углом 45° вниз направо, а линия основания под таким же углом — вниз налево (точка Р3). Получаемые при этом кривые асимптотически приближаются к гиперболам. В областях pH<p/G и рН> >p/G кривые практически совпадают с гиперболами, если они пересекаются в точке с абсциссой p/G и ординатой 1g Со.—0,301.
Рис. Д.39. Логарифмическая pH-диаграмма бензойной кислоты. К5=6,46-10-5 моль/дм3; pKs»4,19; Со==О,01 моль/дм3; pHj относится к раствору эквимоляр'-ной смеси бензойной кислоты с бензоатом натрия.
При этом pH«p/G. В противном случае pH будет зависеть от концентрации кислоты и аниона.
Вершины углов диаграммы связаны двумя диагоналями, описываемыми уравнениями lgCH3o+ =—pH или lgCoH~=pH— —pKtr- Они всегда пересекаются в точке нейтральности pH 7 (независимо от величины Со и /G).
Логарифмическая диаграмма дает значения равновесных концентраций всех компонентов кислотно-основной системы в зависимости от pH.
Пример. Для точки Р2 при pH] получаются следующие концентрации: Сх_=Ю-3’1 моль/дм3=сн3о+; снх = 10-2 моль/дм3; сон- = 10-10’9 моль/дм3. В кислой среде преобладают недиссоциированные молекулы кислот. В соответствии с этим ионы Н3О+ и X- не оказывают существенного влияния на pH, а нонами ОН- можно полностью пренебречь. При pH точки Рз сх”=снх=э = 10-2’3 моль/дм3; сн3о+=10-4'2 моль/дм3; сон- =10-9'3 моль/дм3.
38. Основы количественного химического анализа
125
Из рис. Д.39 можно вывести общие закономерности, важные для кислотно-основного равновесия:
1. В области pH<pKs Снх^>Сх_, т. е. в этой области находится практически только недиссоциированная кислота (наряду с ионами Н3О+). Напротив, в области pH>pKs существует неравенство Сх“^>Снх; это означает, что в растворе находятся только анионы кислоты (наряду с ионами ОН"). Таким образом, pKs разграничивает области недиссоциированной и полностью диссоциированной кислоты. Только при pH»p/G молекулы кислоты и анионы находятся в сравнимых концентрациях. В точке пересечения гипербол Снх = Сх“ и pH = p/G. Это значит, что в растворе, содержащем кислоту и сопряженное с ней основание (т. е. ее соль) в эквимолекулярных количествах, pH = pKs.
2. В водном растворе кислоты существуют следующие равновесия:
НХ + Н2О ч=г: Н3О+ + х-
2Н2О Н3О++ОН“
Из правых частей этих уравнений реакции можно определить-соотношение концентраций ионов Н3О+, X" и ОН-:
сн3о+ — сон"+сх"
Как видно из рис. Д.39, в кислой среде Сх-^Сон^ так что из уравнения (42) получим
сн3о+ сх~ (47)
Это условие выполняется только в точке Р%, отсюда можно сделать вывод, что pH водного раствора кислоты на логарифмической диаграмме определяют как абсциссу точки пересечения (Рг) линий ионов Н3О+ и основания.
3. В водном растворе основания (для рис. Д.39 это раствор бензоата натрия) происходят реакции
X" + Н2О ОН" + НХ
2Н3О ОН" + Н3О+
Откуда
сон~ снх Fgh3o+ (48)
Как видно из рис. Д.39, в щелочной области Снх^£н3о+ , и по уравнению (47) получаем, что
сон~ ~ снх (49)
Это условие выполняется только в точке Рз- Следовательно, pH водного раствора основания (а тем самым и раствора соли, образованной катионом сильного основания и анионом слабой кислоты, например бензоата натрия или ацетата калия) опре-
t < Д. Хцл1ич,еский диализ,.
леляют как а^^с^^тонки^рес^ненвд; (^^давдЙ^ояйнв ОН -И КЙЙ1ОТЫ. пШеЩИШПП Г. ОШИПЕ :!---ПГ.' И’.'-'НП
'''Из р^ссМбтр‘йни‘я'1точж Pi— Pfi .$а р^.1\1Д.39. мо^но ,одела,р> ещ^некото-рЫе'выводы: ’ а.иы ь.•> '•> '> .* '• -
а) При смешеиии 0,1' 1Мбль СНзСООК и 0,!1-1чоль НС1 в воде МЬ'Ьбщего объема 1 дм3 раствора,получают 0,1 М раствор уксусной, кислоты, Содержащий КС1, pH которого определяют в точке Р2, где сх_=т=СнзО+- Это объясняется тем, что ацетат-ионы, являющиеся слабым основанном, полностью протонированы ионами гидрбкоония, обладающими сильнокислотными свойствами., • : б) Раствору'смёси уксусной кислоты и ацетата калий в эквимолярных соотношениях (т. е. слабой кислоты и сопряженного с Ией основания)' соответствует точка,, где снх с.х~, т. е, Р3, поэтому pH этого раствора равно.’ рХ5 уксусной кислоты (независимо от концентрации кислотно-основной пары).
в) При растворении 0,1 моль СН3СООН и 0,1 моль КОН (объем раствора 1 дм3) получают 0,1 М раствор ацетата калия, pH которого определяют в точке /У>, где снх = сон_. Таким образом, слабая уксусная кислота' пол; ностью депротонирована сильным основанием. г д'. . '
г) Для 1 дм3 раствора, содержащего 0,1 моль HCl + 0,1 моль СНаСООН, pH определяют в точке Pi, так как только в этой точке Сн3о+ = снх^ В этом растворе значение pH такое же, как и в растворе-’индивидуальной сильной кислоты. Диссоциация слабой кислоты полностью подавлена. Этот вывод справедлив также для смеси децимолярных растворов гидроксида калия и ацетата калия, pH которой определяется в точке Ре, гд? сон“=сх_«Ц!
л л" !?;1Е? : ' ,
Используя простые соотношения, полученные из данных рис. Д.39, из уравнений (47) и (49) можно вывести полезные .для .практики формулы . приближенного'расчета,, pH ра^дроррв' кислот и оснований. у. ши?;: i
. Водные растворы кислот. Из уравнения (47) следует, что в растворе кислоты Сн3о+ » Сх~. На рис. Д.39 этому соответствует. точка Р%, где ,CxL<CcHx. Тогда уравнение: :(43). гможно.. переписать Как -Д< О ’ .. ';,Г:,?' .
‘ (50)
I , Подставив'уравнения..(47).;м; (50). в выражение;-.закона действующих масс (уравнение . (40).) ,,'получим'Для вычисления'pH' разбавленной кислоты выражение (51), логарифмирование которого дает уравнение (.52)
с2Нзу/С0 = Д5; рН= l/2(p/Cs— 1g Со) (51), (52)' , < ' у j • [ . .
Водные растворы оснований. Выражение для расчета pH растворов оснований получают, используя соотношейия, найденные из правой части рис. Д.39 (рН>7): сх-»снх и Снх1> 1>сн3о+. Вводя их в уравнения (43) и (48), получаем соответственно приближенные равенства
Со » сх~; сон- « снх (53), (54)
«Объединение уравнений (53) и (54) с выражением закона дей
38. Основы количественного химического анализа 12*
ствующих масс (уравнение (40)) дает
сн3о+Со/сон~ = c2h3o+Co/'Kw — (55}
pH =1 /2 (pKs р^ -|- 1g Со) (56}
Общую концентрацию Со можно определить как количество-соли в молях, деленное на конечный объем раствора в кубических дециметрах. В уравнении (56) можно заменить pKs на рКь из соотношения p/Q + pK& = p7G-:
pH = pKw—1 /2 (p/(b — 1g Со) (57}
Уравнение (52) для расчета pH слабых кислот аналогично можно преобразовать в уравнение для расчета рОН слабых оснований, заменив pH на рОН и рК> на рКь:
POH = VJpKfe-lgC0) (58}
Уравнение (58) идентично уравнению (57). Это наглядно видно, если выразить в уравнении (58) величину рОН через pH из условия рН + рОН = р7<№. Уравнение (57) позволяет рассчитывать pH растворов, содержащих соль слабой кислоты и сильного: основания. Такой раствор образуется также в точке4 эквивалентности при титровании слабой кислоты раствором сильного-основания. Значение pH, рассчитанное по уравнению (57), называют показателем титрования рТ. Уравнения (52) и (57) справедливы только при соблюдении условий рис. Д.39, т. е„ для умеренно разбавленных растворов и для растворов кислот и оснований средней силы. В других случаях некоторые из неравенств, взятых из диаграммы, недействительны и формулы для расчета pH усложняются. Такие расчеты можно провести с помощью логарифмических диаграмм pH, построенных для каждого конкретного случая. Для иллюстрации на рис. Д.40 и и Д.41 приведены две логарифмические диаграммы, с помощью которых можно вывести уравнения для более сложных расчетов pH.
В табл. Д.13 приведены формулы для расчета pH одноосновных кислот и границы применимости этих формул. Соответствующие формулы для расчета pH оснований получаются при замене сн3о+ на сон- и на Кь-
Область применения уравнения (52) можно определить при сравнении логарифмической формы этого уравнения и рис. Д.38, на котором уравнением (52) описываются нисходящие прямые с tgcp = —2. Однако эти прямые только в области средних значений pKs могли бы перекрываться кривыми, рассчитанными из уравнения третьей степени. Уравнение (52), таким образом, может быть применено только к сильным и очень слабым кислотам умеренных концентраций. Напротив, уравнение-(S9) применимо почти всегда и может давать отклонения.
Фис. Д.40. Логарифмическая pH-диаграмма для бензойной кислоты с концентрацией Со=Ю-4 моль/дм3.
Фис. Д.41. Логарифмическая pH-диаграмма для кислоты с рК=6,8 и концентрацией Со=10-6’6 моль/дм3.
38. Основы количественного химического анализа
129
Таблица Д.13. Формулы для расчета pH растворов слабых кислот
Границы применимости Формула
Увеличение разбавления сх- > сон- Сх- <S Снх сх- > сон- СХ- « снх Сх- « сон-СХ- « Снх ^Н3О+ = VpH == Х/2p^s 1/а Со сн3о+= |/^ + ДА— c3h3o++^sc2h3o+ — (Kw + KSCO) сНзо+ — — K.wKs = 0
только в области экстремально малых значений концентраций. Уравнение (59) перекрывает область действительных значений уравнения (52) и при /Cs2/4<^/CsC0 переходит в него. Уравнение (45) охватывает области применимости уравнений (52) и (59). При /С1г/С5<с3н3о+ и KsCq^>Kw оно переходит в уравнение (59).
Значительно проще и обычно с достаточной для практики точностью можно определять pH непосредственно из соответствующей логарифмической диаграммы. Точность получаемых при этом значений можно легко проверить, сравнив их со значениями, полученными из кривых рис. Д.37 или Д.38 соответственно. Существенные отклонения от кривых рис. Д.38 наблюдаются только при концентрации кислоты или основания порядка Со=1О_7 моль/дм3. Такое сравнение, кроме того, позволяет определить границы применимости логарифмической pH-диаграммы: например, для сильно разбавленного раствора кислоты с общей концентрацией Со<Ю-7 моль/дм3 получается рН>7! Напротив, диаграмма, приведенная на рис. Д.38, дает возможность сделать однозначные и правильные выводы для растворов таких концентраций.
На рис. Д.40 представлены соотношения концентраций для раствора бензойной кислоты с Со=10-4 моль/дм3. Здесь, так же как на рис. Д.39, в кислой области сх_»сон_, ио в отличие от рис. Д.39 точка Р2 аналогична тачкам Pt и Рз. Она соответствует раствору чистой кислоты, и здесь нельзя пренебречь величиной сх _по сравнению с снх, поскольку они приблизительно равны. При введении условия сх~^>сон~ в уравнение (42) получим Сн3о+=сх~- При этом уравнение (40) переходит в ;!н3о+=^спх; найдя НХ из уравнения (43), получим c2h3o+=Ks(Cq—сНзо+). В итоге получается квадратное уравнение, решение которого имеет вид
<59>
Это уравнение дает хорошие результаты в широкой области значений концентраций.
9—1880
130
Д. Химический анализ
На рис. Д.41 представлена диаграмма для кислот более слабых, чем бен» зойная, с еще меньшей общей концентрацией. При этом нельзя пренебрегать никакими величинами, гак как в области общей кислотности уже Сх_«сон~, а в точке Рг, которая теперь полностью аналогична точкам Pt, Р3 и Сх~=Снх_(Р1 и Р3 не обозначены). В этом случае нужно решать уравнение третьей степени (уравнение (45)) или, проще, иайги его графическое решение по рис. Д.37 или Д.38 соответственно.
С помощью диаграммы 1g с — pH можно также определить : концентрационные соотношения в растворе двух слабых кислот,, например иона аммония и уксусной кислоты (рис. Д.42). На диаграмме различают три области значений pH: pH<p/CSi; p/Cs1<pH<p/C52; pH>p/CS2,. В первой области обе кислоты NH4+ и СН3СООН находятся в недиссоциированном состоянии.
Рис. Д.42. Логарифмическая pH-диаграмма для раствора 0,1 М по уксусной кислоте и 0,1 М по NHi+.
Во второй области в основном существуют более слабая кислота NH4+ и более слабое основание СН3СОО~, а в третьей области почти исключительно находятся основания NH3 и СНзСОО-.
Отсюда ясно, что при кислотно-основном титровании смеси двух слабых кислот происходит их последовательная депротонизация: вначале более сильной кислоты, а затем более слабой. Интересна точка Р. Для нее справедливо условие Сын+,~
38. Основы количественного химического анализа
131
«Ссн+Зсоо~, которое выполняется и для раствора ацетата аммония, так что абсцисса точки Р соответствует значению pH этого раствора. Из рис. Д.42 можно вывести выражение для точки Р, пригодное для расчета pH растворов солей, образованных анионами слабой кислоты и катионами слабого основания:
pH = ^(pKS]+pKS2)
(60)
Оно показывает, что pH такого раствора определяется только значениями констант диссоциации обеих кислот и не зависит от концентрации.
Уравнение (60) можно вывести аналитическим путем; при этом получают еще один интересный вывод: для раствора эквивалеигиых количеств уксусной кислоты и аммиака справедливы выражения
сН3О+сСН3СОО‘7сСН3СООН = KS1 и CH3O+CNH3/CNH4+= ^s2 (61), (62)
Так как в растворе, естественно, устанавливается только одно значение pH, величины Сн3о+ в уравнениях (61) н (62) равны. Решив уравнения относительно Сн3о+ и перемножив их, получим следующее уравнение:
С2н3о+ = CCH3COOHCNH^+ CCH3COO-CNH3 (63)
При смешивании уксусной кислоты с СН3СООН + Н2О аммиаком протекают реакции СН3СОО" + Н3О+ (64)
NH3 + Н2О NH4++OH- (65)
CH3COOH4-NH3 => СН3СОО- + nh4+ (66)
Из данных логарифмической диаграммы, приведенной на рис. Д.42, ясно, что в области, находящейся между значениями pH чистой уксусной кислоты (Pi) и чистого раствора аммиака (Pi), можно пренебречь концентрацией ионов Н3О+ и ОН- по сравнению с концентрацией ацетат-ионов и ионов аммония. Из этого следует вывод, что доля реакций (64) и (65) незначительна и ионы NH4+ и СНзСОО- образуются почти исключительно в результате прямой реакции взаимодействия уксусной кислоты с аммиаком. Но в этом случае для каждой стадии реакции существуют равенства (см. уравнение (66)): Cch3cooh = cnh3и Cch3coo-=cnh+4. При этом уравнение (63) переходит в (67) или в логарифмической форме — в (68):
c2h3o+ = kSi^2; рн = 4-(р^г + р^2) (67)’ (68)
Уравнение (60) можно упростить введением в него константы автопротолиза воды:
pH = 7+4-(pKS1-p^2) (69)
Это означает, что если кислота 1 сильнее основания 2, то раствор соли, образованной катионом этого основания и анионом этой кислоты, имеет кислую реакцию; если основание 2 сильнее кислоты 1, то раствор соли дает щелочную реакцию.
9*
132
Д. Химический анализ
Уравнения (68) и (69) действительны, если только выполняются неравенства Сн3о+<^сх“ и Сон_<^Снх, и недействительны для кислот или оснований, сила которых превышает определенное значение.
Математическое описание логарифмических рН-диаграмм. Кривые, приведенные на диаграммах 1g с — pH, построены на основании уравнений, в которых концентрации снх и Сх- являются функцией pH. Эти зависимости выводят комбинированием уравнений (40) и (43). При решении их относительно снх и затем относительно Сх“ получают следующие уравнения:
„ £qch3o+ Со __________________Со____ (70Y
нх~ ch3o+ + Ks НАДн3о+ ’ 1 + 1O₽H-P*S k '
_________C0Ks________Co_____________Cg (71V Cx " *s + %o+ ~ 1 + cH3O+/^s— 1+10pKs-pH_________V '
Логарифмирование уравнения (70) дает (72), а уравнения (71) — выражение (73):
lg%x = lgCo-lg(l+10pH-pS (72)
lgcx-= lg Со — 1g (1 + КГ*"”») (73)
При графическом изображении этих зависимостей на диаграмме 1g с — pH получаются сильно растянутые изогнутые линии, представленные на рис. Д.39. Их можно найти аппроксимацией двух областей каждой кривой. Для кривой НХ, так называемой линии кислоты, используют:
а) Область pH<p/(s. При этом условии в уравнении (72) 10ph-pks откуда для левой ветви кривой
Ig Снх = 1g Со (74)
Это означает, что линия кислоты в области pHCpKs параллельна оси pH и отстоит от нее на расстояние 1g Со.
б) Область pH>p/Cs. В этом случае 10pH~pKs^>l, и поэтому из уравнения (72) следует
1g снх = —Рн+P^s+1§ Со (75)
Эта функция представлена на диаграмме в виде нисходящей под углом 45° кривой (при одинаковом масштабе по осям ординат и абсцисс), пересекающей верхнюю кривую (уравнение (74)) при pH = pKs.
в) Точку рН = р/О. Из уравнений (72) и (73) получается значение ординаты:
Ig Снх = lg Сх- = lg Со- 1g 2 = lg Со- 0,301 (76)
38. Основы количественного химического анализа
133
Аналогично для линии основания используют:
а) Область pH<p/Cs. Из уравнения (73) следует
lgCx-=pH-p/Cs+lgC0 (77)
Уравнение (77) описывает левую прямолинейную ветвь линии основания, восходящую под углом 45°.
Рис. Д.43. Логарифмическая pH-диаграмма селенистой кислоты (Со= = 0,1 моль/дм3).
pH, относится к селенистой кислоте; рН2 — к раствору гидроселенита щелочного металла; рНз —к раствору селенита щелочного металла; рД’^—к раствору эквимолярной смеси HjSeOa+HSeOa-; PKS — к раствору эквимолярной смеси HSeOs—j-SeOs2-.
б) Область pH>p/Cs. При этом из уравнения (73) получают lgcx- = lgC0 (78)
Уравнение (78) описывает ветвь линии основания, параллель-, ную оси pH.
в) Точку pH = pAs. При этом, как было указано выше, lgcx-=lgC0—0,301 (79)
38.3.1.3. Логарифмические pH-диаграммы многоосновных кислот и многокислотных оснований
На рис. Д.43 представлена логарифмическая диаграмма для Двухосновной селенистой кислоты. Из диаграммы можно сделать следующие выводы.
134
Д. Химический анализ
1. Многоосновная кислота диссоциирует по ступеням. В области pH<pKSi CH2SeO3^CHSeO3- ^CseO32-, Т. е. В ЭТОЙ ОбЛЭСТИ преобладают недиссоциированные молекулы кислоты H2SeO3.
2. В области pKsx<pH<pKs2 преимущественно находится амфолит HSeO3_. Рассматривая равновесия в этой системе по аналогии с системой уксусная кислота — аммиак (реакции (64) — (66)), можно сделать вывод, что из трех протекающих реакций:
HSeO3” + Н2О «=»: SeO32- + Н8О+ (80)
HSeO3- + Н2О =₽=*: H2SeO3 + ОН" (81)
HSeO3" + HScO3“ H2SeO3 + SeO32- (82)
только реакция (82) имеет значение, так как в соответствии с диаграммой для области, находящейся между обоими значениями, рК. Сон-<^Сн2зео3 и для правой части этой области справедливо условие сн3о+<с5ео32-- Следовательно, реакции, записываемые уравнениями (80) и (81), имеют второстепенное значение, а молекулы H2SeO3 и ионы SeO32- образуются преимущественно по реакции диспропорционирования амфолита HSeO3_ (уравнение (82)). Но в этом случае для области между pKSl и pKs2 справедливо условие CH2seo3~c.seo32-- При введении этого уравнения в произведение выражений закона действующих масс
„ СЦ5еО3-Сц3О+ CSeO32-CH3O+
= - с^3 ' CHSeO,-- (83)
получают выражение (84), а при его логарифмировании — уравнение (85):
с2н3о+ = ^Л2; РН=4-^1+Р^ <84)’ (85)
Это уравнение применимо для расчета pH только в растворе амфолита (в рассматриваемом случае гидроселенита натрия) и не пригодно для растворов сильных кислот, например серной. Уравнение (85) можно также применить для расчета pH амфолитов трех- и четырехосновных кислот (например, Н2РО4“, НРО42~ и др.). Даже в тех случаях, когда уравнение (85) неприменимо для расчета pH растворов амфолитов (для очень сильных или очень слабых многоосновных кислот), по уравнению (85) всегда получают значение pH, при котором концентрация амфолита максимальна. Это значение pH называют «изоэлектрической точкой» амфолита.
3. Если величины р/G существенно различаются (больше чем на 4 единицы), то можно не принимать во внимание восходящие ветви кривой на рис. Д.43, так как они соответствуют
38. Основы, количественного химического анализа
135
очень низким значениям концентраций. Тогда полученная диаграмма аналогична диаграмме двух одноосновных кислот с константами диссоциации KS1 и Ksz- Эта аналогия проявляется также в формальной идентичности уравнений (85) и (68).
38.3.1.4. Кислотно-основное титрование
Кривые титрования. Зависимость значений pH, изменяющихся в процессе титрования, от добавляемого количества титранта графически изображается кривыми титрования, причем для простоты часто пренебрегают увеличением объема. На оси ординат
Рис. Д.44. Кривые титрования 0,1 н. растворов кислот 0,1 н. раствором сильного основания.
откладывают значения pH, на оси абсцисс степень оттитровы-вания х = с/с0 (с— количество добавляемого титранта в молях на кубический дециметр титруемого раствора, Со — количество титранта, необходимое в соответствии с уравнением реакции, также в молях на 1 дм3 раствора пробы). Обычно интерес представляет область 0^т^2, причем т=0 соответствует чис
136
Д. Химический анализ
тому титруемому веществу до начала титрования, т=0,5— добавлению половины, т=1 добавлению всего и т=2 добавлению двойного количества титранта, рассчитанного из стехиометрических соотношений. При т = 1 получают точку эквивалентности.
При построении зависимости значений pH от степени оттит-ровывания в ходе титрования кислот различной силы и равной концентрации раствором сильного основания получают кривые, изображенные на рис. Д.44. Из этих кривых можно сделать некоторые важные общие выводы.
1. Чем слабее кислота, тем больше изменение pH при добавлении первых капель раствора основания. Это видно, если на рис. Д.44 нанести значения pH раствора чистой кислоты, полученные из рис. Д.38 или из соответствующей диаграммы Igc— pH. Кривая с pKs = 8 «выпрыгивает» из ординаты при pH 4,5, кривая с p/G = 4 — при pH 2,5, а кривая сильной кислоты — при pH 1.
2. На кривых титрования слабых кислот в точке полунейтрализации (т. е. при т=0,5) наблюдается перегиб, в этой точке pH = pKs. То же самое можно вывести из выражения для закона действующих масс после решения уравнения относительно Сн3о+ и логарифмирования:
pH = p/Cg—lg(cx-/cHX) (86)
При добавлении половины стехиометрического количества раствора основания, т. е. при т=0,5, с достаточным приближением можно считать сх-=сНх и lg(cx-/cHx) =0. При этом уравнение (86) превращается в (pH)i/2 = p/G ((pH)i/2—это pH при т = = 0,5). Возрастающий полого участок кривой, в середине которого находится (pH)i/2, характеризуется тем, что даже при добавлении относительно больших количеств титранта — основания — значение pH изменяется незначительно. Такое поведение называют буферным действием, а соответствующий раствор — буферным раствором.
3. Так называемый показатель титрования рТ, т. е. pH в точке эквивалентности, тем дальше сдвинут в основную- область, чем слабее титруемая кислота. При этом скачок pH в области точки эквивалентности увеличивается с увеличением силы титруемой кислоты. Это явление имеет большое практическое значение: чем меньше скачок титрования, тем менее круто восходит кривая титрования в области точки эквивалентности и тем менее точно можно определить точку перегиба. Поскольку точку эквивалентности при титровании определяют непосредственно по абсциссе точки перегиба на кривой титрования, результаты титрования в целом тем менее точны, чем слабее титруемая кислота. Очень слабые кислоты не титруются в водных средах. Показатель титрования слабой кислоты можно опре
38. Основы количественного химического анализа
137
делить из диаграммы Igc — pH (точка Р5 на рис. Д.39) или из кривой сопряженного с кислотой основания, представленной на рис. Д.38.
Пример. Сопряженное с 0,1 н. кислотой, рК« которой равно 8, 0,1 н., основание имеет рКь=6 (так как pKs+pKb=14) и соответствующее ему значение pH равно 10,5.
Рис. Д.45. Кривые титрования сильных и слабой кислот различной концентрации сильным основанием.
На рис. Д.45 показано влияние исходной концентрации титруемой кислоты на ход кривой титрования слабой или соответственно сильной кислоты. Из рис. Д.45 можно сделать следующие выводы: а) скачок pH в области точки эквивалентности при титровании сильной кислоты тем меньше, чем меньше исходная концентрация кислоты; б) ход кривых титрования слабых кислот в области 0<т<1 в меньшей степени зависит от исходной концентрации кислот, чем ход кривых титрования сильных кислот. Кривые титрования слабых кислот проходят (за исключением кривых для концентраций С0<Кз) через точку, в которой т=0,5; pH = pKs.
Рис. Д.46. Логарифмическая pH-диаграмма и кривая титрования для 0,1 и. раствора муравьиной кислоты.
Рис. Д.47. Логарифмическая pH-диаграмма и кривая титрования для 0,001 и. раствора муравьиной кислоты.
Рис. Д.48. Логарифмическая pH-диаграмма и кривая титрования для 0,1 н. раствора NHiCl или раствора NH3 соответственно.
Рис. Д.49. Логарифмическая pH-диаграмма и кривая титрования для смеси соляной и уксусной кислот (Со=0,1 моль/дм3).
140
Д. Химический анализ
Связь между диаграммами 1gс — pH и кривыми титрования. Логарифмическая диаграмма особенно упрощает построение кривой титрования, как это видно из рис. Д.46 на примере титрования слабой кислоты раствором сильного основания.
На рис. Д.47 и Д.48 представлены другие типы диаграмм — для особенно низких исходных концентраций и для очень слабых кислот соответственно. Такие комбинированные диаграммы дают возможность получить дополнительную информацию
Рис. Д.50. Логарифмическая pH-диаграмма и кривая титрования для раствора двух кислот с рК«г=3,15 и p7G2=6,9 (Со=О,1 моль/дм3).
из диаграмм 1g с—pH. На рис. Д.48 приведена комбинированная диаграмма кислотно-основной пары NH4+/NH3 при концентрации 0,1 моль/дм3. Из формы обеих кривых ясно, что титрование аммиака соляной кислотой характеризуется большей точностью, чем титрование хлорида аммония раствором едкого натра. Так, длина отрезка РзР& (рис. Д.48) является достаточно точной мерой скачка pH при титровании кислоты раствором сильного основания, а длина отрезка Р3Р1 дает величину скачка при титровании сопряженного основания раствором сильной кислоты.
Из диаграммы 1 g с — pH можно также построить кривую дифференцированного титрования кислот в их смеси. На рис. Д.49 приведена диаграмма для раствора, содержащего смесь соляной и уксусной кислот с концентрацией каждой 0,1 моль/дм3, а на рис. Д.50 — диаграмма для раствора смеси двух слабых кислот с p/Gj =3,15 и pKs2 = 6,9. Из рис. Д.50 ясно, что кислоты в их смеси можно оттитровать дифференцированно с достаточ
38. Основы количественного химического анализа
141
ной точностью, если значения их pKs отличаются по меньшей мере на 4 единицы.
На рис. Д.51 приведена комбинированная диаграмма для фосфорной кислоты Н3РО4. При титровании Н3РО4 проявляет свойства двухосновной кислоты, поскольку третья константа
Рис, Д.51. Логарифмическая pH-диаграмма и кривая титрования для фосфор» ной кислоты (Со=0,1 моль/дм3).
диссоциации (рКз3 = 12,66) настолько смещена в основную область, что по третьей ступени кислоту нельзя оттитровать обычным путем.
38.3.1.5. Кислотно-основные индикаторы
Кислотно-основные индикаторы (pH-индикаторы) обычно представляют собой слабые органические кислоты или основания, окраска ионной и молекулярной форм которых различна. Если ионная и молекулярные формы абсорбируют свет в видимой части спектра, такие индикаторы называют двухцветными. Если только одна форма индикатора поглощает свет в видимой части спектра, то такой индикатор называют одноцветным (например, фенолфталеин).
Двухцветные индикаторы. Реакцию нейтрализации индикатора, являющегося кислотой, можно представить следующим образом:
Hind + Н2О т—Ind" +Н3О+ (87)
Логарифмическое выражение закона действующих масс для
142
Д. Химический анализ
этой реакции:
PH = рКнша - Ig - 1g (88)
cInd Tlnd
где р/Снша — показатель индикатора; Диша — константа диссоциации индикатора.
В разбавленном растворе в первом приближении можно считать YHind~Yind~. Тогда каждому значению pH соответствует определенное соотношение <?н ша/сша-. Если принять, что растворы с одинаковой концентрацией Н Ind и Ind" имеют одинаковую интенсивность окраски, которая пропорциональна концентрации, следует ожидать, что в пределах относительно большой области pH окраска ионной формы индикатора непрерывно переходит в окраску недиссоциированной молекулярной формы. При этом было бы невозможно определить значение pH, так как нет резкого изменения окраски. Однако здесь вступают в действие физиологические свойства зрительного аппарата человека: глаз до тех пор воспринимает окраску как чистую, пока содержание второго компонента в 10 раз не превысит содержание первого. Чистую Окраску второго компонента можно видеть, если ее интенсивность примерно в десять раз больше, чем интенсивность окраски первого.
При переходе из кислой в щелочную область значение pH, при котором происходит первое видимое изменение окраски индикатора — кислоты, можно определить на основании уравнения (88) по формуле
(рН)г = p^Hind-lg Ю = р/Сньа-1 (89)
При дальнейшем увеличении pH чистая окраска анионов индикатора возникает при
(pH)2=pKHlnd-lg l/10=ptfHInd4-l (90)
Область перехода окраски индикатора ДрН = 2. При справедливости упомянутых выше предпосылок она расположена симметрично относительно точки перехода окраски, которая получается при CHina = Cina-. Для этого условия pH=p/<Hlnd.
Сказанное справедливо только для двухцветных индикаторов. Как видно из уравнения (88), для этих индикаторов pH перехода окраски зависит только от соотношения снша/сща- и не зависит от общей концентрации индикатора
C0 = cHind-|-clnd- (91)
Двухцветный индикатор позволяет измерить pH оптическим методом на основе закона Ламберта — Бера, в соответствии с которым измеряемая оптическая плотность Е основной формы индикатора Ind- пропорциональна степени диссоциации а сопря
38. 'Основы количественного химического анализа
143
женной кислотной формы индикатора Н Ind, общей концентрации индикатора Со, и толщине поглощающего слоя х:
Е = га.Спх (92)
где е — молярный коэффициент погашения для анионов индикатора. При известных значениях е, Со и х можно из измеренного значения E = lglo/I рассчитать истинную степень диссоциации кислотной формы индикатора. Это осуществляют, вводя степень диссоциации а в выражение закона действующих масс для диссоциации кислотной формы индикатора (уравнение (87,)). Так как Cind-=iaC,0 и cnind=(l—а)Са, получим
„ гща-сн Зо+ а
^Hind = - = сн3о+ тттЕГ (93)
4HInd 1 —
Логарифмирование дает
рН-р/Сн1па=1ёт^Г Л94>
При построении графической зависимости а от разности pH — рДи та, рассчитанной по уравнению (94) для каждого спектро-
Рис. Д.52. Зависимость степени диссоциации слабых кислот от pH.
фотометрически измеренного значения, получают кривую, изображенную на рис. Д.52. Из нее по известной величине показателя индикатора р/Сн ind для каждого значения степени диссоциации можно получить соответствующее значение pH. При переходе из кислой в щелочную среду первое изменение окраски индикатора происходит при соотношении концентраций Ста-/ /сн!па~0,1, а чистую окраску аниона индикатора можно заметить при соотношении Сша-Дн ina ~ 10. Эти соотношения кон-
144
Д. Химический анализ
центрацин соответствуют степени диссоциации «1 = 0,09 и «2 = = 0,91. Из рис. Д.52 видно, что (pH)i—p7<Hind =—1 и (рН)2— —рЛн1п<1= + 1, т. е. интервал перехода окраски индикатора ДрН = 2, как было показано выше. Для индикатора «-нитрофенола (рЛн ind = 7,2) при переходе в щелочную среду первый переход окраски происходит при (pH) [ = 7,2—1=6,2, и чистую окраску аниона можно различить при (рН)2 = 7,2+1 = 8,2.
Рис. Д.53. Схема выбора индикаторов при титровании смеси 0,1 н. растворов хлороводородной и уксусной кислот.
Таким образом, можно определить интервал перехода окраски кислотно-основных индикаторов, наиболее пригодных при проведении титрования кислот различной силы, т. е. можно выбрать индикатор, интервал перехода окраски которого входит в пределы скачка на кривой титрования. Пригодность индикатора для конкретного титрования можно оценить по интервалам перехода окраски индикаторов, обозначенным на кривой титрования (рис. Д.53).
В соответствии с рис. Д.53 0,1 н. растворы сильных кислот можно титровать со многими индикаторами, интервал перехода окраски которых находится в области pH 3—10, в то время как для титрования слабых кислот обычно пригодны лишь некоторые индикаторы, например 0,1 н. раствор уксусной кислоты титруют с фенолфталеином.
38. Основы количественного химического анализа
145>
Одноцветные индикаторы. Уравнение (88) действительно также для одноцветных индикаторов. Снова рассмотрим поведение индикаторов при переходе из кислой в щелочную среду.. В этом случае ун ind~yind-~ 1. Пусть бесцветной формой будет Hind. Такую же концентрацию окрашенной формы Ind”, окраску которой можно увидеть, обозначим с'ша-. Тогда значение pH, при котором происходит первое изменение окраски индикатора, равно
рН = р/Сн1па (CHInd/cInd-) = P^HInd 1g (Со С Ind- (95)
В целом общая концентрация индикатора больше, чем минимальная концентрация наблюдаемой окрашенной формы. Тогда, так как Со/с'ща- 1, получим
pH = p/<Hind—IgCo/c'ind’ (96)
В отличие от уравнения (88) сюда входит общая концентрация индикатора. Поэтому при использовании одноцветных индикаторов концентрация их должна быть постоянной.
В то время как двухцветный индикатор начинает изменять свою окраску при рН = р/<н ind±l, первое изменение окраски одноцветного индикатора происходит при pH<p/<Hind, так как C/lnd~<CCQ.
Солевой эффект. До сих пор мы при расчетах пренебрегали влиянием коэффициентов активности. Однако если проводить титрование в среде конц. НС1 или при определении pH колориметрическим методом, в уравнение (88> необходимо ввести член, учитывающий влияние коэффициентов активности: /5)1114= lg(YHind/yind ), который называют солевой ошибкой. Поскольку электростатическое влияние молекул незначительно, можно с достаточным приближением принять yHind~l- Тогда уравнение (88) переходит в следующее:
PH = p/^HInd — 1g (CHInd/Clnd”) + 1g Ylnd“ (97>
В точке перехода окраски индикатора равенство cHind=Cind~ не означает, что pH=pKHmd, так как здесь
pH < рЛнша (98)
поскольку в растворах с умеренной концентрацией Yind-<1- Это означает,, что переход окраски индикаторов — нейтральных кислот, таких, как крезоловый красный, тимоловый синий, бромкрезоловый зеленый, фенолфталеин и др.,, в растворах солей происходит при более низких значениях pH, а индикаторов — оснований, таких, как а-нафтиловый красный, диметиловый желтый и др., при более высоких значениях pH, чем в растворах, не содержащих солей.
Влияние неводных растворителей на интервал перехода окраски индикатора. Электролиты, растворенные в неводных растворителях, менее диссоциированы, чем в воде, что связано с более низким значением диэлектрической проницаемости неводных растворителей по сравнению с водой и более низкой энергией сольватации иоиов. Таким образом, кислотный индикатор изменил бы свою окраску в спиртовом растворе при большем значении pH, чем в воде.
10—1880
Л 46
Д. Химический анализ
38.3.1.6. Буферные растворы
^Буферными растворами называют растворы, значение pH которых при добавлении умеренного количества сильной кислоты или основания практически не изменяется. Буферные растворы имеют важное значение при проведении работ, связанных с воспроизведением определенных значений pH (например, при калибровке pH-метров, при проведении химических реакций или в биологических процессах), а также для поддержания постоянного pH растворов при действии небольших количеств кислот и оснований, таких, как диоксид углерода и пары кислот из воздуха лаборатории или следовые количества щелочей из стенок стеклянных сосудов.
Буферные растворы можно приготовить растворением эквимолярных количеств слабой или средней силы кислоты и ее соли в воде. Этот раствор соответствует точке полунейтрализации (т=0,5) при титровании слабой кислоты сильным основанием, когда pH раствора равно p/G кислоты (см. рис. Д.44). При логарифмировании выражения закона действующих масс получим формулу для расчета pH буферного раствора:
pH — p/(s—lg(cHX/cx-) (99)
При этом предполагают, что коэффициенты активности кислоты и анионов примерно равны. Таким образом, pH раствора в первую очередь определяется значением pKs н в меньшей степени соотношением концентраций снх/сх--
Поскольку буферный раствор содержит относительно высокие концентрации слабой кислоты и ее соли, можно принять с достаточным приближением, что равновесные концентрации Снх и сх- равны исходной концентрации кислоты Снх и соли ‘Снах. Тогда
pH —p/Q—lg CHX/CNaX (100)
Из уравнения (100) следует, что pH буферного раствора не зависит от разбавления, поскольку (наряду со значением p/G) только соотношение концентраций оказывает влияние на pH. Но эта зависимость является приближенной, поскольку коэффициенты активности зависят от концентрации.
Буферное действие состоит в том, что диссоциация слабой кислоты существенно подавлена высокой концентрацией одноименных анионов. Например, при добавлении основания происходит в основном переход кислоты в соль без образования добавочного большого количества ионов Н3О+ в отличие от «индивидуальных растворов кислот.
38. Основы количественного химического анализа
147
Количественно буферное действие выражают через буферную емкость р:
p = dC/dPH (101>
где dC — количество добавленного сильного основания (в моль/дм-3), вызывающее изменение pH на величину dpHL Обратите в-нимание на то, что 1 /р характеризует крутизну кривой титрования слабой кислоты сильным основанием. Для расчета зависимости буферной емкости от параметров буферной системы исходят из уравнения! (111) и подставляют в него» сх- из уравнения (71). При этом каждое добавляемое количество С сильного основания определяют как
С = Сон cH3O+4“Q-^s/Ws4-CH3O+) (102>
Для вычисления р по уравнению (101) получают вначале отношение dC/dcH3o+:
dC_______Kw____i_______C()KS__ / ] qo-
dcH3O+ c2H3O+ (Ks + СНзо+)2
Из определения p,H=—1§Сн3о+ следует отношение» dpH/dcH3o+ = —М(1/сн3о+), (где М — модуль десятичного логарифма), введение которого в уравнение (103) дает
dC а о о ( । । со^н3о+ \ ..п
__ _ р _ 2,3 ^он-+сНзо+ + + СНзО+)2 J (* 04>
На рисунке Д.54 представлены три функции:
о ___. „ . о С<Л*н3о+ . „ ___
Pi сНз0+, р2- (/<з + сНзО+)2 ’ Рз сон-
аддитивное сложение которых по уравнению (104) дает суммарную функцию (пунктир). Как видно из рис. Д.54, при среднем значении p/Q и при умеренных концентрациях эта функция в основном определяется величиной !р2. Вычисляя с?Р2/с1сн3о+ и приравнивая производную к нулю, получим рНтах = р./С>. Таким образом, максимальное значение буферной емкости соответствует pKs и составляет
Pmax=O,58Go (105>
т. е. буферная емкость прямо пропорциональна общей концентрации.
Функции Pi и Рг отражают чрезвычайно сильное буфериру-Ющее действие сильных кислот или оснований.
Для приготовления буферных растворов желательно рассчитать буферную емкость из навески слабой кислоты и ее соли. Уравнение для расчета выводят из уравнений (70) и 10*
448
Д. Химический анализ
Фис. Д.54. Зависимость буферной емкости р от pH и общей концентрации Со.
(71), при умножении которых получают
C<Msch3o+
Р=2,3 (сон-+%о+ + «^
(106)
(107)
Как видно из рис. Д.54, для области 4<рН<10 можно пренебречь членами Сон- и Сн3о+- Кроме того, можно принять, что Снх = Онх и сх~=Сыах, а также Со = Снх4~^мах- При этом из уравнения (107) следует
С-НХ + C-NaX
(108)
Если буферный раствор содержит эквимолярные количества слабой кислоты и ее соли, буферная емкость имеет максимальное значение, т. е. Снх==Смах- Отсюда
Р = 2,3 (СнХ/2) = О,58Со = 0,58 (CHX-]-CNaX) (Ю9)
Уравнение (109) аналогично уравнению (105). Например, бу
38. Основы количественного химического анализа
149
ферная емкость раствора, содержащего по 0,1 моль/дм3 уксусной кислоты и ацетата натрия, равна ₽ = 2,3 (0,01/0,2) = =0,12 моль/дм3. Таким образом, в соответствии с уравнением (101) только при добавлении 0,12 моль/дм3 сильной кислоты или основания pH буферного раствора изменится на единицу.
S8.3.1.7. Индикаторные ошибки
Систематическая индикаторная ошибка. При кислотно-основном титровании значению pH в точке эквивалентности, так называемому показателю титрования, не всегда соответствует pH перехода окраски индикатора, выражаемого в виде показателя индикатора р/Снша- При этом возникает систематическая индикаторная ошибка F. Ее определяют как разность между количеством С (моль/дм3) титранта, добавляемого до перехода окраски индикатора, и Со титранта, необходимого для достижения точки эквивалентности:
Е = С—Со (ИО)
При этом не учитывают увеличение объема титруемого раствора. Способ нахождения относительной систематической
индикаторной ошибки Fr = — F/Co схематично представлен на рис. Д.55. Можно видеть, что относительная ошибка тем больше, чем больше разность между показателем индикатора и показателем титрования и чем менее резок подъем на кривой в области точки эквивалентности.
Для расчета абсолютной индикаторной ошибки выражают разность С — Со через равновесные концентрации Сн3о+ и Сон-, существующие При pH = p/Giind. При этом ис
Рис. Д.55. Схема, иллюстрирующая
относительную систематическую ииди' каторную ошибку Fr.
ходят из того положения, что на каждой стадии кислотно-
основного титрования количество образующегося основания должно быть равно коли-
честву образующейся кислоты, так как в водных растворах не содержится свободных протонов. Далее, при этом следует
учесть, что вода реагирует как кислота и основание и что добавляемое сильное основание ОН- полностью переходит в сопряженную с ним кислоту Н2О, вновь образовавшееся количе
150
Д. Химический анализ
ство которой можно выразить через количество добавленного основания. Тогда
С-(-сНз0+== сон-4*сх~ (111)
При введении общей концентрации Со слабой кислоты (Со= = сНх+сх~) в уравнение (111) получим
С—Со = F= Дон сн.(о+) снх (И2)
Уравнение (112) справедливо для титрования слабой кислоты сильным основанием. Для оснований в уравнении (112) перед скобкой нужно поставить знак минус и снх заменить равновесной концентрацией сх~. Значение концентраций сон-, Сн3о+ и Снх в точке перехода индикатора лучше взять из логарифмической диаграммы для соответствующей кислоты.
Выражение, более удобное для практических расчетов, получают при замене в уравнении (112) величины снх выражением из уравнения (70) :
с„.о-)-С0 (113)
Для оснований в соответственно преобразованное уравнение
(112) вместо равновесной концентрации сх-вводят выражение из уравнения (71).
На рис. Д.56 приведены зависимости трех членов уравне-_______________________ ния (ИЗ) от значений pH. Из '______________________рисунка следует, что в случае
Рис. Д.56. Зависимость систематической индикаторной ошибки F от интервала перехода окраски индикатора, pH и силы кислот.
Кривая сн3о + относится к титрованию сильных кислот; Со=О,О1 моль/дм3.
сильных кислот ошибка титрования для большой области значений pH сравнительно мало чувствительна к изменениям pH в точке перехода индикатора (вертикальный участок кривой). В случае очень слабых кислот (p/Q = 7) кривая пересекает ось (штриховая линия) под большим углом, что указывает на возникновение большой ошибки при небольшом изменении pH. Точность титрования кислоты при определении конечной точки титрования с помощью индикатора быстро уменьшается с изменением силы кислот.
Титрование сильных кислот. Для случая титрования сильных кислот уравнение (112) можно упростить, поскольку при этом в растворе
38. Основы количественного химического анализа
151
не существует недиссоциированных молекул кислоты НХ, т. е. Снх = 0:
F ~ coi г <-н3о+ 1 Fr~ (<?он- сНз0+)i'C0 {114), (115) При титровании 0,1 и. раствора сильной кислоты сильным основанием в присутствии индикатора метилового оранжевого (рЛн1па = 3,8) по уравнению (115) получаем систематическую индикаторную ошибку:
Fr « 10 ’ -10 « - = —10-2’8 =-1,6-1 О'» = -0,16%
' 1U 10
При титровании с фенолфталеином (р/Сн ind = 8,2) относительная систематическая ошибка равна
pr « ,10 !_-o ilu ~ 10-м = +0,002%
При выборе индикатора для титрования с заданной точностью ориентируются на уравнение [115].
Пример. Нужно определить, в какой области значений pH должен находиться интервал перехода окраски индикатора при титровании 0,01 н. раствора основания с точностью 0,1%.
Если уравнение (115) переписать применительно к основанию и преобразовать, то получим
— (сон--сн3о4 — C0Fr = Ю-2-10-3 = 10-6 моль/дм3
Это соотношение действительно, если си3о+«10-5 моль/дм3, тогда сон~=10-!) моль/дм3 (и этой величиной по сравнению с сн3оь можно пренебречь), а также при сон~«10~5 моль/дм3 (в этом случае сНзо+~ ~10~9 моль/дм3 и этой величиной по сравнению с сОн~ можно пренебречь). Таким образом, для проведения указанного титрования нужно выбрать индикатор, показатель которого находится в интервале pH 5—9.
Определение систематической индикаторной ошибки из диаграммы 1g с—pH. Систематическую индикаторную ошибку можно определить, нанося на диаграмму 1g с—pH титруемой кислоты значение интервала перехода окраски соответствующего индикатора. На рис. Д.57 приведена логарифмическая диаграмма 0,1 н. раствора сильной кислоты и интервалы перехода окраски некоторых индикаторов с допущением, что титрование проводят до первого изменения окраски. При этом отрезок прямой, соответствующий интервалу перехода окраски индикатора, левым концом касается прямых Н3О+ или ОН- соответственно. По уравнению (115) и из диаграммы, приведенной на рис. Д.57, можно рассчитать систематическую относительную индикаторную ошибку при титровании сильных кислот. В кислотной области £’он-<^Сн3о+> а в щелочной области сн3о+<^СоН-> поэтому Fr~—Сн3о+/С'о или FrttCow-ICn. Значение общих концентраций можно взять из диаграммы, так что ошибку легко рассчитать.
152
Д. Химический анализ
Рис. Д.57. Диаграмма для расчета систематической относительной индикаторной ошибки при титровании 0,1 и. раствора сильной кислоты 0,1 н. раствором сильного основания в присутствии различных индикаторов.
1 — 2,4-динитрофенол (интервал перехода окраски 2,0<рН<4,7); 2— метиловый красный (4,4<рН<6,2); 3 — бромтимоловый синий (6,0<рН<;7,6); 4 — фенолфталеин (8,0< <рН<10,1).
Как видно из рис. Д.57, наиболее точные результаты получают при титровании в присутствии индикатора, интервал перехода окраски которого близок к точке нейтральности (pH 7). Кроме того, ясно, что точность определения зависит от окраски, приобретаемой индикатором в результате титрования. Например, при титровании с метиловым красным до получения чистой желто-оранжевой окраски, т. е. до pH 6, индикаторная ошибка уменьшается до ~1О“зо/о по сравнению с ошибкой—
Рис. Д.58. Диаграмма для расчета систематической относительной индикаторной ошибки при титровании 0,1 и. уксусной кислоты 0,1 н. раствором сильного основания в присутствии различных индикаторов.
/ — бромтимоловый синий (интервал перехода окраски 6,0<рН<7,6); 2 — фенолфталеии (8,0<рН< 10,1); 3 — тимолфталеин (9,3<рН<10,6).
38. Основы количественного химического анализа
153
0,04%, получаемой при титровании до pH 4,5 (штриховая линия на рис. Д.57).
Из рис. Д.57 можно также сделать вывод, что точность
титрования уменьшается с уменьшением концентрации кислоты, так как при этом линия Со смещается вниз, подъем становится короче и ошибка Fr больше.
Для слабых кислот действительно уравнение (112): вблизи точки эквивалентности при рН^>рТ, т. е. при титровании до
точки эквивалентности, Снх2>Сн3о+ и Снх^Сон-, так что в этом
случае Fr = —Снх/С0. Отсюда индикаторную ошибку рассчитывают, как указано выше.
Из рис. Д.58 ясно, что точность титрования слабых кислот значительно меньше, чем сильных кислот, и что она уменьшается с понижением силы кислоты: чем слабее кислота, тем больше значение pKs сдвигается вправо, тем короче подъем и тем больше индикаторная ошибка.
Случайная индикаторная ошибка. Если систематическая индикаторная ошибка равна нулю, т. е. достигнуто совпа-
дение значений показателей Рис. Д.59. Схема нахождения случай-титрования и индикатора, то ной индикаторной ошибки ±ДС [212]. всегда остается случайная ин-
дикаторная ошибка, связанная с тем, что при визуальном определении точки перехода окраски индикатора из-за физиологических особенностей зрения значение ее можно определить только с колебаниями ±0,4 единицы. Рис. Д.59 иллюстрирует
влияние этой ошибки. Абсолютное значение случайной ошибки
АС зависит от скачка dpH/dC на кривой титрования в точке эквивалентности. Считая отрезок между pHi и рНг на кривой рис. Д.59 линейным, получим следующую зависимость
d pH/dC = АрН/АС
(П6)
По определению dC/dpH— это буферная емкость, тогда
F=AC=pApH (117)
Из уравнений (104) и (117) для абсолютной ошибки получаем
F= ±2,ЗАрн(Сон~+сн^+Со (кХС)2') (И8)
Уравнение (118) пригодно для расчетов как при титровании
154
Д. Химический анализ
кислот, так и при титровании оснований. Ks всегда можно за-5 менить значениями констант кислотности. Уравнение (118) можно упростить. Применяя уравнения (70) и (71), получим
F= ±2,ЗДрН (Сон-+^+ ) <И9>
\ иНЛ I Л у
Вблизи точки эквивалентности при титровании слабой кислоты сильным' основанием справедливо неравенство снх^сх-, и тогда из уравнения (119) следует
F— — 2,ЗДрН(сНдО+4-снх_1~сон’~) (120)
Для случая титрования слабой кислоты сильным основанием применимо уравнение (111). Поскольку Со=снх + сх- и вблизи точки эквивалентности Сл:С0, из уравнения (111) можно записать
сн3о++сн№сон~ 021)
Подставляя это выражение в уравнение (120), получим
F = ± 2,3 Д pH • 2 (сНд0+ Cfjx) (122)
Подстановка уравнения (121) дает
Г=±2,ЗДрН.2сон- (123)
Как уже упоминалось, при применении визуальной индикации Д pH «0,4. Поэтому приближенно
F« ±2(сн.1О+4~снх) или F«;+2c0H~ (124), (125)
Поскольку при титровании сильной кислоты сильным основанием Снх = 0, из уравнения (124) следует ,
Fж ±2сНзО+ = ±2-10"7 моль/дм® (126)
Относительная ошибка составит
Fr« ±2- 1О-7/Со моль/дм3 (127)
Сильные кислоты или основания можно титровать с большой точностью даже в сильно разбавленных растворах в присутствии индикатора, точка перехода окраски которого лежит вблизи точки нейтральности.
Ошибку при титровании слабой кислоты проще всего рассчитать из уравнения (125). Комбинирование уравнения (57) для показателя титрования с уравнением (125) дает
F - ±2 Уад = ±2 ]/(^ОЖГ (128)
38. Основы количественного химического анализа
155
Относительную ошибку рассчитывают по формуле
Fr = ±2 + 2 ]/^ЖС0) (129)
Таким образом, случайная относительная ошибка тем больше, чем слабее и разбавленнее титруемая кислота.
38.3.1.8. Практическая часть
Приготовление растворов титрантов для кислотно-основного титрования. В ацидиметрии в качестве стандартных растворов титрантов применяют в основном растворы соляной и серной кислот. Серную кислоту, как малолетучее соединение, используют, если необходимо длительное кипячение титруемого раствора с избытком кислоты. Однако при ее применении возможно образование малорастворимых осадков с некоторыми катионами, например сульфатов бария и свинца.
Стандартный раствор соляной кислоты можно приготовить двумя способами: разбавлением кислоты, имеющей постоянную температуру кипения (азеотропа), и стандартизацией разбавленного раствора кислотьв приблизительной концентрации по первичному стандартному веществу.
1. Приготовление стандартного раствора из азеотропа НС1 с водой. Необходимо работать строго по приведенной методике: для получения абсолютно чистой конц. НС1 в аппарате для получения газов к чистой концентрированной соляной кислоте по каплям добавляют концентрированную серную кислоту, не содержащую галогенидов и нитратов. Поток газа промывают солянокислым раствором хлорида меди(1) (для связывания следовых количеств оксида азота и свободного хлора) и пропускают в прокипяченную дистиллированную воду, находящуюся в пропаренной колбе из кварцевого стекла. Кислоту хранят в закрытом сосуде в темноте.
Часть приготовленной соляной кислоты разбавляют до получения 500 см3 кислоты пл. 1,10 г/см3. Ее перегоняют со скоростью 3—4 см3/мин, дистиллат собирают в чистую колбу и время от времени переливают в цилиндр. После отгонки 375 см3 кислотьв следующие 50 см3 кислоты собирают в пропаренную сухую колбу, в которой теперь находится чистый азеотроп соляной кислоты. При сборе этой фракции термометром контролируют постоянство температуры кипения, до и после сбора фракции по барометру определяют давление воздуха и находят среднее из двух значений. Точное содержание кислоты рассчитывают интерполяцией данных из табл. Д.14.
Для приготовления из азеотропа 0,1 н. раствора кислоты, поступают следующим образом. Количество соляной кислоты, необходимое для приготовления 1 дм3 0,1 н. раствора кисло-
156
Д. Химический анализ
Таблица Д.14. Концентрация НС1
в соляной кислоте в зависимости от давления
Давление, мПа Содержание HCI в кислоте, масс. % (в вакууме) Масса кислоты, содержащая 1< моль НС) (при атмосферном давлении), г
103,334 20,173 180,601
102,658 20,197 180,387
101,325 20,221 180,173
99,992 20,245 179,959
98,659 20,269 179,746
97,325 20,293 179,535
ты, рассчитанное по табл. Д. 14, капельной пипеткой переносят в чистую, абсолютно обезжиренную и сухую колбу Эрленмейе-ра (коническую) и взвешивают. Точную массу взвешенной кислоты определяют, вычитая массу взвешенной закрытой колбы. Затем кислоту разбавляют примерно равным объемом воды во избежание потерь НС1, раствор тщательно переносят в мерную колбу вместимостью 1 дм3 и доливают до метки прокипяченной дистиллированной водой при температуре градуировки мерной посуды) (обычно 25°C).
2.Стандартизация 0,1 н. раствора соляной кислоты по тетраборату натрия. Тетраборат натрия (бура) имеет некоторые преимущества перед обычно применяемым карбонатом натрия: большую эквивалентную массу— 190,736 г/моль (у безводного карбоната 52,994 г/моль), возможность простой очистки перекристаллизацией; тетраборат не нужно сушить до получения постоянной массы, он практически негигроскопичен; при титровании с метиловым красным можно наблюдать четкий переход окраски индикатора, так как этот индикатор при комнатной температуре не реагирует на очень слабую борную кислоту.
Приготовление тетрабората натрия как первичного стандартного вещества. Соль Х’агВ-.О?-ЮНгО (ч.д.а.) перекристаллизовывают из дистиллированной воды (50 см3 воды на 15 г соли). Кристаллизацию нужно проводить при температуре не выше 55 °C, так как может образоваться пентагидрат. Кристаллы тщательно отсасывают и промывают два раза водой, затем еще два раза 95%-ным спиртом и наконец два раза эфиром. Для промывания 10 г тетрабората применяют порции промывной жидкости по 5 см3. После каждого промывания кристаллы тщательно отсасывают. Продукт раскладывают тонким слоем и сушат при комнатной температуре в течение 20 ч. При хранении в склянке с притертой пробкой свойства его не изменяются в течение трех-четырех недель.
Для стандартизации титранта три пробы тетрабората натрия титруют в присутствии метилового красного до бледно-ро
38. Основы количественного химического анализа
157
зовой окраски. Для повышения точности определений при проведении титрования готовят раствор сравнения; (растворяют 0,1 г хлорида натрия, 2,2 г борной кислоты и 5 капель метилового красного в 500 см3 воды).
Определение содержания гидро- и пиросульфатов в их смеси. К 100 см3 воды, охлажденной до 0 °C и находящейся в бане со льдом, добавляют 3 капли смешанного индикатора (метиловый красный+бромкрезоловый зеленый) и при перемешивании вносят примерно 100 мг анализируемой смеси; смесь быстро нейтрализуют гидроксидом натрия и по мере протекания реакции оттитровывают ионы НзО+, образующиеся в результате гидролиза пиросульфата. Через каждую минуту в течение часа замечают положение мениска (отсчет времени начинают ,с момента внесения анализируемой смеси в воду).
Строят кривую зависимости расхода объема титранта от времени и рассчитывают из нее содержание гидро- и пиросульфата в пробе. По данным логарифмической кривой скорости-гидролиза рассчитывают период гидролитического полураспада пиросульфата.
Этот опыт можно продолжить и получить интересные результаты. Например, проводя гидролиз пиросульфита при различных температурах, можно рассчитать энергию гидролитической активации связи S—О—S.
Если при титровании анализируемой смеси добавлять метиловый спирт, то скорость гидролиза изменяется в зависимости от количества метанола и количество щелочи, вступающей в реакцию, уже не будет соответствовать стехиометрическому соотношению. Это- обусловлено протеканием следующей-реакции:
S2O;2- + СН3ОН HSO4- + CH3OSO3-
Анализ полифосфата. Примерно 200 г тонкоизмельченного в порошок полифосфата растворяют в 100 см3 воды и раствор кипятят в течение 8 ч. При этом происходит гидролитическое расщепление всех связей Р—О—Р. После охлаждения раствор титруют 0,1 н. раствором щелочи в присутствии фенолфталеина. Расход титранта щ. Затем проводят обратное титрование 0,1 н. раствором кислоты в присутствии смешанного индикатора, при этом расход титранта и2. По объему щ определяют количество в молях сильно- и слабокислотных ОН-групп, по ^2 — только слабокислотных. (Продумайте, как, зная эти объе~ мы, рассчитать содержание фосфора, натрия и сшивающих групп.)
458
Д. Химический анализ
38.3.2. Реакции окисления — восстановления
38.3.2.1. Теоретические основы
Для окислительно-восстановительных реакций можно привести ряд интересных аналогий с реакциями кислотно-основного взаимодействия.
1. В реакциях кислотно-основного взаимодействия происходит обмен протонов, в окислительно-восстановительных — обмен электронов между веществами, участвующими в реакции.
2. Зависимость напряжения ячейки от концентрации описывается математическим выражением, сходным с выражением для pH:
pH = p/Cs-lg-^ и е = Е 0-
t г s о с^_ и ZFM ° (?ох
Восстановитель — донор электронов при этом соответствует кислоте — донору протонов, окислитель — основанию.
3. В системе кислота — основание соотношение концентрации компонентов определяют по величине pH, в реакциях окис-.ления — восстановления соотношение восстановителя и окислителя определяют по величине напряжения ячейки.
4. Шкала кислотности, составленная из величин pKs, соответствует шкале, отражающей окислительно-восстановительную способность элементов значениями стандартных потенциалов Ео, так называемому ряду напряжений.
Наряду с общими признаками реакций обоих типов имеются также и существенные отличия. Так, механизм окислительно-восстановительных реакций значительно сложнее, чем реакций кислотно-основного взаимодействия. Это проявляется в том, что реакции кислотно-основного взаимодействия протекают очень быстро, в то время как реакции окисления — восстановления во многих случаях замедленны, что часто мешает проведению анализа. Небольшая скорость ряда окислительно-восстановительных реакций обусловлена в основном тем, что электронные Переходы часто сопровождаются частичным изменением1 или полным разрушением молекулярной структуры участвующих в реакции частиц. Поэтому окислительно-восстановительные реакции между катионами и анионами часто проходят через стадии обмена лигандов, что, например, имеет место при окислении иодид-ионов ионами железа(III), которое обычно описываются простым уравнением!
2РеЗ++2Г ---► 2Fe2+4-I2
Б действительности ионы железа гидратированы, так что электрон иодид-иона не может непосредственно переходить к иону
38. Основы количественного химического анализа
15®-
железа. Реакция протекает через промежуточные стадии:
[Fe(OH2)e]3+ + I" -> [Fe(OH2)5I]2+ + H2O
[Fe(OH2)sI]2+4-I~ -> [Fe(OH2)4I2]++ H2O
[Fe(OH2)4I2]+ + 2H2O -> [Fe(OH2)el2+4-I2-
2I-2" -» I2 + 2I-
При протекании окислительно-восстановительных реакций атомы лигандов во многих случаях образуют мостики между реагирующими частицами (катионами), что можно показать на. Примере следующих хорошо изученных реакций:
е"
-|2+ f-l+2) г "“х -14+-
[Co(NH3)5ClJ + [Сг(ОН2)6] -> [(NH3)5Co-Cl-Cr(OH2)5] +Н20
-у-* [Co(NH3)5(OH2)] + [Cr(0H2)5Cl] ;
Наряду с галогенид-ионами электронными мостиками могут быть цианид- и сульфат-ионы, молекулы воды и даже сравнительно большие анионы карбоновых и дикарбоновых кислот.
Промежуточные соединения образуются также при окислении молекул диоксида серы хлорат-ионом:
Ох <-) .0 О, о
o\s + io-ci^o -> o>s-o-d^o->.so1 + cior-
Торможение реакций окисления — восстановления происходит также в том случае, когда степени окисления окислителя и восстановителя изменяются неодинаково. При этом переход; электронов невозможен в ходе одной бимолекулярной реакции, а требует протекания ряда бимолекулярных реакций или реакций высшего порядка. В первом случае образуется нейтральный промежуточный продукт, имеющий большой запас энергии, во втором случае необходимо столкновение более чем двух реагирующих частиц, вероятность которого мала. Ускорить реакции в принципе можно катализом или индуцированием.
Катализ. Применяя катализатор, можно увеличить скорость, реакции. Обычно это связано с тем, что в присутствии катализатора путь реакции связан с меньшей энергией активации,, чем это необходимо для протекания прямой реакции. Катализатор принимает участие в электронных переходах, но в ходе-процесса выделяется, так что в результате остается неизменным.
601
Д. Химический анализ
Пример. Реакция между ионами церия и мышьяка
2Се (IV) + As (III) -------------► 2Се (III) + As (V) (130)
протекает очень медленно, так как не является одностадийной бимолекулярной. При добавлении к этой системе ионов Мп (II) скорость реакции существенно возрастает благодаря протеканию следующих промежуточных реакций:
Се (IV) 4- Мп (II) ----► Се (III) + Мп (III) (131)
Се (IV) + Мп (III) -----► Се (III) + Мп (IV) (132)
Mn(IV) + As(III) -------> Мп (II) + As (V) (133)
Каждая из этих реакций является бимолекулярной. При сложении их получаем суммарное уравнение (130).
'Ниже приведены другие примеры каталитических реакций:
Компоненты реакции МпО4“ + H3AsO3
С1О3- + 1-
Се (IV) + H3AsO3 Ce(IV)+Cl~
Н2О24-1~
S2O82- + Fe(II) S2O82~ + Mn(II) l2+N3—
Катализаторы
I-
Os, Ru, V
OsO4, Mn(II), I-
Ag+
MoO32~, wo42-, vo43-
Cu(II)
Ag+
S2-, S2O32-, SCN-
Скорость каталитической реакции часто пропорциональна концентрации катализатора. Реакции такого типа дают возможность количественно определить содержание катализатора по измерению скорости реакции, что используют в последнее время как эффективный метод анализа следовых количеств элементов.
Индуцирование. Если реакция между А и В протекает с заметной скоростью только после добавления компонента С и при этом С участвует в реакции (в отличие от каталитических реакций!), то реакцию между А и В называют индуцированной, а реакцию между А и С — первичной. При этом А является актором, В — акцептором и С — индуктором.
Влияние индуцирующей реакции А + С состоит в том, что свободная энтальпия переносится на систему Лф-В, благодаря чему во второй реакции легче преодолевается барьер энергии •активации. Ниже приведены примеры индуцированных реакций:
Индуцированные реакции
А В
<г2О?2- +Мп(П)—>-Мп(П1)
Н2С2О4 +HgCl2
.MnO4- + Cl-
Первичные реакции
А С
Сг2О?2— +H3AsOs
Н2С2О4 +МпО4-
MnO4- -FFe(II)
38. Основы количественного химического анализа
161
I- + О2 I- + S2O82-, Fe(III), [Fe(CN6]3-, V(V)
Sn(II) + О2 Sn(II) + CrO42-, MnOr, H2O2
H2SO3 + О2 H2SO3 + CrO42-, MnOr, H2O2, s2o82-
Восстановитель + Ог Восстано- витель +Окислитель
В объемном анализе протекание индуцированных реакций в общем нежелательно. Так, например, последняя из приведенных реакций показывает, что реакция окисления — восстановления может индуцироваться окислением восстановителя кислородом воздуха, что приводит к ошибкам титрования.
38.3.2.2. Окислительно-восстановительное титрование
Реакция окисления-—восстановления пригодна для объемного анализа, если она протекает быстро или если существует возможность ускорения замедленной реакции действием катализаторов и подавления мешающих индуцированных реакций. Кроме того, должен существовать индикаторный электрод, правильно отражающий изменения потенциала в зависимости от концентрации электрохимически активных частиц. Математической основой окислительно-восстановительного титрования является уравнение Нернста
Е = Е0—^-1пК
(134)
где Е — напряжение ячейки, В; £0 — стандартный потенциал, В; z — число электронов, переходящих в процессе стехиометрической реакции; F—число Фарадея; К — отношение активностей или (приближенно) концентраций веществ в каждой реакции.
Если система находится в равновесии, то К — это термодинамическая константа равновесия; при этом второй член правой части уравнения (134) становится равным £0 и £ = 0.
Для практических расчетов в уравнение (134) вводят десятичный логарифм и рассчитывают фактор RTIMF при температуре 25°С = 298 К; # = 8,3143 Дж/(К-моль), М = 0,43429 и # = 96485 Кл/моль; RT/MF=0,0590 В, так что можно записать
£ = £0-^^-lgtf (135)
При соединении полуэлемента редокс-пары Fe3+/Fe2+, который получают опустив платиновую пластинку в раствор ио-11—1880
162
Д. Химический анализ
нов железа, со стандартным водородным электродом протекает реакция
2Fe3+ 4- Н2 3=*: 2Fe2+ -f- 2Н+ (136 >
Уравнение (135) в этом случае имеет вид
0,0590 , c2Fe2+c2H3o+
£-£»——2 13 с.р,..рн, <137>
Поскольку для стандартного водородного электрода сНзо+ = = 1 моль/дм3 и /?н2 = 0,1 мПа, получаем окончательно
Е = Е0—0,0590 lg (138)
cFe3+
Таким образом, напряжение ячейки прямо пропорционально логарифму отношения концентраций сРез+ /сре2+- В присутствии второй редокс-системы с большим (т. е. более положительным) потенциалом, чем потенциал системы Fe3+/Fe2+, реакция
Ох + Fe2+ -► Red 4- Fe3+ (139 >
на каждой стадии обмена полностью смещена вправо, и при постепенном добавлении более сильного окислителя ионы железа (II) можно полностью окислить в ионы железа (III). Измеряя изменение потенциала ячейки, можно проследить за изменением соотношения Сре3+/сре2+ в зависимости от добавляемого количества окислителя. Это дало бы возможность осуществить окислительно-восстановительное титрование ионов железа (II). Важнейшим1 условием возможности проведения окислительно-восстановительного титрования является необратимость реакции
Vi Oxj -f- v2 Red2 < » Redx 4- v2 Ox2 (140>
Иначе говоря, величина константы равновесия К должна быть, как можно большей. Ее рассчитывают из- напряжений полуэлементов для обеих редокс-пар:
г- г- 0,0590- , . cV1Redi, ,, ,,,
Ei = Eoi---; (141)
4 с Ох1
Е2 = Е0а—(142) С Ох2
До добавления окислителя Oxi к восстановителю Red2 потенциал Е2 имеет наименьшую величину (отрицательную или положительную), так как на этой стадии cRedg3>cOx2. Потенциал Е1, напротив, при этом имеет наибольшую величину, так как в растворе чистого окислителя Cox1^>CRed1-При добавлении окислителя тотчас же возникает напряжение ячейки АЕ, как раз-
38. Основы количественного химического анализа
163
ность потенциалов полуэлементов Е\ и Е2-.
Л г- г. Г- Г, Г7 0,0590 ,
&Е— Ег Е2 — EOi Ео2 z 1g cqX1 ) "т"
+ Q>059p4g(-^MV2 (143)
Величина ДЕ постоянно уменьшается, так как Ei и Е2 изменяются в противоположном направлении: восстановитель 2 все больше переходит в окислитель 2, при этом Е2 возрастает, но Ei вследствие уменьшения сОХ1 уменьшается. Наконец устанавливается (очень быстро в случае реакций, протекающих без торможения) состояние, при котором Е\—Е2 и тем самым ДЕ = 0. Теперь окислительно-восстановительная реакция протекает в соответствии с уравнением (140) до достижения химического равновесия. Тогда
ДР-Е-Е-Е —Е --°’0590 1g 1 2 01 02
= Eol-Eo2--^-lgtf=O (144)
МК=~„К^В'^: 045)
Обратите внимание на то, что концентрации, входящие в уравнения (141) и (142) в отличие от уравнения, (144) в общем не являются равновесными.
В уравнении (145) Еп2 всегда представляет собой стандартный потенциал восстановителя, a EOi — стандартный потенциал окислителя левой части соответствующего уравнения реакции.
Примеры. 1. Какой из ионов — Fe2+ или Sn2+ — является более сильным восстановителем в кислой среде? Для ответа на этот вопрос нужно решить, в каком направлении протекает реакция
2Fe2+ -)- Sn1+ 3Fe3+ Sn2+ (146)
В ряду напряжений находим величины Ео ге(ш)/ге(П) = 0,771 В и Ео Sn(iv)/Sn(ii) = 0,15 В. Из реакции (146) видно, что Sn4+ является окислителем, а восстановителем — Fe2+. Тогда из уравнения (145) следует, что
2 (0,771 —0,15)
рЕ = - о?О59О-- = 21,05 и К» 10-21 (147)
Таким образом, при 25 °C реакция самопроизвольно протекает, т. е. ионы 8п2+ полностью восстанавливают иоиы Fe3+. При перестановке левой и правой частей уравнения (146) последовательность значений Ео в уравнении (147) также нужно изменить, при этом рК будет отрицательным и вывод не изменится.
2. Можно ли определить содержание Fe(II) в растворе титрованием перманганатом калия? 1
1 1*
164
Д. Химический анализ
Соответствующее уравнение реакции:
МпО4" + 5Fe2+ + 8Н3О+ Mn2+ + 5Fe3+ + 12Н2О (148)
В процессе реакции в соответствии с этим уравнением осуществляется переход пяти электронов, т. е. г=5, тогда
5 (0,771 — 1,52) 0,0590 ——63,5
Получена большая отрицательная величина рТС Таким образом, реакция полностью сдвинута в прямом направлении. Следовательно, перманганат пригоден для титрования ионов Fe2+ (эта реакция протекает, кроме того, без торможения).
В целом показатель окислительно-восстановительной реакции р/(Е можно определить как
AG0 = — RT\nK=— zE0F (149)
pK£=-lgK=-^-£0 (150)
Окислительно-восстановительную реакцию, потенциал которой равен Ео, можно всегда записать как
Ох-5-ze" ->- Red (151)
Только в этом случае знаки рКЕ и AG0 совпадают.
Таким образом, реакция, характеризующаяся большой отрицательной величиной стандартного потенциала Ео, т. е. большой положительной величиной показателя окисления — восстановления рКв (например, Na+4-е-—>Na), сильно сдвинута влево.
При 25 °C уравнение (150) имеет вид
рК£^ —16,95г£0 (В-1) (152)
Показатель окислительно-восстановительной реакции можно использовать так же, как другие показатели равновесия (например, кислотности или растворимости); при этом появляется возможность сложением показателей составляющих реакций получить показатель суммарной реакции и константу ее равновесия. Например, для реакции (148):
МпО4" 4- 8Н+ 4- 5е" -> Мп2+4-4Н2О
Fe2+ --► Fe3+ + е~
МпО4" 4- 5Fe2+ 4- 8Н+ -► Мп2+ 4- 5Fe3+ 4- 4Н2О
рК4 = рК£Мп = — 16,95 -5-1,52 = — 128,8
рК2=—pK£>Fe= 16,95-1 -0,771 = 13,07-5
рК = р/^4- 5рК2 = -128,84-13,07 • 5 = —63,46
38. Основы количественного химического анализа
165
Кривые окислительно-восстановительного титрования. При построении графической зависимости напряжения ячейки от добавляемого количества титранта получают кривые титрования. Рассмотрим титрование восстановителя Rech окислителем Oxi (реакция (140)). Так же, как в случае кислотно-основных равновесий, рассчитывают некоторые характерные точки кривой титрования.
1. Напряжение ячейки для исходного титруемого раствора Red2 (т = 0) получают из уравнения для системы Ox2/Red2:
п П 0,0590 cRed2 п 0,0590 Со~ сОх2
£,=£«----^lg—=£„---------— Ig СОх, (|83)
Приблизительное значение Е2 можно рассчитать, если для Сох2 принять достаточно малое значение, 10~4 моль/дм3. Так как при этом сОх2<С0, уравнение (153) переходит в следующее:
£2 = Е02-^0.1g-^- (154)
2 02 % СОх2 V ’
2. Напряжение ячейки в момент, когда добавлено 50% титранта (т = 0,5). После окисления половинного количества исходного восстановителя Скеб2 = Сох2 и из уравнения (153) получаем Е2 = Ео2. Кривая титрования редокс-пары независимо от общей концентрации Со при т = 0,5 проходит через точку, в которой E = Eq. Это напоминает титрование слабых кислот, когда кривая титрования при т = 0,5 проходит через pH = pKs.
3. Напряжение ячейки в точке эквивалентности Еэ (т = 1) рассчитывают на основе следующих рассуждений. В ходе всего титрования напряжение ячейки Е равно напряжениям полуэлементов Ei и Е2. Тогда и в точке эквивалентности
Еэ1 = Еэ2~Еа (155)
До точки эквивалентности при взаимодействии эквивалентных количеств восстановителя Red2 и окислителя Oxi образуются эквивалентные количества Ох2 и Redi, откуда
^Ох2 = Ср (156)
где Со— общая концентрация титруемой редокс-пары, которая равна количеству титранта, добавленного в точке эквивалентности (в моль/дм3 без учета изменения объема раствора). Если бы реакция проходила до конца, то для точки эквивалентности можно было бы считать Сох1=Скеа2=0. Но тогда в этой точке в соответствии с уравнениями (141) и (142) потенциалы редокс-пары должны были бы составлять Ег=—оо. и Е2= + оо, что в действительности невозможно. Напротив, верно, что
^Oxj cRed2 причем обе концентрации очень малы.
(157)
166
Д. Химический анализ
Уравнения (141) и (142) несколько преобразуют. Для, этого z — число переходящих в процессе стехиометрической реакции электронов заменяют стехиометрическим коэффициентом Vi и электрохимическим эквивалентом zj:
z = v1z1 = v2z2 (158)
Тогда
р р 0,0590 - roi ~ Z1 cOxt (159)
с о 0,0590 cRed, En — Eo„ Ig 2 02 Z2 COx2 (160)
Если в выражение закона действующих масс применительно к
рассматриваемой реакции (161) вводят соотношение концентра-
ций в точке эквивалентности cOx2=CRed1, получают уравнение (162), справедливое только для точки эквивалентности:
цеп, (162)
Уравнение (162) подставляют затем в уравнения (159) и (160)', умножают уравнение (159) на гь а уравнение (160) на z2. Сложение полученных результатов дает
z^-J-z^ = z^+2^- 0,0590?g/K+0,0590 lg/tf
Отсюда с учетом уравнения (155) получают выражение для расчета потенциала в точке эквивалентности:
g _ Zigoi ~Ь 3 zi + гг
Если электрохимические эквиваленты Zi обеих систем, участвующих в реакции окисления — восстановления, равны, т. е. Zi = z2 (симметричная реакция окисления — восстановления), то
= ~2~ (Eoi+^oa)
(163)
(164)
Уравнение (162) позволяет установить соотношение концентраций в точке эквивалентности. Оно показывает, что полнота протекания реакции окисления — восстановления и точность титрования тем больше, чем больше константа равновесия, поскольку, чем больше К> тем больше отношение концентраций cRed Мох и сохг/ске<12в точке эквивалентности и тем; более полнее происходит переход Oxi в Redi и Red2 в Ох2.
4. Напряжение ячейки в момент, когда добавлено количество титранта в два раза больше стехиометрического (т = 2). Титруемый восстановитель Red2 в точке эквивалентности пол
38. Основы количественного химического анализа
167
ностью переходит в Ох2, причем эквивалентное количество окислителя Oxi достаточно полно переходит в Redi. При повторном добавлении такого же количества. Охь какое необходимо для достижения точки эквивалентности, степень оттитровы-вания т = 2 и cox^CRedi и, как следует из уравнения (159), £=£оь
На рис. Д.60 приведена кривая титровании Fe2+ ионами Се4+ (в среде 1 н. НС1О4), на которой отмечены четыре харак
терные напряжения ячейки: Е2, Ео2, Е3 и Е01. Из рис. Д.60 можно сделать общий вывод, что скачок потенциала не зависит от концентрации титруемого вещества и тем больше, чем больше разность EOi—Е02.
Если в уравнение реакции входят также ионы гидроксо-ния, то положение всех точек на кривой титрования зависит от pH.
Подготовка к окислительно-восстановительному титрованию. При необходимости определения общего количества вещества окислительно-восстановительным титрованием его предварительно нужно полностью восстановить или
Рис. Д.60. Кривая окислительно-восстановительного титрования Fe2+ ионами Се4+ (в среде 1 н. хлорной кислоты) .
окислить. Поскольку растворы
титрантов, применяемые в методах окислительно-восстановительного титрования, в большинстве случаев являются окислителями, наибольший интерес представляет количественное восстановление определяемого компонента. Применяемый для это-
го восстановитель и продукт его окисления при последующем титровании не должны оказывать мешающего влияния, или
должна существовать простая возможность их удаления из раствора.
Этим требованиям обычно удовлетворяют металлы. При этом лучше применять металлы с высокой степенью окисления, потенциал которых имеет небольшую отрицательную величину; если использовать металлы, потенциал которых имеет большую отрицательную величину, происходит сильное выделение водорода. Из этих соображений очень хорошим восстановителем является кадмий, который применяют в редукторе Джонса. Редуктор представляет собой стеклянную' трубку, заполненную стружками электролитического кадмия, через которую. медленно протекает восстанавливаемый раствор. В кад-
168
Д'. Химический анализ
Таблица Д.15. Восстановление действием амальгам
Амальгама Zn Амальгама Cd Амальгама Pb Амальгама Bi
Fe(III) —>Fe(II) Fe(III)—>-Fe(II) Fe(III)—>Fe(II) Fe(III)—>Fe(II)
V(V)—^V(II) V(V)—>V(H) V(V)—>V(II) V(V)—^V(IV)
Cr(III)—>Cr(II) — — —
Mo(IV)—►Mo(III) Mo(lV)—^Mo(llI) Mo(VI)—>-Mo(III) Mo (VI)—►Mo (V), Mo(III)
U(VI)—>-U(IV), U(VI) —>U(IV) U (VI)—MJ (IV)
U(III) Ti(IV)—^Ti(III)
W(VI)—>W(III) W(VI)—>-W(V)
миевом редукторе могут проходить следующие реакции: Fe(III) —->Fe(II), Ti(IV)—>Ti(III), Ст (III) —>Cr(II),MoO42-—> I—>-Мо(Ш), С10з~—»C1". Хорошими восстановителями являются жидкие амальгамы различных неблагородных металлов, таких, как цинк, кадмий, олово, свинец и висмут. Эти амальгамы "при самой высокой концентрации ионов водорода не выделяют Н2, так как его выделение на ртути требует высокого перенапряжения. Примеры восстановления амальгамами приведены в табл. Д.15. Поскольку восстановительные свойства амальгам могут существенно изменяться в зависимости от pH, а реакции четко фиксируются, с их помощью можно проводить селективное восстановление, которое позволяет раздельно определять многие окислители в растворе.
Для восстановления Fe(III) обычно применяют хлорид олова (II), избыток которого окисляют хлоридом ртути(II). Образующийся при этом хлорид ртути(I) малорастворим и не разрушается окислителем — титрантом. К летучим восстановителям, избыток которых можно легко удалить кипячением раствора, относятся сероводород и диоксид серы.
Из сильных окислителей в объемном анализе применяют: пероксид водорода, пероксодисульфаты, висмутат натрия (NaBiO3) и перманганат калия. Избыток пероксида водорода и пероксодисульфата можно разрушить кипячением в щелочной среде, перманганата'—нагреванием с соляной кислотой, спиртом или азидом.
Изящным методом количественного окисления или восстановления является метод с использованием электронообменных смол. При их применении посторонние ионы, за исключением ионов гидроксония, не попадают в раствор. Поскольку окислительно-восстановительный потенциал этих обменников зависит также от pH и структуры смолы, его можно варьировать в широких пределах. Были проведены следующие восстановле
38. Основы количественного химического анализа
169
ния на электронообменных смолах: Fe(III)—HFe(II), Cr (VI)—-^Cr(III), Mo (VI)—^Mo (IV) и Mo(III), V (V)t->-t—*V(IV).
38.3.2.3 . Окислительно-восстановительные индикаторы
Окислительно-восстановительные индикаторы представляют собой соединения, окисленная и восстановленная форма которых имеет различную окраску. Обычно это органические соединения, восстановленная форма которых бесцветна. Хотя окислительно-восстановительные индикаторы формально можно сопоставить с кислотно-основными индикаторами (первые фиксируют определенное значение потенциала, вторые — определенное значение pH), необходимо помнить и об их существенных различиях. Поскольку в окислительно-восстановительной реакции обычно участвуют протоны, интервал перехода окраски индикатора зависит от pH. При визуальном титровании с окислительно-восстановительными индикаторами нужно поддерживать постоянное значение pH с помощью буферных растворов. Другое отличие от кислотно-основных индикаторов состоит в-том, что переход окраски окислительно-восстановительных индикаторов обычно необратим.
Если раствор в точке перехода окраски индикатора имеет потенциал Е, а Е01 — стандартный потенциал индикаторной системы, то
£=£о1+4г1падХраУ (165>
ui Red
где аю*, ^/Red — активности окисленной и восстановленной форм индикаторной системы; у — стехиометрический коэффициент.
Уравнение (165) можно преобразовать: £-£«.+4г(1п^-+1п^?Г-'/рН) (166)
\ 4Red TtRed j
где у — коэффициент активности. '
Рассмотрим поведение одноцветного окислительно-восстановительного индикатора. Первое изменение окраски заметно, если концентрация окрашенной окисленной формы равна с*юх. Концентрация восстановленной формы тогда равна Со—с*юх, если Со — общая концентрация индикатора. Тогда уравнение (166) переходит в следующее:
« = WH) (167)
где М — модуль натурального логарифма.
170
Д. Химический инализ
Таким образом, потенциал перехода окраски индикатора зависит а) от специфических свойств индикаторной системы, выраженных через £0»; б) от температуры (£ог также зависит от температуры); в) от интенсивности окраски окисленной формы индикатора, выраженной через с*юх; г) от общей концентрации индикатора Со; д) от электростатических взаимодействий в растворе (вызванных содержанием солей), выраженных через уюх и y/Red; е) от pH раствора.
Ошибка титрования близка нулю, если потенциал перехода •окраски индикатора равен потенциалу в точке эквивалентности. Практически допустимой считают относительную ошибку в10_3 или 0,1%, благодаря чему возможен широкий выбор окислительно-восстановительных индикаторов. При титровании восстановителя 2 окислителем 1 предельные значения потенциалов (в вольтах) получают из следующих уравнений:
Р р । 0,059 • 99,9 р . 0,18 /ico\
Д = — lg-Qj- = £02H—— (168)
Р Р I 0,0э9 1 0,1 р 0,18 п rq\
£1 = £014------— 1g^9T = £oi---(169)
Эти уравнения основаны на том, что значение потенциала
до точки эквивалентности определяется системой титруемого вещества, а после точки эквивалентности — системой титранта. Выбранную область А£ = £2—можно рассчитать из уравнения (170):
А£ = А£о4-О,18 (170)
Z1Z2
Величина скачка потенциала, находящегося в пределах ДЕ, а также количество окислительно-восстановительного индикатора обусловлены в первую очередь разностью стандартных потенциалов, так как второй член уравнения мал.
Специальные окислительно-восстановительные индикаторы. Неорганическими окислительно-восстановительными индикаторами являются системы 1_/12 и 12/1С1. Первую систему, для которой £о = О,535 В, можно применять для индикации реакции Ce(IV) +Sn(II). Избыток Ce(IV) определяют по появлению синей окраски крахмала. Монохлорид иода служит для индикации конца реакций, протекающих в сильнокислой среде, например при титровании раствором йодата. В восстановительной среде из монохлорида иода выделяется свободный иод, тотчас же вызывающий обесцвечивание небольшого избытка окислителя. Иод извлекают тетрахлоридом углерода или хлороформом.
К органическим окислительно-восстановительным индикаторам относится ферроин — трис(о-фенантролин)овый комп-
38. Основы количественного химического анализа
171'
леке железа, имеющий красную окраску в присутствии двухвалентного железа и бледно-синюю в присутствии Fe(III). Потенциал перехода окраски индикатора £=1,20 В. Нитроферроин— нитропроизводное ферроина, изменяет окраску при £ = = 1,25 В. В соответствии с этими значениями £ оба индикатора можно применять только при титровании растворами очень сильных окислителей, например Ce(IV).
При титровании церием(1У) или бихроматом хорошими индикаторами являются дифениламин, дифенилбензидин и дифениламиносульфоновая кислота. Дифениламин имеет особенно важное значение при титровании Fe(II) раствором бихромата. Потенциал перехода его окраски £ = 0,76 В значительно' ниже стандартного потенциала системы Fe(II)/Fe(III), что1 при обычных условиях не дает возможности индицировать точку эквивалентности при титровании Fe(II). Поэтому потенциал системы Fe(II)/Fe(III) смещают в область более отрицательных значений добавлением фосфорной кисло™, образующей с ионами Fe(III) устойчивый комплекс. Следовые количества вольфрама мешают определению конечной точки титрования,, так как делают необратимым переход окраски индикатора.
Лучшими свойствами, чем дифениламин, обладает дифениламиносульфоновая кислота, легко растворимая в воде и не1 чувствительная к присутствию вольфрама. При £=0,83 В происходит четкий переход окраски из бесцветной в красно-фиолетовую.
Трифенилметановые красители, такие, как эриоглауцин, зеленый эрио, сетоглауцин и др. представляют собой двухцветные обратимые окислительно-восстановительные индикаторы. В кислой среде можно наблюдать переход окраски от зеленой к оранжевой, розовой или сине-красной.
К необратимым индикаторам относятся метиловый оранжевый, стифниновая кислота и др. Их обычно применяют при титровании броматом. При избытке бромата образуется свободный бром, окисляющий индикатор.
38.3.2.4 . Практическая часть
Титрование раствором перманганата калия. Перманганат является сильным окислителем; его окислительно-восстановительный потенциал существенно зависит от активности ионов НзО+:
В сильнощелочной среде МпО4”----► МпО42У £0 = 0,54В (171)
В слабощелочной или нейтральной среде
МпО4~----► МпО2, £0 = 0,58В (172)
В кислой среде МпО4~ —► А1п2+, £0= 1,52В (173)
172
Д. Химический анализ
В соответствии с этим эквивалентная масса перманганата в окислительно-восстановительных реакциях может иметь по крайней мере три величины.
Наиболее часто перманганатометрическое титрование проводят в кислой среде. При этом индикатором является сама система: конец титрования определяют по интенсивной окраске перманганат-ионов, окраска которых хорошо различима при концентрации 10-6 моль/дм3. При проведении титрования в очень разбавленных растворах индикацию конечной точки можно сделать более четкой, добавив в титруемый раствор перед концом титрования несколько капель окислительно-восстановительного индикатора, например дифениламин-я-сульфоно-вой кислоты или сульфата трис(о-фенантролин)железа (II) (ферроина).
Приготовление стандартного раствора КМпО^ Раствор перманганата калия нельзя приготовить простым растворением навески соли в воде, так как нельзя получить перманганат калия, свободный от ионов марганца более низких степеней окисления. Особенно мешает оксид Мп (IV), катализирующий разложение перманганата. Из-за высокой окислительной способности соединений Мп(VII) происходит медленное разложение растворов титранта (восстановление пылью), поэтому характеристики раствора необходимо время от времени устанавливать заново. Поскольку под действием света скорость разложения увеличивается, раствор нужно хранить в склянках из темного стекла.
Для приготовления примерно 0,1 н. раствора КМ.ПО4 растворяют 3,2—3,25 г. КМпО4 (ч. д. а.) в 1 дм3 дистиллированной воды. Этот раствор медленно кипятят в накрытом стакане в течение 15—30 мин. После охлаждения раствора до комнатной температуры его фильтруют через фильтр с пористой стеклянной пластинкой, промытый сначала теплой смесью бихромата с азотной или серной кислотой, а затем большим количеством воды, в склянку с пришлифованной пробкой, обработанной аналогично фильтру. Эту операцию проводят для разрушения находящихся в растворе окисляющихся примесей и для удаления образующихся при этом продуктов, например диоксида марганца.
Стандартизацию раствора перманганата проводят по оксиду мышьяка(III) или оксалату натрия. Реакцию окисления | оксида мышьяка (III), которая может протекать очень медленно, ускоряют добавлением' иодида, йодата или хлорида иода. '
Стандартизация раствора КМпО^ по оксиду мышьяка. Оксид высушивают при температуре 105 °C (осторожно, ядовит!). Навеску -~0,25 г переносят в стакан вместимостью 400 см3 и растворяют в 10 см3 чистого холодного раствора едкого нат-
38. Основы количественного химического анализа
173
pa, приготовленного растворением 20 г NaOH в 100 см3 воды. Затем добавляют 100 см3 воды, 10 см'3 HCI и 1 к. 0,0025 М раствора иодида калия и медленно титруют раствором перманганата до появления устойчивой в течение 30 мин слаборозовой окраски. При более высоких требованиях к точности проводят «холостой опыт». Для этого такой же объем раствора гидроксида натрия и иодида в воде, приготовленных как указано выше, титруют раствором перманганата и полученный объем титранта (максимально 0,03 см3) вычитают из полученного ранее значения. Титрование повторяют еще два раза и рассчитывают среднее значение. Три результата титрования должны совпадать с точностью 0,1%.
Титрование по Рейнгардту — Циммерману. Важнейшим применением перманганатометрического титрования является определение Fe(II) в солянокислом растворе по Рейнгардту— Циммерману, так как в лабораториях металлургических и сталелитейных производств анализируемые пробы металлов обычно растворяют в соляной кислоте. С термодинамических позиций хлорид может окисляться перманганатом количественно, как это видно из значений стандартных потенциалов: для МпО4_/Мп2+ Ео=1,52 В; для пары CI2/CH £'о=|1,36 В. Практически реакция проходит только в сильнокислых или концентрированных растворах хлоридов, в противном случае она замедлена. В присутствии ионов Fe(II) реакция индуцируется, так что Fe(II) нельзя титровать перманганатом в солянокислой среде. Эту трудность преодолевают, добавляя к титруемому раствору большое количество соли Мп (II) и фосфорную кислоту.
О механизме этой важнейшей реакции, применяемой в объемном анализе, а также о влиянии добавки марганца(II) до настоящего времени нет общепринятых объяснений.
По одной из гипотез, предложенной Г. Шефферсом, ионы железа (II) частично переводятся перманганатом в неустойчивое соединение с более высокой степенью окисления (больше +3), которое энергично окисляет хлорид-ионы. Ионы марганца(П), добавляемые по Рейнгардту — Циммерману, вступают в реакцию, реагируя вместо ионов железа(II) с перманганатом или вместо хло-рид-ионов с соединениями железа более высокой степени окисления и таким образом препятствуют окислению хлорида.
В соответствии с другими гипотезами при добавлении соли марганца(II) уменьшаются окислительно-восстановительные потенциалы системы Мп (II)/ /Mn(VH), а также промежуточных систем Mn(II)/Mn(IV) и Mn(II)/Mn(III), которые особенно эффективно окисляют хлорид (Ео=1,51 В). Фосфорная кислота связывает Fe(III) в бесцветный комплекс, и окислительно-восстановительный потенциал системы Fe(II)/Fe(III) при этом снижается; тем самым появляется возможность устранить желтую окраску солянокислого раствора Fe(III) и четко определить конечную точку титрования. Кроме того, фосфорная кислота образует фосфатоманганат(Ш), тем самым снижается окислительно-восстановительный потенциал системы Мп(II)/Мп(III).
174
Д. Химический анализ
Титрование по Фольгарду — Вольфу. В основе метода лежит реакция
2МпО4- + ЗМп2+ + 2Н2О -► 5МпО2 -}- 4Н+
Этот метод обычно применяют для анализа сплавов железа. Титруемый солянокислый раствор содержит ионы железа(III), затрудняющие четкость индикации конечной точки титрования. Для устранения их мешающего влияния в раствор вносят оксид цинка, связывающий ионы железа(III). в осадок гидроксида. Кроме того, ионы цинка подавляют адсорбцию ионов Мп(II) осадком диоксида марганца, на которую расходуются неокисленные ионы марганца(II).
Титрование раствором бихромата калия. Титрование основано на восстановлении бихромат-ионов:
СгаО72- + НН+ + бе" -► 2Сг3+ + 7Н2О
Стандартный потенциал системы Сг (VI)/Сг (III) £О=1,36 В. Как видно, его значение ниже, чем для системы Мп (VII)/ /Мп(II), но, несмотря на это, метод имеет ряд преимуществ: из бихромата калия можно приготовить первичный стандартный раствор, который устойчив при хранении. Кроме того, хлориды окисляются бихроматом только в очень сильнокислых растворах и поэтому не мешают определению. Поскольку в. данном случае Fe(II) не оказывает индуцирующего действия, его можно определять в присутствии хлорид-ионов. Точку эквивалентности можно устанавливать потенциометрически или с помощью дифениламиносульфоновой кислоты в качестве окислительно-восстановительного индикатора. Можно также применять внешний индикатор — гексацианоферрат(III) калия.
Титрование солями церия(1¥). Цериметрическое титрование основано на протекании реакции
Се (IV) + ё~ -» Се (III)
Окислительно-восстановительный потенциал солей Се (IV) вследствие склонности ионов Ce(IV) к образованию комплексных соединений существенно зависит от типа анионов. Например, при ан+ = 1 моль/дм3 для сернокислого раствора сульфата церия (IV) Ео=1,44 В, для азотнокислого раствора Се(ЫОз)< £0=1,61 В, для хлорнокислого раствора перхлората церия(Г¥). £о=1,7О В. В хлорнокислом растворе при ан+ = 8 моль/дм3 Е0=1,87 В.
Растворы солей церия (IV) имеют более высокие значения окислительно-восстановительных потенциалов, чем перманганат; они значительно устойчивее, чем раствор перманганата, в окисляют соли Fe(II) также и в присутствии ионов С1~. Другим преимуществом их является однозначность механизма реакции. Конечную точку титрования определяют потенциометрн-
38. Основы количественного химического анализа
175
ческим методом или с помощью индикаторов ферроина или иитроферроина.
Титр растворов солей Се(IV), которые обычно готовят растворением трисульфатоцерата(IV) аммония (NH4)2[Ce(SO4)3J • • 2Н2О, устанавливают по щавелевой кислоте или по оксиду мышьяка (III).
Титрование хлором. Титрование раствором хлора, несмотря на удовлетворительное значение стандартного окислительно-восстановительного потенциала (Е0 = 1,36 В), невозможно вследствие неустойчивости раствора. Для титрования в щелочных средах можно применять раствор гипохлорита. При этом хорошие результаты дает добавление брома, который, окисляясь в гипобромит, .быстрее реагирует с веществами, чем гипохлорит.
Еще лучше применять n-толуолсульфоновую кислоту, так называемый хлорамин Т, поскольку в кислой среде она также является окислителем:
В водных растворах хлорамин Т ведет себя аналогично гипохлориту. Например, его можно с успехом применять вместо хлорной воды в качественном анализе. Титр раствора устанавливают реакцией окисления иодидов в солянокислой среде с последующим титрованием тиосульфатом выделившегося иода. Конечную точку при титровании хлорамином определяют по образованию соединения крахмала с иодом, образующимся при окислении ионов I" избытком первого реагента.
Хлорамин можно применять для проведения следующих определений: As(III)—>-As(V), Sb(III)—>Sb(V), Sn(II)—> —>Sn(IV), N2H4-^N2.
Титрование броматом. Метод броматометрии основан на протекании реакции
ВгО3" + 6Н+-|-бе" -► Вг" + ЗН2О
Возможно также проводить титрование свободным бромом полученным из бромата:
ВгО3" + 5Вг" + 6Н+ -> ЗВг2 + ЗН2О
В качестве окислительно-восстановительных индикаторов применяют метиловый оранжевый, стифниновую кислоту или индигокармин, которые вначале бронируются, а затем необратимо окисляются. Поскольку эти индикаторы могут разрушиться •еще до достижения точки эквивалентности из-за локальных избытков реагента, целесообразно вводить индикатор незадолго
176
Д. Химический анализ
до конца реакции, который определяют предварительным тит-
рованием.
Почти обратимым окислительно-восстановительным индикатором при окислении броматом является а-нафтофлавон, который при бромировании окрашивается в коричневый цвет. Реакцию проводят в 5%-ном растворе соляной кислоты.
Типичным примером броматометрических определений является титрование As (III), Sb (III) и Sn(II). Можно предложить общий метод определения сильных окислителей: раствор окис-
лителя, например хлора, гипохлорита натрия, хлората калия,
пероксида водорода,
пероксодисульфата
калия,
бромата
калия
восстанавливают оксидом мышьяка(III) и избыток его оттитро-
вывают раствором бромата.
Важное значение имеет метод быстрого количественного бромирования оксина (8-оксихинолина) бромид-броматной смесью, так как при этом все ионы металлов, образующие малорастворимые оксинаты Mg(II), Zn(II), Cu(II), Cd(II), Al (III), Fe(III), можно определять объемным методом. Избыток бромид-броматного раствора титруют иодометрически.
Титрование иодом. Стандартный потенциал окислительно-восстановительной системы 12/1_ Во = 0,535 В, т. е. иод являет-
ся лишь слабым окислителем. Поэтому число титрований, проводимых раствором иода, невелико. Поскольку иод малорас-
творим в воде и титр такого раствора достаточно неустойчив, применяют раствор трииодида калия (полученный растворением иода в растворе иодида калия) или иодид-иодатный, или иодид-броматный растворы, которые в кислой среде выделяют иод. Реакцию между иодидом и броматом нужно каталитически ускорять действием молибдена и проводить в сильнокислой среде, так же как реакцию с йодатом.
Точку эквивалентности при титровании иодом можно определить по образованию соединения иода с крахмалом. Харак-
терным примером титрования иодом является определение As(III):
AsO33- + I2+Н2О AsO43-+ 21-+ 2Н+
Поскольку значения стандартных потенциалов обеих систем очень близки (£oi2/i-= 0,53 В; 5oas(V)/as(hd = O,56 В), положение равновесия существенно зависит от pH. Количественное окисление арсенита возможно только при связывании выделяющихся в реакции ионов гидроксония. С другой стороны, значение pH системы не должно быть слишком большим, так как в противном случае установится равновесие Iz+OH'^tH-r ЮН, при котором невозможно определить конечную точку титрования по образованию соединения иода с крахмалом. Для поддержания оптимального pH можно в раствор добавить гидро-
38. Основы количественного химического анализа
177
карбонат натрия (pH 6,5) или систему НгРОс/НРСЬ2- (pH 7), обладающие буферными свойствами.
Примером эффективного применения йодометрического титрования является метод К. Фишера, который используют для определения содержания воды в неводных системах. Коричневый раствор титранта готовят из иода, диоксида серы, пиридина и метанола в мольных соотношениях 1 :3: 10:50. Эта смесь нестабильна в присутствии воды вследствие протекания реакции (Ру-пир идин):
ЗРу + I2 + SO2 + СН3ОН + Н2О -> ЗРуН+ + 2Г + CH3OSO3“
Эта реакция в действительности проходит по стадиям:
SO2 + C5H6N ---► C5H5N.SO2
C5H5N-I2 + H2O --► C5H5NH+ + IOH + I-
IOH + C5H5N-SO2 + C5H5N --> C5H6N.SO3 + C5H6NH+ + r
C5H5N-SO3 + CH3OH —-*• C5H5NH++CH3OSO3-
B присутствии воды окраска раствора Фишера изменяется из коричневой в слабо-желтую. Конечную точку титрования определяют по появлению коричневого окрашивания, обусловленного окраской раствора титранта. Более точным является потенциометрическое установление конца титрования.
Титрование иода. Вследствие низкого значения окислительно-восстановительного потенциала системы I-/12 многие вещества могут восстанавливаться иодидами. К раствору добавляют избыток иодида и выделившийся иод титруют раствором тиосульфата в слабокислой среде. При этом протекает реакция с образованием тетратионат-иона:
I2 + 2S2O32- -j—J, 2Г + 54ОЛ
Раствор титранта нестабилен, так как тиосульфат разлагается при действии слабых кислот (диоксид углерода воздуха) и бактерий.
Характеристики стандартного раствора тиосульфата можно установить по иоду или лучше по иодид-иодатному раствору. Реакцией восстановления с иодидами можно определять такие сильные окислители, как диоксид марганца, пероксид водорода, гипохлорит, хромат, перманганат и т. д.
В некоторых случаях, например при определении диоксида марганца, для того чтобы реакция началась, раствор нужно довольно сильно подкислить. Так как в сильнокислой среде происходит окисление иодида кислородом воздуха, лучше пропускать в раствор иодида хлор, полученный окислением соляной кислоты. Таким образом можно проводить количественное определение некоторых высших оксидов и соединений кислорода, например диоксида свинца, селеновой кислоты, теллуровой кислоты, хлората калия и др. (метод дистилляции Бунзена).
12—1880
178
Д. Химический анализ
Очень точным является йодометрический метод определения меди(П)
,Сц2++21- --> Cul+V2l2
Титрование растворами солей титана(Ш). Редокс-пара Ti (III)/Ti (IV) характеризуется низким значением стандартного окислительно-восстановительного потенциала (Ео=О В), поэтому ее можно применять в качестве сильного восстановителя. Растворы устойчивы при защите от действия воздуха. Раствором хлорида титана(Ш) можно титровать железо(Ш), хроматы, хлораты, перхлораты. При этом в растворе не должна находиться азотная кислота, поскольку она также восстанавливается.
Обычно используют потенциометрическую индикацию конечной точки титрования. В качестве индикаторов при титровании железа применяют роданид-ионы или метиленовый синий, который по достижении точки эквивалентности обесцвечивается.
Титрование растворами соединений Сг(П). Стандартный окислительно-восстановительный потенциал системы Сг(П)/ /Сг(Ш) Ео = —0,41 В. Растворы солей хрома(П) имеют более сильные восстановительные свойства, чем растворы солей титана (III), и дают возможность проводить ряд важных определений обычно с использованием потенциометрической индикации конечной точки титрования.
38.3.3. Реакции комплексообразования
38.3.3.1. Равновесия реакций комплексообразования
Реакции комплексообразования имеют важное значение в аналитической химии. Часто эти реакции сочетаются с кислотно
основными, окислительно-восстановительными и осадительными равновесиями. Вопросы равновесий реакций комплексообразования многократно будут обсуждаться в различных главах книги. Данный раздел посвящен хелатометрии — методу титри-метрического анализа, в котором используются реакции комплексообразования.
Метод комплексонометрии или хелатометрии был предложен в 1945 г. Г. Шварценбахом. Метод основан на том, что органические соединения определенного типа, такие, как нитрилотри-уксусная кислота (НТУ), этилендиаминтетрауксусная кислота (ЭДТА) и т. п., довольно быстро реагируют с ионами металлов с образованием устойчивых хелатных комплексов. Такие соединения названы Шварценбахом комплексонами (в ГДР их называют «хелаплексы»). Так как для определения концентрации ионов металлов были найдены соответствующие цветные инди
каторы, появилась возможность применить этот метод для тит
38. Основы количественного химического анализа
179
рования ионов металлов анионами хелатообразующих реагентов по аналогии с титрованием ионов водорода ионами гидроксила.
Реакции комплексообразования известны давно. Возникает вопрос: почему их не применяли раньше в объемном анализе и в чем их отличие от реакций, предложенных Шварценбахом? Чтобы ответить на этот вопрос, рассмотрим реакцию ионов меди (II) с аммиаком. Реакция протекает ступенчато:
Си(Н2О)42+Ц- NH, Cu(NH3) (Н2О)32+ 4-НаО рКх = 4,13
Cu(NH3) (Н2О)32+ + NH3 Cu(NH3)2(H2O)22+ + Н2О ptf2 = 3,48
Cu(NH3)2(H2O)22+ 4- NH3 Cu(NH3)3(H2O)24 + H2O pK3 = 2,87
Cu(NH3)3(H2O)2++ NH3 =?=> Cu(NH3)42++ H2O ptf4 = 2,11
Рядом с уравнениями реакции приведены значения рК комплексов. К.п — константы устойчивости комплексов определяют как
V __ С(-*ип_
" CCun_!CNHs
где сС11п — равновесная концентрация комплекса
[Cu(NH3)n(H2O)4_n]2+. Значение рД относительно мало, т. е. в каждый момент титрования Си (II) аммиаком в растворе одновременно сосуществуют различные комплексы, концентрация которых различна. В точке эквивалентности образуются не только комплексные ионы [Cu(NH3)4]2+, но и значительные количества соединений с меньшим содержанием NH3. Только при большом избытке аммиака основное количество Си (II) связано в тетрамминокомплекс. Это приводит к тому, что концентрация ионов металла уменьшается постепенно и по кривой титрования нельзя установить конец реакции (рис. Д.61).
Десятичный логарифм константы суммарного равновесия К. равен lg/C = lg/Ci + lgK2+lg^3 + lgK4=12,59. Значение К получают из следующего уравнения:
К = CCu(NH3)42+
C4NH3CCu(H2O)42+
Это соотношение напоминает случай алкалиметрического титрования смеси слабых кислот, концентрации которых близки.
Напротив, величины константы устойчивости комплексов с хелатообразующими лигандами значительно больше (хелатный эффект). Так, для комплексных соединений меди
рК1ря= 18,8 (три= (H2N—С2Н4—)3N)
р^етРа = 22,4 (тетра= (H2N—C2H4)2N—С2Н4—N (С2Н4—NH2)2)
Р^ЭДТА = 18>8
12*
480
Д. Химический анализ
Рис. Д.61. Кривая титрования ионов Си2+ раствором аммиака.
Рис. Д.62. Кривые титрования Си(II) раствором ЭДТА при различных значениях pH (обозначены цифрами на кривых) [64].
Повышенная устойчивость комплекса и способность одной молекулы хелатообразующего лиганда заполнять все координационные места центрального иона приводят к простой стехиометрии реакции комплексообразования:
Си(Н2О)42++ Х4~ CuX2- + 4H2O
где Х4~ — анион лиганда хелатообразующего реагента.
Все сказанное обусловливает резкий скачок величины рА (рА=—lgCA+) в точке эквивалентности при титровании ионов металла А+ реагентом (рис. Д.62, ср. с рис. Д.61).
38.3.3.2. Комплексонометрическое титрование
Эффективная константа устойчивости. Понятие эффективной, или условной, константы устойчивости следует пояснить на примере комплекса двухзарядного иона металла с ЭДТА. ЭДТА представляет собой слабую четырехосновную кислоту, которую обозначим формулой ХН4. В ее водных растворах ус
38. Основы количественного химического анализа
181
танавливаются следующие протолитические равновесия:
ХН4+Н2О ХН3"+Н3О+ pAs = 2,0 (174)
ХН3" + Н2О ХН22- + Н3О+ pAs = 2,67 (175)
ХН22" + Н2О t ХН3~ + Н3О- pAs = 6,16 (176)
ХН3~ +Н2О X4- + Н3О+ pKs = 10,26 (177)
Как видно из уравнений (174)—(177), в области значений pH 3—6 существуют в основном ионы ХН22-. Они реагируют с ионами металлов по следующему уравнению:
А2+ + ХН22- ч=—> АХ2- 4- 2Н+ (178)
Однако в сильнощелочной среде протекает реакция
А2++ X1- <—fe АХ2- (179)
Для последующих описаний введем обозначения:
сх-4, схн3-. схн22—равновесные концентрации свободного, не связанного с ионами металла, ЭДТА, на различных стадиях протонирования
сх — общая концентрация свободного ЭДТА сда+— равновесная концентрация свободных ионов металла
Yi, У2 — комплексообразующий реагент (кроме ЭДТА)
сау —равновесная концентрация комплекса AYi Сд,— общая концентрация металла, не связанного с ЭДТА
а, р — коэффициенты распределения К — константа устойчивости
Ке — эффективная константа устойчивости
В титруемом растворе существует соотношение между различными ступенями протонирования свободного комплексона:
т—4
сх~ Сх«--(-Схнз—гсх1122-4-схн3-4-схн.1:= 2 Схнт*~т = асХ4- (180) ш=0
Из уравнения следует, что только для сильнощелочного раствора а=1, в остальных случаях а>1.
Аналогичное соотношение справедливо для металла, не связанного с комплексоном:
л=л
СА — САг + 4~ CAYj 4- CAY2- • • ~ CAY„ = PCA2+ (1^1)
п=0
Это уравнение основано на том, что катионы в водном растворе обычно существуют в виде комплексов, например с водой, ионами гидроксила или со вспомогательными комплексообразующими реагентами, такими, как аммиак, который добавляют для предотвращения выпадения осадков гидроксидов металлов
182
Д. Химический анализ
в щелочной среде. Коэффициент распределения .0 только в том случае равен единице, когда ион А2+ не образует ни гидроксо-, ни аквакомплексов и в раствор не вводят вспомогательные ре-агенты-комплексообразователи. Обычно 0>1.
Константу устойчивости К комплекса металла с ЭДТА АХ2“ определяют следующим образом (см. уравнение (179)):
К = — (182)
' СА2+СХ4- М '
Если в уравнении (182) сА2+ и Сх4~ заменить выражениями (180) и (181), то получим
К.=-^ (183), (184)
Эффективная константа устойчивости хелатного комплекса Ке определяет вид кривой титрования и отражает зависимость устойчивости комплекса от pH и от присутствия других комплексообразующих реагентов. Уравнение (184) показывает, что в общем случае Ке<К и только в сильнощелочной среде и в отсутствие других реагентов а»0»1, и тем самым Ке = К. Коэффициенты а и 0 взаимозависимы. Например, при титровании часто добавляют соль аммония во избежание выпадения осадков гидроксидов металлов. В этом случае, а также если определяемые металлы образуют гидроксокомплексы, 0 должен был бы возрастать при попытке достигнуть а=1, т. е. при создании высокого pH. Поэтому приходится идти на компромисс. Отсюда ясно, что для эффективного хелатометрического титрования необходимо постоянство условий титрования.
Кривые титрования. Концентрации ионов, образующихся в процессе титрования,.можно рассчитать из следующих соотношений:
Сх = Сх+сАх2-; Са = са+сах2-; ^- = Ке (185), (186), (187)
где Сх — общая концентрация ЭДТА в титруемом растворе; СА— общая концентрация металла в титруемом растворе. Количество не связанных в хелатный комплекс ионов металлов, находящихся в точке эквивалентности, рассчитывают из уравнений (185) — (187) и из соотношений Сх = сА, сх = Са и Са= = Сах2~, справедливых для точки эквивалентности. Отсюда следует уравнение (188), а поскольку до точки эквивалентности cx<SCax2- , из уравнений (185) и (186) выводят соотношение (189): i
= ^а=Са-Сх (188), (189)
38. Основы количественного химического анализа
183
После точки эквивалентности Ca<SX\x2- , тогда из (185)—(187) следует
с СА
А Ле(Сх-СА)
уравнении
(190)
При логарифмировании уравнений (188) — (190) получают три участка на кривой титрования:
до точки эквивалентности: рА = lg сА = 1g (СА—Сх) (191) после точки эквивалентности:
pA = lgcA = lgCA-lgKe-lg(Cx-CA) (192)
в точке эквивалентности: рАэ = 1g сА = 1 /2 (1g Сд—1g Ке) (193)
Кривая титрования на диаграмме рА—С^1СА может быть ориентировочно построена следующим образом (рис. Д.63). Перед началом титрования Сх = 0, отсюда lgCA = lgcA. После
Рис. Д.63. Типичная кривая комплекоонометрического титрования 0,1 и. раствора иона металла (Ке=101в).
добавления СХ = 2СА принимают lgcA==—1g Ке. В точке эквивалентности действительно уравнение (185). Кривая титрования проходит через эти три точки; точный ход кривой можно рассчитать по уравнениям (191) и (192).
Как видно из рис. Д.63, комплексонометрическое титрование тем точнее, чем выше концентрация титруемого иона металла и чем больше эффективная константа устойчивости Ке.
Хелатообразующие соединения. Обычно применяемыми хелатообразующими реагентами являются этилендиаминтетрауксус-ная кислота (ЭДТА), называемая также версеновой кислотой, и нитрилотриуксусная кислота (НТУ). Кроме них, изучена воз
г
184
Д. Химический анализ
можность применения и других комплексообразующих реагентов в качестве титрантов, но они до сих пор не имеют большого значения.
НООС-СН, сщ-соон
>n-ch2-ch2-n<
НООС-СН/ хсн2-соон
ЭДТА
/СН2-СООН n^ch2-cooh хсн2-соон
НТУ
Комплексоны представляют собой многоосновные кислоты средней силы или слабые. Из значений p/Q ЭДТА (2,0; 2,67; 6,16; 10,26) следует, что два протона относительно легко отщепляются, а дальнейшая диссоциация происходит только в щелочной среде.
На основании данных ИК-спектроскопии в настоящее время считают, что два прочно удерживаемых протона образуют внутримолекулярные водородные мостики между карбоксильными группами.
Для нитрилотриуксусной кислоты значения p/Q равны 1,9; 2,5; 9,7 соответственно.
При взаимодействии с указанными реагентами почти исключительно получаются комплексы состава 1:1. При избытке , комплексона такие высокозарядные катионы, как А1(Ш), | Fe(III), Th(IV), образуют также комплексы состава 1 : 2, одна- 1 ко их устойчивость по сравнению с устойчивостью комплексов 1 1 : 1 мала:
А1:1/А1:2= CThx/^ThX« = Ю11 I
I где Кш, /С1:2 — константы устойчивости комплексов состава 1:1 |
и 1 : 2 соответственно; Стих, Стнх2 — равновесные концентрации I комплексов тория.
В кислой среде образуются протонированные комплексы. Их « в титриметрии не используют. Нужно помнить, что двухзаряд- w ные ионы металлов образуют гидроксокомплексы только при Ц рН>10, а ионы металлов с более высокими зарядами, например Al(III), Fe(III), Th(IV), — уже в нейтральной среде. «;
Структура комплекса металла с ЭДТА приведена на | рис. Д.64. s;
38.3.3.3. Установление конечной точки титрования J
Металлоиндикаторы. Быстрому развитию комплексонометрии способствовала возможность применения цветных индикаторов, ; реагирующих на изменение концентрации ионов металлов. Эти ; индикаторы представляют собой органические красители, обра- i зующие с ионами металлов хелатные комплексы, окраска которых отличается от окраски самого красителя в свободном со
38. Основы количественного химического анализа
185
стоянии. Кроме того, условием применимости индикаторов является меньшая устойчивость комплекса индикатора с определяемым металлом по сравнению с комплексонатом металла. В процессе титрования комплекс ионов металлов с индикатором разрушается и образуется прочный комплекс с ЭДТА. В точке эквивалентности все ионы металла (точнее, их количество, соответствующее константам устойчивости комплекса) связаны в комплекс с ЭДТА и окраска раствора изменяется.
Так же как для комплек
Рис. Д.64. Структура комплекса металла с ЭДТА.
Пунктирные линии — это не связи, а обозначения для пояснения геометрии комплекса.
сонов, для индикаторов характерен ряд равновесных состоя-ний—различные ступени протонирования свободного красителя. При этом могут образоваться водородные и гидроксокомп-лексы состава 1:1, 1 : 2 и 2:1.
При выборе индикатора нужно знать константы устойчивости комплексов индикатора и комплексона с ионами металла.
Эриохром черный Т. Наиболее широкое применение нашел эриохром черный Т, так как константы устойчивости комплексов, образуемых этим реагентом, имеют наиболее подходящие для определений значения:
В области 7<рН<12, представляющей наибольший интерес для метода комплексонометрии, группа сульфоновой кислоты полностью диссоциирована. Значения рК отщепления обоих протонов p/G = 6,3 и рКг= 11,6 ограничивают три области:
pH <p/Ci
7<рН< 11 рН>12
Диссоциация не происходит. Соединение окрашено в красный цвет.
Существует амфолит. Ионы окрашены в синий цвет.
Обе ОН-группы диссоциированы. Ионы окрашены в желто-оранжевый цвет.
Поскольку комплексы эриохрома Т с металлами окрашены в красный цвет, при проведении титрования в области 7<рН<
186
Д. Химический анализ
<11 происходит четкое изменение окраски из красной в си-И тою. Красную окраску имеет комплекс индикатора с металла-И ми: Mg, Са, Sr, Y, лантаноидами; Мп, Zn, Cd, Hg, Ga, In, PbJI Al, Fe, Ti, Co, Ni, Си, платиновыми металлами. 1
Щелочные растворы индикатора чувствительны к действию | окислителей, поэтому индикатор добавляют непосредственно 1 перед титрованием и вводят в раствор небольшое количество I аскорбиновой кислоты или гидроксиламина. Поскольку водные | растворы эриохрома Т неустойчивы, обычно его в сухом виде з смешивают с хлоридом натрия (для разбавления). Для этой же цели добавляют раствор триэтаноламина, разбавленный • для снижения вязкости небольшим количеством абсолютного ’ спирта или концентрированным аммиаком. ;
Комплексонометрическое титрование дает неверные результаты, если в применяемой дистиллированной воде находятся ионы металлов. Особенно сказывается на изменении окраски индикатора присутствие меди (например, при использовании медного аппарата для дистилляции). Мешающее влияние оказывают также ионы магния и кальция. Поэтому для комплексонометрического титрования используют только бидистиллат или деионизированную воду, которую проще всего получить, пропуская обычную дистиллированную воду через сильно кислотный катионит в Па+-форме. На колонке диаметром 2 см и длиной 15 см можно очистить за один цикл более 500 дм3 загрязненной дистиллированной воды.
Мурексид также находит широкое применение в качестве металлоиндикатора. Он представляет собой аммониевую соль пурпуровой кислоты
У
Н
ОС
О н .с-к
NH.
Н О
О Н
Мурексид имеет следующие переходы окраски:
pH
<1 2-9 9—11
>11
Окраска
Желтая
Кра сно-фиолетова я Фиолетовая Сине-фиолетовая
Мурексид образует с ионами кальция, никеля, кобальта, меди и редкоземельных металлов устойчивые комплексы, которые можно использовать при титровании. В отличие от эриохрома черного Т, комплексы металлов с мурексидом являются рН-ин-дикаторами. Окраска комплекса металла с мурексидом зависит
38. Основы количественного химического анализа
(87
от металла: с кальцием — красная окраска, с медью — оранжевая, с никелем и кобальтом — желтая.
Поскольку комплексы мурексида с ионами металлов менее устойчивы, чем комплексы эриохрома черного Т, мурексид менее чувствителен к следовым количествам металлов.
Другие металлоиндикаторы. Металлфталеин представляет собой индикатор, образующий окрашенные в красный цвет комплексы с ионами кальция, стронция и бария. Его нельзя применять для титрования тяжелых металлов, так как их комплексы не окрашены или имеют слабую окраску.
Бренцкатехиновый фиолетовый дает возможность титровать Bi(III) и Th(IV), причем в точке эквивалентности происходит четкий переход окраски из желтой в синюю.
Кроме названных, существуют и другие индикаторы специального применения. Так, Fe(III) можно титровать ЭДТА после его перевода в роданидный комплекс. Аналогично можно осуществить титрование Со(II).
Окислительно-восстановительные индикаторы. В некоторых случаях комплексонометрического титрования вблизи точки эквивалентности возникает скачок окислительно-восстановительного потенциала, который можно установить с помощью окислительно-восстановительного индикатора. Например, при титровании Fe(III) в точке эквивалентности происходит резкое уменьшение окислительно-восстановительного потенциала и следовые количества Fe(II), которые всегда находятся в растворе, восстанавливают окисленную форму вариаминового синего Б с образованием бесцветного лейкооснования.
Аналогично можно титровать Zn(II) в присутствии гексацианоферрата (III) с индикатором диметилнафтидином. Этот раствор имеет большую величину окислительно-восстановительного потенциала, так как с [Fe(CN)e|3^ в равновесии находятся только следовые количества [Fe(CN)e]4^, поскольку основное количество этих ионов связано с Zn2+. Вследствие высокого значения окислительно-восстановительного потенциала индикатор окисляется и раствор приобретает красно-фиолетовую окраску. При титровании раствором ЭДТА ионы цинка переходят в хелатный комплекс и в точке эквивалентности в свободном состоянии образуется гексацианоферрат(П). Окислительно-восстановительный потенциал при этом резко уменьшается и индикатор, восстанавливаясь, обесцвечивается.
Другие методы индикации. Наряду с перечисленными методами конечную точку комплексонометрического титрования можно определить также фотометрическими методами, измеряя оптическую плотность раствора. При 225 нм ионы НХ3~ и X4-ЭДТА сильно поглощают свет, а комплексы при этой длине волны бесцветны.
188
Д. Химический анализ
Особенно эффективен амперометрический метод индикации конечной точки титрования. Потенциометрическую индикацию можно проводить следующими способами: а) применяя индикаторный электрод, специфичный к определяемому иону, например Ag+/Ag; б) с помощью платинового электрода в случае обратимой редокс-пары, например Fe(II)/Fe(III); в) применяя электрохимическую систему ЭДТА — Hg(II)— Hg, для которой £=£o+2^Ligw+ (194)
Рассмотрим механизм действия ртутного электрода. Если в растворе, кроме ионов металла А2+, находится ЭДТА, то действительны следующие соотношения:
(195), (196)
Комбинирование уравнений (194) — (196) дает р_________________р । 0,0590 « сд2+ Кдх /] пух
Е - Ео+—— lg — cHgx2- (197)
Концентрация cHgX2- практически постоянна, так как комплекс HgX2- очень устойчив, и в данном случае очень мала (3-10-6). Тогда
Е = Е0' 4—°-059° 1g-(198) 2 Сдх2-
Такнм образом, ртутный электрод реагирует на изменение концентрации ионов А2+.
Кроме того, для индикации можно применять также метод высокочастотного титрования.
38.3.3.4. Способы титрования
Прямое титрование. Буферированный раствор иона металла А2+ титруют стандартным раствором комплексона в присутствии металлоиндикатора. При этом комплексон разрушает комплекс металла с индикатором. В точке эквивалентности резко возрастает рА и окраска раствора изменяется.
Обратное титрование. К титруемому раствору иона металла добавляют точно измеренный избыток стандартного раствора комплексона, непрореагировавший остаток которого титруют раствором Mg(II) или Zn(II).
Этот метод применяют в том случае, если для определения металла нельзя выбрать подходящий индикатор. Если невозможно провести прямое титрование из-за выпадения осадка гидроксида (высокое значение pH), то к анализируемому раствору, содержащему ионы металла, добавляют избыток раствора комплексона и только после этого буферируют и проводят обратное титрование.
38. Основы количественного химического анализа
18&
Вытеснительное титрование. Этот метод можно применять вместо обратного титрования. Для этого к раствору определяемого иона металла добавляют избыток раствора комплексоната магния. Поскольку комплексонат магния обычно менее устойчив, чем комплексные соединения других металлов, происходит вытеснение ионов Mg2+, которые можно определить прямым титрованием раствором ЭДТА в присутствии эриохрома черного Т:
А«+ + MgX3- АХ(л-4)_+ Mg2+
Алкалиметрическое титрование. Если в реакции с ионами металла участвуют ионы ЭДТА ХН3~ или ХН22-, то в процессе комплексообразования происходит выделение ионов водорода. Их можно оттитровать раствором щелочи в присутствии или смешанного индикатора бромкрезоловый зеленый+метиленовый синий, или метилового красного. Недостатком этого способа является необходимость очень точной нейтрализации раствора' перед титрованием, что иногда трудно осуществить из-за протолиза сольватированных ионов металла.
38.3.3.5. Практическая часть
Приготовление стандартного раствора ЭДТА. Обычно для комплексонометрического титрования применяют дигидроэти-лендиаминтетраацетат натрия (хелаплекс) Na2H2C10H12O8N2-•2Н2О (молекулярная масса 372,242 г/моль). Препарат марки ч.д. а. после высушивания до постоянной массы при 80 °C можно применять для приготовления стандартного раствора титранта по его навеске.
Если препарат загрязнен, его очищают следующим образом: к насыщенному при комнатной температуре водному раствору соли (примерно 20 г в 100 см3 воды) медленно добавляют этанол до образования небольшого осадка. Осадок отфильтровывают и отбрасывают. К прозрачному раствору добавляют равный объем этанола, полученный осадок отсасывают, промывают ацетоном, затем эфиром и сушат сначала при комнатной температуре в течение ночи и затем в течение 24 ч при 80 °C.
Для установки характеристик раствора ЭДТА в качестве первичного стандартного вещества применяют карбонат кальция, чистый цинк или висмут и различные двойные соли, например цинковый шенит K2SO4- ZnSO4-6H2O.
Методика стандартизации по карбонату кальция.
Необходимые реактивы: 1. Буферный раствор. Раствор 54 г хлорида аммония в 350 см3 аммиачной воды (rf=0,88 г/см3, ==34%) разбавляют дистиллированной водой до объема 1000 см3.
2. Индикатор эрнохром черный Т. 0,2 г индикатора растворяют в 15 см’ триэтаноламина и 5 см3 этанола. Раствор стабилен в течение месяца.
190
Д. Химический анализ
3. Комплекоонат магния. 3,01 г комплексона и 1,990 г MgSC>4-7H2O растворяют в дистиллированной воде. Раствор доливают до 100 см3. В 2 см3, раствора содержится 0,1 г комплексоната магния.
К 0,25 г карбоната кальция, высушенного при 150—200 °C, добавляют 20—30 см3 воды и по каплям 2 н. раствор НС1 до растворения карбоната. Раствор кипятят в течение ~2 мин для удаления диоксида углерода, охлаждают и разбавляют до объема ~ 100 см3. Добавляют 2 см3 комплексоната магния, 5 см3 буферного раствора и 1—2 капли индикатора и титруют до перехода окраски раствора из винно-красной в синюю.
38.3.4. Реакции осаждения
38.3.4.1. Растворимость
.Между осадком АХ, находящимся под слоем раствора, и его ионами А+ и X- в растворе существует равновесие:
АХ (тв) А+ + X- (199)
Для него можно написать соотношение, называемое произведением растворимости:
aA+ctx_ = ca+cx“Ya+Vx_= Kl° (200)
где а — активности; с — концентрации; у — коэффициенты активности; K°l — термодинамическое произведение растворимо-•сти.
В чистом насыщенном растворе са+ = сх-=А (L— растворимость осадка АХ) и
Ь = (Кд°/Та+Тх-)1/2 (201)
В растворах малорастворимых соединений са+ = Сх-<С1 и уд+ « »ух-« 1. Тогда
(202)
Влияние электролитов на растворимость. В зависимости от типа раствора электролита, добавляемого к насыщенному раствору АХ, различают следующие случаи:
1. Добавляемый электролит не реагирует с ионами осадка и а) не содержит ионов, одноименных с осадком (введение разноименных ионов); б) содержит одноименные с осадком ионы (введение одноименных ионов).
2. Добавляемый электролит реагирует с ионами осадка: а) с образованием слабо диссоциированных соединений; б) с образованием растворимого комплекса; в) с одновременным окислением или восстановлением.
Введение разноименных, ионов. Введение электролита, со-лержащего разноименные с осадком ионы и не реагирующего с
38. Основы количественного химического анализа
191
осадком, повышает растворимость осадка, что связано с увеличением силы ионных взаимодействий. По Дебаю — Хюккелю средние значения коэффициентов активности у± в водных растворах при 25 °C зависят от ионной силы:
lg 0.50851 | Kjx (203)
1 + 0,3281a
где z+, z_ — заряды катионов и анионов соответственно; а — диаметр гидратированных ионов, нм; ц = 0,5 — ионная си-
ла; mt — концентрация ионов, моль/кг растворителя.
С увеличением количества добавляемого электролита ггц ионная сила возрастет, а коэффициенты активности у± уменьшаются. Следовательно, в соответствии с уравнением (201) растворимость L возрастает, так как Kl при постоянной температуре — величина постоянная.
Введение одноименных ионов. Если к насыщенному раствору соединения АХ добавить такое количество соли ВХ, чтобы концентрация ее равнялась Со, т. е. аж с, то справедливы следующие соотношения:
L = сА+; сд+сх~ = KL (204), (205)’
СХ~ = СА+ ~ЬСВ+ ~ СА+ + Q (206)
Комбинируя уравнения (204), (205) и (206), находим
са+ (са+ Н~св+)= Kl (207)
Так как
са+ св+> А ^d^o (208).
Более точное соотношение имеет вид
L = /4Co2+^—ТСо (209>
При этом могут иметь место два случая: а) С^жКс и б) СоЗ> ^KL. В первом случае из уравнения (209) получаем уравнение (210) (так как Cq2<^Kl), во втором— (211):
. ЬжУК—^Со-, (210), (211)>
При увеличении количества добавляемого электролита, содержащего одноименные с осадком ионы и не реагирующего с ним, растворимость осадка должна уменьшаться от значения Ь=УКь до L«0. В действительности L никогда не достигает нулевого значения, так как не существует абсолютно нерастворимых солей. Кроме того, с увеличением концентрации ионов-становятся заметными межионные взаимодействия, что в соответствии с уравнениями (203) и (201) приводит к увеличению.
192
Д. Химический анализ
растворимости. В результате растворимость малорастворимого соединения с увеличением содержания ионов, одноименных с осадком, проходит через более или менее выраженный минимум. Таким образом, избыток осадителя, применяемый в гравиметрическом анализе для уменьшения растворимости осадка, должен быть минимальным, в противном случае растворимость может снова возрасти.
Образование малодиссоциированных соединений. Реакции этого типа имеют значение в количественном и качественном анализах для перевода в раствор малорастворимых соединений. •Обычно при этом образуются слабые кислоты.
Большинство аналитически важных малорастворимых солей образованы двухзарядными катионами и анионами слабых кис--лот или кислот средней силы, например PbS, FeS, СаСгО4, BaSO4 и др. При добавлении к соли такого типа (АХ) сильной кислоты образуются ионы слабых кислот НХ~ или кислоты Н2Х, и для раствора, находящегося над осадком, справедливы следующие выражения:
= KL; =KS1 (212), (213)
СН2Х
= ^ = ^-4-Снх+Сн2х = Ь (214), (215)
^Г1Л
Это четыре независимых уравнения с четырьмя неизвестными, которые можно решить относительно pH. Так как точный -расчет слишком трудоемок и приведенные формулы могут •быть неудобными для практики, рассчитывают приближенные значения.
Из теории кислотно-основных равновесий известно, что на логарифмических диаграммах можно выделить три области pH двухосновных слабых кислот, разграниченные значениями pKsi и pKrf (разд. 38.3.1.3):
При pH<pKS| сн2х » снх~ + сх2- (216)
При pKS1<pH<pKS2 снх- > сн2х + сх2- (217)
При pH>pKS2 сх2- » сн2х + снх- (218)
Эти допущения тем более справедливы, чем больше разность величин pH и pKs- На основании неравенств (216) — (218) из уравнения (215) можно получить три уравнения:
pH < рК,;, сАг+ = сН2х рАЬ1 < pH < pXS2; сА2+ = снх-
PH>p/<S2; сА2+ = сХ2-
(219)
(220)
(221)
38. Основы количественного химического анализа
193
Сочетание уравнений (212) — (214) дает л==сн+V xjfcr
После логарифмирования
lg L = —pH 4- (р/^ 4- р^2—pKL)
(222)
(223), (224)
(225)
igL= —2-рн4-4(р^а-р^); --ГР^ (226)’ (227>
Уравнения (225) и (226) описывают на диаграмме lg L— pH прямые с тангенсом угла наклона —1, —'/г и 0. При построении диаграммы необходимо следить за тем, чтобы три части прямых соприкасались друг с другом при рН = рЛ41 и рН = = pjKs2- На рис. Д.65 приведен пример такой диаграммы.
Рис. Д.65. Зависимость растворимости малорастворимого соединения АХ от значения pH.
и — 1-я и 2-я константы диссоциации слабой кислоты НгХ [212].
*1 2
Построение диаграммы проводят следующим способом.
Сначала в области pH>p/Q, проводят горизонтальную прямую с расстоянием от оси абсцисс Ig L——(уравнение (227)). Затем описывают полуокружность около точки Р2 с радиусом '/zPzPi, которая пересекает вертикаль pXSj в точке Рз. Затем описывают полуокружность около точки с радиусом Р1Р3. Эта полуокружность пересекает ось pH в точке Ps. Наконец, точки Pi, Рз и Рз соединяют прямыми линиями.
Чтобы определить, при какой концентрации кислоты начнет растворяться данный осадок, достаточно одного уравнения (222). Ответ на этот вопрос можно также получить другим пу-
13—1880
194
Д. Химический анализ
тем. Напишем суммарную реакцию растворения осадка в сильной кислоте и выражение закона действующих масс для нее:
АХ + 2Н+ А2+ + н2Х (228)
ск2+сщх1с2‘'еД ~ К (229)-
Из суммарной реакции следует равенство (230), а из уравнения (229) — выражение (231):
с^+ = Сьх = L- L = сн+ УК (230), (231)
Суммарную реакцию (228) можно представить как последовательность стадий с соответствующими константами равновесия: ;
№l)
(1/-М
(.1/^)
(АХ); «=* А2+ 4- х2-X2~ + H+ НХ~
НХ~ + Н+ Н2Х
Константу равновесия суммарной реакции множая константы равновесия отдельных стадий: к Kl Л KS1.Ks2
Подставив выражение (235) в (231), получим уравнение (222).
рн ------
(232)
(233)
(234)
получают, пере-
(235)
Рис. Д.66. Зависимость растворимости некоторых малорастворимых веществ АХ от величины pH [212].
»—га-
38. Основы количественного химического анализа
195
На рис. Д.66 приведены диаграммы некоторых малорастворимых соединений, из которых можно сделать выводы о взаимодействии осадков со слабыми кислотами. При внесении малорастворимого соединения АХ в раствор слабой кислоты HY в зависимости от pH будут протекать следующие реакции:
АХ + HY А2-* + НХ' + Y-
АХ 4- 2HY А^ 4- Н2Х 4- 2Y-
(236)
(237)
Из уравнений (236) и (237) можно получить зависимости са2+=су- и сд2+ = 1/2су- соответственно. Если нанести линии сопряженного основания для слабой кислоты HY на логарифмическую диаграмму кислотно-основного . равновесия (разд. 38.3.1.2), то в точке ее пересечения с прямой амфолита (tgcp = = —1/2) можно определить растворимость осадка в этой кислоте. Для определения влияния слабой кислоты в сильнокислой области кривой растворимости нужно найти точку пересечения участка кривой с большим наклоном (tgcp=— 1) с кривой 1g су---1g 2.
Эти же положения справедливы для такой слабой кислоты, как вода. Точка пересечения прямой растворимости с кривой 1gсон- или IgCoH------lg2 показывает увеличение растворимо-
сти осадка в результате протолиза. Как видно из рис. Д.66, растворимость гидроксида серебра, сульфида железа и сульфида цинка при этом значительно увеличивается.
Образование растворимых комплексов. Во многих случаях малорастворимые осадки растворяются при добавлении электролитов, имеющих одно- или разноименные с осадком ионы, если катион или анион осадка (чаще катион) образует растворимый комплекс с добавленным электролитом. Эта реакция происходит, например, между хлоридом или бромидом серебра и аммиаком, иодидом серебра и цианидом калия, сульфидом мышьяка и гидросульфидом аммония. Поскольку комплексы, образующиеся в результате этих реакций, обычно очень устойчивы, на кривой растворимости не наблюдается ожидаемого минимума.
При растворении галогенида серебра в аммиаке, протекают следующие реакции:
AgCl (тв) ;?=* Ag+ + СГ (Kl)
Ag+ 4- NH3 Ag(NH3)-' (Ko)
Ag(NH3)+4-NH3 Ag(NH3)2+______(Kp)__________
AgCl (tb) 4- 2NH3 ч —* Ag(NH3)2+ + СГ (K = KlKd^Kd)
Применим закон действующих масс к этим реакциям:
_ CQ- cAg(NH3)2+ c2NH3
(238)
(239)
(240)
(241)
(242)
13*
196
Д. Химический анализ
Растворимость хлорида серебра L определится как
= CAg+ + CAg(NH3)+ + CAg(NH3)2+ = ССГ (243)
Поскольку диаммипокомплекс очень устойчив, справедливы соотношения
CAg(XH3)2+ CAg++CAg(NH3)+ И — СсГ == CAg(NH3l2+ (244), (245) Из уравнения (242) получим
= с>:н3 У К — Cnh3 V (246)
Так как изменение концентрации аммиака при комплексообразовании мало, можно принять Cnh3~C0 (Со — общая концентрация NHa) и окончательное уравнение имеет вид
Z, — Со У
(247)
Окислительно-восстановительные реакции осадка. Еще одной возможностью перевода компонентов малорастворимого осадка в раствор является сдвиг равновесия, существующего в системе раствор — осадок, путем окисления или восстановления. Примерами этого могут служить
HgS Hg2++S2-
Hg2+ 4- Zn -> Hg + Zn2+
HgS ,=4= Hg2+ + S2~
S2- + C12 --»- so42- + ci-
Для выяснения вопроса, протекает ли реакция такого типа количественно, ее представляют через отдельные стадии, для каждой из которых записывают константу равновесия и затем их суммируют.
Пример. При восстановлении цинком ионов Hg2+ из малорастэоримого сульфида ртути в раствор могут перейти ионы S2-. Уравнение суммарной реакции имеет вид
HgS (тв) + Zn + 2Н3О+ Hg(I) + Zn2+ + H2S (г) + 2H3O (248)
Составляющие реакции и показатели их
констант равновесия (t=25eC):
HgS (ТВ) =* Hg2+ + S2-; pK1 = pK£ = 52,4 (249)
Zn ч= =£: Zn2+ + 2e~; pK2 = — pKzn = —25,8 (250).
Hg2+ + 2e' += =* Hg(I); рКз= pflHg = — 28,9 (251)
S2- + 2Н3О+ =* H2S + 2H2O; pK4 = — рД, = —19,8 (252)
H2S (В1 одн) «=* H2S(r); PKS = -1 (253)
Для суммарной реакции р^=р/С + р/С + рК?,+р/<4+р/<г, =—23,1.
Поскольку величина показателя равновесия прямо пропорциональна работе основной реакции и имеет одинаковый с ней знак, большая отрицательная величина рЛ' означает, что сульфид ртути при действии цинка в кислой среде полностью может перейти в раствор. p^2 и pKs — показатели окисления-восстановления (разд. 38.3.2.2),
38. Основы количественного химического анализа
197
Влияние температуры на растворимость. Зависимость растворимости от температуры определяют по произведению растворимости (влиянием температуры на величину коэффициентов активности можно пренебречь). Зависимость произведения растворимости от температуры описывается уравнением Вант-Гоффа:
d\nKL/dT — ^H/RT2 (254)
Поскольку энтальпия \Н реакции АВ(тв)=^А+В_ для малорастворимых веществ всегда положительна, Kl и, следовательно, растворимость L возрастают с увеличением температуры.
Влияние неводных растворителей на растворимость. При добавлении к водному раствору соли смешивающегося с водой неэлектролита, например ацетона, спирта и др., растворимость соли уменьшается. Это можно объяснить тем, что молекулы неэлектролита гидратируются, причем с увеличением количества неэлектролита гидратная оболочка ионов разрушается, и в итоге соль выпадает в осадок. Однако некоторые соли растворимы и в органических растворителях. Это происходит в том случае, когда силы межатомных взаимодействий в твердых веществах невелики и преодолеваются даже небольшими энергиями сольватации органического растворителя (например, при растворении перхлората бария в ацетоне) или если ионы твердых веществ особенно легко сольватируются (например, при растворении солей Li+ или перхлората натрия в спирте).
Различное влияние, оказываемое органическими растворителями на неорганические соединения, часто используют в анализе. Например, хлорид лития можно отделить от галогенидов других щелочных металлов экстракцией спиртом или эфиром. Метод количественного определения калия в виде перхлората основан на том, что его растворимость уменьшается при добавлении спирта, а перхлорат натрия при этом переходит в раствор. Хлориды и нитраты щелочноземельных металлов можно разделить смесью спирт+эфир.
38.3.4.2. Образование малорастворимых осадков
К свойствам осадка, позволяющим использовать его для проведения количественных определений, наряду с другими относится возможность выделения осадка в чистом виде и хорошая фильтруемость. Оба условия выполняются только при соблюдении оптимальных условий осаждения (концентрации и температуры растворов, скорости осаждения и т. д.).
Фильтруемость осадка зависит от размеров его частиц, которые в свою очередь определяются соотношением двух факторов: скорости образования зародышей кристаллов и скорости роста кристаллов.
198
Д. Химический анализ
При образовании осадка происходит разделение фаз, поэтому этот процесс подчиняется законам, аналогичным законам конденсации малых капель из парообразной фазы или появлению пузырьков паров при кипении жидкости. Во всех случаях первично образующиеся частицы новой фазы очень малы (<1 нм), а отношение их поверхности к объему и, следовательно, свободная поверхностная энергия велики, т. е. химический потенциал, а также и активность высокодисперсной фазы выше, чем твердой фазы. Иначе говоря, константа равновесия фазовых переходов зависит от степени развития поверхности фаз. Для процесса образования осадка это означает: чем меньше радиус образующихся зародышей кристаллов, тем больше произведение растворимости, и следовательно растворимость. Растворимость Lr зародышей и их радиус г связаны между собой следующим соотношением (по аналогии с уравнением для давления паров малых капель):
<255)
где L — растворимость макроскопических кристаллов; о— поверхностное натяжение твердых тел; V — молярный объем.
Уравнение (255) можно упростить:
In-^ = In L + L^-_L = In( 1 + Lr~L )« -- (256)
Тогда
(Lr—L)/L = 2oV/RTr (257)
Отношение (Lr—L)/L называют относительным пересыщением или относительным увеличением растворимости. Из уравнения (257) можно сделать ряд выводов.
1. Для конкретного осадка (oV=const) относительное увеличение растворимости тем больше, чем меньше радиус зародышей. Частицы с радиусом ri находятся в равновесии с раствором, концентрация которого ЬГ1 (ЬГ1 >Л) (рис. Д.67). Частицы с меньшим радиусом растворимы в этом растворе, частицы той же природы, но с большим радиусом, нерастворимы. Приведенные в литературе данные по растворимости относятся к макроскопическим, хорошо сформировавшимся кристаллическим осадкам (если нет специальных примечаний). Такие данные надо осторожно использовать при анализе веществ, так как при осаждении сначала образуются мелкодисперсные псевдо-аморфные осадки.
2. Для частиц определенного радиуса (r = const) относительное увеличение растворимости пропорционально поверхностному натяжению соответствующих твердых тел. Небольшая величина поверхностного натяжения означает небольшое относи-
38. Основы количественного химического анализа
199
Рис. Д.67. Зависимость растворимости Lt от радиуса частиц г.
тельное повышение растворимости; образовавшиеся зародыши при этом практически не могут расти, так как их растворимость мало отличается от растворимости твердой фазы. Осадки таких соединений состоят из плохо сформировавшихся небольших кристаллов и зачастую аморфны. Малорастворимые соединения с высоким поверхностным натяжением, напротив, обладают высокой относительной растворимостью; их зародыши растут, и в результате образуются хорошо сформировавшиеся, большие кристаллы. Примером соединения с небольшим поверхностным натяжением служит хлорид серебра, соединения с большим поверхностным натяжением — сульфат бария.
Скорость зародышеобразования. Образование больших кристаллов в процессе осаждения происходит следующим образом (рис. Д.67): при образовании первых зародышей кристаллов с радиусом г первоначальная концентрация раствора Lr уменьшается. При этом увеличивается радиус частиц, находящихся с этим раствором в состоянии равновесия. Для образования больших частиц в принципе возможны два пути: а) крупные частицы образуются спонтанно из большого числа частиц с б) частицы больших размеров образуются объединением малых
докритическими радиусами или
частиц с докритическими радиусами.
Первая возможность отпадает, так как существует очень малая вероятность одновременных соударений большого числа ионных пар (примерно 10) в одном и том же месте. Во втором случае всегда необходимы два соударения, что во много раз вероятнее. Отсюда вытекает вывод, что большие зародыши растут за счет малых или за счет растворенного вещества. Этот процесс подобен изотермической дистилляции маленьких капель. В принципе невозможно образование центра кристаллизации в результате соударения двух частиц, так как энергия при этом должна складываться из энергии образования и относительной кинетической энергии обеих соударяющихся частиц, т. е. значение энергии больше, чем нужно для образования связей, поэтому зародыш тотчас же распадается. Зародыш кристалла может образоваться, если избыточная энергия свое
200
Д. Химический анализ
временно расходуется. Это может произойти при введении третьей частицы X, участвующей в соударении, но индифферентной к данной реакции:
А + В + X ---► (АВХ)* --* АВ + X* (258)
где звездочкой обозначено возбужденное состояние. Третьим участником реакции может быть зародыш кристалла постороннего вещества или стенка сосуда.
Как следует из сказанного, тщательно очищенная от посторонних ионов система может обладать значительно большей энергией, чем нужно для достижения равновесного состояния. Примерами такой метастабильной системы являются перегретая и переохлажденная жидкости и пересыщенные растворы.
Размеры растущих кристаллов тем больше, чем меньше образуется первичных зародышей. Количество зародышей, образующихся в единицу времени в единице объема, называют скоростью зародышеобразования. По П. Ваймарну,
2 L' — L /осг..
w~ RT L (259)
где w — скорость зародышеобразования; L' — концентрация раствора в момент образования зародыша; L — концентрация насыщения раствора, находящегося в состоянии равновесия с макроскопическим осадком.
Уравнение (259) описывает влияние различных факторов на процесс осаждения. Число зародышей растет с увеличением пересыщения раствора и уменьшением растворимости осадка. Для получения крупнокристаллических, хорошо фильтруемых осадков пересыщение должно быть мало. Поэтому осадитель нужно добавлять медленно. Кроме того, осаждение проводят при нагревании до температуры кипения, при этом значение L увеличивается и скорость зародышеобразования становится еще меньше.
Эти требования наиболее полно удовлетворяются при проведении гомогенного осаждения. К осаждаемому раствору добавляют соединения, образующие осадитель только в необходимом для проведения реакции количестве. Для осаждения малорастворимого сульфата применяют, например, диметилсульфат или амидосульфат, для осаждения сульфидов — тиоацетамид или тиокарбамид. На этом же принципе основано применение уротропина для осаждения гидроксидов трехвалентных металлов или тиосульфата, нитрита или смеси иодида и йодата для осаждения легко фильтруемого гидроксида алюминия. Во всех этих методах концентрация L пересыщенного раствора должна быть минимальной.
38. Основы количественного химического анализа
201
Иногда пересыщенные растворы очень устойчивы. Это характерно для хорошорастворимых веществ, например квасцов,-нитрата хрома (III) и др. (как следует из уравнения (259) при большом значении L скорость w мала). В этих случаях кристаллизацию вызывают внесением в раствор затравки — маленького кристаллика осаждаемого соединения.
Важно, чтобы осадок при осаждении не загрязнялся посторонними ионами, что может происходить, например, из-за царапин на стенках стаканов или примесей, вносимых с палочкой.
Относительное пересыщение, необходимое для спонтанной кристаллизации, помимо прочих условий, существенно зависит' от формулы соединения. В то время как при осаждении сульфата бария для образования зародыша необходимо как минимум два иона, минимальное число ионов для квасцов и нитрата, хрома равно четырем, а в случае фосфата бария их даже пять, что значительно снижает вероятность зародышеобразования.
Развитию теории процессов зародышеобразования посвящены работы М. Волмера и др.
Скорость роста кристаллов. Величина и структура частиц: осадка зависят не только от скорости их образования, но также от скорости роста кристаллов. Каждый зародыш окружен адгезионным слоем насыщенного раствора. В момент образования первого зародыша раствор пересыщен. Скорость роста зародыша пропорциональна скорости переноса из пересыщенного раствора в насыщенный адгезионный слой. Изменения концентраций пересыщенного раствора в единицу времени dL'fdt определяют из уравнения Нойеса — Нернста:
T=T?<i-L'> <260>
где L — концентрация насыщенного раствора; L' — концентрация пересыщенного раствора; D — коэффициент диффузии;. О — размер поверхности кристалла; 6 — толщина адгезионного слоя (при 25°С~10-3 см); v— объем раствора.
Скорость роста кристаллов и определяется по уравнению (261), из которого следует (262):
u=~klF’ U = k^-(L'-L) (261), (262)
где k — коэффициент пропорциональности.
Таким образом, скорость роста кристаллов пропорциональна абсолютной величине пересыщения. Для образования крупнокристаллических осадков скорость роста кристаллов должна быть значительно больше, чем скорость зародышеобразования. Этому способствует перемешивание, которое, с одной стороны, предотвращает локальное обеднение раствора, окружающего
202
Д. Химический анализ
растущие частицы (локальное уменьшение L'), а с Другой стороны, снижает толщину адгезионного слоя 6.
Уравнение (262) в общем можно применять для качественной оценки некоторых факторов, оказывающих влияние на процесс зародышеобразования, однако для сложных процессов уравнение непригодно. Это объясняется тем, что рост кристаллов определяется не только диффузионными процессами, происходящими в жидкой фазе, но также свойствами структуры растущих кристаллов, как, например, дефектами кристаллической решетки, внедрением в нее ионов из добавляемых растворов и т. д.
38.3.4.3. Коллоиды
Выше было сказано, что малорастворимые осадки, такие, как сульфиды или гидроксиды, при нормальных условиях образуются в тонкодисперсном состоянии. Это осадки с развитой поверхностью, обладающей адсорбционными свойствами. Преимущественно адсорбируются одноименные с ионами осадка ионы, находящиеся в растворе в избытке. Например, поверхность осадка, образующегося из раствора иодида при действии ионов серебра, количество которых меньше теоретического, заряжена отрицательно, так как на поверхности осадка адсорбируются преимущественно иодид-ионы. Заряженные частицы осадка взаимно отталкиваются и не могут расти или растут очень медленно и поэтому остаются в растворе во взвешенном состоянии. При этом образуется коллоидный раствор.
Коллоидными считаются частицы с диаметром 10~7— 10-4 см. Поскольку форма коллоидных частиц различна, для обозначения их размеров имеет смысл предложение Г. Штау-дингера использовать число атомов на частицу. Эта величина колеблется в пределах 103—109.
В аналитической химии образование коллоидных растворов обычно нежелательное явление, так как они затрудняют разделение вследствие высокой адсорбционной способности, и, кроме того, их практически нельзя отфильтровать. Однако в некоторых случаях специально вызывают коллоидообразование, например в осадительном титровании с применением адсорбционных индикаторов, в нефелометрии и др.
Коллоиды, устойчивость которых обусловлена электрическими поверхностными зарядами, называют гидрофобными. Их можно скоагулировать добавлением электролитов. При этом ионы с зарядом, противоположным по знаку поверхностному заряду коллоидных частиц, защищают поверхность, давая возможность коллоидным частицам настолько сблизиться, что начинают действовать адгезионные силы Ван-дер-Ваальса; частицы растут, объединяются и образуют осадок (коагулируют).
38. Основы количественного химического анализа
203
Количество противоионов, необходимое для коагуляции, уменьшается с увеличением заряда ионов.
Скоагулированный гидрофобный коллоид при промывании электролитами часто снова растворяется, т. е. пептизируется. Это особенно характерно для осадков сульфидов и гидроксидов (сульфида никеля, гидроксида алюминия). Во избежание пептизации осадок промывают разбавленными растворами электролитов, обычно нитратом аммония, который легко полностью удалить при последующем прокаливании осадка.
38.3.4.4. Загрязнения осадков
Невозможно получить идеально чистый осадок — он всегда в большей или меньшей степени загрязнен ионами (одноименными или посторонними) и молекулами растворителя. Загрязнение осадков может происходить путем соосаждения и в результате так называемого последующего осаждения. Для проведения исследований в этой важнейшей области аналитической химии особенно успешно применяют радиоактивные атомы.
Соосаждение. Различают следующие типы соосаждения: адсорбцию, окклюзию и образование твердых растворов. Эти три явления в литературе не всегда четко разграничены: обычно нельзя установить, какой из перечисленных процессов вызвал загрязнение конкретного осадка. Поэтому и мы не приводим определений, а обсуждаем взаимосвязь типов соосаждения при образовании осадка.
Адсорбция. Как правило, это основная причина загрязнения осадков. Адсорбция особенно характерна для осадков с большой удельной поверхностью, например гидроксидов и сульфидов. Загрязнение осадков гидроксидов было исследовано И. М. Кольтгофом на примере осаждения гидроксида железа (III) без добавления и с добавлением двухзарядных ионов. Осадок, полученный в отсутствие двухзарядных ионов, он смешивал с раствором, содержащим те же ионы. Оба осадка содержали значительные количества двухзарядных ионов. Тем самым Кольтгоф доказал, что имела место адсорбция, а не окклюзия.
Процессы адсорбции наблюдаются и при осаждении кристаллических осадков, так как каждый осадок после образования зародышей проходит через стадию высокодисперсного состояния. С другой стороны, в этом случае вследствие роста кристаллов могут преобладать такие процессы, как окклюзия и образование твердых растворов.
В принципе любые ионы и молекулы, находящиеся в растворе, могут адсорбироваться, причем адсорбируемость ионов (при постоянной концентрации) уменьшается в следующей последовательности:
204
Д. Химический анализ
а) ионы, одноименные с осадком;
б) посторонние ионы, образующие с ионами кристаллической решетки малорастворимые и слабодиссоциирующие соединения;
в) посторонние ионы, оказывающие существенное деформирующее действие (высокозарядные катионы с малым радиусом) или соответственно легко деформируемые (анионы с большим радиусом);
г) недиссоциированные молекулы.
Из ионов одного вида в первую очередь адсорбируются те, концентрация которых больше.
Примеры. 1. Гидроксид металла, осаждаемый аммиаком, адсорбирует преимущественно ионы гидроксила. При этом образуется первичный адсорбционный слой, заряд которого компенсируется зарядом вторичного адсорбционного слоя, образованного катионами. Об этом следует помнить при разделении ионов трехвалентных и двухвалентных металлов, так как ионы двухвалентных металлов могут адсорбироваться в значительной степени, образуя вторичный адсорбционный слой, как, например, ионы кальция на гидроксиде железа(III). Во избежание соосаждения осаждение нужно проводить при наименьшем значении pH и добавлять большой избыток хлорида аммония. Понижение pH препятствует адсорбции ионов гидроксида, а при введении избытка хлорида аммония ионы аммония, образующие второй адсорбционный слой, могут быть легко удалены прокаливанием. Это явление используют при разделении трех- и двухзарядных катионов уротропиновым методом.
2. При осаждении иодида серебра из раствора иодида калия на поверхности осадка находятся адсорбированные ионы калия и серебра, которые могут обмениваться с ионами Н3О+ при промывании осадка азотиой кислотой. Второй адсорбционный слой образуют нитрат-ионы. При высушивании осадка (при 120 °C) азотная кислота удаляется, так что для галогенидов серебра обычно ие возникает ошибки, связанной с адсорбцией.
Адсорбцию недиссоциированных молекул можно наблюдать на осадках сульфидов. Они содержат большое количество сероводорода, которое тем больше, чем меньше pH при осаждении, так как с уменьшением pH увеличивается концентрация недиссоциированного H2S.
Адсорбция больших деформируемых ионов наблюдается при использовании адсорбционных индикаторов в осадительном титровании (разд. 38.3.4.7).
Окклюзия. При быстром росте частиц, покрытых адсорбционным слоем, как это происходит при образовании зародышей осадка с высоким поверхностным натяжением или при большой скорости осаждения, адсорбированные посторонние ионы или молекулы окружают ионную решетку осадка и затем внедряются в нее. Вследствие этого образуются сильно поврежденные кристаллы с большим запасом энергии. Наиболее известна окклюзия больших количеств маточного раствора пустотами больших кристаллов. Этот вид включений с аналитической точки зрения имеет меньшее значение. Более важна окклюзия посторонних ионов.
38. Основы количественного химического анализа
205
Пример. Окклюзия при осаждении сульфата бария. При медленном добавлении раствора хлорида бария к подкисленному раствору сульфата происходит окклюзия катионов, а при добавлении сульфата к раствору соли бария — преимущественно анионов. Это приводит к тому, что осадки, выпадающие из растворов сульфатов щелочных металлов при добавлении к ним по каплям хлорида бария, всегда содержат сульфат щелочного металла, с чем связана отрицательная ошибка определения. Если же осадитель приливать струей, над осадком в растворе создается избыток хлорида бария и осадок окклюдирует адсорбированные в первом слое ионы бария и щелочных металлов и адсорбированные во втором слое хлорид-ионы. Дефицит, вызванный окклюдированием сульфата щелочного металла, компенсируется хлоридом бария, так что по этому методу получают приблизительно правильные значения.
Сульфат бария, образующийся при осаждении из разбавленной серной кислоты, при медленном добавлении хлорида бария окклюдирует гидросуль-фат-ионы, и при последующем прокаливании осадка получаются триоксид серы и вода. Эту ошибку за счет окклюзии можно уменьшить, если осаждение вести из разбавленного раствора, так как в этом случае концентрация ионов HSO4~ уменьшается в результате их диссоциации и, кроме того, при небольшой концентрации гидросульфат-ионов адсорбция их незначительна.
Образование твердых растворов. Особым видом соосажде-ния является изоморфное замещение ионов кристаллической решетки посторонними ионами или молекулами. Изоморфизм в узком смысле этого слова наблюдается, если определяемый и находящийся в растворе мешающий ионы имеют одинаковые заряд и радиус (с допуском в пределах ±10—15%), а структура обеих соответствующих солей одна и та же. При этом образуются твердые растворы как равновесные системы. Это явление принципиально отличается от окклюзии, которая зависит от кинетических данных и всегда приводит к образованию метастабильных кристаллов с большим запасом энергии. Из сказанного следует, что компоненты твердых растворов нельзя разделить при старении осадка.
Для твердых растворов можно сформулировать закон распределения. Например, бромид- и хлорид-ионы могут изоморфно замещать друг друга в солях серебра:
AgBr ± СП я—fc AgCl ± Вг~ (263)
Для такого процесса закон распределения имеет вид
X = CBr-XAgC!. (264)
CC1~ *AgBr
где К. — коэффициент распределения; с — концентрация в растворе; х— мольные доли AgCl и AgBr в осадке.
Аналитически важным является образование следующих твердых растворов: AgBr — AgCl, BaSO4— BaCrO4, BaSO4— PbSO4, MgNH4PO4 —MgNH4AsO4, MgNH4PO4 — MgKPO4, MnNH4PO4 —ZnNH4PO4, Zn[Hg(SCN)4] — Cu[Hg(SCN)4], BaSO4—SrSO4 и др.
Твердые растворы другого типа возникают при замещении ионных пар, например в случае двух однотипных солей, крис
206
Д. Химический анализ
таллизующихся с образованием решеток одного и того же типа,, когда константы решеток различаются не слишком сильно: BaSO4— КМ11О4, BaSeO4— KMnQ4, BaCrO4 — KMnO4, BaSO4 — KBF4.
Образование твердых растворов — довольно распространенное явление. Оказалось, что единственной предпосылкой для их образования является условие, чтобы внедряющиеся частицы могли найти место в решетке основного соединения. Так, сульфат бария образует твердые растворы с нитратом бария, гидросульфатами аммония, калия, натрия и лития, моногидратом сульфата лития, марганцовой кислотой и даже водой. Вероятно, что и фосфат образует твердый раствор с сульфатом бария.
Пример. Образованием твердого раствора сульфата бария с нитрат-иона-ми объясняется чрезвычайно сильное мешающее влияние, оказываемое нитратами при осаждении сульфата бария. Поэтому перед осаждением их обязательно надо удалить выпариванием с соляной кислотой.
Интересным является образование твердых растворов воды с сульфатом бария, при этом три молекулы воды замещают ионную пару BaSO4. Следует учесть, что при анализе в этом случае воду трудно удалить даже при прокаливании (в отличие от адсорбированной и окклюдированной воды).
Возможность образования твердых растворов зависит не' только от геометрических и энергетических, но и от кинетических факторов, например от скорости установления равновесия распределения. В системе AgBr — AgCl эта скорость неизмеримо велика, а в системе BaSO4 — PbSO4 она мала.
Последующее осаждение. В некоторых случаях выделение малорастворимых осадков возможно только в присутствии осадка другой соли. Так, при отделении кальция от магния действием оксалата можно легко превысить произведение растворимости оксалата магния, но осадок не будет выпадать. Только после осаждения оксалата кальция происходит медленное образование осадка соли магния.
Пересыщение наблюдается также при осаждении сульфида цинка. Это особенно заметно при осаждении сульфида ртути в присутствии ионов цинка, которые не образуют осадка ZnS в отсутствие сульфида ртути. На поверхности осадка HgS адсорбируются сульфид-ионы и ионы цинка, так что в адсорбированном слое произведение концентраций <?zn2+ cs2- значительно больше, чем в растворе, и в соответствии с этим зародыши кристаллов легче образуются на поверхности осадка. В конечном итоге на поверхности сульфида ртути выделяется сульфид цинка. Его нельзя затем полностью вымыть соляной кислотой, что может привести к образованию твердого раствора сульфидов цинка и ртути. Последующее осаждение можно уменьшить-увеличением концентрации соляной кислоты при осаждении, так как при этом ионы гидроксония вытесняют ионы цинка и®
38. Основы количественного химического анализа
207
второго адсорбционного слоя и, кроме того, концентрация суль->фид-ионов в растворе, а также и в пограничном слое уменьшается
Старение. Понятие старения включает все процессы, которые происходят после образования осадка и ведут к уменьшению его запаса энергии. Процессы старения делят на четыре группы: рекристаллизацию, «созревание» по Оствальду, термическое и химическое старение.
Рекристаллизация заключается в том, что по окончании осаждения происходит непрерывный переход ионов из кристаллов в раствор и из раствора в кристаллическую решетку осадка. Не все участки кристалла растворимы в равной степени: ионы, находящиеся на ребрах и в вершинах, в значительно большей степени склонны к переходу в раствор, чем ионы, находящиеся на гранях. Растворенные ионы диффундируют в адгезионный слой насыщенного раствора к тому участку кристалла, где присоединение сопровождается наибольшим энергетическим эффектом. Это происходит в местах с незаполненными элементарными ячейками и незавершенными слоями решетки. В процессе рекристаллизации уменьшается число ребер и вершин кристалла, он становится компактнее, его удельная поверхность уменьшается. При этом также уменьшается количество адсорбируемых посторонних ионов.
Окклюдированные посторонние ионы удаляются из решетки вместе с ее ионами. Адгезионный слой не насыщен посторонними ионами, и поэтому они не выделяются из этого слоя в раствор.
Рекристаллизацию можно считать методом самоочистки осадка. Скорость рекристаллизации увеличивается с ростом температуры (увеличивается растворимость и скорость диффузии в адгезионный слой). Кроме того, сильное влияние оказывают растворитель и вид растворенных и адсорбированных ионов. Особенно прочно адсорбируемые ионы и молекулы, например органических красителей, могут подавить рекристаллизацию.
«Созревание» по Оствальду, представляет собой упорядочение роста кристаллов больших размеров при одновременном растворении мелких кристаллов. Это представление является классической концепцией старения осадков. Согласно последним исследованиям, такой механизм старения не является столь общим, как предполагалось ранее, поскольку установлено, что скорость старения многих малорастворимых осадков не зависит от перемешивания. Интенсивное «созревание» происходит в случае осадков бромида серебра в бромидных растворах и коллоидных осадков хлорида серебра. Поэтому можно принять, -что такой механизм старения верен в случае довольно хорошо растворимых осадков.
208
Д. Химический анализ
Под термическим старением понимают процессы, приводящие к образованию осадка с небольшим запасом энергии без участия растворителя. Суть их заключается в том, что при термической обработке осадка ставшие мобильными компоненты решетки диффундируют с участков с более высокой энергией на участки с меньшей энергией. Эти процессы в соответствии с небольшой скоростью диффузии в твердых телах и высокой энергией решетки обычно становятся заметными только при относительно высокой температуре, часто соответствующей там-мановской температуре релаксации, которая равна примерно половине абсолютной температуры плавления. Однако и при более низких температурах благодаря насыщенным растворам, которые образуются в виде поверхностной пленки при адсорбции влаги воздуха, могут протекать процессы упорядочения, связанные с уменьшением энергии. Например, термическое старение поверхности бромида серебра происходит уже при комнатной температуре, что вызвано высокой подвижностью ионов, обусловленной дефектами решетки. Кристаллы сульфата свинца медленно упорядочиваются при комнатной температуре, если они находятся в атмосфере с 85 %-ной влажностью. Для сульфата бария эффект термического старения наблюдается только при 500 °C.
Старение в ходе химических реакций характерно для гидратов различных солей. Например, оксалат кальция при комнатной температуре выпадает в осадок в виде смеси ди- и тригидрата, которые при выдерживании при повышенной температуре переходят в моногидрат. Эта рекристаллизация способствует интенсивной самоочистке.
Еще один тип старения характерен для малорастворимых гидроксидов, например гидроксида железа(III): при конденсации возникают связи Fe—О—Fe, что приводит к образованию молекул с увеличивающейся длиной цепи и уменьшению растворимости. Такой процесс конденсации может происходить также при старении осадка кремневой кислоты путем многократного выпаривания с азотной кислотой.
38.3.4.5. Осадительное титрование
Реакции, сопровождающиеся образованием малорастворимых соединений и соответствующие требованиям, указанным в разд. 38.2.3.1, можно применять для осадительного титрования. В ходе осадительного титрования, так же как в случае ацидиметрии, в точке эквивалентности происходит резкое изменение величины рМ (рМ =—1g см+), для индикации которого применяют обычно электрометрические или визуальные методы.
Симметричные кривые осадительного титрования. Чтобы проследить за изменением концентрации ионов в процессе оса
38. Основы количественного химического анализа
209
дительного титрования, нужно определить концентрацию ионов X- при их взаимодействии с ионами А+, сопровождающемся образованием малорастворимого соединения АХ.
В начальный момент образования осадка справедливы следующие соотношения:
Произведение растворимости
Закон сохранения массы
(265)
(266)
(267)
са+сх“ — Ках.
ПА = ПАГ^ГПАХ Пх = пх |“«АХ
где Сд+ и Сх---равновесные концентрации ионов А+ и X- в
насыщенном растворе; Пд и Пх — молярные доли А и X, находящихся в растворе; па+, пх—ю же для ионов А+ и X-;
Пах — количество осадка в молях.
Вычитание уравнения (267) из уравнения (266) дает
ПА — rtX = rtA +— ПХ~ (268)
Если Vx — объем исходного раствора X- и vA — добавленный объем раствора А+, то из уравнения (268) следует
--А^”х = = _ (269)
уа + &х va + vx '
Обозначив концентрацию титруемого раствора CQ = tixlvx и концентрацию титранта C — nAjvA и предположив, что обе концентрации равны, т. е. С = С0, из уравнения (269) получим
= (270)
При титровании большого объема исходного раствора X-концентрированным раствором А+ в расчетах кривой до точки эквивалентности можно пренебречь разбавлением раствора. Для этого случая с'х^ид. Тогда
Ca+-cx- = -^2L=C-C0 (271)
где Со — начальная концентрация X, которая остается постоянной при допущении, указанном выше; С — количество добавляемого титранта, моль/дм3.
Практический интерес представляет определение не разности концентраций, а изменения концентрации титруемого или осаждаемого иона. Ее получают, вводя в уравнение (271) величину произведения растворимости:
С-С0 = Са+—(272)
Решение уравнения относительно сл+ имеет вид
са+ = Чг (С-С0 + ]/(С-С0)24-4Ках) (273)
14—1880
210
Д. Химический анализ
Подстановка этого выражения в уравнение (271) дает
Сх- = v2 (со - С + ]/(С0 - С)2 + 4КЛХ) (274)
Если не учитывать разбавления, то степень оттитровывания можно выразить как
т = С/Со (275)
Перед началом титрования т = 0 (С = 0), а в точке эквивалентности т=1 (С = Со). Вводя т в уравнения (273) и (274), получим
Са+/С0 = % (т- 1 +/(1-т)2+4/<ах/С02) (276)
Ог/С0 = Ч (1- т+У(1 -т)2+4/<ах/С02) (277)
По аналогии со степенью диссоциации можно определить степень осаждения:
а = -С°7СХ~ = 1---(278)
Со с©
Перед началом титрования сх-=С0 и а=0; в точке эквивалентности Сх- = 0 (приближенно) и ад=1. Точное значение а в точке эквивалентности получают, вводя величину произведения растворимости:
(279)
На основании уравнения (279) можно сформулировать условия для получения высокой точности при осадительном титровании (ад—>-1): малое значение произведения растворимости осадка и высокая исходная концентрация раствора пробы.
Введение уравнения (278) в (277) приводит к следующему:
а = i/2 (1 +т-]/(1-т)2 + 4/<ах/С02) (280)
Если подставить в уравнение (280) значение т=1, то получится уравнение (279).
На рис. Д.68 графически изображены зависимости, описываемые уравнениями (276), (277) и (280). При этом нисходящие кривые отражают зависимость сх-/Со и ад от т (уравнения (277) и (280)), восходящие — сА+/С0 (уравнение (276)) для трех значений отношения КАх/С02: 0,1, 0,01 и 0,001. Восходящий линейный участок кривой 1 (низшая концентрация Со) до точки изгиба Pi соответствует началу выпадения осадка. Координаты точки изгиба Pi — (CA+/C0)i и Т; можно рассчитать из произведения растворимости и уравнения (276). Ордината .(сА+/С0)1 отражает концентрацию ионов А+ в момент начала осаждения. Из произведения растворимости Ках и начальной
38. Основы количественного химического анализа
211
Рис. Д.68. Зависимость степени осаждения а и отношения концентраций с/Сд от степени оттитровывания т при титровании X- ионами А+ с образованием осадка малорастворимого соединения АХ [212].
/ - Яах/Со2=0,1; 2-Л'ах/С02=О,О1; 3 - KAX/Co2=O,OOi.
концентрации Со ионов Х~ находят ca+j =Ках/С0, и отсюда (Са+/С0)1 = /<ах/С02 (281)
Абсциссу Т] можно вычислить при подстановке уравнения (281) в уравнение (276): т; = Ках/С02. Для Р} получают
(сА+/С0)1 = И — КАХ/С02 = 0,1
Кривые, приведенные на рис. Д.68, показывают, что осаждение тем полнее и степень осаждения тем ближе к единице, чем больше концентрация раствора.
Построение кривых осадительного титрования. Из рис. Д.68 можно вывести два приближенных соотношения: до точки эквивалентности концентрация сд+ добавляемого иона, а после точки эквивалентности концентрация <?х- титруемого иона пренебрежимо малы. Таким образом,
до точки эквивалентности сА+ < сх- (282)
после точки эквивалентности сх- <$( сА+ (283)
Применяя эти неравенства и соотношение т = С/С0, из уравнения (271) получим значения концентрации до точки эквивалентности (284) или, в логарифмической форме, (285):
сх- = Со-С=-Сот+Со; lgcx- = lg(C0—С) (284), (285)
Выразив <?х- через произведение растворимости, находим
1g = -Р^АХ- 1g (Со—С) (286)
Тогда для концентрации сА+ после точки эквивалентности спра
14*
212
Д. Химический анализ
ведливо соотношение (287) (в логарифмической форме — (288)):
Сд+ = С- Со = Сот—Со; lgcA+ = lg(C Cg) (287), (288) Вновь вводим произведение растворимости. Тогда
1g ^х- = -Р^дх ~ 1g (С-Со) (289)
и в точке эквивалентности сх- — сА+ = ]/Ккх (290)
или 1g сх- = 1g сд+ = — ^рДах (291)
Если нанести зависимости, описываемые уравнениями (284) и (287) для общей концентрации Со = О,1 моль/дм3, на диаграмму с — т, то получатся графики, приведенные на рис. Д.69. 06-
Рис. Д.69. Типичная диаграмма осадительного титрования иоиов X- ионами А+ (Z. — растворимость).
ратите внимание на то, что прямые на рис. Д.69 представляют собой фактически спрямленные кривые, построенные для небольшого значения Кдх/Со2, т. е. для малого произведения растворимости и большой исходной концентрации (ср. с рис. Д.68). Для пояснения рассмотрим уравнение (277). Для малого значения K^ICg2 в первом приближении можно считать, что
38. Основы количественного химического анализа
213
4/Сдх/Со2^ (1—т)2, и тогда уравнение (277) переходит в (292):
Сх-/С0 = ч (1 - Т(292)
Решение этого уравнения относительно Сх- дает соотношение (284), которое описывает кривую X- на рис. Д.69.
Для построения кривых титрования значения с, полученные из графика, логарифмируют и в соответствии с уравнениями (285), (286), (288) рассчитывают Igtx- и 1§сд+, причем нужно помнить о том, что ординаты кривой Х~ на рис. Д.69, а представляют собой разность Со—С кривой А+. Так, для расчета кривой титрования А+ (рис. Д.69, б) применяют уравнения: до точки эквивалентности— (293), после точки эквивалентности— (294) и в точке эквивалентности— (291):
1g са+ — —Р^ах 1g (Со—Q = —Р^лх 1g сх~ (293) lgcA+ = lg(C-Со) (294)
Из рассмотрения рис. Д.69, б можно сделать общий вывод: если при осадительном титровании образуется осадок состава АХ, например AgCl, BaSO4, LaPO4, то ход титрования всегда описывается такой кривой, для которой характерно совпадение точек эквивалентности и перегиба и симметричность. Именно поэтому эти кривые осадительного титрования называют симметричными. Скачок pH или соответственно рА при т = 1 тем больше и титрование тем точнее, чем больше начальная концентрация титранта и чем меньше произведение растворимости.
Асимметричные кривые осадительного титрования. Осадительное титрование называют «асимметричным» в том случае, если осадок, образующийся при титровании, имеет состав АтХч, где ш^=п (например, оксалат серебра Ag2C2O4 или хлорид ртути (I) Hg2Cl2). Соответствующие кривые титрования несимметричны и точка их перегиба не совпадает с точкой эквивалентности.
В случае асимметричного осадительного титрования потенциометрическая индикация точки эквивалентности приводит к ошибкам, так как в данном случае точку эквивалентности нельзя находить по перегибу на кривой титрования. Подобную ошибку можно устранить расчетным путем. Например, при титровании ионов X2- ионами А+, когда образуется малорастворимое соединение А2Х, рассчитывают ионную концентрацию Сд+ в точке перегиба как с3д+ =8 Ка2х, а в точке эквивалентности — как с3А+ =2 №а2х (А^х — термодинамическое произведение растворимости).
Раздельное осаждение. Если ионы X- и Y~ образуют с Ионом А+ малорастворимые соединения АХ и AY, то при определенных условиях можно определить их содержание в ходе
214
Д. Химический анализ
одного титрования. При этом Л'ах-СЛ'ау. Примем A=Ag, Х = 1 и У = С1. Ход титрования можно разделить на два этапа:
1. Меньшее произведение растворимости ХАХ превышено,, большее /’Cay еще не достигнуто. Выпадает в осадок соединение АХ. Для этого этапа действительны уравнения (273) и (274) в предположении, что ионы Y~ как бы отсутствуют.
2. Превышено и произведение растворимости /Cay- Образуются осадки обоих соединений АХ и AY. На этом этапе действительны следующие соотношения:
СА+СХ- = ^Ах’> CA+CY~ = -^AY ПХ = пХ.~ пАХ> ПЧ~ nY F nAY ПА = nA+ 4~nAx4‘nAY
(295), (296)
(297), (298)
(299)
Аналогично уравнениям (273) и (274) можно записать
са+— х/г C0X-\-C0Y-j-y(C0X-{-C0Y С)2-;-4(Aax4“/Cay) ] (300)
При сравнении уравнения (300) с уравнением (273) с учетом Хах-сКау можно заметить, что на втором этапе осадительного титрования получается такая концентрация Га+, как будто бы один AY выпадает из раствора исходной концентрации СОх + 4- Coy-
Рис. Д.70. Диаграммы дифференцированного титрования раствора, содержащего 0,06 моль/дм3 I- и 0,04 моль/дм3 С1~, раствором Ag+ [212].
38. Основы количественного химического анализа
215
При этом можно построить кривые титрования аналогично рис. Д.69. Вначале проводят прямую из точки на оси ординат Сох под углом 45°. Она пересекает абсциссу в точке С = Сох, соответствующей первой точке эквивалентности, и показывает уменьшение концентрации ионов Х~ в процессе титрования. В то же время cY- постоянна, что отражает прямая, параллельная оси абсцисс и пересекающая ординату в точке Соу. В точке С = С0Х линия изгибается под углом 45°, что соответствует осаждению ионов Y~, и затем, как это следует из уравнения (300), пересекает ось абсцисс в точке С = Сох + Соу (рис. Д.70).
Из этих прямых можно построить кривые титрования (рис. Д.70, б) точно также, как было указано для рис. Д.69.
38.3.4.6. Ошибки при осадительном титровании
Абсолютная ошибка (см. разд. 38.3.1.7) определяется как разность между количеством титранта С (моль/дм3), израсходованного до точки эквивалентности, и количеством титранта Со, необходимым для достижения точки эквивалентности (уравнение 301). Относительная ошибка титрования определяется соотношением (302):
В = С-С0; Fr=(C-C0)/C0 = C/C0-l (301), (302)
Так как по уравнению (275) С/С0=т (303), то
Аг=т—1 (304)
Наиболее надежно ошибку титрования можно определить экспериментально, проводя большое число анализов стандартного образца. Это особенно необходимо при разработке нового метода анализа. Однако такая работа очень трудоемка, поэтому ошибку титрования рассчитывают из условий равновесия для каждой системы. К рассчитанным минимальным ошибкам прибавляют затем другие систематические ошибки: капельную, ошибку градуировки, температурную и др. Так как этот расчет основан на применении закона действующих масс к водным растворам, в следующем разделе будут приведены некоторые конкретные примеры.
Ошибка титрования при осаждении соединения типа АХ. Осаждение такого соединения происходит в соответствии с уравнением реакции
X- + А+ *=> АХ (305)
где А+ — титрант. Тогда из закона действующих масс получаем
Va + vx са+ + сах Va + Ux -Ог+Сах (306) (307)
где Са, Со — концентрации титранта и исходного раствора со-
216
Д. Химический анализ
ответственно; v — соответствующие объемы; Сд+, сх-, Сдх — равновесные концентрации А+, X- и АХ соответственно.
Вычитание уравнения (307) из уравнения (306) дает
__£л£л_________Соух _
ид + ^х + vx л х
(308)
С учетом разбавления степень оттитровывания определяют следующим образом:
т = САцА/Соих
(309)
Из уравнений (308), (309) и произведения растворимости получается, что
(т— 1) Соих/(иАЦ-их) — сА+—(310)
Вблизи точки эквивалентности можно принять т«1. Тогда из уравнения (309) следует
ил/иХ — С0/СЛ
(311)
Добавим к обеим частям уравнения единицу и проведем преобразования:
(ул+ух)/их = (С0+СЛ)/СЛ (312)
Если ввести это соотношение в уравнение (310) и использовать уравнение (304), можно получить выражение для расчета относительной ошибки титрования X- ионами А+ с учетом разбавления:
Fr=A+co (313)
г САС0 ( А СА+ ) 4 7
Из уравнения (313) можно сделать вывод о том, что относительная ошибка осадительного титрования тем меньше, чем больше концентрация титруемого вещества и титранта и чем ближе к конечной точке титрования находится точка эквивалентности, т. е. чем точнее концентрация сд+ в конечной точке титрования совпадает с концентрацией в точке эквивалентности. Если эти концентрации полностью совпадают, то ошибка титрования равна нулю, так как сА+ =УКдх.
Ошибка при раздельном осадительном титровании компонентов смеси. Как видно из рис. Д.70, первая точка эквивалентности при титровании не наблюдается. Ее можно определить приблизительно как точку пересечения восходящей и параллельной оси абсцисс ветвей кривой при их экстраполяции. Поскольку первая конечная точка титрования не идентична первой точке эквивалентности, при определении иона, который первым образует осадок, возникает ошибка титрования. Для ее расчета используют уравнение (313), где Сд+—концентрация ионов А+ в момент осаждения первого осадка AY. Если
38. Основы количественного химического анализа
217
выразить концентрацию через произведение растворимости AY, уравнение (313) примет вид
Fr = (314)
С\С0 ( Су- ЛДу / 7
где cY---концентрация ионов Y~ в момент осаждения первого
осадка AY (не исходная концентрация Y~).
Пример. Раствор с исходной концентрацией иодид-ионов 0,1 моль/дм3 и концентрацией хлорид-ионов в первой конечной точке титрования 0,1 моль/дм3 титруют 0,1 н. раствором нитрата серебра. Ошибка титрования при определении содержания иодида составляет
0,1 +0,1 / io-10 ю-1в
Fr~' 0,Ь0,1 ( 0,1 ~ IO-1” ‘°’1
Fr = —2-Ю-4 %
(315)
(316)
В общем случае нет необходимости учитывать разбавление раствора в процессе титрования, так как оно не оказывает влияния на порядок ошибки. Тогда расчет проводят по уравнениям (301) и (308) и получают
F=C-C0 = Ca+-cx- = oa+--^- (317)
Уравнение (317) выводят следующим образом: если не учитывать разбавление раствора при титровании, то оа<^(Ох- Тогда уравнение (308) можно переписать следующим образом:
-£^А-Со = -^— С0 = С-С0 = са+-сх- (318) иХ VX
Ошибка в первой конечной точке титрования выразится как
г 1 / KAY
г~ СоХ 1 Соу
Я'дХ г
КАУ C°y
(319)
Сопоставление с уравнением (314) показывает, что в уравнении (319) фактор (Са + Со)/Са, отражающий разбавление, равен единице и что в первой конечной точке вместо равновесной концентрации Су- применяют исходную концентрацию Со.
Ошибка титрования при определении галогенидов по методу Мора. Титрование основано на протекании реакции
Cl' +Ag+AgCl (320)
Конечную точку титрования определяют по красно-коричневой окраске осадка хромата серебра:
CrO42- + 2Ag+ Ag2CrO4 (321)
Относительную ошибку вычисляют по уравнению (313):
F _ Cfig + Cci
Г CAgCci
CAg+
A'AgCl \ cAg+ )
(322)
218
Д. Химический анализ
Можно считать, что в данном случае имеет место раздельное осадительное титрование хлорида и хромата ионами серебра. При введении величины произведения растворимости в уравнение (322) получим
Р — CAg + Ccl ( _ jz сСгО42~ \
Г С^а \ cCrO42- AgC1 ^Ag2CrO4 /
В табл. Д.16 приведены ошибки титрования, рассчитанные для титрования растворов галогенидов различной концентра-
Таблица Д.16. Относительная ошибка определения галогенидов при титровании по методу Мора
Концентрация галогенида, моль/дм3 Fr, % ДЛЯ
С1- Вг- I-
0,1 0,02 0,04 0,04
0,01 0,2 0,4 0,4
0,001 2 4 4
ции 0,1 н. раствором нитрата серебра. Для расчета были использованы следующие значения произведений растворимости: KAgci = 2-10-1°, /CAgBr = 6-10-13 [моль2/(дмЗ)2], KAgacro4 = = 4-10~12 [моль3/(дм3)3]. Концентрация индикатора 0,01 моль/дм3. Как видно из табл. Д.16, при такой концентрации индикатора сантинормальные растворы галогенидов еще можно титровать с удовлетворительной точностью.
Ошибки при определении серебра по Фольгарду. При титровании происходит реакция
Ag+ + SCN" 5=^ AgSCN
Конечную точку титрования устанавливают по красной окраске комплекса, образуемого роданид-ионами с ионами добавляемого железа(III). Экспериментально установлено, что при добавлении 2 см3 насыщенного раствора железо(III) аммонийсульфата на 100 см3 титруемого раствора видимая окраска наблюдается уже при концентрации cScn- = 10-5 моль/дм3. Так как KAgscN= Ю~12, абсолютная ошибка титрования составляет
F = C—C0=cSCN-----cAg+ = Ю-з—ю~7 & Ю'5 ь'оль/дм3
Ошибка при определении цианидов по методу Либиха. Реакция при титровании
2CN- + Ag+ Ag(CN)2- (323}
В конечной точке титрования
Ag(CN)2‘ + Ag+ 5=^ Ag[Ag(CN)J (324}
38. Основы количественного химического анализа
219
В момент образования осадка справедливы следующие условия равновесия:
= К cAg+cAg(CN)r = KL (325), (326)
°Ag(CN)2
C-Ag = cAg++cAg(cN)2~; ^cn = ccn h 2cAg(CN)2- (327), (328) где Ccn — начальная концентрация цианида; CAg — количество добавляемого титранта в моль/дм3 раствора пробы (без учета разбавления).
При этом ошибка титрования составит
F= 2 (CAg—CoAg) (329)
где Со Ag — количество титранта, необходимое для достижения точки эквивалентности. Исходя из уравнения (323), для точки эквивалентности получим соотношение
Q Ag = 1/2GcN (330)
Из уравнений (325)—(329) следует
F = 2CAg CCN = 2 (cAg+ 4"Сдв(сН>2~) ^cn ~
= 2 [(^/Ccn)(/^T+2)+Ccn/(]/^+2)]-CcN (331)
Fr — (2^/С%) (/Ж7+2)-(/ЖГ)/(/Ж+2) (332)
Так как в данном случае К= 10~21 (моль/дм3)2 и Лц = 4-10-12 (моль/дм3)2, то ^K/Kl^.2 и уравнение (332) преобразуется к виду
Fr к 4^/C2cN -*/2 УЖГ (333)
На практике в большинстве случаев 10~2<Ccn<1, поэтому справедливы следующие соотношения:
4/(l/C2cS « -V2 Ж (334), (335)
Ошибка титрования при определении цианидов по методу Либиха практически не зависит от концентрации и может быть снижена до —8-10~4%. Таким образом, титрование характеризуется большой точностью.
По этому методу можно проводить определение цианидов в смеси с галогенидами и роданидами. Конечную точку титрования определяют по образованию галогенида серебра, если его произведение растворимости меньше произведения растворимости цианида серебра. Ошибку титрования рассчитывают следующим образом. Если KAgx — произведение растворимости галогенида серебра, то в момент появления осадка AgX
CAg+ = ^Agx/cx- = KbgxJCx (336)
Сочетание уравнений (336), (325) и (328) дает
cAg(CN)2~ = Vs (Qn ~ VСсцСх) (337)
220
Д. Химический анализ
При подстановке уравнений (336) и (337) в уравнение (331) получают
Fr = 2KAgx/CcNCx - V(K/2KAgx) (Cx/Ccn) (338)
Отсюда для бромида серебра Г,®—3,5-10_6УСвг/Ссм, а для иодида « —2,2-10-Дс7сД.
Чтобы получать удовлетворительные результаты, нужно титровать медленно и при сильном встряхивании, особено в конце титрования, так как в месте прикапывания выпадающий цианид серебра растворяется медленно. Эти трудности можно устранить добавлением определенных количеств аммиачной воды, в которой цианид серебра растворяется быстрее.
Титрование по методу Либиха можно применять для определения содержания серебра в галогенидах серебра. Галогенид серебра растворяют в избытке стандартного раствора цианида калия, избыток которого титруют по методу Либиха.
Ошибки титрования при асимметричном осадительном титровании с потенциометрической индикацией точки эквивалентности. Такое титрование всегда сопровождается ошибкой титрования (см. разд. 38.3.4.4). Если титруют ионы Хт~ ионами А"+ и образуется соединение AmXn то ошибку титро-
вания рассчитывают как
m-j-n _________
Fr = [п (т*-п*)/т*Сх] ]/К^п (т/п)*» (339)
где Сх — исходная нормальность титруемых ионов Хт~, отнесенная к объему раствора в точке эквивалентности; №дтхп — термодинамическое произведение растворимости соединения АтХ«.
Для расчетов при титровании А"+ ионами Хт~ в уравнении (339) пит меняют местами. При этом получают такую же ошибку титрования, как в первом случае, но с обратным знаком. Из уравнения (339) следует, что для соединения типа АХ, т. е. при т = п, ошибка равна нулю. Для соединения типа АХг и А2Х используют формулу
ЕГ=±(3/2С)К1/3 (340)
38.3.4.7. Адсорбционные индикаторы
Конечную точку осадительного титрования можно определить с помощью адсорбционных индикаторов, предложенных Фаянсом. Их действие основано на том, что малорастворимое соединение АХ, выпадающее из водного раствора, в первую очередь адсорбирует ионы, одноименные с осадком и находящиеся в избытке. Например, при титровании раствора иодида натрия раствором нитрата серебра до точки эквивалентности в растворе находятся в избытке иодид-ионы, которые и адсорбируются в первую очередь поверхностью осадка; осадок при этом приобретает от
38. Основы количественного химического анализа
22 Г
рицательный поверхностный заряд. После точки эквивалентности поверхность осадка заряжается положительно из-за избытка в растворе ионов серебра. Этот поверхностный заряд частично компенсируется диффузионным, слоем противоионов, в. данном примере — натрия или нитрат-ионов (разд. 38.3.4.3). Если в растворе находятся анионы и катионы органических красителей, например эозина, флуоресцеина, дихлорфлуорес-цеина и др., они раньше адсорбируются поверхностью осадка,, чем ионы натрия или нитрата, так как образуются малорастворимые соли серебра. Адсорбция оказывает влияние на электронную структуру ионов красителя, и их окраска изменяется. Таким образом, изменение окраски осадка в точке эквивалентности связано с адсорбцией или десорбцией ионов красителя.
Переход окраски в точке эквивалентности тем отчетливее, чем больше красителя адсорбировано поверхностью осадка, количество красителя в свою очередь пропорционально поверхности адсорбирующего вещества, поэтому в осадительном титровании стараются получить мелкодисперсные осадки. С этой' целью к титруемому раствору добавляют вещества, способствующие коагуляции и седиментации малорастворимых соединений, например крахмал.
При применении адсорбционных индикаторов концентрацию-кислот и солей нужно точно поддерживать в соответствии с методикой анализа, так как процесс адсорбции специфичен для определенного вида ионов и поэтому зависит от вида и концентрации посторонних ионов. При высоком содержании в растворе посторонних солей переход окраски индикатора может измениться. Кроме того, красители являются слабыми кислотами или основаниями, следовательно, участвуют в протолитических равновесиях. Анион красителя в сильнокислом растворе-находится в виде недиссоциированной соли, которая в необходимом количестве уже не адсорбируется.
Сравнение адсорбционных индикаторов и обзор индицируемых ими реакций даны в книге: Бишоп Э. Индикаторы. В 2-х. томах. Т. 2. Пер. с англ. — М.: Мир, 1976.
38.3.4.8. Практическая часть
Приготовление стандартного раствора нитрата серебра. Взвешивают 8,494 г нитрата серебра марки ч.д. а., высушенного в. течение 2 ч при 120 °C, переносят в мерную колбу вместимостью 500 см3 и доливают до метки свежей дистиллированной водой (не содержащей галогенидов) при температуре градуировки. Получают раствор точно 0,1000 н. Можно также взвесить примерно 8,5 г нитрата серебра и концентрацию раствора рассчитать из навески.
222
Д. Химический анализ
При приготовлении раствора нитрата серебра из загрязненного, перекристаллизованного AgNO3 характеристики этого раствора нужно установить по первичному стандартному веществу—хлориду натрия. Для этой цели хлорид натрия марки ч.д. а., тонко измельченный в порошок, сушат в течение 2 ч при 350 °C в электрической печи. Около 2,9 г этого продукта растворяют в воде. Полученный ~0,1 н. раствор в мерной колбе доливают до объема 500 см3 водой и рассчитывают его характеристики из навески.
Для титрования по методу Мора готовят раствор индикатора: 4,2 г хромата калия и 0,7 г бихромата калия (обе соли марки ч.д. а) растворяют в 100 см3 воды. Для титрования добавляют по 1 см3 раствора на 50 см3 конечного объема титруемого раствора.
Методика титрования. Отбирают мерной пипеткой 25 см'3 0,1 н. раствора хлорида натрия в фарфоровый ролик, добавляют 1 см3 индикатора (пипеткой вместимостью 1 см3) и медленно титруют раствором нитрата серебра при постоянном вращении ролика до медленного исчезновения окраски в месте добавления капель титранта. Титрант добавляют по каплям до резкого перехода окраски в слабую красно-коричневую, которая при дальнейшем вращении раствора также не должна исчезать. Проводят холостой опыт, титруя смесь 50 см3 воды с 1 см3 индикатора, причем на это должно затрачиваться не более 0,03—0,1 см3 раствора титранта. Это количество вычитают от объема, полученного в основном титровании.
Метод Мора отличается быстротой и точностью. Титруемый раствор должен быть холодным и нейтральным, так как вследствие довольно высокой растворимости хромата серебра переход окраски может не произойти. Кислые растворы нужно нейтрализовать гидрокарбонатом натрия.
Вопросы и задания
1. Какой должна быть концентрация хромата при титровании галогенидов по методу Мора, чтобы ошибка определения была равна нулю?
2. Рассчитайте и постройте кривую потенциометрического осадительного титрования 0,1 М раствора Na3PO4 0,1 М раствором Pb(NO3)2. Чему равна относительная ошибка титрования?
3. Рассчитайте относительную ошибку при титровании 0,01 н. растворов хлорида и бромида 0,01 н. раствором нитрата серебра.
4. Какое влияние на ошибку титрования оказывает увеличение объема в процессе титрования?
5. Рассчитайте относительную ошибку потенциометрического осадительного титрования, при котором образуются малорастворимые соединения типа .АтХ„ прн т^=п, считая, что такое титрование практически возможно.
38. Основы количественного химического анализа
223:
38.3.5. Жидкостная экстракция
Если вещество X растворяется в двух несмешивающихся растворителях А и В, то при встряхивании раствора X в А с чистым растворителем В вещество X распределится между обеими фазами*. Фазовый переход можно выразить в виде химической реакции:
X' ^=±. X" (341)
Применив закон действующих масс, получим
X = ах"1й'^' (342)
где а — активность вещества X в соответствующей фазе; X — константа распределения.
Уравнение (342) представляет собой выражение закона распределения Нернста. В числителе стоит активность вещества в более легкой верхней фазе. Для простоты можно записать йх" =а0 и ах' = аи. В идеальном случае или при очень небольшой концентрации распределяющегося между двумя фазами вещества можно принять с ха. Тогда из уравнения (342) следует
К = с0/си (343)
где с0 и Си — равновесные концентрации определенного соединения. Константа К постоянна только в том случае (при постоянной температуре), если соединение в обеих фазах рассматриваемой области концентраций находится в одной и той же молекулярной форме. Если протекают процессы диссоциации или ассоциации молекул и если вводят в уравнение (вследствие незнания этих равновесий) вместо с0 или си аналитически определяемую общую концентрацию, то отношение Со/си уже не будет постоянным. Концентрацию молекул определенного вида можно в этом случае выразить как функцию общей концентрации вещества в соответствующей фазе, если известны все равновесия и их константы. Но это бывает очень редко, поэтому важное значение для практики имеет коэффициент распределения G (см. уравнение (344)).
При построении взаимной зависимости равновесных концентраций вещества в двух фазах (ca от с«) получают изотерму экстракции. Она представляет собой прямую, если экстрагируемое вещество в обеих фазах находится в одной и той же молекулярной форме или если процессы ассоциации или соответственно диссоциации и их концентрационные зависимости
* В литературе на русском языке метод разделения и концентрирования,, основанный на распределении вещества между двумя жидкими фазами, называется экстракционным методом. Поэтому в данном разделе вместо понятия «распределение» использован термин «экстракция». — Прим. ред.
224
Д'. Химический анализ
идентичны в обеих фазах. По наклону графика определяют величину константы распределения. При несоблюдении указанных условий изотерма отклоняется от прямой. В случае неорганических соединений эти отклонения, вызванные межионными взаимодействиями, могут происходить уже при сравнительно небольших концентрациях.
Эффективного распределения можно добиться, подбирая соответствующую систему растворителей. Результирующая константа распределения, не зависящая от концентрации, является константой вещества аналогично температуре кипения, коэффициенту преломления и др. и может быть применена для характеристики веществ и испытания их на чистоту. В этом случае нужно дополнительно указывать температуру и систему растворителей, например вещество X, К2о°с = О,.3 (уксусноэтило-.вый эфир — вода).
В методах периодической экстракции важное практическое и теоретическое значение имеют соотношение масс вещества, .распределенного в обеих фазах, и объемные соотношения фаз. .Для введения этих величин в выражение закона распределения Нернста его преобразуют:
7<=^.== «о. G G=KV (344),(345)
где то, ти — массы растворенного вещества в верхней и нижней фазах; и0, vu— объемы верхней и нижней фаз; G — коэффициент распределения; V — отношение объемов фаз.
Для характеристики вещества вместо константы распреде-.ления К можно также применять коэффициент распределе-.ния G. В этом случае дополнительно необходимо знать отношение объемов обеих фаз V. Коэффициент распределения позво-.ляет непосредственно найти отношение массовых долей вещества, растворенного в обеих фазах, как это видно из уравнения (346);
где т — общее количество экстрагируемого вещества.
Из коэффициента распределения G получают две важные .для двух растворенных веществ X и Y величины — коэффициент разделения Q и объемный фактор Р:
= P = G^ = ^V2 (347), (348)
Коэффициент разделения Q является мерой селективности системы растворителей при разделении двух веществ X и Y. Разделение легко осуществить, если Q^>1; оно невозможно при >Q = 1.
38. Основы количественного химического анализа
225
Объемный фактор важен для выбора отношения объемов фаз при разделении. Пусть необходимо, например, разделить два вещества X и Y путем распределения между двумя фазами. После первого встряхивания в легкой фазе находятся массовые доли веществ рх и ру, в тяжелой фазе — рх и q--:. Разделение оптимально, если
рх = <7У (349)
Тогда из уравнений (346) и (348) следует
Р = 1; V = 1 //ад? (350), (351)
По уравнению (351) определяют оптимальное отношение объемов фаз, необходимое для разделения.
38.3.5.1 . Однократная экстракция
Наиболее известным способом простой периодической экстракции является встряхивание в делительной воронке. Эту операцию обычно применяют при необходимости экстрагирования вещества из раствора. Эффективность экстракции можно определить по закону распределения Нернста:
К = (352)
mul v0 v ’
(353)
<354>
После второй экстракции
(355>
<356)
После п последовательных операций экстракций в нижней фазе содержится
= <357)
В верхней фазе после первой экстракции содержится mOi = pm, после второй экстракции — mo2 = p9m, после третьей — Щоз = = pmUi = pq2m и после п экстракций — tnan=pqn'xtn.
Если обозначим через <ри долю исходного количества вещества, оставшегося в тяжелой фазе после проведения п операций экстракций, а через <ро — долю вещества, перешедшего при 15—1880
226
Д. Химический анализ
этом в верхнюю фазу, то получим следующие соотношения:
Фи= 9” = (1 ку)п ’ Фо = Р9п 1 (358), (359)
Экстракцию считают полной при —>0. В соответствии с
уравнением (358) полнота экстракции достигается при большой
величине константы распределения, большом отношении объ-
емов V= Vo/Vu и при частом
Рис. Д.71. Зависимость массовой доли экстрагируемого вещества ф,<, оставшейся в нижней фазе после однократной экстракции, от константы распределения К при объемном отношении К=1.
повторении процесса экстракции, т. е. большом п. Так как в приведенных уравнениях п входит в показатель степени, на значение <ри оно оказывает большое влияние, чем V. Отсюда следует, что эффективнее десятикратная экстракция раствором объемом 1 см3, чем однократная объемом 10 см3. Эта закономерность справедлива не только в случае жидкость-жидкостной экстракции, но и при любых случаях распределения вещества между двумя фазами, т. е. при промывании осадков, фракционной перегонке и т. п.
Влияние, оказываемое константой распределения на долю вещества <ри, иллюстрирует
рис. Д.71. Кривую получают, строя зависимость <р„= 1/(1 + KV) от /(. Кривая показывает, что при К=1 и К— 100 экстракция уже после однократного контак-
та фаз происходит количественно.
В табл. Д.17 указано число экстракций, которое необходимо провести для извлечения 99% экстрагируемого вещества, в зависимости от коэффициента распределения. При G=1 достаточно провести семь экстракций. На практике важно найти оптимальное соотношение между объемным отношением и числом
экстракций, так как частое встряхивание, а также испарение большого количества растворителя (для выделения экстрагированного вещества), требует больших затрат времени.
Для разделения двух растворенных веществ простая экстракция непригодна, как будет показано ниже. Если, например, нужно однократной обработкой разделить смесь, содержащую по 1 г веществ X и Y (Gx = 2,0; Gy = 0,2), то после первой экстракции в верхней фазе находится 4/6 г X и 1/6 г Y. Массовое отношение при этом составит X : Y = 4. После второго контактирования фаз в верхней фазе находится 4/6-2/6 = 8/36 г X и
38. Основы количественного химического анализа
Таблица Д.17. Число экстракций, необходимых для извлечения 99% исходного раствореииого вещества из нижней фазы, в зависимости от коэффициента распределений®
0 Q п99% о Q п99%
99 0,01 1 0,66 0,6 9
9 0,1 2 0,43 0,7 13
4 0,2 3 0,25 0,8 21
2,33 0,3 4 0,111 0,9 44
1,5 0,4 5 0,053 0,95 88
1 0,5 7 0,010 0,99 454
а —доля экстрагируемого вещества, остающаяся в нижней фазе после 1-го встряхивания [128, с. 43].
1/6-5/6 = 5/36 г Y, т. е. X : Y=l,6; таким образом, относительно первой верхней фазы разделение неудовлетворительно.
Напротив, хорошие результаты получают при повторном встряхивании первой верхней фазы с чистой нижней. После этого в верхней фазе находится 16/36 г X и 1/36 г Y, и теперь X:Y=16. Этот метод называют «противоточной» экстракцией; он лежит в основе фракционной экстракции.
Периодическая экстракция вещества, имеющего небольшую константу распределения, требует больших затрат времени. В этом случае применяют равномерную простую экстракцию (перфорацию). В методе перфорации одну фазу (подвижную) пропускают через вторую (стационарную). В лаборатории в основном применяют перфораторы Кутчера — Штейделя, Палкина и др. Для эффективного проведения перфорации важно, чтобы капельки подвижной фазы имели как можно меньшие размеры, так как при этом в соответствии с законом Стокса (уравнение (360)) существенно увеличивается полезная поверхность экстрагента и уменьшается скорость переноса капель:
« = 2/e^2(Ps~Рг)Л1 (360)
где г — радиус капель; ps и р; — плотность тяжелой и легкой фаз соответственно; ц— вязкость неподвижной фазы; g — гравитационная постоянная.
Большая поверхность экстрагента и маленькая скорость переноса капель способствуют установлению равновесия. Однако при слишком большой разности плотностей возрастает склонность к образованию эмульсий.
38.3.5.2 . Фракционная экстракция
Простейшим способом фракционной экстракции является метод Крейга. Ниже описана схема этого метода (рис. Д.72). В пяти пробирках находятся равные объемы чистого раствори-15*
Рис. Д.72. Схема фракционной экстракции по методу Крейга.
Число зкстпрак-ций Номер фракций
0 1 2 3 4 5
1 0 р
Ч 0
Ч р
г 0 р-ч Р2
ч2 р-ч 0
ч2 2р-ч Р2
3 0 р-ч2 2р2Ч р3
ч3 Зр-Ч2 Р2Ч 0
ч3 Зрчг Зр2Ч Р3
4 0 РЧ3 Зр2р2 Зр3р Р4
ч4 Зрч3 Зр2Ч2 р3ч 0
4 а 4рр3 6р2р2 4р3Ч Р4
5 ' 0 РЧ4 4р2р3 6р3р2 4р4(Г Р5
ч5 4рр4 бр2ч3 4р3Ч2 Р4Ч 0
ч5 5рр4 10p2(f 10p3q2 5р4Ч Р5
Рис. Д.73. Схема пятиступенчатой экстракции по методу Крейга.
38. Основы количественного химического анализа
229
теля — нижней фазы. Нижнюю фазу в пробирке 0 смешивают с раствором распределяемого вещества в легкой фазе (рис. Д.72,а). После установления равновесия и разделения фаз легкую фазу переводят из пробирки 0 в пробирку 1, тяжелую фазу в пробирке 0 снова встряхивают с чистой верхней фазой (рис. Д.72,б). После установления равновесия верхнюю фазу переводят из пробирки 1 в 2 и из пробирки 0 в пробирку 1, затем
Рис. Д.74. Описание схемы распределения по методу Крейга с помощью треугольника Паскаля.
в пробирку 0 снова добавляют чистую верхнюю фазу (рис. Д.72,в). Процесс продолжают до тех пор, пока во всех пробирках будут содержаться нижняя и верхняя фазы. Содержимое каждой пробирки образует фракцию. Верхняя фаза медленно проходит по ряду пробирок через нижнюю фазу (верхняя фаза — подвижная, нижняя — неподвижная).
По окончании процесса вещество, находившеся вначале в
верхней фазе пробирки 0, распределяется по всем фракциям. Строя зависимость массы находящегося во фракции растворенного вещества от номера фракции, получают кривую распределения. В идеальном случае (линейная изотерма экстракции) можно рассчитать относительные количества вещества во фракциях. На рис. Д.73 представлены относительные количества веществ в отдельных фазах после каждого установления равновесия и переноса фаз. Над штриховой линией обозначено содержание вещества в легкой фазе, под ней — в тяжелой. Количества веществ, находящихся во фракциях, при экстракции по методу Крейга могут быть представлены в виде членов бинома (<7+р)п, где п — число ступеней экстракции.
Числовые факторы (коэффициенты) этих членов и степени Р и q можно найти из треугольника Паскаля (рис. Д.74). Ко
230
Д. Химический анализ
эффициенты в формуле (р + р)" получают последовательным суммированием двух соседних коэффициентов вышестоящей строки. Степени р или q для (q+p)n получают умножением буквенных обозначений вышестоящей строки (без учета цифр!): стоящих слева — на р, стоящих справа — на q. Биномиальные коэффициенты рассчитывают из уравнения
(П \ п(п— 1). . . (ti — Г + 1) п!
~J 1-2-3...г п (п-г)! 1)
Отсчет членов (в данном случае — фракции) начинают с 0. Отдельные члены бинома, т. е. относительное содержание <рг в отдельных фракциях, рассчитывают по формуле
PV-' <Э62>
где г — номер фракции (O^grigS). Выражая р и q через коэффициент G и учитывая, что G — pjq и р + р = 1, получают
= г! (и —г)! (1 + G)n (363)
При построении зависимости рассчитанных таким образом относительных количеств веществ <рг от номера фракции получают теоретическую кривую распределения.
Рис. Д.75. Ход восемнадцатиступенчатой экстракции по методу Крейга для трех веществ с коэффициентами распределения 0,333; 1,0 и 3,0.
На рис. Д.75 приведен ход восемнадцатиступенчатой экстракции по методу Крейга для нескольких веществ с различными коэффициентами распределения G. Из кривых можно сделать следующие выводы:
38. Основы количественного химического анализа
231
а) чем больше G, тем дальше передвигается вещество с верхней фазой;
б) при 0 = 1 ширина кривой распределения максимальна (поскольку, чем шире кривая распределения, тем хуже происходит разделение; величины G = 1 нужно избегать);
в) если для двух веществ Хи Y Gx<l> Gy или Gx>/< < Gy, то X и Y разделить трудно;
г) в соответствии с уравнениями Gi = XiV и G2 = /(2V можно сдвинуть кривые распределения двух веществ на диаграмме распределения, изменяя объемное отношение, причем коэффициент разделения при этом не изменяется;
д) разделение оптимально при минимальном наложении кривых, что наблюдается при симметричном расположении кривых относительно центра диаграммы распределения. В этом случае коэффициенты распределения веществ обратно пропорциональны и справедливы уравнения (350) и (351). Это еще раз подчеркивает, что оптимальные отношения объемов обусловливают высокую четкость разделения.
При больших значениях коэффициента распределения (р, трудно рассчитать из уравнения (363). Для этого лучше использовать формулу для приближенного расчета (которая справедлива при л>25), соответствующую уравнению колоколообразной гауссовой кривой нормального распределения:
-1/ (G4-1) (G-Н)2 . „1
Фг=]/ 2nGn ехр[“ 2nG (Нпах— G] (364)
где Гщах— номер фракции с максимальным количеством вещества. Значение Гтах рассчитывают по уравнению
rmax = nG/(G+l) (365)
Приближение заключается в том, что асимметричную кривую распределения заменяют симметричной колоколообразной кривой. Для массовой доли вещества ершах, находящейся во фракции лШах, из уравнения (364) получают, принимая г=гшах, выражение
фтах = / (G + 1)®/2пОЛ (366)
Из уравнения (366) следует, что максимальные концентрации уменьшаются с увеличением коэффициента распределения, т. е. кривая распределения становится шире. Ширину кривой распределения в этом случае определяют из соотношения
b = 6 VnG/(G+ I)2 (367)
где b — число фракций, содержащих 99,7% вещества.
Относительная ширина равна
b f । (j
*0TH = ~7Г = 6 Г ~ (G + I)2 ( 368)
Хотя абсолютная ширина кривой распределения с увеличением числа операций растет, одновременно уменьшается относительная ширина, которая является определяющей для разделения.
232
Д. Химический анализ
Из сказанного следует, что с увеличением числа ступеней экстракции возрастает возможное число разделяемых компонентов. Метод периодического распределения в этом случае требует больших затрат, в частности на конструирование полуавтоматических установок.
Дальнейшим развитием метода Крейга является метод Мартина и Синджа, представляющий собой очень эффективный метод равномерной экстракции. Его осуществляют в вертикальной стеклянной трубке со стационарной фазой и носителем из инертного материала, пропуская через трубку сверху вниз вначале анализируемый раствор, а затем чистую подвижную фазу. Пленка подвижной фазы, образующаяся в этом случае на носителе, действует как элемент многоступенчатой распределительной батареи. Выходящую подвижную фазу собирают равными порциями и в каждой части определяют содержание разделяемых веществ. При построении зависимости содержания веществ от номера фракции получают характеристическую кривую распределения. Авторы назвали метод распределительной хроматографией. Принципы распределительной хроматографии являются основой хроматографических методов.
Предложены также установки для работы с двумя подвижными противоточными фазами. Такой метод дает высокую эффективность разделения, его называют противоточной экстракцией.
38.3.5.3 . Применение экстракционных методов в неорганической химии
До настоящего времени в неорганической химии в основном применяли методы однократной экстракции. Трудность экстракции неорганических веществ состоит в том, что в обычно применяемом растворителе — воде вещества находятся в виде ионов, которые трудно переходят в органическую фазу. Поэтому необходимо перед распределением переводить ионы веществ в нейтральные комплексы или другие недиссоциирован-ные соединения.
Эффективность экстракции можно иногда существенно повысить добавлением нейтральных солей. Они уменьшают растворимость разделяемых веществ в воде и сдвигают экстракционное равновесие распределения (эффект высаливания).
Для экстракции ионов металлов обычно применяют органические комплексообразующие реагенты, такие, как дитизон, оксихинолин, купферон, ацетилацетон, теноилтрифторацетон и др. Ниже перечислены металлы, которые можно извлекать из растворов в виде дитизонатов или соответственно оксихинолятов. Дитизонаты: Мп, Fe, Со, Ni, Си, Zn, Pd, Ag, Cd, In, Sn, Pt, Au, Hg, Tl, Pb, Bi, Po; оксихиноляты: Al, Sc, Ti, V, Mn, Fe, Co, Ni,
38. Основы количественного химического анализа
233
Си, Zn, Ga, Zr, Мо, Pd, Cd, In, Sn, Tl, Pb, Bi, Th, трансурановые элементы. Купферон также находит широкое применение в в качестве комплексообразующего реагента. Металлы, экстрагируемые этим реагентом, можно расположить в следующие ряды: легко экстрагируемые — Al, Ti, V, Fe, Си, Мо, Sn, Sb, Th, Ра, U; сравнительно легко экстрагируемые — Мп, Со, Ni, Zn, Zr, Nb, In, W, Hg, Bi, Ce.
Некоторые катионы можно экстрагировать в виде галогенидов, роданидов или нитратов. В виде хлоридов можно извлечь диэтиловым эфиром из 6 н. раствора соляной кислоты Fe(III), Au(III), Ga(III), Tl(III), As(III), Sb(III), Sb(V), Mo(VI) и Sn(II), но нельзя извлечь Fe(II), T1(I), As(V) (извлекается в небольшом количестве), Al (III). Аналогично ведут себя бромиды. Наряду с Fe(III) диэтиловым эфиром можно извлечь ряд других металлов в виде роданидных комплексов: Zr, Hf, Be, Zn, Al, Sc, Ga, In, Sn, Ti, V, Mo, U, Fe, Co. Экстракция нитратов имеет особое значение благодаря успешно использующимся методам концентрирования и разделения урана и плутония. Элементы, извлекаемые из раствора 8 н. азотной кислотой, можно расположить в следующий ряд: очень легко извлекаемые — Au, Се, Th, U; легко извлекаемые—Р, Cr, As, Zr, Tl, Bi; умеренно извлекаемые — Be, Al, Sc, V, Mn, Fe, Co, Ni, Cu, Zn, Ga, Ge, Y, Mo, Ag, Cd, In, Sb, La, Hg, Pb.
Можно проводить также экстракцию анионов. Так, сульфат и хлорид с помощью длинноцепочечных аминов (например, ди-октиламина) можно экстрагировать хлороформом. Этот метод пригоден для извлечения анионных комплексов металлов.
Рекомендуемые опыты
Построение изотермы экстракции. Реактивы: бензойная кислота, ч.д.а; 0,1 и. раствор едкого натра; тетрахлорид углерода, насыщенный водой, и вода, насыщенная тетрахлоридом углерода (150 см3 чистого ССЦ энергично встряхивают в течение 30 мин с 300 см3 дистиллированной воды и после полного расслаивания тщательно отделяют).
В чистой (обезжиренной) и высушенной делительной воронке вместимостью 100 см3 точно взвешивают примерно 40 мг бензойной кислоты, затем вносят пипеткой 25 см3 тетрахлорида углерода, насыщенного водой. Для установления равновесия хорошо закрытую делительную воронку в течение примерно 5 мин вращают вокруг поперечной оси (при простом встряхивании можно легко получить устойчивую эмульсию). По завершении распределения фазы тщательно разделяют. Из каждой фазы отбирают пипеткой по 20 см3 раствора в стакан Филлипса и титруют 0,1 н. раствором едкого натра из бюретки вместимостью 5 см3 в присутствии фенолфталеина. Фазу тетрахлори-да углерода перед титрованием нужно смешать с 25 см3 ССЦ, насыщенного водой. Так как экстракционное равновесие устанавливается довольно медленно, смесь в процессе титрования нужно постоянно хорошо перемешивать и раствор едкого натра, особенно вблизи точки эквивалентности, добавлять по каплям.
Аналогичный опыт следует повторить для навесок бензойной кислоты 60, 30, 100 и 120 мг.
234
Д. Химический анализ
Строят изотерму экстракции, нанося зависимость концентрации (в моль/дм3) вещества в верхней фазе (ось ординат) от соответствующих концентраций в нижней фазе.
Разделение Лэ и Sb методом фракционной экстракции. Соединения As(V) и Sb(V) распределяются между диэтиловым эфиром ибн. соляной кислотой или лучше диизопропиловым эфиром и 6,5—8 н. соляной кислотой.
Помимо указанных задач можно по данной теме выполнить следующие работы:
1. Построение кривой фракционного распределения двух соединений (в пробирках).
2. Определение неизвестных значений констант распределения.
3. Построение зависимости некоторых коэффициентов распределения от концентрации (построение изотерм экстракции).
4. Идентификация неизвестного соединения по константе распределения.
5. Построение кривой распределения в зависимости от числа ступеней распределения.
6. Определение скорости экстракции.
7. Сопоставление теоретической и экспериментальной кривых распределения.
38.3.6. Хроматографические методы
38.3.6.1. Общие положения распределительной
хроматографии
Под хроматографией в настоящее время понимают все методы разделения, основанные на распределении веществ между подвижной, обычно равномерно движущейся, и стационарной фазами. Такое определение не означает, что все хроматографические методы основаны на жидкость-жидкостном распределении. В хроматографии часто сочетаются процессы распределения и адсорбции, которые на практике определяются природой неподвижной и подвижной фазы и типом разделяемых соединений.
Метод идеальной распределительной хроматографии, заключающийся в равномерном распределении веществ, предложили Мартин и Синдж (разд. 38.3.5). Процессы адсорбции при этом не имеют значения, так как или не применяют носитель, а используют свободную стационарную фазу, или в качестве носителя применяют абсолютно инертный (стекловидный) материал.
Адсорбционную хроматографию применяют для распределения смесей органических веществ, растворимых в петролейном эфире или подобных ему растворителях; в качестве сорбентов при этом используют активированный уголь, диоксид алюминия или аналогичные носители.
Для теоретического описания хроматографических процессов составляют уравнения, описывающие как распределение, так и адсорбцию. В данной главе мы рассматриваем хроматографию как процесс распределения по Крейгу. Это очень полезно, так как хроматография связана с большим числом ступеней разделения, и поэтому для ее описания можно использовать математический аппарат жидкость-жидкостной экстракции. Выве
38. Основы количественного химического анализа
235
денные соотношения пригодны также для адсорбционной хроматографии и даже для ионного обмена, поскольку активную поверхность адсорбентов можно рассматривать как самостоятельную фазу.
Установление равновесия при периодической экстракции по методу Крейга происходит в распределительных элементах различных установок. В хроматографии за такие элементы распределения приняты «теоретические тарелки». Это понятие можно пояснить следующим образом: если скорость распределения вещества между подвижной и неподвижной фазами в колонке была бы бесконечно большой, фазы находились бы в равновесии в любом поперечном сечении колонки. В действительности вещество распределяется между двумя фазами с конечной скоростью и, кроме того, одна фаза проходит через другую. Прежде чем концентрация вещества в подвижной фазе станет равной равновесной концентрации в поперечном сечении колонки на выходе, подвижная фаза продвинется дальше на отрезок h. Объем hF, где F — поперечное сечение колонки, и является объемом «теоретической тарелки». Буквой h обозначена высота, эквивалентная «теоретической тарелке» (английское название: Hight equivalent of a theoretical plate, сокращенно НЕТР).
Точно так же, как в экстракции по методу Крейга, разделение двух веществ происходит тем эффективнее, чем больше ступеней распределения, четкость хроматографического разделения возрастает с увеличением числа «теоретических тарелок». Это число является характеристикой эффективности хроматографических колонок и зависит от скорости потока подвижной фазы и скорости распределения вещества между фазами, которая в первую очередь зависит от величины поверхности раздела фаз, т. е. от констант колонки (плотность упаковки носителя, размер зерен и пористость).
Для математического описания функции распределения введем следующие обозначения: h — высота, эквивалентная «теоретической тарелке»; Fs — поперечное сечение неподвижной фазы; Fm •—поперечное сечение подвижной фазы; F = FS + Fm; v — объем растворителя, необходимый для развития хроматограммы; К—коэффициент распределения:
___Количество растворенного вещества в г на 1 см3 неподвижной фазы ' Количество растворенного вещества в г на 1 см3 подвижной фазы
G—число распределения; R—коэффициент:
п Участок от старта до места с максимальной концентрацией вещества Участок от старта до фронта растворителя
г — номер тарелки, начиная с 0; VE = h(Fm + RFs) ~vm + Kvs — эффективный объем тарелки; V—отношение объемов (vs/vm).
236
Д. Химический анализ
Понятие эффективного объема требует пояснений. Адсорбционная способность одной тарелки зависит не только от ее геометрического объема, но также и от того, какое количество растворенного вещества переходит в неподвижную фазу. Это количество описывается коэффициентом распределения. Отсюда и вытекает указанное понятие эффективного объема.
В тарелке № 0 находится единица количества вещества. В отличие от экстракции по методу Крейга после достижения равновесия не весь объем подвижной фазы попадает в первую тарелку, а только часть dv, в которой содержится массовая доля pdvlvm. В нулевой фракции остается 1—pdv]vm вещества. Аналогично распределению по Крейгу и в этом случае перенос растворенного вещества можно представить в виде бинома:
[(1— pdv^m)--pdv;vm\n^ 1
(369)
Уравнение (369) целесообразно преобразовать следующим образом:
vm!p = vm (1 +q/p) = vm (14-G) = vm (1+AV) = vm + Kvs = vE (370)
При этом условии уравнение (369) принимает вид
г/ i_jq+_*Li"= 1
VE / 1 VE J
(371)
Этот бином разлагают, как было показано в разд. 38.3.5.2. После п ступеней распределения, т. е. при добавлении п раз по объему dv, образуется /г+1 фракция (номера фракций изменяются от 0 до /г). Относительную массу <рг вещества (в данном случае равную его абсолютному количеству, так как исходят из единичного количества), во фракции г после п распределений находят по уравнению, аналогичному (362):
Если п очень велико и dvfvE очень мало, т. е. при соблюдении обоих условий, которые должны выполняться в хроматографии, уравнение (372) переходит в выражение распределения Пуассона:
ndv
(373)
Подставив ndv — v, получают
(374)
где v/vE— объем подвижной фазы в единицах объема тарелки, применяемый для развития хроматограммы. При большом г (г>> 10) уравнение (374) с учетом приближения Стирлинга
38. Основы количественного химического анализа
237
(375) переходит в уравнение колоколообразной гауссовой кривой (376), которое значительно удобнее для практических расчетов при большом значении г:
П-(2«г)'«^; „ (375), (376)
Если первая производная уравнения (376) равна нулю, то максимум на кривой диаграммы ф— v наблюдается при
(«max = г; Фтах = (377), (378)
Чтобы уяснить смысл уравнений (377) и (378), можно провести такой мысленный эксперимент. Предположим, что в хрома-
Рис. Д.76. Идеальная кривая элюирования.
ci — число «теоретических тарелок» колонки; vE — эффективный объем «теоретической тарелки»; vR — удерживаемый объем; (р —доля элюированного вещества.
тографическую колонку, содержащую вещество в теоретической тарелке № 0, последовательно, маленькими порциями dv вводят чистую подвижную фазу и в то же время непрерывно определяют массу растворенного вещества в тарелке Л. В соответствии с уравнением (377) после г добавлений Ve объемов чистой подвижной фазы в тарелке № г находится максимально возможное для данной тарелки количество вещества Фтах. Это иллюстрирует рис. Д.76. Если г самая нижняя из п+1 тарелок колонки, т. е. г = п, то при прохождении объема nvE=vR чистой подвижной фазы получим максимум вещества. Объем vR называют удерживаемым объемом. Фронт растворителя появляется после добавления ndv чистого растворителя.
Величину R находят из следующего соотношения:
п__ v/(Fm + Kfs)_______Р С379>
К v!(Fm + Fs) Fm+ KFS
Для бумажной хроматографии из практических соображений определяют значения Rf и К:
^Т^Т7-’ <38^(38|>
238
Д. Химический анализ
Частное Fm/Fs равно отношению объемов растворителя и водной фазы на хроматограмме. При известном содержании воды в бумаге можно определить Fm/Fs из соотношения масс сухой бумаги и влажной хроматограммы.
38.3.6.2. Эффективность хроматографической колонки
в газовой хроматографии
Эффективность хроматографической колонки выражают числом «теоретических тарелок» или высотой, эквивалентной «теоретической тарелке». Ван Деемтер с сотрудниками предложил для газовой хроматографии следующее уравнение, названное его именем:
H = 2i.dp~
8 К' d? л2 1 + К' йж
(382)
2vDa и
где Н — высота, эквивалентная «теоретической тарелке»; X — степень неравномерности заполнения колонки сорбентом; dp— диаметр частиц носителя; v •— коэффициент извилистости; и — линейная скорость потока газа; Da — коэффициент диффузии растворенного вещества в газовой фазе; DM — то же в жидкой фазе, df — эффективная толщина пленки неподвижной фазы; К' = КРж1Рг (где К — коэффициент распределения, Рж и Fa — объемные части жидкости и газа в колонке).
Для конкретной колонки уравнение (382) переходит в
Н = А-\-В/и-[-Си (383)
где А, В и С — константы. Величина А отражает влияние вихревой диффузии, В — молекулярной диффузии, С — массопере-дачи.
Под вихревой диффузией, или диффузией рассеяния, понимают распространение (рассеяние) вещества, переносимого подвижной фазой, происходящее во всех направлениях в колонке и обусловленное нерегулярностью расположения каналов между частицами носителя.
Зависимость Н от и представляет собой кривую, изображенную на рис. Д.77. Из ее рассмотрения можно сделать следующий важный вывод: существует скорость потока, при которой колонка характеризуется наименьшей высотой, эквивалентной «теоретической тарелке», или большим числом «теоретических тарелок», т. е. наиболее высокой эффективностью. При очень малой скорости газового потока, т. е. при Си<^А + В/и,
Н — АА-В/и (384)
Это уравнение гиперболы, которая образует левую ветвь на кривой (рис. Д.77). При высокой скорости потока, т. е. при
38. Основы количественного химического анализа
239
В/и<^А + Си, получим уравнение прямой линии
Я = С«+Л (385)
В области, которая отражена левой вертикальной ветвью кривой, т. е. при малом значении и, преобладает диффузия растворенного вещества, которая нарушает образование четких зон адсорбции и тем самым уменьшает эффективность разделения. В области, где и велико (правая линейная часть кривой), большая скорость потока мешает установлению адсорб
ционного и распределительного равновесий, что также оказывает существенное влияние на разделение веществ. В минимуме оба влияния компенсируются. Интересно, что на эффективность хроматографической колонки более существенно влияет малая скорость потока, а не большая.
Из констант В и С следует, что высота, эквивалентная «теоретической тарелке», принимает различные значения для каждой системы растворенное вещество — подвижная фаза — неподвижная фаза.
Эти зависимости, выведенные для газовой хроматографии, можно в основном пере-
Рис. Д.77. Кривая эффективности хроматографической колонки (кривая ван Деемтера).
нести и на жидкостную хроматографию, если учесть, что коэффициент диффузии газов примерно в 104 раз больше, чем жидкостей. Это соответственно уменьшает влияние члена В/u: оно становится заметным только при очень небольшой скорости потока. Кроме того, следует учесть, что в газовой хроматографии скорость потока газа составляет 50—100 см3/мин, а скорость в жидкостной хроматографии 1—5 см3/мин.
38.3.6.3. Положение и вид хроматографических зон
Положение и вид хроматографических зон определяются формой изотерм распределения и адсорбции, скоростью установления равновесия и степенью диффузии растворенного вещества в подвижной фазе.
В отличие от идеального распределения, описываемого линейной изотермой распределения, ход реальной изотермы адсорбции зависит от концентрации. Так как в системе твердое
240
Д. Химический анализ
вещество — жидкость наблюдаются также адсорбция растворителя и сольватация растворенного вещества, на изотерме адсорбции могут наблюдаться максимумы или минимумы. Адсорбционные равновесия в разбавленных растворах можно изобразить кривой, представленной на рис. Д.78. Изогнутая часть этой кривой соответствует эмпирической изотерме адсорбции Фрейндлиха, которая рассчитывается по уравнению
п' = kck' (386)
где п' — количество адсорбированного вещества на 1 г адсорбента; k — коэффициент адсорбции; с — концентрация раствора; k' — константа. Пригодность уравнения можно определить, построив зависимость 1g п' от 1gс. Если получают прямую, то изотерма Фрейндлиха применима. Область высоких концентраций (прямая, параллельная оси абсцисс) не описывается уравнением изотермы Фрейндлиха.
Всю кривую, приведенную на рис. Д.78 (изотерма адсорбции Ленгмюра), описывает уравнение
(387>
где Ь, Сое — константы.
При небольших концентрациях, когда Ьс<^1, п' = схЬс. Это уравнение относится к линейному восходящему участку кривой на диаграмме п'/с. Иначе говоря, уравнение (387) при небольших концентрациях переходит в выражение закона распределения Нернста.
В области больших концентраций Ьс^>1 и п' = сх— это уравнение линии, параллельной оси абсцисс. Из последнего уравнения становится ясным значение сх: это емкость насыщения адсорбента.
Идеальная хроматограмма получается при выполнении следующих условий: линейность изотермы адсорбции, мгновенное установление равновесия, пренебрежимо малая величина диффузии. При этом хроматографическая зона имеет колоколообразную форму гауссовой кривой нормального распределения (упрощенно представленную на рис. Д.79, а). Вещества, характеризующиеся большими величинами коэффициентов распределения, имеют меньшую величину Rf, чем вещества с малыми коэффициентами распределения. Симметричное расширение хроматографических зон обычно обусловлено практически всегда происходящей диффузией, а также затратой определенного времени на установление равновесия (рис. Д.79,б). Нелинейная изотерма адсорбции соответствует получению асимметричных хроматографических зон (рис. 79,в). Если изотерма адсорбции имеет вид, как на рис. Д.78, в хроматографической зоне появляется так называемый «хвост», образование кото-
Рис. Д.78. Изотерма адсорбции Ленгмюра.
Рис. Д.79. Изотермы адсорбции и соответствующие им формы хроматографических зон.
сл — концентрация вещества на адсорбенте; cL — концентрация вещества в растворе; s — пУть, пройденный веществом; L — фроит растворителя [Martin A. J. Р., Endeavour, 6, 21 (1947)].
16—1880
242
Д. Химический анализ
рого вызвано тем, что растворенное вещество сильнее адсорбируется из раствора с более низкой концентрацией (край зоны), чем из более концентрированного раствора. Однако образование более прочных адсорбционных соединений вызывает замедление скорости передвижения зоны.
38.3.6.4. Методы хроматографического анализа
Фронтальный анализ. Хроматографируемый раствор компонентов А, В и С непрерывно подают сверху колонки. Пусть коэффициенты распределения располагаются в последовательности КаЖв>Кс (ср. с определением коэффициентов распределения в хроматографии, разд. 38.3.6.1). Компоненты выходят из колонки ступенчато в последовательности, обратной значениям их коэффициентов распределения (рис. Д.80, а). Этот метод можно применять для оп-
Рис. Д.80. Схемы разделения в различных хроматографических методах.
ределения неизвестного числа п компонентов смеси, но полного разделения веществ не происходит.
Вытеснительный анализ. Колонку, в которой находятся вещества А, В и С, промывают раствором вещества D. Если при этом КцЖаЖвЖс, вещество D вытесняет все три соединения, находившиеся в колонке (рис. Д.80, б).
Так как зоны располагаются непосредственно одна за другой, эффективное разделение веществ не достигается. Вещества можно разделить при последовательном элюировании сначала веществом 1, если соотношение коэффициентов распределения следующее: Лс<К1<Лв, затем веществом 2 при Ав<К2<Ка и наконец веществом 3 при КзЖа-Получается кривая элюирования, представленная на рис. Д.80, в.
Этот метод можно применять в том случае, если соединения 1, 2 и 3 легко можно удалить из элюата или если они не мешают дальнейшей работе.
38. Основы количественного химического анализа
243
Элюентный анализ. В методе элюентного анализа адсорбированные соединения вымывают избытком растворителя с меньшим коэффициентом распределения, чем у адсорбата. При этом в элюате образуются раздельные зоны (рис. Д.80,г).
трубку, снабженную на конце
Рис. Д.81. Схема хроматографической противоточной колонки.
38.3.6.5. Приборы и оборудование
Колоночная хроматография. Основным узлом хроматографической установки является колонка, в простейшем случае представляющая собой стеклянную фриттой и краном. Ее заполняют адсорбентом и пропускают через нее раствор с разделяемыми компонентами. Скорость прохождения раствора регулируют краном. По завершении процесса проявляют хроматограмму, т. е. разделяют зоны, промывая колонку чистым растворителем и собирая элюат на выходе отдельными фракциями.
Для разделения двух веществ, коэффициенты распределения которых сильно отличаются, нужно применять противоточные колонки. Действие их основано на~том, что прочно адсорбируемое вещество проходит в колонке небольшое расстояние, в то время как слабо адсорбируемое вещество быстро выходит из колонки. При этом за небольшой промежуток времени получают концентрированный раствор. Конструкции противоточных колонок различны, схема одной из них приведена на рис. Д.81. Для получения воспроизводимых результатов нужно работать при постоянной температуре, поэтому колонки снабжают специальными рубашками.
Разделение более сложных смесей веществ требует больших аппаратурных затрат. В этом случае применяют автоматические приспособления для подачи растворителей и устройства для сбора фракций.
Основной областью применения колоночной хроматографии является препаративное разделение химически сходных соединений. Этим способом можно, например, разделить а- и р-каро-тины, отличающиеся только положением двойной связи. Инте-
16*
244
Д. Химический анализ
ресным примером из области неорганической химии является разделение оптических изомеров хлорида триэтилендиаминхро-ма(Ш) на колонках, заполненных d- или L-кварцем. Особенно эффективно применение колоночной хроматографии для концентрирования очень разбавленных растворов. При этом для адсорбции следовых количеств веществ требуется совсем немного адсорбента, элюировать из которого концентрируемое вещество можно сравнительно небольшим объемом растворителя. Тот же способ можно использовать для удаления загрязнений, например из растворителя. Для химической идентифика
Рис. Д.82. Блок-схема газового хроматографа.
] — баллон с газом-носителем; 2 — регулятор давления; 3 — вентиль избыточного давления; 4 — манометр; 5 — измеритель расхода газа-носителя; 6 — детектор; 7 — камера сравнения; 8— измерительная камера; 9 — ввод пробы; 10 — газохроматографнческая колонка; 11 — термостат.
ции вещества его можно хроматографировать на колонках с различными наполнителями. Если во всех случаях образуется только одна зона, это с большой вероятностью говорит о том, что мы имеем дело с индивидуальным веществом.
Газовая хроматография. Газовая хроматография требует сложной аппаратуры. Прежде всего необходим детектор для измерения концентрации соединений во фракциях, выходящих из колонки. Действие детекторов может быть основано на различных физических принципах. Чаще всего используют теплопроводность и плотность газов, ионизацию, происходящую при горении, диэлектрическую проницаемость и иногда радиоактивность. Блок-схема газового хроматографа приведена на рис. Д.82.
Газовую хроматографию в основном используют для аналитического разделения смесей летучих компонентов и их идентификации. В этом методе можно достичь такой высокой селективности и чувствительности анализа, которая не достигается в других методах. Так, например, можно полностью разделить нефтяные фракции, содержащие более чем 20 компонентов. Высокотемпературная газовая хроматография дает возможность разделять сложные смеси компонентов с большой разницей
38. Основы количественного химического анализа
245
температур кипения. Газовую хроматографию используют также и в неорганической химии, например для разделения различных летучих хлоридов и ацетилацетонатов металлов.
Если известны удерживаемые объемы или соответственно времена удерживания веществ, их можно идентифицировать методом газовой хроматографии (качественный анализ). Для гомологического ряда органических соединений, например парафинов, спиртов, сложных эфиров и т. д., установлена линейная зависимость между логарифмами значений удерживаемых объемов и числом групп СН2 в молекулах, что также дает возможность провести качественный анализ веществ внутри каждого гомологического ряда.
Точное количественное определение веществ можно провести путем построения градуировочной кривой — зависимости количества вещества от площади под пиком.
Газохроматографический анализ можно сравнительно легко автоматизировать, тогда он становится пригодным для контроля продукции, управления и регулирования в химической промышленности.
Кроме того, метод газовой хроматографии дает возможность получать информацию теоретического характера, например определять коэффициенты и энтальпию адсорбции, исследовать поведение твердых катализаторов.
Важнейшей задачей является увеличение числа разделяемых газов. Развитие препаративной газовой хроматографии даст возможность выделять большие количества чистых летучих веществ из сложной смеси сходных соединений. В этом случае газовая хроматография приблизится к методу фракционной перегонки.
Бумажная хроматография. Метод прост по аппаратуре и чрезвычайно эффективен для аналитических целей, что приводит к его использованию практически во всех химических лабораториях. Для получения бумажной хроматограммы небольшое количество раствора (1—3 мм3) исследуемой смеси наносят в виде пятна на расстоянии ~ 5 см от конца полоски хроматографической бумаги, которую затем этим концом опускают в специальный подвижный растворитель. В зависимости от применяемого метода подвижный растворитель может поступать сверху или снизу (нисходящая и восходящая бумажная хроматография). Время развития хроматограммы составляет, как правило, 8—20 мин*. Бумага может быть расположена горизонтально, как, например, при использовании круглых фильтров. При этом раствор и подвижный растворитель помещаются в центр фильтра, а хроматографические зоны располагаются в виде концентрических кругов. Бумага должна нахо
* В оригинале, видимо, ошибочно указано 8—10 ч. — Прим. ред.
246
Д. Химический анализ
диться в атмосфере насыщенных паров растворителя, например в закрытой стеклянной хроматографической ванне. При определении Rf нужно поддерживать постоянную температуру. Пятна или кольца, появляющиеся на бумаге, проявляют, обрызгивая ее подходящими реактивами. Этот процесс в бумажной хроматографии называют проявлением. В количественной бумажной хроматографии для определения используют следующие приемы: пятно планиметрируют, фотометрируют или вырезают, экстрагируют вещество и раствор после проведения соответствующей обработки фотометрируют.
Бумажная хроматография как метод микроанализа применяется для эффективного разделения сложных смесей компонентов. Пятна неизвестных компонентов идентифицируют, сравнивая с пятнами эталонных образцов, полученных в таких же условиях. Таким способом можно просто и быстро провести анализ неорганических веществ.
Если неизвестное соединение относится к определенному гомологическому ряду (жирные кислоты, аминокислоты, полифосфаты, политионаты и др.), то его можно идентифицировать, сравнивая экспериментальное значение Rf с рассчитанным.
Тонкослойная хроматография. В этом методе в качестве стационарной фазы применяют тонкий слой кизельгура, оксида алюминия, карбоната кальция, целлюлозы и т. п., нанесенный на стеклянную пластинку. Метод сочетает такие преимущества, как небольшое время развития хроматограммы (от нескольких минут до нескольких часов), с поразительным в ряде случаев эффектом разделения.
38.3.7. Методы обмена ।
38.3.7.1. Ионный обмен
В отличие от адсорбции ионный обмен представляет собой химическую реакцию. Одно из реагирующих веществ является нерастворимым полиэлектролитом и образует неподвижную фазу. Ниже приведены уравнения реакций: (388) —для анионного обмена и (389)—для катионного. Рамкой обведены ионы, задерживаемые ионитом.
38. Основы количественного химического анализа
247
Состояние равновесия, как обычно в химических реакциях, определяется свободной энергией ДЕ, которая связана с термодинамической константой равновесия следующим соотношением:
ДЕ = — RTlnR (390)
Если в реакцию обмена вместо иона М+ с объемом с'м вступает ион N+ с объемом гщ (им и Un— объемы гидратированных ионов), то ионит набухает или сжимается, при этом возникает давление набухания л. Произведение л(цм— гщ) выражает свободную энергию процесса обмена. Тогда
RT — л (цм— pN) (391)
Если Е заменить на Кс, то
К = Vm_ (392)
с Ум Ух v ’
где у'м, ‘y'n — коэффициенты активности ионов, адсорбированных ионитом; ум и ум — то же для ионов, находящихся в растворе. Из уравнений (391) и (392) следует:
1пКе=^(0„~«+1п-»— lnJ< • (393)
Уравнение (393) описывает зависимость состояния равновесия от давления набухания л (которое, в свою очередь, зависит от степени сшитости ионита), разности объемов конкурирующих гидратированных ионов и от электростатических взаимодействий, происходящих в растворе и в ионите. Значения л и Vm—Vn можно рассчитать, измеряя давление набухания и объем набухшего ионита, а коэффициенты активности — из полуэм-пирических зависимостей.
Соотношение между концентрациями ионов, адсорбированных ионитом, и ионов, находящихся в растворе, приближенно отражает уравнение, сходное с выражением для изотермы адсорбции Ленгмюра:
Мм
00 (1 + М ом -J- Mn
(394)
где п' — количество ионов М+, адсорбированное единицей массы ионита, моль/дм3; — общая обменная емкость ионита; а — активность ионов в растворе; bi и Ь2 — константы.
Иониты. К неорганическим катионитам относятся алюмосиликаты щелочных металлов — цеолиты (встречающиеся в природе) и пермутиты (искусственные). Эти иониты представляют интерес только для специальных областей. В настоящее
248
Д. Химический анализ
время в лабораториях в качестве ионитов применяют, за редким исключением, высокополимерные органические смолы с ионообменными группами. Каркас смолы обычно получают сополимеризацией стирола и дивинилбензола. Чем больше доля дивинилбензола, тем выше степень сшитости полученной смолы и тем меньше ее растворимость, пористость и способность к набуханию.
Катиониты изготавливают, например, сульфированием полиэфирных смол; при этом образуются полисульфоновые кислоты. Введением СН2С1-групп в бензольное кольцо (хлорметили-рование) с последующим замещением хлорметильных групп ди-или триметиламинами получают сильноосновные аниониты с
группами —NH(CH3)2 или —N(CH3)3 соответственно.
Кроме перечисленных существуют и другие функциональные ионообменные группы: —СООН, —ОН, —SH, —РОаН2 — в катионитах; —NH2, — NH — в анионитах.
Равновесие обмена слабокислотных или соответственно слабоосновных ионитов зависит от pH, что видно из следующих уравнений реакций:
Н+ + Na+ Na+ + Н+ '
(395)
он- + ci- ci- + он-
(396)
Полная обменная емкость характеризуется общим количеством способных к ионному обмену однозарядных ионов ионита (в моль на 1 г сухой массы ионита). Ее можно установить следующим способом: ионит в Na-форме обрабатывают несколько раз 3 н. раствором сильной кислоты до перевода всех ионнообменных групп в Н-форму. Затем ионит промывают водой до нейтральной реакции, пропускают через него концентрированный раствор хлорида натрия и элюат титруют.
Для аналитических работ полную обменную емкость ионита можно не использовать, так как количественный обмен Na+ на Н+ происходит даже тогда, когда в ионите еще находится некоторый избыток ионов Н+. Поэтому вводят понятие «полезная обменная емкость». Она составляет 60—75% общей обменной емкости, и ее можно определить как количество ионов, поглощаемых ионитом при постоянной скорости потока жидкости до проскока ионов. Из всего сказанного ясно, что слабокислотные катиониты обладают наибольшей полезной обменной емкостью в щелочных средах, а слабоосновные аниониты — в кислых. Полезная обменная емкость сильнокислотных или соответственно сильноосновных ионитов не зависит от pH в широкой области значений.
38. Основы количественного химического анализа
249
38.3.7.2. Применение ионного обмена
Различают два варианта проведения ионного обмена: статический и динамический. В статическом методе анализируемый раствор смешивают с ионообменной смолой. Дают установиться равновесию и отфильтровывают ионит. Чтобы реакция прошла количественно, эти операции повторяют. Статический метод анализа применяют только в двух случаях: а) для регенерации ионита при сильном разбухании смолы или выделении из нее газа; б) для растворения малорастворимых осадков с помощью ионного обмена. При этом раствор не загрязняется избытком посторонних ионов:
. СаСО3 + 2 Н+ Са2+ + Н2О + СО2
BaSO4 + 2 Н+ Ва2+ + 2Н++8ОГ
Было предложено использовать этот метод также для получения плавиковой кислоты из фторида кальция.
Для аналитических и препаративных работ наиболее часто применяют динамический метод ионного обмена в колонках. Способы работы такие же, как в колоночной хроматографии (разд. 38.3.6.4). Следует кратко остановиться на некоторых специфичных областях применения ионообменной хроматографии.
Количественный анализ. Ионный обмен является одним из важнейших методов количественного анализа и дает возможность определять ионы, анализ которых другими методами связан с большими трудностями, например ионы натрия:
Н+ + Na+^ Na+ + Н+
Анализируемый раствор пропускают через колонку с ионитом и затем промывают водой до нейтральной реакции элюата. Весь объем элюата титруют раствором щелочи. При определении Na+ в смеси солей и кислот лучше применять метод ионного обмена на сильноосновном анионите в ОН-форме:
2| ОН- + Na+ + СГ+ Н+ + СГ Na+ + ОН- + Н2О + 2 С1~
Уделение мешающих ионов. Ионный обмен можно применять для удаления ионов магния или меди, находящихся в дистиллированной воде и мешающих проведению комплексонометрического титрования, особенно если применяют эриохром
250
Д. Химический анализ
черный Т в качестве индикатора. Эти ионы легко можно уда- 1 лить с помощью катионитов. На этом же принципе основан ме- ' тод деионизации воды: воду пропускают последовательно черев колонки с катионитом и анионитом. Для удаления мешающих ионов применяют также ионообменные колонки типа «mixed-bed», заполненные смесью катионита и анионита в отношении,, обратном их обменным емкостям. Для проведения регенерации иониты необходимо разделить, что можно сделать, используя их различную плотность или различные размеры частиц.
Мнение о том, что деионизованная вода, полученная методом ионного обмена, чище, чем дистиллированная, неверно, так как иониты пропускают многие неионные загрязнения. Кроме того, вода загрязняется из-за того, что матрица ионита еще в течение длительного времени после загрузки выделяет в воду органические соединения. Таким образом, очень низкая электропроводность деионизованной воды не является точным критерием ее чистоты.
Ионообменное концентрирование. Для концентрирования следовых количеств ионов через ионит пропускают большой объем соответствующего раствора. Ионы, поглощенные ионитом, можно затем элюировать относительно небольшими количествами концентрированного раствора электролита. Таким способом можно сконцентрировать очень разбавленные растворы без нагревания.
Существует ряд природных ионитов, специфичных только к определенным ионам. Если бы удалось синтезировать аналогичные иониты с большой емкостью, то появилась бы интересная возможность извлечения редких металлов из морской воды. Уже опубликованы первые работы по синтезу высокоселективных ионитов.
Препаративное применение. С помощью ионитов можно синтезировать нестойкие кислоты или основания из их солей, например гипофосфорную кислоту.
Na++ Н2РО2 4- ГЙ+
Н3РО2 4- Na+
Можно также заменять катионы солей, что особенно ценно в тех случаях, когда для данного катиона нет реакций количественного осаждения. Например, пероксо дисульф ат калия можно перевести в соответствующую соль натрия.
Разделение ионов. Методом элюентного анализа можно разделять ионы, используя их различную способность к полному обмену. Поскольку методика работы такая же, как в методе хроматографического разделения, этот метод называют ионообменной хроматографией.
38. Основы количественного химического анализа
251
Для применения метода нужно знать относительную сорбируемость разделяемых ионов. Для этого существует общее правило, согласно которому в ряду ионов одинакового заряда сорбируемость ионов уменьшается с увеличением их эффективного радиуса, под которым понимают радиус гидратированного иона. Поскольку радиусы гидратированных ионов обычно обратны радиусам ионов в кристаллах, для большинства ионитов существуют следующие сорбционные ряды ионов (в порядке возрастания сродства к иониту):
Li+ < Н+ < Na+ < К+ = NH4+ < Pb+ < Cs+ < Ag+
e2+<Mn2+< Mg2+ = Zn2+<Cu2+ = Ni2+< Со2+< Ca2+<Sr2+ <Pb2+< Ba2+ F" <СГ <Br" <I"
Для ионов с разными зарядами сорбируемость возрастает с увеличением заряда:
Na+ < Са2+ < La3+ < Th4+
В некоторых случаях можно существенно изменить сорбируемость путем комплексообразования, что дает возможность разделять ионы с близкой сорбируемостью. Этот способ применяют, например, при разделении редкоземельных металлов. По уменьшению сорбируемости трехзарядные ионы редкоземельных металлов можно расположить в следующей последовательности:
La > Се > Pr > Nd > Pm > Sm > Eu > Gd > Tb > Dy > > Y > Ho > Er > Tm > Yb > Lu
Таким образом, наиболее медленно перемещается в колонке лантан, наиболее быстро — лютеций. По завершении ионного обмена ионы элюируют комплексообразующим реагентом — цитратом аммония. Тенденция к образованию устойчивых комплексов возрастает с уменьшением радиуса негидратиро-ванного иона. Следовательно, лютеций элюируется наиболее быстро, при этом увеличивается эффективность разделения.
Следует помнить, что в существенной степени поглощаются ионитом только те ионы, которые могут проникать в поры матрицы смолы. Используя это свойство, можно проводить разделение ионов низко- и высокомолекулярных соединений: высокомолекулярные проходят через колонку, низкомолекулярные задерживаются в ней. Аналогично можно использовать различие скоростей обмена.
38.3.7.3. Ионные и молекулярные сита
Все большее значение приобретают синтетические кристаллические алюмосиликаты щелочных металлов в качестве ионных или молекулярных сит (например, типа «цеосорб», изготовляе
252
Д. Химический анализ
мых предприятием Farbenfabrik Wolfen). В этих высокомолекулярных соединениях существуют каналы, радиусы которых различны в зависимости от структуры силикатов, но одинаковы для одного и того же образца силикагеля.
Ионные и молекулярные сита применяют для разделения молекул различных размеров. Так, ионы натрия синтетического файазита обмениваются с ионами аммония, которые можно расположить в следующий ряд в порядке уменьшения сорбируемости: NH4+ > NH3CH3+ > NH3(C2H5) + > NH2(CH3)2+ > >NH (CH3)3+>N (CH3)4+>N (С2Н5)4+.
Полную обменную емкость молекулярные сита приобретают только после нагревания их при 350—400 °C. В каналы, образующиеся после удаления воды, могут проникнуть, например, «зо-парафины, в то время как «-парафины не адсорбируются. Другие силикаты поглощают низкомолекулярные «-парафины, но не сорбируют «зо-парафины и ароматические углеводороды. Для разделения можно использовать также различную скорость адсорбции. Молекулярные сита часто применяют для осушки газов.
Сорбционную способность молекулярных сит можно изменять предварительным введением молекул подходящей величины или с помощью ионного обмена.
38.3.7.4. Электронообменные иониты
Электронообменными ионитами называют высокомолекулярные нерастворимые соединения, в состав которых входят обратимые электронообменные группы. Различают два вида электронообменных ионитов: окислительно-восстановительные иониты и ре-докситы.
Окислительно-восстановительные иониты получают, вводя в обычный ионит ионы обратимой редокс-системы. Например, окислительно-восстановительные иониты образуются при введении в сильнокислотный катионит пар ионов Fe (11)/Fe (111). Sn(II)/Sn(IV), Ce(II)/Се (IV), Ti(III)/Ti(IV) и др.
Более стабильные редокситы (на основе гидрохинона, метиленового синего, тиолов и др.) содержат электронообменные группы непосредственно в матрице смолы.
Электронообменные иониты можно перевести в восстановленную форму действием дитионита натрия или в окисленную действием пероксида водорода. В восстановленной форме их применяют для удаления кислорода, пероксидов и галогенов из водных и других растворов. Ионы обратимой редокс-системы можно также количественно восстанавливать и затем определять методами окислительно-восстановительного титрования. Другие области использования электронообменных смол: выделение из растворов благородных металлов (золота, серебра),
38. Основы количественного химического анализа
253
а также ртути и меди; получение свободного иода и иодоводо-родной кислоты пропусканием раствора иодида калия через сильнокислотный катионит с последующим переводом полученной кислоты в восстановленную форму; получение пероксида водорода путем восстановления кислорода в редоксите. Сильноосновной анионит с поглощенным иодом можно применять как нерастворенный иод.
Рекомендуемые опыты. Количественный анализ смеси сульфата, гидросульфата и дигидромонофосфата методом ионообменной хроматографии.
Реактивы: 0,1 и. раствор гидроксида натрия; 0,1 н. раствор соляной кислоты; сильноосновной анионит SBW (выпускаемый предприятием Farbenfabrik Wolfen), сульфат калия, гидросульфат калия, гидрофосфат калия.
Подготовка ионита. Для проведения анализа анионит переводят в ОН-форму. Для этого необходимое количество коммерческого препарата ионита (обычно в Cl-форме) оставляют на ночь для набухания. Затем его заливают в стакане 1 н. раствором едкого натра, тщательно перемешивают в течение ~ 10 мин и отсасывают на фильтре со стеклянной пористой пластинкой G = 3. Эти операции повторяют до тех пор, пока в фильтрате будут обнаружены лишь следовые количества хлорид-иона. Затем ионитом равномерно заполняют колонку (20X1,5 см), в которой находится NaOH и промывают 1 н. раствором гидроксида натрия до отсутствия следов С1~ в фильтрате. Если ионит обрабатывается первый раз, то необходимо 3—4 дм3 1 н. раствора гидроксида натрия для данного количества ионита. Перевод в ОН-форму нельзя сразу проводить в колонке — ионит при этом сильно набухает, что приводит к увеличению
давления в колонке и к существенному уменьшению скорости капания. Ионит затем промывают прокипяченной дистиллированной водой до нейтральной реакции (при первичной обработке ионита необходимо 8—10 дм3 воды).
Приготовление стандартного раствора. Примерно по 600 мг KaSO4. KHSO4 н КН2РО4 точно отвешивают в мерную колбу вместимостью 100 см3 и растворяют в прокипяченной дистиллированной воде. Раствор в колбе доливают до 100 см3 водой. В 25 см3 этого раствора определяют содержание сначала гндросульфата калия титрованием 0,1 н. раствором гидроксида натрия при pH 4,5 (в присутствии смешанного индикатора бромкрезоловый зеленый + метиловый красный) и затем содержание фосфата титрованием при pH 4,5—9,5 (в присутствии фенолфталеина). Соетветствующие объемы титранта равны Vi и v2 (см3).
Рис. Д.83. Схема простого устройства для поддержания постоянной скорости капания.
/ — мерная колба; 2 — элюируемый раствор;. 3 — слой ионита; 4 — капиллярная трубка.
254
Д. Химический анализ
Проведение ионного обмена. 20 см3 стандартного раствора пипеткой переносят в воронку ионообменной колонки и пропускают ео скоростью капания примерно 15 капель в минуту. Затем ионит промывают прокипяченной водой, пропуская ее с такой же скоростью до получения объема элюата примерно 1 дм3. Элюат титруют 0,1 н. раствором соляной кислоты в присутствии смешанного индикатора (объем титранта v3 см3). Если количество гидросульфата, фосфата или сульфата (в ммоль) обозначить через пнзо4, про4 или nso4 соответственно, то получим пнзо4 = 0,1 vt; Про4 = 0,1 v2; пзо4 = 0,05 (у3—v2—Vi).
Для проведения ионного обмена, промывания колонки до нейтральной реакции промывных вод и элюирования применяют приспособление, схематически изображенное на рис. Д.83. Оно позволяет получить постоянную скорость капания и проводить элюирование в отсутствие экспериментатора (например, в ночное время).
39. ПРИМЕНЕНИЕ ЭЛЕКТРОХИМИЧЕСКИХ МЕТОДОВ
В КОЛИЧЕСТВЕННОМ АНАЛИЗЕ
Развитие количественных методов анализа исторически тесно связано с созданием новой измерительной техники. Так, возможность разложения света1 в спектр обусловила появление разнообразных и чрезвычайно ценных оптических методов анализа, дальнейшая разработка которых продолжается и, в настоящее время. 3 свою очередь, применение этих методов в количественном анализе вызвало необходимость точных электрических способов измерения интенсивности светового потока. Изучение закономерностей электрических процессов и создание точных приборов для измерения силы тока и напряжения стало основой возникновения и развития электрохимических методов анализа. Затем! появились термические методы анализа, основанные на точном измерении температуры* с помощью термоэлементов и термисторов, и радиохимические методы анализа, в которых осуществляется чувствительная регистрация радиоактивных излучений.
В принципе почти все характерные физические свойства элемента или соединения могут быть использованы в методах количественного анализа. Но на практике крайне редко непосредственно измеряют физическое свойство вещества. Обычно определяют изменение физической величины относительно известного стандарта и таким образом находят неизвестное количество вещества. Тем самым измерение физической величины является лишь средством, позволяющим сравнить концентрацию определяемого соединения в анализируемом образце и стандарте.
39. Электрохимические методы в количественном анализе
255
Значение инструментальных методов анализа, как и современных методов разделения (см. гл. 38), постоянно возрастает, что обусловлен^ требованиями науки и производства. Так, например, появилась тенденция использования сырья, содержащего очень небольшие количества целевого продукта, а также извлечения элементов из отходов производства, в которых эти: элементы находятся' в очень небольших количествах. •Кроме того, все шире используются особо чистые вещества1 и композиционные материалы, к которым предъявляются высокие требования, в частности постоянство концентраций компонентов (металлургия, полупроводниковая техника). Постоянно растущая рационализация и автоматизация производств и связанный с этим более быстрый*- выпуск продукции диктуют необходимость использования аналитических методов, обладающих большой чувствительностью, точностью и быстротой. Быстрота анализа — особенно важный фактор, так как все в большей степени контроль готовой продукции заменяют своевременным контролем качества полупродуктов в ходе технологического процесса с целью регулирования процесса в нужном1 направлении. Поэтому анализ, также должен быть по возможности автоматизирован, саморегистрируем, а полученный сигнал должен быть использован для управления процессом.
Если в начале столетия считалось достаточным1 определение десятых долей процента веществ и нормальным проведение анализа в течение ряда дней, то в настоящее время необходимо определять тысячные доли процента в течение нескольких минут. Методы классического анализа даже при самом тщательном совершенствовании техники работы не отвечают таким требованиям. Границы применимости этих методов определяются! точностью аналитических весов в гравиметрии, интервалом перехода окраски индикаторов в объемном анализе и, прежде всего, необходимостью длительных, приводящих к? ошибкам определения методов разделения.
Принципиально большую эффективность новых физических методов можно показать на образном примере, приведенном М. Штакельбергом: один грамм-атом — это пылинка, но заряд величиной в один фарадей может сообщить всему земному шару потенциал 160 миллионов вольт.
Инструментальные методы анализа столь тесно связывают неорганическую, органическую и физическую химию, что невозможно разграничить, например, методы органического и неорганического инструментального анализа. Тем не менее целью этой книги является рассмотрение важнейших методов и принципов анализа неорганических веществ. Новейшие данные по различным областям) анализа можно найти в обзорах, помещаемых каждые два года в апрельских номерах журнала «Analytical Chemistry».
256
Д. Химический анализ
39.1- Электрогравиметрия
Электрогравиметрия — наиболее старый метод электрохимического анализа. Название «электрогравиметрия» более правильно отражает сущность метода, чем применявшийся ранее термин «электролиз». Под электрогравиметрией понимают такой метод анализа, в котором «реагентом» служит избыточное количество электрического тока. Под действием тока определяемый компонент выделяется из раствора в виде осадка, который затем взвешивают. Следовательно, способом определения как таковым в данном методе является гравиметрия.
В раствор электролита погружают два электрода, к которым прикладывают постоянное напряжение. На обоих электродах при этом могут протекать химические реакции: процессы восстановления (присоединение электронов) на отрицательно заряженном катоде и процессы окисления (отдача электронов) на положительно заряженном аноде. При этом число электронов, принимаемых на аноде, должно быть равно числу электронов, отдаваемых на катоде. В соответствии с законами Фарадея количество выделившегося при этом из раствора вещества пропорционально количеству тока, проходящего через цепь.
Для понимания сути процесса нужно рассмотреть вопрос о взаимосвязи напряжения, силы тока и сопротивления. Для проводников первого рода эта зависимость выражается законом Ома: U — iR. При прохождении тока через раствор электролита эта зависимость изменится. Пусть два электрода, изготовленные из одного и того же металла, опущены в водный раствор соли этого металла (например, серебряные электроды в раствор AgNO3) и к ним приложено постепенно увеличивающееся напряжение. Напряжение ек, необходимое для выделения металла на катоде при комнатной температуре, определяют в соответствии с уравнением Нернста:
ек = е°ме/ме+ + 1g <*кМе++*1Ме (397)
где т] — перенапряжение. Напряжение еа, необходимое для растворения металла на аноде, равно
еа = е«ме/ме+ + ^~ lg«aMe++r]Me (398)
До тех пор пока акМе+=«аМе+, ек и еа равны и противоположны по знаку и поэтому взаимно уничтожаются. В этом идеальном случае ток протекает при приложении любого сколь угодно малого напряжения. Электрическая работа затрачивается только на нагревание контура (включая раствор электролита).
39. Электрохимические методы в количественном анализе 257
Закон Ома при этом справедлив. Кривая зависимости силы -тока от напряжения линейно возрастает (рис. Д.84, кривая У).
В действительности при прохождении электролиза вблизи электродов происходит изменение концентрации ионов Ме+, связанное с восстановлением их на катоде (по уравнению де+_р.£-—>-Ме) и с окислением на аноде (Me—>Ме+ + а_). Эти изменения концентраций вблизи электродов нельзя полностью
устранить даже при интенсивном перемешивании раствора в процессе пропускания тока. Они, естественно, тем больше, чем больше сила тока и чем меньше размер электродов. Поэтому
аКме+<йаМе+ и еа и ек уже не равны: еа увеличивается, а ек уменьшается. Разность этих потенциалов противоположна при-
ложенному напряжению и поэтому ее называют противопотен-циалом или концентрационным перенапряжением (Up)- Прило-
женное напряжение должно быть больше потенциала перенапряжения. Если для начальной стадии электролиза соотноше-
ние между величинами можно было = UKn/i (где Rz — сопротивление и«л-—напряжение на клеммах), то теперь его следует заменить на Rz=(UKn—Up)/i.
выразить формулой Rz = ячейки; i — сила тока;
Следовательно, в электрогравиметрии напряжение на клеммах должно расти быстрее, чем ток, проходящий через раствор. На графике (рис. Д.84, кривая 2) наблюдается отклонение от линейности. Поскольку концентрационная поляризация электро-
Рас. Д.84. Вольтамперные кривые.
i —• сила тока; С7 — напряжение; inp—предельный ток; — остаточный ток; Up — поляризационное напряжение; Uz~~ напря-
дов тем сильнее, чем больше ток, это отклонение всегда проявляется с увеличением силы тока. Если при увеличении
напряжения сила тока уже не
женне разложения.
возрастает даже при переме-
шивании раствора электролита, то достигнут так называемый предельный ток. Сила тока в этом случае ограничена скоростью Диффузии ионов к электродам через пограничный слой. Скорость диффузии определяется законом Фика: при постоянной температуре она зависит только от концентрации. Поэтому
вольт-амперная кривая идет в этом случае параллельно оси напряжений (рис. Д.84, кривая <?), сила тока имеет постоянную величину, обозначаемую как inp. Величина его зависит от концентрации разряжающихся ионов, находящихся в растворе. Эту зависимость используют в полярографических методах анализа.
17—1880
258
Д. Химический анализ'
Рассматриваемый до сих пор случай, когда два электрода, выполненные из одного и того же металла, погружены в раствор соли этого металла, может встретиться только при электролитическом рафинировании металлов и не находит применения в электрохимическом анализе. В электрогравиметрии ионы Ме+ должны полностью быть выделены из раствора, и поэтому нельзя использовать электроды из того же металла. Кроме того, материал электродов не должен реагировать с раствором и должен быть устойчивым к агрессивным средам. Обычно используют платину.
Пример. Рассмотрим ход вольт-амперной кривой при электролизе раствора CuSOi с двумя платиновыми электродами. Ток может протекать тогда, когда .напряжение достаточно велико', чтобы на катоде происходил процесс восстановления и одновременно на аноде — процесс окисления. На катоде иоиы Си2+ восстанавливаются до Си, при этом катод покрывается слоем металлической меди. Катодный потенциал можно выразить как
0,059
8к = e°cu/Cu2+ + ——lg “Cu2+ + TlCu (399)
На аноде ионы ОН- окисляются до О2 в соответствии с уравнением
4ОН" ---->- О2 + 2Н2О 4- 4е*
Поэтому анодный потенциал можно выразить уравнениями (400) или (401):
„ 0,059 , Ро,
еа = е0ОН~/О2 +-4— 1g + 1А (400)
еа = е°он~/о2 — 0,059 lgаон~ + -°’^59 1g ро2 + ПО, (401)
Поскольку аон-ян3о+= Ю-14, можно записать
0 059 , еа = е°он~/о2+ 14-0,059—pH-0,059+ —— lgPo2 + T)°2 '
Так как е°он~/О2 = +0,402, окончательно получаем
еа== 1,23-рН-0,059+-^~lgpo2+^02 (402)
Это означает, что анодный потенциал тем больше, чем чище растворитель. Ход вольт-амперной кривой для электролиза иллюстрирует кривая 4 (рис. Д.84). Значительный ток появляется, если напряжение превышает известное минимальное значение, т. е. становится выше напряжения разложения Uz. Точное значение его можно определить экстраполяцией вольт-амперной кривой на ось напряжений. Выше этого значения платиновый катод становится медным электродом, а платиновый анод — кислородным электродом (аналогичным водородному). Начиная с этого момента, равновесное давление кислорода у платинового анода достигает величины 0,1 МПа. Под действием атмосферного давления кислород выделяется из раствора. Выше напряжения разложения ход кривой 4 на большом протяжении аналогичен ходу приведенных на этом же рисунке кривых 2 и 3. Незначительное возрастание тока при наприжении ниже напряжения разложения, так называемый остаточный ток in можно объяснить следующим. На аноде ионы ОН~ могут окисляться
39. Электрохимические методы в количественном анализе 259
уже при более низких значениях напряжения, чем это необходимо для создания ро; = 0,1 мПа. Кислород при этом выделяется из раствора не в виде пузырьков газа, а диффундирует от электрода в раствор. Аналогичные рассуждения можно применить для реакции восстановления Н+ до Н2 на катоде, которая может начаться уже при небольших катодных потенциалах.
В табл. Д.18 приведены значения напряжений разложения Uz, определенные экспериментально для некоторых 1 н. растворов солей. Если сравнить приведенные экспериментальные
Таблица Д.18. Величины напряжений разложения
Соль C/z, в Соль c/z, в
CdSOt 2,03 CdCl2 1,88
C0SO4 1,92 CoCi 1,78
N1SO4 2,09 NiCl2 1,85
Таблица Д.19. Величины перенапряжений
Соль ^гэксп- В ^грасч» В ьи, в
CdSO4 2,03 1,38 0,70
COSO4 1,92 1,52 0,40
NiSO4 2,09 1,49 0,60
данные с расчетными, то можно заметить, что они отличаются друг от друга (табл. Д.19). Это различие может быть вызвано перенапряжением, которое имеет место в реальных катодных и анодных процессах. Перенапряжение складывается из анодной и катодной составляющих. В общем перенапряжение тем больше, чем выше плотность тока (сила тока, отнесенная к величине поверхности электродов). Поэтому перенапряжение меньше при работе с платинированными платиновыми электродами (большая поверхность); при работе с гладкими платиновыми электродами оно возрастает. Кроме того, оно существенно зависит от материала электродов. Для других металлических электродов свойственно, как правило, большее перенапряжение. Для электродов из одного и того же материала перенапряжение также может различаться в зависимости от того, какое вещество выделяется на электроде. Выделение металлов на катоде сопровождается в общем очень небольшим перенапряжением, которым можно пренебречь: например, для Пары Cu2+/Cu при небольшой плотности тока оно составляет 17*
260
Д. Химический анализ
только 0,001 В. Несколько выше перенапряжение только для металлов группы железа (Fe, Со, Ni).
Выделение водорода сопровождается минимальным перенапряжением на платиновом электроде; оно значительно выше для всех других металлических электродов. Особенно большое перенапряжение наблюдается в случае ртутных электродов. ’Поэтому эти электроды особенно эффективны в полярографическом анализе. Только перенапряжением водорода объясняется возможность проведения электролитического выделения неблагородных металлов, таких, как Cd, Zn и др. из водных растворов или растворов кислот.
Величина перенапряжения (табл. Д.19) определяется в основном большой величиной перенапряжения выделения кислорода на аноде (0,4—0,5 В), так как перенапряжения выделения металлов малы. Мы еще не раз остановимся на использовании этого эффекта в электрохимических методах анализа.
Значения потенциалов разложения дают нам сведения только о начале электролитического выделения в 1 н. растворе соли. Так как в ходе процесса происходит уменьшение концентрации ионов вследствие выделения их на катоде и аноде, то потенциалы катода и анода при электролизе также изменяются. Из уравнения (399) следует, что в ходе разрядки катионов катодный потенциал уменьшается (сдвигаясь в сторону отрицательных значений), так как уменьшается активность катионов. Соответственно из уравнения (401) видно, что с потерей заряда ионами ОН~ (аОц- становится меньше) анодный потенциал возрастает (сдвигаясь в сторону положительных значений). Напряжение разложения определяется как
^г = еа—ек (403)
Отсюда следует, что напряжение разложения в процессе электролиза должно увеличиваться. Степень изменения катодного потенциала в процессе электролиза Дек можно получить, исходя из уравнения (399):
lg £у на?„. (404)
* “Си + конеч
Для изменения анодного потенциала из уравнения (402) следует
Деа = 0,059ДрН (405)
Все другие величины в уравнениях (399) и (402) при создании разности потенциалов остаются постоянными.
Для расчета изменения напряжения разложения At/z используют выведенное из уравнений (403), (404) и (405) еле-
39. Электрохимические методы в количественном, анализе
261
дуюшее выражение:
—0,059 (ДрН+4 1g °с^+-ач-) (406)
у aCir+ конеч /
Значение ДрН в процессе электролиза можно рассчитать, исходя из расхода ОН-, которое эквивалентно выделившемуся количеству Си2+. Подкисляя раствор, можно добиться такого положения, чтобы анодный потенциал в процесе проведения анализа изменялся лишь незначительно (почему?). При электрогравиметрическом определении речь идет о «полном» выделении ионов определенного типа, В действительности полного выделения, естественно, добиться невозможно, что следует из уравнения (406). Ведь если бы аси2+ действительно равнялась нулю, то Uz стремилось бы к бесконечности. Бесконечно большое напряжение нельзя получить: при этом прежде всего произошло бы электролитическое разложение растворителя — в случае воды на катоде выделился бы водород. Для аналитических целей обычно достаточным является 99,99%-ное выделение, ошибка определения при этом составляет 0,01%. Тогда в соответствии с уравнением (404) катодный потенциал ионов Ме2+ должен составлять —0,119 В, а ионов Ме+ —0,238 В. Эту область называют зоной осаждения. Предпосылкой для проведения безупречных электрогравиметрических определений является выполнение требования, чтобы в интервал напряжений катода, соответствующий зоне осаждения, не входило значение катодного потенциала других катионов, которые также могут восстанавливаться. Определив предварительно концентрации катионов других металлов, находящихся в растворе, можно, зная величины стандартных потенциалов, рассчитать зону осаждения этих катионов. Избирательное электрогравиметрическое определение возможно, если рассчитанные области не перекрываются или лишь соприкасаются; для надежности разрыв между ними должен быть не менее 100 мВ, тем самым будут исключены посторонние включения в решетку металла, выделяющегося первым. Если концентрация катионов менее активных металлов значительно больше концентрации электрохимически более активных (например, 99% Си и 1% Cd), то Целесообразно расширить зону осаждения первых, если необходимо определить содержание последних с высокой точностью (почему?).
При неудовлетворительном расположении зон осаждения разделение можно осуществить, если зону осаждения сдвинуть Добавлением комплексообразующих веществ. Благодаря различию в значениях констант устойчивости комплексов активность свободных катионов уменьшается не одинаково, что обусловливает разницу в степени смещения зон осаждения в отрицательную область.
262
[[.Химический анализ
Процесс выделения существенно осложняется при выделении водорода на катоде. Катодный потенциал восстановления Н-+ не должен попадать в пределы зоны осаждения металла. Рассчитать его можно по формулам
екна ~ е°н2+0,059 Ig аИз0+ -|-цН2 (407)
еВН2=—0,059рН4-лН2 (408)
Значение потенциала можно легко сдвинуть, изменяя кислотность среды, хотя возможности смещения его в отрицательную область (большие значения pH) ограничены образованием осадков гидроксидов выделяемых катионов. Выпадение гидроксидов можно предотвратить, используя реакции комплексообразования, но все же для получения хороших результатов необходимо принимать защитные меры (так как в результате комплексообразования уменьшается активность катионов металлов и их потенциал также сдвигается в отрицательную область). Сильно отрицательное перенапряжение водорода (т)Н2) на многих металлах по этой причине оказывает благоприятное влияние, поскольку дает возможность проводить электрогравиметрическое определение ряда металлов, как было указано выше. Наконец, следует также учитывать, что потенциал водорода в процессе электролиза сдвигается в сторону положительных значений, так как в растворе возрастает концентрация ионов Н3О+, образующихся эквивалентно количеству выделившегося на катоде металла. Потенциал выделения водорода и по окончании электролиза не должен достигать потенциала зоны осаждения.
Иногда протекание нежелательных побочлых реакций можно уменьшить применением так называемых деполяризаторов. Окислители относятся к катодным деполяризаторам, а восстановители— к анодным. Примером катодных деполяризаторов являются ионы NOs-, которые могут подавлять реакцию выделения Н2 на катоде. Выделение кислорода на аноде можно уменьшить, например применяя гидразин в качестве деполяризатора. Значение окислительно-восстановительного потенциала деполяризатора должно достигаться раньше, чем окислительно-восстановительного потенциала иона, разряжение которого хотят предотвратить, но позже чем выделяемого иона (почему?).
Примеры. При электролизе разбавленного солянокислого раствора РЬС1» на катоде выделяется РЬ, на аноде С12. Окисление хлора (2С1-->С12+2е~) происходит при более низком значении потенциала ( + 1,36 В), чем реакция РЬ2+->РЬ4+ + 2е- ( + 1,46 В). Ион С1~ действует как анодный деполяризатор.
Из азотнокислого раствора РЬ(КтОз)г, напротив, не происходит выделения РЬ на катоде, а на аноде выделяется РЬО2. Ион NOj- легче восстанавливается на катоде, чем РЬ2+, и действует поэтому как катодный деполяризатор.
39. Электрохимические методы в количественном анализе 263
При электролизе раствора CufNOsh преимущественно образуется осадок меди, а ие раствор CuSO4. Деполяризатор NO3- подавляет катодное выделение Нг, что влечет за собой выделение губчатого осадка меди. Но, с другой стороны, целесообразно добавлять раствор мочевины, чтобы разрушать ломы NOa", образующиеся при восстановлении NO3~ в качестве промежуточного продукта: поскольку их потенциал меньше потенциала меди, они могут затруднить ее выделение.
В присутствии ионов железа невозможно' надежно определить содержание Си2+ методом электрогравиметрии. Это также можно объяснить деполяризующим действием. Окисление железа до Fe4+ на аноде и восстановление до Fe2+ на катоде происходит легче, чем восстановление Си2+ до Си. При проведении электролиза в солянокислом растворе с применением платиновых электродов следует опасаться повреждения платины из-за анодного образования СЬ. Добавляя деполяризатор — гидразин, подавляют выделение С12.
Мешающие реакции, протекающие на аноде, можно также устранить, применяя анод из менее благородного металла, чем платина; при этом ионы переходят в раствор до начала нежелательного процесса окисления. Эти ионы нужно сразу выделить в осадок во избежание совместного осаждения с определяемым ионом и искажения результатов. Например, если применяют серебрянный электрод и одновременно добавляют к раствору электролита СГ~, на аноде происходит единственный процесс Ag—>Ag++e_ и образуется малорастворимый осадок AgCl. В раствор необходимо вводить количество ионов С1~, неэквивалентное количеству металла, выделившегося на катоде, а большее (почему?).
Для проведения процесса электролитического выделения вещества можно использовать следующую простую схему (рис. Д.85). Через регулируемое сопротивление R. и амперметр А от источника постоянного тока подают на электроды постоянное напряжение, контролируемое вольтметром V (рис. Д.85); напряжение можно менять. Во избежание ошибок при разделении напряжение, фиксируемое на клеммах, не должно превышать допустимой величины. В конце выделения напряжение на клеммах падает вследствие резкого увеличения напряжения поляризации.
В каждом конкретном случае важно следить за тем, чтобы выделяющийся продукт прочно сцеплялся с катодом и при высушивании и взвешивании не осыпался с него. Благоприятное влияние оказывает перемешивание, так как при этом уменьшается обеднение раствора вблизи катода. Плотность тока также не должна быть слишком высокой. Добавление коллоидов, например желатина, способствует иногда образованию более мелких кристаллов и лучшему удерживанию выделяемого вещества на электроде.
Если электролиз осуществляют при постоянном перемешивании и нагревании раствора, то в этом случае говорят о «быстром электролизе». При таком способе создается большая
264
Д. Химический анализ
Рис. Д.85. Схема установки для электрогравиметрии при постоянной силе тока.
Я — амперметр; Я — регулируемое сопро-тивление; V — вольтметр; ЕЕ — электроды; Z — электролитическая ячейка.
плотность тока и тем самым уменьшается время электролиза (почему?).
Более надежен метод чистого электрогравиметрического разделения с автоматическим регулированием катодного потенциала. Для этого используют схему с контролируемым катодным потенциалом, который измеряют относительно электрода сравнения (например, каломельного электрода), не участвующего в электролитическом процессе; постоянство катодного потенциала достигается потенциостатическим включением установки. Сила тока в процессе электролиза изменяется от на-чального максимального значения до нуля в конце анализа.
Обзор установок для проведения электролиза при контролируемом потенциале приведен в соответствующей литературе.
Сходный эффект можно иногда получить, используя более простые способы, например так называемый внутренний электролиз. В основу этого метода положен принцип цементации металла из его раствора при добавлении другого металла. Отличие заключается только в том, что при
разделении анодного и катодного пространств с помощью диафрагмы (как в известном элементе Даниеля) в процессе внутреннего электролиза получают прочно удерживающиеся на электродах осадки. Путем подбора подходящего металла можно добиться необходимой разности потенциалов по отношению к катоду. Однако только сравнительно небольшие количества веществ можно определять при этом за не слишком большой промежуток времени. Преимущество внутреннего электролиза заключается в том, что с анода в раствор переходит только металл и на аноде не протекают побочные процессы, такие, как выделение С12 или реакция Fe2+—>-Fe3++e~. Метод внутреннего электролиза успешно применяют для определения небольших количеств благородных металлов в сплавах.
Пример. Методика электрогравиметрического разделения и определения Си2+ и Cd2+.
Напряжение, создаваемое свинцовым аккумулятором (2,05В), является достаточным для количественного выделения меди, так как Uz cuso4= 1,49 В (рассчитайте, насколько концентрация ионов Си2+ может быть при этом снижена), но не для выделения Cd2+.
39. Электрохимические методы в количественном анализе '265
Очищенный и взвешенный катод в виде сетки из платиновой проволоки анод из платиновой проволоки погружают в сернокислый раствор сульфатов меди и кадмия. Анод помещают в центр проволочного цилиндра, верхний край которого должен подниматься над уровнем раствора на 1 см. Об Окончании выделения меди судят по отклонению силы тока до минимального остаточного значения (почему?). Окончание выделения можно также контролировать, добавив небольшое количество дистиллированной воды. Тогда оставшийся еще гладким край платинового цилиндра покрывается раствором. Если на нем через некоторое время не образуется красного налета (Си), то сосуд для электролиза (стакан) можно осторожно удалить, опустив его вниз. Затем электроды промывают, подставляя стакан с дистиллированной водой, чтобы полностью погрузить электроды в воду. Через несколько минут его можно удалить. Только теперь можно отключить напряжение (почему?).
Для удаления воды электроды погружают в ацетон и катод затем высушивают на воздухе или на несколько минут помещают его в сушильный шкаф при 100 °C. Заключительное взвешивание позволяет определить количество меди по увеличению массы электрода. Для выделения кадмия анод и покрытый медью катод (гладкая платина легко легируется некоторыми металлами, например Cd, Hg, Sn, Zn, Bi, и очень трудно очищается после этого) снова погружают в электролизуемый раствор. Источниками напряжения служат два последовательно включенных аккумулятора; силу тока 0,5 А устанавливают с помощью регулируемого сопротивления. (Каково должно быть напряжение на клеммах для количественного выделения Cd? Как можно получить такое напряжение в водном растворе?)
Конец электролиза определяют, отобрав несколько капель раствора для проведения пробы на Cd2+ действием ионов S2-. Если при этом не образуется желтый осадок, сосуд для электролиза (какие продукты находятся в нем. после окончания электролиза?) можно заменить на стакан с налитой в него уже использованной дистиллированной водой (почему не со свежей порцией?). Через 15 мин можно промыть анод ацетоном, высушить и взвесить.
Аналогично можно провести разделение и определение меди и цинка.
При электроанализе, например латуни, одновременно с выделением меди на катоде возможно получить на аноде РЬОг из примеси свинца, содержащегося в латуни. В литературе можно найти много других примеров.
Для экспериментального подтверждения теоретических положений рекомендуем провести следующий опыт: снять вольт-амперную кривую при электролизе раствора CuSO4 с применением медных электродов: а) иа холоду; б) на холоду при перемешивании; в) при 60 °C.
Важнейшим методом разделения металлов является их электролитическое выделение на ртутном катоде. Поскольку перенапряжение водорода на ртути превышает 1 В, из раствора можно выделить многие металлы. Однако алюминий, скандий, титан, ванадий, вольфрам и некоторые другие даже и в этих условиях не могут быть выделены, а ионы щелочных и щелочноземельных металлов восстанавливаются только в щелочном растворе. Напротив, железо можно успешно удалить электролитическим путем из переведенного в раствор алюминиевого сплава. Указанный способ можно также применять для очистки растворов урана. Выделение веществ на ртутном катоде чаще всего проводят при контролируемом потенциале, опти
266
Д. Химический анализ
мальное значение которого определяют из предварительно снятой полярограммы для анализируемого раствора. С другой стороны, возникают трудности при точном взвешивании количества вещества, выделившегося на ртутном катоде. Поэтому для таких определений лучше использовать кулонометрические методы.
39.2. Кулонометрия
39.2.1. Основные положения
Кулонометрический анализ во многом сходен с электрогравиметрическим. Однако имеется существенное отличие: в электрогравиметрии электрический ток применяют для осаждения веществ аналогично реагентам классической гравиметрии, поэтому в данном случае используют избыток тока. Определение как таковое проводят взвешиванием. В кулонометрии, напротив, электрический ток применяют аналогично реагентам тит-риметрии, т. е. в количестве, эквивалентном определенному веществу. В этом случае количество тока (как и объем титранта в титриметрическом анализе) является мерой количества определяемого вещества, вступившего в реакцию.
Кулонометрия основана на законах Фарадея, так что ее можно рассматривать как метод, обратный методу, предложенному М. Фарадеем для измерения количества электричества с помощью химического кулонометра. Между количеством вещества и количеством электричества существует следующая зависимость:
G== 96 490п (4°9)
где G — количество вещества, г; М — молекулярная или соответственно атомная масса определяемого вещества; Q — измеренное количество электричества, кулоны; п—число электронов на частицу вещества, участвующих в электродной реакции. Законы Фарадея могут быть применены для количественных кулонометрических определений, если выполняются следующие условия: а) при восстановлении или окислении анализируемого вещества образуется соединение с точно известной степенью окисления элемента; б) реакция протекает со 100%-ным выходом по току; в) если предыдущее условие выполняется при определении нижней границы концентраций с требуемой точностью.
В растворе возможны побочные реакции, которые могут искажать результаты анализа, а именно окисление и восстановление растворителя; восстановление находящегося в раство-
39. Электрохимические методы в количественном анализе 267
ителе кислорода воздуха; окислительно-восстановительные реакции взаимодействия ионов, образующихся в процессе электролиза, с растворителем; восстановление или окисление примесей, находящихся в растворе. Например, для воды: 2Н+4~ , 2е"—^Нг (восстановление) и 4ОН~—>О24-2Н2О+4е“ (окисление). Электродные потенциалы, при которых может происходить выделение кислорода на аноде и водорода на катоде, рассчитывают следующим образом:
eos = е°о2/он 0,059аон-; е°О2/гОН- = 4-0,401В;
аон-= 10-14/аНзО+; е02 = 0,401 + 0,825 — 0,059рН (410)
е02= 1,23—0,059рН; еН2 = —0,059рН (411)
При этом предполагают, что реакции могут идти в небольшой степени с несколько меньшими потенциалами, еще до того как давление газов (О2, Н2) станет равным атмосферному. Уравнения (410) и (411) показывают, что оба процесса в равной степени зависят от pH, поэтому разность потенциалов независимо от значения pH остается постоянной (1,23 В). На практике она увеличивается за счет перенапряжения, например, между
Рис. Д.866. Принципиальная схема установки для гальваиостатической кулонометрии (кулонометрического титрования).
и Ri — регулируемые сопротивления;
А — амперметр; Ег — рабочий электрод;
Е2 — противоэлектрод: Инд. — система индикации.
Рис. Д.86а. Принципиальная схема установки для потенциостатической кулонометрии.
Р — сопротивление потенциометра; V — Устройство для измерения потенциала;
— рабочий электрод; — протнвоэлект-род; Ei — электрод сравнения.
платиновыми электродами на 1,73 В из-за того, что на платине т]о2 = 0,5 В.
Вследствие большого перенапряжения водорода на ртутном катоде (~1,2 В) можно проводить катодное восстановление в значительно более широкой области напряжений, чем на платиновом катоде, со 100%-ным выходом по току (без восста
268
Д. Химический анализ
новления Н+). Напротив, ртутный анод легко окисляется, по-этому нужно использовать платину.
Восстановление кислорода воздуха (О2+2Н2О4-4е~— —ИОН-) можно снизить, работая в токе азота. Растворенный кислород перед началом электролиза необходимо вытеснить из раствора, пропуская азот. Восстановление или окисление примесей, находящихся в растворе, можно устранить, выбирая, контролируя и регулируя необходимые значения электродных потенциалов.
Так же как и в электрогравиметрии, в кулонометрии различают два способа выполнения анализа: кулонометрию при постоянном контролируемом потенциале (потенциостатическая кулонометрия или кулонометрический анализ) и кулонометрию при постоянной силе тока и неконтролируемом потенциале (гальваностатическая кулонометрия).
На рис. Д.86а и Д.866 приведены принципиальные электрические схемы для обоих методов.
39.2.2. Потенциостатическая кулонометрия
Напряжение ячейки Uz оцределяют как
С4=ф1-*2| + ^ (412)
где &1 — потенциал рабочего электрода; е2 — потенциал проти-воэлектрода; i — сила тока; Rl — сопротивление раствора электролита, находящегося между электродами; iRt.— падение напряжения в электролите, находящемся между электродами.
Простейшим способом достижения постоянства потенциала рабочего электрода является применение в качестве противо-электрода неполяризуемого электрода, например насыщенного каломельного. При этом е2 остается постоянным при небольшой силе тока, когда не наблюдается существенных концентрационных изменений. При небольших значениях RL и i можно также пренебречь продуктом реакции и е; остается постоянным 'при постоянной величине Uz.
Этот метод неприменим для определения небольших концентраций веществ, так как из-за необходимости работы при малой силе тока потребовалось бы слишком много времени для их определения. Для разделения анодного и катодного пространства обычно ставят диафрагму, поскольку в растворе могут присутствовать ионы, окисляющиеся на аноде или восстанавливающиеся на катоде без выделения (например, Fe3+/Fe2+). Такие циклические реакции окисления-восстановления могли бы явиться причиной возникновения дополнительного тока, вследствие чего выход по току для данного аналитического определения уже не был бы равен 100%. Благодаря применению диафрагмы RL возрастает примерно с 200 Ом для
39. Электрохимические методы в количественном анализе 269
обычных ячеек до 2000 Ом. Поэтому для поддержания постоянного потенциала необходимы сложные установки.
К электродам Ei и Е2 прикладывают напряжение и изменяют его таким образом, чтобы потенциал рабочего электрода Ei по отношению к постоянному потенциалу электрода сравнения Ез (чаще всего каломельного) всегда имел определенное значение. Регулирование потенциала лучше проводить автоматически. Для этой цели применяют электромеханические или электронные приборы, называемые потенциостатами.
Индикацию конечной точки титрования в потенциостатиче-ской кулонометрии проводят по значению силы тока, которая в перемешиваемом электролите уменьшается по экспоненциальному закону:
it = i0. \Pr-At (413)
где io — начальная сила тока; t—время; .4 = 0,43 DaSjVd (S — поверхность рабочего электрода; V—объем раствора; D — коэффициент диффузии определяемого иона; d—толщина Диффузионного слоя на поверхности электродов; ia — постоянная, значение которой может находиться в интервале 0—1).
Потенциал электрода выбирают таким образом, чтобы величина силы тока, насколько это возможно, совпадала бы с величиной предельного диффузионного тока.
Теоретически для протекания реакции до конца (с = 0; i = 0) потребовалось бы бесконечно большое время. Но практически всегда существует незначительный остаточный ток, вызванный реакциями окисления-восстановления посторонних ионов на рабочем электроде. Поэтому электролиз считают законченным, если сила тока составляет 1/100—1/1000 первоначального значения. Возникающая при этом1 систематическая ошибка оцреде-ления составляет от —1% до —0,1%. Время, за которое достигается это значение, тем меньше, чем больше значение А в уравнении (413). Значение А увеличивают, применяя большую поверхность S рабочего электрода, малый объем растворителя V и сильное перемешивание (уменьшается d).
Количество электричества, эквивалентное количеству определяемого вещества, находят из уравнения
t=o
Существуют различные возможности оцределения количества электричества Q.
1. Построение зависимости ток — время с последующим измерением планиметром площади под кривой, которая является мерой Q. Для точного определения необходимо вычесть долю
Д. Химический анализ
остаточного тока. Его определяют путем холостого опыта, проводя кулонометрию раствора, содержащего все компоненты анализируемого раствора, за исключением определяемого вещества, в тех же условиях, при которых проводилось основное определение.
2. Построение диаграммы lg i—t по небольшому числу экспериментальных данных. При этом время проведения анализа можно существенно уменьшить. Интегрирование уравнения (413) в пределах (=0 и i=oo дает
Q = io/2,3034 (415)
Значение остаточного тока при этом остается пренебрежимо малым. Значение io получают экстраполяцией прямой lg i—t. Поскольку в методе не учитывается влияние остаточного тока, его нельзя применять для определения Q в анализе следовых количеств веществ.
3. Включение при измерениях химического кулонометра (два электрода, опущенные в раствор подходящего электролита) в цепь последовательно с кулонометрической ячейкой. Тогда через кулонометр и ячейку проходят равные количества тока Q. Происходящие при этом взаимодействия веществ (осаждение, окисление-восстановление, выделение газа, изменение окраски раствора) используют для определения Q.
Гравиметрический кулонометр состоит из платинового катода и анода, изготовленного из металла, ноны которого находятся в растворе (например, Ag+ или Си2+). По увеличению массы катода определяют количество тока Q. Применяя гравиметрический кулонометр, можно получить хорошие результаты в тех случаях, когда непригоден метод электрогравиметрии.
В титрацнонном кулонометре происходит анодное окисление или катодное восстановление ионов (например, Ag+, ОН~, Fe2+), количество которых (эквивалентное Q) можно определить титрованием.
В газовом кулонометре определяют количество электричества Q по объему газа, выделяющегося в ходе электролиза, например гремучего газа, образующегося при электролизе воды, или азота, образующегося у анода при электролизе раствора гидразина.
В колориметрическом кулонометре анодным окислением иодид-крахмаль-ного раствора получают иод, образующий с крахмалом окрашенное в интенсивно синий цвет соединение включения. Интенсивность синей окраски, определяемая колориметрическим методом, является мерой количества выделившегося иода и, следовательно, мерой количества электричества Q. Этот кулонометр рекомендуют применять при измерении очень небольших количеств электричества.
4. Применение электронных или электромеханических интеграторов, которые входят в комплект любого кулонометра. Химические кулонометры применяют для градуировки и контроля. Принцип действия электромеханического кулонометра заключается в том, что скорость вращения мотора, вырабатывающего постоянный ток в постоянном магнитном поле, в широ
39. Электрохимические методы в количественном анализе 271
кой области пропорциональна приложенному напряжению. Параллельное подключение сопротивления R цепи, на котором возникает падение напряжения U~iR, обусловливает пропорциональную зависимость между скоростью вращения и силой тока i- Счетчик оборотов градуируют в кулонах или грамм-эквивалентах. Точность определения составляет +0,1%.
Ниже приведены типичные примеры определений методом лотенциостатической кулонометрии.
1. Выделение металлов на катоде. На платиновом катоде выделяются Ag, Си, на ртутном катоде—Bi, Zn, Cd, Со, Ni, РЬ (при использовании перенапряжения водорода). В этих случаях анодом служит обычно серебряная проволока. При избытке С1~ в растворе на аноде образуется только AgCl.
2. Выделение на аноде. Наиболее важным применением этого варианта метода является определение С1~, Вг_, I-, SCN- окислением на серебряном аноде. При совместном присутствии в растворе С1_ и Вг~ даже при самом тщательном выборе потенциала рабочего электрода всегда происходит их совместное выделение, поскольку произведения растворимости AgCl и AgBr различаются незначительно. Для раздельного определения С1~ и Вг~ нужно применять сочетание кулонометрии и электрогравиметрии, так называемый метод кулоногравиметрии. При этом одновременно выделяют оба компонента и определяют необходимое для этого количество электричества Q (кулонометрическим методом) н увеличение массы Д(3 анода (гравиметрическим методом), Содержание обоих компонентов можно определить из следующих уравнений:
ДО = Оа- + ОВг- и Q/96500 = Gcl-/35,46+GBr-/79,92
(Объясните эти уравнения!)
Точное определение содержания компонентов по такой комбинированной методике возможно только при большом различии их атомных масс (почему?) .
3. Изменение степени окисления с образованием в растворе окисленной или восстановленной формы соединения. В этом случае необходимо применять диафрагму для разделения анодного и катодного пространства. Компоненты систем Asni/Asv, CrVI/CrIII/Cr11, Fen/Fenl и т. д. определяют анодным окислением или катодным восстановлением.
4. Кулонометрия электронеактивных веществ. Различают два случая.
а) Кулонометрию можно применить, например, для определения комплексонов, таких, как ЭДТА (анион комплексона сокращенно обозначим X4-), окислением ртути ртутного анода при соответствующем потенциале:
Hg+X4- — 1е~ ----->- HgX2-
При этом Hg2+ переходит в раствор в виде комплекса. Предельная сила тока, обусловленная скоростью диффузии Х4~ к аноду, в соответствии с изменением •концентрации Х4~ к концу реакции становится равной нулю.
б) При определении металлов, для которых нельзя применить метод прямой кулонометрии, например щелочноземельных (Ме2+), к раствору можно добавить известное избыточное количество комплексона и затем определить непрореагировавшее его количество, как описано выше. При повышении -анодного потенциала до необходимого второго значения происходит реакция
Hg + МеХ«~ — 2е~ ----► HgX2~ + Ме2+
и сила тока в конце реакции опять стаиовится равной нулю. Таким образом последовательно (сначала косвенно, а затем прямо) определяют количество Ме2+ в растворе пробы.
т
Д. Химический анализ
Потенциостатической кулонометрии, так же как электрогравиметрии при контролируемом потенциале, присуща высокая селективность. В этом заключается ее преимущество перед гальваностатической кулонометрией. Например, можно определить содержание каждого металла в смеси, если их окислительно-восстановительные потенциалы отличаются только на 0,2 В. Преимущество кулонометрии перед электрогравиметрией состоит в том, что определение в этом методе основано не на образовании осадков веществ в виде подходящей гравиметрической формы, осаждающейся на поверхности электрода в виде прочной пленки (или которую легко можно выделить), а связано с выполнением только вышеуказанных условий, прв которых не требуется выделение соединения в твердом виде. Кроме того, даже в тех случаях, когда для определения веществ можно использовать как кулонометрию, так и электрогравиметрию, при использовании кулонометрии получают значительный выигрыш во времени, связанный с устранением one*: раций высушивания и взвешивания.
39.2.3. Гальваностатическая кулонометрия
Кулонометрию при постоянной силе тока применяют, если необходимо провести высокоселективные определения. По сравнению1 с методом потенциостатической кулонометрии она обладает рядом достоинств: меньшей продолжительностью электролиза и более удобным способом измерения количества электричества, рассчитываемого по формуле Q — it. Небольшую силу тока, которая дает возможность полностью осуществить электролиз растворов с большими концентрациями ионов металлов за удовлетворительное время, можно легко поддерживать постоянной, включив последовательно с кулонометрической ячейкой высокое внешнее сопротивление и применяя высокое напряжение источника питания (батареи). Силу тока определяют по уравнению
I = ^в/(^в+ «д)
где UB~ напряжение батареи (например 100 В); RA — внешнее сопротивление (например 100 кОм); Rz — сопротивление кулонометрической ячейки; RB— внутреннее сопротивление батареи. Значения Rz и RB очень малы по сравнению с RA, их изменением в процессе электролиза можно пренебречь. Поэтому в данном примере силу тока iml мА можно поддерживать с ошибкой ±0,1%. В промежутках между работой вместо кулонометрической ячейки подключают сопротивление, соответствующее сопротивлению Rz, для поддержания постоянства силы тока и тем1 самым термического равновесия в цепи.
39. Электрохимические методы в количественном анализе 273
Наряду с такой простой установкой при более высоких требованиях, особенно при большей силе тока, применяют амперо-статы. В этом случае уменьшение напряжения, связанное с величиной IR на прецизионном сопротивлении, включенном последовательно с электролитической ячейкой, поддерживают постоянным с помощью электромеханического или электронного’ регулятора напряжения, тем самым поддерживается постоянной и сила тока.
Применяя этот метод, можно с высокой точностью определить силу тока 1 мкА в секунду, что соответствует концентрациям веществ порядка 10-11 моль или Ю~9 г. Это означает, что кулонометрия относится к эффективным методам определения микроколичеств веществ.
39.2.3.1. Прямая кулонометрия при постоянной силе тока
Если вещество может выделяться в твердом виде на электроде, например в виде металла, оксида или нерастворимой соли, то существует возможность кулонометрического определения количества тока, необходимого для полного выделения определяемого вещества из раствора. Конечную точку устанавливают при этом по резкому возрастанию потенциала рабочего электрода, которое связано с тем, что из-за необходимости поддержания постоянного значения силы тока по окончании основной реакции должен протекать другой окислительно-восстановительный процесс (обычно разложение воды), сопровождаемый соответствующим увеличением потенциала. Этот метод, можно успешно применять для определения тонких слоев покрытий на проводниках.
Хорошие результаты получают при комбинировании методов гальваностатической и потенциостатической кулонометрии. 'При этом определяемые компоненты селективно выделяют по-тенциостатическим методом и определяют с большой точностью амперостатическим методом при растворении. Таким образом используют специфические преимущества обоих методов.
39.2.3.2. Кулонометрическое титрование
В процессе гальваностатической кулонометрии диффузионный предельный ток уменьшается с уменьшением концентрации растворенных веществ. Поскольку для работы установки необходима постоянная сила тока, должны протекать и другие электродные реакции (других ионов или воды), что обусловливает увеличение электродного потенциала. Эти электродные реакции нарушили бы 1ОО°/о-ный выход по току и сделали бы невозможным кулонометрическое определение веществ.
18—1880
-274
Д. Химический анализ
В принципе можно выбрать такую силу тока в электролитической цепи, чтобы она составляла менее 1 % величины диффузионного предельного тока. В этом случае мешающие реакции начинают протекать только после того, как прореагировало 99% определяемого 'вещества. Погрешность составляет, та-жим образом, менее —1%. Но проведение анализа при небольшой силе тока требует больших затрат времени. Поэтому обычно поступают по-другому: в анализируемый раствор вводят довольно большую концентрацию вспомогательного ре-.агента, окислительно-восстановительный потенциал которого немного больше окислительно-восстановительного потенциала определяемого иона. К началу электролиза определяемый ион опять восстанавливается или окисляется. В соответствии с уменьшением концентрации определяемого иона у поверхности .электродов электродный потенциал снова возрастает, но только -До тех пор, пока его значение не станет равным значению потенциала иона вспомогательного реагента. После этого окис-.ляется или восстанавливается реагент. Поскольку его концентрация! намного больше концентрации определяемого иона, обеспечивается дополнительная подача вещества путем диффузии к поверхности электродов. Электродные потенциалы остаются постоянными (не происходит разложения воды; 100%-ный выход по току), остается постоянным значение Rz, а следовательно, и I. Диффундирующий от электродов вспомогательный реагент, являющийся окислителем или восстановителем, реагирует в растворителе с определяемым ионом, и, таким образом, действует только как посредник.
В этом варианте метода точку эквивалентности можно определить с помощью обычных индикаторов. Вместо индикаторов, которые могут разлагаться при электролизе, лучше использовать электрические методы индикации — потенциометрию или амперометрию. Эти методы дают возможность использовать хронометр, который автоматически включается (с помощью электроуправляемого реле) одновременно с началом шропускания тока при электролизе. Силу тока и время необходимо определять очень точно. Точность анализа существенно зависит от правильности установления точки эквивалентности.
Мы видели, что гальваностатическая кулонометрия — это по -существу кулонометрическое титрование, где используются не титрованные растворы, а количество тока. При этом нет необходимости определять титр раствора и использовать первичные стандарты; устраняется также и ошибка разбавления.
Чтобы продукт катодного восстановления не окислялся на аноде, анодное и катодное пространство разделяют диафрагмой или соединяют солевым мостиком с двумя диафрагмами.
Если условия работы не позволяют получить 100%-ный выход по току или надежно определить конечную точку титро-
39. Э лекгрохимические методы в количественном, анализе
275
ания, то титрант можно генерировать в специальной ячейке, находящейся отдельно. При таком внешнем электролизе раствор электролита проходит около генераторного электрода и затем поступает в ячейку для титрования. Но в этом случае, как и в объемном анализе, возникает ошибка разбавления, которую можно устранить. Ус
тановка для внешней генерации титранта показана на рис. д.87. В качестве генераторных применяют два платиновых электрода. Электролитный раствор протекает около них. У анода образуются растворы окислителей или кислоты, у катода — растворы восстановителей или основания. В зависимости от типа титрования применяют один из потоков жидкостей и отбрасывают другой. При 100%-ном выходе по току действительное содержание компонента можно
рассчитать, зная силу тока на генераторных электродах и время достижения точки эквивалентности, которое определяют, например, амперометрическим методом. Между генераторными электродами на-
Рис. Д.87. Кулонометрическая установка с внешней генерацией титранта.
1 — анализируемый раствор; 2 — индикаторные электроды; 3 — мешалка; 4, 6 —генераторные электроды; 5 — стекловата;
7 — сосуд для вспомогательного реагента.
ходится стекловата для предотвращения смешивания катодной-и анодной жидкости в результате конвекции. Для титрования применяют любые реагенты, которые можно электролитически, генерировать из исходного раствора.
Если при этом одновременно образуется газ, например Н2 на катоде при генерировании ОН" или Ог на аноде при генерировании Н+, то это не мешает определению. Поток жидкости уносит с собой пузырьки газа. Поскольку анодная и катодная жидкости разделены, не нужно ставить диафрагму.
Различными способами кулонометрии можно осуществить практически все классические методы титрования. При этом-устраняется необходимость приготовления стандартных растворов. Можно также использовать неустойчивые растворы титрантов, как, например, Мп3+, СиВг2~ и Sn2+. Кроме того,, кулонометрическое титрование легко автоматизировать. Промышленность выпускает большое количество таких приборов.
Ниже приведены примеры кулонометрического титрования сг применением вспомогательных реагентов.
18*
эль
Д. Химический анализ
Вспомогательный реагент а) Восстановление
Fes+/Fe2+
Ti4+/Ti3+
Cu2+/Cu+
Sn4+/Sn2+
б) Окисление
Се3+/Се4+
Мпг+/МпО4-
в) Осаждение
Ag/Ag+
Hg/Hg?+
Zn/Zn2+
Основная реакция
Се4+— >-Се3+; СгО?-—»-Сг3+
МпО4_- —»Мп24-, 1О4-—»Ю-
Се44-— VO43-- >-Ces+; Fe’+—>-Fe2+ -»VO2+
ВгОз~- VO?-- -»Br-; CrO?--»Cr3+ -»VO24-
Cu2+—>-Cu+; I—>1-
Br—>-Br~ и т. n.
As3+—>-As5+;
Ti3+—>Ti4+
Fe2+—>-Fe3+
As3+—>-As5+;
Fe2+—>-Fe3+;
‘ t. n.
—<-2CO2
H2O2-->"©2 4“ H2O
Титруемое вещество
C1-, Br~, I-
C1-, Br~, I-
s2-
-39.2.4. Техника работы
39.2.4.1. Кулонометрические определения на ртутном катоде Для проведения определения можно применить установку, схема которой изображена на рис. Д.88. Ячейка для проведения электролиза соединена с сосудом, содержащим ртуть, смешанную с водой. Проводником служит платиновая проволока, впаянная в стеклянную трубку. Ячейка для электролиза закрыта крышкой для предотвращения доступа воздуха. С помощью трубки в ячейку подают азот для вытеснения кислорода из анализируемого раствора. Для контроля катодного потенциала применяют в качестве электрода сравнения каломельный электрод. Мешалка обеспечивает перемешивание анализируемого раствора и одновременно ртути. Анодом служит спираль из платиновой проволоки. Если вместо платины применить серебро, то при добавлении С1~ в качестве деполяризатора можно устранить выделение кислорода, мешающее проведению •реакции.
С помощью потенциостата поддерживают постоянное значение оптимальной величины катодного потенциала (на 100— 200 мВ больше величины полярографического потенциала полуволны). В измерительную цепь включают подходящий кулонометр. Для проведения измерений ячейку для электролиза за
39. Электрохимические методы в количественном анализе
277
полняют вспомогательным электролитом (например, 0,5 М раствором КС1), потоком азота вытесняют кислород (в течение примерно 10 мин) и затем, открыв кран, впускают ртуть.
Подвергают электролизу вспомогательный электролит при потенциале, величина которого примерно на 100 мВ меньше величины потенциала, используемого для анализа. Если сила тока через некоторое время становится равной нулю, это означает, что мешающее влияние других компонентов практически устранено.
В ячейку вносят анализируемый раствор и после удаления кислорода снова про-
Рис. Д.88. Установка для кулонометрического анализа на ртутном катоде. 1 — мешалка; 2 — каломельный электрод сравнения; 3 — шланг; Pt — платиновая проволока.
водят электролиз при оптимальном значении потенциала до тех пор, пока сила тока не станет равной нулю. Содержание определяемого вещества можно рассчитать, исходя из реакции, протекающей в кулонометре.
39.2.4.2. Кулонометрический анализ на платиновом аноде
Поскольку из-за образования безводных окислителей, которые снова восстанавливаются на катоде, в цепи может дополнительно расходоваться ток, установку (рис. Д.88) модифицируют— прежде всего ставят диафрагму для разделения анодного и катодного пространства.
39.2.4.3. Кулонометрическое титрование
Для проведения кулонометрического титрования можно использовать установку с диафрагмой. Например, при титровании As3+ электрогенерированным Вг2 целесообразно в качестве электролита применить раствор 0,1 М по H2SO4 и 0,2 М по <NaBr. Конечную точку титрования лучше всего определять методом амперометрии.
Подробную информацию о методах кулонометрии можно найти в литературе [69, 70]*.
* См. также книгу. Зозуля А. 17. Кулонометрический анализ. — М.: Химия, 1968. — Прим перев. _
278
Д. Химический анализ
Вопросы и задания
1. Как в кулонометрии можно генерировать титрант для проведения аци-ди- и алкалиметрического титрования?
2. Выполнимо ли требование 100%-иого выхода по току для процессов,, происходящих одновременно на обеих электродах?
3. Означает ли нагревание раствора электролита в ячейке при электролизе несоблюдение требования 100%-ного выхода по току?
39.3. Постояннотоковая полярография
39.3.1. Общая схема измерительной ячейки
для электрохимических методов анализа
Измерительная ячейка, применяемая в электрохимических методах анализа (кроме осциллометрии), включает два электрода, погруженные в анализируемый раствор. Для описания работы такой ячейки можно предложить общую, так называемую ^эквивалентную схему, в которой абстрактно представлены реальные процессы, происходящие в ячейке. Правильность ее
Рис. Д.89. Принципиальная схема измерительной ячейки для электрохимических методов анализа.
Ei, Ei — электроды; Rit Ri — поляризационные сопротивления электродов; Ct, С2 — емкости электродов; RL — омическое сопротивление раствора электролита, находящегося между электродами; CL — емкость конденсатора, образуемого электродами и раствором, находящимся между ними.
Рис. Д.90. Эквивалентная схема измерительной ячейки для электрохимических методов анализа.
можно установить сравнением полученных выводов и рассчитанных значений с реальной ячейкой.
На электродах Е\ и Е2 возникают поляризационные сопротивления Ri и R2, отличающиеся от омического сопротивления, поскольку зависят от потенциала и включают в себя сопротивления, соответствующие всем видам перенапряжений. Кроме того, электроды можно представить (исходя из теории электрохимического двойного слоя) как конденсаторы с емкостью и С2. Поверхности F этих конденсаторов равны поверхностям электродов, расстояние между «пластинами» конденсаторов d составляет ~10~8 см (порядка диаметров молекул). Параллельно конденсаторам С! и С2 включены сопротивления Ry и R&. Эти системы разделены раствором электролита с омиче
39. Электрохимические методы в количественном анализе 279
ским сопротивлением Rl. К этому надо добавить емкость CL конденсатора, образуемого электродами и раствором, находящимся между ними, имеющего также поверхность F и расстояние между пластинами, соответствующее расстоянию между электродами (раствор электролита между ними играет роль диэлектрика).
Рис. Д.89 и Д.90 иллюстрируют сказанное. Схема, приведенная на рис. Д.90, дает возможность определить условия пригодности измерительной ячейки для решения поставленной задачи. При необходимости измерения, например, электропроводности раствора можно устранить влияние всех параметров, кроме Rl (см. кондуктометрию). Если для решения задачи необходимо использовать поляризационные сопротивления Rt или R2 (например, в полярографии), то значения их по отношению к другим должны быть определены1.
39.3.2. Электрохимические процессы в полярографической ячейке постоянного тока
В полярографии для определения вида и количества компонентов раствора используют описанные выше вольт-амперные кривые. Для этой цели рабочие условия выбирают таким образом, чтобы вольт-амперные кривые являлись практически единственной характеристикой поляризационного сопротивления на рабочем электроде.
'При работе с постоянным током, когда конденсаторы действуют как запорные устройства, эквивалентная схема ячейки упрощается (рис. Д.91). В этом случае имело бы место соотношение U=i(Ri-\-R2-[-RL)- Чтобы ход кривой определялся только величиной Ri, вычисляемой из равенства U=iR}, сопротивления Rl и R2 должны быть очень небольшими по сравнению с R\. Для Rl это достигается тем, что в раствор вносят большое количество полярографически инертного фонового электролита (с концентрацией 0,1 — 1 н.), а для R2— применением неполяризующегося противоэлектрода (электрода 2-го рода, например каломельного или электрода из донной ртути с большой поверхностью). Величина R\ должна быть большой. Поэтому рабочий электрод делают как можно более поляризуемым.
Поляризацией называют отклонение потенциала электрода при прохождении тока от его значения в отсутствие тока:
т1 = ei—е/-о
Причиной поляризации является уменьшение числа реакционноспособных частиц на поверхности электродов, которое тем заметнее, чем меньше при данной силе тока поверхность элект-
280
Д. Химический анализ
Рис. Д.91. Упрощенная эквивалентная схема измерительной ячейки для работы при постоянном токе.
Рис. Д.92. Кривая сила тока — потенциал для поляризуемого электрода в присутствии деполяризатора.
родов. Поляризационное напряжение связано с поляризационным сопротивлением: t] = i/?Kp). Чтобы его значение было большим по сравнению с Rz и Rl, в полярографии выбирают относительно малые рабочие электроды. Можно дать следующее определение: электрод поляризован до тех пор, пока подача внешнего потенциала не вызывает или вызывает только небольшое изменение тока; электрод деполяризован, пока при прохождении тока его потенциал не изменяется или изменяется незначительно.
Зависимость сила тока —
потенциал для поляризуемого электрода показана на рис. Д.92. Вещество, которое в результате электродной реакции вызывает подъем кривой на среднем участке, т. е. деполяризует электрод в этой области, называют деполяризатором.
39.3.3. Ртутный капельный электрод
В качестве поляризуемого рабочего электрода в полярографии используют ртутный капельный электрод. Он имеет небольшую поверхность и, следовательно, высокую плотность тока при малой силе тока (если пренебречь изменением концентрации пробы в результате электролиза), поэтому он легко поляризуется. При добавлении ртути по каплям (удовлетворительное время капания 3—5 с) в каждый момент образуется идеальная электродная поверхность. Другое преимущество электрода—большое перенапряжение водорода на ртути, что дает возможность в нейтральном растворе проводить определение даже щелочных металлов. Этот электрод можно применять в области относительно высоких отрицательных потенциалов. Напротив, его положительная граница, измеренная относительно каломельного электрода, находится при -f-0,45 В. (из-за анодного растворения ртути).
Устройство ртутного капельного электрода таково. Толстостенный стеклянный капилляр (рис. Д.93) длиной ~25 см с толщиной стенок около 5 мм и внутренним диаметром ~0,1 мм соединен толстостенным шлангом длиной около 1 м с резервуаром грушеобразной формы, в котором налита ртуть (рис. Д.94). Положение этого резервуара можно регулировать, передвигая
39. Электрохимические методы в количественном анализе
281
н9
У
Капли Ну
Рис. Д.93. Капилляр ртутного капельного электрода.
Рис. Д.94. Схема ртутного капельного электрода с резервуаром для ртути, положение которого по высоте можно менять (Л — высота столба ртути).
еажим штатива, а скорость капания ртути можно регулировать, меняя разность уровней ртути в резервуаре и в капилляре. Шланг и капилляр должны быть чистыми, в них не должно быть пузырьков воздуха (во избежание закупорки капилляра), применяемая ртуть должна быть очищена вакуумной перегонкой.
39.3.4. Техника работы в постояннотоковой полярографии и построение вольт-амперных кривых
Схема простейшей установки для полярографии приведена на рис. Д.95. Постоянное напряжение 2—4 В (например, от аккумулятора) прилагают к измерительной проволоке потенциометра сопротивлением 10—20 Ом; напряжение, поступающее от потенциометра на полярографическую ячейку, варьируют посредством скользящего контакта. Ток электролитической ячейки измеряют чувствительным гальванометром.
Наиболее изящный способ измерения (обычный для всех промышленных полярографов) основан на использовании следующего принципа: при синхронном вращении барабана Коль-рауша с проволокой потенциометра регулируют напряжение в зависимости от времени, при этом можно выбрать различную скорость вращения (рис. Д.96). Применяя одновремен*
Рис. Д.97. Принципиаль
Рис. Д.95. Принципиальная схема полярографической установки.
В — батарея; Р — проволока потенциометра; G — гальванометр; S — скользящий контакт; Z — ячейка.
Рис. Д.96. Принципиальная схема полярографической установки с барабаном Кольрауша (К).
ная схема ческой
полярографи
установки
анодио-катодной поляри
зацией
ртутного
капаю-
щего электрода.
Рис. Д.98. Схематическое изображение полярографических ячеек.
а — пустая б — ячейки
измерительная ячейка с донной ртутью, над пельным электродом и
с вплавленной контактной платиновой
проволокой
которой находится раствор электролита, ртутным, ка с трубками для пропускания потока азота.
Д.100. Полярограммы, получае
Рис. Д.99. Типы полярограмм.
1 — полярограмма раствора Na2SO4, насыщенного воздухом; 2 — то же прн частичном удалении кислорода; 3 — то же при полном удалении кислорода.
Рис. мне
при увеличении содержания нов таллия в растворе.
ио
39. Электрохимические методы в количественном анализе 283
ло зеркальный гальванометр с фотографической регистрацией .Или электронный ленточный самописец, можно автоматически записывать кривые.
Изменяя начальное напряжение на потенциометре, выбирают различные области напряжений и простым переключением (см. рис. Д.97) меняют потенциал ртутного капельного электрода от положительных значений через нуль до отрицательных значений или наоборот (вращение барабана потенциометра в одну или в другую сторону). Для получения полярограммы дно измерительной ячейки покрывают ртутью (рис. Д.98, а и б) и добавляют в качестве фонового электролита 10 см3 0,1 н. раствора Na2SO4, к которому для подавления нежелательных максимумов добавляют несколько капель 0,5%-ного раствора желатина. Полярограмма для области напряжений от 0 до —2,4 В приведена на рис. Д.99, кривая 1.
Появление двух волн вызвано восстановлением деполяризатора— кислорода; возрастание волны в конце записи обусловлено ионами Na+ фонового электролита (для регистрации суммарной волны не хватило бы диапазона измерения прибора). Кислород в раствор поступает из воздуха. При комнатной температуре в 1 дм3 воды растворяется 8 мг кислорода, что соответствует 0,001 н. раствору О2 (присоединение четырех ^электронов при восстановлении). Волны на полярограмме соответствуют процессам восстановления:
I О2 + 2Н2О + 2е- ->- Н2О2 + 2ОН'
II Н2О2 + 2е“ -► 2ОН-
Таким образом можно определить содержание кислорода в растворе, а также изменение его содержания вследствие биологических процессов, происходящих в воде. Эти реакции мешают проведению большинства аналитических определений; продукты реакции Н2Ог и ОН-, находящиеся у электродов, могут также оказывать влияние на протекание других электродных процессов (например, вызывают осаждение малорастворимых гидроксидов вблизи электродов даже в достаточно кислом растворе). Поэтому кислород воздуха предварительно надо удалить. При работе со щелочными растворами кислород можно 'быстро удалить добавлением нескольких капель свежеприготовленного насыщенного раствора ЫагЭОз. Избыточное количество КагЭОз одновременно служит защитой от кислорода воздуха, так что вместо ячейки (рис. Д.98), можно применять небольшой открытый стакан.
Для нейтральных и кислых растворов такой способ непригоден: образующийся SO2 мог бы оказывать мешающее влияние. Поэтому из таких растворов кислород удаляют, пропуская ток чистого азота или водорода (в течение примерно 10 мин). Для кислых растворов можно применять также СОг, посколь-
284 Д. Химический анализ
ку состав фонового электролита при этом не изменяется и не Я образуются осадки карбонатов. j
Полярограмма 2 на рис. Д.99 приведена для случая, когда 1 кислород частично удален, а вольт-амперная кривая 3 — для 1 фонового электролита Na2SO4, полностью освобожденного от ’ кислорода. При добавлении по каплям нескольких капель насыщенного раствора сульфата таллия наблюдается возрастание высоты волны (рис. Д.100), вызванное присутствием деполяризатора.
39.3.5. Напряжение деполяризации и потенциалы полуволн
Напряжение, при котором сила тока достигает половины значения диффузионного предельного тока, называют напряжени-ем деполяризации. Положение его на вольт-амперной кривой | определяется равенством^ j
^=8а-8к+«Д (416>|
Поскольку произведение IRl пренебрежимо мало из-за высо- ] кого содержания фонового электролита, получаем
1/2=еа—ек (417)
Электродный потенциал ек при катодном восстановлении на. ртутном капельном электроде определяют как
(418)
где Бо — стандартный потенциал соответствующей окислительно-восстановительной пары; i — сила тока; id — предельный диффузионный ток для данных концентраций; D и D'—коэффициенты диффузии окисленной и восстановленной формы соответственно. При выделении металла с образованием амальгамы вместо стандартного потенциала ео в уравнение подставляют несколько измененное его значение.
Поскольку коэффициенты диффузии окисленной и восстановленной форм деполяризатора часто практически равны и, кроме того, в уравнении находятся под знаком радикала, можно принять, что ^DID'=\, и тогда
=. = «.-#147^7 (419>
На высоте полуволны id = 2i, откуда потенциал полуволны равен (е1/2)к = ео. Таким образом, потенциал полуволны не зависит от концентрации и является качественной характеристикой соответствующих ионов. Практически он равен стандартному
39- Электрохимические методы в количественном анализе_285.
кислительно-восстановительному потенциалу, если продукт постановления не образует амальгамы с каплями ртути; в пробивном случае он соответствует стандартному потенциалу Амальгамного электрода.
Полярограммы, например для ионов 11+, полученные с раз-пииными фоновыми электролитами, отличаются величинами напряжений деполяризации Uz- 0,31 В (1 н. КОН); 0,49 В (1 н КС1); 0,69 В (1 н. KaSO4), как видно из рис. Д.101. Это связано с тем, что анодный потенциал еа донной ртути приобре-
ОН"
С1" .
S0?
1
0,5
О
Рис. Д.102. Кривые ток — потенциал, соответствующие вольт-амперным кривым рис. Д.101.
Рис. Д.101. Полярограммы для ионов таллия, полученные с различны* ми фоновыми электролитами.
тает различные значения в зависимости от типа фонового Электролита и его концентрации (напомним, что это неполя-ризуемый электрод). Ионы, поступающие в раствор в результате анодного окисления донной ртути, образуют осадок с анионом фонового электролита; при этом возникают электроды 2-го рода (аналогичные каломельному электроду), потенциал которых определяется произведением растворимости.
Дополнительно к ртутному капельному электроду и электроду из донной ртути можно поместить в раствор стандартный каломельный электрод. Измеренные относительно этого электрода значения еа электрода из донной ртути равны: —0,18 В '(1 н. КОН); ±0 В (1 н. КС1); +0,20 В (1 н. K2SO4). На основании уравнения (417) можно записать
sK = sa — uz (420>
Тогда во всех трех случаях для Т1+ (ei/2)K =—0,49 В относительно стандартного каломельного электрода.
Значения потенциалов полуволн в полярографии определяют относительно потенциала стандартного каломельного электрода. Обычно при этом идут не по пути определения еа и последующего пересчета, а подключают в качестве противоэлект-
286
Д. Химический анализ
рода каломельный электрод к ртутному капельному электроду! ьГогда вольт-амперные кривые (рис. Д.101) переходят в кри! вне ток — потенциал; для одного и того же деполяризатора! они идентичны (рис. Д.102) и не зависят от фонового элект! ролита при условии, что фоновый электролит не образует! комплексов с деполяризатором. Величина потенциала полувол! ны Т1+ в различных фоновых электролитах всегда равна .—0,49 В (из-за отсутствия комплексообразования), поэтому можно добавить Т1+ к раствору другого деполяризатора и, найдя разность напряжений деполяризации этих растворов, по вольт-амперной кривой рассчитать потенциал полуволны!
Рис. Д.103. Полярограммы смеси Т1+ и РЬ2+. а —в 0,1 н. HN03; б — в 1 н. КОН.
другого деполяризатора; при этом отпадает необходимость использования стандартного каломельного электрода.
Если протекают реакции комплексообразования, значения потенциала полуволны сдвигаются в отрицательную область в соответствии с уравнением
'(£1/2)компл ~ (®1/г)своб Ч" («О
где D — коэффициент диффузии свободного иона; D* — коэффициент диффузии комплекса; Кс— константа устойчивости комплекса; сх — концентрация свободного лиганда; Р — число лигандов в комплексе.
Потенциал полуволны тем больше сдвинут в область отрицательных значений, чем больше устойчивость комплекса. Это 'можно использовать в анализе веществ. Если значения потенциалов полуволн двух деполяризаторов настолько близки, что их волны не разделяются и накладываются друг на друга в широкой области значений, то в раствор вводят комплексообразующий реагент, который с одним деполяризатором образует гораздо более прочный комплекс, чем с другим. В результате различного сдвига потенциалов в отрицательную область про-
39. Электрохимические методы в количественном анализе 287~
исходит разделение волн. На рис. Д.103 приведены полярограм-МЫ смеси Т1+ и РЬ2+ в различных средах: а) 0,1 н. HiNO3 (оба; потенциала полуволн равны-----0,50 В); б) 1 н. КОН ((8i/2)ti+ =
!₽— 0,50 В, (ei/2)pb2+=—0,81В). Смещение потенциала для pb2+ связано с образованием комплекса [РЬ(ОН)3]_.
39.3.6. Предельный диффузионный ток и количественная! оценка полярограмм
Дри добавлении индифферентного электролита к анализируемому раствору обычно происходит стабилизация потенциала, донной ртути (образование электрода 2-го рода). В ряде случаев наблюдается сдвиг потенциала полуволны вследствие комплексообразования, причем одновременно с уменьшением разности потенциалов между электродами происходит снижение воздействия электрического поля между электродами; ;(в соответствии с выражением IRl). При этом доля тока, переносимого через раствор ионами деполяризатора при воздействии электрического поля (миграционный ток), очень мала, и !ионы деполяризатора перемещаются к ртутному капельному Электроду почти исключительно за счет диффузии. Если сила; тока достигает такой величины, при которой происходит восстановление этих ионов деполяризатора, которые при данном потенциале электрода могут восстанавливаться, достигая поверхности электрода за счет диффузии, то дальше она уже не увеличивается. Это диффузионный предельный ток, величина которого пропорциональна концентрации деполяризатора в растворе.
При дальнейшем увеличении потенциала электрода можно' силу тока вновь повысить до величины предельного диффузионного тока, используя реакцию другого деполяризатора. Наконец, значительное увеличение тока происходит при разложении растворителя (2Н3О+—*-2Н2О+Н2) или фонового электролита (например, Na+—*Na), но при этом; из-за высоких концентраций уже .нельзя достичь величины предельного тока.
Если используют стационарный гладкий электрод при постоянном значении потенциала, то вследствие обеднения раствора деполяризатором вблизи электродов сила тока снижается примерно пропорционально ~qt~1l* (q — площадь поверхности, t — время электролиза). На капельном электроде поверхность каждой новой капли до ее падения формируется в 'зависимости от времени так, что q^t2^3. Объединение обоих Уравнений дает выражение т. е. сила тока возрастает
по параболической зависимости от .нуля до максимального значения перед отрывом капли. Поверхность под кривой ток — время для отдельной капли соответствует количеству электричества, проходящего через каплю (в кулонах, если силу тока
288
Д. Химический анализ
Рис. Д.104. Кривая ток — время при постоянном потенциале в зависимости от периода роста капли.
•а — без демпфирования; б — с применением демпфирования; в — при выравнивании поверхности.
измеряют в амперах, а время — в секундах) (рис. Д.104, а). При регистрации полярограммы на осциллографе получают Пилообразную кривую. Использование демпфированного гальванометра и параллельно с ним включенного конденсатора для уменьшения флуктуаций тока дает волнообразную кривую )(рис. Д.104, б), которую делят посередине (выравнивание поверхности). На реальной полярографической кривой в отличие от упрощенных кривых, приведенных на рисунках в зависимости от степени демпфирования в большем и меньшем размере наблюдаются осцилляции (рис. Д.105).
Среднее значение диффузионного предельного тока для ртутного капельного электрода можно вычислить по уравнению Ильковича:
= 0,627«РсД1'2т2'3Д6
(422)
Рис. Д.105. Реальная полярограмма, полученная при небольшом демпфировании.
где п — число электронов, обмениваемых частицей деполяризатора с электродом; F — число Фарадея; D — коэффициент диффузии частицы деполяризатора; т—скорость вытекания ртути, г/с; t — период капания, с; с — концентрация, моль/см3. Средняя сила тока составляет 6/7 максимальной величины. Поэтому при оценке можно провести прямую также вдоль верхних частей зубцов, если используют методы сравнения.
В уравнение Ильковича не входит температура, но члены D, т и t зависят от температуры; сила тока id меняется на 1,6% при повышении или понижении комнатной температуры на один градус. Поэтому при проведений точных измерений температура полярографической ячейки должна оставаться постоянной.
Поляризация и образование полярографических волн
£
39. Электрохимические методы в количественном анализе
289
могут быть обусловлены не только диффузией, но и другими факторами. Из уравнения Ильковича выводят критерии для предельного диффузионного тока. Скорость вытекания ртути m из капельного электрода пропорциональна давлению и тем самым разности уровней ртути электрода и мениска ртути в резервуаре: m=kh. Масса капли (mt) при постоянном напряжении независимо от значения h постоянна, поэтому t = k'/h. Вводя это выражение в уравнение Ильковича, получим
id = Ktrfl3?l& = К (kh)2'3 (k'/h)1/e = К' Vh
Пропорциональную зависимость предельного тока от корня квадратного из значения h можно проверить, повторив измерения при различной высоте подъема резервуара со ртутью.
Для использования полярограмм в количественном анализе определяют высоту волны деполяризатора. Наиболее точные результаты получают, если снимают последовательно полярограмму фонового электролита и фона с деполяризатором. Разность значений в области предельного тока деполяризатора (дает величину id (рис. Д.106). Обычно id определяют с доста
Рис. Д.106. Определение значения предельного тока по разности основных токов.
Рис. Д.107. Определение значения предельного тока по расстоянию между экстраполированными прямыми при значениях потенциалов полуволн.
точной точностью, экстраполируя обе прямые и определяя расстояние между ними при значениях потенциалов полуволн (рис. Д.107).
Концентрацию деполяризатора не определяют прямым методом по уравнению' Ильковича, поскольку m и t нельзя измерить с достаточной точностью, а главное, потому, что обычно ‘Неизвестно точное значение D для конкретных условий. Определение концентрации проводят следующими косвенными методами.
19—1880
f
290 Д. Химический анализ
1 1. Метод сравнения. Сравнивают высоту волн анализируе-
мого и стандартного растворов, полученных в одинаковых условиях (постоянство фонового электролита, ячейки, капилляра, t, m, температуры).
1 2. Метод градуировочной кривой. При проведении серийных измерений к точно измеренному объему V фонового электролиза (например, 10 см3) добавляют объемы и стандартного раствора в порядке их возрастания (например, 1, 2, 3 см3 и т. д.). !В полученные полярограммы нужно ввести поправку на объем (полученное значение в каждом случае делят на У+у). Строят (градуировочную кривую, откладывая по оси ординат значение предельного тока с поправкой, а по оси абсцисс — добавляемые ’объемы и. К анализируемой пробе (объемом, например, 1 см3) добавляют равный объем фонового электролита. По аналогично скорректированному значению предельного тока по градуировочной кривой находят объем стандартного раствора, содержащий такое же количество деполяризатора, что и 1 см3 анализируемой пробы, и определяют концентрацию деполяризатора сх- Стандартные растворы и анализируемое вещество должны полярографироваться в строго одинаковых условиях. Поэтому градуировочную кривую целесообразно в каждом случае строить заново. Если она представляет собой прямую, проходящую через начало координат, то для контроля в каждом случае достаточно измерить значение предельного тока только для самой большой из применяемых концентраций.
3. Метод добавок. К известному объему раствора определяемого деполяризатора добавляют определенный объем стандартного раствора. Снимают полярограммы до и после внесения стандартного раствора. Высоту волны корректируют по соответствующему суммарному объему. Разница объемов соответствует увеличению высоты волны, происходящему при добавлении стандартного раствора. Пропорция, составленная относительно высоты первой волны, дает возможность рассчитать сх (выведите формулу для расчета).
Преимуществом этого метода является постоянство условий в процессе определения. Наименьшую ошибку получают, если после добавления стандартного раствора высота волны увеличивается вдвое.
39.3.7. Другие полярографические токи
На вид полярограммы наряду с диффузией могут оказывать влияние и другие процессы!. Если деполяризатор образуется в результате химической реакции в непосредственной близости от электродов или если его подают непрерывно, возникают кинетические токи, как, например, в случае образования форм-
39. Электрохимические методы в количественном анализе
291
альдегида из метиленгликоля:
/ОН
н2с; = н2с=о + Н2О
Х)Н
Метиленгликоль Формальдегид
((полярографически неактивен) (деполяризатор)
Кинетические токи линейно зависят от концентрации. Большое влияние на них оказывает температура (изменение составляет 10—30% на градус); от высоты столба ртути они не зависят.
। Каталитические токи возникают, если электродные процессы связаны с протеканием каталитической электрохимической реакции; обычно они зависят от скорости каталитического выделения водорода. На максимумы волн по-разному влияют высота столба ртути и температура. С увеличением концентрации катализатора каталитические токи приближаются к предельному значению. Каталитические токи можно успешно применять в аналитических целях, если использовать градуировочные кривые, так как в этом случае волны намного больше (например, в 500 раз), чем можно было бы ожидать для диффузионного предельного тока каталитически действующего деполяризатора.
При адсорбции деполяризатора или продукта его электрохимического превращения на электроде возникают адсорбционные токи в виде волн, расположенных на вольт-амперных кривых до или после волны диффузионного тока. Значение адсорбционных токов прямо пропорционально высоте столба ртути. С ростом температуры адсорбционный ток уменьшается и затем исчезает.
Полярографические максимумы представляют собой воспроизводимое увеличение силы' тока сверх ожидаемого значения предельного тока. Различают максимумы 1-го и 2-го рода (рис. Д.108). Они образуются в результате вихревых явлений вокруг капель и перемещения дополнительных количеств деполяризатора к электродам. Причиной возникновения максимумов 1-го рода является разность потенциалов и связанное с ней различие поверхностного натяжения в нижней и верхней частях капли ртути. Образование максимумов 2-го рода обусловлено большой скоростью вытекания ртути из капилляра. Максимумы 1-го рода характерны в основном для разбавленных растворов фонового электролита, максимумы 2-го рода — Для растворов с высокой концентрацией фонового электролита (>0,5 н.). Максимумы первого рода практически не зависят от Л, максимумы второго рода исчезают при малых значениях h (небольшая скорость вытекания ртути).
19*
292
Д. Химический анализ
Оба типа максимумов можно подавить добавлением небольших количеств поверхностно-активных веществ, таких, как желатин, крахмал и др. Аналогичное действие оказывают многие другие высокомолекулярные соединения. Например, уксус, по-лучаемый при брожении, дает две волны восстановления кислорода (1 и 2, кривая б рис. Д.109), а синтетический уксус кривая а — только один ярко выраженный максимум в области 1-й волны. Это можно использовать для их разделения.
Рис. Д.108. Полярограммы с максимумами.
/ — максимум 1-го рода; 2 — максимум 2-го рода.
Рис. Д.109. Полярографические волны восстановления кислорода в синтетическом уксусе (а) и бродильном уксусе (б).
Аналогично можно провести определение суммарного количества загрязнений в воде. Получают полярографический максимум кислорода для чистейшего бидистиллата (0,01 н. по К.С1) и затем полярографируют смеси с увеличивающимся содержанием анализируемой воды до тех пор, пока получаемый максимум не уменьшится наполовину. При анализе воды, сильно загрязненной поверхностно-активными веществами, этот результат получают уже при добавлении небольших количеств воды.
39.3.8. Области применения. Емкостный ток. Дифференциальная полярография
Постояннотоковую полярографию можно применять для количественного определения веществ (катионов, анионов, молекул), которые в рабочей области потенциалов ртутного электрода (до —1,8 В, а при использовании солей тетраалкиламмо-мия в качестве фонового электролита — до —2,6 В) вступают в электрохимическую реакцию на ртути. Для аналитических целей можно применять деполяризаторы в концентрациях 1О2—10-6 н. (оптимальная область 10-3—10-4 н.). Относи-
39. Электрохимические методы в количественном анализе 293
тельная точность определения составляет 1—3%. Разрешающая способность, определяемая как разность потенциалов полуволн двух деполяризаторов, которую при одинаковой высоте волн еще можно различить на полярограмме, составляет jlOO—-150 мВ. При раздельном количественном определении двух деполяризаторов различают два случая (рис. Д.110): $) вещество 1, концентрация которого меньше, легко восстанавливается (тогда Ci : с2«1 : 1000 — предел обнаружения это-(го вещества); б) вещество 1 труднее восстанавливается (тог-
Рис. Д.110. Полярограммы смеси двух веществ с разными концентрациями и различной способностью к восстановлению.
а _ деполяризатор с меньшей концентрацией (/) легче восстанавливается, чем деполяризатор с большей концентрацией (2); б — вещество 1 труднее восстанавливается, чем вещество 2.
да Ci : с2х. 1 : 10). Нижний предел концентраций, еще определяемых полярографическим методом, обусловлен прежде всего емкостным или зарядным током. Он возникает также в чистом растворе фонового электролита даже в отсутствие следов деполяризатора (ионов металлов, кислорода воздуха и т. д.). !В этом случае при работе с обычными ртутными капельными '(Электродами наблюдают примерно линейно возрастающий с ростом напряжения ток порядка 1—2-10~7 А на 1 В. Ток заряжает регулярно образующийся при получении каждой капли ртути конденсатор между поверхностью ртути и слоем электролита на капле, но не способствует протеканию электрохимической реакции, т. е. не является фарадеевым током, а действует как чисто зарядный ток. В случае стационарных электродов ток возникал бы только один раз для зарядки конденсатора в его пограничном слое при каждом приложенном потенциале. В случае капельного ртутного электрода емкостный ток протекает постоянно, и значение его тем больше, чем быстрее скорость капания. Так как диффузионный ток id уменьшается с тюнижением концентрации деполяризатора, в итоге ток в цепи Должен упасть до значения емкостного тока ic. Таким образом, в области низких концентраций деполяризатора (ниже 10~5 -н.) емкостный ток может внести существенную ошибку в измерен
294
Д. Химический анализ
ную суммарную силу тока. При этом ток полярографической ячейки уже больше не определяется исключительно величиной а эквивалентная схема электрохимической ячейки упрощается (рис. Д.1Н) и представляет собой параллельно включенные поляризационное сопротивление R\ и конденсатор Сь
Можно показать на так называемых электрокапиллярных кривых, что при определенном значении потенциала (электро-капиллярный нуль, —0,56 В в растворах хлорида по отношению .к стандартному каломельному электроду) поверхность ртутного капельного электрода не заряжена и, следовательно, кон-
Рис. Д.111. Эквивалентная схема измерительной ячейки постояннотоковой полярографии с учетом емкостного тока ртутного капельного электрода.
Рис. Д. 112. Двойной электрический слой ртутного капельного электрода.
денсатор не образуется, т. е. емкостный ток не протекает. При положительной величине потенциала капля окружена анионами раствора (рис. Д.112), при отрицательном, наоборот, катионами. Из-за деформируемости анионов емкость такого двойного слоя обычно больше, чем в случае катионов. При более положительных потенциалах, чем потенциал в нулевой точке ©лектрокапиллярной кривой, электроны движутся от капельного электрода через внешний контур к. донной ртути; при более отрицательных значениях — в обратном направлении ’(рис. Д.113). Различие в наклоне анодной и катодной ветвей обусловлено различной емкостью. Емкостный ток описывается следующим выражением:
ic = dQ/dt = E*C'dqldt (423)
где Q — заряд; С' — удельная емкость конденсатора ртутной капли (в расчете на 1 см2); Е*— потенциал, определенный относительно электрокапиллярного нуля.
Поскольку для увеличения поверхности отдельной капли q со временем существует зависимость то Так
как то ясно, что для каждой новой капли диффузион-
ный ток растет сначала быстро, а затем медленно; емкостный ток, наоборот, уменьшается сначала быстро, а затем медленно. Если средние значения обоих токов в случае очень разбавленных растворов деполяризаторов равны, то на протяжении
39. Электрохимические методы в количественном анализе
295
существования капли токи изменяются, как указано на рис. Д.П4.
Для исправления искажений на полярограмме, вызываемых емкостным током, можно включить в измерительную цепь компенсатор с программированным линейным возрастанием тока; при этом не принимают во внимание различные отклонения емкостного тока от линейности (см. рис. Д.113) (компенсация емкостного тока). Другая возможность состоит в том, чтобы измерение силы тока проводить для каждой капли только в
Рис. Д.113. Зависимость направления и силы емкостного тока от смещения потенциала ртутного капельного электрода относительно нулевой точки электрокапиллярной кривой.
Рис. Д.114. Зависимость диффузионного предельного ia и емкостного ic токов от времени роста капли ртутного капельного электрода.
Д/ — интервал измерений импульсной полярографии.
конце ее существования, незадолго до падения, автоматически включая измерительное устройство на небольшое время А/ ;(импульсная полярография), так как в этом случае id имеет Максимальное значение, a ic— минимальное (рис. ДЛИ).
Разрешающую и разделяющую' способность постояннотоковой полярографии можно улучшить, применяя метод дифференциальной полярографии (рис. Д.115). Для тока зарядки конденсатора емкостью С, включенного последовательно с гальванометром, существует зависимость ic = dQ[dt=CdVldt. Напряжение V на конденсаторе равно падению потенциала iR на параллельно включенном сопротивлении R, т. е. ic = CRdi]dt. Так как U растет со временем, то di}dt = KdifdU и ic = = CRKdi!dU. Току зарядки ic конденсатора С, который регистрируется гальванометром (С' служит только для демпфирования), соответствует difdU, т. е. отклонение на вольт-амперной кривой (рис. Д.116).
Вместо ступени на полярограмме возникает максимум, пик которого соответствует значению потенциала полуволны, а вы-
296
Д. Химический анализ
Рис. Д.115. Схема установки для дифференциальной полярографии.
Рис. Д.116. Постояннотоковая и соответствующая дифференциальная полярограмма для четырех деполяризаторов.
сота — концентрации деполяризатора. При этом разрешающая! .способность возрастает примерно на 50 мВ. Одновременно ем-| костный ток ртутного капельного электрода, соответствующий! на исходной полярограмме линейному возрастанию кривой, из-| меняется в постоянных пределах и не мешает интерпретации] полярограммы. Подробное описание метода постояннотокововд полярографии приведено в литературе. 1
39.4. Амперометрия jl
39.4.1. Основные положения
В то время как электрогравиметрия, кулонометрия и поляро- |
графия являются электрохимическими методами определения {
содержания вещества, амперометрию применяют для опреде- I ления точки эквивалентности при титровании, т. е. она служит методом индикации. Амперометрия основана на тех же явле- 1 ниях, что и постояннотоковая полярография, поэтому амперо- метрическое титрование называют также поляриметрическим или титрованием по предельному току. Принцип метода заключается в измерении значения постоянного тока, протекающего при постоянном напряжении через раствор электролита между электродами, один из которых поляризуемый, а другой — неполяризуемый, как функции поляризационного сопротивления R\. В отличие от амперометрии в кондуктометрии измеряют значение переменного тока как функции сопротивления электролита RL. Метод амперометрии с двумя поляризуемыми электродами называют «методом конечной точки» («dead-stop»).
39. Электрохимические методы в количественном анализе
297
39 4.2. Амперометрия с одним поляризуемым электродом
В этом методе разность напряжений выбирают таким образом, чтобы по крайней мере для одного из ионов, участвующих в химической реакции (осаждения, комплексообразования или окисления-восстановления), приложенное напряжение находилось в области значений диффузионного предельного тока. Можно добиться также, чтобьи посторонние ионы с большими значениями потенциалов полуволн, находящиеся в растворе и не участвующие в реакциях обмена, не вносили бы изменений р величину электропроводности (в отличие от кондуктометрии). ,В этом состоит существенное преимущество амперометрии по-сравнению с кондуктометрией.
При построении зависимости силы тока от объема добавляемого титранта на кривой титрования получают две прямолинейные ветви, точка пересечения которых является точкой эквивалентности. По измерению- объема удается проводить определение концентрации значительно точнее, чем по высоте полярографической волны.
В зависимости от того, происходит ли на электроде изменение заряда ионов определяемого вещества или реагента и ионов определяемого вещества, или реагента при выбранном
Рис. Д.117. Кривые амперометрического титрования.
fl — определяемые ионы полярографически активны, титрант неактивен; б — определяемые ионы полярографически неактивны, титрант активен; в — ионы определяемого ве* щества и титрант полярографически активны.
напряжении, можно получить три типа кривых титрования, показанных на рис. Д.117. В первом случае электропроводность, обусловленная ионами определяемого вещества, изменяется скачкообразно (рис. Д.Г17, а), во втором — проводящие ток ионы расходуются до достижения точки эквивалентности, затем концентрация их в растворе возрастает (рис. Д.117, б). В третьем случае электропроводность раствора вначале обусловлена ионами определяемого вещества и уменьшается до точки эквивалентности в соответствии с уменьшением его концентрации^ затем на кривой наблюдается подъем, поскольку ионы титран-
298
Д. Химический анализ
Рис. Д.118. Схема простой установки для амперометрического титрования. R — сопротивление; G — гальванометр; Ptr — вращающийся платиновый электрод;
К — каломельный электрод.
та также электропроводки и находятся в избытке (рис Д.117, в). На практике наблюдается некоторое искривление кривых титрования вблизи точки эквивалентности, как и в методе кондуктометрии. Оно тем сильнее, чем более растворимо соединение, выпадающее в осадок, или чем менее устойчив образующийся комплекс.
Для проведения определений можно применять полярографическую аппаратуру. Целесообразно вначале снять полярограмму раствора для установления значения напряжения, соответствующего области диффузионного предельного тока определяемого иона. При амперометрическом титровании это напряжение затем поддерживают постоянным. Однако работу можно выполнять и на упрощенной установке (рис. Д.118). В ней в качестве неполяризуемого электрода применяют каломельный электрод, а в качестве поляризуемого можно использовать ртутный капельный электрод. Однако для амперометрического титрования более подходит вращающийся платиновый электрод, так как перемеши
вание обеспечивает более эффективный контакт анализируемого раствора и титранта. Кроме того, при перемешивании увеличивается сила тока, так как плотность диффузионного предельного тока уменьшается. Наконец, при использовании вращающегося платинового электрода устраняется мешающее влияние емкостных токов.
Амперометрическая индикация точки эквивалентности применима для растворов с предельной концентрацией 10-6 М '(в то время как потенциометрическая — только до 10-4 М). Точность этого метода (±0,1%) превышает точность полярографических методов практически в десять раз. С другой стороны, амперометрию нельзя использовать для проведения . качественного анализа и для раздельного определения компонентов в смесях (отличие от полярографии), но зато можно применить для определения полярографически неактивных ионов (например, титровать ионы SO42- раствором соли свинца).
Амперометрия сравнительно новый метод, но она уже нашла широкое применение. Возможности ее использования шире, чем потенциометрии, так как в амперометрии нет необходимости применять специфичные электроды. Большинство ионов и ’
39. Электрохимические методы в количественном анализе
299
молекул дают полярографические волны, так что нетрудно вы-боать реагенты, с помощью которых можно провести амперометрическую индикацию точки эквивалентности.
Рекомендуемые опыты
1. Два серебряных электрода погружают в раствор NaCl. Анод при этом неполяризуем (почему?). При наложении напряжения 0,1 В протекает только-небольшой остаточный ток. При титровании раствором AgNO3 после точки, эквивалентности на кривой наблюдается резкий подъем.
2. Готовят раствор арсенита (1 н. по НО и 0,05 н. по КВг). К катоду (вращающийся электрод из платиновой проволоки) прилагают напряжение’ q 2_0,3 В относительно насыщенного каломельного электрода. Затем титруют
01 н. раствором КВгОа. Арсенит при этом окисляется в арсенат, и после точки эквивалентности бромид окисляется в бром. Из всех веществ, включая и кислород воздуха, только бром при этом значении потенциала катода дает полярографическую волну. Поэтому лишь после точки эквивалентности происходит линейное возрастание тока.
39.4.3 . Амперометрия с двумя поляризуемыми электродами. Метод конечной точки
к двум электродам из платиновой проволоки, опущенным в хорошо перемешиваемый раствор, прилагают постоянное поляризационное напряжение 10—100 мВ. Пока оба электрода поляризованы, может протекать лишь крайне незначительный остаточный ток. Только в присутствии анодного или катодного деполяризатора возможно увеличение силы тока (порядка нескольких микроампер). Кривую титрования строят в координатах ток — расход титранта, так же как в методе амперометрии с одним поляризуемым электродом.
Поскольку этот метод особенно широко применяется в иодометрии, рассмотрим следующий пример. При титровании раствора 12 в KI на катоде происходит восстановление 12 до I-, а на аноде в эквивалентном количестве I- окисляется в Ь. Поэтому концентрационное соотношение 12/1_ остается в растворе неизменным, чему способствует перемешивание раствора. Только в непосредственной близости к электродам происходит обеднение раствора и дополнительная подача деполяризатора к электродам за счет диффузии. Диффузионная поляризация из-за небольшой силы тока крайне мала, и ею можно пренебречь. Таким образом, оба электрода почти идеально деполяризованы, и возникает ток.
Когда 12 титруют ионами БгОз2-:
I2 + 2S2O32- -> 2Г + S4Oe2-
концентрация иода уменьшается, а поляризация катода увеличивается. В точке эквивалентности в растворе практически нет иода. Ионы S4O62~ при приложенном напряжении на катоде не
300
Д. Химический анализ
могут восстанавливаться, так как система
2SaO32- S4Oe2- + 2е-
йеобратима. Катод в этом случае практически полностью поляризован, существует лишь очень небольшой остаточный ток. Пересечение экстраполированных прямолинейных участков кривой дает точку эквивалентности. При титровании необратимой тиосульфатной системы раствором иода ток возникает после точки эквивалентности.
При титровании обратимой системы титрантом, система которого также обратима, сила тока сначала падает до нуля 1(точка эквивалентности), а затем снова возрастает.
Если к началу титрования раствор не содержит одновременно анодного и катодного деполяризаторов, как в рассмотренном примере (раствор Ь в KI), то титрование происходит иначе. Например, титруют ионы [Fe(CN)6]4^ раствором соли Се4+. До начала титрования в растворе находится только анодный деполяризатор [Fe(CN)6]4~. Катод поляризован, а анод деполяризован, и ток практически не протекает. Только в процессе титрования образуется катодный деполяризатор {[Fe(CN)6]3~ и сила тока возрастает. Можно показать, что сила тока в этом случае может достичь максимума при прохождении реакции на 50%. При полном окислении [Fe(CN)6]4~ до [Fe(CN)6]3~ (в точке эквивалентности) в растворе больше нет анодного деполяризатора, анод поляризован и сила тока снова становится равной нулю.
Между собственно амперометрией и методом конечной точки можно отметить следующие различия.
1. В амперометрии к электродам прилагают напряжение, обеспечивающее протекание диффузионного тока, в методе конечной точки напряжения могут быть значительно меньше (ток служит только индикатором на присутствие деполяризатора в растворе).
2. В методе конечной точки не требуется применять неполяризуемый электрод сравнения. Это является достоинством метода, поскольку метод можно использовать не только для водных растворов. Например, для титрования в неводном растворителе можно применить реактив Фишера. Современные pH-метры поэтому часто снабжены так называемой приставкой для титрования по методу Фишера, с помощью которых можно проводить титрование при напряжениях порядка 10 мВ.
3. Метод конечной точки дает высокую чувствительность определения: можно анализировать, например, 0,01 мкг вещества в 100 см3 раствора.
39. Электрохимические методы в количественном анализе
301
Более подробные сведения о методах амперометрии можно ,найти в литературе*.
39.5. Новые полярографические методы
39.5.1. Переменнотоковая полярография
В этом методе на электроды подают равномерно возрастающее постоянное напряжение (как в классической полярографии) одновременно с переменным синусоидальным напряжением небольшой амплитуды (например, ±10 мВ) и на измерительном устройстве после выпрямления регистрируют только фара-деев ток. Фильтром постоянного тока служит конденсатор. Общая эквивалентная схема электрохимической ячейки (см. рис. Д.90) в этих условиях изменяется следующим образом: сопротивление Р2 неполяризуемого электрода мало. Переменнотоковое сопротивление конденсатора С2 также мало, так как для него
a) Rc = 1/шС; б) С я F/d (424)
где <о=2л/—частота поля; F — поверхность конденсатора; d — расстояние между пластинами конденсатора.
Поскольку значение F велико (донная ртуть), a d мало (молекулярный конденсатор), в упрощенную схему эквивалентной измерительной ячейки для переменнотоковой полярографии (рис. Д. 119) входят параллельно включенные Ry и С] малополяризуемого ртутного капельного электрода.
Если в ячейке находится раствор чистого фонового электролита без деполяризатора, то в области значений потенциалов между анодным растворением ртути на ртутном капельном электроде и катодным разложением фонового электролита Ry'3>Rc1 и электрические свойства ячейки определяются только переменнотоковым сопротивлением Rc конденсатора Су (рис. Д.120 яижняя диаграмма, кривая 1). Для этого «чистого» емкостного тока справедливы выражения
С = /(£,0 и £-/(/)
dQ d(CE) dC dE j dC \
dt ~ dt = E di dt [ C + dE E) ^425^
Первый член (EdC/dt) связан с процессом образования капель и соответствует емкостному току постояннотоковой полярографии. Для небольших значений амплитуды переменного напряжения член dC/dE (в скобках) можно принять за нуль, так что
ic = EdC/dt CdE/dt (426)
Поскольку в современной переменнотоковой полярографии при применении частотных фильтров регистрируют только ток, соответствующий частоте приложенного напряжения, можно пренебречь долей емкостного тока, связанного 'С образованием капель ртути (EdC/dt), тогда получим
ic= CdE/dt (427)
Наименьшее количество деполяризатора, определяемое методом переменно-токовой полярографии, составляет 5-10~5 М, это значение определяется величиной остаточного емкостного тока. Разрешающая и разделяющая способность переменнотоковой полярографии значительно выше, чем постояннотоко-
* См. например книгу: Сангина О. А., Захаров В. А. Амперометрическое нитрование. — М.: Химия, 1979. — Прим перев.
302
Д. Химический анализ
Рис. Д.119. Эквивалентная схема измерительной ячейки переменнотоковой полярографии.
Рис. Д.120. Сопоставление постояннотоковых и переменнотюковых полярограмм.
Вверху, постояннотоковые полярограммы раствора фонового электролита 1 и деполяризатора 2. Переменное напряжение, наложенное на постоянное напряжение в точках а, б и в, вызывает переменный ток (точка б'}. Внизу: переменнотоковые полярограммы фонового электролита Z и деполяризатора 2.
никают максимумы при значениях их
в идеальном случае переменнотоковая полярограмма совпадает с первой производной соответствующей постояннотоковой полярограммы (рис. Д.121), а также с дифференциальной полярограммой. Существенным отличием является очень небольшой максимум в случае необратимого электродного процесса,
вой и близка к достигаемой в методе дифференциальной полярографии.
Применение фазоселективного выпрямителя в переменнотоковой полярографии дает возможность полностью устранить емкостный ток, поскольку он опережает фарадеев ток (остаточный ток, обусловленный электродной реакцией деполяризатора). Ход переменнотоковой полярограммы становится понятным при сопоставлении переменнотоковой полярограммы с постояннотоковой (рис. Д.120). На постояннотоковой полярограмме (верхняя диаграмма) чистому фоновому электролиту соответствует кривая 1 (штриховая линия). Подъем на этой кривой при положительном потенциале ртутного капельного электрода обусловлен анодным растворением ртутя, а при большом отрицательном значении потенциала— выделением катионов фонового электролита. При добавлении к фоновому электролиту деполяризатора ход кривой 2 вначале будет таким же. Вблизи потенциала полуволны деполяризатора возникает волна,, а затем на кривой снова наблюдается горизонтальный участок до значения потенциала разложения фонового электролита. Небольшое переменное напряжение, наложенное на линейно возрастающее постоянное напряжение переменнотоковой полярографии (в точках а, б, в), вызывает в области небольшого возрастания постояннотоковой полярограммы (о и в) незначительное изменение силы тока, ио большое изменение потенциала полуволны в области б, обозначенное б'. Поскольку, как указано выше, протекает только переменный ток, на переменнотоковой полярограмме (нижняя диаграмма) наблюдаются только эти изменения. Для обычных деполяризаторов воз-
потенциалов полуволн. Таким образом,
поскольку малого значения переменного напряжения уже недостаточно для
окисления и восстановления соответствующего количества деполяризатора на электродах. Поэтому применение переменнотоковой полярографии ограничено обратимостью электродных реакций. Однако этот метод имеет то преимуще-
39. Электрохимические методы в количественном анализе
303
Рис. Д.121. Сопоставление постояннотоковой (а) и переменнотоковой (б) полярограмм смеси двух деполяризаторов, потенциалы полуволи которых различаются незначительно.
Рис. Д.122. Полярограмма с тензам-метрическими максимумами (1).
Кривая 2 показывает значительное увеличение основного тока переменнотоковой полярографии.
<ство, что необратимые сопутствующие компоненты мало мешают определению, например О2 нет необходимости полностью удалять.
Для расчета максимальной силы тока в переменнотоковой полярографии ^известно выражение
jmax = kn^SD^f^U^c (428)
где k — константа; п — число обменивающихся электронов;. S — площадь поверхности электродов; D — коэффициент диффузии; U- — амплитуда переменного напряжения; f — частота переменного напряжения.
Чувствительность (высота пика), таким образом, возрастает с увеличением амплитуды переменного напряжения, но одновременно снижается разрешающая и разделяющая способность, что связано с возможностью наложения пиков при близких значениях потенциалов полуволн определяемых веществ. Также нецелесообразно увеличивать частоту, так как поскольку в
•соответствии с уравнением (424а) емкостный ток (ic~f) растет быстрее и при высокой частоте сила тока imax определяется не скоростью диффузии, зависящей от концентрации, а скоростью самих реакций, происходящих на электродах. Поэтому работают обычно в области частот 10—100 Гц.
Преимуществом переменнотоковой полярографии* является возможность •ее применения для исследования адсорбционных процессов, происходящих на ртутном капельном электроде, а также для количественного определения поверхностно-активных веществ, таких, как высшие спирты, жирные кислоты, моющие средства и др. Адсорбция вещества в пограничном слое ртутного •капельного электрода достигает максимума при значении потенциала, соответствующем нулевой точке электрокапиллярной кривой (е«), при котором .двойной электрический слой находится в незаряженном состоянии. В зависимости от знака потенциала происходит притяжение анионов или соответственно катионов фонового электролита, а также в обоих случаях — притяжение диполей растворителя к пограничному слою, причем адсорбция поверх-
* О возможностях новых полярографических методов см. книгу; -Бонд. А. М. Полярографические методы в аналитической химии. — М.: Химия, 4983. — Прим. ред.
304
Д. Химический анализ
Рис. Д.123. Кривые квадратно-волновой полярографии.
Ug — квадратно-волновое напряжение, наложенное на постоянное напряжение; ic^ — быстро исчезающий под действием Up емкостный ток; iF — медленно исчезающий фарадеев переменный ток; tM — интервал проведения измерений.
ностно-активного вещества противодействует этому процессу. Поверхностно-активное вещество вытесняется в идеальном случае при значениях потенциалов, симметрично расположенных относительно ем, а именно, там, где силы электростатического притяжения ионов и адсорбции поверхностно-активного вещества находятся в равновесии. При подаче переменного напряжения эти потенциалы увеличиваются или уменьшаются и попеременно происходят процессы адсорбции и десорбции. Поскольку двойной электрический слой в присут-
ствии адсорбированного поверхностно-активного вещества обладает меньшей емкостью (уменьшение основного-тока, рис. Д.122), чем в обычных условиях (ионы и диполи), происходит сильное изменение переменной емкости с частотой переменного напряжения. Это означает, что в этих областях значительно увеличивается член EdCjdE в уравнении (425), т. е.
емкостный ток. На диаграмме емкостного тока наблюдается поэтому образование двух тензамметрических максимумов (рис. Д.122, кривая 1). Высоту их.
можно использовать для определения концентрации поверхностно-активных веществ, причем зависимость выражается в виде изотермы Ленгмюра. С другой стороны, при определении деполяризаторов методом переменнотоковой полярографии тензамметрические максимумы могут оказывать мешающее влияние, особенно в случае высокой концентрации солей в растворе, способствующих образованию таких максимумов.
Для основного тока, протекающего через ячейку в методе переменнотоковой полярографии, можно на основании вышесказанного записать
i = ip= + + t'c= + lC~ (429).
где!;?——отфильтровываемый фарадеев постоянный ток; —фарадеев переменный ток, являющийся сигналом деполяризатора; — емкостный ток, связанный с периодическим процессом образования капель, устраняемый частотным фильтром; ic_— емкостный переменный ток, обусловленный изменением: заряда двойного электрического слоя (сигнал тензамметрии). При определении деполяризаторов через емкостный переменный ток также часто устраняют, применяя фазоселективный выпрямитель.
39.5.2. Квадратно-волновая полярография
В методе квадратно-волновой полярографии (К.ВП), так же как в переменнотоковой полярографии, применяют равномерно возрастающее напряжение (как в классической полярографии), но вместо синусоидального напряжения одновременно подают квадратно-волновое напряжение Ur с продолжительностью каждого полупериода г. В этот короткий промежуток времени поверхность ртутного капельного электрода можно считать постоянной. Тогда для фарадеева тока, как указывалось в методе постоянноток-овой полярографии, справедливо выражение Емкостный переменный ток выражается как iC— при С, равной емкости двойного электрического-
слоя, и R, равном суммарному омическому сопротивлению полярографиче
39. Электрохимические методы в количественном анализе
305-
ской цепи. При небольшом значении R (прежде всего при большом содержании фонового электролита) двойной электрический слой быстро меняет заряд, так что ic^_ быстрее исчезает, чем (рис. Д.123). Измерение тока в конце полупериода г за время tM дает возможность регистрировать только остаточный ток при устранении ic~. В переменнотоковой полярографии это опережение г'с^ используют для его отделения от iF^_ с помощью фазоселективного выпрямителя. Вместо отделения гс= при помощи частотного фильтра, применяемого в постояннотоковой полярографии, в методе КВП работают по описанному ранее импульсному методу (измерение в конце роста капли), используя небольшое скашивание квадратно-волнового импульса (см. рис. Д. 123).
Так же как в методе переменнотоковой полярографии на полярограммах КВП наблюдаются характерные колоколообразные симметричные максимумы деполяризаторов (рис. Д.124), но с характерными ступенями. Для максимальной силы тока по аналогии с уравнением (428) можно записать
imax = (430)
Для обратимых электродных процессов центрацию деполяризаторов до 10~7 М, а в случае необратимых реакций до 10~6 М. Разрешающая способность метода составляет 40 мВ для обратимых электродных реакций при и=2.
Достоинством КВП по сравнению с постояннотоковой полярографией является значительно большая разделяющая способность. При разнице потенциалов пиков в 200 мВ она составляет 1 : 100 000 и 5000: 1
этот метод позволяет определять кон-
£
(для случаев, рассмотренных в разд. 39.3), т. е. возрастает примерно в 103 раз. Это означает, что существует возможность определения следовых количеств компонента в присутствии 5000-кратного избытка металла, восстанавливаемого при более положительном потенциале, и 100 000-кратного избытка металла, восстанавливаемого при более отрицательном потенциале, что дает возможность определять следовые количества элементов без отделения матрицы.
Рис. Д.124. Квадратно-волиовая полярограмма деполяризатора.
39.5.3. Импульсная полярография
Принцип метода заключается в том, что на электрод равномерно подают постоянное напряжение, а примерно за 1/25 с до конца существования капли подают дополнительный импульс постоянного напряжения, например 50 мВ. Перед подачей дополнительного напряжения ток автоматически компенсируют. Для устранения влияния дополнительного емкостного тока дополнительный фарадеев ток регистрируют только в конце полупериода. В результате компенсации влияние емкостного тока устраняется для всех компонентов раствора, для которых уже достигнуто значение предельного тюка. Более положительные деполяризаторы даже в 10000-кратном избытке при этом не мешают определению. Кроме того, метод пригоден для обратимых и необратимых электродных процессов, поскольку используют импульс постоянного напряжения.
20—1880
306
Д. Химический анализ
39.5.4. Вольтамперометрия
Термином вольтамперометрия определяют совокупность методов, в которых используются вольт-амперные кривые. До сих пор мы рассматривали кривые, характерные для ртутного капельного электрода, т. е. область применения полярографии. В других вольтамперометрических методах используют стационарные электроды, например твердые электроды или висящую каплю ртути.
39.5.4.1. Хроноамперометрия
Если к стационарному поляризуемому рабочему электроду, находящемуся в неперемешиваемом растворе с большим избытком проводящей соли, приложить постоянный потенциал, величина которого достаточна для протекания электродной реакции растворенного деполяризатора, то возникает ток, который можно измерить.
В отсутствие конвекции (т. е. без перемешивания) и миграции (т. е. при избытке фонового электролита) частицы передвигаются к электродам только за счет диффузии. При этом происходит обеднение раствора деполяризатором вблизи электродов и уменьшение силы тока. Линейному увеличению потен-«циала со временем соответствует выражение
8 = 8,+ tdz/dt
(431)
где 8; — начальный потенциал, выбранный таким образом, чтобы обеспечить
протекание соответствующей реакции
Рис. Д.125. Хроноамперометрическая кривая.
i — максимальный фарадеев ток; 1С — емкостный ток.
с пренебрежимо малой скоростью; deldt=v — скорость изменения потенциала, обычно 0,1—2 В/мин. Скорость электрохимической реакции быстро возрастает, и на кривой наблюдается подъем. Одновременно уменьшается концентрация вещества у поверхности электродов и происходит диффузия.
На кривой сначала наблюдается максимум, однако если преобладает обеднение раствора деполяризатором, то минимум, и при дальнейшем протекании электродной реакции сила тока возрастает и возникает следующий максимум (рис. Д.125). Положе-
ние максимума коррелируют с величинами стандартных потенциалов; высота
максимума прямо пропорциональна концентрации:
im = feSn3/2O1/sv1/2c
(432)
Отсюда очевидно, что !т~о1/2.
С другой стороны, для емкостного тока на стационарном электроде справедливо уравнение (427), выведенное из уравнения (425), так как отсутствует изменение поверхности электрода (капель); отсюда следует ic~v и ~iyv. Тем самым получают оптимальную величину скорости увеличения напряжения. Однако большая площадь поверхности S, чем у капельного ртутного электрода дает возможность определять концентрации порядка 10~7М.
При анализе смесей высота второго максимума уже не прямо пропорциональна концентрации. В этом случае для определения компонентов смеси применяют метод сравнения. Две ячейки, заполненные одним и тем же фоно-
39. Электрохимические методы в количественном анализе ЗОТ
Рис. Д.126. Хроноамперометрическая диаграмма нескольких деполяризаторов (i).
При включении второй ячейки в дифференциальную схему получают разность токов Ato; Дл« первого деполяризатора Atj; для второго деполяризатора Д£*
Рис. Д.127. Диаграмма инверсионной вольтамперометрии.
i — максимальный ток растворения
вым электролитом, включают в дифференциальную схему. В одной из этих ячеек находится анализируемая смесь. Определение вещества / проводят относительно ячейки сравнения, заполненной фоновым электролитом. Затем известное количество вещества добавляют в ячейку сравнения и определяют вещество 2, после чего добавляют известное количество второго вещества и т. д. (рис. Д.126).
39.5.4.2. Инверсионная вольтамперометрия
Дальнейшего повышения чувствительности определений достигают в методе инверсионной вольтамперометрии, применяя электролитическое концентрирование вещества на электроде. Собственно определение заключается в растворении ранее выделенного на поверхности электрода вещества (stripping analysis). Повышение чувствительности связано с фактором накопления, который для стационарных электродов может достигать 1000. При концентрировании веществ должна воспроизводимо поддерживаться скорость перемешивания, поскольку только при высокой скорости перемешивания максимумы тока не зависят от этой величины. Значение тока растворения (рис. Д.127) возрастает вначале линейно, а затем стремится к предельному значению. Поэтому выделение с концентрированием проводят не количественно, а только в течение определенного промежутка времени.
Основная область применения инверсионной вольтамперометрии — анализ следовых количеств веществ (до 10~10 М). Ограничением является необходимость проведения холостого опыта (определение примесей) для применяемых фоновых электролитов или реагентов.
39.6. Потенциометрическая индикация точки
эквивалентности
39.6.1. Основные положения
Выше было рассмотрено (разд. 38.3), как при титровании анализируемого раствора изменяется логарифм концентрации или показатель активности ионов, находящихся в растворе, в зависимости от количества добавляемого реагента.
20*
308
Д. Химический анализ
В методе потенциометрии потенциал индикаторного электрод да, погруженного в анализируемый раствор, измеряют относи-* тельно постоянного потенциала электрода сравнения. Потенциал! индикаторного электрода пропорционален логарифму концентра-! ции определяемого иона. Поэтому, измеряя потенциал, можно* проследить за ходом реакции в процессе титрования.
Вспомним основные положения электролиза. Для протекания окислительно-восстановительных процессов на электроде,
например,
Ме2+ ---> Ме3+ -р е~
необходимо приложить к нему потенциал, который измеряют относительно постоянного потенциала электрода сравнения, j Потенциал рассчитывают по уравнению Нернста в зависимо- ! сти от отношения активностей окисленной и восстановленной форм:
Е = Ёод_^1п (433)
1 nF Яме2+ V ;
Для окислительно-восстановительной системы-, содержащей свя- j ванный кислород, например
Мп2+ + 4Н2О -► МпО4~ + 8Н+ + 5е~
уравнение Нернста принимает вид
е= е°Д
RT . ЙМпО4~ й8н+
—— in-----!-----
nF «Мп2+
(434)
При измерении потенциала с помощью соответствующего устройства без прохождения тока через электроды рассчитывают напряжение по уравнениям (433) и (434).
39.6.2. Измерение потенциала
Прохождение тока при измерении потенциала вызвало бы изменение концентрации определяемого иона в анализируемом растворе. Это изменение наиболее сильно проявилось бы вблизи индикаторного электрода. Возникновение концентрационной поляризации привело бы к тому, что измеренное значение потенциала совершенно не соответствовало бы1 концентрации определяемого иона в анализируемом растворе. Поэтому измерение потенциала проводят в отсутствие тока. Наиболее ранним является компенсационный метод с использованием схемы Поггендорфа.
В настоящее время в основном применяют приборы с ламповыми усилителями. Благодаря высокому сопротивлению на входе — до 10'2 Ом — они также дают возможность проводить измерения практически без протекания тока. Отсчет показаний
39. Э лектрохимические методы в количественном, анализе 309
проводят сразу, без дополнительных операций по стабилизации показаний. Подключив самописец, можно автоматически регистрировать результаты измерений.
39.6.3. Электрод сравнения
Потенциал электрода сравнения не должен зависеть от потенциала индикаторного электрода в ходе измерений и должен оставаться постоянным. Каломельный электрод (рис. Д.128) состоит из ртутного электрода, покрытого каломелью (Hg2Cl2);
ПкоподводящаЯ ' проволока
Рис. Д.128. Схема каломельного электрода.
над ртутью находится раствор КС1 определенной концентрации. Через солевой мостик, заполненный раствором электролита (целесообразно применять также КС1), электрод соединяется с анализируемым раствором. Во избежание смешивания электролитов солевой мостик обычно закрыт с двух сторон диафрагмами. Потенциал каломельного электрода (электродный процесс 2Hg—>-Hg22++2e~) определяют по уравнению
Нернста:
„п , 0,059
8 — 8°Hg/Hg22+ Н 2 ан?22+ (435)
Поскольку на электроде находится избыток каломели, значение HHg22+ находят из произведения растворимости:
«нй2’+ = ^/«2сГ
(436)
310
Д. Химический анализ
.Введя эту величину в уравнение (435), получим
,.о । 0,059 . Kl lAv^
е — 6Hg/Hg22+ 4-2— 'а?сг (437)
„ . 0,059 . „ . 0,059 . 1
е—е Hg/Hg22+ 4~ 2 8^ь4-----2 ^cF" (438}
(Гак как Kl — величина постоянная, два первых члена уравнения (438) можно обозначить константой е0', а последним членом можно пренебречь. Тогда потенциал каломельного электрода выразится, как
е==е0' — 0,0591g ас!- (439)
Значение потенциала зависит от константы в0' и активности ионов С1~. Существует три типа каломельных электродов:
Насыщенный каломельный электрод е°=+0,242 В Каломельный электрод, 1 н е°=+0,280 В
Каломельный электрод, 0,1 н е°=+0,334 В
Концентрация (насыщенный, 1 н. и 0,1 н.) относится к раствор РУ КС1. I
Достоинством насыщенного каломельного электрода явля-ется постоянство активности ионов СК при испарении воды^ происходящем при длительном использовании электрода. Од-, нако на значение потенциала этого электрода температура доказывает более сильное влияние, чем в случае других электродов, так как растворимость КС1 зависит от изменения темпе--ратуры.
39.6.4. Окислительно-восстановительное титрование
Принцип индикации в данном случае очень прост. В качестве' индикаторного электрода применяют платиновую1 проволоку^ или пластинку. Электрод погружают в перемешиваемый анализируемый раствор. С помощью солевого мостика соединяют ; раствор с каломельным электродом. Платиновый электрод ] •инертен и приобретает потенциал, определяемый отношением 1 концентраций компонентов титруемой редокс-пары в соответст- | вии с уравнением Нернста. |
Напряжение между индикаторным электродом и электродом | сравнения обусловлено в основном разностью окислителъно-вос- 1 становительных потенциалов обоих полуэлементов. Кроме то- 1 го, на границе раздела анализируемый раствор/раствор электролита в солевом мостике и между растворами электролитов Н солевом мостике и в электроде сравнения возникает диффузионный потенциал. Последний в ходе титрования изменяется
39. Э лектрохимические методы в количественном анализе
311
незначительно, и для построения кривой потенциометрического титрования нужны только разности потенциалов (величину диффузионного потенциала можно не учитывать). Однако диффузионный потенциал необходимо учитывать при определении концентраций окислительной или восстановительной форм соответствующей редокс-пары по измеренному значению потенциала.
Оценку результатов титрования проводят, строя зависимость потенциала измерительной ячейки от расхода титранта. Получают характерную S-образную кривую. Точка ее перегиба (наибольший подъем кривой) обычно является точкой эквивалентности при титровании (рис. Д.129). Небольшие отклонения наблюдаются, например, на асимметричной кривой осадительного титрования. Более точно точку эквивалентности можно определить при построении первой производной AE/AV. Тогда на кривой вместо точки перегиба наблюдается максимум (рис. Д.129). Современные приборы с саморегистра-цией могут автоматически записывать эту кривую. Если работают на простых установках для титрования, титрант добавляют небольшими равными порциями. И в этом случае построение зависимости изменения потенциала от расхода титранта дает дифференциальную кривую, максимум которой соответствует точке эквивалентности.
Часто применяют метод
Рис. Д.129. Кривая потенциометрического титрования (а) и диаграмма Д£/ДУ—V (б).
э—точка эквивалентности.
Рис. Д.130. Ячейка для дифференциальной потенциометрии.
/ — резиновая груша; 2 — капилляр;
Ei — платиновые электроды [212].
312
Д. Химический анализ
титрования до заданного потенциала. Навстречу индикаторной му электроду измерительной ячейки включают потенциал, со ответствующий предварительно измеренному потенциалу точи
эквивалентности, и титруют раствор до тех пор, пока разност! потенциалов станет равной нулю? Можно также включить на
встречу измерительной ячейке с платиновым электродом и ана лизируемым раствором аналогичную измерительную ячейку (
раствором, состав которого такой же, как в точке эквивалент-
ности. В этом случае отпадает необходимость применения кало
Достаточно иметь две ячейки с
мельного электрода.
платино
выми электродами и соединить их солевым мостиком. Метод
титрования до заданного потенциала применяют в основном-при проведении серийных анализов близких по составу растворов.
Все достоинства описанных вариантов объединяет метод, дифференциальной потенциометрии, который дает возможность-прямо определять отношение Д£/ЛУ на очень простой установке-(рис. Д.130). В этом методе отпадает необходимость применения электрода сравнения и солевого мостика. В раствор опускают две платиновые проволоки. Один из электродов находится в 'стеклянной трубке, отверстие которой сильно сужено. С помощью резиновой груши в эту трубку можно засосать анализируемый раствор. Разность потенциалов между электродами -вначале равна нулю-. При добавлении титранта по каплям к анализируемому раствору изменяется отношение концентраций окисленной и восстановленной форм редокс-пары. Из-за узкого отверстия стеклянной трубки титрант туда попадает не сразу, поэтому между анализируемым раствором в трубке и раствором в ячейке возникает разность потенциалов, которую фиксируют. Затем концентрации выравнивают, просасывая через трубку анализируемый раствор до тех пор, пока разность потенциалов снова не станет равной нулю. Добавляют новую порцию титранта, опять регистрируют разность потенциалов и т. д.
1 В точке эквивалентности частное AE/AV приобретает наибольшее значение, по которому можно определить соответствующий ему объем V. Когда титрант добавляют равными порциями AV, построение зависимости AE/AV от V дает дифференциальную кривую, представленную на рис. Д.129, б. Поскольку перед взаимодействием каждой порции титранта с определяемым веществом в трубке остается небольшой объем жидкости, возникает ошибка измерения, которая тем меньше, чем меньше этот объем. Погрешность определения в этом методе доставляет до 0,003%, поэтому его особенно целесообразно применять для анализа разбавленных растворов.
39. Электрохимические методы в количественном анализе
313
39.6.5. Осадительное и комплексометрическое титрование
В основе методов лежит взаимодействие определяемых катионов или анионов с ионами других реагентов с образованием соответствующей редокс-пары. В этом случае можно обычным -способом зафиксировать изменение потенциала в зависимости от изменения концентрации определяемого иона в процессе реакции.
При определении катионов металлов целесообразно использовать металлический индикаторный электрод. Например, серебряный индикаторный электрод можно применять во всех реакциях, где участвуют ионы Ag+. Однако не все металлы пригодны для изготовления индикаторных электродов, так как редокс-потенциал системы часто устанавливается недостаточно быстро и воспроизводимо и, кроме того, многие металлы могут восстанавливать ионы Н+ анализируемых растворов до Нг, т. е. растворяться.
При определении некоторых анионов для создания редокс-.пары можно ввести в анализируемый раствор окисленную форму аниона. Например, осадительное титрование 1~ можно осуществить с платиновым электродом, добавив к раствору небольшое количество Ь.
При определении анионов можно также использовать электроды второго и третьего рода. К электродам второго рода относится, например, описанный выше каломельный электрод. Как было указано, его можно применять для измерения концентрации ионов СИ в растворе, а следовательно, и в качестве индикаторного в осадительном титровании для реакций, протекающих с участием этих ионов.
Электродом третьего рода могла бы служить система типа
Zn/Zn—оксалат/Са—оксалат/Са2+
Электрод реагирует на изменение концентрации ионов кальция, а поскольку она связана с концентрацией оксалат-ионов, косвенно определяется и концентрация ионов Zn2+. Принцип действия электрода такой же, как в электроде Са2+/Са (не применяемом в водных растворах), хотя он служит для индикации редокс-пары Zn/Zn2+.
В табл. Д.20 приведены некоторые реакции осадительного й комплексометрического титрования, редокс-пары и индикаторные электроды.
39.6.6. Кислотно-основное титрование
Изменение pH анализируемого раствора определяют путем потенциометрического измерения pH. Наряду с электродом сравнения в этом случае применяют электрод, потенциал которого
314
Д. Химический анализ
Таблица Д.20. Реакция осадительного и комплексометрического титрования 1 и определяемые редокс-пары 1
Реакция титрования Индикаторный электрод Определяемая 1 редокс — пара
Ag+ + Cl~—>AgCl Ag Ag/Ag+
Ag+ + I-—>-AgI >
Ag+ + Br-—>AgBr » »
Ag++CN-—>AgCN » »
Ag++SCN~—*AgSCN » » с
Ag+ + I~—>-AgI I2+Pt Wi- ' :
T1+ + I-—>TII » »
Hg22+4-2I~—5-Hg2I2 » »
Fe3+4-6F~—->FeF63- Fe2+ + Pt Fe2+/Fe3+
зависит от концентрации ионов ЬР или ОН" в анализируемом растворе. Индицирующими редокс-парами являются Н+/Н2 и о2/он-
Для индикации редокс-пары Н+/На применяют водородный электрод. Во избежание перенапряжения платинированную 'платиновую проволоку (или пластину) насыщают очищенным газообразным водородом и помещают в анализируемый раствор. В соответствии с уравнением Нернста потенциал для реакции Н2—>2Н++2е_ рассчитывают как
s = eV/Ha+§-ln^t. (440)
гН2
где рн2~~ парциальное давление газообразного водорода.
Поскольку £°н+/н2 = 0, для рн2=1 получим
£=+0,0591g ан+ или е=—0,059рН (441), (442)
Отсюда следует, что при изменении pH анализируемого раствора на единицу потенциал измерительной ячейки сдвигается на 59 мВ.
Ацидиметрическое титрование можно также проводить, используя вместо водородного гладкий платиновый электрод в качестве индикаторного. Здесь потенциалобразующей является реакция 4ОН~—>-О2+2Н2О+4е~ и уравнение Нернста имеет вид
п । 0,059 1 РОо /лло\
е -Е О2/ОН~Н 4---1g (443^
‘где ро2 — парциальное давление кислорода воздуха. По достижении равновесия ему соответствует определенная постоян-
39. Электрохимические методы в количественном анализе
315
лая активность растворенного Ог. Константы можно объединить, тогда
е — е0' — 0,0591g аон- (444)
Поскольку «H+aoH- = Kw или йон- = Кда/ан+, введя эти выражения в уравнение (444) и обозначив постоянный логарифмический член вместе с е0/ через е°", получим
е = е°" — 0,059рН (445)
Сравнение уравнений (442) и (445) показывает, что гладкий платиновый электрод в растворе реагирует на изменение pH аналогично водородному электроду. Потенциалы различаются только по абсолютному значению в0". С другой стороны, в этом случае пренебрегают перенапряжением кислорода на платине, «которое привело бы к введению дополнительного члена в уравнение.
На воспроизводимость значения потенциала платинового индикаторного электрода оказывают влияние факторы1, мешающие протеканию электродных процессов, а также изменение активности растворенного Ог. Поэтому относительно дорогой водородный электрод часто заменяют так называемым хингидронным электродом. Для индикации в этом электроде используют другую обратимую редокс-пару, потенциал которой зависит от pH. Хингидрон представляет собой продукт присоединения хинона (Ch) и гидрохинона (СйНг) в молярном отношении 1:1. В водном растворе он диссоциирует с образованием исходных компонентов. На поверхности хингидронного электрода протекает реакция
ChH2 Ch + 2Н+ + 2е“
Уравнение Нернста для данной реакции имеет вид
или
е==ео+_2^1 ig^_+o,O591gaH+ Л иСпН2
При растворении хингидрона йсь/асьн2 = 1» поэтому е= =е°—0,059 pH. Следовательно, потенциал платинового электрода, погруженного в водный раствор хингидрона, так же зависит от pH, как потенциал водородного электрода. Значения Потенциалов различаются только по абсолютной величине. Достоинствами хингидронного электрода по сравнению с водородным являются простота его изготовления, более быстрое установление значения потенциала и относительно высокое значение стандартного потенциала (е° ='4-0,703 В).
316
Д. Химический анализ
Последнее обстоятельство обусловливает возможность его применения даже в тех случаях, когда в анализируемом рас» гворе находятся вещества, восстанавливающиеся на платине под действием водорода, если используют водородный элект-род. Например, с хингидронным электродом можно титровать разбавленную HNO3. Хингидронный электрод нельзя применять в щелочных растворах при рН>8,5: гидрохинон является слабой двухосновной кислотой, которая, начиная с этого значения pH, находится в диссоциированном состоянии, так что зависимость потенциала от pH усложняется. Кроме того, хингидрон в сильнощелочном растворе легко окисляется (даже кислородом воздуха).
Хингидронный электрод можно использовать также в качестве электрода сравнения и в методе титрования до заданного потенциала, поскольку при изменении pH возникает потенциал, который легко можно определить.
Для измерения pH можно применять также оксидные электроды, но только в такой области значений pH, в которой оксид нерастворим. Широкое применение находит сурьмяный электрод. В качестве индикаторного электрода используют сурьмяный стержень. На его поверхности устанавливается равновесие:
2Sb + ЗН2О -> Sb2O3 + 6Н+ + бе"
Из уравнения Нернста в этом случае получают следующую зависимость:
е= £°—0,059рН
Однако зависимость между pH и потенциалом на практике не всегда линейна, поэтому необходимо проводить градуировку для каждой исследуемой системы. Применяя сурьмяный электрод, можно надежно измерить pH только в узкой области значений, из-за этого использование сурьмяного электрода в кислотно-основном титровании нецелесообразно.
Иногда применяют электродную пару из двух различных металлов, на которых с разной скоростью устанавливается потенциал, соответствующий окислительно-восстановительному равновесию в растворе, в который опущены эти электроды. Например, на платине это происходит очень быстро, а на вольфраме особенно медленно. Поскольку при титровании необходимо знать только относительные значения потенциалов, можно использовать оба эти электрода, причем вольфрамовый стержень будет функционировать как электрод сравнения. Такая установка, разумеется, значительно проще и надежнее, чем система с каломельным электродом.
Для измерения pH в широкой области значений (от 1 до 14) й настоящее время применяют так называемый стеклянный
39. Электрохимические методы в количественном анализе 317’
электрод. В стеклянной трубке, продолжением которой является шарообразная стеклянная мембрана, находится рабочий электрод, например серебряная проволока, опущенная в насыщенный водный раствор AgCl, заполняющий электрод. В анализируемом растворе стеклянный электрод приобретает потенциал, значение которого, пропорциональное активности Н3О+, можно измерить относительно постоянного потенциала электрода сравнения. Причиной зависимости потенциала электрода от pH является то, что внешняя и внутренняя поверхности-стеклянной мембраны имеют различный заряд, в результате <чего возникает разность потенциалов, пропорциональная концентрации Н+ в обоих растворах.
Существенное влияние на электрохимическое поведение стеклянного электрода оказывает состав стекол, применяемых для изготовления мембран. В продаже имеются различные типы таких электродов. Для хорошей воспроизводимости показаний электрода мембрану необходимо постоянно хранить в воде. Существенными достоинствами стеклянного электрода являются независимость показаний от присутствия окислителей1 или восстановителей в растворе и отсутствие травления электрода деполяризаторами, что характерно, например, для водородного электрода.
Сравнительно недавно появились стеклянные электроды, реагирующие на изменение активности не только протонов, но и других катионов, например Na+ или К+. Такие электроды, называемые ионселективными*, представляют большой практический интерес. Наряду со стеклянными в качестве ионселек-тивных электродов используют монокристаллы различных неорганических соединений. Особенно большие успехи достигнуты в области создания ионселективных мембран для определе-.‘ния концентрации анионов. Мембраны изготавливают на инертной основе (например, на силиконовом каучуке) с диспергиро-(ванной в ней малорастворимой солью (например, AgCl для измерения концентрации С1~).
Для более детального изучения потенциометрии см. соответствующую литературу.
* Уже сейчас ионометрия занимает значительное место в аналитической-химии и роль ее все возрастает. Gm. книги: Лакшминараянайах Н. Мембранные электроды. Пер. с англ. — Л.: Химия, 1979; Никольский Б. П., Матеро-ва Е. А. Ионселективные электроды. — Л.: Химия, 1980; Камман К. Работа-с ионселективщыми электродами. Пер. с нем./Под ред. О. М. Петрухина.— М.: Мир, 1980. — Прим ред.
.318
Д. Химический анализ
39.7. Кондуктометрия
39.7.1. Общие положения
Кондуктометрия — это метод электрохимической индикации, в котором для нахождения точки эквивалентности используют изменение электропроводности в ходе титрования. Поэтому говорят также о титровании по электропроводности.
' В отличие от электрохимических величин, используемых в других методах индикации, таких, как потенциометрия, амперометрия, вольтамперометрия, суммарная электропроводность электролита аддитивно складывается из электропроводности всех находящихся в растворе ионов независимо от того, принимают они участие в реакции или нет. Поэтому кондуктометрические измерения отражают не конкретные процессы, происходящие при титровании, а изменения, происходящие в растворе в ходе титрования и связанные с вкладом ионов, участвующих в реакции, в суммарную электропроводность всех ионов, находящихся в растворе. При титровании по электропроводности точность определения тем меньше, чем больше в растворе 'концентрация посторонних ионов, не участвующих в реакции. Иначе говоря, наиболее удовлетворительные результаты получаются при титровании растворов с минимальным содержанием посторонних электролитов.
39.7.2. Теоретические основы
Измеряя электропроводность растворов, тем самым измеряют их сопротивление, поскольку электропроводность А связана с сопротивлением R соотношением
1=1/R (446)
'Сопротивление проводника прямо пропорционально его длине I и обратно пропорционально поперечному сечению q:
R = pl/q (447)
Коэффициент пропорциональности р при длине проводника 1 см и поперечном сечении 1 см2 называют удельным сопротивлением. Обратная ему величина — удельная электропроводность х:
х — 1 /р (448)
Из уравнений (447) и (448) следует
n = URq (449)
'Удельная электропроводность измеряется в единицах Ом-1 см-1. Электропроводность разбавленных растворов электролитов
39. Электрохимические методы в количественном анализе 319
зависит от числа ионов, находящихся в растворе (т. е. от их концентрации), числа элементарных зарядов, переносимых каждым ионом (т. е. от заряда ионов), от подвижности ионов (т. е. от скорости, с которой одинаково заряженные ионы под действием постоянного электрического поля перемещаются к электроду). Все эти факторы объединены в понятии эквивалентной ионной электропроводности (Э^), которая и определяет ход кривых кондуктометрического титрования.
Эквивалентная электропроводность ионов зависит от их. концентраций: с разбавлением она увеличивается. Данные,, приведенные в табл. Д.21, получены экстраполяцией на беско-
Таблица Д.21. Эквивалентная электропроводность ионов при бесконечном разбавлении и 25 °C
Катионы эк Анионы Д
н+ 349,8 он- 198
Li+ 38,7 ci- 76,3
Na+ 50,1 Вг- 78,4
К+ 73,5 I- 76,8
NH<+ 73,4 NO3- 71,4
/2 Mg2+ 53,1 СЮ4- 68,0
72 Ca2+ 59,5 HCO3- 44,5
7г Ba2+ 63,6 CH3CO2- 40,9
7г Fe2+ 54 C6H5CO2~ 32,3
i/3 Fe3+ 68 7г СОз2- 70
V2 Cu2+ 54 V2 SO42- 79,8
V2 Pb2+ 73 7г С2О42- 70
нечно большое разбавление при 25 °C. Аномально высокой; электропроводностью обладают ионы Н+ и ОН-. Это объясняется особым механизмом передвижения этих ионов в растворе.. !В ряду щелочных и щелочноземельных металлов электропроводность растет с увеличением ионных радиусов от Li к Cs и от :Ве к Ва. Это связано с тем, что небольшие катионы окружены более прочной гидратной оболочкой, которая перемещается-«вместе с ними при их движении. Эффективное поперечное селение движущихся частиц в этом случае особенно велико, и в-растворителе возникает большая сила трения. Трение уменьшается при повышении температуры из-за уменьшения вязкости растворителя. Повышение температуры на 1 °C вызывает'
320
Д. Химический анализ
увеличение электропроводности примерно на 1—2%. Поэтому при проведении точных кондуктометрических измерений необходимо поддерживать постоянную температуру.
39.7.3. Установки для кондуктометрических измерений
.В кондуктометрии измеряют сопротивление электролитов, находящихся в ячейке. В соответствии с эквивалентной схемой измерительной ячейки для электрохимических методов анали- '! за (см. рис. Д.90) это означает, что определяемой величиной j /является только Rl- Измерение можно провести двумя спо- i •собами.
1. Работа при постоянном токе с двумя неполяризуемыми электродами (например, насыщенными каломельными). В этом случае ток через конденсатор не может проходить и в схеме остаются последовательно включенные 3 сопротивления. Поскольку на неполяризуемых электродах при токе небольшой силы поляризационное сопротивление не меняется, изменение сопротивления контура определяется только значением Rl-Этот метод не находит применения в практике кондуктометрии.
2. Работа при переменном токе. При достаточно большой поверхности электродов (обычно платинированных платиновых пластин) и достаточной частоте (1000 Гц—для разбавленных растворов электролитов, 50 Гц — для концентрированных) на электродах не происходит заметного изменения концентрации и они не поляризуются, так что величинами R1 и можно пренебречь. Аналогично можно пренебречь емкостями Ci и Сг. Это следует из соотношений
Rc = 1 /©С и С ~ Fid
Площадь поверхности F велика, а расстояние d между пластинами «молекулярного конденсатора» крайне мало (порядка радиуса атомов), следовательно, емкость С большая, a Rc мало. Оба внешних контура эквивалентной схемы измерительной -ячейки, таким образом, устраняются, и остаются параллельно включенные Rl и Cl- Из-за сравнительно большого расстояния между электродами Cl мало, и сопротивление Rc этого конденсатора велико по сравнению с Rl- Поэтому ток проходит почти исключительно через Rl, изменения которого и определяют изменение тока. Для измерения сопротивления применяют мостик Уитстона (рис. Д.131). Скользящий контакт передвигают до тех пор, пока нуль-инструмент не укажет отсутствие тока. Это происходит в том случае, когда отношение пленен а и Ь, полученных при делении участка АВ скользящим -контактом, равно отношению неизвестного сопротивления рас-
39. Электрохимические методы в количественном анализе 321
Явора электролита в ячейке Rl к эталонному сопротивлению R: a/b = RL/R (450)
(Отсюда определяют сопротивление раствора электролита в измерительной ячейке:
RL = aRlb (451)
Эталонное сопротивление R целесообразно выбрать таким образом, чтобы, оно соответствовало среднему значению RL. iB этом случае а и b примерно равны, что дает возможность определить их с максимальной точностью (почему?).
Простейшим нуль-инструментом является телефонная трубка. Скользящий контакт передвигают до тех пор, пока произнесенные вполголоса слова не будут с минимальной громкостью звучать в трубке. В современных приборах используются гальванометры переменного тока или электронно-оптические индикаторы настройки, что дает большую точность измерений: можно определить сопротивление 105—106 Ом с точностью до 1%. Существуют даже кондуктоскопы, чувствительность измерения которых настолько велика, что изменение температуры на 1 °C, т. е. изменение электропроводности примерно на 2%, вызывает отклонение стрелки прибора на всю шкалу. Для работы на таких приборах необходимо поддерживать температуру постоянной с точностью 0,005 °C.
Кондуктометрические определения проводят в ячейке для измерения электропроводности (рис. Д.132), в которой находится титруемый раствор. Ячейка обычно представляет собой стеклянный сосуд с двумя неподвижно закрепленными гладкими платиновыми электродами с площадью поверхности 1 — 2 см2. При титровании растворов с небольшой электропроводностью поверхность электродов должна быть большой, а рас-’ ртояние между ними малым, и наоборот. Во избежание поляризации электродов при проведении измерений, их предварительно платинируют. С этой целью тщательно очищенную ячейку заполняют раствором 3 г платинохлороводородной кислоты и 25 мг ацетата свинца в 100 см3 дистиллированной воды. Оба электрода соединяют проводами и включают в цепь в качестве катода, дополнительный платиновый анод помещают между ними. Электролиз проводят при напряжении 4В и максимальной плотности тока 30 мА на 1 см2 поверхности электродов в течение ~10 мин. Затем раствор удаляют, заменяют его .разбавленной H2SO4 и проводят электролиз еще некоторое время. Затем ячейку промывают дистиллированной водой.
Для лучшей сохранности электродов из платинированной рлатины при хранении ячейку всегда следует заполнять дистиллированной водой.
21—1880
322
Д. Химический анализ
Рис, Д.131. Измерительная схема с мостиком Уитстона.
д — ячейка для измерения электропроводности; Я — сопротивление сравнения; N — нуль-инструмент (гальванометр); S — скользящий контакт; АВ— градуированное проволочное сопротивление (30 Ом) длиной 1000 мм.
Рис. Д.132. Схема ячейки для измерения электропроводности.
Для нахождения удельной электропроводности х, опре-деляемой уравнением (449), необходимо измерить R и знать отношение l]q (I — расстояние между электродами; q — поперечное сечение слоя электролита между электродами). Поскольку обе эти величины можно измерить лишь с небольшой точностью, их объединяют (уравнение (452)). Тогда значение х выразится соотношением (453)-
С = l/q; h = C/R (452), (453)
где С — константа ячейки. Ее можно просто и точно определить, измерив сопротивление стандартного раствора с известной удельной электропроводностью (например, для 1 н. КС1 х = 0,09827 Ом-'см-1)-
Для кондуктометрического титрования не требуется точное значение С, поскольку фиксируют только изменение х в процессе титрования, а не ее абсолютное значение. Однако в процессе титрования константа ячейки С должна
оставаться постоянной, поэтому электроды нельзя слишком глубоко погружать в жидкость. Объем раствора в процессе титрования также не должен
существенно увеличиваться. К 50 см3 титруемого раствора нужно добавлять не больше 5 см3 довольно концентрированного раствора титранта. Влияние, оказываемое на константу С объемом жидкости в ячейке, объясняется тем, что ток прохо-
дит не только через раствор с поперечным сечением q, находящийся между электродами.
39.7.4. Кривые кондуктометрического титрования
В качестве примера рассмотрим реакцию нейтрализации НС1 раствором, NaOH:
Н3О+ + Cl" + Na+ + ОН" -> Na++ С1‘ + 2H2q
39. Электрохимические методы в количественном анализе
323
При этом ионы Н+ и ОН- взаимодействуют с образованием воды, не оказывающей влияния на электропроводность раствора. Концентрация ионов С1- остается практически неизменной, если добавленным объемом раствора при титровании можно пренебречь. Поэтому изменение электропроводности до точки эквивалентности обусловлено заменой Н+ на Na+ (рис. Д.133). Поскольку электропроводность ионов Na+ значительно меньше, чем ионов Н+ (см. табл. Д.21), до точки эквивалентности наблюдается уменьшение электропроводности. При построении
Рис. Д.133. Кривая кондуктометрического титрования НС1 раствором NaOH. Общий ход изменения электропроводности в процессе титрования аддитивно складывается нз изменения электропроводности ионов.
зависимости электропроводности от добавляемого объема титранта получают нисходящую прямую, наклон которой тем больше, чем больше разница в электропроводностях ионов, участвующих в реакции. После точки эквивалентности уже не происходит изменения концентрации ионов водорода. Вместо «их возрастает содержание ионов Na+ и ОН-. Ионы ОН- уже 'не вступают в реакцию и вносят существенный вклад в электропроводность раствора. Восходящая прямая после точки эквивалентности обусловлена суммарной электропроводностью ионов Na+ и ОН-.
Точка эквивалентности определяется пересечением двух прямых. Если реакции ионов протекают неколичественно, изменение электропроводности при титровании происходит нелинейно. Поэтому вблизи точки эквивалентности всегда наблюдается 'больший или меньший плавный изгиб кривой (см.рис. Д.134, а), который тем сильнее, чем больше растворимость образующегося осадка в осадительном титровании или чем меньше кон-1станта устойчивости комплекса в комплексометрическом титровании. Если искажения кривых титрования не слишком велики, при оценке ими можно пренебречь. Для этого необходимо, чтобы несколько точек (обычно достаточно пяти) рас-21*
324
Д. Химический анализ
полагались на прямолинейных участках кривой до и после дочки эквивалентности. Точку эквивалентности получают экстраполяцией прямолинейных участков кривой до их пересечения. Поэтому достоинством метода кондуктометрии по сравнению с потенциометрией является то, что не нужно строить всю кривую титрования — достаточно провести несколько измерений.
39.7.5. Области применения кондуктометрии
В ходе кондуктометрического титрования происходит замещение ионов, находящихся в анализируемом растворе и участвующих !в реакции с титрантом, ионами титранта, электропроводность которых больше или меньше электропроводности ионов анализируемого раствора. Этим обусловлено получение восходящих или нисходящих ветвей кривых кондуктометрического титрования. После точки эквивалентности титрант уже не расходуется, поэтому обычно получают восходящие прямые, угол подъема которых зависит от электропроводности титранта. Точность индикации точки эквивалентности определяется углом пересечения прямых: он должен быть возможно более острым, тогда точность определения достигает ±0,3%. Обычная же точность метода до ±1%. Наиболее острый угол пересечения прямых получается при кислотно-основном кондуктометрическом титровании, так как ионы Н+ и ОН- вносят особенно большой вклад в электропроводность раствора (см. табл. Д.21). Наряду с реакциями кислотно-основного взаимодействия в кондуктометрии можно применять многие реакции осаждения и некоторые реакции комплексообразования. В принципе кондуктометрия годится и для индикации точки эквивалентности в окислительно-восстановительном титровании, если оно сопровождается изменением концентрации ионов Н3О+. Но все же лучшие результаты дают в этом случае другие методы индикации.
Кислотно-основное титрование. При титровании сильной кислоты сильным основанием получают кривую, представленную на рис. Д.134, а. При титровании сильной кислоты слабым основанием, например гидроксидом аммония, после точки эквивалентности угол подъема прямой невелик, что связано с диссоциацией образовавшейся соли и подавлением диссоциации избытка основания (буферное действие катионов соли, в данном случае NH4+). Кривая титрования представлена на рис. Д.134, б. В случае титрования сильного основания слабой кислотой кривая будет такой же. Кривые титрования слабого основания (или кислоты) сильной кислотой (или основанием) имеют другой вид. Слабое основание (или кислота) слабо диссоциирует, и поэтому растворы его имеют небольшую электро-
39. Электрохимические методы в количественном анализе
325
Рис, Д.134. Кривые кондуктометрического титрования.
а — сильная кислотаЧ-сильиое основание; б — сильная кислота+слабое основание; в — слабая кислота (или основание)H-сильное основание (или кислота); г —смесь сильной и слабой кислот (оснований)-(-сильное основание (или кислота). э2 — точки эквивалентности; х — электропроводность.
проводность. В процессе нейтрализации образуется хорошо диссоциируемая соль, подавляющая диссоциацию слабой кислоты или основания, и в начале титрования электропроводность немного уменьшается. Затем электропроводность растет за счет образующейся соли. После точки эквивалентности ионы ОН' (или соответственно Н3О+) уже не вступают в реакцию, поэтому электропроводность возрастает значительно меньше (рис. Д.134, в).
Кривая титрования смеси сильной, и слабой кислот (оснований), представляющая сочетание кривых рис. Д.134, а и Д.134, в, изображена на рис. Д.134, г. На практике для дифференцированного титрования кислот (оснований) в их смесях константы диссоциации кислот (оснований) должны существенно различаться. В противном случае на кривых наблюдаются большие плавные изгибы, что не позволяет четко определить точку эквивалентности. (Каким был бы ход кривой титрования слабой кислоты слабым основанием?)
Вытеснительное титрование. Растворы солей, образованных катионами сильных оснований и анионами слабых кислот
326
Д. Химический анализ
(и наоборот), можно титровать сильными кислотами (или основаниями). В качестве примера можно привести титрование хлорида аммония раствором NaOH и ацетата натрия раствором H2SO4. Наклон прямых до точки эквивалентности зависит от соотношения ионных эквивалентных электропроводностей вытесняемого и вытесняющего ионов, в нашем примере 3xNH + и 3xNa+ (лучше, чем К+; почему?) или 5хСНзСОО- и 5j.SOi2-. Если электропроводность вытесняющего иона больше, чем вытесняемого, получают восходящую прямую, в противном случае— нисходящую. После точки эквивалентности прямая круто поднимается.
> Условия вытеснительного титрования такие же, как в прямом титровании смеси сильной и слабой кислот (оснований); константы диссоциации вытесняющей сильной кислоты (основания) и вытесняемой слабой кислоты (основания) должны существенно различаться.
Другим примером вытеснительного титрования является определение ионов СО32. Из этого следует, что при титровании по методу нейтрализации надо применять растворы щелочей, не содержащие карбонатов, иначе могут возникнуть ошибки.
Осадительное титрование. Большое число реакций осаждения нельзя использовать в классическом титриметрическом анализе, что связано с отсутствием подходящих индикаторе: для надежного определения конечной точки титрования. В это( случае наряду с другими электрохимическими методами инди кации хорошие' результаты дает применение кондуктометрии и осадительном титровании катион или анион анализи-: руемого раствора образует осадок с анионом (или соответственно катионом) титранта. Это означает, что катион титран-; та замещает катион анализируемого раствора или происходит: соответствующее замещение аниона. Поэтому до точки эквивалентности получают восходящую или нисходящую прямую в зависимости от электропроводности осаждаемого или добавляемого иона. После точки эквивалентности происходит резкий подъем прямой, поскольку как анионы, так и катионы добавляемого титранта остаются в растворе без изменения, что вызывает увеличение электропроводности.
Изобразите ход кривых кондуктометрического титрования галогенидов С1~, Вг~ и 1~ раствором нитрата или ацетата серебра, а также ход кривой титрования ионов Ag+ растворами хлоридов различных щелочных металлов.
Трудности при проведении осадительного титрования заключаются в том, что осадки иногда имеют не однозначный состав или происходит искажение истинной электропроводности вследствие адсорбции осадками посторонних ионов из раствора. При большом разбавлении эти явления не оказывают
39. Электрохимические методы в количественном анализе
32?
влияния. Применяя современные чувствительные измерительные приборы, можно получить хорошие результаты именно при определении небольших концентраций веществ. Очень малые количества Pb2+, Cd2+, Cu2+, Ag+, Bi3+ можно надежно титровать сероводородной водой. При этом протекает реакция
Ме2+ + H2S + 2Н2О -- MeS + 2Н3О+
При титровании до точки эквивалентности кривая резко возрастает, что связано с образованием ионов Н3О+, обладающих хорошей электропроводностью, и затем идет параллельно оси абсцисс.
В некоторых случаях осадительного титрования состав осадка лишь через некоторое время становится постоянным. Это проявляется в том, что электропроводность после добавления титранта медленно изменяется и только через некоторое время становится постоянной. В качестве примера можно привести осаждение BaSO4. При повышенной температуре конечное значение электропроводности часто устанавливается значительно быстрее, поэтому в таких случаях титрование проводят при постоянной повышенной температуре. Например, ячейку для измерения электропроводности можно обогревать циркулирующими парами постоянно кипящей жидкости. Этот метод применяют для осадительного титрования ионов SO42-раствором ацетата бария (почему не BaCl??).
39.8. Осциллометрия
Осциллометрию часто называют высокочастотным титрованием. Но поскольку этот метод можно применять не только для индикации точки эквивалентности при титровании, но и для прямых измерений концентрации электролитов, исследования кинетики процессов (например, процесса кристаллизации) и др., названию осциллометрия следует отдать предпочтение (по аналогии с названиями потенциометрия, кондуктометрия, амперометрия и др-). Осциллометрия сравнительно новый электрохимический метод анализа. По-видимому, этим объясняется тот факт, что осциллометрии недостаточно уделяют внимания при обучении студентов, и отчасти этим же объясняется медленное внедрение метода в научные исследования и практику. Другая причина заключается, вероятно, в многообразии возможных конструктивных форм измерительных устройств, подробное теоретическое описание которых часто отпугивает исследователей.
Для установления влияния, оказываемого веществом или гомогенной смесью веществ на электрическое поле, определяют в основном электропроводность, диэлектрическую и магнит-
328 Д. Химический анализ
ную проницаемость. При титровании растворов электролитов магнитная и диэлектрическая проницаемости практически остаются постоянными, изменяется только электропроводность, ©то изменение можно определить прямым кондуктометрическим методом, поскольку существует линейная зависимость между электропроводностью и измеряемой величиной Rl- Методом осциллометрии нельзя провести прямое измерение переменнотоковой электропроводности, так называемого адмитанса, или соответственно переменнотокового сопротивления, так называемого импеданса, а также их изменений, но можно косвенно проследить за изменением измеряемой величины, которая свя-вана с электропроводностью более сложной зависимостью, чем в кондуктометрии. При этом анализируемый раствор определенным образом включают в электрический колебательный контур, изменение свойств которого используют для индикации импеданса и его изменений.
Преимуществом метода по сравнению с кондуктометрией является отсутствие гальванического контакта электродов, т. е. электроды не погружают в анализируемый раствор; таким образом, можно сказать, что измерения проводят «без электродов». При этом устраняют основные помехи, вызываемые поляризационными сопротивлениями или емкостными влияниями. Можно также не опасаться, что раствор пробы или образующиеся продукты реакции разрушат поверхность электродов. Это же относится к адсорбции, отравлению и блокировке электродов выделяющимися осадками. Наконец, отпадает необходимость таких важных для кондуктометрии операций, как подготовка, очистка и сохранение электродов. На фоне этих преимуществ, пожалуй, стоит смириться с небольшой и один раз возникающей трудностью, связанной с определением оптимальной области концентраций для проведения титрования на конкретном приборе.
39.8.1. Теоретические основы
Электрический колебательный контур состоит из емкости С и индуктивности L, включенных последовательно или параллельно. В методе осциллометрии применят практически только параллельное включение. Частота f недемпфированного колебательного контура (или частота контура a> — 2nf) определяется величинами С и L и может быть рассчитана из выражения
(1> = 2nf=l/]/CL (454)
При внешнем возбуждении колебательного контура интенсивность колебаний, которую можно измерить, например, как амплитуду тока, достигает максимума, если частота возбуж-
1
39. Электрохимические методы в количественном, анализе_329
пения и собственная частота колебаний контура равны (резонансная частота). В случае идеального колебательного контура амплитуда колебаний при этих условиях должна была бы иметь бесконечно большое значение. В каждый реальный колебательный контур входит омическое сопротивление, которое вызывает потери энергии при прохождении тока, что проявляется в выделении тепла. Поэтому амплитуда может принимать только конечное значение, т. е. должна быть всегда демпфирована. Это снижает эффективность колебательного контура.
Значение и вид емкости и индуктивности, а также омического сопротивления R колебательного контура оказывают решающее влияние на частоту, эффективность действия или демпфирование возбуждаемого извне колебательного контура. Кроме того, следует помнить, что не существует конденсаторов или катушек, совершенно не дающих потерь. Поэтому уравнение (454) для недемпфированного колебательного контура можно видоизменить. Для описания собственной частоты демпфированного колебательного контура емкость С, не дающую потерь, можно заменить, например, так называемой эффективной емкостью Се, которая в свою очередь является функцией to, С и R: Ce = F{<p, С, R). i
Если анализируемая проба находится в конденсаторе колебательного контура, то говорят об измерении с помощью емкостной ячейки. На эффективную емкость такой ячейки оказывают влияние диэлектрическая проницаемость и электропроводность пробы, а следовательно, и резонансная частота и демпфирование колебательного контура. Таким образом, переменнотоковое сопротивление—-импеданс ячейки зависит от диэлектрической проницаемости и электропроводности пробы. Резонансная частота и амплитуда колебаний в колебательном -контуре отражают изменение импеданса.
Анализируемую пробу можно также внести в катушку колебательного контура, при этом создается индуктивная ячейка. Вместо эффективной емкости в ней возникает эффективная индуктивность. Диэлектрическая проницаемость пробы практически не имеет значения, нужно учитывать лишь магнитную проницаемость. Если с помощью .емкостной ячейки можно проследить за изменением как электропроводности, так и диэлектрической проницаемости, то с помощью индуктивной ячейки — за изменением электропроводности и магнитной проницаемости.
В осциллометрии в основном используют количественную зависимость импеданса от электропроводности и диэлектрической проницаемости или соответственно от магнитной проницаемости, причем не имеет значения, проводят ли измерение импеданса или его изменений через частоту, амплитуду или другим способом.
330
Д. Химический анализ
Емкостные ячейки находят значительно большее применение в практике, чем индуктивные. Поэтому возможности метода рассмотрим только на их примере. На рис. Д.135 показана схема «безэлектродной» емкостной ячейки. Проводящая внешняя обкладка 1 и фиктивная антиобкладка 2 — противоположная ей поверхность жидкости — образуют с находящейся между ними стенкой сосуда (диэлектрик) конденсатор С\. Аналогично на противоположной стороне находится, конденсатор Сч,. Между С?! и Сг включен конденсатор Cl, который образу-
Рис. Д.135. Схема осциллометри- Рис. Д.136. Эквивалентная схема изме-т ческой емкостной ячейки. рительной емкостной ячейки. 1
ется обеими антиобкладками и раствором пробы, в стакан! (диэлектрик); параллельно с CL включено омическое сопротивч ление Rl раствора диэлектрика. При этом получаем общую эквивалентную схему измерительной ячейки (см. рис. Д.90), иную, чем в кондуктометрии, полярографии и других электро-аналитических методах, в которых электроды находятся в непосредственном контакте с электролитом. Там символами и были обозначены поляризационные сопротивления, а в осциллометрии это омические сопротивления стеклянных стенок сосуда. Так как Rt и R% значительно больше, чем Rc± и Rc„ (при высокой частоте), электропроводность практически обусловлена только значениями С! и С2 (из-за последовательного включения их можно объединить в С). Таким образом, получается упрощенная эквивалентная схема измерительной ячейки (рис. Д.136).
Очевидно, что определяемая величина Rl совпадает с величиной переменнотокового сопротивления ячейки, но не измеряется отдельно, как в кондуктометрии.
39. Электрохимические методы в количественном анализе
331
39.8.2. Характеристические кривые
{Как было указано в начале раздела, в методе осциллометрии для индикации концентрации электролита в анализируемом растворе используют величину переменного тока, например пе-1ременнотоковое сопротивление — импеданс Z— или переменно-токовую электропроводность—адмитанс G, являющийся величиной, обратной импедансу.
Поскольку данный метод связан с применением поля высокой частоты', обязательно следует обратить внимание на то, что величина переменного тока всегда слагается из величины активной, или действительной, составляющей и реактивной, 'или мнимой, составляющей, например:
Z = R-\-jX и G-Gw~\-jGB
где R— активное сопротивление; X — реактивное сопротивление; Gw и GB — активная и реактивная электропроводности соответственно; / — мнимая единица (/=У—1).
Обычно измеряют или реактивную или активную составляющую. Зависимость активной составляющей от концентрации электролита иная, чем для реактивной составляющей: в первом случае получают колоколообразную кривую, во втором— ^S-образную. Если при постоянном напряжении измеряют, например, активную составляющую электропроводности емкостной ячейки, то получают
п_________со2ДдСг____ /лееч
1 + (iPRi} (С + С/.)2
Для реактивной составляющей
с _ соС + w>R2CCl (С + CL) ив- 1 +ю2Ль2(С+С/)2
При построении графической зависимости активной составля-
ющей Gw от омической электропроводности 1/Т?£ кривая имеет
(456)
Рис. Д.137. Характеристическая кривая активной составляющей как функции удельной электропроводности.
Рис. Д.138. Характеристическая кривая активной составляющей как функции логарифма удельной элек-тропров одности.
Реактивная составляющая
Кривые титрования
Характеристическая кривая
Активная составляющая
Чувствительность титрования
Рис. Д.139. Оптимальные рабочие области, чувствительность и кривые осциллометрического титрования при измерении реактивной и активной составляющих электропроводности.
Рис. Д.140. Зависимость характеристических кривых реактивной составляющей электропроводности от частоты.
Рис. Д.141. Зависимость характеристических кривых активной составляющей электропроводности от частоты.
39. Электрохимические методы в количественном анализе 333
вид, показанный на рис. Д.137. Зависимость Gw от lg I/Rl — симметричная колоколообразная кривая — представлена на рис. Д.138. Аналогичный, вид имеют зависимости активной составляющей от удельной электропроводности х, пропорциональной омической электропроводности, и от концентрации электролитов с (в предположении, что соединение полностью диссоциировано) или от логарифмов этих величин. Напротив, можно показать, что зависимость реактивной составляющей от логарифма омической электропроводности или соответственно логарифма удельной электропроводности представляет собой S-образную кривую.
Оптимальную чувствительность измерений получают при максимальном изменении измеряемой величины в зависимости от удельной электропроводности. Для обоих типов кривых это имеет место в областях с наибольшей крутизной, т. е. в области их точки перегиба. При этом нужно помнить, что колоколообразная кривая имеет две точки перегиба; иначе говоря, для методов, в которых используется активная составляющая, существуют две области оптимальных измерений (численные значения в них противоположны по знаку). S-образная кривая имеет только одну точку перегиба. Таким образом, измерения реактивной составляющей могут быть проведены только в одной оптимальной области электропроводности. На рис. Д.139 представлена взаимосвязь между показаниями прибора, удельной электропроводностью пробы, оптимальной рабочей областью и чувствительностью, а также типичные кривые титрования. Из кривых, приведенных на рис. Д.139, можно сделать вывод, что при одинаковых параметрах приборов оптимальная область при измерении реактивной составляющей находится посередине между двумя рабочими областями метода активной составляющей. Положение указанных областей зависит от параметров приборов, поэтому перед проведением измерений нужно один раз снять характеристическую кривую для определения оптимальной рабочей области. В общем в обоих методах, повышая рабочую частоту, можно охватить также и область более высоких значений электропроводности, т. е. область более высоких концентраций электролитов, как это видно из рис. Д.140 и Д.141.
39.8.3. Влияние различных параметров на ход характеристических кривых
Как было показано, ход характеристических кривых реактивной и активной составляющей определяется удельной электропроводностью пробы. Поэтому при одинаковых значениях диэлектрической проницаемости природа электролитов не оказывает существенного влияния. Если же рассматривать интере
334
Д. Химический анализ
сующую аналитика зависимость показаний прибора от кон- центрации электролита, то можно заметить, что наблюдаемый в обоих методах характерный ход кривых зависит от различных факторов. При замене на оси абсцисс 1g х на 1gс получают кривые, отражающие влияние различных факторов, связанных с с и х, таких, как ионная подвижность и степень диссоциации. Оба параметра меняются с температурой. На рис. Д.142 в качестве примера приведены характеристические кривые реактивной составляющей
для различных водных раство- , ров электролитов при 25 °C и | частоте 34 МГц (на оси абс- 1 цисс откладывают значения I 1gс). Концентрационные кри- 1 вые раствора НС1 из-за особенно большой подвижности иона Н+ сильнее сдвинуты в область более низких концен- j траций по сравнению с кривы- 1 ми сходных по силе электро- л литов КС1, NaCI, ВаС1г и j LaCh, ионная подвижность- 1 которых меньше. Ионы гидро- 1 ксила занимают промежуточ- | ное положение между Н+ и 1 С1~, поэтому кривая NaOH .1 располагается между кривыми 1 НС1 и NaCI. Характеристиче- 1 напротив, значительно сдвинута |
Рис. Д.142. Зависимость характеристических кривых реактивной составляющей электропроводности от концентрации электролитов.
/ — НС1; г —NaOH; 3 — КС1; 4 — NaCI, BaCI2, LaCl3; 5 — CdSO4.
ская кривая раствора CdSO4,
в область более высоких концентраций. Из рис. Д.142 видно, ] что CdSO4, константа диссоциации которого примерно равна 1 0,5, значительно более слабый электролит, чем другие указанные вещества. Аналогичные результаты можно получить при построении зависимости активной составляющей от концентрации для других электролитов.
В обоих методах измерения чувствительность индикации зависит от максимальной крутизны характеристической кривой и определяется высотой кривой, которая пропорциональна ак-тивной составляющей, определяемой по уравнению (457), и ре- I активной составляющей, определяемой по уравнению (458): :
Для активной составляющей высота характеристической кривой возрастает с увеличением частоты, для реактивной составляющей— она не зависит от частоты (если применяют ячейки без параллельно включенной емкости), что показано также на рис. Д.140 и Д.141.
39. Э лектрохимические методы в количественном анализе Зээ
При большой величине емкости С чувствительность измерений повышается и в том и в другом методе, что видно из уравнений (457) и (458). Поскольку (где Kv — ем-
кость в вакууме), очевидно, что в обоих случаях при большом значении диэлектрической проницаемости высота характеристических кривых уменьшается, по возрастает при небольших значениях е. Это иллюстрируют два примера, приведенные на |>ис. Д.143 и Д.144. Для практических целей важно, что изменение измеряемой величины во всей области значений электро
lg ре
Рис. Д.143. Зависимость характеристических кривых активной составляющей электропроводности от диэлектрической проницаемости раствора.
Рис. Д.144. Зависимость характеристических кривых реактивной составляющей электропроводности от диэлектрической проницаемости раствора.
проводности возрастает с уменьшением диэлектрической проницаемости. Сравнивая результаты, полученные для различных растворителей, можно видеть, что при равных изменениях электропроводности чувствительность измерений возрастает с уменьшением диэлектрической проницаемости. Однако использовать эту зависимость можно лишь ограниченно, так как с уменьшением диэлектрической проницаемости, как правило, уменьшается степень диссоциации электролита и часто одновременно снижается электропроводность, так что чувствительность резко падает. Поэтому использование в осциллометрии йеводных растворов с низкими значениями диэлектрической проницаемости не всегда дает преимущества по сравнению с водными растворами. Но если чувствительность кондуктометрии существенно уменьшается в каждом случае при замене воды неводными растворителями с низкими значениями диэлектрической проницаемости, в которых соответствующий электролит менее электропроводен, то для осциллометрии это характерно в меньшей степени, что объясняется указанными выше закономерностями.
336 Д, Химический анализ
39.8.4. Области применения осциллометрии
Мы не будем рассматривать здесь различные типы измерю тельных ячеек и приборов, выпускаемых промышленностью, и технику работы на них — для этого существуют специальные руководства. Типы кривых осциллометрического титрования в основном сходны с кондуктометрическими. Но в осциллометрии ветви кривых линейны только в том случае, если измерения проводят в области перегиба характеристических кривых и не происходит слишком сильных изменений электропроводности. В противном случае на кривых в большей или меньшей степени возникают плавные изгибы. При проведении измерений в выбранной оптимальной рабочей области получают такую же, а иногда даже большую точность измерений, чем в кондуктометрии. Поэтому области применения осциллометрии и кондуктометрии совпадают, иногда осциллометрия даже более предпочтительна. Это происходит в тех случаях, когда важны такие преимущества осциллометрии, как возможность «безэлектродных измерений» и увеличение чувствительности с уменьшением диэлектрической проницаемости. Осциллометрию используют для индикации кислотно-основного, осадительного и комплексометрического титрования различных типов, а также при титровании агрессивных растворов и в неводных средах.. Юна пригодна и для решения различных кинетических проблем при исследовании процессов кристаллизации, растворения (например, гидраргиллита в алюминатном щелоке), омыления, этерификации, полимеризации, самоокисления и т. д. Метод осциллометрии находит применение в фазовом анализе, например при изучении процесса плавления, затвердевания, фазового обмена, расслоения, для построения диаграмм состояния и т. д. Особенно важным является использование осциллометрии для контроля и регулирования процессов производства. Этот метод пригоден для неразрушающего анализа ряда продуктов или содержимого ампул.
Рекомендуемые опыты
1. Построение характеристических кривых для водных растворов НС1, NaC104, Ва(СЮ4)2 и CdSO4.
а) К точно отмеренному объему воды в измерительной ячейке при перемешивании добавляют как можно меньшими порциями 0,1 М стандартный раствор одного из перечисленных веществ и при постоянном перемешивании проводят измерения. По объему титранта и титру раствора рассчитывают концентрации веществ, учитывая разбавление раствора при титровании. Затем строят кривую завнсимостн результатов измерений от концентрации.
б) Кондуктометрическим методом определяют зависимость удельной электропроводности растворов указанных веществ от их нормальной концентрации, так же как в п. а). Для этого достаточно провести несколько измерений (8—10). Откладывая по оси абсцисс значения удельной электропровод-
39. Электрохимические методы в количественном анализе 337
ности, строят диаграмму, на которую наносят также и кривые, полученные в п. а).
в) Аналогично пп. а) и б) строят характеристические кривые зависимости измеряемых величин от концентрации и от удельной электропроводности' для растворов NaClOi н Ba(C10i)2 в метаноле и ацетоне. При этом исходят из возможно более концентрированных растворов (примерно 1 М).
На основании полученных данных делают выводы о влиянии подвижности ионов, степени диссоциации и диэлектрической проницаемости на полученные результаты.
2. Проведение ацидиметрического и аргентометрического титрования с осциллометрической индикацией точки эквивалентности.
39.9. Титрование в неводных средах
Если появление первых исследований химических реакций в-, неводных растворах относится к началу столетия, то бурное развитие теории и практики титрования в неводных средах наблюдается лишь в последние два десятилетия. Это находит отражение в быстро растущем числе публикаций. Следует отметить, что препаративное применение растворителей предшествовало их использованию в аналитических целях; оно стимулировало разработку различных теорий кислот и оснований применительно к неводным средам, расплавам солей, а также реакциям кислотно-основного взаимодействия, протекающим в отсутствие растворителей. Развитие теории в свою очередь послужило основой аналитических исследований.
Успехи органической химии привели к синтезу многих новых органических растворителей с большим диапазоном разнообразных свойств, а с развитием лабораторной техники появилась возможность работать с новыми неорганическими растворителями при повышенных и пониженных температурах и бед доступа влаги. Все это позволило в некоторых случаях заменить воду, являющуюся до сих пор универсальным растворителем. Особенно часто воду заменяют другими растворителями при кислотно-основном титровании. Причинами служат плохая растворимость некоторых веществ в воде, что особенно характерно для многих органических соединений; мешающее влияние гидролиза, например, при титровании кислот в присутствии хлоридов или соответственно ангидридов кислот; нивелирующий эффект растворителя, из-за которого невозможно' •проводить дифференцированное титрование сильных кислот или оснований в их смесях; высокая полярность воды, что' исключает возможность диффренцированного титрования карбоновых кислот в их смесях. Применению неводных растворителей способствовало также создание чувствительных и надежных инструментальных методов индикации точки эквивалентности.
Первоначально неводные среды использовали в основном для ацидиметрического титрования органических кислот и ос
22— 1ЕЕО
338
Д. Химический анализ
нований, а также для прямого определения функциональных органических групп. Широкое применение неводные растворители нашли в анализе фармацевтических препаратов. Все успешнее и чаще применяют их также при определении неорганических кислот и оснований. Наряду с этим началась разработка методов окислительно-восстановительного и осадительного титрования в неводных средах. Особенно много новых возможностей появляется в осадительном титровании, так как изменение растворимости неорганических веществ в неводных растворителях по сравнению с водой дает возможность проводить анализ таких соединений, о которых нельзя было и думать при титровании в водной среде. Таким образом удалось существенно расширить область осадительного титрования.
Теория и практика титрования в неводных растворителях в данной книге описана довольно кратко, подробные сведения можно найти в литературе.
39.9.1. Растворители
Растворители, применяемые для неводного титрования, принято делить на четыре основные группы. Эти группы не всегда достаточно четко разграничены, что, как правило, бывает, когда многообразные явления пытаются уложить в определенную схему.
К первой группе, которая стоит несколько особняком, относят инертные (нейтральные) апротонные растворители, в которых нет легко отдаваемых протонов. Диэлектрическая проницаемость их значительно ниже, чем воды, например порядка 1—5. К этой группе относятся, например, бензол, хлорбензол, хлороформ и Др.
Вторая группа охватывает амфотерные (амфипротные) растворители. Подобно воде, они слабо диссоциируют, однако их ионное произведение обычно значительно меньше. Как и вода, они легко отдают протоны, но также легко и принимают их. Диэлектрическая проницаемость их больше, чем растворителей первой группы, но обычно меньше,, чем воды. В качестве примера растворителей этой группы можно назвать спирты.
К третьей группе относят протогенные (кислотные) растворители. Они в основном диссоциированы в большей степени, чем вода, и ионное произведение их выше. Растворители этой группы имеют большую диэлектрическую проницаемость, чем растворители первой группы. Существенным отличием от растворителей второй группы является очень слабо выраженный амфипротный характер. Протогенные растворители очень легко отдают протоны, но принимают их с большим трудом. К протогенным растворителям относятся карбоновые кисло-
39. Электрохимические методы в количественном анализе
339-
ггы, такие, как муравьиная и уксусная, а также безводные жидкие галогеноводороды.
В четвертой группе находятся протофильные (основные) растворители. Они менее диссоциированы, чем вода, и имеют меньшее, а часто и значительно меньшее, ионное произведение. Диэлектрическая проницаемость такая же, как растворителей второй и третьей групп. Эти растворители, так же как растворители третьей группы, обладают слабо выраженными амфотерными свойствами, однако в отличие от них являются хорошими акцепторами' и очень плохими донорами протонов. К этой группе растворителей относятся амины, например этилендиамин, бутиламин и т. д., а также жидкий гидразин или жидкий аммиак.
39.9.2. Кислотность в неводных растворителях
Для характеристики кислотности растворителей Шварценбах предложил ввести нормальный кислотный потенциал. По аналогии с уравнением Нернста для окислительно-восстановительных процессов нормальный кислотный потенциал для соответствующей кислотно-основной пары выражают следующим уравнением:
р — рО I in йкисл °кисл ° КИСЛЧ р 1X1 п
* “осн
Тогда Е^кисл можно измерить как потенциал погруженного в раствор платинового электрода, насыщенного водородом, если активности кислоты и сопряженного с ней основания равны. Это всегда имеет место в- случае чистого растворителя. Растворители, таким образом, можно расположить в порядке возрастания нормальных кислотных потенциалов. Однако при измерении этих потенциалов появляются значительные ошибки, связанные с наличием диффузионных потенциалов на границе раздела растворителей, включенных в измерительную ячейку. Для их устранения электрод сравнения, применяемый при изменении е°кИсл, необходимо заполнять тем же растворителем,, какой находится в измерительной ячейке. Нормальный потенциал каждого обратимого к ионам электрода зависит от энергии сольватации соответствующих ионов, различной для разных растворителей. Поэтому В. А. Плесков предложил в качестве стандарта использовать малополяризуемые ионы с возможно большими диаметрами, такие, как Rb+ или Cs+. Энергия сольватации этих ионов мала и почти не меняется при переходе от растворителя к растворителю. Применив стандартный рубидиевый электрод, Плесков показал, что константы диссоциации сильных кислот в муравьиной кислоте в 10® раз больше, а в безводном' гидразине в 1016 раз меньше, чем в во
22*
340
Д. Химический анализ
де при одинаковых условиях. Однако и такой способ не свободен от ошибок.
В общем было показано, что электрометрические измерения концентрации сольватированных протонов в неводных средах пока еще нельзя проводить с такой же точностью, как измерения в воде. Поэтому на практике большее значение имеет предложенное Л. Гамметом эмпирическое соотношение. Как •меру кислотности он предложил использовать величину Но, рассчитываемую по уравнению1
— Р^нв+ ~Ь Св/Снв+
Для определения диссоциации окрашенных веществ (В) в различных растворителях применяют оптические методы. При этом степень протонирования Св/Снв+ окрашенного вещества в соответствующем растворителе связывают с известной для него в воде величиной рК. Таким образом, речь идет об измерении pH с помощью индикаторов. Применяя набор индикаторов, "можно провести измерения в широкой области. Чтобы отношение значений рК отдельных индикаторов при смене растворителей оставалось постоянным, химические свойства индикаторов должны быть как можно более близкими.
Несмотря на трудности проведения абсолютных измерений и сравнения значений,'полученных в различных растворителях, концепцию pH можно формально перенести на неводные растворители. Рассмотрим диссоциацию растворителя LH:
2LH ===> LH2+ + L-
Ионное произведение и значение pH для него равны1 соответственно
aLH2+ = ^LH
pH = — lg aLH2+ = lg aL-—lg KLH
Тогда величину pH в точке нейтральности определяют из соотношения
Рнп = —г/218 Кнь
Следовательно, значения pH в точке нейтральности для растворителей, характеризующихся неравными константами диссоциации Кнь не будут совпадать и растворы кислот или оснований одинаковой активности в разных растворителях могут иметь разные величины pH относительно точки нейтральности. Например, ионное произведение уксусной кислоты равно примерно 10-13 моль2/(дм3)2, поэтому шкала pH уксусной кислоты составляет 13 единиц и точка нейтральности находится при pH 6,5. Ионное произведение этанола ~ 10-20 моль2/(дм3)2, т. е. шкала pH этанола составляет 20 единиц; точка нейтраль
39. Электрохимические методы в количественном анализе 341
ности находится при pH 10. Для апротонных недиссоциирован-ных растворителей шкала pH соответственно должна быть бесконечно большой.
39.9.3. Нивелирование и протолиз
При растворении кислот и оснований в амфипротных растворителях, т. е. растворителях, которые могут являться как донорами, так и акцепторами протонов, протекает реакция кислотно-основного взаимодействия, которая по теории Бренстеда приводит к частичной нейтрализации. Это можно описать следующими уравнениями реакций:
SH + LH S’+ LH2+ (I)
B + LH 4=4= BH++L- (II)
где LH — растворитель; SH — кислота; В — основание. Если речь идет о сильных в данном растворителе кислотах или основаниях, то равновесие обеих реакций практически полностью сдвинуто в прямом направлении. Вместо сильной кислоты SH образуется катион лиония ЬН2+, а вместо сильного основания В — анион лиата L-. Катион лиония — наиболее сильная кислота в соответствующем растворителе, а анион лиата — наиболее сильное основание. Таким образом, силу кислоты определяют при растворении вещества, исходя из кислотных свойств катиона лиония. Аналогичные рассуждения применимы и в случае оснований, сила которых определяется основными свойствами анионов лиата. Эти положения впервые были даны А. Ганчем, а эффект назван нивелированием. Эффект нивелирования позволяет, например, объяснить давно известный факт выделения одного и того же количества тепла (13,6 ккал/моль) при нейтрализации водных растворов сильных кислот и сильных оснований различной силы. При нейтрализации таких растворов ионы лиония и лиата всегда реагируют с образованием молекул недиссоциированного растворителя:
LH2+ + L- 2LH (для воды Н3О+ + ОН- < -Ь 2Н2О)
Для дифференцирования сильных кислот нельзя применять растворитель, обладающий более сильноосновными свойствами, чем вода, в которой равновесие (I) сдвинуто вправо, а нужно использовать растворитель, обладающий сильнокислотными свойствами и являющийся плохим акцептором протонов, например уксусную, кислоту. Для дифференцированного определения сильных оснований необходимо, наоборот, использовать возможно более сильноосновные растворители. Нивелирующий эффект существенно ограничивает границы применения растворителей, так как при этом можно определить лишь общее
342
Д. Химический анализ
содержание кислот или оснований, но невозможно установить силу различных компонентов.
При нейтрализации образуется раствор соли. Если использовать символы, примененные в уравнениях (I) и (II), можно сказать, что соль содержит ионы S" и ВН+. Соль протежируется растворителем в соответствии с уравнениями реакций
S-+ LH SH + L" (III).
ВН+ + LH «= В + LH2+ (IV)
Для количественного протекания реакции нейтрализации необходимо, чтобы это равновесие было сдвинуто в обратную сторону. Процессы! (III) и (IV) называют также сольволизом (в случае воды протолизом).
Таким образом, каждый амфипротный растворитель применим только для кислоты или основания вполне определенной силы. В противном случае происходит эффект нивелирования или сольволиз. Чем меньше константа диссоциации растворителя, тем' большее число соединений можно в нем определить. С этой точки зрения лучшими растворителями для кислотноосновного титрования должны быть инертные апротонные не-диссоциированные растворители первой группы. Однако эти растворители обычно очень слабо полярны, поэтому растворимость и диссоциация солей в них часто затруднена. Это приводит к незначительной электропроводности растворов и затрудняет электрометрическую индикацию точки эквивалентности.
Исходя из констант равновесия процессов (I) и (II), определяют константу кислотности кислоты или константу основности Кв основания:
„ as“aLH2+ . „ °вн+
ь «SH ь «в
По аналогии с pH отрицательные десятичные логарифмы значений этих констант обозначают pKs или р/<в соответственно. Положение равновесия реакций (I) и (II) зависит, как было указано, от растворителя. Это значит, что рК одной и той же кислоты или одного и того же основания различно в зависимости от растворителя. Было показано, что отношение констант диссоциации или разность значений рК кислот или оснований при переходе от одного растворителя к другому примерно остается неизменным, если кислоты или основания химически родственны. Эмпирически можно определить кислотность растворителя в виде определенной шкалы потенциалов. Предпосылкой успешного потенциометрического титрования, как известно, является достаточно большой скачок потенциала в области точки эквивалентности. При титровании раствора наиболее сильных кислот растворами наиболее сильных оснований в данном растворителе получают максимальный скачок потенциала.
39. Электрохимические методы в количественном анализе
343
При этом скачок потенциала между так называемым потенциалом полунейтрализации (перегиб кривой титрования) и потенциалом, находящимся на расположенном выше горизонтальном участке кривой, служит мерой шкалы потенциалов данного растворителя и оценивается1 с помощью системы! электродов. Потенциал полунейтрализации данной кислоты или ос-
-ЮООн
-500
->500
1
О
>1000
Рис. Д.145. Шкалы потенциалов, измеренные со стеклянным и каломельным электродами в 12 различных растворителях.
Растворители расположены в порядке возрастания их основности: / — трифторуксусная кислота; 2 — уксусная кислота; 3 — хлорбензол; 4 —ацетон; 5 — ацетонитрил; 6 — метанол; 7 — ызо-пропанол; 5 —вода; 9 — диметилформамнд; /0 —пиридин; 11 — бутиламин; 12 — этилендиамии.
нования изменяется незначительно при переходе от одного растворителя к другому, если используют растворители, не обладающие нивелирующим1 эффектом в отношении исследуемых кислот или оснований. На рис. Д.145 приведены шкалы потенциалов, т. е. относительные шкалы кислотности, измеренные с применением стеклянного и каломельного электродов для 12 различных растворителей. Растворители расположены в порядке возрастания их основности: кислые растворители группы 3 (трифторуксусная и уксусная кислоты), инертные растворители группы 1 (хлорбензол, ацетон, ацетонитрил), амфотерные растворители группы 2 (метанол, нзо-пропанол), вода и основные растворители группы 4 (диметилформамнд, пиридин, бутиламин и этилендиамин). Шкалы потенциалов кислых растворителей характеризуются небольшой протяженностью и сильно сдвинуты в «кислую область», так как применяемые кислоты в этой области лишь слабо нивелированы. Инертные
344
Д. Химический анализ
растворители имеют большую протяженность шкалы потенциалов. Это связано с тем, что ни в отношении кислот, ни в Ртношении оснований в этой области нивелирующий эффект не проявляется. Амфотерные растворители в отличие от инертных имеют с обеих сторон четкие пределы шкалы потенциалов. 'Предел шкалы потенциалов основных растворителей в «основной области» определяется основностью гидроксида трибутил-аммония.
39.9.4. Влияние диэлектрической проницаемости
'Большой интерес представляет рассмотрение влияния диэлектрических свойств растворителя на процессы диссоциации и протонирования. По теории Бренстеда возможны^ три случая: SH+ + LH S + LH2+ (а)
SH + LH S- + LH2+ (б)
SH" -Г LH S2- 4- LH2+ (b)
Они соответствуют реакциям катионной кислоты SH+ (а), Нейтральной кислоты SH (б) и анионной кислоты SH- (в). В случае (а) между компонентами реакции не происходит кулоновского взаимодействия, величина которого зависит от диэлектрической проницаемости (ДП) растворителя. В случае (б) и еще в большей степени в случае (в) эти взаимодействия проявляются сильнее; они тем больше, чем меньше значение ДП. Равновесие обеих реакций тем сильнее сдвинуто в прямом направлении, чем больше значение ДП, и наоборот. Из этого следует, что константы кислотности катионных кислот зависят не от значения ДП, а только от основности растворителя. Напротив, кислотность нейтральных и еще в большей степени анионных кислот в растворителях с низким значением ДП меньше, чем в растворителях с высокой ДП, если допустить, 1что кислотность растворителя не изменяется. Если катионная и нейтральная кислоты, находящиеся в смеси, из-за сходства кислотных свойств титруются совместно, то при переходе к фастворителю с другим значением ДП становится возможным 'их дифференцированное титрование. Это правило применимо также и при титровании кислоты по двум ступеням диссоциации. Если растворитель характеризуется низким значением ДП, то кислоту можно нейтрализовать последовательно по жаждой ступени диссоциации, в то время как в растворителе с высокой ДП происходит нейтрализация по двум ступеням одновременно (рис. Д.146). Растворители с небольшими значениями ДП обладают большой склонностью к образованию ассоциатов различных типов, в связи с чем двухступенчатые процессы могут быть кажущимися. Образование растворенными частицами ассоциатов и взаимодействие их с растворите-
39. Электрохимические методы в количественном анализе
345
леи оказывает на структурные и другие специфические свойства веществ влияние, характер которого еще полностью не выяснен.
Незначительная диссоциация сильных электролитов в растворителях с низкими значениями ДП приводит к особенностям в их поведении, которые подробно исследованы на примере уксусной кислоты. Реакцию очень сильной кислоты SH с растворителем LH можно записать следующим образом:
SH-J-LH <7-^: (LH2+-S-) LH2+ + S-
В растворе существует преимущественно средний продукт, который не может образоваться в растворителях с высокой ДП, например в воде. Таким образом, мы получаем две константы равновесия: константу ионизации для первой ступени реакции Л1 и константу диссоциации для второго процесса Kd‘-
fl(LH2+-S~) aSH
KD
aS~ аШ2+ a(LH2+-S-)
Их можно объединить в константу К- Аналогичные зависимо-
сти существуют и для оснований. Для сильных кислот и оснований К практически идентично Ко. Для слабых кислот и оснований K — KiKo-На основании этой зависимости можно предсказать влияние растворителя на кислотность или основность соединений.В среде безводной уксусной кислоты ацетаты, имеющие различные значения Ко, могут диссоциировать как электролиты разной силы, что находит отражение в дифференциации их основных свойств и возможности их раздельного титрования. Так, например, смесь ацетатов калия, натрия и лития в среде смешанного растворителя уксусная кислота + хлороформ (1 : 2С оттитровать раствором НС1О4 в
Рис. Д.146. -Кривые потенциометрического титрования равных количеств янтарной кислоты в среде смешанного растворителя изо-пропанол+вода переменного состава раствором гидроксида тетрабутиламмония.
) можно потенциометрически диоксане с индикаторным стек-
лянным электродом и каломельным электродом сравнения. Первый скачок потенциала соответствует нейтрализации более диссоциированного ацетата калия, следующий — суммы ацетатов остальных щелочных металлов. Ошибка титрования до
346
Д. Химический анализ
—2%. Для перевода в ацетаты галогениды щелочных металлов перед титрованием обрабатывают ацетатом ртути, фториды — ацетатом кальция, сульфаты — ацетатом бария.
39.9.5.
Области применения титрования в неводных средах
39.9.5.1. Кислотно-основное титрование
В первую очередь следует назвать анализ смесей кислот, дифференцированное титрование которых в воде обычно невозможно, так как их значения рК различаются менее чем на четыре единицы (условие для раздельного титрования кислот в воде). Сильные кислоты в воде имеют практически одинаковую силу из-за нивелирующего эффекта воды. Если вместо воды в качестве растворителя взять, например, уксусную кислоту, кислоты в смеси можно оттитровать дифференцированно. Примером может служить дифференцированное титрование азотной и хлорной кислот. В воде невозможно также оттитровать серную кислоту по двум ступеням диссоциации, их всегда титруют суммарно. Однако при потенциометрическом титровании серной кислоты в изо-бутаноле раствором гидроксида тетрабутиламмония происходит последовательная нейтрализация обеих ступеней. Другим' примером является последовательное титрование муравьиной и серной кислоты в метаноле.
Описаны методики дифференцированного титрования серной и соляной, а также серной и азотной кислот в ацетонитриле или в ацетоне. В последнем случае установлено, что небольшие количества воды не мешают титрованию сильных кислот, но оказывают влияние на титрование слабых. При титровании смесей серной и соляной кислот в среде уксусной кислоты раствором ацетата лития можно последовательно оттитровать все три протона: сначала отщепляющегося по первой ступени диссоциации серной кислоты, затем протона соляной кислоты и наконец, отщепляющегося по второй ступени серной кислоты
Соли неорганических кислот в неводных растворителях про являют кислые, основные или нейтральные свойства в зависи мости от их анионов, катионов и применяемого в каждом кон -кретпом случае растворителя. Была сделана попытка распо ложить ионы в ряд в соответствии с уменьшением их кислот ных свойств: Mg2+>Ca2+>Sr2+>Ba2+>Li+>-Na+>>NH4+ = = K+>Rb и C104->I->Br->Cl->N03-.
Для сравнения были взяты соли с разными катионами и одинаковыми анионами, а также соли с разными анионами, но Ю одним и тем же катионом. Исходя из этих данных, соли ам мония и щелочных металлов и различных органических кислот
39. Электрохимические методы в количественном анализе 347
можно титровать в среде уксусной кислоты раствором хлорной кислоты. Соли в этом случае проявляют свойства оснований. С другой стороны, аммонийные соли неорганических кислот можно титровать также в среде этилендиамина или диметил-формамида раствором метилата натрия в смеси метанола и бензола; при этом они проявляют кислотные свойства. Другой метод основан на реакции обмена солей неорганических кислот в неводных средах с серной кислотой: нерастворимые сульфаты выпадают в осадок, а кислоты после их извлечения можно титровать последовательно. Для определения сульфатов их обрабатывают в среде уксусной кислоты раствором ацетата бария, взятым в избытке, и полученный раствор титруют раствором хлорной кислоты потенциометрическим методом.
К другой группе можно отнести часто применяемые титри-,метрические методы, основанные на использовании кислотноосновного взаимодействия в более широком смысле, в соответствии с положениями более новых теорий кислот и оснований, ©то можно пояснить на нескольких примерах. Соединения А1С1з, А1Вг3 и SnCl4 можно титровать в среде тионилхлорида раствором пиридина в тионилхлориде. При этом титрование проводят в присутствии индикаторов кристаллического фиолетового или малахитового зеленого. В других работах описано титрование SnCl4 или TiCl4 в таких растворителях, как РОС1з, SOCb, SO2CI2, AsCU, ацетилхлорид или бензоилхлорид, растворами таких оснований, как хинолин, пиколин или диметил-анилин. Тетрахлорид олова можно оттитровать в хлороформе, тетрахлориде углерода, бензоле или нитробензоле диоксаном, применяемым как основание, с термометрической индикацией точки эквивалентности. Возможно также титрование органических азотсодержащих оснований растворами А1С13 или 6пС14 в среде ацетонитрила с осциллометрической индикацией точки эквивалентности. В бензоле определяют многие кислородсодержащие основания: спирты, кетоны, простые и сложные эфиры, применяя в качестве титранта SnCl4.
При титровании в неводных средах можно использовать также типично «анионотропные» взаимодействия, происходящие, например, при титровании SbCI5, А1С13, SbCl4, SnCl4 или ‘TiCI4 в среде SOCI2 раствором хлорида тетраэтиламмония. При этом образуются высококоординированные анионные хлорокомплексы, фиксируемые потенциометрическим методом. Другие реакции этого типа: взаимодействие TiCl4 в среде РОС13 с раствором хлорида тетраэтиламмония, при котором образуются TiClg~ и Tide2-, а точка эквивалентности устанавливается кондуктометрическим методом1; титрование хлорида тетраэтиламмония в среде SOCI2 раствором SnCl4 или FeCl3 или SbCl5 в среде POCI3 с образованием FeCl4~ или соответственно SbCle-.
348
Д. Химический анализ
Здесь следовало бы упомянуть также титрование, например раствором LiCl, Zn2+ или Cd2+, в кетонах с образованием1 три-или тетрагалогенокомплексов.
39.9.5.2. Методы окислительно-восстановительного титрования
Число работ, относящихся к этой области неводного титрования, значительно меньше. Это объясняется тем, что многие растворители разрушаются при действии сильных окислителей или восстановителей. Если удалось выбрать устойчивый к реакциям окисления — восстановления растворитель, его нужно очень тщательно очистить от примесей, которые могут окисляться или восстанавливаться. Кроме того, оказалось, что методы окислительно-восстановительного титрования смесей в неводных средах не имеют принципиальных преимуществ перед методами их определения в таком доступном’ растворителе, как вода. Несмотря на указанные недостатки, исследования продолжают и в этом направлении. Можно привести некоторые примеры.
В среде безводной уксусной кислоты при использовании в качестве титрантов брома, хромовой кислоты, перманганата калия или трихлорида титана проводят титрование мышьяка, сурьмы, ртути, селена, железа, титана, таллия, бромидов, йодидов, иода и пероксида водорода, г также органических соединений, таких, как резорцин, гидрохинон, бренцкатехин, тетрахлоргидрохинон, /г-хинон, тетрахлорхинон, /г-аминофенол или дифениламин. Точку эквивалентности определяют потенциометрическим методом.
При титровании целого ряда веществ в уксусной кислоте можно использовать также такие сравнительно новые титранты, как монохлорид иода или тетраацетат свинца. Определение иодида в присутствии хлорида и бромида проводят титрованием в среде уксусной кислоты раствором С1О2 в качестве титранта. В серии окислительно-восстановительных титрований в среде уксусной кислоты некоторых окислителей (бром, хромовая кислота, перманганат калия, монохлорид иода, бромат калия и иодат калия) были апробированы в качестве титрантов такие соединения, как дитионат натрия, ацетат ванадила, трихлорид мышьяка или хлорид олова (II).
При потенциометрическом титровании раствором трихлорида титана можно определить в среде диметилформамида бром, иод, пентахлорид сурьмы, СиС12 и ЕеС1з. Для этих целей успешно применяют также раствор дихлорида хрома. Для проведения окислительно-восстановительного титрования в среде уксусной кислоты и ацетонитрила успешно используют раство
39. Электрохимические методы в количественном анализе 349>-
ры Се(IV). В безводном метаноле оказалось возможным определить сумму Ti (III) и Ti (II) титрованием раствором FeCI3.
Перечисление этих методов, конечно, можно продолжить, причем можно было бы назвать особенно большое число методов определения различных органических веществ. Такие методы можно найти в литературе, приведенной в списке.
89.9.5.3. Титрование в расплавах солей
К расплавленным солям как реакционной среде для проведения химических взаимодействий исследователи в настоящее время проявляют повышенный интерес. Многие работы посвящены вопросам использования таких растворителей в препаративных целях. Однако в последнее время описан также целый ряд примеров титрования в этих средах. В расплавах солей, имеющих-небольшую электропроводность, таких, как HgCl2, HgBr2, SbCl.3 и AsBr3, можно определить точку эквивалентности методом кондуктометрии. Для расплавов нитратов щелочных металлов, обладающих высокой электропроводностью, этот метод использовать нельзя. В этих случаях применяют потенциометрию, амперометрию, осциллометрию и термометрию. Кислотно-основное титрование в расплавах можно объяснить с позиций концепции «ионо-тропии». В расплавах проводят также окислительно-восстановительное и осадительное титрование. Приведем некоторые практические примеры.
Оксиды хрома, молибдена и вольфрама растворяют в расплавленном нитрате калия и титруют пероксидом натрия. Определение точки эквивалентности проводят с платииовым индикаторным электродом и кислородным-электродом сравнения. В эвтектическом расплаве нитратов лития и калия можно проводить потенциометрическое и амперометрическое титрование галогенидов и цианидов электролитически полученными ионами серебра. Измерения проводят при 150 °C с серебрянными электродами. В то время как цианиды в расплавах нестабильны, галогениды можно определить с погрешностью 1—2%. Применяя в качестве индикаторного стеклянный электрод, можно в аналогичных условиях получить еще лучшие результаты. Метод потенциометрического титрования в расплаве KNO3 растворами оснований К2СО3, Na2C>2, NaHCO3 и КОН можно успешно применить для определения К2СГ2О7.
89.9.5.4. Осадительное титрование
Ряд определений методами осадительного титрования, проводимых в водных растворах, можно осуществить при добавлении к воде смешивающегося с ней растворителя (для уменьшения растворимости осадка) или при использовании чистого) неводного растворителя в качестве среды для титрования. Примером второго случая может служить титрование сульфата в среде уксусной кислоты раствором ацетата бария или титрование галогенидов и роданидов в среде метанола раствором нитрата серебра.
Не так давно появились работы, связанные с применением осадительного титрования в неводных растворах в тех случаях, когда его нельзя применить в водной среде. При этом исходят из изменения растворимости солей в неводных растворителях по сравнению с растворимостью в воде. Титрант и титруемое вещество должны быть хорошо растворимы в выбранном растворителе, а их ионы должны реагировать с образованием малорастворимого в данном растворителе соединения. Таким способом можно, например, оттитровать в среде уксусной кислоты хлориды, бромиды и роданиды раствором нитрата кадмия; при этом в уксуснокислой среде в отличие от воды образуются нерастворимые хлориды, бромид и роданид кадмия. Аналогично титруют
350
Д. Химический анализ
нитраты в среде уксусной кислоты уксуснокислым раствором ацетата свинца, при этом в осадок выпадает нитрат свинца. В этом случае для индикаций точки эквивалентности применяют метод амперометрии. Если титрование галогенидов в неводной среде отличается от соответствующего титрования в воде только применением другого титранта (раствора нитрата кадмия вместо нитрата серебра), то при титровании нитратов особенно отчетливо про-
Рис. Д.147. Кривые осциллометрического титрования 0,206 мг-экв KSbFe раствором LiCl в различных растворителях.
1 — ацетон; 2 — бутанол; 3 — метил-ызо-бу-тилкетон.
являются возможности осадительного титрования в неводных средах, так как нитраты в воде нельзя оттитровать этим методом.
Растворимые в кетонах соли щелочных и щелочноземельных металлов можно титровать раствором хлорида лития в кетонах, при этом в осадок выпадают нерастворимые в кетонах хлориды щелочных или соответственно щелочноземельных металлов. Особенно хорошие результаты дает использование осциллометрии для индикации точки эквивалентности. Однако ход осциллограммы нельзя объяснить на основе различия в подвижностях ионов, как в случае водных растворов. Из-за низкого значения диэлектрической проницаемости растворителя растворы солей диссоциированы неполностью, и поэтому ход осциллограммы в значительной степени определяется различием степени диссоциации соединений. При титровании солей натрия электропроводность раствора до точки эквивалентности может уменьшаться или возрастать в зависимости от того, является ли образующееся соединение более электропроводным, чем соответствующая соль лития, или менее электропроводным. При титровании одной и той же соли в различных растворителях это влияние диссоциации также может сказаться, что видно из рис. Д.147. Часть диаграммы после точки эквивалентности имеет обычно лишь не-
значительный наклон в зависимости от реализующихся в растворе концентрационных соотношений, так как появляющийся в этом случае в избытке хлорид лития слабо диссоциирован и поэтому вносит лишь очень малый вклад в значение электропроводности. Возможно также проводить раздель-
ное титрование некоторых щелочных и щелочноземельных металлов растворами хлоридов. Например, в ацетоне можно раздельно оттитровать натрий и барий, калий и барий и с нечетким разделением стронций и калий, а также натрий и кальций. Пример такого титрования приведен на рис. Д.148.
39.9.6.
Особенности титрования в неводных растворителях
Наряду с обычными разработанными для водных растворов дравидами проведения точного титрования при титровании в «неводных средах приходится сталкиваться с некоторыми осо
39. Электрохимические методы в количественном анализе 351
бенностями, которые необходимо принимать во внимание. Важнейшие из них следует кратко перечислить. Например, коэффициент расширения растворов титрантов в органических растворителях часто значительно больше (почти в шесть раз!), чем водных растворов титрантов. Поскольку при титровании в воде при 10 °C уже возникает ошибка 0,23%, следует выяснить, какую опасность таит в себе этот источник ошибок. Устойчи-
Рис. Д.148. Кривые осциллометрического титрования смеси Ва12 (0,332мг-экв) с различным количеством KI в ацетоне раствором LiCl.
1 — 0,061 мг-экв KI; 2 — 0,122 мг-экв K.I; 3 — 0,305 мг-экв, KI.
вость характеристик титрованных неводных растворов также хуже, чем водных. Это обычно связано с большей летучестью неводных растворителей и более быстрым поглощением СОг из воздуха. Далее, необходимо принять во внимание тот факт, что градуированная на выливание мерная посуда, например пипетки, была градуирована применительно к водным растворам. При использовании других растворителей вследствие иных, чем у воды, поверхностного натяжения и вязкости могут возникнуть существенные ошибки, которые лучше устранить, градуируя посуду применительно к конкретным условиям работы.
Вопросы осушки и очистки применяемых растворителей, а также техника работы без доступа воздуха рассмотрены в литературе, приведенной в конце книги.
•'352
Д. Химический анализ
Рекомендуемые опыты
1. Титрование смеси серной и азотной кислот. Точно взвешивают при-мерно 10 мг-экв смеси концентрированных кислот, затем осторожно разбавляют, используя 10 см3 воды и дают установиться температуре смеси. Смесь ацетоном смывают в мерную колбу вместимостью 100 см3 и доливают ацетоном до метки. Перемешивание надо проводить осторожно, обращая внимание на установление температуры. 10 ом3 этого раствора в стакане разбавляют 40 см3 ацетона и титруют 0,1 н. бензольным раствором гидроксида трибу-тилметиламмоння. Точку эквивалентности можно определить потенциометрическим титрованием с платиновым и графитовым электродами или визуально. В последнем случае добавляют по 2 капли 0,2%-ного раствора нейтрального красного или 0,1%-ного раствора фенолфталеина в абсолютном метаноле.
Первая точка эквивалентности соответствует суммарной нейтрализации азотной кислоты и H2SOi по первой ступени диссоциации, вторая — нейтрализации суммарного количества кислот.
Отдельно определяют расход раствора титранта на титрование 50 см3 ацетона в присутствии фенолфталеина и вносят соответствующую поправку при титровании смеси кислот. Результаты определения количеств H2SO4 и HNO3 в анализируемой смеси выражают в массовых процентах.
2. Осадительное титрование щелочных н щелочноземельных металлов. Приготавливают небольшое количество насыщенного раствора (2—3 М) безводного L1CI в абсолютном этаноле. Разбавлением этого раствора чистым (нли высушенным и перегнанным) ацетоном в мерной колбе получают примерно 0,1 М раствор титранта. Содержание С1~ в воде определяют аргентометрическим методом.
Кроме того, приготавливают примерно 0,2 н. ацетоновые растворы KI и Ва12. Небольшой объем (несколько см3) этих растворов, содержащих 0,1— 0,3 мг-экв указанных веществ, доливают в стаканах ацетоном до объема 100 см3 и титруют раствором титранта. Индикацию точки эквивалентности лучше проводить осциллометрическим методом, но можно также применить и менее чувствительный кондуктометрический метод. В конце титруют смесь обоих компонентов.
40.
СПЕКТРОСКОПИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
40.1. Общие основы и краткий обзор методов
'Спектроскопия занимает ведущее положение среди современных инструментальных методов анализа. В спектральных методах используют различные формы взаимодействия электромагнитного излучения с веществом для определения структуры соединений, свойств атомов и молекул, для качественного обнаружения и количественного анализа веществ. В этой главе дан краткий обзор спектроскопических методов анализа и подробно рассмотрены наиболее важные из них.
Структура спектра молекулы вещества определяется ее внутренней энергией. Энергия молекулы в свою очередь складывается из трех составляющих: энергии движения электронов, энергии колебания атомов и энергии вращения молекулы:
^общ £эл + £коЛ + £вр
40. Спектроскопические методы анализа 353
При поглощении молекулой электромагнитного излучения могут происходить возбуждение электронов, колебательные и вращательные переходы. При этом энергия электронов изменяется довольно значительно по сравнению с колебательной, а вращательная — в меньшей степени:
> ^КОЛ > Евр
При изменении вращательной энергии возникают спектральные линии, расположенные в длинноволновой инфракрасной и в микроволновой областях (Х>50 000 нм). Изменение колебательной энергии обычно связано с одновременным изменением энергии вращения. При этом получают колебательно-вращательный спектр (X от 1000 до 50 000 нм). Изменения энергии электронов связаны с двумя другими составляющими энергии, поэтому полосатый электронный спектр особенно сложен. Он охватывает видимую и ультрафиолетовую области (X от 50 до 1000 нм). Теоретическая интерпретация этих спектров дана в разд. 6.1.
Колебательно-вращательный спектр называют также инфракрасным спектром. Такие спектры очень разнообразны, особенно в случае свободных молекул (в газах при уменьшенном давлении). Разрешающая способность обычного спектрального прибора слишком мала для разделения индивидуальных линий, вызванных вращательными переходами. При повышении давления или при конденсировании фаз эти линии исчезают, так как продолжительность существования отдельного вращательного состояния настолько сильно изменяется при соударениях молекул, что наблюдается уширение и перекрывание линий. Спектры в ближней инфракрасной области 1(Х от 1000 до 50 000 нм) обусловлены колебаниями атомов. При этом, различают колебания вдоль валентных связей атомов (валентные) и колебания с изменением валентных углов '(деформационные). Колебания возникают, если поглощение электромагнитного излучения связано с изменением1 направления и величины дипольного момента молекул. Поэтому молекулы, состоящие, например, из двух атомов, не могут давать инфракрасные спектры. Симметричные валентные колебания молекул СОг также нельзя возбудить абсорбцией света. Отдельные группы атомов в молекулах больших размеров дают специфические полосы поглощения, которые практически не зависят от строения остальной части молекулы. Этот факт используют для идентификации таких групп. В симметричных молекулах колебания одинаковых групп энергетически равноценны и поэтому вызывают появление одной полосы поглощения. По такому упрощению1 ПК-спектра можно сделать вывод о строении поглощающих излучение молекул. ПК-спектроско-23—1880
354
Д. Химический анализ
пию используют в основном в количественном анализе органических веществ.
Электронные полосатые спектры расположены в видимой и ультрафиолетовой областях. При поглощении света возбуждаться могут только внешние, сравнительно непрочно связанные с ядром атома электроны. Как и в случае колебательновращательного спектра, при повышении давления газа происходит наложение колебательных и вращательных переходов. Если энергия возбуждения достаточна для диссоциации молекулы, то в полосатом спектре наблюдается уширение линий. На использовании электронных полосатых спектров основаны очень важные для количественного анализа методы колориметрии и фотометрии, которые будут подробно рассмотрены ниже.
Если возбужденная молекула при излучении возвращается на значительно более низкий колебательный энергетический уровень, возникает спектр флуоресценции. По правилу Стокса длина волны флуоресценции больше длины волны возбуждающего излучения или, по крайней мере, имеет такую же величину. Продолжительность возбужденного состояния при этом составляет примерно 10-8 с, поэтому флуоресценция наблюдается практически одновременно с возбуждением. Иногда излучение может быть значительно более длительным (в пределах нескольких секунд). Такое явление называют фосфоресценцией. При достаточно большом давлении газа или в конденсированной фазе электроны в возбужденном состоянии из-за взаимных столкновений часто переходят на самый нижний энергетический колебательный уровень, прежде чем произойдет излучение энергии. Спектр флуоресценции характеризует колебательную структуру основного состояния электронов, спектры поглощения преимущественно отражают колебательную структуру возбужденного состояния. Поэтому полосы флуоресценции часто являются зеркальным отражением полос поглощения.
При когерентном, рассеянии света молекулами, описываемом законом Рэлея (см. уравнение (467)), часть энергии излучения переходит в энергии вращательного и колебательного состояния молекул. Поэтому в спектре рассеянного света наряду с частотой линии возбуждающего света наблюдаются линии с большими и меньшими частотами, соответствующие выделению и поглощению энергии молекулами. Поскольку при комнатной температуре преобладает основное колебательное состояние, происходит только поглощение энергии. Линии получаемого таким образолг спектра комбинационного рассеяния 1(КР) часто значительно сдвинуты по сравнению с линиями падающего на вещество света в сторону больших длин волн. В то время как ИК-спектр связан с изменением дипольного момента молекул, появление линий в КР-спектре связано с изменением поляризуемости молекул. Поэтому линии спектра
40. Спектроскопические методы анализа 355
.комбинационного рассеяния можно получить в том случае, когда не возникает ПК-спектр, например для двухатомных молекул газов. Таким образом, методы дополняют друг друга. Области применения К.Р- и ПК-спектроскопии аналогичны.
। Для малорастворимых твердых веществ можно получить отражательный спектр. При интенсивном измельчении твердого вещества уменьшается часть светового потока, отражающаяся от его поверхности, а большая часть падающего света проникает в глубь вещества. Эта доля частично поглощается, а частично, после многократного отражения снова диффузно выделяется через поверхность вещества наружу. При таком •«внутреннем» отражении ослабляются участки спектра, связанные с абсорбцией света молекулами. Для дальнейшего уменьшения поверхностного отражения порошкообразное вещество можно смешать с веществом, индифферентным в используемой спектральной области (белый стандарт), и получить известную аналогию с раствором вещества. Отражательная спектроскопия пригодна также для получения спектров поглощения малорастворимых веществ. Этот метод применяют в основном при исследовании состава красок и строения неорганических твердых соединений. Абсорбция света окрашенными катионами зависит от различных факторов: от координационного числа, симметрии молекулы и межатомных расстояний в кристаллической решетке соединения. По изменению' абсорбции можно сделать выводы об изменениях, происходящих в решетке соединения при включении посторонних ионов.
40.2. Поглощение света
и закон Бугера — Ламберта — Бера
Спектроскопические методы используют в химии для определения строения вещества, для качественного и количественного анализа. Для решения первых двух задач снимают спектры поглощения или излучения в сравнительно большом диапазоне длин волн. Для проведения количественного анализа, напротив, выбирают небольшой участок спектра и, сравнивая интенсивности излучения анализируемого вещества и стандартов по градуировочной кривой, определяют количество какого-либо компонента.
При прохождении монохроматического светового потока с интенсивностью I через поглощающее свет гомогенное вещество интенсивность его ослабляется в каждом слое вещества dx на постоянную величину. Поэтому
dljdx =—al и dljl =—adx (459)
При интегрировании получаем, что при прохождении светового потока через слой с конечной толщиной х начальная интенсив
23*
356
Д. Химический анализ
ность /0 уменьшается до значения /. Отсюда
In I/Io = —ах; I = 1^~ах (460), (461)
Часто это выражение приводят в логарифмической форме, тогда из уравнения (460) следует
lg I/Io — —а'х (462)
где o'—коэффициент поглощения, связанный с а (а'=0,434а).
Величину 1g ///0 называют оптической плотностью, поглощением (экстинкцией) поглощающего вещества Е*, а отношение/До— прозрачностью или пропусканием раствора 6.
В итоге можно написать
1g /До — lg = Е = — а'х (463)
Эту закономерность первым сформулировал Бугер, а позже Ламберт дал ее математическое выражение.
На поглощение существенное влияние оказывают длина волны падающего света и природа поглощающего свет вещества. При постоянной длине волны падающего света оптическая плотность Е является постоянной для данного слоя вещества. Как следует из уравнения (463), оптическая плотность прямо пропорциональна толщине слоя, в то время как коэффициент поглощения а' не зависит от толщины слоя абсорбирующего вещества.
В смесях, в которых лишь некоторые компоненты поглощают свет данной длины волны, коэффициент поглощения может быть пропорционален концентрации с абсорбирующего вещества (закон Бера):
а' — ес (464)
где е — молярный коэффициент погашения; если с=1 моль/дм3, то а' = Е. ?
Закон Бера справедлив, если молекулы поглощающего вещества не взаимодействуют между собой или с молекулами растворителя (т. е. не происходят процессы диссоциации, ассоциации, сольватации, конденсации, полимеризации), что зависит ®т концентрации веществ и оказывает влияние на свето-поглощение. Для равновесного состояния системы
А + Н2О В поглощение раствора складывается из двух составляющих:
Е — еАСА4“ЕвСв
Поэтому оптическая плотность уже не пропорциональна изме
* В литературе обычно принято называть светопоглощением величину 1g (7о/Л. — Прим, перев.
40. Спектроскопические методы анализа 357
нению концентрации. Пропорциональность могла бы иметь место только при равенстве коэффициентов поглощения обоих компонентов (изобестическая точка). Отклонения от закона могут быть вызваны также немонохроматичностью падающего света. Для очень разреженных газов ошибки обусловлены появлением в спектре вращательных линий и перекрыванием их при росте концентрации. Следует подчеркнуть, что независимо от указанных мешающих влияний закон Бера справедлив для сильно разбавленных растворов. Но если концентрация раствора с<10“2 моль/дм^, ошибка определения ниже границы недостоверности результатов прецизионных светоэлектрических измерений (0,01%). Это связано с тем, что коэффициент преломления среды также зависит от концентрации. При необходимости количественного определения больших концентраций веществ надо построить предварительно градуировочную кривую для серии растворов определяемого вещества с известными концентрациями. При объединении уравнения (463) и /464) получают выражение закона Ламберта — Бера:
1g///0 = £ = —есх или lg/0// = ecx (465), (466)
Можно показать также, что этот закон справедлив и для не слишком мутных коллоидных растворов, в которых тиндале-во рассеяние света мало. Если при работе с такими растворами наблюдается отклонение от закона Ламберта — Бера, следует изменить число и размеры коллоидных частиц. Таким образом, закон Ламберта — Бера можно при определенных условиях применить для определения концентраций коллоидных растворов.
При прохождении света через коллоидные растворы часть света рассеивается под определенным углом. Это явление называют эффектом Фарадея — Тиндаля. Рэлей показал, что оно имеет место в том случае, когда свет попадает на частицу, размер которой мал по сравнению с длиной волны падающего света. Рэлей установил следующую закономерность, которую используют для определения концентраций веществ в тин-далиметре:
I = Io const (zw2/X4) sin2 а (467)
где ?о — интенсивность первичного излучения, падающего на объект; г — число коллоидных частиц; и — их объем; Л. — длина волны света; а—угол отклонения первичного излучения, при котором измерена интенсивность /.
40.3. Абсорбционная спектрофотометрия
При практическом .использовании приведенных выше закономерностей необходимо учитывать поглощение света средой, в которой находится определяемый компонент, а также стенками
Я
358
Д. Химический анализ
применяемого сосуда (кюветы). Это осуществляют, сравнивая интенсивность светового потока, прошедшего через анализируемый раствор с интенсивностью светового потока, прошедшего при таких же условиях через аналогичный сосуд с чистым растворителем (/о). Большинство фотоэлектрических приборов в настоящее время имеет две шкалы: шкалу показаний в процентах степени пропускания, т. е. (/До) -100, и шкалу показаний в 1g /о /I-
Рис. Д.149. Зависимость поглощения от длины волны (кривая поглощения).
Рис. Д.150. Зависимость поглощения от концентрации.
Как было указано, закон Ламберта — Бера справедлив только для монохроматического света. Различные молекулы и группы атомов имеют специфичные полосы поглощения. Для определения характеристического поглощения частиц одного вида в растворе измеряют оптическую плотность при разных длинах волн и получают так называемую кривую поглощения (рис. Д.149). Длину волны, при которой на кривой наблюдается максимум, обозначают как Xmax, а измеренное при этом поглощение как Дтах. Количественное определение, проведенное при /-max, характеризуется большой селективностью и чувствительностью.
Для определения применимости закона Ламберта — Бера в условиях анализа поступают следующим образом: при ^тах измеряют оптическую плотность ряда растворов с различной концентрацией определяемого компонента и представляют ее как функцию концентрации (рис. Д.150). Линейная зависимость указывает, что к данной системе закон Ламберта — Бера применим. Эту диаграмму в дальнейшем можно использовать как градуировочный график для определения неизвестной концентрации частиц. По углу наклона прямой можно определить величину молярного коэффициента погашения.
40. Спектроскопические методы анализа
359
40.3.1. Спектрофотометры
Устройство спектрофотометра определяется областью спектра, в которой проводят измерения. Но основными узлами всех приборов являются следующие: источник света, монохроматор, ячейки для раствора пробы и раствора сравнения, детектор. Свет от источника излучения попадает через узкую щель в монохроматор. Здесь излучение направляется с помощью вогнутого зеркала на призму или решетку, где разлагается в спектр. Выходная щель выделяет пучок монохроматического света определенной длины волны. Нужная длина волны создается поворотом призмы или решетки. Монохроматический свет проходит далее через кюветы, которые должны пропускать свет выбранной длины волны, не разрушаться применяемыми растворителями и веществами и быть совершенно идентичными. Энергию излучения, прошедшего через раствор, обычно преобразуют в детекторе в электрический сигнал, который можно усилить.
Простые спектрофотометры обычно делают однолучевыми. Рассмотрим порядок работы на приборах этого типа. Вначале устанавливают выбранную длину волны. Затем компенсируют темновой ток, т. е. показания прибора, появляющиеся до включения оптической части прибора. Кювету с раствором сравнения устанавливают на пути светового потока и, открыв шторку, пропускают через раствор монохроматический свет. Проводят компенсацию, выводя стрелку гальванометра на нуль. Это означает, что для раствора сравнения установлено 100% пропускания. Затем раствор сравнения заменяют исследуемым раствором и стрелку гальванометра опять выводят на нуль. Теперь значение оптической плотности можно определить по шкале. Поскольку предварительно поглощение чистого растворителя было скомпенсировано, измеренную величину можно рассматривать как оптическую плотность анализируемой пробы.
Для построения кривой поглощения меняют длины волн и при каждой длине волны компенсируют поглощение чистого растворителя, так как оно также зависит от частоты падающего света. При проведении измерений в большом диапазоне длин волн и с большой частотой измерений построение кривой требует значительных затрат времени. Этого можно избежать, применяя двухлучевые спектрофотометры, в которых монохроматический свет делится на два потока одинаковой интенсивности. Один из них проходит через раствор сравнения, другой — через анализируемый раствор, после чего световые потоки попадают на два не связанных друг с другом детектора. Возникает сигнал разбаланса, который подается на сервомотор, управляющий движением оптического клина. Клин перемешается на пути светового потока, падающего на раствор
360
Д. Химический анализ
сравнения, до выравнивания интенсивностей обоих потоков. Диапазон перемещения клина можно согласовать со шкалой пропускания или поглощения прибора и зафиксировать изменение этих величин как функцию длины волны.
Можно упомянуть о применении спектрофотометрии при решении аналитических проблем органической химии. Для этих целей можно работать в инфракрасной, ультрафиолетовой, а также в видимой части спектра.
40.3.2. Фотометрическая индикация точки эквивалентности при титровании
Во многих классических методах анализа индикация точки эквивалентности проводится по характерным изменениям окраски. При этом можно использовать изменение окраски добавляемых индикаторов или самих компонентов, участвующих в реакции. В последнем случае окраску может иметь определяемый компонент или титрант, окрашивающий титруемый раствор тогда, когда реакция закончена и титрант находится в избытке.
В спектрофотометрии устраняются субъективные ошибки, зависящие от наблюдателя, измерение проводится объективно и точно. Спектрофотометры дают возможность записать ход кривой титрования и найти точку эквивалентности. Надежные результаты получаются и в случае визуально трудно различаемой окраски. Возможность фотометрической индикации точки эквивалентности не только в видимой части спектра существенно расширяет границы применимости метода.
В индикаторном варианте метода вначале снимают кривые поглощения для обеих окрашенных форм индикатора. В фотометре устанавливают оптимальную длину волны, выбранную по максимуму одной из кривых, причем выбирают такую длину волны, при которой поглощение второй формы индикатора минимально. При проведении титрования в этом случае наблюдается резкое увеличение или уменьшение поглощения в точке эквивалентности в зависимости от того, длина волны какой формы индикатора была установлена в приборе.
Если в ходе кислотно-основного титрования измерить значение оптической плотности в зависимости от концентрационного соотношения обеих форм индикатора и затем построить график зависимости Е от объема раствора титранта, то можно получить прямую, пересекающую ось абсцисс в точке эквивалентности.
В безиндикаторном варианте метода длину волны выбирают по максимуму поглощения определяемого компонента или титранта, тогда при титровании до точки эквивалентности происходит линейное возрастание или уменьшение оптической плот-
40. Спектроскопические методы анализа
361
пости, а после точки эквивалентности поглощение постоянно. Точку эквивалентности определяют как точку пересечения двух прямых аналогично, например, кондуктометрическому титрованию. Как правило, следует вводить поправку на разбавление раствора в процессе титрования, но при работе с концентрированными растворами титранта этой поправкой можно пренебречь.
Применяемые растворители должны обладать хорошей пропускающей способностью в спектральной рабочей области прибора. Ошибка, связанная с присутствием в растворе посторонних веществ, тем больше, чем меньше рабочая длина волны. Преимуществом метода является возможность использования его для серийных определений в одинаковых условиях. Метод фотометрического титрования достаточно полно описан в литературе. В нем можно использовать не только реакции кислотноосновного взаимодействия, но и окисления-восстановления, комплексообразования и осаждения. Метод осадительного фотометрического титрования называют также гетерометрией.
40.4. Колориметрия
Буквальный перевод слова колориметрия — измерение окрашивания. Концентрацию окрашенного вещества определяют, сравнивая интенсивность его окрасок в двух смешанных фазах (обычно растворах), в одной из которых концентрация вещества известна. Растворитель в обеих фазах должен быть одним и тем же.
Дальнейшее развитие метода привело к возможности количественного определения компонентов мутных коллоидных растворов и суспензий. Если при этом измеряют ослабление интенсивности света, прошедшего через коллоидный раствор, т. е. мутность раствора, метод называют турбидиметрией. Можно измерять и интенсивность рассеянного света. Для этой цели используют прибор, действие которого основано на эффекте Тиндаля — так называемый тиндалиметр. Этот метод называют нефелометрией.
Интенсивность флуоресценции, возникающей при облучении некоторых веществ, используют в методе флуориметрии.
Все перечисленные методы можно рассматривать как варианты колориметрии. В литературе, однако, это не является общепринятым, и термин колориметрия все еще применяют для обозначения методов определения, связанных со сравнением оптических свойств растворов одного и того же вещества, но разной концентрации. Фотометрами называют все приборы, снабженные так называемыми серыми клиньями, измерительными диафрагмами или фотоэлементами. Такие приборы не имеют принципиальных различий, различаясь лишь техникой
362
Д. Химический анализ
измерений. Колориметрию можно рассматривать как упрощем ный вариант описанного выше метода абсорбционной спектрсЗ фотометрии. i
40.4.1. Техника проведения измерений
В простейшем случае в визуальных колориметрах или нефелометрах сравнивают окраску или мутность исследуемого раствора и ряда растворов сравнения с известными концентрациями. Условия измерений, естественно, должны быть одинаковыми.
Поскольку в некоторых случаях нельзя получить устойчивые стандартные растворы, содержащие анализируемый компонент, готовят стандартные растворы других окрашенных веществ с идентичными оптическими свойствами или применяют окрашенные стекла.
Расчеты проводят, исходя из закона Ламберта — Бера (уравнение (466)). Для двух растворов одного и того же вещества в одном и том же растворителе с равной степенью пропускания 1/10 или с равной оптической плотностью Е справедливо следующее соотношение:
cvx{ — сгхг (468)
(Коэффициент молярного погашения е — величина постоянная для конкретного вещества.) Если известна концентрация раствора сравнения Ci и толщина поглощающего слоя %i этого раствора, то, меняя толщину х2 поглощающего слоя анализируемого раствора, можно добиться равенства оптических плотностей обоих растворов и, следовательно, можно рассчитать с2 по уравнению (468).
Если подаваемый снизу свет проходит через два цилиндрических сосуда с растворами, а наблюдение осуществляют сверху, то толщину поглощающего слоя можно менять, выливая раствор из градуированного цилиндрического сосуда через находящийся внизу кран до тех пор, пока пропускание света в цилиндрах не уравняется. Этот принцип с некоторыми усовершенствованиями лежит в основе схемы погружного колориметра Дюбоска (рис. Д.151). В нем сосуды с определяемым и стандартным растворами можно передвигать вертикально, фиксируя их положение по измерительной шкале. Стеклянные по-гружатели, представляющие собой массивные плоскопараллельные пришлифованные стеклянные стержни или стеклянные цилиндры с закрытым торцом, погружают на различную глубину в растворы, меняя тем самым толщину поглощающего слоя. В колориметре с клином (колориметр Аутенрита — Кенигсбер-гера) раствор сравнения находится в клинообразной кювете. Через нее пропускают свет параллельно основной поверхности. Поднимая или опуская клин, можно варьировать толщину по-
40. Спектроскопические методы анализа
363
глотающего слоя и фиксировать это изменение. Во всех перечисленных приборах оба световых луча получают от одинаковых источников света; после прохождения через раствор лучи объединяются в одном окуляре, где каждый из них освещает половину поля зрения окуляра. При работе добиваются одинаковой освещенности обеих половин.
В приборах другого типа (фотометр) раствор сравнения не нужен. Свет от источника излучения пропускают через ослабля-
ющее световой поток устройство и, регистрируя его, добиваются совпадения его интенсивности с интенсивностью света, прошедшего через анализируемый раствор. Для ослабления светового потока сравнения можно использовать так называемый серый раствор, поглощающий всегда одинаковую часть проходящего через него видимого света (какой бы длины волны этот свет ни был), аналогично действующий серый клин или регулируемую диафрагму (в фотометре Пульфриха). При этом, разумеется, нужно строить градуировочную кривую зависимости поглощения от концентрации анализируемого раствора.
Перечисленные приборы
Рис. Д.151. Схема погружного колориметра Дюбоска.
1 — окуляр; 2 — зеркало; 3 — вертикально перемещаемые сосуды для растворов; 4 — луч света; 5 — измерительная шкала.
можно использовать с незначительными усовершенствованиями как тиндалиметры. Нужно только обеспечить возможность боковой подачи падающего света. В погружных колориметрах это достигается зачернением дна стеклянных цилиндров с растворами.
Визуальное наблюдение (уравнивание освещенности участков поля зрения окуляра) дает точность определения ~1%. Применение фотоэлектроколориметров повышает точность определения до 0,1%. Эти приборы не очень дорогие, поэтому их широко применяют в последнее время. Достоинствами фотоэлектроколориметров являются возможность их использования для серийных анализов (при визуальных наблюдениях быстро наступает усталость глаз), пригодность для автоматизации и непрерывного контроля производства. Применение фотоэлементов в приборах дает возможность работать в ультрафиолетовой и инфракрасной областях спектра.
364
Д. Химический анализ
Рассмотрим некоторые принципиальные схемы фотоэлект! роколориметров (рис. Д.152). На рис. Д.152,а приведена схем! фотометра с непосредственным отсчетом показаний. Сопротиа ление R служит для регулирования чувствительности прибора! Кювету, заполненную вначале чистым растворителем, помеща* ют в прибор и выводят стрелку гальванометра в крайнее положение по шкале. Затем проводят градуировку прибора, заполняя кювету последовательно растворами определяемого ве-
Рис. Д.152. Схемы основных типов фотоэлектроколориметров (см. текст).
L — источник света; Z — фотоэлемент; К — кювета с раствором, поглощающим излучение; G — гальванометр; 7? — сопротивление; В — батарея,- 5 — зеркало; D— диафрагма; М — модулятор.
щества известных концентраций. При этом по шкале регистрирующего прибора (гальванометра) определяют результаты измерений: по линейной шкале-—степень пропускания, по логарифмической шкале — непосредственно оптическую плотность. Градуировочную кривую строят в координатах Е — с. По ней можно определить концентрацию раствора анализируемого вещества.
В отличие от рассмотренной схемы в фотоэлектроколориметре (рис. Д.152, б) гальванометр применен в качестве нуль-инст-румента. С помощью потенциометра сопротивлений напряжение батареи регулируют таким образом, чтобы оно компенсировало напряжение фотоэлемента, а гальванометр при этом показывал нуль. Такая схема дает возможность использовать более чувствительный гальванометр, снижает «усталость» фотоэлемента, связанную с проведением бестоковых измерений. Значения измеряемых величин определяют по сопротивлению.
40. Спектроскопические методы анализа
365
Недостатками обеих схем является зависимость результатов измерений от флуктуаций интенсивности источника света и длины волны падающего света.
Фотоэлектроколориметр, схема которого представлена на рис. Д.152, в, снабжен двумя кюветами, двумя фотоэлементами и одним источником света и может работать как по принципу прямого отсчета, так и по компенсационному методу. Компенсацию можно осуществить, как в предыдущей схеме, или с помощью серого клина, диафрагмы или же изменением расстояния от источника света до фотоэлементов. Достоинством схемы является независимость результатов измерений от флуктуаций интенсивности источника света, а недостатком — необходимость применения двух фотоэлементов, которые никогда не бывают полностью идентичными. Независимость показаний прибора от изменений интенсивности падающего света связана с тем, что в отличие от первых двух схем здесь предусмотрено одновременное измерение поглощения раствора сравнения и анализируемого раствора.
Наиболее удачной является четвертая схема (рис. Д.152,г). Свет от источника излучения через систему зеркал с помощью модулятора попеременно подают на две кюветы и затем на фотоэлемент. При одинаковом поглощении света растворами в обеих кюветах на фотоэлемент попадает постоянный поток света, при разном поглощении — переменный. В этом случае его нужно преобразовать в постоянный с помощью устройства, ослабляющего световой поток (диафрагма). Фотоэлемент служит нуль-индикатором и поэтому не оказывает влияния на точность измерений. Такой метод называют методом с модуляцией светового потока.
40.4.2. Практическое применение колориметрии
Неокрашенные анализируемые вещества с помощью химических реакций переводят в окрашенные соединения, после чего их можно определять колориметрическими методами. Вследствие высокой чувствительности колориметрию целесообразно применять при определении содержания следовых количеств элементов, хотя точность определения часто невысока. Как видно из рис, Д.150, чувствительность определения зависит от значения молярного коэффициента погашения. Поэтому молярный коэффициент погашения окрашенного соединения должен быть как можно большим.
Одним из известных примеров практического применения колориметрии является определение следовых количеств марганца в железных рудах или сплавах на основе железа или других металлов. Большинство соединений других ионов так
366
Д. Химический анализ
же можно перевести в окрашенные соединения. При этом часто возникают проблемы специфичности определений, так как не только определяемый компонент, но и ионы других соединений, присутствующих в растворе, могут образовывать окрашенные соединения. Лишь в отдельных случаях существует возмож-<| ность выбора специфичного реагента. Поэтому необходимо со-? здавать такие условия, чтобы реакция была специфичной на определяемый ион, или удалять из пробы мешающие определению ионы. Первые колориметрические анализы проводили, используя избирательные реагенты, образующие окрашенные соединения с определяемыми ионами, например реактив Несслера для обнаружения аммиака, нингидрин для определения а-аминокислот и молибденовую жидкость для анализа фосфатов (образование молибденовой сини фосформолибденовой кислоты) .
Другие реакции имеют более широкий диапазон применения. Например, малорастворимая в воде хлораниловая кислота, растворы которой интенсивно поглощают свет в зеленой области спектра, образует осадки с такими катионами, как кальций, стронций, барий и цирконий. Уменьшение оптической плотности раствора при образовании осадков можно использовать для определения катионов. Этот реагент пригоден и для колориметрического определения анионов. Например, малорастворимый хлоранилат бария в присутствии следовых количеств сульфата переходит в нерастворимый в воде сульфат бария, а эквивалентное количество хлораниловой кислоты переходит в раствор. Содержание ее можно определить по увеличению светопоглоще-ния раствора. Аналогично можно проводить анализ хлоридов и фторидов в растворе, используя хлоранилаты ртути или лантана.
Диэтилдитиокарбаминат диэтиламмония
[(C2H5)2NCSS]~[(C2H5)2NH2]+ реагирует с ионами многих металлов, например с Zn, Си, Pb, Hg, Ag, Ni, Со, Al, Мп, Сг и Mg с образованием окрашенных комплексных соединений. Водный раствор соединений металлов встряхивают с раствором реагента в хлороформе при pH 7,5—8. При этом Zn, Си, Cd, Pb, Hg, Ag, Ni и Co переходят в хлороформ, в то время как ионы остальных металлов остаются в водной фазе. Фазы разделяют. Используя амфотерные свойства соединений цинка, его можно извлечь из хлороформа действием щелочного раствора цитрата натрия и затем определить колориметрическим методом.
Специально для колориметрического анализа переходных металлов синтезирован ряд специфических органических комплексообразующих реагентов. Общеизвестным реагентом на никель является диметилглиоксим. Для определения меди оказался пригоден оксалил-Н,Н-бис(Н'-циклогексилиденгидразид). Другими наиболее часто применяемыми реагентами являются
40. Спектроскопические методы анализа
367
8-оксихинолин, дитизон, рубеановодородная кислота, дитиол и нитрозо-/?-соль.
Более подробные сведения о рассмотренных в этом разделе методах можно найти в литературе.
Рекомендуемые опыты
1. Проводят фотометрическое титрование сильной кислоты раствором сильного основания в воде. Строят кривые поглощения кислотной и основной форм применяемого индикатора, например нейтрального красного. Для обеих форм при выбранном значении /.тах строят градуировочную кривую зависимости поглощения стандартных растворов от концентрации. Определяют соотношение концентраций обеих форм индикатора в ходе титрования по ряду измеренных значений оптической плотности раствора и строят зависимость этих значений от объема титранта.
2. Железо(Ш) образует с роданид-ионом комплексный ион, окрашенный в интенсивно красный цвет, что дает возможность проводить колориметрическое определение Fe3+, Для построения градуировочной кривой готовят основной раствор: точно взвешивают 50 г железных квасцов, переносят в мерную колбу вместимостью 100 см3, добавляют примерно 5 см3 концентрированной соляной кислоты марки ч.д.а. (не содержащей железа) для подавления протолиза и доливают дистиллированной водой до метки.
Готовят 1 М раствор роданида аммония, растворяя соль марки ч.д.а. в 250 см3 воды.
Разбавлением основного раствора в колбах вместимостью 100 см3 готовят серию стандартных растворов известных концентраций, добавляя в каждую колбу по 5 см3 конц. НС1. Отбирая, например, по 10 см3 каждого' стандартного раствора, смешивают его с 10 см3 раствора роданида аммония, тщательно перемешивают и точно через 5 мин измеряют оптическую плотность раствора. В качестве раствора сравнения используют 1 М раствор роданида аммония, не содержащий ионов железа. Строят градуировочную прямую зависимости оптической плотности от концентрации железа. После измерения поглощения анализируемого раствора по градуировочной прямой находят концентрацию железа в пробе.
40.5. Флуориметрия
Причины возникновения флуоресценции были кратко описаны выше. Использование этого явления в анализе основано на способности некоторых веществ флуоресцировать под действием ультрафиолетового излучения. Число таких веществ невелико, так что метод вначале в основном применяли для анализа органических веществ и в меньшей степени для неорганических. В последние годы метод успешно применяют и для чувствительного определения многих неорганических соединений.
Анализ выполняют на простых приборах, называемых флуориметрами. В приборе находится источник излучения, с помощью которого можно возбудить флуоресцентное свечение молекул вещества. Исследуемый раствор помещают в кювету из кварцевого стекла. Поскольку источником возбуждения обычно служит ультрафиолетовое излучение, применять кюветы из обычного стекла нельзя, так как они не пропускают ультрафио-
368
Д. Химический анализ
летовых лучей. Излучение флуоресценции измеряют под пря-( мым углом к направлению возбуждающего излучения. Такой же принцип используется в установках для нефелометрии, поэтому описанные приборы можно использовать в обоих методах анализа. Различие заключается только в следующем: при проведении нефелометрических определений на вещество попадает видимый свет, а ультрафиолетовое излучение задерживают светофильтром, в то время как в методе флуориметрии^ для возбуждения флуоресценции применяют ультрафиолетовое
излучение, а длинноволновое излучение задерживают светофильтром. В последнем случае на пути излучения флуоресценции перед детектором помещают еще один светофильтр, не пропускающий ультрафиолетовое излучение, и прибор показывает интенсивность более длинноволновой флуоресценции.
На интенсивность флуоресценции оказывает влияние ряд факторов. Но если проводят измерения в одних и тех же условиях и на одном и том же приборе, то по градуировочной кривой (зависимость интенсивности излучения от концентрации) можно определить содержание вещества, как и в колориметрии. Для очень разбавленных растворов зависимость между интенсивностью флуоресценции и концентрацией определяемого вещества прямолинейна.
Дальнейшим развитием флуориметрии стал метод спектро-флуориметрии. Взаимосвязь этих методов такая же, как методов колориметрии и абсорбционной спектрофотометрии. В методе спектрофлуориметрии используют более сложное оборудование, что дает возможность регистрировать спектр флуоресценции. Кроме того, можно селективно выбрать определенную длину волны флуоресценции.
Зависимость интенсивности флуоресценции F от концентрации выражается формулой
F = (70-7)<p
(469)
где /о — интенсивность возбуждающего света; I — интенсивность прошедшего через раствор света; ф — квантовый выход флуоресценции (часть энергии поглощенного излучения преобразуется в тепловую энергию, прежде чем произойдет флуоресценция) .
Применение закона Ламберта — Бера дает
Г=70(1—е~£“)ф
(470)
Для очень разбавленных растворов, используемых в методе флуориметрии, зависимость принимает вид
F~(]oecx)q> (471)
Из этого выражения можно сделать два очень важных для количественного анализа вывода; значения 70 и х можно поддер
40. Спектроскопические методы анализа
369
живать постоянными, а е и <р остаются постоянными, если концентрация вещества в растворе изменяется незначительно и среда одна и та же. При этих условиях F линейно зависит от с, и, кроме того, с увеличением /0 растет F, т. е. чувствительность флуориметрии линейно возрастает с увеличением интенсивности возбуждающего излучения. В этом отношении флуо-риметрия имеет преимущество по сравнению с колориметрией и абсорбционной спектрофотометрией, где проводят измерение разности I—/о- Поэтому флуориметрию часто можно применять для определения таких концентраций веществ, которые нельзя определить методом абсорбционной спектрофотометрии.
40.5.1. Применение флуориметрии
Не касаясь широких возможностей применения метода флуориметрии в анализе органических веществ, кратко рассмотрим ее значение в неорганическом анализе. Прекрасным примером использования флуориметрии в анализе неорганических веществ является определение следовых количеств бора, например в стали. Пробу растворяют, бор переводят в борную кислоту, которую затем отгоняют с метанолом обычным способом. Затем борную кислоту нейтрализуют гидроксидом натрия. Метанол испаряют и остаток растворяют в этаноле. К спиртовому раствору добавляют бензоин и точно через две минуты измеряют интенсивность флуоресценции. Линейная зависимость интенсивности флуоресценции от концентрации соблюдается до содержания бора примерно 100 мкг в 50 см3 раствора. При более высоких концентрациях возрастание интенсивности флуоресценции становится меньше. Следует учесть, что в лабораторном стекле содержится бор, поэтому следует применять кварцевую и платиновую посуду.
Аналогичные чувствительные флуориметрические методы анализа предложены и для других веществ. Можно привести еще один пример: соли урана выпаривают с азотной кислотой и затем сплавляют с фторидом натрия. Расплав затвердевает в стекловидную массу, которую флуориметрируют. Чувствительность определения урана в этом случае составляет 0,005 мкг урана в 1 г твердой пробы.
40.6. Эмиссионный спектральный анализ
40.6.1. Принцип метода, чувствительность и точность
Эмиссионный спектральный анализ позволяет проводить качественное обнаружение и количественное определение всех металлов и ряда неметаллов. Преимуществом метода являются его быстрота и чувствительность определения при крайне незначительном расходе анализируемого вещества.
При проведении анализа небольшую пробу анализируемого вещества переводят в парообразное состояние и возбуждают. Возникающее излучение (эмиссию) разлагают в спектр, который регистрируют на фотопластинке или фотоэлектрическим способом. По положению спектральных линий можно идентифицировать элементы, содержащиеся в анализируемой пробе, а по интенсивности линий определить количественные соотно
24—1880
370
Д. Химический анализ
шения элементов. Следует отметить, что идентификация спектра неизвестной пробы, содержащей ряд элементов, представляет собой сложную задачу и требует больших затрат времени по сравнению с другими методами анализа. Расшифровка спектров значительно облегчается при проведении серийных количественных анализов проб одинакового качественного состава, что очень часто встречается в практике аналитического контроля на производстве.
Чувствительность обнаружения разных элементов неодинакова. Кроме того, она, естественно, связана с чувствительностью используемого прибора. Обычно этот метод можно применить для обнаружения концентраций веществ порядка 10~2%, а часто даже менее 10_3 %.
Для обеспечения правильности количественных определений необходимо применять точные фотометрические методы измерения интенсивности линий спектра. При этом минимальная погрешность определения обычно составляет ±2%. Часто получают большую погрешность определения ±5% и даже , ±10%.
Пример. Если истинное содержание компонента анализируемой пробы равно 30%, то при использовании спектрографического метода, дающего погрешность 10%, мы получим результат 27-—33%. Однако при содержании определяемого компонента 0,030% получим доверительный интервал 0,027— 0,033% н можем при этом достоверно' определить, что содержание компонента составляет 0,03%.
Из рассмотренного примера ясно, что эмиссионный анализ следует применять при определении следовых количеств элементов в пробе. Это справедливо также для всех методов, обладающих высокой чувствительностью и сравнительно небольшой точностью определения (см., например, полярографию).
40.6.2. Возбуждение излучения
Наиболее простым и давно применяемым источником возбуждения эмиссии является пламя, его использовали еще в ручном спектроскопе при проведении качественного анализа. В настоящее время пламя применяют для точных количественных определений содержания щелочных и щелочноземельных металлов в растворе в методе фотометрии пламени. Поскольку температура в зонах пламени неодинакова, возбуждающая способность этих зон также различна. Количественная опенка интенсивности излучения возможна только при работе с очень равномерным пламенем, при исключительно равномерном распределении анализируемого раствора в пламени и использовании для возбуждения одной и той же зоны пламени.
Значительно более сильное возбуждение происходит в дуговом разряде, температура которого достигает 5000°C. При
40. Спектроскопические методы анализа
371
этих условиях практически все вещества переходят в парообразное состояние. За исключением небольшого числа элементов (галогены, сера), атомы в дуговом разряде переходят в возбужденное состояние с последующим излучением света. Электрический дуговой разряд возникает между угольными электродами, на один из которых нанесена анализируемая проба, под действием постоянного или переменного тока. Для изготовления электродов применяют уголь высокой чистоты во избежание мешающего влияния примесей. Влияние этих примесей, естественно, надо учитывать при получении конечного результата.
Значительно большую энергию возбуждения дает конденсированная искра. Включение в колебательный контур индуктивного и емкостного элементов обусловливает возможность получения искрового разряда. Размеры конденсатора и катушки самоиндукции, а также соотношение процессов разрядки и заряжения конденсатора определяют энергию искрового разряда и тем самым интенсивность спектральных линий и долю атомов, существующих в высоковозбужденном состоянии.
При расшифровке спектров можно различить линии, возникающие при возбуждении электронейтральных атомов, однократно ионизированных атомов (первичный ионный спектр) или двукратно ионизированных атомов (вторичный ионный спектр). Для возбуждения спектров нейтральных атомов достаточно энергии дугового разряда, поэтому эти спектры раньше упрощенно называли дуговыми. В отличие от них спектры цонов, обычно возбуждаемые действием конденсированной искры, называли искровыми спектрами. Имея определенные формулы серий (см. разд. 5.1.3), можно установить взаимосвязь атомных и ионных спектров, описываемую спектроскопическим законом смещения А. Зоммерфельда и В. Косселя, который гласит, что спектр, испускаемый нейтральными атомами какого-либо элемента, подобен спектру, испускаемому однократно ионизированными атомами элемента, стоящего за ним в Периодической системе, а также спектру, испускаемому двукратно ионизированными атомами элемента, стоящего через один элемент за ним в Периодической системе.
40.6.3. Разложение излучения в спектр и регистрация спектра
Разложение излучения в спектр проводят в спектральных приборах. В данной главе невозможно подробно остановиться на конструкции всех выпускаемых промышленностью приборов, которые можно отнести к двум группам: призменные спектральные приборы и приборы с диффракционными решетками. В основе такой классификации лежит способ разложения излучения
24*
372
Д. Химический анализ
в спектр: с помощью призмы или пропусканием его через щели диффракционной решетки с последующей интерференцией. Применяемые в спектрографах второго типа диффракционные решетки могут быть пропускающими и отражательными. Приборы обоих типов равноценны по эффективности действия.
Спектры регистрируют на фотопластинках. Четкость почернения отдельных линий спектра зависит от свойств светочувствительной эмульсии, от способов проявления, фиксирования и сушки фотопластинок. Поскольку все эти параметры трудно поддерживать постоянными, для количественной оценки спектры анализируемых веществ регистрируют на пластинке совместно со спектрами эталонов.
Почернение фотопластинки (S) определяют фотометрирова-нием. Оно выражается логарифмом отношения интенсивности светового потока, прошедшего через незасвеченную часть пластинки (/о) к интенсивности светового потока, прошедшего через зачерненную часть пластинки (/):
S-lg/0// (472)
Чтобы по значению почернения сделать надежные выводы о количественном содержании вещества, необходимо между плотностью почернения и интенсивностью светового потока (Г) наличие прямой пропорциональной зависимости:
S = fl' (473)
При графическом построении этой зависимости оказывается, что прямая пропорциональность обычно наблюдается только для интервала значений S 0,5—2. Почернения аналитических линий должны находиться в этой области.
Вместо фотопластинок все большее применение для регистрации спектров находят фотоэлементы, фотоэлектронные умножители или полупроводниковые элементы. Также и в этих случаях, естественно, должна соблюдаться линейная зависимость между величиной сигнала и интенсивностью спектральной линии в широкой области значений.
40.6.4. Значение последних линий в спектре
Поскольку спектры многих элементов состоят из большого числа линий, при анализе многокомпонентных проб получаются спектры с едва заметным различием линий отдельных компонентов. Поэтому при расшифровке спектра следует ограничиться наблюдением нескольких наиболее заметных линий, остальными линиями можно пренебречь. Для количественных определений применяют так называемые последние линии. Это наиболее интенсивные линии излучения атома данного элемента, которые последними исчезают в спектре при постепенном
40. Спектроскопические методы анализа
373
уменьшении концентрации элемента в пробе. Последние линии определяют для каждого случая анализа экспериментальным путем, поскольку их появление зависит от способа испарения пробы, вида источника света, светочувствительности пластинки и типа матрицы.
Для идентификации спектров в продаже имеются специальные наборы стандартов, позволяющие идентифицировать спектры 50 элементов. Эти стандарты содержат такое количество элемента, что в его спектре возникает только одна или две последние линии. Количественное определение элементов в анализируемой пробе значительно упрощается при фотографировании искрового спектра такого образца рядом со спектром определяемого вещества. Подробности метода описаны в специальной литературе.
40.7. Фотометрия пламени
Фотометрию пламени в узком смысле можно рассматривать как метод эмиссионной спектроскопии. Окрашивание пламени, возникающее, например, при внесении летучих солей щелочных и щелочноземельных металлов в пламя, издавна используют для целей качественного анализа. Но визуальным методом можно определить окрашивание пламени только в видимой части спектра и невозможно разложить смешанную окраску на составные цвета, а интенсивность окраски можно оценить лишь очень приблизительно. В фотометрии пламени измеряют интенсивность излучения и при определенных условиях используют зависимость ее от концентрации веществ, вызывающих окрашивание пламени.
При распылении анализируемого раствора в пламя происходят следующие процессы. Растворитель испаряется, и образуется аэрозоль твердые частицы — газ. Затем происходит частичное испарение частиц и диссоциация их на нейтральные атомы. Некоторые атомы вступают в реакции с другими компонентами, находящимися в пламени. Часть атомов в пламени возбуждается; при возвращении в исходное невозбужденное состояние атомы излучают свет.
Температура пламени ниже температуры дугового и искрового разряда, поэтому вероятность перехода электронов на более высокий энергетический уровень мала и интенсивность соответствующих спектральных линий невелика. В пламени, как правило, получают линейчатые спектры. Обычно в спектре появляются только резонансные и основные линии (соответствующие электронным переходам с первого возбужденного уровня на основной), которые являются наиболее интенсивными. Это и есть последние линии спектра. При подводе большого количества энергии к атому электроны могут даже удалиться из
374
Д. Химический анализ
атома. При этом образуются положительно заряженные ионы) которые также могут возбуждаться. Однако их спектр отличается от спектра возбужденного нейтрального атома. Температура пламени хотя обычно и достаточна для образования ионов, все же не вызывает их возбуждения.
Кроме линий, соответствующих возбуждению нейтральных атомов, в пламенных спектрах часто наблюдаются линии и полосы молекул и радикалов. При этом возможны наложения спектров, обусловленных процессами возбуждения электронов, и спектров, связанных с изменениями колебательного и вращательного движения атомов в молекулах. Возникающие при этом полосатые спектры уже нельзя разрешить самыми чувствительными приборами. Иногда их можно использовать для целей количественного анализа (например, интенсивную полосу излучения радикала СаОН или молекулы СаО).
Само пламя также дает излучение. Например, голубое окрашивание водородно-кислородного пламени вызвано возникающим при горении радикалом ОН. Пламена углеводородов обладают интенсивным свечением, обусловленным наличием радикалов ОН, СН, НСО и с2.
Температура пламени светильный газ — воздух составляет около 1700°C. Такой температуры достаточно для возбуждения атомов примерно 15 элементов и в первую очередь щелочных и щелочноземельных металлов. Таким образом, область применения пламенной фотометрии ограничена температурой пламени. Поэтому предложено использовать целый ряд пламен (табл. Д.22). Наивысшую температуру имеет пламя смеси дициан-(-кислород. Однако из-за ядовитости дициана и других его недостатков это пламя применяют сравнительно редко. С помощью высокотемпературных пламен можно возбудить
Таблица Д.22. Температура пламен
Газовая смесь
Температу-
ра, °C
Водород+воздух 2100
Водород+кислород 2750
Ацетилен + воздух 2150
Ацетилен+кислород 3100
Метан+воздух 1950
Метан-(-кислород 2730
Этилен+воздух 1895
Пропан-(~воздух 1925
Пропан 4-кислород 2775
Светильный газ + воздух 1840
Светильный газ+кислород 2000
Дициан-|-кислород 5000
40. Спектроскопические методы анализа
375
эмиссию атомов свыше 50 элементов Периодической системы и определить их методом фотометрии пламени.
Существуют пламенные фотометры двух типов. Простейший состоит из приспособления для распыления раствора в пламя, фильтра излучения и фотоэлемента, соединенного с гальванометром (рис. Д.153). С помощью этого прибора можно определять щелочные и щелочноземельные металлы с погрешностью ±2%. Высокая точность анализа объясняется тем, что атомы
5 6
7 8
Рис. Д. 153. Принципиальная схема простого пламенного фотометра.
/ — анализируемый раствор; 2—подача кислорода нли сжатого воздуха; 3 — подача горючего газа; 4 — смесительная камера; 5 — зеркало; 6 — пламя; 7 — линза; 8 — фильтр;
9 — фотоэлемент; 10 — прибор для измерения силы тока.
этих элементов легко возбуждаются в пламени и их характеристические линии четко разделяются в спектрах при использовании специальных фильтров. Пламенную фотометрию особенно часто применяют для определения щелочных и щелочноземельных металлов в биологических растворах, пищевых продуктах и удобрениях.
Для пламенно-фотометрического определения элементов, возбуждение которых происходит в высокотемпературных пламенах, а также для повышения точности определения необходимо применять более сложные приборы. В этом случае необходимо, чтобы в определенной области длин волн не происходило наложения линий определяемого элемента и линий других элементов. Для такого разрешения фильтров недостаточно, его можно достичь только с помощью монохроматора. Кроме того, из-за небольшой интенсивности получаемых линий следует применять детектирующее устройство с усилителем.
376
Д. Химический анализ
Правильная количественная оценка интенсивности излучения зависит от целого ряда факторов, на которые необходимо обращать внимание. Прежде всего это влияние свойств раствора. При увеличении вязкости и поверхностного натяжения распыление раствора в пламя происходит хуже, а интенсивность излучения уменьшается, н наоборот. Чем больше теплота испарения растворителя, тем сильнее он охлаждает пламя. Это особенно характерно для водных растворов. Органические растворители, напротив, имеют незначительную теплоту испарения и при сгорании выделяют дополнительное количество энергии. Поэтому при их использовании температура пламени всегда выше, чем в случае водных растворов, что значительно повышает интенсивность эмиссии. Особенно эффективно сочетание экстракции ионов органическими растворителями с последующим определением методом фотометрии пламени. Таким образом определяют железо в сплавах не на железной основе или в известняке. Следовые количества железа экстрагируют из подкисленного раствора ацетилацетоном. Интенсивность излучения железа в присутствии ацегилацетона значительно выше, чем в водном растворе.
Интенсивность излучения элемента зависит от зоны пламени, так как температура, а также распределение различных атомов, ионов, молекул и радикалов по зонам пламени неодинаково. Наконец, состояние химических равновесий процессов, протекающих в зоне пламени, также влияет на интенсивность излучения. Эти процессы очень сложны — в них участвуют продукты сгорания и остатки молекул горючих газов, продукты диссоциации и горения растворителя и растворенных в нем веществ.
Растворенные вещества могут мешать определению по двум причинам. Одна из них — так называемый анионный эффект. В его основе лежит образование малолетучих и слабодиссоциированных соединений между анионами и определяемыми катионами, что оказывает существенное влияние иа концентрацию возбуждаемых в пламени атомов. Хорошо известно влияние боратов, сульфатов, фосфатов, а также алюминия, образующего термически устойчивые алюминаты, на точность количественного анализа щелочноземельных металлов. Напротив, хлориды и нитраты определению щелочноземельных металлов не мешают. Иногда и между катионами происходят специфические взаимодействия, вызывающие уменьшение или увеличение интенсивности излучения. Влияние катионов можно устранить, добавляя предварительно большой избыток мешающих определению компонентов, так что изменение их содержания в анализируемом растворе прн этом уже не будет иметь значения. С другой стороны, чувствительность обнаружения определяемого элемента при этом снижается.
Интенсивность излучения при характеристической частоте в правильно выбранной области длин волн пропорциональна концентрации элементов с учетом поправки на фон. Фоновое излучение возникает из-за присутствия в пламени других возбужденных частиц, излучающих в области частот, выбранных для проведения анализа. Так как при этом происходит возбуждение не только свободных атомов или ионов, но и радикалов и возникновение вращательных и колебательных спектров недиссоциированных молекул, излучение фона наблюдается во всех областях частот. Поэтому нужно определять интенсивность излучения фона в условиях, в которых проводят анализ пробы, в отсутствие определяемого компонента и величину интенсивности фона вычитать при получении конечных результатов. Концентрацию определяемого компонента находят по градуировочной кривой, отображающей зависимость интенсивности излучения от концентрации.
40. Спектроскопические методы анализа 377
Основная причина отклонения от линейной зависимости — самопоглоще-ние, которое особенно сильно влияет на интенсивность резонансных линий. Атомы возбуждаются в горячей части пламени, однако их излучение поглощается атомами этого же вещества, находящимися в невозбуждеином состоянии во внешней, более холодной части пламени. Степень самопоглощения зависит от толщины более холодной зоны пламени. С увеличением диаметра пламени, который определяется конструкцией горелки, возрастает отклонение градуировочного графика от линейности.
Содержание компонентов можно определить не только по градуировочному графику, но и другими способами. В так называемом методе добавок к анализируемому раствору добавляют точно известное количество определяемого компонента с концентрацией, близкой к его концентрации в анализируемом растворе. Измеряя интенсивность излучения анализируемого раствора с добавкой и без добавки, можно после внесения поправки на излучение фона с помощью простой пропорции рассчитать неизвестную концентрацию определяемого компонента. Это возможно лишь в том случае, если зависимость интенсивности от концентрации в данной области линейна. Помехи можно исключить, если они не связаны с концентрацией определяемого компонента.
В методе внутреннего стандарта к анализируемому раствору добавляют известное количество другого элемента. Интенсивность его излучения сравнивают с интенсивностью излучения определяемого элемента. Для количественного расчета необходимо знать фактор пропорциональности f. Его вычисляют по значениям интенсивностей, измеренным при известных концентрациях обоих элементов:
А /4 = ЛтА-г
В методе устраняются помехи от присутствия посторонних элементов, поскольку они оказывают одинаковое влияние на оба элемента. То же относится и к влиянию различных температур пламени и способа распыления раствора в пламя.
Все помехи можно надежно устранить, если приготовить ряд стандартных растворов такого же состава, как анализируемые, с предполагаемым набором концентраций определяемого компонента и построить градуировочную кривую. Однако при этом необходимо провести полный анализ пробы, что целесообразно только в случае серийных анализов, как, например, при определении кальция и магния в цементе.
Предел обнаружения методом фотометрии пламени составляет 0,002—5 мкг/см3. Для щелочных металлов этот метод наиболее чувствителен из всех существующих методов их определения, за исключением радиохимических. Это справедливо также для кальция и стронция, если отсутствует анионный эффект. Определению меди, серебра, галлия, индия и таллия почти не мешают другие компоненты, поэтому фотометрию пламе-
378
Д. Химический анализ
ни рекомендуют применять для их определения. Воспроизводи-! мость результатов при использовании эффективно действую-1 щих приборов лучше чем 0,5%. 1
40.8. Атомно-абсорбционная спектрофотометрия
В методе фотометрии пламени измеряют интенсивность излучения атомов, возбужденных в пламени, поэтому более правильно было бы называть этот метод атомно-эмиссионной спектрофотометрией. Но можно измерять и поглощение (абсорбцию) излучения свободными атомами, находящимися в пламени в невозбужденном состоянии. Такой метод называют атомно-абсорбционной спектрофотометрией и используют его для определения концентрации атомов путем определения поглощения излучения. Таким образом, оба метода дополняют друг друга. Между находящимися в пламени возбужденными атомами и атомами в основном состоянии существует следующее соотношение:
(474)
где Л/а — число возбужденных атомов; No — число атомов, находящихся в основном невозбужденном состоянии; ga и g0 — статистические веса обоих состояний; Еа — энергия возбуждения. Для доли возбужденных частиц а поэтому можно написать
a = -gbe-£a/^ (475)
Эта доля очень мала и увеличивается только при высоких температурах и небольших значениях Еа (большая длина волны возбуждающего излучения). В табл. Д.23 приведены значения а для резонансных линий ряда элементов при различных температурах. Наиболее яркие резонансные линии для большинства элементов соответствуют длинам волн <600 нм. При температуре пламени ~ 3000 °C значение а все еще очень мало.
Таблица Д.23. Значения величины а для некоторых резонансных линий при различных температурах
Резонансные линнн, нм Температура, К
2000 3000 4000 5000
Cs 852,1 4,5- К)”4 7,2-10-3 3,0-10-2 6,8-Ю-2
Na 589,0 9,9-10-6 5,9-10-4 4,4-Ю-3 1,5-10-2
Са 422,7 1,2-IO’7 3,7-Ю~5 6, (И (Г* 3,3-10~3
Zn 213,9 7,3-10-16 5,6-1О-10 1,5-Ю-7 4,3-Ю*6
40. Спектроскопические методы анализа
379
В еще большей степени это характерно для атомов, обладающих высокой энергией возбужденного состояния. Таким образом, можно считать, что Na с изменением температуры изменяется по экспоненциальному закону, a No остается практически постоянным (из-за небольшого значения Na). При пропускании через пламя излучения определенной интенсивности и с характеристической для определяемого элемента резонансной частотой излучение поглощается невозбужденными атомами определяемого элемента пропорционально их концентрации и независимо от температуры пламени. Условием этого является совпадение частоты падающего излучения с частотой характеристического резонансного излучения поглощающих атомов или незначительное расхождение между ними. Чем больше диапазон частот возбуждающего излучения, тем ниже чувствительность метода.
Если используют источники непрерывного излучения, такие, как вольфрамовые лампы, то даже при выделении излучения определенной частоты с помощью эффективно действующего монохроматора возникают заметные ошибки определения, связанные с немонохроматичностью падающего излучения. Поэтому для каждого элемента необходим специальный источник излучения, что является недостатком метода. Но с другой стороны, это же обусловливает меньшее влияние посторонних элементов, чем в методах эмиссионного анализа.
В качестве источников излучения, специфичных для атомов различных элементов, обычно применяют газоразрядные трубки с полым катодом. Цилиндрический полый катод изготавливают из элемента, резонансное излучение которого должно быть возбуждено; работу проводят при напряжении 400 В и силе тока 100 мА. В качестве материала катода иногда используют сплавы, тогда получают резонансные частоты излучения ряда элементов в одной трубке; например, сплавы меди, цинка и свинца можно использовать для одновременного определения этих трех элементов. Однако при этом существует возможность изменения состава сплавов на поверхности катода из-за неравномерного испарения и, как следствие, изменение интенсивности излучения наиболее летучего компонента.
На рис. Д.154 приведена принципиальная схема установки атомно-абсорбционного анализа. Для увеличения поглощения обычно применяют вытянутое в длину пламя. Резонансное характеристическое излучение определяемого элемента возбуждают с помощью источника света. После этого излучение попадает в пламя, проходит через монохроматор и регистрируется. Чувствительность метода зависит от частоты резонансного характеристического излучения, а также в значительной степени от интенсивности возбуждающего резонансного излучения.
380
Д. Химический анализ
В методе атомно-абсорбционной спектрофотометрии используется поглощение излучения атомами, находящимися в пламени в невозбужденном состоянии, в отличие от фотометрии пламени, где необходимо термическое возбуждение атомов. Поэтому атомную абсорбцию можно использовать для определения содержания таких элементов, излучение которых нельзя возбудить в пламени, что является преимуществом метода. Область применения метода атомно-абсорбционной спектрофотометрии тем самым значительно шире, чем фотометрии пламени. По-
Рис. Д.154. Принципиальная схема атомно-абсорбционного спектрофотометра. 1 — анализируемый раствор; 2 —подача кислорода или сжатого воздуха; 3 — подача горючего газа; 4 — смесительная камера; 5 — спектрофотометр; 6 — пламя; 7 — кварцевое окно; 8 — полый катод.
скольку концентрация невозбужденных атомов в пламени гораздо больше, чем возбужденных, можно было бы предположить, что методы, связанные с измерением абсорбции, принципиально чувствительнее, чем методы, основанные на измерении эмиссии. Однако это неверно, так как в последних непосредственно измеряют сигнал, а в первых — разность интенсивностей. Проведенные исследования показали, что пламенно-фотометрический метод определения щелочных и щелочноземельных металлов значительно чувствительнее атомно-абсорбционного определения, правда, доля возбужденных частиц в пламени достаточно велика. В других случаях нельзя дать однозначного ответа о преимуществах того или иного метода, что подтверждают данные табл. Д.24.
Концентрация свободных атомов в невозбужденном состоянии не меняется существенно при появлении какой-то доли возбужденных атомов, и, казалось бы, температура пламени не должна оказывать на нее влияния, однако косвенно это влияние проявляется в том, что состояние равновесия процессов диссоциации соединений в пламени зависит от температуры. Поэтому все факторы, оказывающие влияние на кон-
40. Спектроскопические методы анализа
381
Таблица Д.24. Сравнение чувствительности методов атомно-абсорбционной и эмиссионной спектроскопии®
Чувствительность ААС по сравнению с ЭС
выше равная ниже
Au 0,5 Ag 0,1 Ba 5,0
Cd 0,05 Co 0,5 Ca 0,1
Mg 0,01 Cr 0,2 К 1 0,1
Mo 1,0 Cu 0Д Li 0,05
Pb 1,0 Fe 0,5 Na 0,05
Pd 1,0 Ni 0,3
Pt 2,0 Ti 0,1
Sn 5,0
Zn 0,03
а Приведены концентрации элементов в водных растворах (в мкг/см3), при которых происходит 1%-иое поглощение. ААС — атомио-абсорбционная спектроскопия; ЭС — эмиссионная спектроскопия.
центрацию свободных атомов в пламени, действуют так же, как в методе фотометрии пламени, например также наблюдается анионный эффект.
Если атом, находящийся в парообразном состоянии, излучает после поглощения резонансного излучения, то возникает атомная флуоресценция. На использовании этого явления основан метод атомно-флуоресцентной спектрофотометрии.
Более подробные сведения об атомно-абсорбционном анализе можно найти в литературе*.
Рекомендуемые опыты
1. Определение содержания натрия и кальция в водопроводной воде.
а) Приготовление стандартных растворов. Сначала готовят 0,1 М растворы NaCl и СаС12. Применяют реактивы марки ч.д.а. Для приготовления раствора СаС12 можно растворить СаСОз в соляной кислоте. Путем разбавления 0,1 М растворов готовят стандартные растворы следующих концентраций:
NaCl 5-10~3М; 2-10~3М; 10"3 М; 5-(10~4 М; 2-10~4 М; 10~4 М
СаС12 IO-2 М; 5-10-3 М; 2-103-М; 10-'1 М; 5-10-4М
Растворение ведут в деминерализованной воде, которую сохраняют в сосуде из пластмассы.
б) Выполнение измерений. Для определения натрия используют линию 589 нм, а для кальция — полосу поглощения оксида кальция при 624 нм.
* См. например книгу: Львов Б. В. Атомно-абсорбционный анализ.—М.: Наука, 1966. — Прим, перев.
382
Д. Химический анализ
Пламя водородно-кислородное, турбулентное; скорость испарения 2 см3 водного раствора в минуту.
Строят градуировочный график зависимости интенсивности излучения от концентрации. Затем измеряют интенсивность излучения при обеих длинах волн для неразбавленной водопроводной воды и по графику определяют содержание Na и Са.
При высокой жесткости воды большое содержание кальция мешает определению натрия. Однако такое влияние аддитивно и линейно связано с концентрацией кальция, поэтому можно использовать следующий прием. Измеряют излучение раствора соли кальция при 624 нм и при 589 нм (линия натрия). Соотношение интенсивностей излучения при этих длинах волн постоянно. Затем измеряют излучение анализируемого раствора при 624 нм, определяют долю эмиссии кальция при длине волны 589 нм и вычитают ее из ранее измеренной интенсивности, при этом получают интенсивность излучения натрия.
2. Определение концентрации сульфат-ионов, вызывающей помехи при определении содержания кальция.
Измеряют излучение кальция при длинах волн 624 нм (молекулярная полоса) и 423 нм (атомная полоса), распыляя в пламя 10-2 М раствор СаС12 в смесн с растворами H2SO4 с концентрацией 10-3, 10 2, 10-1 и 2-10~1 М. Строят зависимость интенсивности излучения от содержания сульфата в растворе.
Мешающее влияние сульфат-ионов становится заметным, когда их содержание в десять раз превышает содержание кальция в растворе. Интенсивности излучения больше при длине волны 624 нм, но симбатны при обеих длинах волн. Из диаграммы можно, определить избыточное количество сульфат-ионов, которое оказывает мешающее влияние при определении кальция. Сульфат-ионы также можно предварительно удалить с помощью ионного обмена.
41. РАДИОХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Многие химические проблемы можно решить с помощью радиохимических методов исследования. Некоторые из этих проблем интересны для аналитических целей, например определение растворимости малорастворимых осадков, исследование соосаждения и адсорбции. В данной главе рассматриваются только радиохимические методы определения и индикации.
Основы явлений радиоактивности и радиоактивного распада были рассмотрены выше (разд. 4). Частота распада любого радиоактивного нуклида характеризуется определенной константой, так называемым периодом полураспада, который характеризует время, необходимое для того, чтобы исходное количество радиоактивных нуклидов уменьшилось вдвое. Периоды полураспада для известных радиоактивных нуклидов лежат в широком диапазоне — от миллионных долей секунды до миллионов лет. Предельные значения периодов полураспада можно измерить лишь косвенным методом, поэтому их не используют в аналитических целях. Поскольку на каждый анализ затрачивается определенное время, нуклиды с весьма коротким временем жизни за время аналитических операций могут практи
41. Радиохимические методы анализа
383
чески полностью распасться. Поэтому в анализе можно использовать нуклиды с периодом полураспада не менее нескольких часов. Существует ориентировочное правило, согласно которому продолжительность анализа должна не более чем в 10 раз превышать период полураспада применяемого изотопа. Применение нуклидов со слишком большим временем жизни создает трудности, так как частотность их распада мала. В целом периоды полураспада применяемых нуклидов находятся в пределах 1000 лет.
41.1. Техника работы с радиоактивными веществами
Обычно в радиохимических методах применяют вещества с небольшой радиоактивностью. При этом используют обычную технику проведения химических работ с учетом двух особенностей: необходимости защиты от вредных для здоровья излучений и необходимости работы с очень малыми концентрациями веществ.
Поражение радиоактивным излучением может происходить при попадании радиоактивных веществ в организм или при внешнем его облучении. Прежде всего возможность поражения возникает при работе с долгоживущими нуклидами, а также тогда, когда соответствующие вещества могут накапливаться в организме. Так, например, 90Sr, накапливаясь в костях, препятствует образованию в крови красных кровяных шариков. Особенно опасно воздействие у-излучения. Напротив, а- и |3-ча-стицы легко поглощаются и поэтому имеют небольшую длину пробега. Если работа с веществами, активность которых лежит в области порядка милликюри, ведется в стеклянных сосудах, то вредное действие этих частиц уже сводится к минимуму. Труднее осуществить защиту от нейтронного излучения. Его можно ослабить слоем парафина или воды толщиной 10—15 см. В общем интенсивность любого излучения обратно пропорциональна квадрату расстояния от источника излучения до облучаемого объекта. Поэтому работу проводят на максимально возможном удалении от источника излучения и за возможно более короткий промежуток времени.
При проведении анализа часто применяют экстремально малые количества активных веществ, например порядка 10~12 моль/дм3. При этом приходится сталкиваться с тем, что поведение одного и того же вещества в растворе может быть различным в зависимости от его концентрации. В случае экстремально малых концентраций радиоактивных веществ в растворе может, например, не выпасть осадок, хотя произведение концентраций ионов реагирующих веществ превышает произведение растворимости осадка, или ионы из раствора могут полностью абсорбироваться стенками сосуда. Для устранения
384
Д. Химический анализ
трудностей, связанных с поведением предельно разбавленных растворов, содержащих радиоактивные изотопы, применяют так называемые носители, т. е. активный изотоп в значительной степени разбавляют неактивным изотопом этого же элемента. Это означает, что, хотя активный изотон используется в экстремально низкой концентрации, химическое поведение раствора определяется значительно более высокой концентрацией носителя.
41.2. Методы обнаружения регистрации излучений
В аналитической химии используют три основных метода обнаружения и регистрации излучений: а) электрическое детектирование ионизации газов под действием излучения; б) измерение светового излучения, возникающего при облучении некоторых веществ; в) прямую регистрацию излучений фотографическим методом. Последний из перечисленных методов по существу применяется только для определения характера распределения радиоактивных веществ по поверхности твердых тел, таких, как минералы или биологические объекты.
Рис. Д.155. Электрическая схема детектора для измерения ионизации газов под действием излучения.
Наибольшее значение имеет первый метод. Все виды ядер-ных излучений, кроме нейтронного, ионизируют атомы вещества, через которые проходят. Поскольку образующиеся в газах ионы находятся в свободном состоянии и легкоподвижны, то в этом случае излучение легко может быть измерено. Принцип измерения иллюстрирует рис. Д.155. Металлическая трубка диаметром ~2 см и длиной ~10 см закрыта с одной стороны, на торцевой стороне трубки находится узкое окно, сделанное из материала, пропускающего излучение. Трубка заполнена газом. Для этого обычно применяют газовые смеси, например ар
41. Радиохимические методы анализа
385
гон+1% этанола или гелий + аргон+этилформиат. При этом органические компоненты добавляют для поглощения образующихся фотонов, которые являются источником мешающей вторичной эмиссии электронов. Такой же эффект наблюдается при использовании небольших количеств галогенов. Давление газа в трубке счетчика составляет примерно 0,1 мПа. Внутри трубки по ее оси расположена металлическая проволока. Она служит анодом, а металлические стенки трубки — катодом. Анод обычно через конденсатор С соединен с решеткой усилителя. На выходе трубки находится регистрирующее или счетное уст-
ройство. При прохождении через торец цилиндра постоянного излучения, которое может ионизировать молекулы газа в цилиндре, и повышении прилагаемого напряжения от нуля до нескольких тысяч вольт можно получить зависимости такого типа, как представленные на рис. Д.156. При приложении небольших напряжений образующиеся ионы под действием слабого электрического поля ускоряются мало. При этом основ-
Рис. Д.156. Зависимость показаний счетчика от напряжения детектора, используемая для детектирования ионизирующего излучения.
ное количество ионов реком-
бинируется в молекулы, не доходя до электродов. С увеличением приложенного напряжения растет число ионов, попадающих на электроды. В этой области (7) показания прибора пропорциональны напряжению. При дальнейшем увеличении напряжения достигается состояние насыщения, при котором все ионизированные молекулы газа попадают на электроды прежде, чем произойдет их рекомбинация. Область насыщения. (2) возникает при напряжении свыше 100 В. Если дальше увеличи-чить напряжение, показания прибора возрастают (область 3), так как ускорение ионов теперь настолько велико, что они са-
ми ионизируют молекулы, сталкиваясь с ними на пути к электродам. Если все образовавшиеся вторичные ионы достигают электродов прежде, чем произойдет их рекомбинация, то на кривой получают вторую область насыщения (4). Новый подъем (5) на кривой наблюдается при резком увеличении вторичной ионизации, вызывающей образование и распространение плазмы в области проволочных электродов. Если плазма стабилизирована по всей длине проволоки, на кривой получают площадку (6). Показания прибора можно рассматривать как функцию только ионизирующего излучения на участках 2, 4 и 6, расположенных параллельно оси абсцисс; здесь показания при
25—1880
386
Д. Химический анализ
бора практически не зависят от изменения напряжения. Эти области поэтому можно использовать для измерения излучения. Область 2 называют областью ионизационной камеры, 4— областью пропорционального счетчика, 6 — областью работы счетчика Гейгера.
Описанные процессы ионизации газов вызываются только заряженными частицами. Однако счетчик Гейгера можно применять также для измерения у-излучения. Попадая на стенки счетчика, оно вызывает эмиссию вторичных электронов. Каждая рабочая область или соответственно каждый тип счетчика имеет свои достоинства и недостатки. Для работы ионизационной камеры необходимы небольшие напряжения, но при этом возникают слабые токи, и поэтому необходимо использовать большое усиление или чувствительный регистрирующий прибор. Ионизационные камеры применяют в основном при измерении излучений большой интенсивности или при работе с сильно ионизирующим а-излучением.
Если применяют пропорциональный счетчик, то получают, как описано выше, значительное самопроизвольное увеличение числа ионов (в 103—104 раз). Поэтому в этом случае нет необходимости в большом внешнем усилении. Счетчики такого типа имеют очень высокую разрешающую способность и могут измерять до 105 отдельных импульсов в минуту. С другой стороны, оптимальная область напряжения узка, так что необходим хорошо регулируемый источник напряжения. Для работы счетчика Гейгера — Мюллера почти не требуется внешнее усиление, но его скорость счета значительно ниже: надежно можно регистрировать примерно 104 импульсов в минуту. Кроме того, с помощью такого счетчика нельзя различать энергии ионизирующих излучений, поэтому он постепенно теряет свое значение.
Все шире используются сцинтилляционные счетчики. Радиоактивное излучение в этом случае направляют на кристаллы стильбена, антрацена или иодида натрия; эффективность счета возрастает при активации этих веществ следовыми количествами иодидов тяжелых металлов, например иодидом таллия. Индуцированную световую вспышку можно с помощью фотоумножителя превратить в электрический сигнал, используемый для измерения. Применяют также жидкие и газообразные сцинтилляторы.
Поскольку электрические сигналы получают -в виде импульсов, их нужно считать с помощью специального устройства. Электромеханические счетчики могут регистрировать только до 100 импульсов в минуту. Для большей скорости счета применяют, например, электронные выключатели, которые передают только каждый второй импульс. Последовательным включением таких элементов можно получить любые факторы пересчета (уменьшения).
41. Радиохимические методы анализа
387
Нейтроны представляют собой электронейтральные частицы, поэтому они не ионизируют газы. Для регистрации нейтронов можно применить счетчик с трифторидом бора. При этом нейтроны реагируют с бором с образованием Li и а-частиц, которые считают, как указано выше. В сцинтилляционные счетчики также можно ввести соединения бора.
Энергии различных видов излучения могут существенно различаться, причем каждый нуклид характеризуется определенной энергией. Нуклид, испускающий а- и р-частицы, можно обнаружить, применяя стандартные поглотители, например листы фольги различных металлов с известной толщиной. Толщина слоя фольги, необходимая для снижения активности излучения вдвое, может служить мерой энергии излучения нуклида. Эту величину можно определить по градуировочному графику. Можно также применять описанные выше счетчики, сортирующие импульсы излучения в соответствии с их энергией. Самописцы при этом регистрируют число импульсов в минуту как функцию энергии частиц. Счетчики можно также использовать как спектрометры. Созданы также нейтронные спектрометры, которые позволяют определять ряд элементов по измерению поглощения нейтронов.
41.3. Оценка измерений
В отсутствие радиоактивной пробы счетчики всегда показывают небольшое излучение. Оно вызвано естественной радиоактивностью окружающей среды, а также частично воздействием космических лучей; такое излучение называется фоном. Влияние естественной радиоактивности уменьшают до 15—20 импульсов в минуту, применяя защитные свинцовые экраны. При оценке результатов измерений надо вводит^ поправку на радиоактивность фона.
Радиоактивный распад имеет статистическую природу и подчиняется закону вероятности. Поэтому измеренные значения надежны лишь при большом числе импульсов, что дает возможность провести статистически правильную оценку полученных результатов. Если период полураспада излучающего изотопа значительно больше, чем продолжительность эксперимента, то значение стандартного отклонения рассчитывают как квадратный корень из числа подсчитанных импульсов, так что результат можно представить как п±У«. Излучение фона оценивают аналогично и при оценке результатов измерения его вычитают.
Пример. Если излучение пробы составляет, например, 20 000 импульсов за 20 мин, а излучение фона — 400 импульсов за 10 мин, то число импульсов в минуту составит (1000±7) — (40±2), и окончательный результат измерений
25*
388
Д. Химический анализ
можно представить как 960±7. Недостоверность, таким образом, равна ±0,7%. При измерении излучения пробы в течение 1 мин получим (1000± ±32) — (40±2) =960±32, т. е. недостоверность более 3%. Эти факторы необходимо учитывать и выбирать условия измерения в зависимости от конкретной задачи. Но в каждом случае необходимо подсчитывать не менее 10 000 импульсов. Тогда существует 95%-ная вероятность того, что истинное значение находится между 9800 и 10 200.
Другие ошибки возникают, например, при большой частоте поступления импульсов на счетчик, из-за чего счетчик уже не разделяет два последовательных импульса. В этом случае также необходимо применять корректирующую формулу, которую можно найти в специальной литературе. Необходимо учесть также, что обычно измеряют только часть излучения пробы; исключение составляют счетчики, внутрь которых вводят пробу. Следовательно, геометрия установки оказывает большое влияние на измерения. Это же важно и при поглощении излучения пробой (если проба имеет большую толщину) или растворителем. Поэтому при исследовании растворов часто также применяют погружные счетчики.
Более подробные сведения о рассмотренных закономерностях приведены в ряде специальных монографий.
41.4. Аналитическое применение
Применяя явление радиоактивности в аналитических целях, следует различать использование излучения, возникающего при распаде нуклидов, и использование самих радиоактивных нуклидов.
Возникающее при радиоактивном распаде у-излучение соответствует рентгеновскому излучению такой же длины волны и поэтому не дает никаких дополнительных преимуществ. В аналитических целях может быть использовано поглощение а- и р-частиц другими веществами. При этом, например, можно поместить источник а-частиц в ионизационную камеру и пропустить через нее поток анализируемого газа. При постоянном давлении газы по-разному поглощают излучаемые частицы, поэтому полученные данные являются функцией состава газа. Путем сравнения с результатами, полученными для газовой смеси известного состава, находят точные значения количеств компонентов.
р-Излучение абсорбируется в первую очередь электронами пробы. Поскольку электронная плотность водорода по меньшей мере в два раза больше, чем у других элементов, явление абсорбции p-излучения можно использовать при определении содержания водорода в углеводородах.
Гораздо большее значение имеют другие аналитические методы, основанные на простом и высокочувствительном обнару-
41. Радиохимические методы анализа
389
женин радиоактивных нуклидов. Среди этих методов в первую очередь следует назвать активационный анализ. Многие элементы превращаются в радиоактивные нуклиды при бомбардировке их частицами, обладающими большой энергией, такими, как нейтроны, протоны, дейтроны или а-частицы. Ядра многих атомов поглощают нейтроны, при этом образуются изотопы с большей массой, которые зачастую нестабильны и распадаются с возникновением вторичной эмиссии. Период полураспада, вид и энергия этого излучения определяются природой элементов и могут служить их характеристикой. Большинство элементов состоит из смесей изотопов. В количественном отношении в элементе преобладает один изотоп, а активностью других изотопов можно пренебречь.
Для предварительно активированной анализируемой пробы строят кривые распада — зависимость логарифма активности от времени. Наклон графика зависит от периода полураспада изотопа. При анализе смесей элементов эти кривые достаточно сложны и аддитивно складываются из кривых распада отдельных компонентов. Для оценки в этом случае используют наиболее долгоживущий нуклид. Последнее значение активности на кривой распада относится к распаду наиболее долгоживущего нуклида, так как на этом этапе компоненты с коротким временем жизни уже не вносят существенного вклада в измерения. Можно также определить содержание элемента с наименьшим периодом полураспада, если вычесть долю излучения наиболее долгоживущего компонента и т. д.
Чувствительность активационного анализа зависит от источника возбуждения и периода полураспада образовавшихся радиоактивных нуклидов и может достигать 1СН2 г. С другой стороны, она определяется также природой содержащихся в анализируемой пробе элементов, которые можно активировать. Источники потока нейтронов с небольшой плотностью, такие, как смесь бериллия и радия, активируют лишь немногие элементы, но позволяют определять их с большой точностью. Источниками потока нейтронов большой плотности являются ядерные реакторы.
Пример. Определение следовых количеств фосфора в иоде имеет важное техническое значение при получении кремния высокой чистоты, применяемого в качестве полупроводника, из тетраиодида кремния. Так же как фосфор, под существует в природе только в виде одного изотопа. Изотоп ксенона, образующийся при облучении иода нейтронами, распадается с периодом полураспада 25 мин, а образующийся из фосфора изотоп серы — с периодом полураспада 14,3 сут. Через 24 ч после облучения активность иода составляет 10~!1 исходной величины, и на фоне активности фосфора ею можно пренебречь.
Для иллюстрации эффективности метода можно упомянуть о возможности его использования для решения проблем крими
390
Д. Химический анализ
налистики. Например, с помощью активационного анализа можно идентифицировать преступника по волосу, найденному на месте преступления. Распределение следовых количеств различных элементов в волосе зависит от генетических факторов, окружающей среды, а также от профессии.
Метод изотопного разбавления — другой важный аналитический метод, основанный на использовании явления радиоактивности. Например, если соединение невозможно выделить в чистом виде, то его нельзя количественно определить классическими методами анализа. Если же в анализируемую смесь ввести следовое количество радиоактивного изотопа определяемого компонента и тщательно смешать, то даже при неполном отделении определяемого компонента можно определить его содержание в анализируемой пробе. Обозначим количество определяемого компонента в граммах в анализируемой пробе через W, а дополнительно введенное в пробу количество этого вещества в радиоактивной форме через w (его активность обозначим как А). После тщательного смешивания выделяют g грамм чистого компонента или соединения этого компонента, имеющего активность В. Необходимые расчеты можно провести по уравнениям
(W-\-w)lg= А/В или W' = g(4/B)— w
Если добавляемое вещество обладает большой активностью, величиной w можно пренебречь.
Можно легко показать преимущества использования этого метода для проведения различных количественных определений. Например, используя классические методы анализа, трудно проанализировать смесь ниобия и тантала. Однако можно выделить из смеси чистую фракцию, содержащую ниобий и тантал, правда, с плохим выходом. Эффективность разделения, т. е. долю выделенных металлов от их исходного количества в анализируемой смеси, можно легко определить указанным способом. В литературе описаны Другие примеры подобных определений.
В методах активационного анализа и изотопного разбавления явление радиоактивности используют непосредственно для определения веществ. Кроме того, существует еще одна область использования радиоактивных изотопов — применение их для индикации точки эквивалентности при титровании. Метод радиометрического титрования впервые был применен в 1941 г. В ходе титрования измеряют радиоактивность раствора. Точку эквивалентности можно определить так же, как, например, в методе кондуктометрического титрования, по пересечению двух прямых. Существенным преимуществом радиометрического титрования по сравнению с другими методами индикации точки эквивалентности является тот факт, что численное значение измеряемого свойства может быть любым и достаточно большим даже при очень малых концентрациях благодаря введению в
391
41. Радиохимические методы анализа i „
определяемое вещество соответствующего количества активного нуклида. На индикатор не оказывают влияния окраска, мутность или агрессивность раствора. Метод можно автоматизировать.
Необходимым условием для применения радиоактивных индикаторов является разделение фаз. В процессе осадительного титрования это происходит самопроизвольно, в других методах нужно проводить дополнительные операции. Для автоматизации метода разделение должно осуществляться непрерывно.
Многие реакции образования осадков не используют в титриметрии, так как нет адекватных визуальных или инструментальных методов для индикации точки эквивалентности. По сравнению с кислотно-основным титрованием метод визуальной индикации точки эквивалентности при осадительном титровании имеет тот недостаток, что для каждого конкретного случая необходим свой индикатор. Для радиометрического титрования специальный индикатор не нужен — он содержится в титруемом веществе или в титранте. Например, перед титрованием в титруемое вещество можно ввести активный нуклид и фиксировать уменьшение его активности в процессе титрования до точки эквивалентности, как меру уменьшения его концентрации. Можно использовать также обратный метод, в котором применяют радиоактивный титрант — при его избытке в растворе активность резко возрастает. Если радиоактивные нуклиды находятся и в титранте, и в определяемом веществе, то активность раствора в точке эквивалентности минимальна.
Отделение осадка перед каждым измерением активности проводят фильтрованием, центрифугированием или флотацией. При фильтровании, осуществляемом, например, отсасыванием через пористую стеклянную пластинку, после добавления каждой порции титранта измеряют активность части раствора, и эту часть раствора вносят опять в колбу для титрования. Недостатком этого метода является возможность забивания пор стеклянной фильтрующей пластинки мелкодисперсным осадком. Этот недостаток устраняют, заменяя фильтрование центрифугированием. Равные количества анализируемого раствора вносят в несколько центрифужных пробирок, добавляют различные количества титранта и доливают растворителем до равных объемов (0,5—1 см3). После отделения осадка центрифугированием равные порции раствора, находящегося над осадком, пипеткой наносят на фильтровальную бумагу и после высушивания измеряют активность.
В методе флотации исследуют содержащую воздух пену, поглощающую твердые частицы и поднимающую их наверх, или органическую фазу, не смешивающуюся с водой. При этом твердые частицы собираются на поверхности раздела фаз. Поскольку проводят измерение активности водного раствора, целесооб
392
Д, Химический анализ
разно использовать находящуюся в нижней части сосуда органическую фазу, более тяжелую, чем вода.
Если радиометрическую индикацию применяют в процессах комплексометрического титрования, разделение фаз нужно проводить по-другому, а именно удалять из водного раствора продукт или продукты реакций. Для этой цели пригодная экстракция растворителем, полностью или частично не смешивающимся с водой. Принцип этого метода был описан в гл. 38. В то время как точность осадительного титрования зависит от произведения растворимости, точность комплексометрического титрования определяется устойчивостью образовавшихся комплексов. При использовании хороших комплексообразующих реагентов устойчивость комплексов всегда чрезвычайно высока. С другой стороны, как было указано выше, радиометрическая индикация высокочувствительна. Все это обеспечивает экстремально высокую чувствительность комплексометрического титрования (при условии, что константа распределения комплексного соединения между фазами имеет подходящую величину), которая может конкурировать с чувствительностью активационного анализа. В описанном методе можно использовать также изменение активностей обеих фаз.
Другой способ фазовых разделений, применяемый с недавнего времени, состоит в следующем: в титруемый раствор добавляют малорастворимое твердое радиоактивное вещество. Его нужно подобрать таким образом, чтобы оно реагировало только с избыточным количеством титранта и растворялось в отсутствие определяемого иона в растворе. В этом случае после точки эквивалентности активность раствора возрастает. Чувствительность этого метода определяется устойчивостью комплекса, образующегося при титровании, а также растворимостью и радиоактивностью осадка индикатора. Для разделения фаз можно использовать ионный обмен.
Такой способ можно использовать в некоторых случаях и в окислительно-восстановительном титровании. В раствор, содержащий ион, который можно определить редокс-титрованием, вносят жидкую амальгаму с активным компонентом. В процессе титрования сначала должен окисляться определяемый ион, а затем компонент амальгамы, который переходит в раствор после точки эквивалентности и тем самым вызывает увеличение активности.
Для радиометрической индикации можно также использовать явления рассеяния или поглощения p-излучения атомами титруемого раствора или титранта.
42. Методы термического анализа
393
42. МЕТОДЫ ТЕРМИЧЕСКОГО АНАЛИЗА
42.1 Термогравиметрия
Взвешивание можно применять не только для определения неизвестной массы веществ, но и для исследования химических реакций, если в ходе реакции увеличивается или уменьшается количество вещества, которое в условиях работы находится в. газообразном состоянии и поэтому не мешает взвешиванию. Если о ходе химической реакции, протекающей при повышенной температуре, судят по изменению массы исследуемого вещества, определяемой взвешиванием, то такой метод исследования называют термогравиметрией.
В термогравиметрии цель взвешивания иная по сравнению с гравиметрией, где задача заключается в как можно более точном определении массы вещества; в данном случае фиксируется изменение массы в процессе реакции.
Массу исходных веществ и конечного продукта можно с достаточной точностью определить на обычных аналитических весах. Для надежной регистрации изменения массы во времени и. получения на основании проведенных измерений сведений о ходе химической реакции, необходимы весы специального типа, которые в зависимости от цели применения должны удовлетворять нескольким дополнительным требованиям. Поскольку реакции в основном протекают непрерывно, даже если скорость их переменна, изменение массы также нужно регистрировать непрерывно и по возможности получать эти данные без временных задержек. Обычные аналитические (рычажные) весы пригодны для взвешивания в очень ограниченных пределах. Применение весов с низко расположенным центром тяжести (см. гл. 38) и оптическое увеличение амплитуды стрелки весов (принцип световой стрелки, используемый также в зеркальных гальванометрах) значительно расширяет возможности метода. Некоторые термовесы сконструированы по этому принципу.
В других типах гермовесов используют принцип компенсации, когда отклонение стрелки весов полностью компенсировано. Практически нецелесообразно компенсировать разницу масс на чашках весов с помощью разновесов — это потребовало бы больших затрат времени. Кроме того, часто бывает необходимо проводить измерение в закрытой системе или в атмосфере определенного газа. В этом случае особенно нерационально применять обычные весы с разновесами. Компенсацию проводят, прикладывая электромагнитное поле определенной силы. Это, например, можно сделать следующим образом: ферромагнитный металлический стержень помещают в стеклянную трубку с обмоткой и подвешивают на коромысло весов. При прохождении электрического тока через обмотку на металлический стержень действует определенная сила. Величину ее регулируют, варьируя силу така, которая и служит мерой массы вещества.
394
Д. Химический анализ
Рис. Д.157. Термовесы с расположенной сбоку электропечью.
Бюкс с исследуемым веществом помещают в электрическую печь. Его нагревают в соответствии с программированным (обычно линейным) изменением температуры, например со скоростью 50—100 °C в час. Температуру пробы вещества измеряют термоэлементом и одновременно фиксируют изменение массы. При перегреве происходит конвекция газов и, если печь находится под весами, возникающие тепловые потоки вносят искажения в измерения массы. Поэтому бюкс с нагреваемой пробой обычно помещают в стороне или выше коромысла весов (рис. Д.157).
Другие аппаратурные трудности связаны с качественным и количественным составом га
Рис. Д.158. Схема термовесов Мак-Байна и Бакра.
зовой фазы. При необходимости проведения реакции в среде определенного газа (например, Н2, СО или О2) или при пониженном давлении аппаратура должна быть герметичной, что трудно достичь из-за больших размеров рычажных весов. Агрессивные газы, такие, как Cl2, НС1 или SO2, выделяющиеся при разложении веществ, разрушают металлы. В этих случаях вместо металлических деталей применяют оборудование из кварцевого стекла. Наряду с относительно сложными рычажными весами часто работают с весами, в которых ис
42. Методы термического анализа
395
пользованы силы упругости, возникающие при эластичной деформации веществ. К такому типу относятся предложенные Мак-Байном и Бакром в 1926 г. весы (рис. Д.158) со спиральной пружиной из кварцевого стекла и подсоединенным к ней указателем. На пружину подвешивают чашечки из кварцевого стекла с исследуемым веществом. Эту простую систему можно поместить в трубку из кварцевого стекла, обогреваемую снаружи электропечью. Трубку легко вакуумировать или заполнить любым газом. Помимо оптической фиксации отклонения указателя, используют электронное устройство для точных измерений. Результаты можно автоматически регистрировать.
Примеры.
1. На рис. Д.159 приведены термоправиметрические кривые оксалатов кальция (а) и магния (б) и, отмечены некоторые характерные точки. С повышением температуры СаС2О4-Н2О сначала теряет воду (в интервале 100—
Рис. Д.159. Термогравиметрические кривые оксалата кальция (а) и оксалата магния (б).
Рис. Д.160. Термогравиметрические кривые нитрата серебра (а), нитрата меди- Cu(NO3)2-6H2O (б) и их смеси (в).
200 °C) и переходит в безводную соль. Оксалат кальция устойчив до температуры 400 °C. Выше этой температуры происходит разложение соли на карбонат кальция и оксид углерода:
СаС2О4 ---> СаСО3 СО
При температуре ~700°С происходит разложение карбоната кальция:
СаСО3 ----»- СаО + СО2
При 900 °C разложение заканчивается. Указанные температуры практически не зависят от условий эксперимента (скорости нагревания, природы применяемого газа), а также от степени измельчения и толщины слоя анализируемого вещества.
Термическое разложение оксалата магния (рис. Д.159, б) протекает по-другому. Сначала также происходит отщепление кристаллизационной воды. В интервале 240—400 °C оксалат устойчив; при дальнейшем повышении тем-
396
Д. Химический анализ
пературы он разлагается, но с образованием не карбоната, а оксида: MgC2O4 -----------------------------г- MgO+CO + COa
Из данных диаграмм можно сделать важные выводы для гравиметрического определения Са2+ и Mg2+ при осаждении их в виде оксалатов. Можно так выбрать области температуры прокаливания оксалатов, чтобы при этом получался нужный конечный продукт. Различный ход термогравиграмм CaCaOi и MgCsO4 дает возможность определять компоненты смеси при их совместном осаждении. При прокаливании и взвешивании полученного осадка при 500 °C
получают массу осадка, состоящего из СаСОз и MgO. После заключительного прокаливания этой же смеси при 900 °C при взвешивании получают массу MgO+CaO. Путем несложных расчетов можно определить содержание в смеси обоих компонентов.
2. На рис. Д.160 приведены термогравиметрические кривые нитрата серебра AgNOa (а), нитрата меди Си(МОз)2-6Н2О (б) и смеси обоих компонентов. Можно аналогично проследить за ходом иагрева-
Рис. Д.162. Схема дифференциальных термовесов де Кейзера.
Рис. Д.161. Термогравиметрическая (ТГ) и соответствующая ей дифференциальная термогравиметрическая (ДТГ) диаграммы.
ния, соблюдая известную осторожность, так как компоненты смеси при нагревании могут реагировать между собой. В этом случае разложение происходит ие так, как можно было бы ожидать, исходя из диаграмм индивидуальных веществ. Но с другой стороны, как раз в результате таких исследований можно получить новые ценные сведения.
Масса некоторых веществ изменяется не столь резко, как в рассмотренных выше идеальных примерах. Часто ступени на диаграмме невозможно разграничить. В таких случаях хороший эффект дает построение наряду с термогравиметрической кривой (ТГ), дифференциальной термогравиметрической кри
42. Методы термического анализа 397
вой (ДТГ), представляющей собой первую производную ТГ. В области наиболее резких изменений массы (резкие подъемы на ТГ) на ДТГ наблюдаются четкие минимумы, как можно видеть на рис. Д.161.
В некоторых типах термовесов кривые ДТГ получают из кривых ТГ с помощью электронных преобразователей. Термовесы других типов снабжены особыми измерительными устройствами. Интересный принцип положен в основу действия дифференциальных термовесов де Кейзера (рис. Д.162), позволяющих сразу получить ДТГ. На коромысле весов с обеих сторон подвешивают платиновые тигли одинаковой массы. Оба тигля заполняют равными количествами одного и того же исследуемого вещества. Весы при этом находятся в равновесии. Включают печи и регулируют нагрев таким образом, чтобы температура одной постоянно превышала температуру другой на некоторую определенную величину (А?) (например, на 5 или 10°C). Разложение пробы вещества происходит прежде всего в печи с более высокой температурой. Поэтому в процессе нагревания при каждой температуре весы фиксируют соответствующую разность масс. В этом случае измеряемые величины регистрируют световым лучом, который отражается от зеркала на коромысле весов и попадает на барабан с фотобумагой.
42.2. Термический анализ и дифференциальный термический анализ
Резкое изменение энергии межатомных или межмолекулярных связей в веществе сопровождается обычно выделением или поглощением тепла. Это означает, что температура анализируемой пробы может существенно отличаться от температуры окружающей среды. Изменение температуры может происходить, например, при выделении летучих компонентов, прохождении реакций между твердыми веществами, изменении кристаллической структуры веществ и плавлении. Величина разности температур зависит от поглощения или выделения тепла и некоторых экспериментальных параметров. Эту зависимость можно использовать для качественного и количественного анализа веществ.
На рис. Д.163 приведены типы диаграмм, полученных тремя основными методами термоанализа. В первом случае (кривая а) при нагревании пробы вещества в печи с возможно более равномерной скоростью нагрева зависимость температуры анализируемой пробы от времени должна представлять собой прямую, угол наклона которой определяется скоростью нагрева. Такая же зависимость наблюдается, если не происходит превращения вещества. Между печью и пробой возникает при
398
Д. Химический анализ
этом постоянная разность температур. Теплообмен между ними
пропорционален величине разности температур.
Если же в пробе происходит реакция, то выделение (экзотермическая реакция) или поглощение (эндотермическая реакция) тепла вызывает значительное отклонение прямой от линейности. В первом случае кривая изгибается вверх, т. е. температура пробы изменяется быстрее, чем наблюдалось бы при
Рис. Д.163. Типичные кривые термического (а, б) и дифференциального термического (в) анализа.
такой же скорости нагрева в отсутствие реакции. Для эндотермической реакции картина обратная. Хотя тепло от источника поступает к пробе, проба нагревается слабо, так как энергия затрачивается на эндотермическую реакцию и ее не хватает на нагревание пробы. По окончании процесса, чему соответствует вертикальный участок штриховой линии на рис. Д.163, а, поступающее тепло снова затрачивается только на нагревание пробы. Так как тепловой поток пропорционален возникающей разности температур и эта разность возрастает в ходе реакции, количество подводимого тепла нужно увели-. чить. Температура пробы по-Лэтому очень быстро возраста-
ет в течение небольшого промежутка времени, а затем кривая опять имеет нормальный линейный вид.
Недостатком этого метода является необходимость использования сравнительно малочувствительных регистрирующих приборов для измерений в широкой области значений температуры. Таким образом, могут остаться незамеченными небольшие термические эффекты. Трудности, возникающие при этом, можно сравнить с трудностями метода термогравиметрии, при котором в большой области температур происходит медленное изменение массы вещества. И в том и другом случае целесообразно применять дифференциальные методы. При проведении измерений в ходе реакций (или при разделении фаз), происходящих в области небольших интервалов температуры, такой проблемы не существует. Основная область применения метода в настоящее время— термический анализ (ТА) сплавов.
Кривую (рис. Д.163, б) получают, проводя измерения через определенные промежутки времени, в течение которых температура пробы изменяется на определенное небольшое значение, например на 1—2°C. Это время постоянно, если пробу нагревают с определенной скоростью и не происходит никаких реакций.
42. Методы термического анализа
399
Линейному участку диаграммы (рис. Д.163, а) соответствует параллельный оси х участок кривой б, расстояние которого от оси абсцисс определяется скоростью нагрева.
При выделении или поглощении тепла в ходе реакции временной интервал уменьшается (экзотермическая реакция) или увеличивается (эндотермическая реакция). Это приводит к положительным или отрицательным отклонениям от основной линии. По окончании реакции наблюдается изгиб кривой в другую сторону и затем возвращение к основной линии.
Такой метод применяют редко, так как для автоматической регистрации изменений dt/dT требуются очень сложные приборы. Кроме того, метод имеет много недостатков, общих с описанным выше первым методом. Например, при попытке определения температурного интервала протекания реакции при медленном взаимодействии веществ всегда получают более узкую область температур, чем в действительности, так как в обоих случаях (кривые а и б) практически не учитываются изменения разности температур пробы и окружающей среды, хотя эти изменения, всегда имеющие место при протекании реакций, модулируют величину теплового потока.
Третья диаграмма (рис. Д.163, в) получена методом дифференциального термического анализа (ДТА). На рис. Д. 164 показан принцип действия установки ДТА. В системе, которую можно нагревать с линейным программированием температуры, симметрично расположены три сосуда одинаковой вместимости. Один из них заполнен анализируемым веществом, два других — инертным веществом, не подвергающимся термическим превращениям (как правило, а-А^Оз). В каждый сосуд введен термоэлемент. Термоэлемент, измеряющий температуру анализируемого вещества, соединен с термоэлементом, измеряющим температуру инертного вещества, таким образом, что термонапряжение гасится, если температуры их равны. При возникновении разности температур между пробой и инертным веществом соответствующую разность напряжений можно заметить по регистрирующему прибору. Одновременно можно зафиксировать температуру системы, которую третий термоэлемент преобразует в напряжение.
При строго симметричном расположении трех сосудов и при одинаковых условиях подвода тепла температуры их равны. В этом случае можно зафиксировать нулевую или основную линию с небольшим отклонением от нуля (если, например, анализируемое вещество имеет неодинаковую с инертным веществом теплопроводность). При изменении фазовых состояний или при протекании реакции разложения, связанной с подводом или отдачей тепла, получают пики, соответствующие эндотермическому или экзотермическому процессам (рис. Д.165). Температуры пиков качественно характеризуют соответствующие процессы. Площадь пика является мерой количества тепла, выделяющегося в процессе реакции, а также (после соответствую-
400
Д. Химический анализ
Рис. Д.164. Схема установки для дифференциального термического анализа.
1 — сосуд с пробой анализируемого вещества; 2 и 3 — сосуды с инертным веществом;
4— система с контролируемым изменением температуры; 5 — контакты подсоединения термоэлементов.
щей градуировки) мерой количества вступающего в реакцию вещества или компонента анализируемой смеси.
С другой стороны, эти кривые сравнительно трудно использовать, так как условия подвода тепла оказывают существенное влияние на вид и положение пиков. Для получения пропорциональной зависимости площади под пиком ДТА от количества тепла реакции необходимо выполнять следующие требования: а) сосуды для пробы и стандарта должны быть одинаковой формы и находиться в одинаковых температурных условиях; б) теплопроводность и теплоемкость пробы и инертного вещества должны быть одинаковы; в) в пробах не должны возникать температурные градиенты. Первое условие можно выполнить, тщательно подготавливая аппаратуру- Труднее соблюдать второе требование. Теплоемкость и теплопроводность анализируемой пробы и инертного вещества, естественно, различны. Кроме того, эти параметры изменяются в самой анализируемой пробе вследствие происходящих в ней превращений. При смешивании анализируемого вещества с инертным компонентом в определенном
соотношении можно говорить об известном приближении к идеализированному состоянию, так как анализируемое вещество сильно разбавляется. При этом изменение теплоемкости и теплопроводности будет пренебрежимо мало. Третье условие вообще невыполнимо. Температурный градиент всегда возникает в пробе. Он оказывает несущественное влияние на результаты, пока значение его постоянно. Из этого следует важнейший вывод: количественную оценку можно проводить только по диаграммам ДТА, снятым на одной и той же аппаратуре и при одинаковой линейной скорости нагрева.
Метод ДТА применяют для анализа почв, глины, руд и минерален, карбонатов, гидратов, гидроксидов, формиатов, оксалатов и других неорганических соединений, а также шерсти, шелка, целлюлозы, крахмала, сахаров и искусственных волокон различного вида.
Для эффективного сравнения результатов, полученных методами ТГ, ДТГ н ДТА, были предложены специальные при-
42. Методы термического анализа
401
боры, к которым относится, например, дериватограф (рис. Д.166). Пробу нагревают в тигле; определяемое взвешиванием изменение массы пробы с помощью светового указателя фиксируется на фотобумаге в виде термогравиметрической кривой (ТГ). На другом плече коромысла весов подвешен постоянный магнит с катушкой, имеющей очень большое число витков. При изменении массы пробы катушка движется под действием по* стоянного магнита. Скорость ее движения и, следовательно, ин
•44 Т
ДТ=О
-ДТ
Эндотермическая область
Рис. Д.165. Кривая ДТА с эндотермическим пиком при температуре 7\ и экзотермическим пиком при температуре Т?.
дуцированное напряжение пропорциональны скорости разложения пробы. Значение напряжения регистрируется на той же фотобумаге, что и кривая ТГ, в виде соответствующей кривой ДТГ. В тигель с пробой, висящий на коромысле весов, кроме того, помещают термоэлемент. Второй термоэлемент находится в тигле с инертным веществом. При дифференциальном включении обоих элементов можно одновременно получить кривые ТГ, ДТГ и ДТА. На рис. Д.167 в качестве примера приведены результаты дериватографического исследования смеси магнезита и доломита.
42.3. Термометрическое титрование
В описанных методах термогравиметрии и дифференциального термического анализа масса или температура исследуемой системы исследовалась как функция температуры среды. В отличие от этого в методе термометрического титрования изучают зависимость температуры анализируемой системы от объема добавляемого титранта. Таким образом, два первых метода являются методами определения, последний — методом индикации точки эквивалентности.
При проведении термометрического титрования титрант добавляют в анализируемый раствор из термостатированной бю-
26—1880
Рис. Д.166. Схема дериватографа.
/ — тигель с анализируемой пробой; 2 — тигель с инертным веществом; 3 — фарфоровая трубка; 4 — термоэлемент; 5 — электропечь; 6 — некрученые электропровода; 7 —весы; 5 —катушка; 9 — магнит; 10 — ДТГ-гальванометр; 11 — /'-гальванометр; 12— ДТА-гальва-иометр; 13 — лампа; 14 — оптическая щель; 15 — барабан для фотобумаги; 16 — фотобумага.
Показания гальванометра
’W
Изменение массы
MgC03+CaMg(C03j2
620 640. Й
\ 1 810
10
Mg0+CaMg[C03)2
40
МдО+СаО 5(7
МдО+СаСО3 30
< 41-880 \soo
.900960.
840 {
400 600 800 1000 °C
Рис. Д.167. Дифференциально-термоаналитическая (ДТА), дифференциальнотермогравиметрическая (ДТГ) и термогравиметрическая (ТГ) кривые для смеси магнезита и доломита.
Кривые получены; 1 — на приборе для проведения дифференциального термического анализа; 2 —с применением термовесов, снабженных устройством для получения дифференциальной термогравиграммы; 3 — на дериватографе. На кривых 1 и 2 видны изменения формы кривой, фазовые сдвиги и различия температур соответствующих пиков. Кривые 3 хорошо совпадают во всех трех случаях.
42. Методы термического анализа
403
ретки по возможности в адиабатических условиях; титруемый раствор находится в термоизолированном сосуде типа сосуда Дьюара. Температуру раствора в сосуде Дьюара измеряют чувствительным устройством. Полученные кривые температура— объем (или, если раствор титранта добавляют непрерывно, кривые температура — время) похожи на кривые титрования других методов. Поскольку все реакции происходят с экзо-или эндотермическим изменением энтальпии, которое можно измерить, метод термометрического титрования нашел широкое применение в аналитической химии и особенно в тех случаях,
Рис. Д.168. Идеализированная кривая термометрического титрования.
Рис. Д.169. Экспериментальная кривая термометрического титрования.
когда другие методы дают небольшую точность или вообще непригодны. Методы термометрического титрования могут быть использованы при проведении реакций нейтрализации, осаждения, комплексообразования и окисления-восстановления как в водных, так и в неводных средах и в расплавах.
В первых вариантах термометрического титрования раствор титранта добавляли порциями и температуру измеряли термометром Бекмана, что служило источником ошибок. Сравнительно большие затраты времени, обусловленные несовершенством техники измерения (а также медленным установлением показаний термометра), вызывали большие или меньшие отклонения от адиабатических условий, что приводило к частичному выравниванию температур пробы и окружающей среды, и, естественно, вело к возникновению субъективных ошибок. В современных установках эти ошибки сведены к минимуму. Для титрования используют автоматические бюретки, для измерения температуры — быстрые и крайне чувствительные термисторы, показания регистрируют с помощью самописца. При этом измерение температуры проводят с точностью ±0,0002 °C.
26*
404
Д. Химический анализ
На рис. Д.168 приведена идеализированная кривая термометрического титрования для экзотермического процесса. На практике получают кривые типа изображенной на рис. Д.169. Искривление обусловлено неполнотой протекания реакции. Такие кривые обрабатывают методом экстраполяции. Небольшой наклон участка АВ вызван частичным выравниванием температуры пробы и окружающей среды. Наклон участка CD обусловлен отчасти этой же причиной, а отчасти разностью температур титруемого раствора и титранта.
Если известна калориметрическая константа ячейки Q, то теплоту реакции АН можно определить по уравнению
АН = —QAT/X
где % — число молей вещества, вступивших в реакцию обмена.
Реакции, используемые в термометрическом титровании, должны протекать быстро. При титровании небольших объемов может возникнуть ошибка, связанная с теплотой испарения. При работе с разбавленными растворами выделяющейся тепловой энергии недостаточно, чтобы вызвать большие изменения температуры. В таких случаях необходимо особенно хорошее соответствие температур анализируемого раствора и титранта. Обычно применяют 1 н. растворы титрантов, в то время как концентрация анализируемого раствора должна быть по меньшей мере 10~3 н.
Термометрическое титрование сильных кислот и оснований дает такие же хорошие результаты, как и другие методы титрования. Преимущества термометрического титрования особенно отчетливо проявляются при титровании слабых кислот: результаты значительно лучше, чем в других методах титрования. Это объясняется тем, что теплота нейтрализации при взаимодействии сильных и слабых кислот с растворами сильных оснований различается менее чем на 50%.
Теплоту нейтрализации кислот растворами сильных оснований (Д/7п°) можно разложить на две составляющие, а именно теплоту ионизации ДЯг° и теплоту нейтрализации сильных (полностью ионизированных) кислот (56,5 кДж/моль):
—ДДП° = — ДЯг<>+56,5
С другой стороны, энтальпия ионизации является аддитивной функцией свободной энергии F и энтропийного фактора TS. Поэтому
ДД{° =
где &Fi°=—RT In Kt.
42. Методы термического анализа
405
Величина AFf в значительной степени компенсируется произведением TASi°, так что AHt° в ряду кислот изменяется значительно в меньшей степени, чем AF? или AS,0.
Так, например, кривые термометрического титрования HCI и
НзВО.з (Л^Ю-10) не отличаются друг от друга (рис. Д.170). Теплота нейтрализации НС1 составляет —56,6, а борной кислоты —42,7 кДж/моль. Борная
Рис. Д.170. Кривые термометрического титрования НС1 (а) и Н3ВО3 (б). По, 0,004 моль кислот титруют раствором NaOH.
кислота Н3ВОз при этом реагирует как одноосновная. Напротив, кривые их потенциометрического титрования совершенно различны. При титровании соляной кислоты в точке эквивалентности наблюдается большой скачок потенциала, а в случае НгВО3 не происходит скачка потенциала, пригодного для индикации точки эквивалентности.
Подробности метода и примеры применений можно найти в специальной литературе.
Следует упомянуть о разновидности метода термометрии — энтальпиметрии. В этом методе к анализируемому раствору добавляют избыток реагента и по количеству выделившегося тепла судят о количестве анализируемого вещества. Преимуществами метода являются возможность выполнения анализа, даже если реакции не протекают до конца, а также отсутствие необходимости приготовления растворов титрантов с точно известными характеристиками. Но с другой стороны, необходимо проводить дорогостоящую градуировку оборудования, так что метод применяют при проведении технических серийных анализов, например доменных шлаков.
406
Д. Химический анализ
43. МЕТОДЫ И ПРОБЛЕМЫ АНАЛИЗА
СЛЕДОВЫХ КОЛИЧЕСТВ
43.1. Задачи анализа следовых количеств
В количественном анализе веществ различают следующие диапазоны содержаний определяемых компонентов:
Содержание Компоненты
100—10% основные
10-1% примеси
1—1 о-3 % мнллиследовые
ю~3—ю-6% микроследовые
ю-6—ю-9% наноследовые
10~9— ю-12% пикоследовые
В зависимости от того, с какими навесками проб работают, говорят об анализе макроколичеств (100—1 г), милликоличеств (1 —10~3 г) или микроколичеств (10~3—10~е г).
С задачами аналитической химии следовых количеств сталкиваются в следующих случаях: когда пробы для анализа достаточно, но в ней содержатся небольшие количества определяемых компонентов, и когда анализируют пробы, содержащие сравнительно высокие концентрации определяемых компонентов, но количество пробы ограничено из-за ее ценности или малодоступности. Задачи первого рода встречаются значительно чаще. Развитие аналитической химии в обоих направлениях, т. е. решение указанных задач определения малых содержаний компонентов или анализа небольших проб, чем бы ни вызывалась постановка подобного рода задач — практическими нуждами или особенностями метода, в котором по необходимости имеют дело с пробами небольшого объема (например, в искровой масс-спектроскопии), — представляет важную проблему. Еще одна особенность анализа следовых количеств состоит в том, что, чем меньше содержание определяемого компонента в пробе, тем в большей степени проявляется негомогенность его распределения в твердом материале. Поэтому определение следовых количеств элементов в небольших пробах характеризуется экстремально большими величинами случайного разброса получаемых результатов.
Еще в прошлом столетии разработаны методы, позволяющие с высокой воспроизводимостью определять содержание основных компонентов и примесей в больших количествах пробы. Если результат аналитического определения выразить через среднее арифметическое и доверительный интервал как х±Дх (см. разд. 44.8), а доверительный интервал представить как Ax = st(PJ)/ynA
то высокая воспроизводимость классических методов определе
43. Методы и проблемы анализа следовых количеств
407
ния означает, что результаты характеризуются незначительным случайным разбросом (т. е. малым значением стандартного отклонения s, например 0,1%). При этом можно ограничиться небольшим числом параллельных определений пА (например, 3), однако получение каждого результата связано с большими затратами времени (несколько дней). Инструментальные методы анализа характеризуются значительно меньшей воспроизводимостью. Поскольку затраты времени на проведение параллельных определений в этом случае значительно меньше, то при большом числе опытов за короткий промежуток времени можно уменьшить значение Дх.
Классические методы не применяются для анализа следовых количеств элементов. Если нужно определить содержание элемента, например порядка 10-4%, что соответствует 1 г на 1 т или 1 млн-1 (1 часть на 1 млн.), то это означает, что нужно определить одну массовую единицу в миллионе массовых единиц или один атом в миллионе атомов. Такой анализ можно сравнить с розыском приезжего в городе с миллионным населением. Следовые количества порядка 10-7% соответствуют 1 млрд."1 (1 часть на 1 млрд.), и их определение можно сравнить с обнаружением четырех инопланетян, затерявшихся среди населения земли. На практике встречаются задачи обнаружения веществ в количествах значительно меньших, чем 1 млрд-1.
Вопросы анализа следовых количеств рассмотрены в литературе.
43.2. Определение следовых количеств элементов в биохимии и геохимии
Термин «анализ следовых количеств» впервые возник при биологических исследованиях. К концу прошлого столетия уже были известны основные компоненты тканей живых организмов — углеводы, белки и жиры, а при анализе растений были обнаружены 10 важнейших элементов: С, О, Н, N, S, Р, К, Са, Mg, Fe. Позже были найдены также следовые количества других элементов, не всегда присутствующих в живых тканях, таких, как В, Со, Си, Мп, Mo, Zn. В организмах животных редко встречаются бор или марганец, но важным элементом является селен. Заметное влияние на жизненно важные процессы оказывают также Sn, Ti, V, Cr, <Ni и другие элементы, находящиеся в тканях живых организмов в следовых количествах. Практически невозможно указать, какие из них наиболее важны, поскольку влияние, оказываемое элементами на жизнедеятельность растений или животных, различно. Такие важнейшие элементы, как В, Си, Mo, Zn, Se, Ст, находясь в избытке, могут стать для организма ядом. Особенно ядовиты кадмий и серебро даже в следовых количествах. Поэтому очень важно контролировать содержание следовых количеств элементов в воздухе, воде, почве, растениях и в организмах животных и людей.
В табл. Д.25 показано распределение следовых количеств элементов в природных материалах. Содержание элементов в почве отличается от содержания их в минералах, являющихся источниками получения элементов. С одной стороны, это вызвано различной устойчивостью пород к выветриванию,
408
Д. Химический анализ
Таблица Д.25. Содержание следовых количеств элементов в природных материалах, млн-1
Элемент Минералы Почва Ткани растений Ткани животных
Мп 500 300 80 1
Си 60 20 10 10
Sr 120 120 80 0,1
Li 60 70 0,5 1,2
В 10 15 20 0,3
Мо 5 0,5 1 0,8
а с другой стороны, различной устойчивостью элементов к вымыванию из почвы под действием атмосферных осадков. Известно, что калий и натрий, примерно в равных количествах входят в состав минералов как примеси, но калий задерживается в почве, а натрий накапливается в морской воде. Одни элементы, например В и Мо, накапливаются в растениях, в которые они переходят из почвы, содержание других, например лития, при этом значительно уменьшается. Организмы животных не нуждаются в таких элементах, как Мп, В, Sr; некоторые элементы проходят через организм без изменения, например Мо, Си, а литий накапливается в организме. Сейчас хорошо известно важнейшее влияние лития на организмы животных и иа психическое со-стояне людей.
43.3. Анализ следовых количеств элементов и охрана окружающей среды
Земная кора по-прежнему остается источником следовых количеств элементов, поступающих в почву, растения и в организмы животных, но уже все большее влияние на круговорот следовых количеств элементов, например Fe, Си, Pb, Ni, Zn, Cd, Hg, оказывает промышленность. В сточных водах и воздухе содержатся повышенные концентрации многих элементов. Табл. Д.26 дает представление об элементах, содержащихся в атмосфере.
В то время как изменение естественного содержания элементов в воде (реках, грунтовых водах) можно легко установить и не использовать эту воду для питья, при загрязнении атмосферы это сделать гораздо труднее: воздушные массы быстро и беспорядочно перемещаются и, кроме того, все дышат окружающим воздухом. Поэтому контроль чистоты атмосферы — важнейшая задача аналитической химии следовых количеств.
Установлено, что содержащиеся в воздухе в виде аэрозолей твердые частицы представляют собой в основном SiO2 и силикаты Fe, Al, Са, Mg, поступающие в воздух из земной коры, а также карбонаты и хлориды Na и Mg из морской воды. В состав и тех и других частиц входят также в значительных количествах такие следовые элементы, как Си, Мп, V, Ti, Cd, Со,
43. Методы и проблемы анализа следовых количеств
409
Таблица Д.26. Количество металлов, попадающих в течение года в атмосферу (США, 1976 г.)
Металл Количество
т/год %
РЬ 200 000 30
Zn 160 000 24
Ti 80 000 12
Mg 75 000 11
V 20 000 3
Мп 18 000 3
Другие 100 000 15
Итого: 660 000 100
Zn, Ni, Cr, Pb. Общая концентрация аэрозолей составляет 20— 150 мкг/м3. Химический состав аэрозолей зависит от географических условий, источника загрязнения атмосферы, слоя атмосферы и погодных условий (направление ветра, атмосферные осадки) (табл. Д.27).
Таблица Д.27. Содержание следовых количеств элементов в атмосфере (мг/м3) в различных географических районах
Элемент США (Чикаго) Северная Атлантика Южный ПОЛЮС
А1 2 175 8—370 0,57
Fe 13 800 3,4—220 0,84
V 18,1 0,06—14 0 0015
Сг ИЗ 0,07—1,1 0,1—64 0,0053
РЬ 35-380 0,63
Se 3,8 0,09—0,4 0,0056
В аэрозолях, твердые частицы которых попали в воздух из морской воды, можно отметить значительное накопление многих следовых элементов. Если за элемент сравнения принять алюминий, являющийся одним из основных компонентов земной коры, и обозначить отношение следовых количеств элементов к содержанию алюминия как х/А1, то для аэрозолей океанического происхождения, находящихся, например, над. Северной Атлантикой, получим следующее увеличение содержания ряда элементов: Fe—1,4; Мп — 2,6; Сг—11; Zn— 100; Cd — 730; Pb — 2200; Se — в 10 000 раз. Особенно опасные для здоровья элементы больше других накапливаются в воздухе. Атмосферные осадки, содержащие эти элементы, пора
410
Д. Химический, анализ
жают окружающую среду, загрязняя флору и поверхностные воды. Следует помнить о воздействии, оказываемом ими на человеческий организм. Наименьшие частицы аэрозолей (< 1 ммк), аккумулирующие наиболее ядовитые элементы, могут глубоко проникать в легкие и оттуда попадать в кровь и ткани организма. В настоящее время наряду с издавна известными путями проникновения ядовитых веществ в организм через органы пищеварения следует учитывать возможность попадания ядовитых веществ в организм через органы дыхания.
Отклонения в содержании следовых количеств элементов в организме человека оказывают влияние на его состояние здоровья, рост, самочувствие и работоспособность. Раньше для контроля содержания этих элементов в организме служил анализ крови и мочи. Однако при этом наблюдались значительные колебания в результатах анализа, связанные со временем приема пищи, содержанием металлов в атмосфере рабочих помещений и другими причинами. В настоящее время известно, что в волосах происходит накопление следовых элементов из организма с увеличением их концентрации от миллиардных до миллионных долей, т. е. в 1000 раз. Следовые элементы задерживаются белками, которые реагируют на изменение содержания следовых элементов в организме человека в процессе всего цикла роста волос. Поэтому в целом волоске определяют среднее содержание следовых элементов, а при исследовании небольших его сегментов — накопление элемента за отдельные промежутки времени.
Для проведения анализа пригодны методы, обладающие наибольшей эффективностью. В первую очередь это нейтронноактивационный анализ или атомно-абсорбционная спектроскопия. О высокой эффективности инструментальных методов определения следовых количеств элементов свидетельствует тот факт, что для определения изменяющегося во времени содержания элемента исследуют кусочки волоса длиной всего несколько миллиметров. При определении влияния загрязнения окружающей среды на людей необходимо использовать средние данные, характерные для большинства населения, чтобы уменьшить отклонения, связанные с индивидуальными особенностями. С другой стороны, именно индивидуальные особенности представляют интерес для криминалистов. Если преступник оставляет на месте преступления волос, то по этому волосу его можно обнаружить. При этом используют зависимости, существующие между содержанием следовых количеств элементов в волосах и режимом питания, местом жительства, возрастом, полом и расой преступника.
43.4. Аналитическая химия особо чистых веществ
К аналитической химии особо чистых веществ относится анализ следовых количеств элементов в области концентраций <10~8%. В промышленности вещество считается технически чистым, если свойства вещества, важные для его применения,
43. Методы и проблемы анализа следовых количеств
411
при повторной очистке вещества больше не изменяются. В аналитической химии под чистым веществом понимают вещество, в котором с помощью соответствующих эффективных методов анализа загрязнения уже не обнаруживаются. Из сказанного ясно, что с развитием требований к свойствам веществ, к технологии их очистки и эффективности инструментальных методов анализа степень чистоты веществ все более сдвигается в область меньших концентраций загрязнений.
В начале столетия вещество считалось чистым, если оно содержало меньше 0,1% примесей. Ни науке, ни практике (за редким исключением) еще не были нужны особо чистые вещества. Даже в тридцатых годах нашего сто-ления проблема чистоты веществ не стояла особенно остро. Содержание примесей ворядка 1 млн.-1 в то время представляло интерес только* для геохимиков, оперирующих такими числами при описании распределения химических элементов в земной коре. Металлурги считали вещество достаточно чистым, если в нем содержалось не более чем 0,01 % примесей.
Стремительный рост производства особо чистых веществ в пятидесятых годах был вызван развитием ядерной энергетики, которой потребовались материалы и вещества с необычно высокой для того времени степенью чистоты, например уран, содержащий не более 100 млрд.-1 или даже 10 млрд.-1 примесей. Еще более высокие требования к чистоте веществ стали предъявляться в последние десятилетия в. связи с развитием электронной промышлеяности. Чистота полупроводниковых материалов, таких, как Ge или Si, должна быть в 1000 раз больше, чем указанные для урана значения. Содержание некоторых примесей в них не должно превышать 10~2 млрд.-1 (10-12%). Аналогичные требования предъявляют к химическим реактивам, применяемым в ходе обработки полупроводниковых элементов, таким, как кислоты, соли и органические растворители.
Приведенное выше определение понятия особо чистых веществ дано исходя из исторического процесса развития науки и техники. С химических же позиций особо чистым являлось бы вещество, не содержащее атомов или молекул посторонних веществ. В этом соединении, кроме того, ни один из компонентов не должен был бы находиться в избытке. Однако доказано, что большинство соединений имеет нестехиометрический состав и что отклонения от стехиометрии часто оказывают значительное влияние на свойства твердых веществ, например для полупроводников AIIIBV или AIIBVI. Задачей аналитической химии особо чистых веществ в данном случае является не только определение экстремально низких следовых количеств посторонних веществ, но и определение крайне малого (Ю-5%) отклонения содержаний обоих основных компонентов от оптимального теоретического соотношения. Вторую задачу решает так называемая прецизионная аналитическая химия, одним из основных методов которой является высокоэффективный вариант кулонометрии.
Многие свойства веществ, такие, как оптические или электрические, не только существенно зависят от присутствия посторонних компонентов, но и значительно изменяются при очень небольшом варьировании концентраций следовых количеств
412
Д. Химический анализ
примесей. Для решения такой трудной задачи необходимо объединение методов аналитической химии следовых количеств и прецизионной аналитической химии. Результаты анализов одной и той же пробы на содержание следовых количеств веществ, выполненных в разных лабораториях, часто существенно различаются (иногда даже на порядок). Например, в семи лабораториях проводили определение содержания углерода в образце молибдена и получили следующие тщательно проверенные средние значения: 5; 11; 10; 16; 21; 10 и 9 млн-1. Идеальным условием определения следовых количеств элементов в пробе является их равномерное распределение в ней, как, например, в гомогенной жидкой или газообразной фазе; в этом случае ошибка анализа определяется только правильностью и воспроизводимостью метода. Анализ твердых веществ усложняется неравномерностью распределения в них следовых количеств элементов. В этом случае проба может быть неоднородной по чистоте, и, следовательно, не представительной. В целом вероятность неравномерного распределения следовых количеств элементов возрастает с уменьшением их содержания.
Все вышесказанное можно продемонстрировать на примере исследования неравномерности, распределения следовых количеств элементов в чистом металлическом марганце, полученном электролизом. В табл. Д.28 приведены результаты анализа (в млн-1) стружек марганца диаметром 0,5—2 см и толщиной 2 мм. Были отобраны две пробы массами 500 и 10 г соответственно. Проба массой 500 г была предварительно гомогенизирована измельчением в порошок. По результатам 16 измерений каждой пробы рассчитаны стандартные отклонения ssoo и ею. Отношение sio/ssoo показывает, что при работе со слишком маленькой пробой (10 г), состав которой не является представительным, увеличиваются значения случайного разброса. Для содержания железа эта величина, например, колеблется в пределах 1—3 (рис. Д.171).
При установлении зависимости относительного стандартного отклонения от массы пробы при определении 10 млн-1 цинка методом атомной абсорбции с пламенной атомизацией пробы оказалось, что стандартное отклонение приближается к предельному для данного метода значению 1,5%, только если масса пробы превышает 100 г (рис. Д.172).
Таблица Д.28. Неравномерность распределения следовых количеств элементов в металлическом марганце3
Элемент Количество в 500 г, млн"’ S5<3Q 5j0 S)o/S5DQ
Со 2,1 1,8 9,0 5,0
Си 5,9 3,7 49 13,2
Fe 12,1 2,2 57 25,9
Ni 4,8 3,9 9,3 2,4
Pb 2,5 1,6 6,1 3,8
Zn 4,6 9,4 17 1,8
а Цифры при обозначении стандартного отклонения показывают массу пробы.
43. Методы и проблемы анализа следовых количеств
413
Рис. Д.171. Графическое представление результатов определения содержания Fe в Мп.
/ — из гомогенизированной пробы массой 500 г; 2 — из отдельной пробы массой 10 г.
Рис. Д.172. Зависимость стандартного отклонения Доти от массы пробы при определении содержания Zn в марганце Методом атомно-абсорбционной спектроскопии.
При обсуждении вопроса о возможности получения абсолютно чистых веществ необходимо учесть следующее. При проведении очистки вещества, например методом зонной плавки, в основном достигается лишь вполне определенная предельная степень чистоты. Технические и временные затраты возрастают, как и в любых других методах, с ростом требований к степени чистоты и для получения абсолютно чистых веществ должны быть бесконечно большими. Кроме того, материал сосудов, в которых получают и хранят чистые вещества, всегда неизбежно является источником их загрязнений. Получение и
414
Д. Химический анализ
очистка природных изотопов некоторых элементов (40К, 87Rb,a 130Те) осложняется их радиоактивным распадом или превра-Я щениями, происходящими под влиянием космического излуче-Я ния. .1
В соответствии с существующей в настоящее время теоре-Я тической концепцией получение абсолютно чистых веществ Я (т. е. совершенно не содержащих примесей) принципиально Ц возможно, но только в очень небольшой области концентраций Я для достаточно большой пробы чистого вещества и за более я или менее ограниченный промежуток времени. Для контроля чистоты необходимы особо чувствительные методы анализа. Я Применение методов ультрамикроанализа позволяет осущест- и вить «мечту» аналитиков — обнаружение отдельных атомов в Я матрице вещества. Одним из таких методов является лазерная л спектроскопия. Вещество испаряют и атомы селективно возбуждают действием лазерного излучения в узкой области частот. Возбужденный атом затем ионизируется вторичными фотонами. Число испускаемых при этом свободных электронов фиксируют пропорциональным счетчиком. С помощью эффективно действующей лазерной установки можно ионизировать все атомы определяемого вещества. Метод, основанный на использовании этого явления, называют резонансной ионизационной спектро- < скопией (РИС). Например, можно определять отдельные ато- ' мы цезия. В другом варианте метода — оптически насыщенной ’ нерезонансной эмиссионной спектроскопии (ОНРЭС) — изме- •' ряют интенсивность флуоресцентного излучения возбужденных атомов. Чтобы отличить излучение определяемых элементов от излучения других компонентов пробы, длины волн флуоресценции сдвигают воздействием других атомов или молекул. Этим методом также можно определять отдельные атомы вещества, например натрия.
43.5. Методы аналитической химии следовых количеств
В данном разделе мы попытались выбрать из существующих и разрабатываемых в настоящее время методов анализа те методы, которые можно успешно применять в анализе следовых количеств элементов. Следует учесть, что любой выбор не всегда объективен, так как зависит от субъективной оценки метода и прежде всего от постановки задачи. Для решения задач анализа следовых количеств элементов наиболее пригодны следующие методы*:
* В аналитической химии микроколичеств большое значение имеют методы, основанные на предварительном концентрировании определяемых микроэлементов и последующем анализе концентрата физическими, прежде всего,
43. Методы и проблемы анализа следовых количеств 415
Абсорбционная спектрофотометрия растворов (АСР) Флуориметрии
Инверсионная вольтамперометрия
Дифференциальная импульсная полярография Ионометрия
Атомно-абсорбционная спектроскопия (ААС) Атомно-флуоресцентная спектроскопия (АФС) Эмиссионная спектроскопия (ЭС)
Рентгено-флуоресцентная спектроскопия (РФС)
Комбинированная фотоэлектронная спектроскопия и электронная оже-спектроскопия
Искровая масс-спектроскопия (ИМС)
Газовая хроматография (ГХ) Активационный анализ (АА) Кинетические методы
Метод абсорбционной спектрофотометрии растворов начали применять в анализе следовых количеств элементов в 50-е годы. С помощью этого метода можно обнаружить до 0,1 мкг/см3 вещества. Однако при определении возникает много помех, в том числе и матричный эффект. Желательно применять для определения каждого элемента специальный реагент.
В методе флуориметрии измеряют не поглощение света веществом, а интенсивность длинноволновой эмиссии. Поэтому в отличие от АСР в этом методе с увеличением интенсивности облучения увеличивается чувствительность, что приводит также к значительному снижению предела обнаружения. В этом случае увеличивается также селективность. Метод модифицируют, меняя растворитель, pH и реагенты, а также длины волн флуоресценции и возбуждающего света.
Хорошие результаты получаются и с помощью некоторых электрохимических методов. Но их применение еще находится в стадии разработки, например внедрение в практику ионселек-тивных электродов. Иногда на эти м'ётоды оказывают существенное влияние условия определения и матричный эффект. Часто селективность их недостаточна для определения отдельных элементов при совместном присутствии. В постояннотоковой полярографии предел обнаружения составляет 1 мкг/см3, селективность мала; в переменнотоковой полярографии при том же пределе обнаружения селективность лучше; в квадратноволновой полярографии,' импульсной полярографии и дифференциальной импульсной полярографии предел обнаружения 0,01 мкг/см3, а селективность повышается в порядке перечисления методов. При сочетании постояннотоковой полярографии с
спектрохимическими методами. См. книги: Золотов Ю. А., Кузьмин Н. Л4. Концентрирование микроэлементов. — М.: Химия, 1982; Спектроскопические методы определения следов элементов./Под ред. Дж. Вайнфорднера. Пер. с англ. — М.: Мир, 1979. — Прим. ред.
416
Д. Химический анализ
каталитическими или кинетическими методами предел обнару- Я жения может достигать 10-5 мкг/см3. Однако проблемы селек-тивности этих методов также достаточно сложны. Я
В случае простой инверсионной вольтамперометрии с при- || менением ртутного капельного электрода можно обнаруживать Я до 10-3 мкг/см3 при небольшой селективности. При переходе к дифференциальным импульсам предел обнаружения достигает 10-5 мкг/см3 при хорошей селективности. Если к тому же за-меняют ртутный капельный электрод слоем ртути, нанесенным на стеклографит, чувствительность обоих методов возрастает ; в 100 раз, в то время как селективность не изменяется.
В атомной спектроскопии различают методы, основанные на использовании явлений эмиссии, абсорбции и флуоресценции. Источниками возбуждения эмиссии могут быть дуга, искра, пламя (существует также хемилюминесценция, основанная на использовании энергии химической реакции горения веществ). Все большее значение приобретают методы, основанные на возбуждении эмиссии высокочастотной плазмой и прежде всего индуктивно связанной плазмой (ИСП). Сравнение показывает, что возбуждение эмиссии с использованием дуги и искры не может составить конкуренцию современным методам (табл.
Таблица Д.29. Пределы обнаружения элементов (мкг/см3) методами U
атомной спектроскопии И
Элемент Источник возбуждения эмиссии Атомизатор в методах абсорбции
искра пламя (CN)2 + n2o плазма пламя печь
Ва 0,4 0,02 0 0001 0,008 0,00015
В 0,1 30 0,003 0,7 0,15
Cd 4 0,8 0,003 0,002 0,000003
Мп 0,08 0,005 0,0006 0,002 0,00001
Р - — 0,02 50 0,03
Ti 0,4 0,2 0,002 0,04 0,0005
W ,— — 0,02 1 —
V 4 0,01 0,002 0,04 0,0001
Zr 2 — 0,004 0,35 —
и — 10 0,04 30 —
Д.29). Можно также считать, что высокочастотная плазма успешно конкурирует с другими источниками возбуждения эмиссии (например, таким «экзотическим» пламенем, как смесь '(CN)2 + N2O). Этот метод имеет также преимущества перед методом пламенной атомно-абсорбционной спектроскопии (ПААС). Но предел обнаружения ААС с электротермическими источниками атомизации (например, с применением графито
43. Методы и проблемы анализа следовых количеств 417
вых кювет) значительно ниже, чем дает ИСП, за исключением таких элементов, которые практически невозможно определить ААС из-за образования трудно испаряющихся оксидов (С1, Zr, W и др.). Методы, указанные в табл. Д.29, обладают сравнимой точностью (2—3%). Точность неудовлетворительна только при работе с графитовыми кюветами (5—10%), но в них зато можно работать с малыми количествами пробы. Абсолютный предел обнаружения методами ААС и АФС составляет Ю-10—10-14 г. При анализе растворов часто используют объемы до 1 мм3, поэтому предел обнаружения составляет 10-1— Ю-5 мкг/см3. Методы ПААС и ААС уже применяют в серийных анализах, все больше внедряется в производство и плазменно-эмиссионный анализ; использование метода атомнофлуоресцентной спектроскопии все еще остается редким.
Рентгено-флуоресцентная спектроскопия (РФС) приобретает все большее значение в анализе следовых количеств элементов. В качестве источника возбуждения используют обычную рентгеновскую трубку или чаще радиоактивные изотопы. Этот метод относится к неразрушающим и позволяет определять содержание многих элементов; это обеспечило ему прочное положение при проведении серийных анализов твердых веществ. Предел обнаружения элементов во многих случаях составляет >10 млн-1. Но и в этом методе необходимо применять эталоны. В сочетании с химическими методами концентрирования (например, с осаждением с малорастворимыми сульфидами) дает хорошие результаты при анализе жидких или растворенных проб: во многих случаях можно снизить предел обнаружения на несколько порядков, если удается взять для анализа достаточно большую пробу (например, при анализе родниковой, речной, морской воды на содержание следовых количеств элементов).
Особое место занимают такие методы анализа поверхностей, как комбинированная фотоэлектронная спектроскопия или электронная оже-спектроскопия. Эти методы позволяют установить распределение элементов в слоях твердых тел, а также проводить градиентный анализ по глубине. Это физические методы исследования структуры, но с их помощью можно с рчень большой чувствительностью определить следовые количества элементов. Однако из-за высокой стоимости оборудования и необходимости высококвалифицированного обслуживающего персонала методы все еще применяют только в специализированных лабораториях.
Искровая масс-спектроскопия также требует высококвалифицированного обслуживания и очень дорогого оборудования. Этим методом проводят анализ твердой микропробы. Экстремально малый объем пробы делает проблематичным ее представительность и правильность получаемых результатов вслед
27—1880
418
Д. Химический анализ
ствие микронегомогенности пробы. Кроме того, необходимы эталоны, изготовление которых иногда очень сложно, например при анализе тугоплавких металлов. Поэтому метод медленно внедряется в практику количественного анализа.
Газовую хроматографию также можно применять в анализе следовых количеств элементов. Многие элементы, например А1, Cr, Be, Zn, Ga, In, Си и др., образующие летучие и термически достаточно устойчивые комплексы, можно селективно обнаружить и количественно определить. Для анализа можно применять такие комплексные соединения, как ацетилацетонаты, фторированные диэтилдитиокарбаминаты и в первую очередь фторированные <р-дикетонаты. Последние термически очень устойчивы, и, кроме того, электронный детектор особенно чувствителен к фторированным соединениям. При этом абсолютный предел обнаружения равен 10~13 г. Из-за небольшого объема анализируемой пробы при работе с растворами предел обнаружения в этом случае такой же, как в ААС.
Активационный анализ занимает значительное место в аналитической химии следовых количеств элементов. Он относится к наиболее чувствительным аналитическим методам; преимуществом его является возможность проведения неразрушающего анализа. В то же время реальные возможности метода определяются соотношением значений поперечных сечений захвата ядерных реакций изотопов определяемых элементов и элементов матрицы и периодов полураспада соответствующих нуклидов. Эффективность активационного анализа зависит также от видов применяемого возбуждения: нейтронами, заряженными частицами и фотонами. Поэтому часто становится необходимой предварительная радиохимическая подготовка пробы, например частичное растворение матрицы.
Сходная ситуация существует при использовании кинетических методов, которые основаны на том, что при прохождении многих реакций окисления-восстановления возникают кинетические помехи, обусловливающие очень медленное протекание реакций. Реакции можно ускорить, вводя в качестве катализаторов различные группы элементов. При этом в определенных пределах увеличение скорости реакции пропорционально концентрации следовых количеств элементов — катализаторов, что можно использовать для их количественного определения. Добавляя подходящие комплексообразующие реагенты, часто можно добиться увеличения селективности. Но, несмотря на это, небольшая селективность является недостатком этого высокочувствительного и относительно редко применяемого метода.
Выбор метода определения для решения конкретной аналитической задачи осуществляют, принимая во внимание следующие факторы: а) предел обнаружения; б) требуемое и име-
43. Методы и проблемы анализа следовых количеств 419
ющееся количество вещества для анализа (пробы); в) трудоемкость анализа и затраты времени на его проведение; г) имеющуюся аппаратуру или расходы на ее приобретение; д) требования к квалификации аналитика, простоту и надежность метода. Кроме того, следует учесть, насколько часто будет использоваться метод.
Для оптимизации анализа существуют сложные теоретические выкладки, в которые, однако, включены также субъективные оценки, так что в результате можно получить различные данные. С другой стороны, на практике всегда проводят оптимизацию по методу «проб и ошибок». Поэтому в лабораториях, где проводят анализ следовых количеств элементов, обычно получают средние значения из большого числа определений при соблюдении всех требований. Реальная ситуация в лабораториях часто такова, что приобретенные однажды приборы и разработанные методы из соображений экономии и просто по инерции применяют и тогда, когда они уже устарели. Нужно только, чтобы это отставание от современного уровня развития не превышало 10 лет.
В 1974 г. ИЮПАК провел опрос сотрудников ряда лабораторий о методах определения следовых количеств элементов в высокочистых химических реактивах. На основании 200 ответов методы были расположены в соответствии с полученными статистическими данными и частотой их использования в лабораториях (табл. Д.30). В ряде лабораторий используют все методы, в большинстве применяют несколько методов, и только немногие лаборатории специального назначения используют в работе один метод. Как видно из табл. Д.30, атомная абсорбция с пламенной или электротермической атомизацией, а также определение щелочных металлов методом фотометрии пламени занимают первое место, за ними вплотную следуют метод абсорбционной спектрофотометрии растворов; несколько реже
Таблица Д.30. Частота использования методов анализа следовых количеств элементов в чистых химических реактивах
Метод
Количество лабораторий, применяющих метод, %
Атомно-абсорбционная спектроскопия и фотометрия пламени
Абсорбционная спектрофотометрия растворов
Эмиссионная спектроскопия с возбуждением дугой или искрой
Полярография
Активационный анализ
Рентгено-флуоресцентная спектроскопия
Масс-спектроскопия
Другие методы (электрохимические, хроматографические)
48,5
41,5
23
18,5
14
13
9
20
27*
420
Д. Химический анализ
Таблица Д.31. Частота использования (в %) основных методов для определения некоторых элементов в чистых химических реактивах
Элемент Метода
ААС н ФП АСР эс АА Пол РФС
Со 30 22 27 10 и
Bi —- — 50 20 30 —
Сг 23 21 29 8 и 8
Си 33 19 15 9 15 8
Fe 32,5 32,5 16 7 4 8
Мп 35 20 19 5 11 10
К 84 — — 16 .— —
Na 78 — — 22 —— —
Мо 13 32 30 8 11 6
W — 38 31 31 — —
Si — 45 42 13 — —
а Полные названия даны в начале разд. 43.5. Пол — полярография; ФП—фотометрия пламени.
используется эмиссионная спектроскопия с дуговыми или ис-кровыми источниками возбуждения. В табл. Д.31 приведены данные аналогичного опроса о методах определения отдельных элементов. В этом случае промежуточное положение занимают методы эмиссионной спектроскопии с искровым, дуговым, а также лазерным возбуждением, а доминируют атомно-абсорбционная спектроскопия и эмиссионный анализ с возбуждением высокочастотной плазмой.
43.6. Методы разделения и концентрирования
в аналитической химии следовых количеств
При проведении операций разделения и концентрирования преследуют две цели: во-первых, увеличение концентрации следовых количеств элементов для последующего анализа и, во-вторых, отделение следовых количеств определяемых элементов от основных компонентов (матрицы) и от следовых количеств других элементов. При концентрировании, проводимом для достижения первой цели, нужно ориентироваться на предел обнаружения, достигаемый данным методом. При отделении следовых количеств определяемых элементов следует исходить из селективности метода определения, т. е. ориентироваться на возможность мешающего действия других элементов.
Методы выделения и концентрирования следовых (милли-, микро- или нанограммовых) количеств элементов из большой массы матрицы должны отличаться высокой селективностью.
43. Методы и проблемы анализа следовых количеств 421
Классические методы осаждения и фильтрования пригодны лишь в том случае, если в осадок выпадает только соединение выделяемого элемента, так как при осаждении основного компонента пришлось бы иметь дело с большими поверхностями фильтра и осадка. Кроме того, часть раствора, содержащего следовые количества элементов, включалась бы при этом в осадок за счет окклюзии. Для выделения следовых количеств элементов и концентрирования используют хроматографические методы, электролиз, соосаждение, адсорбцию и экстракцию.
Под хроматографией в данном случае понимают все методы, в которых поток подвижной жидкой или газообразной фазы взаимодействует со стационарной фазой. К этим методам относятся следующие:
1. Адсорбционная хроматография. Для сорбции и повторного выделения в раствор комплексных соединений металлов, находящихся в следовых количествах в органических растворителях, используют носители, например А12О3, CaSO4, СаСОз, MgO и др. В аналитической химии следовых количеств веществ в основном применяют метод тонкослойной хроматографии.
2. Распределительная хроматография. На поверхности носителя (например, SiO2 или целлюлозы) удерживается пленка жидкой фазы, через которую проходит другая, не смешивающаяся с ней фаза. Наиболее часто применяют бумажную хроматографию.
3. Ионообменная хроматография. С ее помощью можно отделять мешающие определению элементы или, наоборот, определяемые элементы при прохождении анализируемого раствора через ионообменную колонку. Если определяемый элемент затем выделить в небольшой объем растворителя, можно сконцентрировать следовые количества элемента до легко измеримых концентраций, и поэтому такой способ концентрирования приобретает все большее значение при анализе следовых количеств элементов. Четкость разделения элементов, сорбируемых ионообменной смолой, можно увеличить, применяя при элюировании комплексообразующие реагенты. Особенно эффективным вариантом метода является использование комплексообразующих ионообменных смол. Эти смолы содержат активные группы, способные к образованию специфичных комплексов с определяемыми ионами, которые задерживаются смолой. При этом происходит эффективное разделение.
4. Газовая хроматография. Следовые количества газообразных компонентов пробы, находящихся в газовом потоке, разделяются в ходе адсорбции твердым носителем или распределяются между газообразной фазой и жидкой фазой, находящейся на твердом носителе. Этот метод связан с детектированием, поэтому его часто относят к методам определения (см. разд. 43.5).
422
Д. Химический анализ
Электролиз можно применять для выделения следовых количеств элементов, стоящих в ряду напряжений дальше, чем элемент матрицы или другие мешающие определению компоненты. При этом можно провести кулонометрическое определение с одновременным электрохимическим отделением или выделить элемент электрохимически или химически, а затем применить другие методы анализа. После выделения следовых количеств элементов на проволоке из инертного тугоплавкого металла их можно определить эмиссионными методами, внося проволоку, например, в пламя. Электролиз можно также применить для отделения матрицы, если металл матрицы стоит в ряду напряжений дальше, чем элемент, содержащийся в следовых количествах. Такие выделения обычно осуществляют, проводя восстановление на ртутном катоде. Преимуществом использования ртутного катода по сравнению с электролитическим осаждением является то, что не происходит адсорбции следовых количеств элемента, т. е. определяемый элемент практически полностью остается в растворе, не содержащем ионов металла матрицы. Но с другой стороны, при этом не достигается концентрирование определяемого элемента.
Выделение в осадок следовых количеств элемента — сложная задача. Применение соосаждения ограничено растворимостью веществ, явлениями коллоидообразования и трудностями, возникающими в связи с ними при фильтровании, а также проблемой выделения и дальнейшей переработки столь малых количеств осадка. Перед осаждением вводят специальный коллектор, который в отличие от матрицы не мешает при последующих операциях и при осаждении увлекает с собой следовые количества элементов. Например, проводят осаждение в виде сульфидов, используя в качестве коллектора Hg2+ или As3+, которые затем испаряются при нагревании, а в остатке концентрируются следовые количества определяемых элементов. Действие коллектора основано на образовании смешанных кристаллов, соединений, ионном обмене, адсорбции и других явлениях, например зародышеобразовании. Наряду с сульфидами коллекторами могут служить галогениды серебра, Ре(ОН)з, МпО2-хН2О и др.
Большое значение имеет, например, метод выделения и концентрирования, основанный на обменном осаждении. Этот метод применяют при анализе воды (питьевой, талой, речной, морской) ,на содержание следовых количеств Hg, Ag, Си, Bi, Pb, Cd, Sn, As, Se, Те, Zn, Co и Ni. Пробу воды (от 0,1 до 6 дм3; pH 3—6) просасывают через гомогенный слой свежеосажденных сульфидов (ZnS, MnS, CuS, PbS и др.), находящийся на мембранном фильтре (нитрат целлюлозы или политетрафторэтилен с диаметром пор <1 мкм). Толщина слоя сульфида 300—400 нм. При этом из раствора практически полностью выделяются элементы (за исключением мышьяка), образующие малорастворимые сульфиды, величина произведения растворимости которых меньше, чем для сульфида обменного слоя (табл. Д.32).
43. Методы и проблемы анализа следовых количеств
423
Таблица Д.32. Произведения растворимости сульфидов некоторых элементов
Сульфид Сульфид Kl
B12S3 1,6-10-72 PbS 3,4-10~28
HgS 3-10-54 CoS 1,9-IO-22
AgaS 1,6-ю-49 ZnS 6,9- 10-2e
CuS 8-Ю-43 NiS 1-10~26
As2Sa 8-Ю-29 FeS 4-10-1’
CdS 7-10-28 MnS 7- 10-ie
Таблица Д.ЗЗ. Результаты анализа воды методами РФС и ААС
Анализируемый объект Количество Hg, мкг/см3 Количество Cd, мкг/см3
РФС ААС РФС ААС
Питьевая вода (1) ^0,2 0,003 0,6 0,6
Питьевая вода (2) ^0,2 0,004 0,3 0,6
Талая вода — —- 0,5 0,5
Речная вода 0,2 0,17 sC0,l —
Морская вода (береговая полоса) 1,9 — 0,75 —
После обмена можно провести определение элементов или методом рентгено-флуоресцентной спектроскопии (РФС) прямо на фильтре или после растворения осадка в небольшом количестве смеси кислот (600 мм3; 80 °C; НО: : HNOa : Н2О= 1 : 1 : 2) методом атомно-абсорбционной спектроскопии (ААС) с графитовой кюветой. При таком концентрировании предел обнаружения может иметь порядок 1 нг/см3. В табл. Д.ЗЗ приведены результаты анализа воды.
Адсорбция. С учетом различных механизмов действия (адсорбции, образования соединений с отделяемыми компонентами и т. д.) в качестве коллекторов были исследованы галогениды и другие малорастворимые соли серебра. Известно, что свежие осадки AgX имеют поверхностный заряд, знак которого зависит от заряда ионов, участвующих в реакции осаждения и находящихся в избытке, т. е. отрицательный при избытке ионов X", сорбированных осадком, и положительный при избытке Ag+. Осадок AgX — эффективный сорбент: наряду с ионами элементов на его поверхности могут удерживаться ионы различных молекул и полярные вещества; на этом принципе основано, в частности, применение адсорбционных индикаторов в аргентометрическом титровании по методу Фаянса. При этом катионы находятся на поверхности отрицательно заряженных осадков, анионы — на поверхности положительно заряженных.
424
Д. Химический анализ
Осадки солей серебра в каждом конкретном случае сорбируют лишь небольшую группу элементов. Селективность адсорбции можно целенаправленно изменить, связывая ионы металлов в катионные или анионные хелатные комплексы. Поэтому образование осадка AgX особенно эффективно для отделения следовых количеств элементов в сочетании с такими реакциями. Хорошо изучена сорбция хелатов 1,10-фенантроли-на и его аналогов:
Следовые количества ионов элементов, которые не сорбируются или плохо сорбируются осадками AgX, образуют с 1,10-фенантролином и подобными комплексообразующими реагентами катионные хелаты, сорбируемые отрицательно заря-
Таблица Д.34. Элементы, сорбируемые в виде
1,10-фенантролиновых комплексов солями серебра*
Элемент Соль
AgCl AgBr Agl AgSCN
Fe(II) + 4- +
Fe(III) + — + +
Ni(II) + — ' + +
Co(II) +
Cu(II) + + 4-
Pb(II) + +
Bi (III) + +
Tl(III) + +
Au(III) +
Hg(II) +
Zn(II) + + + +
Pd(II) + + + +
V(IV)
Mn(II) —- — .— —•
U(VI) —* — — —
а Знак плюс соответствует сорбированию более 95%, знак минус — сорбированию менее 5%.
женными осадками галогенидов серебра в присутствии избытка галогенид-ионов в растворе. В табл. Д.34 приведены результаты, полученные при выделении ряда элементов этим методом.
Осадки AgCl или AgBr сорбируют фенантролиновые хелатные комплексы только после точки эквивалентности; напротив,
43. Методы и проблемы анализа следовых количеств 425
при выпадении осадков Agl и AgSCN заметная сорбция происходит, даже если в растворе еще находятся ионы серебра. В этом случае и после точки эквивалентности сорбция происходит количественно. При образовании осадка AgCl не связанные в комплекс ионы Fe2+ практически полностью находятся в растворе (рис. Д.173).
Рис. Д.173. Зависимость содержании Ре2+-иоиов в растворе от количества титранта NaX при, осаждении AgCl или AgSCN.
з — точка эквивалентности при осаждении AgCl.
Сорбция хелатов осадками AgCl или AgBr —• процесс обратимый; при добавлении избытка Ag+ происходит десорбция. Напротив, в случае Agl и AgSCN сорбция необратима. Поэтому в растворе уже до достижения точки эквивалентности заметно падает содержание свободных (несорбированных) катионных хелатных комплексов металлов. Катионные комплексы металлов с фенантролином или его аналогами образуют с ионами I- или SCN- малорастворимые соединения. Но и в области концентраций, где не достигается произведение растворимости, адсорбция достаточно заметна, так что оба эффекта — сорбция и осаждение — накладываются.
Если в растворе идут конкурирующие реакции хелатообразования, то степень сорбции уменьшается. К таким реакциям относятся, например, реакции образования прочных галогено-комплексов, а также разрушения хелатов кислотами. Например, фенантролиновый комплекс Zn в среде 0,05 н. H2SO4 практически не сорбируется осадками AgBr и Agl, а в почти нейтральной среде сорбируется на 95%. Если в образовании хелатных комплексов принимают участие отрицательно заряженные лиганды, как, например, ионы ЭДТА, то образуется
426
Д. Химический анализ
отрицательно заряженный комплекс, который сорбируется осадком AgX при избытке Ag+ и десорбируется при избытке Х~. Поскольку ионы АР+, Сг3+, Мп2+ и щелочноземельных элементов не сорбируются малорастворимой солью серебра, металлы после переведения в раствор или их соли можно очищать описанным методом от следовых количеств примесей, указанных в табл. Д.34 (но для этого нужен AgNOs высокой чистоты). Переведение следовых количеств элементов из осадка AgX в раствор (например, из Agl) можно осуществить действием 65 %-ной HNO3. При этом достигается коэффициент обогащения 103, предел обнаружения 0,01 млн-1 и стандартное отклонение 5%.
При отделении следовых количеств элементов собственно адсорбционным методом в качестве коллектора применяют чаще всего активированный уголь. Следовые количества элементов при этом связывают в комплекс, например, действием ксан-тогената калия при оптимальной величине pH 6,5—7. Затем раствор пробы фильтруют через маленький бумажный фильтр, в котором находится слой активированного угля. Этот слой получают, нанося 10 см3 водной суспензии предварительного активированного угля на фильтр. Для определения методом ААС с пламенем в качестве источника излучения следовые количества элементов, сконцентрированных на угле (коэффициент обогащения до 2-Ю3), можно перевести в раствор обработкой кислотой.
Основным методом отделения и концентрирования следовых количеств веществ является экстракция (см. гл. 38). Многие элементы можно перевести в соединения, малорастворимые в воде, но хорошо растворимые в органических растворителях. Применяя различные лиганды для комплексообразования, меняя pH водного раствора, степень окисления экстрагируемых катионов и применяя различные растворители для экстракции, можно варьировать степень извлечения и концентрирования. В идеальном случае извлекаемый элемент при встряхивании (для увеличения поверхности раздела фаз с целью ускорения достижения равновесного состояния) полностью переходит в органическую фазу, в то время как мешающие определению элементы остаются в водной фазе. Таким образом удается отделить следовые количества элементов от больших количеств других элементов (матрицы) и сконцентрировать их.
В анализе следовых количеств элементов экстракция (или любое выделение как таковое) считается полной, если степень извлечения определяемого элемента составляет 95%. Возникающие при этом систематические ошибки стараются устранить, проводя экстракцию эталонных добавок из такой же матрицы и при одинаковых условиях эксперимента. При оптимальных соотношениях элемента, комплексообразующего реагента и
43. Методы и проблемы анализа следовых количеств 427
растворителя можно показать, что даже при так называемой полной экстракции можно провести еще одно экстрагирование и снизить концентрацию извлекаемого элемента до 0,1 нг/см3. Встряхивание обычно проводят вручную. При автоматизации процесса экстракции, т. е. применении механического перемешивания и разделения, удается снизить результаты холостого опыта, улучшить воспроизводимость, а также понизить предел обнаружения. Необходимое при этом увеличение затрат, прежде всего при работе с радиоактивными веществами (для выполнения мероприятий по технике безопасности), вполне оправдано.
Используемые при экстракции следовых количеств элементов комплексные соединения можно условно разделить на две группы. К первой группе относятся прежде всего ацидокомп-лексы (например, HFeCB), нитраты и тиоцианаты (например,. [Fe(SCN)g] 3“), многие из которых можно извлечь сложными и простыми эфирами и кетонами из сильнокислых растворов. Однако селективность экстрагирования в этом случае практически нельзя изменить. Ко второй группе относятся почти исключительно хелаты (в том числе внутрикомплексные соединения, в которых ион металла соединяется несколькими связями с атомами реагента (О, S, N) с образованием нескольких циклов. Хелаты часто удовлетворяют указанным в начале раздела условиям, так как обладают малой растворимостью в воде, хорошо растворяются в органических растворителях и т. д. Для дальнейшего увеличения селективности экстракции применяют такие комплексообразующие реагенты, как фториды, цианиды, цитраты, тартраты, амины, а также ЭДТА в водном растворе, служащие для маскировки компонентов, которые не должны извлекаться при экстракции. Часто используют следующие комплексообразующие реагенты: р-дикетоны, 8-оксихинолин и его производные, оксимы, дифенилтиокарбазон (дитизон), диэтилдитиокарбамат (ДДТК), пирролидиндитиокарбамат и многие другие. В качестве растворителей применяют хлороформ, эфиры, кетоны и, чаще всего, метилизобутилкетон (МИБК).
Экстракционное концентрирование хорошо сочетается с определением следовых количеств элементов методом пламенной ААС. Точно отмеряют объем, например 200 см3, анализируемой пробы воды (например, морской) или водного раствора исследуемой матрицы (типа Na2SO4, Na2WO4 и др.). При встряхивании насыщают раствор МИБК, затем отделяют от МИБК и переносят в мерную колбу вместимостью 250 см3. Для стабилизации pH в колбу добавляют 20 см3 10%-noro раствора цитрата аммония, служащего вспомогательным комплексообразующим реагентом и для повышения содержания солей. Раствор цитрата аммония одновременно является 3%-ным по диэтил-дитиокарбаминату диэтиламмония (ДДТК ДА); его предварительно трехкратно обрабатывают МИБК для удаления следовых количеств металлов. Через 30 мин (время установления равновесия реакции комплексообразования) к пробе добавляют точно 20 см3 чистого МИБК, встряхивают 5 мин в колбе,, выдерживают до разделения фаз и осторожно доливают раствор до метки
43. Методы и проблемы анализа следовых количеств
429
бидистиллированной водой, предварительно очищенной трехкратной экстракцией ДДТК ДА/МИБК. Все растворы предварительно насыщаются МИБК, так что объем при добавлении растворов существенно не меняется. Все растворы, кроме пробы, перед проведением анализа очищают экстракцией от следовых количеств соответствующих металлов. Следовые количества элементов (например, Go, Pb, Zn, Ni, Си, Cd)L, содержащиеся первоначально в 200 см3 пробы, теперь находятся в 20 см3 МИБК (концентрирование 10: 1) и могут быть непосредственно из этого раствора определены методом фотометрии пламени.
Описано применение автоматической системы для многоэлементной экстракции (Шубигер и др.), например для разделения ионов радиоактивных изотопов Hg(II), Cu(II), Mo(VI), Cd(Il), As(V), Sb(V), Fe(III) и Co(II), экстрагируемых в виде комплексов с ДДТК. Анализом управляют централизованно, включая такие операции, как регулирование pH и проведение реакции окисления-восстановления. Органические растворители, применяемые в этом методе, должны быть тяжелее воды. Анализ в данном случае выполняется быстрее и устраняется воздействие облучений. Повторяя процессы экстракции и реэкстракции и используя метод нейтронного активационного анализа, для ряда матриц можно получить большую селективность и чувствительность определения следовых количеств веществ, чем это достигается с помощью неразрушающей •у-спектрометрии с высокой разрешающей способностью.
Методика анализа. Примерно по 100 мкг солей указанных выше элементов растворяют в 40 см3 0,01 м НСЮ4 (pH раствора 2,0). Добавляют 30 см3 1,7-10~3 М раствора В1(ДДТК)з в СНС13. Прн этом Си и Hg извлекаются примерно на 97%. В экстракт переходит менее 0,4% Cd2+ и менее 0,1% Со и Fe. На второй ступени из водной фазы экстрагируют еще около 2,5% Си и Hg, промывая ее 30 см3 СНС1з. На третьей ступени экстрагируют Cd и Мо, добавляя 1,7-10-3 М раствора 2п(ДДТК)г в СНС13. Добавление комплексных соединений Bi или Zn с ДДТК повышает селективность экстракции за счет различия в константах устойчивости комплексов.
Мышьяк и сурьму можно экстрагировать только в виде ионов As(III) и Sb(III). Поэтому их восстанавливают, добавляя 4 см3 10 М НС1 (для создания pH 0) и 10 см3 раствора, содержащего 0,2 г аскорбиновой кислоты и 1 г KI. Теперь на 4-й ступени экстракции экстрагируют Sb3+ и As3'+, добавляя 30 см3 1,7-10~3 М раствора 2п(ДДТК)2 в СН3С1. На пятой ступени полностью экстрагируют эти металлы при помощи 30 см3 СНС13. Затем устанавливают pH 5 и экстрагируют Сог+ и Fel3+ 30 см3 раствора ДДТК в СНС13. При проведении экстракции вручную степень извлечения достигает 99,9%, при использовании автоматики — только 99%. Приблизительно до 1% элементов содержится в «чужих» фракциях. Если не промывать водные фазы после экстракции чистым растворителем, то теряется примерно до 3% металлов и степень извлечения снижается до 97%. Результаты анализа приведены в табл. Д.35. На проведение экстракции или установление pH затрачивают 10 мин, на нагревание — 8 мин, на восстановление'—17 мин и на охлаждение—10 мин. Для пробы, описанной в примере, проводят регулирование pH 2 раза, 4 экстракции и одно восстановление. Таким образом, продолжительность всего анализа 120 мин.
430
Д. Химический анализ
44. ТЕОРИЯ ИЗМЕРЕНИЙ В АНАЛИТИЧЕСКОЙ ХИМИИ
44.1. Основы теории измерений
Задача аналитической химии как научной дисциплины — получение информации об исследуемых вещественных системах, а именно: о природе составных частей (качественный анализ), о числе составных частей (количественный анализ), о пространственном строении и распределении составных частей (структурный анализ), об изменении во времени перечисленных выше характеристик (анализ процессов). Кроме того, аналитическая химия включает развитие й оценку методов анализа, необходимых для получения указанной информации.
Информация, получаемая в результате аналитических исследований, отражает взаимодействия веществ или вещества с энергией (например, химические реакции, поглощение света), а часто сочетание взаимодействий обоих типов. Получение информации о природе и числе составных частей можно назвать компонентной аналитической химией, причем под компонентами вещественной системы подразумевают как химические элементы, так и соединения.
Большинство аналитических методов, применяемых в компонентной аналитической химии, дают информацию и о качественном, и о количественном составе пробы. Если обозначить через z величину, характеризующую природу составных частей, а через у величину, характеризующую их количество, то в качестве примера можно привести постояннотоковую полярограмму (рис. Д.174) и спектр, полученный в пламени (рис. Д.175). Таким образом, речь в данном случае идет о получении двухмерной аналитической информации. Превращение ее в одномерную в случае фотометрии пламени дало бы точки на оси z для качественного параметра (в данном случае для длин волн) и колоколообразную кривую распределения интенсивности эмиссии (количественный параметр) для определенного значения z (рис. Д.176, а и б). Такую одномерную аналитическую информацию используют в качественном анализе, например, при проведении классического разделения или при применении селективных цветных реакций, когда нужно получить сведения только об отсутствии или присутствии какого-либо элемента; а также в количественном анализе, когда нужно только установить, какое количество определенного элемента вступило в реакцию. Не будем останавливаться на рассмотрении вопросов получения и обработки информации о структуре вещества, поскольку это не входит в задачи данной книги.
При исследовании изменений, происходящих в пробе во времени (аналитическая химия процессов), получают трехмерную аналитическую информацию (рис. Д.177). Если считать
Рис. Д.174. Постояннотоковая полярограмма.
Л, нм
Рис. Д.175. Спектр железа в пламени.
431
432
Д. Химический анализ
zi zz z3 zi
-*----x—*—Мт---»- Z
Рис. Д.176. Положение максимума характеристической эмиссии на шкале длин волн (а) и интенсивность эмиссии (характеристического сигнала) при различных концентрациях (б).
Рис. Д.177. Трехмерное представление происходящих во времени изменений интенсивности различных сигналов.
фиксированной одну из интересующих исследователя величин, то получается двухмерная аналитическая информация (рис. Д.178).
Целью компонентного анализа является возможно более точное определение качественного и количественного состава исследуемого объекта или материала (например, рудных отвалов; чугуна при выпуске из доменной печи; содержимого нефтяных танкеров и т. д.). Естественно, невозможно обработать или исследовать весь исследуемый объект. Для анализа нужно отобрать пробу, которая бы достаточно полно воспроизводила или представляла состав анализируемого материала. Также очень важно, чтобы по возможности вся информация, содержащаяся в материале или в исследуемом объекте, была представлена в пробе. Чем больше объем пробы, тем лучше выполняется это требование; идеальным случаем является идентичность объемов пробы и исследуемого объекта. Однако на практике в целях экономии затрат материала и времени отбирают возможно меньшие объемы проб. Поэтому особое внимание следует уделять тщательности обработки пробы и точному соответствию способа отбора пробы имеющимся методикам. Это особенно важно при отборе проб негомогенных материалов.
После отбора пробы следуют операции: подготовка пробы к анализу (измельче-
44. Теория измерений в аналитической химии
433
ние, растворение или отделение определяемого компонента от других, например отделение следовых количеств элементов от основного компонента); проведение измерений выбранным методом и оценка полученных результатов (например, по градуировочной прямой и др.), что является источником аналитической информации. Из этой последовательности операций легко увидеть разницу важных для аналитической химии понятий. Так, принцип анализа сводится к методу измерения аналитического сигнала (например, полярографическому), метод анализа включает уже подготовку пробы + измерение+оценку ре-
Рис. Д.178. Двухмерное представление происходящих во времени изменений интенсивности определенного сигнала.
зультатов, а способ выполнения анализа—весь ход анализа исследуемого объекта, в том числе отбор пробы, подготовку пробы, измерение, оценку результатов вплоть до получения аналитической информации.
К способам выполнения анализа обычно предъявляют следующие требования. Информация должна быть получена за возможно меньший промежуток времени /д; он должен быть значительно меньше времени, за которое заметно изменяется состав исследуемого объекта. Получение информации должно быть связано с минимумом экспериментальных затрат (число сотрудников, приборы, реактивы). Информация не должна быть неверной, т. е. источником сигнала, соответствующего Za, должен быть только компонент А. Таким образом, метод должен быть селективным или специфичным по отношению к А, посторонние сигналы других компонентов не должны оказывать влияния на получаемую информацию. Информация должна быть воспроизводимой (повторяемой), т. е. не должно быть большой величины случайного разброса результатов измерений. Для определения малых содержаний веществ (например, в аналитической химии следовых количеств, особо чистых веществ) нужно применять высокочувствительные методы анализа.
28—1880
434
Д. Химический анализ
44.2. Виды, источники и характеристики ошибок определения
Ошибки (погрешности) классифицируют на систематические и случайные. Их наложение, обычно наблюдаемое на практике, дает суммарную ошибку определения. Взаимосвязь ошибок подтверждена надежными статистическими данными; как правило, большое число малых систематических ошибок приводит к увеличению случайной ошибки. Систематической ошибкой называют направленное отклонение полученных значений от теоретического. Таким образом, систематическая ошибка всегда имеет знак и на результаты измерений она оказывает одинаковое влияние: получаемые результаты или постоянно занижены, или постоянно завышены. Систематическая ошибка характеризует правильность* результата. Случайные ошибки определяют его точность* и воспроизводимость*. На гауссовой кривой нормального распределения случайные ошибки располагаются около наиболее часто встречающегося (наиболее вероятного) значения, которое обычно является средним арифметическим.
Сказанное можно пояснить на примере попадании выстрелов в мишень (рнс. Д.179). Стрелок А стреляет метко (небольшие случайные ошибки), стрелок Б — дает промахи (большие случайные ошибки). Винтовка А при стрельбе не дает отклонений (нет систематической ошибки: идеальный случай), винтовка Б дает отклонения направо вверх (систематическая ошибка). В идеальном случае все выстрелы из винтовки А должны попасть в центр мишени. Выстрелы стрелка А из винтовки А попадают в мишень с небольшим разбросом около центра, выстрелы стрелка Б из винтовки А — с большим разбросом. Стрелок Б должен стрелять значительно чаще, чтобы попасть в центр мишени. Из винтовки Б стрелок А вряд ли сможет попасть в центр мишени. Разброс попаданий винтовки Б тем отчетливее, чем больше выстрелов произведено. Систематическая ошибка очевидна. Стрелок Б из винтовки Б может случайно попасть в центр мишени, но это маловероятное событие. При оценке очень большого числа выстрелов (большего, чем у стрелка А) систематическая ошибка винтовки Б становится очевидной.
В аналитической химии это означает, что нужно выявить причины систематических ошибок (неверных данных) и устранить их, так как сами систематические ошибки нельзя устранить многократным повторением анализа, их можно при этом только выявить. Случайные ошибки, напротив, нельзя устранить. Их мож
* В литературе на русском языке для характеристики качества анализа рекомендуется использовать количественно определяемые понятия «правильность» и «воспроизводимость», а термин «точность» оставлен как качественное понятие, характеризующее правильность и воспроизводимость метода одновременно. «Чувствительность» определяется как тангенс угла наклона градуировочного графика. (См. Термины, определения и обозначения метрологических характеристик анализа вещества. — ЖАХ, 1975, т. 30, № 10, с. 2058.) Между тем в данной книге под чувствительностью понимается то тангенс угла наклона графика, то минимально определяемое количество вещества. Однако смысл термина ясен из контекста и потому не изменен прн переводе. — Прим. ред.
44. Теория измерений в аналитической химии
435
но только свести к минимуму; сделать это нужно обязательно. Чем больше случайный разброс метода анализа, тем больше определений нужно провести и усреднить для получения результата измерений, наиболее близкого к истинному, или соответственно тем меньше вероятность близости единичного результата измерений к истинному значению.
ЛВ ББ
Рис. Д.179. Картина попаданий в мишень.
АА — систематическая ошибка отсутствует, малая случайная ошибка; БА — систематиче-ская ошибка отсутствует, большая случайная ошибка; ДБ — значительная систематическая ошибка, случайная ошибка отсутствует; ББ — значительная систематическая ошибка, большая случайная ошибка.
Ошибки могут возникнуть на всех этапах выполнения анализа. При отборе пробы, например, появляются случайные ошибки, связанные с негомогенностью вещества в случае гетерогенных фаз или градиента концентраций в случае гомогенных фаз. Этих ошибок нельзя полностью избежать даже при правильном отборе пробы. Неверный отбор пробы, напротив, приводит к неизбежному преобладанию отдельных компонентов в пробе и возникновению систематической ошибки. В процессе подготовки пробы к анализу ее обычно растворяют, иначе могут возникнуть систематические ошибки вследствие преобладания отдельных компонентов в пробе. При этом случайными ошибками можно пренебречь, 28*
436
Д. Химический анализ
а возможность появления систематических ошибок мала (за- Л грязнения от реактивов и материала сосудов). Методы разделе- ] ния, применяемые при подготовке пробы к анализу, могут слу- 1 жить источником больших систематических и случайных ошибок, •, связанных с неполнотой разделения (вследствие осаждения, ча- 4 стичного растворения, соосаждения, а также зависимости этих , процессов от температуры). :
Проведение самих измерений неизбежно связано с возникно- 1 вением случайных ошибок при получении сигнала (например, из-за температурных колебаний в методе фотометрии пламени), а также при превращении полученного сигнала в электрический сигнал, его усилении и преобразовании (шумы). Эти случайные ошибки неизбежно сказываются на результатах, как правило занижая их. Систематические ошибки
измерении и оценки результатов можно устранить точным соблюдением условий работы и правильной градуировкой. В принципе ошибки измерений и оценки результатов измерений должны быть меньше, чем ошибки самого аналитического метода. При работе с растворами не сложно установить, дает ли данный ошибки: значение, полученное
Рис. Д.180. Влияние различных видов систематических ошибок на график зависимости у—х.
1 — аддитивные; . 2 мультипликативные; 3 — нелинейно связанные с измеряемой величиной; 4 — теоретическая зависимость.
метод анализа систематические
в результате анализа, сравнивают с теоретическим, поскольку известно соотношение компонентов в приготовленном стандартном растворе. По-другому обстоит дело в случае твердых веществ, когда сложно приготовить стандарт с гомогенным составом. Правильность метода анализа для часто встречающихся в практике гетерогенных объектов можно считать доказанной только в том случае, если найденное значение совпадает с результатом, полученным другим методом анализа, по возможности совершенно отличным от первого. При расхождении результатов решить вопрос о том, какой из методов дает систематическую ошибку, можно только с привлечением нескольких (возможно большего числа) методов анализа.
Различают следующие виды систематических ошибок. Аддитивные (постоянные) ошибки возникают, если, например, не учитывают расход реагентов на холостую пробу, матрицу и др. Причиной появления ошибок другого вида — мультипликативных
44. Теория измерений в аналитической химии 437
является, например, неверный титр, неправильно проведенная градуировка. К третьему виду относятся ошибки, нелинейно связанные с измеряемой величиной. Причины их появления многообразны и должны быть определены в каждом конкретном случае. Влияние этих видов ошибок показано на рис. Д.180.
Метод анализа, полностью свободный от систематических ошибок, никогда не бывает не свободен от случайных. Выявление случайных ошибок можно провести статистическим описанием вероятности их появления при проведении определенного числа анализов пробы.
Ошибки можно выразить в абсолютных единицах с размерностью определяемых величин (мг, моль, моль/дм3 и т. д.) и относительных (обычно в %). Например, при определении Сг в сплаве получен результат 63,5+0,1 %; абсолютная ошибка составляет + 0,1%, а относительная — 0,1/63,5-100= + 0,16% Сг.
Абсолютная ошибка наглядно отражает надежность аналитического метода; относительная ошибка (поскольку отнесена к измеряемой единице) —эффективность метода анализа.
44.3. Случайный разброс результатов измерений
При проведении анализа происходит случайный разброс результатов измерений, который подчиняется законам математической статистики. Значение случайного разброса определяет воспроизводимость результатов. Это справедливо как для качественного параметра z (длина волны, потенциал полуволны и т. д.), для которого случайный разброс обычно наименьший, так и для количественного параметра у (интенсивность эмиссии, оптическая плотность раствора, диффузионный ток и т. д.).
Если обозначить через п число опытов, а число контрольных измерений через f=n—1 (f также называют числом степеней свободы), то стандартное отклонение о для бесконечного числа измерений (п—>-оо) можно рассчитать по уравнению (476):
J/ ----п-----гдеиа = ^~ (476), (477)
— среднее арифметическое результатов всех измерений. Таким образом, вычисляют среднее арифметическое, находят разности между ним и каждым результатом единичных измерений уг, возводят разности в квадрат, суммируют, делят на число опытов и извлекают квадратный корень. Соответственно можно рассчитать также стандартное отклонение для z(oz).
На практике всегда проводят ограниченное число определений. Тогда
/'ZWi-l}2 - ^У{ ,.7Q. ,.7Пч
„ где У = (478), (479)
438
Д. Химический анализ
Таким образом, о и ц характеризуют в идеале стандартное отклонение и среднее арифметическое совокупности результатов всех измерений, так называмой генеральной совокупности, а Sy м у или соответственно sz и z — стандартное отклонение и среднее арифметическое результатов измерений, полученных на практике для небольшого числа опытов, так называемой выборки результатов измерений из их генеральной совокупности.
Генеральная совокупность и выборка в известном смысле соотносятся между собой так же, как исследуемый объект и анализируемая проба. Так же как проба должна представительно отражать состав материала, выборка должна представительно отражать генеральную совокупность результатов измерений. Это достигается оптимальной величиной выборки (числом опытов п). Значения стандартного отклонения s и среднего арифметического, например, у, рассчитанные для ограниченного числа определений, называют оценочными величинами для <т и ц генеральной совокупности. Проще можно рассчитать более грубые оценочные величины: для стандартного отклонения— это так называемый диапазон значений ^ = г/тах — —г/min, представляющий собой разность между наибольшим и наименьшим результатом выборки, а для среднего арифметического — так называемое серединное значение или медиану у. Если результаты измерений расположить в порядке возрастания, то при нечетном числе измерений медиану определяют как центральный результат, при четном числе измерений — как среднее арифметическое двух средних результатов выборки. При небольшом числе измерений на медиану не оказывают влияния отдельные случайные ошибки результатов больше или меньше среднего, так как она определяется только средним (или двумя средними) результатами. Но по этой же причине при большом числе измерений (п>10) медиана непригодна, нужно рассчитывать среднее арифметическое.
Для химико-аналитических определений плотность вероятностей результатов измерений в большинстве случаев описывается функцией Гаусса:
Н(у) = —I е“^М0)а (480)
<т у 2л
Это распределение частот называют нормальным распределением. На рис. Д.181 приведена кривая нормального распределения; положение кривой зависит от ц (максимум кривой распределения соответствует у = ц), а ширина — от о (чем больше о, тем более широкой и пологой получается колоколообразная кривая). Точки перегиба колоколообразной кривой расположены при i/i = ц—о и у2=р + о. Ордината (частота значе-
Рис. Д.181. Кривая нормального распределения результатов измерения. W|, W2 —точки перегиба.
Рис. Д.182. Гауссов интеграл ошибок и части площади под колоколообразной кривой (в процентах), получаемые при интегрировании в различных пределах.
440
Д. Химический анализ
ний z/) только при z/=p±oo становится равной нулю. Однако на практике она пренебрежимо мала уже при г/ = ц±3о.
Функцию Гаусса при ff=l, и us (у—ix)/o можно
преобразовать в следующее выражение:
я(й=-Йае’Л’ (481>
Интегрирование функции Гаусса в пределах ±оо дает гауссов интеграл ошибок (рис. Д.182):
+°°
FQ/H-Ufe-^du (482)
У 2л J , —00
Если принять площадь под колоколообразной кривой за единицу, то при интегрировании получим: 68,3% площади в пределах ±1 о; 90%— для ±1,64 о; 95%—для ±1,96о; 99% — для ±2,58 о и 99,7% —для ±3о. Полученные значения площадей отражают статистическую вероятность Р, равную для приведенных примеров 0,683; 0,90; 0,95; 0,99 и 0,997 соответственно. Разность между единицей и Р обозначают через а — степень надежности: а + Р=1 или в процентах а + Р = 100%.
При данном о значение Р одновременно определяет вероятность нахождения отдельных результатов измерений в заданных пределах интегрирования р.±ио или соответственно «степень надежности» нахождения этих результатов вне заданных пределов.
Кривую распределения частот можно построить не только для отдельных результатов измерений, но и для средних результатов нескольких параллельных определений. Обозначим через па число параллельных определений для каждого измерения и через т число групп измерений. Тогда общее число результатов измерений будет п — па>п- Можно показать, что среднее арифметическое отдельных средних значений (пг из каждых па) равно среднему арифметическому всех отдельных значений.
Однако стандартное отклонение средних значений уменьшается в ~!па раз:
= (483)
Кривая распределения частот для средних результатов измерений острее, чем кривая распределения, построенная для отдельных результатов измерений, так как самые большие и самые малые значения отдельных результатов при усреднении накладываются, оказывая соответствующее влияние на кривую распределения.
44, Теория измерений в аналитической химии 441
Поскольку рабочие затраты пропорциональны Па, а точность пропорциональна 1/Упд, с увеличением Па затраты увеличиваются. Поэтому обычно ограничиваются проведением двух-трех параллельных опытов.
Частоту распределения результатов измерений, получаемую на практике, наиболее просто можно изобразить в виде точек на линейной шкале (одномерное изображение) (рис. Д.183,б).
6
У
Рис. Д.183, Двухмерное (а) и одномерное (б) представление распределения частот результатов измерений.
Она характеризуется плотностью распределения точек. Более наглядно двухмерное изображение в виде гистограмм. Для построения гистограммы значения yt располагают в порядке их возрастания. Диапазон значений ^ = z/max—*/ипп делят на k классов. Оптимальное число классов находят из соотношения k^^n. Таким образом, при 50 результатах измерений целесообразно делить значения на 7 классов. Можно рекомендовать диапазон 20>&>5. При небольшой протяженности класса могут выпасть отдельные важные значения в распределении частот. Напротив, в случае больших классов случайные колебания искажают представления о характере распределения. Протяженность класса можно вычислить по формуле d — RJk. При этом результаты измерений могут соответствовать гистограммам с протяженностью от d\ до dk. Число результатов измере-
442
Д. Химический анализ
Рис. Д.184. Кривые нормального распределения, построенные для различных значений случайных ошибок.
ний в отдельных классах отражает высота гистограммы (рис. Д.183,а). Наибольшая высота соответствует группе наиболее часто встречающихся результатов измерений. На числовой прямой ей соответствует участок с наибольшей плотностью точек. При п—*оо идеальным предельным случаем гистограмм является колоколообразная кривая, ширина которой определяется значением случайной ошибки (рис. Д.184).
Единичную систематическую ошибку найти по гистограмме нельзя. Две различные систематические ошибки могут проявиться в виде расщепления максимума или появления выступа на гистограмме. Результирующее распределение представ-
Рис. Д.185. Результирующая кривая распределения частот при наличии систематической ошибки.
а — небольшая разность средних значений; б — большая разность средних значений. При различном числе измерений двух признаков на кривой образуются выступы.
44. Теория измерений в аналитической химии
443
ляет собой сумму двух распределений, расположенных около двух различных средних значений (рис. Д.185). Для того чтобы это различие было заметно, нужно иметь не меньше 30 результатов измерений для каждого из двух признаков. Поэтому более целесообразно применять другие методы. Однако по гистограмме можно установить симметричность или асимметричность кривой распределения частот. Если колоколообразная кривая имеет неправильную форму, то это связано или с отклонениями в условиях эксперимента, или с неверным делением оси абсцисс. Неправильность кривой можно иногда устранить. Если искажение появилось при анализе следовых количеств веществ или в результате очень большой случайной ошибки (например, в полуколичественном спектральном анализе), то для всех расчетов можно применять логарифмы результатов измерения. Вместо среднего арифметического при этом часто используют среднее геометрическое.
Рис. Д.186. Среднее значение ц и стандартное отклонение о генеральной совокупности, представленные на «сетке вероятностей».
Интегральную кривую (рис. Д.182) гауссовою интеграла ошибок (уравнение (482)) можно преобразовать в прямую, откладывая по оси ординат значения гауссовою интеграла. В этом случае говорят о «сетке вероятностей». По оси абсцисс можно откладывать как линейные, так и логарифмические единицы. В продаже имеется соответствующая миллиметровая бумага. Для оценки результаты измерений располагают упорядоченно в соответствии с их численными значениями, абсциссу делят на классы и определяют число результатов измерений в каждом классе. Затем рассчитывают суммарную частоту (абсолютную и в процентах для каждого класса), например 1-й класс —
Рис. Д.187. Одинаковые стандартные отклонения (01 = 02) в случае двух различных средних значений ра и р2, представленные на «сетке вероятностей».
Рис. Д.188. Неодинаковые стандартные отклонения (0^02) в случае двух различных средних значений (р1=#Р-2)> представленные на «сетке вероятностей»
44. Теория измерений в аналитической химии '445
1 результат, 2-й класс — 2 результата, 3-й класс — 8 результатов или для 1-го класса 1 результат, для 2-го—3 результата, для 3-го—II результатов и г. Д.
Если на специальной миллиметровой бумаге по оси ординат отложить процентные доли всех результатов измерений, значения которых ниже предельного значения у,-, а по оси абсцисс — значения у,, то при нормальном распределении вероятностей от 10 до 90% полученных точек расположатся иа прямой. При построении зависимости в логарифмических координатах прямую получают в случае нормального логарифмического распределения. Угол подъема прямой тем больше, чем меньше о (колоколообразная кривая с острой вершиной). Значению 50% на оси ординат соответствует на оси абсцисс величина ц генеральной совокупности, т. е. ее среднее значение. Точкам перегиба колоколообразной кривой при pi ± 1 ст соответствуют суммарные частоты 15,9 и 84,1%. Исходя из этих значений ординат, на оси абсцисс получают отрезок ц±о, т. е. 2<т, и отсюда легко находят сг — стандартное отклонение генеральной совокупности (рис. Д.186). То, что на гистограмме отражается в виде двух вершин, т. е. значений из двух генеральных совокупностей (Ц1¥=Ц2) (ср. рнс. Д.185), на. «сетке вероятностей» представлено в виде двух прямых. При равенстве стандартных отклонений генеральных совокупностей (oi = o2) углы подъема прямых одинаковы (рис. Д.187). В противном случае (oi#=G2) получают две прямые с различными углами подъема (рис. Д.188).
44.4. Закон распространения ошибок и корреляция результатов измерения
Результат измерения у часто складывается из отдельных результатов по более или менее простому закону. Случайный разброс его значения складывается соответственно из случайных разбросов отдельных значений. В этом случае справедлив закон распространения ошибок:
J (484)
2) У — У а У в J
При нахождении суммы или разности квадраты абсолютных ошибок отдельных измерений складываются, давая квадрат абсолютной ошибки суммарного значения измерения. Для аналитических целей наиболее важным является нахождение разности, например, при взвешивании или при отсчете показаний по бюретке:
3) У УаУв 1 ( \ _/ ®уа । ( ®ув \
os-wJvd ( ’
При расчете произведения или частного квадраты относительных ошибок отдельных измерений складываются, давая квадрат относительной ошибки суммарного значения измерения. Для аналитических целей более важно нахождение частного, например, при измерениях с внутренним стандартом. Последовательно получаемые результаты измерения сигнала у, т. е. У\, У2, Уз---Уп, сопровождаются случайным разбросом. Это же справедливо и для серии результатов измерения сигнала у', не
446
Д. Химический анализ
зависящего от у (другой элемент или другой параметр z этого же элемента). Если же одновременно или с небольшим интервалом по времени получают пары результатов измерений с очень близкими значениями — г/ь у/; уг, у2' и т. д., то это часто приводит к завышению yi, занижению у2 и т. д. (например, вследствие температурных колебаний, колебаний степени всасывания или распыления раствора пробы в фотометрии пламени, а также в большей или меньшей степени к аналогичным изменениям у/, у г' и т. д. При этом наблюдают более или менее тесную взаимосвязь колебаний значений пары результатов
Рис. Д.189. Корреляция результатов измерений (а) и отсутствие корреля ции (б).
измерений, называемую корреляцией. При нанесении на диаграмму пар значений у и у' получают эллипс. Чем больше корреляция, тем эллипс сильнее вытянут и приближается к прямой и наоборот. В отсутствие корреляции получают круг, в центре которого находится максимум частот (рис. Д.189).
Поскольку можно ожидать случайного разброса значений у около его среднего арифметического у и значений у' около у', то у и у' можно принять за центр системы координат. Преимущество такого способа заключается в возможности сделать определенные выводы о наличии корреляции по подсчету точек. Если во всех четырех квадрантах находится примерно одинаково большое число точек, то корреляция отсутствует. Если большинство точек находится в квадрантах I и III, корреляция существует, и она тем больше, чем меньше точек расположено в квадрантах II и IV (рис. Д.190). Если число точек больше во втором и четвертом квадрантах, то наблюдается обратная корреляция (отрицательная). Такая корреляция не встречается в рассматриваемых в данной главе методах ана-
44. Теория измерений в аналитической химии 441
1
литической химии, хотя такой случай имеет место при корреляции свойств и состава, например электропроводности с загрязнениями в чистых металлах.
Степень корреляции точно описывается коэффициентом корреляции г:
Г_ (486
У^^-уГ^н'-у'У
Значение коэффициента колеблется в пределах —1 <<<•+!.
Рис. Д.190. Оценка корреляции при подсчете пар результатов измерений (точки в квадрантах).
Отрицательное значение коэффициента отражает обратную корреляцию, положительное—-прямую. При г = 0 корреляция отсутствует (круг на рис. Д.189).
При небольшом числе значений найденный коэффициент корреляции г, естественно, является грубой оценкой корреляции генеральной совокупности. Две точки всегда можно соединить прямой; при этом коэффициент корреляции имеет максимальное значение 1. Однако этот результат не является истинным. Только при использовании третьего значения можно получить какую-то ясность. Поэтому для корреляции с числом степеней свободы f=n—2 действительно число пар измерений п. Если третье значение располагается на этой же прямой, т. е. коэффициент корреляции г остается равным единице, можно предполагать наличие корреляции. В противном случае оно не подтверждает корреляцию. Но все же и это может оказаться случайностью. Надежным доказательством существования корре-
448
Д. Химический анализ
Таблица Д.36. Значение коэффициента корреляции г в зависимости от статистической вероятности Р и числа степеней свободы f
f=fl — 2 ^ = 0,95 Р=0,99 | f = n — 2 Р = 0,95 Р = 0,99
1 1,00 1,00 8 0,63 0,77
2 0,95 0,99 9 0,60 0,74
3 0,88 0,96 10 0,58 0,71
4 0,81 0,92 15 0,48 0,61
5 0,75 0,87 20 0,42 0,54
6 0,71 0,83 50 0,27 0,35
7 0,67 0,80 100 0,20 0,25
ляции может служить только сравнение с табличными величинами (табл. Д.36): \r\>r(P,f). Это означает, что абсолютное значение коэффициента корреляции, рассчитанное по уравнению (486), должно быть больше 0,95 для корреляции четырех пар результатов измерений (/ = 2) с 95 %-ной вероятностью или больше 0,96 при п = 5 (/ = 3) с 99%-ной вероятностью. Такие высокие значения коэффициента корреляции трудно получить. Поэтому для надежного подтверждения корреляции необходимо провести достаточное большое число измерений. Например, для 10 пар результатов измерений (/ = 8) |г| =0,7 означает, что можно получить надежную корреляцию с вероятностью >95%, но недостоверную при Р>99%.
44.5. Условия регистрации сигналов
Информация о качественном составе образца, которую мы получаем при анализе пробы, находит свое выражение в константах вещества (например, потенциал полуволн в полярографии, длины волн резонансных линий в атомно-эмиссионной спектроскопии, величина Rf в бумажной хроматографии и т. п.). Во многих методах инструментального анализа измерения проводят в интервале —>z2, т. е. от нижней до верхней границы значений, и появляющиеся сигналы записывают (рис. Д.174 и Д.175). При этом часто получают колоколообразную кривую, которая приближенно описывается функцией Лоренца или Гаусса (газовая хроматография, дифференциальный термический анализ, атомная спектроскопия и т. д.). В методах, дающих интегральную S-образную кривую, например в постояннотоковой полярографии, осуществляя дифференцирование при помощи определенной схемы, также можно получить аналогичную колоколообразную кривую. И наоборот, интегрирование колоколообразной кривой приводит к кривой S-образной формы. Координата максимума сигнала колоколообразной кривой или
44. Теория измерений в аналитической химии 449
точка перегиба интегральной кривой соответствуют Zi. Определение Zi проводят обычно путем вспомогательных графических приемов.
В общем случае максимум на кривой легче найти, чем точку перегиба кривой, и в качественном анализе лучше использовать дифференциальные кривые, а не интегральные. Ширину сигнала колоколообразной формы принято характеризовать
0,35 мм
0,01 о,ог о,os
0,10 0,15 0,20 0,25 0,30
Рнс. Д.191. Разрешение дублета линии Na (589,6 и 589,0 нм) в зависимости от ширины щели (обозначена числами под пиками).
значением его полуширины, т. е. той ширины, при которой интенсивность сигнала составляет половину максимального значения. Полуширину обозначают как Azi/г. Вследствие несовершенства применяемой аппаратуры получаемое на практике значение полуширины сигнала превышает «естественное» его значение. Чем больше реальное значение полуширины, тем больше опасность взаимного наложения сигналов (рис. Д.191). Минимально допустимое расстояние между двумя разделяемыми сигналами Azmin пропорционально Azj/2. Таким образом, разрешающая способность уменьшается с увеличением полуширины. Следовательно, получения полностью разрешенных (изолированных) сигналов, интенсивности которых не оказывают взаимного мешающего влияния, можно ожидать только в том случае, если максимумы сигналов удалены один от другого примерно на две с половиной полуширины. В других случаях для получения правильных количественных результатов необ
29—1880
450
Д. Химический анализ
ходимо учитывать вклад соседнего сигнала в измеряемый сигнал.
При регистрации сигналов в интервале от zx дог2 необходимо, чтобы продолжительность регистрации интервала ti, полуширина Дг]/2 и случайная ошибка для значений ординат принимали наименьшее из возможных значение. Для указанных трех величин справедливо следующее выражение:
M^1/2)42 = * (487)
Константа К в каждом конкретном случае иная, т. е. ее значения не совпадают для квадратно-волновой полярографии и, например, фотометрии пламени и т. д., но постоянна для данной установки. Это означает, что добиться минимального значения одновременно для всех трех величин нельзя, в каждом случае нужно находить наилучшее решение. Этого можно достичь следующими приемами.
1. Увеличение продолжительности регистрации сигналов. При быстрой регистрации (tA—ниш) получают нечеткие сигналы с плохим разрешением и (или) с небольшой точностью.
2. Уменьшение полуширины. Для качественного анализа необходимо, чтобы Azj/2—ниш; это возможно при увеличении продолжительности регистрации, хотя одновременно ухудшается точность.
3. Уменьшение случайной ошибки. В количественном анализе стремятся получить о,/—нпш, но при этом во избежание наложения сигналов Azi/2 не должна быть слишком большой. Регистрацию сигналов следует проводить очень медленно.
Высокую разрешающую способность (наименьшая Дг1/2) можно получить, повышая так называемую четкость сигнала. При регистрации сигналов, имеющих форму кривой Лоренца или Гаусса, полуширину можно уменьшить, если вместо основной функции записывать ее вторую производную, осуществляя двукратное дифференцирование при помощи электронной схемы (рис. Д.192). Для функции Лоренца отношение полуширины Д31/2 основной функции и ее второй производной составляет 1/(1/3), а для функции Гаусса — 1/( 1/2). При получении второй производной Azi/2 уменьшается, таким образом, на 1/3 или на 1/2 соответственно. Теоретически допустимо усиливать четкость сигнала, получая производные более высокого порядка с одновременным увеличением интенсивности сигналов.
Так как tA и К не изменяются, из уравнения 487 следует: (Az1/2)4a% = (Az"1/2)4oV, о^» = oy (Azi/2/Az"1/2)2
Иначе говоря, если Az"i/2= 1/3Azi/2, то = 9оу. Это означает увеличение случайной ошибки для интенсивности сигнала в де
44. Теория измерений в аналитической химии
451
вять или соответственно в четыре раза. Таким образом, увеличение четкости сигнала связано с квадратичными потерями точности. Это ограничивает применимость выбранных методов в качественном анализе, так как очень четкие сигналы нельзя уже однозначно отделить от сигнала матрицы из-за очень большого разброса их интенсивностей.
Рис. Д.192. Схема усиления четкости сигнала.
В случае функции Гаусса при записи второй производной вместо основной функции полуширина сигнала уменьшается наполовину.
Систематические ошибки измерения могут искажать значение параметра z,, применяемого' для получения информации о качественном составе веществ. Например, в полярографии при определении потенциала полуволны могут быть получены неправильные значения напряжения ячейки, потенциала электрода сравнения, диффузионного потенциала и т. д. Ситуацию в таких случаях можно улучшить добавлением стандарта с определенным известным значением zCt, например ионов Т1+, значение потенциала полуволны которых. —0,49 В, измеренное относительно насыщенного каломельного электрода, не зависит от фонового электролита. Координаты стандартного сигнала используют также в методах оптической атомной эмиссионной спектроскопии, ЯМР и т. д.
При достаточно малом удалении аналитического сигнала от стандартного линейное интерполирование можно выполнять по формуле Zi/zCr = Zi'/z'cT, где Zi —искомая координата сигнала; гст — теоретическая координата сигнала стандарта; Zi и г'ст — измеренные координаты сигналов. Поскольку z^z^Zijz'^, систематическую ошибку можно значительно уменьшить. Оценку
29*
452
Д. Химический анализ
случайных ошибок проводят в соответствии с законом распространения ошибок.
Пусть zCT — значение параметра, определенное без ошибок, тогда по уравнению (485) находим:
(<WZ02 = lZi У 4-(°'гсЛ'ст)2
Если искомый и стандартный сигналы расположены близко друг к другу, но разрешимы, т. е. z/лгУ, можно принять для их стандартных отклонений, что ог-£»ог-Ст. Отсюда
Таким образом, при значительном уменьшении систематической ошибки относительная случайная ошибка увеличивается по крайней мере в }'2 раз. При необходимости этот фактор можно уменьшить многократным повторением измерений и нахождением среднего результата.
44.6. Интервал измерения и предел обнаружения
Интервал измерения сигнала у определяется верхней у% и нижней границами yi. Часто абсолютная величина оу случайной ошибки для всего интервала измерений постоянна (например, капельная ошибка бюретки 0,03 см3) или близка к постоянной (как для аналитических весов). При выполнении обычных технических измерений стремятся получить относительную ошибку 0,1%, т. е. оу/г/2 = 10-3. При этом для обнаружения сигнала нижней границы интервала измерений с вероятностью более 99% необходимо, чтобы он был по крайней мере в три раза больше случайной ошибки г/1^3оу; в этом случае относительная ошибка для г/i равна oy/Pi=0,33 (33%). Из изложенного следует, что при постоянном значении сту интервал измерения у охватывает два-три порядка (о:7 = уг • 1 СГ3 и оу =ух • 0,33). Точность определения у вблизи нижней границы интервала измерения неудовлетворительна.
В некоторых методах анализа относительная ошибка оу/у постоянна во всем интервале измерений. Поэтому эти методы особенно эффективны для исследования небольших количеств пробы, например в анализе следовых количеств веществ.
Пример. Выполнен анализ двух сплавов на содержание меди. Получены следующие результаты:
1. Абсолютная ошибка постоянна и равна 0,1 (80±0,1)% Си соответствует ±0,125% отн. (5±0,1) % Си соответствует ±2°/о отн.
2. Относительная ошибка постоянна и равна 1 % (80±0,8) % Си соответствует ±0,8 абс.
(5±0,05)% Си соответствует ±0,05 абс.
44. Теория измерений в аналитической химии
453
Исходя из у\ = 3ъу и г/] = ЗорДля среднего_из результатов
большого числа измерений, а также <5~<5уЦпа— стандартного отклонения среднего значения из пА параллельных измерений, получим
Ут 3+/ V пА
Таким образом, минимально обнаруживаемую интенсивность сигнала у\ (с вероятностью >99%) при получении среднего из Пл параллельных определений можно уменьшить в ^пА раз. Реализовать это можно, используя электронную вычислительную технику в сочетании с автоматическими анализаторами. Например, Пд = 100 означает уменьшение нижнего предела обнаружения в 1/10 раза.
С учетом сигнала фона ув и его разброса аналитический сигнал вычисляют как у\=ув + 3вв или в виде среднего значения:
Ут Ув+3°b/V пл (489)
При измерении сигнала определенной таким образом величины у\ (говорят также о обнаруживаемом минимуме) снова получается случайный разброс: 50% значений находятся выше j/i, 50%—ниже. Поэтому для получения надежных результатов анализа определяют предел обнаружения: Уе — У\ + 3<уу и при ов~Оу находят (рис. Д.193)
Ув = УвЛ-^в (49°)
При этом содержание компонента в пробе можно определить с вероятностью 99,8%;
Р=0,997/2 + 0,5=0,998
При наличии сигнала фона градуировочный график выражается уравнением y = yB + bx. Так как y = yB + 3oB, то Ьх = 6ов, отсюда
x£=6oB/i> (491)
Это выражение для предела обнаружения. Следовательно, предел обнаружения не зависит от фона, а только от стандартного отклонения, которое нужно определять из большого числа результатов измерений (п~10) и от чувствительности метода Ь.
Полученное таким образом значение предела обнаружения может характеризовать эффективность методов анализа следовых количеств веществ. Однако при этом не учитываются другие важные параметры, такие, как селективность, размер пробы, продолжительность анализа и затраты.
454
Д. Химический анализ
Рис. Д.193. Взаимосвязь между сигналом фона у в, сигналом t/t, определенным с 99,7%-пой вероятностью (при содержании хк, равном обнаруживаемому минимуму), и сигналом ув, соответствующем пределу обнаружения хв при большой (bi) и малой (&2) чувствительности метода.
Часто применимость метода для целей обнаружения оценивают, исходя из идеализированного предела обнаружения, который в принципе может быть получен в данном методе анализа.
44.7. Методы измерений в количественном анализе
Практическая работа обычно связана с измерением разностей. Например, массу в гравиметрии находят как разность результатов двух взвешиваний — тигля с осадком и пустого тигля, т. е. У = Ул—Ув. При титровании измерение объема титранта, затрачиваемого до точки эквивалентности, связано с ошибками двух отсчетов по бюретке — в точке эквивалентности и в начале титрования. Ошибки отдельных стадий в этом случае суммируются в соответствии с законом распространения ошибок (уравнение (484)): о2у='О2Ул+о2ув. Выражение (Д^о2^справедливо и для гравиметрии, поскольку масса тигля намного больше массы осадка, т. е. Уа^Ув, и для объемных методов анализа, поскольку
44, Теория измерений в аналитической химии 455
ошибка отсчета показаний по бюретке не зависит от заполнения бюретки. Отсюда следует; ау~(>УАу2, и для относительной случайной ошибки с у = ул—ув
(492)
У У а-У в v ’
Это означает, что при измерении разности случайная ошибка возрастает по сравнению со случайной ошибкой отдельного измерения в у 2 раз, но ее относительное значение тем меньше, чем больше измеряемая разность. Увеличить разность в объемном анализе можно, максимально увеличив объем титранта, идущий на титрование до точки эквивалентности. В гравиметрии этого сделать нельзя, поскольку было бы необходимо без потерь собрать, высушить и прокалить большой, объемистый осадок. Небольшая относительная ошибка определения в этом случае связана только с высокой точностью отдельных результатов измерения. Этому требованию вполне удовлетворяют аналитические весы.
Метод градуировки. Целью количественного анализа является определение содержания какого-либо элемента или соединения х. Поэтому необходимо точно знать функциональную зависимость между измеряемой величиной у и содержанием х (рис. Д.194). Желательно, чтобы эта зависимость не была многозначной (а). В случае двузначной зависимости, например для активной составляющей метода осциллометрии, нужно определить, в какой области должно находиться значение у для получения правильных результатов для х (б). Даже однозначная функциональная зависимость не всегда является идеальной (в), так как при наличии кривизны функции существует сильная зависимость чувствительности измерений от содержания компонента. Такая ситуация возникает, например, при подавлении максимумов первого рода в постояннотоковой полярографии при определении содержания примесей поверхностно-активных веществ в воде. В таких случаях используют специальные приемы, например измеряют объем пробы, при добавлении которого сигнал уменьшается наполовину. Фиксируют значение у и определяют х при соответствующем разбавлении пробы. Как правило, для аналитических определений необходимо наличие однозначной линейной функциональной зависимости (г). Тогда градуировочный график можно описать уравнением у = ув+Ьх. При х = 0, т. е. в отсутствие определяемого компонента, у = ув, поэтому ув называют сигналом фона. Причинами возникновения сигнала фона могут служить примеси определяемых компонентов в реактивах и растворителе, а также наложение сигналов, перекрывающих сигналы определяемых компонентов. Сигнал фона стараются в каждом конкретном случае уменьшить (при-
456
Д. Химический анализ
Рис. Д.194. Графики зависимостей между измеряемой величиной у и содержанием х.
а — многозначная; б — двузначная; в — однозначная, но не линейная; е — линейная с фоном.
менением чистых реактивов, аппаратуры с высокой разрешающей способностью и т. д.). При невозможности устранения сигналы фона, полученные при постоянных условиях, вычитают из основного сигнала. Но в этом случае у снова определяют как разность двух значений, и, если эти значения близки (значение сигнала фона велико), относительная случайная ошибка становится большой, как было показано выше. Поэтому сигнал фона перед вычитанием нужно уменьшить до минимальных размеров. Наилучшие результаты получают, когда градуировочный график описывается уравнением у = Ьх (рис. Д.195). Коэффициент b
44. Теория измерений в аналитической химии
457
называют также коэффициентом регрессии, потому что обычно его значение определяют, рассчитывая регрессию. Величину b рассматривают как характеристику чувствительности метода: чем больше Ь, тем больше интенсивность сигнала у для данного содержания х.
При одной и той же абсолютной ошибке измерения абсолютная ошибка определения количества ах уменьшается с увеличением чувствительности метода. Напротив, относительная
Рис. Д.195. Линейный градуировочный график в отсутствие фона.
При большой чувствительности Ь\ при постоянном <зу (стандартное отклонение сигнала) стандартное отклонение результата анализа ох меньше, чем ах , полученное при меньшей чувствительности &2-
ошибка не зависит от чувствительности:
= но (3yiy=ejxix
Чувствительность b инструментальных методов анализа определяется фактором пересчета показаний прибора (обычно в единицах шкалы) на содержание вещества; в гравиметрии — это обратная величина стехиометрического гравиметрического фактора (й=1//). Чем меньше /, тем больше чувствительность метода и тем меньше абсолютная ошибка гравиметрического определения количества вещества х. В объемных методах анализа фактору f соответствует эквивалентная концентрация сэ применяемого титранта. Чтобы ошибка определения была невелика, а чувствительность метода высока, эта величина должна быть как можно меньшей, что способствует получению интенсивного сигнала у. Однако при этом начинает сказываться эффект разбавления, что приводит к систематическим ошибкам определения, поэтому следует выбирать оптимальную величину сэ.
458
Д. Химический анализ
Таким образом, для количественной оценки результатов измерений желательно знать значение Ь. Поскольку стехиометрический гравиметрический фактор можно рассматривать как достаточно постоянную величину, то гравиметрию можно считать абсолютным методом. В объемных же методах анализа необходима градуировка, так как эти методы связаны с определением концентрации титрантов. Градуировка необходима также для всех инструментальных методов количественного анализа.
В идеальном случае, когда у = Ьх, достаточно было бы однократного определения величины b измерением у при известном х при условии, что чувствительность остается постоянной во всем диапазоне измерений при градуировке и проведении анализа. Однако на практике этого не бывает, поэтому проводят серию измерений эталонных и анализируемых проб. Поскольку •содержание компонентов в анализируемых пробах может быть различным, эталонные пробы готовят с широким диапазоном содержаний определяемых компонентов и таким образом получают на градуировочной кривой ряд точек.
Случайные ошибки градуировки и самого анализа входят в общую ошибку метода анализа:
(493>
где пе — число эталонных проб; пА— число параллельных определений; уА — величина сигнала отдельного результата анализа; ум — величина сигнала, соответствующего середине интервала измерений; сг& — стандартное отклонение для чувствительности Ь; Оу — стандартное отклонение сигнала; ах — стандартное отклонение полученного значения х.
Следовательно, увеличивается с увеличением <ху и О(, и обратно пропорционально чувствительности Ь.
Получение точного результата (ох мало) возможно, еслиоу мало, а b велика; кроме того, измеряемая величина уА должна находиться в середине интервала измерений уА~Ум- При выполнении последнего условия уравнение (493) переходит в следующее:
Если общее число измерений п = пА + пЕ, то можно провести большое число измерений эталонных проб и пе'Д>па- Отсюда следует, что ошибки уменьшаются в раз:
о =-^-----tU- для абсолютной ошибки,
b V"A
----L- для относительной ошибки
Х У V«A
459
44. Теория измерений в аналитической химии
Если Пе = Па, то получим
ff, = ----для абсолютной ошибки,
х b у/п
<1Х Щ 2 „ ,
—- = — -----=^ для относительной ошибки
х У уп
В зависимости от числа анализируемых проб (много или одна) выбирают один из приведенных вариантов.
Условием применения описанного метода является постоянство чувствительности Ъ для всех последовательно исследуемых проб. Однако иногда это условие не выполняется — чувствительность может изменяться во времени. В таких случаях рекомендуется выбор второго варианта метода, т. е. при каждом анализе нужно проводить измерение новой эталонной пробы. На величину Ъ также может оказать влияние смена измерительной ячейки (например, в полярографии). Особое внимание следует обращать на присутствие в анализируемой пробе наряду с определяемым компонентом большого избытка других компонентов, т. е. на возникновение переменного матричного эффекта.
Вместо описанного метода градуировки можно использовать так называемый метод стандартных добавок. При этом исходят из прямой пропорциональной зависимости. Сигнал фона должен отсутствовать (й = 0). Раствор исследуемой пробы делят на равные части, в которые вводят эталонные добавки в порядке возрастания их концентрации. Если происходит разбавление, нужно вносить поправку на изменение концентрации. Если же в эталонном растворе определяемый компонент находится в значительно большей (например, в 100 раз) концентрации, чем в исследуемой пробе, разбавлением раствора можно пренебречь. Достоинством такого способа является отсутствие разбавления матрицы пробы в растворе, однако необходимы специальные устройства для точного дозирования малых объемов. Из уравнений у\ = Ьх и y2 = b (х + xz) получим у\/х=уг/(х + хг), а искомое содержание х составит
Применение закона распространения ошибок дает следующую формулу:
(zt \ 2 / гг \ 2
х ) \ У )
X \ %
1+^ (1-0 xz /
(495)
где г — коэффициент корреляции. Чем больше коэффициент корреляции, тем более строго коррелируют уг и у2 благодаря быстрому проведению последовательных измерений и тем меньше случайная ошибка результата измерения. Аналогичное влияние
460
Д. Химический анализ
оказывает введение возможно большей стандартной добавки хг^>х. Однако ввиду заданности интервала концентраций не следует вводить слишком большую стандартную добавку (х2<бх).
Если сопоставить метод градуировочных кривых, преобразуя уравнение (494) введением х2Ь2 = у2 и fiA = n£=l, с методом стандартных добавок, то получатся следующие соотношения:
1 = (1+х/хгГ (1 - Г)
Отсюда следует г = 0,75 при x = xz и г=0,30 прихг=5х. Только тогда, когда коэффициент корреляции превышает приведенные значения, метод стандартных добавок по своей точности превосходит метод градуировочных кривых.
Точность обоих методов, естественно, увеличивается при проведении большего числа измерений (больше двух). Точность метода стандартных добавок повышается, если использовать
Рис. Д.196. Иллюстрация метода внутреннего стандарта.
{/ — сигнал пробы; уст — сигнал стандарта; и пст — стандартные отклонения; д—отношение сигналов определяемого компонента и внутреннего стандарта; oq — стандартное отклонение.
стандартные добавки различной величины и затем экстраполировать полученные результаты к хг = 0. Преимуществом этого приема является устранение матричного эффекта.
В заключение следует сказать о методе внутреннего стандарта. Внутренний стандарт добавляют к эталонным и анализируемым пробам в строго одинаковом количестве. Его сигнал не должен накладываться на сигнал определяемого компонента, но должен находиться близко от него и согласованно изменяться под действием различных факторов, т. е. аналитический и
44. Теория измерений в аналитической химии
461
стандартный сигналы должны коррелироваться. Получают отношение q сигнала у определяемого компонента к сигналу уст добавленного внутреннего стандарта (рис. Д.196) и используют градуировочную функцию y/yCT = g = f (х), а в идеальном случае q — bx.
Для успешной корреляции измерение у и уст желательно проводить с возможно меньшим промежутком времени и лучше всего одновременно (двухканальные приборы). Применение закона распространения ошибок в этом случае дает следующую формулу:
\ __ ( Ту \ \ _2г
Q / \ У ) \ Уст / УУст
Принимая, что в широких пределах (Уу~(5:/п и при соответствующем количестве добавленного внутреннего стандарта у~ ~уст, получим для интенсивности сигнала выражения
= 1/2(1—
q У г
Уменьшение случайной ошибки aqlq<Gyly происходит в случае, когда У2(1—г)<1, т. е. при г>0,5. Для у^Д/ст этот предел повышается.
44.8. Оценка методов анализа и результатов измерений
Как было показано в разд. 44.3, при измерении какого-либо параметра различными аналитическими методами происходит небольшой, но неизбежный случайный разброс результатов. При оценке результатов измерений, например, методами, приведенными в разд. 44.7, этот разброс тем или иным образом сказывается на результатах анализа. Из данных по случайному разбросу результатов анализа эталонной пробы можно определить случайный разброс, или точность, метода анализа, а из отклонения среднего значения от известного теоретического найти правильность, или систематическую ошибку, метода. Если аналогично оценить операции отбора пробы и подготовки ее к анализу, то можно сделать соответствующие выводы о методе анализа в целом. Эти выводы имеют особенно важное значение для аналитической практики, но на их получение тратится много времени, поскольку необходимо осуществить весь ход анализа. Часто соответствующие рекомендации касаются только принципа проведения анализа или в лучшем случае собственно метода
462
Д. Химический анализ
анализа, что не позволяет объективно оценить возможности метода в конкретном случае.
Теория измерения в различных вариантах дает отличную возможность оценить с высокой статистической вероятностью результаты и методы анализа, особенно при большом числе результатов, на основе математической обработки экспериментальных данных. При этом, с одной стороны, оценивают точность и правильность методик и методов анализа и, с другой стороны, информацию, заложенную в результатах анализа (т. е. обобщения и практические выводы, которые можно сделать из полученных результатов).
Разброс результатов анализа (х), а также аналитических сигналов (у) в их генеральной совокупности оценивают величиной о. Грубой оценочной величиной для о является диапазон значений 7? = хгпах—-Xmin, т. е. разность между наибольшим и наименьшим результатами анализа серии идентичных проб или идентичных материалов, полученными с использованием одних и тех же методов анализа. Значительно более точной оценочной величиной является стандартное отклонение
s
(497)
S-><7 ПрИ П->ОО.
Чтобы s являлось надежной оценочной величиной для <т, необходимо большое число опытов. Это достигается анализом серии проб аналогично серийным анализам на производстве. Необходимым условием является использование одного и того же метода анализа идентичных материалов со сходным содержанием определяемого компонента. Например, в образцах латуни различных марок сходного состава можно определять содержание цинка, проводя 3, 2 и 4 параллельных определений. Для расчета стандартного отклонения s существует следующая формула:
Или в общем виде:
(499)
Разность п—т, где п — общее число определений и т — число проб, представляет собой число степеней свободы f. При одинаковом числе параллельных определений для каждой пробы п== = тпА. Если рассчитаны величины стандартных отклонений для
44. Теория измерений в аналитической химии
463
каждой пробы Si, s2, «з и т. д., то для получения величины суммарного стандартного отклонения поступают следщощим образом. Из уравнения (497) находят s2(n—l)=S(xz—х)2, т. е.
sl2 (Я1 — i) 4~ s22 (Я2 — 1) ~Ь ~Ь s'2-m (flj — 1) ^2 п — m
(500)
Для парных определений, часто осуществляемых_на практике, при Па = 2 из уравнения (500) и соотношений х=(х'+х")/2; х'—х=х'—(х'+х")/2= (V—х")/2 следует
где х/ и Xj" — результаты парных определений; f~m — число парных определений. При определении содержания одного и того же компонента в одной и той же пробе или в одном и том же материале можно сравнить стандартные отклонения двух аналитических методик или методов анализа. Для этого определяют и сравнивают два стандартных отклонения Si и s2. Если s( больше, чем s2, то это ещё не означает, что точность первого метода хуже, чем второго, поскольку все значения 5г и s2 и т. д., вычисляемые при небольшом числе измерений, редко бывают надежны. При большем числе измерений можно получить обратный результат, но в то же время едва заметное расхождение стандартных отклонений становится отчетливее, а существовавшее четкое расхождение постепенно сглаживается. Для получения статистически обоснованных выводов при сравнении этих величин применяют так называемый F-критерий, определяемый как S|2/s22 = F. В числителе обычно находится большее из двух стандартных отклонений, поэтому F всегда больше 1. Рассчитанное значение F сравнивают с табличной величиной F(P, fi, fz) в зависимости от статистической вероятности Р (например, 0,95 или 0,99) и числа степеней свободы = —1 стандартного отклонения s(, а также /2 = н2—1 для s2. При F> >F(P, ft, f2) расхождение между величинами стандартных отклонений S] и s2 считается значимым с вероятностью Р. Если F<F(P, fi, f%) это еще не означает безусловного отсутствия расхождения при высокой вероятности (95% или 99%); можно лишь сделать вывод, что из полученных данных нельзя этого определить. Тогда можно или довольствоваться полученным результатом, или увеличить число определений для установления значимости расхождения. Отсутствие расхождения доказать таким способом нельзя.
Следовательно, табличные данные F(P, f,, f2) (табл. Д.37) коррелируют, или, лучше сказать, учитывают ненадежность вывода о том, что si>s2, т. е. также Oi>o2, обусловленную И S2=#<T2.
464
Д. Химический анализ
Таблица Д.37. Статистические значения F-критерия3
Л i
h 1 2 3 4 ~ 1 ""л 5
1 161 200 216 225 230
2 18,51 19,00 19,16 19,25 19,30
3 10,13 9,55 9,28 9,12 9,01
4 7,71 6,94 6,59 6,39 6,26
5 6,61 5,79 5,41 5,19 5,05
6 5,99 5,14 4,76 4,53 4,39
7 5,59 4,74 4,35 4,12 3,97
8 5,32 4,46 4,07 3,84 3,68
9 5,12 4,26 3,86 3,63 3,48
10 4,96 4,10 3,71 3,48 3,33
1 4052 4999 5403 5625 5764
2 98,49 99,00 99,17 99,25 99,30
3 34,12 30,81 29,46 28,71 28,24
4 21,20 18,00 16,69 15,98 15,52
5 16,26 13,27 12,06 11,39 10,97
6 13,74 10,92 9,78 9,15 8,75
7 12,25 9,55 8,45 7,85 7,46
8 11,26 8,65 7,59 7,01 6,63
9 10,56 8,02 6,99 6,42 6,06
10 10,04 7,56 6,55 5,99 5,64
а Верхняя часть таблицы относится к Р=0,95; нижияя часть—к Р=0,99.
Пример. Пусть получено по 4 значения стандартных отклонений, т. е. fi = f2 = 3. Тогда с вероятностью 95% справедливо условие Si2/s22>F (0,95; 3; 3) =9,28, 5i/s2>1'9,28.
Значение st должно быть по крайней мере в 3 раза больше s2, чтобы при данном числе результатов (по 4) с 95%-ной вероятностью можно было говорить о значимости расхождения. Иначе говоря, при st~3s2 существует примерно' 5% степени надежности для вывода, что для двух сравниваемых методов 014^02. Если из соотношения st«3s2 двадцать раз делают выводы о том, что 01>а2, то один из этих выводов неверен.
Чтобы с 99%-ной вероятностью сделать вывод о том, что од>а2 при F{P, fi, fz) =29,46, должно соблюдаться условие Si«5,5 s2.
Из таблицы ясно, что при малом стандартном отклонении s2 целесообразно увеличить число получаемых значений, так как F (0,95; А = 3; /2 = 3)=9,28; F (0,95; /, = 5; /2 = 3)=9,01, но F (0,95; fi = 3; f2 = 5) = 5,41 и т. д. Данные, используемые для обнаружения значимости расхождений методов и методик анализа, можно использовать также для ориентировочного решения вопроса о том, связан ли разброс значений, получаемых при решении одних и тех же задач одними и теми же методами, но-
44. Теория измерений в аналитической химии 465
разными исследователями, с различной точностью методов или является случайным (из-за небольшого числа определений).
Аналогично тому как s, рассчитанное для ограниченного числа определений п, со статистической надежностью приближенно отражает стандартное отклонение генеральной совокупности (о), арифметическое среднее х=(2хг)/п также только со статистической надежностью приближенно (оценочная величина) отражает среднее генеральной совокупности (ц). При получении из двух серий мало различающихся значений Х] и х2 можно говорить или о случайном различии двух выборок из одной генеральной совокупности (т. е. несмотря на то, что xi>x2, pi = 1x2), или о действительно различных генеральных совокупностях щ>р2 (что обусловлено систематической ошибкой или различием проб). Для решения этого вопроса при незначительном расхождении Xi и х2 необходимо учесть стандартные отклонения первой (si) и второй (s2) серий измерений. Это также необхо
Рис. Д.197. /-Распределение результатов анализа с уменьшением числа степеней свободы (оо; 5; 1) и увеличением пределов интегрирования (1,96; 2,57;
12,7) для 95%-ной вероятности Р.
30—1880
466
Д. Химический анализ
димо для решения вопроса о размерах области ненадежных результатов х и о величине интервала надежных значений х. В качестве грубого приближения используют интервалы X]±Si и x2±s2. Если эти интервалы не налагаются, можно говорить о расхождении (р^рг), но его нельзя считать доказанным.
Вспомним, что ± означает вероятность 68,3% или соответственно степень надежности 31,7%. Так как с увеличением числа параллельных определений Па уменьшается случайный разброс средних значений, то соотношение ц±о/па привело бы к XzEs/Упл. Однако для небольшого числа определений Нормальное распределение Гаусса строго выполнимо только при п->оо, а при меньшем п плотность распределения можно определить лишь с отклонениями. Эти отклонения, а также вероятность отклонения х от истинного значения в зависимости от числа измерений подчиняются так называемому /-распределению, представляющему собой модифицированное симметричное распределение.
Максимумы частот гауссова и /-распределения находятся при одном и том же значении абсциссы. Высота и ширина кривых /-распределения в отличие от кривых нормального распределения зависят от числа степеней свободы f соответствующего стандартного отклонения. Это означает, что расхождение при данной
Таблица Д.38. Пределы интегрирования для /-распределения
f 0,75 0,90 0,95 0,99
1 2,41 6,31 12,7 63,7
2 1,60 2,92 4,30 9,92
3 1,42 2,35 3,18 5,84
4 1,34 2,13 2,78 4,60
5 1,30 2,01 2,57 4,03
6 1,27 1,94 2,45 3,71
7 1,25 1,89 2,36 3,50
8 1,24 1,86 2,31 3,36
9 1,23 1,83 2,26 3,25
10 1,22 1,81 2,23 3,17
11 1,21 1,80 2,20 3,11
12 1,21 1,78 2,18 3,05
13 1,20 1,77 2,16 3,01
14 1,20 1,76 2,14 2,98
15 1,20 1,75 2,13 2,95
20 1,18 1 ,73 2,09 2,85
25 1,18 1,71 2,06 2,79
30 1,17 1,70 2,04 2,75
40 1,17 1,68 2,02 2,70
60 1,16 1,67 2,00 2,66
120 1,16 1,66 1,98 2,62
00 1,15 1,64 1,96 2,58
44. Теория измерений в аналитической химии 467
вероятности тем больше, чем меньше число измерений (рис. Д.197). Значения /-распределения (пределы интегрирования и) приведены в табл. Д.38. Можно видеть, что для /=оо существуют определенные пределы р,±ио, ограничивающие 75%, 90%, 95% или 99% площади гауссовой колоколообразной кривой (см. также разд. 44.3). Но поскольку из-за ненадежности значения s при небольшом f колоколообразная кривая шире, чем кривая /-распределения (при вероятности Р), пределы расширяются, например для вероятности 95% вместо р,±1,96ст при f-^-оо они становятся уже ± 12,7 s для f=l или соответственно ±4,30 s для /=2. В общем виде получаем для рЩст/упл генеральной совокупности при выборке х±/(Р, f) s/Упл
t(P,f) s/V^= Ьх (502)
где Ах представляет собой так называемый доверительный интервал среднего арифметического х при заданной вероятности Р. В табл. Д.38 приведены значения /(Р, f) в зависимости от числа степеней свободы f—n—1 стандартного отклонения s. Число определений Па может быть равно п, но может быть и меньше. Последнее происходит тогда, когда значение суммарного стандартного отклонения рассчитывают из большого числа определений, проведенных в идентичных условиях с применением соответствующих методов, а х определяют из небольшого числа Па вновь проведенных измерений.
Если имеется очень надежное значение стандартного отклонения s (s — о), вычисленное при п>100, то при x+uo/Упд находят доверительный интервал причем и=1,64 (Р = 0,90),
u== 1,96 (Р = 0,95) и и = 2,58 (Р = 0,99). При делении на ]пА получают сначала быстро, а затем медленно уменьшающиеся величины. В случае Па = \ также можно определить доверительный интервал (в то время как при п=1 его определить нельзя). На практике часто используются Р = 0,997 (и~3) и получают при s = o и па = 1 так называемую область рассеяния с разбросом ±3s.
Не рекомендуется использовать так называемую «среднюю квадратичную ошибку арифметического среднего», рассчитанную по формуле
] /*2 (xi ~ Ч2
|/ "и (503)
Как видно из уравнения (497), этому выражению соответствует отношение s/y'n. Такая ошибка идентична доверительному интервалу с t(P, f) = l, что происходит прн н Р= 0,683. Статистическая вероятность в этом случае равна 68,3% и степень надежности 31,7%. При небольшом числе определений, характерных обычно для практики, степень надежности была бы еще больше и при f=l (т. е. при м=2) составляла бы уже 50%, чему соответствует _Р=0,50, как видно из таблиц значений t(P, f).
30*
1
-2? a.
Рис. Д.198. Оценка верхней и нижней границ доверительного интервала (а) и одной из границ (б).
44. Теория измерений в аналитической химии
469
Доверительный интервал принято указывать как х±Ахр, т. е. с обозначением статистической вероятности Р. Указывая доверительный интервал, устанавливают число значащих цифр для х, которое определяется интервалом Дх. До сих пор мы оценивали и нижнюю и верхнюю границы доверительного интервала х±Дх. На практике часто вопрос рассматривают «односторонне», т. е. оценивают одну из границ, например нижнюю, когда интересует, отвечает ли объект по содержанию компонента принятой норме (металл в руде или др.), или верхнюю, когда необходимо знать, не превышают ли допустимый уровень количества нежелательных или вредных компонентов. В этих случаях вероятность Р или степень надежности составляют: Р=0,5+ + Р/2 и а=100—Р, т. е. Р>Р и а<м (рис. Д.198). Для одинакового разброса и одинакового числа определений степень надежности уменьшается, а вероятность правильной оценки увеличивается.
Пример. Никель определили осаждением диметилглиоксимом. В табл. Д.39 приведены результаты, полученные тремя экспериментаторами. Первый и второй добавляли при осаждении большой избыток осадителя (неверно/), третий правильно проводил анализ, но юн готовил новый раствор NiSCh взвешива-
Таблица Д.39. Результаты определения никеля осаждением диметилглиоксимом по данным трех исследователей
п Количество Ni, мг/см3
1 2 3
1 9,23 9,26 9,80
2 9,24 9,29 9,82
3 9,24 9,35 9,83
4 9,30 9,29 9,80
5 9,27 — 9,84
6 9,29 — 9,82
7 — — 8,83
8 — — 9,83
X 9,2617 9,2975 9,8213
S 0,0293 0,0377 0,0146
нием навески и растворением ее в определенном объеме воды. Среднее арифметическое первого и второго определений идентичны, близки и величины стандартных отклонений. Значение х, полученное третьим экспериментатором, больше, а 5, напротив, меньше. Выясним, являются эти различия характерными для данного определения вследствие вышеназванных причин или случайными, обусловленными небольшим числом определений. Конечно, у аналитиков устают глаза, что приводит к ошибке, но тем реже, чем опытнее аналитик. Полученную в сериях опыта информацию можно объективно оценить только методами математической статистики. Во всех случаях примем Р= = 0,95.
470
Д. Химический анализ
По данным первого экспериментатора, пА = 6; Упл=2,45; п=пА; [=5; t(P, 0=2,57, и отсюда ДХ1 = 0,0307. Результат х±Дх, равный 9,2617± ±0,0307, связан только с усталостью экспериментатора. Как показывает Дх, для х вторая цифра после запятой уже не является значащей. Целесообразно выразить интервал как (9,26±0,03) мг iNi/см3. По данным второго экспериментатора, пл —4; Умд=2; п=пА; f=3; t(P, f) =3,18, и отсюда Дх2=0,060. Этот доверительный интервал, таким образом, в два раза больше, чем ДХ] из-за небольших значений пА, f, а также и s. Результат можно представить как х2=9,30±0,06 мг Ni/см3. Сопоставление результатов показывает, что доверительные интервалы xt и х2 накладываются: верхняя граница X]=Xi + + ДХ1 = 9,29; нижняя граница х2—х2—Дх2 = 9,24. Таким образом, в данном случае речь идет об одном и том же содержании у, генеральной совокупности, которое только из-за разброса результатов и небольшого числа определений представляют в виде xt=H=x2.
По данным третьего экспериментатора, пА = 8; Умл = 2,88; п=пА; f=7' t(P, 0=2,36; Дх3=0,012; х3=9,82±0,01.
Значение Дх3 составляет только ‘/в от Дх2 прежде всего потому, что пл = 8 и f—7, но также и вследствие малого значения s12. Эта величина и ее разброс около х3 явно не совпадают с Si и s2 и их разбросом около Xi и х2.
Из третьей серии значений находят взаимосвязь между числом определений п, оценочной величиной — медианой х и средним арифметическим х для величины у, генеральной совокупности, а также диапазоном значений R и стандартным отклонением s для стандартного отклонения о генеральной совокупности. Кроме того, устанавливают зависимость величин Уп, t(P, f) и доверительного интервала Дх от п (табл. Д.40).
Таблица 40. Взаимосвязь статистических характеристик на примере результатов одной серии измерений
Измеренные значения п f X X R S Vn Z (Р0,9Б= f) ЛХ0,95
9,80 1 0 9,80 9,80 1
9,82 2 1 9,81 9,81 0,02 0,0141 1,414 12,71 0,127
9,83 3 2 9,82 9,817 0,03 0,0153 1,732 4,30 0,038
9,83 4 3 9,825 9,820 0,03 0,0141 2 3,18 0,0225
9,80 5 4 9,82 9,816 0,03 0,0152 2,236 2,78 0,0189
9,84 6 5 9,825 9,820 0,04 0,0167 2,45 2,57 0,0176
9,82 7 6 9,82 9,820 0,04 0,0153 2,646 2,45 0,0141
9 83 8 7 9,825 9,821 0,04 0,0146 2,83 2,36 0,0122
Оказалось, что численные значения медианы х и арифметического среднего х хорошо совпадают. С ростом числа определений п стандартное отклонение s изменяется в пределах 0,014—0,017. Диапазон значений R вначале больше (0,02) и затем еще немного возрастает. Величина R менее пригодна для оценки о генеральной совокупности, чем стандартное отклонение s. Значение }'п вначале резко, а затем медленнее возрастает, a t(P, f) вначале резко, а затем медленнее уменьшается. Величины Уп и t(P, f) с ростом числа определений п оказывают влияние на доверительный интервал Дх, уменьшая его, т. е. уменьшая тем самым ненадежное случайное отклонение х от у. При переходе от двух определений к восьми s изменяется незначительно, Ум удваивается, ЦР, f) уменьшается примерно в пять раз, а Дх — примерно-в 10 раз.
Обратимся к данным табл. Д.39. Для сравнения стандартных отклонений нужно использовать F-критерий. Для двух первых серий измерений по
44. Теория измерений в аналитической химии
471
лучим s2>si, S22/S12=1,66, F (0,95; fi = 3; f2=5)=5,41 и для F (0,99) даже 12,06. Число 1,66 показывает, что расхождение между стандартными отклонениями s2 и St несмотря на их отношение 2 : 1 нельзя считать значимым с вероятностью 95%, т. е. и дальнейшие измерения нельзя считать надежными. Напротив, отношение s22/s-3 = 6,72 при сравнении с F(0,95; f=3; /2=7)=4,35 и F(0,99)=8,45 показывает, что расхождение между стандартными отклонениями является значимым с вероятностью значительно большей 95%, но меньшей 99%, которую обычно считают критерием значимости расхождения стандартных отклонений генеральных совокупностей (о2><Хз). Дальнейшие измерения могут быть также приведены к этому результату.
Для надежного определения расхождения средних значений используют /-критерий, рассчитываемый по следующей формуле:
t = 1*1~Д I.1/ (504)
Иначе говоря, получают абсолютное значение разности сравниваемых средних значений и делят ее на величину стандартного отклонения, рассчитанного для результатов обеих серий определений (в соответствии с уравнениями (498) и (500)) по формулам
s=-|/ (505) s 1/~ sl2 (»1 — 1) + s22 (п2 — 1)
V п — 2
Полученный результат затем умножают на ^nin2/ (п\ + п2), где «1 и «2 — числа результатов определений в обеих сериях. Кроме того, п = Н1 + п2, т = 2. Рассчитанное значение сравнивают с соответствующим /-критерием (табл. Д.38), причем нужно использовать f=ni + «2 — 2 (см. уравнение (505)). Если />/(Р, f), расхождение между обоими средними результатами генеральных совокупностей значимо с вероятностью Р (рчТ^рД-
В рассмотренном примере с использованием данных табл. Д.39 получим при сравнении xt и x2(|xi—х2| =0,0358) следующие значения: Si,2 = 0,0327; ______ 0,0358
3'6• 4/10= 1,55, / = 0-Q327 ‘ 1,55«1,7, в то время как t(P, f) при Р=0,95 и f=8 равно 2,31, а при Р=0,99 даже 3,36. Таким образом, эти результаты действительно не доказывают расхождения между средними значениями. По выраже-Дх
нию 2,31 = Q Qggy • 1>55 рассчитывают, каким должен быть Дх, чтобы при данном разбросе значений и числе определений можно было говорить о значимости расхождений. При Р=0,95 получают Дх>0,049; при Р=0,99—Дх> >0,071. При сравнении средних значений определений, проведенных в первой и третьей серии или соответственно во второй и третьей, в обоих случаях с 99%-ной вероятностью установлена значимость расхождений, т. е. p,i'»p,2< <Цз-
472
Д. Химический анализ
/-Критерий можно также использовать для сравнения среднего значения х с теоретическим значением хтеор:
/ = J. ДГ I (506)
Из выражения т/пдъ/^гц + пя) (ср. уравнение (504)) при пА = = П\ и получают (хтеор— величина, определенная, можно сказать, бесконечное число раз).
В уравнении (506) пА— число параллельных опытов, из которых определено х. Значение s рассчитано из результатов измерений при f=nA—1 в соответствии с уравнением (497).
При f) расхождение между результатом анализа и
теоретическим значением можно рассматривать как значимое с вероятностью Р, в то время как для t<t(P, f) разность |х—хтеор| можно оценить как случайную. Такую оценку результатов применяют для решения вопроса, соответствует или нет величина х теоретической величине хтеор для этого же соединения. Однако только по одной формуле этот вопрос можно и не решить. Другими критериями являются молекулярная масса, валентность, свойства вещества и др. характеристики. Но в любом случае, если f>t(P, f), решение однозначно: состав соединения можно считать неверным с вероятностью Р.
E.I. ОБОРУДОВАНИЕ ЛАБОРАТОРИЙ И ЛАБОРАТОРНАЯ ТЕХНИКА
45. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
45.1. Стекло
Использование стекла в качестве материала в химических лабораториях обусловлено в основном следующими его свойствами: прозрачностью, химической и термической устойчивостью, легкоплавкостью, пластичностью в жидком состоянии, а также стабильностью стекловидного состояния. В табл. Е.1. указаны свойства и области применения наиболее употребительных сортов лабораторного стекла*; температурой стеклования (Тст) называют температуру, при которой вязкость стекла равна 1012 пуаз. Ниже температуры стеклования стёкла находятся в твердом состоянии, при нанесении на них царапин образуются трещины; выше температуры перехода стёкла существуют в пластичном состоянии.
Экспериментальные работы в лаборатории требуют знания основ стеклодувного дела. Для формования стекла в стеклодувных работах выбирают узкую область температур, в которой стекло находится в пластичном состоянии. В этом состоянии для стекла характерны высокая вязкость и большое поверхностное натяжение. Очень важно правильно выбирать температуру формования стекла. Работу со стеклом ведут в пламени ручной паяльной горелки или горелки Теклю.
Стеклянные трубки часто необходимо резать. На лабораторных трубках обычных диаметров достаточно сделать один надрез острым ножом для резания стекла в месте раздела. Затем трубку разламывают, растягивая ее вдоль оси (рис. Е.1). Острые края трубок оплавляют. Стеклянные трубки небольшого диаметра можно легко согнуть, нагревая их при вращении в пламени горелки с насадкой типа «ласточкин хвост» или в не слишком горячем пламени стеклодувной горелки. Трубку нагревают до тех пор, пока она не согнется под действием собственной тяжести. Чтобы согнуть трубку под большим углом, ее закрывают с одного конца и, немного растягивая, нагревают в месте сгиба, затем при поддувании сгибают (рис. Е.2). Если трубку сгибать без поддувания, в ней образуются трещины, изгиб выравнива-
* О сортах стекла, выпускаемого в СССР, см. книгу: Стекло. Справочник.— М.: Стройиздат, 1973. — Прим, перев.
473
Таблица ЕЛ. Свойства и применение различных сортов лабораторного стекла
Применение Стеклянные приборы (эксикаторы), установки для получения газа, капельные воронки; стеклянная тара для твердых веществ Тонкостенная стеклянная посуда (стаканы, колбы, чашки, для выпаривания), установки из стекла Стеклянные сосуды больших размеров, трубки; из-за незначительного теплового расширения непригодно для изготовления толстостенных сосудов (склянок для отсасывания) Толстостенные сосуды Трубки для сжигания, трубки для прокаливания, трубки для работы под давлением и др. Части установок, тигли, лодочки
Свойства стекла Небольшая устойчивость к перепаду температур; следовые количества щелочей и кремневой кислоты из стекла переходят в раствор Хорошая устойчивость к перепаду температур Химически менее устойчиво, чем йенское термостойкое стекло марки «G» Термическая и механическая устойчивость такая же, как у йенского термостойкого стекла марок «G» н «D» Термически очень устойчиво См. йенское термостойкое стек- ло марки G Термически очень устойчиво, в том числе при резком перепаде температур; другие свойства см. разд. Е.45.3.
Коэффициент линейного расширения в области температур, °C w S S о .. g § J. I I 1112 сч <М <м <м <м
о е сч СО со СО 00 со со со сч ю 00
Температура стеклования, °C со 4? о 40 1-~ <3 о о чз СО <М <М о 40 40 40 чо t- U0 О 40 )
Сорт стекла 0) » гь <D г , г « <U 1 г чл - °Е®3 с =к га о га ° S? И Ьй О X Д. и о и ОО й О О- с сх »S h 5 »S £ Я H »s н E с Л S v g 03 gco S £ О * « ’§ <U * g- S О J. О H KsSsSssS Q. § S SsSscoKg
45. Материалы и оборудование
475
Рис. Е.1. Правильное разламывание Рис. Е.2. Способ сгибания стеклян-стеклянной трубки. ной трубки.
Тпубка закрыта с одной стороны. Стрелки обозначают место нагревания.
ется; устранить дефекты можно повторным нагреванием с поддувом. Описание стеклодувных работ можно найти в книге П. И. Воскресенского*.
45.2. Стеклянное оборудование
Обычная лабораторная посуда — пробирки, стаканы, колбы, воронки— хорошо известна. Посуда специального назначения рассматривается при описании техники работы, а также в главах по качественному и количественному анализу и синтезу препаратов.
Отдельные элементы установок можно соединить с помощью просверленных корковых и резиновых пробок, стеклянных трубок и резиновых шлангов или шлангов из полимерных материалов. При проведении экспериментальных работ удобно пользоваться стандартными шлифами, состоящими из керна и муфты. В зависимости от формы поверхности различают плоские, цилиндрические, шаровые и конические шлифы (рис. Е.З). Плоские шлифы применяют для соединения деталей большого диаметра (например, в эксикаторах), цилиндрические — преимущественно в колбах с пришлифованными капиллярами (для предотвращения вскипания жидкости) и мешалками (типа K.PG, представляющими собой точно калиброванные стеклянные трубки). Шаровые шлифы в лаборатории находят ограниченное применение, несмотря на свое преимущество — шарнирную подвижность; для соединения этих шлифов необходимо использовать металлические зажимы (рис. Е.4). Невысокие шаровые шлифы с большим радиусом называют чечевицеобразными. Шаровые шлифы уже широко используются в стеклянном оборудовании на опытных установках. В химических лабораториях в основном применяют конические шлифы. Имеющееся в продаже стеклян-
* Воскресенский П. И. Техника лабораторных работ. — М.: Химия, 1973. — Прим, перев.
Рис. Е.З. Различные типы шлифов.
а — плоский; б — цилиндрический; в — шаровой; г — конический.
Рис. Е.4. Схема уплотнения шарового шлифа при помощи зажима.
Рис. Е.5. Продольный разрез кониче- Рис. Е.6. Способ уплотнения стан-
ского шлифа для керна и муфт. дартных шлифов с помощью пружин
и хомутиков.
45. Материалы и оборудование
477
ное оборудование оснащено в настоящее время стандартными шлифами, размеры которых стандартизированы в соответствии с техническими условиями TGL 14972. Наиболее распространены конические шлифы с конусностью 1: 10, имеющие следующие соотношения между диаметром d и высотой шлифа h (рис. Е.5): 5/13, 7,5/16; 10/19, 12,5/21; 14,5/23; 19/26; 24/29; 29/32; 34,5/35; 45/40; 60/46; 70/50; 85/55; 100/60. В соответствии с ранее применявшимися стандартами шлифы и сейчас еще часто характеризуют только размером диаметра верхней части. Примерно 80% всего лабораторного оборудования оснащено шлифами 29/32 (прежнее обозначение NS29). Для посуды малых размеров применяют шлифы преимущественно 14,5/23, а для больших— 45/40. Шлифы соединяют пружинами и хомутиками (рис. Е.6) или просто резинками, а также пружинами, натянутыми на впаянные крючочки. В вакуумных установках используют также шлифы с более крутым углом и конусностью 1 :5, с размерами 60/46, 75/52 и 90/57. При соединении шлифов, изготовленных из стекол, значительно различающихся коэффициентами расширения (см. табл. Е.1), при температуре выше 200 °C внешний шлиф (муфта) может лопнуть, поэтому лучше использовать шлифы из одинаковых сортов стекла.
Надежность стандартных шлифов зависит от их правильной обработки. Поверхность шлифов необходимо тщательно защищать от цараиин и перед соединением протирать чистой, мягкой тряпочкой. На самый широкий участок керна наносят тончайший слой смазки (силиконовой или смазки для кранов) в виде кольца шириной 5 мм и шлифы при легком надавливании слегка проворачивают.
При заклинивании нужно осторожно постучать по шлифу деревянным предметом или осторожно нагреть внешний шлиф (муфту) по возможности с одновременным охлаждением керна. Склеившиеся шлифы в отдельных случаях можно разъединить нагреванием, если клеящее вещество легко плавится и при нагревании в пламени не разрушает стекло. Однако в большинстве случаев шлифы при этом заклиниваются еще сильнее. Иногда помогает применение химических веществ (кислот) или жидкостей с высокой поверхностной активностью (керосин). Наилуч-шие результаты получаются, если из установки выкачать воздух, заполнить ее растворителем, обычно водой под небольшим давлением (например, подключив установку к водоструйному насосу) и оставить стоять до растворения веществ, склеивших шлифы.
Надписи на стеклянную посуду наносят карандашами для стекла различных цветов и чернилами для стекла (например, разбавленной плавиковой кислотой). На стеклянной посуде часто имеются специально вытравленные места, на которых можно делать надписи обычным карандашом.
478
E.I. Оборудование лабораторий и лабораторная техника
45.3. Керамика и кварц
Из-за устойчивости к перепаду температур и высокой температуры размягчения твердый фарфор применяют главным образом для изготовления тиглей, чашек для выпаривания и трубок. Фарфор, покрытый глазурью, выдерживает температуру 1200 °C, неглазурованный фарфор — почти 1400 °C. Большие толстостенные фарфоровые сосуды нельзя нагревать ни на открытом пламени (даже на асбестовой сетке с проволочным каркасом), ни на песчаной бане. Фарфор мало устойчив к действию щелочных веществ и сильных восстановителей, например электрохимически активных металлов. Для работы при повышенных температурах применяют некоторые специальные сорта фарфора с высоким содержанием А12О3 (массу Пифагора, К.-массу, пиродур и др.). Надписи на фарфоровом тигле можно выцарапать железной иглой на наружной поверхности дна.
Особенно устойчивыми материалами являются спекшиеся оксиды. Спекшееся стекло, рекристаллизованный корунд с содержанием А120з>99,5°/о (товарные названия дегуссит AL 23, хи-локс 972, алсинт, пурокс, кавенит S) применяют для изготовления тиглей, лодочек, ступок и выпускают также в виде сырого продукта. Изделия из спекшегося стекла можно применять до 1950 °C; они устойчивы к перепаду температур и разрушаются только под действием щелочных и фтор содержащих расплавов.
Для особых целей изготавливают тигли и трубки спеканием следующих материалов: оксида магния (пористый, рабочая температура 2300°C); оксида циркония (плотный, рабочая температура до 2300°C); шпинели MgO-Al2O3 (плотный, рабочая температура до 1950°C); нитрида бора (плотный, рабочая температура 1980 °C).
Плавление чистого кварца дает прозрачное кварцевое стекло (см. табл. Е.1). Используя тонкоизмельченный исходный материал, получают спекшийся с одной или с обеих сторон плавленый кварц (витрозил, гомозил, ротозил). Достоинствами кварцевого стекла являются отличная устойчивость к перепаду температур, прозрачность, проницаемость для УФ-лучей и относительно хорошая химическая стойкость. Фтороводородная кислота, кипящая фосфорная кислота и щелочные расплавы разрушают кварц. Внешние загрязнения, например следы пота (NaCl), остающиеся при прикосновении к кварцевым сосудам, могут при температурах выше 1000 °C значительно ускорить переход метастабильного стекловидного состояния в кристаллический тридемит, так называемое «расстекловывание». Этот процесс ведет к быстрому разрушению кварцевых изделий, поэтому посуду из кварцевого стекла необходимо перед нагреванием тщательно очищать спиртом или ацетоном. Кроме того, следует
45. Материалы и оборудование
479
по возможности избегать длительного нагревания изделий из кварцевого стекла в области температурного перехода в триде-мит, т. е. при 870 °C, в присутствии минерализаторов.
45.4. Металлы и графит
Для изготовления тиглей, лодочек, чашек и т. д., используемых в лабораториях, применяют химически стойкие металлы или металлы, имеющие высокую температуру плавления (табл. Е.2). Платина, пожалуй, наиболее широко применяемый для изготовления аппаратуры благородный металл, обладает и тем и другим свойствами. При легировании платины родием или иридием улучшается не только ее механическая прочность, но и химическая стойкость. Максимальная температура применения плати-нородиевого сплава с содержанием 10% Rh достигает 1700°C.
Графит (часто электрографит) по некоторым свойствам не уступает тугоплавким металлам и значительно дешевле. К недостаткам графита относятся его небольшая механическая прочность и неформуемость. Сравнительно недавно начали применять также аппаратуру (тигли, лодочки, чашки, стаканы, трубки) из стеклографита и графита, полученного пиролизом. К преимуществам стеклографита относится его устойчивость к действию расплавов гидроксидов щелочных металлов, галогенидов щелочных металлов, фторидов, KHSO4, а также Вг2, НС1, H2F2, HNO3, HNO3 + H2O2, HNO3 + KCIO3, H2SO4, хромовой кислоты. Изделия из стеклографита не имеют пор; для работы в вакууме их легко очистить от газов; они устойчивы на воздухе до 600 °C, в отсутствие окислителей их можно нагревать до 3000 °C. Очищают изделия из графита и стеклографита кипячением в воде или в кислотах с последующим прокаливанием при 2000 °C в атмосфере инертного газа. Рекомендуют также обработку током хлора.
45.5. Пластмассы
Посуда (пипетки, мерные колбы, цилиндры для встряхивания и др.), а также трубки и шланги из полиэтилена (миратена, лу-полена) не разрушаются при 100 °C под действием HF любых концентраций, конц. НС1, 30%-ной HNO3 и 50%-кого раствора NaOH. Полиэтилен взаимодействует с галогенами, конц. H2SO4 (начиная с 40 °C) и силиконовыми маслами. В органических растворителях, особенно в углеводородах и хлоропроизводных углеводородов, полиэтилен набухает; выше 65 °C он растворим в толуоле.
Политетрафторэтилен (хейдефлон, тефлон, хостафлон TF) наряду с высокой химической стойкостью обладает хорошей
Таблица Е.2. Физические и химические свойства некоторых металлов и графита
S О со » Ю о M X
га та е? схе и £ с-S ®. О га Я
га X Jog “ 2
в- S х я S <D а Я о « 9S >. О га H c0
то « и 5 й S 2 о S к 2 га в s ч ш о
и XS о га га ® н О <u
о ю д со ° я
со п °- ’S . >»
2 ю CD О ю
ф S о «в s LO дО
я о га S СХ га
о 3 о сд4 »s н 5 м ° g 3 §
я S X X га 3 (V о о га S’ 2 3 га я _ о га х ю Ч S со я вы мета.
2 к g м га £ о S и га Я о С-, я 2 5 га >» со га сх ь о га 2 я ° К Е W с- га с? Е-. s
га ю CD со 2 о vo
2 И со _г со га со СХ X >»\о 3 со 5 »я сх * о to
О НЕ» Е о я е< С чя
иаль-мпе-при-я, °C о о ООО о о о о о о
д £ <5 X о о ООО о о о о ю о о
3 н СХ- о см ю см О О — со 00 о ю
««>•? со см см — — СМ — см
та та S
та
Е о о —s О О —Ю
О Ю С*“^ ('""О о О О О ~
см со со со ь- t- со со
X СМ СМ о — ~ о —
о
та м »н О W 05 <Я о О 14
X ) ) Ю 1 1 1 1 Ю 1 ( I
о о см о о о о см о о о см
ф * Ч W4 ив _ к ““
я
я ф со со СО со СО СО —I СО СО СО СО со
cd ~ СО — — •—-и •—-Ч W—Ч •—-ч СО
та • СО
ч V
и
о
о о о о г- LO О О О О О - (*"*)
о о О со Ь- СП о О О ь- со о
О 00 *4^4 1*^*4 ^w^4 Ю Tf Tt< LD — Q СО
я со Tt< см со CM Tt< Tt< CM CM CM Tf-
A А
К
и
о CJ
co о СК
с О Ю СО co CD co о о co я о
ю ~ оо со ю 00 CO Tt< co co co
со со CD Ю Tt< О b~ Tf- C7> CD О 00
СО см см — ~ CM — — о
га
га
я
ф
то •е
о £ СО <D Н U- Z □ J3 Ы □ О 0, >E X < < со
В скобках указана температура в °C.
480
45. Материалы и оборудование 481
термической устойчивостью (до 250 °C). В лаборатории применяют стаканы, чашки, бутылки, трубки, пластины и фольгу из политетрафторэтилена. Благодаря хорошим фрикционным свойствам он пригоден для изготовления уплотнений и подшипников, например в мешалках. Тефлон не разрушается при действии плавиковой кислоты, царской водки или дымящейся азотной кислоты на холоду и при кипячении, абсолютно устойчив к действию хлора; концентрированная серная кислота действует на тефлон только при температурах выше 300 °C. Тефлон быстро разрушается при действии фтора при повышенной температуре (выше 150°C), а также щелочных металлов и Na2O2 при нагревании. Изделия из политрифторхлорэтилена (экафлувина, хос-тафлона), обладающего почти такой же химической стойкостью, как тефлон, сохраняют форму при нагревании только примерно до 190 °C и применяются для тех же целей, что и тефлоновые изделия.
Для соединения стеклянных трубок и для транспорта газов и жидкостей используют резиновые и пластмассовые шланги. Многочисленные органические растворители, галогены, галогено-водороды и диоксид серы, а также некоторые кислоты, например азотная и серная, разрушают резиновые шланги. Преимуществом пластмассовых шлангов по сравнению с резиновыми является их прозрачность и химическая устойчивость. Но, с другой стороны, органические растворители вымывают из пластмассовых шлангов пластификаторы, при этом шланги становятся твердыми и хрупкими. Перед натягиванием на соединяемые трубки пластмассовые шланги размягчают, погружая концы шланга в горячую воду.
45.6. Лабораторное оборудование
Обычное лабораторное оборудование — штативы и принадлежности к ним (муфты, лапки), штативы для пробирок, держатели для пипеток и бюреток, асбестовые сетки с проволочным каркасом, щипцы, пинцеты, обжимающие устройства для пробок, зажимы для шлангов, лабораторные ложки и шпатели, пробки из корки или резины, шланги и т. Д. — выпускается различного вида в зависимости от назначения и описано в специальной литературе и каталогах. Специальное лабораторное оборудование описано в разд. 46.1.1 и гл. 38 (весы), 47. Детали из чистого железа целесообразно покрывать алюминиевой бронзой или печным лаком. Резьба муфт и зажимов всегда должна быть смазана. При закреплении стеклянных приборов в лапках штатива между металлической лапкой и прибором помещают тщательно подогнанную резиновую или корковую прокладку.
45.7. Вспомогательные средства для лабораторных работ
Для герметичного соединения частей установок из различных материалов и уплотнения шлифов в лабораторных установках и для работы в вакууме применяют различные замазки. Замазки должны иметь низкую упругость паров и не быть хрупкими.
31—1880
482 E.I. Оборудование лабораторий и лабораторная техника
Промышленностью выпускаются термопластичные замазки, размягчающиеся при высоких температурах — пицеин (несколько сортов) и белый сургуч. Пластичный при 50—140 °C пицеин представляет собой каучуковую замазку. Для работ в вакууме пицеин применим лишь до 60 °C из-за высокой упругости паров (13,3-10-3 Па при 20°C). Пицеин растворим в бензоле, толуоле, бензине и тетрахлориде углерода. Белый сургуч, упругость паров которого составляет 133,3-10-3 Па при 20 °C, применим до температуры — 50 °C; при 70 °C он размягчается, при 100 °C становится жидким. Белый сургуч растворим в спирте.
Из необратимо затвердевающих замазок промышленность выпускает только полимерные, которые обычно можно приготовить самим. Например, замазку из жидкого стекла готовят, смешивая вязкое жидкое стекло с тонко-измельченным тальком, полевым шпатом или асбестом до получения густой кашицеобразной массы. Наносят замазку в места уплотнения и оставляют для затвердевания на несколько часов; затем посуду сушат в сушильном шкафу. Замазка выдерживает действие достаточно высоких температур и обладает химической стойкостью.
При работе в глубоком вакууме применяют «жяровые смазки» с низкой упругостью паров, из которых можно назвать имеющиеся в продаже смазку Рамзая и апиезоновые смазки. Для смазывания кранов, шлифов и т. д. в зависимости от химических требований применяют силиконовые смазки, вазелин и хлоростойкие смазки для кранов.
45.8. Очистка оборудования
Порядок и чистота на рабочем месте, а также чистота оборудования являются основным условием всех работ в химической лаборатории. Стеклянную и фарфоровую посуду лучше всего очищать ершиком и использовать для очистки поверхностно-активные моющие вещества. Не рекомендуется применять песок для механической очистки стеклянного оборудования, так как при этом легко повредить поверхность стекла.
Особенно эффективное средство очистки стекла и фарфора — смесь бихромат+серная кислота, так называемая хромовая смесь, которую готовят растворением 20—30 г тонкоизмельчен-ного Na2Cr2O7 или КгСг2О7 в 1 дм3 конц. H2SO4. Очищающая способность этой очень агрессивной жидкости красно-коричневого цвета основана преимущественно на ее окислительном действии. Безводная хромовая смесь может реагировать с органическими веществами даже со взрывом, о чем нужно помнить при обработке сосудов с неизвестным содержимым. При разбавлении хромовая смесь теряет свои свойства, поэтому перед ее употреблением надо дать стечь каплям воды с очищаемой посуды, предварительно вымытой водой. Толстостенные сосуды с хромовой смесью лучше всего держать закрытыми. Если моющая смесь окрашена в зеленый цвет, значит, хром восстановлен [Cr(VI)->Cr (III)]; такая смесь уже непригодна для работы. Для очистки шлифов от находящейся на них смазки вместо хромовой смеси лучше использовать органические растворители, такие, как бензин, бензол или тетрахлорид углерода.
46. Нагревание и охлаждение.
483
В заключение всех операций по очистке посуды ее тщательно промывают водопроводной, а затем дистиллированной водой. Посуду высушивают на сушильной доске, нагреванием в пламени горелки или в сушильном шкафу.
Градуированные сосуды, например бюретки, нельзя нагревать; их сушат, пропуская через них чистый, не содержащий пыли воздух. Не следует промывать сосуды спиртом и эфиром, так как эти растворители обычно недостаточно чисты и при их испарении снова может выделиться смазка. При работе с микроколичествами веществ лучше всего на рабочем месте приготовить большой сосуд с водой для ополаскивания микрооборудования тотчас же после употребления.
Методы очистки посуды из других материалов, применяемой в лаборатории, нужно выбирать в соответствии со свойствами конкретных материалов. Платиновый тигель, например, можно очистить, нагревая его с чистой конц. НС1 или HNO3 (но не с их смесью), если недостаточно кипячения с водой. Еще более эффективное действие оказывает расплавленный K2S2O7 или сплав трех частей карбоната натрия с одной частью тетрабората натрия. Часто положительный эффект дает очистка платиновых сосудов чистым морским песком.
46. НАГРЕВАНИЕ И ОХЛАЖДЕНИЕ.
ИЗМЕРЕНИЕ И РЕГУЛИРОВАНИЕ ТЕМПЕРАТУРЫ
46.1. Источники нагревания и охлаждения
46.1.1. Нагревательные приборы
В качестве источников тепла в химических лабораториях используют пламя светильного газа и электрические спиральные нагреватели, обеспечивающие точное регулирование температуры. Сжигание смеси газ + воздух в основном осуществляют в горелке Бунзена, подачу воздуха к которой можно регулировать. Газ можно смешать максимально с 60% воздуха. Смесь горит пламенем, в котором можно различить внутренний конус, окрашенный в голу’бо-ватый цвет, и внешнее несветящееся пламя. Внутренний конус — восстановительная зона (температура 350—550 °C) состоит из смеси несгоревшего газа и воздуха. Газ сгорает полностью только на краю внутреннего восстановительного конуса (около 1500 °C) и во внешнем несветящемся пламени — за счет поступающего снаружи воздуха.
В горелке Теклю, в которую можно подавать больше воздуха, чем в горелку Бунзена, количество подаваемого воздуха регулируют вращением дискообразного винта. Температура при этом достигает 1700 °C. Пламя горелок Бунзена и Теклю можно расширить, надевая на устье горелки щелевые насадки, что рекомендуют делать для равномерного нагревания лодочек с веществом или при сгибании стеклянных трубок.
К стеклодувным горелкам через специальную трубку подводят сжатый воздух или кислород, что позволяет повысить температуру пламени до 2000 °C. Необходимо следить за тем, чтобы перед подачей воздуха горелка была зажжена.
31*
484
E.I. Оборудование лабораторий и лабораторная техника
В бесцветном пламени горелки Бунзена можно нагреть маленький тигель высотой примерно 40 мм приблизительно до 700 °C, на горелке Теклю до 800 °C и в пламени стеклодувной горелки — до 900—950 °C.
Для равномерного распределения тепла при нагревании колб, чашек и т. д. применяют асбестовые сетки, воронки Бабо, воздушные, водяные, масляные, песчаные или металлические бани, нагреваемые газом ’или с электронагревом; также следует упомянуть металлические блоки, чаще всего из алюминия, с гнездом для тигля и отверстием для термометра.
Электронагрев по сравнению с газовым обладает рядом преимуществ, к которым относятся возможность лучшего- регулирования и поддержания постоянной температуры, чистота рабочего места в условиях эксперимента, возможность свободного выбора размеров обогревателя. В качестве электрических источников тепла со спиральными нагревательными элементами на практике наиболее широко применяют сушильные шкафы, плитки и печи (так называемые муфельные печи), печи с карборундовыми (силитовыми) стержнями и инфракрасные лампы. Для изготовления нагревательных элементов применяют хромникелевую ленту для температур до 1000 °C или ленту из кантала (сплава Fe, Сг, А1 и Со). Силитовые печи конструируют с нагревательными стержнями из спекшегося карбида кремния и обычно поставляют в комплекте с устройствами для включения и регулирования температуры. Они могут давать температуру до 1300 °C.
46.1.2. Охлаждение и охлаждающие средства
Для охлаждения до 0 °C смешивают лед (лучше в виде кусочков размером с горошину) с водой до образования кашицеобразной массы. Для получения более низких температур применяют смесь 1 части льда с 0,3 частями NaCl (до —21 °C) или с 1,43 частями СаСЬ-бНгО (до —55°C).
При использовании в качестве охлаждающего средства сухого льда, т. е. твердого диоксида углерода, большие блоки сухого льда измельчают в полотняном мешке и измельченный лед вносят деревянной ложкой маленькими порциями при помешивании в сухой ацетон, находящийся в сосуде Дьюара (стеклянный сосуд с двойными стенками, пространство между которыми вакуумировано, и с зеркальной внутренней поверхностью). При атмосферном давлении в сосуде Дьюара можно достичь температуры —78 °C.
46.2. Измерение температуры
При измерении температуры ртутными термометрами необходимо добиться теплового равновесия между термометром и веществом, температуру которого измеряют. Теплоемкость шарика термометра должна быть как можно меньше в отличие от теплоемкости вещества.
Для измерения небольших перепадов температуры, например понижения температуры замерзания или повышения температуры кипения, применяют термометр Бекмана, с помощью которого можно измерить температуру в интервале 5 °C. Его шкала имеет деления 1/50 или 1/100 °C, а тысячную долю градуса можно еще достаточно надежно определить по шкале с помощью лупы. Для измерения температуры в различных диапазонах часть ртути отделена в петлеобразное расширение, которое снабжено вспомогательной шкалой. Температуру, измеренную термометром Бекмана, нужно определять по сравнению с измеренной другим термометром.
Капилляры термометров для измерения низких температур заполнены окрашенной жидкостью, такой, как изопентан (диапазон измерений от +35 до —195 °C), н-пентан (до —130 °C), амиловый спирт (до —110 °C) или толуол (до —90 °C).
Для измерения температур в интервале 300—1600 °C применяют -металлические термопары (медь — константан до 600 °C, железо — константан до
47. Методы работы в лаборатории 485
900 °C, хромникель — никель до 1100 °C и платинародий—платина до 1600°C). В названии термопар на первом месте называют положительный элемент. Толщина проволочных термоэлементов составляет 0,3—0,5 мм. Для защиты от химических или механических повреждений термоэлементы помещают в защитные трубки из стекла, кварца или фарфора. Между местом контакта термоэлементов и их концами возникает термоэлектродвижущая сила, которая только в том случае не искажает измеряемых значений температуры, если используют так называемые компенсирующие провода или если место контакта находится в условиях постоянной температуры (при 20 °C или при 0°C для технических измерений). Компенсирующие провода изготовляют из сплавов, термоэлектродвижущая сила которых совпадает с термо-э.д.с. соответствующего термоэлемента.
Дополнительное преимущество термопар — возможность передачи измеряемого значения температуры на регулирующий прибор, находящийся на расстоянии от нагреваемого объекта.
46.3. Регулирование температуры
Для регулирования степени нагрева электроприборов в области температур примерно до 500 °C применяют контактные термометры. В этих термометрах одна контактная проволочка впаяна в сосуд со ртутью, а другая — вольфрамовая— при помощи вращающегося магнита может быть установлена в положение, обеспечивающее необходимую температуру. По достижении установленной температуры мениск столба ртути касается проволоки, срабатывает реле, обычно приводящееся в действие ртутным опрокидным выключателем, и электронагреватель отключается.
Для регулирования температур ниже 400 °C применяют биметаллические регуляторы в форме полос или спиралей. При более высоких температурах (до 1200 °C) можно использовать стержневые регуляторы, обеспечивающие точность регулирования ±5°C. Для регулирования температуры с большей точностью, в том числе и температур выше 1200 °C, применяют термопары в комплекте с регуляторами с падающей дужкой.
47. МЕТОДЫ РАБОТЫ В ЛАБОРАТОРИИ
Описанные в этой части методы работы следует применять для решения конкретных препаративных задач, описанных в части E.IL Для лучшего овладения техникой работ в лаборатории рекомендуем совместно использовать материал частей E.I и E.IL
47.1. Измельчение
Для переведения твердых веществ в порошкообразное состояние, т. е. для увеличения их поверхности, применяют ступки с пестиками, имеющими шишкообразное утолщение. Чаще всего в лабораториях пользуются фарфоровыми ступками. Твердые материалы после грубого измельчения обычно растирают в агатовой ступке или в ступке из спекшегося корунда. Для измельчения твердых веществ применяют также стальную ступку (рис. Е.7), называемую «алмазной». Она представляет собой толстостенный стальной цилиндр с навинчивающейся нижней крышкой, в кото
486
E.I. Оборудование лабораторий и лабораторная техника
рой имеется углубление для вещества. Пробу измельчают, ударяя молотком по стальному стержню, тщательно пригнанному к стенкам полого цилиндра.
Вместо трудоемкого растирания веществ вручную можно использовать мельницы, в которых пестик, снабженный передвижным плоским грузом и приводимый в действие электромотором, давит на вращающуюся' ступу. В центробежных мельницах измельчение осуществляется шарами, которые под действием
центробежной силы ударяются о стенки барабана, вращающегося вокруг оси и защищенного от попадания пыли. Для измельчения больших количеств веществ, используемых в препаративных работах, применяют шаровые мельницы. Шары различного
размера должны заполнять
Рис. Е.7. Разрез стальной ступки. примерно 55% объема мельницы и быть покрыты измельчаемым материалом. Фарфоровый сосуд, заполненный измельчаемым веществом и шарами, закрывают пришлифованной крышкой и приводят во вращение с помощью электромотора.
После измельчения необходимо просеиванием отделить неиз-
мельченные крупные частицы.
47.2. Растворение и перемешивание
Как было показано в разд. 33.3, при растворении вещества при определенной температуре образуется насыщенный раствор, в котором существует равновесие между нерастворившимся веществом, так называемым донным осадком (если растворяют твердое вещество в жидкости), и ионами растворившегося вещества. Скорость растворения определяется преимущественно площадью поверхности вещества и перепадом концентраций. Площадь поверхности увеличивают путем измельчения, а перепад концентраций— перемешиванием или встряхиванием раствора: он возрастает при перемещении концентрированного раствора к границе раздела твердое вещество/жидкость. Поскольку скорость реакции увеличивается при повышении температуры, растворение ускоряют нагреванием.
Обычно растворение проводят в стаканах, колбах Эрленмей-ера или бутылях. Во многих случаях для перемешивания применяют простейшие мешалки — оплавленные и закругленные на концах стеклянные палочки; для длительного перемешивания используют мешалки с электромотором (лопастные, пропеллерные, центрифужные), обеспечивающие эффективное перемеши-
47. Методы работы в лаборатории
487
ванне. Мешалка должна быть правильно отцентрована, что способствует равномерному вращению. Для этого мешалку пропускают через просверленное в пробке отверстие или помещают ее в толстостенную стеклянную трубку и затем закрепляют в лапках штатива. Пробку или направляющую трубку необходимо смазать глицерином. При перемешивании легко испаряющихся веществ в закрытом сосуде мешалку снабжают ртутным затвором; можно использовать мешалки типа KPG, применяемые также при работе в вакууме, так как направляющая трубка и ось таких мешалок хорошо пришлифованы. Для перемешивания не очень больших объемов жидкостей удобны магнитные мешалки, представляющие собой маленькие магнитные стержни, запаянные в стекло или в пластмасу. Мешалка приводится во вращение действием сильного постоянного магнита, расположенного под сосудом.
При необходимости длительного перемешивания жидкости, например при растворении, смешивании, экстракции, используют электрические устройства для встряхивания, в которых закрепляют сосуды с жидкостью, плотно закрытые пришлифованными пробками. Жидкость перемешивают, вращая или раскачивая сосуд.
Перечисленные методы применяют для смешивания любых фаз, начиная от приготовления растворов и получения коллоидных систем — эмульсий и суспензий — и кончая приготовлением взвесей и грубодисперсных смесей.
Приготовление растворов — наиболее распространенная в лабораториях операция. Посуда, применяемая для измерения объемов растворов, а также расчеты при приготовлении растворов указаны в разд. 38.2.3.2.
47.3. Разделение веществ
47.3.1. Выпаривание
Выпариванием растворителя можно выделить из раствора нелетучее растворенное вещество. Сравнительно небольшие количества растворителя можно выпарить на часовом стекле или в чашке; при нагревании этот процесс ускоряется. Негорючие жидкости выпаривают в чашках (по возможности под тягой), нагревая их на асбестовой сетке пламенем газовой горелки. Для удаления небольших количеств легко воспламеняющихся растворителей по соображениям техники безопасности пользуются инфракрасными лампами. Большие количества горючих или ценных растворителей после отгонки собирают. Для получения хорошо кристаллизующегося продукта (см. разд. 47.3.2) нагреванием удаляют только основное количество растворителя, т. е.
488 E.I. Оборудование лабораторий и лабораторная техника
раствор сначала упаривают, и затем (обычно после фильтрования) оставляют стоять при комнатной температуре без перемешивания для медленной кристаллизации.
47.3.2. Кристаллизация
При проведении кристаллизации из растворов при медленном снижении температуры выделяются большие кристаллы. Однако такие кристаллы могут содержать растворитель в виде включений. Поэтому лучше всего проводить кристаллизацию так, чтобы образовались одинаковые кристаллы средних размеров. Часто, несмотря на пересыщение раствора, кристаллизации вещества не происходит. В таких случаях кристаллизацию можно вызвать только введением затравки или потиранием стенок сосуда стеклянной палочкой. Для получения индивидуальных больших кристаллов необходимо в качестве зародышей вводить кристаллики соответствующего соединения. Если их подвесить в растворе на нити из перлона, то они быстро растут, образуя хорошо сформированные кристаллы (например, октаэдры в случае квасцов). По окончании процесса кристаллы небольшим количеством растворителя смывают на воронку для удаления с их поверхности маточного раствора, а затем высушивают.
Для проведения простого эксперимента по выращиванию кристаллов, в котором успешно можно использовать квасцы, нитрат калия, дихромат калия и сульфат меди, готовят насыщенный прн 80 °C раствор вещества, фильтруют и помещают его для медленного охлаждения в ячейку с термоизоляцией (изоляционным материалом может быть древесная шерсть, опилки, кизельгур) или в термос.
Многократная перекристаллизация, т. е. многократное повторение процессов растворения и кристаллизации при использовании каждый раз минимального количества свежей порции растворителя, приводит к увеличению чистоты кристаллизата (ср. препараты, описанные в части Е.П). Условием очистки является более высокая растворимость удаляемых примесей по сравнению с растворимостью основного вещества, в связи с чем примеси остаются в маточном растворе. Схема на рис. Е.8а поясняет процесс очистки. Этот вид перекристаллизации, естественно, сопровождается значительными потерями очищаемого вещества, так как маточный раствор обычно отбрасывают.
Фракционная кристаллизация (рис. Е.86) значительно более экономична, так как в этом случае для растворения загрязненного кристаллизата применяют уже использованный растворитель. Начало процесса, а также операции растворения, указанные в левой части схемы (получение Хь Х2, Х4), соответствуют простой перекристаллизации, проводимой каждый раз с испарением маточных растворов (Li, L3, Lg). Из маточного раствора
47. Методы работы в лаборатории
489
.Lm
Lm Lm
Повышение чистоты Увеличение содержания вещества А примесей В в маточном
растворе
Рис. Е.8а. Схема перекристал- Рис. Е.86. Схема фракционной кристаллиза-лизации. цик.
Lm — растворитель; АВ — загрязненное вещество; А — менее растворимое вещество; В — более растворимые примеси;, Хь ...» Хз вещество А с уменьшающимся содержанием примесей В; Li, ...» L3 — маточный раствор, насыщенный примесями В.
Li наряду с сильно загрязненным маточным раствором L3 выделяется кристаллизат Х3. Его растворяют в L3 (маточный раствор кристаллизата Xi), при охлаждении выделяется более чистый кристаллизат Х3. Эти операции можно продолжать до получения вещества необходимой чистоты. Фракционная кристаллизация— наиболее старый метод, долгое время остававшийся почти единственным методом разделения редкоземельных металлов.
47.3.3. Сорбция и ионный обмен
Сорбционные процессы традиционно делятся на адсорбцию (см. гл. 14.1), т. е. захват газов или жидкостей поверхностью твердых веществ, и абсорбцию, т. е. поглощение молекул веществ всем объемом жидких или твердых веществ с образованием раствора или соединения.
Сорбцию применяют в основном для очистки и разделения веществ. Специальная аппаратура, используемая для этих целей, была описана в соответствующих разделах. Важное значение имеют такие адсорбенты, как активированный уголь и силикагель (см. табл. Е.З). Большую роль играют сорбционные процессы при осаждении и промывании осадков, имеющих большую поверхность, а также при загрязнении этих осадков примесями (см. разд. 38.3.4).
Адсорбционную хроматографию можно применять и в препаративных и аналитических целях (разд. 38.3.6). Из большого числа адсорбентов для аналитических целей наиболее важны специальные сорта бумаги, а для препаративных работ — оксид алюминия со стандартизованными характеристиками. Каждая установка для хроматографических разделений приспособлена
490 E.I. Оборудование лабораторий и лабораторная техника
для конкретных целей эксперимента, но независимо от этого всегда состоит из трех основных частей (рис. Е.9): резервуара с хроматографируемым раствором, колонки с адсорбентом и сосуда для сбора элюата. Эта же установка может быть использована и в методе ионообменной хроматографии, основанном на использовании специфичного обмена ионами между ионообмен-ником и раствором.
47.3.4. Декантация, центрифугирование, фильтрование
Декантацию для разделения твердых веществ и жидкостей можно применять, если твердые вещества, характеризующиеся высокой плотностью, легко осаждаются из раствора. При этом раствор, находящийся над осадком, становится прозрачным.
Рис. Е.9. Схема установки для ионообменной хроматографии.
/ — резервуар для раствора; 2—адсорбент или ионообменная смола; 3 —насадка для колонки; -/ — пробка из ваты; 5 — склянка для сбора элюата.
Жидкость сливают (деканти-руют), наклоняя сосуд. При определенном навыке декантацию можно проводить из обычных стаканов или установленных наклонно колб Эр-ленмейера. Простейший способ декантирования без потерь, важного для целей количественного анализа, заключается в том, что стеклянную! палочку прижимают к краю стакана и по ней сливают жидкость.
При работе с медленно или неполностью осаждающимися осадками для ускорения седиментации используют центробежную силу. Применение центрифуг особенно целесообразно для разделения небольших количеств твердых частиц и жидкостей. Для работы со смесями используют центрифуги с тарированными путем взвешивания пробирками. В лаборатории в основном применяют ручную центрифугу, снабженную планкой с двумя гильзами для пробирок. В электроцентрифугах центрифужные пробирки при вращении принимают горизонталь-
47. Методы работы в лаборатории 491
ное положение относительно оси ротора (радиальная центрифуга) или находятся в гильзах, жестко закрепленных под постоянным углом к.оси ротора (угловые центрифуги). Осадок, получаемый при центрифугировании, отделяют от основной массы находящейся над ним жидкости декантацией или отсасыванием пипеткой. Полное разделение твердой и жидкой фаз осуществляют фильтрованием. Фильтрующее приспособление выбирают в соответствии с количеством осадка и объемом жидкости, а также в зависимости от конкретной задачи. Например, при необходимости отделения небольших количеств твердого вещества от большого объема жидкости или при проведении количественных определений берут фильтр меньшего размера, чтобы сконцентрировать небольшое количество осадка в малом объеме. Если необходимо удалить из раствора небольшие примеси твердых веществ для дальнейшей работы с жидкостью, то фильтр выбирают в соответствии с объемом жидкости; для быстрого отфильтровывания большого объема жидкости применяют фильтр с большой поверхностью, например складчатый.
Бумажный фильтр обычно помещают в стеклянную воронку. Это, как правило, круги различной величины из бумаги с различными размерами пор (2—6 нм). Для проведения фильтрования круглые фильтры складывают вчетверо в конус (рис. Е.10); край фильтра должен находиться ниже края воронки. Перед фильтрованием бумажный фильтр увлажняют и прижимают к стенкам воронки.
Скорость фильтрования можно увеличить, используя «висящий» фильтр. Для этой цели при втором складывании бумажный фильтр перегибают не точно пополам, а делают одну часть больше на 1—3 мм. Затем фильтр расправляют таким образом, чтобы конусность фильтра была больше конусности воронки. Тогда только верхний край бумажного фильтра будет полностью соприкасаться со стеклом воронки (рис. Е.11), и образуется большая свободная фильтрующая поверхность. Тот же принцип действия использован в аналитических воронках из йенского стекла для быстрого фильтрования. Внутренняя поверхность конуса этих воронок в нижней части остается свободной, так что бумажный фильтр прочно прилегает к верхнему краю воронки, в то время как большая часть фильтра находится в свободно подвешенном состоянии.
Более быстро, чем через гладкие бумажные фильтры, происходит фильтрование через складчатые фильтры, которые только ребрами касаются стенок воронки и при равном диаметре имеют большую фильтрующую поверхность, чем обычные фильтры. Складчатые фильтры применяют в основном, если для последующей работы нужен раствор, свободный от осадка или от мути. Если же для работы нужен осадок, то используют гладкие фильтры, с которых осадок легче удаляется.
♦92______E.I. Оборудование лабораторий и лабораторная техника
Р® Е.10. Способ складывания гладкого бумажного фильтра.
Р’с- Е.11. Положение «висящего» фильтра в воронке.
Р0С- Е.12. Склянка для отсасывания воронкой Бюхнера.
При проведении препаративных работ для отфильтровывания кристаллических, но не мелкодисперсных осадков применяют воронки Бюхнера (рис. Е.12). Эти фарфоровые фильтры с помощью резиновой пробки или резинового конуса вставляют в склянку для отсасывания, которую можно вакуумировать, применяя водоструйный насос. Фильтрование ускоряется благодаря разрежению, образующемуся под фильтром. Для проведения фильтрования на фарфоровую пластину с отверстиями кладут бумажный фильтр, который во влажном состоянии при отсасывании плотно прилегает к пластине. При работе с сильно кислыми и щелочными жидкостями, разъедающими бумажные фильтры, рекомендуют применять пластинки из пористого стекла. Они выпускаются четырех типов, в зависимости от размеров пор: G1 (крупнопористые), G4 (мелкопористые), остальные с промежуточными размерами пор. Пластинки следует выбирать в зависимости от размера частиц осадка.
По принципу работы воронки Бюхнера действует также склянка для отсасывания с фильтрующей пластинкой Витта (рис. Е.13). Фильтрование осадков, чувствительных к действию воздуха или влаги, проводят на установке, позволяющей проводить фильтрование без доступа воздуха (рис. Е.14). Для фильтрования горячих растворов, которые нельзя охлаж-
47. Методы работы в лаборатории
493
Рис. Е.14. Схема иутч-фильтра с пластинкой из пористого стекла для фильтрования без доступа воздуха.
Рис. Е.13. Схема установки для отсасывания с пластинкой Витта (фильтрующим тиглем).
дать также и в процессе фильтрования, применяют воронки или фильтры для горячего фильтрования, имеющие двойные стенки. Через рубашку воронки или фильтра пропускают горячую воду или водяной пар.
В полумикрометоде часто применяют фильтрование под давлением по Барберу. Жидкость, отделяемую от твердых частиц, при этом продавливают через фильтровальную трубочку. Фильтрующим материалом служит химически чистая вата, а для фильтрования агрессивных жидкостей используют асбестовое волокно, плотно набитое в стеклянную трубочку. Для ручной подачи воздуха можно самим изготовить воздушный насос (см. рис. Д.16).
47.3.5. Перегонка
Для разделения веществ все более широко применяют перегонку. Этот метод основан на переводе вещества в парообразное состояние в колбе для перегонки и конденсации паров в холодильнике. Дистиллат собирают в приемнике (рис. Е.15). Сосудами для перегонки служат колбы с горлами различной длины и боковой трубкой, которая должна быть расположена высоко по отношению к колбе при перегонке низкокипящих веществ и низко, т. е. ближе к колбе, в случае высококипящих. Термометр, измеряющий температуру кипения, размещают таким образом,
494
Е.1. Оборудование лабораторий и лабораторная техника
чтобы шарик ртути находился чуть ниже боковой трубки и полностью омывался парами перегоняемого вещества. Для предотвращения задержки кипения перед нагреванием в колбу помещают стеклянные бусины, кусочки глины или стеклянные капилляры.
При перегонке жидкостей с температурой кипения до 120 °C охлаждение ведут проточной водой; при перегонке веществ с более высокой температурой кипения рекомендуется прекратить
Рис. Е.15. Установка для перегонки.
/ — колба для перегонки; 2 —термометр; 3 — холодильник Либнха; 4 — форштос; 5 —приемник дистиллата; 6 — подача охлаждающей воды.
Рис. Е.16. Виды насадок для фракционной перегонки.
подачу воды в рубашку холодильника, так как иначе холодильник легко может лопнуть. При температурах кипения выше 150 °C можно применять воздушный холодильник, т. е. длинную охлаждающую трубку без рубашки.
Разделение компонентов смесей с близкими температурами кипения проводят путем фракционной перегонки, т. е. многократным повторением процесса перегонки. В случае низкокипя-щих веществ можно применять фракционирующие насадки (рис. Е.16), которые увеличивают расстояние, проходимое парами вещества до холодильника. При этом пары высококипящих компонентов смеси конденсируются в насадке и в виде конденсата стекают обратно в колбу, в то время как низкокипящие компоненты отгоняются. Насадки для фракционной перегонки действуют по принципу ректификации и дефлегмации. В случае ректификации речь идет о постоянном обмене компонентов и теплообмене между газовой и жидкой фазами. Дефлегмация заключается в разделении веществ частичной конденсацией их на охлаждаемых поверхностях.
Рис. Е.17. Схема установки с колонкой Видмера для фракционной перегонки.
Рис. Е.18. Схема установки для ректификации.
/ — круглодонная колба с капилляром для предотвращения вскипания жидкости; 2 — ректификационная колонна; 3 — головка колонны, в которой происходит разделение жидкости на конденсат (флегму) и дистиллат; 4 — термометр; 5 — приемник (на рис. изображена насадка Аншютца — Тиле для смены приемников фракций без отключения вакуума); 6 — вакуум-шланг.
Рис. ЕЛ9. Возгонка в стеклянном стакане с осаждением конденсата на стенках охлаждаемой круглодонной колбы. / — вещество; 2 — конденсат; <? —охлаждающая вода.
Рис. Е.20. Возгонка между двумя часовыми стеклами с плоскими пришлифованными краями.
496 E.I. Оборудование лабораторий и лабораторная техника
Разделение в колонке Видмера (рис. Е.17) основано на двух принципах: увеличении пути, проходимого парами, и предварительном нагревании или соответственно ректификации по противоточному способу (рис. Е.18).
Перегонку веществ, разлагающихся при кипении при атмосферном давлении, проводят при пониженном давлении (см. разд. 47.5.1).
47.3.6. Возгонка
Возгонку (сублимацию) также можно применять для разделения компонентов смесей, если в смеси одновременно присутствуют и сублимируемые, и нелетучие компоненты. Возгонка протекает вследствие постоянного нарушения равновесия в системе твердое вещество — пар, причем в охлаждаемой части установки
Рис. Е.21. Схема установки для горизонтальной возгонки в токе инертного газа.
сублимируемое вещество снова переходит из парообразного в твердое состояние.
Скорость возгонки зависит от температуры, давления, а также от расстояния между нагреваемой и охлаждаемой поверхностями. Для увеличения скорости возгонку можно проводить в токе инертного газа, уносящего пары вещества к охлаждаемой поверхности. Для проведения возгонки предложены разнообразные устройства (рис. Е.19—Е.21; о возгонке в вакууме см. разд. 47.5.1).
Можно проводить возгонку соединений аммония, иода (см. разд. 35.5.1) и хлорида ртути(II) (сублимата), причем простейшее устройство для возгонки состоит из двух часовых стекол (рис. Е.20). Кристаллы сублимата часто имеют характерную форму.
47.3.7. Экстракция и распределение
Бинарную смесь твердых веществ можно разделить, извлекая один из компонентов избирательно действующим растворителем. С этой целью смесь веществ многократно обрабатывают растворителем при интенсивном встряхивании и затем раствор отделяют от остатка декантацией или фильтрованием. Для непрерывного проведения процесса созданы экстракторы, из которых наиболее известен экстрактор Сокслета (рис. Е.22). Вещество в тонкоизмельченном, но не порошкообразном состоянии поме-
47. Методы работы в лаборатории
497
щают в патрон из фильтровальной бумаги, или в сосуд из спекшегося стекла (фильтрующая свеча), или в фильтрующий тигель, вносят в экстракционный сосуд и подсоединяют к нему обратный холодильник. Пары растворителя, нагретого до кипения в круглодонной колбе, поднимаются вверх по боковой труб-
ке экстракционного сосуда, конденсируются в обратном холодильнике, и растворитель по каплям попадает на патрон. Экстракт накапливается в экстракционном сосуде, поднимаясь до сгиба сифонной трубки, и сбрасывается в колбу с растворителем, где происходит постепенное накопление экстрагируемого компонента.
Если имеется раствор смеси веществ с различной растворимостью в органических растворителях, то разделение можно осуществить, добавляя к раствору несмешивающийся с ним растворитель, в котором хорошо растворяется одно из веществ. Наиболее простым способом распределения веществ между несмешивающи-мися жидкостями является встряхивание в делительной воронке (рис. Е.23, а) или в делительной трубочке (рис. Е23, б). Встряхивание повторяют, добавляя вместо исходного раствора новые порции раствори-
Рис. Е.22. Схема экстракционного аппарата Сокслета.
1 — колба с растворителем; 2 — экстрактор с патроном; 3 — сифонная трубка; 4 — боковая трубка; 5—обратный холодильник.
теля; можно также дать возможность растворителю равномерно проходить через раствор в соответствующей установке (рис. Е.24). Этот способ называют перфорацией. Многократное последовательное повторение операций простого разделения приводит к фракционному распределению (см. разд. 38.3.5.2), благодаря которому можно в несколько раз увеличить эффективность разделения.
47.3.8. Высушивание
Под высушиванием обычно понимают удаление воды. Однако в лабораторной практике высушивание в более широком смысле означает удаление остатков растворителей; разумеется, в неор-
32—1880
498
E.I. Оборудование лабораторий и лабораторная техника
Рис. Е.23. Сосуды для проведения экстракции — распределения веществ между двумя растворителями.
а — делительная воронка для больших количеств веществ; б — делительная трубочка для малых количеств.
Рис. Е.24. Схема перфоратора.
1 — колба для испарения подвижной фазы и для сбора экстрагируемого компонента; 2 — неподвижная фаза, содержащая экстрагируемый компонент; 3—подвижная фаза, стекающая обратно в колбу для испарения при насыщении экстрагируемым компонентом; 4 — воронкообразная стеклянная трубка (а) нлн насадка с пластинкой из пористого стекла (б) для диспергирования подвижной фазы в неподвижную; 5 — обратный холодильник.
ганической химии наиболее распространенным растворителем является вода.
Твердые негигроскопичные вещества, устойчивые на воздухе, сушат самым простым способом на листах фильтровальной бумаги или на глиняных тарелках при комнатной температуре. При нагревании (например, в сушильном шкафу) или в вакууме этот процесс можно ускорить. Особенно быстро и эффективно вещества высушивают с помощью осушителей, помещаемых в эксикаторы или в пистолеты для сушки, при атмосферном давлении или в вакууме. В пистолетах для сушки (рис. Е.25) можно сушить небольшие количества вещества в вакууме при одновременном постоянном нагревании. Колбу установки заполняют жидкостью (например, водой, толуолом, ксилолом), температура кипения которой соответствует необходимой температуре сушки. В обратном холодильнике происходит конденсация обогревающей жидкости. Вещество помещают в лодочку и вносят ее во внутренний обогреваемый сосуд с двойными стенками. Осушитель помещают в похожий на колбу сосуд, снабженный
47. Методы работы в лаборатории
499'
краном и закрывающий горло внутреннего сосуда. Твердые
вещества также часто сушат, пропуская над ними поток сухого воздуха или хорошо высушенного инертного газа. О высушивании воздуха и других газов см. разд. 47.4.2.
Жидкости легко можно обезводить, вводя в них подходящие
осушители. Смесь тщательно перемешивают при встряхивании
и после длительного контакта с регоняют. Многие осушители (например, СаС12 или Na2SO4) при нагревании снова выделяют поглощенную ими воду, так что перегонку следует проводить только после их отфильтровывания. Осушители выбирают таким образом, чтобы они не реагировали с осушаемой жидкостью и не растворялись в ней. В табл. Е.З дан обзор свойств наиболее распространенных осушителей.
Наряду с образованием гидратов для обезвоживания используют также явления адсорбции, например, в так называемых молекулярных ситах. В кристаллической решетке молекулярных сит алю-мосиликат-ионы расположены в виде сшитых колец, в результате чего образуются многочисленные трубчатые поры. Воду, находящуюся в этих
осушителем фильтруют или пе-
Рис. Е.25, Схема пистолета для сушки.
1 — колба с обогревающей жидкостью; 2 — сушильная камера с двойными стенками; 3 — колба с осушителем; 4 — отвод к вакууму; 5 —обратный холодильник.
порах, можно удалить нагреванием без изменения структуры алюмосиликатного каркаса. Высушивание газов и жидкостей на молекулярных ситах значительно эффективнее, чем с помощью пентоксида фосфора или перхлората магния. Дополни-
тельным преимуществом молекулярных сит является то, что они не расплываются после поглощения воды. Способность молекулярных сит адсорбировать воду сохраняется до 150°C. При более высоких температурах молекулярные сита могут
регенерироваться.
Высушивание с химическим разложением воды применяют, например, при обезвоживании эфиров, основанном на реакции образования гидроксида натрия при введении прессованного натрия:
H2O + Na ---------------------------► NaOH + ’/aHj,
32*
09 О
СО
X к сЗ И 3
2
3 С?
со со
I о
сч
09 о
сч
аблица Е.З. Свойства наиболее распространенных осушителей
СО
S а
О
еч со о~ о „°О
дО нч еч .Z еч ► «дг сз
Z . .о о и _£?£ ечС^ Z о ад X
« - * о гте . С ) X сг>
аде XU- га
X
X л
х н л X
о о Q
о к О
с=(\о
о о е X
X О
о X о <и
о X
К к 3 s § 3 к 03 X о X «V Г 3 X U <5 о
й 2 О
►а сз ю
га s га а, —
1 1 Органические вещества и газы, содержащие при-| меси органических ве- ществ NH3, HF, НС1, HBr, HaS, Cl2, Br2, эфир, ацетон Юз, ^2» N0, кислоты, ) альдегиды, кетоиы !
20, Нз,1 УГ-, X гие
Z X 3
X о X к( X о о X к( ►а со СЗ < U? z’^5 N20; п сх X X сх. X X о
0) X 0) X &> и X zou К( о о ео Ж га ёг X X «I X X X СЗ X
о X о X X о с аОД Оси о и 0) С? ео * О 5® 5 Z < о X- ч
Количество воды в миллиграммах, содержащейся в 1 дм3 осушенного воздуха при 25 °C.
47. Методы работы в лаборатории
501
47.4. Получение газов и работа с ними
47.4.1. Получение газов
В лабораториях используют магистральный газ и газы, хранящиеся в баллонах, применение которых будет рассмотрено ниже. Кроме того, для получения газов применяют следующие методы: а) нагревание твердых веществ или жидкостей; б) действие жидкостей на твердые или жидкие вещества. В первом методе используют как реакции термического разложения твердых веществ, например
2HgO----► 2Hg + O2f
так и реакции взаимодействия порошкообразных твердых веществ, например
2NH4C1 + Са(ОН)2 -► СаС12 + 2Н2О + 2NH3(
В зависимости от необходимой температуры и образующихся продуктов реакции проводят в ретортах, длинногорлых круглодонных колбах илй в колбах для перегонки, а при высоких температурах — в запаянных с одной стороны трубках из тугоплавкого стекла. Для вытеснения газа из жидкости раствор нагревают в длинногорлой колбе. Колбу укрепляют наклонно для конденсации растворителя в горле или снабжают ее обратным холодильником.
Значительно чаще в лабораториях получают газы по второму методу. Для небольших количеств газов применяют трубку для отсасывания (рис. Е.26), а для больших —длинногорлую колбу с боковой отводной трубкой, снабженную капельной воронкой для реакционной жидкости. Отводная трубка капельной воронки должна быть оттянута на конце. Чтобы при взаимодействии двух жидкостей получить равномерный ток газа, трубку воронки погружают в пробирку, находящуюся на дне сосуда, верхний конец которой находится выше уровня жидкости в сосуде. Можно также добиться равномерного прикапывания жидкости из воронки, соединив Т-образной трубкой верхнее отверстие капельной воронки с газоотводной трубкой реакционной колбы.
Для получения газов путем взаимодействия твердых веществ с жидкостями, протекающего без нагревания, например
СаСО3 + 2НС1 ->- СаС12 + Н2О + CO2f
применяют аппарат Киппа (рис. Е.27). Твердое вещество вносят через тубус 4 в средний шар 2, заполняя его самое большее наполовину. Если вещество мелкораздробленно, в суженную часть прибора кладут резиновое или асбестовое кольцо с отверстиями 5 для предотвращения ссыпания вещества в нижнюю часть прибора 3. В тубус вставляют трубку с краном и в воронку 1
502 E.I. Оборудование лабораторий и лабораторная техника
наливают такое количество жидкости, чтобы она наполовину заполнила нижнюю часть прибора; затем кран закрывают и дают жидкости подняться до половины воронки 1. Если теперь открыть кран, то жидкость поднимается в средний шар, контактирует с твердым веществом, в результате чего происходит выделение газа. Через некоторое время кран закрывают, при этом
Рис. Е.27. Аппарат Киппа для получения газов.
/ — воронка; 2 — средний шар; 3 — нижняя часть прибора: 4— тубус; 5 — кольцо с отверстиями.
Рис. Е.26. Трубка для отсасывания с насаженной капельной воронкой.
жидкость передавливается в нижнюю часть установки и в во-ронку под действием возрастающего в шаре 2 давления газа, так что выделение газа прекращается. Если снова открыть кран, то накопившийся газ выделяется, кислота снова будет подниматься в средний шар до тех пор, пока давление газа не уменьшится.
47.4.2. Очистка и высушивание газов
Газы, как промышленные, так и полученные лабораторным путем, в большей или меньшей степени загрязнены сопутствующими веществами, а газы, выделенные из водных растворов веществ, содержат влагу. Способы очистки наиболее часто приме
47. Методы работы в лаборатории
503
няемых в лаборатории газов, будут подробно описаны в части E.II.
Очистку или высушивание газов жидкими реагентами проводят в склянках для промывания газов, которые одновременно используют как счетчики пузырьков при определении скорости прохождения газа. Прямую или U-образную трубку или колонку, в которую можно поместить большее количество твердого осушителя, заполняют осушителем в виде гранул или зерен, например СаС12, и закрывают с обеих сторон пробками из стекловаты. Порошкообразные осушители, такие, как пентоксид фосфора, смешивают со стеклянными бусинами или глиняными черепками, используемыми в качестве носителей для предотвращения спекания осушителя при взаимодействии с проходящим газом. Наиболее часто применяемые осушители представлены в табл. Е.З.
Часто в лаборатории для осушки газов применяют метод глубокого охлаждения. При охлаждении влажного газа воду выделяют в твердом или жидком состоянии в зависимости от температуры охлаждения. Например, давление паров льда при —50 °C равно всего 4 Па. При вымораживании действием охлаждающей смеси СО2 +ацетон или жидкого воздуха можно получить еще более низкие температуры и тем самым добиться эффективного высушивания.
Для очистки и осушки газов применяют также молекулярные сита, которые хорошо адсорбируют воду, а также молекулы других полярных веществ, таких, как H2S, NH3, SO2, CS2 и СО2.
47.4.3. Хранение газов в баллонах
Газы, нашедшие наиболее широкое применение, поступают в продажу в стальных баллонах в сжиженном (Cl2, NH3, SO2, СО2) или в сжатом (под большим давлением) состоянии (Н2, О2, N2, СО). Ацетилен растворяют под давлением в ацетоне. Поскольку газы в баллонах находятся под высоким давлением, следует соблюдать особые меры предосторожности: баллоны с газами необходимо защищать от нагревания, от встряхивания или падения их страхуют, прикрепив цепью к стене или неподвижному столу. Баллоны лучше хранить в специальных стойках, одновременно пригодных для простой и безопасной их транспортировки. Чтобы не спутать, баллоны с наиболее часто употребляемыми газами окрашивают в определенные цвета (табл. Е.4).
Стальные баллоны снабжены запорными вентилями. Во избежание несчастных случаев вентили баллонов, заполненных горючими газами, снабжены левой резьбой. Чтобы при отборе газа регулировать скорость потока, необходимо подсоединить редуктор; кроме того, между редуктором и установкой нужно
Таблица ЕД. Маркировка стальных баллонов и рабочее давление хранящихся в них газов
Данные для СССР составлены переводчиком.
47. Методы работы в лаборатории 505
подключить предохранительное устройство. В простейшем случае это может быть Т-образная трубка, отверстие которой погружено в ртуть, воду или другую жидкость, используемую в качестве затвора для данного газа.
Запас газа в стальных баллонах, снабженных манометрами, можно установить по давлению, указанному на манометре, если речь идет о сжатом газе и ацетилене; в случае сжиженных газов манометр показывает только давление паров жидкости независимо от степени заполнения баллона.
47.4.4. Измерение количества газа
Скорость газового потока можно оценить, как было указано выше, по числу пузырьков газа в газопромывной склянке. Количество газа измеряют с помощью дифференциального манометра (рис. Е.28) или имеющегося в продаже ротаметра, действие которого основано на перемещении вращающегося поплавка в градуированной трубке при прохождении газового потока.
Рис. Е.28. Схема дифференциального манометра.
1 — капилляр для прохождения газового потока (скорость потока пропорциональна разности давлений на входе и выходе капилляра); 2 — кран для перекрывания капилляра; 3 — штуцер для заполнения U-образной трубки манометрической жидкостью; 4 — U-образные манометрические трубки. В зависимости от газа, количество которого замеряют, U-образную трубку примерно иаполовииу заполняют конц. H2SO4, парафиновым маслом, ртутью или подкрашенной водой. Перепад давлений можно определить по шкале (не изображенной на рисунке) по разности уровней столбов манометрической жидкости в U-об-разной трубке.
Рис. Е.29. Схема водоструйного насоса.
/ — вода; 2 —воздух; 3 — воздушно-водяная смесь. Давление подпора в верхнем сопле обусловливает засасывание воздуха в водоструйный насос и выброс воздушно-водяной смеси наружу через нижиюю трубку.
5 06
E.I. Оборудование лабораторий и лабораторная техника
47.5. Работы, проводимые под давлением,
отличающемся от атмосферного
47.5.1. Вакуумирование
При использовании водоструйного насоса (рис. Е.29) для вакуумирования сосудов получают вакуум 1,3-103—2,1 • 103 Па (давление паров воды) при скорости откачивания 0,5—2 м3/ч. Более высокого вакуума (до 1,3-10—3 Па) добиваются, применяя масляные роторные насосы различных типов, а также парортут-ный и ртутный диффузионный насосы. Последние работают только при создании форвакуума <4-103 Па, например, масляным насосом. Вещества с более временно загрязняющие насос,
высоким давлением паров, однонеобходимо предварительно удалить, пропуская газ через поглотительную колонку или охлаждаемую ловушку. Проще применять так называемые газобалластные насосы, которые засасывают легко конденсирующиеся газы и пары без их конденсации, оказывающей вредное воздействие на насос. Поэтому эти насосы широко используют в вакуумной перегонке, при высушивании в вакууме и т. п.
Давление 0,1-103—26,7-10® Па измеряют манометром, закрытым с одной стороны (рис. Е.ЗО); по его подвижной шкале определяют разность давлений столбов ртути. Для измерения вакуума <130 Па применяют манометр Мак-Ле-ода, по градуированному капилляру которого можно сра-
зу определить давление. Скорость откачивания значительно снижается, если трубки и отверстия кранов узки, поэтому диаметры должны быть не менее 15 мм для трубок и 10 мм для кранов. На нижнюю и верхнюю половину пробок и шлифов кранов перед употреблением необходимо нанести вакуум-смазку в виде тонкого кольца.
Благодаря понижению температуры кипения веществ в вакууме можно проводить перегонку веществ, разлагающихся при атмосферном давлении при температуре кипения. Схема установки для вакуумной перегонки приведена на рис. Е.31. Очень
Рис. Е.31. Схема установки для вакуумной перегонки.
J —водяная баня; 2 — колба для перегонки; 3 — капилляр для перегонки; 4 — насадка КлаЙзена; 5 — термометр; 6 — холодильник Либиха; 7 — вакуум-форштос; S —вакуум-шланг; 9 — «паук» для присоединения трех приемных колб, который может поворачиваться в вакуум-форштосе; 10— колбы-приемники отдельных фракций днстиллата; 11— манометр; 12 — склянка Вульфа с краном для впуска воздуха в установку; 13 — отвод к вакуумному насосу.
Рис. Е.32. Схема прибора для возгонки в вакууме.
I — ввод охлаждающей воды; 2 — охлаждаемая трубка для сублимата; 3сублимируемое вещество; 4 —отвод к вакуумному насосу.
508 E.I. Оборудование лабораторий и лабораторная техника
важно использовать капилляры, предотвращающие перегревание жидкости. Капилляр вытягивают при нагревании в пламени до такой длины, чтобы его конец почти касался дна колбы. Диаметр капилляра должен быть таким, чтобы при продувании воздуха в стаканчик с эфиром образовывались лишь мелкие отдельные пузырьки. Равномерное кипение жидкости обеспечивают, погружая колбу в жидкостную баню. Прибор для проведения возгонки в вакууме с водяным охлаждением изображен на рис. Е.32.
В вакууме значительно ускоряется и процесс высушивания^ для этой цели рекомендуют применять вакуум-эксикаторы и пистолеты для сушки. Предложены специальные методы, позволяющие исключить доступ воздуха и влаги.
47.5.2. Применение повышенного давления
Достаточно большая скорость реакции обычно достигается только при температурах, при которых создается значительное давление паров разделяемых веществ. Поэтому в лаборатории часто приходится проводить работы, связанные с применением повышенного давления. Следует особенно подчеркнуть необходимость соблюдения правил техники безопасности при проведении работ с повышенным давлением (защитные очки!).
Толстостенные стеклянные сосуды выдерживают небольшое избыточное давление до 40-103 Па. Их можно медленно нагре-
Рис. Е.ЗЗ. Схема печи с электрообогревом для нагревания запаянных трубок, вать на кипящей водяной бане, обернув тряпкой (для защиты от осколков), или также медленно охлаждать. Для работы при более высоких давлениях (до 2-108 Па) применяют специальные трубки из стекла марки «дюран» или «супремакс», которые можно нагревать примерно до 300 °C (минимальное время нагрева 4 ч). После внесения вещества трубки запаивают. Запаивание надо проводить таким образом, чтобы трубка заканчивалась толстостенным капилляром длиной несколько сантиметров. Затем трубку помещают в железную защитную трубку и в наклонном положении нагревают в так называемой бронированной
48. Правила техники безопасности
509
печи (рис. Е.ЗЗ). После охлаждения печи рабочую трубку вместе с защитной вынимают из печи и капилляр трубки нагревают до размягчения на пламени горелки Теклю. При этом под действием внутреннего давления стенки капилляра раздуваются и газообразные продукты могут улетучиваться (помнить об опасности взрыва!).
Для работы под давлением применяют также шариковую трубку, состоящую из нескольких одинаковых стеклянных шаров, соединенных между собой возможно более короткими цилиндрическими отрезками трубок с одинаковой толщиной стенок. Такие шариковые трубки из стекла «дюробакс» выдерживают нагревание при 500 °C в течение нескольких суток при давлении внутри трубки 6-108 Па.
Значительно более высокие давления можно создать в стальных автоклавах.
47.6. Ведение рабочего журнала
При проведении любых химических работ необходимо составлять отчеты. В них нужно приводить все данные, необходимые для точного воспроизведения выполненной работы. При составлении отчета нельзя полагаться на свою память, необходимо уже в ходе работы в лаборатории делать записи в лабораторном журнале, записывать методики работы и возможные отклонения от них, протоколировать все данные, необходимые для отчета, а также особые наблюдения в ходе эксперимента. В заглавии каждого отчета указывают вид и номер задания и дату работы. При проведении препаративных работ необходимо после краткой записи методики, уравнений реакции, литературных источников дать описание собственного эксперимента, из которого можно получить сведения о количестве, концентрациях и чистоте (или изготовителе) исходных веществ, температуре и продолжительности реакции, операциях очистки, выходе (абсолютное количество и процент от теоретического выхода) и исследуемых свойствах конечного продукта (внешний вид, чистота, температура кипения, температура плавления и т. д.).
В отчете о качественном анализе указывают внешний вид вещества, результаты предварительных испытаний, растворимость, присутствие нераство-рившегося остатка, приемы перевода вещества в растворимое состояние, появление осадков и окраски, ход обнаружений отдельных компонентов и четкие выводы.
В отчетах о количественных определениях приводят методику определения (сущность метода, уравнения реакций и литературные источники) и точное описание собственных работ с указанием навесок анализируемого вещества, количеств, концентрации и чистоты применяемых реактивов, титра и расхода титрованных растворов, применяемых индикаторов, используемых фильтров, продолжительности и температуры высушивания и прокаливания осадков, результатов взвешивания и общего расчета результатов анализа, а также ошибок анализа.
48. ПРАВИЛА ТЕХНИКИ БЕЗОПАСНОСТИ
Работа в химических лабораториях требует особых мер предосторожности. Перечислим некоторые наиболее важные меры по предотвращению несчастных случаев и смягчению их последствий.
510 E.I. Оборудование лабораторий и лабораторная техника
Порядок на рабочем месте. Соблюдение чистоты и порядка на рабочем месте и в лаборатории является обязательным условием точной работы химика. Использованное оборудование сразу же по окончании работы надо очистить и поставить на место. Все склянки для хранения химических веществ должны быть снабжены четкими и нестирающимися надписями. Рабочее место можно оставить, только убедившись, что все сетевые краны закрыты, а все электроприборы (плитки) и освещение выключены. Перед каждым опытом следует продумать, какие реакции могут произойти, и принять необходимые меры предосторожности. При проведении химических работ не разрешается оставаться одному в лаборатории. Работающие с большими количествами горючих жидкостей, опасных, ядовитых или взрывоопасных веществ должны находиться под особым наблюдением, в таких случаях необходимо информировать о характере работы соседа по рабочему месту и лаборанта.
Обращение со стеклянным оборудованием. Многочисленные травмы возникают из-за неправильного обращения со стеклом. Вставляя стеклянные трубки в пробки или натягивая шланги на стеклянные трубки, палочки или термометры, необходимо защищать руку, обернув ее полотенцем, чтобы не пораниться, если сломается стекло. Целесообразно предварительно смочить стекло водой или лучше глицерином, что значительно уменьшает трение между стеклом и резиной. Поврежденное стеклянное оборудование нужно уничтожить, а если возможно'—отремонтировать перед употреблением. Пробирки с отбитыми краями надо выбрасывать, концы стеклянных палочек и трубок должны быть оплавлены или отшлифованы. В каждой лаборатории должна находиться аптечка со средствами для оказания первой помощи.
Работа с едкими или ядовитыми веществами. Растворы таких веществ нельзя засасывать в пипетку ртом, а только с помощью груши (пипетки с насадкой). Концентрированные кислоты разбавляют, приливая при энергичном перемешивании кислоту к воде, а не наоборот. Брызги горячих кислот или щелочей часто вызывают ожоги, а концентрированная фтороводородная кислота и безводный HF к тому же и очень трудно залечиваемые раны. Во избежание отравления запрещается использовать для химических работ посуду (бутылки, стаканы), применяемую для продуктов питания или, наоборот, еду или питье помещать в лабораторную посуду. Запрещается принимать пищу на рабочем месте.
При попадании в глаза брызг кислот или щелочей глаза промывают сначала большим количеством воды, затем раствором бикарбоната натрия или разбавленным водным раствором борной кислоты. Эти растворы, снабженные четкими надписями, должны быть в каждой лаборатории, использование их для других целей недопустимо.
Работа с ядовитыми, едкими или сильно дымящими газами. Такие работы проводят под тягой с хорошей вытяжкой. Длительное воздействие даже небольших концентраций ядовитых газов дает тяжелые последствия. Особую опасность представляют газы без запаха (СО, СО2, пары Hg) или газы, действующие на обоняние (Cl2, H2S, HCN, СОС12, NO2), так как их нельзя распознать при длительном воздействии или при больших концентрациях. Сероводород так же ядовит, как и синильная кислота. Работы, связанные с образованием вредных для здоровья газов, необходимо проводить в противогазе. Противогазы могут быть снабжены фильтрами, специфичными к определенным веществам.
Фильтры классифицируют на следующие группы.
Гриппа А. Маркировка коричневая; защищают от паров муравьиной кислоты, бензола, хлороформа, уксусной кислоты, формальдегида, сероуглерода, сульфурилхлорида, тетрахлорида углерода.
Группа В. Маркировка серая; защищают от газов, выделяющихся при горении, брома, хлора, галогеноводородных кислот, иода, газообразных иит-розосоединений, трихлорида фосфора, фосгена, дымящих кислот, азотной кислоты, соляной кислоты, сульфурилхлорида.
Группа О. Маркировка серо-красная; применяют при работе с арсином и фосфином.
48. Правила техники безопасности
511
Группа G. Маркировка голубая; для работы с синильной кислотой и пылеобразным цианидом калия.
Группа Е. Маркировка желтая; защищают от диоксида серы.
Группа М. Маркировка желто-зелеиая; применяют при работе с аммиаком и сероводородом.
Группа L. Маркировка желто-красная; для работы с сероводородом.
Группа R. Маркировка желто-коричневая; для работы с сероводородом.
Группа К. Маркировка зеленая; для работы с аммиаком.
Группа СО. Маркированы черным кольцом; только для однократного применения при работе с оксидом углерода. Нужно учесть, что фильтры через определенное время теряют пригодность.
Для быстрого оказания помощи при несчастных случаях необходимо, чтобы несколько противогазов и фильтров всегда висели на четко обозначенных местах.
Работа с открытой ртутью требует особой тщательности из-за опасности хронического отравления парами ртути. Во избежание растекания ртути приборы ставят в плоские поддоны (фотографические кюветы). Ртуть, попавшую на пол, необходимо собрать или, если это невозможно, обезвредить, превратив ее в амальгаму действием цинка или олова; можно посыпать ртуть иодидом углерода. Пыль и пары большинства других металлов (например, РЬ, Cd, Zn, Be), а также летучие соединения тяжелых металлов (оксиды, карбонилы, металлоорганические соединения) также ядовиты.
Все работы под давлением, в вакууме, с пожароопасными и взрывчатыми веществами следует проводить, надевая защитные очки. Это необходимо и при работе с сосудом Дьюара. Защитные очки не должны иметь оправу из целлулоида (пожароопасны).
Работы при повышенном давлении в автоклавах или специальных трубках необходимо проводить в отдельных помещениях, соблюдая действующие там правила. Баллоны со сжатыми газами должны быть надежно закреплены, чтобы не падали, или храниться в горизонтальном положении. Работа в вакууме также требует соблюдения мер предосторожности: при первоначальном вакуумировании эксикатора его нужно обернуть полотенцем или лучше поместить в защитный чехол, сплетенный из проволоки (опасность взрыва). В эксикатор с осушителем — серной кислотой — нужно поместить наполнители (стеклянные шарики, кольца Рашига) так, чтобы слой наполнителя поднимался над уровнем серной кислоты.
Многие соединения и смеси взрывоопасны: галогеноксиды, галогенокислородные кислоты и их соли, азиды и .ацетилиды тяжелых металлов, нитраты амминокомплексов тяжелых металлов, бертолетово гремучее серебро Ag3N, смеси порошкообразного Mg и серы, порошкообразного алюминия с Na2SOi (при нагревании), а также с оксидами (применяемые для получения металлов. по Гольдшмидту), если соответствующие оксиды не полностью обезвожены (В20з) или являются оксидами металлов не той степени окисления, некоторая должна быть (МпО2 вместо МпзСЬ). Взрывчатыми являются также все смеси сильных окислителей (нитратов, хлоратов, перхлоратов, перманганата, оксида хрома (VI), хромовой смеси и т. д.) с легко воспламеняемыми или органическими веществами, даже с пылью.
Смеси всех горючих газов (Н2, H2S, AsH3, углеводородов, светильного газа) с воздухом или кислородом взрывоопасны. Перед зажиганием таких газов, получаемых в газогенераторах, необходимо постоянно проверять аппаратуру на отсутствие кислорода воздуха пробой на гремучий газ. Белый фосфор, щелочные металлы, а также многие металлы в тонкодисперсном состоянии самовоспламеняются на воздухе и поэтому наиболее огнеопасны. Горючие органические растворители нельзя напревать на пламени горелок, открытых электронагревательных приборах или вблизи них, а также вблизи таких источников искр, как выключатели, электромоторы и др.
Для тушения пожаров в лаборатории наиболее пригоден пенный углекислотный огнетушитель; несколько хуже тетрахлоруглеродный огнетушитель из-за образования в пламени ядовитого фосгена. Оба типа огнетушителей
512 E.I. Оборудование лабораторий и лабораторная техника
нельзя применять для гашения горящего натрия, калия и магния. Горящий металл засыпают песком. Ни в коем случае нельзя применять огнетушители для тушения загоревшейся на человеке одежды! Пламя сбивают водой или заворачивают человека в плотную ткань (одеяло, покрывало).
Электрические приборы, находящиеся под сетевым напряжением (220 В), должны удовлетворять соответствующим требованиям. Кабели и т. п. нельзя подвергать химическому или другому воздействию (нагревание в печах), вызывающему коррозию.
Регулярно необходимо проводить обучение персонала лаборатории правилам техники безопасности на основе применяемого в ГДР стандарта TGL 30582/01—03*, в котором изложены правила техники безопасности при работе в химических лабораториях, в том числе правила работы с химическими и взрывчатыми веществами, под давлением и в вакууме, охраны здоровья, пожаробезопасности, а также регламентированы защитная рабочая одежда и защитные средства. Кроме того, разработаны специальные регламенты по технике безопасности (ASAO), которые важно знать во избежание несчастных случаев и причинения материального ущерба. Эти регламенты постепенно заменяют на TGL. Разработан также ряд инструкций, в том числе инструкции по работе с цианидами щелочных металлов, фтороводородом, фосфором и бензолом. Постановление о порядке работы с ядовитыми веществами от 7.4.1977 (GB1, DDR I № 10, с. 103) четко регламентирует работу с ядами. Прн обучении правилам техники безопасности это постановление необходимо изучать.
* В СССР действующими нормативами являются ГОСТы. См.: Система стандартов безопасности труда. Гос. ком. СССР по стандартам.—М.: Стан-дартгиз, 1979. О правилах техники безопасности при работе в лаборатории см. книгу: Макаров Г. В., Стрельчук Н. А., Кушелев В. П. и др. Охрана труда в химической промышленности.—М.: Химия, 1977. — Прим, перев.
E.II. ПРАКТИКУМ
ПО ПРЕПАРАТИВНОЙ ХИМИИ
49. ТЕХНИКА РАБОТЫ
49.1. Общие указания к проведению
химико-препаративных работ
49.1.1. Реакции в растворах
В препаративной химии многие реакции проводят в растворах, особенно в водных, применяя воду для растворения веществ и разбавления растворов. Простейшим оборудованием для этого являются стеклянные стаканы и круглодонные колбы. Не требуют особых аппаратурных затрат способы получения препаратов, основанные на реакциях осаждения. К ним относятся сливание растворов двух реагирующих веществ (см. разд. 49.2, препараты 4 и 7), дополнительная обработка препарата, полученного при сливании растворов (препарат 8), кристаллизация препаратов из растворов, например при длительном стоянии (препараты 2, 9), нагревании (препарат 1), охлаждении (препараты 3, 5), упаривании (препараты 6, 10) или пропускании водяного пара (препарат 18). Для ускорения реакции часто применяют нагревание (см. разд. 46.1). При проведении реакций, протекающих с выделением тепла, реакционную смесь следует охлаждать в соответствии с температурой разложения или испарения компонентов смеси (см. разд. 46.1.2).
Для лучшего прохождения реакции растворы необходимо перемешивать (разд. 47.2). Кратковременное перемешивание можно осуществлять вручную, используя в качестве мешалки стеклянные палочки с закругленными оплавленными концами. При длительном прохождении реакции, когда один из компонентов медленно добавляют, например из капельной воронки (рис. Е.26; препараты 37, 52, 53, 59, 66, 91, 129), применяют механическое перемешивание (препараты 29—34, 49). Для точного дозирования и равномерного добавления по каплям используют бюретку.
Сложные препаративные работы осуществляют в многогор-лых колбах со стандартными шлифами (см. разд. 45.2). Наряду с мешалкой в горла колбы могут быть помещены и другие устройства: капельные воронки, трубки для подвода газов, холодильники, термометры и т. д.
Работу с агрессивными веществами, разъедающими стекло и фарфор, ведут в платиновых сосудах (препараты 44—47).
Газообразные компоненты реакций получают в газогенераторах (разд. 47.4, препараты 143, 159, 161—164, 172) и подают в растворы через газоподводящие трубки (препараты 16—28, 47,
33—1880
514 Е.П. Практикум no препаративной химии
60, 61, 63, 66, 85, 89, 109—111, 113, 124, 168, 173). Использование клапана Бунзена (препарат 17) позволяет очень просто устранить влияние атмосферного воздуха на растворы, выделяющие газ в процессе реакции. Если при введении газа в раствор образуется малорастворимое вещество, следует применять широкую трубку для подвода газа, которая не будет засоряться осадком. Горючие или ядовитые газы, находящиеся в избытке или образующиеся как побочные продукты, необходимо обезвредить, удаляя их через тягу (препараты 109, 113, 171) или пропуская через поглотитель.
Рис. Е.34. Схема выделения препарата из раствора.
Во избежание испарения реакционной среды при температуре кипения или при работе с легколетучими компонентами реакционную колбу снабжают обратным холодильником (разд. 47.3.5, препараты 38—43, 59, 61, 62, 65, 66, 72, 90, 117), а в случае высококипящих растворителей достаточно применить удлиненную трубку (препарат 39).
Препарат, образующийся при проведении реакций в растворах, может а) остаться в растворе; б) выкристаллизоваться при остывании или охлаждении раствора; в) из-за малой растворимости сразу выпасть в осадок; г) быть летучим.
Если препарат остается в растворе, то выделить его можно одним из способов, схематично указанных на рис. Е.34. Очень часто вещество необходимо сначала отфильтровать от малорастворимых продуктов побочной реакции или остатков непрореагировавших исходных веществ (разд. 47.3.4). Для дальнейшего выделения вещества возможны следующие пути.
Кристаллизация. Очень простым способом является кристаллизация веществ из упаренных пересыщенных растворов, в то время как перегонка (разд. 47.3.5) и экстракция (разд. 47.3.7) связаны с использованием более сложной аппаратуры.
Упаривание растворов (разд. 47.3.1). используют при получении твердого вещества, если растворитель можно удалить испарением. Если в качестве растворителя применялась не вода,
49. Техника работы
515
а горючее или ценное вещество, упаривание растворов проводят в установке для перегонки, отделяя или улавливая при этом растворитель (препараты 70, 170). После упаривания и охлаждения вещество выделяется из насыщенного раствора в кристаллическом виде (разд. 47.3.2). Если при упаривании раствора выделяются твердые примеси, их нужно отфильтровать из горячего раствора перед охлаждением (разд. 47.3.4; препарат 13).
Кристаллизацию веществ проводят в кристаллизаторах — низких цилиндрических стеклянных сосудах, маточный раствор из которых можно легко удалить декантацией (разд. 47.3.4). Дополнительное количество основного вещества, но более загрязненного другими продуктами реакции, можно выкристаллизовать упариванием и охлаждением маточного раствора. В этом случае хороший эффект дает повторная кристаллизация или фракционная кристаллизация (разд. 47.3.2). Если кристаллизующееся вещество имеет низкий температурный коэффициент растворимости, то кристаллизацию нужно проводить при пониженной температуре, например помещая кристаллизатор в холодильник или в охлаждающую смесь (разд. 46.1.2; препараты 5, 6, 28). Если вещество, получаемое упариванием, при этом разлагается, упаривание нужно проводить при пониженном давлении (препараты 49—51, 86, 94). Для этой цели применяют установку для перегонки (рис. Е.15) с простым вакуумным алонжем; вакуум создают с помощью водоструйного насоса. Упаривание растворов веществ, разлагающихся при комнатной или немного повышенной температуре, целесообразно проводить в эксикаторе (разд. 47.3.8) с подходящим осушителем (табл. Е.З), который можно еще вакуумировать (препараты 4— 7, 9, 13, 14, 49, 50). Для получения индивидуальных кристаллов больших размеров существуют специальные методы выращивания кристаллов (разд. 47.3.2).
Осадок отделяют от маточного раствора декантацией или фильтрованием. В общем случае для фильтрования в препаративных работах применяют склянку для отсасывания с воронкой Бюхнера (рис. Е.12; препараты 10, 43, 168). Для агрессивных растворов всегда используют стеклянные или фарфоровые нутч-фильтры. Осадок, находящийся на фильтре, очищают от остатков маточного раствора водой. Вещества, хорошо растворимые в воде, промывают небольшим количеством спирта.
Осадок необходимо высушить, т. е. освободить от находящегося в нем растворителя. Для высушивания на воздухе обычно бывает достаточно поместить влажное вещество, особенно если оно промыто этанолом, на уложенную в несколько слоев фильтровальную бумагу или на глиняные тарелочки. Более быстрое и интенсивное удаление влаги происходит при нагревании вещества инфракрасными лампами на плитках (разд. 46.1.1) или в сушильном шкафу. Высушивание веществ с применением по-
33*
516 ЕЛ, Практикум по препаративной химии
глотителей влаги и без доступа воздуха проводят в эксикаторах (препараты 39, 40, 48), а также в нагреваемом пистолете для сушки (рис. Е.25; препарат 52), которые можно также вакуумировать (препараты 21, 78, 85),
Высушенные вещества через воронку для порошкообразных веществ ссыпают в сосуды для хранения препаратов (толстостенные пробирки) или в склянки. Препараты, чувствительные-к действию воздуха или влаги, хранят в запаянных стеклянных ампулах.
Экстрагирование (разд. 47.3.7; препараты 67—72). Для экстрагирования веществ, находящихся в водных растворах, их встряхивают с определенным растворителем, который не смешивается с водой и растворяет в первую очередь извлекаемое вещество. Этот простой метод разделения (разд. 38.3.5.1) проводят периодически в делительной воронке (рис. Е.23; препарат 67). Разработаны также установки (аппарат Сокслета, см. рис. Е.22; препараты 68, 71, 72, 138 и перфоратор, рис. Е.24) для непрерывной экстракции и для проведения экстракции при повышенной температуре.
Если препарат выкристаллизовывается при остывании или охлаждении растворов, то операции, выполняемые в этом случае, в основном будут теми же, что и в первом случае, не применяют только упаривание растворов (препарат 98). Осадок отделяют (разд. 47.3.4), очищают (разд. 47.3.2) и путем охлаждения увеличивают количество получаемого вещества. Необходимо также дополнительно обрабатывать маточный раствор (разд. 47.3.8). Высушивание проводят, как описано в разд. 47.3.8.
Рассмотрим третий случай, когда препарат малорастворим и выпадает в осадок. Большое число препаратов получают из растворов с помощью реакций осаждения, т. е. используя малую растворимость синтезируемых веществ. Существуют различные способы проведения реакций осаждения: взаимодействие жидких или растворенных компонентов (препараты 95, 90, 180); введение газов в реакционную жидкость (препарат 20); добавление веществ, уменьшающих растворимость одного из компонентов, уже находящихся в растворе (препарат 95); переведение растворенного компонента в малорастворимый аддукт с его последующим разложением; диффузия (препараты 179, 181).
Осадок собирают на фильтре, промывают и высушивают (разд. 47.3.8). Эти операции могут быть дополнены операциями очистки веществ: перекристаллизацией из неводных растворителей (препараты 42, 98); экстракцией (препарат 84); возгонкой (препарат 78).
Если препарат является летучим соединением, то его отделяют от других соединений, находящихся в растворе, перегонкой (разд. 47.3.5; препараты 54—61, 115, 160, 167). Часто эту операцию проводят уже в ходе синтеза, что оказывает положи
49. Техника работы
517
тельное влияние на выход продукта. Выше уже были описаны установки для простой и фракционной перегонки (рис. Е.15 и Е.17 соответственно), ректификации (рис. Е.18; препараты 63, 108, 116, 117, 121, 171, 173), вакуумной перегонки (разд. 47.5.1 и рис. Е.31; препараты 62—66, 84, 93, 119, 120, 129, 138) и рассмотрена техника выполнения этих операций. Дистиллат собирают в приемники (препарат 131), дополнительно охлаждаемые в случае необходимости (разд. 46.1.2); дальнейшую очистку препарата проводят фракционной перегонкой.
49.1.2. Реакции в расплавах
Реакции в расплавах, применяемые для синтеза препаратов, можно разделить на три большие группы.
1. Реакции взаимодействия веществ в расплавах (препараты 101—105). Большинство таких реакций проводят при температурах ниже 400 °C в сосудах из простого или закаленного стекла. Так же, как для описанных выше методов, проводимых преимущественно в водных растворах, методы получения и очистки многих препаратов, синтезируемых в расплавах, требуют повышенных аппаратурных затрат. При этом применяют следующие методы: использование газов (гл. 47.4); образование конденсата; экстракцию (разд. 47.3.7; препарат 138); перегонку (препарат 167; также при пониженном давлении, разд. 47.5.1); возгонку (препарат 79); использование пониженного давления (препарат 107).
Синтезы препаратов часто необходимо проводить в атмосфере сухого воздуха (разд. 47.3.8; препарат 147) или в атмосфере инертного газа (разд. 47.4).
2. Термические методы (препараты 135—137). Методы описаны в разд. 50.1.1.3.
3. Использование солевых расплавов в качестве реакционной среды (см. табл. Е.11; препараты 166, 167). Синтезы в расплавах солей, используемых в качестве инертных неводных реакционных сред, требуют небольших затрат, так как большинство таких реакций можно проводить при 1000 °C в закрытых тиглях.
49.1.3. Реакции с участием твердой фазы
1. Реакции взаимодействия твердых веществ с газами (препараты 114—116, 121, 131, 132, 156, 169, 75). Твердые вещества, которые должны находиться в возможно более тонкодисперсном состоянии (разд. 47.1), взаимодействуют с газами при повышенных температурах. Типовая установка для проведения таких синтезов состоит из кварцевой или фарфоровой трубки, находящейся в трубчатой электропечи с регулируемой температурой. В реакционной трубке находится лодочка с порошкообразным ве
518 E.II. Практикум, no препаративной химии
ществом. Трубка с обоих концов закрыта пробками, через отверстия в которых проходят трубки для подвода и отвода вводимого (по возможности из стальных баллонов; разд. 47.4.3) газа. Продукт реакции образуется при определенной температуре часто уже в виде сублимата, находящегося на некотором расстоянии от лодочки (препараты 79—82). Техника работы во многих случаях усложняется необходимостью проведения синтеза в атмосфере сухого воздуха (разд. 47.5.1; препараты 115, 116, 121), в атмосфере инертного газа (препарат 114) или в вакууме.
Полученное вещество часто необходимо отделять от исходных компонентов. Для этого применяют следующие операции: экстракцию (разд. 47.3.7); возгонку (разд. 47.3.6), в том числе возгонку в потоке газа и в атмосфере сухого воздуха; перегонку (разд. 47.3.5; препараты 108, 115), в том числе в атмосфере сухого воздуха и фракционную перегонку (разд. 47.3.5; препараты 116, 121).
2. Реакции между твердыми веществами (препараты 106, 176, 177). Эти реакции редко применяют в препаративной химии, так как в результате образуются продукты спекания, которые лишь в исключительных случаях имеют однозначно стехио-метричный состав. Таким препаратом является Cdl2:Pb (препарат 100)—люминофор, светящийся в ультрафиолетовом свете. К числу реакций взаимодействия твердых веществ относятся также топохимические реакции (препарат 15).
3. Реакции разложения (препараты 141, 142, 152—154, 160). В препаративных целях можно использовать пиролиз. При этом происходит термическое разложение исходных соединений с образованием твердого вещества и газа. Для полноты протекания реакции образующийся газ необходимо удалять из сферы реакции. С этой целью синтез проводят в открытых тиглях, применяют вакуум или пропускают поток инертного газа. Получение элементов в свободном состоянии термическим разложением веществ см. табл. Е.10.
Некоторые оксиды образуются из соответствующих гидратов или карбонатов при температурах ниже температуры кипения воды, поэтому их можно синтезировать нагреванием водных суспензий.
49.1.4. Возгонка
Возгонку (разд. 47.3.6 и 35.5.1; препараты 78—84, 155) можно применять для разделения и очистки веществ, например имеющихся в продаже препаратов, а также для смесей, отдельные компоненты которых при нагревании переходят из твердой сразу в газообразную фазу. Сублимированный компонент конденсируют на охлаждаемой поверхности, для чего разработаны спе
49. Техника работы
519
циальные установки (рис. С.ЗЗ, Е.19, Е.20), а также устройства для возгонки в токе инертного газа (рис. Е.21; препараты 81— 83, 108) и в вакууме (рис. Е.32; препарат 138).
49.1.5. Работа без доступа воздуха
Все работы с веществами, склонными к гидролизу, необходимо проводить в атмосфере сухого воздуха (препараты 91, 92, 129, 130, 173, 180). В связи с этим к аппаратуре для синтеза предъявляются высокие требования. Все части аппаратуры, имеющие открытые отверстия, например алонж установки для перегонки (рис. Е.15), должны быть защищены от непосредственного контакта с воздухом с помощью осушительных средств (разд. 47.3.8 и 47.4.2). Синтез веществ, чувствительных к действию влаги или не содержащих гидратной воды, можно проводить в токе газа (препарат 27) или в среде неводного растворителя (препараты 80—88, 90, 93, 94). Препарат в этом случае выделяют, отгоняя растворитель.
Реакции галогенирования (препараты 78, 81, 82, 92, 114— 116, 118, 121), а также гидрирования (препарат 170), имеющие важное значение для препаративных работ, обычно нужно проводить без доступа воздуха и влаги. С этой целью аппаратуру часто промывают потоком хорошо высушенного инертного газа (разд. 47.4.2); газы, участвующие в реакции, также должны быть предварительно тщательно высушены.
49.1.6. Электролиз
В зависимости от типа растворителей, применяемых при электролизе, электролитические методы (см. разд. 36.2.1.5) можно разделить на: электролиз в водных (препараты 73—76, 139) и неводных (препарат 77) растворах, а также в расплавах (препарат 140). Материал для изготовления электролизера нужно выбирать в соответствии с агрессивностью электролита и образующихся веществ. Работа с вращающимися электродами (препарат 75), диафрагмами, мешалками, без доступа воздуха, в атмосфере инертных газов (препараты 76, 77) и т. д. требует значительных технических и аппаратурных затрат.
49.2. Препаративные синтезы в растворах
49.2.1. Реакции простого обмена
Препаративные синтезы в стаканах, колбах и чашках 1. Карбонат кадмия CdCO3
CdCl2 + (NH4)2CO3 + 4NH3 ->' [Cd(NH3)4]CO3 + 2NH4C1
[Cd(NH3)4]CO3 -> CdCO3 + 4NH3
520 ЕЛ. Практикум по препаративной химии
К раствору CdCh сразу приливают весь объем раствора (NH4)2CO3 и такое количество раствора гидроксида аммония, чтобы образовавшийся осадок перешел в раствор. Затем жидкость нагревают в открытом сосуде на водяной бане. СбСОз выделяется в виде блестящих кристаллов.
Белый порошок или ромбоэдрические кристаллы, малорастворимые в воде; разлагается при 357 °C.
2. Сульфат аммония-хрома(Ш), аммонийхромовые квасцы, додекагидрат NH4Cr(SO4)2* 12Н2О
12 г дихромата аммония растворяют в воде и смешивают с 7 г этанола и 100 см3 2М серной кислоты ( — 20 г конц. H2SO4). Восстановление дихромата следует проводить при комнатной температуре во избежание образования комплексного иона [Cr(H2O)2(SO4)2]-. Через несколько часов восстановление заканчивается образованием сине-фиолетового раствора, после чего аммонийхромовые квасцы выделяются в виде красно-фиолетовых октаэдров.
3. Трииодид калия, моногидрат К1з’Н2О
К14-12 --► К13
В насыщенный при нагревании раствор К1 добавляют теоретически рассчитанное количество 12 и смесь после растворения иода охлаждают до О°С, после чего выкристаллизовывается К1з-Н2О.
Темно-коричневые гигроскопические призмы; плавится в закрытой трубке при 38 °C; при 225 °C выделяется иод и образуется KI.
4, Пероксид серебра Ag2O2
2AgNO3 + 4КОН + 2ЦМпО4 --> Ag2O2 + 2K2MnO4 + 2KNO3 4- 2H2O
Смешивают растворы AgNO3 и КМпО4 и добавляют к смеси избыток КОН. Выпавший осадок отфильтровывают через стеклянный фильтрующий тигель и промывают ледяной водой до тех пор, пока фильтрат не станет бесцветным; осадок высушивают в течение 2 ч при температуре <100 °C и затем на 24 ч оставляют в эксикаторе над Р4Ою- Таким образом получают безводные препараты с содержанием до 60% Ag2O2.
Серо-черный порошок; разлагается выше 100 °C на Ag и О2.
5. Пероксомонохромат калия КзСгО8
Смешивают 60 см3 3%-ного раствора Н2О2, 5 см3 30%-ного раствора Н2О2 и 5 см3 50%-ного раствора КОН; выдерживают смесь в охлаждающем средстве (дед+поваренная соль) до образования густой кашицеобразной массы. Затем добавляют 5 г тонко-измельченного в порошок КгСгО4 и выдерживают массу в колбе
49. Техника работы
521
Эрленмейера в указанной охлаждающей смеси в течение еще 2 ч. После этого в колбе снова появляется жидкость, а пероксомонохромат выделяется в виде мелких красно-коричневых октаэдров. Кристаллы отфильтровывают через фильтр с пластинкой из пористого стекла, промывают спиртом и эфиром и затем высушивают в эксикаторе.
6. Дигидроарсенат натрия, моногидрат NaH2AsO4«H2O
H3AsO4 + NaOH --► NaH2AsO4 + H2O
5M. раствор H3AsO4 нейтрализуют гидроксидом натрия по метиловому оранжевому и упаривают раствор до образования кристаллов. Выделившийся при охлаждении NaH2AsO4-H2O перекристаллизовывают из насыщенного при 100 °C раствора при помешивании и охлаждении до 0 °C.
Мелкие кристаллы; в 100 г Н2О при 100 °C растворяется 75,3 г препарата.
7. Имидобис(сульфат) калия HN(SO3K)2
N(SO3K)3-2H2O HN(SO3K)2 + KHSO4 + Н2О
38 г нитридотрис(сульфата) калия смачивают 16 см3 2%-ной H2SO4 и оставляют стоять на 24 ч. Кашицеобразную массу отсасывают и промывают 60 см3 ледяной воды. Затем вещество перекристаллизовывают из смеси 60 см3 Н2О с 10 см3 конц. NH4OH. Выпавшие при охлаждении раствора кристаллы промывают ледяной водой, спиртом и эфиром и высушивают в эксикаторе над H2SO4.
Зернистые кристаллические друзы или блестящие листочки, малорастворимые в холодной воде; кипящая вода омыляет вещество с образованием амидосульфата.
8. Монотиофосфат натрия, додекагидрат Na3PO3S«12H2O
P2S5 + 6NaOH -->- 2Na3PO2S2 + H2S 2H,0
Na3PO2S2 + H2O -<- Na3PO3S + H,S
В умеренно концентрированный раствор NaOH медленно вносят P2S5 до получения соотношения 6 NaOH : 1 P2S3. Для уменьшения разогревания смеси, особенно в конце реакции, реакционный сосуд охлаждают холодной водой. Дитиофосфат осаждают из раствора спиртом, при этом полисульфиды остаются в растворе. Смесь тиофосфатов натрия сразу промывают спиртом, снова растворяют в воде (получается примерно 30%-ный раствор) и нагревают 10—15 мин, при этом все высшие тиофосфаты гидролизуются до монотиофосфата. Раствор фильтруют, смешивают со спиртом (Vs,' объема раствора) и сразу охлаждают льдом во избежание дальнейшего гидролиза. Соль еще раз перекристаллизовывают указанным способом при 50 °C.
522 E.II. Практикум no препаративной химии
Тонкие шестиугольные листочки; легко выветриваются на воздухе, хорошо растворяются в горячей воде; ГПл = 60°С.
9. Хлорохромат калия К[СгО3С1]
K2Cr2O7 + 2НС1 -► 2К[СгО3С1] + Н2О
50 г тонкоизмельченного в порошок КгСг2О7 растворяют при нагревании до 70 °C в смеси 65 см3 конц. НС1 с 50 см3 Н2О. Раствор фильтруют через воронку для горячего фильтрования. Через 1—2 сут образовавшиеся кристаллы отсасывают, перекристаллизовывают из ледяной уксусной кислоты и высушивают в вакуум-эксикаторе над H2SO4.
Оранжевые блестящие кристаллические иглы.
10. Иодат калия КЮ3
KI + 2КМпО4 + Н2О ---► КЮ3 + 2МпО2 + 2КОН
В колбе вместимостью 1 дм3 растворяют 30 г КМпО4 в 750 см3 горячей дистиллированной воды и добавляют концентрированный водный раствор иодида калия /15 г КД). Смесь нагревают на кипящей водяной бане около 30 мин и удаляют избыток перманганата медленным добавлением спирта (при этом раствор обесцвечивается). После фильтрования фильтрат подкисляют уксусной кислотой и упаривают на водяной бане до начала кристаллизации. Выделившиеся кристаллы КЮ3 быстро отсасывают на воронке Бюхнера, промывают 15 см3 спирта и высушивают при 60 °C. Маточный раствор еще раз упаривают до начала кристаллизации.
Кубические кристаллы; в 100 г НгО при 20 °C растворяется 8,1 г препарата; ТПл=560°С.
11. Хлорид нитропентамминокобальта(Ш) [Co(NH3)5(NO2)]C12 [Co(NH3) 6С1]С12+ NaNO2 —> [Со (NH3) 5 (NO2) ]С12+ NaCl
В колбе Эрленмейера, закрытой часовым стеклом, растворяют 20 г [Co(NH3)5C1]C12 (см. препарат 69) в смеси 200 см3 Н2О и 50 см3 10%-ного раствора NH4OH при нагревании на водяной бане и при частом взбалтывании. Смесь фильтруют, фильтрат охлаждают и слабо подкисляют разбавленной НС1, добавляют 25 г кристаллического NaNO2 и нагревают на водяной бане до тех пор, пока первоначально образовавшийся красный осадок не перейдет полностью в раствор. К холодному буро-желтому раствору, из которого в большом количестве выделяется кристаллический осадок, добавляют (сначала осторожно) 250 см3 конц. НС1. После охлаждения осадок отфильтровывают, промывают сначала разбавленной в два раза конц. НС1, затем отмывают кислоту спиртом и высушивают препарат на воздухе.
49. Техника работы
523
Буро-желтые кристаллы; в 100 г Н2О при 20 °C растворяется 2,9 г препарата.
12. Нитрозилпентацианоферрат(Ш) натрия, дигидрат (нитропруссид натрия) Na2 [Fe(CN)5NO] «2Н2О
40 г тонкоизмельченного в порошок гексацианоферрата(II) ка* лия растворяют в стеклянном стакане в 60 см3 воды и добавляют при перемешивании 64 см3 HNO3 (р=1,24 г/см3). Раствор умеренно нагревают на водяной бане. Реакция закончена, если при действии раствора FeSO4 на отобранную из раствора пробу образуется не берлинская лазурь, а осадок темно-зеленого цвета. Через 1—2 сут раствор нейтрализуют добавлением Na2CO3 (избегать избытка!), затем нагревают до кипения, фильтруют и выпаривают фильтрат. К охлажденному раствору добавляют примерно равный объем спирта, при этом выпадает в осадок KNO3. После выпаривания спирта из фильтрата выделяются темно-красные кристаллы нитропруссида натрия. Кристаллы промывают небольшим количеством холодной воды и высушивают между листами фильтровальной бумаги.
13. Гексацианохромат(Ш) калия Кз [Cr(CN)s]
Сг(СН3СОО)3 + 6KCN -► K3[Cr(CN)6] + ЗСН3СООК
Для получения исходного ацетата хрома(III) смешивают 17 г СгОз или 25 г К2Сг2О7 с 70 см3 НС1 (45 см3 конц. НС1+ 25 см3 Н2О); смесь при нагревании восстанавливают, добавляя порциями этанол (в общей сложности 25 см3). В кипящий раствор добавляют NH4OH в очень небольшом избытке; выпавший Сг(ОН)3 отфильтровывают из горячего раствора через складчатый фильтр, промывают осадок несколько раз горячей водой и затем растворяют его в небольшом количестве разбавленной уксусной кислоты. Полученный раствор ацетата выпаривают почти досуха для удаления избытка уксусной кислоты. Остаток растворяют в 150 см3 Н2О, фильтруют и фильтрат приливают к кипящему раствору, содержащему 100 г KCN в 200 см3 Н2О (под тягой!). Образовавшийся темно-красный раствор выпаривают на водяной бане, при этом в большинстве случаев выделяется буро-черная масса, которую отфильтровывают. При дальнейшем концентрировании на стенках сосуда образуются светло-желтые кристаллы. Фильтрату дают охладиться и кристаллы отсасывают. Из маточного раствора получают еще несколько фракций кристаллов. Кристаллы перекристаллизовывают два-три раза из воды и высушивают над H2SO4.
Светло-желтые, моноклинные кристаллы; в 100 г Н2О при 20 °C растворяется 30,96 г препарата; разлагается выше 150 °C.
14. Триоксалатоферрат(Ш) калия, тригидрат K3[Fe(C2O4)3]» • ЗН2О
Fe(OH)3 + ЗКНС2О4 ► K3[Fe(C2O4)3] + ЗН2О
524 E.II. Практикум no препаративной химии
35 г FeSO4-7H2O растворяют в 100 см3 Н2О и при кипении окисляют действием HNO3. После разбавления раствора до 2 дм3 осаждают гидроксид железа действием NH3 и промывают осадок методом декантации в течение нескольких суток. Затем осадок отфильтровывают и еще раз промывают горячей водой. Гидроксид железа медленно вносят в горячий раствор, содержащий 44 г гидрооксалата калия в 100 см3 Н2О. Как только гидроксид железа перестанет растворяться, его отфильтровывают л фильтрат концентрируют на водяной бане для кристаллизации препарата. Выделившиеся изумрудно-зеленые кристаллы промывают сначала небольшим количеством воды, затем спиртом и высушивают над конц. H2SO4.
Соль чувствительна к действию света, поэтому ее следует оберегать от прямого воздействия солнечных лучей. Fe(III) восстанавливается под действием света, при этом в свободном виде выделяется СО2:
2K3[Fe(C2O4)3] -> 2K2[Fe(C2O4)2] + К2С2О4 + СО2
Реакцию можно использовать для измерения количества света.
15. Пирофорный гидрат оксида железа(Ш) РегО3«пН2О
20 г FeSO4-7H2O (ч.д.а.) нагревают в течение 40—60 мин в колбе Кьельдаля с 200 см3 чистой —70 %-ной серной кислоты до кипения, причем парам дают возможность улетучиваться, чтобы немного сконцентрировать кислоту. По окончании реакции большие кристаллы в форме табличек размером до 2 мм собирают на стеклянном фильтре, промывают небольшим количеством воды, несколько раз ацетоном и высушивают в вакууме. Кристаллы затем вносят при сильном перемешивании в 200 см3 приблизительно 2 М раствора NH4OH (или в соответственно меньшее количество более концентрированного раствора). Через 10—15 мин реакция заканчивается; осадку дают осесть, сливают маточный раствор и 4—5 раз промывают осадок методом декантации. Наконец, гидратированный оксид, который еще имеет внешнюю форму исходных кристалликов сульфата железа, переносят на нутч-фильтр, промывают несколько раз водой и высушивают ацетоном и эфиром.
Для получения препарата, проявляющего максимальный пирофорный эффект, высушенный эфиром продукт осторожно нагревают в течение 30 мин при 300 °C. При нагревании до 350 °C начинается самопроизвольная кристаллизация, сопровождающаяся пирофорным эффектом и приводящая к образованию a-Fe2O3.
Для получения препарата используют топохимическую реакцию. В таких реакциях взаимодействие происходит иа поверхности твердого вещества, т. е. локально. Для протекания топохимических реакций необходимы следующие условия: твердое исходное вещество не должно растворяться слишком быстро, чтобы взаимодействие происходило на границе раздела фаз; образующий-
49. Техника работы
525
•ся на поверхности твердого вещества продукт реакции должен оставаться достаточно пористым, чтобы отдельные кристаллики могли полностью вступить в реакцию. Этим условиям часто удовлетворяют сульфаты трехвалентных металлов. Пирофорные гидратированные оксиды образуют также Сг, Nb, Sc, Та, Ti и Zr.
Реакции с использованием газов
А. Воздух
16. Гексанитрокобальтат(Ш) натрия Na3 [Co(NO2)6]
2Co(N03)2 + 12NaNO2 + 2СН3СООН + 1!2О2 -►
---► 2Na3[Co(NO2)6] + 4NaNO3 + 2CH3COONa + Н2О
150 г NaNO2 растворяют в 150 см3 воды и охлаждают раствор до 50—60 °C; при этом выделяется некоторое количество исходного NaNO2. К раствору добавляют 50 г Co(NO3)2-6H2O, затем порциями при встряхивании 50 см3 50%-ной СН3СООН и в течение 0,5 ч пропускают через раствор сильный поток воздуха. Через 2 ч отфильтровывают коричневый осадок; фильтрат должен быть прозрачным. Осадок смешивают с 50 см3 воды, нагретой до 70—80 °C; раствор отфильтровывают и присоединяют к предыдущему прозрачному фильтрату. Ко всему объему фильтрата (около 500 см3) добавляют 250 см3 96%-ного спирта, оставляют выпавший осадок на несколько часов, затем отфильтровывают его на нутч-фильтре, отсасывают досуха, промывают 4 раза порциями по 25 см3 спирта, затем 2 раза эфиром и высушивают на воздухе. Целесообразно переосадить препарат спиртом. Чистый препарат должен растворяться в воде с образованием прозрачного раствора. Для осаждения соли к раствору добавляют из промывалки спирт, после чего раствор сильно взбалтывают, чтобы осадок не был слишком мелкодисперсным.
Желтый кристаллический порошок, хорошо растворимый в воде.
Б. Кислород
17. Родохромхлорид [(NH3)5Cr(OH)Cr(NH3)5]С13»Н2О
60 г измельченного в порошок К2Сг2О7 при перемешивании в колбе вместимостью 2,5 дм3 обливают 200 см3 конц. НС1 и 75 см3 спирта. Полученный зеленый раствор соли хрома (III) еще теплым (температура ниже 50 °C) восстанавливают металлическим цинком без доступа воздуха, для чего горло колбы закрывают так называемым клапаном Бунзена. Он представляет собой вставленную в отверстие пробки стеклянную трубку, на верхний конец которой натянут прорезанный с одной стороны кусочек резинового шланга. Резиновый шланг затыкают стеклянной палочкой. Раствор голубого цвета вливают в смесь 500 г NH4C1 с 750 см3 конц. NH4OH, которую хорошо охлаждают, бросая в нее кусочки льда или поместив ее в лед. После декантации раствора с нерастворившегося NH4C1 создают условия
526
E.IL Практикум no препаративной химии
для быстрого окисления, вводя Ог и сильно встряхивая раствор. При этом жидкость окрашивается в красный цвет и выделяется обильный осадок родохромхлорида. Отфильтрованную соль промывают вначале смесью двух объемов НгО с одним объемом' конц. НС1, а затем один раз холодной водой. Родохромхлорид растворяют в холодной воде и раствор вливают в охлажденную-смесь двух объемов НС1 с одним объемом воды, причем препарат осаждается почти полностью. Осадок сначала промывают разбавленной в два раза НС1, затем отмывают от кислоты спиртом и высушивают на воздухе (в темноте).
Аналогично можно получить родохромбромид и -нитрат. .
Бледно-красный кристаллический порошок.
В. Водяной пар
18. Гидрат оксида железа(Ш) (игольчатая железная руда, гетит) a-FeO(OH)
Fe(NO3)3 -I- 3NH3 + 2H2O -<- FeO(OH) + 3NH4NO3
405 г Ре(ЬЮз)3-9Н2О растворяют в 1 дм3 Н2О и раствор на холоду медленно добавляют при перемешивании к раствору NH4OH (60 г NH3 в 100 см3 Н2О). Выпавший осадок гидроксида несколько раз промывают методом декантации холодной водой” общим объемом —4 дм3 при сильном перемешивании и декантируют. Аморфный гидроксид перемешивают с таким количеством-конц. КОН, чтобы смесь стала 2 н. по КОН. После стояния в течение 3—4 ч с последующим двухчасовым пропусканием водяного пара осадок полностью превращается в желто-красный a-FeO(OH). Избыток КОН удаляют путем обработки хлоридом аммония, добавленным в несколько большем количестве, чем теоретически рассчитанное. Перед высушиванием в вакуум-экси-каторе препарат еще раз тщательно промывают водой. При нагревании в вакууме или в потоке сухого воздуха образуется исключительно a-Fe2O3.
Г. Диоксид углерода
19. Перманганат(УП) бария Ва(МпО4)2
2КМпО4 + Ba(NO3)2 4- Ва(ОН)2 -► 2ВаМпО4 + V2O2 + 2KNO3 + Н2О
ЗВаМпО4 + 2СО2 --* Ва(МпО4)2 + 2ВаСО3 + МпО2
50 г КМпО4 и 50 г Ва(ЫОз)г растворяют в 1 дм3 кипящей водьт и обрабатывают 10 г Ва(ОН)2-8Н2О. Смесь нагревают при частом перемешивании на водяной бане почти до полного прекращения выделения кислорода, затем добавляют еще 10 г Ва(ОН)2-8Н2О и так повторяют до тех пор, пока жидкость не-обесцвечивается полностью. Объем жидкости поддерживают постоянным, добавляя воду. После того как малорастворимый ВаМпО4 осядет на дно вместе с некоторым количеством МпОг и?
49. Техника работы
527
ВаСОз, жидкость сливают, осадок многократно промывают кипящей водой (несколько куб. дециметров) путем декантации, кипятят с разбавленным раствором Ва(ОН)2 и еще раз тщательно промывают кипящей водой.
Осадок взмучивают в 1 дм3 воды и полностью разлагают, пропуская в течение нескольких часов одновременно СО2 и перегретый водяной пар. Затем дают осадку осесть, отсасывают жидкость через стеклянный фильтр и упаривают раствор до тех пор, пока при охлаждении еще будут выделяться почти черные кристаллы. Перманганаты других металлов могут быть получены взаимодействием Ва(МпО4)2 с соответствующими сульфатами.
Д. Аммиак
20. Арсенат аммония, тригидрат (NH4)3AsO4-3H2O
H3AsO4 + 3NH3 --► (NH4)3AsO4
Возможно более концентрированный водный раствор мышьяковой кислоты (препарат 37) насыщают газообразным NH3.
Ромбические кристаллические листочки.
Е. Сероводород
21. Тионитрозилпентацианоферрат(П) калия К4 [Fe(CN)sNOS] Нитрозилпентацианоферрат(Ш) натрия (препарат 12) высушивают при 105 °C, растворяют в холодном абсолютном метаноле и смешивают с раствором, полученным насыщением сероводородом раствора КОН в абсолютном метаноле. Сразу же выпадает микрокристаллический темно-синий осадок, который промывают абсолютным метанолом. Соль высушивают над СаС12 в вакууме. Препарат растворяется в воде с образованием сине-фиолетового раствора.
22. Тритиокарбонат бария BaCS3
Половину объема раствора Ва(ОН)2 (приготовленного растворением 32 г Ва(ОН)2 в 100 см3 теплой воды) насыщают сероводородом и затем смешивают с другой частью раствора и с 8 г CS2. После перемешивания полученного раствора в закрытой склянке в течение 0,5 ч образуется желтый осадок BaCS3. Осадок отфильтровывают через складчатый фильтр, промывают небольшим количеством воды, затем спиртом, разбавленным водой (1:1), и наконец чистым спиртом. При добавлении спирта к фильтрату можно выделить в осадок еще некоторое количество тритиокарбоната бария. Препарат высушивают при 60 °C.
При нагревании разлагается на BaS + CSa.
Тиофосфаты щелочных металлов, например тетратиоортофосфат натрия, можно получить в водном растворе по реакции
3Na2S-9H2O 4- P2S5 -> 2Na3PS4-8H2O + 11Н2О
528 ЕЛ. Практикум no препаративной химии
Тиофосфаты более тяжелых металлов могут быть получены только путем сплавления компонентов. Так, удалось получить ряд тетратиоортофосфатов по следующим реакциям:
3MnS 4- P2S5 -► Mn3P2S8
3FeS4-P2S5 ---> Fe3P2S8
SbCl3 + P2S3 -► SbPS4 + PSCl3
Ж. Хлор
23. Дигидропероксоиодат натрия Na3H2IO6
NaIO3 + 4NaOH + Cl2 --► Na3H2IO6 + 2NaCl + H2O
Для получения исходного NaIO3 в колбе вместимостью 5 дм® растворяют 62,5 г NaC103 в 250 см3 Н2О при 45 °C и подкисляют раствор 1 см3 конц. HNO3. Добавляют 50 г 12 и закрывают горло колбы перевернутым стаканом. Смесь нагревают до 50— 70 °C при постоянном перемешивании. Если реакция протекает слишком бурно, колбу погружают в холодную воду. Окончание реакции контролируют по исчезновению окраски иода. В колбу добавляют 70 г твердого NaOH и, если необходимо, 50—100 см3 воды. Смесь нагревают до кипения и через широкую стеклянную трубку пропускают в раствор сильную струю хлора. Через 10—15 мин С12 не будет поглощаться, так как раствор уже нейтрализован. Чтобы перевести образовавшийся одновременно в небольшом количестве Na2H3IO6 в малорастворимый Na3H2IO3, раствор слабо подщелачивают NaOH. Смесь охлаждают и фильтруют; осадок промывают небольшим количеством холодной воды и высушивают при ПО °C.
По этой же методике можно получить КЮ-,.
24. Сульфат гидразиния (2+) [N2H6]SO4
2NH3 + NaOCl + H2SO4 --->- [N2H6]SO4 + NaCl + H2O
К смеси 165 см3 конц. NH4OH с 35 см3 Н2О, помещенной в колбу Эрлейнмейера вместимостью 750 см3, добавляют 35 см3 раствора столярного клея, приготовленного размачиванием 2 г клея в небольшом количестве воды с последующим растворением в горячей воде (вместо столярного клея можно применять раствор комплексона). Кроме того, получают NaOCl, пропуская хлор в 100 см3 2 н. раствора NaOH, охлаждаемого льдом, до тех пор, пока масса раствора не увеличится на 6 г. Увеличение массы несколько меньше, чем требуется для получения рассчитанного по уравнению реакции количества NaOCl, чтобы избежать избытка свободного хлора. Раствор гипохлорита вливают в раствор NH4OH, быстро нагревают до кипения и упаривают примерно наполовину. Затем смесь охлаждают сначала водой, затем льдом и при охлаждении приливают конц. H2SO4 до прекращения образования осадка. После фильтрования белые кри
49. Техника работы
529
сталлы сульфата гидразиния перекристаллизовывают из минимально возможного количества воды.
Толстые блестящие таблички или призмы. 7’ПЛ = 254°С.
3. Хлороводород
25. Хлорид гексааквахрома(Ш) [Сг(Н2О)6] С1з
[Cr(H2O)6] (NO3)3 + ЗНС1 -> [Сг(Н2О)6]С13 + 3HNO3
В раствор, полученный растворением 100 г нитрата хрома(III) Сг(ХО3)з-9Н2О в 100 см3 Н2О, приливают 100 см3 38%-ной НС1 и пропускают при охлаждении льдом сильную струю НС1 до прекращения выделения [Сг (Н2О)б] С1з. Кристаллическую кашицеобразную массу быстро отсасывают на большом нутч-фильтре с пластинкой из пористого стекла, промывают небольшим количеством дымящей НС1, растворяют в 100 см3 воды и снова при охлаждении льдом осаждают газообразным НС1. По окончании осаждения находящийся над кристаллами зеленоватый раствор сливают и путем трехкратного смешивания с ацетоном отмывают серо-синий осадок от основного количества захваченной им НС1 и зеленого хлорида, которые затем окончательно удаляют на нутч-фильтре с пластинкой из пористого стекла промыванием небольшими количествами ацетона. Промывание заканчивают после того, как фильтрат становится бесцветным. Ацетон удаляют промыванием абсолютным эфиром. В эксикаторе над H2SO4 соль освобождают от эфира и следов влаги.
Сине-серые кристаллы; на воздухе сильно расплываются; растворяются в воде с образованием сине-фиолетового* раствора, хорошо растворимы в спирте.
И. Диоксид серы
26. Тетраиодомеркурат(П) меди(1) Cu2HgI4
2CuSO4 + K2HgI4 + SO2 + 2H2O --> Cu2HgI4 + K2SO4 + 2H2SO4
Смесь 4,5 г Hgl2 c 3,3 г KI растворяют в 25 см3 воды. Профильтрованный раствор обрабатывают раствором 5 г CuSO4-•5Н2О в 15 см3 воды и в смесь пропускают SO2. При этом выпадает светло-красный осадок, который отсасывают, промывают водой и высушивают при 100 °C. Препарат можно перекристаллизовывать из горячей соляной кислоты.
При нагревании препарата до 70 °C происходит изменение окраски из красной в шоколадно-коричневую (термокраска).
27. Дитионат бария BaS2O6-2H2O
MnO2 -р 2SO2 ---> MnS2O6
MnS2Oe + Ва(ОН)2 -► BaS2O6 + Мп(ОН)2
34—1880
530 Е.П. Практикум no препаративной химии
Готовят суспензию 50 г тонко измельченного пиролюзита в 250 см3 воды. В суспензию, охлаждаемую ледяной водой, пропускают SO2 до тех пор, пока основное количество МпО2 не вступит в реакцию. Температура суспензии не должна превышать 10 °C. Отфильтрованный раствор разбавляют до 1,5 дм3 и при 30 °C добавляют к нему при перемешивании раствор, содержащий 200 г Ва(ОН)2-8Н2О; при этом осаждается Мп(ОН)2. Смесь нагревают до 75 °C и добавляют раствор Ва(ОН)2 до щелочной реакции; выдерживают при 75 °C в течение 30 мин, фильтруют в горячем состоянии и промывают осадок 300 см3 теплой воды, содержащей Ва(ОН)2. Промывные воды и фильтрат объединяют, проверяют щелочность среды (при необходимости добавляют раствор Ва(ОН)2), избыток Ва(ОН)2 удаляют действием СО2 и нагревают раствор до кипения. Фильтрат упаривают на водяной бане до начала кристаллизации. После охлаждения раствора отсасывают выделившиеся кристаллы BaS2O6-2H2O и высушивают их между листами фильтровальной бумаги. Добавив этанол к маточному раствору, можно выделить еще некоторое количество дитионата.
При нагревании препарата до 120 °C происходит выделение кристаллизационной воды; выше 140 °C соль разлагается с образованием BaSO4.
К. Триоксид диазота
28. Нитрозилгидросульфат (NO)HSO4
N2O3 + 2H2SO4 --► 2(NO)HSO4 + H2O
В 35 см3 конц. H2SO4, находящейся в колбе Эрленмейера, пропускают N2O3 (см. препарат 37). Конец трубки для подвода газа погружают в H2SO4 на глубину 1 см. Реакционный сосуд охлаждают, погрузив его в ледяную воду. Образуется густая кашицеобразная кристаллическая масса (NO)HSO4, которую быстро отсасывают через стеклянный нутч-фильтр, промывают ледяной уксусной кислотой и тетрахлоридом углерода и высушивают на глиняных тарелках в эксикаторе над Р4Ою-
7ПЛ=73,5° (плавится с разложением).
Препаративные синтезы с использованием мешалок, бюреток и капельных воронок
29. Тетрагидропероксоиодат бария Ва3Н4(10б)2
2Na3H2IOe 4- 3Ba(NO3)2 -► Ba3H4(IOe)3 + 6NaNO3
К раствору, полученному растворением 50 г Na3H2IO6 (см. препарат 23) в 200 см3 воды, добавляют 2 см3 конц. HNO3, нагревают до кипения и смешивают с горячим водным раствором, содержащим 72 г Ba(NO3)2- Смесь кипятят в течение 1,5—2 ч ;при энергичном перемешивании и затем нейтрализуют, добавляя
49. Техника работы
531
Ва(ОН)2. После охлаждения смеси выкристаллизовывается Ва3Н4(1О6)2, который многократно промывают путем декантации горячей водой.
30. Хлорид олова(П), безводный (дегидратированный) SnCl2 SnCl2-2H2O + 2(СН3СО)2О ----------► SnCl2 + 4СН3СООН
В стакан вместимостью 300 см3 помещают 102 г уксусного ангидрида (99—100%-ного) и добавляют при перемешивании 113 г кристаллического БпСЬ-ЗНгО. Обезвоживание начинается сразу же, сопровождаясь сильным выделением тепла, так что (СН3СО)гО временами начинает кипеть (работать под тягой!). Одновременно выделяется безводная соль в виде мелких белых кристаллов. Через 1,5 ч смесь отсасывают, два раза промывают соль порциями по 50 см3 абсолютного эфира и высушивают в вакуум-эксикаторе.
Белое кристаллическое вещество с жирным блеском; 7’ПЛ = 247ОС; ТКип = = 606 °C.
31. Амидосерная кислота H2NSO3H
CO(NH2)2 + H2S2O7 -<- 2H2NSO3H + со2
100 г мочевины при перемешивании механической мешалкой вносят в течение 45 мин в 560 г 100%-ной H2SO4; смесь необходимо хорошо охлаждать и следить за тем, чтобы температура не поднималась выше 40 °C. Затем добавляют еще 309 г олеума (65% SO3) и оставляют стоять при 42—45 °C на 16 ч. Кристаллизат отсасывают на стеклянном нутч-фильтре, промывают сначала концентрированной, а затем 50%-ной H2SO4 и наконец холодным метанолом. Если конечный продукт недостаточно чист, его растворяют в кипящей воде, раствор фильтруют и, охлаждая льдом, снова вызывают кристаллизацию.
32. Тетраацетат свинца(1У) РЬ(СН3СОО)4
РЬ3О4 + 8СН3СООН --► Pb(CH3COOH)4 -I- 2РЬ(СН3СОО)3 + 4Н2О
В трехгорлую колбу, снабженную термометром и мешалкой, помещают 150 г ледяной уксусной кислоты, затем добавляют 60— 65 г тонко измельченного и высушенного при 200 °C РЬ3О4, который до этого сохранялся в эксикаторе над Р4Ою; каждый раз оставляют раствор стоять до обесцвечивания, указывающего на то, что РЬ3О4 (сурик) прореагировал полностью. Поддерживают температуру реакционной смеси 55—65 °C. Теплую смесь фильтруют; после охлаждения тетраацетат выкристаллизовывается в виде белых игл, в то время как ацетат свинца (II) остается в маточном растворе. Кристаллы отсасывают и для очистки растворяют в 20 см3 ледяной уксусной кислоты. При охлаждении выделяются чистые кристаллы РЬ(СН3СОО)4. Слабо-розовая или чисто-коричневая окраска кристаллов вызвана при-34*
532 E.II. Практикум no препаративной химии
месью в них РЬО2, образующегося в результате гидролитического разложения тетраацетата, поэтому при проведении синтеза должно быть полностью исключено влияние влаги воздуха.
Бесцветные призматические кристаллы, очень чувствительные к действию влаги; ГПл=175—180°C (плавится с разложением). Применяют в органической химии как окислитель.
33. Дигидрогипофосфат натрия, гексагидрат Na2H2P2O6-6H2O
В открытую круглодонную колбу, хорошо охлаждаемую подводимой снаружи водой, наливают 1 дм3 воды, добавляют 100 г красного фосфора. К полученной суспензии в течение 5 ч при энергичном перемешивании по каплям добавляют раствор, содержащий 170 г NaC102 в 350 см3 Н2О, после чего отфильтровывают не вступивший в реакцию фосфор. После добавления 10— 12 г активированного угля смесь оставляют стоять на ночь. Затем отфильтровывают активированный уголь и в прозрачном растворе добавлением 15%-ного NaOH устанавливают pH 5,4 (контролируют стеклянным электродом). При этом в осадок выделяются соединения примесей металлов. Для более полного осаждения смесь кипятят, фильтруют и затем оставляют в холодильнике на ночь. Выделившиеся кристаллы отсасывают, промывают ледяной водой и высушивают на воздухе. Выход около 120 г. Для очистки соль один или несколько раз перекристаллизовывают из воды.
Моноклинные кристаллы в форме табличек, устойчивые на воздухе при -обычной температуре; при небольшом нагревании теряют кристаллизационную воду. Обезвоженная при 100 °C соль плавится при 250 °C и разлагается при красном калении с образованием воспламеняющегося фосфина.
34. Тетраметафосфат натрия, тетрагидрат Na4P4Oi2«4H2O
CuSO4 + 2NaOH ---► CuO + Na2SO4 + H2O
2CuO + 4H3PO4 ---► Cu2P4O12 + 6H2O
Cu2P4O12 2Na2S --> Na4P4O12 + 2CuS
При 80 °C свежеосажденный CuO постепенно вносят небольшими порциями в 76,9 %-ную НзРО4, взятую с избытком 5% против теоретически рассчитанного количества. При этом образуется густая масса, которая окрашивается после стояния в течение ночи в чистый светло-голубой цвет. Для удаления свободной воды массу нагревают в фарфоровой чашке на горелке Бунзена сначала слабо, а затем в течение нескольких часов при температуре не выше 430 °C. Образовавшиеся мелкие кристаллы тетраметафосфата меди восемь раз промывают горячей водой; промывная вода должна иметь нейтральную реакцию.
72 г Cu2P40i2 небольшими порциями добавляют при энергичном перемешивании к раствору, содержащему 78 г Na2S>9H2O в 750 см3 воды, освобожденной от кислорода. Отфильтрованный
49. Техника работы
533
раствор упаривают на 7з его объема, фильтруют и выделяют осадок, добавляя спирт или ацетон. После перекристаллизации осадка получают белые кристаллы Na4P40i2-ЮН2О. Из маточного раствора можно выделить еще некоторое количество соли.
Десятиводный тетраметафосфат при нагревании до 40 °C теряет шесть молекул воды. Тетрагидрат имеет две полиморфные модификации с точкой перехода 54 °C; переход обратим. При 100 °C образуется ангидрид, который около 400 °C превращается в триметафосфат.
35. Вольфрамат кальция CaWO4 (люминофор)
Свежеосажденную вольфрамовую кислоту растворяют в 25% -ном растворе NH4OH. Образовавшемуся паравольфрамату аммония дают выкристаллизоваться и два раза перекристаллизовывают его из горячей воды. Затем в один и тот же сосуд медленно сливают из бюреток раствор, содержащий 25 г паравольфрамата аммония, 20 см3 конц. NH4OH и 1 дм3 воды, и раствор хлорида кальция, содержащий 23 г соли в 1 дм3 воды. При этом в точно стехиометрическом количестве образуется зернистый и легко фильтруемый осадок, после высушивания его прокаливают при 1000 °C в кварцевом тигле в течение 1—2 ч.
Под действием коротковолновых ультрафиолетовых лучей CaW04 люми-несцирует; излучение имеет максимум при 460 цм (синее свечение). Аналогично могут быть получены другие люминофоры — вольфраматы цинка, кадмия и магния.
36. Бромат калия КВгО3
6КОН + ЗВг2 --► KBrOg + 5КВг ф ЗН2О
62 г КОН растворяют в равном количестве воды и к раствору (под тягой!) по каплям осторожно добавляют 80 г брома. Сначала появляется устойчивая желтая окраска, а затем выпадают мелкие кристаллы КВгО3. Раствор при этом разогревается; после охлаждения раствора кристаллы отфильтровывают и перекристаллизовывают из 130 см3 кипящей воды.
В 100 г НаО при 20 °C растворяется 6,9 г КВгОз; разлагается выше 435 °C.
В 100 г Н2О при 20 °C растворяется 65,6 г КВг. 7’Пд= 741,8 °C; 7*кжп= = 1383 °C.
37. Мышьяковая кислота, полугидрат H3AsO4’V2H2O
В колбе со шлифом нагревают 100 г Аэ20з со 100 см3 конц. HNO3 (р=1,38 г/см3), которую медленно приливают из капельной воронки. Образующиеся оксиды азота лучше всего улавливать концентрированной серной кислотой, в результате чего получается нитрозилсерная кислота. Когда образование оксидов азота прекращается, жидкость сливают с нерастворившегося остатка и выпаривают досуха. Остаток растворяют в небольшом
534 E.II. Практикум no препаративной химии
количестве воды, раствор фильтруют через фильтр с пластинкой из пористого стекла и снова упаривают до тех пор, пока погруженный в жидкость термометр не покажет 130 °C. Раствору мышьяковой кислоты, имеющему на холоду консистенцию меда,, дают кристаллизоваться в холодильнике (в эксикаторе над. H2SO4). Выделяются прозрачные кристаллы состава H3AsO4-• V2H2O. Кристаллизация ускоряется после введения затравки.
Крупные прозрачные гигроскопичные кристаллы. Полугидрат при нагревании выше 250—300 °C теряет воду и переходит в As2O5.
Препаративные синтезы с использованием обратных холодильников
38. Иодид кадмия Cdb
Cd -J- I2 -► Cdl2
Кадмиевую стружку смешивают с эквивалентным количеством 12 и, добавив воду, кипятят с обратным холодильником в течение примерно 2 ч. После обесцвечивания жидкости ее фильтруют и упаривают на водяной бане. Кристаллы Cdl2 сушат в течение 24 ч при 100—150 °C в вакуум-эксикаторе над P4Oi0.
Бесцветные блестящие гексагональные листочки,. В 100 см3 Н2О при 18°С растворяется 85 г Cdl2, при 100°С—128 г. Т’Пл=387°С; 7’КИп=787°С.
39. Йодноватая кислота НЮ3
3I2 + I0HNO3 --> 6Н1О3 + 10NO + 2Н2О
В круглодонной колбе, снабженной обратным воздушным холодильником, нагревают 32 г 1г со 170 г конц. HNO3 при 70— 80 °C до тех пор, пока раствор будет иметь лишь слабо-желтую окраску. Раствор упаривают на водяной бане, обрабатывают несколько раз небольшими количествами воды и снова упаривают. Остаток нагревают с конц. HNO3 на водяной бане. При охлаждении из раствора выкристаллизовывается НЮ3. Для увеличения выхода продукта реакционный сосуд охлаждают льдом. Кристаллы Н1О3 высушивают над твердым КОН в эксикаторе.
В 100 г Н2О при 0 °C растворяется 286 г препарата, при 80 °C —378,1 г. В 100 г HNO3 при 25 °C растворяется 141 г. Плавится при ПО °C с переходом в HI3O8.
40. Иодид олова(П) Snl2
Sn + 2HI --► Snl2 + Н2
Чистое олово помещают в колбу со шлифом и обливают конц. HI, взятой в избытке. После присоединения обратного холодильника смесь нагревают в течение продолжительного времени до кипения, пока весь металл не перейдет в раствор и не начнут выделяться красные кристаллы SnK. Продукт, основная
49. Техника работы
535
масса которого выпадает после охлаждения, отсасывают на стеклянном нутч-фильтре и для очистки перекристаллизовывают из спирта. Под конец его высушивают в вакуум-эксикаторе над Р4О10.
Красное кристаллическое вещество. В 100 см3 воды при 19,8 °C растворяется 0,96 г препарата.
41. Сульфурилхлорид SO2CI2
2HSO3C1 --► SO2C12 + H2SO4
150 г хлоросерной кислоты (препарат 89) кипятят с 1 г Hg или 1,5 г HgSO4 в течение 1,5—3 ч, причем температура в обратном холодильнике должна быть 70 °C. Пары SO2CI2 конденсируются в холодильнике.
Бесцветная жидкость, немного дымит на воздухе и гидролизуется с образованием H2SOi и НС1. 7’пл=—54,1 °C; ТКип=69,2°С.
42. Ацетат бора В(СН3СОО)3
В(ОН)3 + 3(СН3СО)2О -► В(СН3СОО)3 + 3CH3COOH
Медленно нагревают одну весовую часть Н3ВО3 с пятью весовыми частями уксусного ангидрида на водяной бане в круглодонной колбе, снабженной обратным холодильником. Когда температура достигнет 60 °C, Н3ВО3 начинает бурно реагировать с уксусным ангидридом и переходит в раствор. Раствор при этом вскипает. После охлаждения при стоянии в течение нескольких часов ацетат бора почти полностью выкристаллизовывается в виде белых игл. Продукт перекристаллизовывают из уксусного ангидрида и промывают абсолютным эфиром.
Очень чувствителен к действию влаги. 7’пл=147—148 °C.
43. Гидроксид хрома(1П), гидрат Сг(ОН)3*пНгО
[2СгО3+ЗС2Н5ОН ---•- 2Сг(ОН)3+ЗСН3СНО
К раствору, полученному растворением 160 г СгО3 в 2 дм3 НгО, добавляют при энергичном перемешивании 80 см3 этанола порциями по 10 см3 через каждые 5 мин (осторожно, работать под тягой!). Оставляют стоять на 4 ч и снова таким же способом добавляют еще 80 см3 этанола. Реакционную смесь затем кипятят в течение 16 ч с обратным холодильником; во избежание выбросов смесь необходимо помешивать. После этого высокодисперсный темно-коричневый осадок отсасывают на воронке Бюхнера диаметром 24 см и, не промывая, высушивают при 110 °C. Дополнительные количества препарата могут быть выделены из фильтрата путем упаривания. Кроме того, содержащую хром жидкость, являющуюся отходом в данном случае, можно приме
536 Е.П. Практикум no препаративной химии
нять вместо воды в качестве растворителя для обработки следующей порции.
Черный блестящий порошок. Окраска обусловлена небольшим содержанием в препарате оксидов хрома более высокой степени окисления.
Препаративные синтезы в платиновой посуде ,
44. Тетрафтороантимонат(Ш) калия K[SbF4]'
К2СО3 + Sb3O3 + 8HF -»- 2K[SbF4] 4- СО2 + 4Н2О
Смесь К2СО3 и Sb2O3 в стехиометрических соотношениях растворяют в избыточном количестве HF. Раствор выпаривают досуха. Остаток растворяют в воде и оставляют при комнатной температуре для кристаллизации. Кристаллы образуются в виде тонких листочков.
45. Тетрафтороборат калия K[BF4]
- Н3ВО3 4- 4HF+ КОН -> K[BF4] 4Н2О
К 25 г 40 %-ной фтороводородной кислоты в охлаждаемой льдом платиновой чашке прибавляют 6,2 г Н3ВО3. Затем смесь оставляют стоять в течение 6 ч при комнатной температуре, после чего снова охлаждают льдом и при помешивании добавляют 5 М раствор КОН до изменения окраски метилового оранжевого. При этом K[BF4] выпадает в виде кристаллов. Маточный раствор декантируют, кристаллы промывают водой путем декантации и высушивают в вакууме.
Белая кристаллическая соль, не гигроскопична. Т„л=530°С. В 100 см* НгО при 20 °C растворяется 0,45 г препарата, при 100 °C — 6,3 г.
46. Фторид железа(Ш), гидрат FeF3.4,5H2O
Fe(OH)3 + 3HF -FeF3 4- ЗН2О
Из раствора FeCl3 действием аммиака осаждают гелеобразный гидроксид железа и промывают его до удаления хлорид-ионов. В платиновой чашке осадок обрабатывают избыточным количеством 40%-ной HF и концентрируют раствор на водяной бане. При охлаждении выкристаллизовывается розовая соль, с которой удаляют маточный раствор, отжимая ее между листами фильтровальной бумаги; препарат промывают холодной водой и высушивают в эксикаторе.
47. Ортофосфорная кислота Н3РО4
Р 4- 3HNO3 -> Н3РО4 4- 2NO2 4- NO
В круглодонную колбу помещают 250 г HNO3 (р= 1,2 г/см3), осторожно порциями вносят в нее 20 г красного фосфора и нагревают до полного его окисления. Затем раствор упаривают в фарфоровой чашке и нагревают. Присутствующий в растворе мышьяк осаждают, пропуская в разбавленный раствор H2S.
49. Техника работы
537
Упариванием можно получить раствор Н3РО4 необходимой концентрации.
Поскольку конц. Н3РО4 разрушает фарфор при повышенной температуре, упаривание концентрированных растворов проводят в платиновой чашке. Нагреванием до 150 °C получают сиропообразную жидкость, которая затвердевает при охлаждении. Иногда бывает необходимо добавить в качестве затравки кристаллик кислоты.
Твердые расплывающиеся на воздухе ромбические кристаллы в форме призм. Т'пл = 42°С.
49.2.2. Препаративные синтезы при пониженном давлении
48. Селенит натрия, пентагидрат Na2SeO3*5H2O
H,SeO3 + 2NaOH --> Na,SeO3 4- 2H,0
Чистый SeO2 растворяют в небольшом количестве Н2О и раствор, тщательно предохраняя от попадания пыли, упаривают на водяной бане до начала кристаллизации. Полученный раствор обрабатывают точно стехиометрическим количеством гидроксида натрия, не содержащего карбонатов, упаривают при комнатной температуре в вакууме над СаС12 и для начала кристаллизации раствора потирают стенки сосуда стеклянной палочкой. Кристаллы отсасывают на стеклянном фильтре и высушивают в эксикаторе на глиняной тарелочке.
Белые иглообразные кристаллы, устойчивы во влажном воздухе; теряют воду при 40 °C; 100 г водного раствора содержит при 20 °C около 68 г Na2SeO3-5H2O.
49. Ортопериодная кислота H5IO6
Ва3Н4(Ю6)2 + 6HNO3 -► 2Н5Ю„ + 3Ba(NO3)2
50 г ВазН4(Юб)2 смачивают 40 см3 воды и смешивают с 100 см3 бесцветной азотной кислоты (р= 1,42 г/см3). Смесь нагревают до 60—70 °C в течение 1 ч при помешивании, затем охлаждают до 30—40 °C и отфильтровывают от выделившегося Ba(NO3)2 через фильтр с пластинкой из пористого стекла. Осадок промывают конц. HNO3 при постоянном помешивании до полного удаления перйодата. Фильтраты объединяют и упаривают при 60— 70 °C в вакууме, создаваемом водоструйным насосом (если при этом снова начнет выкристаллизовываться Ba(NO3)2, то его необходимо отфильтровать и затем продолжать упаривание), до тех пор, пока не начнет выделяться Н5Юб. При охлаждении образуются блестящие кристаллы. В связи со склонностью орто-периодной кислоты к образованию пересыщенных растворов, раствор приходится оставлять на длительное время до начала кристаллизации. Кристаллы отсасывают и высушивают при
538
Е.11. Практикум no препаративной химии
50 °C в вакууме. Из маточного раствора можно упариванием получить вторую порцию кристаллов.
50. Фосфиновая кислота Н3РО2
1 моль Ва(Н2РО2)2-Н2О растворяют в воде и добавляют к раствору 1 моль H2SO4 в виде 30%-ного раствора. После осаждения BaSO4 раствор, находящийся над осадком, декантируют и затем упаривают сначала на воздухе, а затем в вакууме. Образовавшуюся маслянистую жидкость кристаллизуют, охлаждая ее до 0 °C или вводя затравку.
Безводная НзРО2 кристаллизуется в виде бесцветных листочков; хороша растворима в воде. Ti:.,= 26,5 °C.
51. Тритионат натрия, тригйдрат Na2S3O6-3H2O
2Na2S2O3 + 4Н2О2 -► Na2S3O6 + Na2SO4 + 4Н2О
62 г Na2S2O3 растворяют в —50 см3 Н2О. При длительном перемешивании к раствору добавляют 30%-ный Н2Ог, причем необходимо тщательно охлаждать раствор, поддерживая постоянно температуру в пределах 0—10 °C. В течение небольшого промежутка времени после добавления 52 см3 30%-ного Н^О2 раствор остается нейтральным и не мутнеет при подкислении. При сильном охлаждении в осадок почти полностью выпадает образующийся Na2SO4, и его отделяют от маточного раствора сильным отсасыванием. Сразу же после фильтрования раствор упаривают в вакууме до образования сиропа, из которого можно выделить кристаллический тритионат, потирая палочкой стенки сосуда. При очень длительном выпаривании препарат загрязняется сульфатом натрия.
Бесцветные призмы или таблички, хорошо растворимые в воде.
52. Сульфат титанила TiOSO4
TiCl4 + H2SO4 + Н2О -► TiOSO4 + 4HCl
Чистый TiCl4 вливают по каплям при длительном энергичном перемешивании в рассчитанное количество 50%-ной H2SO4. Приливание по каплям регулируют таким образом, чтобы перед очередным добавлением осадок, образовавшийся в качестве промежуточного продукта, полностью растворялся. После добавления 3/4 рассчитанного количества TiCl4 образуется вязкий раствор желтоватого цвета. Этот раствор разбавляют на '/5 водой и приливают по каплям оставшийся TiCl4. Полученный раствор, содержащий значительное количество НО, выпаривают досуха на водяной бане, остаток растирают в порошок, после чего в течение нескольких дней его очищают от НС1, высушивая при 80—100 °C в пистолете для сушки под давлением >100 Па.
49. Техника работы
539
Очень гигроскопичен; медленно растворяется в воде; при упаривании водного раствора образуется дигидрат сульфата титанила.
53. Дисульфатопероксотитанат( IV) калия, тригидрат K2[TiO2(SO4)2] -ЗН2О
TiO2 + H2O2+H2SO4 + K2SO4 -* K2[TiO2(SO4)2] + 2Н2О
5 г водного диоксида титана растворяют при нагревании в 10 см3 конц. H2SO4, раствор разбавляют трехкратным объемом воды и охлаждают до —10 °C. Затем готовят второй раствор, смешивая водный раствор 8,6 г K2SO4 с 15 см3 30%-ного Н2О2. В этот раствор, охлажденный до 0 °C, из капельной воронки при постоянном встряхивании приливают раствор сульфата титана. Оставляют стоять на холоду в течение 0,5 ч и осаждают продукт добавлением примерно 1 дм3 ледяного ацетона, предварительно обработанного пероксидом водорода до появления окраски при внесении его в раствор сульфата титана (спирт благодаря восстановлению может обусловить пониженное содержание активного кислорода). Выпавший осадок отделяют на нутч-фильтре и промывают ледяным абсолютным эфиром до тех пор, пока фильтрат перестанет обесцвечивать раствор перманганата. После многочасового высушивания в глубоком вакууме при возможно более низкой температуре получают желто-красный дисульфатопероксотитанат калия.
49.2.3. Синтезы с перегонкой
Перегонка при атмосферном давлении
54. Трифторид мышьяка AsF3
CaF2 + H2SO4 ->- CaSO4 + 2HF
As2O3 + 6HF --> 2AsF3 + 3H2O
Смесь 100 г As2O3 co 120 г CaF2, высушенную при 150 °C, обрабатывают 250 см3 конц. H2SO4 в колбе для перегонки. После многократного встряхивания AsF3 отгоняют. Очистку AsF3 проводят кипячением с обратным холодильником в присутствии NaF и повторной перегонкой на колонке длиной 30 см.
Ткип=58°С.
55. Гидразин, моногидрат N2H4-H2O
[N2H6]SO4 + 2КОН -► N2H4-H2O+K2SO4+H2O
Смешивают 50 г сухого сульфата гидразиния с таким же количеством порошкообразного КОН, добавляют 2,5 см3 воды и перегоняют образующийся гидрат гидразина с нисходящим холодильником. Сначала перегонку проводят без нагревания, однако к концу реакции для полного ее завершения необходимо •смесь сильно нагреть. Гидрат гидразина, в котором еще содер
540
E.II. Практикум no препаративной химии
жится вода, очищают фракционной перегонкой. Чистый гидрат гидразина перегоняется в интервале 117—119 °C. Из первых потоков снова получают сульфат гидразиния.
56. Гидразин, безводный N2H4
n2h4-h2o —> n2h4 + h2o
В колбу для фракционной перегонки с пришлифованной пробкой и пришлифованным термометром помещают 20 г моногидрата гидразина и 20 г NaOH в гранулах. Можно пользоваться либо пришлифованным к колбе холодильником, либо применять колбу с непосредственно припаянной к ней охлаждающей трубкой достаточной длины. Колбу очень медленно нагревают на масляной бане, чтобы гидразин начал кипеть только через 2 ч. К этому моменту весь NaOH должен уже раствориться в жидкости. Температура масляной бани должна быть ~ 120 °C. После начала перегонки колбу продолжают медленно нагревать, пока температура масляной бани не достигнет ~ 150 °C. При этом отгоняется чистый безводный гидразин.
Маслянистая, сильно дымящая жидкость. Разъедает пробку, каучук и другие органические вещества. Затвердевает при 0°С и кипит при 113,5 °C. В склянках с притертыми пробками может сохраняться в течение неограниченно долгого времени. Горючее вещество. Смешивается с водой и спиртами.
57. Триметилборат В(ОСН3)3
Н3ВО3 + 3CH3OH ---► B(OCHg)g + ЗН2О
Метиловый эфир борной кислоты получают нагреванием Н3ВОэ с СН3ОН в присутствии конц. НС1 или H2SO4 в. качестве водоотнимающего средства. Для отделения образовавшегося сложного эфира от избытка исходного спирта реакционную смесь оставляют стоять над СаС12. При этом образуются два слоя; нижний отбрасывают, а из верхнего путем фракционной перегонки получают чистый В(ОСН3)3.
Прозрачная как вода жидкость, горит зеленым пламенем: ГКжп=68,7°С.
58. Хлорид хромила СгОгС12
К2СгО4 + 2NaCl + 2H2SO4 -> СгО2С12 4- Na2SO4 + K2SO4 + 2Н2О
Хорошо высушенную смесь 50 г NaCl с 80 г К2СГО4 вносят в колбу с пришлифованной пробкой. Колбу через холодильник соединяют с пришлифованной колбочкой, снабженной газоотводной трубкой. К смеси добавляют порциями 150 г дымящей H2SO4. Когда первоначально бурная реакция начнет протекать более спокойно, колбу осторожно нагревают до тех пор, пока не прекратится отгонка СгО2С12. Сырой продукт очищают путем повторной перегонки из сухой аппаратуры с пришлифован
49. Техника работы
541:
ными частями; чистый СгОгСБ сразу собирают в высушенные стеклянные ампулы и.запаивают.
Темно-красная, на влажном воздухе сильно дымящая жидкость. Ее сле-дует хранить в запаянных стеклянных сосудах в темноте. 7’КЖП=117°С; Тпл = —96,5 °C. С горючими неорганическими и органическими веществами СгО2С12 может реагировать со взрывом. Растворим в хлорангидридах неорганических кислот и в органических жидкостях, таких, как POCls, СС1*, СНС1з, С6Н6.
59. Тетраэтилсиликат (этиловый эфир ортокремневой кислоты) Si(OC2H5)4
SiCl4 + 4С2Н5ОН -► Si(OC2H5)4 + 4НС1
К 40 г SiCl4, помещенного в колбу, снабженную длинным обратным холодильником, добавляют по каплям свежеперегнан-ный абсолютный этанол в количестве в 1,1 раза большем, чем теоретически рассчитанное. При этом выделяется НС1. Охлаждения не требуется. Смесь кипятят с обратным холодильником в течение некоторого времени до прекращения выделения НС! и сырой продукт подвергают фракционной перегонке с дефлегматором. Тетраэтилсиликат быстро перегоняется при температуре 168—169 °C. Более высококипящие эфиры мета- и поли-кремневой кислот остаются на дне колбы.
Бесцветная жидкость, не смешивается с водой, медленно подвергается гидролизу. 7’кип=165°С.
60. Хлорид мышьяка(Ш) AsCls
As2O3 + 6HC1 --► 2AsCl3 + ЗН2О
As2O3 перегоняют из конц. НС1 при пропускании хлороводорода через раствор.
Бесцветная маслянистая жидкость, дымит на воздухе, чрезвычайно ядовита. Т„л ——16,2 °C; ниже этой температуры существует в виде бесцветных кристаллов с перламутровым блеском. Ткип—130,2 °C. Растворяется в воде и НС1. Растворяет иодиды щелочных металлов, серу, фосфор и масла.
61. Тионилхлорид SOCh и фосфорилхлорид POCI3
PC15 + SO2 —> SOC12 + РОС13
В круглодонную колбу, снабженную обратным холодильником, помещают 25 г РС15 и пропускают через него сухой SO2. Конец холодильника закрывают хлоркальциевои трубкой. После растворения всего PCI5 продукты реакции разделяют многократной фракционной перегонкой.
Собирают следующие фракции: а) до 82 °C — SOC12; б) до 02 °C — SOC12-pPOCl3; в) до 105 °C — POC13+SOC12; г) до 115 °C — РОС13. Вторую и третью фракции снова разгоняют на 4 фракции и полученные дистиллаты присоединяют к первой или четвертой фракциям соответственно (таким образом 2-к>
542
E.U. Практикум no препаративной химии
и 3-ю фракции ликвидируют). В конце еще раз перегоняют первую (7’Кип = 76—79 °C) и четвертую (Ткип=106—109 °C) фракции.
SOCh: 7’пл=—104,5 °C; ТКип=77°С; бесцветная, .сильно преломляющая свет жидкость с запахом, похожим на запах SO2.
POCI3: Тпл=1>72°С; 7’кип= 108,7 °C; бесцветная, сильно преломляющая свет жидкость, дымящая на влажном воздухе.
Перегонка при пониженном давлении
62. Селеновая кислота H2SeO4
В 100 см3 дистиллированной воды растворяют 150 г SeO2 и медленно добавляют 500 г 30%-ного Н2О2. Смесь кипятят с обратным холодильником1 в течение 12 ч, пропуская через нее О2. Для концентрирования раствора H2SeO4 воду отгоняют в вакууме. При достижении температуры 150 °C при 1,5—2,0-•103 Па образуется H2SeO4 85—90%-ной концентрации.
63. Дисерадихлорид S2C12
2S + С12 -► S2C12
Колбу для фракционной перегонки, соединенную через холодильник Либиха с охлажденным приемником (склянка для отсасывания), наполовину заполняют серой. После вытеснения хлором воздуха колбу осторожно нагревают почти до температуры плавления серы. S2C(2 отгоняется при постоянном пропускании С12 и стекает в приемник в виде темно-красной маслянистой жидкости. Хлорирование прекращают до того, как вся сера вступит в реакцию. Сырой продукт разгоняют на фракции в колбе для фракционной перегонки (вначале над оставшейся серой). После добавления новой порции серы перегонку повторяют еще раз при 1,6-103 Па и 39—40°C.
Тяжелая желтая жидкость с неприятным запахом. ТКЯп=137°С. Поража- ет слизистую оболочку. Получение S2CI2 необходимо проводить только под тягой с хорошей вытяжкой. »
64. Тиофосфорилхлорид PSC13
РС1а +S --► PSCI3
В круглодонной или конической колбе, снабженной насадкой .Аншютца и обратным холодильником, нагревают до кипения на водяной бане 100 г РС13 с 24 г измельченной в порошок серы. Конец холодильника закрывают хлоркальциевой трубкой. Как только смесь начинает бурно кипеть, к ней добавляют 3—5 г тонкоизмельченного безводного А1С13. При сильном и даже бурном кипении сера быстро растворяется в жидкости. В некоторых случаях реакционную колбу приходится немного охлаждать. К концу реакции, заканчивающейся через 5— 10 мин, жидкость приобретает оранжево-желтую окраску. Затем’кипение жидкости в колбе прекращается; это указывает,
49. Техника работы
543
что весь РС13 превратился в PSC13. После этого охладившуюся жидкость смешивают в большой делительной воронке со значительным количеством воды и осторожно встряхивают, стараясь избежать образования устойчивой эмульсии. После перехода в раствор А1С13 и возможных примесей (PCI5, Н3РО3 и НС1) продукт реакции немедленно обесцвечивается и выделяется PSC13, образуя нижний слой. Его отделяют, высушивают хлоридом кальция и перегоняют.
Бесцветная подвижная жидкость, дымящая на воздухе, с резким запахом, который ослабевает при разбавлении. Пары раздражают глаза, вызывая слезотечение. Водой на холоду медленно (при нагревании быстро) разлагается,, образуя НС1, H2S и Н3РО4. 7„п=— 35°C; ТКИп=125°С.
65. Триэтилалюминат А1(ОС2Н3)3
Al + ЗС2Н6ОН --> А1(ОС2Н5)а + з/2Н2
В круглодонную колбу, снабженную обратным холодильником,, помещают алюминиевые стружки или опилки, заливают их абсолютным этанолом (соотношение 27:276) и к смеси добавляют 0,1—0,25 части HgCl2 и следовые количества иода. Через-: несколько секунд начинается выделение водорода, которое постепенно усиливается. Если реакция начинается не сразу, колбу осторожно нагревают при встряхивании на водяной бане. Если алюминий и в этом случае не вступает в реакцию, то его необходимо быстро протравить разбавленным раствором NaOH. Этот раствор затем удаляют повторным промыванием абсолютным спиртом путем декантации. Когда реакция замедлится, смесь нагревают на водяной бане еще в течение нескольких часов до тех пор, пока серая кашица триэтилалюми-ната не вспучится и не станет сухой и слоистой. После этого-отгоняют избыток спирта на масляной бане при 210—220 °C. Темный жидкий остаток быстро переливают в колбу Клайзе-на и перегоняют в вакууме с коротким воздушным холодильником большого диаметра, нагревая колбу светящимся пламенем'. Прозрачный жидкий дистиллат переливают в склянку, которую плотно закрывают притертой пробкой. Препарат затвердевает в виде белоснежной массы.
Тпл=130°С; Ткип=210—220°С при 1,3-10’ Па.
66. Диэтилсульфит SO(OC2H5)2
SOC12 + 2С2Н5ОН --> SO(OC2H5)2 + 2НС1
К 100 см3 свежеперегнаного SOC12, находящегося в трехгорлой/ колбе, снабженной газоотводной трубкой, капельной воронкой' и обратным холодильником’, по каплям добавляют 120 см3 этанола. Одновременно через колбу пропускают аргон или азот. По окончании реакции смесь нагревают с обратным холодильником в течение 3 ч. Затем проводят фракционную перегонку
- 544 ЕЛ. Практикум по препаративной химии
под вакуумом под давлением1 1,3-102 Па. Фракция, отгоняющаяся при 50—55 °C, представляет собой бесцветную, ароматичную жидкость и содержит диэтилсульфит. Эту фракцию можно перегнать еще раз для дальнейшей очистки.
ГКИп = 158°С при атмосферном давлении.
49.2.4. Синтезы с применением экстракции
67. Додекавольфраматокремневая кислота
50 г Na2WO4-2H2O растворяют на холоду в 400 см3 воды. К раствору добавляют по каплям около 27 см3 6 М НС1 до нейтральной реакции по лакмусу. Образующийся в качестве промежуточного продукта белый осадок при перемешивании растворяется. К полученному раствору добавляют избыток свеже-осажденного гидрата кремневой кислоты. (Имеющийся в продаже силикат натрия растворяют в минимально возможном количестве холодной Н2О и нейтрализуют его по каплям по .лакмусу концентрированной НС1. Через ’Д ч добавляют небольшой избыток кислотьг, декантируют жидкость, промывают осадок 1—2 раза холодной водой и снова декантируют.) Смесь кипятят в течение 2 ч, причем поддерживают кислую реакцию раствора, добавляя небольшими порциями НС1 до тех пор, пока из отфильтрованного раствора пробы при добавлении разбавленной соляной кислоты не перестанет выпадать осадок .гидрата вольфрамовой кислоты. После этого жидкость отделяют от нерастворившегося SiO2 фильтрованием и встряхивают раствор с эфиром’ и конц. НС1. Для этого как можно более кОн-щентрированный раствор кислоты в делительной воронке интенсивно перемешивают с эфиром, объем которого составляет Приблизительно '/з объема воронки. Затем к раствору добавляют небольшими порциями охлажденную льдом конц. НС1 (37%-ную), не содержащую железа, избегая нагревания жидкости. Продукт присоединения додекавольфрамовой кислоты к эфиру после этого выделяется в виде тяжелых маслянистых капель, образуя третью нижнюю фазу в делительной воронке, которую легко можно слить. Если при добавлении новой порции НС1 маслянистые капли уже не появляются, образование гетерополикислоты можно считать законченным. Масло обрабатывают приблизительно равным объемом воды, а эфир испаряют просасыванием чистого сухого воздуха. Оставшийся прозрачный водный раствор свободной гетерополикислоты ставят в вакуум-эксикатор до начала кристаллизации над конц.
H2SO4, затем над твердым КОН для поглощения адсорбированной НС1. При получении свободной гетерополикислоты це-.лесообразно применять соляную кислоту, так как она легче
49. Техника работы
545
удаляется из продукта присоединения эфира, чем H2SO4 или HNO3.
Препарат кристаллизуется при комнатной температуре в виде бесцветных блестящих правильных кристаллов. Легко растворяется в воде. Тпл = 53 С.
68. Амидосульфат калия NH2SO3K
N(SO3K)3-2H2O -->- NH2SO3K + 2KHSO4
/60 г нитридотрис(сульфата) калия кипятят с 300 см3 воды в течение 75 мин. Затем раствор нейтрализуют 20 г К2СО3 и вышаривают досуха. Остаток обрабатывают в аппарате Сокслета 80%-ным спиртом в течение 46 ч. Из спиртового раствора получают при охлаждении амидосульфат.
Бесцветные кристаллы, хорошо растворимые в воде.
69. Хлорид хлоропентамминокобальта(1П) Co[(NH3)5C1] С|2
20 г карбоната кобальта растворяют в минимально возможном количестве НС1, смешивают с 250 см3 10%-ного раствора NH3 и 50 г (NH4)2CO3 в 250 см3 НгО и окисляют смесь в течение 3 ч пропусканием сильной струи воздуха. После добавления 150 г NH4C1 смесь упаривают на водяной бане до кашицеобразной консистенции и затем при перемешивании подкисляют соляной кислотой до прекращения выделения СО2. Добавляя 1NH3, создают слабощелочную- среду, затем добавляют еще 10 см3 конц. NH4OH и смесь, разбавленную до объема 400— 1500 см3, нагревают на водяной бане в течение 1 ч. После добавления к смеси 200 см3 конц. НС1 и нагревания в течение-!*/2—3/4 ч выделяется хлорид хлоропентамминокобальта(III). Осадок после охлаждения отсасывают и промывают разбавленной НС1.
Для очистки продукт ио частям обрабатывают 2% -ным раствором NH4OH (400 см3), если нужно, при нагревании, и соль выделяют в осадок из фильтрата, нагревая фильтрат с 300 см3 конц. НС1 в течение 3/4 ч на водяной бане. По охлаждении осадок отсасывают и промывают разбавленной НС1 и спиртом.
Фиолетово-красные ромбические кристаллы; в 100 г Н2О при 17,5 °C растворяется 0,13 г препарата.
70. Хлорид гидроксиламмония [NH3OH]C1
HON(SO3K)2 + 2Н2О --► [NH3OH]HSO4 + K2so4
[NHaOH]HSO4 + HCI --► [NH3OH]C1 + H2SO4
Смесь 40 г KNO2 и 50 г KNO3 растворяют в 100 см3 ледяной воды и добавляют 750 г мелко измельченного чистого льда. Через раствор пропускают SO2 до насыщения, при этом начинает выделяться HON(SO3K)2. Затем осадок отсасывают, про-35—1880
546 Е.П. Практикум no препаративной химии
,мывают ледяной водой, растворяют в 500 см3 0,5 М НС1 и ки-’пятят в течение 2 ч. При дальнейшем кипении осаждают образовавшуюся H2SO4, добавляя ВаСЬ до прекращения образования осадка. После фильтрования раствор выпаривают досуха. Из сухого остатка экстрагируют хлорид гидроксиламмо-’пия абсолютным этанолом. При выпаривании спиртового экстракта препарата остаются белые кристаллы, которые можно перекристаллизовать еще раз из небольшого количества воды. 71. Тиоантимонат цинка Zn3 [SbS4]2
3ZnCl2 + 2NaaSbS4 -»- Zn3[SbS4]2 4- 6NaCl
Раствор 25 г соли Шлиппе (см. препарат 103) в 75 см3 Н2О смешивают с раствором, содержащим 10,6 г ZnCI2 (22,5 г ZnSO4-7H2O) в 50 см3 Н2О, Желтый осадок несколько раз промывают горячей водой и центрифугируют. Осадок высушивают сначала при 80 °C, а затем при 100 °C. Препарат, окрашенный в оранжевый цвет, измельчают; в нем содержится юкбло 60% свободной серы, которую экстрагируют сероуглеродом в аппарате Сокслета.
Оранжевый порошок, при 200 °C разлагается на ZnS + Sb2Ss с выделением серы и обесцвечивается.
Соответствующая кадмиевая соль окрашена в оранжево-красный цвет; соль ртути (II) имеет цвет охры.
72. Хлорид ванадия, безводный VC13
2V0O5 4- 6S2C1, -► 4VCI, + 5SO„ + 7S
48 г тонко измельченного чистого V2O5 нагревают с 40 см3 'S2C12 (препарат 63), предохраняя от действия влаги воздуха, ъ течение 8 ч в колбе с обратным холодильником при температуре кипения S2CI2, постоянно перемешивая смесь. Сливают избыток S2C12 вместе с растворенной в нем серой и промывают образовавшийся VC13 сухим сероуглеродом. Загрязняющие препарат летучие компоненты можно удалить, нагревая1 его под вакуумом до 120—150 °C, или многократной обработкой сероуглеродом в аппарате Сокслета. Остаточное содержание серы в полученном мелкокристаллическом VC13 после тщательной очистки составляет -~0,2%.
Фиолетовые кристаллы, очень гигроскопичны.
49.2.5. Синтезы путем электролиза
73. Сульфат кобальта(Ш), октадекагидрат Co2(SO4)3- 18Н2О
Для анодного окисления ионов Со(П) в пористый глиняный стакан вместимостью 120 см-3 помещают раствор, полученный растворением 24 г CoSO4-7H2O в 75 см/3 теплой 8 М Нг5О4. Анодом служит цилиндр из листовой пластины (высота 4 см,
49. Техника работы
547
;ширина 12 см). Подводку тока осуществляют при помощи припаянной платиновой проволоки. Вокруг глиняного стакана помещают в качестве катода медный цилиндр высотой 8 см с подводкой к нему тока. Католитом служит 8 М H2SO4. Электролизер охлаждают льдом и водой. После охлаждения анолита до 30 °C начинают электролиз и продолжают его в течение 12 ч. Вязкую темно-синюю суспензию быстро фильтруют через стеклянный фильтр и высушивают, размазывая осадок платиновым шпателем по глиняной тарелке.
Сине-зеленые блестящие листочки. В сухой атмосфере при нагревании разлагаются.
74. Пероксодисульфат калия KzSzOg
2HSO4- --► S2O82~ + 2Н+ + 2е-
(Электролитом служит насыщенный при 10 °C раствор KHSO4, находящийся в стакане большого размера. В этот стакан погружен стеклянный цилиндр диаметром 3—3,5 см, в котором укреплен анод так, чтобы его нижний конец находился на уровне нижнего края цилиндра (рис. Е.35). Анод представляет собой платиновую проволоку диаметром 0,3 мм, вплавленную в стеклянную трубку. Конец платиновой проволоки дол-
Платпиновый. электрод
Рис. Е.35. Схема установки для электролитического получения пероксодисульфатов.
жен на 0,5 см выступать из трубки. Платиновую проволоку с помощью капли ртути соединяют с токоподводящим проводом, вводимым сверху в стеклянную трубку. В качестве катода .применяют или свинцовую ленту шириной 1 см, обмотанную вокруг стеклянной трубки, или свинцовую трубку, через которую' для охлаждения подается как можно более холодная вода. Вся система помещена в смесь льда с поваренной солью, через которую пропускают также и воду для охлаждения 35*
548
Е.П. Практикум no препаративной химии
свинцовой трубки. Электролиз проводят при температуре не более 10 °C. Плотность анодного тока должна составлять 10 А/см2. При указанных размерах анода сила тока приблизительно равна 1 А. Через несколько минут после начала пропускания тока на аноде выделяется относительно мало растворимый K2S2O8. Электролиз проводят в течение — 2 ч, затем отсасывают кристаллы на холоду, промывают их небольшим объемом ледяной воды, затем спиртом и эфиром и высушивают в эксикаторе.
В 100 г Н2О растворяется при 0 °C 1,62 г; при 10 °C — 2,60 г; при 20 °C — 4,49 г препарата.
75. Пероксодифосфат калия К4Р2О8
ЗНдРОд --► Н4Р2О8 HgPO.j -j- Н2О 02
Смесь 302,2 г КН2РО4, 198 г КОН, 120 г KF и 0,355 г К2СгО4 ^растворяют в 1 дм3 воды. 215 см3 этого раствора подвергают электролизу в большой платиновой чашке, являющейся анодом, с изогнутой быстро вращающейся платиновой проволокой в качестве катода. Плотность анодного тока должна составлять 0,02—0,03 А/см2. В электролите поддерживают температуру ниже 14 °C. Через 3 ч отключают ток и оставляют смесь .на ночь при комнатной температуре. При этом вначале образующаяся соль кислоты Н3РО5 разлагается с регенерацией ортофосфата и выделением некоторого количества К4Р2О8, в то время как одновременно получающаяся соль К4Р2О8 не разлагается. После повторного электролиза, проводимого в течение 2 ч, смесь снова оставляют стоять на ночь. Наконец, электролиз проводят третий раз в течение 1 ч и снова оставляют смесь на ночь. Раствор упаривают на водяной бане при помешивании, пропуская одновременно струю воздуха над поверхностью раствора таким образом, чтобы температура раствора не превышала 80 °C. Трехкратной перекристаллизацией получают 96,4—99,8%-ный препарат.
76. Гексахлоромолибдат(Ш) калия Кз [МоС16]
2МоО3 + 6НС1 + 6КС1 + бе- ->- 2К3[МоС16] + 6ОН~
Раствор 20 г Н2МоО4-Н2О в 150 см3 конц. НС1 и 50 см3 дистиллированной воды восстанавливают электрическим током плотностью около 1—2 А/дм2 в течение нескольких часов при охлаждении водой и пропускании СОг до образования красного соединения трехвалентного молибдена. В качестве материала для катода можно применять гладкую платину, Н§или амальгамированный свинец. Угольный анод погружают в 15%-ную НС1, отделенную от катодной жидкости глиняным цилиндром. Восстановленный раствор как можно быстрее упаривают на открытом пламени до 90 см3, насыщают газообраз
49. Техника работы
549
ным НС1 и обрабатывают 10%-ным освобожденным от воздуха раствором, содержащим 15—20 г КС1. Затем раствор упаривают при 70 °C под уменьшенным давлением до начала кристаллизации, фильтруют и при охлаждении льдом насыщают газообразным HCL Отделенные отсасыванием кристаллы промывают конц. НС1, спиртовым раствором НС1, затем спиртом и высушивают в вакууме.
Кирпично-красные кристаллы, хорошо растворимые в воде.
77. Хлорид цинка ZnCL
Zn-j-2CuCl -> Z1'C124-2Cu
6,7% -ный раствор CuCl в чистом сухом^ ацетонитриле (многократно перегнанным над Р4О10) подвергают электролизу при комнатной температуре с платиновым катодом и ццрковым анодом при напряжении на клеммах 12 В. Одновременно пропускают поток абсолютно сухого азота. Электролиз заканчивают, когда выделяющаяся на катоде медь начинает приобретать серый оттенок (выделение цинка). Растворитель выпаривают в вакууме, затем осторожным нагреванием разрушают сольват ZnCl2 с ацетонитрилом.
Этим методом могут быть получены ZnBr2 и Znl2 при использовании соответствующих солей меди, а при работе с кадмиевым анодом — также CdBr2 и Cdb.
Бесцветные очень гигроскопичные кристаллы. Тпл=313°С; ТКип=7320С.
49.3. Синтезы, основанные на возгонке
78. Иодид висмута ВП3
Bicla4-3HI -► Bil3 + ЗНС1
‘На раствор BiCl3 в соляной кислоте действуют концентрированной иодоводородной кислотой. Осадок отфильтровывают на нутч-фильтре из пористого стекла и отмывают от хлорид-ио-нов конц. HI. Кристаллы высушивают в вакууме над Р4Ою. При этом в конце высушивания их нагревают почти до температуры плавления и затем при сильном нагревании подвергают возгонке.
Темные кристаллы с металлическим блеском. ТПл немного выше 400 °C.
79. Хлорид селена(1У) SeCl4
Se + 2С12 --► SeCl4
Реакцию взаимодействия элементарного селена с С12 проводят в удлиненной стеклянной трубке после вытеснения из нее воздуха. При полном поглощении газообразного хлора масса вначале довольно сильно разогревается, но затем ее приходится 36—1880
550
E.II. Практикум no препаративной химии
подогревать. SeCl4 возгоняют по направлению к концу трубки, чтобы по окончании реакции можно было выплавить оставшийся продукт с той части трубки, где началось выделение SeCl4. Плотную корку сублимата SeCl4 можно отделить легким постукиванием по трубке после нагревания пламенем горелки.
80. Хлорид цинка ZnCl2
Zn + 2НС1 --► ZnCl2 + Н2
Для получения безводного ZnCl2 наивысшей чистоты цинк хлорируют сухим НС1 при температуре около 700 °C в кварцевой лодочке, помещенной в тугоплавкую стеклянную трубку. При этой температуре образование и возгонка ZnCl2 протекают с достаточной скоростью. Сублимированный хлорид собирается в холодной части установки. Нагревания выше 700 °C следует -избегать, так как в противном случае вместе с хлоридом отгоняются значительные количества металла, что заметно по-окрашиванию бесцветного сублимата. При необходимости хлорид можно подвергнуть вторичной возгонке в атмосфере НС1.
Бесцветные очень гигроскопичные мелкие кристаллы. ТПл=313°С; Ткяп=-= 732 °C.
81. Хлорид висмута BiCl3
2Bi + ЗС12 -► 2BiCl3
Лодочку свисмутом нагревают в трубке из тугоплавкого стекла в электрической печи. При помощи двухходового крана можно пропускать через эту трубку чистый азот или хлор. Сначала вытесняют азотом воздух, высушивают реакционную трубку, пропуская при нагревании азот, затем переключают кран на подачу хлора и повышают температуру до начала реакции. BiCis возгоняется, осаждаясь на холодных стенках трубки, которую целесообразно охлаждать или охлаждающей .рубашкой или влажной фильтровальной бумагой. Приблизительно через час образование BiCl3 заканчивается; тогда хлор вытесняют азотом и препарат быстро переносят в сосуд для хранения.
Бесцветные кристаллы, расплывающиеся на воздухе, гидролизуются с образованием BiOCl. ТПл=233°С; Ткип = 4470С.
82. Бромид железа(П) FeBr2
Fe + 2HBr --► FeBr2-i-H2
|Восстановленное водородом железо наивысшей чистоты помещают в лодочку из неглазурованного фарфора, которую ставят в фарфоровую трубку, и нагревают в токе совершенно сухого азота, содержащего НВг, приблизительно до 800 °C, так чтобы образующийся FeBr2 сразу возгонялся. Пропуская сухой азот, сублимат переводят в хорошо закрывающиеся сосуды.
49. Техника работы
551
1Эта операция облегчается, если внутрь реакционной трубки, в ее концевую часть, плотно вставить вторую трубку для сбора FeBr2.
Гигроскопичные кристаллы от светло-желтого до темно-коричневого цвета. ТПЛ=684°С.
83. Гексахлороантимонат нитрозила NO[SbCle]
NOC1 + SbCl5 --> NO[SbCl6]
33 г NOC1 растворяют в сухом СС14, раствор охлаждают смесью лед-фповаренная соль и при тщательнейшей защите от влаги воздуха прибавляют к нему по каплям раствор, содержащий 91 г SbCl5 в небольшом количестве СС14. Выделившийся продукт отфильтровывают, промывают небольшим' количеством СС1|4 и высушивают в вакууме над Р40ю.
Желтые кристаллы, могут возгоняться в потоке СО2 при 150 °C. Тпл= = 170 °C (в закрытой трубке); препарат чувствителен к влаге воздуха.
84. Фосфорнитридхлорид (PNCl2)n
PC1S + NH4C1 --► PNC12 + 4HC1
62 г PCls тщательно смешивают с 50—100 г NH4C1„ защищая от влаги воздуха, и вносят смесь в стеклянную трубку, закрытую с одной стороны (длина 50 см, диаметр 5 см). Смесь покрывают еще одним слоем; NH4C1 толщиной 7 см. Трубку в вертикальном положении помещают в масляную баню, при этом слой NH4C1 должен находиться над поверхностью жидкости. Верхний конец трубки соединяют с промывной склянкой, заполненной H2SO4, через которую может улетучиваться образующийся НС1. Масляную баню нагревают в течение 4—6 ч при 145—160 °C, конец реакции определяют по прекращению выделения НС1.
Из продукта реакции петролейным эфиром экстрагируют тример, частично сублимированный в более холодной части реакционной трубки, а также тетрамер при температуре кипения 50—70°C; высшие гомологи при этом будут находиться в не-растворившемся остатке. Растворитель выпаривают и (PNC12)3 отделяют от (PNC12)4 перегонкой в вакууме (на масляной бане при 140°С).
(PNC12)3 образует ромбические кристаллы. 7'Пл=114°С; Ткпп=127°С (при 1,7-103 Па) или 256 °C (при 105 Па). В 100 г эфира растворяется 46,4 г, в 100 г бензола — 55 г, в 100 г СС14 — 38,9 г, в 100 г петролейного эфира — 27,9 г тримера.
(PNChji образует тетрагональные кристаллы. ТПл= 123,5°C; Ткип=188°С (при 1,7-Ю3 Па) или 328,5°С (при 105 Па). В 100 г эфира растворяется 12,4 г, в 100 г бензола — 21,42 г, в 100 г СС14—16,6 г, в 100 г петролейного эфира — 8,4 г тетрамера.
Фосфорнитридхлориды обладают ароматичным запахом; вредно действуют на органы дыхания.
36* к
552 E.II. Практикум no препаративной химии
49.4. Синтезы в атмосфере сухого воздуха
85. Сульфат ванадила(1У), тригидрат VOSO4»3H2O
V2O5 + H2SO4 + S02 --► 2VOSO4 + H20
‘Пентаоксид ванадия обрабатывают при нагревании серной кислотой, разбавленной равным количеством воды, и смешивают со щавелевой кислотой до тех пор, пока закончится выделение СО2, a V(V) полностью' не восстановится до степени окисления (IV), и раствор приобретет темно-синий цвет; это можно осуществить также, пропуская через смесь газ-восстановитель— SO2 или H2S. После фильтрования раствор сильно упаривают и оставляют стоять на холоду, выделяются синие кристаллы; их отфильтровывают, промывают абсолютным спиртом и высушивают над силикагелем в вакууме. Кристаллы гигроскопичны, поэтому их хранят без доступа воздуха.
86. Иодид олова(1У) Snl4
Sn -|- 2I2 -► Snl4
В круглодонной колбе с притертой пробкой смешивают олово в виде порошка (1 весовая часть) с чистейшим CS2 (6 весовых частей) и добавляют постепенно 4 весовые части иода, открывая каждый раз на короткое время притертую пробку. При работе с большими количествами смеси необходимо при добавлении иода охлаждать колбу льдом. Образовавшийся бурокрасный раствор затем отсасывают без доступа влаги воздуха, отделяя от избытка олова, и выпаривают досуха в вакууме, создаваемом водоструйным насосом, с защитой от паров воды.
Оранжево-красные игольчатые гигроскопичные кристаллы. ТПл= 143,5 °C; 7'кип=340 °C.
87. Хлорид титанила(1У) ТЮС12
3TiCl4+As2O3 ---► 3TiOCl2 Ч- 2AsCl3
As2O3 смешивают с избыточным количеством TiCl4. Реакция протекает с энергичным выделением тепла и практически до конца, если удается предотвратить спекание твердого продукта. Получают вещество желтоватого цвета, которое можно очистить от избытка TiCl4 и образовавшегося AsCl3 отсасыванием без доступа влаги воздуха и тщательным промыванием абсолютно сухим СС14. Избыточное количество ССЦ отгоняют в вакууме при комнатной температуре. Полученный продукт обычно содержит следы мышьяка.
Бледно-желтый кристаллический гигроскопичный порошок.
88. Тетраацетат кремния Si(CH3COO)4
SiCl4 -ь 4(СНаСО)2О -Н Si(CH3COO)4 + 4CH3COCI
49. Техника работы
553
К смеси безводной уксусной кислоты с уксусным ангидридом добавляют тетрахлорид кремния в количестве несколько меньшем, чем необходимо по теоретическому расчету, и нагревают на водяной бане с обратным холодильником, снабженным хлоркальциевой трубкой. По окончании выделения хлороводорода смеси дают охладиться. Через несколько дней тетраацетат кремния выделяется из раствора в виде белых кристаллов. После охлаждения до О °C жидкость, находящуюся над кристаллами, сливают и кристаллы промывают чистым спиртом.
Очень гигроскопичен. Гпл=110°С; разлагается выше 170 °C; 7УЯП=148°С (при 8-102 Па).
89. Хлоросерная кислота HSO3C1
SO3 + НС1 --► HSO3C1
'Хорошо высушенную колбу, снабженную термометром и обратным холодильником, заполняют серной кислотой с возможно большим содержанием SO3. Отверстие холодильника закрывают хлоркальциевой трубкой. При комнатной температуре через трубку, доходящую до дна колбы, пропускают НС1 до тех пор, пока газ уже не будет поглощаться. Содержимое колбы перегоняют в токе НС1 без доступа влаги воздуха. Фракцию, полученную при 145—160 °C, подвергают фракционной перепонке и при 151—152 °C отгоняют хлоросерную кислоту.
Тпл=—80 °C; ТКИ„=152°С.
90. Иодид алюминия АП3
2А1 + 312 --> 2AlIg
3,5 г листового алюминия в колбе с обратным холодильником ^нагревают с раствором 20 г высушенного и возогнанного иода в 80 см3 CS2. По окончании реакции смесь фильтруют, из .фильтрата отгоняют на водяной бане основное количество С5г-.{Осторожно! Не применять открытого пламени горелки!) Концентрат затем охлаждают, при этом выделяется кристаллический АП3. Кристаллы отфильтровывают, промывают гексаном и высушивают при 100 °C. Продукт окрашен в слабый желтобурый цвет.
Кристаллы в форме листочков; препарат чувствителен к действию влаги. При нагревании на воздухе разлагается с выделением 12 и AI2O3. Растворяется в CS2, спирте и эфире. Гпл= 191 °C; ГКЯп=382°С.
91. Гексафторофосфат(У) тетрахлорофосфония [РСЦ][РР6]
[РС14] [РС16] + 2AsF3 -> [РС14] [PF6] + 2AsCl3
Для получения препарата необходима следующая аппаратура: трехгорлая колба, снабженная мешалкой типа KPG и капель-
554
ЕЛ. Практикум по препаративной химии
„ой воронкой, а также осушительная трубка, заполненная Р4О10. 25 г PCI5 растворяют в трехгорлой колбе в 150 см3 AsCl3. К раствору при перемешивании медленно добавляют по каплям 10 см3 AsF3, при этом [РСЦ] [PF6] выпадает в осадок. При разогревании колбы необходимо охлаждать ее снаружи водой. Появление густого тумана PF5 указывает на окончание реакции:
3[РСЦ] [PF,] + 4AsF3 -► 6PF5 + 4AsCl3
Для полноты осаждения '[РСЦ] [PFg] колбу охлаждают льдом. Продукт отсасывают через трубку Шленка с пористой фильтрующей пластинкой, подсоединенную к колбе через изогнутый отвод. Между насосом и отсасывающим устройством следует подключить охлаждаемую ловушку. Препарат, представляющий собой белый порошок, высушивают в вакууме.
92. Гексафтороарсенат(У) тетрахлороарсония [АвСЦ] [AsF6]
2AsF3 + 2СЦ ► [AsC14] [AsF6]
Через смесь 20 см3 AsF3 с 200 см3 AsCl3, находящуюся в охлаждаемой льдом колбе, пропускают хлор. AsCl3 служит для образования суспензии и переноса хлора. Газообразные продукты, выделяющиеся в ходе реакции, необходимо удалять, пропуская их сначала через подсоединенную к колбе осушительную трубку, заполненную Р4Ою (под тягой!). Продукт отделяют фильтрованием без доступа влаги воздуха.
93. Триметиларсенат(У) AsO(OCH3)3
AsCl3 + ЗСН3ОН + 3NH3 --г- As(OCH3)3 + 3NH4C1
As(OCH3)3 + Br2 -► {AsBr2(OCH3)3}
{AsBr2(OCH3)3} 4- As(OCH3)3 {AsBr(OCH3)4} + AsBr(OCH3)2 {AsBr(OCH3)4} ----------► AsO(OCH3)3 + CH3Br
В колбу вместимостью 5 дм3 вносят 3 дм3 абсолютного метанола и 600 г AsCl3. Затем в раствор через широкую трубку пропускают NH3 до насыщения (в течение 10—12 ч). Раствор охлаждают льдом из-за сильного разогревания смеси в ходе реакции. Выпавший в осадок NH4C1 отсасывают через фильтр с пластинкой из пористого стекла (типа G4) без доступа влаги воздуха (под тягой!). Фильтрат, содержащий эфир, переносят в трехгорлую колбу. Избыток метанола отгоняют на длинной колонке с насадкой из колец Рашига (под тягой), нагревая колбу на плитке. Из-за образования осадка NH4C1 кипение задерживается, поэтому в процессе перегонки нужно непрерывно перемешивать раствор с помощью мешалки (типа KPG). Из раствора, находящегося в колбе для перегонки, при охлаждении выпадает в осадок еще некоторое количество NH4CI.
49. Техника работы
555
Отфильтрованный раствор затем еще раз перегоняют на колонке длиной 20 см с елочной насадкой.
Температура кипения триметилового эфира мышьяковистой кислоты 120—125 °C.
К As(OCH3)3 при перемешивании и охлаждении льдом добавляют по каплям бром до появления красного окрашивания раствора. Бромированный эфир оставляют стоять на ночь. Затем его подвергают вакуум-перегонке на длинной колонке с елочной насадкой. Перед вакуум-насосом подсоединяют ловушку, заполненную смесью сухого льда с этанолом. Нагревание проводят на масляной бане с силиконовым маслом. Триметиловый эфир мышьяковой кислоты перегоняется при 75 °C и (4— 6)-102 Па.
94. Литийалюминийгидрид ЫА1Н4
4LiH + A!Ct3 —* LiAlH4 + 3LiCl
2.7 г безводного А1С13 смешивают с 4 г тонкоизмельченного LiH, взятого в шестикратном избытке (препарат 170), в атмосфере сухого азота в небольшой круглодонной колбе со шлифом. При охлаждении жидким воздухом на поверхности смеси конденсируется 15 см3 эфира (безводного и не содержащего пероксидов), применяемого в- качестве среды. После извлечения колбы из охлаждающего сосуда начинается реакция, в ходе которой происходит постепенное разогревание смеси. Скорость реакции можно регулировать, время от времени охлаждая колбу. Через несколько минут реакция заканчивается; нерастворимый в эфире LiCl отфильтровывают. Эфир, содержащийся в фильтрате, удаляют при атмосферном давлении; получается густой сиропообразный раствор, из которого остаточное количество эфира можно удалить длительным выдерживанием в вакууме при нагревании до 70° С. Остаток представляет собой LiAlH4 в виде белого порошка.
При получении больших количеств продукта, чтобы избежать бурного начала реакции, вводят в реакционную смесь небольшое количество LiAlH4, полученного по описанному выше методу (можно в виде эфирного раствора). Благодаря этому вначале очень интенсивно протекающая реакция замедляется.
Литийалюминийгидрид — энергичный гидрирующий агент относится к важнейшим вспомогательным веществам, применяемым в синтезе органических и неорганических препаратов. Например, хлорид бора и тетрахлорид кремния количественно реагируют с LiAlHi с образованием диборана или соответственно моносилана:
4ВС1а -ф 3LiAlH4 -► 2(ВН3)а + 3LiAICl4
SiCl4 + LiAlH4 --->- SiH4-J-LiAlCl4
556 E.II. Практикум no препаративной химии
49.5 . Препаративные синтезы в неводных растворителях
Неводные растворители могут быть использованы прежде всего для получения безводных соединений, особенно таких, которые при обезвоживании склонны вступать в реакции гидролиза или разложения. Особым классом неводных сред, в которых можно проводить химические реакции, являются расплавы солей.
95. Тетрабромоантимонат(Ш) тетраэтил аммония [N(C2H5)4] [SbBr4]
SbBr3+ [Ь1(С2Нв)4]Вг -> [N(C2Hb)4] [SbBr4]
Препарат получают при взаимодействии эквимолекулярных количеств растворов' SbBr3 и [N(C2Hs)4]Br в ацетонитриле.
Ацетонитрил, необходимый для синтеза, должен быть тщательно осушен. Для этого его подвергают фракционной перегонке и фракцию, кипящую при температуре 80,5—81,5 “0, кипятят с обратным холодильником в течение нескольких часов над СаН2. После повторной фракционной перегонки с обратным холодильником ацетонитрил кипятят над Р4Ою. Эту операцию и кипячение над СаН2 можно повторить. Осушенный CHaCN хранят над СаН2.
44 г SbBr3 в трехгорлой колбе растворяют в 25 см3 ацетонитрила. К раствору при энергичном перемешивании добавляют из капельной воронки, укрепленной в горле колбы, раствор, содержащий 25,5 г [N (С2Н5)4]Вг в 200 см3 CH3CN. При этом образуется желтый осадок. Раствор с осадком перемешивают еще в течение нескольких часов, вначале при небольшом нагревании на водяной бане, а затем без нагревания. При добавлении еще 50 см3 CH3GN почти весь осадок растворяется; раствор фильтруют и отбрасывают нерастворившийся остаток. При добавлении к прозрачному фильтрату обезвоженного эфира выпадает мелкокристаллический, пастельно-желтый осадок. Кристаллы отсасывают и высушивают в вакууме. Образовавшийся продукт переосаждают еще два раза, как описано выше, и затем нагревают в течение нескольких часов при ~40°С в вакууме, создаваемом масляным насосом, для удаления находящегося на веществе растворителя.
Мелкие кремово-желтые кристаллы, малогигроскопнчны; в воде легко гидролизуются с образованием SbOBr.
96. Хлорид меди(П), безводный СиС12
Си(СН3СОО}2+ 2СН3СОС1 ---► СиС12 4-.2(СН3СО)2О
В колбу аппарата Сокслета помещают безводную уксусную кислоту, к которой добавлено небольшое количество уксусного ангидрида. После заполнения экстрактора медной стружкой в него впускают воздух и кипятят растворитель. Через 1—2 ч образуется раствор, насыщенный ацетатом меди. Полученный
49. Техника работы
557
раствор охлаждают до 35 °C, сливают раствор с осадка путем декантации и осаждают препарат действием рассчитанного количества ацетилхлорида при 40—50 °C. Расчет можно проводить, исходя из того, что растворимость ацетата меди(II) в уксусной кислоте при 35 °C составляет 20 г/дм?. Осадок промывают горячей безводной уксусной кислотой или холодным уксусным ангидридом'. В конце промывную жидкость заменяют безводным эфиром. Осадок высушивают при 120 °C.
Желтая расплывающаяся масса. Тпл=630°С; ТКИП=655°С.
97. Нитрат меди(П) Си(МОз)2
Cu + 2N2O4 -»- Cu(NO8)2 + 2NO
10 г тонкой медной ленты, поверхность которой должна быть полностью очищена от оксида, растворяют в смеси 20 см3 жидкого N2O4 с 20 см3 осушенного этилацетата. Синтез проводят в закрытой с одного конца стеклянной трубке, на другой конец которой насажена осушительная! трубка, заполненная Р4Ою. При 16 °C реакция заканчивается через 4 ч. При дальнейшем добавлении N2O4 в осадок полностью выпадает Cu(NO3)2-N2O4 в виде сине-зеленых мелких кристаллов, которые отфильтровывают и промывают N2O4. Для удаления присоединившегося N2O4 соль медленно нагревают до 120 °C в стеклянной трубке при разрежении (на масляной бане). Безводная соль образуется в виде синего аморфного порошка. Возгонкой в вакууме при 200 °C с холодильником типа «охлаждающий палец» выделяют Cu(NO3)2 в виде синих иглообразных кристаллов.
Хлорирование металлов
98. Хлорид кобальта(П) СоС12
В колбе со шлифом вместимостью 300 см3, снабженной длинным обратным холодильником (с хлоркальциевой трубкой), мешалкой и газоподводящей трубкой, при перемешивании готовят суспензию 5,8 г порошкообразного кобальта в 60 см3 безводного этанола. Через суспензию пропускают хлор. Если тепла реакции недостаточно, чтобы нагреть этанол до кипения, колбу нагревают на водяной бане. Продолжительность реакции 140 мин. После фильтрования этанольный раствор ставят в эксикатор над конц. H2SO4. При этом образуются светло-синие кристаллы продукта присоединения СоС12-2С2Н5ОН, которые перекристаллизовывают в атмосфере сухого азота из абсолютного этанола. Аддукт разрушают медленным нагреванием до 100 °C в вакууме в течение приблизительно 4 ч; при этом образуется синий порошкообразный СоС12. Выход препарата 84%.
.558
ЕЛ. Практикум по препаративной химии
Очень чувствителен к действию влаги, при поглощении воды светло-голубая окраска переходит в темно-розовую. ТПл=740°С; возгоняется при 1053 °C.
Аналогично могут быть получены NiCh, МпС12, СгС13 (через аддукты с эфиром).
99. Комплексные фторидные соли аммония
Комплексные фторидные соли аммония могут быть получены реакцией обмена бромидов металлов с фторидом аммония в среде метанола. Для этого готовят суспензию 0,1 моль порошкообразного металла в 100 см3 абсолютного метанола и порциями по 1 см3 прикапывают к ней избыточное количество брома (при получении FeBr2 добавлять стехиометрическое количество брома!). Реакционную колбу, которую снабжают капельной воронкой, мешалкой типа KPG и хлоркальциевой трубкой, в ходе бромирования необходимо охлаждать. Раствор фильтруют и добавляют к фильтрату, помещенному в полиэтиленовый сосуд, тщательно перемешанный насыщенный раствор фторида аммония примерно в трехкратном по отношению к фильтрату объеме. Образующийся продукт отфильтровывают и промывают на фильтре холодным метанолом до исчезновения в фильтрате бромид-ионов (проба с раствором AgNOs). Препарат окончательно промывают эфиром и высушивают при 60 °C.
NHJZnFs], белый; NHJCdF.aJ, белый; NH^CoFaJ, розовый; NH4[CuF3], светло-синий; NH4[MnF3], светло-розовый; (N Н4) s[FeFl8], красновато-желтый; NH4[BiF4), белый; (NH4)2[NiF4], желтый; (NH4) з[А1Р6], белый; (NH4)3[InFeJ, белый; (NH4)a[GeFeL белый; (NH4)2[SnFe], белый; (NH4)2[TiFe], белый: (NH4) slZrFv], белый.
49.6. Реакции в твердой фазе и в расплавах
100. Иодид кадмия — люминофор, активированный свинцом Cdl2: РЬ
Cdl2 смешивают с 10 масс. % РЬ12, растирая вещества в ступке до порошкообразного состояния, и в течение короткого промежутка времени нагревают до 150—200 °C. Под действием коротковолнового ультрафиолетового излучения образующиеся смешанные кристаллы (Cd, Pb)I2 испускают золотисто-желтое свечение (эмиссия наблюдается в основном в области длин волн 570—635 нм).
101. Диамминотетрароданохромат(Ш) аммония, моногидрат (соль Рейнеке) NH4[Cr(SCN)4(NH3)2] • Н2О
34 г тонкоизмельченного (NH4)2Cr2O7 вносят в 200 г расплавленного в фарфоровой чашке (нагревать осторожно!) NH4SCN при медленном перемешивании. Твердый продукт реакции после охлаждения растирают в порошок и несколько раз выщелачивают небольшим количеством холодной воды. Остаток со
49. Техника работы
559
держит соль Рейнеке, которую для отделения от примеси соли Морланда выщелачивают водой, нагретой до 50 °C. Соль Морланда, представляющая собой гуанидиновую соль кислоты Рейнеке (диамминотетрароданохромат(Ш)гуанидиния), остается на фильтре.
Рубииово-красиые блестящие светочувствительные кристаллы в форме листочков. Применяется в качестве реактива для количественного определения меди.
102. Натриево-вольфрамовая бронза Na^WO3
Сплавляют 60—80 г смеси Na2WO4 с WO3 (в молярном соотношении 2:1). К расплаву постепенно добавляют 30 г оловянной фольги и смесь оставляют в расплавленном состоянии на 1—2 ч. Затем для обеспечения хорошей кристаллизации получаемой бронзы очень медленно охлаждают тигель с расплавом. Охлажденный сплав измельчают и кипятят в фарфоровой чашке попеременно с гидроксидом натрия и соляной кислотой. Бронза образуется в виде золотисто-желтых или красно-желтых кубиков с металлическим блеском, максимальная длина ребер которых достигает 5 мм.
Вольфрамовые бронзы представляют собой интенсивно окрашенные чрезвычайно реакционноспособные вещества, обладающие хорошей электропроводностью.
Натриево-вольфрамовые бронзы Nax\VO3, где х колеблется в пределах 0,95—0,30, кристаллизуются с образованием кубической решетки по типу пе-ровксита. Они получаются из NaWO3 при уменьшении в нем числа ионов натрия, при котором происходит переход соответствующего числа ионов W(V) в степень окисления(VI) (для выравнивания заряда). Цвет бронз изменяется от золотисто-желтого (х=0,93) через оранжево-красный (х=0,64) и красно-фиолетовый (х=0,46) до темно-фиолетового (х=0,32).
103. Тиоантимонат натрия, наногидрат (соль Шлиппе) Na3SbS4-9H2O
Тщательно перемешанную смесь 36 г Sb2S3, 43 г безводного Na2SO4 и 16 г просеянного порошка древесного угля вносят в глиняный тигель, заполняя его наполовину, покрывают смесь небольшим количеством порошкообразного угля, нагревают до начала спокойного плавления и затем еще в течение 10 мин. Расплав выливают на железную пластинку и после охлаждения измельчают в порошок. В течение получаса порошок кипятят с 7 г серного цвета и 300 cmi3 воды. Фильтрат после добавления небольшого количества NaOH упаривают в фарфоровой чашке до начала кристаллизации. Перекристаллизацию проводят из воды, содержащей небольшое количество NaOH. Препарат высушивают в вакуум-эксикаторе над СаО, смоченном несколькими каплями раствора сульфида аммония.
Светло-желтые легко выветривающиеся иа воздухе кристаллы в форме тетраэдров.
560
E.II. Практикум no препаративной химии
104. Оксихлорид железа(111) FeOCl
FeCl3-6H2O +5FeCl3 -► 6FeOCl + 12HC1
10 г РеС1з-6Н2О сплавляют на водяной бане в короткогорлой круглодонной колбе из йенского стекла с 35 г возогнанного FeCl3. Реакция, уравнение которой приведено выше, идет при нагревании массы до 250 °C и не выше 300 °C. Лучше всего открытую' колбу погрузить довольно глубоко в масляную баню, нагретую до 250 °C так, чтобы влага не могла конденсироваться в горле колбы. Через 60—80 мин реакция заканчивается, что можно заметить по прекращению выделения НС1. Массу, превратившуюся в твердую красную лепешку, после охлаждения растирают в порошок; чтобы очистить ее от избытка FeCl3, в течение короткого времени промывают значительным количеством холодной воды, потом ацетоном и наконец высушивают в вакууме.
Небольшие красные иглы. Если в качестве исходного материала был взят возогнанный FeCl3 и реакционную массу не нагревали до высокой температуры, то препарат не содержит Fe2O3. Выше 300°C происходит реакция диспропорционирования с образованием оксида и хлорида.
105. Феррат(У1) бария, моногидрат BaFeO4-H2O
В колбочке из тугоплавкого стекла -нагревают сухую смесь 10 г порошка железа с 20 г KNO3 до окончания реакции. (Осторожно! Синтез вести в закрытом вытяжном шкафу!) Затем смесь охлаждают, обрабатывают 50 см3 воды (лучше ледяной), быстро отфильтровывают через стеклянный нутч-фильтр и тотчас же осаждают раствором ВаС12. После долгого стояния отфильтровывают осадок через плотный фильтр. Осадок промывают водой, спиртом и эфиром и высушивают в вакуум-эксикаторе.
Красный нестойкий кристаллический порошок.
106. Фосфат бора ВРО4
Н3ВО3 + (NH4)2HPO4 -► ВРО4 + 2NH3 + ЗН2О
В фарфоровом тигле смешивают кристаллическую Н3ВО3 с (NH4)2HPO4 в таком соотношении, чтобы Н3ВО3 была в избытке. Осторожным нагреванием удаляют воду и NH3 Затем вещество выдерживают в электрической печи при 700—800 °C в течение 5 сут. После этого образуются кристаллы двойного оксида размером- до 30 мкм. Двойной оксид получают в виде мелкокристаллического белого порошка путем обработки большим количеством кипящей воды из охлажденного расплава. Под микроскопом можно рассмотреть отдельные кристаллы, по Форме похожие на тетраэдры.
49. Техника работы
561
107. Хлорид лантана(Ш) LaCI3
La2O3 + 6NH4C1 -► 2LaCl3+6NH3+ЗН2О
10 r La2O3 смешивают с 20 г чистого МН4С1, мелко растирая их в ступке. Смесь осторожно нагревают при длительном помешивании в фарфоровой чашке на пламени горелки Бунзена. Реакция протекает несколько часов, о ее окончании можно судить по полному растворению продукта реакции в воде. Для удаления избытка NH4C1 реакционную смесь вносят в закрытую с одного конца стеклянную трубку длиной около 30 см. При разрежении трубку со смесью медленно нагревают в электрической печи до 300 °C, так чтобы1 NH4CI конденсировался в той части трубки со шлифом, которая выходит из печи. Такую термическую обработку надо проводить в течение нескольких часов, после чего чистый LaCl3 (при осторожной работе препарат не содержит оксихлорида) можно охладить в вакууме.
Белый очень гигроскопичный порошок.
108. Фторид бора BF3
6K[BF4J + В2О3 + 6H2SO4 -► 8BF3 + 6KHSO4 + 3H2O
Смесь 80 г высушенного или, лучше, плавленого K[BF4] и 30 г В2О3 медленно .нагревают в открытой с одной стороны, наклонно поставленной железной трубке длиной 40 см и диаметром 3 см. Железная трубка закрыта фланцем^ (с медным кольцом в качестве уплотнения), через отверстие которого проходит другая железная трубка диаметром около 10 мм. К этой трубке присоединена осушительная трубка, заполненная стеклянной ватой для защиты от пыли. За этой трубкой помещают стеклянную или кварцевую газовую ловушку, погруженную в жидкий кислород; она служит сосудом для конденсации. В конце установки находится осушительная трубка, наполненная свежеобезвоженным KF. Полученный BF3 можно очистить многократной фракционной перегонкой.
Бесцветный газ с удушливым запахом; дымят во влажном воздухе. Тпл=—128 °C; 7Уип~—101 °C.
49.7. Препаративные синтезы с использованием газов
109. Рубеановодородная кислота C2H4N2S2
Дициан (CN)2 реагирует с сероводородом с образованием ру-беановодородной кислоты (C2H4N2S2):
S=C—NH, Н—S— C=NH
(CN)2 + 2H2S = I « I
s=c—NH2 H—S—c=nh
В концентрированный раствор CUSO4 пропускают NH3 до рас
562 E.II. Практикум no препаративной химии
творения образовавшегося вначале Си(ОН)2. Затем на холоду при перемешивании к раствору добавляют KCN до исчезновения синей окраски. Следует избегать избытка KCN. Если образуется осадок, то его отделяют фильтрованием и через фильтрат пропускают сильную струю H2S. Раствор сначала окрашивается в желтый цвет, и затем сразу же выделяются оранжево-желтые кристаллы* рубеановодородной кислоты. В процессе осаждения реакционный сосуд следует хорошо охлаждать. Кристаллический осадок быстро отсасывают, промывают водой и перекристаллизовывают из спирта или из уксусной кислоты.
Плохо растворяется в воде, растворяется в спирте. 74.4=200 °C (плавится с разложением).
110. Хлорид ванадия(1П), гексагидрат, ¥С1з«6Н2О
V2O3 + 6НС1 --► 2VCI3 4- ЗН2О
7,5 г V2O3, полученного при восстановлении V2O5 водородом, растворяют в 200 см3 конц. НС1 при длительном кипячении. Упаривают полученный раствор до 50 см3, охлаждают до температуры —104—20 °C и насыщают его газообразным HCL Выпадающий в осадок УС1д-6Н2О отсасывают на фильтре с пластинкой из пористого стекла, растворяют еще раз в небольшом количестве воды и при охлаждении снова осаждают действием газообразного НС1.
Зеленые гигроскопичные кристаллы.
111. Хлорит натрия, тригидрат NaC102*3H20
2КС1О3 + 2H2SO4 4- Н2С2О4 --> 2С1О2 4- 2СО2 4- 2Н2О 4- 2KHSO4
2С1О24-Ва(ОН)24-Н2О2 -----> Ва(С1О2)2 4-2Н2О 4-О2
Ва(С1О2)2 4- Na2SO4 -> 2NaC102 4- BaSO4
В колбе со шлифом вместимостью 1 дм3 смешивают 24,5 г КС1О3 с 20 г щавелевой кислоты, 21,6 г H2SO4 (11,8 см3 H2SO4 пл. 1,84) и 80 см3 Н2О2. Реакционную смесь нагревают на водяной бане, улавливая выделяющиеся СЮ2 и СО2 в 200 см3 воды, находящейся в охлаждаемой льдом* колбе Эрленмейера.. Желто-оранжевый раствор встряхивают до обесцвечивания с 40 г твердого Ва(ОН)2-8Н2О и 12 г 30%-ного Н2О2. Образовавшийся ВаСО3 отсасывают, кипящий фильтрат смешивают с твердым Na2SO4 для осаждения Ва2+ в виде BaSO4. Осадок BaSO4 отфильтровывают, раствор упаривают на водяной бане до образования кристаллов NaC102-3H20.
Белые кристаллы в форме листочков. Кристаллы, обезвоженные над КОН в эксикаторе, взрываются при ударе.
112. Хромат(У) бария Ва3(СгО4)2
2ВаСгО4 4- ВаСО3 -> Ва3(СгО4)2 4- СО2 4- 72О2
49. Техника работы
563-
Тщательно смешивают 1 моль ВаСгО4 и 0,5 моль ВаСО3 и нагревают смесь до 1000 °C в потоке азота, не содержащего кислород. Смесь массой приблизительно 2 г достаточно нагревать в течение 4 ч; при этом получают Ва3(СгО4)2 высокой чистоты.
Темно-зеленый микрокристаллический порошок. Под действием воды постепенно разлагается. Хорошо растворим в разбавленных кислотах (при этомв происходит реакция диспропорционирования с образованием Сг(Ш) и Cr(VI)).
113. Дигидрофосфинит бария, моногидрат Ва(Н2РО2)2-Н2О
8Р + ЗВа(ОН)2 + 6Н2О -► ЗВа(Н2РО2)2 + 2РН3
120 г кристаллического Ва(ОН)2 растворяют в 1,2 дм3 Н2О и нагревают в течение 4 ч с 30 г белого фосфора в круглодонной колбе, снабженной длинной стеклянной трубкой, конец которой направлен в вытяжной шкаф для отвода образующейся самовоспламеняющейся смеси фосфинов. После полного растворения фосфора в раствор пропускают СО2 для удаления избыточного количества Ва(ОН)2. Осадок отфильтровывают и промывают горячей водой. Раствор и промывные воды упаривают до половины объема, фильтруют еще раз и продолжают упаривать до начала кристаллизации. После добавления небольшого количества спирта раствору дают остыть. Выделившиеся кристаллы отсасывают, а маточный раствор снова упаривают до кристаллизации. Объединяют обе порции продукта, и перекристаллизовывают его из горячей воды.
Бесцветные кристаллы в форме призм с перламутровым блеском. Нерастворимы в спирте, легко растворимы в воде: в 100 г холодной воды растворяется 28,6 г, а в 100 г кипящей — 33,3 г соли.
114. Хлорид хрома(Ш), безводный СгС13
2Сг + ЗС12 -► 2СгС13
Измельченный до крупнозернистого порошка металлический хром (10—20 г) помещают в фарфоровую или кварцевую (можно из плавленого кварца) трубку длиной 50 см и внутренним диаметром 3 см, которую нагревают в горизонтальном положении пламенем паяльной горелки. Важно, чтобы трубка предварительно была очищена от следов воздуха пропусканием через нее сильного потока совершенно сухого хлора (в течение по меньшей мере 0,5 ч). Только после этого повышают температуру, насколько возможно, дают охладиться в течение 1—2 ч и затем вытесняют хлор сухим СО2. В трубке образуется СгС13 в виде блестящих фиолетовых листочков. Для предотвращения закупорки трубки в результате сильного увеличения объема вещества необходимо исходный хром распределить по всей длине трубки.
564
E.II. Практикум no препаративной химии
115. Бромид мышьяка(Ш) AsBr3
2As + 3Br2 -► 2AsBr3
В трубку из йенского стекла помещают лодочку с порошкообразным мышьяком. Через трубку пропускают поток сухого азота, который был насыщен парами брома при пропускании через промывную склянку с бромом. Трубка должна быть (наклонена к приемнику и соединена с ним форштоссом, уплотненным в трубке асбестовой бумагой. Трубку нагревают до тех пор, пока не начнется реакция. Продукт реакции перегоняют из приемника над порошкообразным мышьяком.
Бесцветные расплывающиеся кристаллы в форме ромбических призм; гидролизуются водой. ГПЛ=31,2°С; Т’Кип=221 °C.
116. Бромид кремния(1У) SiBr4
Si 2Br2 ----► SiBr4
Через кварцевую трубку с кремнием пропускают поток СОг, который был насыщен парами брома при пропускании через промывную склянку с бромом. Кремний, находящийся в фарфоровой лодочке, нагревают до красного каления. Если используют так называемый аморфный кремний, оптимальная температура нагрева составляет 650°C. Пары образующегося SiBr4 конденсируют в приемнике, охлаждая его смесью льда с поваренной солью. Скорость подачи брома регулируют таким образом, чтобы SiBr4, конденсирующийся в приемнике, не был загрязнен свободным бромом. Присутствующий все же избыток брома удаляют перегонкой, а его остаточные следы — добавлением порошкообразной меди. Продукт подвергают фракционной перегонке без доступа влаги воздуха, отдельно собирают фракцию с 7'Кип=153°С и помещают ее в склянку для хранения препаратов.
Прозрачная, бесцветная жидкость.
117. Сульфурилхлорид SO2C12
SO2 + C12 --► SO2C12
Реакцию проводят в шариковом холодильнике; часть объема шариков неплотно заполняют сухим активированным углем. Пробками из стеклянной ваты предотвращают высыпание угля. Холодильник укрепляют на склянке для отсасывания, отводная трубка которой закрыта хлоркальциевой трубкой. Оба газа (SO2 и С12) высушивают, пропуская через серную кислоту, и смешивают их через тройник, подсоединенный к верхней части холодильника. В обратный холодильник подают охлаждающую воду и пропускают через него сильный поток газов. В результате энергичной реакции образуется SO2C12, капли
49. Техника работы
565
которого после насыщения активированного угля стекают в склянку для отсасывания. Для очистки от избытка хлора препарат встряхивают с Hg. Затем его перегоняют и собирают фракцию, отгоняющуюся при 68—70 °C.
118. Нитрозилхлорид NOC1
2NO+ С12 --> 2NOC1
Для синтеза нитрозилхлорида полностью осушенные и чистые газы (NO и С12) смешивают в Т-образной трубке, к которой подсоединяют длинную стеклянную трубку, заполненную зер-неным активированным углем (рис. Е.36). Предварительно из
Рис. Е.36. Схема установки для каталитического синтеза NOC1 или СОС12.
угля полностью удаляют адсорбированную воду, нагревая его в потоке азота с последующим охлаждением в вакууме. Устанавливают температуру 40—50 °C, охлаждая стеклянную трубку в печи с терморегулятором; повышение или понижение температуры значительно ухудшает выход продукта. Оба газа вводят медленно; образовавшийся нитрозилхлорид сжижают в U-образной трубке, охлаждаемой смесью льда с поваренной солью, или в другом подходящем для конденсации сосуде. При сжижении с применением охлаждающей смеси не конденсируются такие примеси, как воздух, диоксид углерода или не вступившие в реакцию исходные газы.
Газ желто-красного цвета; реагирует е веществами, применяемыми для смазки кранов. Гкип=—6,5°С. Жидкий NOCI сильно поражает кожу.
119. Хлорид селенила(1¥) SeOCl2
SeCl44-SeO2 --> 2SeOCl2
Через суспензию селена в СС14 пропускают газообразный хлор. Сначала С12 растворяется в ССЦ, но затем образуется Se2Cl2, который, в свою очередь, хорошо растворяет селен, существенно повышая тем самым скорость реакции. При дальнейшем хлорировании образуется малорастворимый хлорид селена (IV), который выделяется в виде белого твердого вещества. В суспензию хлорида селена(IV) в СС14 вводят рассчитанное коли
566
E.II. Практикум no препаративной химии
чество SeO2. Образующийся при этом SeOCl2 растворяется в СС14, и его легко можно выделить вакуумной перегонкой.
Дымящая на влажном воздухе гигроскопичная жидкость соломенно-желтого цвета. 7Ул=110С; ГКИп=179°С (кипит с частичным разложением).
120. Дисерадибромид S2Br2
S„C 12 + 2НВг -► S,Br, + 2НС1
Реакцию обмена проводят в приборе, схема которого изображена на рис. Е.37. При добавлении по каплям Вг2 к тетрагидронафталину образуется НВг (препарат 129). Для удаления следов элементарного брома полученную кислоту пропускают последовательно через промывную склянку с тетрагидронафта-
Рис. Е.37. Схема установки для получения БзВгг.
/ — капельная воронка с Вг2; 2 — склянка с тетрагидронафталином; 3 — LJ-образная трубка с красным фосфором; 4 — хлоркальцневая трубка; 5 — двухгорлая колба с S2CI2; о — отвод газа в тягу.
лином, U-образную трубку, заполненную глиняными черепками и влажным красным- фосфором, и две хлоркальциевые трубки (для высушивания-). Газ, подготовленный таким образом, пропускают при комнатной температуре в двухгорлую кол-бу вместимостью 250 см-3, в которой находится около 30 г S2C12 (препарат 63), так, чтобы он хорошо перемешивал содержимое колбы. О ходе обменной реакции, протекающей с небольшим выделением тепла, можно судить по постепенному появлению темно-красного окрашивания маслянистой жидкости. Реакция заканчивается через 1—2 ч, что определяют по качественной пробе на содержание хлора в продукте. В процессе реакции избыток НВг и образовавшийся НС1 через хлоркальциевую трубку направляют в вытяжной шкаф.
Темно-красная маслянистая, не смачивающая стенок стеклянных сосудов, жидкость. ГпЛ=—46 °C. Диссоциирует при нагревании на элементы, поэтому может быть перегнана без разложения только в глубоком вакууме. Разлагается при действии воды на НВт, SO2 и S. Растворяется в CS2, СС14 и СвН
4д. Техника работы
567
121. Трихлорсилан SiHCU
Si + 3HCI --* SiHCI3 + H2
Фарфоровую лодочку заполняют тонкоизмельченным кремнием, очищенным кипячением с НС1 и разбавленной HF, и смешанным с ~10% СнС12. Лодочку ставят в стеклянную трубку из йенского стекла, один конец которой соединен с форштос-сом и холодильником, а другой — с трубкой для подвода газа. Конец холодильника должен доходить до середины приемника. Установку предварительно тщательно прогревают. Хлористый водород получают по реакции между NaCl и H2SOi без добавления соляной кислоты, так как газ должен быть абсолютно сухим. Устанавливают в печи, в которую помещают трубку с лодочкой, температуру 300 °C и медленно пропускают через трубку поток хлороводорода. Приемник охлаждают смесью ацетона с сухим льдом. Сухой продукт перегоняют непосредственно из приемника, причем сначала отгоняется растворенный НС1, a SiHCl3 перегоняется при 36,5 °C. Тщательной фракционной перегонкой первого погона можно получить SiH2Cl2. Выход SiHCl3 составляет ~50%. Для увеличения выхода SiH2Cl2 рекомендуют добавлять водород к хлороводороду (в соотношении Н2: НС] = 4 : 1).
Прозрачная как вода, подвижная горючая жидкость (синоним — силико-хлороформ). Очень летуча; гидролизуется водой; дымит на воздухе. Тпл = = —!34°С; 7’1;ИП=35,5°С.
49.8. Получение коллоидных систем
В принципе любые вещества могут быть получены в виде коллоидных систем. При этом различают два основных препаративных метода: конденсацию, при которой молекулы растворенных веществ объединяются в коллоидные частицы, и дисперсию, при которой грубодисперсные материалы раздробляются до коллоидных частиц.
49.8.1. Конденсационные методы
122. Золь серебра
К раствору 0,2—1 см3 0,1 М нитрата серебра в 500 см3 воды по каплям прибавляют ~1 см'3 разбавленного раствора гидрата гидразина. Раствор окрашивается в зеленовато-серый цвет и обычно быстро становится мутным.
123. Золь серы
Раствор, содержащий 50 г Na2S2O3-7H2O в 30см3 Н2О, по каплям добавляют к 70 г конц. H2SO4, охлаждая кислоту льдом (под тягой!). Выделяется Н2О и SO2 и образуется вязкая реакционная масса, к которой добавляют 30 см3 Н2О. Смесь на
568
Е.П. Практикум no препаративной химии
гревают в течение 10 мин при 80 °C. Образовавшуюся прозрачную желтую жидкость фильтруют через стеклянную вату и оставляют до охлаждения; при этом снова выделяется твердая сера. Остаток, который можно дополнительно очистить центрифугированием, применяют для приготовления золя серы с частицами размером 10 4 см. Для этого разбавляют небольшие количества остатка большим объемом воды и получают прозрачную, очень устойчивую систему. При удалении избытка солей (особенно Na2SO4) диализом постепенно выделяется сера. 124. Золь As2S3
Кипятят в воде в течение 0,5 ч тонкоизмельченный порошок As2O3 с последующим отфильтровыванием остатка. 20 см3 полученного почти насыщенного раствора H3AsO3 разбавляют водой до 100 см3 и пропускают через раствор H2S до насыщения. Образующийся прозрачный желтый коллоидный раствор тонкодиснерсного As2S3 устойчив при нагревании, но очень чувствителен к присутствию электролитов.
При растворении новых порций As2O3 в коллоидном растворе и повторном пропускании через него H2S можно получить золь, содержащий свыше 35% As2S3.
49.8.2. Дисперсионные методы
125. Золь меди с защитным коллоидом
Смешивают 2 см3 1 %-ного раствора CuSO4, 2 см3 5%-шого раствора NH4OH, 1—4 см3 0,4%-ного водного раствора гуммиарабика, 100 см3 воды и 2 см3 1 %-ного раствора гидрата гидразина. Смесь осторожно нагревают почти до кипения в колбе Эрленмейера, которую ставят на проволочную сетку. Следует избегать встряхивания раствора. Через 1 —10 ми.н раствор приобретает слабую красную окраску, которая затем быстро становится интенсивной. При правильном ходе синтеза получают ярко-красный прозрачный золь меди. Если получается коллоид зеленого цвета или мутный со смешанной окраской, это означает, что гидрат гидразина уже не оказывает необходимого действия.
126 Золь V2O5
1 г NH4VO3 растирают в ступке с небольшим количеством воды и прибавляют при перемешивании пестиком 10 см3 2 М раствора НС1. Образовавшийся красный осадок V2O5 переносят на фильтр, используя находящуюся вад осадком жидкость; дают жидкости стечь и промывают осадок водой. Прозрачный и желтый вначале фильтрат через некоторое время стекает в виде красноватой мутной жидкости. После этого фильтрование прекращают, осадок смывают с фильтра водой из про-мывалки в колбу Эрленмейера и разбавляют приблизительно до 100 см3. Частицы осадка постепенно диспергируют в жид
49. Техника работы
5G9
кость. Через несколько часов получают прозрачный оранжево-красный золь.
Золь V2O5 относится к оптически анизотропным веществам; при длительном стоянии из него выделяются субмикроскопические кристаллы стержневидной формы, что вызывает явление двойного лучепреломления. Оно проявляется в виде образования полос при встряхивании золя. Для золя V2O5 образование полос можно наблюдать через несколько недель после его получения.
127. Золь кремневой кислоты
20 г кристаллического силиката натрия растворяют в 70 см3 горячей воды; раствор, если необходимо, фильтруют и дают ему охладиться. Этот раствор прибавляют по каплям при перемешивании к смеси 20 см3 конц. НС1 с 40 см3 Н2О до появления неисчезающей розовой окраски фенолфталеина в месте его внесения в раствор. Для отделения ионов от коллоидных частиц прозрачный коллоидный раствор помещают в диализатор, который в простейшем случае состоит из ячейки для диализа, подвешенной в стакане с дистиллированной водой. Воду, находящуюся во внешней части установки, меняют несколько раз в день, каждый раз до отрицательной реакции пробы на ионы С1~.
49.9. Препаративные синтезы при низких температурах
128. Монохлорид иода IC1
Во взвешенной колбе вместимостью 50 см3, погруженной в охлаждающую смесь (СОг+ацетон), конденсируют около 30 см3 сухого хлора. К смеси добавляют примерно половину эквивалентного количества 12, которое ориентировочно оценивают по объему хлора. После прибавления иода реакционная смесь затвердевает. Охлаждающую смесь удаляют, при этом избыточный хлор испаряется через горло колбы, закрытое хлоркальциевой трубкой. Затем колбу с содержимым взвешивают. После вычитания массы пустой колбы и добавленного иода находят массу израсходованного хлора. Если его масса больше, чем требуется по теоретическому расчету для образования IC1, то образуется 1С1,3. Соразмерно избыточному количеству хлора добавляют эквивалентное количество иода. Закрытую стеклянную пробкой колбу оставляют стоять при комнатной температуре на 24 ч.
Затем сырой продукт подвергают особому виду «перекристаллизации». Жидкий IC1 охлаждают до тех пор, пока ~80% всей массы не затвердеет, после чего жидкость сливают. «Перекристаллизацию» проводят от двух до трех раз.
Красно-бурая жидкость, довольно устойчивая. Разъедает пробки, резину и прежде всего поражает кожу (осторожно!).
37—1880
570
ЕЛ. Практикум по препаративной химии.
129. Бромоводород НВг
,СН2. ,СНВг
сн2 СНВг
[ Н I +4Вгг—* Г J I +4НБ1Г Ч>А. сн, >НВг
' 4 СНг,-'-' хНВг
В круглодонную колбу -с пришлифованной капельной воронкой и газоотводной трубкой помещают тетрагидронафталин, к которому добавлено немного чистых железных опилок, и медленно приливают по каплям бром. Перед применением тетрагидронафталин высушивают обезвоженным^ Na^SC^ и перегоняют (7’кип = 207 °C; давление паров при 15 °C 40 Па). Поскольку вначале синтез проводят при охлаждении, круглодоннуку колбу погружают в водяную баню, которую при замедлении реакции можно нагреть до 30—40 °C. Образовавшийся газ для очистки от незначительных количеств брома пропускают через-промывную склянку, заполненную тетрагидронафталином (также предварительно высушенным и перегнанным), а для поглощения оставшихся следов влаги — через ловушку, охлаждединую до —60°C. Можно выморозить продукт жидким воздухом в следующей ловушке и по окончании реакции запаять ее и отделить от установки для получения газа.
130. Амид натрия NaNHb
Na + NH3 --► NaNH2 + 7^
5 г металлического натрия, не содержащего оксида, в виде кусочков величиной с горошину помещают в ампулу в атмосфере инертного газа (рис. Е.38). К оттянутому концу ампулы подсоединяют шланг, который вначале перекрыт зажимом. Сосуд Дьюара заполняют жидким аммиаком, приоткрыв кран бал-
Рис. Е.38. Схема установки для получения NaNHj.
/ — ампула с кусочками натрия; 2 — сосуд Дьюара7 наполовину заполненный жидким ам* миаком; 3—U-образная трубка с ртутным затвором.
49. Техника работы
571
лона с аммиаком и пропуская1 через сосуд слабый поток газа в течение примерно 1 мин. Следует принять меры предосторожности, чтобы в случае возможного взрыва не повредить сосуд Дьюара. Затем кран баллона с аммиаком открывают и впускают в сосуд Дьюара примерно 150 см3 жидкого аммиака. К NH3 добавляют кристаллик Fe(NO3)3-9H2O размером с рисовое зерно. Затем сосуд Дьюара герметично закрывают пробкой, в которую вставлены стеклянные трубки, как показано на рис. Е.38. Более узкая трубка ведет к U-образной трубке, в которой находится небольшое количество ртути. Это дает возможность выделения аммиака без доступа воздуха в систему. Слегка наклоняя ампулу, к аммиаку добавляют натрий с такой скоростью, чтобы аммиак умеренно вскипал. Эту операцию заканчивают приблизительно через 0,5 ч, при этом голубая окраска жидкого аммиака исчезает. Отсоединяют пустую ампулу и закрывают резиновой пробкой трубку, ведущую к ампуле. Установку оставляют стоять до тех пор, пока через 1—-2 сут аммиак не улетучится из сосуда Дьюара. Сосуд Дьюара открывают в боксе с инертным газом и извлекают NaNH2 с помощью шпателя. Препарат помещают в плотно закрывающийся пришлифованный сосуд. При проведении всех операций необходимо исключить доступ воздуха и влаги, так как частично окисленный продукт (желтая поверхность!) при трении или нагревании может взорваться.
Белая кристаллическая масса. Тпл=155аС.
131. Никель, карбонил никеля Ni(CO)4
NiC2O4 --► NiO + СО + СО2
NiO + CO ► Ni+CO2 Ni + 4CO Ni(CO)4
Для получения никеля в высокодисперсном состоянии растворяют 30 г Ni(NO3)2-6H2O в горячей воде и суспендируют в полученный раствор примерно 10—20 г кизельгура. При кипячении и перемешивании к суспензии добавляют горячий раствор, содержащий 13 г щавелевой кислоты (Н2С2О4-2Н2О). Образовавшийся1 осадок, состоящий из NiC2O4 и кизельгура, после отфильтровывания и тщательного промывания высушивают при 120 °C до получения твердой массы. После добавления нескольких капель конц. H2SO4 вещество еще раз высушивают.
Полученную массу помещают в тугоплавкую трубку длиной 50 см и диаметром приблизительно 2,5 см и после вытеснения из трубки воздуха слабым потоком СО в течение примерно 1,5 ч нагревают ее до 450 °C и выдерживают при этой температуре в течение 0,5 ч. При этом: NiC2O4 полностью превращается в Ni, находящийся в кизельгуре в1 высокодисперсном 37*
572 E.II. Практикум no препаративной химии
состоянии. Дают массе охладиться до 100 °C,, пропуская слабый поток СО. К реакционной трубке подсоединяют ловушку, охлаждаемую жидким воздухом. При увеличении скорости, пропускания СО в охлаждаемой ловушке концентрируется Ni(CO)4 (чрезвычайно ядовит; все операции проводить в вытяжном шкафу!).
Бесцветная жидкость. Тпл=—25°С; 7’КИп=430С. Ni(CO)4 быстро окисляется на воздухе; при зажигании горит ярко светящимся пламенем. При-пропускании через трубку, нагретую до* 2ОО‘°С, образует блестящее .никелевое зеркало.
50. МЕТОДЫ СИНТЕЗА ПРЕПАРАТОВ
В гл. 49 были приведены общие сведения о технике эксперимента, применяемой в синтезе сравнительно простых препаратов. Данная глава посвящена описанию более сложных методов синтеза; для получения указанных в этой главе препаратов, необходимо использовать сложные приемы работы или сочетание нескольких простых приемов. Подробные методики в сочетании с данными, приведенными в таблицах, дают возможность синтезировать большое число препаратов, не описанных в этой главе. Экспериментатор самостоятельно должен определить условия синтеза препаратов, исходя из физических и химических свойств веществ, используемых в реакции.
50.1. Методы получения элементов*
50.1.1. Реакции восстановления
Восстановление водородом
При восстановлении оксидов водородом наряду с металлом образуются только пары воды1:
МеПО + Н2 --► Me + Н2О
Этот метод применим для получения металлов, не реагирующих с водой и не способных к образованию солеподобных гидридов. Поскольку восстановление происходит уже при сравнительно низких температурах, существует возможность получения металлов в высокодисперсном состоянии. Поэтому такой метод применяют премущественно для получения пирофоров,, а также катализаторов. В табл. Е.6 дан обзор реакций, применяемых в лаборатории для получения металлов.
132. Никель из оксида никеля (катализатор гидрирования по Сабатье)
Исходным соединением служит Ni, который получают нагреванием Ni(NO3)2-6H2O в тигле в, течение 6 ч при 1000—1100 °C.
50. Методы синтеза препаратов
573
Таблица Е.6. Металлы, получаемые восстановлением водородом
Металл Уравнение реакции Температура восстановления, *С
Со COC2O4+H2—^Со+НгО+СО+СОг 500
Си CuO4-H2—^Си+НгО 250—300
Fe 2Fe(OH)3+3H2—>-2Fe+6H2O (пирофорное Fe, применяемое для восстаиовлеиия) 350
Ge ОеОг-рЗНг— 600
Mo МоОз 4" ЗН2—>-Мо+ЗН2О Начальная — 500;
последующая — 1000
Ni №О+Н2—»-Ni+H2O (ср. препарат 132) 300—400
Re 2NH4ReO4+7H2—>-2Re+2NH3+8H2O Начальная — 500; последующая — 1000
V 2VC13 + ЗН2—*2V+6HC1 900
W WO3+3H2—>-W+3H2O 800—1000
Для восстановления NiO переносят в фарфоровую лодочку и помещают ее в середину реакционной трубки. Один конец трубки присоединяют к аппарату Киппа, в котором получают водород, так чтобы водород поступал в трубку через осушитель— пентаоксид фосфора. Другой конец трубки соединяют со стеклянной трубочкой, имеющей шарообразное расширение и служащей приемником чувствительного к действию воздуха металлического порошка. Трубочку с никелем можно запаять. Для нагревания применяют трубчатые электропечи с терморегулятором. Перед нагреванием реакционной трубки через установку в течение нескольких минут пропускают поток сухого Н2 для вытеснения из нее воздуха. Для предотвращения взрыва проверяют наличие свободного кислорода в установке пробой на гремучий газ. Затем проводят нагревание в трубчатой печи. NiO восстанавливают в токе сухого водорода при 300—400°C. Через 15 ч нагревание прекращают и установке дают охладиться в потоке водорода. Получаемый таким способом порошкообразный никель применяется в качестве катализатора при гидрировании.
Пирофорные свойства металла существенно зависят от условий восстановления. Для получения никеля, самопроизвольно проявляющего пирофорные свойства, восстановление следует проводить при ~155 °C в течение примерно 35 ч; в качестве исходного вещества в этом случае лучше всего применять свежеосажденный хорошо высушенный гидроксид никеля.
574 E.1I. Практикум no препаративной химии
Таблица Е.7. Металлы, получаемые восстановлением углеродом или цианидами
Металл Уравнение реакции Условия получения
Bi Bi2O3 +8KCN—>-2Bi+3KOCN Нагревание в фарфоровом тигле
Pb 2PbO-pC—>-2Pb+CO2 Восстановление древесным углем при 800—900 °C
Sb Sb2O5+5KCN—->-2Sb+5KOCN Нагревание равных количеств KCN и Sb2Os до температуры плавления сурьмы
Sn SnO2+C—^Sn+CO2 Нагревание смеси 40 г SnO2 с 6,5 г древесного угля при 900— 1000 °C
Sn ' SnO2+2KCN—>Sn+2KOCN См. препарат 133
Восстановление углеродом и цианидами (табл. Е.7)
133. Олово из оловянного камня (см. также разд. 36.2.1)
В ходе реакций окисления — восстановления цианид калия окисляется в цианат:
SnOa + 2KCN --► Sn + 2KOCN
Для получения препарата сплавляют в течение ~0,5 ч равные количества тонкоизмельченного оловянного камня и порошкообразного KCN в фарфоровом тигле, который ставят перед воздуходувкой. По охлаждении зёрна олова обрабатывают водой для растворения находящегося на нем расплава.
Восстановление металлами
Многие элементы можно получить взаимодействием их соединений с металлами (табл. Е.8).
Этот метод часто применяют в лаборатории для получения металлов, причем в качестве исходных соединений используют растворы солей металлов, а также их оксиды, сульфиды или галогениды, которые восстанавливают более электрохимически активными металлами.
134. Обработка отходов серебра
Аммиачные растворы серебра нельзя долго хранить, так как при хранении в осадок может выпасть взрывчатый нитрид Ag3N. Раствор, находящийся1 над отходами серебра, лучше всего смешать с соляной кислотой до прекращения образования осадка AgCl. Для получения серебра осадок отфильтровывают, промывают, добавляют НС1 (1 : 1) при помешивании и восстанавливают палочками цинка в фарфоровой чашке. После восстановления всего галогенида серебра серебряный
50. Методы синтеза препаратов
575
шлам отмывают горячей водой от кислоты и ионов цинка и отделяют фильтрованием.
Термические методы
Термические методы часто применяют в лаборатории для синтеза металлов. Термические реакции — это реакции взаимодействия твердых веществ, сопровождающиеся значительным выделением тепла. Наилучшим восстановителем, применяемым в этих методах, является алюминий; этот метод называется алюминотермией. Для того чтобы реакция началась, применяют зажигательную смесь, далее реакция протекает без дополнительного подвода тепла извне. Вещества вступают в реакцию в основном' в стехиометрических количествах. Но при этом следует учесть, что, с одной стороны, реакция должна протекать без взрыва, что обеспечивают применением подходящего разбавителя (обычно — избыток одного из компонентов), а с другой стороны, образующегося в ходе реакции тепла должно хватить для переведения всей реакционной массы в расплавленное состояние. В качестве флюса часто применяют СаРг. Температура основной реакции может повыситься, если в системе протекает параллельная реакция. Например, при добавлении порошкообразной серы и соответствующем увеличении содержания алюминия количество выделяющегося тепла увеличивается вследствие протекания экзотермической реакции
2А1 + 3S --> A12S3
Приготовление зажигательной смеси. Смешивают в склянке для порошкообразных веществ при встряхивании 5 г пероксида бария с 7 г порошкообразного магния. Ни в коем случае нельзя готовить смесь растиранием в ступке! Для поджигания в смесь вставляют длинную магниевую ленту.
Применение зажигательной смеси. Синтез проводят в маленьком глиняном тигле высотой 10—15 см, который лучше поставить в цветочный горшок, заполненный песком. В глиняный тигель помещают реакционную смесь и засыпают ее зажигательной смесью с магниевой лентой. Все материалы должны быть совершенно сухими. Работать лучше на воздухе в сухую погоду и только в защитных очках. Для того чтобы реакция началась, с некоторого расстояния поджигают магниевую ленту. При восстановлении металл собирается на дне тигля преимущественно в виде зерен. Тигель нужно затем только разбить и зерна целевого продукта отделить от шлака механическим путем.
135. Хром Сг
Сг2О3 + 2А1 -»- А12О3 + 2Сг; ДЯ = —3,85-105 Дж/моль
70 г хорошо высушенного оксида хрома (III) смешивают с 33 г мелкозернистого алюминия и 25 г тонкоизмельченного дихромата калия и вносят в глиняный тигель, на дно которого предварительно иасыпают 10 г CaF2. Расход зажигательной смеси 20 г.
Таблица Е.8. Элементы, получаемые восстановлением металлами
Элемент Уравнение реакции Условия получения
Реакции в водной среде
Ag 2Ag+ + Zn—>2Ag +Zn2+ Обработка отходов серебра (см. препарат 134)
Pt (NHOdCPtCle] +2Zn—>Pt +2NH4Cl+2ZnCl2 Обработка отходов платины
Н2 2HC1+Zn—>H2+ZnCl2 В аппарате Киппа
Реакции взаимодействия твердых, жидких и газообразных веществ
Щелочные металлы (Li, Na, К, Rb, Cs) 2Me2CrO4+Zr—>-4Me + Zr(CrO4)2 (получение чистых щелочных металлов) Восстановление хроматов (дихроматов, молибдатов и вольфраматов) щелочных металлов порошкообразным ^глубоком ВакУУме в кваРИев°й трубке при 700—
В В в (А1В12) Сг Щелочноземельные металлы (Be, Mg, Са, Sr, Ba) н2 d2 Hg Мп Mo Nb Ta B2O3-|-6Na—>-2B + 3Na2O (получение аморфного бора) BaOs+SMg—>2B+3MgO (получение аморфного продукта, содержащего 86% В) В20д+2А1—>2В+А120з (получение кристаллического А1В12) Сг20з+ 2А1—->2Сг+А12О3 ЗМеО+2Л1—>ЗМе+Л12О3 4Н2О+ 3Fe—>4H2+Fe3O4 2D2O+2Na—>D2+2NaOD HgS + Fe—>Hg+FeS ЗМп3О4+ 8A1 —>9Mn + 4 A12O3 8MoO2+4A1—>3Mo+2A12O3 K2NbF7+5Na—>Nb+2KF+5NaF ) K2TaF7+5Na—>-Ta + 2KF+5NaF / Прокаливание в никелевом тигле с добавлением NaCl и КС1 Термический метод с применением магния в качестве восстановителя (препарат 137) Термический метод (алюминотермия) Термический метод (см. препарат 135) Алюминотермии в глубоком вакууме при 1000—1050 °C (установку следует открывать в атмосфере СО2 или аргона) Пропускание водяного пара над раскаленным докрасна железом Глубокий вакуум, охлаждение жидким воздухом Восстановление железными стружками при нагревании Термический метод (см. препарат 136) Термический метод (алюминотермия) Восстановление натрием или кальцием в толстостенном стальном сосуде с привинчивающейся крышкой при температуре красного каления
P (светло-красный) Редкоземельные металлы Si Si Si Ti, Zr, Hf (Th) V W Nb2O3+5Ca—^-2Nb + 5CaO ) Ta2O3+5Ca—>-2Ta+5CaO > 2PBr3+3Hg—*2P+3HgBr2 2CeCl3+3Ca—>-2Ce+3CaCl2 LaCls+ЗК—>La + 3KCl SiCl4+2Zn—>Si+2ZnCl2 SiO2+2Mg—>-Si+2MgO (получение Si с выходом 95—98%) 3SiO2+ 4А1—>-3Si + 2A12O3 MeO2+2Ca—>-Me+2CaO (аналогично для тории) Na2MeFe+4Na—>-Me+6NaF MeCl4+4Na—>-Me+4NaCl (аналогично для тория) MeCl4+2Mg—>-Me+2MgCl2 V2O5+ 5Ca—»2 V+ 5CaO WO34-3Zn—>\V+3ZnO Восстановление 30%-иым избытком кальция с добавлением СаС12 в качестве флюса в атмосфере аргона в сварной железной трубке при 1000—1100 °C Нагревание при встряхивании до 100—170 °C в трубке для проведения реакций с последующей экстракцией (см. препарат 138) Нагревание смеси в трубчатой печи при 650—750 °C; для лучшего отделения королька металла от шлака добавляют вещество, реагирующее с кальцием с высоким тепловым эффектом (иод) Ступенчатое восстановление в глубоком вакууме при многократном добавлении калия в установке из стекла «супремакс» Восстановление газообразного SiCl4 парами цинка в кварцевой трубке электрической трубчатой печи Восстановление в тигле; зажигание смеси при нагревании пламенем стеклодувной горелки или с помощью зажигательных таблеток Поджигание смесн 90 г хорошо высушенного и тонко измельченного песка, 100 г порошкообразного алюминия и il20 г серы Реакция в сварном железном баллоне при '1000 °C Реакция в сварном баллоне при 1200 °C Реакция в прочно закрытом стальном баллоне при температуре красного каления Реакция в атмосфере аргона Нагревание смеси 90 г V2O3 и 450 г кальция с добавкой 150 г безводного СаС12 в закрытом железном баллоне при 900—950 °C Нагревание смеси WO3 с трехкратным количеством цинка (см. уравнение реакции) в фарфоровом тигле при 500—520 °C до воспламенении ч
578 E.II. Практикум no препаративной химии
136. Марганец Мп
ЗМп3О4 + 8А1 --► 4AlsO34-9Mn; ДЯ = —2.04-106 Дж/моль
В качестве исходного вещества для получения марганца термическим методом применяют Мп3О4, так как при использовании МпО2 реакция протекает со взрывом. Вначале 80 г МпО2 нагревают в тигельной печи до 800—900°C в течение 30 мин. После охлаждения получившийся Мп3О4 измельчают в порошок, смешивают с 20 г мелкозернистого алюминия и вносят смесь в тигель, в котором находится 10 г CaF2. Расход зажигательной смеси 20 г.
137. Бор В (восстановление В2О3 магнием)
Шамотный тигель высотой 20 см и диаметром 16 см обмазывают пастой из прокаленного оксида магния и спекшегося хлорида магния и высушивают в сушильном шкафу. В тигель вносят смесь НО г В2О3, 115 г магниевых стружек (не порошка) и 94 г порошка серы. Реакцию' инициируют при помощи зажигательной смеси (1,5 г ВаО2 и 0,2 г порошкообразного магния растирают в пасту с раствором коллодия и наносят на магниевую ленту). После охлаждения продукт реакции в течение 8 сут выщелачивают водой и разбавленной НС1. Остаток многократно обрабатывают при нагревании плавиковой и соляной кислотами, промывают водой и высушивают в вакууме при 100 °C.
138. Красный фосфор Р
2РВг3 + 3Hg -► 2Р + 3HgBr2
В трубку для проведения реакций в расплавах помещают 55 г Hg и 51 г РВг3 (оба реактива в возможно более чистом и сухом состоянии) и нагревают при 100 °C в течение 2 сут при непрерывном встряхивании. Необходимо следить за тем, чтобы ртуть не задерживалась в кончике трубки. Затем трубку нагревают в течение 1 сут в печи при 130°С и на следующий день повышают температуру до 170 °C. Полученный продукт осторожно измельчают, шесть раз обрабатывают абсолютным эфиром в экстракционном аппарате (каждый раз по 2 ч) и после высушивания нагревают 1—2 раза в вакууме для возгонки образовавшейся HgBr2 в атмосфере сухого диоксида углерода, не содержащего кислорода. Остаток содержит приблизительно 87% фосфора, Hg2Br2 и HgBr2. Этот продукт снова помещают в трубку для проведения реакций под давлением длиной около 0,5 м, добавляют по 2 капли РВг3 на каждые 2 г вещества и после вакуумирования трубку запаивают. Трубку вставляют в печь концом, содержащим вещество, а другой конец на */з оставляют вне печи. В течение 1 сут трубку осторожно нагревают до 230—-240 °C и затем дают ей охладиться. В выступающем из печи конце трубки осаждается вновь по
50. Методы синтеза препаратов 579*
лученный HgBr2 в виде кристаллов. Продукт реакции отделяют в экстракционном аппарате от главной массы находящегося в избытке РВгз путем шестичасового экстрагирования при трехкратной смене эфира; после этого отгоняют еще сохранившийся в нем HgBr2 в атмосфере СО2 под давлением (3—4) • •103 Па и при температуре 250 °C. Черно-коричневый остаток приобретает при охлаждении красную окраску (похожую на., цвет киновари).
Светло-красный фосфор не является особой аллотропической модификацией; это разновидность обыкновенного красного фосфора (см. разд. 35.7.1), от которого он отличается меньшей величиной частиц. Светло-красный фос--фор нерастворим в эфире и CS2. Он не окрашивается в темный цвет ни : жидким аммиаком, ни раствором аммиака в воде. Температура вспышки на воздухе приблизительно 300 °C. Во влажном воздухе светло-красный фосфор-медленно окисляется.
50.1.2. Электролитическое получение элементов
139. Таллий, получение электролизом раствора T12SO4 Металлический таллий можно достаточно полно выделить из насыщенного сернокислого раствора сульфата таллия(I). Катод слегка покрывают смазкой для того, чтобы выделившийся металл легко можно было снять, и помещают на дно электролизера. Катод изготавливают из медного листа в виде кольца шириной 4,5 см с поверхностью 100 см2. Поскольку токоподводящий провод проходит через электролит, его 'нужно заключить в стеклянную трубку. Между катодом и находящимся в верхней части электролита анодом вращается лопастная мешалка для предотвращения короткого замыкания в цепи из-за возможного роста кристаллов таллия от катода к аноду. Анодом служат две платиновые пластинки по 7—8 см2, горизонтально укрепленные в верхней части электролита по обеим сторонам от оси мешалки. Электролиз проводят при напряжении <3,5 В и силе тока 1,3—1,5 А, что соответствует катодной плотности тока 0,13—0,15 А/см2. Таллий выделяется в виде блестящих, крупных листочков и игл. Электролиз заканчивают, когда значительно усиливается выделение водорода, а при действии соляной кислоты на пробу электролита не происходит выпадения осадка. Ток отключают, вынимают мешалку и кислый раствор быстро заменяют водой. Затем кристаллы таллия соскабливают с катода стеклянным шпателем, обминают их под водой, затем хорошо высушивают между листами фильтровальной бумаги и плавят под слоем1 цианида калия. Продукт лучше всего хранить в растворе гидроксида таллия в запаянной трубке.
140. Литий, получение электролизом расплава LiCl
В фарфоровом тигле высотой 6—8 см и шириной 9 см сплава
Таблица Е.9. Элбменты, получаемые электролитическим путем
Уравнение реакции Электролит Анод Катод Условия электролиза
« Из водных растворов
Ag+ + e-—>-Ag 10%-ный раствор AgNO3 для получения серебра наивысшей степени чистоты Свеча с фильтрующей пластинкой из пористого стекла, заполненная неочищенным серебром Ag 1,39 В
Cd2++2e-—э-Cd Сернокислый раствор CdSO4 Pt Pt 0,1—0,3 А/см2
HCrO4-+7H++6e-—> 1 > Сг+4H 2О Раствор, в 1 дм3 которого содержатся 240 г СгОз, 3 г Cr2(SO4)3- 12Н2О, 9 г Сг(ОН)3-ЗН2О РЬ Си 0,1 А/см2
Fe2++2e~—>-Fe 0,01—0,02 н. по НС1 раствор электролита, в 1 дм3 которого содержатся 800 г FeCl2-4H2O и '1,5—2,0 г А1С13-6Н2О Fe Сталь V2A марки 0,1—1,5 А/см2
Ga3+ + 3e~—>-Ga Разбавленный раствор гидроксида галлия в NaOH Pt Pt 3—4 В; 1 А
2H++2e~—»H2 15%-ный раствор КОН Ni или Pt Ni или Pt 0,5 А/см2
1п3++Зе-—>In Сернокислый раствор In2(SO4)3 Pt In или Al 0,01 А/см2
Mn2++2e_—>-Mn Катодный электролит: раствор (pH 6—8), РЬ , Сталь 0,2—0,3 А/см2 (диафрагма)
4ОН-—>О2+2Н2О + 4е- в 1 дм5 которого соДержа+ся 300 г MnSO4-4H2O и 100 г (NH4)2SO4. Анодный электролит: раствор (<5% H2SO4), в 1дм которого содержится 100 г (NH4)2SO4 Насыщенный раствор гидроксида бария Pt Pt 0,3 А/см2
Т1+ + е-—>Т1 Сернокислый раствор T12SO4 Pt Си См. препарат 139
Zn2+ + 2e-—>Zn Раствор ZnSO4 ( — 50 г Zn/дм4). В анодном пространстве находится суспензия ZnO Pt Al, Pt 0,01—0,03 А/см2 (диафрагма)
Из расплавов 15 В; 200 А; 950 °C
А13+ + 3е~—=»-А1 Йаствор А12О3 в расплавленном криолите в графитовом тигле Графит Графитовый тигель
Са2+ + 2е-—>-Са Расплав 100 г СаС12 и 16,5 г CaF2 Графит Fe 8 В; 8 А; 800 °C
2F-—>F2+2e- Расплавленный KF-3HF (или KHF2) Никель фит) (гра- Медный или магниевый ти- 100 °C; 10 В; 4—5 А (200—300 °C)
гель
Li++e-—>Li Расплавленная смесь LiCl+KCl (1 :’1) (или LiBr+LiCl) Графит Железная проволока См. препарат 140
Та5+ + 5е-—э-Та Расплавленная смесь КгТаР7, Та2О3, КС1 и KF (аналогично для Nb) Уголь или Мо Никелевый тигель (1,6 А/см2
(Nb5+ + 5e~—»Nb)
582
ЕЛ. Практикум по препаративной химии
ляют на горелке Теклю 30 г высушенного LiCl с 30 г сухого* КС1 и проводят электролиз расплава. Анодом служит погруженный в расплав крепкий кусок угля из дуговой лампы размером 0,8 см. Катод — прочная железная проволока длиной 0,3 см, вставленная в стеклянную трубку диаметром 2 см. Сверху стеклянную трубку закрывают пробкой, через которую1 и проходит железная проволока. Нижнюю (открытую) часть стеклянной трубки погружают в расплав на глубину примерно 1 см. Железная проволока погружена на 0,3 см глубже. Включают ток и, регулируя сопротивление, создают силу тока 6— 10 А (показания амперметра). Для этого необходимо напряжение 7—12 В (от 3 до 6 последовательно включенных аккумуляторов). Металлический литий собирается в пространстве-между железной проволокой и стеклянной трубкой и оказывается достаточно защищенным стеклянной оболочкой от распыления его внутри электролита.
50.1.3. Синтезы, основанные на реакциях разложения
Для проведения синтеза, основанного на разложении соединений (табл. Е.10), вещество переводят или в нестабильное в условиях синтеза соединение, например
МпО2 + 4НС1 --> МпС14+2Н2О
МпС14---> МпС12 + С12
или изменяют условия* реакции (давление и температуру) таким образом, чтобы создать нестабильную область, в которой вещество не находится в равновесном состоянии с продуктами своего разложения, например при выделении чистого никеля по карбонильному методу (см. препарат 131).
Применение катализатора значительно ускоряет установление равновесия (гл. 15). Каталитическое разложение, однако;, лишь ограниченно применяется в препаративной неорганической химии (например, для получения абсолютно чистого азота из аммиака (табл. Е.10)). При менее высоких требованиях к чистоте продукта азот получают более простым путем из нитрита аммония.
141. Азот N2
NH4NO2 ---► N2 + 2Н2О
К насыщенному на холоду раствору NaNO2 или KNO2, нагретому на водяной бане, добавляют по каплям из капельной воронки насыщенный на холоду раствор (NH4)2SO4 и регулируют выделение N2, меняя скорость прибавления раствора (NH4)2SO4. В азоте могут содержаться примеси N2O и NO, а также небольшие количества кислорода и диоксида углерода. NO лучше всего удалять промыванием газа раствором
50. Методы синтеза препаратов
583
Таблица Е.10. Элементы, получаемые разложением их соединений
Элементы Уравнения реакций Примечания
Щелочные металлы и Na Щелочноземельные металлы .и N2 MeN3—>-Me+3/2N2 (Me=Na, K, Rb, Cs) Me(Ns)2—>-Me+3N2 (Me=Ca, Sr, Ba) Разложение азидов в вакууме для получения металлов наивысшей степени чистоты или чистого азота (опасность взрыва!)
М2 2 N H3—*• N 2 4" 3 H 2 NH4NO2—^No+2H20 Каталитическое разложение при 1000 °C и атмосферном давлении (см. препарат 141)
Ni Ni(CO)4—>Ni+4CO Получение никеля высокой чистоты (препарат 131)
NiC2O4—*-Ni +2CO2 (NiC2O4—>NiO+CO-f- +CO2, NiO + CO—>Ni+CO2) .Реакция протекает в две стадии
10KMnO4—>6O2+ 4- ЗКгМпС^-^КгОЧ" Термическое разложение при 200—240 °C
+ 7MnO2 2КС10з—>O2+KC1O4+ Н-КС1 Термическое разложение при 400 °C (в присутствии или без катализатора)
:Н2О2—> О2+Н2 Каталитическое разложение 30 %-ного Н2О2 (в присутствии платины или МпО2)
Pt (и платиновые металлы) (NH4)2PtCI6—>Pt+ + 2NH3+2HC14-2C12 H2PtCl6—>-Pt+2HCl + +2C12 Получение платинированного асбеста (см. препарат 142)
Ti, Zr, Hf, Th, V, Mb, Ta X 'MeIx==M.e+ 2~ I2 Метод наращивания для получения металлов высокой чистоты
сульфата железа(II), пропуская газ через ‘несколько промывных склянок, с последующим высушиванием натронной известью. Затем газ пропускают через трубку длиной 60 см над раскаленной медью; при этом оставшиеся еще оксиды азота количественно разлагаются на элементы, а кислород одновременно связывается в виде СиО.
142. Платинированный асбест
Чистый асбест смачивают концентрированным раствором гексахлороплатината и осаждают платину в волокнах асбеста до-
584
E.I1. П рантикум по препаративной химии
давлением концентрированного раствора NH4CI. При этом осаждается (NH4)2PtCl6. Затем отжимают избыток жидкости, асбест высушивают и нагревают его до каления, в результате чего гексахлороплатинат разлагается с образованием мелкодисперсной металлической платины. Обычно получают асбест с 10%-ным содержанием платины.
50.1.4. Синтезы с применением окислителей
Для получения галогенов окисляют галогеноводороды НХ сильными окислителями. Часто для этого сначала в реакционном сосуде выделяют галогеноводород в свободном состоянии из его солей. Из-за большой реакционной способности галогенов при их получении следует по возможности избегать, применения резиновых шлангов.
143. Хлор С12
В колбу помещенного под тягу аппарата для получения газов вносят в равных количествах NaCl и тонкоизмельченный пиролюзит. При повышенных требованиях к чистоте продукта применяют МпО2-хН2О, полученный осаждением, так как природный пиролюзит зачастую загрязнен сульфидом. (Как природный, так и синтетический продукт содержат карбонаты.) Из. вертикально укрепленной на колбе капельной воронки, конец; которой глубоко опущен в. реакционную колбу, добавляют наполовину разбавленную конц. Н2ВО4. Газоотводную трубку соединяют со склянкой для промывания газа, заполненной на '/б высоты конц. H2SO4; склянка одновременно служит счетчиком пузырьков. Для получения потока хлора, не содержащего НС1, в качестве первой промывной жидкости применяют воду, и только затем H2SO4. Для полного высушивания хлор-можно пропустить еще над пентаоксидом фосфора. Изменяя скорость прикапывания H2SO4 и немного нагревая реакционную колбу, можно регулировать выделение С12.
Желто-зелеиый газ с резким запахом; поражает поверхность кожи с опасностью для жизни. ТКИп=—34 °C.
144. Бром Вг2
6КВг -Ь K2Cr2O, + 7H2SO4-► ЗВт2 4- Cr„(SO4)3 4- 4K2SO4 + 7Н2О
Синтез проводят обычно в установке для перегонки (рис. Е. 15).. Поскольку бром очень реакционноспособен, применяют аппаратуру на шлифах. Растертый в мелкий порошок КВг и порошкообразный К2Сг2О7 помещают в колбу для перегонки, снабженную пришлифованной насадкой, и смешивают с конц.. H2SO4 (примерно 100 см3 96%-ной H2SO4 на 75 г твердой реакционной смеси). После этого колбу быстро подсоединяют к. нисходящему холодильнику Либиха и собирают отгоняющийся Вг2 в приемник. Неконденсирующиеся пары направляют в
50. Методы синтеза препаратов 585-
тягу. При прекращении реакции, вначале бурно протекающей, остаток Вг2 отгоняют, осторожно нагревая реакционную колбу.
Темио-коричиевая жидкость. Вызывает болезненные раны на коже. Красно-коричневые пары брома сильно поражают слизистую оболочку и опасны даже при большом разбавлении. ТКЯП = 58,8°С; Таа=—7,3 "С.
50.1.5. Специальные методы синтеза
Способы очистки веществ
145. Очистка сжиженного водорода
Водород получают электролизом или разложением водяного газа. Электролитический водород содержит лишь небольшую примесь воздуха (<0,1% О2, ~0,2% N2) и во многих случаях пригоден для лабораторных целей. Водород, получаемый из водяного газа, напротив, содержит значительные количества СО, СО2, О2 и N2, а также случайные примеси AsH3 и Fe(CO)5. Диоксид углерода поглощают гидроксидом калия или натронной известью, AsH3— насыщенным раствором КМпО4. Для удаления О2 газообразный водород пропускают над платинированным асбестом (препарат 142), нагретым до 200—400 °C; при этом одновременно разлагается Fe(CO)s. Для удаления О2 можно применять также носитель катализатора,, содержащий медь, и мелкодиспергированный высокоактивный палладий, нанесенный на у-А12О3. Удаление СО лучше всего проводить вымораживанием жидким воздухом. Для очистки водорода от всех примесей, особенно от кислорода, используют селективную диффузию через палладиевую трубку при 350 °C, благодаря чему достигается высокая чистота водорода. Во избежание накопления остатков газов их непрерывно вытесняют из трубки слабым потоком водорода и сжигают. При температуре ~150 °C палладий образует хрупкую, непроницаемую для водорода фазу, поэтому при нагревании и охлаждении палладиевую трубку нужно хорошо вакуумировать.
Водород, хранящийся в стальных баллонах, как и полученный другим путем, всегда содержит влагу и должен быть высушен над Р4О10 или на молекулярных ситах. Для осушки водорода нельзя применять конц. H2SO4, так как даже при небольшом повышении температуры она восстанавливается с образованием H2S.
Проба на гремучий газ. При всех операциях, связанных с использованием водорода, существует опасность взрыва гремучего газа. Поэтому любую установку, заполненную водородом, перед иагреваиием или поджиганием выходящего газа испытывают «а отсутствие воздуха. С этой целью над концом трубки на выходе из установки подвешивают пробирку, перевернутую открытым концом вниз. Через 30 с пробирку убирают, закрывают ее отверстие большим пальцем и, поднеся к пламени, открывают отверстие. Находящийся в пробирке газ ие должен вспыхивать со свистом и звуком хлопка, а должен спокойно гореть едва заметным пламенем.
38—1880
586
E.II. Практикум no препаративной химии
146. Очистка ртути
Поскольку ртуть легко растворяет другие металлы с образованием амальгам, ее необходимо очистить от примесей металлов. От грязи и механических примесей ртуть очищают обычным фильтрованием через гладкий сухой фильтр, в дне которого •сделано маленькое отверстие. Все металлы (кроме благородных) можно удалить из ртути, окисляя их воздухом или HNO3. С этой целью ртуть помещают в склянку для отсасывания, размер которой выбирают таким, чтобы дно было покрыто слоем ртути толщиной 1—2 см. Затем! приливают ~3 М раствор HNO3 и закрывают склянку плотно прилегающей к ее горлу просверленной резиновой пробкой, через отверстие которой проходит стеклянная трубка, доходящая до дна склянки. Подсоединяют отвод к водоструйному насосу и через ртуть пропускают поток воздуха, приводящий ее в движение. В азотной кислоте наряду с электрохимически активными металлами растворяется также небольшое количество ртути, однако все металлы, стоящие в ряду напряжений перед ртутью, первыми растворяются в кислоте. Через 24 ч раствор сливают, промывают ртуть водой, сушат листами фильтровальной бумаги и затем фильтруют, как описано выше. Полученная таким способом ртуть по чистоте пригодна для очень многих целей.
Получение сплавов
Наиболее простым лабораторным способом получения сплавов является сплавление металлических компонентов. Исходным материалом служат маленькие кусочки металлов, металлические стружки или порошки. В случае легко окисляемых металлов по возможности применяют кусочки металлов, которые легко можно освободить от оксидной пленки обтачиванием, обработкой напильником или наждачной бумагой. Чистую поверхность можно получить, также травлением кислотами. Для сплавления лучше использовать крупные куски металлов, так как при этом на стенках сосуда задерживается совсем мало вещества, но в то же время значительно усложняется гомогенизация расплава, особенно если компоненты сплава существенно различаются по плотности или температуре плавления. Порошкообразный металл и стружку промышленного изготовления многократно очищают с помощью смазочных веществ, которые можно удалить действием органических растворителей. Загрязняющие металлы растворители и влагу перед получением сплава нужно удалить.
147. Амальгама натрия
6,9 г аккуратно разрезанного на куски натрия (для амальга-мы с содержанием 2 масс.% натрия) или 10 г (для амальгамы .с содержанием 3 масс. % натрия) помещают в круглодонную
50. Методы синтеза препаратов____________________587"
трехгорлую колбу вместимостью 250 смР. Через оба боковых горла колбы пропускают трубки для ввода и вывода азота. В среднее горло вставляют капельную воронку с 340 г (25 см3) ртути. После тщательного заполнения колбы азотом к натрикг добавляют 10 см3 Hg и колбу осторожно нагревают на открытом пламени до тех пор, пока не начнется реакция. Затем медленно приливают остальное количество ртути, при этом достаточно лишь слегка подогревать колбу. Когда вся ртуть будет перенесена в колбу, еще горячую расплавленную амальгаму выливают на чистую плитку и разбивают ее на куски (под' тягой), пока она еще теплая и хрупкая. Если берут только' 1 масс. % натрия, то получают жидкую амальгаму.
Синтез смешанных соединений галогенов
148. Трихлорид иода 1С13
В колбу Эрленмейера вместимостью 250 см3 насыпают 10 г тонкоизмельченного КСЮ3. Сверху насыпают слой 200 г порошкообразного иода. Затем приливают 10 см3 воды и в течение 30—45 мин добавляют небольшими порциями из капельной воронки при непрерывном помешивании 375 см3 конц. НС1. Температура реакции не должна превышать 40 °C .Охлажденный раствор фильтруют через фильтр с пластинкой из пористого стекла. Кристаллы 1С13 перекристаллизовывают ,из спирта и высушивают в вакууме над СаС12. Получают рыхлый оранжевый порошок или длинные желтые иглы. 1С13 очень летуч уже при комнатной температуре, и поэтому его нужно хранить в плотно закрывающихся склянках. (Осторожно! Поражает кожу!)
149. Монобромид иода IBr
К кристаллическому иоду, находящемуся в колбе со шлифами, снабженной капельной воронкой и трубкой для подвода газа, по каплям добавляют бром, в количестве немного большем эквивалентного. При этом масса нагревается, расплывается; при охлаждении (если необходимо, при встряхивании) она частично затвердевает. Для удаления свободного брома через колбу пропускают при 50 °C поток СО2. При охлаждении буро-красная жидкость затвердевает с образованием твердой компактной массы. Пары* имеют красно-бурую окраску; в значительной степени диссоциированы.
Черно-бурые кристаллы с запахом, похожим на запах брома. Т’Пл=40— 45 °C; ТКИП=119°С.
50.2. Методы получения оксидов
50.2.1. Оксиды металлов
Реакции окисления
1. Окисление металлов кислородом:
4Си Ог-----> 2Си2О'
38
588
Е.П. Практикум по препаративной химии
2. Окисление соединений металлов кислородом: TiCl4 + O2-------------------> TiO2 + 2Cl2
(температура реакции 650—750 °C)
3. Окисление металлов или их соединений оксидами, кислородсодержащими кислотами или их солями:
2Rb + HgO ----► Rb2o + Hg
Sn 4- 2HNO3 -► ₽-SnO2 + NO2 + NO + H2O
5NaN3 + NaNO3 ---- 3Na2O + 8N2
2T1NO3 + 6KOH + 2C12 --► T12O3 + 2KNO3 + 4KC1 + 3H2O
Эффективным окислителем являются ионы СЮ~, а также РЬО2 (препарат 150).
150. Оксид свинца(1У) РЬО2
2РЬ(СН3СОО)2 + Са(ОС1)2 + 4NaOH -►
---► 2РЬО2 + СаС12 + 4CH3COONa + 2Н2О
20 г ацетата свинца растворяют в 50 см3 воды и смешивают с раствором Юг NaOH в 90 см3 Н2О. К смеси добавляют 100 см3 профильтрованного раствора, содержащего 25 г технического гипохлорита кальция в 200 см3 воды. Раствор нагревают до исчезновения запаха уксусной кислоты. РЬО2 выделяется в виде коричневого осадка. Если при добавлении новых порций раствора гипохлорита кальция снова образуется осадок, окисление продолжают, как описано выше. Темный мелкокристаллический препарат несколько раз промывают горячей водой. Затем осадок смешивают с 50 см3 3 М раствора HNO3 для удаления плюмбатов, которые образуются в небольшом количестве, а также РЬ(ОН)2. После многократного промывания горячей водой методом декантации осадок отсасывают и высушивают в вакуум-эксикаторе над фосфорным ангидридом.
Темно-коричневый микрокристаллический порошок; при нагревании выше 345 °C разлагается с выделением Оз и с образованием РЬ3О4 и РЬО.
Гидролиз
2SbCl3 + ЗН2О ---Sb2O3 + 6НС1
Sn(SO4)2 + 2Н2О -> SnO2 + 2H2SO4
151. Оксид олова(П) SnO
SnCl2 + Na2CO3 + H2O -> Sn(OH)2 + 2NaCl -f- CO2
Sn(OH)2 --► SnO + H2O
SnCl2-2H2O растворяют в небольшом количестве горячей конц. НС1 и приливают раствор Na2CO3 до тех пор, пока жидкость не приобретет щелочную реакцию (проба по фенолфталеину). Выделившийся Sn(OH)2 нагревают в маточном растворе в те
50. Методы синтеза препаратов
589
чение 2—3 ч; при этом образуется SnO. Продукт промывают водой и высушивают при 110 °C.
Темно-синее кристаллическое вещество с металлическим блеском; при нагревании на воздухе выше 220 °C окисляется с образованием SnOj.
Термическое разложение
1. Пероксиды:
ВаО2 --->• ВаОд-1/2О2 (при 1100 °C)
2. Гидроксиды. Термическое разложение гидроксидов или оксигидратов металлов — еще один важный метод получения оксидов металлов. В некоторых случаях разложение гидроксида с выделением воды происходит уже при комнатной температуре; к таким соединениям относятся AgOH, Hg(OH)2, CuOH и Н2СгО4. При несколько более высокой температуре, например в кипящей воде, разлагаются'Sn(ОН)2 (ср. с предыдущим препаратом) и ТЮОН. Многие гидроксиды разлагаются только при высоких температурах, например Mg(OH)2, Sr(OH)2 и Ва(ОН)2 (~800°С), Си(ОН)2 (600—700°С), Т1О2-•aq (800—1000 °C).
3. Оксигалогениды:
ZrOCl2-8H2O --► ZrO2 + 2НС1 + 7Н2О
Аналогично получают НЮ2.
4. Иодаты:
2Ва(1О3)2 -► 2ВаО -{- 212 + 5О2
5. Карбонаты::
МеСО3 ---► МеО-(-СО2
Термическое разложение карбонатов также является технологически важным методом получения оксидов (например, обжиг известняка). Для количественного протекания реакции необходимо или непрерывно удалять образующийся СО2 (поток воздуха, открытый тигель), или выбирать такую температуру разложения, при которой давление СО2 превышало бы атмосферное давление. Условия термического разложения ряда карбонатов: Li2CO3 — 700 °C (вакуум); ВеСО3— 900°C; MgCO3* •Mg(OH)2 — 600°C; СаСО3 — 800°C; SrCO3— 1300°C (Н2); ВаСО3 — 950—1150 °C (глубокий вакуум); СоСОз — 350 °C (глубокий вакуум; до СоО), 700 °C (до Со3О4); NiCO3 — 1000—1100 °C.
6. Нитраты:
2Me(NO3)2 -»- 2МеО 4NO2 + О2
(Ме = Ва, Со, Си, Mg, Ni, Sr)
В подобных реакциях могут участвовать металлы со степенью окисления +IV (Мп, Th).
590
ЕЛ. Практикум по препаративной химии
152. Оксид марганца(1У), пиролюзит 0-МпО2
Mn(NO3)2 --► МпО2 4- 2NO2
Mn(NO3)2-6H2O разлагают на воздухе при нагревании приблизительно до 190 °C, растирают в порошок, кипятят с азотной кислотой (конц. HNO3, разбавленная в отношении 1:6) и нагревают в потоке воздуха до 450—500 °C. При температуре выше 530°C и атмосферном давлении МпО2 разлагается с выделением О2.
7. Сульфаты:
MeiiSO4 --► MeilO-f-SO3; 2MeiiSO4 -->- SO2 4- (MeiH0)2S04
(MeIHO)2SO4---► Me2niO3 + SO3
Для получения кристаллического оксида проводят разложение сульфата в расплаве, используемом в качестве растворителя. Для этого применяют расплавы фторидов, хлоридов или сульфатов щелочных металлов (Li, Na, К). Предварительно высушенную смесь исходных конпонентов (табл. Е.11) помещают в фарфоровый или (в случае фторидов) платиновый тигель и плавят в течение 2—4 ч при 1000 °C в закрытом тигле. После охлаждения содержимое тигля обрабатывают водой для удаления растворителя и фильтрованием отделяют оксид металла. Выход оксида составляет 60—90%.
8. Оксалаты:
МеС2О4 ---► МеО + СО2 4- СО
(Ме=Ве, Mg, Са, Fe(Fe2O3), Си, Zn, Cd)
Эта реакция характерна лишь для некоторых оксалатов, например для перечисленных выше или Th (ThO2), и протекает только при проведении разложения в открытых сосудах на воздухе. В остальных случаях идут побочные реакции образования карбонатов и металлов. Одновременно выделяющийся оксид углерода создает восстановительную среду, что приводит к получению оксидов более низких степеней окисления. 153. Оксид цинка ZnO
ZnC2O4 --► ZnO 4- СО2 4- СО
Для получения оксалата цинка, применяемого для разложения, растворяют 32,6 г ZnCl2-l,5H2O в 200 см>3 Н2О и добавляют 2,5 см3 2 М раствора НС1. Одновременно готовят второй раствор, содержащий 31,3 г (NH4)2C2O4-Н2О в 220 см3 воды с 2,5 см3 2 М раствора NH4OH. Оба раствора нагревают до 70 °C, после чего раствор оксалата аммония тонкой струей приливают к раствору соли цинка при постоянном помешивании. Осадок после отстаивания промывают методом, декантации до отрицательной реакции на ионы С1“ в промывных водах. Затем осадок переносят в нутч-фильтр и отсасывают досуха. Пе-
50. Методы синтеза препаратов
591
Таблица Е.11. Реагенты для получения и свойства некоторых оксидов металлов
Оксид металла Исходное вещество (А) Растворитель (В) A/В, x моль/моль Цвет и форма частиц оксида металла
СеО2 Ce(SO4)2 NaCl 0,1 Белые блестящие нити и иглы
СоО COSO4 Na2SO4 0,03 Серо-черные листочки
C0S04 KF 0,1 Темно-зеленые хорошо сформировавшиеся кристаллики
СГ2О3 Сг2(5О4)з NaCl 0,1 Зеленый кристаллический порошок
СиО C11SO4 KF 0,1 Черные иглы
Ре20з FeSO4 Na2SO4 0,03 Темно-красные листочки
Fe2 (804)3 Na2SO4 0,03 Темно-красные листочки
FeSO4 NaCl 0,1 Крупные темно-красные кристаллы в форме листочков
FeSO4 LiCl 0,1 Темно-зеленые листочки
Fe2(SO4)3 LiCl 0,1 Черные листочки
FeSO4 KF 0,1 Крупные черные кристаллы в форме листочков
ЪйгОз La2 (504)3 NaCl 0,1 Белые иглы и листочки
Мп20з MnSO4 Na2SO4 0,03 Темно-красный кристаллический порошок
MnSO4 NaCl ' 0,1 ) Темно-красные игольчатые, > частично хорошо сформиро-
MnSO4 LiCl 0,1 J вавшиеся кристаллики
NiO NiSO4 Na2SO4 0,03 Зеленые иглы и листочки
NiSO4 KF 0,1 Длинные зеленые иглы
NiSO4 LiCl 0,1 Иглы от темно-зеленого до черного цвета
NiSO4 L12SO4 0,2 Иглы от темно-зеленого до черного цвета
TiO2 TiOSO4 Na2SO4 0,03 Желтоватый кристаллический порошок
реносят ZnC2O4-2H2O в плоскую чашку и постепенно в течение 6 ч нагревают в сушильном шкафу до 240 °C; выдерживают соль при этой температуре еще в течение 12 ч, при этом соль почти полностью теряет кристаллизационную воду. Безводный оксалат нагревают затем в течение 4 ч при 400 °C для получения ZnO.
Белый тонкий порошок; при нагревании обратимо окрашивается в желтый цвет; в 100 г Н2О растворяется при комнатной температуре 3-10-4 г ZnO. Возгоняется при 1800 °C.
592 Е.11. Практикум- no препаративной химии
9. Аммонийные соли кислородсодержащих кислот
154. Оксид ванадия(V) V2O5
2NH4VO3 ----- V2O8 + 2NH3 + Н2О
V2O5 особой чистоты получают путем прокаливания NH4VO3 при 500—550°C. Следовые количества азота, обычно еще сохраняющиеся в препарате, можно удалить нагреванием в потоке влажного кислорода в течение 18 ч при 530—570 °C.
Порошок красного или оранжево-красного цвета; в 100 г НаО растворяется 7-Ю-2 г V2O5. 7’пл = 674°С.
Реакции с кислотами
В этом случае речь в общем идет о выделении оксидов в свободном виде из солей кислородсодержащих кислот при добавлении к ним минеральных кислот:
Na2[Sn(OH)e] + 2НС1 ->- SnO2 + 2NaCl + 4Н2О
Na2WO4 + 2НС1 ---- WO3 4-2NaCl + Н2О
Na2Cr2O7 H2SO4 1 * 2CrO3 Na2SO4 -f- H2O
155. Оксид молибдена(У1) MoO3
(NH4)2MoO4 + 2HNO3 --> H2MoO4 + 2NH4NO3
MoO3-H2O ---► MoO3 + H2O
При добавлении к кипящему раствору чистого перекристаллизованного молибдата аммония нагретой до кипения конц. HNO3 в осадок выпадает МоОз-НгО. После многочасового отстаивания зернистый осадок отфильтровывают на воронке Бюхнера, промывают и высушивают в течение 16—20 ч при температуре выше 150 °C. Препарат можно очистить возгонкой в кварцевой трубке при 780 °C.
Белый порошок; при нагревании обратимо окрашивается в желтый цвет. Сублимат состоит из блестящих бесцветных кристаллических крупинок. 7пл = = 795 °C; 7’квО=1155°С.
Реакции восстановления
Если металл может иметь несколько степеней окисления, то его низшие .оксиды можно получить из высших путем восстановления. При восстановлении водородом можно, регулируя температуру, получать оксид металла определенной степени окисления. При 800—900 °C, например, протекает реакция
WO3 + H2----► WO2 + H2O
Аналогично получают МоО2 при 500°C; РезО4 из Fe2O3 при 400 °C; УгОз (препарат *156). Водород в момент выделения при взаимодействии Zn с НС1 восстанавливает WO3 и МпО2:
2WO3 4- 1/2Н2 -► HW2Oe (вольфрамовая синь)
МпО2 + Н2 ---► МпО + Н2О
50. Методы синтеза препаратов
593
Аналогично получают молибденовую синь (см. также разд. 36.12). Целесообразно также использовать восстановление высших оксидов порошкообразных металлов:
3TiO2 + Ti --» 2Ti3O3; TiO2 + Ti -► 2TiO (1600°C)
V2O3 + V ----► 3VO; 11МоО3 Mo 3Mo4Ou
В последней реакции при 580 °C образуется у-оксид молибдена (вакуум). Аналогично получают у-оксид вольфрама.
156. Оксид ванадия(Ш) V2O3
V2OS 4- 2Н2 ► V2O3 2Н2О
V2Os восстанавливают в потоке Н2 сначала в течение 3 ч при 600°C (не выше, так как V2O5 плавится при 658°C), а затем еще в течение 6 ч при 900—1000 °C.
Восстановление можно осуществить также действием NH3, который разлагается при повышенной температуре:
2NH3 —> N2 + ЗН2
Черный порошок; окисляется на воздухе е образованием VaOs. Тая= •=1970 °C.
157. Оксид меди(1) С112О
4Cu(CH3COO)2 + N2H4 + 2Н2О -► 2Си2О + 8СН3СООН 4- N2
К 50 см3 концентрированного раствора ацетата меди добавляют 3—5 см3 2%-ного раствора гидрата гидразина. Раствор окрашивается в зеленый цвет, выделяется азот, и при стоянии выпадает желтый или желто-оранжевый осадок CU2O. Осадок промывают сначала водой, затем спиртом и эфиром. Следует избегать избытка гидразина при восстановлении; в противном случае может образоваться металлическая медь.
Желтый порошок, нерастворимый в воде. 7’ПЛ = 1232°С.
50.2.2. Оксиды неметаллов
Окисление
1. Окисление кислородом и озоном
Общая схема реакции; элементф-Ог—>оксид. Так получают СО2, Н2О, NO, O2F2, Р4О10, SO2, SeO2. Например:
ЗВг2 + 4О3 —> 6ВгО2
2СЮ2 + 2О3 --> С12О6 + 2О2
2. Действие окислителей:
Se + 4HNO, -->- H2SeO, + 4NO2 + Н,0
H2SeO3 -->SeO2+H2O
2Те + 9HNO3 -> Te2O3(OH)NO3 + 8NO2 Д 4H2O
594
E.II. Практикум no препаративной химии
Te2O3(OH)NO3 --> 2ТеО2 + HNOS
2F2 + 2NaOH --► 0F2 4- 2NaF 4- H20
2C12 4- 2HgO -► C12O 4- HgO • HgCl2
Гидролиз
2AsCl3 4- 3H2O --► As2O3 4- 6HC1
2HSbCle 4- 5H2O -► Sb2O5 4- 12HC1
158. Триоксид диазота N2O3
2(NO)HSO4 4- H2O --► N2O3 4- 2H2SO4
Поскольку для получения N2O3 нет необходимости применять чистую нитрозилсерную кислоту, SO2 пропускают в дымящую HNO3 до образования жидкой кашицы, к которой в колбе с пришлифованной капельной воронкой по каплям приливают воду.
Газообразный N2O3 при —20 °C конденсируется в зеленую жидкость; растворяется в бензоле, толуоле и хлороформе с образованием растворов си* него цвета. 7™=—103°C.
Дегидратация:
4HNO3 4- Р4О10 ---» 2NaO6 4- 4НРО3
4НС1О44-Р4О10 ---->- 2С12О7 4-4НРО3
2КС1О3 4- 2H2SO4 4- Н2С2О4-► 2СЮа 4- 2СОа 4- 2Н2О 4- 2KHSO4
(Образующийся в процессе реакции СО3 разбавляет взрывоопасный С1О2.)
С3Н4О4 (малоновая кислота) 4- Р4О10-► С,О2 (диоксид триуглерода) 4- 4HPOS
H2SO3 ----> SO2 4- НаО
ЗНЮ3 -----► НЮ3-12О8 4-Н2О
НЮ3-12О6 + НЮ3 ----->- 2I2OS 4- Н2О
-* ТеО3 4- ЗН2О (300—320 °C)
ТеО2 4- г/2Оа 4- ЗН2О (600 °C)
Н2ТеОв
159. Моноксид углерода СО
а) НССЮН --► СО4-Н2О
Прибор для получения газа (круглодонная колба вместимостью 1 дм3 с притертой капельной воронкой и газоотводной трубкой), заполненный на 2/3 конц. Н3РО4, нагревают на водяной бане приблизительно до 80 °C; затем приливают по каплям муравьиную кислоту. Для удаления примесей (СО2, воздуха, паров кислот, паров воды) газ пропускают последовательно через 50%-ный раствор КОН, щелочной раствор
50. Методы синтеза препаратов
595
Na2S2O4 (25 г Na2S2O4, 125 см3 Н20 и 20 см3 70%-кого КОН), над КОН, СаС12 и Р4Ою.
б) Н2С2О4-2Н2О -► СО + СО2 + ЗН2О
Смесь 100 г дигидрата щавелевой кислоты с 275 см3 конц. H2SO4 осторожно нагревают в круглодонной колбе до начала < выделения газа, не допуская, чтобы это выделение было слишком бурным. Наряду с СО в равных количествах образуется СО2, который поглощают в двух промывных склянках со 100 см3 50%-ного раствора КОН. Затем очищают газ так, как было указано в методе а).
Бесцветный ядовитый газ, не имеющий запаха. ТПл=—205,1° С; ТКжп= =—191,5°C. Горит голубым пламенем; смесь СО (12,5%) с воздухом взрывоопасна.
Термическое разложение
160. Диоксид азота NO2, N2O4
Pb(NO3)2 -» 2NOa-f-РЬО-f-VjOg
250 г мелкоизмельченного Pb(NO3)2 высушивают при 120 °C в течение нескольких суток в сушильном шкафу. Подготовленную таким образом соль нагревают в стальной трубке, закрытой с одного конца. Выделяющиеся пары осушают в трубке, заполненной Р4Ою, и конденсируют в сосуде для конденсации при —15 °C. Вследствие агрессивности газа установка должна быть снабжена стеклянными шлифами. Примесь N2O3 можно удалить только перегонкой в потоке кислорода и повторным высушиванием, в трубке, заполненной Р4Ою. (Необходимо избегать применения СаС12 в качестве осушителя, так как это приводит к образованию NOC1.) Для хранения жидкого Ы2О4 его перегоняют из колбы со шлифами в препаративную склянку с суженным горлом, охлаждаемую смесью льда с ^поваренной солью. Диоксид азота можно также поглотить конц. H2SO4 и снова выделить из раствора нагреванием.
Тпл=—10,8°C; ГКИП=21,2°С.
Протолиз
161. Диоксид углерода СОа
СаСО3 + 2НС1 -->- СО2 + СаС12 4- Н2О
Газ получают при взаимодействии кусочков мрамора с НС1 в аппарате Диппа (рис. Е.27).
Средства для очистки СО2 указаны в табл. Е.12. Диоксид углерода можно хранить над водой, насыщенной СО2, над конц. H2SO4 и Hg.
596
ЕЛ. Практикум по препаративной химии
Таблица Е.12. Средства для очистки СО2
Примесь Средство
02 Пары кислот H2S Раствор Сг(СН3СОО)2 или VOSO4 Кусочки КНСО3 Пемза, пропитанная CuSO4; 1 М раствор КМпО4; 1 М. раствор К2Сг2О/
Н2О О2, СО Конц. H2SO4 Активированная нагретая до 200°C смесь Cu4-Cu2O
Бесцветный газ, не имеющий запаха. Тпл =—56,7°C (при 105 Па). Возгоняется при —78,48 °C.
162. Диоксид серы SO2 (см. препарат 165)
NaHSO3 + H2SO4 -> SO2 4- NaHSO4 4- H2O
Газообразный SO2 получают, добавляя по каплям конц. H2SO< к 40%-ному раствору NaHSO3. Целесообразно вести синтез в возможно большей склянке для отсасывания с насаженной капельной воронкой. Газ очищают, пропуская последовательно через две промывные склянки с конц. H2SO4.
Реакции окисления-восстановления
163. Оксид диазота N2O
HNO, 4- NH2SO,H -► N2O 4- H2SO4 4- H2O
Реакцию целесообразно проводить в колбе с пришлифованной газоотводной трубкой, нижний конец которой заполнен слоем стеклянной ваты для улавливания увлекаемых N2O паров кислоты. 8 г амидосерной кислоты нагревают в колбе с 20 см3 прокипяченной 73%-ной HNO3 на небольшом пламени гррелки до начала выделения газа. При слишком бурном прохождении реакции необходимо охлаждать колбу. Для очистки выделяющегося газа от примесей его последовательно пропускают над твердым карбонатом калия, через промывную склянку с концентрированным раствором сульфата железа(II) (для удаления NO) и раствор КОН (1 : 1).
164. Моноксид азота NO
а) Концентрированный раствор NaNO2 приливают по каплям к концентрированному раствору FeSO4, разбавленному равным объемом НС1 (1: 1). Образуется очень чистый NO; высшие оксиды азота можно удалить промыванием 5 М раствором КОН и 90%-ной H2SO4, влагу — действием конц. H2SO4 и Р4Ою.
50. Методы синтеза препаратов
597
б) К обезжиренной медной стружке или листовой меди добавляют HNO3 (пл. 1,1 —1,2) (если необходимо, охлаждают) и после вытеснения воздухом бурых паров NO2 улавливают бесцветный NO. Его можно высушить над H2SO4 и Р4О10. Реакцию проводят в аппарате Киппа.
Т’нИП=—151,8 °C; 7'пл=—163,7 °C. Сохранять только над Hg в качестве жидкостного затвора.
165. Диоксид серы SO2 (см. препарат 162)
2H2SO4 + Cu -► SO2 + CuSO4 + 2Н2О
В круглодонную колбу вместимостью 200 см3 помещают 20 г Си (в виде стружек, нарезанных на токарном станке или тонких листовых полос) и 50 см3 конц. H2SO4. Содержимое колбы нагревают на горелке Бунзена и выделяющийся SO2 направляют в трехгорлую склянку Вульфа. Газоподводящая трубка должна доходить до дна склянки Вульфа, на 74 заполненной конц. H2SO4. Во втором1 горле склянки укрепляют газоотводную трубку; через среднее горло вводят длинную вертикальную предохранительную трубку, доходящую почти до-дна склянки. Для получения 1 дм3 необходимо около 30 г Си.
50.3. Методы получения сульфидов
50.3.1. Синтез сульфидов из элементов
В табл. Е.13 приведены условия, синтеза из элементов наиболее часто применяемых препаратов.
166. Сульфид кадмия, кристаллический CdS
3 г металлического кадмия (порошкообразного или электролитического кристаллического) сплавляют в фарфоровом тигле с 15 г К2СО3, 3 г Ыа2СОа и 8 г серы. Приблизительно через 2 ч расплавленную массу охлаждают и выщелачивают водой. Под микроскопом видны небольшие четко ограниченные золотисто-желтые блестящие гексагональные столбики или пластинки CdS.
Сульфиды неметаллов также можно синтезировать из элементов. При этом в зависимости от весовых соотношений реагирующих компонентов образуются различные побочные соединения. Например, при взаимодействии серы с хлором могут образоваться S2C12 (препарат 63), SC12 и SC14. Реакция между фосфором и серой в зависимости от количества реагирующих элементов приводит к образованию следующих сульфидов фосфора: P4S3, P4S5, P4S7 и P2Ss-
167. Пентасульфид дифосфора P2S5
100 г сухого красного фосфора тщательно смешивают с 260 г серы. Из круглодонной колбы вместимостью 1 дм3 вытесняют
Таблица Е.13. Сульфиды, синтезируемые из элементов
Сульфид Уравнение реакции Условия получения
Ag2S 2Ag+S—>-Ag2S Пропускание паров серы над Ag в кварцевой трубке при 250 °C в потоке азота
A12S3 2A1 + 3S—>-Al2S3 Поджигание смеси 3,4 г А1 с 6 г порошковой серы в глиняном тигле с помощью магниевой ленты
CdS Cd + S—>-CdS См. препарат 166
CoS Co + S—>-CoS Нагревание тонко измельченной смеси эквивалентных количеств кобальта и серы в вакуумированной запаянной кварцевой трубке в течение 2—3 сут при 650’С
•Cu2S 2Cu + S—>-Cu2S Сплавление меди с серой в глубоком вакууме t
’FeS Fe+S—>-FeS Нагревание смеси стехиометрических количеств компонентов в глубоком вакууме в течение ~ 24 ч до 1000 °C
'6a2S3 2Ga+3S—>-Ga2S3 Пропускание паров серы" над нагретым Ga в запаянной с одного конца кварцевой трубке
K2S 2K+S—>K2S | В среде жидкого NH3 в специаль-
'N.a2S 2Na + S—>-Na2S J ном приборе для работы с глубоким вакуумом
Na2S3 2Na+3S—>-Na2S3 В среде кипящего толуола
MoS2 Mo+2S—>-MoS2 Нагревание расчетных количеств Мо и S в железной трубке
P2S3 2P+5S—>-P2Ss См. препарат 167
ReS2 Re+2S—>ReS2 Нагревание смеси в запаянной кварцевой трубке до 980—1000 °C в течение 18 ч
SnS Sn+S—>-SnS Нагревание олова с избытком серы в закрытой с одного конца фарфоровой трубке до ~ 900 °C
T12S 2T1+S—>-Tl2S Сплавление стехиометрических количеств Т1 и S под давлением в трубке, заполненной Н2
WS2 W+2S—>WS2 Нагревание расчетных количеств W и S в атмосфере азота в запаянной кварцевой трубке до 800 °C в течение 24 ч
50. Методы синтеза препаратов 599'
диоксидом углерода воздух и вносят туда ложку смеси фосфора с серой. Колбу осторожно нагревают на горелке Бунзена до окончания реакции. Затем горелку удаляют и добавляют следующую порцию смеси, при необходимости снова нагревая реакционный сосуд. После охлаждения колбу разбивают и собирают серый, немного гигроскопичный неочищенный продукт. Для очистки препарат перегоняют из маленькой ши-рокогорлой реторты без тубуса; первые порции дистиллата отбрасывают. Основную массу дистиллата собирают в сухую колбу. Колбу разбивают, затвердевшее желтое аморфное вещество освобождают от осколков стекла и хранят в плотно закрывающейся банке.
В целях безопасности к началу синтеза следует приготовить песок и поддон, чтобы при случайном повреждении стеклянного сосуда можно было немедленно загасить пламя.
Светло-желтое вещество; раствориется в NaOH с образованием тиофосфатов натрия. Тпл=276°С; ТКип = 514 °C.
50.3.2. Получение сульфидов действием сероводорода
Простейший метод синтеза сульфидов тяжелых металлов заключается в осаждении их из растворов солей металлов под действием сероводорода, основанном на использовании малой растворимости сульфидов тяжелых металлов в воде (табл. Е.14). При этом получают соединения в мелкодисперсном, зачастую даже в коллоидном состоянии, в то время как при проведении реакции в газовой фазе часто образуются кристаллические сульфиды.
Хорошо растворимые в воде сульфиды щелочных и щелочноземельных металлов в виде гидратов получают взаимодействием гидроксидов металлов с H2S.
168. Сульфид ртути(П), красная модификация HgS
Hg(CH3COO)2 + H2s -► HgS + 2CH3COOH
35 г Hg(CH3COO)2 и 25 г NH4SCN растворяют в 10 см3 горячей ледяной уксусной кислоты. В горячий раствор пропускают равномерный поток H2S до полного осаждения. Затем уксусную кислоту медленно испаряют (осторожно, HCN!), при этом черный осадок становится красным. Ледяная уксусная кислота должна находиться в растворе до полного перехода HgS в красную модификацию-; перегревания следует избегать. На этой, последней, стадии реакции пасту непрерывно перемешивают. При несоблюдении такой меры предосторожности получается продукт матово-красного или коричневого цвета. После окончательного удаления- кислоты и охлаждения препарата смесь отфильтровывают на воронке Бюхнера, прибавив к ней'
Таблица Ё.14. Сульфиды, синтезируемые по реакции с Й2§
Сульфид Уравнение реакции Условия получения
AS2S5 2H3AsO44- 5H2S—*-As2S3+8H2O Осаждение из водного раствора, подкисленного конц. НС1
BaS, CaS, SrS МеСО3+H2S—>MeS 4- H2O+CO2 Нагревание карбоната в потоке H2S4-H2 до ~900 °C
CdS (кристаллический) Cd + H2S—>CdS + H2 Взаимодействие H2S и паров кадмия при ~ 800 °C
CdS CdSO4+H2S—>CdS + H2SO4 Осаждение из сернокислого водного раствора
CoS Co(NO3)2+H2S—»-CoS+2HNO3 Осаждение из аммиачного водного раствора без доступа воздуха (в потоке N2 или СО2)
Cr2S3 (кристаллический) 2CrCl3+3H2S—>Cr2S3+6HCl См. препарат 169
GeSa GeO2+ 2H2S—>GeS2-}-2H2O Пропускание сильного потока H2S в раствор GeO2 в 6 М НС1
GeS (кристаллический) GeO2+ H2S 4- H2—э-GeS 4- 2H2O Пропускание смеси H2S и Н2 над нагретым GeO2 и возгонка GeS
HgS (черный) HgS (красный) HgCl24-H2S—>HgS + 2HCl Hg (CH3COO) 2+ H2S—»-HgS 4-2CH3COOH См. препарат 168
La2S3 (и сульфиды других редкоземельных металлов) 2LaCl34-3H2S—>-La2S34-6HCl Пропускание чистого H2S над LaCl3 при 500— 700 °C в течение ~115 ч с последующим нагреванием в потоке H2S при 800—1000 °C в течение нескольких часов _
0881—68
MgS Mg4-H2S—->MgS4-Hz
MnS MnClz4-HzS—->MnS4-2HCl
(NH4)2S5 2NH34-H2S4-4S—>(NH4)2S3
NaHS NaOC2H6 4- H2S—>N all S 4- C2H6OH
Na2S-2H2O 2NaOH 4- H2S—>-NazS 2H2O
NiS •NiCl2+H2S—>NiS4-2HCl
PtS2 H2PtCl64-2H2S—>PtS2+6HCI
Re2S? 2KReO44-7H2S4-2HCl—>Re2S74-2KCl+8H
TiS2 TiCl4+H2S—-»-TiS24-4HCl
v2s3 V2O34-8H2S—>V2S34- 3H2O
ZnS ZnSO44- H2S—>-ZnS 4- H2SO4
Нагревание магния в потоке H2S (скорость 8 см3/мин), сначала при 580 °C, затем при более низкой температуре (скорость H2S 15 см3/мин)
Осаждение из Кипящего аммиачйого водного раствора
Пропускание H2S без доступа боздуха в Суспензию серы в растворе NH4OH
Внесение Натрия в абсолютный спирт и пропускание H2S до насыщения
Выпарийание насыщенного раствора H2S, смешанного с эквивалентным количеством NaOH
Осаждение из насыщенного водного раствора, содержащего NH4C1, без доступа воздуха
Осаждение при Нагревании из солянокислого водного раствора
ОсажДение из соляйокисЛого водного раствора
Взаимодействие газообразных веществ в трубке, нагретой До температуры красного каления
Пропускание H2S над V2O3 при 750 °C
Осаждение из водного раствора в присутствии ацетатного буфера (pH 2-^3)
602 Е.11. Практикум, no препаративной химии
200 см3 воды. Осадок затем1 промывают и высушивают между толстыми слоями фильтровальной бумаги.
Исходный ацетат ртути (II) получают растворением HgO в 50 %-ной СНзСООН при нагревании на водяной бане с последующим фильтрованием и охлаждением фильтрата льдом. Выделившиеся кристаллы отсасыв'ают.
169. Сульфид хрома(Ш) Сг25з
2СгС13 + 3HaS -► Cr2S3 + 6НС1
Безводный СгС13 (см. препарат 114) нагревают в фарфоровой лодочке, помещенной в трубку из тугоплавкого стекла или, .лучше, в кварцевую, до температуры красного каления. Через трубку пропускают поток сероводорода, высушенного в осушительной колонке с СаС12. Через 2 ч дают трубке охладиться в потоке H2S и получают Cr2S3 в виде черных, похожих на графит, блестящих кристаллов.
50.4. Методы получения водородсодержащих
соединений
50.4.1. Синтезы из элементов
170. Гидрид лития ЫН
Li + 1/гНг -► LiH
Литий реагирует с водородом при температуре выше 440 °C с образованием гидрида; при 600—630 °C реакция протекает очень бурно. Поскольку литий и гидрид лития выщелачивают кремний из стекла и фарфора, а пары гидрида при температуре синтеза создают значительное давление, при проведении реакции следует соблюдать особые меры предосторожности. Лучше всего синтез проводить в фарфоровой трубке, облицованной внутри на протяжении всей обогреваемой зоны листовым никелем. Литий гидрируют в лодочке из листового железа, полученного электролизом. Для1 полной очистки железных и никелевых частей установки от оксидов ее вместе с лодочкой нагревают до 800 °C в потоке чистого сухого водорода (водород, полученный электролизом, пропускают над паллади-рованным асбестом при 300 °C, СаС12 и Р4Ою). После охлаждения литий очищают парафиновым маслом, промывают безводным эфиром, помещают в железную лодочку, поверхность которой полностью очищена от оксидов, и во влажном состоянии как можно быстрее вносят в установку. Вакуумируют, нагревают до 200°C для удаления остатка растворителя, пропускают через установку поток водорода и продолжают нагревание. При 440 °C начинается поглощение водорода, которое ;энергично протекает при 600—630°C. В этот момент устанав
50. Методы синтеза препаратов
603
ливают давление водорода 400—500 мм> рт. ст., затем быстро нагревают установку до 700 °C. Эту температуру не следует превышать, так как в противном случае гидрид лития возгоняется. Реакция заканчивается за 10—20 мин. Дают установке охладиться в потоке водорода и затем вытесняют водород сухим воздухом. Продукт удаляют из лодочки заостренным стальным шпателем.
Белое, очень твердое и хрупкое вещество. При взаимодействии с водой •бурно выделяет Hg, образуя LiOH. Тпл=680°С.
50.4.2. Реакции взаимодействия с водородсодержащими соединениями
В табл. Е.15 приведен обзор некоторых реакций, используемых для получения гидридов, в которых участвуют такие доноры водорода, как Н2О, кислоты НХ или основания МеОН.
171. Бромоводород НВг (гидролитическое получение)
2Р + ЗВг2 --► 2РВг3
РВг3 + ЗН2О --► Н3РО3 + ЗНВг
При гидролитическом получении бромоводорода из трибромида фосфора в качестве исходных веществ лучше всего брать красный фосфор и бром и проводить обе приведенные выше реакции непосредственно в процессе синтеза НВг. В колбу с пришлифованной капельной воронкой и газоотводной трубкой вносят 5 см3 дистиллированной воды и 3 г красного фосфора. Газоотводная трубка соединена с U-образной трубкой, которую для удаления непрореагировавшего Вг2 из выделяющегося газа заполняют влажным красным фосфором, нанесенным на стеклянные шарики или на глиняные черепки. Для начала реакции из капельной воронки в колбу медленно добавляют 30 г брома при охлаждении ледяной водой, слегка встряхивая колбу. После добавления всего количества Вг2 отгоняют образующийся НВг, осторожно нагревая колбу. Бромоводород полностью поглощают небольшим количеством воды, находящейся в приемнике-абсорбере; при этом трубка подвода газа по возможности не должна быть погружена в воду.
Получение безводного бромоводорода описано в методике синтеза препарата 129.
Бесцветный ядовитый газ с резким запахом. Водный раствор НВг можно сконцентрировать фракционной перегонкой до содержания кислоты 48%.
172. Хлороводород НС1
2NH4C1 + H2SO4 --> 2НС1 4- (NH4)aSO4
Хлороводород лучше всего получать взаимодействием NH4C1 (в виде кусочков размером с орех) с конц. H2SO4 при комнатной температуре в аппарате Киппа. При этом образуется су-
39*
Таблица Е.15. Реакции, используемые для синтеза
водородсодержащих соединений
Водородсодержащее соединение Уравнение реакции Условия синтеза
Реакции с водой
НВг РВг3+ЗН2О—►ЗНВг+НзРОз См. препарат 171
HI Р13 + ЗН2О_,ЗШ + Н3РО3 Внесение фосфора в суспензию иода в воде (бурная реакция)
сгн2 СаС2+2Н2О—>-С2Н2Ч-Са (ОН) 2 Сопровождается сильным выделением тепла
РНз Са3Р 2~Р 6Н2О—>2PH3-f- ЗСа (ОН)2 Можно использовать коммерческий препарат Са3Р4
РН3 А1Р + ЗН2О—*РНз+А1 (ОН)з В среде разбавленной H2SO4
H2S Mg(SH)2+2H2O—>2H2S+Mg(OH)2 Нагревание концентрированного раствора Mg(SH)2
H2Se Al2Se3+6H2O—>-3H2Se+2Al (ОН)3 Гидролиз в колбе для получения газа при добавлении воды по каплям
Реакции с кислотами
НВг KBr+H2SO4—*HBr+KHSO4 Добавление разб. H2SO4 к охлажденному раствору КВг, фильтрование и перегонка реакционной смеси (получается раствор НВг, кипящий при постоянной температуре
НС1 2NH4CI + H2SO4—>-2НС1+ (NH4)2SO4 См. препарат 172 -
HF CaF2-f-H2SO4—>2HF4-CaSO4 Сильное нагревание смеси в железном автоклаве на воздушной бане (дистиллят собирают в медный сосуд, охлаждаемый смесью льда с NaCI)
HI H2S + I2—>2HI+ S См. препарат 173
HCN 2NaCN+H2SO4—*2HCN + Na2SO4 Добавление по каплям водного раствора NaCN (100 г/120 см3 Н2О) в нагретую до 90 °C смесь 100 г конц. H2SO4, 40 см3 Н20 и 2 г FeSO4 и отгонка (Ткип = 26,5 °C), (под тягой!)
HN3 NaN3+H2SO4—>HN3+NaHSO4 Работать только с растворами н в строгом соответствии с методикой синтеза (возможен взрыв!)
H2S FeS + 2HC1—>H2S + FeCl2
Н2Те Al2Te34- 6HC1—>ЗН2Те4-2А1С13 Внесение А12Те3 в 4 М НС1 в потоке N2
SbH3 Mg3Sb2+6HCl—>-2SbH3+3MgCl2 Внесение порошкообразного Mg3Sb2 в охлажденную разбавленную НС1 (р = 1,06)
Реакции с основаниями
NH3 2NH4Cl+Ca(OH)2—>2NH3+CaCl2+2H2O
РН3 PH4I + KOH—>ph3+ki+h2o Добавление по каплям разбавленного раствора КОН (1:2) к РН41 без подвода тепла
РНз 4P + 3KOH +3H2O—>PH3+3KH2PO2 Диспропорционирование белого фосфора при внесении его в разбавленный раствор КОН
606
E.II. Практикум no препаративной химии
хой газообразный НС1. При использовании NaCl вместо NH4C1 синтез проводят в установке для получения газов при периодическом нагревании.
Сильный и равномерный поток хлороводорода пропускают
через смесь 1 объема конц.
H2SO4, находящейся в рядом
Рис. Е.39. Схема прибора для получения постоянного потока газообразного НС1.
1— ситовая пластинка; 2 — кран; 3 — пришлифованный колпачок; 4 — отводная трубка.
соляной кислоты с 1 объемом стоящей установке (рис. Е.39).
Суженную к нижнему концу стеклянную трубку диаметром 5 см наполняют инертным материалом слоем 20—25 см (галькой или стеклянными шариками), который удержи-• вают в трубке с помощью ситовой пластинки 1. Внизу находится кран 2, снабженный двухколенной трубкой и предназначенный для слива отработанной жидкости. Верхнее колено расположено ниже уровня пластинки с отверстиями. В пришлифованную крышку 3 вставлены две капельные воронки для соляной и серной кислот, снабженные счетчиками капель для точной регулировки подачи кислот. Для обеспечения тщательного смешивания кислот сливные трубки воронок располагают рядом над поверхностью наполнителя. Меняя скорость подачи кислот, можно регулировать поток газа. Воздух, вначале находящийся в установке, можно вытеснить через отводную трубку 4, закрыв кран 2 и тем самым заполнив пространство над наполнителем смесью кислот.
Высушенный хлороводород — бесцветный газ с резким запахом; очень хорошо растворяется в НгО, растворение сопровождается сильным выделением тепла.
173. Иодоводород HI
H2S + I2 -► 2HI + S
Суспензию 120 г иода в 150 см3 воды энергично перемешивают в широкогорлом сосуде вместимостью 500 см?, закрытом
50. Методы синтеза препаратов 607
пробкой с тремя отверстиями (для газоподводящей трубки, погруженной в жидкость, газоотводной трубки и мешалки). Мешалка должна быть хорошо пригнана к стенкам сосуда. В суспензию пропускают поток H2S, скорость прохождения которого регулируют в зависимости от степени поглощения газа раствором. Небольшой избыток H2S из реакционного сосуда через газоотводную трубку направляют в тягу или к поверхности содержащегося в соответствующей склянке NaOH (газоотводная трубка не должна быть погружена в гидроксид натрия). Приблизительно через 1 ч раствор почти совершенно обесцвечивается и одновременно выделяются значительные количества серы. С крупных частиц серы раствор декантируют, а мелкодисперсные отделяют фильтрованием через стеклянный фильтр. Содержащийся еще в растворе сероводород удаляют кратковременным кипячением. Проба раствора не должна давать реакции на ион S2-.
Раствор перегоняют из колбы вместимостью 250 см3; во избежание бурного вскипания в колбу помещают эффективно действующие кипятильники и собирают фракцию, перегоняющуюся при 125—127 °C. Выход составляет ~90%, считая на введенный в реакцию Ь.
Полученный продукт содержит 57% HI; р=1,70 г/см3. Т’кип=126°С (при 760 мм рт. ст.). Кислота сильно дымит на воздухе. Водные растворы HI следует хранить в хорошо закрывающихся темных склянках, пробки которых целесообразно заливать парафином. Можно рекомендовать еще одну меру защиты от окисления: перед закупориванием склянок вытеснить воздух из пространства над уровнем жидкости инертным газом.
Бесцветный газ с резким запахом; иа воздухе окрашивается в коричневый цвет вследствие выделения иода. 7’пл=—50,8 °C; ТКип=—35,5 °C.
50.5. Отдельные группы веществ
50.5.1. Бориды, карбиды, нитриды, силициды
Для характеристики этих соединений (см. также разд. 35.8.1) необходимо провести их классификацию, что полезно также и для препаративных работ.
1. Соединения металлического характера. В целом металлическая проводимость уменьшается в следующей последовательности: металл>карбид>нитрид>борид. К этой группе относятся соединения элементов побочных подгрупп четвертой, пятой и шестой групп периодической системы. Все они характеризуются высокой химической устойчивостью, твердостью и являются тугоплавкими соединениями (например, температуры плавления HfC 3890°C; ZrN 2985°C).
608
E.II. Практикум no препаративной химии
2. Соединения' с решеткой алмаза. Свойства соединений этой и предыдущей групп аналогичны, однако рассматриваемые соединения не обладают металлической проводимостью. В эту группу входят A1N, В4С, BiN, SiC.
3. Солеобразные соединения. Их получают из наиболее электроположительных металлов. Как правило, они бесцветны и образуются в кристаллическом состоянии. Карбиды разлагаются водой или кислотами с образованием следующих продуктов: а) СН4 — из ВегС, А14С3; б) С2Н2— из ацетилидов общей формулы) Ме2[С2, МепСг и Ме2ш(С2)з (где Me1 = щелочные металлы; Me11 = щелочноземельные металлы, Ag, Си, Hg, Cd, Zn, редкоземельные металлы; Меи1=А1), в случае щелочньгх металлов— также и Ме'НСг; в) С3Н4 (аллилы)—из М^2Сз; г) смесь углеводородов — из карбидов металлов группы железа, U2C3.
Методы получения соединений с решеткой алмаза можно разделить на пять групп:
а) синтез из элементов, включая алюминотермию; б) реакции с гидридами:
3Mg 4- 2NH3 --► Mg3N2 4- ЗН2 (аналогично Zn3N2)
2CuCl 4- С2Н2 -► Cu2C2 + 2НС1
в) разложение амидов:
3Cd(NHa)2 -> Cd3N2 + 4NH3
г)' взаимодействие с оксидами:
СаО 4- ЗС--► СаС2 4- СО
2ТЮ2 4- 4С 4- Na -► 2TiN 4- 4СО
д) выделение из газовой фазы.
Металл или галогенид металла реагирует' с галогенидами бора, углерода или кремния в атмосфере водорода (методы наращивания), для получения нитридов в пары галогенидов металлов вводят аммиак.
174. Борид алюминия А1В!2
Черные очень твердые и устойчивые кристаллы (см. табл. Е.8).
175. Нитрид алюминия A1N
Al-HAN, ---- A1N
Порошок алюминия особой чистоты обезжиривают эфиром и высушивают или нагревают в потоке азота до 150 °C, затем его насыпают в никелевую лодочку, помещают в трубку из кварца или твердого фосфора и нагревают в электрической печи в потоке очищенного азота. Образование нитрида на поверхности алюминия начинается уже ниже 650 °C, но основная реакция, сопровождающаяся разогревом реакционной массы,
50. Методы синтеза препаратов
609
начинается только при 820 °C. К этому времени необходимо усилить поток азота, чтобы избежать понижения давления. По окончании основной реакции дают массе охладиться в потоке азота. Поскольку продукт еще содержит включения металла, его измельчают в порошок и подвергают вторичной обработке азотом при 1100—1200 °C в течение 1—2 ч. Получают почти белый продукт с содержанием азота лишь немного ниже теоретического.
Медленно гидролизуется на влажном воздухе с выделением аммиака. Тпл = 2150—2200 °C.
176. Нитрид бора BN
В2О3 + NHaCONHa ---»- 2BN + СОа + 2НаО
1 весовую часть плавленого тонко измельченного В2О3 хорошо перемешивают с 1,5—2 весовыми частями мочевины и нагревают в закрытом крышкой фарфоровом тигле (под конец до светло-красного каления). Образовавшуюся массу растирают и промывают водой, к которой добавляют несколько капель соляной кислоты. Затем смесь отсасывают и нитрид бора высушивают в сушильном шкафу.
Белый легкий порошок; в кипящей воде медленно гидролизуется. 7’Пл> >2800 °C.
177. Силицид магния Mg2Si
4Mg + SiO2 --► Mg2Si -4- 2MgO
Осажденную кремневую кислоту обезвоживают нагреванием до светло-красного каления в течение нескольких часов. Кремневая кислота должна содержать менее 0,5% щелочи. Хорошо растертую в порошок безводную кислоту тщательно смешивают с двойным количеством порошкообразного магния. Затем 100 г этой смеси поджигают в железном тигле вместимостью около 1 дм3, глубоко погруженном в большой сосуд с холодной водой. Реакция, сопровождаютцаяся разогреванием смеси до ослепительно белого свечения, быстро распространяется по всей массе. Сразу после начала реакции тигель закрывают крышкой с прикрепленной к ней газоподводящей трубкой и пропускают через эту трубку сильный поток водорода. После охлаждения продукт реакции в виде сплавленной лепешки можно вытряхнуть из тигля.
Довольно твердые и очень хрупкие бледно-синие кристаллы. Соединение устойчиво к щелочам; разлагается кислотами с образованием силанов и водорода.
610___________E.II. Практикум no препаративной химии__
50.5.2. Хелаты, клатраты
178. Ацетилацетонат ванадила [VO(acac)2]
Раствор 5 г V2O5 в 12 см3 Н2О смешивают с 9 см3 конц. H2SO4 и 25 см3 этанола. Через некоторое время (1—2 ч) зеленая окраска меняется на синюю, после* чего не вступивший в реакцию V2O5 отфильтровывают и фильтрат смешивают с 13 см3 ацетилацетона. Раствор затем необходимо нейтрализовать, для чего при помешивании медленно добавляют раствор карбоната натрия (20 г безводной соли в 125 см3 Н2О). Продукт отфильтровывают, .высушивают на воздухе и перекристаллизовывают из хлороформа.
179. Аддукт ацетилацетоната Na—Ni с диоксаном
Некоторые ацетилацетонаты металлов способны образовывать аддукты общей формулы М[М1[(СН3—СО—СН—СО—СНз)з— —О—СН2—СН2—О—СН2—СН2, которые получают из ацетил-ацетонатов СО—СН—СО—СН3)3 (M' = Na, К;
M" = Mg, Мп, Fe, Со, Ni, Zn) при присоединении к ним1 диоксана (СН2СН2О)2. Для получения аддуктов Na—Ni смешивают 100 см3 Н2О с 15 см3 1 н. NaOH в стеклянном стакане, добавляют 2 см3 ацетилацетона и перемешивают до завершения реакции. Затем к смеси добавляют 5 см3 1 М. раствора Ni(NO3)2-6Н2О. Раствор ацетилацетоната NaNi(CH3—СО—СН—СО— —СН3)з ( зеленого цвета) переносят в чашку и> ставят ее в эксикатор, на дне которого находится стакан со 100 см3 диоксана. Пары диоксана постепенно диффундируют в раствор ацетилацетоната, и аддукт начинает медленно выпадать в осадок. Поскольку образуется мало зародышей кристаллов, призматические сине-зеленые кристаллы аддукта достигают размера 1 см.
180. Гидрохлорид трис(ацетилацетонато)кремнийхлорида
В двухгорлой колбе вместимостью' 250 см3, снабженной капельной воронкой и обратным холодильником, на конце которого находится хлоркальциевая трубка, растворяют 15 г SiCl4 в 40 см3 абсолютного бензола. Изредка встряхивают колбу, в нее по каплям добавляют смесь 9 см3 ацетилацетона с 20 см3 абсолютного бензола и нагревают раствор с обратным холодильником в течение 30 мин до выпадения белого осадка (после исчезновения желтого маслянистого слоя). Осадок быстро отфильтровывают и несколько раз промывают эфиром. Вещество затем сушат в вакуум-эксикаторе.
Для стабилизации комплексного катиона можно синтезировать тетрахлороферрат(Ш) [Si(acac)3] [FeCl4]. Для этого 1,3г безводного FeCl3 вносят в раствор, содержащий 3 г [Si(acac)3] С1-НС1 в 25 см3 хлороформа. Смесь слегка встряхивают до прекращения выделения хлороводорода. Раствор отделяют от непрореагировавшего FeCl3 фильтрованием и к фильтрату мед-
Литература
611
денно добавляют 50 см3 эфира. Через некоторое время выделяются желто-зеленые кристаллы, которые после отфильтровывания несколько раз промывают сухим эфиром.
181. Дицианоамминобензолникель(П)
К давно известным клатратам относятся соединения Ni(CN)2-• NH3 с различными ароматическими углеводородами. Кристал* лы Ni(CN)2-NH3-C6H6 получают перемешиванием водного раствора Ni(CN)2-NH3 с бензолом в большом высоком бюксе, закрытом пришлифованной крышкой.
Для получения Ni(GN)2-NH3 растворяют 12 г NiCl2-6H2O в 50 см3 Н2О, смешивают с раствором, содержащим- 7 г KCN в 30 см3 НгО, и после тщательного перемешивания добавляют примерно 35 см3 30%-ного раствора NH4OH (устанавливают pH 8—9). После перемешивания с бензолом начинается диффузная реакция и в течение нескольких недель на дне сосуда выделяются кристаллы клатрата Ni(CiN)2-NHs-С6Н6 в виде прямоугольных пластинок размером 5X5X1 мм3.
Литература
I. Алимарин И. П., Архангельская В. Н. Качественный полумикроанализ.— М. — Л.: Госхимиздат, 1952.
2. Антропов Л. И. Теоретическая электрохимия. — М.: Высшая школа, 1969.
3. Берка А., Вултерин Я., Зыка Я. Новые редокс-методы в аналитической химии. Пер. с чешек. — М.: Химия, 1968.
4. Боуэн Г., Гиббонс Д. Радиоактивационный анализ. Пер. с англ. — М.: Атомиздат, 1968.
5. Бродский А. И. Химия изотопов. — М.: Изд-во АН СССР, 1957.
6. Использование радиоактивности при химических исследованиях/Под ред. А. Валя, Н. Боннера. Пер. с англ. — М.: ИЛ, 1954.
7. Волькенштейн М. В. Строение и физические свойства молекул. — М.-Л.: Изд-во АН СССР, 1955.
8. Гайтлер В. Элементарная квантовая механика. Пер. с англ. — М.: ИЛ, 1948.
9. Галюс 3. Теоретические основы электрохимического анализа. Пер. с польск. — М.: Мир, 1974.
10. Гейдон А. Спектроскопия пламен. Пер. с англ. — М.: ИЛ, 1959.
И. Гейровский Я-, Кута Я- Основы полярографии. Пер. с чешек. — М.: Мир, 1965.
12. Гельферих Ф. Иониты. Основы ионного обмена. Пер. с нем.—М.: ИЛ, 1962.
13. Гильтнер В. Практика потенциометрических анализов. Пер. с нем. — М.: ОНТИ, 1936.
14. Глесстон С. Введение в электрохимию. Пер. с англ. — М.: ИЛ, 1951.
15. Глесстон С„ Лейдлер К-, Эйринг Г. Теория абсолютных скоростей реакций: Кинетика химических реакций, вязкость, диффузия и электрохимические явления. Пер. с англ. — М.: ИЛ, 1948.
16. Гутман В. Химия координационных соединений в неводных растворах. Пер. с англ. — М.: Мир, 1971.
17. Давтян О. К- Квантовая химия. — М.: Высшая школа, 1962.
612
Литература
18. Даниэльс Ф., Олберти Р. Физическая химия. Пер. с англ. — М.: Мир, 1978.
19. Данцер К., Тан Э., Мольх Д. Аналитика. Пер. с нем. — М.; Химия, 1981,
20. Дей М., Селбин Дж. Теоретическая неорганическая химия. Пер. с англ. — М.: Химия, 1969.
21. Денеш И. Титрование в неводных средах. Пер. с аигл. — М.: Мир, 1971.
22. Доерфель К. Статистика в аналитической химии. Пер. с нем. — М.: Мир, 1969.
23. Зеель Ф. Строение атома и химическая связь. Введение в современную теорию химической связи на основе наглядных представлений. Пер. с нем. — М.: ИЛ, 1958.
24. Иванчев Г. Дитизон и его применение. Пер. с нем.—М.: ИЛ, 1961.
25. Даплан Б. Я. Импульсная полярография. — М.: Химия, 1978.
26. Кейлеманс А. Хроматография газов. Пер. с англ. — М.: ИЛ, 1959.
27. Кендлин Дж., Тейлор К, Томпсон Д. Реакции координационных соединений переходных металлов. Пер. с англ. — М.: Мир, 1970.
28. Киселева Е. В., Каретников Г. С., Кудряшов И. В. Сборник примеров и задач по физической химии. — М.: Высшая школа, 1976.
29. Кольтгоф И. М„ Лайтинен Г. А. Определение концентрации водородных ионов и электротитрование. Колориметрическое и потенциометрическое определение pH. Пер. с англ. — М.: ИЛ, 1947.
30. Кольтгоф И. М., Лингейн Дж. Дж. Полярография. Пер. с англ. — М.-Л.: Госхимиздат, 1948.
31. Кольтгоф И. М., Стенгер В. А. Объемный анализ. В 3-х томах. Пер. с аигл. — М.-Л.г Госхимиздат. Т. 1, 1950; т. 2, 1952; т. 3, 1961.
32. Кольтгоф И. М., Фурман И. X. Потенциометрическое титрование. Пер. с англ. — М.-Л.: Химтеоретиздат, 1935.
33. Коттон Ф. А., Уилкинсон Дж. Основы неорганической химии. Пер. с англ. — М.: Мир, 1979.
34. Коулсон Ч. Валентность. Пер. с аигл. — М.: Мир, 1965.
35. Крешков А. П., Быкова Л. Н., Казарян Н. А. Кислотно-основное титрование в неводных растворах. — М.: Химия, 1967.
36. Крюкова Т. А., Синякова С. И., Арефьева Т. В. Полярографический анализ.— М.: Госхимиздат, 1959.
37. Лайтинен Г. А. Химический анализ. Пер. с аигл. — М.: Химия, 1966.
38. Латимер В. М. Окислительные состояния элементов и их потенциалы в водных растворах. Пер. с англ. — М.: ИЛ, 1954.
39. Лейдлер К- Кинетика органических реакций. Пер. с англ. — М.: Мир, 1966.
40. Мюллер Г., Гнаук Г. Газы высокой чистоты. Пер. с нем. — М.: Мир, 1968.
41. Некрасов Б. В. Основы общей химии. В 2-х томах. — М.: Химия, 1973.
42. Некрасов Б. В. Учебник общей химии. — М.: Химия, 1972.
43. Неводные растворители/Под ред. Т. Баддингтона. Пер. с англ. — М.: Химия, 1971.
44. Паулинг Л. Природа химической связи. Пер. С англ. — М.-Л.: Госхимиздат, 1947.
45. Полинг Л. Общая химия. Пер. с аигл. — М.: Мир, 1974.
46. Полярография. Проблемы и перспективы/Под ред. Я- П. Страдыня, С. Г. Майрановского. — Рига: «Зинатне», 1977.
47. Рао Ч. Электронные спектры в химии. Пер. с англ. — М.: Мир, 1964.
48. Реми Г. Курс неорганической химии. В 2-х томах. Пер. с нем. — М.: Мир. Т. 1, 1972; т. 2, 1974.
49. Рогинский С. 3. Теоретические основы изотопных методов изучения химических реакций. — М.: Изд-во АН СССР, 1956.
50. Руководство по аналитической химии. Пер. с нем. — М.: Мир, 1975.
51. Руководство по препаративной неорганической химии/Под ред. Г. Брауе-ра. Пер. с нем. — М.: ИЛ, 1956.
Литература
613
52. Семенов Н. Н. О некоторых проблемах химической кинетики и реакционной способности. — М.: Изд-во АН СССР, 1958.
53. Синтезы неорганических соединений. В 3-х томах/Под ред. У. Джолли. Пер. с англ. — М.; Мир. Т. 1, 1966; т. 2, 1967; т. 3, 1979.
54. Справочник химика. В 6-ти томах/Под ред. Б. П. Никольского. — Л.-М.: Госхимиздат, 1962—68.
55. Тейлор Д. Нейтронное излучение и активационный анализ. Пер. с англ.— М.: Атомиздат, 1965.
56. Тредвелл Ф. Курс аналитической химии. В 2-х томах. Пер. с ием. — М.-Л.: Госиздат, 1931.
57. Уотерс У. Механизм окисления органических соединений. Пер. с англ. — М.; Мир, 1966.
58. Уэллс А. Строение неорганических веществ. (Структурная неорганическая химия.) Пер. с англ. — М.: ИЛ, 1949.
59. Фридлендер Г., Кеннеди Дж., Миллер Дж. Ядериая химия и радиохимия. Пер. с англ. — М.; Мир, 1967.
60. Херинг Р. Хелатообразующие июнообменники. Пер. с ием. — М.: Мир, 1971.
61. Хроматография в тонких слоях/Под ред. Э. Шталя. Пер. с нем. — М.: Мир, 1965.
62. Шарло Г. Методы аналитической химии. Количественный анализ неорганических соединений. Ч. 1—2. Пер. с франц. — М.: Химия, 1969.
63. Швабе К. Основы техники измерения pH. Пер. с нем.'—М.: ИЛ, 1962.
64. Шварценбах Г., Флашка X. Комплексонометрическое титрование. Пер. с ием. — М.; Химия, 1970.
65. Шефер Г. Химические транспортные реакции. (Транспорт неорганических веществ через газовую фазу и его применение.) Пер. с нем. — М.: Мир, 1964.
66. Шпольский Э. В. Атомная физика. В 2-х томах. — М.-Л.: Гостехиздат, 1974.
67. Шульце Г. Металлофизика. Пер. с нем. — М.: Мир, 1971.
68. Эйринг Г., Уолтер Дж.,'Кимбалл Дж. Квантовая химия. Пер. с аигл. — М.: ИЛ, 1948.
69. Юинг Г. В. Инструментальные методы химического анализа. Пер. с англ. — М.: Госатомиздат, 1963.
70. Abresch К-, Claassen J. Die coulometrische Analyse. Weinheim/Bergstr., Verlag Chemie, 1961.
71. Ackermann G. Einfiihrung in die qualitative anorganische Halbmikroanaly-se. 5. Aufl. Leipzig, VEB Deutscher Verlag fiir Grundstoffindustrie, 1968.
72. Adams D. M., Raynor J. B. Advanced practical inorganic chemistry. New York, John Wiley, 1965.
73. Angeger E. V., Ebert H. Technische Kunstgriffe bei physikalischen Unter-suchungen. 13. Aufl. Braunschweig, Verlag Friedr. Vieweg, 1964.
74. Barnard J. A., Chayen R. Modern methods of chemical analysis. London, McGraw-Hill, 1965.
75. Barrow G. M. The structure of molecules. New York/Amsterdam, W. A. Benjamin, 1964.
76. Barthel J. Thermometric titrations. New York, John Wiley, 1975.
77. Baumann R. P. Absorption spectroscopy. New York, John Wiley, 1962.
78. Bayer F., Wagner G. Gasanalyse. 3. Aufl. Stuttgart, Ferdinand Enke Verlag, 1960.
79. Berl W. G. Physikalische Methoden in der chemischen Analyse. Frank-furt/M., Akademische Verlagsgesellschaft, 1966.
SO. Bitirich H.-L, Heberland D., last G. Leitfaden der chemischen Kinetik. Berlin, VEB Deutscher Verlag der Wissenschaften, 1973.
81. Blumental G„ Hartung H. Stochiometrie auf der Grundlage des modernen Molbegriffs. Leipzig, Akademische Verlagsgesellschaft Geest & Portig, 1974.
614
Литература
82. Bock R. Aufschlufimethoden der anorganischen und organischen Chemie. Weinheim/Bergstr., Verlag Chemie, 1972.
83. Bock R. Methoden der analytischen Chemie. Eine Einftihrung. Bd. 1. Tren-nungsmethoden. Weinheim/Bergstr., Verlag Chemie, 1974.
84. Bockris J., Reddy О. M-, Reddy A. K. N. Modern electrochemistry. V. 1—2. New York, Plenum Press, 1973.
85. Brdicka R. Grundlagen der physikalischen Chemie. 14. Aufl. Berlin, VEB Deutscher Verlag der Wissenschaften, 1977.
86. Chariot G. Colorimetric determination of elements. Principles and methods. Amsterdam/New York, Elsevier, 1964.
87. Chariot G., Badoz-Lambling J., Tremillon B. Electrochemical reactions. Amsterdam/New York, Elsevier, 1962.
88. Christen H. R. Grundlagen der allgemeinen und anorganischen Chemie.
4. Aufl. Frankfurt/M., Diesterweg-Salle Verlag, 1973.
89. Cook G. B., Dungan J. F. Modern radiochemical practice. London, Oxford. University Press, 1952.
90. Cramer F. Papierchromatographie. 5. Aufl. Weinheim/Bergstr., Verlag Chemie, 1962.
91. Cremer E„ Pahl M. Kinetik der Gasreaktionen. Berlin, Walter de Gruyter, 1961.
92. Cruse K., Huber R. Hochfrequenztitration. Weinheim/Bersgtr., Verlag Chemie, 1957.
93. Dean J. A. Flame photometry. London, McGraw-Hill, 1960.
94. Denbigh K. Prinzipien des chemischen Gleichgewichts. Darmstadt, Verlag' Dietrich Steinkopff, 1959.
95. Dorain P. B. Symmetric und anorganische Strukturchemie, Braunschweig,. Friedr. Vieweg; GmbH/Berlin, Akademie-Verlag, 1972.
96. Durrant Ph. J., Durrant B. Introduction to advanced inorganic chemistry. 2nd ed. London, Longmans, 1970; 1972.
97. Duval C. Inorganic thermogravimetric analysis. 2nd. London, Longmans, 1970.
98. Eckschlager K- Fehler bei chemischen Analysen. 2. Aufl. Leipzig, Akademi-sche Verlagsgesellschaft Geest & Portig, 1965.
99. Eckstein H. J. Spurenanalyse in hochschmelzenden Metallen. Leipzig, VEB-Deutscher Verlag fiir Grundstoffindustrie, 1970.
100. Emeleus H. J., Anderson J. S. Modern aspects of inorganic chemistry.
3rd ed. London, Routledge & Kegan, 1960.
101. Emeleus H. J., Sharpe A. G. Modern aspects of inorganic chemistry. 4th ed. London, Routledge & Kegan, 1973.
102. Engels S. Anorganische Festkorperreaktionen. Berlin, Akademie-Verlag, 1980.
103. Erdey L. Theorie und Praxis der gravimetrischen Analyse. 2 Bde. Budapest, Akademiai Kiad6, 1964.
104. Erdey-Cruz T. Kinetik der Elektrodenprozesse. Budapest, Akademiai Kiado, 1978.
105. Eucken A., Wicke E. Grundrifi der physikalischen chemie. 10. Aufl. Leipzig, Akademische Verlagsgesellschaft Geest & Portig, 1959.
106. Falkenhagen H. Theorie der Elektrolyte. Leipzig, S. Hirzel Verlag, 1971.
107, Feigl F. Spot tests in inorganic analysis. 6th ed. Amsterdam/New York, Elsevier, 1972.
108. Felber W. Laborpraxis. Leipzig, VEB Deutscher Verag fiir Grundstoffindustrie, 1971.
109. Forker W. Elektrochemische Kinetik. Berlin, Akademie-Verlag, 1966.
110. Fresenius C. R. Anleitung zur qualitativen chemischen Analyse. Braunschweig, Verlag Friedr. Vieweg, 1969.
111. Fresenius W., Zander G. Handbuch der analytischen Chemie. Berlin/Gottin-gen/Heidelberg, Springer, ab 1940.
\
\______Литература __________ 615
112. Friedrichs F. Das Gias im chemischen Laboratorium. 3. Aufl. Berlin/Gottin-gen/Heidelberg, Springer, 1960.
113. Fromherz H. Physikahsch-chemisches Rechnen in Wissenschaft und Technik. Nachdruck der 3. Aufl. Weinheim/Bergstr. Verlag Chemie, 1973.
114. Frost A. A., Pearson p. G. Kinetik und Mechanismen homogener chemischer Reaktionen. 2. Aufl. Weinheim/Bergstr., Verlag Chemie, 1973.
115. Geilmann W. Bilder zur qualitativen Mikroanalyse anorganischer Stoffe.
3. Aufl. Weinheim/Bersgtr., Verlag Chemie, 1960.
116. Gould E. S. Inorganic reactions and structure. 3rd ed. New York, Holt, Rinehart and Winston, 1963.
117. Gray H. B„ Haight G. P. Basis principles of chemistry. New York/Amsterdam, W. A. Benjamin, 1967.
118. Gregg S. I. Oberflachenchemie fester Stoffe. Berlin, VEB Verlag Technik, 1958, S. 186.
119. Grubitsch H. Anorganisch-praparative Chemie. Wien, Springer, 1950.
120. Gutmann V. Halogen chemistry. V. 1—3 London/New York, Academic Press, 1967.
121. Gutmann V. Chemische Funktionslehre. Wien/New York, Springer, 1971.
122. Gutmann V., Hengge E. Allgemeine und anorganische Chemie. 2- Aufl. Weinheim/Bergstr., Verlag Chemie, 1975.
123. Hais I. M., Macek K. Handbuch der papierchromatographie. 2. Aufl. Jena, VEB Gustav Fischer Verlag, 1963.
124. Hagg G. Die theoretischen Grundlagen der analytischen Chemie. Ba-sel/Stuttgart, Birkhauser-Verlag, 1962.
125. Hagg G. General and inorganic chemistry. Stockholm. Almquist & Wiksell, 1969.
126. Handbuch der Gaschromatographie. Hrsg. Leibnitz E. Struppe H. G.
2. Aufl. Leipzig, Akademische Verlagsgesellschaft Geest & Portig, 1971.
127. Havemann p. Einftihrung in die chemische Thermodynamik. Berlin, VEB Deutscher Verlag der Wissenschaften, 1957.
128. Hecker E. Verteilungsverfahren im Laboratorium. Weinheim/Bergstr., Ver-lag Chemie, 1955.
129. Heide K,. Dynamische thermische Analysenmethoden. Leipzig, VEB Deutscher Verlag fiir Grundstoffindustrie, 1975.
130. Hennig F. Temperaturmessung. Leipzig, Johann Ambrosium Barth, 1951.
131. Herbst J. Glastechnische Praxis. Leipzig, VEB Deutscher Verlag fiir Grundstoffindustrie, 1962,
132. Herrmann p., Alkemade C. Th. J. Flammenphotometrie. Berlin/Gottin-gen/Heidelberg, Springer, 1960.
133. Heslop p. B„ Jones K. Inorganic Chemistry. A guide to advanced study. Amsterdam/New York, Elsevier, 1976.
134. Heyrovsky J., Zuman P. Einftihrung in die praktische Polarographie. Berlin, VEB Verlag Technik, 1959.
135. Hofmann U„ piidorff W. Anorganische Chemie. 21. Aufl. Braunschweig, Verlag Friedr. Vieweg, 1973.
136. Holemann A. F., Wiberg E. Lehrbuch der anorganischen Chemie 81.—90. Aufl. Berlin, Walter de Gruyter, 1976.
137. Huber W. Methoden der Analyse in der Chemie. Bd. I. Titrationen in nichtwafirigen Losungsmitteln. Frankfurt/M., Akademische Verlagsgesellschaft, 1964.
138. Huheey J. E. Inorganic chemistry. Principles of structure and bonding. 2nd ed New York/Evanston/San Francisco/London, Harper & Row, 1978.
139. Inorganic Synthesis. Serie. New York, McGraw-Hill, 1939—1980.
140. Ives D. J. G., Janz G. J. Reference electrodes. New York, Academic Press, 1961.
141. lander G. Die Chemie in wasserahnlichen Losungsmitteln. Berlin/Gottin-gen/Heidelberg, Springer, 1949.
142. lander G., Blasius E. Lehrbuch der analytischen und praparativen anorganischen Chemie. 8—10. Aufl. Leipzig, S. Hirzel Verlag, 1973.
614
Литература
82. Bock R. AufschluSmethoden der anorganischen und organischen Chemie. Weinheim/Bergstr., Verlag Chemie, 1972.
83. Bock R. Methoden der analytischen Chemie. Eine Einfiihrung. Bd. 1. Tren-nungsmethoden. Weinheim/Bergstr., Verlag Chemie, 1974.
84. Bockris J,, Reddy О. M„ Reddy A. K. N. Modern electrochemistry. V. 1—2. New York, Plenum Press, 1973.
85. Brdicka R. Grundlagen der physikalischen Chemie. 14. Aufl. Berlin, VEB Deutscher Verlag der Wissenschaiten, 1977.
86. Chariot G. Colorimetric determination of elements. Principles and methods. Amsterdam/New York, Elsevier, 1964.
87. Chariot G., Badoz-Lambling J., Tremillon B. Electrochemical reactions. Amsterdam/New York, Elsevier, 1962.
88. Christen H. R, Grundlagen der allgemeinen und anorganischen Chemie.
4. Aufl. Frankfurt/M., Diesterweg-Salle Verlag, 1973.
89. Cook G. B., Dungan J. F. Modern radiochemical practice. London, Oxford University Press, 1952.
90. Cramer F. Papierchromatographie. 5. Aufl. Weinheim/Bergstr., Verlag Chemie, 1962.
91. Cremer E., Pahl M. Kinptik der Gasreaktionen. Berlin, Walter de Gruyter, 1961.
92. Cruse K., Huber R. Hochfrequenztitration. Weinheim/Bersgtr., Verlag Chemie, 1957.
93. Dean I. A. Flame photometry. London, McGraw-Hill, 1960.
94. Denbigh K. Prinzipien des chemischen Gleichgewichts. Darmstadt, Verlag' Dietrich Steinkopff, 1959.
95. Dorain P. B. Symmetric und anorganische Strukturchemie, Braunschweig,. Friedr. Vieweg; GmbH/Berlin, Akademie-Verlag, 1972.
96. Durrant Ph. J., Durrant B. Introduction to advanced inorganic chemistry. 2nd ed. London, Longmans, 1970; 1972.
97. Duval C. Inorganic thermogravimetric analysis. 2nd. London, Longmans, 1970.
98. Eckschlager !(. Fehler bei chemischen Analysen. 2. Aufl. Leipzig, Akademi-sche Verlagsgesellschaft Geest & Portig, 1965.
99. Eckstein H. J. Spurenanalyse in hochschmelzenden Metallen. Leipzig, VEB-Deutscher Verlag fiir Grundstoffindustrie, 1970.
100. Emel£us H. J., Anderson J. S. Modern aspects of inorganic chemistry.
3rd ed. London, Routledge & Kegan, 1960.
101. Emeleus H. J., Sharpe A. G. Modern aspects of inorganic chemistry. 4th ed. London, Routledge & Kegan, 1973.
102. Engels S. Anorganische Festkorperreaktionen. Berlin, Akademie-Verlag, 1980.
103. Erdey L. Theorie und Praxis der gravimetrischen Analyse. 2 Bde. Budapest, Akademiai Kiado, 1964.
104. Erdey-Cruz T. Kinetik der Elektrodenprozesse. Budapest, Akademiai Kiado, 1978.
105. Eucken A., Wicke E. Grundrifi der physikalischen chemie. 10. Aufl. Leipzig, Akademische Verlagsgesellschaft Geest & Portig, 1959.
106. Falkenhagen H. Theorie der Elektrolyte. Leipzig, S. Hirzel Verlag, 1971.
107. Feigl F. Spot tests in inorganic analysis. 6th ed. Amsterdam/New York, Elsevier, 1972.
108. Felber W. Laborpraxis. Leipzig, VEB Deutscher Verag fiir Grundstoffindustrie, 1971.
109. Forker W. Elektrochemische Kinetik. Berlin, Akademie-Verlag, 1966.
110. Fresenius C. R. Anleitung zur qualitativen chemischen Analyse. Braunschweig, Verlag Friedr. Vieweg, 1969.
111. Fresenius W„ Jander G. Handbuch der analytischen Chemie. Berlin/Gottin-gen/Heidelberg, Springer, ab 1940.
Литература
615
112. Friedrichs F. Das Gias im chemischen Laboratorium. 3. Aufl. Berlin/Gottin-gen/Heidelberg, Springer, 1960.
113. Fromherz H. Physikalisch-chemisches Rechnen in Wissenschaft und Technik. Nachdruck der 3. Aufl. Weinheim/Bergstr. Verlag Chemie, 1973.
114. Frost A. A., Pearson R. G. Kinetik und Mechanismen homogener chemischer Reaktionen. 2. Aufl. Weinheim/Bergstr., Verlag Chemie, 1973.
115. Geilmann ll7. Bilder zur qualitativen Mikroanalyse anorganischer Stoffe.
3. Aufl. Weinheim/Bersgtr., Verlag Chemie, 1960.
116. Gould E. S. Inorganic reactions and structure. 3rd ed. New York, Holt, Rinehart and Winston, 1963.
117. Gray H. B., Haight G. P. Basis principles of chemistry. New York/Amsterdam, W. A. Benjamin, 1967.
118. Gregg S. J. Oberflachenchemie fester Stoffe. Berlin, VEB Verlag Technik, 1958, S. 186.
119. Grubitsch H. Anorganisch-praparative Chemie. Wien, Springer, 1950.
120. Gutmann V. Halogen chemistry. V. 1—3 London/New York, Academic Press, 1967.
.121. Gutmann V. Chemische Funktionslehre. Wien/New York, Springer, 1971.
122. Gutmann V., Hengge E. Allgemeine und anorganische Chemie. 2. Aufl. Weinheim/Bergstr., Verlag Chemie, 1975.
123. Hais I. M., Macek K. Handbuch der papierchromatographie. 2. Aufl. Jena, VEB Gustav Fischer Verlag, 1963.
124. Hagg G. Die theoretischen Grundlagen der analytischen Chemie. Ba-sel/Stuttgart, Birkhauser-Verlag, 1962.
125. Hagg G. General and inorganic chemistry. Stockholm. Almquist & Wiksell, 1969.
126. Handbuch der Gaschromatographie. Hrsg. Leibnitz E. Struppe H. G.
2. Aufl. Leipzig, Akademische Verlagsgesellschaft Geest & Portig, 1971.
127. Havetnann R. Einfiihrung in die chemische Thermodynamik. Berlin, VEB Deutscher Verlag der Wissenschaften, 1957.
128. Hecker E. Verteilungsverfahren im Laboratorium. Weinheim/Bergstr., Verlag Chemie, 1955.
129. Fleide K. Dynamische thermische Analysenmethoden. Leipzig, VEB Deutscher Verlag fiir Grundstoffindustrie, 1975.
130. Hennig F. Temperaturmessung. Leipzig, Johann Ambrosium Barth, 1951.
131. Herbst J. Glastechnische Praxis. Leipzig, VEB Deutscher Verlag fiir Grundstoffindustrie, 1962.
132. Herrmann R., Alkemade C. Th. J. Flammenphotometrie. Berlin/Gottin-gen/Heidelberg, Springer, 1960.
133. Heslop R. B., Jones A. Inorganic Chemistry. A guide to advanced study. Amsterdam/New York, Elsevier, 1976.
134. Heyrovsky J., Zuman P. Einfiihrung in die praktische Polarographie. Berlin, VEB Verlag Technik, 1959.
135. Hofmann U., Rildorff W. Anorganische Chemie. 21. Aufl. Braunschweig, Verlag Friedr. Vieweg, 1973.
136. Holemann A. F., Wiberg E. Lehrbuch der anorganischen Chemie 81.—90. Aufl. Berlin, Walter de Gruyter, 1976.
137. Huber W. Methoden der Analyse in der Chemie. Bd. I. Titrationen in nichtwafirigen Losungsmitteln. Frankfurt/M., Akademische Verlagsgesellschaft, 1964.
138. Huheey J. E. Inorganic chemistry. Principles of structure and bonding. 2nd ed New York/Evanston/San Francisco/London, Harper & Row, 1978.
139. Inorganic Synthesis. Serie. New York, McGraw-Hill, 1939—1980.
140. Ives D. I. G., Janz G. I. Reference electrodes. New York, Academic Press, 1961.
141. Jander G. Die Chemie in wasserahnlichen Losungsmitteln. Berlin/Gottin-gen/Heidelberg, Springer, 1949.
142. Jander G„ Blasius E. Lehrbuch der analytischen und praparativen anorganischen Chemie. 8—10. Aufl. Leipzig, S. Hirzel Verlag, 1973.
616________________________________Литература_________д________________________
/
143. lander G., Pfundt О. Die konduktometrische Mafianalyse u. a. Anwendun-gen der Leitfahigkeitmessungen auf dem chemischen Gebiet unter besonde-rer Beriicksichtigung der visuellen Methoden. Stuttgart, Ferdinand Enke Verlag, 1945.
144. Jander J., Lafrenz Ch. Wasserahnliche Losungsmittel (Chemische Taschen-biicher, Bd. 3). Weinheim/Bersgtr., Verlag Chemie, 1968.
145. Jesring H. Electrosorptionsanalyse mit der Wechselstrompolarographie. Berlin, Akademie-Verlag, 1974.
146. Jerschkewitz H. G. Analytische Chemie. Leipzig, Teubner Verlag, 1961.
147. Johnson K. R., Eichler E., Okelley G. D. Technique of inorganic chemistry. V. Il in: Nuclear chemistry. New York, Interscience, 1963.
148. Jordan J. Handbook of analytical chemistry. Ed. L. Meites. New York, McGraw-Hill, 1963.
149. Kauzmann W. Quantum chemistry. New York, Academic Press, 1957.
150. Кейпе H. Bilderatlas zur qualitativen anorganischen Mikroanalyse. Leipzig, VEB Deutscher Verlag fiir Grundstoffindustrie, 1973.
151. Kleber W. Einfiihrung in die Kristallographie. 13. Aufl. Berlin, VEB Verlag Technik, 1977.
152. Kletnenc A. Die Behandlung und Reindarstellung von Gasen. 2. Aufl. Wien, Springer, 1948.
153. Klikorka J., Klazar J., Zastera A., Horak J. Einfiihrung in die praparative anorganische Chemie. Leipzig, Akademische Verlagsgesellschaft Geest & Portig, 1963.
154. Koch 0. G. Koch-Dedic G. A. Handbuch der Spurenanalyse. Berlin/Heidel-berg/New York, Springer, 1974.
155. Kolditz L. Anorganischen Chemie. In 2 Teilen. Berlin, VEB Deutscher Verlag der Wissenschaften, 1980.
156. Kolrausch F. Praktische Physik. Bd. I. 21. Aufl. Stuttgart, B. G. Teubner Verlagsgesellschaft, 1960.
157. Kolthoff I. M. Konduktometrische Titration. Dresden und Leipzig, Verlag Theodor Steinkopf, 1923.
158. Kolthoff I. M., Belcher R. Volumetric Analysis. Vol. III. Oxidation — Reduction Reactions. New York, Interscience, 1957.
159. Kortum G. Einfiihrung in die chemische Thermodynamik. 6. Aufl. Weinheim/Bergstr., Verlag Chemie, 1972.
160. KortUrn G. Kolorimetrie, Photometrie und Spektrometrie (Anleitungen fur die chemische Laboratoriumpraxis, Bd. 2). 4. Aufl. Berlin/Gottingen/Heidel-berg, Springer, 1962.
161. Kortiim G. Lehrbuch der Elektrochemie. 5. Aufl. Weinheim/Bergstr., Verlag Chemie, 1972.
162. Kraft G. Die elektrochemischen Methoden der Chemischen Analyse. Elektro-metrie. Frankfurt/M., Umsehau-Verlag, 1962.
163. Kraft G., Fischer J. Indikation von Titrationen. Berlin, Walter Gruyter, 1972.
164. Krugers J., Keuletnans A. J. M. Practical instrumental analysis. Amster-dam/London/New York, Elsevier, 1965.
165. Kucharski J., Safarik L. Titrations in non-aqueous solvents. Amsterdam/Lon-don/New York, Elsevier, 1965.
166. Lagowski J. H. Modern inorganic chemistry. New York, Marcel Dekker, 1973.
167. Landolt-Bornstein. Zahlenwerte und Funktionen aus Physik, Chemie, Astro-nomie, Geophysik und Technik (19 Teilbande). 6. Aufl. Berlin/Heidel-berg/New York, Springer, 1950—1974.
168. Landsberg R., Bartelt H. Elektrochemische Reaktionen und ProzeSe. Berlin, VEB Deutscher Verlag der Wissenschaften, 1977.
169. Lange B. Kolorimetrische Analyse. 6. Aufl. Weinheim/Bergstr., Verlag Chemie, 1970.
170. Laporte H. Messung, Erzeugung und Konstanthaltung hoher bis tiefer Tem-neraturen. Leipzig, VEB Fachbuchverlag, 1961.
Литература
617
171. Lederer Е., Lederer М. Chromatography. 2. Aufl. Amsterdam/London/New York, Elsevier, 1960.
172. Lehrwerk Chemie fiir Universitaten und Hochschulen. Herausgeberkollektiv. Leipzig, VEB Deutscher Verlag fiir Grundstoffindustrie. a) Struktur und Bindung — Atome und Molekiile. 3. Aufl., 1973; 6) Structure und Bindung— Aggregierte Systeme und Stoffsystematik, 1973; в) Struktu-raufklarung— Spektroskopie und Rontgenbeugung, 1973; r) Chemische Thermodynamik, 1973; д) Elektrolytgleichgewichte und Elektrochemie, 1974; e) Chemische Kinetik, 1973; ж) Reaktionsverhalten und Syntheseprin-zipien, 1973.
173. Lewis G. N. Valence and structure of atoms and molecules. New York, Chemical Catalog Comp., 1923.
174. Lieser К. H. Einfiihrung in die Kernchemie (Kernchemie in Einzeldarstel-lungen, Bd. 1). Weinheim/Bergstr., Verlag Chemie. 1969.
175. Lingerie J. J. Electroanalytical chemistry. New York, Interscience, 1958.
176. Lux H. Anorganisch-chemische Experimentierkunst. 3. Aufl. Leipzig, Johann Ambrosius Barth Verlag, 1970.
177. Lux H. Praktikum der quantitativen anorganischen Analyse. 7. Aufl. (Neuauflage Stock, Stabler). Miinchen, J. F. Bergmann Verlag., 1979.
178. Mahr C. Anorganisches Grundpraktikum. 5. Aufl. Weinheim/Bersgtr., Verlag Chemie, 1973.
179. Mavrodineanu R., Beiteux H. Flame spectroscopy. New York, John Wiley, 1965.
180. Meehatn E. J. Optical methods of analysis. New York, Interscience, 1964.
181. Meites L. Polarographic techniques. New York, Interscience, 1955.
182. Milner G. W. C. The principles and applications of polarography. London, Longmans, 1957.
183. Moenke H. Atomspektroskopische Spurenanalyse. Leipzig, Akademische Verlagsgesellschaft Geest & Portig, 1974.
184. Neeb R. Inverse Polarographie und Voltametrie. Berlin. Akademie-Verlag, 1969.
185. Okdc A. Qualitative analytische Chemie. Leipzig, Akademische Verlagsgesellschaft Geest & Portig, 1960.
186. Pauling L. Chemie —Eine Einfiihrung. 8. Aufl. Weinheim/Bersgtr., Verlag Chemie, 1969.
187. Phillips C. S. G., Williams R. L P. Inorganic chemistry. Vol. 1,2. Oxford, Clarendon Press, 1965/66.
188. Pimentel G. C., McClellan A. L. The hydrogen bond. San Francisco/London, Freeman and Comp., 1960.
189. Pinta M. Modern methods for trace element analysis. Ann. Arbor Mich., Ann. Arbor Science Publischers, 1978.
190. Prause H. J. Die Arbeitsschutztechnik bei der Herstellung und Verarbeitung-chemischen Erzeugnisse. Berlin, VEB Verlag Technik, 1959.
191. Pribil R, Komplexometrie. Leipzig, VEB Deutscher Verlag fiir Grundstoffindustrie: Bd. I. Prinzipen und Grundbestimmungen, 1963; Bd. II. Analyse von Legierungen, 1962; Bd. III. Analyse von anorganischen Rohstoffen und Erzeugnissen, 1962; Bd. IV. Pharmazeutische und organische Analyse, 1966.
192. Prigogine I., Defay R. Chemische Thermodynamik. Leipzig, VEB Deutscher Verl.ag fiir Grundstoffindustrie, 1962.
193. Proszt I., Cieleszky V., Gyorbiro K. Polarographie. Budapest, Akademiai Kiadd, 1967.
194. Prutton C. F., Maron S. H. Fundamental principles of physical chemistry. New York, Macmillan, 1948.
195. Pungor E. Oscillometry and conductometry. London, Pergamon Press, 1964.
196. Pungor E. Flame photometry theorie. London, Van Nostrand Reinhold, 1967.
40—1880
€18
Литература
,197. Qualitative Schnellanalyse der Kationen und Anionen nach Chariot. Hrsg. Koster E., Pflugmacher A. 5. Aufl. Berlin, Walter de Gruyter, 1969.
198. Radley J. A., Grant J. Fluorescence analysis in ultraviolet light. London, Chapman & Hall, 1954.
199. Rauscher K., Voigt J., Wilke I., Wilke К.-Th. Chemische Tabellen und Re-chentafeln fiir die analytische Praxis. 5. Aufl. Leipzig, VEB Deutscher Verlag fiir Grundstoffindustrie, 1972.
200. Rexer R„ Witzmann H., Lange W. Reinstoffe in Wissenschaft und Technik, Berlin, Akademie-Verlag, 1963.
201. Rodicker H., Geier G. Physikalisch-chemische Untersuchungsmethoden. Bd. I. Elektrochemische Mefimethoden. Leipzig, VEB. Deutscher Verlag fiir Grundstoffindustrie, 1962.
202. Schay G. Theoretische Grundlagen der Gaschromatographie. 2. Aufl. Berlin, VEB Deutscher Verlag der Wissenschaften, 1961.
203. Schilt A. A., Jaselskis B. Ultraviolet und visible spectrophotometry. In: Kolthoff I. M., Elving P. J. Treatise on analytical chemistry. V. 5, Part 1. New York, Interscience, 1964, p. 2943—3055.
204. Schleicher A. Elektroanalytische Schnellmethoden. Stuttgart, Ferdinand Enke Verlag, 1947.
205. Schmalzried H. Festkorperreaktionen. Chemie des festen Zustandes. Wein heim/Bergstr., Verlag Chemie, 4971.
206. Schmalzried H., Navrotsky A. Festkorpersthermodynamik, Chemie des festei Zustandes. Weinheim, Verlag Chemie, 1975. [
207. Schneller H., Einfiihrng in die angewandte spektrochemische Analyse. Ber lin, VEB Verlag Technik, 1953.
.208 . Scholander A., Scholander H. Handledning i kvalitativ oorganisk analys i halvmikroskala. Stockholm, 1949.
209. Schreiter W. Seltene Metalle. Bde. I—III. Leipzig, VEB Deutscher Verlag fiir Grundstoffindustrie, 1961/1963.
210. Schwabe K. Physikalische Chemie. Bd. 1,2 (Elektrochemie). Berlin, Akademie-Verlag, 1973, 1974.
211. Schwarzenbach G. Allgemeine und anorganische Chemie. Leipzig, VEB Georg Thieme Verlag, 1950.
212. Seel F. Grundlagen der analytischen Chemie. 6. Aufl. Weinheim/Bersgtr., Verlag Chemie, 1976.
213. SeidellA. Solubilities of inorganic and organic substances. 4th ed. New York, Van Nostrand Reinhold, 1955.
214. Seith W„ Ruthard K. Chemische Spektranalyse. Berlin/Gottingen/Heidel-berg, Springer-Verlag, 1958.
215. Shriver D. F. The ambident nature of cyanide. In: Structure and bonding.
Vol. I. Berlin/Heidelberg/Gottingen/New York, Springer-Verlag, 1966, p. 32.
216. Sillen L. G., Lange P. W., Gabrielson С. O. Problems in physical chemistry. New York, Prentice-Hall, 1952.
217. Souci S. W., Thies H. Praktikum der qualitativen Analyse. 6. Aufl. Munchen, Bergmann, 1960.
218. Spice J. E. Chemische Bindung und Struktur. Leipzig, Akademische Verlagsgesellschaft Geest & Porting, 1970.
219. Sutton L. E. Chemische Bindung und Molekiilstruktur. Berlin/Gottin-gen/Heidelberg, Springer-Verlag, 1961.
220. Sykes A. G. Kinetics of inorganic reactions. 2nd ed. New York, Pergamon Press, 1970.
221. Stackelberg M. Polarographische Arbeitsmethoden. Nachdruck. Berlin, Walter de Gruyter, 1960.
222. Telle W. Chemische Laboratoriumsgerate. 2. Aufl. Leipzig, VEB Deutscher Verlag fiir Grundstoffindustrie, 1969.
223. Trzebiatowski W. Lehrbuch der anorganischen Chemie. 7.. Aufl. Berlin, VEB Deutscher Verlag der Wissenschaften, 1976.
Литература
619
224. Ulich И„ Jost W., Troe J. Kurzes Lehrbuch der physikalischen Chemie. 18. Aufl. Darmstadt, Dietrich Steinkopf Verlag, 1973.
225. Ullmanns Encyclopedic der technischen Chemie. Hrsg. W. Foerst, Bd. 2/1. Anwendung physikalischer und physikalisch-chemischer Methoden im Laboratorium. Miinchen/Berlin, Verlag Urban & Schwarzenberg, 1961.
226. Vetter K.-J. Elektrochemische Kinetik. Berlin/Gottingen/ Heidelberg, Springer-Verlag, 1961.
227. Vogt E. Physikalische Eigenschaften der Metalle. Bd. I. Leipzig, Akademische Verlagsgesellschaft Geest & Portig, 1958.
228. Volmer M. Kinetik der Phasenbildung. Dresden u. Leipzig, Theodor Stein-kopff Verlag, 1939.
229. Wast R. C., Astle M. J. CRC Handbook of chemistry and physics. Cleveland (Ohio), Chemical Rubber Publ. Comp., 1978.
230. Weisz H. Microanalysis by the ring oven technique. 2nd ed. New York, Pergamon Press, 1970.
231. Welz B. Atomabsorptionsspektroskopie. Weinheim, Verlag Chemie, 1975.
232. Wendlandt W. W. Thermal methods of analysis. New York. Interscience, 1964.
233. Willard H. H„ Merrit L. L., Dean J. A. Instrumental Methods of analysis. New York, Van Nostrand Reinhold, 1951.
234. Wilson C. L., Wilson D. W„ Strouts C. R. N. Comprehensive analytical chemistry. Vol. II A. Electrical methods. Amsterdam/London/New York, Elsevier, 1964.
235. Winsch G. Optische Analysenmethoden zur Bestimmung anorganischer Stoffe. Berlin, Walter de Gruyter, 1978.
40
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Адмитанс. 328. См. также Осциллометрия
Адсорбенты 489
Адсорбция 203, 423. См. также Изотерма адсорбции
Азота синтез 582
диоксид 595
моноксид 596
Актор 160
Акцептор 160
Ализарин S 15
Алюминий
бромид 608
иодид 553
нитрид 608
Алюминотермия 575
Амальгамы как восстановители 168
Аммоний
обнаружение 78 сл.
синтез арсената 527
— диамминотетрароданохрома-та(Ш) 558'
•— комплексных фторидных солей 558
Аммонийхромовые квасцы 520
Амперометрия 296 сл.
с одним поляризуемым электродом 297
с двумя поляризуемыми электродами 299
Амфолиты 134
Анализ
активационный 389 гравиметрический 106 сл. капельный 8, 90
— на бумаге 90, 91
— на пластинке 32, 33 качественный 7 сл.
— макроанализ 7
— микроанализ 7, 90
— полумикроанализ 8 сл. количественный 7, 98 сл.
— источники ошибок 98
— практика 101
— теоретические основы 120 сл.
компонентный 431
особо чистых веществ 410 сл. ? полуколичественный 97 процессов 430
следовых количеств 406 сл.
— методы 414 сл.
спектроскопический 352 сл.
стандартных образцов 99
структурный 430
термический 393 сл.
— дифференциальный 397
титриметрический 111 сл.
хроматографический 242 сл.
— аппаратура и применение 243
— вытеснительный 242
— сталей 86
— фронтальный 242
— элюентный 243
эмиссионный спектральный 369 сл.
— по последним линиям 372
Анионы
групповые реакции 54
обнаружение иона ацетата 56
•---бората 48, 56
---- бромида 58, 60
----гексацианоферрата 65
----иодида 58, 60
---- карбоната 56
----нитрата 56, 62
----нитрита 56, 62 ,, •
— — оксалата 64
---- пероксида 66
---- роданида 56
— — силиката 56
----сульфата 60
----сульфида 60
----сульфита 60
----тиосульфата 60
----- фосфата 63
---- фторида 58
----хлорида 58, 60
---- цианида 58
Ацетилацетон, применение в экстракции 232
Ацетилацетонат Na — Ni, аддукт с диоксаном 610
Предметный указатель
621
Баллоны для хранения газов 503
Барий
дигидрофосфинит 563
дитионат 529
перманганат 526
сульфат, осаждение 205
тетрагидропероксоиодат 530
тритиокарбонат 527
феррат(VI) 560
хромат(У) 562
Бекмана термометр 484
Бера закон 356
Берлинская лазурь 65
Бора синтез 578
ацетат 535
нитрид 609
фосфат 560
фторид 561
Бориды 607
Бренцкатехиновый фиолетовый 187
Бромкрезоловый зеленый 145
Бромводород
безводный, синтез 570
гидролитическое получение 603
Бром тимоловый синий 150, 152
Бронза вольфрамовая 559
Бунзена метод дистилляции 177
Буферная емкость 147, 148, 149
Буферные растворы 136, 146, 169
Вакуумирование 506
Ванадий(Ш)
хлорид, безводный 546
хлорид, гексагидрат 562
оксид 593
Ванадий (V)
обнаружение 48
оксид 592
— золь 568
Ванадила ацетилацетонат 610
Ванадия(IV) сульфат 552
Ван-Деемтера уравнение 238, 239
Вариаминовый синий Б 187
Вероятность статистическая 440, 448,
463, 467, 471, 472
Весы аналитические 101 сл.
Взвешивание 101 сл.
Видмера колонка для фракционной перегонки 495
Висмут
иодид 549
хлорид 550
Водород сжиженный, очистка 585
Водородсодержащие соединения 602 сл.
Возгонка 496, 507, 549
Вольт-амперные кривые 257, 281, 282
Вольтамперометрия 306 сл.
инверсионная 307
Вольфрам, обнаружение 47
Вольфрамовая синь 47, 48
Вспомогательные средства для лабораторных работ 481 сл.
Выпаривание 487
Высушивание 497
Газы
высушивание 502
измерение количества 505
использование в синтезах 525 сл., 561 сл.
очистка 502
получение 501
Гаусса кривая нормального распределения 231, 237, 240, 434 метод двойного взвешивания 103 функция 438
Гауссов интеграл ошибок 440
Гексааквахрома(Ш) хлорид 529
Гексацианоферрат(Ш), окислительно-восстановительный индикатор 187
Гейгера — Мюллера счетчик 386
Генеральная совокупность 438
Гепариновая проба 37, 43
Гетерометрия 361
Гидразин 539, 540
Гидразиния сульфат 528
Гидроксиды, термическое разложение 589
Гидроксиламмония хлорид 545
Гистограммы 441
Горелки 26, 38, 483
Гравиметрическая форма 107
Гравиметрия 106 сл.
основные операции 109
Графит 479
Декантация 490
Деполяризатор 262, 284, 292, 300
Дериватограф 402
Детектор в газовой хроматографии
244
Дефлегмация 494
Диазот
оксид 596
триоксид 594
Диметилглиоксим 14
Диметнлнафтидин 187
Диметиловый желтый 145
2,4-Динитрофенол 152
Дноктиламин 233
622
Предметный указатель
Дисерадибромид 566
Дисерадихлорид 5-12
Дисперсионные методы синтеза 568
Дитизон 83, 232, 367
Дифениламин 16, 171
Дифеиилбензлдин 171
Дифосфора пентасульфид 597
Диффузия
вихревая 238
коэффициент 201, 269, 288
молекулярная 238
Дихлорфлуоресцеин 221
Дициацоамминобензолникель(П) 611
Диэлектрическая проницаемость не-
водных растворителей 344
Диэтиламмонийдиэтилдитиокарбамн-нат 366
Диэтилсульфит 543
Дьюара сосуд 484, 403
Дюбоска колориметр 363
Емкость ионитов 248
Железо (III)
бромид 550
гидрат оксида 526
---пирофорный 524
оксихлорид 560
определение гравиметрическое 107 фторид 536
Закон распространения ошибок 108, 445
Замазки 481, 482
Зародышеобразование 199 сл.
Значение R-, 86, 87, 89, 235, 237, 240
Зона осаждения металлов 261
Измельчение 485
Измерений теория 430 сл.
диапазон значений 438
методы 454
— внутреннего стандарта 460
— градуировки 455
— стандартных добавок 459
оценка результатов 461
Изотерма адсорбции 240
Ильковича уравнение 422
Импеданс 328, 329. См. также Осцил-
лометрия
Индигокармин 175
Индикаторы
адсорбционные 220
кислотно-основные 141 сл.
— интервал перехода окраски 1421 144
— оптическая плотность 142 !
— показатель 142
окислительно-восстановительные
169, 170
— в комплексонометрии 187
— необратимые 171
радиоактивные 391
Индуктор 160
Индуцирование реакций окисления—-восстановления 160
Интервал
доверительный 406, 467. 469
измерения 452
Информация аналитическая 430, 432, 433
Иод
монобромид 587
монохлорид 569
трихлорид 587
Иодаты, термическое разложение 589
Иодоводород 606
Иониты 247, 248
электронообменные 252
Ионная сила 191
Ионные сита 251
Ионный обмен 246
применение в качественном анализе 33, 73
— в количественном анализе 249
— для деионизации воды 250, 251
— для концентрирования 250
— препаративное 250
Кадмий иодид 534 — люминофор, активированный свинцом 558
карбонат 519
сульфид кристаллический 597
Калий, обнаружение 78
амидосульфат 545
бромат 533
гексахлормолибдат(Ш) 548 гексацианохромат(Ш) 523 дисульфатопероксотитанат(IV) 539 имидо (бис) сульфат 521 иодат 522 перйодат 528
перманганат, щелочной раствор 112
— приготовление стандартного раствора 172
пероксодисульфат 547 пероксодифосфат 548 пероксохромат 520 тетрафтороантимонат(Ш) 536
Предметный указатель
623
Са-
тетрафтороборат 536
тионитрозилпентацнаноферрат(П)
527
тринодид 520
триоксалатоферрат(Ш) 523 хлорохромат 522
Кальция вольфрамат (люминофор) 533
Карбиды 607
Карбонаты, термическое разложение 589
Катализ в реакциях окисления—восстановления 159, 160
Катализатор гидрирования по батье 572
Катионы
группа аммиака 68, 74
— карбоната аммония 68, 76
— сероводорода 68, 71, 72
— соляной кислоты 68, 69
— сульфида аммония 68, 75
— щелочных металлов, аммония, магния 68, 73, 77, 78, 79
групповые реакции 66 разделение
— без применения газообразного сероводорода 80
— бессероводородные методы 81
— хроматографическое, подгруппы меди 89
Керамика 478
Киппа аппарат для получения газов 501, 502
Кислота
амидосерная 531
дифениламиносульфоновая 171, 172, 174
додекавольфраматокремневая 544 йодноватая 534
кремневая, золь 569
ортопериодиая 537 рубеановодородная 14, 61, 367 селеновая 542
соляная, приготовление и стандартизация растворов 155, 156
фосфиновая 538
хлоросерная 553
этилендиамиитетрауксусная, приготовление растворов 184, 189
Кислотность неводных растворителей 339, 343
Клатраты 610
Кобальта (II) хлорид 537
Кобальта (III) сульфат 546
Коллоиды
образование 202
методы синтеза 567, 568
Колориметрия 361 сл. См. также Ко-
лориметры, Спектрофотометрия
Колориметры 362, 363
Комплексоны 178, 184
Кондуктометрия 318 сл.
Кремний (IV)
бромид 564 третраацетат 552
Кристаллизация фракционная 488
Концентрирование 420
Крейга метод фракционной экстракции 227 сл.
Критерий
К 463 сл.
t 466 сл.
Кулонометрия 266 сл. См. также Кулонометры
вспомогательные реагенты 275, 276 гальваностатическая 272 определение количества электричества 269, 270, 271
потенциостатическая 268 применение 271, 276, 277 с внешней генерацией тиранта 275 способы выполнения анализа 268
Кулонометры, типы 270, 271
Константа
кислотности 121
устойчивости комплекса 179
— — эффективная 180
Купферон 232
Ламберта — Бера закон 142, 357, 358, 362, 368
Лантана(Ш) хлорид 561
Литий, обнаружение 78 гидрид 602 получение электролизом расплава 579
Литийалюминийгидрид 554 Логарифмическая рН-диаграмма бензойной кислоты 124, 128 математическое описание 132 многоосновных кислот, оснований 133 сл.
муравьиной кислоты 138
одноосновных кислот, оснований 123 сл.
селенистой кислоты 133 смеси слабых кислот 130, 140 — соляной и уксусной кислот 139 — хлорида аммония и раствора аммиака 139
фосфорной кислоты 141
Лоренца функция 450
Магний, обнаружение 77 силицид 609
’ 624
Предметный указатель
Максимумы полярографические 291, 292
Манометры 505, 506
Марганец 578
Марганец(1У), оксид 590
Медиана 470
Медь(1), оксид 593
Медь(11)
золь, с защитным коллоидом 568
нитрат 557
тетраиодомеркурат 529
хлорид 556
Металлоиндикаторы 184
Металлы
восстановители 574, 576
материалы для лабораторного оборудования 479
оксиды 587
щелочные и щелочноземельные, обнаружение 85
Метиловый оранжевый 144, 150, 151,
171
Метод
изотопного разбавления 390
кольцевой печи 93
конечной точки 299
регистрации излучений 384
Микрокамера газовая 29
Микрокристаллоскопия 32
Молекулярные сита 251, 252
Молибден (VI)
обнаружение 47
оксид 592
Мурексид 186 '
Мышьяк(Ш), обнаружение 47
бромид 564
сульфид, золь 568
трифторид 539
хлорид 541
Мышьяк (V), восстановление 68
Натрий
обнаружение 77, 78
синтез амальгамы 586
— амида 570
— гексайитрокобальтата(Ш) 525
— дигидроарсената 521
— дигидрогипофосфата 532
— дигидропероксоиодата 528
— монотиофосфата 521
— нитрозилпентацианоферрата(Ш) (нитропруссид) 523
— селенита 537
— тетраметафосфата 532
— тиоантимоната 559
— тритионата 538
— хлорита 562
а-Нафтиламин 15, 21
Неметаллов оксиды 593
Нернста закон распределения 223,
224, 225, 240
уравнение 161 сл., 308, 314, 315
Нейтральный красный 150
Нефелометрия 361
Нивелирование 341 сл. См. также
Растворители неводные
Никель
из оксида никеля 572
карбонил 571
Нитраты, термическое разложение
589
Нитриды 607
Нитрозил
гексахлороантимонат 551
гидросульфат 530
хлорид 565
Нитропентамминокобальта хлорид
522
Нитроферроин 171 а-Нитрозо-р-нафтол 14
Нойеса — Нернста уравнение 201
Оборудование лабораторное 481 сл.
Озоление ПО
Окислители, синтезы с применением
584
Окклюзия 204
Оксалаты, термическое разложение
Оксигалогениды, термическое разложение 589
8-Оксихинолин 14, 367
Олово(П), обнаружение 47
иодид 534, 552
окисление 68
оксид 588
получение из оловянного камня 574
хлорид 531
Органические реагенты 11 классификация 13 сл.
Осадки
загрязнения 203
получение 197
промывание 109
старение 207
Осаждение
гомогенное 200
последующее 206
раздельное 213 сл.
Осушители 499, 500
Осциллометрия 327 сл.
характеристические кривые 331
Открываемый минимум 10
Охлаждающие средства 484
Предметный указатель
5
Ошибки
абсолютные 437
в гравиметрических определениях 108
индикаторные систематические 149
— случайные 153
относительные 437
систематические 434
— аддитивные 436
— мультипликативные 436
случайные 434
титрования 115, 215
Паскаля треугольник 229 Пептизация 203 Перегонка 493 фракционная 494
Перенапряжение 256, 257, 267. См. также Электрогравиметрия
Период полураспада 382
Перлы 41, 42
Пероксиды, термическое разложение 589
Перфорация 497
Пистолет для сушки 499
Пламена 374
Погашения молярный коэффициент 356
Поглощение света веществом 356.
См. также Спектрофотометрия Полумикроанализ оборудование 23 сл. реактивы 18 сл.
техника 17 сл.
Полуширина сигнала 447 сл. Поляризация 257, 279 Полярограммы, количественная оценка 289
Полярография дифференциальная 292 импульсная 305 квадратно-волновая 304 переменнотоковая 301 постояннотоковая 278 токи адсорбционные 291 — емкостный 294 — каталитические 291 — кинетические 290
Посуда лабораторная мерная 112 — градуировка 112 — очистка 482 пластмассовая 479 платиновая для синтезов 536 стеклянная 475 фарфоровая 478
Потенциал полуволны 284. См. также
Полярограммы, Полярография Потенциометрия 307 сл.
Правила техники безопасности 509
Предел обнаружения 452
Предельная концентрация 10
Предельное отношение 11
Приборы нагревательные 483
Проба
на восстановление в пламени прн помощи паяльной трубки 36
на окрашивание пламени 38 нагреванием в калильной трубочке
43
с серной кислотой 44
отбор 432
переведение в растворимое состояние сплавлением 51
растворение 49
сплавление с металлическим натрием 45
Произведение растворимости 190
Протолиз 341
Равновесие
кислотИо-основное, расчеты 120 сл. комплексообразования 178
Радиохимические методы анализа
382 сл.
Распределения константа 223 коэффициент 224 сл., 235
Растворение 486
Растворимость 190. См. также Произведение растворимости
влияние неводных растворителей
197
— температуры 197
— электролитов 190
Растворители неводные 338
Растворы твердые, образование 205
Реакции
обнаружения групповые 8
— селективные 9
— специфические 9
синтеза в растворах 513
— в расплавах 517
— с участием твердой фазы 517
чувствительность абсолютная 10
Редокситы 252
Резонансные линии 373, 378
Результаты измерений, корреляция
445
Родохромхлорид 525
Ртути (II) сульфид 599
Ртуть, очистка 525
Свинец(1У)
оксид 588 тетраацетат 531
626
Предметный указатель
Селена хлорид 549
Селенила(1У) хлорид 565
Сера
диоксид 596, 597
золь 567
Серебро
золь 567
нитрат, стандартный раствор 221, 222
отходы, обработка 574
пероксид 520
Сероводород, получение по Зеелю 28
Сигнал аналитический 433, 445
матрицы 451
регистрация 448
фона 436, 453
Силициды 607
Содовая вытяжка 52
Сокслета аппарат для экстракции 497
Соосаждение 203
на коллекторах 422
Сорбция 489
хелатов 424
Спектр
излучения в пламени 38
колебательно-вращательный 353
комбинационного рассеяния 354
отражательный 355
флуоресценции 354
электронный, полосатый 354
эмиссионный, регистрация 372
Спектроскоп ручной 40
Спектрофлуориметрия 368
Спектрофотометрия
абсорбционная 357, 415
атомно-абсорбционная 378 сл.
— схема установки 380
атомно-флуоресцентная 381
Спектрофотометры 359 сл.
Среднее арифметическое результатов измерений 438
Стандарт первичный 117
Стандартизация растворов титрантов
117, 156, 172, 189, 190, 222
Стекло лабораторное 473—478
Стеклографит 479
Степень надежности результатов 440
Стехиометрический коэффициент 117
Сурьма
восстановление 68
обнаружение 47
Сульфаты, термическое разложение
Сульфурилхлорид 535, 564
Таллий 579
Тетрахлороарсония гексафтороарсенат (V) 554
Тетрахлорофосфония гексафторофосфат 553
Тетраэтиламмония тетрабромоантн-монат 556
Тетраэтилсиликат 541
Температуры измерение 484
регулирование 485
Термовесы 394, 396
Термогравиметрия 393 сл. •
Термопары 484
Термоэлементы 399, 485
Тионилхлорид 541
Титанила
сульфат 538
хлорид 552
Титрование
амперометрическое 297
— кривые 297
в неводных средах 348—350
в расплавах 349
высокочастотное 327
комплексонометрическое 180 сл.
— кривые 183
— способы 188, 189
кондуктометрическое 323 сл.
— кривые 323
кривые титрования сильных и слабых кислот 137 сл.
кулонометрическое 273
окислительно-восстановительное 161
— кривые 165, 166
— раствором бромата 175
---- дихромата калия 174
— — иода 176
----соединений хрома (II) 178
---- тиосульфата 177
— солями титана(Ш) 178
----церия (IV) 174
— Рейнгардта—Циммермана 173
— Фольгарда — Вольфа 174
— хлором 175
осадительное 208
— кривые титрования асимметричные 213
-------симметричные 208
— определение галогенидов по Мо-
РУ 217
----серебра по Фольгарду 218
----цианидов по Либиху 218, 220
потенциометрическое 310, 313
радиометрическое 322, 391
термометрическое 401
фотометрическое 360
Триметиларсенат(У) 554
Триметилборат 540
Трихлорсилан 567
Триэтилалюминат 543
Турбидиметрия 361
Предметный указатель
62/
Углерод диоксид 595 как восстановитель 574 монооксид 594
Уран, обнаружение 48
Фарадея — Тиндаля эффект 357
Фенолфталеин 141, 144, 150
Ферроин 170, 171
Фика закон 257
Фильтрование 490
без доступа воздуха, установка 493
под давлением по Барберу 493
Фильтры 491, 492
Флуориметрии 361 сл.
Фосфор красный 578
Фосфортрихлорид 541
Фосфорнитридхлорид 551
Фотометрия пламени 370 сл.
Фотоэлектроколориметры 361—364
Хелатообразующие реагенты 13, 183
Хелаты, синтез 610
Хлор, синтез 584
Хлороводород 603
Хлоропентамминокобальта хлорид
545
Хром, получение 575
Хром (III)
гидроксид 535
хлорид 563
сульфид 602
Хроматографические зоны 240
колонки 235, 238
Хроматография 234
адсорбционная 489, 421, 234
бумажная 85, 245
газовая 244, 245, 421, 418
колоночная 243
распределительная 232, 421
тонкослойная 87, 246
Хромила хлорид 540
Хроноамперометрия 306
Центрифуги 491
Центрифугирование 30, 490
Цинк
оксид 590
тиоантимонат 546
хлорид 549, 550
Шлифы стандартные 475 сл.
Эквивалентная схема электрохимической ячейки 278, 280, 294, 302, 330
Экстракционное концентрирование 427
Экстракция 223, 496, 497
жидкостная 223 многоэлементиая 429 объемный фактор 224, 225 однократная 225
применение в неорганической химии 232
— в определении следовых количеств веществ 426
противоточная 232 фракционная 227
Электрогравиметрия 256 сл.
Электрография 91
Электроды
водородный 314
ионселективные 317
каломельный насыщенный 310
металлические 313
платиновый 310, 314, 315
ртутный капельный 280
сравнения 309
стеклянный 316
сурьмяный 316 хингидронный 315
Электрокапиллярные кривые 294 Электролиз 422, 519, 579 сл.
Электропроводность эквивалентная 319, 331
Элементы, методы получения 572 восстановлением 572 окислением 584 термические 575 электролитические 579
Энтальпиметрия 405
Эозин 16
Эриохром черный Т 185
Ячейки измерительные 278 кондуктометрическая 322 осциллометрические 330
v<vz/цл znAXllllli
Д. ХИМИЧЕСКИЙ АНАЛИЗ................................................. 7
37. Качественный анализ............................................. 7
37.1. Общая часть.................................................... 7
37.1.1. Историческое развитие и задачи качественного анализа . 7
37.1.2. Общие реакции в качественном анализе.................... 8
37.1.3. Чувствительность аналитических реакций.................. 9
37.1.4. Органические реагенты в анализе......................... 11
37.1.5. Техника полумикроанализа.................................17
37.2. Практическая часть..............................................34
37.2.1. Ход качественного химического анализа....................34
37.2.2. Другие способы разделения катионов.......................80
37.2.3. Экспресс-анализ..........................................84
37.3. Применение физико-химических методов в качественном анализе 85
37.3.1. Хроматографические методы................................85
37.3.2. Микроанализ на фильтровальной бумаге.....................90
38. Основы количественного химического анализа.......................98
38.1. Общие положения.................................................98
38.2. Основы практики количественного химического анализа ... 101
38.2.1. Взвешивание.............................................101
38.2.2. Гравиметрия.............................................106
38.2.3. Титриметрнческий (объемный) анализ......................111
38.3. Теоретические основы количественного химического анализа . 120
38.3.1. Кислотно-основные равновесия............................120
38.3.2. Реакции окисления — восстановления......................158
38.3.3. Реакции комплексообразования...........................178
38.3.4. Реакции осаждения.......................................190
38.3.5. Жидкостная экстракция..................................223
38.3.6. Хроматографические методы...............................234
38.3.7. Методы обмена..........................................246
39. Применение электрохимических методов в количественном анализе 254
39.1. Электрогравиметрия.............................................256
39.2. Кулонометрия...................................................266
39.2.1. Основные положения...................................266
39.2.2. Потенциостатическая кулонометрия.......................268
39.2.3. Гальваностатическая кулонометрия.......................272
39.2.4. Техника работы........................................ 276
39.3. Постояниотоковая полярография..................................278
39.3.1. Общая схема измерительной ячейки для электрохимических методов анализа ............................................... 278
39.3.2. Электрохимические процессы в полярографической ячейке постоянного тока...............................................279
628
Содержание 62У-
39.3.3. Ртутный капельный электрод............................280'
39.3.4. Техника работы в постояннотоковой полярографии и построение вольт-амперных кривых..............................281
39.3.5. Напряжение деполяризации и потенциалы полуволи . . 284
39.3.6. Предельный диффузионный ток и количественная оценка полярограмм....................................................287
39.3.7. Другие полярографические токи........................ 290'
39.3.8. Области применения. Емкостный ток. Дифференциальная полярография.............................................. 292'
39.4. Амперометрия..............................................296
39.4.1. Основные положения.................................296
39.4.2. Амперометрия с одним поляризуемым электродом . . . 297
39.4.3. Амперометрия с двумя поляризуемыми электродами. Метод конечной точки ........................................... 299
39.5. Новые полярографические методы............................301
39.5.1. Переменнотоковая полярография......................301
39.5.2. Квадратно-волновая полярография....................304
39.5.3. Импульсная полярография............................305
39.5.4. Вольтамперометрия................................. 306<
39.6. Потенциометрическая индикация точки эквивалентности . . . 307
39.6.1. Основные положения.................................307
39.6.2. Измерение потенциала...............................308
39.6.3. Электрод сравнения.................................309
39.6.4. Окислительно-восстановительное титрование..........310
39.6.5. Осадительное и комплексонометрическое титрование . . 313
3'9.6.6. Кислотно-основное титрование......................313
39.7. Кондуктометрия............................................318
39.7.1. Общие положения....................................318
39.7.2. Теоретические основы...............................318
39.7.3. Установки для кондуктометрических измерений . . . 320
39.7.4. Кривые кондуктометрического титрования............... 322:
39'.7.5. Области применения кондуктометрии.....................324
39.8. Осциллометрия.................................................327
39.8.1. Теоретические основы ..................................328
39.8.2. Характеристические кривые..............................331
39.8.3. Влияние различных параметров на ход характеристических кривых.........................................................333
39.8.4. Области применения осциллометрии..................... 336'
39.9. Титрование в неводных средах..................................337
39.9.1. Растворители...........................................338
39.9.2. Кислотность в неводных растворителях................. 339'
39.9.3. Нивелирование и протолиз...............................341
39.9.4. Влияние диэлектрической проницаемости..................344
39.9.5. Области применения титрования в неводных средах . . 346
39.9.6. Особенности титрования в неводных растворителях . . 350.
40. Спектроскопические методы анализа..............................352
40.1. Общие основы и краткий обзор методов........................ 352'
40.2. Поглощение света и закон Бугера — Ламберта — Бера. . . , 355
40.3. Абсорбционная спектрофотометрия...............................357
40.3.1. Спектрофотометры ......................................359
40.3.2. Фотометрическая индикация точки эквивалентности при титровании.....................................................366
40.4. Колориметрия...............................................361
40.4.1. Техника проведения измерений...........................362
40.4.2. Практическое применение колориметрии...................365
40.5. Флуориметрия ............................................. 367~
40.5.1. Применение флуориметрии....................... . . 369»
-630
Содержание
40.6. Эмиссионный спектральный анализ..............................369
40.6Л. Принцип метода, чувствительность и точность . . . 369
40.6.2. Возбуждение излучения.................................370
40.6.3. Разложение излучения в спектр и регистрация спектра . 371
40.6.4. Значение последних линий в спектре....................372
40.7. Фотометрия пламени...........................................373
40.8. Атомно-абсорбционная спектрофотометрия.......................378
41. Радиохимические методы анализа................................382
41.1. Техника работы с радиоактивными веществами...................383
41.2. Методы обнаружения и регистрации излучений...................384
41.3. Оценка измерений.............................................387
41.4. Аналитическое применение.....................................388
42. Методы термического анализа...................................393
42.1. Термогравиметрия ............................................393
42.2. Термический анализ и дифференциальный термический анализ . 397
42.3. Термометрическое титрование..................................401
43. Методы и проблемы анализа следовых количеств..................406
43.1. Задачи анализа следовых количеств............................406
43.2. Определение следовых количеств элементов в биохимии и геохимии 407
43.3. Анализ следовых количеств элементов и охрана окружающей среды..............................................................408
43.4. Аналитическая химия особо чистых веществ.....................410
43.5. Методы аналитической химии следовых количеств .... 414
43.6. Методы разделения и концентрирования в аналитической химии следовых количеств.................................................420
44. Теория измерений в аналитической химии........................430
44.1. Основы теории измерений..................................430
44.2. Виды, источники и характеристики ошибок определения . . 434
44.3. Случайный разброс результатов измерений..................437
44.4. Закон распространения ошибок и корреляция результатов измерения .............................................................445
44.5. Условия регистрации сигналов.............................448
44.6. Интервал измерения и предел обнаружения..................452
44.7. Методы измерений в количественном анализе................454
44.8. Оценка методов анализа и результатов измерений .... 461
E.I. ОБОРУДОВАНИЕ ЛАБОРАТОРИЙ И ЛАБОРАТОРНАЯ ТЕХНИКА............................................................473
45. Материалы и оборудование......................................473
45.1. Стекло.......................................................473
45.2. Стеклянное оборудование......................................475
45.3. Керамика и кварц.............................................478
45.4. Металлы и графит............................................479
45.5. Пластмассы.............................................. . 479
45.6. Лабораторное оборудование....................................481
45.7. Вспомогательные средства для лабораторных работ .... 481
45.8. Очистка оборудования.........................................482
46. Нагревание и охлаждение. Измерение и регулирование температуры 483
-46.1. Источники нагревания и охлаждения...........................483
46Л.Т. Нагревательные приборы 483
Содержание 631
46.1.2 . Охлаждение и охлаждающие средства..................484
46.2 . Измерение температуры....................................484
46.3 . Регулирование температуры.............................. 485-
47. Методы работы в лаборатории..................................485
47.1. Измельчение..................................................485
47.2. Растворение и перемешивание..................................486
47.3. Разделение веществ...........................................487
47.3.1. Выпаривание...........................................487
47.3.2. Кристаллизация........................................488
47.3.3. Сорбция и ионный обмен................................489
47.3.4. Декантация, центрифугирование, фильтрование .... 49q
47.3.5. Перегонка............................’................493
47.3.6. Возгонка..............................................493
47.3.7. Экстракция и распределение............................496
47.3.8. Высушивание...........................................497
47.4. Получение газов и работа с ними..............................501
47.4.1. Получение газов . . . . '.................. . . 501
47.4.2. Очистка и высушивание газов...........................502
47.4.3. Хранение газов в баллонах.............................503
47.4.4. Измерение количества газа.............................505
47.5. Работы, проводимые под давлением, отличающемся от атмосферного- ............................................................ 506
47.5.1. Вакуумирование........................................506
47.5.2. Применение повышенного давления.......................508
47.6. Ведение рабочего журнала.....................................509
48. Правила техники безопасности...................................... 509-
E.II. ПРАКТИКУМ ПО ПРЕПАРАТИВНОЙ ХИМИИ..........513
49. Техника работы ................................................513
49.1. Общие указания к проведению химико-препаративных работ . 513
49.1.1. Реакции в растворах...................................513
49.1.2. Реакции в расплавах...................................517
49.1.3. Реакции с участием твердой фазы.......................517
49.1.4. Возгонка..............................................518
49.il.5 . Работа без доступа воздуха..........................519
49.1.6. Электролиз............................................519
49.2. Препаративные синтезы в растворах...................... . . 519
49.2.1. Реакции простого обмена...........................519
49.2.2. Препаративные синтезы при пониженном давлении . . 537
49.2.3. Синтезы с перегонкой..............................539
49.2.4. Синтезы с применением экстракции..................544
49.2.5. Синтезы путем электролиза.........................546
49.3. Синтезы, основанные на возгонке...........................546
49.4. Синтезы в атмосфере сухого воздуха........................552
49.5. Препаративные синтезы в неводных растворителях .... 556
49.6. Реакции в твердой фазе и в расплавах......................558
49.7. Препаративные синтезы с использованием газов.............561
49.8. Получение коллоидных систем.............................. 567"
49.8.4. Конденсационные методы................................567
49.8.2. Дисперсионные методы................................ 568
49.9. Препаративные синтезы при низких температурах .... 569’
40.6.
<632
Содержание
40.7 .
40.8 . .
41. Pi
41.1. '
41.2. 1
41.3. (
41.4. 1
42. Mt
42.1. I
42.2. I
42.3. T
43. Me
43.1. 3.
43.2. О
43.3. A cp
43.4. Ai
43.5. M
43.6. M сл
44. Teo]
44.1. Oc
44.2. Bn
44.3. Сл
44.4. За:
pen
44.5. Ус.
44.6. Ин
44.7. Me
44.8. Ощ
E.I. ОБС ТЕХНИК
45. Мате:
45.1. Orel
45.2. Сты
45.3. Кер
45.4. Мет
45.5. Пла
45.6. Лаб
45.7. Вспс
45.8. Очи<
-46. Натре
-46.1. Исто
46.4/
50. Методы синтеза препаратов................................
50.1. Методы получения элементов.............................
50.1.1. Реакции восстановления...........................
50.1.2. Электролитическое получение элементов ....
50.1.3. Синтезы, основанные на реакциях разложения
50.1.4. Синтезы с применением окислителей................
50.1.5. Специальные методы синтеза.......................
50.2. Методы получения оксидов...............................
50.2.1. Оксиды металлов..................................
50.2.2. Оксиды неметаллов................................
50.3. Методы получения сульфидов.............................
50.3.1. Синтез сульфидов из элементов....................
50.3.2. Получение сульфидов действием сероводорода .
50.4. Методы получения водородсодержащих соединений
50.4.4. Синтезы из элементов.............................
50.4.2. Реакции взаимодействия с водородсодержащими соедине ниями ..................................................
50.5. Отдельные группы веществ...............................
50.5.1. Бориды, карбиды, нитриды, силициды...............
50.5.2. Хелаты, клатраты . ,.............................
Литература...................................................
Предметный указатель.........................................
Герд Блументаль, Зигфрид Энгельс, Ингомар Фиц и др.
АНОРГАНИКУМ.
Т. 2
Научный редактор Н. А. Васина
Мл. научный редактор Н. П. Власова
Художник Б. П. Кузнецов
Художественный редактор М. Н. Кузьмина
Технический редактор Г, Б. Алюлина
Корректор Е. Г. Литвак
ИБ № 3695
Сдано в набор 28.12.83. Подписано к печати 25.05.84.
Формат 60X90716. Бумага типографская № 2.
Гарнитура литературная. Печать высокая. Объем 19,75 бум. л.
Уел. печ. л.39.50. Уел. кр.-отт. 39,75. Уч.-изд. л. 41,11.
Изд. № 3/2560. Тираж 8400 экз. Зак. 1880. Цена 2 р. 40 к.
ИЗДАТЕЛЬСТВО «МИР»
Москва, 1-й Рижский пер., 2.
Московская типография № 11 Союзполиграфпрома
•при Государственном комитете СССР по делам издательств, полиграфии и книжной торговли.
Москва, .113105, Нагатинская ул., д. 1.